9-аминометилминоциклиновые соединения и их применение - RU2760186C2
Код документа: RU2760186C2
Чертежи






Описание
ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка испрашивает приоритет в соответствии с § 119(e) Раздела 35 Свода законов США по дате подачи предварительных заявок на патент США №62/370527, поданной 3 августа 2016 года; 62/514479, поданной 2 июня 2017 года; и 62/532454, поданной 14 июля 2017 года, содержание каждой из которых полностью включено в настоящую заявку посредством ссылки.
УРОВЕНЬ ТЕХНИКИ
Развитие тетрациклиновых антибиотиков является прямым результатом системного скрининга образцов почвы, собранных в различных регионах, на наличие микроорганизмов, из которых можно получать бактерицидные и/или бактериостатические композиции. Первое из указанных новых соединений было описано в 1948 году под названием хлортетрациклин. Спустя два года был получен окситетрациклин. При выявлении химической структуры этих соединений была подтверждена их схожесть и подготовлена аналитическая основа для получения в 1952 году третьего члена указанной группы - тетрациклина. Новое семейство миноциклиновых соединений, не содержащих присоединенную к кольцу метальную группу, которая присутствовала в ранних тетрациклинах, было получено в 1957 году и стало общедоступным в 1967 году; и миноциклин начали применять в 1972 году.
Более поздние исследования были направлены на разработку новых композиций тетрациклиновых антибиотиков, обладающих эффективностью при различных условиях терапии и способах введения. Также были изучены новые аналоги тетрациклинов, которые продемонстрировали равную или даже более высокую эффективность по сравнению с предложенными изначально миноциклиновыми соединениями. Примеры включают патенты США №2980584; 2990331; 3062717; 3165531; 3454697; 3557280; 3674859; 3957980; 4018889; 4024272; и 4126680. В указанных патентах приведена типовая подборка разнообразных фармацевтически активных композиций тетрациклина и аналогов тетрациклина.
Впоследствии, вскоре после разработки и начала использования, было обнаружено, что тетрациклины обладают высокой фармакологической эффективностью в отношении риккетсий; ряда грамположительных и грамотрицательных бактерий; и агентов, вызывающих венерическую лимфогранулему, конъюнктивит с включениями и пситтакоз. Таким образом, тетрациклины стали называть антибиотиками «широкого спектра». После того, как были установлены их противомикробная активность in vitro, эффективность в отношении экспериментальных инфекций и фармакологические свойства, тетрациклины, в целом как класс, стали широко применять для терапевтических задач.
Тем не менее, указанное повсеместное применение тетрациклинов при серьезных и незначительных болезнях и заболеваниях непосредственно поспособствовало возникновению устойчивости к указанным антибиотикам даже у крайне чувствительных к их действию комменсальных и патогенных штаммов бактерий (например, pneumococci и Salmonella). Появление устойчивых к тетрациклинам организмов привело к общему ограничению использования композиций тетрациклина и аналогов тетрациклина в качестве назначаемых антибиотиков. Кроме того, избыточное использование других антибактериальных агентов привело к появлению штаммов бактерий с множественной лекарственной устойчивостью (МЛУ).
В последнее десятилетие появление грамположительных бактерий с множественной лекарственной устойчивостью к разнообразным антибиотикам стало огромной проблемой при проведении лечения. Два пути развития привели к появлению спектра грамположительных организмов, и эффективность лечения инфекций, вызываемых этими организмами, с применением доступных в настоящее время антибиотиков стала еще более низкой. Первым является приобретение устойчивости к ванкомицину у штамма Enterococcus (spp.) и последующий перенос указанных элементов устойчивости на Staphylococcus aureus. Несмотря на то, что устойчивый к ванкомицину Staphylococcus aureus не приобрел эпидемиологическую значимость, сам факт его существования вызывает обеспокоенность, так как ванкомицин выбирают при инфекциях, вызываемых устойчивыми грамположительными патогенами.
Вторым важным путем развития является появление внебольничного устойчивого к метициллину Staphylococcus aureus (MRSA). Указанные штаммы все чаще приобретают множественную лекарственную устойчивость. Во многих регионах инфекции MRSA являются основными спорадическими стафилококковыми инфекциями, проявляющимися во внебольничных условиях. Эти штаммы также связаны с многочисленными вспышками локализованных (на коже и кожных структурах) и инвазивных (бактериемических) инфекций.
Помимо общей потребности в эффективных антибактериальных агентах для лечения бактериальных инфекций также существует конкретная потребность в способах терапии с применением пероральных антибиотиков.
По сравнению с в.в. введением терапия с применением пероральных антибиотиков может быть предпочтительной вследствие отсутствия необходимости посещения и/или пребывания в стационаре, таким образом, уменьшаются общие затраты на лечение, ограничивается появление у пациента вторичных инфекций в больничных условиях и увеличивается доступность лечения в тех регионах, где больницы доступны в меньшей степени или недоступны, в частности, в отдаленных или экономически неразвитых регионах или областях.
К сожалению, вследствие появления устойчивости к антибиотикам использование старых агентов приводит к увеличению частоты посещения больниц, что в свою очередь повышает вероятность инфицирования пациентов другими бактериями.
Таким образом, сохраняется необходимость в новом эффективном пероральном антибактериальном агенте, в частности, предназначенном только для перорального режима введения, для лечения, например, бактериальной инфекции кожи или кожных структур, такой как ABSSSI. Только ABSSSI является причиной более чем 750000 случаев госпитализации в год (на основании последних данных, полученных в 2011 году), что соответствует увеличению на 17,3% госпитализированных пациентов с ABSSSI за период от 2005 до 2011 года.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Согласно одному из аспектов изобретения предложен способ лечения субъекта-человека, нуждающегося в лечении бактериальной инфекции кожи или кожных структур, включающий пероральное введение субъекту эффективного количества 9-[(2,2-диметилпропиламино)метил]миноциклина (также известного как омадациклин или ОМС) или его соли с обеспечением лечения указанного субъекта где 9-[(2,2-диметилпропиламино)метил]миноциклин вводят один раз в день в виде пероральной дозы 450 мг или 600 мг в течение 5 дней или более.
В определенных вариантах реализации пероральную дозу 450 мг вводят один раз в день в течение 5 или более дней подряд.
В определенных вариантах реализации пероральную дозу 600 мг вводят один раз в день в течение 5 или более дней подряд.
Согласно родственному аспекту изобретения предложен способ лечения субъекта-человека, нуждающегося в лечении бактериальной инфекции кожи или кожных структур, включающий пероральное введение субъекту эффективного количества 9-[(2,2-диметилпропиламино)метил]миноциклина или его соли с обеспечением лечения указанного субъекта, в котором 9-[(2,2-диметилпропиламино)метил]миноциклин вводят один раз в день в виде пероральной дозы 300 мг в течение 5, 6, 7 или 8 дней подряд.
Согласно другому родственному аспекту изобретения предложен способ лечения субъекта-человека, нуждающегося в лечении бактериальной инфекции кожи или кожных структур, включающий пероральное введение субъекту эффективного количества 9-[(2,2-диметилпропиламино)метил]миноциклина или его соли с обеспечением лечения указанного субъекта, в котором 9-[(2,2-диметилпропиламино)метил]миноциклин вводят в виде пероральной нагрузочной дозы и затем один раз в день вводят пероральные дозы по 300-600 мг (например, 300 мг, 450 мг или 600 мг) в течение 5 дней или более.
В определенных вариантах реализации пероральная нагрузочная доза состоит по существу/состоит из одной пероральной дозы 600 мг 9-[(2,2-диметилпропиламино)-метил]миноциклина, которую вводят за 24 часа перед первой из вводимых раз в день пероральных доз по 300 мг, 450 мг или 600 мг.
В определенных вариантах реализации пероральная нагрузочная доза состоит по существу/состоит из одной пероральной дозы 450 мг 9-[(2,2-диметилпропиламино)-метил]миноциклина, которую вводят за 24 часа перед первой из вводимых раз в день пероральных доз по 300 мг, 450 мг или 600 мг.
В определенных вариантах реализации пероральная нагрузочная доза состоит по существу/состоит из двух пероральных доз по 300 мг 9-[(2,2-диметилпропиламино)-метил]миноциклина, которые вводят за 12 часов и 24 часа, соответственно, перед первой из вводимых раз в день пероральных доз по 300 мг, 450 мг или 600 мг.
В определенных вариантах реализации пероральная нагрузочная доза состоит по существу/состоит из двух пероральных доз по 450 мг 9-[(2,2-диметилпропиламино)-метил]миноциклина, которые вводят за 12 часов и 24 часа, соответственно, перед первой из вводимых раз в день пероральных доз по 300 мг, 450 мг или 600 мг.
В определенных вариантах реализации пероральная нагрузочная доза состоит по существу/состоит из двух пероральных доз по 600 мг 9-[(2,2-диметилпропиламино)-метил]миноциклина, которые вводят за 24 часа и 48 часов, соответственно, перед первой из вводимых раз в день пероральных доз по 300 мг, 450 мг или 600 мг.
В определенных вариантах реализации пероральная нагрузочная доза состоит по существу/состоит из двух пероральных доз по 450 мг 9-[(2,2-диметилпропиламино)-метил]миноциклина, которые вводят за 24 часа и 48 часов, соответственно, перед первой из вводимых раз в день пероральных доз по 300 мг, 450 мг или 600 мг.
Согласно другому аспекту в изобретении предложен способ лечения субъекта-человека, нуждающегося в лечении бактериальной инфекции кожи или кожных структур, включающий пероральное введение субъекту эффективного количества 9-[(2,2-диметилпропиламино)метил]миноциклина или его соли с обеспечением лечения указанного субъекта, в котором 9-[(2,2-диметилпропиламино)метил]миноциклин вводят перорально в дозе примерно 450 мг в день в течение двух дней подряд, затем в дозе примерно 300 мг в день в течение 5 дней или более.
В определенных вариантах реализации бактериальная инфекция кожи или кожных структур представляет собой раневую инфекцию, панникулит/рожу, крупный гнойник, фурункулез/чирей, бородавку, стафилококковый синдром ошпаренной кожи (ССОК) или эктиму.
В определенных вариантах реализации бактериальная инфекция кожи или кожных структур представляет собой острую бактериальную инфекцию кожи и кожных структур (ABSSSI), такую как внебольничная ABSSSI.
В определенных вариантах реализации общая площадь поверхности поражения ABSSSI смежных тканей составляет 75 см2 или более.
В определенных вариантах реализации ABSSSI включает раневую инфекцию, панникулит/рожу и/или крупный гнойник.
В определенных вариантах реализации бактериальная инфекция кожи или кожных структур возникает в результате повреждения кожи, включая, но не ограничиваясь ими, травму, хирургическую процедуру или в.в. введение лекарственного средства.
В определенных вариантах реализации бактериальная инфекция кожи или кожных структур возникает в результате сосудистой недостаточности или отека.
В определенных вариантах реализации субъекту-человеку вводят 9-[(2,2-диметилпропиламино)метил]миноциклин натощак.
В определенных вариантах реализации 9-[(2,2-диметилпропиламино)метил]-миноциклин вводят один раз в день (например, каждую пероральную дозу вводят с интервалом примерно 24 часа).
В определенных вариантах реализации продолжительность лечения субъекта составляет примерно до 14 дней включительно, примерно до 10 дней включительно, примерно до 9 дней включительно, примерно до 8 дней включительно или примерно до 7 дней включительно, примерно до 5 дней включительно, с обеспечением лечения указанного субъекта.
В определенных вариантах реализации субъекта лечат в течение 7-10 дней.
В определенных вариантах реализации субъекта лечат в течение 8 дней.
В определенных вариантах реализации соль представляет собой тозилатную соль.
В определенных вариантах реализации бактериальная инфекция кожи или кожных структур заведомо или предположительно вызвана грамположительными патогенами. Например, грамположительные патогены могут включать Staphylococcus aureus, Staphylococcus lugdunensis, вид Streptococcus, Streptococcus agalactiae, Streptococcus mitis, вид Enterococcus (Enterococcus faecalis (такие как VRE или VSE) или Enterococcus faecium (такие как VRE или VSE)), группу Streptococcus anginosus (S. anginosus, S. constellatus и S. intermedius, то есть, бета-, альфа- или негемолитические), Streptococci группы вириданс (VGS), Clostridium perjringens, Finegoldia magna или их комбинацию. Staphylococcus aureus может представлять собой устойчивый к метициллину Staphylococcus aureus (MRSA) или чувствительный к метициллину Staphylococcus aureus (MSSA). Вид Streptococcus может включать группу Streptococcus anginosus. Вид Streptococcus может включать бета-гемолитические Streptococci или S. anginosus. Вид Streptococcus может включать негемолитические Streptococci или S. intermedius. Вид Streptococcus может включать альфа-гемолитические Streptococci или S. constellatus. Вид Enterococcus может включать Enterococcus faecalis (VSE). Вид Streptococcus может включать Streptococcus pyogenes.
В определенных вариантах реализации бактериальная инфекция кожи или кожных структур заведомо или предположительно вызвана грамотрицательными патогенами. Например, грамотрицательные патогены могут включать Enterobacter cloacae, Escherichia coli, Klebsiella pneumoniae, Prevotella denticola, Prevotella melaninogenica или их комбинацию.
В определенных вариантах реализации бактериальная инфекция кожи или кожных структур заведомо или предположительно вызвана любым из приведенных выше грамположительных и грамотрицательных патогенов или их комбинацией.
В определенных вариантах реализации нежелательные явления (НЯ) в желудочно-кишечном тракте (ЖКТ) являются преимущественно слабыми. В определенных вариантах реализации нежелательные явления (НЯ) со стороны ЖКТ, связанные с лечением, не приводят к прекращению терапии.
В определенных вариантах реализации значение AUC0-24 после введения двух первых доз 9-[(2,2-диметилпропиламино)метил]миноциклина или его соли составляет примерно 10000 нг*ч/мл.
В определенных вариантах реализации каждую дозу 9-[(2,2-диметилпропиламино)метил]миноциклина или его соли вводят в виде 150 мг таблеток.
В определенных вариантах реализации способ имеет показатель клинической эффективности примерно 70-100%. Например, показатель клинической эффективности может составлять примерно 80-100%, примерно 79-98%, примерно 79-94%, примерно 84-98%, примерно 80-88% или примерно 84-89%.
В определенных вариантах реализации показатель клинической эффективности, который отмечают через 48-72 часа после введения первой пероральной дозы, составляет примерно 79-94% или примерно 84-89% или примерно 87,5%. В определенных вариантах реализации ABSSSI состоит по существу из раневой инфекции, и показатель клинической эффективности составляет примерно 84-94% или примерно 89%. В определенных вариантах реализации ABSSSI состоит по существу из панникулита/рожи, и показатель клинической эффективности составляет примерно 74-84% или примерно 79%. В определенных вариантах реализации ABSSSI состоит по существу из крупного гнойника, и показатель клинической эффективности составляет примерно 90-98% или примерно 94%.
В определенных вариантах реализации показатель клинической эффективности представляет собой показатель общей клинической эффективности, отмечаемый примерно через 7-14 дней после введения последней дозы, и составляет примерно 79-98%, примерно 75-95%, примерно 79-89%, 84%, примерно 95-100% или примерно 98%. В определенных вариантах реализации ABSSSI состоит по существу из раневой инфекции, и показатель общей клинической эффективности составляет примерно 80-85% или примерно 82-83%. В определенных вариантах реализации ABSSSI состоит по существу из панникулита/рожи, и показатель общей клинической эффективности составляет примерно 85-91%, примерно 87-88% или примерно 88%. В определенных вариантах реализации ABSSSI состоит по существу из крупного гнойника, и показатель общей клинической эффективности составляет примерно 80-88% или примерно 84%.
Согласно родственному аспекту в изобретении предложен способ лечения субъекта-человека, нуждающегося в лечении бактериальной инфекции кожи или кожных структур, включающий введение субъекту эффективного количества 9-[(2,2-диметилпропиламино)метил]миноциклина или его соли с обеспечением лечения указанного субъекта, в котором 9-[(2,2-диметилпропиламино)метил]миноциклин вводят внутривенно (в.в.) в дозе примерно 150 мг в день в течение двух дней подряд, а затем перорально в дозе примерно 300 мг в день в течение 5 дней или более. С учетом установленной биоэквивалентности пероральной и в.в. доз 9-[(2,2-диметилпропиламино)метил]миноциклина 150 мг в. в. дозу рассматривают как биоэквивалентную 450 мг пероральной дозе. Таким образом, считается, что альтернативный вариант реализации согласно настоящему аспекту изобретения включен в объем настоящего изобретения.
Следует понимать, что любой вариант реализации может быть объединен с любыми одним или более другими вариантами реализации, включая варианты реализации, описанные только в примерах или только для одного аспекта изобретения, если комбинация не является неудовлетворительной или явным образом исключена.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На ФИГ. 1 показана кривая зависимости концентрация в плазме-время для одной пероральной 300 мг дозы омадациклина.
На ФИГ. 2 показаны данные РК для режима лечения, в котором 100 мг омадациклина вводят в.в. BID (два раза в день, с интервалом 12 часов) в течение 1 дня, затем 100 мг в.в. в день.
На ФИГ. 3 показаны данные РК для режима лечения, в котором 450 мг омадациклина вводят п.о. QD (один раз в день) в течение 2 дней, затем 300 мг п.о. в день.
На ФИГ. 4 показаны данные РК для режима лечения, в котором 600 мг омадациклина вводят п.о. QD (один раз в день) в течение 2 дней, затем 300 мг п.о. в день.
На ФИГ. 5 показаны кривые зависимости концентрации в плазме омадациклина от времени после перорального введения. Зависимость средней (± СКО) концентрации в плазме омадациклина от времени показана для дозы омадациклина (300, 450 или 600 мг) в популяции для анализа фармакокинетики. Пероральные дозы омадациклина вводили в момент времени 0 в каждый из 5 последовательных дней в каждый из 3 периодов. Образцы крови собирали для анализа РК в день 1 (левый график) и день 5 (правый график). Данные объединяли по дозам омадациклина для всех субъектов, независимо от периода, когда им давали конкретную дозу.
На ФИГ. 6 показано, что омадациклин имел статистически не меньшую эффективность (предел 10%) по сравнению с линезолидом в отношении раннего клинического ответа (ECR) в популяции mITT (модифицированная популяция с назначенным лечением) (см. пары столбцов слева) (первичный конечный критерий оценки FDA); и в отношении клинической эффективности на стадии РТЕ (оценка после лечения/терапии) в популяции mITT РТЕ (см. среднюю пару столбцов) и популяции СЕ-РТЕ (подходящая для клинической оценки популяция на стадии РТЕ) (см. правую пару столбцов) (комбинированные первичные конечные критерии оценки ЕМА).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
1. Обзор
Омадациклин (9-[(2,2-диметилпропиламино)метил]миноциклин) обладает очень высокой активностью in vitro в отношении большинства грамположительных патогенов. Например, было показано, что он является эффективным при лечении осложненных инфекций кожи и кожных структур (см. патент США №9265740, содержание которого включено в настоящую заявку посредством ссылки). Он также обладает активностью в отношении атипических патогенов (например, Legionella spp.) и некоторых анаэробных и грамотрицательных патогенов. Лекарственное средство является активным в отношении штаммов, в которых проявляются оба механизма устойчивости к тетрациклинам, а также штаммов, которые являются устойчивыми к доступным в настоящее время антибиотикам, включая метициллин, ванкомицин, эритромицин и ципрофлоксацин. Также была продемонстрирована активность омадациклина в отношении большинства распространенных патогенов ABSSSI, включая изоляты, устойчивые к стандартам здравоохранения.
Омадациклин был разработан для в.в. и п.о. (перорального) введения. На момент подачи заявки более 1000 субъектов получили один или оба указанных состава в рамках завершенных клинических исследований.
В завершенных исследованиях 1 фазы изучали однократное введение в.в. доз до 600 мг и п.о. доз до 600 мг. Было изучено введение нескольких в.в. доз по 100 мг один раз в день и 200 мг один раз в день в течение периода до 14 и 7 дней подряд, соответственно. Было изучено введение нескольких п.о. доз по 200 мг один раз в день и 300 мг один раз в день в течение периода до 10 дней подряд. Кроме того, также было изучено введение нескольких п.о. доз по 450 мг один раз в день и 600 мг один раз в день в течение 5 дней подряд.
В исследовании 2 фазы 219 субъектам с осложненными инфекциями кожи и кожных структур (cSSSI) вводили омадациклин (n=111) или линезолид (n=108); лечение начинали с в.в. введения (100 мг в день), затем по решению исследователя переходили на п.о. терапию (200 мг в день). В указанном исследовании общая продолжительность изучаемого способа лечения в среднем составляла 10 и максимально 20 дней.
В досрочно прекращенном по решению спонсора исследовании 3 фазы со схожим дизайном 140 субъектам с cSSSI вводили омадациклин (n=68) или линезолид (n=72) в среднем в течение 10 и максимально в течение 20 дней в случае омадациклина и 22 дней в случае линезолида. Пациентам сначала вводили в.в. исследуемое лекарственное средство омадациклин (100 мг в день), а затем переходили на пероральную терапию по решению исследователя (300 мг в день). Ожидаемая продолжительность в.в. введения составляла 4-7 дней; ожидаемая общая продолжительность лечения (в.в. и перорального) составляла до 14 дней. Исследование было двойным слепым в фазе в.в. введения и слепым для эксперта в фазе перорального введения.
В завершенном недавно исследовании 3 фазы, в котором сравнивали омадациклин и линезолид при лечении взрослых с ABSSSI субъектам начинали проводить терапию с использованием 2 доз омадациклина по 100 мг в.в. каждые 12 часов (q12h), затем вводили 100 мг в.в. каждые 24 часа (q24h) или 600 мг линезолида в.в. q12h. Субъектов могли переводить на пероральную терапию (300 мг омадациклина q24h или 600 мг линезолида q12h) по прошествии как минимум 3 дней в.в. терапии; общая продолжительность лечения составляла 7-14 дней. Результаты исследования показали, что омадациклин имел не меньшую эффективность по сравнению с линезолидом. В популяции первичного анализа FDA (определенной как субъекты ITT, у которых отсутствовали какие-либо грамотрицательные патогены во время скрининга, всего N=627) для сравнения омадациклина и линезолида, соответственно, клиническая эффективность, определенная по уменьшению размера поражения через 48-72 часа после введения первой дозы, составляла 84,8% и 85,5% (95% доверительный интервал [ДИ]: -6,3, 4,9); клиническая эффективность по результатам оценки клинического ответа исследователем через 7-14 дней после введения последней дозы составляла 86,1% и 83,6% (ДИ: -3,2, 8,2). Омадациклин хорошо переносился: нежелательные явления, возникшие в ходе лечения (НЯВЛ) отмечались в 48,3% случаев по сравнению с 45,7%; серьезные НЯВЛ в 3,4% случаев по сравнению с 2,5%, и досрочное прекращение исследования из-за НЯВЛ в 1,9% случаев по сравнению с 2,2% у субъектов, которым вводили омадациклин и линезолид, соответственно.
Во всех описанных выше исследованиях, тем не менее, в режиме введения требуется период в.в. введения омадациклина, что делает необходимым посещение и/или пребывание в стационаре или требует временных затрат на посещение амбулаторного центра для проведения инфузии. Для разработки эффективного исключительно перорального режима введения, в котором устранена необходимость посещения стационара, что, таким образом, является особенно желательным при лечении внебольничных бактериальных инфекций кожи или кожных структур (например, ABSSSI или cSSSI), был предложен начальный исключительно пероральный режим введения, в котором субъекту-человеку, нуждающемуся в лечении бактериальной инфекции кожи или кожных структур, вводят омадациклин перорально в дозе, сравнимой с дозой, используемой в недавно завершенном исследовании 3 фазы: т.е. примерно 300 мг BID (два раза в день, по 300 мг с интервалом примерно 12 часов) в течение одного дня, затем дозу примерно 300 мг в день всего в течение 7-14 дней. Следует отметить, что приведенном выше исследовании 1 фазы было продемонстрировано, что пероральная доза 300 мг состава омадациклина в виде таблетки является биоэквивалентом (на основании AUC в сыворотке) 100 мг в.в. дозы.
Было показано, что прием пищи значительно влияет на эффективность омадациклина, то есть употребление пищи в значительной степени изменяет пероральную биодоступность вводимой перорально 300 мг дозы омадациклина. См. пример 1. В исследовании РК у здоровых добровольцев было показано, что по сравнению с введением дозы натощак биодоступность уменьшалась на 15%-17% при приеме пищи, не содержащей молочные продукты, за 4 часа перед введением дозы, на 40%-42% при приеме пищи, не содержащей молочные продукты, за 2 часа перед введением дозы, и на 59%-63% при приеме пищи, содержащей молочные продукты, за 2 часа перед введением дозы. Таким образом, эффект от приема пищи был более выраженным при употреблении пищи с высоким содержанием жиров незадолго до введения дозы и при включении молочных продуктов в рацион. С учетом этого результата, пероральный омадациклин следует вводить по меньшей мере через 6 часов после приема пищи для достижения максимальной биодоступности пероральной дозы, предназначенной для обеспечения терапевтической эффективности.
Указанный эффект от приема пищи создает значительные трудности для соблюдения больным схемы лечения, в частности, если в один день требуется вводить две пероральные дозы начале режима лечения.
Предварительные результаты исследования 1 фазы показали, что пероральные режимы введения 300 мг QD (q24h) и 450 мг QD (q24h) имели схожие и предпочтительные профили переносимости. Кроме того, результаты PK и моделирование PK показали, что режим введения 2 доз по 450 мг п.о. QD (q24h) и затем 300 мг п.о. доз QD (q24h) обеспечивает практически такие же стационарные концентрации в пределах того же временного интервала, что и режим введения, в котором начинают вводить 2 дозы по 300 мг п.о. q12h, затем 300 мг п.о. QD (q24h). В обоих режимах в первые 2 дня вводят всего 900 мг перорального омадациклина. См. пример 2.
Таким образом, согласно одному из аспектов в изобретении предложен способ лечения субъекта-человека, нуждающегося в лечении бактериальной инфекции кожи или кожных структур.
В 1-м варианте реализации способ включает пероральное введение субъекту эффективного количества 9-[(2,2-диметилпропиламино)метил]миноциклина или его соли с обеспечением лечения указанного субъекта, где 9-[(2,2-диметилпропиламино)метил]-миноциклин вводят один раз в день в виде пероральной дозы 450 мг или 600 мг в течение 5 дней или более.
Во 2-м варианте реализации способ включает пероральное введение субъекту эффективного количества 9-[(2,2-диметилпропиламино)метил]миноциклина или его соли с обеспечением лечения указанного субъекта, где 9-[(2,2-диметилпропиламино)метил]-миноциклин вводят один раз в день в виде пероральной дозы 300 мг в течение 5, 6, 7 или 8 дней подряд.
В 3-м варианте реализации вводимая один раз в день пероральная доза согласно 1-му варианту реализации составляет 450 мг, и ее вводят в течение 5 или более дней подряд.
В 4-м варианте реализации вводимая один раз в день пероральная доза согласно 1-му варианту реализации составляет 600 мг, и ее вводят в течение 5 или более дней подряд.
В 5-м варианте реализации способ включает пероральное введение субъекту эффективного количества 9-[(2,2-диметилпропиламино)метил]миноциклина или его соли с обеспечением лечения указанного субъекта, где 9-[(2,2-диметилпропиламино)метил]-миноциклин вводят в виде пероральной нагрузочной дозы (например, дозы, превышающей вводимую один раз в день пероральную 300 мг дозу), затем один раз в день вводят пероральные дозы 300-600 мг (например, 300 мг, 450 мг или 600 мг) в течение 5 дней или более.
В 6-м варианте реализации пероральная нагрузочная доза согласно 5-му варианту реализации состоит по существу/состоит из одной пероральной дозы 600 мг 9-[(2,2-диметилпропиламино)метил]миноциклина, которую вводят за 24 часа перед первой из вводимых один раз в день пероральных доз по 300, 450 или 600 мг.
В 7-м варианте реализации пероральная нагрузочная доза согласно 5-му варианту реализации состоит по существу/состоит из одной пероральной дозы 450 мг 9-[(2,2-диметилпропиламино)метил]миноциклина, которую вводят за 24 часа перед первой из вводимых один раз в день пероральных доз по 300, 450 или 600 мг.
В 8-м варианте реализации пероральная нагрузочная доза согласно 5-му варианту реализации состоит по существу/состоит из двух пероральных доз по 300 мг 9-[(2,2-диметилпропиламино)метил]миноциклина, которые вводят за 12 часов и 24 часа, соответственно, перед первой из вводимых один раз в день пероральных доз по 300 мг, 450 мг или 600 мг.
В 9-м варианте реализации пероральная нагрузочная доза согласно 5-му варианту реализации состоит по существу/состоит из двух пероральных доз 450 мг 9-[(2,2-диметилпропиламино)метил]миноциклина, которые вводят за 12 часов и 24 часа, соответственно, перед первой из вводимых один раз в день пероральных доз по 300 мг, 450 мг или 600 мг.
В 10-м варианте реализации пероральная нагрузочная доза согласно 5-му варианту реализации состоит по существу/состоит из двух пероральных доз 600 мг 9-[(2,2-диметилпропиламино)метил]миноциклина, которые вводят за 24 часа и 48 часов, соответственно, перед первой из вводимых один раз в день пероральных доз по 300 мг, 450 мг или 600 мг.
В 11-м варианте реализации пероральная нагрузочная доза согласно 5-му варианту реализации состоит по существу/состоит из двух пероральных доз по 450 мг 9-[(2,2-диметилпропиламино)метил]миноциклина, которые вводят за 24 часа и 48 часов, соответственно, перед первой из вводимых один раз в день пероральных доз по 300 мг, 450 мг или 600 мг.
В 12-м варианте реализации способ включает пероральное введение субъекту эффективного количества 9-[(2,2-диметилпропиламино)метил]миноциклина (омадациклина) или его соли с обеспечением лечения указанного субъекта, где 9-[(2,2-диметилпропиламино)метил]миноциклин вводят перорально в дозе примерно 450 мг в день в течение двух дней подряд, затем в дозе примерно 300 мг в день в течение 5 дней или более, например, в течение всего периода лечения в 7-14 дней.
При использовании в настоящем описании «бактериальная инфекция кожи и кожных структур» представляет собой инфекцию кожи и связанных мягких тканей, таких как рыхлая соединительная ткань и слизистые мембраны. Патоген, участвующий в бактериальной инфекции кожи и кожных структур, представляет собой один из видов бактерий. Указанные инфекции часто необходимо лечить антибиотиками, такими как предложенный 9-[(2,2-диметилпропиламино)метил]миноциклин.
