Новый эффективный аминогликозидный антибиотик против бактерий с множественной лекарственной резистентностью - RU2751634C2
Код документа: RU2751634C2
Описание
Перекрестная ссылка на родственную заявку
[0001]
По настоящей заявке испрашивается приоритет согласно патентной заявке Японии № 2015-151250 (дата подачи: 30 июля 2015), которая является первичной заявкой, заявленной в Японии. Все содержание данной первичной заявки включено в настоящее описание посредством ссылки.
Уровень техники изобретения
Область техники, к которой относится изобретение
[0002]
Настоящее изобретение относится к новым аминогликозидным антибиотикам и содержащей их фармацевтической композиции.
Уровень техники
[0003]
Аминогликозидные антибиотики обладают, аналогично бета-лактаму и хинолонам, антибактериальной активностью как против грамположительных, так и против грамотрицательных бактерий. Однако, в настоящее время существуют лекарственные средства, включая антибактериальные средства, приведенные выше, обладающие широким спектром воздействия, справляющимся с резистентными к антибиотикам бактериями. Как описано ниже, разработка таких медицинских препаратов также сталкивается с трудностями.
[0004]
В последнее время имеется быстрый рост случаев инфекционных заболеваний, вызванных метициллин-резистентной бактерией Staphylococcus aureus (называемой "MRSA", как указано далее) как в Японии, так и за рубежом. MRSA создает клинические проблемы, как причинная бактерия, вызывающая в результате серьезные инфекционные заболевания, и были проведены исследования по использованию терапевтических средств против таких инфекционных заболеваний.
[0005]
Как сообщалось, (S)-1-N-(4-амино-2-гидроксибутирил)дибекацин (арбекацин), который получают ацилированием аминогруппы в положении 1 дибекацина (тип аминогликозидов) с аминогидроксимасляной кислотой (HABA), эффективен против резистентной к метициллину Staphylococcus aureus (MRSA) (непатентный документ 1). На самом деле, арбекацин использовался в качестве чудодейственного средства против MRSA инфекции в Японии, начиная с конца 1990.
[0006]
В то же время, арбекацин использовался в качестве терапевтического средства для воздействия на MRSA в течение более чем 20 лет, и появление резистентной к арбекацину MRSA вызывает ряд вопросов в клинической практике.
[0007]
Кроме того, в последнее время увеличилось число бактерий с множественной лекарственной резистентностью, включая не только грамположительные бактерии, такие MRSA, но и грамотрицательные бактерии, такие как Escherichia coli, Klebsiella pneumoniae, Serratia, Акинетобактерия, Pseudomonas aeruginosa. Среди таких бактерий многие обладают резистентностью в отношении общепринятых аминогликозидных антибиотиков, бета-лактамных антибиотиков и новых хинолоновых антибиотиков и часто вызывают некупирующиеся инфекционные заболевания.
[0008]
Что касается грамотрицательных бактерий с множественной лекарственной резистентностью, таких как Escherichia coli с множественной лекарственной резистентностью и ацинетобактер (Acinetobacter) с множественной лекарственной резистентностью, сообщалось, что эффективным является (S)-1-N-(4-амино-2-гидроксибутирил)-6'-N-гидроксиэтилсизомицин (плазомицин), который продуцируется из сизомицина (тип аминогликозидных антибиотиков), путем ацилирования аминогруппы в положении 1 сизомицина с аминогидроксимасляной кислотой (HABA) и алкилирования аминогруппы в положении 6' сизомицина (патентный документ 1).
[0009]
Однако плазомицин не является эффективным в отношении резистентных метилазу-продуцирующих грамотрицательных бактерий, показывая при этом эффективность в отношении некоторых грамотрицательных бактерий с множественной лекарственной резистентностью. Кроме того, их основная противомикробная активность и безопасность являются недостаточной.
[0010]
Кроме того, описано, что апрамицин умеренно эффективен в отношении карбапенем-резистентных грамотрицательных бактерий, в отношении которых, как было обнаружено, большинство аминогликозидных антибиотиков являются неэффективными (непатентный документ 2). Раскрыто соединение, полученное химической модификацией гидроксильной группы в положениях 5, 6 или 6" апрамицина (патентные документы 2, 3 и 4). Раскрыто соединение, полученное химической модификацией аминогруппы в положении 1 или 4" апрамицина (патентные документы 5 и 6). Однако ни одно из указанных соединений четко не выявлено относительно их эффективности против резистентных бактерий.
Документы известного уровня
Патентный документ
[0011]
Патентный документ 1: WO 2009/067692
Патентный документ 2: Публикация патентной заявки, не прошедшей экспертизу № 57-72998
Патентный документ 3: Публикация патентной заявки, не прошедшей экспертизу № 57-72999
Патентный документ 4: Патент США 4379917
Патентный документ 5: Патент США 4424345
Патентный документ 6: Патент США 4360665
Непатентный документ
[0012]
Непатентный документ 1: Kondo, S. et al., Journal of Antibiotics, Vol. 26, pp. 412-415, 1973
Непатентный документ 2: J Antimicrob Chemother, Vol. 66, pp. 48-53, 2011
Сущность изобретения
[0013]
Настоящее изобретение предназначено для обеспечения нового аминогликозидного антибиотика, который эффективен как против грамположительных, так и против грамотрицательных бактерий, особенно против грамотрицательных и грамположительных бактерии с множественной лекарственной резистентностью.
[0014]
Авторы настоящего изобретения, в результате тщательного исследования производных апрамицина, типа аминогликозидных антибиотиков, обнаружили соединения, обладающие антибактериальной активностью как против грамположительных, так и против грамотрицательных бактерий. Эти соединения оказались также эффективными против резистентных бактерий, таких как MRSA, и грамотрицательных бактерий с множественной лекарственной резистентностью. Настоящее изобретение основано на этих выводах.
[0015]
Таким образом, настоящее изобретение включает следующие объекты.
(1) Соединение, представленное общей формулой (I), или его фармацевтически приемлемая соль или сольват:
[Химическая формула 1]

где
R1 представляет собой атом водорода или гидроксильную группу,
R2 представляет собой атом водорода или аминогруппу,
R3 представляет собой атом водорода, атом галогена, гидроксильную группу или аминогруппу,
R4 представляет собой атом водорода, атом галогена, гидроксильную группу или аминогруппу,
где R1 и R4 могут вместе образовывать двойную связь,
R5 представляет собой атом водорода, гидроксильную группу или аминогруппу,
R6 представляет собой атом водорода, гидроксильную группу или аминогруппу,
R7 представляет собой атом водорода, гидроксильную группу или аминогруппу,
R8 представляет собой атом водорода, гидроксильную группу или аминогруппу,
R9 и R10, каждый независимо, представляют собой атом водорода, C1-6алкильную группу, амино-C1-6алкильную группу, гуанидино-C1-6алкильную группу, амино-C3-7циклоалкильную группу, амино-C3-7циклоалкил-C1-6алкильную группу, амидиногруппу, азетидиногруппу, необязательно замещенную C1-6алкильной группой, глицильную группу, саркозильную группу, L-аланильную группу, D-аланильную группу, L-серильную группу, D-серильную группу, β-аланильную группу, L-изосерильную группу или D-изосерильную группу; и
R11 представляет собой атом водорода, гидроксильную группу или атом фтора,
за исключением случаев, когда
(i) R1, R4, R5, R8 и R11 представляют собой гидроксильные группы, R2, R3, R6, R7, R9 и R10 представляют собой атомы водорода (апрамицин),
(ii) R5, R8 и R11 представляют собой гидроксильные группы, R1, R2, R3, R4, R6, R7, R9 и R10 представляют собой атомы водорода (5,6-дидезоксиапрамицин),
(iii) R1, R5, R8 и R11 представляют собой гидроксильные группы, R2, R3, R4, R6, R7, R9 и R10 представляют собой атомы водорода (5-дезоксиапрамицин),
(iv) R1, R4, R5 и R8 представляют собой гидроксильные группы, R2, R3, R6, R7, R9, R10 и R11 представляют собой атомы водорода (6"-дезоксиапрамицин),
(v) R1, R4, R5, R8 и R11 представляют собой гидроксильные группы, R2, R3, R6 и R7 представляют собой атомы водорода, один из R9 или R10 представляет собой атом водорода, другой представляет собой этильную группу или 2-аминоэтильную группу.
(2) Соединение по пункту (1), представленное общей формулой (I-1), или его фармацевтически приемлемая соль или сольват:
[Химическая формула 2]
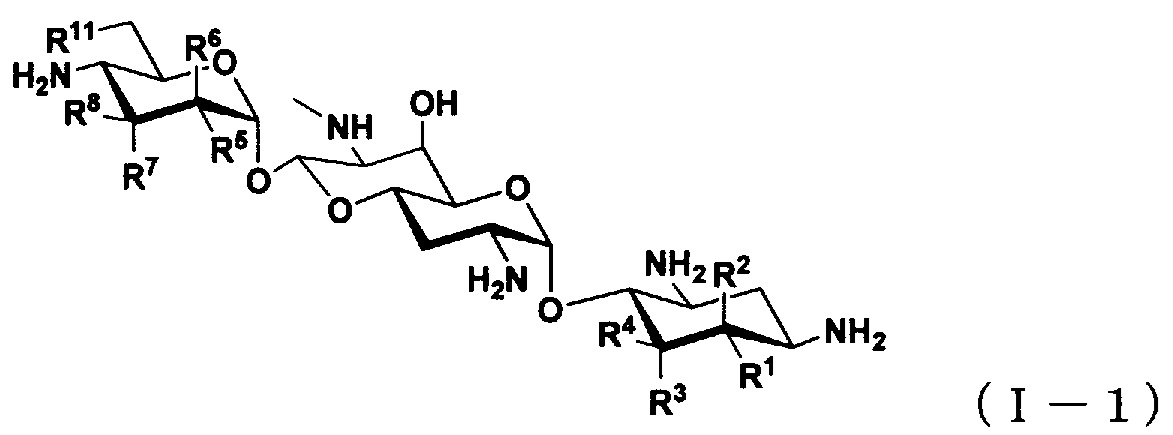
где
R1 представляет собой атом водорода или гидроксильную группу,
R2 представляет собой атом водорода или аминогруппу,
R3 представляет собой атом водорода, атом галогена, гидроксильную группу или аминогруппу,
R4 представляет собой атом водорода, атом галогена или аминогруппу,
где R1 и R4 могут вместе образовывать двойную связь,
R5 представляет собой атом водорода, гидроксильную группу или аминогруппу,
R6 представляет собой атом водорода, гидроксильную группу или аминогруппу,
R7 представляет собой атом водорода, гидроксильную группу или аминогруппу,
R8 представляет собой атом водорода, гидроксильную группу или аминогруппу; и
R11 представляет собой атом водорода, гидроксильную группу или атом фтора,
за исключением случаев, когда
(i) R5, R8 и R11 представляют собой гидроксильные группы, R1, R2, R3, R4, R6 и R7 представляют собой атомы водорода (5,6-дидезоксиапрамицин),
(ii) R1, R5, R8 и R11 представляют собой гидроксильные группы, R2, R3, R4, R6 и R7 представляют собой атомы водорода (5-дезоксиапрамицин),
(iii) R1, R4, R5 и R8 представляют собой гидроксильные группы, R2, R3, R6, R7 и R11 представляют собой атомы водорода (6"-дезоксиапрамицин).
(3) Соединение по пункту (1), представленное общей формулой (I-2), или его фармацевтически приемлемая соль или сольват:
[Химическая формула 3]
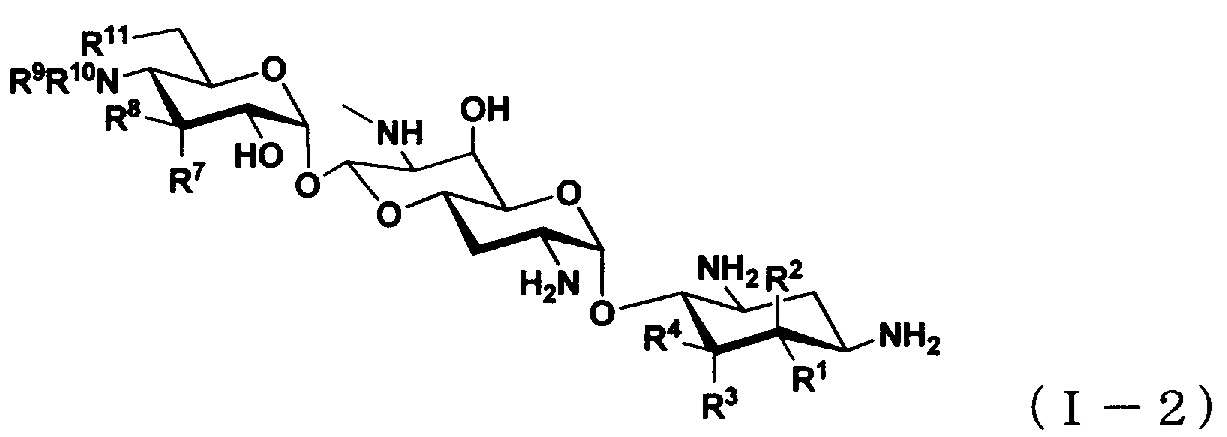
где
R1 представляет собой атом водорода или гидроксильную группу,
R2 представляет собой атом водорода или аминогруппу,
R3 представляет собой атом водорода, атом галогена, гидроксильную группу или аминогруппу,
R4 представляет собой атом водорода, атом галогена или аминогруппу,
где R1 и R4 могут вместе образовывать двойную связь,
R7 представляет собой атом водорода, гидроксильную группу или аминогруппу,
R8 представляет собой атом водорода, гидроксильную группу или аминогруппу,
R9 представляет собой атом водорода, C1-6алкильную группу или амино-C1-6алкильную группу,
R10 представляет собой C1-6алкильную группу, амино-C1-6алкильную группу, гуанидино-C1-6алкильную группу, амино-C3-7циклоалкильную группу, амино-C3-7циклоалкил-C1-6алкильную группу, амидиногруппу, азетидиногруппу, необязательно замещенную C1-6алкильной группой, глицильную группу, саркозильную группу, L-аланильную группу, D-аланильную группу, L-серильную группу, D-серильную группу, β-аланильную группу, L-изосерильную группу или D-изосерильную группу; и
R11 представляет собой атом водорода или гидроксильную группу.
(4) Соединение по пункту (1), представленное общей формулой (I-3), или его фармацевтически приемлемая соль или сольват:
[Химическая формула 4]

где
R9 представляет собой атом водорода, C1-6алкильную группу или амино-C1-6алкильную группу,
R10 представляет собой метильную группу, C3-6алкильную группу, амино-C3-6алкильную группу, гуанидино-C1-6алкильную группу, амино-C3-7циклоалкильную группу, амино-C3-7циклоалкил-C1-6алкильную группу, амидиногруппу, азетидиногруппу, необязательно замещенную C1-6алкильной группой, глицильную группу, саркозильную группу, L-аланильную группу, D-аланильную группу, L-серильную группу, D-серильную группу, β-аланильную группу, L-изосерильную группу или D-изосерильную группу.
(5) Соединение по пункту (1), представленное общей формулой (I-4), или его фармацевтически приемлемая соль или сольват:
[Химическая формула 5]

где
R1 представляет собой атом водорода или гидроксильную группу,
R2 представляет собой атом водорода или аминогруппу,
R3 представляет собой атом водорода, атом галогена, гидроксильную группу или аминогруппу,
R4 представляет собой атом водорода, атом галогена или аминогруппу; и
где R1 и R4 могут вместе образовывать двойную связь,
за исключением случаев, когда
(i) R1, R2, R3 и R4 представляют собой атомы водорода (5,6-дидезоксиапрамицин),
(ii) R1представляет собой гидроксильную группу, и R2, R3 и R4 представляют собой атомы водорода (5-дезоксиапрамицин).
(6) Соединение по пункту (1), представленное общей формулой (I-5), или его фармацевтически приемлемая соль или сольват:
[Химическая формула 6]

где
R5 представляет собой атом водорода, гидроксильную группу или аминогруппу,
R6 представляет собой атом водорода, гидроксильную группу или аминогруппу,
R7 представляет собой атом водорода, гидроксильную группу или аминогруппу,
R8 представляет собой атом водорода, гидроксильную группу или аминогруппу; и
R11 представляет собой атом водорода, гидроксильную группу или атом фтора,
за исключением случаев, когда
(i) R5, R8 и R11 представляют собой гидроксильные группы, R6 и R7 представляют собой атомы водорода (апрамицин),
(ii) R5 и R8 представляют собой гидроксильные группы, и R6, R7 и R11 представляют собой атомы водорода (6"-дезоксиапрамицин).
(7) Соединение по пункту (1), или его фармацевтически приемлемая соль или сольват, где данное соединение представляет собой:
4"-N-метилапрамицин,
4"-N-(3-аминопропил)апрамицин,
4"-N-((1-аминоциклопентил)метил)апрамицин,
4"-N-(1,3-диаминопропан-2-ил)апрамицин,
4"-N,N-бис(2-аминоэтил)апрамицин,
4"-N-(цис-1,4-4-аминоциклогексил)апрамицин,
4"-N-(транс-1,4-4-аминоциклогексил)апрамицин,
4"-N-(азетидин-3-ил)апрамицин,
4"-N-(1-метилазетидин-3-ил)апрамицин,
4"-дезамино-4"-гуанидиноапрамицин,
4"-N-гуанидиноэтилапрамицин,
5-эпиапрамицин,
5-дезокси-5-эпи-5-фторапрамицин,
6-дезокси-5-эпиапрамицин,
5,6-дидезокси-5-фторапрамицин,
5-амино-5-дезокси-5-эпиапрамицин,
5-амино-5-дезоксиапрамицин,
6-амино-5,6-дидезокси-5,6-диэпи-5-фторапрамицин,
5-амино-5,6-дидезоксиапрамицин,
2"-амино-2"-дезокси-2",3"-диэпиапрамицин,
3"-амино-3"-дезоксиапрамицин,
3"-эпиапрамицин,
2",3"-диэпиапрамицин,
6"-дезокси-6"-фторапрамицин,
3",6"-дидезоксиапрамицин,
5,6"-дидезоксиапрамицин,
5,3"-дидезоксиапрамицин,
3"-дезокси-5-эпиапрамицин,
5,3"-дидезокси-5-эпи-5-фторапрамицин,
6,3"-дидезокси-5-эпиапрамицин,
5,6,3"-тридезоксиапрамицин,
5-амино-5,3"-дидезокси-5-эпиапрамицин,
5,2"-дидезокси-5,3"-диэпи-5-фторапрамицин,
5,3"-диэпиапрамицин,
6,6"-дидезокси-5-эпиапрамицин,
5-ено-5,6,6"-тридезоксиапрамицин,
5,6,6"-тридезоксиапрамицин,
5-дезокси-4"-N-метилапрамицин,
4"-N-(2-аминоэтил)-5-дезоксиапрамицин,
4"-N-(3-аминопропил)-5-дезоксиапрамицин,
5-дезокси-4"-N-(1,3-диаминопропан-2-ил)апрамицин,
4"-дезамино-5-дезокси-4"-гуанидиноапрамицин,
5-эпи-4"-N-метилапрамицин,
4"-N-(2-аминоэтил)-5-эпиапрамицин,
4"-N-(3-аминопропил)-5-эпиапрамицин,
4"-N-(1,3-диаминопропан-2-ил)-5-эпиапрамицин,
4"-деазмино-5-эпи-4"-гуанидиноапрамицин,
4"-дезамино-5-дезокси-5-эпи-5-фтор-4"-гуанидиноапрамицин,
5,6-дидезокси-4"-N-метилапрамицин,
4"-N-(2-аминоэтил)-5,6-дидезоксиапрамицин,
4"-N-(3-аминопропил)-5,6-дидезоксиапрамицин,
4"-N-(1,3-диаминопропан-2-ил)-5,6-дидезоксиапрамицин,
4"-дезамино-5,6-дидезокси-4"-гуанидиноапрамицин,
6-дезокси-5-эпи-4"-N-метилапрамицин,
4"-N-(2-аминоэтил)-6-дезокси-5-эпиапрамицин,
4"-N-(3-аминопропил)-6-дезокси-5-эпиапрамицин,
4"-дезамино-6-дезокси-5-эпи-4"-гуанидиноапрамицин,
4"-N-(1,3-диаминопропан-2-ил)-5,6"-дидезоксиапрамицин,
4"-дезамино-5,6"-дидезокси-4"-гуанидиноапрамицин,
4"-дезамино-5,3"-дидезокси-4"-гуанидиноапрамицин,
4"-N-глицилапрамицин,
4"-N-саркозилапрамицин,
4"-N-(L-аланил)апрамицин,
4"-N-(D-аланил)апрамицин,
4"-N-(L-серил)апрамицин,
4"-N-(D-серил)апрамицин,
4"-N-(β-аланил)апрамицин,
4"-N-(L-изосерил)апрамицин,
5-эпи-4"-N-глицилапрамицин,
5-эпи-4"-N-саркозилапрамицин,
4"-N-(L-аланил)-5-эпиапрамицин,
5-эпи-4"-N-(L-серил)апрамицин,
4"-N-(β-аланил)-5-эпиапрамицин,
5-эпи-4"-N-(L-изосерил)апрамицин,
5-эпи-4"-N-(D-изосерил)апрамицин,
6-дезокси-5-эпи-4"-N-глицилапрамицин,
6-дезокси-5-эпи-4"-N-саркозилапрамицин,
4"-N-(β-аланил)-6-дезокси-5-эпиапрамицин,
6-дезокси-5-эпи-4"-N-(L-изосерил)апрамицин,
5-амино-4"-дезамино-5-дезокси-5-эпи-4"-гуанидиноапрамицин,
5-амино-5-дезокси-5-эпи-4"-N-глицилапрамицин,
5-амино-5-дезокси-5-эпи-4"-N-(L-изосерил)апрамицин,
4"-дезамино-3"-дезокси-5-эпи-4"-гуанидиноапрамицин,
4"-дезамино-5,3"-дидезокси-5-эпи-5-фтор-4"-гуанидиноапрамицин или
2"-дезокси-5,3"-диэпиапрамицин.
(8) Фармацевтическая композиция, содержащая соединение по любому из пунктов (1)-(7) или его фармацевтически приемлемую соль или сольват.
(9) Фармацевтическая композиция по пункту (8) для применения при профилактике или лечении инфекционного заболевания.
(10) Фармацевтическая композиция по пункту (8) или (9), где инфекционным заболеванием является сепсис, инфекционный эндокардит, дерматологические инфекции, инфекции областей хирургического вмешательства, ортопедические инфекции областей хирургического вмешательства, респираторные инфекции, инфекции мочевыводящих путей, энтеральные инфекции, перитонит, менингит, офтальмологические инфекции или отоларингологические инфекции.
(11) Фармацевтическая композиция по любому из пунктов (8)-(10), где инфекционное заболевание вызвано резистентной к метициллину Staphylococcus aureus (MRSA), Staphylococcus aureus, Escherichia coli, Klebsiella pneumoniae или Pseudomonas aeruginosa.
(12) Соединение по любому из пунктов (1)-(7) или его фармацевтически приемлемая соль или сольват для применения в терапии.
(13) Соединение по любому из пунктов (1)-(7) или его фармацевтически приемлемая соль или сольват для применения при профилактике или лечении инфекционного заболевания.
(14) Применение соединения по любому из пунктов (1)-(7) или его фармацевтически приемлемой соли или сольвата при изготовлении лекарственного средства для профилактики или лечения инфекционного заболевания.
(15) Применение соединения по любому из пунктов (1)-(7) или его фармацевтически приемлемой соли или сольвата для профилактики или лечения инфекционного заболевания.
(16) Применение по пункту (15), где его применяют в комбинации вместе с другими медицинскими средствами (например, антибиотиками).
(17) Способ профилактики или лечения инфекционного заболевания, включающий введение терапевтически эффективной дозы соединения по любому из пунктов (1)-(7) или его фармацевтически приемлемой соли или сольвата животному, включая человека.
(18) Противомикробное средство, содержащее соединение по любому из пунктов (1)-(7) или его фармацевтически приемлемую соль или сольват.
[0016]
Соединение настоящего изобретения или его фармацевтически приемлемая соль или сольват полезны в широком антибактериальном спектре против грамположительных бактерий и грамотрицательных бактерий. Также, они полезны с точки зрения антибактериальной активности в отношении грамположительных и грамотрицательных бактерий с множественной лекарственной резистентностью, которые не поддаются лечению имеющимися в настоящее время антибиотиками. Особенно, они полезны при профилактике или лечении ряда инфекционных заболеваний, вызванных MRSA или грамотрицательными бактериями с множественной лекарственной резистентностью.
Подробное описание настоящего изобретения
[0017]
Настоящее изобретение будет конкретно разъяснено следующим образом.
[0018]
Определения
В соединении настоящего изобретения атом галогена означает атом фтора, атом хлора, атом брома или атом йода.
[0019]
В соединении настоящего изобретения C1-6алкильная группа означает алкильную группу с прямой или разветвленной цепью, имеющей 1-6 атомов углерода. Например, алкильные группы включают метильную группу, этильную группу, н-пропильную группу, изопропильную группу, н-бутильную группу, изобутильную группу, трет-бутильную группу, втор-бутильную группу, н-пентильную группу, изопентильную группу, 2-метилбутильную группу, неопентильную группу, 1-этилпропильную группу, н-гексильную группу, 4-метилпентильную группу, 3-метилпентильную группу, 2-метилпентильную группу, 1-метилпентильную группу, 3,3-диметилбутильную группу, 2,2-диметилбутильную группу, 1,1-диметилбутильную группу, 1,2-диметилбутильную группу, 1,3-диметилбутильную группу, 2,3-диметилбутильную группу, 2-этилбутильную группу и тому подобное.
[0020]
В соединении настоящего изобретения амино-C1-6алкильная группа означает приведенную выше C1-6алкильную группу, 1-3 атома водорода которой замещены аминогруппой(ами), и положение замещения особенно не ограничено. Например, амино-C1-6алкильные группы включают аминометильную группу, аминоэтильную группу, аминопропильную группу, аминобутильную группу, аминопентильную группу, аминогексильную группу, 1,3-диаминопропальную группу и тому подобное.
[0021]
В соединении настоящего изобретения гуанидино-C1-6алкильная группа означает приведенную выше C1-6алкильную группу, в которой 1-2 атома водорода замещены (a) гуанидиногруппой(ами), и положение замещения особенно не ограничено. Например, гуанидино-C1-6алкильные группы включают гуанидинометильную группу, гуанидиноэтильную группу, гуанидинопропильную группу и тому подобное.
[0022]
В соединении настоящего изобретения амино-C3-7циклоалкильная группа означает циклическую алкильную группу, имеющую 3-7 атомов углерода, в которой 1-2 атома водорода замещены аминогруппой(ами), и положение замещения особенно не ограничено. Амино-C3-7циклоалкильные группы включают аминоциклопропильную группу, аминоциклобутильную группу, аминоциклопентильную группу, аминоциклогексильную группу, аминоциклогептильную группу и тому подобное.
[0023]
В соединении настоящего изобретения амино-C3-7циклоалкил-C1-6алкильная группа означает приведенную выше C1-6алкильную группу, замещенную приведенными выше амино-C3-7циклоалкильными группами. Амино-C3-7циклоалкил-C1-6алкильные группы включают аминоциклопропилметильную группу, аминоциклобутилметильную группу, аминоциклопентилметильную группу, аминоциклогексилметильную группу и тому подобное.
[0024]
В соединении настоящего изобретения азетидиногруппа, необязательно замещенная C1-6алкилом, означает азетидиногруппу, незамещенную или замещенную C1-6алкильной группой, приведенной выше. Азетидиногруппы, замещенные C1-6алкилом, включают N-метилазетидиногруппу, N-этилазетидиногруппу, N-пропилазетидиногруппу, N-изопропилазетидиногруппу и тому подобное.
[0025]
В соединении настоящего изобретения "необязательно замещенный" означает, что может быть замещен 1 или более заместителями или может быть незамещенным.
[0026]
Аминогликозидный антибиотик
Соединение настоящего изобретения представляет собой соединение, представленное приведенными выше общими формулами (I), (I-1), (I-2), (I-3), (I-4) или (I-5), или его фармацевтически приемлемую соль или их сольват.
[0027]
В одном из вариантов осуществления R9 и R10 в приведенной выше общей формуле (I), каждый независимо, представляет собой атом водорода, C1-6алкильную группу, амино-C1-6алкильную группу, гуанидино-C1-6алкильную группу, амино-C3-7циклоалкильную группу, амино-C3-7циклоалкил-C1-6алкильную группу, амидиногруппу, азетидиногруппу, необязательно замещенную C1-6алкильной группой.
[0028]
В одном из вариантов осуществления R10 в приведенной выше общей формуле (I-2) представляет собой C1-6алкильную группу, амино-C1-6алкильную группу, гуанидино-C1-6алкильную группу, амино-C3-7циклоалкильную группу, амино-C3-7циклоалкил-C1-6алкильную группу, амидиногруппу или азетидиногруппу, необязательно замещенную C1-6алкильной группой.
[0029]
В одном из вариантов осуществления R10 в приведенной выше общей формуле (I-3) представляет собой метильную группу, C3-6алкильную группу, амино-C3-6алкильную группу, гуанидино-C1-6алкильную группу, амино-C3-7циклоалкильную группу, амино-C3-7циклоалкил-C1-6алкильную группу, амидиногруппу или азетидиногруппу, необязательно замещенную C1-6алкильной группой.
[0030]
Соединение настоящего изобретения может быть представлено в виде соли. Соль включает, например, фармацевтически приемлемую нетоксичную соль. Конкретные примеры соли включают галоидоводородные соли, такие как фтористоводородная соль, хлористоводородная соль, бромистоводородная соль и йодистоводородная соль; соли неорганических кислот, такие как сульфат, нитрат, фосфат, перхлорат и карбонат; карбоксилаты, такие как ацетат, трихлорацетат, трифторацетат, гидроксиацетат, лактат, цитрат, тартрат, оксалат, бензоат, манделат, бутират, малеат, пропионат, формат и малат; соли аминокислот, такие как аргининат, аспартат и глутамат; сульфонаты, такие как метансульфонат, паратолуолсульфонат, и предпочтительные примеры включают соли неорганических кислот, такие как сульфат и тому подобное.
[0031]
Соединение настоящего изобретения могут быть представлены в виде сольвата. Предпочтительные сольваты включают гидрат и этанолсольват.
[0032]
Способ получения аминогликозидного антибиотика
Соединения настоящего изобретения могут быть получены согласно следующим способам A-U, но, не ограничиваясь указанными способами.
[0033]
Способ A
Способ A представляет собой путь получения соединения, представленного общей формулой (A4), включающий введение заместителя в положение 4" апрамицина и последующее удаление защиты. Стадии представлены следующим образом. В дополнение, стадии A1-A3 осуществляют согласно способу, описанному в US2013/0165395 A1.
[Химическая формула 7]

