1,5-бензотиазепины и их применение в качестве антигиперлипидемических средств - RU2302414C2
Код документа: RU2302414C2
Описание
Описание
Данное изобретение относится к производным бензотиазепина или их фармацевтически приемлемым солям, сольватам, сольватам таких солей и пролекарствам. Данные бензотиазепины обладают ингибирующей активностью в отношении транспорта желчной кислоты в подвздошной кишке (ileal bile acid transport - IBAT), поэтому представляют интерес для лечения заболеваний, связанных с состояниями гиперлипемии, и полезны в способах лечения теплокровных животных, таких как человек. Изобретение также относится к способам получения указанных производных бензотиазепина, фармацевтическим композициям, содержащим данные соединения, и к их применению в производстве лекарственных средств, предназначенных для ингибирования IBAT в организме теплокровного животного, такого как человек.
Хорошо известно, что состояния гиперлипемии, связанные с повышенными концентрациями общего холестерина и холестерина липопротеинов низкой плотности, являются главными факторами риска для сердечно-сосудистого атеросклероза (см., например, «Coronary Heart Disease: Reducing the Risk; a Worldwide View» Assman G., Garmena R., Cullen P. et al.; Circulation 1999, 100, 1930-1938, «Diabetes and Cardiovascular Desease: A Statement for Healthcare Professionals from the American Heart Association» Grundy S., Benjamin I., Burke G., et al.; Circulation, 1999, 100, 1134-46). Установлено, что нарушение циркуляции желчных кислот в полости кишечника снижает уровень холестерина. Разработанные ранее терапевтические методы снижения концентрации холестерина включают, например, лечение ингибиторами HMG-CoA-редуктазы, предпочтительно, статинами, такими как симвастатин (simvastatin) и флувастатин (fluvastatin), или лечение веществами, связывающими желчную кислоту, такими как смолы. Широко применяемыми веществами, связывающими желчную кислоту, являются, например, холестирамин (cholestyramine) и холестипол (cholestipol). Один из предложенных ранее методов терапевтического лечения («Bile Acids and Lopoprotein Metabolism: a Renaissance for Bile Acids in the Post Statin Era» Angelin B., Eriksson M., Rudling M; Current Opinion on Lipidology, 1999, 10, 269-74) включает лечение веществами, обладающими ингибирующим действием в отношении IBAT.
Повторная абсорбция желчной кислоты из желудочно-кишечного тракта является нормальным физиологическим процессом, который протекает, главным образом, в подвздошной кишке по механизму IBAT. Ингибиторы IBAT могут использоваться в лечении гиперхолестеринемии (см., например, публикацию «Interaction of bile acids and cholesterol with nonsystemic agents having hypocholesterolaemic properties», Biochemica et Biophysica Acta, 1210 (1994) 255-287). Таким образом, подходящие соединения, обладающие такой ингибирующей активностью в отношении IBAT, полезны и при лечении болезненных состояний, связанных гиперлипемии. Соединения, обладающие такой ингибирующей активностью в отношении IBAT, были описаны в литературе (см., например, соединения, описанные в WO 93/16055, WO 94/18183, WO 94/181884, WO 96/05188, WO 96/08484, WO 96/16051, WO 97/33882, WO 98/38182, WO 99/35135, WO 98/40375, WO 99/35153, WO 99/64409, WO 99/64410, WO 00/01687, WO 00/47568, WO 00/61568, WO 01/68906, DE 19825804, WO 00/38725, WO 00/38726, WO 00/38727, WO 00/38728, WO 00/38729, WO 01/68906 и ЕР 0864582.
Дополнительный аспект данного изобретения относится к применению соединений данного изобретения при лечении дислипидемических состояний и расстройств, таких как гиперлипемия, гипертриглицеридемия, гипербеталипопротеинемия (высокий уровень содержания LDL), гипербеталипопротеинемия (высокий уровень содержания VLDL), гиперхиломикронемия, гиполипопротеинемия, гиперхолестеринемия, гиперлипопротеинемия и гипоальфалипопротеинемия (низкое содержания HDL). Кроме того, ожидается, что эти соединения будут полезны для профилактики и лечения различных клинических состояний, таких как атеросклероз, сужение артерий, аритмия, гипертромботические состояния, сосудистая дисфункция, эндотелиальная дисфункция, сердечная недостаточность, коронарные болезни сердца, сердечно-сосудистые заболевания, инфаркт миокарда, стенокардия, периферийные сосудистые заболевания, воспаление сердечно-сосудистых тканей, таких как сердце, клапан, сосудистая сеть, артерии и вены, аневризмы, стеноз, рестеноз, сосудистые бляшки, сосудистые жировые прожилки, лейкоцитарная, моноцитарная и/или макрофагоцитарная инфильтрация, уплотнение внутренней оболочки сосудов (intimital thickening), медиальное истончение сосудов (medial thinning), инфекционная и хирургическая травма и сосудистый тромбоз, удар и временные приступы ишемии.
Данное изобретение основано на открытии того факта, что некоторые производные бензотиазепина неожиданно ингибируют IBAT. Ожидается, что данные свойства представляют интерес для лечения заболеваний, связанных с состояниями гиперлипидемии.
Соответственно, данное изобретение предлагает соединение формулы (I):

где
RV и RWнезависимо выбраны из водорода или С1-6 алкила;
R1 и R2 независимо выбраны из С1-6алкила;
Rx и Ry независимо выбраны из водорода или С1-6алкила, или один из Rx и Ry представляет собой водород или С1-6алкил, а другой представляет собой гидроксил или С1-6алкокси;
Rz выбран из галогена, нитро, циано, гидроксила, амино, карбокси, карбамоила, меркапто, сульфамоила, С1-6алкила, С2-6алкенила, С2-6алкинила, С1-6алкокси, С1-6 алканоила, С1-6алканоилокси, N-(С1-6алкил)амино, N,N-(С1-6алкил)2амино, С1-6алканоиламино, N-(С1-6алкил)карбамоила, N,N-(С1-6алкил)2карбамоила, С1-6алкилS(O)a, где а принимает значение от 0 до 2, С1-6алкоксикарбонила, С1-6алкоксикарбониламино, уреидо, N'-(С1-6алкил)уреидо, N-(С1-6алкил)уреидо, N',N'-(С1-6алкил)2уреидо, N'-(С1-6алкил)-N-(С1-6алкил)уреидо, N',N'-(С1-6алкил)2 -N-(С1-6алкил)уреидо, N-(С1-6алкил)сульфамоила и N,N-(С1-6алкил)2сульфамоила;
V принимает значение от 0 до 5;
один из R4 и R5 представляет собой группу формулы (IA):

R3 и R6 и второй из R4 и R5 независимо выбраны из водорода, галогена, нитро, циано, гидроксила, амино, карбокси, карбамоила, меркапто, сульфамоила, С1-4алкила, С2-4алкенила, С2-4алкинила, С1-4алкоксигруппы, С1-4алканоила, С1-4алканоилокси, N-(C1-4алкил)амино, N,N-(C1-4алкил)2амино, С1-4алканоиламино, N-(С1-4алкил)карбамоила, N,N-(С1-4алкил)2карбамоила, С1-4алкилS(O)a, где а принимает значение от 0 до 2, С1-4алкоксикарбонила, N-(C1-4алкил)сульфамоила и N,N-(C1-4алкил)2сульфамоила; где R3 и R6 и второй из R4 и R5 могут быть необязательно замещены по углероду одним или несколькими R16;
D представляет собой -О-, -N(Ra)-, -S(O)b- или -CH(Ra)-; где Ra представляет собой водород или С1-6алкил, и b принимает значение от 0 до 2;
кольцо А представляет собой арил или гетероарил; где кольцо А является необязательно замещенным одним или несколькими заместителями, выбранными из R17;
R7 представляет собой водород, С1-4 алкил, карбоциклил или гетероциклил; где R7 является необязательно замещенным одним или несколькими заместителями, выбранными из R18;
R8 представляет собой водород или С1-4алкил;
R9 представляет собой водород или С1-4алкил;
R10 представляет собой водород, С1-4 алкил, карбоциклил или гетероциклил; где R10 является необязательно замещенным одним или несколькими заместителями, выбранными из R19;
R11 представляет собой карбокси, сульфо, сульфино, фосфоно, тетразолил, -P(O)(ORc)(ORd), -P(O)(OH)(ORc), -P(O)(OH)(Rd) или
-P(O)(ORc)(Rd), где Rc и Rd независимо выбраны из С1-6алкила; либо R11 представляет собой группу формулы (IB):

где:
Х представляет собой -N(Rq)-, -N(Rq)C(O)-, -O- и -S(O)a; где а принимает значение от 0 до 2, и Rq представляет собой водород или С1-4алкил;
R12 представляет собой водород или С1-4 алкил;
R13 и R14 независимо выбраны из водорода, С1-4алкила, карбоциклила, гетероциклила или R23; где указанные С1-4алкил, карбоциклил или гетероциклил могут быть независимо необязательно замещенными одним или несколькими заместителями, выбранными из R20;
R15 представляет собой карбокси группу, сульфо, сульфино, фосфоно, тетразолил, -P(O)(ORe)(ORf), -P(O)(OH)(ORе),
-P(O)(OH)(Re) или -P(O)(ORe)(Rf), где Re и Rf независимо выбраны из С1-6алкила; или R15 представляет собой группу формулы (IC):
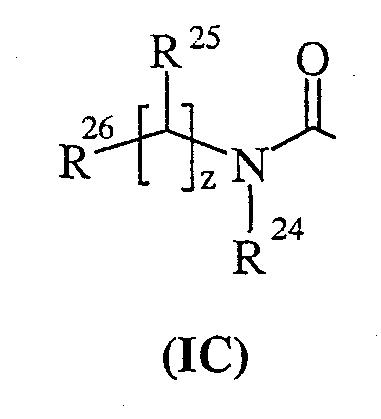
где
R24 выбран из водорода или С1-4 алкила;
R25 выбран из водорода, С1-4алкила, карбоциклила, гетероциклила или R27; где указанные С1-4алкил, карбоциклил или гетероциклил могут быть независимо необязательно замещенными одним или несколькими заместителями, выбранными из R28;
R26 выбран из карбокси, сульфо, сульфино, фосфоно, тетразолила, -P(O)(ORg)(ORh), -P(O)(OH)(ORg), -P(O)(OH)(Rg) или
-P(O)(ORg(Rh), где Rg и Rhнезависимо выбраны из С1-6алкила;
р принимает значение от 1 до 3; где значения R13 могут быть одинаковыми или разными;
q принимает значение от 0 до 1;
r принимает значение от 0 до 3; где значения R14 могут быть одинаковыми или разными;
m принимает значение от 0 до 2; где значения R10 могут быть одинаковыми или разными;
n принимает значение от 1 до 3; где значения R7 могут быть одинаковыми или разными;
z принимает значение от 0 до 3; где значения R25 могут быть одинаковыми или разными;
R16, R17 и R18 независимо выбраны из галогена, нитро, циано, гидроксила, амино, карбокси, карбамоила, меркапто, сульфамоила, С1-4алкила, С2-4алкенила, С2-4алкинила, С1-4 алкокси, С1-4алканоила, С1-4алканоилокси, N-(C1-4алкил)амино, N,N-(C1-4алкил)2амино, С1-4алканоиламино, N-(С1-4 алкил)карбамоила, N,N-(С1-4алкил)2карбамоила, С1-4алкилS(O)a, где а принимает значение от 0 до 2, С1-4алкоксикарбонила, N-(C1-4 алкил)сульфамоила и N,N-(C1-4алкил)2сульфамоила; где R16, R17 и R18 могут быть независимо необязательно замещенными по углероду одним или несколькими R21;
R19, R20, R23, R27 и R28 независимо выбраны из галогена, нитро, циано, гидроксила, амино, карбокси, карбамоила, меркапто, сульфамоила, С1-4алкила, С2-4алкенила, С2-4алкинила, С1-4алкоксигруппы, С1-4алканоила, С1-4алканоилокси, N-(C1-4алкил)амино, N,N-(C1-4алкил)2амино, С1-4алканоиламино, N-(С1-4алкил)карбамоила, N,N-(С1-4алкил)2карбамоила, С1-4алкилS(O)a, где а принимает значение от 0 до 2, С1-4алкоксикарбонила, N-(C1-4алкил)сульфамоила, N,N-(C1-4алкил)2сульфамоила, карбоциклила, гетероциклила, сульфо, сульфино, амидино, фосфоно, P(O)(ORa)(ORb), -P(O)(OH)(ORa), -P(O)(OH)(Ra) или -P(O)(ORa)(Rb), где Ra и Rb независимо выбраны из С1-6алкила; и где R19, R20, R23, R27 и R28 могут быть независимо необязательно замещенными по углероду одним или несколькими R22;
R21 и R22 независимо выбраны из галогена, гидроксила, циано, карбамоила, уреидо, амино, нитро, карбокси, карбамоила, меркапто, сульфамоила, трифторметила, трифтометокси, метила, этила, метокси, этокси, винила, аллила, этинила, метоксикарбонила, формила, ацетила, формамидо, ацетиламино, ацетокси, метиламино, диметиламино, N-метилкарбамоила, N,N-диметилкарбамоила, метилтио, метилсульфинила, мезила, N-метилсульфамоила и N,N-диметилсульфамоила;
или его фармацевтически приемлемая соль, сольват, сольват такой соли или пролекарство.