В 13-м варианте реализации бактериальная инфекция кожи или кожных структур согласно любому из 1-12 вариантов реализации представляет собой раневую инфекцию, панникулит/рожу, крупный гнойник, фурункулез/чирей, бородавку, стафилококковый синдром ошпаренной кожи (ССОК) или эктиму.
В 14-м варианте реализации бактериальная инфекция кожи или кожных структур согласно любому из 1-12 вариантов реализации представляет собой острую бактериальную инфекцию кожи и кожных структур (ABSSSI), которая чаще всего включает раневую инфекцию, панникулит/рожу и крупный гнойник.
В США ABSSSI является причиной примерно 10% случаев госпитализации и 3,4-3,8 миллиона случаев посещения отделений скорой помощи в год. Анализ больничных листов показывает, что в 74% случаев ABSSSI назначают эмпирическое лечение с использованием антибиотиков, имеющих активность в отношении устойчивого к метициллину Staphylococcus aureus (MRSA).
В 15-м варианте реализации ABSSSI согласно 14-му варианту реализации представляет собой внебольничную ABSSSI.
В 16-м варианте реализации внебольничная ABSSSI согласно 15-му варианту реализации вызвана внебольничной инфекцией MRSA.
В 17-м варианте реализации общая площадь поверхности поражения ABSSSI смежных тканей согласно любому из 14-16 вариантов реализации составляет 75 см2 или более.
В 18-м варианте реализации ABSSSI согласно любому из 14-17 вариантов реализации включает раневую инфекцию, панникулит/рожу и/или крупный гнойник.
В 19-м варианте реализации бактериальная инфекция кожи или кожных структур (например, ABSSSI) согласно 14-му варианту реализации представляет собой инфекцию, возбуждаемую или возникающую во время госпитализации.
В 20-м варианте реализации ABSSSI согласно любому из 1-19 вариантов реализации представляет собой умеренную или тяжелую форму ABSSSI.
В 21-м варианте реализации бактериальная инфекция кожи или кожных структур согласно любому из 1-20 вариантов реализации является результатом (или возникает в результате) повреждения кожи, включая, но не ограничиваясь ими, травму, хирургическую процедуру или в.в. введение лекарственного средства.
В 22-м варианте реализации бактериальная инфекция кожи или кожных структур согласно любому из 1-20 вариантов реализации является результатом (или возникает в результате) сосудистой недостаточности или отека.
В 23-м варианте реализации субъекту-человеку согласно любому из 1-22 вариантов реализации вводят 9-[(2,2-диметилпропиламино)метил]миноциклин перорально натощак.
Например, субъект-человек, которому вводят омадациклин, не принимает пищу, антациды или поливитамины, содержащие поливалентные катионы (например, алюминий, магний, кальций, висмут, железо или цинк), или напитки за исключением воды по меньшей мере в течение 6 часов перед введением каждой дозы; и после введения каждой дозы не принимает пищу в течение 2 часов, не употребляет молочные продукты, антациды или поливитамины, содержащие поливалентные катионы (например, алюминий, магний, кальций, висмут, железо или цинк) в течение 4 часов.
В 24-м варианте реализации 9-[(2,2-диметилпропиламино)метил]миноциклин согласно любому из 1-23 вариантов реализации вводят один раз в день (например, каждую пероральную дозу вводят с интервалом примерно 24 часа). Например, субъект-человек может принимать пероральную дозу омадациклина утром натощак по меньшей мере через 6 часов после приема пищи.
В 25-м варианте реализации субъекту согласно любому из 1-24 вариантов реализации проводят лечение в течение периода примерно до 14 дней включительно, примерно до 13 дней включительно, примерно до 12 дней включительно, примерно до 11 дней включительно, примерно до 10 дней включительно, примерно до 9 дней включительно, примерно до 8 дней включительно или примерно до 7 дней включительно, примерно до 6 дней включительно, примерно до 5 дней включительно или примерно до 4 дней включительно, с обеспечением лечения указанного субъекта.
В 26-м варианте реализации субъекта согласно 25-му варианту реализации лечат в течение 7-10 дней.
В 27-м варианте реализации субъекта согласно 25-му варианту реализации лечат в течение 8 дней.
В 28-м варианте реализации соль согласно любому из 1-27 вариантов реализации представляет собой тозилатную соль 9-[(2,2-диметилпропиламино)метил]-миноциклина/омадациклина.
В 29-м варианте реализации бактериальная инфекция кожи или кожных структур согласно любому из 1-28 вариантов реализации заведомо или предположительно вызвана грамположительными патогенами.
В 30-м варианте реализации грамположительные патогены согласно 29-му варианту реализации могут включать (без ограничений) Staphylococcus aureus, Staphylococcus lugdunensis, вид Streptococcus, Streptococcus agalactiae, Streptococcus mitis, вид Enterococcus (Enterococcus faecalis (такие как VRE или VSE) или Enterococcus faecium (такие как VRE или VSE)), группу Streptococcus anginosus (S. anginosus, S. constellatus и S. intermedius, то есть бета-, альфа- или негемолитические), Streptococci группы вириданс (VGS), Clostridiumperfringens, Finegoldia magna или их комбинацию.
В 31-м варианте реализации Staphylococcus aureus согласно 30-му варианту реализации представляет собой устойчивый к метициллину Staphylococcus aureus (MRSA) или чувствительный к метициллину Staphylococcus aureus (MSSA).
В 32-м варианте реализации вид Streptococcus согласно 30-му варианту реализации включает группу Streptococcus anginosus.
В 33-м варианте реализации вид Streptococcus согласно 30-му варианту реализации включает бета-гемолитические Streptococci или S. anginosus.
В 34-м варианте реализации вид Streptococcus согласно 30-му варианту реализации включает негемолитические Streptococci или S. intermedius.
В 35-м варианте реализации вид Streptococcus согласно 30-му варианту реализации включает альфа-гемолитические Streptococci или S. constellatus.
В 36-м варианте реализации вид Enterococcus согласно 30-му варианту реализации включает Enterococcus faecalis (VSE).
В 37-м варианте реализации вид Streptococcus согласно 30-му варианту реализации включает Streptococcus pyogenes.
В 38-м варианте реализации бактериальная инфекция кожи или кожных структур согласно любому из 1-28 вариантов реализации заведомо или предположительно вызвана грамотрицательными патогенами.
В 39-м варианте реализации грамотрицательные патогены согласно 38-му варианту реализации включают Enterobacter cloacae, Escherichia coli, Klebsiella pneumoniae, Prevotella denticola, Prevotella melaninogenica или их комбинацию.
В 40-м варианте реализации нежелательные явления (НЯ) со стороны ЖКТ, возникшие в ходе лечения (если они были), согласно любому из 1-39 вариантов реализации преимущественно являются слабыми.
В 41-м варианте реализации нежелательные явления (НЯ) со стороны ЖКТ, возникшие в ходе лечения (если они были), согласно любому из 1-40 вариантов реализации не приводят к прекращению терапии.
В 42-м варианте реализации способ согласно любому из 1-41 вариантов реализации имеет более высокий показатель клинической эффективности по сравнению с линезолидом (N-[[3-(3-фтор-4-морфолинофенил)-2-оксооксазолидин-5-ил]метил]-ацетамид), где линезолид вводят перорально в дозе 600 мг каждые 12 часов.
В 43-м варианте реализации согласно любому из 1-42 вариантов реализации у субъекта-человека достигается эффективный ранний клинический ответ (ECR) (например, через 48-72 часа после введения первой дозы), определенный как выживаемость при уменьшении размера поражения на 20% или более по сравнению со значением, измеренным до начала лечения (без проведения какой-либо вспомогательной антибактериальной терапии).
В 44-м варианте реализации согласно любому из 1-43 вариантов реализации эффективный клинический ответ (CR) достигается у субъекта-человека после завершения лечения, где эффективный клинический ответ определен как выживаемость при устранении или улучшении одного или более признаков или симптомов инфекции до такого уровня, при котором дальнейшая антибактериальная терапия не требуется.
В 45-м варианте реализации согласно любому из 1-44 вариантов реализации AUC0-24 после введения двух первых доз 9-[(2,2-диметилпропиламино)метил]-миноциклина или его соли составляет примерно 9000 нг*ч/мл, 9500 нг*ч/мл, 10000 нг*ч/мл или 10500 нг*ч/мл или находится в диапазоне между любыми двумя из указанных выше значений.
В 46-м варианте реализации согласно любому из 1-45 вариантов реализации каждую дозу 9-[(2,2-диметилпропиламино)метил]миноциклина или его соли вводят в виде 150 мг таблеток. То есть, вводят 3 таблетки в случае 450 мг пероральной дозы, 2 таблетки в случае 300 мг пероральной дозы.
В 47-м варианте реализации способ согласно любому из 1-46 вариантов реализации имеет показатель клинической эффективности примерно 70-100%.
В 48-м варианте реализации показатель клинической эффективности согласно 47-му варианту реализации составляет примерно 80-100%, примерно 79-98%, примерно 79-94%, примерно 84-98%, примерно 80-88% или примерно 84-89%.
В 49-м варианте реализации показатель клинической эффективности согласно 47ому или 48-му вариантам реализации, который отмечают через 48-72 часа после введения первой пероральной дозы, составляет примерно 79-94% или примерно 84-89% или примерно 87,5%.
В 50-м варианте реализации ABSSSI согласно 49-му варианту реализации состоит по существу из раневой инфекции, и показатель клинической эффективности составляет примерно 84-94% или примерно 89%.
В 51-м варианте реализации ABSSSI согласно 49-му варианту реализации состоит по существу из панникулита/рожи, и показатель клинической эффективности составляет примерно 74-84% или примерно 79%.
В 52-м варианте реализации ABSSSI согласно 49-му варианту реализации состоит по существу из крупного гнойника, и показатель клинической эффективности составляет примерно 90-98% или примерно 94%.
В 53-м варианте реализации показатель клинической эффективности согласно 47-му или 48ому вариантам реализации представляет собой показатель общей клинической эффективности, отмечаемый примерно через 7-14 дней после введения последней дозы, и составляет примерно 79-98%), примерно 75-95%), примерно 79-89%), 84%, примерно 95-100% или примерно 98%.
В 54-м варианте реализации ABSSSI согласно 53-му варианту реализации состоит по существу из раневой инфекции, и показатель общей клинической эффективности составляет примерно 80-85% или примерно 82-83%.
В 55-м варианте реализации ABSSSI согласно 53-му варианту реализации состоит по существу из панникулита/рожи, и показатель общей клинической эффективности составляет примерно 85-91%, примерно 87-88% или примерно 88%.
В 56-м варианте реализации ABSSSI согласно 53-му варианту реализации состоит по существу из крупного гнойника, и показатель общей клинической эффективности составляет примерно 80-88% или примерно 84%.
В 57-м варианте реализации согласно любому из 1-56 вариантов реализации 9-[(2,2-диметилпропиламино)метил]миноциклин или его соль имеет более высокую эффективность по сравнению с линезолидом при лечении бактериальной инфекции кожи или кожных структур (например, ABSSSI). Линезолид представляет собой N-[[3-(3-фтор-4-морфолинофенил)-2-оксооксазолидин-5-ил]метил]ацетамид, реализуемый под торговой маркой ZYVOX™. В определенных вариантах реализации линезолид вводят перорально в дозе 600 мг или внутривенно в дозе 600 мг каждые 12 часов.
В 58-м варианте реализации согласно любому из 1-57 вариантов реализации 9-[(2,2-диметилпропиламино)метил]миноциклин или его соль вводят совместно с фармацевтически приемлемым носителем.
В 59-м варианте реализации фармацевтически приемлемый носитель согласно 58ому варианту реализации подходит для перорального введения.
В 60-м варианте реализации согласно любому из 1-59 вариантов реализации способ имеет показатель клинической эффективности, например, определенный по уменьшению размера поражения через 48-72 часа после введения первой дозы, составляющий примерно 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90% или более.
В 61-м варианте реализации согласно любому из 1-60 вариантов реализации способ имеет показатель клинической эффективности, например, клиническую эффективность, определенную на основании оценки исследователем клинического ответа через 7-14 дней (например, 7-10 дней или 7, 8, 9, 10 дней) после введения последней дозы, составляющий примерно 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% или более.
В 62-м варианте реализации согласно любому из 1-61 вариантов реализации у субъекта-человека имеется сопутствующая бактериемия.
В 63-м варианте реализации согласно любому из 1-62 вариантов реализации субъект-человек имеет ожирение (например, ИМТ≥30).
В 64-м варианте реализации согласно любому из 1-63 вариантов реализации субъект-человек имеет слабое или умеренное нарушение функции почек.
В 65-м варианте реализации согласно любому из 1-64 вариантов реализации субъект-человек имеет нарушение функции печени.
В 66-м варианте реализации согласно любому из 1-65 вариантов реализации возраст субъекта-человека составляет более 45 лет, более 50 лет, более 55 лет, более 60 лет, более 65 лет, более 70 лет или более 75 лет.
В 67-м варианте реализации согласно любому из 1-66 вариантов реализации субъекту-человеку вводят в.в. лекарственные средства.
Дополнительные подробности различных вариантов реализации описаны ниже.
2. Определения
«ABSSSI» или «острую бактериальную инфекцию кожи и кожных структур» также иногда называют инфекцией кожи и мягких тканей (SSTI). Она является одним из типов инфекции кожи и связанных мягких тканей, таких как рыхлая соединительная ткань и слизистые мембраны. FDA Министерства здравоохранения и социального обеспечения США опубликовало в октябре 2013 года документ под названием «Руководство для промышленности. Острые бактериальные инфекции кожи и кожных структур: Разработка лекарственных средств для лечения» («Руководство», содержание которого включено в настоящую заявку посредством ссылки) для помощи спонсорам в клинической разработке лекарственных средств для лечения ABSSSI. В Руководстве ABSSSI определена как панникулит/рожа, раневая инфекция и крупный кожный гнойник с измеренной площадью поверхности 75 см2 или более. В Руководстве не рассматривают менее тяжелые кожные инфекции, такие как импетиго и небольшой кожный гнойник, а также инфекции, для которых требуются более сложные режимы лечения, такие как инфекции, возникающие в результате укусов животных или насекомых, некротический фасциит, инфекции при синдроме диабетической стопы, инфекции в пролежневой язве, мионекроз и гангренозная эктима, или инфекции размером менее 75 см2, для которых может требоваться введение антибиотиков.
Таким образом, в определенных вариантах реализации ABSSSI при использовании в настоящем документе включает панникулит/рожу, раневую инфекцию и крупный кожный гнойник. В определенных вариантах реализации панникулит/рожа, раневая инфекция и/или крупный кожный гнойник имеет минимальную площадь поверхности поражения примерно 75 см2. Размер поражения может быть измерен по площади покраснения, отека или уплотнения.
В определенных вариантах реализации, в случае ABSSSI, которая поражает определенные участки поверхности тела, такие как лицо, или детей, подходящих и доступных для клинического исследования 3 фазы, ABSSSI может включать поражения с площадью поверхности менее 75 см2, такой как примерно 70 см2, 65 см2, 60 см2, 55 см2, 50 см2, 45 см2 или примерно 40 см2.
В определенных вариантах реализации ABSSSI может поражать определенные участки тела, например, на лице, рядом с ушами или на руках, где поражения могут быть даже меньше 40 см2, но при этом все равно могут быть вылечены антибиотиком.
В определенных других вариантах реализации ABSSSI не включает поражения с площадью поверхности менее 75 см2.
Размер поддающейся лечению инфекции кожи и кожных структур может быть измерен по общей площади поверхности смежных тканей. Способы оценки размера поражения в общем случае включают, но не ограничиваются ими, следующие: (1) умножение значения измеренной вручную длины на значение перпендикулярной длине ширины; (2) цифровую планиметрию; и (3) полученные на компьютере изображения. Например, общая площадь поверхности смежных пораженных тканей может быть вычислена как произведение максимальной длины (от одного края до другого) и максимальной ширины (измеренной перпендикулярно длине), определенных, например, с использованием линейки для измерения ран.
В определенных вариантах реализации ABSSSI при использовании в настоящем документе включает панникулит/рожу, раневую инфекцию, крупный кожный гнойник, а также менее тяжелые кожные инфекции, такие как импетиго, и небольшой кожный гнойник (например, имеющий площадь поражения менее 75 см2). В другом варианте реализации ABSSSI не включает менее тяжелые кожные инфекции, такие как импетиго и небольшой кожный гнойник.
«Пораженная ткань» определена как ткань, на которой присутствует одно или более из следующего: покраснение, отек или уплотнение.
«Раневая инфекция» включает инфекцию, характеризующуюся истечением гноя из раны и покраснением, отеком и/или уплотнением окружающих тканей. В определенных вариантах реализации наименьшее расстояние, на которое распространяется покраснение, отек и/или уплотнение окружающих тканей относительно периферических границ раны, составляет по меньшей мере 5 см.
«Панникулит/рожа» включает диффузную кожную инфекцию, характеризующуюся распространением участков покраснения, отека и/или уплотнения.
«Крупный гнойник» включает инфекцию, характеризующуюся скоплением гноя внутри слоя дермы или более глубокого слоя кожи и покраснением, отеком и/или уплотнением окружающих тканей. В определенных вариантах реализации наименьшее расстояние, на которое распространяется покраснение, отек и/или уплотнение окружающих тканей относительно периферической границы гнойника, составляет по меньшей мере 5 см.
Распространенные бактериальные патогены, вызывающие ABSSSI, включают, но не ограничиваются ими: Streptococcus pyogenes и Staphylococcus aureus, включая устойчивый к метициллину S. aureus, а также менее распространенные источники включают другие виды Streptococcus, Enterococcus faecalis или грамотрицательные бактерии.
В определенных вариантах реализации ABSSSI представляет собой внебольничную ABSSSI, которая (в отличие от внутрибольничной ABSSSI) развивается у индивидуума, который не сталкивается или сталкивается в незначительной степени с системой здравоохранения. В противоположность этому, внутрибольничная ABSSSI развивается у лиц, проживающих в учреждениях для оказания долгосрочного ухода или недавно посещавших больницу или другое медицинское учреждение.
«Нежелательное явление (НЯ)» включает любое неблагоприятное, побочное или незапланированное явление в виде признаков, симптомов, заболевания или лабораторных или физиологических показателей, возникающих у индивидуума, которому дают дозу или совокупность доз предложенного 9-аминометилминоциклинового соединения (9-[(2,2-диметилпропиламино)метил]-миноциклин) во время лечения.
Термин «эффективное количество» в настоящем документе включает количество омадациклина, требуемое для лечения бактериальной инфекции. Например, эффективное количество описывает эффективный уровень, достаточный для достижения целевого терапевтического эффекта за счет уничтожения бактерий и/или подавления роста бактерий. Предпочтительно бактериальная инфекция излечивается, когда уничтожается патоген (например, бактерия).
«Условия натощак» при использовании в настоящем описании означают, что субъект, которого лечат, не употребляет пищу, антациды или поливитамины, содержащие поливалентные катионы (например, алюминий, магний, кальций, висмут, железо или цинк), или напитки за исключением воды по меньшей мере в течение 6 часов перед введением каждой дозы; и после введения каждой дозы не употребляет пищу в течение 2 часов, не употребляет молочные продукты, антациды или поливитамины, содержащие поливалентные катионы (например, алюминий, магний, кальций, висмут, железо или цинк) в течение 4 часов.
«Фурункулез/чирей» включает глубокую бактериальную инфекцию волосяных фолликул.
«Бородавка» включает объединение нескольких фурункулов.
«Стафилококковый синдром ошпаренной кожи (ССОК)» включает образование красных нарывов на коже, вызванное экзотоксинами токсигенных штаммов бактерий Staphylococcus aureus.
«Эктима» включает крустозную эрозию или изъязвление (глубокая форма импетиго), вызванные Streptococcus pyogenes и/или Staphylococcus aureus.
«Клинический ответ» в случае ABSSSI может быть определен на основании уменьшения в процентах размера поражения через 48-72 часа по сравнению с исходным уровнем, измеренного у пациентов, которые не получали вспомогательную терапию и остались живыми. Клинический ответ у пациента в указанном временном интервале в общем случае определен как уменьшение размера поражения на 20 процентов или более по сравнению с исходным уровнем.
Наличие системного воспалительного ответа может быть определено по меньшей мере по одному из следующего: повышенному уровню белых кровяных телец (WBC) (например, 10000 клеток/мм3 или более) или лейкопении (например, 4000 клеток/мм3 или менее); повышенному уровню незрелых нейтрофилов (например, 15% палочкоядерных форм или более) независимо от общего уровня периферических белых кровяных телец (WBC); поражению лимфатической системы, например, лимфангиту или лимфаденопатии, поблизости от рассматриваемого инфицированного участка и в области, которая позволяет предположить истечение гноя из указанного участка; жару или гипотермии, определяемым при температуре выше чем примерно 38,0°С [100,4°F] или ниже 36,0°С [95,5°F].
Термин «лечение» или «способ лечения» относится к ослаблению, устранению или снижению одного или более симптомов нарушения, например, бактериальной инфекции кожи или кожных структур (например, ABSSSI), подвергающегося лечению. В определенных вариантах реализации термин «лечение» включает уничтожение бактерий, связанных с инфекцией, подвергающейся лечению.
Термин «профилактика» обозначает предупреждение или уменьшение риска бактериальной инфекции.
Термин «устойчивость» или «устойчивый» относится к стандартам антибиотиков/организмов, таким как определено Институтом клинических и лабораторных стандартов (CLSI) и/или Управлением по контролю за продуктами питания и лекарственными средствами (FDA).
«Фармацевтически приемлемый носитель» включает вещества, которые можно вводить совместно с 9-[(2,2-диметилпропиламино)метил]миноциклином или его солью и которые позволяют осуществлять лекарственному соединению свою предполагаемую функцию, например, излечение или предупреждение бактериальной инфекции кожи или кожных структур. Подходящие фармацевтически приемлемые носители включают, но не ограничиваются ими, воду, солевые растворы, спирт, растительные масла, полиэтиленгликоли, желатин, лактозу, амилозу, стеарат магния, тальк, кремниевую кислоту, вязкий парафин, парфюмерное масло, моноглицериды и диглицериды жирных кислот, сложные эфиры нефтяных жирных кислот, гидроксиметилцеллюлозу, поливинилпирролидон и т.д. Фармацевтические препараты можно стерилизовать и при желании смешивать со вспомогательными агентами, например, смазывающими веществами, консервантами, стабилизаторами, увлажнителями, эмульгаторами, солями для регулирования осмотического давления, буферами, красителями, вкусоароматическими и/или ароматическими веществами и т.д., которые не воздействуют отрицательно на активные соединения согласно настоящему изобретению.
В определенных вариантах реализации фармацевтически приемлемые (инертные) носители имеют форму таблеток, капсул, пастилок, формованных пастилок, леденцов, порошков, распыляемых составов, кремов, бальзамов, суппозиториев, желе, гелей, паст, лосьонов, мазей, водных суспензий, инъекционных растворов, эликсиров, сиропов и т.д. Указанные носители включают твердые разбавители или наполнители, стерильные водные среды и различные нетоксичные органические растворители и т.д. Кроме того, в пероральные фармацевтические композиции можно добавлять подходящие подсластители и/или вкусоароматические добавки. В общем случае терапевтически эффективный омадациклин согласно настоящему изобретению содержится в указанных лекарственных формах в концентрации в диапазоне от примерно 5,0% до примерно 70% по массе.
В случае перорального введения можно применять таблетки, содержащие различные вспомогательные вещества, такие как микрокристаллическая целлюлоза, цитрат натрия, карбонат кальция, фосфат дикальция и глицин, совместно с различными разрыхлителями, такими как крахмал (и предпочтительно кукурузный, картофельный или тапиоковый крахмал), альгиновая кислота и определенные силикатные комплексы, совместно с гранулирующими связывающими веществами, такими как поливинилпирролидон, сахароза, желатин и аравийская камедь. Кроме того, смазывающие агенты, такие как стеарат магния, лаурилсульфат натрия и тальк, часто крайне полезны для получения таблеток. Твердые композиции схожего типа также можно применять в качестве наполнителей в желатиновых капсулах; предпочтительные материалы для указанной задачи также включают лактозу или молочный сахар, а также высокомолекулярные полиэтиленгликоли.
В случае, когда требуется получение водных суспензий и/или эликсиров для перорального введения, активный ингредиент можно объединять с различными подсластителями или вкусоароматическими добавками, окрашивающими веществами или красителями и, при желании, с эмульгаторами и/или суспендирующими агентами, а также совместно с такими разбавителями, как вода, этанол, пропиленгликоль, глицерин и различные их комбинации.
В определенных вариантах реализации 9-[(2,2-диметилпропиламино)метил]-миноциклин или его соль получают в виде таблетки совместно со вспомогательными веществами, включая лактозы моногидрат, микрокристаллическую целлюлозу, стеарилфумарат натрия, кросповидон, коллоидный диоксид кремния, бисульфит натрия, поливиниловый спирт, диоксид титана, тальк, глицерилмонокаприлокапрат, лаурилсульфат натрия и/или желтый оксид железа.
Помимо лечения субъекта-человека способы лечения согласно настоящему изобретению также являются полезными при ветеринарном использовании, например, для лечения домашнего скота, такого как крупный рогатый скот, овцы, козы, коровы, свиньи и т.д.; птицы, такой как курица, утки, гуси, индейки и т.д.; лошадей; и домашних животных, таких как собаки и кошки. Кроме того, омадациклин и его соли можно применять для лечения субъектов, не являющихся животными, таких как растения.
Изобретение в целом было описано выше, и в приведенных ниже примерах дополнительно проиллюстрировано (но не ограничено) изобретение, описанное в настоящем документе.
ПРИМЕРЫ
Пример 1 Влияние приема пищи на биодоступность омадациклина у здоровых добровольцев
Омадациклин (ОМС, 9-[(2,2-диметилпропиламино)метил]миноциклин) является лучшим среди аминометилциклиновых антибиотиков, который характеризуется улучшенной противомикробной активностью in vitro (Honeyman et al, Antimicrob Agents Chemother. 59:7044-7053, 2015). Его активность in vitro продемонстрирована для широкого спектра грамположительных и грамотрицательных аэробов, множества анаэробов и атипических патогенов, включая Legionella spp.и Mycoplasma spp. (Macone et al, Antimicrob Agents Chemother. 58:1127-1135, 2014; Dubois et al, In vitro activity of omadacycline against Legionella pneumophila. Обзор, представленный на 55ой конференции ICAAC, San Diego, СА, 17-21 сентября 2015 года; Kim et al, Activity and efficacy of omadacycline against Clostridium difficile. Обзор, представленный на 2016 ECCMID, Amsterdam, the Netherlands). У пациентов с осложненной инфекцией кожи и кожных структур для омадациклина наблюдались клиническая эффективность и переносимость, сравнимые с линезолидом (Noel, et al, Antimicrob Agents Chemother. 56:5650-5654, 2012; Noel et al, Safety and efficacy of PTK 0796 (omadacycline) as treatment of complicated skin and soft tissue infection (cSSTI). Стенд, представленный на 23ем Европейском конгрессе по клинической микробиологии и инфекционным заболеваниям, 31 марта - 3 апреля 2012 года, London, UK).
По результатам исследований 3 фазы был сделан вывод о возможности применения омадациклина в рамках пероральной и внутривенной (в.в.) монотерапии у пациентов с острой бактериальной инфекцией кожи и кожных структур (ABSSSI).
В процессе разработки пероральные составы омадациклина прошли путь от использования свободного основания в капсулах до различных таблеток и солевых составов для оптимизации пероральной доступности и улучшения переносимости. В таблетированном составе, используемом в актуальном исследовании 3 фазы, содержится тозилатная соль омадациклина, для которой была показана абсолютная биодоступность 34,5% при введении натощак. Первичной задачей настоящего исследования являлась оценка относительной биодоступности отдельной пероральной 300 мг дозы омадациклина (которую вводили в виде таблетированного состава для исследования 3 фазы) в различные моменты времени после приема пищи здоровыми взрослыми субъектами.
Результаты данного исследования показали, что прием пищи влиял на пероральную биодоступность отдельной 300 мг дозы ОМС.
Вкратце, исследование представляло собой рандомизированное открытое перекрестное исследование 1 фазы с 4 периодами введения. Перед введением в день 1 периода 1 субъектов случайным образом распределяли в одну из четырех групп с различными последовательностями введения (см. таблицу 1). В день 1 каждого периода субъектам вводили одну пероральную дозу 300 мг омадациклина (2×150 мг таблетки) в различные моменты времени после приема пищи. Период вымывания между периодами введения составлял по меньшей мере 5 дней. Заключительное посещение для завершения исследования происходило через 6-10 дней после введения последней дозы омадациклина.
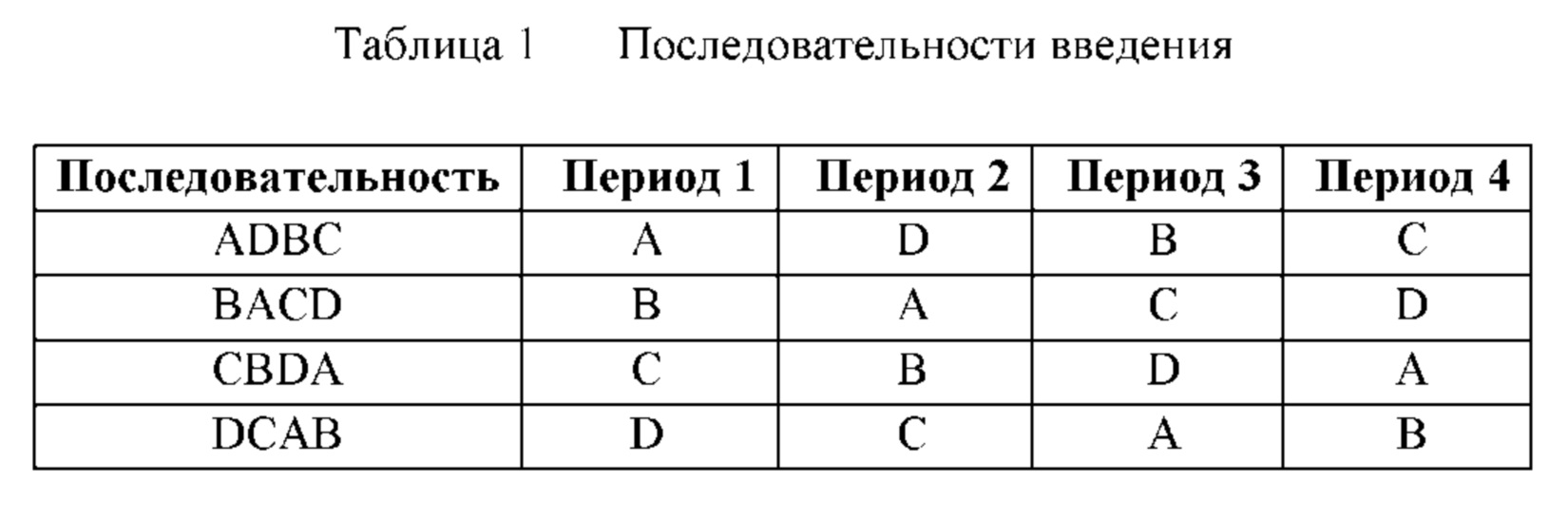
А: субъекты не принимали пищу ночью (пищу или напитки за исключением воды по меньшей мере в течение 6 часов перед введением дозы); принимали обычную пищу с высоким содержанием жиров (без молочных продуктов) через 3 часа после введения дозы
В: принимали обычную пищу с высоким содержанием жиров (без молочных продуктов) за 4 часа перед введением дозы
С: принимали обычную пищу с высоким содержанием жиров (без молочных продуктов) за 2 часа перед введением дозы
D: принимали обычную пищу с высоким содержанием жиров за 2 часа перед введением дозы
Пища с высоким содержанием жиров (примерно 50% от общей энергетической ценности пищи) и высококалорийная пища (примерно от 800 до 1000 калорий) удовлетворяли рекомендациям Руководства Управления США по контролю за продуктами питания и лекарственными средствами и обеспечивали примерно 150, 250 и 500-600 калорий от белков, углеводов и жиров, соответственно (Руководство FDA, 2002). Указанную пищу следовало принимать в течение 20 минут. Введение дозы для способов лечения В, С и D проводили относительно завершения приема пищи. Во все 4 периода введения субъекты не принимали пищу или напитки за исключением воды в течение по меньшей мере 3 часов после введения дозы и не принимали молочные продукты, антациды или поливитамины в течение 4 часов после введения дозы.
Всего были отобраны 32 субъекта, которым проводили введение по меньшей мере в течение одного периода. Общий средний возраст составлял 32,3 года, диапазон составлял от 21 года до 50 лет; 47% были мужчинами (таблица 2). Один субъект был исключен из исследования из-за положительного результата скрининга на алкоголь на исходном уровне во время периода 3, и ему не проводили способы лечения А и D. Один субъект попросил о досрочном завершении, и ему не проводили способы лечения В и С. Данные РК были доступны для 31 субъекта для каждого способа введения.