[0034]
Стадия A4
Стадия A4 представляет собой путь получения соединения, представленного общей формулой (A4), алкилированием или амидированием аминогруппы в положении 4" соединения, представленного формулой (A3), с последующим удалением защиты. Данную стадию проводят путем взаимодействия различных кетонов с соединением формулы (A3) и восстанавливающим агентом в присутствии кислоты для моноалкилирования, путем взаимодействия различных альдегидов с соединением формулы (A3) и восстанавливающим агентом в присутствии кислоты для диалкилирования, и путем взаимодействия с амидиновым реакционноспособным реагентом в присутствии основания для амидирования.
[0035]
Восстанавливающие агенты, используемые на данной стадии, включают борогидрид натрия, цианоборогидрид натрия и боран-2-метилпиридиновый комплекс, и предпочтительно цианоборогидрид натрия. Используемые растворители включают метанол, этанол, изопропиловый спирт, диоксан, воду или их смешанный растворитель, и предпочтительно смешанный растворитель из метанола и диоксана. Реагенты, используемые при амидировании, включают 1,3-бис(трет-бутоксикарбонил)-2-(трифторметансульфонил)гуанидин (реагент Гудмана), N,N'-ди-(трет-бутоксикарбонил)тиомочевину, трет-бутил-(Z)-(((трет-бутоксикарбонил)имино)(1H-пиразол-1-ил)метил)карбамат и тому подобное, и предпочтительно реагент Гудмана, и основанием предпочтительно является триэтиламин. Все реакции проводят при реакционной температуре от 10°C до 90°C за время реакции от 1 до 24 часов.
[0036]
Бензилоксикарбонильная группа может быть удалена путем взаимодействия водорода и катализатора каталитического восстановления. Используемые катализаторы каталитического восстановления включают палладий на углероде, палладиевую чернь, гидроксид палладия, оксид палладия и тому подобное, и предпочтительно палладий на углероде. Используемые растворители особенно не ограничиваются, если не вовлечены в данную реакцию, и предпочтительно представляют собой метанол, этанол, тетрагидрофуран, диоксан или смешанный растворитель из такого органического растворителя и воды. Температура реакции составляет от 10°C до 30°C, и время реакции обычно составляет 1-24 часа. Циклический карбамат может быть удален основным гидролизом. Основания включают гидроксид натрия и гидроксид калия. Температура реакции составляет от 20°C до 110°C, и время реакции составляет от 0,5-48 часов.
[0037]
Стадия A5
Стадия A5 представляет собой путь получения соединения, представленного формулой (A5), введением бензильной группы для моноалкилирования аминогруппы в положении 4'' соединения формулы (A3). Данную стадию проводят путем взаимодействия соединения, представленного формулой (A3), с бензальдегидом и борогидридом натрия в присутствии основания. Используемые растворители на стадии A5 включают метанол, тетрагидрофуран, диоксан и их смешанный растворитель, и предпочтительно метанол. Температура реакции составляет от 10°C до 20°C, и время реакции составляет 1-2 часа.
[0038]
Стадия A6
Стадия A6 представляет собой путь получения соединения, представленного общей формулой (A4), алкилированием бензилированной аминогруппы в положении 4'' соединения формулы (A5) с последующим удалением защиты. Данную стадию проводят путем взаимодействия различного типа альдегидов с соединением формулы (A5) и восстанавливающим агентом в присутствии кислоты.
[0039]
Используемые растворители на данной стадии включают тетрагидрофуран, диоксан, метанол и их смешанный растворитель. Восстанавливающие агенты включают цианоборогидрид натрия и боран-2-метилпиридиновый комплекс. Удаление защитной бензильной группы, бензилоксикарбонильной группы и циклического карбамата осуществляют в условиях, аналогично приведенным выше для стадии A4.
[0040]
Способ B
Способ B представляет собой путь получения соединения, представленного формулами (B5) и (B7), химической модификацией положения 5 соединения, полученного освобождением гидроксильной группы только в положении 5 апрамицина и последующим удалением защиты. Стадии представлены следующим образом.
[Химическая формула 8]
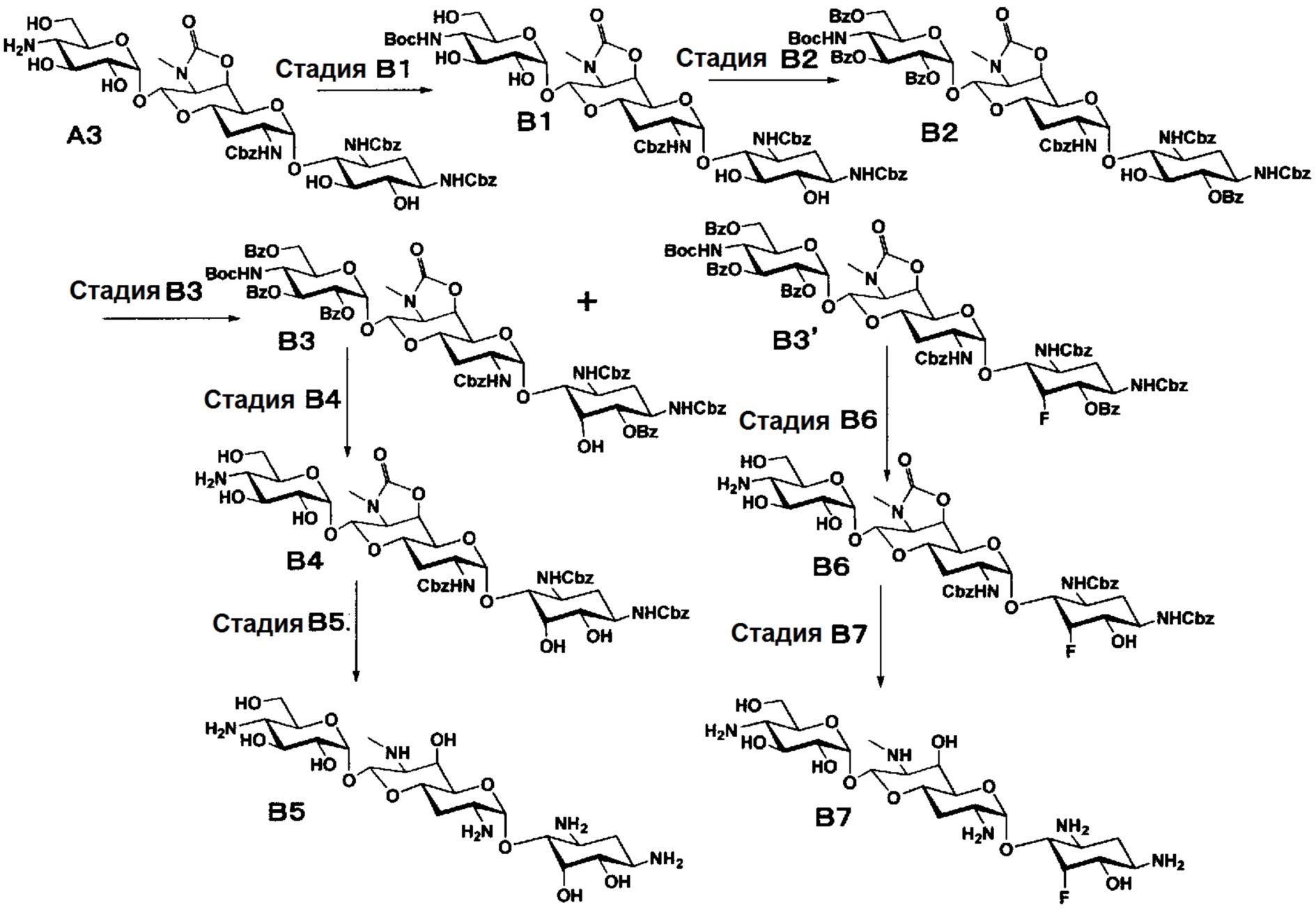
[0041]
Стадия B1
Стадия B1 представляет собой путь получения соединения, представленного формулой (B1), введением трет-бутоксикарбонильной группы в аминогруппу в положении 4'' соединения, представленного формулой (A3). Данную стадию проводят путем взаимодействия соединения формулы (A3) с ди-трет-бутилдикарбонатом в присутствии основания.
[0042]
Используемые растворители на данной стадии включают воду, N,N-диметилформамид, тетрагидрофуран, диоксан и их смешанный растворитель, и предпочтительно смешанный растворитель из воды и N,N-диметилформамида. Используемые основания могут включать гидроксид натрия, карбонат калия, карбонат натрия, бикарбонат калия, бикарбонат натрия, триэтиламин и тому подобное, и предпочтительно триэтиламин. Температура реакции составляет от 0°C до 40°C, и время реакции составляет 1-3 часа.
[0043]
Стадия B2
Стадия B2 представляет собой путь получения соединения, представленного формулой (B2), селективным введением бензоильной защитной группы в гидроксильную группу в положениях 6, 2", 3" и 6" соединения, представленного формулой (B1). Данную стадию проводят путем взаимодействия соединения формулы (B1) с бензоилхлоридом в присутствии основания.
[0044]
Используемые растворители на данной стадии включают пиридин, N,N-диметилформамид, метиленхлорид, хлороформ, 1,2-дихлорэтан и тому подобное, и предпочтительно пиридин. Используемые основания включают триэтиламин, пиридин, 4-диметиламинопиридин и тому подобное, и предпочтительно пиридин. Температура реакции составляет от 0°C до 30°C, и время реакции составляет 1-5 часов.
[0045]
Стадия B3
Стадия B3 представляет собой путь получения соединения, представленного формулами (B3) и (B3'), эпимеризацией или эпифторированием гидроксильной группы в положении 5 соединения, представленного формулой (B2). Данную стадию проводят путем взаимодействия соединения, представленного формулой (B2), с трифторидом диэтиламиносеры (DAST).
[0046]
Используемые растворители на данной стадии включают толуол, метиленхлорид, хлороформ, 1,2-дихлорэтан и тому подобное, и предпочтительно метиленхлорид, Температура реакции составляет от -5°C до 5°C, и время реакции составляет 1-5 часов.
[0047]
Стадия B4
Стадия B4 представляет собой путь получения соединения, представленного формулой (B4), путем удаления бензоильной группы трет-бутоксикарбонильной группы соединения, представленного формулой (B3). Данную стадию проводят путем взаимодействия соединения формулы (B3) с основанием для удаления гидроксилзащитной группы, и взаимодействия полученного в результате соединения с кислотой для удаления аминозащитной группы в положении 4''.
[0048]
Используемые растворители на стадии удаления гидроксилзащитной группы включают метанол, этанол, изопропиловый спирт, трет-бутиловый спирт, метиленхлорид, хлороформ и их смешанный растворитель, и предпочтительно смешанный растворитель из метанола и хлороформа. Используемые основания включают карбонат калия, карбонат натрия, гидроксид калия, гидроксид натрия, метоксид натрия, этоксид натрия, трет-бутоксид калия и тому подобное, и предпочтительно метоксид натрия. Температура реакции составляет от 0°C до 30°C, и время реакции составляет 1-5 часов.
[0049]
Используемые растворители на стадии удаления аминозащитной группы в положении 4'' включают этилацетат, метиленхлорид, ацетонитрил, ацетон, метанол и подобные, и предпочтительно метанол. Используемые кислоты включают п-толуолсульфоновую кислоту, метансульфоновую кислоту, уксусную кислоту, трифторуксусную кислоту и тому подобное, и предпочтительно трифторуксусную кислоту. Температура реакции составляет обычно от 0°C до 50°C, и время реакции составляет 1-5 часов.
[0050]
Стадия B5
Стадия B5 представляет собой путь получения соединения, представленного формулой (B5), путем удаления бензилоксикарбонильной группы и циклического карбамата соединения, представленного формулой (B4). Бензилоксикарбонильная группа может быть удалена путем взаимодействия водорода и катализатора, каталитически восстанавливающего водород. Используемые катализаторы, каталитически восстанавливающие водород, включают палладий на углероде, палладиевую чернь, гидроксид палладия, оксид палладия и тому подобное, и предпочтительно палладий на углероде. Используемые растворители особенно не ограничиваются, если не вовлечены в данную реакцию, и предпочтительно представляют собой метанол, этанол, тетрагидрофуран, диоксан или смешанный растворитель из этих органических растворителей и воды. Температура реакции составляет от 10°C до 30°C, и время реакции обычно составляет 1-24 часа. Циклический карбамат может быть удален основным гидролизом. Основания включают гидроксид натрия и гидроксид калия. Температура реакции составляет от 90°C до 110°C, и время реакции составляет от 0,5 до 1 часа.
[0051]
Стадия B6
Стадия B6 представляет собой путь получения соединения, представленного формулой (B6), путем удаления бензоильной группы и трет-бутоксикарбонильной группы соединения, представленного формулой (B3'). Удаление защитной группы осуществляют в условиях, аналогично приведенным выше для стадии B4.
[0052]
Стадия B7
Стадия B7 представляет собой путь получения соединения, представленного формулой (B7), путем удаления бензилоксикарбонильной группы и циклического карбамата соединения, представленного формулой (B6). Удаление защитной группы осуществляют в условиях, аналогично приведенным выше для стадии B5.
[0053]
Способ C
Способ C представляет собой путь получения соединения, представленного формулами (C6), (C8) и (C11), сначала путем введения уходящей группы в положение 5 апрамицина, и затем получая 6-дезокси-5-эпи, 6-дезокси-5-фтор и 5-азидо-6-дезокси производные, с последующим удалением защиты. Стадии представлены следующим образом.
[Химическая формула 9]

[0054]
Стадия C1
Стадия C1 представляет собой путь получения соединения, представленного формулой (C1), введением метансульфонильной группы в гидроксильную группу в положении 5 соединения, представленного формулой (B2). Данную стадию проводят путем взаимодействия соединения формулы (B2) с метансульфонилхлоридом в присутствии основания.
[0055]
Используемые растворители на данной стадии включают пиридин, метиленхлорид, хлороформ, 1,2-дихлорэтан и тому подобное, и предпочтительно метиленхлорид. Используемые основания включают триэтиламин, пиридин, 4-диметиламинопиридин и тому подобное, и предпочтительно 4-диметиламинопиридин. Температура реакции составляет от 0°C до 30°C, и время реакции составляет 1-2 часа.
[0056]
Стадия C2
Стадия C2 представляет собой путь получения соединения, представленного формулой (C2), сначала путем удаления бензоильной группы соединения, представленного формулой (C1), и одновременного выполнения ангидридизации (эпоксидирования) положений 5 и 6 с последующим введением бензоильной защитной группы в гидроксильные группы в положениях 2", 3" и 6". Данную стадию проводят путем взаимодействия соединения, представленного формулой (C1), с основанием и дальнейшего взаимодействия с бензоилхлоридом в присутствии основания.
[0057]
Используемые растворители на стадии дебензоилирования и ангидридизации включают метанол, этанол, метиленхлорид, хлороформ, 1,2-дихлорэтан и тому подобное, и предпочтительно хлороформ. Используемые основания включают карбонат калия, карбонат натрия, гидроксид калия, гидроксид натрия, метоксид натрия, этоксид натрия, трет-бутоксид калия и тому подобное, и предпочтительно метоксид натрия. Температура реакции составляет от 0°C до 30°C, и время реакции составляет 1-5 часов.
[0058]
Бензоилирование осуществляют в условиях, аналогично приведенным выше для стадии B2.
[0059]
Стадия C3
Стадия C3 представляет собой путь получения соединения, представленного формулой (C3), путем раскрытия эпоксида соединения, представленного формулой (C2). Данную стадию проводят путем взаимодействия соединения, представленного формулой (C2), с йодидом натрия в присутствии забуференного кислотой раствора. Используемые растворители на данной стадии включают ацетон, N,N-диметилформамид, тетрагидрофуран, диоксан и тому подобное, и предпочтительно ацетон. Используемые забуференные кислотой растворы включают 5% раствор ацетата натрия-уксусной кислоты и тому подобное. Температура реакции составляет от 60°C до 100°C, и время реакции составляет 1-6 часов.
[0060]
Стадия C4
Стадия C4 представляет собой путь получения соединения, представленного формулой (C4), путем восстановления йодного соединения, представленного формулой (C3). Данную стадию проводят путем взаимодействия соединения, представленного формулой (C3), с гидридом трибутилолова в присутствии 2,2'-азобис(изобутиронитрила).
[0061]
Используемые растворители на данной стадии включают толуол, тетрагидрофуран, диоксан и тому подобное, и предпочтительно диоксан. Температура реакции составляет от 60°C до 100°C, и время реакции составляет 3-8 часов.
[0062]
Стадия C5
Стадия C5 представляет собой путь получения соединения, представленного формулой (C5), путем удаления бензоильной группы и трет-бутоксикарбонильной группы соединения, представленного формулой (C4). Удаление защитной группы осуществляют в условиях, аналогично приведенным выше для стадии B4.
[0063]
Стадия C6
Стадия C6 представляет собой путь получения соединения, представленного формулой (C6), путем удаления бензилоксикарбонильной группы и циклического карбамата соединения, представленного формулой (C5). Удаление защитной группы осуществляют в условиях, аналогично приведенным выше для стадии B5.
[0064]
Стадия C7
Стадия C7 представляет собой путь получения соединения, представленного формулой (C7), эпифторированием положения 5 соединения, представленного формулой (C4). Эпифторирование осуществляют в условиях, аналогично приведенным выше для стадии B3.
[0065]
Стадия C8
Стадия C8 представляет собой путь получения соединения, представленного формулой (C8), путем удаления защитной группы соединения, представленного формулой (C7). Удаление защитной группы осуществляют в условиях, аналогично приведенным выше для стадий B4 и B5.
[0066]
Стадия C9
Стадия C9 представляет собой путь получения соединения, представленного формулой (C9), метансульфонилированием гидроксильной группы в положении 5 соединения, представленного формулой (C4). Метансульфонилирование осуществляют в условиях, аналогично приведенным выше для стадии C1.
[0067]
Стадия C10
Стадия C10 представляет собой путь получения соединения, представленного формулой (C10), азидированием в положении 5 соединения, представленного формулой (C9). Данную стадию проводят путем взаимодействия соединения, представленного формулой (C9), с азидом натрия. Используемые растворители на данной стадии включают ацетон, N,N-диметилформамид, тетрагидрофуран, диоксан и тому подобное, и предпочтительно N,N-диметилформамид. Температура реакции составляет от 60°C до 100°C, и время реакции составляет 1-6 часов.
[0068]
Стадия C11
Стадия C11 представляет собой путь получения соединения, представленного формулой (C11), путем удаления защитной группы соединения, представленного формулой (C10). Удаление защитной группы осуществляют в условиях, аналогично приведенным выше для стадий B4 и B5.
[0069]
Способ D
Способ D представляет собой путь получения соединения, представленного формулой (D2), азидированием соединения, представленного формулой (C1), в положении 5 с последующим восстановлением и удалением защиты. Стадии представлены следующим образом.
[Химическая формула 10]

[0070]
Стадия D1
Стадия D1 представляет собой путь получения соединения, представленного формулой (D1), азидированием положения 5 соединения, представленного формулой (C1). Азидирование осуществляют в условиях, аналогично приведенным выше для стадии C10.
[0071]
Стадия D2
Стадия D2 представляет собой путь получения соединения, представленного формулой (D2), путем удаления защитной группы соединения, представленного формулой (D1). Удаление защитной группы осуществляют в условиях, аналогично приведенным выше для стадий B4 и B5.
[0072]
Способ E
Способ E представляет собой путь получения соединения, представленного формулой (E3), путем хлорирования положения 5 соединения, представленного формулой (B2), по способу B, с последующим азидированием и удалением защиты. Стадии представлены следующим образом.
[Химическая формула 11]

[0073]
Стадия E1
Стадия E1 представляет собой путь получения соединения, представленного формулой (E1), путем хлорирования положения 5 соединения, представленного формулой (B2). Данную стадию проводят путем взаимодействия соединения формулы (B2) с сульфурилхлоридом в присутствии основания.
[0074]
Используемые растворители на данной стадии включают пиридин, метиленхлорид, хлороформ, 1,2-дихлорэтан и тому подобное, и предпочтительно метиленхлорид. Используемые основания включают триэтиламин, пиридин, 4-диметиламинопиридин и тому подобное, и предпочтительно 4-диметиламинопиридин. Температура реакции составляет от 0°C до 30°C, и время реакции составляет 1-2 часа.
[0075]
Стадия E2
Стадия E2 представляет собой путь получения соединения, представленного формулой (E2), азидированием положения 5 соединения, представленного формулой (E1). Азидирование осуществляют в условиях, аналогично приведенным выше для стадии C10.
[0076]
Стадия E3
Стадия E3 представляет собой путь получения соединения, представленного формулой (E3), путем удаления защитной группы соединения, представленного формулой (E2). Удаление защитной группы осуществляют в условиях, аналогично приведенным выше для стадий B4 и B5.
[0077]
Способ F
Способ F представляет собой путь получения соединения, представленного формулой (F3), азидированием в положении 6 соединения, представленного формулой (C2), которое представляет собой общее промежуточное соединение в способе C, с последующим фторированием в положении 5 и удалением защиты. Стадии представлены следующим образом.
[Химическая формула 12]

[0078]
Стадия F1
Стадия F1 представляет собой путь получения соединения, представленного формулой (F1), путем раскрытия эпоксида соединения, представленного формулой (C2), для преобразования эпоксида в азид и гидроксильную группу. Данную стадию проводят путем взаимодействия соединения, представленного формулой (C2), с азидом натрия в присутствии хлорида аммония.
[0079]
Используемые растворители на данной стадии включают ацетон, N,N-диметилформамид, тетрагидрофуран, диоксан и тому подобное, и предпочтительно N,N-диметилформамид. Температура реакции составляет от 60°C до 100°C, и время реакции составляет 1-6 часов.
[0080]
Стадия F2
Стадия F2 представляет собой путь получения соединения, представленного формулой (F2), путем фторирования положения 5 соединения, представленного формулой (F1). Фторирование осуществляют в условиях, аналогично приведенным выше для стадии B3.
[0081]
Стадия F3
Стадия F3 представляет собой путь получения соединения, представленного формулой (F3), путем удаления защитной группы соединения, представленного формулой (F2). Удаление защитной группы осуществляют в условиях, аналогично приведенным выше для стадий B4 и B5.
[0082]
Способ G
Способ G представляет собой путь получения соединения, представленного формулами (G7) и (G8), сначала путем введения уходящей группы в положении 3ʺ соединения, представленного формулой (G3) (в котором только имеющаяся в положении 3ʺ гидроксильная группа присутствует в свободном состоянии), полученного из апрамицина на 4 стадии, затем путем получения 3"-азид-3"-дезокси и 2"-азид-2", 3"-диэпи-2"-дезокси производных через 2",3"-ангидро промежуточное соединение, с последующим проведением удаления защиты. Стадии представлены следующим образом.
[Химическая формула 13]

[0083]
Стадия G1
Стадия G1 представляет собой путь получения соединения, представленного формулой (G1), введением защитных групп в гидроксильные группы в положениях 5 и 6 соединения, представленного формулой (A1). Данную стадию проводят путем взаимодействия соединения, представленного формулой (A1), с 1,1-диметоксициклогексаном в присутствии кислоты. Используемые растворители на данной стадии включают N,N-диметилформамид, метиленхлорид, хлороформ, 1,2-дихлорэтан, этилацетат и тому подобное, и предпочтительно N,N-диметилформамид. Используемые кислоты включают п-толуолсульфоновую кислоту, п-толуолсульфонат пиридиния, камфорсульфоновую кислоту, хлористоводородную кислоту и тому подобное, и предпочтительно п-толуолсульфоновую кислоту. Температура реакции составляет от 20°C до 60°C, и время реакции составляет 1-8 часов.
[0084]
Стадия G2
Стадия G2 представляет собой путь получения соединения, представленного формулой (G2), путем включения положений 6' и 7', и положений 4" и 6" соединения, представленного формулой (G1), в циклические карбаматы. Преобразование в циклический карбамат осуществляют в условиях, аналогично приведенным выше для стадии A2.
[0085]
Стадия G3
Стадия G3 представляет собой путь получения соединения, представленного формулой (G3) селективным введением бензоильной защитной группы в гидроксильную группу в положении 2" соединения, представленного формулой (G2). Введение бензоильной защитной группы осуществляют в условиях, аналогично приведенным выше для стадии B2.
[0086]
Стадия G4
Стадия G4 представляет собой путь получения соединения, представленного формулой (G4), введением бензилсульфонильной группы в гидроксильную группу в положении 3" соединения, представленного формулой (G3). Данную стадию проводят путем взаимодействия соединения формулы (G3) с бензилсульфонилхлоридом в присутствии основания. Используемые растворители на данной стадии включают пиридин, метиленхлорид, хлороформ, 1,2-дихлорэтан и тому подобное, и предпочтительно пиридин. Используемые основания включают триэтиламин, пиридин, 4-диметиламинопиридин и тому подобное, и предпочтительно пиридин. Температура реакции составляет от -20°C до комнатной температуры, и время реакции составляет от 0,5 до 1 часа.
[0087]
Стадия G5
Стадия G5 представляет собой путь получения соединения, представленного формулой (G5), путем удаления бензоильной группы соединения, представленного формулой (G4), и одновременного проведения ангидридизации (эпоксидирования) в положениях 2" и 3". Данную стадию проводят путем взаимодействия соединения, представленного формулой (G4), с основанием.
[0088]
Используемые растворители при проведении ангидридизации включают метанол, этанол, метиленхлорид, хлороформ, 1,2-дихлорэтан и тому подобное, и предпочтительно хлороформ. Используемые основания включают карбонат калия, карбонат натрия, гидроксид калия, гидроксид натрия, метоксид натрия, этоксид натрия, трет-бутоксид калия и тому подобное, и предпочтительно метоксид натрия. Температура реакции составляет от 0°C до 30°C, и время реакции составляет 1-5 часов.
[0089]
Стадия G6
Стадия G6 представляет собой путь получения соединения, представленного формулами (G6) и (G6'), путем раскрытия эпоксида соединения, представленного формулой (G5), для преобразования эпоксида в азид и гидроксильную группу. Азидирование осуществляют в условиях, аналогично приведенным выше для стадии F1.
[0090]
Стадия G7
Стадия G7 представляет собой путь получения соединения, представленного формулой (G7), путем удаления защитной группы соединения, представленного формулой (G6). Данную стадию проводят путем удаления гидроксилзащитной группы через кислотный гидролиз соединения, представленного формулой (G6), и затем путем удаления аминозащитной группы через каталитическое восстановление и щелочной гидролиз полученного соединения. Используемые кислоты в кислотном гидролизе включают 1 N хлористоводородную кислоту, 1 N серную кислоту, 80% водный раствор уксусной кислоты, 80% водный раствор муравьиной кислоты и тому подобное, и предпочтительно 80% водный раствор уксусной кислоты. Температура реакции составляет от 30°C до 80°C, и время реакции составляет 1-3 часа. Удаление аминозащитной группы осуществляют в условиях, аналогично приведенным выше для стадии B5.
[0091]
Стадия G8
Стадия G8 представляет собой путь получения соединения, представленного формулой (G8), путем удаления защитной группы соединения, представленного формулой (G6'). Удаление защитной группы осуществляют в условиях, аналогично приведенным выше для стадии G7.
[0092]
Способ H
Способ H представляет собой путь получения соединения, представленного формулой (H3), сначала путем введения уходящей группы в положение 3" соединения, представленного формулой (G3) (в котором только имеющаяся в положении 3" гидроксильная группа присутствует в свободном состоянии), полученного из апрамицина на 4 стадии, затем путем инверсии гидроксильной группы в положении 3", с последующим проведением удаления защиты. Стадии представлены следующим образом.
[Химическая формула 14]

[0093]
Стадия H1
Стадия H1 представляет собой путь получения соединения, представленного формулой (H1), введением трифторметансульфонильной группы в гидроксильную группу в положении 3" соединения, представленного формулой (G3). Данную стадию проводят путем взаимодействия соединения формулы (G3) с трифторметансульфоновым ангидридом в присутствии основания.
Используемые растворители на данной стадии включают пиридин, метиленхлорид, хлороформ, 1,2-дихлорэтан и тому подобное, и предпочтительно метиленхлорид. Используемые основания включают триэтиламин, пиридин, 4-диметиламинопиридин и тому подобное, и предпочтительно пиридин. Температура реакции составляет от -10°C до 5°C, и время реакции составляет от 0,5 до 1 часа.
[0094]
Стадия H2
Стадия H2 представляет собой путь получения соединения, представленного формулой (H2), эпимеризацией гидроксильной группы в положении 3" и преобразованием положения 4" вместе с положением 4" в циклический карбамат в соединении, представленном формулой (H1). Эпимеризацию на данной стадии проводят путем взаимодействия соединения, представленного формулой (H1), с ацетатом цезия с последующей обработкой основанием. Используемые растворители на данной стадии включают диоксан, N,N-диметилформамид, 1,2-диметоксиэтан и тому подобное, и предпочтительно N,N-диметилформамид. Температура реакции составляет от 50°C до 80°C. Время реакции составляет 1-3 часа.
Используемые основания для преобразования в циклический карбамат включают карбонат калия, карбонат натрия, гидроксид калия, гидроксид натрия, метоксид натрия, этоксид натрия, трет-бутоксид калия и тому подобное, и предпочтительно метоксид натрия. Температура реакции составляет от 0°C до 30°C, и время реакции составляет 1-3 часа.
[0095]
Стадия H3
Стадия H3 представляет собой путь получения соединения, представленного формулой (H3), путем удаления защитной группы соединения, представленного формулой (H2). Удаление защитной группы осуществляют в условиях, аналогично приведенным выше для стадии G7.
[0096]
Способ I
Способ I представляет собой путь получения соединения, представленного формулой (I3), путем диаксиального расщепления эпоксида соединения, представленного формулой (G5), для получения 2",3"-диэпи производного, и последующего удаления защиты, где соединение (G5) получено из апрамицина на 6 стадии. Стадии представлены следующим образом.
[Химическая формула 15]

[0097]
Стадия I1
Стадия I1 представляет собой путь получения соединения, представленного формулой (I1), путем преобразования положений 4" и 6" соединения, представленного формулой (G5), в циклический карбамат. Преобразование в циклический карбамат осуществляют в условиях, аналогично приведенным выше для стадии A2.
[0098]
Стадия I2
Стадия I2 представляет собой путь получения соединения, представленного формулой (I2), путем диэпимеризации в положениях 2" и 3" через кислотный гидролиз соединения, представленного формулой (I1). Используемые кислоты для кислотного гидролиза включают 1 N хлористоводородную кислоту, 1 N серную кислоту, 80% водный раствор уксусной кислоты, 80% водный раствор муравьиной кислоты и тому подобное, и предпочтительно 80% водный раствор уксусной кислоты. Температура реакции составляет от 30°C до 80°C, и время реакции составляет 1-3 часа.
[0099]
Стадия I3
Стадия I3 представляет собой путь получения соединения, представленного формулой (I3), путем удаления бензилоксикарбонильной группы и циклического карбамата соединения, представленного формулой (I2). Удаление защитной группы осуществляют в условиях, аналогично приведенным выше для стадии B5.
[0100]
Способ J
Способ J представляет собой путь получения соединения, представленного формулой (J4), путем фторирования положения 6" соединения, представленного формулой (A1), полученного из апрамицина на 3 стадии, с последующим удалением защиты. Стадии представлены следующим образом.
[Химическая формула 16]

[0101]
Стадия J1
Стадия J1 представляет собой путь получения соединения, представленного формулой (J1), введением защитных групп в гидроксильные группы в положениях 5, 6 и положениях 2", 3" соединения, представленного формулой (A1). Данную стадию проводят путем взаимодействия соединения, представленного формулой (A1), с 1,1-диметоксициклогексаном в присутствии кислоты.
[0102]
Используемые растворители на данной стадии включают N,N-диметилформамид, метиленхлорид, хлороформ, 1,2-дихлорэтан, этилацетат и тому подобное, и предпочтительно N,N-диметилформамид. Используемые кислоты включают п-толуолсульфоновую кислоту, п-толуолсульфонат пиридиния, камфорсульфоновую кислоту, хлористоводородную кислоту и тому подобное, и предпочтительно п-толуолсульфоновую кислоту. Данную реакцию проводят при температуре от 40°C до 60°C, при пониженном давлении от 20 до 40 Торр (мм рт.ст.), и время реакции составляет 1-8 часов.
[0103]
Стадия J2
Стадия J2 представляет собой путь получения соединения, представленного формулой (J2), путем преобразования положений 6' и 7' соединения, представленного формулой (J1), в циклический карбамат. Преобразование в циклический карбамат осуществляют в условиях, аналогично приведенным выше для стадии A2.
[0104]
Стадия J3
Стадия J3 представляет собой путь получения соединения, представленного формулой (J3), путем фторирования положения 6" соединения, представленного формулой (J2). Фторирование осуществляют в условиях, аналогично приведенным выше для стадии B3.
[0105]
Стадия J4
Стадия J4 представляет собой путь получения соединения, представленного формулой (J4), путем удаления защитной группы соединения, представленного формулой (J3). Удаление защитной группы осуществляют в условиях, аналогично приведенным выше для стадии G7.
[0106]
Способ K
Способ K представляет собой путь получения соединения, представленного формулой (K4), введением бензилсульфонильной группы в гидроксильную группу в положении 6" и йодированием положений 3" и 6" соединения, представленного формулой (G5), полученного из апрамицина на 6 стадии, с последующим восстановлением и удалением защиты. Стадии представлены следующим образом.
[Химическая формула 17]

[0107]
Стадия K1
Стадия K1 представляет собой путь получения соединения, представленного формулой (K1), введением бензилсульфонильной группы в гидроксильную группу в положении 6" соединения, представленного формулой (G5). Введение бензоилсульфонильной группы осуществляют в условиях, аналогично приведенным выше для стадии G4.
[0108]
Стадия K2
Стадия K2 представляет собой путь получения соединения, представленного формулой (K2), путем раскрытия эпоксида соединения, представленного формулой (K1), для преобразования эпоксида в йодид и гидроксильную группу, и последующего преобразования бензилсульфонилоксигруппы в положении 6" в йодид. Йодирование осуществляют в условиях, аналогично приведенным выше для стадии C3.
[0109]
Стадия K3
Стадия K3 представляет собой путь получения соединения, представленного формулой (K3), путем восстановления йодидов соединения, представленного формулой (K2). Восстановление осуществляют в условиях, аналогично приведенным выше для стадии C4.
[0110]
Стадия K4
Стадия K4 представляет собой путь получения соединения, представленного формулой (K4), путем удаления защитной группы соединения, представленного формулой (K3). Удаление защитной группы осуществляют в условиях, аналогично приведенным выше для стадии G7.
[0111]
Способ L
Способ L представляет собой путь получения соединения, представленного формулой (L5), путем селективного замещения гидроксильной группы хлором в положении 6 соединения, представленного формулой (E1), полученного из апрамицина на 6 стадии, с последующим удалением защиты после восстановления. Стадии представлены следующим образом.
[Химическая формула 18]

[0112]
Стадия L1
Стадия L1 представляет собой путь получения соединения, представленного формулой (L1), путем удаления бензоильной группы соединения, представленного формулой (E1). Удаление бензоильной группы осуществляют в условиях, аналогично приведенным выше для стадии G5.
[0113]
Стадия L2
Стадия L2 представляет собой путь получения соединения, представленного формулой (L2), путем селективного замещения гидроксигрупп хлором в положении 6" соединения, представленного формулой (L1). Данную стадию проводят путем взаимодействия соединения, представленного формулой (L1), с трифенилфосфином и тетрахлоридом углерода. Используемые растворители на данной стадии включают диоксан, N,N-диметилформамид, пиридин, тетрагидрофуран и тому подобное, и предпочтительно N,N-диметилформамид. Температура реакции составляет от 40°C до 90°C, и время реакции составляет 1-6 часов.
[0114]
Стадия L3
Стадия L3 представляет собой путь получения соединения, представленного формулой (L3), путем восстановления хлорных групп в положениях 5 и 6" соединения, представленного формулой (L2). Восстановление осуществляют в условиях, аналогично приведенным выше для стадии C4.
[0115]
Стадия L4
Стадия L4 представляет собой путь получения соединения, представленного формулой (L4), путем удаления трет-бутоксикарбонильной группы в положении 4" соединения, представленного формулой (L3). Используемые растворители на данной стадии включают этилацетат, метиленхлорид, ацетонитрил, ацетон, метанол и тому подобное, и предпочтительно метанол. Используемые кислоты включают п-толуолсульфоновую кислоту, метансульфоновую кислоту, уксусную кислоту, трифторуксусную кислоту и тому подобное, и предпочтительно трифторуксусную кислоту. Температура реакции составляет от 0°C до 50°C, и время реакции составляет 1-2 часа.
[0116]
Стадия L5
Стадия L5 представляет собой путь получения соединения, представленного формулой (L5), путем удаления защитной группы соединения, представленного формулой (L4). Удаление защитной группы осуществляют в условиях, аналогично приведенным выше для стадии G7.
[0117]
Способ M
Способ M представляет собой путь получения соединения, представленного формулами (M7), (M9) и (M10), сначала путем получения 3"-дезокси производного через соединение, представленное формулой (G5), которое получено на 6 стадии из апрамицина, и преобразования его в 5-OH производное, и затем преобразования 5-OH производного в 5-дезокси, 5-эпи и 5-эпифторитное производные, с последующим проведением удаления защиты. Стадии представлены следующим образом.
[Химическая формула 19]

[0118]
Стадия M1
Стадия M1 представляет собой путь получения соединения, представленного формулой (M1), путем раскрытия эпоксида соединения, представленного формулой (G5), и его преобразования в йодид и гидроксильную группу. Йодирование осуществляют в условиях, аналогично приведенным выше для стадии C3.
[0119]
Стадия M2
Стадия M2 представляет собой путь получения соединения, представленного формулой (M2), путем восстановления йодного соединения, представленного формулой (M1). Восстановление осуществляют в условиях, аналогично приведенным выше для стадии C4.
[0120]
Стадия M3
Стадия M3 представляет собой путь получения соединения, представленного формулой (M3), путем бензоилирования гидроксигрупп в положениях 2" и 6" соединения, представленного формулой (M2). Бензоилирование осуществляют в условиях, аналогично приведенным выше для стадии B2.
[0121]
Стадия M4
Стадия M4 представляет собой путь получения соединения, представленного формулой (M4), путем проведения селективного бензоилирования в положении 6 соединения, представленного формулой (M3), после удаления циклогексилиденовой группы в положениях 5, 6. Используемые кислоты для удаления циклогексилиденовой группы включают 1 N хлористоводородную кислоту, 1 N серную кислоту, 80% водный раствор уксусной кислоты, 80% водный раствор муравьиной кислоты и тому подобное, и предпочтительно 80% водный раствор уксусной кислоты. Температура реакции составляет от 30°C до 80°C, и время реакции составляет 1-3 часа. Бензоилирование осуществляют в условиях, аналогично приведенным выше для стадии B2.
[0122]
Стадия M5
Стадия M5 представляет собой путь получения соединения, представленного формулой (M5), путем хлорирования положения 5 соединения, представленного формулой (M4). Хлорирование осуществляют в условиях, аналогично приведенным выше для стадии E1.
[0123]
Стадия M6
Стадия M6 представляет собой путь получения соединения, представленного формулой (M6), путем восстановления хлорной группы в положении 5 соединения, представленного формулой (M5). Восстановление осуществляют в условиях, аналогично приведенным выше для стадии C4.
[0124]
Стадия M7
Стадия M7 представляет собой путь получения соединения, представленного формулой (M7), путем удаления защитной группы соединения, представленного формулой (M6). Данную стадию проводят путем удаления гидроксилзащитной группы соединения, представленного формулой (M6), через обработку основанием с последующим удалением аминозащитной группы через каталитическое восстановление и щелочной гидролиз полученного соединения. Удаление гидроксилзащитной группы можно проводить в условиях, аналогично приведенным выше для стадии B4, и удаление аминозащитной группы можно проводить в условиях аналогично приведенным на стадии B5.
[0125]
Стадия M8
Стадия M8 представляет собой путь получения соединения, представленного формулами (M8) и (M8'), эпимеризацией или эпифторированием гидроксильной группы в положении 5 соединения, представленного формулой (M4). Данную стадию осуществляют в условиях, аналогично приведенным выше для стадии B3.
[0126]
Стадия M9
Стадия M9 представляет собой путь получения соединения, представленного формулой (M9), путем удаления защитной группы соединения, представленного формулой (M8). Удаление защитной группы осуществляют в условиях, аналогично приведенным выше для стадии M7.
[0127]
Стадия M10
Стадия M10 представляет собой путь получения соединения, представленного формулой (M10), путем удаления защитной группы соединения, представленного формулой (M8'). Удаление защитной группы осуществляют в условиях, аналогично приведенным выше для стадии M7.
[0128]
Способ N
Способ N представляет собой путь получения соединения, представленного формулами (N5), (N7) и (N9), путем получения 5-эпи-6-дезокси, 5,6-дидезокси и 5-эпиамино производных из соединения, представленного формулой (M4), которое получают из апрамицина на 10 стадии, с последующим проведением удаления защиты. Стадии представлены следующим образом.
[Химическая формула 20]

