1,4-бензотиазепин-1,1-диоксид, фармацевтическая композиция, понижающая уровень липидов в крови, способ лечения клинического состояния млекопитающего, способ получения соединения - RU2156245C2
Код документа: RU2156245C2
Чертежи
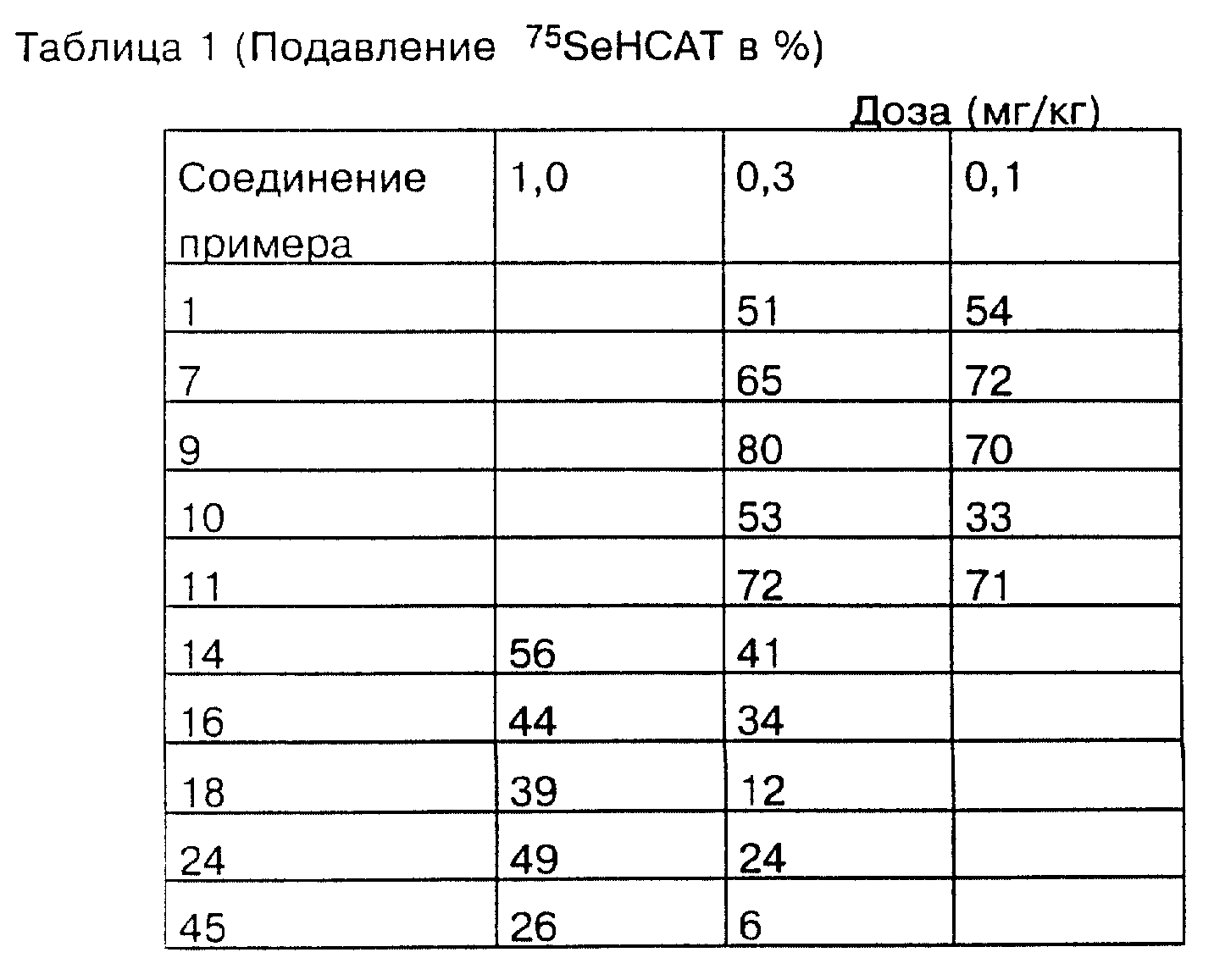


Описание
Изобретение относится к новым гиполипидемическим соединениям, к способам их получения и необходимым для этого промежуточным соединениям и к содержащим их фармацевтическим составам и их применению в медицине, в частности, для профилактики и лечения таких гиперлипидемических состояний, как атеросклероз.
Гиперлипидемия часто связана с повышенными концентрациями в плазме крови липопротеинового холестерина низкой (ЛHП) и очень низкой (ЛОHП) плотности. Эти концентрации могут быть понижены ослаблением всасывания желчных кислот из кишечника. Одним из способов, которым этого можно достичь, является подавление активного всасывания желчных кислот в подвздошной кишке. Такое подавление стимулирует превращение холестерина в желчную кислоту печенью, а увеличение в результате этого потребности в холестерине приводит к соответственному ускорению удаления ЛHП и ЛОHП холестерина из плазмы или сыворотки крови.
Был обнаружен новый класс гетероциклических соединений, снижающих концентрацию ЛHП и ЛОHП холестерина в плазме или сыворотке крови и, следовательно, применимых в качестве гиполипидемических средств. Уменьшая концентрацию холестерина и сложного холестеринового эфира в плазме, соединения согласно изобретению замедляют развитие атеросклеротических поражений и уменьшают частоту коронарных сердечных и связанных с ними заболеваний. Последние определяют как сердечные заболевания, связанные с повышенной концентрацией холестерина и сложного холестеринового эфира в плазме или сыворотке крови.
В данном описании гиперлипидемическое состояние определено как любое состояние, при котором общая концентрация холестерина (ЛHП + ЛОHП) в плазме или сыворотке крови превышает 240 мг/дл (6.21 ммолей/л) (J. Amer. Med. Assn. 256, 20. 2849-2858 (1986)).
В заявке N WO 93/16055 описаны соединения формулы (0)
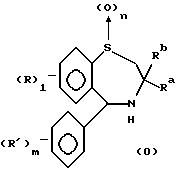
в которой l - целое число от 0 до 4;
m - целое число от 0 до 5;
n - целое число от 0 до 2;
R и R' - атомы или функциональные группы, произвольно выбранные из группы, содержащей галоген, нитрогруппу, фенилалкоксил, C1-4алкоксил, C1-6алкил, -SO3R'', -CO2R'' и -O(CH2)p SO3P'', где p - целое число от 1 до 4, а R'' - водород или C1-6алкил группа, в котором указанные фенилалкоксил-, алкоксил- и алкил- произвольно замещены одним или более атомами галогена;
Ra - прямоцепной C1-6алкил и
Rb - прямоцепной C2-6алкил;
и их соли, сольваты и физиологически активные производные, пригодные в качестве гиполипидемических средств.
Мы открыли группу соединений с более высокой гиполипидемической активностью in vivo в сравнении с конкретными примерами из заявки N WO 93/16055.
Согласно изобретению предложены соединения формулы (I):
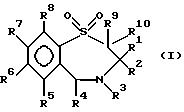
в которой R1 - прямоцепной C1-6алкил,
R2 - прямоцепной C1-6алкил,
R3 - водород или группа OR11, где R11 - водород, произвольно замещенный C1-6алкил, или C1-6алкилкарбонил;
R4 - пиридил или произвольно замещенный фенил;
R5, R6, R7 и R8 - одинаковы или различны и каждый выбран из группы, содержащей водород, галоген, циан, R15-ацетилид, OR15, произвольно замещенный C1-6алкил, COR15, CH(OH)R15, S(O)nR15, P(O)(OR15)2, OCOR15, OCF3, OCN, SCN, NHCN, CH2OR15, CHO, (CH2)pCN, CONR12R13, (CH2)pCO2R15, (CH2)pNR12R13, CO2R15, NHCOCF3, NHSO2R15, OCH2OR15, OCH=CHR15, O(CH2CH2O)nR15, O(CH2)pSO3R15, O(CH2)pNR12R13 и O(CH2)pN+R12R13R14, где p - целое число от 1 до 4, n- целое число от 0 до 3, a R12, R13, R14 и R15 произвольно выбраны из группы, содержащей водород и произвольно замещенный C1-6алкил;
или R6 и R7 связаны в виде группы
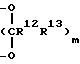
в которой R12 и R13 идентичны определенным ранее и m = 1 или 2; и
R9 и R10, одинаковы или различны и каждый представлен водородом или C1-6алкилом; и их соли, сольваты или физиологически активные производные, при условии, если R3 - водород, то либо R7 - не водород, либо, по меньшей мере, два из радикалов R5, R6, R7 и R8 не являются водородом; и их соли, сольваты и физиологически активные производные.
Если R4 - замещенный фенил, возможны от одного до пяти, предпочтительно один или два одинаковых или различных заместителей, выбранных каждый из группы, содержащей галоген, гидроксил, нитрогруппу, фенил-C1-6алкоксил, C1-6алкоксил, произвольно замещенный C1-6алкил, S(O)nR15, CO2R15, O(CH2CH2O)nR15, O(CH2)p SO3R15, O(CH2)pNR12R13 и O(CH2)pN+R12R13R14, где p, n, R12 , R13, R14 и R15 идентичны определенным ранее.
Кроме того, согласно изобретению предложены соединения формулы (I), в которой:
R1 и
R2 - прямоцепной C1-6алкил;
R3 - водород или гидроксил;
R4 - незамещенный фенил;
R5 - водород:
R9
и R10 - водород; и
R7 - выбранный из группы, состоящей из галогена, гидроксила, C1-6алкоксила, произвольно замещенного C1-6алкила, - S(O)nR15, -OC(O)R15 и CH2OR15, где R15 - водород или C1-6алкил; и R6 и R8 - произвольно выбраны из группы,
состоящей из водорода и групп, указанных в определении R7; или
R8 - водород, a R6 и R7 связаны в виде группы -O-(CH2)m-O-, в которой m =
1 или 2; и их соли, сольваты и физиологически активные производные.
Предпочтительны соединения формулы (I), в которых R8 - водород, a R6 и R7 - C1-6алкоксил, более конкретно метоксил.
В число предпочтительных соединений формулы (I) входят соединения формул (II), (III), (IV) или (IVa):
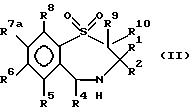
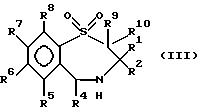
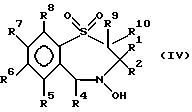
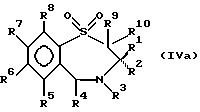
в которых R1 -R10 определены ранее, a R7a выбран из группы, содержащей галоген, циан, R15-ацетилид, OR15, произвольно замещенный C1-6алкил, COR15, CH(OH)R15, S(O)nR15, P(O)(OR15)2, OCOR15, OCF3, OCN, SCN, NHCN, CH2OR15, CHO, (CH2)pCN, CONR12R13, (CH2)pCO2R15, (CH2)pNR12R13, CO2R15, NHCOCF3, NHSO2R15, OCH2OR15, OCH=CHR15, O(CH2CH2O)pR15, O(CH2)p SO3R15, O(CH2)pNR12R13 и O(CH2)pN+R12R13R14, где n, p, R12 , R13, R14 и R15 идентичны определенным ранее при условии, что в соединениях формулы (III) по меньшей мере два из радикалов R5 - R8 не являются водородом, и их соли, сольваты и физиологически активные производные.
Если один или более радикалов R3-R8 или R11-R14 являются замещенным C1-6алкилом или содержат эту группу, то заместители могут быть одинаковы или различны и каждый из них выбран из группы, содержащей гидроксил, галоген, C1-6алкил, C1-6 алкоксил, COR16, нитрил, CO2R16, SO3R16, NR17R18, N+R17R18R19, где R16-R19 одинаковы или различны и каждый представлен водородом или C1-6алкилом.
В качестве R1 пригодны метил, этил или н-пропил, предпочтительно этил, а в качестве R2-метил, этил, н-пропил, н-бутил или н-фенил, предпочтительно н-бутил.
R5 - предпочтительно водород.
Пригодны R7 и R7a, выбранные из группы, состоящей из OR15, S(O)nR15, OCOR15, OCF3, OCN, SCN, CHO, OCH2OR15, OCH=CHR15, O(CH2CH2O)nR15, O(CH2)pSO3R15, O(CH2)pNR12R15 и O(CH2)p; N+R12R13R14, где p - целое число от 1 до 4, n - целое число от 0 до 3 и R12, R13, R14 и R15 - произвольно выбраны из группы, состоящей из водорода и произвольно замещенного C1-6алкила. R7 и R7a - предпочтительно OR15.
В качестве R9 и R10 пригодны водород, метил или этил, предпочтительно водород для обоих.
В качестве R4 пригодны пиридил или фенил, произвольно замещенные предпочтительно в 4-й и/или 3-й позиции заместителем, выбранным из группы, содержащей галоген, метил, этил, метоксил, этоксил, трифторметил, гидроксил, карбоксил или O(CH2)3 SO3H. Предпочтительный R4 - незамещенный фенил.
В соединениях формулы (II) один или два, предпочтительно один из радикалов R5, R6 и R8 не должен быть водородом и каждый из них выбран из группы, содержащей произвольно замещенный фтором C1-4алкил, C1-4алкоксил, галоген и гидроксил. Лучше, если каждый из них выбран из группы, содержащей метил, метоксил, гидроксил, трифторметил и галоген. Предпочтительно, когда R6 - метоксил или бром, a R5 и R8 - водород. R7a выбран из группы, содержащей произвольно замещенный фтором C1-4алкил, C1-4алкоксил, галоген и гидроксил. Лучше, когда R7a - метоксил, гидроксил или трифторметил, но предпочтителен метоксил.
В соединениях формулы (III) по меньшей мере один, а предпочтительно два из радикалов R5 - R8 должны быть водородом. Предпочтительно, чтобы по меньшей мере один из радикалов R6 и R7 не был водородом. Если ни один из R5 - R8 не водород, то они выбраны из группы, содержащей произвольно замещенный фтором C1-4алкил, C1-4алкоксил, галоген и гидроксил, лучше всего из группы, содержащей метил, метоксил, гидроксил, трифторметил или хлор, а наиболее предпочтителен метоксил.
В соединениях формулы (IV) два, три или четыре из радикалов R5 - R8 должны быть водородом, остальные же выбраны из группы, содержащей произвольно замещенный фтором C1-4алкил, C1-4алкоксил, галоген и гидроксил, лучше всего из группы, содержащей метил, метоксил, гидроксил, трифторметил или хлор. Наиболее предпочтителен метоксил.