Образцы крови для оценки фармакокинетики (PK) омадациклина собирали перед введением дозы (до введения дозы) и через 0,5, 1, 1,5, 2, 2,5, 3, 4, 6, 8, 12, 16 и 24 часа после введения дозы о время каждого периода. Параметры РК включали: площадь под кривой зависимости концентрации в плазме от времени (AUC) от момента времени 0 до 24 часов после введения дозы (AUC0-24); AUC от момента времени 0 до последней поддающейся количественной оценке концентрации (AUC0-t); AUC от момента времени 0, экстраполированная на бесконечность (AUC0-inf); максимальную (пиковую) наблюдаемую концентрацию в плазме (Cmax); время достижения Cmax (Tmax); терминальный период полувыведения (Т1/2); константу скорости терминальной элиминации (λz).
Оценку безопасности и переносимости проводили на основании: нежелательных явлений (НЯ); определения основных показателей жизнедеятельности в различные моменты времени в течение 24 часов после введения дозы каждый период введения; и клинических лабораторных исследований в течение 24 часов после введения дозы каждый период введения.
Для статистического анализа, отдельно определенные параметры РК омадациклина обобщали при помощи описательной статистики. Среднее геометрическое определяли для AUC и Cmax. Параметры РК оценивали при помощи некомпартментного анализа с использованием Phoenix® WinNonlin® (Pharsight Corp, St. Louis, Missouri) версии 6.2.1. Доверительные интервалы (ДИ) для сравнения исследуемых способов введения (после приема пищи: способы В, С и D) со сравнительным способом введения (натощак: способ А) определяли для AUC0-24, AUC0-t, AUC0-inf и Cmax. Отсутствие влияния приема пищи определяли, если 90% ДИ отношения средних геометрических, определенных для исследуемого способа и контроля (В/А, С/А или D/A), не выходил за пределы критерия интервалов от 80% до 125% в случае AUC0-24, AUC0-t, AUC0-inf и Cmax. Для Tmax использовали критерий знаковых рангов Уилкоксона. р≤0,05 принимали за статистически значимые различия.
В линейной модели со смешанными эффектами, где способ введения, последовательность и период использовали в качестве фиксированных эффектов, а субъектов, сгруппированных по последовательности введения, использовали в качестве случайного эффекта, проводили подстановку преобразованных в натуральный логарифм параметров РК для оценки эффектов и 90% доверительных интервалов (ДИ) для сравнения условий после приема пищи и натощак.


