Гликолипидные соединения и их применение в лечении опухолей - RU2719486C2
Код документа: RU2719486C2
Чертежи

Описание
Изобретение относится к новым гликолипидным соединениям и фармацевтическим композициям, включающим указанные гликолипиды, и к способам получения указанных гликолипидов. Изобретение также относится к указанным гликолипидам для применения в лечении опухолей и способам лечения опухолей с использованием указанных гликолипидов.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Основной причиной смерти пациентов с раком с солидными опухолями является рецидив рака после операции, поскольку множественные метастазы являются неоперабельными и/или рефрактерными к любой терапии. Большинство таких пациентов расцениваются, как имеющие терминальный рак. Поскольку для них недоступно лечение, множество из таких пациентов умирают в течение недель или нескольких месяцев после обнаружения метастатических опухолевых поражений.
Опухоли развиваются у раковых пациентов, так как иммунная система не может определить опухолевые клетки, как клетки, которые должны быть уничтожены. Опухолевые клетки экспрессируют аутологичные опухолевые антигены у большей части раковых пациентов. Такие аутологичные опухолевые антигены могут проявлять противоопухолевый иммунный ответ. Опухолевые клетки, или мембраны опухолевых клеток должны интернализироваться антиген-представляющими клетками с целью индукции развития противоопухолевого иммунного ответа. Однако иммунная система у раковых пациентов демонстрирует "игнорирование" в отношении опухолевых антигенов, что ассоциировано с ранним развитием опухоли "скрытым" образом, так что они являются «невидимыми» для антиген-представляющих клеток (Pardoll D M. Clin. Immunol. 2000; 95:S44-49; и Dunn G P et al. Nat Immunol 2002; 3: 991-8).
Кроме того, микроокружение опухоли и локальное цитокиновое окружение часто являются супрессивными в отношении иммунной функции и могут активно индуцировать анергию и смерть иммунных клеток (Malmberg K J. Cancer Immunol. Immunother. 2004; 53: 879-92; Lugade A A et al. J. Immunol. 2005; 174: 7516-23). Эффективное лечение таких метастатических поражений требует двух компонентов:
1. Деструкция поражений, которые являются достаточно большими для визуальной детекции или посредством визуализирующей технологии, и
2. Индукция защитного противоопухолевого иммунного ответа против опухолевых антигенов.
Такой иммунный ответ приводит к иммуно-опосредованной детекции, регрессу и/или деструкции микрометастазов, которые не могут быть определены на глаз и не определяются визуализацией.
Индукция защитного противоопухолевого иммунного ответа требует поглощения опухолевых клеток или клеточных мембран антиген-представляющими клетками и их транспортировки в дренирующие лимфатические узлы, где антиген-представляющие клетки обрабатывают молекулы опухолевых антигенов. Большинство таких опухолевых антигенов являются специфическими для каждого пациента. Иммуногенные пептиды опухолевых антигенов представляются антиген-представляющими клетками в ассоциации с молекулами MHC класса I или класса II для активации опухоль-специфических CD8+ и CD4+ T клеток, соответственно. Только после того, как такие Т клетки активируются обработанными и представленными пептидами опухолевых антигенов, такие лимфоциты могут пролиферировать, покидать лимфатические узлы, циркулировать в организме, искать и разрушать метастатические опухолевые клетки, экспрессирующие опухолевые антигены. Кроме того, только после того как они активируются, Т клетки хелперы могут обеспечивать помощь В клеткам в продукции антител против опухолевых антигенов. Однако, так как опухолевые клетки естественно изменяются, чтобы быть "невидимыми" для антиген-представляющих клеток, развивающиеся опухолевые метастазы обычно игнорируются иммунной системой в той степени, что метастазирующие опухолевые клетки могут пролиферировать даже в лимфатических узлах. Следовательно, осуществление эффективного противоопухолевого иммунного ответа требует эффективного нацеливания на опухолевые клетки антиген-представляющих клеток.
Необходимы композиции и способы для внесения соединений в опухоль, например, посредством нехирургических или хирургических методов, в таких условиях, что соединение встроится в мембраны опухолевых клеток, и натуральные антитела будут взаимодействовать с введенным соединением. Считают, что такое взаимодействие будет индуцировать локальное воспаление с регрессом и/или деструкцией опухоли и нацеливание на опухолевые клетки и/или мембраны опухолевых клеток антиген-представляющих клеток. Такой процесс обеспечит защитный иммунный ответ в организме хозяине против опухолевых клеток, экспрессирующих опухолевые антигены в микрометастазах, которые не могут быть обнаружены визуально или путем визуализации и, следовательно, не могут быть удалены резекцией.
В US 2006/251661 описаны способы введения натуральных гликолипидных соединений в опухолевые поражения, которые индуцируют локальную экспрессию в опухоли (α-Gal эпитопов, которые взаимодействуют с натуральным анти-Gal антителом.)
Следовательно, существует необходимость в обеспечении альтернативных гликолипидных соединений, которые способны доставляться непосредственно в опухоль с целью активации иммунного ответа против опухоли.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В соответствии с первым аспектом изобретения обеспечивают гликолипидное соединение, выбираемое из соединения по формуле (I), (II) и (III) или его фармацевтически приемлемой соли:

(III).
В соответствии с дополнительным аспектом изобретения обеспечивают фармацевтическую композицию, включающую гликолипидное соединение, выбираемое из соединения по формуле (I), (II) и (III) или его фармацевтически приемлемой соли, как определено в настоящем описании.
В соответствии с дополнительным аспектом изобретения обеспечивают гликолипидное соединение, выбираемое из соединения по формуле (I), (II) и (III) или его фармацевтически приемлемой соли, как определено в настоящем описании, или фармацевтическую композицию, как определено в настоящем описании для применения в лечении опухоли.
В соответствии с дополнительным аспектом изобретения обеспечивают фармацевтическую композицию, включающую гликолипидное соединение, выбираемое из соединения по формуле (I), (II) и (III) или его фармацевтически приемлемой соли, как определено в настоящем описании, в комбинации с одним или более дополнительными терапевтическими средствами.
В соответствии с дополнительным аспектом изобретения обеспечивают способ лечения опухоли у пациента, включающий:
a) обеспечение:
i) пациента, имеющего по меньшей мере одну опухоль, которая включает множество раковых клеток, имеющих клеточную поверхность; и
ii) гликолипидного соединения, выбираемого из соединения по формуле (I), (II) и (III) или его фармацевтически приемлемой соли, или фармацевтическая композиция, как определено в настоящем описании;
b) введение указанного гликолипида или композиции в опухоль.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
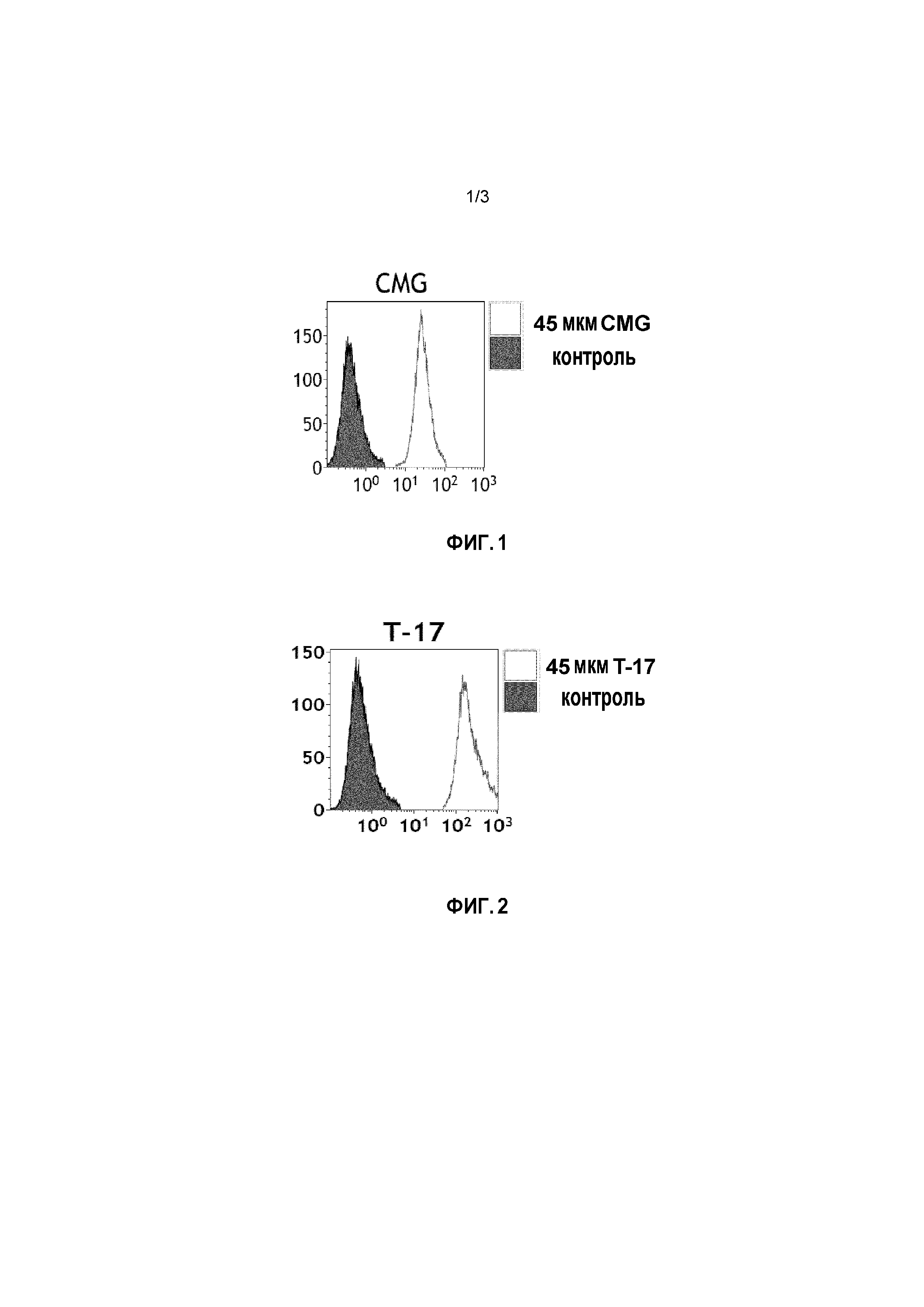
Фиг. 1: Данные, полученные из анализа включения анти-Gal для соединения по формуле (I), как получено в настоящем описании в примере 1 (Galili-CMG2-DOPE).
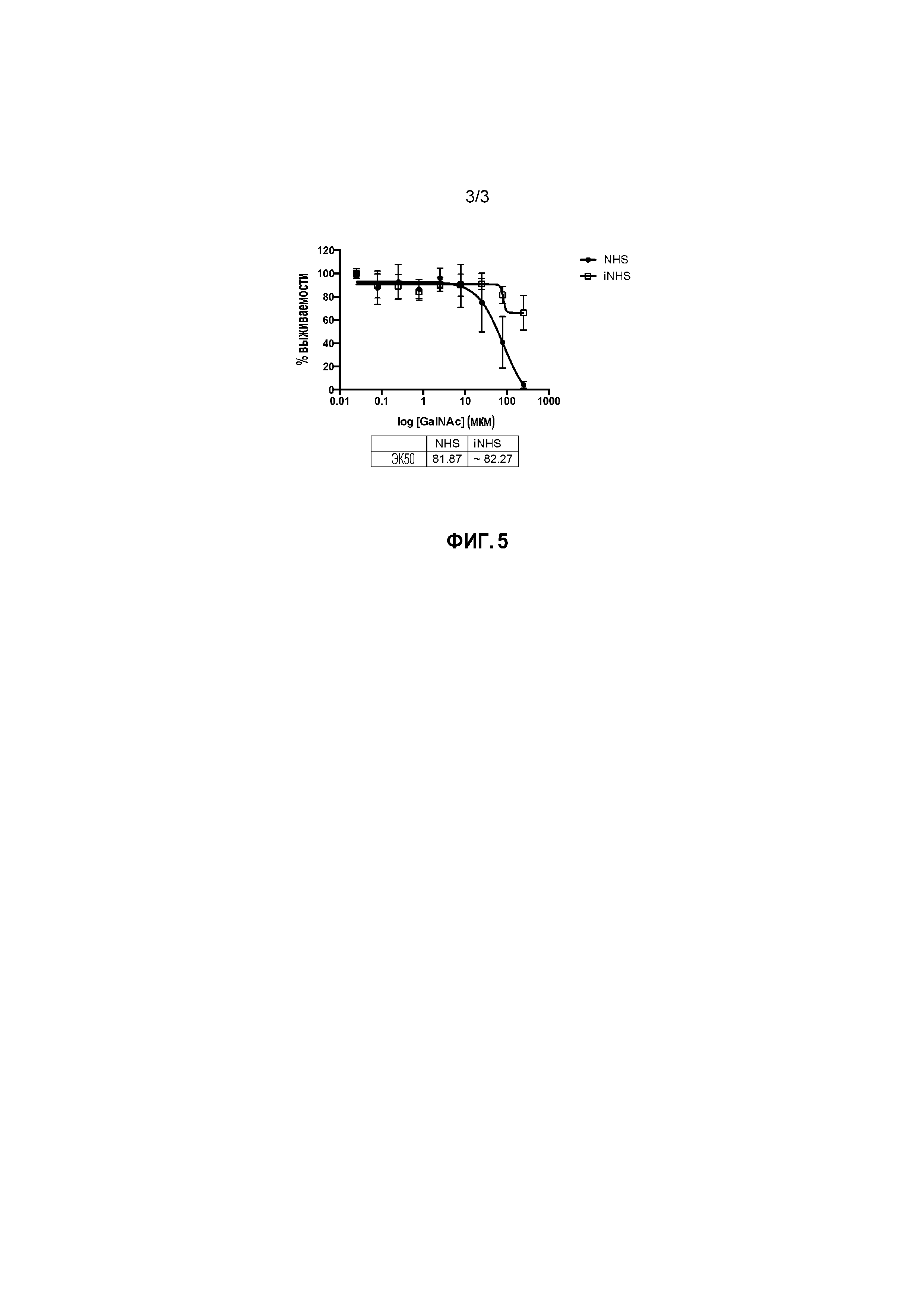
Фиг. 2: Данные, полученные из анализа включения анти-Gal для соединения по формуле (II), как получено в настоящем описании в примере 2 (Galili-T17 DOPE).
Фиг. 3: Данные, полученные из анализа комплементзависимой цитотоксичности для соединения по формуле (I), как получено в примере 1 (Galili-CMG2-DOPE).
Фиг. 4: Данные, полученные из анализа комплементзависимой цитотоксичности для соединения по формуле (II), как получено в настоящем описании в примере 2 (Galili-T17 DOPE).
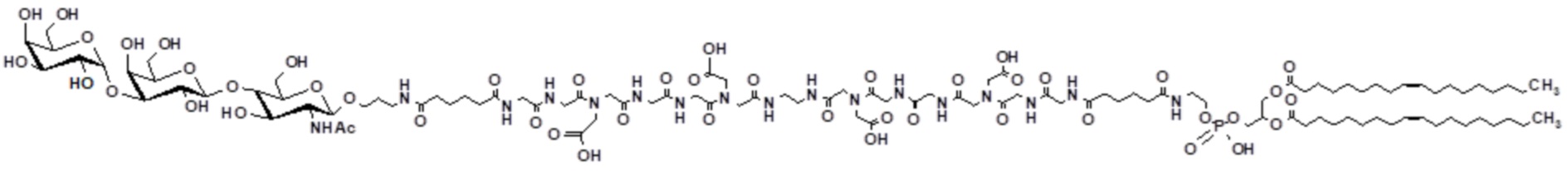
Фиг. 5: Данные, полученные из анализа комплементзависимой цитотоксичности для соединения по формуле (III), как получено в настоящем описании в примере 3 (GalNAc-Gal-GlcNAc-Ad-DOPE).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В соответствии с первым аспектом изобретения обеспечивают гликолипидное соединение, выбираемое из соединения по формуле (I), (II) и (III) или его фармацевтически приемлемой соли, как определено ранее.
Изобретение, описанное в настоящем описании, обеспечивает гликолипиды (т.е. соединения по формуле (I), (II) и (III)) которые способны встраиваться в клеточную мембрану опухолевых клеток в обработанной опухоли. Считают, что присутствие гликолипидов по изобретению приводит к деструкции или регрессу опухоли опосредованными иммунитетом воспалительными процессами, которые индуцированы взаимодействием между натуральными анти-Gal антителами, присутствующими у пациента, и α-Gal эпитопом соединений по формуле (I) и (II) (полученных, как описано в настоящем описании в примерах 1 и 2, соответственно). Более того, такое лечение превращает обработанную опухоль в вакцину, которая оказывает системный защитный противоопухолевый иммунный ответ, который предотвращает развитие отдаленных метастазов посредством иммунной деструкции метастатических опухолевых клеток.
В добавление к антителам к α-Gal, человеческая сыворотка также содержит антитела к другим углеводам. Линейный трисахарид типа 2 группы крови A (эпитоп GalNAc(1-3-Gal-β-4GlcNAc, GalNAc) представляет собой один такой гликан, который может распознаваться натуральными антителами в человеческой сыворотке (von Gunten, S. et al. (2009) J. Allergy Clin. Immunol. 123, 1268-76.e15; и Bovin (2013) Biochemistry (Moscow) 78(7), 786-797). Такие антитела также могут иметь способность индуцировать уничтожение опухолевых клеток, меченных гликолипидами, содержащими эпитоп GalNAc. Гликолипидное соединение по формуле (III) (полученное как описано в настоящем описании в примере 3) представляет собой гликолипид, содержащий эпитоп GalNAc, который синтезировали, чтобы оценить, могут ли антитела, присутствующие в человеческой сыворотке, селективно распознавать клетки, меченные таким гликолипидом, и стимулировать лизис меченных клеток, опосредованный комплементом.
Изобретение, описанное в настоящем описании, включает вариант терапевтического лечения, который включает, без ограничения, доставку в опухоль специфического гликолипида, относящегося к соединениям по формуле (I), (II) и III), который несет эпитоп α-Gal или GalNAc и, следовательно, может называться как ʺα-Gal гликолипидʺ или ʺGalNAc гликолипидʺ. α-Gal или GalNAc гликолипид вставляется в наружную оболочку клеточной мембраны опухолевых клеток, в поражении, которое лечат. Присутствие α-Gal ил GalNAc гликолипидов в опухолевом поражении достигает двух целей:
1. Опосредованная иммунитетом деструкция опухолевых поражений посредством воспалительного процесса, который индуцируется в опухолевом поражении в результате взаимодействия между натуральным анти-Gal или анти-GalNAc антителом и эпитопами α-Gal или GalNAc α-Gal или GalNAc гликолипидов, встроенных в мембраны опухолевых клеток; и
2. Эффективное поглощение антиген-представляющими клетками опухолевых клеток и мембран опухолевых клеток с встроенными α-Gal или GalNAc гликолипидами и, следовательно, экспрессирующими эпитопы α-Gal или GalNAc, которые связываются in situ с анти-Gal или анти-GalNAc антителами, таким образом преобразуя леченные опухолевые поражения в аутологичную опухолевую вакцину.
Хотя нет необходимости понимать механизм изобретения считают, что такое поглощение приводит к эффективному иммунному ответу против опухолевых антигенов, присутствующих на или внутри опухолевых клеток, экспрессирующих эпитопы α-Gal или GalNAc. Дополнительно считают, что такой иммунный ответ может приводить к иммунитет-опосредованной деструкции метастатических опухолевых клеток, которые не экспрессируют эпитопы α-Gal или GalNAc, но экспрессируют опухолевый антиген.
Изобретение предусматривает введение посредством инъекции или любыми другими средствами в опухоли соединений, которые индуцируют экспрессию эпитопа α-Gal или GalNAc на клетках в обработанной опухоли. Такое введение α-Gal или GalNAc гликолипидов решает следующие задачи:
1. Связывание натурального анти-Gal или анти-GalNAc антитела с эпитопами α-Gal или GalNAc α-Gal или GalNAc гликолипидов может приводить к локальной активации комплемента, посредством этого создавая хемотаксические факторы, включая, без ограничения, C5a и C3a. Такие хемотаксические факторы индуцируют обширную миграцию в опухолевые ткани антиген-представляющих клеток, таких как, без ограничения, дендритные клетки и макрофаги.
2. Липидные хвосты α-Gal или GalNAc гликолипидов будут спонтанно вставляться в мембраны опухолевых клеток в обработанном поражении, приводя к экспрессии эпитопов α-Gal или GalNAc на опухолевых клетках. Считают, что связывание эпитопов Анти-Gal или анти-GalNAc индуцирует регресс и/или деструкцию опухолей, включающих опухолевые клетки.
3. Опсонизация мембран опухолевых клеток анти-Gal или анти-GalNAc нацеливает их на эффективное поглощение антиген-представляющими клетками, которые мигрируют в опухоль. Миграция таких антиген-представляющих клеток направляется посредством пептидов отщепления хемотаксического комплемента, которые образуются после связывания анти-Gal или анти-GalNAc с α-Gal или GalNAc гликолипидами в обработанной опухоли.
Без связи с каким-либо определенным механизмом, считают, что Fc часть молекул анти-Gal или анти-GalNAc IgG, связанных с мембраной опухолевых клеток, связывается с рецепторами Fc-гамма (Fc(γ) на антиген-представляющих клетках и индуцирует потребление опухолевых клеток антиген-представляющими клетками. Сходная индукция потребления может возникать в результате взаимодействия между отложениями C3b компонента комплемента на анти-Gal или анти-GalNAc связывающих опухолевых клетках и рецепторах C3b на антиген-представляющих клетках. Такое анти-Gal или анти-GalNAc опосредованное нацеливание на опухолевые мембраны антиген-представляющих клеток позволяет эффективный транспорт аутологичных опухолевых антигенов в дренирующие лимфатические узлы, и обработку и презентацию иммуногенных опухолевых антигенных пептидов антиген-представляющими клетками в лимфатических узлах.
Следовательно, внутриопухолевая инъекция α-Gal или GalNAc гликолипидов преобразует обработанные опухолевые поражения в in situ аутологичную опухолевую вакцину, которая доставляет опухолевые антигены в иммунную систему, посредством этого обеспечивая противоопухолевый иммунный ответ. Такой иммунный ответ способ индуцировать регресс опухоли, включающий деструкцию отдельных опухолевых клеток или мелких агрегатов опухолевых клеток (т.е., например, микрометастаз). Такие микрометастазы обычно не определяются или визуально или путем визуализации и недоступны для обычного хирургического или лучевого лечения (т.е. они нерезектабельны из-за мелкого размера). Следовательно, настоящий метод имеет дополнительное преимущество, что он способен лечить микрометастазы, которые обычно не определяются или визуально или путем визуализации и недоступны для обычных хирургических и лучевых методик.
Определения
Ссылки в настоящем описании на термин ʺсоединение по формуле (I)ʺ относятся к специфическому примеру α-Gal гликолипида, который состоит из функционального (F), спейсерного (S) и липидного (L) компонента и может быть использован для вставки в клеточные мембраны, так что клетки несут на поверхности функциональный (F) компонент. Функциональный (F) компонент соединения по формуле (I) представляет собой трисахаридную группу: Gal-α1-3-Gal-β1-4GlcNAc (т.е. α-Gal эпитоп). Спейсерный (S) компонент состоит из двух CMG групп и липидным (L) компонентом является DOPE. Ссылки на соединение по формуле (I) в настоящем описании также включают ʺGalili-CMG2-DOPEʺ и ʺCMGʺ, которые могут быть использованы взаимозаменяемо. Структура соединения по формуле (I) представляет собой, как показано ранее. Соединение по формуле (I) может быть получено в соответствии с подробной методикой синтеза, описанной в настоящем описании для примера 1.
Ссылки в настоящем описании на термин ʺсоединение по формуле (II)ʺ относятся к специфическому примеру α-Gal гликолипида, который состоит из функционального (F), спейсерного (S) и липидного компонента (L) и может быть использован для вставки в клеточные мембраны так, чтобы клетки демонстрировали функциональный (F) компонент на поверхности. Функциональный (F) компонент соединения по формуле (II) представляет трисахаридную группу из: Gal-α1-3-Gal-β1-4GlcNAc (т.е. α-Gal эпитоп). Спейсерный (S) компонент состоит из T17 группы и липидным (L) компонентом является DOPE. Ссылки на соединение по формуле (II) в настоящем описании также включают ʺGalili-T17 DOPEʺ и ʺT17ʺ, которые могут быть использованы взаимозаменяемо. Структура соединения по формуле (II) показана ранее. Соединение по формуле (II) может быть получено в соответствии с подробной процедурой синтеза, описанной в настоящем описании для примера 2. Считают, что тримерное соединение по формуле (II) содержит примеси димерного соединения по формуле (II)a:
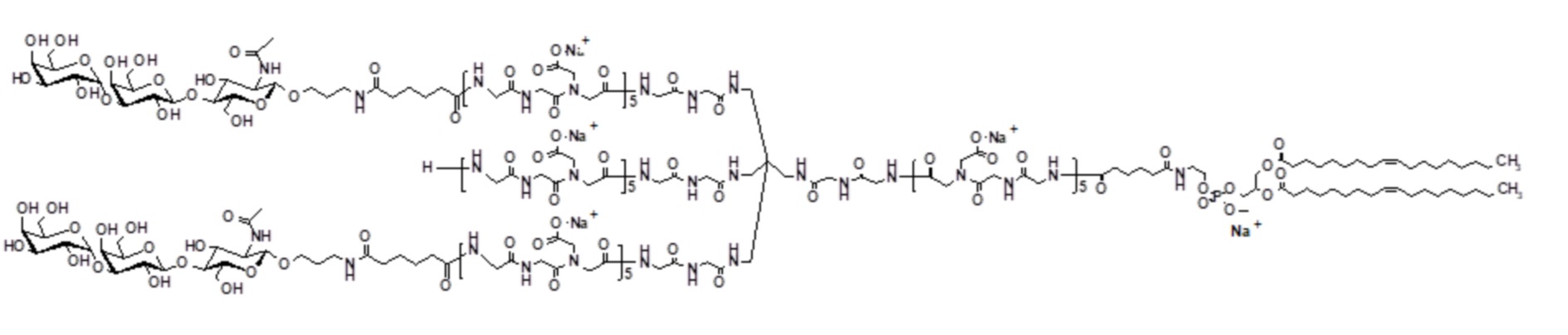
Следовательно, ссылки в настоящем описании на термины ʺсоединение по формуле (II)ʺ, ʺGalili-T17 DOPEʺ и ʺT17ʺ относятся к смеси соединений по формуле (II) и (II)a.
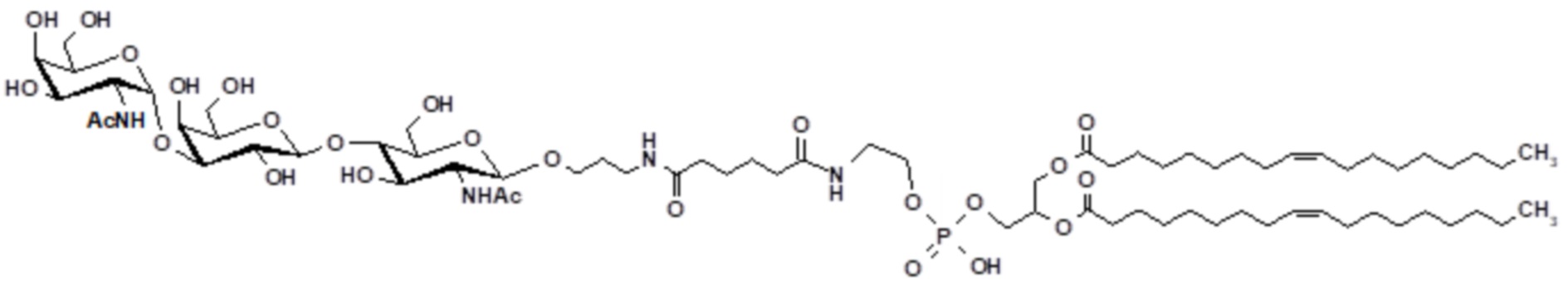
Ссылки в настоящем описании на термин ʺсоединение по формуле (III)ʺ относятся к специфическому примеру GalNAc гликолипида, который состоит из функционального (F), спейсерного (S) и липидного (L) компонента и могут быть использованы для вставки в клеточные мембраны так, что клетки демонстрируют функциональный (F) компонент на его поверхности. Функциональный (F) компонент соединения по формуле (I) представляет собой трисахаридную группу: GalNAc-α1-Gal-β1-4GlcNAc (т.е. GalNAc эпитоп). Спейсерный (S) компонент включает O(CH2)3NH группу и липидный (L) компонент представляет собой DOPE. Ссылки на соединение по формуле (III) в настоящем описании также включают ʺGalNAc-Gal-GlcNAc-Ad-DOPEʺ и ʺGalNAcʺ которые могут быть использованы взаимозаменяемо. Структура соединения по формуле (III) показана ранее в настоящем описании. Соединение по формуле (III) может быть получено в соответствии с подробной методикой синтеза, описанной в настоящем описании для примера 3.
В одном варианте осуществления изобретения гликолипидное соединение выбирают из соединения по формуле (I). В альтернативном варианте осуществления изобретения гликолипидное соединение выбирают из соединения по формуле (II). В альтернативном варианте осуществления изобретения гликолипидное соединение выбирают из соединения по формуле (I) и (II). В альтернативном варианте осуществления изобретения, гликолипидное соединение выбирают из соединения по формуле (III).
Ссылки в настоящем описании на термин ʺDOPEʺ относятся к фосфатидилэтаноламину (PE), имеющему химическое наименование 1,2-диолеил-sn-глицеро-3-фосфоэтаноламин.
Соединения по формуле (I), (II) и (III) могут существовать в форме солей, например, аддитивных солей кислот или в определенных случаях солей органических оснований, таких как карбоксилат, сульфонат и фосфатные соли. Все такие соли находятся в рамках настоящего изобретения и ссылки на соединения по формуле (I), (II) и (III) включают солевые формы соединений.
Соли по настоящему изобретению могут быть синтезированы из исходного соединения, которое содержит основной компонент, обычными химическими методами, такими как методы, описанные в Pharmaceutical Salts: Properties, Selection, and Use, P. Heinrich Stahl (Editor), Camille G. Wermuth (Editor), ISBN: 3-90639-026-8, Hardcover, 388 pages, August 2002. Обычно, такие соли могут быть получены путем реакции основных форм таких соединений с соответствующим основанием или кислотой в воде или в органическом растворителе, или в смеси двух; обычно используют неводную среду, такую как простой эфир, этилацетат, этанол, изопропанол или ацетонитрил.
Аддитивные соли кислот (моно- или дисоли) могут быть получены с большим множеством кислот, и неорганических и органических. Примеры аддитивных солей кислот включают моно- или дисоли, образованные с кислотой, выбираемой из группы, состоящей из уксусной, 2,2-дихлоруксусной, адипиновой, альгиновой, аскорбиновой (например, L-аскорбиновой), L-аспарагиновой, бензолсульфоновой, бензойной, 4-ацетаминобензойной, бутаноевой, (+) камфорной, камфор-сульфоновой, (+)-(1S)-камфор-10-сульфоновой, капровой, капроевой, каприловой, коричной, лимонной, цикламовой, додецилсерной, этан-1,2-дисульфоновой, этансульфоновой, 2-гидроксиэтансульфоновой, муравьиной, фумаровой, галактаровой, гентизовой, глюкогептоновой, D-глюконовой, глюкуроновой (например, D-глюкуроновой), глютамовой (например, L-глютамовой), (-оксоглютаровой, гликолевой, гиппуровой, галогенводородных (например, бромистоводородная, соляная, йодистоводородная), изетионовой, молочной (например, (+)-L-молочной, (±)-DL-молочной), лактобионовой, малеиновой, яблочной, (-)-L-яблочной, малоновой, (±)-DL-миндальной, метансульфоновой, нафталан-2-сульфоновой, нафталан-1,5-дисульфоновой, 1-гидрокси-2-нафтоевой, никотиновой, азотной, олеиновой, оротовой, щавелевой, пальмитиновой, памоевой, фосфорной, пропионовой, пировиноградной, L-пироглютамовой, салициловой, 4-аминосалициловой, себациновой, стеариновой, янтарной, серной, дубильной, (+)-L-виннокаменной, тиоциановой, п-толуолсульфоновой, ундециленовой и валериановой кислоты, а также ацилированных аминокислот и катионообменных смол.
Одна определенная группа солей состоит из солей, образованных из уксусной, соляной, йодистоводородной, фосфорной, азотной, серной, лимонной, молочной, янтарной, малеиновой, яблочной, изетионовой, фумаровой, бензолсульфоновой, толуолсульфоновой, метансульфоновой, (мезилат), этансульфоновой, нафталансульфоновой, валериановой, уксусной, пропаноевой, бутаноевой, малоновой, глюкуроновой и лактобионовой кислот. Одной определенной солью является соль гидрохлорид. Другой определенной солью является соль гидросульфат, также известная как гемисульфатная соль. В дополнительном варианте осуществления изобретения соль выбирают из натрия и калия или включают амин противоион.
Когда соединения по формуле (I), (II) и (III) содержат аминовую функциональную группу, они могут образовывать соли четвертичного аммония, например, путем реакции с алкилирующим агентом в соответствии со способами, хорошо известными специалисту. Такие четвертичные соединения аммония находятся в рамках формулы (I).
Соединения по изобретению могут содержать одиночный или множественные противоионы, в зависимости от pKa кислоты, из которой образуется соль. Например, пример 1 содержит 4 кислых группы и пример 2 содержит 20 кислых групп, следовательно, каждое из указанных соединений хорошо приспособлено для включения множества противоионов.
Солевыми формами соединений по изобретению обычно являются фармацевтически приемлемые соли, и примеры фармацевтически приемлемых солей обсуждаются в Berge et al., 1977, "Pharmaceutically Acceptable Salts," J. Pharm. Sci., Vol. 66, pp. 1-19. Однако соли, которые не являются фармацевтически приемлемыми, также могут быть получены в промежуточных формах, которые могут затем быть преобразованы в фармацевтически приемлемые соли. Такие нефармацевтически приемлемые солевые формы, которые могут быть применимыми, например, в очистке или отделении соединений по изобретению, также являются частью изобретения.
Термин "эпитоп α-Gal", как используется в настоящем описании, относится к любой молекуле или части молекулы с концевой структурой, включающей Galα1-3Galβ1-4GlcNAc-R, Galα1-3Galβ1-3GlcNAc-R, или любую углеводородную цепь с концевым Galα1-3Gal на невосстановленном конце. α-галактозил (также называемый как ʺальфа-Galʺ или ʺα-Galʺ) эпитоп, т.е., галактозил-альфа-1,3-галактозил-бета-1,4-N-ацетилглюкозамин описан в Galili, U. and Avila, J.L., Alpha-Gal and Anti-Gal, Subcellular Biochemistry, Vol. 32, 1999. Исследования кссенотрансплантации определили, что люди дают иммунный ответ на α-галактозил эпитоп, который сам по себе обычно не обнаруживают у людей, но обнаруживают у других животных и множества микроорганизмов.
Термин ʺGalNAc эпитопʺ, как используется в настоящем описании, относится к любой молекуле или части молекулы, с концевой структурой, включающей GalNAcα1-Gal-β1-4GlcNAc или любую углеводородную цепь с концевым GalNAcα1-3-Gal на невосстановленном конце.
Термин "гликолипиды", как используется в настоящем описании, относится к любой молекуле с по меньшей мере одной углеводородной цепью, связанной с церамидом, цепочкой жирной кислоты, или любым другим липидом. Альтернативно, гликолипид может называться как гликосфинголипид.
Термин ʺанти-Galʺ, как используется в настоящем описании, относится к натуральным антителам, которые связываются с эпитопом α-Gal.
Термин ʺанти-GalNAcʺ, как используется в настоящем описании, относится к натуральным антителам, которые связываются с эпитопом GalNAc.
Термин "α-1,3-галактозилтрансфераза", как используется в настоящем описании, относится к любому ферменту, способному синтезировать эпитопы α-Gal.
Термин "анти-Gal связывающий эпитоп", как используется в настоящем описании, относится к любой молекуле или части молекулы, которая способна связывать, in vivo или in vitro, натуральное анти-Gal антитело.
Термин "анти-GalNAc связывающий эпитоп", как используется в настоящем описании, относится к любой молекуле или части молекулы, которая способна связывать, in vivo или in vitro, натуральное анти-GalNAc антитело.
Термин "неоперабельный", как используется в настоящем описании, относится к любой части органа или структуры тела, которая не может быть хирургически удалена. Например, "неоперабельной опухолью" может быть опухоль, физически недосягаемая обычными хирургическими методиками, опухоль, когда ее удаление не улучшает общее раковое заболевание или существование пациента, или опухоль, когда ее удаление может быть вредным для жизненноважных органов.
Термин "мембран-связанный", как используется в настоящем описании, относится к любой молекуле, которая стабильно прикреплена к, или связана с фосфолипидным бислоем. Такое прикрепление или связь могут включать силы, включая, без ограничения, ионные связи, ковалентные связи, гидрофобные связи или силы Ван дер Ваальса и др. Например, белок, включающий участок гидрофобной аминокислоты, может вставляться в фосфолипидный бислой мембраны, или молекула, которая содержит липидный хвост, может встраиваться сама по себе в фосфолипидный бислой клеток и становиться захваченной. Липидный компонент α-Gal или GalNAc содержащих гликолипидов по изобретению используют для вставки в клеточные мембраны опухоли для создания опухоли, несущей эпитоп α-Gal или GalNAc на клеточной поверхности.
Термин "подгруппа", как используется в настоящем описании, относится к специализированной группе, меньшей по количеству, чем вся группа. Например, пациент может иметь множество неоперабельных солидных опухолей. Из этого множества подгруппа может быть достижима нехирургическими технологиями, тогда как другая подгруппа может быть недостижима нехирургическими технологиями.
Термин "достижимый", как используется в настоящем описании, относится к способности лечить солидную опухоль нехирургическими методиками. Такие методики включают, без ограничения, инъекцию в кожу или инъекцию посредством эндоскопии, бронхоскопии, цистоскопии, колоноскопии, лапароскопии, катетеризации или местного нанесения посредством лосьона, мази или порошка. Например, солидная опухоль яичника может быть достижима лаапароскопией. В другом примере солидная опухоль толстой кишки может быть достижима колоноскопией.
Термин "введение", как используется в настоящем описании, относится к любому способу переноса соединения в ткани и впоследствии в клетки в указанной ткани. Такие методы внесения могут включать, без ограничения, вирусные векторы, ретровирусные векторы, аденовирусные векторы, биобалистики, липофекцию и множество коммерчески доступных ДНК векторов, известных в области техники. Альтернативно, соединение может быть размещено по соседству с клеткой, так что соединение включается в клетку посредством физиологических механизмов (т.е., например, гидрофобных взаимодействий или активного транспорта). Один метод внесения включает инъекцию, где соединение помещают непосредственно в межклеточное пространство в инъецируемой ткани. Такая инъекция может быть возможной, когда часть органа, образование (т.е. например, солидная опухоль), или полость тела являются "достижимыми".
Термин "в", как используется в настоящем описании, относится к успешному проникновению молекулы через или внутрь клеточной мембраны. Например, вирусный вектор может быть введен в клетки солидной опухоли в условиях, так что опухолевая клетка трансфицируется. В другом примере гликолипид может быть внесен в опухолевые клетки в условиях, так что гликолипид становится встроенным в клеточную мембрану из липидного бислоя.
Термин "регресс", "представляет собой по меньшей мере частично уменьшенный в размерах" или "уменьшенный", как используется в настоящем описании, относится к уменьшению роста образования, такого как, например, солидная опухоль. Такое уменьшение может быть определено по уменьшению измеренных параметров, таких как, без ограничения, диаметр, масса (т.е. вес), или объем. Уменьшение никаким образом не определяет, что размер полностью уменьшен, только что измеренный параметр является количественно меньшим, чем предыдущее измерение.
Термин "деструкция", как используется в настоящем описании, относится к полному клеточному распаду физической опухоли, такой как, например, солидная опухоль. Такая деструкция может включать внутриклеточный апоптоз, уничтожение клеток, опосредованное Т клетками, цитолиз, опосредованный комплементом, и/или макрофагальный фагоцитоз, такой что физическая опухоль полностью поглощается и удаляется из организма. Термин ʺдеструкция опухолиʺ относится к уменьшению опухоли до такой степени, что она более не определяется диагностическими средствами.
Термин ʺлечениеʺ, ʺтерапияʺ и ʺлечитьʺ, все используемые в настоящем описании, относятся к процедуре, которая приводит к по меньшей мере частичному уменьшению размера или уменьшению в размере физической опухоли, такой как, например, солидная опухоль.
Термин "меньше, чем все", как используется в настоящем описании, относится к подгруппе группы. В контексте одного варианта осуществления настоящего изобретения предусматривается лечение менее чем всех опухолей у пациента. Иными словами в одном варианте осуществления изобретения нет необходимости лечить каждую опухоль посредством внесения эпитопа α-Gal или GalNAc (например, посредством внесения α-Gal или GalNAc содержащих гликолипидов по изобретению); скорее внесение подгруппе приводит к иммунному ответу во всех опухолях (включая те, которые непосредственно не лечат). Таким образом можно достичь общего уменьшения множества физических опухолей, таких как, например, метастазы солидной опухоли. Такое уменьшение может быть определено по уменьшению измеренных параметров, таких как, без ограничения, количество. Уменьшение совсем не указывает, что параметр уменьшается до нуля, только что измеренный параметр становится количественно меньшим, чем предшествующее измерение.
Термин "рост", как используется в настоящем описании, относится к любой ткани или органу, который включает клеточную массу, расцененную как представляющая патологическую пролиферацию. Такой рост может быть раковым, нераковым, злокачественным или незлокачественным. Если рост включает рак, он может быть опухолевым.
Термин ʺопухольʺ, как используется в настоящем описании, относится к патологической массе ткани, которая возникает в результате патологического роста или деления клеток. Такие опухоли могут быть солидными (т.е. масса клеток в определенном органе, ткани или железе, такой как брюшина, печень, поджелудочная железа, легкое, мочевой пузырь, предстательная железа, матка, шейка матки, влагалище, молочная железа, кожа, головной мозг, лимфатические узлы, голова и шея, желудок, тонкая кишка, толстая кишка или яичники) или несолидными (т.е. жидкостные опухоли, которые развиваются в крови, такие как лейкоз).
Термин "субъект", как используется в настоящем описании, относится к любому организму, в котором может развиваться опухоль. Такие организмы включают, без ограничения, млекопитающих, людей, не приматов млекопитающих, полуобезьян и обезьян Нового Мира и др.
Термин "молекула", как используется в настоящем описании, относится к наименьшей частице композиции, которая сохраняет все свойства композиции и состоит из одного или более атомов. Такие один или более атомов расположены так, что молекула может взаимодействовать (т.е. ионно, ковалентно, нековалентно и др.) с другими молекулами с образованием прикреплений и/или ассоциаций. Например, молекула может иметь один или более атомов, расположенных для обеспечения способности взаимодействия с анти-Gal или анти-GalNAc антителом.
Методики синтеза
Как обсуждается в настоящем описании ранее, подробная процедура синтеза для соединений по формуле (I), (II) и (III) описана в настоящем описании в примерах 1, 2 и 3, соответственно.
Следовательно, в соответствии с дополнительным аспектом изобретения обеспечивают способ для получения соединения по формуле (I), как определено в настоящем описании, который включает реакцию соединения по формуле (21), как описано в примере 1, схема VI с соединением по формуле (20), как описано в примере 1, схема VI. Такой процесс обычно включает применение подходящего основания, такого как триметиламин, и воздействие подходящих условий реакции, таких как перемешивание в течение 24 ч при комнатной температуре.
В соответствии с дополнительным аспектом изобретения обеспечивают способ получения соединения по (II), как определено в настоящем описании, который включает реакцию соединения по формуле (28), как описано в примере 2, схема VII с соединением по формуле (29), как описано в примере 2, схема VII. Такой способ обычно включает применение подходящего основания, такого как триметиламин и подвергается подходящим условиям реакции, таким как перемешивание в течение 24 ч при комнатной температуре.
В соответствии с дополнительным аспектом изобретения обеспечивают способ получения соединения по формуле (III), как определено в настоящем описании, который включает реакцию соединения по формуле (5), как описано в примере 3, схема III с соединением по формуле (8), как описано в примере 3, схема III. Такой способ обычно включает применение подходящего основания, такого как триметиламин, и воздействие подходящих условий реакции, таких как перемешивание в течение 2 ч при комнатной температуре.
Натуральное анти-Gal антитело, α-Gal эпитоп, и отторжение трансплантата
Считают, что анти-Gal является натуральным антителом, которое может присутствовать у всех людей, составляя 0,1-2% иммуноглобулинов сыворотки (Bovin N.V., Biochemistry (Moscow), 2013; 78(7):786-797, Galili et al. J. Exp. Med. 1984; 160: 1519-31, и Hamadeh R M et al. Clin. Diagnos. Lab. Immunol. 1995; 2:125-31). Исследования представляют данные, показывающие, что анти-Gal антитела могут взаимодействовать с α-Gal эпитопами на поверхности клеток или свободными гликолипидами и гликопротеинами. (Galili U et al. J. Exp. Med. 1985, 162: 573-82, и Galili U. Springer Semin Immunopathol. 1993; 15: 155-171). Дополнительно сообщают, что анти-Gal антитело может образовываться на протяжении жизни в результате антигенной стимуляции бактериями желудочно-кишечной флоры (Galili U et al. Infect. Immun. 1988; 56: 1730-37).
α-Gal эпитоп может быть обильно биосинтезирован на гликолипидах и гликопротеинах посредством фермента гликозилирования α1,3 галактозилтрансферазой в аппарате Гольджи клеток не-приматных млекопитающих, прообезьян и обезьян Нового Мира (Galili U et al. Biol. Chem. 1988; 263; 17755-62). Наоборот, люди, высшие обезьяны и обезьяны Старого Мира не имеют α-Gal эпитопов, но продуцируют натуральные анти-Gal антитела в очень больших количествах (Galili U et al. Proc. Natl. Acad. Sci. USA 1987, 84: 1369-73). На основании последовательности псевдогена α1,3галактозилтрансферазы у обезьян и высших приматов считают, что ген α1,3галактозилтрансферазы инактивировался у предков приматов Старого Мира приблизительно 20 миллионов лет назад (Galili U, Swanson K. Proc. Natl. Acad. Sci. USA 1991; 88: 7401-04). Предполагают, что такое эволюционное событие было ассоциировано с появлением инфекционного микробного агента, эндемичного для Старого Мира (т.е. существующей Европы, Азии и Африки), который был вредным для приматов и который экспрессировал α-Gal эпитопы. Приматы могут продуцировать анти-Gal в качестве защитного антитела против таких потенциально вредных агентов, только после того, как они попадают под селективное давление для инактивации гена α1,3галактозилтрансферазы и следовательно, теряют иммунную толерантность к α-Gal эпитопу (Galili U, Andrews P. J. Human Evolution 29:433-42, 1995).
Сильная защитная активность натурального анти-Gal антитела эволюционно рассматривалась у людей и обезьян. Это может быть получено из исследований ксенотрансплантаций органов свиней, экспрессирующих α-Gal эпитопы. Так как клетки множества млекопитающих, включая свиней, экспрессируют α-Gal эпитопы, органы от свиней, трансплантированные людям или обезьянам Старого Мира, отторгаются из-за in vivo связывания анти-Gal антитела такими эпитопами. Трансплантация свиных тканей людям или обезьянам Старого Мира приводит к авидному анти-Gal связыванию с α-Gal эпитопами на in vivo трансплантате и последующей индукции отторжения трансплантата. Васкуляризированные трансплантаты (например, свиное сердце) подвергаются быстрому отторжению (называемому сверхбыстрое отторжение) у обезьян в течение 30-60 минут главным образом в результате связывания молекул анти-Gal антител с α-Gal эпитопами на эндотелиальных клетках свиней, активации комплемента, лизису эндотелиальных клеток и коллапсу сосудистого русла (Collins B H et al. J. Immunol. 1995; 154: 5500-10). Кроме того, большая часть деструкции клеток ксенотрансплантата в экстравазальной области опосредована связыванием анти-Gal IgG с α-Gal эпитопами на различных клетках. Такое связывание приводит к зависимому от антител опосредованному клетками цитолизу (ADCC) с последующим связыванием Fc части анти-Gal IgG со связанными с клеткой Fcγ рецепторами на гранулоцитах, макрофагах и NK клетках.
Анти-Gal опосредованная деструкция ксенотрансплантатов может отслеживаться со свиным хрящом (аваскулярная ткань кссенотрансплантата), трансплантированным макакам резус (т.е. макакам, которые натурально продуцируют анти-Gal антитела). Исследования показали, что связывание анти-Gal с α-Gal эпитопами в свиной ткани приводит к индукции обширной воспалительной реакции, которая приводит к постепенной деструкции ткани в течение 2 месяцев (Stone K R et al. Transplantation 1998, 65: 1577-83). Связывание анти-Gal с α-Gal эпитопами на поверхности хряща и гликопротеинами внеклеточного матрикса дополнительно их опсонизирует (т.е. образует с ними иммунные комплексы) и следовательно, нацеливает на них антиген-представляющие клетки посредством связывания Fc части иммуно-комплексированного анти-Gal с рецепторами Fcγ на антиген-представляющих клетках. Антиген представляющие клетки, в свою очередь, транспортируют такие свиные гликопротеины в дренирующие лимфатические узлы, где они активируют множество Т клеток, специфических для множества свиных ксенопептидов. Такие активированные T клетки впоследствии мигрируют в трансплантированный имплант хряща и составляют приблизительно 80% инфильтрирующих мононуклеарных клеток. Такой воспалительный ответ, главным образом опосредованный анти-Gal взаимодействием с α-Gal эпитопами, может находиться под воздействием мониторинга иммунного ответа на трансплантат свиного хряща, из которого α-Gal эпитопы были удалены ферментативной обработкой (например, с использованием рекомбинантной α-галактозидазы). α-галактозидаза разрушает α-Gal эпитопы на гликопротеинах хряща посредством отщепления (гидролиза) концевой α-галактозильной единицы. В отсутствие α-Gal эпитопов на гликопротеинах свиного хряща, не существует анти-Gal связывания с трансплантатом и, следовательно, не возникает эффективного транспорта, опосредованного антиген представляющими клетками. Это демонстрируется отсутствием значительной T клеточной инфильтрации в трансплантате.
Настоящее изобретение предусматривает использование иммунологического потенциала натуральных анти-Gal антител, продемонстрированного при отторжении трансплантата свиного хряща, для регресса и/или деструкции опухолевых повреждений, обработанных для отображения α-Gal эпитопов и для нацеливания на мембраны опухолевых клеток антиген-представляющих клеток посредством анти-Gal антител. Считают, что такая обработка будет преобразовывать опухолевые повреждения в in situ аутологичные опухолевые вакцины, которые вызывают системный защитный иммунный ответ против метастатических опухолевых клеток, посредством механизмов, сходных с таковыми, наблюдаемыми при отторжении свиного хряща у обезьян. Дополнительно считают, что связывание молекул анти-Gal IgG с опухолевыми клетками, экспрессирующими α-Gal эпитопы, будет нацеливать на мембраны опухолевых клеток антиген-представляющие клетки для обеспечения защитного противоопухолевого иммунного ответа против аутологичных опухолевых антигенов, экпрессируемых на опухолевых клетках в обрабатываемом повреждении и также эспрессируется на метастатических опухолевых клетках.
Фармацевтические композиции
В соответствии с дополнительным аспектом изобретения обеспечивают фармацевтическую композицию, включающую гликолипидное соединение, выбираемое из соединения по формуле (I), (II) и (III) или его фармацевтически приемлемой соли, как описано в настоящем описании.
В соответствии с дополнительным аспектом изобретения обеспечивают гликолипидное соединение, выбираемое из соединения по формуле (I), (II) и (III) или его фармацевтически приемлемой соли, как определено в настоящем описании, или фармацевтической композиции, как определено в настоящем описании для применения в лечении опухоли.
В одном варианте осуществления изобретения опухолью является солидная опухоль, миелома или лимфома. В дополнительном варианте осуществления изобретения опухолью является солидная опухоль. В альтернативном варианте осуществления изобретения опухолью является несолидная опухоль.
В одном варианте осуществления изобретения опухолью является опухоль, происходящая из органа, выбираемого из брюшины, печени, легкого, мочевого пузыря, предстательной железы, матки, шейки матки, влагалища, костного мозга, молочной железы, кожи, головного мозга, лимфатических узлов, головы и шеи, желудка, тонкой кишки, толстой кишки, почки, яичек и яичников.
В одном варианте осуществления изобретения опухоль включает первичную опухоль и/или метастазы. В дополнительном варианте осуществления изобретения опухоль включает первичную опухоль. В альтернативном варианте осуществления изобретения опухоль включает вторичную опухоль.
В одном варианте осуществления изобретения опухоль включает клетки меланомы, саркомы, глиомы или карциномы. В дополнительном варианте осуществления изобретения опухоль включает клетки меланомы или карциномы или метастазы.
Композиция может быть получена в виде водного гликолипидного препарата, включающего гликолипидное соединение по формуле (I), (II) или (III), где указанный препарат включает мицеллы гликолипида.
В одном варианте осуществления изобретения композиция дополнительно включает один или более фармацевтически приемлемых носителя(ей), разбавителя(ей) и/или вспомогательного веществ(а). Носитель, разбавитель и/или вспомогательное вещество должны быть ʺфармацевтически приемлемымиʺ в смысле совместимости с другими ингредиентами композиции и не вредными для реципиента. Специалист в области техники понимает аспекты фармацевтической композиции, которые проиллюстрированы, например, в Remington: The Science and Practice of Pharmacy; Pharmaceutical Press; 22nd Edition; Allen, Loyd V. Ed. 2012, London, UK.
Композиция по изобретению может быть получена путем комбинации гликолипидного соединения по формуле (I), (II) или (III) со стандартными фармацевтическими носителями или разбавителями в соответствии с обычными методиками, хорошо известными в области техники. Такие методики могут включать смешивание, гранулирование и прессование или растворение ингредиентов, как указано для желаемого препарата.
В одном варианте осуществления изобретения фармацевтическая композиция также может содержать деоксихолат, или другие мягкие детергенты, которые могут увеличивать проникновение гликолипидов в клеточные мембраны.
Фармацевтические композиции по изобретению могут быть рецептированы для введения посредством любого пути и включают таковые в форме, адаптированной для перорального, местного или парентерального введения млекопитающим, включая людей.
Следовательно, в одном варианте осуществления изобретения композиция предназначена для введения посредством инъекции. В альтернативном варианте осуществления изобретения композицией является местная композиция, такая как местная мазь, местный лосьон или местный раствор.
В одном варианте осуществления изобретения композицию вводят в одной дозе или множестве доз, например в виде множества доз. В дополнительном варианте осуществления изобретения множество доз вводят одновременно (т.е. в одном случае). В дополнительном альтернативном варианте осуществления изобретения множество доз вводят последовательно (т.е. в два или более отдельных случаев, например, во время отдельного лечения).
Когда введение является последовательным (т.е. в отдельные случаи), композицию можно вводить, когда соответствующее количество времени проходит между введениями, например, 3 дня, 5 дней, неделя, две недели, месяц, 2 месяца, 3 месяца, 6 месяцев или 12 месяцев.
Для парентерального введения жидкие стандартные лекарственные формы получают с использованием композиции и стерильного носителя, такого как вода. В получении растворов композиция может быть растворена в воде для инъекций и стерилизована фильтрацией до заполнения в подходящий флакон или ампулу и герметизирована.
Композиции могут находиться в форме таблеток, капсул, порошков, гранул, пастилок, кремов или жидких препаратов, таких как пероральные или стерильные парентеральные растворы или суспензии.
Местные композиции по настоящему изобретению могут быть представлены в виде, например, мазей, кремов или лосьонов, глазных мазей или глазных и ушных капель, пропитанных повязок и аэрозолей, и могут содержать соответствующие удобные добавки, такие как консерванты и увлажнители в мазях и кремах.
Композиции также могут содержать совместимые удобные носители, такие как кремовые или мазевые основания, и этанол или олеиловый спирт для лосьонов.
Комбинации
Понимают, что соединение по изобретению можно вводить в виде единственного терапевтического средства или можно вводить в комбинированной терапии с одним или более соединениями (или терапией) для лечения опухоли.
Следовательно, в соответствии с дополнительным аспектом изобретения обеспечивают фармацевтическую композицию, включающую гликолипидное соединение, выбираемое из соединения по формуле (I), (II) и (III) или его фармацевтически приемлемой соли, как определено в настоящем описании в комбинации с одним или более дополнительными терапевтическими средствами.
Для лечения опухоли соединение по изобретению может быть преимущественно использовано в комбинации с одним или более другими медицинскими агентами, более предпочтительно с одним или более противораковыми агентами или добавками (поддерживающие средства в лечении) в противораковой терапии.
Примеры других терапевтических средств или терапии, которые могут вводиться вместе (или одновременно или с различными временными интервалами) с соединениями по изобретению, включают, без ограничения:
* ингибиторы топоизомеразы I;
* антиметаболиты;
* средства, нацеленные на тубулин;
* средства, связывающие ДНК, и ингибиторы топоизомеразы II;
* алкилирующие агенты;
* моноклональные антитела;
* антигормоны;
* ингибиторы трансдукции сигнала;
* ингибиторы протеасом;
* ДНК метилтрансферазы;
* цитокины и ретиноиды;
* терапия, нацеленная на хроматин;
* лучевая терапия; и
* другие терапевтические или профилактические средства.
Определенные примеры противораковых средств или добавок (или их соли), включают, без ограничения, любые из средств, выбираемые из групп (i)-(xlvi), и необязательно группы (xlvii), ниже:
(i) соединения платины, например, цисплатин (необязательно в комбинации с амифостином), карбоплатин или оксалиплатин;
(ii) таксановые соединения, например, паклитаксел, связанные с белком частицы паклитаксела (АбраксанTM), доцетаксел, кабазитаксел или ларотаксел;
(iii) ингибиторы топоизомеразы I, например, соединения камптотецина, например, камптотецин, иринотекан (CPT11), SN-38, или топотекан;
(iv) ингибиторы топоизомеразы II, например, противоопухолевые эпиподофиллотоксины или производные подофиллотоксина, например, этопозид или тенипозид;
(v) алкалоиды винка, например, винбластин, винкристин, липосомальный винкристин (Onco-TCS), винорелбин, виндесин, винфлунин или винвесир;
(vi) производные нуклеозидов, например, 5-фторурацил (5-FU, необязательно в комбинации с лейковорином), гемцитабин, капецитабин, тегафур, UFT, S1, кладрибин, цитарабин (Ara-C, цитозина арабинозид), флударабин, клофарабин, или неларабин;
(vii) антиметаболиты, например, клофарабин, аминоптерин или метотрексат, азацитидин, цитарабин, флоксуридин, пентостатин, тиогуанин, тиопурин, 6-меркаптопурин, или гидроксимочевина (гидроксикарбамид);
(viii) Алкилирующие средства, такие как азотистые иприты или нитрозомочевина, например, циклофосфамид, хлорамбуцил, кармустин (BCNU), бендамустин, тиотепа, мелфалан, треосульфан, ломустин (CCNU), алтретамин, бусульфан, дакарбазин, эстрамустин, фотемустин, ифосфамид (необязательно в комбинации с месной), пипоброман, прокарбазин, стрептозоцин, темозоломид, урацил, мехлорэтамин, метилциклогексилхлорэтилнитрозомочевина, или нимустин (ACNU);
(ix) антрациклины, антрацендионы и связанные лекарственные средства, например, даунорубицин, доксорубицин (необязательно в комбинации с дексразоксаном), липосомальные композиции доксорубицина (например, Caelyx™, Myocet™, Doxil™), идарубицин, митоксантрон, эпирубицин, амсакрин, или валрубицин;
(x) эпотилоны, например, иксабепилон, патупилон, BMS-310705, KOS-862 и ZK-EPO, эпотилон A, эпотилон B, дезоксиэпотилон B (также известный как эпотилон D или KOS-862), аза-эпотилон B (также известный как BMS-247550), аулималид, изолаулималид, или лейтеробин;
(xi) ингибиторы ДНК метилтрансферазы, например, темозоломид, азацитидин или децитабин;
(xii) антифолаты, например, метотрексат, пеметрексед натрия или ралтитрексед;
(xiii) цитотоксические антибиотики, например, актиномицин D, блеомицин, митомицин C, дактиномицин, карминомицин, дауномицин, левамизол, пликамицин или митрамицин;
(xiv) тубулин-связывающие средства, например, комбрестатин, колхицины или нокодазол;
(xv) ингибиторы трансдукции сигнала, такие как ингибиторы киназы (например, ингибиторы EGFR (рецептор эпителиального фактора роста), ингибиторы VEGFR (рецептора сосудистого эндотелиального фактора роста), ингибиторы PDGFR (рецептора тромбоцитарного фактора роста), MTKI (ингибитора многоцелевой киназы), ингибиторы Raf, ингибиторы mTOR, например, иматиниб мезилат, эрлотиниб, гефитиниб, дазатиниб, лапатиниб, ддовотиниб, акситиниб, нилотиниб, вандетаниб, ваталиниб, пазопаниб, сорафениб, сунитиниб, темсиролимус, эверолимус (RAD 001), или вемурафениб (PLX4032/RG7204);
