Циклические пуриновые динуклеотиды в качестве модуляторов sting - RU2722019C2
Код документа: RU2722019C2
Чертежи
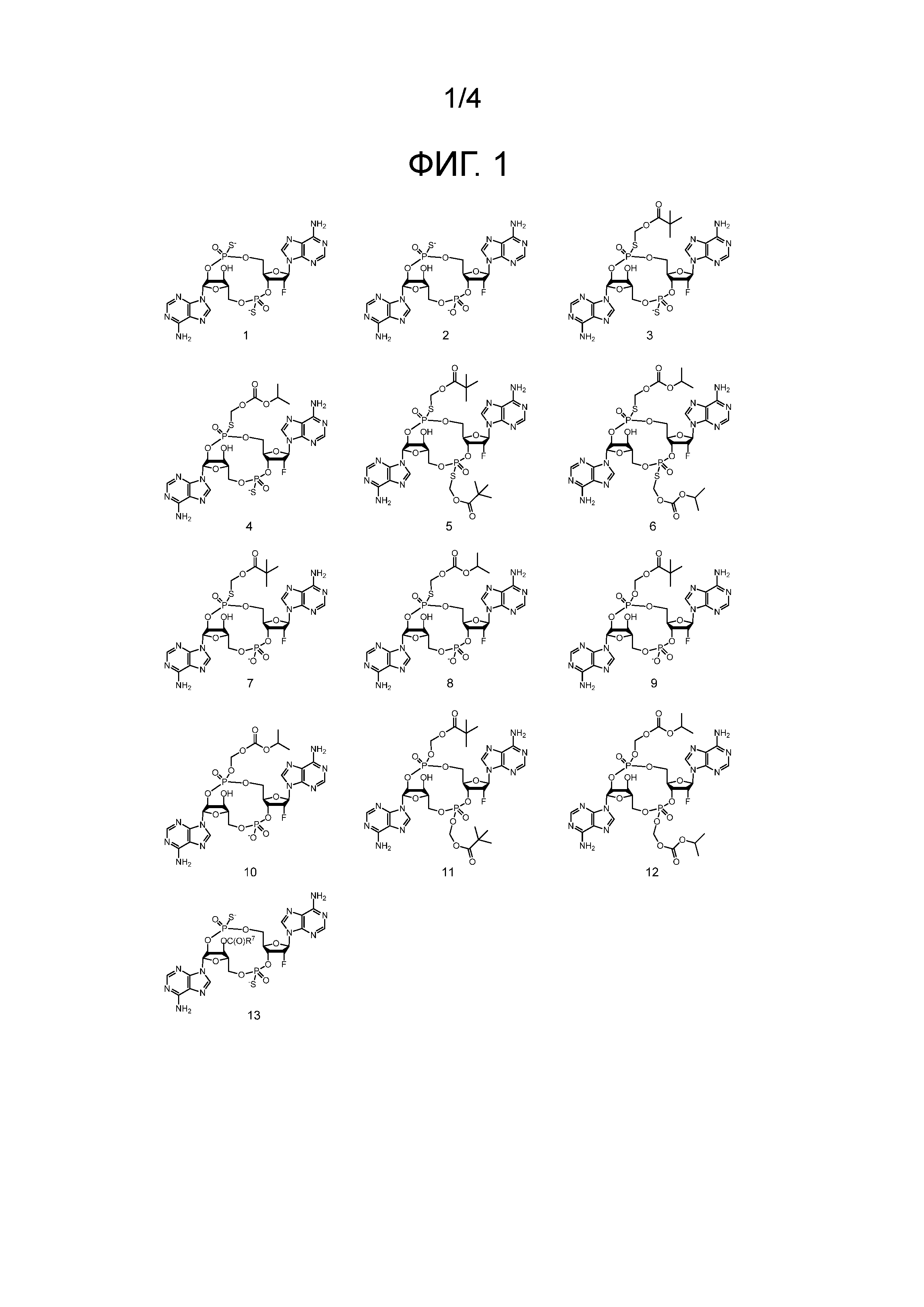
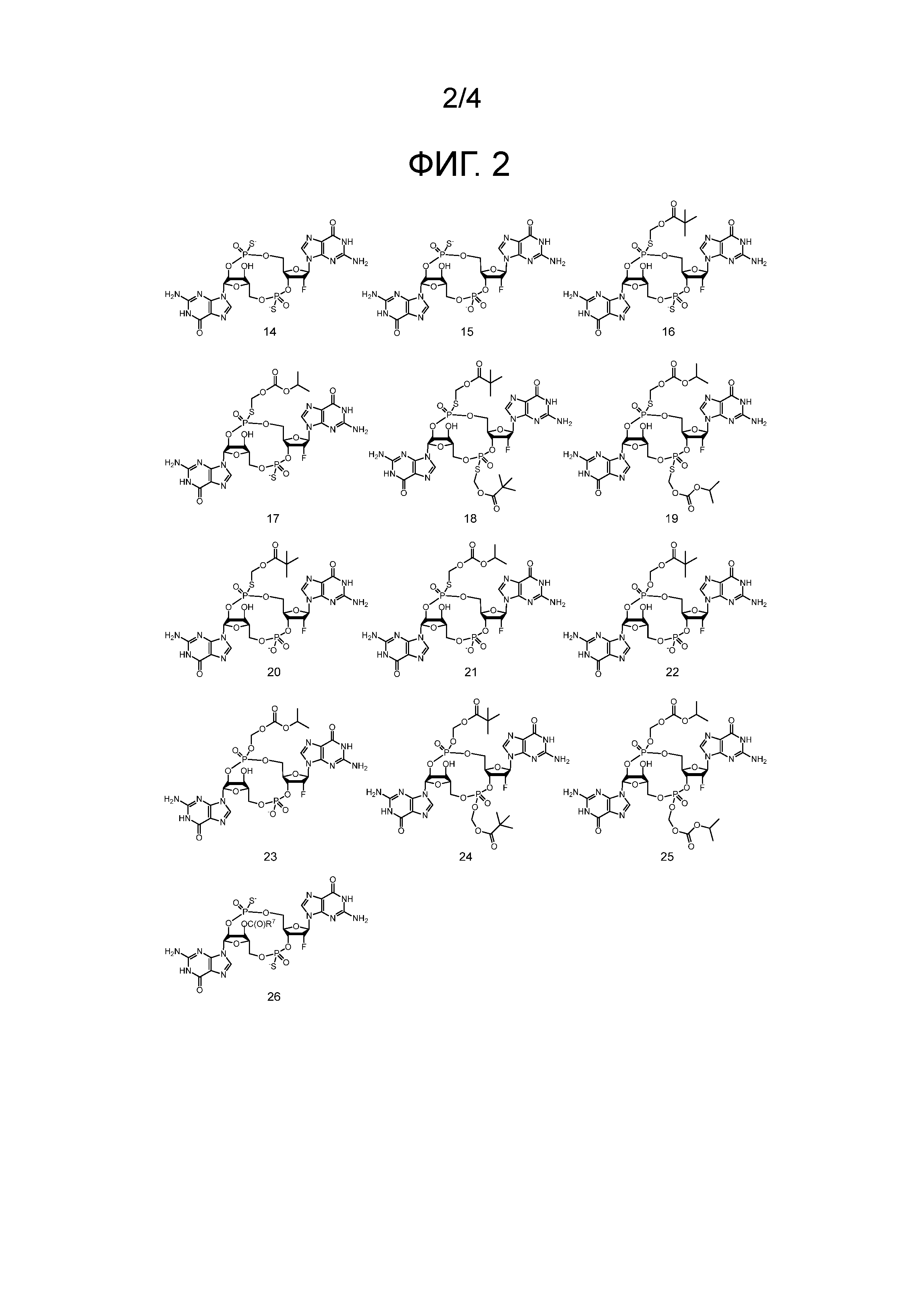
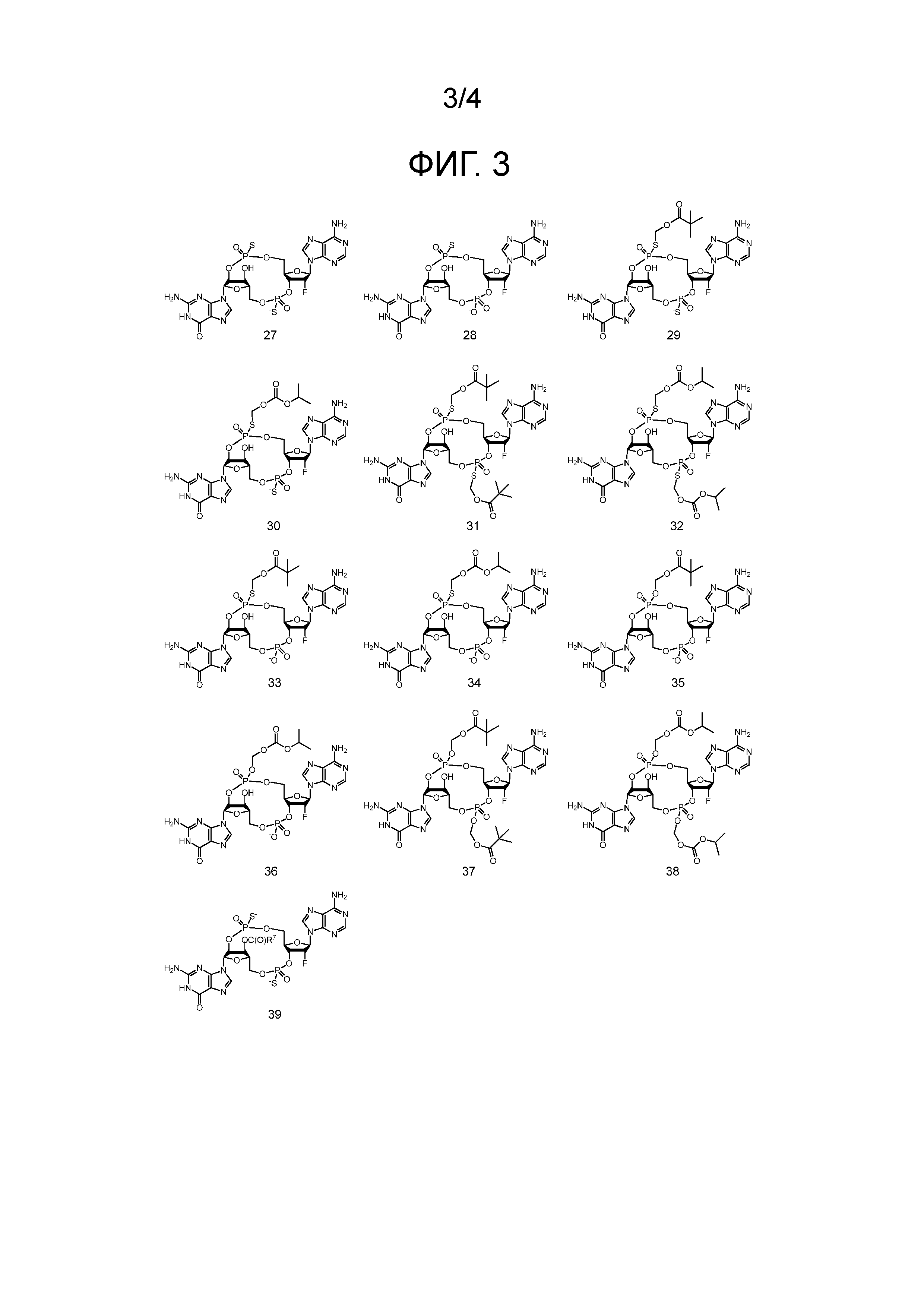
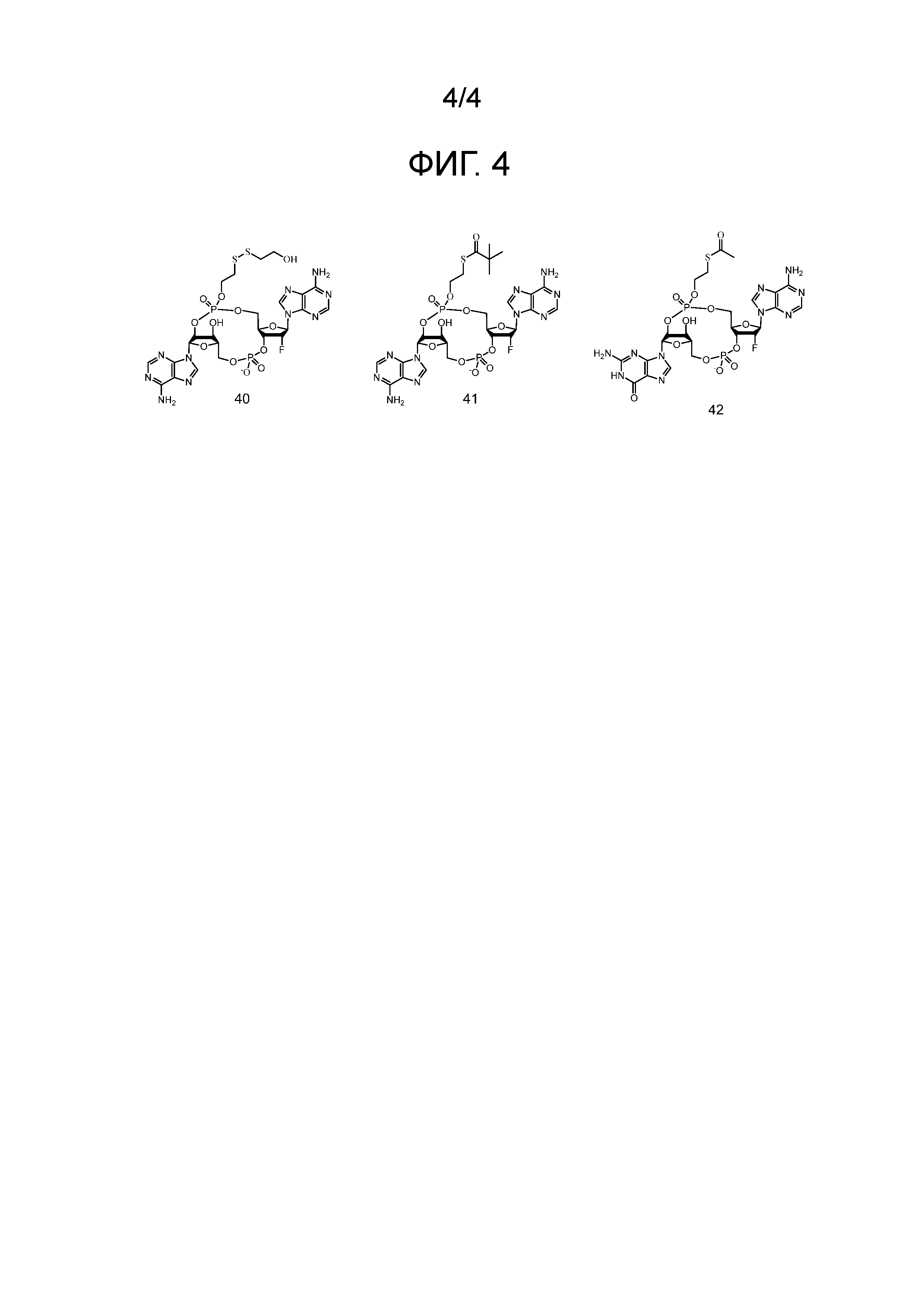
Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к соединениям, композициям, комбинациям и лекарственным средствам, содержащим упомянутые соединения, и к способам их получения. Настоящее изобретение также относится к применению упомянутых соединений, комбинаций, композиций и лекарственных средств при лечении заболеваний, при которых модулирование STING (стимулятор генов интерферонов) является благоприятным, например, воспаления, аллергических и аутоиммунных заболеваний, инфекционных заболеваний, инфекции, вызванной вирусом иммунодефицита человека (HIV), AIDS, злокачественной опухоли, предраковых синдромов и в качестве иммуногенной композиции или адъювантов вакцин.
Предпосылки создания изобретения
Позвоночные постоянно находятся под угрозой инвазии микроорганизмов и развили механизмы иммунной защиты для удаления инфекционных патогенов. У млекопитающих, такая иммунная система включает две ветви: врожденный иммунитет и адаптивный иммунитет. Система врожденного иммунитета представляет собой первую линию защиты, которая инициируется паттерн-распознающими рецепторами (PRR), которые обнаруживают лиганды у патогенов, а также повреждают ассоциированные молекулярные паттерны (Takeuchi O. et al, Cell, 2010: 140, 805-820). Было идентифицировано растущее число таких рецепторов, включая Toll-подобные рецепторы (TLR), лецитиновые рецепторы С-типа, рецепторы, подобные индуцируемому ретиноевой кислотой гену I (RIG-I), и NOD-подобные рецепторы (NLR), а также рецепторы двухцепочечной ДНК. Активация PRR приводит к усилению экспрессии генов, участвующих в воспалительном ответе, включая интерфероны 1 типа, провоспалительные цитокины и хемокины, которые супрессируют репликацию патогена и способствуют адаптивному иммунитету.
Адаптерный белок STING (стимулятор генов интерферонов), также известный как TMEM 173, MPYS, MITA и ERIS, был идентифицирован в качестве центральной сигнальной молекулы в ответе врожденного иммунитета на цитоплазматические нуклеиновые кислоты (Ishikawa H and Barber G N, Nature, 2008: 455, 674-678; WO2013/1666000). Активация STING влечет за собой положительную регуляцию сигнальных путей IRF3 и NFκB, что приводит к индукции интерферона-β и других цитокинов. STING крайне важен для ответов на цитоплазматическую ДНК, берущую свое происхождение от патогена или хозяина, и на необычные нуклеиновые кислоты, называемые циклическими динуклеотидами (CDN).
CDN были впервые идентифицированы в качестве бактериальных вторичных мессенджеров, отвечающих за регуляцию целого ряда ответов в прокариотической клетке. Бактериальные CDN, такие как c-di-GMP, представляют собой симметричные молекулы, характеризующиеся двумя 3',5'-фосфодиэфирными связями.
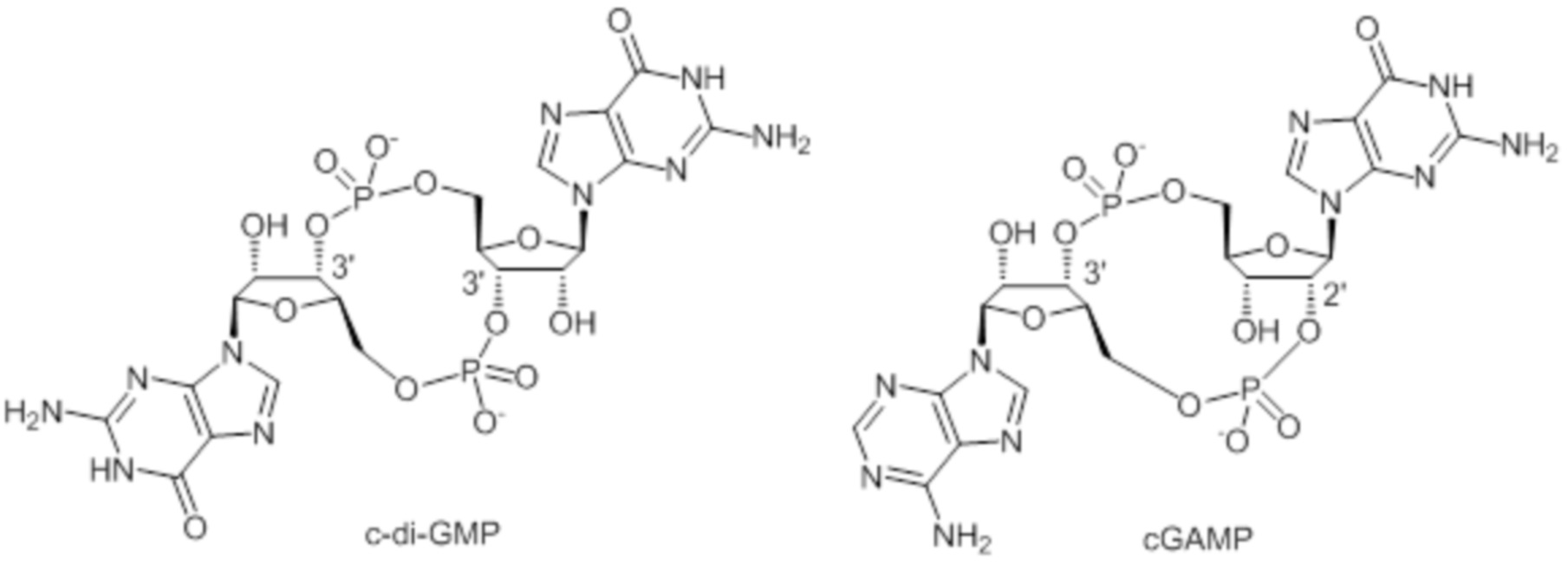
Непосредственное активирование STING бактериальными CDN было недавно подтверждено посредством рентгенокристаллографии (Burdette D L and Vance R E, Nature Immunology, 2013: 14, 19-26). Соответственно, бактериальные CDN и их аналоги привлекают интерес в качестве потенциальных адъювантов вакцин (Libanova R. et al, Microbial Biotechnology 2012: 5, 168-176; WO2007/054279, WO2005/087238).
Недавно был исследован ответ на цитоплазматическую ДНК, и было показано, что он включает формирование посредством фермента, называемого циклической GMP-AMP-синтазой (cGAS, ранее известной как C6orf150 или MB21D1), новой сигнальной CDN-молекулы млекопитающих, идентифицированной как cGAMP, которая затем активирует STING. В отличие от бактериальных CDN, cGAMP представляет собой несимметричную молекулу, характеризующуюся смешанными 2',5'- и 3',5'-фосфодиэфирными связями (Gao P et al, Cell, 2013: 153, 1-14). Взаимодействие cGAMP со STING было также продемонстрировано посредством рентгенокристаллографии (Cai X et al, Molecular Cell, 2014: 54, 289-296).
Интерферон был впервые описан как вещество, которое способно защищать клетки от вирусной инфекции (Isaacs & Lindemann, J. Virus Interference. Proc. R. Soc. Lon. Ser. B. Biol. Sci. 1957: 147, 258-267). У человека, интерфероны 1 типа представляют собой семейство родственных белков, кодируемых генами в хромосоме 9 и кодирующих, по меньшей мере, 13 изоформ интерферона альфа (IFNα) и одну изоформу интерферона бета (IFNβ). Рекомбинантный IFNα представляет собой первое одобренное биологическое лекарственное средство и стал важным терапевтическим средством при вирусных инфекциях и при злокачественных опухолях. Помимо прямой противовирусного действия в отношении клеток, известно, что интерфероны являются сильными модуляторами иммунного ответа, действуя на клетки иммунной системы.
Введение низкомолекулярного соединения, которое могло бы стимулировать ответ со стороны врожденного иммунитета, включая активацию интерферонов 1 типа и других цитокинов, могло бы стать важной стратегией лечения или профилактики заболеваний человека, включая вирусные инфекции. Такой тип иммуномодулирующей стратегии перспективен для определения соединений, которые могут быть пригодны не только при инфекционных заболеваниях, но и при злокачественной опухоли (Zitvogel, L., et al., Nature Reviews Immunology, 2015 15(7), p 405-414), аллергических заболеваниях (Moisan J. et al, Am. J. Physiol. Lung Cell Mol. Physiol., 2006: 290, L987-995), других воспалительных состояниях, таких как синдром раздраженного кишечника (Rakoff-Nahoum S., Cell., 2004, 23, 118(2): 229-41), и в качестве адъювантов вакцин (Persing et al. Trends Microbiol. 2002: 10(10 Suppl), S32-7 и Dubensky et al., Therapeutic Advances in Vaccines, published on-line Sept. 5, 2013).
Аллергические заболевания ассоциированы с Th2-смещенным иммунным ответом на аллергены. Th2-ответы ассоциированы с повышенными уровнями IgE, который посредством своих эффектов на тучные клетки усиливает гиперчувствительность к аллергенам, приводя к видимым симптомам, например, к аллергическому риниту и астме. У здоровых индивидуумов иммунный ответ на аллергены более сбалансирован смешанным Th2/Th1-ответом и ответом регуляторных Т-клеток. Было показано, что индукция интерферонов 1 типа приводит к местному снижению цитокинов Th2 типа и усиливает Th1/Treg-ответы. В этом контексте, индукция интерферонов 1 типа, например, посредством активации STING, может быть благоприятна при лечении аллергических заболеваний, таких как астма и аллергический ринит (Huber J.P. et al J Immunol 2010: 185, 813-817).
Напротив, усиленная и пролонгированная продукция IFN 1 типа ассоциирована с целым рядом хронических инфекций, включая Mycobacteria (Collins et al, CHM 2015; Wassermann et al., CHM 2015; Watson et al., CHM 2015), Franciscella (Storek et al., JI 2015; Jin et al., JI 2011), Chlamydia (Prantner et al., JI 2010; Barker et al., Mbio 2013; Zhang et al., JI 2014), Plasmodium (Sharma et al., Immunity 2011) и HIV (Herzner et al., Nat Immunol 2015; Nissen et al., Clin Exp Immunol 2014; Gao et al., Science 2013; Lahaye et al, Science 2013;) (обзор в Stifter and Feng, JI 2014). По аналогии, избыточная продукция интерферона 1 типа обнаруживается среди пациентов с комплексными формами аутоиммунного заболевания. Генетические данные у людей и подтверждение по исследованиям на животных моделях поддерживает гипотезу, что ингибирование STING приводит к снижению уровня интерферона 1 типа, который обуславливает аутоиммунное заболевание (Crow YJ,et al., Nat. Genet. 2006; 38917-920, Stetson DB, et al., Cell 2008; 134; 587-598). Поэтому, ингибиторы STING обеспечивают лечение пациентам с хронической продукцией интерферона 1 типа и провоспалительных цитокинов, ассоциированной с инфекциями или системными аутоиммунными заболеваниями. Аллергические заболевания ассоциированы с Th2-смещенным иммунным ответом на аллергены.
Было показано, что при инкубации с PBMC человека соединения, которые связываются со STING и действуют в качестве агонистов, индуцируют интерфероны 1 типа и другие цитокины. Соединения, которые индуцируют интерфероны человека, могут быть применимы при лечении различных заболеваний, например, при лечении аллергических заболеваний и других воспалительных заболеваний, например, аллергического ринита и астмы, при лечении инфекционных заболеваний, предраковых синдромов и злокачественной опухоли, и также могут быть применимы в качестве иммуногенной композиции или адъюванта вакцин. Соединения, которые связываются со STING, могут действовать как антагонисты и могут быть применимы при лечении, например, аутоиммунного заболевания.
Предполагается, что направленное воздействие на STING активирующими или ингибирующими средствами может являться многообещающим подходом для лечения заболеваний, при которых модулирование пути IFN 1 типа является благоприятным, включая воспаление, аллергические и аутоиммунные заболевания, инфекционные заболевания, злокачественную опухоль, предраковые синдромы, и в качестве иммуногенных композиций или адъювантов вакцин.
В международных патентных заявках WO2014/093936, WO2014/189805, WO2013/185052, U.S.2014/0341976, WO2015/077354, PCT/EP2015/062281 и GB1501462.4 раскрыты определенные циклические динуклеотиды и их применение для индукции иммунного ответа.
Целью настоящего изобретения является предоставление дополнительных циклических динуклеотидов, применимых для лечения злокачественной опухоли.
Краткое описание сущности изобретения
Настоящее изобретение относится к соединениям в соответствии с формулой (I):
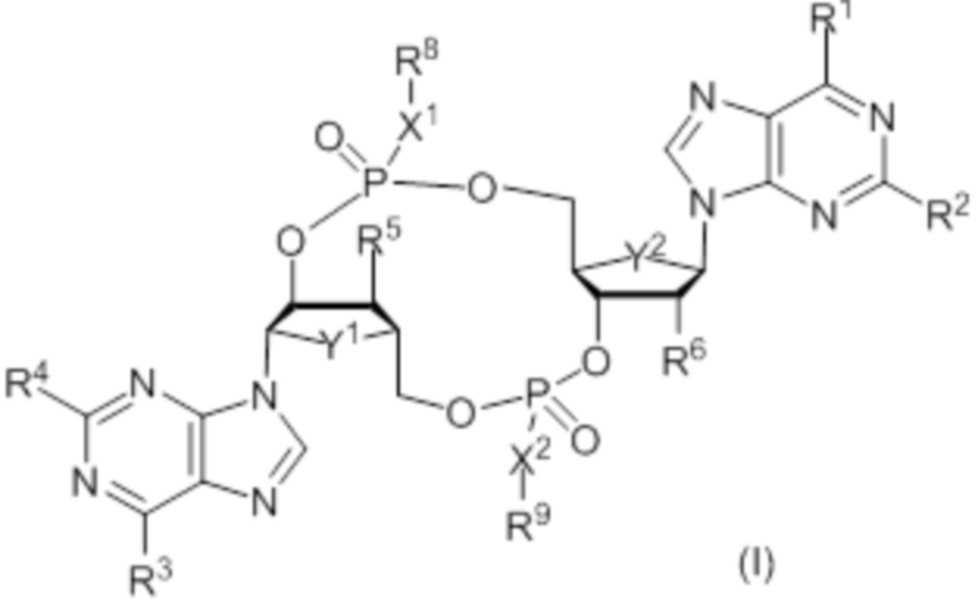
в которой значения Y1, Y2, X1, X2, R1, R2, R3, R4, R5, R6, R8 и R9 определены ниже, и их фармацевтически приемлемым солям.
Согласно дополнительному аспекту настоящего изобретения, предусмотрена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и один или более фармацевтически приемлемых эксципиентов.
Согласно дополнительному аспекту настоящего изобретения, предусмотрено соединение формулы (I) или его фармацевтически приемлемая соль для применения в терапии.
Согласно дополнительному аспекту настоящего изобретения, предусмотрено соединение формулы (I) или его фармацевтически приемлемая соль для применения при лечении заболевания, при котором модулирование STING является благоприятным.
Согласно дополнительному аспекту настоящего изобретения, предусмотрено соединение формулы (I) или его фармацевтически приемлемая соль для применения при лечении воспаления, аллергических и аутоиммунных заболеваний, инфекционных заболеваний, злокачественной опухоли, предраковых синдромов, и в качестве иммуногенной композиции или адъюванта вакцин.
Согласно дополнительному аспекту настоящего изобретения, предусмотрен способ лечения у субъекта заболевания, при котором модулирование STING является благоприятным, включающий в себя введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно дополнительному аспекту настоящего изобретения, предусмотрен способ лечения у субъекта воспаления, аллергических и аутоиммунных заболеваний, инфекционных заболеваний и злокачественной опухоли, включающий в себя введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно дополнительному аспекту настоящего изобретения, предусмотрено применение соединения формулы (I) или его фармацевтически приемлемой соли при производстве лекарственного средства для применения при лечении заболевания, при котором модулирование STING является благоприятным.
Согласно дополнительному аспекту настоящего изобретения, предусмотрено применение соединения формулы (I) или его фармацевтически приемлемой соли при производстве лекарственного средства для применения при лечении воспаления, аллергических и аутоиммунных заболеваний, инфекционных заболеваний, предраковых синдромов и злокачественной опухоли.
Согласно дополнительному аспекту, предусмотрена комбинация, включающая в себя соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, одно дополнительное терапевтическое средство.
Согласно дополнительному аспекту настоящего изобретения, предусмотрена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, одно дополнительное терапевтическое средство и один или более фармацевтически приемлемых эксципиентов.
Согласно дополнительному аспекту, предусмотрена комбинация, включающая в себя соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, одно дополнительное терапевтическое средство для применения в терапии.
Согласно дополнительному аспекту, предусмотрена комбинация, включающая в себя соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, одно дополнительное терапевтическое средство для применения при лечении заболевания или состояния, при котором модулирование STING является благоприятным.
Согласно дополнительному аспекту, предусмотрена комбинация, включающая в себя соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, одно дополнительное терапевтическое средство для применения при лечении воспаления, аллергических и аутоиммунных заболеваний, инфекционных заболеваний, предраковых синдромов и злокачественной опухоли.
Согласно дополнительному аспекту настоящего изобретения, предусмотрен способ лечения у субъекта заболевания или состояния, при котором модулирование STING является благоприятным, включающий в себя введение терапевтически эффективного количества комбинации, включающей в себя соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, одно дополнительное терапевтическое средство.
Согласно дополнительному аспекту настоящего изобретения, предусмотрен способ лечения у субъекта воспаления, аллергических и аутоиммунных заболеваний, инфекционных заболеваний и злокачественной опухоли, включающий в себя введение терапевтически эффективного количества комбинации, включающей в себя соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, одно дополнительное терапевтическое средство.
Согласно дополнительному аспекту настоящего изобретения, предусмотрена иммуногенная композиция или адъювант вакцин, содержащие соединение формулы (I) или его фармацевтически приемлемую соль.
Согласно дополнительному аспекту настоящего изобретения, предусмотрена композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и одно или несколько иммуностимулирующих средств.
Согласно дополнительному аспекту настоящего изобретения, предусмотрена иммуногенная композиция, содержащая антиген или антигенную композицию и соединение формулы (I) или его фармацевтически приемлемую соль.
Согласно дополнительному аспекту настоящего изобретения, предусмотрена иммуногенная композиция, содержащая антиген или антигенную композицию и соединение формулы (I) или его фармацевтически приемлемую соль, для применения при лечении или профилактике заболевания.
Согласно дополнительному аспекту настоящего изобретения, предусмотрено применение соединения формулы (I) или его фармацевтически приемлемой соли при производстве иммуногенной композиции, содержащей антиген или антигенную композицию, для лечения или профилактики заболевания.
Согласно дополнительному аспекту настоящего изобретения, предусмотрен способ лечения или профилактики заболевания, включающий в себя введение субъекту-человеку, страдающему заболеванием или предрасположенному к нему, иммуногенной композиции, содержащей антиген или антигенную композицию и соединение формулы (I) или его фармацевтически приемлемую соль.
Согласно дополнительному аспекту настоящего изобретения, предусмотрена иммуногенная или вакцинная композиция, содержащая антиген или антигенную композицию и соединение формулы (I) или его фармацевтически приемлемую соль, для применения при лечении или профилактике заболевания.
Согласно дополнительному аспекту настоящего изобретения, предусмотрена иммуногенная композиция, содержащая антиген или антигенную композицию и соединение формулы (I) или его фармацевтически приемлемую соль, для применения при лечении или профилактике заболевания.
Согласно дополнительному аспекту настоящего изобретения, предусмотрено применение соединения формулы (I) или его фармацевтически приемлемой соли при производстве иммуногенной или вакцинной композиции, содержащей антиген или антигенную композицию, для лечения или профилактики заболевания.
Согласно дополнительному аспекту настоящего изобретения, предусмотрен способ лечения или профилактики заболевания, включающий в себя введение субъекту-человеку, страдающему заболеванием или предрасположенному к нему, вакцинной композиции, содержащей антиген или антигенную композицию и соединение формулы (I) или его фармацевтически приемлемую соль.
Согласно дополнительному аспекту, предусмотрен способ лечения HIV-инфекции у человека, характеризующегося наличием инфекции или подвергающегося риску ее возникновения, путем введения человеку терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно дополнительному аспекту, предусмотрен способ лечения AIDS-инфекции у человека, характеризующегося наличием инфекции, путем введения человеку терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно дополнительному аспекту, предусмотрен способ лечения HIV-инфекции у человека путем введения человеку терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Краткое описание чертежей
Фигура 1
На фигуре 1 отражены структуры соединений 1-13, где значение R7 определено в формуле (I).
Фигура 2
На фигуре 2 отражены структуры соединений 14-26, где значение R7 определено в формуле (I).
Фигура 3
На фигуре 3 отражены структуры соединений 27-39, где значение R7 определено в формуле (I).
Фигура 4
На фигуре 4 отражены структуры соединений 40-42.
Подробное описание изобретения
Настоящее изобретение относится к новым соединениям формулы (I):
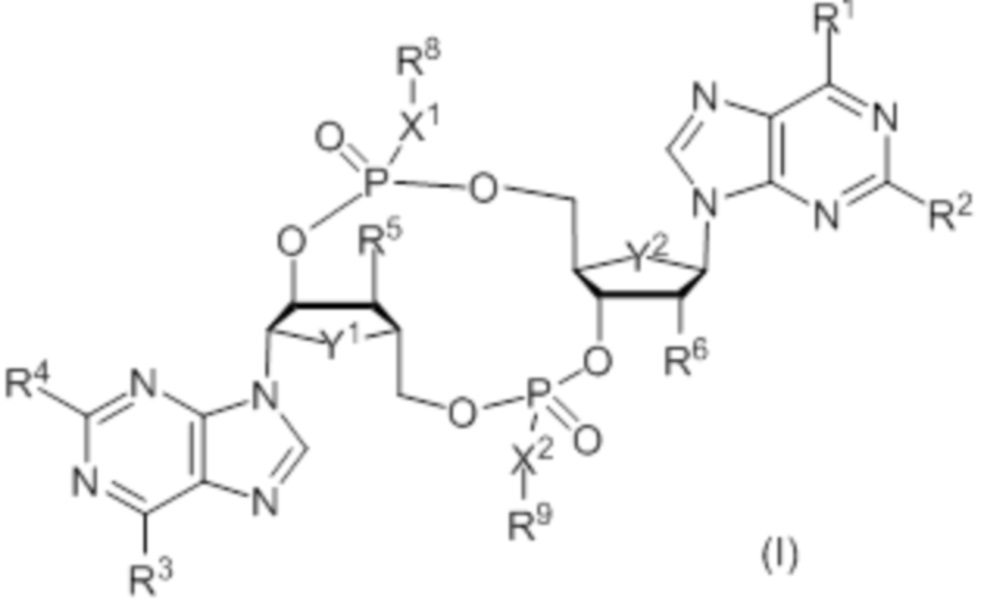
где:
Y1 и Y2 независимо представляют собой CH2 или O;
X1 и X2 независимо представляют собой S или O;
R1 представляет собой OH, и R2 представляет собой NH2, или R1 представляет собой NH2, и R2 представляет собой H;
R3 представляет собой OH, и R4 представляет собой NH2, или R3 представляет собой NH2, и R4 представляет собой H;
R5 выбирают из: F, OH и OC(O)R7;
R6 выбирают из: F, OH и OC(O)R7;
при условии: если ни R5, ни R6 не представляют собой F, то, по меньшей мере, один из Y1 и Y2 представляет собой CH2; и
R8 и R9 независимо выбирают из: H, CH2OC(O)R7, CH2OCO2R7, CH2CH2SC(O)R7 и CH2CH2SSCH2R7;
при условии: если X1 и X2 оба представляют собой O, то, по меньшей мере, один из R8 и R9 не представляет собой H;
где R7 выбирают из: арила, гетероарила, гетероциклоалкила, циклоалкила, C1-20алкила и C1-20алкила, замещенного 1-5 заместителями, независимо выбранными из: арила, циклоалкила, гидрокси и F;
и их фармацевтически приемлемым солям.
Соответственно требованиям к соединениям формулы (I), по меньшей мере, один из Y1 и Y2 представляет собой O. Соответственно требованиям к соединениям формулы (I), Y1 и Y2 оба представляют собой O.
Соответственно требованиям к соединениям формулы (I), по меньшей мере, один из X1 и X2 представляет собой S. Соответственно требованиям к соединениям формулы (I), X1 представляет собой S. Соответственно требованиям к соединениям формулы (I), X1 и X2 оба представляют собой S.
Соответственно требованиям к соединениям формулы (I), R1 представляет собой NH2, и R2 представляет собой H.
Соответственно требованиям к соединениям формулы (I), R5 представляет собой OH.
Соответственно требованиям к соединениям формулы (I), R6 представляет собой F.
Соответственно требованиям к соединениям формулы (I), если один из R8 и R9 не представляет собой H, то он представляет собой CH2CH2SC(O)C1-6алкил.
Соответственно требованиям к соединениям формулы (I), если один из R8 и R9 не представляет собой H, то он представляет собой CH2CH2SSC1-4алкилOH.
Соответственно требованиям к соединениям формулы (I), если X1 представляет собой S, то R8 и R9 представляют собой H.
Соответственно требованиям к соединениям формулы (I), если X2 представляет собой S, то R8 и R9 представляют собой H.
Соответственно требованиям к соединениям формулы (I), oодин из R8 и R9 представляет собой H.
Соответственно требованиям к соединениям формулы (I), если X1 и X2 представляют собой O, то один из R8 и R9 представляет собой H.
Соответственно требованиям к соединениям формулы (I), R7 представляет собой C12-18алкил.
Соответственно требованиям к соединениям формулы (I), R7 выбирают из: C1-20алкила и C1-20алкила, замещенного 1-5 заместителями, независимо выбранными из: арила, циклоалкила и F.
Соответственно требованиям к соединениям формулы (I), R7 представляет собой C1-20алкил.
Соответственно требованиям к соединениям формулы (I), R7 представляет собой трет-бутил.
Соответственно требованиям к соединениям формулы (I), R7 представляет собой изопропил.
Примеры соединений согласно настоящему изобретению включают в себя соединения, отраженные на фигурах 1, 2, 3 и 4.
Соединения формулы (I) могут находиться в форме соли.
В число соединений формулы (I) включены соединения формулы (II):

где:
Y11 и Y12 независимо представляют собой CH2 или O;
X11 представляет собой S;
X12 представляет собой O;
R11 представляет собой OH, и R12 представляет собой NH2, или R11 представляет собой NH2, и R12 представляет собой H;
R13 представляет собой OH, и R14 представляет собой NH2, или R13 представляет собой NH2, и R14 представляет собой H;
R15 выбирают из: F, OH и OC(O)R17;
R16 выбирают из: F, OH и OC(O)R17;
при условии: если ни R15, ни R16 не представляют собой F, то, по меньшей мере, один из Y11 и Y12 представляет собой CH2; и
R18 и R19 независимо выбирают из: H, CH2OC(O)R17, CH2OCO2R17, CH2CH2SC(O)R17 и CH2CH2SSCH2R17;
где R17 выбирают из: арила, гетероарила, гетероциклоалкила, циклоалкила, C1-20алкила и C1-20алкила, замещенного 1-5 заместителями, независимо выбранными из: арила, циклоалкила, гидрокси и F;
и их фармацевтически приемлемые соли.
Соответственно требованиям к соединениям формулы (II), по меньшей мере, один из Y11 и Y12 представляет собой O. Соответственно требованиям к соединениям формулы (I), Y11 и Y12 оба представляют собой O.
Соответственно требованиям к соединениям формулы (II), R11 представляет собой NH2, и R12 представляет собой H.
Соответственно требованиям к соединениям формулы (II), R15 представляет собой OH.
Соответственно требованиям к соединениям формулы (II), R16 представляет собой F.
Соответственно требованиям к соединениям формулы (II), если один из R18 и R19 не представляет собой H, то он представляет собой CH2CH2SC(O)C1-6алкил.
Соответственно требованиям к соединениям формулы (II), если один из R18 и R19 не представляет собой H, то он представляет собой CH2CH2SSC1-4алкилOH.
Соответственно требованиям к соединениям формулы (II), R18 и R19 представляют собой H.
Соответственно требованиям к соединениям формулы (II), один из R18 и R19 представляет собой H.
Соответственно требованиям к соединениям формулы (II), R17 представляет собой C12-18алкил.
Соответственно требованиям к соединениям формулы (II), R17 выбирают из: C1-20алкила и C1-20алкила, замещенного 1-5 заместителями, независимо выбранными из: арила, циклоалкила и F.
Соответственно требованиям к соединениям формулы (II), R17 представляет собой C1-20алкил.
Соответственно требованиям к соединениям формулы (II), R17 представляет собой трет-бутил.
Соответственно требованиям к соединениям формулы (II), R17 представляет собой изопропил.
Соответственно требованиям, соединения формулы (II) находятся в форме фармацевтически приемлемой соли.
В число соединений формулы (I) включены соединения формулы (III):
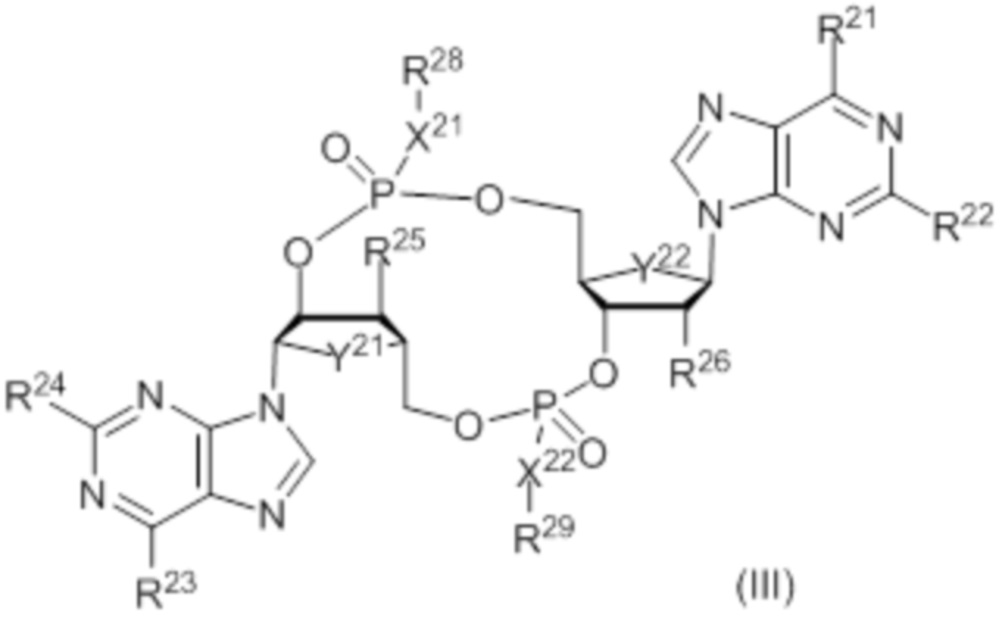
где:
Y21 и Y22 независимо представляют собой CH2 или O;
X21 представляет собой O;
X22 представляет собой S;
R21 представляет собой OH, и R22 представляет собой NH2, или R21 представляет собой NH2, и R22 представляет собой H;
R23 представляет собой OH, и R24 представляет собой NH2, или R23 представляет собой NH2, и R24 представляет собой H;
R25 выбирают из: F, OH и OC(O)R27;
R26 выбирают из: F, OH и OC(O)R27;
при условии: если ни R25, ни R26 не представляют собой F, то, по меньшей мере, один из Y21 и Y22 представляет собой CH2; и
R28 и R29 независимо выбирают из: H, CH2OC(O)R27, CH2OCO2R27, CH2CH2SC(O)R27 и CH2CH2SSCH2R27;
где R27 выбирают из: арила, гетероарила, гетероциклоалкила, циклоалкила, C1-20алкила и C1-20алкила, замещенного 1-5 заместителями, независимо выбранными из: арила, циклоалкила, гидрокси и F;
и их фармацевтически приемлемые соли.