Анализ РК показал, что у 31 субъекта, для которых проводили анализ РК, значения AUC0-inf, AUC0-t и AUC0-24 натощак составляли 10,2, 7,2 и 7,2 мкг*ч/мл, соответственно, и Cmax составляла 0,6 мкг/мл. Для всех периодов введения средний Т1/2 находился в диапазоне от 13,5 до 13,8 часа, и медианное Tmax находилось в диапазоне от 2,5 до 2,9 часа. Нежелательные явления, связанные с лечением, или клинически значимые изменения лабораторных показателей или основных показателей жизнедеятельности не наблюдали. См. таблицу 3.
Значительное уменьшение системного уровня омадациклина наблюдали для всех трех способов введения (способы введения В, С и D) по сравнению со способом введения А (ФИГ. 1 и таблица 4).


Влияние приема пищи было более выраженным при приеме пищи с высоким содержанием жиров незадолго до введения дозы и при включении молочных продуктов в рацион. По сравнению с дозой, вводимой натощак, уровень омадациклина (Cmax и AUC) были понижены на 15%-17% в случае приема пищи, не содержащей молочные продукты, за 4 часа перед введением дозы; были понижены на 40%-42% в случае приема пищи, не содержащей молочные продукты, за 2 часа до введения дозы; и были понижены на 59%-63% в случае приема пищи, содержащей молочные продукты, за 2 часа перед введением дозы. Изменчивость системного уровня омадациклина в группе субъектов была схожей для способов лечения А, В и С (CV 22,4-29,2%)) в случае Cmax и AUC. В противоположность этому, в способе D CV для указанных параметров составлял 42,6-44,4%.
Что касается безопасности и переносимости, то два субъекта отмечали НЯ, возникшие в ходе лечения (один сообщал о тошноте, один сообщал о сонливости); оба явления имели слабую интенсивность, и их рассматривали как не связанные с исследуемым лекарственным средством. Ни один субъект не закончил досрочно исследование по причине НЯ, и ни один субъект не отмечал серьезные НЯ (СНЯ). Незначительное увеличение частоты сердечных сокращений относительно исходного уровня (медианное значение от 8 до 10 уд./мин через 4-6 часов после введения дозы) наблюдали для способа введения А (т.е. в группе с наивысшим уровнем омадациклина). Во всех других группах медианное изменение частоты сердечных сокращений относительно исходного уровня составляло ≤3 уд./мин во все моменты времени, когда проводили измерения. Значительные изменения кровяного давления не наблюдали. Клинически значимые изменения клинических лабораторных показателей отсутствовали.
Результаты показывают, что одна пероральная доза омадациклина хорошо переносилась. Введение 300 мг дозы через 2-4 часа после приема пищи понижало биодоступность по сравнению с введением натощак. Таким образом, предпочтительно вводимый раз в день пероральный омадациклин следует принимать по меньшей мере через 6 часов после приема пищи.
Пример 2 Рандомизированное двойное слепое перекрестное исследование 1 фазы с 3 периодами введения для оценки безопасности, переносимости и фармакокинетики после введения нескольких доз омадациклина или плацебо здоровым взрослым субъектам
Первичной задачей настоящего исследования являлась оценка и сравнение фармакокинетики (PK) 300, 450 и 600 мг доз перорального омадациклина, вводимых ежедневно в течение 5 дней. Вторичной задачей исследования являлась оценка безопасности и переносимости нескольких доз омадациклина у здоровых взрослых субъектов.
Для лечения ABSSSI предполагаемая терапевтическая дневная пероральная доза (исключая какие-либо нагрузочные дозы) составляла 300 мг. Для возможных будущих исследований или в случае введения нагрузочной дозы перорального состава можно использовать дневную дозу, превышающую 300 мг, для достижения концентрации омадациклина, достаточной для борьбы с целевыми бактериями в рассматриваемых органах/тканях. В одном проводимом ранее клиническом исследовании проводили оценку отдельных пероральных доз омадациклина до 600 мг, но в исследованиях не изучали несколько дневных доз более 300 мг. Настоящее исследование было разработано для получения данных о безопасности, переносимости и фармакокинетике (линейность и пропорциональность дозе) нескольких пероральных доз омадациклина при введении дневных доз, превышающих 300 мг. Группы плацебо были включены в качестве контроля для минимизации потенциального смещения выборки при оценке переносимости.
В настоящем исследовании для введения выбирали несколько дневных пероральных доз по 300, 450 и 600 мг омадациклина или плацебо. Наименьшая доза в 300 мг, которую оценивали в исследовании нескольких доз, хорошо переносилась; указанную дневную дозу также изучали в исследованиях 3 фазы при ABSSSI. Отдельные пероральные дозы до 600 мг вводили в виде капсул здоровым взрослым субъектам в 1 клиническом исследовании ранней фазы, было определено, что они имеют приемлемый профиль безопасности. Наблюдали некоторое увеличение частоты НЯ со стороны ЖКТ для пероральных доз 400 мг или более, хотя эти явления были, как правило, слабыми (ни одно не было тяжелым), но существует возможность того, что некоторые из указанных явлений могли быть связаны с пероральным составом. Согласно ожиданиям несколько дневных доз до 600 мг в виде конечного оптимизированного таблетированного состава омадациклина имели приемлемый профиль безопасности, но было важно определить это в небольшом тщательно контролируемом исследовании 1 фазы перед оценкой указанных доз более крупных клинических исследованиях.
Таким образом, исследование было разработано как рандомизированное двойное слепое перекрестное исследование 1 фазы с 3 периодами введения здоровым взрослым субъектам. Исследование включало период скрининга (от -21 до -2 дня), 3 периода для определения исходного уровня (день -1 для каждого периода), 3 периода введения (от 1 до 6 дня для каждого периода) и посещение для завершения исследования (через 6-10 дней после введения последней дозы исследуемого лекарственного средства в период 3). Период вымывания между введением последней дозы одном периоде и введением первой дозы следующем периоде составлял по меньшей мере 5 дней. Субъектов размещали в центре исследования в день -1 периода 1 и выпускали в день 6 периода 3 после завершения отбора проб крови, отбора проб мочи и оценки безопасности через 24 часа. Субъекты возвращались в центр исследования через 6-10 дней после введения последней дозы исследуемого лекарственного средства в период 3 в рамках посещения для завершения исследования.
Выбор субъектов
Здоровые некурящие субъекты мужского и женского пола были доступны для участия в исследовании, если их возраст составлял от 18 до 55 лет (включительно), масса тела составляла ≥ 50 кг, индекс массы тела составлял от 18 до 30 кг/м2 (включительно), если они удовлетворяли всем критериям включения во время скрининга (который проводили за 21 день перед введением в период 1) и на исходном уровне (день -1) в период 1 и предоставляли подписанное информированное согласие. Состояние здоровья определяли на основании истории перенесенных заболеваний, клинических лабораторных показателей, основных показателей жизнедеятельности (пероральная температура, систолическое кровяное давление, диастолическое кровяное давление и частота сердечных сокращений), электрокардиограммы (ЭКГ) в 12 отведениях и медицинского осмотра во время скрининга. Критерии включения включали способность проглотить до 4 таблеток подряд.
Субъектов отстраняли от участия в исследовании в случае предшествующего лечения омадациклином, недавнего использования других исследуемых лекарственных средств; отклонений ЭКГ; неспособности переносить пероральные лекарственные средства; беременности или грудного кормления; использования табачных продуктов, назначаемых лекарственных средств, травяных добавок или безрецептурных лекарственных препаратов или приема пищи или напитков, содержащих ксантин (например, кофеин), в течение указанного периода времени перед началом исследования; потери/донорства крови; низкого уровня гемоглобина; высокого уровня креатинина или азота мочевины в крови; непроходимости мочевых путей/затрудненного мочеиспускания; положительного результата теста на алкоголь или наркотики; повышенной чувствительности или аллергии на какой-либо тетрациклин; признаков заболевания печени или повреждения печени; серьезной болезни за 2 недели до начала исследования; какого-либо запланированного медицинского вмешательства, которое может препятствовать исследованию; или истории заболеваний или медицинских состояний, указанных в протоколе исследования.
Дизайн исследования
От 1 до 5 дня каждого периода субъектам проводили один раз в день через 6 часов после приема пищи один из следующих способов лечения (омадациклин или плацебо) согласно схеме рандомизации:
A. 300 мг омадациклина (2×150 мг таблетки)
АР. Плацебо для 300 мг омадациклина (2 × таблетки с плацебо)
B. 450 мг омадациклина (3×150 мг таблетки)
BP. Плацебо для 450 мг омадациклина (3 × таблетки с плацебо)
C. 600 мг омадациклина (4×150 мг таблетки)
СР. Плацебо для 600 мг омадациклина (4 × таблетки с плацебо)
Все дозы исследуемого лекарственного средства вводили утром, при этом за 6 часов до введения дозы не допускался прием пищи или напитков за исключением воды. Субъекты не должны были принимать пищу или напитки за исключением воды в течение по меньшей мере 2 часов после введения дозы и не должны были принимать молочные продукты, антациды или поливитамины в течение 4 часов после введения дозы.
Перед началом введения субъекты проходили скрининг для определения доступности для исследования за 21 день перед стартом введения в период 1. Затем субъекты посещали клинический центр за день до начала введения (день -1 периода 1) для определения показателей на исходном уровне. Перед началом введения в день 1 периода 1 до 30 субъектов (24 омадациклин, 6 плацебо) случайным образом распределяли в 1 из 3 групп с различными последовательностями введения по схеме латинских квадратов, как показано в следующей таблице:

Примерно десять субъектов случайным образом распределяли в группу, в которой вводили каждую последовательность. Плацебо вводили 2 субъектам из каждой группы в качестве контроля для оценки переносимости. Субъекты, которых определяли в группу, в которой вводили омадациклин, получали омадациклин во время всех 3 периодов и во всех исследуемых дозировках. Субъекты, которых определяли в группу плацебо, получали плацебо во время всех 3 периодов. Проводили заслепление исследователей и субъектов относительно введения субъекту омадациклина или плацебо.
Оценка результатов исследования
1. Фармакокинетика в плазме
Несколько проб крови для анализа фармакокинетики (PK) омадациклина собирали в конкретные моменты времени в течение 24 часов после введения в день 1 и день 5 каждого периода. В частности, образцы крови для оценки PK омадациклина собирали у всех субъектов в следующие моменты времени: перед введением дозы (до введения) и через 0,5, 1, 1,5, 2, 2,5, 3, 4, 6, 8, 12, 16 и 24 часа после введения дозы день 1 и день 5 в каждый период. Образец крови, полученный через 24 часа после введения в день 1, собирали перед введением дозы день 2 каждого периода.
Параметры PK для некомпартментного анализа определяли в дни 1 и 5 каждого периода по результатам концентрации омадациклина в плазме и данных о фактическом времени отбора проб с использованием Phoenix® WinNonlin® (Certara, Princeton, New Jersey) версии 6.2.1., включая площадь под кривой зависимости концентрации в плазме от времени (AUC) от момента времени 0 до 24 часов после введения дозы (AUC0-24), AUC от момента времени 0 до последней поддающейся количественной оценке концентрации (АUСпосл), максимальную наблюдаемую концентрацию в плазме (Cmax), время до достижения максимальной наблюдаемой концентрации в плазме (Tmax), терминальный период полувыведения (Т1/2), константу скорости терминальной элиминации (λz) и коэффициент нарастания (Rac) AUC0-24 и Cmax.
Субъектов, которым вводили омадациклин и у которых по меньшей мере один параметр РК был доступен для оценки, включали в популяцию для анализа РК; тем не менее, субъекты могли быть исключены из популяции РК, если они пропускали введение дозы, имели диарею или тошноту в период до достижения удвоенного медианного значения Tmax, включительно.
2. Фармакокинетика в моче
Пробы мочи собирали у подгруппы субъектов в указанные интервалы в день 5 периода 2 и в день 1 и день 5 периода: перед введением дозы, от 0 до 4, от 4 до 8, от 8 до 12 и от 12 до 24 часов после введения дозы. Пробу в интервале 12-24 часа в день 1 собирали перед введением дозы день 2. Пробы мочи собирали только у подгруппы субъектов, так как анализ PK в моче был добавлен после внесения поправки в протокол исследования уже после его начала.
Следующие параметры PK в моче определяли по результатам концентрации омадациклина в моче и данных об интервале сбора проб с использованием SAS версии 9.2: почечный клиренс (CLr), фракцию дозы, которая выводится с мочой в неизменном виде, в период от 0 до 24 часов после введения (Fe0-24), и количество лекарственного средства, выводимого с мочой в неизменном виде, через 24 часа после введения дозы (Aet1-t2). Также вычисляли дополнительные параметры Ае0-4, Ае4-8, Ae8-12, Ae12-24 и Ае0-24.
3. Безопасность и переносимость
Оценка безопасности включала отслеживание нежелательных явлений (НЯ), результаты клинических лабораторных исследований, определение основных показателей жизнедеятельности, результаты электрокардиограммы (ЭКГ) в 12 отведениях и результаты медицинского осмотра. Всех случайным образом распределенных по группам субъектов, которым вводили по меньшей мере одну дозу любого исследуемого лекарственного средства (омадациклин или плацебо), включали в популяцию для анализа безопасности. Нежелательные явления были классифицированы по предпочтительным терминам и классам систем органов при помощи MedDRA версии 17.1.
Безопасность и переносимость оценивали путем отслеживания и фиксации НЯ, результатов клинических лабораторных исследований (гематология, химия сыворотки и анализ мочи), определения основных показателей жизнедеятельности (пероральная температура, систолическое кровяное давление, диастолическое кровяное давление и ЧСС), результатов ЭКГ в 12 отведениях и результатов медицинского осмотра.
Статистический анализ исследования фармакокинетики:
Отдельные значения концентрации в плазме и моче и данные об изменении времени отмечали в бланках для данных. Данные о концентрации в плазме и моче обобщали по дню и моменту времени или интервалу для каждого способа введения с использованием описательной статистики (число субъектов, среднее, СКО, коэффициент вариации [CV], медианное значение, минимальное и максимальное значение). Концентрации ниже предела количественного обнаружения (BLQ) принимали за ноль в обобщенных данных описательной статистики концентрации в плазме и моче. Профили зависимости средней и отдельной концентрации в плазме от времени приведены на фигурах с линейными и полулогарифмическими шкалами.
Параметры PK для некомпартментного анализа определяли по результатам концентрации в плазме и данных о фактическом времени отбора проб с использованием Phoenix® WinNonlin® (Certara, Princeton, New Jersey) версии 6.2.1 или более поздних версий. Параметры РК в моче определяли по результатам концентрации в моче и данных об интервале сбора пробы с использованием SAS версии 9.2 или более поздних версий. Все дополнительные статистические исследования проводили при помощи программного обеспечения SAS® (SAS Institute, Сагу, North Carolina) версии 9.2.
Для анализа PK значения BLQ принимали за ноль с тем исключением, что значение BLQ между 2 концентрациями, для которых можно было проводить вычисления, отмечали как пропущенное. Пропущенные концентрации пропускали при вычислении параметров PK. Если после нескольких последовательных концентраций BLQ следовали концентрации, для которых можно было проводить вычисления, в терминальной фазе, то указанные концентрации, следующие после концентраций BLQ, отмечали как пропущенные.
Отдельные параметры PK отмечали в бланках для данных. Описательную статистику (число субъектов, среднее, СКО, CV, медианное, минимальное и максимальное значения) вычисляли для оценочных параметров РК после введения в день 1 и день 5 каждого периода (например, AUC0-24, АUСпосл, Cmax, Tmax, Т1/2 и Rac [только в день 5] по результатам концентрации в плазме; CLr, Fe0-24 и Ае0-24 по результатам концентрации в моче). Средние геометрические были включены при определении AUC0-24, АUСпосл и Cmax.
В линейной модели со смешанными эффектами (SAS PROC MIXED), где последовательность (1А, 2А и 3А) способов введения (А, В и С) и период введения использовали в качестве фиксированных эффектов, а субъектов, сгруппированных по последовательности введения, использовали в качестве случайного эффекта, проводили подстановку преобразованных в натуральный логарифм и нормированных по дозе параметров РК AUC0-24/доза, АUСпосл/доза и Cmax/доза после введения в день 1 и день 5 каждого периода для оценки эффектов и построения 90% доверительных интервалов (ДИ). Оценочные точки и 90% ДИ для различий по логарифмической шкале экспонировали для получения оценочных значений отношений средних геометрических и соответствующих 90% ДИ по исходной шкале. Учет множественности сравнений не проводили.
Линейность доз для всех 3 дозировок оценивали путем подстановки значений Cmax, АUСпосл и AUC0-24 омадациклина после введения доз день 1 и день 5 в степенную модель (10): ln(PK)=а+b × ln(доза) + ошибка, где PK соответствует параметру PK, а соответствует свободному члену, и b соответствует коэффициенту наклона. Оценочные значения коэффициента наклона b отмечали совместно с соответствующими 2-сторонними 90% ДИ.
Для статистического анализа накопления омадациклина в линейной модели со смешанными эффектами, где день использовали в качестве фиксированного эффекта, и субъекта использовали в качестве случайного эффекта, проводили подстановку преобразованных в натуральный логарифм значений Cmax и AUC0-24 для построения 90% ДИ для дня 5 по сравнению с днем 1 (для каждой дозировки по отдельности).
Результаты
а. Демографические данные, характеристики на исходном уровне и распределение исследуемых субъектов
Среди 33 субъектов, отобранных для исследования, 26 вводили омадациклин и 7 вводили плацебо. Демографические данные и характеристики на исходном уровне в целом были схожими в группах, в которых вводили омадациклин и плацебо (таблица 2-1) и для всех последовательностей введения омадациклина (данные не показаны). Основная часть субъектов в исследовании были белыми (57,6%) мужчинами (81,8%). Общий средний возраст субъектов составлял 36,9 лет, были включены возраста от 21 до 55 лет.