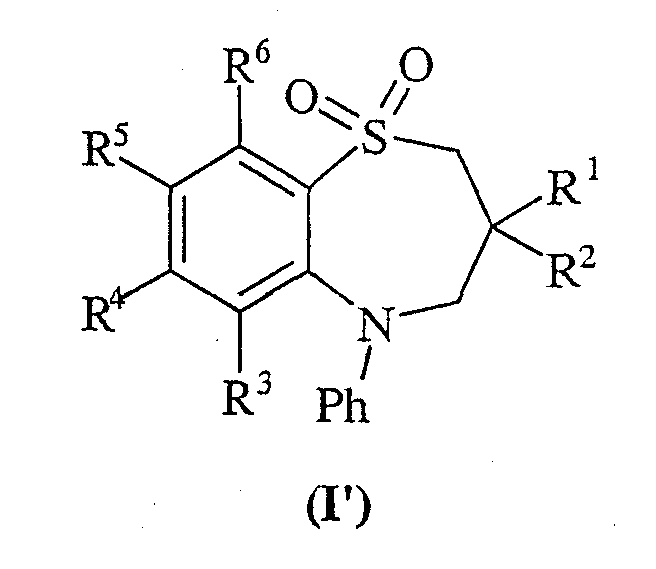
В соответствии с еще одним аспектом данного изобретения, предложено соединение формулы (I'):
где
R1 и R2 независимо выбраны из С1-6алкила;
один из R4 и R5 представляет собой группу формулы (IA'):

R3 и R6 и второй из R4 и R5 независимо выбраны из водорода, галогена, нитро, циано, гидроксила, амино, карбокси, карбамоила, меркапто, сульфамоила, С1-4 алкила, С2-4алкенила, С2-4алкинила, С1-4алкоксигруппы, С1-4алканоила, С1-4алканоилокси, N-(C1-4алкил)амино, N,N-(C1-4 алкил)2амино, С1-4алканоиламино, N-(С1-4алкил)карбамоила, N,N-(С1-4алкил)2карбамоила, С1-4алкилS(O)a, где а принимает значение от 0 до 2, С1-4алкоксикарбонила, N-(C1-4алкил)сульфамоила и N,N-(C1-4алкил)2сульфамоила; где R3 и R6 и второй из R4 и R5 могут быть необязательно замещенными по углероду одним или несколькими R16;
Кольцо А представляет собой арил или гетероарил; где кольцо А является необязательно замещенным одним или несколькими заместителями, выбранными из R13;
R7 представляет собой водород, С1-4 алкил, карбоциклил или гетероциклил; где R7 является необязательно замещенным одним или несколькими заместителями, выбранными из R18;
R8 представляет собой водород, С1-4алкил, карбоциклил или гетероциклил; где R8 является необязательно замещенным одним или несколькими заместителями, выбранными из R15;
R9 представляет собой представляет собой карбокси, сульфо, сульфино, фосфоно, тетразолил, -P(O)(ORc)(ORd), -P(O)(OH)(ORc),
-P(O)(OH)(Rd) или -P(O)(ORc )(Rd), где Rc и Rd независимо выбраны из С1-6алкила; или R9 представляет собой группу формулы (IB');

где
R10 представляет собой водород, С1-4 алкил, карбоциклил или гетероциклил; где R10 является необязательно замещенным одним или несколькими заместителями, выбранными из R16;
R11 представляет собой карбокси, сульфо, сульфино, фосфоно, -P(O)(ORe)(ORf), -P(O)(OH)(ORe), -P(O)(OH)(Re) или -P(O)(ORe)(Rf), где Re и Rf независимо выбраны из С1-6алкила;
р принимает значение от 1 до 3; где значения R10 могут быть одинаковыми или разными;
m принимает значение от 0 до 2; где значения R8 могут быть одинаковыми или разными;
n принимает значение от 1 до 3; где значения R7 могут быть одинаковыми или разными;
R12, R13 и R14 независимо выбраны из галогена, нитро, циано, гидроксила, амино, карбокси, карбамоила, меркапто, сульфамоила, С1-4алкила, С2-4алкенила, С2-4алкинила, С1-4алкоксигруппы, С1-4алканоила, С1-4алканоилокси, N-(C1-4алкил)амино, N,N-(C1-4алкил)2амино, С1-4алканоиламино, N-(С1-4алкил)карбамоила, N,N-(С1-4алкил)2карбамоила, С1-4алкилS(O)a, где а принимает значение от 0 до 2, С1-4алкоксикарбонила, N-(C1-4алкил)сульфамоила и N,N-(C1-4алкил)2сульфамоила; где R12, R13 и R14 могут быть независимо необязательно замещенными по углероду одним или несколькими R17;
R15 и R16 независимо выбраны из галогена, нитро, циано, гидроксила, амино, карбокси, карбамоила, меркапто, сульфамоила, С1-4алкила, С2-4алкенила, С2-4алкинила, С1-4алкоксигруппы, С1-4алканоила, С1-4алканоилокси, N-(C1-4алкил)амино, N,N-(C1-4алкил)2амино, С1-4алканоиламино, N-(С1-4алкил)карбамоила, N,N-(С1-4алкил)2карбамоила, С1-4алкилS(O)a, где а принимает значение от 0 до 2, С1-4алкоксикарбонила, N-(C1-4алкил)сульфамоила, N,N-(C1-4алкил)2сульфамоила, сульфо, сульфино, амидино, фосфоно,
-P(O)(ORa)(ORb), -P(O)(OH)(ORa), -P(O)(OH)(Ra) или -P(O)(ORa)(Rb), где Ra и Rb независимо выбраны из С1-6алкила; и где R15 и R16 могут быть независимо необязательно замещенными по углероду одним или несколькими R18;
R17 и R18 независимо выбраны из галогена, гидроксила, циано, карбамоила, уреидо, амино, нитро, карбокси, карбамоила, меркапто, сульфамоила, трифторметила, трифтометокси, метила, этила, метокси, этокси, винила, аллила, этинила, метоксикарбонила, формила, ацетила, формамидо, ацетиламино, ацетокси, метиламино, диметиламино, N-метилкарбамоила, N,N-диметилкарбамоила, метилтио, метилсульфинила, мезила, N-метилсульфамоила и N,N-диметилсульфамоила;
или его фармацевтически приемлемая соль, сольват, сольват такой соли или пролекарство.