Соответственно требованиям к соединениям формулы (III), по меньшей мере, один из Y21 и Y22 представляет собой O. Соответственно требованиям к соединениям формулы (III), Y21 и Y22 оба представляют собой O.
Соответственно требованиям к соединениям формулы (III), R21 представляет собой NH2, и R22 представляет собой H.
Соответственно требованиям к соединениям формулы (III), R25 представляет собой OH.
Соответственно требованиям к соединениям формулы (III), R26 представляет собой F.
Соответственно требованиям к соединениям формулы (III), если один из R28 и R29 не представляет собой H, то он представляет собой CH2CH2SC(O)C1-6алкил.
Соответственно требованиям к соединениям формулы (III), если один из R28 и R29 не представляет собой H, то он представляет собой CH2CH2SSC1-4алкилOH.
Соответственно требованиям к соединениям формулы (III), R28 и R29 представляют собой H.
Соответственно требованиям к соединениям формулы (III), один из R28 и R29 представляет собой H.
Соответственно требованиям к соединениям формулы (III), R27 представляет собой C12-18алкил.
Соответственно требованиям к соединениям формулы (III), R27 выбирают из: C1-20алкила и C1-20алкила, замещенного 1-5 заместителями, независимо выбранными из: арила, циклоалкила и F.
Соответственно требованиям к соединениям формулы (III), R27 представляет собой C1-20алкил.
Соответственно требованиям к соединениям формулы (III), R27 представляет собой трет-бутил.
Соответственно требованиям к соединениям формулы (III), R27 представляет собой изопропил.
Соответственно требованиям, соединения формулы (III) находятся в форме фармацевтически приемлемой соли.
В число соединений формулы (I) и соединений формулы (II) включены соединения формулы (IV):
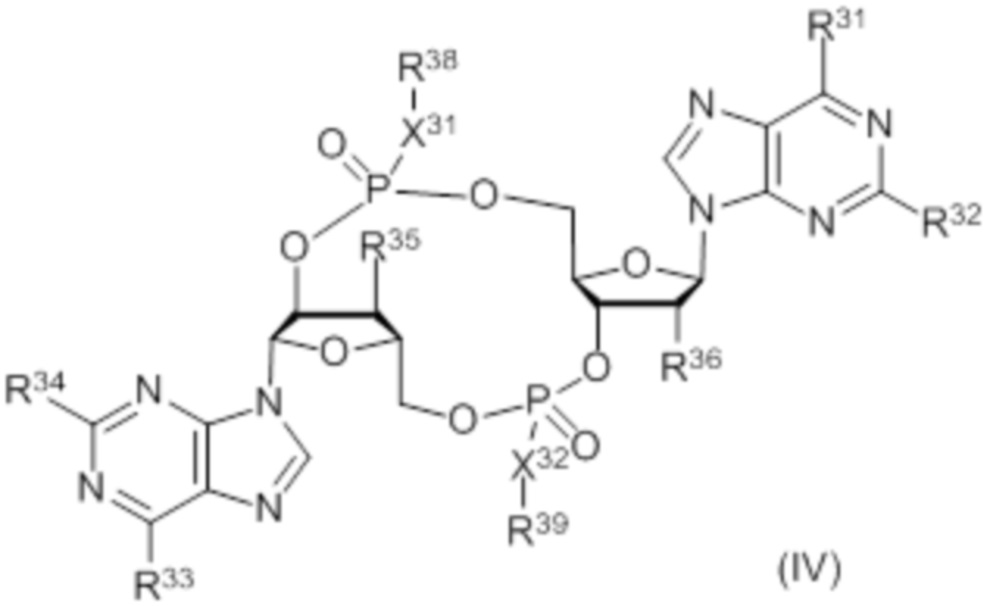
где:
X31 представляет собой S;
X32 представляет собой O;
R31 представляет собой OH, и R32 представляет собой NH2, или R31 представляет собой NH2, и R32 представляет собой H;
R33 представляет собой OH, и R34 представляет собой NH2, или R33 представляет собой NH2, и R34 представляет собой H;
R35 выбирают из: F, OH и OC(O)R37;
R36 выбирают из: F, OH и OC(O)R37;
при условии: по меньшей мере, один из R35 и R36 представляет собой F; и
R38 и R39 независимо выбирают из: H, CH2OC(O)R37, CH2OCO2R37, CH2CH2SC(O)R37 и CH2CH2SSCH2R37;
где R37 выбирают из: арила, гетероарила, гетероциклоалкила, циклоалкила, C1-20алкила и C1-20алкила, замещенного 1-5 заместителями, независимо выбранными из: арила, циклоалкила, гидрокси и F;
и их фармацевтически приемлемые соли.
Соответственно требованиям к соединениям формулы (IV), R31 представляет собой NH2, и R32 представляет собой H.
Соответственно требованиям к соединениям формулы (IV), R35 представляет собой OH.
Соответственно требованиям к соединениям формулы (IV), R36 представляет собой F.
Соответственно требованиям к соединениям формулы (IV), если один из R38 и R39 не представляет собой H, то он представляет собой CH2CH2SC(O)C1-6алкил.
Соответственно требованиям к соединениям формулы (IV), если один из R38 и R39 не представляет собой H, то он представляет собой CH2CH2SSC1-4алкилOH.
Соответственно требованиям к соединениям формулы (IV), R38 и R39 представляют собой H.
Соответственно требованиям к соединениям формулы (IV), один из R38 и R39 представляет собой H.
Соответственно требованиям к соединениям формулы (IV), R37 представляет собой C12-18алкил.
Соответственно требованиям к соединениям формулы (IV), R37 выбирают из: C1-20алкила и C1-20алкила, замещенного 1-5 заместителями, независимо выбранными из: арила, циклоалкила и F.
Соответственно требованиям к соединениям формулы (IV), R37 представляет собой C1-20алкил.
Соответственно требованиям к соединениям формулы (IV), R37 представляет собой трет-бутил.
Соответственно требованиям к соединениям формулы (IV), R37 представляет собой изопропил.
Соответственно требованиям, соединения формулы (IV) находятся в форме фармацевтически приемлемой соли.
В число соединений формулы (I) и соединений формулы (III) включены соединения формулы (V):
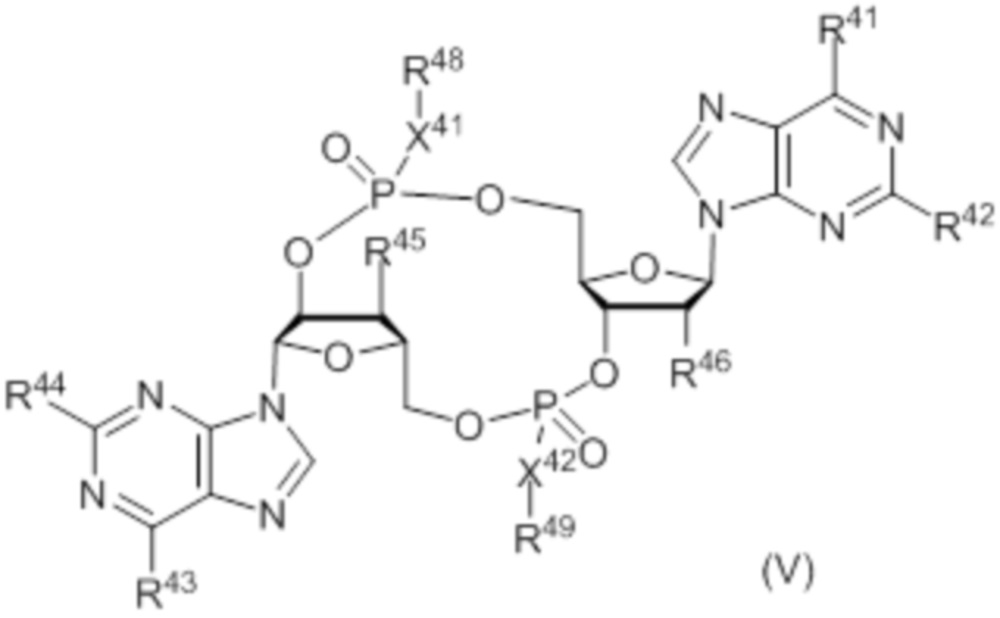
где:
X41 представляет собой O;
X42 представляет собой S;
R41 представляет собой OH, и R42 представляет собой NH2, или R41 представляет собой NH2, и R42 представляет собой H;
R43 представляет собой OH, и R44 представляет собой NH2, или R43 представляет собой NH2, и R44 представляет собой H;
R45 выбирают из: F, OH и OC(O)R47;
R46 выбирают из: F, OH и OC(O)R47;
при условии: по меньшей мере, один из R45 и R46 представляет собой F; и
R48 и R49 независимо выбирают из: H, CH2OC(O)R47, CH2OCO2R47, CH2CH2SC(O)R47 и CH2CH2SSCH2R47;
где R47 выбирают из: арила, гетероарила, гетероциклоалкила, циклоалкила, C1-20алкила и C1-20алкила, замещенного 1-5 заместителями, независимо выбранными из: арила, циклоалкила, гидрокси и F;
и их фармацевтически приемлемые соли.
Соответственно требованиям к соединениям формулы (V), R41 представляет собой NH2, и R42 представляет собой H.
Соответственно требованиям к соединениям формулы (V), R45 представляет собой OH.
Соответственно требованиям к соединениям формулы (V), R46 представляет собой F.
Соответственно требованиям к соединениям формулы (V), если один из R48 и R49 не представляет собой H, то он представляет собой CH2CH2SC(O)C1-6алкил.
Соответственно требованиям к соединениям формулы (V), если один из R48 и R49 не представляет собой H, то он представляет собой CH2CH2SSC1-4алкилOH.
Соответственно требованиям к соединениям формулы (V), R48 и R49 представляют собой H.
Соответственно требованиям к соединениям формулы (V), один из R48 и R49 представляет собой H.
Соответственно требованиям к соединениям формулы (V), R47 представляет собой C12-18алкил.
Соответственно требованиям к соединениям формулы (V), R47 выбирают из: C1-20алкила и C1-20алкила, замещенного 1-5 заместителями, независимо выбранными из: арила, циклоалкила и F.
Соответственно требованиям к соединениям формулы (V), R47 представляет собой C1-20алкил.
Соответственно требованиям к соединениям формулы (V), R47 представляет собой трет-бутил.
Соответственно требованиям к соединениям формулы (V), R47 представляет собой изопропил.
Соответственно требованиям, соединения формулы (V) находятся в форме фармацевтически приемлемой соли.
В число соединений формулы (I) включены соединения формулы (VI):
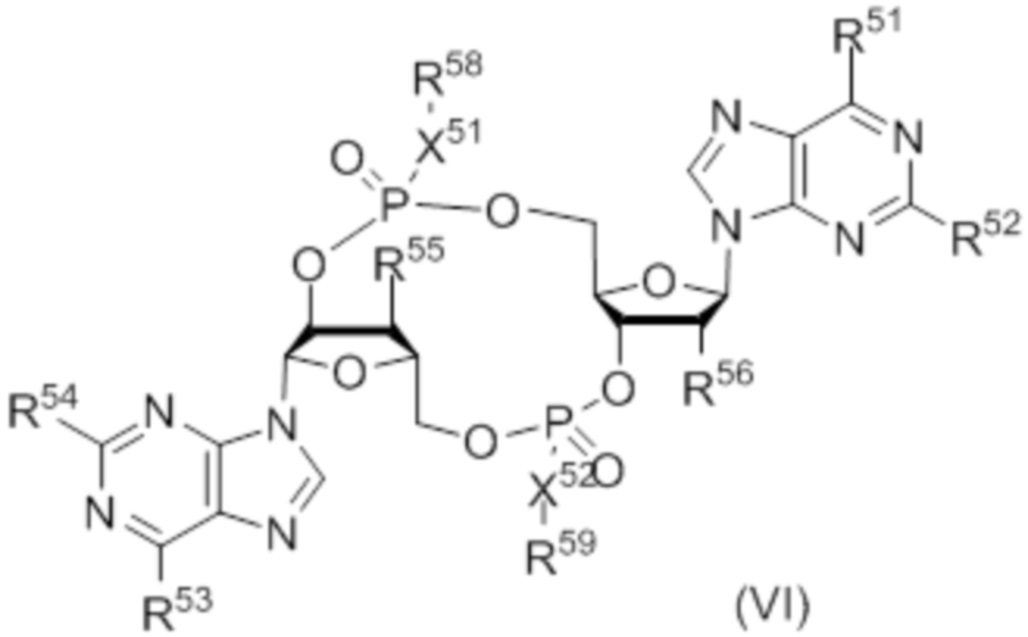
где:
X51 представляет собой O;
X52 представляет собой O;
R51 представляет собой OH, и R52 представляет собой NH2, или R51 представляет собой NH2, и R52 представляет собой H;
R53 представляет собой OH, и R54 представляет собой NH2, или R53 представляет собой NH2, и R54 представляет собой H;
R55 выбирают из: F, OH и OC(O)R47;
R56 представляет собой F;
R58 и R59 независимо выбирают из: H, CH2OC(O)R57, CH2OCO2R57, CH2CH2SC(O)R57 и CH2CH2SSCH2R57;
где R57 выбирают из: арила, гетероарила, гетероциклоалкила, циклоалкила, C1-20алкила и C1-20алкила, замещенного 1-5 заместителями, независимо выбранными из: арила, циклоалкила, гидрокси и F;
при условии, что, по меньшей мере, один из R58 и R59 не представляет собой H.
и их фармацевтически приемлемые соли.
Соответственно требованиям к соединениям формулы (VI), R51 представляет собой NH2, и R52 представляет собой H.
Соответственно требованиям к соединениям формулы (VI), R55 представляет собой OH.
Соответственно требованиям к соединениям формулы (VI), если один из R58 и R59 не представляет собой H, то он представляет собой CH2CH2SC(O)C1-6алкил.
Соответственно требованиям к соединениям формулы (VI), если один из R58 и R59 не представляет собой H, то он представляет собой CH2CH2SSC1-4алкилOH.
Соответственно требованиям к соединениям формулы (VI), один из R58 и R59 представляет собой H.
Соответственно требованиям к соединениям формулы (VI), R57 представляет собой C12-18алкил.
Соответственно требованиям к соединениям формулы (VI), R57 выбирают из: C1-20алкила и C1-20алкила, замещенного 1-5 заместителями, независимо выбранными из: арила, циклоалкила и F.
Соответственно требованиям к соединениям формулы (VI), R57 представляет собой C1-20алкил.
Соответственно требованиям к соединениям формулы (VI), R57 представляет собой трет-бутил.
Соответственно требованиям к соединениям формулы (VI), R57 представляет собой изопропил.
Соответственно требованиям к соединениям формулы (VI) находятся в форме фармацевтически приемлемой соли.
В число соединений формулы (I) включены:
(1R,6R,8R,9R,10R,15R,17R,18R)-8,17-бис(6-амино-9H-пурин-9-ил)-9-фтор-18-гидрокси-3,12-дисульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-дион;
(1R,6R,8R,9R,10R,15R,17R,18R)-8,17-бис(6-амино-9H-пурин-9-ил)-9-фтор-18-гидрокси-3,12-дисульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-дион, изомер 1;
(1R,6R,8R,9R,10R,15R,17R,18R)-8,17-бис(6-амино-9H-пурин-9-ил)-9-фтор-18-гидрокси-3,12-дисульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-дион, изомер 2;
(1R,6R,8R,9R,10R,15R,17R,18R)-8,17-бис(6-амино-9H-пурин-9-ил)-9-фтор-12,18-дигидрокси-3-сульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-дион;
(1R,6R,8R,9R,10R,15R,17R,18R)-8,17-бис(6-амино-9H-пурин-9-ил)-9-фтор-12,18-дигидрокси-3-сульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-дион, изомер 1;
(1R,6R,8R,9R,10R,15R,17R,18R)-8,17-бис(6-амино-9H-пурин-9-ил)-9-фтор-12,18-дигидрокси-3-сульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-дион, изомер 2;
(1R,6R,8R,9R,10R,15R,17R,18R)-17-(2-амино-6-оксо-6,9-дигидро-1H-пурин-9-ил)-8-(6-амино-9H-пурин-9-ил)-9-фтор-12,18-дигидрокси-3-сульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-дион;
(1R,6R,8R,9R,10R,15R,17R,18R)-17-(2-амино-6-оксо-6,9-дигидро-1H-пурин-9-ил)-8-(6-амино-9H-пурин-9-ил)-9-фтор-12,18-дигидрокси-3-сульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-дион, изомер 1;
(1R,6R,8R,9R,10R,15R,17R,18R)-17-(2-амино-6-оксо-6,9-дигидро-1H-пурин-9-ил)-8-(6-амино-9H-пурин-9-ил)-9-фтор-12,18-дигидрокси-3-сульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-дион, изомер 2;
(1R,6R,8R,9R,10R,15R,17R,18R)-17-(2-амино-6-оксо-6,9-дигидро-1H-пурин-9-ил)-8-(6-амино-9H-пурин-9-ил)-9-фтор-18-гидрокси-3,12-дисульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-дион;
(1R,6R,8R,9R,10R,15R,17R,18R)-17-(2-амино-6-оксо-6,9-дигидро-1H-пурин-9-ил)-8-(6-амино-9H-пурин-9-ил)-9-фтор-18-гидрокси-3,12-дисульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-дион, изомер 1; и
(1R,6R,8R,9R,10R,15R,17R,18R)-17-(2-амино-6-оксо-6,9-дигидро-1H-пурин-9-ил)-8-(6-амино-9H-пурин-9-ил)-9-фтор-18-гидрокси-3,12-дисульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-дион, изомер 2;
и их фармацевтически приемлемые соли.
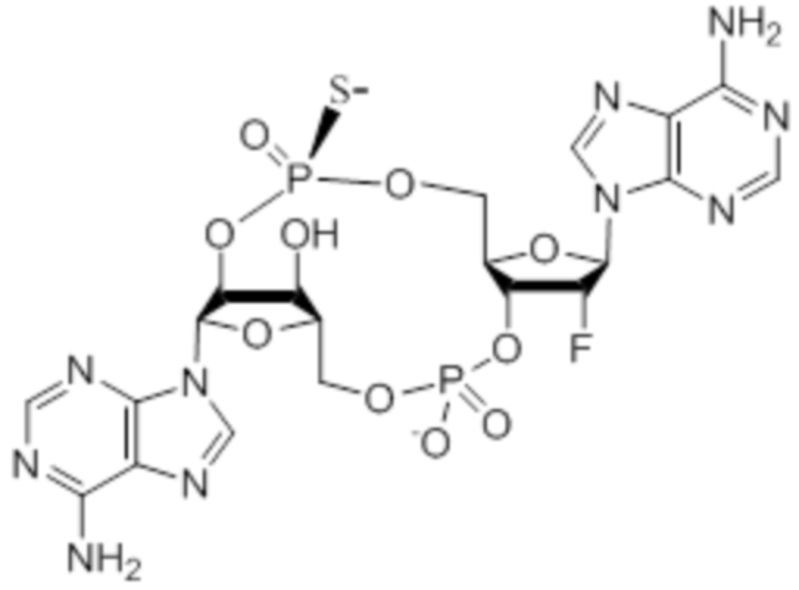
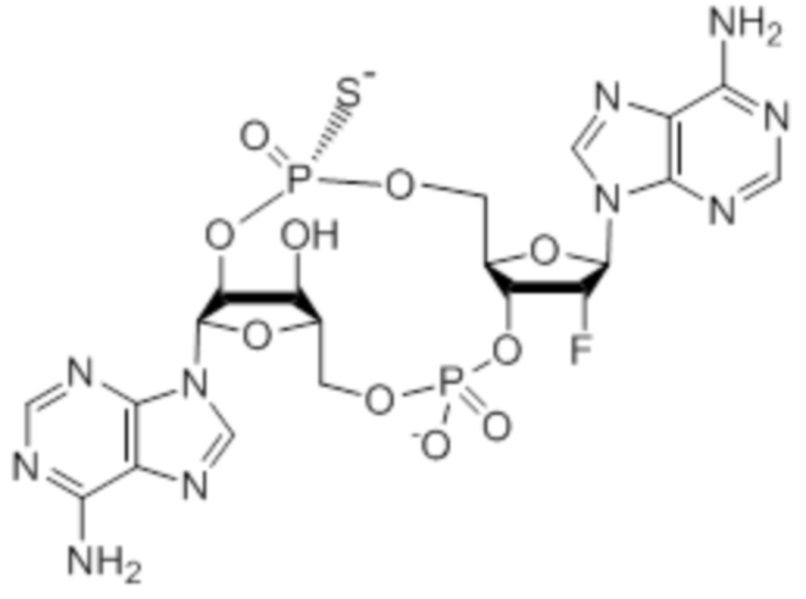
Следует понимать, что соединение 2 представляет собой смесь изомеров, указанную ниже.
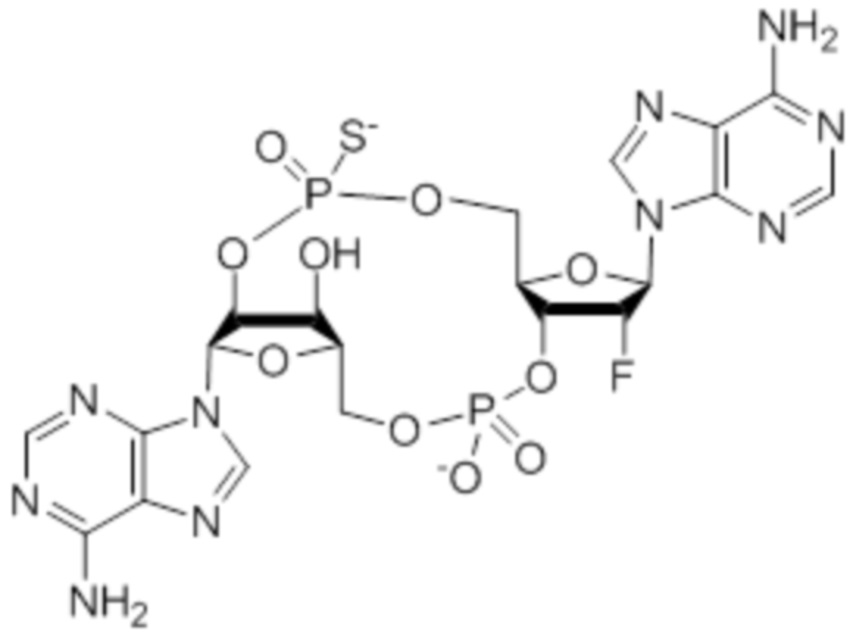
Изомеры соединения 2 представляют собой:
Следует понимать, что соединение 28 представляет собой смесь изомеров, указанную ниже.
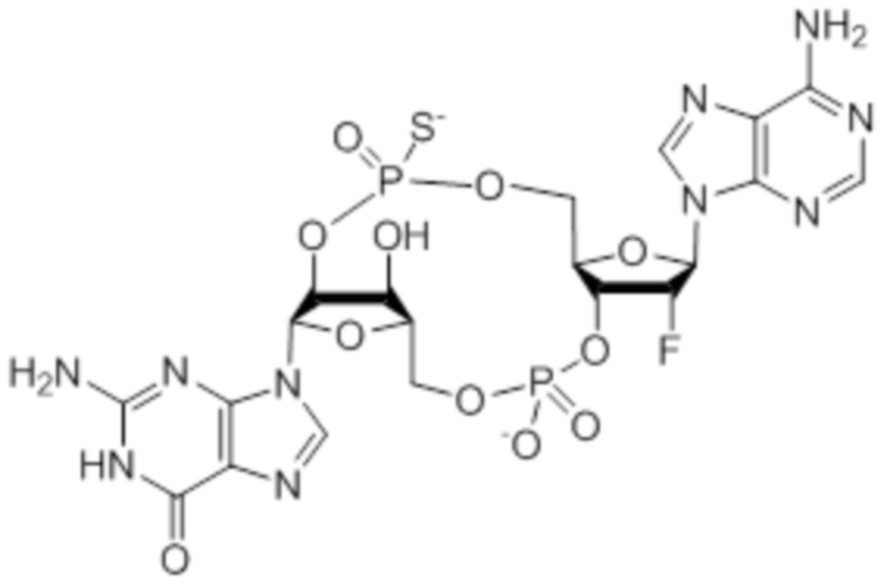
Изомеры соединения 28 представляют собой:


Следует понимать, что соединения, описываемые, например, структурой:
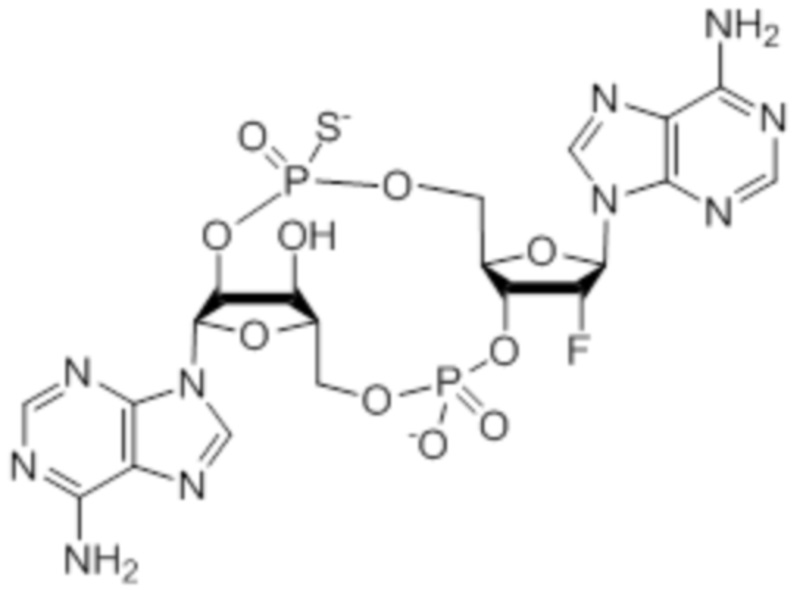
также существуют в протонированной форме, такой как:

которая представляет собой то же самое соединение.
Следует понимать, что соединения, описываемые, например, структурой:
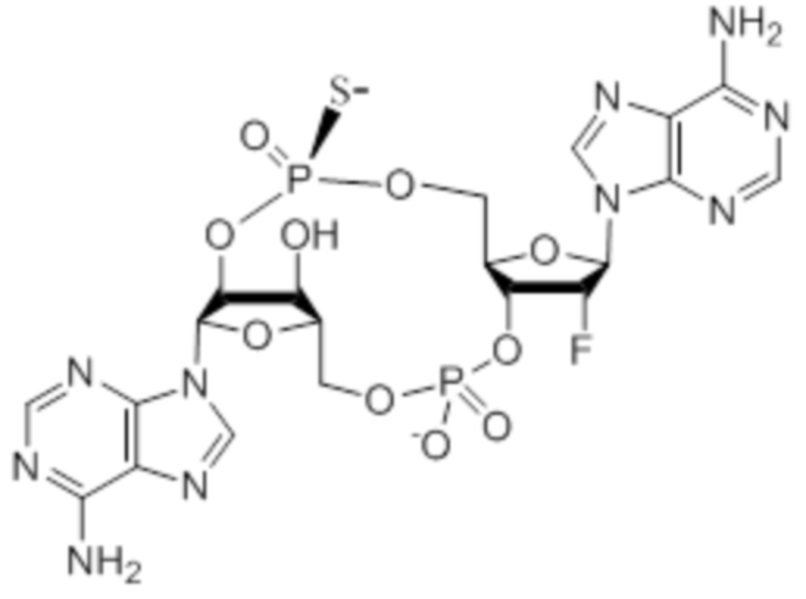
также существуют в протонированной форме, такой как:
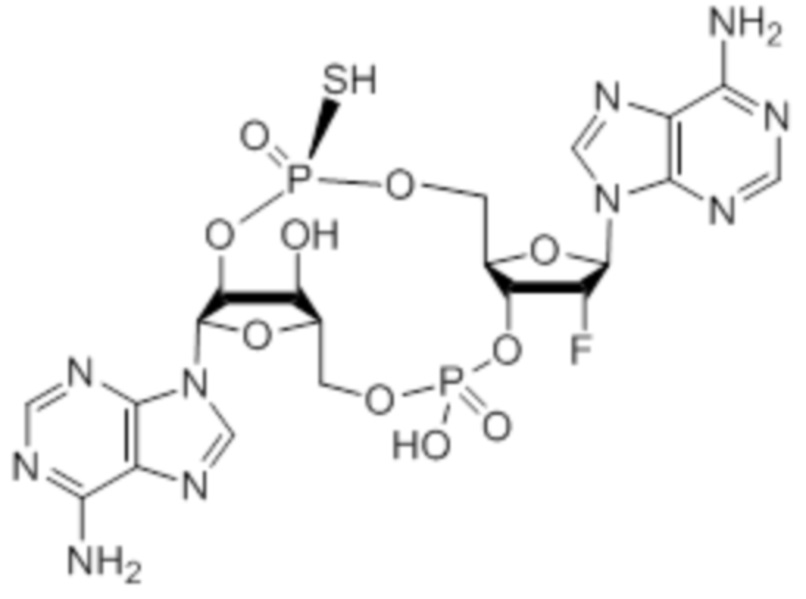
которая представляет собой то же самое соединение.
Следует понимать, что соединения, описываемые, например, структурой:
также существуют в протонированной форме, такой как:

которая представляет собой то же самое соединение.
Обычно, соли согласно настоящему изобретению являются фармацевтически приемлемыми солями. Соли, охватываемые термином «фармацевтически приемлемые соли», относятся к нетоксичным солям соединений согласно настоящему изобретению.
Соли, включая фармацевтически приемлемые соли, легко получаемы специалистами в данной области техники.
Типичные представители фармацевтически приемлемых кислотно-аддитивных солей включают в себя без ограничения 4-ацетамидобензоат, ацетат, адипат, альгинат, аскорбат, аспартат, бензолсульфонат (безилат), бензоат, бисульфат, битартрат, бутират, кальция эдетат, камфорат, камфорсульфонат (камзилат), капрат (деканоат), капроат (гексаноат), каприлат (октаноат), циннамат, цитрат, цикламат, диглюконат, 2,5-дигидроксибензоат, дисукцинат, додецилсульфат (эстолат), эдетат (этилендиаминтетраацетат), эстолат (лаурилсульфат), этан-1,2-дисульфонат (эдизилат), этансульфонат (эзилат), формиат, фумарат, галакторат (муцинат), гентизат (2,5-дигидроксибензоат), глюкогептонат (глюцептат), глюконат, глюкуронат, глутамат, глутарат, глицерофосфат, гликолат, гексилрезорцинат, гиппурат, гидрабамин (N,N'-ди(дегидроабиэтил)этилендиамин), гидробромид, гидрохлорид, гидройодид, гидроксинафтаноат, изобутират, лактат, лактобионат, лаурат, малат, малеат, малонат, манделат, метансульфонат (мезилат), метилсульфат, муцинат, нафтален-1,5-дисульфонат (нападизилат), нафтален-2-сульфонат (напзилат), никотинат, нитрат, олеат, пальмитат, пара-аминобензолсульфонат, пара-аминосалицилат, памоат (эмбонат), пантотренат, пектинат, персульфат, фенилацетат, фенилэтилбарбитурат, фосфат, полигалактуронат, пропионат, пара-толуолсульфонат (тозилат), пироглютамат, пируват, салицилат, себацинат, стеарат, субацетат, сукцинат, сульфамат, сульфат, таннат, тартрат, теоклат (8-хлортеофиллинат), тиоцианат, триэтиодид, ундеканоат, ундециленат и валерат.
Типичные представители фармацевтически приемлемых кислотно-аддитивных солей включают в себя без ограничения соли алюминия, 2-амино-2-(гидроксиметил)-1,3-пропандиола (TRIS, трометамина), аргинина, бенэтамина (N-бензилфенэтиламина), бензатина (N,N'-дибензилэтилендиамина), бис(2-гидроксиэтил)амина, висмута, кальция, хлорпрокаина, холина, клемизола (1-пара-хлорбензил-2-пирролидин-1'-илметилбензимидазола), циклогексиламина, дибензилэтилендиамина, диэтиламина, диэтилтриамина, диметиламина, диметилэтаноламина, допамина, этаноламина, этилендиамина, L-гистидина, железа, изохинолина, лепидина, лития, лизина, магния, меглумина (N-метилглюкамина), пиперазина, пиперидина, калия, прокаина, хинина, хинолина, натрия, стронция, трет-бутиламина и цинка.
Настоящее изобретение включает в свой объем все возможные стехиометрические и нестехиометрические формы соединений формулы (I).
Соединения согласно настоящему изобретению могут существовать в твердой или жидкой форме. В твердой форме, соединения согласно настоящему изобретению могут существовать в большом разнообразии вариантов твердого состояния, варьирующих от полностью аморфного до полностью кристаллического. Термин «аморфный» относится к состоянию, при котором вещество утрачивает дальний порядок структуры на молекулярном уровне и, в зависимости от температуры, может обладать физическими свойствами твердого вещества или жидкости. Обычно, такие вещества не характеризуются отличительными рентгеновскими дифрактограммами и, хотя и обладают свойствами твердого вещества, с формальной точки зрения описываются как жидкость. При нагревании, происходит изменение свойств твердого вещества на свойства жидкого вещества, которое обычно характеризуется изменением состояния второго рода («стеклование»). Термин «кристаллический» относится к твердой фазе, при которой вещество имеет строго упорядоченную внутреннюю структур на молекулярном уровне и характеризуются отличительными рентгеновскими дифрактограммами с определенными пиками. Такие вещества, будучи значительно нагретыми, также обладают свойствами жидкости, но изменение состояния от твердого до жидкого обычно характеризуется фазовым переходом первого рода («плавление»).
Соединения согласно настоящему изобретению могут обладать способностью к кристаллизации в различных формах, т.е. характеристикой, известной как полиморфизм («полиморфы»). Полиморфизм, как правило, возникает в виде ответа на изменения температуры или давления, или и того и другого, и также может являться результатом изменений процесса кристаллизации. Полиморфы можно различать по различным известным из уровня техники физическим характеристикам, таким как рентгеновские дифрактограммы, растворимость и точка плавления.
Соединения формулы (I) могут существовать в сольватированной и несольватированной формах. Используемый в настоящем документе термин «сольват» относится к комплексу с переменной стехиометрией, образованному растворенным веществом (согласно настоящему изобретению, соединением формулы (I) или его солью) и растворителем. Применительно к настоящему изобретению, такие растворители не могут влиять на биологическую активность растворенного вещества. Специалисту в данной области техники следует понимать, что фармацевтически приемлемые сольваты могут формироваться кристаллическими соединениями, когда в процессе кристаллизации молекулы растворителя встраиваются в кристаллическую решетку. Встроившиеся молекулы растворителя могут представлять собой молекулы воды и отличные от воды молекулы, такие как молекулы этанола, изопропанола, DMSO, уксусной кислоты, этаноламина и этилацетата. Кристаллическую структуру со встроенными молекулами воды обычно называют «гидратами». Гидраты включают в себя стехиометрические гидраты, а также композиции, содержащие различные количества воды.
Также следует отметить, что соединения формулы (I) могут формировать таутомеры. «таутомеры» относятся к соединениям, которые представляют собой взаимозаменяемые структурные формы конкретного соединения, и которые варьируют по смещению атомов водорода и электронов. Таким образом, две структуры могут находиться в равновесии за счет движения π-электронов и атомов (обычно, H). Например, енолы и кетоны являются таутомерами, поскольку они легко преобразуются друг в друга при обработке кислотой или основанием. Следует понимать, что все таутомеры и смеси таутомеров соединений согласно настоящему изобретению охватываются объемом соединений согласно настоящему изобретению. В качестве примеры и для ясного понимания, в соединениях формулы (I), если R1 или R3 представляет собой OH, то соединения будут формировать кето-таутомер (=O).
Хотя аспекты каждой переменной величины были в общих чертах перечислены выше по отдельности для каждой переменной величины, настоящее изобретение включает соединения, в которых несколько аспектов в формуле (I) выбирают из каждого из аспектов, перечисленных выше. Поэтому, предполагается, что настоящее изобретение включает все комбинации аспектов для каждой переменной величины.
Нативные молекулы CDN могут быть чувствительны к деградации фосфодиэстеразами, которые присутствуют в крови, на поверхности клетки хозяина или в клетках хозяина, например, в антиген-презентирующих клетках, которые поглощают вакцинные составы, содержащие указанные нативные молекулы CDN. Конкретными примерами являются эктонуклеотидазы, такие как CD39, CD73 и ENPP1, которые находятся на стороне плазматической мембраны клеток, обращенной к плазме, многие из которых, как известно, разрушают нуклеотиды, например, CD39 и ENPP1 обе преобразуют ATP до AMP. Недавно было выяснено, что ENPP1 вносит основной вклад в деградацию CDN, содержащих 2'-5'-фосфодиэфирную связь (Li, L., et al., 2014, Nature Chemical Biology, 10(12), p. 1043-1048). В результате такой деградации может быть снижена эффективность CDN, обладающих агонистической активностью в отношении STING, приводя к меньшему уровню индуцированной экспрессии ключевой молекулы врожденного иммунитета (например, IFNβ) и, следовательно, ослаблению адъювантной способности. Настоящее изобретение описывает два различных и дополняющих друг друга подхода, которые могут быть использованы для усиления и поддержания активности описанных новых CDN. Как более детально описано в последующих разделах, таковыми являются замена кислорода на серу в несвязывающих положениях фосфодиэфира и применение стратегии получения пролекарства для усиления проникновения в клетку и защиты CDN от деградации.
Один аспект настоящего изобретения относится к стереохимически определенным диастереоизомерам циклических пуриновых моно- и дитиодифосфатных динуклеотидов, которые индуцируют активацию STING-зависимой TBK1, и к способам их получения и применения.
Настоящее изобретение относится к способам получения эффективного агониста STING, способного к праймированию и поддержанию T-клеточного ответа на опухолевые антигены, отдельно или в комбинации с другими иммуноонкологическими агентами, и к способам получения адъювантных композиций. Указанные композиции состоят из одного или нескольких циклических пуриновых динуклеотидов формулы (I), где содержащиеся в композиции циклические пуриновые динуклеотиды представляют собой по существу чистые отдельные монотиофосфатные диастереоизомеры или дитиофосфатные диастереоизомеры, способы их производства и способы из применения для стимуляции иммунного ответа у животного. Целью и отдельного средства, и вакцинного состава, является предоставление комбинации антигенов и адъювантов, способной индуцировать генерацию значительной популяции T-клеток памяти и/или B-клеток для быстрой реакции с патогеном, опухолевой клеткой, и т. д., несущими интересующий антиген.
Тиофосфаты (также называемые фосфотиоатами) представляют собой вариант нормальных нуклеотидов, в которых один из атомов кислорода, не являющийся мостиковым и присоединенный к атому фосфора, заменен атомов серы. Фосфотиоатная связь по своему существу является хиральной. Специалисту в данной области техники будет понятно, что тиофосфаты в такой структуре могут существовать в R или S форме. Таким образом, для каждого атома фосфора возможны Rp и Sp формы. В каждом случае, предпочтительными являются по существу чистые диастереоизомеры указанных молекул. Примеры таких CDN-тиофосфатных молекул представлены в настоящем документе на фиг. 1-4.
Используемый в настоящем документе термин «пролекарство» относится к модификации рассматриваемых соединений, при которой модифицированное соединение преобразуется в организме (например, в клетке-мишени или в органе-мишени) обратно в немодифицированную форму посредством ферментативных и неферментативных реакций. Во многих случаях, пролекарственная форма является неактивной или значительно менее активной по сравнению с исходной формой, не являющейся пролекарством, рассматриваемых соединений. Согласно определенным вариантам осуществления, гидрокси на одной рибозе содержит пролекарственную уходящую группу (соединения 13, 26 и 39). Согласно другим вариантам осуществления, пролекарственная форма претерпевает дериватизацию по одному или по обоим из фосфатов и/или тиофосфатов (соединения 3-12, 16-25 и 29-38). Пролекарства могут изменять физико-химические, биофармацевтические и фармакокинетические свойства лекарств. Причинами разработки пролекарств обычно являются плохая растворимость в воде, химическая нестабильность, плохая пероральная биодоступность, неспособность к проникновению через гематоэнцефалический барьер и высокий пресистемный метаболизм, ассоциированный с исходным лекарством. Пригодные пролекарственные фрагменты описаны, например, в "Prodrugs and Targeted Delivery," J. Rautico, Ed., John Wiley & Sons, 2011. Пролекарства фосфатов, которые имеют особую значимость применительно к настоящему изобретению, описаны в Wiemer, A.J. и Wiemer, D.F. ʺProdrugs of Phosphonates and Phosphates: Crossing the Membrane Barrierʺ in Topics of Current Chemistry, (2015) V360, 115-160.
Предпочтительные циклические пуриновые динуклеотиды согласно настоящему изобретению включают в себя пролекарственные дифосфатные CDN (таких как соединения 9, 10, 11 и 12 на фигуре 1; соединения 22, 23, 24 и 25 на фигуре 2; соединения 35, 36, 37 и 38 на фигуре 3 и соединения 40, 41 и 42 на фигуре 4), не являющиеся пролекарствами монотиофосфаты (такие как соединения 2, 15 и 28 на фигурах 1-3), не являющиеся пролекарствами дитиофосфаты (такие как соединения 1, 14 и 27 на фигурах 1-3) и пролекарственные формы монотиофосфатов (такие как соединения 7, 8, 20, 21, 33 и 34 на фигурах 1-3) и дитиофосфатов (такие как соединения 3-6, 16-19 и 29-32 на фигурах 1-3).
Определения
Используемый в настоящем документе термин «соединение согласно настоящему изобретению» включает все сольваты, комплексы, полиморфы, меченые радиоактивным изотопом производные, таутомеры, стереоизомеры и оптические изомеры соединений формулы (I) и их солей.
Используемые в настоящем документе конкретные соединения согласно настоящему изобретению обозначаются в числовом виде в соответствии с обозначением на фигурах. Например, соединение 2 представляет собой соединение на фигуре 1 с цифрой «2» под ним, и соединение 27 представляет собой соединение на фигуре 3 w с цифрой «27» под ним. Дополнительные обозначения «a» и «b», и т. д., соответствуют изомеру 1 и изомеру 2, и т. д., соответственно. Например, «соединение 2a» представляет собой изомер 1 соединения 2, «соединение 27b» представляет собой изомер 2 соединения 27.
Если не определено иное, то обозначение «изомер» или «диастереоизомер» представляет собой указание на порядок, в котором указанное соединение элюируется с делительной колонки в указанных условиях. Определенное соединение с меньшим временем удерживания при проведении LCMS обозначается как «изомер 1» или «диастереоизомер 1», а определенное соединение с большим временем удерживания при проведении LCMS обозначается как «изомер 2» или «диастереоизомер 2», и т. д.
Используемый в настоящем документе термин «эффективное количество» означает, количество лекарства или фармацевтического средства, которое вызывает со стороны ткани, системы, животного или человека биологический или медицинский эффект, который является искомым, например, для исследователя или врача. Более того, термин «терапевтически эффективное количество» означает любое количество в сравнении с соответствующим субъектом, не получающим такое количество, приводит к улучшению лечения, заживлению, предупреждению или уменьшению интенсивности заболевания, нарушения или побочного эффекта, или к снижению скорости развития заболевания или нарушения. Указанный термин также включает в свой объем количества, эффективные для усиления нормальной физиологической функции.
Термин «профилактика» включает предупреждение и относится к мере или способу, которые направлены скорее на предупреждение, чем на исцеление или лечение заболевания. Профилактика относится к снижению риска возникновения или развития заболевания, при котором у субъекта, который может быть подвержен действию болезнетворного средства, или у субъекта, предрасположенного к заболеванию до его начала, не возникает, по меньший мере, один клинический симптом заболевания.
Используемый в настоящем документе термин «фармацевтически приемлемый» относится к соединениям, веществам, композициям и лекарственным формам, которые в рамках здравого медицинского суждения подходят для применения в контакте с тканями людей и животных без возникновения чрезмерной токсичности, раздражения или другой проблемы или осложнения, соизмеримыми с приемлемым соотношением риска и пользы.
«Используемые в настоящем документе фармацевтически приемлемые наполнители» включают в себя все разбавители, носители, связующие вещества, глиданты и другие компоненты фармацевтических композиций, вместе с которыми вводят соединение согласно настоящему изобретению.
«Алкил» относится к углеводородной цепи, содержащей определенное число «кольцевых атомов». Например, C1-C6алкил относится к алкильной группе, содержащей от 1 до 6 кольцевых атомов. Например, C12-C18алкил относится к алкильной группе, содержащей от 12 до 18 кольцевых атомов. Например, C1-C20алкил относится к алкильной группе, содержащей от 1 до 20 кольцевых атомов. Алкильные группы могут быть насыщенными, ненасыщенными, неразветвленными или разветвленными. Типичные представители разветвленных алкильных групп имеют одно, два или три разветвления. Пример алкила включает метил, этил, этилен, пропил (н-пропил и изопропил), бутен, бутил (н-бутил, изобутил и трет-бутил), пентил и гексил.
Если не определено иное, то «циклоалкил» относится к насыщенной или ненасыщенной неароматической углеводородной кольцевой системе, содержащей от 3 до 7 кольцевых атомов. Циклоалкильные группы представляют собой моноциклические или бициклические кольцевые системы. Например, C3-C7циклоалкил относится к циклоалкильной группе, содержащей от 3 до 7 кольцевых атомов. Используемые в настоящем документе примеры циклоалкила включают в себя циклопропил, циклобутил, циклопентил, циклогексил, циклобутенил, циклопентенил, циклогексенил, циклогептил и спирогептан.
«Арил» относится к ароматическому углеводородному кольцу. Арильные группы представляют собой моноциклические, бициклические и трициклические кольцевые системы, содержащие всего от 5 до 14 кольцевых атомов, где, по меньшей мере, одна кольцевая система является ароматической, и где каждое кольцо в системе содержит от 3 до 7 кольцевых атомов, такие фенил, нафталин, тетрагидронафталин и бифенил. Подходящим арилом является фенил.
