Расширенная адоптивная клеточная терапия - RU2703438C2
Код документа: RU2703438C2
Чертежи



















































































































































































































































































































































































































































































Описание
Область изобретения
Настоящее изобретение относится к области наук о жизни и медицины. В частности, изобретение относится к терапии злокачественных опухолей человека. Более конкретно, настоящее изобретение относится к онколитическим аденовирусным векторам отдельно или вместе с терапевтическими композициями для терапевтического применения и терапевтических способов для злокачественных опухолей. В одном из аспектов настоящее изобретение относится к отдельному введению адоптивной клеточной терапевтической композиции и онколитических аденовирусных векторов. Кроме того, настоящее изобретение относится к фармацевтическому набору и фармацевтической композиции, в которых используют онколитические аденовирусные векторы.
Предпосылки изобретения
Для лечения злокачественных опухолей постоянно разрабатывают новую терапию. Адоптивная клеточная терапия (ACT) представляет собой эффективный подход для лечения злокачественной опухоли, а также для лечения других заболеваний, таких как инфекции и реакция трансплантат против хозяина. Перенос адоптивных клеток представляет собой пассивный перенос выращенных ex vivo клеток, наиболее часто клеток иммунного происхождения, в организм-хозяин с целью перенести иммунологическую функциональность и характеристики трансплантата. Перенос адоптивных клеток может быть аутологическим, как обычно бывает в адоптивной T-клеточной терапии, или аллогенным, что типично для лечения инфекций или реакции трансплантат против хозяина. Клинически обычные варианты осуществления этого подхода включают перенос или способствующих развитию иммунитета или толерогенных клеток, таких как лимфоциты, пациентам для того, чтобы или усиливать иммунитет против вирусов и злокачественной опухоли или способствовать толерантности в условиях аутоиммунного заболевания, такого как диабет I типа или ревматоидный артрит.
В отношении терапии злокачественных опухолей, подход ACT разработан в 1980-х небольшим числом групп, работающих в США, одной из ведущих групп является Steven Rosenberg с коллегами, работающие в NCI. Адоптивный перенос аутологических инфильтрирующих опухоль лимфоцитов (TIL) или генетически модифицированных мононуклеарных клеток периферической крови использовали для успешного лечения пациентов с запущенными солидными опухолями, такими как меланома, а также пациентов с CD19-экспрессирующими гематологическими злокачественными новообразованиями. При ACT наиболее широко используемыми типами клеток являются T-клетки, иногда отсортированные по CD8+, но другие вариации включают CD4+ клетки, NK-клетки, δ-γ T-клетки, регуляторные T-клетки и мононуклеарные клетки периферической крови. Клетки могут быть немодифицированными, например, как в TIL терапии, или генетически модифицированными. Существует два общих пути для достижения генетического нацеливания T-клеток на опухолеспецифичные мишени. Один заключается в переносе T-клеточного рецептора с известной специфичностью (TCR терапия) и с совпадающим лейкоцитарным антигеном человеческого типа (HLA, известный как главный комплекс гистосовместимости у грызунов). Другой заключается в модификации клеток искусственными молекулами, такими как химерные антигенные рецепторы (CAR). Этот подход не зависит от HLA и более гибок по отношению к молекулам-мишеням. Например, можно использовать одноцепочечное антитело, и CAR также может содержать костимуляторные домены. Однако мишени CAR клеток должны быть на мембране клеток-мишеней, тогда как модификации TCR могут использовать внутриклеточные мишени.
В первые десять лет разработка ACT была сосредоточена на TIL. TIL находят в опухолях, что подсказывает, что опухоли запускают иммунный ответ у организма-хозяина. Эту так называемую опухолевую иммуногенность опосредуют опухолевые антигены. Эти антигены отличают опухоль от здоровых клеток, тем самым предоставляя иммунологический стимул.
Например, в US2003194804 A1 описан способ усиления реактивности T-клеток в отношении опухолевых клеток за счет применения TIL. В US2003194804 A1 T-клетки подвергают воздействию определенного агента и повторно вводят пациенту. Агент способен снижать или предотвращать экспрессию или взаимодействие эндогенного Notch или Notch-лиганда в T-клетке.
В US5126132 описан способ лечения злокачественной опухоли, где используют эффективное количество аутологических TIL и цитокина.
Diaz RM et al. (Cancer Res. 2007 Mar 15;67(6):2840-8) описывают увеличение уровней циркулирующих специфичных к опухолевому антигену T-клеток при использовании терапии переносом адоптивных T-клеток в комбинации с внутриопухолевой виротерапией вирусом везикулярного стоматита. При терапии переносом адоптивных T-клеток Diaz et al. использовали клетки OT1, т.е. искусственную моноклональную клеточную линию.
Даже не смотря на то, что в ранних испытаниях ACT имели место существенные примеры эффектов лечения и даже излечения, большинство пациентов не испытывали эффекта, и многие пациенты испытывали тяжелые побочные эффекты. Во время первых двух десятилетий адоптивной клеточной терапии, безопасность переноса клеток per se была в целом хорошей, но значительная токсичность и даже смертность была связана с сопутствующим лечением, используемым для того, чтобы усиливать терапию, включая предварительную химиотерапию и лучевую терапию, а также IL-2, используемый после переноса. Предварительное лечение используют для того, чтобы убивать супрессорные клетки, такие как регуляторные T-клетки и супрессоры миелоидного происхождения, в организме-хозяине для того, чтобы модулировать микроокружение опухоли и «создать пространство» для трансплантата. IL-2 используют после переноса для того, чтобы снижать анергию трансплантата и чтобы он разрастался.
В отношении эффективности есть простор для усовершенствования. В целом, гарантированы увеличенная специфичность клеточной терапии и достаточная ее способность уничтожать опухоли. В частности, при ACT известного уровня техники перенесенные клетки не способны переходить в опухоли, и даже если это происходит, они часто быстро становятся анергическими или иным образом неспособными убивать опухолевые клетки или разрастаться, что ведет к быстрому снижению числа клеток. Кроме того, злокачественные опухоли часто осуществляют понижающую регуляцию лейкоцитарного антигена человека (HLA) - известного как главный комплекс гистосовместимости у животных - в опухолевых клетках, таким образом, приводя к неспособности T-клеток к убийству, поскольку HLA необходим для презентирования опухолевых эпитопов T-клеточному рецептору.
Настоящее изобретение предусматривает эффективные инструменты и способы для терапевтических средств против злокачественных опухолей с применением переноса адоптивных клеток.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Цель настоящего изобретения состоит в том, чтобы предоставить простые способы и инструменты для преодоления вышеуказанных проблем неэффективной, небезопасной и непредсказуемой терапии злокачественных опухолей. Более конкретно, изобретение относится к новым способам и средствам для клеточной терапии. Целей изобретения достигают с помощью вирусных векторов, способов и приспособлений, которые отличаются тем, что заявлено в независимых пунктах формулы изобретения. Конкретные варианты осуществления изобретения раскрыты в зависимых пунктах формулы изобретения.
В настоящей заявке описаны конструкция рекомбинантных вирусных векторов, способы, связанные с вирусными векторами, и их применение на опухолевых клеточных линиях, животных моделях и у пациентов со злокачественными опухолями.
Изобретение основано на идее объединения онколитических аденовирусных векторов, кодирующих цитокины или аденовирусные векторы, с адоптивными клеточными терапевтическими средствами для лечения злокачественных опухолей новым и патентоспособным путем. Изобретение основано на неожиданных эффектах, т.е. следующих усовершенствованиях адоптивной T-клеточной терапии: i) рекрутирование перенесенных клеток в опухоль, ii) размножение перенесенных клеток в опухоли, iii) расширенная реактивность перенесенных клеток в опухоли (фиг. 20). В действительности, указанная комбинация вирусных векторов и цитокинов с адоптивными клеточными терапевтическими средствами дает более эффективные результаты на большем числе мишеней, чем можно предположить. Эффекты указанной комбинации вирусных векторов, содержащих цитокиновый трансген, с переносом адоптивных клеток являются синергическими по сравнению с эффектами только вирусных векторов, содержащих цитокиновый трансген, или только переноса адоптивных клеток.
Дополнительная цель настоящего изобретения состоит в том, чтобы предоставить комбинацию инфильтрирующих опухоль лимфоцитов (TIL) и трансгенного (получаемого из доставляемого вирусом трансгена) интерлейкина-2 (IL-2) для лечения злокачественного новообразования у человека. Приведенных выше и других целей и преимуществ по настоящему изобретению достигают с помощью способа лечения злокачественного новообразования у человека, который включает введение эффективного количества TIL и IL-2, с предварительной химиотерапией и/или лучевой терапией или без них, пациенту, страдающему злокачественной опухолью, чтобы вызывать регрессию или стабилизацию злокачественной опухоли.
Настоящее изобретение относится к способу лечения злокачественной опухоли у субъекта, где способ включает отдельное введение адоптивной клеточной терапевтической композиции и онколитических (способных к репликации в опухоли, но не в нормальных клетках) аденовирусных векторов, кодирующих по меньшей мере один цитокин, субъекту.
Настоящее изобретение дополнительно относится к онколитическому аденовирусному вектору, который кодирует по меньшей мере один цитокин, вместе с отдельной адоптивной клеточной терапевтической композицией для применения при лечении злокачественной опухоли.
Настоящее изобретение дополнительно относится к применению онколитического аденовирусного вектора, который кодирует по меньшей мере один цитокин, вместе с отдельной адоптивной клеточной терапевтической композицией при изготовлении лекарственного средства для лечения злокачественной опухоли у субъекта.
Настоящее изобретение также относится к онколитическому аденовирусному вектору для применения для увеличения эффективности адоптивной клеточной терапии или T-клеточной терапии у субъекта.
Также настоящее изобретение относится к применению онколитического аденовирусного вектора при изготовлении лекарственного средства для увеличения эффективности T-клеточной терапии у субъекта.
Также настоящее изобретение относится к способу увеличения эффективности адоптивной клеточной терапии или T-клеточной терапии у субъекта посредством введения онколитического аденовирусного вектора нуждающемуся в этом субъекту.
Настоящее изобретение также относится к фармацевтическому набору, который содержит адоптивную клеточную терапевтическую композицию и онколитические аденовирусные векторы, которые кодируют по меньшей мере один цитокин, где адоптивную клеточную терапевтическую композицию формулируют в первом составе и онколитические аденовирусные векторы, которые кодируют по меньшей мере один цитокин, формулируют во втором составе.
Кроме того, настоящее изобретение относится к онколитическому аденовирусному вектору, который содержит
1) остов нуклеиновой кислоты аденовируса серотипа 5 (Ad5), который содержит 5/3 химерный выступ волокна;
2) промотор E2F1 для опухолеспецифичной экспрессии E1A;
3) делецию 24 п. о. (D24) в Rb-связывающей константной области 2 аденовирусного E1;
4) делецию вирусных рамок считывания gp19k и 6.7k в последовательности нуклеиновой кислоты; и
5) последовательность нуклеиновой кислоты, которая кодирует по меньшей мере один цитокиновый трансген вместо удаленного gp19k/6.7k в области E3, что ведет к ассоциированному с репликацией контролю экспрессии трансгена под управлением вирусного промотора E3, где цитокин выбирают из группы, состоящей из интерферона α, интерферона β, интерферона γ, C5a комплемента, IL-2, TNFα, CD40L, IL-12, IL-23, IL-15, IL-17, CCL1, CCL11, CCL12, CCL13, CCL14-1, CCL14-2, CCL14-3, CCL15-1, CCL15-2, CCL16, CCL17, CCL18, CCL19, CCL19, CCL2, CCL20, CCL21, CCL22, CCL23-1, CCL23-2, CCL24, CCL25-1, CCL25-2, CCL26, CCL27, CCL28, CCL3, CCL3L1, CCL4, CCL4L1, CCL5 (RANTES), CCL6, CCL7, CCL8, CCL9, CCR10, CCR2, CCR5, CCR6, CCR7, CCR8, CCRL1, CCRL2, CX3CL1, CX3CR, CXCL1, CXCL10, CXCL11, CXCL12, CXCL13, CXCL14, CXCL15, CXCL16, CXCL2, CXCL3, CXCL4, CXCL5, CXCL6, CXCL7, CXCL8, CXCL9, CXCL9, CXCR1, CXCR2, CXCR4, CXCR5, CXCR6, CXCR7 и XCL2.
Кроме того, настоящее изобретение относится к онколитическому аденовирусному вектору серотипа 3 (Ad3), который содержит: делецию в области E3 и опухолеспецифичный промотор для экспрессии трансгена вместо удаленной области E3.
Кроме того, настоящее изобретение относится к фармацевтической композиции, которая содержит онколитический вектор по изобретению.
Также настоящее изобретение относится к способу лечения злокачественной опухоли у субъекта, где способ включает введение онколитического аденовирусного вектора по настоящему изобретению нуждающемуся в этом субъекту. Также настоящее изобретение относится к онколитическому аденовирусному вектору по настоящему изобретению для применения при лечении злокачественной опухоли.
Также настоящее изобретение относится к применению онколитического аденовирусного вектора по настоящему изобретению при изготовлении лекарственного средства для лечения злокачественной опухоли у субъекта.
Преимуществами приспособлений по настоящему изобретению являются расширенный терапевтический эффект и сниженные побочные эффекты. Тяжелые нежелательные явления, вплоть до смертей, предотвращены за счет увеличения эффективности, и антисупрессорные эффекты подхода авторов изобретения могут снижать необходимость предварительной химиотерапии и/или лучевой терапии, используемых в способах известного уровня техники для того, чтобы «создать пространство» для перенесенных клеток и снизить опухолевую иммуносупрессию. Также тяжелые нежелательные явления, вплоть до смертей, предотвращены, поскольку отдельное добавление IL-2, используемого в способах известного уровня техники для размножения и поддержания перенесенных клеток после их переноса пациенту, не требуется, если вирус его продуцирует во время репликации в опухоли. Также локальное продуцирование в опухоли может усиливать искомые эффекты IL-2 (стимуляция и размножение трансплантата), при этом снижая системное воздействие, которое является причиной нежелательных явлений. Настоящее изобретение предусматривает избирательное лечение при меньшей токсичности или повреждении здоровых тканей.
Также настоящее изобретение предусматривает неожиданные терапевтические эффекты за счет: i) Предоставление сигналов направленной миграции в опухоль, например, посредством инъекции вирусных векторов, которые содержат рекомбинантные цитокины в опухоли. Вирусная инъекция ведет к образованию цитокинов, релевантных для этого эффекта (в реакции на связывание вируса с патоген-ассоциированными рецепторами опознавания молекулярного паттерна), но значительно более выраженных эффектов можно достичь посредством дополнительного продуцирования наиболее релевантного цитокина в качестве трансгена из вируса. ii) Снижение толерантности посредством усиления сигналов опасности. Вирусная инъекция per se позволяет достигать этого посредством связывания с патоген-ассоциированными рецепторами опознавания молекулярного паттерна, но эффект можно усиливать посредством дополнительного продуцирования цитокина в качестве трансгена из вируса. iii) Индукция экспрессии HLA. Вирусная инфекция увеличивает экспрессию HLA, поскольку клетки пытаются презентировать вирусные эпитопы для установления противовирусного T-клеточного ответа. Неожиданно, это можно использовать для того, чтобы усиливать T-клеточную терапию против опухолевых эпитопов, которая требует, чтобы работал HLA. Эффект вируса, оказываемый на HLA, отчасти опосредован цитокинами; продуцирование указанного цитокина из вируса, таким образом, может повышать экспрессию HLA также в близлежащих опухолевых клетках в неожиданном варианте осуществления изобретения. iv) Индукция размножения клеток посредством устранения иммуносупрессии, опосредованного и присутствием вируса per se (также через патоген-ассоциированные рецепторы опознавания молекулярного паттерна), но расширенного с помощью продуцирования цитокинов (фиг. 48). Таким образом, этот подход позволяет решать критические проблемы, в настоящее время препятствующие адоптивной клеточной терапии.
КРАТКОЕ ОПИСАНИЕ ФИГУР
Далее изобретение описано более подробно посредством конкретных вариантов осуществления со ссылкой на приложенные фигуры, на которых:
На фиг. 1 представлено, что лечение с применением Ad5/3 химерного онколитического аденовируса увеличивало секрецию цитокинов и хемокинов в опухолях B16-OVA. Интерферон-γ может вызывать повышающую регуляцию экспрессии HLA (MHC) класса I, таким образом создавая фенотип опухолевой клетки, который могут эффективно распознавать TIL. Различные индуцируемые IFN-γ хемокины (такие как RANTES, MIP-1α и MCP-1) в рекрутирование клеток иммунной системы, что может способствовать активации и пролиферации TIL. Также направленная миграция TIL может быть расширена посредством повышающей регуляции этих хемокинов.
На фиг. 2 представлен контроль опухолевого роста после множества инъекций 5/3 химерного онколитического аденовируса с переносом адоптивных клеток или без него. Лечение только аденовирусом (A) оказывало небольшой эффект на опухолевый рост B16-OVA по сравнению с лечением PBS. Адоптивный перенос 500 000 (B) или 2 000 000 (C) опухолеспецифичных OT-I лимфоцитов в комбинации с вирусными инъекциями вел к значительному контролю опухолевого роста. Слабый терапевтический эффект, оказываемый на опухолевый рост при использовании только аденовируса или OT-I клеток в комбинации с PBS, подчеркивает главные недостатки терапии онколитическими вирусами и переносом адоптивных клеток, используемых в качестве отдельных средств, что подтверждает цель изобретения повысить эффективность адоптивной клеточной терапии с применением аденовируса.