[0129]
Стадия N1
Стадия N1 представляет собой путь получения соединения, представленного формулой (N1), введением метансульфонильной группы в гидроксильную группу в положении 5 соединения, представленного формулой (M4). Введение метансульфонильной группы осуществляют в условиях, аналогично приведенным выше для стадии C1.
[0130]
Стадия N2
Стадия N2 представляет собой путь получения соединения, представленного формулой (N2), сначала путем удаления бензоильной группы соединения, представленного формулой (N1), и одновременного проведения ангидридизации (эпоксидирования) в положениях 5 и 6, и затем введения бензоильной защитной группы в гидроксильную группу в положениях 2" и 6". Эпоксидирование и бензоилирование осуществляют в условиях, аналогично приведенным выше для стадии C2.
[0131]
Стадия N3
Стадия N3 представляет собой путь получения соединения, представленного формулой (N3), путем раскрытия эпоксида соединения, представленного формулой (N2), для преобразования эпоксида в йодид и гидроксильную группу. Данную стадию осуществляют в условиях, аналогично приведенным выше для стадии C3.
[0132]
Стадия N4
Стадия N4 представляет собой путь получения соединения, представленного формулой (N4), путем восстановления йодида в положении 6 соединения, представленного формулой (N3). Восстановление осуществляют в условиях, аналогично приведенным выше для стадии C4.
[0133]
Стадия N5
Стадия N5 представляет собой путь получения соединения, представленного формулой (N5), путем удаления защитной группы соединения, представленного формулой (N4). Удаление защитной группы осуществляют в условиях, аналогично приведенным выше для стадии M7.
[0134]
Стадия N6
Стадия N6 представляет собой путь получения соединения, представленного формулой (N6), введением бензилсульфонильной группы в гидроксильную группу в положении 5 соединения, представленного формулой (N3), и затем добавлением воды, с последующей реакцией элиминирования. Введение бензилсульфонильной группы осуществляют в условиях, аналогично приведенным выше для стадии G4. Реакционная температура после добавления воды составляет от 40°C до 90°C, и время реакции составляет 1-5 часов.
[0135]
Стадия N7
Стадия N7 представляет собой путь получения соединения, представленного формулой (N7), путем удаления защитной группы соединения, представленного формулой (N6), и восстановления двойной связи. Данную стадию осуществляют в условиях, аналогично приведенным выше для стадии M7.
[0136]
Стадия N8
Стадия N8 представляет собой путь получения соединения, представленного формулой (N8), азидированием положения 5 соединения, представленного формулой (N1). Азидирование осуществляют в условиях, аналогично приведенным выше для стадии C10.
[0137]
Стадия N9
Стадия N9 представляет собой путь получения соединения, представленного формулой (N9), путем удаления защитной группы соединения, представленного формулой (N8). Удаление защитной группы и преобразование азидной группы в аминогруппу осуществляют в условиях, аналогично приведенным выше для стадии M7.
[0138]
Способ O
Способ O представляет собой путь получения соединения, представленного формулой (O5), из соединения, представленного формулой (I1). Стадии представлены следующим образом.
[Химическая формула 21]
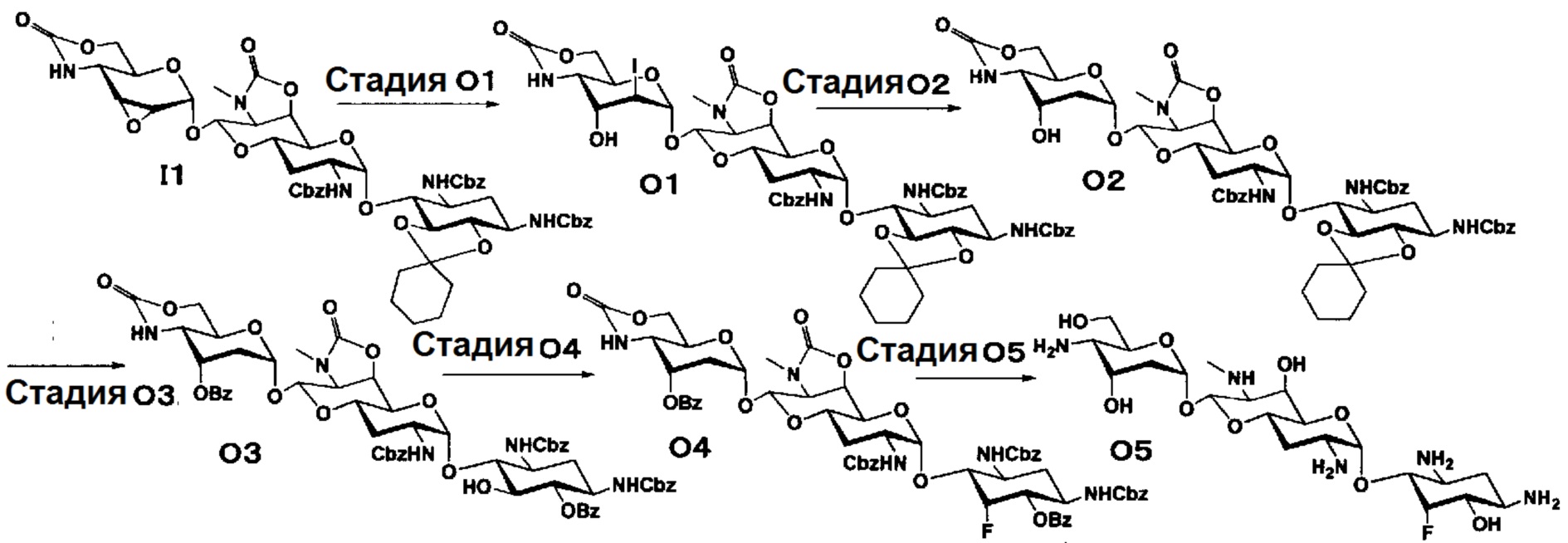
[0139]
Стадия O1
Стадия O1 представляет собой путь получения соединения, представленного формулой (O1), путем раскрытия эпоксида соединения, представленного формулой (I1), для преобразования эпоксида в йодид и гидроксильную группу. Данную стадию осуществляют в условиях, аналогично приведенным выше для стадии C3.
[0140]
Стадия O2
Стадия O2 представляет собой путь получения соединения, представленного формулой (O2), путем восстановления йодида в положении 2" соединения, представленного формулой (O1). Восстановление осуществляют в условиях, аналогично приведенным выше для стадии C4.
[0141]
Стадия O3
Стадия O3 представляет собой путь получения соединения, представленного формулой (O3), путем проведения селективного O-бензоилирования в положениях 6 и 3" соединения, представленного формулой (O2), после удаления циклогексилиденовой группы в положениях 5 и 6. Удаление циклогексилиденовой группы и бензоилирование осуществляют в условиях, аналогично приведенным выше для стадии M4.
[0142]
Стадия O4
Стадия O4 представляет собой путь получения соединения, представленного формулой (O4), путем эпифторирования гидроксильной группы в положении 5 соединения, представленного формулой (O3). Данную стадию осуществляют в условиях, аналогично приведенным выше для стадии B3.
[0143]
Стадия O5
Стадия O5 представляет собой путь получения соединения, представленного формулой (O5), путем удаления защитной группы соединения, представленного формулой (O4). Удаление защитной группы осуществляют в условиях, аналогично приведенным выше для стадии M7.
[0144]
Способ P
Способ P представляет собой путь получения соединения, представленного формулой (P4), путем инверсии положения 5 соединения, представленного формулой (H2), полученного из апрамицина на 5 стадии. Стадии представлены следующим образом.
[Химическая формула 22]

[0145]
Стадия P1
Стадия P1 представляет собой путь получения соединения, представленного формулой (P1), путем удаления циклогексилиденовой группы в положениях 5 и 6 соединения, представленного формулой (H2), и последовательной селективной защиты гидроксильных групп в положениях 6-, 2"- и 6" бензоильными группами. Удаление циклогексилиденовой группы и бензоилирование осуществляют в условиях, аналогично приведенным выше для стадии M4.
[0146]
Стадия P2
Стадия P2 представляет собой путь получения соединения, представленного формулой (P2), введением метансульфонильной группы в свободную гидроксильную группу в положении 5 соединения, представленного формулой (P1). Метансульфонилирование осуществляют в условиях, аналогично приведенным выше для стадии C1.
[0147]
Стадия P3
Стадия P3 представляет собой путь получения соединения, представленного формулой (P3), путем инверсии положения 5 соединения, представленного формулой (P2). Реакция достигается путем взаимодействия соединения, представленного формулой (P2), с ацетатом цезия. Используемые растворители на данной стадии включают диоксан, N,N-диметилформамид, 1,2-диметоксиэтан и тому подобное, и предпочтительно N,N-диметилформамид. Температура реакции составляет от 80°C до 100°C. Время реакции составляет 3-6 часов.
[0148]
Стадия P4
Стадия P4 представляет собой путь получения соединения, представленного формулой (P4), путем удаления защитной группы соединения, представленного формулой (P3). Удаление защитной группы осуществляют в условиях, аналогично приведенным выше для стадии M7.
[0149]
Способ Q
Способ Q представляет собой путь получения соединения, представленного формулой (Q4), путем селективного хлорирования гидроксильной группы в положения 6" соединения, представленного формулой (C4), полученного из апрамицина на 9 стадии, с последующим восстановлением и удалением защиты. Стадии представлены следующим образом.
[Химическая формула 23]

[0150]
Стадия Q1
Стадия Q1 представляет собой путь получения соединения, представленного формулой (Q1), путем удаления бензоильной группы соединения, представленного формулой (C4). Удаление бензоильной группы осуществляют в условиях, аналогично приведенным выше для стадии L1.
[0151]
Стадия Q2
Стадия Q2 представляет собой путь получения соединения, представленного формулой (Q2), путем селективного хлорирования гидроксигруппы в положении 6" соединения, представленного формулой (Q1). Хлорирование осуществляют в условиях, аналогично приведенным выше для стадии L2.
[0152]
Стадия Q3
Стадия Q3 представляет собой путь получения соединения, представленного формулой (Q3), путем восстановления хлорной группы в положении 6" соединения, представленного формулой (Q2). Восстановление осуществляют в условиях, аналогично приведенным выше для стадии L3.
[0153]
Стадия Q4
Стадия Q4 представляет собой путь получения соединения, представленного формулой (Q4), путем удаления защитной группы соединения, представленного формулой (Q3). Удаление защитной группы осуществляют в условиях, аналогично приведенным выше для стадий L4 и B5.
[0154]
Способ R
Способ R представляет собой путь получения соединения, представленного формулой (R5), путем селективного хлорирования гидроксильной группы в положении 6" соединения, представленного формулой (C3), полученного из апрамицина на 8 стадии через 5,6-дидезокси-5-еновое производное, с последующим восстановлением и удалением защиты, и получением соединения, представленного формулой (R6), путем гидрирования положений 5 и 6 данного соединения. Стадии представлены следующим образом.
[Химическая формула 24]

[0155]
Стадия R1
Стадия R1 представляет собой путь получения соединения, представленного формулой (R1), путем бензилсульфонилирования гидроксильной группы в положения 5 соединения, представленного формулой (C3), и затем добавлением воды с последующей реакцией элиминирования. Данную стадию осуществляют в условиях, аналогично приведенным выше для стадии N6.
[0156]
Стадия R2
Стадия R2 представляет собой путь получения соединения, представленного формулой (R2), путем удаления бензоильной группы соединения, представленного формулой (R1). Данную стадию проводят путем взаимодействия соединения, представленного формулой (R1), с основанием. Удаление бензоильной группы осуществляют в условиях, аналогично приведенным выше для стадии G5.
[0157]
Стадия R3
Стадия R3 представляет собой путь получения соединения, представленного формулой (R3), путем селективного хлорирования гидроксильной группы в положении 6" соединения, представленного формулой (R2). Хлорирование осуществляют в условиях, аналогично приведенным выше для стадии L2.
[0158]
Стадия R4
Стадия R4 представляет собой путь получения соединения, представленного формулой (R4), путем восстановления хлорной группы в положении 6" соединения, представленного формулой (R3). Восстановление осуществляют в условиях, аналогично приведенным выше для стадии L3.
[0159]
Стадия R5
Стадия R5 представляет собой путь получения соединения, представленного формулой (R5), путем удаления трет-бутоксикарбонильной группы, бензилоксикарбонильной группы и циклического карбамата соединения, представленного формулой (R4). Удаление трет-бутоксикарбонильной группы осуществляют в условиях, аналогично приведенным выше для стадии L4. Удаление бензилоксикарбонильной группы достигается путем взаимодействия с металлическим натрием в жидком аммиаке. Температура реакции составляет от -70°C до -30°C, и время реакции обычно составляет 1-2 часа. Циклический карбамат может быть удален основным гидролизом. Используемые основания включают гидроксид натрия и гидроксид калия. Данную реакцию осуществляют при температуре от 90°C до 110°C, и время реакции обычно составляет от 0,5 до 1 часа.
[0160]
Стадия R6
Стадия R6 представляет собой путь получения соединения, представленного формулой (R6), путем гидрирования положений 5 и 6 соединения, представленного формулой (R5). Гидрирование достигается путем взаимодействия водорода и катализатора, каталитически восстанавливающего водород. Используемые катализаторы каталитического восстановления для гидрирования включают палладий на углероде, палладиевую чернь, гидроксид палладия, оксид палладия и тому подобное, и предпочтительно оксид палладия. Используемым растворителем предпочтительно является вода. Температура реакции составляет от 10°C до 30°C, и время реакции обычно составляет 1-2 часа.
[0161]
Способ S
Способ S представляет собой путь получения соединения, представленного общей формулой (S1), введением заместителя в аминогруппу в положении 4" соединения, представленного общей формулой (S), и последующим удалением защиты. Стадии представлены следующим образом.
[Химическая формула 25]

[0162]
Стадия S1
Стадия S1 представляет собой путь получения соединения, представленного общей формулой (S1), алкилированием или амидированием аминогруппы в положении 4" соединения, представленного общей формулой (S), с последующим удалением защиты. Данную стадию осуществляют в условиях, аналогично приведенным выше для стадии A4.
[0163]
Стадия S2
Стадия S2 представляет собой путь получения соединения, представленного общей формулой (S2), путем предварительного введения бензильной группы в аминогруппу соединения, представленного общей формулой (S), для моноалкилирования аминогруппы в положении 4". Введение бензильной группы осуществляют в условиях, аналогично приведенным выше для стадии A5.
[0164]
Стадия S3
Стадия S3 представляет собой путь получения соединения, представленного общей формулой (S1), путем алкилирования аминогруппы в положении 4" соединения, представленного общей формулой (S2), с последующим удалением защиты. Данную стадию осуществляют в условиях, аналогично приведенным выше для стадии A6.
[0165]
Способ T
Способ T представляет собой путь получения соединения, представленного общей формулой (T2), введением заместителя в аминогруппу в положении 4" соединения, представленного формулой (R1), полученного из апрамицина на 9 стадии, и последующим удалением защиты. Стадии представлены следующим образом.
[Химическая формула 26]

[0166]
Стадия T1
Стадия T1 представляет собой путь получения соединения, представленного формулой (T1), путем удаления бензоильной группы и трет-бутоксикарбонильной группы соединения, представленного формулой (R1). Удаление защитной группы осуществляют в условиях, аналогично приведенным выше для стадии B4.
[0167]
Стадия T2
Стадия T2 представляет собой путь получения соединения, представленного общей формулой (T2), алкилированием или амидированием свободной аминогруппы в положении 4" соединения, представленного формулой (T1), с последующим удалением защиты. Данную стадию осуществляют в условиях, аналогично приведенным выше для стадии A4.
[0168]
Стадия T3
Стадия T3 представляет собой путь получения соединения, представленного формулой (T3), путем предварительного введения бензильной группы в аминогруппу соединения, представленного формулой (T1), для моноалкилирования аминогруппы в положении 4". Введение бензильной группы осуществляют в условиях, аналогично приведенным выше для стадии A5.
[0169]
Стадия T4
Стадия T4 представляет собой путь получения соединения, представленного общей формулой (T2), путем алкилирования бензилированной аминогруппы в положении 4" соединения, представленного формулой (T3), с последующим удалением защитных бензильных групп. Данную стадию осуществляют в условиях, аналогично приведенным выше для стадии A6.
[0170]
Способ U
Способ U представляет собой путь получения соединения, представленного общей формулой (U4), сначала путем получения свободного аминопроизводного в положении 4" в 3 стадии, с использованием соединение, представленное формулой (M6), полученное из апрамицина на 12 стадии, и введения заместителя в аминогруппу, с последующим удалением защиты. Стадии представлены следующим образом.
[Химическая формула 27]
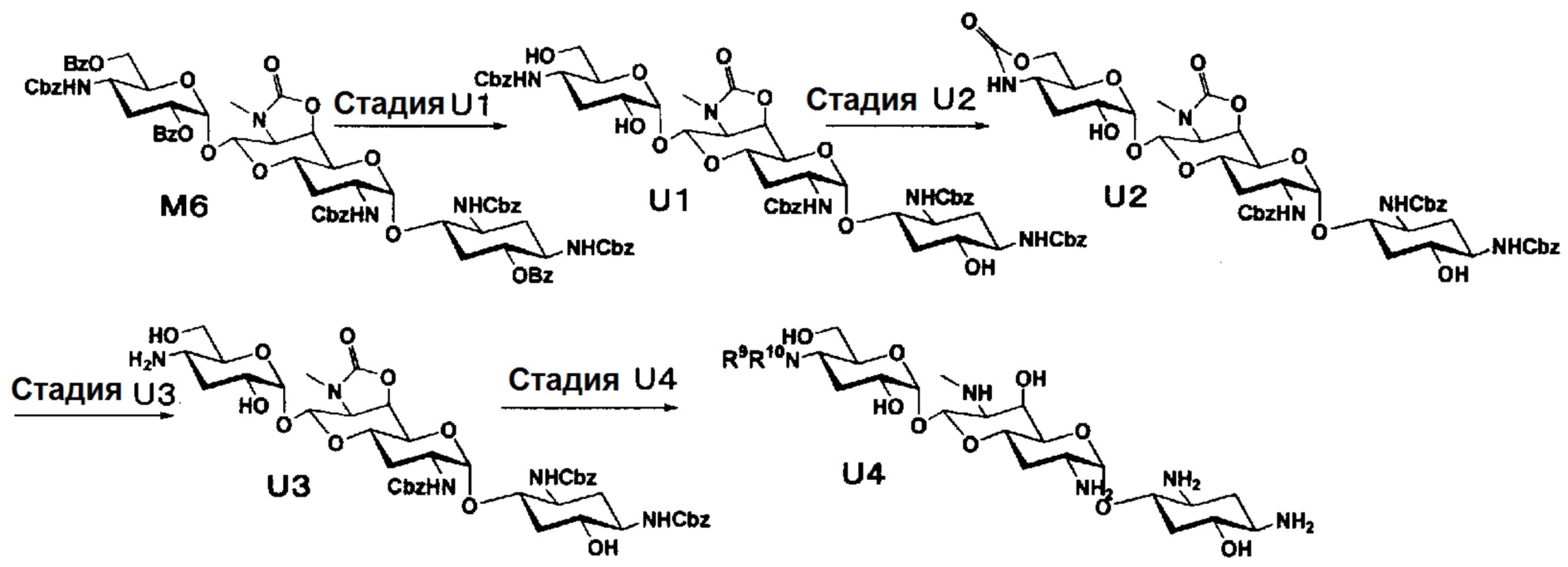
[0171]
Стадия U1
Стадия U1 представляет собой путь получения соединения, представленного формулой (U1), путем удаления бензоильной группы соединения, представленного формулой (M6). Удаление бензоильной группы осуществляют в условиях, аналогично приведенным выше для стадии L1.
[0172]
Стадия U2
Стадия U2 представляет собой путь получения соединения, представленного формулой (U2), путем преобразования положений 4" и 6" соединения, представленного формулой (U1), в циклический карбамат. Преобразование в циклический карбамат осуществляют в условиях, аналогично приведенным выше для стадии A2.
[0173]
Стадия U3
Стадия U3 представляет собой путь получения соединения, представленного формулой (U3), путем гидролиза циклического карбамата в положениях 4" и 6" соединения, представленного формулой (U2), и освобождения аминогруппы в положении 4" и гидроксильной группы в положении 6". Удаление карбамата осуществляют в условиях, аналогично приведенным выше для стадии A3.
[0174]
Стадия U4
Стадия U4 представляет собой путь получения соединения, представленного общей формулой (U4), алкилированием или амидированием аминогруппы в положении 4" соединения, представленного формулой (U3), с последующим удалением защиты. Данную стадию осуществляют в условиях, аналогично приведенным выше для стадии A4.
[0175]
Способ V
Способ V представляет собой путь получения соединения, представленного общей формулой (V1), путем амидирования аминогруппы в положении 4" соединения, представленного общей формулой (V), и последующего удаления защиты. Стадии представлены следующим образом.
[Химическая формула 28]

[0176]
Стадия V1
Стадия V1 представляет собой путь получения соединения, представленного общей формулой (V1), путем ацилирования аминогруппы в положении 4" соединения, представленного общей формулой (V), с последующим удалением защиты. Данную стадию проводят путем взаимодействия соединения общей формулы (V) с различными активными сложными эфирами защитных аминокислот в присутствии основания, с последующим удалением защиты.
[0177]
Используемые активные сложные эфиры на данной стадии включают N-гидроксиамины, S-алкилы, S-фенилы и тому подобное, и среди N-гидроксиаминов предпочтителен N-гидроксисукцинимидный эфир. Основанием предпочтительно является триэтиламин. Все реакционные температуры находятся в диапазоне от 10°C до 30°C, и время реакции составляет 1-24 часа.
[0178]
Удаление трет-бутоксикарбонильной и п-метоксибензилоксикарбонильной групп можно проводить в условиях, аналогично приведенным выше для стадии L4. Удаление бензилоксикарбонильной группы и циклического карбамата осуществляют в условиях, аналогично приведенным выше для стадии A4.
[0179]
Способ W
Способ W представляет собой путь получения соединения, представленного общей формулой (W2), введением заместителя в аминогруппу в положении 4" соединения, представленного формулой (D1), после удаления защитных групп, за исключением бензилоксикарбонильной группы соединения, с последующим удалением защиты. Стадии представлены следующим образом.
[Химическая формула 29]

[0180]
Стадия W1
Стадия W1 представляет собой путь получения соединения, представленного формулой (W1), путем удаления бензоильной группы, трет-бутоксикарбонильной группы и циклического карбамата соединения, представленного формулой (D1). Бензоильная группа и циклический карбамат могут быть удалены основным гидролизом. Используемые основания включают гидроксид натрия и гидроксид калия. Данную реакцию осуществляют при температуре от 10°C до 100°C, и время реакции обычно составляет от 0,5 до 16 часов. Удаление трет-бутоксикарбонильной группы осуществляют в условиях, аналогично приведенным выше для стадии L4.
[0181]
Стадия W2
Стадия W2 представляет собой путь получения соединения, представленного общей формулой (W2), путем ацилирования или амидирования аминогруппы в положении 4" соединения, представленного общей формулой (W1), с последующим удалением защиты. Амидирование и удаление защиты на данной стадии можно проводить в условиях, аналогично приведенным выше для стадии A4, и ацилирование осуществляют в условиях, аналогично приведенным выше для стадии V1.
[0182]
Способ X
Способ X представляет собой путь получения соединения, представленного общей формулой (X4), путем использования соединения, представленного общей формулой (X), в условиях, аналогично приведенным в способе U. Стадии представлены следующим образом.
[Химическая формула 30]

[0183]
Стадия X1
Стадия X1 представляет собой путь получения соединения, представленного формулой (X1), путем удаления бензоильной группы соединения, представленного общей формулой (X). Удаление бензоильной группы осуществляют в условиях, аналогично приведенным выше для стадии L1.
[0184]
Стадия X2
Стадия X2 представляет собой путь получения соединения, представленного формулой (X2), путем преобразования положений 4" и 6" соединения, представленного формулой (X1), в циклический карбамат. Преобразование в циклический карбамат осуществляют в условиях, аналогично приведенным выше для стадии A2.
[0185]
Стадия X3
Стадия X3 представляет собой путь получения соединения, представленного формулой (X3), путем гидролиза циклического карбамата в положениях 4" и 6" соединения, представленного формулой (X2), и освобождения аминогруппы в положении 4" и гидроксильной группы в положении 6". Удаление карбамата осуществляют в условиях, аналогично приведенным выше для стадии A3.
[0186]
Стадия X4
Стадия X4 представляет собой путь получения соединения, представленного общей формулой (X4), алкилированием или амидированием аминогруппы в положении 4" соединения, представленного формулой (X3), с последующим удалением защиты. Данную стадию осуществляют в условиях, аналогично приведенным выше для стадии A4.
[0187]
Способ Y
Способ Y представляет собой путь получения соединения, представленного формулой (Y3), путем использования соединения, представленного формулой (O3), в условиях, аналогично приведенным в способе P. Стадии представлены следующим образом.
[Химическая формула 31]

[0188]
Стадия Y1
Стадия Y1 представляет собой путь получения соединения, представленного формулой (Y1), введением метансульфонильной группы в свободную гидроксильную группу в положении 5 соединения, представленного формулой (O3). Метансульфонилирование осуществляют в условиях, аналогично приведенным выше для стадии C1.
[189]
Стадия Y2
Стадия Y2 представляет собой путь получения соединения, представленного формулой (Y2), путем инверсии положения 5 соединения, представленного формулой (Y1). Данную реакцию осуществляют в условиях, аналогично приведенным выше для стадии P3.
[0190]
Стадия Y3
Стадия Y3 представляет собой путь получения соединения, представленного формулой (Y3), путем удаления защитной группы соединения, представленного формулой (Y2). Удаление защитной группы осуществляют в условиях, аналогично приведенным выше для стадии M7.
[0191]
Соединения настоящего изобретения и приведенные выше соединения, полученные на стадиях их получения, могут быть очищены и выделены общепринятыми способами очистки. Для очистки и выделения могут быть использованы способы, такие как, например, способ жидкостного разделения, способ дистилляции, техника сублимации, способ осаждения, способ кристаллизации, колоночная хроматография с нормальной и обращенной фазой с использованием силикагеля в качестве упаковочного материала, колоночная хроматография с использованием ионообменной смолы, такой как Amberlite CG-50, Dowex 50W X 2 или CM-sephadex C-25 и тому подобное, колоночная хроматография с использованием целлюлозы и тому подобное, способ препаративной тонкослойной хроматографии или высокоэффективной жидкостной хроматографии и тому подобное. Кроме того, соединения, полученные на приведенных выше стадиях получения, также могут быть получены соответствующим образом для использования на последующей стадии без дальнейшего выделения или очистки.
[0192]
Применение аминогликозидного антибиотика
Соединение настоящего изобретения или его фармацевтически приемлемая соль или сольват обладают широким антибактериальным спектром против различных грамположительных бактерий и грамотрицательных бактерий среди патогенности бактерий. Кроме того, соединение настоящего изобретения или его фармацевтически приемлемая соль или сольват обладает превосходной антибактериальной активностью против бактерий, вызывающих инфекционные заболевания (MRSA, Staphylococcus aureus, Escherichia coli, Klebsiella pneumonia, Pseudomonas aeruginosa и тому подобное), поэтому могут использоваться в качестве антибактериального средства.
[0193]
Таким образом, в соответствии с другими вариантами осуществления настоящего изобретения, предоставлено антибактериальное средство, содержащее соединение по настоящему изобретению. Кроме того, в соответствии с другим вариантом осуществления настоящего изобретения, предоставлено применение соединения настоящего изобретения или его фармацевтически приемлемой соли или сольвата для получения антибактериального средства.
[0194]
Как указано выше, соединение настоящего изобретения или его фармацевтически приемлемая соль или сольват могут с пользой применяться в качестве противомикробного средства или лекарственного средства для профилактики или лечения инфекционных заболеваний. Поэтому, в соответствии с другим вариантом осуществления настоящего изобретения, предоставлен способ профилактики или лечения инфекционных заболеваний, включающий введение терапевтически эффективного количества соединения настоящего изобретения или его фармацевтически приемлемой соли или сольвата животным, включая людей. Целевыми инфекционными заболеваниями предпочтительно являются бактериальные инфекционные заболевания, включающие, например, сепсис, инфекционный эндокардит, дерматологические инфекции, инфекции областей хирургического вмешательства, ортопедические инфекции областей хирургического вмешательства, респираторные инфекции, инфекции мочевыводящих путей, энтеральные инфекции, перитонит, менингит, офтальмологические инфекции или отоларингологические инфекции, и предпочтительно гнойные кожные заболевания, вторичные инфекции, вызванные ожогом/хирургическими разрезами, пневмонией, эндобронхиальными инфекциями, туберкулезом, пиелонефритом, энтеритом (включая пищевые отравления), конъюнктивитом, средним отитом или подобными. Целевыми животными для профилактики или лечения предпочтительно являются млекопитающие, и более предпочтительно люди. Также, доза соединения настоящего изобретения или его фармацевтически приемлемой соли или сольвата соответствующим образом определяется специалистом в данной области в зависимости от пути введения, видов патогенов, возраста, пола и массы тела пациента и тяжести заболевания. В случае перорального введения человеку, например, соединение настоящего изобретения может быть введено для взрослого в дозе от 0,1 до 1000 мг/кг/день, и в случае внутривенного введения, оно может быть введено в дозе от 0,01 до 100 мг/кг/день на взрослого.
[0195]
В соответствии с дополнительным вариантом осуществления настоящего изобретения предоставлено следующее.
(1) Соединение настоящего изобретения или его фармацевтически приемлемая соль или сольват для применения в терапии.
(2) Соединение настоящего изобретения или его фармацевтически приемлемая соль или сольват для применения при профилактике или лечении инфекционного заболевания.
(3) Применение соединения настоящего изобретения или его фармацевтически приемлемой соли или сольвата при изготовлении лекарственного средства для профилактики или лечения инфекционного заболевания.
(4) Применение соединения настоящего изобретения или его фармацевтически приемлемой соли или сольвата для профилактики или лечения инфекционного заболевания.
[0196]
Соединение настоящего изобретения или его фармацевтически приемлемая соль или сольват обладает антибактериальной активностью против грамположительных и грамотрицательных бактерий с множественной лекарственной резистентностью, которые не поддаются лечению имеющимися в настоящее время антибиотиками. Соединение настоящего изобретения или его фармацевтически приемлемая соль или сольват особенно полезны для профилактики или лечения серьезных инфекционных заболеваний, вызванных MRSA или грамотрицательными бактериями с множественной лекарственной резистентностью, и тому подобное.
[0197]
Соединение настоящего изобретения или его фармацевтически приемлемая соль или сольват может быть введено животному в виде фармацевтической композиции, содержащей фармацевтически приемлемые добавки, при желании. Поэтому, в соответствии с другим вариантом осуществления настоящего изобретения, предоставлена композиция, в частности фармацевтическая композиция, содержащая соединение настоящего изобретения или его фармацевтически приемлемую соль или сольват.
[0198]
Фармацевтическая композиция настоящего изобретения может быть введена посредством перорального или парентерального пути введения (например, внутривенной инъекцией, внутримышечной инъекцией, подкожным введением, ректальным введением, чрескожным введением, местным глазным введением, легочным введением) всем животным, включая людей, в зависимости от типов патогенов, заболеваний и природы пациента. Поэтому, для фармацевтического компонента настоящего изобретения можно подобрать подходящее формулирование в зависимости от путей введения. Такие формулированные составы, например, могут быть скорректированы для парентеральных инъекций, главным образом используемых для внутривенного введения, внутримышечных инъекций и тому подобное; для перорального средства, такого как пероральные капсулы, таблетки, гранулы, порошки, пилюли, тонкие гранулы, сиропы, пастилки и тому подобное; для внешних препаратов для парентерального введения, таких как мази, капли для глаз, ушные капли, назальные капли, глазные мази, кожно-слизистый абсорбенты, дерматологические средства, вдыхаемые летучие вещества, суппозитории и тому подобное; для других сухих порошковых или распыляемых аэрозольных составов, и тому подобное.
[0199]
Приведенный выше сформулированный состав может быть получен с использованием добавок, таких как эксципиенты, агенты наполнители, связующие, смачивающие агенты, дезинтегрирующие агенты, поверхностно-активные вещества, лубриканты, диспергирующие агенты, буферирующие агенты, консерванты, солюбилизаторы, антисептики, ароматизаторы, анальгетики, стабилизаторы и тому подобное, в рамках обычной процедуры. Конкретные примеры доступных нетоксичных добавок включают солюбилизаторы и солюбилизирующие агенты (дистиллированная вода для инъекций, солевой раствор, этанол, глицерин, пропиленгликоль, кукурузное масло, кунжутное масло и тому подобное), которые могут представлять собой водные растворы или составы для разведения перед применением для парентеральных инъекций, глазные капли, ушные капли и капли в нос; регуляторы pH (аддитивные соли неорганических кислот: тринатрийортофосфат, бикарбонат натрия и тому подобное; соли органических кислот: цитрат натрия и тому подобное, соли органических оснований: L-лизин, L-аргинин и тому подобное); придающие изотоничность агенты (хлорид натрия, глюкоза, глицерин и тому подобное); буферирующие агенты (хлорид натрия, бензалконийхлорид, цитрат натрия и тому подобное); поверхностно-активные вещества (моноолеат сорбитана, полисорбат 80 и тому подобное); диспергирующие агенты (D-маннитол и тому подобное); стабилизаторы (антиоксиданты: аскорбиновая кислота, сульфит натрия, пиросульфит натрия и тому подобное, хелатирующие агенты: лимонная кислота, винная кислота и тому подобное). Кроме того, соответствующее формулирование компонентов в виде мазей, кремов и пластырей для глазной мази, кожно-слизистых абсорбентов и дерматологических средств включают белый вазелин, макрогол, глицерин, жидкий парафин, хлопчатобумажную ткань и тому подобное. Кроме того, жидкие вдыхаемые летучие вещества включают регуляторы pH (цитрат натрия, гидроксид натрия и тому подобное), придающие изотоничность агенты (хлорид натрия, бензалконийхлорид, цитрат натрия и тому подобное) и буферирующие агенты (хлорид натрия, бензалконийхлорид, цитрат натрия и тому подобное), и порошковые ингаляторы включают лактозу и тому подобное в качестве носителя. Также, перорально вводимые средства и суппозитории включают эксципиенты (лактоза, D-маннитол, кукурузный крахмал, кристаллическая целлюлоза и тому подобное), дезинтегрирующие агенты (карбоксиметилцеллюлоза, кальцийкарбоксиметилцеллюлоза и тому подобное), связующие (гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, поливинилпирролидон и тому подобное), лубриканты (стеарат магния, тальк и тому подобное), покрывающие агенты (шеллак очищенный, гидроксипропилметилцеллюлоза, сахароза, оксид титана и тому подобное), пластификаторы (глицерин, полиэтиленгликоль и тому подобное), субстраты (масло какао, полиэтиленгликоль, твердый жир и тому подобное), и тому подобное.
[0200]
Также, при рассмотрении вопроса повышения эффективности соединения настоящего изобретения для профилактики или лечения инфекционных заболеваний, к фармацевтической композиции настоящего изобретения может быть добавлен отличный от соединения настоящего изобретения один или более пригодных с клинической точки зрения существующих антибиотиков (например, β-лактамные антибиотики (карбапенемы, цефалоспорины, цефамицины, пенициллины), гликопептидные антибиотики, ансамицинные антибиотики, аминогликозидные антибиотики, хинолоновые антибиотики, монобактамные антибиотики, макролидные антибиотики, тетрациклиновые антибиотики, хлорамфениколовые антибиотики, линкомициновые антибиотики, стрептограминовые антибиотики, оксазолидиноновые антибиотики, фосфомицины, новобиоцины, циклосерины, моеномицины и тому подобное). Альтернативно, соединение настоящего изобретения может быть введено совместно с приведенными выше антибиотиками в живые организмы. Кроме того, при рассмотрении вопроса расширения или повышения эффективности фармацевтической композиции настоящего изобретения против грамотрицательных бактерий и бактерий с множественной лекарственной резистентностью в отношении доступных в настоящее время антибиотиков, фармацевтическая композиция настоящего изобретения может также включать подающий лекарство насос (эффлюксный насос) для ингибирования или ингибитор существующего антибактериально разлагающего фермента (β-лактамаза и тому подобное), и может вводиться в живой организм вместе с этими ингибиторами. Кроме того, при рассмотрении улучшения профилактического или терапевтического эффектов в отношении инфекционных заболеваний, фармацевтическая композиция настоящего изобретения может быть использована в комбинации с соединениями, обладающими антибактериальной активностью (например, лекарственными средствами для лечения осложнений), и настоящее изобретение также включает такой вариант осуществления.
Примеры
[0201]
Настоящее изобретение объясняется в деталях с помощью примеров, но не ограничивается указанными примерами.
[0202]
Пример 1: Синтез 4"-N-бензил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонилапрамицина (A5) и 4"-N-метилапрамицина (A4-a)
[Химическая формула 32]

