Дедифференцированные программируемые стволовые клетки моноцитарного происхождения и их получение и применение - RU2333243C2
Код документа: RU2333243C2
Чертежи















Описание
Данное изобретение относится к дедифференцированным программируемым взрослым стволовым клеткам, полученным из моноцитов человека, а также их получению и применению для получения клеток и тканей тела. Согласно особенно предпочтительному варианту осуществления данного изобретения эти клетки являются аутологичными стволовыми клетками человека, т.е. эта клетка моноцитарного происхождения происходит из пациента, который подлежит лечению стволовой клеткой, полученной из этой исходной клетки, и/или клетками тела, продуцируемыми из этой стволовой клетки.
В существующем состоянии данной области термин «стволовые клетки» обозначает клетки, которые (а) имеют способность самообновления и (b) способность образования, по меньшей мере, одного и, часто, ряда типов специализированных клеток вследствие их способности асимметричного деления (см. Donovan P.J., Gearhart J., Nature 414: 92-97 (2001)). Термин «плюрипотентные» означает стволовые клетки, которые могут быть по существу дифференцированы во все возможные типы клеток тела человека и животного. Такие стволовые клетки могли быть до сих пор получены только из эмбриональной ткани или эмбриональной карциномы (опухоли яичка) (см. Donovan P.J., Gearhart J., loco citato). Применение эмбриональных стволовых клеток было предметом широкого публичного обсуждения, в частности, в Германии и считается крайне проблематичным. Наряду с этическими и юридическими проблемами, связанными с эмбриональными стволовыми клетками, терапевтическое применение таких клеток также сталкивается с трудностями. По своей природе эмбриональные стволовые клетки получают из организмов-доноров, которые являются гетерологичными относительно потенциальных реципиентов дифференцированных клеток или дифференцированной ткани (далее называемых соматическими клетками-мишенями или соматической тканью-мишенью), развивающихся из этих клеток. Таким образом, ожидается, что такие клетки-мишени будут запускать немедленную иммунологическую реакцию у потенциальных реципиентов в форме отторжения.
Стволовые клетки могут быть также выделены из различных тканей взрослых, т.е. из дифференцированных индивидуумов. Такие стволовые клетки называют в существующем состоянии данной области «мультипотентными взрослыми стволовыми клетками». В организме они играют роль в регенерации и гомеостазе тканей. Существенное различие между эмбриональными плюрипотентными стволовыми клеткам и взрослыми мультипотентными стволовыми клетками заключается в количестве дифференцированных тканей, которые могут быть получены из соответствующих клеток. Предположительно это обусловлено тем фактом, что плюрипотентные стволовые клетки происходят из сперматозоидов или из клеток, которые продуцируют сперматозоиды, тогда как взрослые мультипотентные стволовые клетки происходят из тела или сомы взрослых индивидуумов (Donovan P.J., Gearhart J., loco citato, Page 94), которые не способны к образованию сперматозоидов.
Однако реальные проблемы, связанные с получением и использованием взрослых стволовых клеток, заключаются в редкости этих клеток. Так, в костном мозге, стволовые клетки присутствуют только в отношении 1:10000, в периферической крови 1:250000 и в печени в отношении 1:100000. Таким образом, получение таких стволовых клеток является очень дорогим и напряженным для пациента. Кроме того, генерирование больших количеств клеток, требующихся для клинической терапии, вряд ли до сих пор возможно с разумными расходами.
Это идет в разрез с постоянно увеличивающейся потребностью в возможностях лечения разрушенной ткани в форме «конструирования ткани» или в виде клеточной терапии, в рамках которой должны восполняться клетки кожи, мышц, сердечной мышцы, печени, островковые клетки, нервные и нейронные клетки, клетки кости и хряща, эндотелиальные и жировые клетки.
В этой связи, поддающееся предвидению развитие профиля возраста и заболеваний населения в западном мире является критическим, ведущим к ожиданию решительного перелома в следующие 10 лет в секторе здоровья и охраны здоровья западно-европейского населения, в том числе населения США и Канады. Только в Федеральной Республике Германии демографическое развитие предполагает 21%-ный рост населения в возрастной группе 45-64 лет и 26%-ный рост в возрастной группе более 65 лет. Это должно неизбежно привести к изменению в структуре пациентов и спектре заболеваний, требующих лечения. Предсказуемо, что заболевания сердечно-сосудистой системы (высокое давление, инфаркт миокарда), сосудистые заболевания, обусловленные артериосклерозом и метаболическими заболеваниями, метаболические заболевания, такие как сахарный диабет, заболевания, связанные с метаболизмом печени, заболевания почек, а также заболевания скелетной системы, вызываемые связанной с возрастом дегенерацией, и дегенеративные заболевания головного мозга, вызываемые потерями нервных и глиальных клеток, будут встречаться чаще и требовать новаторских концепций лечения.
Эти факты объясняют масштабное национальное и международное исследования и рост усилий со стороны вовлеченных в эти исследования специалистов для получения стволовых клеток, которые могут быть программируемыми для развития в дифференцированные клетки, типичные для тканей (печени, кости, хряща, мышц, кожи и т.д.).
Таким образом, проблема, лежащая в основе данного изобретения, состоит в создании доступных взрослых стволовых клеток, генерирование которых не приводит ни к каким этическим и/или юридическим (правовым) проблемам, которые являются легко доступными для планируемого терапевтического применения в количествах, требуемых для этого, и при позволительных затратах на получение и которые, при использовании в качестве «клеточных терапевтических средств», не дают побочных вредных действий, не говоря уже о клеточном отторжении и индукции опухолей, в частности злокачественных опухолей, у рассматриваемого пациента.
Согласно данному изобретению эта проблема решается получением дедифференцированных программируемых клеток из моноцитов человека, которые, для целей данного изобретения, называются далее «стволовыми клетками». Термин «дедифференцировка» известен лицам с квалификацией в данной области, см. Weissman I.L., Cell 100: 157-168, Fig. 4 (2000). Он означает регресс взрослой, уже специализированной (дифференцированной) клетки тела в статус стволовой клетки, т.е. клетки, которая, в свою очередь, может быть превращена (программирована) в ряд типов клеток. Неожиданно было продемонстрировано, что способ данного изобретения приводит к дедифференцировке моноцитов. Стволовые клетки, полученные этим путем, могут быть превращены (программированы) в большое число различных клеток-мишеней/тканей-мишеней, как видно из примеров. Стволовые клетки данного изобретения экспрессируют, кроме поверхностного антигена CD14, характерного для дифференцированных моноцитов, по меньшей мере один, предпочтительно два или три, типичный маркер плюрипотентности CD90, CD117, CD123 и CD135. Особенно предпочтительным образом стволовые клетки, продуцируемые в соответствии с данным изобретением, экспрессируют поверхностный антиген CD14, а также четыре маркера плюрипотентности CD90, CD117, CD123 и CD135, см. пример 2, таблицу 1. Предпочтительно стволовые клетки данного изобретения экспрессируют моноцит-специфический антиген CD14 и по меньшей мере один маркер плюрипотентности, выбранный из группы, состоящей из CD117, CD123 и CD135. Более предпочтительно стволовые клетки данного изобретения несут антиген CD14 в комбинации, по меньшей мере, с маркерами плюрипотентности CD123 и/или CD135. Менее чем 3%, предпочтительно менее чем 1%, стволовых клеток данного изобретения экспрессируют антиген CD34. Наиболее предпочтительно ни одна из стволовых клеток данного изобретения не экспрессирует антиген CD34. Таким образом, впервые сделаны доступными взрослые стволовые клетки, которые могут быть в пределах короткого времени перепрограммированы предпочтительно в аутологичные ткани.
Генерирование стволовых клеток в соответствии с данным изобретением является совершенно безвредным для пациента и - в случае аутологичного использования - сравнимо с получением собственной крови в качестве донорской крови. Количество стволовых клеток (108-109 клеток), требуемое для обычных случаев терапии (см. выше), может быть сделано доступным с приемлемыми расходами в пределах 10-14 дней после взятия крови. Кроме того, клеточный продукт, предназначенный для этого лечения, в случае аутологичного использования, не приводит к какой-либо иммунологической проблеме в виде отторжения клеток, так как клетки и реципиент предпочтительно являются генетически идентичными.
Стволовые клетки данного изобретения оказались также безопасными в экспериментах на животных и в культуре в отношении возникновения злокачественности, т.е. был получен результат, который только и мог ожидаться вследствие моноцитарного происхождения клетки, из которой происходят стволовые клетки в соответствии с данным изобретением.
Основные стадии способа данного изобретения для получения дедифференцированных программируемых стволовых клеток, происходящих из моноцитов человека, включают в себя:
(а) выделение моноцитов из крови человека;
(b) размножение моноцитов в подходящем культуральном сосуде, содержащем среду для культуры клеток, которая содержит колониестимулирующий фактор макрофагов (далее называемый M-CSF);
(с) культивирование моноцитов в присутствии интерлейкина-3 (IL-3); и
(d) получение дедифференцированных программируемых стволовых клеток человека отделением этих клеток от культуральной среды.
Согласно особенно предпочтительному варианту осуществления этого способа M-CSF и IL-3 одновременно добавляют в среду культуры клеток на стадии b).
Однако можно также добавлять сначала только M-CSF в среду культуры клеток на стадии b), чтобы заставить моноциты размножаться, и затем добавлять IL-3 в среду культуры клеток.
Наконец, способ на стадии b) может проводиться таким образом, что моноциты сначала размножают в среде культуры кулеток, содержащей только M-CSF, затем эту среду отделяют от клеток и затем используют вторую среду культуры клеток, которая содержит IL-3.
Согласно предпочтительному варианту осуществления данного изобретения культуральную среду стадии b) отделяют от клеток, прикрепленных ко дну культурального сосуда, и дедифференцированные программируемые стволовые клетки получают отделением клеток от дна и выделением этих клеток.
Согласно предпочтительному варианту осуществления данного изобретения клетки дополнительно культивируют в присутствии соединения серы. Это культивирование проводят в отдельной стадии способа, которая следует за стадией b), описанной выше. Однако она может также проводиться на стадии b) дополнительным добавлением соединения серы в культуральную среду, предпочтительно уже в начале культивирования.
Способ данного изобретения неожиданно приводит к дедифференцировке моноцитов, при которой взрослые стволовые клетки, происходящие из этой дедифференцировки, наряду с поверхностным антигеном CD14, типичным для дифференцированных моноцитов, экспрессируют также, по меньшей мере, один или несколько, предпочтительно все, маркеры плюрипотентности CD90, CD117, CD123 и CD135 (см. таблицу 1). Предпочтительно стволовые клетки данного изобретения экспрессируют мембраносвязанный моноцит - специфический поверхностный антиген CD14 и по меньшей мере один из маркеров плюрипотентности, выбранный из группы, состоящей из CD117, CD123 и CD135. Более предпочтительно стволовые клетки данного изобретения несут антиген CD14 в комбинации, по меньшей мере, с маркером плюрипотентности CD123 и/или CD135. Менее чем 3%, предпочтительно менее чем 1%, стволовых клеток данного изобретения экспрессируют антиген CD34. Наиболее предпочтительно ни одна из стволовых клеток данного изобретения не экспрессирует антиген CD34. Экспрессия соответствующих маркеров (поверхностных антигенов) может быть доказана с использованием коммерчески доступных антител, специфических в отношении соответствующих подлежащих детектированию антигенов, с использованием стандартных процедур иммуноанализа, см. пример 2.
Поскольку клетки, во время процесса размножения и дедифференцировки, прикрепляются ко дну соответствующей культуральной чашки, необходимо отделять эти клетки от культуральной среды из стадии b) и отделять их от дна после завершения дедифференцировки. Согласно предпочтительному варианту осуществления данного изобретения супернатант культуры клеток отбрасывают перед отделением клеток, прикрепившихся ко дну, и затем эти прикрепившиеся клетки предпочтительно промывают свежей культуральной средой. После промывания свежую культуральную среду снова добавляют к клеткам, прикрепившимся ко дну, и затем следует стадия освобождения клеток от дна (см. пример 13).
Согласно предпочтительному варианту осуществления данного изобретения клетки приводят в контакт с биологически хорошо переносимым органическим растворителем в конце стадии с) и перед стадией d). Биологически хорошо переносимым органическим растворителем может быть спирт с 1-4 атомами углерода, причем предпочтительным является применение этанола.
В следующем варианте осуществления в конце стадии с) и перед стадией d) клетки приводят в контакт с паровой фазой биологически хорошо переносимого органического растворителя.
Кроме того, это отделение проводят также механически, но предпочтительным является ферментативный способ отделения, например, с использованием трипсина.