«Гетероарил» относится к моноциклическому ароматическому 4-8-членному кольцу, содержащему от 1 до 7 атомов углерода и от 1 до 4 гетероатомов, при условии, что если число атомов углерода равно 3, то ароматическое кольцо содержит, по меньшей мере, два гетероатома. Гетероарильные группы, содержащие более одного гетероатома, могут содержать различные гетероатомы. Примеры гетероарила включают в себя пирролил, пиразолил, имидазолил, оксазолил, изоксазолил, тиазолил, изотиазолил, фуранил, фуразанил, тиенил, триазолил, пиридинил, пиримидинил, пиридазинил, пиразинил, триазинил, тетразинил.
«Гетероциклоалкил» относится к насыщенному или ненасыщенному неароматическому кольцу, содержащему от 4 до 12 кольцевых атомов, из которых от 1 до 11 атомов являются атомами углерода, и от 1 до 6 атомов являются гетероатомами. Гетероциклоалкильные группы, содержащие более одного гетероатома, могут содержать различные гетероатомы. Гетероциклоалкильные группы представляют собой моноциклические кольцевые системы или моноциклическое кольцо, конденсированное с арильным кольцом или с гетероарильным кольцом, содержащим от 3 до 6 кольцевых атомов. Примеры гетероциклоалкила включают в себя пирролидинил, тетрагидрофуранил, дигидрофуранил, пиранил, тетрагидропиранил, дигидропиранил, тетрагидротиенил, пиразолидинил, оксазолидинил, оксетанил, тиазолидинил, пиперидинил, гомопиперидинил, пиперазинил, морфолинил, тиаморфолинил, 1,3-диоксоланил, 1,3-диоксанил, 1,4-диоксанил, 1,3-оксатиоланил, 1,3-оксатианил, 1,3-дитианил, 1,3-оксазолидин-2-он, гексагидро-1H-азепин, 4,5,6,7,тетрагидро-1H-бензимидазол, пиперидинил, 1,2,3,6-тетрагидропиридинил и азетидинил.
Если не определено иное, то «гетероатом» относится к атому азота, серы или кислорода.
Композиции
Хотя представляется возможным, что для применения в терапии соединение согласно настоящему изобретению можно вводить в виде исходного химического вещества, соединение согласно настоящему изобретению можно доставлять в виде активного ингредиента в фармацевтической композиции. Такие композиции могут быть приготовлены способом, хорошо известным в области фармацевтики, и содержать, по меньшей мере, одно активное соединение. Соответственно, настоящее изобретение дополнительно относится к фармацевтическим композициям, содержащим соединение согласно настоящему изобретению и один или более фармацевтически приемлемых эксципиентов. Эксципиент(ы) должен(ны) быть приемлемыми с точки зрения совместимости с другими ингредиентами композиции и безвредным для реципиента. В соответствии с другим аспектом настоящего изобретения, также предусмотрен способ приготовления фармацевтической композиции, включающей в себя средство или его фармацевтически приемлемые соли с одним или более фармацевтически приемлемыми эксципиентами. Фармацевтическая композиция может быть предназначена для применения при лечении и/или профилактике любого из состояний, описанных в настоящем документе.
Как правило, соединение согласно настоящему изобретению вводят в фармацевтически эффективном количестве. Действительно вводимое количество соединения будет обычно определяться лечащим врачом в свете соответствующих обстоятельств, включая подлежащее лечению состояние, выбранный путь введения, конкретное вводимое соединение, возраст, вес, ответ индивидуального пациента, тяжесть симптомов пациента и т. п.
Фармацевтические композиции могут быть представлены в стандартных лекарственных формах, содержащих предопределенное количество активного ингредиента в стандартной форме. Термин «стандартные лекарственные формы» относится к физически дискретным единицам, подходящим в качестве стандартных дозировок для субъектов-людей и другим млекопитающих, причем каждая единица содержит предопределенное количество активного вещества, рассчитанное для получения желаемого терапевтического эффекта, в сочетании с подходящим фармацевтическим эксципиентом, основой или носителем. Обычные стандартные лекарственные формы включают в себя предварительно наполненные, предварительно отмеренные ампулы или шприцы с жидкими композициями или пилюли, таблетки, капсулы или т. п. в случае твердых композиций.
Предпочтительные стандартные лекарственные композиции представляют собой формы, содержащие суточную дозу или субдозу, или ее подходящую фракцию, активного ингредиента. Поэтому, такие стандартные дозы можно вводить однократно или несколько раз в сутки. Такие фармацевтические композиции могут быть приготовлены посредством любого из способов, хорошо известных в области фармацевтики.
Фармацевтические композиции могут быть адаптированы для введения посредством любого подходящего пути, например, посредством перорального (включая буккальный или сублингвальный), ректального, ингаляционного, интраназального, местного (включая буккальный, сублингвальный или чрескожный), вагинального или инъекционного (включая подкожный, внутримышечный, парентеральный, внутривенный или внутрикожный) пути. Такие композиции могут быть приготовлены посредством любого из способов, известных в области фармацевтики, например, путем комбинирования активного ингредиента с носителем(ями) или эксципиентом(ами).
В дополнение к вышеописанным путям введения при лечении злокачественных опухолей, фармацевтические композиции могут быть адаптированы для введения посредством внутриопухолевой или периопухолевой инъекции. Ожидается, что внутриопухолевая или периопухолевая инъекция соединения согласно настоящему изобретению непосредственно в единичную солидную опухоль или в прилегающие к ней области вызовет иммунный ответ, который может атаковать или разрушить клетки злокачественной опухоли по всему организму, по существу уменьшая, а в некоторых случаях полностью удаляя, злокачественную опухоль у больного субъекта. Активация таким способом иммунной системы для устранения опухолей в уделенной области общеизвестна как абскопальный эффект и была продемонстрирована на животных при многих лечебных воздействиях (van der Jeught, et al., Oncotarget, 2015, 6(3), 1359-1381). Дополнительным преимуществом местного или внутриопухолевого или периопухолевого введения является способность достигать эквивалентной эффективности при многократно меньших дозах, тем самым минимизируя или устраняя побочные эффекты, которые могут наблюдаться при более высоких системных дозах (Marabelle, A., et al., Clinical Cancer Research, 2014, 20(7), p1747-1756).
Фармацевтические композиции, адаптированные для перорального введения могут быть представлены в виде дискретных единиц, таких как капсулы или таблетки; порошки или гранулы; растворы или суспензии в водных или неводных жидкостях; съедобные пены или кремы; или жидкие эмульсии типа «масло-в-воде» или жидкие эмульсии типа «вода-в-масле».
Например, для перорального введения в форме таблетки или капсулы, активный лекарственный компонент может быть сочетан с пероральным, нетоксичным фармацевтически приемлемым инертным эксципиентом, таким как этанол, глицерин, вода и т. п. Порошки приготавливают путем измельчения соединения до подходящего размера частиц и смешивания с аналогично приготовленным фармацевтическим эксципиентом, таким как съедобный углевод, например, крахмал или маннит. Также могут присутствовать вкусоароматизаторы, консерванты, средства, способствующие диспергированию и красители.
Капсулы получают путем приготовления порошковой смеси, как описано выше, и заполнения формованных желатиновых капсул. эксципиенты, включая глиданты и смазки, такие как коллоидный кремний, тальк, стеарат магния, стеарат кальция или твердый полиэтиленгликоль, можно добавлять в порошковую смесь перед процедурой заполнения. Разрыхлитель или солюбилизатор, такой как агар-агар, карбонат кальция или карбонат натрия, также можно добавлять для улучшения биодоступности лекарственного средства после введения капсулы.
Кроме того, если это желательно или необходимо, то в состав смеси также могут быть включены эксципиенты, включая подходящие связующие вещества, глиданты, смазки, подсластители, вкусоароматизаторы, разрыхлители и красители. Подходящие связующие вещества включают в себя крахмал, желатин, природные сахара, такие как глюкоза или бета-лактоза, кукурузные подсластители, природные и синтетические камеди, такие как акация, трагакант, или альгинат натрия, карбоксиметилцеллюлозу, полиэтиленгликоль, воски и т. п. Смазки, используемые в таких лекарственных формах, включают в себя олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и т. п. Разрыхлители включают в себя без ограничения крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь и т. п. Таблетки составляют, например, путем приготовления порошковой смеси, гранулирования или комкования, добавления смазки и разрыхлителя и прессования в таблетки. Порошковую смесь приготавливают путем смешивания подходящим образом измельченного соединения с разбавителем или основой, как описано выше, и необязательно, со связующим веществом, таким как карбоксиметилцеллюлоза, альгинат, желатин или поливинилпирролидон, с раствором замедлителя, таким как парафин, с ускорителем всасывания, таким как четвертичная соль и/или со средством для абсорбции, таким как бентонит, каолин или фосфат дикальция. Порошковая смесь может быть гранулирована путем увлажнения посредством связующего вещества, такого как сироп, крахмальная паста, акадийское клейкое вещество или растворами целлюлозных или полимерных веществ и продавлена через сито. В качестве альтернативы, гранулирования порошковую смесь можно обработать в машине для таблеток, и в результате получить комки неровной формы, разбитые на гранулы. Гранулы могут быть смазаны для профилактики слипания в таблеткообразные штампы посредством добавления стеариновой кислоты, стеарата, талька или минерального масла. Смазанную смесь часто прессуют в таблетки. Соединения согласно настоящему изобретению также могут быть сочетаны со свободно текучим инертным носителем и спрессованы в таблетки непосредственно без прохождения этапов гранулирования или комкования. Может быть предусмотрено прозрачное или непрозрачное защитное покрытие, состоящее из герметичного покрытия из шеллака, покрытия из сахара или полимерного вещества и отшлифованного покрытия из воска. Красители могут быть добавлены в такие покрытия для отличия различных лекарственных форм.
Пероральные жидкости, такие как раствор, суспензии, сиропы и эликсиры могут быть приготовлены в стандартной лекарственной форме так, чтобы данное количество содержало предопределенное количество соединения. Сиропы могут быть приготовлены путем растворения соединения в подходящем образом вкусоароматизированном водном растворе, тогда как эликсиры приготавливают посредством использования нетоксичной спиртовой основы. Суспензии могут быть составлены путем диспергирования соединения в нетоксичной основе. Также могут быть добавлены солюбилизаторы и эмульгаторы, такие как этоксилированные изостеариловые спирты и простые эфиры полиоксиэтиленсорбита, консерванты, вкусоароматические добавки, такие как масло перечной мяты или натуральные подсластители или сахарин или другие искусственные подсластители, и т. п.
При необходимости стандартные лекарственные композиции для перорального введения могут быть микроинкапсулированы. Композиция также может быть приготовлена для пролонгирования или замедления высвобождения, как например, посредством покрытия или заливки конкретного вещества в полимерах, воске или т. п.
Соединения согласно настоящему изобретению также можно вводить в форме липосомальных систем доставки, таких как маленькие однослойные везикулы, большие однослойные везикулы и многослойные везикулы. Липосомы могут быть сформированы из ряда фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины. Соединения согласно настоящему изобретению также можно вводить в форме системы доставки наночастиц, для которых существует множество композиций и способов приготовления. Как полимерные наночастицы, так и подходящим образом составленные и калиброванные липосомы, представляют собой особенно предпочтительные составы при лечении злокачественных опухолей и, в частности, для доставки соединений согласно настоящему изобретению, поскольку они предпочтительно нацелены на опухоль и лимфоузлы. Такие составы направленного действия обладают несколькими потенциальными преимуществами, а именно: защита соединений согласно настоящему изобретению от разрушения, увеличение количества активного средства в области действия и минимизация нежелательных потенциальных побочных эффектов, и в результате, чрезмерного системного воздействия (Cai, Shuang et al., 2011 Advanced Drug Delivery Reviews, 2011, V63, p. 901-908). Потенциальная польза таких подходов в составлении CDN-агониста STING была продемонстрирована для составов, действующих непосредственно на опухоли (Nakumura, T. et al., Journal of Controlled Release, 2015, V216, p. 149-157) и при применении в качестве адъюванта (Hanson, M. et al., Journal of Clinical Investigation, 2015, V125(6), p. 2532-2546). Кроме того, существует множество путей введения (внутриопухолевый, подкожный, внутривенный, интраперитонеальный и внутримышечный) наночастиц и липосомальных составов, которые могут быть особенно полезны для соединений согласно настоящему изобретению. Более конкретно, молекулы согласно настоящему изобретению, аналогичные естественным CDN молекулам, могут быть чувствительны к разрушению посредством фосфодиэстераз, которые присутствуют внутри или на клетках хозяина, например, внутри антиген-презентирующих клеток. Эффективность соединения согласно настоящему изобретению может быть снижена за счет такого разрушения, приводя к более низкой индуцированной экспрессии ключевой молекулы врожденного иммунитета (например, IFN-бета). Следовательно, такое разрушение может обеспечивать более слабую эффективность, измеренную посредством высвобождения IFN-бета из PBMC, или сниженную эффективность вакцины, определенную магнитудой измеренного антиген-специфического иммунного ответа.
Фармацевтические композиции, адаптированные для чрескожного введения, могут быть представлены в виде дискретных пластырей, предназначенных для сохранения плотного контакта с эпидермисом реципиента в течение продолжительного периода времени.
Фармацевтические композиции, адаптированные для местного введения, могут быть составлены в виде мазей, кремов, суспензий, лосьонов, порошков, растворов, паст, гелей, спреев, аэрозолей и масел.
Для лечения глаз или других наружных тканей, например, полости рта или кожи, композиции предпочтительно наносят в виде местной мази или крема. При составлении в виде мази, активный ингредиент может быть использован либо вместе с парафиновой, либо с водорастворимой основой для мази. В качестве альтернативы, активный ингредиент может быть составлен в виде крема вместе с основой для крема типа «масло-в-воде» или основой типа «вода-в-масле».
Фармацевтические композиции, адаптированные для местного введения в глаза, включают в себя глазные капли, где активный ингредиент растворен или суспендирован в подходящем носителе, особенно в водном растворителе.
Фармацевтические композиции, адаптированные для местного введения в полости рта, включают в себя леденцы, пастилки и ополаскиватели для полости рта.
Фармацевтические композиции, адаптированные для ректального введения могут быть представлены в виде суппозиториев или в виде клизм.
Лекарственные формы для назального или ингаляционного введения могут быть удобным образом составлены в виде аэрозолей, растворов, суспензий, капель, гелей и сухих порошков.
Композиции для интраназального введения включают в себя водные композиции, вводимые в полость носа посредством капель или посредством помпы под давлением. С этой целью, подходящие композиции содержат воду в качестве разбавителя или носителя. Композиции для введения в легкие или полость носа могут содержать один или более эксципиентов, например, одно или несколько средств, способствующих суспендированию, один или более консервантов, одно или более поверхностно-активных веществ, одно или более средств, регулирующих тоничность, один или более сорастворителей, и могут включать в себя компоненты для контроля рН композиции, например, буферную систему. Дополнительно, композиции могут содержать другие эксципиенты, такие как антиоксиданты, например, метбисульфит натрия, и средства, маскирующие вкус. Композиции также можно вводить в полость носа или другие области респираторного тракта посредством небулайзера.
Интраназальные композиции могут позволить доставлять соединение(я) формулы (I) или (а) его фармацевтически приемлемую соль(и) во все области полостей носа (ткань мишень) и дополнительно, могут позволить соединению(ям) формулы (I) или (а) его фармацевтически приемлемой соли(ям) сохранять контакт с тканью мишенью в течение более длительного периода времени. Подходящий режим дозирования для интраназальных композиций будет представлять собой для пациента медленный вдох через нос после того, как полость носа была очищена. В процессе ингаляции композиция будет вводиться в одну ноздрю, тогда как другая будет зажата вручную. Эта процедура затем повторяют для другой ноздри. Как правило, производят один или два впрыскивания в каждую ноздрю посредством вышеуказанной процедуры один, два или три раза в сутки каждый день, в идеале однократно в сутки. Особый интерес представляют собой интраназальные композиции, подходящие для введения однократно в сутки.
Средство(а), способствующее суспендированию, будучи включенным в состав, как правило, будет присутствовать в количестве приблизительно от 0,1 до 5% (по массе), как например, от 1,5% до 2,4% (по массе), относительно общей массы композиции. Примеры фармацевтически приемлемых средств, способствующих суспендированию, включают в себя без ограничения Авицел®(микрокристаллическая целлюлоза и карбоксиметилцеллюлоза натрия), карбоксиметилцеллюлозу натрия, вигум, трагакант, бентонит, метилцеллюлозу, ксантановую камедь, карбопол и полиэтиленгликоли.
Композиции для введения в легкие или полость носа могут содержать один или более эксципиентов, и могут быть защищены от микробной или грибковой контаминации и роста путем включения в состав одного или нескольких консервантов. Примеры фармацевтически приемлемых противомикробных средств или консервантов включают в себя без ограничения соединения четвертичного аммония (например, бензалкония хлорид, бензетония хлорид, цетримид, цетилпиридиния хлорид, лауралкония хлорид и миристилпиколиния хлорид), ртуть-содержащие средства (например, фенилмеркурнитрат, фенилмеркурацетат и тимеросал), спиртовые средства (например, хлорбутанол, фенилэтиловый спирт и бензиловый спирт), антибактериальные сложные эфиры (например, сложные эфиры парагидроксибензоевой кислоты), хелатирующие средства, такие как динатрия эдетат (EDTA) и другие противомикробные средства, такие как хлоргексидин, хлоркрезол, сорбиновая кислота и ее соли (такие как сорбат калия) и полимиксин. Примеры фармацевтически приемлемых противогрибковых средств и консервантов включают в себя без ограничения бензоат натрия, сорбиновую кислоту, пропионат натрия, метилпарабен, этилпарабен, пропилпарабен и бутилпарабен. Консервант(ы), если они включены в состав, могут присутствовать в количестве от 0,001 до 1% (по массе), как например, от 0,015% до 0,5% (по массе), относительно общей массы композиции.
Композиции (например, в которых, по меньшей мере, одно соединение представляет собой суспензию) могут включать в себя одно или несколько поверхностно-активных веществ, чья функция облегчать растворение частиц лекарственного средства в водной фазе композиции. Например, используемое количество поверхностно-активного вещества представляет собой количество, которое не вызывает пенообразование в процессе перемешивания. Примеры фармацевтически приемлемых поверхностно-активных веществ включают в себя жирные спирты, сложные эфиры и простые эфиры, такие как полиоксиэтилен (20) сорбитанмоноолеат (Полисорбат 80), простые эфиры макрогола и полоксамеры. Поверхностно-активное вещество может присутствовать в количестве приблизительно от 0,01 до 10% (по массе), как например от 0,01 до 0,75% (по массе), например, приблизительно 0,5% (по массе) относительно общей массы композиции.
Одно или несколько средств для регулирования тоничности могут быть включены в состав для достижения тоничности с жидкостями тела, например, жидкостями для назальной полости, приводя к снижению уровня раздражения. Примеры фармацевтически приемлемых средств для регулирования тоничности включают в себя без ограничения хлорид натрия, декстрозу, ксилит, хлорид кальция, глюкозу, глицерин и сорбит. Средство, регулирующее тоничность, если присутствует, может быть включено в состав в количестве от 0,1 до 10% (по массе), как например от 4,5 до 5,5% (по массе), например, приблизительно 5,0% (по массе) относительно общей массы композиции.
Композиции согласно настоящему изобретению могут быть забуференными посредством добавления подходящих буферных средств, таких как цитрат натрия, лимонная кислота, трометамол, фосфаты, такие как динатрия фосфат (например, додекагидрат, гептагидрат, дигидрат и безводные формы) или фосфат натрия или их смеси.
Буферные средства, если присутствуют, могут быть включены в состав в количестве от 0,1 до 5% (по массе), например, от 1 до 3% (по массе) относительно общей массы композиции.
Примеры средств, маскирующих вкус, включают в себя сукралозу, сахарозу, сахарин или его соль, фруктозу, декстрозу, глицерин, кукурузный сироп, аспартам, ацесульфам-К, ксилит, сорбит, эритрит, глицирризинат аммония, тауматин, неотам, маннит, ментол, эвкалиптовое масло, камфору, природные вкусоароматизирующие средства, искусственные вкусоароматизирующие средства и их сочетания.
Один или более сорастворителей могут быть включены в состав, чтобы способствовать растворению лекарственного(ых) средства(ств) и/или других эксципиентов. Примеры фармацевтически приемлемых сорастворителей включают в себя без ограничения пропиленгликоль, дипропиленгликоль, этиленгликоль, глицерин, этанол, полиэтиленгликоли (например, PEG300 или PEG400) и метанол. Согласно одному варианту осуществления, растворитель представляет собой пропиленгликоль.
Сорастворитель(и), если присутствует, может быть включен в состав в количестве от 0,05 до 30% (по массе), как например от 1 до 25% (по массе), например, от 1 до 10% (по массе) относительно общей массы композиции.
Композиции для ингаляционного введения включают в себя водные, органические или водно/органические смеси, сухой порошок или кристаллические композиции, вводимые через респираторный тракт посредством помпы под давлением или ингалятора, например, ингаляторов резервуара сухого порошка, порошковых ингаляторов стандартной дозы, предварительно дозированных многодозовых порошковых ингаляторов, назальных ингаляторов или ингаляторов с аэрозолем под давлением, небулайзеров или инсуффляторов. С этой целью, подходящие композиции содержат воду в качестве разбавителя или носителя и могут доставляться с общепринятыми эксципиентами, такими как буферные средства, средства для регулирования тоничности и т. п. Водные композиции также можно вводить в полость носа или в другие области респираторного тракта посредством небулайзера. Такие композиции могут представлять собой водные растворы или суспензии или аэрозоли, доставляемые посредством упаковок под давлением, как например, посредством дозированного ингалятора с использованием подходящего разжиженного пропеллента.
Композиции для местного введения в полость носа (например, при лечении ринита) или в легкие включают в себя композиции аэрозоля под давлением и водные композиции, доставляемые в полости носа посредством помпы под давлением. Композиции, которые находятся не под давлением, и подходят для местного введения в полости носа, представляют собой особый интерес. С этой целью, подходящие композиции содержат воду в качестве разбавителя или носителя. Водные композиции для введения в легкие или полость носа могут быть доставлены вместе с общепринятыми эксципиентами, такими как буферные средства, средства для регулирования тоничности и т. п. Водные композиции также можно вводить в полость носа посредством небулайзера.
Жидкостной диспенсер может быть обычным образом использован для доставки жидкой композиции в полости носа. Жидкая композиция может быть водной или неводной, но, как правило, водной. Такой жидкостной диспенсер может иметь сопло-дозатор или распределяющее отверстие, через которое измеренная доза жидкой композиции распределяется после надавливания на механизм помпы жидкостного диспенсера. Такие жидкостные диспенсеры обычно предоставляются вместе с резервуаром множества измеренных доз жидкой композиции, дозы распределяются после последовательного приведения помпы в действие. Сопло-дозатор или отверстие могут быть конфигурированы для введения внутрь ноздрей пользователя для распределения спрея жидкой композиции в назальной полости. Жидкостной диспенсер вышеуказанного типа описан и проиллюстрирован в международной патентной заявке с публикационным номером WO 2005/044354 (Glaxo Group Limited). У диспенсера есть полость, в которой находится устройство нагнетания жидкости, имеющее помпу под давлением, установленную на контейнер для содержания жидкой композиции. Полость имеет, по меньшей мере, один рычаг для надавливания пальцем, который подвижен вовнутрь, относительно полости, для движения контейнера вверх в полости посредством замка, что вызывает сжатие помпы и выброс измеренной дозы композиции из трубки помпы через назальное сопло полости. Согласно одному варианту осуществления, жидкостной диспенсер представляет собой обычный тип, проиллюстрированный в Фигурах 30-40 WO 2005/044354.
Водные композиции, содержащие соединение формулы (I) или его фармацевтически приемлемую соль также могут быть доставлены посредством помпы, как раскрыто в международной патентной заявке с публикационным номером WO2007/138084 (Glaxo Group Limited), например, как раскрыто в ее ссылке на фигуры 22-46, или как раскрыто в патентной заявке Великобритании с номером GB0723418.0 (Glaxo Group Limited), например, как раскрыто в ее ссылке на Фигуры 7-32. Помпа может управляться рычагом, как раскрыто в Фигурах 1-6 заявки GB0723418.0.
Сухие порошковые композиции для местной доставки в легкие посредством ингаляции могут быть представлены, например, в капсулах или картриджах, например, из желатина, или блистерах, например, из ламинированной алюминиевой фольги, для использования в ингаляторе или инсуффляторе. Порошковые смешанные композиции обычно содержат смесь порошка для ингаляции, содержащую соединение формулы (I) или его фармацевтически приемлемую соль и подходящую основу для порошка (вещества носителя/разбавителя/эксципиента), такой как моно-, ди- или полисахариды (например, лактоза или крахмал). В дополнение к лекарству и носителю, сухие порошковые композиции также могут включать в себя дополнительный эксципиент (например, тройное средство, такое как сложный эфир сахара, например, октаацетат целлобиозы, стеарат кальция или стеарат магния).
Согласно одному варианту осуществления, композиция, подходящая для ингаляционного введения, может быть включена в состав множества запечатанных дозированных контейнеров, предусматривающих упаковку(и) лекарственного средства, помещенного внутрь подходящего ингаляционного устройства. Контейнеры можно разорвать, расслоить или иным образом поочередно открыть, и дозы сухой порошковой композиции вводят путем ингаляции через мундштук ингаляционного устройства, как известно в данной области техники. Упаковка лекарственного средства может принимать вид различных форм, например, форму диска или вытянутой полоски. Примеры ингаляционных устройств представляют собой DISKHALER™ и DISKUSTM устройства производства GlaxoSmithKline.
Композиции в виде сухого ингаляционного порошка также могут быть предусмотрены в виде объемного резервуара в ингаляционном устройстве, причем устройство предусмотрено с дозирующим механизмом для измерения дозы композиции, поступающей из резервуара в ингаляционный канал, где измеренную дозу пациент может ингалировать, проводя ингаляцию через мундштук устройства. Примеры коммерческих устройств такого типа представляют собой TURBUHALER™ (AstraZeneca), TWISTHALER™ (Schering) и CLICKHALER™ (Innovata).
Дополнительный способ доставки для композиции в виде сухого ингаляционного порошка осуществляется для измеренных доз композиции в капсулах (одна доза на капсулу), которые затем помещаются внутрь ингаляционного устройства, обычно пациентом по мере необходимости. Устройство имеет средства для перфорации, прокалывания или открытия капсулы иным образом, так что доза может быть доставлена внутрь легких пациентов, когда они проводят ингаляцию через мундштук устройства. В качестве торговых примеров таких устройств могут быть упомянуты ROTAHALER™ (GlaxoSmithKline) и HANDIHALER™ (Boehringer Ingelheim.)
Композиции аэрозоля под давлением, подходящие для ингаляции могут представлять собой как суспензию, так и раствор, и могут содержать соединение формулы (I) или его фармацевтически приемлемую соль и подходящий пропеллент, такой как фторуглерод или водородсодержащий хлорфторуглерод или их смеси, в частности, гидрофторалканы, особенно 1,1,1,2-тетрафторэтан, 1,1,1,2,3,3,3-гептафтор-н-пропан или их смесь. Аэрозольная композиция может необязательно содержать дополнительные композиционные эксципиенты, хорошо известные в данной области техники, такие как поверхностно-активные вещества, например, олеиновая кислота, лецитин или олигомолочная кислота или ее производное, например, как описано в WO 94/21229 и WO 98/34596 (Minnesota Mining and Manufacturing Company) и сорастворители, например, этанол. Композиции под давлением будут обычно храниться в канистре (например, в алюминиевой канистре), закрытой клапаном (например, калибровочным клапаном) и помещенной внутрь механизма с рычагом, обеспеченным мундштуком.
Фармацевтические композиции, адаптированные для вагинального введения, могут быть представлены в виде составов пессариев, тампонов, кремов, гелей, паст, пен или спреев.
Фармацевтические композиции, адаптированные для инъекционного введения, включают в себя водные и неводные стерильные инъекционные растворы, которые могут содержать антиоксиданты, буферы, бактериостатики и растворы, которые придают композиции изотоничность с кровью предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут включать в себя средства, способствующие суспендированию и загустители. Композиции могут быть представлены в однодозовых или мультидозовых контейнерах, например, в запечатанных ампулах и флаконах, и могут храниться в замороженно-высушенном (лиофилизированном) состоянии, требуя лишь добавления стерильного жидкого носителя, например, воды для инъекций, непосредственно перед использованием. Инъекционные растворы и суспензии, приготовленные для немедленного приема, могут быть приготовлены из стерильных порошков, гранул и таблеток.
Следует понимать, что в дополнение к ингредиентам, конкретно упомянутым выше, композиции могут включать в себя другие средства, общепринятые в данной области техники, с учетом типа состава, более конкретно, например, средства, подходящие для перорального введения, могут включать в себя вкусоароматизаторы.
Антисмысловые или интерферирующие молекулы РНК можно вводить нуждающимся в этом млекопитающим. В качестве альтернативы, можно вводить конструкты, включающие в себя такие молекулы. Такие молекулы и конструкты можно использовать для препятствия экспрессии интересующего белка, например, гистондеметилазы, а потому модифицируя деметилирование гистонов. Обычно доставка осуществляется посредством известных в данной области техники способов.
Антисмысловые или интерферирующие молекулы РНК могут быть доставлены in vitro в клетки или in vivo, например, в опухоли млекопитающего. Можно использовать без ограничения любые пути доставки, включая внутривенный, внутримышечный, интраперитонеальный, интраартериальный, местную доставку в процессе хирургического вмешательства, эндоскопический, подкожный и пероральный. Векторы могут быть выбраны для желаемых свойств для любого конкретного применения. Векторы могут быть вирусными или плазмидными. С этой точки зрения применимы аденовирусные векторы. Для контроля транскрипции ингибирующих полинуклеотидных молекул можно использовать тканеспецифические, специфические в отношении клеточного типа или иным образом регулируемые промоторы. Также можно использовать невирусные носители, такие как липосомы или наносферы.
Соединения формулы (I) и их фармацевтически приемлемые соли также могут быть включены в состав композиции для применения в качестве адъюванта для модулирования активности вакцины. Такие композиции могут содержать антитело(а) или фрагмент(ы) антитела или антигенный компонент, включая без ограничения белок, ДНК, живые или мертвые бактерии и/или вирусы или вирусоподобные частицы, вместе с одним или несколькими компонентами с адъювантной активностью, включая без ограничения соли алюминия, масляные и водные эмульсии, белки теплового шока, препараты липида А и производные, гликолипиды, другие агонисты TLR, такие как CpG DNA или аналогичные средства, цитокины, такие как GM-CSF или IL-12, или аналогичные средства.
Терапевтически эффективное количество средства будет зависеть от целого ряда факторов, включая, например, возраст и вес субъекта, конкретное требующее лечения состояние и его тяжесть, природу состава и путь введения, и будет в конечном итоге оставлено на усмотрение лечащего врача или ветеринара. В частности, подлежащий лечению субъект представляет собой млекопитающее, в частности, человека.
Средство можно вводить в суточной дозе. Такое количество может быть представлено в однократной дозе в сутки или, чаще всего, в виде ряда (как, например, в виде двух, трех, четырех, пяти или шести раз) субдоз в сутки, так чтобы общая суточная доза была такой же.
Соответственно, количество соединения согласно настоящему изобретению, вводимое в соответствии с настоящим изобретением, будет представлять собой количество, выбранное от 0,01 мг до 1 г в сутки (вычислено для свободного соединения или соединения в отличном от соли виде).
Соединения формулы (I) и их фармацевтически приемлемые соли можно применять по отдельности или в комбинации с другими терапевтическими средствами. Соединения формулы (I) и их фармацевтически приемлемые соли и другое фармацевтически активное средство(а) можно вводить вместе или по отдельности, и при введении по отдельности введение может осуществляться одновременно или последовательно в любом порядке, посредством любого удобного пути в отдельных или комбинированных фармацевтических композициях.
Для достижения желаемого комбинированного терапевтического эффекта выбирают количества соединения(ий) формулы (I) или их фармацевтически приемлемая соль(и) и другого фармацевтически активного средства(в) и соответствующее время введения. Соединения согласно настоящему изобретению и дополнительное терапевтическое средство(а) можно применять в комбинации посредством одновременного введения в одной фармацевтической композиции, содержащей оба соединения. В качестве альтернативы, комбинация может быть введена по отдельности в отдельных фармацевтических композициях, каждая из которых включает одно из соединений, в последовательности, где, например, соединение согласно настоящему изобретению вводится первым, а другое средство вводят вторым, и наоборот. Такое последовательное введение может быть близким по времени (например, одновременным) или разделено по времени. Кроме того, не имеет значения, вводят ли соединения в одной и той же лекарственной форме, например, одно соединение можно вводить местно, а другое соединение можно вводить перорально.
Комбинации могут быть представлены в виде комбинированного набора. Под используемыми в настоящем документа терминами «комбинированный набор» или «набор частей» подразумевается фармацевтическая композиция или композиции, которые используют для введения комбинации в соответствии с настоящим изобретением. Если оба соединения вводят одновременно, то комбинированный набор может содержать оба соединения в одной фармацевтической композиции, такой как таблетка, или в отдельных фармацевтических композициях. Если соединения вводят не одновременно, то комбинированный набор будет содержать каждое соединение либо в отдельных фармацевтических композициях в одной упаковке, либо в отдельных фармацевтических композициях в отдельных упаковках.
Комбинированный набор также может быть снабжен инструкцией, такой как инструкции по дозированию и введению. Такие инструкции по дозированию и введению могут относиться к типу, который предоставляется для врача, например, в виде формуляра готовой лекарственной формы, или они могут относиться к типу, который предоставляется врачом, такой как инструкции для пациента.
Если комбинацию вводят по отдельности последовательным образом, где одно соединение вводят первым, а другое вторым, или наоборот, то такое последовательное введение может быть близким по времени или разделенным по времени. Например, предусмотрены варианты, когда после введения первого средства другое средство вводят через несколько минут или через несколько десятков минут, и когда после введения первого средства другое средство вводят через несколько часов или через несколько суток, где интервал времени не ограничен. Например, одно средство можно вводить однократно в сутки, а другое средство можно вводить 2 или 3 раза в сутки, или одно средство можно вводить один раз в неделю, а другое средство можно вводить однократно в сутки и т. п.
Специалисту в данной области будет очевидно, что при необходимости для оптимизации активности и/или стабильности и/или физических характеристик, таких как растворимость, терапевтического ингредиента, другой терапевтический ингредиент(ы) могут быть использованы в форме солей, например, в виде солей щелочных металлов или аминов или в виде кислотно-аддитивных солей, или пролекарств, или в виде сложных эфиров, например сложных эфиров низших алкилов, или в виде сольватов, например, гидратов. Также будет очевидно, что при необходимости терапевтические ингредиенты могут быть использованы в оптически чистой форме.
В случае комбинации в одной и той же композиции, следует понимать, что два соединения должны быть стабильны и совместимы друг с другом и другими компонентами композиции и могут быть включены в состав для введения. При введении по отдельности, они могут быть соответствующим образом доставлены в виде любой удобной композиции, способом, известным для таких соединений в данной области техники.
Если соединение формулы (I) используется в комбинации со вторым терапевтическим средством, активным в отношении того же заболевания, состояния или нарушения, то доза каждого соединения может отличаться от дозы, используемой для соединения при отдельном применении. Подходящие дозы будут легко оценены специалистами в данной области техники.
Согласно одному варианту осуществления, млекопитающее в способах и применениях настоящего изобретения представляет собой человека.
Настоящее изобретение также относится к фармацевтической композиции, содержащей от 0,5 до 1000 мг соединения формулы (I) или его фармацевтически приемлемой соли и от 0,5 до 1000 мг фармацевтически приемлемого эксципиента.
Соединения согласно настоящему изобретению применимы для лечения заболеваний, при которых модулирование STING является благоприятным. Такие заболевания включают в себя воспаление, аллергические и аутоиммунные заболевания, инфекционные заболевания, злокачественные опухоли и предраковые синдромы.
Соединения формулы (I) и их фармацевтически приемлемые соли также могут быть применимы в качестве модуляторов иммунного ответа, используемые как по отдельности, так и в комбинации в качестве адъюванта для лечения заболеваний, при которых модулирование STING является благоприятным.
Согласно одному аспекту, заболевание или состояние представляет собой воспалительные, аллергические или аутоиммунные нарушения.
Ассоциированные аутоиммунные заболевания включают в себя без ограничения системную красную волчанку, псориаз, инсулин-зависимый сахарный диабет (IDDM), дерматомиозит, вирус иммунодефицита человека (HIV), AIDS и синдром Шегрена (SS).
Воспаление представляет собой группу сосудистых, клеточных и неврологических ответов на повреждение. Воспаление может характеризоваться как движение воспалительных клеток, таких как моноциты, нейтрофилы и гранулоциты в ткани. Это обычно ассоциировано со сниженной функцией эндотелиального барьера и отеком в тканях. Воспаление может быть классифицировано как острое, так и хроническое. Острое воспаление представляет собой первоначальный ответ организма на вредные стимулы и достигается повышенным движением плазмы и лейкоцитов из крови в поврежденные ткани. Каскад биохимических событий распространяет и формирует воспалительный ответ, вовлекая местную сосудистую систему, иммунную систему и различные клетки внутри поврежденной ткани. Длительное воспаление, известное как хроническое воспаление, приводит к нарастающему изменению в типах клеток, которые присутствуют в очаге воспаления, и характеризуются одновременной деструкцией и заживлением ткани после воспалительного процесса.
Если это происходит как часть иммунного ответа на инфекцию или как острый ответ на повреждение, то воспаление может быть полезным и в норме самоограничивающимся. Однако, в различных условиях воспаление может быть вредным. Такая ситуация включает запуск чрезмерного воспаления в ответ на инфекционные агенты, что может вести к значительному повреждению органа или гибели (например, в случае сепсиса). Кроме того, хроническое воспаление обычно разрушительно и лежит в основе большого количества хронических заболеваний, вызывая тяжелое и необратимое повреждение тканей. В таких условиях иммунный ответ часто направлен против собственных тканей (аутоиммунитет), хотя хронические ответы на чужеродные инвазии тоже могут вести к второстепенному повреждению собственных тканей.
Поэтому, целью противовоспалительной терапии является снижение такого воспаления, ингибирование аутоиммунитета (если он присутствует) и развития физиологического процесса заживления и восстановления ткани.
Средства можно применять для лечения воспаления любой ткани и органов организма, включая скелетно-мышечное воспаление, сосудистое воспаление, нейрональное воспаление, воспаление пищеварительной системы, воспаление глаз, воспаление репродуктивной системы и другое воспаление, как приведено в примерах ниже.
Скелетно-мышечное воспаление относится к воспалительному состоянию скелетно-мышечной системы, в частности к таким состояниям, действующим на скелетные суставы, включая суставы руки, запястья, локтя, плеча, нижней челюсти, позвоночника, шеи, бедра, колена, щиколотки и стопы, и к состояниям, действующим на ткани, соединяющие мышцы и кости, такие как сухожилия. Примеры скелетно-мышечного воспаления, которое можно лечить соединения согласно настоящему изобретению включают в себя артрит (включая, например, остеоартрит, ревматоидный артрит, псориатический артрит, анкилозирующий спондилит, острый и хронический инфекционный артрит, артрит, ассоциированный с подагрой и псевдоподагрой и ювенильный идиопатический артрит), тендинит, синовит, теносиновит, бурсит, фиброзит (фибромиалгия), эпикондилит, миозит и остеит (включая, например, болезнь Паджета, остеит лобковой кости и кистозный фиброзный остеит).
Воспаление глаз относится к воспалению любой структуры глаза, включая глазные веки. Примеры воспаления глаз, которые можно лечить соединениями согласно настоящему изобретению, включают в себя блефарит, блефарохалазию, конъюнктивит, дакриоаденит, кератит, сухой кератоконъюнктивит (сухой глаз), склерит, трихиаз и увеит.
Примеры воспаления нервной системы, которые можно лечить соединениями согласно настоящему изобретению, включают в себя энцефалит, синдром Гийена-Барре, менингит, нейромиотонию, нарколепсию, рассеянный склероз, миелит и шизофрению.
Примеры воспаления сосудистой или лимфатической системы, которые можно лечить соединениями согласно настоящему изобретению, включают в себя артросклероз, артрит, флебит, васкулит и лимфангит.
Примеры воспалительных состояний пищеварительной системы, которые можно лечить соединениями согласно настоящему изобретению, включают в себя холангит, холецистит, энтерит, энтероколит, гастрит, гастроэнтерит, воспалительное заболевание кишечника (такое как болезнь Крона и язвенный колит), илеит и проктит.
Примеры воспалительных состояний репродуктивной системы, которые можно лечить соединениями согласно настоящему изобретению, включают в себя цервицит, хориоамнионит, эндометрит, эпидидимит, омфалит, оофорит, орхит, сальпингит, тубоовариальный абсцесс, уретрит, вагинит, вульвит и вульводинию.
Средства можно применять для лечения аутоиммунных состояний, имеющих воспалительный компонент. Такие состояния включают в себя острую диссеменированную универсальную алопецию, болезнь Бехчета, болезнь Чагаса, синдром хронической усталости, вегетативную дистонию, энцефаломиелит, анкилозирующий спондилит, апластическую анемию, гнойный гидраденит, аутоиммунный гепатит, аутоиммунный оофорит, целиакию, болезнь Крона, сахарный диабет 1 типа, гиганто-клеточный артериит, синдром Гудпасчера, болезнь Грейвса, синдром Гийена-Барре, болезнь Хашимото, пурпуру Шенлейн-Геноха, болезнь Кавасаки, системную красную волчанку, микроскопический колит, микроскопический полиартерит, смешанное заболевание соединительной ткани, рассеянный склероз, миастению гравис, синдром опсоклонус-миоклонус, неврит зрительного нерва, атрофическая форма аутоиммунного тиреоидит, пузырчатку, узловой полиартериит, полимиалгию, ревматоидный артрит, синдром Рейтера, синдром Шегрена, височный артериит, гранулематоз Вегенера, тепловую аутоиммунную гемолитическую анемию, интерстициальный цистит, болезнь Лайма, очаговую склеродермию, псориаз, саркоидоз, склеродермию, язвенный колит и витилиго.
Средства можно применять для лечения опосредованной Т-клетками гиперчувствительности, имеющей воспалительный компонент. Такие состояния включают в себя контактную гиперчувствительность, контактный дерматит (включая дерматит, вызванный сумахом), крапивницу, кожные аллергии, респираторные аллергии (поллиноз, аллергический ринит) и глютен-чувствительную энтеропатию (целиакию).
Другие воспалительные состояния, которые можно лечить средствами, включают в себя, например, аппендицит, дерматит, дерматомиозит, эндокардит, фиброзит, гингивит, глоссит, гепатит, гнойный гидраденит, ирит, ларингит, мастит, миокардит, нефрит, отит, панкреатит, паротит, перикардит, перитонит, фарингит, плеврит, пневмонит, простатит, пиелонефрит и стоматит, реакцию отторжения трансплантата (с вовлечением органов, таких как почки, печень, сердце, легкое, поджелудочная железа (например, островковые клетки), костный мозг, роговица, тонкий кишечник, кожные аллографты, кожные гомографты и ксенографты сердечных клапанов, сывороточная реакция и реакция трансплантат против хозяина), острый панкреатит, хронический панкреатит, острый респираторный дистресс-синдром, синдром Сезари, врожденную гиперплазию надпочечников, негнойный тиреоидит, гиперкальциемию, ассоциированную со злокачественной опухолью, пузырчатку, герпетиформный буллезный дерматит, тяжелую мультиформную эритему, эксфолиативный дерматит, себорейный дерматит, сезонный или постоянный аллергический ринит, бронхиальную астму, контактный дерматит, атопический дерматит, реакции гиперчувствительности к лекарствам, аллергический конъюнктивит, кератит, опоясывающий герпес с поражением глаз, ирит и иридоциклит, хориоретинит, неврит оптического нерва, симптоматический саркоидоз, химиотерапию скоротечного или диссеминированного легочного туберкулеза, идиопатическую тромбоцитопеническую пурпуру у взрослых, вторичную тромбоцитопению у взрослых, приобретенную (аутоиммунную) гемолитическую анемию, лейкоз и лимфомы у взрослых, острый лейкоз у детей, регионарный энтерит, аутоиммунный васкулит, рассеянный склероз, хроническую обструктивную болезнь легких, реакцию отторжения трансплантата солидного органа, сепсис. Предпочтительные варианты лечения включают в себя лечение реакции отторжения трансплантата, ревматоидного артрита, псориатического артрита, рассеянного склероза, сахарного диабета 1 типа, бронхиальной астмы, воспалительного заболевания кишечника, системной красной волчанки, псориаза, хронического заболевания легких и воспаления, сопровождающего инфекционные состояния (например, сепсис).
Согласно дополнительному аспекту настоящего изобретения, предусмотрено соединение формулы (I) или его фармацевтически приемлемая соль для применения при лечении воспаления, аллергии и аутоиммунного заболевания.
Согласно дополнительному аспекту настоящего изобретения, предусмотрен способ лечения воспаления, аллергии и аутоиммунного заболевания, включающий в себя введение нуждающемуся в этом человеку терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно дополнительному аспекту настоящего изобретения, предусмотрено применение соединения формулы (I) или его фармацевтически приемлемой соли в производстве лекарственного средства для лечения воспаления, аллергии и аутоиммунного заболевания.
Согласно одному аспекту, подлежащее лечению заболевание представляет собой бронхиальную астму.
Соединения формулы (I) или их фармацевтически приемлемые соли можно применять в комбинации с одним или несколькими другими средствами, которые могли бы быть полезными для профилактики или лечения аллергического заболевания, воспалительного заболевания, аутоиммунного заболевания, например, с антигенной иммунотерапией, антигистаминами, стероидами, NSAID, бронходилятаторами (например, бета-2-агонисты, адренергические агонисты, антихолинергические средства, теофиллин), метотрексатом, модуляторами лейкотриенов и аналогичными средствами; терапией моноклональными антителами, такой как анти-IgE, анти-TNF, анти-IL-5, анти-IL-6, анти-IL-12, анти-IL-1 и аналогичными средствами; рецепторной терапией, например, этанерцептом и аналогичными средствами; антиген-неспецифической иммунотерапией (например, интерферон или другие цитокины/хемокины, модуляторы рецепторов цитокинов/хемокинов, агонисты или антагонисты цитокинов, агонисты TLR и аналогичные средства).