В соответствии с еще одним аспектом данного изобретения, предложено соединение формулы (I''):

где
R1 и R2 независимо выбраны из С1-6алкила;
один из R4 и R5 представляет собой группу формулы (IA''):

R3 и R6 и второй из R4 и R5 независимо выбраны из водорода, галогена, нитро, циано, гидроксила, амино, карбокси, карбамоила, меркапто, сульфамоила, С1-4алкила, С2-4алкенила, С2-4алкинила, С1-4алкоксигруппы, С1-4алканоила, С1-4алканоилокси, N-(C1-4алкил)амино, N,N-(C1-4алкил)2амино, С1-4алканоиламино, N-(С1-4алкил)карбамоила, N, N-(С1-4алкил)2карбамоила, С1-4алкилS(O)a, где а принимает значение от 0 до 2, С1-4алкоксикарбонила, N-(C1-4алкил)сульфамоила и N, N-(C1-4алкил)2сульфамоила; где R3 и R6 и второй из R4 и R5 могут быть необязательно замещенными по углероду одним или несколькими R16;
Кольцо А представляет собой арил или гетероарил; где кольцо А является необязательно замещенным одним или несколькими заместителями, выбранными из R17;
R7 представляет собой водород, С1-4 алкил, карбоциклил или гетероциклил; где R7 является необязательно замещенным одним или несколькими заместителями, выбранными из R18;
R8 представляет собой водород или С1-4алкил;
R9 представляет собой водород или С1-4алкил;
R10 представляет собой водород, С1-4 алкил, карбоциклил или гетероциклил; где R10 является необязательно замещенным одним или несколькими заместителями, выбранными из R19;
R11 представляет собой карбокси, сульфо, сульфино, фосфоно, -P(O)(ORc)(ORd), -P(O)(OH)(ORc), -P(O)(OH)(Rd) или -P(O)(ORc)(Rd), где Rc и Rd независимо выбраны из С1-6алкила; или R11 представляет собой группу формулы (IB'');
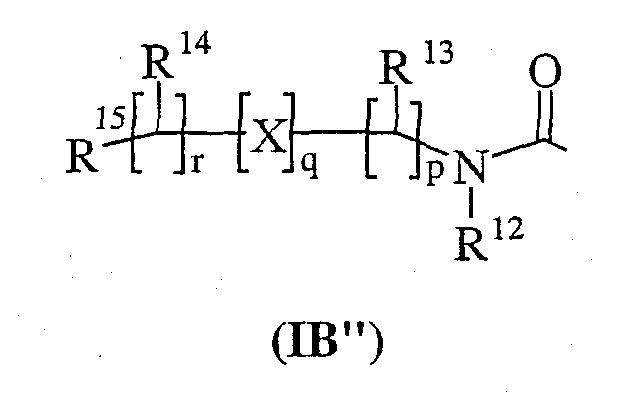
где
Х представляет собой -N(Rq)-, -N(Rq)C(O)-, -O- и -S(O)a; где а принимает значение от 0 до 2, и Rq представляет собой водород или С1-4алкил;
R12 представляет собой водород или С1-4 алкил;
R13 и R14 независимо выбраны из водорода, С1-4алкила, карбоциклила или гетероциклила; где R13 и R14 могут быть независимо необязательно замещенными одним или несколькими заместителями, выбранными из R20;
R15 представляет собой карбокси, сульфо, сульфино, фосфоно, -P(O)(ORe)(ORf), -P(O)(OH)(ORe), -P(O)(OH)(Re) или -P(O)(ORe)(Rf), где Re и Rf независимо выбраны из С1-6алкила;
р принимает значение от 1 до 3; где значения R13 могут быть одинаковыми или разными;
q принимает значение от 0 до 1;
r принимает значение от 1 до 3; где значения R14 могут быть одинаковыми или разными;
m принимает значение от 0 до 2; где значения R10 могут быть одинаковыми или разными;
n принимает значение от 1 до 3; где значения R7 могут быть одинаковыми или разными;
R16, R17 и R18 независимо выбраны из галогена, нитро, циано, гидроксила, амино, карбокси, карбамоила, меркапто, сульфамоила, С1-4алкила, С2-4алкенила, С2-4алкинила, С1-4алкоксигруппы, С1-4алканоила, С1-4алканоилокси, N-(C1-4алкил)амино, N,N-(C1-4алкил)2амино, С1-4алканоиламино, N-(С1-4алкил)карбамоила, N, N-(С1-4алкил)2карбамоила, С1-4алкилS(O)a, где а принимает значение от 0 до 2, С1-4алкоксикарбонила, N-(C1-4алкил)сульфамоила и N, N-(C1-4алкил)2сульфамоила; где R16, R17 и R18 могут быть независимо необязательно замещенными по углероду одним или несколькими R21;
R19 и R20 независимо выбраны из галогена, нитро, циано, гидроксила, амино, карбокси, карбамоила, меркапто, сульфамоила, С1-4алкила, С2-4 алкенила, С2-4алкинила, С1-4алкоксигруппы, С1-4алканоила, С1-4алканоилокси, N-(C1-4алкил)амино, N,N-(C1-4алкил)2амино, С1-4алканоиламино, N-(С1-4алкил)карбамоила, N,N-(С1-4алкил)2карбамоила, С1-4алкилS(O)a, где а принимает значение от 0 до 2, С1-4алкоксикарбонила, N-(C1-4алкил)сульфамоила, N,N-(C1-4алкил)2сульфамоила, карбоциклила, гетероциклила, сульфо, сульфино, амидино, фосфоно, -P(O)(ORa )(ORb), -P(O)(OH)(ORa), -P(O)(OH)(Ra) или -P(O)(ORa)(Rb), где Ra и Rb независимо выбраны из С1-6алкила; и где R19 и R20 могут быть независимо необязательно замещенными по углероду одним или несколькими R22;
R21 и R22 независимо выбраны из галогена, гидроксила, циано, карбамоила, уреидо, амино, нитро, карбокси, карбамоила, меркапто, сульфамоила, трифторметила, трифтометокси, метила, этила, метокси, этокси, винила, аллила, этинила, метоксикарбонила, формила, ацетила, формамидо, ацетиламино, ацетокси, метиламино, диметиламино, N-метилкарбамоила, N,N-диметилкарбамоила, метилтио, метилсульфинила, мезила, N-метилсульфамоила и N, N-диметилсульфамоила;
или его фармацевтически приемлемая соль, сольват, сольват такой соли или пролекарство.
Следует понимать, что если далее в описании и в формуле изобретения, делается ссылка на соединение формулы (I), то это относится также к соединениям формулы (I') и соединениям формулы (I'').
Кроме того, квалифицированному специалисту должно быть ясно, что система нумерации в соединениях формулы (I) и соединения формулы (I') различается. Система нумерации, используемая далее, относится к соединениям формулы (I), но следует понимать, что эти формулировки также применимы и к соответствующим значениям формулы (I').
В данном описании термин «алкил» включает как прямые, так и разветвленные алкильные группы, но ссылки на отдельные алкильные группы, такие как «пропил» являются специфическими только для группы с прямой цепью. Например, термин «С1-6алкил» включает С1-4алкил, С1-3алкил, пропил, изопропил и трет-бутил. Однако ссылки на отдельные алкильные группы, такие как «пропил», являются специфическими только для группы с прямой цепью, а ссылки на отдельные алкильные группы с разветвленной цепью, такие как «изопропил», являются специфическими только для группы с разветвленной цепью. Это относится и к другим радикалам, например, термин «фенилС1-6алкил» будет включать фенилС1-4алкил, бензил, 1-фенилэтил и 2-фенилэтил. Термин «галоген» относится к фтору, хлору, брому и йоду.
В том случае, когда необязательные заместители выбраны из «одной или нескольких» групп, следует понимать, что это определение включает все заместители, которые выбраны из одной из указанных групп, или заместители, которые выбраны из двух или нескольких из указанных групп.
«Гетероарил» представляет собой полностью ненасыщенное моно- или бициклическое кольцо, содержащее 3-12 атомов, из которых, по меньшей мере, один атом выбран из атомов азота, серы или кислорода, и если не указано другого условия, данное кольцо может присоединяться через атом углерода или азота. Предпочтительно, термин «гетероарил» относится к полностью ненасыщенному моноциклическому кольцу, содержащему 5 или 6 атомов углерода, или бициклическому кольцу, содержащему 9 или 10 атомов, из которых, по меньшей мере, один атом выбран из атомов азота, серы или кислорода, и если не указано другого условия, указанное кольцо может присоединяться через атом углерода или азота. В еще одном аспекте данного изобретения термин «гетероарил» относится к полностью ненасыщенному моноциклическому кольцу, содержащему 5 или 6 атомов, или бициклическому кольцу, содержащему 8, 9 или 10 атомов, из которых, по меньшей мере, один атом выбран из азота, серы или кислорода, и если не указано другого условия, указанное кольцо может присоединяться через атом углерода или азота. Примерами и подходящими значениями термина «гетероарил» являются тиенил, изоксазолил, имидазолил, пирролил, тиадиазолил, изотиазолил, триазолил, пиранил, индолил, пиримидил, пиразинил, пиридазинил, пиридил и хинолил. Предпочтительно, термин «гетероарил» относится к тиенилу или индолилу.
«Арил» представляет собой полностью ненасыщенное моно- или бициклическое углеродное кольцо, которое содержит 3-12 атомов. Предпочтительно, «арил» представляет собой моноциклическое кольцо, содержащее 5 или 6 атомов, или бициклическое кольцо, содержащее 9 или 10 атомов. Подходящие значения для термина «арил» включают фенил или нафтил. В частности, «арил» представляет собой фенил.
«Гетероциклил» представляет собой насыщенное, частично насыщенное или ненасыщенное моно- или бициклическое кольцо, содержащее 3-12 атомов, из которых, по меньшей мере, один атом выбран из азота, серы или кислорода, данное кольцо, если не указано другого условия, может присоединяться через атом углерода или азота, и в указанном кольце группа -СН2- может необязательно замещаться на группу -С(О)- или кольцевой атом серы может необязательно подвергаться окислению с образованием S-оксидов. Предпочтительно, «гетероциклил» представляет собой насыщенное, частично насыщенное или ненасыщенное моно- или бициклическое кольцо, содержащее 5 или 6 атомов, из которых, по меньшей мере, один атом выбран из азота, серы или кислорода, данное кольцо, если не указано другого условия, может присоединяться через атом углерода и азота, и в указанном кольце группа -СН2- может необязательно заменяться группой -С(О)- или кольцевой атом серы может необязательно подвергаться окислению с образованием S-оксида(ов). Примерами и подходящими значениями термина «гетероциклил» являются тиазолидинил, пирролидинил, пирролинил, 2-пирролидонил, 2,5-диоксопирролидинил, 2-бензоксазолинонил, 1,1-диоксотетрагидротиенил, 2,4-диоксоимидазолидинил, 2-оксо-1,3,4-(4-триазолинил), 2-оксазолидинонил, 5,6-дигидроурацинил, 1,3-бензодиоксолил, 1,2,4-оксадиазолил, 2-азабицикло[2.2.1]гептил, 4-тиазолидонил, морфолино, 2-оксотетрагидрофуранил, тетрагидрофуранил, 2,3-дигидробензофуранил, бензотиенил, тетрагидропиранил, пиперидил, 1-оксо-1,3-дигидроизоиндолил, пиперазинил, тиоморфолино, 1,1-диоксотиоморфолино, тетрагидропиранил, 1,3-димоксоланил, гомопиперазинил, тиенил, изоксазолил, имидазолил, пирролил, тиадиазолил, изотиазолил, 1,2,4-триазолил, 1,3, 4-триазолил, пиранил, индолил, пиримидил, тиазолил, пиразинил, пиридазинил, пиридил, 4-пиридонил, хинолил и 1-изохинолонил.
«Карбоциклил» представляет собой насыщенное, частично насыщенное или ненасыщенное моно- или бициклическое углеродное кольцо, которое содержит 3-12 атомов; и в указаннолм кольце группа -СН2- может быть необязательно замещена группой -С(О). Предпочтительно, «карбоциклил» представляет собой моноциклическое кольцо, содержащее 5 или 6 атомов, или бициклическое кольцо, содержащее 9 или 10 атомов. Подходящие значения термина «карбоциклил» включают циклопропил, циклобутил, 1-оксоциклопентил, циклопентил, циклопентенил, циклогексил, циклогексенил, фенил, нафтил, тетралинил, инданил или 1-оксоинданил. В частности, «карбоциклил» представляет собой циклопропил, циклобутил, 1-оксоциклопентил, циклопентил, циклопентенил, циклогексил, циклогексенил, фенил или 1-оксоинданил.