В соединениях формулы (IVa) по меньшей мере один, а предпочтительно два из радикалов R5 - R8 должны быть водородом. Предпочтительно, чтобы по меньшей мере один из радикалов R6 и R7 не был водородом. Если ни один из R5 - R8 не водород, то они выбраны из группы, содержащей произвольно замещенный фтором C1-4алкил, C1-4алкоксил, галоген и гидроксил, лучше всего из группы, содержащей метил, метоксил, гидроксил, трифторметил или хлор. Наиболее предпочтительно, когда R1 -н-бутил, R2-этил, R3, R5, R8, R9 и R10 - водород, R4 - пиридил или произвольно замещенный фенил, а R6 и R7- метоксил.
Предпочтительные соединения формулы (I) выбраны из группы, содержащей:
(3R, 5R)-3-бутил-3-этил-2,3,4,5-тетрагидро-7,8-диметокси-5- фенил-1,4-бензотиазепин-1,
1-диоксиды;
(3R, 5R)-3-бутил-3-этил-2,3,4,5-тетрагидро-7,8-диметокси- 5-фенил-1,4-бензотиазепин-4-ол-1,1-диоксид;
(±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-7,
8-диметокси-5-фенил- 1,4-бензотиазепин-1,1-диоксид;
(±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-7,8-диметокси-5- фенил-1,4-бензотиазепин-4-ол-1,1-диоксид;
(3R,
5R)-7-бром-3-бутил-3-этил-2,3,4,5-тетрагидро-8-метокси-5-фенил- 1,4-бензотиазепин-1,1-диоксид;
(3R, 5R)-7-бром-3-бутил-3-этил-2,3,4,5-тетрагидро-8-метокси-5-фенил- 1,4-бензотиазепин-4-ол-1,
1-диоксид;
(3R, 5R)-3-бутил-3-этил-2,3,4,5-тeтparидpo-5-фeнил-1,4-бeнзoтиaзeпин- 7,8-диол-1,1-диоксид;
(3R, 5R)-3-бутил-3-этил-2,3,4,5-тетрагидро-8-метокси-5-фенил-1,
4- бензотиазепин-7-ол-1,1-диоксид;
(3R, 5R)-3-бутил-3-этил-2,3,4,5-тетрагидро-7-метокси-5-фенил-1,4- бензотиазепин-8-ол-1,1-диоксид;
(±)-транс-3-бутил-3-этил-2,3,4,
5-тетрагидро-8-метокси-5-фенил-1,4- бензотиазепин-1,1-диоксид;
(±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-5-фенил-1,4- бензотиазепин-8-ол-1,1-диоксид;
(±
)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-5-фенил-1,4- бензотиазепин-4,8-диол-1,1-диоксид;
(±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-5-фенил-1,4- бензотиазепин-1,1-диоксид;
(±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-8-метил-5-фенил-1,4- бензотиазепин-1,1-диоксид;
(±)-транс-2-((3-бутил-3-этил-2,3,4,5-тетрагидро-8-метокси-5- фенил-1,
4-бензотиазепин-7-ил)метокси)этанол-S,S-диоксид;
(±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-8-гидрокси-5- фенил-1,4-бензотиазепин-7-карбальдегид-1,1-диоксид;
(±
)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-5-фенил-1,4- бензотиазепин-8-тиол-1,1-диоксид;
(±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-5-фенил-1,4- бензотиазепин-8-сульфокислота-1,
1-диоксид;
(7R, 9R)-7-бутил-7-этил-6,7,8,9-тетрагидро-9-фенил-1,3-диоксоло (4,5H)(1,4)-бензотиазепин-5,5-диоксид;
(±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-8,
0- диметокси-5-фенил-1,4-бензотиазепин-1,1-диоксид:
(3R, 5R)-3-бутил-3-этил-5-(4-фторфенил)-2,3,4,5-тетрагидро-7,8- диметокси-1,4-бензотиазепин-4-ол-1,1-диоксид;
(±
)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-8-метокси-5-фенил- 1,4-бензотиазепин-7-метанол-S,S-диоксид;
(3R, 5R)-3-бутил-3-этил-2,3,4,5-тетрагидро-8-метокси-7-нитро-5-фенил- 1,4-бензотиазепин-1,
1-диоксид;
(±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-8-метокси-7- (метоксиметил)-5-фенил-1,4-бензотиазепин-1,1-диоксид;
(3R, 5R)-3-бутил-3-этил-2,3,4,5-тетрагидро-5-фенил-1,
4-бензотиазепин- 7,8-диил-диацетат-1,1-диоксид;
(8R, 10R)-8-бутил-8-этил-2,3,7,8,9,10-гексагидро-10-1,4- диоксоно(2,3-H)(1,4)-бензотиазепин-6,6-диоксид;
(3R, 5R)-3-бутил-7,
8-диэтокси-2,3,4,5-тетрагидро-5-фенил-1,4- бензотиазепин-1,1-диоксид;
(±)-транс-3-бутил-8-этокси-3-этил-2,3,4,5-тетрагидро-5- фенил-1,4-бензотиазепин-1,1-диоксид;
(±
)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-8-изопропокси-5- фенил-1,4-бензотиазепин-1,1-диоксид-гидрохлорид;
(±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-5-фенил-1,
4- бензотиазепин-8-карбальдегид-1,1-диоксид;
3,3-диэтил-2,3,4,5-тетрагидро-7,8-диметокси-5-фенил-1,4- бензотиазепин-1,1-диоксид;
3,3-диэтил-5-(4-фторфенил)-2,3,4,
5-тетрагидро-8-метокси-1,4- бензотиазепин-1,1-диоксид:
3,3-диэтил-2,3,4,5-тетрагидро-8-метокси-5-фенил-1,4-бензотиазепин- 1,1-диоксид;
3,3-диэтил-2,3,4,5-тетрагидро-5-фенил-1,
4-бензотиазепин-4,8-диол- 1,1-диоксид;
(RS)-3,3-диэтил-2,3,4,5-тетрагидро-4-гидрокси-7,8-диметокси-5-фенил- 1,4-бензотиазепин-1,1-диоксид;
(±)-транс-3-бутил-8-этокси-3-этил-2,
3,4,5-тетрагидро-5-фенил- 1,4-бензотиазепин-4-ол-1,1-диоксид;
(±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-8-изопропокси-5- фенил-1,4-бензотиазепин-4-ол-1,1-диоксид;
(±
)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-7,8,9-триметокси-5- фенил-1,4-бензотиазепин-4-ол-1,1-диоксид;
(3R, 5R)-3-бутил-3-этил-2,3,4,5-тетрагидро-5-фенил-1,4-бензотиазепин- 4,7,8-триол-1,
1-диоксид;
(±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-4,7,8-триметокси-5- фенил-1,4-бензотиазепин-1,1-диоксид;
(±)-транс-3-бутил-3-этил-5-фенил-2,3,4,5-тетрагидро-7,
8- диметокси-1,4-бензотиазепин-4-ил-ацетат-S,S-диоксид;
3,3-диэтил-2,3,4,5-тетрагидро-5-фенил-1,4-бензотиазепин-8-ол-1,1- диоксид;
3,3-диэтил-2,3,4,5-тетрагидро-7-метокси-5-фенил-1,
4-бензотиазепин- 8-ол-1,1-диоксид;
3,3-дибутил-2,3,4,5-тетрагидро-5-фенил-1,4-бензотиазепин-8- ол-1,1-диоксид;
(±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-1,
1-диоксо-5- фенил-1,4-бензотиазепин-8-ил-гидросульфат;
(±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-1,1-диоксо-5- фенил-1,4-бензотиазепин-8-ил-дигидрофосфат;
3,3-диэтил-2,3,4,
5-тетрагидро-1,1-диоксо-5-фенил-1,4-бензотиазепин- 8-ил-гидросульфат;
3,3-диэтил-2,3,4,5-тетрагидро-1,1-диоксо-5-фенил-1,4-бензотиазепин- 8-ил-дигидрофосфат;
(±
)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-1,1-диоксо-5- фенил-1,4-бензотиазепин-8-ил-аспартат; и
3,3-диэтил-2,3,4,5-тетрагидро-1,1-диоксо-5-фенил-1,4-бензотиазепин- 8-ил-аспартат.
Для применения в медицине особенно удобны фармацевтически приемлемые соли благодаря их лучшей растворимости в воде в сравнении с исходными (т.е. в форме оснований) соединениями. Очевидно, что такие соли должны содержать фармацевтически приемлемые анион или катион. В число подходящих фармацевтически приемлемых кислых солей присоединения для соединений согласно изобретению входят соли таких неорганических кислот, как соляная, бромистоводородная, фосфорная, метафосфорная, азотная, сульфоновая и серная, и таких органических кислот, как уксусная, бензолсульфоновая, бензойная, лимонная, этансульфоновая, фумаровая, глюконовая, гликолевая, изотионовая, молочная, мясомолочная, малеиновая, яблочная, метансульфоновая, янтарная, п-толуолсульфоновая, винная и трифторуксусная. Для медицинских целей особенно предпочтительны хлориды. В число подходящих фармацевтически приемлемых основных солей присоединения для соединении согласно изобретению входят соли аммония и таких щелочных металлов, как натрий и калий, и соли таких щелочноземельных металлов, как магний и кальций.
Соли согласно изобретению с фармацевтически неприемлемым анионом могут служить промежуточными соединениями для получения или очистки фармацевтически приемлемых солей и/или для нетерапевтических целей, например для применения in vitro.
Термин "физиологически активное производное" здесь означает любое физиологически активное производное соединения согласно изобретению, например сложный эфир, который после введения животному (например, человеку) способствует (прямо или косвенно) возникновению такого соединения или его активного метаболита.
Дальнейшим объектом изобретения являются "пролекарства" соединений согласно изобретению. Такие пролекарства могут в процессах обмена веществ in vivo образовывать соединения согласно изобретению. Эти пролекарства могут быть или не быть активны сами по себе.
Соединения согласно изобретению могут существовать также в различных, например, аморфных и кристаллических полиморфных видах. Все полиморфные разновидности соединений согласно изобретению включены в его объем и являются его дальнейшими объектами.
Используемый здесь термин "алкил" означает, если не обусловлено иное, одновалентный радикал с прямой или разветвленной цепью. Аналогично, термин "алкоксил" относится к одновалентному радикалу с прямой или разветвленной цепью, который атомом кислорода соединен с породившим его остатком молекулы. Термин "фенилалкоксил" означает одновалентную фенильную группу, соединенную с двухвалентным C1-6алкенилом, который как таковой атомом кислорода соединен с породившим его остатком молекулы.
Соединения формулы (I) существуют в форме, когда углеродные центры -C(R1)(R2)- и -CHR4-хиральны. Изобретение включает каждый возможный существенно свободный, т.е. соединенный менее чем с 5% любых других возможных изомеров, оптический изомер и его смеси с одним или более оптическим изомером в любых пропорциях, включая рацемические смеси.
Для нужд описания абсолютные хиральности вышеупомянутых углеродных центров даны в порядке: -C(R1)(R2)-, затем -CHR4-.
В случаях неопределенности абсолютной стереохимии при -C(R1)(R2)- и -CHR4 - соединения согласно изобретению описывают в терминах относительных положений заместителей R1/R2 и H/R4. Так, соединения, в которых более крупный, т. е. более массивный из заместителей R1 и R2, и заместитель R4 расположены по одну сторону тиазепинового кольца, обозначены здесь "цис", а предпочтительные соединения, в которых эти же заместители расположены по разные стороны кольца, обозначены "транс". Специалисту очевидно, что каждое из цис- и транс-соединений согласно изобретению может существовать в двух энантиомерных формах, индивидуально обозначаемых "(+)-" или "(-)-" соответственно направлению вращения плоскости поляризации света при его прохождении через образец соединения. Цис- или транс-соединения согласно изобретению, в которых отдельные энантиомеры не разделены, обозначены здесь префиксом "(+-)-".
Дальнейшими объектами изобретения
служат:
(а) соединения формулы (I) и их фармацевтически приемлемые соли, сольваты и физиологически активные производные, используемые как терапевтические агенты, в частности, для профилактики
и лечения клинических состояний, при которых показано подавление всасывания желчной кислоты, например, таких гиперлипидемических состояний, как атеросклероз;
(б) фармацевтические композиции,
содержащие соединения формулы (I) или одну из фармацевтически приемлемых солей, сольватов или физиологически активных производных наряду с по меньшей мере одним фармацевтически приемлемым носителем и,
при необходимости, одним или более другими физиологически активными агентами;
(в) применение соединения формулы (I) или его фармацевтически приемлемых соли, сольвата и физиологически
активного производного в производстве медикаментов для профилактики и лечения клинического состояния, при котором показано подавление всасывания желчной кислоты, например, таких гиперлипидемических
состояний, как атеросклероз;
(г) способ подавления всасывания желчных кислот в кишечнике животного (например, человека), включающий введение животному соединения формулы (I), его
фармацевтически приемлемых соли, сольвата или физиологически активного производного в количестве, необходимом для эффективного подавления всасывания желчной кислоты;
(д) способ уменьшения
концентрации ЛHП и ЛОHП холестерина в плазме или сыворотке крови животного (например, человека), который включает введение животному соединения формулы (1), его фармацевтически приемлемых соли,
сольвата или физиологически активного производного в количестве, необходимом для эффективного снижения количества холестерина;
(е) способ уменьшения концентрации холестерина и сложного
холестеринового эфира в плазме или сыворотке крови животного (например, человека), который включает введение животному соединения формулы (I), его фармацевтически приемлемых соли, сольвата или
физиологически активного производного в количестве, необходимом для эффективного снижения концентрации холестерина и сложного холестеринового эфира;
(ж) способ усиления выведения желчных
кислот у животного (например, человека) с фекалиями, который включает введение животному соединения формулы (I), его фармацевтически приемлемых солей, сольватов или физиологически активных производных
в количестве, необходимом для эффективного
усиления выведения желчных кислот с фекалиями;
(з) способ профилактики или лечения клинического состояния у животных (например, человека), при
котором показано подавление всасывания желчной кислоты, например, такого гиперлипидемического состояния, как атеросклероз, включающий введение животному соединения формулы (I), его фармацевтически
приемлемых соли, сольвата или физиологически активного производного в терапевтически эффективном количестве;
(и) способ уменьшения частоты коронарных сердечных и связанных с ними заболеваний
у животных (например, человека), включающий введение соединения формулы (I), его фармацевтически приемлемых соли, сольвата или физиологически активного производного в количестве, необходимом для
эффективного снижения вероятности коронарных сердечных и связанных с ними заболеваний.
(к) способ понижения концентрации холестерина в плазме или сыворотке крови животного (например,
человека), который включает введение соединения формулы (I) в количестве, необходимом для эффективного понижения концентрации холестерина;
(л) способы получения соединений формулы (I) (а
также их солей, сольватов и физиологически активных производных, которые здесь определены):
(м) новые промежуточные химические соединения, используемые при получении соединений формулы (I);
(н) соединения, синтез которых описан далее в примерах 1-13.
Все имеющиеся далее ссылки на "соединение(я) формулы (I) относятся к определенным ранее соединению(ям) (I), его(их) солям, сольватам или физиологически активным производным.