(xvi) ингибиторы Аврора киназы, например, AT9283, барасертиб (AZD1152), TAK-901, MK0457 (VX680), сенисертиб (R-763), данусертиб (PHA-739358),алисертиб (MLN-8237), или MP-470;
(xvii) ингибиторы CDK, например, AT7519, росковитин, селициклиб, алвоцидиб (флавопиридол), динациклиб (SCH-727965), 7-гидрокси-стауроспорин (UCN-01), JNJ-7706621, BMS-387032 (также известные как SNS-032), PHA533533, PD332991, ZK-304709, или AZD-5438;
(xviii) ингибиторы PKA/B и ингибиторы пути PKB (akt), например, AT13148, AZ-5363, семафор, ингибиторы SF1126 и MTOR, такие как аналоги рапамицина, AP23841 и AP23573, ингибиторы кальмодулина (ингибиторы транслокации forkhead), API-2/TCN (трицирибин), RX-0201, энзастаурин HCl (LY317615), NL-71-101, SR-13668, PX-316, или KRX-0401 (перифозин/NSC 639966);
(xix) ингибиторы Hsp90, например, AT13387, гербимицин, гелданамицин (GA), 17-аллиламино-17-дезметоксигелданамицин (17-AAG) например, NSC-330507, Kos-953 и CNF-1010, 17-диметиламиноэтиламино-17-деметоксигелданамицина гидрохлорид (17-DMAG) например, NSC-707545 и Kos-1022, NVP-AUY922 (VER-52296), NVP-BEP800, CNF-2024 (BIIB-021 пероральный пурин), ганетеспиб (STA-9090), SNX-5422 (SC-102112) или IPI-504;
(xx) моноклональные антитела (неконъюгированные или конъюгированные с радиоизотопами, токсинами или другими агентами), производные антител и связанные агенты, такие как анти-CD, анти-VEGFR, анти-HER2 или анти-EGFR антитела, например, ритуксимаб (CD20), офатумаб (CD20), ибритумомаб тиуксетан (CD20), GA101 (CD20), тозитумомаб (CD20), эпратузумаб (CD22), линтузумаб (CD33), гемтузумаб озогамицин (CD33), алемтузумаб (CD52), галиксимаб (CD80), трастузумаб (HER2 антитело), пертузумаб (HER2), трастузумаб-DM1 (HER2), эртумаксомаб (HER2 и CD3), цетуксимаб (EGFR), панитумумаб (EGFR), неситумумаб (EGFR), нимотузумаб (EGFR), бевацизумаб (VEGF), ипилимумаб (CTLA4), катумаксумаб (EpCAM и CD3), абаговомаб (CA125), фарлетузумаб (фолатный рецептор), элотузумаб (CS1), деносумаб (RANK лиганд), фигитумумаб (IGF1R), CP751,871 (IGF1R), мапатумумаб (TRAIL рецептор), metMAB (met), митумомаб (GD3 ганглиозид), наптумомаб эстафенатокс (5T4), или силтуксимаб (IL6);
(xxi) антагонисты рецепторов эстрогена или селективные модуляторы рецепторов эстрогена (SERM) или ингибиторы синтеза эстрогена, например, тамоксифен, фулвестрант, торемифен, дролоксифен, фазлодекс, или ралоксифен;
(xxii) ингибиторы ароматазы и связанные лекарственные средства, такие как экземестан, анастрозол, летразол, тестолактон аминоглютетимид, митотан или ворозол;
(xxiii) антиандрогены (т.е. антагонисты рецепторов андрогенов) и связанные агенты, например, бикалутамид, нилутамид, флутамид, ципротерон, или кетоконазол;
(xxiv) гормоны и их аналоги, такие как медроксипрогестерон, диэтилстильбэстрол (также известный как диэтилстильбоэстрол) или октреотид;
(xxv) стероиды, например, дромостанолона пропионат, мегестрол ацетат, нандролон (деканоат, фенпропионат), флуоксиместрон или госсипол,
(xxvi) стероидные ингибиторы цитохрома P450 17альфа-гидроксилазы-17,20-лиазы (CYP17), например, абиратерон;
(xxvii) агонисты или антагонисты гонадотропин рилизинг гормона (GnRAs) например, абареликс, госерелин ацетат, гистрелин ацетат, лейпролид ацетат, трипторелин, бусерилин, или дезлорелин;
(xxviii) глюкокортикоиды, например, преднизон, преднизолон, дексаметазон;
(xxix) дифференцирующие средства, такие как ретиноиды, рексиноиды, витамин D или ретиноевая кислота и средства, блокирующие метаболизм ретиноевой кислоты (RAMBA) например, аккутан, алитретиноин, бексаротен или третиноин;
(xxx) ингибиторы фарнезилтрансферазы, например, типифарниб;
(xxxi) терапия, нацеленная на хроматин, такая как ингибиторы гистондеацетилазы (HDAC), например, бутират натрия, субероиланилид гидроксамидная кислота (SAHA), депсипептид (FR 901228), дациностат (NVP-LAQ824), R306465/ JNJ-16241199, JNJ-26481585, трихостатин A, вориностат, хламидоцин, A-173, JNJ-MGCD-0103, PXD-101, или апицидин;
(xxxii) ингибиторы протеасом, например, бортезомиб, карфилзомиб, CEP-18770, MLN-9708, или ONX-0912;
(xxxiii) фотодинамические лекарственные средства, например, порфимер натрия или темопорфин;
(xxxiv) противораковые средства из морских животных, такие как трабектидин;
(xxxv) лекарственные средства, меченные радиоактивным изотопом, например, изотопом, излучающим бета частицы (например, йод-131, иттрий-90) или изотоп, излучающий альфа частицы (например, висмут-213 или актиний-225) например, ибритумомаб или йода тозитумомаб;
(xxxvi) ингибиторы теломеразы, например, теломестатин;
(xxxvii) ингибиторы матриксных металлопротеиназ, например, батимастат, маримастат, приностат или метастат;
(xxxviii) рекомбинантные интерфероны, такие как интерферон-γ и интерферон α) и интерлейкины (например, интерлейкин 2), например, альдезлейкин, денилейкин дифтитокс, интерферон альфа 2a, интерферон альфа 2b, или пегинтерферон альфа 2b;
(xxxix) селективные модуляторы иммунного ответа, например, талидомид или леналидомид;
(xl) терапевтические вакцины, такие как сипулейцел-T (Provenge) или ОнкоВекс;
(xli) цитокин-активирующие средства включают пицибанил, ромуртид, сизофиран, вирулизин или тимозин;
(xlii) триоксид мышьяка;
(xliii) ингибиторы рецепторов, связанных с G-белками (GPCR) например, альтрасентан;
(xliv) Ферменты, такие как L-аспарагиназа, пегаспаргаза, расбуриказа или пегадемаза;
(xlv) ингибиторы репарации ДНК, такие как ингибиторы PARP, например, олапариб, велапариб, инипариб, INO-1001, AG-014699, или ONO-2231;
(xlvi) агонисты рецептора смерти (например, TNF-связанный апоптоз, индуцирующий лиганд рецептора (TRAIL)), такой как мапатумумаб (ранее HGS-ETR1), конатумумаб (ранее AMG 655), PRO95780, лексатумумаб, дуланермин, CS-1008, апомаб или рекомбинантные лиганды TRAIL, такие как рекомбинантный человеческий лиганд TRAIL/Apo2;
(xlvii) Профилактические средства (добавки); т.е. средства, которые уменьшают или облегчают некоторые эффекты, ассоциированные с химиотерапевтическими средствами, например,
-противорвотные средства,
-средства, которые предотвращают или уменьшают длительность нейтропении, ассоциированной с химиотерапией, и предотвращают осложнения, которые возникают из-за сниженного уровня тромбоцитов, эритроцитов или лейкоцитов, например, интерлейкин-11 (например, опрелвекин), эритропоэтин (EPO) и его аналоги (например, дарбэпоэтин альфа), аналоги колоний-стимулирующего фактора, такие как гранулоцитарно-макрофагальный колоний стимулирующий фактор (GM-CSF) (например, саграмостим), и гранулоцитарный колоний стимулирующий фактор (G-CSF) и их аналоги (например, филграстим, пегфилграстим),
-средства, которые ингибируют резорбцию кости, такие как деносумаб или бисфосфонаты, например, золендронат, золендроновая кислота, памидронат и ибандронат,
-средства, которые подавляют воспалительные ответы, такие как дексаметазон, преднизон и преднизолон,
-средства, используемые для снижения уровня в крови гормона роста и IGF-I (и других гормонов), у пациентов с акромегалией или другими редкими гормон-продуцирующими опухолями, такие как синтетические формы гормона соматостатина, например, октреотида ацетат,
-антидот к лекарственным средствам, которые снижают уровень фолиевой кислоты, такой как лейковорин или фолиновая кислота,
-средства от боли, например, опиаты, такие как морфин, диаморфин и фентанил,
-нестероидные противовоспалительные средства (НПВС), такие как ингибиторы COX-2, например, целекоксиб, эторикоксиб и лумиракоксиб,
-средства от мукозита, например, палифермин,
-средства для лечения побочных эффектов, включая анорексию, кахексию, отеки или эпизоды тромбоэмболии, такие как мегестрола ацетат.
В одном определенном варианте осуществления изобретения фармацевтическая композиция дополнительно включает одни или более системных ингибиторов подавления иммунной системы. Примеры подходящих системных ингибиторов подавления иммунной системы описаны в US 2012/263677 и включают анти-CTLA-4, анти-PD-1 и анти-PD-L1 антитела.
В еще одном варианте осуществления изобретения один или более системных ингибиторов подавления иммунной системы выбирают из анти-PD-1 антител.
В дополнительном варианте осуществления изобретения фармацевтическая композиция дополнительно включает один или более усилителей стимуляции иммунной системы. Примеры подходящих усилителей стимуляции иммунной системы описаны в US 2012/263677 и включают подходящие неспецифические цитокины, такие как интерлейкин-1, -2, или -6 (IL-1, IL-2 или IL-6) и альдезлейкин; интерферон альфа или гамма (IFN-α и IFN-γ), интерферон альфа-2b и пегилированный интерферон (включая пегилированный интерферон альфа-2a и пегилированный интерферон альфа-2b); гранулоцитарный макрофагальный колоний стимулирующий фактор (GM-CSF, молграмостим или сарграмостим); вакцины дендритных клеток и другие аллогенные или аутологичные терапевтические раковые вакцины, включая внутриочаговые вакцины, содержащие онколитический герпес вирус кодирующий GM-CSF (OncoVex®) или плазмиду, кодирующую человеческий лейкоцитарный антиген-B7, и бета-2 микроглобулиновый агент, созданный для экспрессии аллогенных MHC класс I антигенов (Алловектин-7®); и антитела к специфическим опухолевым антигенам. В еще одном варианте осуществления изобретения один или более усилителей стимуляции иммунной системы выбирают из IL-2 и интерферона гамма.
Каждое из соединений, присутствующих в комбинации по изобретению, можно давать в отдельно варьирующихся схемах лечения и посредством различных путей. Например, гликолипидные соединения по изобретению предназначены для введения непосредственно в опухоль, тогда как системные ингибиторы подавления иммунной системы, например, как анти-PD-1 антитела, обычно доставляются системно, т.е. посредством внутривенной инъекции. Как таковая, позология каждого из двух или более агентов может различаться: каждый можно вводить в одно и то же время или в различные моменты времени. Специалист в области техники понимает из своего обычного знания схем лечения и комбинированной терапии для применения. Например, соединение по изобретению может быть использовано в комбинации с одним или более другими агентами, которые вводят в соответствии с их существующей комбинированной схемой.
Способы лечения
В соответствии с дополнительным аспектом изобретения обеспечивают способ лечения опухоли у пациента, включающий:
a) обеспечение:
i) пациента, имеющего по меньшей мере одну опухоль, которая включает множество раковых клеток, имеющих клеточную поверхность; и
ii) гликолипидного соединения, выбираемого из соединения по формуле (I), (II) и (III) или его фармацевтически приемлемой соли или фармацевтически приемлемой композиции, как определено в настоящем описании; и
b) введение указанного гликолипида или композиции в опухоль.
В одном варианте осуществления изобретения гликолипид или фармацевтическая композиция индуцируют иммунный ответ на опухоль, посредством этого леча опухоль.
В одном варианте осуществления изобретение обеспечивает способ индукции иммунного ответа на опухоль у пациента, включающий:
a) введение пациенту, имеющему по меньшей мере одну опухоль, эффективного количества гликолипидного соединения, выбираемого из соединения по формуле (I), (II) и (III) или его фармацевтически приемлемой соли, как определено в настоящем описании, для индукции иммунного ответа на по меньшей мере одну опухоль.
В одном варианте осуществления изобретение обеспечивает способ лечения опухоли у пациента, включающий:
a) введение пациенту, имеющему по меньшей мере одну опухоль, эффективного количества гликолипидного соединения, выбираемого из соединения по формуле (I), (II) и (III) или его фармацевтически приемлемой соли, или фармацевтической композиции, как определено в настоящем описании, для индукции иммунного ответа на по меньшей мере одну опухоль,
где индукция иммунного ответа на опухоль приводит к уменьшению опухоли, таким образом леча опухоль у пациента.
В одном варианте осуществления изобретения композиция дополнительно включает по меньшей мере один системный ингибитор подавления иммунной системы.
В одном варианте осуществления изобретения по меньшей мере один системный ингибитор подавления иммунной системы выбирают из анти-CTLA-4, анти-PD-1 и анти-PD-L1 антител.
В одном варианте осуществления изобретения, способ повторяют 1-5 раз до того, как опухоль уменьшится в размере.
В одном варианте осуществления изобретения, способ повторяют 1-5 раз до того, как опухоль станет операбельной.
В одном варианте осуществления изобретения гликолипид или фармацевтическую композицию вводят в первичную опухоль и индуцируют иммунный ответ, который является эффективным в лечении по меньшей мере одной вторичной опухоли, которые возникают из первичной опухоли.
В одном варианте осуществления изобретения гликолипид или фармацевтическую композицию вводят в первичную опухоль, и индуцируют иммунный ответ, который является эффективным в уменьшении размера по меньшей мере одной вторичной опухоли, которая развивается из первичной опухоли.
В одном варианте осуществления изобретения, способ дополнительно включает хирургическое удаление опухоли после индукции иммунного ответа на опухоль.
В одном варианте осуществления изобретения, способ дополнительно включает хирургическое удаление опухоли после введения гликолипида или фармацевтической композиции.
В одном варианте осуществления изобретения, хирургическое удаление опухоли проводят между около 1-21 днями после введения гликолипида или фармацевтической композиции.
В одном варианте осуществления изобретения, хирургическое удаление опухоли проводят между 1-14 днями после введения гликолипида или фармацевтической композиции.
В одном варианте осуществления изобретения, хирургическое удаление опухоли проводят между около 1-7 днями после введения гликолипида или фармацевтической композиции.
В одном варианте осуществления изобретения, хирургическое удаление опухоли проводят между около 7-14 дней после введения гликолипида или фармацевтической композиции.
В одном варианте осуществления изобретения, хирургическое удаление опухоли проводят между около 14-21 дней после введения гликолипида или фармацевтической композиции.
Способ по изобретению допускает введение гликолипидного соединения по изобретению с целью отображения α-Gal или GalNAc эпитопа на поверхности раковой клетки.
В одном варианте осуществления изобретения, способ дополнительно включает отображение мембран связанного α-Gal или GalNAc эпитопа на указанной опухолевой клетке.
В одном варианте осуществления настоящее изобретение предусматривает метод лечения пациента, включающий:
a) обеспечение:
i) пациента, имеющего эндогенные анти-Gal или анти-GalNAc антитела и множество нерезектабельных опухолей, где по меньшей мере подгруппа указанных опухолей является доступной для процедуры, выбираемой из группы, состоящей из прямой инъекции, инъекции посредством эндоскопии, бронхоскопии, цистоскопии, колоноскопии, лапароскопии и катетеризации,
ii) гликолипидного соединения или фармацевтической композиции, как определено в настоящем описании; и
b) внутриопухолевой инъекции указанного гликолипидного соединения или композиции с использованием указанной методики.
В одном варианте осуществления изобретения α-Gal или GalNAc эпитоп гликолипидных соединений по изобретению становится опсонизированным. В одном варианте осуществления изобретения опсонизированный α-Gal или GalNAc эпитоп индуцирует продукцию аутологичной вакцины против указанной опухоли посредством нацеливания опухолевых клеток и клеточных мембран на антиген-представляющие клетки.
В одном варианте осуществления изобретения, пациентом является человек или мышь. В одном варианте осуществления изобретения пациентом является человек. В альтернативном варианте осуществления изобретения пациентом является мышь.
В соответствии с другим аспектом изобретения обеспечивают способ внесения гликолипидных соединений по изобретению в опухоль у мыши, включающий:
a) обеспечение:
i) мыши, (1) не имеющие гена α1,3галактосилтрансферазы, (2) имеющие анти-Gal антитела, и (3) имеющие по меньшей мере одну опухоль, включающую множество раковых клеток, имеющих клеточную поверхность;
ii) гликолипидного соединения, выбираемого из соединения по формуле (I) и (II) или его фармацевтически приемлемой соли; и
b) внесение указанного гликолипида в по меньшей мере одну из указанных опухолей для отображения α-Gal эпитопа на поверхности клетки на раковых клетках.
Анти-Gal нацеливание аутологичных опухолевых вакцин на антиген-представляющие клетки
Было показано, что α-Gal эпитопы могут быть вставлены in vitro в мембраны опухолевых клеток посредством инкубации опухолевых клеток с α-Gal гликолипидами. Совместная инкубация опухолевых клеток или мембран опухолевых клеток с такими α-Gal гликолипидами приводит к их спонтанной in vitro вставке в мембраны опухолевых клеток и экспрессии α-Gal эпитопов на таких клеточных мембранах. Опухолевые клетки, созданные для экспрессии α-Gal эпитопов посредством различных методов молекулярной биологии с геном α1,3галактосилтрансферазы исследовали в качестве аутологичных опухолевых вакцин. После их внутрикожной инъекции, натуральные анти-Gal IgG антитела связываются in situ в месте вакцинации с α-Gal эпитопами на мембране вакцинирующих опухолевых клеток и нацеливают вакцину на антиген-представляющие клетки. Хотя нет необходимости понимать механизм изобретения, считают, что связывание Fc части комплексованного анти-Gal с рецепторами Fcγ на антиген-представляющих клетках индуцирует эффективное поглощение опсонизированных мембран вакцинирующих опухолевых клеток в антиген-представляющие клетки. Следовательно, нехарактеризованные опухолевые антигены аутологичной опухоли также интернализируются в антиген-представляющие клетки. После транспорта вакцинирующих аутологичных опухолевых мембран в дренирующие лимфатические узлы антиген-представляющие клетки обрабатывают и представляют пептиды опухолевых антигенов для активации опухоль-специфических цитотоксических и хелперных T клеток (т.е., CD8+ и CD4+ T клеток, соответственно).
Доказательство принципа эффективности опухолевых вакцин, экспрессирующих α-Gal эпитопы, получали в исследованиях на экспериментальной мышиной модели, иммунизированной клетками меланомы, экспрессирующими α-Gal эпитопы и сенсибилизированными теми же клетками меланомы, которые, однако, не имеют α-Gal эпитопов (LaTemple D C et al. Cancer Res. 1999, 59: 3417-23, и Deriy L et al. Cancer Gene Therapy 2005; 12: 528-39). Мышами, используемыми в этих исследованиях были мыши, нокаутированные в отношении гена α1,3галактосилтрансферазы (т.е. эти мыши не имеют α-Gal эпитопа и могут продуцировать анти-Gal антитела). Мышей иммунизировали клетками меланомы, созданными для экспрессии α-Gal эпитопов, отражающих эффективную иммунную защиту против сенсибилизации теми же опухолевыми клетками, которые однако не имеют α-Gal эпитопов. Наоборот, мыши, иммунизированные опухолевыми клетками, не имеющими α-Gal эпитопов, не проявляли иммунный ответ в отношении сенсибилизации живыми опухолевыми клетками, не имеющими α-Gal эпитопов.
α-Gal гликолипиды в противоопухолевой терапии
Настоящее изобретение предусматривает лечение пациентов с солидными опухолевыми массами. Определенные варианты осуществления настоящего изобретения предусматривают новое иммунотерапевтическое лечение раковых пациентов, имеющее целью иммунизировать отдельного пациента против его или ее собственных опухолевых поражений посредством конверсии собственной опухоли пациента в аутологичную опухолевую вакцину (см. патент США No. 5879675, включенный в настоящее описание в виде ссылки). Например, в патенте '675 описана in vitro обработка опухолевых клеток и/или клеточных мембран. При инъекции таких клеток пациенту вакцина нацеливается посредством анти-Gal антитела на APC и оказывает защитный иммунный ответ против аутологичного опухолевого антигена. В отличие от настоящего изобретения, однако, в '675 патенте не указано: i) in vivo внутриопухолевое лечение для индукции воспаления, регресса и/или деструкции опухоли посредством натурального анти-Gal антитела; или ii) отображение α-Gal эпитопов на опухолевых клетках in vivo с последующим внутриопухолевым введением α-Gal гликолипидов у раковых пациентов.
В одном варианте осуществления настоящего изобретения α-Gal гликолипиды могут быть доставлены в опухолевое поражение, включающее опухолевые клетки, посредством нехирургической внутриопухолевой инъекции (т.е. например, посредством эндоскопии, катетеризации или подобного), или посредством любого другого метода для in vivo введения в опухоли α-Gal гликолипидов или анти-Gal связывающих эпитопов на различных молекулах.
Считают что постхирургический рецидив метастазов, рефрактерных к химиотерапии, является наиболее частой причиной смерти у пациентов с солидными опухолями. О высокой частоте таких рецидивирующих метастазов (80%) сообщают у пациентов с карциномами поджелудочной железы и яичника и в некоторой степени с другими солидными опухолями, такими как меланома и карцинома колоректальная, легкого и молочной железы. Многие из таких пациентов с рецидивами расцениваются как имеющие терминальное заболевание, поскольку для них недоступно лечение, и они умирают в течение недель или месяцев после определения метастазов.
В одном варианте осуществления настоящее изобретение предусматривает терапевтический способ регресса и/или деструкции опухолевых метастазов путем использования того факта, что все люди естественно продуцируют анти-Gal антитела в виде приблизительно 1% иммуноглобулинов. Иммунологический потенциал анти-Gal антител может быть направлен на регресс и/или разрушение любых опухолевых повреждений и преобразовывать их в in situ аутологичную опухолевую вакцину посредством внутриопухолевой инъекции гликолипидов, несущих α-Gal эпитоп (т.е. гликолипидные соединения по формуле (I) или (II)).
Следовательно, изобретение, описанное в настоящем описании, может индуцировать регресс и/или деструкцию леченных опухолевых поражений. Следовательно, в одном варианте осуществления изобретения обработанная опухоль подвергается регрессу. В альтернативном варианте осуществления изобретения обработанная опухоль разрушается.
В дополнительном варианте осуществления изобретения опухоль (т.е. которая несет α-Gal эпитоп) подвергается регрессу, где указанную опухоль выбирают из меланомы или метастазов в органы, такие как метастазы в печень. В дополнительном варианте осуществления изобретения опухоль (т.е. которая несет α-Gal эпитоп) разрушается, где указанную опухоль выбирают из меланомы или органных метастазов, таких как метастазы в печень.
В одном варианте осуществления изобретения стадия введения вызывает регресс вторичной опухоли у пациента в результате конверсии обработанной опухоли в аутологичную опухолевую вакцину. В дополнительном варианте осуществления изобретения указанную вторую опухоль выбирают из меланомы или метастазов в печень.
В одном варианте осуществления изобретения стадия введения вызывает деструкцию вторичной опухоли у пациента. В дополнительном варианте осуществления изобретения указанную вторичную опухоль выбирают из меланомы или метастазов в печень.
Множество α-Gal гликолипидов спонтанно встраивается в мембраны опухолевых клеток, так как гидрофобный (т.е. липофильный) липидный хвост α-Gal гликолипидов является более стабильной энергетической формой при вставке в наружный листок липидного бислоя клеточной мембраны при сравнении с окруженным водой мицеллярным ядром. Спонтанная вставка (включение) других типов гликолипидов, называемых ганглиозиды, в клеточные мембраны ранее была продемонстрирована (Kanda S et al. J Biochem. (Tokyo). 1982; 91: 1707-18, и Spiegel S et al. J. Cell Biol. 1985; 100: 721-26). Ожидают, что вставка α-Gal гликолипидов в мембраны опухолевых клеток приведет к de novo отображению α-Gal эпитопов на поверхности мембран клеток. Экспрессия α-Gal эпитопа может облегчать анти-Gal антитело опосредованную регрессию и/или деструкцию опухолевых клеток посредством таких механизмов, которые включают, без ограничения, цитолиз, опосредованный комплементом (CDC), и антител-зависимый, опосредованный клетками цитолиз (ADCC), и также может приводить к некрозу опухоли. На анти-Gal опсонизированную мембрану опухолевых клеток будут эффективно нацеливаться антиген-представляющие клетки, таким образом преобразуя обработанные опухолевые поражения в аутологичные опухолевые вакцины. Такая аутологичная вакцина затем будет стимулировать иммунную систему реагировать против опухолевых антигенов, приводя к дополнительному регрессу и/или деструкции опухолевых клеток, экспрессирующих такие антигены в других опухолевых повреждениях и/или микрометастазах леченного пациента.
В одном варианте осуществления изобретения пациента лечили, ранее хирургически удалив опухоль.
В альтернативном варианте осуществления изобретения пациента не лечили ранее, хирургически удаляя опухоль, т.е. способ, описанный в настоящем описании, может быть проведен как неоадьювантная терапия за несколько недель до резекции первичной опухоли. В одном варианте осуществления изобретения инъекция в опухоль гликолипидов по изобретению уменьшает размер опухоли и преобразует обработанную опухоль в аутологичную опухолевую вакцину. Хотя такая опухоль в конечном счете резецируется, считают, что перед ее резекцией обработанная опухоль будет оказывать иммунный ответ против микрометастаз, которые имеют те же опухолевые антигены.
Механизмы анти-Gal антител регрессии и/или деструкции опухоли
Хотя нет необходимости понимать механизм изобретения считают, что регресс и/или деструкция опухолевого поражения вводимыми α-Gal гликолипидами может иметь биохимическую и физиологическую основу.
В одном варианте осуществления изобретения способ дополнительно включает индукцию внутриопухолевого воспаления.
Внутриопухолевая инъекция может приводить к локальному разрыву капилляров, ассоциированных с опухолью, таким образом обеспечивая доступ натуральных молекул анти-Gal IgM и анти-Gal IgG антител к внутренней части опухоли. Анти-Gal антитела затем будут способны взаимодействовать с α-Gal эпитопами на α-Gal гликолипидных мицеллах, или отдельных α-Gal гликолипидных молекулах, посредством этого индуцируя локальную активацию комплемента и создание расщепляющих хемотаксических факторов комплемента C5a и C3a. Более того, C3b становится ковалентно отложенным на целевых клетках. Активация комплемента затем инициирует локальный воспалительный процесс, облегчающий внутриопухолевую миграцию гранулоцитов, моноцитов, макрофагов и дендритных клеток посредством de novo продуцируемых C5a и C3a хемотактических факторов в леченные опухолевые поражения. Воспалительный процесс может быть дополнительно усилен в результате вставки α-Gal гликолипидов в клеточные мембраны, вызывая анти-Gal активацию эндотелиальных клеток (Palmetshofer A et al. Transplantation. 1998; 65: 844-53; Palmetshofer A et al. Transplantation. 1998; 65: 971-8). Активация эндотелиальных клеток и общее повреждение опухолевых клеток может приводить к локальной продукции дополнительных провоспалительных цитокинов и хемокинов. Такие локально секретируемые цитокины и хемокины индуцируют дополнительную миграцию макрофагов, дендритных клеток и последующую миграцию лимфоцитов в повреждение, в которое вводят α-Gal гликолипиды. Такая клеточная миграция опосредована рецепторами к провоспалительным цитокинам и хемокинам на антиген-представляющих клетках и на лимфоцитах (Cravens P D and Lipsky P E Immunol. Cell Biol. 2002; 80: 497-505). Такая исходная индукция воспалительного ответа позволяет иммунной системе преодолевать общее отсутствие способности определять "скрытую природу" развивающихся опухолевых поражений. Такое воспаление также позволяет иммунной системе преодолевать иммуносупрессивное микроокружение в солидных опухолях, которое индуцируется локальным цитокиновым окружением, и которое обычно предотвращают пенетрацию лимфоцитов в опухоль (Malmberg K J. Cancer Immunol. Immunother. 2004; 53: 879-92; Lugade A A et al. J. Immunol. 2005; 174:7516-23).
Деструкция опухолевых клеток развивается посредством анти-Gal связывания с α-Gal гликолипидами, вставленными в клеточные мембраны. α-Gal гликолипиды, вводимые в опухоль, могут спонтанно встраиваться во внешний слой фосфолипидного бислоя мембран опухолевых клеток. Последующее связывание анти-Gal IgM и/или анти-Gal IgG с α-Gal эпитопами на вставленных α-Gal гликолипидах индуцируют регресс и/или деструкцию обработанной опухоли посредством комплемент-зависимого цитолиза (CDC). Связывание анти-Gal IgG молекул с такими α-Gal эпитопами также облегчает зависимый от антител клеточный цитолиз (ADCC) опухолевых клеток.
В одном варианте осуществления изобретения опухоль подвергается регрессу и/или деструкции посредством комплемент зависимого цитолиза (CDC).
В одном варианте осуществления изобретения опухоль подвергается регрессу и/или деструкции посредством клеточного цитолиза, зависимого от антител (ADCC).
При комплеммент-зависимом цитолизе считают, что связывание молекул анти-Gal IgG и/или IgM с опухолевыми клетками, экспрессирующими α-Gal эпитопы (из-за вставки α-Gal гликолипидов) активируют систему комплемента. Впоследствии C5b-9 мембраноатакующий комплекс комплемента образуется в результате такой активации комплемента, затем "протыкает" отверстия в мембранах опухолевых клеток, приводя к лизису опухолевых клеток. Такой комплемент-зависимый цитолиз сходным образом обнаруживают, когда лизируют эпителиальные клетки свиней, что приводит к сверхострому отторжению трансплантата (Collins B H et al. J. Immunol. 1995; 154: 5500-10,). В ADCC эффекторными клетками являются гранулоциты, макрофаги и NK клетки. Такие клетки привлекаются к повреждению, из-за воспалительного процесса, индуцированного анти-Gal. Они связываются посредством их рецепторов Fcγ (FcγR) с Fc частью анти-Gal IgG молекул, которые связываются с α-Gal гликолипидом, вставленным в мембрану опухолевых клеток. При прикреплении к опухолевым клеткам такие эффекторные клетки секретируют их гранзимные пузырьки в контактных областях мембраны, создавая отверстия в мембране опухолевых клеток, таким образом индуцируя деструкцию таких опухолевых клеток. Эффективность анти-Gal IgG в индукции ADCC деструкции клеток, экспрессирующих α-Gal эпитопы, была продемонстрирована с ксенотрансплантированными свиными клетками связыванием анти-Gal посредством их α-Gal эпитопов (Galili, U. Immunol. Today 1993, 14: 480-82). Сходный анти-Gal опосредованный ADCC процесс развивается, когда опухолевые клетки связывают анти-Gal посредством α-Gal эпитопов, экспрессируемых на их клеточной поверхностной мембране (Tanemura M et al. J. Clin. Invest. 2000; 105: 301-10).
Потребление мембран опухолевых клеток антиген-представляющими клетками может приводить к индукции защитного иммунного ответа против аутологичных опухолевых антигенов с целью регресса и/или разрушения микрометастазов, резистентных к химиотерапии. Анти-Gal IgG антитело, связанное с α-Gal эпитопами на гликолипидах α-Gal, встроенных в мембрану, или C3b, отложенных на целевых клетках, посредством анти-Gal зависимой активации комплемента стимулирует антиген-представляющие клетки интернализировать клеточные мембрны, экспрессирующие опухолевые антигены (т.е. например, антигены, ассоциированные с опухолью, TAA). Интернализированные опухолевые антигены затем могут переноситься антиген-представляющими клетками из леченного опухолевого поражения в дренирующие лимфатические узлы. Такие опухолевые антигены затем могут быть обработаны антиген-представляющими клетками и представлены, как иммуногенные опухолевые пептиды, которые активируют опухоль-специфические T клетки. Такой процесс приводит к индукции системного защитного противоопухолевого иммунного ответа (т.е. например, аутологичной опухолевой вакцины). Следовательно, опухолевые поражения, в которые вводили α-Gal гликолипиды, в конечном счете преобразовывались в in situ аутологичные опухолевые вакцины, которые проявляли иммунный ответ против микрометастаз, экспрессирующих опухолевые антигены, как таковые, в обработанных опухолевых поражениях.
В качестве варианта клинического лечения гликолипиды можно вводить в раковые поражения различными методами, включая, без ограничения, внутрикожную инъекцию (т.е., например, в опухоль меланому); эндоскопическую инъекцию (т.е. например в колоректальные кишечные метастазы); лапароскопическую инъекцию (т.е. например, в внутрибрюшные метастазы карциномы яичника, толстой кишки, желудка, печени или поджелудочной железы (например, на брюшине или в печени)); чрескожную инъекцию иглой под визуализацией (т.е., например, в опухоли легких); бронхоскопическую инъекцию (т.е., например, в опухоли легкого); колоноскопическую инъекцию; или цистоскопическую инъекцию (т.е., например, в карциномы мочевого пузыря).
Следовательно, в одном варианте осуществления изобретения введение включает процедуру, включая, без ограничения инъекцию, инъекцию под визуализацией, эндоскопию, бронхоскопию, цистоскопию, колоноскопию, лапароскопию и катетеризацию.
В одном варианте осуществления изобретения введение включает нехирургическую инъекцию в опухоль. Например, введение включает процедуру, выбираемую из: внутрикожной инъекции, чрескожной инъекции под визуализацией, эндоскопической инъекции, бронхоскопической инъекции, цистоскопической инъекции, колоноскопической инъекции и лапароскопической инъекции.
В одном варианте осуществления изобретения гликолипид по изобретению вводят в фармацевтически приемлемом растворе (т.е. стерильном растворе), выбираемом из группы, включая, без ограничения, фосфатный буферный раствор (PBS), солевой раствор, другие водные растворы или другие вспомогательные вещества, обычно считаемые безопасными (GRAS). В одном варианте осуществления изобретения раствор гликолипидов также может содержать деоксихолат или другие мягкие детергенты, которые могут увеличивать проникновение гликолипидов в клеточные мембраны.
В одном варианте осуществления настоящее изобретение предусматривает внутриопухолевую инъекцию гликолипидов по изобретению в первичные опухоли в качестве неоадьювантной терапии, проводимой до оперативного удаления опухоли. В одном варианте осуществления изобретения быстрый воспалительный ответ, индуцируемый пре-хирургической инъекцией гликолипида, приводит к уменьшению размера опухолевого поражения, а также преобразованию ее в in situ аутологичную опухолевую вакцину. Хотя нет необходимости понимать механизм изобретения, считают, что иммунный ответ на обработанную опухоль может в конечном счете помочь индуцировать иммунную деструкцию микрометастаз, которые не определяются в момент хирургической резекции первичных опухолей. Дополнительно считали, что дохирургическое введение может помочь в предотвращении рецидива заболевания из-за иммунологической деструкции микрометастазов, резистентных к обычной адьювантной терапии (т.е., например, химиотерапии и облучению) и которые экспрессируют опухолевые антигены, как первичная опухоль. Такую неоадьювантную терапию можно вводить для любой солидной опухоли или лимфомы, в которые можно ввести непосредственно, или посредством визуализации или посредством любого другого известного метода.
В соответствии с дополнительным аспектом изобретения обеспечивают набор, включающий фармацевтическую композицию, как определено в настоящем описании, и необязательно инструкции для использования указанного набора в соответствии с методом, описанном в настоящем описании.
В одном варианте осуществления изобретения набор дополнительно включает устройство для доставки, такое как устройство для доставки в опухоль.
Все публикации и патенты, цитированные в настоящей спецификации, включены в настоящее описание в виде ссылки, как будто каждая отдельная публикация или патент были специфически и индивидуально указаны для включения в виде ссылки и включены в настоящее описание в виде ссылки для раскрытия и описания методов и/или материалов в сочетании с цитированными публикациями. Цитирование любой публикации предназначено для его раскрытия до даты публикации и не должно расцениваться, как вероятность того, что настоящее описание не предназначено для предшествования такой публикации посредством предшествующего описания.
Следующие примеры предназначены только в качестве иллюстративных примеров вариантов осуществления изобретения. Они не должны расцениваться, как ограничивающие настоящее изобретение.
Материалы и методы
Ацетон, бензол, хлороформ, этилацетат, метанол, o-ксилен, толуол, 2-пропанол и o-ксилен были от Chimmed (Russian Federation). Ацетонитрил был от Cryochrom (Russian Federation). DMSO, DMF, CF3COOH, Et3N, N,N'-дициклогексилкарбодиимид и N-гидроксисукцинимид были от Merck (Germany). N-метилморфолин (NMM), 2-малеимидопропионовая кислота и дисукцинидилкарбонат поставляли от Fluka. Гидрохлорид диметилового эфира иминодиуксусной кислоты был от Reakhim (Russian Federation). Тетраамин (H2N-CH2)4C x 2H2SO4 синтезировали, как описано Litherland and Mann (1938) The amino-derivatives of pentaerythritol Part I. Preparation Journal of the Chemical Society, 1588-95.
Dowex 50X4-400 и Sephadex LH-20 были от Amersham Biosciences AB (Sweden). Силикагель 60 был от Merck (Germany). Тонкослойную хроматографию проводили с использованием алюминиевых листов силикагеля 60 F254 (Merck, 1.05554) с определением посредством обжига после погружения в 7% H3PO4 или нингидрин.
1H спектры ЯМР записывали при 30°C с помощью прибора Bruker WM 500 MГц или спектрометра Bruker DRX-500 с использованием сигнала остаточных протонов растворителя в качестве контроля ([D6]DMSO, 2500 ч/млн; [D2]H2O, 4750 ч/млн; CD3OD).
Пример 1: Получение соединения по формуле (I) ʺGalili-CMG2-DOPEʺ
Получение 3-трифторацетамидопропил-3,4-ди-O-ацетил-2,6-ди-O-бензил-α-D-галактопиранозил-(1→3)-2,4-ди-O-ацетил-6-O-бензил-β-D-галактопиранозил-(1→4)-2-ацетамидо-3-O-ацетил-6-O-бензил-2-деокси-β-D-глюкопиранозид (3) (Схема I)
Акцептор гликозила (3-трифторацетамидопропил)-2-ацетамидо-3-O-ацетил-6-O-бензил-2-деокси-4-O-(2,4-ди-O-ацетил-6-O-бензил-β-D-галактопиранозил)-β-D-глюкопиранозид (2) получали в соответствии со способом, описанным в публикации Pazynina et al (2008). Смесь акцептора гликозила 2 (500 мг, 0,59 ммоль), тиогалактопиранозида 1 (576 мг, 1,18 ммоль), NIS (267 мг, 1,18 ммоль), безводный CH2Cl2 (25 мл) и молекулярные сита 4 Α (500 мг) перемешивали при -45°C в течение 30 мин в атмосфере Ar. Затем добавляли раствор TfOH (21 мкл, 0,236 ммоль) в безводном CH2Cl2 (0,5 мл). Реакционную смесь перемешивали в течение 2 ч при -45°C и температуру затем увеличивали до -20°C в течение 4 ч. Смесь выдерживали при -20°C в течение ночи. Затем избыточные количества тиогалактопиранозида 1 (144 мг, 0,295 ммоль), NIS (66 мг, 0,295 ммоль) и добавляли TfOH (5 мкл, 0,06 ммоль), и перемешивание продолжали при -20°C в течение 2 ч до возможности медленного нагревания до к.т. (1 ч). Насыщенный водный раствор Na2S2O3 затем добавляли и смесь фильтровали. Фильтрат разводили CHCl3 (300 мл), промывали H2O (2×100 мл), сушили фильтрацией через хлопковую вату и концентрировали. Гельфильтрация на LH-20 (CHCl3-MeOH) давала продукт 3 (600 мг, 80%), в виде белой пены.
1Н ЯМР (700 МГц, CDCl3, характерные сигналы), δ, ч/млн: 1,78-1,82 (м, 4H, CHCHC, OC(O)CH3), 1,84-1,90 (м, 1H, CHCHC), 1,91, 1,94, 1,97, 1,98, 2,06 (5 с, 5×3Н, 4 OC(O)CH3, NH(O)CH3), 3,23-3,30(м, 1H, NCHH), 3,59-3,65 (м, 1H, NCHH), 4,05 (м, 1H, H-2I), 4,33 (д, 1H, Дж1,2 7,55, H-1I), 4,40 (д, 1H, Дж 12,04, PhCHH), 4,42 (д, 1H, Дж1,2 8,07, H-1II), 4,45 (д, 1H, Дж 11,92, PhCHH), 4,48 (д, 1H, Дж 12,00, PhCHH), 4,50 (д, 1H, Дж 12,00, PhCHH), 4,52 (д, 1H, Дж 12,04, PhCHH), 4,54 (д, 1H, Дж 12,00, PhCHH), 4,57 (д, 1H, Дж 12,00, PhCHH), 4,64 (д, 1H, Дж 11,92, PhCHH), 4,99 (дд ≈ т, 1H, Дж 8,24, H-2II), 5,08-5,13 (м, 2H, H-3I, H-3III), 5,23 (д, 1H, Дж1,2 3,31, H-1III), 5,46 (д, 1H, Дж3,42,25, H-4II), 5,54 (д, 1H, Дж3,4 3,11, H-4III), 7,20-7,40 (м, 20H, ArH); 7,49-7,54 (м, 1H, NHC(O)CF3), Фр 0,4 (PhCH3-AcOEt, 1:2),
Получение 3-аминопропил-α-D-галактопиранозил-(1→3)-β-D-галактопиранозил-(1→4)-2-ацетамидо-2-деокси-β-D-глюкопиранозида (5) (Схема I)
Продукт 3 (252 мг, 0,198 ммоль) деацетилировали в соответствии с Zemplen (8 ч, 40°C), нейтрализовали AcOH и концентрировали. ТЖХ (CH3Cl-MeOH, 10:1) анализ полученного продукта показал два пятна: главное пятно с Фр 0,45, и другое на стартовой линии (позитивное пятно нингидрина), которое было показателем частичной потери трифторацетила. Следовательно, продукт был N-трифторацетилирован посредством обработки CF3COOMe (0,1 мл) и Et3N (0,01 мл) в MeOH (10 мл) в течение 1 ч, концентрирован и подвергнут колоночной хроматографии на силикагеле (CHCl3-MeOH, 15:1) для получения продукта 4 в виде белой пены (163 мг, 77%), Фр 0,45 (CH3Cl-MeOH, 10:1). Продукт 4 подвергали гидрогенолизу (200 мг Pd/C, 10 мл MeOH, 2 ч), фильтровали, N-дефторацетилировали (5% Et3N/H2O, 3 ч) и концентрировали. Катионообменная хроматография на Dowex 50X4-400 (H+) (элюция с помощью 5% водного аммиака) давала продукт 5 (90 мг, 98%) в виде белой пены.
1Н ЯМР (D2O, характерные сигналы), δ, ч/млн: 1,94-1,98 (м, 2H, CCH2C), 2,07 (с, 3H, NHC(O)CH3), 3,11 (м, Дж 6,92, 2H, NCH2), 4,54 и 4,56 (2д, 2H, Дж1,2 8,06, Дж1,2 7,87, H-1I и H-1II), 5,16 (д, 1H, Дж1,2 3,87, H-1III), Фр 0,3 (EtOH-BuOH-Py-H2O-AcOH; 100:10:10:10:3),
Схема I
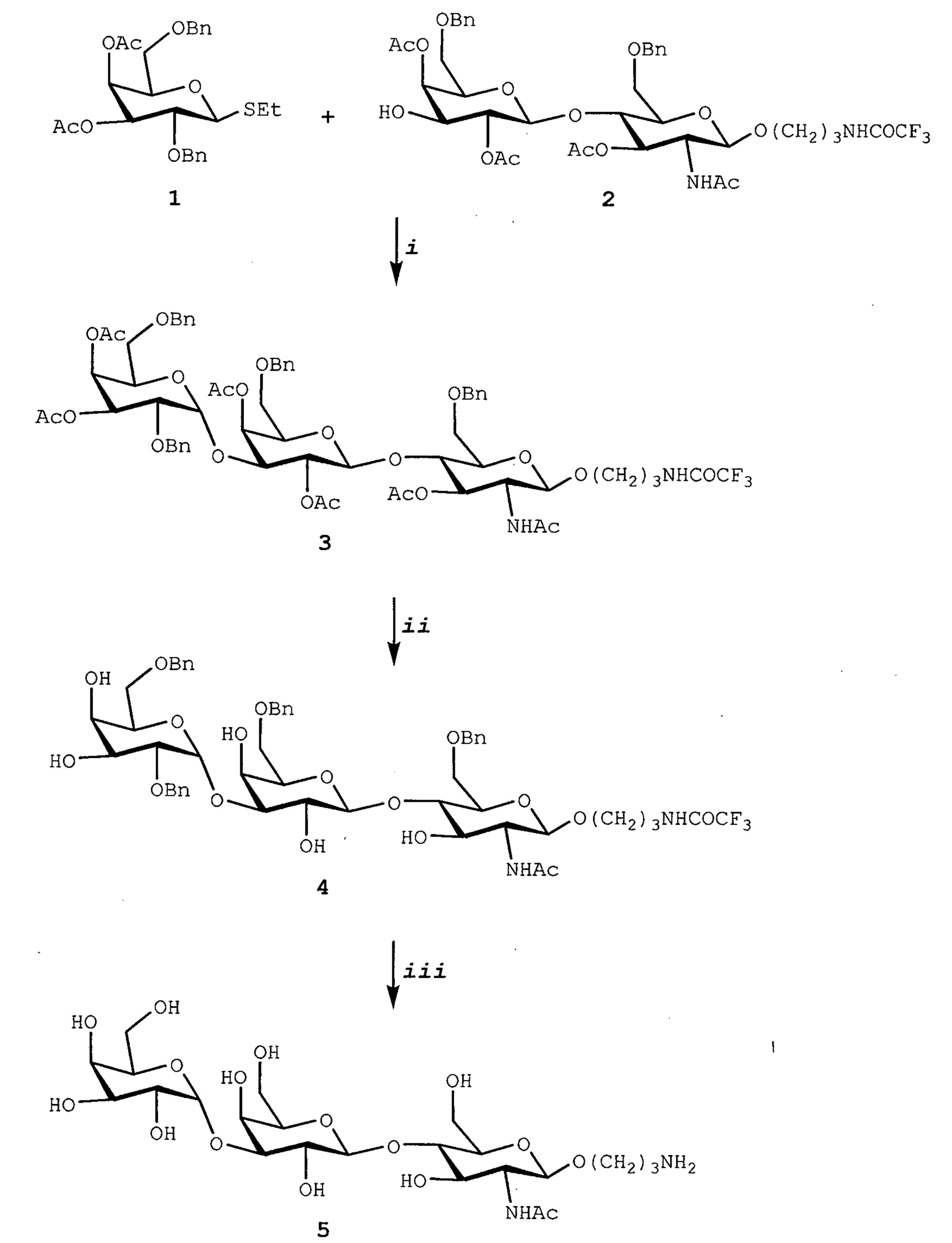
Получение {[2-(2-трет-бутоксикарбониламиноацетиламино)ацетил]метоксикарбонилметиламино}уксусной кислоты метилового эфира (8) (Схема II)
N-Метилморфолин (11,0 мл, 0,1 моль) добавляли к перемешиваемой суспензии Boc-глицилглицина (23,2 г, 0,1 моль) в 150 мл метиленхлорида, раствор охлаждали до -15°C и изобутилхлорформат (13,64 г, 0,1 моль) добавляли в течение 10 мин. Затем 1-гидроксибензотриазол и раствор (метоксикарбонилметиламино)уксусной кислоты метилового эфира (7) (16,1 г, 0,1 моль) в 50 мл DMF добавляли к реакционной смеси при той же температуре. Полученную смесь перемешивали в течение 30 мин при 0°C, затем в течение 2 ч при температуре окружающей среды и выпаривали досуха. Остаток растворяли в 200 мл метиленлорида и промывали 100 мл 0,5 M HCl и 200 мл 2% водн. NaHCO3. Растворители выпаривали в вакууме, и остаток очищали колоночной хроматографией на силикагеле (3% MeOH в CHCl3) для получения чистого целевого соединения (34,08 г, 91%) в виде бесцветного стекла. ТЖХ: Фр=0,40 (5% MeOH в CHCl3), Фр=0,49 (7:1 (об/об) хлороформ/метанол).
1H ЯМР (500 МГц, [D6]DMSO, 30°C) δ, ч/млн: 7,826 (т, J=5,1 Гц, 1H; NHCO), 6,979 (т, Дж=5,9 Гц, 1H; NHCOO), 4,348 и 4,095 (с, 2H; NCH2COO), 3,969 (д, Дж=5,1 Гц, 2H; COCH2NH), 3,689 и 3,621 (с, 3H; OCH3), 3,559 (д, Дж=5,9 Гц, 2H; COCH2NHCOO), 1,380 (с, 9H; C(CH3)3), Фр 0,49 (7:1 (об/об) хлороформ/метанол),
Получение {[2-(2-трет-бутоксикарбониламиноацетиламино)ацетил]метоксикарбонилметиламино}уксусной кислоты (9) (Схема II)
0,2 M водный NaOH (325 мл) добавляли к перемешиваемому раствору {[2-(2-третбутоксикарбониламиноацетиламино)ацетил]метоксикарбонилметиламино}уксусной кислоты метилового эфира (8)(24,42 г, 65,12 ммоль) в метаноле (325 мл), реакционную смесь выдерживали в течение 15 мин при температуре окружающей среды, подкисляли уксусной кислотой (5 мл) и выпаривали досуха. Колоночная хроматография остатка на силикагеле (метанол-этилацетат 1:1) давала целевое соединение в виде Na-соли (20,44 г), которое растворяли в смеси метанол/вода/пиридин (20:10:1, 350 мл) и пропускали через ионообменную колонку (Dowex 50X4-400, пиридиновая форма, 300 мл) для удаления катионов Na. Колонку промывали той же смесью, элюат выпаривали и сушили в вакууме для получения чистого целевого соединения (20,15 г, 86%) в виде белого твердого вещества. ТЖХ: Фр= 0,47 (iPrOH/ этилацетат/вода 4:3:1).
1H ЯМР (500 МГц, [D6]DMSO, 30°C), смесь цис- и транс- конформеров N-карбоксиметилглицинового блока c.3:1. Основной конформер; δ, ч/млн: 7,717 (т, Дж=5 Гц, 1H; NHCO), 7,024 (т, Дж=5,9 Гц, 1H; NHCOO), 4,051 (с, 2H; NCH2COOCH3), 3,928 (д, Дж=5 Гц, 2H; COCH2NH), 3,786 (с, 2H; NCH2COOH), 3,616 (с, 3H; OCH3), 3,563 (д, Дж=5,9 Гц, 2H; COCH2NHCOO), 1,381 (с, 9H; C(CH3)3) ч/млн; минорный конформер, δ=7,766 (т, Дж=5 Гц, 1H; NHCO), 7,015 (т, Дж=5,9 Гц, 1H; NHCOO), 4,288 (с, 2H; NCH2COOCH3), 3,928 (д, Дж=5 Гц, 2H; COCH2NH), 3,858 (с, 2H; NCH2COOH), 3,676 (с, 3H; OCH3), 3,563 (д, Дж=5,9 Гц, 2H; COCH2NHCOO), 1,381 (с, 9H; C(CH3)3), Фр 0,47 (4:3:1 (об/об/об) i-PrOH/этилацетат/вода)
Получение {[2-(2-трет-бутоксикарбониламиноацетиламино)ацетил]метоксикарбонилметиламино}уксусной кислоты N-оксисукцинимидного сложного эфира (Boc-Gly2(MCM)GlyOSu) (10) (Схема II)
N,N'-Дициклогексилкарбодиимид (14,03 г, 68,10 ммоль) добавляли к ледяному перемешиваемому раствору {[2-(2-трет-бутоксикарбониламиноацетиламино)ацетил]метоксикарбонилметиламино}уксусной кислоты (26,40 г, 73,13 ммоль) и N-гидроксисукцинимида (8,70 г, 75,65 ммоль) в DMF (210 мл). Смесь перемешивали в течение 30 мин при 0°C, затем в течение 2 ч при температуре окружающей среды. Осажденную N,N'-дициклогексилмочевину отфильтровывали, промывали DMF (80 мл). Фильтрат и промывные воды концентрировали, и остаток перемешивали с Et2O (500 мл) в течение 1 ч. Эфирный экстракт декантировали и остаток концентрировали для получения целевого соединения в виде белой пены (32,57 г, 97%). ТЖХ: Фр=0,71 (ацетон/уксусная кислота 40:1).
1H ЯМР (500 МГц, DMSO[D6], 30°C), смесь цис- и транс- конформеров N-карбоксиметилглицинового блока c. 3:2.
Главный конформер; δ, ч/млн: 7,896 (т, J=5,1 Гц, 1H; NHCO), 6,972 (т, J=5,9 Гц, 1H; NHCOO), 4,533 (с, 2H; NCH2COON), 4,399 (с, 2H; NCH2COOCH3), 3,997 (д, Дж=5,1 Гц, 2H; COCH2NH), 3,695 (с, 3H; OCH3), 3,566 (д, дж=5,9 Гц, 2H; COCH2NHCOO), 1,380 (с, 9H; C(CH3)3), минимальный конформер; δ, ч/млн: 7,882 (т, Дж=5,1 Гц, 1H; NHCO), 6,963 (т, Дж=5,9 Гц, 1H; NHCOO), 4,924 (с, 2H; NCH2COON), 4,133 (с, 2H; NCH2COOCH3), 4,034 (д, Дж=5,1 Гц, 2H; COCH2NH), 3,632 (с, 3H; OCH3), 3,572 (д, Дж=5,9 Гц, 2H; COCH2NHCOO), 1,380 (с, 9H; C(CH3)3),
Фр 0,71 (40:1 (об/об) ацетон/уксусная кислота).
Схема II