Примером групп «С1-6 алканоилокси» и «С1-4алканоилокси» является ацетоксигруппа. Примеры групп «С1-6алкоксикарбонил» и «С1-4алкоксикарбонил» включают метоксикарбонил, этоксикарбонил, н- и трет-бутоксикарбонил. Примеры групп «С1-6алкокси» и «С1-4алкокси» включают метокси, этокси и пропокси. Примеры групп «С1-6алканоиламино» и «С1-4 алканоиламино» включают формамидо, ацетамидо и пропиониламино. Примеры групп «С1-6алкилS(O)a, где а принимает значение от 0 до 2» и «С1-4алкилS(O)a, где а принимает значение от 0 до 2» включают метилтио, этилтио, метилсульфинил, этилсульфйинил, мезил и этилсульфонил. Примеры групп «С1-6алканоил» и «С1-4алканоил» включают С1-3алканоил, пропионил и ацетил. Примеры групп «N-(С1-6алкил)амино» и «N-(С1-4алкил)амино» включают метиламино и этиламино. Примеры групп «N,N-(C1-6алкил)2амино» и «N,N-(C1-4алкил)2амино» включают ди-N-метиламино, ди-(N-этил)амино и N-этил-N-метиламино. Примерами групп «С2-6алкенил» и «С2-4алкенил» являются винил, аллил и 1-пропенил. Примерами групп «С2-6алкинил» и «С2-4алкинил» являются этинил, 1-пропинил и 2-пропинил. Примерами групп «N-(C1-6алкил)сульфамоил» и «N-(C1-4алкил)сульфамоил» являются N-(C1-3алкил)сульфамоил, N-(метил)сульфамоил и N-(этил)сульфамоил. Примерами групп «N-(C1-6алкил)2сульфамоил» и «N-(C1-4алкил)2сульфамоил» являются N,N-(диметил)сульфамоил и N-(метил)-N-(этил)сульфамоил. Примерами групп «N-(C1-6алкил)карбамоил» и «N-(C1-4алкил)карбамоил» являются метиламинокарбонил и этиламинокарбонил. Примерами групп «N,N-(C1-6алкил)2карбамоил» и «N,N-(C1-4алкил)2карбамоил» являются диметиламинокарбонил и метилэтиламинокарбонил. Примерами группы «С1-6алкоксикарбониламино» являются этоксикарбониламино и трет-бутоксикарбониламино. Примерами групп «N'-(C1-6алкил)уреидо» являются N'-метилуреидо и N'-этилуреидо. Примерами группы «N-(C1-6алкил)уреидо» являются N-метилуреидо и N-этилуреидо. Примерами группы «N-(C1-6алкил)2уреидо» являются N', N'-диметилуреидо и N'-метил-N'-этилуреидо. Примерами группы «N'-(C1-6алкил)-N-(C1-6алкил)уреидо являются N'-метил-N-метилуреидо и N'-пропил-N-метилуреидо. Примерами группы «N', N'-(C1-6алкил)2-N-(C1-6алкил)уреидо» являются N',N'-диметил-N-метилуреидо и N'-метил-N'-этил-N-пропилуреидо.
Подходящая фармацевтически приемлемая соль соединения данного изобретения представляет собой, например, кислотно-аддитивную соль соединения данного изобретения, которое обладает достаточной основностью, например, кислотно-аддитивную соль неорганической или органической кислоты, например, хлористоводородной, бромистоводородной, серной, фосфорной, трифторуксусной, лимонной или малеиновой кислоты. Кроме того, подходящей фармацевтически приемлемой солью соединения данного изобретения, которая обладает достаточной кислотностью, является соль щелочного металла, например, натриевая или калиевая, соль щелочно-земельного металла, например, кальциевая или магниевая соль, аммониевая соль или соль с органическим основанием, которое образует физиологически приемлемый катион, например, соль с метиламином, диметиламином, триэтиламином, пиперидином, морфолином или трис(2-гидроксиэтил)амином.
Соединения формулы (I) могут вводиться в форме пролекарства, которое разлагается в организме человека или животного с получением соединения формулы (I). Примеры пролекарств включают сложные эфиры соединения формулы (I), способные подвергаться гидролизу in vivo, и амиды соединения формулы (I), способные подвергаться гидролизу in vivo.
Способный подвергаться гидролизу сложный эфир соединения формулы (I), содержащего карбоксильную или гидроксильную группу, представляет собой, например, фармацевтически приемлемый сложный эфир, который гидролизуется в организме человека или животного с получением исходной кислоты или спирта. Подходящие фармацевтически приемлемые сложные эфиры соединения, содержащего карбоксильную группу, включают сложные С1-6алкоксиметильные эфиры, например, сложный метоксиметильный эфир, сложные С1-6алканоилоксиметильные эфиры, например, сложные пивалоилоксиметильный, фталидиловый эфиры, сложные С3-8циклоалкилоксикарбонилоксиС1-6алкильные эфиры, например, сложный 1-циклогексилкарбонилоксиэтильный эфир; сложные 1,3-диоксолен-2-онилметильные эфиры, например, 5-метил-1,3-диоксолен-2-онилметильный эфир; и сложные С1-6алкоксикарбонилоксиэтильные эфиры, например, 1-метоксикарбонилоксиэтильный эфир, и они могут быть получены на любой карбоксильной группе соединений данного изобретения.
Способный гидролизоваться in vivo сложный эфир соединения формулы (I), содержащего гидроксильную группу, включает сложные неорганические эфиры, такие как сложные фосфатные эфиры и простые α-ацилоксиалкильные эфиры, а также родственные им соединения, которые в качестве конечного продукта гидролиза in vivo, протекающего с расщеплением сложного эфира, дают исходную гидроксильную группу. Примеры простых α-ацилоксиалкильных эфиров включают ацетоксиметокси и 2, 2-диметилпропионилокси-метоксиэфиры. Выбор групп, образующих способный гидролизоваться in vivo сложный эфир с гидроксильной группой, включает алканоил, бензоил, фенилацетил и замещенный бензоил и фенилацетил, алкоксикарбонил (с получение сложных алкилкарбонатных эфиров), диалкилкарбамоил и N-(диалкиламиноэтил)-N-алкилкарбамоил (с получением карбаматов), диалкиламиноацетил и карбоксиацетил. Примеры заместителей на бензоиле включают морфолино и пиперазино, присоединенные через кольцевой атом азота и метиленовую группу в положение 3 или 4 бензольного кольца.
Подходящим примером способного подвергаться гидролизу in vivo амида соединения (I), содержащего карбоксильную группу, является, например, N-C1-6алкил- или N,N-ди-С1-6 алкиламид, такой как N-метил-, N-этил-, N-пропил, N,N-диметил-, N-этил-N-метил- или N,N-диэтиламид.
Некоторые соединения формулы (I) могут содержать хиральные центры и/или центры геометрической изомерии (Е- или Z-изомеры), и следует понимать, что изобретение включает все возможные диастереоизомеры и геометрические изомеры, которые обладают ингибирующей активностью в отношении IBAT.
Изобретение относится к любому и всем таутомерным формам соединений формулы (I), которые обладают ингибирующей активностью в отношении IBAT.
Следует также понимать, что некоторые соединения формулы (I) могут существовать как в сольватированной, так и в несольватированной формах, таких как, например, гидратные формы. И следует понимать, что изобретение включает все сольватированные формы, которые обладают ингибирующей активностью в отношении IBAT.
Далее представлены предпочтительные значения R1, R2, R3, R4, R5 и R6. Данные значения могут использоваться в подходящих случаях в любом из определений, пунктов формулы изобретения или воплощений.
Предпочтительно RV и RW оба представляют собой водород.
Предпочтительно, R1 и R2 независимо выбраны из С1-4алкила.
Более предпочтительно, R1 и R2 независимо выбраны из этила или бутила.
Более предпочтительно, R1 и R2 независимо выбраны из этила, пропила или бутила.
В частности, в одном аспекте данного изобретения, R1 и R2 оба представляют собой бутил.
В еще одном аспекте данного изобретения, R1 и R2 оба представляют собой пропил.
В еще одном аспекте данного изобретения один из R1 и R2, в частности, представляет собой этил, а второй представляет собой бутил.
Предпочтительно, Rx и Ry независимо выбраны из водорода или С1-6алкила.
Более предпочтительно, Rx и Ry оба представляют собой водород.
Предпочтительно, Rz выбран из галогена, амино, С1-6алкила, С1-6алкоксикарбониламино или N'-(С1-6алкил)уреидо.
Более предпочтительно, Rz выбран из хлора, амино, трет-бутила, трет-бутоксикарбониламино или N-(трет-бутил)уреидо.
Предпочтительно, v принимает значение 0 или 1.
В одном аспекте данного изобретения, более предпочтительно v принимает значение 0.
В одном аспекте данного изобретения, более предпочтительно, v принимает значение 1.
В одном аспекте данного изобретения R4 предпочтительно представляет собой группу формулы (IA) (которая представлена выше).
В другом аспекте данного изобретения R5 предпочтительно представляет собой группу формулы (IA) (которая представлена выше.)
Предпочтительно, R3 и R6 представляют собой водород.
Предпочтительно, второй из R4 и R5, который не является группой формулы (IA), выбран из галогена, С1-4алкокси или С1-4алкилS(O), где а принимает значение от 0 до 2; и данный R4 или R5 может быть необязательно замещенным по углероду одним или несколькими R16; где R16 независимо выбран из гидроксила и N,N-(C1-4алкил)2амино.
Более предпочтительно, второй из R4 и R5, который не является группой формулой (IA), выбран из брома, метокси, изопропокси, метилтио, этилтио, изопропилтио или мезила; где указанный R4 или R5 может быть необязательно замещенным по атому углерода одним или несколькими R16; и где R16 независимо выбран из гидроксила или N,N-диметиламино.
В частности, второй из R4 и R5, который не является группой формулы (IA), выбран из брома, метокси, изопропокси, метилтио, этилтио, изопропилтио, 2-гидроксиэтилтио, 2-(N,N-диметиламино)этилтио или мезила.
Точнее, второй из R4 и R5, который не является группой (IA), представляет собой метилтио.
Предпочтительно, второй из R4 и R5, который не является группой формулы (IA), выбран из водорода, галогена, С1-4алкокси или С1-4алкилS(O)a, где а принимает значение от 0 до 2; где указанный R4 или R5 может быть необязательно замещенным по углероду одним или несколькими R16; и где R16 независимо выбран из гидроксила, карбокси и N,N-(C1-4алкил)2амино.
Более предпочтительно, второй из R4 или R5, который не является группой формулы (IA), выбран из водорода, брома, метокси, изопропокси, метилтио, этилтио, изопропилтио или мезила; где указанный R4 или R5 может быть необязательно замещенным по углероду одним или несколькими R16; и где R16 независимо выбран из гидроксила, карбокси и N,N-диметиламино.
В частности, второй из R4 и R5, который не является группой (IA), выбран из водорода, брома, метокси, изопропокси, метилтио, карбоксиметилтио, этилтио, изопропилтио, 2-гидроксиэтилтио, 2-(N,N-диметиламино)этилтио или мезила.
В другом аспекте данного изобретения, более предпочтительно, второй из R4 и R5, который не является группой формулы (IA), выбран из водорода, хлора, брома, метокси, изопропокси, метилтио, этилтио или изопропилтио; где указанный R4 или R5 может быть необязательно замещенным по углероду одним или несколькими R16;и гдеR16независимо выбран из гидроксила, карбокси и N,N-диметиламино.
В еще одном аспекте данного изобретения, второй из R4 и R5, который не является группой формулы (IA), выбран, в частности, из водорода, хлора, брома, метокси, изопропокси, метилтио, карбоксиметилтио, этилтио, изопропилтио, 2-гидроксиэтилтио или 2-(N,N-диметиламино)этилтио.
В еще одном аспекте данного изобретения, второй из R4 и R5, который не является группой формулы (IA), более точно, представляет собой бром или хлор.
В еще одном аспекте данного изобретения, второй из R4 или R5, который не является группой формулы (IA), более точно, представляет собой метокси.
В одном аспекте данного изобретения, предпочтительно, кольцо А представляет собой арил.
В другом аспекте данного изобретения, предпочтительно, кольцо А представляет собой гетероарил.
Когда кольцо А представляет собой арил, предпочтительно, кольцо А представляет собой фенил.
Когда кольцо А представляет собой гетероарил, предпочтительно, кольцо А представляет собой тиенил или индолил.