Дедифференцированные программируемые стволовые клетки, полученные таким образом, плавающие свободно в среде, могут либо непосредственно переноситься в процесс перепрограммирования, либо выдерживаться в культуральной среде в течение нескольких дней; в последнем случае предпочтительно к среде добавляют цитокин или LIF (фактор ингибирования лейкоза) во избежание преждевременной потери программируемости (см. Donovan P.J., Gearhart J. loco citato, page 94). Наконец, эти клетки могут быть подвергнуты глубокому замораживанию с целью хранения без потери программируемости.
Стволовые клетки данного изобретения отличаются от плюрипотентных стволовых клеток эмбрионального происхождения, известных до сих пор, и от известных взрослых стволовых клеток из различных тканей тем, что, наряду с мембраносвязанным моноцит-специфическим поверхностным антигеном CD14, они несут, по меньшей мере, один маркер плюрипотентности из группы, состоящей из CD90, CD117, CD123 и CD135, на их поверхности. Предпочтительно стволовые клетки данного изобретения несут мембраносвязанный моноцит-специфический антиген CD14 и по меньшей мере один из маркеров плюрипотентности, выбранный из группы, состоящей из CD117, CD123 и CD135. Более предпочтительно стволовые клетки данного изобретения несут антиген CD14 в комбинации, по меньшей мере, с маркером плюрипотентности CD123 и/или CD135. Менее чем 3%, предпочтительно менее чем 1%, стволовых клеток данного изобретения экспрессируют антиген CD34. Наиболее предпочтительно ни одна из стволовых клеток данного изобретения не экспрессирует антиген CD34.
Стволовые клетки, получаемые способом данного изобретения, могут быть перепрограммированы в любые клетки тела. Способы перепрограммирования стволовых клеток известны в существующем уровне данной области, см., например, Weissman I.L., Science 287: 1442-1446 (2000) и Insight Rewier Articles Nature 414: 92-131 (2001), и справочник «Methods of Tissue Engineering», Eds. Atala A., Lanza R.P., Academic Press, ISBN 0-12-436636-8; Library of Congress Catalog Card No. 200188747.
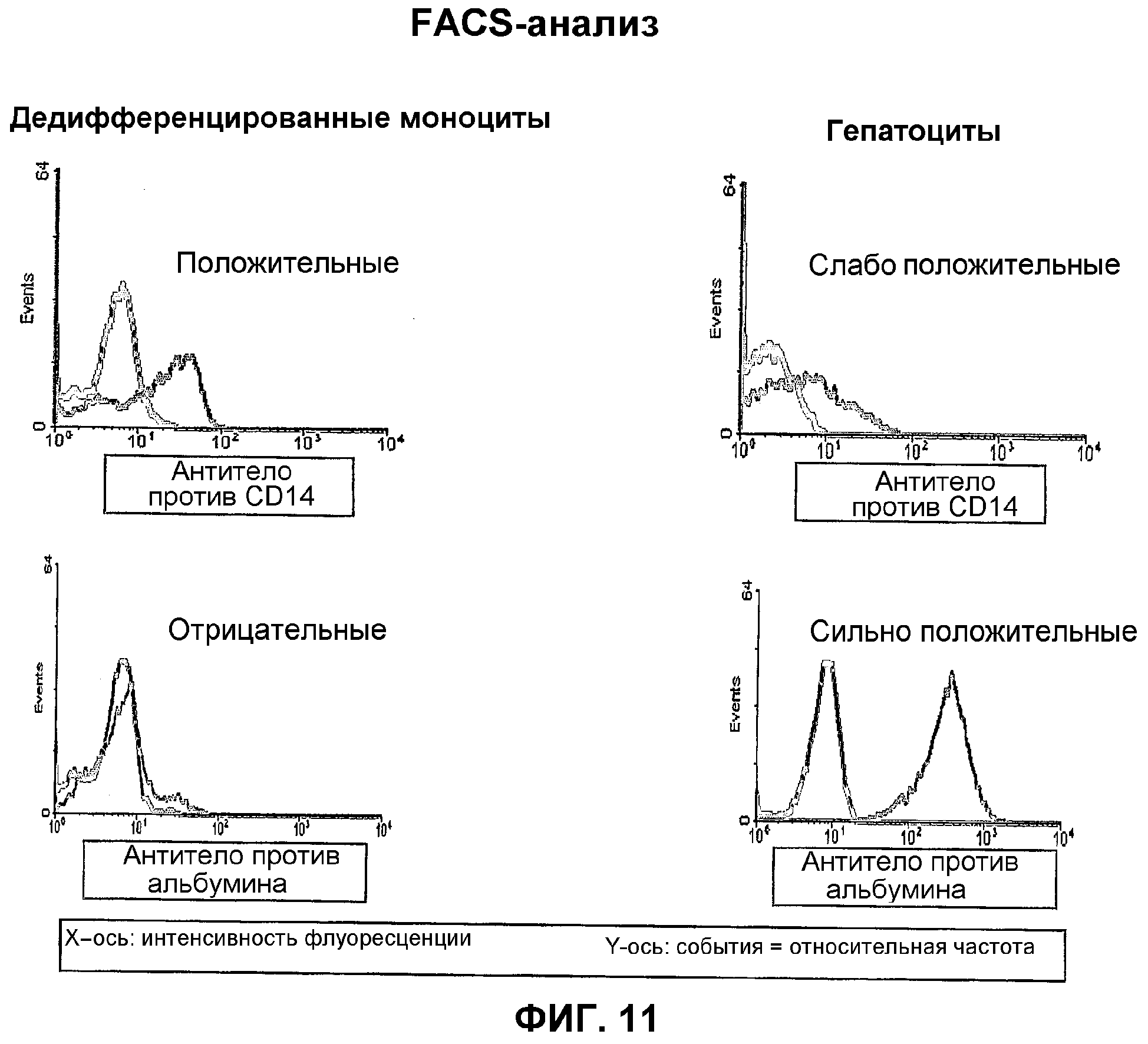
Дифференцированные выделенные соматические клетки-мишени и/или ткань-мишень, полученные перепрограммированием стволовых клеток в соответствии с данным изобретением, несут, кроме того, мембраносвязанный маркер CD14 дифференцировки моноцитов. Кроме того, менее чем 3%, предпочтительно менее чем 1%, этих соматических клеток-мишеней и/или этих тканей-мишеней данного изобретения экспрессируют антиген CD34. Наиболее предпочтительно ни одна из этих клеток или тканей не экспрессирует антиген CD34. Как показано в примере 11, гепатоциты, которые получают из стволовых клеток в соответствии с данным изобретением, экспрессируют поверхностный маркер CD14, который является типичным для моноцитов, тогда как в то же самое время они продуцируют белок альбумин, который является типичным для гепатоцитов. Таким образом, гепатоциты, происходящие из стволовых клеток данного изобретения, могут быть отличены от природных гепатоцитов. Таким же образом, мембраносвязанный поверхностный маркер CD14 детектировали на продуцирующих инсулин клетках, которые происходили из стволовых клеток данного изобретения (пример 9).
В одном варианте осуществления данного изобретения дедифференцированные программируемые стволовые клетки используют для продуцирования in vitro клеток-мишеней и ткани-мишени (см. примеры). Таким образом, дифференцированные, выделенные клетки ткани, которые получены дифференцировкой (перепрограммированием) стволовых клеток в соответствии с данным изобретением и которые несут мембраносвязанный поверхностный антиген CD14, являются также предметом данного изобретения.
Стволовые клетки данного изобретения, предпочтительно, просто и достоверно дифференцируются in vitro в желаемые клетки-мишени, такие как, например, адипоциты (см. пример 6), нейроны и глиальные клетки (см. пример 3), эндотелиальные клетки (см. пример 5), кератиноциты (см. пример 8), гепатоциты (см. пример 7) и островковые клетки (островки Лангерганса, см. пример 9) посредством выращивания этих стволовых клеток в среде, которая содержит супернатант культуральной среды, в которой инкубируются соответствующие клетки-мишени и/или их фрагменты (см. примеры 6-8). Этот супернатант называется далее «кондиционированной клетками-мишенями средой».
Таким образом, для дифференцировки (перепрограммирования) дедифференцированных стволовых клеток в соответствии с данным изобретением может быть использована следующая процедура, в которой
а) ткань, которая содержит желаемые клетки-мишени или которая состоит из желаемых клеток-мишеней, измельчают;
b) получают клетки ткани (клетки-мишени) и/или их фрагменты;
с) клетки-мишени и/или их фрагменты инкубируют в подходящей культуральной среде;
d) супернатант этой культуральной среды собирают во время инкубирования и после инкубирования в качестве кондиционированной клетками-мишенями среды; и
е) для перепрограммирования/дифференцировки дедифференцированных стволовых клеток в желаемые клетки-мишени или ткань-мишень эти стволовые клетки выращивают в присутствии кондиционированной клетками-мишенями среды.
В качестве культуральной среды могут быть использованы стандартные среды для культуры клеток (см. примеры). Эти среды обычно содержат факторы роста, такие как, например, эпидермальный фактор роста.
Инкубирование клеток-мишеней и/или их фрагментов («осадка клеток») может проводиться на протяжении 5-15, предпочтительно 10 дней. Супернатант, т.е. кондиционированную клетками-мишенями среду, удаляют в каждом случае после 2-4 дней и заменяют свежей средой. Полученные таким образом супернатанты могут быть отфильтрованы при стерильных условиях по отдельности или в объединенном виде и храниться при приблизительно -20°С или использоваться сразу для программирования стволовых клеток. Как показано выше, программирование стволовых клеток в желаемые клетки-мишени проводят выращиванием стволовых клеток в присутствии среды, кондиционированной соответствующими клетками-мишенями (см. примеры). Эта среда для выращивания предпочтительно дополнительно содержит специфический для клеток-мишеней фактор, такой как, например, «фактор роста гепатоцитов» или «фактор роста кератиноцитов» (см. примеры).
В одном варианте осуществления данного изобретения дедифференцированные программируемые стволовые клетки данного изобретения используют per se для приготовления фармацевтической композиции для получения in vivo клеток-мишеней и ткани-мишени.
Такие фармацевтические препараты могут содержать стволовые клетки данного изобретения, суспендированные в физиологически хорошо переносимой среде. Подходящими средами являются, например, ЗФР (забуференный фосфатом солевой раствор) или физиологический солевой раствор с 20% раствором альбумина человека и т.п.
Эти фармацевтические препараты содержат жизнеспособные дедифференцированные программируемые стволовые клетки в соответствии с данным изобретением, которые имеют на их поверхности поверхностный маркер CD14 и по меньшей мере один или более из маркеров мультипотентных стволовых клеток CD90, CD117, CD123, и/или CD135, в количестве по меньшей мере 1, 2, 3, 4, 5, 6, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49 или 50%, предпочтительно 60 или 70%, особенно предпочтительно 80 или 90% и очень предпочтительно 100%, относительно общего количества клеток, присутствующих в препарате, и необязательно дополнительные фармацевтически хорошо переносимые адъюванты и/или вещества-носители.
Препараты стволовых клеток могут также содержать жизнеспособные дедифференцированные программируемые стволовые клетки в соответствии с данным изобретением, которые имеют на их поверхности поверхностный маркер CD14 и по меньшей мере один или более из маркеров мультипотентных стволовых клеток CD90, CD117, CD123, и/или CD135, в количестве по меньшей мере 1, 2, 3, 4, 5, 6, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58 или 59%, предпочтительно, по меньшей мере, 60%, относительно общего количества клеток, присутствующих в препарате; предпочтительными являются суспензии клеток в среде для культуры клеток или среде для транспорта хорошо переносимой клетками, такой как, например, ЗФР или RPMI и т.д., или глубоко замороженные препараты клеток в подходящей среде для хранения, такой как RPMI, с 50% раствором альбумина человека и 10% ДМСО.
Количество жизнеспособных клеток и, следовательно, доля их в композициях, описанных выше, может быть определено оптически с использованием «способа вытеснения красителя трипанового синего», так как жизнеспособные клетки могут быть отличены оптически от нежизнеспособных клеток с использованием этого красителя.
Как правило, для клинического применения неважно, если некоторые из клеток, присутствующих в фармацевтическом препарате, не удовлетворяют критериям дедифференцированных программируемых стволовых клеток данного изобретения, при условии, что присутствует достаточное количество функциональных стволовых клеток. Однако можно также элиминировать не-дедифференцированные клетки при помощи способов, известных в существующем состоянии данной области, на основе поверхностных маркеров, типичных для дедифференцированных клеток данного изобретения, в таких препаратах, так что они содержат желаемые клетки по существу в чистом виде. Одним примером подходящего способа является «Immuno magnetic bead sorting» («Иммуносортинг с использованием магнитных гранул»), см. Romani et al., J. Immunol. Methods 196: 137-151 (1996).