На фиг. 3 представлено, что аденовирусные инъекции индуцируют направленную миграцию T-клеток в опухоли и увеличивают пролиферацию адоптивно перенесенных T-клеток в опухоли. A) Количество адоптивно перенесенных CD8+ CFSE+ клеток возрастало в опухолях и снижалось в крови, опустошая лимфатические узлы и селезенку мышей, которые получали лечение Ad, в сутки 1 после лечения, предположительно, из-за направленной миграции лимфоцитов. Общее количество CD8+ T-клеток оставалось высоким в опухолях, которые лечили аденовирусом, на всем протяжении эксперимента, что намекает на резистентность этих клеток к вредному опухолевому микроокружению и/или увеличенную пролиферацию CD8+ T-клеток. B) На сутки 6 и 14 клеточная пролиферация OT-I усиливалась в опухолях, которые лечили Ad, по сравнению с группой PBS, что наблюдали в виде большей фракции клеток, которая подвергалась клеточному делению (двигались в направлении M7), чем в группе PBS. Следовательно, лечение онколитическим аденовирусом индуцирует направленную миграцию и пролиферацию адоптивно перенесенных TIL. C) Пример того, как выполняли пропускание CFSE-положительных клеток. M0 обозначает клетки, которые не делились, M7 показывает клетки, которые делились достаточно для того, чтобы разводить CFSE ниже пределов, поддающихся обнаружению (больше чем 7 раз).
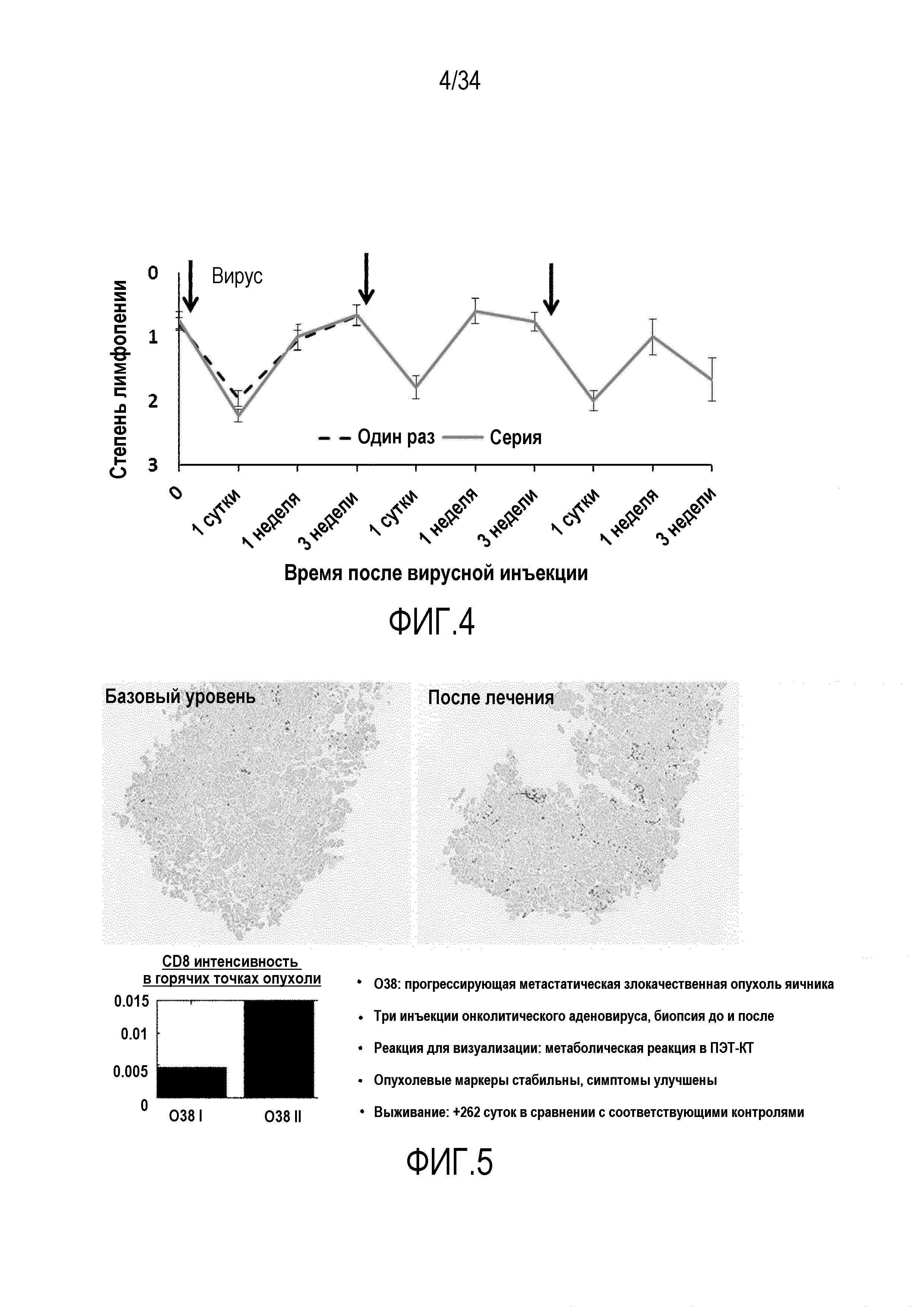
На фиг. 4 представлены данные для людей, которых лечили онколитическим аденовирусом, на сутки, указанные стрелками. Вирусная инъекция в опухоль вела к снижению лимфоцитов в крови, что отражает их направленную миграцию в опухоли.
На фиг. 5 представленные данные о том, что инъекция онколитического аденовируса в опухоль пациента-человека со злокачественной опухолью вызывала приток CD8+ T-клеток. Внутриопухолевая инъекция онколитического аденовируса ведет к накоплению CD8+ T-клеток в опухоли, которую оценивали по игольным биопсиям до и после лечения.
На фиг. 6 представлены результаты аденовирусных инъекции в комбинации с адоптивным переносом T-клеток. Мышам с подкожными опухолями B16-Ova адоптивно переносили 5×105OT1 лимфоцитов интраперитонеально и опухоли оставляли без лечения или инъецировали PBS или Ad5/3 (см. «Материалы и способы» в «Примерах»). В иммуносупрессорной модели B16-Ova, схожей с меланомой человека, адоптивный перенос OT1 клеток против Ova дает немного. Добавление вирусных инъекций значительно повышает эффективность (a). Возрастают CD8+ T-клетки (b). Эти клетки не являются T-клетками против Ova (c).
На фиг. 7 представлено существенное увеличение числа «естественных» противоопухолевых T-клеток из-за адоптивного переноса и вирусной инъекции. Мышам с подкожными опухолям B16-Ova адоптивно переносили 5×105OT1 лимфоцитов интраперитонеально и опухоли оставляли без лечения или инъецировали PBS или Ad5/3 (см. «Материалы и способы» в «Примерах»). Действия по адоптивному переносу + вирусные инъекции действуют как катализатор для размножения «других» T-клеток в опухоли и регионарных лимфатических узлах. (a) Trp2 CD8+ клетки в месте опухоли. (b) CD8+ клетки против gp100 в месте опухоли.
На фиг. 8 представлены активированные CD8+ клетки в опухоли и экспрессия TIM-3 в опухоли на сутки 14. Мышам с подкожными опухолями B16-Ova адоптивно переносили 5×105OT1 лимфоцитов интраперитонеально, и опухоли оставляли без лечения или инъецировали PBS или Ad5/3 (см. «Материалы и способы» в «Примерах»). Иммуносупрессия при иммунотерапии: увеличение числа T-клеток не является достаточным, если иммуносупрессию не устраняют. Имеют место больше активированных T-клеток в опухолях, которые лечили вирусами, и меньше иммуносупрессии.
На фиг. 9 представлено, что увеличение противоопухолевых T-клеток и снижение иммуносупрессии ведет к системному иммунитету против опухолевых антигенов. Мышам с подкожными опухолям B16-Ova адоптивно переносили 5×105 OT1 (a) или 2×106 (b) OT1 лимфоцитов интраперитонеально, и опухоли оставляли без лечения или инъецировали PBS или Ad5/3 (см. «Материалы и способы» в «Примерах»). Антигенная презентация усилена с помощью вируса: T-клетки работают лучше. Результатом является системный иммунитет против нескольких опухолевых эпитопов. (a) Экспрессия костимулиряторных молекул на дендритных клетках (CD11c+ CD80+ CD86+) в опухоли на сутки 14. (b) IFNg ELISPOT со спленоцитами на сутки 14.
На фиг. 10 представлено распределение OTI T-клеток после вирусной инъекции: тенденция к направленной миграции, но ее не достаточно для того, чтобы объяснить эффективность. (a) диаграмма, (b) животная модель, (c) опухоль.
На фиг. 11 показано, что подъем иммуносупрессии может индуцировать размножение клеток. Опухоли, которые лечили аденовирусом, содержали больше опухолеспецифичных лимфоцитов (OT-I клетки). В опухолях, которые лечили PBS, OTI клетки задерживались в фазе M5 (левая стрелка), тогда как в группе Ad они продолжали пролиферировать (правая стрелка).
На фиг. 12 представлена эффективность рекомбинантных цитокинов (без вируса) в комбинации с OT1 клетками.
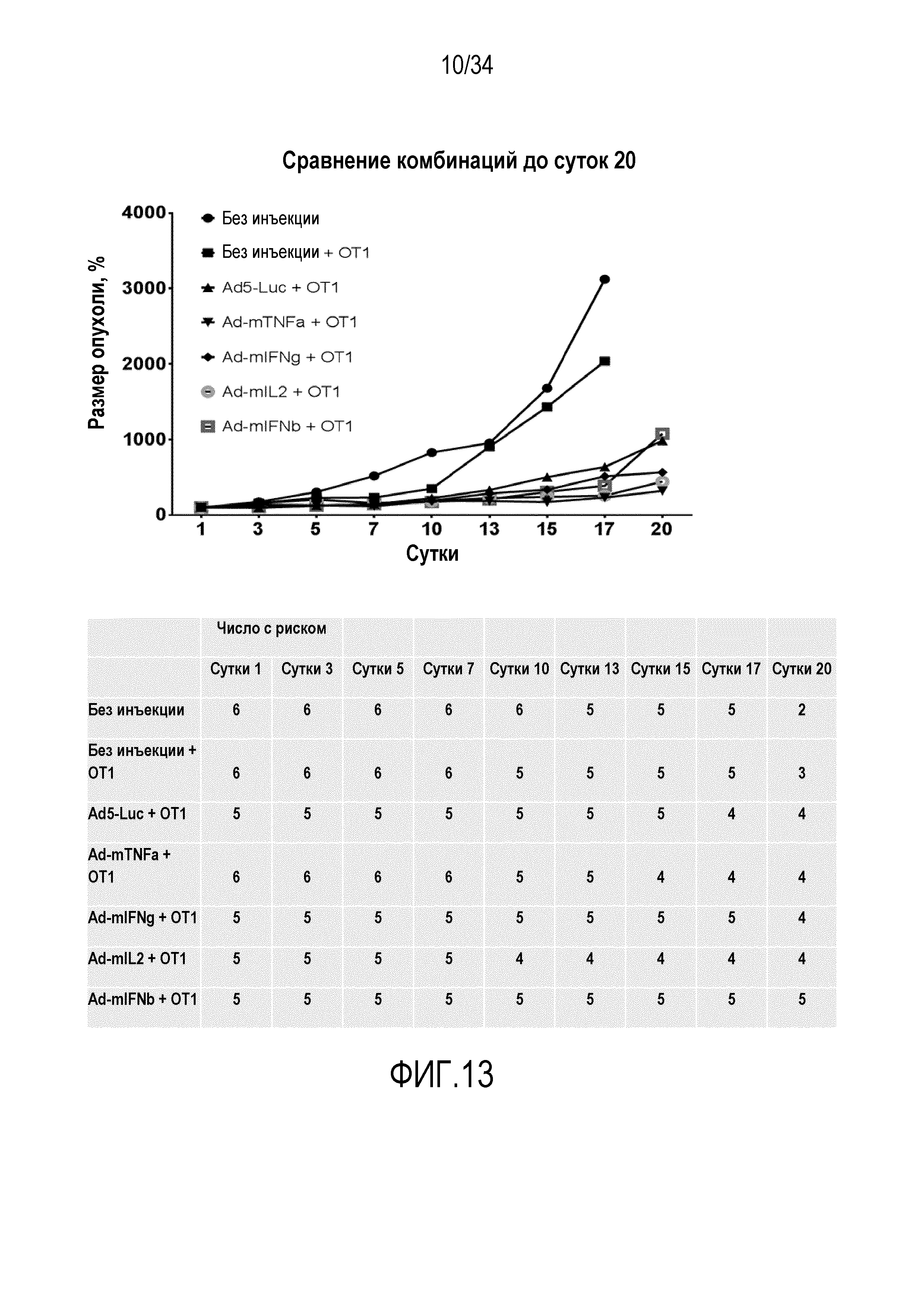
На фиг. 13 представлена противоопухолевая эффективность аденовирусов, вооруженных цитокинами, в комбинации с адоптивным переносом T-клеток. C57BL/6 мышей, несущих подкожные опухоли меланомы B16-OVA, лечили 1,5×106 CD8+ обогащенных OT-1 T-клеток интраперитонеально на сутки 1. Кодирующие цитокин аденовирусы или контрольный вирус Ad5-Luc1 инъецировали внутрь опухоли на сутки 1 и еженедельно после этого (1×109 вирусных частиц на опухоль). Объем опухоли вычисляли, как описано ранее (Bramante et al. Serotype chimeric oncolytic adenovirus coding for GM-CSF for treatment of sarcoma in rodents and humans. Int J Cancer. 24 декабря 2013 года) и размеры опухоли представляли в виде процентной доли, соответствующе суткам 1, которые соответствуют 100%. Фиг. «Число с риском»: число животных, оставшихся в каждой экспериментальной группе в заданный момент времени. Животных умерщвлял человек, когда опухоли преодолевали максимальный приемлемый размер или когда становились видны какие-либо признаки боли или дистресса.
На фиг. 14 представлены эффекты различных вирусов, оказываемые на размер опухоли.
На фиг. 15 представлены превосходные результаты аденовирусных векторов, которые содержат трансген mTNFa, в комбинации с OT1 T-клетками в виде уменьшения размера опухоли.
На фиг. 16 представлены превосходные результаты аденовирусных векторов, которые содержат трансген mIL3, в комбинации с OT1 T-клетками, в виде уменьшения размера опухоли.
На фиг. 17 представлена схема экспрессирующих C5a или TNF-α онколитических аденовирусов. Показаны некоторые важные признаки вирусов, включая место, куда вставляют трансгены.
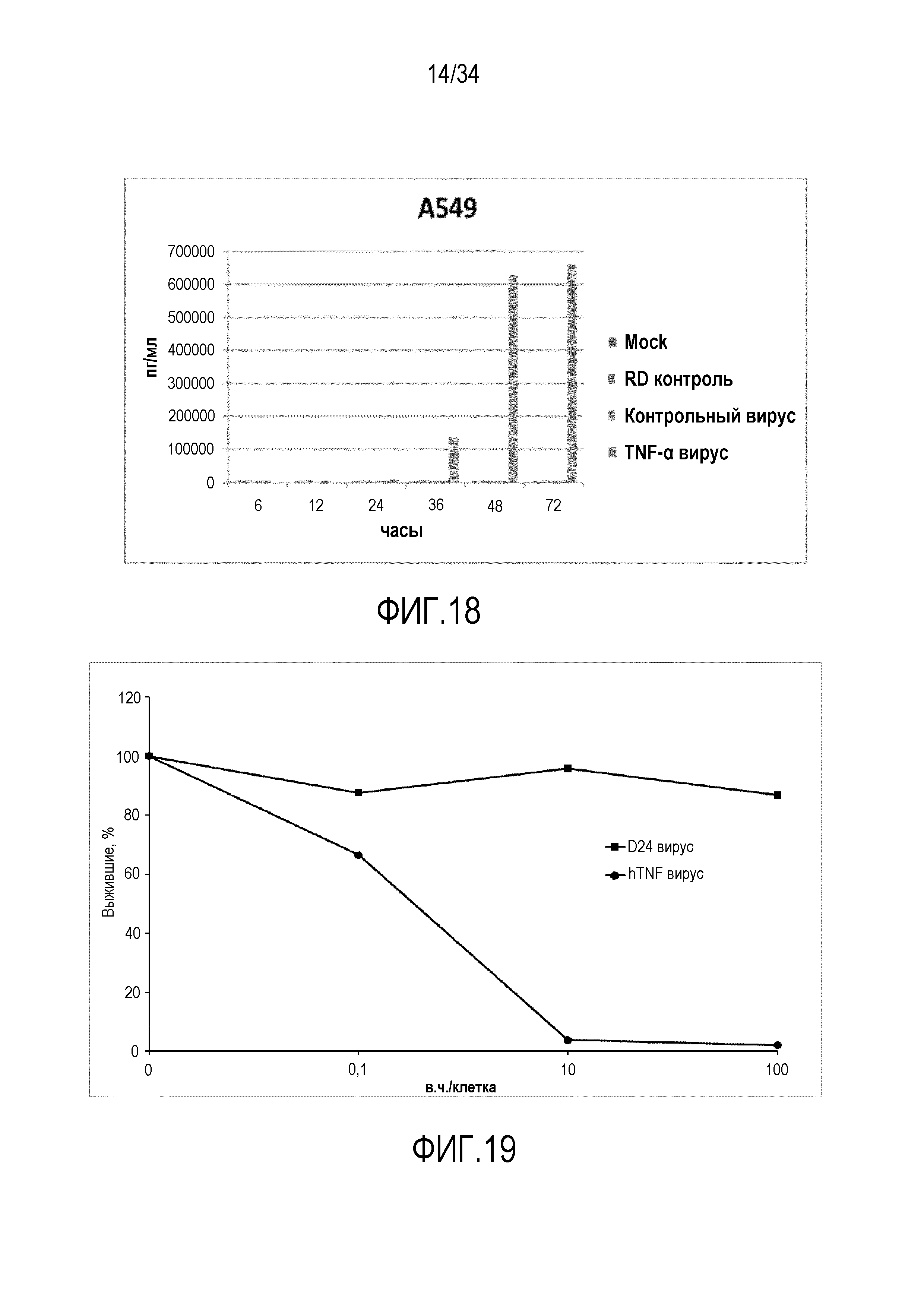
На фиг. 18 представлена экспрессия TNF-α с помощью онколитического аденовируса в клетках A549. Клетки инфицировали с применением 10 в. ч./клетка, среды собирали в указанные моменты времени и ELISA использовали для того, чтобы оценивать количество TNF-α в средах. Вирус индуцирует экспрессию и секрецию TNF-α инфицированными клетками.
На фиг. 19 представлена биологическая активность TNF-α, полученного посредством онколитического вооруженного TNF-α онколитического аденовируса. В этом анализе супернатант от инфицированных клеток использовали для того, чтобы стимулировать TNF-чувствительные клетки WEHI-13VAR, что подтверждает, что онколитический аденовирус управляет экспрессией функциональных цитокинов.
На фиг. 20 представлено дозозависимое уничтожение клеток злокачественной опухоли человека с помощью онколитического аденовируса. Как ожидали, в нечувствительных к TNF-α допускающих онколизис опухолевых клетках A549 или PC3 человека не наблюдали разницы между невооруженным контрольным вирусом и экспрессирующим TNF-α онколитическим аденовирусом, поскольку лишь онколизиса достаточно для того, чтобы уничтожать клетки. Однако поскольку человек TNF-α частично активен в клетках мыши, которые не допускают онколизис посредством аденовируса человека, TNF-α вносил вклад в более сильную цитотоксичность вируса, которую наблюдали в клетках B16-OVA мыши по сравнению с невооруженным вирусом. Вирус с дефектом репликации демонстрирует незначительную способность к уничтожению клеток.