Предпочтительно, кольцо А представляет собой арил или гетероарил; где кольцо А является необязательно замещенным одним или несколькими заместителями, выбранными из R17; где
R17 выбран из галогена, гидроксила или С1-4алкила; где R17 может быть необязательно замещен по углероду одним или несколькими R21; где
R21 выбран из галогена.
Предпочтительно, D представляет собой -О- или -S-.
В одном аспекте данного изобретения, более предпочтительно, D представляет собой -О-.
В другом аспекте данного изобретения, более предпочтительно, D представляет собой -S-.
Более предпочтительно, кольцо А представляет собой фенил, тиенил или индолил; где кольцо А является необязательно замещенным одним или несколькими заместиттелями, выбранными из галогена, гидроксила или трифторметила.
В частности, кольцо А выбрано из фенила, 4-гидроксифенила, тиен-2-ила, 4-трифторметилфенила, 3-гидроксифенила, 2-фторфенила, 2,3-дигидроксифенила или индол-3-ила.
Более конкретно, кольцо А представляет собой фенил.
В другом аспекте данного изобретения, предпочтительно, кольцо А представляет собой арил или гетероарил; где кольцо А является необязательно замещенным одним или несколькими заместителями, выбранными из R17; где
R17 выбран из галогена, гидроксила, С1-4алкила или С1-4алкокси; где R17 может быть необязательно замещенным по углероду одним или несколькими R21; где
R21 выбран из галогена.
В еще одном аспекте данного изобретения, предпочтительно, кольцо А представляет собой фенил, тиенил или индолил; где кольцо А является необязательно замещенным одним или несколькими заместителями, выбранными из галогена, гидроксила, метокси или трифторметила.
В еще одном аспекте данного изобретения кольцо А, в частности, выбрано из фенила, 4-гидроксифенила, 4-метоксифенила, тиен-2-ила, 4-трифторметилфенила, 3-гидроксифенила, 2-фторфенила, 2,3-дигидроксифенила или индол-3-ила.
В еще одном аспекте данного изобретения кольцо А, в частности, выбрано из фенила, 4-гидроксифенила, 4-метоксифенила, тиен-2-ила, 4-трифторметилфенила, 3-гидроксифенила, 2-фторфенила, 4-фторфенила, 2,3-дигидроксифенила или индол-3-ила.
Предпочтительно, R7 представляет собой водород, С1-4алкил или карбоциклил.
Более предпочтительно, R7 представляет собой водород, метил или фенил.
В частности, R7 представляет собой водород.
В одном аспекте данного изобретения, предпочтительно, R8 представляет собой водород.
В другом аспекте данного изобретения, предпочтительно, R8 представляет собой С1-4 алкил.
В еще одном аспекте данного изобретения, более предпочтительно, R8 представляет собой водород или метил.
В одном аспекте данного изобретения, предпочтительно, R9 представляет собой водород.
В другом аспекте данного изобретения, предпочтительно, R9 представляет собой С1-4алкил.
В еще одном аспекте данного изобретения, более предпочтительно, R9 представляет собой водород или метил.
Предпочтительно, R10 представляет собой водород.
В одном аспекте данного изобретения, предпочтительно, R11 представляет собой карбокси, сульфо, сульфино, фосфоно,
-P(O)(ORc)(ORd), -P(O)(OH)(ORc), -P(O)(OH)(Rd) или -P(O)(ORc)(Rd), где Rc и Rd независимо выбраны из С1-6алкила.
В другом аспекте данного изобретения, предпочтительно, R11 представляет собой группу формулы (IB) (которая представлена выше).
Предпочтительно, R11 представляет собой карбокси,
-P(O)(OH)(ORc) или группу формулы (IB) (которая представлена выше).
Более предпочтительно, R11 представляет собой карбокси,
-P(O)(OH)(OEt) или группу формулы (IB) (которая представлена выше).
В еще одном аспекте данного изобретения, предпочтительно, R11 представляет собой карбокси, сульфо, -P(O)(OH)(ORc), где Rc выбран из С1-4алкила, или группу формулы (IB) (которая представлена выше).
Предпочтительно, Х представляет собой -NH- или -NHC(O)-.
Более предпочтительно, Х представляет собой -NHC(O)-.
В одном аспекте данного изобретения, предпочтительно, R12 представляет собой водород.
В другом аспекте данного изобретения, предпочтительно, R12 представляет собой С1-4алкил.
В еще одном аспекте данного изобретения, более предпочтительно, R12 представляет собой водород или метил.
Предпочтительно, R13 представляет собой водород, С1-4алкил или карбоциклил; где R13 является необязательно замещенным одним или несколькими заместителями, выбранными из R20; где
R20 представляет собой гидроксил.
Более предпочтительно, R13 представляет собой водород, метил или фенил; где R13 является необязательно замещенным одним или несколькими заместителями, выбранными из R20; и где
R20 представляет собой гидроксил.
В частности, R13 представляет собой водород, гидроксиметил или фенил.
Более предпочтительно, R13 представляет собой водород или гидроксиметил.
В другом аспекте данного изобретения, предпочтительно, R13 представляет собой водород, С1-4алкил или карбоциклил; где R13 является необязательно замещенным одним или несколькими заместителями, выбранными из R20; и где
R20 представляет собой гидроксил, карбокси, карбоциклил или амино; где R20может быть необязательно замещенным по углероду одним или несколькими R22;
R22 представляет собой гидроксил.
В другом аспекте данного изобретения, более предпочтительно, R13 представляет собой водород, метил, этил, бутил или фенил; где R13 является необязательно замещенным одним или несколькими заместителями, выбранными из R20; где
R20 представляет собой гидроксил, карбокси, фенил или амино; где R20может быть необязательно замещенным по углероду одним или несколькими R22; где
R22 представляет собой гидроксил.
В еще одном аспекте данного изобретения, в частности, R13 представляет собой водород, гидроксиметил, 4-аминобутил, 2-карбоксиэтил, 4-гидроксибензил или фенил.
В еще одном аспекте данного изобретения, предпочтительно, R13 представляет собой водород, С1-4 алкил или карбоциклил; где R13 является необязательно замещенным одним или несколькими заместителями, выбранными из R20; где
R20 представляет собой гидроксил, карбокси, карбоциклил, гетероциклил или амино; где R20 может быть необязательно замещенным по углероду одним или несколькими R22;
R22 представляет собой гидроксил.
В еще одном аспекте данного изобретения, более предпочтительно, R13 представляет собой водород, метил, этил, бутил или фенил; где R13является необязательно замещенным одним или несколькими заместителями, выбранными из R20; где
R20 представляет собой гидроксил, карбокси, фенил, имидазолил или амино; где R20 может быть необязательно замещенным по углероду одним или несколькими R22;
R22 представляет собой гидроксил.
В еще одном аспекте данного изобретения, в частности, R13 представляет собой водород, гидроксиметил, 4-аминобутил, 2-карбоксиэтил, 4-гидроксибензил, имидазол-5-илметил или фенил.
В еще одном аспекте данного изобретения, предпочтительно, R13 представляет собой водород, С1-4 алкил, карбоциклил или R23; где R13 является необязательно замещенным одним или несколькими заместителями, выбранными из R20; где
R20 представляет собой гидроксил, С1-4алкилS(O)a, где а принимает значение 0, С1-4алкокси, амино, карбоциклил, гетероциклил или меркапто; где R20 может быть независимо необязательно замещенным по углероду одним или несколькими R22;
R22 выбран из гидроксила; и
R23 представляет собой карбокси.
В еще одном аспекте данного изобретения, более предпочтительно, R13 представляет собой водород, метил, этил, бутил, фенил или R23; где R13 является необязательно замещенным одним или несколькими заместителями, выбранными из R20; где
R20 представляет собой гидроксил, метилтио, метокси, амино, имидазолил или меркапто; где R20 может быть независимо необязательно замещенным по углероду одним или несколькими R22;
R22 выбран из гидроксила; и
R23 представляет собой карбокси.
В еще одном аспекте данного изобретения R13, в частности, представляет собой водород, карбокси, гидроксиметил, меркаптометил, метоксиметил, метилтиометил, 2-метилтиоэтил, 4-аминобутил, 4-гидроксибензил, имидазол-5-илметил или фенил.
В еще одном аспекте данного изобретения R13, точнее, представляет собой метилтиометил, метилсульфинилметил или метилсульфонилметил.
Предпочтительно, R14 представляет собой водород.
В еще одном аспекте данного изобретения, предпочтительно, R14 выбран из водорода, С1-4алкила или карбоциклила; где указанный С1-4алкил или карбоциклил может быть необязательно замещенным одним или несколькими заместителями, выбранными из R20; и
R20представлялет собой гидроксил.
В еще одном аспекте данного изобретения, более предпочтительно, R14 выбран из водорода, метила или фенила; где указанный метил или фенил может быть является необязательно замещенным одним или несколькими заместителями, выбранными из R20; и
R20 представляет собой гидроксил.
В еще одном аспекте данного изобретения R14, в частности, представляет собой водород, фенил или гидроксиметил.
R15, в частности, представляет собой карбокси или сульфо.
В одном аспекте данного изобретения R15, точнее, представляет собой карбокси.
В другом аспекте данного изобретения R15, точнее, представляет собой сульфо.
Предпочтительно, R15 представляет собой карбокси, сульфо,
-P(O)(ORe)(ORf), -P(O)(OH)(ORе), -P(O)(OH)(Re) или -P(O)(ORe)(Rf), где Re и Rf независимо выбраны из C1-4алкила.
Более предпочтительно, R15 представляет собой карбокси, сульфо, -P(O)(ORe)(ORf), -P(O)(OH)(ORе), -P(O)(OH)(Re) или
-P(O)(ORe)(Rf), где Re и Rf независимо выбраны из метила или этила.
Предпочтительно, R15 представляет собой карбокси, сульфо,
-P(O)(OEt)(OEt), -P(O)(OH)(OEt), -P(O)(OH)(Me) или
-P(O)(OEt)(Me).
Предпочтительно, R15 представляет собой карбокси, сульфо, фосфоно, -P(O)(ORe)(ORf), -P(O)(OH)(ORе), -P(O)(OH)(Re) или
-P(O)(ORe)(Rf), где Re и Rf независимо выбраны из С1-4алкила, или R15 представляет собой группу формулы (IC) (которая представлена выше).
R15 более предпочтительно представляет собой карбокси, сульфо, фосфоно, -P(O)(ORe)(ORf), -P(O)(OH)(ORе), -P(O)(OH)(Re) или -P(O)(ORe)(Rf), где Re и Rf независимо выбраны из метила или этила, или R15 представляет собой группу формулы (IC) (которая представлена выше).
R15 предпочтительно представляет собой карбокси, сульфо, фосфоно, P(O)(OEt)(OEt), -P(O)(Ot-Bu)(Ot-Bu), -P(O)(OH)(OEt),
-Р(О)(Он)Ме) или -P(O)(OEt)(Me), или R15 представляет собой группу формулы (IC) (которая представлена выше).
В одном аспекте данного изобретения R15 предпочтительно представляет собой группу формулы (IC) (которая представлена выше).
В другом аспекте данного изобретения, предпочтительно, R15 не является группой формулы (IC) (которая представлена выше).
В следующем аспекте данного изобретения, предпочтительно, R15 представляет собой карбокси.