Кроме того, стволовые клетки имеют способность спонтанной дифференцировки in vivo посредством прямого контакта с группой клеток специфического типа в клетки этого типа. Способы получения ткани с использованием клеток, которые могут быть передифференцированными («конструирование тканей»), известны в данной области. Например, Wang, X. et al. («Liver repopulation and correction of metabolic liver disease by transplanted adult mouse pancreatic cells». Am. J. Pathol. 158 (2): 571-579 (2001)) показали, что даже некоторые взрослые клетки поджелудочной железы мышей способны превращаться, у мышей с FAH-(фумароилацетоацетатгидролаза)-недостаточностью, в гепатоциты, которые могут полностью компенсировать данный метаболический дефект в этих животных. Другим примером являются эксперименты Lagasse et al., «Purified hematopoietic stem cells can differentiate into hepatocytes in vivo», Nature Medicine, 6 (11): 1229-1234 (2000). Эти авторы показали, что гемопоэтические стволовые клетки из костного мозга были способны после переноса in vivo в мышей с FAH-недостаточностью превращаться в гепатоциты, которые после этого могли компенсировать данный метаболический дефект; см. также обзор Grompe M., «Therapeutic Liver Repopulation for the Treatment of Metabolic Liver Diseases». Hum. Cell, 12: 171-180 (1999).
Особенно предпочтительными формами применения для in vivo-дифференцировки дедифференцированных стволовых клеток в соответствии с данным изобретением являются инъекция, инфузия или имплантация стволовых клеток в одну ассоциацию специфических клеток в теле, чтобы позволить этим стволовым клеткам дифференцироваться в ней посредством прямого контакта с этой ассоциацией клеток в клетки данного клеточного типа. Для инъекции или инфузии эти клетки могут вводиться в ЗФР (забуференном фосфатом солевом растворе).
Предпочтительными примерами релевантных показаний в этой связи являются цирроз печени, недостаточность поджелудочной железы, острая или хроническая почечная недостаточность, пониженное гормональное функционирование, инфаркт миокарда, легочная эмболия, инсульт и кожное повреждение.
Поэтому предпочтительным вариантом осуществления данного изобретения является использование дедифференцированных программируемых стволовых клеток для получения различных фармацевтических композиций для лечения цирроза печени, недостаточности поджелудочной железы, острой или хронической почечной недостаточности, пониженного гормонального функционирования, инфаркта миокарда, легочной эмболии, инсульта и кожного повреждения.
Для терапевтического применения клеток-мишеней, получаемых из стволовых клеток данного изобретения, доступны ряд концепций (см. выше Science 287: 1442-1446 (2000) и Nature 414: 92-131 (2001)).
Следующее предпочтительное применение относится к инъекции дедифференцированных стволовых клеток данного изобретения в перитонеум (брюшину), так что они дифференцируют в ней перитонеальные клетки вследствие влияния окружающих их клеток в. В случае перитонеального диализа пациентов с почечной недостаточностью, эти клетки могут принимать на себя функцию почки посредством их полупроницаемой мембраны и отдавать независимо от почки отработанные вещества в перитонеум, откуда они могут быть удалены посредством диализа.
Таким образом, эти дифференцированные, выделенные, соматические клетки-мишени и/или ткань-мишень, которые образуются посредством перепрограммирования стволовых клеток и характеризуются мембраносвязанным антигеном CD14, также являются предметом данного изобретения. Эти соматические клетки-мишени и/или ткань-мишень, предпочтительно, включают в себя адипоциты, нейроны и глиальные клетки, эндотелиальные клетки, кератиноциты, гепатоциты и островковые клетки.
Однако эти клетки могут также вводиться непосредственно в орган, который должен быть восстановлен. Введение может проводиться через матриксные конструкции, которые покрыты соответствующими дифференцированными клетками или клетками, способными к дифференцировке. Эти матриксные конструкции являются, как правило, биодеградируемыми, так что они исчезают из тела, в то время как заново введенные клетки растут вместе с присутствующими клетками. С этой точки зрения рассматриваются, например, клеточные, предпочтительно, аутологичные трансплантаты островковых клеток, гепатоцитов, жировых клеток, кожных клеток, мышц, сердечных мышц, нервов, костей, эндокринных клеток и т.д. для восстановления, например, после частичной хирургической резекции органа, для восстановления, например, после травмы или для поддерживающего применения, например, в случае отсутствующей или недостаточной функции органа.
Стволовые клетки данного изобретения и клетки-мишени, полученные из них, могут быть также использованы для покрытия имплантируемых материалов, для увеличения биосовместимости. Таким образом, имплантируемые материалы, которые покрыты дедифференцированными, программируемыми стволовыми клетками или соматическими клетками-мишенями и/или тканью-мишенью, являются также предметом данного изобретения. Согласно одному варианту осуществления данного изобретения этими имплантируемыми материалами являются протезы. В особенно предпочтительных вариантах осуществления данного изобретения эти протезы являются клапанами сердца, протезами сосудов, протезами костей и суставов.
Имплантируемые материалы могут быть также искусственными и/или биологическими материалами-носителями, которые содержат дедифференцированные программируемые стволовые клетки или клетки-мишени. В этой связи материалы-носители могут быть сумками или камерами для имплантации в тело человека.
В одном варианте данного изобретения такую сумку, содержащую островковые клетки, которые являются дифференцированными соматическими клетками данного изобретения, используют для получения фармацевтической конструкции для применения в качестве искусственной камеры-депо для доставки инсулина.
Согласно следующему варианту осуществления данного изобретения сумку или камеру, содержащую адипоциты, которые являются дифференцированными соматическими клетками в соответствии с данным изобретением, используют для получения искусственного полимера, заполненного адипоцитами, в качестве фармацевтической конструкции для конструирования молочной железы после хирургического вмешательства и в случае других показаний пластической и/или косметической коррекции.
Кроме того, системы полупроницаемых камер-депо, содержащих эндокринные клетки очень широко варьирующегося происхождения, могут быть использованы in vivo для лечения эндокринных, метаболических или гемостатических нарушений. Примерами таких эндокринных клеток являются клетки, которые продуцируют тироксин, стероиды, ADH (антидиуретический гормон), альдостерон, мелатонин, серотонин, адреналин, норадреналин, TSH (тиреотропин), LH (лютеинизирующий гормон), FSH (фолликулостимулирующий гормон), лептин, холецистокинин, гастрин, инсулин, глюкагон или факторы свертывания.
Таким образом, имплантируемые материалы, которые являются системами полупроницаемых камер-депо, содержащими выделенные дифференцированные соматические клетки-мишени данного изобретения, также являются предметом данного изобретения. Эти полупроницаемые камеры-депо используют в различных вариантах данного изобретения для приготовления фармацевтической конструкции для лечения in vivo эндокринных, метаболических мили гемостатических нарушений.
Кроме того, клетки-мишени, полученные из стволовых клеток в соответствии с данным изобретением, могут быть использованы в качестве культур клеток в биореакторах вне тела, например, для проведения реакций детоксикации. Эта форма применения особенно предпочтительна в случае острых условий, например, в случае острой печеночной недостаточности в качестве биореактора гепатоцитов.
Получение вышеописанных конструкций и проведение соответствующего терапевтического способа уже было описано много раз в данной области, см., например, обзор Lalan S., et al. «Tissue engineering and its potential impact on surgery», World J. Surg. 25: 1458-1466 (2001); Nasseri B.A., et al., «Tissue engineering: an evolving 21st-century science to provide replacement for reconstruction and transplantation», Surgery 130: 781-784 (2001) и Fuchs J.R., et al. «Tissue engineering: a 21st century solution to surgical reconstruction», Ann. Thorac. Surg. 72: 577-591 (2001).
Наконец, плюрипотентные стволовые клетки данного изобретения открывают широкое поле для трансгенной модификации и терапии. Согласно предпочтительному варианту осуществления данного изобретения дедифференцированные программируемые стволовые клетки per se или соматические клетки-мишени и/или ткань-мишень, в конечном счете дифференцированные из них, трансфицируют одним или несколькими генами. Таким путем один или несколько генов, которые необходимы для поддержания метаболизма некоторых органов, таких как, например, печень или почки, восстанавливаются и/или поддерживаются или повторно вводятся. Например, стволовые клетки или происходящие из них гепатоциты могут быть трансфицированы геном FAH (фумароилацетоацетатгидролаза). В модели мыши с FAH-недостаточностью инъекция внутрь селезенки 1000 FAH-положительных донорских гепатоцитов была достаточной для полного повторного «заселения» печени после 6-8 недель и полной компенсации в отношении метаболического дефекта, ведущего к циррозу печени (см. Grompe M., et al., Nat. Genet. 12: 266 ff. (1996)).
Таким образом, трансфекцией стволовых клеток или соответствующих клеток-мишеней, полученных из этих стволовых клеток программированием (например, гемопоэтических клеток, гепатоцитов, клеток яичника, мышечных клеток, нервных клеток, нейронов, глиальных клеток, хрящевых или костных клеток и т.д.), «генами устойчивости к множественным лекарственным средствам» была сделана доступной продолжительная радикальная химиотерапия в случае злокачественных заболеваний посредством восстановления соответствующих гемопоэтических клеток или устойчивости к облучению, которые могут быть получены.
Данное изобретение подробно объясняется следующим образом.
Исходным материалом для способа данного изобретения являются моноциты из крови человека. Предпочтительно они являются аутологичными моноцитами, т.е. моноцитами, которые происходят из крови пациента, который должен лечиться стволовыми клетками данного изобретения или клетками-мишенями, полученными из них.
Для получения моноцитов кровь может быть сначала, после стандартной обработки антикоагулянтом известным образом, предпочтительно центрифугированием, разделена на плазму, и лейкоциты, и эритроциты. После центрифугирования плазма должна находиться в супернатанте; под ней лежит слой, который содержит все лейкоциты. Этот слой называют также «лейкоцитной пленкой». Под ним лежит фаза, содержащая эритроциты (гематокрит).
Затем «лейкоцитную пленку» выделяют и разделяют для получения моноцитов, например, центрифугированием с использованием известного процесса. В соответствии с предпочтительным вариантом способа слой «лейкоцитной пленки» наносят в виде покрытия на среду для разделения лимфоцитов (например, Ficoll Hypaque) и центрифугируют. Последующим центрифугированием и промыванием получают фракцию моноцитов из этой крови (см. пример 1).
Примерами альтернативных способов получения моноцитов из цельной крови являются «Сортинг клеток с активацией флуоресценции» (FACS), «Сортинг с иммуно-магнитными гранулами» (см. Romani et al., J. Immunol. Methods 196: 137-151 (1996) и «Активированный магнитом сортинг клеток» (MACS) или так называемый «Способ розеткообразования» (см. Gmelig-Meyling F., et al., «Simplified procedure for the separation of human T and non-T cells», Vox Sang. 33: 5-8 (1977)).
Согласно данному изобретению моноциты могут быть получены из любой выделенной крови человека, и эта кровь может также происходить из таких органов, как селезенка, лимфатические узлы или костный мозг. Получение моноцитов из органов рассматривается, в частности, в тех случаях, когда отделение моноцитов из крови человека, например, в случае анемии или лейкоза, не является возможным или может дать недостаточные количества, и в случае аллогенного применения, если, в рамках удаления множественных органов, селезенка является доступной в качестве источника для выделения моноцитов.
Для получения достаточного количества стволовых клеток в соответствии с данным изобретением сначала необходимо размножить моноциты. Для этой цели могут быть использованы среды для выращивания, подходящие для моноцитов, где согласно данному изобретению указанная среда содержит M-CSF (колониестимулирующий фактор макрофагов). M-CSF (также называемый CSF-1) получают из моноцитов, фибробластов и эндотелиальных клеток. Концентрация M-CSF в культуральной среде может быть равна 2-20 мкг/л, предпочтительно 4-6 мкг/л и особенно предпочтительно 5 мкг/л.
На моноцитах M-CSF связывается со специфическим с-Fms-рецептором (также называемым CSF-1R), который присутствует исключительно на поверхности моноцитов и связывает только M-CSF (Sherr C.J., et al., Cell 41 (3): 665-676 (1985)). Так как специфическое взаимодействие между M-CSF и этим рецептором индуцирует деление моноцитов, среда, в которой культивируют моноциты, содержит M-CSF или его аналог, который может связываться с этим рецептором и активировать его. Другие факторы роста, такие как GM-CSF (колониестимулирующий фактор гранулоцитов-моноцитов) и G-CSF (колониестимулирующий фактор гранулоцитов), являются непригодными, так как вследствие отсутствия аффинности в отношении c-Fms-рецептора они неспособны индуцировать деление моноцитов.
В особенно предпочтительном варианте осуществления этого способа M-CSF и IL-3 добавляют одновременно в среду культуры клеток на стадии b) этого способа. Концентрация IL-3 в среде может быть равна 0,2-1 мкг/л, предпочтительно 0,3-0,5 мкг/л и особенно предпочтительно 0,4 мкг IL-3 на литр.
