Новые активаторы рецепторов витамина d и способы их получения - RU2535448C2
Код документа: RU2535448C2
Чертежи




Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к новым производным витамина D и способам получения указанных соединений. Упомянутые новые соединения могут применяться в качестве лекарственных средств для лечения ряда заболеваний, включая, но без ограничения, заболевания костей, сердечно-сосудистое заболевание, гиперпаратиреоз, иммунные расстройства, пролиферативное заболевание, почечное заболевание и тромбоз.
Предпосылки создания изобретения
Открытие того, что витамин D3 является предшественником функционально активного гормона, 1,25-дигидроксивитамина D3, было сделано более 30 лет назад. Последующие исследования привели к современному пониманию того, что витамин D3 образуется из 7-дегидрохолестерина в коже после воздействия на нее ультрафиолетового света, далее модифицируется под действием витамин-D3-25-гидроксилазы в печени и затем под действием 25-дигидроксивитамин-D3-1α-гидроксилазы (CYP27B1) в почках с образованием активного гормона 1,25-дигидроксивитамина D3 (кальцитриол, коммерчески доступен под торговым названием CALCIJEX от Abbott Laboratories, Abbott Park, IL). Кальцитриол реализует свою функцию связыванием с рецептором витамина D (сокращенно называемым далее «VDR» (vitamine D receptor)), ядерным рецептором. Связывание кальцитриола с VDR активирует рецептор для привлечения кофакторов и образования комплекса, который связывается с элементами ответа витамина D в промоторной области целевых генов для регулирования генной транскрипции. Путь передачи сигнала витамина D схематически представлен на Фигуре 1.
В течение последних трех десятилетий большинство исследований в области VDR было направлено на объяснение биохимической роли кальцитриола, например, в минеральном гомеостазе, который включает в себя регулирование паратиреоидного гормона, кишечную кальциевую и фосфатную абсорбцию и костный метаболизм. Как показано на Фигуре 2, 1α-гидроксилаза (CYP27B1) в почке отвечает за продуцирование активного метаболита 1α,25-дигидроксивитамина D3 (кальцитриола), который последовательно связывается с VDR и, в конечном счете, проявляет свое физиологическое действие, включая модулирование транспорта кальция в кишечнике и мобилизацию кальция в костях, регуляцию синтеза паратиреоидного гормона (РТН) и снижение уровня регуляции CYP27B1 через механизм обратной связи. В свою очередь, РТН стимулирует CYP27B1, повышает ресорбцию кальция и снижает ресорбцию фосфатов в почке. Посредством координированных функций РТН и кальцитриола поддерживается постоянное содержание в организме кальция и фосфора. Кальцитриол окисляется с помощью CYP24 (24-гидроксилазы) до метаболитов, которые выводятся из организма. VDR обнаружен в более чем 30 тканях и помимо его функции контроля продуцирования РТН и минерального гомеостаза может обладать и другими эффектами.
В результате проведенных исследований было разработано много новых аналогов кальцитриола, некоторые из которых обладают сниженным гиперкальцемическим действием, и некоторые аналоги, такие как парикальцитол (коммерчески доступен под торговым названием ZEMPLAR от Abbott Laboratories, Abbott Park, IL) и доксеркальциферол (коммерчески доступен под торговым названием HECTOROL от Genzyme, Cambridge, MA) в настоящее время продаются в качестве лекарственных средств для лечения гиперпаратироза, вторичного по отношению к хроническому заболеванию почек (chronic kidney disease - CKD). Кроме того, несколько VDR модуляторов в продаже для лечения псориаза и остеопороза.
Кроме того, поскольку VDR широко распространен в органах и тканях организма, он, вероятно, вовлечен в ряд болезненных состояний. Результаты многочисленных доклинических исследований подтверждают, что VDR модуляторы могут быть полезными для лечения различных заболеваний, включая сердечно-сосудистые заболевания (cardiovascular diseases - CVD), иммунные расстройства, тромбоз, связанный с онкологией, и т.п.
В частности, некоторые факты подтверждают представление о том, что VDR играет важную роль в регулировании сердечно-сосудистой физиологии, иммунной системы и других биофизиологических систем в организме человека. Однако предклинические данные позволили сделать вывод о том, что, по меньшей мере, некоторые активаторы рецепторов витамина D (далее в описании сокращенно взаимозаменяемо называются также «VDRA» (Vitamin D Receptor activators)) и/или аналоги витамина D, особенно в более высоких дозах, могут вызывать гиперкальциемию, которая связана с кальцинозом сосудов, инфарктом миокарда, сердечной недостаточностью, кардиомиопатией и инсультами. В связи с этим, медицинское сообщество не только не одобряет применение таких соединений в качестве терапевтических средств для лечения сердечно-сосудистого заболевания, но в значительной степени рекомендует ограничивать их применение.
Аналогично, хотя в настоящее время некоторые VDRA и/или аналоги витамина D используются для лечения псориаза, иммунных расстройств, их применение ограничено, поскольку они тесно связаны с гиперкальциемическими побочными эффектами.
В последних публикациях сравнивается выживание пациентов с хронической почечной недостаточностью, проходивших гемодиализ, которые получали лечение кальцитриолом или парикальцитолом (Teng, M. et al. N. Engl. J. Med., 2003, 349, 446-456). Было показано значительное превосходство выживания пациентов, принимающих парикальцитол, по сравнению с пациентами, принимающими кальцитриол. Хотя уровни содержания кальция и фосфористых соединений повышались в меньшей степени у пациентов, принимающих лечение парикальцитолом, исследование не показало, обусловлено ли преимущество выживания при применении парикальцитола улучшением дисбаланса минеральных веществ, либо оно является результатом действия терапевтического лечения специфическим витамином D. Кроме того, выживаемость не была связана дозой активатора рецептора витамина D и не зависела от исходных уровней содержания в сыворотке кальция, фосфора или паратиреоидного гормона, подтверждая, что причина более низкой заболеваемости не может быть тесно связана с содержанием этих маркеров заболеваний. Фактически, действительный механизм полезного действия не был определен. Однако, поскольку сердечно-сосудистое заболевание является причиной смерти большинства пациентов, получающих диализ, выживание пациентов может повышаться в результате воздействия парикальцитола на сердечно-сосудистую систему.
Другие исследования (Salusky, I.B.; Goodman, W.G. Nephrology, Dialysis and Transplantation, 2002, 17, 336-339) показали, что терапевтическое лечение с помощью активаторов рецепторов витамина D может действительно ухудшать выживаемость пациентов с хронической почечной недостаточностью вследствие побочных эффектов, таких как кальциноз сердечно-сосудистой системы. Это привело медицинское сообщество к ограничению применения активаторов рецептора витамина D для терапевтического лечения.
Терапия, альтернативная применению активаторов рецептора витамина D, обеспечена кальцимиметиками, такими как цинакальцет (Sensipar®, Amgen). Цинакальцет в отличие от активаторов рецептора витамина D снижает уровни содержания паратиреоидного гормона посредством повышения чувствительности чувствительного к кальцию рецептора паращитовидной железы. Однако существуют ограничения и в таком терапевтическом подходе. Повышенная чувствительность и тяжелая гипокальцемия являются наиболее известными противопоказаниями. Необходим подбор дозы для назначения оптимального терапевтического лечения. Некоторые врачи-клиницисты предлагали одновременно введение и VDRA в качестве подхода к лечению вторичного гиперпаратиреоза.
Введение фармакологического терапевтического активатора рецептора витамина D традиционно включает подбор дозы для регулировки уровней содержания в сыворотке паратиреоидного гормона и/или кальция. Передозировка контролируется для того, чтобы исключить токсическое действие. Следовательно, может быть полезно разработать активаторы рецептора витамина D, обладающие полезным действием, таким как снижение уровней содержания паратиреоидного гормона в широком интервале доз при хроническом заболевании почек, но ограниченным влиянием на повышение уровней содержания кальция в плазме и значительно расширяющие терапевтическое окно. Оказывается также, что преимущество выживания, возможно, связано с улучшенным состоянием сердечно-сосудистой системы. Разумеется, преклинические исследования продемонстрировали желательное улучшение, которое показано с помощью сердечно-сосудистых маркеров. Улучшения в данных аспектах терапевтического лечения с помощью активаторов рецептора витамина D предоставляют благоприятную возможность расширить применение терапии активаторов рецептора витамина D.
Производные витамина D представляют собой сложные молекулы, и их синтез может быть проблематичным. Например, для синтеза соединений с искусственной 20S стереохимией необходим способ эпимеризации С20 центра (как обозначено в соответствии с системой нумерации витамина D и показано на формуле (I)), а также способ разделения двух полученных изомеров, который, как правило, представляет собой хроматографию. Таким образом, в синтезе данных соединений должен включать эпимеризацию в мягких условиях и химический способ разделения изомеров.
Аналогично, для описанного синтеза колец А, содержащих 2-метиленовый фрагмент (систему нумерации см. выше), необходимо только шесть стадий, но общий выход является неудовлетворительным, и нет никаких кристаллических промежуточных продуктов для гарантированной очистки.
Кроме того, конечное связывание кольца А с кольцом C/D обычно протекает с небольшим выходом; более совершенная методика связывания сделала бы производные витамина D более доступными.
Сущность изобретения
Изобретение относится к активаторам рецептора витамина D, композициям, включающим указанные соединения, способам их получения и промежуточным продуктам, полученным указанными способами. Один аспект настоящего изобретения относится к соединению формулы (I)

или его фармацевтически приемлемой соли или пролекарству, где
атом углерода, к которому присоединен Х, может иметь R или S конфигурацию;
X представляет собой -CH2OR1, -CH2OC(O)R2, -CR3R4-(CH2)m-CR5R6-CR7(CH3)2 или OR8;
Y1 и Y2 каждый представляет собой водород или они вместе образуют метиленовую группу;
Y3 и Y4 каждый представляет собой водород или они вместе образуют метиленовую группу;
Z1 представляет собой фтор, гидроксильную группу или гидроксиметил;
Z2 представляет собой фтор или гидроксильную группу;
R1 представляет собой водород, алкил или арил;
R2 представляет собой алкил, алкиламино, алкилкарбонилоксиалкил или гидроксиалкил;
R3 и R4 независимо представляют собой водород или алкоксигруппу, при условии, что одновременно они оба не являются алкоксигруппой;
R5 и R6 независимо представляют собой водород или алкил;
R7 представляет собой водород, алкоксигруппу или гидроксильную группу;
R8 представляет собой -CH2CH2C(CH3)2OH; и
m равно 1, 2 или 3.
Другой аспект настоящего изобретения относится к фармацевтическим композициям, включающим в себя соединения согласно настоящему изобретению. Такие композиции могут вводиться в соответствии со способом согласно настоящему изобретению, обычно как часть схемы лечения для лечения или предупреждения состояний и расстройств, связанных с активностью рецептора витамина D, в частности у млекопитающих.
Еще один аспект настоящего изобретения относится к способу селективного модулирования активности рецептора витамина D. Способ может применяться для лечения, предупреждения или лечения и предупреждения состояний и расстройств, связанных с активностью рецептора витамина D у млекопитающих. Точнее, способ применим для лечения состояний и расстройств, связанных с почечным заболеванием, вторичного гиперпаратиреоза, связанного с хроническим заболеванием почек, остеопороза, остеомаляции, остеодистрофии, образования тромбов, ренин-ангиотензиновой системы, миокардиальной гипертрофии, гипертензии, аутоиммунных расстройств, подавления иммунитета, отторжения трансплантата, артрита, рассеянного склероза, псориаза, воспалительной болезни кишечника, диабета 1 типа или системной красной волчанки, злокачественных опухолей прямой кишки, предстательной железы, молочной железы, лейкоза или саркомы Капоши.
Соединения, композиции, включающие в себя указанные соединения, способы применения соединений и способы получения соединения, а также промежуточные продукты, полученные в таких способах, также описаны в данном изобретении.
Краткое описание рисунков
На фигуре 1 представлена схема пути передачи сигнала витамина D в организме человека.
На фигуре 2 представлена схема, иллюстрирующая роль витамина D в минеральном гомеостазе.
На фигуре 3 представлена последовательность in vitro и/или vivo анализов, проводимых на соединениях согласно настоящему изобретению для оценки их биологической активности.
Подробное описание изобретения
Определение терминов
Термины, используемые в данном описании, имеют следующие значения.
Термин «алкенил», когда используется в данном описании, означает углеводород с прямой или разветвленной цепью, содержащей от 2 до 10 атомов углерода и, по меньшей мере, одну двойную связь, образованную посредством удаления двух атомов водорода. Типичные примеры алкенила включают, но без ограничения, этенил, 2-пропенил, 2-метил-2-пропенил, 3-бутенил, 4-пентенил, 5-гексенил, 2-гептенил, 2-метил-1-гептенил и 3-деценил.
Термин «алкенилен» означает двухвалентную углеводородную группу с прямой или разветвленной цепью, содержащей от 2 до 10 атомов углерода и, по меньшей мере, одну двойную связь. Типичные примеры алкенилена включают, но без ограничения, -CH=CH-, -CH=CH2CH2- и -CH=C(CH3)CH2-.
Термин «алкенилокси», когда используется в данном описании, означает алкенильную группу, которая определена выше, присоединенную к фрагменту основной молекулы через атом кислорода. Типичные примеры алкенилокси включают, но без ограничения, аллилокси, 2-бутенилокси и 3-бутенилокси.
Термин «алкокси», когда используется в данном описании, означает алкильную группу, которая определена в описании, присоединенную к фрагменту основной молекулы через атом кислорода. Типичные примеры алкоксигрупп включают, но без ограничения, метокси, этокси, пропокси, 2-пропокси, бутокси, трет-бутокси, пентилокси и гексилокси.
Термин «алкоксиалкокси», когда используется в данном описании, означает алкоксигруппу, которая определена в описании, присоединенную к фрагменту основной молекулы через другую алкоксигруппу, которая определена в описании. Типичные примеры алкоксиалкоксигруппы включают, но без ограничения, трет-бутоксиметокси, 2-этоксиэтокси, 2-метоксиэтокси и метоксиметокси.
Термин «алкоксиалкоксиалкил», когда используется в данном описании, означает алкоксиалкоксигруппу, которая определена в описании, присоединенную к фрагменту основной молекулы через алкильную группу, которая определена в описании. Типичные примеры алкоксиалкоксиалкила включают, но без ограничения, трет-бутоксиметоксиметил, этоксиметоксиметил, (2-метоксиэтокси)метил и 2-(2-метоксиэтокси)этил.
Термин «алкоксиалкил», когда используется в данном описании, означает алкоксигруппу, которая определена в описании, присоединенную к фрагменту основной молекулы через алкильную группу, которая определена в описании. Типичные примеры алкоксиалкила включают, но без ограничения, трет-бутоксиметил, 2-этоксиэтил, 2-метоксиэтил и метоксиметил.
Термин «алкоксикарбонил», когда используется в данном описании, означает алкоксигруппу, которая определена в описании, присоединенную к фрагменту основной молекулы через карбонильную группу, которая определена в описании. Типичные примеры алкоксикарбонила включают, но без ограничения, метоксикарбонил, этоксикарбонил и трет-бутоксикарбонил.
Термин «алкоксикарбонилалкил», когда используется в данном описании, означает алкоксикарбонильную группу, которая определена в описании, присоединенную к фрагменту основной молекулы через алкильную группу, которая определена в описании. Типичные примеры алкоксикарбонилаклила включают, но без ограничения, 3-метоксикарбонилпропил, 4-этоксикарбонилбутил и 2-трет-бутоксикарбонилэтил.
Термин «алкоксисульфонил», когда используется в данном описании, означает алкоксигруппу, которая определена в описании, присоединенную к фрагменту основной молекулы через сульфонильную группу, которая определена в описании. Типичные примеры алкоксисульфонила включают, но без ограничения, метоксисульфонил, этоксисульфонил и пропоксисульфонил.
Термин «алкил», когда используется в данном описании, означает углеводородную группу с прямой или разветвленной цепью, содержащей от 1 до 10 атомов углерода. Типичные примеры алкила включают, но без ограничения, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, изопентил, неопентил, н-гексил, 3-метилгексил, 2,2-диметилпентил, 2,3-диметилпентил, н-гептил, н-октил, н-нонил и н-децил.
Термин «алкиламино», когда используется в данном описании, означает алкильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через группу -N(H). Типичные примеры алкиламино включают, но без ограничения, метиламино, циклопропиламино и трет-бутиламино.
Термин «алкилкарбонил», когда используется в данном описании, означает алкильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через карбонильную группу, которая определена в описании. Типичные примеры алкилкарбонила включают, но без ограничения, ацетил, 1-оксопропил, 2,2-диметил-1-оксопропил, 1-оксобутил и 1-оксопентил.
Термин «алкилкарбонилалкил», когда используется в данном описании, означает алкилкарбонильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через алкильную группу, которая определена в данном описании. Типичные примеры алкилкарбонилалкила включают, но без ограничения, 2-оксопропил, 3,3-диметил-2-оксопропил, 3-оксобутил и 3-оксопентил.
Термин «алкилкарбонилокси», когда используется в данном описании, означает алкилкарбонильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через атом кислорода. Типичные примеры алкилкарбонилокси включают, но без ограничения, ацетилокси, этилкарбонилокси и трет-бутилкарбонилокси.
Термин «алкилкарбонилоксиалкил», когда используется в данном описании, означает алкилкарбонилоксигруппу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через алкильную группу, которая определена в данном описании. Типичные примеры алкилкарбонилоксиалкила включают, но без ограничения, ацетоксиметил, ацетоксиэтил и пивалоилоксиметил.
Термин «алкилен» означает двухвалентную группу, полученную из прямой или разветвленной углеводородной цепи, содержащей от 1 до 10 атомов углерода. Типичные примеры алкилена включают, но без ограничения, -CH2-, -CH(CH3)-, -C(CH3)2-, -CH2CH2-, -CH2CH2CH2-, -CH2CH2CH2CH2- и -CH2CH(CH3)CH2-.
Термин «алкилсульфинил», когда используется в данном описании, означает алкильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через сульфинильную группу, которая определена в данном описании. Типичные примеры алкилсульфинила включают, но без ограничения, метилсульфинил и этилсульфинил.
Термин «алкилсульфинилалкил», когда используется в данном описании, означает алкилсульфинильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через алкильную группу, которая определена в данном описании. Типичные примеры алкилсульфинилалкила включают, но без ограничения, метилсульфинилметил и этилсульфинилметил.
Термин «алкилсульфонил», когда используется в данном описании, означает алкильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через сульфонильную группу, которая определена в данном описании. Типичные примеры алкилсульфонила включают, но без ограничения, метилсульфонил и этилсульфонил.
Термин «алкилсульфонилалкил», когда используется в данном описании, означает алкилсульфонильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через алкильную группу, которая определена в данном описании. Типичные примеры алкилсульфонилалкила включают, но без ограничения, метилсульфонилметил и этилсульфонилметил.
Термин «алкилтио», когда используется в данном описании, означает алкильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через атом серы. Типичные примеры алкилтиогруппы включают, но без ограничения, метилтио, этилтио, трет-бутилтио и гексилтио.
Термин «алкилтиоалкил», когда используется в данном описании, означает алкилтиогруппу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через алкильную группу, которая определена в данном описании. Типичные примеры алкилтиоалкила включают, но без ограничения, метилтиометил и 2-(этилтио)этил.
Термин «алкинил», когда используется в данном описании, означает углеводородную группу с прямой или разветвленной цепью, содержащей от 2 до 10 атомов углерода и, по меньшей мере, одну тройную углерод-углеродную связь. Типичные примеры алкинила включают, но без ограничения, ацетиленил, 1-пропинил, 2-пропинил, 3-бутинил, 2-пентинил и 1-бутинил.
Термин «алкинилен» означает двухвалентную группу, полученную из углеводородной группы с прямой или разветвленной цепью, включающей от 2 до 10 атомов углерода, и содержащую, по меньшей мере, одну тройную связь. Типичные примеры алкинилена включают, но без ограничения, -C≡C-, -CH2C≡C-, -CH(CH3)CH2C≡C-, -C≡CCH2- и -C≡CCH(CH3)CH2-.
Термин «алкинилокси», когда используется в данном описании, означает алкинильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через атом кислорода. Типичные примеры алкинилоксигруппы включают, но без ограничения, 2-пропинилокси и 2-бутинилокси.
Термин «арил», когда используется в данном описании, означает фенил, бициклический арил или трициклический арил. Бициклический арил представляет собой нафтил, фенил, конденсированный с циклоалкилом, или фенил, конденсированный с циклоалкенилом. Типичные примеры бициклического арила включают, но без ограничения, дигидроинденил, инденил, нафтил, дигидронафталинил и тетрагидронафталинил. Трициклический арил представляет собой антрацен, фенантрен или бициклический арил, конденсированный с циклоалкилом, бициклический арил, конденсированный с циклоалкенилом, или бициклический арил, конденсированный с фенилом. Типичные примеры трициклического арила включают, но без ограничения, азуленил, дигидроантраценил, флуоренил и тетрагидрофенантренил
Арильные группы согласно изобретению могут быть замещенными и содержать 1, 2, 3, 4 или 5 заместителей, независимо выбранных из алкенила, алкоксигруппы, алкоксиалкоксигруппы, алкоксиалкоксиалкила, алкоксиалкила, алкоксикарбонила, алкоксикарбонилалкила, алкила, алкилкарбонила, алкилкарбонилалкила, алкилкарбонилоксигруппы, алкилсульфинила, алкилсульфинилалкила, алкилсульфонила, алкилсульфонилалкила, алкилтиогруппы, алкилтиоалкила, алкинила, карбоксильной группы, карбоксиалкила, цианогруппы, цианоалкила, формила, формилалкила, галогена, галогеналкила, гидроксильной группы, гидроксиалкила, меркаптогруппы, нитрогруппы, -NZ7Z8 и (NZ9Z10)карбонила.
Термин «арилалкокси», когда используется в данном описании, означает арильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через алкоксигруппу, которая определена в данном описании. Типичные примеры арилалкокси включают, но без ограничения, 2-фенилэтокси, 3-нафт-2-илпропокси и 5-фенилпентилокси.
Термин «арилалкоксикарбонил», когда используется в данном описании, означает арилалкоксигруппу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через карбонильную группу, которая определена в данном описании. Типичные примеры арилалкоксикарбонила включают, но без ограничения, бензилоксикарбонил и нафт-2-илметоксикарбонил.
Термин «арилалкил», когда используется в данном описании, означает арильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через алкильную группу, которая определена в данном описании. Типичные примеры арилалкила включают, но без ограничения, бензил, 2-фенилэтил, 3-фенилпропил и 2-нафт-2-илэтил.
Термин «арилалкилтио», когда используется в данном описании, означает арилалкильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через атом серы. Типичные примеры арилалкилтиогруппы включают, но без ограничения, 2-фенилэтилтио, 3-нафт-2-илпропилтио и 5-фенилпентилтио.
Термин «арилкарбонил», когда используется в данном описании, означает арильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через карбонильную группу, которая определена в данном описании. Типичные примеры арилкарбонила включают, но без ограничения, бензоил и нафтоил.
Термин «арилокси», когда используется в данном описании, означает арильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через атом кислорода. Типичные примеры арилоксигруппы включают, но без ограничения, фенокси, нафтилокси, 3-бромфенокси, 4-хлорфенокси, 4-метилфенокси и 3,5-диметоксифенокси.
Термин «арилоксиалкил», когда используется в данном описании, означает арилоксигруппу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через алкильную группу, которая определена в данном описании. Типичные примеры арилоксиалкила включают, но без ограничения, 2-феноксиэтил, 3-нафт-2-илоксипропил и 3-бромфеноксиметил.
Термин «арилтио», когда используется в данном описании, означает арильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через атом серы. Типичные примеры арилтиогруппы включают, но без ограничения, фенилтио и 2-нафтилтио.
Термин «арилтиоалкил», когда используется в данном описании, означает арилтиогруппу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через алкильную группу, которая определена в данном описании. Типичные примеры арилтиоалкила включают, но без ограничения, фенилтиометил, 2-нафт-2-илтиоэтил и 5-фенилтиометил.
Термин «азидо», когда используется в данном описании, означает -N3 группу.
Термин «карбонил», когда используется в данном описании, означает -C(O)- группу.
Термин «карбокси», когда используется в данном описании, означает -CO2H группу.
Термин «карбоксиалкил», когда используется в данном описании, означает карбоксильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через алкильную группу, которая определена в данном описании. Типичные примеры карбоксиалкила включают, но без ограничения, карбоксиметил, 2-карбоксиэтил и 3-карбоксипропил.
Термин «циано», когда используется в данном описании, означает -CN группу.
Термин «цианоалкил», когда используется в данном описании, означает цианогруппу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через алкильную группу, которая определена в данном описании. Типичные примеры цианоалкила включают, но без ограничения, цианометил, 2-цианоэтил и 3-цианопропил.
Термин «циклоалкенил», когда используется в данном описании, означает циклический углеводород, содержащий от 3 до 8 атомов углерода и, по меньшей мере, одну двойную углерод-углеродную связь, образованную в результате удаления двух атомов водорода. Типичные примеры циклоалкенила включают, но без ограничения, 2-циклогексен-1-ил, 3-циклогексен-1-ил, 2,4-циклогексадиен-1-ил и 3-циклопентен-1-ил.
Термин «циклоалкил», когда используется в данном описании, означает моноциклическую, бициклическую или трициклическую кольцевую систему. Типичными примерами моноциклических кольцевых систем являются насыщенные циклические углеводородные группы, содержащие от 3 до 8 атомов углерода. Такие примеры моноциклических кольцевых систем включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил. Типичными примерами бициклических кольцевых систем являются моноциклические кольцевые системы, содержащие мостиковые связи, в которых два соседних или несоседних атома углерода моноциклического кольца соединены алкилeновыми мостиками, включающими от одного до трех дополнительных атомов углерода. Типичные примеры бициклических кольцевых систем включают, но без ограничения, бицикло[3.1.1]гептан, бицикло[2.2.1]гептан, бицикло[2.2.2]октан, бицикло[3.2.2]нонан, бицикло[3.3.1]нонан и бицикло[4.2.1]нонан. Типичные примеры трициклических кольцевых систем включают бициклическую кольцевую систему, в которой два несоседних атома углерода бициклического кольца соединены связью или алкилeновым мостиком, содержащим от одного до трех атомов углерода. Такие примеры трициклических кольцевых систем включают, но без ограничения, трицикло[3.3.1.03,7]нонан и трицикло[3.3.1.13,7]декан (адамантан).
Циклоалкильные группы согласно изобретению являются необязательно замещенными и содержат 1, 2, 3, 4 или 5 заместителей, выбранных из группы, включающей алкенил, алкоксигруппу, алкоксиалкоксигруппу, алкоксиалкил, алкоксикарбонил, алкоксисульфонил, алкил, алкилкарбонил, алкилкарбонилоксигруппу, алкилсульфонил, алкилтио, алкилтиоалкил, алкинил, карбокси, цианогруппу, формил, галогеналкоксигруппу, галогеналкил, галоген, гидроксильную группу, гидроксиалкил, меркаптогруппу, оксогруппу, -NZ7Z8 и (NZ9Z10)карбонил.
Термин «циклоалкилалкил», когда используется в данном описании, означает циклоалкильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через алкильную группу, которая определена в данном описании. Типичные примеры циклоалкилалкила включают, но без ограничения, циклопропилметил, 2-циклобутилэтил, циклопентилметил, циклогексилметил и 4-циклогептилбутил.
Термин «циклоалкилкарбонил», когда используется в данном описании, означает циклоалкильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через карбонильную группу, которая определена в данном описании. Типичные примеры циклоалкилкарбонила включают, но без ограничения, циклопропилкарбонил, 2-циклобутилкарбонил и циклогексилкарбонил.
Термин «циклоалкилокси», когда используется в данном описании, означает циклоалкильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через атом кислорода, который определен в данном описании. Типичные примеры циклоалкилокси включают, но без ограничения, циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси, циклогептилокси и циклооктилокси.
Термин «циклоалкилтио», когда используется в данном описании, означает циклоалкильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через атом серы, который определен в данном описании. Типичные примеры циклоалкилтиогруппы включают, но без ограничения, циклопропилтио, циклобутилтио, циклопентилтио, циклогексилтио, циклогептилтио и циклооктилтио.
Термин «этилендиокси», когда используется в данном описании, означает -О(CH2)2-O- группу, в которой атомы кислорода этилендиоксигруппы присоединены к фрагменту основной молекулы через атом углерода, образуя 5-членное кольцо, или атомы кислорода этилендиоксигруппы присоединены к фрагменту основной молекулы через два соседних атома углерода, образуя шестичленное кольцо.
Термин «формил», когда используется в данном описании, означает -C(O)H группу.
Термин «формилалкил», когда используется в данном описании, означает формильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через алкильную группу, которая определена в данном описании. Типичные примеры формилалкила включают, но без ограничения, формилметил и 2-формилэтил.
Термин «галоген», когда используется в данном описании, означает -Cl, -Br, -I или -F.
Термин «галогеналкокси», когда используется в данном описании, означает, по меньшей мере, один атом галогена, который определен в данном описании, присоединенный к фрагменту основной молекулы через алкоксигруппу, которая определена в данном описании. Типичные примеры галогеналкоксигруппы включают, но без ограничения, хлорметокси, 2-фторэтокси, трифторметокси и пентафторэтокси.
Термин «галогеналкил», когда используется в данном описании, означает, по меньшей мере, один атом галогена, который определен в данном описании, присоединенный к фрагменту основной молекулы через алкильную группу, которая определена в данном описании. Типичные примеры галогеналкила включают, но без ограничения, хлорметил, 2-фторэтил, трифторметил, пентaфторэтил, и 2-хлор-3-фторпентил.
Термин «гетероарил», когда используется в данном описании, означает моноциклический гетероарил или бициклический гетероарил. Моноциклический гетероарил представляет собой 5- или 6-членный цикл, который содержит, по меньшей мере, один гетероатом, выбранный из группы, включающей атом азота, атом кислорода и атом серы. 5-членный цикл содержит две двойных связи, и 6-членный цикл содержит три двойных связи. 5- или 6-членный гетероарил присоединен к фрагменту основной молекулы через любой атом углерода или любой, способный содержать заместители атом азота в гетероарильном цикле при условии, что сохраняется приемлемая валентность. Типичные примеры моноциклического гетероарила включают, но без ограничения, фурил, имидазолил, изоксазолил, изотиазолил, оксадиазолил, оксазолил, пиридинил, пиридазинил, пиримидинил, пиразинил, пиразолил, пирролил, тетразолил, тиадиазолил, тиазолил, тиенил, триазолил и триазинил. Бициклический гетероарил состоит из моноциклического гетероарила, конденсированного с фенилом, моноциклического гетероарила, конденсированного с циклоалкилом, моноциклического гетероарила, конденсированного с циклоалкенилом, или моноциклического гетероарила, конденсированного с моноциклическим гетероарилом. Бициклический гетероарил присоединен к фрагменту основной молекулы через любой атом углерода или любой способный замещаться атом азота в бициклическом гетероариле при условии, что сохраняется приемлемая валентность. Типичные примеры бициклического гетероарила включают, но без ограничения, азаиндолил, бензимидазолил, бензофуранил, бензоксадиазолил, бензоизоксазол, бензоизотиазол, бензоксазол, 1,3-бензотиазолил, бензотиофенил, циннолинил, фуропиридин, индолил, индазолил, изобензофуран, изоиндолил, изохинолинил, нафтиридинил, оксазолопиридин, хинолинил, хиноксалинил и тиенопиридинил.
Гетероарильные группы согласно изобретению являются необязательно замещенными и могут содержать 1, 2, 3 или 4 заместителя, независимо выбранные из группы, включающей алкенил, алкоксигруппу, алкоксиалкокси, алкоксиалкил, алкоксикарбонил, алкоксикарбонилалкил, алкоксисульфонил, алкил, алкилкарбонил, алкилкарбонилалкил, алкилкарбонилокси, алкилтио, алкилтиоалкил, алкинил, карбокси, карбоксиалкил, циано, цианоалкил, формил, галогеналкокси, галогеналкил, галоген, гидроксильную группу, гидроксиалкил, меркапто, нитро, -NZ7Z8 и (NZ9Z10)карбонил. Гетероарильные группы согласно изобретению, которые являются замещенными и содержат гидроксильную группу в качестве заместителя, могут существовать в форме таутомеров. Гетероарильные группы согласно изобретению включают все таутомеры, в том числе неароматические таутомеры.
Термин «гетероарилалкокси», когда используется в данном описании, означает гетероарильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через алкоксигруппу, которая определена в данном описании. Типичные примеры гетероарилалкокси включают, но без ограничения, фур-3-илметокси, 1H-имидазол-2-илметокси, 1H-имидазол-4-илметокси, 1-(пиридин-4-ил)этокси, пиридин-3-илметокси, 6-хлорпиридин-3-илметокси, пиридин-4-илметокси, (6-(трифторметил)пиридин-3-ил)метокси, (6-(циано)пиридин-3-ил)метокси, (2-(циано)пиридин-4-ил)метокси, (5-(циано)пиридин-2-ил)метокси, (2-(хлор)пиридин-4-ил)метокси, пиримидин-5-илметокси, 2-(пиримидин-2-ил)пропокси, тиен-2-илметокси и тиен-3-илметокси.
Термин «гетероарилалкил», когда используется в данном описании, означает гетероарильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через алкильную группу, которая определена в данном описании. Типичные примеры гетероарилалкила включают, но без ограничения, фур-3-илметил, 1H-имидазол-2-илметил, 1H-имидазол-4-илметил, 1Н-(пиридин-4-ил)этил, пиридин-3-илметил, 6-хлорпиридин-3-илметил, пиридин-4-илметил, (6-(трифторметил)пиридин-3-ил)метил, (6-(циано)пиридин-3-ил)метил, (2-(циано)пиридин-4-ил)метил, (5-(циано)пиридин-2-ил)метил, (2-(хлор)пиридин-4-ил)метил, пиридин-5-илметил, 2-(пиримидин-2-ил)пропил, тиен-2-илметил и тиен-3-илметил.
Термин «гетероарилалкилкарбонил», когда используется в данном описании, означает гетероарилалкильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через карбонильную группу, которая определена в данном описании.
Термин «гетероарилалкилтио», когда используется в данном описании, означает гетероарилалкильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через атом серы. Типичные примеры гетероарилалкилтио включают, но без ограничения, фур-3-илметилтио, 1H-имидазол-2-илметилтио, 1H-имидазол-4-илметилтио, пиридин-3-илметилтио, 6-хлорпиридин-3-илметилтио, пиридин-4-илметилтио, (6-(трифторметил)пиридин-3-ил)метилтио, (6-(циано)пиридин-3-ил)метилтио, (2-(циано)пиридин-4-ил)метилтио, (5-(циано)пиридин-2-ил)метилтио, (2-(хлор)пиридин-4-ил)метилтио, пиримидин-5-илметилтио, 2-(пиримидин-2-ил)пропилтио, тиен-2-илметилтио и тиен-3-илметилтио.
Термин «гетероарилкарбонил», когда используется в данном описании, означает гетероарильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через карбонильную группу, которая определена в данном описании. Типичные примеры гетероарилкарбонила включают, но без ограничения, фур-3-илкарбонил, 1H-имидазол-2-илкарбонил, 1H-имидазол-4-илкарбонил, пиридин-3-илкарбонил, 6-хлорпиридин-3-илкарбонил, пиридин-4-илкарбонил, (6-(трифторметил)пиридин-3-ил)карбонил, (6-(циано)пиридин-3-ил)карбонил, (2-(циано)пиридин-4-ил)карбонил, (5-(циано)пиридин-2-ил)карбонил, (2-(хлор)пиридин-4-ил)карбонил, пиримидин-5-илкарбонил, пиримидин-2-илкарбонил, тиен-2-илкарбонил и тиен-3-илкарбонил.
Термин «гетероарилокси», когда используется в данном описании, означает гетероарильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через атом кислорода. Типичные примеры гетероарилоксигруппы включают, но без ограничения, фур-3-илокси, 1H-имидазол-2-илокси, 1H-имидазол-4-илокси, пиридин-3-илокси, 6-хлорпиридин-3-илокси, пиридин-4-илокси, (6-(трифторметил)пиридин-3-ил)окси, (6-(циано)пиридин-3-ил)окси, (2-(циано)пиридин-4-ил)окси, (5-(циано)пиридин-2-ил)окси, (2-(хлор)пиридин-4-ил)окси, пиримидин-5-илокси, пиримидин-2-илокси, тиен-2-илокси- и тиен-3-илокси-.
Термин «гетероарилоксиалкил», когда используется в данном описании, означает гетероарилоксигруппу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через алкильную группу, которая определена в данном описании. Типичные примеры гетероарилоксиалкила включают, но без ограничения, пиридин-3-илоксиметил и 2-хинолин-3-илоксиэтил.
Термин «гетероарилтио», когда используется в данном описании, означает гетероарильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через атом серы. Типичные примеры гетероарилтиогруппы включают, но без ограничения, пиридин-3-илтио и хинолин-3-илтио.
Термин «гетероарилтиоалкил», когда используется в данном описании, означает гетероарилтиогруппу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через алкильную группу, которая определена в данном описании. Типичные примеры гетероарилтиоалкила включают, но без ограничения, пиридин-3-илтиометил и 2-хинолин-3-илтиоэтил.
Термин «гетероцикл» или «гетероциклический», когда используется в данном описании, означает моноциклический гетероцикл, бициклический гетероцикл или трициклический гетероцикл. Моноциклический гетероцикл представляет собой 3-, 4-, 5-, 6- или 7-членный цикл, содержащий, по меньшей мере, один гетероатом, независимо выбранный из группы, включающей атомы O, N и S. 3- или 4-членный цикл содержит 1 гетероатом, выбранный из группы, включающей атомы O, N и S. 5-членные циклы не содержат двойной связи или содержат одну двойную связь и один, два или три гетероатома, выбранные из группы, включающей атомы O, N и S. 6- или 7- членный цикл не содержит двойных связей или содержит одну или две двойные связи и один, два или три гетероатома, выбранные из группы, включающей атомы O, N и S. Моноциклический гетероцикл присоединен к фрагменту основной молекулы через любой атом углерода или любой атом азота, который содержится в моноциклическом гетероцикле. Типичные примеры моноциклического гетероцикла включают, но без ограничения, азетидинил, азепанил, азиридинил, диазепанил, 1,3-диоксанил, 1,3-диоксоланил, 1,3-дитиоланил, 1,3-дитианил, имидазолинил, имидазолидинил, изотиазолинил, изотиазолидинил, изоксазолинил, изоксазолидинил, морфолинил, оксадиазолинил, оксадиазолидинил, оксазолинил, оксазолидинил, пиперазинил, пиперидинил, пиранил, пиразолинил, пиразолидинил, пирролинил, пирролидинил, тетрагидрофуранил, тетрагидротиенил, тиадиазолинил, тиадиазолидинил, тиазолинил, тиазолидинил, тиаморфолинил, 1,1-диоксидотиаморфолинил (тиоморфолинсульфон), тиопиранил и тритианил. Бициклический гетероцикл представляет собой 5- или 6-членный моноциклический гетероцикл, конденсированный с фенильной группой, 5- или 6-членный моноциклический гетероцикл, конденсированный с циклоалкилом, 5- или 6-членный моноциклический гетероцикл, конденсированный с циклоалкенилом, или 5- или 6-членный моноциклический гетероцикл, конденсированный с моноциклическим гетероциклом. Бициклический гетероцикл присоединен к фрагменту основной молекулы через любой атом углерода или любой атом азота, находящийся в бициклическом гетероцикле. Типичные примеры бициклического гетероцикла включают, но без ограничения, 1,3-бензодиоксолил, 1,3-бензодитиолил, 2,3-дигидро-1,4-бензодиоксинил, бензодиоксолил, 2,3-дигидро-1-бензофуранил, 2,3-дигидро-1-бензотиенил, хроменил и 1,2,3,4-тетрагидрохинолинил. Трициклический гетероцикл представляет собой бициклический гетероцикл, конденсированный с фенилом, бициклический гетероцикл, конденсированный с циклоалкилом, бициклический гетероцикл, конденсированный с циклоалкенилом, или бициклический гетероцикл, конденсированный с моноциклическим гетероциклом. Трициклический гетероцикл присоединен к фрагменту основной молекулы через любой атом углерода или любой атом азота трициклического гетероцикла. Типичные примеры трициклического гетероцикла включают, но без ограничения, 2,3,4,4a,9,9a-гексагидро-1H-карбазолил, 5a,6,7,8,9,9a-гексагидродибензо[b,d]фуранил и 5a,6,7,8,9,9a-гексагидродибензо[b,d]тиенил.
Гетероциклы согласно настоящему изобретению являются необязательно замещенными и могут содержать 1, 2, 3 или 4 заместителя, независимо выбранных из группы, включающей алкенил, алкоксигруппу, алкоксиалкокси, алкоксиалкил, алкоксикарбонил, алкоксикарбонилалкил, алкоксисульфонил, алкил, алкилкарбонил, алкилкарбонилалкил, алкилкарбонилокси, алкилтио, алкилтиоалкил, алкинил, карбокси, карбоксиалкил, циано, цианоалкил, формил, галогеналкокси, галогеналкил, галоген, гидроксильную группу, гидроксиалкил, меркаптогруппу, оксогруппу, -NZ7Z8 и (NZ9Z10)карбонил.
Термин «гетероциклоалкокси», когда используется в данном описании, означает гетероциклическую группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через алкоксигруппу, которая определена в данном описании. Типичные примеры гетероциклоалкоксигруппы включают, но без ограничения, 2-пиридин-3-илэтокси, 3-хинолин-3-илпропокси и 5-пиридин-4-илпентилокси.
Термин «гетероциклоалкил», когда используется в данном описании, означает гетероцикл, который определен в данном описании, присоединенный к фрагменту основной молекулы через алкильную группу, которая определена в данном описании. Типичные примеры гетероциклоалкила включают, но без ограничения, пиперидин-4-илметил, пиперазин-1-илметил, 3-метил-1-пирролидин-1-илбутил, (1R)-3-метил-1-пирролидин-1-илбутил, (1S)-3-метил-1-пирролидин-1-илбутил.
Термин «гетероциклоалкилкарбонил», когда используется в данном описании, означает гетероциклоалкил, который определен в данном описании, присоединенный к фрагменту основной молекулы через карбонильную группу, которая определена в данном описании. Типичные примеры гетероциклоалкилкарбонила включают, но без ограничения, пиперидин-4-илметилкарбонил, пиперазин-1-илметилкарбонил, 3-метил-1-пирролидин-1-илбутилкарбонил, (1R)-3-метил-1-пирролидин-1-илбутилкарбонил, (1S)-3-метил-1-пирролидин-1-илбутилкарбонил.
Термин «гетероциклоалкилтио», когда используется в данном описании, означает гетероциклалкильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через атом серы. Типичные примеры гетероциклалкилтио включают, но без ограничения, 2-пиридин-3-илэтилтио, 3-хинолин-3-илпропилтио и 5-пиридин-4-илпентилтио.
Термин «гетероциклокарбонил», когда используется в данном описании, означает гетероцикл, который определен в данном описании, присоединенный к фрагменту основной молекулы через карбонильную группу, которая определена в данном описании.
Термин «гетероциклокарбонилалкил», когда используется в данном описании, означает гетероциклокарбонил, который определен в данном описании, присоединенный к фрагменту основной молекулы через алкильную группу, которая определена в данном описании.
Термин «гетероциклокси», когда используется в данном описании, означает гетероциклическую группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через атом кислорода. Типичные примеры гетероциклоксигруппы включают, но без ограничения, пиридин-3-илокси и хинолин-3-илокси.
Термин «гетероциклоксиалкил», когда используется в данном описании, означает гетероциклоксигруппу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через алкильную группу, которая определена в данном описании. Типичные примеры гетероциклоксиалкила включают, но без ограничения, пиридин-3-илоксиметил и 2-хинолин-3-илоксиэтил.
Термин «гетероциклтио», когда используется в данном описании, означает гетероциклическую группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через атом серы. Типичные примеры гетероциклтиогруппы включают, но без ограничения, пиридин-3-илтио и хинолин-3-илтио.
Термин «гетероциклотиоалкил», когда используется в данном описании, означает гетероциклотиогруппу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через алкильную группу, которая определена в данном описании. Типичные примеры гетероциклотиоалкила включают, но без ограничения, пиридин-3-илтиометил и 2-хинолин-3-илтиоэтил.
Термин «гидроксильная группа», когда используется в данном описании, означает -OH группу.
Термин «гидроксиалкил», когда используется в данном описании, означает, по меньшей мере, одну гидроксильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через алкильную группу, которая определена в данном описании. Типичные примеры гидроксиалкила включают, но без ограничения, гидроксиметил, 2-гидроксиэтил, 3-гидроксипропил, 2,3-дигидроксипентил и 2-этил-4-гидроксигептил.
Термин «защитная группа гидроксильной группы» или «O-защищающая группа» означает заместитель, который защищает гидроксильные группы от нежелательных взаимодействий в процессе синтеза. Примеры защитных групп гидроксильной группы включают, но без ограничения, группы замещенных простых метиловых эфиров, например, метоксиметил, бензилоксиметил, 2-метоксиэтоксиметил, 2-(триметилсилил)этоксиметил, бензил- и трифенилметил; группы простых тетрагидропираниловых эфиров; группы замещенных простых этиловых эфиров, например, 2,2,2-трихлорэтил и трет-бутилэтил; группы простых силиловых эфиров, например, триэтилсилил, триметилсилил, трет-бутилдиметилсилил и трет-бутилдифенилсилил; группы циклических ацеталей и кеталей, например, метиленацеталь, ацетонид и бензилиденацеталь; группы сложных циклических ортоэфиров, например, метоксиметилен; группы циклических карбонатов; и группы циклических боронатов. Традиционно используемые защитные группы гидроксильной группы описаны в публикации T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 3rd Edition, John Wiley & Sons, New York (1999).
Термин «низший алкенил», когда используется в данном описании, означает подгруппу алкенильных групп, которые определены в данном описании, и означает алкенильную группу, содержащую от 2 до 4 атомов углерода. Примерами низшего алкенила являются этенил, пропенил и бутенил.
Термин «низшая алкоксигруппа», когда используется в данном описании, относится к подгруппе алкоксигрупп, которые определены в данном описании, и означает низшую алкильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через атом кислорода, который определен в данном описании. Типичные примеры низшей алкоксигруппы включают, но без ограничения, метоксигруппу, этоксигруппу, пропоксигруппу, 2-пропоксигруппу, бутоксигруппу и трет-бутоксигруппу.
Термин «низший алкил», когда используется в данном описании, относится к подгруппе алкильных групп, которые определены в данном описании, и означает углеводородную группу с прямой или разветвленной цепью, содержащей от 1 до 4 атомов углерода. Примеры низшего алкила включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил и трет-бутил.
Термин «низший алкилтио», когда используется в данном описании, относится к подгруппе алкилтиогрупп и означает низшую алкильную группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через атом серы. Типичные примеры низшей алкилтиогруппы включают, но без ограничения, метилтиогруппу, этилтиогруппу и трет-бутилтиогруппу.
Термин «низший алкинил», когда используется в данном описании, относится к подгруппе алкинильных групп, которые определены в данном описании, и означает алкинильную группу, содержащую от 2 до 4 атомов углерода. Примеры низшего алкинила включают этинил, пропинил и бутинил.
Термин «низший галогеналкокси», когда используется в данном описании, относится к подмножеству галогеналкоксигрупп, которые определены в данном описании, и означает галогеналкоксигруппу с прямой или разветвленной цепью, содержащей от 1 до 4 атомов углерода. Типичные примеры низшей галогеналкоксигруппы включают, но без ограничения, трифторметокси, трихлорметокси, дихлорметокси, фторметокси и пентафторэтокси.
Термин «низший галогеналкил», когда используется в данном описании, представляет собой подгруппу галогеналкильных групп, которые определены в данном описании, и означает галогеналкильную группу с прямой или разветвленной цепью, содержащей от 1 до 4 атомов углерода. Типичные примеры низшего галогеналкила включают, но без ограничения, трифторметил, трихлорметил, дихлорметил, фторметил и пентафторэтил.
Термин «меркапто», когда используется в данном описании, означает -SH группу.
Термин «меркаптоалкил», когда используется в данном описании, означает меркаптогруппу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через алкильную группу, которая определена в данном описании. Типичные примеры меркаптоалкила включают, но без ограничения, 2-меркаптоэтил и 3-меркаптопропил.
Термин «метилендиокси», когда используется в данном описании, означает -OCH2O- группу, в которой атомы кислорода метилендиоксигруппы присоединены к фрагменту основной молекулы через два соседних атома углерода.
Термин «защитная группа атома азота», когда используется в данном описании, означает группы, предназначенные для защиты аминогруппы от нежелательных взаимодействий в процессе синтеза. Предпочтительные защитные группы атома азота включают ацетил, бензоил, бензил, бензилоксикарбонил (Cbz), формил, фенилсульфонил, трет-бутоксикарбонил (Boc), трет-бутилацетил, трифторацетил и трифенилметил (тритил).
Термин «нитро», когда используется в данном описании, означает -NO2 группу.
Термин "NZ7Z8», когда используется в данном описании, означает две группы, Z7 и Z8, которые присоединяются к фрагменту основной молекулы через атом азота. Z7 и Z8 независимо выбраны из группы, включающей водород, алкил, алкилкарбонил, алкоксикарбонил, арил, арилалкил, формил и (NZ11Z12)карбонил. В некоторых случаях осуществления данного изобретения Z7 и Z8 вместе с атомом азота, к которому они присоединены, образуют гетероцикл. Типичные примеры NZ7Z8 включают, но без ограничения, амино, метиламино, ацетиламино, ацетилметиламино, фениламино, бензиламино, азетидинил, пирролидинил и пиперидинил.
Термин «NZ9Z10», когда используется в данном описании, означает две группы, Z9 и Z10, которые присоединены к фрагменту основной молекулы через атом азота. Z9 и Z10 независимо выбраны из группы, включающей водород, алкил, арил и арилалкил. Типичные примеры NZ9Z10 включают, но без ограничения, амино, метиламино, фениламино и бензиламино.
Термин "NZ11Z12», когда используется в данном описании, означает две группы, Z11 и Z12, которые присоединены к фрагменту основной молекулы через атом азота. Z11 и Z12 независимо выбраны из группы, включающей водород, алкил, арил и арилалкил. Типичные примеры NZ11Z12 включают, но без ограничения, амино, метиламино, фениламино и бензиламино.
Термин «(NZ9Z10)карбонил», когда используется в данном описании, означает NZ9Z10 группу, которая определена в данном описании, присоединенную к фрагменту основной молекулы через карбонильную группу, которая определена в данном описании. Типичные примеры (NZ9Z10)карбонила включают, но без ограничения, аминокарбонил, (метиламино)карбонил, (диметиламино)карбонил и (этилметиламино)карбонил.
Термин «оксо», когда используется в данном описании, означает фрагмент =O.
Термин «сульфинил», когда используется в данном описании, означает -S(O)- группу.
Термин «сульфонил», когда используется в данном описании, означает -SO2- группу.
Термин «таутомер», когда используется в данном описании, означает сдвиг протона от одного атома соединения к другому атому этого соединения, где две или несколько структурно отличных соединений находятся в равновесии по отношению друг к другу.
Соединения согласно изобретению
Соединения согласно изобретению могут быть представлены формулой (I), которая описана в разделе «Сущность изобретения».
В одном варианте осуществления изобретения соединения согласно изобретению могут быть представлены формулой (I), где X представляет собой -CR3R4-(CH2)m-CR5R6-CR7(CH3)2; Y1 и Y2 каждый представляют собой водород или вместе образуют метиленовую группу; Y3 и Y4 каждый представляют собой водород или вместе образуют метиленовую группу; Z1 представляет собой фтор, гидроксильную группу или гидроксиметил; Z2 представляет собой фтор или гидроксильную группу; R3 и R4 независимо представляют собой водород или алкоксигруппу; R5 и R6 независимо представляют собой водород или алкил; R7 представляет собой водород, алкоксигруппу или гидроксильную группу; и m равно 1 или 2.
В другом варианте осуществления изобретения соединения согласно изобретению могут быть представлены формулой (I), где X представляет собой -CH2OC(O)R2; Y1 и Y2 каждый представляет собой водород; Y3 и Y4 вместе образуют метиленовую группу; Z1 представляет собой гидроксильную группу; Z2 представляет собой гидроксильную группу; и R2 представляет собой алкил, алкиламино, алкилкарбонилоксиалкил или гидроксиалкил.
В еще одном варианте осуществления изобретения соединения согласно изобретению могут быть представлены формулой (I), где X представляет собой -CH2OR1; Y1 и Y2 каждый представляет собой водород; Y3 и Y4 вместе образуют метиленовую группу; Z1 представляет собой гидроксильную группу; Z2 представляет собой гидроксильную группу; и R1 представляет собой водород, алкил или арил.
В еще одном варианте осуществления изобретения соединения согласно изобретению могут быть представлены формулой (I), где X представляет собой -OR8; Y1 и Y2 каждый представляет собой водород; Y3 и Y4 вместе образуют метиленовую группу; Z1 представляет собой гидроксильную группу; Z2 представляет собой гидроксильную группу; и R8 представляет собой -CH2CH2C(CH3)2OH.
В еще одном варианте осуществления изобретения соединения согласно изобретению могут быть представлены формулой (I), где X представляет собой -OR8; Y1 и Y2 каждый представляет собой водород; Y3 и Y4 вместе образуют метиленовую группу; Z1 представляет собой гидроксиметил; Z2 представляет собой гидроксильную группу; и R8 представляет собой -CH2CH2C(CH3)2OH.
Конкретные варианты осуществления изобретения, которые рассматриваются как часть изобретения, включают, но без ограничения, соединения формулы (I), их соли или пролекарства, например:
(2S)-2-[(1R,3R,7E,17β)-1,3-дигидрокси-2-метилен-9,10-секоэстра-5,7-диен-17-ил]пропилпивалат;
(1R,3R,7E,17β)-17-[(1S)-2-гидрокси-1-метилэтил]-2-метилен-9,10-секоэстра-5,7-диен-1,3-диол;
(2R)-2-[(1R,3R,7E,17β)-1,3-дигидрокси-2-метилен-9,10-секоэстра-5,7-диен-17-ил]пропилпивалат;
(2S)-2-[(1R,3R,7E,17β)-1,3-дигидрокси-2-метилен-9,10-секоэстра-5,7-диен-17-ил]пропил-2,2-диметилбутаноат;
(2S)-2-[(1R,3R,7E,17β)-1,3-дигидрокси-2-метилен-9,10-секоэстра-5,7-диен-17-ил]пропил-трет-бутилкарбамат;
(2S)-2-[(1R,3R,7E,17β)-1,3-дигидрокси-2-метилен-9,10-секоэстра-5,7-диен-17-ил]пропил-2-(ацетилокси-)-2-метилпропаноат;
(1R,3R,7E)-2-метилен-17-[(1R,4S)-1,4,5-триметилгексил]-9,10-секоэстра-5,7-диен-1,3-диол;
(1R,3R,7E,17β)-17-[(1S)-1-(3-гидрокси-3-метилбутокси)этил]-2-метилен-9,10-секоэстра-5,7-диен-1,3-диол;
(1R,3R,7E)-17-[(1R,4S)-1,4,5-триметилгексил]-9,10-секоэстра-5,7-диен-1,3-диол;
(1S,3R,5Z,7E,24R)-22,25-диметокси-9,10-секоэргоста-5,7,10-триен-1,3-диол;
(1R,3R,7E,17β)-17-[(1S,4R)-2,5-диметокси-1,4,5-триметилгексил]-9,10-секоэстра-5,7-диен-1,3-диол;
(1R,3R,7E,17β)-2-метилен-17-[(1S)-1-метил-2-феноксиэтил]-9,10-секоэстра-5,7-диен-1,3-диол;
(1R,3S,5Z,7E,17β)-3-фтор-17-[(1R)-5-гидрокси-1,5-диметилгексил]-2-метилен-9,10-секоэстра-5,7-диен-1-ол;
(2S)-2-[(1R,3R,7E,17β)-1,3-дигидрокси-2-метилен-9,10-секоэстра-5,7-диен-17-ил]пропил-2-гидрокси-2-метилпропаноат;
(1R,3R,7E,17β)-17-[(1R,4R)-5-гидрокси-1,4,5-триметилгексил]-9,10-секоэстра-5,7-диен-1,3-диол;
(1R,3R,5E,7E,17β)-17-[(1R)-5-гидрокси-1,5-диметилгексил]-3-(гидроксиметил)-2-метилен-9,10-секоэстра-5,7-диен-1-ол;
(1R,3R,5E,7E,17β)-3-(гидроксиметил)-17-[(1S)-1-(3-гидрокси-3-метилбутокси)этил]-2-метилен-9,10-секоэстра-5,7-диен-1-ол; и
(1R,3S,5E,7E,17β)-3-фтор-17-[(1R)-5-гидрокси-1,5-диметилгексил]-2- метилен-9,10-секоэстра-5,7-диен-1-ол.
Соединения согласно настоящему изобретению могут существовать в форме стереоизомеров, в которых присутствуют асимметричные или хиральные центры. Такие стареоизомеры обозначаются как «R» или «S» в зависимости от конфигурации заместителей относительно хирального элемента. Термины «R» и «S», используемые в данном описании, относятся к конфигурациям, определенным в публикации IUPAC 1974 Recommendations for Section E, Fundamental Stereochemistry, Pure Appl. Chem., 1976, 45: 13-30. Данное изобретение охватывает различные стереоизомеры и их смеси и те, которые конкретно включенные в область настоящего изобретения. Стереоизомеры включают энантиомеры и диастереомеры и смеси энантиомеров или диастереомеров. Индивидуальные стереоизомеры соединений согласно изобретению могут быть синтезированы из коммерчески доступных исходных веществ, которые содержат асимметрические или хиральные центры, или могут быть получены из рацемических смесей с последующим разделением с помощью методов разделения, которые хорошо известны специалисту в данной области техники. Примерами таких методов разделения являются (1) присоединение смеси энантиомеров к хиральным вспомогательным веществам, разделение полученной смеси диастереомеров перекристаллизацией или хроматографией и необязательное отделение оптически чистого продукта от вспомогательного вещества, как описано в публикации Furniss, Hannaford, Smith, and Tatchell, "Vogel's Textbook of Practical Organic Chemistry", 5th Edition (1989), Longman Scientific & Technical, Essex CM20 2JE, England; (2) прямое разделение смеси оптических энантиомеров на хиральных хроматографических колонках; или (3) методы фракционной перекристаллизации.
Таким образом, еще один вариант осуществления данного изобретения относится к способу получения (1R,3aR,4S,7aR)-1-[(1R)-2-гидрокси-1-метилэтил]-7a-метилоктагидро-1H-инден-4-ола, включающему:
(a) взаимодействие витамина D2 с озоном в метаноле и пиридине при температуре примерно -70°С для получения (2S)-2-[(1R,3aR,7aR)-7a-метил-4-оксооктагидро-1H-инден-1-ил]пропаналя;
(b) взаимодействие (2S)-2-[(1R,3aR,7aR)-7a-метил-4-оксооктагидро-1H-инден-1-ил]пропаналя с примерно от 0,05 до 0,30 эквивалентами основания, выбранного из пирролидина или пиперидина, в растворителе, выбранном из трет-бутилметилового эфира, хлороформа, дихлорметана, изопропилацетата, этилацетата, толуола или метанола, при температуре окружающей среды или при температуре, близкой к температуре окружающей среды, в инертной атмосфере в течение примерно от 10 до 24 часов; добавление примерно 0,1 дополнительного эквивалента основания с последующим непрерывным перемешиванием в течение дополнительных от 24 до 120 часов с получением смеси (2R)-2-[(1R,3aR,7aR)-7a-метил-4-оксооктагидро-1H-инден-1-ил]пропаналя и (2S)-2-[(1R,3aR,7aR)-7a-метил-4-оксооктагидро-1H-инден-1-ил]пропаналя в соотношении примерно от 1:1 до 2:1;
(c) взаимодействие смеси (2R)-2-[(1R,3aR,7aR)-7a-метил-4-оксооктагидро-1H-инден-1-ил]пропаналя и (2S)-2-[(1R,3aR,7aR)-7a-метил-4-оксооктагидро-1H-инден-1-ил]пропаналя с борогидридом натрия в смеси трет-бутилметилового эфира или ацетонитрила и протонсодержащего растворителя, выбранного из метанола, этанола и н-пропанола, при температуре примерно от 0 до 15°С с последующим медленным нагреванием до комнатной температуры в течение примерно от 0,5 до 3 часов, с получением смеси (1R,3aR,4S,7aR)-1-[(1R)-2-гидрокси-1-метилэтил]-7a-метилоктагидро-1H-инден-4-ола и (1R,3aR,4S,7aR)-1-[(1S)-2-гидрокси-1-метилэтил]-7a-метилоктагидро-1H-инден-4-ола в соотношении примерно от 1:1 до 2:1; содержание (R)-изомера повышают хроматографической очисткой; и
(d) взаимодействие смеси (1R,3aR,4S,7aR)-1-[(1R)-2-гидрокси-1-метилэтил]-7a-метилоктагидро-1H-инден-4-ола и (1R,3aR,4S,7aR)-1-[(1S)-2-гидрокси-1-метилэтил]-7a-метилоктагидро-1H-инден-4-ола с от 1 до 3 мольными эквивалентами винилацетата и от 15 до 300 процентами (масс.) фермента, выбранного из липазы AK или липазы PS, в растворителе, выбранном из трет-бутилметилового эфира, ацетонитрила, толуола или изопропилацетата, при температуре в интервале примерно от 5 до 50°С в течение примерно от 4 до 7 часов и при температуре от 0 до 15°С в течение примерно от 2 до 15 часов для получения (1R,3aR,4S,7aR)-1-[(1R)-2-гидрокси-1-метилэтил]-7a-метилоктагидро-1H-инден-4-ола и нежелательного изомера в форме ацетата, который отделяют хроматографическим методом.
Еще один вариант осуществления данного изобретения относится к способу сочетания фосфиноксида кольца А формулы (II) с кетоном кольца С/D формулы (III), включающему:
(а) смешивание фосфиноксида кольца А формулы (II) с примерно 1,4 эквивалентами кетона кольца С/D формулы (III) в толуоле и затем выпаривание летучих растворителей; данный процесс повторяют второй раз; где Y1 и Y2 каждый представляет собой водород или вместе образуют метиленовую группу; Y3 и Y4 каждый представляет собой водород или вместе образуют метиленовую группу; Z5 представляет собой фтор, -O-(защитную группу гидроксила) или -CH2O-(защитную группу гидроксила); Z6представляет собой фтор или -O-(защитную группу гидроксила); X1 представляет собой -CH2OR1, -CH2OC(O)R2, -CR3R4-(CH2)m-CR5R6-CR7a(CH3)2 или OR8a; R1 представляет собой водород, алкил или арил; R2 представляет собой алкил, алкиламино, алкилкарбонилоксиалкил или гидроксиалкил; R3 и R4 независимо представляют собой водород или алкоксигруппу, при условии, что одновременно они оба не являются алкоксигруппой; R5 и R6 независимо представляют собой водород или алкил; R7a представляет собой водород, алкоксигруппу, гидроксильную группу или защищенную гидроксильную группу; R8a представляет собой -CH2CH2C(CH3)2OH или -CH2CH2C(CH3)2OSi(CH3)3; и m равно 1, 2 или 3.