В другом аспекте данного изобретения, предпочтительно, R15представляет собой сульфо.
В следующем аспекте данного изобретения, предпочтительно, R15представляет собой -Р(О)(ОН)(OEt).
В еще одном аспекте данного изобретения, предпочтительно, R15представляет собой -Р(О)(ОН)(ОМе).
В еще одном аспекте данного изобретения, предпочтительно, R15представляет собой -Р(О)(OEt)(Me).
В одном аспекте данного изобретения, предпочтительно, R24 представляет собой водород.
В другом аспекте данного изобретения, предпочтительно, R24 представляет собой С1-4 алкил.
Предпочтительно, R25 представляет собой водород.
Предпочтительно, R26 представляет собой карбокси.
Предпочтительно, р принимает значение 1 или 2; и значения R13 могут быть одинаковыми или разными.
В одном аспекте данного изобретения, более предпочтительно, р принимает значение 1.
В другом аспекте данного изобретения, более предпочтительно, р принимает значение 2; и значения R13 могут быть одинаковыми или разными.
В еще одном аспекте данного изобретения, более предпочтительно, р принимает значение 3, и значения R13 могут быть одинаковыми или разными.
В одном аспекте данного изобретения, предпочтительно, q принимает значение 0.
В еще одном аспекте данного изобретения, предпочтительно, q принимает значение 1.
В одном аспекте данного изобретения, предпочтительно, r принимает значение 0.
В одном аспекте данного изобретения, более предпочтительно, r принимает значение 1.
В другом аспекте данного изобретения, более предпочтительно, r принимает значение 2, и значения R14 могут быть одинаковыми или разными.
В еще одном аспекте данного изобретения, более предпочтительно, r принимает значение 3; и значения R14 могут быть одинаковыми или разными.
Предпочтительно, m принимает значение 0.
В другом аспекте данного изобретения, предпочтительно, m принимает значение 0 или 1.
Предпочтительно, n принимает значение 1.
В другом аспекте данного изобретения, предпочтительно, n принимает значение 1 или 2.
Предпочтительно, z принимает значение 1.
Группа формулы (IA') представляет собой группу, в которой R7 представляет собой водород, метил или фенил, n принимает значение 1, кольцо А представляет собой фенил, тиенил или индолил; где кольцо А является необязательно замещенным одним или несколькими заместителями, выбранными из галогена, гидроксила или трифторметила, m принимает значение 0, и R9 представляет собой карбокси, -Р(О)(ОН)(ORc) или группу формулы (IB).
Группа формулы (IA) представляет собой группу, в которой:
D представляет собой -О- или -S-;
кольцо А представляет собой фенил, тиенил или индолил; где кольцо А является необязательно замещенным одним или несколькими заместителями, выбранными из галогена, гидроксила, метокси или трифторметила;
R7 представляет собой водород, метил или фенил;
R8 представляет собой водород или метил;
R9 представляет собой водород или метил;
R10 представляет собой водород;
m принимает значение от 0 до 2, и значения R10 могут быть одинаковыми или разными; и
R11 представляет собой карбокси, -Р(О)(OH)(OEt) или группу формулы (IB) (которая представлена в пункте 1);
Группа формулы (IB') представляет собой группу, в которой R10 представляет собой водород, гидроксиметил или фенил, р принимает значение 1 или 2; и значения R10 могут быть одинаковыми или разными, и R11 представляет собой карбокси или сульфо.
Группа формулы (IB) представляет собой группу, в которой:
R12 представляет собой водород или метил;
R13 представляет собой водород, метил, этил, бутил, фенил или R23; где R13 является необязательно замещенным одним или несколькими заместителями, выбранными из R20; R20 представляет собой гидроксил, метилтио, метокси, амино, имидазолил или меркапто; где R20 может быть независимо необязательно замещенным по углероду одной или несколькими гидроксильными группами; R23 представляет собой карбокси;
Х представляет собой -NH- или -NHC(O)-;
R14 выбран из водорода, метила или фенила; где указанный метил или фенил может быть необязательно замещенным одним или несколькими заместителями, выбранными из гидроксила;
R15 представляет собой карбокси, сульфо, фосфоно, -Р(О)(ORe)(ORf), -P(O)(OH)(ORe), -P(O)(OH)(Re) или -P(O)(ORe)(Rf), где Re и Rf независимо выбраны из метила или этила, или R15 представляет собой группу формулы (IC) (которая представлена в пункте 1);
p принимает значение от 1 до 3, и значения R13 могут быть одинаковыми или разными;
q принимает значение от 0 до 1; и
r принимает значение от 0 до 3, и значения R14 могут быть одинаковыми или разными.
Группа формулы (IC) представляет собой группу, в которой
R24 представляет собой водород;
R25 представляет собой водород;
R26 представляет собой карбокси; и
z принимает значение 1;
или его фармацевтически приемлемая соль, сольват, сольват такой соли или пролекарство.
Таким образом, в еще одном аспекте данного изобретения предложено соединение формулы (I'), которая представлена выше, где
R1 и R2 независимо выбраны из этила или бутила;
R3 и R6 представляет собой водород;
R4 выбран из галогена, С1-4алкоксигруппы или С1-4алкилS(O)a, где а принимает значение от 0 до 2; и R4 может быть необязательно замещенным по углероду одним или несколькими R16; где R16 независимо выбран из гидроксила и N,N-(C1-4алкил)2амино;
R5 представляет собой группу формулы (IA');
кольцо А представляет собой арил или гетероарил; где кольцо А является необязательно замещенным одним или несколькими заместителями выбранными из R17; где
R17 выбран из галогена, гидроксила или С1-4алкила; где R17 может быть необязательно замещенным по углероду одним или несколькими R21; где
R21 выбран из галогена;
R7 представляет собой водород, С1-4алкил или карбоциклил;
R11представляет собой карбокси, -Р(О)(ОН)(ORc) или группу формулы (IB') (которая представлена выше);
R13 представляет собой водород, С1-4алкил или карбоциклил; где R13является необязательно замещенным одним или несколькими заместителями, выбранными из R20; где
R20 представляет собой гидроксил;
R15 представляет собой карбокси или сульфо;
р принимает значение 1 или 2; и значения R13 могут быть одинаковыми или разными;
m принимает значение 0; и
n принимает значение 1;
или его фармацевтически приемлемая соль, сольват, сольват такой соли или пролекарство.
Таким образом, в дополнительном аспекте данного изобретения предложено соединение формулы (I'), которая представлена выше, где:
R1 и R2 оба представляют собой бутил или один из R1 и R2 представляет собой этил, а второй представляет собой бутил;
R4 представляет собой метилтио;
R5 представляет собой группу формулы (IA') (которая представлена выше);
R3 и R6 представляют собой водород;
кольцо А представляет собой фенил;
R7представляет собой водород;
R11 представляет собой группу формулы (IB') (которая представлена выше);
R13 представляет собой водород или гидроксиметил;
R15 представляет собой карбокси или сульфо;
р принимает значение 1 или 2; и значения R13 могут быть одинаковыми или разными;
m принимает значение 0;
n принимает значение 1;
или его фармацевтически приемлемая соль, сольват, сольват такой соли или пролекарство.
Таким образом, в еще одном дополнительном аспекте данного изобретения предложено соединение формулы (I''), которая представлена выше, где:
R1 и R2 независимо выбраны из этила или бутила;
R3 и R6 представляют собой водород;
R4 выбран из галогена, С1-4алкокси или С1-4алкилS(O)a, где а принимает значение от 0 до 2; где R4может быть необязательно замещенным по углероду одним или несколькимиR16; и где R16 независимо выбран из гидроксила и N,N-(C1-4алкил)2амино;
R5 представляет собой группу формулы (IA'');
кольцо А представляет собой арил или гетероарил; где кольцо А является необязательно замещенным одним или несколькими заместителями, выбранными из R17;
R7 представляет собой водород, С1-4алкил или карбоциклил;
R8 представляет собой водород или метил;
R9 представляет собой водород или метил;
R11 представляет собой карбокси, -Р(О)(ОН)(ORc) или группу формулы (IB'')(которая представлена выше);
Х представляет собой -NH- или -NHC(O)-;
R12 представляет собой водород или метил;
R13 представляет собой водород, С1-4алкил или карбоциклил; где R13 является необязательно замещенным одним или несколькими заместителями, выбранными из R20;
R14 представляет собой водород;
R15 представляет собой карбокси или сульфо;
R17 выбран из галогена, гидроксила, С1-4алкил или С1-4алкокси; где R17 может быть необязательно замещенным по углероду одним или несколькими R21;
R20 представляет собой гидроксил, карбокси, карбоциклил или амино; где R20 может быть необязательно замещенным по углероду одним или несколькими R22;
R21 выбран из галогена;
R22 представляет собой гидроксил;
р принимает значение от 1 до 3; и значения R13 могут быть одинаковыми или разными,
q принимает значение от 0 до 1;
r принимает значение от 0 до 3; и значения R14 могут быть одинаковыми или разными; и если q принимает значение 1, r не принимает значение 0;
m принимает значение от 0 до 2; и
n принимает значение от 1 до 3;
или его фармацевтически приемлемая соль, сольват, сольват такой соли или пролекарство.
Таким образом, в еще одном дополнительном аспекте данного изобретения предложено соединение формулы (I), которая представлена выше, где
RV и RWоба представляют собой водород;
R1 и R2 независимо выбраны из С1-6алкила;
Rx и Ry оба представляют собой водород;
Rz выбран из галогена, амино, С1-6алкила, С1-6алкоксикарбониламино или N'-(С1-6алкил)уреидо;
v принимает значение 0 или 1;
R3 и R6 представляют собой водород;
один из R4 и R5 представляет собой группу формулы (IA) (которая представлена выше), а второй выбран из водорода, галогена, С1-4алкоксигруппы или С1-4алкилS(O)a, где а принимает значение от 0 до 2; указанный R4 или R5 может быть необязательно замещенным по углероду одним или несколькими R16; где R16 независимо выбран из гидроксила, карбокси и N,N-(С1-4алкил)2амино;
D представляет собой -О- или -S-;
R7 представляет собой водород, метил или фенил;
R8 представляет собой водород или метил;
кольцо А представляет собой арил или гетероарил; и кольцо А является необязательно замещенным одним или несколькими заместителями, выбранными из R17; где R17 выбран из галогена, гидроксила, С1-4алкила или С1-4алкокси; и где R17 может быть необязательно замещенным по углероду одним или несколькими R21; где R21 выбран из галогена;
R9 представляет собой водород или метил;
R10 представляет собой водород;
R11 представляет собой карбокси, -P(O)(OH)(ORc), где Rc выбран из С1-4алкила или группы формулы (IB) (которая представлена выше);
R12 представляет собой водород или метил;
Х представляет собой -NH- или -NHC(O);
R13 представляет собой водород, С1-4алкил, карбоциклил или R23; где R13 является необязательно замещенным одним или несколькими заместителями, выбранными из R20; где R20 представляет собой гидроксил, С1-4алкилS(O)a, где а принимает значение 0, С1-4алкокси, амино, карбоциклил, гетероциклил или меркапто; где R20 может быть независимо необязательно замещенным по углероду одним или несколькими R22; R22 выбран из гидроксила; и R23 представляет собой карбокси;
R14 выбран из водорода, С1-4алкила или карбоциклила; где указанный С1-4алкил или карбоциклил могут быть необязательно замещенными одним или несколькими заместителями, выбранными из R20; и R20 представляет собой гидроксил;
R15 представляет собой карбокси, сульфо, фосфоно, -P(O)(ORe)(ORf), -P(O)(OH)(ORе), -P(O)(OH)(Re) или -P(O)(ORe)(Rf), где Re и Rf независимо выбраны из С1-4алкила; или R15 представляет собой группу формулы (IC)(которая представлена выше);
R24 представляет собой водород;
R25 представляет собой водород;
R26 представляет собой карбокси;
р принимает значение от 1 до 3; где значения R13 могут быть одинаковыми или разными;
q принимает значение от 0 до 1;
r принимает значение от 0 до 3; где значения R14 могут быть одинаковыми или разными;
m принимает значение от 0 до 2; где значения R10 могут быть одинаковыми или разными;
n принимает значение от 1 до 2; где значения R7 могут быть одинаковыми или разными;
z принимает значение от 0 до 1; где значения R25 могут быть одинаковыми или разными;
или его фармацевтически приемлемая соль, сольват, сольват такой соли или пролекарство.