Однако можно сначала добавлять в среду для культуры клеток только M-CSF на стадии b), а IL-3 добавлять только после этого.
В следующем варианте осуществления культуральный сосуд первоначально содержит среду для культуры клеток, содержащую только M-CSF, которую после отделения клеток заменяют второй средой для культуры клеток, содержащей IL-3.
Согласно предпочтительному варианту осуществления данного изобретения клетки на стадии b) этого способа дополнительно культивируют в присутствии соединения серы, например меркаптосоединения, в котором, по меньшей мере, одна углеводородная группа связана с серой, и указанная углеводородная группа (группы) может быть замещена одной или несколькими функциональными группами. Меркаптосоединения определяются как соединения, которые имеют, по меньшей мере, одну меркаптогруппу (-SH), которая связана с углеводородной группой. Посредством дополнительного применения такого соединения серы может быть увеличено количество стволовых клеток, полученных дедифференцировкой клеток моноцитарного происхождения, которые экспрессируют один или более маркеров стволовых клеток CD90, CD117, CD123 и CD135.
Эта функциональная группа (или группы) является, предпочтительно, гидроксильной и/или аминогруппой. В особенно предпочтительном варианте осуществления этим соединением серы является 2-меркаптоэтанол. Согласно следующему предпочтительному варианту осуществления соединением серы является диметилсульфоксид (ДМСО).
Количество используемого соединения серы может находиться в диапазоне от приблизительно 4 до приблизительно 200 мкмоль/л в расчете на серу. Предпочтительным является количество 100 мкмоль/л.
При использовании 2-меркаптоэтанола культуральная среда должна содержать приблизительно от 3 мкл до приблизительно 13 мкл, предпочтительно 7 мкл 2-меркаптоэтанола на л.
Обработка IL-3 и, необязательно, соединением серы может проводиться одновременно с размножением моноцитов культивированием с M-CSF или после размножения моноцитов культивированием с M-CSF, причем предпочтительными являются одновременные размножение и обработка IL-3 и, необязательно, соединением серы. Размножение и дедифференцировка, взятые вместе, должны продолжаться не более чем 10 дней, а обработка IL-3 и, необязательно, соединением серы должна проводиться на протяжении по меньшей мере 3 и максимально 10 дней, предпочтительно на протяжении 6 дней.
Таким образом, согласно данному изобретению в случае культивирования моноцитов в культуральной среде, которая одновременно содержит M-CSF, IL-3 и, предпочтительно, меркаптосоединение, продолжительность культивирования до отделения этих клеток от дна культурального сосуда равна по меньшей мере 3 и максимально 10 дням, предпочтительно 5-8 дням и, в частности, предпочтительно 6 дням.
В предпочтительном варианте осуществления способ данного изобретения проводят таким образом, что моноциты на стадии b) сначала размножают в среде, содержащей только M-CSF, размножение в такой культуральной среде может иметь место на протяжении периода по меньшей мере 2, предпочтительно 3 и особенно предпочтительно 4 дней с максимальной продолжительностью 7 дней, а последующее культивирование в присутствии IL-3 и, необязательно, меркаптосоединения может иметь место на протяжении дополнительных 3 дней. Но, предпочтительно, в таком случае культивирование в среде, содержащей только M-CSF, будет продолжаться максимально 4 дня с последующим культивированием в присутствии IL-3 и, необязательно, меркаптосоединения на протяжении периода 3, 4, 5 или 6 дней.
Для одновременного проведения размножения и дедифференцировки, как описано в примерах 2 и 13, моноциты после выделения переносят в среду, которая содержит как M-CSF, так и IL-3, а также, предпочтительно, соединение серы, в частности, меркаптоэтанол или ДМСО.
Вследствие своих адгезивных свойств моноциты и стволовые клетки, полученные из них во время этого способа, прикрепляются ко дну соответствующего культурального сосуда. Согласно предпочтительному варианту данного изобретения культуральную среду после стадии с) отделяют от клеток, прикрепившихся ко дну культурального сосуда, и затем отбрасывают. После этого предпочтительно следует промывание клеток, прикрепившихся ко дну, культуральной средой, и затем эти клетки покрывают свежей культуральной средой (см. пример 13).
На этой стадии в качестве культуральной среды может быть использована среда для размножения и дедифференцировки, описанная выше, а также стандартная среда для культуры клеток, например, RPMI.
Согласно следующему предпочтительному варианту данного изобретения эти клетки приводят в контакт с биологически хорошо переносимым органическим растворителем в конце стадии с) и перед стадией d) для увеличения количества стволовых клеток, плавающих свободно в среде, в конце этого способа. Количество растворителя может находиться в диапазоне от 10 мкл до 1 мл. Предпочтительно им является спирт с 1-4 атомами углерода, причем особенно предпочтительным является добавление этанола. Согласно особенно предпочтительному варианту эти клетки приводят в контакт с паровой фазой ранее определенного биологически хорошо переносимого органического растворителя, предпочтительно с парами этанола (см. пример 2). Время подвергания действию этого органического растворителя, особенно предпочтительно паров этанола, должно составлять 4-12 часов, предпочтительно 8-10 часов.
Способ данного изобретения проводят предпочтительно в культуральных сосудах, поверхность которых была предварительно покрыта фетальной телячьей сывороткой (ФТС) (см. пример 2). Альтернативно может быть также использована АВ-сыворотка человека доноров-мужчин. Покрытие ФТС может проводиться нанесением ФТС на поверхность культуральных сосудов перед использованием и после экспонирования в течение нескольких, в частности, 2-12 часов и особенно предпочтительно 7 часов, и удалением ФТС, не прикрепившейся к поверхности, подходящим образом.
Если обработка органическим растворителем имеет место после стадии с), необязательно после замены культуральной среды, эти клетки уже становятся до некоторой степени отделенными от дна в этой стадии способа. Дополнительное отделение может выполняться механическим образом, например, тонким клеточным скребком, шпателем или кончиком пипетки (см. пример 13).
Согласно предпочтительному варианту данного способа полное отделение проводят обработкой подходящим ферментом, например, трипсином (см. пример 2). Эти клетки могут быть подвергнуты действию раствора трипсина (0,1-0,025 г/л, предпочтительно 0,05 г/л) в течение 2-10 минут при 35-39°С, предпочтительно при 37°С, в присутствии СО2.
Затем активность трипсина блокируют стандартным способом, и теперь свободно плавающие дедифференцированные программируемые стволовые клетки могут быть получены стандартными способом, например центрифугированием, и в одном варианте суспендированы в подходящей культуре клеток в конце стадии d). Теперь они являются доступными, суспендированными в подходящей среде, например, в RPMI 1640 или DMEM, для немедленной дифференцировки в желаемые клетки-мишени. Однако они могут также выдерживаться в этой среде в течение нескольких дней. В предпочтительном варианте эта среда содержит цитокин или фактор LIF (фактор ингибирования лейкоза), см. Nature 414: 94 (2001, Donovan P.J., Gearhart J., loco citato), если эти клетки должны храниться в культуре в течение более продолжительного времени, чем приблизительно 48 часов, в виде дедифференцированных программируемых стволовых клеток. В среде, содержащей такие факторы, стволовые клетки могут храниться в течение, по меньшей мере, 10 дней в виде дедифференцированных программируемых стволовых клеток.
В предпочтительном варианте эти клетки суспендируют для более длительного хранения в жидкой среде и затем подвергают глубокому замораживанию. Протоколы для глубокого замораживания живых клеток известны в данной области, см. Griffith M., et al. «Epithelial Cell Culture, Cornea, in Methods of Tissue Engineering», Atala A., Lanza R.P., Academic Press 2002, Chapter 4, Pages 131-140. Предпочтительной суспензионной средой для глубокого замораживания стволовых клеток в соответствии с данным изобретением является ФТС-содержащая DMEM, см. пример 2.
Далее данное изобретение представлено и описано со ссылкой на примеры.
Если нет указаний в примерах, состав сред и использованные вещества являются следующими:
1. Раствор пенициллина/стрептомицина:
10000 единиц пенициллина в виде натриевой соли пенициллина G и 1000 мкг стрептомицина в виде сульфата стрептомицина на мл физиологического раствора хлорида натрия (0,9% NaCl).
2. Трипсин-ЭДТА
0,5 г трипсина и 0,2 г ЭДТА (4 Na)/л.
3. Инсулин
Человеческий, рекомбинантный, продуцируемый в E. coli, приблизительно 28 единиц/мг.
4. RPMI 1640 (1х, жидкая (11875)) содержит L-глутамин.
Среды RPMI (Roswell Park Memorial Institute) 1640 являются обогащенными препаратами, которые могут быть широко использованы для клеток млекопитающих.
Ссылка: Moore G.E., et al., J.A.M.A. 199: 519 (1967).
5. ЗФР (забуференный фосфатом солевой раствор Дульбекко) см. J. Exp. Med. 98: 167 (1954):
6. 2-Меркаптоэтанол
Качество - для синтеза; содержание > 98%, Плотность 1,115-1,116, см., например, Momo J., et al., J. Am. Chem. Soc. 73: 4961 (1951).
7. Ficoll-Hypaque
Среда для разделения лимфоцитов (сополимеризат сахароза/эпихлоргидрин Mg 400000; плотность 1,077, доведенная диатриазоатом натрия).
8. Ретиноевая кислота
Кислотная форма витамин А (С20Н28О2), 300 мкл в 1,5 мл ЗФР, что соответствует 1 мМ. В качестве среды для программирования нейронов и глиальных клеток используют 150 мкл на 10 мл среды (что соответствует 10-6 М).
9. DMEM
Модифицированная по способу Дульбекко среда Игла (с высоким содержанием глюкозы), см. Dulbecco, R. et al., Virology 8: 396 (1959); Smith, J.D. et al., Virology 12: 158 (1960); Tissue Culture Standards Committee, In Vitro 6: 2 (1993)
10. L-Глутамин
Жидкость: 29,2 мг/мл
11. Коллагеназа типа II
См. Rodbell, M. et al., J. Biol. Chem. 239: 375 (1964).
12. Интерлейкин-3 (IL-3)
Рекомбинантный IL-3 человека из E. coli (Yang Y.C. et al., Cell 47: 10 (1986)); содержит 133 аминокислотных остатка, представляющий собой зрелый IL-3, и 134 аминокислотных остатка, представляющий собой метионильную форму, в соотношении приблизительно 1:2; рассчитанная мол. масса приблизительно 17,5 кДа; удельная активность 1 × 103 Е/мкг; (R&D Catalogue No. 203-IL)
13. Колониестимулирующий фактор макрофагов (M-CSF)
Рекомбинантный M-CSF человека из E. coli; содержит в виде мономера (18,5 кДа) 135 аминокислотных остатков, в том числе N-концевой метионин; присутствует в виде гомодимера с молекулярной массой 37 кДа; (Sigma Catalogue No. M 6518)
14. Антитела
Антитела, используемые в этих примерах против антигенов CD14, CD31, CD90, CD117, CD123, CD135, являются коммерчески доступными. Их получали из следующих источников:
CD14: DAKO, Monoclonal Mouse Anti-Human CD14, Monocyte, Clone TUK4, Code No. M 0825, Lot 036, Edition 02.02.01;
CD31: PharMingen International, Monoclonal Mouse Anti-Rat CD31 (PECAM-1), Clone TLD-3A12, Catalogue No. 22711D, 0,5 мг;
CD90: Biozol Diagnostica, Serotec, Mouse Anti-Human CDw90, Clone No. F15-42-1, MCAP90, Batch No. 0699;
CD117: DAKO, Monoclonal Mouse Anti-Human CD117, c-kit, Clone No. 104D2, Code M 7140, Lot 016, Edition 04.05.00;
CD123: Research Diagnostics Inc., Mouse Anti-human CD123 antibodies, Clone 9F5, Catalogue No. RDI-CD123-9F5;
CD135: Serotec, Mouse Anti-Human CD135, MCA1843, Clone No. BV10A4H2.
Пример 1
Выделение моноцитов из цельной крови
Во избежание свертывания крови и для кормления клеток 450 мл цельной крови в 3-камерном комплекте-сумке смешивали с 63 мл стабилизирующего раствора, который содержал на каждый литр Н2О 3,27 г лимонной кислоты, 26,3 г цитрата тринатрия, 25,5 г декстрозы и 22,22 г дигидрофосфата натрия. Величина рН была равна 5,6-5,8.
Затем для разделения компонентов крови проводили «резкое центрифугирование» этой смеси при 4000 об/мин в течение 7 минут при 20°С. Это приводило к тройному расслоению корпускулярных и некорпускулярных компонентов. Затем вставлением этого комплекта сумок в пресс, предназначенный для этой цели, эритроциты спрессовывали в нижнюю сумку, плазму спрессовывали в верхнюю сумку, а «лейкоцитная пленка» оставалась в средней сумке и она содержала объем приблизительно 50 мл.