[0203]
Пример 1-(i): Синтез 4"-N-бензил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонилапрамицина (A5)
Раствор, полученный добавлением 15 мл триэтиламина и 6 мл бензальдегида к раствору 20,4 г (21 ммоль) соединения, представленного формулой (A3), описанному в патенте США 2013/0165395A1, растворенного в 200 мл метанола, перемешивали при комнатной температуре в течение 2 часов. Затем, после добавления 1,6 г NaBH4, полученную в результате смесь подвергали взаимодействию при комнатной температуре в течение 10 минут. Полученный реакционный раствор концентрировали при пониженном давлении и промывали водой. После сушки, полученный в результате остаток промывали изопропиловым эфиром, что давало 21,2 г (95%) указанного в заголовке соединения (A5) в виде белого твердого вещества.
[0204]
МС (ESI) m/z: 1081 (M+Na)+.
[0205]
Пример 1-(ii): Синтез 4"-N-метилапрамицина (A4-a)
Смесь, полученную добавлением 0,1 мл 37% раствора формалина и 10 мг NaBH3CN к раствору 550 мг (0,51 ммоль) соединения (A5) примера 1-(i), растворенного в 10 мл смеси 10% уксусная кислота-метанол, подвергали взаимодействию при комнатной температуре в течение 13 часов. После завершения реакции, смесь концентрировали при пониженном давлении и промывали водой. После сушки, остаток растворяли в 5,2 мл 50% водного 1,4-диоксана, и к полученному раствору добавляли 0,5 мл уксусной кислоты и палладиевую чернь, и каталитическое восстановление проводили в атмосфере водорода при комнатной температуре в течение 10 часов. После завершения реакции, реакционную смесь нейтрализовали добавлением NH4OH и, после фильтрования, концентрировали при пониженном давлении. После сушки, остаток растворяли в 2,5 мл воды, и полученную в результате смесь нагревали до 110°C, к которой добавляли 2,5 мл 1 N водного гидроксида калия. Полученную смесь подвергали взаимодействию в течение 2 часов. После завершения реакции, реакционную смесь нейтрализовали добавлением 1 N водн. HCl при охлаждении льдом и очищали ионообменной хроматографией (CG50), что давало 152 мг (54%) указанного в заголовке соединения (A4-a).
[0206]
МС (ESI) m/z: 554 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 2,77 (6H, с, 4"-NMe и 7'-NMe), 5,36 (1H, д, H-1') и 5,68 (1H, д, H-1").
[0207]
Пример 2: Синтез 4"-N-(3-аминопропил)апрамицина (A4-b)
[Химическая формула 33]

[0208]
Указанное в заголовке соединение (A4-b) [87,1 мг (46%)] получали способом, аналогично примеру 1-(ii), с использованием 333 мг (0,32 ммоль) соединения (A5) примера 1-(i) и 80 мг 3-[(бензилоксикарбонил)амино]пропиональдегида.
[0209]
МС (ESI) m/z: 597 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,91-2,05 (3H, м, 4"-NH2Pr(β) и H-3' акс), 2,94-3,09 [6H, м, H-1 и 7' и 4"-NH2Pr(α, γ)], 5,28 (1H, д, H-1") и 5,67 (1H, д, H-1').
[0210]
Пример 3: Синтез 4"-N-((1-аминоциклопентил)метил)апрамицина (A4-c)
[Химическая формула 34]

[0211]
Смесь, полученную добавлением 80 мг N-Boc-2-аминоацетальдегида и 10 мг NaBH3CN к раствору 300 мг (0,30 ммоль) соединения (A5) примера 1-(i), растворенного в 6 мл смеси 10% уксусная кислота-метанол, подвергали взаимодействию при комнатной температуре в течение 16 часов. После завершения реакции, смесь концентрировали при пониженном давлении, и остаток растворяли в 10 мл раствора 90% ТФУК-MeOH. Полученную в результате смесь подвергали взаимодействию при комнатной температуре в течение 2 часов. Полученный реакционный раствор концентрировали при пониженном давлении и промывали водой. Остаток растворяли в 10 мл 50% водного 1,4-диоксана, и к полученному раствору добавляли 0,5 мл уксусной кислоты и палладиевую чернь, и каталитическое восстановление проводили в атмосфере водорода при комнатной температуре в течение 10 часов. После завершения реакции, смесь нейтрализовали добавлением NH4OH и, после фильтрования, концентрировали при пониженном давлении. После сушки, остаток растворяли в 2,5 мл воды, и полученную в результате смесь нагревали до 110°C, к которой добавляли 2,5 мл 1 N водного гидроксида калия. Полученную смесь подвергали взаимодействию в течение 2 часов. После завершения реакции, реакционную смесь нейтрализовали добавлением 1 N водн. HCl при охлаждении льдом и очищали ионообменной хроматографией (CG50), что давало 87,5 мг (46%) указанного в заголовке соединения (A4-c).
[0212]
МС (ESI) m/z: 637 (M+1)+;1H ЯМР (ТФУК соль, 500 МГц, D2O): δ 1,98 (1H, кв, J=12 Гц, H-3' акс), 2,33 (1H, дт, J=4,5, 4,5 и 12 Гц, H-3' экв), 2,45 (1H, дт, J=4, 4 и 12,5 Гц, H-2 экв), 2,74 (3H, с, NCH3), 2,90 (1H, слегка ушир. т, J=10 Гц, H-4"), 3,16 (1H, д, J=14 Гц), 3,22 (1H, д, J=14 Гц), 3,32 (1H, дд, J=3 и 8,5 Гц, H-7'), 3,71 (1H, дд, J=2,5 и 10 Гц, H-5'), 4,51 (1H, т, J=2,5 Гц, H-6'), 5,16 (1H, д, J=8,5 Гц, H-8'), 5,39 (1H, д, J=4 Гц, H-1") и 5,68 (1H, д, J=3,8 Гц, H-1').
[0213]
Пример 4: Синтез 4"-N-(1,3-диаминопропан-2-ил)апрамицина (A4-d)
[Химическая формула 35]

[0214]
Указанное в заголовке соединение (A4-d) [80,6 мг (53%)] получали способом, аналогично примеру 1-(ii), с использованием 250 мг (0,26 ммоль) соединения, представленного формулой (A3), описанному в патенте США 2013/0165395A1, и 115 мг 1,3-дибензилоксикарбониламиноацетона.
[0215]
МС (ESI) m/z: 612 (M+1)+;1H ЯМР (ТФУК соль, 500 МГц, D2O): δ 1,81 (1H, кв, J=12,5 Гц, H-2 акс), 1,98 (1H, кв, J=12 Гц, H-3' акс), 2,33 (1H, дт, J=4, 4 и 12 Гц, H-3' экв), 2,45 (1H, дт, J=4, 4 и 12,5 Гц, H-2 экв), 2,66 (1H, т, J=10,5 Гц, H-4 "), 2,73 (3H, с, NCH3), 3,31 (1H, дд, J=3 и 8,5 Гц, H-7'), 4,51 (1H, т, J=~3 Гц, H-6'), 5,15 (1H, д, J=8,5 Гц, H-8'), 5,37 (1H, д, J=4 Гц, H-1") и 5,67 (1H, д, J=3,8 Гц, H-1').
[0216]
Пример 5: Синтез 4"-N,N-бис(2-аминоэтил)апрамицина (A4-e)
[Химическая формула 36]

[0217]
Указанное в заголовке соединение (A4-e) [74,3 мг (44%)] получали способом, аналогично примеру 3, с использованием 260 мг (0,27 ммоль) соединения, представленного формулой (A3), описанного в патенте США 2013/0165395A1, и 127 мг N-Boc-2-аминоацетальдегида.
[0218]
МС (ESI) m/z: 626 (M+1)+;1H ЯМР (ТФУК соль, 500 МГц, D2O): δ 1,81 (1H, кв, J=12,5 Гц, H-2 акс), 1,98 (1H, кв, J=12 Гц, H-3' акс), 2,33 (1H, дт, J=4,5, 4,5 и 12 Гц, H-3' экв), 2,45 (1H, дт, J=4,5, 4,5 и 12,5 Гц, H-2 экв), 2,73 (3H, с, NCH3), 2,75 (1H, т, J=10,5 Гц, H-4"), 3,27 (1H, ддд, J=4,5, 10 и 12,5 Гц, H-1), 3,30 (1H, дд, J=3 и 8,5 Гц, H-7'), 4,51 (1H, т, J=2,5 Гц, H-6'), 5,15 (1H, д, J=8,5 Гц, H-8'), 5,36 (1H, д, J=4 Гц, H-1") и 5,67 (1H, д, J=3,8 Гц, H-1').
[0219]
Пример 6: Синтез 4"-N-[(1S,4S)-4-(трет-бутоксикарбонил)аминоциклогексил]-4"-N-бензил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонилапрамицина (A3-a), 4"-N-[(1R,4R)-4-(трет-бутоксикарбонил)аминоциклогексил]-4"-N-бензил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонилапрамицина (A3-b) и 4"-N-(цис-1,4-4-аминоциклогексил)апрамицина (A4-f)
[Химическая формула 37]

[0220]
Пример 6-(i): Синтез 4"-N-[(1S,4S)-4-(трет-бутоксикарбонил)аминоциклогексил]-4"-N-бензил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонилапрамицина (A3-a) и 4"-N-[(1R,4R)-4-(трет-бутоксикарбонил)аминоциклогексил]-4"-N-бензил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонилапрамицина (A3-b)
Раствор, полученный добавлением 85,2 мг 4-(трет-бутоксикарбонил)аминоциклогексанона и 10 мг NaBH3CN к раствору 260 мг (0,27 ммоль) соединения, представленного формулой (A3), растворенного в 5 мл смеси 10% уксусная кислота-метанол, подвергали взаимодействию при комнатной температуре в течение 16 часов. Полученный реакционный раствор концентрировали при пониженном давлении, и осадок, образованный добавлением насыщенного раствора бикарбоната натрия, отфильтровывали. Полученное в результате твердое вещество очищали колоночной хроматографией на силикагеле (хлороформ: метанол=10:1), что давало 122 мг (36%) указанного в заголовке соединения (A3-a) и 97,1 мг (31%) указанного в заголовке соединения (A3-b).
[0221]
МС (ESI) m/z: (A3-a), 1187 (M+Na)+; (A3-b), 1187 (M+Na)+.
[0222]
Пример 6-(ii): Синтез 4"-N-(цис-1,4-4-аминоциклогексил)апрамицина (A4-f)
Раствор, полученный растворением 110 мг (0,095 ммоль) указанного в заголовке соединения (A3-a) примера 6-(i), растворенного в 1 мл 90% ТФУК-MeOH, подвергали взаимодействию при комнатной температуре в течение 2 часов. Полученный реакционный раствор концентрировали при пониженном давлении и растворяли в 1 мл 50% смеси 1,4-диоксан-вода, и к данной смеси добавляли 0,1 мл уксусной кислоты и палладиевую чернь. Затем, полученную в результате смесь подвергали каталитическому восстановлению в атмосфере водорода при комнатной температуре в течение 10 часов. После завершения реакции, смесь нейтрализовали добавлением NH4OH и, после фильтрования, концентрировали при пониженном давлении. После сушки, остаток растворяли в воде (1 мл) и нагревали до 110°C, и добавляли 1 N водный гидроксид калия (0,5 мл). Полученную в результате смесь подвергали взаимодействию в течение 2 часов при той же самой температуре, как приведено выше. После завершения реакции, реакционную смесь нейтрализовали добавлением 1 N водн. HCl при охлаждении льдом и очищали ионообменной хроматографией (CG50), что давало 34,5 мг (52%) указанного в заголовке соединения (A4-f).
[0223]
МС (ESI) m/z: 737 (M+1)+;1H ЯМР (ТФУК соль, 500 МГц, D2O): δ 2,34 (1H, дт, J=4,5, 4,5 и 11,5 Гц, H-3' экв), 2,46 (1H, дт, J=4, 4 и 12,5 Гц, H-2 экв), 2,76 (3H, с, NCH3), 3,34 (1H, дд, J=3 и 8,5 Гц, H-7'), 3,40 (1H, т, J=10 Гц, H-4"), 3,95 (1H, т, J=10 Гц, H-3"), 4,53 (1H, слегка ушир. т, J=~3 Гц, H-6'), 5,18 (1H, д, J=8,5 Гц, H-8'), 5,46 (1H, д, J=4 Гц, H-1") и 5,68 (1H, д, J=3,8 Гц, H-1').
[0224]
Пример 7: Синтез 4"-N-(транс-1,4-4-аминоциклогексил)апрамицина (A4-g)
[Химическая формула 38]

[0225]
Указанное в заголовке соединение (A4-g) [26,8 мг (50%)] получали способом, аналогично примеру 6-(ii), с использованием 90,1 мг (0,077 ммоль) указанного в заголовке соединения (A3-b) примера 6-(i).
[0226]
МС (ESI) m/z: 737 (M+1)+;
1H ЯМР (ТФУК соль, 500 МГц, D2O): δ 1,83 (1H, кв, J=12,5 Гц, H-2 акс), 1,99 (1H, кв, J= 12 Гц, H-3' акс), 2,46 (1H, дт, J=4, 4 и 12,5 Гц, H-2 экв), 2,75 (3H, с, NCH3), 3,33 (1H, дд, J=3 и 8,5 Гц, H-7'), 3,38 (1H, т, J=10 Гц, H-4"), 4,52 (1H, слегка ушир. т, J=~2,5 Гц, H-6'), 5,18 (1H, д, J=8,5 Гц, H-8'), 5,45 (1H, д, J=4 Гц, H-1") и 5,69 (1H, д, J=3,8 Гц, H-1').
[0227]
Пример 8: Синтез 4"-N-(азетидин-3-ил)-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонилапрамицина (A3-c) и 4"-N-(азетидин-3-ил)апрамицина (A4-h)
[Химическая формула 39]

[0228]
Пример 8-(i): Синтез 4"-N-(азетидин-3-ил)-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонилапрамицина (A3-c)
Раствор, полученный добавлением 74,5 мг 1-Boc-3-азетидинона и 10 мг NaBH3CN к раствору 300 мг (0,29 ммоль) соединения, представленного формулой (A3), растворенного в 6 мл смеси 10% уксусная кислота-метанол, подвергали взаимодействию при комнатной температуре в течение 16 часов. После завершения реакции, смесь концентрировали при пониженном давлении, и остаток растворяли в 5 мл раствора 90% ТФУК-MeOH, и полученную в результате смесь подвергали взаимодействию при комнатной температуре в течение 2 часов. Полученный реакционный раствор концентрировали при пониженном давлении, и осадок, образованный при добавлении к остатку насыщенного водного раствора бикарбоната натрия, отфильтровывали, и осадок после фильтрования сушили при пониженном давлении, что давало 284 мг (90%) указанного в заголовке соединения (A3-c) в виде белого твердого вещества.
[0229]
МС (ESI) m/z: 1045 (M+Na)+.
[0230]
Пример 8-(ii): Синтез 4"-N-(азетидин-3-ил)апрамицина (A4-h)
Смесь, полученную добавлением 0,2 мл уксусной кислоты и палладиевой черни к раствору 105 мг (0,1 ммоль) указанного в заголовке соединения (A3-c) примера 8-(i), растворенного в 2 мл 50% смеси 1,4-диоксан-вода, подвергали каталитическому восстановлению в атмосфере водорода при комнатной температуре в течение 10 часов. После завершения реакции, смесь нейтрализовали добавлением NH4OH и, после фильтрования, концентрировали при пониженном давлении. Остаток растворяли в воде (1 мл) и нагревали до 110°C, и добавляли 1 N раствор водного гидроксида калия (1 мл). Полученную в результате смесь подвергали взаимодействию в течение 2 часов при этой же температуре. После завершения реакции, реакционную смесь нейтрализовали добавлением 1 N водн. HCl при охлаждении льдом и очищали ионообменной хроматографией (CG50), что давало 36,2 мг (61%) указанного в заголовке соединения (A4-h).
[0231]
МС (ESI) m/z: 595 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 2,75 (3H, с, NMe), 3,5-3,75 (5H, м, азетидин), 5,51 (1H, д, J=3,5 Гц, H-1") и 5,73 (1H, д, J=3 Гц, H-1').
[0232]
Пример 9: Синтез 4"-N-(1-метилазетидин-3-ил)апрамицина (A4-i)
[Химическая формула 40]

[0233]
Указанное в заголовке соединение (A4-i) [33,2 мг (42%)] получали путем процесса удаления защиты, аналогично примеру 1-(ii), с использованием 130 мг (0,13 ммоль) указанного в заголовке соединения (A3-c) примера 8-(i).
[0234]
МС (ESI) m/z: 609 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 2,25 (3H, с, NMe), 2,75 (3H, с, NMe), 5,53 (1H, д, J=3,5 Гц, H-1") и 5,77 (1H, д, J=3 Гц, H-1').
[0235]
Пример 10: Синтез 4"-дезамино-4"-гуанидиноапрамицина (A4-j)
[Химическая формула 41]

[0236]
Раствор, полученный добавлением 0,16 мл триэтиламина и 420 мг 1,3-бис(трет-бутоксикарбонил)-2-(трифторметансульфонил)гуанидина (реагент Гудмана) к раствору 303 мг (0,31 ммоль) соединения, представленного формулой (A3), растворенного в 6,7 мл смешанного раствора метиленхлорид:метанол (10:1), подвергали взаимодействию при 40°C в течение 48 часов. После завершения реакции, реакционный раствор концентрировали при пониженном давлении и промывали водой. После сушки, смесь растворяли в 6 мл 90% ТФУК-MeOH, и полученную в результате смесь подвергали взаимодействию при комнатной температуре в течение 1 часа. После завершения реакции, смесь концентрировали при пониженном давлении. Остаток растворяли в 5,4 мл 50% водного 1,4-диоксана и 0,5 мл уксусной кислоты, и добавляли палладиевую чернь, и полученную в результате смесь подвергали каталитическому восстановлению в атмосфере водорода при комнатной температуре в течение 10 часов. После завершения реакции, смесь нейтрализовали добавлением NH4OH и, после фильтрования, концентрировали при пониженном давлении. Остаток растворяли в 1 мл воды, и добавляли 1 мл 1 M водн. KOH, нагретого до 105°C, и полученную смесь подвергали взаимодействию в течение 15 минут. После завершения реакции, смесь нейтрализовали добавлением 1 N HCl при охлаждении льдом и, после фильтрования, концентрировали при пониженном давлении. Полученный в результате остаток очищали ионообменной хроматографией (CG50), что давало 85 мг (47%) указанного в заголовке соединения (A4-j).
[0237]
МС (ESI) m/z: 582 (M+1)+;1H ЯМР (ТФУК соль, 500 МГц, D2O): δ 1,81 (1H, кв, J=13 Гц, H-2 акс), 1,99 (1H, кв, J=12 Гц, H-3' акс), 2,33 (1H, дт, J=4,5, 4,5 и 12 Гц, H-3' экв), 2,45 (1H, дт, J=4, 4 и 13 Гц, H-2 экв), 2,74 (3H, с, NCH3), 3,32 (1H, дд, J=3 и 8,5 Гц, H-7'), 3,51 (1H, т, J=10 Гц, H-4"), 4,52 (1H, т, J=3 Гц, H-6'), 5,17 (1H, д, J=8,5 Гц, H-8'), 5,44 (1H, д, J=4 Гц, H-1") и 5,68 (1H, д, J=3,8 Гц, H-1'),13C ЯМР (DCl-D2O, 125 МГц): δ 157,52 (C=NH).
[0238]
Пример 11: Синтез 4"-N-(2-аминоэтил)-4"-N-бензил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонилапрамицина (A5-a) и 4"-N-гуанидиноэтилапрамицина (A4-k)
[Химическая формула 42]

[0239]
Пример 11-(i): Синтез 4"-N-(2-аминоэтил)-4"-N-бензил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонилапрамицина (A5-a)
Указанное в заголовке соединение (A5-a) [644 мг (89%)] получали способом, аналогично примеру 8-(i), с использованием 684 мг (0,66 ммоль) указанного в заголовке соединения (A5) примера 1-(i) и 100 мг N-Boc-2-аминоацетальдегида.
[0240]
МС (ESI) m/z: 1123 (M+Na)+.
[0241]
Пример 11-(ii): Синтез 4"-N-гуанидиноэтилапрамицина (A4-k)
Указанное в заголовке соединение (A4-k) [96,8 мг (55%)] получали способом, аналогично примеру 10, с использованием 300 мг (0,27 ммоль) указанного в заголовке соединения (A5-a) примера 11-(i) и 120 мг N,N'-ди-Boc-N"-трифлилгуанидин (реагент Гудмана).
[0242]
МС (ESI) m/z: 625 (M+1)+;1H ЯМР (ТФУК соль, 500 МГц, D2O): δ 1,81 (1H, кв, J=12,5 Гц, H-2 акс), 1,98 (1H, кв, J=12 Гц, H-3' акс), 2,32 (1H, дт, J=4, 4 и 12 Гц, H-3' экв), 2,45 (1H, дт, J=4, 4 и 12,5 Гц, H-2 экв), 2,74 (3H, с, NCH3), 3,27 (1H, ддд, J=4, 10,5 и 12,5 Гц, H-1), 3,32 (1H, дд, J=3 и 8,5 Гц, H-7'), 3,37 (1H, т, J=10 Гц, H-4"), 4,52 (1H, т, J=3 Гц, H-6'), 5,16 (1H, д, J=8,5 Гц, H-8'), 5,43 (1H, д, J=4 Гц, H-1") и 5,67 (1H, д, J=3,8 Гц, H-1'),13C ЯМР (ТФУК соль, 125 МГц): δ 157,52 (C=NH).
[0243]
Пример 12: Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонилапрамицина (B1), 6,2",3",6"-тетра-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонилапрамицина (B2), 6,2",3",6"-тетра-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5-эпиапрамицина (B3), 6,2",3",6"-тетра-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5-дезокси-5-эпи-5-фторапрамицина (B3'), 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5-эпиапрамицина (B4) и 5-эпиапрамицина (B5)
[Химическая формула 43]

[0244]
Пример 12-(i): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонилапрамицина (B1)
Раствор, полученный добавлением 13 мл триэтиламина и 8,5 г Boc2O к раствору 29,0 г (30 ммоль) соединения, представленного формулой (A3), растворенного в 200 мл раствора ТГФ, подвергали взаимодействию при 60°C в течение 5 часов. После завершения реакции, смесь концентрировали при пониженном давлении при добавлении конц. водного аммиака, и полученный в результате остаток промывали водой. После сушки получали 31,3 г (98%) указанного в заголовке соединения (B1) в виде светло-коричневого твердого вещества.
[0245]
МС (ESI) m/z: 1090 (M+Na)+.
[0246]
Пример 12-(ii): Синтез 6,2",3",6"-тетра-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонилапрамицина (B2)
Раствор, полученный добавлением 24,9 мл (5,5 экв.) бензоилхлорида при охлаждении льдом к раствору 41,9 г (39 ммоль) указанного в заголовке соединения (B1) примера 12-(i), растворенного в 220 мл пиридина, подвергали взаимодействию при охлаждении льдом в течение 35 минут. После завершения реакции, реакционную смесь концентрировали при пониженном давлении при добавлении воды, и полученный в результате остаток разбавляли этилацетатом. Органический слой последовательно промывали 5% водн. KHSO4, 5% водн. NaHCO3 и насыщенным раствором соли, сушили над Na2SO4 и концентрировали при пониженном давлении, что давало 55,4 г (96%) указанного в заголовке соединения (B2) в виде светло-желтого твердого вещества.
[0247]
МС (ESI) m/z: 1507 (M+Na)+;1H ЯМР (ДМСО-d6, 400 МГц): δ 1,15 (9H, м, трет-Bu), 3,66 (1H, т, H-5), 4,53 (2H, м, H-6"), 5,21 (1H, дд, H-2"), 5,63 (1H, д, H-1") и 5,84 (1H, т, H-3").
[0248]
Пример 12-(iii): Синтез 6,2",3",6"-тетра-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5-эпиапрамицина (B3) и 6,2",3",6"-тетра-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5-дезокси-5-эпи-5-фторапрамицина (B3')
Раствор, полученный добавлением 2,4 мл DAST при охлаждении льдом к раствору 16,5 г (11 ммоль) указанного в заголовке соединения (B2) примера 12-(ii), растворенного в 90 мл метиленхлорида, подвергали взаимодействию при комнатной температуре в течение 1 часа. После завершения реакции, реакционный раствор последовательно промывали насыщенным раствором бикарбоната натрия и водой, и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (хлороформ:метанол=25:1), что давало 9,59 г (58%) указанного в заголовке соединения (B3) и 5,29 г (31,9%) указанного в заголовке соединения (B3').
[0249]
МС (ESI) m/z: (B3), 1507 (M+Na)+; (B3'), 1509 (M+Na)+;1H ЯМР (ДМСО-d6, 400 МГц): (B3), δ 5,40 (1H, ушир. с, H-5) и 5,63 (1H, д, H-1"); (B3'), δ 5,61 (1H, д, H-1") и 5,99 (1H, ушир. д, H-5).
[0250]
Пример 12-(iv): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5-эпиапрамицина (B4)
Раствор, полученный добавлением 0,35 мл раствора 5Н. NaOMe-метанол к раствору 2,47 г (1,7 ммоль) указанного в заголовке соединения (B3) примера 12-(iii), растворенного в 24 мл MeOH, подвергали взаимодействию при комнатной температуре в течение 2 часов. После завершения реакции, реакционный раствор нейтрализовали добавлением 1 N HCl при охлаждении льдом, концентрировали при пониженном давлении и промывали водой. Полученное твердое вещество промывали изопропиловым эфиром, и остаток растворяли в 18 мл раствора 90% ТФУК-MeOH, и полученную смесь подвергали взаимодействию при комнатной температуре в течение 2 часов. Полученный реакционный раствор концентрировали при пониженном давлении, и остаток промывали изопропиловым эфиром, что давало 1,72 г (93% в виде соли ТФУК) указанного в заголовке соединения (B4) в виде бесцветного твердого вещества.
[0251]
МС (ESI) m/z: 990 (M+Na)+.
[0252]
Пример 12-(v): Синтез 5-эпиапрамицина (B5)
Указанное в заголовке соединение (B5) [203 мг (74%)] получали способом, аналогично примеру 8-(ii), с использованием 550 мг (0,51 ммоль в виде соли ТФУК) указанного в заголовке соединения (B4) примера 12-(iv).
[0253]
МС (ESI) m/z: 540 (M+Na)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 4,53 (1H, т, H-5), 5,33 (1H, д, H-1') и 5,67 (1H, д, H-1").
[0254]
Пример 13: Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5-дезокси-5-эпи-5-фторапрамицина (B6) и 5-дезокси-5-эпи-5-фторапрамицина (B7)
[Химическая формула 44]
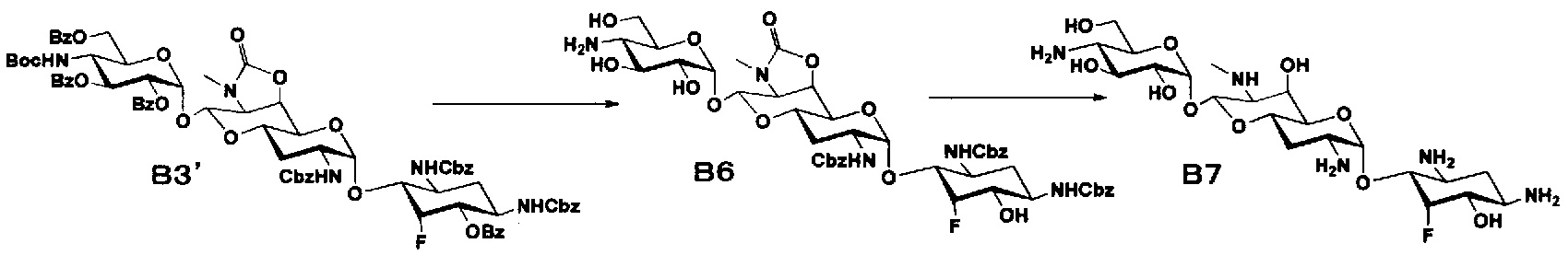
[0255]
Пример 13-(i): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5-дезокси-5-эпи-5-фторапрамицина (B6)
Указанное в заголовке соединение (B6) [1,49 г (94% в виде соли ТФУК)] получали способом, аналогично примеру 12-(iv), с использованием 12 мл метанольного раствора указанного в заголовке соединения (B3') [2,18 г (1,5 ммоль)] примера 12-(iii).
[0256]
МС (ESI) m/z: 992 (M+Na)+.
[0257]
Пример 13-(ii): Синтез 5-дезокси-5-эпи-5-фторапрамицина (B7)
Указанное в заголовке соединение (B7) [188 мг (49%)] получали способом, аналогично примеру 12-(v), с использованием 766 мг (0,71 ммоль в виде соли ТФУК) указанного в заголовке соединения (B6) примера 13-(i).
[0258]
МС (ESI) m/z: 542 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 5,33 (1H, д, H-1'), 5,39 (1H, дт, H-5) и 5,67 (1H, д, H-1").
[0259]
Пример 14: Синтез 6,2",3",6"-тетра-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5-O-мезилапрамицина (C1), 5,6-ангидро-2",3",6"-три-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5-эпиапрамицина (C2), 2",3",6"-три-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-6-дезокси-5,6-диэпи-6-йодапрамицина (C3), 1,3,2'-трис-N-(бензилоксикарбонил)-2",3",6"-три-O-бензоил-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-6-дезокси-5-эпиапрамицина (C4), 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-6-дезокси-5-эпиапрамицина (C5) и 6-дезокси-5-эпиапрамицина (C6)
[Химическая формула 45]
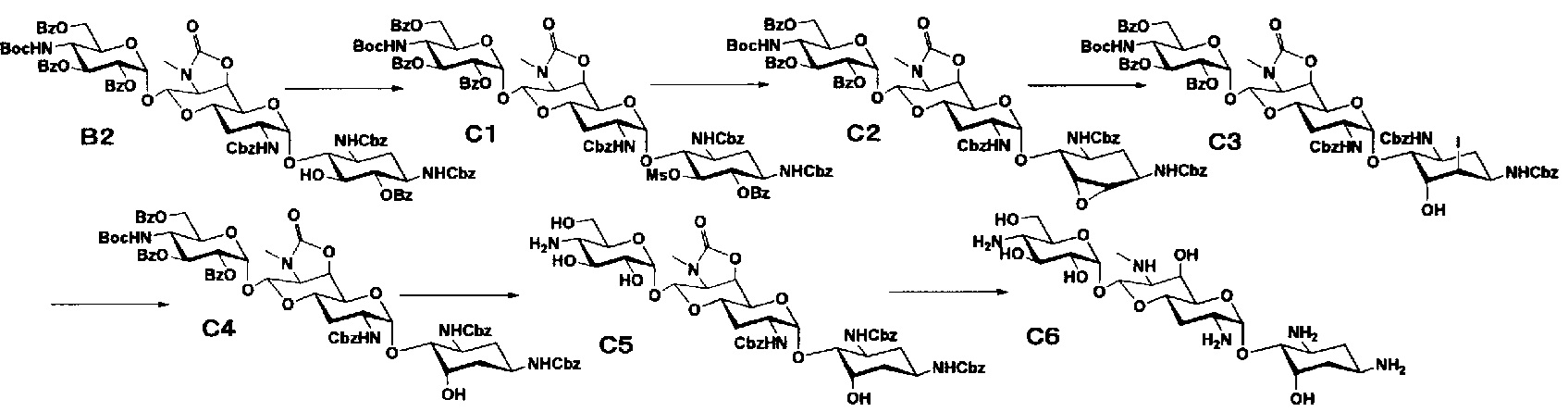
[0260]
Пример 14-(i): Синтез 6,2",3",6"-тетра-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5-O-мезилапрамицина (C1)
Раствор, полученный добавлением 1,25 г 4-диметиламинопиридина и 0,33 мл мезилхлорида при охлаждении льдом к раствору 4,16 г (2,8 ммоль) указанного в заголовке соединения (B2) примера 12-(ii), растворенного в 21 мл метиленхлорида, подвергали взаимодействию при комнатной температуре в течение 2 часов. Полученный реакционный раствор последовательно промывали водой, 10% водным раствором бисульфата калия, насыщенным раствором бикарбоната натрия и водой. Затем полученную смесь концентрировали при пониженном давлении, что давало 4,31 г (98%) указанного в заголовке соединения (C1) в виде светло-желтого твердого вещества.
[0261]
МС (ESI) m/z: 1584 (M+Na)+.
[0262]
Пример 14-(ii): Синтез 5,6-ангидро-2",3",6"-три-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5-эпиапрамицина (C2)
Раствор, полученный добавлением 2,7 мл раствора 5Н. NaOMe-метанол к раствору 4,28 г (2,7 ммоль) указанного в заголовке соединения (C1) примера 14-(i), растворенного в 20 мл метанола, подвергали взаимодействию при комнатной температуре в течение 1 часа. После завершения реакции, реакционный раствор нейтрализовали добавлением 1 N HCl при охлаждении льдом, концентрировали при пониженном давлении и промывали водой. Полученное твердое вещество промывали изопропиловым эфиром и растворяли в 20 мл пиридина. К полученной смеси добавляли 1,58 мл бензоилхлорида при охлаждении льдом, и полученную в результате смесь подвергали взаимодействию при охлаждении льдом в течение 35 минут. К полученному реакционному раствору добавляли воду, и остаток, полученный после концентрирования при пониженном давлении, разбавляли этилацетатом. Органический слой последовательно промывали водой, 10% водным раствором бисульфата калия, насыщенным раствором бикарбоната натрия и водой. Затем смесь концентрировали при пониженном давлении, что давало 3,60 г (98%) указанного в заголовке соединения (C2) в виде светло-желтого твердого вещества.
[0263]
МС (ESI) m/z: 1384 (M+Na)+.
[0264]
Пример 14-(iii): Синтез 2",3",6"-три-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-6-дезокси-5,6-диэпи-6-йодапрамицина (C3)
Раствор, полученный добавлением 1,2 г йодида натрия и 87 мг ацетата натрия, растворенного в 1,7 мл уксусной кислоты, к раствору 3,68 г (2,7 ммоль) указанного в заголовке соединения (C2) примера 14-(ii), растворенного в 14 мл ацетона, кипятили с обратным холодильником в течение 6 часов. К остатку, полученному концетрированием реакционного раствора, добавляли этилацетат, и, после промывки водой, органический слой концентрировали, что давало 3,70 г (92%) указанного в заголовке соединения (C3) в виде бесцветного твердого вещества.
[0265]
МС (ESI) m/z: 1512 (M+Na)+.
[0266]
Пример 14-(iv): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-2",3",6"-три-O-бензоил-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-6-дезокси-5-эпиапрамицина (C4)
Раствор, полученный добавлением 64 мг AIBN и 1,5 мл гидрида трибутилолова к раствору 3,50 г (2,4 ммоль) указанного в заголовке соединения (C3) примера 14-(iii), растворенного в 15 мл диоксана, подвергали взаимодействию в атмосфере N2 при 80°C в течение 1,5 часов. Полученный реакционный раствор концентрировали при пониженном давлении, и, после промывки изопропиловым эфиром, полученный в результате остаток сушили при пониженном давлении, что давало 2,19 г (67%) указанного в заголовке соединения (C4) в виде бесцветного твердого вещества.
[0267]
МС (ESI) m/z: 1386 (M+Na)+;1H ЯМР (ДМСО-d6, 400 МГц): δ 1,28-1,51 (11H, м, H-6 акс, H-2 акс, трет-Bu), 1,83-1,98 (3H, м, H-6 экв, H-2 экв, H-3' экв), 4,82 (1H, д, H-1') и 5,14 (1H, д, H-1").
[0268]
Пример 14-(v): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-6-дезокси-5-эпиапрамицина (C5)
Раствор, полученный добавлением 0,3 мл раствора 5Н. NaOMe-метанол к раствору 2,01 г (1,5 ммоль) указанного в заголовке соединения (C4) примера 14-(iv), растворенного в 20 мл метанола, подвергали взаимодействию при комнатной температуре в течение 2 часов. Полученный реакционный раствор нейтрализовали добавлением 1 N HCl при охлаждении льдом и концентрировали при пониженном давлении, и остаток промывали водой и затем промывали изопропиловым эфиром. Полученное твердое вещество растворяли в 10 мл раствора 90% ТФУК-MeOH, и полученную смесь подвергали взаимодействию при комнатной температуре в течение 2 часов. Полученный реакционный раствор концентрировали при пониженном давлении, и остаток промывали изопропиловым эфиром, что давало 1,43 г (90% в виде соли ТФУК) указанного в заголовке соединения (C5) в виде бесцветного твердого вещества.
[0269]
МС (ESI) m/z: 974 (M+Na)+.
[0270]
Пример 14-(vi): Синтез 6-дезокси-5-эпиапрамицина (C6)
Указанное в заголовке соединение (C6) [115 мг (47%)] получали способом, аналогично примеру 8-(ii), с использованием 500 мг (0,47 ммоль в виде соли ТФУК) указанного в заголовке соединения (C5) примера 14-(vi).
[0271]
МС (ESI) m/z: 546 (M+Na)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,70 (1H, ддд, H-6 акс), 2,31-2,41 (2H, м, H-2 экв и H-6 экв), 4,64 (2H, м, H-6' и H-5), 5,32 (1H, д, H-1') и 5,68 (1H, д, H-1").
[0272]
Пример 15: Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,6-дидезокси-5-фторапрамицина (C7) и 5,6-дидезокси-5-фторапрамицина (C8)
[Химическая формула 46]