Согласно дополнительному аспекту, предусмотрена комбинация, включающая в себя соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, одно дополнительное терапевтическое средство, применимое для лечения аллергического заболевания, воспаления или аутоиммунного заболевания.
Согласно дополнительному аспекту, предусмотрена комбинация, включающая в себя соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, одно дополнительное терапевтическое средство, применимое для лечения аллергического заболевания, воспаления или аутоиммунного заболевания для применения в терапии.
Согласно дополнительному аспекту, предусмотрена комбинация, включающая в себя соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, одно дополнительное терапевтическое средство, применимое для лечения аллергического заболевания, воспаления или аутоиммунного заболевания для применения при лечении аллергического заболевания, воспаления или аутоиммунного заболевания.
Согласно дополнительному аспекту, предусмотрено использование комбинации, включающей в себя соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, одно дополнительное терапевтическое средство, применимое для лечения аллергического заболевания, воспаления или аутоиммунного заболевания, при производстве лекарственного средства для лечения аллергического заболевания, воспаления или аутоиммунного заболевания.
Согласно дополнительному аспекту, предусмотрен способ лечения аллергического заболевания, воспаления или аутоиммунного заболевания, включающий в себя введение нуждающемуся в этом человеку терапевтически эффективного количества комбинации, включающей в себя соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, одно дополнительное терапевтическое средство, применимое для лечения аллергического заболевания, воспаления или аутоиммунного заболевания.
Согласно дополнительному аспекту, предусмотрена фармацевтическая композиция, содержащая комбинацию, включающую в себя соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, одно дополнительное терапевтическое средство, применимое для лечения аллергического заболевания, воспаления или аутоиммунного заболевания, и один или более фармацевтически приемлемых эксципиентов.
Согласно одному аспекту, заболевание, подлежащее лечению такой комбинацией, представляет собой бронхиальную астму.
Согласно одному аспекту, подлежащее лечению заболевание или состояние представляет собой злокачественную опухоль.
Примеры злокачественных опухолей, при которых соединения формулы (I) или их фармацевтически приемлемые соли или сольваты могут обладать потенциально благоприятными противоопухолевыми эффектами включают в себя без ограничения злокачественные опухоли легких, костей, поджелудочной железы, кожи, головы, шеи, матки, яичников, желудка, толстого кишечника, молочной железы, пищевода, тонкого кишечника, кишечника, эндокринной системы, щитовидной железы, паращитовидной железы, надпочечников, уретры, предстательной железы, полового члена, яичек, мочеточника, мочевого пузыря, почек или печени; ректальную злокачественную опухоль; злокачественную опухоль анальной области; карциномы фаллопиевых труб, эндометрия, шейки майки, влагалища, вульвы, лоханки почки, почечно-клеточную карциному; саркому мягких тканей; миксому; рабдомиому; фиброму; липому; тератому; холангиокарциному; гепатобластому; ангиосаркому; гемангиому; гепатому; фибросаркому; хондросаркому; миелому; хронический и острый лейкоз; лимфоцитарную лимфому; первичную ЦНС лимфому; новообразования ЦНС; опухоли оси позвоночника; сквамозно-клеточные карциномы; синовиальную саркому; злокачественные мезотелиомы плевры; глиому ствола головного мозга; аденому гипофиза; бронхиальную аденому; хондроматозную гамартому; мезотелиому; болезнь Ходжкина, или сочетание одной или нескольких из вышеупомянутых злокачественных опухолей.
Соответственно, настоящее изобретение относится к способу лечения или облегчения тяжести злокачественных опухолей, выбранных из группы, состоящей из опухолей головного мозга (глиом), глиобластом, астроцитом, мультиформной глиобластомы, синдрома Банаяна-Зонана, болезни Каудена, болезни Лермитта-Дюкло, опухоли Вильмса, саркомы Юинга, рабдомиосаркомы, эпендимомы, медуллобластомы, опухолей головы и шеи, почек, печени, меланомы, опухолей яичников, поджелудочной железы, аденокарциномы, протоковой аденокарциномы, аденосквамозной карциномы, ацинозно-клеточной карциномы, глюкагономы, инсулиномы, опухолей предстательной железы, саркомы, остеосаркомы, гиганто-клеточной опухоли кости, опухолей щитовидной железы, лимфобластного Т-клеточного лейкоза, хронического миелогенного лейкоза, хронического лимфоцитарного лейкоза, волосато-клеточного лейкоза, острого лимфобластного лейкоза, острого миелогенного лейкоза, хронического нейтрофильного лейкоза, острого лимфобластного Т-клеточного лейкоза, плазмацитомы, иммунобластного крупноклеточного лейкоза, лейкоза клеток мантии, множественной миеломы, мегакариобластного лейкоза, множественной миеломы, острого мегакариоцитарного лейкоза, промиелоцитарного лейкоза, эритролейкоза, злокачественной лимфомы, лимфомы Ходжкина, неходжкиновской лимфомы, лимфобластной Т-клеточной лимфомы, лимфомы Беркита, фолликулярной лимфомы, нейробластомы, злокачественной опухоли мочевого пузыря, злокачественной опухоли уротелия, злокачественной опухоли вульвы, злокачественной опухоли шейки матки, злокачественной опухоли эндометрия, злокачественной опухоли почки, мезотелиомы, злокачественной опухоли пищевода, злокачественной опухоли слюнных желез, гепатоцеллюлярного рака, злокачественной опухоли желудка, злокачественной опухоли носоглотки, злокачественной опухоли щеки, злокачественной опухоли ротовой полости, GIST (желудочно-кишечной стромальной опухоли) и злокачественной опухоли яичек.
Соответственно, настоящее изобретение относится к способу лечения или облегчения тяжести предраковых синдромов у млекопитающих, включая людей, где предраковые синдромы выбирают из интраэпителиальной цервикальной неоплазии, моноклональной гаммапатии неустановленной этиологии (MGUS), миелодиспластического синдрома, апластической анемии, заболеваний шейки матки, невуса кожи (премеланома), интраэпителиальной (внутрипротоковой) неоплазии предстательной железы (PIN), протоковой карциномы in situ (DCIS), полипов толстой кишки и тяжелого гепатита или цирроза.
Соединения согласно настоящему изобретению также можно применять в качестве адъювантов для улучшения иммунного ответа, возникающего на любой данный антиген, и/или снижения реактогенности/токсичности у нуждающегося в этом пациента, в частности у людей. В этой связи, соединение согласно настоящему изобретению можно применять в комбинации с вакцинными композициями для модифицирования, в особенности, усиления, иммунного ответа, например, путем увеличения уровня или длительности защиты и/или позволяя снизить антигенную дозу.
Соединения формулы (I) и их фармацевтически приемлемые соли можно применять в комбинации с одной или несколькими вакцинами или иммуногенными антигенами, применимыми для профилактики или лечения вирусных инфекций. Такие вакцины или иммуногенные антигены включают в себя без ограничения белки или частицы, полученные из патогенов, такие как аттенуированные вирусы, вирусные частицы и вирусные белки, обычно используемые в качестве иммуногенных веществ. Примеры вирусов и вирусных антигенов включают в себя без ограничения полиовирусы, Coronaviridae и коронавирусы, риновирус (все подтипы), аденовирусы (все подтипы), вирусы гепатита A, гепатита B, гепатита С, гепатита D, папилломавирус человека (включая все подтипы), вирусы бешенства, Т-клеточный лимфотропный вирус человека (все подтипы), вирус краснухи, вирус эпидемического паротита, вирус Коксаки A (все подтипы), вирус Коксаки B (все подтипы), энтеровирусы человека, герпесвирусы, включая цитомегаловирус, вирус Эпштейн-Барра, герпесвирус человека (все подтипы), вирус простого герпеса, вирус ветряной оспы, вирус иммунодефицита человека (HIV) (все подтипы), AIDS, вирус Эпштейн-Барра, реовирусы (все подтипы), филовирусы, включая вирус, вызывающий «марбургскую болезнь», и вирус Эбола (все штаммы), аренавирусы, включая вирус лимфоцитарного хориоменингита, вирус Ласса, вирус Хунина и вирус Мачупо, арбовирусы, включая вирус западного Нила, вирусы Денге (все серотипы), вирус Зика, вирус колорадской клещевой лихорадки, вирус Синдбис, тогавирусы, флавивирусы, буньявирусы, реовирусы, рабдовирусы, ортомиксовирусы, поксвирусы, включая ортопоксвирус (вирус оспы человека, вирус оспы обезьян, вирус осповакцины, вирус коровьей оспы), ятапоксвирусы (танапоксвирус, онкогенный вирус Яба обезьян), парапоксвирус, поксвирус контагиозного моллюска, желтую лихорадку, хантавирусы, включая вирус Хантаан, вирус Сеула, вирус Добрава-Белград, вирус Син Номбре, вирус Пуумала и Добрава-подобный вирус Сааремаа, вирусы парагриппа человека и вирусы гриппа (все типы), вирусы гриппа H1N1 и свиного гриппа, респираторный синтициальный вирус (все подгруппы), ротавирусы, включая ротавирусы человека A-E, ротавирус жвачных, ротавирус резус-макак, полиомавирусы, включая вирус обезьян 40, вирус JC, вирус BK, колтивирусы, вирус Eyach, кальцивирусы и парвовирусы, включая депендовирус, парвовирус и эритровирус.
Соединения согласно настоящему изобретению также могут быть применимы для лечения одного или нескольких заболеваний, поражающих млекопитающих, которые характеризуются пролиферацией клеток в области нарушений, ассоциированных с неоваскуляризацией и/или сосудистой проницаемостью, включая пролиферативные нарушения кровеносных сосудов, включая артрит (ревматоидный артрит) и рестеноз; фиброзные нарушения, включая цирроз печени и атеросклероз; пролиферативные нарушения мезангиальных клеток включают в себя гломерулонефрит, диабетическую нефропатию, злокачественный нефросклероз, синдромы тромботической микроангиопатии, пролиферативные ретинопатии, реакцию отторжения трансплантата и гломерулопатии; и метаболические нарушения, включая псориаз, сахарный диабет, хроническое заживление ран, воспаление и нейродегенеративные заболевания.
Согласно дополнительному аспекту настоящего изобретения, предусмотрено соединение формулы (I) или его фармацевтически приемлемая соль для применения при лечении болезненного состояния, выбранного из группы, состоящей из HIV, HBV, HCV, гриппа, кожных бородавок, рассеянного склероза, аллергического воспаления, и в качестве адъюванта.
Zhijian Chen - Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing - Nature Immunology (2016), 17, 1142-1149.
Seng-Ryong Woo - STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors (2014), 41, 830-842.
Jenny P.-Y. Ting - NLRX1 Sequesters STING to Negatively Regulate the Interferon Response, Thereby Facilitating the Replication of HIV-1 and DNA Viruses - Cell Host and Microbe (2016), 19, 515-528.
Zhijian Chen - Pivotal Roles of cGAS-cGAMP Signaling in Antiviral Defense and Immune Adjuvant Effects - Science (2013), 341, 1390-1394.
Nuchsupha Sunthamala - E2 Proteins of High Risk Human Papillomaviruses Down-Modulate STING and IFN-κ Transcription in Keratinocytes - PLoS (2014), 9, 1-11.
Guo, H., et al. (2016). NLRX1 Sequesters STING to Negatively Regulate the Interferon Response, Thereby Facilitating the Replication of HIV-1 and DNA Viruses. Cell host & microbe 19, 515-528.
Gao, D., et al. (2013). Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341, 903-906.
Guo, F., et al. (2015). Sting agonists induce an innate antiviral immune response against hepatitis B virus. Antimicrobial Agents and Chemotherapy 59, 1273-1281.
Dansako, H., et al. (2016). The cyclic GMP-AMP synthetase-STING signaling pathway is required for both the innate immune response against HBV and the suppression of HBV assembly. FEBS J 283, 144-156.
Chang, J., et al. (2015). Treatment of chronic hepatitis B with pattern recognition receptor agonists: Current status and potential for a cure. Antiviral Research 121, 152-159.
Li, X.D., et al. (2013). Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341, 1390-1394.
Carroll, E.C., et al. (2016). The Vaccine Adjuvant Chitosan Promotes Cellular Immunity via DNA Sensor cGAS-STING-Dependent Induction of Type I Interferons. Immunity 44, 597-608.
Wang, J., et al. (2016). Natural STING Agonist as an "Ideal" Adjuvant for Cutaneous Vaccination. J Invest Dermatol 136, 2183-2191.
Holm, C.K., et al. (2016). Influenza A virus targets a cGAS-independent STING pathway that controls enveloped RNA viruses. Nat Commun 7, 10680.
Shirey, K.A., et al. (2011). The anti-tumor agent, 5,6-dimethylxanthenone-4-acetic acid (DMXAA), induces IFN-beta-mediated antiviral activity in vitro and in vivo. J Leukoc Biol 89, 351-357.
Nitta, S., et al. (2013). Hepatitis C virus NS4B protein targets STING and abrogates RIG-l-mediated type I interferon-dependent innate immunity. Hepatology 57, 46-58.
Sunthamala, N., et al. (2014). E2 proteins of high risk human papillomaviruses down-modulate STING and IFN-kappa transcription in keratinocytes. PLoS One 9, e91473.
Lau, L, et al. (2015). DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 350, 568-571.
Kidd, P. (2003). Th1/Th2 balance: the hypothesis, its limitations и implications for health and disease. Altern Med Rev 8, 223-246.
Huang, L, et al. (2013). Cutting edge: DNA sensing via the STING adaptor in myeloid dendritic cells induces potent tolerogenic responses. J Immunol 191, 3509-3513.
Lemos, H., et al. (2014). Activation of the STING adaptor attenuates experimental autoimmune encephalitis. J Immunol 192, 5571-5578.
Согласно дополнительному аспекту настоящего изобретения, предусмотрено соединение формулы (I) или его фармацевтически приемлемая соль для применения при лечении злокачественной опухоли и/или предраковых синдромов.
Согласно дополнительному аспекту настоящего изобретения, предусмотрен способ лечения злокачественной опухоли, включающий в себя введение нуждающемуся в этом человеку терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно дополнительному аспекту, предусмотрено применение соединения формулы (I) или его фармацевтически приемлемой соли при производстве лекарственного средства для лечения злокачественной опухоли и/или предраковых синдромов.
Согласно одному варианту осуществления, соединение согласно настоящему изобретению можно применять с другими терапевтическими способами лечения злокачественной опухоли. В частности, предусмотрены антинеопластическая терапия, комбинированная терапия с другим химиотерапевтическим средством, гормоном, антителом, а также, хирургические и/или лучевые способы лечения, отличные от упомянутых выше способов.
Согласно одному варианту осуществления, дополнительная противораковая терапия представляет собой хирургическое вмешательство и/или радиотерапию.
Согласно одному варианту осуществления, дополнительная противораковая терапия представляет собой, по меньшей мере, одно дополнительное антинеопластическое средство.
Согласно дополнительному аспекту, предусмотрена комбинация, включающая в себя соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, одно антинеопластическое средство.
Согласно дополнительному аспекту, предусмотрена комбинация, включающая в себя соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, одно антинеопластическое средство, для применения в терапии.
Согласно дополнительному аспекту, предусмотрена комбинация, включающая в себя соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, одно антинеопластическое средство, для применения при лечении злокачественной опухоли и/или предраковых синдромов.
Согласно дополнительному аспекту, предусмотрено применение комбинации, включающей в себя соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, одно антинеопластическое средство, при производстве лекарственного средства для лечения злокачественной опухоли и/или предраковых синдромов.
Согласно дополнительному аспекту, предусмотрен способ лечения злокачественной опухоли, включающий в себя введение нуждающемуся в этом человеку терапевтически эффективного количества комбинации, включающей в себя соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, одно антинеопластическое средство.
Согласно дополнительному аспекту, предусмотрена фармацевтическая композиция, включающая в себя комбинацию, включающую в себя соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, одно дополнительно терапевтическое средство, в частности, по меньшей мере, одно антинеопластическое средство, и один или более фармацевтически приемлемых носителей, разбавителей и эксципиентов.
В комбинации может быть использовано любое антинеопластическое средство, которое обладает активностью в отношении чувствительной подлежащей лечению опухоли. Обычно применимые антинеопластические средства включают в себя без ограничения антимикротубулярные средства, такие как дитерпеноиды и алкалоиды барвинка; координационные комплексы платины; алкилирующие средства, такие как мустарген, оксазафосфорины, алкилсульфонаты, нитрозомочевины и тиазены; антибиотики, такие как антрациклины, актиномицины и блеомицины; ингибиторы топоизомеразы II, такие как эпиподофиллотоксины; антиметаболиты, такие как аналоги пурина и пиримидина и антифолатные соединения; ингибиторы топоизомеразы I, такие как каптотецины; гормоны и аналоги гормонов; ингибиторы сигнальных путей трансдукции; ингибиторы нерецепторного тирозинового ангиогенеза; иммунотерапевтические средства; проапоптотические средства; ингибиторы сигнального пути клеточного цикла; иммуно-онкологические средства и иммуностимулирующие средства.
Антимикротубулярные и антимитотические средства:
Антимикротубулярные и антимитотические средства представляют собой фазо-специфические средства, активные в отношении микротрубочек опухолевых клеток в процессе М или митотической фазы клеточного цикла. Примеры антимикротубулярных средств включают в себя без ограничения дитерпеноиды и алкалоиды барвинка.
Дитерпеноиды, которые получают из природных источников, представляют собой фазо-специфические противораковые средства, которые действуют в G2/M фазы клеточного цикла. Считается, что дитерпеноиды стабилизируют β-тубулиновую субъединицу микротрубочек путем связывания с этим белком. Поэтому считается, что разборка белка ингибируется с остановкой митоза и последующей клеточной смертью. Примеры дитерпеноидов включают в себя без ограничения паклитаксел и его аналог, доцетаксел.
Паклитаксел, 4,10-диацетат-2-бензоат-13-эфир 5β,20-эпокси-1,2α,4,7β,10β,13α-гексагидрокситакс-11-ен-9-она с (2R,3S)-N-бензоил-3-фенилизосерином, представляет собой природный продукт дитерпена, выделенный из дерева тиса тихого Taxus brevifolia, и является коммерчески доступным в виде инъекционного раствора ТАКСОЛ®. Он является представителем таксанового семейства терпенов. Паклитаксел был утвержден для клинического применения при лечении резистентного рака яичников в США (Markman et al., Yale Journal of Biology and Medicine, 64:583, 1991; McGuire et al., Ann. lntem, Med., 111:273,1989) и для лечения рака молочной железы (Holmes et al., J. Nat. Cancer Inst., 83:1797,1991.) Он является потенциальным кандидатом для лечения новообразований кожи (Einzig et. al., Proc. Am. Soc. Clin. Oncol., 20:46) и карцином головы и шеи (Forastire et. al., Sem. Oncol., 20:56, 1990). Соединение также демонстрирует потенциал для лечения поликистозного заболевания почек (Woo et. al., Nature, 368:750. 1994), рака легких и малярии. Лечение пациентов паклитакселом приводит к супрессии костного мозга (множества клеточных линий, Ignoff, R.J. et. al, Cancer Chemotherapy Pocket Guide, 1998), связанной с длительностью дозирования в сверхпороговой концентрации (50 нМ) (Kearns, C.M. et. al., Seminars in Oncology, 3(6) p.16-23, 1995).
Доцетаксел, 4-ацетат-2-бензоат-13-эфир 5β-20-эпокси-1,2α,4,7β,10β,13α-гексагидрокситакс-11-ен-9-она с N-трет-бутиловым сложным эфиром (2R,3S)-N-карбокси-3-фенилизосерина, тригидратом, является коммерчески доступным в виде инъекционного раствора ТАКСОТЕР®. Доцетаксел показан для лечения рака молочной железы. Доцетаксел представляет собой полусинтетическое производное паклитаксела q.v., полученное с использованием природного предшественника, 10-деацетилбаккатина III, экстрагированного из иголок тиса тихого.
Алкалоиды барвинка представляют собой фазо-специфические антинеопластические средства, полученные из растения барвинка. Алкалоиды барвинка действуют в М фазу (митоз) клеточного цикла путем специфического связывания с тубулином. Следовательно, связанная молекула тубулина неспособна полимеризоваться в микротрубочки. Считается, что митоз останавливается в метафазу, с последующей клеточной гибелью. Примеры алкалоидов барвинка включают в себя без ограничения винбластин, винкристин и винорелбин.
Винбластин, винкалейкобластина сульфат, коммерчески доступен как ВЕЛБАН® в виде инъекционного раствора. Хотя он имеет возможное применение в качестве второй линии терапии различных солидных опухолей, в первую очередь он показан для лечения рака яичек и различных лимфом, включая болезнь Ходжкина; и лимфоцитарных и гистиоцитарных лимфом. Миелосупрессия является дозо-лимитирующим побочным эффектом винбластина.
Винкристин, винкалейкобластина 22-оксосульфат, коммерчески доступен как ОНКОВИН® в виде инъекционного раствора. Винкристин показан для лечения острых лейкозов и также нашел свое применение в схемах лечения ходжкиновских и неходжкиновских злокачественных лимфом. Алопеция и неврологические эффекты представляют собой самые распространенные побочные эффекты винкристина, и возникает менее распространенная миелосупрессия и воспаление слизистой оболочки желудочно-кишечного тракта.
Винорелбин, 3',4'-дидегидро-4'-деокси-C'-норвинкалейкобластина [R-(R*,R*)-2,3-дигидроксибутандиоат (1:2) (соль)], коммерчески доступен в виде инъекционного раствора винорелбина тартрата (НАВЕЛБИН®), представляет собой полусинтетический алкалоид барвинка. Винорелбин показан в качестве единственного средства или в комбинации с другими химиотерапевтическими средствами, такими как цисплатин, для лечения различных солидных опухолей, в частности, немелкоклеточного рака легких, запущенного рака молочной железы и гормонорезистентного рака предстательной железы. Миелосупрессия является самым распространенным дозо-лимитирующим побочным эффектом винорелбина.
Координационные комплексы платины:
Координационные комплексы платины представляют собой нефазовые специфические противораковые средства, которые взаимодействуют с ДНК. Платиновые комплексы проникают в опухолевые клетки, подвергаются гидратированию и формируют межцепочечные и внутрицепочечные связи с ДНК, вызывая побочные биологические эффекты в опухоли. Примеры координационных комплексов платины включают в себя без ограничения оксалиплатин, цисплатин и карбоплатин.
Цисплатин, цисдиамминдихлорплатина, коммерчески доступен как ПЛАТИНОЛ® в виде инъекционного раствора. Цисплатин в первую очередь показан для лечения метастатического рака яичек и яичников и запущенного рака мочевого пузыря.
Карбоплатин, [1,1-циклобутандикарбоксилат(2-)-O,O']диамин платины, коммерчески доступен как ПАРАПЛАТИН® в виде инъекционного раствора. Карбоплатин в первую очередь показан как первая и вторая линия терапии запущенной карциномы яичника.
Алкилирующие средства:
Алкилирующие средства представляют собой нефазные противораковые специфические средства и сильные электрофильные средства. Обычно, алкилирующие средства формируют ковалентные связи, путем алкилирования, с ДНК через нуклеофильные фрагменты молекулы ДНК, такие как фосфатные, аминогруппы, сульфгидрильные, гидроксильные, карбоксильные и имидазольные группы. Функционирование таких фрагментов алкилирования нуклеиновых кислот ведет к клеточной гибели. Примеры алкилирующих средств включают в себя без ограничения мустаргены, такие как циклофосфамид, мелфалан и хлорамбуцил; алкилсульфонаты, такие как бусульфан; нитрозомочевины, такие как кармустин; и триазены, такие как дакарбазин.
Циклофосфамид, 2-[бис(2-хлорэтил)амино]тетрагидро-2H-1,3,2-оксазафосфорин 2-оксида моногидрат, коммерчески доступен в виде инъекционного раствора или таблеток, как ЦИТОКСАН®. Циклофосфамид показан в качестве отдельного средства или в комбинации с другими химиотерапевтическими средствами для лечения злокачественных лимфом, множественной миеломы и лейкозов.
Мелфалан, 4-[бис(2-хлорэтил)амино]-L-фенилаланин, коммерчески доступен в виде инъекционного раствора или таблеток как АЛКЕРАН®. Мелфалан показан для паллиативного лечения множественной миеломы и эпителиальной карциномы яичников, не подлежащей резекции. Супрессия костного мозга представляет собой самый распространенный дозо-лимитирующий побочный эффект мелфалана.
Хлорамбуцил, 4-[бис(2-хлорэтил)амино]бензолбутановая кислота, коммерчески доступен как таблетки ЛЕЙКЕРАН®. Хлорамбуцил показан для паллиативного лечения хронического лимфатического лейкоза и злокачественных лимфом, таких как лимфосаркома, гигантофолликулярная лимфома и болезнь Ходжкина.
Бусульфан, 1,4-бутандиолдиметансульфонат, коммерчески доступен в виде таблеток МИЛЕРАН®. Бусульфан показан для паллиативного лечения хронического миелогенного лейкоза.
Кармустин, 1,3-[бис(2-хлорэтил)-1-нитрозомочевина, коммерчески доступен в виде отдельных флаконов лиофилизированного вещества как BiCNU®. Кармустин в качестве отдельного средства или в комбинации с другими средствами показан для паллиативного лечения опухолей головного мозга, множественной миеломы, болезни Ходжкина и неходжкиновских лимфом.
Дакарбазин, 5-(3,3-диметил-1-триазено)-имидазол-4-карбоксамид, коммерчески доступен в виде отдельных флаконов вещества как DTIC-Dome®. Дакарбазин показан для лечения метастатической злокачественной меланомы и, в комбинации с другими средствами, для второй линии лечения болезни Ходжкина.
Антинеопластические антибиотики:
Антинеопластические антибиотики представляют собой нефазовые специфические средства, которые связываются или встраиваются в ДНК. Обычно, такое действие приводит к стабильным ДНК комплексам или разрыву цепочек, что нарушает обычное функционирование нуклеиновых кислот и ведет к клеточной гибели. Примеры антинеопластических антибиотиков включают в себя без ограничения актиномицины, такие как дактиномицин, антроциклины, такие как даунорубицин и доксорубицин; и блеомицины.
Дактиномицин, также известный как Актиномицин D, коммерчески доступен в виде инъекционной формы как КОСМЕГЕН®. Дактиномицин показан для лечения опухоли Вильмса и рабдомиосаркомы.
Даунорубицин, (8S-цис-)-8-ацетил-10-[(3-амино-2,3,6-тридеокси-α-L-ликсо-гексопиранозил)окси]-7,8,9,10-тетрагидро-6,8,11-тригидрокси-1-метокси-5,12-нафтаценедиона гидрохлорид, коммерчески доступен в виде липосомальной инъекционной формы как ДАУНОКСОМ® или в виде инъекционной формы как ЦЕРУБИДИН®. Даунорубицин показан для индукции ремиссии при лечении острого нелимфоцитарного лейкоза и запущенной, ассоциированной с HIV, саркомы Капоши.
Доксорубицин, (8S,10S)-10-[(3-амино-2,3,6-тридеокси-α-L-ликсо-гексопиранозил)окси]-8-гликолоил-7,8,9,10-тетрагидро-6,8,11-тригидрокси-1-метокси-5,12-нафтаценедиона гидрохлорид, коммерчески доступен в виде инъекционной формы как РУБЕКС® или АДРИАМИЦИН RDF®. Доксорубицин в первую очередь показан для лечения острого лимфобластного лейкоза и острого миелобластного лейкоза, но также представляет собой применимый компонент при лечении некоторых солидных опухолей и лимфом.
Блеомицин, смесь цитотоксических гликопептидных антибиотиков, выделенная из штамма Streptomyces verticillus, коммерчески доступен как БЛЕНОКСАН®. Блеомицин показан в качестве отдельного средства или в комбинации с другими средствами в качестве паллиативного лечения сквамозно-клеточной карциномы, лимфом и карцином яичек.
Ингибиторы топоизомеразы II:
Ингибиторы топоизомеразы II включают в себя без ограничения эпиподофиллотоксины.
Эпиподофиллотоксины представляют собой фазо-специфические антинеопластические средства, получаемые из растения мандрагоры. Эпиподофиллотоксины обычно воздействуют на клетки в S и G2 фазах клеточного цикла путем формирования тройного комплекса с топоизомеразой II и ДНК, вызывая разрывы цепочек ДНК. Разрывы цепочек накапливаются и вызывают клеточную гибель. Примеры эпиподофиллотоксинов включают в себя без ограничения этопозид и тенипозид.
Этопозид, 4'-деметилэпиподофиллотоксин-9-[4,6-0-(R)-этилиден-β-D-глюкопиранозид], коммерчески доступен в виде инъекционного раствора или капсул как ВЕПЕЗИД® и общеизвестен как VP-16. Этопозид показан в качестве отдельного средства или в комбинации с другими химиотерапевтическими средствами для лечения злокачественных опухолей яичек и немелкоклеточного рака легких.
Тенипозид, 4'-деметил-эпиподофиллотоксин-9-[4,6-0-(R)-тенилиден-β-D-глюкопиранозид], коммерчески доступен в виде инъекционного раствора как ВУМОН® и общеизвестен как VM-26. Тенипозид показан в качестве отдельного средства или в комбинации с другими химиотерапевтическими средствами для лечения острого лейкоза у детей.
Антиметаболические неопластические средства:
Антиметаболические неопластические средства представляют собой фазо-специфические антинеопластические средства, которые действуют в S фазу (синтез ДНК) клеточного цикла путем ингибирования синтеза ДНК или путем ингибирования синтеза оснований пурина или пиримидина, таким образом, ограничивая синтез ДНК. Следовательно, S-фаза не осуществляется и затем следует гибель клетки. Примеры антиметаболических неопластических средств включают в себя без ограничения фторурацил, метотрексат, цитарабин, меркаптопурин, тиогуанин и гемцитабин.
5-фторурацил, 5-фтор-2,4-(1H,3H)пиримидиндион, коммерчески доступен как фторурацил. Введение 5-фторурацила ведет к ингибированию синтеза тимидилата, и также встраивается внутрь как РНК, так и ДНК. Результатом, как правило, является клеточная гибель. 5-Фторурацил показан в качестве отдельного средства или в комбинации с другими химиотерапевтическими средствами для лечения карцином молочной железы, толстого кишечника, прямой кишки, желудка и поджелудочной железы. Другие аналоги фторпиримидина включают в себя 5-фтордеоксиуридин (флоксуридин) и 5-фтордеоксиуридина монофосфат.
Цитарабин, 4-амино-1-β-D-арабинофуранозил-2-(1H)-пиримидинон, коммерчески доступен как ЦИТОЗАР-U® and общеизвестен как Ara-C. Считается, что цитарабин демонстрирует клеточно-фазовую специфичность в S-фазу путем ингибирования элонгации ДНК цепи, посредством терминального встраивания цитарабина в растущую ДНК цепь. Цитарабин показан в качестве отдельного средства или в комбинации с другими химиотерапевтическими средствами для лечения острого лейкоза. Другие аналоги цитидина включают в себя 5-азацитидин и 2',2'-дифтордеоксицитидин (гемцитабин).
Меркаптопурин, 1,7-дигидро-6H-пурин-6-тиона моногидрат, коммерчески доступен как ПУРИНЕТОЛ®. Меркаптопурин демонстрирует клеточно-фазовую специфичность в S-фазу, ингибируя синтез ДНК, посредством еще не определенного механизма. Меркаптопурин показан в качестве отдельного средства или в комбинации с другими химиотерапевтическими средствами для лечения острого лейкоза. Применимым аналогом меркаптопурина является азатиоприн.
Тиогуанин, 2-амино-1,7-дигидро-6H-пиурин-6-тион, коммерчески доступен как ТАБЛОИД®. Тиогуанин демонстрирует клеточно-фазовую специфичность в S-фазу, ингибируя синтез ДНК, посредством еще не определенного механизма. Тиогуанин показан в качестве отдельного средства или в комбинации с другими химиотерапевтическими средствами для лечения острого лейкоза. Другие аналоги пурина включают в себя пентостатин, эритрогидроксинониладенин, флударабина фосфат и кладрибин.
Гемцитабин, 2'-деокси-2',2'-дифторцитидина моногидрохлорид (β-изомер), коммерчески доступен как ГЕМЗАР®. Гемцитабин демонстрирует клеточно-фазовую специфичность в S-фазу и, посредством блокирования развития клеток на границе G1/S. Гемцитабин в комбинации с цисплатином показан для лечения локально запущенного немелкоклеточного рака легких и отдельно для лечения локально запущенного рака поджелудочной железы.
Метотрексат, N-[4[[(2,4-диамино-6-птеридинил)метил]-метиламино]бензоил]-L-глутаминовая кислота, коммерчески доступен как метотрексат натрия. Метотрексат демонстрирует клеточно-фазовые эффекты, особенно в S-фазу, ингибируя синтез, репарацию и/или репликацию ДНК посредством ингибирования редуктазы дигидрофолиевой кислоты, которая необходима для синтеза пуриновых нуклеотидов и тимидилата. Метотрексат показан в качестве отдельного средства или в комбинации с другими химиотерапевтическими средствами для лечения хориокарциномы, менингеального лейкоза, неходжкиновской лимфомы и карцином молочной железы, головы, шеи, яичников и мочевого пузыря.
Ингибиторы топоизомеразы I:
Камптотецины, включая камптотецин и производные камптотецина, доступны или находятся в процессе разработки в качестве ингибиторов топоизомеразы I. Считается, что цитотоксическая активность камптотецинов связана с ее активностью, ингибирующей топоизомеразу I. Примеры камптотецинов включают в себя без ограничения иринотекан, топотекан и различные оптические формы 7-(4-метилпиперазинометилен)-10,11-этилендиокси-20-камптотецина, описанные ниже.
Иринотекана гидрохлорид, (4S)-4,11-диэтил-4-гидрокси-9-[(4-пиперидинопиперидино)карбонилокси]-1H-пирано[3',4',6,7]индолизино[1,2-b]хинолин-3,14(4H,12H)-диона гидрохлорид, коммерчески доступен в виде инъекционного раствора КАМПТОЗАР®. Иринотекан представляет собой производное камптотецина, которое связывается, вместе с его активным метаболитом SN-38, с комплексом топоизомераза I - ДНК. Считается, что цитотоксичность возникает в результате двухцепочечных разрывов, не подлежащих репарации, вызванных взаимодействием топоизомеразы I: ДНК: иринотекана или SN-38 тройного комплекса с репликационными ферментами. Иринотекан показан для лечения метастатического рака толстого кишечника и прямой кишки.
Топотекана гидрохлорид, (S)-10-[(диметиламино)метил]-4-этил-4,9-дигидрокси-1H-пирано[3',4',6,7]индолизино[1,2-b]хинолин-3,14-(4H,12H)-диона моногидрохлорид, коммерчески доступен в виде инъекционного раствора ГИКАМТИН®. Топотекан представляет собой производное камптотецина, которое связывается с комплексом топоизомераза I - ДНК и предотвращает повторное лигирование одноцепочечных разрывов, вызванных топоизомеразой I в ответ на деформацию кручения молекулы ДНК. Топотекан показан в качестве второй линии лечения метастатической карциномы яичников и мелкоклеточного рака легких.
Гормоны и аналоги гормонов:
Гормоны и аналоги гормонов представляют собой применимые соединения для лечения злокачественных опухолей, при которых существует взаимосвязь между гормоном(ами) и ростом и/или недостатком роста злокачественной опухоли. Примеры гормонов и аналогов гормонов, применимых при лечении злокачественной опухоли, включают в себя без ограничения адренокортикостероиды, такие как преднизон и преднизолон, которые применимы при лечении злокачественной лимфомы и острого лейкоза у детей; аминоглутетимид и другие ингибиторы ароматазы, такие как анастрозол, летразол, воразол и эксеместан, применимые при лечении адренокортикальной карциномы и гормон-зависимой карциномы молочной железы, содержащей рецепторы эстрогена; прогестрины, такие как мегестрола ацетат, применимые при лечении гормон-зависимого рака молочной железы и карциномы эндометрия; эстрогены, эстрогены и антиэстрогены, такие как фулвестрант, флутамид, нилутамид, бикалутамид, ципротерона ацетат и 5α-редуктазы, такие как финастерид и дутастерид, применимые для лечения карциномы предстательной железы и доброкачественной гипертрофии предстательной железы; антиэстрогены, такие как тамоксифен, торемифен, ралоксифен, дролоксифен, йодоксифен, а также селективные модуляторы эстрогеновых рецепторов (SERMS), такие как описано в патенте США № 5,681,835, 5,877,219, и 6,207,716, применимые для лечения гормон-зависимой карциномы молочной железы и других чувствительных злокачественных опухолей; и гонадотропин-высвобождающий гормон (GnRH) и его аналоги, которые стимулируют высвобождение лютеинизирующего гормона (LH) и/или фолликулостимулирующего гормона (FSH) для лечения карциномы предстательной железы, например, LHRH агонисты и антагонисты, такие как гозерелина ацетат и лупролид.
Ингибиторы сигнальных путей трансдукции:
Ингибиторы сигнальных путей трансдукции представляют собой такие ингибиторы, которые блокируют или ингибируют химический процесс, который вызывает внутриклеточные изменения. Используемое в настоящем документе такое изменение представляет собой клеточную пролиферацию или дифференциацию. Ингибиторы сигнальной трансдукции, применимые согласно настоящему изобретению, включают в себя ингибиторы рецепторных тирозинкиназ, нерецепторных тирозинкиназ, блокаторы домена SH2/SH3, сериновые/треониновые киназы, фосфатидилинозитол-3 киназы, миоинозитол сигнальные и Ras онкогены.
Некоторые протеинтирозинкиназы катализируют фосфорилирование специфических тирозильных остатков в различных белках, вовлеченных в регулирование клеточного роста. Такие протеинтирозинкиназы можно в широком смысле классифицировать на рецепторные или нерецепторные киназы.
Рецепторные тирозинкиназы представляют собой трансмембранные белки, содержащие внеклеточный лиганд-связывающий домен, трансмембранный домен и домен тирозинкиназы. Рецепторные тирозинкиназы вовлечены в регулирование клеточного роста и в целом называются рецепторами фактора роста. Было продемонстрировано, что неприемлемая или неконтролируемая активация многих таких киназ, например, т. е., аберрантная активность фактора роста киназ, например, посредством оверэкспресии или мутации, приводит к неконтролируемому клеточному росту. Соответственно, аберрантная активность таких киназ связана со злокачественным ростом ткани. Следовательно, ингибиторы таких киназ могут обеспечивать способы лечения злокачественных опухолей. Рецепторы фактора роста включают в себя, например, эпидермальный рецептор фактора роста (EGFr), тромбоцитарный рецептор фактора роста (PDGFr), erbB2, erbB4, ret, сосудистый эндотелиальный рецептор фактора роста (VEGFr), тирозинкиназу с иммуноглобулино-подобными и гомологичными доменами эпидермального фактора роста (TIE-2), рецептор инсулинового фактора роста - I (IGFI), макрофагальный колоний-стимулирующий фактор (cfms), BTK, ckit, cmet,рецепторы фактора роста фибробластов (FGF), Trk рецепторы (TrkA, TrkB и TrkC), рецепторы эфрина (eph) и RET протоонкогены. Некоторые ингибиторы рецепторов роста находятся в процессе разработки и включают в себя антагонисты лигандов, антитела, ингибиторы тирозинкиназ и антисмысловые олигонуклеотиды. Рецепторы фактора роста и средства, которые ингибируют функцию рецептора фактора роста, описаны, например, в Kath, John C., Exp. Opin. Ther. Patents (2000) 10(6):803-818; Shawver et al DDT Vol 2, No. 2 February 1997; и Lofts, F. J. et al, ʺGrowth factor receptors as targetsʺ, New Molecular Targets for Cancer Chemotherapy, ed. Workman, Paul and Kerr, David, CRC press 1994, London.
Тирозинкиназы, которые не являются рецепторными киназами фактора роста, называют нерецепторными тирозинкиназами. Нерецепторные тирозинкиназы применимые согласно настоящему изобретению которые представляют собой мишени и или потенциальные мишени для противораковых лекарств, включают в себя cSrc, Lck, Fyn, Yes, Jak, cAbl, FAK (киназа фокальной адгезии), тирозинкиназу Брутона и Bcr-Abl. Такие нерецепторные киназы и средства, которые ингибируют функцию нерецепторных тирозинкиназ, описаны в Sinh, S. and Corey, S.J., (1999) Journal of Hematotherapy and Stem Cell Research8 (5): 465-80; и Bolen, J.B., Brugge, J.S., (1997) Annual review of Immunology. 15: 371-404.
Блокаторы SH2/SH3 домена представляют собой средства, которые разрушают SH2 или SH3 доменную связь в ряде ферментов или адапторных белков, включая PI3-K p85 субъединицу, семейство Src киназ, адапторные молекулы (Shc, Crk, Nck, Grb2) и Ras-GAP. SH2/SH3 домены, в качестве мишеней для противораковых лекарств, обсуждались в Smithgall, T.E. (1995), Journal of Pharmacological and Toxicological Methods. 34(3) 125-32.
Ингибиторы сериновых/треониновых киназ, включая блокаторы каскада MAP киназ, которые включают в себя блокаторы Raf киназ (rafk), митогенных или внеклеточно регулируемых киназ (MEK) и внеклеточно регулируемых киназ (ERK); и блокаторы представителей семейства протеинкиназ С, включая блокаторы PKC (альфа, бета, гамма, эпсилон, мю, лямбда, йота, зета). Представители семейства IkB киназ (IKKa, IKKb), семейства РКВ киназ, семейства akt киназ и TGF бета рецепторных киназ. Такие сериновые/треониновые киназы и их ингибиторы описаны в Yamamoto, T., Taya, S., Kaibuchi, K., (1999), Journal of Biochemistry. 126 (5) 799-803; Brodt, P, Samani, A., and Navab, R. (2000), Biochemical Pharmacology, 60. 1101-1107; Massague, J., Weis-Garcia, F. (1996) Cancer Surveys. 27:41-64; Philip, P.A., and Harris, A.L. (1995), Cancer Treatment and Research. 78: 3-27, Lackey, K. et al Bioorganic and Medicinal Chemistry Letters, (10), 2000, 223-226; патенте США № 6,268,391; и Martinez-Iacaci, L., et al, Int. J. Cancer (2000), 88(1), 44-52.
Ингибиторы представителей семейства фосфатидилинозитол-3-киназ, включая блокаторы PI3-киназ, ATM, DNA-PK и Ku, также применимы согласно настоящему изобретению. Такие киназы обсуждались в Abraham, R.T. (1996), Current Opinion in Immunology. 8 (3) 412-8; Canman, C.E., Lim, D.S. (1998), Oncogene 17 (25) 3301-3308; Jackson, S.P. (1997), International Journal of Biochemistry and Cell Biology. 29 (7):935-8; и Zhong, H. et al, Cancer res, (2000) 60(6), 1541-1545.
Применимыми согласно настоящему изобретению также являются ингибиторы миоинозитол-сигнального пути, такие как блокаторы фосфолипазы С и аналоги миоинозитола. Такие сигнальные ингибиторы описаны в Powis, G., and Kozikowski A., (1994) New Molecular Targets for Cancer Chemotherapy ed., Paul Workman and David Kerr, CRC press 1994, London.
Другая группа ингибиторов сигнальных путей трансдукции представляет собой ингибиторы Ras онкогена. Такие ингибиторы включают в себя ингибиторы фарнесилтрансферазы, геранил-геранил трансферазы и CAAX протеаз, а также антисмысловых олигонуклеотидов, рибозимов и иммунотерапии. Было показано, что такие ингибиторы блокируют активацию ras в клетках, содержащих мутантный ras дикого типа, действуя, таким образом, в качестве антипролиферативных средств. Ингибирование ras онкогена обсуждалось в Scharovsky, O.G., Rozados, V.R., Gervasoni, S.I. Matar, P. (2000), Journal of Biomedical Science. 7(4) 292-8; Ashby, M.N. (1998), Current Opinion in Lipidology. 9 (2) 99-102; and BioChim. Biophys. Acta, (19899) 1423(3):19-30.
Как упомянуто выше, антагонисты антител к связыванию лиганда с рецепторной киназой также могут служить в качестве ингибиторов сигнальной трансдукции. Эта группа ингибиторов сигнальных путей трансдукции включает применение гуманизированных антител к внеклеточному лиганд-связывающему домену рецепторных тирозинкиназ. Например, Imclone C225 EGFR специфическое антитело (смотри Green, M.C. et al, Monoclonal Antibody Therapy for Solid Tumors, Cancer Treat. Rev., (2000), 26(4), 269-286); герцептин ® erbB2 антитело (смотри Tyrosine Kinase Signalling in Breast cancer:erbB Family Receptor Tyrosine Kinases, Breast cancer Res., 2000, 2(3), 176-183); и 2CB VEGFR2 специфическое антитело (смотри Brekken, R.A. et al, Selective Inhibition of VEGFR2 Activity by a monoclonal Anti-VEGF antibody blocks tumor growth in mice, Cancer Res. (2000) 60, 5117-5124).
Антиангиогенные средства:
(i) Также могут быть применимы антиангиогенные средства, включая ингибиторы ангиогенеза нерецепторных MEK. Антиангиогенные средства, которые ингибируют эффекты сосудистого эндотелиального фактора роста (например, анти-сосудистое эндотелиальное антитело клеточного фактора роста бевацизумаб [Авастин™]), и соединения, которые действуют посредством других механизмов (например, линомид, ингибиторы функции αvβ3 интегрина, эндостатин и ангиостатин);
Иммунотерапевтические средства:
Средства, используемые в иммунотерапевтических схемах лечения, также могут быть применимы в комбинации с соединениями формулы (I). Иммунотерапевтические подходы, включают в себя, например, подходы ex-vivo и in-vivo для увеличения иммуногенности клеток опухоли пациента, такие как трансфекция цитокинами, такими как интерлейкин 2, интерлейкин 4 или гранулоцитарный-макрофагальный колоний-стимулирующий фактор, подходы для снижения Т-клеточной анергии, подходы с использованием иммунных клеток, подвергшихся трансфекции, таких как цитокин-трансфицированные дендритные клетки, подходы с использованием цитокин-трансфицированных опухолевых клеточных линий и подходы с использованием антиидиотипических антител.
Проапоптотические средства:
Средства, используемые в проапоптотических схемах лечения (например, bcl-2 антисмысловые олигонуклеотиды), также можно применять в комбинации согласно настоящему изобретению.