Получение CMG(2) диамина (16) (Схемы III и IV)
Раствор этилендиамина (11) (808 мг, 13,47 ммоль) и Et3N (1,87 мл, 13,5 ммоль) в DMSO (5 мл) добавляли к перемешиваемому раствору Boc-Gly2-(MCM)Gly-OSu (10) (15,42 г, 33,68 ммоль) в DMSO (50 мл). Реакционную смесь перемешивали в течение 30 мин при температуре окружающей среды и подкисляли уксусной кислотой (1,2 мл), затем фракционировали с колонкой Sephadex LH-20 (объем колонки 1200 мл, элюент - MeOH/вода 2:1+0,2% AcOH). Фракции, содержащие соединение Boc2MCMG (12), комбинировали, растворители выпаривали, и остаток концентрировали в вакууме. Продукт дополнительно очищали хроматографией на колонке из силикагеля с использованием 2-пропанол/этилацетат/воды (2:6:1) в качестве элюента. Фракции, содержащие чистый Boc2MCMG (12), комбинировали, растворители выпаривали, и остаток сушили в вакууме для получения целевого Boc2MCMG (12) в виде бесцветной пены (8,41 г, 84%). ТЖХ: Фр= 0,48 (iPrOH/этилацетат/вода 2:3:1).
1H ЯМР (500 МГц, [D6]DMSO, 30°C), смесь конформеров ~3:2: 8,166, 8,125, 7,917 и 7,895 (м, всего 2H; 2 CONHCH2), 7,793 (м, 2H; NHCH2CH2NH), 7,001 (бр, т, 2H; 2 NHCOO), 4,277-3,893 (всего 12H; 2 CH2COO, 4 NCH2CO), 3,690 и 3,635 (с, всего 6H; 2 COOCH3), 3,567 (д, Дж=5,8 Гц, 4H; 2 CH2NHCOO), 3,131 (м, 4H; NHCH2CH2NH), 1,379 (с, 18H; 2 C(CH3)3) ч/млн,
МС, м/з: 769 [M+Na], 785 [M+K].
Трифторуксусную кислоту (25 мл) добавляли к перемешиваемому раствору Boc2MCMG (12) (4,88 г, 6,535 ммоль) в метиленхлориде (25 мл) и раствор выдерживали в течение 1 ч при температуре окружающей среды. Затем реакционную смесь концентрировали, и остаток выпаривали три раза с безводным MeOH (50 мл), затем остаток экстрагировали три раза Et2O (100 мл) для удаления следов трифторуксусной кислоты. Полученный осадок (в виде белого твердого вещества) сушили для получения 5,06 г (~100%) MCMG (13) в виде бис-трифторуксусной соли. ТЖХ: Фр= 0,23 (этанол/вода/пиридин/уксусная кислота 5:1:1:1).
1H ЯМР (500 МГц, D2O, 30°C), смесь конформеров ~5:4: 4,400-4,098 (всего 12H; 2 CH2COO, 4 NCH2CO), 3,917 (с, 4H; 2 COCH2NH2), 3,829 и 3,781 (с, всего 6H; 2 COOCH3), 3,394 (м, 4H; NHCH2CH2NH) ч/млн.
МС, м/з: 547 [M+H], 569 [M+Na], 585 [M+K].
Раствор Boc-Gly2-(MCM)Gly-OSu (10) (7,79 г, 16,994 ммоль) в DMSO (17 мл) и Et3N (2,83 мл, 20,4 ммоль) добавляли к перемешиваемому раствору MCMG (13) (5,06 г, 6,796 ммоль) в DMSO (13 мл). Реакционную смесь после перемешивания в течение 2 ч при температуре окружающей среды подкисляли уксусной кислотой (4,0 мл) и фракционировали колоночной хроматографией Sephadex LH-20 (объем колонки 1200 мл, элюент - MeOH/вода 2:1+0,2% AcOH). Фракции, содержащие чистый Boc2MCMG (14), комбинировали, растворители выпаривали, и остаток сушили в вакууме для получения целевого Boc2MCMG (14) в виде бесцветной пены (8,14 г, 97%). ТЖХ: Фр= 0,25 (iPrOH/этилацетат/вода 2:3:1).
1H ЯМР (500 МГц, [D6]DMSO, 30°C), смесь конформеров: 8,393-7,887 (всего 6H; 6 CONHCH2), 7,775 (м, 2H; NHCH2CH2NH), 6,996 (бр, т, 2H; 2 NHCOO), 4,299-3,730 (всего 28H; 4 CH2COO, 10 NCH2CO), 3,691 и3,633 (с, всего 12H; 4 COOCH3), 3,564 (д, Дж=5,8 Гц, 4H; 2 CH2NHCOO), 3,129 (м, 4H; NHCH2CH2NH), 1,380 (с, 18H; 2 C(CH3)3) ч/млн,
МС, м/з: 1256 [M+Na], 1271 [M+K].
Boc2MCMG (14) (606 мг, 0,491 ммоль) растворяли в CF3COOH (2 мл) и раствор выдерживали в течение 30 мин при к.т. Трифторуксусную кислоту выпаривали в вакууме и остаток экстрагировали три раза Et2O (растирание с 25 мл Et2O с последующей фильтрацией) для удаления остаточного CF3COOH и полученный белый порошок сушили в вакууме. Порошок растворяли в 4 мл воды и затем лиофилизировали. Выход MCMG (15) (соль TFA) оценивали как количественный (актуальная масса была больше чем теоретическая на ~ 10% из-за стабильности гидратов). ТЖХ: Фр=0,21 (этанол/вода/пиридин/уксусная кислота 5:1:1:1).
1H ЯМР (500 МГц, [D2]H2O, 30°C), смесь конформеров: 4,430-4,014 (всего 28H; 4 CH2COO, 10 NCH2CO), 3,911 (с, 4H; 2 COCH2NH2), 3,823 и 3,772 (с, всего 12H; 4 COOCH3), 3,386 (м, 4H; NHCH2CH2NH) ч/млн,
МС, м/з: 1034 [M+H], 1056 [M+Na].
К раствору MCMG (15) (~0,49 ммоль) в воде (20 мл) добавляли Et3N (0,5 мл) и раствор выдерживали в течение 15 ч при к.т. Реакционную смесь выпаривали досуха и остаток обессаливали на колонке Sephadex LH-20 (два метода):
Метод A. Остаток растворяли в воде (3 мл) и раствор обессаливали на колонке Sephadex LH-20 (объем колонки 250 мл, элюент - MeOH/вода 1:1+0,05 M пиридинацетат). Фракции, содержащие CMG (16), контаминированные солями, комбинировали отдельно, выпаривали и остаток обессаливали снова. Комбинированные фракции, содержащие чистый CMG (16), выпаривали до ~4 мл объема и лиофилизировали. Выход CMG (16) (внутренняя соль) составил 431 мг (90%).
Метод B. Остаток растворяли в воде (3 мл) и раствор обессаливали на колонке Sephadex LH-20 (объем колонки 250 мл, элюент - MeOH/вода 1:1+1% конц. водн. NH3). Фракции, содержащие чистый CMG (16), выпаривали до объема ~4 мл и лиофилизировали. Остаток (аммониевая соль CMG (16)) растворяли в смеси iPrOH/воды 1:1 (10 мл), добавляли Et3N (0,2 мл) и раствор выпаривали досуха. Такую процедуру повторяли дважды; остаток растворяли в 4 мл воды и лиофилизировали. Выход ди-Et3N соли CMG (16) составил 549 мг (95%).
ТЖХ: Фр=0,50 (iPrOH/MeOH/ацетoнитрил/вода 4:3:3:4+3% конц. водн. NH3), или Фр=0,43 (iPrOH/EtOH/MeOH/вода 1:1:1:1, 0,75M NH3).
1H ЯМР CMG (16) внутренняя соль (500 МГц, [D2]H2O, 30°C), смесь конформеров: 4,328-4,006 (всего 28H; 4 CH2COO, 10 NCH2CO), 3,907 (с, 4H; 2 COCH2NH2), 3,381 (м, 4H; NHCH2CH2NH) ч/млн.
МС, м/з: 977 [M+H], 999 [M+Na], 1015 [M+K].
Схема III