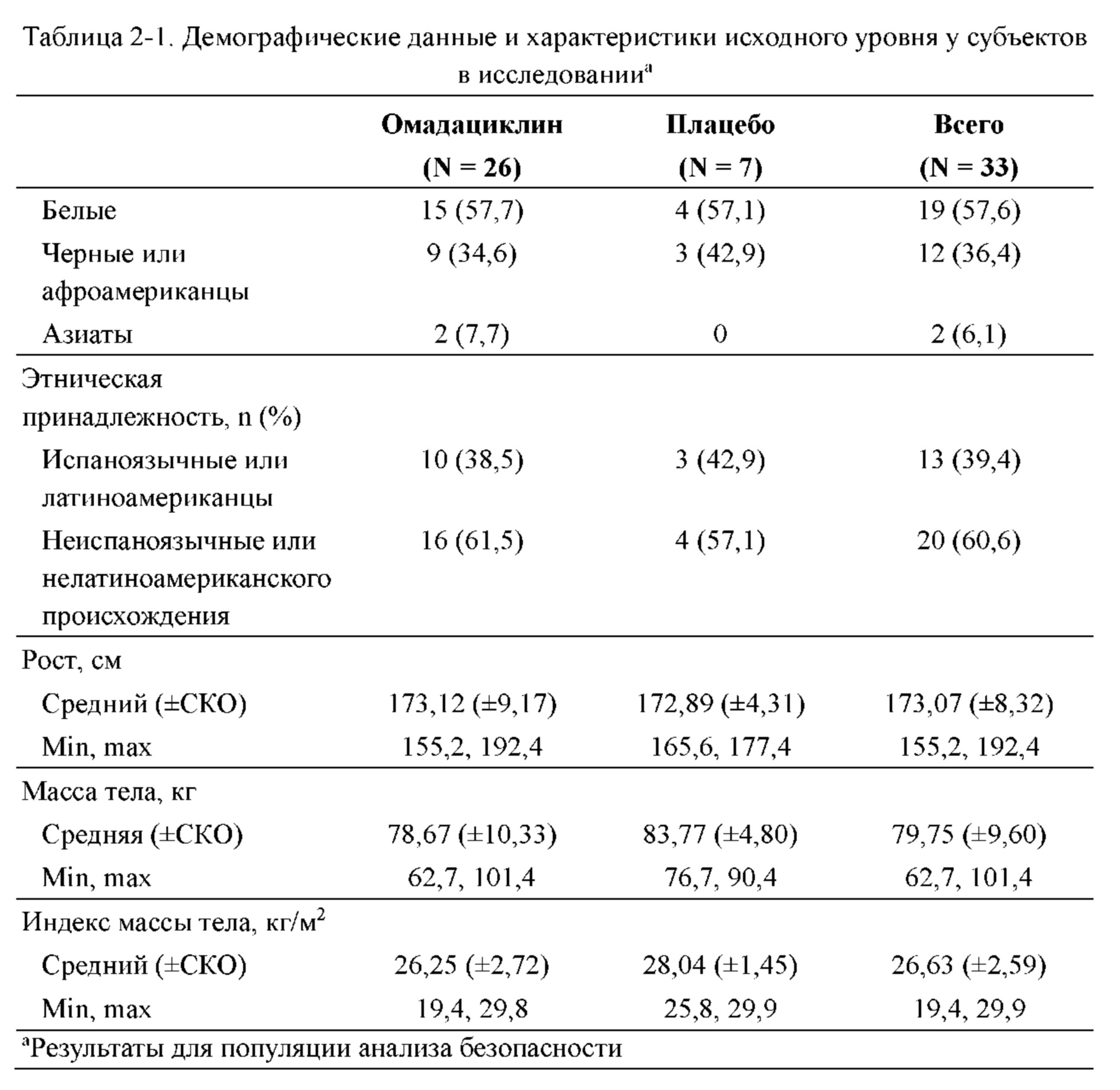
Всем 33 субъектам вводили по меньшей мере одну дозу исследуемого лекарственного средства (омадациклин или плацебо), и их включали в популяцию для анализа безопасности. Двадцать пять из 26 субъектов, которым вводили омадациклин (96,2%), были включены в популяцию для анализа PK (один субъект был исключен из этой популяции из-за рвоты после введения дозы). Четыре субъекта, которым вводили омадациклин (15,4%), и один субъект, которому вводили плацебо (14,3%), досрочно завершили исследование; указанные случаи досрочного завершения исследования были вызваны нежелательными явлениями, возникшими в ходе лечения (НЯВЛ) у 4 субъектов (см. ниже); кроме того, один субъект, которому вводили омадациклин, стал недоступен для последующего наблюдения. Таким образом, 22 субъекта получали все 5 доз по 300, 450 и 600 мг омадациклина, и 6 субъектам вводили все 5 доз плацебо в периоды 1, 2 и 3. Указанных субъектов рассматривали как завершивших исследование.
b. Фармакокинетика в плазме
Предварительные результаты исследования РК после завершения дня 1 и дня 5 приведены ниже.


Основываясь на данных фармакокинетики из данного и других исследований 1 фазы, были созданы модели PK для следующих режимов введения, и результаты приведены на ФИГ. 2-4.
В частности, на ФИГ. 2 показаны данные PK для режима введения, в котором 100 мг омадациклина вводили в.в. BID (два раза в день с интервалом 12 часов) в течение 1 дня, затем 100 мг в.в./день).
На ФИГ. 3 показаны данные PK для режима введения, в котором 450 мг омадациклина вводили п.о. QD (один раз в день) в течение 2 дней, затем 300 мг п.о./день.
На ФИГ. 4 показаны данные PK для режима введения, в котором 600 мг омадациклина вводили п.о. QD (один раз в день) в течение 2 дней, затем 300 мг п.о./день.
Очевидно, что AUC0-24 для режимов введения, показанных на ФИГ. 2 и 3, были практически идентичными.
Более подробный анализ данных приведен ниже.
Для всех исследуемых дозировок омадациклина в день 1 и день 5 каждого из 5-дневных периодов введения средний концентрации омадациклина в плазме достигали максимального значения через 2,5 часа после введения дозы (Tmax), и уровень омадациклина поддавался измерению в плазме до 24 часов после введения дозы (время последнего отбора проб) (ФИГ. 5 и таблица 2-2).

Общий уровень омадациклина (AUC0-24 и АUСпосл) и максимальная концентрация (Cmax) увеличивались при увеличении дозы омадациклина (300/450/600 мг) в день 1 и день 5 и были более высокими в день 5 по сравнению с днем 1 для соответствующих доз (ФИГ. 5 и таблица 2-2). Средний период полувыведения омадациклина из плазмы (Т1/2) был схожим для 3 исследуемых дозировок и находился в диапазоне от 13,03 до 13,66 часа в день 1 и от 15,49 до 16,83 часа в день 5 (таблица 2-2). Изменчивость системного уровня омадациклина в группе субъектов была низкой и схожей для всех трех исследуемых дозировок, где коэффициенты вариации (CV) находились в диапазоне от 23,2% до 26,6% для Cmax, AUC0-24 и АUСпосл в день 1 и от 25,0% до 27,1% для Cmax, AUC0-24 и АUСпосл в день 5 (таблица 2-2).
Несмотря на то, что значения AUC0-24, АUСпосл и Cmax увеличивались при увеличении дозы омадациклина, наблюдаемое увеличение уровня было более низким по сравнению с изменением дозы для обоих дней, когда проводили анализ (таблицы 2-2 и 2-3).

Статистический анализ показал, что при увеличении дозы от 300 мг до 600 мг уровень омадациклина (на основании значения нормированной по дозе AUC0-24) в день 1 составлял 76% от значения, ожидаемого при точной пропорциональности уровня и дозы (таблица 2-3); в день 5 наблюдаемое увеличение уровня омадациклина составляло 88% от ожидаемого (таблица 2-3). Анализ значений Cmax также продемонстрировал, что концентрации омадациклина изменялись линейно в зависимости от дозы, но в меньшей степени по сравнению с дозой, в указанном исследовании (таблицы 2-2 и 2-3).
Статистический анализ также показал накопление омадациклина в плазме после введения один раз в день в течение 5 дней подряд. В зависимости от дозы отношение накопления в день 5 и день 1 находилось в диапазоне от 1,40 до 1,62 в случае AUC0-24 и от 1,24 до 1,35 в случае Cmax (данные не показаны). Указанные факты согласуются с продолжительным периодом полувыведения омадациклина из плазмы.
Приведенные выше данные показывают, что средние концентрации омадациклина достигали максимального значения через 2,5 часа и оставались на поддающемся измерению уровне до 24 часов (последний исследуемый момент времени) для всех дозировок омадациклина (300, 450 и 600 мг). В день 5 средний стационарный уровень (AUC0-24) у субъектов, которым вводили 300 мг омадациклина, составлял 9267 нг⋅ч/мл, что согласуется с результатами предыдущих исследований перорального введения 300 мг дозы. Значения AUC0-24 и Cmax оба увеличивались при увеличении дозы практически пропорционально дозе, хотя иногда и в меньшей степени (74% - 88% от ожидаемого). Это наблюдали как в день 1, так и в день 5 введения. Благодаря относительно продолжительному периоду полувыведения (средний = ~13 ч в день 1, ~16 ч в день 5) омадациклин накапливался в плазме в течение 5 последовательных дней введения. Таким образом, для всех исследуемых дозировок системный уровень в день 5 был на ~50% выше по сравнению с днем 1. Указанный уровень накопления также согласуется с фактами, наблюдаемыми при введении нескольких доз в.в или пероральных составов омадациклина один раз в день в проводившихся ранее фармакологических исследованиях.
Что касается системного уровня, то в настоящем исследовании было показано, что концентрация омадациклина в плазме в день 1 после введения 450 мг дозы была схожей с концентрацией в день 5 после введения 300 мг дозы (средняя AUC0-24=8976,5 и 9267,2 нг⋅ч/мл, соответственно). Для тех показаний, при которых режим терапевтического лечения включает пероральное введение 300 мг дневной дозы, полученные данные обосновывают стратегию использования начальной пероральной «нагрузочной дозы» в 450 мг один раз в день в течение 1-2 дней и последующее введение 300 мг один раз в день. Указанная стратегия потенциально может устранять потребность во в.в. стадии введения и в настоящее время проходит оценку в текущем испытании 3 фазы введения исключительно перорального омадациклина пациентам с ABSSSI (Clinical Trials.gov ID, NCT02877927).
с. Фармакокинетика в моче
Так как сбор проб мочи и анализ РК были добавлены в протокол исследования в рамках поправок уже после начала исследования, была проведена оценка ограниченного числа проб (образцы от 9 субъектов в день 5 периода 2 и образцы от 8 субъектов в день 1 и день 5 в период 3). Несмотря на то, что размер выборки был слишком мал, чтобы проводить обоснованное сравнение групп, в которых вводили различные дозы омадациклина, результаты анализа, тем не менее, дали общее представление о частичном почечном клиренсе омадациклина и выведении с мочой.
Для всех доз омадациклина средняя фракция дозы, выводимой в неизменном состоянии с мочой, через 0-24 часа после введения дозы (Fe0-24) находилась в диапазоне от ~5% до ~7% в день 1 и от ~7% до ~9% в день 5. Почечный клиренс (CLr) находился в диапазоне от 2,8 до 4,2 л/ч в день 1 и от 2,4 до 3,3 л/ч в день 5 (таблица 2-4).

Анализ PK в моче в подгруппе субъектов дал предварительное представление о частичном почечном клиренсе и выведении с мочой омадациклина. В день 5 в зависимости от дозировки от ~7% до ~9% вводимой пероральной дозы выводилось с мочой в неизменном виде через 24 часа. Это составляет от примерно 20% до 25% от всасываемой дозы, так как известно, что абсолютная биодоступность таблетированного состава, используемого в настоящем исследовании, составляла 35%. Наличие неизменного омадациклина в моче позволяет предположить, что он может быть полезным при инфекциях мочевыводящих путей, что в настоящее время является предметом изучения.
d. Безопасность и переносимость
В целом, 12 из 33 субъектов из популяции для анализа безопасности отмечали всего 36 НЯВЛ во время исследования (таблица 2-5).