Естественно, что количество соединения формулы (I), требуемое для получения желаемого биологического эффекта, зависит от многих факторов, например от выбранной для использования конкретной разновидности соединения, способа введения и клинического состояния реципиента. Суточная доза обычно составляет от 0,3 мг до 100 мг (типичная доза - от 3 мг до 50 мг) на кг массы тела, например 3-10 мг/кг•сутки. Например, доза для внутривенного введения может составлять от 0,3 мг до 1,0 мг/кг, и ее удобно вводить, вливая от 10 до 100 нг/кг в минуту. Пригодные для этого жидкости для вливания могут содержать, например, от 0,1 нг до 10 мг, обычно от 1 нг до 10 мг/мл. Единичные дозы могут содержать, например, от 1 мг до 10 г активного соединения. Так, например, ампулы для инъекций могут содержать от 1,0 до 100 мг, а такие единичные дозы составов для перорального введения, как таблетки или капсулы, могут содержать от 1,0 до 1000 мг, обычно от 10 до 600 мг. В случае применения фармацевтически приемлемых солей приведенные массы следует относить к массе иона бензотиазепина в составе соли.
Для профилактики или лечения упомянутых ранее состояний соединения формулы (I) могут быть использованы per se, но предпочтительно их применение с приемлемым носителем, т.е. в форме фармацевтического состава. Конечно, носитель должен быть приемлемым в смысле совместимости с другими ингредиентами состава и безвредным для реципиента. Он может быть твердым, или жидким, или суспензией, а его композиции с соединением предпочтительно изготовлять в виде единичных доз, например таблеток, которые могут содержать от 0,05 до 95% по массе активного соединения. Могут присутствовать другие фармакологически активные компоненты, включая другие соединения формулы (I). Фармацевтические составы согласно изобретению могут быть приготовлены любым применяемым в фармацевтике способом, состоящим по существу в смешивании компонентов.
Фармацевтические составы согласно изобретению включают составы для перорального, ректального, локального, трансбуккального (т.е. под язык) и парентерального (например, подкожно, внутримышечно, внутрикожно или внутривенно) введения, хотя выбор наиболее подходящего способа в каждом конкретном случае зависит от вида и тяжести состояния и конкретного соединения формулы (I), используемого для его лечения.
Составы в энтеросолюбильной оболочке и составы в такой оболочке с регулируемым высвобождением активного ингредиента также включены в объем изобретения. Для изготовления оболочки пригодны ацетофталат целлюлозы, поливинилацетофталат, фталат гидроксипропилметилцеллюлозы и анионные полимеры метакриловой кислоты и ее метилового эфира.
Фармацевтические составы для перорального введения могут быть представлены в таких формах, как капсулы, крахмальные облатки, лепешки или таблетки, содержащие каждая определенное количество соединения формулы (I) в виде порошка или гранул, раствора, или водной или неводной суспензий, или в виде масляноводной или водомасляной эмульсии. Как было указано, такие составы могут быть приготовлены любым применяемым в фармацевтике способом, включающим смешивание активного соединения с носителем (который может содержать один или более вспомогательных ингредиентов). Обычно составы приготовляют до получения однородной и хорошо перемешанной смеси активного соединения с жидким, или тонко измельченным, или тем и другим носителями с последующим (если это необходимо) формованием. Например, таблетка может быть изготовлена прессованием или укладкой в форму порошкообразного или гранулированного соединения совместно (при необходимости) с одним или более вспомогательными ингредиентами. Прессованные таблетки изготовляют таблетированием на соответствующей установке соединения, взятого в сыпучем виде, например в виде порошка или гранул, и если необходимо, смешанного со связующим или смазывающим веществом, инертным разбавителем и/или поверхностно- активным(и)/диспергирующим(и) агентом(ами). Формованные таблетки изготовляют формованием на соответствующей установке порошкообразного соединения, смоченного инертным жидким разбавителем.
Фармацевтические составы для трансбуккального введения включают содержащие соединения формулы (I) лепешки на базе корригента вкуса (обычно сахарозы с аравийской камедью или трагаканта) и пастилки на инертной базе (такой, как желатин с глицерином или сахароза с аравийской камедью).
Фармацевтические составы для парентерального введения обычно включают стерильные водные препараты соединения формулы (I), предпочтительно изотоничные крови предполагаемого реципиента. Эти препараты предпочтительно вводить внутривенно, хотя их можно также вводить путем подкожных, внутримышечных или внутрикожных инъекций. Такие препараты удобно готовить, смешивая соединение с водой с последующей стерилизацией и доведением до изотоничности с кровью. Составы для инъекций согласно изобретению обычно содержат от 0,1 до 5% (по массе) действующего начала.
Фармацевтические составы для ректального введения предпочтительно изготавливать в виде содержащих единичную дозу суппозиториев. Их можно получать, смешивая соединения формулы (I) с одним или более обычных твердых носителей, например маслом какао, и затем формуя смесь.
Фармацевтические составы для локального введения предпочтительно применять в виде мази, крема, лосьона, пасты, геля, распыляемого раствора, аэрозоля или масла. В качестве носителей пригодны вазелин, ланолин, полиэтиленгликоль, спирты и сочетания двух или более перечисленных веществ. Такие составы обычно содержат от 0,1 до 15%, например от 0,5 до 2% (по массе), действующего начала.
Возможно также трансдермальное введение. Фармацевтические составы для него можно применять в виде отдельных наклеек, приспособленных для длительного тесного контакта с эпидермисом реципиента. Такие наклейки содержат необходимое действующее начало в буферном (если это необходимо) водном растворе, или растворенное/диспергированное в клеящем агенте, или диспергированное в полимере. Подходящая концентрация действующего начала составляет примерно от 1 до 35%, предпочтительно примерно от 3 до 15%. В особых случаях перенос активного соединения с наклейки может быть осуществлен электро- или ионофорезом, как это описано, например, в Pharmaceutical Research, 3(6), 318, (1986).
Соединения согласно изобретению могут быть получены обычными известными специалистам способами, аналогичными известным в этой области.
Например, соединения формулы (I), в которых R3 - водород, можно получить окислением соответствующего соединения формулы (V)
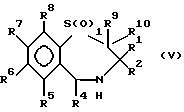
в которой R1 - R10 идентичны указанным ранее, а I равно 0 или 1. Окисление может быть проведено пероксидом, например перекисью водорода, в присутствии трифторуксусной кислоты при умеренной температуре, например от -20 до 50oC, предпочтительно от -10 до 10oC. Соединение формулы (V), в котором I=1, может быть приготовлено из соответствующего соединения, где I = 0, частичным окислением пероксидом, как это описано выше.
Соединения формулы (V) могут быть получены восстановлением иминовой связи соединения формулы (VI)
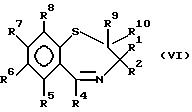
в которой R1-R10 идентичны указанным ранее, например, такими соединениями бора, как бораны, в подходящем растворителе, например, таком эфире, как тетрагидрофуран, или каталитической гидрогенизацией с использованием в качестве катализатора такого палладия, как 10% Pd/C, при умеренной температуре, например от - 20 до 100oC, предпочтительно от -10 до 50oC.
Соединения формулы (VI), как они определены здесь, а также их каждый возможный оптический изомер, который существенно свободен, т.е. связан менее чем с 5% любого другого(их) оптического(их) изомера(ов), и смеси одного или более изомеров в любых пропорциях рассматриваются как элементы новизны и служат следующим объектом изобретения.
Соединения формулы (VI) могут быть получены замыканием цикла в соединениях формулы (VII)
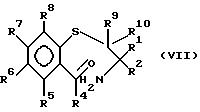
в которой R1 - R8 идентичны указанным ранее, отгонкой азеотропа или выдерживанием с обратным холодильником в присутствии такого подходящего осушителя, как молекулярные сита, в подходящем растворителе, например 2,6-лутидине, в присутствии такой кислоты, как HCl.
Соединения формулы (VII) можно
получить реакцией соединения формулы (VIII)
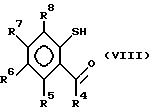
в которой R4-R8 идентичны указанным ранее, с соответствующим образом замещенным азиридином обычно в полярном растворителе, например метаноле.
Соединения формулы (VII) могут быть также получены реакцией
соединения формулы (IX)
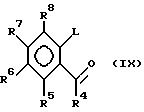
в которой R4-R8 идентичны указанным ранее, a L - подходящая отщепляемая группа, например галоген, с соединением формулы HSC(R9)(R10)C(R1)(R2)NH2, где R1, R2, R9 и R10 идентичны указанным выше.
Соединения формулы (IX) можно получить реакцией соответствующей кислоты с соединением формулы R4H, где R4 идентичен вышеуказанному, т.е. реакцией Фриделя-Крафтса с использованием, как правило, хлорида алюминия.
Соединения формулы (VIII) могут быть получены реакцией соединения формулы (X)
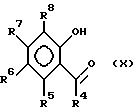
в которой R5 - R8 идентичны указанным ранее, с таким ненуклеофильным основанием, как гидрид натрия, с последующей обработкой полученной соли N, N-диметилтиокарбамоилхлоридом и пиролизом полученного O-арилдиалкилтио-карбамата (например, в таком высококипящем растворителе, как тетрадекан при температуре около 255oC) и гидролизом (например, таким сильным основанием, как KOH) до S-арилдиалкилтиокарбамата.
Альтернативным способом получения соединений формулы (VIII) является реакция соединений формулы (IX) с гидросульфидом натрия (NaSH).
Вышеупомянутые исходные материалы могут быть куплены или приготовлены способами, известными специалистам в данной области или описанными в химической литературе; например, азиридины могут быть получены из соответствующих 2-замещенных 2-аминоэтанолов.
Соединения формулы (V), в которой одна или более групп R5-R8-галогены, могут быть превращены в соединения формулы (V), в которой R5-R8 представляют другие группы, способами, известными специалистам или описанными в литературе.
Соединения формулы (I), в которой R3-OH, можно получить из соответствующих соединений формулы (I), в которой R3-H, окислением, например м-хлорпербензойной кислотой.
Соединения формулы (I), существенно свободные от других оптических изомеров, могут быть получены либо хиральным синтезом с использованием, например, таких соответствующих исходных хиральных веществ, как азиридин, либо разделением продуктов ахирального синтеза, например хиральной ВЭЖХ, или классическим разделением хиральными кислотами.
Произвольное превращение соединения формулы (I) или соединения формулы (I) с основным заместителем в соответствующую кислую соль присоединения может быть осуществлено реакцией с раствором соответствующей (например, одной из указанных выше) кислоты. Произвольное превращение соединения формулы (I) с кислотным заместителем в соответствующую основную соль можно осуществить реакцией с раствором соответствующего основания, например гидроксида натрия. Произвольное превращение в такое физиологически активное производное, как сложный эфир, может быть проведено способами, известными специалистам или описанными в литературе.
Кроме того, соединения формулы (I) могут быть превращены в другие соединения формулы (I) стандартными описанными в литературе и известными специалистам способами, например алкилированием гидроксильной группы.
С иллюстративными целями далее приведены примеры, поясняющие изобретение, но не ограничивающие его объем.
Пример синтеза 1. Получение (3R,5R)-3-бутил-3-этил-2,3,4,5- тетрагидро-7,8-диметокси-5-фенил-1,4-бензотиазепин-1,1-диоксида
(а) гидрохлорид
этил-2-аминобутирата
Суспензию 2-аминомасляной кислоты (100 г, Aldrich) в 300 г абсолютного этанола перемешивают при 0oC в атмосфере азота, прикапывают 120,8 г тионилхлорида,
перемешивают при 0oC в течение ночи и затем постепенно подогревают до комнатной температуры. Полученную белую суспензию нагревают с обратным холодильником в течение 3 ч, оставляют остыть в
течение 10 мин и вливают в 600 мл охлажденного диэтилэфира при помешивании вручную. Суспензию фильтруют и твердое вещество высушивают, получая 150 г целевого продукта в виде белого порошка.1H ЯМР согласуется с ожидаемой структурой.
(б) этил-2-бензилиденаминобутират
Раствор 149,6 г продукта стадии (а), 74,3 г сульфата магния и 246 мл триэтиламина в 1500 мл
дихлорметана перемешивают при комнатной температуре в атмосфере азота и прикапывают бензальдегид (94,9 г, Aldrich). Смесь перемешивают при комнатной температуре в течение 3 ч, затем фильтруют.
Фильтрат концентрируют, перетирают с диэтилэфиром, фильтруют и концентрируют, получая 174 г целевого продукта в виде желтого маслянистого вещества.1H ЯМР согласуется с ожидаемой
структурой.
(в) (±)-этил-2-бензилидинаминобутират
Гидрид натрия (32,5 г 60%-й дисперсии в масле) и 700 мл N,N-диметилформамида (ДМФ) перемешивают при комнатной
температуре в атмосфере азота, прикапывают раствор 178,1 г продукта стадии (б) в ДМФ, перемешивают при комнатной температуре в течение 2 ч, прикапывают раствор 149,5 г бутилиодида в ДМФ и выдерживают
при помешивании в течение еще 2 ч для завершения реакции. Реакционную смесь вливают в ледяную смесь 560 мл воды, 300 мл диэтилэфира и 120 г хлорида аммония. Полученный органический слой сушат над
карбонатом калия, затем концентрируют, получая 220 г целевого продукта в виде коричневого маслянистого вещества.
(г) (±)-этил-2-амино-2-этилгексаноат
223,0 г продукта
стадии (в) обрабатывают петролейным эфиром и 421 мл 10%-й (по массе) соляной кислоты, перемешивая при комнатной температуре в течение 2 ч и отстаивают. Водный слой дважды экстрагируют петролейным
эфиром и затем охлаждают с этилацетатом в солеледяной ванне. В смесь добавляют зерна гидроксида натрия до достижения pH 10 водяного слоя. Этот слой дважды экстрагируют этилацетатом, экстракты
смешивают и сушат над карбонатом калия, затем концентрируют и перегоняют под вакуумом, получая целевой продукт в виде бесцветного маслянистого вещества.1H ЯМР согласуется с ожидаемой
структурой.
(д) (R)- 2-амино-2-этилгексанойная кислота
Взвесь эстеразы свиной печени (0,1 г, Sigma-Aldrich-Fluka) в воде добавляют к водному раствору 100 г продукта стадии
(г). После этого 1N водным раствором NaOH доводят pH смеси до 9,7 и поддерживают это значение, добавляя 1N раствор NaOH. После добавления определенного количества 1N водного раствора NaOH (83 г в
течение 10 ч) смесь промывают диэтилэфиром для удаления непрореагировавшего (S)-этил-2-амино-2-этилгексаноата. Оставшуюся водную фазу упаривают под вакуумом, получая белое твердое вещество, содержащее
требуемое соединение и его натриевую соль.