(b) растворение смеси фосфиноксида кольца А формулы (II) и кетона кольца C/D формулы (III) в тетрагидрофуране при температуре примерно от -80 до -65°С;
(с) медленное добавление основания, такого как бис(триметилсилил)амид лития, при непрерывном перемешивании в течение от 15 до 30 минут с последующим нагреванием до температуры примерно от -10 до 10°С и перемешиванием в течение дополнительных от 15 до 30 минут при данной температуре с получением соединений формулы (IV)
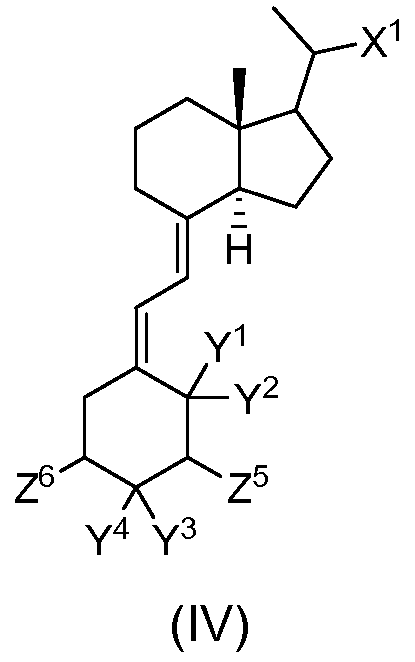
Таким образом, еще один вариант осуществления данного изобретения относится к соединениям формулы (V), применимым для получения соединений формулы (I), где Y1a и Y2a каждый представляет собой водород; Y3 и Y4 каждый представляет собой водород или вместе образуют метиленовую группу; Z3 представляет собой фтор, гидроксильную группу, гидроксиметил, -O-(защитную группу гидроксильной группы) или -CH2O-(защитную группу гидроксильной группы); и Z4 представляет собой фтор, гидроксильную группу или -O-(защитную группу гидроксильной группы). Предпочтительные защитные группы гидроксильной группы выбраны из трет-бутил(диметил)силила и трет-бутил(дифенил)силила.

Конкретные варианты осуществления изобретения, рассматриваемые как часть изобретения, включают, но без ограничения, соединения формулы (V), например:
(3R,5R)-3-{[трет-бутил(диметил)силил]окси}-5-{[трет-бутил(дифенил)силил]окси}-4-метиленциклогексанон;
(3R,5R)-3-{[трет-бутил(диметил)силил]окси}-5-({[трет-бутил(диметил)силил]окси}метил)-4-метиленциклогексанон; или
(3R,5R)-3,5-бис{[трет-бутил(дифенил)силил]окси}-4-метиленциклогексанон.
Способы изобретения
Соединения и композиции согласно изобретению могут применяться для модулирования действий рецепторов витамина D. В частности, соединения и композиции согласно изобретению могут применяться для лечения или предупреждения расстройств, опосредуемых рецепторами витамина D. Обычно, такие расстройства могут облегчаться селективным модулированием рецептора витамина D у млекопитающего, предпочтительно введением одного соединения или одной композиции или сочетанием соединения или композиции с другим активным ингредиентом, например, как части схемы лечения.
Кроме того, изобретение относится к способу лечения или предупреждения состояний, расстройств или недостаточностей, опосредуемых рецепторами витамина D, где состояние, расстройство или недостаточность выбрано(а) из группы, включающей болезни костей, сердечно-сосудистое заболевание, гиперпаратиреоз, иммунологические недостаточности, пролиферативное заболевание, почечное заболевание и тромбоз, который включает введение терапевтически приемлемого количества соединения формулы (I)