На фиг. 21 представлено, что лучевая терапия проявляет синергизм с экспрессирующим TNF-α вирусом. A) схема режима лечения в этом эксперименте. Излучение (XRT) представляло собой облучение всего организма в дозе 2×2 Гр, и вирус использовали по 1×108 в. ч./опухоль, где каждая «голая» мышь несла два ксенотрансплантата A549. B) TNF-α вирус скрывает более высокую противоопухолевую активность, чем невооруженный родительский вирус. Поскольку вирус с дефектом репликации (RD) не уничтожает клетки A549 в культуре (фиг. 20), противоопухолевый эффект, который обеспечивает RD вирус in vivo, вероятно обусловлен врожденными иммунными ответами, в том числе цитокинами, естественными киллерными клетками и макрофагами, которые вызваны вирусными инъекциями. C) Экспрессирующий TNF-α вирус вызывает более выраженные противоопухолевые эффекты в комбинации с клинически релевантными дозами внешнего облучения пучком, что подтверждает клиническую трансплантируемость вооруженных цитокинами вирусов. Что важно, эти и предыдущие эксперименты показывают, что экспрессирующий TNF-α онколитический аденовирус способен к репликации и уничтожению клеток, что доказывает, что TNF-α не проявляется противовирусных эффектов против аденовируса.
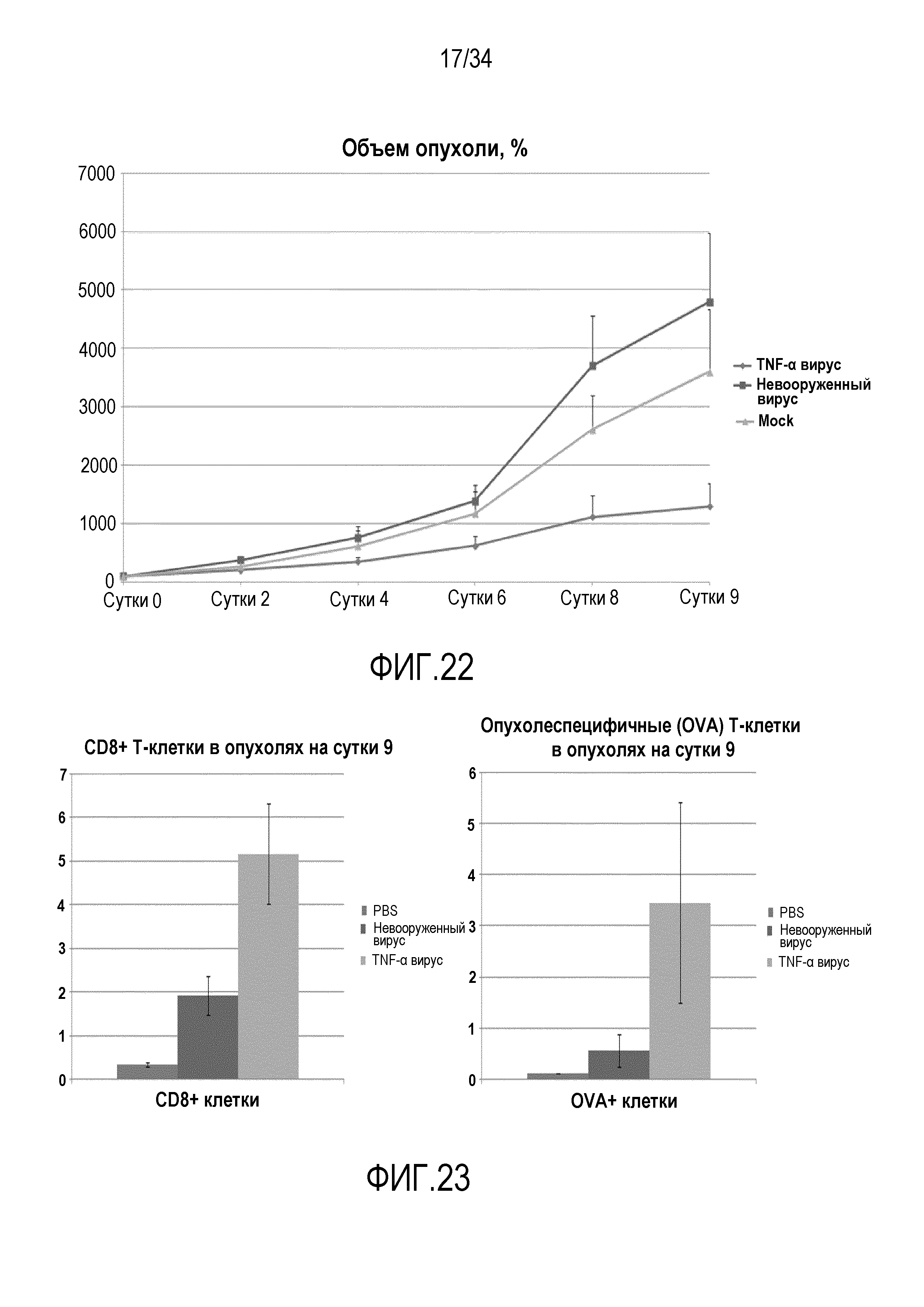
На фиг. 22 представлена противоопухолевая эффективность кодирующего hTNF-α аденовируса на опухолях B16-OVA. Этот эксперимент аналогичен тому, который изображен на фиг. 26 с применением C5a вируса, и демонстрирует, что экспрессия TNF-α дает более значительное терапевтическое преимущество по сравнению с невооруженным вирусом.
На фиг. 23 представлена расширенная индукция/экспансия опухолеспецифичных CD8+ T-клеток в опухолях, которые лечили вирусом, вооруженным цитокином (II). Опухоли в эксперименте, изображенном на фиг. 20, иссекали и обрабатывали для проточного цитометрического анализа, подобно эксперименту 11. Более высокую индукцию/число OVA-специфичных CD8+ T-клеток обнаруживали в опухолях, которые лечили TNF-кодирующим вирусом по сравнению с невооруженным контрольным вирусом, что вместе с C5a данными указывает на то, что рационально выбранные цитокины, экспрессируемые с помощью онколитического аденовируса, вместе с воспалением, вызванным вирусом, создают уникальное окружение опухоли, которое надежно поддерживает экспансию и активацию опухолеспецифичных T-клеток предположительно и в сравнении с фиг. 2B, C также с адоптивно перенесенными T-клетками.
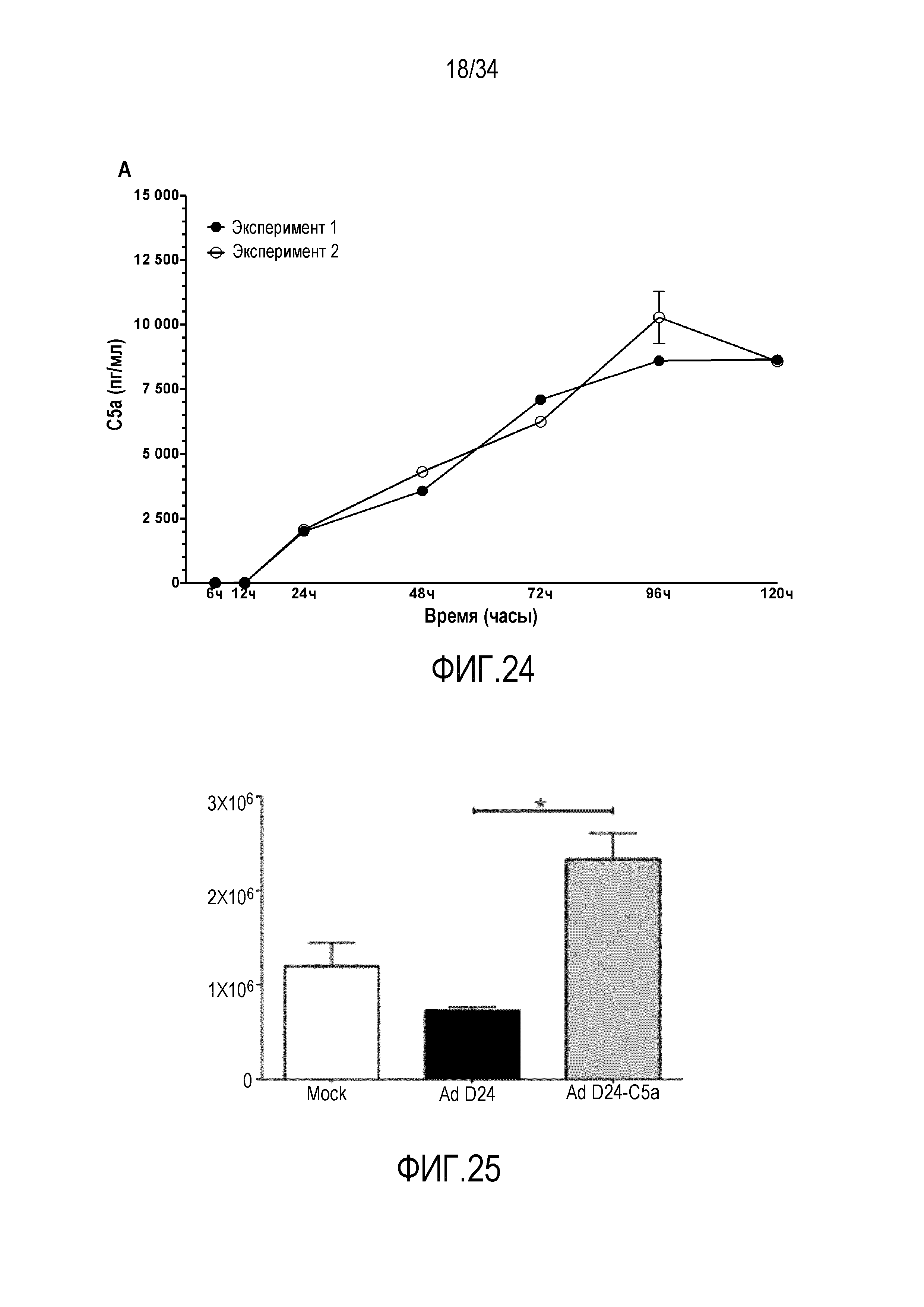
На фиг. 24 представлена экспрессия C5a в клетках A549. Клетки инфицировали 10 в. ч./клетка, среды собирали в указанные моменты времени и ELISA использовали для того, чтобы оценивать количество C5a в средах. Представлены результаты двух отдельных экспериментов.
На фиг. 25 представлены результаты анализа хемотаксиса in vitro. Количество моноцитов THP1 человека, проходящих через полупроницаемую мембрану в нижнюю камеру, которых привлекали хемокины в тестовых супернатанта, определяли количественно в соответствии с инструкциями производителя (набор Millipore QCM). C5a-экспрессирующий вирус высвобождает более сильные хемоаттрактивные факторы из инфицированных клеток, чем контрольный вирус. Результаты говорят в пользу применения вооруженного цитокинами вируса вместо невооруженного вируса.
На фиг. 26 представлена противоопухолевая эффективность AdD24-C5a in vivo. Развившиеся подкожные опухоли инъецировали на сутки 0, 2 и 4 по 1×109 в. ч. каждого вируса или 50 мкл PBS и объемы опухолей измеряли с помощью штангенциркуля. C5a-экспрессирующий вирус дает превосходный контроль опухоли по сравнению с контрольным вирусом. Поскольку аденовирус не реплицируется или не убивает клетки мыши, т.е. он не является цитолитическим в этой модели (Young AM et al. Mol Ther. 2012 Sep;20(9):1676-88, PMID: 22735379), в этих результатах недостаточно отражена надежная способность вооруженного цитокинами вируса усиливать иммунологические противоопухолевые эффекты, что решительно подтверждает идею его применения для увеличения эффективности адоптивной клеточной терапии.
На фиг. 27 представлена расширенная индукция/экспансия опухолеспецифичных CD8+ T-клеток в опухолях, которые лечили вирусом, вооруженным цитокином (I). Опухоли в эксперименте, представленном на фиг. 26, иссекали и обрабатывали для проточного цитометрического анализа. Отдельные клеточные суспензии окрашивали антителами против CD8 и пентамером против TCR против ova. C5a-экспрессия с помощью нецитолитического аденовируса индуцирует более высокие количества OVA-специфичных CD8 T-клеток в опухолях по сравнению с контрольными вирусами, невооруженным аденовирусом или вирусом, экспрессирующим антагонист C5a, что подтверждает применение вооруженного цитокинами вируса для увеличения числа адоптивно перенесенных T-клеток в опухолях. Также см. эксперимент 13.
На фиг. 28 представлена схема адаптивного T-клеточного ответа.
На фиг. 29 представлено, что адаптивно перенесенные T-клетки действуют в качестве катализатора («искры») для уже существующих T-клеток.
На фиг. 30 представлено, что адаптивная «искра» ведет к увеличению «естественных» противоопухолевых T-клеток.
На фиг. 31 представлен способ переноса адоптивных клеток.
На фиг. 32 показано, что с применением TILT технологии по настоящему изобретению можно избегать токсичного предварительного лечения (химиотерапия + лучевая терапия) и последующего лечения (IL-2 системно). (Механизмы технологии TILT см. на фиг. 48.)
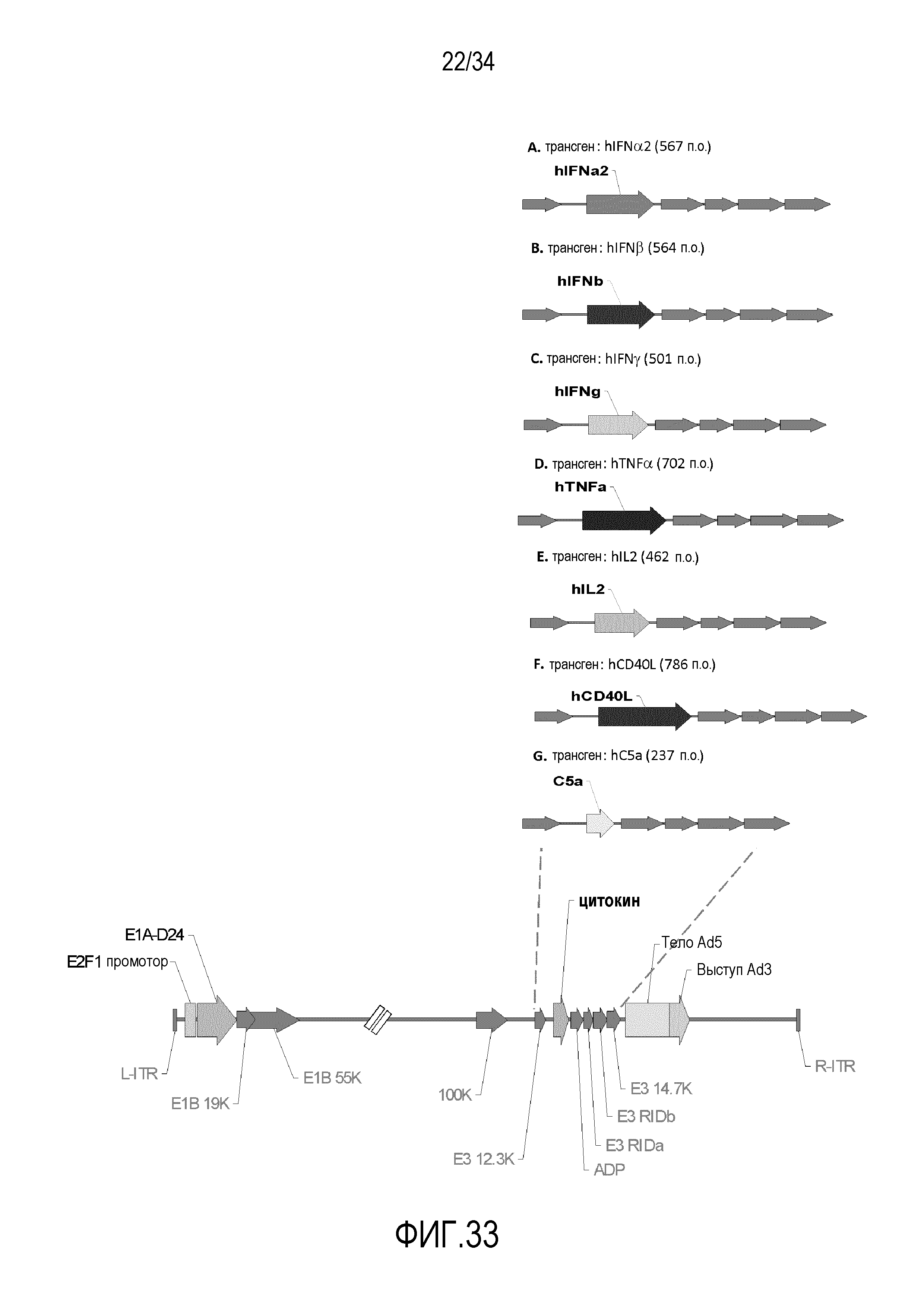
На фиг. 33 представлена схема новых вирусных конструкций, экспрессирующих один цитокин. Вирусный остов представляет собой аденовирус человека серотипа 5, помимо выступа волокна, который взят из серотипа 3. И отдельные и двойные трансгены находятся под транскрипционным контролем промотор E3 вируса. Оба трансгена помещают в удаленную область E3 для gp19k и 6.7k. Удаляют 24 аминокислоты белка E1A («D24») в константной области 2, что создает дефект связывания Rb. Экспрессия E1A находится под управлением промотора E2F. Некоторые генные области вируса представлены для справки.
На фиг. 34 представлена схема новых вирусных конструкций, экспрессирующих два цитокина. В одной версии, «сайт шунтирования рибосомы»/«сайт пропуска рибосомы»/«цис-действующий элемент гидролазы» (CHYSEL) помещают в виде слияний с сохранением рамки считывания между каждым цитокином. Цитокиновые вставки синтезируются в виде одного полипротеина, который проходит котрансляционное расщепление, давая два цитокина, результатом чего также являются несколько дополнительных аминокислот на 3'-конце первого цитокина и один пролин на 5' последнего цитокина, IL-2). В другой версии элемент IRES разделяет два цитокина, что ведет к синтезу цитокинов без дополнительных аминокислот.
На фиг. 35 представлена нуклеотидная и аминокислотная последовательности 2A.
На фиг. 36 представлена технология внутривенной доставки аденовируса TILT Biotherapeutics. Описанные выше аденовирусы TILT следует вводить пациентам внутрь опухоли для того, чтобы усиливать T-клеточную терапию (помечено 4a, 5a). Однако, не до всех опухоли могут быть достигнуты через внутриопухолевый путь. Таким образом, авторы настоящего изобретения разработали носитель для доставки на основе Ad3, который может достигать опухолей через внутривенный путь (помечено 4b, 5b).
На фиг. 37 представлена структура вектора Ad3-hTERT-E3del-CMV-CD40L. Нуклеотидная последовательность вирусного вектора Ad3-hTERT-E3del-CMV-CD40L представлена в SEQ ID № 30.