(е) (R)-2-амино-2-этилгексан-1-ол
20 г продукта стадии (д) добавляют к эквивалентному 1,5 молям алюмогидрида лития его 1М раствору в
тетрагидрофуране, смесь выдерживают с обратным холодильником в течение 3 ч и перемешивают при комнатной температуре в течение 16 ч. Реакционную смесь охлаждают примерно до 0oC, затем гасят
водой и 1N водным раствором NaOH. Полученное твердое вещество диспергируют в дополнительном количестве воды. Суспензию подогревают до 50oC в течение 5 мин, охлаждают до комнатной
температуры, добавляют 100 мл диэтилэфира, перемешивают и фильтруют. Диэтилэфирный слой отделяют, сушат и концентрируют в вакууме, получая целевой продукт в виде маслянистого вещества (выход 82%).1H ЯМР согласуется с ожидаемой структурой.
(ж) (R)-2-амино-2- этилгексилгидродсульфат
20 г продукта стадии (е) растворяют в 170 мл дихлорметана и обрабатывают 26,8
г хлорсульфоновой кислоты. Реакционную смесь перемешивают при комнатной температуре в течение 17 ч. Большую часть растворителя отгоняют, а полученную суспензию разбавляют ацетоном, фильтруют и
высушивают, получая твердое белое вещество.1H ЯМР согласуется с ожидаемой структурой.
(з) 2-гидрокси-4,5-диметоксибензальдегид
1,0 М раствор трихлорида бора (210
мл, Aldrich) в дихлорметане добавляют к раствору бензоилхлорида (30,1 г, Aldrich) в 350 мл бензола. Затем добавляют раствор 3,4-диметоксифенола (30,0 г, Aldrich) в 130 мл бензола, реакционную смесь
перемешивают при комнатной температуре в течение 2,5 ч, добавляют 55 мл 50%-го раствора NaOH и вновь перемешивают в течение 15 мин. Органические слои отделяют, сушат и концентрируют в вакууме.
Полученный остаток в течение 40 мин перетирают с 1N раствором NaOH и отфильтровывают. Щелочной водный фильтрат нейтрализуют концентрированной HCl, получая целевой продукт в виде желтого твердого
вещества (25,9 г) с tпл. 104-105oC.1H ЯМР согласуется с ожидаемой структурой.
(и) O-(2-бензоил-4,5-диметоксифенил)-N,N-диэтилтиокарбамат
Триэтиламин (106,3 г, Aldrich), 4-диметиламинопиридин (6,3 г, Aldrich) и 86,4 г диэтилтиокарбамоила добавляют к 130,4 г продукта стадии (з) и смесь вносят в 1 л диоксана. Реакционную смесь при
перемешивании выдерживают с обратным холодильником в течение 22 ч, охлаждают до комнатной температуры и фильтруют. Фильтрат концентрируют в вакууме и добавляют 600 мл 1N HCl и 500 мл диэтилэфира.
Смесь оставляют на 45 мин, затем фильтруют. Осадок тщательно промывают диэтилэфиром и высушивают в вакуумсушильном шкафу, получая целевой продукт в виде желтого твердого вещества (120,5 г) с tпл. 94-95oC.1H ЯМР согласуется с ожидаемой структурой.
(к) S-(2-бензоил-4,5-диметоксифенил)-N,N-диэтилтиокарбамат
Суспензию 106,3 г продукта
стадии (и) в 250 мл тетрадекана нагревают до температуры во всей массе 250oC и выдерживают в течение 25 мин. Реакционную смесь охлаждают в ледяной ванне. Растворитель отцеживают, а остаток
перетирают с диэтилэфиром, получая в результате целевой продукт в виде бежевого твердого вещества (43,4 г) с tпл 114-116oC.1H ЯМР согласуется с ожидаемой
структурой.
(л) 2-меркапто-4,5-диметоксибензофенон
48,8 г гранулированного гидроксида калия медленно добавляют к раствору 85,0 г продукта стадии (к) 1 л смеси 1:1 метанола и
тетрагидрофурана. После трехчасовой выдержки с обратным холодильником реакционную смесь охлаждают до комнатной температуры и концентрируют в вакууме. Полученный остаток перетирают с 1N HCl, затем
экстрагируют EtOAc. Органический слой отделяют и последовательно промывают 1N HCl (2х250 мл) и IN раствором NaOH (3х400 мл). Щелочные водные слои смешивают и нейтрализуют концентрированной HCl,
получая целевой продукт в виде золотистого твердого вещества (54,8 г).1H ЯМР согласуется с ожидаемой структурой.
(м) (H)-2-(2-амино-2-этилгексилтио)-4,
5-диметоксибензофенон
48,8 г продукта стадии (ж) растворяют в 250 мл воды и к этому раствору добавляют 54,2 г продукта стадии (л) в 300 мл бутилацетата. Реакционную смесь перемешивают,
нагревают до температуры во всей массе 93oC и прикапывают раствор 18,9 г NaOH в 250 мл воды, после чего перемешивают в течение еще 25 мин при температуре 93oC и затем охлаждают
до комнатной температуры. Органический слой отделяют, сушат и концентрируют, получая целевой продукт в виде твердого оранжевокоричневого вещества с tпл 75- 78oC.1H
ЯМР согласуется с ожидаемой структурой.
(н) (3R)-3-бутил-3-этил-2,3-дигидро-7,8-диметокси-5-фенил-1,4- бензотиазепин
78,0 г продукта стадии (м) растворяют в 400 мл 2,
6-лутидина, добавляют 0,70 г п-толуолсульфоновой кислоты и реакционную смесь выдерживают с обратным холодильником, используя ловушку Дина-Старка, в течение 22 ч. За это время растворитель покидает
аппарат и его заменяют свежим. Реакционную смесь концентрируют в вакууме и остаток обрабатывают 300 мл 5%-го раствора NaHCO3 и 300 мл EtOAc. Слой EtOAc отделяют, промывают рассолом, сушат и
концентрируют в вакууме, получая темно-красное маслянистое вещество. Хроматография на силикагеле с элюированием смесью 4:1 гексана и EtOAc дает 64,1 г целевого продукта в виде светло-коричневого
маслянистого вещества.1H ЯМР согласуется с ожидаемой структурой.
(о) (3R, 5R)-3-бутил-3-этил-2,3,4,5-тетрагидро-7,8-диметокси-5- фенил 1,4-бензотиазепин.
1 М раствор диборана в 200 мл тетрагидрофурана добавляют к раствору 64,0 г продукта стадии (н) в 350 мл тетрагидрофурана. Реакционную смесь перемешивают при комнатной температуре в течение 17 ч, добавляют 150 мл 6N HCl и раствор концентрируют d вакууме для удаления тетрагидрофурана. Водный остаток ощелачивают 50%-м раствором NaOH и экстрагируют EtOAc. Слой EtOAc отделяют, сушат и концентрируют в вакууме, получая маслянистое вещество, которое хроматографируют на силикагеле, элюируя смесью 85:15 гексана и EtOAc, и получают 25,5 г целевого продукта в виде бежевого твердого вещества с tпл 64-66oC.1H ЯМР согласуется с ожидаемой структурой.
(п) (3R, 5R)-3-бутил-3-этил-2,3,4,5-тетрагидро-7,8-диметокси- 5-фенил-1, 4-бензотиазепин-1,1 -диоксид.
Раствор 25,5 г продукта стадии (о) в 125 мл трифторуксусной кислоты добавляют к 18,8 г 30%-й H2O2 в 100 мл той же кислоты. Реакционную смесь перемешивают при комнатной температуре в течение 17 ч, затем вливают 800 мл воды, после чего добавляют 50%-й раствор NaOH до достижения pH 10. Затем реакционную смесь расслаивают EtOAc и перемешивают в течение 1 ч. Органический слой отделяют, сушат и концентрируют в вакууме, получая твердый остаток, который перекристаллизовывают из EtOH, получая 18,5 г целевого продукта с tпл 148-149oC.1H ЯМР согласуется с ожидаемой структурой.
Анализ. Рассчитано: C - 66,16: H - 7,48; N - 3,35; S - 7,68; найдено: C - 66,01; H - 7,56: N - 3,31: S - 7,74.
1H ЯМР (ДМСО-d6),δ; 0,74-0,86 (6H, m); 1,07-1,39 (4H, m); 1,39-2,20 (4H, m); 3,33 (2H, q); 3,44 (3H, s); 3,83 (3H, s); 5,92 (1H, d); 6,11 (1H, s); 7,33-7,48 (6H, m).
Пример синтеза 2. Получение (3R,5R)-3-бутил-3-этил-2,3,4,5- тетрагидро-7,8-диметокси-5-фенил-1,4-бензотиазепин-4-ол-1,1-диоксида
Оксон (146,7 г,
Aldrich) в 550 мл воды добавляют к раствору 18,4 г продукта стадии (о) примера 1 в 500 мл MeOH. Реакционную смесь перемешивают при комнатной температуру в течение 17 ч, затем осторожно ощелачивают
50%-м раствором NaOH. Гетерогенную смесь расслаивают EtOAc и перемешивают в течение 1 ч. Органический слой отделяют, сушат и концентрируют в вакууме, получая розовое твердое вещество. Хроматография на
силикагеле с элюированием смесью 65: 35 гексана и EtOAc дает 6,7 г конечного продукта в виде белого твердого вещества с tпл 174-175oC.
Анализ. Рассчитано: C - 63, 72; H - 7,21: N - 3,23; S - 7,39; найдено: C - 63,81; H - 7,22; N - 3,19; S - 7,47.
1H ЯМР (ДМСО-d6) δ ; 0,77- 0,90 (6H, m); 1,10-2,17 (8H, m); 3,27-3,45 (5H, m); 3,84 (3H, s); 6,14 (1H, s); 6,38 (1H, s); 7,30-7,53 (5H, m); 7,97 (1H, s).
Пример синтеза 3. Получение (±)-транс-3-бутил-3-этил-2,3,4,5- тетрагидро-7,
8-диметокси-5-фенил-1,4-бензотиазепин-1,1-диоксида
(а) (±)-2-амино-2-этилгексан-1-ол
22,2 г алюмогидрида лития добавляют к 450 мл безводного диэтилэфира в атмосфере азота,
прикапывают 129 г продукта стадии (г) примера 1, разбавленного 40 мл диэтилэфира, реакционную смесь выдерживают с обратным холодильником в течение 1 ч и после охлаждения до комнатной температуры
прикапывают 23 мл 1М раствора гидроксида натрия и затем деионизированную воду. Полученную суспензию фильтруют и фильтрат концентрируют, получая 87,9 г целевого продукта в виде бесцветного маслянистого
вещества.1H ЯМР согласуется с ожидаемой структурой.
(б) (±)-2-бутил-2-этилазиридин.
150 г ацетонитрила и 20,0 г продукта стадии (а) смешивают в атмосфере азота, охлаждают до 2-3oC и, поддерживая температуру ниже 10oC, прикапывают хлорсульфоновую кислоту (16,0, Aldrich). Охлаждение прекращают и суспензию перемешивают 80 мин при комнатной температуре. Реакционную смесь концентрируют в вакууме и дистиллируют с 50 мл воды. Добавляют 55,2 г 50%-го водного раствора гидроксида натрия и 50 мл воды и смесь дистиллируют при атмосферном давлении. Органический слой дистиллята отбирают и сушат над твердым гидроксидом калия, получая 12,8 г целевого продукта.1H ЯМР согласуется с ожидаемой структурой.
(в) (±)-3-бутил-3-этил-2,3-дигидро-7,8-диметокси-5-фенил-1,4- бензоти-азепин;
14,7 г продукта стадии (л) примера 1 в 50 мл 2,6-лутидина добавляют к раствору 6,5 г продукта
стадии (б) данного примера в 200 мл 2,6-лутидина, перемешивают в течение 1 ч, добавляют концентрированную HCl и затем выдерживают с обратным холодильником с ловушкой Дина-Старка в течение 17 ч.
Реакционную смесь концентрируют в вакууме, а остаток разделяют между 5%-м раствором NaHCO3 и EtOAc. Органический слой отделяют, сушат и концентрируют, получая маслянистое вещество, которое
хроматографируют на силикагеле, элюируя смесью 7: 3 гексана и EtOAc, и получают 12,0 г целевого продукта в виде маслянистого вещества.1H ЯМР согласуется с ожидаемой структурой.
(г) (±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-7,8-диметокси- 5-фенил-1,4-бензотиазепин-1,1-диоксид;
Требуемый продукт получают согласно стадиям (о)-(п) примера 1 с
использованием продукта стадии (в) данного примера, получая белое твердое вещество с tпл 146-147oC.
Анализ (0,50 H2O). Рассчитано: C - 64,54; H - 7,35; N - 3,24; S - 7,40; найдено: C - 64,76; H - 7,56; N - 3,28; S - 7,52.
1H ЯМР (ДМСО-d6), δ , 0,74-0,86 (6H, m); 1,07-1,39 (4H, m); 1,40-2,20 (4H, m); 3,33 (2H, q); 3,44 (3H, s); 3,83 (3H, s); 5,92 (1H, d): 6,11 (1H, s); 7,30-7,48 (6H, m).
Пример синтеза 4. Получение (±)-транс-3-бутил-3- этил-2,3,4,5-тетрагидро-7,
8-диметокси-5-фенил-1,4-бензотиазепин- 4-ол-1,1-диоксида
Оксон (7,3 г, Aldrich) в 100 мл воды добавляют к раствору 1,7 г продукта стадии (г) примера 3 в 100 мл MeOH. Реакционную смесь
перемешивают при комнатной температуру в течение 17 ч и добавляют воду и EtOAc. После перемешивания в течение 1 ч органический слой отделяют, сушат и концентрируют, получая пенообразное вещество.
Хроматография на силикагеле с элюированием смесью 4:1 гексана и EtOAc дает 1,2 г целевого продукта в виде белого твердого вещества tпл 172-174oC.
Анализ. Рассчитано: C - 63,72; H - 7,21: N - 3,23; S - 7,39; найдено: C - 63,79; H - 7,26; N-3,18; S - 7,47.
1H ЯМР (ДМСО-d6), δ ; 0,78-0,90 (6H, m); 1,14- 2,14 (8H, m); 3,27-3,41 (5H, m); 3,84 (3H, s); 6,13 (1H, s); 6,37 (1H, s); 7,34-7,53 (5H, m); 7,96 (1H, s).