В соответствии с другим аспектом данного изобретения, предпочтительными соединениями данного изобретения являются любые соединения из примеров или их фармацевтически приемлемые соли, сольваты, сольваты таких солей или пролекарства.
В одном аспекте данного изобретения предложено соединение формулы (I), выбранное из примеров 8, 9, 46, 56, 59, 60, 61, 62, 66 и 69 или его фармацевтически приемлемая соль, сольват, сольват такой соли или пролекарство.
В другом аспекте данного изобретения предложено соединение формулы (I), которое представляет собой соединение примера 73, 74, 95, 96, 97, 98, 99 и 100 или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство.
В еще одном аспекте данного изобретения предпочтительные соединения данного изобретения представляют собой любое соединение примеров 43, 50, 51 и 52 или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство.
В еще одном дополнительном аспекте данного изобретения предпочтительные соединения данного изобретения представляют собой любые соединения примеров 43, 46, 50, 51, 56, 58, 59, 61, 62, 63, 69, 81, 83, 85, 94, 97, 98, 108, 109, 110, 111 или 117.
Предпочтительными аспектами данного изобретения являются аспекты, которые относятся к соединению формулы (I) или его фармацевтически приемлемой соли.
Другой аспект данного изобретения предлагает способ получения соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства, данный способ (где, если не указано другого условия, изменяемые группы принимают значения, определенные в формулы (I)) включает:
Способ 1): окисление бензотиазепина формулы (II):
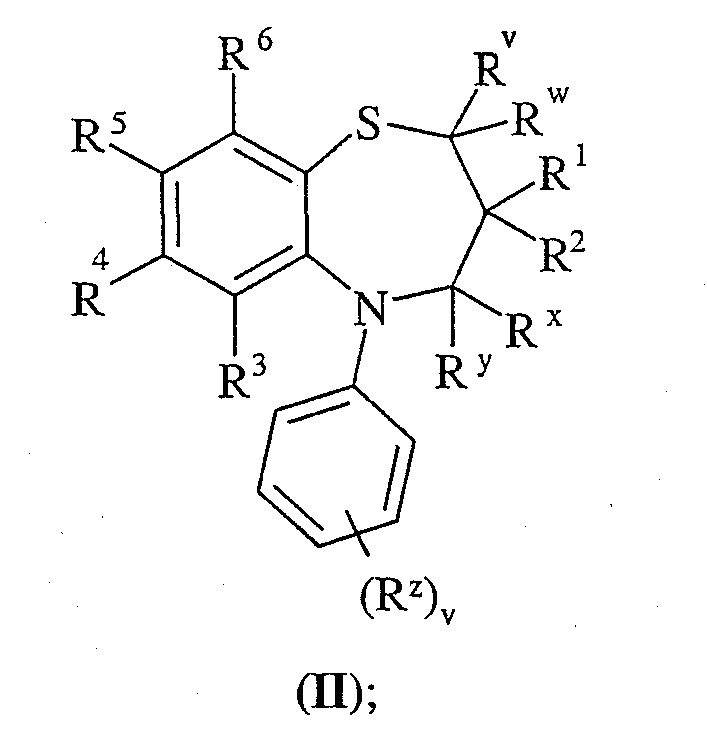
Способ 2): для соединений формуы (I), где D представляет собой -О-, -NRa или -S-; взаимодействие соединения формулы (IIIa) или (IIIb):

с соединением формулы (IV):

где L представляет собой замещаемую группу;