или его фармацевтически приемлемой соли или пролекарства, где атом углерода, к которому присоединен Х, может иметь R или S конфигурацию; X представляет собой -CH2OR1, -CH2OC(O)R2, -CR3R4-(CH2)m-CR5R6-CR7(CH3)2 или OR8; Y1 и Y2 каждый представляет собой водород или вместе образуют метиленовую группу; Y3 и Y4 каждый представляет собой водород или вместе образуют метиленовую группу; Z1 представляет собой фтор, гидроксильную группу или гидроксиметил; Z2 представляет собой фтор или гидроксильную группу; R1 представляет собой водород, алкил или арил; R2 представляет собой алкил, алкиламино, алкилкарбонилоксиалкил или гидроксиалкил; R3 и R4 независимо представляют собой водород или алкоксигруппу, при условии, что одновременно оба не являются алкоксигруппой; R5 и R6 независимо представляют собой водород или алкил; R7 представляет собой водород, алкоксигруппу или гидроксильную группу; R8 представляет собой -CH2CH2C(CH3)2OH; и m равно 1, 2 или 3.
Изобретение также относится к способу лечения или предупреждения состояния или расстройства, опосредуемого рецептором витамина D, который включает стадию введения соединения формулы (I), где состояние или расстройство выбрано из почечного заболевания и вторичного гиперпаратиреоза, связанного с хронической болезнью почек.
Изобретение также относится к способу лечения или предупреждения состояния или расстройства, опосредуемого рецептором витамина D, который включает стадию введения соединения формулы (I), где состояние или расстройство выбрано из заболеваний костей, связанных с остеопорозом, остеомаляцией и остеодистрофией.
Изобретение также относится к способу лечения или предупреждения состояния или расстройства, опосредуемого рецептором витамина D, который включает стадию введения соединения формулы (I), где состояние или расстройство выбрано из сердечно-сосудистых заболеваний, связанных с образованием тромбов, ренин-ангиотензиновой системой, миокардиальной гипертрофией и гипертензией.
Изобретение также относится к способу лечения или предупреждения состояния или расстройства, опосредуемого рецептором витамина D, который включает стадию введения соединения формулы (I), где состояние или расстройство выбрано из иммунологических расстройств, связанных с аутоиммунными нарушениями, иммуносупрессией, отторжением трансплантата, артритом, рассеянным склерозом, псориазом, воспалительной болезнью кишечника, диабетом 1 типа и системной красной волчанкой.
Изобретение также относится к способу лечения или предупреждения состояния или расстройства, опосредуемого рецептором витамина D, который включает стадию введения соединения формулы (I), где состояние или расстройство выбраны из рака толстой кишки, рака предстательной железы, рака молочной железы, лейкоза и саркомы Капоши.
Соединения для способа согласно изобретению, включая, но без ограничения, соединения, указанные в примерах или конкретно указанные иным образом, могут модулировать рецепторы витамина D и зачастую обладают сродством к указанным рецепторам. Соединения согласно изобретению в качестве активаторов рецепторов витамина D могут применяться для лечения или предупреждения ряда заболеваний или состояний, опосредуемых рецепторами витамина D.
Например, было показано, что активаторы рецепторов витамина D играют значительную роль в снижении уровней содержания паратиреоидного гормона (Hudson, J. Q. The Annals of Pharmacotherapy, 2006, 40, 1584-1593). Сами по себе активаторы рецепторов витамина D подходят для лечения состояний и расстройств, связанных с хроническим заболеванием почек. Некоторые активаторы витамина D не повышают функциональную активность интестинальных рецепторов витамина D, ограничивая таким образом кальцемический и гиперфосфатемический эффекты, а также связанные с ними побочные эффекты (Slatopolsky, E.; Finch, J.; Ritter, C; Takahashi, F. American Journal of Kidney Disease, 1998, 4, S40-S47). Исследования показали, что лечение с помощью активаторов рецепторов витамина D снижает развитие почечного заболевания (Agarwal, R.; Acharya, M.; Tian, J.; Hippensteel, R.L.; Melnick, J.Z.; Qiu, P.; Williams, L.; Batlle, D. Kidney International, 2005, 68, 2823-2828 и Schwarz, U.; Amann, K.; Orth, S.R.; Simonaviciene, A.; Wessels, S.; Ritz, E. Kidney International, 1998, 53, 1696-1705).
Кроме того, было показано, что активаторы рецепторов витамина D вовлечены в скелетный и минеральный гомеостаз. Эти активаторы рецепторов имеют большое значение для интестинальной абсорбции кальция и его последующей анаболической активности на кости (Hendy, G.N.; Hruska, K.A.; Methew, S.; Goltzman, D. Kidney International, 2006, 69, 218-223). Некоторые агонисты показали способность селективно лечить болезни костей со сниженным влиянием на супрессию паратиреоидного гормона (Shevde, N. K.; Plum, L. A.; Clagett-Dame, M.; Yamamoto, H.; Pike, J. W.; DeLuca, H. F. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 13487-13491; Uchiyama, Y.; Higuchi, Y.; Takeda, S.; Masaki, T.; Shira-Ishi, A.; Sato, K.; Kubodera, N.; Ikeda, K.; Ogata, E. Bone, 2002, 4, 582-588; Shiraishi, A.; Higashi, S.; Ohkawa, H.; Kubodera, N.; Hirasawa, T.; Ezawa, I.; Ikeda, K.; Ogata, E. Calcified Tissue International, 1999, 65, 311-316).
Считалось, что активаторы рецепторов витамина D обладают рядом влияний на многие факторы системы кровообращения. Система рецепторов витамина D играет важную роль в поддержании противотромботического гомеостаза (Aihara, K.; Azuma, H.; Akaike, M.; Ikeda, Y.; Yamashita, M.; Sudo, T.; Hayashi, H.; Yamada, Y.; Endoh, F.; Fujimura, M.; Yoshida, T.; Yamaguchi, H.; Hashizume, S.; Kato, M.; Yoshimura, K.; Yamamoto, Y.; Kato, S.; Matsumoto, T. J. Biol. Chem., 2004, 279, 35798-35802). Было показано, что активаторы рецепторов витамина D изменяют экспрессию и активность белков, важных для коагуляции, таких как тромбомодулин, фактор ткани и ингибитор 1 активатора плазминогена, что позволяет лечить атеросклеротические заболевания (Beer, T. M.; Venner, P.M.; Ryan, C. W.; Petrylak, D. P.; Chatta, G.; Ruether, J. D.; Chi, K. N.; Curd, J. G.; DeLoughery, T. G. British Journal of Haematology, 2006, 135, 392-394 и Ohsawa, M.; Koyama, T.; Yamamoto, K.; Hirosawa, S.; Kamei, S.; Kamiyama, R. Circulation, 2000, 102, 2867-2872). Ренин-ангиотензиновая система II является центральной в стабилизации кровяного давления, и повышенные уровни ренина приводят к гипертонии и кардиальной гипертрофии. Активаторы рецептора витамина D напрямую подавляют транскрипцию генов ренина в рецепторе витамина D в механизме, зависимом от рецепторов витамина D, предоставляя механизм контроля этой системы (Li, Y.C; Qiao, G.; Uskokovic, M.; Xiang, W.; Zheng, W.; Kong, J. Journal of Steroid Biochemistry & Molecular Biology, 2004, 89-90, 397-392). Пациенты с хронической почечной недостаточностью, получающие поддерживающий гемодиализ, зачастую страдают сердечно-сосудистыми осложнениями, среди которых ишемическая болезнь сердца как результат гипертрофии левого желудочка, является наиболее распространенной. Гиперпаратиреоз также наносит ущерб здоровью, и даже частичное регулирование с помощью активатора рецептора витамина D приводит к регрессии миокардиальной гипертрофии без изменений в других гемодинамических параметрах (Park, C.W.; Oh, Y.S.; Shin, Y.S.; Kim, C-M.; Kim, Y.-S.; Kim, S.Y.; Choi, E.J.; Chang, Y.S.; Bang, B.K. American Journal of Kidney Diseases, 1999, 33, 73-81).
Рецептор витамина D экспрессирован в большинстве типов клеток иммунной системы и, в частности, в модулирующих ответы Т лимфоцитов. В настоящее время активаторы рецептора витамина D используются для местного лечения псориаза. Опыты на животных моделях позволили предположить, что активаторы рецепторов витамина D могут быть полезными в лечении артрита, аутоиммунного диабета, экспериментального аллергического энцефаломиелита, воспалительной болезни кишечника или системной красной волчанки, что позволяет относить возможность их терапевтического применения и для лечения людей (Adorini, L. Cellular Immunology, 2005, 233, 115-124).
Активаторы витамина D оказывают влияние на ряд сигнальных путей, задействованных в развитии рака. Они четко, хотя с большой неоднородностью, ответственны за противопролиферативное, противоангиогенное и продифференцированное действие в широком спектре раковых заболеваний, опосредуемых через геномные и негеномные механизмы (Deeb, K.K.; Trump, D.L.; Johnson, C.S. Nature Reviews Cancer, 2007, 7, 684-700). Роль метаболизма витамина D, как оказывается, важна в регулировании клеточной пролиферации клеток в предстательной железе (Lou, Y.-R.; Qiao, S.; Talonpoika, R.; Syvala, H.; Tuohimaa, P. Journal of Steroid Biochemistry and Molecular Biology, 2004, 92, 317-3250). Существует связь между подавлением аутокринных факторов роста IL-6 и IL-8 активаторами рецептора витамина D и развитием саркомы Капоши (Masood, R.; Nagpal, S.; Zheng, T.; Cai, J.; Tulpule, A.; Smith, D.L.; Gill, P. S. Blood, 2000, 96, 3188-3194). Аналоги витамина D оказывают дифференциирующее влияние на лейкозные клетки (James, S.Y.; Williams, M.A.; Newland, A.C; Colston, K.W. Gen. Pharmaс, 1999, 32, 143-154).
Фактические уровни доз активных ингредиентов в фармацевтических композициях согласно настоящему изобретению могут изменяться таким образом, чтобы получить количество активного(ых) соединения(ий), которое эффективно для достижения желательного терапевтического ответа у конкретного пациента, в конкретной композиций и конкретного способа введения. Выбранный уровень дозировки будет зависеть от активности конкретного соединения, способа введения, тяжести состояния, подлежащего лечению, а также состояния и предыдущей истории болезни пациента, подлежащего лечению. Однако квалифицированный специалист в данной области техники может назначать лечение соединением с начальных доз, более низких, чем необходимо для достижения желательного терапевтического эффекта, и постепенно повышать дозировку до достижения желательного эффекта.
Когда одно из соединений согласно изобретению используется в указанных выше или других способах лечения, указанное соединение в терапевтически эффективном количестве может применяться в чистой форме или, когда такие формы существуют, в форме фармацевтически приемлемой соли, сложного эфира, амида или пролекарства. Альтернативно, соединение может вводиться в форме фармацевтической композиции, содержащей соединение согласно изобретению и один или несколько фармацевтически приемлемых носителей. Фраза «терапевтически эффективное количество» соединения согласно изобретению означает количество соединения, достаточное для лечения расстройств с подходящим приемлемым соотношением «польза/риск», применимое при любом медицинском лечении. Однако следует представлять, что общая суточная доза применения соединений и композиций согласно изобретению будет определяться лечащим врачом в рамках обычного медицинского назначения. Конкретный уровень терапевтически эффективной дозы для любого конкретного пациента будет зависеть от ряда различных факторов, включая расстройство, подлежащее лечению, и тяжесть указанного расстройства; активность конкретного используемого соединения; конкретную применяемую композицию; возраст, массу тела, общее состояние здоровья, пол и рацион питания пациента; время введения, способ введения и скорость экскреции конкретного используемого соединения; продолжительность лечения; лекарственные средства, используемые совместно, или лекарственные средства, применение которых случайно совпадает с применением конкретного используемого соединения; и аналогичные факторы, хорошо известные в области медицины. Например, специалист в данной области техники может назначать лечение соединением с начальных доз, более низких, чем необходимо для достижения желательного терапевтического эффекта, и постепенно повышать дозировку до достижения желательного эффекта.
Общая суточная доза соединений согласно настоящему изобретению, вводимая человеку или животному, находится в интервале от примерно 0,01 мкг до примерно 150 мг. Более предпочтительные дозы могут находиться в интервале от примерно 0,010 мкг до примерно 10 мг. Если желательно, эффективная суточная доза может разделяться на несколько доз для более удобного введения. Следовательно, композиции разовой дозы могут содержать такие количества или их дольные единицы, которые могли бы составить суточную дозу.
Способы получения соединений согласно изобретению
Соединения согласно изобретению могут быть лучше представлены в сочетании с приведенными схемами синтеза и способами, которые иллюстрируют методы, с помощью которых могут быть получены соединения согласно изобретению.
Аббревиатуры, которые использовались в описаниях схем и примерах, имеют следующие значения: 9-BNN означает 9-борабицикло[3.3.1]нонан; ВСА означает бицинхониновую кислоту; BUN означает мочевинный азот крови; CASMC означает клетки гладких мышц коронарной артерии; DAST означает трифторид (диэтиламино)серы; DIBAL означает гидрид диизобутилалюминия; DMEM означает минимально чувствительную среду Игла в модификации Дульбекко; DTT означает дитиотреитол; Et2AlCl означает хлорид диэтилалюминия; ГХ означает газовую хроматографию; НОАс означает уксусную кислоту; ВЭЖХ означает высокоэффективную жидкостную хроматографию; LiHMDS означает бис(триметилсилил)амид лития; MsCl означает метансульфонилхлорид; NBT означает нитросиний тетразолий; nBuLi означает н-бутиллитий; Ncor1 означает корепрессор 1 ядерного рецептора; РА% означает площадь пика, выраженную в процентах; PBS-T означает фосфатно-буферный раствор, содержащий TWEEN 20; PCR (ПЦР) означает цепную реакцию полимеразы; PMA означает форбол-12-миристат-13-ацетат; PPAR означает пероксисомный пролифератор-активируемый рецептор; PPNS означает п-толуолсульфонат пиридиния; psi (фунтов на кв. дюйм) означает давление, выраженное в фунтах на квадратный дюйм; PVDF означает поливинилидинфторид; RPMI означает среду, разработанную Институтом имени Парка Розуэлла; SDS-PAGE означает электрофорез белков в полиакриламидном геле в присутствии додецилсульфата натрия; TBAF означает фторид тетра-н-бутиламмония; TBDPS-Cl означает хлорид трет-бутилдифенилсилила; TBS-Cl означает хлорид трет-бутилдиметилсилила; TBS-имидазол означает трет-бутилдиметилсилилимидазол; TBS-OTf означает трифторметансульфонат трет-бутилдиметилсилила; TES-Cl означает хлорид триэтилсилила; TMP означает 2,2,6,6-тетраметилпиперидин; Tris означает трисгидроксиметиламинометан; TWEEM 20 означает полиоксоэтиленсорбитанмонолаурат; VDR означает рецептор витамина D;% масс. означает процент по массе.
Соединения формулы (II) могут быть получены способами химического синтеза, примеры которых представлены ниже. Следует представлять, что порядок стадий, приведенный в данных способах, может изменяться, конкретно указанные реагенты и условия реакций также могут изменяться, фрагменты, способные подвергаться нежелательному взаимодействию, могут защищаться от нежелательного взаимодействия, и защита может удаляться, когда это необходимо. Некоторые группы могут быть замещенными, как описано в данном изобретении, что должно быть понятно специалисту в данной области техники. Типичные методики представлены, но без ограничения только ими, на схемах 1-7.
Схема 1
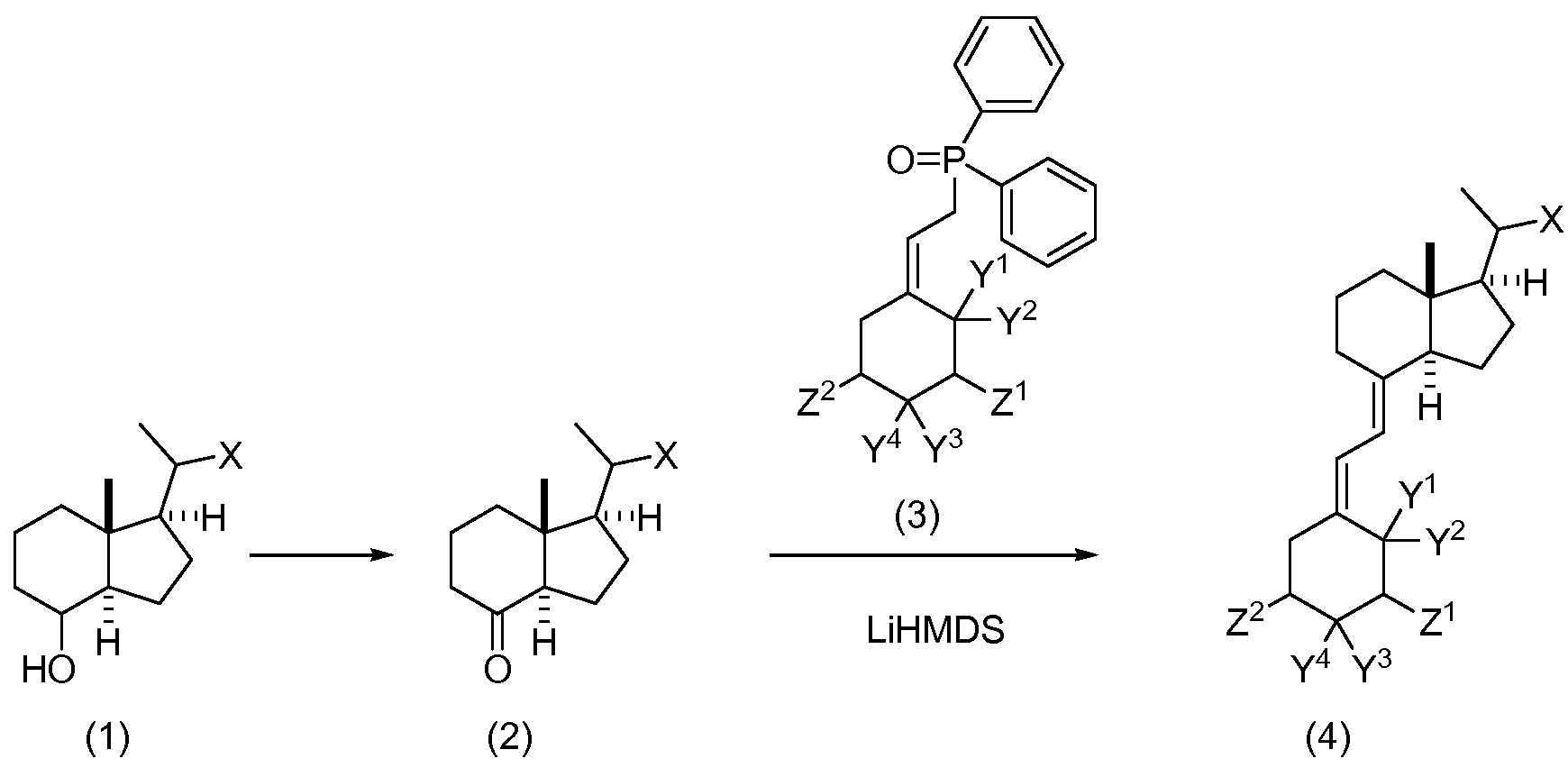
Как показано на схеме 1, соединения формулы (1), где Х принимает значения, определенные в описании, может подвергаться окислению такими окислителями, как хлорхромат пиридиния на оксиде алюминия или дихлорам пиридиния с п-толуолсульфоном пиридиния, в растворителе, таком как дихлорметан, при температуре в интервале от 0°С до 30°С в течение от 12 до 48 часов с получением соединений формулы (2). Соединения формулы (2) могут подвергаться взаимодействию с анионами соединений формулы (3). Анионы формулы (3) получают взаимодействием соединений формулы (3) с основаниями, такими как бис(триметилсилил)амид лития или диизопропиламид лития, в растворителе, таком как тетрагидрофуран или диоксан, при температуре в интервале от -78°С до 0°С в течение от 1 до 24 часов.
Схема 2

Соединения формулы (5) являются примерами соединений (1), (2) и (4), представленными на схеме 1, где Х представляет собой -СН2ОН и функционально присоединенная к нижней части С-цикла группа, обозначенная (

Например, соединения формулы (5) могут подвергаться этерификации взаимодействием с хлорангидридом кислоты формулы R10C(O)Cl, где R10 представляет собой алкильную группу, в присутствии основания, такого как пиридин, в растворителе, таком как дихлорметан, при температуре в интервале от 0°С до 30°С в течение от 6 до 48 часов для получения сложных эфиров формулы (6). Альтернативно, соединения формулы (5) могут подвергаться этерификации взаимодействием с карбоновой кислотой формулы R10CO2Н, где R10 представляет собой гидроксиалкильную группу, в присутствии трифенилфосфина и ди-трет-бутилазодикарбоксилата в растворителе, таком как толуол, при температуре в интервале от 30 до 100°С в течение от 1 до 24 часов.
Карбаматы формулы (7) могут быть получены из соединений формулы (5) под действием изоцианата R11NCO, где R11 представляет собой алкильную группу, в смеси растворителей, таких как диметилформамид и толуол, при температуре в интервале от 50 до 110°С в течение от 1 до 5 дней.
Соединения формулы (5) могут подвергаться окислению до альдегидов формулы (8) окислителем, таким как перрутенат тетра-н-пропиламмония и N-метилморфолиноксид, в присутствии 4Å молекулярных сит в растворителе, таком как дихлорметан, в течение от 1 до 24 часов при температуре в интервале от 10 до 30°С.
Соединения формулы (5) также могут превращаться в простые эфиры формулы (9) в условиях реакции Митсунобу. Например, соединения формулы (5) могут подвергаться взаимодействию с фенолом в присутствии трифенилфосфина и ди-трет-бутилазодикарбоксилата в растворителе, таком как толуол, при температуре в интервале от 30 до 100°С в течение от 0,5 до 24 часов.
Сложные эфиры формулы (6) могут иногда служить в качестве защитных групп гидроксильного фрагмента соединений формулы (5). Удаление защиты для получения незащищенной гидроксильной группы может достигаться восстановлением с помощью таких реагентов, как алюмогидрид лития, в растворителях, таких как эфир или тетрагидрофуран, при исходной температуре приблизительно -78°С в течение от 5 до 30 минут с последующим с медленным нагреванием до температуры в интервале от 0 до 25°С в течение от 0,5 до 6 часов.
Схема 3

Альдегиды формулы (10), где R12 представляет собой защитную силильную группу, такую как трет-бутилдиметилсилил или трет-бутилдифенилсилил, могут подвергаться взаимодействию с сульфонами формул (11) и (14) в условиях, описанных в публикации Kutner, A.; et al. Journal of Organic Chemistry 1988, 53, 3450-3457, с получением соединений формул (12) и (15), соответственно.
Соединения формулы (12) далее подвергались превращению в соединения формулы (13) обработкой избытком метилйодида в присутствии гидрида натрия в растворителе, таком как тетрагидрофуран, при температуре окружающей среды или близкой к температуре окружающей среды в течение от 6 до 48 часов.
Соединения формулы (15), где R13 представляет собой водород или триметилсилильную группу, подвергают восстановлению водородом в присутствии катализатора, такого как платина на углероде, в растворителе, таком как уксусная кислота, или в присутствии катализатора, такого как палладий на углероде, в растворителе, таком как этилацетат, с получением соединений формулы (16).
Схема 4
Соединения формулы (17) подвергали восстановлению до соединений формулы (18) в присутствии водорода и катализатора, такого как платина на углероде, в растворителе, таком как уксусная кислота.
Схема 5

Соединения формулы (19) могут подвергаться превращению в соединения формулы (20) в соответствии со следующей последовательностью реакций. Сначала гидроксильная группа в соединениях формулы (19) защищается посредством образования соответствующего трет-бутилдиметилсилилового эфира при выдерживании в хлориде трет-бутилдиметилсилила в присутствии имидазола в растворителе, таком как диметилфорамид, при температуре окружающей среды или близкой к температуре окружающей среды в течение приблизительно 4 часов и затем при повышенной температуре в интервале от 30 до 80°С в течение дополнительного периода времени в интервале от 8 до 48 часов. Полученный алкен затем подвергается обработке 9-борабицикло[3.3.1]нонаном в растворителе, таком как тетрагидрофуран, в течение от 2 до 24 часов при температуре в интервале от 35 до 60°С. Последующая обработка при температуре 0°С или близкой к 0°С водным гидроксидом натрия и пероксидом водорода приводит к получению соединений формулы (20).
Соединения формулы (20) подвергаются превращению в соединения формулы (22) в соответствии со следующей последовательностью реакций. Гидроксильная группа соединений формулы (20) подвергается алкилированию соединениями формулы (21) при обработке основанием, таким как гидрид натрия, в растворителе, таком как тетрагидрофуран, при температуре образования флегмы или близкой к ней, в течение от 0,5 до 8 часов. После охлаждения до комнатной температуры обработка алюмогидридом натрия в течение от 2 до 16 часов стереоселективно раскрывает эпоксид с получением соответствующего третичного карбинола. Последовательность получения соединений формулы (22) завершается удалением защитной трет-бутилдиметилсилильной группы под действием фторида тетра-н-бутиламмония в тетрагидрофуране при температуре в интервале от 60 до 80°С с течение от 8 до 24 часов.
Схема 6

Последовательность синтеза, представленная на схеме 6, описывает получение кольца А и введение функциональности с присоединением ее к части кольца C/D в соответствии с методикой, представленной на схеме 1, как показано с помощью соединений формулы (31). Стереохимию определяли, исходя из (R)-карбон-оксида (23). Восстановление по Луче (Luche) достигали объединением (23) с CeCl3 в метаноле при 0°С. После охлаждения до -20°С медленно добавляли борогидрид натрия и смеси дали нагреться до комнатной температуры. Гидроксильную группу, образованную таким образом, защищали в форме трет-бутилдифенилсилилового эфира обработкой трет-бутилдифенилсилилхлоридом и имидазолом в диметилформамиде при температуре окружающей среды в течение от 48 до 100 часов с получением соединения (24).
Изопропилиденовую группу соединения (24) подвергали окислению озоном при -70°С в присутствии бикарбоната натрия в смеси дихлорметана и метанола. Перегруппировку Криге (Criegee) проводили на промежуточном продукте с п-нитробензоилхлоридом в смеси дихлорметана и пиридина при начальной температуре реакции <5°С, которая поддерживалась в течение 1 часа, и затем при повышении температуры до температуры окружающей среды, которая поддерживалась в течение от 8 до 24 часов. Гидролиз промежуточного ацетата основанием, таким как карбонат калия, в смеси воды и метанола проводили при температуре окружающей среды в течение от 1 до 6 часов. Обработка высвобожденного гидроксильного фрагмента триэтилсилилхлоридом в присутствии имидазола в диметилформамиде при температуре окружающей среды в течение от 8 до 24 часов приводила к получению соединения (25).
Эпоксид (25) раскрывается при обработке 2,2,6,6-тетраметилпиперидином и н-бутиллитием в присутствии хлорида диэтилалюминия в растворителе, таком как толуол, при 0°С и последующим осторожным нагреванием в течение нескольких часов до температуры окружающей среды с получением аллилового спирта (26).
Аллиловый спирт (26) может подвергаться превращению в соответствующий трет-бутилдиметилсилиловый эфир при обработке трет-бутилдиметилсилилтрифторметансульфонатом в присутствии основания, такого как 2,6-лутидин, в растворителе, таком как дихлорметан, при 0°С. Селективное удаление защитной триэтилсилильной группы достигается обработкой п-толуолсульфонатом пиридиния в этаноле при температуре окружающей среды в течение 1-5 часов. Высвобожденная гидроксильная группа после этого может подвергаться окислению до соответствующего кетона с помощью реагента Десса-Мартина (1,1,1-трис(ацетилокси)-1,1-дигидро-1,2-бензойодоксол-3-(1Н)-он) в присутствии основания, такого как гидрокарбонат натрия, в растворителе, таком как дихлорметан, при 0°С с получением соединения (27).
Кетон (27) может подвергаться превращению в α,β-незамещенный сложный эфир (28) посредством взаимодействия (27) с этилтриметилсилилацетатом и н-бутиллитием в растворителе, таком как тетрагидрофуран, при -78°С в течение от 4 до 8 часов. Группа трет-бутилдиметилсилилового эфира селективно удаляется обработкой концентрированной соляной кислотой в метаноле при температуре окружающей среды в течение от 8 до 24 часов с получением смеси алкенов, которая может хроматографически разделяться с получением (28).
Гидроксильная группа соединения (28) может подвергаться взаимодействию с трифторидом(диэтиламино)серы в дихлорметане при -78°С в течение от 20 минут до 120 минут с получением фторида (29).
α,β-Ненасыщенный сложный эфир (28) может подвергаться восстановлению гидридом диизобутилалюминия в смеси дихлорметана и толуола при -78°С в течение от 10 до 60 минут с получением соответствующего аллилового спирта. Последующая обработка трифосгеном в присутствии триэтиламина при 0°С в течение от 30 до 60 минут с последующим медленным нагреванием до температуры окружающей среды в течение от 1 до 3 часов приводит к получению аллилхлорида (30).
Аллилхлорид (30) может подвергаться обработке литиевым анионом дифенилфосфина лития в тетрагидрофуране при -60°С в течение от 1 до 4 часов. Окисление до соответствующего дифенилфосфиноксида достигается выдерживанием на воздухе или обработкой водным пероксидом водорода в дихлорметане при 0°С в течение от 0,5 до 3 часов с получением соединения (31).
Схема 7

(S)-Карвон-оксид (32) может подвергаться селективному восстановлению с помощью L-Selectride® при температуре -78°С в течение от 5 до 12 часов в растворителе, таком как тетрагидрофуран, с получением спирта (33). Полученный продукт может подвергаться превращению в соответствии с последовательностью реакций, представленной на схеме 6, с получением аллилового спирта (34).
Аллиловый спирт (34) может подвергаться превращению в соответствующий мезилат обработкой метансульфонилхлоридом в присутствии диметиламинопиридина в дихлорметане при 0°С в течение от 3 до 8 часов. Последующая обработка йодидом натрия в присутствии гидрокарбоната натрия и сульфита натрия в ацетоне приводит к получению аллилйодида (35).
Соединение (36) может быть получено из соединения (35) обработкой индием в присутствии формальдегида в смеси тетрагидрофурана и воды.
Селективная защита первичной гидроксильной группы соединения (36) может достигаться обработкой трет-бутилдиметилсилилимидазолом в дихлорметане при температуре окружающей среды в течение от 24 до 48 часов. После этого окисление вторичной гидроксильной группы может проводиться с помощью окислителя, такого как реагент Десса-Мартина, в растворителе, таком как дихлорметан, при температуре окружающей среды в течение от 1 до 8 часов, с получением кетона (37).
Кетон (37) может подвергаться превращению в дифенилфосфиноксид (38) в соответствии с последовательностью реакций, представленной на схеме 6.
Соединения и способы согласно настоящему изобретению далее будут объяснены с помощью представленных примеров, которые, как подразумевается, предназначены только для иллюстрации, а не для ограничения области данного изобретения. Далее будут описаны типичные способы синтеза этих новых соединений согласно настоящему изобретению. В сущности, примеры демонстрируют новые способы синтеза, применимые для получения широкого спектра производных витамина D, включая новые и патентоспособные соединения согласно настоящему изобретению. Таким образом, согласно некоторым вариантам осуществления настоящего изобретения, были получены фрагменты целевых производных витамина D, фрагменты были дополнительно модифицированы, когда это было желательно, и затем, в конечном итоге, соединены для получения желательных производных витамина D.
ПРИМЕРЫ

Пример 1
(2S)-2-[(1R,3R,7E,17β)-1,3-дигидрокси-2-метилен-9,10-секоэстра-5,7-диен-17-ил]пропилпивалат
Пример 1A
[2-((3R,5R)-3,5-бис{[трет-бутил(дифенил)силил]окси}-4-метиленциклогексилиден)этил](дифенил)фосфиноксид
Указанное в заголовке соединение получали в соответствии с методиками, описанными в WO98/41500 (DeLuca и Sicinski), с их незначительным изменением.
Пример 1B
(1R,3aR,4S,7aR)-1-[(1S)-2-гидрокси-1-метилэтил]-7a-метилоктагидро-1H-инден-4-ол
Указанное в заголовке соединение получали из витамина D2 в соответствии с методиками, описанными в публикации Sardina et al. J. Org. Chem. 1986, 51 (8), 1264-1269.
Пример 1C
(2S)-2-[(1R,3aR,4S,7aR)-4-гидрокси-7a-метилоктагидро-1H-инден-1-ил]пропилпивалат
Соединение примера 1B (1,57 г, 6,7 ммоль) растворяли в 10 мл дихлорметана и 5 мл пиридина; раствор охлаждали до 0°С и по каплям в течение 5 минут добавляли 0,94 мл пивалоилхлорида. Полученную смесь перемешивали при 0°С в течение 4 часов, затем нагревали до температуры окружающей среды в течение ночи. Реакцию гасили водой, и смесь концентрировали в вакууме на бане, поддерживая температуру ниже температуры окружающей среды. Сырой продукт распределяли между эфиром и 0,5N водной HCl; органическую фазу промывали 0,5N водной HCl, затем раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме; остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 10% до 20%), получая указанное в заголовке соединение (1,25 г, 63%).
Пример 1D
(2S)-2-[(1R,3aR,7аR)-7a-метил-4-оксооктагидро-1H-инден-1-ил]пропилпивалат
Соединение примера 1C (1,1 г, 3,7 ммоль) растворяли в 5 мл дихлорметана и охлаждали до 0°С; добавляли 4,1 г дихромата пиридиния с последующим добавлением 30 мг п-толуолсульфоната пиридиния. Полученную смесь перемешивали в течение 8 часов при 0°С; добавляли еще 1,8 г дихромата пиридиния и 20 мг п-толуолсульфоната пиридиния и смеси давали возможность нагреться до температуры окружающей среды в течение ночи. Смесь разбавляли эфиром, фильтровали через рыхлый слой диатомовой земли и промывали эфиром. Фильтрат промывали 1N водной HCl и затем фильтровали через рыхлый слой силикагеля. Сырой продукт очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 10% до 15%), получая указанное в заголовке соединение (1,0 г, 94%).
Пример 1E
(2S)-2-[(1R,3R,7E,17β)-1,3-дигидрокси-2-метилен-9,10-секоэстра-5,7-диен-17-ил]пропилпивалат
Примечание: представленную далее последовательность синтеза проводили в затемненном вытяжном шкафу. Соединение примера 1A (64 мг, 0,077 ммоль) объединяли с соединением примера 1D (32 мг, 0,11 ммоль) и полученную смесь подвергали азеотропной сушке, дважды перегоняя с 1 мл толуола. Добавляли тетрагидрофуран (2 мл) и раствор охлаждали до -78°С. К смеси по каплям двумя порциями добавляли раствор бис(триметилсилил)амида лития (0,6M в тетрагидрофуране; 0,40 мл), получая желтовато-оранжевую окраску, которая исчезает в течение 20 минут. Раствор нагревали до 0°С и перемешивали при данной температуре в течение 20 минут. Реакцию гасили добавлением 1 мл 1N водного раствора NH4Cl и полученную смесь экстрагировали этилацетатом. Органический экстракт промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 0% до 6%). Продукт (61 мг) растворяли в 2 мл 1N раствора фторида тетра-н-бутиламмония в тетрагидрофуране; добавляли 0,25 мл уксусной кислоты, смесь нагревали до 70°С и выдерживали при данной температуре в течение ночи. Реакционную смесь распределяли между этилацетатом и водой. Органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 25% до 40%), получая указанное в заголовке соединение (21 мг).
1H ЯМР (500 МГц, CD2Cl2) δ м.д. 6,33 (д, J=11,0 Гц, 1H), 5,90 (д, J=11,3 Гц, 1H), 5,06 (д, J=5,8 Гц, 2H), 4,35-4,50 (м, 2H), 4,04 (дд, J=10,7, 3,4 Гц, 1H), 3,79 (дд, J=10,7, 7,0 Гц, 1H), 2,82 (с, 1H), 2,76 (дд, J=13,1, 4,3 Гц, 1H), 2,55 (дд, J=13,1, 4,0 Гц, 1H), 2,40-2,50 (м, 1H), 2,01 (дд, J=12,4, 3,8 Гц, 2H), 1,90 (с, 1H), 1,65-1,78 (м, 4H), 1,32-1,40 (м, 4H), 1,24-1,33 (м, 1H), 1,23-1,34 (м, 3H), 1,13-1,23 (м, 9H), 0,96-1,06 (м, 3H), 0,58 (с, 3H); MС (+DCI) m/z 448 (M+NH4)+.

Пример 2
(1R,3R,7E,17β)-17-[(1S)-2-гидрокси-1-метилэтил]-2-метилен-9,10-секоэстра-5,7-диен-1,3-диол
Пример 2A
(2S)-2-((1R,3R,7E,17β)-1,3-бис{[трет-бутил(дифенил)силил]окси}-2-метилен-9,10-секоэстра-5,7-диен-17-ил)пропилпивалат
Примечание: представленную далее последовательность синтеза проводили в затемненном вытяжном шкафу. Соединение примера 1A (64 мг, 0,077 ммоль) объединяли с соединением примера 1D (32 мг, 0,11 ммоль) и полученную смесь дважды подвергали азеотропной сушке с 1 мл толуола. Добавляли тетрагидрофуран (2 мл) и раствор охлаждали до -78°С. К смеси по каплям двумя порциями добавляли раствор бис(триметилсилил)амида лития (0,6M в тетрагидрофуране; 0,40 мл), что приводило к появлению желтовато-оранжевой окраски, которая исчезает в течение 20 минут. Раствор нагревали до 0°С и перемешивали в течение 20 минут. Реакцию гасили добавлением 1 мл 1N водного раствора NH4Cl и полученную смесь экстрагировали этилацетатом. Органический экстракт промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 0% до 6%).
Пример 2B
(1R,3R,7E,17β)-17-[(1S)-2-гидрокси-1-метилэтил]-2-метилен-9,10-секоэстра-5,7-диен-1,3-диол
Примечание: представленную далее последовательность синтеза проводили в затемненном вытяжном шкафу. Соединение примера 2A (70 мг, 0,77 ммоль) растворяли в 1,5 мл эфира и охлаждали до -78°С. Добавляли раствор алюмогидрида лития в тетрагидрофуране (1,0 M, 0,62 мл); смесь перемешивали в течение 10 минут, затем смесь нагревали до 0°С в течение 50 минут. Реакцию гасили осторожным добавлением этилацетата с последующим добавлением раствора соли Рошелля (Rochelle's salt). После перемешивания в течение 15 минут полученную смесь экстрагировали этилацетатом; органический экстракт промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме. Навеску полученного продукта (19 мг) объединяли с 1,5 мл 1N раствора фторида тетра-н-бутиламмония в тетрагидрофуране и 0,2 мл уксусной кислоты и смесь выдерживали при 70°С в течение ночи. Реакционную смесь распределяли между этилацетатом и водой. Органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 50% до 66%). Фракции, содержащие продукт, объединяли и концентрировали в вакууме, получая указанное в заголовке соединение (4 мг).1H ЯМР (400 МГц, CD2Cl2) δ м.д. 6,31 (д, J=11,2 Гц, 1H), 5,88 (д, J=11,2 Гц, 1H), 5,03-5,06 (м, 2H), 4,37-4,45 (м, 2H), 3,60 (дд, J=10,4, 3,2 Гц, 1H), 3,33 (дд, J=10,4, 6,8 Гц, 1H), 2,78-2,87 (м, 1H), 2,74 (дд, J=13,2, 4,3 Гц, 1H), 2,53 (дд, J=13,2, 4,1 Гц, 1H), 2,24-2,32 (м, 2H), 1,96-2,06 (м, 2H), 1,78-1,94 (м, 2H), 1,60-1,74 (м, 3H), 1,44-1,60 (м, 4H), 1,28-1,40 (м, 4H), 1,03 (д, J=6,5 Гц, 3H), 0,56 (с, 3H); MС (+DCI) m/z 364 (M+NH4)+.