Пример синтеза 5. Получение (3R,5R)-7-бром-3-бутил-3-этил-2,3,4,
5- тетрагидро-8-метокси-5-фенил-1,4-бензотиазепин-1,1-диоксида
(а) O-(2-бензоил-5-метоксифенил)диметилтиокарбамат
Гидрид натрия (8,8 г, Aldrich) медленно добавляют к раствору
2- гидрокси-4-метоксибензофенона (50,0 г, Aldrich) в 300 мл диметилформамида, прикапывают 43,0 г гексаметилфосфорамида и перемешивают при комнатной температуре в течение 2 ч, добавляют
диметилтиокарбамоилхлорид (37,0 г, Aldrich) и перемешивают при 50oC в течение ночи. Реакционную смесь вливают в 300 мл деионизированной воды и экстрагируют смесью 1:4 петролейного эфира и
хлороформа. Органический слой промывают 10%-м гидроксидом натрия, затем рассолом и концентрируют, получая целевой продукт в виде желтого твердого вещества (40,0 г).1H ЯМР согласуется с
ожидаемой структурой.
(б) S-(2-бензоил-5-метоксифенил)диметилтиокарбамат
97,4 г продукта стадии (а) суспендируют в 500 мл тетрадекана и нагревают до температуры 255o
C во всей массе в течение 30 мин. После охлаждения до комнатной температуры реакционную смесь хроматографируют на силикагеле, элюируя смесью 7:3 гексана и этилацетата, и получают 65,0 г целевого
продукта в виде светло-коричневого твердого вещества с tпл 95-97oC.1H ЯМР согласуется с ожидаемой структурой.
(в) 2-меркапто-4-метоксибензофенон
20,0 г гранулированного гидроксида калия медленно добавляют к раствору 28,0 г продукта стадии (б) в 800 мл смеси 1:1 метанола и тетрагидрофурана. После четырехчасовой выдержки с обратным
холодильником реакционную смесь охлаждают до комнатной температуры, добавляют дихлорметан и раствор экстрагируют 5%-й соляной кислотой. Органический слой сушат и концентрируют. Хроматография на
силикагеле с элюированием смесью 99:1 гексана и этилацетата дает 17,1 г целевого продукта в виде оранжевого маслянистого вещества.1H ЯМР согласуется с ожидаемой структурой.
(г) (R)-2-(2-амино-2-этилгексилтио)-4-метоксибензофенон
Это соединение получают по стадии (м) примера 1, используя 46,4 г продукта стадии (в) этого примера и 44,6 г продукта стадии (ж)
примера 1. Концентрирование органического слоя дает 66,5 г требуемого продукта в виде красного маслянистого вещества.1H ЯМР согласуется с ожидаемой структурой.
(д)
(3R)-3-бутил-3-этил-2,3-дигидро-8-метокси-5-фенил-1,4- бензотиазепин
Это соединение получают по стадии (н) примера 1, используя 66.5 г продукта стадии (г) данного примера. Хроматография на
силикагеле с элюированием смесью
9:1 гексана и EtOAc дает 54,5 г целевого продукта в виде желтого маслянистого вещества.1H ЯМР согласуется с ожидаемой структурой.
(е) (3R,
5R)-3-бутил-3-этил-2,3,4,5-тетрагидро-8-метокси-5-фенил-1,4- бензотиазепин
Это соединение получают по стадии (о) примера 1, используя 66,5 г продукта стадии (д) этого примера. Хроматография
на силикагеле с элюированием смесью 9: 1 гексана и EtOAc дает 22,8 г целевого продукта в виде оранжевого маслянистого вещества.1H ЯМР согласуется с ожидаемой структурой.
(ж) (3R, 5R)-7-бром-3-бутил-3-этил-2,3,4,5- тетрагидро-8-метокси-5-фенил-1,4-бензотиазепин
18,6 г брома добавляют к раствору 10,4 г продукта стадии (е) в 150 мл ледяной уксусной кислоты,
перемешивают при комнатной температуре в течение 2 ч, отгоняют под вакуумом уксусную кислоту, добавляют еще 100 мл уксусной кислоты и концентрируют под вакуумом. Полученный остаток растворяют в EtOAc
и промывают метабисульфитом натрия и 1N раствором NaOH. Органический слой отделяют, сушат и концентрируют в вакууме, получая коричневое маслянистое вещество, превращаемое далее в гидрохлорид раствором
HCl в эфире. Выпавший осадок отфильтровывают, промывают эфиром и обрабатывают 1N раствором NaOH и EtOAc, получая 8,9 г целевого продукта в виде оранжевого маслянистого вещества.1H ЯМР
согласуется с ожидаемой структурой.
(з) (3R,5R)-7-бром-3-бутил-3-этил-2,3,4,5-тетрагидро-8- метокси-5-фенил-1,4-бензотиазепин-1,1-диоксид
Это соединение получают по стадии
(п) примера 1, используя 8,2 г продукта стадии (ж) этого примера. Хроматография на силикагеле с элюированием смесью 4:1 гексана и EtOAc дает пену, из которой после перетирания с эфиром получают 5,0 г
конечного продукта в виде белого твердого вещества с tпл 132-134oC.
Анализ. Рассчитано: C - 56,65; H - 6,05; N - 3,00; Br - 17,13; S - 6,87, найдено: C - 56,71; H - 6,01; N - 2,94; Br - 17,07: S - 6,95.
1H ЯМР (ДМСО-d6), δ ; 0,64-0,81 (6H, m); 0,97-1,19 (4H, m); 1,22-1,50 (2H, m); 1,69-1,78 (1H, m); 1,98-2,06 (1H, m); 2,67 (1H, d,); 3,39 (2H, q,); 3,92 (3H, s); 5,88 (1H, d); 6,63 (1H, s); 7,29-7,43 (5H, m); 7,55(1H, s).
Пример синтеза 6. Получение (3R,5R)-7-бром-3-бутил-3-этил-2,3,4,
5- тетрагидро-8-метокси-5-фенил-1,4-бензотиазепин-4-ол-1,1-диоксида
Метахлорпербензойную кислоту (57-86%, 0,90 г, Aldrich) в 50 мл CH2Cl2 добавляют в раствор 2,4 г
продукта стадии (з) примера 5 в 50 мл CH2Cl2, перемешивают при комнатной температуре в течение 1 ч, добавляют 100 мл 5%-го раствора NaHCO3 и перемешивают в течение 30
мин. Органический слой отделяют, сушат и концентрируют в вакууме, получая пенообразное вещество. Хроматография на силикагеле с элюированием смесью 9:1 гексана и EtOAc дает пенообразное вещество, из
которого после перетирания с эфиром получают 1,3 г конечного продукта в виде белого твердого вещества с tпл 202-204oC.
Анализ. Рассчитано: C - 54,77; H - 5,85; N - 2,90; Br - 16,56; S - 6,65; найдено: C - 54,92; H - 5,90; N - 2,85; Br - 16,65; S - 6,75.
1H ЯМР (ДМСО-d6), δ ; 0,75-0,86 (6H, m); 1,05-1,41 (5H, m); 1, 43-1,64 (1H, m); 1,66-1,79 (1H, m); 1,83-2,49 (1H, m); 3,46 (2H, S); 3,93 (3H, s); 6,33 (1H, s); 6,67 (1H, s); 7,30-7,50 (6H, m); 8,07 (1H, s).
Пример синтеза 7. Получение (3R,
5R)-3-бутил-3-этил-2,3,4,5- тетрагидро-5-фенил-1,4-бензотиазепин-7,8-диол-1,1-диоксида;
5,0 г продукта стадии (п) примера 1 растворяют в 36 мл ледяной уксусной кислоты и 36 мл 48%-го раствора
HBr и перемешивают с обратным холодильником 2 ч. Реакционную смесь вливают в смесь воды и льда и ощелачивают 50% раствором NaOH до pH 7. От смеси отфильтровывают твердое вещество, которое
хроматографируют на силикагеле, элюируя смесью 3:2 гексана и EtOAc, и получают 1,6 г конечного продукта в виде твердого белого вещества с tпл 117- 118oC.
Анализ (0,30 H2O). Рассчитано: C - 63,87; H - 7,04; N - 3,55; S - 8,12; найдено: C - 63,86; H - 7,09; N - 3,51: S - 8,18.
1H ЯМР (ДМСО-d6), δ ; 0,76 (3H, t); 0,81 (3H, t); 1,08-2,41 (8H, m); 3,24 (2H, q); 5,83 (1H, d); 6,03 (1H, s); 7,31-7,42 (6H, m); 9,60 (3H, bs).
Пример синтеза 8. Получение (3R,5R)-3-бутил-3-этил-2,3,4,
5- тетрагидро-8-метокси-5-фенил-1,4-бензотиазепин-7-ол-1,1-диоксида
Хроматография продуктов реакции из примера 7 дает смеси, которые соединяют и повторно хроматографируют, элюируя толуолом и
смесью 95:5 толуола и EtOAc, и получают 0,29 г конечного продукта в виде твердого белого вещества с tпл 155-156oC.
Анализ. Рассчитано: C - 65,48; H - 7,24; N - 3, 47; S - 7,95; найдено: C - 65,58; H - 7,28; N - 3,43; S - 8,03.
1H ЯМР (ДМСО-d6), δ ; 0,76 (3H, t): 0,81 (3H, t); 1,18-2,04 (8H, m); 3,28 (2H, q); 3,82 (3H, s); 5,85 (1H, d); 6,09 (1H, s); 7,31-7,45 (6H, m); 9,43 (1H, s).
Пример синтеза 9. Получение (3R,5R)-3-бутил-3-этил- 2,3,4,5-тетрагидро-7-метокси-5-фенил-1,4-бензотиазепин-8-ол-1,1
- диоксида
Хроматография реакционной смеси из примера 7 дает конечные соединения примеров 7 и 8. Хроматография смеси из примера 8 дает 0,35 г еще одного конечного продукта в виде твердого
белого вещества с tпл 165-166oC.
Анализ. Рассчитано: C - 65,48; H - 7,24; N - 3,47: S - 7,95; найдено: C - 65,32; H - 7,28: N - 3,49; S - 8,00.
1H ЯМР (ДМСО-d6), δ ; 0,77 (3H, t); 0,81 (3H, t); 1,11- 2,08 (8H, m); 3,29 (2H, q); 3,44 (3H, s); 5,86 (1H, d); 6,06 (1H, s); 7,32-7,43 (6H, m); 9,73 (1H, s).
Пример синтеза 10. Получение (±)-транс-3-бутил-3- этил-2,3,4,5-тетрагидро-8-метокси-5-фенил-1,4-бензотиазепин-1,1 -диоксида
(а)
O-(2-бензоил-5-метоксифенил)диметилтиокарбамат
Гидрид натрия (8,8 г, Aldrich) медленно добавляют к раствору 2- гидрокси-4-метоксибензофенона (50,0 г, Aldrich) в 300 мл диметилформамида,
прикапывают 43,0 г гексаметилфосфорамида и перемешивают при комнатной температуре в течение 2 ч, добавляют диметилтиокарбамоилхлорид (37,0 г, Aldrich) и перемешивают при 50oC в течение
ночи. Реакционную смесь вливают в 300 мл деионизированной воды и экстрагируют смесью 1:4 петролейного эфира и хлороформа. Органический слой промывают 10%-м гидроксидом натрия, затем рассолом и
концентрируют, получая целевой продукт в виде желтого твердого вещества (40,0 г).1H ЯМР согласуется с ожидаемой структурой.
(б)
S-(2-бензоил-5-метоксифенил)диметилтиокарбамат
97,4 г продукта стадии (а) суспендируют в 500 мл тетрадекана и нагревают до температуры 255oC во всей массе в течение 30 мин. После
охлаждения до комнатной температуры реакционную смесь хроматографируют на силикагеле, элюируя смесью 7:3 гексана и этилацетата, и получают 65,0 г целевого продукта в виде светло-коричневого твердого
вещества с tпл 95-97oC.1H ЯМР согласуется с ожидаемой структурой.
(в) 2-меркапто-4-метоксибензофенон
20,0 г гранулированного гидроксида
калия медленно добавляют к раствору 28,0 г продукта стадии (б) в 800 мл смеси 1:1 метанола и тетрагидрофурана. После четырехчасовой выдержки с обратным холодильником реакционную смесь охлаждают до
комнатной температуры, добавляют дихлорметан и раствор экстрагируют 5%-й соляной кислотой. Органический слой сушат и концентрируют. Хроматография на силикагеле с элюированием смесью 99:1 гексана и
этилацетата дает 17,1 г целевого продукта в виде оранжевого маслянистого вещества.1H ЯМР согласуется с ожидаемой структурой.
(г) гидрохлорид этил-2-аминобутирата
Суспензию 2-аминомасляной кислоты (100 г, Aldrich) в 300 г абсолютного этанола перемешивают при 0oC в атмосфере азота и прикапывают 120,8 г тионилхлорида, реакционную массу при 0o
C перемешивают в течение ночи и затем постепенно нагревают до комнатной температуры. Полученную белую суспензию нагревают с обратным холодильником в течение 3 ч, остужают в течение 10 мин, вливают в
600 мл охлажденного диэтилэфира при перемешивании вручную, фильтруют и твердое вещество высушивают, получая 150 г целевого продукта в виде белого порошка.1H ЯМР согласуется с ожидаемой
структурой.
(д) этил-2-бензилиденаминобутират
Раствор 149,6 г продукта стадии (а), 74,3 г сульфата магния и 246 мл триэтиламина в 1500 мл дихлорметана перемешивают при
комнатной температуре в атмосфере азота и прикапывают бензальдегид (94,9 г, Aldrich). Смесь перемешивают при комнатной температуре в течение 3 ч и фильтруют. Фильтрат концентрируют, перетирают с
диэтилэфиром, фильтруют и концентрируют, получая 174 г целевого продукта в виде желтого маслянистого вещества.1H ЯМР согласуется с ожидаемой структурой.
(е) (± )-этил-2-бензилиденамино-2-этилгексаноат.
32,5 г 60%-й дисперсии гидрида натрия в масле и 700 мл N,N-диметилформамида (ДМФ) перемешивают при комнатной температуре в атмосфере азота и прикапывают раствор 178,1 г продукта стадии (д) в ДМФ, перемешивают при комнатной температуре в течение 2 ч, прикапывают раствор 149,5 г бутилиодида в ДМФ и проводят реакцию в течение еще 2 ч при перемешивании. Реакционную смесь вливают в ледяную смесь 560 мл воды, 300 мл диэтилэфира и 120 г хлорида аммония. Полученный органический слой сушат над карбонатом калия и концентрируют, получая 220 г целевого продукта в виде коричневого маслянистого вещества.
(ж) (±)-этил-2-амино-2-этилгексаноат
223,0 г продукта стадии (е) экстрагируют петролейным эфиром и 421 мл
10%-й (по массе) соляной кислоты и перемешивают при комнатной температуре в течение 2 ч. Водный слой дважды экстрагируют петролейным эфиром, затем охлаждают с этилацетатом в солеледяной ванне,
добавляют гранулы гидроксида натрия до достижения pH 10, затем дважды экстрагируют этилацетатом. Соединенные экстракты сушат над карбонатом калия, затем концентрируют и дистиллируют под вакуумом,
получая целевой продукт в виде бесцветного маслянистого вещества.1H ЯМР согласуется с ожидаемой структурой.