Свежеполученную «лейкоцитную пленку» количеством 50 мл затем делили на 2 порции по 25 мл каждая, каждую из которых затем покрывали 25 мл среды для разделения Ficoll-Hypaque, которая была заранее введена в две пробирки Falcon на 50 мл.
Эту смесь центрифугировали без торможения в течение 30 минут при 2500 об/мин. После этого эритроциты и мертвые клетки, все еще присутствовавшие в «лейкоцитной пленке», находятся под фазой Фиколла, тогда как лейкоциты, в том числе моноциты, отделяются в виде белой межфазной поверхности на Фиколле.
Затем белую межфазную поверхность осторожно собирали пипеткой и смешивали с 10 мл забуференного фосфатом солевого раствора (ЗФР).
Затем эту смесь центрифугировали с торможением три раза в течение 10 минут при 1800 об/мин; супернатант собирали пипетированием после каждого центрифугирования и наливали свежий ЗФР.
Осадок клеток, собранный на дне центрифужного сосуда (пробирки Falcon), содержал фракцию мононуклеарных клеток, т.е. моноцитов.
Пример 2
Размножение и дедифференцировка моноцитов
Культивирование и размножение моноцитов, с одной стороны, и дедифференцировку этих клеток, с другой стороны, проводили в одну стадию в питательной среде следующего состава:
Эта питательная среда дополнительно содержала 2,5 мкг/500 мл M-CSF и 0,2 мкг/500 мл интерлейкина-3 (IL-3).
Моноциты, выделенные в примере 1, переносили в 5 камер 6-камерного (луночного) планшета (диаметр каждой лунки 30 мм) в количестве приблизительно 105 клеток на камеру в каждом случае и заполняли в каждом случае вышеуказанной питательной средой. Этот 6-луночный планшет предварительно заполняли чистой неинактивированной ФТС, и эту ФТС декантировали спустя приблизительно 7 часов для получения таким образом покрытого ФТС планшета. Количество клеток для точной дозы на лунку определяли в соответствии с известным способом, см. Hay R.J. «Cell Quantification and Characterisation» in Methods of Tissue Engineering, Academic Press 2002, Chapter 4, Pages 55-84.
6-Луночный планшет закрывали крышкой и выдерживали в течение 6 дней в термостате при 37°С. Клетки оседали на дно камер после 24 часов. Каждый второй день супернатант извлекали пипетированием и камеры 6-луночного планшета опять заполняли 2 мл свежей питательной среды. На 6-й день 2 мл 70% этанола вводили в 6-ю камеру 6-луночного планшета, которая оставалась свободной, планшет опять закрывали и выдерживали еще в течение 10 часов при 37°С в термостате.
Затем 1 мл раствора трипсина, разбавленного 1:10 ЗФР, вводили пипетированием в каждую из камер планшета, которая содержала клетки. Закрытый планшет помещали на 5 минут в термостат при 37°С и 5% СО2.
Затем активность трипсина блокировали добавлением 2 мл среды RPMI 1640 в каждую из лунок. Весь супернатант в каждой из камер (1 мл трипсина + 2 мл среды) извлекали пипетированием, объединяли в пробирке Falcon на 15 мл и центрифугировали в течение 10 минут при 1800 об/мин. Затем супернатант выбрасывали, а осадок смешивали со свежей средой RPMI 1640 (2 мл/105 клеток).
Эта клеточная суспензия могла быть непосредственно использована для дифференцировки в различные клетки-мишени.
Альтернативно после центрифугирования и выбрасывания содержащего трипсин супернатанта клетки смешивали с ДМСО/ФТС в качестве среды для замораживания и подвергали глубокому замораживанию при концентрации 106/мл.
Среда для замораживания содержала 95% ФТС и 5% ДМСО. В каждом случае приблизительно 106 клеток помещали в 1 мл среды и охлаждали в следующих стадиях:
30 минут на льду;
2 часа при -20°С в предварительно охлажденных боксах Styropor;
24 часа при -80°С в боксах Styropor;
хранение в пробирках в жидком азоте (N2) при -180°С.
Для иммуногистохимического фенотипирования клеточной популяции дедифференцированных программируемых стволовых клеток моноцитарного происхождения, полученных в соответствии с описанным выше способом, в каждом случае брали 105 клеток и фиксировали в виде цитоспинового клеточного мазка на предметных стеклах для последующего гистохимического окрашивания (Watson P. «A slide centrifuge; An apparatus for concentrating cells in suspension on microscope slide». J. Lab. Clin. Med., 68: 494-501 (1966)). После этого клетки могли быть окрашены с использованием способа, описанного Cordell J.L., et al. (Литература, см. ниже), комплексом APAAP red. Если нет других указаний, добавляемое первичное антитело разбавляли 1:100 ЗФР и в каждом случае использовали 200 мкл раствора этой концентрации антител. В качестве первичных антител против эпитопов клеточных антигенов, перечисленных в таблице 1, использовали моноклональные антитела. Фиг.6 показывает окрашенные цитоспиновые клеточные мазки и соответствующее подтверждение маркеров стволовых клеток CD90, CD117, CD123 и CD135.
Литература, относящаяся к способу окрашивания:
Cordell J.L., et al., «Immunoenzymatic labeling of monoclonal antibodies using immune complexes of alkaline phosphatase and monoclonal anti-alkaline phosphatase (APAAP complexes)». J. Histochem. Cytochem. 32: 219-229 (1984).
Литература, относящаяся к маркерам:
CD14
Ferrero E., Goyert S.M. «Nucleotide sequence of the gene encoding the monocyte differentiation antigen, CD14». Nucleic Acids Res. 16: 4173-4173 (1988).
CD31
Newman P.J., Berndt M.C., Gorski J., White J.C. II, Lyman S., Paddock C., Muller W.A. «PECAM-1(CD31) cloning and relation to adhesion molecules of the immunoglobulin gene superfamily». Science 247: 1219-1222 (1990).
CD90
Seki T., Spurr N., Obata F., Goyert S., Goodfellow P., Silver J. «The human thy-1 gene: structure and chromosomal location». Proc. Natl. Acad. Sci. USA 82: 6657-6661 (1985).
CD117
Yarden Y., Kuang W.-J., Yang-Feng T., Coussels L., Munemitsu S., Dull T.J., Chen E., Schlessinger J., Francke U., Ullrich A. «Human proto-oncogene c-kit: a new cell surface receptor tyrosine kinase for an unidentified ligand». EMBO J. 6: 3341-3351 (1987).
CD123
Kitamura T., Sato N., Arai K., Miyajima A. «Expression cloning of the human IL-3 receptor cDNA reveals a shared beta subunit for the human IL-3 and GM-CSF receptors». Cell 66: 165-1174 (1991).
CD135
Small D., Levenstein M., Kim E., Carow C., Amn S., Rockwell P., Witte L., Burrow C., Ratajazak M.Z., Gewirtz A.M., Civin C.I.
«STK-1, the human homolog of Flk-2/Flt-3, is selectively expressed in CD34+ human bone marrow cells and is involved in the proliferation of early progenitor/stem cells». Proc. Natl. Acad. Sci. USA 91: 459-463 (1994).
Показанная градация соответствует положительности детектируемого антигена, который становится видимым с 4 по 9 день после культивирования моноцитов в соответственно указанной среде, и ее проводили сравнением под микроскопом соответствующих окрашиваний цитоспиновых препаратовок с отрицательным контролем (окрашиванием, наблюдаемым без первичных антител):
+ явная цветная реакция клеток с первичным антителом;
++ сильная цветная реакция клеток с первичным антителом.
Оценивали только цитоспиновые препараты, которые имели более 70% жизнеспособных клеток с типичной морфологией стволовых клеток (см. фиг.6). Менее 1% этих клеток экспрессировали антиген CD34.
Пример 3
Получение нейронов и глиальных клеток из взрослых стволовых клеток
Получение нейронов и глиальных клеток проводили в чашках Петри с диаметром 100 мм. Для приготовления чашек Петри 5 мл чистой фетальной телячьей сыворотки (ФТС) вводили в каждую чашку так, чтобы было покрыто дно. Спустя 7 часов часть ФТС, не прикрепившуюся к дну чашки Петри, удаляли пипетированием. Приблизительно 106 клеток, полученных в соответствии с примером 2, вводили в одну из приготовленных чашек Петри и добавляли 10 мл питательной среды следующего состава:
Эта питательная среда содержала дополнительно ретиноевую кислоту в количестве 1х10-6 М/500 мл.
Перепрограммирование/дифференцировка используемых стволовых клеток в нейроны и глиальные клетки проводили в течение 10 дней, причем среду заменяли с интервалами приблизительно 3 дня. После этого периода клетки были большей частью прикрепившимися ко дну камеры и могли быть отделены от дна чашки кратковременной трипсинизацией аналогично процедуре, описанной ранее для стволовых клеток.
Пример 4
Доказательство образования нейронных клеток-предшественников, нейронов и глиальных клеток
Для последующей иммуногистохимической характеристики клеток-мишеней, индуцированных дедифференцированными программируемыми стволовыми клетками, стволовые клетки, образуемые из моноцитов (105 клеток/покровное стекло), наносили на стеклянные покровные стекла (20 мм × 20 мм), которые помещали на дно 6-луночных планшетов (диаметр камер 30 мм) и культивировали с питательной средой (2 мл на планшет). После дифференцировки соответствующих клеток-мишеней их фиксировали следующим образом: после удаления питательной среды (супернатанта) культивируемые клетки-мишени фиксировали добавлением 2 мл метанола, который действовал на протяжении 10 минут. Затем этанол удаляли пипетированием и луночные планшеты промывали два раза ЗФР (2 мл в каждом случае). После этого клетки могли быть окрашены комплексом АРААР red с использованием способа, описанного Cordell J.L., et al., «Immunoenzymatic labeling of monoclonal antibodies using immune complexes if alkaline phosphatase and monoclonal anti-alkaline phosphatase (APAAP complexes)», J. Histochem. Cytochem. 32: 219-229 (1994). Если нет другого указания, добавляемое первичное антитело разбавляли 1:100 ЗФР и в каждом случае 200 мкл этой концентрации антител пипетировали в каждую из 6 лунок.
Клетки-предшественники нейронов детектировали окрашиванием клеток антителом против антигена S100, см. среднюю фотографию фиг.1 (х200).
Нейроны детектировали по специфической экспрессии синаптофизина МАР2 (ассоциированного с микротрубочками белка 2) или нейрофиламента 68 с использованием соответствующих специфических антител (первичного антитела, разбавленного 1:300 ЗФР), правая фотография фиг.1 (х200).
Глиальные клетки, такие как, например, астроциты, идентифицировали детектированием GFAP (ассоциированного с глиями фибриллярного белка) (с первичным антителом, разбавленным 1:200 ЗФР), левая фотография фиг.1 (х200).
Разделение нейронов и глиальных клеток проводили с использованием антител, специфических против МАР2 (нейроны) или GFAP (глиальные клетки), при помощи MACS (Активированного магнитом сортинга клеток) в соответствии со способом, описанным, например, в Carmiol S., «Cell Isolation and Selection», Methods of Tissue Engineering, Academic Press 2002, Chapter 2, Pages 19-35.
Эти типы клеток, визуализированные окрашиванием, показаны на фиг.1.
Пример 5
Получение эндотелиальных клеток из дедифференцированных взрослых стволовых клеток моноцитарного происхождения
Для культивирования эндотелиальных клеток в качестве матрикса использовали Matrigel® (Beckton and Dickinson, Heidelberg, DE). Этот матрикс состоит из фибронектина, ламинина и коллагенов I и IV.
Замороженный матрикс медленно оттаивали при 4°С в холодильнике на протяжении периода 12 часов. Во время этого периода его состояние изменялось, т.е. первоначально твердый матрикс становился губчато-жидким. В этом состоянии его вводили в 48-луночный планшет (с диаметром лунки 10 мм) таким образом, что дно каждой лунки было покрыто.
После нанесения планшет выдерживали в течение 30 минут при комнатной температуре пока гель не отверждался на дне в виде прикрепленного слоя.
Затем приблизительно 1х102 клеток на лунку инкубировали на Matrigel® с добавлением питательной среды (как описано в примере 2).
Спустя 4-5 дней появились первые тяжи трубчатых клеток, которые развивались спустя 6-8 дней в трехмерные сетки клеток. На этих клетках могли быть идентифицированы эндотелиальные маркеры CD31 и фактор VIII с использованием соответствующих специфических первичных антител (200 мкл, в каждом случае разбавленных 1:100 ЗФР).