[0273]
Пример 15-(i): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,6-дидезокси-5-фторапрамицина (C7)
Указанное в заголовке соединение (C7) [995 мг (92%)] получали способом, аналогично примеру 12-(iii) и (iv), с использованием 1,07 г (0,08 ммоль) указанного в заголовке соединения (C4) примера 14-(iv).
[0274]
МС (ESI) m/z: 1388 (M+Na)+.
[0275]
Пример 15-(ii): Синтез 5,6-дидезокси-5-фторапрамицина (C8)
Раствор, полученный добавлением 0,13 мл 5Н. смеси NaOMe-метанол к раствору 844 мг (0,62 ммоль) указанного в заголовке соединения (C7) примера 15-(i), растворенного в 8,4 мл метанола, подвергали взаимодействию при комнатной температуре в течение 2 часов. После завершения реакции, реакционный раствор нейтрализовали добавлением 1 N HCl при охлаждении льдом и концентрировали при пониженном давлении, и остаток промывали водой и затем промывали изопропиловым эфиром. Остаток растворяли в 5 мл раствора 90% ТФУК-MeOH, и полученную смесь подвергали взаимодействию при комнатной температуре в течение 2 часов. После концентрирования реакционного раствора при пониженном давлении, образовавшийся в результате остаток промывали изопропиловым эфиром и растворяли в 10 мл смеси 50% диоксан-вода, и смесь, полученную добавлением 0,5 мл уксусной кислоты и палладиевой черни к полученному раствору, подвергали каталитическому восстановлению в атмосфере водорода при комнатной температуре в течение 10 часов. После завершения реакции, смесь нейтрализовали добавлением NH4OH, и, после фильтрования, фильтрат концентрировали. Остаток растворяли в воде (3 мл) и нагревали до 110°C, и добавляли 1 N водный раствор гидроксида калия (1 мл). Полученную смесь подвергали взаимодействию в течение 2 часов при этой же температуре. После завершения реакции, реакционную смесь нейтрализовали добавлением 1 N водн. HCl при охлаждении льдом и очищали ионообменной хроматографией (CG50), что давало 244 мг (63%) указанного в заголовке соединения (C8).
[0276]
МС (ESI) m/z: 526 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,85 (1H, дддд, H-6 акс), 2,64 (1H, м, H-6 экв), 5,04 (1H, дддд, H-5), 5,48 (1H, д, H-1') и 5,70 (1H, д, H-1").
[0277]
Пример 16: Синтез 5-азид-1,3,2'-трис-N-(бензилоксикарбонил)-6,2",3",6"-тетра-O-бензоил-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5-дезокси-5-эпиапрамицина (D1) и 5-амино-5-дезокси-5-эпиапрамицина (D2)
[Химическая формула 47]
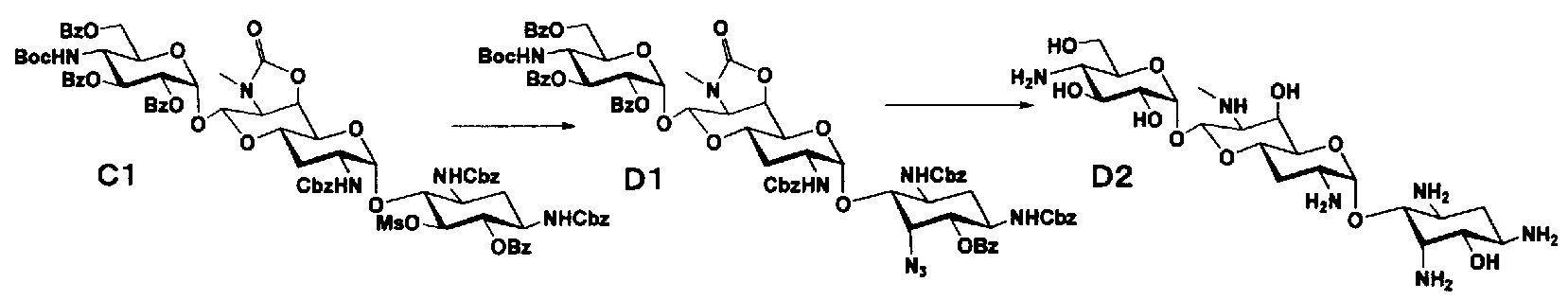
[0278]
Пример 16-(i): Синтез 5-азид-1,3,2'-трис-N-(бензилоксикарбонил)-6,2",3",6"-тетра-O-бензоил-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5-дезокси-5-эпиапрамицина (D1)
Раствор, полученный добавлением 30,1 мг NaN3 к раствору 330 мг (0,21 ммоль) указанного в заголовке соединения (C1) примера 14-(i), растворенного в 4 мл ДМФА, подвергали взаимодействию при 100°C в течение 6 часов. После чего, полученный реакционный раствор концентрировали при пониженном давлении, и остаток промывали водой, полученный остаток очищали колоночной хроматографией на силикагеле (система растворителей, CHCl3:MeOH=30:1), что давало 264 мг (83%) указанного в заголовке соединения (D1) в виде светло-желтого твердого вещества.
[0279]
МС (ESI) m/z: 1531 (M+Na)+.
[0280]
Пример 16-(ii): Синтез 5-амино-5-дезокси-5-эпиапрамицина (D2)
Указанное в заголовке соединение (D2) [47,6 мг (52%)] получали способом, аналогично примеру 15-(ii), с использованием 260 мг (0,17 ммоль) указанного в заголовке соединения (D1) примера 16-(i).
[0281]
МС (ESI) m/z: 539 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 3,93-4,05 (5H, м, H-2", -5', -3", -5 и -5"), 5,36 (1H, д, H-1') и 5,74 (1H, д, H-1").
[0282]
Пример 17: Синтез 6,2",3",6"-тетра-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5-хлор-5-дезокси-5-эпиапрамицина (E1), 5-азид-1,3,2'-трис-N-(бензилоксикарбонил)-6,2",3",6"-тетра-O-бензоил-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5-дезоксиапрамицина (E2) и 5-амино-5-дезоксиапрамицина (E3)
[Химическая формула 48]

[0283]
Пример 17-(i): Синтез 6,2",3",6"-тетра-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5-хлор-5-дезокси-5-эпиапрамицина (E1)
Раствор получали добавлением 400 мл пиридина и затем 0,17 мл (2,1 экв.) сульфурилхлорида при охлаждении льдом к раствору 1,49 г (1,0 ммоль) указанного в заголовке соединения (B2) примера 12-(ii) в 15 мл метиленхлорида. Спустя 5 минут, полученный в результате раствор возвращали обратно к комнатной температуре, и полученную смесь подвергали взаимодействию в течение 1,5 часов. После добавления MeOH к полученному реакционному раствору при охлаждении льдом, смесь концентрировали при пониженном давлении, и полученный остаток разбавляли этилацетатом. Органический слой последовательно промывали водн. Na2SO3, водн. NaCO3 и насыщенным раствором соли, сушили над Na2SO4 и концентрировали при пониженном давлении, что давало 1,1 г (98%) указанного в заголовке соединения (E1) в виде светло-желтого твердого вещества.
[0284]
МС (ESI) m/z: 1523 (M+Na)+.
[0285]
Пример 17-(ii): Синтез 5-азид-1,3,2'-трис-N-(бензилоксикарбонил)-6,2",3",6"-тетра-O-бензоил-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5-дезоксиапрамицина (E2)
Указанное в заголовке соединение (E2) [264 мг (83%)] получали способом, аналогично примеру 16-(i), с использованием 330 мг (0,21 ммоль) указанного в заголовке соединения (E1) примера 17-(i).
[0286]
МС (ESI) m/z: 1531 (M+Na)+.
[0287]
Пример 17-(iii): Синтез 5-амино-5-дезоксиапрамицина (E3)
Указанное в заголовке соединение (E3) [47,6 мг (52%)] получали способом, аналогично примеру 15-(ii), с использованием 260 мг (0,17 ммоль) указанного в заголовке соединения (E2) примера 17-(ii).
[0288]
МС (ESI) m/z: 539 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 3,93-4,05 (5H, м, H-2", -5', -3", -5 и -5"), 5,36 (1H, д, H-1') и 5,74 (1H, д, H-1").
[0289]
Пример 18: Синтез 6-азид-1,3,2'-трис-N-(бензилоксикарбонил)-6,2",3",6"-тетра-O-бензоил-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5-дезокси-5,6-диэпи-5-эпиапрамицина (F1), 6-азид-1,3,2'-трис-N-(бензилоксикарбонил)-6,2",3",6"-тетра-O-бензоил-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5,6-дидезокси-5,6-диэпи-5-фторапрамицина (F2) и 6-амино-5,6-дидезокси-5,6-диэпи-5-фторапрамицина (F3)
[Химическая формула 49]

[0290]
Пример 18-(i): Синтез 6-азид-1,3,2'-трис-N-(бензилоксикарбонил)-6,2",3",6"-тетра-O-бензоил-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5-дезокси-5,6-диэпи-5-эпиапрамицина (F1)
Раствор, полученный добавлением 43 мг NH4Cl и 72 мг NaN3 к раствору 980 мг (0,72 ммоль) указанного в заголовке соединения (C2) примера 14-(ii), растворенного в 4 мл ДМФА, подвергали взаимодействию при 100°C в течение 2 часов. Полученный реакционный раствор концентрировали при пониженном давлении, и остаток промывали водой. Остаток очищали колоночной хроматографией на силикагеле (система растворителей, CHCl3:MeOH=30:1), что давало 778 мг (77%) указанного в заголовке соединения (F1) в виде светло-желтого твердого вещества.
[0291]
МС (ESI) m/z: 1427 (M+Na)+.
[0292]
Пример 18-(ii): синтез 6-азид-1,3,2'-трис-N-(бензилоксикарбонил)-6,2",3",6"-тетра-O-бензоил-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5,6-дидезокси-5,6-диэпи-5-фторапрамицина (F2)
Указанное в заголовке соединение (F2) [442 мг (60%)] получали способом, аналогично примеру 12-(iii) и (iv), с использованием 730 мг (0,52 ммоль) указанного в заголовке соединения (F1) примера 18-(i).
[0293]
МС (ESI) m/z: 1429 (M+Na)+.
[0294]
Пример 18-(iii): Синтез 6-амино-5,6-дидезокси-5,6-диэпи-5-фторапрамицина (F3)
Указанное в заголовке соединение (F3) [96,5 мг (63%)] получали способом, аналогично примеру 15-(ii), с использованием 400 мг (0,28 ммоль) указанного в заголовке соединения (F2) примера 18-(ii).
[0295]
МС (ESI) m/z: 541 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 3,90-4,01 (5H, м, H-2", -5', -3", -6 и -5 "), 5,37 (1H, д, H-1'), 5,51 (1H, м, H-5) и 5,71 (1H, д, H-1").
[0296]
Пример 19: Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-6,2",3",6"-тетра-O-бензоил-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-6-дезокси-5-эпи-5-O-мезилапрамицина (C9), 5-азид-1,3,2'-трис-N-(бензилоксикарбонил)-6,2",3",6"-тетра-O-бензоил-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5,6-дидезоксиапрамицина (C10) и 5-амино-5,6-дезоксиапрамицина (C11)
[Химическая формула 50]

[0297]
Пример 19-(i): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-6,2",3",6"-тетра-O-бензоил-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-6-дезокси-5-эпи-5-O-мезилапрамицина (C9)
Указанное в заголовке соединение (C9) [403 мг (85%)] получали способом, аналогично примеру 14-(i), с использованием 450 мг (0,33 ммоль) указанного в заголовке соединения (C4) примера 14-(iv).
МС (ESI) m/z: 1464 (M+Na)+.
[0298]
Пример 19-(ii): Синтез 5-азид-1,3,2'-трис-N-(бензилоксикарбонил)-6,2",3",6"-тетра-O-бензоил-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5,6-дидезоксиапрамицина (C10)
Указанное в заголовке соединение (C10) [342 мг (88%)] получали способом, аналогично примеру 16-(i), с использованием 401 мг (0,28 ммоль) указанного в заголовке соединения (C9) примера 19-(i).
[0299]
МС (ESI) m/z: 1411 (M+Na)+.
[0300]
Пример 19-(iii): Синтез 5-амино-5,6-дидезоксиапрамицина (C11)
Указанное в заголовке соединение (C11) [54,2 мг (88%)] получали способом, аналогично примеру 15-(ii), с использованием 342 мг (0,25 ммоль) указанного в заголовке соединения (C10) примера 19-(ii).
[0301]
МС (ESI) m/z: 523 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,47-1,64 (2H, м, H-2 акс и H-6 акс), 2,32-2,46 (2H, м, H-2 экв и H-6 экв), 3,22-3,33 (2H, м, H-1 и H-5), 3,43 (1H, дт, H-2'), 3,52 (1H, т, H-4), 5,42 (1H, д, H-1') и 5,76 (1H, д, H-1").
[0302]
Пример 20: Синтез 1,3,2',7',4"-пентакис-N-(бензилоксикарбонил)-5,6-O-циклогексилиденапрамицина (G1), 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-5,6-O-циклогексилиденапрамицина (G2), 2"-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-5,6-O-циклогексилиденапрамицина (G3), 2"-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-3"-O-бензилсульфонил-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-5,6-O-циклогексилиденапрамицина (G4), 2",3"-ангидро-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,6-O-циклогексилиден-3"-эпиапрамицина (G5), 2"-азид-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4",N-6"-O-карбонил-5,6-O-циклогексилиден-2"-дезокси-2",3"-диэпиапрамицина (G6), 3"-азид-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-5,6-O-циклогексилиден-3"-дезоксиапрамицина (G6') и 2"-амино-2"-дезокси-2",3"-диэпиапрамицина (G7)
[Химическая формула 51]

[0303]
Пример 20-(i): Синтез 1,3,2',7',4"-пентакис-N-(бензилоксикарбонил)-5,6-O-циклогексилиденапрамицина (G1)
Раствор, полученный добавлением 1,0 г моногидрата п-толуолсульфоновой кислоты и 20 мл 1,1-диметоксициклогексана к раствору 20,0 г (16,5 ммоль) соединения, представленного формулой (A1), растворенного в 100 мл ДМФА, подвергали взаимодействию при 60°C в течение 4 часов. Полученный реакционный раствор нейтрализовали добавлением триэтиламина, и остаток, полученный концентрированием при пониженном давлении, разбавляли этилацетатом. Органический слой промывали водой и концентрировали, и остаток растворяли в 200 мл диоксана. Раствор, полученный добавлением 100 мл 20% водной уксусной кислоты к полученному раствору, подвергали взаимодействию при комнатной температуре в течение 18 часов. Полученный реакционный раствор концентрировали при пониженном давлении, и остаток кристаллизовали из метанола, что давало 17,7 г (83%) указанного в заголовке соединения (G1).
[0304]
МС (ESI) m/z: 1312 (M+Na)+.
[0305]
Пример 20-(ii): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-5,6-O-циклогексилиденапрамицина (G2)
Указанное в заголовке соединение (G2) [12,2 г (92%)] в виде бесцветного твердого вещества получали способом, аналогично примеру 1-(ii), с использованием 16,0 г (12,4 ммоль) указанного в заголовке соединения (G1) примера 20-(i).
[0306]
MS(ESI)m/z: 1096 (M+Na)+.
[0307]
Пример 20-(iii): Синтез 2"-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-5,6-O-циклогексилиденапрамицина (G3)
Раствор, полученный добавлением 2 мл (1,5 экв.) бензоилхлорида к раствору 12,0 г (11,3 ммоль) указанного в заголовке соединения (G2) примера 20-(ii), растворенного в 60 мл пиридина, обрабатывали способом, аналогично примеру 12-(ii), что давало 12,7 г (96%) указанного в заголовке соединения (G3) в виде бесцветного твердого вещества.
[0308]
МС (ESI) m/z: 1200 (M+Na)+.
[0309]
Пример 20-(iv): Синтез 2"-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-3"-O-бензилсульфонил-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-5,6-O-циклогексилиденапрамицина (G4)
Раствор, полученный добавлением 2,85 г бензилсульфонилхлорида при (-10)- (0°C) к раствору 12,0 г (10,2 ммоль) указанного в заголовке соединения (G3) примера 20-(iii), растворенного в 60 мл пиридина, подвергали взаимодействию при той же самой температуре, как приведено выше, в течение 1 часа. После добавления воды к реакционному раствору, полученный концентрированием при пониженном давлении остаток разбавляли этилацетатом. Органический слой последовательно промывали 5% водн. KHSO4, 5% водн. NaHCO3 и насыщенным раствором соли, сушили над Na2SO4 и концентрировали при пониженном давлении, что давало 12,9 г (93%) указанного в заголовке соединения (G4) в виде светло-желтого твердого вещества.
[0310]
МС (ESI) m/z: 1377 (M+Na)+.
[0311]
Пример 20-(v): Синтез 2",3"-ангидро-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,6-O-циклогексилиден-3"-эпиапрамицина (G5)
Раствор, полученный добавлением 27 мл (3 экв) раствора 1 N NaOBn-бензиловый спирт к раствору 12,5 г (9,2 ммоль) указанного в заголовке соединения (G4) примера 20-(iv), растворенного в 100 мл хлороформа, подвергали взаимодействию при комнатной температуре в течение 1 часа. Полученный реакционный раствор, после добавления к нему воды, нейтрализовали добавлением 1 N хлористоводородной кислоты, и органический слой промывали водой и концентрировали при пониженном давлении. Полученный в результате осадок, после добавления к нему изопропилового эфира, отфильтровывали и сушили, что давало 10,1 г (94%) указанного в заголовке соединения (G5).
[0312]
МС (ESI) m/z: 1186 (M+Na)+.
[0313]
Пример 20-(vi): Синтез 2"-азид-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-5,6-O-циклогексилиден-2"-дезокси-2",3"-диэпиапрамицина (G6) и 3"-азид-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-5,6-O-циклогексилиден-3"-дезоксиапрамицина (G6')
Указанные в заголовке соединения (G6) [452 мг (21%)] и (G6') [1,16 г (53%)] в виде бесцветных твердых веществ получали способом, аналогично примеру 18-(i), с использованием 2,05 г (1,8 ммоль) указанного в заголовке соединения (G5) примера 20-(v).
[0314]
МС (ESI) m/z: (G6), 1229 (M+Na)+, (G6'), 1229 (M+Na)+.
[0315]
Пример 20-(vii): Синтез 2"-амино-2"-дезокси-2",3"-диэпиапрамицина (G7)
Раствор, полученный растворением 402 мг (0,33 ммоль) указанного в заголовке соединения (G6) примера 20-(vi) в 80% водной уксусной кислоте, подвергали взаимодействию при 80°C в течение 0,5 часов. После завершения реакции, смесь концентрировали при пониженном давлении, и остаток растворяли в 10 мл смеси 50% диоксан-вода. Смесь, полученную добавлением 0,5 мл уксусной кислоты и палладиевой черни к полученному раствору, подвергали каталитическому восстановлению в атмосфере водорода при комнатной температуре в течение 10 часов. После завершения реакции, смесь нейтрализовали добавлением NH4OH, и, после фильтрования, фильтрат концентрировали при пониженном давлении. Остаток растворяли в воде (3 мл) и нагревали до 110°C, и добавляли 1 N раствор водного гидроксида калия (3 мл). Полученную в результате смесь подвергали взаимодействию в течение 2 часов при этой же температуре. После завершения реакции, реакционную смесь нейтрализовали добавлением 1 N водн. HCl при охлаждении льдом и очищали ионообменной хроматографией (CG50), что давало 66,5 мг (37%) указанного в заголовке соединения (G7).
[0316]
МС (ESI) m/z: 539 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 3,28 (1H, дд, J=3,5 и 9,5 Гц, H-4"), 4,18 (1H, дд, J=3,5 и 4 Гц, H-3"), 3,34 (1H, дд, J=2 и 4 Гц, H-2"), 5,31 (1H, д, J=2 Гц, H-1") и 5,38 (1H, д, J=3,5 Гц, H-1').
[0317]
Пример 21: Синтез 3"-амино-3"-дезоксиапрамицина (G8)
[Химическая формула 52]
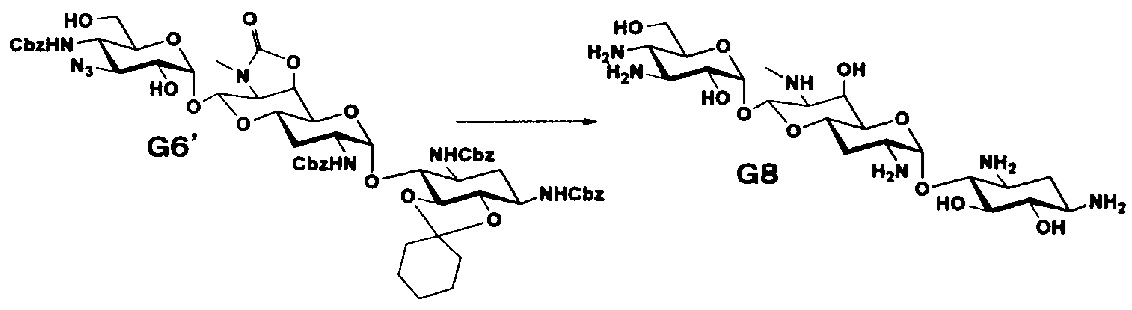
[0318]
Указанное в заголовке соединение (G8) [125 мг (51%)] получали способом, аналогично примеру 20-(viii), с использованием 551 мг (0,46 ммоль) указанного в заголовке соединения (G6') примера 20-(vi).
[0319]
МС (ESI) m/z: 539 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 3,18 (1H, т, 10 Гц, H-3"), 3,76 (1H, дд, J=4 и 10 Гц, H-2"), 5,42 (1H, д, J=3,5 Гц, H-1') и 5,60 (1H, д, J=4 Гц, H-1").
[0320]
Пример 22: Синтез 2"-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-5,6-O-циклогексилиден-3"-трифторметансульфонилапрамицина (H1), 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,3"-O-карбонил-5,6-O-циклогексилиден-3"-эпиапрамицина (H2) и 3"-эпиапрамицина (H3)
[Химическая формула 53]

[0321]
Пример 22-(i): Синтез 2"-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-5,6-O-циклогексилиден-3"-трифторметансульфонилапрамицина (H1)
Раствор, полученный добавлением 2 мл пиридина и 0,95 мл трифторметансульфонового ангидрида при охлаждении льдом к раствору 4,55 г (3,87 ммоль) указанного в заголовке соединения (G3) примера 20-(iii) в 50 мл метиленхлорида, подвергали взаимодействию при охлаждении льдом в течение 1 часа. Полученный реакционный раствор последовательно промывали 10% водн. KHSO4, 5% водн. NaHCO3 и водой, с последующим концентрированием при пониженном давлении, что давало 4,99 г (98%) указанного в заголовке соединения (H1) в виде светло-желтого твердого вещества.
[0322]
МС (ESI) m/z: 1332 (M+Na)+.
[0323]
Пример 22-(ii): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,3"-O-карбонил-5,6-O-циклогексилиден-3"-эпиапрамицина (H2)
Раствор, полученный добавлением 2,33 г ацетата цезия к раствору 4,67 г (3,57 ммоль) указанного в заголовке соединения (H1) примера 22-(i), растворенного в 25 мл ДМФА, подвергали взаимодействию при 80°C в течение 3 часов. К реакционной смеси добавляли этилацетат, и полученную в результате смесь промывали водой и концентрировали при пониженном давлении. Остаток растворяли в 30 мл хлороформа, и добавляли 1 мл 5Н. раствора NaOMe-метанол, и полученную в результате смесь подвергали взаимодействию при комнатной температуре в течение 1 часа. Полученный реакционный раствор, после нейтрализации 1 N хлористоводородной кислотой, концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле (система растворителей, CHCl3:MeOH=30:1), что давало 2,75 г (72%) указанного в заголовке соединения (H2).
[0324]
МС (ESI) m/z: 1096 (M+Na)+.
[0325]
Пример 22-(iii): Синтез 3"-эпиапрамицина (H3)
Указанное в заголовке соединение (H3) [115 мг (52%)] получали способом, аналогично примеру 20-(viii), с использованием 440 мг (0,41 ммоль) указанного в заголовке соединения (H2) примера 22-(ii).
[0326]
МС (ESI) m/z: 540 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 4,18 (1H, т, J=3 Гц, H-3"), 5,32 (1H, д, J=3,5 Гц, H-1') и 5,46 (1H, д, J=4,5 Гц, H-1").
[0327]
Пример 23: Синтез 2",3"-ангидро-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-5,6-O-циклогексилиденапрамицина (I1), 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-2",3"-диэпиапрамицина (I2) и 2",3"-диэпиапрамицина (I3)
[Химическая формула 54]

[0328]
Пример 23-(i): Синтез 2",3"-ангидро-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-5,6-O-циклогексилиденапрамицина (I1)
Указанное в заголовке соединение (I1) [1,38 г (93%)] получали способом, аналогично примеру 1-(ii), с использованием 1,63 г (1,40 ммоль) указанного в заголовке соединения (G5) примера 20-(v).
[0329]
МС (ESI) m/z: 1078 (M+Na)+.
[0330]
Пример 23-(ii): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-2",3"-диэпиапрамицина (I2)
Раствор, полученный растворением 622 мг (0,58 ммоль) указанного в заголовке соединения (I1) примера 23-(i) в 80% водной уксусной кислоте, подвергали взаимодействию при 80°C в течение 0,5 часов. После чего, полученный реакционный раствор концентрировали при пониженном давлении, образовавшийся в результате остаток промывали изопропиловым эфиром и сушили, что давало 548 мг (95%) указанного в заголовке соединения (I2).
[0331]
МС (ESI) m/z: 1016 (M+Na)+.
[0332]
Пример 23-(iii): Синтез 2",3"-диэпиапрамицина (I3)
Указанное в заголовке соединение (I3) [226 мг (68%)] получали способом, аналогично примеру 8-(ii), с использованием 600 мг (0,60 ммоль) указанного в заголовке соединения (I2) примера 23-(ii).
[0333]
МС (ESI) m/z: 540 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 3,27 (1H, дд, J=3 и 10 Гц, H-4"), 4,05~4,18 (3H, м, H-4', -2", -3"), 5,38 (1H, д, J=3,5 Гц, H-1') и 5,40 (1H, д, J=4,5 Гц, H-1").
[0334]
Пример 24: Синтез 1,3,2',7',4"-пентакис-N-(бензилоксикарбонил)-5,6:2",3"-ди-O-циклогексилиденапрамицина (J1), 1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,6:2",3"-ди-O-циклогексилиденапрамицина (J2), 1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,6:2",3"-ди-O-циклогексилиден-6"-дезокси-6"-фторапрамицина (J3) и 6"-дезокси-6"-фторапрамицина (J4)
[Химическая формула 55]

[0335]
Пример 24-(i): Синтез 1,3,2',7',4"-пентакис-N-(бензилоксикарбонил)-5,6:2",3"-ди-O-циклогексилиденапрамицина (J1)
Раствор, полученный добавлением 250 мг моногидрата п-толуолсульфоновой кислоты и 5 мл 1,1-диметоксициклогексана к раствору 5,0 г (4,13 ммоль) соединения, представленного формулой (A1), растворенного в 25 мл ДМФА, подвергали взаимодействию при пониженном давлении при 60°C в течение 4 часов. Полученный реакционный раствор нейтрализовали добавлением триэтиламина, и остаток, полученный концентрированием при пониженном давлении, разбавляли этилацетатом. Органический слой промывали водой, и полученный концентрированием остаток растворяли в 50 мл диоксана. Смесь, полученную добавлением к полученному раствору 25 мл 20% водной уксусной кислоты, подвергали взаимодействию при комнатной температуре в течение 18 часов. Полученный реакционный раствор концентрировали, и остаток промывали изопропиловым эфиром, и сушили, что давало 5,55 г (98%) указанного в заголовке соединения (J1).
[0336]
МС (ESI) m/z: 1392 (M+Na)+.
[0337]
Пример 24-(ii): Синтез 1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,6:2",3"-ди-O-циклогексилиденапрамицина (J2)
Указанное в заголовке соединение (J2) [4,61 г (93%)] получали способом, аналогично примеру 1-(ii), с использованием 5,40 г (3,94 ммоль) указанного в заголовке соединения (J1) примера 24-(i).
[0338]
МС (ESI) m/z: 1284 (M+Na)+.
[0339]
Пример 24-(iii): Синтез 1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,6:2",3"-ди-O-циклогексилиден-6"-дезокси-6"-фторапрамицина (J3)
Указанное в заголовке соединение (J3) [896 мг (92%)] получали способом, аналогично примеру 12-(iii) и (iv), с использованием 977 мг (0,77 ммоль) указанного в заголовке соединения (J2) примера 24-(ii).
[0340]
МС (ESI) m/z: 1286 (M+Na)+.
[0341]
Пример 24-(iv): Синтез 6"-дезокси-6"-фторапрамицина (J4)
Указанное в заголовке соединение (J4) [133 мг (55%)] получали способом, аналогично примеру 20-(viii), с использованием 565 мг (0,45 ммоль) указанного в заголовке соединения (J3) примера 24-(iii).
[0342]
МС (ESI) m/z: 542 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 3,85-4,05 (2H, м, H-6"), 5,32 (1H, д, J=4,5 Гц, H-1') и 5,46 (1H, д, J=4 Гц, H-1").
[0343]
Пример 25: Синтез 2",3"-ангидро-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-6"-O-бензилсульфонил-7'-N,6'-O-карбонил-5,6-O-циклогексилиден-3"-эпиапрамицина (K1), 1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,6-O-циклогексилиден-3",6"-дидезокси-3",6"-дийодапрамицина (K2), 1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,6-O-циклогексилиден-3",6"-дидезоксиапрамицина (K3) и 3",6"-дидезоксиапрамицина (K4)
[Химическая формула 56]

[0344]
Пример 25-(i): Синтез 2",3"-ангидро-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-6"-O-бензилсульфонил-7'-N,6'-O-карбонил-5,6-O-циклогексилиден-3"-эпиапрамицина (K1)
Указанное в заголовке соединение (K1) [926 мг (96%)] получали способом, аналогично примеру 20-(iv), с использованием 850 мг (0,73 ммоль) указанного в заголовке соединения (G5) примера 20-(v).
[0345]
МС (ESI) m/z: 1340 (M+Na)+.
[0346]
Пример 25-(ii): Синтез 1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,6-O-циклогексилиден-3",6"-дидезокси-3",6"-дийодапрамицина (K2)
Указанное в заголовке соединение (K2) [889 мг (93%)] получали способом, аналогично примеру 14-(iii), с использованием 900 мг (0,68 ммоль) указанного в заголовке соединения (K1) примера 25-(i).
[0347]
МС (ESI) m/z: 1424 (M+Na)+.
[0348]
Пример 25-(iii): Синтез 1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,6-O-циклогексилиден-3",6"-дидезоксиапрамицина (K3)
Указанное в заголовке соединение (K3) [645 мг (92%)] получали способом, аналогично примеру 14-(iv), с использованием 850 мг (0,61 ммоль) указанного в заголовке соединения (K2) примера 25-(ii).
[0349]
МС (ESI) m/z: 1172 (M+Na)+.
[0350]
Пример 25-(iv): Синтез 3",6"-дидезоксиапрамицина (K4)
Указанное в заголовке соединение (K4) [155 мг (59%)] получали способом, аналогично примеру 20-(viii), с использованием 600 мг (0,52 ммоль) указанного в заголовке соединения (K3) примера 25-(iii).
[0351]
МС (ESI) m/z: 508 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,47 (3H, д, CH3-6"), 1,99 (1H, кв, H-3" акс), 2,27 (1H, дт, H-3" экв), 5,31 (1H, д, H-1') и 5,72 (1H, д, H-1").
[0352]
Пример 26: Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5-хлор-5-дезокси-5-эпиапрамицина (L1), 1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5,6"-дихлор-5,6"-дидезокси-5-эпиапрамицина (L2), 1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5,6"-дидезоксиапрамицина (L3), 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,6"-дидезоксиапрамицина (L4) и 5,6"-дидезоксиапрамицина (L5)
[Химическая формула 57]