(з) (±)-2-амино-2-этилгексан-1-ол
22,2 г
алюмогидрида лития добавляют к 450 мл безводного диэтилэфира в атмосфере азота, прикапывают 129 г продукта стадии (ж), разбавленного 40 мл диэтилэфира. Реакционную смесь выдерживают с обратным
холодильником в течение 1 ч, охлаждают до комнатной температуры и прикапывают 23 мл 1М раствора гидроксида натрия, затем деионизированную воду. Полученную суспензию фильтруют и фильтрат концентрируют,
получая 87,9 г целевого продукта в виде бесцветного маслянистого вещества.1H ЯМР согласуется с ожидаемой структурой.
(и) (±)-2-бутил-2-этилазиридин
150 г
ацетонитрила и 20,0 г продукта стадии (з) смешивают в атмосфере азота, охлаждают до 2-3oC и, поддерживая температуру ниже 10oC, прикапывают хлорсульфоновую кислоту (16,0,
Aldrich). Охлаждение прекращают и суспензию перемешивают 80 мин при комнатной температуре. Реакционную смесь концентрируют в вакууме и дистиллируют с 50 мл воды. Добавляют 55,2 г 50% водного раствора
гидроксида натрия и 50 мл воды и смесь дистиллируют при атмосферном давлении. Органический слой дистиллята отбирают и сушат с твердым гидроксидом калия, получая 12,8 г целевого продукта.1H
ЯМР согласуется с ожидаемой структурой.
(к) (±)-3-бутил-3-этил-8-метокси-5-фенил-2,3-дигидробензотиазепин
55,2 г продукта стадии (и) в 100 мл 2,6-лутидина добавляют к
раствору 118,5 г продукта стадии (в) в 400 мл 2,6-лутидина. Реакционную смесь перемешивают в течение 1 ч, добавляют п-толуолсульфоновую кислоту, выдерживают с обратным холодильником с ловушкой
Дина-Старка в течение 17 ч, концентрируют в вакууме и остаток экстрагируют 5%-м раствором NaHCO3 и EtOAc. Органический слой отделяют, сушат и концентрируют, получая маслянистое вещество,
которое хроматографируют на силикагеле, элюируя смесью 85:15 гексана и EtOAc, и получают 124,3 г целевого продукта в виде оранжевого маслянистого вещества.1H ЯМР согласуется с ожидаемой
структурой.
(л) (±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-8- метокси-5-фенил-1,4-бензотиазепин
1 М раствор диборана в 40 мл тетрагидрофурана добавляют к раствору 12,
3 г продукта стадии (к) в 150 мл тетрагидрофурана. Реакционную смесь перемешивают при комнатной температуре в течение 17 ч, вводят 50 мл 6N HCl и раствор концентрируют в вакууме. Остаток ощелачивают
50% раствором NaOH и экстрагируют EtOAc. Слой EtOAc отделяют, сушат и концентрируют в вакууме, получая маслянистое вещество, которое хроматографируют на силикагеле, элюируя гексаном и затем толуолом,
и получают 4,9 г целевого продукта в виде желтого маслянистого вещества.1H ЯМР согласуется с ожидаемой структурой.
(м) (±)-транс-3-бутил-3-этил-2,3,4,
5-тетрагидро-8-метокси-5- фенил-1,4-бензотиазепин-1,1-диоксид
Раствор 4,9 г продукта стадии (л) в 50 мл трифторуксусной кислоты добавляют к 30% H2O2 в 50 мл
трифторуксусной кислоты. Реакционную смесь перемешивают при комнатной температуре в течение 17 ч, вливают в 200 мл деионизованной воды и добавляют гранулы NaOH до pH 14. Затем реакционную смесь
выдерживают при 45oC в течение 1 ч и экстрагируют дихлорметаном. Органический слой отделяют, сушат и концентрируют, получая маслянистое вещество, которое хроматографируют на силикагеле,
элюируя смесью 9:1 гексана и EtOAc, и получают 4,9 г конечного продукта в виде твердого белого вещества с tпл 123-125oC.
Анализ. Рассчитано: C - 68,18; H - 7,54; N - 3,61; S - 8,27; найдено: C - 68,19; H - 7,49: N - 3,55; S - 8,35.
1H ЯМР (ДМСО-d6), δ ; 0,73-0,85 (6H, m, CH3); 1,07- 1,47 (4H, m, CH2); 1,48-2,20 (4H, m, CH2); 2,48-2,53 (1H, d, NH); 3,51 (2H, q, CH2SO2); 3,84 (3H, s, OMe); 5,90 (1H, d, CHPh); 6,50 (1H, d, ArH); 7,09-7,20 (1H, m, Ar); 7,32-7, 48 (6H, m, ArH).
Пример синтеза 11. Получение (±)-транс-3-бутил-3-этил-2,3,4,5-тетра-гидро-5-фенил-1,4- бензотиазепин-8-ол-1,1-диоксида
Это соединение получают по
процедуре примера 7, используя 4,8 г продукта стадии (м) примера 10. Хроматография на силикагеле с элюированием смесью 4:1 гексана и EtOAc дает 1,8 г конечного продукта в виде твердого белого вещества
с tпл 130-132oC.
Анализ. Рассчитано: C - 67,53; H - 7,28; N - 3,75; S - 8,58; найдено: C - 67,26; H - 7,21: N - 3,76; S - 8,65.
1H ЯМР (ДМСО-d6), δ ; 0,70-0,86 (6H, m); 0,96-1,23 (4H, m); 1,25-1,49 (1H, m); 1,66-1,75 (1H, m); 1,98-2,07 (1H, m); 2,40 (1H, d); 3,33 (2H, q); 5,82 (1H, d): 6,35(1H, d); 6,77-6,80 (1H, m); 7,24-7,38 (6H, m); 10,0 (1H, s).
Пример синтеза 12. Получение (±)-транс-3-бутил-3- этил-2,3,4,5-тетрагидро-5-фенил-1,4-бензотиазепин-4,8-диол-1,1- диоксида
1,0 г
продукта из примера 11 растворяют в 100 мл дихлорметана, охлаждают до 0oC и добавляют м-хлорпербензойную кислоту (0,55 г, 57-86%, Aldrich). Реакционную смесь перемешивают в течение 5 ч при
температуре ледяной ванны и нейтрализуют избыток кислоты 5%-м раствором NaHCO3. Органический слой отделяют, сушат и концентрируют в вакууме. Полученный остаток хроматографируют на
силикагеле, элюируя смесью гексана и EtOAc и получая 0,68 г конечного продукта в виде бледно-желтого твердого вещества с tпл 213-214oC.
Анализ. Рассчитано: C - 64,76; H - 6,99; N - 3,60; S - 8,23; найдено: C - 64,86; H - 7,03: N - 3,63: S - 8,31.
1H ЯМР (ДМСО-d6), δ ; 0,77-0,89 (6H, m); 1,09-1,64 (6H, m); 1,68-2, 03 (2H, m); 3,36 (2H, q); 6,30 (1H, s); 6,44 (1H, d); 6,82-6,87 (1H, m); 7,27- 7,49 (6H, m); 7,89 (1H, s); 10,0 (1H, s).
Пример синтеза 13. Получение (±)-транс-3-бутил-3-этил-2,
3,4,5-тетрагидро-8-метил-5-фенил- 1,4-бензотиазепин-1,1-диоксида
(а) 3-метилфенилбензоат
Раствор бензоилхлорида (32,5 г, Aldrich) в 200 мл эфира прикапывают в перемешиваемый раствор
м-крезола (25,0 г, Aldrich) и триэтиламина (27,2 г, Aldrich) в 500 мл эфира, затем перемешивают при комнатной температуре в течение 1 ч и фильтруют. Эфирный фильтрат промывают насыщенным раствором
NaHCO3 и водой и сушат над Na2SO4. Эфирный слой отделяют, сушат и концентрируют в вакууме, получая 104,0 г целевого продукта в виде белого твердого вещества с tпл 45-47oC.1H ЯМР согласуется с ожидаемой структурой.
(б) 2-гидрокси-4-метилбензофенон
48 г продукта стадии (а) расплавляют (при 70oC)
и порциями добавляют 30,2 г хлорида алюминия. Реакционную смесь в течение 5 мин нагревают до 200oC, затем охлаждают до комнатной температуры. Полученное твердое вещество размалывают в
порошок и медленно вносят в смесь 800 мл концентрированной HCl и льда. Смесь экстрагируют эфиром и промывают экстракт водой. Эфирный слой отделяют, сушат и концентрируют. Полученный остаток
хроматографируют на силикагеле, элюируя толуолом, и получают 39 г целевого продукта в виде желтого маслянистого вещества.1H ЯМР согласуется с ожидаемой структурой.
(в)
(±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-8-метил-5- фенил-1,4-бензотиазепин-1,1-диоксид
Продукт стадии (б) превращают в конечный продукт, следуя стадиям (а) - (м) примера 10, и
получают его в виде твердого белого вещества с tпл 121-122oC.
Анализ. Рассчитано: C - 71,12; H- 7,87: N - 3,77; S - 8,63; найдено: C -71,23; H - 7,94; N - 3,67; S - 8,74.
1H ЯМР (ДМСО-d6), δ ; 0,77-0,82 (6H, m); 1,16-2,07 (8H, m); 2,36 (3H, s); 3,37 (2H, q); 5,92 (1H, d); 6,47 (1H, d); 7,27-7,39 (6H, m); 10,0 (1H, s).
Пример синтеза 14. Получение (±)-транс-3-бутил-3- этил-2,3,4,5-тетрагидро-8-метокси-5-фенил-1,4-бензотиазепин-7- карбальдегид-1,1 -диоксида
а) (±
)-7-Бромо-3-бутил-3-этил-2,3-дигидро-8-метокси-5-фенил- 1,4-бензотиазепин
2,3-Дихлор-5,6-дециан-1,4-бензохинон (16,9 г) добавляют непосредственно к раствору продукта (как рацемата) (30,2 г)
стадии (ж) примера 5 в 300 мл бензола. Реакционную смесь перемешивают с обратным холодильником в течение 3 ч и охлаждают до комнатной температуры. Добавляют 1N раствора NaOH (200 мл), перемешивают в
течение 30 мин. Затем органический слой отделяют, промывают рассолом и 1N раствора NaOH. Бензоловый слой отделяют, сушат и концентрируют, получая маслянистое вещество, которое растворяют в генсане,
фильтруют и концентрируют, получая 25,8 г целевого продукта в виде красного маслянистого вещества.1H ЯМР согласуется с ожидаемой структурой.
б) (±
)-3-бутил-7-карбальдегид-3-этил-2,3-дигидро-8-метокси- 5-фенил-1,4-бензотиазепин
49,0 раствора 1,6 М н-бутиллития добавляют к ледяному раствору 25,8 г продукта стадии (а) в 500 мл гексана.
Реакционную смесь и 9 г 4-формилморфолина перемешивают в течение 25 мин. Реакционную смесь снимают с ледяной бани и перемешивают при комнатной температуре в течении 2,5 ч, затем быстро охлаждают в 250
мл насыщенного раствора NH4Cl и перемешивают в течение 1 ч. Органический слой отделяют, сушат и концентрируют, получая 26,9 г красного маслянистого вещества. Хроматография на силикагеле с
элюированием смесью 85: 15 гексана и EtOAc дает 13,9 г целевого продукта в виде оранжевого маслянистого вещества.1H ЯМР согласуется с ожидаемой структурой.
в) (±
)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-8-метокси-5- фенил- 1,4-бензотиазепин-7-карбальдегид
Этиленгликоль (9,3 г) и пиридин п-толуолсульфонат (1,3 г) добавляют к раствору продукта (19,0)
стадии (б) в 250 мл бензола и эту смесь выдерживают с обратным холодильником с ловушкой Дина-Старка в течение 17 ч. Реакционную смесь охлаждают до комнатной температуры и обрабатывают в течение 15 мин
водным раствором NaHCO3 (150 мл). Органический слой отделяют, сушат и концентрируют, получая 19,7 г густого желто-оранжевого маслянистого вещества.1H ЯМР согласуется с
производным диохолана. Это маслянистое вещество обрабатывают B2H6 согласно стадии (0) примера 1, получая 3,5 г целевого продукта в виде оранжевого маслянистого вещества.1H ЯМР согласуется с ожидаемой структурой.
(г) (±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-8-метокси-5-фенил- 1,4-бензотиазепин-7-карбальдегид-1,1-диоксид:
3,5 г
продукта стадии (в) растворяют в 60 мл смеси t-бутанола и тетрагидрофурана (пропорция 1: 4) и добавляют 3,4 г N-метилморфолин-N-оксида и затем 5 мл 2,5%-ного (по массе) раствора 0s04 в
2-метил-2-пропаноле. Реакционную смесь помешивают при комнатной температуре в течение 17 ч, затем разбавляют 250 мл EtOAc. Органический слой отделяют, промывают 1 N NaOH (2х150 мл) и рассолом, сушат и
концентрируют, получая в результате маслянистое вещество, которое перетирают в диэтиловом эфире и получают 3,10 г требуемого продукта в виде твердого белого вещества с темп. плавл. 127-128o
C.
Результаты анализа:
Расчетные данные: C - 66,48; H - 7,03; N - 3,37; S - 7,72.
Получено: C - 66,26; H - 7,04; N - 3,30; S - 7,82.
1H ЯМР (ДМСО-d6), δ ; 0,73- 0,86 (6H, m); 1,07-2,05 (8H, m); 2,65 (1H, d); 3,50 (2H, q); 4,03 (3H, s); 5,91 (1H, s); 6,92 (1H, s); 7,33-7,48 (5H, m); 7,74 (1H, s); 10,28 (1H, s).
Пример синтеза 15. Получение (±)-транс-2-((3-бутил-3-этил- 2,3,4,5- тетрагидро-8-метокси-5-фенил-1,4-бензотиазепин-7- ил)метокси)этанол-1,1-диоксида
Хроматография
продукта стадии (в) примера 14 дает 2,3 г соответствующего сульфидного соединения требуемого продукта в виде маслянистого вещества. Данные H1 ЯМР находятся в согласии с ожидаемой
структурой. Это маслянистое вещество затем обрабатывают согласно процедуре примера 1 (п) и получают в результате 0,65 г требуемого продукта в виде твердого белого вещества с темп. плавл. 83-85oC.
Результаты анализа:
Расчетные данные: C - 65,05; H - 7,64; N - 3,03; S - 6,95.
Получено: C - 64,82; H - 7,72; N - 2,99: S - 6,91.
1H ЯМР (ДМСО-d6), δ ; 0,74-0,86 (6H, m); 1,07-2,14 (8H, m); 2,52(1 H, d); 3,35 (4H, m); 3,41 (2H, q); 3,87 (3H, s); 4,39 (2H, s); 4,54 (1H, t); 5,91 (1H, d); 6,64 (1H, s); 7,29-7,45 (5H, m); 7,51 (1H, s).