Ингибиторы сигнального пути клеточного цикла
Ингибиторы сигнального пути клеточного цикла ингибируют молекулы, вовлеченные в контролирование клеточного цикла. Семейство протеинкиназ, называемое циклин-зависимые киназы (CDK) и их взаимодействие с семейством белков, называемых циклины, контролирует прохождение эукариотической клетки жизненного цикла. Координированная активация и инактивация различных циклин/CDK комплексов необходима для нормального прохождения клеточного цикла. Некоторые ингибиторы сигнального пути клеточного цикла находятся в процессе разработки. Например, примеры циклин-зависимых киназ, включая CDK2, CDK4 и CDK6, и их ингибиторы описаны, например, в Rosania et al, Exp. Opin. Ther. Patents (2000) 10(2):215-230.
Согласно одному варианту осуществления, комбинация согласно настоящему изобретению включает соединение формулы I или его соль или сольват и, по меньшей мере, одно антинеопластическое средство, выбранное из антимикротубулярных средств, координационных комплексов платины, алкилирующих средств, антибиотиков, ингибиторов топоизомеразы II, антиметаболитов, ингибиторов топоизомеразы I, гормонов и аналогов гормонов, ингибиторов сигнальных путей трансдукции, ингибиторов ангиогенеза нерецепторных тирозиновых МЕК, иммунотерапевтических средств, проапоптотических средств и ингибиторов сигнального пути клеточного цикла.
Согласно одному варианту осуществления, комбинация согласно настоящему изобретению включает соединение формулы I или его соль или сольват и, по меньшей мере, одно антинеопластическое средство, которое представляет собой антимикротубулярное средство, выбранное из дитерпеноидов и алкалоидов барвинка.
Согласно дополнительному варианту осуществления, по меньшей мере, одно антинеопластическое средство представляет собой дитерпеноид.
Согласно дополнительному варианту осуществления, по меньшей мере, одно антинеопластическое средство представляет собой алкалоид барвинка.
Согласно одному варианту осуществления, комбинация согласно настоящему изобретению включает соединение формулы I или его соль или сольват и, по меньшей мере, одно антинеопластическое средство, которое представляет собой координационный комплекс платины.
Согласно дополнительному варианту осуществления, по меньшей мере, одно антинеопластическое средство представляет собой паклитаксел, карбоплатин или винорелбин.
Согласно дополнительному варианту осуществления, по меньшей мере, одно антинеопластическое средство представляет собой карбоплатин.
Согласно дополнительному варианту осуществления, по меньшей мере, одно антинеопластическое средство представляет собой винорелбин.
Согласно дополнительному варианту осуществления, по меньшей мере, одно антинеопластическое средство представляет собой паклитаксел.
Согласно одному варианту осуществления, комбинация согласно настоящему изобретению включает соединение формулы I или его соли или сольваты и, по меньшей мере, одно антинеопластическое средство, которое представляет собой ингибитор сигнальных путей трансдукции.
Согласно дополнительному варианту осуществления, ингибитор сигнальных путей трансдукции представляет собой ингибитор киназ рецептора фактора роста VEGFR2, TIE2, PDGFR, BTK, erbB2, EGFr, IGFR-1, TrkA, TrkB, TrkC или c-fms.
Согласно дополнительному варианту осуществления, ингибитор сигнальных путей трансдукции представляет собой ингибитор сериновых/треониновых киназ rafk, akt или PKC-zeta.
Согласно дополнительному варианту осуществления, ингибитор сигнальных путей трансдукции представляет собой ингибитор нерецепторных тирозиновых киназ, выбранных из src семейства киназ.
Согласно дополнительному варианту осуществления, ингибитор сигнальных путей трансдукции представляет собой ингибитор c-src.
Согласно дополнительному варианту осуществления, ингибитор сигнальных путей трансдукции представляет собой ингибитор Ras онкогена, выбранный из ингибиторов фарнезилтрансферазы и геранилгеранилтрансферазы.
Согласно дополнительному варианту осуществления, ингибитор сигнальных путей трансдукции представляет собой ингибитор сериновых/треониновых киназ, выбранных из группы, состоящей из PI3K.
Согласно дополнительному варианту осуществления, ингибитор сигнальных путей трансдукции представляет собой двойной ингибитор EGFr/erbB2, например, N-{3-хлор-4-[(3-фторбензил)-окси]фенил}-6-[5-({[2-(метансульфонил)этил]амино}метил)-2-фурил]-4-хиназолинамин (структура ниже):
Согласно одному варианту осуществления, комбинация согласно настоящему изобретению включает соединение формулы I или его соль или сольват и, по меньшей мере, одно антинеопластическое средство, которое представляет собой ингибитор сигнальных путей клеточного цикла.
Согласно дополнительному варианту осуществления, ингибитор сигнальных путей клеточного цикла представляет собой ингибитор CDK2, CDK4 или CDK6.
Иммуностимулирующие средства:
Используемый в настоящем документе термин «иммуностимулирующее средство» относится к любому средству, которое может стимулировать иммунную систему. Используемые в настоящем документе иммуностимулирующие средства включают в себя без ограничения адъюванты вакцин, такие как агонисты Toll-подобных рецепторов, блокаторы контрольной точки Т-клеток, такие как mAbs и до PD-1 и CTL4 и агонисты контрольной точки Т-клеток, такие как агонисты mAbs и до OX-40 и ICOS.
Дополнительные примеры дополнительного активного ингредиента или ингредиентов (антинеопластическое средство) для применения в комбинации с соединением формулы (I) согласно настоящему изобретению или для совместного введения с ним представляют собой анти-PD-L1 средства.
Анти-PD-L1-антитела и способы их получения известны в данной области техники.
Такие антитела к PD-L1 могут быть поликлональными или моноклональными, и/или рекомбинантными и/или гуманизированными.
Примеры антител к PD-L1 раскрыты в:
Патенте США № 8,217,149; 12/633,339;
Патенте США № 8,383,796; 13/091,936;
Патенте США № 8,552,154; 13/120,406;
Патентной публикации США № 20110280877; 13/068337;
Патентной публикации США № 20130309250; 13/892671;
WO 2013019906;
WO 2013079174;
Заявке США № 13/511,538 (поданной 7 августа 2012 года), которая представляет собой национальную фазу международной заявки США № PCT/US10/58007 (подана в 2010 году);
и
Заявке США № 13/478,511 (подана 23 мая 2012 года).
Дополнительные примеры антитела к PD-L1 (также называемые как CD274 или B7-H1) и способы их применения раскрыты в патенте США № 7,943,743; US 20130034559, WO 2014055897, патенте США № 8,168,179; и патенте США № 7,595,048. Антитела к PD-L1 находятся в процессе разработки в качестве иммуномодулирующих средств для лечения злокачественных опухолей.
Согласно одному варианту осуществления, антитело к PD-L1 представляет собой антитело, раскрытое в патенте США № 8,217,149. Согласно другому варианту осуществления, анти-PD-L1 антитело содержит CDR антитела, раскрытого в патенте США № 8,217,149.
Согласно другому варианту осуществления, антитело к PD-L1 представляет собой антитело, раскрытое в заявке США № 13/511,538. Согласно другому варианту осуществления, анти-PD-L1 антитело содержит CDR антитела, раскрытого в заявке США № 13/511,538.
Согласно другому варианту осуществления, антитело к PD-L1 представляет собой антитело, раскрытое в заявке США № 13/478,511. Согласно другому варианту осуществления, анти-PD-L1 антитело содержит CDR антитела, раскрытого в заявке США № 13/478,511.
Согласно одному варианту осуществления, анти-PD-L1 антитело представляет собой BMS-936559 (MDX-1105). Согласно другому варианту осуществления, анти-PD-L1 антитело представляет собой MPDL3280A (RG7446). Согласно другому варианту осуществления, анти-PD-L1 антитело представляет собой MEDI4736.
Дополнительные примеры дополнительного активного ингредиента или ингредиентов (антинеопластическое средство) для применения в комбинации с соединением формулы (I) согласно настоящему изобретению или для совместного введения с ним представляют собой антагонист PD-1.
Термин «антагонист PD-1» означает любое химическое соединение или биологическую молекулу, которая блокирует связывание PD-L1, экспрессирующегося на клетке злокачественной опухоли, с PD-1, экспрессирующимся на иммунной клетке (Т-клетке, В-клетке или NKT-клетке) и, предпочтительно, также блокирует связывание PD-L2, экспрессирующегося на клетке злокачественной опухоли, с PD-1, экспрессирующимся на иммунной клетке. Альтернативные названия или синонимы для PD-1 и его лигандов включают в себя: PDCD1, PD1, CD279 и SLEB2 для PD-1; PDCD1L1, PDL1, B7H1, B7-4, CD274 и B7-H для PD-L1; и PDCD1L2, PDL2, B7-DC, Btdc и CD273 для PD-L2. Согласно некоторым вариантам осуществления аспектов или вариантам осуществления настоящего изобретения, в которых лечению подлежит индивидуум-человек, антагонист PD-1 блокирует связывание PD-L1 человека с PD-1 человека, и предпочтительно, блокирует связывание обоих PD-L1 и PD-L2 человека с PD-1 человека. Аминокислотные последовательности PD-1 человека можно найти в NCBI Locus №: NP_005009. Человеческие PD-L1 и PD-L2 аминокислотные последовательности можно найти в NCBI Locus №: NP_054862 и NP_079515, соответственно.
Антагонисты PD-1, применимые в любом из аспектов согласно настоящему изобретению, включают в себя моноклональное антитело (mAb) или его антиген-связывающий фрагмент, который специфически связывается с PD-1 или PD-L1, и предпочтительно, специфически связывается с PD-1 человека или PD-L1 человека. mAb может представлять собой антитело человека, гуманизированное антитело или химерное антитело, и может включать в себя константную область антитела человека. Согласно некоторым вариантам осуществления, константную область антитела человека выбирают из группы, состоящей из константных областей IgG1, IgG2, IgG3 и IgG4, и согласно предпочтительным вариантам осуществления, константную область антитела человека представляет собой константную область IgG1 или IgG4. Согласно некоторым вариантам осуществления, антиген-связывающий фрагмент выбирают из группы, состоящей из Fab, Fab'-SH, F(ab')2, scFv и Fv фрагментов.
Примеры mAb, которые связываются с PD-1 человека, и применимы в различных аспектах и вариантах осуществления настоящего изобретения, описаны в US7488802, US7521051, US8008449, US8354509, US8168757, WO2004/004771, WO2004/072286, WO2004/056875 и US2011/0271358.
Специфические mAb против PD-1 человека, применимые в качестве антагониста PD-1 в любом из аспектов или вариантов осуществления настоящего изобретения, включают в себя: MK-3475, гуманизированное IgG4 mAb со структурой, описанной в WHO Drug Information, Vol. 27, No. 2, pages 161-162 (2013) и содержащее тяжелую и легкую цепь аминокислотных последовательностей, продемонстрированных в фигуре 6; ниволумаб, IgG4 mAb человека со структурой, описанной в WHO Drug Information, Vol. 27, No. 1, pages 68-69 (2013) и содержащее тяжелую и легкую цепь аминокислотных последовательностей, продемонстрированных в фигуре 7; гуманизированные антитела h409A11, h409A16 и h409A17, которые описаны в WO2008/156712, и AMP-514, которые разрабатываются в Medimmune.
Другие антагонисты PD-1, применимые в любых аспектах и вариантах осуществления настоящего изобретения, включают в себя иммуноадгезин, который специфически связывается с PD-1, и предпочтительно, специфически связывается с PD-1 человека, например, рекомбинантный белок, содержащий внеклеточную или PD-1-связывающую часть PD-L1 или PD-L2, слитую с константной областью, такой как Fc область молекулы иммуноглобулина. Примеры иммуноадгезионных молекул, которые специфически связываются с PD-1, описаны в WO2010/027827 или WO2011/066342. Специфические рекомбинантные белки, применимые в качестве антагонистов PD-1 для способа лечения, лекарственных средств и применений согласно настоящему изобретению, включают в себя AMP-224 (также известный как B7-DCIg), который представляет собой рекомбинантный белок PD-L2-FC и связывается с PD-1 человека.
Другие примеры mAb, которые связываются с PD-L1 человека и применимы для способа лечения, лекарственных средств и применений согласно настоящему изобретению, описаны в WO2013/019906, W02010/077634 A1 и US8383796. Специфические mAb против PD-L1 человека, применимые в качестве антагониста PD-1 для способа лечения, лекарственных средств и применений согласно настоящему изобретению, включают в себя MPDL3280A, BMS-936559, MEDI4736, MSB0010718C.
КЕЙТРУДА/пембролизумаб представляет собой анти-PD-1-антитело, коммерчески доступное от Merck для лечения рака легкого. Аминокислотная последовательность пембролизумаба и способы применения раскрыты в патенте США № 8,168,757.
Опдиво/ниволумаб представляет собой полноразмерное моноклональное антитело человека, коммерчески доступное от Bristol Myers Squibb, направленное против негативного иммунорегуляторного поверхностного PD-1 рецептора клеток человека (программируемая гибель-1 или программируемая клеточная гибель-1/PCD-1) с иммуностимулирующей активностью. Ниволумаб связывается и блокирует активацию PD-1, Ig суперсемейство трансмембранных белков, посредством его лигандов PD-L1 и PD-L2, приводя к активации Т-клеток и клеточно-опосредованных иммунных ответов в отношении опухолевых клеток или патогенов. Активированный PD-1 отрицательно регулирует Т-клеточную активацию и эффекторную функцию посредством супрессии P13k/Akt пути активации. Другие названия для ниволумаба включают в себя BMS-936558, MDX-1106 и ONO-4538. Аминокислотная последовательность для ниволумаба и способов применения и приготовления раскрыта в патенте США № 8,008,449.
Дополнительные примеры дополнительного активного ингредиента или ингредиентов (антинеопластическое средство) для применения в комбинации с соединением формулы (I) согласно настоящему изобретению или для совместного введения с ним представляют собой иммуномодуляторы.
Используемый в настоящем документе термин «иммуномодуляторы» относится к любому веществу, включая моноклональные антитела, которое воздействует на иммунную систему. В качестве иммуномодуляторов можно рассматривать ICOS связывающие белки согласно настоящему изобретению. Иммуномодуляторы можно применять в качестве антинеопластических средств для лечения злокачественных опухолей. Например, иммуномодуляторы включают в себя без ограничения анти-CTLA-4 антитела, такие как ипилимумаб (ЕРВОЙ), и анти-PD-1 антитела (Опдиво/ниволумаб и Кейтруда/пембролизумаб). Другие иммуномодуляторы включают в себя без ограничения антитела к OX-40, антитела к PD-L1, антитела к LAG3, антитела к TIM-3, антитела к 41BB и антитела к GITR.
Ервой (ипилимумаб) представляет собой полноразмерное CTLA-4 антитело человека коммерчески доступное посредством Bristol Myers Squibb. Белковая структура ипилимумаба и способы применения описаны в патенте США № 6,984,720 и 7,605,238.
CD134, также известный как OX40, является представителем суперсемейства TNFR-рецепторов, который в противоположность CD28 не экспрессируется на покоящихся интактных Т-клетках конститутивно. OX40 представляет собой вторичную костимулирующую молекулу, экспрессирующуюся через 24-72 часа после активации; ее лиганд, OX40L, также не экспрессируется на покоящихся антиген-презентирующих клетка, но экспрессируется после их активации. Экспрессия OX40 зависит от полной активации Т-клеток; без CD28 экспрессия OX40 отсрочена и составляет вчетверо более низкие уровни. Антитела к OX-40, рекомбинантные белки OX-40 и способы их применения описаны в патентах США №: US 7,504,101; US 7,758,852; US 7,858,765; US 7,550,140; US 7,960,515; WO2012027328; WO2013028231.
Используемый в настоящем документе термин «Toll-подобный рецептор» (или «TLR») относится к представителю семейства белков Toll-подобных рецепторов или к его фрагменту, который чувствителен к продукту микроорганизмов и/или инициирует адаптивный иммунный ответ. Согласно одному варианту осуществления, TLR активирует дендритную клетку (DC). Toll-подобные рецепторы (TLR) представляют собой семейство рецепторов, распознающих паттерны, которые первоначально были определены как сенсоры врожденной иммунной системы, которые распознают микробные патогены. TLR распознают индивидуальные структуры в микробах, часто называемые «PAMP» (патоген-ассоциированные молекулярные паттерны). Лиганд, связавшийся с TLR, вызывает каскад внутриклеточных сигнальных путей, которые индуцируют продукцию факторов, вовлеченных в процесс воспаления и иммунный ответ. У людей были определены десять TLR. TLR, которые экспрессируются на поверхности клеток, включают в себя TLR-I, -2, -4, -5 и -6, тогда как, TLR-3, -7/8 и -9 экспрессируются вместе с ER компартментом. Человеческие субпопуляции DC могут быть определены на основании индивидуальных паттернов экспрессии TLR. В качестве примера, при стимулировании миелоидная или «общепринятая» субпопуляция DC (mDC) экспрессирует TLR 1-8 и приводит к каскаду активации маркеров (например, CD80, CD86, MHC I и II класса, CCR7), провоспалительных цитокинов и хемокинов. Результатом такой стимуляции и конечной экспрессии является примирование антиген-специфических CD4+ и CD8+ T-клеток. Такие DC требуют усиленной способности к поглощению антигенов и презентируют их Т-клеткам в подходящей форме. Напротив, плазмацитоидная субпопуляция DC (pDC) после активации экспрессирует только TLR7 и TLR9, с конечной активацией NK-клеток, а также Т-клеток. Поскольку гибнущие опухолевые клетки могут отрицательно воздействовать на функцию DC, было предположено, что активирование DC с TLR агонистами может быть благоприятным для инициирования противоопухолевого иммунитета в иммунотерапевтическом подходе для лечения злокачественной опухоли. Также было предположено, что успешное лечение злокачественной молочной железы с использованием облучения и химиотерапии требует активации TLR4.
Агонисты TLR, известные в данной области техники и находящие свое применение в настоящем изобретении, включают в себя без ограничения следующие: Pam3Cys, агонист TLRI/2; CFA, агонист TLR2; MALP2, агонист TLR2; Pam2Cys, агонист TLR2; FSL-I, агонист TLR-2; Hib-OMPC, агонист TLR-2; полирибозиновая: полирибоцитидиновая кислота (поли I:C), агонист TLR3; полиаденозин-полиуридиловая кислота (поли AU), агонист TLR3; полиинозиновая-полицитидиловая кислота, стабилизированная поли-L-лизином и карбоксиметилцеллюлозой (Хилтонол), агонист TLR3; бактериальный флагеллин агонист TLR5; имихимод, агонист TLR7; резихимод, агонист TLR7/8; локсорибин, агонист TLR7/8; и неметилированный CpG динуклеотид (CpG-ODN), агонист TLR9.
Дополнительные агонисты TLR, известные в данной области техники и находящие свое применение в настоящем изобретении, дополнительно включают в себя без ограничения аминоалкилглюкозаминидфосфаты (AGP), которые связываются с TLR4 рецептором и известны как применимые в качестве адъювантов вакцин и иммуностимулирующих средств для стимулирования продукции цитокинов, активирования макрофагов, усиления врожденного иммунного ответа и увеличения продукции антител в иммунизированных животных. Примером встречающегося в природе агониста TLR4 является бактериальная LPS. Примером полусинтетического агониста TLR4 является монофосфориллипид А (MPL). AGP и их иммуномодулирующие эффекты посредством TLR4 раскрыты в патентных публикациях, таких как WO 2006/016997, WO 2001/090129, и/или патенте США № 6,113,918, и были сообщены в литературе. Дополнительные AGP производные раскрыты в патенте США № 7,129,219, патенте США № 6,525,028 и патенте США № 6,911,434. Определенные AGP действуют как агонисты TLR4, тогда как другие определены в качестве антагонистов TLR4.
В дополнение к иммуностимулирующим средствам, описанным выше, композиции согласно настоящему изобретению могут дополнительно содержать одно или несколько дополнительных веществ, которые, благодаря их адъювантной природе, могут действовать как стимуляторы иммунной системы для ответа на антигены злокачественных опухолей, присутствующие на инактивированной опухолевой клетке(ах). Такие адъюванты включают в себя без ограничения липиды, липосомы, инактивированные бактерии, которые индуцируют врожденный иммунитет (например, инактивированные или аттенуированные I/ster/a моноцитогены), композиции, которые опосредуют врожденную иммунную активацию посредством (NOD)-подобных рецепторов (NLR), рецепторов RLR, подобных индуцируемому ретиноевой кислотой гену I (RIG)-I, и/или лецитиновых рецепторов C-типа (CLR). Примеры PAMP включают в себя липопротеины, липополипептиды, пептидогликаны, зимозан, липополисахарид, порины нейссерий, флагеллин, профиллин, галактоцерамид, мурамил-дипептид. Пептидогликаны, липопротеины и липотейхоевые кислоты представляют собой компоненты Грамположительной клеточной стенки. Липополисахариды экспрессируются большинством бактерий, с MPL, представляющим собой один пример. Флагеллин относится к структурному компоненту бактериального жгутика, который секретируется патогенной и условно-патогенной бактерией. rt.-галактозилцерамид (rt.-GalCer) представляет собой активатор Т-клеток естественных киллеров (NKT). Мурамил-дипептид представляет собой биоактивный пептидогликановый мотив, общий для всех бактерий.
Благодаря их адъювантным свойствам, агонисты TLR предпочтительно используют в комбинации с другими вакцинами, адъювантами и/или иммуномодуляторами, и могут быть объединены в различных комбинациях. Таким образом, согласно определенным вариантам осуществления, в настоящем документе описаны соединения формулы (I), которые связываются со STING и индуцируют STING-зависимую активацию TBKI и инактивированные опухолевые клетки, которые экспрессируют и секретируют один или более цитокинов, которые стимулируют индукцию, рекрутинг и/или созревание DC, и как описано в настоящем документе, могут быть введены вместе с одним или несколькими агонистами TLR для терапевтических целей.
Дополнительные примеры дополнительного активного ингредиента или ингредиентов (антинеопластическое средство) для применения в комбинации или совместного введения с соединением формулы (I) согласно настоящему изобретению представляют собой антитела к ICOS.
Обладающие агонистической активностью CDR мышиных антител к ICOS человека представлены в PCT/EP2012/055735 (WO 2012/131004). Антитела к ICOS также раскрыты в WO 2008/137915, WO 2010/056804, EP 1374902, EP1374901 и EP1125585.
Индолеамин-2,3-диоксигеназа 1 (IDO1) представляет собой ключевой иммуносупрессивный фермент, который модулирует противоопухолевый иммунный ответ, посредством усиления генерации регуляторных Т-клеток и блокирования активации эффекторных Т-клеток, способствуя тем самым опухолевому росту и позволяя раковым клеткам избежать иммуноконтроля (Lemos H,,et al., Cancer Res. 2016 Apr 15;76(8):2076-81), (Munn DH, et at., Trends Immunol. 2016 Mar;37(3):193-207). Дополнительные активные ингредиенты (антинеопластические средства) для применения в комбинации или совместного введения с соединениями формулы (I) согласно настоящему изобретению представляют собой ингибиторы IDO. Эпакадостат, ((Z)-N-(3-бром-4-фторфенил)-N'-гидрокси-4-[2-(сульфамоиламино)этиламино]-1,2,5-оксадиазол-3-карбоксамидин), представляет собой высоко эффективный и селективный пероральный ингибитор фермента IDO1, который изменяет опухоль-ассоциированную иммуносупрессию и восстанавливает эффективные противоопухолевые иммунные ответы. Эпакадостат раскрыт в патенте США № 8,034,953.
Дополнительные примеры дополнительного активного ингредиента или ингредиентов (антинеопластическое средство) для применения в комбинации или совместного введения с соединением формулы (I) согласно настоящему изобретению представляют собой ингибиторы CD73 и антагонисты аденозина A2a и A2b.
Согласно одному аспекту подлежащее лечению заболевание представляет собой инфекционное заболевание, например, вызванное бактерией или вирусом.
Согласно дополнительному аспекту настоящего изобретения, предусмотрено соединение формулы (I) или его фармацевтически приемлемая соль для применения при лечении инфекционного заболевания.
Согласно дополнительному аспекту, предусмотрен способ лечения инфекционного заболевания, включающий в себя введение нуждающемуся в этом человеку терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Согласно дополнительному аспекту, предусмотрено применение соединения формулы (I) или его фармацевтически приемлемой соли при производстве лекарственного средства для лечения инфекционного заболевания.
Согласно одному варианту осуществления, соединение согласно настоящему изобретению может применяться с другими терапевтическими способами лечения инфекционного заболевания. В частности, предусмотрены противовирусные и антибактериальные средства.
Соединения формулы (I) и их фармацевтически приемлемые соли можно применять в комбинации с одним или несколькими средствами, применимыми для профилактики или лечения бактериальных и вирусных инфекций. Примеры таких средств включают в себя без ограничения: ингибиторы полимераз, такие как ингибиторы, раскрытые в WO 2004/037818-A1, а также ингибиторы, раскрытые в WO 2004/037818 и WO 2006/045613; JTK-003, JTK-019, NM-283, HCV-796, R-803, R1728, R1626, а также ингибиторы, раскрытые в WO 2006/018725, WO 2004/074270, WO 2003/095441, US2005/0176701, WO 2006/020082, WO 2005/080388, WO 2004/064925, WO 2004/065367, WO 2003/007945, WO 02/04425, WO 2005/014543, WO 2003/000254, EP 1065213, WO 01/47883, WO 2002/057287, WO 2002/057245 и аналогичные средства; ингибиторы репликации, такие как ацикловир, фамцикловир, ганцикловир, цидофовир, ламивудин и аналогичные средства; ингибиторы протеаз, такие как ингибиторы HIV-протеазы саквинавир, ритонавир, индинавир, нелфинавир, ампренавир, фосампренавир, бреканавир, атазанавир, типранавир, палинавир, лазинавир, и ингибиторы HCV-протеазы BILN2061, VX-950, SCH503034; и аналогичные средства; ингибиторы обратной транскриптазы нуклеозида и нуклеотида, такие как зидовудин, диданозин, ламивудин, залцитабин, абакавир, ставидин, адефовир, адефовира дипивоксил, фозивудин, тодоксил, эмтрицитабин, аловудин, амдоксовир, элвуцитабин и аналогичные средства; ингибиторы обратной транскриптазы ненуклеозидов (включая средство, имеющее антиокислительную активность, такое как иммунокал, олтипраз и т. п.), такие как невирапин, делавирдин, эфавиренз, ловирид, иммунокал, олтипраз, каправирин, TMC-278, TMC-125, этравирин и аналогичные средства; ингибиторы проникновения в клетку, такие как энфувиртид (T-20), T-1249, PRO-542, PRO-140, TNX-355, BMS-806, 5-Helix и аналогичные средства; ингибиторы интегразы, такие как L-870,180 и аналогичные средства; ингибиторы почкования, такие как PA-344 и PA-457, и аналогичные средства; ингибиторы хемокиновых рецепторов, такие как викривирок (Sch-C), Sch-D, TAK779, маравирок (UK-427,857), TAK449, а также, таковые, раскрытые в WO 02/74769, WO 2004/054974, WO 2004/055012, WO 2004/055010, WO 2004/055016, WO 2004/055011 и WO 2004/054581, и аналогичные средства; ингибиторы нейраминидазы, такие как CS-8958, занамивир, оселтамивир, перамивир и аналогичные средства; блокаторы ионных каналов, такие как амантадин или римантадин и аналогичные средства; и интерферирующие РНК и антисмысловые олигонуклеотиды, такие как ISIS-14803 и аналогичные средства; противовирусные средства с неопределенным механизмом действия, например, средства, раскрытые в WO 2005/105761, WO 2003/085375, WO 2006/122011, рибавирин, и аналогичные средства. Соединения формулы (I) и их фармацевтически приемлемые соли также можно применять в комбинации с одним или несколькими другими средствами, которые могут быть применимы для профилактики или лечения вирусных инфекций, например, с иммунотерапией (например, интерферон или другие цитокины/хемокины, модуляторы рецепторов цитокино/хемокинов, агонисты или антагонисты цитокинов и аналогичные средства); и терапевтическими вакцинами, антифибротическими средствами, противовоспалительными средствами, такими как кортикостероиды или NSAID (нестероидные противовоспалительные средства) и аналогичными средствами.
Согласно дополнительному аспекту, предусмотрена комбинация, включающая в себя соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, одно дополнительное терапевтическое средство, применимое для лечения инфекционного заболевания.
Согласно дополнительному аспекту, предусмотрена комбинация, включающая в себя соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, одно дополнительное терапевтическое средство, применимое для лечения инфекционного заболевания, для применения в терапии.
Согласно дополнительному аспекту, предусмотрена комбинация, включающая в себя соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, одно дополнительное терапевтическое средство, применимое для лечения инфекционного заболевания, для применения при лечении инфекционного заболевания.
Согласно дополнительному аспекту, предусмотрена комбинация, включающая в себя соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, одно дополнительное терапевтическое средство, применимое для лечения инфекционного заболевания, при производстве лекарственного средства для лечения инфекционного заболевания.
Согласно дополнительному аспекту, предусмотрен способ лечения инфекционного заболевания, включающий в себя введение нуждающемуся в этом человеку терапевтически эффективного количества комбинации, включающей в себя соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, одно дополнительное терапевтическое средство, применимое для лечения инфекционного заболевания.
Согласно дополнительному аспекту, предусмотрена фармацевтическая композиция, включающая в себя комбинацию, включающую в себя соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, одно дополнительное терапевтическое средство, применимое для лечения инфекционного заболевания, и один или более фармацевтически приемлемых эксципиентов.
Согласно дополнительному аспекту, предусмотрена композиция, включающая в себя соединение формулы (I) или его фармацевтически приемлемую соль и одно или несколько иммуностимулирующих средств.
Таким образом, также предусмотрены иммуногенная композиция или адъювант вакцин, содержащие соединение формулы (I) или его фармацевтически приемлемую соль.
Дополнительно предусмотрена иммуногенная композиция, включающая в себя антиген или антигенную композицию и соединение формулы (I) или его фармацевтически приемлемую соль.
Дополнительно предусмотрена вакцинная композиция, содержащая антиген или антигенную композицию и соединение формулы (I) или его фармацевтически приемлемую соль.
Дополнительно предусмотрен способ лечения или профилактики заболевания, включающий в себя введение субъекту-человеку, страдающему от заболевания или предрасположенному к нему, иммуногенной композиции, включающей в себя антиген или антигенную композицию и соединение формулы (I) или его фармацевтически приемлемую соль.
Дополнительно предусмотрен способ лечения или профилактики заболевания, включающий в себя введение субъекту-человеку, страдающему от заболевания или предрасположенному к нему, вакцинной композиции, содержащей антиген или антигенную композицию и соединение формулы (I) или его фармацевтически приемлемую соль.
Дополнительно предусмотрено использование соединения формулы (I) или его фармацевтически приемлемой соли при производстве иммуногенной композиции, содержащей антиген или антигенную композицию, для лечения или профилактики заболевания.
Дополнительно предусмотрено использование соединения формулы (I) или его фармацевтически приемлемой соли при производстве вакцинной композиции, содержащей антиген или антигенную композицию, для лечения или профилактики заболевания.
Следует понимать, что представленные в заявке соединения могут быть изображены с использованием разных общепринятых способов. Например, следующие два соединения считаются эквивалентными по химической структуре и стереохимии.
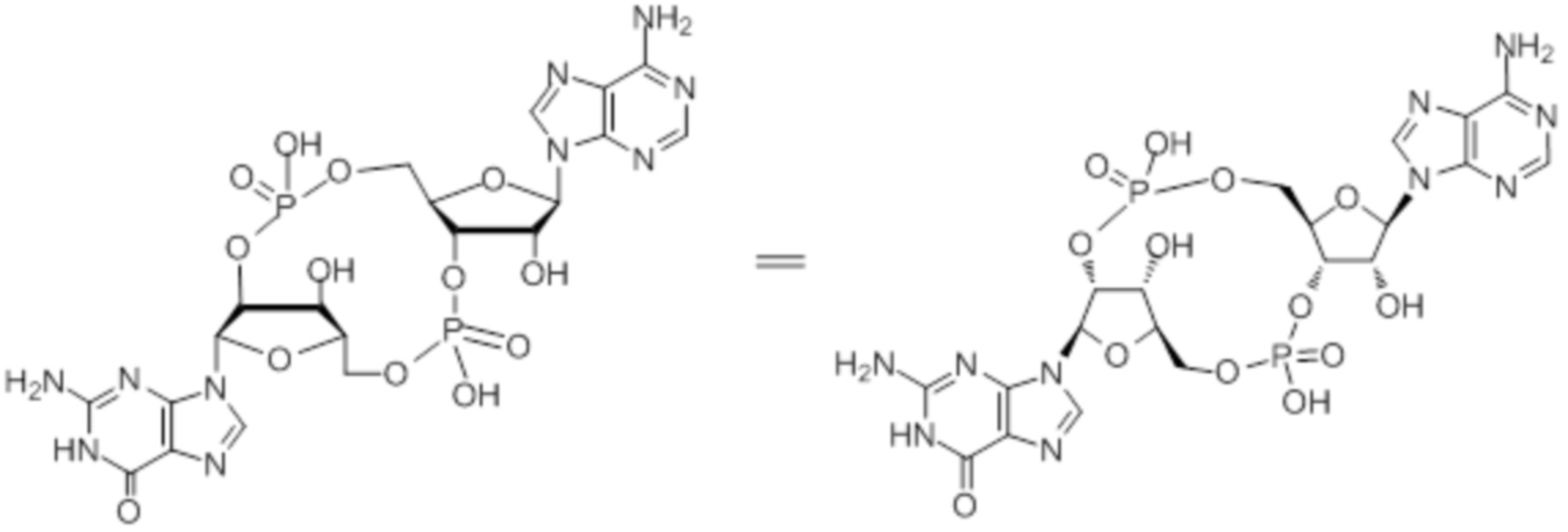
Получение соединений и примеры

Соединения формулы (I), где значения Y1, Y2, X1, X2, R1, R2, R3, R4, R5, R6, R8 и R9 определены выше в настоящем документе, могут быть получены способами, известными в области органического синтеза, как представлено ниже на схемах и в Примерах. Для всех способов следует хорошо понимать, что в случае необходимости для чувствительных и реакционноспособных групп в соответствии с общими принципами химии могут применяться защитные группы. Защитные группы используют в соответствии со стандартными способами органического синтеза (P. G. M. Wuts and T. W. Green (2007) Greene's Protective Groups in Organic Synthesis, 4th edition, John Wiley & Sons). Такие группы удаляют на удобной стадии синтеза соединения с использованием способов, хорошо знакомых специалистам в данной области техники. Выбор способов, а также условий проведения реакций и порядка их проведения, должны соответствовать получению соединений формулы (I).
Соединения формулы (I) и их соли могут быть получены посредством методологии, описанной далее в настоящем документе, составляющей дополнительные аспекты настоящего изобретения.
Соответственно, предложен способ получения соединения формулы (I), в которой R5 представляет собой OC(O)R7, и R6 представляет собой F, и Y1 и Y2 оба представляют собой O, X1 и X2 оба представляют собой S-, как представлено формулой (IIa), где значения R1, R2, R3 и R4 определены выше в настоящем документе для соединения формулы (I). Способ включает ацилирование соединения формулы (IIIa):

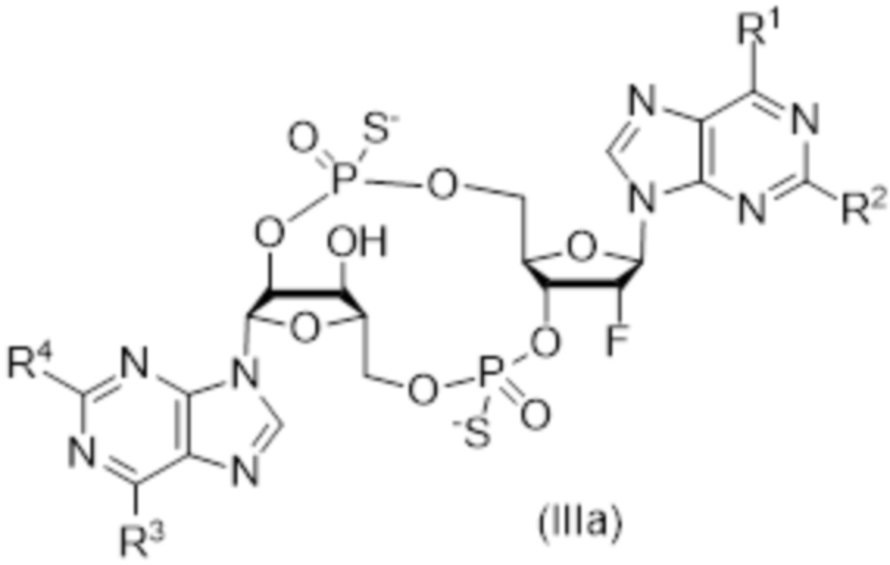
где значения R1, R2, R3, R4 и R7 определены выше в настоящем документе для соединения формулы (IIa), при необходимости, с последующим получением соли полученного тем самым соединения.
Пример 1: Соединение формулы (IIIa) и миристиновый ангидрид в подходящем растворителе, например, в диметилформамиде (DMF), в присутствии основания, такого как пиридин, перемешивают при комнатной температуре или нагревают при подходящей температуре, например, при 60°C, в течение походящего периода времени, например, в течение 2-48 часов. Продукт формулы (IIa) выделяют путем удаления летучих веществ и, при необходимости, очистки.
Также предложен способ получения соединения формулы (I), в которой R5 представляет собой OH, и R6 представляет собой F, и Y1 и Y2 оба представляют собой O, X1 и X2 оба представляют собой S, и R8 и R9 оба представляют собой CH2OC(O)tBu, как представлено формулой (IVa), где значения R1, R2, R3 и R4 определены выше в настоящем документе для соединения формулы (I). Способ включает введение карбонилоксиметильной группы в соединение формулы (IIIa):
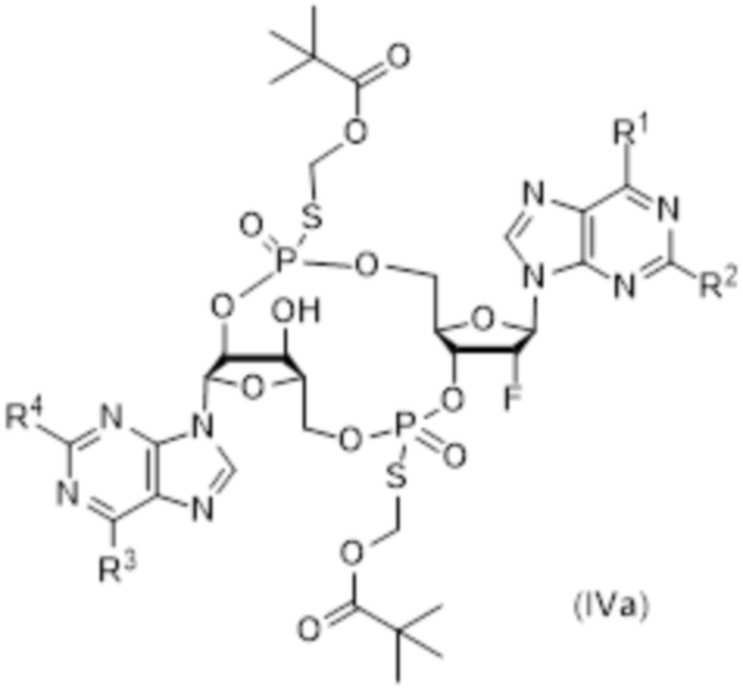
Пример 2: Соединение формулы (IIIa) и хлорметилпивалат (POM-Cl) в подходящем растворителе, например, в диметилформамиде (DMF), в присутствии основания, такого как Et3N, перемешивают при комнатной температуре в течение походящего периода времени, например, в течение 48 часов. Продукт формулы (IVa) выделяют путем удаления летучих веществ и, при необходимости, очистки.
Соединение формулы (IIIa) может быть получено путем снятия защиты с соединения формулы (Va):

где значения R1, R2, R3 и R4 определены выше в настоящем документе для соединения формулы (IIIa), и P1 представляет собой подходящую защитную группу, такую как трет-бутилдиметилсилилокси (TBDMS), при необходимости, с последующим получением соли полученного тем самым соединения.
Пример 3: Соединение формулы (Va) в подходящем растворителе, например, в пиридине, нагревают при подходящей температуре, например, при 50°C, а затем обрабатывают смесью триэтиламина тригидрофторида и триэтиламина в течение походящего периода времени, например, в течение 2-3 часов. Продукт формулы (IIIa) выделяют путем осаждения посредством добавления растворителя, например, ацетона, или путем удаления летучих веществ и, при необходимости, очистки.
Соединение формулы (Va) может быть получено путем снятия защиты с соединения формулы (VIa):
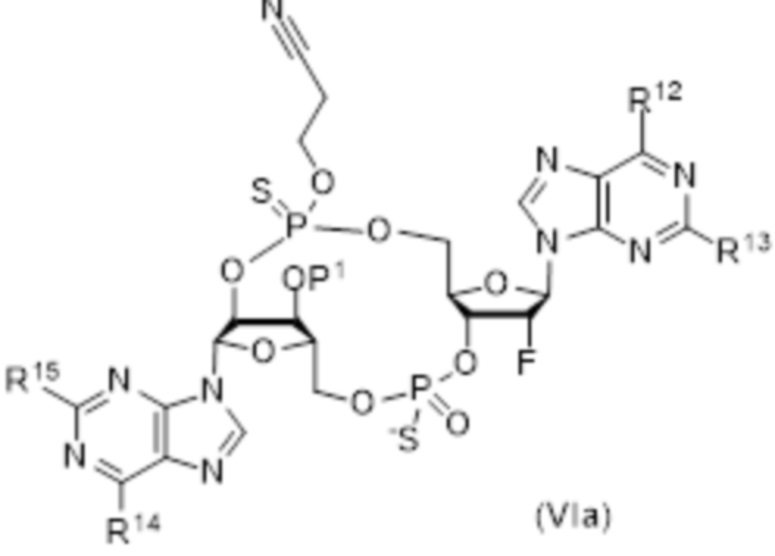
где P1 представляет собой защитную группу, определенную для соединения формулы (Va), и значения R12, R13, R14 и R15 определены как:
R12 представляет собой OH, и R13 представляет собой NHCOiPr, или R12 представляет собой NHBz, и R13 представляет собой H;
R14 представляет собой OH, и R15 представляет собой NHCOiPr, или R14 представляет собой NHBz, и R15 представляет собой H;
Пример 4: Соединение формулы (VIa) растворяют в подходящей смеси, например, в метиламине в метаноле или в водном аммиаке в метаноле, и нагревают при подходящей температуре, например, при 50-55°C, в течение походящего периода времени, например, в течение 2-72 часов. Продукт формулы (Va) выделяют путем удаления растворителя и, при необходимости, очистки.
Соединение формулы (VIa) может быть получено путем осуществления взаимодействия соединения формулы (VIIa):
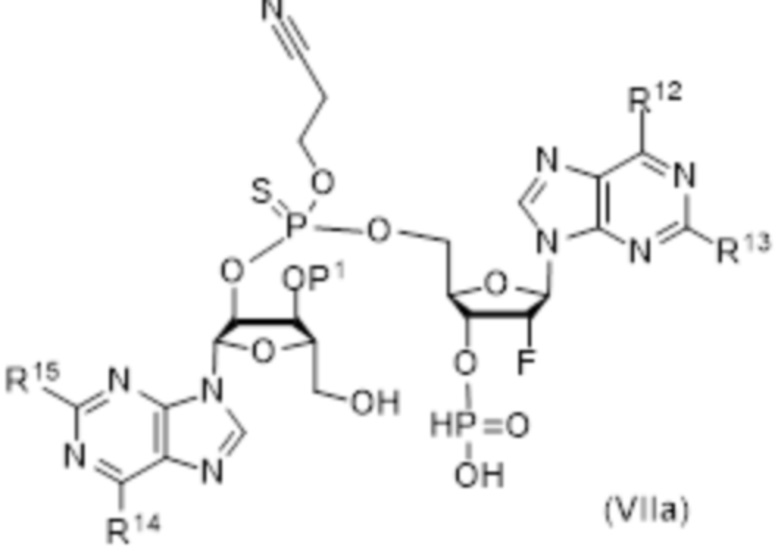
где значения P1, R12, R13, R14 и R15 определены выше в настоящем документе для соединения формулы (VIa).
Пример 5: Соединение формулы (VIIa) растворяют в подходящем растворителе, например, в пиридине, и обрабатывают подходящим агентом реакции сочетания, например, 2-хлор-5,5-диметил-1,3,2-диоксафосфоринан-2-оксидом, и перемешивают при подходящей температуре, например, при 20°C, в течение походящего периода времени, например, в течение 0,5-2 часов. Реакционную смесь гасят добавлением подходящего растворителя, например, воды, а затем добавляют сульфитирующий агент, например, 3H-бензо[c][1,2]дитиол-3-он, и перемешивают при подходящей температуре, например, при 20°C, в течение походящего периода времени, например, в течение 5-10 минут. Реакционную смесь гасят добавлением подходящего растворителя, например, водного раствора NaHCO3. Продукт формулы (VIa) экстрагируют подходящим органическим растворителем, например, EtOAc. Продукт формулы (VIa) выделяют путем удаления растворителя и, при необходимости, очистки.