[0353]
Пример 26-(i): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5-хлор-5-дезокси-5-эпиапрамицина (L1)
Раствор, полученный добавлением 3 мл смеси 5Н. NaOMe-метанол к раствору 100 мг (0,067 ммоль) указанного в заголовке соединения (E1) примера 17-(i), растворенного в 2 мл метанола, подвергали взаимодействию при комнатной температуре в течение 1 часа. Полученный реакционный раствор нейтрализовали добавлением 1 N HCl и концентрировали при пониженном давлении, и остаток промывали водой. Остаток затем промывали изопропиловым эфиром и сушили, что давало 65,9 мг (91%) указанного в заголовке соединения (L1) в виде бесцветного твердого вещества.
[0354]
МС (ESI) m/z: 1108 (M+Na)+.
[0355]
Пример 26-(ii): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5,6"-дихлор-5,6"-дидезокси-5-эпиапрамицина (L2)
Раствор, полученный добавлением 1,1 мл пиридина, 6,7 мл тетрахлорида углерода и 1,81 г трифенилфосфина к раствору 1,50 г (1,38 ммоль) указанного в заголовке соединения (L1) примера 26-(i), растворенного в 30 мл ТГФ, подвергали взаимодействию при 50°C в течение 2 часов. Полученный реакционный раствор концентрировали при пониженном давлении, и остаток растворяли в хлороформе. Органический слой последовательно промывали 5% водн. KHSO4, 5% водн. NaHCO3 и насыщенным раствором соли, и сушили над Na2SO4 с последующим концентрированием. Остаток очищали колоночной хроматографией на силикагеле (система растворителей, CHCl3:ацетон=1:1), что давало 1,10 г (72%) указанного в заголовке соединения (L2) в виде бесцветного твердого вещества.
[0356]
МС (ESI) m/z: 1126 (M+Na)+.
[0357]
Пример 26-(iii): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5,6"-дидезоксиапрамицина (L3)
Указанное в заголовке соединение (L3) [184 мг (98%)] получали способом, аналогично примеру 14-(iv), с использованием 200 мг (0,18 ммоль) указанного в заголовке соединения (L2) примера 26-(ii).
[0358]
МС (ESI) m/z: 1058 (M+Na)+.
[0359]
Пример 26-(iv): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,6"-дидезоксиапрамицина (L4)
Указанное в заголовке соединение (L4) [147 мг (79% в виде соли ТФУК)] получали способом, аналогично примеру 14-(vi), с использованием 184 мг (0,17 ммоль) указанного в заголовке соединения (L3) примера 26-(iii).
[0360]
МС (ESI) m/z: 936 (M+1)+.
[0361]
Пример 26-(v): Синтез 5,6"-дидезоксиапрамицина (L5)
Указанное в заголовке соединение (L5) [19,0 мг (67%)] получали способом, аналогично примеру 8-(ii), с использованием 91,1 мг (0,087 ммоль в виде соли ТФУК) указанного в заголовке соединения (L4) примера 26-(iv).
[0362]
МС (ESI) m/z: 508 (M+1)+;1H ЯМР (DCl-D2O, 500 МГц): δ 1,27 (3H, д, CH3-6"), 1,42 (1H, кв, H-5 акс), 2,61 (1H, ддд, H-5 экв), 5,29 (1H, д, H-1') и 5,37 (1H, д, H-1").
[0363]
Пример 27: Синтез 1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,6-O-циклогексилиден-3"-дезокси-3"-йодапрамицина (M1), 1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил -5,6-O-циклогексилиден-3"-дезоксиапрамицина (M2), 2",6"-ди-O-бензоил-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,6-O-циклогексилиден-3"-дезоксиапрамицина (M3), 5,2",6"-три-O-бензоил-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-3"-дезоксиапрамицина (M4), 5,2",6"-три-O-бензоил-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5-хлор-5,3"-дидезокси-5-эпиапрамицина (M5), 5,2",6"-три-O-бензоил-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,3"-дидезоксиапрамицина (M6) и 5,3"-дидезоксиапрамицина (M7)
[Химическая формула 58]

[0364]
Пример 27-(i): Синтез 1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,6-O-циклогексилиден-3"-дезокси-3"-йодапрамицина (M1)
Указанное в заголовке соединение (M1) [10,2 г (93%)] получали способом, аналогично примеру 14-(iii), с использованием 9,92 г (8,53 ммоль) указанного в заголовке соединения (G5) примера 20-(v).
[0365]
МС (ESI) m/z: 1314 (M+Na)+.
[0366]
Пример 27-(ii): Синтез 1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил -5,6-O-циклогексилиден-3"-дезоксиапрамицина (M2)
Указанное в заголовке соединение (M2) [8,08 г (90%)] получали способом, аналогично примеру 14-(iv), с использованием 10,0 г (7,74 ммоль) указанного в заголовке соединения (M1) примера 27-(i).
[0367]
МС (ESI) m/z: 1188 (M+Na)+.
[0368]
Пример 27-(iii): Синтез 2",6"-ди-O-бензоил-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,6-O-циклогексилиден-3"-дезоксиапрамицина (M3)
Указанное в заголовке соединение (M3) [11,2 г (97%)] в виде бесцветного твердого вещества получали способом, аналогично примеру 12-(ii), с использованием 50 мл пиридинового раствора указанного в заголовке соединения (M2) [9,80 г (8,4 ммоль)] примера 27-(ii) и 4 мл (3 экв.) бензоилхлорида.
[0369]
МС (ESI) m/z: 1396 (M+Na)+.
[0370]
Пример 27-(iv): Синтез 5,2",6"-три-O-бензоил-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-3"-дезоксиапрамицина (M4)
Раствор, полученный растворением 11,0 г (8 ммоль) указанного в заголовке соединения (M3) примера 27-(iii) в 50 мл 80% водной уксусной кислоты, подвергали взаимодействию при 80°C в течение 30 минут. Полученный реакционный раствор концентрировали, и, после разбавления остатка этилацетатом, органический слой нейтрализовали добавлением NaHCO3, и затем промывали водой и концентрировали. Затем раствор, полученный растворением остатка в 50 мл пиридина, и добавлением к полученной смеси 3,7 мл (4 экв.) бензоилхлорида при охлаждении льдом, подвергали взаимодействию при охлаждении льдом в течение 35 минут. Полученный реакционный раствор, после добавления воды, концентрировали, и остаток разбавляли этилацетатом. Органический слой последовательно промывали 5% водн. KHSO4, 5% водн. NaHCO3 и водой, и концентрировали, что давало 11,1 г (99%) указанного в заголовке соединения (M4) в виде светло-желтого твердого вещества.
[0371]
МС (ESI) m/z: 1420 (M+Na)+.
[0372]
Пример 27-(v): Синтез 5,2",6"-три-O-бензоил-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5-хлор-5,3"-дидезокси-5-эпиапрамицина (M5)
Указанное в заголовке соединение (M5) [904 мг (90%)] получали способом, аналогично примеру 17-(i), с использованием 1,00 г (0,71 ммоль) указанного в заголовке соединения (M4) примера 27-(iv).
[0373]
МС (ESI) m/z: 1438 (M+Na)+.
[0374]
Пример 27-(vi): Синтез 5,2",6"-три-O-бензоил-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,3"-дидезоксиапрамицина (M6)
Указанное в заголовке соединение (M6) [762 мг (89%)] получали способом, аналогично примеру 14-(iv), с использованием 880 мг (0,62 ммоль) указанного в заголовке соединения (M5) примера 27-(v).
[0375]
МС (ESI) m/z: 1404 (M+Na)+.
[0376]
Пример 27-(vii): Синтез 5,3"-дидезоксиапрамицина (M7)
Раствор, полученный добавлением 0,2 мл смеси 5Н. NaOMe-метанол к раствору 750 мг (0,54 ммоль) указанного в заголовке соединения (M6) примера 27-(vi), растворенного в 10 мл метанола, подвергали взаимодействию при комнатной температуре в течение 2 часов. После завершения реакции, реакционный раствор нейтрализовали добавлением 1 N хлористоводородной кислоты и концентрировали при пониженном давлении. Остаток растворяли в 10 мл 50% водного 1,4-диоксана. Смесь, полученную добавлением 0,5 мл уксусной кислоты и палладиевой черни, подвергали каталитическому восстановлению в атмосфере водорода при комнатной температуре в течение 10 часов. Полученный реакционный раствор нейтрализовали добавлением NH4OH и фильтровали, и фильтрат концентрировали. Остаток растворяли в 5 мл воды и нагревали до 110°C, и добавляли 5 мл раствора 1 N водного гидроксида калия. Полученную в результате смесь нагревали в течение 0,5 часов. Полученную реакционную смесь нейтрализовали добавлением 1 N водн. HCl при охлаждении льдом и очищали ионообменной хроматографией (CG50), что давало 121 мг (44%) указанного в заголовке соединения (M7).
[0377]
МС (ESI) m/z: 508 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,66 (1H, кв, H-5 акс), 1,98 (1H, кв, H-3" акс), 2,30 (1H, дт, H-3" экв), 2,68-2,75 (4H, м, H-5 экв и 7'-NMe), 5,30 (1H, д, H-1') и 5,69 (1H, д, H-1").
[0378]
Пример 28: Синтез 5,2",6"-три-O-бензоил-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-3"-дезокси-5-эпиапрамицина (M8), 5,2",6"-три-O-бензоил-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,3"-дидезокси-5-эпи-5-фторапрамицина (M8') и 3"-дезокси-5-эпиапрамицина (M9)
[Химическая формула 59]

[0379]
Пример 28-(i): Синтез 5,2",6"-три-O-бензоил-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-3"-дезокси-5-эпиапрамицина (M8) и 5,2",6"-три-O-бензоил-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,3"-дидезокси-5-эпи-5-фторапрамицина (M8')
Указанные в заголовке соединения (M8) [1,12 г (56%)] и (M8') [445 мг (22%)] получали способом, аналогично примеру 12-(iii), с использованием 2,01 г (1,43 ммоль) указанного в заголовке соединения (M4) примера 27-(iv).
[0380]
МС (ESI) m/z: (M8), 1420 (M+Na)+; (M8'), 1422 (M+Na)+.
[0381]
Пример 28-(ii): Синтез 3"-дезокси-5-эпиапрамицина (M9)
Указанное в заголовке соединение (M9) [78,6 мг (52%)] получали способом, аналогично примеру 27-(vii), с использованием 410 мг (0,29 ммоль) указанного в заголовке соединения (M8) примера 28-(i).
[0382]
МС (ESI) m/z: 524 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,95 (1H, кв, J=12,5 Гц, H-3" акс), 2,27 (1H, дт, J=4 и 12,5 Гц, H-3" экв), 4,51 (1H, т, J=2,5 Гц, H-5), 5,21 (1H, д, J=3,5 Гц, H-1') и 5,51 (1H, J=4 Гц, д, H-1").
[0383]
Пример 29: Синтез 5,3"-дидезокси-5-эпи-5-фторапрамицина (M10)
[Химическая формула 60]

[0384]
Указанное в заголовке соединение (M10) [70,5 мг (50%)] получали способом, аналогично примеру 27-(vii), с использованием 380 мг (0,27 ммоль) указанного в заголовке соединения (M8') примера 28-(i).
[0385]
МС (ESI) m/z: 526 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,95 (1H, кв, J=12,5 Гц, H-3" акс), 2,30 (1H, дт, J=4 и 12,5 Гц, H-3" экв), 5,28 (1H, д, J=3,5 Гц, H-1'), 5,35 (1H, ушир. д, J=55 Гц, H-5) и 5,51 (1H, д, J=4 Гц, H-1").
[0386]
Пример 30: Синтез 5,2",6"-три-O-бензоил-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-3"-дезокси-5-O-мезилапрамицина (N1), 5,6-ангидро-2",6"-ди-O-бензоил-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-3"-дезокси-5-эпиапрамицина (N2), 2",6"-ди-O-бензоил-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-6,3"-дидезокси-5,6-диэпи-6-йодапрамицина (N3), 2",6"-ди-O-бензоил-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-6,3"-дидезокси-5-эпиапрамицина (N4), и 6,3"-дидезокси-5-эпиапрамицина (N5)
[Химическая формула 61]

[0387]
Пример 30-(i): Синтез 5,2",6"-три-O-бензоил-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-3"-дезокси-5-O-мезилапрамицина (N1)
Указанное в заголовке соединение (N1) [2,31 г (97%)] получали способом, аналогично примеру 14-(i), с использованием 2,25 г (1,61 ммоль) указанного в заголовке соединения (M4) примера 27-(v).
[0388]
МС (ESI) m/z: 1498 (M+Na)+.
[0389]
Пример 30-(ii): Синтез 5,6-ангидро-2",6"-ди-O-бензоил-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-3"-дезокси-5-эпиапрамицина (N2)
Указанное в заголовке соединение (N2) [1,46 г (82%)] получали способом, аналогично примеру 14-(ii), с использованием 2,02 г (1,40 ммоль) указанного в заголовке соединения (N1) примера 30-(i).
[0390]
МС (ESI) m/z: 1298 (M+Na)+.
[0391]
Пример 30-(iii): Синтез 2",6"-ди-O-бензоил-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-6,3"-дидезокси-5,6-диэпи-6-йодапрамицина (N3)
Указанное в заголовке соединение (N3) [1,37 г (92%)] получали способом, аналогично примеру 14-(iii), с использованием 1,35 г (1,06 ммоль) указанного в заголовке соединения (N2) примера 30-(ii).
[0392]
МС (ESI) m/z: 1426 (M+Na)+.
[0393]
Пример 30-(iv): Синтез 2",6"-ди-O-бензоил-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-6,3"-дидезокси-5-эпиапрамицина (N4)
Указанное в заголовке соединение (N4) [331 мг (88%)] получали способом, аналогично примеру 14-(iv), с использованием 417 мг (0,29 ммоль) указанного в заголовке соединения (N3) примера 30-(iii).
[0394]
МС (ESI) m/z: 1300 (M+Na)+.
[0395]
Пример 30-(v): Синтез 6,3"-дидезокси-5-эпиапрамицина (N5)
Указанное в заголовке соединение (N5) [66,8 мг (55%)] получали способом, аналогично примеру 27-(vii), с использованием 310 мг (0,24 ммоль) указанного в заголовке соединения (N4) примера 30-(iv).
[0396]
МС (ESI) m/z: 508 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,37 (1H, кв, J=12,5 Гц, H-6 акс), 1,62 (1H, т, J=12,5 Гц, H-6 экв), 1,93 (1H, кв, J=12,5 Гц, H-3" акс), 2,33 (1H, дт, J=4 и 12,5 Гц, H-3" экв), 4,57 (2H, ушир. с, H-5 и H-6'), 5,24 (1H, д, J=3 Гц, H-1') и 5,50 (1H, д, J=3,5 Гц, H-1").
[0397]
Пример 31: Синтез 2",6"-ди-O-бензоил-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,6,3"-тридезокси-5-еноапрамицина (N6) и 5,6,3"-тридезоксиапрамицина (N7)
[Химическая формула 62]

[0398]
Пример 31-(i): Синтез 2",6"-ди-O-бензоил-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,6,3"-тридезокси-5-еноапрамицина (N6)
Раствор, полученный добавлением 200 мг бензилсульфонилхлорида при (-10)-(0°C) к раствору 952 мг (0,67 ммоль) указанного в заголовке соединения (N3) примера 30-(iii), растворенного в 5 мл пиридина, подвергали взаимодействию при той же самой температуре, как приведено выше, в течение 1 часа. Затем к полученному реакционному раствору добавляли 0,5 мл воды, и полученную смесь нагревали при 80°C в течение 2 часов. Полученный реакционный раствор концентрировали, и образовавшийся в результате добавления воды осадок отфильтровывали. Затем осадок очищали колоночной хроматографией на силикагеле (система растворителей, CHCl3:MeOH=30:1), что давало 578 мг (67%) указанного в заголовке соединения (N6).
[0399]
МС (ESI) m/z: 1282 (M+Na)+.
[0400]
Пример 31-(ii): Синтез 5,6,3"-тридезоксиапрамицина (N7)
Указанное в заголовке соединение (N7) [81,3 мг (61%)] получали способом, аналогично примеру 27-(vii), с использованием 480 мг (0,27 ммоль) указанного в заголовке соединения (N6) примера 31-(i).
[0401]
МС (ESI) m/z: 492 (M+1)+.1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,60 (1H, кв, J=12,5 Гц, H-6 акс), 1,65 (1H, кв, J=12 Гц, H-5 акс), 1,95 (1H, кв, J=12,5 Гц, H-3" акс), 2,20-2,32 (2H, м, H-6eq и H-3" экв), 2,29 (1H, м, H-6 экв), 5,37 (1H, д, J=3,6 Гц, H-1') и 5,69 (1H, д, J=3,9 Гц, H-1").
[0402]
Пример 32: Синтез 5-азид-2",6"-ди-O-бензоил-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,3"-дидезокси-5-эпиапрамицина (N8) и 5-амино-5,3"-дидезокси-5-эпиапрамицина (N9)
[Химическая формула 63]

[0403]
Пример 32-(i): Синтез 5-азид-2",6"-ди-O-бензоил-1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,3"-дидезокси-5-эпиапрамицина (N8)
Указанное в заголовке соединение (N8) [375 мг (70%)] получали способом, аналогично примеру 16-(i), с использованием 552 мг (0,37 ммоль) указанного в заголовке соединения (N1) примера 30-(i).
[0404]
МС (ESI) m/z: 1445 (M+Na)+.
[0405]
Пример 32-(ii): Синтез 5-амино-5,3"-дидезокси-5-эпиапрамицина (N9)
Указанное в заголовке соединение (N9) [66,8 мг (55%)] получали способом, аналогично примеру 27-(vii), с использованием 322 мг (0,23 ммоль) указанного в заголовке соединения (N8) примера 32-(i).
[0406]
МС (ESI) m/z: 523 (M+1)+; 1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,95 (1H, кв, J=12,5 Гц, H-3" акс), 2,30 (1H, дт, J=4 и 12,5 Гц, H-3" экв), 3,93-4,05 (5H, м, H-2", -5', -3", -5 и -5"), 5,36 (1H, д, J=3,6 Гц, H-1') и 5,73 (1H, д, J=3,9 Гц, H-1").
[0407]
Пример 33: Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-2"-дезокси-2",3"-диэпи-5,6-O-циклогексилиден-3"-йодапрамицина (O1), 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-2"-дезокси-5,6-O-циклогексилиден-3"-эпиапрамицина (O2), 6,3"-ди-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-2"-дезокси-3"-эпиапрамицина (O3), 6,3"-ди-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-5,2"-дидезокси-5,3"-диэпи-5-фторапрамицина (O4) и 5,2"-дидезокси-5,3"-диэпи-5-фторапрамицина (O5)
[Химическая формула 64]
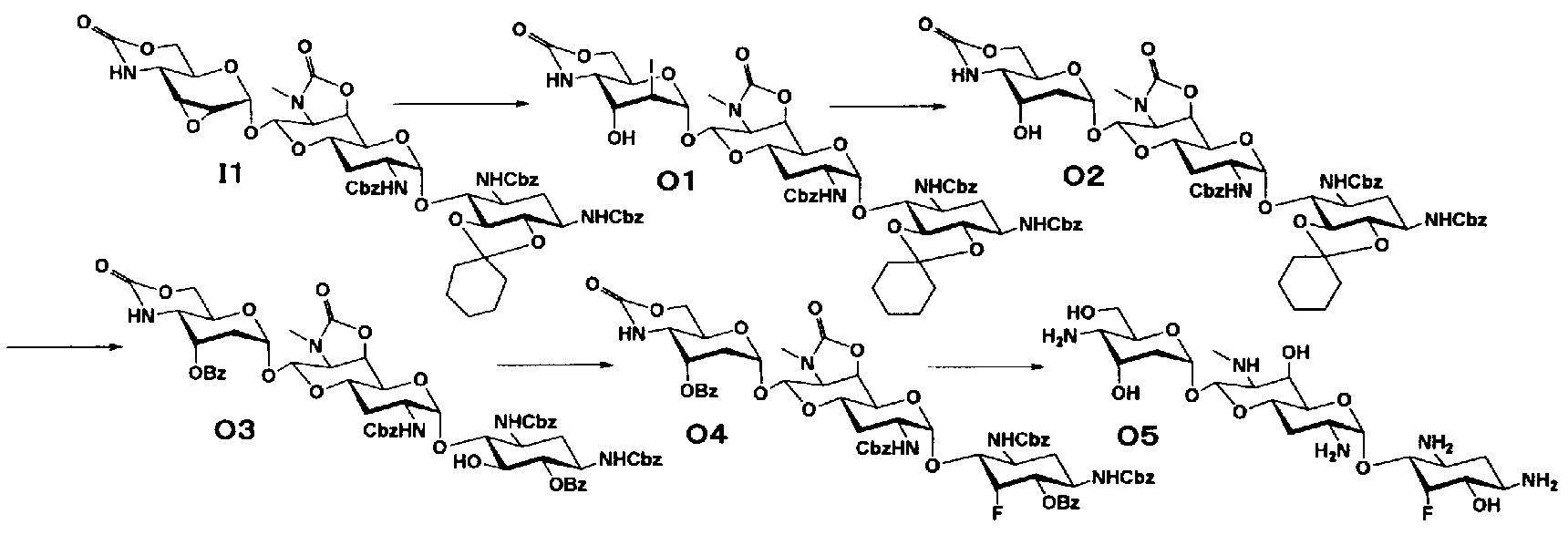
[0408]
Пример 33-(i): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-2"-дезокси-2",3"-диэпи-5,6-O-циклогексилиден-3"-йодапрамицина (O1)
Указанное в заголовке соединение (O1) [5,70 г (91%)] получали способом, аналогично примеру 14-(iii), с использованием 5,60 г (5,30 ммоль) указанного в заголовке соединения (I1) примера 23-(i).
[0409]
МС (ESI) m/z: 1206 (M+Na)+.
[410]
Пример 33-(ii): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-2"-дезокси-5,6-O-циклогексилиден-3"-эпиапрамицина (O2)
Указанное в заголовке соединение (O2) [4,94 г (99%)] получали способом, аналогично примеру 14-(iv), с использованием 5,55 г (4,70 ммоль) указанного в заголовке соединения (O1) примера 33-(i).
[0411]
МС (ESI) m/z: 1080 (M+Na)+.
[0412]
Пример 33-(iii): Синтез 6,3"-ди-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-2"-дезокси-3"-эпиапрамицина (O3)
Указанное в заголовке соединение (O3) [5,09 г (94%)] получали способом, аналогично примеру 27-(iv), с использованием 4,85 г (4,59 ммоль) указанного в заголовке соединения (O2) примера 33-(ii).
[0413]
МС (ESI) m/z: 1208 (M+Na)+.
[0414]
Пример 33-(iv): Синтез 6,3"-ди-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-5,2"-дидезокси-5,3"-диэпи-5-фторапрамицина (O4)
Указанное в заголовке соединение (O4) [332 мг (33%)] получали способом, аналогично примеру 12-(iii) и (iv), с использованием 1,00 г (0,84 ммоль) указанного в заголовке соединения (O3) примера 33-(iii).
[0415]
МС (ESI) m/z: 1210 (M+Na)+.
[0416]
Пример 33-(v): Синтез 5,2"-дидезокси-5,3"-диэпи-5-фторапрамицина (O5)
Указанное в заголовке соединение (O5) [48,5 мг (37%)] получали способом, аналогично примеру 27-(vii), с использованием 300 мг (0,25 ммоль) указанного в заголовке соединения (O4) примера 33-(iv).
[0417]
МС (ESI) m/z: 526 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 2,30-2,40 (1H, м, H-2" акс), 2,37 (1H, дт, H-2" экв), 4,30 (1H, дд, H-3"), 5,31 (1H, д, H-1'), 5,35 (1H, д, H-5) и 5,60 (1H, д, H-1").
[0418]
Пример 34: Синтез 6,2",6"-три-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,3"-O-карбонил-3"-эпиапрамицина (P1), 6,2",6"-три-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,3"-O-карбонил-3"-эпи-5-O-мезилапрамицина (P2), 5-O-ацетил-6,2",6"-три-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,3"-O-карбонил-5,3"-диэпиапрамицина (P3) и 5,3"-диэпиапрамицина (P4)
[Химическая формула 65]

[0419]
Пример 34-(i): Синтез 6,2",6"-три-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,3"-O-карбонил-3"-эпиапрамицина (P1)
Указанное в заголовке соединение (P1) [3,02 г (54%)] получали способом, аналогично примеру 27-(iv), с использованием 2,60 г (2,41 ммоль) указанного в заголовке соединения (H2) примера 22-(ii).
[0420]
МС (ESI) m/z: 1328 (M+Na)+.
[0421]
Пример 34-(ii): Синтез 6,2",6"-три-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,3"-O-карбонил-3"-эпи-5-O-мезилапрамицина (P2)
Указанное в заголовке соединение (P2) [3,05 г (95%)] получали способом, аналогично примеру 14-(i), с использованием 2,92 г (2,24 ммоль) указанного в заголовке соединения (P1) примера 34-(i).
[0422]
МС (ESI) m/z: 1406 (M+Na)+.
[0423]
Пример 34-(iii): Синтез 5-O-ацетил-6,2",6"-три-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,3"-O-карбонил-5,3"-диэпиапрамицина (P3)
Раствор, полученный добавлением 745 мг ацетата цезия к раствору 1,47 г (1,06 ммоль) указанного в заголовке соединения (P2) примера 34-(ii), растворенного в 15 мл ДМФА, подвергали взаимодействию при 90°C в течение 5 часов. К реакционному раствору добавляли этилацетат, и полученную смесь дважды промывали водой и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле (система растворителей, CHCl3:MeOH=40:1), что давало 1,07 г (75%) указанного в заголовке соединения (P3).
[0424]
МС (ESI) m/z: 1370 (M+Na)+.
[0425]
Пример 34-(iv): Синтез 5,3"-диэпиапрамицина (P4)
Указанное в заголовке соединение (P4) [168 мг (48%)] получали способом, аналогично примеру 27-(vii), с использованием 886 мг (0,66 ммоль) указанного в заголовке соединения (P3) примера 34-(iii).
[0426]
МС (ESI) m/z: 540 (M+1)+;
1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,92 (1H, кв, H-2" акс), 4,18 (1H, т, H-3"), 4,48 (1H, т, H-5), 5,32 (1H, д, H-1') и 5,46 (1H, д, H-1").
[0427]
Пример 35: Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-6-дезокси-5-эпиапрамицина (Q1), 1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-6"-хлор-6,6"-дидезокси-5-эпиапрамицина (Q2), 1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-6,6"-дидезокси-5-эпиапрамицина (Q3) и 6,6"-дидезокси-5-эпиапрамицина (Q4)
[Химическая формула 66]

[0428]
Пример 35-(i): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-6-дезокси-5-эпиапрамицина (Q1)
Раствор, полученный добавлением 0,3 мл смеси 5Н. NaOMe-метанол к раствору 2,01 г (1,5 ммоль) указанного в заголовке соединения (C4) примера 14-(iv), растворенного в 20 мл MeOH, подвергали взаимодействию при комнатной температуре в течение 2 часов. Полученный реакционный раствор, после нейтрализации 1 N HCl при охлаждении льдом, концентрировали при пониженном давлении, и остаток промывали водой. Остаток затем промывали изопропиловым эфиром и сушили при пониженном давлении, что давало 1,45 г (92%) указанного в заголовке соединения (Q1) в виде бесцветного твердого вещества.
[0429]
MS(ESI)m/z: 1074 (M+Na)+.
[0430]
Пример 35-(ii): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-6"-хлор-6,6"-дидезокси-5-эпиапрамицина (Q2)
Указанное в заголовке соединение (Q2) [804 мг (87%)] получали способом, аналогично примеру 26-(ii), с использованием 965 мг (0,86 ммоль) указанного в заголовке соединения (Q1) примера 35-(i).
[0431]
МС (ESI) m/z: 1092 (M+Na)+.
[0432]
Пример 35-(iii): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-6,6"-дидезокси-5-эпиапрамицина (Q3)
Указанное в заголовке соединение (Q3) (706 мг (93%)) получали способом, аналогично примеру 14-(iv), с использованием 785 мг (0,73 ммоль) указанного в заголовке соединения (Q2) примера 35-(ii).
[0433]
МС (ESI) m/z: 1058 (M+Na)+.
[0434]
Пример 35-(iv): Синтез 6,6"-дидезокси-5-эпиапрамицина (Q4)
Указанное в заголовке соединение (Q4) (143 мг (41%)) получали способом, аналогично примеру 6-(iii), с использованием 702 мг (0,68 ммоль) указанного в заголовке соединения (Q3) примера 35-(iii).
[0435]
МС (ESI) m/z: 508 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,32 (1H, кв, J=12,5 Гц, H-6 акс), 1,43 (3H, д, H-6"), 1,52 (1H, т, J=12,5 Гц, H-6 экв), 4,49 (2H, ушир. с, H-5 и H-6'), 5,16 (1H, д, J=3,5 Гц, H-1') и 5,47 (1H, д, J=3,5 Гц, H-1").
[0436]
Пример 36: синтез 2",3",6"-три-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5,6-дидезокси-5-еноапрамицина (R1), 1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5,6-дидезокси-5-еноапрамицина (R2), 1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-6"-хлор-5-ено-5,6,6"-тридезоксиапрамицина (R3), 1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5-ено-5,6,6"-тридезоксиапрамицина (R4) и 5-ено-5,6,6"-тридезоксиапрамицина (R5)
[Химическая формула 67]

[0437]
Пример 36-(i): Синтез 2",3",6"-три-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5,6-дидезокси-5-еноапрамицина (R1)
Указанное в заголовке соединение (R1) (2,24 г (82%)) получали способом, аналогично примеру 31-(i), с использованием 3,01 г (2,02 ммоль) указанного в заголовке соединения (C3) примера 14-(iii).
[0438]
МС (ESI) m/z: 1368 (M+Na)+.
[0439]
Пример 36-(ii): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5,6-дидезокси-5-еноапрамицина (R2)
Указанное в заголовке соединение (R2) (1,51 г (98%)) получали способом, аналогично примеру 26-(i), с использованием 2,02 г (1,50 ммоль) указанного в заголовке соединения (R1) примера 36-(i).
[0440]
МС (ESI) m/z: 1056 (M+Na)+.
[0441]
Пример 36-(iii): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-6"-хлор-5-ено-5,6,6"-тридезоксиапрамицина (R3)
Указанное в заголовке соединение (R3) (1,22 г (85%)) получали способом, аналогично примеру 26-(ii), с использованием 1,40 г (1,36 ммоль) указанного в заголовке соединения (R2) примера 36-(ii).
[0442]
МС (ESI) m/z: 1074 (M+Na)+.
[0443]
Пример 36-(iv): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-4"-N-(трет-бутоксикарбонил)-7'-N,6'-O-карбонил-5-ено-5,6,6"-тридезоксиапрамицина (R4)
Указанное в заголовке соединение (R4) [976 мг (91%)] получали способом, аналогично примеру 14-(iv), с использованием 1,10 г (1,05 ммоль) указанного в заголовке соединения (R3) примера 36-(iii).
[0444]
МС (ESI) m/z: 1040 (M+Na)+.
[0445]
Пример 36-(v): Синтез 5-ено-5,6,6"-тридезоксиапрамицина (R5)
Смесь, полученную добавлением 500 мг металлического натрия и раствора 1,00 г (0,98 ммоль) указанного в заголовке соединения (R4) примера 36-(iv), растворенного в 5 мл ТГФ, к 50 мл жидкого аммиака при -50°C, подвергали взаимодействию при той же самой температуре, как приведено выше, в течение 0,5 часов. MeOH добавляли к полученному реакционному раствору до тех пор, пока цвет раствора не исчез, и концентрировали. Смесь, полученную добавлением 10 мл воды к остатку, нагревали при 110°C в течение 0,5 часов. После завершения реакции, реакционную смесь нейтрализовали добавлением 1 N водн. HCl при охлаждении льдом, и очищали ионообменной хроматографией (CG50), что давало 186 мг (39%) указанного в заголовке соединения (R5).
[0446]
МС (ESI) m/z: 490 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,44 (3H, д, H-6"), 5,25 (1H, д, H-1'), 5,51 (1H, д, H-1") и 6,03 (2H, с, H-5 и H-6).
[0447]
Пример 37: Синтез 5,6,6"-тридезоксиапрамицина (R6)
[Химическая формула 68]

[0448]
Смесь, полученную добавлением оксида палладия к 10 мл водного раствора 100 мг (0,20 ммоль) указанного в заголовке соединения (R5) примера 36-(v), подвергали каталитическому восстановлению в атмосфере водорода при комнатной температуре в течение 3 часов. После фильтрования, реакционный раствор очищали ионообменной хроматографией (CG50), что давало 92,1 мг (92%) указанного в заголовке соединения (R6).
[0449]
МС (ESI) m/z: 492 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,46 (3H, д, H-6"), 1,42-1,67 (3H, м, H-2 акс, -6 акс и -5 акс), 2,25 (1H, м, H-6 экв), 2,41-2,52 (2H, м, H-3' экв и -5 экв), 5,34 (1H, д, H-1') и 5,70 (1H, д, H-1").
[0450]
Пример 38: Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5-дезоксиапрамицина (S-a), 4"-N-бензил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5-дезоксиапрамицина (S2-a) и 5-дезокси-4"-N-метилапрамицина (S1-a)
[Химическая формула 69]

[0451]
Пример 38-(i): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5-дезоксиапрамицина (S-a)
Указанное в заголовке соединение (S-a) [939 мг (93% в виде соли ТФУК)] получали способом, аналогично примеру 14-(iv) и примеру 12-(v), с использованием 1,46 г (0,97 ммоль) указанного в заголовке соединения (E1) примера 17-(i).
[0452]
МС (ESI) m/z: 974 (M+Na)+.
[0453]
Пример 38-(ii): Синтез 4"-N-бензил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5-дезоксиапрамицина (S2-a)
Указанное в заголовке соединение (S2-a) [209 мг (95%)] в виде бесцветного твердого вещества получали способом, аналогично примеру 1-(iv), с использованием 221 мг (0,21 ммоль в виде соли ТФУК) указанного в заголовке соединения (S-a) примера 38-(i).
[0454]
МС (ESI) m/z: 1064 (M+Na)+.
[0455]
Пример 38-(iii): Синтез 5-дезокси-4"-N-метилапрамицина (S1-a)
Указанное в заголовке соединение (S1-a) [38 мг (47%)] получали способом, аналогично примеру 1-(v), с использованием 150 мг (0,15 ммоль) указанного в заголовке соединения (S2-a) примера 38-(ii).
[0456]
МС (ESI) m/z: 538 (M+H)+;
1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,65 (1H, кв, H-5 акс), 2,64-2,79 (7H, м, H-5 экв, 7'-NMe и 4"-NMe), 5,29 (1H, д, H-1') и 5,67 (1H, д, H-1").
[0457]
Пример 39: Синтез 4"-N-(2-аминоэтил)-5-дезоксиапрамицина (S1-b)
[Химическая формула 70]

[0458]
Указанное в заголовке соединение (S1-b) [34 мг (72%)] получали способом, аналогично примеру 3, с использованием 96 мг (0,09 ммоль) указанного в заголовке соединения (S2-a) примера 38-(ii) и 18 мг N-Boc-2-аминоацетальдегида.
[0459]
МС (ESI) m/z: 567 (M+1)+;
1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,66 (1H, кв, H-5 акс), 2,68-2,78 (4H, м, H-5 экв и 7'-NMe), 2,92 (1H, т, H-4"), 3,01-3,13 [5H, м, H-1 и 4"-NH2Et(β, α)], 5,30 (1H, д, H-1') и 5,69 (1H, д, H-1").
[0460]
Пример 40: Синтез 4"-N-(3-аминопропил)-5-дезоксиапрамицина (S1-c)
[Химическая формула 71]