Схема IV

Получение H2N-CMG(16)-DOPE (20) (Схема V)
К интенсивно перемешиваемому раствору CMG (16) (425 мг, 0,435 ммоль внутренней соли) в смеси i-PrOH/воды (i-PrOH/вода 3:2, 10 мл) 1 M водн. раствор NaHCO3 (0,435 мл, 0,435 ммоль) и затем добавляли раствор DOPE-Ad-OSu (16) (211 мг, 0,218 ммоль) в дихлорэтане (0,4 мл). Реакционную смесь перемешивали в течение 2 ч и затем подкисляли 0,2 мл AcOH и выпаривали до минимального объема при 35°C. Твердый остаток сушили в вакууме (твердая пена) и затем тщательно экстрагировали смесью CHCl3/MeOH (CHCl3/MeOH 4:1, несколько раз 10 мл, ТЖ контроль). Экстрагированный остаток состоял из непрореагировавшего CMG(2) и соли (около 50% CMG (16) восстанавливали путем обессаливания комбинированного остатка и фракций после хроматографии на силикагеле в соответствии с методикой, описанной в CMG (16) синтез.). Комбинированные экстракты CHCl3/MeOH (раствор CMG (16)-Ad-DOPE амин, DOPE-Ad-CMG (16)-Ad-DOPE, N-оксисукцинимид и некоторый CMG (16)) выпаривали в вакууме и сушили. Полученную смесь разделяли на колонке из силикагеля (2,8×33 см, ~ 200 мл силикагеля в CHCl3/MeOH 5:1). Смесь размещали на колонку в смеси MeOH/CHCl3/вода (MeOH/CHCl3/вода 6:3:1+0,5% пиридина) и компоненты элюировали в пошаговом тройном градиенте композиции MeOH/CHCl3/вода от 6:3:1 до 6:2:1 и затем до 6:2:2 (все с 0,5% пиридина). DOPE-Ad-CMG(16)-Ad-DOPE элюировали первым (Фр=0,75, MeOH/CHCl3/вода 3:1:1), с последующим желательным DOPE-Ad-CMG(16) амином (Фр=0,63, MeOH/CHCl3/вода 3:1:1), последним элюировали CMG (16) (Фр=0,31, MeOH/CHCl3/вода 3:1:1). Фракции, содержащие чистый CMG(16)-Ad-DOPE амин (20), комбинировали и выпаривали досуха. Для удаления любых низкомолекулярных примесей и растворенного силикагеля остаток растворяли в смеси PrOH/вода 1:2 (2 мл), и пропускали через колонку Sephadex LH-20 (объем колонки 130 мл, элюент - iPrOH/вода 1:2+0,25% пиридина). Фракции, содержащие чистый CMG(16)-Ad-DOPE амин (20) комбинировали и выпаривали (~ 20% 2-пропанола добавляли для предотвращения пенообразования) досуха, остаток растворяли в воде (~4 мл) и лиофилизировали. Выход CMG(16)-Ad-DOPE амина (20) составил 270 мг (68% на DOPE-Ad-OSu или 34% на CMG(16)).
1H ЯМР (500 МГц, [D2]H2O/[D4]CH3OH 2:1, 30°C): 5,505 (м, 4H; 2 CH2CH=CHCH2), 5,476 (м, 1H; OCH2CHCH2O), 4,626 (дд, Джгем=11,6 Гц, 1H; OCHCHCH2O), 4,461-4,084 (всего 37H; 4 CH2COO, 11 NCH2CO, OCHCHCH2O, OCH2CH2N), 4,002 (с, 2H; COCH2NH2), 3,573 (м, 4H; NHCH2CH2NH), 2,536-2,463 (м, всего 8H; 4 CH2CO), 2,197 (м, 8H; 2 CH2CH=CHCH2), 1,807 (м, 8H; 4 CH2CH2CO), 1,480 (м, 40H; 20 CH2), 1,063 (~т, Дж≈6 Гц, 6H; 2 CH3) ч/млн,
МС, м/з: 1831 [M+H].
Получение Galili-CMG(2)-DOPE (22) (Схема VI)
К перемешиваемому раствору соединения 21 (66 мг, 0,079 ммоль) в сухом DMSO (6 мл) добавляли 15 мкл Et3N и порошкообразный H2N-CMG(2)-DOPE (20) (95 мг, 0,0495 ммоль) в 3 частях. Смесь перемешивали в течение 24 ч при комнатной температуре и затем подвергали колоночной хроматографии (Sephadex LH-20, i-PrOH-H2O, 1:2, 0,5 об% Py, 0,25 об% AcOH) для получения сырого соединения 22 в форме Py-соли; Соединение лиофилизировали из воды два раза, затем снова растворяли в 10 мл воды, добавляли водный раствор NaHCO3 (50 мM) до pH 6,5 для получения соединения 22 в форме Na-соли и раствор подвергали лиофилизации. Выход соединения 22 (Na-соли) составил 114 мг (86% на основании NH2-CMG2-DE), Фр 0,6 (i-PrOH-MeOH-MeCN-H2O, 4:3:6:4).1Н ЯМР (Фиг. 4) (700 МГц, D2O-CD3OD, 1:1 (об/об), 40°C; выбранные сигналы) δ, ч/млн: 1,05 (т, Дж 7,03 Гц, 6H; 2 CH3), 1,40-1,58 (м, 40H; 20 CH2), 1,73-1,87 (м, 12H; 2х-COCH2CH2CH2CH2CO и 2х -COCH2CH2-), 1,90-1,99 (м, 2H; OCH2CH2CH2N), 2,15-2,25 (м, 11H; 2х -CH2CH=CHCH2-, NHC(O)CH3), 2,39-2,59 (2м, всего 12H, 2х-COCH2CH2CH2CH2CO- и 2х-COCH2CH2-) 4,63 (дд, 1H, Дж 2,51, Дж 12,20, C(O)OCHHCHOCH2O-), 4,67 и 4,69 (2дх1H, Дж1,2 7,81, Дж1,2 7,95, H-1I, H-1II), 5,30 (д, 1H, Дж1,2 3,88, H-1III), 5,42-5,46 (м, 1H, -OCH2-CHO-CH2O-), 5,49-5,59 (м, 4H, 2х-CH=CH-); MALDI TOF масс-спектр, М/З 2567 (M+Na); 2583 (M+K); 2589 (MNa+Na); 2605 (MNa+K); 2611 (MNa2+Na),
Схема V