НЯВЛ были отмечены 38,5% субъектов, которым давали омадациклин, и 28,6% субъектов, которым давали плацебо. Самые частые НЯВЛ были классифицированы как расстройства желудочно-кишечного тракта (ЖКТ). Наиболее часто отмечаемыми НЯВЛ являлись тошнота, которую наблюдали у ≤7,7% субъектов из групп, в которых вводили 300 и 450 мг дозу омадациклина, и 16,7% в группе 600 мг. Все НЯВЛ, отмечаемые в настоящем исследовании, были слабыми или умеренными. Во время исследования не сообщалось о серьезных НЯВЛ (СНЯ). Четыре субъекта отмечали НЯВЛ, которые приводили к досрочному прекращению исследования, включая по одному субъекту в каждой из групп, в которых вводили омадациклин, и 1 субъекта в группе плацебо.
Клинически значимые отклонения в результатах анализа основных показателей жизнедеятельности, медицинского осмотра, ЭКГ, гематологии или анализа мочи отсутствовали. Химический анализ сыворотки показал, что медианное изменение концентрации аланинаминотрансферазы (АЛТ) между исходным уровнем и днем 5 каждого периода введения составляло -2,0, 5,0 и 19,5 Ед/л у субъектов, которым вводили 300, 450 и 600 мг омадациклина, соответственно. Соответствующие изменения в группе плацебо находились в диапазоне от -5,0 до -1,0 Ед/л. Существенные изменения медианного уровня аспартатаминотрансферазы (ACT), билирубина или других химических параметров сыворотки отмечены не были. Максимальное отдельное значение АЛТ составляло 150 Ед/л (в 2,7 раза выше верхнего предела нормы [ВПН]), которое наблюдали у субъекта, которому сначала вводили 450 мг омадациклина в период 1, затем 300 мг в период 2, после чего тот завершил исследование из-за изменений профиля ферментов печени; уровень билирубина у данного субъекта оставался в норме во все оцениваемые моменты времени.
Данные анализа PK в плазме указывают на то, что повышенный системный уровень лекарственного средства может быть достигнут при увеличении количества вводимого омадациклина на дозу при пероральном введении один раз в день, но увеличение уровня происходит не пропорционально дозе. Кроме того, увеличение вводимого количества омадациклина сверх определенного значения вероятно может вызывать нежелательные эффекты с точки зрения безопасности и переносимости. Несмотря на то, что введение нескольких доз по 300, 450 и 600 мг во всех случаях в целом хорошо переносилось в настоящем исследовании (все НЯВЛ были слабыми или умеренными), между дозами наблюдали некоторые различия. Частота НЯВЛ, связанных с лечением, не увеличивалась при увеличении дозы омадациклина от 300 до 450 мг (15,4% и 8,3%), но указанные явления происходили более часто при введении 600 мг (25,0%). В классе наиболее частых НЯВЛ, расстройств ЖКТ, тошноту отмечали по меньшей мере на 9% чаще в случае 600 мг дозы по сравнению с более низкими дозами, и только в 2 случаях отмечали диарею при введении 600 мг. Кроме того, химический анализ сыворотки показал небольшое, но значимое зависящее от дозы увеличение медианных концентраций АЛТ. С учетом того, что ни одно отдельное значение АЛТ не превосходило ВПН более чем в 3 раза, повышение медианной АЛТ для дозы 600 мг позволяет предположить увеличенную вероятность более значительного повышения уровня трансаминазы в сыворотке для указанной дозы. На основании полученных данных для тех случаев, при которых пероральная доза более 300 мг может быть предпочтительной, доза в 450 мг была определена как пероральная доза, которая наиболее вероятно может обеспечивать повышенный уровень омадациклина при благоприятном профиле безопасности и переносимости.
В целом, в настоящем исследовании 1 фазы изучали фармакокинетику (PK) и безопасность/переносимость нескольких пероральных доз омадациклина более 300 мг. Согласно перекрестному дизайну с использованием 3 периодов здоровых взрослых субъектов случайным образом распределяли в группы, в которых вводили омадациклин (300, 450 и 600 мг дозы различных последовательностях; n=26) или плацебо (n=7) один раз в день в течение 5 дней подряд в рамках одного периода. В плазме максимальная концентрация и общий уровень омадациклина увеличивались при увеличении дозы, но в меньшей степени по сравнению с дозой (от 74% до 88% от ожидаемого значения). Кинетика накопления омадациклина в плазме была схожей для различных доз; уровень в день 5 был на ~50% выше по сравнению с днем 1. Концентрация омадациклина в плазме в день 1 при введении 450 мг была схожей с концентрацией в день 5 при введении 300 мг. Анализ РК в моче показал частичный почечный клиренс и выведение с мочой неизменного омадациклина. Все дозы целом хорошо переносились. Полученные результаты обосновывают введение один раз в день 450 мг перорального омадациклина в рамках режима исключительно перорального введения, такого как введение один раз в день 450 мг перорального омадациклина (в виде одной или двух доз) в качестве нагрузочной дозы перед переходом к введению один раз в день более низкой 300 мг дозы перорального омадациклина, или в рамках режима введения, в котором один раз в день вводят 450 мг перорального омадациклина на протяжении всего курса лечения.
Пример 3 Рандомизированное двойное слепое многоцентровое исследование 3 фазы для сравнения безопасности и эффективности перорального омадациклина и перорального линезолида при лечении взрослых субъектов с острой бактериальной инфекцией кожи и кожных структур (ABSSSI)
9-[(2,2-диметилпропиламино)метил]миноциклин, также известный как омадациклин (ОМС), первый аминометилциклиновый антибиотик, представляет собой полусинтетическое производное класса тетрациклинов. Тетрациклины, как класс, используют уже примерно 70 лет. Они хорошо переносятся и эффективны при лечении различных бактериальных инфекций.
Для омадациклина продемонстрирована активность в отношении наиболее распространенных патогенов ABSSSI, включая устойчивый к метициллину Staphylococcus aureus (MRSA). Омадациклин оценивали в исследовании 2 фазы у 219 субъектов с осложненной инфекцией кожи и кожных структур (cSSSI) и досрочно завершенном по решению спонсора исследовании 3 фазы у 140 субъектов с cSSSI. Омадациклин хорошо переносился и имел эффективность, схожую с общепринятым препаратом сравнения (линезолидом).
Согласно недавно завершенному глобальном исследовании 3 фазы у 645 субъектов, в котором сравнивали безопасность и эффективность внутривенного (в.в.) и перорального (п.о.) омадациклниа и в.в. и п.о. линезолида при лечении взрослых субъектов с ABSSSI, омадациклин имел не меньшую эффективность по сравнению с линезолидом и хорошо переносился.
I. ДИЗАЙН ИССЛЕДОВАНИЯ
Было проведено исследование 3 фазы, разработанное для демонстрации безопасности и эффективности п.о. омадациклина по сравнению с п.о. линезолидом при лечении взрослых субъектов с ABSSSI. В частности, исследование было разработано для демонстрации того, что омадациклин, вводимый перорально (п.о.) в течение 7-14 дней, имеет не меньшую (10%) эффективность по сравнению с линезолидом, вводимым перорально в течение 7-14 дней при лечении взрослых субъектов с ABSSSI, которая заведомо или предположительно вызвана грамположительными патогенами. Кроме того, исследование было разработано для демонстрации того, что омадациклин является безопасным при лечении взрослых субъектов с ABSSSI; и того, что наблюдаемый клинический ответ связан с нейтрализацией выявленного патогена, вызывающего ABSSSI. Наконец, была проведена оценка фармакокинетики (PK) перорального омадациклина у указанных взрослых субъектов.
Эксперимент был разработан как рандомизированное (1:1), двойное слепое, двойное плацебо-контролируемое исследование 3 фазы с использованием активного препарата сравнения, в котором сравнивали омадациклин и линезолид при лечении взрослых субъектов с ABSSSI, которая была заведомо или предположительно вызвана грамположительным(-и) патогеном(-ами). Отобранные для участия субъекты, имеющие крупные гнойники, могли составлять до 30% от числа субъектов, участвовавших в рандомизации. Число отобранных для участия субъектов, которые принимали одну дозу разрешенного антибиотика краткосрочного действия за 72 часа перед рандомизацией было ограничено 25% от количества субъектов, участвовавших в рандомизации. Рандомизация субъектов была стратифицирована во всех группах по типу инфекции (раневая инфекция, панникулит/рожа или крупный гнойник) и прописанному ранее разрешенному антибиотику краткосрочного действия (принимался или нет).
Исследование состояло из 3 определенных в протоколе фаз: скрининг, двойное слепое введение и период последующего наблюдения. Все оценки для скрининга, за исключением посева крови, проводили за 24 часа перед рандомизацией. Посев крови собирали за 24 часа перед введением первой дозы исследуемого препарата. Субъектов, которые удовлетворяли критериям включения и не удовлетворяли критериям исключения, случайным образом распределяли по группам, и вводили первую дозу исследуемого препарата (омадациклина или линезолида) в центре через 4 часа после рандомизации.
Исследование было разработано согласно руководству FDA США и Европейского медицинского агентства (ЕМА) по разработке противомикробных лекарственных средств для лечения ABSSSI, а также руководствам Американского сообщества по инфекционным заболеваниям (IDSA) и обновленной в 2012 году информации от консорцима по биомаркерам фонда Национальных институтов здравоохранения.
Лекарственное средство сравнения, линезолид, было одобрено по всему миру для лечения ABSSSI, вызванной грамположительными патогенами, и имеет приемлемый и хорошо определенный профиль безопасности. Линезолид можно вводить перорально, и он одобрен регулирующими органами для лечения ABSSSI, вызванной грамположительными патогенами, включая MRSA.
Субъекты участвовали в исследовании в течение примерно 30 дней. После скрининга доступных для исследования субъектов случайным образом распределяли по группам, в которых в течение 7-14 дней вводили п.о. омадациклин или линезолид. Посещение центра для последующего наблюдения проводили примерно через 7-14 дней после введения последней дозы исследуемого препарата, и телефонный опрос в период последующего наблюдения проводили примерно через 30-37 дней после введения первой дозы исследуемого препарата.
Для того, чтобы быть допущенным до рандомизации в настоящем исследовании, субъект, помимо подписания информированного согласия, должен был удовлетворять всем из следующих ключевых критериев: (1) мужчина или женщина 18 лет или старше; (2) наличие рассматриваемой инфекции кожи и кожных структур. Для задач клинического исследования все рассматриваемые пораженные участки имели общую площадь поверхности смежных пораженных тканей 75 см2 или более, которую вычисляли как произведение максимальной длины (от одного края до другого) и максимальной ширины (измеренной перпендикулярно длине), определенных исследователем с использованием линейки для измерения ран. Пораженная ткань была определена как ткань, в которой явным образом присутствует одно или более из следующего: покраснение, отек или уплотнение; (3) рассматриваемые инфекции были классифицированы как: раневая инфекция - инфекция, характеризующаяся истечением гноя из раны и покраснением, отеком и/или уплотнением окружающих тканей, распространяющимся по меньшей мере на 5 см от периферических границ раны; панникулит/рожа - диффузная кожная инфекция, характеризующаяся распространением покраснения, отека и/или уплотнения; крупный гнойник - инфекция, характеризующаяся скоплением гноя внутри дермы или более глубокого слоя кожи и покраснением, отеком и/или уплотнением окружающих тканей, распространяющимся по меньшей мере на 5 см от периферических границ гнойника; (4) подтвержденный системный воспалительный ответ за 24 часа перед рандомизацией, определяемый ОДНИМ из следующего: повышенным уровнем белых кровяных телец (WBC) (10000 клеток/мм3 или более) или лейкопенией (4000 клеток/мм3 или менее); повышенным уровнем незрелых нейтрофилов (например, 15% палочкоядерных форм или более) независимо от общего уровня периферических WBC; поражением лимфатической системы, например, лимфангитом или лимфаденопатией, поблизости от рассматриваемого инфицированного участка и в области, которая позволяет предположить истечение из указанного участка; жаром или гипотермией, определяемым исследователем (температура выше 38,0°С [100,4°F] или ниже 36,0°С [95,5°F]).
Во время фазы скрининга определяли возможность включения и характеристики на исходном уровне (на основе тщательного изучения анамнеза и медицинского осмотра, уделялось особое внимание анамнезу, касающемуся исследуемой инфекции) для каждого субъекта. Субъекты допускались до скрининга, если у них присутствовали признаки и симптомы ABSSSI. Например, во время скрининга определяли тип ABSSSI: раневая инфекция, панникулит/рожа или крупный гнойник, как определено в критериях включения. Для возможности исследования общая площадь поверхности смежных пораженных тканей должна составлять 75 см2 или более (т.е. площадь покраснения, отека и/или уплотнения окружающих тканей).
Также отмечали анатомическое расположение первичного инфицированного участка. У субъектов с несколькими несмежными инфицированными участками наиболее тяжело пораженный участок выявляли во время скрининга и принимали за первичный участок поражения. Отмечали наличие и природу любых инородных тел (например, из древесины, металла, пластика и т.д.) в инфицированном участке. Записывали следующую информацию, касающуюся первичного инфицированного участка: наличие лимфоденопатии поблизости от первичного пораженного участка; и наличие лимфангита поблизости от первичного пораженного участка; истечение из первичного пораженного участка и его описание (серозное/серозно-кровянистое, серозно-гнойное или гнойное). Также проводили полуколичественное описание (отсутствует, слабый, умеренный, тяжелый) следующих признаков инфекции: болезненность, отек, покраснение и уплотнение.
Оценку размера поражения во время скрининга проводили за 4 часа перед рандомизацией. Площадь поверхности поражения вычисляли путем умножения максимальной длины поражения от одного края до другого на максимальную ширину (перпендикулярную максимальной длине), включая поражение смежных тканей (покраснение, отек и/или уплотнение). Измерения, которые исследователь проводил при помощи линейки, можно было использовать для фиксации размера поражения.
Кроме того, каждого субъекта просили сообщать определенные результаты или параметры, связанные со статусом заболевания и эффективностью лечения, такие как числовая оценочная шкала боли (0-10) в первичном участке поражения ABSSSI; опросник оценки состояния здоровья SF-36v2® Health Survey (который рассматривали в качестве общего опросника оценки состояния здоровья, так как его можно использовать у субъектов различных возрастов (18 лет и старше), с различными заболеваниями и из групп, в которых проводят различное лечение, в отличие от специфических опросников оценки состояния здоровья, которые относятся к конкретному состоянию или заболеванию). Кроме того, во время скрининга проводили анализ мочи тест-полосками.
Субъектов, которые удовлетворяли критериям включения и не удовлетворяли критериям исключения, случайным образом распределяли по группам, и вводили первую дозу исследуемого препарата в центре за 4 часа перед рандомизацией.
Субъектов случайным образом (1:1) распределяли в 1 из следующих 2 групп:
• Исследуемая терапия: омадациклин, 450 мг п.о. каждые 24 часа (q24h), 2 дозы, затем 300 мг п.о. q24h. Общая продолжительность лечения от 7 до 14 дней.
• Сравнительная терапия: линезолид, 600 мг п.о. каждые 12 часов (q12h). Общая продолжительность лечения от 7 до 14 дней.
В исследовании использовали двойной слепой двойной плацебо-контролируемый дизайн, применяли таблетки плацебо для омадациклина, имеющие такой же размер и форму, что и активные таблетки с омадациклином, и таблетки плацебо, заключенные в такие же капсулы, что и активные таблетки с линезолидом. Для сохранения двойного заслепления субъектам из обеих групп вводили одинаковое количество таблеток.
Период двойного слепого введения согласно дизайну настоящего эксперимента составлял от 7 до 14 дней. В следующей таблице приведен фактический уровень исследуемого лекарственного средства в популяции для анализа безопасности:


Указанные выше значения в процентах для продолжительности терапии определяли для субъектов, которым вводили по меньшей мере одну дозу. Продолжительность введения исследуемого лекарственного средства в днях = дата введения последней дозы - дата введения первой дозы + 1.
Все дозы исследуемого препарата принимали с водой. Для нечетных доз требовалось введение натощак из-за влияния приема пищи на пероральный омадациклин. Указанные требования к введению натощак учитывались при определении времени дня для приема субъектами доз.
II. ЭФФЕКТИВНОСТЬ И ОЦЕНКА
Субъектов оценивали во время 2 посещений после завершения лечения: во время посещения РТЕ (оценка после терапии), через 7-14 дней после заключительного дня проведения исследуемой терапии у субъекта, и для оценки в период последующего наблюдения, через 30-37 дней после введения первой дозы. Конечную оценку в период последующего наблюдения можно было проводить при помощи телефонного разговора или другой интерактивной технологии у субъектов, для которых была показана клиническая эффективность и у которых отсутствовали НЯ или клинически значимые отклонения лабораторных показателей или ЭКГ, которые отмечались во время или после посещения РТЕ. В иных случаях, данную оценку проводили при личном посещении центра исследования.
Анализ эффективности проводили на основании следующего перечня ключевых способов оценки:
• Клиническая оценка инфицированного участка
• Оценка пораженного участка
• Оценка необходимости проведения дополнительных хирургических процедур
• Микробиологическая оценка инфекции
• Оценка клинического ответа исследователем
• Оценка смертности от любых причин
Каждый из указанных способов более подробно описан ниже.
a. Клиническая оценка инфицированного участка
Клиническую оценку инфицированного участка проводили во время скрининга и каждого запланированного периода оценки за исключением оценки в период последующего наблюдения. Клиническую оценку инфицированного участка проводили через 48-72 часа после введения первой дозы. Изучали первичный инфицированный участок, собирали следующую информацию: истечение из первичного пораженного участка и его описание (серозное/серозно-кровянистое, серозно-гнойное или гнойное); и полуколичественное описание (отсутствует, слабый, умеренный, тяжелый) следующих признаков инфекции - болезненность; отек; покраснение; и уплотнение.
b. Оценка размера поражения
Измерение размера поражения линейкой проводили во время скрининга и каждого запланированного периода оценки за исключением оценки в период последующего наблюдения.
Площадь поверхности поражения вычисляли путем умножения максимальной длины от одного конца пораженного участка до другого на максимальную ширину (перпендикулярную максимальной длине), включая поражение смежных тканей (покраснение, отек и/или уплотнение). Измерения, которые исследователь проводил при помощи линейки, использовали для фиксации размера поражения.
с. Микробиологическая оценка инфицированного участка
Во время посещения для скрининга собирали материал из инфицированного участка и передавали в микробиологическую лабораторию центра исследования для окрашивания по Граму и выращивания бактерий. Отмечали тип отправленного образца. Лабораторные отчеты об окрашивании по Граму включали полуколичественное описание числа полиморфонуклеарных лейкоцитов при малом увеличении (т.е. 100×) и описание наблюдаемых бактерий. Также проводили посев крови для оценки сопутствующей бактериемии.
При наличии ответа на терапию в инфицированном участке повторный отбор культуры может быть клинически нецелесообразным, и/или материал для выращивания может отсутствовать. Во время посещения EOT и/или РТЕ образцы культуры из инфицированных участков и образцы для окрашивания по Граму собирали только у субъектов, у которых получали клинически неэффективные результаты и которым требовалось альтернативное антибактериальное лечение исследуемой инфекции.
Все образцы, отправляемые в лабораторию в центре исследования, оценивали на наличие аэробных и, при необходимости, анаэробных культур. Результаты исследования культур включали полуколичественное описание организмов в планшетах с первичной культурой и выявление всех изолятов на уровне родов и видов. Исследование чувствительности к линезолиду проводили на месте при помощи стандартного способа, выбранного в лаборатории. Результаты данного исследования использовали совместно с клиническими показателями для помощи в регулировании терапии.
Все изоляты бактерий, выявленные в образцах из инфицированного участка или в крови, отправляли в Центральную лабораторию для подтверждения рода и вида и проведения стандартизированного исследования минимальной ингибирующей концентрации (МИК) омадациклина, линезолида и группы одобренных в настоящее время антибиотиков.
Если подтверждалось наличие грамотрицательных или анаэробных микроорганизмов или микроорганизмов, не чувствительных к линезолиду, то решение о продолжении или прекращении введения исследуемого препарата и изменении режима антибактериального лечения принимали на основании клинической оценки исследователя и отмечали в первичных документах.
d. Оценка клинических результатов
Оценку клинических результатов проводили на стадии оценки раннего клинического ответа (на программном уровне), EOT и РТЕ, как описано ниже.
i) Оценка исследуемой инфекции на стадии оценки раннего клинического ответа
Формальное определение ответа на терапию на стадии оценки раннего клинического ответа (через 48-72 часов после введения первой дозы исследуемого препарата) проводили на программном уровне с использованием измеренных значений размера поражения.
«Клиническая эффективность» в рамках оценки раннего клинического ответа была определена как удовлетворение всех 3 из следующих пунктов: субъект был живым; размер первичного поражения уменьшался на 20% или более по сравнению с измеренным во время скрининга без проведения какой-либо альтернативной (вспомогательной) антибактериальной терапии; и субъект не удовлетворял каким-либо критериям клинической неэффективности и неопределенной эффективности (определения см. ниже).
«Клиническую неэффективность» определяли в случае удовлетворения любого из приведенных ниже критериев: размер первичного поражения не уменьшался на 20% или более по сравнению с измеренным во время скрининга; прием исследуемого препарата прекращали в связи с определением недостаточного ответа инфекции, в результате чего требовалась альтернативная (вспомогательная) антибактериальная терапия; субъекту проводили антибактериальную терапию, которая может являться эффективной в отношении исследуемой инфекции, при лечении инфекции, отличной от исследуемой; у субъекта проявились НЯ, при которых требовалось прекращение введения исследуемого препарата перед проведением оценки раннего клинического ответа и была необходима альтернативная (вспомогательная) антибактериальная терапия; или гибель перед проведением оценки раннего клинического ответа.
Статус «неопределенной эффективности» присваивали, если клинический ответ на исследуемый препарат невозможно было надлежащим образом определить из-за того, что: субъект не наблюдался во время оценки раннего клинического ответа по причине отзыва информированного согласия, недоступности в период последующего наблюдения, или по другим причинам.
ii) Клиническая оценка исследуемой инфекции на стадии EOT
Во время посещения EOT (в день введения последней дозы исследуемого препарата или в течение 2 дней после ее введения) определяли клинический статус инфекции, как описано ниже.
«Клиническая эффективность» во время оценки в конце лечения (EOT) была определена при удовлетворении следующих пунктов: субъект был живым; и инфекция в достаточной степени подавлялась, в результате чего дальнейшая антибактериальная терапия не требовалась (у указанных субъектов могли сохраняться некоторые остаточные изменения, связанные с инфекцией, для которых требовалось вспомогательное лечение (т.е. не требующее использования антибиотиков), например, перевязка исцеляющейся раны, санация неинфицированной ткани (т.е. некротической).
Клиническая неэффективность была определена при удовлетворении любого из приведенных ниже критериев, указывали первичную причину клинической неэффективности: прием исследуемого препарата прекращали в связи с определением недостаточного ответа инфекции, в результате чего требовалась альтернативная (вспомогательная) антибактериальная терапия; субъекту проводили антибактериальную терапию, которая может являться эффективной в отношении исследуемой инфекции, при лечении инфекции, отличной от исследуемой; или у субъекта проявились НЯ, при которых потребовалось прекращение введения исследуемого препарата перед завершением запланированного режима введения исследуемого препарата и была необходима альтернативная (вспомогательная) антибактериальная терапия; незапланированное обширное хирургическое вмешательство (т.е. процедуры, которые обычно не проводят в центре исследования), направленное на исследуемую инфекцию; гибель субъекта перед оценкой; или прочее.
Статус «неопределенной эффективности» присваивали, если клинический ответ на исследуемый препарат невозможно было надлежащим образом определить. Указаны все доступные варианты: Субъект не наблюдался во время оценки EOT по причине отзыва информированного согласия, недоступности в период последующего наблюдения, или по иной причине.
iii) Клиническая оценка исследуемой инфекции на стадии РТЕ
Во время посещения для оценки после проведения терапии РТЕ (через 7-14 дней после заключительного дня проведения исследуемой терапии у субъекта) отмечали ОДИН из следующих результатов, касающихся первичной исследуемой инфекции:
«Клиническая эффективность» во время оценки после проведения терапии была определена при удовлетворении следующих пунктов: субъект был живым; инфекция в достаточной степени подавлялась, в результате чего дальнейшая антибактериальная терапия не требовалась (у указанных субъектов могли сохраняться некоторые остаточные изменения, связанные с инфекцией, для которых требовалось вспомогательное лечение (т.е. не требующее использования антибиотиков), например, перевязка исцеляющейся раны, санация неинфицированной ткани (т.е. некротической).
«Клиническая неэффективность» была определена при удовлетворении какого-либо из приведенных ниже критериев, указывали первичную причину клинической неэффективности: требовалось дополнительное лечение инфекции с использованием альтернативной (вспомогательной) антибактериальной терапии; субъекту проводили антибактериальную терапию в период от EOT до РТЕ, которая могла быть эффективной в отношении исследуемой инфекции, при лечении инфекции, отличной от исследуемой; незапланированное обширное хирургическое вмешательство (т.е. процедуры, которые обычно не проводят в центре исследования), направленное на исследуемую инфекцию, в период от EOT до РТЕ; гибель субъекта перед оценкой; и прочее.
Статус «неопределенной эффективности» присваивали, если клинический ответ на исследуемый препарат невозможно было надлежащим образом определить. Указаны все доступные варианты: Субъект не наблюдался во время оценки РТЕ по причине отзыва информированного согласия, недоступности в период последующего наблюдения, или по иной причине.
III. ИССЛЕДОВАНИЯ ФАРМАКОКИНЕТИКИ
Данные PK анализировали при помощи популяционной модели PK. Образцы крови для анализа PK собирали выборочным способом в рамках популяционной модели PK. Число образцов и схема отбора проб отличались у различных субъектов. У субъекта собирали до 4 проб крови между днями 2 и 3. Кровь собирали путем венепункции на новом участке или с использованием канюли, предназначенной исключительно для данной задачи. Отмечали дату и время введения всех доз исследуемых препаратов и отбора проб для PK. Данные субъекта, номер пробы и время сбора пробы с точностью до минуты немедленно отмечали на пробирке для сбора проб. Пробирку центрифугировали при 1500 ×g в течение 10 минут; выделенную плазму переносили в виде 2 равных аликвот в предварительно помеченные пробирки; и пробирки замораживали при -70°С в течение 60 минут после отбора проб. Время заморозки образца записывали с точностью до минуты.
Образцы для оценки омадациклина анализировали специфическим чувствительным и валидированным способом жидкостной хроматографии/тандемной масс-спектрометрии (ЖХ/МС/МС).
IV. ОТСЛЕЖИВАНИЕ БЕЗОПАСНОСТИ
Нежелательное явление (НЯ) представляет собой любое неблагоприятное, побочное или незапланированное явление в виде признаков, симптомов, заболевания или лабораторных или физиологических показателей, возникающих у индивидуума, которому вводят исследуемый препарат, или во время клинического исследования. Явление не обязательно должно иметь причинную связь с исследуемым препаратом или клиническим исследованием. НЯ включает, но не ограничивается ими, следующее: любое клинически значимое ухудшение уже существующего состояния; НЯ, возникающие при передозировке исследуемого препарата независимо от того, является она случайной или намеренной; передозировка представляет собой дозу, превышающую ту, что указана в протоколе; НЯ, возникающие в результате ошибочного употребления (например, использования в неклинических целях) исследуемого препарата; и НЯ, которые связаны с прекращением использования исследуемого препарата.
Тяжесть (или интенсивность) НЯ классифицировали при помощи следующих критериев:
Слабая: Указанные явления, как правило, были временными, требовали минимального лечения или не требовали его вовсе и не влияли на повседневную активность субъекта.
Умеренная: Указанные явления приводили к незначительным неудобствам или проблемам, которые требовали терапевтического вмешательства. Умеренные явления могут в некоторой степени влиять на обычную деятельность, но при этом не имеют значительного или постоянного риска нанесения вреда.
Тяжелая: Указанные явления нарушали обычную повседневную деятельность субъекта и могли требовать проведения системной лекарственной терапии или другого способа лечения. Тяжелые явления, как правило, приводили к потере трудоспособности.
Серьезное нежелательное явление (СНЯ) представляет собой НЯ, которое: приводит к смерти; представляет угрозу для здоровья (см. ниже); требует госпитализации или продления текущей госпитализации (см. ниже); приводит к длительной или значительной инвалидности или потере трудоспособности (см. ниже); приводит к врожденной аномалии или пороку развития; кроме того, важные медицинские явления, которые могут не приводить к смерти, представлять угрозу для жизни или требовать госпитализации, могут рассматриваться как СНЯ, если согласно результатам надлежащей медицинской оценки они могут подвергать опасности субъекта и могут требовать медикаментозного или хирургического вмешательства для предотвращения любого одного (1) из результатов, перечисленных выше в данном определении. Примеры указанных явлений включают аллергический бронхоспазм, требующий интенсивного лечения в реанимации или на дому, дискразию крови или конвульсии, которые не приводят к госпитализации, или развитие лекарственной зависимости или злоупотребление лекарственным средством.
Угроза жизни относится к риску немедленной гибели при возникновении явления у сообщившего. Угрожающее жизни явление не включает явление, которое при протекании в более тяжелой форме могло бы вызывать смерть, но фактически не привело к риску немедленной гибели.
Госпитализация представляет собой официальное посещение лечебного учреждения. Госпитализация или продление госпитализации является критерием отнесения НЯ к серьезным; тем не менее, ее, как таковую, не рассматривают как СНЯ. В отсутствие НЯ исследователь не отмечал госпитализацию или продление госпитализации как СНЯ. Это происходило в следующих ситуациях: если госпитализация или продление госпитализации требовались для процедуры, требуемой по протоколу; если госпитализация или продление госпитализации являлись частью обычной процедуры, используемой в центре (например, удаление стента после хирургии); и если госпитализация проводилась по причине уже существующего состояния, которое при этом не усугубилось.
Инвалидность определена как значительное нарушение способности индивидуума осуществлять нормальные жизненные функции. При возникновении каких-либо сомнений о соответствии представленной информации СНЯ, информацию указывали как СНЯ.
НЯ и СНЯ у субъекта записывались и отмечались, начиная от подписания информированного согласия и до момента конечной оценки в период последующего наблюдения. Субъектам проводили инструктаж по фиксации НЯ и СНЯ во время указанного периода. Сообщения о смерти в течение 30 дней после последнего контакта с субъектом отправляли спонсору, собирали дополнительную информацию, касающуюся причины смерти, и фиксировали ее.
Последующее наблюдение НЯ и СНЯ у субъектов проводили в соответствии с медицинскими показаниями до тех пор, пока явления не исчезали, состояние не возвращалось к исходному уровню, или, в случае хронического нарушения, до стабилизации состояния.
НЯ определяли на основании признаков или симптомов, обнаруженных во время медицинского осмотра и клинической оценки субъекта. Если НЯ требовало хирургической или диагностической процедуры, то как НЯ отмечали болезнь, делающую необходимой процедуру, но не саму процедуру. Смерть отмечали как результат НЯ. Любые непредвиденные риски для субъектов отмечались незамедлительно.
Для определения связи с исследуемым препаратом проводили оценку причинно-следственной связи (т.е. наличия обоснованной вероятности того, что исследуемый препарат явился причиной явления) для всех НЯ и СНЯ. Взаимосвязь была охарактеризована при помощи следующей классификации:
• Не связано: при указанном статусе предположительно отсутствовала взаимосвязь исследуемого препарата и рассматриваемого явления. Явление может быть объяснено при помощи других факторов, таких как исходное медицинское состояние, сопутствующая терапия или несчастный случай, между исследуемым препаратом и явлением отсутствовала убедительная временная или биологическая взаимосвязь.
• Связано: при указанном статусе предположительно имелась определенная причинно-следственная связь между введением исследуемого препарата и НЯ, или имелась обоснованная вероятность того, что явление было вызвано исследуемым лекарственным средством, и другие состояния (протекающая болезнь, прогрессирование/проявление болезненного состояния или реакция на сопутствующее лекарственное средство) вероятно не могли объяснить это явление.
Также оценивали возможную связь НЯ и СНЯ с протоколом. Нежелательное явление, связанное с протоколом, представляло собой явление, которое не было связано с исследуемым препаратом, но рассматривалось исследователем или медицинским наблюдателем (или уполномоченным сотрудником) как связанное с условиями исследования, т.е. связанное уже с тем фактом, что субъект принимал участие в исследовании. Например, НЯ, связанное с протоколом, может представлять собой неблагоприятное явление, связанное с медицинской процедурой, требуемой по протоколу.
Тяжесть (или интенсивность) НЯ классифицировали с использованием следующих критериев:
• Слабая: Указанные явления, как правило, были временными, требовали минимального лечения или не требовали его вовсе и не влияли на повседневную активность субъекта.
• Умеренная: Указанные явления приводили к незначительным неудобствам или проблемам, которые требовали терапевтического вмешательства. Умеренные явления могут в некоторой степени влиять на обычную деятельность, но при этом не имеют значительного или постоянного риска нанесения вреда.
• Тяжелая: Указанные явления нарушали обычную повседневную деятельность субъекта и могли требовать проведения системной лекарственной терапии или другого способа лечения. Тяжелые явления, как правило, приводили к потере трудоспособности.
Изменения тяжести НЯ отмечали как новое явление для того, чтобы можно было проводить оценку продолжительности явления на каждом уровне интенсивности.
Определенные протоколом результаты лабораторных исследований безопасности анализировали в рамках специфических лабораторных анализов безопасности. Дополнительные результаты лабораторных исследований, полученные в другие моменты времени, могут быть доступны в рамках стандартной клинической практики. Во время исследования все отклонения лабораторных показателей отмечали как НЯ, только если их рассматривали как клинически значимые, если они выходили за рамки ожидаемых значений с учетом оценки субъекта на исходном уровне и клинического течения болезни, и если они точно не являлись частью других диагностированных НЯ.
Кожную инфекцию, которая являлась критерием допуска субъекта для участия в исследовании («определяющая допуск ABSSSI»), рассматривали как уникальную, так как данные, связанные с прогрессированием указанной инфекции, собирали в рамках анализа эффективности. Таким образом, усугубление или прогрессирование определяющей допуск ABSSSI отмечали как показатель клинической неэффективности (в рамках оценки эффективности), но не как НЯ, если только усугубление/прогрессирование также не удовлетворяло критериям для серьезного НЯ (в этом случае явление также отмечали как СНЯ). В противоположность этому, любые новые или вторичные инфекции, которые исследователь рассматривал как отличные от определяющей допуск ABSSSI (например, вторичные кожные гнойники в другой анатомической области) отмечали как НЯ во всех случаях независимо от того, были они несерьезными или серьезными.
V. АНАЛИЗ ДАННЫХ
Анализ всех данных удовлетворял требованиям Международной конференции по гармонизации технических требований к регистрации фармацевтических препаратов для использования у человека (ICH-E9) и регламентирующих документов и стандартов спонсора. Статистический анализ проводили при помощи программного обеспечения Statistical Analysis Software (SAS).
Подготовку SAP, включающих разделы, приведенные ниже, и командные оболочки с макетами таблиц, чертежей и списков (TFL), проводили перед началом исследования. В указанном плане были определены популяции для анализа, отмечены все условия обработки данных и указаны статистические способы для анализа безопасности и эффективности. Из-за различных регламентирующих требований к выбору первичного показателя эффективности и статистического анализа были подготовлены 2 отдельных SAP (по одному для FDA и ЕМА). В приведенных ниже разделах указаны общая структура и подход к проведению анализа.
Инференциальный статистический анализ первичных и вторичных результатов проводили, как указано ниже. Использовали описательную статистику, включая количество и уровень в процентах для категориальных переменных и количество, среднее, среднеквадратическое отклонение (СКО), медианное, минимальное и максимальное значение для непрерывных переменных. Все сравнения проводили для омадациклина и линезолида. Также допускалось проведение предварительного анализа. Получали списки данных для отдельных субъектов.
Анализ популяций
Для проведения различных анализов эффективности и безопасности был определен ряд популяций для анализа субъектов, приведенных ниже:
Популяция с назначенным лечением (ITT) включала всех субъектов, прошедших рандомизацию.
Популяция для анализа безопасности включала всех субъектов, прошедших рандомизацию, которым вводили исследуемый препарат.
Модифицированная популяция с назначенным лечением (mITT) включая всех субъектов, прошедших рандомизацию, у которых отсутствовал(-и) грамотрицательный(-е) патоген, вызывающий(-е) инфекцию, во время скрининга.
Модифицированная популяция с назначенным лечением для микробиологического анализа (micro-mITT) включала субъектов из популяции mITT, у которых имелся по меньшей мере 1 грамположительный патоген, вызывающий инфекцию, во время скрининга (например, бактериальный патоген, выявленные в посеве крови или после выращивания микробиологического образца, полученного из участка первичной ABSSSI на исходном уровне).
Популяция для проведения клинической оценки (СЕ) (СЕ-РТЕ и СЕ-ЕОТ) включала всех субъектов mITT, которым вводили по меньшей мере одну дозу исследуемого препарата, которые имели определяющую допуск ABSSSI, у которых исследователь проводил оценку клинического ответа во время посещения PTE/EOT, отсутствовал неопределенный клинический ответ, и которые удовлетворяли специфическим критериям, связанным с требуемой оценкой.
Популяция для микробиологической оценки (ME) включала субъектов из популяции СЕ, у которых имелся по меньшей мере 1 грамположительный патоген, вызывающий инфекцию, во время скрининга (т.е. всех субъектов из популяций micro-mITT и СЕ-РТЕ/ЕОТ).
Различные популяции субъектов, участвовавших в исследовании, такие как определено выше, приведены ниже:


Распределение субъектов в популяции ITT приведено ниже:


[1] Субъекты, которые завершили исследуемое лечение.
[2] Субъекты, которые завершили исследование (т.е. получили по меньшей мере 5 одну дозу исследуемого препарата и совершили посещения EOT, РТЕ и в период последующего наблюдения).
Распределение субъектов в популяции mITT приведено ниже:


[1] Субъекты, которые завершили исследуемое лечение.
[2] Субъекты, которые завершили исследование (т.е. получили по меньшей мере 5 одну дозу исследуемого препарата и совершили посещения EOT, РТЕ и в период последующего наблюдения).
Распределение субъектов в популяции СЕ-РТЕ приведено ниже:


[1] Субъекты, которые завершили исследуемое лечение.
[2] Субъекты, которые завершили исследование (т.е. получили по меньшей мере 5 одну дозу исследуемого препарата и совершили посещения EOT, РТЕ и в период последующего наблюдения).
Демографические данные и характеристики на исходном уровне в популяции для анализа безопасности приведены ниже:



Демографические данные и характеристики на исходном уровне в популяции mITT приведены ниже:




Участки первичной инфекции ABSSSI на исходном уровне в популяции mITT приведены ниже:


[1] Фактический тип инфекции, указанный в эИРК.
Род и вид патогенных организмов в участке ABSSSI или посеве крови на исходном уровне в популяции micro-mITT приведены ниже:


В приведенной выше таблице значения в процентах указаны для числа субъектов в каждой группе. Субъектов, у которых один патоген выделяли из нескольких участков, учитывали только один раз при описании данного патогена. Субъектов, у которых один патоген выявляли в посеве крови и культуре первичной ABSSSI, учитывали только один раз. MRSA и MSSA рассматривали как различные патогены; VRE и VSE рассматривали как различные патогены. При указании числа Staphylococcus aureus у субъекта, субъектов, имеющих одновременно MRSA и MSSA, учитывали только один раз. При указании числа каждого вида Enterococcus у субъекта, субъектов, имеющих одновременно VRE и VSE, учитывали только один раз. Показаны только типовые или наиболее распространенные роды/виды.
Первичный анализ эффективности
Для всех анализов эффективности данные субъекта анализировали в рамках группы, в которую был случайным образом определен субъект. В случае первичного анализа для FDA и ЕМА анализ субъектов проводили в рамках страты, в которую был случайным образом определен субъект.
Ранний клинический ответ мог иметь статус клинической эффективности, клинической неэффективности и неопределенной эффективности (что определено выше). Неопределенный ответ учитывали в знаменателе при вычислении доли субъектов в процентах, имеющих статус клинической эффективности, в популяции mITT, и, таким образом, его учитывали как клиническую неэффективность при первичном анализе для FDA.
Переменная эффективности «оценка исследователем клинического ответа РТЕ» была определена как результат оценки исследователем клинического ответа во время посещения РТЕ и имела значения клинической эффективности, клинической неэффективности и неопределенной эффективности (определено выше) для популяции mITT и клинической эффективности и клинической неэффективности для популяции СЕ. Субъектам, имеющим клинически неэффективный ответ на стадии EOT, присваивали статус клинической неэффективности на стадии РТЕ. Неопределенный ответ учитывали в знаменателе при вычислении доли субъектов в процентах, имеющих статус клинической эффективности, в популяции mITT, и, таким образом, его учитывали как клиническую неэффективность при первичном анализе для ЕМА.
Для демонстрации того, что омадациклин имеет не меньшую эффективность по сравнению с линезолидом при лечении взрослых субъектов с ABSSSI, проводили оценку следующей гипотезы посредством анализа показателей клинической эффективности:
Нулевую гипотезу и альтернативную гипотезу для конечного показателя раннего клинического ответа проверяли в популяции mITT следующим образом:


где показатель клинической эффективности для режима введения омадациклина обозначен как θТ, а для линезолида как θС,
Δ представляет собой предел не меньшей эффективности (NI) и составляет 0,10.
Схожие нулевая и альтернативная гипотезы могут быть предложены при Δ 0,10 и для конечного критерия оценки РТЕ. В случае конечного критерия оценки раннего клинического ответа (FDA) использовали подход с 2-сторонним 95% доверительным интервалом (ДИ) для проведения различий между показателями клинической эффективности (методом точечной оценки различий: уровень ответа в группе омадациклина минус уровень ответа в группе линезолида) для изучения NI в группе омадациклина по сравнению с группой линезолида в популяции mITT. 95% ДИ вычисляли при помощи методики нерасслоенной выборки, предложенной Миеттиненом и Нурминеном (Miettinen and Nurminen). Считалось, что омадациклин имел не меньшую эффективность по сравнению с линезолидом, если нижняя граница ДИ превышала -0,10 (т.е. -10%).
Для первичного анализа эффективности во время оценки исследователем клинического ответа на стадии РТЕ (ЕМА) в популяциях mITT и СЕ использовали подход с 2-сторонним 95% ДИ для проведения различий между показателями клинической эффективности (методом точечной оценки различий: уровень ответа в группе омадациклина минус уровень ответа в группе линезолида) для изучения NI в группе омадациклина по сравнению с группой линезолида. 95% ДИ вычисляли по методике расслоенной выборки (для факторов рандомизации со стратификацией), предложенной Миеттиненом и Нурминеном. Считалось, что омадациклин имел не меньшую эффективность по сравнению с линезолидом, если нижняя граница ДИ превышала -0,10 (т.е. -10%).
Ранний клинический ответ и оценку исследователем клинического ответа на стадии РТЕ изучали отдельно, и они не являлись комбинированными первичными критериями оценки. Вероятность одобрения неэффективного лекарственного средства по результатам определения эффективности на стадии РТЕ составляла 2,5% независимо от результатов раннего клинического ответа, и наоборот. Поправка может требоваться только в том случае, если улучшение по меньшей мере 1 конечного критерия оценки приводит к одобрению в целом. Кроме того, введение поправки показателя альфа для комбинированных первичных критериев оценки эффективности для ЕМА (популяции mITT и СЕ) не требуется, так как для определения NI, NI должен быть показан в обеих популяциях. Таким образом, поправку на множественные критерии оценки не проводили.
Показатели раннего клинического ответа через 48-72 часа после введения первой дозы исследуемых препаратов (омадациклин или линезолид) в популяции mITT приведены ниже:


В приведенной выше таблице ДИ = доверительный интервал; различия представляли собой наблюдаемые различия показателей ранней клинической эффективности в группах омадациклина и линезолида. 95% ДИ строили по методике Миеттинена и Нурминена без стратификации. Значения в процентах вычисляли на основании количества субъектов в каждой группе.
Показатели ранней клинической эффективности (через 48-72 часа) в популяции mITT для омадациклина и линезолида изображены на ФИГ. 6. См. левую пару столбцов. Данные показывают, что наблюдаемое 5,0% различие показателя клинической эффективности не выходит за рамки 10% предела статистической не меньшей эффективности, которые составляют от -0,2% до 10,3% при 95% ДИ (доверительный интервал), и, таким образом, требуемое значение первичного критерия оценки эффективности (для одобрения FDA) достигается.
Зависимость раннего клинического ответа через 48-72 часа после введения первой дозы исследуемых препаратов (омадациклин или линезолид) от патогена, присутствующего на исходном уровне, в участке ABSSSI или посеве крови, в популяции micro-mITT приведена ниже:


В приведенной выше таблице N1 = число субъектов из популяции micro-mITT, имеющих данный патоген на исходном уровне, включенных в конкретную группу; n = число субъектов в конкретной категории. Значения в процентах определяли в пересчете на N1. Субъектов, у которых один патоген выделяли из нескольких участков, учитывали только один раз при описании данного патогена. Субъектов, у которых один патоген выявляли в посеве крови и культуре первичной ABSSSI, учитывали только один раз. Значения в процентах определяли в пересчете на количество субъектов, имеющих указанный патоген. Показаны только типовые или наиболее распространенные роды/виды.
Если нулевая гипотеза о не меньшей эффективности опровергалась в случае раннего клинического ответа в популяции mITT, и наблюдаемый уровень эффективного ответа для омадациклина превышал наблюдаемый уровень для линезолида, то проводили формальный статистический анализ для подтверждения более высокой эффективности. Если нижний предел 2-стороннего 95% ДИ различия способов лечения превышал 0%, то омадациклин рассматривали как более эффективный по сравнению с линезолидом.
Первичный показатель эффективности оценивали отдельно для всех факторов стратификации по типу инфекции и назначению допустимой антибактериальной терапии за 72 часа перед рандомизацией по группам. Для каждой страты по типу инфекции и каждой страты, касающейся проведения предшествующей терапии, вычисляли 2-сторонний 95% ДИ для наблюдаемого различия уровня раннего клинического ответа в популяции mITT. Дополнительный анализ подгрупп первичного показателя эффективности можно проводить в рамках описательного анализа.
Результаты указанных отдельных оценок по всем факторам стратификации приведены ниже.
В частности, ранний клинический ответ через 48-72 часа после введения первой дозы исследуемых препаратов (омадациклин или линезолид) в зависимости от типа инфекции в популяции mITT приведен ниже:
Ниже в таблице ДИ = доверительный интервал; различие представляет собой наблюдаемое различие показателя ранней клинической эффективности в группах омадациклина и линезолида. 95% ДИ для каждого типа инфекции строили по методике Миеттинена и Нурминена без стратификации. Значения в процентах указаны в пересчете на число субъектов в каждой группе для каждого типа инфекции. [1] Фактический тип инфекции = Тип инфекции, записанный в эИРК.