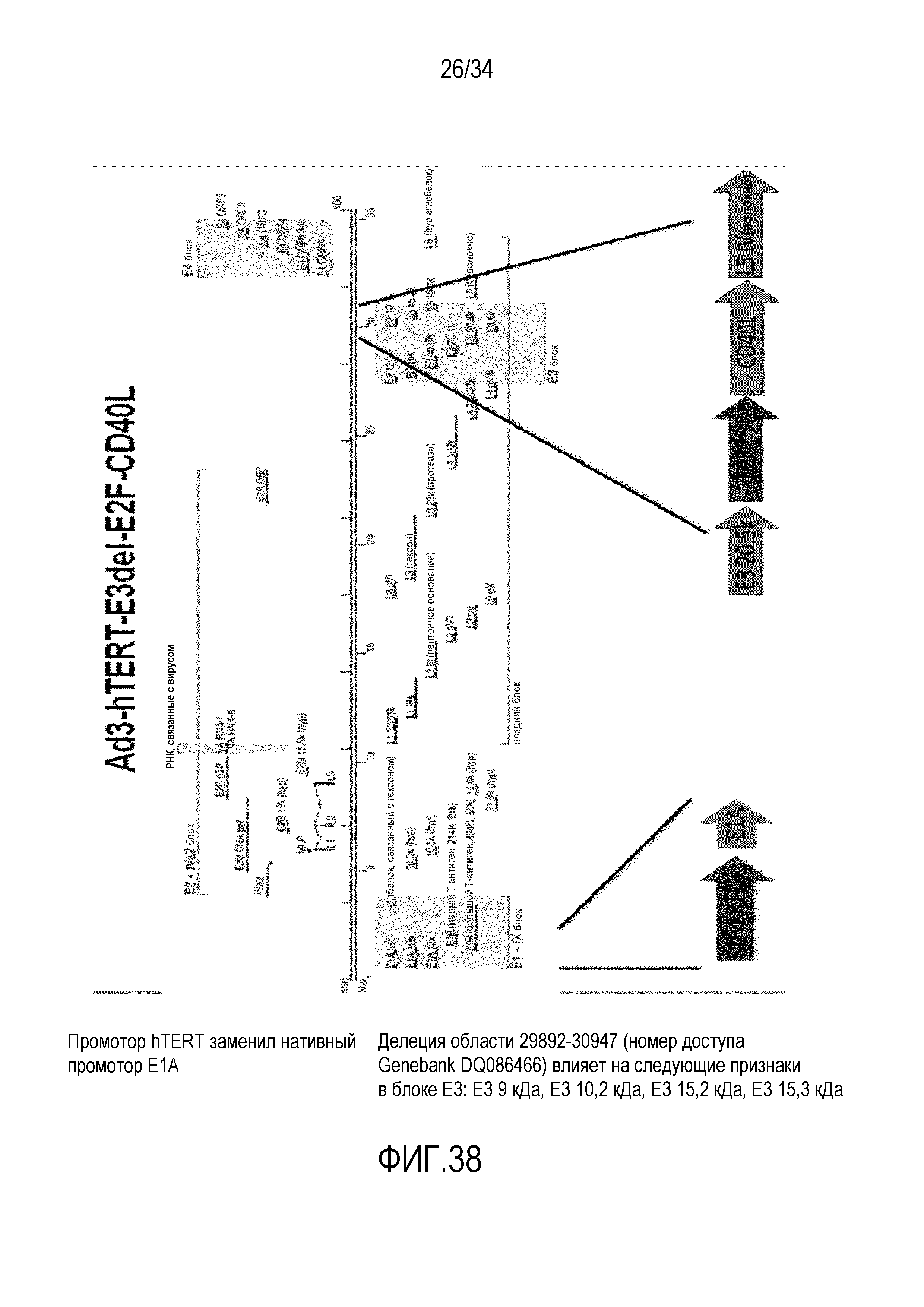
На фиг. 38 представлена структура вектора Ad3-hTERT-E3del-E2F-CD40L. Нуклеотидная последовательность вирусного вектора Ad3-hTERT-E3del-E2F-CD40L представлена в SEQ ID № 31.
На фиг. 39 представлен агарозный гель с вектором pWEA-Ad3-hTERT-CMV-CD40L, разрезанным рестрикционными ферментами. Правильный рестрикционный анализ клонированных вирусных векторов подтверждает правильную ДНК последовательность вируса.
На фиг. 40 представлен агарозный гель с вектором pWEA-Ad3-hTERT-E2F-CD40L, разрезанным рестрикционными ферментами. Правильный рестрикционный анализ клонированных вирусных векторов подтверждает правильную ДНК последовательность вируса.
На фиг. 41 представлена функциональность векторов E2F-CD40L и CMV-CD40L in vitro. На вертикальной оси логарифмическая шкала выхода относительного визуального титра TCID50(БОЕ/мл). На горизонтальной оси сутки после инфекции (d). Показано, что вирусы функциональны и способны инфицировать по меньшей мере некоторые опухолевые клеточные линии. Разведения вируса не выполняли в соответствии с титрами в. ч. Прогрессивная TCID50: вновь полученные вирусы сначала тестировали на прогрессивную TCID50 для того, чтобы определять, обладают ли они онколитическими свойствами. После девяти (9) суток инкубации инфекции становились видны во всех культуральных планшетах с клетками A549, что указывало на то, что все новые вирусы были функциональны. Во время следующих суток инфекции продолжали распространяться в соответствии с пипетированным количеством вируса на клетку. Обнаруживали небольшие различия в количестве и скорости лизиса клеток.
На фиг. 42-44 показано, что все онколитические вирусы серотипа 3 демонстрировали значительно (P<0,05) более выраженную способность убивать клетки, чем нереплицирующийся контрольный вирус Ad3eGFP в клетках злокачественной опухоли легких A549, клетках злокачественной опухоли предстательной железы PC3-MM2 и клетках злокачественной опухоли яичника SKOV3. Можно видеть незначительные различия между онколитическими Ad3 вирусами, что подсказывает, что все вирусные конструкции полностью функциональны и что делеция области E3, вставленные промоторы (CMV или E2F) или вставленный трансген (CD40L) не могут влиять на онколитическую активность in vitro.
На фиг. 45 представлена противоопухолевая эффективность вирусов на основе Ad3 in vivo: ортотопическая модель интраперитонеальной злокачественной опухоли яичников. Ad3-hTERT-E3del-E2F-CD40L обладал наилучшей противоопухолевой эффективностью. ELISA подтверждал высвобождение CD40L в кровоток.
На фиг. 46 представлено терапевтическое окно онколитического аденовируса, кодирующего мышиный CD40L у иммунокомпетентных мышей. Доза 5: 1×1011 в. ч./мышь; доза 4: 3×1010в. ч./мышь; доза 3: 1×1010 в. ч./мышь; доза 2: 1×109 в. ч./мышь; доза 1: 1×108 в. ч./мышь; Положительный контроль (доза 2 внутрь опухоли.) При использовании дозы 5 67% мышей имели признаки печеночной токсичности. Доза 4 позволяет достигать хорошей опухолевой трансдукции после внутривенной доставки, без признаков печеночной токсичности.
На фиг. 47 представлено высвобождение ферментов печени у мышей, которых лечили через внутривенный путь онколитическим аденовирус, который кодирует мышиный CD40L у иммунокомпетентных мышей. Значительная печеночная токсичность отсутствовала, как измеряли по высвобождению ферментов печени, в каких-либо группах внутривенного лечения (доза 1-5). Последний столбец обозначает дозу 2, которую вводили внутрь опухоли. Однако, при дозе 5 имела место печеночная токсичность при визуальном исследовании → доза 4 представляет собой максимальную переносимую дозу для внутривенной доставки. (Дозы от имитатора до дозы 2 представлены в виде столбцов слева направо)
На фиг. 48 представлены механизмы усиления адоптивной клеточной терапии посредством экспрессирующего два цитокина вируса. Вирусная инфекция и врожденное восприятие вирусных частиц индуцируют сигналы опасности, которые включают повышающую регуляцию молекул HLA/MHC класса I на клетках злокачественной опухоли, активацию и созревание антигенпредставляющих клеток и секрецию цитокинов рекрутирования клеток иммунной системы. Дополнительное усиление сигналов опасности происходит посредством гибели опухолевой клетки от онколитических вирусов, при которой также высвобождаются опухолевые антигены и возрастает распознавание опухолевой ткани иммунной системой. Вирусы экспрессируют два цитокина: рекрутирующий T-клетки цитокин привлекает адоптивно перенесенные T-клетки в опухоль, и цитокин роста T-клеток, в конкретном варианте осуществления интерлейкин 2, увеличивает и поддерживает их пролиферацию.
На фиг. 49 представлена схема эксперимента по направленной миграции с применением рекомбинантных мышиных цитокинов. Несущим B16-OVA самкам мышей C57BL/6 адоптивно переносят 2,0×106 обогащенных CD8a+ OT-I лимфоцитов (квадрат) интраперитонеально на сутки 0, и лечат их внутриопухолевыми инъекциями рекомбинантных мышиных цитокинов (треугольники) в рабочие дни. Мониторинг опухолевого роста осуществляют и регистрируют три раза в неделю (круги), используя электронные штангенциркули. Мышей умерщвляют (X) в два различных момента времени (SAC1 и SAC2), собирают опухоли и анализируют образцы с применением OT-I qPCR и FACS анализа T-клеток.
На фиг. 50 представлена схема эксперимента по направленной миграции с применением аденовирусов, кодирующих мышиные цитокины. Несущим B16-OVA самкам мышей C57BL/6 адоптивно переносят 2,0×106 обогащенных CD8a+ OT-I лимфоцитов (квадрат) интраперитонеально на сутки 0, и лечат внутриопухолево с применением аденовирусов, вооруженных различными мышиными цитокинами (красные треугольники) в рабочие дни. Мониторинг опухолевого роста осуществляют и регистрируют три раза в неделю (круги) с применением электронных штангенциркулей. Мышей умерщвляют (X) в два различных момента времени (SAC1 и SAC2), собирают опухоли и анализируют образцы с применением OT-I qPCR и FACS анализа T-клеток.
На фиг. 51 представлена схема эксперимента по направленной миграции с применением радиоактивно меченного111In OT-I клеток и ОФЭКТ/КТ визуализации. Несущим B16-OVA самкам мышей C57BL/6 внутрь опухоли инъецируют 1e9 в. ч. 5/3 химерного вируса (треугольники) шесть суток подряд. Первая группа мышей получает адоптивный перенос 2,0×106 обогащенных CD8a+ меченных оксином индия OT-I лимфоцитов (квадрат) внутривенно на сутки 0, а другая группа мышей на сутки 7. Накопление OT-I клеток в опухолях количественно определяют посредством ОФЭКТ/КТ визуализации (круги). Мышей умерщвляют (X) в два различных момента времени (SAC1 и SAC2), собирают опухоли и измеряют их конечную радиоактивность.
Подробное описание изобретения
Адоптивная клеточная терапия
Основной подход по настоящему изобретению состоит в разработке лечения для пациентов со злокачественными опухолями, используя перенос иммунных лимфоцитов, которые способны реагировать на злокачественную опухоль и уничтожать ее. Выделенные инфильтрирующие опухоль лимфоциты выращивают в культуре до большого числа и вливают пациенту. В настоящем изобретении аденовирусные векторы, которые кодируют по меньшей мере один цитокин, используют для увеличения эффекта лимфоцитов. Отдельным введениям адоптивной клеточной терапевтической композиции и аденовирусных векторов часто предшествует миелоаблятивная или немиелоаблятивная предварительная химиотерапия и/или лучевая терапия. Лечение адоптивной клеточной терапией предназначено для того, чтобы снижать или устранять злокачественную опухоль у пациента (фиг. 21).
Данное изобретение относится к терапии с применением адоптивной клеточной терапевтической композиции, например, инфильтрирующих опухоль лимфоцитов, лимфоцитов с модифицированным TCR или лимфоцитов с модифицированным CAR. Данное изобретение, в частности, относится к T-клеточной терапии, а также к другой адоптивной терапии, такой как терапия естественными киллерными клетками или терапия другими клетками. В действительности, в соответствии с настоящим изобретением адоптивная клеточная терапевтическая композиция может содержать немодифицированные клетки, такие как в TIL терапии, или генетически модифицированные клетки. Существует два обычных пути для того, чтобы достичь генетического нацеливания T-клеток на опухолеспецифичные мишени. Один заключается в переносе T-клеточного рецептора с известной специфичностью (TCR терапия) и с лейкоцитарным антигеном человека совпадающего типа (HLA, известен как главный комплекс гистосовместимости у грызунов). Другой заключается в модификации клеток искусственными молекулами, такими как химерные антигенные рецепторы (CAR). Этот подход не зависит от HLA и более гибок по отношению к молекулам-мишеням. Например, можно использовать одноцепочечные антитела, а CAR также могут содержать костимуляторные домены. Однако, мишени CAR клеток должны находиться на мембране клеток-мишеней, тогда как модификации TCR могут использовать внутриклеточные мишени.
Как используют в настоящем документе «адоптивная клеточная терапевтическая композиция» относится к какой-либо композиции, которая содержит клетки, подходящие для переноса адоптивных клеток. В одном из вариантов осуществления изобретения адоптивная клеточная терапевтическая композиция содержит клетки того типа, который выбран из группы, состоящей из инфильтрирующих опухоль лимфоцитов (TIL), лимфоциты с модифицированным TCR (т.е. гетерологичным T-клеточным рецептором) и лимфоциты с модифицированным CAR (т.е. химерным антигенным рецептором). В другом варианте осуществления изобретения адоптивная клеточная терапевтическая композиция содержит клетки того типа, который выбран из группы, состоящей из T-клеток, CD8+ клеток, CD4+ клеток, NK-клеток, δ-γ T-клеток, регуляторных T-клеток и мононуклеарных клеток периферической крови. В другом варианте осуществления TIL, T-клетки, CD8+ клетки, CD4+ клетки, NK-клетки, δ-γ T-клетки, регуляторные T-клетки или мононуклеарные клетки периферической крови образуют адоптивную клеточную терапевтическую композицию. В одном конкретном варианте осуществления изобретения адоптивная клеточная терапевтическая композиция содержит T-клетки. Как используют в настоящем документе «инфильтрирующие опухоль лимфоциты» или TIL относятся к белым клеткам крови, которые покинули кровоток и мигрировали в опухоль. Лимфоциты можно подразделять на три группы, содержащие B-клетки, T-клетки и естественные киллерные клетки. В другом конкретном варианте осуществления изобретения адоптивная клеточная терапевтическая композиция содержит T-клетки, которые модифицированы мишенеспецифичными химерными антигенными рецепторами или, в частности, отобранными T-клеточными рецепторами. Как используют в настоящем документе «T-клетки» относятся к CD3+ клеткам, в том числе CD4+ хелперным клеткам, CD8+ цитотоксическим T-клеткам и γδ T-клеткам.
В дополнение к подходящим клеткам, адоптивная клеточная терапевтическая композиция, используемая в настоящем изобретении, может содержать какие-либо другие средства, такие как фармацевтически приемлемые носители, буферы, эксципиенты, адъюванты, добавки, антисептики, наполнители, стабилизаторы и/или загустители и/или какие-либо компоненты, которые обычно встречаются в соответствующих продуктах. Выбор подходящих ингредиентов и подходящих способов изготовления для формулирования композиций относится к общим знаниям специалиста в данной области.
Адоптивная клеточная терапевтическая композиция может быть в какой-либо форме, такой как твердая, полутвердая или жидкая форма, подходящая для введения. Состав можно выбирать из группы, состоящей из, но не ограничиваясь этим, растворов, эмульсий, суспензий, таблеток, пеллетов и капсул. Композиции не ограничены определенным составом, и вместо этого композицию можно формулировать в каком-либо известном фармацевтически приемлемом составе. Фармацевтические композиции можно получать посредством любых стандартных процессов, известных в данной области.
Вирусные векторы
Онколитические аденовирусные векторы, используемые в настоящем изобретении, могут представлять собой любые аденовирусные векторы, подходящие для лечения человека или животного. В одном из вариантов осуществления изобретения аденовирусные векторы представляют собой векторы из человеческих вирусов, и их можно выбирать из группы, состоящей из векторов Ad5, Ad3 и Ad5/3. В другом варианте осуществления вектор представляет собой вектор Ad5 или Ad5/3.
Как используют в настоящем документе, «онколитический аденовирусный вектор» относится к аденовирусному вектору, способному инфицировать и убивать клетки злокачественной опухоли посредством избирательной репликации в опухоли по сравнению с нормальными клетками.
Векторы можно модифицировать каким-либо образом, известным в данной области, например, посредством удаления, вставки, введения мутаций или модификаций каких-либо вирусных участков. В отношении репликации, векторы делают обладающими специфичностью к опухолям. Например, аденовирусный вектор может содержать модификации в E1, E3 и/или E4, такие как инсерция опухолеспецифичных промоторов (например, для контроля E1), делеции областей (например, константная область 2 E1, как используют в «D24», E3/gp19k, E3/6.7k) и инсерции трансгенов. Кроме того, можно модифицировать области выступа волокна в векторе. В одном из вариантов осуществления изобретения аденовирусный вектор представляет собой Ad5/3, который содержит Ad5 остов нуклеиновой кислоты и Ad3 выступ волокна или Ad5/3 химерный выступ волокна.
Как используют в настоящем документе, выражение «остов нуклеиновой кислоты аденовируса серотипа 5 (Ad5)» относится к геному Ad5. Аналогичным образом, «остов нуклеиновой кислоты аденовируса серотипа 3 (Ad3)» относится к геному Ad3. «Ad5/3 вектор» относится к химерному вектору, который имеет части как Ad5 вектора, так и Ad3 вектора. В конкретном варианте осуществления изобретения, капсидная модификация вектора представляет собой Ad5/3 химеризм. Как используют в настоящем документе, «Ad5/3 химерный выступ волокна» относится к химеризму, где часть с выступом волокна взята из Ad серотипа 3, а остальная часть волокна взята из Ad серотипа 5. В частности, в одном из вариантов осуществления конструкция имеет выступ волокна из Ad3, тогда как остальная часть генома взята из Ad5 (см. фиг. 17, 33 и 34).
Один подход для создания опухолеспецифичного онколитического аденовируса состоит в конструировании делеции 24 пар оснований (D24), которая затрагивает константную область 2 (CR2) в E1. В аденовирусе дикого типа CR2 отвечает за связывание клеточного Rb опухолевого супрессора/регуляторного белка клеточного цикла для индукции синтетической (S) фазы, т.е. фазы синтеза или репликации ДНК. Для взаимодействия между pRb и E1A необходимы восемь аминокислот с 121 до 127 из консервативной области белка E1A, которые удаляют в настоящем изобретении. Вектор по настоящему изобретению содержит делецию нуклеотидов, соответствующих аминокислотам 122-129 вектора согласно Heise C. et al. (2000, Nature Med 6, 1134-1139). Известно, что вирусы D24 обладают пониженной способностью преодолевать контрольную точку G1-S и эффективно реплицируются только в клетках, где это взаимодействие не является обязательным, например, в опухолевых клетках, обладающих дефектом пути Rb-p16, которые включают большинство, если не все опухоли человека (см. фиг. 17, 33 и 34).