Соединение формулы (VIIa) может быть получено путем осуществления взаимодействия соединения формулы (VIIIa) с соединением формулы (IXa):
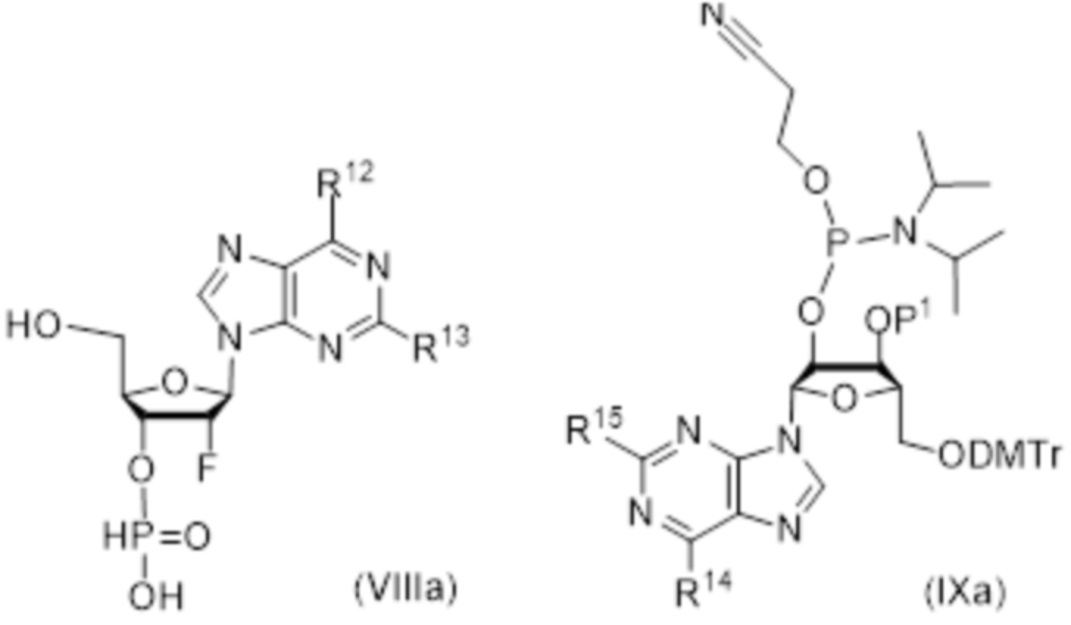
где значения P1, R12, R13, R14 и R15 определены выше в настоящем документе для соединения формулы (VIIa), и DMTr представляет собой защитную 4,4-диметокситритильную группу.
Пример 6: Соединение формулы (IXa) в подходящем растворителе, например, в ацетонитриле, в присутствии молекулярных сит обрабатывают раствором соединения формулы (VIIIa), растворенного в подходящем растворителе, например, в ацетонитриле, и перемешивают при подходящей температуре, например, при 20°C, в течение походящего периода времени, например, в течение 0,5-2 часов. Добавляют раствор подходящего сульфитирующего агента, например, N,N-диметил-N'-(3-тиоксо-3H-1,2,4-дитиазол-5-ил)формимидамида (DDTT), и перемешивают смесь при подходящей температуре, например, при 20°C, в течение походящего периода времени, например, в течение 0,5-1 часа. После упаривания растворителя, остаток растворяют в подходящем растворителе, например, в смеси дихлорметана и воды, и обрабатывают подходящим реагентом, например, дихлоруксусной кислотой, и перемешивают при подходящей температуре, например, при 20°C, в течение походящего периода времени, например, в течение 15 минут. Раствор, содержащий продукт формулы (VIIa), получают путем добавления подходящего растворителя, например, пиридина, и концентрирования путем упаривания.
Соединение формулы (VIIIa) может быть получено путем осуществления взаимодействия соединения формулы (Xa)
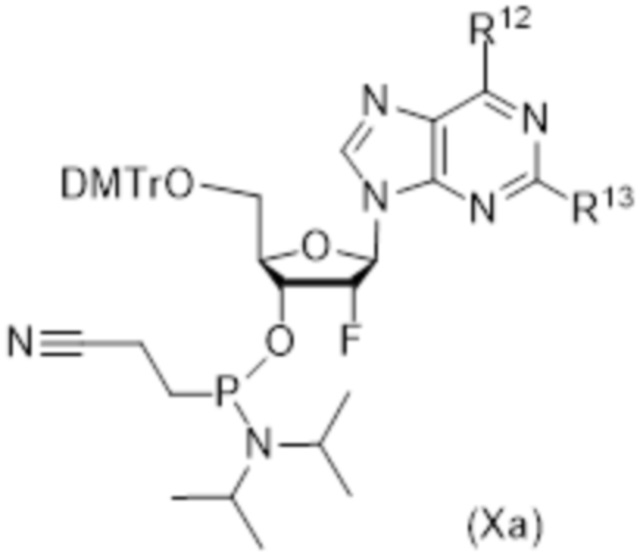
где значения R12 и R13 определены выше в настоящем документе для соединения формулы (VIIIa), и DMTr представляет собой защитную 4,4-диметокситритильную группу.
Пример 7: Соединение формулы (Xa), растворенное в подходящей смеси, например, в ацетонитриле с содержанием воды, обрабатывают трифторацетатом пиридиния и перемешивают при подходящей температуре, например, при 20°C, в течение походящего периода времени, например, в течение 1-5 минут. Затем, добавляют трет-бутиламин, и перемешивают смесь при подходящей температуре, например, при 20°C, в течение походящего периода времени, например, в течение 10 минут. Продукт выделяют путем упаривания растворителя, последующего растворения в подходящем растворителе, например, в дихлорметане с содержанием воды, обработки дихлоруксусной кислотой и перемешивания при подходящей температуре, например, при 20°C, в течение походящего периода времени, например, в течение 15 минут. Концентрированный раствор продукта формулы (VIIIa) в ацетонитриле получают путем добавления пиридина с последующей азеотропной перегонкой смеси с безводным ацетонитрилом.
Фосфорамидиты формулы (IXa) и (Xa) либо известны из литературы, либо коммерчески доступны у таких поставщиков, как Sigma, Chemgenes и CarboSynth, либо могут быть получены известными способами.
Пример 8 - Соединение 1b
(1R,6R,8R,9R,10R,15R,17R,18R)-8,17-бис(6-амино-9H-пурин-9-ил)-9-фтор-18-гидрокси-3,12-дисульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-диона бисаммониевая соль
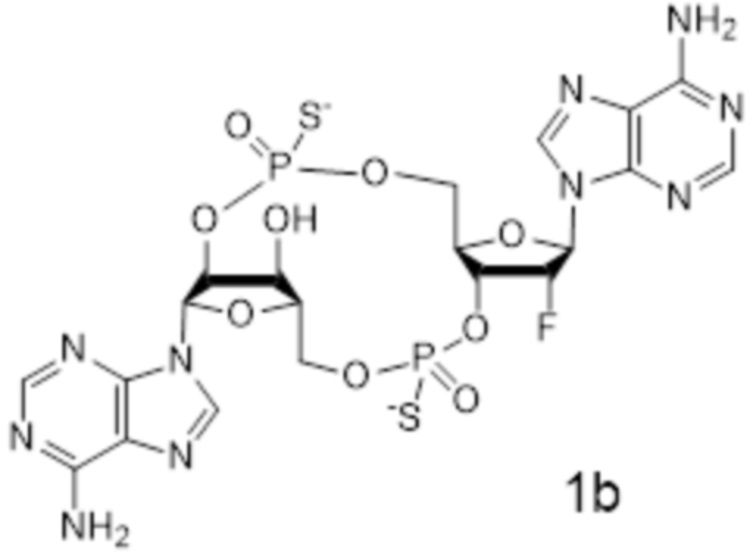
Промежуточный продукт 1: (2R,3R,4R,5R)-5-(6-бензамидо-9H-пурин-9-ил)-2-((бис(4-метоксифенил)(фенил)метокси)метил)-4-фтортетрагидрофуран-3-ила гидрофосфонат
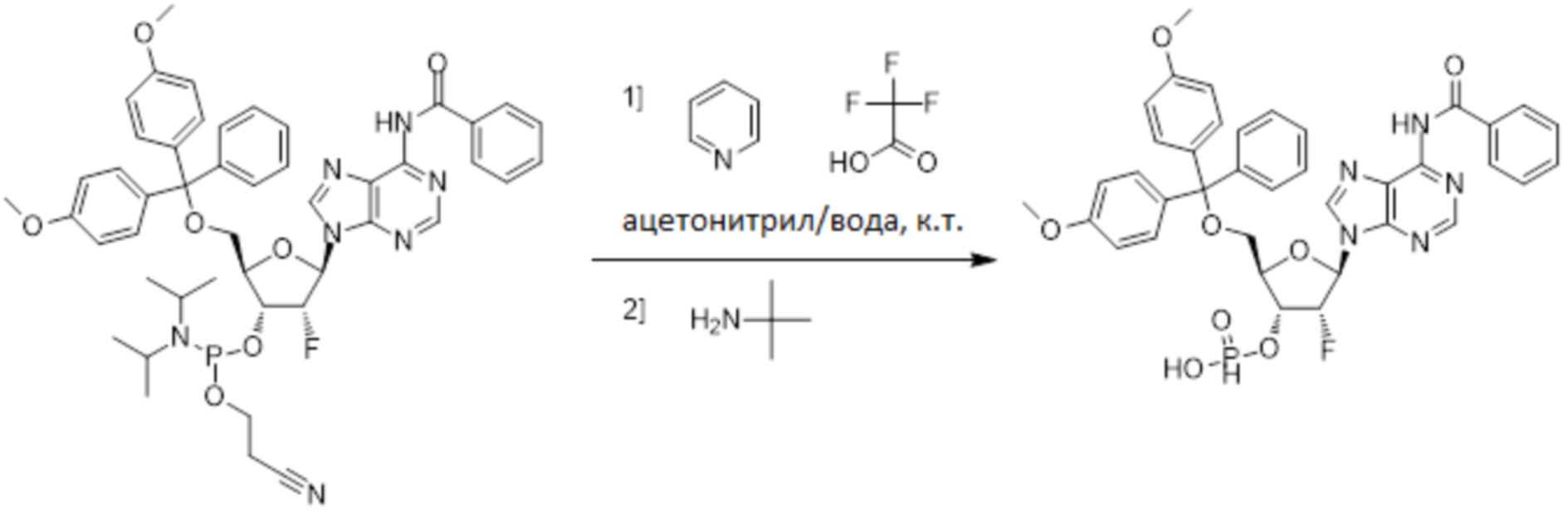
К раствору (2R,3R,4R,5R)-5-(6-бензамидо-9H-пурин-9-ил)-2-((бис(4-метоксифенил)(фенил)метокси)метил)-4-фтортетрагидрофуран-3-ил-(2-цианоэтил)диизопропилфосфорамидита (2190 мг, 2,5 ммоль) в ацетонитриле (15 мл) и воде (0,090 мл, 5,00 ммоль) при комнатной температуре добавляли пиридин-2,2,2-трифторацетат (579 мг, 3,00 ммоль). Смесь перемешивали в течение 1 минуты, после чего методом LCMS обнаруживали полное преобразование до первого промежуточного продукта, m/z (M+H)=793,3. Затем, добавляли 2-метилпропан-2-амин (13,14 мл, 125 ммоль), и перемешивали смесь в течение 10 минут, после чего методом LCMS обнаруживали расходование первого промежуточного продукта.
Смесь концентрировали в условиях вакуума с получением белой пены. Затем, пену растворяли в ацетонитриле (20 мл) и концентрировали. Эту операцию повторяли еще 1 раз. Неочищенное вещество растворяли в дихлорметане (10 мл) и очищали двумя порциями методом хроматографии (силикагель, элюирование градиентом 0-30% метанола в дихлорметане). Целевые фракции объединяли и упаривали с получением двух отдельных белых твердых веществ, которые затем растворяли в дихлорметане, объединяли и упаривали с получением указанного в заголовке соединения (780 мг, 1,055 ммоль, выход 42,2%) в виде белого твердого вещества. LCMS m/z 740,4 (M+H).
Промежуточный продукт 2: (2R,3R,4R,5R)-5-(6-бензамидо-9H-пурин-9-ил)-4-фтор-2-(гидроксиметил)тетрагидрофуран-3-ила гидрофосфонат
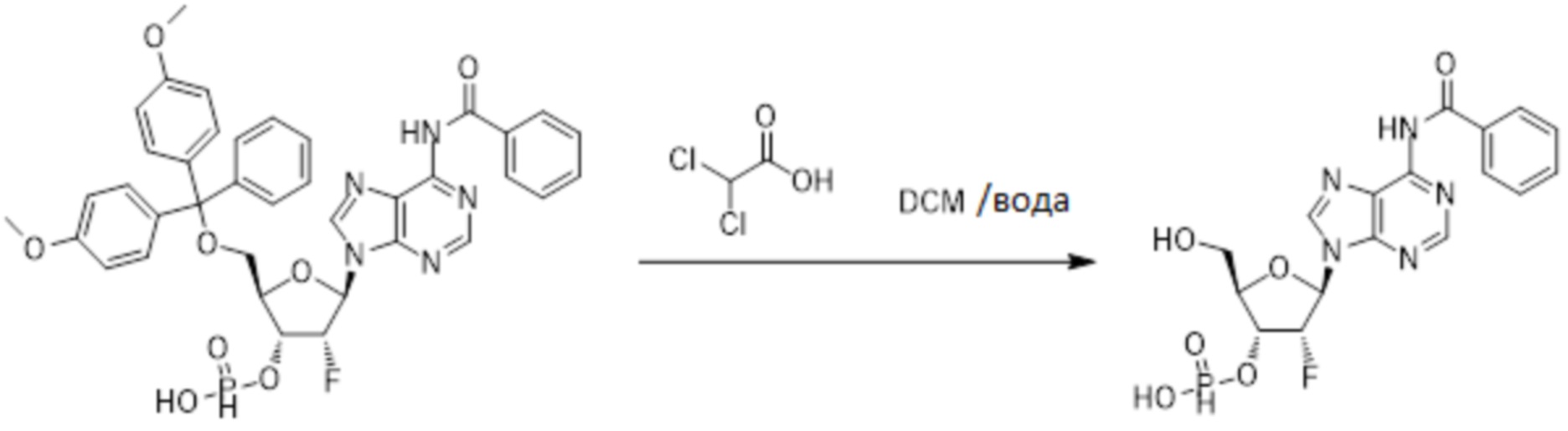
Промежуточный продукт 2 получали, в общем, в соответствии с методикой, представленной ниже. В других Примерах могут применяться небольшие модификации, например, описанные для промежуточного продукта 2.
К раствору гидрофосфоната (2R,3R,4R,5R)-5-(6-бензамидо-9H-пурин-9-ил)-2-((бис(4-метоксифенил)(фенил)метокси)метил)-4-фтортетрагидрофуран-3-ила (775 мг, 1,048 ммоль) в дихлорметане (20 мл) и воде (0,189 мл, 10,48 ммоль) при комнатной температуре добавляли 2,2-дихлоруксусную кислоту (0,655 мл, 8,38 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут. Затем, реакционную смесь гасили добавлением пиридина (1,356 мл, 16,76 ммоль) и концентрировали в условиях вакуума с получением бесцветного масла. Вещество хранили в атмосфере азота при 4°C. После хранения при 4°C, вещество отверждалось с получением содержащего примеси указанного в заголовке соединения в виде восковидного белого твердого вещества, которое использовали без дополнительной очистки. LCMS m/z 438,3 (M+H).
Промежуточный продукт 3: гидрофосфонат (2R,3R,4R,5R)-5-(6-бензамидо-9H-пурин-9-ил)-2-((((((2R,3R,4R,5R)-2-(6-бензамидо-9H-пурин-9-ил)-4-((трет-бутилдиметилсилил)окси)-5-(гидроксиметил)-тетрагидрофуран-3-ил)окси)(2-цианоэтокси)фосфоротиоил)окси)-метил)-4-фтортетрагидрофуран-3-ила
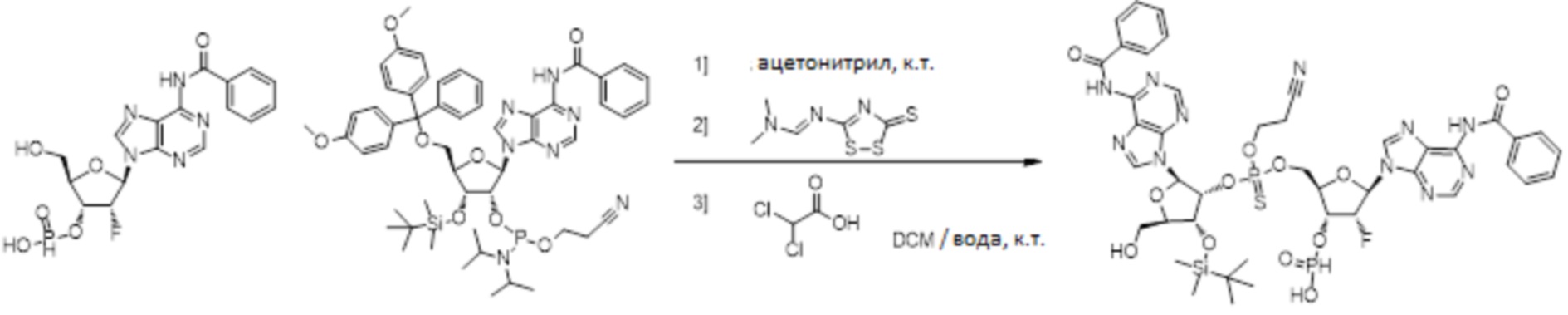
Промежуточный продукт 3 получали, в общем, в соответствии с методикой, представленной ниже. В других Примерах могут применяться небольшие модификации, например, описанные для промежуточного примера 3.
Полученный выше содержащий примеси твердый гидрофосфонат (2R,3R,4R,5R)-5-(6-бензамидо-9H-пурин-9-ил)-4-фтор-2-(гидроксиметил)тетрагидрофуран-3-ила (458 мг, 1,047 ммоль) подвергали азеотропной перегонке с безводным ацетонитрилом (3×20 мл). После последнего концентрирования, в колбе оставляли ~10 мл ацетонитрила. При комнатной температуре (2R,3R,4R,5R)-2-(6-бензамидо-9H-пурин-9-ил)-5-((бис(4-метоксифенил)(фенил)метокси)-метил)-4-((трет-бутилдиметилсилил)окси)тетрагидрофуран-3-ил-(2-цианоэтил)диизопропилфосфорамидит (1345 мг, 1,361 ммоль) сушили путем азеотропной перегонки с безводным ацетонитрилом (3×20 мл). После последнего концентрирования, в колбе оставляли ~5 мл ацетонитрила, и добавляли 3Å молекулярные сита (~40 зерен). Раствор оставляли отстаиваться с молекулярными ситами при комнатной температуре в течение 1 часа.
К осушенной суспензии гидрофосфоната (2R,3R,4R,5R)-5-(6-бензамидо-9H-пурин-9-ил)-4-фтор-2-(гидроксиметил)-тетрагидрофуран-3-ила (458 мг, 1,047 ммоль) в ацетонитриле (10 мл) при комнатной температуре в атмосфере азота шприцем добавляли осушенный раствор (2R,3R,4R,5R)-2-(6-бензамидо-9H-пурин-9-ил)-5-((бис(4-метоксифенил)(фенил)метокси)метил)-4-((трет-бутилдиметилсилил)окси)тетрагидрофуран-3-ил-(2-цианоэтил)-диизопропилфосфорамидита (1345 мг, 1,361 ммоль) в ацетонитриле (5 мл). Раствор из оранжевого становился светло-желтым. Смесь перемешивали в атмосфере азота при комнатной температуре в течение 1 часа, после чего методом LCMS обнаруживали минимальное остаточное количество исходного вещества. Затем, к реакционной смеси добавляли (E)-N,N-диметил-N'-(3-тиоксо-3H-1,2,4-дитиазол-5-ил)формимидамид (237 мг, 1,152 ммоль), и перемешивали смесь при комнатной температуре в течение 30 минут. Растворитель упаривали в условиях вакуума, остаток переносили в дихлорметан (40 мл) и воду (0,189 мл, 10,47 ммоль), а затем добавляли 2,2-дихлоруксусную кислоту (1,037 мл, 12,57 ммоль). Смесь перемешивали при комнатной температуре в течение 10 минут, после чего методом LCMS обнаруживали формирование целевого продукта. Реакционную смесь гасили добавлением пиридина (10 мл, 124 ммоль), а затем концентрировали смесь в условиях вакуума с получением содержащего примеси указанного в заголовке соединения в виде оранжевого масла. LCMS: m/z 1054 (M+H). Продукт хранили в атмосфере азота при 4°C и использовали на следующей стадии без дополнительной очистки.
Промежуточный продукт 4: N-{9-[(1R,6R,8R,9R,10R,15R,17R,18R)-17-(6-бензамидо-9H-пурин-9-ил)-18-[(трет-бутилдиметилсилил)окси]-3-(2-цианоэтокси)-9-фтор-12-оксо-12-сульфанил-3-сульфанилиден-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-8-ил]-9H-пурин-6-ил}бензамид

Раствор неочищенного (2R,3R,4R,5R)-5-(6-бензамидо-9H-пурин-9-ил)-2-((((((2R,3R,4R,5R)-2-(6-бензамидо-9H-пурин-9-ил)-4-((трет-бутилдиметилсилил)окси)-5-(гидроксиметил)тетрагидрофуран-3-ил)окси)(2-цианоэтокси)фосфоротиоил)окси)метил)-4-фтортетрагидрофуран-3-ила гидрофосфоната (1104 мг, 1,047 ммоль) в пиридине (~20 мл) и упаривали, а затем повторно растворяли в пиридине (20 мл) и концентрировали приблизительно до 10 мл. К этому раствору в атмосфере азота одной порцией добавляли 2-хлор-5,5-диметил-1,3,2-диоксафосфинан-2-оксид (677 мг, 3,67 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 30 минут, после чего методом LCMS обнаруживали расходование исходного вещества, а затем гасили реакционную смесь путем добавления воды (0,660 мл, 36,7 ммоль). Затем, добавляли 3H-бензо[c][1,2]дитиол-3-он (264 мг, 1,571 ммоль), и перемешивали смесь при комнатной температуре в течение 5 минут, после чего выливали на раствор бикарбоната натрия (4400 мг, 52,4 ммоль) в воде (160 мл). Эту смесь перемешивали в течение 5 минут, затем добавляли EtOAc (150 мл), и перемешивали смесь дополнительно в течение 10 минут. Раствор переносили в делительную воронку, и отделяли водный слой от органического. Водный слой дополнительно экстрагировали EtOAc (150 мл), затем объединенные органические слои сушили (Na2SO4), фильтровали и упаривали в условиях вакуума до оранжевого масла. Вещество хранили в атмосфере азота при 4°C в течение ночи.
После нагревания до комнатной температуры, неочищенное вещество разбавляли толуолом (20 мл) и упаривали для удаления избытка пиридина. Неочищенный продукт очищали методом хроматографии (силикагель, ISCO Teledyne Gold, 80 г). Проводили элюирование градиентом 0-10% метанола в дихлорметане в течение 15 минут, а затем изократически элюировали 10% метанола в дихлорметане в течение 5 мин. Затем, увеличивали градиент до 10-20% метанола в дихлорметане в течение 15 минут, затем изократически элюировали 20% метанола в дихлорметане в течение 10 минут. Целевые фракции объединяли и упаривали в условиях вакуума с получением указанного в заголовке соединения (570 мг, 0,342 ммоль, выход 32,6%) в виде желтого твердого вещества.
Методом LCMS обнаруживали четыре изомера [m/z 1068,5 (M+H)] в соотношении 8:4:2:1 со значениями времени удерживания 1,13, 1,23, 1,18 и 1,08 минут, соответственно. Продукт хранили в атмосфере азота при 4°C и использовали на следующей стадии без дополнительной очистки.
Промежуточный продукт 5: (1R,6R,8R,9R,10R,15R,17R,18R)-8,17-бис(6-амино-9H-пурин-9-ил)-18-[(трет-бутилдиметилсилил)-окси]-9-фтор-3,12-дисульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-диона бисаммониевая соль
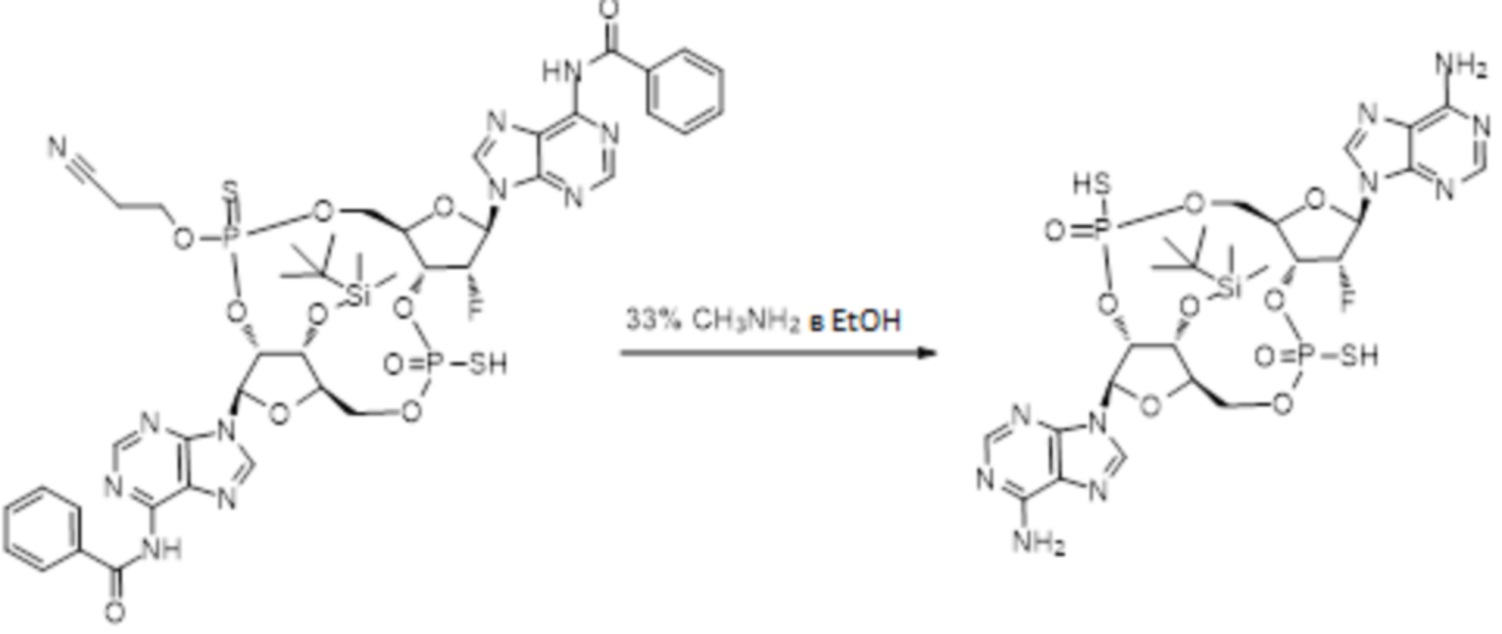
Раствор полученного выше N-{9-[(1R,6R,8R,9R,10R,15R,17R,18R)-17-(6-бензамидо-9H-пурин-9-ил)-18-[(трет-бутилдиметилсилил)окси]-3-(2-цианоэтокси)-9-фтор-12-оксо-12-сульфанил-3-сульфанилиден-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-8-ил]-9H-пурин-6-ил}бензамида (570 мг, 0,534 ммоль) в метиламине (33 масс.% в этаноле) (25 мл, 201 ммоль) перемешивали при комнатной температуре в течение 50 минут, после чего методом LCMS обнаруживали расходование исходных веществ. Реакционную смесь концентрировали в условиях вакуума до оранжевого остатка. Вещество хранили в атмосфере азота при 4°C в течение ночи.
Вещество оставляли нагреваться до комнатной температуры и растворяли в смеси метанол/DMSO (всего 4 мл). Порцию очищали методом обращенно-фазовой HPLC (10-90% ацетонитрила в воде (с 0,1% NH4OH в качестве модификатора), колонка Gemini C18 50×30 мм, 47 мл/мин, градиент 8 минут, УФ-обнаружение=214 нм). Другую порцию очищали методом обращенно-фазовой HPLC (10-50% ацетонитрила в воде (с 0,1% NH4OH в качестве модификатора), колонка Gemini C18 50×30 мм, 47 мл/мин, градиент 8 минут, УФ-обнаружение=214 нм).
Фракции, полученные в результате двух очисток, объединяли и концентрировали с получением трех изомерных продуктов:
Изомер 1 указанного в заголовке соединения в виде бисаммониевой соли с неопределенной точной стереохимией двух фосфорных центров (6 мг, чистота согласно LCMS=70%; 4,99 мкмоль, выход 0,936%) в виде не совсем белой смолы, LCMS m/z 807,2 (M+H), tRET=0,68 мин.
Изомер 2 указанного в заголовке соединения в виде бисаммониевой соли с неопределенной точной стереохимией двух фосфорных центров (64 мг, чистота согласно LCMS=22%, 0,017 ммоль, выход 3,14%) в виде бесцветной смолы, LCMS m/z 807,2 (M+H), tRET=0,80 мин.
Изомер 3 указанного в заголовке соединения в виде бисаммониевой соли с неопределенной точной стереохимией двух фосфорных центров (26 мг, чистота согласно LCMS=50%, 0,015 ммоль, выход 2,90%) в виде белого твердого вещества, LCMS m/z 807,2 (M+H), tRET=0,92 мин.
Согласно определенной по хроматограмме площади пика (УФ при 214 нм), выходящий последним изомер, изомер 3 указанного в заголовке соединения, представлял собой основной продукт, который использовали на следующей стадии снятия защиты.
Пример 8: (1R,6R,8R,9R,10R,15R,17R,18R)-8,17-бис(6-амино-9H-пурин-9-ил)-9-фтор-18-гидрокси-3,12-дисульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-диона бисаммониевая соль
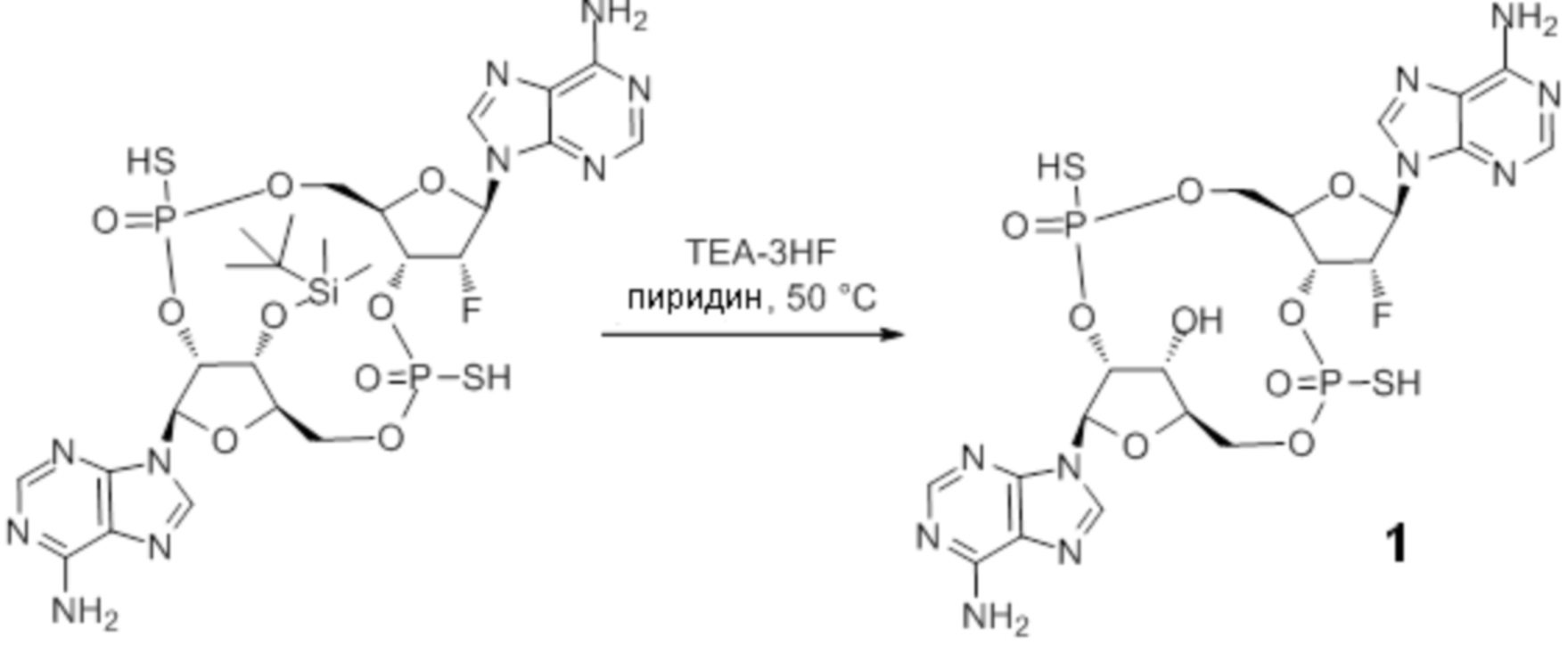
Изомер 3 (1R,6R,8R,9R,10R,15R,17R,18R)-8,17-бис(6-амино-9H-пурин-9-ил)-18-[(трет-бутилдиметилсилил)окси]-9-фтор-3,12-дисульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-диона (25 мг, 0,031 ммоль), полученный на предыдущей стадии, суспендировали в пиридине (0,5 мл) и триэтиламине (0,5 мл). Смесь перемешивали и нагревали до 50°C, затем добавляли триэтиламина тригидрофторид (0,5 мл, 3,07 ммоль), и перемешивали смесь при 50°C дополнительно в течение 2 часов, после чего методом LCMS обнаруживали полное расходование исходного вещества и преобразование до целевого продукта.
Смесь оставляли охлаждаться до комнатной температуры, затем добавляли ацетон (~10 мл), упаривали растворитель, и хранили вещество в атмосфере азота при 4°C в течение ночи.
Неочищенный остаток очищали методом обращенно-фазовой HPLC (0-20% ацетонитрил в воде (0,1% NH4OH), колонка Gemini 50×30 мм, 47 мл/мин, градиент 8 минут, УФ-обнаружение при 214 нм). Целевые фракции объединяли и упаривали с получением указанного в заголовке соединения в виде бисаммониевой соли (2,3 мг) в виде единственного стереоизомера с неопределенной точной стереохимией по двум фосфорным центрам. Продукт получали в виде белого твердого вещества. LCMS m/z 693,1 (M+H).1H-ЯМР (600 МГц, DMSO-d6 с одной каплей D2O): δ м.д. 8,69 (с, 1H), 8,33 (с, 1H), 8,14-8,17 (м, 1H), 8,12 (ушир. с, 1H), 6,24 (ушир. дд, J=14,5, 3,2 Гц, 1H), 6,08 (ушир. д, J=8,3 Гц, 1H), 5,71-5,86 (м, 1H), 5,22 (ушир. т, J=8,7 Гц, 2H), 4,49-4,54 (м, 1H), 4,32 (ушир. с, 1H), 4,16 (ушир. с, 1H), 4,08-4,15 (м, 2H), 4,03-4,06 (м, 1H), 4,00-4,03 (м, 1H), 3,73 (ушир. с, 1H).13C-ЯМР (150 МГц, DMSO-d6 с одной каплей D2O): δ м.д. 156,1, 155,9, 153,2, 152,9, 150,4, 149,1, 119,1, 118,4, 90,6, 85,3, 83,6, 83,0, 80,6, 77,7, 71,6, 71,1, 67,2, 63,3.31P-ЯМР (162 МГц, DMSO-d6 с одной каплей D2O) δ м.д. 53,84 и 49,04.
Отмечается, что в Примере 8 получалось соединение 1b - изомер 2. По причине малого масштаба реакции, соединение 1a - изомер 1 - не выделяли. Изомер 1 и изомер 2 соединения 1 получали в Примерах 8a и 8b, представленных ниже.
Примеры 8a и 8b - соединения 1a и 1b
(1R,6R,8R,9R,10R,15R,17R,18R)-8,17-бис(6-амино-9H-пурин-9-ил)-9-фтор-18-гидрокси-3,12-дисульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-диона бисаммониевая соль
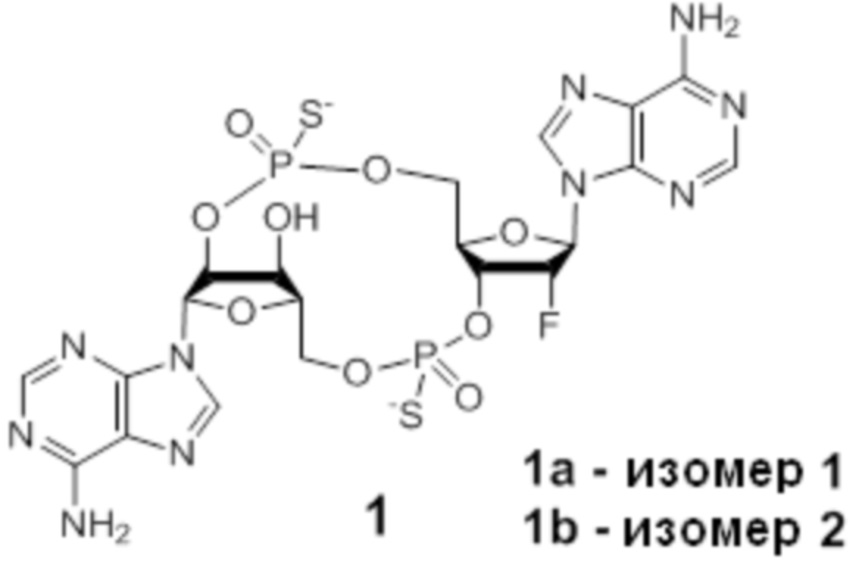
Промежуточный продукт 1: (2R,3R,4R,5R)-5-(6-бензамидо-9H-пурин-9-ил)-2-((бис(4-метоксифенил)(фенил)метокси)метил)-4-фтортетрагидрофуран-3-ила гидрофосфонат
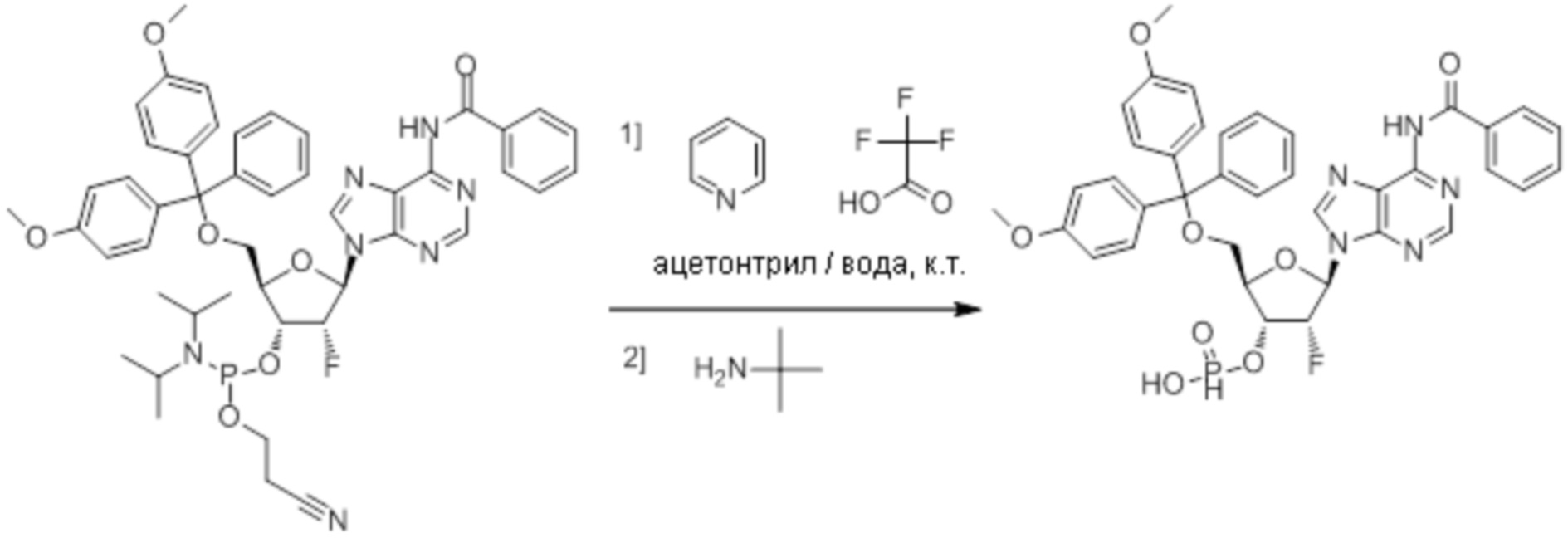
К раствору (2R,3R,4R,5R)-5-(6-бензамидо-9H-пурин-9-ил)-2-((бис(4-метоксифенил)(фенил)метокси)метил)-4-фтортетрагидрофуран-3-ила (2-цианоэтил)диизопропилфосфорамидита (10 г, 11,42 ммоль) в ацетонитриле (65 мл) и воде (0,411 мл, 22,83 ммоль) при комнатной температуре добавляли пиридин-2,2,2-трифторацетат (2,65 г, 13,70 ммоль). Смесь перемешивали в течение 1 минуты, после чего методом LCMS обнаруживали полное преобразование до первого промежуточного продукта с m/z 793,7 (M+H). Затем, добавляли 2-метилпропан-2-амин (60,0 мл, 571 ммоль), и перемешивали смесь в течение 15 минут, после чего методом LCMS обнаруживали расходование первого промежуточного продукта.
Смесь концентрировали в условиях вакуума до белой пены. Затем, пену растворяли в ацетонитриле и концентрировали (50 мл). Эту операцию повторяли еще 1 раз. Неочищенное вещество растворяли в дихлорметане и очищали методом хроматографии (силикагель, ISCO RediSep, 120 г силикагеля), элюируя градиентом 0-30% метанола в дихлорметане. Целевые фракции объединяли и упаривали в условиях вакуума с получением указанного в заголовке соединения (4,8 г, 6,49 ммоль, выход 56,8%) в виде белого твердого вещества. LCMS m/z 740,3 (M+H).
Промежуточный продукт 2: (2R,3R,4R,5R)-5-(6-бензамидо-9H-пурин-9-ил)-4-фтор-2-(гидроксиметил)тетрагидрофуран-3-ила гидрофосфонат
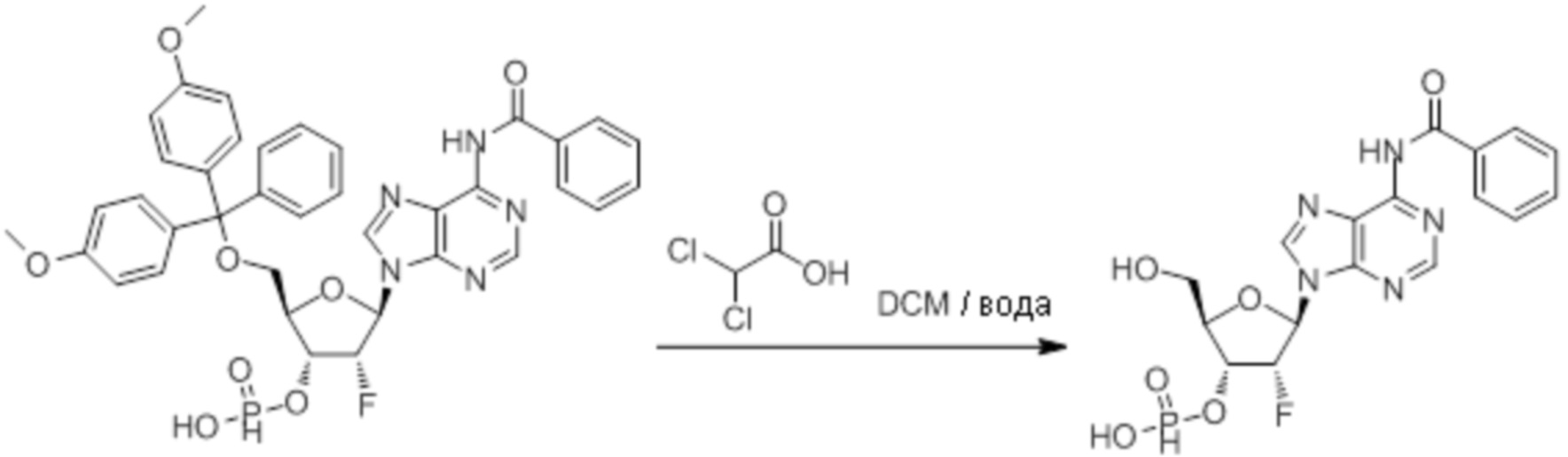
Промежуточный продукт 2 получали, в общем, в соответствии с методикой, представленной ниже. В других Примерах могут применяться небольшие модификации, например, описанные для промежуточного примера 2.
К раствору (2R,3R,4R,5R)-5-(6-бензамидо-9H-пурин-9-ил)-2-((бис(4-метоксифенил)(фенил)метокси)метил)-4-фтортетрагидрофуран-3-ила гидрофосфоната (4,8 г, 6,49 ммоль) в дихлорметане (100 мл) и воде (1,169 мл, 64,9 ммоль) при комнатной температуре добавляли 2,2-дихлоруксусную кислоту (4,06 мл, 51,9 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут. Затем, реакционную смесь гасили добавлением пиридина (8,40 мл, 104 ммоль) и концентрировали в условиях вакуума с получением содержащего примеси указанного в заголовке соединения в виде бесцветного масла. Вещество сразу использовали в полученном виде на следующей стадии. Конечную массу не определяли. LCMS m/z 438,3 (M+H).
Промежуточный продукт 3: (2R,3R,4R,5R)-5-(6-бензамидо-9H-пурин-9-ил)-2-((((((2R,3R,4R,5R)-2-(6-бензамидо-9H-пурин-9-ил)-4-((трет-бутилдиметилсилил)окси)-5-(гидроксиметил)-тетрагидрофуран-3-ил)окси)(2-цианоэтокси)фосфоротиоил)окси)-метил)-4-фтортетрагидрофуран-3-ила гидрофосфонат

Промежуточный продукт 3 получали, в общем, в соответствии с методикой, представленной ниже. В других Примерах могут применяться небольшие модификации, например, описанные для промежуточного примера 2.
Поученный выше содержащий примеси твердый гидрофосфонат (2R,3R,4R,5R)-5-(6-бензамидо-9H-пурин-9-ил)-4-фтор-2-(гидроксиметил)тетрагидрофуран-3-ила (2,84 г, 6,49 ммоль) подвергали азеотропной перегонке с безводным ацетонитрилом (3×60 мл). После последнего концентрирования, в колбе оставалось ~20 мл ацетонитрила. При комнатной температуре, (2R,3R,4R,5R)-2-(6-бензамидо-9H-пурин-9-ил)-5-((бис(4-метоксифенил)(фенил)метокси)-метил)-4-((трет-бутилдиметилсилил)окси)тетрагидрофуран-3-ила (2-цианоэтил)диизопропилфосфорамидит (8,00 г, 8,10 ммоль) сушили путем азеотропной перегонки с безводным ацетонитрилом (3×60 мл). После последнего концентрирования, в колбе оставалось ~30 мл ацетонитрила, и добавляли 3A молекулярные сита (~40 зерен). Раствор оставляли отстаиваться с молекулярными ситами при комнатной температуре в течение 1 часа.