[0461]
Указанное в заголовке соединение (S1-c) [62,5 мг (53%)] получали способом, аналогично примеру 1-(v), с использованием 200 мг (0,2 ммоль в виде соли ТФУК) указанного в заголовке соединения (S2-a) примера 38-(i) и 48 мг 3-[(бензилоксикарбонил)амино]пропиональдегида.
[0462]
МС (ESI) m/z: 581 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,91-2,05 [2H, м, 4"-NH2Pr(β) и H-3' акс], 2,65-2,78 (4H, м, H-5 экв и 7'-NMe), 2,88 (1H, т, H-4"), 2,94-3,09 [6H, м, H-1, -7' и 4"-NH2Pr(α, γ)], 3,63 (1H, дд, H-6), 5,28 (1H, д, H-1') и 5,67 (1H, д, H-1").
[0463]
Пример 41: Синтез 4"-N-(1,3-диаминопропан-2-ил)-5-дезоксиапрамицина (S1-d)
[Химическая формула 72]

[0464]
Указанное в заголовке соединение (S1-d) [70,5 мг (59%)] получали способом, аналогично примеру 1-(v), с использованием 190 мг (0,2 ммоль в виде соли ТФУК) указанного в заголовке соединения (S-a) примера 38-(i) и 90 мг 1,3-бис[(бензилоксикарбонил)амино]пропан-2-она.
[0465]
МС (ESI) m/z: 596 (M+1)+;1H ЯМР (DCl-D2O, 500 МГц): δ 1,45 (1H, кв, J=12 Гц, H-5 акс), 1,75 (1H, кв, J=12,5 Гц, H-2 акс), 2,01 (1H, кв, J=12 Гц, H-3' акс), 2,35 (1H, дт, J=4,5, 4,5 и 12 Гц, H-3' экв), 2,45 (1H, дт, J=4, 4 и 12,5 Гц, H-2 экв), 2,67 (1H, т, J=10 Гц, H-4"), 2,75 (3H, с, NCH3), 3,34 (1H, дд, J=3 и 8,5 Гц, H-7'), 4,55 (1H, т, J=3 Гц, H-6'), 5,13 (1H, д, J=8,5 Гц, H-8'), 5,35 (1H, д, J=3,8 Гц, H-1') и 5,38 (1H, д, J=4 Гц, H-1").
[0466]
Пример 42: Синтез 4"-дезамино-5-дезокси-4"-гуанидиноапрамицина (S1-e)
[Химическая формула 73]

[0467]
Указанное в заголовке соединение (S1-e) [76,2 мг (45%)] получали способом, аналогично примеру 10, с использованием 290 мг (0,3 ммоль в виде соли ТФУК) указанного в заголовке соединения (S-a) примера 38-(i) и 310 мг реагента Гудмана.
[0468]
МС (ESI) m/z: 566 (M+1)+;1H ЯМР (DCl-D2O, 500 МГц): δ 1,76 (1H, кв, H-5 акс), 2,46 (1H, ддд, H-5 экв), 5,36 (1H, д, H-1') и 5,45 (1H, д, H-1"),13C ЯМР (DCl-D2O, 125 МГц): δ 157,52 (C=NH).
[0469]
Пример 43: Синтез 4"-N-бензил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5-эпиапрамицина (S2-b) и 5-эпи-4"-N-метилапрамицина (S1-f)
[Химическая формула 74]

[0470]
Пример 43-(i): Синтез 4"-N-бензил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5-эпиапрамицина (S2-b)
Указанное в заголовке соединение (S2-b) [2,34 г (96%)] в виде бесцветного твердого вещества получали способом, аналогично примеру 1-(iv), с использованием 2,52 г (2,3 ммоль в виде соли ТФУК) указанного в заголовке соединения (B4) примера 12-(v).
[0471]
МС (ESI) m/z: 1080 (M+Na)+.
[0472]
Пример 43-(ii): Синтез 5-эпи-4"-N-метилапрамицина (S1-f)
Указанное в заголовке соединение (S1-f) [113 мг (72%)] получали способом, аналогично примеру 1-(v), с использованием 320 мг (0,30 ммоль) указанного в заголовке соединения (S2-b) примера 43-(i) и 0,1 мл 37% формалина.
[0473]
МС (ESI) m/z: 554 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 2,77 (6H, с, 4"-NMe и 7'-NMe), 4,55 (1H, т, H-5), 5,35 (1H, д, H-1') и 5,68 (1H, д, H-1").
[0474]
Пример 44: Синтез 4"-N-(2-аминоэтил)-5-эпиапрамицина (S1-g)
[Химическая формула 75]

[0475]
Указанное в заголовке соединение (S1-g) [94,5 мг (51%)] получали способом, аналогично примеру 3, с использованием 342 мг (0,32 ммоль) указанного в заголовке соединения (S2-b) примера 43-(i) и 52 мг N-Boc-2-аминоацетальдегида.
[0476]
МС (ESI) m/z: 583 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 3,02-3,14 (4H, м, 4"-NH2Et(β, α)), 4,57 (1H, м, H-5), 5,34 (1H, д, H-1') и 5,70 (1H, д, H-1").
[0477]
Пример 45: Синтез 4"-N-(3-аминопропил)-5-эпиапрамицина (S1-h)
[Химическая формула 76]

[0478]
Указанное в заголовке соединение (S1-h) [87,1 мг (48%)] получали способом, аналогично примеру 1-(v), с использованием 333 мг (0,31 ммоль) указанного в заголовке соединения (S2-b) примера 43-(i) и 80 мг 3-[(бензилоксикарбонил)амино]пропиональдегида.
[0479]
МС (ESI) m/z: 597 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,98 [2H, м, 4"-NH2Pr(β)], 2,92-3,08 (5H, м, H-7' и 4"-NH2Pr(α, γ)), 4,65 (1H, м, H-5), 5,33 (1H, д, H-1') и 5,66 (1H, д, H-1").
[0480]
Пример 46: Синтез 4"-N-(1,3-диаминопропан-2-ил)-5-эпиапрамицина (S1-i)
[Химическая формула 77]
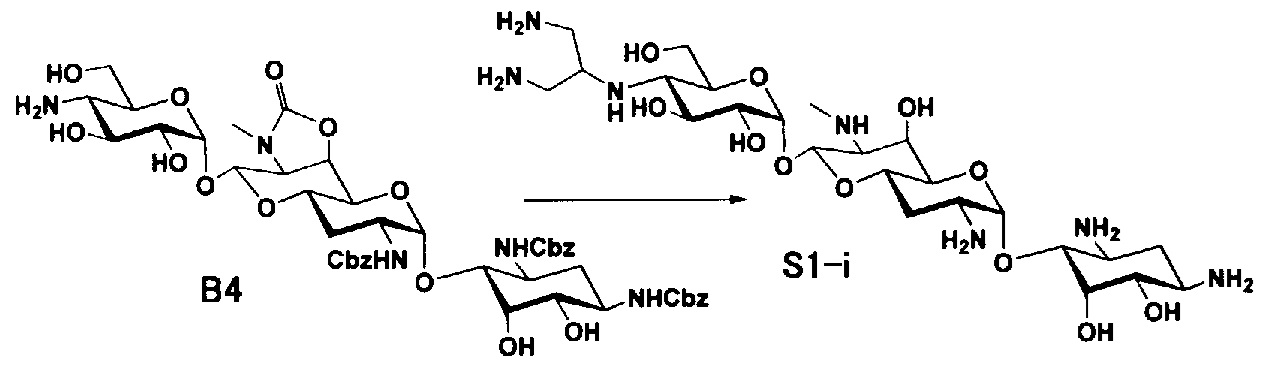
[0481]
Указанное в заголовке соединение (S1-i) [73,4 мг (54%)] получали способом, аналогично примеру 1-(v), с использованием 250 мг (0,23 ммоль в виде соли ТФУК) указанного в заголовке соединения (B4) примера 12-(v) и 90 мг 1,3-бис[(бензилоксикарбонил)амино]пропан-2-она.
[0482]
МС (ESI) m/z: 596 (M+1)+;1H ЯМР (ТФУК соль, 500 МГц, D2O): δ 1,70 (1H, кв, J=12,5 Гц, H-2 акс), 2,03 (1H, кв, J=12 Гц, H-3" акс), 2,36 (1H, дт, J=4,5, 4,5 и 12 Гц, H-3' экв), 2,43 (1H, дт, J=4,5, 4,5 и 12,5 Гц, H-2 экв), 2,65 (1H, т, J=10 Гц, H-4"), 2,73 (3H, с, NCH3), 3,29 (1H, дд, J=3 и 8,5 Гц, H-7"), 3,95 (1H, дд, J=2,5 и 11 Гц, H-4), 4,46 (1H, т, J=2,5 Гц, H-5 экв), 4,50 (1H, т, J=3 Гц, H-6'), 5,16 (1H, д, J=8,5 Гц, H-8') и 5,37 (2H, д, J=4 Гц, H-1' и H-1").
[0483]
Пример 47: Синтез 4"-дезамино-5-эпи-4"-гуанидиноапрамицина (S1-j)
[Химическая формула 78]

[0484]
Указанное в заголовке соединение (S1-j) [65,8 мг (43%)] получали способом, аналогично примеру 10, с использованием 285 мг (0,26 ммоль в виде соли ТФУК) указанного в заголовке соединения (B4) примера 12-(v) и 273 мг реагента Гудмана.
[0485]
МС (ESI) m/z: 550 (M+1)+;1H ЯМР (ТФУК соль, 500 МГц, D2O): δ 1,71 (1H, кв, J=12,5 Гц, H-2 акс), 2,05 (1H, кв, J=12 Гц, H-3' акс), 2,38 (1H, дт, J=4,5, 4,5 и 12 Гц, H-3' экв), 2,46 (1H, дт, J=4,5, 4,5 и 12,5 Гц, H-2 экв), 2,75 (3H, с, NCH3), 3,31 (1H, дд, J=3 и 8,5 Гц, H-7'), 3,52 (1H, т, J=10 Гц, H-4"), 4,47 (1H, слегка ушир. т, J=~2,5 Гц, H-5), 4,51 (1H, слегка ушир. т, J=~3 Гц, H-6'), 5,19 (1H, д, J=8,5 Гц, H-8'), 5,39 (1H, д, J=3,8 Гц, H-1') и 5,45 (1H, д, J=4 Гц, H-1").
[0486]
Пример 48: Синтез 4"-дезамино-5-дезокси-5-эпи-5-фтор-4"-гуанидиноапрамицина (S1-k)
[Химическая формула 79]
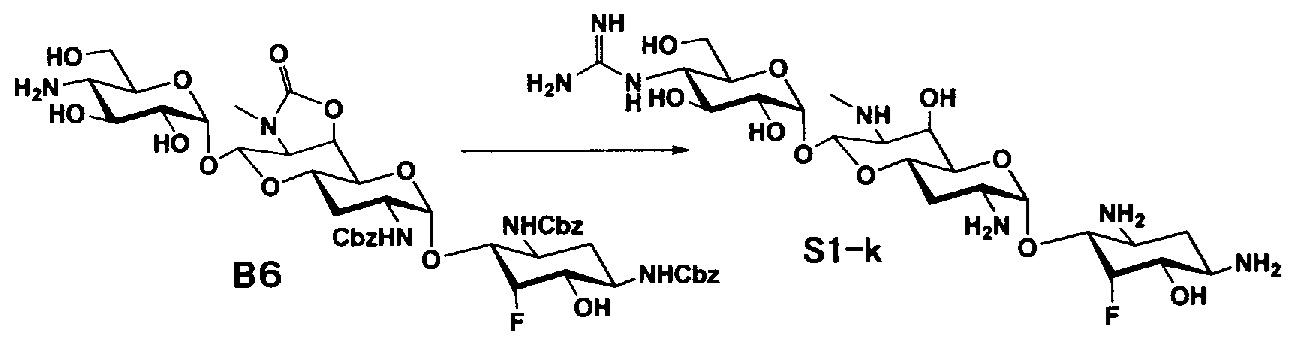
[0487]
Указанное в заголовке соединение (S1-k) [77,1 мг (45%)] получали способом, аналогично примеру 10, с использованием 305 мг (0,32 ммоль в виде соли ТФУК) указанного в заголовке соединения (B6) примера 13-(i) и 280 мг реагента Гудмана.
[0488]
МС (ESI) m/z: 552 (M+1)+;1H ЯМР (ТФУК соль, 500 МГц, D2O): δ 1,80 (1H, кв, J=12,5 Гц, H-2 акс), 2,05 (1H, кв, J=12 Гц, H-3' акс), 2,38 (1H, дт, J=4,5, 4,5 и 12 Гц, H-3' экв), 2,51 (1H, дт, J=4,5, 4,5 и 12,5 Гц, H-2 экв), 2,74 (3H, с, NCH3), 3,32 (1H, дд, J=2,5 и 8,5 Гц, H-7'), 3,52 (1H, т, J=10 Гц, H-4"), 4,14 (1H, ддд, J=~1,5, 11 и 26 Гц, H-4), 4,52 (1H, слегка ушир. т, J=~3 Гц, H-6'), 5,35 (1H, слегка ушир. дт, J=~2,~2 и 52 Гц, H-5), 5,19 (1H, д, J=8,5 Гц, H-8') и 5,43-5,57 (2H, H-1' и H-1").
[0489]
Пример 49: Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,6-дидезокси-5-еноапрамицина (T1), 4"-N-бензил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,6-дидезокси-5-еноапрамицина (T3) и 5,6-дидезокси-4"-N-метилапрамицина (T2-a)
[Химическая формула 80]
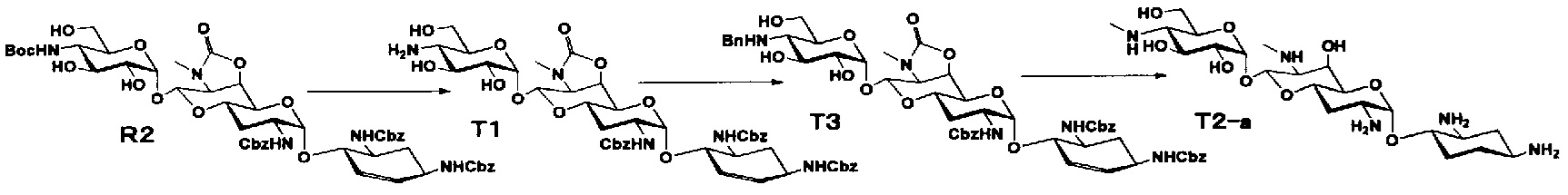
[0490]
Пример 49-(i): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,6-дидезокси-5-еноапрамицина (T1)
Указанное в заголовке соединение (T1) [2,58 г (94% в виде соли ТФУК)] получали способом, аналогично примеру 14-(vi), с использованием 3,50 г (2,6 ммоль) указанного в заголовке соединения (R2) примера 36-(ii).
[0491]
МС (ESI) m/z: 956 (M+Na)+.
[0492]
Пример 49-(ii): Синтез 4"-N-бензил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,6-дидезокси-5-еноапрамицина (T3)
Указанное в заголовке соединение (T3) [1,38 г (92%)] в виде бесцветного твердого вещества получали способом, аналогично примеру 1-(iv), с использованием 1,46 г (1,3 ммоль в виде соли ТФУК) указанного в заголовке соединения примера 49-(i).
[0493]
МС (ESI) m/z: 1046 (M+Na)+.
[0494]
Пример 49-(iii): Синтез 5,6-дидезокси-4"-N-метилапрамицина (T2-a)
Указанное в заголовке соединение (T2-a) [97,3 мг (62%)] получали способом, аналогично примеру 1-(v), с использованием 310 мг (0,30 ммоль) указанного в заголовке соединения (T3) примера 49-(ii) и 0,1 мл 37% формалина.
[0495]
МС (ESI) m/z: 522 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,42-1,67 (3H, м, H-2 акс, -6 акс и -5 акс), 2,25 (1H, м, H-6 экв), 2,41-2,52 (2H, м, H-3' экв и -5 экв), 2,75 (6H, с, 4"-NMe и 7'-NMe), 5,32 (1H, д, H-1') и 5,71 (1H, д, H-1").
[0496]
Пример 50: Синтез 4"-N-(2-аминоэтил)-5,6-дидезоксиапрамицина (T2-b)
[Химическая формула 81]

[0497]
Указанное в заголовке соединение (T2-b) [96,5 мг (61%)] получали способом, аналогично примеру 3, с использованием 300 мг (0,29 ммоль) указанного в заголовке соединения (T3) примера 49-(ii) и 50 мг N-Boc-2-аминоацетальдегида.
[0498]
МС (ESI) m/z: 551 (M+1)+;
1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,43-1,67 (3H, м, H-2 акс, -6 акс и -5 акс), 2,25 (1H, м, H-6 экв), 2,39-2,51 (2H, м, H-3' экв и -5 экв), 3,02-3,14 [4H, м, 4"-NH2Et(α, β)], 5,32 (1H, д, H-1') и 5,70 (1H, д, H-1").
[0499]
Пример 51: Синтез 4"-N-(3-аминопропил)-5,6-дидезоксиапрамицина (T2-c)
[Химическая формула 82]

[0500]
Указанное в заголовке соединение (T2-c) [88,2 мг (54%)] получали способом, аналогично примеру 1-(v), с использованием 303 мг (0,29 ммоль) указанного в заголовке соединения (T3) примера 49-(ii) и 80 мг 3-[(бензилоксикарбонил)амино]пропиональдегида.
[0501]
МС (ESI) m/z: 565 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,43-1,67 (3H, м, H-2 акс, -6 акс и -5 акс), 2,25 (1H, м, H-6 экв), 2,39-2,50 (2H, м, H-3' экв и H-5 экв), 2,92-3,08 [5H, м, H-7' и 4"-NH2Pr(α, γ)], 5,31 (1H, д, H-1') и 5,70 (1H, д, H-1").
[0502]
Пример 52: Синтез 4"-N-(1,3-диаминопропан-2-ил)-5,6-дидезоксиапрамицина (T2-d)
[Химическая формула 83]

[0503]
Указанное в заголовке соединение (T2-d) [76,4 мг (47%)] получали способом, аналогично примеру 1-(v), с использованием 301 мг (0,29 ммоль в виде соли ТФУК) указанного в заголовке соединения (T3) примера 49-(i) и 100 мг 1,3-бис[(бензилоксикарбонил)амино]пропан-2-она.
[0504]
МС (ESI) m/z: 580 (M+1)+;1H ЯМР (ТФУК соль, 500 МГц, D2O): δ 1,33 (1H, слегка ушир. дкв, J=~3,5,~12,~12 и ~12 Гц, H-5 акс), 1,52 (1H, дкв, J=3, 13, 13 и 13 Гц, H-6 акс), 1,72 (1H, кв, J=12 Гц, H-2 акс), 2,00 (1H, кв, J=12 Гц, H-3' акс), 2,15 (1H, м, H-6 экв), 2,34 (1H, дт, J=4, 4 и 12 Гц, H-3' экв), 2,42 (2H, м, H-2 экв и H-5 экв), 2,67 (1H, т, J=10 Гц, H-4"), 2,75 (3H, с, NCH3), 3,34 (1H, дд, J=3 и 8,5 Гц, H-7'), 4,54 (1H, т, J=3 Гц, H-6'), 5,16 (1H, д, J=8,5 Гц, H-8'), 5,34 (1H, д, J=4 Гц, H-1') и 5,38 (1H, д, J=4 Гц, H-1").
[0505]
Пример 53: Синтез 4"-дезамино-5,6-дидезокси-4"-гуанидиноапрамицина (T2-e)
[Химическая формула 84]
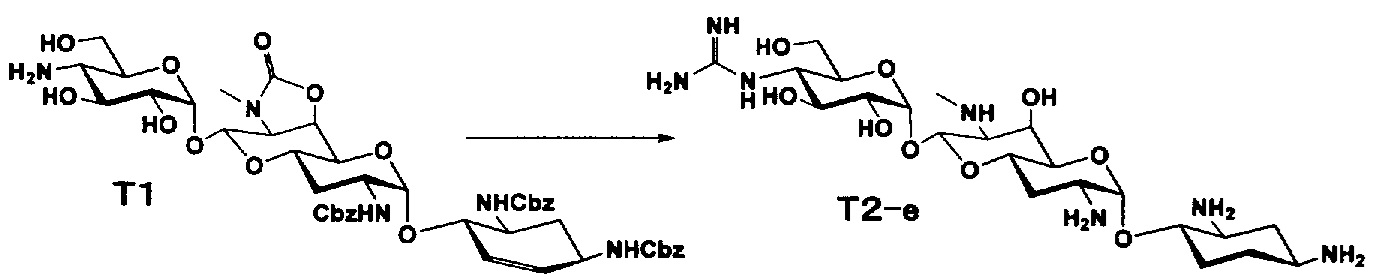
[0506]
Указанное в заголовке соединение (T2-e) [61,3 мг (43%)] получали способом, аналогично примеру 10, с использованием 275 мг (0,26 ммоль в виде соли ТФУК) указанного в заголовке соединения (T3) примера 49-(i) и 270 мг реагента Гудмана.
[0507]
МС (ESI) m/z: 550 (M+1)+.
[0508]
Пример 54: Синтез 4"-N-бензил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-6-дезокси-5-эпиапрамицина (S2-c) и 6-дезокси-5-эпи-4"-N-метилапрамицина (S1-l)
[Химическая формула 85]

[0509]
Пример 54-(i): Синтез 4"-N-бензил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-6-дезокси-5-эпиапрамицина (S2-c)
Указанное в заголовке соединение (S2-c) [1,63 г (92%)] в виде бесцветного твердого вещества получали способом, аналогично примеру 1-(iv), с использованием 1,78 г (1,7 ммоль в виде соли ТФУК) указанного в заголовке соединения (C5) примера 14-(iv).
[0510]
МС (ESI) m/z: 1064 (M+Na)+.
[0511]
Пример 54-(ii): Синтез 6-дезокси-5-эпи-4"-N-метилапрамицина (S1-l)
Указанное в заголовке соединение (S1-l) [105 мг (67%)] получали способом, аналогично примеру 1-(v), с использованием 300 мг (0,29 ммоль) указанного в заголовке соединения (S2-c) примера 54-(i) и 0,1 мл 37% формалина.
[0512]
МС (ESI) m/z: 538 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,72 (1H, ддд, H-6 акс), 2,35-2,43 (2H, м, H-2 экв и H-6 экв), 2,75 (6H, с, 4"-NMe и 7'-NMe), 4,67 (1H, м, H-5), 5,34 (1H, д, H-1') и 5,70 (1H, д, H-1").
[0513]
Пример 55: Синтез 6-дезокси-4"-N-(2-аминоэтил)-5-эпиапрамицина (S1-m)
[Химическая формула 86]

[0514]
Указанное в заголовке соединение (S1-m) [87,0 мг (53%)] получали способом, аналогично примеру 3, с использованием 302 мг (0,29 ммоль) указанного в заголовке соединения (S2-c) примера 54-(i) и 52 мг N-Boc-2-аминоацетальдегида.
[0515]
МС (ESI) m/z: 566 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,70 (1H, ддд, H-6 акс), 2,32-2,41 (2H, м, H-2 экв и 6 экв), 3,02-3,14 [4H, м, 4"-NH2Et(α,β)], 4,62-4,68 (2H, м, H-6' и H-5), 5,24 (1H, д, H-8'), 5,32 (1H, д, H-1'), 5,68 (1H, д, H-1").
[0516]
Пример 56: Синтез 6-дезокси-4"-N-(3-аминопропил)-5-эпиапрамицина (S1-n)
[Химическая формула 87]
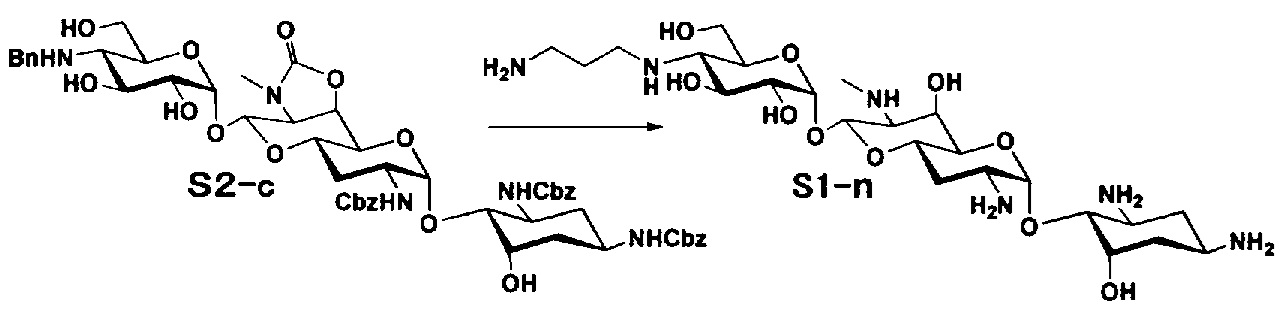
[0517]
Указанное в заголовке соединение (S1-n) [79,1 мг (47%)] получали способом, аналогично примеру 1-(v), с использованием 303 мг (0,29 ммоль) указанного в заголовке соединения (S2-c) примера 54-(i) и 83 мг 3-[(бензилоксикарбонил)амино]пропиональдегида.
[0518]
МС (ESI) m/z: 581 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,68 (1H, ддд, H-6 акс), 1,92-1,98 (2H, м, 4"-NH2Pr(β)), 2,31-2,40 (2H, м, H-2 экв и -6 экв), 2,92-3,08 (5H, м, H-7' и 4"-NH2Pr(α, γ)), 4,65 (1H, м, H-5), 5,30 (1H, д, H-1') и 5,66 (1H, д, H-1").
[0519]
Пример 57: Синтез 4"-дезамино-6-дезокси-5-эпи-4"-гуанидиноапрамицина (S1-o)
[Химическая формула 88]

[0520]
Указанное в заголовке соединение (S1-o) [67,5 мг (46%)] получали способом, аналогично примеру 10, с использованием 285 мг (0,26 ммоль в виде соли ТФУК) указанного в заголовке соединения (C5) примера 14-(vi) и 273 мг реагента Гудмана.
[0521]
МС (ESI) m/z: 566 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,74 (1H, ддд, H-6 акс), 2,36-2,42 (2H, м, H-2 экв и -6 экв), 4,68 (1H, м, H-5), 5,35 (1H, д, H-1') и 5,75 (1H, д, H-1"),13C ЯМР (25% ND3-D2O, 125 МГц): δ 158,3 (C=NH).
[0522]
Пример 58: Синтез 4"-N-(1,3-диаминопропан-2-ил)-5,6"-дидезоксиапрамицина (S1-p)
[Химическая формула 89]

[0523]
Указанное в заголовке соединение (S1-p) [18,1 мг (39%)] получали способом, аналогично примеру 1-(v), с использованием 83,9 мг (0,081 ммоль в виде соли ТФУК) указанного в заголовке соединения (L4) примера 26-(iv) и 57 мг 1,3-бис[(бензилоксикарбонил)амино]пропан-2-она.
[0524]
МС (ESI) m/z: 580 (M+1)+;1H ЯМР (DCl-D2O, 500 МГц): δ 1,22 (3H, д, J=6 Гц, CH3-6"), 1,45 (1H, кв, J=12 Гц, H-5 акс), 1,75 (1H, кв, J=12,5 Гц, H-2 акс), 2,00 (1H, кв, J=12 Гц, H-3' акс), 2,38 (1H, т, J=10 Гц, H-4"), 2,45 (1H, дт, J=4, 4 и 12,5 Гц, H-2 экв), 2,67 (1H, дт, J=4,5, 4,5 и 12 Гц, H-5 экв), 2,75 (3H, с, NCH3), 3,34 (1H, дд, J=2,5 и 8,5 Гц, H-7'), 4,55 (1H, т, J=2,5 Гц, H-6'), 5,13 (1H, д, J=8,5 Гц, H-8'), 5,32 (1H, д, J=4 Гц, H-1") и 5,35 (1H, д, J=3,8 Гц, H-1').
[0525]
Пример 59: Синтез 4"-дезамино-5,6"-дидезокси-4"-гуанидиноапрамицина (S1-q)
[Химическая формула 90]

[0526]
Указанное в заголовке соединение (S1-q) [12,2 мг (23%)] получали способом, аналогично примеру 10, с использованием 100 мг (0,095 ммоль в виде соли ТФУК) указанного в заголовке соединения (L4) примера 26-(iv) и 81,8 мг реагента Гудмана.
[0527]
МС (ESI) m/z: 550 (M+1)+;1H ЯМР (DCl-D2O, 500 МГц): δ 1,21 (3H, д, H-6"), 1,78 (1H, кв, H-5 акс), 2,45 (1H, ддд, H-5 экв), 5,35 (1H, д, H-1') и 5,38 (1H, д, H-1"),13C ЯМР (DCl-D2O, 125 МГц): δ 157,41 (C=NH).
[0528]
Пример 60: Синтез 1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,3"-дидезоксиапрамицина (U1), 1,3,2'-три-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-5,3"-дидезоксиапрамицина (U2), 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,3"-дидезоксиапрамицина (U3) и 4"-дезамино-5,3"-дидезокси-4"-гуанидиноапрамицина (U4-a)
[Химическая формула 91]

[0529]
Пример 60-(i): Синтез 1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,3"-дидезоксиапрамицина (U1)
Указанное в заголовке соединение (U1) [1,09 г (97%)] получали способом, аналогично примеру 14-(v), с использованием 1,45 г (1,05 ммоль) указанного в заголовке соединения (M6) примера 27-(vii).
[0530]
МС (ESI) m/z: 1092 (M+Na)+.
[0531]
Пример 60-(ii): Синтез 1,3,2'-три-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-5,3"-дидезоксиапрамицина (U2)
Указанное в заголовке соединение (U2) [866 мг (96%)] получали способом, аналогично примеру 1-(ii), с использованием 1,00 г (0,94 ммоль) указанного в заголовке соединения (U1) примера 60-(i) и 45 мг NaH.
[0532]
МС (ESI) m/z: 984 (M+Na)+.
[0533]
Пример 60-(iii): Синтез 1,3,2'-три-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,3"-дидезоксиапрамицина (U3)
Указанное в заголовке соединение (U3) [713 мг (92%)] получали способом, аналогично примеру 1-(iii), с использованием 801 мг (0,83 ммоль) указанного в заголовке соединения (U2) примера 60-(ii).
[0534]
МС (ESI) m/z: 958 (M+Na)+.
[0535]
Пример 60-(iv): Синтез 4"-дезамино-5,3"-дидезокси-4"-гуанидиноапрамицина (U4-a)
Указанное в заголовке соединение (U4-a) [174 мг (45%)] получали способом, аналогично примеру 10, с использованием 735 мг (0,70 ммоль в виде соли ТФУК) указанного в заголовке соединения (U3) примера 60-(iii) и 550 мг реагента Гудмана.
[0536]
МС (ESI) m/z: 550 (M+Na)+;1H ЯМР (ТФУК соль, 500 МГц, D2O): δ 1,46 (1H, кв, J=12 Гц, H-5 акс), 1,73 (1H, кв, J=12,5 Гц, H-2 акс), 1,84 (1H, кв, J=12 Гц, H-3" акс), 2,00 (1H, кв, J=12 Гц, H-3' акс), 2,15 (1H, дт, J=4, 4 и 12 Гц, H-3" экв), 2,36 (1H, дт, J=4,5, 4,5 и 12 Гц, H-3' экв), 2,46 (1H, дт, J=4, 4 и 12,5 Гц, H-2 экв), 2,67 (1H, дт, J=4,5, 4,5 и 12 Гц, H-5 экв), 2,77 (3H, с, NCH3), 3,32 (1H, дд, J=3 и 8,5 Гц, H-7'), 4,52 (1H, слегка ушир. т, J=~2,5 Гц, H-6'), 5,22 (1H, д, J=8,5 Гц, H-8'), 5,33 (1H, д, J=4 Гц, H-1") и 5,35 (1H, д, J=3,8 Гц, H-1').
[0537]
Пример 61: Синтез 4"-N-глицилапрамицина (V1-a)
[Химическая формула 92]

[0538]
Раствор, полученный добавлением 0,16 мл триэтиламина и 122 мг N-гидроксисукцинимидного эфира N-(трет-бутоксикарбонил)глицина к раствору 300 мг (0,31 ммоль) соединения, представленного формулой (A3), растворенного в 2 мл ДМФА, подвергали взаимодействию при комнатной температуре в течение 8 часов. После завершения реакции, реакционную смесь концентрировали при пониженном давлении и растворяли в 1-бутаноле с последующей промывкой водой. После чего, органический слой концентрировали при пониженном давлении, концентрированный органический слой обрабатывали способом, аналогично примеру 10, что давало 131 мг (71%) указанного в заголовке соединения (V1-a).
[0539]
МС (ESI) m/z: 597 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,58 (1H, кв, H-2 акс), 2,03 (1H, кв, J=12 Гц, H-3' акс), 2,34 (1H, дт, H-3' экв), 2,50 (1H, дт, H-2 экв), 2,75 (3H, с, NCH3), 3,62 (2H, с, CH2 (глицил)), 5,28 (1H, д, H-8'), 5,50 (1H, д, H-1') и 5,75 (1H, д, H-1").
[0540]
Пример 62: Синтез 4"-N-саркозилапрамицина (V1-b)
[Химическая формула 93]

[0541]
Указанное в заголовке соединение (V1-b) [125 мг (66%)] получали способом, аналогично примеру 61, с использованием 300 мг (0,31 ммоль) соединения, представленного формулой (A3), и 122 мг N-гидроксисукцинимидного эфира N-(трет-бутоксикарбонил)саркозина.
[0542]
МС (ESI) m/z: 611 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,58 (1H, кв, H-2 акс), 2,05 (1H, кв, H-3' акс), 2,33 (1H, дт, H-3' экв), 2,51 (1H, дт, J=4, 4 и 12,5 Гц, H-2 экв), 2,75 (3H, с, 7'-NCH3), 2,65 (3H, с, NCH3(саркозил)), 3,60 и 3,64 (каждый 1H, каждый д, CH2(саркозил)), 5,29 (1H, д, H-8'), 5,52 (1H, д, H-1') и 5,76 (1H, д, H-1").
[0543]
Пример 63: Синтез 4"-N-(L-аланил)апрамицина (V1-c)
[Химическая формула 94]

[0544]
Указанное в заголовке соединение (V1-c) [121 мг (64%)] получали способом, аналогично примеру 61, с использованием 300 мг (0,31 ммоль) соединения, представленного формулой (A3), и 125 мг N-гидроксисукцинимидного эфира N-(трет-бутоксикарбонил)-L-аланина.
[0545]
МС (ESI) m/z: 611 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,58 (1H, кв, H-2 акс), 1,65 (3H, д, C-CH3(аланил)), 2,04 (1H, кв, H-3' акс), 2,35 (1H, дт, H-3' экв), 2,50 (1H, дт, H-2 экв), 2,76 (3H, с, 7'-NCH3), 3,83-3,89 (1H, м, CH(аланил)), 5,27 (1H, д, H-8'), 5,50 (1H, д, H-1') и 5,75 (1H, д, H-1").
[0546]
Пример 64: Синтез 4"-N-(D-аланил)апрамицина (V1-d)
[Химическая формула 95]