В альтернативном способе этот ожиженный матрикс наносили на протез сосуда, который затем покрывали дедифференцированными программируемыми взрослыми стволовыми клетками в соответствии с примером 2. Спустя приблизительно 6 дней мог быть идентифицирован газон эндотелиальных клеток, который покрывал этот протез кольцевым образом.
Эти эндотелиальные клетки, визуализированные окрашиванием соответствующими эндотелий-специфическими антителами (см. выше), показаны на фиг.2. На средней фотографии эти клетки показаны после 5 дней инкубирования на Matrigel®. Первые трубчатые тяжи объединяют отдельные агрегаты клеток. Темнокоричневые окрашенные клетки экспрессируют антиген CD31 (увеличение х200 с желтым фильтром). Спустя 8 дней имеется увеличивающееся образование трехмерных сетчатых структур (окрашивание антителом против антигена CD31, х200 с желтым фильтром). После 12 дней вновь дифференцированные CD31+-клетки, которые культивировали на Matrigel®, образуют подобную сосуду трехмерную трубку со стенкой многослойной структуры, которая уже морфологически напоминает сосуд. Можно видеть, что теперь почти все эти клетки экспрессируют антиген CD31 (окрашивание на CD31, х400, голубой фильтр), правая фотография.
Пример 6
Получение жировых клеток (адипоцитов)
А: Для программирования/дифференцировки взрослых стволовых клеток в соответствии с примером 2 в жировые клетки сначала готовили кондиционированную среду. Для этой цели 20 г аутологичной жировой ткани, т.е. жировой ткани из того же самого человека-донора, из крови которой происходили также моноциты, обрабатывали следующим образом.
Сначала жировую ткань измельчали в чашке Петри, и кусочки измельченной ткани пропускали через сито (с диаметром отверстий 100 мкм).
Полученную таким образом суспензию затем переносили в чашку Петри с диаметром 100 мм и добавляли 10 мл среды DMEM, содержащей 30 мг коллагеназы типа II. Эту смесь оставляли на приблизительно 60 минут при комнатной температуре (22°С ± 2°С), чтобы дать возможность коллагеназе подействовать на жировые клетки.
Затем эту смесь переносили в пробирки Falcon на 50 мл, и эти пробирки центрифугировали в течение 10 минут при 1800 об/мин.
После центрифугирования супернатант выбрасывали, а клеточный осадок, состоящий из адипоцитов и клеток-предшественников, помещали в 8 мл среды следующего состава и инкубировали в чашках Петри (с диаметром 100 мм) в течение 10 дней при 37°С в термостате:
Раствор инсулина содержал 18 мг инсулина (Sigma 1-0259), растворенного в 2 мл смеси уксусная кислота-вода (состоящей из 40 мл Н2О и 0,4 мл ледяной уксусной кислоты). Раствор инсулина разбавляли 1:10 смесью уксусная кислота-вода.
Во время инкубирования на протяжении 10 дней кондиционированная жировыми клетками среда (FCCM) образовывала супернатант. В каждом случае после 2-4 дней супернатант заменяли свежей питательной средой. FCCM, полученную во время каждой смены среды, подвергали стерильному фильтрованию и хранили при -20°С. Затем 10 мл FCCM, описанной выше, вводили в чашку Петри (с диаметром 100 мм) вместе с приблизительно 106 стволовых клеток в соответствии с примером 2. Первые клетки-предшественники, содержащие вакуоли жира, становились видимыми спустя 4 дня (фиг.3А). Спустя 6 дней появлялись одиночные адипоциты, которые могли быть окрашены Судановым красным (фиг. 3В и С). Спустя 10 дней наблюдали типичную агрегацию и образование скоплений этих клеток, которые на этой стадии можно было уже наблюдать макроскопически в виде жировой ткани (фиг.3D).
Таким образом, жировые клетки, визуализированные окрашиванием на фиг.3А-3D, очень сильно отличаются от контролей 3Е и 3F: фиг.3Е показывает клетки моноцитарного происхождения, которые культивировали в питательной среде (указанной в примере 2) в течение 6 дней, но без добавления IL-3 и 2-меркаптоэтанола к этой питательной среде. После этого следовало добавление FCCM. Эти клетки были неспособны дифференцироваться в жировые клетки. Фиг.3F показывает клетки, которые культивировали в течение 6 дней с полной средой (в соответствии с примером 2) и которые затем обрабатывали еще в течение 6 дней питательной средой вместо FCCM (в соответствии с примером 2). Таким образом, FCCM содержит компоненты, которые являются необходимыми для обеспечения сигнала для дифференцировки в жировые клетки.
Окрашивание клеток Судановым красным на фиг.3А, В, С и D выполняли в соответствии со способом, описанным Patrick Jr., C.W., et al., «Epithelial Cell Culture: Breast» в Methods of Tissue Engineering, Academic Press 2002, Chapter 4, Pages 141-149.
В: Кроме фенотипирования жировых клеток окрашиванием Судановым красным проводили молекулярно-биологическую характеристику этих жировых клеток на уровне мРНК для проверки, подвергается ли генетическая программа этих жировых клеток, после соответствующего программирования используемой кондиционированной жировыми клетками средой, соответствующему изменению и могут ли быть идентифицированы типичные транскрипты информационной рибонуклеиновой кислоты (мРНК), описанные для жировых клеток, в жировых клетках, программированных из программируемых моноцитов. Две мРНК-последовательности, типичные для метаболизма жировых клеток, из выделенных проб РНК из дедифференцированных программируемых стволовых клеток моноцитарного происхождения амплифицировали с использованием полимеразной цепной реакции (ПЦР) и в параллельной тест-смеси, амплифицировали из мРНК программированных жировых клеток, а именно мРНК «активированного пролиферацией пероксисом рецептора гамма» (PPARG) (Tontonoz P., et al. «Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid activated transcription factor», Cell 79: 1147-1156 (1994), кодовый номер доступа банка генов NM_005037) и мРНК «лептина (гомолога ожирения, мышь)» (Zhang Y., et al. «Positional cloning of the mouse obese gene and its human homologue», Nature 372: 425-432 (1994), кодовый номер доступа банка генов NM_000320).
Выделение РНК, необходимое для этой цели, способ обратной транскрипции и условия ПЦР-амплификации желаемых мРНК-последовательностей были такими же, как подробно описанные в данной области, см. Ungefroren H., et al., «Human pancreatic adenocarcinomas express Fas and Fas ligand yet are resistant to Fas-mediated apoptosis», Cancer Res. 58: 1741-1749 (1998).
Для этой цели соответствующие праймеры, продуцируемые для ПЦР-амплификации, выбирали таким образом, что прямой и обратный праймеры связываются с мРНК-последовательностями, гомологичные районы которых в хромосомном гене лежат в двух различных экзонах и отделены друг от друга большим интроном. Таким образом может гарантироваться то, что полученный фрагмент амплификации происходит из мРНК, содержащейся в данной клетке, а не из последовательности, присутствующей в хромосомной ДНК. В частности, следующие праймерные последовательности были выбраны для PPAR-γ и для лептина:
PPAR-γ: прямой праймер: 265-288 (соответствующий последовательности гена в экзоне 1), обратный праймер: 487-465 (соответствующий последовательности гена в экзоне 2), это приводит к фрагменту амплификации 487-265 п.н. = 223 п.н., см. фиг.3G. Как также показано на фиг.3G, следы транскрибированной PPAR-γ-специфической мРНК могут быть уже идентифицированы в программируемой стволовой клетке и в линии опухолевых клеток HL-60 (линии клеток промиелоидного лейкоза человека), хотя со значительно более узкими сигнальными полосами, чем в самих жировых клетках. В противоположность этому специфический для жировых клеток белок лептин мог быть детектирован в жировых клетках, происходящих из программируемых стволовых клеток, на уровне мРНК с помощью ПЦР с обратной транскриптазой.
Программируемые стволовые клетки (прогр. стволовые клетки), используемые в качестве контроля, и линии опухолевых клеток HL-60 человека, Панк-1 и WI-38, не транскрибируют лептин. В качестве отрицательных контролей одновременно определяли все пробы без добавления обратной транскриптазы (жировая клетка/-ОТ) и Н2О-пробы. Посредством идентификации гена «домашнего хозяйства» GAPDH в положительных контролях подтверждается, что соответствующие стадии ПЦР-амплификации проводились надлежащим образом в отдельных смесях.
Пример 7
Получение печеночных клеток (гепатоцитов)
А: Для программирования дедифференцированных программируемых стволовых клеток моноцитарного происхождения в соответствии с примером 2 в клетки печени сначала готовили кондиционированную среду. Для этой цели 40 г ткани печени человека обрабатывали следующим образом.
Сначала ткань печени промывали несколько раз в ЗФР для, по существу, удаления эритроцитов. Затем эту ткань измельчали в чашке Петри и инкубировали с раствором для диссоциации в течение приблизительно 45 минут при комнатной температуре. Раствор для диссоциации состоял из 40 мл ЗФР (забуференного фосфатом солевого раствора), 10 мл раствора трипсина, разбавленного 1:10 ЗФР, и 30 мг коллагеназы типа II (Rodbel M., et al., J. Biol. Chem. 230: 375 (1964)). После 45-минутного инкубирования кусочки ткани пропускали через сито (см. пример 6).
Затем эту смесь переносили в пробирки Falcon на 50 мл, заполняли до 50 мл ЗФР и центрифугировали в течение 10 минут при 1800 об/мин.
После центрифугирования супернатант отбрасывали и осадок клеток, содержащий клетки печени, снова промывали 50 мл ЗФР и центрифугировали. Полученный таким образом супернатант опять отбрасывали, а осадок клеток помещали в 25 мл среды следующего состава и инкубировали в сосудах для культуры клеток (с объемом 250 мл) в течение 10 дней при 37°С в термостате.
Эта питательная среда содержала, кроме того, 5 мкг (10 нг/мл) эпидермального фактора роста (Pascall I.C. et al., J. Mol. Endocrinol. 12: 313 (1994)). Состав раствора инсулина был такой же, какой описан в примере 6.
Во время инкубирования, длящегося 10 дней, образовывалась кондиционированная клетками печени среда (LCCM) в виде супернатанта. Этот супернатант заменяли свежей питательной средой после 2-4 дней соответственно. Соответствующую LCCM, полученную во время смены среды в каждом случае, подвергали стерильному фильтрованию (фильтр с размерами пор 0,2 мкм) и хранили при -20°С.
Затем 1х106 дедифференцированных стволовых клеток культивировали с 10 мл среды следующего состава в чашке Петри (с диаметром 100 мм) или культуральной колбе.
Фактор роста гепатоцитов (Kobayashi Y. et al., Biochem. Biophys. Res. Commun. 220: 7 (1996)) использовали в концентрации 40 нг/мл. Спустя несколько дней можно было наблюдать морфологические изменения в направлении плоских, многоугольных моно- или диплоидных клеток (фиг.4А). Спустя 10-12 дней гепатоциты, возникающие из дедифференцированных стволовых клеток, могли быть идентифицированы иммуногистохимическим детектированием печень-специфического антигена альфа-фетопротеина (Jacobsen G.K. et al., Am. J. Surg. Pathol. 5: 257-66 (1981)), как показано на фиг.4В и 4С.
В: Кроме фенотипирования гепатоцитов иммуногистохимической идентификацией альфа-фетопротеина проводили молекулярно-биологическую характеристику этих гепатоцитов на уровне мРНК для проверки, подвергается ли генетическая программа стволовых клеток, после соответствующего программирования используемой кондиционированной клетками печени средой, соответствующему изменению и могут ли быть идентифицированы типичные транскрипты информационной рибонуклеиновой кислоты (мРНК), описанные в качестве типичных для клеток печени, в гепатоцитах, возникающих из стволовых клеток данного изобретения. Для этой цели исследовали присутствие пяти различных мРНК-последовательностей, типичных для гепатоцитов, при помощи полимеразной цепной реакции (ПЦР) в пробах выделенной РНК из дедифференцированных программируемых стволовых клеток моноцитарного происхождения и в параллельной тест-пробе из клеток печени, полученных программированием этих стволовых клеток. В частности, исследовали присутствие мРНК альбумина Homo sapiens (Lawn R.M., et al., «The sequence of Human serum albumin cDNA and its expression in E. coli». Nucleic Acids Res. 9: 6103-6114 (1981), кодовый номер доступа банка данных: NM-000477), мРНК альфа-фетопротеина (Morinaga T., et al., «Primary structures of human alpha-fetoprotein and its mRNA». Proc. Natl. Acad. Sci. USA 80: 4604-4608 (1983), кодовый номер доступа банка данных: V01514), мРНК карбамилфосфатсинтетазы I человека (Haraguchi Y., et al. «Cloning and sequence of a cDNA encoding human carbamyl phosphate synthetase I: molecular analysis of hyperammonemia». Gene 107: 335-340 (1991), кодовый номер доступа банка данных: D90282), мРНК фактора свертывания II Homo sapiens (Тромбин, F2) (Degen S.J. et al. «Characterization of the complementary deoxyribonucleic acid and gene coding for human prothrombin». Biochemistry 22: 2087-2097 (1983), кодовый номер доступа банка данных: NM-000506), мРНК фактора свертывания VII Homo sapiens (сывороточного ускорителя превращения протромбина, F7) (NCBI Annotation Project. Direct Submission, 06-Feb-2002, National Center for Biotechnology Information, NIH, Bethesda, MD 20894, USA, кодовый номер доступа банка данных: МХ-027508).