Также возможно заменять эндогенный вирусный промотор E1A, например, на опухолеспецифичный промотор. В конкретном варианте осуществления изобретения промотор hTERT используют вместо эндогенного вирусного промотора E1A.
Область E3 не обязательна для репликации вируса in vitro, но E3 белок играет важную роль в регуляции иммунного ответа организма-хозяина, т.е. в ингибировании и врожденного и специфического иммунного ответа. Делеция gp19k/6.7k в E3 относится к делеции 965 пар оснований из аденовирусной области E3A. В получаемой аденовирусной конструкции удалены оба гена gp19k и 6.7k (Kanerva A et al. 2005, Gene Therapy 12, 87-94). Известно, что продукт гена gp19k связывается с молекулами главного комплекса гистосовместимости I и секвестрирует их (MHC1, известный как HLA1 у человека) в эндоплазматическом ретикулуме, а также предотвращает распознавание инфицированных клеток цитотоксическими T-лимфоцитами. Поскольку многие опухоли дефицитны по HLA1/MHC1, делеция gp19k увеличивает избирательность опухоли к вирусам (устранение вируса происходит быстрее, чем для вируса дикого типа из нормальных клеток, но для опухолевых клеток разницы не существует). Белки 6.7k экспрессированы на клеточных поверхностях и участвуют в понижающей регуляции рецептора 2 TNF-связанного индуцирующего апоптоз лиганда (TRAIL) (см. фиг. 17, 33 и 34).
Обе эти делеции предоставляют неожиданное преимущество в отношении данного изобретения. Поскольку авторы изобретения пытаются восстановить экспрессию HLA/MHC для представления опухолевых эпитопов адоптивно перенесенным T-клеткам, экспрессия белка gp19k является контрпродуктивной, и фактически для повышающей регуляции HLA/MHC необходима делеция gp19k. В отношении 6.7k, поскольку вариант осуществления данного изобретения состоит в получении TNFα из вируса, и одна из его противоопухолевых активностей направлена против опухолевого проапоптозного эффекта (оказываемого как на трансдуцированные, так и на нетрансдуцированные фоновые клетки), присутствие 6.7k является контрпродуктивным.
В одном из вариантов осуществления изобретения цитокиновый трансген или трансгены помещают в область делеции gp19k/6.7k в E3, под управлением промотора E3. Это ограничивает экспрессию трансгена теми опухолевыми клетками, которые допускают репликацию вируса и последующую активацию промотора E3. Промотор E3 может представлять собой какой-либо экзогенный (например, промотор CMV или E2F) или эндогенный промотор, известный в данной области, в частности, эндогенный промотор E3. Несмотря на то, что активация промотора E3 главным образом происходит посредством репликации, возникает некоторая экспрессия, когда экспрессируют E1. Поскольку избирательность вирусов типа D24 возникает после экспрессии E1 (когда E1 не способен связывать Rb), эти вирусы экспрессируют E1 также в трансдуцированных нормальных клетках. Таким образом, критически важно регулировать также экспрессию E1, чтобы ограничивать опухолевыми клетками опосредованную промотором E3 экспрессию трансгена.
В другом варианте осуществления изобретения E3 gp19k/6.7k содержится в векторе, но одна или несколько других областей E3 удалены (например, E3 9 кДа, E3 10,2 кДа, E3 15,2 кДа и/или E3 15,3 кДа).
В конкретном варианте осуществления изобретения онколитический аденовирусный вектор основан на остове нуклеиновой кислоты аденовируса серотипа 5 (Ad5), содержащем 5/3 химерный выступ волокна, и содержит следующее: промотор E2F1 для опухолеспецифичной экспрессии E1A, делеция 24 п. о. (D24) в Rb-связывающей константной области 2 аденовирусного E1, делеция последовательности нуклеиновой кислоты вирусных рамок считывания gp19k и 6.7k, с инсерцией трансгена в удаленную область, что ведет к ассоциированному с репликацией контролю экспрессии трансгена под управлением вирусного промотора E3, и последовательность нуклеиновой кислоты, которая кодирует по меньшей мере один цитокиновый трансген вместо удаленных аденовирусных генов gp19k/6.7k в области E3 (фиг. 17). В одном из вариантов осуществления изобретения аденовирусный вектор основан на аденовирусе человека. (см. фиг. 17, 33 и 34)
В другом конкретном варианте осуществления изобретения онколитический аденовирусный вектор основан на остове нуклеиновой кислоты аденовируса серотипа 3 (Ad3) и содержит следующее: делеция в области E3, и опухолеспецифичный промотор (например, CMV или E2F) для экспрессии трансгена (например, CD40L) вместо удаленной области E3. В одном из вариантов осуществления изобретения аденовирусный вектор основан на аденовирусе человека. (См. фиг. 37 и 38, соответствующие нуклеотидные последовательности вирусных векторов Ad3-hTERT-E3del-CMV-CD40L и Ad3-hTERT-E3del-E2F-CD40L представлены в SEQ ID №№ 30 и 31)
Точные функции белков ранней области (E3) аденовируса 3 не известны. В целом, не похоже, чтобы в аденовирусах они ослабляли репликацию, когда их удаляют, и, похоже, они влияют на противовирусную реакцию организма-хозяина на аденовирусы (Wold et al., 1999). E3 генома аденовируса человека имеет наивысший уровень генетического разнообразия среди шести видов (A-F) аденовирусов, встречающихся у человека. Это разнообразие генетического содержимого, в первую очередь, расположено между высоко консервативными открытыми рамками считывания (ORF) E3-gp19K и E3-RIDα, в которых закодированы видоспецифичные массивы генов (Burgert and Blusch, 2000).
E3-gp19K модулирует опосредованное цитотоксическими T-клетками уничтожение инфицированных вирусом клеток. Это происходит посредством блокирования транспорта MHC класса I в плазматическую мембрану и ингибирования формирования комплекса TAP-MHC класса I (Andersson et al., 1985; Andersson et al., 1987; Burgert and Kvist, 2002, Bennet et al., 1999).
Таким образом, в одном аспектов изобретения важная молекула E3-gp19K содержится в аденовирусном векторе для того, чтобы делать репликацию вируса более скрытной и дать больше времени для онколизиса и его положительных эффектов. Также сохранение E3-gp19K может снижать индукцию цитотоксических T-клеток против аденовирусов, что ведет к большему количеству противоопухолевых T-клеток.
В одном из вариантов осуществления изобретения онколитический аденовирусный вектор основан на остове нуклеиновой кислоты аденовируса серотипа 3 (Ad3) и содержит следующее: промотор (например hTERT) для опухолеспецифичной экспрессии E1A, делеция в области E3 (например, делеция, которая затрагивает E3 9 кДа, E3 10,2 кДа, E3 15,2 кДа и E3 15,3 кДа) и опухолеспецифичный промотор (например, CMV или E2F) для экспрессии трансгена (например, CD40L) вместо удаленной области E3. В одном из вариантов осуществления изобретения в векторе остов нуклеиновой кислоты полностью представляет собой аденовирус серотипа 3. В одном из вариантов осуществления изобретения в вирусах Ad3 delE3 удалены следующие признаки: E3 9 кДа, E3 10,2 кДа, E3 15,2 кДа, E3 15,3 кДа, и, кроме того, на их место вставлен CD40L с промотором (CMV или E2F). Эти вирусы индуцируют апоптоз опухолевых клеток и запускают несколько иммунных механизмов, включая ответ T-хелперов 1 типа (TH1), который ведет к активации цитотоксических T-клеток и снижению иммуносупрессии.
Цитокины участвуют в иммунном ответе, действуя через различные механизмы, включая рекрутирование T-клеток в направлении опухоли. Нуклеотидная последовательность, кодирующая цитокиновый трансген, может быть от какого-либо животного, такого как человек, примат, крыса, мышь, хомяк, собака или кошка, но, в частности, ее кодирует последовательность человека. Нуклеотидную последовательность, кодирующую трансген, можно модифицировать для того, чтобы усовершенствовать ее эффекты, или не модифицировать, т.е. использовать дикий тип.
Конкретные варианты осуществления настоящего изобретения включают вирусные векторы, которые кодируют по меньшей мере один цитокин. Цитокины, используемые в настоящем изобретении, можно выбирать из любых известных цитокинов в данной области. В одном из вариантов осуществления изобретения цитокин выбирают из группы, состоящей из интерферона α, интерферона β, интерферона γ, C5a комплемента, IL-2, TNFα, CD40L, IL-12, IL-23, IL-15, IL-17, CCL1, CCL11, CCL12, CCL13, CCL14-1, CCL14-2, CCL14-3, CCL15-1, CCL15-2, CCL16, CCL17, CCL18, CCL19, CCL19, CCL2, CCL20, CCL21, CCL22, CCL23-1, CCL23-2, CCL24, CCL25-1, CCL25-2, CCL26, CCL27, CCL28, CCL3, CCL3L1, CCL4, CCL4L1, CCL5, CCL6, CCL7, CCL8, CCL9, CCR10, CCR2, CCR5, CCR6, CCR7, CCR8, CCRL1, CCRL2, CX3CL1, CX3CR, CXCL1, CXCL10, CXCL11, CXCL12, CXCL13, CXCL14, CXCL15, CXCL16, CXCL2, CXCL3, CXCL4, CXCL5, CXCL6, CXCL7, CXCL8, CXCL9, CXCL9, CXCR1, CXCR2, CXCR4, CXCR5, CXCR6, CXCR7 и XCL2. В конкретном варианте осуществления изобретения цитокин представляет собой IL-2 или TNFα. В другом варианте осуществления изобретения цитокин или цитокины выбирают из группы хемокинов, состоящей из CCL1, CCL11, CCL12, CCL13, CCL14-1, CCL14-2, CCL14-3, CCL15-1, CCL15-2, CCL16, CCL17, CCL18, CCL19, CCL19, CCL2, CCL20, CCL21, CCL22, CCL23-1, CCL23-2, CCL24, CCL25-1, CCL25-2, CCL26, CCL27, CCL28, CCL3, CCL3L1, CCL4, CCL4L1, CCL5, CCL6, CCL7, CCL8, CCL9, CCR10, CCR2, CCR5, CCR6, CCR7, CCR8, CCRL1, CCRL2, CX3CL1, CX3CR, CXCL1, CXCL10, CXCL11, CXCL12, CXCL13, CXCL14, CXCL15, CXCL16, CXCL2, CXCL3, CXCL4, CXCL5, CXCL6, CXCL7, CXCL8, CXCL9, CXCL9, CXCR1, CXCR2, CXCR4, CXCR5, CXCR6, CXCR7 и XCL2.
Вирусные векторы по изобретению могут кодировать один, два, три, четыре, пять или больше цитокинов. В одном из вариантов осуществления изобретения онколитический аденовирусный вектор кодирует два или более цитокинов, наиболее конкретно два. Эти два цитокина могут представлять собой какие-либо известные цитокины, например, включая в качестве неограничивающих примеров, те, которые перечислены выше, с добавлением GMCSF. Два цитокина могут представлять собой различные цитокины. В одном из вариантов осуществления изобретения онколитический аденовирусный вектор кодирует какие-либо два или более цитокинов, выбранных из группы цитокинов, состоящей из интерферона α, интерферона β, интерферона γ, C5a комплемента, GMCSF, IL-2, TNFα, CD40L, IL-12, IL-23, IL-15, IL-17, CCL1, CCL11, CCL12, CCL13, CCL14-1, CCL14-2, CCL14-3, CCL15-1, CCL15-2, CCL16, CCL17, CCL18, CCL19, CCL19, CCL2, CCL20, CCL21, CCL22, CCL23-1, CCL23-2, CCL24, CCL25-1, CCL25-2, CCL26, CCL27, CCL28, CCL3, CCL3L1, CCL4, CCL4L1, CCL5, CCL6, CCL7, CCL8, CCL9, CCR10, CCR2, CCR5, CCR6, CCR7, CCR8, CCRL1, CCRL2, CX3CL1, CX3CR, CXCL1, CXCL10, CXCL11, CXCL12, CXCL13, CXCL14, CXCL15, CXCL16, CXCL2, CXCL3, CXCL4, CXCL5, CXCL6, CXCL7, CXCL8, CXCL9, CXCL9, CXCR1, CXCR2, CXCR4, CXCR5, CXCR6, CXCR7 и XCL2, или онколитический аденовирусный вектор кодирует IL-2 и цитокин или цитокины, выбранные из группы цитокинов, состоящей из интерферона α, интерферона β, интерферона γ, C5a комплемента, GMCSF, TNFα, CD40L, IL-12, IL-23, IL-15, IL-17, CCL1, CCL11, CCL12, CCL13, CCL14-1, CCL14-2, CCL14-3, CCL15-1, CCL15-2, CCL16, CCL17, CCL18, CCL19, CCL19, CCL2, CCL20, CCL21, CCL22, CCL23-1, CCL23-2, CCL24, CCL25-1, CCL25-2, CCL26, CCL27, CCL28, CCL3, CCL3L1, CCL4, CCL4L1, CCL5, CCL6, CCL7, CCL8, CCL9, CCR10, CCR2, CCR5, CCR6, CCR7, CCR8, CCRL1, CCRL2, CX3CL1, CX3CR, CXCL1, CXCL10, CXCL11, CXCL12, CXCL13, CXCL14, CXCL15, CXCL16, CXCL2, CXCL3, CXCL4, CXCL5, CXCL6, CXCL7, CXCL8, CXCL9, CXCL9, CXCR1, CXCR2, CXCR4, CXCR5, CXCR6, CXCR7 и XCL2. В конкретном варианте осуществления изобретения цитокины представляют собой IL-2 и TNFα. Другой цитокин функционирует посредством привлечения и активации T-клеток и снижения опухолевой иммуносупрессии, тогда как IL-2 индуцирует размножение T-клеточного трансплантата. Таким образом, продуцирование IL-2 происходит локально в опухоли, где он нужен, вместо системной инъекции, как типично выполняют при T-клеточной терапии, что может вызывать побочные эффекты, и, следовательно, с помощью этого варианта осуществления можно предотвращать главную проблему терапии известного уровня техники (т.е. токсичность системного IL-2).
Сигналы опасности, предоставляемые посредством репликации онколитического вируса и активации патоген-ассоциированных рецепторов опознавания молекулярного паттерна с помощью вирусной ДНК, вместе с действием трансгена(трансгенов) могут снижать опухолевую иммуносупрессию до такой степени, что предварительную терапию можно не использовать. Следовательно, можно избегать основной проблемы известного уровня техники, токсичности из-за предварительной химиотерапии и лучевой терапии.
В одном из вариантов осуществления изобретения вирусный вектор содержит участок внутренней посадки рибосомы (IRES) или, необязательно, сайт шунтирования рибосомы 2A между двумя трансгенами. Таким образом, IRES или сайт шунтирования рибосомы 2A может находиться между какими-либо цитокинами, такими как IL-2 и какой-либо другой цитокин, выбранный из вышеперечисленной группы цитокинов. Как используют в настоящем документе «IRES» относится к нуклеотидной последовательности, которая делает возможной инициацию трансляции в середине последовательности информационной РНК при синтезе белка. IRES может быть из какого-либо вируса, но в одном из вариантов осуществления изобретения IRES взят из вируса энцефаломиокардита (EMCV). Как используют в настоящем документе, «сайт шунтирования рибосомы 2A» относится к сайту трансляции инициации, в котором рибосомы физически пропускают части 5'-нетранслируемой области для того, чтобы достичь инициирующего кодона. И IRES и A2 позволяют вирусам продуцировать два трансгена с одного промотора (промотора E3).
Общие схемы вирусных геномов, которые можно использовать в настоящем изобретении, представлены на фиг. 17, 33, 34, 37 и 38. Нуклеотидные последовательности вирусных векторов, содержащих трансгены C5a, hCD40L, hIFNa2, hIFNb1, hIFNg1, hIL-2 или TNFα, представлены в SEQ ID №№ 1-7, соответственно (Ad5/3-E2F-D24-трансген). Нуклеотидные последовательности вирусных векторов, содержащих CD40L, также представлены в SEQ ID №№ 30 и 31 (Ad3-hTERT-E3del-CMV-CD40L и Ad3-hTERT-E3del-E2F-CD40L). Кроме того, нуклеотидные последовательности вирусных векторов, которые содержат два трансгена, один представляет собой IL-2 и другой представляет собой C5a, CD40L, IFNa2, IFNb, IFNg, GMCSF или TNFα, представлены в SEQ ID №№ 8-21 (SEQ ID № 8 C5a-2A-IL-2, SEQ ID № 9 IFNa-2A-IL-2, SEQ ID № 10 TNFα-2A-IL-2, SEQ ID № 11 CD40L-2A-IL-2, SEQ ID № 12 IFNb-2A-IL-2, SEQ ID № 13 GMCSF-2A-IL-2, SEQ ID № 14 IFNg-2A-IL-2, SEQ ID № 15 C5a-IRES-IL-2, SEQ ID № 16 IFNa-IRES-IL-2, SEQ ID № 17 TNFα-IRES-IL-2, SEQ ID № 18 CD40L-IRES-IL-2, SEQ ID № 19 IFNb-IRES-IL-2, SEQ ID № 20 GMCSF-IRES-IL-2, SEQ ID № 21 IFNg-IRES-IL-2) (Ad5/3-E2F-D24-трансген-IRES/2A-трансген).
Вкратце, ключевые преимущества по настоящему изобретению, в котором используют вирусные векторы, содержащие по меньшей мере один цитокиновый трансген: i) цитокины и вирус per se являются причиной сигнала опасности, который рекрутирует T-клетки и другие клетки иммунной системы в опухоли, ii) цитокины индуцируют T-клеточную пролиферацию как в опухоли, так и в регионарных лимфоидных органах, iii) цитокины и вирус per se способны индуцировать T-клетки (как адоптивный T-клеточный трансплантат, так и естественные врожденные противоопухолевые T-клетки) к размножению в опухоли, iv) цитокин и/или вирус индуцирует повышающую регуляцию антигенпрезентирующих молекул (HLA) на клетках злокачественной опухоли, что делает их чувствительными к распознаванию и уничтожению T-клетками, и v) цитокины и репликация вируса благоприятно изменяют опухолевое микроокружение посредством снижения иммуносупрессии и клеточной анергии.
Вирусные векторы, используемые в настоящем изобретении, также могут содержать другие модификации, нежели описанные выше. Какие-либо дополнительные компоненты или модификации в настоящем изобретении использовать можно при желании, но это не обязательно.
Инсерция экзогенных элементов может усиливать эффекты векторов в клетках-мишенях. Применение экзогенной ткани или опухолеспецифичных промоторов является обычным в рекомбинантных векторах, и их также можно использовать в настоящем изобретении.
Вкратце, настоящее изобретение раскрывает, что репликация онколитического вируса позволяет рекрутировать T-клетки и индуцировать сигналы опасности в опухоли, что снижает иммуносупрессию и клеточную анергию. Эти эффекты опосредованы через патоген-ассоциированные рецепторы опознавания молекулярного паттерна, эволюционно консервативный механизм индукции иммунитета, не подверженный толерантности. Настоящее изобретение также раскрывает, что дополнительным эффектом онколитической платформы, способной к репликации в опухолях, но не в нормальных клетках, является самостоятельная амплификация в опухоли. Кроме того, онколитический эффект per se может увеличивать общий противоопухолевый эффект у человека.