Пример синтеза 16. Получение (±)-транс-3-бутил-3-этил- 2,3,4,5-тетрагидро-8-гидрокси-5-фенил-1,4-бензотиазепин-7- карбальдегид-1,1-диоксида
2,0 г продукта стадии (г) примера 14 добавляют к 20 мл ледяной уксусной кислоты и 20 мл 48%-й HBr и подогревают при 150oC в течение 24 ч. Реакционную смесь концентрируют в вакууме и
разделяют между диэтиловым эфиром и 5%-й NaHCO3. Органический слой отделяют, сушат и концентрируют, получая 0,85 г требуемого продукта в виде светло-коричневого твердого вещества с темп.
плавл. 158-159oC.
Результаты анализа:
Расчетные данные: C - 65,81; H - 6,78; N - 3,49; S - 7,99.
Получено: C - 65,63; H - 7,04: N - 3,32: S - 7, 74.
1H ЯМР (ДМСО-d6), δ ; 0,72-0,85 (6H, m); 1,07- 2,05 (8H, m); 2,58 (1H, d); 3,46 (2H, q); 5,85 (1H, d); 6,83 (1H, s); 7,34-7,47 (5H, m); 7,70 (1H, s); 10,25 (1H, s); 11,33 (1H, широкий s).
Пример синтеза 17. Получение (±)-транс-3-бутил-3-этил-2,3,4,5- тетрагидро-5-фенил-1,4-бензотиазепин-8-тиол-1,1-диоксида
Продукт
примера 11 обрабатывают, следуя процедуре стадий (к) - (л) примера 1, и получают требуемый продукт в виде твердого белого вещества с темп. плавл. 108-110oC.
Результаты
анализа:
Расчетные данные: C - 64,75; H - 6,99: N - 3,60; S - 16,46.
Получено: C - 64,83; H - 7,03; N - 3,56; S - 16,54.
1H ЯМР (ДМСО-d6 ), δ ; 0,70-0,81 (6H, m); 1,05-2,06 (8H, m); 2,54 (1H, d); 3,37 (2H, q); 5,85 (1H, d); 6,06 (1H, широкий s); 6,40 (1H, d); 7,26-7,40 (6H, m); 7,90 (1H, s).
Пример синтеза 18.
Получение (±)-транс-3-бутил-3-этил- 2,3,4,5-тетрагидро-8-гидрокси-5-фенил-1,4-бензотиазепин-8- сульфокислото-1,1-диоксида
5,3 г продукта примера 17 растворяют в 13 мл ДМСО, затем
добавляют 0,3 мл воды и 0,2 мл 48%-ой HBr. Реакционную смесь подогревают при 120oC в течение 4 ч до удаления дистиллята, затем охлаждают до комнатной температуры, разбавляют 1N NaOH и
фильтруют через фильтр из пористого стекла. Фильтрат закисляют 1N HCl, полученное твердое вещество фильтруют и высушивают, получая в результате 1,6 г требуемого продукта в виде бежевого твердого
вещества с темп. плавл. > 295oC.
Результаты анализа:
Расчетные данные: C - 57,64; H - 6,22; N - 3,20; S - 14,65.
Получено: C - 57,48: H - 6, 19; N - 3,25; S - 14,73.
1H ЯМР (ДМСО-d6), δ ; 0,82-0,95 (6H,m); 1,32-2,06 (8H, m); 2,54 (1H, d); 3,93 (2H, q); 4,70 (1H, широкий s); 6,23 (1H, s); 6,93 (1H, d); 7,60 (6H, широкий s); 7,84 (1H, d); 8,30 (1H, s).
Пример синтеза 19. Получение (7R,9R)-7-бутил-7-этил-6,7,8,9-тетрагидро- 9-фенил-1,3-диоксоло(4,5-H)(1,4)-бензотиазепин-5,
5-диоксида
0,74 г продукта примера 7 растворяют в 5 мл N,N-диметилформамида. К реакционной смеси добавляют 0,50 г карбоната натрия и 0,47 г бромхлорэтана и перемешивают при 110oC в
течение 2 ч. Смесь фильтруют через броунмиллерит, промывают EtOAc, и фильтрат высушивают, и концентрируют, получая в результате маслянистое вещество. Хроматография на силикагеле с использованием для
экстрагирования смеси гексан-EtOAc (пропорция 1:1) дает в результате 0,68 г требуемого продукта в виде твердого белого вещества с темп. плавл. 71-73oC.
Результаты
анализа:
Расчетные данные: C - 65,81: H - 6,78: N - 3,49; S - 7,99.
Получено: C - 65,89; H - 6,80; N - 3,50; S - 8,09.
1H ЯМР (ДМСО-d6), δ ; 0,71-0,85 (6H, m); 1,05-2,12 (8H, m); 2,49 (1H, d); 3,25 (2H, q); 3,42 (2H,s); 5,91 (1H, d); 6,06 (1H, s); 7,27-7,41 (6H, m).
Пример синтеза 20. Получение (±
)-транс-3-бутил-3-этил- 2,3,4,5-тетрагидро-8,9-диметокси-5-фенил-1,4-бензотиазепин-1,1-диоксида
(а) 2-гидрокси-3,4-метоксибензальдегид
21,81 хлорида алюминия добавляют шпателем в
ледяной раствор бензоилхлорида (22,1 г) и 1,2,3-триметоксибензола (25,0 г) в 250 мл 1,2-дихлорэтана. Реакционную смесь перемешивают при 0 - 5oC в течение 3 ч, затем подогревают с обратным
холодильником в течение 2 ч, вливают в 100 мл ледяной концентрированной HCl и перемешивают в течение 30 мин, после чего экстрагируют диэтиловым эфиром. Органический слой отделяют, сушат и
концентрируют, получая 23,0 г твердого вещества. Хроматография на силикагеле с использованием для экстрагирования смеси толуол-EtOAc (пропорция 9:1) дает в результате 18,0 г требуемого продукта в виде
твердого белого вещества с темп. плавл. 127-128oC. Данные H1 ЯМР находятся в согласии с ожидаемой структурой
(б) (±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-8,
9- диметокси-5-фенил-1,4-бензотиазепин-1,1-диоксид
Продукт стадии (а) превращают в требуемый, следуя процедурам стадий (а) - (м) примера 10. Конечный продукт выделяют в виде белого твердого
вещества с темп. плавл. 142-144oC
Результаты анализа:
Расчетные данные: C - 66,16: H- 7,48; N - 3,35; S - 7,68.
Получено: C - 66,03; H - 7,53; N - 3,28; S - 7,77.
1H ЯМР (ДМСО-d6), δ : 0,64 (3H, t); 0,81 (3H, t); 0,87-2,08(8H,m); 2.42(1H, d): 3,73(2H, q); 3,75(3H,S): 3,79 (3H, s); 5,50 (1H, d); 6,05 (1H, d); 6,97 (1H, d); 7,27-7,41 (5H, m).
Пример синтеза 21. Получение (3R,5R)-3-бутил-3-этил-5-(4-фторфенил)- 2,3,4,5-тетрагидро-7,8-диметокси-1,4-бензотиазепин-4-ол-1,1-диоксида
(a)
2-гидрокси-4,5-диметокси-4'-(фторбензофенон
1М раствор трихлорида бора (142 мл) в дихлорметане добавляют к раствору 4-фторбензоилхлорида (16,8 г) в бензоле. Затем добавляют 20 г 3,
4-диметоксифенола в 100 мл бензола и перемешивают реакционную смесь в течение 2 ч при комнатной температуре, после чего смесь вливают в ледяную воду и оставляют для перемешивания на 15 мин, затем
добавляют 500 мл 1N HCl и перемешивают при комнатной температуре в течение 17 ч.
Реакционную смесь экстрагируют EtOAc, отделяют EtOAc, концентрируют и сушат, получая 41,7 г требуемого
продукта в виде оранжевого твердого вещества. Данные H1 ЯМР находятся в согласии с ожидаемой структурой
(б) (3R, 5R)-3-бутил-3-этил-5-(4-фторфенил)-2,3,4,5-тетрагидро-7,
8-диметокси- 1,4-бензотиазепин-4-ол-1,1-диоксида
Продукт стадии (а) превращают в требуемый, следуя процедурам стадий (а) - (п) примера 1 и процедуре примера 2. Конечный продукт выделяют в
виде белого твердого вещества с темп. плавл. 170-171oC
Результаты анализа:
Расчетные данные: C - 61,18; H - 6,70; N - 3,10; S-7,10.
Получено: C - 61,28; H - 6,78; N - 2,99; S - 7,27.
1H ЯМР (ДМСО-d6), δ ; 0,75-0,85 (6H, m); 1,07-2,04 (8H, m); 3,35 (2H, q); 3,42 (3H, s); 3,81 (3H, s); 6,07 (1H, s); 6,33 (1H, s); 7,22 (2H, t); 7,39 (1H, s); 7,40- 7,50 (2H, m); 7,96(1H, s).
Примеры синтеза 22-54
Для каждого из нижеследующих примеров применен способ приготовления, аналогичный
приведенным в примере 1 и других примерах, или химические способы, известные специалистам. Во всех случаях данные H1 ЯМР и элементного анализа находятся в согласим с ожидаемой
структурой.
(2) (±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-8-метокси- 5-фенил-1,4-бензотиазепин-7-метанол-S,S-диоксид, темп. плавления 122-123oC
(23) (3R,
5R)-3-бутил-3-этил-2,3,4,5-тетрагидро-8-метокси-7-нитро- 5-фенил-1,4-бензотиазепин-1,1-диоксид (0,40 гидрат), темп. плавления 122-123oC
(24) (±)-транс-3-бутил-3-этил-2,3,4,
5-тетрагидро-8-метокси- 7-(метоксиметил)-5-фенил-1,4-бензотиазепин-1,1-диоксид, темп. плавления 118-119oC
(25) (±)-транс-7-бром-3-бутил-3-этил-2,3,4,5- тетрагидро-5-фенил-1,
4-бензотиазепин-8-ол-1,1-диоксид (0,40 гидрат), темп. плавления 137-138oC
(26) (±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-7,8,9- триметокси-5-фенил-1,4-бензотиазепин-1,
1-диоксид, темп. плавления 169-170oC
(27) (3R,5R)-3-бутил-3-этил-2,3,4,5-тетрагидро-5-фенил- 1,4-бензотиазепин-7,8-диил-диацетат-1,1-диоксид (0,40 гидрат), темп. плавления
79-81oC
(28) (8R, 10R)-8-бутил-8-этил-2,3,7,8,9,10-гексагидро-10-1,4- диоксоно(2,3-H)(1,4)-бензотиазепин-6,6-диоксид, темп. плавления 82oC
(29) (3R,
5R)-3-бутил-7,8-диэтокси-2,3,4,5-тетрагидро-5- фенил-1,4-бензотиазепин-1,1-диоксид (0,20 гидрат), темп. плавления 110-111oC
(30) (±)-транс-3-бутил-8-этокси-3-этил-2,3,4,
5-тетрагидро- 5-фенил-1,4-бензотиазепин-1,1-диоксид, темп. плавления 45-54oC
(31) (±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-8- (метилтио)-5-фенил- 1,4-бензотиазепин-1,
1-диоксидгидрохлорид, темп. плавления 194-197oC
(32)
(±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-8- изопропокси-5-фенил-1,4-бензотиазепин-1,1-диоксидгидрохлорид, темп.
плавления 178-181oC
(33) (±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-5-фенил- 1,4-бензотиазепин-8-карбальдегид-1,1-диоксид, темп. плавления 165- 170oC
(34) 3,3-диэтил-2,3,4,5-тетрагидро-1,1-диоксо-5-фенил-1,4- бензотиазепин-8-ил-аспартат
(35) 3,3-диэтил-2,3,4,5-тетрагидро-7,8-диметокси-5-фенил- 1,4-бензотиазепин-1,1-диоксид, темп. плавления
163-164oC
(36) 3,3-диэтил-5-(4-фторфенил)-2,3,4,5-тетрагидро-8- метокси-1,4-бензотиазепин-1,1-диоксид, темп. плавления 101-103oC
(37) 3,3-диэтил-2,3,4,
5-тетрагидро-8-метокси-5-фенил-1,4- бензотиазепин-1,1-диоксид, темп. плавления 132-133oC
(38) 3,3-диэтил-2,3,4,5-тетрагидро-5-фенил-1,4- бензотиазепин-4,8-диол-1,1-диоксид, темп.
плавления 225-227oC
(39) (RS)-3,3-диэтил-2,3,4,5-тетрагидро-4-гидрокси-7,8- диметокси-5-фенил-1,4-бензотиазепин-1,1-диоксид, темп. плавления 205-206oC
(40)
(±)-транс-3-бутил-8-этокси-3-этил-2,3,4,5-тетрагидро- 5-фенил-1,4-бензотиазепин-4-ол-1,1-диоксид, темп. плавления 149- 150oC
(41) (±)-транс-3-бутил-3-этил-2,3,4,
5-тетрагидро-8- изопропокси-5-фенил-1,4-бензотиазепин-4-ол-1,1 -диоксид, темп. плавления 109-115oC
(42) (±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-7,8,
9- триметокси-5-фенил-1,4-бензотиазепин-4-ол-1,1-диоксид, темп. плавления 84-96oC
(43) (3R, 5R)-3-бутил-3-этил-2,3,4,5-тетрагидро-5-фенил-1,4- бензотиазепин-4,7,8-триол-1,1-диоксид,
темп. плавления 215-220oC
(44) (±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-4,7,8- триметокси-5-фенил-1,4-бензотиазепин-1,1-диоксид, темп. плавления 169-187oC
(45) (±)-транс-3-бутил-3-этил-5-фенил-2,3,4,5-тетрагидро- 7,8-диметокси-1,4-бензотиазепин-4-ил-ацетат-S,S-диоксид, темп. плавления 154-156oC
(46) 3,3-диэтил-2,3,4,
5-тетрагидро-6-фенил-1,4- бензотиазепин-8-ол-1,1-диоксид, темп. плавления 177-178oC
(47) 3,3-диэтил-2,3,4,5-тетрагидро-7-метокси-5-фенил-1,4- бензотиазепин-8-ол-1,1-диоксид
(48) 3,3-дибутил-2,3,4,5-тетрагидро-5-фенил-1,4-бензотиазепин- 8-ол-1,1-диоксид
(49) (±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-1,1- диоксо-5-фенил-1,
4-бензотиазепин-8-ил-гидросульфат, темп. плавления 196,5-200oC
(50) (±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-1,1- диоксо-5-фенил-1,4-бензотиазепин-8-ил-дигидрофосфат,
темп. плавления 154-156oC
(51) 3,3-диэтил-2,3,4,5-тетрагидро-1,1-диоксо-5-фенил-1,4- бензотиазепин-8-ил-гидросульфат
(52) 3,3-диэтил-2,3,4,5-тетрагидро-1,1
-диоксо-5-фенил-1,4- бензотиазепин-8-ил-дигидрофосфат
(53) (±)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-1,1- диоксо-5-фенил-1,4-бензотиазепин-8-ил-аспартат
Биологический тест.