Пример 3
(2R)-2-[(1R,3R,7E,17β)-1,3-дигидрокси-2-метилен-9,10-секоэстра-5,7-диен-17-ил]пропилпивалат
Пример 3A
(2S)-2-[(1R,3aR,7aR)-7a-метил-4-оксооктагидро-1H-инден-1-ил]пропаналь
Витамин D2 (30,04 г, 75,7 ммоль) растворяли в 1050 мл метанола (35 мл/г) и 10,5 мл пиридина (1% метанола). Полученный раствор продували азотом, затем раствор охлаждали до -70°С. В процессе охлаждения из раствора выпадает в осадок некоторое количество витамина D2. Через охлажденную, хорошо размешанную взвесь барботировали озон, взвесь постепенно превращалась в раствор, барботирование продолжали до тех пор, пока метанольный раствор не приобретет голубой цвет избыточного озона. Избыток озона удаляли из раствора потоком азота. К смеси в течение 13 минут добавляли триметилфосфит (77 мл, 652,8 ммоль), в процессе чего температура возрастала от -75,1 до -69,5°С. Охлаждающую баню удаляли и реакционную смесь нагревали до комнатной температуры (1,5 часа). Анализ ГХ (масс./масс.) реакционной смеси показывает содержание 71,9% кето-альдегида примера 3A. Растворитель удаляли отгонкой на роторном испарителе, получая 98,03 г густого масла. Масло переносили в делительную воронку, используя 500 мл насыщенного раствора соли и 500 мл трет-бутилметилового эфира. После разделения слоев водный слой экстрагировали дополнительным количеством (2×500 мл) трет-бутилметилового эфира. Объединенные органические экстракты сушили над сульфатом натрия и концентрировали, получая 50,20 г масла. ГХ анализ показывает 75,13% указанного в заголовке кетоальдегида и пять примесей с содержанием от 1,27 до 4,97% (без учета триметилфосфата). Масло растворяли в 500 мл метанола и отгоняют его на роторном испарителе. Процедуру повторяют с еще с 500 мл метанола, 300 мл н-бутанола и 500 мл метанола, получая 57,04 г масла. Масло растворяли в 500 мл трет-бутилметилового эфира и промывали (3×100 мл) насыщенным водным раствором гидрокарбоната натрия. Промывные растворы объединяли и снова экстрагировали 200 мл трет-бутилметилового эфира. Эмульсию на поверхности раздела экстрагировали этилацетатом. Органические растворы сушили над сульфатом натрия и концентрировали, получая 24,40 г указанного в заголовке сырого кетоальдегида из трет-бутилметилового эфира и 1,53 г указанного в заголовке сырого кетоальдегида из этилацетата.1H ЯМР показывает, что полученный продукт является сравнительно чистым, поэтому его используют без дополнительной очистки в стадии эпимеризации, описанной далее. ГХ показывает приблизительно 4 PA% триметилфосфата, оставшегося в сыром кетоальдегиде.1H ЯМР (400 МГц, CDCl3) δ 0,69 (с, 3H, 18-H3), 1,17 (д, J=6,86 Гц, 3H, 21-H3), 2,47 (дд, J=7,41, 11,66 Гц, 1H, 14-H), 9,60 (д, J=2,88 Гц, 1H, 22-H);13C ЯМР (100 МГц, CDCl3) δ 13,18 (CH3), 13,80 (CH3), 19,82 (CH2), 24,18 (CH2), 26,70 (CH2), 38,82 (CH2), 41,11 (CH2), 49,26, 50,13 (C-13), 51,60, 61,10, 203,35 (C-22), 210,35 (C-8); ГХ: (колонка: RTX-5, 1,5 мкм, 30 м × 0,53 мм ID; температура на входе 110°С, детектор 250°С; программа сушильной печи: нагревание от 100 до 250°С при 6°С/минута, затем нагревание до 275°С при 10°C/минута и поддерживание температуры 275ºC в течение 5 минут; колонка: давление на выходе 10 фунтов на квадратный дюйм (0,689 бар) при 40°С; расщепленный поток: 36 мл/минута, продувка; 10 мл/минута) время удержания для кетоальдегида примера 3A: ~16,5 минут.
Пример 3B
(2R)-2-[(1R,3aR,7aR)-7a-метил-4-оксооктагидро-1H-инден-1-ил]пропаналь (3B)
и (2S)-2-[(1R,3aR,7aR)-7a-метил-4-оксооктагидро-1H-инден-1-ил]пропаналь (3A)
Сырой кетоальдегид (пример 3A, 25,93 г, 75,73 ммоль, теоретическое количество) растворяли в 155 мл трет-бутилметилового эфира и помещали в атмосферу азота. Добавляли пирролидин (0,65 мл, 7,9 ммоль), раствор перемешивали при комнатной температуре и контролировали ГХ. Через 15,5 часов соотношение эпимеров составляет 0,3:1 (3B:3A). Добавляли еще 0,65 мл пирролидина. Через 24 часа соотношение эпимеров равно 1,3:1. Спустя еще двадцать четыре часа соотношение эпимеров равно 1,79:1. Раствор перемешивали в течение дополнительных 72 часов. После этого соотношение эпимеров равно 1,86:1. Реакцию проводили непосредственно до восстановления.1H ЯМР (400 МГц, CDCl3) δ 0,64 (с, 3H, 18-H3), 1,08 (д, J=6,86 Гц, 3H, 21-H3), 2,49 (дд, J=6,79, 11,73 Гц, 1H, 14-H), 9,56 (д, J=4,80 Гц, 1H, 22-H); ГХ: (колонка: RTX-5, 1,5 мкм, 30 м × 0,53 мм ID; температура на входе 110°С, детектор 250°С; программа сушильной печи: нагревание от 100 до 250°С при 6°С/минута, затем нагревание до 275°С при 10°C/минута и удерживание в течение 5 минут 275ºC; колонка: давление на выходе 10 фунтов на квадратный дюйм (0,689 бар) при 40°С; расщепленный поток: 36 мл/минута, продувка: 10 мл/минута), время удержания для эпимерного кетоальдегида (3B): ~15,9 минут.
Пример 3C
(1R,3аR,4S,7аR)-1-[(1R)-2-гидрокси-1-метилэтил]-7a-метилоктагидро-1H-инден-4-ол (3C') и
(1R,3аR,4S,7аR)-1-[(1S)-2-гидрокси-1-метилэтил]-7a-метилоктагидро-1H-инден-4-ол (3C")
Раствор трет-бутилметилового эфира кетоальдегидных эпимеров (75,73 ммоль, теоретическое количество) из реакции эпимеризации, описанной выше (155 мл трет-бутилметиловый эфир), охлаждали на ледяной бане, затем разбавляли 225 мл метанола. Раствор выдерживали при 0ºC в атмосфере азота. К реакционному раствору порциями добавляли раствор борогидрида натрия (10,3 г, 272,6 ммоль). После добавления охлаждающую баню удаляли, позволяя реакционной смеси нагреться до комнатной температуры. После нагрева реакционной смеси до комнатной температуры (один час) реакционную смесь снова охлаждали до 0°С. В течение 10 минут добавляли 160 мл 1N раствора HCl и реакционную смесь нагревали до комнатной температуры. Растворитель удаляли в вакууме на роторном испарителе. Остаток распределяли между 100 мл насыщенного раствора соли и 200 мл этилацетата. Значение рН доводили до 2 добавлением 1N раствора HCl. Слои разделяли, и водный слой снова экстрагировали (2×100 мл) этилацетатом. Органические слои объединяли, промывали насыщенным раствором соли и сушили над сульфатом натрия. Раствор концентрировали на роторном испарителе, затем переупаривали с метиленхлоридом. Остаток оставляли при пониженном давлении, используя вакуумный насос, в результате получали 16,6 г масла.1H ЯМР образца показывает, что соотношение эпимеров равно 1,84:1 (3C':3C"). Смесь диолов хроматографировали на двух колонках (330 г Biotage 65M™, элюирование с градиентом: этилацетат:гексан).
Первая фракция: 7,06 г, содержащие 93,73 PA% диола 3C', 2,65 PA% диола 3C" и 1,67% (1S,3aR,4S,7аR)-1-[(1S)-1-гидроксиэтил]-7a-метилоктагидро-1H-инден-4-ола.
Вторая фракция: 4,66 г, содержащие 21,31 PA% диола 3C', 77,22 PA% диола 3C" и 1,47% неизвестных примесей.
Диол 3C:1H ЯМР (400 МГц, CDCl3) δ 0,96 (с, 3H, 18-H3), 0,96 (д, J=6,72 Гц, 3H, 21-H3), 3,45 (дд, J=6,86, 10,57 Гц, 1H, 22-H), 3,71 (дд, J=3,57, 10,70 Гц, 1H, 22-H), 4,08 (уш.д, кв, J=2,70 Гц, 1H, 8-H);13C ЯМР (100 МГц, CDCl3) δ 14,24 (CH2), 16,90 (CH2), 17,76, 22,64, 26,84, 33,80, 37,65 (CH2), 40,00, 41,85 (C-13), 52,64 (CH2), 53,00 (CH2), 66,75 (C-8), 69,27 (C-22); ГХ: (колонка: RTX-5, 1,5 мкм, 30 м × 0,53 мм ID; температура на входе 110°С, детектор 250°С; программа сушильной печи: нагревание от 100 до 250°С при 6°С/минута, затем нагревание до 275°С при 10°C/минута и удерживание в течение 5 минут температуры 275ºC; колонка: давление на выходе 10 фунтов на квадратный дюйм (0,689 бар) при 40°С; расщепленный поток: 36 мл/минута, продувка: 10 мл/минута), время удержания для диола 3C: ~17,3 минут.
Пример 3D
(1R,3aR,4S,7aR)-1-[(1R)-2-гидрокси-1-метилэтил]-7a-метилоктагидро-1H-инден-4-ол
Диольную смесь из описанной выше стадии (пример 3C':3C", 1,86:1 12,2 г, содержание: 93% обоих эпимеров), винилацетат (14,8 г, 3 экв.) и Lipase AK (lot# 56-678-AS, 3,0 г, 25 масс.%, Amano Enzyme USA, Elgin, IL) в трет-бутилметиловом эфире (40 мл, 32,8 мл/г диола) смешивают при 9°С в течение 6,5 часов, затем при 6°С в течение 15,5 часов.
Соотношение синтетический диол (3D)/синтетический ацетат/природный ацетат согласно ЯМР: 62,1%/2,5%/35,4%, согласно ГХ: 59,2%/2,7%/36,9%.
Сырой продукт: 53,97% синтетического диола;
39,24% природного ацетата и синтетического ацетата.
Полученную смесь очищали на колонке Biotage 65M™. Полученный искусственный диол затем перекристаллизовывают из смеси гексан-трет-бутилметиловый эфир (95:5), получая указанное в заголовке соединение (6,395 г, 67,6%).
Пример 3E
(2R)-2-[(1R,3aR,4S,7аR)-4-гидрокси-7a-метилоктагидро-1H-инден-1-ил]пропилпивалат
Соединение примера 3D (0,58 г, 2,7 ммоль) растворяли в 4 мл дихлорметана и 2 мл пиридина; раствор охлаждали до 0°С и к полученной смеси по каплям в течение 5 минут добавляли 0,39 мл пивалоилхлорида. Полученную смесь перемешивали при 0°С в течение 2,5 часов, затем реакцию гасили водой и смесь концентрировали в вакууме, охлаждая реакционную смесь на бане до температуры ниже температуры окружающей среды. Сырой продукт распределяли между эфиром и 0,5N водной HCl; органическую фазу промывали 0,5N водной HCl, затем раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме; остаток (0,96 г) используют далее без дополнительной очистки.
Пример 3F
(2R)-2-[(1R,3aR,7аR)-7a-метил-4-оксооктагидро-1H-инден-1-ил]пропилпивалат
Сырой продукт примера 3E (0,96 г) растворяли в 19 мл дихлорметана и охлаждали до 0°С; добавляли 3,1 г дихромата пиридиния и затем 22 мг п-толуолсульфоната пиридиния. Полученную смесь перемешивали в течение 1,5 часов при 0°С; к смеси добавляли еще 1,3 г дихромата пиридиния и 20 мг п-толуолсульфоната пиридиния и смеси давали возможность нагреться до температуры окружающей среды в течение ночи. Смесь разбавляли эфиром, фильтровали через рыхлый слой диатомовой земли и промывали эфиром. Фильтрат промывали 1N водной HCl и затем фильтровали через рыхлый слой силикагеля. Сырой продукт очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 8% до 15%). Фракции, содержащие указанное в заголовке соединении, объединяли и концентрировали в вакууме (0,77 г, 96%, 2 стадии).
Пример 3G
(2R)-2-[(1R,3R,7E,17β)-1,3-дигидрокси-2-метилен-9,10-секоэстра-5,7-диен-17-ил]пропилпивалат
Примечание: представленную далее последовательность синтеза проводили в затемненном вытяжном шкафу. Соединение примера 1A (30 мг, 0,036 ммоль) объединяли с соединением примера 3F (23 мг, 0,08 ммоль) и полученную смесь дважды подвергали азеотропной сушке с 1 мл толуола. К полученной смеси добавляли тетрагидрофуран (2 мл) и раствор охлаждали до -78°С. К смеси по каплям тремя порциями добавляли раствор бис(триметилсилил)амида лития (1M в тетрагидрофуране; 0,33 мл), что приводило к появлению желтовато-оранжевой окраски, которая исчезает в течение 30 минут. Раствор нагревали до 0°С и перемешивали при данной температуре в течение 30 минут. Реакцию гасили добавлением 1 мл 1N водного раствора NH4Cl и полученную смесь экстрагировали этилацетатом. Органический экстракт промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток очищали хроматографией на Analogix IntelliFlash 280™, (элюирование с градиентом: этилацетат в гексанах - от 0% до 4%). Продукт (22 мг) растворяли в 1,5 мл 1N фторида тетра-н-бутиламмония в тетрагидрофуране; добавляли 0,2 мл уксусной кислоты, смесь нагревали до 70°С и выдерживали при данной температуре в течение ночи. Реакционную смесь распределяли между этилацетатом и водой. Органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток очищали хроматографией на Analogix IntelliFlash 280™, (элюирование с градиентом: этилацетат в гексанах - от 10% до 38%). Фракции, содержащие продукт, объединяли и концентрировали в вакууме, получая указанное в заголовке соединение (5,7 мг).1H ЯМР (500 МГц, CD2Cl2) δ м.д. 6,33 (д, J=11,2 Гц, 1H), 5,89 (д, J=11,2 Гц, 1H), 5,04-5,07 (м, 2H), 4,38-4,48 (м, 2H), 4,15 (дд, J=10,7, 3,5 Гц, 1H), 3,82 (дд, J=10,8, 6,7 Гц, 1H), 2,80-2,85 (м, 1H), 2,77 (дд, J=13,1, 4,5 Гц, 1H), 2,52-2,57 (м, 1H), 2,24-2,32 (м, 2H), 2,02-2,08 (м, 1H), 1,88-1,97 (м, 1H), 1,83-1,87 (м, 1H), 1,74-1,81 (м, 1H), 1,64-1,73 (м, 4H), 1,58-1,64 (м, 1H), 1,47-1,56 (м, 3H), 1,28-1,41 (м, 3H), 1,19 (с, 9H), 0,96 (д, J=6,6 Гц, 3H), 0,59 (с, 3H); MС (+ESI) m/z 448 (M+NH4)+.

Пример 4
(2S)-2-[(1R,3R,7E,17β)-1,3-дигидрокси-2-метилен-9,10-секоэстра-5,7-диен-17-ил]пропил 2,2-диметилбутаноат
Пример 4A
(2S)-2-((1R,3R,7E,17β)-1,3-бис{[трет-бутил(дифенил)силил]окси}-2-метилен-9,10-секоэстра-5,7-диен-17-ил)пропан-1-ол
Примечание: представленную далее последовательность синтеза проводили в затемненном вытяжном шкафу. Соединение примера 2A (70 мг, 0,77 ммоль) растворяли в 1,5 мл эфира и охлаждали до -78°С. К смеси добавляли раствор алюмогидрида лития в тетрагидрофуране (1,0 M, 0,62 мл); смесь перемешивали в течение 10 минут и затем нагревали до 0ºC в течение 50 минут. Реакцию гасили осторожным добавлением этилацетата с последующим добавлением раствора соли Рошелля. После перемешивания в течение 15 минут смесь экстрагировали этилацетатом; органический экстракт промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, получая указанное в заголовке соединение (74 мг).
Пример 4B
(2S)-2-[(1R,3R,7E,17β)-1,3-дигидрокси-2-метилен-9,10-секоэстра-5,7-диен-17-ил]пропил-2,2-диметилбутаноат
Примечание: представленную далее последовательность синтеза проводили в затемненном вытяжном шкафу. Соединение примера 4A (29 мг, 0,03 ммоль) растворяли в 2,5 мл 1,2-дихлорэтана; к раствору добавляли триэтиламин (120 мкл) и полученный раствор охлаждали до 0°С. К смеси добавляли 2,2-диметилбутирилхлорид (90 мг), затем каталитическое количество 4-диметиламинопиридина и смесь медленно нагревали до температуры окружающей среды. Спустя 2 часа растворители удаляли в вакууме, и остаток распределяли между этилацетатом и 1N водной HCl. Органический экстракт промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток очищали хроматографией на Analogix IntelliFlash 280™, (элюирование с градиентом: этилацетат в гексанах - от 0% до 6%). Продукт (34 мг) растворяли в 1,5 мл 1N раствора фторида тетра-н-бутиламмония в тетрагидрофуране; добавляли 0,2 мл уксусной кислоты, смесь нагревали до 70°С и выдерживали при данной температуре в течение ночи. Реакционную смесь распределяли между этилацетатом и водой. Органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 30% до 45%). Фракции, содержащие указанное в заголовке соединение, объединяли и концентрировали в вакууме (4,7 мг).1H ЯМР (500 МГц, CD2Cl2) δ м.д. 6,33 (д, J=11,2 Гц, 1H), 5,90 (д, J=11,2 Гц, 1H), 5,05-5,07 (м, 2H), 4,38-4,48 (м, 2H), 4,04 (дд, J=10,6, 3,2 Гц, 1H), 3,79 (дд, J=10,7, 6,9 Гц, 1H), 2,80-2,86 (м, 1H), 2,76 (дд, J=13,2, 4,5 Гц, 1H), 2,55 (дд, J=13,3, 4,0 Гц, 1H), 2,25-2,33 (м, 2H), 1,98-2,08 (м, 2H), 1,83-1,96 (м, 1H), 1,63-1,77 (м, 5H), 1,51-1,63 (м, 2H), 1,56 (кв, J=7,5 Гц, 2H), 1,31-1,48 (м, 4H), 1,14 (с, 6H), 1,04 (д, J=6,6 Гц, 3H), 0,84 (т, J=7,5 Гц, 3H), 0,58 (с, 3H); MС (+DCI) m/z 462 (M+NH4)+.
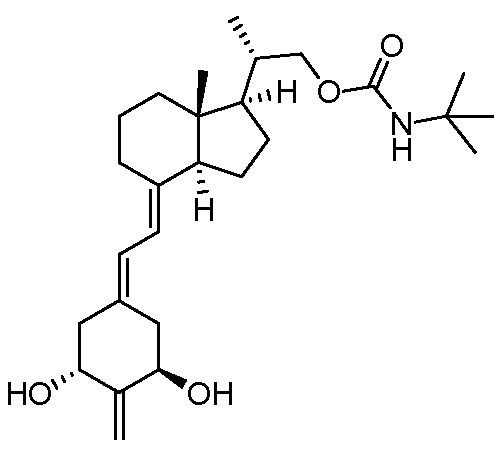
Пример 5
(2S)-2-[(1R,3R,7E,17β)-1,3-дигидрокси-2-метилен-9,10-секоэстра-5,7-диен-17-ил]пропил-трет-бутилкарбамат
Примечание: представленную далее последовательность синтеза проводили в затемненном вытяжном шкафу. Соединение примера 4A (20 мг, 0,02 ммоль) растворяли в 1 мл смеси 1:1 толуол:диметилформамид; тремя порциями в течение трех дней добавляли трет-бутилизоцианат (0,2 мл), выдерживая смесь при температуре 100-105°С. Растворители удаляли в вакууме и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 0% до 20%). Полученный продукт растворяли в 1,5 мл 1N раствора фторида тетра-н-бутиламмония в тетрагидрофуране, добавляли 0,2 мл уксусной кислоты, смесь нагревали до 70°С и выдерживали при данной температуре в течение ночи. Реакционную смесь распределяли между смесью этилацетат/гексаны (3:2) и водой. Органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 25% до 45%). Фракции, содержащие продукт, объединяли и концентрировали в вакууме, получая указанное в заголовке соединение (1,7 мг).1H ЯМР (500 МГц, CD2Cl2) δ м.д. 6,33 (д, J=11,3 Гц, 1H), 5,89 (д, J=11,3 Гц, 1 H), 5,06 (д, J=6,4 Гц, 2H), 4,66 (с, 1H), 4,34-4,49 (м, 2H), 3,95-4,06 (м, 1H), 3,63-3,77 (м, 1H), 2,70-2,91 (м, 2H), 2,55 (дд, J=13,4, 4,0 Гц, 2H), 2,20-2,38 (м, 5H), 1,94-2,05 (м, 2H), 1,83-1,96 (м, 2H), 1,52-1,62 (м, 2H), 1,33-1,47 (м, 4H), 1,26-1,33 (м, 9H), 0,98-1,06 (м, 3H), 0,57 (с, 3H).

Пример 6
(2S)-2-[(1R,3R,7E,17β)-1,3-дигидрокси-2-метилен-9,10-секоэстра-5,7-диен-17-ил]пропил-2-(ацетилокси)-2-метилпропаноат
Пример 6A
[2-((3R,5R)-3,5-бис{[трет-бутил(диметил)силил]окси}-4-метиленциклогексилиден)этил](дифенил)фосфиноксид
Указанное в заголовке соединение получают в соответствии с методикой, описанной в WO98/41500 (DeLuca и Sicinski).
Пример 6B
(2S)-2-((1R,3R,7E,17β)-1,3-бис{[трет-бутил(диметил)силил]окси}-2-метилен-9,10-секоэстра-5,7-диен-17-ил)пропилпивалат
Примечание: представленную далее последовательность синтеза проводили в затемненном вытяжном шкафу. Соединение примера 6A (64 мг, 0,077 ммоль) объединяли с соединением примера 1D (32 мг, 0,11 ммоль) и полученную смесь дважды подвергали азеотропной сушке с 1 мл толуола. К смеси добавляли тетрагидрофуран (2 мл) и полученный раствор охлаждали до -78°С. К смеси по каплям двумя порциями добавляли раствор бис(триметилсилил)амида лития (0,6 M в тетрагидрофуране; 0,40 мл), в результате смесь приобретала желтовато-оранжевое окрашивание, которое исчезает в течение 20 минут. Раствор нагревали до 0°С и перемешивали при данной температуре в течение 20 минут. Реакцию гасили добавлением 1 мл 1N водного раствора NH4Cl и полученную смесь экстрагировали этилацетатом. Органический экстракт промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 0% до 6%).
Пример 6C
(2S)-2-((1R,3R,7E,17β)-1,3-бис{[трет-бутил(диметил)силил]окси}-2-метилен-9,10-секоэстра-5,7-диен-17-ил)пропан-1-ол
Примечание: представленную далее последовательность синтеза проводили в затемненном вытяжном шкафу. Соединение примера 6B (70 мг, 0,77 ммоль) растворяли в 1,5 мл эфира и охлаждали до -78°С. К полученному раствору добавляли раствор алюмогидрида лития в тетрагидрофуране (1,0 M, 0,62 мл); смесь перемешивали в течение 10 минут, затем нагревали до 0°С в течение 50 минут. Реакцию гасили осторожным добавлением этилацетата с последующим добавлением раствора соли Рошелля. После перемешивания в течение 15 минут полученную смесь экстрагировали этилацетатом; органический экстракт промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, получая указанное в заголовке соединение (74 мг).
Пример 6D
(2S)-2-[(1R,3R,7E,17β)-1,3-дигидрокси-2-метилен-9,10-секоэстра-5,7-диен-17-ил]пропил-2-(ацетилокси)-2-метилпропаноат
Примечание: представленную далее последовательность синтеза проводили в затемненном вытяжном шкафу. Соединение примера 6C (31 мг, 0,054 ммоль) растворяли в 2 мл 1,2-дихлорэтана; к полученному раствору добавляли триэтиламин (75 мкл, избыток) с последующим добавлением 62 мкл ацетоксиизобутирилхлорида и каталитического количества 4-диметиламинопиридина. Спустя 2 часа растворители удаляли в вакууме, и остаток распределяли между эфиром и 1N водной HCl. Органический экстракт промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 0% до 8%). Продукт (40 мг) растворяли в 2 мл 1N раствора фторида тетра-н-бутиламмония в тетрагидрофуране и перемешивали при температуре окружающей среды в течение 2,5 часов. Реакционную смесь распределяли между смесью этилацетат/гексаны (3:1) и водой. Органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 30% до 50%), получая указанное в заголовке соединение (3,6 мг).1H ЯМР (400 МГц, CD2Cl2) δ м.д. 6,33 (д, J=11,0 Гц, 1H), 5,89 (д, J=11,4 Гц, 1H), 5,06 (д, J=4,6 Гц, 2H), 4,34-4,49 (м, 2H), 4,00-4,13 (м, 1H), 3,85 (дд, J=10,7, 6,8 Гц, 1H), 2,70-2,90 (м, 2H), 2,55 (дд, J=13,2, 4,0 Гц, 1H), 2,22-2,38 (м, 2H), 1,96-2,08 (м, 5H), 1,84-1,93 (м, 2H), 1,65-1,76 (м, 4H), 1,55-1,62 (м, 2H), 1,55-1,60 (м, 1H), 1,49-1,57 (м, 6H), 1,30-1,46 (м, 3H), 0,97-1,06 (м, 3H), 0,58 (с, 3H); MС (+DCI) m/z 492 (M+NH4)+.

Пример 7
(1R,3R,7E)-2-метилен-17-[(1R,4S)-1,4,5-триметилгексил]-9,10-секоэстра-5,7-диен-1,3-диол
Пример 7A
(1R,3aR,4S,7aR)-7a-метил-1-[(1R,2E,4R)-1,4,5-триметилгекс-2-енил]октагидро-1H-инден-4-ол
Указанное в заголовке соединение получают в соответствии с методиками, описанными в публикации Toh and Okamura, J. Org. Chem. 1983, 48, 1414, но используя витамин D2 в качестве исходного сырья. В этом случае 20,6 г (52 ммоль) витамина D2 превращают в 4,75 г целевого продукта (общий выход последовательности из четырех стадий равен 33%).
Пример 7B
(1R,3aR,4S,7aR)-7a-метил-1-[(1R,4S)-1,4,5-триметилгексил]октагидро-1H-инден-4-ол
Соединение примера 7A (400 мг, 1,4 ммоль) растворяли в 20 мл уксусной кислоты и гидрируют, используя в качестве катализатора 10% платину на углероде. После продувки азотом и фильтрации для удаления катализатора растворитель удаляли в вакууме, и остаток распределяли между этилацетатом и насыщенным водным раствором гидрокарбоната натрия. Органическую фазу промывали насыщенным раствором соли и сушили над сульфатом натрия. Растворители удаляли в вакууме, и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 5% до 30%), получая указанное в заголовке соединение (390 мг).
Пример 7C
(1R,3aR,7аR)-7a-метил-1-[(1R,4S)-1,4,5-триметилгексил]октагидро-4Н-инден-4-он
Соединение примера 7B (66 мг, 0,25 ммоль) растворяли в 5 мл дихлорметана и охлаждали до 0°С; добавляли 370 мг дихромата пиридиния и 2 мг п-толуолсульфоната пиридиния и полученную смесь встряхивают при температуре окружающей среды в течение 4 часов. Реакционную смесь фильтровали сначала через рыхлый слой диатомовой земли, затем через рыхлый слой силикагеля, промывая этилацетатом. Объединенный фильтрат концентрировали и очищали хроматографией на Analogix IntelliFlash 280™ (элиюрование: смесь 10% этилацетат в гексанах), получая указанное в заголовке соединение (56 мг).
Пример 7D
(1R,3R,7E)-2-метилен-17-[(1R,4S)-1,4,5-триметилгексил]-9,10-секоэстра-5,7-диен-1,3-диол
Примечание: представленную далее последовательность синтеза проводили в затемненном вытяжном шкафу. Соединение примера 6A (20 мг, 0,034 ммоль) объединяли с соединением примера 7C (19 мг, 0,068 ммоль) и полученную смесь с дважды подвергали азеотропной сушке с 1 мл толуола. Добавляли тетрагидрофуран (1 мл) и раствор охлаждали до -78°С. К раствору по каплям двумя порциями добавляли раствор бис(триметилсилил)амида лития (1M в тетрагидрофуране; 0,30 мл), что приводило к появлению желтовато-оранжевой окраски, которая исчезает в течение 20 минут. Раствор нагревали до 0°С и перемешивали при данной температуре в течение 20 минут. Реакцию гасили добавлением 1 мл 1N водного раствора хлорида аммония и полученную смесь экстрагировали этилацетатом. Органический экстракт промывали насыщенным раствором соли и сушили над сульфатом натрия. Растворители удаляли в вакууме, и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование: 2% этилацетат в гексанах). Продукт (10 мг) растворяли в 0,3 мл 1N раствора фторида тетра-н-бутиламмония в тетрагидрофуране и перемешивали в течение ночи при температуре окружающей среды. Реакционную смесь распределяли между смесью этилацетат/гексаны (3:1) и водой. Органическую фазу промывали насыщенным раствором соли и сушили над сульфатом натрия. Растворители удаляли в вакууме и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование: 30% этилацетат в гексанах), получая указанное в заголовке соединение (6 мг).1H ЯМР (400 МГц, CD2Cl2) δ м.д. 6,33 (д, J=11,4 Гц, 1H), 5,88 (д, J=11,0 Гц, 1H), 5,06 (д, J=4,3 Гц, 2H), 4,34-4,51 (м, 2H), 2,83 (д, J=4,3 Гц, 2H), 2,78 (д, J=4,6 Гц, 2H), 2,55 (дд, J=13,2, 4,0 Гц, 1H), 2,21-2,43 (м, 4H), 1,97-2,08 (м, 2H), 1,82-1,99 (м, 2H), 1,52-1,63 (м, 3H), 1,31-1,47 (м, 4H), 1,18-1,34 (м, 3H), 0,83-0,93 (м, 6H), 0,73-0,83 (м, 6H), 0,55 (с, 3H); MС (DCI+) m/z 432 (M+NH4)+.

Пример 8
(1R,3R,7E,17β)-17-[(1S)-1-(3-гидрокси-3-метилбутокси)этил]-2-метилен-9,10-секоэстра-5,7-диен-1,3-диол
Пример 8A
(1Z,3аR,4S,7aS)-1-этилиден-7a-метилоктагидро-1Н-инден-4-ол
Указанное в заголовке соединение получают в соответствии с методикой, описанной в публикации Daniewski and Liu, J. Org. Chem. 2001, 66(2), 626-628.
Пример 8B
Трет-бутил-{[(1Z,3аR,4S,7aS)-1-этилиден-7а-метилоктагидро-1H-инден-4-ил]окси}диметилсилан
Соединение примера 8A (0,74 г, 4,1 ммоль) растворяли в 4 мл диметилформамида; к раствору добавляли 0,4 г имидазола и затем 0,7 г трет-бутилдиметилсилилхлорида. Полученную смесь перемешивали при температуре окружающей среды в течение 4 часов; добавляли равные порции трет-бутилдиметилсилилхлорида и имидазола, смесь нагревали до 60°С и выдерживали при данной температуре в течение ночи. Добавляли воду; смесь перемешивали при температуре окружающей среды в течение 30 минут и затем экстрагировали эфиром. Органическую фазу промывали последовательно 1N водной H3PO4, водой и раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 0% до 10%). Указанное в заголовке соединение выделяют загрязненным некоторым количеством непрореагировавшего исходного вещества (0,31 г, 42%).
Пример 8C
(1S)-1-((1S,3aR,4S,7aR)-4-{[трет-бутил(диметил)силил]окси}-7a-метилоктагидро-1H-инден-1-ил)этанол
Соединение примера 8B (1,9 г, 6,4 ммоль) переносили в раствор 9-борабицикло[3.3.1]нонана (9-BBN) (0,5 M в тетрагидрофуране; 28 мл); смесь нагревали до 45°С, выдерживали при указанной температуре в течение 5 часов, затем охлаждали до 0°С и гасили добавлением смеси 8 мл 2N раствора NaOH и 4 мл 30% раствора пероксида водорода. Полученный раствор нагревали до температуры окружающей среды и перемешивали в течение ночи. Реакционную смесь экстрагировали этилацетатом; органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 10% до 90%), получая указанное в заголовке соединение (количественный выход).
Пример 8D
3-(Бромметил)-2,2-диметилоксиран
Указанное в заголовке соединение получают в соответствии с методикой, описанной Shimizu с соавторами в публикации Org. Proc. Res. & Dev. 2005, 9, 278-287.
Пример 8E
4-{[(1S)-1-((1S,3aR,4S,7aR)-4-{[трет-бутил(диметил)силил]окси}-7a-метилоктагидро-1H-инден-1-ил)этил]окси}-2-метилбутан-2-ол
Соединение примера 8C (1,1 г, 3,5 ммоль) растворяли в 5 мл тетрагидрофурана; добавляли гидрид натрия (0,28 г 60% масляной дисперсии) и смесь перемешивали при температуре окружающей среды в течение 10 минут до прекращения выделения газа. К смеси добавляли соединение примера 8D (1,2 г) и смесь кипятили с обратным холодильником в течение 60 минут. Смесь охлаждали до температуры окружающей среды и по каплям добавляли 6,5 мл 1N раствора алюмогидрида лития в тетрагидрофуране (внимание: после непродолжительного периода индукции реакция протекает с энергичным выделением теплоты). Смесь перемешивали в течение 2 часов, по мере чего выделение теплоты постепенно снижается, затем реакцию гасили добавлением 10 мл этилацетата (результатом является более экзотермическая реакция). Стандартная обработка Файзера (Fieser) приводила к получению желеобразной массы, которую смешивали с твердым Na2SO4 в течение 2 часов, затем фильтровали через рыхлый слой диатомовой земли, промывая этилацетатом. Объединенные промывные растворы концентрировали в вакууме, и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 10% до 80%). Более полярной фракцией является регенерированный исходный спирт; смесь менее полярных фракций снова хроматографировали на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 0% до 40%), получая указанное в заголовке соединение (0,80 г, 57%).
Пример 8F
(1S,3аR,4S,7aS)-1-[(1S)-1-(3-гидрокси-3-метилбутокси)этил]-7a-метилоктагидро-1H-инден-4-ол
Соединение примера 8E (230 мг, 0,58 ммоль) растворяли в 8 мл 1N раствора фторида тетра-н-бутиламмония в тетрагидрофуране, нагревали до 80°С и выдерживали при данной температуре в течение ночи. Реакционную смесь распределяли между эфиром и водой; органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 0% до 50%), получая указанное в заголовке соединение (140 мг, 85%).
Пример 8G
(1S,3aR,7аR)-1-[(1S)-1-(3-гидрокси-3-метилбутокси)этил]-7a-метилоктагидро-4H-инден-4-он
Соединение примера 8F (70 мг, 0,25 ммоль) растворяли в 1,5 мл дихлорметана; к раствору добавляли 350 мг дихромата пиридиния и 15 мг п-толуолсульфоната пиридиния и полученную смесь перемешивали в течение ночи при температуре окружающей среды. Растворители удаляли в вакууме; остаток переносили в этилацетат и фильтровали через диатомовую землю, промывая этилацетатом. Фильтрат концентрировали, и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 10% до 40%), получая указанное в заголовке соединение (65 мг, 73%).
Пример 8H
(1R,3R,7E,17β)-17-[(1S)-1-(3-гидрокси-3-метилбутокси)этил]-2-метилен-9,10-секоэстра-5,7-диен-1,3-диол
Примечание: представленную далее последовательность синтеза проводили в затемненном вытяжном шкафу. Соединение примера 6A (50 мг, 0,088 ммоль) объединяли с соединением примера 8G (32 мг, 0,09 ммоль) и полученную смесь дважды подвергали азеотропной сушке с 1 мл толуола. Добавляли тетрагидрофуран (1,5 мл) и раствор охлаждали до -78°С, к полученной смеси по каплям двумя порциями добавляли раствор бис(триметилсилил)амида лития (1M раствор в тетрагидрофуране; 0,20 мл), что приводило к появлению желтовато-оранжевой окраски, которая исчезает в течение 20 минут. Раствор нагревали до 0ºC и перемешивали при данной температуре в течение 15 минут. Реакцию гасили добавлением 1 мл 1N водного раствора NH4Cl и полученную смесь экстрагировали этилацетатом. Органический экстракт промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме; остаток растворяли в 1,5 мл 1N раствора фторида тетра-н-бутиламмония в тетрагидрофуране и перемешивали в течение ночи при температуре окружающей среды. Реакционную смесь распределяли между этилацетатом и водой. Органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 10% до 100%), получая указанное в заголовке соединение (18 мг).1H ЯМР (500 МГц, CD2Cl2) δ м.д. 6,32 (д, J=11,3 Гц, 1H), 5,90 (д, J=11,3 Гц, 1 H), 5,06 (д, J=7,0 Гц, 2H), 4,33-4,50 (м, 2H), 3,75-3,90 (м, 1H), 3,41-3,55 (м, 2H), 3,26 (дд, J=7,9, 6,1 Гц, 1H), 2,69-2,89 (м, 2H), 2,55 (дд, J=13,1, 4,0 Гц, 1H), 2,21-2,39 (м, 2H), 1,96-2,04 (м, 3H), 1,89 (дд, J=15,4, 2,9 Гц, 2H), 1,50-1,65 (м, 6H), 1,31 (д, J=4,3 Гц, 1H), 1,14-1,27 (м, 12H), 0,54 (с, 3H); MС(+ESI) m/z 441 (M+Na)+.

Пример 9
(1R,3R,7E)-17-[(1R,4S)-1,4,5-триметилгексил]-9,10-секоэстра-5,7-диен-1,3-диол
Пример 9A
[2-((3R,5R)-3,5-бис{[трет-бутил(диметил)силил]окси}циклогексилиден)этил](дифенил)фосфин-оксид
Указанное в заголовке соединение получают в соответствии с методиками, описанными в EP 516410 B1 (DeLuca et al.).
Пример 9B
(1R,3R,7E)-17-[(1R,4S)-1,4,5-триметилгексил]-9,10-секоэстра-5,7-диен-1,3-диол
Примечание: представленную далее последовательность синтеза проводили в затемненном вытяжном шкафу. Соединение примера 9A (20 мг, 0,034 ммоль) объединяли с соединением примера 7C (20 мг, 0,07 ммоль) и полученную смесь дважды подвергали азеотропной сушке с 1 мл толуола. Добавляли тетрагидрофуран (1 мл) и раствор охлаждали до -78ºC. К полученной смеси по каплям двумя порциями добавляли раствор бис(триметилсилил)амида лития (1M раствор в тетрагидрофуране; 0,20 мл), что приводило к появлению желтовато-оранжевого окрашивания, которое исчезает в течение 20 минут. Раствор нагревали до 0ºC и перемешивали при данной температуре в течение 20 минут. Реакцию гасили добавлением 1 мл 1N водного раствора NH4Cl и полученную смесь экстрагировали этилацетатом. Органический экстракт промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, остаток растворяли в 1 мл 0,5N раствора фторида тетра-н-бутиламмония в тетрагидрофуране и полученный раствор перемешивали в течение ночи при температуре окружающей среды. Реакционную смесь распределяли между смесью этилацетат/гексаны (3:1) и водой. Органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 50% до 60%), получая указанное в заголовке соединение (6 мг).1H ЯМР (500 МГц, CD2Cl2) δ м.д. 6,28 (д, J=11,3 Гц, 1H), 5,86 (д, J=11,3 Гц, 1H), 3,90-4,15 (м, 2H), 2,61-2,85 (м, 2H), 2,45 (дд, J=13,3, 3,5 Гц, 1H), 2,11-2,26 (м, 2H), 1,98-2,05 (м, 4H), 1,81-1,94 (м, 4H), 1,72-1,79 (м, 1H), 1,61-1,72 (м, 4H), 1,29-1,36 (м, 3H), 1,22-1,38 (м, 7H), 0,76-0,89 (м, 9H), 0,54 (с, 3H); MС (+ESI) m/z 420 (M+NH4)+.