[0547]
Указанное в заголовке соединение (V1-d) [115 мг (61%)] получали способом, аналогично примеру 61, с использованием 300 мг (0,31 ммоль) соединения, представленного формулой (A3), и 125 мг N-гидроксисукцинимидного эфира N-(трет-бутоксикарбонил)-D-аланина.
[0548]
МС (ESI) m/z: 611 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,58 (1H, кв, H-2 акс), 1,65 (3H, д, Me(аланил)), 2,04 (1H, кв, H-3' акс), 2,35 (1H, дт, H-3' экв), 2,50 (1H, дт, H-2 экв), 2,76 (3H, с, 7'-NCH3), 3,83-3,89 (1H, м, CH(аланил)), 5,27 (1H, д, H-8'), 5,50 (1H, д, H-1') и 5,75 (1H, д, H-1").
[0549]
Пример 65: Синтез 4"-N-(L-серил)апрамицина (V1-e)
[Химическая формула 96]

[0550]
Указанное в заголовке соединение (V1-e) [128 мг (66%)] получали способом, аналогично примеру 61, с использованием 300 мг (0,31 ммоль) соединения, представленного формулой (A3), и 138 мг N-гидроксисукцинимидного эфира N-(трет-бутоксикарбонил)-L-серина.
[0551]
МС (ESI) m/z: 627 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,58 (1H, кв, H-2 акс), 2,03 (1H, кв, H-3' акс), 2,35 (1H, дт, H-3' экв), 2,50 (1H, дт, H-2 экв), 2,75 (3H, с, 7'-NCH3), 4,13-4,20 (2H, м, CH2(серил)), 4,30 (1H, т, CH(серил)), 5,28 (1H, д, H-8'), 5,50 (1H, д, H-1') и 5,76 (1H, д, H-1").
[0552]
Пример 66: Синтез 4"-N-(D-серил)апрамицина (V1-f)
[Химическая формула 97]

[0553]
Указанное в заголовке соединение (V1-f) [122 мг (63%)] получали способом, аналогично примеру 61, с использованием 300 мг (0,31 ммоль) соединения, представленного формулой (A3), и 138 мг N-гидроксисукцинимидного эфира N-(трет-бутоксикарбонил)-D-серина.
[0554]
МС (ESI) m/z: 627 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,57 (1H, кв, H-2 акс), 2,03 (1H, кв, H-3' акс), 2,34 (1H, дт, H-3' экв), 2,50 (1H, дт, H-2 экв), 2,76 (3H, с, 7'-NCH3), 4,13-4,20 (2H, м, CH2(серил)), 4,30 (1H, т, CH(серил)), 5,28 (1H, д, H-8'), 5,50 (1H, д, H-1') и 5,76 (1H, д, H-1").
[0555]
Пример 67: Синтез 4"-N-(β-аланил)апрамицина (V1-g)
[Химическая формула 98]

[0556]
Указанное в заголовке соединение (V1-g) [120 мг (63%)] получали способом, аналогично примеру 61, с использованием 300 мг (0,31 ммоль) соединения, представленного формулой (A3), и 125 мг N-гидроксисукцинимидного эфира N-(трет-бутоксикарбонил)-β-аланина.
[0557]
МС (ESI) m/z: 611 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,58 (1H, кв, H-2 акс), 2,03 (1H, кв, H-3' акс), 2,35 (1H, дт, H-3' экв), 2,50 (1H, дт, H-2 экв), 2,65 (2H, т, CH2(β-аланил)), 2,75 (3H, с, 7'-NCH3), 3,17 (2H, т, CH2(β-аланил)), 5,28 (1H, д, H-8'), 5,50 (1H, д, H-1') и 5,75 (1H, д, H-1").
[0558]
Пример 68: Синтез 4"-N-(L-изосерил)апрамицина (V1-h)
[Химическая формула 99]

[0559]
Указанное в заголовке соединение (V1-h) [105 мг (54%)] получали способом, аналогично примеру 61, с использованием 300 мг (0,31 ммоль) соединения, представленного формулой (A3), и 158 мг N-гидроксисукцинимидного эфира N-(п-метоксибензилоксикарбонил)-L-изосерина.
[0560]
МС (ESI) m/z: 627 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,57 (1H, кв, H-2 акс), 2,03 (1H, кв, H-3' акс), 2,35 (1H, дт, H-3' экв), 2,50 (1H, дт, H-2 экв), 2,75 (3H, с, 7'-NCH3), 3,20 (1H, дд, CH2(изосерил)), 3,33 (1H, дд, CH2(изосерил)), 4,55 (1H, т, CH(изосерил)), 5,27 (1H, д, H-8'), 5,52 (1H, д, H-1') и 5,76 (1H, д, H-1").
[0561]
Пример 69: Синтез 5-эпи-4"-N-глицилапрамицина (V1-i)
[Химическая формула 100]

[0562]
Указанное в заголовке соединение (V1-i) [76,1 мг (64%)] получали способом, аналогично примеру 61, с использованием 200 мг (0,20 ммоль) соединения, представленного формулой (B4), и 88,2 мг N-гидроксисукцинимидного эфира N-(трет-бутоксикарбонил)глицина.
[0563]
МС (ESI) m/z: 597 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,35 (1H, кв, H-2 акс), 1,99 (1H, кв, H-3' акс), 2,25 (1H, дт, H-3' экв), 2,34 (1H, дт, H-2 экв), 2,64 (3H, с, 7'-NCH3), 3,63 (2H, с, CH2(глицил)), 4,53 (1H, т, H-5), 5,18 (1H, H-8'), 5,25 (1H, д, H-1') и 5,67 (1H, д, H-1").
[0564]
Пример 70: Синтез 5-эпи-4"-N-саркозилапрамицина (V1-j)
[Химическая формула 101]

[0565]
Указанное в заголовке соединение (V1-j) [81,5 мг (65%)] получали способом, аналогично примеру 61, с использованием 200 мг (0,20 ммоль) соединения, представленного формулой (B4), и 95,2 мг N-гидроксисукцинимидного эфира N-(трет-бутоксикарбонил)саркозина.
[0566]
МС (ESI) m/z: 611 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,40 (1H, кв, H-2 акс), 2,04 (1H, кв, H-3' акс), 2,30 (1H, дт, H-3' экв), 2,43 (1H, дт, H-2 экв), 2,64 (3H, с, 7'-NCH3), 2,70 (3H, с, NCH3(саркозил)), 3,57 и 3,62 (каждый 1H, каждый д, CH2(саркозил)), 4,56 (1H, т, H-5), 5,22 (1H, д, J=8,5 Гц, H-8'), 5,32 (1H, д, H-1') и 5,69 (1H, д, H-1").
[0567]
Пример 71: Синтез 4"-N-(L-аланил)-5-эпиапрамицина (V1-k)
[Химическая формула 102]

[0568]
Указанное в заголовке соединение (V1-k) [121 мг (64%)] получали способом, аналогично примеру 61, с использованием 200 мг (0,20 ммоль) соединения, представленного формулой (B4), и 96,3 мг N-гидроксисукцинимидного эфира N-(трет-бутоксикарбонил)-L-аланина.
[0569]
МС (ESI) m/z: 611 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,39 (1H, кв, H-2 акс), 1,65 (3H, д, CH3(аланил)), 2,03 (1H, кв, H-3' акс), 2,31 (1H, дт, H-3' экв), 2,43 (1H, дт, H-2 экв), 2,65 (3H, с, 7'-NCH3), 3,85-3,90 (1H, м, CH(аланил)), 4,53 (1H, т, H-5), 5,21 (1H, д, H-8'), 5,31 (1H, д, H-1') и 5,67 (1H, д, H-1").
[0570]
Пример 72: Синтез 5-эпи-4"-N-(L-серил)апрамицина (V1-l)
[Химическая формула 103]
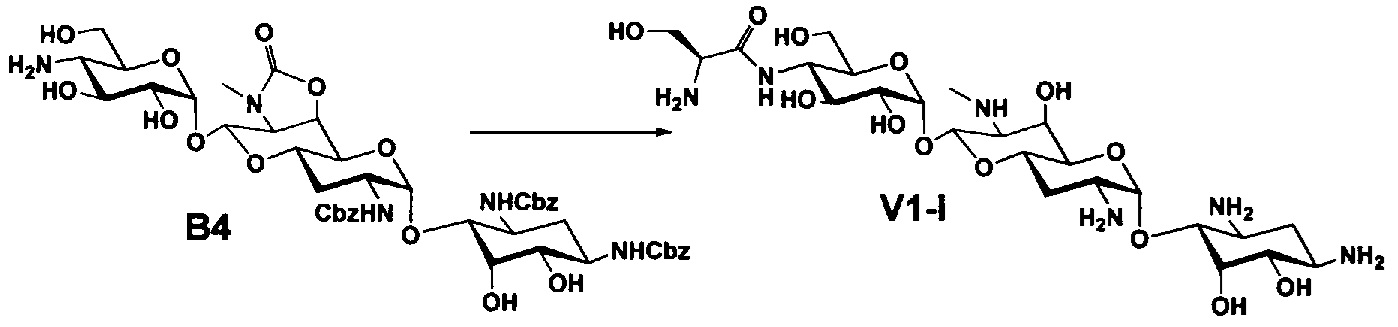
[0571]
Указанное в заголовке соединение (V1-l) [83,4 мг (65%)] получали способом, аналогично примеру 61, с использованием 200 мг (0,31 ммоль) соединения, представленного формулой (B4), и 92,0 мг N-гидроксисукцинимидного эфира N-(трет-бутоксикарбонил)-L-серина.
[0572]
МС (ESI) m/z: 627 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,39 (1H, кв, H-2 акс), 2,03 (1H, кв, H-3' акс), 2,31 (1H, дт, H-3' экв), 2,43 (1H, дт, H-2 экв), 2,65 (3H, с, 7'-NCH3), 4,13-4,20 (2H, м, CH2(серил)), 4,30 (1H, т, CH(серил)), 4,55 (1H, т, H-5), 5,21 (1H, д, H-8'), 5,30 (1H, д, H-1') и 5,68 (1H, д, H-1").
[0573]
Пример 73: Синтез 4"-N-(β-аланил)-5-эпиапрамицина (V1-m)
[Химическая формула 104]

[0574]
Указанное в заголовке соединение (V1-m) [79,6 мг (65%)] получали способом, аналогично примеру 61, с использованием 200 мг (0,20 ммоль) соединения, представленного формулой (B4), и 95,5 мг N-гидроксисукцинимидного эфира N-(трет-бутоксикарбонил)-β-аланина.
[0575]
МС (ESI) m/z: 611 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,35 (1H, кв, H-2 акс), 1,99 (1H, кв, H-3' акс), 2,25 (1H, дт, H-3' экв), 2,38 (1H, дт, H-2 экв), 2,64 (3H, с, 7'-NCH3), 2,67 (2H, т, CH2(β-аланил)), 3,15 (2H, т, CH2(β-аланил)), 5,16 (1H, д, H-8'), 4,50 (1H, т, H-5), 5,25 (1H, д, H-1') и 5,63 (1H, д, H-1").
[0576]
Пример 74: Синтез 5-эпи-4"-N-(L-изосерил)апрамицина (V1-n)
[Химическая формула 105]

[0577]
Указанное в заголовке соединение (V1-n) [77,5 мг (62%)] получали способом, аналогично примеру 61, с использованием 200 мг (0,20 ммоль) соединения, представленного формулой (B4), и 105 мг N-гидроксисукцинимидного эфира N-(п-метоксибензилоксикарбонил)-L-изосерина.
[0578]
МС (ESI) m/z: 627 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,34 (1H, кв, H-2 акс), 1,98 (1H, кв, H-3' акс), 2,24 (1H, дт, H-3' экв), 2,37 (1H, дт, H-2 экв), 2,61 (3H, с, 7'-NCH3), 3,08 (1H, дд, CH2(изосерил)), 3,33 (1H, дд, CH2(изосерил)), 4,43 (1H, т, CH(изосерил)), 4,51 (1H, т, H-5), 5,15 (1H, д, H-8'), 5,24 (1H, д, H-1') и 5,65 (1H, д, H-1").
[0579]
Пример 75: Синтез 6-дезокси-5-эпи-4"-N-глицилапрамицина (V1-o)
[Химическая формула 106]

[0580]
Указанное в заголовке соединение (V1-o) [77,5 мг (74%)] получали способом, аналогично примеру 61, с использованием 170 мг (0,18 ммоль) соединения, представленного формулой (C5), и 79,4 мг N-гидроксисукцинимидного эфира N-(трет-бутоксикарбонил)глицина.
[0581]
МС (ESI) m/z: 581 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,48 (1H, кв, H-2 акс), 1,72 (1H, кв, H-6 акс), 2,07 (1H, кв, H-3' акс), 2,37 (1H, дт, H-3' экв), 2,40 (1H, дт, H-6 экв), 2,48 (1H, дт, H-2 экв), 2,63 (3H, с, 7'-NCH3), 3,72 (2H, с, CH2(глицил)), 4,69 (1H, дд, H-5), 5,26 (1H, д, H-8'), 5,35 (1H, д, H-1') и 5,74 (1H, д, H-1").
[0582]
Пример 76: Синтез 6-дезокси-5-эпи-4"-N-саркозилапрамицина (V1-p)
[Химическая формула 107]

[0583]
Указанное в заголовке соединение (V1-p) [70,6 мг (66%)] получали способом, аналогично примеру 61, с использованием 170 мг (0,18 ммоль) соединения, представленного формулой (C5), и 85,5 мг N-гидроксисукцинимидного эфира N-(трет-бутоксикарбонил)саркозина.
[0584]
МС (ESI) m/z: 611 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,48 (1H, кв, H-2 акс), 1,72 (1H, кв, H-6 акс), 2,08 (1H, кв, H-3' акс), 2,37 (1H, дт, H-3' экв), 2,40 (1H, дт, H-6 экв), 2,48 (1H, дт, H-2 экв), 2,68 (3H, с, 7'-NCH3), 2,73 (3H, с, NMe(саркозил)), 3,63 и 3,67 (каждый 1H, каждый д, CH2(саркозил)), 4,65 (1H, дд, H-5), 5,26 (1H, д, H-8'), 5,35 (1H, д, H-1') и 5,75 (1H, д, H-1").
[0585]
Пример 77: Синтез 4"-N-(β-аланил)-6-дезокси-5-эпиапрамицина (V1-q)
[Химическая формула 108]

[0586]
Указанное в заголовке соединение (V1-q) [72,1 мг (67%)] получали способом, аналогично примеру 61, с использованием 170 мг (0,18 ммоль) соединения, представленного формулой (C5), и 86,0 мг N-гидроксисукцинимидного эфира N-(трет-бутоксикарбонил)-β-аланина.
[0587]
МС (ESI) m/z: 595 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,48 (1H, кв, H-2 акс), 1,73 (1H, кв, H-6 акс), 2,08 (1H, кв, H-3' акс), 2,37 (1H, дт, H-3' экв), 2,42 (1H, дт, H-6 экв), 2,48 (1H, дт, H-2 экв), 2,70 (3H, с, 7'-NCH3), 2,73 (2H, т, CH2(β-аланил)), 3,18 (2H, т, CH2(β-аланил)), 4,69 (1H, дд, H-5), 5,26 (1H, д, H-8'), 5,37 (1H, д, H-1') и 5,77 (1H, д, H-1").
[0588]
Пример 78: Синтез 6-дезокси-5-эпи-4"-N-(L-изосерил)апрамицина (V1-r)
[Химическая формула 109]

[0589]
Указанное в заголовке соединение (V1-r) [70,5 мг (64%)] получали способом, аналогично примеру 61, с использованием 170 мг (0,18 ммоль) соединения, представленного формулой (C5), и 94,5 мг N-гидроксисукцинимидного эфира N-(п-метоксибензилоксикарбонил)-L-изосерина.
[0590]
МС (ESI) m/z: 611 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,48 (1H, кв, H-2 акс), 1,73 (1H, кв, H-6 акс), 2,08 (1H, кв, H-3' акс), 2,37 (1H, дт, H-3' экв), 2,42 (1H, дт, H-6 экв), 2,48 (1H, дт, H-2 экв), 2,74 (3H, с, 7'-NCH3), 3,20 (1H, дд, CH2(изосерил)), 3,45 (1H, дт, CH2(изосерил)), 4,54 (1H, кв, CH(изосерил)), 4,69 (1H, дд, H-5), 5,26 (1H, д, H-8'), 5,37 (1H, д, H-1') и 5,77 (1H, д, H-1").
[0591]
Пример 79: Синтез 5-азид-1,3,2'-трис-N-(бензилоксикарбонил)-5-дезокси-5-эпиапрамицина (W1) и 5-амино-4"-дезамино-5-дезокси-5-эпи-4"-гуанидиноапрамицина (W2-a)
[Химическая формула 110]

[0592]
Пример 79-(i): Синтез 5-азид-1,3,2'-трис-N-(бензилоксикарбонил)-5-дезокси-5-эпиапрамицина (W1)
Раствор, полученный добавлением 3,4 мл 4Н. водного раствора NaOH к раствору 1,31 г (0,87 ммоль) указанного в заголовке соединения (D1) примера 16-(i), растворенного в 20 мл 1,4-диоксана, подвергали взаимодействию при комнатной температуре в течение 2 часов. Полученный реакционный раствор нейтрализовали добавлением 2Н. HCl, концентрировали при пониженном давлении, и остаток промывали водой и затем промывали изопропиловым эфиром. Полученное твердое вещество растворяли в 10 мл раствора 90% ТФУК-MeOH, и полученную смесь подвергали взаимодействию при комнатной температуре в течение 2 часов. Полученный реакционный раствор концентрировали при пониженном давлении, и остаток промывали изопропиловым эфиром и сушили, что давало 937 мг (90% в виде соли ТФУК) указанного в заголовке соединения (W1) в виде бесцветного твердого вещества.
[0593]
МС (ESI) m/z: 967 (M+1)+.
[0594]
Пример 79-(ii): Синтез 5-амино-4"-дезамино-5-дезокси-5-эпи-4"-гуанидиноапрамицина (W2-a)
Указанное в заголовке соединение (W2-a) [45,5 мг (37%)] получали способом, аналогично примеру 10, с использованием 254 мг (0,21 ммоль в виде соли 2ТФУК) указанного в заголовке соединения (W1) примера 79-(i) и 253 мг реагента Гудмана.
[0595]
МС (ESI) m/z: 581 (M+1)+;1H ЯМР (ТФУК соль, 500 МГц, D2O): δ 1,82 (1H, кв, J=12,5 Гц, H-2 акс), 2,03 (1H, кв, J=12 Гц, H-3' акс), 2,40 (1H, дт, J=4,5, 4,5 и 12 Гц, H-3' экв), 2,53 (1H, дт, J=4,5, 4,5 и 12,5 Гц, H-2 экв), 2,74 (3H, с, NCH3), 3,30 (1H, дд, J=3 и 8,5 Гц, H-7'), 3,52 (1H, т, J=10 Гц, H-4"), 3,58 (1H, дд, J=2,5 и 10 Гц, H-5'), 3,80 (1H, т, J=10 Гц, H-3"), 4,04 (1H, дд, J=4 и 11 Гц, H-6), 4,18 (1H, т, J=4 Гц, H-5), 4,23 (1H, дд, J=4 и 11 Гц, H-4), 4,54 (1H, слегка ушир. т, J=2,5 Гц, H-6'), 5,19 (1H, д, J=8,5 Гц, H-8'), 5,41 (1H, д, J=3,8 Гц, H-1') и 5,44 (1H, д, J=4 Гц, H-1").
[0596]
Пример 80: Синтез 5-амино-5-дезокси-5-эпи-4"-N-глицилапрамицина (W2-b)
[Химическая формула 111]

[0597]
Указанное в заголовке соединение (W2-b) [40,1 мг (34%)] получали способом, аналогично примеру 61, с использованием 254 мг (0,21 ммоль в виде соли 2ТФУК) указанного в заголовке соединения (W1) примера 79-(i) и 90,0 мг N-гидроксисукцинимидного эфира N-(трет-бутоксикарбонил)глицина.
[0598]
МС (ESI) m/z: 596 (M+1)+;1H ЯМР (ТФУК соль, 500 МГц, D2O): δ 1,81 (1H, кв, J=12,5 Гц, H-2 акс), 2,03 (1H, кв, J=12 Гц, H-3' акс), 2,40 (1H, дт, J=4,5, 4,5 и 12 Гц, H-3' экв), 2,54 (1H, дт, J=4,5, 4,5 и 12,5 Гц, H-2 экв), 2,75 (3H, с, NCH3), 3,31 (1H, дд, J=3 и 8,5 Гц, H-7'), 3,95 (1H, дт, J=4,5, 4,5 и 11 Гц, H-4'), 4,04 (1H, дд, J=4 и 11 Гц, H-6), 4,14 (1H, дд, J=4 и 11 Гц, H-4), 4,18 (1H, т, J=4 Гц, H-5), 4,54 (1H, слегка ушир. т, J=~2,5 Гц, H-6'), 5,20 (1H, д, J=8,5 Гц, H-8'), 5,41 (1H, д, J=3,8 Гц, H-1') и 5,44 (1H, д, J=4 Гц, H-1").
[0599]
Пример 81: Синтез 5-амино-5-дезокси-5-эпи-4"-N-(L-изосерил)апрамицина (W2-c)
[Химическая формула 112]

[0600]
Указанное в заголовке соединение (W2-c) [46,6 мг (49%)] получали способом, аналогично примеру 61, с использованием 254 мг (0,21 ммоль в виде соли 2ТФУК) указанного в заголовке соединения (D2) примера 79-(i) и 105 мг N-гидроксисукцинимидного эфира N-(п-метоксикарбонил)-L-изосерина.
[0601]
МС (ESI) m/z: 626 (M+1)+;1H ЯМР (ТФУК соль, 500 МГц, D2O): δ 1,87 (1H, кв, J=12,5 Гц, H-2 акс), 2,03 (1H, кв, J=12 Гц, H-3' акс), 2,42 (1H, дт, J=4,5, 4,5 и 12 Гц, H-3' экв), 2,57 (1H, дт, J=4,5, 4,5 и 12,5 Гц, H-2 экв), 2,75 (3H, с, NCH3), 4,42 (1H, дд, J=4 и 8 Гц, COCH(OH)), 4,56 (1H, слегка ушир. т, J=~3 Гц, H-6'), 5,20 (1H, д, J=8,5 Гц, H-8'), 5,42 (1H, д, J=4 Гц, H-1') и 5,44 (1H, д, J=4 Гц, H-1").
[0602]
Пример 82: Синтез 1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-3"-дезокси-5-эпиапрамицина (X1-a), 1,3,2'-три-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-3"-дезокси-5-эпиапрамицина (X2-a), 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-3"-дезокси-5-эпиапрамицина (X3-a) и 4"-дезамино-3"-дезокси-5-эпи-4"-гуанидиноапрамицина (X4-a)
[Химическая формула 113]

[0603]
Пример 82-(i): Синтез 1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-3"-дезокси-5-эпиапрамицина (X1-a)
Указанное в заголовке соединение (X1-a) [1,08 г (95%)] получали способом, аналогично примеру 14-(v), с использованием 1,47 г (1,05 ммоль) указанного в заголовке соединения (M8) примера 28-(i).
[0604]
МС (ESI) m/z: 1108 (M+Na)+.
[0605]
Пример 82-(ii): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-3"-дезокси-5-эпиапрамицина (X2-a)
Указанное в заголовке соединение (X2-a) [891 мг (96%)] получали способом, аналогично примеру 1-(ii), с использованием 1,03 г (0,95 ммоль) указанного в заголовке соединения (X1-a) примера 82-(i) и 45 мг NaH.
[0606]
МС (ESI) m/z: 1000 (M+Na)+.
[0607]
Пример 82-(iii): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-3"-дезокси-5-эпиапрамицина (X3-a)
Указанное в заголовке соединение (X3-a) [881 мг (93% в виде соли ТФУК)] получали способом, аналогично примеру 1-(iii), с использованием 870 мг (0,89 ммоль) указанного в заголовке соединения (X2-a) примера 82-(ii).
[0608]
МС (ESI) m/z: 974 (M+Na)+.
[0609]
Пример 82-(iv): Синтез 4"-дезамино-3"-дезокси-5-эпи-4"-гуанидиноапрамицина (X4-a)
Указанное в заголовке соединение (X4-a) [201 мг (47%)] получали способом, аналогично примеру 10, с использованием 800 мг (0,75 ммоль в виде соли ТФУК) указанного в заголовке соединения (X3-a) примера 82-(iii) и 600 мг реагента Гудмана.
[0610]
МС (ESI) m/z: 566 (M+H)+;1H ЯМР (ТФУК соль, 500 МГц, D2O): δ 1,69 (1H, кв, J=12,5 Гц, H-2 акс), 1,82 (1H, кв, J=12 Гц, H-3" акс), 2,10 (1H, кв, J=12 Гц, H-3' акс), 2,12 (1H, дт, J=4, 4 и 12 Гц, H-3" экв), 2,35 (1H, дт, J=4, 4 и 12 Гц, H-3' экв), 2,42 (1H, дт, J=4, 4 и 12,5 Гц, H-2 экв), 2,74 (3H, с, NCH3), 3,29 (1H, дд, J=2,5 и 8,5 Гц, H-7'), 4,44 (1H, слегка ушир. т, J=~2 Гц, H-5), 4,49 (1H, слегка ушир. т, J=~2,5 Гц, H-6'), 5,19 (1H, д, J=8,5 Гц, H-8'), 5,30 (1H, д, J=3,5 Гц, H-1") и 5,36 (1H, д, J=4 Гц, H-1').
[0611]
Пример 83: Синтез 1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,3"-дидезокси-5-эпи-5-фторапрамицина (X1-b), 1,3,2'-три-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-5,3"-дидезокси-5-эпи-5-фторапрамицина (X2-b), 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,3"-дидезокси-5-эпи-5-фторапрамицина (X3-b) и 4"-дезамино-5,3"-дидезокси-5-эпи-5-фтор-4"-гуанидиноапрамицина (X4-b)
[Химическая формула 114]

[0612]
Пример 83-(i): Синтез 1,3,2',4"-тетракис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,3"-дидезокси-5-эпи-5-фторапрамицина (X1-b)
Указанное в заголовке соединение (X1-b) [544 мг (96%)] получали способом, аналогично примеру 14-(v), с использованием 722 мг (0,52 ммоль) указанного в заголовке соединения (M8') примера 28-(i).
[0613]
МС (ESI) m/z: 1110 (M+Na)+.
[0614]
Пример 83-(ii): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-5,3"-дидезокси-5-эпи-5-фторапрамицина (X2-b)
Указанное в заголовке соединение (X2-b) [451 мг (92%)] получали способом, аналогично примеру 1-(ii), с использованием 500 мг (0,46 ммоль) указанного в заголовке соединения (X1-b) примера 83-(i) и 22 мг NaH.
[0615]
МС (ESI) m/z: 1002 (M+Na)+.
[0616]
Пример 83-(iii): Синтез 1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-5,3"-дидезокси-5-эпи-5-фторапрамицина (X3-b)
Указанное в заголовке соединение (X3-b) [438 мг (91%в виде соли ТФУК)] получали способом, аналогично примеру 1-(iii), с использованием 440 мг (0,45 ммоль) указанного в заголовке соединения (X2-b) примера 83-(ii).
[0617]
МС (ESI) m/z: 976 (M+Na)+.
[0618]
Пример 83-(iv): Синтез 4"-дезамино-5,3"-дидезокси-5-эпи-5-фтор-4"-гуанидиноапрамицина (X4-b)
Указанное в заголовке соединение (X4-b) [105 мг (50%)] получали способом, аналогично примеру 10, с использованием 400 мг (0,37 ммоль в виде соли ТФУК) указанного в заголовке соединения (X3-b) примера 83-(iii) и 600 мг реагента Гудмана.
[0619]
МС (ESI) m/z: 568 (M+H)+;1H ЯМР (ТФУК соль, 500 МГц, D2O): δ 1,76 (1H, кв, J=12,5 Гц, H-2 акс), 1,81 (1H, кв, J=12 Гц, H-3" акс), 2,02 (1H, кв, J=12 Гц, H-3' акс), 2,12 (1H, дт, J=4, 4 и 12 Гц, H-3" экв), 2,35 (1H, дт, J=4,5, 4,5 и 12 Гц, H-3' экв), 2,47 (1H, дт, J=4, 4 и 12,5 Гц, H-2 экв), 2,74 (3H, с, NCH3), 3,30 (1H, дд, J=3 и 8,5 Гц, H-7'), 4,10 (1H, кажущийся дд, J=11 и 26 Гц, H-4), 4,49 (1H, слегка ушир. т, J=~2,5 Гц, H-6'), 5,19 (1H, д, J=8,5 Гц, H-8'), 5,30 (1H, д, J=3,5 Гц, H-1"), 5,32 (1H, кажущийся д, J=52 Гц, H-5) и 5,43 (1H, д, J=4 Гц, H-1').
[0620]
Пример 84: Синтез 6,3"-ди-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-2"-дезокси-3"-эпи-5-O-мезилапрамицина (Y1), 5-O-ацетил-6,3"-ди-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-2"-дезокси-5,3"-диэпиапрамицина (Y2) и 2"-дезокси-5,3"-диэпиапрамицина (Y3)
[Химическая формула 115]

[0621]
Пример 84-(i): Синтез 6,3"-ди-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-2"-дезокси-3"-эпи-5-O-мезилапрамицина (Y1)
Указанное в заголовке соединение (Y1) [1,05 г (99%)] получали способом, аналогично примеру 14-(i), с использованием 1,00 г (0,84 ммоль) указанного в заголовке соединения (O3) примера 33-(iii).
[0622]
МС (ESI) m/z: 1286 (M+Na)+.
[623]
Пример 84-(ii): Синтез 5-O-ацетил-6,3"-ди-O-бензоил-1,3,2'-трис-N-(бензилоксикарбонил)-7'-N,6'-O-карбонил-4"-N,6"-O-карбонил-2"-дезокси-5,3"-диэпиапрамицина (Y2)
Указанное в заголовке соединение (Y2) [732 мг (79%)] получали способом, аналогично примеру 34-(iii), с использованием 955 мг (0,76 ммоль) указанного в заголовке соединения (Y1) примера 84-(i).
[0624]
МС (ESI) m/z: 1250 (M+Na)+.
[0625]
Пример 84-(iii): Синтез 2"-дезокси-5,3"-диэпиапрамицина (Y3)
Указанное в заголовке соединение (Y3) [77,5 мг (45%)] получали способом, аналогично примеру 27-(vii), с использованием 400 мг (0,33 ммоль) указанного в заголовке соединения (Y2) примера 84-(ii).
[0626]
МС (ESI) m/z: 524 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,38 (1H, кв, H-2 акс), 1,83 (1H, кв, J=12 Гц, H-3' акс), 2,15 (1H, дт, H-2 экв), 2,21-2,27 (1H, м, H-2" акс), 2,28 (1H, дт, H-3' экв), 3,08 (1H, дд, H-2" экв), 4,18 (1H, т, H-3"), 4,31 (1H, кв, H-3"), 4,53 (1H, т, H-5), 5,19 (1H, д, J=8,5 Гц, H-8'), 5,07 (1H, д, H-1"), 5,28 (1H, д, H-1') и 5,63 (1H, д, H-1").
[0627]
Пример 85: Синтез 5-эпи-4"-N-(D-изосерил)апрамицина (V1-s)
[Химическая формула 116]

[0628]
Указанное в заголовке соединение (V1-s) [22,5 мг (18%)] и указанное в заголовке соединение (V1-n) [20,8 мг (17%)] примера 74 получали способом, аналогично примеру 61, с использованием 200 мг (0,20 ммоль) соединения, представленного формулой (B4), и 105 мг N-гидроксисукцинимидного эфира N-(трет-бутоксикарбонил)-DL-изосерина.
[0629]
МС (ESI) m/z: 627 (M+1)+;1H ЯМР (25% ND3-D2O, 500 МГц): δ 1,33 (1H, кв, H-2 акс), 1,97 (1H, кв, H-3' акс), 2,23 (1H, дт, H-3' экв), 2,36 (1H, дт, H-2 экв), 2,61 (3H, с, 7'-NCH3), 3,03 (1H, дд, CH2(изосерил)), 3,23 (1H, дд, CH2(изосерил)), 4,20 (1H, т, H-4"), 4,44 (1H, дд, CH(изосерил)), 4,49 (1H, т, H-5), 5,13 (1H, д, H-8'), 5,23 (1H, д, H-1') и 5,63 (1H, д, H-1").
[0630]
Пример тестирования 1
Антибактериальная активность
Используя в качестве иллюстрации соединения нового аминогликозидного антибиотика по настоящему изобретению, описанного в примерах, измеряли минимальную ингибирующую концентрацию (MIC, мкг/мл) для анализа различных штаммов бактерий, применяя чашечный метод определения микроорганизмов на агаре, в соответствии со способом, описанным в the Japan Society of Chemotherapy. Результаты представлены в таблицах 1-6.
[0631]
[0632]
[0633]
[0634]
[0635]
[0636]
[0637]
Приведенные в таблицах 1-6 результаты показали, что соединения настоящего изобретения обладают антибактериальной активностью как против грамположительных, так и против грамотрицательных бактерий. Также было продемонстрировано, что соединения настоящего изобретения обладают сильной противомикробной активностью против резистентных штаммов или малочувствительных штаммов Staphylococcus aureus, Klebsiella pneumoniae, Акинетобактерий, Serratia и Pseudomonas aeruginosa, которые являются резистентными или малочувствительными к существующим антибиотикам, таким как арбекацин (ABK), амикацин (AMK) и гентамицин (GM).
Реферат
Настоящее изобретение относится к соединению, представленному общей формулой (I), или к его фармацевтически приемлемой соли, обладающим антибактериальной активностью, к фармацевтической композиции на их основе, противобактериальному средству, к применению заявленных соединений и к способу профилактики или лечения бактериального инфекционного заболевания.,где R1представляет собой атом водорода или гидроксильную группу, R2представляет собой атом водорода или аминогруппу, R3представляет собой атом водорода, атом галогена, гидроксильную группу или аминогруппу, R4представляет собой атом водорода, атом галогена или аминогруппу, где R1и R4могут вместе образовывать двойную связь, R5представляет собой атом водорода или гидроксильную группу, R6представляет собой атом водорода, гидроксильную группу или аминогруппу, R7представляет собой атом водорода или гидроксильную группу, R8представляет собой атом водорода, гидроксильную группу или аминогруппу, R9и R10, каждый независимо, представляют собой атом водорода, C1-6алкильную группу, амино-C1-6алкильную группу, гуанидино-C1-6алкильную группу, амино-C3-7циклоалкильную группу, амино-C3-7циклоалкил-C1-6алкильную группу, амидиногруппу, азетидиногруппу, необязательно замещенную C1-6алкильной группой, глицильную группу, саркозильную группу, L-аланильную группу, D-аланильную группу, L-серильную группу, D-серильную группу, β-аланильную группу, L-изосерильную группу или D-изосерильную группу; и R11представляет собой атом водорода, гидроксильную группу или атом фтора, за исключением случаев, когда (ii) R5, R8и R11представляют собой гидроксильные группы, R1, R2, R3, R4, R6, R7, R9и R10представляют собой атомы водорода, (iii) R1, R5, R8и R11представляют собой гидроксильные группы, R2, R3, R4, R6, R7, R9и R10представляют собой атомы водорода, (a) R1и R4вместе образуют двойную связь, R5, R8и R11представляют собой гидроксильные группы, и R2, R3, R6, R7, R9и R10представляют собой атомы водорода, (b) R1и R4вместе образуют двойную связь, R8и R11представляют собой гидроксильные группы, и R2, R3, R5, R6, R7, R9и R10представляют собой атомы водорода, (c) R1, R2, R3, R4, R5, R6, R7, R9и R10представляют собой атомы водорода, и R8и R11представляют собой гидроксильные группы. 5 н. и 9 з.п. ф-лы, 6 табл., 86 пр.
Формула




Комментарии