Общий клинический ответ во время посещения РТЕ, основанный на результатах оценки исследователем, в популяциях mITT и СЕ-РТЕ приведен ниже:

ДИ = доверительный интервал; различие представляет собой наблюдаемое различие показателей общей клинической эффективности на стадии РТЕ между группами омадациклина и линезолида. [1] 95% ДИ строили на основании методики Миеттинена и Нурминена без стратификации. [2] 95% ДИ корректировали по типу инфекции и предшествующему назначению антибиотиков на основании методики Миеттинена и Нурминена со стратификацией с использованием критерия Кохрана-Мантеля-Гензеля в качестве весов страты. В случае [2] подгруппы, которым ранее назначали антибиотики, объединяли в одну группу. Объединение по типу инфекции не проводили. Общий клинический ответ на стадии РТЕ определяли по результатам оценки исследователем во время посещений EOT и РТЕ. Значения в процентах указаны в пересчете на количество субъектов в каждой группе.
Показатели общей клинической эффективности/ответа во время посещения РТЕ, определенные на основании оценки исследователем популяции mITT РТЕ и популяции СЕ-РТЕ, для омадациклина и линезолида также изображены на ФИГ. 6. См. среднюю (mITT РТЕ) и правую (СЕ-РТЕ) пары столбцов. Данные показывают, что наблюдаемое 3,3% различие показателя общего клинического ответа в популяции mITT РТЕ не выходит за рамки 10% предела статистически не меньшей эффективности от -2,2% до 9,0% (со стратификацией) с 95% ДИ (доверительный интервал); и наблюдаемое 2,3% различие показателя общего клинического ответа в популяции СЕ-РТЕ не выходит за рамки 10% предела статистически не меньшей эффективности от -0,5% до 5,8% (со стратификацией) с 95% ДИ (доверительный интервал). Таким образом, требуемые значения комбинированных первичных критериев оценки эффективности (для одобрения ЕМА) достигаются.
Общий клинический ответ во время посещения РТЕ, основанный на оценке исследователем, в зависимости от типа инфекции в популяции mITT приведен ниже:


ДИ = доверительный интервал; различие представляет собой наблюдаемое различие показателя общей клинической эффективности на стадии РТЕ между группами омадациклина и линезолида. 95% ДИ для каждого типа инфекции строили на основании методики Миеттинена и Нурминена без стратификации. Значения в процентах указаны в пересчете на количество субъектов в каждой группе, имеющих каждый тип инфекции.
[1] Фактический тип инфекции = Тип инфекции, указанный в эИРК.
Общая клиническая эффективность во время посещения РТЕ, основанная на оценке исследователем, в зависимости от патогена, присутствующего на исходном уровне в участке ABSSSI или в посеве крови, в популяции micro-mITT приведена ниже:


N1 = Количество субъектов из популяции micro-mITT, имеющих данный патоген на исходном уровне, включенных в конкретную группу, n = число субъектов в конкретной категории. Значения в процентах указаны в пересчете на N1. Субъектов, у которых один патоген выделяли из нескольких участков, учитывали только один раз при описании данного патогена. Субъектов, у которых один патоген выявляли в посеве крови и культуре первичной ABSSSI, учитывали только один раз. Значения в процентах определяли в пересчете на количество субъектов, имеющих указанный патоген.
Были проведены дополнительные анализы и анализы чувствительности первичных показателей эффективности (ранний клинический ответ и оценка исследователем клинического ответа на стадии РТЕ) (данные не показаны). Анализ чувствительности включал: проведение скорректированного анализа первичных показателей эффективности с учетом рандомизированных страт и отдельно с учетом страт, к которым фактически принадлежали субъекты, и проведение анализа, в котором всех субъектов, имеющих статус неопределенной эффективности, рассматривали как субъектов со статусом клинической эффективности.
Вторичный анализ эффективности
Для каждой группы вычисляли количество и долю субъектов, которым присваивали статусы клинической эффективности, клинической неэффективности и неопределенной эффективности по результатам оценки исследователем на стадии РТЕ в популяциях mITT и СЕ (по определению субъекты, имевшие неопределенный ответ, были исключены из популяции СЕ). 2-сторонний нескорректированный 95% ДИ строили для наблюдаемых различий показателей клинической эффективности по методике Миеттинена и Нурминена. В случае оценки исследователем клинического ответа на стадии РТЕ в популяциях mITT и СЕ 2-сторонний 95% ДИ использовали исключительно для описания, и на его основании не делали выводы о NI.
Количество и доля субъектов в каждой группе, имеющих каждую категорию ответа в случае раннего клинического ответа, были указаны для популяции micro-mlTT. Вычисляли количество и долю субъектов, которым исследователь присваивал статус клинической эффективности или клинической неэффективности во время посещения РТЕ, из популяции ME. Двухсторонний нескорректированный 95% ДИ строили для наблюдаемых различий показателей клинической эффективности по методике Миеттинена и Нурминена.
Количество и доля субъектов, имеющих эффективный ранний клинический ответ и статус клинической эффективности по результатам оценки исследователем клинического ответа на стадии РТЕ, в зависимости от патогена (включая вызывающие инфекцию грамотрицательные патогены и MRSA) были определены в популяциях micro-mITT и ME.
Анализ дополнительных переменных эффективности
Для подтверждения данных об эффективности, полученных на основании первичных и вторичных показателей были проведены дополнительные анализы эффективности. ДИ определяли для описания, но на их основании не делали выводы о NI.
Вычисляли количество и долю субъектов, которым присваивали статус ранней клинической эффективности, клинической неэффективности и неопределенной эффективности через 48-72 часа после введения первой дозы исследуемого препарата, в популяции ITT. 2-сторонний нескорректированный 95% ДИ строили для наблюдаемых различий показателя клинической эффективности по методике Миеттинена и Нурминена.
Для каждой группы вычисляли количество и долю субъектов, которым присваивали статус клинической эффективности, клинической неэффективности и неопределенной эффективности по результатам оценки исследователем на стадии EOT в популяциях mITT и СЕ (по определению субъекты, имевшие неопределенный ответ, были исключены из популяции СЕ). 2-сторонний нескорректированный 95% ДИ строили для наблюдаемых различий показателей клинической эффективности по методике Миеттинена и Нурминена.
Были получены сводные показатели описательной статистики, включая изменение относительно исходного уровня, если это было доступно, клинических признаков и симптомов (болезненность, отек, покраснение, уплотнение и истечение), полное разрешение клинических признаков и симптомов, изменение температуры, размеров поражения ABSSSI (включая абсолютное и процентное уменьшение площади поражения, например, от 0 до <5%, от 5 до <10%, от 10 до <20%, >20% и т.д.) и системных признаков на момент посещения для исследования.
Смертность от любых причин (АСМ) через 15 и 30 дней после введения первой дозы исследуемого препарата была приведена для популяции ITT. Для данного анализа субъектов, которые были недоступны в период последующего наблюдения, рассматривали как умерших. Для АСМ вычисляли 2-сторонний нескорректированный 95% ДИ для наблюдаемых различий уровня смертности.
Микробиологические показатели для отдельных субъектов и отдельных патогенов были определены для популяций micro-mITT и ME по время посещений EOT и РТЕ. Были построены двухсторонние нескорректированные 95% ДИ для определения различий уровня благоприятных микробиологических показателей у каждого субъекта.
Был проведен анализ соответствия раннего клинического ответа и оценки исследователем клинического ответа на стадии РТЕ на выборке для анализа mITT.
Измеряемые показатели безопасности
Переменные безопасности включали частоту НЯ, изменение основных показателей жизнедеятельности, параметры ЭКГ и результаты лабораторных исследований, полученные в ходе исследования. Для анализа безопасности субъектов исследовали в соответствии с фактически проведенным лечением.
Сводные таблицы были представлены для всех нежелательных явлений, возникших в ходе лечения (НЯВЛ). НЯВЛ определено как НЯ, которое проявилось во время или после введения первой дозы исследуемого препарата. НЯ были классифицированы в соответствии с медицинским словарем регуляторной деятельности (MedDRA) и описаны путем указания количества и доли субъектов, имеющих каждое НЯВЛ в каждой группе, по классам систем органов (SOC) и предпочтительным терминам (РТ). В дополнительных табличных данных предложено описание по SOC и РТ субъектов, отмечающих СНЯ, тяжелые НЯВЛ, НЯВЛ, определенно связанные с исследуемым препаратом, НЯВЛ, приводящие к прекращению приема исследуемого препарата, и НЯВЛ, представляющие особый интерес.
Обзор НЯ в популяции для анализа безопасности приведен ниже:

Значения в процентах указаны для популяции для анализа безопасности. НЯВЛ определено как НЯ, возникшее после введения первой дозы активного исследуемого препарата.
Описание нежелательных явлений, возникших в ходе лечения (НЯВЛ), приводящих к прекращению приема исследуемого лекарственного средства, по системам классов органов и предпочтительным терминам в популяции для анализа безопасности приведено ниже:

Классификацию по системам классов органов и предпочтительным терминам проводили на основании MedDRA версии 17.1. Значения в процентах указаны для популяции для анализа безопасности. НЯВЛ определено как НЯ, возникшее после введения первой дозы активного исследуемого препарата. Если субъект имел более одного НЯВЛ, которое было отнесено к одной категории MedDRA, его учитывали только один раз.
Описание наиболее частых (≥2%) нежелательных явлений, возникших в ходе лечения (НЯВЛ), по предпочтительным терминам в популяции для анализа безопасности приведено ниже:

Классификация по предпочтительным терминам была основана на MedDRA версии 17.1. Значения в процентах указаны для популяции для анализа безопасности. НЯВЛ было определено как НЯ, возникшее после введения первой дозы активного исследуемого препарата. Если субъект имел более одного НЯВЛ, которое было отнесено к одной категории MedDRA, его учитывали только один раз.
Описание наиболее частых нежелательных явлений со стороны ЖКТ, возникших в ходе лечения (НЯВЛ), в популяции для анализа безопасности приведено ниже:
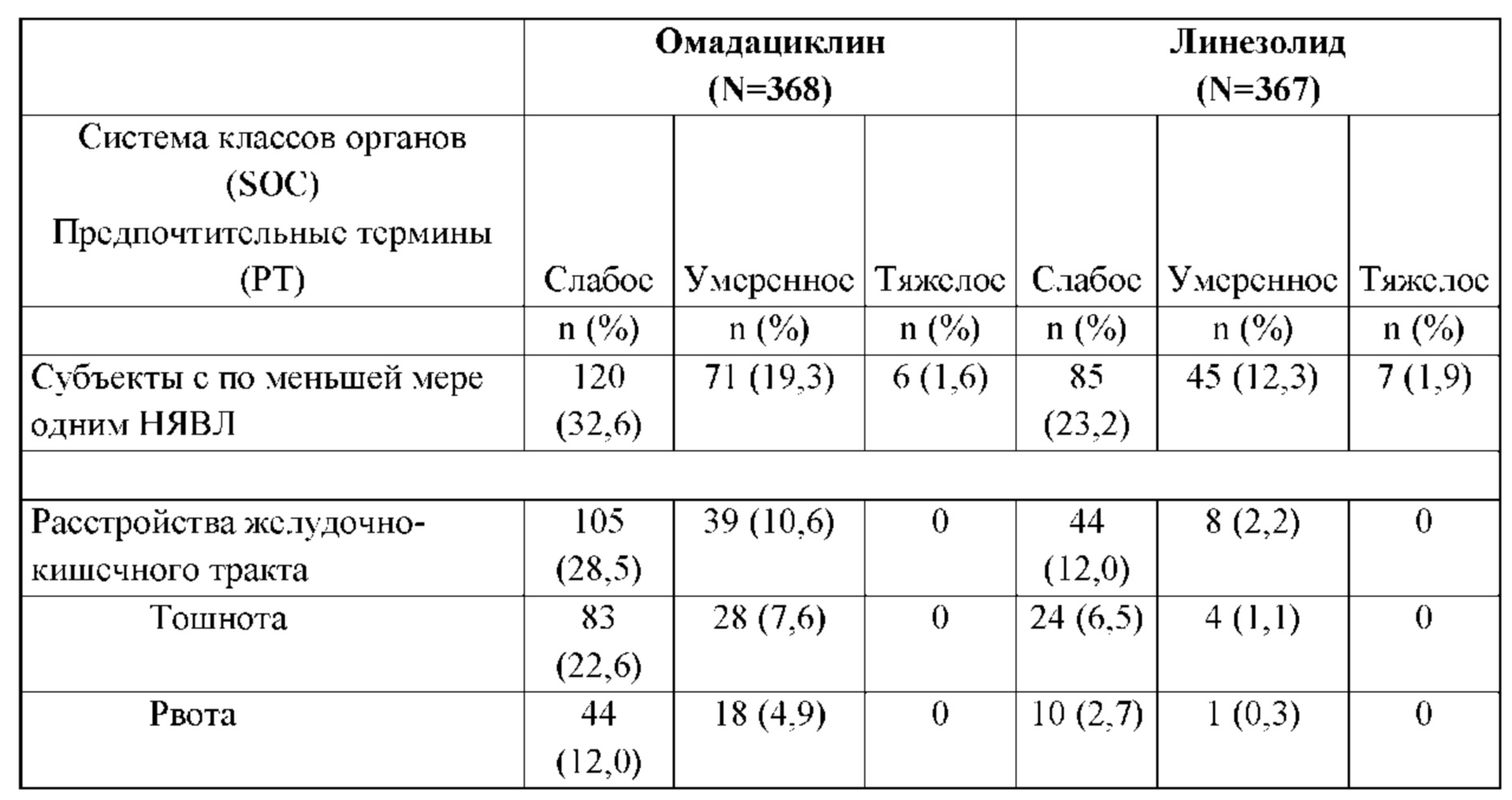

Классификацию по системам классов органов и предпочтительным термином проводили на основании MedDRA версии 17.1. Значения в процентах указаны для популяции для анализа безопасности. НЯВЛ было определено как НЯ, возникшее после введения первой дозы активного исследуемого препарата. Если субъект имел более одного НЯВЛ, отнесенного к одной категории MedDRA, его учитывали только один раз по наиболее тяжелому явлению. НЯВЛ с неуказанной тяжестью не показаны.
Проводили описательный анализ следующих переменных: основные показатели жизнедеятельности (систолическое и диастолическое КД, пульс, температура тела, частота дыхания), включая изменение от скрининга до данного посещения; клинически значимые основные показатели жизнедеятельности (удовлетворяющие заранее определенным критериям, указанным в SAP) на момент данного посещения. Были перечислены субъекты со значимыми данными основных показателей жизнедеятельности.
Данные ЭКГ (RR интервал, PR интервал, QRS интервал, QTc, корригированный QTc по формуле Базетта [QTcB] и корригированный QTc по формуле Фредерика [QTcF]) были определены в рамках описательной статистики во время каждой запланированной оценки и для наиболее плохого значения после скрининга. Также указаны изменения относительно скрининга во время каждого посещения, когда получали ЭКГ в 12 отведениях. Анализ для исключения выбросов проводили на основании наихудшего значения после скрининга.
Проводили описательный анализ следующих переменных: переменные лабораторные показатели во время данного посещения; изменение относительно скрининга переменных лабораторных показателей во время данного посещения; и клинически значимые лабораторные показатели (удовлетворяющие заранее определенным критериям, указанным в SAP) во время данного посещения.
Получали списки лабораторных данных для отдельных субъектов. В этих списках помечали значения, удовлетворяющие заранее определенным критериям клинической значимости.
Фармакокинетика
Анализ популяции PK проводили для описания параметров PK. Выборку данных популяции PK, включающую субъектов, у которых концентрация омадациклина, поддающаяся обнаружению, была определена 1 раз или более, получали с учетом даты и времени введения дозы и отбора проб крови, а также определения всех биоаналитических показателей и исходной информации о субъекте. Если дата и время фактического отбора пробы крови или введения дозы отсутствовали, то результаты соответствующего определения биоаналитических показателей на основе концентрации для анализа PK были исключены из всех анализов. Концентрацию омадациклина ниже предела количественного определения учитывали как отсутствующую в сводной статистике и при расчете параметров PK.
Переменные, включая возраст (лет), массу тела (кг), пол и расу/этническую принадлежность, а также другие ковариаты, которые заранее были определены как значимые, были включены в базу данных для популяции PK. В зависимости от субъектов из выборки для анализа популяции использовали описательную статистику указанных переменных во время скрининга. Выбросы могут быть исключены из анализа. Их определяли с использованием графика рассеяния зависимости наблюдаемой концентрации от указанного времени после введения дозы. Распределение числа образцов, взятых у каждого субъекта и поданных для проведения анализа на основе модели, было внесено в таблицы. Кроме того, проводили вычисления в рамках простой описательной статистики в случае концентрации в образцах в зависимости от дня или недели исследования.
Что касается моделирования популяции PK, то результаты исследований 1 фазы указывали на то, что PK омадациклина была линейной, и после внутривенной инфузии профили зависимости концентрация в плазме-время имели распределение согласно 3- камерной модели. Таким образом, в качестве возможной структурной модели PK можно использовать 3-камерную модель с вводением нулевого порядка в случае в.в. инфузии и первого порядка в случае п. о. введения. Указанная PK модель включает следующие параметры: клиренс, объем распределения, биодоступность и скорость всасывания. Связанные популяционные модели представляют собой нелинейные модели со смешанными эффектами. В популяционной модели вводят дополнительные случайные эффекты и ковариаты параметров PK для выявления различий между индивидуумами и аналогий при наблюдении одного субъекта. При проведении популяционного моделирования описанные ранее структурные модели PK учитывали в первую очередь. Изначально рассматривалась модель остаточных ошибок, в которой были объединены аддитивная ошибка и пропорциональная ошибка. Упрощения (например, уменьшение случайных эффектов или альтернативная модель остаточных ошибок) можно вводить, если при диагностике модели предполагается ложная конвергенция. Дополнительные ковариаты изучают графическим методом (пол, раса/этническая принадлежность, возраст) в рамках диагностики модели, некоторые из них могут сохраняться в конечной модели, а некоторые другие в конкурентной модели для получения оценочных значений предположительно незначимых эффектов. Графики рассеяния зависимости наблюдаемых концентраций от оценочных по популяции и отдельных оценочных концентраций также используют в рамках общей оценки общего качества подстановки. Во время моделирования следовали общим принципам, описанным FDA.
Вычисляли и обобщали отдельные измеряемые в соответствии с моделью показатели уровня лекарственного средства в стационарном состоянии (площадь под кривой [AUC]0-24,ss, время достижения максимальной концентрации в плазме [Tmax,ss], максимальная концентрация в плазме [Cmax,ss]).
Взаимосвязь уровня омадациклина и ответа (эффективность и безопасность) будут изучаться в соответствии с полученными данными. Популяционную модель PK планируется использовать для вычисления AUC у отдельных субъектов и затем возможных предельных значений AUC/МИК.
Реферат
Изобретение относится к области медицины и представляет собой способ лечения острой бактериальной инфекции кожи или кожных структур (ABSSSI) у взрослого субъекта-человека. Способ лечения осуществляют путем введения пероральной нагрузочной дозы в виде двух доз по 450 мг 9-[(2,2-диметил-пропиламино)метил]миноциклина, которые вводят за 24 часа и 48 часов, соответственно, перед первой из вводимых раз в день пероральных доз по 300 мг, 450 мг или 600 мг в течение 5 или более дней. Дозы вводят в виде таблеток по 150 мг. Технический результат заключается в разработке усовершенствованного способа лечения острой бактериальной инфекции кожи или кожных структур (ABSSSI) у взрослого субъекта-человека на основе перорального антибактериального агента – омадациклина. 27 з.п. ф-лы, 6 ил., 30 табл.
Комментарии