Пример 10
(1S,3R,5Z,7E,24R)-22,25-диметокси-9,10-секоэргоста-5,7,10-триен-1,3-диол
Пример 10A
[(2Z)-2-((3S,5R)-3,5-бис{[трет-бутил(диметил)силил]окси}-2- метиленциклогексилиден)этил](дифенил)фосфин-оксид
Указанное в заголовке соединение получают в соответствии с методиками, описанными в публикации Radinov еt al., J. Org. Chem. 2002, 67(5), 1580-1587.
Пример 10B
(3S)-2,3-диметил-4-(фенилсульфонил)бутан-2-ол
Указанное в заголовке соединение получают в соответствии с методиками, описанными в публикации Kutner et al., J. Org. Chem. 1988, 53, 3450-3457.
Пример 10C
Триметил-{[(2S)-1,1,2-триметил-3-(фенилсульфонил)пропил]окси}силан
Соединение примера 10B (0,95 г, 3,9 ммоль) растворяли в 3 мл диметилформамида; в полученный раствор добавляли 1 мл 1-(триметилсилил)имидазола, полученную смесь перемешивали в течение ночи при температуре окружающей среды, затем нагревали до 80ºC и выдерживали при указанной температуре в течение 8 часов. Сырую смесь распределяли между этилацетатом и водой; органическую фазу промывали раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 0 до 20%), получая указанное в заголовке соединение (1,18 г, 96%).
Пример 10D
(2S)-2-((1R,3aR,4S,7aR)-4-{[трет-бутил(диметил)силил]окси}-7a-метилоктагидро-1H-инден-1-ил)пропилпивалат
Соединение примера 1B (5,05 г, 24 ммоль) растворяли в 35 мл дихлорметана и 15 мл пиридина; раствор охлаждали до 0ºC и к смеси по каплям в течение 5 минут добавляли 3,3 мл пивалоилхлорида. Полученную смесь перемешивали при 0ºC в течение 4 часов и затем реакционную смесь нагревали до температуры окружающей среды в течение 30 минут. Реакцию гасили водой, и смесь концентрировали в вакууме, поддерживая температуру ниже температуры окружающей среды с помощью охлаждающей бани. Сырой продукт распределяли между эфиром и 0,5N водной HCl; органическую фазу промывали 0,5N водной HCl, затем раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме; остаток растворяли в 15 мл диметилформамида. К раствору добавляли имидазол (2,0 г) и трет-бутилдиметилсилилхлорид (4,0 г), полученную смесь перемешивали при температуре окружающей среды в течение 4 дней, затем нагревали до 60ºC и выдерживали при данной температуре в течение 4 часов. Растворители удаляли в вакууме; остаток распределяли между этилацетатом и 1,5 N водной HCl. Органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 0 до 60%), получая указанное в заголовке соединение (6,45 г, 66%).
Пример 10E
(2S)-2-((1R,3aR,4S,7аR)-4-{[трет-бутил(диметил)силил]окси}-7a-метилоктагидро-1H-инден-1-ил)пропан-1-ол
Соединение примера 10D (6,45 г, 16 ммоль) растворяли в 30 мл эфира и охлаждали до -30ºC; к смеси добавляли 5 мл 1N раствора алюмогидрида лития в эфире таким образом, чтобы поддерживать постоянную температуру и умеренную скорость выделения газа. Смесь перемешивали при -30ºC в течение 10 минут и затем ее нагревали до 0ºC в течение 1 часа. Реакцию гасили осторожным добавлением этилацетата (осторожно! энергичное выделение газа) и перемешивали в течение 10 минут, затем раствором соли Рошелля. Полученную смесь распределяли между этилацетатом и водой; органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 5 до 30%).
Пример 10F
(2S,5R)-2-((1R,3aR,4S,7aR)-4-{[трет-бутил(диметил)силил]окси}-7a-метилоктагидро-1H-инден-1-ил)-5,6-диметил-6-[(триметилсилил)окси]гептан-3-ол
Соединение примера 10E (200 мг, 0,61 ммоль) растворяли в 2 мл дихлорметана; добавляли 100 мг 4Å молекулярных сит, затем 15 мг перрутената тетрапропиламмония и 75 мг 4-метилморфолин-N-оксида. Смесь перемешивали в течение 2 часов при температуре окружающей среды и затем фильтровали через диатомовую землю для удаления твердых веществ. Сырой продукт очищали хроматографией на силикагеле с использованием Analogix IntelliFlash™ 40 (элюирование с градиентом: этилацетат в гексанах - от 5% до 30%). Соединение примера 10C (200 мг, 0,63 ммоль) подвергали реакции связывания с данным альдегидом в соответствии с методикой реакции связывания, описанной в публикации Kutner et al. in J. Org. Chem. 1988, 53, 3450-3457. Указанный в заголовке продукт, полученный в результате реакции десульфуризации без отщепления, выделяют хроматографией с использованием Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 0 до 50%).
Пример 10G
трет-бутил({(1R,3aR,4S,7aR)-1-[(1S,4R)-2,5-диметокси-1,4,5-триметилгексил]-7a-метилоктагидро-1Н-инден-4-ил}окси)диметилсилан
Соединение примера 10F (50 мг, 0,1 ммоль) растворяли в 0,5 мл сухого тетрагидрофурана; добавляли 20 мг NaH (60% масляная дисперсия; в избытке) и затем (после прекращения выделения газа) 0,1 мл (избыток) йодметана. Полученную смесь перемешивали в течение ночи при температуре окружающей среды, затем гасили водой и экстрагировали этилацетатом. Органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 0 до 20%), получая указанное в заголовке соединение (43 мг, 94%).
Пример 10H
(1R,3aR,4S,7aR)-1-[(1S,4R)-2,5-диметокси-1,4,5-триметилгексил]-7a-метилоктагидро-1H-инден-4-ол
Соединение примера 10G (42 мг, 0,093 ммоль) растворяли в 1,5 мл 1N раствора фторида тетра-н-бутиламмония в тетрагидрофуране, нагревали и выдерживали в течение ночи при 80ºC. Реакционную смесь распределяли между этилацетатом и водой; органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 0 до 25%), получая указанное в заголовке соединение (27 мг, 86%).
Пример 10I
(1R,3aR,7аR)-1-[(1S,4R)-2,5-диметокси-1,4,5-триметилгексил]-7a-метилоктагидро-4H-инден-4-он
Соединение примера 10H (27 мг, 0,079 ммоль) растворяли в 1 мл дихлорметана; добавляли 150 мг дихромата пиридиния и 10 мг п-толуолсульфоната пиридиния и полученную смесь перемешивали в течение ночи при температуре окружающей среды. Добавляли диатомовую землю (~500 мг) и растворители удаляли в вакууме. Остаток переносили в этилацетат и наносят на рыхлый слой диоксида кремния. Элюирование с градиентом (этилацетат в гексанах, от 50% до 100%) и концентрирование объединенного элюата приводило к получению продукта, который используют в следующих стадиях синтеза без дополнительной очистки.
Пример 10J
(1S,3R,5Z,7E,24R)-22,25-диметокси-9,10-секоэргоста-5,7,10-триен-1,3-диол
Примечание: представленную далее последовательность синтеза проводили в затемненном вытяжном шкафу. Соединение примера 10A (20 мг, 0,035 ммоль) объединяли с соединением примера 10I (12 мг, 0,036 ммоль) в 1 мл толуола; растворитель удаляли в вакууме для полной сушки реагентов. Остаток переносили в 1,5 мл сухого тетрагидрофурана и охлаждали до -78ºC. К полученной смеси по каплям добавляли раствор бис(триметилсилил)амида лития (1,0 M в тетрагидрофуране; 0,1 мл), что приводило к появлению желтовато-оранжевой окраски, которая исчезает в течение 20 минут. К смеси добавляли дополнительные 0,05 мл бис(триметилсилил)амида лития; полученную смесь перемешивали при -78ºC в течение 1 часа, затем нагревали до 0ºC и перемешивали при данной температуре в течение 30 минут. Реакцию гасили добавлением 1 мл 1N водного раствора NH4Cl и полученную смесь экстрагировали этилацетатом. Органический экстракт промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток растворяли в 1 мл 1N раствора фторида тетра-н-бутиламмония в тетрагидрофуране. После перемешивания в течение ночи при температуре окружающей среды реакционную смесь распределяли между этилацетатом и водой. Органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 10% до 100%, с последующей полной заменой растворителя на 100% этилацетат), получая указанное в заголовке соединение (1,2 мг).1H ЯМР (400 МГц, CD2Cl2) δ м.д. 6,37 (д, J=11,0 Гц, 1 H), 6,03 (д, J=11,4 Гц, 1H), 4,38 (дд, J=4,3 Гц, 1H), 4,09-4,24 (м, 2H), 3,25-3,34 (м, 3H), 3,17-3,21 (м, 1H), 3,07-3,16 (м, 3H), 2,86 (с, 1H), 2,55 (д, J=3,1 Гц, 1H), 2,27 (дд, J=13,3, 6,3 Гц, 1H), 1,59-1,79 (м, 7H), 1,42-1,54 (м, 4H), 1,18-1,34 (м, 5H), 1,01-1,13 (м, 7H), 0,81-1,00 (м, 9H), 0,51-0,63 (м, 3H).

Пример 11
(1R,3R,7E,17β)-17-[(1S,4R)-2,5-диметокси-1,4,5-триметилгексил]-9,10-секоэстра-5,7-диен-1,3-диол
Примечание: представленную далее последовательность синтеза проводили в затемненном вытяжном шкафу. Соединение примера 9A (20 мг, 0,035 ммоль) объединяли с соединением примера 10I (12 мг, 0,036 ммоль) в 1 мл толуола; растворитель удаляли в вакууме для полной сушки реагентов. Остаток переносили в 1,5 мл сухого тетрагидрофурана и охлаждали до -78ºC. К полученной смеси по каплям добавляли раствор бис(триметилсилил)амида лития (1,0 M в тетрагидрофуране; 0,1 мл), что приводило к появлению желтовато-оранжевой окраски, которая исчезает через 20 минут. К смеси добавляли еще 0,05 мл бис(триметилсилил)амида лития; полученную смесь перемешивали при -78ºC в течение 1 часа, затем нагревали до 0ºC и перемешивали при данной температуре в течение 30 минут. Реакцию гасили добавлением 1 мл 1N водного раствора NH4Cl и полученную смесь экстрагировали этилацетатом. Органический экстракт промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток растворяли в 1 мл 1N раствора фторида тетра-н-бутиламмония в тетрагидрофуране. После перемешивания в течение ночи при температуре окружающей среды реакционную смесь распределяли между этилацетатом и водой. Органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 10% до 100%, далее 100% этилацетата), получая указанное в заголовке соединение (4,4 мг).1H ЯМР (400 МГц, CD2Cl2) δ м.д. 6,29 (д, J=11,4 Гц, 1H), 5,87 (д, J=11,4 Гц, 1H), 4,02 (д, J=26,7 Гц, 1H), 3,29 (с, 3H), 3,17-3,27 (м, 1H), 3,12 (с, 3H), 2,75-2,90 (м, 1H), 2,63-2,73 (м, 1H), 2,44 (д, J=3,7 Гц, 2H), 2,18 (д, J=5,5 Гц, 2H), 1,82-2,03 (м, 4H), 1,61-1,70 (м, 2H), 1,60-1,81 (м, 7H), 1,34-1,59 (м, 6H), 1,19-1,40 (м, 3H), 1,07 (с, 6H), 0,82-0,96 (м, 3H), 0,56 (с, 3H).
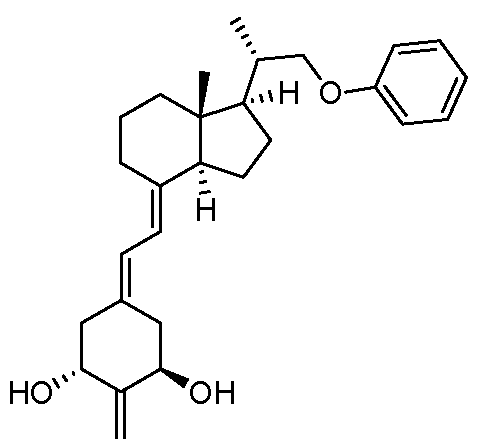
Пример 12
(1R,3R,7E,17β)-2-метилен-17-[(1S)-1-метил-2-феноксиэтил]-9,10-секоэстра-5,7-диен-1,3-диол
Примечание: представленную далее последовательность синтеза проводили в затемненном вытяжном шкафу. Соединение примера 6C (16 мг, 0,028 ммоль) объединяли с 8 мг фенола и 20 мг трифенилфосфина в 2 мл толуола с последующим добавлением 18 мг ди-трет-бутилазодикарбоксилата. Смесь продували аргоном, нагревали до 85ºC и выдерживали при данной температуре в течение 4 часов. После удаления растворителей в вакууме остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 0% до 5%). Продукт (6 мг) растворяли в 1,5 мл 1N раствора фторида тетра-н-бутиламмония в тетрагидрофуране. После перемешивания в течение 3 часов при температуре окружающей среды реакционную смесь распределяли между этилацетатом и водой. Органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 25% до 45%), получая указанное в заголовке соединение (4 мг).1H ЯМР (500 МГц, CD2Cl2) δ м.д. 7,20-7,37 (м, 2H), 6,76-7,01 (м, 3H), 6,34 (д, J=11,0 Гц, 1H), 5,90 (д, J=11,3 Гц, 1 H), 5,06 (д, J=5,5 Гц, 2H), 4,32-4,54 (м, 2H), 3,94 (дд, J=3,1 Гц, 1H), 3,69 (дд, J=7,3 Гц, 1H), 2,69-2,94 (м, 2H), 2,55 (дд, J=13,4, 4,0 Гц, 2H), 2,19-2,39 (м, 3H), 1,98-2,14 (м, 4H), 1,81-1,99 (м, 3H), 1,64-1,77 (м, 2H), 1,49-1,66 (м, 2H), 1,27-1,35 (м, 1H), 0,85-1,02 (м, 3H), 0,61 (с, 3H); MС (+DCI) m/z 440 (M+NH4)+.

Пример 13
(1R,3S,5Z,7E,17β)-3-фтор-17-[(1R)-5-гидрокси-1,5-диметилгексил]-2-метилен-9,10-секоэстра-5,7-диен-1-ол
Пример 13A
трет-бутил-{[(1R,2R,4R,6R)-4-изопропенил-1-метил-7-оксабицикло[4.1.0]гепт-2-ил]окси}дифенилсилан
(R)-Карвон-оксид (106 г, 640 ммоль, полученный в соответствии с методикой получения энантиомера публикации Klein and Ohloff, Tetrahedron 1963, 19, 1091-1099), растворяли в 120 мл метанола и полученный раствор добавляли к раствору CeCl3 (гептагидрат; 119 г, 320 ммоль) в 1,5 л метанола, предварительно охлажденному до 0ºC. Колбу ополаскивали 60 мл метанола, и смесь охлаждали до -20ºC. К смеси в течение часа добавляли раствор NaBH4 (2M в триглиме, 175 мл). После перемешивания в течение 30 минут при -20ºC к смеси добавляли 720 мл воды и смеси давали возможность нагреться до температуры окружающей среды. Органический растворитель удаляли в вакууме; к остатку добавляли 800 мл этилацетата и затем достаточное количество 2N водной HCl (~100 мл) для доведения значения pH раствора до ~5,5. Водную фазу декантировали, органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Раствор концентрировали в вакууме с получением раствора сырого спирта в триглиме. К раствору добавляли диметилформамид (300 мл) и затем имидазол (70 г, 1000 ммоль) и трет-бутилдифенилсилилхлорид (236 г, 1000 ммоль). Смесь перемешивали при температуре окружающей среды в течение 84 часов, затем через 24 часа добавляли еще 50 мл диметилформамида для улучшения растворимости. Смесь охлаждали на ледяной бане, добавляли 30 мл воды и перемешивание продолжали в течение 30 минут. Реакционную смесь распределяли между гептаном и водой; органическую фазу промывали последовательно водой и раствором соли и сушили над MgSO4. После концентрирования в вакууме сырой продукт очищали флэш-хроматографией (элюирование с градиентом: дихлорметан в гексанах - от 1:5 до 1:4), получая указанное в заголовке соединение (206 г, 79%).
Пример 13B
(1R,3R,5R,6R)-5-{[трет-бутил(дифенил)силил]окси}-6-метил-7-оксабицикло[4.1.0]гептан-3-ол
Смесь соединения примера 13A (34 г, 92% (масс.), 77 ммоль) и NaHCO3 (3,3 г, 39 ммоль) в 300 мл дихлорметана и 60 мл метанола охлаждали до <-70ºC и обрабатывают озоном (7-8 фунтов на кв. дюйм (0,4823-0,5512 бар), 90 вольт, 4 slpm) до тех пор, пока не наблюдали стойкое голубое окрашивание. После продувки азотом для удаления окрашивания реакционную смесь нагревали до ~0°C, затем фильтровали через бумажный фильтр и концентрировали. Смесь обрабатывают бензолом (2×100 мл), добавляли 300 мл дихлорметана и 60 мл пиридина и охлаждали до <5ºC. К смеси добавляют п-нитробензоилхлорид (22,2 г, 120 ммоль) и перемешивали в течение 1 часа, затем охлаждающую баню удаляли и реакционную смесь перемешивали при температуре окружающей среды в течение ночи. Полученную суспензию концентрировали в вакууме до получения густой взвеси, затем разбавляли этилацетатом и твердые вещества удаляли фильтрацией. Фильтрат промывали 250 мл насыщенного водного раствора гидрокарбоната натрия, затем 100 мл каждой из 2N HCl, 1N HCl и 2 N HCl и далее 200 мл 1N HCl, 200 мл насыщенного водного раствора гидрокарбоната натрия и раствором соли. Органический слой сушили над MgSO4, фильтровали и концентрировали, получая сырой сложный эфир. Навеску данного продукта (20,6 г, 32 ммоль) растворяли в 115 мл метанола; добавляли 15 мл воды и затем 11,0 г K2CO3. Спустя 2 часа реакционную смесь гасили 6,5 мл уксусной кислоты и затем концентрировали в вакууме. Остаток обрабатывают 100 мл воды и затем экстрагировали последовательно 150 мл и 100 мл этилацетата. Объединенные органические слои промывали 100 мл насыщенного водного раствора гидрокарбоната натрия, затем 100 мл 10% водного раствора NaCl и далее 100 мл 20% водного раствора NaCl. Этилацетатный слой сушили над MgSO4, фильтровали и концентрировали. Хроматография (Isco CombiFlash® system, Analogix RS 300 г колонка, этилацетат:дихлорметан - 1:9 в течение 5 минут, затем изменение до 12:88 в течение 35 минут и элюирование растворителем указанного состава в течение 10 минут) приводило к получению указанного в заголовке соединения (10,3 г, 91%(масс.), 76%).
Пример 13C
трет-Бутил-({(1R,2R,4R,6R)-1-метил-4-[(триэтилсилил)окси]-7-оксабицикло[4.1.0]гепт-2-ил}окси)дифенилсилан
Соединение примера 13B (16,0 г, 42 ммоль) растворяли в 20 мл диметилформамида; к полученному раствору добавляли 7,1 г имидазола (100 ммоль, 2,5 экв.) и затем 10,5 мл хлортриэтилсилана. Полученную смесь перемешивали в течение ночи при температуре окружающей среды, затем разбавляли водой и экстрагировали этилацетатом. Органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Смесь концентрировали в вакууме, остаток очищали флэш-хроматографией (элюирование с градиентом: дихлорметан в гексанах - от 50% до 100%), получая указанное в заголовке соединение (14,0 г, 90%).
Пример 13D
(1R,3R,5R)-3-{[трет-бутил(дифенил)силил]окси}-2-метилен-5-[(триэтилсилил)окси]циклогексанол
2,2,6,6-Тетраметилпиперидин (47 мл) растворяли в 250 мл толуола; добавляли н-бутиллитий (2,5 M в гексанах, 113 мл) и полученную смесь перемешивали в течение 40 минут. Добавляли хлорид диэтилaлюминия (1,0 M в гексанах, 289 мл) и перемешивание продолжают в течение 1 часа. Раствор охлаждали до 0ºC; соединение примера 13C (34,9 г, 70 ммоль) растворяли в 100 мл толуола и добавляли к смеси в течение 5 минут. Смесь перемешивали в течение 3 часов, медленно нагревая до температуры окружающей среды. Реакцию гасили добавлением насыщенного водного раствора NH4Cl; после перемешивания в течение 5 минут к реакционной смеси добавляли 2N водный раствор HCl для доведения значения рН до ~1,5. Полученную смесь экстрагировали этилацетатом; органическую фазу промывали последовательно водой и раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме для получения указанного в заголовке соединения, которое используют в последующих стадиях без дополнительной очистки.
Пример 13E
(3R,5R)-3-{[трет-бутил(диметил)силил]окси}-5-{[трет-бутил(дифенил)силил]окси}-4-метиленциклогексанон
Соединение примера 13D (8,5 г, 17 ммоль) растворяли в 50 мл дихлорметана; к раствору добавляли 4,4 мл 2,6-лутидина и полученный раствор охлаждали до 0ºC. Добавляли трет-бутилдиметилсилилтрифторметансульфонат (5,9 мл) и полученную смесь перемешивали в течение 1 часа. Реакцию гасили добавлением насыщенного водного раствора NH4Cl, растворители удаляли в вакууме и остаток распределяли между этилацетатом и водой. Органическую фазу промывали 1N водной HCl, затем раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме; остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 2% до 5%). Продукт (9,3 г) растворяли в 75 мл этанола; добавляли п-толуолсульфонат пиридиния (390 мг) и смесь перемешивали при температуре окружающей среды в течение 1 часа. Растворители удаляли в вакууме; остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 9% до 17%). Навеску полученного продукта (5,8 из 6,8 г) растворяли в 30 мл дихлорметана; добавляли 3,0 г NaHCO3 и смесь охлаждали до 0ºC. Добавляли реагент Десса-Мартина (5,9 г) и перемешивание продолжают в течение 2 часов. Растворители удаляли в вакууме, и остаток распределяли между этилацетатом и водой. Органическую фазу промывали 1N водной HCl, затем раствором соли и сушили над Na2SO4. Органическую фазу пропускают через рыхлый слой силикагеля и концентрировали в вакууме, получая указанное в заголовке соединение (5,5 г, 71%).
Пример 13F
Этил-(2E)-((3R,5R)-3-{[трет-бутил(дифенил)силил]окси}-5-гидрокси-4-метиленциклогексилиден)ацетат
Раствор диизопропиламина (1,2 мл, 9,4 ммоль) в 10 мл тетрагидрофурана охлаждали до -78ºC; к раствору добавляли н-бутиллитий (2,5 M в гексанах, 3,6 мл) и спустя 5 минут этилтриметилсилилацетат (1,7 мл). Полученный раствор перемешивали при -78ºC в течение 30 минут и затем в течение 5 минут добавляли раствор соединения примера 13E (2,0 г, 4 ммоль) в 10 мл тетрагидрофурана. Перемешивание продолжают в течение 4 часов. Реакцию гасили добавлением насыщенного водного раствора NH4Cl; смесь нагревали до температуры окружающей среды и распределяли между этилацетатом и водой. Органическую фазу промывали 1N водной HCl и раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме; остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 5% до 10%). Продукт (1,9 г) растворяли в 17 мл этанола; добавляют 1,3 мл концентрированной HCl и полученный раствор перемешивали в течение ночи при температуре окружающей среды. Растворители удаляли в вакууме, остаток переносили в насыщенный водный раствор гидрокарбоната натрия и экстрагировали этилацетатом. Органическую фазу концентрировали в вакууме и очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: дихлорметан в ацетонитриле - от 2% до 3%). Указанное в заголовке соединение выделяют в форме более медленно элюирующегося вещества (0,62 г).
Пример 13G
Этил-(2Z)-((3R,5S)-3-{[трет-бутил(дифенил)силил]окси}-5-фтор-4-метиленциклогексилиден)ацетат
Соединение примера 13F (96 мг, 0,22 ммоль) растворяли в 2 мл дихлорметана, и полученный раствор охлаждали до -78ºC. К смеси добавляли трифторид(диэтиламино)серы (DAST, 0,12 мл); смесь перемешивали в течение 30 минут, затем гасили добавлением насыщенного водного раствора гидрокарбоната натрия. Смесь нагревали до температуры окружающей среды; органическую фазу сушили над Na2SO4. Растворители удаляли в вакууме; остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование: 10% этилацетат в гексанах), получая указанное в заголовке соединение (42 мг, 44%).
Пример 13H
(2Z)-2-((3R,5S)-3-{[трет-бутил(дифенил)силил]окси}-5-фтор-4-метиленциклогексилиден)этанол
Соединение примера 13G (40 мг, 0,09 ммоль) растворяли в 1 мл смеси дихлорметан/толуол (1:1) и полученную смесь охлаждали до -78ºC. К смеси добавляли гидрид диизобутилалюминия (1M в гексанах, 0,36 мл) и полученную смесь перемешивали в течение 20 минут. Реакцию гасили добавлением воды; смесь нагревали до температуры окружающей среды и затем подкисляют до pH~2 добавлением концентрированной HCl. Полученную смесь экстрагировали этилацетатом; органические фракции промывали раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме; остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование: 17% этилацетат в гексанах), получая указанное в заголовке соединение (25 мг, 68%).
Пример 13I
[(2Z)-2-((3R,5S)-3-{[трет-бутил(дифенил)силил]окси}-5-фтор-4-метиленциклогексилиден)этил](дифенил)фосфин-оксид
Соединение примера 13H (180 мг, 0,44 ммоль) растворяли в 2 мл гексанов; добавляли 85 мг (0,28 ммоль) трифосгена и смесь охлаждали до 0ºC. К полученной смеси по каплям в течение 2 минут добавляли триэтиламин (0,22 мл, 1,5 ммоль); реакционную смесь перемешивали в течение 30 минут и затем нагревали до температуры окружающей среды в течение 1 часа. Добавляли дополнительное количество гексанов (3 мл) и реакционную смесь промывали последовательно охлажденной 3% водной HCl, водой и раствором соли. Органическую фазу сушили над Na2SO4; растворители удаляли в вакууме, получая сырой аллилхлорид.
Дифенилфосфин (0,72 г, 7 экв.) растворяли в 2 мл тетрагидрофурана, и раствор охлаждали до 0ºC. Добавляли раствор н-бутиллития (2,5M в гексанах) и смесь перемешивали в течение 5 минут. Полученный ранее промежуточный аллилхлорид растворяли в 1 мл тетрагидрофурана и охлаждали до -60ºC. Анионовый раствор добавляли в течение 5 минут и полученную смесь перемешивали при -60ºC в течение 1 часа. Реакцию гасили водой; смесь нагревали до температуры окружающей среды и экстрагировали этилацетатом. Органическую фазу промывали 1N водной HCl и раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме; остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 40% до 50%), получая указанное в заголовке соединение (200 мг, 76%).
Пример 13J
(1R,3aR,7аR)-1-{(1R)-1,5-диметил-5-[(триметилсилил)окси]гексил}-7a-метилоктагидро-4H-инден-4-он
Указанное в заголовке соединение получают в соответствии с методиками, описанными в публикации Kiegiel, Wovkulich, Uskokovic; Tetrahedron Lett. 1991, 32(43), 6057.
Пример 13K
(1R,3S,5Z,7E,17β)-3-фтор-17-[(1R)-5-гидрокси-1,5-диметилгексил]-2-метилен-9,10-секоэстра-5,7-диен-1-ол
Примечание: представленную далее последовательность синтеза проводили в затемненном вытяжном шкафу. Соединение примера 13I (20 мг, 0,034 ммоль) объединяли с соединением примера 13J (12 мг, 0,056 ммоль) в 1 мл толуола; растворитель удаляли в вакууме для сушки реагентов. Остаток переносили в 1 мл сухого тетрагидрофурана и охлаждали до -78ºC. К смеси по каплям добавляли раствор бис(триметилсилил)амида лития (1,0M в тетрагидрофуране; 0,1 мл), что приводило к появлению желтовато-оранжевой окраски, которая исчезает через 20 минут. Полученную смесь перемешивали при -78ºC в течение 1 часа, затем нагревали до 0ºC и перемешивали при данной температуре в течение 30 минут. Реакцию гасили добавлением 1 мл 1N водного раствора NH4Cl и полученную смесь экстрагировали этилацетатом. Органический экстракт промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток растворяли в 1 мл 1N фторида тетра-н-бутиламмония в тетрагидрофуране. После перемешивания в течение ночи при температуре окружающей среды реакционную смесь распределяли между этилацетатом и водой. Органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 20% до 50%), получая указанное в заголовке соединение (6 мг).1H ЯМР (500 МГц, CD2Cl2) δ м.д. 6,25-6,41 (м, 1H), 5,98-6,13 (м, 1H), 5,16-5,26 (м, 2H), 4,91 (д, 1H), 4,02-4,22 (м, 1H), 2,87-3,05 (м, 1H), 2,60 (дд, J=12,8, 4,6 Гц, 1H), 2,42-2,53 (м, 3H), 2,40 (д, J=15,3 Гц, 1H), 2,13-2,32 (м, 4H), 2,00 (с, 1H), 1,80-1,95 (м, 2H), 1,66-1,79 (м, 2H), 1,55-1,66 (м, 4H), 1,27-1,45 (м, 6H), 1,10-1,25 (м, 9H), 0,91-1,01 (м, 3H); MС (+DCI) m/z 436 (M+NH4)+.

Пример 14
(2S)-2-[(1R,3R,7E,17β)-1,3-дигидрокси-2-метилен-9,10-секоэстра-5,7-диен-17-ил]пропил-2-гидрокси-2-метилпропаноат
Соединение примера 6C (20 мг, 0,034 ммоль) объединяли с 11 мг (3 экв.) 2-гидроксиизомясляной кислоты и 25 мг (2,8 экв.) трифенилфосфина в 1,5 мл толуола; к смеси добавляли 22 мг (2,8 экв.) ди-трет-бутилазодикарбоксилата, полученный раствор продували аргоном, нагревали до 70ºC и выдерживали при данной температуре в течение 1,5 часа. Смесь концентрировали и очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 5% до 12%). Сырой полупродукт (21 мг из полученных 25 мг) растворяли в 2 мл 1N раствора фторида тетра-н-бутиламмония в тетрагидрофуране. После перемешивания в течение 3 часов при температуре окружающей среды реакционную смесь распределяли между этилацетатом и водой. Органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 45% до 68%), получая указанное в заголовке соединение (5,1 мг).1H ЯМР (500 МГц, CD2Cl2) δ м.д. 6,33 (д, J=11,3 Гц, 1H), 5,90 (д, J=11,3 Гц, 1H), 5,06 (д, J=5,8 Гц, 1H), 4,36-4,52 (м, 2H), 4,16 (дд, J=10,7, 3,4 Гц, 1H), 3,92 (дд, J=10,7, 7,0 Гц, 1H), 2,83 (дд, J=12,1, 3,8 Гц, 1H), 2,76 (дд, J=13,1, 4,6 Гц, 1H), 2,55 (дд, J=13,1, 4,0 Гц, 1H), 2,21-2,35 (м, 2H), 2,02-2,11 (м, 2H), 1,95-2,04 (м, 2H) 1,85-1,94 (м, 1H), 1,64-1,81 (м, 4H), 1,49-1,65 (м, 4H), 1,37-1,45 (м, 9H), 1,04 (д, J=6,7 Гц, 3H), 0,58 (с, 3H); MС (+DCI) m/z 364 (M+NH4)+.

Пример 15
(1R,3R,7E,17β)-17-[(1R,4R)-5-гидрокси-1,4,5-триметилгексил]-9,10-секоэстра-5,7-диен-1,3-диол
Пример 15A
трет-Бутил-(диметил)-[((1R,3aR,4S,7aR)-7a-метил-1-{(1R,2E,4R)-1,4,5-триметил-5-[(триметилсилил)окси]гекс-2-енил}октагидро-1H-инден-4-ил)окси]силан и (3R,4E,6R)-6-((1R,3aR,4S,7aR)-4-{[трет-бутил(диметил)силил]окси}-7a-метилоктагидро-1H-инден-1-ил)-2,3-диметилгепт-4-ен-2-ол
Соединение примера 10E (200 мг, 0,61 ммоль) растворяли в 2 мл дихлорметана; к раствору добавляли 100 мг 4Å молекулярных сит, затем 15 мг перрутената тетрапропиламмония и 75 мг 4-метилморфолин-N-оксида. Смесь перемешивали в течение 2 часов при температуре окружающей среды и фильтровали через диатомовую землю для удаления твердых веществ. Сырой продукт очищали хроматографией на силикагеле, используя Analogix IntelliFlash™ 40 (элюирование с градиентом: этилацетат в гексанах - от 5% до 30%). Соединение примера 10C (200 мг, 0,63 ммоль) объединяли с полученным альдегидом в соответствии с методикой реакции связывания, описанной в публикации Kutner et al., J. Org. Chem. 1988, 53, 3450-3457. Указанный в заголовке продукт выделяют в виде смеси триметилсилилового эфира (фракция A) и гидроксила (фракция B) после хроматографической очистки с использованием Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 0% до 50%). Полученные продукты объединяли и использовали в следующей стадии.
Пример 15B
(1R,3aR,4S,7aR)-1-[(1R,2E,4R)-5-гидрокси-1,4,5-триметилгекс-2-енил]-7a-метилоктагидро-1H-инден-4-ол
Смешанные фракции примера 15A растворяли в 2 мл 1N раствора фторида тетра-н-бутиламмония в тетрагидрофуране; полученный раствор перемешивали при температуре окружающей среды в течение 2 часов, нагревали до 60ºC и выдерживали при данной температуре в течение ночи, затем нагревали до 80ºC и выдерживали при данной температуре в течение 3 часов. Реакцию гасили водой и экстрагировали этилацетатом. Органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 10% до 40%), получая указанное в заголовке соединение (72 мг).
Пример 15C
(1R,3aR,4S,7аR)-1-[(1R,4R)-5-гидрокси-1,4,5-триметилгексил]-7a-метилоктагидро-1H-инден-4-ол
Соединение примера 15B (89 мг) растворяли в уксусной кислоте и гидрировали над 10% платиной на углероде. После продувки смеси азотом ее фильтровали через рыхлый слой диатомовой земли для удаления катализатора и концентрировали в вакууме, получая продукт, который используют в следующей стадии без дополнительной очистки.
Пример 15D
(1R,3aR,7аR)-7a-метил-1-{(1R,4R)-1,4,5-триметил-5-[(триметилсилил)окси]гексил}октагидро-4H-инден-4-он
Соединение примера 15C (73 мг, 0,25 ммоль) растворяли в 1 мл дихлорметана; к раствору добавляли 150 мг дихромата пиридиния и 10 мг п-толуолсульфоната пиридиния и полученную смесь перемешивали в течение ночи при температуре окружающей среды. Растворители удаляли в вакууме; остаток переносили в этилацетат и фильтровали через рыхлый слой диатомовой земли. Растворители удаляли в вакууме, и остаток растворяли в 1 мл диметилформамида. К полученному раствору добавляли 1-(триметилсилил)имидазол (100 мг), раствор нагревали до 50ºC и выдерживали при данной температуре в течение 3 часов. Растворители удаляли в вакууме и остаток очищали хроматографией на Analogix IntelliFlash 280™, (элюирование с градиентом: этилацетат в гексанах - от 10% до 40%), получая указанное в заголовке соединение (72 мг, 79%).
Пример 15E
(1R,3R,7E,17β)-17-[(1R,4R)-5-гидрокси-1,4,5-триметилгексил]-9,10-секоэстра-5,7-диен-1,3-диол
Примечание: представленную далее последовательность синтеза проводили в затемненном вытяжном шкафу. Соединение примера 9A (57 мг, 0,1 ммоль) объединяли с соединением примера 15D (36 мг, 0,1 ммоль) в 1 мл толуола; растворитель удаляли в вакууме для тщательной сушки реагентов. Остаток переносили в 1,5 мл сухого тетрагидрофурана и охлаждали до -78ºC. К полученной смеси по каплям добавляли раствор бис(триметилсилил)амида лития (1,0M в тетрагидрофуране; 0,1 мл), что приводило к появлению желтовато-оранжевой окраски, которая исчезает через 20 минут. К смеси добавляли дополнительное количество бис(триметилсилил)амида лития (0,05 мл); полученную смесь перемешивали при -78ºC в течение 1 часа, затем нагревали до 0ºC и перемешивали при данной температуре в течение 30 минут. Реакцию гасили добавлением 1 мл 1N водного раствора NH4Cl и полученную смесь экстрагировали этилацетатом. Органический экстракт промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме и остаток растворяли в 1 мл 1N раствора фторида тетра-н-бутиламмония в тетрагидрофуране. Реакционную смесь выдерживали в при 50ºC в течение 5 часов, затем распределяли между этилацетатом и водой. Органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 0% до 100%, затем 100% этилацетат), получая указанное в заголовке соединение (13 мг).1H ЯМР (500 МГц, CD2Cl2) δ м.д. 6,28 (д, J=11,3 Гц, 1H), 5,86 (д, J=11,3 Гц, 1H), 3,90-4,15 (м, 2H), 2,75-2,87 (м, 1H), 2,75-2,86 (м, 1H), 2,67 (дд, J=13,1, 4,0 Гц, 1H), 2,45 (дд, J=13,1, 3,4 Гц, 1H), 2,10-2,26 (м, 2H), 1,97-2,06 (м, 2H) 1,81-1,94 (м, 2H), 1,43-1,81 (м, 9H), 1,23-1,39 (м, 3H), 1,06-1,20 (м, 8H), 0,81-1,00 (м, 9H), 0,55 (с, 3H); MС (+DCI) m/z 436 (M+NH4)+.