К осушенной суспензии (2R,3R,4R,5R)-5-(6-бензамидо-9H-пурин-9-ил)-4-фтор-2-(гидроксиметил)тетрагидрофуран-3-ила гидрофосфоната (2,84 г, 6,49 ммоль) в ацетонитриле (40 мл) при комнатной температуре в атмосфере азота шприцем добавляли предварительно осушенный раствор (2R,3R,4R,5R)-2-(6-бензамидо-9H-пурин-9-ил)-5-((бис(4-метоксифенил)(фенил)метокси)метил)-4-((трет-бутилдиметилсилил)окси)тетрагидрофуран-3-ила (2-цианоэтил)диизопропилфосфорамидита (8,00 г, 8,10 ммоль) в ацетонитриле (30 мл). Смесь перемешивали в атмосфере азота при комнатной температуре в течение 1 часа, после чего методом LCMS обнаруживали минимальное остаточное количество исходного вещества. Затем, к реакционной смеси добавляли (E)-N,N-диметил-N'-(3-тиоксо-3H-1,2,4-дитиазол-5-ил)формимидамид (1,467 г, 7,14 ммоль), и перемешивали смесь при комнатной температуре в течение 45 минут. Растворитель упаривали в условиях вакуума, остаток поглощали хлорметаном (100 мл) и водой (1,170 мл, 64,9 ммоль), а затем добавляли 2,2-дихлоруксусную кислоту (6,43 мл, 78 ммоль). Смесь перемешивали при комнатной температуре в течение 10 минут, после чего методом LCMS обнаруживали формирование целевого продукта. Реакционную смесь гасили добавлением пиридина (12,61 мл, 156 ммоль), а затем концентрировали смесь в условиях вакуума с получением содержащего примеси указанного в заголовке соединения в виде оранжевого масла. Методом LCMS обнаруживали формирование двух изомеров с двумя перекрывающими пиками между 0,98-1,04 мин. Вещество в полученном виде сразу переносили на следующую стадию. Конечную массу образца не определяли. LCMS m/z 1054,6 (M+H).
Промежуточный продукт 4: N-{9-[(1R,6R,8R,9R,10R,15R,17R,18R)-17-(6-бензамидо-9H-пурин-9-ил)-18-[(трет-бутилдиметилсилил)окси]-3-(2-цианоэтокси)-9-фтор-12-оксо-12-сульфанил-3-сульфанилиден-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-8-ил]-9H-пурин-6-ил}бензамид
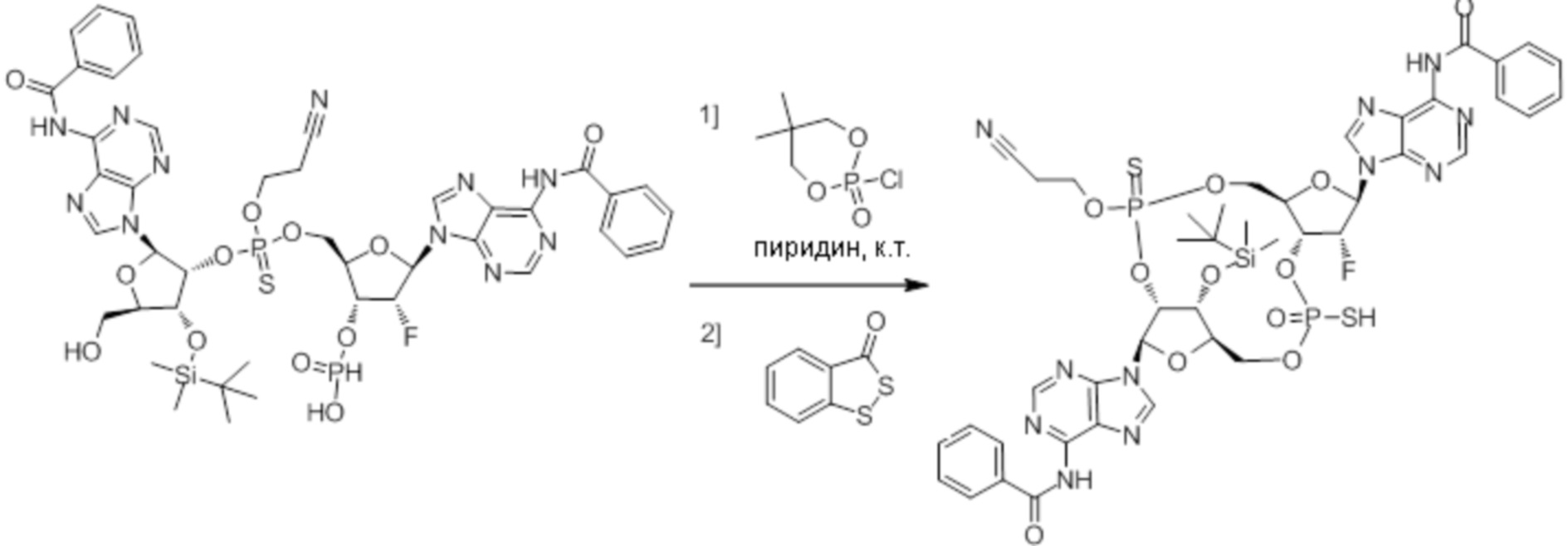
Раствор неочищенного (2R,3R,4R,5R)-5-(6-бензамидо-9H-пурин-9-ил)-2-((((((2R,3R,4R,5R)-2-(6-бензамидо-9H-пурин-9-ил)-4-((трет-бутилдиметилсилил)окси)-5-(гидроксиметил)тетрагидрофуран-3-ил)окси)(2-цианоэтокси)фосфоротиоил)окси)метил)-4-фтортетрагидрофуран-3-ила гидрофосфоната (6,84 г, 6,49 ммоль) подвергали азеотропной перегонке из пиридина (3×50 мл), оставляя пиридин (40 мл) после последнего концентрирования. К этому раствору в атмосфере азота одной порцией добавляли 2-хлор-5,5-диметил-1,3,2-диоксафосфинан-2-оксид (4,19 г, 22,71 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 30 минут, после чего методом LCMS обнаруживали расходование исходного вещества, а затем реакционную смесь гасили добавлением воды (4,09 мл, 227 ммоль). Затем, добавляли 3H-бензо[c][1,2]дитиол-3-он (1,638 г, 9,73 ммоль), и перемешивали смесь при комнатной температуре в течение 5 минут, после чего выливали в раствор бикарбоната натрия (27,3 г, 324 ммоль) в воде (400 мл). Смесь перемешивали в течение 10 минут, затем добавляли EtOAc (200 мл), и перемешивали смесь дополнительно в течение 10 минут. Раствор переносил в делительную воронку, и разделяли водный слой от органического. Водный слой дополнительно экстрагировали EtOAc (2×200 мл), затем объединенные органические слои сушили (Na2SO4), фильтровали и упаривали в условиях вакуума до оранжевого масла. Вещество хранили в атмосфере азота при 4°C до использования на следующей стадии.
После нагревания до комнатной температуры, неочищенное вещество разбавляли толуолом (50 мл) и упаривали для удаления избытка пиридина. Эту операцию повторяли еще 2 раза. Неочищенный продукт очищали методом хроматографии (силикагель, ISCO Teledyne Gold, 220 г силикагеля). Проводили элюирование градиентом 0-10% метанола в дихлорметане в течение 15 минут с последующим изократическим элюированием 10% метанолом в дихлорметане в течение 5 минут. Затем, проводили элюирование повышенным градиентом 10-20% метанола в дихлорметане в течение 15 минут с последующим изократическим элюированием 20% метанолом в дихлорметане в течение 5 минут. Целевые фракции объединяли и упаривали в условиях вакуума с получением оранжевого масла. Вещество хранили в атмосфере азота при 4°C в течение ночи.
После нагревания до комнатной температуры, вещество снова очищали методом хроматографии (силикагель, ISCO Teledyne Gold, 120 г силикагеля). Проводили элюирование градиентом 0-10% метанола в дихлорметане в течение 15 минут с последующим изократическим элюированием 10% метанолом в дихлорметане в течение 5 минут. Затем, проводили элюирование повышенным градиентом 10-20% метанола в дихлорметане в течение 15 минут с последующим изократическим элюированием 20% метанолом в дихлорметане в течение 5 минут. Целевые фракции объединяли и упаривали в условиях вакуума до содержащего примеси указанного в заголовке соединения (2,81 г) в виде желтого твердого вещества. Методом LCMS обнаруживали присутствие двух основных диастереоизомеров со значениями времени удерживания 1,09, 1,18 минут, соответственно. Также наблюдали два минорных изомера. Продукт хранили в атмосфере азота при 4°C до использования на следующей стадии без дополнительной очистки. LCMS m/z 1068 (M+H).
Промежуточный продукт 5: (1R,6R,8R,9R,10R,15R,17R,18R)-8,17-бис(6-амино-9H-пурин-9-ил)-18-[(трет-бутилдиметилсилил)-окси]-9-фтор-3,12-дисульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-диона биаммониевая соль

К раствору полученного выше N-{9-[(1R,6R,8R,9R,10R,15R,17R,18R)-17-(6-бензамидо-9H-пурин-9-ил)-18-[(трет-бутилдиметилсилил)окси]-3-(2-цианоэтокси)-9-фтор-12-оксо-12-сульфанил-3-сульфанилиден-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-8-ил]-9H-пурин-6-ил}бензамида (1,0 г, 0,936 ммоль) в этаноле (5 мл) при комнатной температуре добавляли метиламин (33 масс.% в этаноле) (20 мл, 161 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут, после чего методом LCMS обнаруживали расходование исходных веществ. Реакционную смесь концентрировали в условиях вакуума до рыжеватого остатка. Вещество переносили в DMSO и очищали методом хроматографии (обращенно-фазовая на силикагеле, RediSep Gold C18, 30 г). Проводили элюирование 100% водой (с 0,1% NH4OH в качестве модификатора) в течение 3 о.к., затем градиентом 0-15% ацетонитрила в воде (с 0,1% NH4OH в качестве модификатора) в течение 3 о.к., затем градиентом 15-25% ацетонитрила в воде (с 0,1% NH4OH в качестве модификатора) в течение 6 о.к., затем градиентом 25-90% ацетонитрила в воде (с 0,1% NH4OH в качестве модификатора) в течение 3 о.к., а затем изократически элюировали 90% ацетонитрила в воде (с 0,1% NH4OH в качестве модификатора) в течение 4 о.к.
Полученные при очистке фракции объединяли, и выделяли два основных изомерных продукта:
Изомер 2 указанного в заголовке соединения в виде бисаммониевой соли с неопределенной точной стереохимией двух фосфорных центров (28 мг, чистота согласно LCMS=54%; 0,018 ммоль, выход 1,921%) в виде бесцветного остатка, LCMS m/z 807,1 (M+H), tRET=0,80 мин.
Изомер 3 указанного в заголовке соединения в виде бисаммониевой соли с неопределенной точной стереохимией двух фосфорных центров (46 мг, чистота согласно LCMS=70%, 0,038 ммоль, выход 4,09%) в виде желтого остатка, LCMS m/z 807,2 (M+H), tRET=0,91 мин.
Примеры 8a и 8b: бисаммониевая соль (1R,6R,8R,9R,10R,15R,17R,18R)-8,17-бис(6-амино-9H-пурин-9-ил)-9-фтор-18-гидрокси-3,12-дисульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-диона
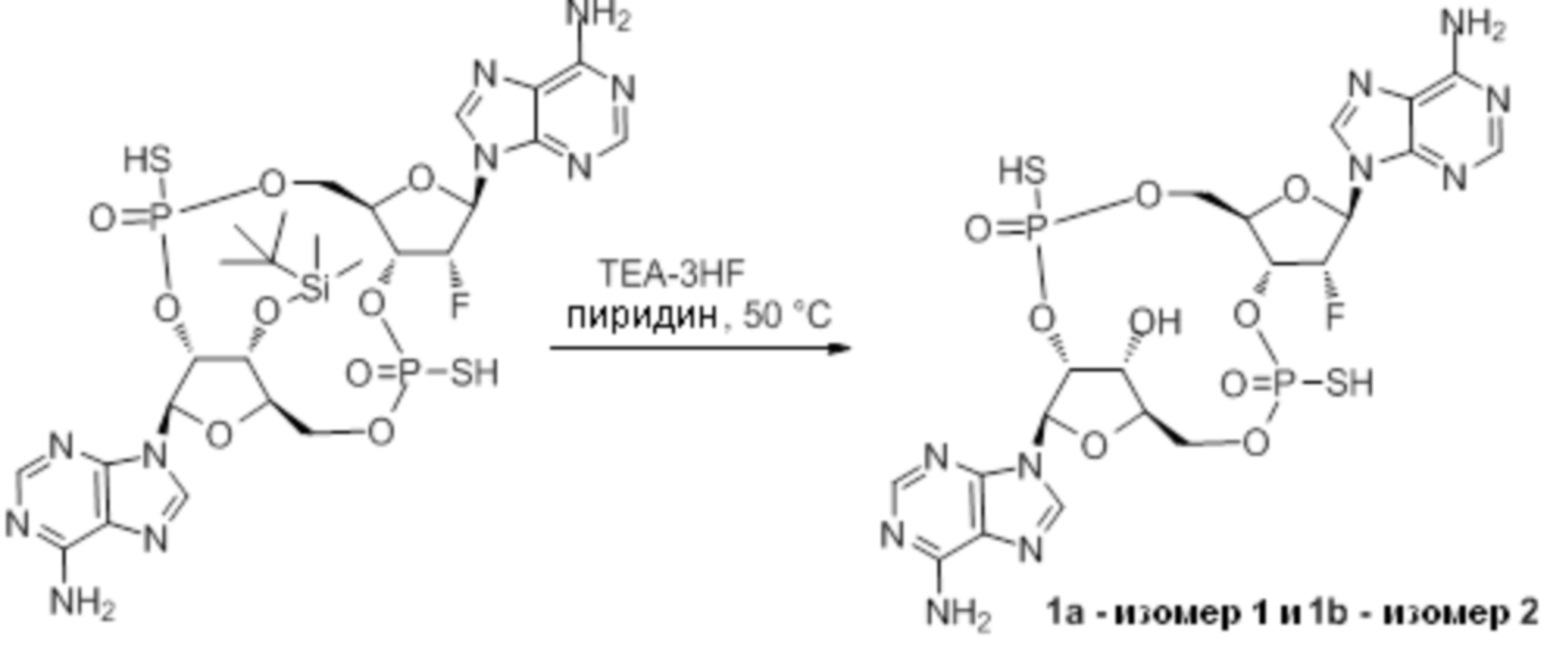
Изомер 2 промежуточного продукта 5, (1R,6R,8R,9R,10R,15R,17R,18R)-8,17-бис(6-амино-9H-пурин-9-ил)-18-[(трет-бутилдиметилсилил)окси]-9-фтор-3,12-дисульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-дион (25 мг, 0,031 ммоль), полученный на предыдущей стадии, суспендировали в пиридине (0,5 мл) и триэтиламине (0,5 мл). Смесь перемешивали и нагревали до 50°C, затем добавляли триэтиламина тригидрофторид (0,5 мл, 3,07 ммоль), и перемешивали смесь при 50°C в течение 2 часов, после чего методом LCMS обнаруживали полное расходование исходного вещества и преобразование до целевого продукта. Реакционную смесь упаривали в условиях вакуума, и хранили колбу в атмосфере азота при 4°C в течение ночи.
После нагревания до комнатной температуры, остаток переносили в воду (~5 мл), и формировался осадок. Твердые вещества отделяли из смеси, и корректировали фильтрат доo pH=10 с использованием гидроксида аммония. Раствор очищали методом обращенно-фазовой HPLC (0-5% ацетонитрил в воде (с 0,1% NH4OH в качестве модификатора), колонка Gemini C18 50×30 мм, 40 мл/мин, градиент 7 минут, УФ-обнаружение при 214 nm). Собранные твердые вещества переносили в воду (~2 мл) и метанол (~0,5 мл), и добавляли гидроксид аммония до pH=10. Раствор очищали методом обращенно-фазовой HPLC (0-5% ацетонитрил в воде (с 0,1% NH4OH в качестве модификатора), колонка Gemini C18 50×30 мм, 40 мл/мин, градиент 7 минут, УФ-обнаружение при 214 nm).
Целевые фракции из обоих процессов очистки объединяли, и дегазировали в условиях вакуума с получением бесцветного остатка. Остаток переносили в воду (2 мл) и лиофилизировали в течение ночи с получением указанного в заголовке соединения (Example 8a, 7 мг) в виде бисаммониевой соли как единственного диастереоизомера с неопределенной точной стереохимией по двум фосфорным центрам. Продукт получали в виде белого твердого вещества. LCMS m/z 693,3 (M+H). tRET=0,29 мин.1H-ЯМР (400 МГц, DMSO-d6 с одной каплей D2O) δ м.д. 8,59-8,62 (м, 1H), 8,39-8,41 (м, 1H), 8,17 (с, 1H), 8,13 (с, 1H), 6,19-6,27 (м, 1H), 6,05-6,10 (м, 1H), 5,70-5,87 (м, 1H), 5,24-5,36 (м, 1H), 5,14-5,23 (м, 1H), 4,31-4,36 (м, 1H), 4,23-4,29 (м, 1H), 4,15-4,23 (м, 1H), 4,11-4,15 (м, 1H), 3,98-4,08 (м, 1H), 3,82-3,91 (м, 1H), 3,66-3,72 (м, 1H).31P-ЯМР (162 МГц, DMSO-d6 с одной каплей D2O) δ м.д. 53,66, 55,91.19F-ЯМР (376 МГц, DMSO-d6 с одной каплей D2O) δ м.д. -206,48.
Изомер 3 промежуточного продукта 5, (1R,6R,8R,9R,10R,15R,17R,18R)-8,17-бис(6-амино-9H-пурин-9-ил)-18-[(трет-бутилдиметилсилил)окси]-9-фтор-3,12-дисульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-дион (175 мг, 0,217 ммоль), полученный из другой партии (чистота согласно LCMS=50%), суспендировали в пиридине (1 мл) и триэтиламине (1 мл). Смесь перемешивали и нагревали до 50°C, затем добавляли триэтиламина тригидрофторид (1 мл, 6,14 ммоль), и перемешивали смесь при 50°C в течение 3 часов, после чего методом LCMS обнаруживали полное расходование исходного вещества и преобразование до целевого продукта. Смесь оставляли охлаждаться до комнатной температуры, затем добавляли ацетон (~10 мл), и формировался мелкий осадок. Осадок собирали путем вакуум-фильтрования с получением серого остатка, который отбрасывали. Фильтрат упаривали в условиях вакуума, и хранили колбу в атмосфере азота при 4°C в течение ночи.
После нагревания до комнатной температуры, остаток переносили в метанол (~6 мл) и очищали методом обращенно-фазовой HPLC (0-15% ацетонитрил в воде (с 0,1% NH4OH в качестве модификатора), колонка Gemini C18 50×30 мм, 47 мл/мин, градиент 8 минут, УФ-обнаружение при 214 нм). Целевые фракции объединяли и дегазировали в условиях вакуума с получением белого твердого вещества, все еще содержащего некоторое количество примесей.
Затем, твердое вещество очищали с использованием препаративной колонки HILIC (Luna HILIC, 5 мкм, 21×250 мм, 20 мл/мин, УФ-обнаружение при 254 нм), элюируя изократической смесью 20% водного формиата аммония и 80% ацетонитрила. Целевые фракции объединяли и упаривали в условиях 90% вакуума, затем переносили в воду и ацетонитрил, и добавляли 5 капель гидроксида аммония до pH =10. Вещество замораживали и лиофилизировали в течение ночи. Эту операцию повторяли еще 2 раза с получением указанного в заголовке соединения (пример 8b, 12 мг) в виде бисаммониевой соли как единственного диастереоизомера с неопределенной точной стереохимией по двум фосфорным центрам. Продукт получали в виде белого твердого вещества. LCMS m/z 693,3 (M+H), tRET=0,37 мин.1H-ЯМР (600 МГц, DMSO-d6 с одной каплей D2O) δ м.д. 8,50-9,29 (м, 1H), 8,43 (ушир. с, 1H), 8,16 (с, 1H), 7,73-8,11 (м, 1H), 6,26 (ушир. д, J=14,4 Гц, 1H), 6,15 (ушир. д, J=7,2 Гц, 1H), 5,72 (с, 1H), 5,29-5,41 (м, 1H), 5,17-5,29 (м, 1H), 4,27-4,46 (м, 2H), 4,21 (ушир. с, 1H), 4,01-4,15 (м, 1H), 3,86-3,91 (м, 1H), 3,81-3,86 (м, 1H), 3,77 (ушир. д, J=10,6 Гц, 1H).31P-ЯМР (162 МГц, DMSO-d6 с одной каплей D2O) δ м.д. 54,27, 49,69.19F-ЯМР (376 МГц, DMSO-d6 с одной каплей D2O) δ м.д. -204,90 (ушир.)
Примеры 9a и 9b - соединения 2a и 2b
(1R,6R,8R,9R,10R,15R,17R,18R)-8,17-бис(6-амино-9H-пурин-9-ил)-9-фтор-12,18-дигидрокси-3-сульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-диона бисаммониевая соль
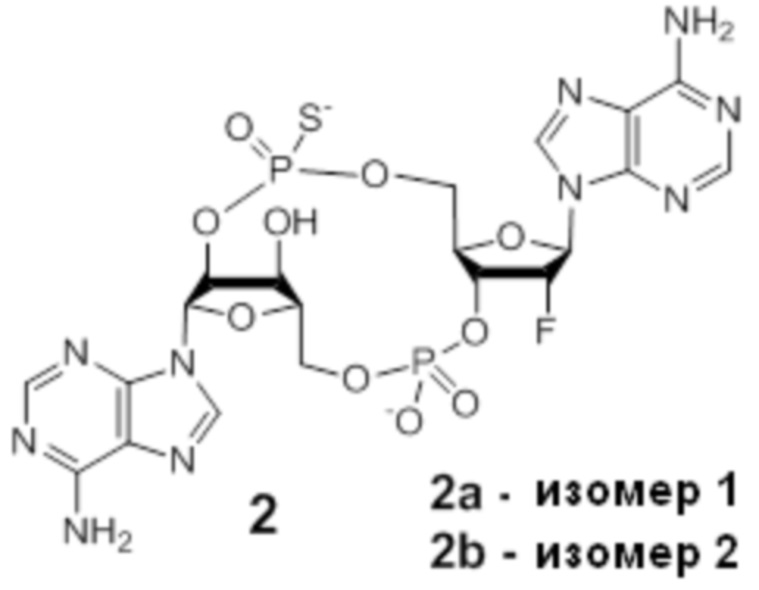
Промежуточный продукт 2: (2R,3R,4R,5R)-5-(6-бензамидо-9H-пурин-9-ил)-4-фтор-2-(гидроксиметил)тетрагидрофуран-3-ила гидрофосфонат

Промежуточный продукт 2 получали, в общем, в соответствии с методикой, представленной ниже. В других Примерах могут применяться небольшие модификации, например, описанные для промежуточного примера 2.
К находящемуся при комнатной температуре раствору (2R,3R,4R,5R)-5-(6-бензамидо-9H-пурин-9-ил)-2-((бис(4-метоксифенил)(фенил)метокси)метил)-4-фтортетрагидрофуран-3-ила (2-цианоэтил)диизопропилфосфорамидита (4,01 г, 4,6 ммоль) в ацетонитриле (35 мл) и воде (0,165 мл, 9,1 ммоль) добавляли пиридин-2,2,2-трифторацетат (1,06 г, 5,5 ммоль). Реакционную смесь перемешивали в течение 10 минут, а затем добавляли чистый трет-бутиламин (24,2 мл, 228 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут, а затем концентрировали в условиях пониженного давления. Полученную белую пену подвергали азеотропной перегонке из ацетонитрила (2×), затем сухой осадок переносили в смесь дихлорметана (100 мл) и воды (0,82 мл, 45,7 ммоль) и обрабатывали чистой 2,2-дихлоруксусной кислотой (3,01 мл, 36,5 ммоль). Спустя 30 минут, реакционную смесь гасили добавлением пиридина (5,91 мл, 73,1 ммоль), а затем концентрировали в условиях пониженного давления до маслянистой суспензии. Вещество подвергали азеотропной перегонке из ацетонитрила (3×), затем переносили в безводный ацетонитрил (60 мл) и концентрировали приблизительно до объема 20 мл с получением содержащего примеси указанного в заголовке соединения в виде светло-оранжевой суспензии. LCMS m/z 437,9 (M+H). Эту смесь использовали на следующей стадии без дополнительной очистки.
Промежуточный продукт 3: (2R,3R,4R,5R)-5-(6-бензамидо-9H-пурин-9-ил)-2-((((((2R,3R,4R,5R)-2-(6-бензамидо-9H-пурин-9-ил)-4-((трет-бутилдиметилсилил)окси)-5-(гидроксиметил)-тетрагидрофуран-3-ил)окси)(2-цианоэтокси)фосфоротиоил)окси)-метил)-4-фтортетрагидрофуран-3-ила гидрофосфонат
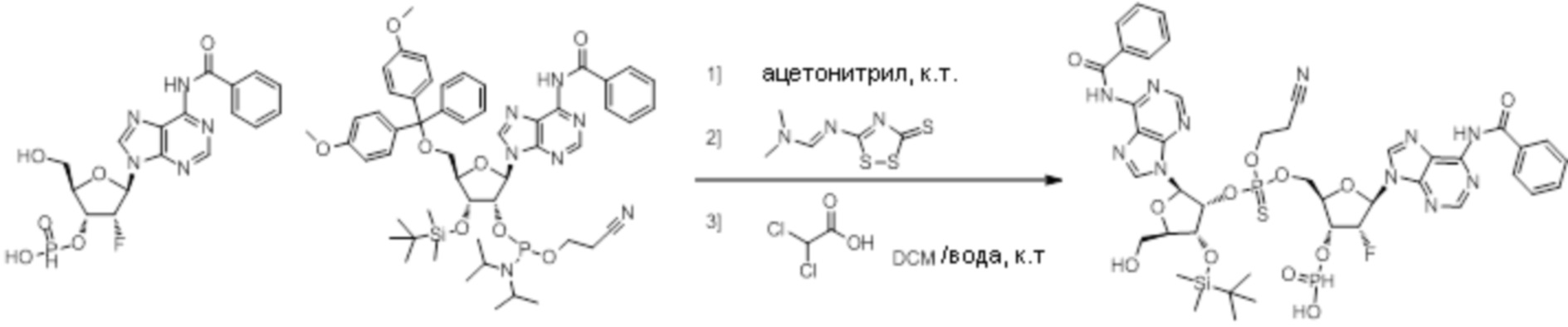
Промежуточный продукт 3 получали, в общем, в соответствии с методикой, представленной ниже. В других Примерах могут применяться небольшие модификации, например, описанные для промежуточного примера 3.
(2R,3R,4R,5R)-2-(6-Бензамидо-9H-пурин-9-ил)-5-((бис(4-метоксифенил)(фенил)метокси)метил)-4-((трет-бутилдиметилсилил)-окси)тетрагидрофуран-3-ила (2-цианоэтил)диизопропилфосфорамидит (5,9 г, 5,9 ммоль) подвергали азеотропной перегонке из ацетонитрила (2×), затем переносили в 40 мл безводного ацетонитрила, концентрировали приблизительно наполовину, а затем хранили в атмосфере азота с 3Å молекулярными ситами. Спустя 1 час, раствор добавляли в атмосфере азота к предварительно полученной неочищенной смеси (промежуточный продукт 2) (2R,3R,4R,5R)-5-(6-бензамидо-9H-пурин-9-ил)-4-фтор-2-(гидроксиметил)тетрагидрофуран-3-ила гидрофосфоната (2,0 г, 4,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, затем обрабатывали 3-(диметиламинометилиден)-амино)-3H-1,2,4-дитиазол-3-тионом (1,03 г, 5,0 ммоль), перемешивали в течение 30 минут и концентрировали в условиях пониженного давления. Остаток переносили в смесь дихлорметана (60 мл) и воды (0,823 мл, 45,7 ммоль), а затем обрабатывали 2,2-дихлоруксусной кислотой (4,5 мл, 54,8 ммоль). Смесь перемешивали при комнатной температуре в течение 15 минут, после чего гасили добавлением пиридина (25 мл, 309 ммоль) и концентрировали в условиях пониженного давления. Маслянистый концентрат подвергали азеотропной перегонке из пиридина, затем переносили в безводный пиридин (60 мл) и концентрировали в условиях пониженного давления приблизительно до 20 мл с получением содержащего примеси указанного в заголовке соединения в виде темно-оранжевого масла. LCMS m/z 1054,2 (M+H). Эту смесь использовали на следующей стадии без дополнительной очистки.
Промежуточный продукт 6: N-{9-[(1R,6R,8R,9R,10R,15R,17R,18R)-17-(6-бензамидо-9H-пурин-9-ил)-18-[(трет-бутилдиметилсилил)окси]-3-(2-цианоэтокси)-9-фтор-12-гидрокси-12-оксо-3-сульфанилиден-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-8-ил]-9H-пурин-6-ил}бензамид
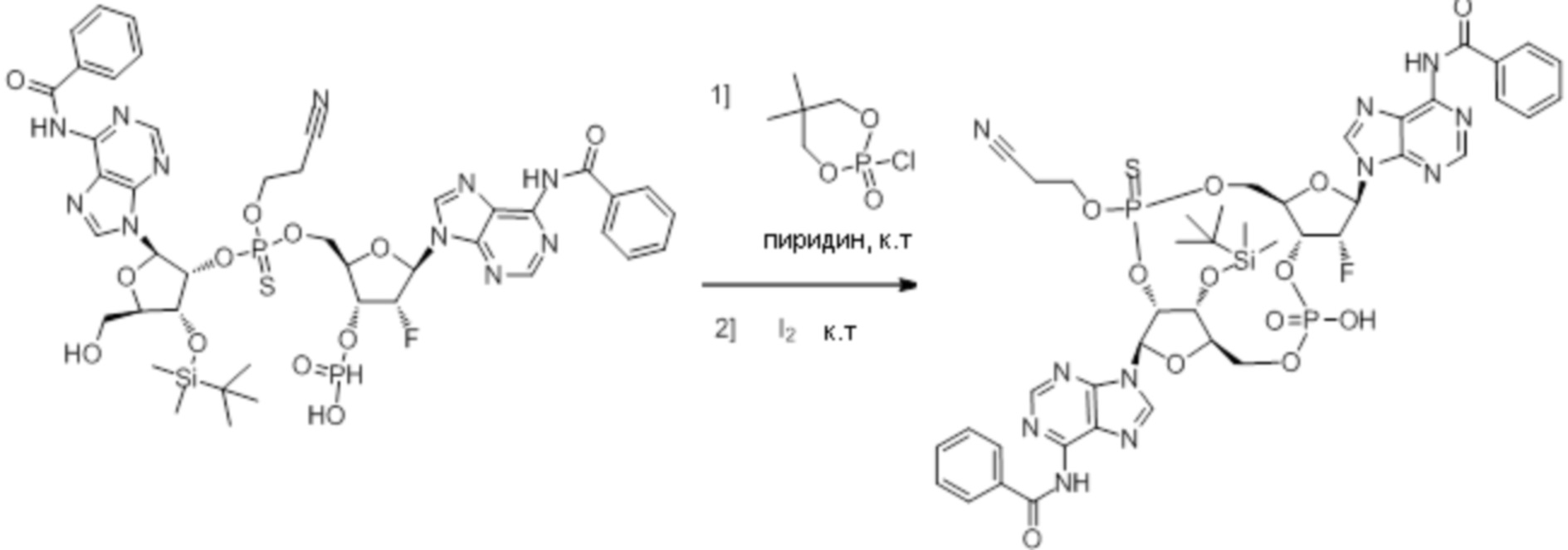
К неочищенному раствору (2R,3R,4R,5R)-5-(6-бензамидо-9H-пурин-9-ил)-2-((((((2R,3R,4R,5R)-2-(6-бензамидо-9H-пурин-9-ил)-4-((трет-бутилдиметилсилил)окси)-5-(гидроксиметил)-тетрагидрофуран-3-ил)окси)(2-цианоэтокси)фосфоротиоил)окси)-метил)-4-фтортетрагидрофуран-3-ила гидрофосфоната (промежуточный продукт 3, 4,8 г, 4,6 ммоль) в пиридине (20 мл) в атмосфере азота добавляли 2-хлор-5,5-диметил-1,3,2-диоксафосфинан-2-оксид (3,0 г, 16,0 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 30 минут, затем гасили добавлением воды (2,9 мл, 160 ммоль), а затем йодом (1,5 г, 5,9 ммоль). Спустя 10 минут, смесь вливали в раствор бисульфита натрия (0,95 г, 9,1 ммоль) в воде (300 мл). Спустя 5 минут, реакционную смесь обрабатывали порциями твердым бикарбонатом натрия (19,2 г, 229 ммоль). Полученную рыжеватую суспензию экстрагировали EtOAc (3×200 мл), затем экстракты промывали насыщенным водным бикарбонатом натрия, сушили над Na2SO4 и концентрировали до получения масла. Затем, для очистки продукта из разных партий использовали один из двух следующих способов очистки.
Способ A: Масло подвергали азеотропной перегонке из толуола для удаления избытка пиридина, а затем очищали методом хроматографии на силикагеле (Biotage 100 г), элюируя повышающимися градиентами: 0-10% MeOH в DCM (10 мин), 10% MeOH в DCM (10 минут), 10-20% MeOH в DCM (10 мин) и, в завершение, 20-40% MeOH в DCM (10 мин). Представляющие интерес фракции, идентифицированные методом LCMS, объединяли и концентрировали с получением содержащего примеси указанного в заголовке соединения (1,03 г, 0,979 ммоль) в виде светло-оранжевого твердого вещества. Методом LCMS обнаруживали два изомера [m/z 1052,3 (M+H)] в приблизительном соотношении 1:1 со значениями времени удерживания 1,00, 1,09 минут, соответственно. Продукт хранили в атмосфере азота при 4°C и использовали на следующей стадии без дополнительной очистки.
Method B: Масло подвергали азеотропной перегонке из толуола для удаления избытка пиридина, а затем очищали методом хроматографии на силикагеле (Teledyne ISCO Gold 120 г), элюируя повышающимися градиентами: 100% DCM (5 мин), 0-10% MeOH в DCM (5 мин), 10% MeOH в DCM (10 мин) и 10-40% MeOH в DCM (20 мин). Представляющие интерес фракции объединяли и концентрировали с получением смеси диастереоизомеров в приблизительном соотношении 1:1 в виде темно-желтого твердого вещества. Смесь диастереоизомеров разделяли методом обращенно-фазовой HPLC (колонка Gemini C-18 30×50 мм; 45-60% CH3CN с 0,1% TFA/вода с 0,1% TFA), прогон 12 мин, обнаружение при 214 нм. Представляющие интерес фракции объединяли, обрабатывали насыщенным водным бикарбонатом натрия, а затем концентрировали для удаления ацетонитрила. Затем, водные концентраты экстрагировали EtOAc. Осушенные (над Na2SO4) экстракты концентрировали с получением отдельных диастереоизомеров.
Диастереоизомер 1 указанного в заголовке соединения (42 мг) в виде белого твердого вещества с неопределенной точной стереохимией хиральных фосфорных центров. LCMS m/z 1052,7 (M+H), tRET=1,00 мин.
Диастереоизомер 2 указанного в заголовке соединения (43 мг) в виде белого твердого вещества с неопределенной точной стереохимией хиральных фосфорных центров. LCMS m/z 1052,7 (M+H). tRET=1,09 мин.
Примеры 9a и 9b: (1R,6R,8R,9R,10R,15R,17R,18R)-8,17-бис(6-амино-9H-пурин-9-ил)-9-фтор-12,18-дигидрокси-3-сульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-диона бисаммониевая соль
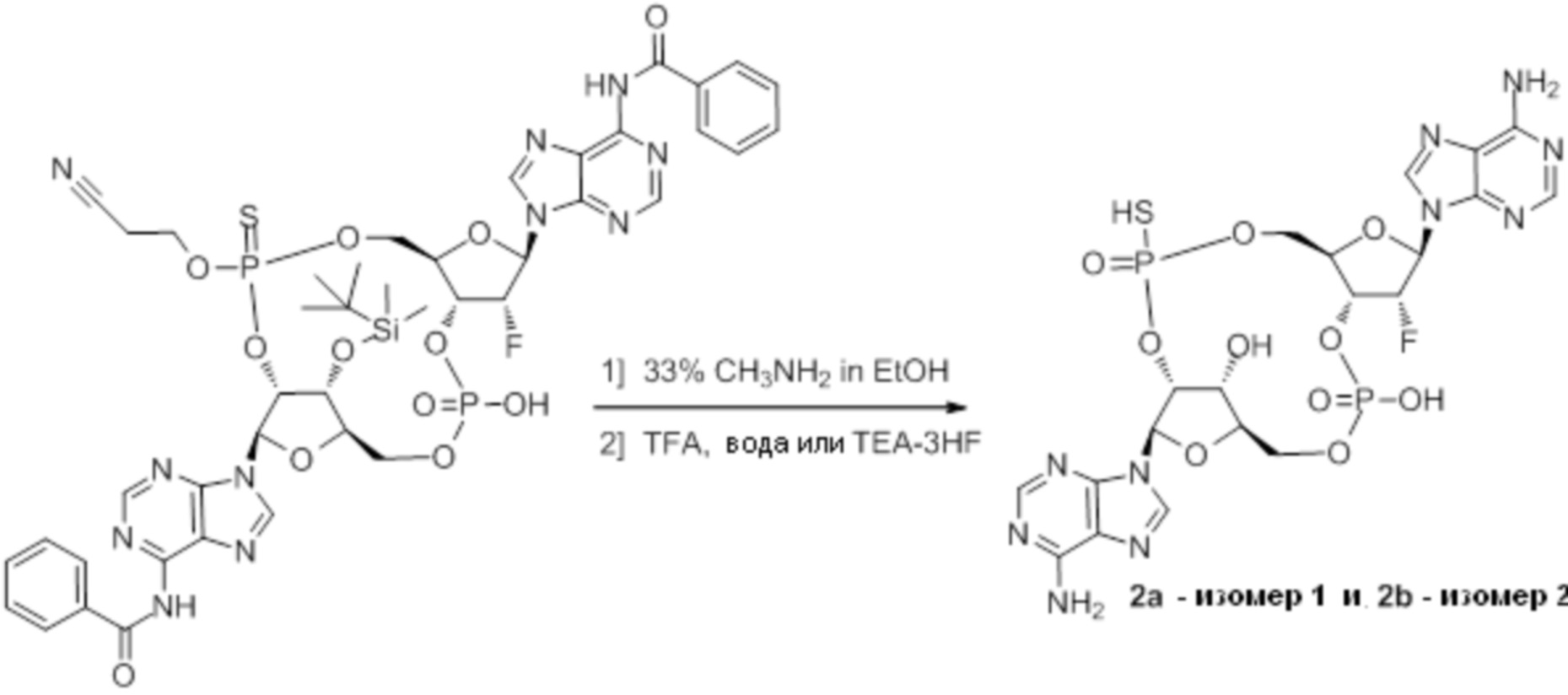
раствор промежуточного продукта 6 (очищенного способом A), N-{9-[(1R,6R,8R,9R,10R,15R,17R,18R)-17-(6-бензамидо-9H-пурин-9-ил)-18-[(трет-бутилдиметилсилил)окси]-3-(2-цианоэтокси)-9-фтор-12-гидрокси-12-оксо-3-сульфанилиден-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-8-ил]-9H-пурин-6-ил}бензамида (1,03 г, 0,979 ммоль) в 33 масс.% метиламина в EtOH (40 мл, 321 ммоль) перемешивали при комнатной температуре в течение 4 часов, после чего методом LCMS обнаруживали полное преобразование исходного вещества и наличие целевого O-TBS-защищенного промежуточного продукта. Для промежуточного продукта обнаруживали только один слегка уширенный пик, указывая на присутствие, по меньшей мере, одного диастереоизомера. Реакционную смесь концентрировали до темно-оранжевого остатка, который очищали методом обращенно-фазовой HPLC (колонка Gemini C-18 30×50 мм; 10-60% ацетонитрил с 0,1% TFA/вода с 0,1% TFA), градиент 12 минут, обнаружение при 254 нм. Представляющие интерес фракции объединяли и концентрировали в условиях пониженного давления. В этот момент времени, методом LCMS обнаруживали утрату силил-защитной группы. Водную фазу дополнительно концентрировали приблизительно до 3-5 мл, а затем добавляли метанол (25 мл). Полученную суспензию фильтровали, твердые вещества промывали MeOH, затем диэтиловым эфиром, а затем сушили аспирированием с получением 75 мг содержащего примеси целевого продукта-дитрифторацетата в виде белого твердого вещества. LCMS m/z 677,2 (M+H).
Продукт затем очищали методом препаративной хроматографии (Luna HILIC 3u: 4,6×150 мм; изократическое элюирование 20% 30 мМ водного HCO2NH4+80% CH3CN). Представляющие интерес фракции объединяли и концентрировали до остатка, который лиофилизировали из воды (5 мл) и 3 капель гидроксида аммония). Для удаления остаточного формиата аммония, процесс лиофилизирования повторяли еще 4 раза с получением указанного в заголовке соединения (Пример 9a, 50 мг) в виде бисаммониевой соли как единственного диастереоизомера с неопределенной точной стереохимией хиральных фосфорных центров. Указанный в заголовке продукт получали в виде белого твердого вещества. LCMS m/z 677,6 (M+H). tRET=0,11 мин.1H-ЯМР (600 МГц, DMSO-d6 с одной каплей D2O): δ м.д. 8,48 (с, 1H), 8,39 (с, 1H), 8,17 (с, 1H), 8,12 (с, 1H), 6,23 (дд, J=15,1, 3,0 Гц, 1H), 6,09 (д, J=8,3 Гц, 1H), 5,69 (ушир. дт, J=52,1, 3,4 Гц, 1H), 5,16 (ддд, J=8,1,6,6, 4,2 Гц, 1H), 5,02-5,10 (м, 1H), 4,35 (д, J=4,2 Гц, 1H), 4,24 (ушир. с, 1H), 4,11-4,18 (м, 1H), 4,09 (ушир. с, 1H), 3,97 (ушир. д, J=10,6 Гц, 1H), 3,89-3,95 (м, 1H), 3,72 (ушир. д, J=12,5 Гц, 1H).13C-ЯМР (150 МГц DMSO-d6, с одной каплей D2O): δ м.д. 156,0, 155,8, 153,0, 152,8, 150,3, 148,9, 119,1, 118,4, 92,4, 85,4, 84,0, 83,3, 81,0, 77,9, 72,3, 71,4, 65,9, 62,6.31P-ЯМР (162 МГц, DMSO-d6, с одной каплей D2O) δ м.д. 55,67 и -2,51.19F-ЯМР (376 МГц, DMSO-d6, с одной каплей D2O) δ м.д. -205,16.
Отмечается, что пример 9a может быть также получен с использованием методики, описанной ниже для Примера 9b.
Раствор диастереоизомера 2 промежуточного продукта 6 (очищенного способом B), N-{9-[(1R,6R,8R,9R,10R,15R,17R,18R)-17-(6-бензамидо-9H-пурин-9-ил)-18-[(трет-бутилдиметилсилил)окси]-3-(2-цианоэтокси)-9-фтор-12-гидрокси-12-оксо-3-сульфанилиден-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-8-ил]-9H-пурин-6-ил}бензамида (43 мг, 0,041 ммоль), в 33 масс.% метиламине в EtOH (8,0 мл, 64,3 ммоль) перемешивали при комнатной температуре в течение 30 минут, а затем концентрировали. Полученный темно-оранжевый остаток переносили в безводный пиридин (0,50 мл) и триэтиламин (0,50 мл), нагретые 50°C, а затем обрабатывали триэтиламина тригидрофторидом (0,50 мл, 3,07 ммоль). Спустя 1 час, реакция завершалась. Смесь охлаждали и концентрировали до темного масла, которое переносили в воду (7,5 мл) и гидроксид аммония (10 капель). Полученную суспензию (pH~3) фильтровали, твердые вещества растворяли в смеси воды (2 мл) и гидроксила аммония (1 мл), а затем очищали методом обращенно-фазовой HPLC (колонка Gemini C-18: 30×50 мм; 0-10% ацетонитрил в воде с 0,1% NH4OH; 214 нм). Представляющие интерес фракции объединяли и концентрировали в условиях пониженного давления до влажного остатка, который переносили в воду (5 мл) и 5 капель гидроксида аммония, а затем лиофилизировали с получением указанного в заголовке соединения (Пример 9b, 7,0 мг) в виде бисаммониевой соли как единственного диастереоизомера с неопределенной точной стереохимией хиральных фосфорных центров. Продукт получали в виде белого твердого вещества. LCMS m/z 677,2 (M+H), tRET=0,32 мин.1H-ЯМР (600 МГц, DMSO-d6 с одной каплей D2O): δ м.д. 8,65-9,35 (м, 1H), 8,44 (ушир. с, 1H), 7,77-8,29 (м, 2H), 6,25 (ушир. д, J=14,4 Гц, 1H), 6,11-6,19 (м, 1H), 5,53-5,73 (м, 1H), 5,18-5,44 (м, 1H), 4,96-5,08 (м, 1H), 4,40-4,54 (м, 1H), 4,33 (ушир. с, 2H), 4,23-4,30 (м, 1H), 4,16 (ушир. с, 1H), 3,98-4,06 (м, 1H), 3,81 (ушир. с, 1H).31P-ЯМР (162 МГц, DMSO-d6 с одной каплей D2O) δ м.д. 49,48 и -2,94.19F-ЯМР (376 МГц, DMSO-d6 с одной каплей D2O) δ м.д. -206,44 (ушир.).