Схема VI

Пример 2: Получение соединения по формуле (II) ʺGalili-T17 DOPEʺ
Получение 3-трифторацетамидопропил-3,4-ди-О-ацетил-2,6-ди-O-бензил-α-D-галактопиранозил-(1→3)-2,4-ди-О-ацетил-6-O-бензил-β-D-галактопиранозил-(1→4)-2-ацетамидо-3-О-ацетил-6-O-бензил-2-деокси-β-D-глюкопиранозида (3) (Схема I)
Акцептор гликозила (3-трифторацетамидопропил)-2-ацетамидо-3-O-ацетил-6-O-бензил-2-деокси-4-O-(2,4-ди-O-ацетил-6-O-бензил-β-D-галактопиранозил)-β-D-глюкопиранозид (2) получали в соответствии со способом, описанным в публикации Pazynina et al (2008) Russian Journal of Bioorganic Chemistry 34(5), 625-631. Смесь акцептора гликозила 2 (500 мг, 0,59 ммоль), тиогалактопиранозида 1 (576 мг, 1,18 ммоль), NIS (267 мг, 1,18 ммоль), безводного CH2Cl2 (25 мл) и молекулярного сита 4 Α (500 мг) перемешивали при -45°C в течение 30 мин в атмосфере Ar. Затем добавляли раствор TfOH (21 мкл, 0,236 ммоль) в безводном CH2Cl2 (0,5 мл). Реакционную смесь перемешивали в течение 2 ч при -45°C и температуру затем увеличивали до -20°C в течение 4 ч. Смесь выдерживали при -20°C в течение ночи. Затем добавляли избыточные количества тиогалактопиранозида 1 (144 мг, 0,295 ммоль), NIS (66 мг, 0,295 ммоль) и TfOH (5 мкл, 0,06 ммоль) и перемешивание продолжали при -20°C в течение 2 ч позволяя медленно нагреться до к.т. (1 ч). Насыщенный водный раствор Na2S2O3 затем добавляли и смесь фильтровали. Фильтрат разводили CHCl3 (300 мл), промывали H2O (2×100 мл), сушили фильтрацией через хлопковую вату и концентрировали. Гель фильтрация на LH-20 (CHCl3-MeOH) давала продукт 3 (600 мг, 80%), в виде белой пены.
1Н ЯМР (700 МГц, CDCl3, характерные сигналы), δ, ч/млн: 1,78-1,82 (м, 4H, CHCHC, OC(O)CH3), 1,84-1,90 (м, 1H, CHCHC), 1,91, 1,94, 1,97, 1,98, 2,06 (5 с, 5×3Н, 4 OC(O)CH3, NH(O)CH3), 3,23-3,30(м, 1H, NCHH), 3,59-3,65 (м, 1H, NCHH), 4,05 (м, 1H, H-2I), 4,33 (д, 1H, Дж1,2 7,55, H-1I), 4,40 (д, 1H, Дж 12,04, PhCHH), 4,42 (д, 1H, Дж1,2 8,07, H-1II), 4,45 (д, 1H, Дж 11,92, PhCHH), 4,48 (д, 1H, Дж 12,00, PhCHH), 4,50 (д, 1H, Дж 12,00, PhCHH), 4,52 (д, 1H, Дж 12,04, PhCHH), 4,54 (д, 1H, Дж 12,00, PhCHH), 4,57 (д, 1H, Дж 12,00, PhCHH), 4,64(д, 1H, Дж 11,92, PhCHH), 4,99 (дд ≈ т, 1H, Дж 8,24, H-2II), 5,08-5,13 (м, 2H, H-3I, H-3III), 5,23 (д, 1H, Дж1,2 3,31, H-1III), 5,46 (д, 1H, Дж3,4 2,25, H-4II), 5,54 (д, 1H, Дж3,4 3,11, H-4III), 7,20-7,40 (м, 20H, ArH); 7,49-7,54 (м, 1H, NHC(O)CF3), Фр 0,4 (PhCH3-AcOEt, 1:2).
Получение 3-аминопропил-α-d-галактопиранозил-(1→3)-β-д-галактопиранозил-(1→4)-2-ацетамидо-2-деокси-β-d-глюкопиранозида (5) (Схема I)
Продукт 3 (252 мг, 0,198 ммоль) деацетилировали в соответствии с Zemplen (8 ч, 40°C), нейтрализовали AcOH и концентрировали. ТЖХ (CH3Cl-MeOH, 10:1) анализ полученного продукта показал два пятна: главное пятно с Фр 0,45, и другое на стартовой линии (нингидрин позитивное пятно), которое было показателем частичной потери трифторацетила. Следовательно, продукт был N-трифторацетилирован посредством обработки CF3COOMe (0,1 мл) и Et3N (0,01 мл) в MeOH (10 мл) в течение 1 ч, концентрировали и подвергали колоночной хроматографии на силикагеле (CHCl3-MeOH, 15:1) для получения продукта 4 в виде белой пены (163 мг, 77%), Фр 0,45 (CH3Cl-MeOH, 10:1). Продукт 4 подвергали гидрогенолизу (200 мг Pd/C, 10 мл MeOH, 2 ч), фильтровали, N-дефторацетилировали (5% Et3N/H2O, 3 ч) и концентрировали. Катионообменная хроматография на Dowex 50X4-400 (H+) (элюция с 5% водным аммиаком) давала продукт 5 (90 мг, 98%) в виде белой пены.
1Н ЯМР (D2O, характерные сигналы), δ, ч/млн: 1,94-1,98 (м, 2H, CCH2C), 2,07 (с, 3H, NHC(O)CH3), 3,11 (м, Дж 6,92, 2H, NCH2), 4,54 и 4,56 (2д, 2H, Дж1,2 8,06, Дж1,2 7,87, H-1I и H-1II), 5,16 (д, 1H, Дж1,2 3,87, H-1III), Фр 0,3 (EtOH-BuOH-Py-H2O-AcOH; 100:10:10:10:3),
Схема I
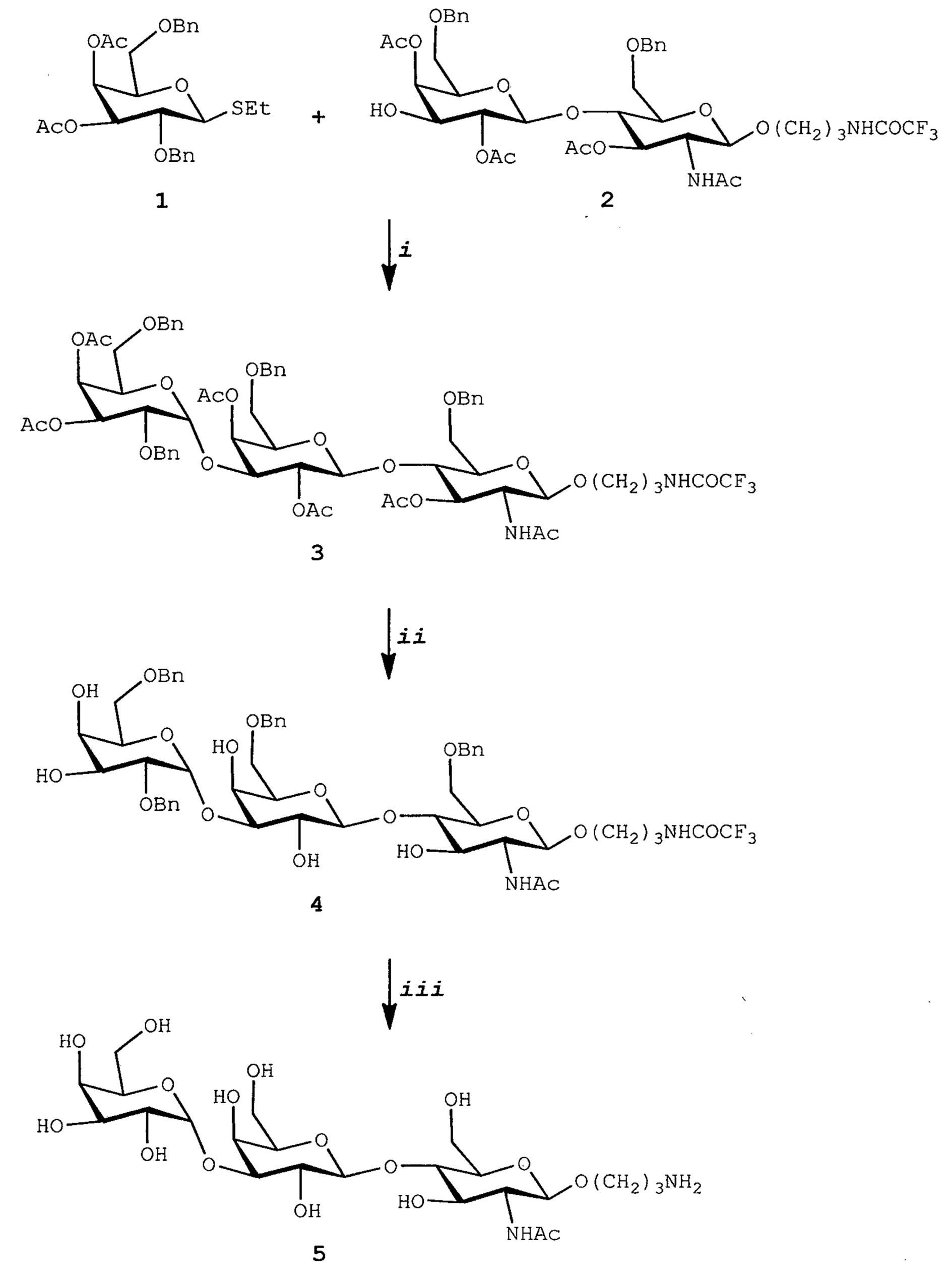
Получение (CF3COOH·H-Gly2-NHCH2)4C (9) (Схема II)
Тетраамин (H2N-CH2)4C (7) синтезировали в соответствии со способом, описанном в публикации Litherland and Mann (1938) The amino-derivatives of pentaerythritol Part I. Preparation Journal of the Chemical Society, 1588-95. К перемешиваемому раствору тетраамина 7 (500 мг, 1,52 ммоль) в смеси 1M водного NaHCO3 (18,2 мл) и i-PrOH (9 мл), Boc-GlyGlyNos (6) (4012 мг, 12,18 ммоль) добавляли (образование CO2, вспенивание). Реакционную смесь перемешивали в течение 30 мин, затем 6 мл 1M водного NaHCO3 добавляли и смесь перемешивали в течение ночи. Осадок (Boc-Gly2-HNCH2)4C (8) фильтровали, промывали тщательно смесью метанол/вода (1:1, 20 мл) и сушили в вакууме. Выход 1470 мг (98%), белое твердое вещество.
1H ЯМР (500 МГц, [D6]DMSO, 30°C) δ, ч/млн: 8,491 (т, Дж=5,6 Гц, 1H; NHCO),7,784 (т, Дж=6,6 Гц, 1H; C-CH2-NHCO), 6,858 (т, Дж=6 Гц, 1H; NHCOO), 3,696 (д, Дж=5,6 Гц, 2H; COCH2NH), 3,675 (д, Дж=6 Гц, 2H; COCH2NHCOO), 2,685 (д, Дж=6,6 Гц, 2H; C-CH2NH), 1,375 (с, 9H; C(CH3)3,
(Boc-Gly2-HNCH2)4C (8) (1450 мг, 1,466 ммоль) растворяли в CF3COOH (5 мл) и раствор держали в течение 2 ч при комнатной температуре. Трифторуксусную кислоту удаляли в вакууме и остаток три раза экстрагировали (CH3CH2)2O (легкое перемешивание с 30 мл (CH3CH2)2O в течение 30 мин, с последующим декантированием) для удаления остаточного CF3COOH. Твердый остаток сушили в вакууме, растворяли в минимальном объеме воды и пропускали через колонку Sephadex LH-20 и элюировали водой. Фракции, содержащие продукт 9, комбинировали, выпаривали до c. 5 мл и лиофилизировали. Выход 1424 мг (93%), белое твердое вещество. ТЖХ: Фр 0,5 (этанол/конц. NH3; 2:1 (об/об)).
1H ЯМР (500 МГц, [D2]H2O, 30°C) δ, ч/млн: 4,028 (с, 2H; COCH2NH), 3,972 (с, 2H; COCH2NH), 2,960 (с, 2H; C-CH2NH).
Схема II

Получение {[2-(2-трет-бутоксикарбониламиноацетиламино)ацетил]метоксикарбонилметиламино}уксусной кислоты метилового эфира (11) (Схема III)
К перемешиваемому раствору (метоксикарбонилметиламино)уксусной кислоты метилового эфира гидрохлорида (10) (988 мг, 5 ммоль) в DMF (15 мл) добавляли Boc-GlyGlyNos (6) (3293 мг, 10 ммоль) и добавляли (CH3CH2)3N (3475 мкл, 25 ммоль). Смесь перемешивали в течение ночи при комнатной температуре и затем разводили o-ксиленом (70 мл) и выпаривали. Хроматография с испарительной колонкой на силикагеле (упакованной в толуоле и элюированной этилацетатом) давала сырой продукт. Сырой продукт растворяли в хлороформа и промывали последовательно водой, 0,5 M NaHCO3 и насыщенным KCl. Экстракт хлороформа выпаривали, и продукт очищали на колонке из силикагеля (упакованной в хлороформ и элюированной 15:1 (об/об) хлороформ/метанол). Выпаривание фракций и сушка в вакууме остатка давала бесцветный густой сироп продукта 11. Выход 1785 мг, (95%). ТЖХ: Фр=0,49 (7:1 (об/об) хлороформ/метанол).
1H ЯМР (500 МГц, [D6]DMSO, 30°C) δ, ч/млн: 7,826 (т, Дж=5,1 Гц, 1H; NHCO), 6,979 (т, Дж=5,9 Гц, 1H; NHCOO), 4,348 и 4,095 (с, 2H; NCH2COO), 3,969 (д, Дж=5,1 Гц, 2H; COCH2NH), 3,689 и 3,621 (с, 3H; OCH3), 3,559 (д, Дж=5,9 Гц, 2H; COCH2NHCOO), 1,380 (с, 9H; C(CH3)3),
Получение {[2-(2-трет-бутоксикарбониламиноацетиламино)ацетил]метоксикарбонилметиламино}уксусной кислоты (12) (Схема III)
К перемешиваемому раствору 11 (1760 мг, 4,69 ммоль) в метаноле (25 мл) добавляли 0,2 M водного NaOH (23,5 мл), и раствор держали в течение 5 мин при комнатной температуре. Затем раствор подкисляли уксусной кислотой (0,6 мл) и выпаривали досуха. Колоночная хроматография остатка на силикагеле (упакованная в этилацетат и элюированная 2:3:1 (об/об/об) i-PrOH/этилацетат/вода) давала восстановленный 11 (63 мг, 3,4%) и целевое соединение 12 (1320 мг). Промежуточный продукт затем растворяли в смеси метанол/вода/пиридин (20:10:1, 30 мл) и пропускали через ионообменную колонку (Dowex 50X4-400, пиридиновая форма, 5 мл) для удаления остаточных катионов натрия. Затем колонку промывали той же смесью растворителей, элюент выпаривали, остаток растворяли в смеси хлороформ/бензол (1:1, 50 мл) и затем выпаривали и сушили в вакууме. Выход продукта 12 составил 1250 мг (74%), белое твердое вещество. ТЖХ: Фр 0,47 (4:3:1 (об/об/об) i-PrOH/этилацетат/вода).
1H ЯМР (500 МГц, [D6]DMSO, 30°C), смесь цис- и транс- конформеров N-карбоксиметилглицинового блока c.3:1. Главный конформер; δ, ч/млн: 7,717 (т, Дж=5 Гц, 1H; NHCO), 7,024 (т, Дж=5,9 Гц, 1H; NHCOO), 4,051 (с, 2H; NCH2COOCH3), 3,928 (д, Дж=5 Гц, 2H; COCH2NH), 3,786 (с, 2H; NCH2COOH), 3,616 (с, 3H; OCH3), 3,563 (д, Дж=5,9 Гц, 2H; COCH2NHCOO), 1,381 (с, 9H; C(CH3)3) ч/млн; минимальный конформер, δ=7,766 (т, Дж=5 Гц, 1H; NHCO), 7,015 (т, Дж=5,9 Гц, 1H; NHCOO), 4,288 (с, 2H; NCH2COOCH3), 3,928 (д, Дж=5 Гц, 2H; COCH2NH), 3,858 (с, 2H; NCH2COOH), 3,676 (с, 3H; OCH3), 3,563 (д, Дж=5,9 Гц, 2H; COCH2NHCOO), 1,381 (с, 9H; C(CH3)3),
Получение {[2-(2-трет-Бутоксикарбониламиноацетиламино)ацетил]метоксикарбонилметиламино}уксусной кислоты N-оксисукцинимид сложного эфира (Boc-Gly2(MCMGly)Nos) (13) (Схема III)
К ледяному перемешиваемому раствору 12 (1200 мг, 3,32 ммоль) и N-гидроксисукцинимида (420 мг, 3,65 ммоль) в DMF (10 мл) добавляли N,N`-дициклогексилкарбодиимид (754 мг, 3,65 ммоль). Смесь перемешивали при 0°C в течение 30 мин, затем в течение 2 часов при комнатной температуре. Осадок N,N`-дициклогексилмочевины фильтровали, промывали DMF (5 мл), и фильтраты выпаривали до минимального объема. Остаток затем перемешивали с (CH3CH2)2O (50 мл) в течение 1 часа и эфирный экстракт удаляли декантированием. Остаток сушили в вакууме, обеспечивая сложный эфир 13 (1400 мг, 92%) в виде белой пены. ТЖХ: Фр 0,71 (40:1 (об/об) ацетон/уксусная кислота).
1H ЯМР (500 МГц, [D6]DMSO, 30°C), смесь цис- и транс- конформеров N-карбоксиметилглицинового блока c. 3:2.
Главный конформер; δ, ч/млн: 7,896 (т, Дж=5,1 Гц, 1H; NHCO), 6,972 (т, Дж=5,9 Гц, 1H; NHCOO), 4,533 (с, 2H; NCH2COON), 4,399 (с, 2H; NCH2COOCH3), 3,997 (д, Дж=5,1 Гц, 2H; COCH2NH), 3,695 (с, 3H; OCH3), 3,566 (д, Дж=5,9 Гц, 2H; COCH2NHCOO), 1,380 (с, 9H; C(CH3)3),
Минимальный конформер; δ, ч/млн: 7,882 (т, Дж=5,1 Гц, 1H; NHCO), 6,963 (т, Дж=5,9 Гц, 1H; NHCOO), 4,924 (с, 2H; NCH2COON), 4,133 (с, 2H; NCH2COOCH3), 4,034 (д, Дж=5,1 Гц, 2H; COCH2NH), 3,632 (с, 3H; OCH3), 3,572 (д, Дж=5,9 Гц, 2H; COCH2NHCOO), 1,380 (с, 9H; C(CH3)3),
Сложный эфир 11 (1380 мг) растворяли в DMSO для обеспечения объема 6 мл и использовали в виде 0,5 M раствора (хранили при -18°C).
Схема III