Пример 16
(1R,3R,5E,7E,17β)-17-[(1R)-5-гидрокси-1,5-диметилгексил]-3-(гидроксиметил)-2-метилен-9,10-секоэстра-5,7-диен-1-ол
Пример 16A
(1R,2R,4R,6S)-4-изопропенил-1-метил-7-оксабицикло[4.1.0]гептан-2-ол
(S)-Карвон-оксид (75,7 г, 455 ммоль; полученный в соответствии с методикой, описанной в публикации Klein and Ohloff, Tetrahedron 1963, 19, 1091-1099) растворяли в 750 мл тетрагидрофурана и охлаждали до -78ºC. К раствору в течение 71 минуты добавляли L-Selectride® (1M в тетрагидрофуране, 708 мл, 708 ммоль). После перемешивания в течение 4 часов реакцию гасили дополнительными 250 мл метанола и оставляли на ночь для нагревания до температуры окружающей среды. Смесь кипятят с обратным холодильником (примерно 63ºC) в течение 7 часов, затем охлаждали до температуры окружающей среды и перемешивали в течение ночи (16,5 часов включая охлаждение). Смесь концентрировали в вакууме и переупаривали с 250 мл метанола. Сырой продукт промывали дважды 300 мл воды. Объединенные промывные водные растворы экстрагировали (3×250 мл) трет-бутилметиловым эфиром. Экстракты трет-бутилметилового эфира и еще 500 мл трет-бутилметилового эфира добавляли к сырому продукту. Раствор трет-бутилметилового эфира промывали водой (3×100 мл), 275 мл раствора соли и затем сушили над MgSO4. После фильтрации раствор концентрировали в вакууме, растворяли в 450 мл тетрагидрофурана и охлаждали до 3ºC. К раствору добавляли 300 мл 10% NaOH раствора, затем 300 мл 30% раствора H2O2. Полученный раствор перемешивали в течение ночи при температуре окружающей среды. После разделения образовавшихся двух слоев к органическому слою добавляли 500 мл тетрагидрофурана. Раствор тетрагидрофурана затем промывали (2×250 мл) 18% водным раствором NaHSO3. Тетрагидрофуран удаляли в вакууме и к концентрату добавляли 1000 мл трет-бутилметилового эфира. Раствор трет-бутилметилового эфира промывали (3×100 мл) водой, затем 300 мл раствора соли и сушили над MgSO4. MgSO4 отфильтровывали, и фильтрат упаривали досуха в высоком вакууме. Продукт очищали на колонке с силикагелем (Silica Gel 60, 2 кг, элюирование с градиентом: смесь гексаны:трет-бутилметиловый эфир - от 4:1 до 13:7), получая указанное в заголовке соединение (11,1 г, 14,5%).
Пример 16B
трет-Бутил-{[(1S,2R,4S,6S)-4-изопропенил-1-метил-7-оксабицикло[4.1.0]гепт-2-ил]окси}диметилсилан
Соединение примера 16A (11,0 г, 65 ммоль) в 66 мл диметилформамида обрабатывали 5,37 г (1,2 экв.) имидазола с последующей обработкой 11,2 г (1,14 экв.) хлоридом трет-бутилдиметилсилила. Реакционную смесь перемешивали в течение 20 часов при температуре окружающей среды. Полученную смесь смешивали с 200 мл воды и образовавшиеся слои разделяли. Водный слой экстрагировали (3×120 мл) трет-бутилметиловым эфиром. Органические слои объединяли и промывали 10% водным раствором NaCl (2×120 мл). Раствор трет-бутилметилового эфира сушили над MgSO4, фильтровали и концентрировали в вакууме. Продукт очищали на колонке с силикагелем (Silica Gel 60, 2 мг, элюирование с градиентом: смесь гексаны:трет-бутилметиловый эфир - от 19:1 (8 л) до 9:1 (4 л)), получая указанное в заголовке соединение (16,2 г, 91%).
Пример 16C
(1S,3S,5R,6S)-5-{[трет-бутил(диметил)слил]окси}-6-метил-7-оксабицикло[4.1.0]гепт-3-илацетат
Соединение примера 16B (14,0 г, 50 ммоль) растворяли в 405 мл дихлорметана и 85 мл метанола; к полученному раствору добавляли 2,15 г (25,6 ммоль, 0,52 экв.) NaHCO3. Смесь охлаждали до температуры ниже -70ºC и через полученную смесь барботировали O3 до тех пор, пока смесь не приобретет голубую окраску (примерно 35 минут). Избыток O3 удаляли барботированием N2 через реакционную смесь при температуре ниже -70ºC до исчезновения окраски. Реакционную смесь нагревали до температуры окружающей среды; твердый NaHCO3 отфильтровывали и промывали 60 мл дихлорметана. Объединенные фильтрат и промывной раствор концентрировали до вязкого масла, которое переупаривали с 425 мл бензола для удаления остатков метанола. Полученное масло растворяли в 400 мл дихлорметана и 80 мл пиридина. Раствор охлаждали до -10ºC и к полученному раствору в течение 10 минут добавляли 4-нитробензоилхлорид (11,6 мг, 61,4 ммоль, 1,24 экв.), растворенный в 80 мл дихлорметана. В процессе добавления температуру реакционной смеси поддерживали на уровне ниже -6ºC. Реакционную смесь охлаждали до температуры ниже -10ºC и перемешивали при данной температуре в течение ночи. Реакционную смесь нагревали до 44ºC и перемешивали в течение 3 часов. После охлаждения до температуры окружающей среды реакционную смесь концентрировали в вакууме, и остаток растворяли в 600 мл этилацетата. Этилацетатный раствор промывали водой (4×100 мл) и затем концентрировали до густой взвеси, которую растирали со 100 мл гексанов. Твердые вещества отфильтровывали и промывали гексанами (3×65 мл). Объединенные гексановые фильтраты и промывной раствор концентрировали и фильтровали. К фильтрату добавляли 400 мл гексанов; полученный раствор промывали водой (3×100 мл), затем 100 мл 10% раствора NaCl, сушили над MgSO4, фильтровали и концентрировали. Полученный сырой продукт очищали хроматографией на силикагеле (Silica Gel 60, 2 кг, элюирование: смесь гексаны:трет-бутилметиловый эфир = 9:1 (24,0 л)), получая указанное в заголовке соединение (8,35 г, 56%).
Пример 16D
(1S,3S,5R,6S)-5-{[трет-бутил(диметил)силил]окси}-6-метил-7-оксабицикло[4.1.0]гептан-3-ол
Соединение примера 16C (9,20 г, 30,6 ммоль) растворяли в 105 мл метанола. К раствору добавляли K2CO3 (10,1 г, 72,1 ммоль, 2,35 экв.) и полученную реакционную смесь перемешивали при температуре окружающей среды в течение 2 часов. К реакционной смеси добавляли 6,60 мл (115 ммоль, 3,77 экв.) уксусной кислоты; после перемешивания в течение 10 минут твердые вещества отфильтровывали и промывали 40 мл метанола. Объединенные фильтраты и промывные растворы упаривали до остатка, который смешивали со 106 мл воды. Слои разделяли, и водный слой экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали раствором соли (2×45 мл), сушили над MgSO4, фильтровали и концентрировали, получая указанное в заголовке соединение (7,72 г, 98%).
Пример 16E
трет-Бутил(диметил)-({(1S,2R,4S,6S)-1-метил-4-[(триэтилсилил)окси]-7-оксабицикло[4.1.0]гепт-2-ил}окси)силан
Соединение примера 16D (4,71 г, 18,2 ммоль) и имидазол (1,76 г, 25,6 ммоль, 1,4 экв.) растворяли в 36 мл диметилформамида. К полученному раствору добавляли хлортриэтилсилан (3,34 г, 21,9 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 2 часов. К реакционной смеси добавляли 125 мл воды. Получают два слоя жидкостей, отделяют примерно 6 г верхнего слоя. Затем нижний водный слой экстрагировали трет-бутилметиловым эфиром (3×100 мл). Объединенные органические слои промывали 125 мл 10% водного раствора NaCl, сушили над MgSO4, фильтровали и концентрировали в вакууме, получая указанное в заголовке соединение (6,71 г, 99%).
Пример 16F
(1S,3R,5S)-3-{[трет-бутил(диметил)силил]окси}-2-метилен-5-[(триэтилсилил)окси]циклогексанол
Тетраметилпиперидин (9,66 г, 67,7 ммоль) растворяли в 76 мл бензола и охлаждали до температуры ниже 0ºC. К бензольному раствору добавляли н-бутиллитий (2,5M в гексане, 27,5 мл, 68,8 ммоль), поддерживая температуру ниже 2,5ºC. Полученный раствор перемешивали, поддерживая температуру ниже 0ºC. К реакционной смеси добавляли Et2AlCl (1,8M в толуоле, 39 мл, 70,2 ммоль), поддерживая температуру смеси ниже 3ºC, с последующей промывкой 5,0 мл бензола. Полученный раствор перемешивали при температуре ниже 0ºC в течение 30 минут. К реакционной смеси при температуре ниже 1ºC в течение 10 минут добавляли соединение примера 16E (6,36 г, 17,1 ммоль), растворенное в 25 мл бензола. 5,0 мл бензола используют в качестве промывного раствора. Реакционную смесь перемешивали при температуре ниже 0ºC в течение 2,7 часов. Реакцию гасили, реакционную смесь выливали в 441 г 17,2% (масс.) раствора NH4Cl (охлажденного до температуры ниже 5ºC). К смеси медленно добавляли 104 г 10% (масс.) раствора HCl, и значение pH доводили до 2,0. Слои разделяли, и водный слой экстрагировали этилацетатом (4×100 мл). Объединенные органические слои промывали водой (3×100 мл), затем 100 мл 7,8% (масс.) раствора NaHCO3 и 100 мл раствора соли, после чего сушили над MgSO4. Фильтрация с последующим упариванием фильтрата в вакууме приводило к получению указанного в заголовке соединения (6,13 г, 96%).
Пример 16G
трет-Бутил({(1R,5S)-2-(йодметил)-5-[(триэтилсилил)окси]циклогекс-2-ен-1-ил}окси)диметилсилан
Соединение примера 16F (2,76 г, 7,4 ммоль) и 4- диметиламинопиридин (1,35 г, 11,1 ммоль) растворяли в 74 мл дихлорметана, и полученный раствор охлаждали до 0ºC. К раствору добавляли метансульфонилхлорид (0,72 мл, 9,25 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 4,5 часов. После разбавления смеси дополнительным количеством дихлорметана реакционную смесь дважды промывали 10% NaCl, затем сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток растворяли в 74 мл ацетона и к раствору добавляли NaHCO3 (0,5 г), Na2SO3 (0,5 г) и NaI (4,5 г, 30 ммоль). Реакционную смесь нагревали до 55ºC и выдерживали при указанной температуре в течение 90 минут, затем перемешивали в течение ночи при температуре окружающей среды. Смесь разбавляли 500 мл трет-бутилметилового эфира, промывали (2×250 мл) смесью (8:1:1) 10% NaCl:1N NaHCO3:1M Na2SO3, затем раствором соли. Полученный раствор сушили над MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле (300 мл SiO2, элюирование: смесь 1:1 дихлорметан:гексаны) приводила к получению указанного в заголовке соединения (2,52 г, 70%).
Пример 16H
(1S,3R,5R)-3-{[трет-бутил(диметил)силил]окси}-5-(гидроксиметил)-4-метиленциклогексанол
Соединение примера 16G (2,5 г, 5,2 ммоль) растворяли в 20 мл воды и 30 мл тетрагидрофурана. К полученному раствору добавляли порошкообразный индий (0,90 г), затем формальдегид (37% водный раствор, 1,6 мл, 21 ммоль). К реакционной смеси добавляли две дополнительные порции индия (0,3 г каждый) для завершения реакции. Реакционную смесь разбавляли 180 мл воды и экстрагировали этилацетатом (2×250 мл). Объединенные органические слои промывали последовательно 5% NaHCO3 и раствором соли, затем сушили над MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле (200 мл SiO2, элюирование с градиентом: смесь этилацетат:гексаны от 1:1 до 3:1) приводила к получению указанного в заголовке соединения (0,97 г, 68%).
Пример 16I
(1S,3R,5R)-3-{[трет-бутил(диметил)силил]окси}-5-({[трет-бутил(диметил)силил]окси}метил)-4-метиленциклогексанол
Соединение примера 16H (574 мг, 2,1 ммоль) растворяли в 35 мл дихлорметана и добавляли трет-бутилдиметилсилилимидазол (0,54 мл, 2,8 ммоль). После перемешивания в течение ночи реакционную смесь разбавляли дихлорметаном и дважды промывали 10% NaCl, затем сушили над MgSO4, фильтровали и концентрировали в вакууме. После повторного проведения реакции с использованием 340 мг исходного диола объединенную смесь сырых продуктов очищали хроматографией на силикагеле (200 мл SiO2, элюирование с градиентом: этилацетат:гексаны от 1:4 до 1:0), получая указанное в заголовке соединение (940 мг), загрязненное небольшим количеством региоизомерного бис-TBS эфира.
Пример 16J
(3R,5R)-3-{[трет-бутил(диметил)силил]окси}-5-({[трет-бутил(диметил)силил]окси}метил)-4-метиленциклогексанон
Соединение примера 16I (0,93 г, 2,4 ммоль) растворяли в 30 мл дихлорметана и обрабатывали 1,02 г (2,4 ммоль) периодинана Десса-Мартина. Спустя 2,5 часа реакционную смесь разбавляли дополнительным количеством дихлорметана и промывали 100 мл смеси 4:1 1M NaHCO3:1M Na2SO3, затем 100 мл смеси 9:1 20% NaCl:1M NaHCO3. Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме, затем очищали хроматографией на силикагеле (200 мл SiO2; элюирование: смесь этилацетат:гексаны 1:4), получая указанное в заголовке соединение (794 мг, 85%).
Пример 16K
Этил-(2E)-[(3R,5R)-3-{[трет-бутил(диметил)силил]окси}-5-({[трет-бутил(диметил)силил]окси}метил)-4-метиленциклогексилиден]ацетат
н-Бутиллитий (2,5M в гексанах, 1,6 мл, 4 ммоль) добавляли в раствор диизопропиламина (0,56 мл, 4 ммоль) в 5 мл тетрагидрофурана при -78ºC. Спустя 30 минут добавляли этилтриметилсилилацетат (0,73 мл, 4 ммоль). После перемешивания в течение 30 минут к смеси добавляли раствор соединения примера 16J (787 мг, 2 ммоль) в 8 мл тетрагидрофурана с последующим добавлением 2 мл тетрагидрофурана, полученных после споласкивания колбы. Спустя 2 часа реакцию гасили добавлением 10 мл насыщенного водного раствора NH4Cl. После нагревания до температуры окружающей среды смесь выливали в 100 мл 20% раствора NaCl, затем экстрагировали трет-бутилметиловым эфиром (2×100 мл). Объединенные экстракты трет-бутилметилового эфира сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле (300 мл SiO2, элюирование с градиентом: дихлорметан:гексаны от 1:1 до 1:0), получая 538 мг (58% выход) смеси (~4:1) изомеров указанного в заголовке соединения (538 мг, 58%).
Пример 16L
(2E)-2-[(3R,5R)-3-{[трет-бутил(диметил)силил]окси}-5-({[трет-бутил(диметил)силил]окси}метил)-4-метиленциклогексилиден]этанол
Соединение примера 16K (589 мг, 1,3 ммоль) растворяли в 20 мл толуола и 10 мл дихлорметана и охлаждали на бaне со смесью сухой лед/ацетон. В раствор по каплям добавляли гидрид диизобутилалюминия (1M в гексанах, 5,8 мл, 5,8 ммоль). Спустя 1 час реакцию гасили добавлением 20 мл 20% водного раствора тартрата натрия-калия. К полученной смеси добавляли 25 мл воды и затем 6 мл 2N раствора HCl. Полученную смесь экстрагировали дихлорметаном (×3). Объединенные экстракты промывали 10% водным раствором NaCl, сушили над MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле (70 мл SiO2, элюирование: дихлорметан) приводила к получению указанного в заголовке соединения (294 мг, 55%), загрязненного ~20% олефинового изомера.
Пример 16M
{(2E)-2-[(3R,5R)-3-{[трет-бутил(диметил)силил]окси}-5-({[трет-бутил(диметил)силил]окси}метил)-4-метиленциклогексилиден]этил}(дифенил)фосфин-оксид
н-Бутиллитий (2,5M в гексанах, 0,62 мл, 1,55 ммоль) добавляли в раствор дифенилфосфина (0,30 мл, 1,73 ммоль) в 3 мл тетрагидрофурана при 0ºC, получая раствор дифенилфосфида лития.
Соединение примера 16L (241 мг, 0,58 ммоль) растворяли в 6,2 мл тетрагидрофурана при 0ºC. Добавляли н-бутиллитий (2,5M в гексанах, 0,24 мл, 0,61 ммоль) с последующим добавлением раствора толуолсульфонилхлорида (124 мг, 0,65 ммоль) в 3 мл тетрагидрофурана. К смеси по каплям добавляли раствор дифенилфосфида лития до тех пор, пока раствор не приобретет стойкое красное окрашивание. После перемешивания в течение дополнительного 1 часа реакцию гасили водой и затем концентрировали в вакууме. Остаток растворяли в 12 мл дихлорметана и добавляли 10% водный раствор пероксида водорода (3 мл) при 0ºC. Спустя 1 час реакционную смесь выливали в охлажденный 1M раствор Na2SO3 и дважды экстрагировали дихлорметаном. Объединенные органические слои промывали 20% водным раствором NaCl, сушили над MgSO4, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле (120 мл SiO2 (элюирование с градиентом: смесь этилацетат:гексаны от 1:4 до 1:1) приводила к получению указанного в заголовке соединения (250 мг, 72%).
Пример 16N
(1R,3R,5E,7E,17β)-17-[(1R)-5-гидрокси-1,5-диметилгексил]-3-(гидроксиметил)-2-метилен-9,10-секоэстра-5,7-диен-1-ол
Примечание: представленную далее последовательность синтеза проводили в затемненном вытяжном шкафу. Соединение примера 16M (40 мг, 0,07 ммоль) объединяли с соединением примера 13J (32 мг, 0,09 ммоль) в 1 мл толуола; растворитель удаляли в вакууме для тщательной сушки реагентов. Остаток переносили в 1,5 мл сухого тетрагидрофурана и охлаждали до -78ºC. К смеси по каплям добавляли раствор бис(триметилсилил)амида лития (1,0 M в тетрагидрофуране; 0,1 мл), что приводило к появлению желтовато-оранжевой окраски, которая исчезает через 20 минут. К смеси добавляли дополнительные 0,05 мл бис(триметилсилил)амида лития; полученную смесь перемешивали при -78ºC в течение 1 часа, затем нагревали до 0ºC и перемешивали при данной температуре в течение 30 минут. Реакцию гасили добавлением 1 мл 1N водного раствора NH4Cl и полученную смесь экстрагировали этилацетатом. Органический экстракт промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток растворяли в 1 мл 1N фторида тетра-н-бутиламмония в тетрагидрофуране. После нагрева до 50ºC и выдерживания при данной температуре в течение 4,5 часов реакционную смесь распределяли между этилацетатом и водой. Органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 10% до 100%, далее 100% этилацетат), получая указанное в заголовке соединение (10,2 мг).1H ЯМР (500 МГц, CD2Cl2) δ м.д. 6,20 (д, J=11,3 Гц, 1H), 6,04 (д, J= 11,0 Гц, 1H), 5,10 (с, 1H), 4,88 (с, 1H), 4,21 (дд, J=7,6, 4,6 Гц, 1H), 3,49-3,72 (м, 2H), 2,59-2,73 (м, 1H), 2,57 (дд, J=12,8, 4,6 Гц, 1H), 2,43-2,52 (м, 2H), 2,31 (дд, J=13,6, 6,6 Гц, 1H), 2,23 (дд, J=12,8, 7,9 Гц, 1H), 2,06-2,18 (м, 2H), 1,81-1,93 (м, 1H), 1,66-1,77 (м, 2H), 1,55-1,66 (м, 6H), 1,44-1,54 (м, 2H), 1,31-1,44 (м, 5H), 1,22-1,31 (м, 3H), 1,13-1,22 (м, 9H), 0,92-0,97 (м, 3H); MС (+DCI) m/z 449 (M+NH4)+.

Пример 17
(1R,3R,5E,7E,17β)-3-(гидроксиметил)-17-[(1S)-1-(3-гидрокси-3-метилбутокси)этил]-2-метилен-9,10-секоэстра-5,7-диен-1-ол
Примечание: представленную далее последовательность синтеза проводили в затемненном вытяжном шкафу. Соединение примера 16M (40 мг, 0,07 ммоль) объединяли с соединением примера 8G (35 мг, 0,1 ммоль) в 1 мл толуола; растворитель удаляли в вакууме для тщательной сушки реагентов. Остаток переносили в 1,5 мл сухого тетрагидрофурана и охлаждали до -78ºC. К смеси по каплям добавляли раствор бис(триметилсилил)амида лития (1,0M в тетрагидрофуране; 0,1 мл), что приводило к появлению желтовато-оранжевой окраски, которая исчезает через 20 минут. Добавляли дополнительное количество 0,05 мл бис(триметилсилил)амида лития; полученную смесь перемешивали при -78ºC в течение 1 часа, затем нагревали до 0ºC и перемешивали при данной температуре в течение 30 минут. Реакцию гасили добавлением 1 мл 1N водного раствора NH4Cl, и полученную смесь экстрагировали этилацетатом. Органический экстракт промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток растворяли в 1 мл 1N раствора фторида тетра-н-бутиламмония в тетрагидрофуране. Смесь нагревали до 50ºC и выдерживали при данной температуре в течение 5 часов, затем распределяли между этилацетатом и водой. Органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 10% до 100%, затем 100% этилацетат), получая указанное в заголовке соединение (5 мг).1H ЯМР (500 МГц, CD2Cl2) δ м.д. 6,26 (д, J=11,3 Гц, 1H), 5,86 (д, J=11,3 Гц, 1H), 5,11 (с, 1H), 4,91 (с, 1H), 4,20 (дд, J=7,9, 4,9 Гц, 1H), 3,74-3,90 (м, 1H), 3,49-3,68 (м, 2H), 3,42-3,51 (м, 2H), 3,40-3,52 (м, 2H), 3,26 (дд, J=7,9, 6,1 Гц, 1H), 3,26 (дд, J=7,9, 6,1 Гц, 1H), 2,82 (дд, J=12,2, 4,0 Гц, 1H), 2,63-2,70 (м, 1H), 2,58 (дд, J=12,8, 4,6 Гц, 1H), 2,32-2,46 (м, 2H), 2,22 (дд, J=12,4, 8,4 Гц, 2H), 1,98-2,08 (м, 2H), 1,63-1,76 (м, 6H), 1,53-1,64 (м, 6H), 1,10-1,24 (м, 9H); MС (+DCl) m/z 450 (M+NH4)+.

Пример 18
(1R,3S,5E,7E,17β)-3-фтор-17-[(1R)-5-гидрокси-1,5-диметилгексил]-2-метилен-9,10-секоэстра-5,7-диен-1-ол
Пример 18A
Метил-(2Z)-((3R,5R)-3-{[трет-бутил(дифенил)силил]окси}-5-гидрокси-4-метиленциклогексилиден)ацетат
Раствор диизoпропиламина (1,2 мл, 9,4 ммоль) в 10 мл тетрагидрофурана охлаждали до -78°C; добавляли н-бутиллитий (2,5M в гексанах, 3,6 мл) и спустя 5 минут добавляли метилтриметилсилилацетат (1,7 мл). Полученный раствор перемешивали при -78ºC в течение 30 минут и затем в течение 5 минут добавляли раствор соединения примера 13E (2,0 г, 4 ммоль) в 10 мл тетрагидрофурана. Перемешивание продолжали в течение 4 часов. Реакцию гасили добавлением насыщенного водного раствора NH4Cl; смесь нагревали до температуры окружающей среды и распределяли между этилацетатом и водой. Органическую фазу промывали 1N водной HCl и раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме; остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 5% до 10%). Продукт (1,9 г) растворяли в 17 мл этанола; добавляли 1,3 мл концентрированной HCl и полученный раствор перемешивали в течение ночи при температуре окружающей среды. Растворители удаляли в вакууме, остаток переносили в насыщенный водный раствор гидрокарбоната натрия и экстрагировали этилацетатом. Органическую фазу концентрировали в вакууме и очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: дихлорметан в ацетонитриле - от 2% до 3%), получая указанное в заголовке соединение (0,76 г).
Пример 18B
Метил-(2E)-((3R,5S)-3-{[трет-бутил(дифенил)силил]окси}-5-фтор-4-метиленциклогексилиден)ацетат
Соединение примера 18A (600 мг, 0,23 ммоль) растворяли в 2,4 мл дихлорметана, и полученный раствор охлаждали до -78ºC. К раствору добавляли трифторид(диэтиламино)серы (DAST, 0,6 мл); смесь перемешивали в течение 30 минут и затем гасили добавлением насыщенного водного раствора гидрокарбоната натрия. Смесь нагревали до температуры окружающей среды; органическую фазу сушили над Na2SO4. Растворители удаляли в вакууме; остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 5% до 7%), получая указанное в заголовке соединение и некоторые смешанные фракции.
Пример 18C
(2E)-2-((3R,5S)-3-{[трет-бутил(дифенил)силил]окси}-5-фтор-4-метиленциклогексилиден)этанол
Соединение примера 18B (130 мг, 0,3 ммоль) растворяли в 1 мл смеси дихлорметан/толуол (1:1) и охлаждали до -78ºC. К полученному раствору добавляли гидрид диизобутилалюминия (1M в гексанах, 0,7 мл, 2,5 экв.) и смесь перемешивали в течение 30 минут. Реакцию гасили добавлением метанола с последующим добавлением насыщенного водного раствора NH4Cl; смесь нагревали до температуры окружающей среды и экстрагировали этилацетатом. Органические фракции промывали раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме; остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование: 15% раствор этилацетата в гексанах), получая указанное в заголовке соединение (111 мг, 95%).
Пример 18D
[(2E)-2-((3R,5S)-3-{[трет-бутил(дифенил)силил]окси}-5-фтор-4-метиленциклогексилиден)этил](дифенил)фосфин-оксид
Соединение примера 18C (110 мг, 0,27 ммоль) растворяли в 2 мл гексанов; к раствору добавляли 54 мг (0,18 ммоль) трифосгена, и смесь охлаждали до 0ºC. К смеси по каплям в течение 2 минут добавляли триэтиламин (0,14 мл, 0,95 ммоль); реакционную смесь перемешивали в течение 30 минут и затем нагревали до температуры окружающей среды в течение 1 часа. К смеси добавляли дополнительное количество гексанов (3 мл), и реакционную смесь промывали последовательно охлажденным 3% водным раствором HCl, водой и раствором соли. Органическую фазу сушили над Na2SO4; растворители удаляли в вакууме.
Дифенилфосфин (0,51 г) растворяли в 2 мл тетрагидрофурана, и раствор охлаждали до 0ºC. К полученному раствору добавляли раствор н-бутиллития (2,5M в гексанах, 0,85 мл), и смесь перемешивали в течение 5 минут. В тоже время упомянутый выше промежуточный аллилхлорид растворяли в 1 мл тетрагидрофурана и охлаждали до -60ºC. Анионовый раствор добавляли в течение 5 минут и полученную смесь перемешивали при -60ºC в течение 1 часа. Реакцию гасили водой; смесь нагревали до температуры окружающей среды и экстрагировали этилацетатом. Органическую фазу промывали 1N водным раствором HCl и раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме; остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 40% до 50%), получая указанное в заголовке соединение (160 мг, 76%).
Пример 18E
(1R,3S,5E,7E,17β)-3-фтор-17-[(1R)-5-гидрокси-1,5-диметилгексил]-2-метилен-9,10-секоэстра-5,7-диен-1-ол
Примечание: представленную далее последовательность синтеза проводили в затемненном вытяжном шкафу. Соединение примера 18D (15 мг, 0,026 ммоль) объединяли с соединением примера 13J (13 мг, 0,06 ммоль) в 1 мл толуола; растворитель удаляли в вакууме для тщательной сушки реагентов. Остаток переносили в 1 мл сухого тетрагидрофурана и охлаждали до -78ºC. К полученному раствору по каплям добавляли раствор бис(триметилсилил)амида лития (1,0M в тетрагидрофуране; 0,1 мл), что приводило к появлению желтовато-оранжевой окраски, которая исчезает через 20 минут. Полученную смесь перемешивали при -78ºC в течение 1 часа, затем нагревали до 0ºC и перемешивали при данной температуре в течение 30 минут. Реакцию гасили добавлением 1 мл 1N водного раствора NH4Cl, и полученную смесь экстрагировали этилацетатом. Органический экстракт промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток растворяли в 1 мл 1N раствора фторида тетра-н-бутиламмония в тетрагидрофуране. После перемешивания в течение ночи при температуре окружающей среды реакционную смесь распределяли между этилацетатом и водой. Органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворители удаляли в вакууме, и остаток очищали хроматографией на Analogix IntelliFlash 280™ (элюирование с градиентом: этилацетат в гексанах - от 20% до 27%), получая указанное в заголовке соединение (4 мг).1H ЯМР (500 МГц, CD2Cl2) δ м.д. 6,32 (д, J=11,3 Гц, 1H), 6,08 (д, J=11,3 Гц, 1H), 5,14-5,25 (м, 2H), 4,75-5,01 (м, 1H), 2,88 (дд, J=13,1, 4,6 Гц, 1H), 2,61-2,75 (м, 1H), 2,36-2,55 (м, 3H), 2,13-2,27 (м, 3H), 2,00 (с, 2H), 1,81-1,91 (м, 2H), 1,67-1,79 (м, 2H), 1,56-1,67 (м, 4H), 1,45-1,56 (м, 6H), 1,35-1,45 (м, 2H), 1,11-1,26 (м, 9H), 0,84-0,92 (м, 3H).