Выделение РНК, необходимое для этого способа ПЦР с обратной транскрипцией, и условия ПЦР-амплификации желаемых мРНК-последовательностей были такими же, как подробно описанные в данной области, см. Ungefroren H., et al., «Human pancreatic adenocarcinomas express Fas and Fas ligand yet are resistant to Fas-mediated apoptosis», Cancer Res. 58: 1741-1749 (1998).
Соответствующие праймеры для ПЦР-амплификации выбирали таким образом, что прямой и обратный праймеры связываются с мРНК-последовательностями, гомологичные районы которых в хромосомном гене лежат в двух различных экзонах и отделены друг от друга большим интроном. Таким образом может гарантироваться то, что полученный фрагмент амплификации происходит из мРНК, содержащейся в данной клетке, а не из последовательности, присутствующей в хромосомной ДНК.
Были выбраны последовательности праймеров, указанные ниже; результаты соответствующих ПЦР-анализов воспроизведены на фиг.4D. Дедифференцированные программируемые стволовые клетки данного изобретения обозначены как «прогр. стволовые клетки», а гепатоциты, полученные программированием этих стволовых клеток, обозначены как «прогр. гепатоцит».
Альфа-фетопротеин: прямой праймер: 1458-1478 (соответствующий последовательности гена в экзоне 1), обратный праймер: 1758-1735 (соответствующий последовательности гена в экзоне 2), это приводит к фрагменту амплификации 1758-1458 п.н. = 391 п.н., см. фиг.4D.
Как показано на фиг.4, программируемая стволовая клетка (прогр. стволовая клетка), которая сама не содержит идентифицируемых специфических мРНК-транскриптов для альфа-фетопротеина, может быть программирована в гепатоцит (прогр. гепатоцит), который содержит этот мРНК-транскрипт (положительная полоса с молекулярной массой 301 п.н.). Это объясняет также иммуногистохимическую детектируемость альфа-фетопротеина, показанную на фиг.4В и 4С. Положительные контроли, а именно ткань печени человека и ткань линии опухолевых клеток человека НерG2, также транскрибируют альфа-фетопротеин-специфическую мРНК, как подтверждают полосы 301 п.н.
Альбумин: прямой праймер: 1450-1473 (соответствующий последовательности гена в экзоне 1), обратный праймер: 1868-1844 (соответствующий последовательности гена в экзоне 2), это приводит к фрагменту амплификации 1868-1450 п.н. = 419 п.н., см. фиг.4D.
Фиг.4D показывает следы транскрибированной альбумин-специфической мРНК уже в программируемой стволовой клетке, тогда как гепатоциты, полученные программированием стволовых клеток, и нормальная ткань печени, а также линии опухолевых клеток человека НерG2, которые обе использовали в качестве положительных контролей, в сильной степени экспрессируют эту мРНК, как можно видеть по явным полосам.
Карбамилфосфатсинтетаза I: прямой праймер: 3135-3157 (соответствующий последовательности гена в экзоне 1), обратный праймер: 4635-4613 (соответствующий последовательности гена в экзоне 2), это приводит к фрагменту амплификации 4635-3135 п.н. = 1500 п.н., см. фиг.4D.
Карбамилфосфатсинтетаза I представляет фермент, специфический для гепатоцитов, который играет важную роль в метаболизме мочевины в «цикле мочевины». Эта детоксицирующая функция гарантируется функционированием гепатоцитов. Как показывает фиг.4D, как в гепатоцитах, генерируемых из программируемых стволовых клеток, так и в положительных контролях (ткани печени человека и линии опухолевых клеток HepG2) могут быть идентифицированы полосы мРНК (1500 п.н.), специфические для карбамилфосфатсинтетазы I. Несколько более слабая экспрессия этих полос мРНК для программируемых гепатоцитов (прогр. гепатоцит) обусловлена отсутствием субстрата, доступного в культуральной чашке.
Фактор свертывания II: прямой праймер: 1458-1481 (соответствующий последовательности гена в экзоне 1), обратный праймер: 1901-1877 (соответствующий последовательности гена в экзоне 2), это приводит к фрагменту амплификации 1901-1458 п.н. = 444 п.н., см. фиг.4D.
Это также гепатоцит-специфический белок, который может быть детектирован только в программируемом гепатоците (прогр. гепатоцит) и в положительном контроле из ткани печени человека на уровне мРНК по полосе экспрессии 444 п.н., тогда как программируемая стволовая клетка (прогр. стволовая клетка) не обнаруживает этой полосы, т.е. этот ген не переносится в нее, как можно видеть на фиг.4D.
Фактор свертывания VII: прямой праймер: 725-747 (соответствующий последовательности гена в экзоне 1), обратный праймер: 1289-1268 (соответствующий последовательности гена в экзоне 2), это приводит к фрагменту амплификации 1289-725 п.н. = 565 п.н., см. фиг.4D.
Как и в случае фактора свертывания II, этот белок также транскрибируется в программируемых гепатоцитах (прогр. гепатоцит) и в положительном контроле (ткань печени человека) (см. полосы при 565 п.н.), хотя слабее, чем фактор свертывания II. Ни программируемая стволовая клетка, ни отрицательный контроль (Н2О) не обнаруживают этой специфической полосы мРНК.
Глицеринальдегиддегидрогеназа: этот ген, также называемый «геном домашнего хозяйства», может быть детектирован в любой эукариотической клетке и служит в качестве контроля, проводится ли ПЦР-амплификация правильно во всех пробах; его определяют параллельно и он происходит от добавления некоторого количества РНК из соответствующих проб клеток.
В качестве негативного контроля пробы Н2О одновременно определяли во всех тестах. Если Н2О не загрязнена РНК, амплифицированный продукт не образуется во время ПЦР и нет детектируемой полосы (таким образом, этот вариант служит в качестве противоконтроля).
Пример 8
Получение клеток кожи (кератиноцитов)
Для программирования дедифференцированных стволовых клеток моноцитарного происхождения в соответствии с примером 2 в клетки кожи сначала готовили кондиционированную среду. Для этой цели 1-2 кв. см. полной кожи человека обрабатывали следующим образом.
Материал кожи сначала освобождали от подкожного слоя в стерильных условиях. Затем эту ткань промывали в целом 10 раз ЗФР в стерильном контейнере энергичным встряхиванием. После 2-го промывания ткань опять освобождали от отсоединившихся остатков соединительной ткани.
Затем материал кожи помещали в чашку Петри с диаметром 60 мм, смешивали с 3 мл раствора трипсина, разбавленного 1:10 ЗФР, и нарезали на небольшие кусочки (приблизительно 0,1-1 мм3). После этого 3 мл раствора трипсина, разбавленного 1:1000 ЗФР, опять добавляли к этой смеси и смесь инкубировали при 37°С в течение 60 минут при прерывистом встряхивании.
Затем более крупным частицам давали осесть, и супернатант, содержащий кератиноциты, выливали и центрифугировали при 800 об/мин в течение 5 минут. Полученный теперь супернатант извлекали пипетированием, а осадок клеток помещали в 3 мл среды следующего состава и инкубировали в чашках Петри (с диаметром 100 мм) в течение 15 дней в термостате при 37°С.
Эта питательная среда содержала 5 мкг эпидермального фактора роста (точное описание см. в примере 7) и 5 мг гидрокортизона (Ref. Merck Index: 12, 4828).
Во время 15-дневного периода инкубирования кондиционированная кератиноцитами среда КССМ образовывалась в виде супернатанта. Этот супернатант заменяли свежей питательной средой после 2-4 дней в каждом случае. КССМ, полученную во время каждой смены среды, подвергали стерильному фильтрованию и хранили при -20°С.
Затем 1х106 дедифференцированных стволовых клеток культивировали с 10 мл среды следующего состава в чашке Петри (с диаметром 100 мм) или культуральной колбе.
Фактор роста кератиноцитов использовали в концентрации 25 нг/мл, как описано Finch et al., Gastroenterology 110: 441 (1996).
Через несколько дней можно было наблюдать морфологическое изменение в этих клетках. Спустя 6 дней могли быть детектированы кератиноцит-специфические антигены, цитокератин 5 и 6, которые оба связывались использованным первичным антителом (Exp. Cell. Res. 162: 114 (1986)) (фиг.5А). Спустя 10 дней уже имело место клеточное сцепление явно более крупных клеток в культуре, которое позволяло идентифицировать видимую комбинацию в виде ткани конфлюентных клеток (фиг.5В).
Пример 9
Получение инсулинпродуцирующих клеток из дифференцированных программированных стволовых клеток
Получение инсулинпродуцирующих клеток проводили в культуральных сосудах с плоскими стенками и объемом приблизительно 250 мл (сосудах для культуры клеток Т75). Приблизительно 5 × 106 клеток, полученных в соответствии с примером 13, суспендировали в приблизительно 5 мл культуральной среды, указанной ниже (среды для дифференцировки для инсулинпродуцирующих клеток), и после введения в эти сосуды смешивали с дополнительным количеством (15 мл) культуральной среды. Для дифференцировки этих клеток сосуды инкубировали в горизонтальном положении в термостате при 37°С и 5% СО2.
Культуральная среда (модифицированная согласно Rameya V.K. et al., Nature Medicine, 6 (3), 278-282 (2000)):
Эта питательная среда дополнительно содержала эпидермальный фактор роста в количестве 10 нг/мл и фактор роста гепатоцитов в количестве 20 нг/мл.
В пределах первого часа клетки прикрепляются ко дну культурального сосуда. Дифференцировку стволовых клеток подвергали мониторингу относительно продуцирования инсулина. Для этой цели культуральную среду сменяли с интервалами приблизительно 2-3 дня, каждый раз собирали клеточный супернатант и замораживали при -20°С. Клетки, прикрепившиеся ко дну культурального сосуда, могли быть отделены трипсинизацией, как описано в примере 2.
Содержание инсулина супернатанта, собранного в различное время, измеряли при помощи ELISA (твердофазного иммуноферментного анализа) для инсулина человека (Bruhn H.D., Fölsch U.R. (Eds.), Lehrbuch der Labormedizin: Grundlagen, Diagnostik, Klinik Pathobiochemie [Textbook of Laboratory Medicine, Principles, Diagnosis, Clinical Pathobiochemistry] (1999), Page 189) и сравнивали с контрольным опытом, т.е. определением показателя среды. Эти результаты, представленные на фиг.8, показывают, что эти клетки достигали максимального уровня продукции инсулина после 14 дней в культуре. Количества инсулина, продуцируемые клетками, обработанными в ходе дифференцировки, увеличивались после 14 дней до 3 мкЕ/мл, тогда как в контрольной среде инсулин не детектировался. Столбцы на фиг.8 обозначают, каждый, три отдельные величины, каждая из которых определена из трех независимых отдельных экспериментов.
После определения продуцирования инсулина в депрограммированных стволовых клетках, которые были дифференцированы в инсулинпродуцирующие клетки в соответствии с данным изобретением, определяли долю инсулинпродуцирующих клеток, которые все еще экспрессировали моноцит-специфический поверхностный антиген CD14 также спустя 3 недели после проведения дедифференцировки. Было обнаружено, что на большой части этих клеток (приблизительно 30-40%) моноцит-специфический антиген CD14 был детектируемым также после 3 недель.
Пример 10
Альтернативный способ получения гепатоцитов из дедифференцированных программируемых стволовых клеток
В качестве альтернативы использованию кондиционированной гепатоцитами среды (LCCM), описанному в примере 7, дифференцировку стволовых клеток в гепатоциты индуцировали питательной средой (На), указанной ниже. Получение гепатоцитов из стволовых клеток, в свою очередь, происходило в культуральных сосудах с плоскими стенками и объемом приблизительно 250 мл (культуральные сосуды Т75). Приблизительно 5 × 106 клеток, полученных в соответствии с примером 13, вводили в приблизительно 5 мл усовершенствованной культуральной среды, указанной ниже (На, среда для дифференцировки для гепатитов), и после введения в сосуды смешивали с дополнительным количеством (15 мл) культуральной среды. Для дифференцировки этих клеток сосуды инкубировали в горизонтальном положении в термостате при 37°С и 5% СО2.