Злокачественные опухоли
Рекомбинантные векторы по настоящему изобретению способны к репликации в опухолевых клетках. В одном из вариантов осуществления изобретения векторы способны к репликации в клетках, которые имеют дефекты в пути Rb, в частности, в пути Rb-p16. Эти дефективные клетки включают все опухолевые клетки животных и человека. Как используют в настоящем документе, «дефекты в пути Rb» относится к мутациям и/или эпигенетическим изменениям в каких-либо генах или белках пути. Из-за этих дефектов опухолевые клетки чрезмерно экспрессируют E2F и, таким образом, связывание Rb с E1A CR2, которое обычно необходимо для эффективной репликации, не является обязательным. Кроме того, избирательность опосредована промотором E2F, который активируется только в присутствии свободного E2F, как видно на клетках с дефективным путем Rb/p16. В отсутствие свободного E2F, не происходит транскрипция E1A и вирус не реплицируется. Наличие E2F промотора является важным для того, чтобы предотвращать экспрессию E1A в нормальных тканях, что может вызывать токсичность как непосредственно, так и опосредованно через разрешение экспрессии трансгена с промотора E3.
Настоящее изобретение относится к подходам для лечения злокачественной опухоли у субъекта. В одном из вариантов осуществления изобретения субъектом является человек или животное, в частности животное или пациент-человек, более конкретно, человек или животное, страдающее злокачественной опухолью.
Подход можно использовать для того, чтобы лечить любые злокачественные опухоли или опухоли, в том числе, как озлокачествленные, так и доброкачественные опухоли, как первичные опухоли, так и метастазы могут быть мишенями этого подхода. В одном из вариантов осуществления изобретения злокачественная опухоль отличается инфильтрирующими опухоль лимфоцитами. Инструменты по настоящему изобретению явно представляют интерес для лечения метастатических солидных опухолей, отличающихся инфильтрирующими опухоль лимфоцитами. В другом варианте осуществления T-клеточный трансплантат модифицирован опухоле- или тканеспецифическим T-клеточным рецептором химерного антигенного рецептора.
Как используют в настоящем документе, термин «лечение» или «лечить» относится ко введению по меньшей мере онколитических аденовирусных векторов или по меньшей мере онколитических аденовирусных векторов и адоптивной клеточной терапевтической композиции субъекту, предпочтительно, субъекту млекопитающему или человеку, в целях, которые включают не только полное излечение, но также профилактику, уменьшение интенсивности или облегчение нарушений или симптомов, связанных со злокачественной опухолью или опухолью. Терапевтический эффект можно оценивать посредством мониторинга симптомов пациента, опухолевых маркеров в крови или, например, размера опухоли или длительности выживаемости пациента.
В другом варианте осуществления изобретения злокачественную опухоль выбирают из группы, состоящей из назофарингеальной злокачественной опухоли, синовиальной злокачественной опухоли, гепатоклеточной злокачественной опухоли, злокачественной опухоли почки, злокачественной опухоли соединительной ткани, меланомы, злокачественной опухоли легких, злокачественной опухоли кишечника, злокачественной опухоли ободочной кишки, злокачественной опухоли кишки, злокачественной опухоли толстой кишки, злокачественной опухоли головного мозга, злокачественной опухоли горла, злокачественной опухоли ротовой полости, злокачественной опухоли печени, злокачественной опухоли кости, злокачественной опухоли поджелудочной железы, хориокарциномы, гастриномы, феохромоцитомы, пролактиномы, T-клеточного лейкоза/лимфомы, невромы, болезни фон Гиппеля-Линдау, синдрома Золингера-Эллисона, злокачественной опухоли надпочечника, злокачественной опухоли ануса, злокачественной опухоли желчных протоков, злокачественной опухоли мочевого пузыря, злокачественной опухоли мочеточника, злокачественной опухоли головного мозга, олигодендроглиомы, нейробластомы, менингиомы, опухоли спинного мозга, злокачественной опухоли кости, остеохондромы, хондросаркомы, саркомы Юинга, злокачественной опухоли неизвестной первичной локализации, карциноида, карциноида желудочно-кишечного тракта, фибросаркомы, злокачественной опухоли молочной железы, болезни Педжета, злокачественной опухоли шейки матки, злокачественной опухоли толстой кишки, злокачественной опухоли прямой кишки, злокачественной опухоли пищевода, злокачественной опухоли желчного пузыря, злокачественной опухоли головы, злокачественной опухоли глаза, злокачественной опухоли шеи, злокачественной опухоли почки, опухоли Вильмса, злокачественной опухоли печени, саркомы Капоши, злокачественной опухоли предстательной железы, злокачественной опухоли легких, злокачественной опухоли яичек, болезни Ходжкина, неходжкинской лимфомы, злокачественной опухоли ротовой полости, злокачественной опухоли кожи, мезотелиомы, множественной миеломы, злокачественной опухоли яичников, эндокринной злокачественной опухоли поджелудочной железы, глюкагономы, злокачественной опухоли поджелудочной железы, злокачественной опухоли паращитовидной железы, злокачественной опухоли пениса, злокачественной опухоли гипофиза, саркомы мягких тканей, ретинобластомы, злокачественной опухоли тонкой кишки, злокачественной опухоли желудка, злокачественной опухоли тимуса, злокачественной опухоли щитовидной железы, трофобластической злокачественной опухоли, пузырного заноса, злокачественной опухоли матки, злокачественной опухоли эндометрия, злокачественной опухоли влагалища, злокачественной опухоли вульвы, акустической невромы, фунгоидного микоза, инсулиномы, карциноидной синдромы, соматостатиномы, злокачественной опухоли десны, злокачественной опухоли сердца, злокачественной опухоли губы, злокачественной опухоли мозговых оболочек, злокачественной опухоли рта, злокачественной опухоли нерва, злокачественной опухоли неба, злокачественной опухоли околоушной железы, злокачественной опухоли брюшины, злокачественной опухоли глотки, плевральной злокачественной опухоли, злокачественной опухоли слюнной железы, злокачественной опухоли языка и злокачественной опухоли миндалины.
Прежде чем классифицировать пациента человека или животное в качестве подходящего для терапии по настоящему изобретению, клиницист может обследовать пациента. На основании результатов, отклоняющихся от нормы и указывающих на опухоль или злокачественную опухоль, клиницист может предложить пациенту лечение по настоящему изобретению.
Фармацевтическая композиция
Фармацевтическая композиция по изобретению содержит вирусные векторы по изобретению по меньшей мере одного типа. Кроме того, композиция может содержать по меньшей мере два, три или четыре различных вектора. В дополнение к вектору, фармацевтическая композиция также может содержать другие терапевтически эффективные средства, какие-либо другие средства, такие как фармацевтически приемлемые носители, буферы, эксципиенты, адъюванты, добавки, антисептики, наполнители, стабилизаторы и/или загустители и/или какие-либо компоненты, которые обычно встречаются в соответствующих продуктах. Выбор подходящих ингредиентов и подходящих способов изготовления для формулирования композиций относится к общим знаниям специалиста в данной области.
Фармацевтическая композиция может быть в какой-либо форме, такой как твердая, полутвердая или жидкая форма, подходящей для введения. Состав можно выбирать из группы, состоящей из, но не ограничиваясь этим, растворов, эмульсий, суспензий, таблеток, пеллетов и капсул. Композиции по данному изобретению не ограничены определенным составом, и композицию можно формулировать в каком-либо известном фармацевтически приемлемом составе. Фармацевтические композиции можно получать посредством любых стандартных процессов, известных в данной области.
В одном из вариантов осуществления изобретения вирусный вектор или фармацевтическая композиция выполняет функцию носителя in situ для рекрутирования T-клеток, усиливающего их терапевтический эффект и позволяющего им размножаться в опухоли.
Фармацевтический набор по настоящему изобретению содержит адоптивную клеточную терапевтическую композицию и онколитические аденовирусные векторы, кодирующие по меньшей мере один цитокин. Адоптивную клеточную терапевтическую композицию формулируют в первом составе и онколитические аденовирусные векторы, которые кодируют по меньшей мере один цитокин, формулируют во втором составе. В другом варианте осуществления изобретения первый и второй составы предназначены для одновременного или последовательного, в любом порядке, введения субъекту.
Введение
Вектор или фармацевтическую композицию по изобретению можно вводить какому-либо эукариотическому субъекту, выбранному из группы, состоящей из растений, животных и человека. В конкретном варианте осуществления изобретения субъектом является человек или животное. Животное можно выбирать из группы, состоящей из комнатных животных, домашних животных и продуктивных животных.
Любой стандартный способ можно использовать для введения вектора или композиции субъекту. Путь введения зависит от состава или формы композиции, заболевания, местоположения опухолей, пациента, сопутствующих патологий и других факторов.
В одном из вариантов осуществления изобретения отдельное введение(введения) адоптивной клеточной терапевтической композиции и онколитических аденовирусных векторов, которые кодируют по меньшей мере один цитокин, субъекту проводят одновременно или последовательно, в любом порядке. Как используют в настоящем документе, «отдельное введение» или «отдельное» относится к ситуации, когда адоптивная клеточная терапевтическая композиция и онколитические аденовирусные векторы представляют собой два различных продукта или композиции, отличающихся друг от друга.
Только одно введение адоптивной клеточной терапевтической композиции и онколитических аденовирусных векторов, которые кодируют по меньшей мере один цитокин по изобретению, или только онколитических или нецитолитических вирусных векторов может иметь терапевтические эффекты. Может иметь место какой-либо период между введениями, например, в зависимости от пациента и типа, степени или местоположения злокачественной опухоли. В одном из вариантов осуществления изобретения имеет место период времени от одной минуты до четырех недель, в частности, от 1 до 10 суток, более конкретно от 1 до 5 суток, между последовательным введением адоптивной клеточной терапевтической композиции и онколитических аденовирусных векторов, которые кодируют по меньшей мере один цитокин, и/или имеют место несколько введений адоптивной клеточной терапевтической композиции и онколитических аденовирусных векторов. Число раз введения адоптивной клеточной терапевтической композиции и онколитических аденовирусных векторов также может различаться во время периода лечения. Онколитические аденовирусные векторы или фармацевтические или адоптивные клеточные композиции можно вводить, например, от 1 до 10 раз в первые 2 недели, 4 недели, ежемесячно или во время периода лечения. В одном из вариантов осуществления изобретения введение векторов или каких-либо композиций выполняют от трех до семи раз в первые 2 недели, затем на 4 неделе и затем раз в месяц. В конкретном варианте осуществления изобретения введение выполняют четыре раза в первые 2 недели, затем на 4 неделе и затем раз в месяц. Длительность периода лечения может варьировать и, например, может длиться от 2 до 12 месяцев или больше.
В конкретном варианте осуществления изобретения адоптивную клеточную терапевтическую композицию и онколитические аденовирусные векторы вводят в те же сутки и после этого онколитические аденовирусные векторы вводят каждую неделю, две недели, три недели или каждый месяц во время периода лечения, который может длиться, например, от 1 до 6 или 12 месяцев или больше.
В одном из вариантов осуществления изобретения введение онколитического вируса проводят посредством внутриопухолевой, внутриартериальной, внутривенной, внутриплевральной, интравезикулярной, внутриполостной или перитонеальной инъекции или перорального введения. Также возможна какая-либо комбинация введений. Подход может давать системную эффективность несмотря на местную инъекцию. Адоптивную клеточную терапевтическую композицию можно вводить внутривенно или внутрь опухоли. В одном из вариантов осуществления введение адоптивной клеточной терапевтической композиции и/или онколитических вирусных векторов, которые кодируют по меньшей мере один цитокин, проводят посредством внутриопухолевой, внутриартериальной, внутривенной, внутриплевральной, интравезикулярной, внутриполостной или перитонеальной инъекции или перорального введения. В конкретном варианте осуществления изобретения TIL или T-клетки вводят внутривенно, а вирусные векторы внутрь опухоли и/или внутривенно. Стоит отметить, что вирус доставляют в опухоль отдельно от введения T-клеток; вирус не используют для того, чтобы модифицировать T-клеточный трансплантат ex vivo. В сущности, вирус модифицирует опухоль таким образом, что T-клеточный трансплантат может работать лучше.
Эффективная доза векторов зависит от по меньшей мере от субъекта, нуждающегося в лечении, типа опухоли, местоположения опухоли и стадии опухоли. Доза может варьировать, например, приблизительно от 1×108 вирусных частиц (в. ч.) приблизительно до 1×1014 в. ч., в частности, приблизительно от 5×109 в. ч. приблизительно до 1×1013 в. ч. и более конкретно приблизительно от 8×109 в. ч. приблизительно до 1×1012 в. ч. В одном из вариантов осуществления онколитические аденовирусные векторы, которые кодируют по меньшей мере один цитокин, вводят в количестве от 1×1010 до 1×1014 вирусных частиц. В другом варианте осуществления изобретения доза находится в диапазоне приблизительно от 5×1010 до 5×1011 в. ч..
Количество перенесенных клеток также зависит от пациента, но типичные количества находятся в диапазоне от 1×109 до 1×1012 клеток на инъекцию. Число инъекций также варьирует, но типичные варианты осуществления включают 1 или 2 раунда лечения с интервалом в несколько (например, 2-4) недель.
Любое другое лечение или комбинацию лечений можно использовать в дополнение к терапии по настоящему изобретению. В конкретном варианте осуществления способ или применение по изобретению дополнительно включает применение у субъекта параллельной или последовательной лучевой терапии, моноклональных антител, химиотерапии или других лекарственных средств против злокачественных опухолей или вмешательств (в том числе, хирургических).
Термины «лечить» или «повышать», а также слова, происходящие от них, как используют в настоящем документе, не обязательно подразумевают 100% или полное лечение или повышение. Точнее, существуют различные степени, которые специалист в данной области признает в качестве имеющих потенциальный эффект или терапевтический эффект. В этом отношении данные патентоспособные способы могут обеспечивать какое-либо количество повышения эффективности T-клеточной терапии или какую-либо степень лечения или предотвращения заболевания.
На фиг. 28-32, 36 и 48 проиллюстрированы способы и механизмы по настоящему изобретению.
Специалисту в данной области очевидно, что с развитием технологий патентоспособную идею можно будет реализовать различными способами. Изобретение и его варианты осуществления не ограничены примерами, описанными выше, и могут варьировать в пределах объема формулы изобретения.
ПРИМЕРЫ
Материалы и способы
Животная модель B16-OVA: экспрессирующие овальбумин клетки B16 (B16-OVA) поддерживали в RPMI, 10% FBS, 5 мг/мл G418, 20 мМ L-глутамин, 1× растворе Pen/Strep (GIBCO). Иммунокомпетентным самкам мышей C57BL/6 в возрасте 4-7 недель имплантировали подкожно 2,5×105 клеток B16-OVA в 50 мкл RPMI, 0% FBS, в правый бок, по одной опухоли на мышь. Приблизительно через 10 суток после имплантации опухоли (когда опухоли становились пригодными для инъекций, минимум ~3 мм в диаметре), мышей делили на группы и лечили в определенных экспериментах на шестые последовательные сутки внутриопухолевыми инъекциями или 50 мкл PBS или 1×109 вирусных частиц (в. ч.) онколитического аденовируса в 50 мкл PBS. В других экспериментах три инъекции делали в сутки 0, 2 и 4. Поскольку мышиные клетки не чувствительны к аденовирусу человека, множество внутриопухолевых вирусных инъекций использовали для того, чтобы имитировать индуцированное репликацией вируса воспаление, (Blair et al., 1989).
Адоптивный перенос: на первые сутки i.t. лечения мыши также получали посредством адоптивного переноса в интраперитонеальную полость от 5×105 до 2×106 покоившихся в течение ночи обогащенных CD8a и разросшихся спленоцитов от мышей C57BL/6-Tg(TcraTcrb)1100Mjb/J (OT-1) в возрасте 4-8 недель, которые генетически сконструированы для того, чтобы иметь только овальбумин (OVA)-специфичные CD8 T-клеточные рецепторы, в 100 мкл RPMI, 0% FBS. Обогащение CD8a осуществляли с помощью микробусин с мышиным CD8a (Ly-2) за 5 суток до переноса, по инструкциям производителя (Miltenyi Biotech, USA, № по каталогу 130-049-401). Обогащенные клетки увеличивались в числе в течение пяти суток в среде для лимфоцитов (RPMI, 10% FBS, 20 мМ L-глутамин, 1× раствор Pen/Strep, 15 мМ HEPES, 50 мкМ 2-меркаптоэтанол, 1 мМ пируват натрия) в присутствии рекомбинантного мышиного IL-2 (160 нг/мл) и растворимого антитела против CD3ε мыши (0,3 мкг/мл, Abcam, клон 145-2C11).
Обработка тканей для проточной цитометрии: Мышей умерщвляли и селезенки, дренирующие лимфатические узлы и опухоли собирали в от 1 до 10 мл RPMI, 10% FBS, и кровь собирали посредством терминального сердечного кровотечения в плевральную полость и переносили с помощью одноразового шприца в ЭДТА-содержащие микропробирки для центрифуги и обрабатывали для анализа: твердые ткани грубо диссоциировали с помощью скальпеля и растирали в 10 мл одноразовом стерильном наконечнике пипетки в от 5 до 10 мл лизирующего буфера ACK (150 мМ NH4Cl, 10 мМ KHCO3, 0,1 мМ ЭДТА, pH 7,2) и инкубировали при комнатной температуре (RT) в течение ~20 минут, после чего клетки осаждали центрифугированием на 1200 об/мин 5 мин +4°C, после чего клетки ресуспендировали в от 1 до 10 мл RPMI, 10% FBS, в зависимости от оценочного количества клеток, и пропускали через 40 мкм стерильный фильтр для того, чтобы создавать раствор отдельных клеток. В определенных экспериментах опухолевую ткань вместо этого обрабатывали непосредственно после нарезания скальпелем (перед добавлением ACK) в общем объеме 1 мл протеазного коктейля (RPMI с добавлением коллагеназы типа A, H или P, Roche, по 1 мг/мл и бензоназы, конечная концентрация 125 Ед/мл, Sigma, E1014-25KU) в течение 1-2 часов при 37°C, 5% CO2, после чего добавляли 10 мл лизирующего буфера ACK и клетки обрабатывали, как указано выше. 200 мкл цельной крови пипетировали в 5 мл лизирующего буфера ACK и обрабатывали, как указано выше. Клетки или инкубировали в течение ночи при 37°C, 5% CO2, или анализировали непосредственно с помощью иммуноокрашивания и проточной цитометрии.
Обработка тканей для цитокинового анализа: мышей умерщвляли и кусочки опухолей ~2-10 мм3 замораживали в 2 мл микропробирках для центрифуги на сухом льду и хранили при -80°C. Кусочки опухоли взвешивали и добавляли 200 мкл ледяного PBS. Кусочки гомогенизировали с помощью ротора Tissue Master 125, добавляли 1× коктейль ингибиторов протеаз (Sigma) и BSA в конечной концентрации 0,1% и пробирки держали на льду. Опухолевый гомогенат центрифугировали 2000 об/мин 10 мин +4°C и супернатант анализировали с применением цитокиновых гранул CBA Flex Set (BD, USA) на BD FACSArray, по инструкциям производителя.