Получение {CF3COOH·H-[Gly2(MCMGly)]Gly2-NHCH2}4C (15) (Схема IV)
К перемешиваемому раствору (CF3COOH·H-Gly2-HNCH2)4C (9) (277 мг, 0,265 ммоль) в DMSO (2 мл) сложный эфир 11 (1,591 ммоль, 3,18 мл 0,5 M раствора в DMSO) и добавляли (CH3CH2)3N (295 мкл, 2,121 ммоль). Смесь перемешивали в течение ночи при комнатной температуре, подкисляли 150 мкл AcOH и растворитель удаляли в вакууме (лиофилизация). Осадок экстрагировали три раза (CH3CH2)2O (легкое перемешивание с 20 мл (CH3CH2)2O в течение 30 мин с последующим декантированием). Твердый осадок растворяли в минимальном объеме ацетона и фракционировали на колонке из силикагеля (упакованной в ацетоне и элюировали ацетоном, 20:2:1 (об/об/об) ацетон/метанол/вода и 15:2:1 (об/об/об) ацетон/метанол/вода). Выбранные фракции выпаривали, и остаток сушили в вакууме. Выход чистого {Boc-[Gly2(MCMGly)]Gly2-NHCH2}4C (14) составил 351 мг (68%), белое твердое вещество. ТЖХ: Фр 0,38 (15:2:1 (об/об/об) ацетон/метанол/вода).
1H ЯМР (500 МГц, [D6]DMSO, 30°C), смесь цис- и транс- конформеров N-карбоксиметилглициновой единицы в цепи c. 3:2.
Главный конформер; δ, ч/млн: 8,593 (т, Дж=5 Гц, 1H; NHCO), 8,335 (т, Дж=5,4 Гц, 1H; NHCO), 7,821 (т, Дж=6,4 Гц, 1H; C-CH2-NHCO), 7,786 (т, Дж=5,1 Гц, 1H; NHCO), 6,993 (т, Дж=6 Гц, 1H; NHCOO), 4,139 (с, 2H; NCH2CO), 4,074 (с, 2H; NCH2COO(CH3)), 3,985 (д, Дж=5 Гц, 2H; COCH2NH), 3,887 (д, Дж=5,4 Гц, 2H; COCH2NH), 3,726 (д, Дж=5,1 Гц, 2H; COCH2NH), 3,634 (с, 3H; OCH3), 3,567 (д, Дж=6 Гц, 2H; COCH2NHCOO), 2,686 (широкий, д, Дж=6,4 Гц, 2H; C-CH2NH), 1,379 (с, 9H; C(CH3)3),
Минимальный конформер; δ, ч/млн: 8,511 (т, Дж=5 Гц, 1H; NHCO), 8,158 (т, Дж=5,4 Гц, 1H; NHCO), 7,821 (т, Дж=6,4 Гц, 1H; C-CH2-NHCO), 7,786 (т, Дж=5,1 Гц, 1H; NHCO), 6,993 (т, Дж=6 Гц, 1H; NHCOO), 4,292 (с, 2H; NCH2CO), 3,998 (с, 2H; NCH2COOCH3), 3,954 (д, Дж=5 Гц, 2H; COCH2NH), 3,826 (д, Дж=5,4 Гц, 2H; COCH2NH), 3,715 (д, Дж=5,1 Гц, 2H; COCH2NH), 3,692 (с, 3H; OCH3), 3,567 (д, Дж=6 Гц, 2H; COCH2NHCOO), 2,686 (широкий, д, Дж=6,4 Гц, 2H; C-CH2NH), 1,379 (с, 9H; C(CH3)3),
{Boc-[Gly2(MCMGly)]Gly2-NHCH2}4C (14) (330 мг, 0,168 ммоль) растворяли в CF3COOH (2 мл) и раствор держали в течение 40 мин при комнатной температуре.
Трифторуксусную кислоту выпаривали в вакууме, остаток экстрагировали три раза с (CH3CH2)2O (легкое перемешивание с 20 мл (CH3CH2)2O в течение 30 мин с последующим декантированием) для удаления остаточного CF3COOH, и затем сушили в вакууме. Выход {CF3COOH·H-[Gly2(MCMGly)]Gly2-NHCH2}4C (15) составил 337 мг (99%), белого твердого вещества.
1H ЯМР (500 МГц, [D2]H2O, 30°C), смесь цис- и транс- конформеров N-карбоксиметилглицинового блока в цепи c. 11:10.
Главный конформер; δ, ч/млн: 4,370 (с, 2H; NCH2CO), 4,265 (с, 2H; NCH2COOCH3), 4,215 (с, 2H; COCH2NH), 4,138 (с, 2H; COCH2NH), 3,968 (с, 2H; COCH2NH), 3,919 (с, 2H; COCH2NH2+), 3,775 (с, 3H; OCH3), 2,914 (с, 2H; C-CH2NH),
минимальный конформер; δ, ч/млн: 4,431 (с, 2H; NCH2CO), 4,241 (с, 2H; NCH2COOCH3), 4,239 (с, 2H; COCH2NH), 4,074 (с, 2H; COCH2NH), 3,960 (с, 2H; COCH2NH), 3,919 (с, 2H; COCH2NH2+), 3,829 (с, 3H; OCH3), 2,914 (с, 2H; C-CH2NH).
Схема IV
Получение {CF3COOH • H-[Gly2(MCMGly)]2Gly2-NHCH2}4C (Схема V)
К перемешиваемому раствору (CF3COOH·H-[Gly2(MCMGly)]Gly2-HNCH2)4C (15) (272 мг, 0,135 ммоль) в DMSO (2 мл) сложный эфир (13) (0,809 ммоль, 1,62 мл 0,5 M раствора в DMSO) и добавляли (CH3CH2)3N (112 мкл, 0,809 ммоль). Смесь перемешивали в течение ночи при комнатной температуре, подкисляли 70 мкл AcOH и растворитель удаляли в вакууме (лиофилизация). Остаток экстрагировали три раза с (CH3CH2)2O (легкое перемешивание с 15 мл (CH3CH2)2O в течение 30 мин с последующим декантированием). Твердый остаток растворяли в минимальном объеме смеси 7:1 (об/об) ацетон/метанол и фракционировали на колонке из силикагеля (упакованной в ацетоне и элюированной 7:1 (об/об) ацетон/метанол, 10:2:1 (об/об/об), 9:2:1 (об/об/об), 8:2:1 (об/об/об) ацетон/метанол/вода). Выбранные фракции выпаривали, и остаток сушили в вакууме. Выход чистого {Boc-[Gly2(MCMGly)]2Gly2-NHCH2}4C (16) составил 279 мг (71%), белое твердое вещество. ТЖХ: Фр 0,42 (8:2:1 (об/об/об) ацетон/метанол/вода).
1H ЯМР (500 МГц, [D6]DMSO, 30°C), смесь конформеров двух N-карбоксиметилглициновых блоков на цепь, δ, ч/млн: 8,604, 8,519, 8,397, 8,388, 8,346, 8,211, 8,200, 8,167, 8,034, 8,024, 7,925, 7,912, 7,819 и 7,773 (т, 6H; 6 NHCO), 6,992 (т, Дж=5,9 Гц, 1H; NHCOO), 4,302-3,723 (18H; 2 NCH2CO, 2 NCH2COOCH3, 5 COCH2NH), 3,692, 3,689 и 3,632 (с, 6H; 2 OCH3), 3,566 (д, Дж=5,9 Гц, 2H; COCH2NHCOO), 2,686 (широкий, д, 2H; C-CH2NH), 1,380 (с, 9H; C(CH3)3).
{Boc-[Gly2(MCMGly)]2Gly2-NHCH2}4C (16) (269 мг, 91,65 мкмоль) растворяли в CF3COOH (2 мл) и раствор держали в течение 40 мин при комнатной температуре. Трифторуксусную кислоту выпаривали в вакууме, остаток экстрагировали три раза с (CH3CH2)2O (легкое перемешивание с 15 мл (CH3CH2)2O в течение 30 мин с последующим декантированием) для удаления остаточного CF3COOH, и затем сушили в вакууме. Выход {CF3COOH·H-[Gly2(MCMGly)]2Gly2-NHCH2}4C составил 270 мг (98%), белое твердое вещество.
1H ЯМР (500 МГц, [D2]H2O, 30°C), смесь конформеров по двум N-карбоксиметилглициновым блокам на цепь, δ, ч/млн: 4,441-3,963 синглеты, 18H; 2 NCH2CO, 2 NCH2COOCH3, 5 COCH2NH), 3,920 (с, 2H; COCH2NH2+), 3,833, 3,824, 3,780 и 3,773 (с, 6H; 2 OCH3), 2,918 (с, 2H; C-CH2NH).
Получение {CF3COOH • H-[Gly2(MCMGly)]3Gly2-NHCH2}4C (Схема V)
К перемешиваемому раствору (CF3COOH·H-[Gly2(MCMGly)]2Gly2-HNCH2)4C (175 мг, 58,5 мкмоль) в DMSO (2 мл) сложный эфир 13 (0,351 ммоль, 0,702 мл 0,5 M раствор в DMSO) и добавляли (CH3CH2)3N (49 мкл, 0,351 ммоль). Смесь перемешивали в течение ночи при комнатной температуре, подкисляли 30 мкл AcOH и растворитель выпаривали в вакууме (лиофилизация). Остаток растворяли в минимальном объеме смеси 1:1 (об/об) ацетoнитрила/воды и фракционировали на колонке Sephadex LH-20 (элюировали 1:1 (об/б) ацетoнитрила/воды). Выбранные фракции выпаривали, и остаток сушили в вакууме. Выход чистого {Boc-[Gly2(MCMGly)]3Gly2-NHCH2}4C составил 279 мг (71%), белое твердое вещество. ТЖХ: Фр 0,42 (8:2:1 (об/об/об) ацетон/метанол/вода). Фракции, содержащие {Boc-[Gly2(MCMGly)]3Gly2-NHCH2}4C комбинировали, выпаривали до объема c. 2 мл и лиофилизировали. Начальный выход составил 215 мг (94%). Дополнительная очистка на колонке из силикагеля (упакованной в ацетонитриле и элюированной с 4:5:2 (об/об/об) i-PrOH/ацетoнитрил/вода) давала 169 мг Boc-[Gly2(MCMGly)]3Gly2-NHCH2}4C (выход 74%, белое твердое вещество). ТЖХ: Фр 0,45 (4:5:2 (об/об/об) i-PrOH/ацетонитрил/вода).
1H ЯМР (500 МГц, [D6]DMSO, 30°C), смесь конформеров по трем N-карбоксиметилглициновым блокам на цепь, δ, ч/млн: 8,594-7,772 (триплеты, вместе 8H; 8 NHCO), 6,989 (т, Дж=5,6 Гц, 1H; NHCOO), 4,303-3,722 (26H; 3 NCH2CO, 3 NCH2COOCH3, 7 COCH2NH), 3,692 и 3,632 (с, 9H; 3 OCH3), 3,565 (д, Дж=5,6 Гц, 2H; COCH2NHCOO), 2,687 (широкий, д, 2H; C-CH2NH), 1,380 (с, 9H; C (CH3)3).
{Boc-[Gly2(MCMGly)]3Gly2-NHCH2}4C (146 мг, 37,36 мкмоль) растворяли в CF3COOH (1 мл) и раствор держали в течение 40 мин при комнатной температуре. Трифторуксусную кислоту выпаривали в вакууме, остаток экстрагировали три раза (CH3CH2)2O (легкое перемешивание с 10 мл (CH3CH2)2O в течение 30 мин с последующим декантированием) для удаления остаточного CF3COOH, и затем сушили в вакууме. Выход {CF3COOH·H-[Gly2(MCMGly)]3Gly2-NHCH2}4C составил 147 мг (99%), белое твердое вещество.
1H ЯМР (500 МГц, [D2]H2O, 30°C), смесь конформеров по трем N-карбоксиметилглициновым блокам на цепь, δ, ч/млн: 4,446-3,964 (синглеты, 26H; 3 NCH2CO, 3 NCH2COOCH3, 7 COCH2NH), 3,924 (с, 2H; COCH2NH2+), 3,836, 3,828, 3,824, 3,783, 3,778 и 3,773 (с, 9H; 3 OCH3), 2,919 (с, 2H; C-CH2NH).
Получение {CF3COOH • H-[Gly2(MCMGly)]4Gly2-NHCH2}4C (Схема V)
К перемешиваемому раствору (CF3COOH·H-Gly2(MCMGly)]3-HNCH2)4C (68 мг, 17,16 мкмоль) в DMSO (1 мл) сложный эфир 13 (0,137 ммоль, 0,275 мл 0,5 M раствора в DMSO) и добавляли (CH3CH2)3N (14,3 мкл, 0,103 ммоль). Смесь перемешивали в течение ночи при комнатной температуре, подкисляли 100 мкл AcOH и растворитель удаляли в вакууме (лиофилизация). Остаток растворяли в минимальном объеме смеси 1:1 (об/об) ацетoнитрил/вода (0,25% AcOH) и фракционировали на колонке Sephadex LH-20 (элюировали с 1:1 (об/об) ацетoнитрила/вода (0,25% AcOH)). Фракции, содержащие {Boc-[Gly2(MCMGly)]4Gly2-NHCH2}4C, комбинировали, выпаривали до объема c. 2 мл и лиофилизировали. Выход составил 81 мг (96%), белое твердое вещество. ТЖХ: Фр 0,24 (4:5:2 (об/об/об) i-PrOH/ацетoнитрил/вода).
1H ЯМР (500 МГц, [D6]DMSO, 30°C), смесь конформеров по четырем N-карбоксиметилглициновым блокам на цепь, δ, ч/млн: 8,590-7,773 (триплеты, 10H; 10 NHCO), 6,989 (т, Дж=5,6 Гц, 1H; NHCOO), 4,303-3,722 (34H; 4 NCH2CO, 4 NCH2COOCH3, 9 COCH2NH), 3,691 и 3,631 (с, 12H; 4 OCH3), 3,565 (д, Дж=5,6 Гц, 2H; COCH2NHCOO), 2,684 (широкий, д, 2H; C-CH2NH), 1,379 (с, 9H; C(CH3)3).
{Boc-[Gly2(MCMGly)]4Gly2-NHCH2}4C (74 мг, 15,16 мкмоль) растворяли в CF3COOH (1 мл) и раствор держали в течение 40 мин при комнатной температуре. Трифторуксусную кислоту выпаривали в вакууме, остаток экстрагировали три раза с (CH3CH2)2O (легкое перемешивание с 10 мл (CH3CH2)2O в течение 30 мин с последующим декантированием) для удаления остаточного CF3COOH, и затем сушили в вакууме. Выход {CF3COOH·H-[Gly2(MCMGly)]4Gly2-NHCH2}4C составил 72 мг (96%), белое твердое вещество.
1H ЯМР (500 МГц, [D2]H2O, 30°C), смесь конформеров по четырем N-карбоксиметилглициновым блокам на цепь, δ, ч/млн: 4,446-3,964 (синглеты, 34H; 4 NCH2CO, 4 NCH2COOCH3, 9 COCH2NH), 3,925 (с, 2H; COCH2NH2+), 3,836, 3,829, 3,827, 3,822, 3,783, 3,779, 3,777 и 3,772 (с, 12H; 4 OCH3), 2,919 (с, 2H; C-CH2NH),
Получение {CF3COOH • H-[Gly2(MCMGly)]5Gly2-NHCH2}4C (23) (Схема V)
К перемешиваемому раствору (CF3COOH·H-Gly2(MCMGly)]4-HNCH2)4C (16,8 мг, 3,403 мкмоль) в DMSO (1 мл) сложный эфир 13 (27,2 мкмоль, 63 мкл 0,5 M раствора в DMSO) и добавляли (CH3CH2)3N (3 мкл, 21,6 мкмоль). Смесь перемешивали в течение ночи при комнатной температуре, подкисляли 100 мкл AcOH и растворитель выпаривали в вакууме (лиофилизация). Остаток растворяли в минимальном объеме смеси 1:1 (об/об) ацетoнитрила/воды (0,25% AcOH) и фракционировали на колонке Sephadex LH-20 (элюированной с 1:1 (об/об) ацетoнитрила/воды (0,25% AcOH)). Фракции, содержащие {Boc-[Gly2(MCMGly)]5Gly2-NHCH2}4C (22), комбинировали, выпаривали до объема c. 1 мл и лиофилизировали. Выход составил 19 мг (95%), белое твердое вещество. ТЖХ: Фр 0,15 (4:3:2 (об/об/об) i-PrOH/ацетoнитрил/вода).
1H ЯМР (500 МГц, [D6]DMSO, 30°C), смесь конформеров пяти N-карбоксиметилглициновых блоков на цепь, δ, ч/млн: 8,595-7,772 (триплеты, 12H; 12 NHCO), 6,989 (т, Дж=5,6 Гц, 1H; NHCOO), 4,303-3,723 (42H; 5 NCH2CO, 5 NCH2COOCH3, 11 COCH2NH), 3,692 и 3,631 (с, 15H; 5 OCH3), 3,565 (д, Дж=5,6 Гц, 2H; COCH2NHCOO), 2,686 (широкий, д, 2H; C-CH2NH), 1,380 (с, 9H; C(CH3)3).
{Boc-[Gly2(MCMGly)]5Gly2-NHCH2}4C (22) (19 мг, 3,25 мкмоль) растворяли в CF3COOH (0,5 мл) и раствор держали в течение 40 мин при комнатной температуре. Трифторуксусную кислоту выпаривали в вакууме, остаток экстрагировали три раза с (CH3CH2)2O (легкое перемешивание с 5 мл (CH3CH2)2O в течение 30 мин с последующим декантированием) для удаления остаточного CF3COOH, и затем сушили в вакууме. Выход {CF3COOH·H-[Gly2(MCMGly)]5Gly2-NHCH2}4C (23) составил 20 мг (99%), белое твердое вещество.
1H ЯМР (500 МГц, [D2]H2O, 30°C), смесь конформеров по пяти N-карбоксиметилглициновым блокам на цепь, δ, ч/млн: 4,446-3,965 (синглеты, 42H; 5 NCH2CO, 5 NCH2COOCH3, 11 COCH2NH), 3,924 (с, 2H; COCH2NH2+), 3,835, 3,829, 3,827, 3,825, 3,823, 3,783, 3,779, 3,777 и 3,773 (с, 15H; 5 OCH3), 2,919 (с, 2H; C-CH2NH),
Получение [CF3COOH+H-(Gly2CMGly)5Gly2-NHCH2]4C, Et3N-соль (24) (Схема V)
К раствору продукта 23 (463 мг, 0,07835 ммоль) в воде (26 мл), добавляли Et3N (523 мкл, 3,761 ммоль) и раствор выдерживали в течение 18 ч при к.т. После выпаривали остаток лиофилизировали в вакууме. Выход продукта 24 составил 587 мг (98%), белое твердое вещество. ТЖХ: Фр 0,39 (1:2:1 (об/об/об) CHCl3/MeOH/вода).
1H ЯМР (600 МГц, [D2]H2O, 30°C) δ, ч/млн: 4,309-3,919 (176 H; 20 NCH2CO, 20 NCH2COOH, 48 COCH2NH), 3,226 (кв, 120 H, Дж=7,3 Гц; 60 NCH2CH3), 2,964 (широкий, с, 8 H; 4 C-CH2NH), 1,305 (т, 180 H, Дж=7,3 Гц; 60 NCH2CH3).
MALDI TOF масс-спектр, М/З: 5174, M+H; 5196, M+Na.
Схема V

Получение активированного 1,2-O-диолеоил-sn-глицеро-3-фосфатидилэтаноламина (DE-Ad-OSu) (27) (Схема VI)
К раствору бис(N-гидроксисукцинимидил)адипата (25) (70 мг, 205 мкмоль) в сухом N,N-диметилформамиде (1,5 мл), добавляли 1,2-O-диолеоил-sn-глицеро-3-фосфатидилэтаноламин (7) (40 мкмоль) в хлороформе (1,5 мл), с последующим триэтиламином (7 мкл). Смесь держали в течение 2 ч при комнатной температуре, затем нейтрализовали уксусной кислотой и частично концентрировали в вакууме. Колоночная хроматография (Sephadex LH-20, 1:1 хлороформ-метанол, 0,2% уксусная кислота) остатка давала продукт 27 (37 мг, 95%) в виде бесцветного сиропа.
Схема VI

1H ЯМР (CDCl3/CD3OD, 2:1) 5,5 (м, 4H, 2х(-CH=CH-), 5,39 (м, 1H, -OCH2-CHO-CH2O-), 4,58 (дд, 1H, Дж=3,67, Дж=11,98, -CCOOHCH-CHO-CH2O-), 4,34 (дд, 1H, Дж=6,61, Дж=11,98, -CCOOHCH-CHO-CH2O-), 4,26 (м, 2H, PO-CH2-CH2-NH2), 4,18 (м, 2H, -CH2-OP), 3,62 (м, 2H, PO-CH2-CH2-NH2), 3,00 (с, 4H, ONSuc), 2,8 (м, 2H, -CH2-CO (Ad), 2,50 (м, 4H, 2х(-CH2-CO), 2,42 (м, 2H, -CH2-CO (Ad), 2,17 (м, 8H, 2х(-CH2-CH=CH-CH2-), 1,93 (м, 4H, COCH2CH2CH2CH2CO), 1,78 (м, 4H, 2х(COCH2CH2-), 1,43, 1,47 (2 бс, 40H, 20 CH2), 1,04 (м, 6H, 2 CH3), Фр 0,5 (хлороформ-метанол-вода, 6:3:0,5.
Получение [H-(Gly2CMGly)5Gly2-NHCH2]3[DE-CO(CH2)4CO-(Gly2CMGly)5Gly2-NHCH2]C, Na, Et3N-соли (28) (Схема VI)
К перемешиваемому раствору продукта 24 (522 мг, 0,06821 ммоль) в смеси вода/2-пропанол (16 мл, 2:3) 1M NaHCO3 (547 мкл, 0,547 ммоль) и добавляли раствор DE-Ad-OSu (27) (66,1 мг, 0,06821 ммоль) в дихлороэтане (368 мкл) и раствор перемешивали в течение 1,5 ч при к.т. После подкисления AcOH (94 мкл) раствор выпаривали и остаток сушили в вакууме. Высушенную смесь растворяли в 3 мл вода/MeOH (15:1) и помещали на колонку с обращенной фазой C18 (~45 мл фазы промывали 75% MeOH и затем вода/MeOH 15:1). Вещества элюировали последовательно водой/MeOH (15:1-50 мл; 9:1-50 мл; 7,5:2,5-50 мл; 1:1-50 мл; 2,5:7,5-100 мл). Непрогреагировавший 24 элюировали водой/MeOH 15:1 (Na соль по данным ЯМР, 116 мг, 30,8% восстановления) и водой/MeOH 9:1 (Et3N соль по ЯМР данным, 63 мг, 13,6% восстановления). Цель (H-CMG5)3C(CMG5-Ad-DE) (28) элюировали водой/MeOH 1:1. Выход чистого лиофилизированного продукта 28 составил 135 мг (25,5% на (24)), белое твердое вещество. ТЖХ (1:2:1 (об/об/об) MeOH/этил ацетат/вода): 24 Фр 0,06; 28 Фр 0,17.
(H-CMG5)3C(CMG5-Ad-DE) Na1(Et3N)20 (28):1H ЯМР (700 МГц, [D2]H2O/[D4]CH3OH 2:1 (об/об), 30°C) δ, ч/млн: 5,561 (м, 4 H; 2 цис CH-CH DE), 5,454 (м, 1 H; OCH2-CH(OCO)CH2O DE), 4,629 (дд, 1 H, Дж=12,3 Гц/2 Гц; OCH2-CH(OCO)CHOCO DE), 4,462-4,057 (181 H; 20 NCH2CO, 20 NCH2COOH, 48 COCH2NH, OCH2-CH(OCO)CHOCO DE, OCH2CH2NH DE), 3,597 (т, 2 H, Дж= 5 Гц; OCH2CH2NH DE), 3,226 (кв, 102 H, Дж=7,3 Гц; 51 NCH2CH3), 3,099 (широкий, с, 8 H; 4 C-CH2NH), 2,557, 2,532, 2,522 и 2,456 (триплеты, всего 8 H; 4 CO-CH2CH2), 2,203 (~дд, 8 H, Дж=12 Гц/5,8 Гц; 2 CH2-CH=CH-CH2 DE), 1,807 и 1,783 (мультиплеты, 8 H; 4 CO-CH2CH2), 1,526 и 1,475 (перекрывающиеся м и т, всего 193 H; м, 20 CH2 DE; т, Дж=7,3 Гц, 51 NCH2CH3), 1,063 (т, 6 H, Дж= 7 Гц; 2 CH3 DE).
MALDI TOF масс спектр, М/З: 6028, M+H; 6050, M+Na.
Схема VII