Среда для дифференцировки для гепатоцитов (На) (модифицированная согласно Schwarz et al., «Multipotent adult progenitor cells from bone marrow differentiate into functional hepatocyte-like cells», J. Clin. Invest. 10 (109), 1291-1302 (2002)):
Эта питательная среда содержала также фактор-4 роста фибробластов (FGF-4) в количестве 3 нг/мл.
В пределах первого часа клетки прикрепляются ко дну культурального сосуда. Дифференцировку стволовых клеток подвергали мониторингу относительно продуцирования альбумина. Для этой цели культуральную среду сменяли с интервалами приблизительно 2-3 дня, каждый раз собирали клеточный супернатант и замораживали при -20°С. Клетки, прикрепившиеся ко дну культурального сосуда, могли быть отделены трипсинизацией, как описано в примере 2.
Содержание альбумина супернатанта, собранного в различное время, измеряли при помощи ELISA (твердофазного иммуноферментного анализа) для альбумина человека (согласно протоколу Bethyl Laboratories Inc. и согласно Schwarz et al., loco citato) и сравнивали со слепым опытом, т.е. определением показателя среды. Результаты, представленные на фиг.9, показывают, что продуцирование альбумина этими клетками во время периода 14-28 дней в культуре оставалось приблизительно постоянным. Измерение проводили в дни 0 (контрольный показатель среды), 14, 21, 28 и 30 относительно времени добавления среды На. Величины, определенные в каждом случае, были равны приблизительно 5 нг/мл, 450 нг/мл, 425 нг/мл, 440 нг/мл и 165 нг/мл. Столбцы на фиг.9 обозначают, каждый, три отдельные величины, каждая из которых определена из трех независимых отдельных экспериментов.
Пример 11
Определение коэкспрессии альбумина и моноцит-специфического антигена CD14 в гепатоцитах, полученных из дедифференцированных стволовых клеток
Определение коэкспрессии альбумина и моноцит-специфического антигена CD14 в гепатоцитах, полученных из дедифференцированных стволовых клеток, проводили, с одной стороны, двойным окрашиванием (А) и, с другой стороны, при помощи FACS-анализа (В).
А) Стволовые клетки данного изобретения, дифференцированные в гепатоциты в соответствии с примером 10, культивировали на покровных стеклах в 6-луночном планшете и фиксировали метанолом, как описано в примере 4. Затем проводили двойное окрашивание для детектирования одновременной экспрессии антигена CD14 (маркера фенотипа моноцитов), с одной стороны, и альбумина (печень-специфического маркера), с другой стороны.
Для этой цели эти клетки сначала инкубировали, как описано в примере 4, с первичным антителом против альбумина человека (антителом морской свинки против альбумина человека) в разведении 1:50 в ЗФР. Затем после стадии промывания клетки инкубировали в течение 45 минут со вторым антителом, мышиным антителом против иммуноглобулина крысы, которое связывается с антителами морской свинки, также в разведении 1:50 в ЗФР. Затем проводили процесс окрашивания в соответствии с примером 4 с использованием способа Cordell J.L., et al., (loco citato) с комплексом АРААР red.
Затем для второй стадии окрашивания клетки инкубировали с первичным антителом, мышиным антителом против CD14 человека, и после стадии промывания в соответствии с примером 4 окрашивали с использованием набора АВС Streptavidin KIT of Vectastain (Vector) по способу Hsu S.M., et al., «The use of antiavidin antibody and avidin-biotin-peroxidase complex in immunoperoxidase technics», Am. J. Clin. Pathol. 75 (6): 816-821 (1981) с DAB-комплексом (коричневым) (Vector Laboratories).
Затем проводили контрастное окрашивание гемалауном, как описано в примере 4, с последующей заливкой в глицериновый желатин Кайзера.
Результаты показаны на фиг.10. Эта фигура показывает экспрессию антигена CD14 в виде коричневого цвета, которая медленно уменьшается параллельно морфологическому превращению клеток в гепатоциты, тогда как экспрессия альбумина в виде красного цвета увеличивается с увеличением созревания гепатоцитов. Фотография № 4 на фиг.10 показывает эти клетки после трехнедельной стимуляции кондиционированной гепатоцитами средой.
В) Параллельно с двойным мечением стволовые клетки, дифференцированные в гепатоциты в соответствии с данным изобретением, подвергали FACS-анализу (клеточному сортингу с активацией флуоресценции).
Стволовые клетки, дифференцированные в гепатоциты в соответствии с данным изобретением согласно примеру 19, сначала собирали механическим отделением этих клеток от культурального сосуда при помощи клеточного скребка (скрепера). Эти клетки осторожно вымывали из сосуда ЗФР и дважды промывали, каждый раз в 10 мл раствора ЗФР. Для этой цели суспензии клеток в растворе ЗФР вводили в центрифужную пробирку на 15 мл и осаждали при 1600 об/мин. Полученный осадок клеток разбавляли ЗФР таким образом, что в 100 мкл ЗФР присутствовали точно 1 × 105 клеток.
Затем 10 мкл каждого из FITC-меченых анти-CD14-антител (BD Pharmingen) или FITC-меченых анти-альбумин-антител (Beckman) и FITC-меченых неспецифических мышиных антител против IgG1 человека добавляли к этой суспензии клеток. После периода инкубирования 20 минут эти клетки дважды ресуспендировали в 500 мл ЗФР и, каждый раз, осаждали в течение 5 минут при 1600 об/мин и затем, наконец, помещали в 200 мкл ЗФР. После ресуспендирования этих клеток измеряли флуоресценцию при помощи проточного цитометра BD FACScalibur от компании BD Biosciences (Franklin Lakes, NJ) (см. Bruhn H.D., Fölsch U.R. (Eds.), Lehrbuch der Labormedizin: Grundlagen, Diagnostik, Klinik Pathobiochemie [Textbook of Laboratory Medicine, Principles, Diagnosis, Clinical Pathobiochemistry], 395-403 (1999); и Holzer U. et al., «Differential antigen sensitivity and costimulatory requirments in human Th1 and Th2 antigen-specific CD4(+) cells with similar TCR avidity», J. Immunol. 170 (3): 1218-1223 (2003)). Оценку результатов проводили с использованием программы Microsoft WinMDI со ссылкой на Marquez M.G., et al., «Flow cytometric analysis of intestinal intra-epithelial lymphocytes in a model of immunodeficiency in Wistar rats», Cytometry 41 (2): 115-122 (2000).
Результаты FACS-анализа представлены на фиг.11. Эта фигура показывает экспрессию CD14 (верхний ряд) и антигена альбумина (нижний ряд), которую измеряли в дедифференцированных моноцитах (левый столбец) и в стволовых клетках, дифференцированных в гепатоциты в соответствии с изобретением (правый столбец). В дедифференцированных моноцитах можно было обнаружить сильную экспрессию CD14, но не экспрессию альбумина, тогда как в гепатоцитах, развившихся из дедифференцированных моноцитов, была детектируемой слабая экспрессия CD14 и очень сильная экспрессия альбумина.
Пример 12
Применение in vivo дедифференцированных программируемых стволовых клеток моноцитарного происхождения
Для выяснения до какой степени программируемые стволовые клетки in vivo после инъекции через воротную вену в печень генетически идентичного животного-реципиента подвергаются специфической дифференцировке через источники сигналов, присутствующие в печени, печени самок крыс LEW сначала обрабатывали ретрорсином для ингибирования гепатоцитов, присутствующих в печени (клетки паренхимы печени) в отношении их активности к пролиферации (см. Lacone E., et al., «Long-term, near-total liver replacement by transplantation of isolated hepatocytes in rats treated with retrosine». Am. J. Path. 153: 319-329 (1998)).
Для этой цели крысы LEW получали 30 мг пирролизидинового алкалоида ретрорсина, инъецируемого внутрибрюшинно два раза в пределах 14 дней. Затем проводили 80% резекцию печеней, обработанных таким образом, с последующим введением 5 × 105 программируемых стволовых клеток в 1 мл ЗФР в воротную вену остатка печени. Стволовые клетки получали, как описано в примере 2, из моноцитов самцов крыс LEW. Спустя пять дней после введения стволовых клеток проводили пункционную биопсию (трепанобиопсию) печени для гистологической оценки печени и для детектирования типов клеток, дифференцированных из стволовых клеток, посредством in situ-гибридизации с использованием флуоресценции (FISH) с Y-хромосома-специфическими зондами, как подробно описано в Hoebee B. et al., «Isolation of rat chromosome-specific paint probes by bivariate flow sorting followed by degenerate oligonucleotide primed-PCR», Cytogenet. Cell. Genet. 66: 277-282 (1994).
Фиг.7А показывает Y-хромосома-положительные (красные точки в ядре клетки) гепатоциты, происходящие из стволовых клеток самцов LEW, на 5-й день после инъекции в портальную вену в предобработанные ретросином подвергнутые 80%-ной резекции печени самок-реципиентов животных. Селективное удаление той же самой печени на 25 день после инъекции стволовых клеток показывает дифференцировку этих стволовых клеток в гепатоциты, эндотелиальные клетки и эпителии желчных протоков (фиг.7В). В этой временной точке печень уже достигала ее нормального размера и >90% этих клеток имели Y-хромосому. Из этих данных можно сделать вывод, что инъецированные сингенные программируемые стволовые клетки моноцитарного происхождения способны in vivo выполнять полное восстановление печени с нормальной метаболической функцией. Фиг.7С показывает, в этой связи, кривые выживаемости Каплана-Мейера (n=4 на группу) обработанных стволовыми клетками крыс-реципиентов в сравнении с необработанными крысами-реципиентами после введения ретрорсина и 80%-ной резекции печени.
Функциональные параметры метаболизма билирубина и аммиака (NH3) доказывают полную метаболическую функциональность долгосрочно выживающих обработанных стволовыми клетками животных (фиг.7D и 7Е).
Пример 13
Размножение и дедифференцировка моноцитов в колбах для культуры ткани
Культивирование и размножение моноцитов, с одной стороны, и дедифференцировку этих клеток, с другой стороны, в крупном масштабе проводили в культуральных сосудах в той же самой среде, которую использовали также для культивирования в луночных планшетах (см. пример 2). Питательная среда содержала 2,5 мкг/500 мл M-CSF и 0,2 мкг/500 мл интерлейкина-3 (IL-3).
Моноциты, выделенные в примере 1, переносили на дно культуральных сосудов, имеющих объем 250 мл и плоские стенки (сосуды для культуры клеток Т75). Приблизительно 10 × 106 клеток переносили в каждый из сосудов, и в сосуды наливали 20 мл указанной выше питательной среды. Определение данного количества клеток для точного введения на колбу проводили в соответствии с известными процедурами, см. Hay R.J., «Cell Quantification and Characterization» в Methods of Tissue Engineering, Academic Press (2002), Chapter 4, pages 55-84.
Сосуды для культуры клеток инкубировали в термостате при 37°С в течение 6 дней. Спустя 24 часа клетки оседали на дно сосудов. Супернатант удаляли каждый второй день, и сосуды наполняли 20 мл свежей питательной среды.
На 6 день сосуды промывали дважды 10 мл ЗФР, каждый, после того как питательную среду предварительно удаляли пипетированием из этих сосудов. Таким образом удаляли все клетки, которые не прикрепились ко дну сосудов. Затем клетки, растущие прикрепленными ко дну сосудов, удаляли со дна сосудов стерильным клеточным скребком. После этого отделенные клетки удаляли из сосудов промыванием ЗФР, объединяли в пробирке Falcon на 50 мл и центрифугировали при 1800 об/мин в течение 10 минут. После этого супернатант отбрасывали, и осадок ресуспендировали в свежей среде RPMI 1640 (2 мл/105 клеток).
Данная суспензия клеток могла использоваться непосредственно для дифференцировки в различные клетки-мишени.
Альтернативно эти клетки смешивали с ДМСО/ФТС, в качестве среды для замораживания, после центрифугирования и подвергали глубокому замораживанию при концентрации 106 клеток/мл.
Среда для замораживания содержала 95% ФТС и 5% ДМСО. Приблизительно 106 клеток помещали в 1 мл этой среды и охлаждали с использованием следующих стадий:
30 минут на льду;
2 часа при -20°С в предварительно охлажденном боксе styropor;
24 часа при -80°С в styropor;
хранили в пробирках в жидком азоте (N2) при -180°С.
Реферат
Изобретение относится к области биотехнологии и может быть использовано для получения взрослых дедифференцированных, программируемых стволовых клеток из моноцитов человека. Моноциты культивируют в культуральной среде, которая содержит M-CSF и IL-3. Полученные дедифференцированные, программируемые стволовые клетки используют для получения фармацевтических композиций и для получения клеток-мишеней и ткани-мишени. Изобретение позволяет быстро и доступно получить безопасный для пациента материал для заместительной клеточной терапии. 20 н. и 19 з.п. ф-лы, 11 ил., 1 табл.

Комментарии