Эксперименты, подтверждающие изобретение
Эксперимент 1 (цитокины и хемокины, индуцированные с помощью внутриопухолевой аденовирусной инъекции)
Чтобы изучать, может ли аденовирусная инфекция вести к экспрессии цитокинов и хемокинов, авторы изобретения инъецировали мышам, несущим подкожные опухоли B16-OVA, внутрь опухоли или PBS или 5/3 химерный онколитический аденовирус на сутки 0, 1, 2, 3, 4 и 5. Опухоли от трех мышей на группу лечения извлекали и обрабатывали для цитокинового анализа на сутки 0 (до вирусной инъекции=базовый контроль), и от трех других мышей на каждый момент времени в сутки 6, 10, 14 и 18.
Что важно, результаты демонстрировали индуцированное вирусом увеличение секреции IFN-γ и последующую повышающую регуляцию индуцируемых IFN-γ хемокинов RANTES, MIP-1α и MCP-1 на сутки 10 (фиг. 1).
Эти находки важны для увеличения терапевтической эффективности адоптивной клеточной терапии.
На основании этих данных лечение онколитическим вооруженным цитокинами аденовирусом ведет к благоприятному изменению опухолевого микроокружения, увеличенному хемотаксису адоптивно перенесенных клеток иммунной системы и расширенному распознаванию опухолевых клеток цитотоксическими CD8+ T-клетками.
Эксперимент 2 (опосредованное аденовирусом улучшение адоптивной T-клеточной терапии)
Чтобы изучать влияние лечения аденовирусом на адоптивную T-клеточную терапию, мышиные опухоли меланомы B16-OVA лечили 5/3 химерным онколитическим аденовирусом отдельно или в комбинации с адоптивным переносом опухолеспецифичных OT-I клеток и сравнивали с мышами, которые получали внутриопухолевые инъекции PBS. Результаты этих независимых экспериментов, резюмированные на фиг. 2, показывают, с одной стороны, что вирусные инъекции сами по себе (стоит помнить, что аденовирус человека не способен к продуктивной репликации в клетках мыши) вели к незначительному контролю опухолевого роста, который длился до суток 14 после лечения и уменьшался после этого (фиг. 2A). С другой стороны, когда мышам, которых лечили, адоптивно переносили 5×105 или 2×106 OT-I клеток, статистически значимые различия между группами PBS и Ad получали в двух отдельных экспериментах (фиг. 2B и 2C, соответственно).
Таким образом, присутствие вируса в опухоли оказывало выраженный усиливающий эффект на адоптивную клеточную терапию. Шесть внутриопухолевых вирусных инъекций по 1×109 в. ч. каждая у авторов изобретения давали в комбинации с адоптивным переносом OT-I клеток равную или превосходящую противоопухолевую эффективность по сравнению с тем, о чем сообщали Song et al. (2011, Mol Ther) для одной внутримышечной инъекции 1×1010 в. ч. OVA-экспрессирующего аденовируса с дефектом репликации (Ad-OVA), смешанного с равным количеством аденовируса, совместно экспрессирующего A20-специфичную РНК в виде короткой шпильки и секреторную форму флагеллина, который стимулирует Toll-подобный рецептор 5 (Ad-shAF) в модели меланомы B16.OVA (Song XT et al. Mol Ther. 2011 Jan;19(1):211-7, PMID: 20959814). В свете этих результатов новый аспект данного изобретения состоит в том, чтобы направлять вирусную инъекцию в опухоль, где авторы изобретения могут достичь, даже с применением невооруженного вируса, контроля опухоли, который превосходит мультиммунофункциональный вооруженный вирус, вводимый внутримышечно.
Эксперимент 3 (опосредованные аденовирусом изменения в качестве и количестве в популяциях клеток иммунной системы in vivo)
Чтобы изучать направленную миграцию и пролиферацию адоптивно перенесенных клеток из эксперимента 2, OT-I клетки окрашивали ex vivo 5 мкМ карбоксифлуоресцеинсукцинимидиловым эфиром (CFSE). Этот флуоресцентный краситель для окрашивания клеток разводится при каждом клеточном делении и, следовательно, позволяет отслеживать пролиферацию лимфоцитов посредством проточной цитометрии, анализируя ~1/2 дольное снижение интенсивности сигнала флуоресценции при каждом клеточном делении (вплоть до 7 делений, здесь помечено M0-7). На сутки 1 после переноса результаты показывали индуцированное вирусом накопление перенесенных клеток OT-I (CD8+ CFSE+ дважды положительная популяция) в опухолях, сопутствующее уменьшению этих клеток в крови (фиг. 3A). В более поздние моменты времени также общее количество CD8+ T-клеток выглядело более высоким в опухолях, которые лечили вирусами, по сравнению с опухолями, в которые инъецировали PBS, и на сутки 14 общее количество CD8+ T-клеток возрастало в лимфоидных органах мышей, которых лечили вирусами.
Количества делений OT-I клеток в различные моменты времени представлены на фиг. 3B. Поскольку статус пролиферации OT-I клеток был одинаковым в обеих группах на сутки 1, различия в количестве CD8+ клеток в различных органах были обусловлены индуцированной аденовирусом направленной миграцией клеток иммунной системы. Однако в более поздние моменты времени ситуация менялась, и увеличение OT-I клеток в опухоли, которую лечили аденовирусом, было обусловлено увеличенной пролиферацией лимфоцитов. На сутки 6 большинство OT-I клеток в опухолях, которые лечили PBS, задерживалось в фазе M5, тогда как перенесенные клетки в группе аденовируса продолжали пролиферировать (деления M6-M7). Эти данные подсказывают, что онколитическая виротерапия или нецитолитическая вирусная инфекция ведут к расширенной направленной миграции и пролиферации адоптивно перенесенных лимфоцитов через нарушение подавления иммунитета в опухолях, которое привлекает клетки иммунной системы, что вносит вклад в активацию CD8+ клеток, и/или через определенные другие важные механизмы, которые помогают преодолеть анергию T-клеток.
В качестве подтверждения находок авторов изобретения в животных моделях, они наблюдали временное снижение количества лимфоцитов крови во время первых суток после введения онколитического вируса пациентам с запущенными злокачественными опухолями (фиг. 4), что говорит о мобилизации циркулирующих T-клеток в ответ на острую аденовирусную инфекцию в опухоли.
Кроме того, в подтверждение того, что аденовирусная инфекция рекрутирует T-клетки в опухоли, авторы изобретения обнаруживали повышенные количества CD8+ T-клеток в тканевых срезах биоптатов опухолей после лечения по сравнению с тем, что было до (фиг. 5).
На фиг. 6 представлены результаты аденовирусных инъекций в комбинации с адоптивным переносом T-клеток.
На фиг. 7 показано существенное увеличение числа «естественных» противоопухолевых T-клеток из-за адоптивного переноса и вирусной инъекции.
На фиг. 8 представлены активированные CD8+ клетки в опухоли и экспрессия TIM-3 в опухоли на сутки 14.
На фиг. 9 показано, что увеличение противоопухолевых T-клеток и снижение иммуносупрессии ведет к системному иммунитету против опухолевых антигенов.
На фиг. 10 представлено распределение OTI T-клеток после вирусной инъекции.
На фиг. 11 показано, что устранение иммуносупрессии может индуцировать размножение клеток.
На фиг. 12 представлена эффективность рекомбинантных цитокинов (без вируса) в комбинации с OT1 клетками.
Эксперимент 4 (Адоптивно перенесенные T-клетки + вооруженный мышиными цитокинами Ad5 аденовирус):
Модель:
C57BL/6 с B16-OVA (0,25×10e6 клеток на животное)
Группы:
Без инъекции
Ad5-Luc
Ad5-CMV-mTNFa
Ad5-CMV-mIFNg
Ad5-CMV-mIL-2
Ad5-CMV-mIFNb1
Без инъекции + OT1
Ad5-Luc + OT1
Ad5-CMV-mTNFa + OT1
Ad5-CMV-mIFNg + OT1
Ad5-CMV-mIL-2 + OT1
Ad5-CMV-mIFNb1 + OT1
Ad5 вектор представляет собой вектор из нерепликативного аденовируса человека, кодирующего трансген мыши. Конструкции получали с применением технологии AdEasy (Agilent Inc); кассета с трансгеном (управляемым CMV промотором) находится в удаленной области E1 (см., например, Diaconu I et al. Cancer Res. 2012 May 1;72(9):2327-38).
Размер группы:
N=7, 12×7=84 (+ дополнительные 20%=100)
Режим лечения:
OT1 клетки: 2×10e6 на животное интраперитонеально на сутки 1
Вирусные инъекции: 1×10e9 вирусных частиц (OD260) на сутки 1 и еженедельно после этого
Конечная точка:
Объем опухоли (измеряли каждые 2 суток в течение первой недели и затем каждые 3 суток)
Сбор опухолей и селезенок, когда мыши погибали или были умерщвлены; для FACS и/или ELISPOT (фокус на анализах, наиболее релевантных согласно данным Siri).
Наилучшими трансгенами в комбинации с T-клеточной терапией являются TNFα и IL-2 (фиг. 13). В подтверждение данных, те же цитокины использовали в эксперименте без вируса.
На фиг. 14 представлены результаты влияния различных вирусов (без T-клеточной терапии) на размер опухоли (фиг. 14).
На фиг. 15 представлены превосходные результаты T-клеточной терапии в комбинации с вектором Ad5-CMV-mTNFα.
На фиг. 16 представлены превосходные результаты T-клеточной терапии в комбинации с вектором Ad5-CMV-mIL-2.
Новые вирусные конструкции
Следующие новые вирусные конструкции представлены в виде примеров технологии, предложенной авторами изобретения:
C5a и TNF-α экспрессирующие онколитические вирусы
Авторы изобретения создавали новые онколитические Ad5/3 аденовирусы, несущие активную часть компонента комплемента C5a или TNF-α человека в качестве трансгенов вместо областей генов 6.7k/gp19 (фиг. 17).
Эксперимент 5 (экспрессия трансгена с C5a-кодирующего аденовирусного вектора):
Для того чтобы подтверждать, в качестве обоснования идеи, что онколитические аденовирусы способны экспрессировать выбранные цитокины, предложенные для усиления адоптивной клеточной терапии, авторы изобретения инфицировали клетки человека A549 в культуре аденовирусом, кодирующим C5a, по 10 в. ч./клетка (фиг. 24) и оценивали уровни C5a в супернатанте клеточной культуры в различные моменты времени после инфекции с помощью ELISA. Результаты действительно валидируют предположение и подтверждают создание проприетарных аденовирусных конструкций, несущих выбранные цитокины.
Эксперимент 6 (эффект, оказываемый на миграцию моноцитов новыми аденовирусными векторами)
Авторы изобретения тестировали способность C5a рекрутировать моноциты с применением анализа хемотаксиса in vitro: клетки A549 инфицировали или аденовирусом, экспрессирующим C5a, или невооруженным контрольным вирусом (10 в. ч./клетка - инфекционные единицы между вирусами схожи), или обрабатывали в PBS, и среды собирали через 48 ч после инфекции и использовали для того, чтобы рекрутировать моноцитарную клеточную линия человека THP1 в анализе хемотаксиса в планшетах Transwell по инструкциям производителя (Millipore QCM, № по каталогу ECM512). Результаты показывают значительно более сильное привлечение моноцитов супернатантом от инфицированных C5a-экспрессирующим вирусом клеток, чем с помощью среды от неинфицированных клеток или клеток, инфицированных невооруженным контрольным вирусом (фиг. 25).
Эксперимент 7 (противоопухолевая эффективность вооруженного C5a аденовируса):
Для того чтобы оценивать противоопухолевую активность C5a в контексте нецитолитической опухолевой инфекции, авторы изобретения лечили развившиеся опухоли B16-OVA у мышей C57BL/6 на сутки 0, 2 и 4 с применением или PBS, или C5a-экспрессирующего или невооруженного контрольного вируса. Результаты показывают сильный противоопухолевый эффект C5a-экспрессирующего вируса (фиг. 26).
Эксперимент 8 (противоопухолевая T-клеточная экспансия, увеличенная с помощью C5a вируса):
Для того чтобы оценивать, связано ли наблюдаемое увеличение противоопухолевой эффективности C5a-экспрессирующего вируса (фиг. 24) с T-клетки, в опухолях анализировали посредством проточной цитометрии овальбумин-специфичные CD8+ T-клетки, обнаруживаемые с помощью окрашивания APC-конъюгированным пентамером, специфичным к TCR, распознающим MHC I, нагруженному иммунодоминантным овальбуминовым пептидом SIINFEKL (ProImmune, USA). В действительности, опухоли в группе C5a вируса содержали значительно большую фракцию опухолеспецифичных CD8 T-клеток, чем опухоли, в которые инъецировали контрольный вирус или PBS (фиг. 27).
Эксперимент 9 (экспрессия трансгена с TNF-α-кодирующего аденовируса)
Подобно C5a вирусу (фиг. 24 и эксперимент 5), авторы изобретения тестировали способность hTNF-α-экспрессирующего онколитического аденовируса опосредовать секрецию предпочтительного трансгена. Результаты подтверждают экспрессию (фиг. 18).
Эксперимент 10 (биологический эффект экспрессируемого трансгена сохраняется в онколитическом аденовирусе)
Для того чтобы оценивать, сохраняет ли экспрессируемый аденовирусом трансген свои биологические эффекты, безвирусный супернатант (пропущенный через фильтр 100 кДа) от клеток A549, инфицированных контрольным невооруженным вирусом или экспрессирующим TNF-α вирусом (варьирует в. ч./клетка, 72 ч после инъекции) применяли к клеткам WEHI-13VAR (ATCC CRL-2148), которые чувствительны к TNF-α, и у этих клеток оценивали жизнеспособность через 72 часа после воздействия супернатанта (например, Espevik T et al. J Immunol Methods. 1986; 95(1): 99-105, описан способ). Результаты показывают, что TNF-α, экспрессированный с онколитического аденовируса, сохраняет выраженные биологические эффекты (фиг. 19).
Эксперимент 11 (онколитические цитокин-экспрессирующие вирусы сохраняют способность уничтожать клетки in vitro):
Поскольку TNF-α может иметь противовирусные эффекты, важно подтверждать, что онколитический эффект аденовируса, экспрессирующего TNF-α, сохраняет свою способность инфицировать и уничтожать клетки злокачественной опухоли. Несколько клеточных линий злокачественных опухолей в культуре инфицировали экспрессирующими TNF-α или контрольными вирусами и оценивали жизнеспособность с помощью анализа CelltiterGlo AQ MTS по инструкциям производителя (Promega, USA). Результаты показывают, что вирус является онколитическим in vitro (фиг. 19-20).
Эксперимент 12 (синергия между лучевой терапией и онколитическим вирусом, экспрессирующим TNF α):
Авторы изобретения лечили голых мышей, несущих подкожные ксенотрансплантаты A549, внутриопухолево с применением вирусов с сопутствующим фокусированным внешним облучением пучком (XRT) или без него (фиг. 21). RD обозначает вирус с дефицитом репликации, а невооруженный вирус представляет собой онколитический вирус без TNFα.
Эксперимент 13 (увеличенная противоопухолевая эффективность экспрессирующего TNF-α аденовируса у иммунокомпетентных организмов-хозяев):
Для того чтобы тестировать, может ли онколитический аденовирус, который не реплицируется в мышиных клетках, быть способным вызывать противоопухолевые эффекты in vivo у иммунокомпетентных мышей, мышам с развившимися опухолями B16.OVA инъецировали внутрь опухоли экспрессирующий TNF-α или невооруженный контрольный вирус или PBS, подобно тому, как на фиг. 26. Результаты показывают более выраженный общий контроль опухоли с применением экспрессирующего TNF-α вируса по сравнению с контролями (фиг. 22), что подсказывает, что TNF-α человека частично активен у мышей, и подтверждает идею о вооружении вирусов для того, чтобы достигать более выраженных противоопухолевых эффектов.
Эксперимент 14 (противоопухолевая T-клеточная экспансия, увеличенная с помощью TNFα-вируса):
Подобно эксперименту 11, авторы изобретения хотели тестировать, связан ли наблюдаемый противоопухолевый эффект экспрессирующего TNF-α вируса с индукцией опухолеспецифичного цитолитического T-клеточного ответа. Авторы изобретения извлекали опухоли и обрабатывали их для проточного цитометрического анализа, как в эксперименте 11. Результаты (фиг. 23) в действительности подтверждают, что экспрессия TNF-α также облегчает экспансию опухолеспецифичных T-клеток в месте опухоли, что явно свидетельствует в пользу предложенной технологии.
На фиг. 28-32, 36 и 48 проиллюстрированы способы и механизмы по настоящему изобретению.
Эксперимент 15 (эксперимент по комбинированию с двумя различными аденовирусными векторами и OT1(Ad-mTNFa/Ad-mIL-2 + OT1)):
Модель:
C57BL/6 с B16-OVA (0,25×10e6 клеток на животное)
Группы:
Ad5-CMV-mTNFa (1×10e9 в. ч.)
Ad5-CMV-mIL-2 (1×10e9 в. ч.)
Ad5-CMV-mTNFa + Ad5-CMV-mIL-2 (0,5 + 0,5×10e9 в. ч.)
Ad5-CMV-mTNFa + OT1
Ad5-CMV-mIL-2 + OT1
Ad5-CMV-mTNFa + Ad5-CMV-mIL-2 + OT1
Ad5Luc1 + OT1
Без инъекции (имитатор-имитатор)
Ad5 вектор представляет собой вектор из нерепликативного аденовируса человека, кодирующий трансген мыши. Конструкции получали с применением технологии AdEasy (Agilent Inc); кассета с трансгеном (под управлением CMV промотора) находится в удаленной области E1 (см., например, Diaconu I et al. Cancer Res. 2012 May 1;72(9):2327-38).