Получение Galili-T-17-DE (30)(Схема VII)
Соединение 28 (4,3 мг, 5 мкмоль) и Et3N (0,5 мкл) в H2O (0,75 мл) добавляли к перемешиваемому раствору соединения 29 (5 мг, 6 мкмоль) в сухом DMSO (0,3 мл) в 3 порциях в течение 1,5 ч. Смесь перемешивали в течение 24 ч при комнатной температуре и затем подвергали колоночной хроматографии (Sephadex LH-20, MeOH-H2O, 3:7) для получения сырого продукта 30. Продукт лиофилизировали из воды, остаток растворяли в 3 мл воды, водный раствор NaHCO3 (10 мM) добавляли до pH 6,5 и раствор лиофилизировали для получения 3,7 мг соединения 30 в виде Na-соли.
1H ЯМР (700 МГц, D2O/CD3OD, 2:1 (об/об), выбранные химические сдвиги) δ, ч/млн: 1,06 (т, Дж 7,03 Гц, CH3 DE), 1,28-1,61 (м, CH2 DE), 1,71-1,88 (м, -COCH2CH2CH2CH2CO и -COCH2CH2-), 1,90-1,99 (м, OCH2CH2CH2N), 2,13-2,27 (м, -CH2CH=CHCH2-, NHC(O)CH3), 2,35-2,58 (м, COCH2CH2CH2CH2CO- и -COCH2CH2-), 2,93-3,24 (широкий, с, 8 H; 4 C-CH2NH), 4,63 (дд, Дж 2,49, Дж 12,32, C(O)OCHHCHOCH2O-), 4,67 и 4,70 (2д, Дж1,2 7,81, Дж1,2 7,95, H-1I, H-1II), 5,30 (д, Дж1,2 3,92, H-1III), 5,42-5,47 (м, -OCH2-CHO-CH2O-), 5,52-5,58 (м, 4H, 2х-CH=CH-), MALDI TOF масс-спектр, М/З: 8188 (M+Na); 8204 (M+K); 8226 (MNa+K),
Пример 3: Получение соединения по формуле (III) ʺGalNAc-Gal-GlcNAc-Ad-DOPEʺ
Получение 3-аминопропил 2-ацетамидо-2-деокси-α-D-галактопиранозил-(1→3)-β-D-галактопиранозил-(1→4)-2-ацетамидо-2-деокси-β-D-глюкопиранозида (5) (Схема I)
Гликозилхлорид 3,4,6-три-O-ацетил-2-азидо-2-дезокси-β-D-галактопиранозилхлорида (1) получали в соответствии со способом, описанным в публикации Paulsen et al (1978) Darstellung selektiv blockierter 2-azido-2-desoxy-d-gluco-und-d-galactophyranosylhalogenide: Reaktivitat und 13C-NMR-Spektren Carbohydrate Research, 64, 339-364. Гликозильный акцептор (3-трифторацетамидопропил)-2-ацетамидо-3-O-ацетил-6-O-бензил-2-деокси-4-O-(2,4-ди-O-ацетил-6-O-бензил-β-D-галактопиранозил)-β-D-глюкопиранозид (2) получали в соответствии со способом, описанным в публикации Pazynina et al (2008) Russian Journal of Bioorganic Chemistry 34(5), 625-631.
Раствор гликозильного акцептора (420 мг, 0,5 ммоль), тритрат серебра (257 мг, 1,0 ммоль), тетраметилмочевину (120 мкл, 1,0 ммоль) и свеже кальцинированные молекулярные сита 4 Α в сухом дихлорметане (20 мл), перемешивали при комнатной температуре в темноте в течение 30 мин. Другую порцию сита 4 Α добавляли и добавляли раствор гликозилхлорида (350 мг, 1,0 ммоль) в сухом дихлорметане (3 мл). Смесь перемешивали в течение 20 ч при комнатной температуре. Смолу обрабатывали и промывали метанолом (4×10 мл), затем растворитель выпаривали. Хроматография на силикагеле (элюция 5-7% изопропанолом в хлороформе) давала 407 мг (70%) продукта 3 в виде смеси аномеров (α/β=3,0 что определяли по1H-ЯМР спектроскопии).
Раствор продукта 3 (407 мг, 0,352 ммоль) в метаноле (30 мл) подвергали гидрогенолизу над 400 мг 10% Pd/C в течение 16 ч. Затем смолу фильтровали, промывали метанолом (4×10 мл) и продукт концентрировали в вакууме. Сухой остаток ацетилировали со смесью 2:1 пиридина-уксусного ангидрида (6 мл) при 20°C в течение 16 ч, реагенты ко-выпаривали с толуолом. Двухстадийная хроматография на силикагеле (элюция с 10% изопропанолом в этилацетате и с 5-10% метанолом в хлороформе) приводила к 160 мг (42%) продукта 4 и 39 мг (10%) продукта 4β.
Раствор 2 M метилата натрия в метаноле (200 мкл) добавляли к раствору продукта 4 (160 мг, 0,149 ммоль) в сухом метаноле (4 мл). Раствор выпаривали после 1 ч, добавляли 4 мл воды и раствор держали в течение 16 ч до хроматографирования на колонке Dowex-H+ (элюция с 1 M аммония). Элюат выпаривали, лиофилизировали для получения 87,2 мг (91%) 3-аминопропилтрисахарида (5).
1H ЯМР спектр записывали на спектрометре Bruker BioSpin GmbH при 303K. Химические сдвиги (δ) для характерных протонов представлены в ч/млн с использованием HOD (4,750), CHCl3 (δ 7,270) в виде ссылки. Константы связывания (J) представлены в Гц. Сигналы в1H ЯМР спектров распределены с использованием методики спин-спин расцепления (двойной резонанс) и 2D-1H,1H-COSY эксперименты.
Значения оптического вращения измеряли на цифровом полариметре Perkin Elmer 341 при 25°C. Масс спектры регистрировали на спектрометре MALDI-TOF Vision-2000 с использованием дигидроксибензойной кислоты в качестве матрицы.
4:1H-ЯМР (700 МГц, CDCl3): 1,759-1,834 (м, 1H, CH сп); 1,853-1,927 (м, 1H, CH сп); 1,972, 1,986, 1,996, 2,046, 2,053, 2,087, 2,106, 2,115, 2,130, 2,224 (10с, 10×3H, COCH3); 3,222-3,276 (м, 1H, NCH сп); 3,544-3,583 (м, 1H, OCH сп); 3,591-3,661 (м, 2H, NCH сп, H-5a); 3,764 (дд ≈ т, 1H, H-4a, Дж 8,8); 3,787 (дд, 1H, H-3b, Дж3,4 3,7, Дж2,39,9); 3,836 (шир, т, 1H, H-5b, Дж 7,3); 3,882-3,920 (м, 1H, OCH сп); 3,950 (дд, 1H, H-6'c, Дж6',6'' 10,6, Дж5,6' 5,2); 4,009 (ддд, 1H, H-2a, Дж1,2 7,9, Дж2,3 10,0, Дж2,NH 9,0); 4,076-4,188 (м, 5H, H-6'a, H-6'b, H-6ʺb, H-5c, H-6ʺc); 4,415 (д, 1H, H-1a, Дж1,2 7,9); 4,443 (д, 1H, H-1b, Дж1,2 7,9); 4,529 (дд, 1H, H-6ʺa, Дж6',6'' 12,0, Дж5,6ʺ 2,5); 4,548 (ддд, 1H, H-2c, Дж1,2 3,4, Дж2,3 11,6, Дж2,NH 9,4); 4,893 (дд, 1H, H-3c, Дж3,4 3,1, Дж2,3 11,6); 5,021 (д, 1H, H-1c, Дж1,2 3,4); 5,039-5,075 (м, 2H, H-3a, H-2b); 5,339 (дд ≈ д, 1H, H-4b, Дж 2,9); 5,359 (дд, 1H, H-4c, Дж3,4 2,7, Дж4,50,9); 5,810 (д, 1H, NHAc a, Дж2,NH 9,0); 6,184 (д, 1H, NHAc c, Дж2,NH 9,4); 7,310-7,413 (м, 1H, NHCOCF3 сп), Фр 0,31 (EtOAc-iPrOH, 10:1), МС, м/з рассчитанное для [C43H60N3F3O25]H+: 1076,35, обнаруженный 1076,
4β:1H-ЯМР (700 МГц, CDCl3): 1,766-1,832 (м, 1H, CH сп); 1,850-1,908 (м, 1H, CH сп); 1,923, 1,969, 1,982, 2,059, 2,071, 2,099 (2), 2,120, 2,136, 2,148 (10с, 10×3H, COCH3); 3,230-3,289 (м, 1H, NCH сп); 3,521 (ддд, 1H, H-2c, Дж1,2 8,2, Дж2,3 11,2, Дж2,NH 7,8); 3,548-3,591 (м, 1H, OCH сп); 3,591-3,648 (м, 2H, NCH сп, H-5a); 3,743 (дд ≈ т, 1H, H-4a, Дж 8,6); 3,795 (шир, т, 1H, H-5b, Дж 6,5); 3,852 (дд, 1H, H-3b, Дж3,4 3,6, Дж2,3 9,9); 3,873-3,923 (м, 2H, H-5c, OCH сп); 4,002 (ддд, 1H, H-2a, Дж1,2 8,0, Дж2,3 9,5, Дж2,NH 8,9); 4,039 (дд, 1H, H-6'b, Дж6',6'' 11,6, Дж5,6' 6,9); 4,087-4,144 (м, 3H, H-6'a, H-6ʺb, H-6'c); 4,160 (дд, 1H, H-6ʺc, Дж6',6'' 11,2, Дж5,6ʺ 6,0); 4,409, 4,417 (2д ≈ т, 2×1H, H-1a, H-1b, Дж 7,6); 4,519 (дд, 1H, H-6ʺa, Дж6',6'' 11,8, Дж5,6ʺ 2,5); 4,992 (д, 1H, H-1c, Дж1,2 8,2); 5,043 (дд, 1H, H-3a, Дж3,4 8,6, Дж2,3 9,5); 5,066 (дд, 1H, H-2b, Дж1,2 8,0, Дж2,3 9,8); 5,350 (дд ≈ д, 1H, H-4c, Дж 3,2); 5,372 (дд ≈ д, 1H, H-4b, Дж 3,4); 5,399 (д, 1H, NHAc c, Дж2,NH 7,8); 5,449 (дд, 1H, H-3c, Дж3,4 3,4, Дж2,3 11,3); 5,856 (д, 1H, NHAc a, Дж2,NH 8,9); 7,361-7,466 (м, 1H, NHCOCF3 сп), Фр 0,24 (EtOAc-iPrOH, 10:1), МС, м/з рассчитанная для [C43H60N3F3O25]H+: 1076,35, обнаруженная 1076,
5:1H-ЯМР (700 МГц, D2O): 1,924-2,002 (м, 2H, CH2 сп); 2,060, 2,064 (2с, 2×3H, NCOCH3); 3,102 (м ≈ т, 2H, NCH2 сп, Дж 6,8); 3,592-3,644 (м, 1H, H-5a); 3,655 (дд, 1H, H-2b, Дж1,2 7,9, Дж2,3 9,9); 3,702 (шир, дд, 1H, H-5b, Дж5,6' 3,8, Дж5,6ʺ 8,2, Дж4,5≤1); 3,713-3,815 (м, 9H); 3,846 (дд, 1H, H-6'a, Дж6',6'' 12,3, Дж5,6' 5,3); 3,984-4,062 (м, 4H, OCH сп, H-6ʺa, H-4b, H-3c); 4,123 (дд ≈ д, 1H, H-4c, Дж 2,9); 4,206 (шир, т, 1H, H-5c, Дж 6,3); 4,248 (дд, 1H, H-2c, Дж1,2 3,6, Дж2,3 11,0); 4,542 (2д ≈ т, 2H, H-1a, H-1b, Дж 7,4); 5,100 (д, 1H, H-1c, Дж1,2 3,5), Фр 0,55 (MeOH-1M водн, Py-AcOH, 5:1), МС, м/з рассчитанные для [C25H45N3O16]H+: 644,28; обнаруженные 644, [α]546 нм +128 (c 0,3; MeCN-H2O, 1:1),
5β:1H-ЯМР (700 МГц, D2O): 1,938-1,991 (м, 2H, CH2 сп); 2,055, 2,062 (2с, 2×3H, NCOCH3); 3,100 (м ≈ т, 2H, NCH2 сп, Дж 6,9); 3,610 (дд, 1H, H-2b, Дж1,2 7,9, Дж2,3 9,9); 3,603-3,636 (м, 1H, H-5a); 3,682 (шир, дд, 1H, H-5b, Дж5,6' 4,9, Дж5,6ʺ 7,8, Дж4,5≤1); 3,693-3,826 (м, 11H); 3,842 (дд, 1H, H-6'a, Дж6',6'' 12,1, Дж5,6' 5,2); 3,934-3,972 (м, 2H, H-4b, H-2c); 4,012 (дд, 1H, H-6ʺa, Дж6',6'' 12,2, Дж5,6ʺ 2,0); 4,023-4,057 (м, 1H, OCH сп); 4,175 (дд ≈ д, 1H, H-4c, Дж 2,9); 4,478 (д, 1H, H-1b, Дж1,2 7,9); 4,531 (д, 1H, H-1a, Дж1,2 8,1); 4,638 (д, 1H, H-1c, Дж1,2 8,4), Фр 0,48 (MeOH-1M водн, Py-AcOH, 5:1), МС, м/з рассчитанные для [C25H45N3O16]H+: 644,28; обнаруженные 644, [α]546 нм +6 (c 0,3; MeCN-H2O, 1:1),
Схема I

Получение активированного 1,2-O-диолеоил-sn-глицеро-3-фосфатидилэтаноламина (DOPE-Ad-ONSu) (8) (Схема II)
К раствору бис(N-гидроксисукцинимид)адипата (6) (70 мг, 205 мкмоль) в сухом N,N-диметилформамиде (1,5 мл), добавляли 1,2-O-диолеоил-sn-глицеро-3-фосфатидилэтаноламин (7) (40 мкмоль) в хлороформе (1,5 мл), с последующим триэтиламином (7 мкл). Смесь держали в течение 2 ч при комнатной температуре, затем нейтрализовали уксусной кислотой и частично концентрировали в вакууме. Колоночная хроматография (Sephadex LH-20, 1:1 хлороформ-метанол, 0,2% уксусная кислота) остатка давала продукт 8 (37 мг, 95%) в виде бесцветного сиропа.
1H ЯМР спектры получали на спектрометре Bruker DRX-500. Химические сдвиги представлены в ч/млн (δ) относительно CD3OD. ТЖХ проводили на планшетах из силикагеля 60 F254 (Merck) с соединениями, определяемыми по окрашиванию 8% фосфорной кислотой в воде с последующим нагреванием более 200°C.
8:1H ЯМР (CDCl3/CD3OD, 2:1) 5,5 (м, 4H, 2х(-CH=CH-), 5,39 (м, 1H, -OCH2-CHO-CH2O-), 4,58 (дд, 1H, Дж=3,67, Дж=11,98-CCOOHCH-CHO-CH2O-), 4,34 (дд, 1H, Дж=6,61, Дж=11,98, -CCOOHCH-CHO-CH2O-), 4,26 (м, 2H, PO-CH2-CH2-NH2), 4,18 (м, 2H, -CH2-OP), 3,62 (м, 2H, PO-CH2-CH2-NH2), 3,00 (с, 4H, ONSuc), 2,8 (м, 2H, -CH2-CO (Ad), 2,50 (м, 4H, 2х(-CH2-CO), 2,42 (м, 2H, -CH2-CO (Ad), 2,17 (м, 8H, 2х(-CH2-CH=CH-CH2-), 1,93 (м, 4H, COCH2CH2CH2CH2CO), 1,78 (м, 4H, 2х(COCH2CH2-), 1,43, 1,47 (2 ш, 40H, 20 CH2), 1,04 (м, 6H, 2 CH3), Фр 0,5 (хлороформ-метанол-вода, 6:3:0,5.
Схема II

Получение GalNAcα1-3Galβ1-4GlcNAc-Ad-DOPE (9) (Схема III)
К раствору продукта 8 (33 мкмоль) в N,N-диметилформамиде (1 мл), добавляли 30 мкмоль 3-аминопропилтрисахарида 5 и 5 мкл триэтиламина (Et3N). Смесь перемешивали в течение 2 ч при комнатной температуре. Колоночная хроматография на силикагеле (CH2Cl2-EtOH-H2O; 6:5:1) давала 81% выход конструкта 9.
9:1H ЯМР (700 МГц, CDCl3-CD3OD, 1:1 об/об, выбранный), δ, ч/млн: 1,05 (т, 6H, Дж 7,05, 2 CH3), 1,39 -1,55 (м, 40H, 20 CH2), 1,75-1,84 (м, 8H, COCH2CH2CH2CH2CO и 2хCOCH2CH2-), 1,84-1,96 (м, 2H, O-CH2CH2CH2-NH), 2,15-2,22 (м, 14H, 2х(-CH2-CH=CH-CH2-), 2х NHC(O)CH3), 2,34-2,46 (м, 4H, 2х-CH2-CO), 2,36-2,44 (м, 4H, 2х-CH2-CO), 3,29-3,34 (м, 1H, -CH2-CHH-NH), 4,17-4,20 (м, 2H, -CHO-CH2OP-), 4,34-4,39 (м, 2H, -CH2OPO-CH2-CH2), 4,57 (д, 1H, Дж1,2 8,39, H-1I), 4,50 (дд, 1H, Дж 3,78, Дж 10,82, -C(O)OCHHCHOCH2O-), 4,58- 4,61 (м, 2H, H-1II, C(O)OCHHCHOCH2O-), 5,15 (д, 1H, Дж1,2 3,76, H-1III), 5,38-5,42 (м, 1H, -OCH2-CHO-CH2O-), 5,47-5,53 (м, 4H, 2х-CH=CH-), Фр 0,5 (CH2Cl2-EtOH-H2O; 6:5:1).
Схема III
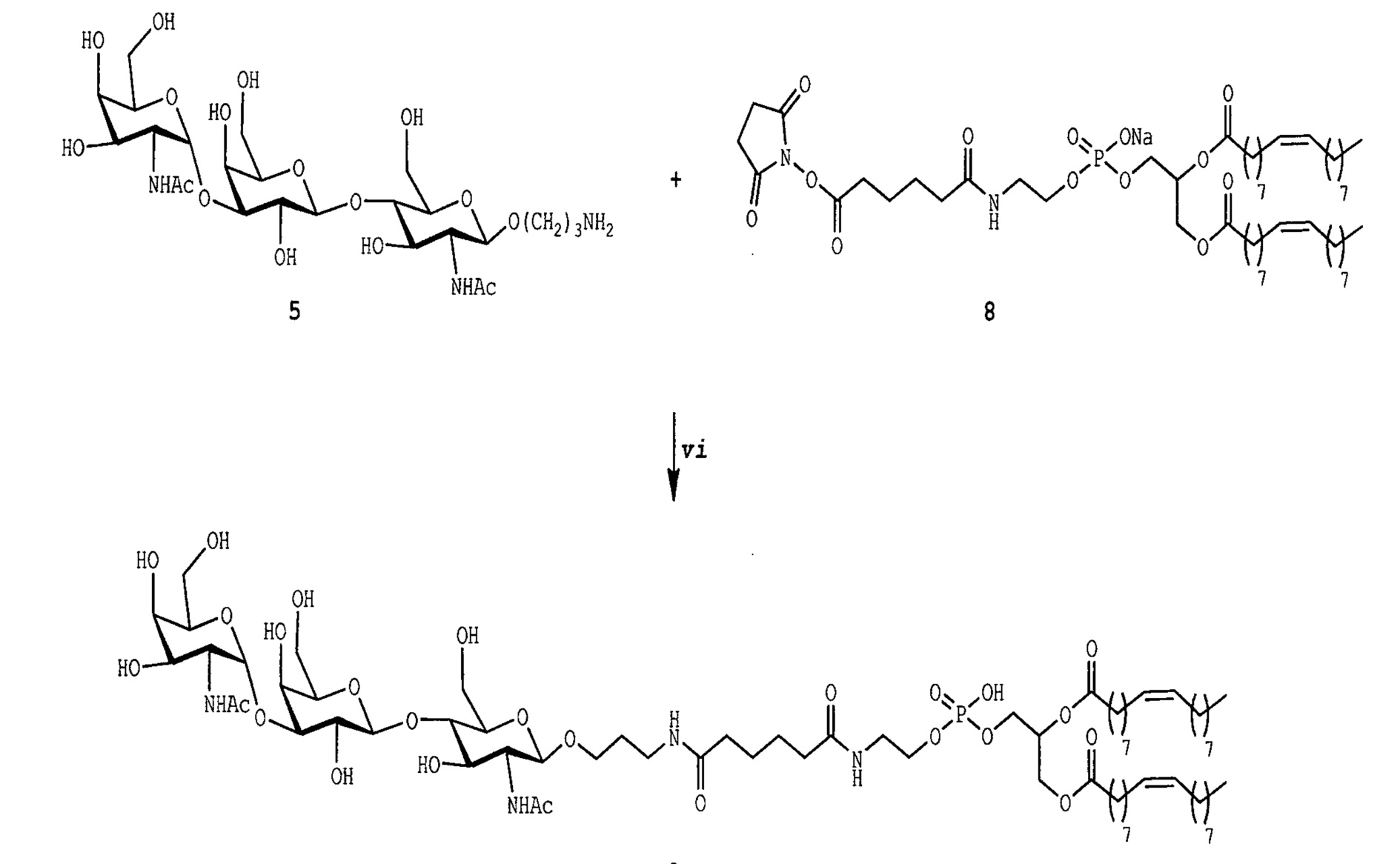
БИОЛОГИЧЕСКИЕ ДАННЫЕ
Анализ включения анти-Gal
Клетки CHO-K1 собирали с чашек для культур клеток, подсчитывали и ресуспендировали в PBS до плотности клеток 5×106 клеток/мл. Каждый гликолипид серийно разводили в PBS в девяти 1,5 мл центрифужных пробирках так, что конечный объем в пробирках составил 100 мкл. К каждой пробирке добавляли 100 мкл суспензии клеток CHO-K1 и пробирки инкубировали в течение 1 часа при 37°C. Через час клетки осаждали центрифугированием при 400 g в течение 3 минут и ресуспендировали в 500 мкл PBS+0,1% BSA. Это повторяли дважды для промывки клеток. После финальной промывки клетки ресуспендировали в 100 мкл моноклонального IgG1 анти-Gal, разведенного 1:8 в PBS+0,1% BSA. Пробирки инкубировали на льду в течение 30 минут. После 30 минут клетки осаждали центрифугированием при 400 g в течение 3 минут и ресуспендировали в 500 мкл PBS+0,1% BSA. Это повторяли дважды для промывки клеток. После финальной промывки клетки ресуспендировали в 100 мкл FITC-конъюгированного мышиного античеловеческого IgG (Biolegend) и пробирки инкубировали на льду в течение 30 минут. После 30 минут клетки осаждали центрифугированием при 400 g в течение 3 минут и ресуспендировали в 500 мкл PBS+0,1% BSA. Это повторяли дважды для промывки клеток. После финальной промывки клетки ресуспендировали в 200 мкл PBS+0,1% BSA содержащих 2,5 мкл 7-AAD (Biolegend). После 5 минут инкубации на льду клетки анализировали на поточном цитометре Cytomics FC500 (Beckman Coulter). Мертвые клетки исключали из анализа.
Соединения, как получено в настоящем описании, как пример 1 (Galili-CMG2-DOPE) и пример 2 (Galili-T17 DOPE), тестировали в анализе включения анти-gal и результаты видны на фиг. 1 и 2. Полученные результаты демонстрируют, что соединение, как получено в настоящем описании в примере 1 (Galili-CMG2-DOPE), которое является анти-Gal гликолипидом, имеющим CMG спейсер между альфа-Gal сахаром и частью одиночного липида молекулы включает в плазматическую мембрану клеток CHO-K1 и представляет альфа-Gal эпитоп для распознавания анти-Gal антителами (см. фиг. 1). Результаты также демонстрируют, что соединение, как получено в настоящем описании, как пример 2 (Galili-T17 DOPE), которое является смесью гликолипидов, имеющих одиночную липидную часть, прикрепленную к двум или трем альфа-Gal сахарам разветвленными CMG линкерами, включают в плазматическую мембрану клеток CHO-K1 и захватывают больше анти-Gal антител, чем эквивалентная концентрация одиночной альфа-Gal молекулы примера 1.
Анализ комплемент-зависимой цитотоксичности
Клетки CHO-K1 собирали из чашек культур клеток, подсчитывали и ресуспендировали в PBS до плотности клеток 5×106 клеток/мл. Каждый гликолипид серийно разводили в PBS в девяти 1,5 мл центрифужных пробирках так, что конечный объем пробирок составил 100 мкл. К каждой пробирке добавляли 100 мкл суспензии клеток CHO-K1 и пробирки инкубировали в течение 1 часа при 37°C. Через один час пробирки помещали на лед в течение 5 минут и затем клетки промывали 3 раза 500 мкл ледяного PBS. Клетки ресуспендировали в конечном объеме 250 мкл ледяного PBS и 50 мкл аликвоты переносили в дупликаты ячеек 96 луночного планшета. К каждой ячейке, содержащей клетки 50 мкл 100% нормального или теплоинактивированного (30 минут при 56°C) комплемента человеческой сыворотки (Innovative Research, добавляли так, чтобы конечная концентрация человеческой сыворотки составила 50%. Планшет инкубировали при 37°C в течение 1 часа, после чего выживаемость клеток измеряли с использованием считывания реагента CellTiter-Glo (Promega) на счетчике планшетов EnVision (Perkin Elmer).
Соединения, как получено в настоящем описании, как пример 1 (Galili-CMG2-DOPE), пример 2 (Galili-T17 DOPE) и пример 3 (GalNAc-Gal-GlcNAc-Ad-DOPE), тестировали в анализе комплемент-зависимой цитотоксичности и результаты могут быть видны в таблице 1 ниже и фиг. 3-5.
Таблица 1: Результаты анализа комплемент-зависимой цитотоксичности
Полученные результаты демонстрируют, что клетки CHO-K1, меченные соединением, как получено в настоящем описании в примере 1 (Galili-CMG2-DOPE; т.е. одиночной альфа-Gal CMG молекулой), лизировали комплементом человеческой сыворотки (см. фиг. 3). Результаты также демонстрируют, что клетки CHO-K1, меченные соединением, как получено в настоящем описании в примере 2 (Galili-T17 DOPE; т.е. димерная/трехмерная альфа-Gal молекула), были более подвержены лизису комплементом человеческой сыворотки, чем клетки, инкубируемые с той же концентрацией одиночной альфа-Gal молекулы (т.е. соединение, как получено в настоящем описании в примере 1 (Galili-CMG2-DOPE). Результаты также демонстрируют, что клетки CHO-K1, меченные соединением, как получено в настоящем описании в примере 3 (GalNAc-Gal-GlcNAc-Ad-DOPE; т.е. гликолипидная молекула, которая имеет GalNAc альфа сахарный антиген), лизировали комплементом человеческой сыворотки.
Реферат
Изобретение относится к пригодному в медицине гликолипидному соединению, выбранному из соединения формулы (I), (II) и (III) или его фармацевтически приемлемой соли:(I)(II)(III). Предложены новые эффективные соединения для индукции защитного протиоопухолевого иммунного ответа и фармацевтическая композиция на их основе для лечения опухолей. 2 н. и 9 з.п. ф-лы, 5 ил., 1 таб., 3 пр.
Формула

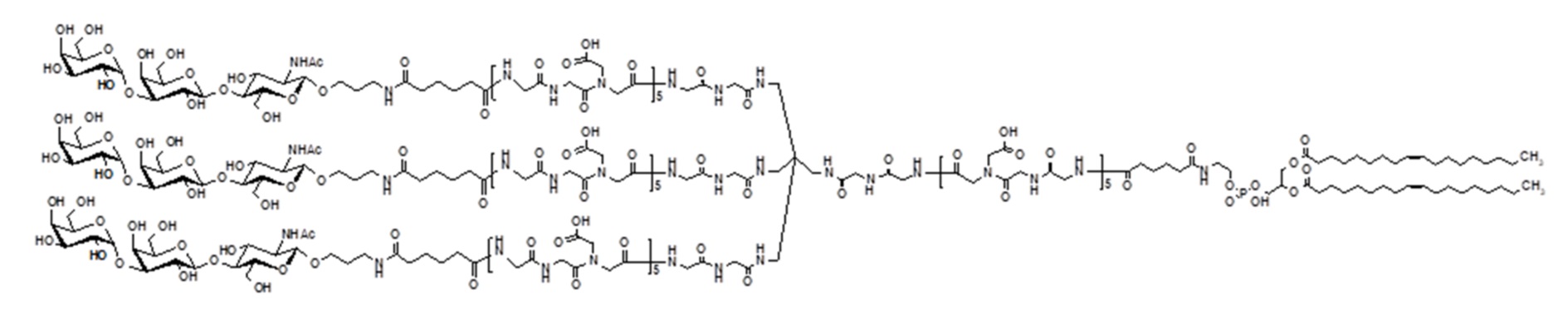
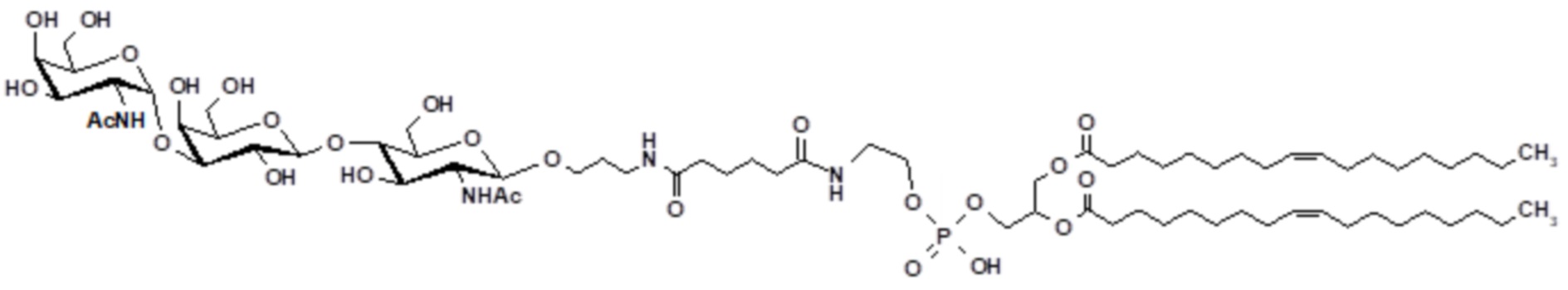
Комментарии