Пример 19
(3R,5R)-3,5-Бис{[трет-бутил(дифенил)силил]окси}-4-метиленциклогексанон
Пример 19A
(1R,4R,6R)-4-изопропенил-1-метил-7-оксабицикло[4.1.0]гептан-2-он
(R)-Карвон подвергали эпоксидированию в соответствии со способом, описанным в публикации E. Klein, G. Ohloff, Tetrahedron, 1963, 11, 1091-1099. Таким образом, H2O2 (31%, 95 мл, 836 ммоль, 1,3 экв.) добавляли в раствор (L)-карвона (100 мл, 640 ммоль) в 650 мл метанола при <5ºC. После охлаждения до <0ºC к смеси добавляли 6N раствор NaOH (10,5 мл, 63 ммоль, 0,1 экв.). Температуру реакционной смеси поддерживали на уровне <5ºC. Спустя 5 часов реакционную смесь разбавляли 650 мл воды, затем гасили 0,5N раствор KH2PO4 (250 мл) и 325 мл 1 М-ного раствора Na2SO3, поддерживая температуру на уровне <25ºC. Реакционную смесь экстрагировали трет-бутилметиловым эфиром (2×750 мл). Объединенные экстракты трет-бутилметилового эфира промывали 500 мл 20% водного раствора NaCl, затем 500 мл 10% раствора NaCl и 500 мл 25% раствора NaCl с получением прозрачного органического слоя, который сушили над MgSO4, фильтровали и концентрировали, затем переупаривали с 100 мл метанола. Анализ (ГХ) полученного раствора (101,3 г, 96%) показывает, что указанное в заголовке соединение содержит 5% минорного изомера.1H ЯМР (400 МГц, CDCl3) δ м.д. 1,40 (с, 3H), 1,70 (с, 3H), 1,89 (ддд, J=14,75, 11,11, 1,17 Гц, 1H), 2,02 (дд, J=17,56, 11,66 Гц, 1H), 2,31-2,41 (м, 1H), 2,58 (ддд, J=17,56, 4,67, 1,37 Гц, 1H), 2,65-2,76 (м, 1H), 3,44 (дд, J=3,09, 1,03 Гц, 1H), 4,70 (д, J=0,69 Гц, 1H), 4,75-4,80 (м, 1H).
Пример 19B
(1S,2R,4S,6R)-4-изопропенил-1-метил-7-оксабицикло[4.1.0]гептан-2-ол
Раствор CeCl3.7H2O (128 г, 343 ммоль, 0,5 экв.) в 1,5 л метанола охлаждали до <0ºC. Добавляли соединение примера 19A (47% (масс.) раствор в метаноле, 242 г, 685 ммоль) и промывали в 60 мл метанола. После охлаждения до температуры <-20ºC, в течение 23 минут добавляли NaBH4 (2M в триглиме, 190 мл, 380 ммоль, 0,55 экв.), поддерживая температуру <-20ºC. После перемешивания в течение дополнительных 25 минут реакционную смесь гасили 720 мл воды. Метанол удаляли отгонкой и добавляли 800 мл изопропилацетата. К смеси добавляли 2N раствор HCl (100 мл) для доведения значения pH смеси до ~5,5, после чего слои разделяли, и водный слой экстрагировали 400 мл изопропилацетата. Объединенные изопропилацетатные слои промывали 500 мл 5% раствора NaCl, затем 525 мл смеси 10% NaCl:10% NaHCO3(19:1), затем 500 мл 20% раствора NaCl. Органический раствор затем сушили над MgSO4, фильтровали и концентрировали, получая 193 г масла, которое, как определяют с помощью анализа, содержит 54,7% (масс.) (105,6 г масла, 92% выход) указанного в заголовке продукта.
Пример 19C
трет-Бутил{[(1R,2R,4R,6R)-4-изопропенил-1-метил-7-оксабицикло[4.1.0]гепт-2-ил]окси}дифенилсилан
К раствору соединения примера 19B (54,7% (масс.), 18,4 г, 60 ммоль) и имидазола (6,94 г, 102 ммоль, 1,7 экв.) в 100 мл диметилформамида добавляли TBDPS-Cl (23,4 мл, 90 ммоль, 1,5 экв.). Реакционную смесь перемешивали в течение 90 часов и затем охлаждали на бане со смесью лед/вода. Добавляли воду (3 мл), охлаждающую баню удаляли, и реакционную смесь перемешивали в течение 15 минут. Реакционную смесь переносили в делительную воронку со 110 мл гептана и 55 мл воды, встряхивали, оставляли для получения слоев и образовавшие слои разделяли. К водному слою добавляли дополнительное количество воды (25 мл), затем полученную смесь дополнительно экстрагировали гептаном (2×50 мл). Объединенные гептановые экстракты промывали 10% раствором NaCl (2×100 мл), затем сушили над MgSO4, фильтровали и концентрировали в вакууме.
Хроматография (Isco CombiFlash® system, Analogix RS300 300 г, колонка, элюирование с градиентом: смесь гексан:CH2Cl2 - 80:20 в течение 10 минут, затем 65:35 в течение 25 минут, затем 60:40 в течение 10 минут), приводила к получению 26,1 г масла, которое, согласно анализу, содержит 83% (масс.) относительно стандарта (21,6 г, 89% выход) указанного в заголовке соединения.
Пример 19D
(1R,3R,5R,6R)-5-{[трет-бутил)(дифенил)силил]окси}-6-метил-7-оксабицикло[4.1.0]гептан-3-ол
Смесь соединения примера 19C (34 г, 92% (масс.), 77 ммоль) и NaHCO3 (3,3 г, 39 ммоль, 0,5 экв.) в 300 мл CH2Cl2 и 60 мл метанола охлаждали до температуры <-70ºC и обрабатывали озоном (7-8 фунтов на кв. дюйм (0,4823-0,5512 бар), 90 вольт, 4 slpm) до тех пор, пока раствор не приобретет стойкую голубую окраску. Раствор продували азотом до исчезновения голубой окраски, реакционную смесь нагревали до ~0ºC, затем фильтровали через бумагу и концентрировали.
После отгонки с бензолом (2×100 мл) добавляли 300 мл CH2Cl2 и 60 мл пиридина, и реакционную смесь охлаждали до <5ºC. К смеси добавляли п-нитробензоилхлорид (22,2 г, 120 ммоль, 1,5 экв.) и реакционную смесь перемешивали в течение 1 часа, затем охлаждающую баню удаляли и реакционную смесь перемешивали при температуре окружающей среды в течение ночи. Полученную суспензию концентрировали на роторном испарителе до густой взвеси, которую затем разбавляли этилацетатом, и твердые вещества удаляли фильтрацией. Фильтрат промывали 1М раствором NaHCO3 (250 мл), затем 2N раствором HCl (100 мл), 1N раствором HCl (100 мл), 2N раствором HCl и, наконец, 1N раствором HCl (200 мл), 1 молярным раствором NaHCO3(200 мл) и 20% раствором NaCl (200 мл). Органический слой сушили над MgSO4, фильтровали и концентрировали, получая 49,6 г промежуточного ацетата в виде масла.
К раствору ацетата (20,6 г, 32 ммоль, теоретическое количество) в 115 мл метанола и 15 мл воды добавляли 11,0 г K2CO3. Спустя 2 часа реакционную смесь гасили 6,5 мл HOAc, затем концентрировали. Остаток обрабатывают 100 мл воды, затем экстрагировали 150 мл этилацетата и 100 мл этилацетата. Объединенные этилацетатные слои промывали 1 молярным раствором NaHCO3(100 мл), 10% раствором NaCl (100 мл) и 20% раствором NaCl (100 мл). Этилацетатный слой сушили над MgSO4, фильтровали и концентрировали.
Хроматография (Isco CombiFlash® system, Analogix RS300 300 г, колонка, элюирование с градиентом: смесь этилацетат:CH2Cl2 - 10:90 в течение 5 минут, затем от 10:90 до 12:88 в течение 35 минут и 12:88 в течение 10 минут) приводила к получению 10,3 г масла, которое, как показывает анализ, содержит 91% (масс.) (76% выход) указанного в заголовке соединения.1H ЯМР (400 МГц, CDCl3) δ м.д. 1,12 (с, 9H), 1,16 (с, 3H), 1,64-1,74 (м, 1H), 1,87 (дт, J=14,10, 4,48 Гц, 1H), 2,09-2,18 (м, 2H), 3,04 (т, J=1,92 Гц, 1H), 3,72-3,90 (м, 2H), 4,14-4,25 (м, 1H), 7,33-7,52 (м, 6H), 7,61-7,78 (м, 4H).
Пример 19E
трет-бутил[((1R,2R,4R,6R)-4-{[трет-бутил(диметил)силил]окси}-1-метил-7-оксабицикло[4.1.0]гепт-2-ил)окси]дифенилсилан
К раствору соединения примера 1D (8,6 г, 20,46 ммоль) и имидазола (2,37 г, 34,86 ммоль) в диметилформамиде (80 мл) при комнатной температуре одной порцией добавляли TBS-Cl (4,6 г, 30,51 ммоль). Реакционную смесь перемешивали в течение 1 часа, после чего ВЭЖХ показывала содержание исходного вещества <1%. Реакционную смесь выливали в воду (150 мл) и водный слой экстрагировали трет-бутилметиловым эфиром (3×50 мл). Объединенные органические слои промывали раствором соли (50 мл), сушили над Na2SO4 и концентрировали в вакууме до масла. Сырой продукт очищали хроматографией (элюирование: смесь 5% этилацетат/гептан), получая 11,48 г указанного в заголовке соединения (99% выход, образец содержит остаточное количество этилацетата) в виде масла.1H ЯМР (400 МГц, CDCl3): δ -0,20 (с, 3H), -0,16 (с, 3H), 0,73 (с, 9H), 1,09 (с, 9H), 1,19-1,30 (м, 1 H), 1,43 (с, 3H), 1,51-1,65 (м, 2H), 2,04 (с, этилацетат), 2,17-2,32 (M, 1H), 3,05 (с, 1H), 3,36-3,46 (м, 1H), 3,90-3,97 (дд, J=10,63, 5,97 Гц, 1H), 4,09-4,14 (кв, этилацетат), 7,32-7,45 (м, 6H), 7,63-7,70 (м, 4H) м.д.
Пример 19F
(1R,3R,5R)-5-{[трет-бутил(диметил)силил]окси}-3-{[трет-бутил(дифенил)силил]окси}-2-метиленциклогексанол
К раствору 2,2,6,6-тетраметилпиперидина (4,55 г, 32,2 ммоль) в бензоле (40 мл) при -5ºC по каплям в течение 20 минут (-10
Пример 19G
[((3R,5R)-3,5-бис{[трет-бутил(дифенил)силил]окси}-4-метиленциклогексил)окси](третбутил)диметилсилан
К раствору соединения 19F (0,65 г, 1,31 ммоль) в диметилформамиде (6 мл) при комнатной температуре добавляли имидазол (0,31 г, 4,58 ммоль) и TBDPS-Cl (1,08 г, 3,92 ммоль), и реакционную смесь перемешивали в течение 3 дней. Реакционную смесь выливают в воду (50 мл) и экстрагировали трет-бутилметиловым эфиром (3×50 мл). Объединенные слои органических экстрактов промывали водой (2×) и раствором соли (2×), сушили над MgSO4 и концентрировали до масла. Сырой продукт очищали хроматографией (элюирование: 5% смесь эфир/гексан), получая 0,92 г (95%) указанного в заголовке соединения в виде бесцветного масла.1H ЯМР (400 МГц, CDCl3) δ -0,16 (с, 6H), 0,74 (с, 9H), 0,92 (с, 9H), 1,12 (с, 9H), 1,17 (ддд, J=13,07, 10,94, 2,47 Гц, 1H), 1,30 (кв., J=11,43 Гц, 1H), 1,68-1,77 (м, 1H), 1,81-1,90 (м, 1H), 3,90-4,02 (м, 1H), 4,38 (т, J=2,74 Гц, 1H), 4,63-4,72 (м, 2H), 5,23-5,30 (м, 1H), 7,28-7,44 (м, 12H), 7,54-7,59 (м, 4H), 7,65-7,73 (м, 4H).
Пример 19H
(3R,5R)-3,5-бис{[трет-бутил(дифенил)силил]окси}-4-метиленциклогексанол
К суспензии соединения примера 19G (1,05 г, 1,43 ммоль) в этаноле (8 мл) добавляли концентрированную HCl (104 мкл, 1,28 ммоль) в этаноле (2 мл), и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь выливают в 1M NaHCO3 (35 мл) и экстрагировали трет-бутилметиловым эфиром (3×35 мл). Объединенные органические слои промывали раствором соли (35 мл), сушили над Na2SO4 и концентрировали в вакууме до масла. Сырой продукт очищали хроматографией на силикагеле (элюирование: смесь 5% этилацетат/гексан), затем сушили в вакууме при комнатной температуре в течение одной недели, получая указанное в заголовке соединение (0,74 г, 83%) в виде твердого белого вещества;1H ЯМР (400 МГц, CDCl3) δ м.д. 1,01 (с, 9H), 1,02 (с, 9H), 1,60-1,85 (м, 4H), 2,46 (с, 1H), 3,96-4,06 (м, 1H), 4,63 (дд, J=6,79, 4,05 Гц, 1H), 4,74 (т, J=5,35 Гц, 1H), 4,85 (с, 1H), 4,91 (с, 1H), 7,27-7,45 (м, 12H), 7,56-7,67 (м, 8H);13C ЯМР (100 МГц, CDCl3) δ 19,5, 19,6, 27,2, 27,3, 44,6, 67,0, 70,4, 107,9, 127,21, 127,24, 127,3, 129,33, 129,35, 129,5, 132,6, 133,2, 133,4, 133,9, 135,44, 135,48, 135,54, 135,6, 149,9; МСВР (ЕSI) [MNa+] C39H48O3Si2: вычислено 643,3034, найдено 643,3022. Элементный анализ: C39H48O3Si2. Вычислено C 75,43, H 7,79. Найдено: C 75,50, H 7,97.
Пример 19I
(3R,5R)-3,5-бис{[трет-бутил(дифенил)силил]окси}-4-метиленциклогексанон
К раствору соединения примера 19H (0,50 г, 0,805 ммоль) в CH2Cl2 (7 мл) добавляли периодинан Десса-Мартина (0,376 г, 0,886 ммоль), и полученную суспензию перемешивали при комнатной температуре в течение 3,5 часов. Реакционную смесь разбавляли CH2Cl2 (10 мл) и промывали смесью 1M NaHCO3:1M Na2SO3 (3:1, 10 мл), а затем смесью насыщенный раствор соли:1M NaHCO3 (3:1½, 10 мл). Органический слой сушили над Na2SO4 и упаривали в вакууме до твердого белого вещества. Сырой продукт очищали хроматографией на силикагеле (5% этилацетат/гексан), затем сушили в вакууме при комнатной температуре в течение одной недели, получая указанное в заголовке соединение (0,49 г, 98%) в виде твердого белого вещества:1H ЯМР (400 МГц, CDCl3) δ м.д. 0,99 (с, 18 H), 2,29-2,49 (м, 4H), 4,74-4,80 (м, 2H), 5,17 (т, J=1,07 Гц, 2H), 7,29-7,36 (м, 8H), 7,36-7,45 (м, 4H), 7,54-7,63 (м, 8H);13C ЯМР (100 МГц, CDCl3) δ 19,5, 27,2, 51,2, 70,9, 109,2, 127,3, 127,4, 129,5, 129,6, 132,7, 133,1, 135,40, 135,43, 148,6, 205,8; МСВР (ESI) [M+Na+] C39H46O3Si2: вычислено 641,2878, найдено 641,2869; [M+NH4+] вычислено 636,3324, найдено 636,3313. Элементный анализ: C39H46O3Si2. Вычислено C 75,68, H 7,49. Найдено C 75,48, H 7,59.
Композиции согласно изобретению
Изобретение предоставляет также фармацевтические композиции, включающие терапевтически эффективное количество соединения формулы (I) в сочетании с фармацевтически приемлемым носителем. Композиции включают соединения согласно изобретению, введенные в препарат вместе с одним или несколькими нетоксичными фармацевтически приемлемыми носителями. Фармацевтические композиции могут изготавливаться в твердой или жидкой форме для перорального введения, для парентеральной инъекции или для ректального введения.
Термин «фармацевтически приемлемый носитель», когда используется в данном описании, означает нетоксичный инертный твердый, полутвердый или жидкий наполнитель, разбавитель, инкапсулирующий материал или вспомогательную добавку любого типа. Некоторые примеры материалов, которые могут служить фармацевтически приемлемыми носителями, представляют собой сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлозу и ее производные, такие как натрийкарбоксиметилцеллюлоза, этилцеллюлоза и ацетат целлюлозы; порошкообразный трагакант; солод; желатин; тальк; масло какао и воск для свечей; масла, такие как арахисовое масло, хлопковое масло, подсолнечное масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; гликоли, такие как пропиленгликоль; сложные эфиры, такие как этилолеат и этилтаурат; агар; буферные агенты, такие как гидроксид магния и гидроксид алюминия; альгиновую кислоту; апирогенную воду; изотонический раствор соли; раствор Рингера; этиловый спирт и фосфатно-буферные растворы; кроме того, в композиции могут присутствовать другие нетоксичные совместимые добавки, такие как добавки, повышающие скольжение, например, натрийлаурилсульфат и стеарат магния, а также красители, добавки для контролируемого высвобождения действующего вещества, добавки для получения покрытия, подсластители, вкусовые добавки и отдушки, консерванты и антиоксиданты, в соответствии с решением специалиста в области получения препаратов.
Фармацевтические композиции согласно настоящему изобретению могут вводиться людям и другим млекопитающим перорально, ректально, парентерально, интрацистернально, интравагинально, интраперитонеально, местно (в форме порошков, мазей или капель), буккально или в форме спрея для перорального или назального введения. Термин «парентерально», когда используется в данном описании, относится к способу введения, включающему внутривенное, внутримышечное, интраперитонеальное, интрастернальное, подкожное, внутрисуставное введение или вливание.
Фармацевтические композиции для парентеральной инъекции включают фармацевтически приемлемые стерильные водные и неводные растворы, дисперсии, суспензии или эмульсии и стерильные порошки для получения из них стерильных растворов или дисперсий для инъекций. Примеры подходящих водных и неводных носителей, разбавителей, растворов или сред включают воду, этанол, многоатомные спирты (пропиленгликоль, полиэтиленгликоль, глицерин и т.п. и их приемлемые смеси), растительные масла (такие как оливковое масло) и органические сложные эфиры, которые могут вводиться инъекцией, такие как этилолеат, или их приемлемые смеси. Подходящая текучесть композиции может поддерживаться, например, применением покрытия, такого как лецитин, сохранением нужного размера частиц в случае дисперсий и применением поверхностно-активных веществ.
Указанные композиции могут также содержать адъюванты, такие как консерванты, смачивающие агенты, эмульгаторы и дисперсанты. Предупреждение действия микроорганизмов может гарантироваться различными противобактериальными и фунгицидными средствами, например, парабенами, хлорбутанолами, фенолом, сорбиновой кислотой и т.п. Может быть желательно включить также изотонические добавки, например, сахара, хлорид натрия и т.п. Пролонгированная абсорбция фармацевтической формы для инъекции может быть получена применением добавок, замедляющих абсорбцию, например, моностеарата алюминия и желатина.
В некоторых случаях для того, чтобы продлить действие лекарственного средство, требуется замедлить абсорбцию лекарственного средства из подкожной или внутримышечной инъекции. Это может достигаться применением жидкой суспензии кристаллического или аморфного материала, плохо растворимого в воде. Скорость абсорбции лекарственного средства может зависеть от скорости его растворения, которая, в свою очередь, может зависеть от размера кристаллов и типа кристаллической формы. Альтернативно, парентерально вводимая лекарственная форма может вводиться при растворении или суспендировании лекарственной формы в масляной среде.
Помимо активных соединений суспензии могут содержать суспендирующие добавки, например, этоксилированные изостеариловые спирты, полиоксиэтиленсорбит, сложные сорбитанэфиры, микрокристаллическую целлюлозу, метагидроксид алюминия, бентонит, агар-агар, трагакант и их смеси.
Если нужно и для более эффективного распределения, композиции согласно изобретению могут вводиться в системы с медленным высвобождением или целевой доставкой действующего вещества, такие как полимерные матрицы, липосомы и микросферы. Они могут стерилизоваться, например, фильтрацией через бактериальный фильтр или посредством введения стерилизующих добавок в форме твердых стерильных композиций, которые могут растворяться в стерильной воде или в некоторой другой стерильной среде для инъекции непосредственно перед применением.
Формы депо для инъекций получают формированием микроинкапсулируемых матриц лекарственного средства в полимерах, способных разлагаться биологическим способом, таких как полилактид-полигликолид. В зависимости от соотношения лекарственного средства и полимера и природы конкретного применяемого полимера, скорость высвобождения лекарственного средства может контролироваться. Примеры других способных разлагаться биологическим способом полимеров включают сложные поли(ортоэфиры) и поли(ангидриды). Депо-препараты для инъекций также получают включением лекарственного средства в липосомы или микроэмульсии, которые совместимы с тканями организма.
Препараты для инъекций могут стерилизоваться, например, фильтрацией через бактериальный фильтр или введением стерилизующих добавок в форме стерильных твердых композиций, которые могут растворяться или диспергироваться в стерильной воде или другой среде для инъекции непосредственно перед применением.
Препараты для инъекций, например, стерильные растворы или масляные суспензии для инъекции, могут приготавливаться в соответствии со способами, известными в данной области техники, с использованием подходящих диспергирующих или смачивающих агентов. Стерильный препарат для инъекции также может представлять собой стерильный раствор, суспензию или эмульсию для инъекции в нетоксичном парентерально приемлемом разбавителе или растворителе, такой как раствор в 1,3-бутандиоле. Приемлемыми разбавителями и растворителями, которые могут применяться, являются вода, раствор Рингера, раствор Фармакопеи США (USP) и изотонический раствор хлорида соли. Кроме того, стерильные нелетучие масла традиционно применяются в качестве растворителя и суспензионной среды. Для этой цели могут применяться любые легкие нелетучие масла, включая синтетические моно- или диглицериды. Кроме того, в препаратах для инъекций применяются жирные кислоты, такие как олеиновая кислота.
Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых лекарственных формах одно или несколько соединений согласно изобретению смешиваются, по меньшей мере, с одним инертным фармацевтически приемлемым носителем, таким как цитрат натрия или гидрофосфат кальция, и/или а) наполнителями или добавками, расширяющими диапазон свойств препарата, такими как крахмалы, лактоза, сахароза, глюкоза, маннит и салициловая кислота; b) связующими веществами, такими как карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидинон, сахароза и гуммиарабик; с) увлажнителями, такими как глицерин; d) дезинтегрирующими добавками, такими как агар-агар, карбонат кальция, картофельный крахмал или крахмал тапиоки, альгиновая кислота, некоторые силикаты и карбонат натрия; е) добавками, замедляющими растворение, такими как парафин; f) ускорителями абсорбции, такими как четвертичные аммониевые соединения; g) смачивающими агентами, такими как цетиловый спирт и глицеринмоностеарат; h) абсорбентами, такими как каолин и бентонитовая глина; и i) добавками, повышающими скольжение, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, натрийлаурилсульфат, и их смесями. В случае капсул, таблеток и пилюль лекарственная форма может также включать буферные агенты.
Твердые композиции аналогичного типа также могут применяться в качестве наполнителей в мягких и твердых желатиновых капсулах с твердыми наполнителями, включая лактозу или молочный сахар, а также высокомолекулярные полиэтиленгликоли.
Твердые лекарственные препараты в форме таблеток, драже, капсул, пилюль и гранул могут быть получены с покрытиями и оболочками, такими как энтеросолюбильные и другие покрытия, хорошо известные в области производства фармацевтических препаратов. Они могут необязательно содержать опалесцирующие добавки и также могут быть представлены в виде композиции, которая высвобождает активный(ые) ингредиент(ы) только или предпочтительно в определенной части кишечного тракта замедленным (отложенным) образом. Примеры материалов, применимых для замедленного высвобождения активного ингредиента, могут включать полимерные вещества и воски.
Композиции для ректального или вагинального введения предпочтительно представляют собой свечи, которые могут быть получены смешиванием соединений согласно изобретению с подходящими нераздражающими носителями, такими как масло какао, полиэтиленгликоль или воск для свечей, которые являются твердыми при температуре окружающей среды, но становятся жидкими при температуре тела и, следовательно, плавятся в прямой кишке или вагинальной полости и высвобождают активное соединение.
Жидкие лекарственные формы для перорального введения включают фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Помимо активных соединений жидкие лекарственные формы могут содержать инертные разбавители, традиционно используемые в данной области техники, такие как, например, вода и другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, хлопковое масло, арахисовое масло, кукурузное масло, масло проростков пшеницы, оливковое масло, касторовое масло и кунжутное масло), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные сорбитанэфиры жирных кислот и их смеси.
Помимо инертных разбавителей композиции для перорального введения могут также включать адъюванты, такие как смачивающие агенты, эмульгаторы и суспендирующие добавки, подстастители, вкусовые добавки и отдушки.
Лекарственные формы для местного или чрезкожного введения соединений согласно данному изобретению включают масла, пасты, кремы, лосьоны, гели, порошки, растворы, спреи, лекарственные формы для ингаляции или «заплатки». Целевое соединение согласно изобретению смешивается в стерильных условиях с фармацевтически приемлемым носителем и любыми необходимыми консервантами или маслами, когда это может потребоваться. Подразумевается, что глазные препараты, ушные капли, глазные мази, порошки и растворы также относятся к области данного изобретения.
Мази, пасты, кремы и гели могут содержать, помимо активного соединения согласно данному изобретению, животные и растительные жиры, масла, воски, парафины, крахмал, трагакант, производные целлюлозы, полиэтиленгликоли, силиконы, бентониты, салициловую кислоту, тальк и оксид цинка или их смеси.
Порошки и спреи могут содержать, помимо соединений согласно данному изобретению, лактозу, тальк, салициловую кислоту, гидроксид алюминия, силикаты кальция и порошкообразные полиамиды или смеси перечисленных веществ. Спреи могут дополнительно содержать традиционно используемые пропелленты, такие как хлорфторуглеводороды.
Соединения согласно изобретению также могут вводиться в форме липосом. Как известно в данной области техники, липосомы обычно получают из фосфолипидов или других липидных соединений. Липосомы образуют моно- или мультиламелярные гидратированные жидкие кристаллы, которые диспергируются в жидкой среде. Любой нетоксичный физиологически приемлемый и метаболизируемый липид, способный образовывать липосомы, может применяться. Композиции согласно настоящему изобретению в форме липосом могут содержать, помимо соединений согласно изобретению, стабилизаторы, консерванты и т.п. Предпочтительные липиды представляют собой природные и синтетические фосфолипиды и фосфатидилхолины (лецитины), используемые раздельно или вместе.
Способы получения липосом известны в данной области техники (см., например, Prescott, Ed., Methods in Cell Biology, Volume XIV, Academic Press, New York, N. Y., (1976), p. 33 et seq.).
Лекарственные формы для местного введения соединений согласно данному изобретению включают порошки, спреи, мази и лекарственные формы для ингаляции. Активное соединение смешивается в стерильных условиях с фармацевтически приемлемым носителем и любыми необходимыми консервантами, буферными добавками или пропеллентами. Подразумевается, что глазные препараты, глазные мази, порошки и растворы относятся к области данного изобретения. Водные жидкие композиции согласно изобретению также являются подходящими.
Соединения согласно изобретению могут применяться в форме фармацевтически приемлемых солей, полученных из органических или неорганических кислот. Термин «фармацевтически приемлемые соли», когда используется в данном описании, включает соли и цвиттерионы соединений формулы (I), которые в пределах нормального медицинского заключения подходят для применения в контакте с тканями людей и низших животных без избыточной токсичности, раздражения, аллергической реакции и т.п., обладают подходящим приемлемым соотношением «польза/риск» и эффективны для планируемого применения.
Термин «фармацевтически приемлемая соль» относится к солям, которые в пределах нормального медицинского назначения подходят для применения в контакте с тканями людей и низших животных без избыточной токсичности, раздражения, аллергической реакции и т.п. и обладают подходящим приемлемым соотношением «польза/риск». Фармацевтически приемлемые соли хорошо известны в данной области техники. Указанные соли могут быть получены in situ в процессе конечного выделения и очистки соединений согласно изобретению или отдельно взаимодействием свободной основной функциональной группы с подходящей органической кислотой.
Типичные кислотно-аддитивные соли включают, но без ограничения, ацетат, адипат, альгинат, цитрат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, камфорат, камфорсульфонат, цитрат, диглюконат, этансульфонат, глицерофосфат, гептаноат, гексаноат, фумарат, гидрохлорид, гидробромид, гидройодид, гидроксибутират, 2-гидроксиэтансульфонат (изотионат), лактат, малат, малеат, метансульфонат, никотинат, 2-нафталинсульфонат, оксалат, памоат, пектинат, персульфат, фенилацетат, 3-фенилпропионат, пикрат, пивалат, пропионат, салицилат, сукцинат, сульфат, тартрат, тиоцианат, фосфат, глутамат, карбонат, п-толуолсульфонат и ундеканоат.
Кроме того, основные азотсодержащие группы могут подвергаться кватернизации, например, низшими алкилгалогенидами, такими как метил-, этил-, пропил- и бутилхлориды, -бромиды и -йодиды; диалкилсульфатами, такими как диметил-, диэтил, дибутил- и диамилсульфаты; галогенидами с длинными цепями, такими как децил-, лаурил, миристил- и стеарилхлориды, -бромиды и -йодиды; арилалкилгалогенидами, такими как бензил- и фенэтилбромиды и др. таким образом получают водо- или маслорастворимые или диспергируемые продукты.
Основно-аддитивные соли могут быть получены in situ в процессе конечного выделения и очистки соединений согласно изобретению или отдельно взаимодействием фрагмента, содержащего группу карбоновой кислоты, с подходящим основанием, таким как гидроксид, карбонат или гидрокарбонат с фармацевтически приемлемым катионом металла, катионом аммония или органическим первичным, вторичным или третичным амином. Фармацевтически приемлемые соли включают, но без ограничения, соли катионов щелочных или щелочно-земельных металлов, такие как соли лития, натрия, калия, кальция, магния и алюминия и т.п., а также нетоксичные катионы четвертичного аммония и аминных катионов, включая аммоний, тетраметиламмоний, тетраэтиламмоний, метиламин, диметиламин, триметиламин, триэтиламин, диэтиламин и этиламин. Другие типичные органические амины, применимые для получения основно-аддитивных солей, включают этилендиамин, этаноламин, диэтаноламин, пиперидин и пиперазин.
Термин «фармацевтически приемлемый сложный эфир», когда используется в данном описании, относится к сложным эфирам соединений согласно изобретению, которые гидролизуются in vivo и включают сложные эфиры, которые легко разлагаются в организме человека, предоставляя исходное соединение или его соль. Примеры фармацевтически приемлемых нетоксичных сложных эфиров согласно изобретению включают сложные C1-C6 алкиловые эфиры и C5-C7 циклоалкиловые эфиры, при этом предпочтительны сложные C1-C4 алкиловые эфиры. Сложные эфиры соединений формулы (I) могут быть получены в соответствии с традиционными способами. Фармацевтически приемлемые сложные эфиры могут образовываться с гидроксильными группами взаимодействием соединения, которое содержит гидроксильную группу, с кислотой и алкилкарбоновой кислотой, такой как уксусная кислота, или с кислотой и арилкарбоновой кислотой, такой как бензойная кислота. В случае соединений, содержащих группы карбоновой кислоты, фармацевтически приемлемые сложные эфиры получают из соединений, содержащих группы карбоновой кислоты, взаимодействием соединения с основанием, таким как триэтиламин и алкилгалогенид или алкилтрифлат, например, с метилйодидом, метилтрифлатом, бензилйодидом или циклопентилйодидом. Они также могут быть получены взаимодействием соединения с кислотой, такой как соляная кислота, и спиртом, таким как этанол или метанол, или со спиртом и агентом связывания, таким как гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (1-(3-dimehylaminopropyl)-3-ethylcarbodiimide - EDAC) или 1,3-дициклогексилкарбодиимид (1,3-dicycloheylcarbodiimide - DCC).
Термин «фармацевтически приемлемый амид», когда используется в данном описании, относится к нетоксичным амидам согласно настоящему изобретению, полученным из аммиака, первичных С1-С6 алкиламинов и вторичных С1-С6 диалкиламинов. В случае вторичных аминов, амин также может быть в форме 5- или 6-членного гетероцикла, содержащего один атом азота. Амиды, полученные из аммиака, вторичные С1-С3алкиламиды и вторичные С1-С3 диалкиламиды являются предпочтительными. Амиды соединений формулы (I) могут быть получены в соответствии с традиционными способами. Фармацевтически приемлемые амиды могут быть получены из соединений, содержащих первичные или вторичные аминогруппы, взаимодействием соединения, которое содержит аминогруппу, с алкилангидридом, арилангидридом, ацилгалогенидом или ароилгалогенидом. В случае соединений, содержащих группы карбоновых кислот, фармацевтически приемлемые сложные эфиры получают из соединений, содержащих группы карбоновых кислот, взаимодействием указанного соединения с основанием, таким как триэтиламин, дегидратирующим агентом, таким как дициклогексилкарбодиимид или карбонилдиимидазол, и алкиламином или диамиламином, например, с метиламином, диэтиламином или пиперидином. Они также могут быть получены взаимодействием соединения с кислотой, такой как серная кислота, и алкилкарбоновой кислотой, такой как уксусная кислота, или с кислотой и арилкарбоновой кислотой, такой как бензойная кислота в условиях дегидратирования, таких как добавление молекулярных сит. Композиция может содержать соединение согласно изобретению в форме фармацевтически приемлемого пролекарства.
Термин «фармацевтически приемлемое пролекарство» или «пролекарство», когда используется в данном описании, относится к таким пролекарствам соединений согласно изобретению, которые в пределах обычного медицинского заключения подходят для применения в контакте с тканями людей и низших животных без избыточной токсичности, раздражения, аллергической реакции и т.п., с подходящим приемлемым соотношением «польза/риск» и эффективны для планируемого применения. Пролекарства согласно изобретению могут быстро трансформироваться in vivo в исходное соединение формулы (I), например, гидролизом в крови. Исчерпывающий обзор пролекарств предоставлен в публикациях T. Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems, V. 14 of the A.C.S. Symposium Series; Edward B. Roche, ed., Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press (1987).
Изобретение также относится к фармацевтически приемлемым соединениям, которые при введении пациенту, нуждающемуся в таком введении, могут полностью превращаться in vivo в соединения формулы (I).
Способ применения
Фактические уровни доз активных ингредиентов в фармацевтических композициях согласно изобретению могут изменяться таким образом, чтобы получить количество активного(ых) соединения(й), которое эффективно для достижения желательного терапевтического ответа для конкретного пациента, композиции и способа введения. Выбранная дозировка будет зависеть от активности конкретного соединения, способа введения, тяжести состояния, подлежащего лечению, а также состояния и предшествующей истории болезни пациента, подлежащего лечению. Однако квалифицированный специалист в данной области техники может назначать лечение соединением с начальных доз, более низких, чем необходимы для достижения желательного терапевтического эффекта, и постепенно повышать дозировку до достижения желательного эффекта.
При применении в указанных выше или других способах лечения терапевтически эффективное количество одного из соединений согласно изобретению может применяться в чистой форме или, когда такие формы существуют, в форме фармацевтически приемлемой соли. Альтернативно, соединение может вводиться в форме фармацевтической композиции, содержащей соединение согласно изобретению в сочетании с одним или несколькими фармацевтически приемлемыми носителями. Фраза «терапевтически эффективное количество» соединения согласно изобретению означает количество соединения, достаточное для лечения расстройств с подходящим приемлемым соотношением «польза/риск», применимое при любом медицинском лечении. Однако следует представлять, что общая суточная доза применения соединений и композиций согласно изобретению будет определяться лечащим врачом в рамках обычного медицинского назначения. Конкретный уровень терапевтически эффективной дозы для любого конкретного пациента будет зависеть от различных факторов, включая расстройство, подлежащее лечению, и тяжесть указанного расстройства; активность конкретного применяемого соединения; конкретную применяемую композицию; возраст, массу тела, общее состояние здоровья, пол и рацион питания пациента; время введения, способ введения и скорость выведения из организма конкретного применяемого соединения; продолжительность лечения; лекарственные средства, используемые совместно, или лекарственные средства, применение которых случайно совпадает с применением конкретного используемого соединения; и аналогичные факторы, хорошо известные в области медицины. Например, специалист в данной области техники может назначать лечение соединением с начальных доз, более низких, чем необходимо для достижения желательного терапевтического эффекта, и постепенно повышать дозировку до достижения желательного эффекта.
Соединения согласно изобретению могут вводиться сами по себе, в сочетании с одним или несколькими другими соединениями согласно изобретению или в сочетании (т.е. совместно вводиться) с одним или несколькими дополнительными фармацевтическими средствами. Комбинированная терапия включает введение одного лекарственного препарата, содержащего одно или несколько соединений согласно изобретению и одно или несколько дополнительных фармацевтических средств, а также введение соединений согласно изобретению и каждого дополнительного фармацевтического средства в его собственном, отдельном лекарственном препарате. Например, соединение формулы (I) и одно или несколько дополнительных фармацевтических средств могут вводиться пациенту в одной лекарственной композиции для перорального введения, содержащей фиксированное соотношение активных ингредиентов, такой как таблетка или капсула; или каждое фармацевтическое средство может вводиться в отдельных лекарственных препаратах для перорального введения.
Когда используются отдельные лекарственные препараты, соединение согласно изобретению и одно или несколько дополнительных фармацевтических средств могут вводиться по существу в одно и то же время (например, одновременно) или в разное время (например, последовательно).
Общая суточная доза соединений согласно изобретению, вводимая человеку или животному, находится в интервале от примерно 0,01 мкг до примерно 150 мг. Более предпочтительные дозы могут находиться в интервале от примерно 0,01 мкг до примерно 10 мг. Если нужно, эффективная суточная доза может делиться на несколько доз для удобного введения. Следовательно, композиции разовой дозы могут содержать такие количества или их дольные единицы, которые могли бы составить суточную дозу.
Определение биологической активности
Биологическая активность соединений оценивалась для выявления соединений, которые могут обладать желательными биохимическими характеристиками, с использованием парикальцитола в качестве образца сравнения. На фигуре 3 представлена схема скрининга активных соединений. Активность соединений оценивали сначала in vitro с использованием метода связывания c ядерным рецептором витамина D (метод гена-репортера) и HL-60 дифференциации. Соединения согласно изобретению оценивали в соответствии с их эффективностью в сравнении с парикальцитолом. Соединения, которые, как установлено в результате одного или нескольких из указанных анализов, обладали эффективностями до 100 раз меньшими, чем парикальцитол, были выбраны для дельнейшего тестирования.
Далее соединения оценивались в опытах на нормальных мышах для определения кальцемического действия и в опытах на подходящих клеточных линиях для определения влияния на сердечно-сосудистые биомаркеры, в частности на ингибитор активатора плазминогена 1 (plasminogen activator inhibitor-1 - PAI-1), тромбомодулин, тромбоспондин-1 и ренин в кардиомиоцитах. Кроме того, оценивалась их рениновая активность в отношении As4.1 клеток. Цель исследования состояла в выявлениях соединений, кальцемическая активность которых в опытах на мышах меньше кальцемической активности парикальцитола, а также соединений, эффективность которых в опытах с использованием биомаркеров больше эффективности парикальцитола.
Для определения терапевтического индекса соединения были далее испытаны на модели почечной недостаточности крыс с определением их способности снижения PTH и кальцемии. Цель данного исследования состояла в выявлении соединений, терапевтический индекс (ТИ, TI) которых выше ТИ парикальцитола, по меньшей мере, в три раза, а также соединений, которые вносят вклад в кальциноз аорты (или мягкой ткани) в меньшей степени, чем парикальцитол.
Наконец, модель гипертрофии левого желудочка в условиях in vivo использовалась для определения возможности применения соединений для лечения сердечно-сосудистого заболевания. Одновременно с кальцинозом сердечно-сосудистой системы развивается гипертония. Уплотнение миокарда левого желудочка приводило к тому, что для поддерживания нормального кровотока в сосудистой сети сердце должно работать с большей нагрузкой. Желательной была эффективность, превосходящая эффективность парикальцитола.
Каждый из этих способов определения активности далее описан более подробно, но квалифицированному специалисту в данной области они должны быть знакомы как стандартные способы определения активности.
VDR связывание
Связывание выбранных соединений согласно изобретению с VDR in vitro оценивали в соответствии со следующей методикой. Очищенный рекомбинантный ядерный рецептор витамина D человека полной длины (коммерчески доступен от PanVera/Invitrogen (Carlsbad, CA) Part Number P2190) разбавляли VDR-связывающим буфером (50 мМ Tris, (pH 7,5), 5 мМ DTT, 300 мМ KCl, 0,01% TWEEN 20). Непосредственно перед испытанием [3H]-кальцитриол (1α,25-дигидрокси[23,24(n)-3H]холекальциферол, Amersham Biosciences, Piscataway, NJ; код продукта TRK588-5UCl) разбавляли в связывающем буфере в силиконизированной пробирке Эппендорфа. Каждое испытываемое соединение затем последовательно разбавляли связывающим буфером до концентраций, подходящих для опыта, как известно специалисту в данной области техники. Разбавленное опытное соединение или позитивный контроль (парикальцитол, коммерчески доступен как ZEMPLAR от Abbott Laboratories, Illinois) добавляли в каждую лунку 96-луночного микротитровального планшета (Wallac Isoplate Part 1450-516, коммерчески доступен от Perkin-Elmer Co., Boston, MA) с последующим добавлением [3H]-кальцитриола (конечная концентрация 1 нМ).
Затем в каждую лунку добавляли разбавленный исходный раствор VDR (100 нг/лунка). Конечный опытный объем составлял 100 мкл. Заполненный планшет инкубировали в течение ночи при 4ºC с осторожным встряхиванием. После 18 часов инкубирования в каждую лунку добавляли 20 мкл шариков силиката иттрия для SPA-анализа (scintillation proximity assay), покрытых агглютинином проростков пшеницы (коммерчески доступный от Amersham Biosciemces, Piscataway, NJ; Part # RPNQ0270) и повторно суспендированных в связывающем буфере (200 мкг/лунка), и планшет инкубировали при комнатной температуре в течение 2 часов с осторожным встряхиванием. После этого связанный [3Н]-кальцитриол подсчитывали с использованием Packard Top-Count. Неспецифическое связывание определяли как количество единиц в минуту [3Н]-кальцитриола, связанного в присутствии избытка (10 мкМ) парикальцитола.
Метод гена-репортера VDRE
Активность соединений также оценивали изучением методом гена-репортера элемента, реагирующего на витамин D (Vitamin D Response Element - VDRE). Клетки человеческой эмбриональной почки (Human Embryonic Kidney cells - HEK клетки) со стабильной экспрессией VDR выращивали в 96-луночных планшетах при ~4×105 клеток/мл (100 мк л/лунка) в DMEM среде, содержащей 10% фетальной телячьей сыворотки. Клетки трансфектировали 0,2 мкг/лунка конструкта «VDRE-люцифераза-репортер» (от Y.Li, университет Чикаго) и затем обрабатывали опытными соединениями в указанных концентрациях в течение 24 часов. Люциферазную активность определяли в соответствии с инструкциями производителя (Promega, Madison, WI). Для каждого образца определяли силу сигнала, и полученные значения повышения уровня сигнала показаны в таблице 2.