Размер группы:
N=9
100 по порядку
Режим лечения:
OT1 клетки: 1,5×10e6 на животное интраперитонеально на сутки 1 (то же количество, что в предыдущем эксперименте, не 2×10e6)
Вирусные инъекции: для отдельных средств: 1×10e9 вирусных частиц (OD260) на сутки 1 и еженедельно после этого; для комбинации: 0,5×10e9 в. ч. + 0,5×10e9 в. ч. на сутки 1 и еженедельно после этого
Конечная точка: объем опухоли (измеряли каждые 2 суток в течение первой недели и затем каждые 3 суток)
Дополнительные эксперименты, подтверждающие изобретение
Несколько экспериментов на животных подтверждают изобретение. Сначала авторы изобретения проводили скрининг оптимальных кандидатов цитокинов для того, чтобы комбинировать с адоптивным переносом T-клеток, с применением рекомбинантных мышиных форм цитокинов (фиг. 49). Цитокин(цитокины) выбирали из следующей группы: интерферон α, интерферон β, интерферон γ, комплемент C5a, GMCSF, IL-2, TNFα, CD40L, IL-12, IL-23, IL-15, IL-17, CCL1, CCL11, CCL12, CCL13, CCL14-1, CCL14-2, CCL14-3, CCL15-1, CCL15-2, CCL16, CCL17, CCL18, CCL19, CCL19, CCL2, CCL20, CCL21, CCL22, CCL23-1, CCL23-2, CCL24, CCL25-1, CCL25-2, CCL26, CCL27, CCL28, CCL3, CCL3L1, CCL4, CCL4L1, CCL5, CCL6, CCL7, CCL8, CCL9, CCR10, CCR2, CCR5, CCR6, CCR7, CCR8, CCRL1, CCRL2, CX3CL1, CX3CR, CXCL1, CXCL10, CXCL11, CXCL12, CXCL13, CXCL14, CXCL15, CXCL16, CXCL2, CXCL3, CXCL4, CXCL5, CXCL6, CXCL7, CXCL8, CXCL9, CXCL9, CXCR1, CXCR2, CXCR4, CXCR5, CXCR6, CXCR7 и XCL2. Общая схема генома вируса, содержащего цитокиновый трансген или два трансгена, представлена на фиг. 33 и 34. На фиг. 35 представлены нуклеотидные и аминокислотные последовательности 2A. Нуклеотидные последовательности вирусных векторов, которые содержат трансгены C5a, hCD40L, hIFNa2, hIFNb1, hIFNg1, hIL-2 или TNFa, представлены в SEQ ID №№ 1-7, соответственно (Ad5/3-E2F-D24-трансген). Кроме того, нуклеотидные последовательности вирусных векторов, которые содержат два трансгена, один представляет собой IL-2 и другой представляет собой C5a, CD40L, IFNa2, IFNb, IFNg, GMCSF или TNFa, представлены в SEQ ID №№ 8-21 (SEQ ID № 8 C5a-2A-IL-2, SEQ ID № 9 IFNa-2A-IL-2, SEQ ID № 10 TNFa-2A-IL-2, SEQ ID № 11 CD40L-2A-IL-2, SEQ ID № 12 IFNb-2A-IL-2, SEQ ID № 13 GMCSF-2A-IL-2, SEQ ID № 14 IFNg-2A-IL-2, SEQ ID № 15 C5a-IRES-IL-2, SEQ ID № 16 IFNa-IRES-IL-2, SEQ ID № 17 TNFa-IRES-IL-2, SEQ ID № 18 CD40L-IRES-IL-2, SEQ ID № 19 IFNb-IRES-IL-2, SEQ ID № 20 GMCSF-IRES-IL-2, SEQ ID № 21 IFNg-IRES-IL-2) (Ad5/3-E2F-D24-трансген-IRES/2A-трансген).
Несколько наилучших кандидатов выбирали для экспериментов с комбинацией цитокин/вирус, где режим приблизительно остается тем же, и все мыши получают интраперитонеальную инъекции CD8a+ обогащенных OT-I лимфоцитов и внутриопухолевое лечение выбранным цитокином в смеси с аденовирусом. Кроме того, проводили отдельный эксперимент по направленной миграции, используя находящиеся у авторов изобретения аденовирусы с дефицитом репликации, которые кодируют или мышиные цитокины или цитокины человека с подтвержденной активностью у мышей (фиг. 50). RD обозначает вирус с дефицитом репликации. На основании этих экспериментов финальный кандидат-цитокин или кандидаты-цитокины можно выбирать и дополнительно анализировать, даже в клинике.
Результаты экспериментов показывают, что a) вирусная инъекция в опухоли ведет к расширенной направленной миграции T-клеток в опухоль, b) вирусная инъекция ведет к усиленной экспрессии MHC1 в опухолях, c) происходит активация сигналов опасности, что ведет к меньшей толерантности и иммуносупрессии, d) T-клетки размножаются в опухоли после вирусных инъекций. Важно, что добавление цитокина в виде трансгена усиливает каждый из этих эффектов. Стоит отметить, что двойные трансгены дополнительно усиливают эффект. Таким образом, внутриопухолевая инъекция вооруженного цитокином онколитического аденовируса усиливает эффект переноса адоптивных клеток синергическим образом больше, чем можно достигнуть с применением отдельно или вирусных векторов или переноса адоптивных клеток.
Для исследования направленной миграции T-клеток и биораспространения после адоптивного переноса, проводили эксперимент по ОФЭКТ/КТ визуализации (фиг. 51). CD8a+ обогащенные OT-I лимфоциты радиоактивно метили с применением111In и адоптивно переносили мышам-реципиентам.
Поскольку время полужизни оксина индия относительно невелико (2,83 суток), максимальный период наблюдения для визуализации ограничен 7-ю сутками. Из-за этого ограничения клетки метили двумя партиями и переносили мышам в два различных момента времени. Данные визуализации от первой партии охватывают события направленной миграции в сутки 0-7, тогда как вторая партия позволяла авторам изобретения наблюдать в опухолях события во время суток 8-14 после вируса.
Онколитические Ad3 вирусы (фиг. 37-40, SEQ ID №№ 30 и 31 (Ad3-hTERT-E3del-CMV-CD40L и Ad3-hTERT-E3del-E2F-CD40L))
Стратегия клонирования:
1. Конструирование Ad3 3'-концевой плазмиды, содержащей соответствующую экспрессирующую кассету, эта плазмида содержит 3'ITR из Ad3 генома, область E3 от 29,892 до 30,947 из Ad3 генома заменяли на экспрессирующую кассету. (Примечание: авторы изобретения использовали преимущество участка рестрикции EcoRI в Ad3 геноме близко к 3'-концу)
2. Конструирование Ad3 5'-концевой плазмиды, эта плазмида содержит 5'ITR и hTERT-E1. (Примечание: авторы изобретения использовали преимущества уникального участка рестрикции NotI в геноме Ad3 и участка рестрикции NheI близко к 5'-концу)
3. Конструирование pWEA-Ad3-hTERT-CMV-CD40L и pWEA-Ad3-hTERT-E2F-CD40L. (примечание: Авторы изобретения использовали преимущество фаговой системы упаковки)
Конструирование Ad3 3'-концевой плазмиды, содержащей соответствующую экспрессирующую кассету:
1. ПЦР амплификация промотора E2F, прямой праймер: 5'AAAttaattaatggtaccatccggacaaagc3' (SEQ ID № 22), обратный праймер 5' TTTgctagcggcgagggctcgatcc3' (SEQ ID № 23). Клонировали в TA вектор pGEM-T (Promega)→pGemT-E2F
2. ПЦР амплификация фрагмента CD40L, прямой праймер: 5'TAGCTGCTAGCATGATCGAAACATACAAC3' (SEQ ID № 26), обратный праймер: 5'GTCAATTTGGGCCCTCAGAGTTTGAGTAAGCCAA3' (SEQ ID № 27). Клонировали в pGEM-T→pGemT-CD40L.
3. Ad3 3'-концевую плазмиду авторов изобретения, содержащую CMV-GFP(pWEA-Ad3-3'-конец-CMVGFP), расщепляли с применением NheI/ApaI для того, чтобы удалять GFP, pGemT-CD40L расщепляли с применением NheI/ApaI → pWEA-Ad3-3'-конец-CMV-CD40L.
4. Промотор CMV в pWEA-Ad3-3'-конец-CMV-CD40L заменяли на промотор E2F (pGemT-E2F расщепляли с применением PacI/NheI) →pWEA-Ad3-3'-конец-E2F-CD40L
Конструирование Ad3 5'-концевой плазмиды, содержащей hTERT-E1:
1. ПЦР амплификация 5'-конца Ad3 генома из pKBS2-hTERT (плазмида из публикации Ad3-hTERT-E1A), прямой праймер 5' gtcagtttaaacttaggccggccctatctatataatataccttatagatggaatgg3' (SEQ ID № 28), обратный праймер 5' CTTCATCAGCAGCTAGCAGCATAGAATCAG3' (SEQ ID № 29). Клонировали в pGem-T→pGemT-Ad3-5'-конец-hTERT.
2. Плазмиду pWEA-Ad3 (которая содержит полноразмерный геном ad3), расщепляли с применением FseI/NotI, фрагмент 13,2 т. п. о., который содержит 5'-конец генома Ad3, клонировали в модифицированный вектор из pBluescript KS(-) (участки рестрикции между SacI и XbaI модифицировали как SacI-PmeI-MluI-FseI-SalI-NotI-XbaI) → pBS-Ad3-5'-конец
3. pBS-Ad3-5'-конец плазмиды расщепляли с применением PmeI/NheI, фрагмент ~800 п. о., который содержит 5'ITR, заменяли на соответствующий фрагмент PmeI/NheI из pGemT-Ad3-5'-конец-hTERT → pBS-Ad3-5'-конец-hTERT
pWEA-Ad3-hTERT-CMV-CD40L и pWEA-Ad3-hTERT-E2F-CD40L:
1. Плазмиду pWEA-Ad3-hTERT-E2F- расщепляли с применением EcoRI для того, чтобы удалять 3'-концевой геном и лигировать с соответствующим фрагментом, содержащим экспрессирующую кассету из pWEA-Ad3-3'-конец-CMV-, pWEA-Ad3-3'-конец-CMV-CD40L и pWEA-Ad3-3'-конец-E2F-CD40L
2. Продукт лигирования упаковывали в фаги с применением экстракта для упаковки Gigapack III plus (Stratagen) и размножали (штамм Xl1 blue)
Функциональность Ad3 вирусов тестировали in vitro и результаты представлены на фиг. 41. Все новые вирусы были функциональны и способны инфицировать опухолевые клеточные линии.
Вирусы также тестировали на CHO-K7, но они не оказывали эффект на жизнеспособность этих клеток в течение TCID50. Вероятно, это обусловлено отсутствием человекоподобного десмоглеина-2 на поверхности этих клеток хомяка.
Результаты AD3 векторов in vivo
Все эксперименты на животных одобрены Experimental Animal Committee из University of Helsinki и местным правительством южной Финляндии. Часто осуществляли мониторинг состояния здоровья мышей, и умерщвляли их как только отмечали признаки боли или дистресса. Использовали самок мышей с тяжелым комбинированным иммунодефицитом Fox Chase (Charles River).
Ортотопическую модель перитонеально диссеминированной злокачественной опухоли яичников создавали посредством инъекции 5×10e6 клеток SKOV3-luc интраперитонеально в 300 мл чистой модифицированной по способу Дульбекко среды Игла мышам с тяжелым комбинированным иммунодефицитом (n=5 на группу). После 3 суток выполняли неинвазивную визуализацию мышей и лечили их интраперитонеально посредством инъекции PBS или 109 в. ч. В PBS на мышь. Мышей визуализировали на сутки 3, 7, 14, 21 и 25 с применением IVIS 100 (Xenogen, Alameda, CA) для того, чтобы оценивать число опухолевых клеток у мышей. Для биолюминесцентной визуализации 150 мг/кг D-люциферина (Promega) инъецировали интраперитонеально и регистрировали через 10 мин при времени экспозиции 10 с, 1 единица диафрагмы, средней группировке и открытом фильтре. Во время визуализации мыши находились под анестезией газообразным изофлураном. Изображения накладывали с применением Living Image 2.50 (Xenogen). Общий поток (фотоны/с) измеряли посредством очерчивания областей, представляющих интерес, вокруг перитонеальной области мыши. Фон вычитали.
На фиг. 45 представлена противоопухолевая эффективность вирусов на основе Ad3 in vivo.
Анализ пролиферации клеток с MTS (фиг. 42-44)
В сутки 1 105 клеток на лунку (A549, PC3-MM2 или SKOV3-luc) высевали в 96-луночные планшеты в 100 мкл среды для выращивания (GM), которая содержала 5% FBS. На сутки 2 монослой промывали один раз GM, содержащей 5% FBS. Затем клетки инфицировали различными вирусами в дозах 100, 10, 1, 0,1 и 0 вирусных частиц на клетку. После этого клетки инкубировали в течение одного часа на качалке и затем промывали в GM. После добавления новой 5% GM, клетки оставляли в инкубаторе и GM заменяли каждые четвертые сутки. Тест прекращали с помощью добавления реактива mts (Promega) после того, как цитопатический эффект одного из тестируемых вирусов достигал 100% при наивысшей концентрации. После двух часов инкубации измеряли поглощение с применением фильтра 490 нм. Затем вычитали фон и анализировали результаты.
Терапевтическое окно онколитического аденовируса, который кодирует мышиный CD40L, у иммунокомпетентных мышей
У иммунокомпетентных животных вирусные геномы присутствовали в опухолях после внутривенных инъекций (фиг. 46). Мышей Albino C57 инокулировали подкожно клетками B16-ova мыши и лечили внутривенно 5 различными вирусными дозами вируса на основе Ad5, который кодирует CD40L мыши (см. эксперименты 4 и 15). Опухоли 3 животных на группу собирали и хранили при -80°C. Экстрагировали общую ДНК и изучали нагрузку вирусной ДНК с применением количественной ПЦР. Число копий вируса E4 нормализовали по геномной ДНК с применением праймеров к B-актину мыши. На фиг. 46 каждая пиктограмма представляет одну опухоль; горизонтальная линия обозначает медиану для группы. Имитатор: n=5; доза 5: n=4; доза 4: n=4; доза 3: n=4; доза 2: n=6; доза 1: n=2; доза 2 i.t.: n=4. доза 5: 1×1011 в. ч./мышь; доза 4: 3×1010в. ч./мышь; доза 3: 1×1010 в. ч./мышь; доза 2: 1×109 в. ч./мышь; доза 1: 1×108 в. ч./мышь; положительный контроль (доза 2 внутрь опухоли).
При дозе 5, 67% мышей имели признаки печеночной токсичности. Доза 4 позволяла достигать хорошей опухолевой трансдукции после внутривенной доставки, без признаков печеночной токсичности.
Результаты эксперимента с высвобождением ферментов печени представлены на фиг. 47. Эксперимент с высвобождением ферментов печени осуществляли следующим образом.Все протоколы для животных просматривали и одобряли Experimental Animal Committee из University of Helsinki и местное правительство южной Финляндии. Самкам мышей Аlbino C57 в возрасте от трех до четырех недель (Harlan Laboratories, The Netherlands) инъецировали 2,5×105 клеток B16-ova подкожно в оба бока и рандомизировали среди 7 групп (3 мыши/группа). Ad5/3 CMV-mCD40L вирус, разведенный в фосфатно-солевом буфере (PBS), инъецировали внутривенно по 108-1011вирусных частиц (в. ч.)/мышь (доза 1 - доза 5). Одна группа лечения получала дозу 2 (109 в. ч./клетка) внутрь опухоли в качестве положительного контроля. Животных анестезировали перед любыми процедурами и ежедневно осуществляли мониторинг состояния здоровья. Через 48 ч после вирусной инъекции мышей умерщвляли и кровь собирали посредством прокола сердца. Сыворотку отделяли центрифугированием образцом крови на скорости 5000 об/мин в течение 10 минут. Уровни аланинаминотрансферазы (ALT) и аспартатаминотрансферазы (AST) (Ед/л) в образцах сыворотки определяли количественно в University of Helsinki Clinical Chemistry Core с применением клинического химического анализатора Siemens ADVIA 1650. Гемолитические образцы исключали из анализа, поскольку сывороточный гемолиз мог мешать анализам (ложные высокие уровни ALT и AST). Столбцы показывают средние значения + SEM.
Литературные источники
Blair GE, Dixon SC, Griffiths SA, Zajdel ME. Restricted replication of human adenovirus type 5 in mouse cell lines. Virus Res. 1989 Dec;14(4):339-46.
Ekkens MJ, Shedlock DJ, Jung E, Troy A, Pearce EL, Shen H, Pearce EJ. Th1 and Th2 cells help CD8 T-cell responses. Infect Immun. 2007 May;75(5):2291-6.
Kratky W, Reis e Sousa C, Oxenius A, Spörri R. Direct activation of antigen-presenting cells is required for CD8+ T-cell priming and tumor vaccination. Proc Natl Acad Sci U S A. 2011 Oct 18;108(42):17414-9.
Lugade AA, Sorensen EW, Gerber SA, Moran JP, Frelinger JG, Lord EM. Radiation-induced IFN-gamma production within the tumor microenvironment influences antitumor immunity. J Immunol. 2008 Mar 1;180(5):3132-9.
Propper DJ, Chao D, Braybrooke JP, Bahl P, Thavasu P, Balkwill F, Turley H, Dobbs N, Gatter K, Talbot DC, Harris AL, Ganesan TS. Low-dose IFN-gamma induces tumor MHC expression in metastatic malignant melanoma. Clin Cancer Res. 2003 Jan;9(1):84-92.
Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004 Feb;75(2):163-89.
Street D, Kaufmann AM, Vaughan A, Fisher SG, Hunter M, Schreckenberger C, Potkul RK, Gissmann L, Qiao L. Interferon-gamma enhances susceptibility of cervical cancer cells to lysis by tumor-specific cytotoxic T cells. Gynecol Oncol. 1997 May;65(2):265-72.
Литературные источники для вирусных конструкций
Blair GE, Dixon SC, Griffiths SA, Zajdel ME. Restricted replication of human adenovirus type 5 in mouse cell lines. Virus Res. 1989 Dec;14(4):339-46.
Ekkens MJ, Shedlock DJ, Jung E, Troy A, Pearce EL, Shen H, Pearce EJ. Th1 and Th2 cells help CD8 T-cell responses. Infect Immun. 2007 May;75(5):2291-6.
Kratky W, Reis e Sousa C, Oxenius A, Spörri R. Direct activation of antigen-presenting cells is required for CD8+ T-cell priming and tumor vaccination. Proc Natl Acad Sci U S A. 2011 Oct 18;108(42):17414-9.
Lugade AA, Sorensen EW, Gerber SA, Moran JP, Frelinger JG, Lord EM. Radiation-induced IFN-gamma production within the tumor microenvironment influences antitumor immunity. J Immunol. 2008 Mar 1;180(5):3132-9.
Propper DJ, Chao D, Braybrooke JP, Bahl P, Thavasu P, Balkwill F, Turley H, Dobbs N, Gatter K, Talbot DC, Harris AL, Ganesan TS. Low-dose IFN-gamma induces tumor MHC expression in metastatic malignant melanoma. Clin Cancer Res. 2003 Jan;9(1):84-92.
Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004 Feb;75(2):163-89.
Street D, Kaufmann AM, Vaughan A, Fisher SG, Hunter M, Schreckenberger C, Potkul RK, Gissmann L, Qiao L. Interferon-gamma enhances susceptibility of cervical cancer cells to lysis by tumor-specific cytotoxic T cells. Gynecol Oncol. 1997 May;65(2):265-72.
Реферат
Настоящее изобретение относится к биотехнологии. В частности, изобретение относится к терапии злокачественных опухолей человека. Более конкретно, настоящее изобретение относится к онколитическим аденовирусным векторам отдельно или вместе с терапевтическими композициями для терапевтического применения и к способам терапии злокачественных опухолей. В одном из аспектов настоящее изобретение относится к отдельному введению адоптивной клеточной терапевтической композиции и онколитических аденовирусных векторов. Кроме того, настоящее изобретение относится к фармацевтическому набору и фармацевтической композиции, в которых используют онколитические аденовирусные векторы. 3 н. и 2 з.п. ф-лы, 51 ил., 15 пр.
Комментарии