Подавление всасывания желчной кислоты in vitro.
Подавление всасывания кишечной желчной кислоты с веществами, усиливающими экскрецию, или вспомогательное подвздошное шунтирование используют как эффективное средство снижения концентрации ЛHП в плазме крови. Другой способ снижения всасывания желчной кислоты заключается в подавлении системы транспорта активного всасывания желчной кислоты в подвздошной кишке. Экспериментально выявлено, что такое подавление, определенное по выделению желчных кислот с фекалиями, приводит к гиперхолестеринемии1.
(1) Lewis, М.C.; Brieaddy, L.E.; and Root, C. Effects of 2164U90 on lleal Bile Acid Absorption and Serum Cholisterol in Rats and Mice. J. Lipid. Research., 1995, 36, 1098-1105.
Выделение
желчных кислот с фекалиями
Самцов крыс линии Спрейк-Доли массой 220 - 260 г поместили в отдельные клетки и кормили обычной казуариной хвощевидной. Крыс разделили на 6 экспериментальных групп
по 10-12 особей в течение двух дней, в 9:00 и 15:30 крысам вводили перорально через зонд, дозами 1 мл/100 г массы тела, опытные соединения в виде суспензии в 0,5%-ной метилцеллюлозе. Контрольная
группа получала только 0,5%-ной метилцеллюлозу. На второй день после утреннего введения дозы крысам ввели перорально малое количество (1,3 нмоль) таурина 23, 25 -75Se-гомохолевой кислоты
(75SeHCAT) в 1,0 мл солевого раствора.
75SeHCAT - это синтетический гамма-излучающий аналог, поглощаемый системой поглощения желчной кислоты в подвздошной кишке и сходный с таурохолевой кислотой. Его с некоторого времени используют в клиниках в качестве показателя поглощения желчной кислоты в подвздошной кишке.
(1) Galatola, G. ; Jazrawi, R. P. ; Bridges, C.; Joseph, A.E.A. and Northfield, T.C. Direct Measurement of First-Pass lleal Clearance of a Bile Acid in Humans. Gastroenterology. 1991, 100, 1100-1105.
(2) Ferraris, R.; Galatoa, G.; Barlotta, A; Pellerito, R.; Fracchia. M.; Cottino, F. and De La Pierre. M. Measurement of Bile Acid Half-Life Using [75Se] HCAT in Health and Intestinal Diseases. Dig. Dis. Sci. 1992, 37, 225-232. После введения75SeHCAT фекалии собирали в течение суток. Содержание75SeHCAT в фекалиях определяли счетчиком гамма-излучения "Packard Auto-Gamma 5000 Series". Характерные данные о подавлении75SeHCAT в процентах представлены в табл. 1.
Для сравнения, наиболее активное соединение, конкретно описанное в Международной патентной заявке WO 93/16055, в этом тесте подавляло 9%75SeHCAT при дозе 1,0 мг/кг.
Примеры фармацевтических составов
В приводимых далее примерах активным
соединением может быть любое соединение формулы (I) и/или его фармацевтически приемлемая соль, сольват или физиологически активное производное, а предпочтительно - (3R,5R)-3-бугил-3-этил-2,3,4,
5-тетрагидро-7,8-диметокси- 5-фенил-1,4-бензотиазепин-1,1-диоксид или одно из соединений согласно примерам синтеза 2-53.
(1) Составы таблеток
Приведенные ниже составы А и Б
(табл. 2 и 3) могут быть приготовлены влажным гранулированием ингредиентов (а) - (в) и (а) - (г) совместно с раствором поливинилпирролидона ("повидона") с последующим добавлением стеарата магния и
прессованием. Во всех примерах с твердыми веществами количество ингредиентов указано в мг/таблетку (или соответственно мг/капсулу, пессарий или суппозиторий. ). При этом знаком (*) отмечены
препараты согласно Британской фармакопее, а знаком (**) - Фармакопее США.
Состав В
(а) активный ингредиент - 100
(б) лактоза - 200
(в)
крахмальный гликолят натрия - 20
(г) повидон - 15
(д) стеарат магния - 5 - --- - 500
Нижеследующие составы Г и Д могут быть приготовлены непосредственным прессованием смеси
ингредиентов. Лактоза в состав Д пригодна для непосредственного прессования.
Состав Г
(а) активный ингредиент - 250
(б) стеарат магния - 4
(в)
предварительно желатинированный крахмал NF15 - 146 - --- - 400
Состав Д
(а) активный ингредиент - 250
(б) стеарат магния - 5
(в) лактоза - 145
(г) авицель
- 100 - --- - 500
Состав Е (с регулируемым высвобождением)
(а) активный ингредиент - 500
(б) гидроксипропилметилцеллюлоза (Methocel K4M Premium) - 112
(в)
лактоза* - 53
(г) повидон В.Р.С. - 28
(д) стеарат магния - 7 - --- - 700
Состав может быть приготовлен влажным гранулированием ингредиентов (а) - (в) с раствором
повидона с последующим добавлением стеарата магния и прессованием.
Состав Ж (таблетки в энтеросолюбильной оболочке)
Таблетки состава В в энтеросолюбильной оболочке могут быть
изготовлены нанесением на них покрытий из таких "энтерополимеров", как ацетофталат целлюлозы, поливинилацетофталат, фталат гидроксипропилметилцеллюлозы или анионные полимеры метакриловой кислоты и ее
эфира (Eudragit L), в количестве 25 мг на таблетку. За исключением Eudragit L они должны включать 10% (от массы используемого полимера) пластификатора для предотвращения растрескивания оболочки при
введении или хранении. Подходящими пластификаторами могут быть диэтилфталат, трибутилцитрат и триацетин.
Состав 3 (таблетки в энтеросолюбильной оболочке с регулируемым высвобождением).
Таблетки состава E в энтеросолюбильной оболочке могут быть изготовлены нанесением на них покрытий из таких "энтерополимеров", как ацетофталат целлюлозы, поливинилацетофталат, фталат гидроксипропилметилцеллюлозы или анионные полимеры метакриловой кислоты и ее эфира (Eudragit L), в количестве 50 мг на таблетку. За исключением Eudragit L они должны включать 10% пластификатора (от массы используемого полимера) для предотвращения растрескивания оболочки при введении или хранении. Подходящими пластификаторами могут быть диэтилфталат, трибутилцитрат и триацетин.
(II) Составы капсул
Состав А
Капсулы могут быть изготовлены смешиванием ингредиентов вышеприведенного состава Г и заполнением этой смесью двухсекционных
желатиновых оболочек. Состав Б (infra) можно изготовить подобным способом.
Состав Б
(а) активный ингредиент - 250
(б) лактоза* - 143
(в)
крахмальный гликолят натрия - 25
(д) стеарат магния - 2 - --- - 420
Состав В
(а) активный ингредиент - 250
(б) Macrogol 4000 BP - 350 - --- - 600
Капсулы
могут быть изготовлены расплавлением Macrogol 4000 BP, диспергированием активного ингредиента в расплаве и заполнением двухсекционных твердых желатиновых оболочек.
Состав Г
(а) активный ингредиент - 250
(б) лецитин - 100
(в) арахисовое масло - 100 - --- - 450
Капсулы могут быть изготовлены диспергированием активного ингредиента в лецитине и
арахисовом масле и заполнением мягких эластичных желатиновых оболочек.
Состав Д (капсула с регулируемым высвобождением)
(а) активный ингредиент - 250
(б)
микрокристаллическая целлюлоза - 125
(в) лактоза BP - 125
(г) этилцеллюлоза - 13 - --- - 513
Состав для капсул с регулируемым высвобождением может быть изготовлен экструзией
смеси ингредиентов (а)-(в), сферодизацией и высушиванием экструдата. Высушенные шарики покрывают оболочкой (г), регулирующей высвобождение, и заполняют ими двухсекционные твердые желатиновые
капсулы.
Состав Е (энтеросолюбильная капсула)
(а) активный ингредиент - 250
(б) микрокристаллическая целлюлоза - 125
(в) лактоза ВР - 125
(г)
ацетатфталат целлюлозы - 50
(д) диэтилфталат - 5 - --- - 555
Состав для энтеросолюбильных капсул может быть изготовлен экструзией смеси ингредиентов (а)-(в), сфероидизацией и
высушиванием экструдата. Высушенные шарики покрывают энтеросолюбильной оболочкой (г) с пластификатором (д) и заполняют ими двухсекционные твердые желатиновые капсулы.
Состав Ж
(энтеросолюбильная капсула с регулируемым высвобождением)
Энтеросолюбильные капсулы с составом Ж с регулируемым высвобождением могут быть изготовлены покрытием шариков таким "энтерополимером",
как ацетофталат целлюлозы, поливинилацетофталат, фталат гидроксипропилметилцеллюлозы или анионные полимеры метакриловой кислоты и ее эфира (Eudragit L) в количестве 50 мг/капс. За исключением
Eudragit L они должны содержать 10% пластификатора (от массы используемого полимера) для исключения растрескивания оболочки при введении или хранении. Подходящими пластификаторами могут быть
диэтилфталат, трибутилцитрат и триацетин.
(III) Состав для внутривенного вливания (на 1 ампулу)
(а) активный ингредиент - 0,200 г
(б) Стерильный апирогенный
фосфатный буфер - 10 мл
Активный ингредиент растворяют в большей части фосфатного буфера при 35-40oC, затем доводят до нужного объема и фильтруют через стерильный микропористый
фильтр в стерильные 10 мл ампулы (типа 1), которые герметизируют стерильной пробкой и запечатывают снаружи.
(IV) Состав для внутримышечных инъекций (на 1 ампулу)
(а) активный
ингредиент - 0,20 г
(б) бензиловый спирт - 0,10
(в) гликофурол 75 - 1,45 г
(г) вода для инъекций - добавить до 3,00 мл
Активный ингредиент растворяют в гликофуроле,
вводят и растворяют бензиловый спирт и добавляют воду до 3 мл. Смесь фильтруют через стерильный микропористый фильтр и запечатывают в стерильные 3 мл ампулы (типа 1).
(V) Состав для
сиропа
(а) активный ингредиент - 0,25 г
(б) раствор сорбита - 1,50 г
(в) глицерин - 1,00 г
(г) бензоат натрия - 0,005 г
(д) корригент вкуса - 0,0125 мл
(е) очищенная вода - до 5,0 мл
Бензоат натрия растворяют в части очищенной воды и добавляют раствор сорбита, вводят и растворяют активный ингредиент. Полученный раствор смешивают с
глицерином и доводят очищенной водой до нужного объема.
(VI) Состав для суппозитория
(а) активный ингредиент - 250
(б) твердый жир (Witepsol H15-Dynamit Nobel)
- 1770 - --- - 2020
Пятую часть Witepsol H15 расплавляют в лодочке на пару не более чем при 45oC. Активный ингредиент просеивают через 2001 м сито из нержавеющей стали и при
помешивании добавляют к расплаву, пользуясь аппаратом Сильверсона с режущей головкой. Поддерживая температуру смеси 45oC, к суспензии добавляют оставшийся Witepsol H15 с перемешиванием для
получения однородной смеси. Всю суспензию затем протирают через 2501 м сито из нержавеющей стали и при постоянном перемешивании остужают до 40oC. При температуре 38-40oC 2,
02-граммовыми порциями смеси заполняют подходящие пластмассовые отливочные формы, после чего суппозиториям дают остыть до комнатной температуры.
(VII) Состав для пессария
(а)
активный ингредиент - 250
(б) безводная декстроза - 380
(в) картофельный крахмал - 363
(г) стеарат магния - 7 - --- - 1000
Вышеприведенные ингредиенты смешивают и
изготавливают пессарии прессованием смеси.
(VIII) Трансдермальный состав
Активный ингредиент - 200 мг
Спирт** - 0,1 мл
Гидроксиэтилцеллюлоза
Активный ингредиент и спирт желируют с гидроксиэтилцеллюлозой и упаковывают в приспособление для трансдермального введения, имеющее площадь 10 см2.
Реферат
Изобретение относится к новым 1,4-бензотиазепин-1,1-диоксидам формулы II, где R1, R2 представляют собой прямоцепные C1-6алкильные группы; R4 представляет собой незамещенный фенил; R5 и R8 представляют собой водород; R6 представляет собой метокси или бром; R7a представляет собой метокси, гидрокси или трифторметил; R9 и R10 представляют собой водород, солям, сольватам или физиологически функциональным производным, а также к способу их получения. Кроме того, раскрыты фармацевтическая композиция на основе указанных соединений, которая понижает уровень липидов в крови, и способ лечения клинического состояния млекопитающего. Изобретение может быть использовано в медицине, в частности, для профилактики и лечения таких гиперлипидемических состояний, как атеросклероз. 4 c. и 5 з.п. ф-лы, 3 табл.
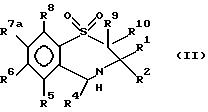
Формула
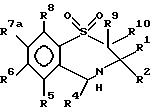
где R1 представляет собой прямоцепную С1-6алкильную группу;
R2 представляет собой прямоцепную С1-6алкильную группу;
R4 представляет собой незамещенный фенил;
R5 и R8 представляют собой водород;
R6 представляет собой метокси или бром;
R7а представляет собой метокси, гидрокси или трифторметил;
R9 и R10 представляет собой водород,
или его соль, сольват или физиологически функциональное производное.
(3R, 5R)-3-бутил-3-этил-2,3,4,5-тетрагидро-7,8-диметокси-5-фенил-1,4-бензотиазепин 1, 1-диоксида;
(+-)-транс-3-бутил-3-этил-2,3,4,5-тетрагидро-7,8-диметокси-5-фенил-1,4-бензотиазепин 1,1-диоксида;
(3R, 5R)-7-бром-3-бутил-3-этил-2,3,4,5-тетрагидро-8-метокси-5-фенил-1, 4-бензотиазепин 1,1-диоксида;
(3R, 5R)-3-бутил-3-этил-2,3,4,5-тетрагидро-7-метокси-5-фенил-1,4-бензотиазепин-8-ол 1,1-диоксида и
3,3-диэтил-2,3,4,5-тетрагидро-7,8-диметокси-5-фенил-1, 4-бензотиазепин 1,1-диоксида,
или его соли, сольвата или физиологически функционального производного.
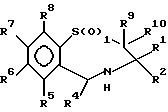
где R7 представляет собой R7а ;
I равно 0 или 1,
и возможно (б) разделяют смесь полученных таким образом изомеров и/или превращают образованное соединение формулы II в его соответствующую соль, сольват или физиологически функциональное производное.
Комментарии