HL-60 дифференциация
Клетки промиелоцитарного лейкоза человека (human promyelocytic leukemia (HL60)) были получены из American Type Culture Collection (ATCC Cat. # CCL-240, Manassas, Virginia). Клетки выдерживали в среде RPMI 1640 (коммерчески доступна от Invitrogen, Carlsbad, CA), содержащей 10% фетальной телячьей сыворотки (коммерчески доступна от Invitrogen, Carlsbad, CA) при 37ºC с 5% CO2. Клетки пересевали еженедельно, не допуская конфлюенса >90%. Для анализа клетки вносили в 200 мкл среды в концентрации 5×105 клеток на лунку. Испытываемые соединения разбавляли в среде до концентраций в интервале от 10-6до 10-10 М и затем добавляли в подходящие лунки. После этого клетки инкубировали в течение четырех дней при 37ºC в атмосфере с 5% СО2. После инкубирования среду аспирировали из каждой лунки, в каждую лунку добавляли 75 мкл раствора NBT (нитросиний тетразолий:200 нг/мл РМА (форбол-12-миристат-13-ацетат) и 2 мг/мл NBT (нитросиний тетразолий) в дистиллированной Н2О) и далее дополнительно инкубировали в течение 2 часов при 37ºC. После инкубирования в каждую лунку добавляли 150 мкл лизисного буфера (225 мл диметилформамида, 67,5 г SDS). Исходный раствор РМА (2 мг/мл) этанола добавляли в каждую лунку, и планшет оставляли на 4 часа при комнатной температуре. Анализировали дифференциацию HL-60 клеток. Влияние каждого соединения на рост клеток определяли как функцию времени; спектральную поглощательную способность в каждой лунке определяли при 570 нм. В таблице 3 представлены значения ЕС50 выбранных соединений.

Ренин мРНК в As4.1
As4.1 клетки (АТСС, Manassas, VA) выращивали в среде Игла с минимальной чувствительностью, модифицированной по Дульбекко (Dulbecco's Modified Eagle Medium - DMEM) (коммерчески доступна от Invitrogen, Carlsbad, CA) с 10% фетальной телячьей сыворотки при 37ºC в увлажненной атмосфере 5% СО2-95% воздух. Клетки трансфектировали pcDNA-hVDR плазмидой (предоставленной Yan Chun Li, University of Chicago) с помощью LIPOFECTAMINE™ 2000 (Invitrogen, Carlsbad, CA). siRNA (малые интерферирующие РНК) трансфектировали pcDNA-hVDR с помощью LIPOFECTAMINE™ 2000 в соответствии с руководством производителя. Через двадцать четыре часа после трансфектирования клетки обрабатывают испытываемыми соединениями.
Полимеразную цепную реакцию (ПЦР) обратной транскрипции в реальном времени проводили с помощью системы обнаружения ПЦР в реальном времени MyiQ (коммерчески доступна от BioRad, Hercules, CA). Конечный объем каждого образца составлял 25 мкл, в котором содержалось 100 нг кДНК, 0,4 мМ каждого из праймеров прямой и обратной ПЦР и 0,1 мМ TaqMan™ зонда. Использовались TaqMan™ зонды, 5'-меченные репортерным 6-карбоксифлюоресцеином (FAM) и 3'-меченные гасителем флуоресценции тетраметилродамином (tetramethylrhodamine - TAMRA); праймеры и зонды получены от Applied Biosystems (Foster City, CA). Температурные условия: стадия продолжительностью 5 минут при 95ºC с последующими 45 циклами - 1 минута при 60ºC и 15 минут при 95ºC. Данные собирали в течение каждой длительной фазы ПЦР реакции и анализировали с помощью прилагающегося пакета программ (BioRad, Hercules, CA). Пороговые циклы определяли для каждого гена. Выбранные значения IC50 представлены в таблице 4.
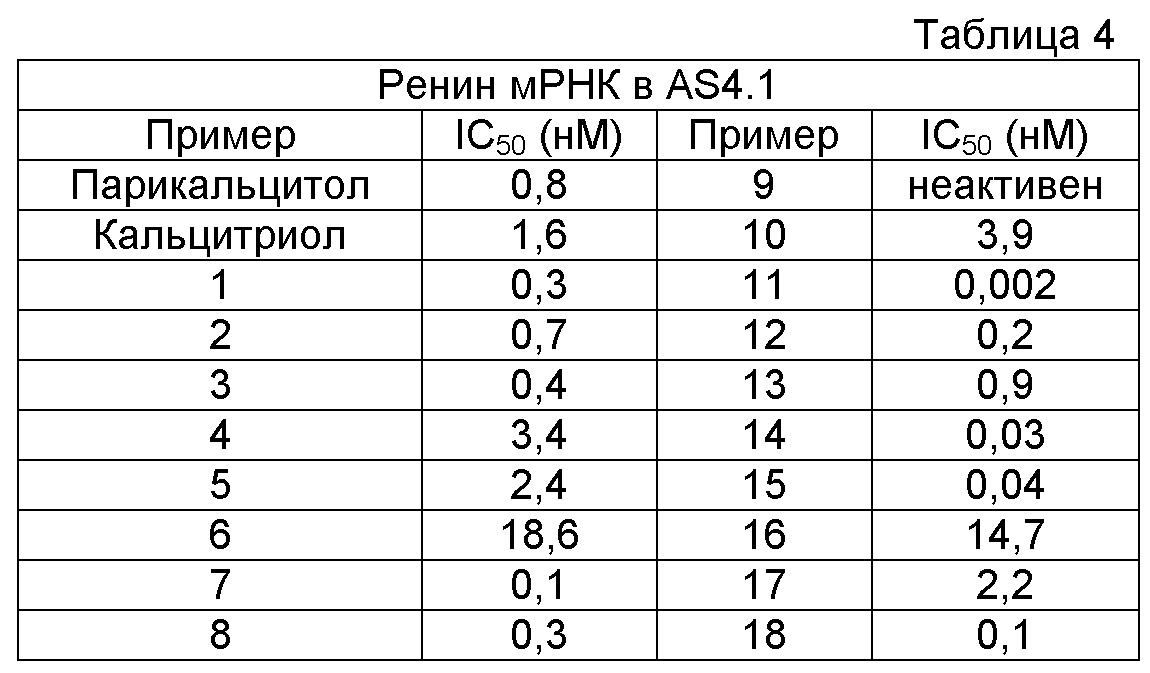
Сердечно-сосудистые биомаркеры в клетках гладких мышц
Первичную культуру клеток гладких мышц коронарной артерии (coronary artery smooth muscle cells - CASMC) (коммерчески доступна от Cambrex, Walkersville, MD) выращивали в ростовой среде клеток гладких мышц SmGM-2 (коммерчески доступна от Lonza Bioscience), содержащей 5,5 мМ глюкозы, 5% FBS, 50 мкг/мл гентамицина, 50 нг/мл амфотерицина-В, 5 мкг инсулина, 2 нг/мл рекомбинантного фактора роста фибробластов человека (human recombinant fibroblast growth factor - hFGF) при 37ºC в увлажненной атмосфере состава 5% СО2- 95% воздуха. Клетки выращивали до >80% конфлюэнтности и использовали в пяти пересевах.
ПЦР обратной транскрипции в реальном времени проводили с помощью системы обнаружения ПЦР в реальном времени MyiQ (BioRad, Hercules, CA). Каждый образец, конечный объем которого составлял 25 мкл, содержал 100 нг кДНК, 0,4 мМ каждого из праймеров прямой и обратной ПЦР и 0,1 мМ TaqMan™ зонда для заданного гена (Applied Biosystems, Foster City, CA). Температурные условия: стадия продолжительностью 5 минут при 95ºC с последующими 40 циклами: 60ºC в течение 1 минуты и 95ºC в течение 15 минут. Данные собирали в течение каждой длительной фазы ПЦР реакции и анализировали с помощью прилагающегося пакета программ (BioRad, Hercules, CA). Пороговые циклы определяли для каждого гена.
SDS PAGE и Western Blot анализ: клетки (1×106 клеток на образец) или препараты клеточных экстрактов солюбилизировали в образце SDS PAGE буфера (Invitrogen, Carlsbad, CA) и в каждом образце определяли содержание белка с помощью анализа на белок Pierce (Rockford, IL). Образцы снова растворяли в SDS PAGE буфере с использованием 12% NuPAGE геля (Invitrogen, Carlsbad, CA) и электрофоретически трансфектировали в PVDF мембрану для вестерн-блоттинга. Мембрану блокировали в течение 1 часа при 25ºC с 5% обезжиренным сухим молоком в PBS-T и затем инкубировали с анти-остеопротегериновым поликлональным антителом кролика (1:100 разбавление, Santa Cruz Biotechnology, Santa Cruz, CA), моноклональным антителом ингибитора-1 активатора плазминогена мыши (anti-plasminogen activator inhibitor-1 - PAI-1) (1000 кратное разбавление, Santa Cruz Biotechnology, Santa Cruz, CA), моноклональным антителом к тромбоспондину-1 (anti-thrombospondin-1 - THBS1) (2000-кратное разбавление, Calbiochem, La Jolla, CA), моноклональным антителом к тромбомодулину (anti-thrombomodulin - TM) мыши (2000-кратное разбавление, Santa Cruz Biotechnology, Santa Cruz, CA), моноклональным антителом к VDR мыши (1:500 разбавление), моноклональным антителом к PPARγ мыши (Santa Cruz Biotechnology, Santa Cruz, CA) (1:200-кратное разбавление), поликлональным антителом к NFkB кролика (1:200-кратное разбавление, Cell Signaling Technology, Danvers, MA) или поликлональным антителом к Ncor-1 кролика (1:200-кратное разбавление, Abeam Inc., Cambridge, MA) в PBS-T в течение ночи при 4ºC. Мембрану промывали PBS-T и инкубировали с меченным пероксидазой хрена вторичным антителом против мыши (для VDR и PPARγ) или против кролика (для остеопротегерина, р65 субъединица NFkB и Ncor1) в течение 1 часа при 25ºC. После этого мембрану инкубировали с реагентом обнаружения (SuperSignal WestPico, Pierce, Rockford, IL). Специфические линии визуализировали воздействием на бумагу пленок Kodak BioMax. Интенсивность линий количественно определяли с помощью Quantity One (BioRad, Hercules, CA). Выбранные значения IC50 и ЕС50 представлены в таблице 5.
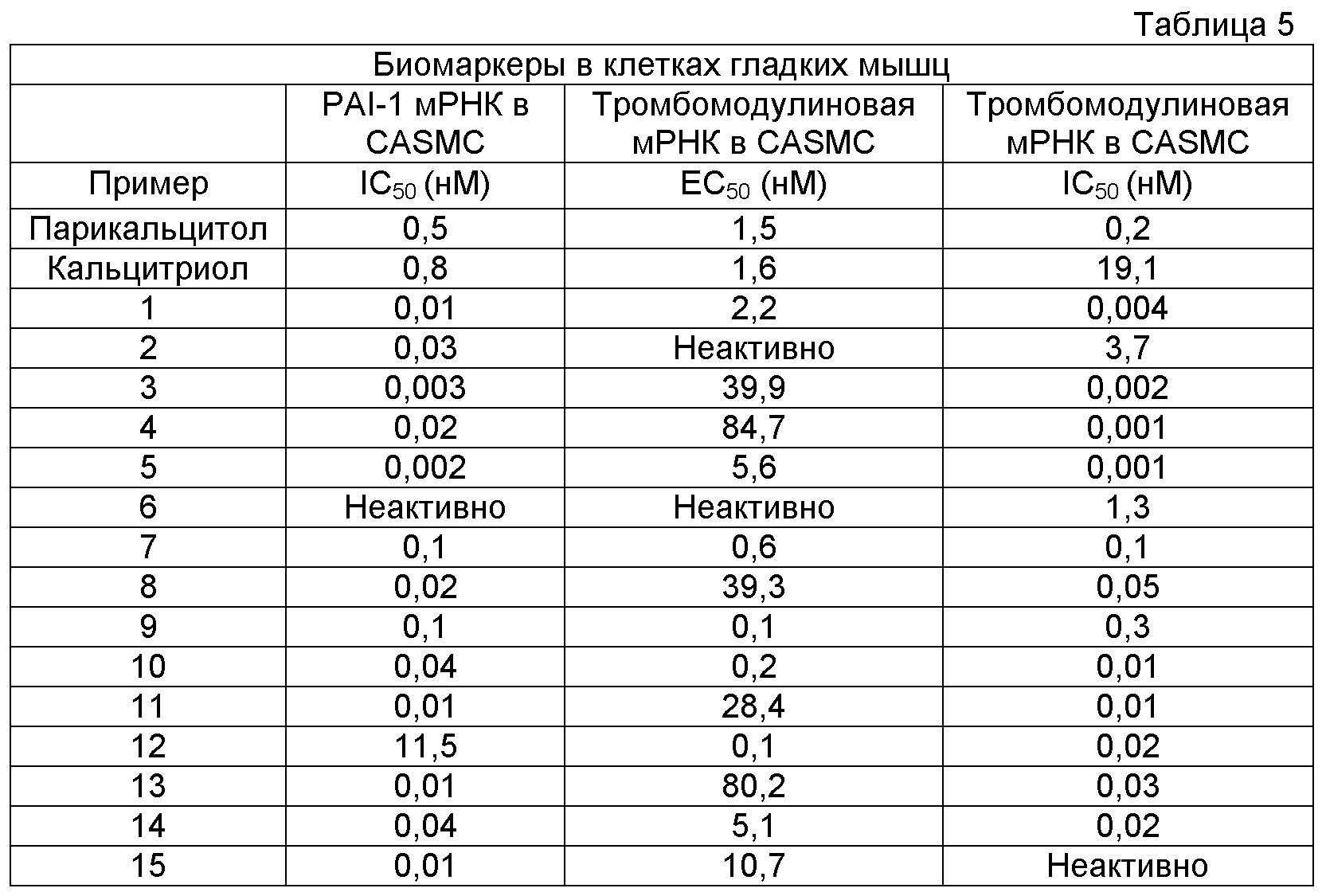
Модель кальцемии нормальной мыши
Самцы мыши С57BL6 были получены от Jackson Laboratories. Мыши адаптировались в течение четырех дней, в процессе чего их содержали на нормальном рационе питания (D10001, Research Diets Inc.). Лечение начинали с разбавителя (20% этанол/30% пропиленгликоль/50% вода, 0,05 мл, s.c.) или испытываемого соединения, которые вводили в дозах в интервале трех единиц логарифмов доз. Дозирование животным проводили раз в день в течение 3 дней. Через двадцать четыре часа после введения последней дозы животных анестезировали смесью кетамин/ксилазин (100/18 мг/кг) и забивали со спуском крови пункцией сердца для измерения РТН и других конечных показателей.
Количественное определение содержания в сыворотке РТН и минеральных веществ: кальция (Са), фосфора (Pi), креатинина и BUN - проводили с использованием Abbott Aeroset® (Abbott Laboratories, Abbott Park, IL). Содержание ионизированного кальция (iCa) определяли с использованием i-STAT® system (Abbott Laboratories, East Windsor, NJ). Сывороточный РТН определяли с использованием набора для определения содержания паратиреоидного гормона (РТН) мыши ЕIA (от ALPCO/Immutopics, Inc. (Windham, NH)). Полученные данные представлены в таблице 6. График, представленный на фигуре 4, показывает зависимость доза-эффект. Примечательно то, что соединение примера 1 приводило к приблизительно 30% снижению содержания РТН в интервале, по меньшей мере, трех единиц логарифма дозы без одновременного повышения уровня содержания кальция в сыворотке до значений более 1 мг/dL. Указанные данные подтвердили возможность более широкого терапевтического окна по сравнению с двумя другими испытанными соединениями.
Анализ данных: Для каждой группы вычисляли «среднее значение ± среднеквадратическая ошибка». Для оценки различий между группами, обработанными разбавителем и обработанными лекарственным средством, использовали способ AVONA с последующим тестом Даннетта. *р<0,05 = статистическая значимость.

Кальциноз аорты у крыс с уремией
Крысы с 5/6 нефрэктомией: самцы крыс Sprague-Dawley с 5/6 NX (~200 г) получали из Charles River. Нефрэктомию проводили стандартной двухстадийной хирургической операцией иссечения. Примерно через 2 недели после нефрэктомии крысам начали давать корм, вызывающий гиперфосфатаземию (фосфор 0,9% и кальций 0,6%), для индуцирования вторичного гиперпаратиреоза (secondary hyperparathyroidism - SHPT). После содержания в течение четырех недель на специальном рационе питания крысам давали разбавитель (5% этанол/95% пропиленгликоль; 0,4 мл/кг; i.p.) или испытываемое соединение - 3 раза в неделю в течение 41 дней. В 0, 13 и 41 дни производили забор крови (через 24 часа после дозировки).
Для снижения до минимума отклонений, вызванных различием в болезненном состоянии, 5/6 NX крыс обследовали после кормления в течение 4 недель кормом с высоким содержанием фосфора (перед введением лекарственного средства или разбавителя) и при обследовании крыс применяли следующие критерии включения/исключения:
- Сывороточный креатинин = 0,8-2,0 (симулированный контроль = 0,43±0,01 мг/дл; n=20);
- Исключение: сывороточный кальций ≤8,5 мг/дл (контроль (имитация лечения) = 10,05±0,05 мг/дл; n=20);
- Исключение: сывороточный фосфор >12 мг/дл (контроль (имитация лечения) = 7,09±12 мг/дл; n=20);
- Исключение: iPTH <400 пг/мл; n=20).
Количественное определение содержания РТН и минеральных веществ в сыворотке: содержание РТН в сыворотке количественно определяли с использованием набора интактного определения содержания РТН у крыс ELISA (ALPO/Immutopics, Inc., Windham, NH); концентрации в сыворотке кальция, фосфора, креатинина и BUN определяли с использованием Abbott Aeroset® (Abbott, Abbott Park, IL); содержание в крови ионизированного кальция определяли с использованием переносного клинического анализатора i-STAT® (Abbott Laboratories, East Windsor, NJ).
Аорты иссекали, отделяли от посторонней ткани, взвешивали, превращали в золу при высокой температуре, разбавляли в кислотном буфере и анализировали для определения общего содержания кальция с помощью клинического анализатора Aeroset® (Abbott Laboratories, Abbott Park, IL).
Анализ данных: для каждой группы вычисляли «среднее значение ± среднеквадратическая ошибка». Для оценки различий между соответствующими днями в группах имитированного лечения, принимающих разбавитель, и принимающих VDRA, использовали способ AVONA с последующим тестом Даннетта. * р<0,05 = статистическая значимость относительно соответствующего дня; #p<0,05 относительно 5/6 NX.
Соединение примера 1 не влияло на кальциноз аорты при < 100 мг/кг.
Исследование 1: Цель исследования заключалась в сравнительном определении влияния соединения примера 1 на содержание в сыворотке РТН, ионизованного Са++, общего Са++и фосфора в модели 5/6 NX крысы.
Способы: 5/6 NX крысы выдерживались на нормальном рационе питания (Teklad 8640; 0,9% фосф. и 1,1% Ca++) в течение 4 недель до исследования и в течение всего уремического исследования, которое продолжалось в течение 6 недель. 5/6 нефрэктомированным крысам вводили разбавитель и соединение примера А-1 в дозах 10, 30 или 100 мкг/кг. Дозировку производили i.p., 3х/неделя в течение 2 недель. Забор крови производили на 0 и 13 дни для определения концентраций РТН, ионизованного Са++, общего Са++и фосфора. Статистический анализ производили с использованием парных t-тестов для сравнения показателей в 0 день относительно показаний в 13 день; статистическая значимость достигалась при p< 0,05.
Результаты: Обработка соединением примера 1 в дозах 30 и 100 мг/кг приводила к снижению РТН на 26% и 78%, соответственно. Содержание ионизованного и общего Са++ повышалось при обработке соединением примера 1 в дозе 100 мг/кг. Соединение примера 1 не влияло на уровни содержания фосфора; однако содержание фосфора снижалось в группах ложного лечения, принимавших разбавитель, 5/6 NX крыс.
Исследование 2: Исследование проводили для определения возможности снижения гипертрофии левого желудочка (left ventricular hypertrophy - LVH) у 5/6 NX крыс при лечении их соединением примера 1 (1, 10 и 100 мг/кг)
Методы: Исследования, проведенные ранее, показали, что 5/6 NX крыса является гипертензивной и в ее организме наблюдается гипертрофия левого желудочка. Поэтому для исследования 5/6 NX крыс с 2 недельной уремией получили от Charles River и кормили кормом TD04151 (0,6% Ca и 0,9%P) в течение 4 недель перед исследованием. 5/6 NX крысам вводили соединение примера 1 (1, 10 и 100 мг/кг) или разбавитель (95% ПЭГ/5% EtOH) 3х/неделя в течение шести недель. На 0, 13 и 41 дни у крыс производили забор крови (через 24 часа после введения дозы). В конце 6-ти недельного периода лечения крыс анестезировали изофлураном и записывали эхокардиограмму. Массу левого желудочка определяли в соответствии с рекомендациями Американского Общества Эхокардиографии (American Society of Echocardiography) с использованием метода куба (cube method). После эвтаназии проводили непосредственное определение массы левого желудочка. Примечание: Для сравнения в данное исследование были включены крысы, получающие разбавитель (ложное лечение), и 5/6 NX крысы (3х/неделя в течение шести недель) из предыдущих исследований. Полученные данные представляли собой средние значения ± среднеквадратичное отклонение. Данные анализировали с использованием ANOVA и, когда отмечено, t-теста. *Показывает р< 0,05 по сравнению с контролем (симулированное лечение).
Результаты: Различий в массе левого желудочка у 5/6 NX крыс, получавших лечение соединением примера 1 (1, 10 и 100 мг/кг, 3х/нед. в течение 6 недель), и у 5/6 NX крыс, получавших разбавитель (ложное лечение), обнаружено не было. Не было различий и в сердечной деятельности у крыс различных групп. Не наблюдалось обострения LVH при лечении соединением примера 1 и по сравнению с контрольными группами.
Способы изобретения
Соединения и композиции согласно изобретению применимы для модуляции действия рецепторов витамина D. В частности, соединения и композиции согласно изобретению могут применяться для лечения и предупреждения расстройств, опосредуемых рецепторами витамина D. Обычно такие расстройства могут облегчаться селективным модулированием рецептора витамина D у млекопитающего, предпочтительно введением соединения или композиции согласно изобретению одного или в сочетании с другим активным лекарственным средством, например, как части программы лечения.
Соединения согласно изобретению, включая, но без ограничения, соединения, описанные в конкретных примерах, обладают сродством к рецепторам витамина D. В качестве активаторов рецепторов витамина D соединения согласно изобретению могут применяться для лечения и предупреждения ряда заболеваний или состояний, опосредуемых рецептором витамина D.
Например, было показано, что активаторы рецептора витамина D играют важную роль в снижении уровней содержания паратиреоидного гормона (Hudson, J. Q. The Annals of Pharmacotherapy, 2006, 40, 1584-1593). По существу, активаторы рецепторов витамина D подходят для лечения состояний и расстройств, связанных с хронической болезнью почек. Некоторые активаторы рецепторов витамина D не повышают функциональной активности кишечных рецепторов витамина D, что ограничивает их кальцемический и гиперфосфатемический эффекты, а также связанные с ними побочные эффекты (Slatopolsky, E.; Finch, J.; Ritter, C; Takahashi, F. American Journal of Kidney Disease, 1998, 4, S40-S47). Исследования показали, что терапевтическое лечение активаторами рецепторов витамина D снижает прогрессирование болезни почек (Agarwal, R.; Acharya, M.; Tian, J.; Hippensteel, R.L.; Melnick, J.Z.; Qiu, P.; Williams, L.; Batlle, D. Kidney International, 2005, 68, 2823-2828; Schwarz, U.; Amann, K.; Orth, S.R.; Simonaviciene, A.; Wessels, S.; Ritz, E. Kidney International, 1998, 53, 1696-1705).
Кроме того, было показано, что активаторы рецепторов витамина D вовлечены в скелетный и минеральный гомеостаз. Такие активаторы рецепторов имеют большое значение для интестинальной абсорбции кальция и последующей анаболической активности на кости (Hendy, G.N.; Hruska, K. A.; Methew, S.; Goltzman, D. Kidney International, 2006, 69, 218-223). Было показано, что некоторые агонисты могут применяться для селективного лечения болезней костей со сниженным воздействием на супрессию паратиреоидного гормона (Shevde, N. K.; Plum, L. A.; Clagett-Dame, M.; Yamamoto, H.; Pike, J. W.; DeLuca, H. F. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 13487-13491; Uchiyama, Y.; Higuchi, Y.; Takeda, S.; Masaki, T.; Shira-Ishi, A.; Sato, K.; Kubodera, N.; Ikeda, K.; Ogata, E. Bone, 2002, 4, 582-588; Shiraishi, A.; Higashi, S.; Ohkawa, H.; Kubodera, N.; Hirasawa, T.; Ezawa, I.; Ikeda, K.; Ogata, E. Calcified Tissue International, 1999, 65, 311-316).
Активаторы рецепторов витамина D были задействованы в существующих неблагоприятных эффектах на многие характеристики кровеносной системы. Система рецепторов витамина D играет важную роль в поддержании противотромботического гомеостаза (Aihara, K.; Azuma, H.; Akaike, M.; Ikeda, Y.; Yamashita, M.; Sudo, T.; Hayashi, H.; Yamada, Y.; Endoh, F.; Fujimura, M.; Yoshida, T.; Yamaguchi, H.; Hashizume, S.; Kato, M.; Yoshimura, K.; Yamamoto, Y.; Kato, S.; Matsumoto, T.J. Biol. Chem., 2004, 279, 35798-35802). Было показано, что активаторы рецептора витамина D изменяют экспрессию и активность белков, важных для коагуляции, таких как тромбомодулин, фактор ткани и ингибитор активатора плазминогена 1, что обеспечивает возможность лечения атеросклеротических заболеваний (Beer, T. M.; Venner, P.M.; Ryan, C.W.; Petrylak, D.P.; Chatta, G.; Ruether, J.D.; Chi, K.N.; Curd, J.G.; DeLoughery, T.G. British Journal of Haematology, 2006, 135, 392-394$; Ohsawa, M.; Koyama, T.; Yamamoto, K.; Hirosawa, S.; Kamei, S.; Kamiyama, R. Circulation, 2000, 102, 2867-2872). Система ренин-ангиотензин II является центральной в регулировании кровяного давления, и повышенные уровни ренина приводят к гипертензии и сердечной гипертрофии. Активаторы рецептора витамина D напрямую подавляют транскрипцию гена ренина в механизме, зависимом от рецепторов витамина D, обеспечивая механизм контроля данной системы (Li, Y.C; Qiao, G.; Uskokovic, M.; Xiang, W.; Zheng, W.; Kong, J. Journal of Steroid Biochemistry & Molecular Biology, 2004, 89-90, 397-392). Пациенты с хронической болезнью почек, получающие поддерживающий гемодиализ, зачастую страдают сердечно-сосудистыми осложнениями, из которых наиболее распространенным является ишемическая болезнь сердца как результат гипертрофии левого желудочка. Гиперпаратиреоз также вносит свой вклад, и даже частичный контроль активатора рецептора витамина D приводит к регрессии гипертрофии миокарда без изменения других гемодинамических параметров (Park, C. W.; Oh, Y. S.; Shin, Y. S.; Kim, C-M.; Kim, Y.-S.; Kim, S. Y.; Choi, E. J.; Chang, Y. S.; Bang, B. K. American Journal of Kidney Diseases, 1999, 33, 73-81).
Рецептор витамина D экспрессирован на большинстве типов клеток иммунной системы и, в частности, на клетках, модулирующих ответы Т лимфоцитов. В настоящее время активаторы рецепторов витамина D используются для местного лечения псориаза. Испытания на животных моделях подтверждают, что активаторы рецепторов витамина D могут применяться для лечения артрита, аутоиммунного диабета, экспериментального аллергического энцефаломиелита, воспалительной болезни кишечника и системной красной волчанки, что предполагает возможность их терапевтического применения в терапевтическом лечении людей (Adorini, L. Cellular Immunology, 2005, 233, 115-124).
Ряд путей проведения сигнала, вовлеченных в рак, находится под влиянием активаторов рецепторов витамина D. Они заметно, хотя и со значительной неоднородностью, ответственны за антипролиферативный, противоангиогенный и продифференционный эффекты в широком спектре раковых заболеваний, проводимых через геномный и негеномный механизмы (Deeb, K.K.; Trump, D.L.; Johnson, C.S. Nature Reviews Cancer, 2007, 7, 684-700). Роль метаболизма витамина D, оказывается, имеет большое значение для регулирования клеточной пролиферации клеток в предстательной железе (Lou, Y.-R.; Qiao, S.; Talonpoika, R.; Syvala, H.; Tuohimaa, P. Journal of Steroid Biochemistry and Molecular Biology, 2004, 92, 317-3250). Существует связь между подавлением аутокринных факторов роста IL-6 и IL-8 активаторами рецепторов витамина D и развитием саркомы Капоши (Masood, R.; Nagpal, S.; Zheng, T.; Cai, J.; Tulpule, A.; Smith, D.L.; Gill, P.S. Blood, 2000, 96, 3188-3194). Аналоги витамина D распространяют дифференциирующий эффект на лейкозные клетки (James, S.Y.; Williams, M.A.; Newland, A.C; Colston, K.W. Gen. Pharmaс., 1999, 32, 143-154).
Фактические уровни доз активных ингредиентов в фармацевтических композициях согласно изобретению могут изменяться таким образом, чтобы получить количество активного(ых) соединения(ий), которое эффективно для достижения желательного терапевтического ответа для конкретного пациента, композиций и способа введения. Выбранный уровень дозировки будет зависеть от активности конкретного соединения, способа введения, тяжести состояния, подлежащего лечению, а также состояния и предшествующей истории болезни пациента, подлежащего лечению. Однако квалифицированный специалист в данной области техники может назначать лечение соединением с начальных доз, более низких, чем необходимы для достижения желательного терапевтического эффекта, и постепенно повышать дозировку до достижения желательного эффекта.
При применении в описанных выше или других способах лечения терапевтически эффективное количество одного из соединений согласно изобретению может применяться в чистой форме или, когда такие формы существуют, в форме фармацевтически приемлемой соли, сложного эфира, амида или пролекарства. Альтернативно, соединение может вводиться в виде фармацевтической композиции, содержащей указанное соединение в сочетании с одним или несколькими фармацевтически приемлемыми носителями. Фраза «терапевтически эффективное количество» соединения согласно изобретению означает количество, достаточное для лечения расстройств, с подходящим приемлемым соотношением «польза/риск», применимое для любого медицинского лечения. Однако следует представлять, что общая суточная доза применения соединений и композиций согласно изобретению будет определяться лечащим врачом в рамках обычного медицинского назначения. Конкретный уровень терапевтически эффективной дозы для любого конкретного пациента будет зависеть от различных факторов, включая расстройство, подлежащее лечению, и тяжесть указанного расстройства; активность конкретного применяемого соединения; конкретную применяемую композицию; возраст, массу тела, общее состояние здоровья, пол и рацион питания пациента; время введения, способ введения и скорость экскреции из организма конкретного применяемого соединения; продолжительность лечения; лекарственные средства, используемые совместно, или лекарственные средства, применение которых случайно совпадает с применением конкретного используемого соединения; и аналогичные факторы, хорошо известные в области медицины. Например, специалист в данной области техники может назначать лечение соединением с начальных доз, более низких, чем необходимо для достижения желательного терапевтического эффекта, и постепенно повышать дозировку до достижения желательного эффекта.
Общая суточная доза соединений согласно данному изобретению, вводимая человеку или низшему животному, находится в интервале от примерно 0,01 мкг до примерно 150 мг. Более предпочтительные дозы могут находиться в интервале от примерно 0,010 мкг до примерно 10 мг. Если нужно, эффективная суточная доза может делиться на несколько доз для введения. Следовательно, композиции разовой дозы могут содержать такие количества или их дольные единицы, которые могли бы составить суточную дозу.
Соединения согласно изобретению представляют собой активаторы рецепторов витамина D, которые модулируют функцию рецепторов витамина D посредством изменения активности рецептора или передачи сигнала. Таким образом, введение терапевтически эффективного количества соединения формулы (I) млекопитающему предоставляет способ селективного модулирования действия рецепторов витамина D.
Кроме того, введение терапевтически эффективного количества соединения формулы (I) млекопитающему предоставляет способ лечения или предупреждения состояния или расстройства, выбранного из группы, включающей болезнь почек, вторичный гиперпаратиреоз, связанный с хронической болезнью почек, остеопороз, остеомаляцию, остеодистрофию, образование тромбов, ренин-ангиотензиновую систему, миокардиальную гипертрофию, гипертензию, аутоиммунные расстройства, иммуносуппрессию, отторжение трансплантата, артрит, рассеянный склероз, псориаз, воспалительную болезнь кишечника, диабет 1 типа и системную красную волчанку, рак толстой кишки, рак предстательной железы, рак молочной железы, лейкоз и саркому Капоши. Более предпочтительно, введение терапевтически эффективного количества соединения формулы (I) предоставляет способ лечения вторичного гиперпаратиреоза, гипертензии и миокардиальной гипертрофии.
Соединения, идентифицированные способами, описанными выше, могут вводиться в форме одного фармацевтического средства или в сочетании с одним или несколькими другими фармацевтическими средствами, где такое сочетание не вызывает нежелательных побочных эффектов.
Подразумевается, что представленное выше подробное описание и сопровождающие его примеры приведены только для иллюстрации изобретения и не должны рассматриваться как ограничения области изобретения, которая определена исключительно прилагаемой формулой изобретения и ее эквивалентами. Различные изменения и модификации описанных вариантов осуществления изобретения будут очевидны специалисту в данной области техники. Такие изменения и модификации, включая, но без ограничений, химические структуры, заместители, производные, промежуточные продукты, способы синтеза, препараты и/или способы применения изобретения, могут осуществляться без выхода за пределы объема и области настоящего изобретения.
Реферат
Настоящее изобретение относится к новым соединениям формулы (I) или их фармацевтически приемлемым солям, используемым для лечения или предупреждения расстройств, опосредуемых рецепторами витамина D, а также к фармацевтической композиции, содержащей данные соединения.В формуле (I) атом углерода, к которому присоединен X, имеет S или R конфигурацию, X представляет собой -CHOC(O)R, Yи Yкаждый представляет собой водород, Yи Yвместе образуют метиленовую группу, Zпредставляет собой фтор, гидроксильную группу или гидроксиметил, Zпредставляет собой фтор или гидроксильную группу, Rпредставляет собой Cалкил, Cалкиламино, CалкилкарбонилоксиCалкил или гидроксиCалкил. 3 н. и 2 з.п. ф-лы, 4 ил., 6 табл., 19 пр.
Формула
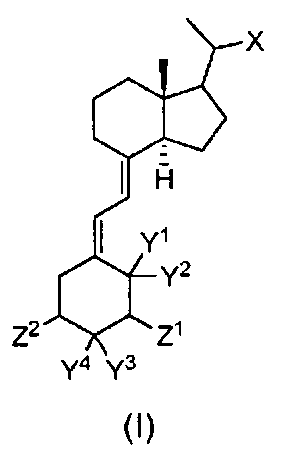
или его фармацевтически приемлемая соль, где
атом углерода, к которому присоединен X, имеет S или R конфигурацию;
X представляет собой -CH2OC(O)R2;
Y1 и Y2 каждый представляет собой водород;
Y3 и Y4 вместе образуют метиленовую группу;
Z1 представляет собой фтор, гидроксильную группу или гидроксиметил;
Z2 представляет собой фтор или гидроксильную группу;
R2 представляет собой C1-10алкил, C1-10алкиламино, C1-10алкилкарбонилоксиC1-10алкил или гидроксиC1-10алкил.
X представляет собой -CH2OC(O)R2;
Y1 и Y2 каждый представляет собой водород;
Y3 и Y4 вместе образуют метиленовую группу;
Z1 представляет собой гидроксильную группу;
Z2 представляет собой гидроксильную группу; и
R2 представляет собой C1-10алкил, C1-10алкиламино, C1-10алкилкарбонилоксиалкил или гидроксиC1-10алкил.
X представляет собой -СН2ОС(О)R2;
Y1 и Y2 каждый представляет собой водород;
Y3 и Y4 вместе образуют метиленовую группу;
Z1 представляет собой гидроксильную группу;
Z2 представляет собой гидроксильную группу; и
R2 представляет собой C1-10алкил.
(2S)-2-[(1R,3R,7Е,17β)-1,3-дигидрокси-2-метилен-9,10-секоэстра-5,7-диен-17-ил]пропилпивалат;
(2R)-2-[(1R,3R,7Е,17β)-1,3-дигидрокси-2-метилен-9,10-секоэстра-5,7-диен-17-ил]пропилпивалат;
(2S)-2-[(1R,3R,7Е,17β)-1,3-дигидрокси-2-метилен-9,10-секоэстра-5,7-диен-17-ил]пропил-2,2-диметилбутаноат;
(2S)-2-[(1R,3R,7E,17β)-1,3-дигидрокси-2-метилен-9,10-секоэстра-5,7-диен-17-ил]пропил-трет-бутилкарбамат;
(2S)-2-[(1R,3R,7Е,17β)-1,3-дигидрокси-2-метилен-9,10-секоэстра-5,7-диен-17-ил]пропил-2-(ацетилокси-)-2-метилпропаноат;
(2S)-2-[(1R,3R,7Е,17β)-1,3-дигидрокси-2-метилен-9,10-секоэстра-5,7-диен-17-ил]пропил-2-гидрокси-2-метилпропаноат
или его фармацевтически приемлемую соль.
Комментарии