Примеры 10a и 10b - соединения 28a и 28b
(1R,6R,8R,9R,10R,15R,17R,18R)-17-(2-амино-6-оксо-6,9-дигидро-1H-пурин-9-ил)-8-(6-амино-9H-пурин-9-ил)-9-фтор-12,18-дигидрокси-3-сульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-дион

Промежуточный продукт 7: (2R,3R,4R,5R)-5-(6-бензамидо-9H-пурин-9-ил)-2-((((((2R,3R,4R,5R)-4-((трет-бутилдиметилсилил)-окси)-5-(гидроксиметил)-2-(2-изобутирамидо-6-оксо-1H-пурин-9(6H)-ил)тетрагидрофуран-3-ил)окси)(2-цианоэтокси)фосфоротиоил)-окси)метил)-4-фтортетрагидрофуран-3-ила гидрофосфонат
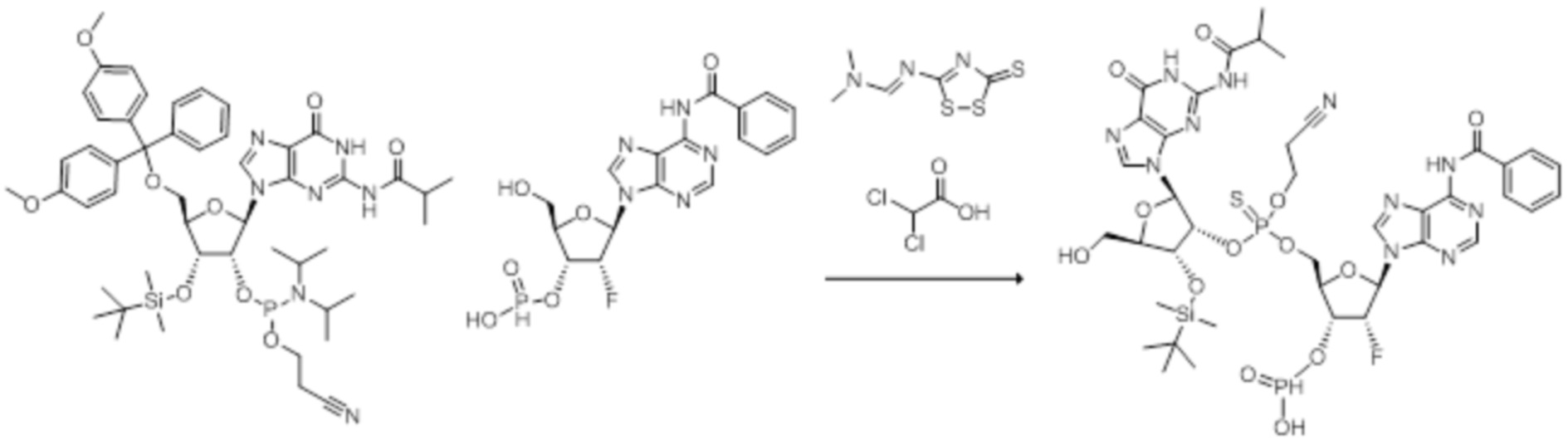
(2R,3R,4R,5R)-5-((бис(4-метоксифенил)(фенил)метокси)метил)-4-((трет-бутилдиметилсилил)окси)-2-(2-изобутирамидо-6-оксо-1H-пурин-9(6H)-ил)тетрагидрофуран-3-ила (2-цианоэтил)-диизопропилфосфорамидит (4,0 г, 4,12 ммоль) трижды подвергали азеотропной перегонке с ацетонитрилом (20 мл). После последнего концентрирования, в реакционной колбе оставляли ~15 мл ацетонитрила, и добавляли к прозрачному раствору 3Å молекулярные сита (~20 зерен). Раствор оставляли отстаиваться с молекулярными ситами в атмосфере азота в течение ~1 часа.
В отдельной круглодонной колбе, к ранее приготовленной неочищенной смеси (промежуточный продукт 2) гидрофосфоната (2R,5R)-5-(6-бензамидо-9H-пурин-9-ил)-4-фтор-2-(гидроксиметил)-тетрагидрофуран-3-ила (1,5 г, 3,43 ммоль) в ацетонитриле (10 мл) шприцем добавляли предварительной осушенный полученный выше (2R,3R,4R,5R)-5-((бис(4-метоксифенил)(фенил)метокси)метил)-4-((трет-бутилдиметилсилил)окси)-2-(2-изобутирамидо-6-оксо-1H-пурин-9(6H)-ил)тетрагидрофуран-3-ила (2-цианоэтил)-диизопропилфосфорамидит в ацетонитриле (~15 мл). После 30 минут перемешивания при комнатной температуре, добавляли (E)-N,N-диметил-N'-(3-тиоксо-3H-1,2,4-дитиазол-5-ил)формимидамид (DDTT) (780 мг, 3,80 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут, а затем упаривали ацетонитрил в условиях вакуума. Затем, к остатку добавляли дихлорметан (DCM) (50 мл) и воду (650 мкл), а затем добавляли 2,2-дихлоруксусную кислоту (3,5 мл, 42,4 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут, после чего гасили добавлением пиридина (20 мл). Смесь концентрировали в условиях вакуума с получением содержащего примеси указанного в заголовке соединенияв виде оранжевого масла. LCMS m/z 1036,2 (M+H). Неочищенный продукт использовали на следующей стадии без дополнительной очистки.
Промежуточный продукт 8: N-{9-[(1R,6R,8R,9R,10R,15R,17R,18R)-18-[(трет-бутилдиметилсилил)-окси]-3-(2-цианоэтокси)-9-фтор-12-гидрокси-17-[2-(2-метилпропанамидо)-6-оксо-6,9-дигидро-1H-пурин-9-ил]-12-оксо-3-сульфанилиден-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-8-ил]-9H-пурин-6-ил}бензамид
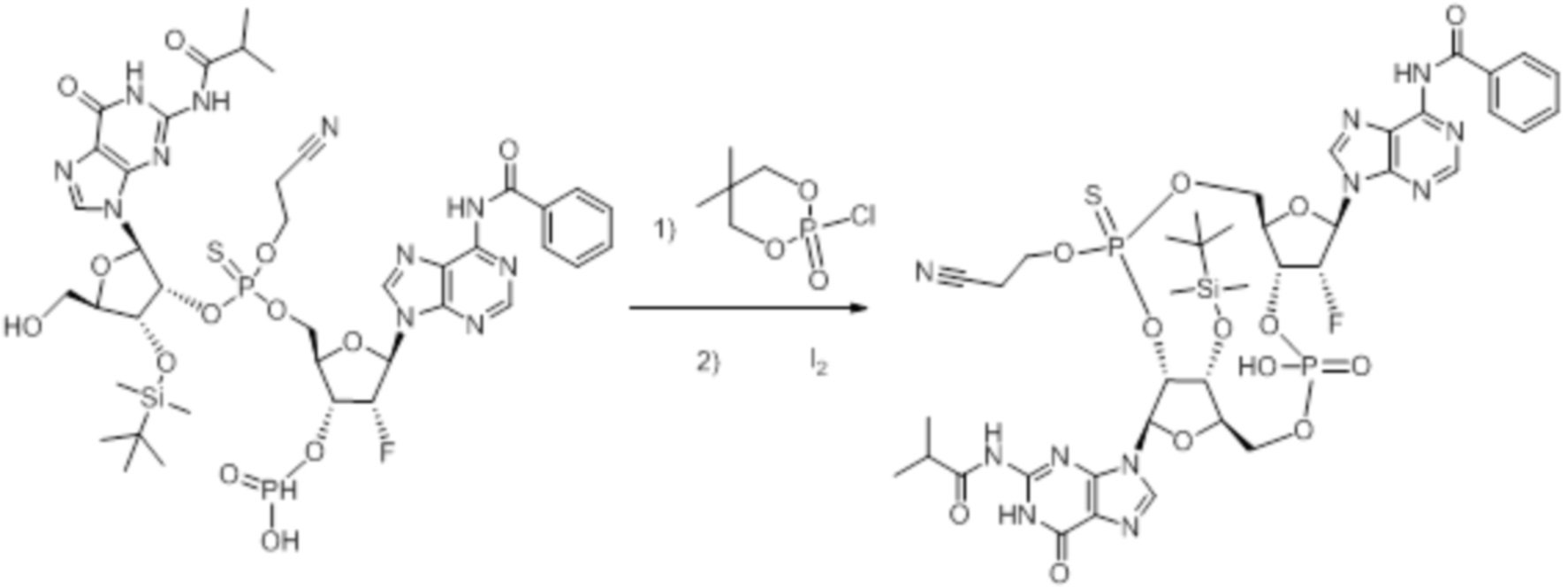
К неочищенному раствору (2R,3R,4R,5R)-5-(6-бензамидо-9H-пурин-9-ил)-2-((((((2R,3R,4R,5R)-4-((трет-бутилдиметилсилил)-окси)-5-(гидроксиметил)-2-(2-изобутирамидо-6-оксо-1H-пурин-9(6H)-ил)тетрагидрофуран-3-ил)окси)(2-цианоэтокси)фосфоротиоил)-окси)метил)-4-фтортетрагидрофуран-3-ила гидрофосфоната (промежуточный продукт 7, 3,55 г, 3,43 ммоль) в пиридине (60 мл) добавляли 2-хлор-5,5-диметил-1,3,2-диоксафосфинан-2-оксид (DMOCP) (2,2 г, 11,92 ммоль), и перемешивали смесь в атмосфере азота при комнатной температуре в течение 30 минут. Реакционную смесь гасили добавлением воды (2,2 мл, 10 эквив. к DMOCP), а затем добавляли йод (1,2 г, 4,73 ммоль). Смесь перемешивали в течение 10 минут, затем вливали в раствор бисульфита натрия (NaHSO3) (1,0 г, 9,61 ммоль) в воде (400 мл). После 5 минут перемешивания, порциями медленно добавляли бикарбонат натрия (NaHCO3) (14,4 г, 171 ммоль) в виде твердого вещества (Осторожно: выделение газа). Продукт экстрагировали смесью диэтиловый эфир/EtOAc (1/1, 2×300 мл), объединенные экстракты сушили над сульфатом натрия, фильтровали и концентрировали в условиях вакуума. Избыток пиридина удаляли путем концентрирования с толуолом (2×100 мл). Неочищенное вещество очищали методом хроматографии на силикагеле (колонка 100 г) с использованием градиентного 0-20% MeOH в DCM, а затем изократического 20% MeOH в DCM, элюирования до выхода всего целевого продукта с колонки. Целевые фракции объединяли и концентрировали с получением двух изомерных продуктов:
Изомер 1 указанного в заголовке соединения, более полярный, в виде содержащей примеси смеси (1,39 г, чистота согласно LCMS ~33% с содержанием ~28% изомера 2). LCMS m/z 1034,1 (M+H), tRET=0,98 мин.
Изомер 2 указанного в заголовке соединения, менее полярный, в виде содержащей примеси смеси (230 мг, чистота согласно LCMS ~ 33%). LCMS m/z 1034,2 (M+H), tRET=1,09 мин.
Промежуточные продукты 9a и 9b: (1R,6R,8R,9R,10R,15R,17R,18R)-17-(2-амино-6-оксо-6,9-дигидро-1H-пурин-9-ил)-8-(6-амино-9H-пурин-9-ил)-18-[(трет-бутилдиметилсилил)окси]-9-фтор-12-гидрокси-3-сульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-дион
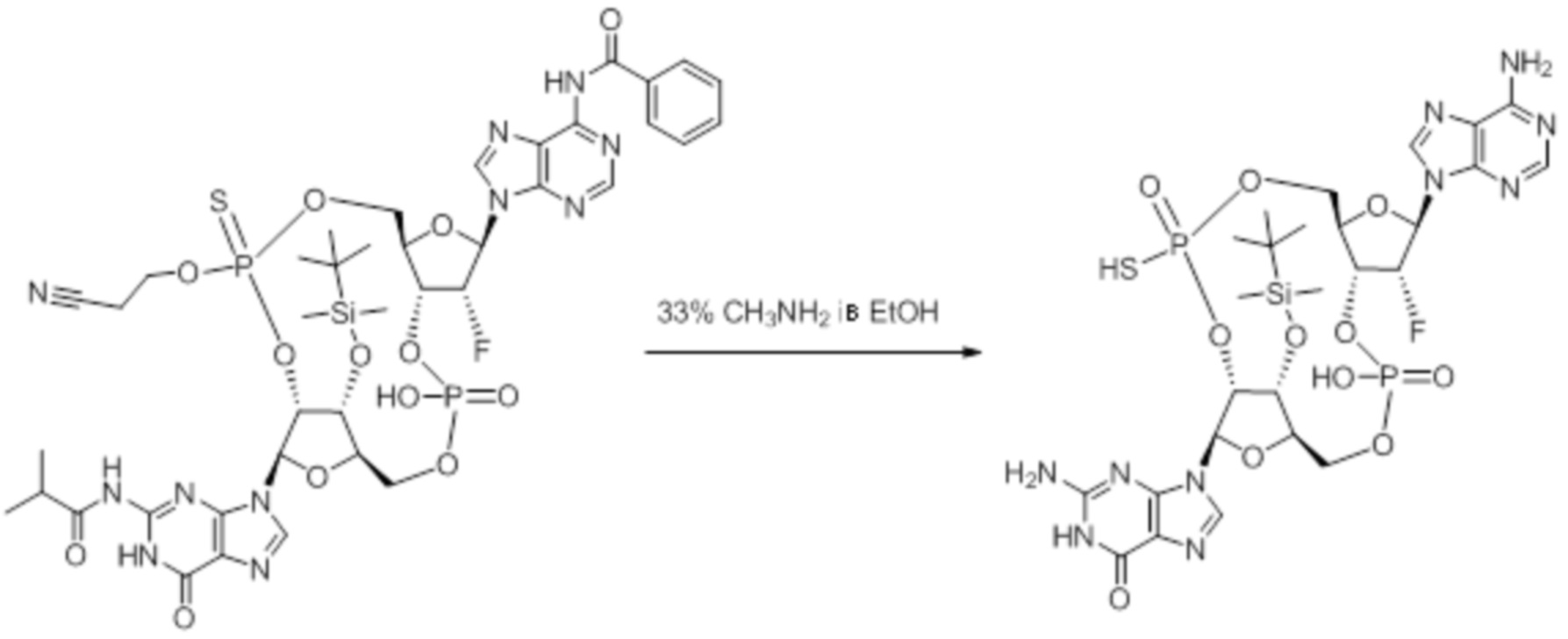
Изомер 1 промежуточного продукта 8, N-{9-[(1R,6R,8R,9R,10R,15R,17R,18R)-18-[(трет-бутилдиметилсилил)окси]-3-(2-цианоэтокси)-9-фтор-12-гидрокси-17-[2-(2-метилпропанамидо)-6-оксо-6,9-дигидро-1H-пурин-9-ил]-12-оксо-3-сульфанилиден-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-8-ил]-9H-пурин-6-ил}бензамид (1,39 г, 1,34 ммоль), перемешивали в метанамине (33 масс.% в EtOH) (10,0 мл, 80 ммоль) в атмосфере азота при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали, и очищали остаток методом обращенно-фазовой HPLC с использованием градиента 10-50% ACN в H2O (0,1% TFA) с получением изомера 1 указанного в заголовке соединения (промежуточный продукт 9a, с примесями, 280 мг) в виде рыжеватого твердого вещества. LCMS m/z 807,1 (M+H), tRET=0,80 мин.
Следуя методике получения промежуточного продукта 9a, изомер 2 промежуточного продукта 8, N-{9-[(1R,6R,8R,9R,10R,15R,17R,18R)-18-[(трет-бутилдиметилсилил)-окси]-3-(2-цианоэтокси)-9-фтор-12-гидрокси-17-[2-(2-метилпропанамидо)-6-оксо-6,9-дигидро-1H-пурин-9-ил]-12-оксо-3-сульфанилиден-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-8-ил]-9H-пурин-6-ил}бензамид (230 мг, 0,22 ммоль), перемешивали в метанамине (33 масс.% в EtOH) (2,0 мл, 16 ммоль) в атмосфере азота при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали, и очищали остаток методом обращенно-фазовой HPLC с использованием градиента 10-50% ACN в H2O (0,1% TFA), с получением изомера 2 указанного в заголовке соединения (промежуточный продукт 9b, с примесями, 60 мг) в виде рыжеватого твердого вещества. LCMS m/z 807,1 (M+H), tRET=0,85 мин.
Примеры 10a и 10b: (1R,6R,8R,9R,10R,15R,17R,18R)-17-(2-амино-6-оксо-6,9-дигидро-1H-пурин-9-ил)-8-(6-амино-9H-пурин-9-ил)-9-фтор-12,18-дигидрокси-3-сульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-дион
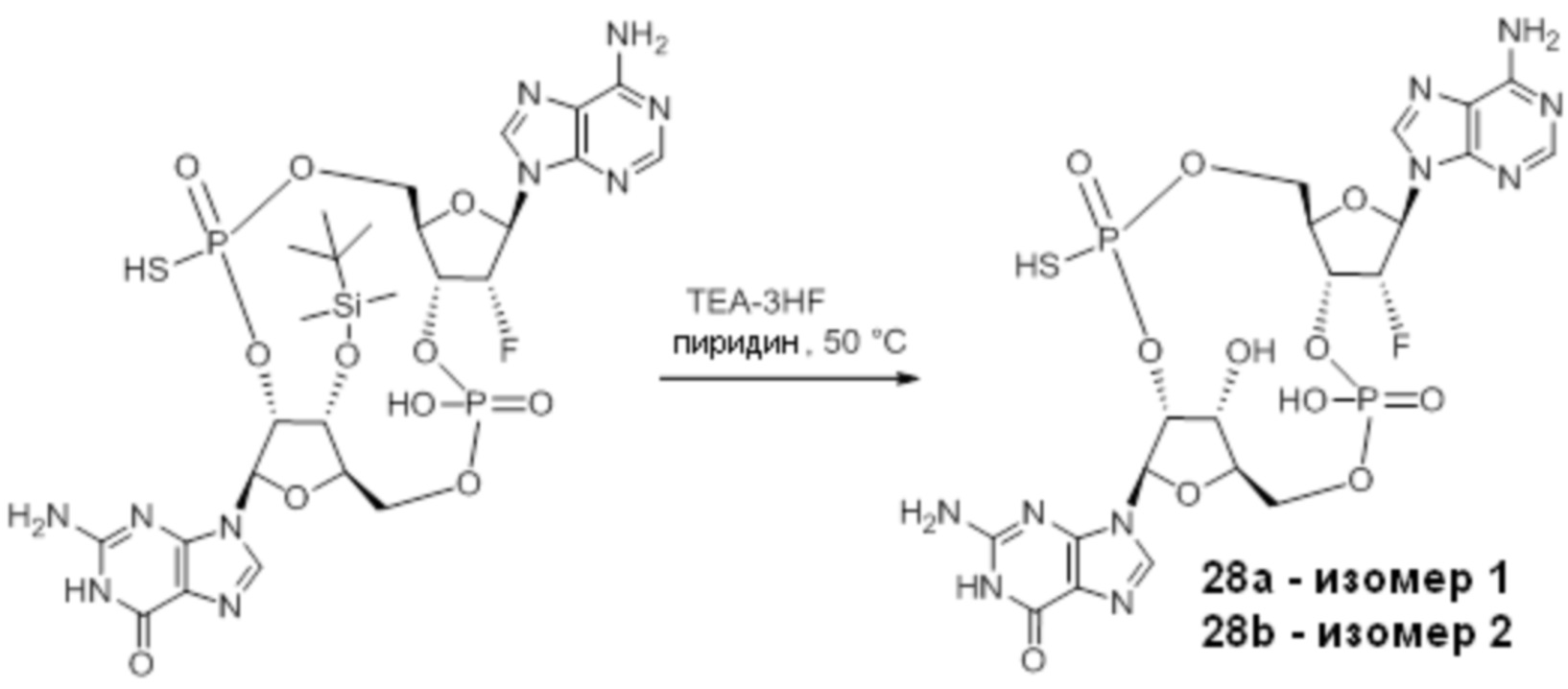
К суспензии изомера 1 (1R,6R,8R,9R,10R,15R,17R,18R)-17-(2-амино-6-оксо-6,9-дигидро-1H-пурин-9-ил)-8-(6-амино-9H-пурин-9-ил)-18-[(трет-бутилдиметилсилил)окси]-9-фтор-12-гидрокси-3-сульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-диона (промежуточный продукт 9a, 280 мг, 0,35 ммоль) в пиридине (2 мл) и триэтиламине (2 мл) при 50°C добавляли триэтиламина тригидрофторид (1,5 мл, 9,21 ммоль). Смесь перемешивали при 50°C в течение 4 часов. Методом LCMS обнаруживали некоторое количество все еще не израсходованного исходного вещества, и оставляли реакционную смесь перемешиваться при комнатной температуре в течение 16 часов. После этого, реакционную смесь разбавляли ацетоном (~25 мл), и формировался осадок. После 15 минут перемешивания, реакционную смесь фильтровали. Твердое вещество промывали ацетоном (~10 мл) и сушили. Фильтрат концентрировали в условиях вакуума, а затем добавляли толуол для удаления оставшегося пиридина. Неочищенный фильтрат и отфильтрованное твердое вещество по отдельности очищали методом обращенно-фазовой HPLC с использованием градиента 0-20% ACN в H2O (0,1% NH4OH) и объединяли. На спектрах19F-ЯМР наблюдали остаточное количество TFA, присутствующей в образцах. После второй очистки методом обращенно-фазовой HPLC с использованием градиента 0-10% ACN в H2O (0,1% NH4OH) получали указанное в заголовке соединение (Пример 10a, 4 мг) в виде бисаммониевой соли как единственного диастереоизомера с неопределенной точной стереохимией хиральных фосфорных центров. Продукт получали в виде белого твердого вещества. LCMS m/z 693,0 (M+H). tRET=0,11 мин.1H-ЯМР (600МГц, DMSO-d6 с одной каплей D2O): δ м.д. 8,30 (с, 1H), 8,19 (с, 1H), 7,97 (с, 1H), 6,26 (дд, J=15,9, 2,3 Гц, 1H), 5,86 (д, J=8,3 Гц, 1H), 5,59-5,76 (м, 1H), 5,30 (ушир. с, 1H), 5,06 (ушир. д, J=15,5 Гц, 1H), 4,35 (д, J=3,8 Гц, 1H), 4,25 (ушир. с, 1H), 4,02-4,07 (м, 1H), 4,01-4,13 (м, 2H), 3,88-3,99 (м, 2H).19F-ЯМР (376 МГц, DMSO-d6 с одной каплей D2O) δ м.д. -203,83.31P-ЯМР (162 МГц, DMSO-d6 с одной каплей D2O) δ м.д. 55,73, -2,66.
Следуя методике получения Примера 10a, из изомера 2 (1R,6R,8R,9R,10R,15R,17R,18R)-17-(2-амино-6-оксо-6,9-дигидро-1H-пурин-9-ил)-8-(6-амино-9H-пурин-9-ил)-18-[(трет-бутилдиметилсилил)окси]-9-фтор-12-гидрокси-3-сульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-диона (промежуточный продукт 9b, 60 мг, 0,35 ммоль) получали указанное в заголовке соединение (Пример 10b, 7 мг) в виде бисаммониевой соли как единственного диастереоизомера с неопределенной точной стереохимией хиральных фосфорных центров. Продукт получали в виде белого твердого вещества. LCMS m/z 693,0 (M+H), tRET=0,37 мин.1H-ЯМР (400МГц, DMSO-d6 с одной каплей D2O): δ м.д. 8,50 (ушир. с, 1H), H) 8,22 (ушир. с, 1H), 7,94 (ушир. с, 1H), 6,35 (д, J=14 Гц, 1H), 5,87 (д, J=7,86 Гц, 1H), 5,54-5,67 (м, 1H), 4,98 (ушир. д, J=15,5 Гц, 1H), 4,39 (ушир. с, 1H), 4,33 (д, J=6,84 Гц, 1H), 4,24 (ушир. с, 1H), 4,13 (ушир. с, 1H), 4,00-4,10 (м, 2H), 3,89-3,98 (м, 2H).19F-ЯМР (376 МГц, DMSO-d6 с одной каплей D2O) δ м.д. -205,00.31P-ЯМР (162 МГц, DMSO-d6 с одной каплей D2O) δ м.д. 49,15, -2,90.
Примеры 11a и 11b - соединения 27a и 27b
(1R,6R,8R,9R,10R,15R,17R,18R)-17-(2-амино-6-оксо-6,9-дигидро-1H-пурин-9-ил)-8-(6-амино-9H-пурин-9-ил)-9-фтор-18-гидрокси-3,12-дисульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-диона бисаммониевая соль
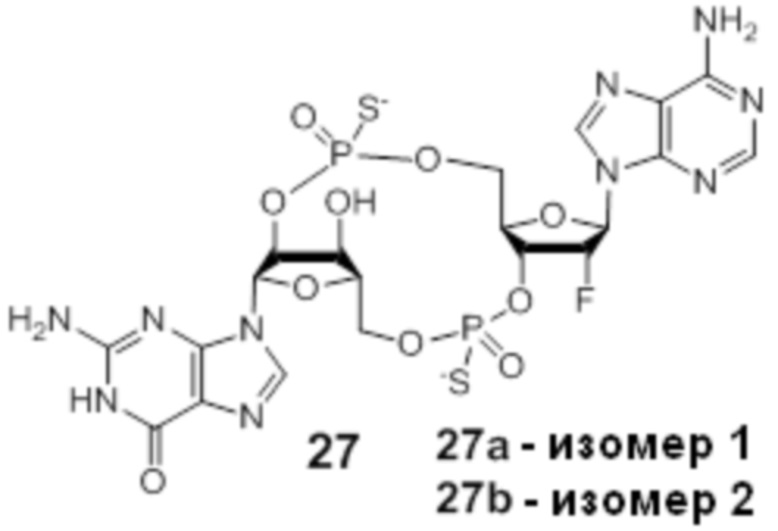
Промежуточный продукт 10: N-{9-[(1R,6R,8R,9R,10R,15R,17R,18R)-18-[(трет-бутилдиметилсилил)-окси]-3-(2-цианоэтокси)-9-фтор-17-[2-(2-метилпропанамидо)-6-оксо-6,9-дигидро-1H-пурин-9-ил]-12-оксо-12-сульфанил-3-сульфанилиден-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-8-ил]-9H-пурин-6-ил}
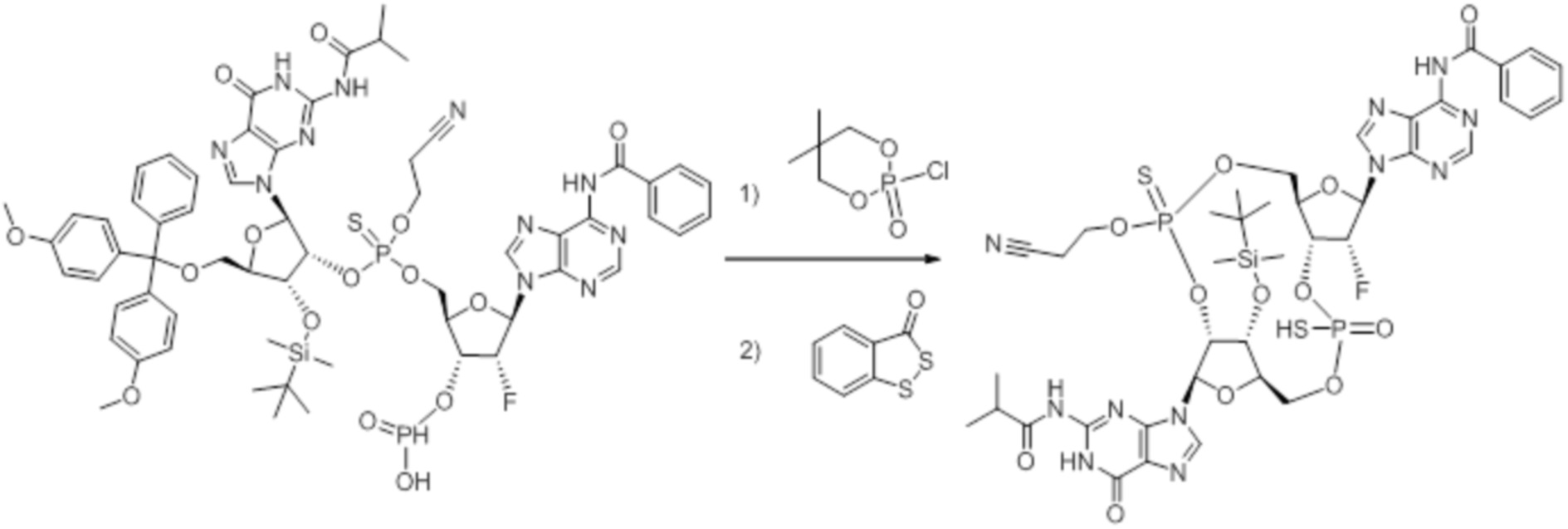
К неочищенному раствору (2R,5R)-5-(6-бензамидо-9H-пурин-9-ил)-2-((((((2R,5R)-4-((трет-бутилдиметилсилил)окси)-5-(гидроксиметил)-2-(2-изобутирамидо-6-оксо-1H-пурин-9(6H)-ил)тетрагидрофуран-3-ил)окси)(2-цианоэтокси)фосфоротиоил)окси)-метил)-4-фтортетрагидрофуран-3-ила гидрофосфоната (промежуточный продукт 7, 2,58 г, 2,49 ммоль) в пиридине (50 мл) добавляли 2-хлор-5,5-диметил-1,3,2-диоксафосфинан-2-оксид (DMOCP) (1,70 г, 9,21 ммоль), и перемешивали смесь в атмосфере азота при комнатной температуре в течение 30 минут. Затем, реакционную смесь гасили добавлением воды (1,6 мл, 89 ммоль), а затем добавляли 3H-бензо[c][1,2]дитиол-3-он (660 мг, 3,92 ммоль). Смесь перемешивали в течение 10 минут, затем вливали в химический стакан с водой (350 мл) и бикарбонатом натрия (NaHCO3) (10 г, 119 ммоль). Желтую взвесь перемешивали в течение 10 минут, а затем переносили в делительную воронку. Продукт экстрагировали смесью диэтиловый эфир/EtOAc (1/1, 2×300 мл). Объединенные экстракты сушили над сульфатом натрия, фильтровали и концентрировали в условиях вакуума. Неочищенное вещество очищали методом хроматографии на силикагеле (колонка 100 г), элюируя 0-10% MeOH в DCM, а затем выдерживания при 10% MeOH в DCM. Целевые фракции объединяли и концентрировали с получением содержащего примеси указанного в заголовке соединения (1,1 г) в виде рыжеватого твердого вещества. Согласно LCMS, из смеси выбивали два основных продукта с выходом ~77%. LCMS m/z 1050,1 (M+H), tRET=1,09 и 1,20 мин, соответственно.
Промежуточный продукт 11: (1R,6R,8R,9R,10R,15R,17R,18R)-17-(2-амино-6-оксо-6,9-дигидро-1H-пурин-9-ил)-8-(6-амино-9H-пурин-9-ил)-18-[(трет-бутилдиметилсилил)окси]-9-фтор-3,12-дисульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-дион
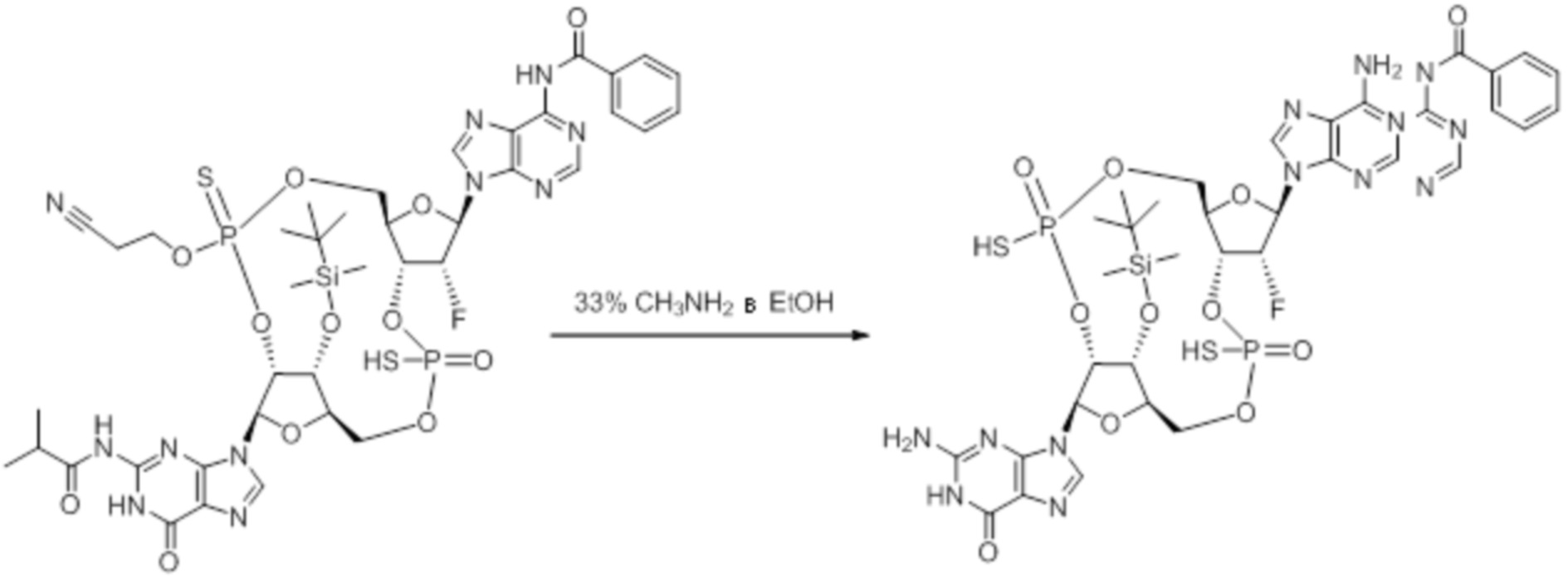
Содержащий примеси N-{9-[(1R,6R,8R,9R,10R,15R,17R,18R)-18-[(трет-бутилдиметилсилил)окси]-3-(2-цианоэтокси)-9-фтор-17-[2-(2-метилпропанамидо)-6-оксо-6,9-дигидро-1H-пурин-9-ил]-12-оксо-12-сульфанил-3-сульфанилиден-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-8-ил]-9H-пурин-6-ил}бензамид (промежуточный продукт 10, 0,77 г, 0,73 ммоль) перемешивали в метанамине (33 масс.% в EtOH) (10,0 мл, 80 ммоль) в атмосфере азота при комнатной температуре в течение 2 часов. Методом LCMS неочищенной смеси обнаруживали 4 изомера ((M+H)+=823) с tRET=0,74, 0,83, 0,90 и 0,94 мин, в грубом соотношении 20:3:13:19 (однако соотношение может существенно измениться вследствие возможных перекрываний с пиками примесей). Летучие вещества удаляли в условиях вакуума. Неочищенное вещество очищали методом обращенно-фазовой HPLC с использованием градиента 10-60% ACN в H2O (0,1% TFA). Разделяли два основных изомера.
Изомер 1 указанного в заголовке соединения (120 мг, чистота 64% согласно LCMS+8% продукта без TBS-защиты) в виде рыжеватого твердого вещества с неопределенной точной стереохимией двух фосфорных центров. LCMS m/z 823,1 (M+H). tRET=0,74 мин.
Изомер 2 указанного в заголовке соединения (130 мг, чистота 50% согласно LCMS+18% продукта без TBS-защиты) в виде рыжеватого твердого вещества с неопределенной точной стереохимией двух фосфорных центров. LCMS m/z 823,1 (M+H). tRET=0,96-1,00 мин в виде уширенного пика с хвостом.
Примеры 11a и 11b: (1R,6R,8R,9R,10R,15R,17R,18R)-17-(2-амино-6-оксо-6,9-дигидро-1H-пурин-9-ил)-8-(6-амино-9H-пурин-9-ил)-9-фтор-18-гидрокси-3,12-дисульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-диона бисаммониевая соль
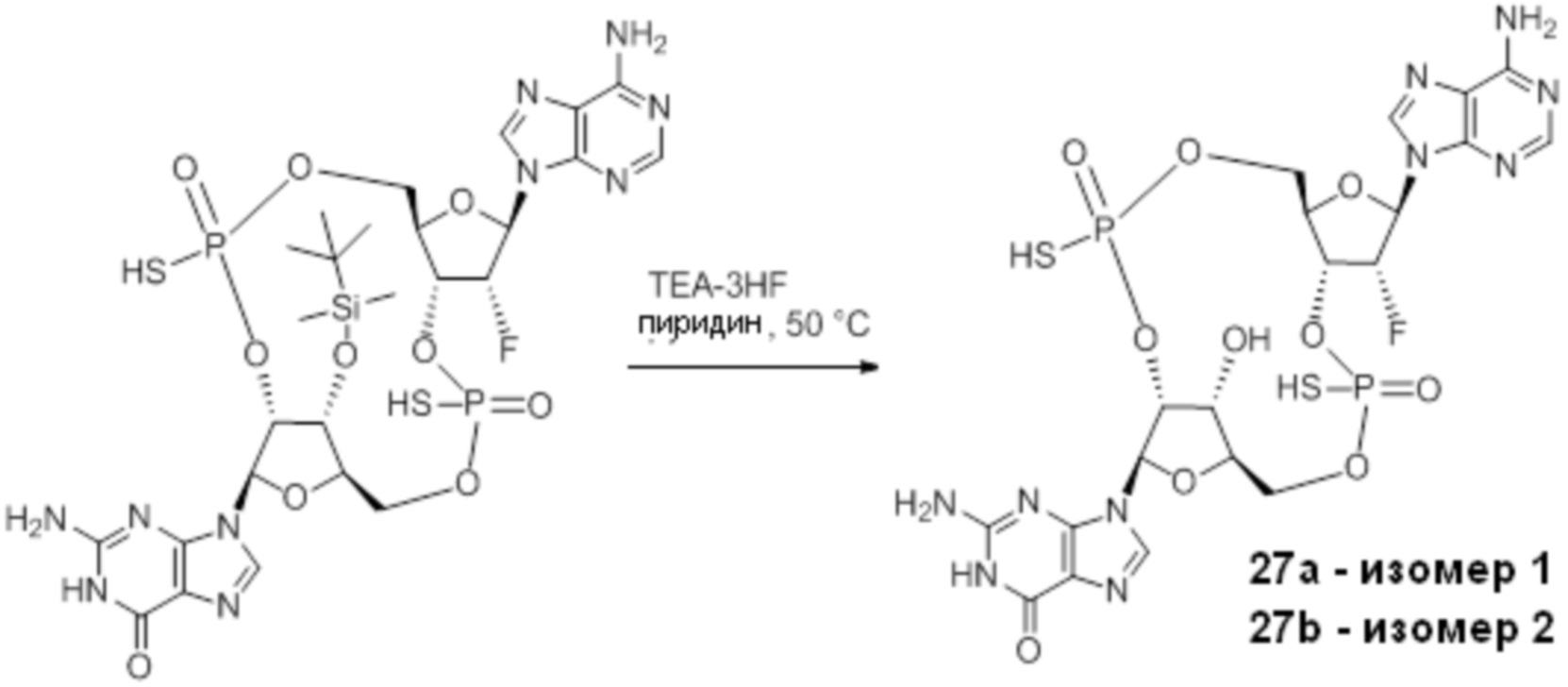
К суспензии изомера 1 промежуточного продукта 11, (1R,6R,8R,9R,10R,15R,17R,18R)-17-(2-амино-6-оксо-6,9-дигидро-1H-пурин-9-ил)-8-(6-амино-9H-пурин-9-ил)-18-[(трет-бутилдиметилсилил)окси]-9-фтор-3,12-дисульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-диона (120 мг, 0,146 ммоль), в пиридине (2 мл) и триэтиламине (2 мл) при 50°C добавляли триэтиламина тригидрофторид (700 мкл, 4,30 ммоль), смесь перемешивали и нагревали при 50°C в течение 4 часов. Методом LCMS обнаруживали, что все еще оставалось некоторое количество исходного вещества. Реакционную смесь оставляли перемешиваться в течение 16 часов при комнатной температуре. Затем, добавляли ацетон (~25 мл), и в осадок выпадало твердое вещество. Смесь оставляли при комнатной температуре на ~30 минут, фильтровали и промывали ацетоном. Согласно LCMS, отфильтрованное твердое вещество не содержало целевого продукта, и его отбрасывали. Фильтрат, содержащий целевой продукт, концентрировали в условиях вакуума, и добавляли толуол для удаления оставшегося пиридина. Остаток очищали методом обращенно-фазовой HPLC с использованием градиента 0-10% ACN в H2O (0,1% NH4OH) с получением продукта, который не был очень чистым и, предположительно, был загрязнен трифторацетатом. Поэтом, твердое вещество поглощали ~2 мл воды и добавляли несколько капель 30% водного NH4OH. Затем, очищали методом обращенно-фазовой HPLC с использованием градиента 0-10% ACN в H2O (0,1% NH4OH) с получением указанного в заголовке соединения (Пример 11a, 13 мг) в виде бисаммониевой соли как единственного диастереоизомера с неопределенной точной стереохимией двух фосфорных центров. Продукт получали в виде белого твердого вещества. LCMS m/z 708,9 (M+H). tRET=0,17 мин.1H-ЯМР (600 МГц, DMSO-d6 с одной каплей D2O): δ м.д. 8,31 (с, 1H), 8,21 (с, 1H), 8,11 (ушир. с, 1H), 6,25 (дд, J=15,1, 2,6 Гц, 1H), 5,84 (д, J=8,3 Гц, 1H), 5,68 (д, J=51,7 Гц, 1H), 5,27-5,37 (м, 1H), 5,16-5,25 (м, 1H), 4,32 (д, J=4,2 Гц, 1H), 4,26 (ушир. с, 1H), 4,01-4,17 (м, 3H), 3,90-3,96 (м, 1H), 3,81 (ушир. д, J=11,7 Гц, 1H).19F-ЯМР (376 МГц, DMSO-d6 с одной каплей D2O) δ м.д. -205,30 (ушир.).31P-ЯМР (162 МГц, DMSO-d6 с одной каплей D2O) δ м.д. 55,77, 54,01.
Следуя методике получения Примера 11a, за исключением того, что при первой очистке использовали градиент 0-20% ACN в H2O (0,1% NH4OH), из изомера 2 промежуточного продукта 11, (1R,6R,8R,9R,10R,15R,17R,18R)-17-(2-амино-6-оксо-6,9-дигидро-1H-пурин-9-ил)-8-(6-амино-9H-пурин-9-ил)-18-[(трет-бутилдиметилсилил)окси]-9-фтор-3,12-дисульфанил-2,4,7,11,13,16-гексаокса-3λ5,12λ5-дифосфатрицикло[13.2.1.06,10]октадекан-3,12-диона (130 мг, 0,158 ммоль), получали указанное в заголовке соединение (Пример 11b, 16 мг) в виде бисаммониевой соли как единственного диастереоизомера с неопределенной точной стереохимией двух фосфорных центров. Продукт получали в виде белого твердого вещества. LCMS m/z 708,9 (M+H). tRET=0,42 мин.1H-ЯМР (600МГц, DMSO-d6 с одной каплей D2O): δ м.д. 8,22 (ушир. с, 1H), 8,18 (с, 1H), 8,05 (ушир. с, 1H), 6,27 (дд, J=15,3, 2,1 Гц, 1H), 5,82 (ушир. д, J=8,3 Гц, 1H), 5,60 (д, J=49,9 Гц, 1H), 5,27-5,46 (м, 1H), 5,12-5,27 (м, 1H), 4,42-4,59 (м, 1H), 4,30 (ушир. с, 1H), 4,14 (ушир. д, J=2,3 Гц, 1H), 4,11 (ушир. д, J=5,7 Гц, 2H), 4,06 (ушир. д, J=9,1 Гц, 1H), 3,82 (ушир. д, J=11,0 Гц, 1H).19F-ЯМР (376 МГц, DMSO-d6 с одной каплей D2O) δ м.д. -205,05.31P-ЯМР (162 МГц, DMSO-d6 с одной каплей D2O) δ м.д. 53,85, 47,48.
Сокращения
В последующем перечне представлены определения конкретных сокращений, использованных в настоящем документе. Следует понимать, что перечень не является исчерпывающим, а значения тех сокращений, которые не определены ниже в этом документе, будут хорошо известны специалистам в данной области техники.
DCM - дихлорметан
DMF - N,N-диметилформамид
DMSO - диметилсульфоксид
DMTr - диметокситритил
THF - тетрагидрофуран
EtOAc - этилацетат
MeOH - метанол
EtOH - этанол
MeCN - ацетонитрил
HCl - соляная кислота
HPLC - высокоэффективная жидкостная хроматография
MDAP - контролируемая по массе автопрепаративная HPLC
SPE - твердофазная экстракция
MeOH - метанол
TBDMS - трет-бутилдиметилсилил
TBME - трет-бутилметиловый эфир
TFA - трифторуксусная кислота
DIPEA - N,N-диизопропилэтиламин
Номенклатура
Соединения именовали на основании структуры с использованием программного средства, содержащегося либо в Chem Draw (CambridgeSoft), либо в Marvin Sketch (ChemAxon).
Пример 12 - Инъецируемая композиция
Инъецируемую форму для введения соединения согласно настоящему изобретению получают путем перемешивания 1,7 масс.% соединения № 2 в 0,9% солевом растворе.
Метод анализа
Соединения тестировали с использованием метода анализа связывания со STING, сходного с описанным Li et al. (Nature Chemical Biology, 10, 1043-1048, (2014)).
Биологическая активность
Соединения согласно настоящему изобретению тестировали с использованием метода анализа связывания со STING, сходного с описанным Li et al. (Nature Chemical Biology, 10, 1043-1048, (2014)). Соединения согласно настоящему изобретению тестировали с использованием анализа связывания методом резонансного переноса энергии флуоресценции (FRET). Li et al. проводили анализ связывания методом сцинтилляционного анализа сближения (SPA).
Активность соединений согласно настоящему изобретению в отношении STING представлена ниже в Таблице 1.
Таблица 1
Хотя предпочтительные варианты осуществления настоящего изобретения проиллюстрированы выше, следует понимать, что настоящее изобретение не ограничивается точными инструкциями, раскрытыми в настоящем документе, и что сохраняется право на все модификации, подпадающие под объем последующей формулы изобретения.
Реферат
Изобретение относится к соединению, которое может быть использовано в фармацевтической промышленности, формулы:,или его фармацевтически приемлемой соли. Предложено новое соединение, эффективное в качестве модулятора STING. 2 з.п. ф-лы, 11 пр., 1 табл., 4 ил.
Формула
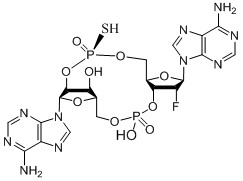
Комментарии