Способ 3): взаимодействие кислоты формулы (Va) или (Vb):

или ее активированного производного с амином формулы (VI):
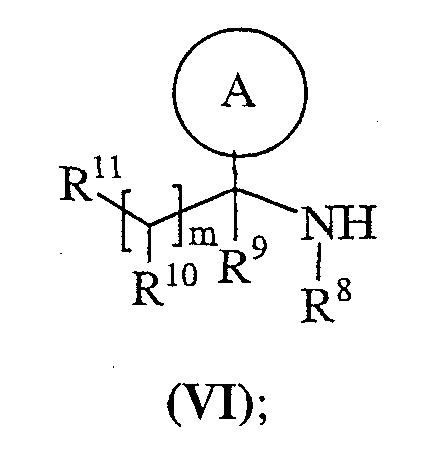
Способ 4): для соединений формулы (I), где R11 представляет собой группу формулы (IB); взаимодействие соединения формулы (I), где R11 представляет собой карбокси, с амином формулы (VII):
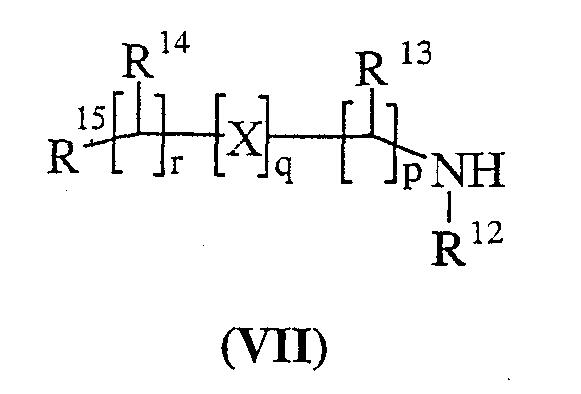
Способ 5) : для соединений формулы (I), где R11 представляет собой карбокси; удаление защитной группы в соединении формулы (VIIIa):
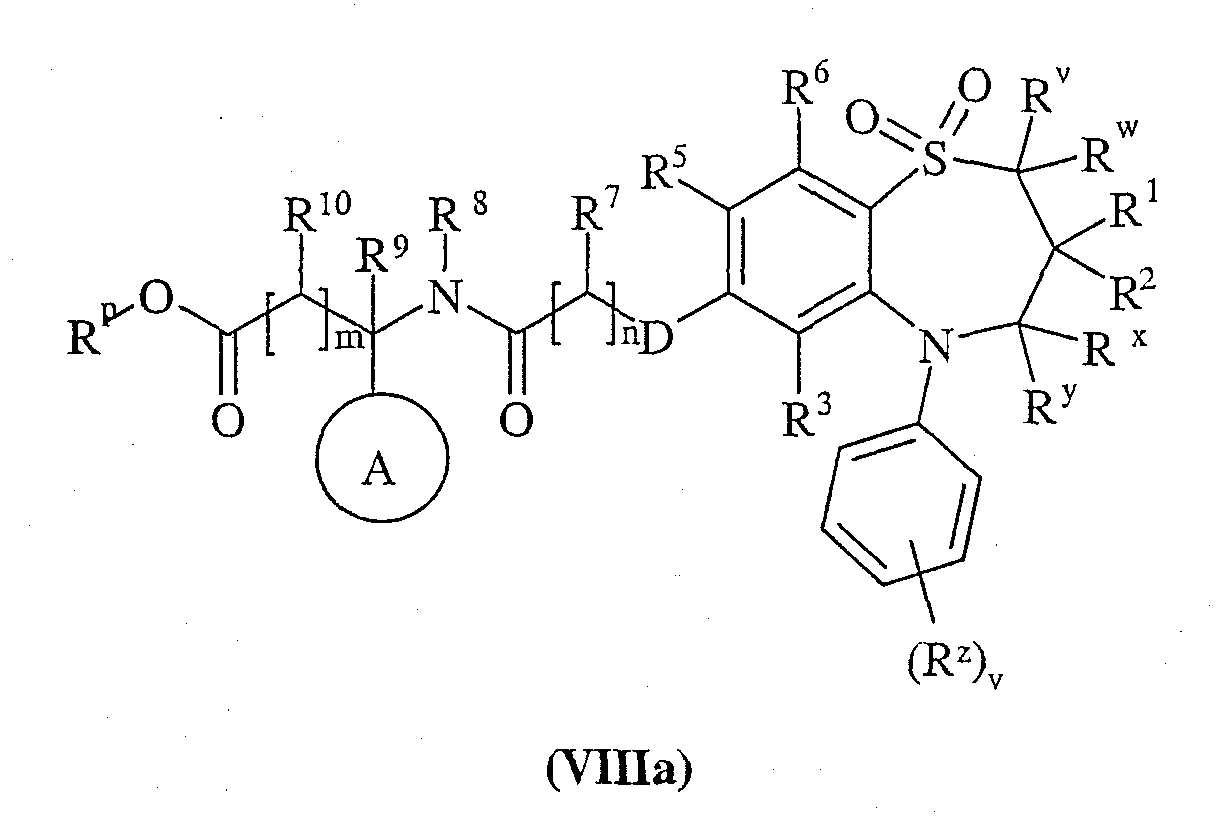
или (VIIIb):

где Rр представляет собой С1-4алкил;
Способ 6): для соединений формулы (I), где R11 представляет собой группу формулы (IB) и R15 представляет собой карбокси, удаление защитной группы соединения формулы (IXa):

или (IXb):
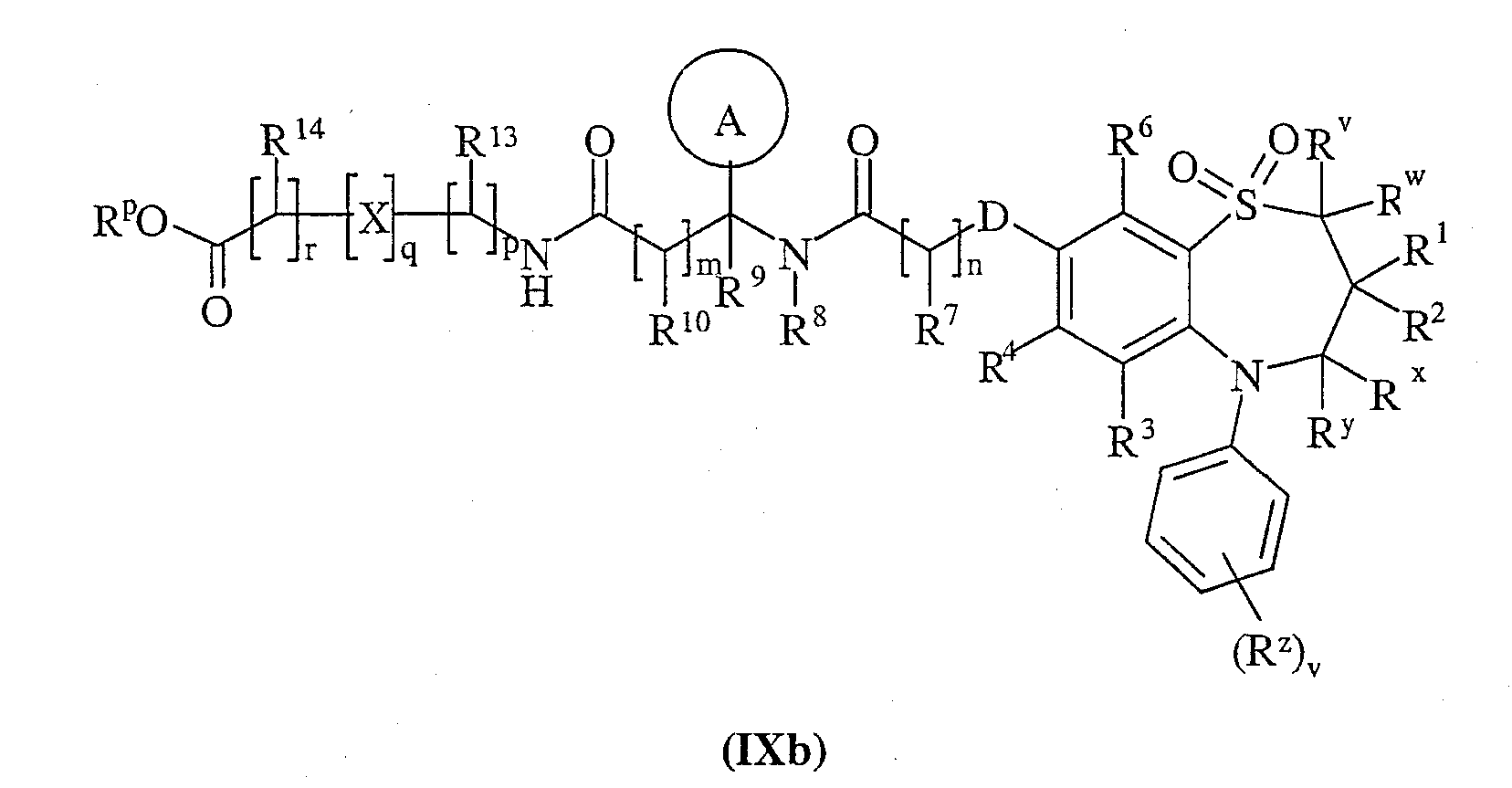
где Rp представляет собой С1-4 алкил;
Способ 7): для соединений формулы (I), где один из R4 и R5 независимо выбран из С1-4алкилтио, необязательно замещенного по углероду одним или несколькими R16; взаимодействие соединения формулы (Xa) или (Xb):
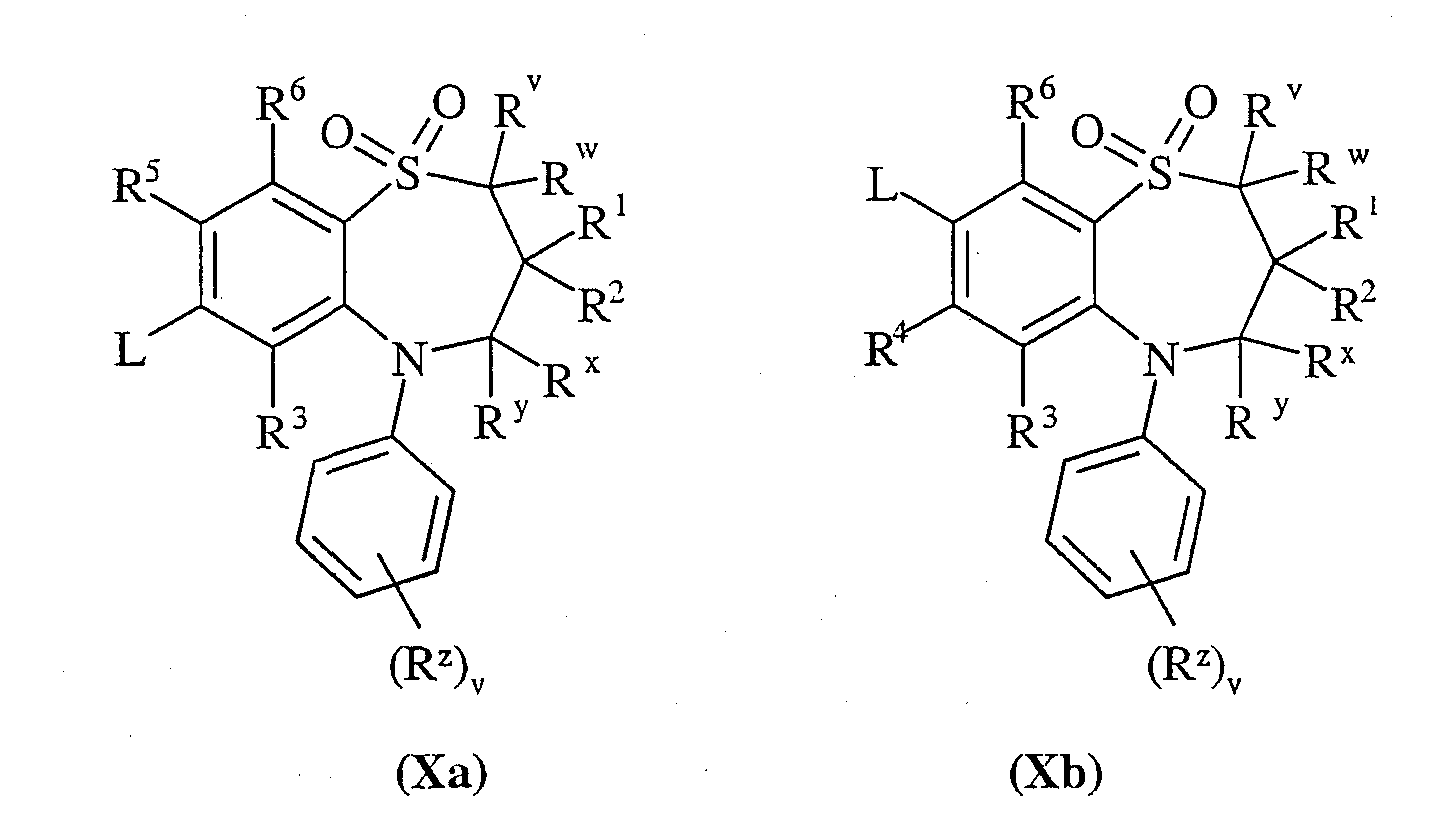
где L представляет собой замещаемую группу, с тиолом формулы (XI):

где Ry представляет собой С1-4алкилтио, необязательно замещенный по углероду одним или несколькими R16;
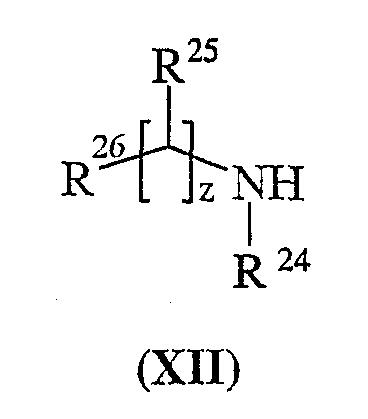
Способ 8): для соединений формулы (I), где R15 представляет собой группу формулы (IC), взаимодействие соединения формулы (IXa) или (IXb), где Rp представляет собой водород, с соединением формулы (XII):
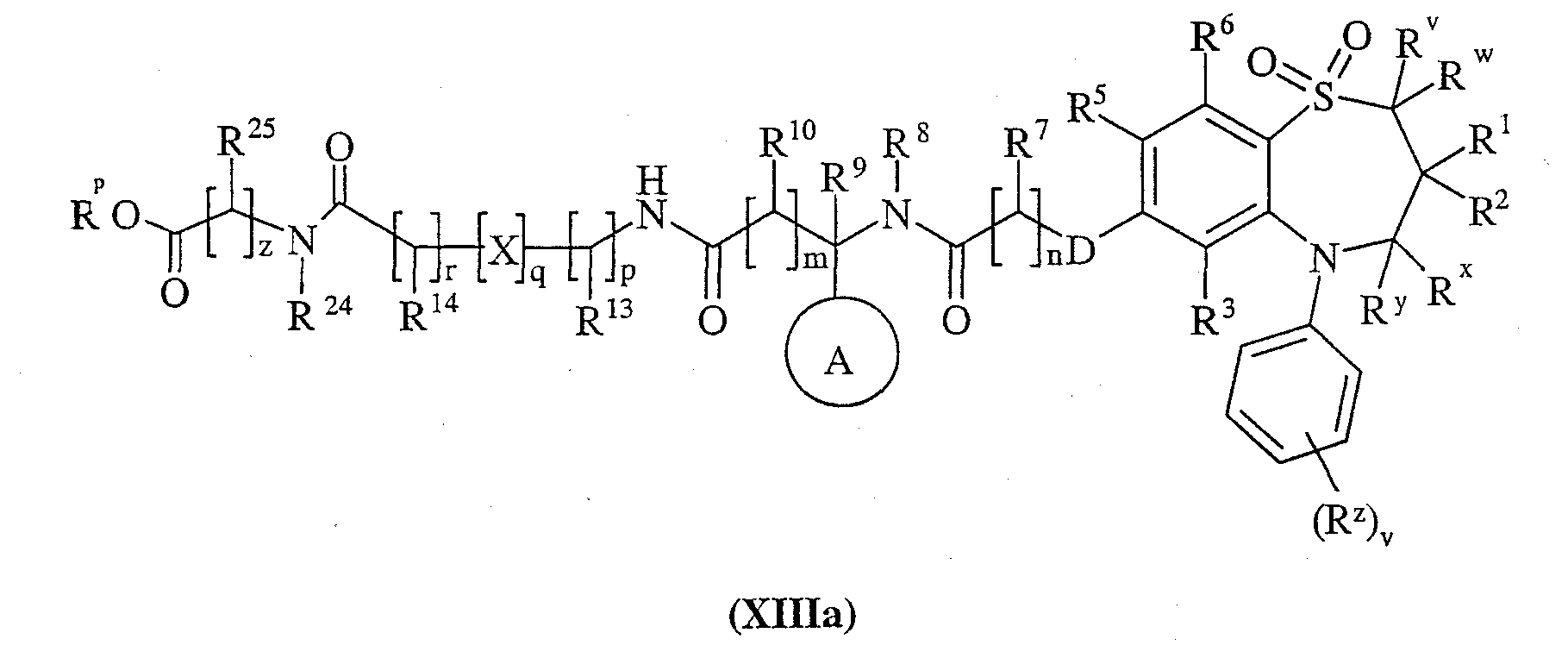
Способ 9) для соединений формулы (I), где R11 представляет собой группу формулы (IB), R15 представляет собой группу формулы (IC) и R26 представляет собой карбокси; удаление защитной группы из соединения формулы (XIIIa):
или (XIIIb):
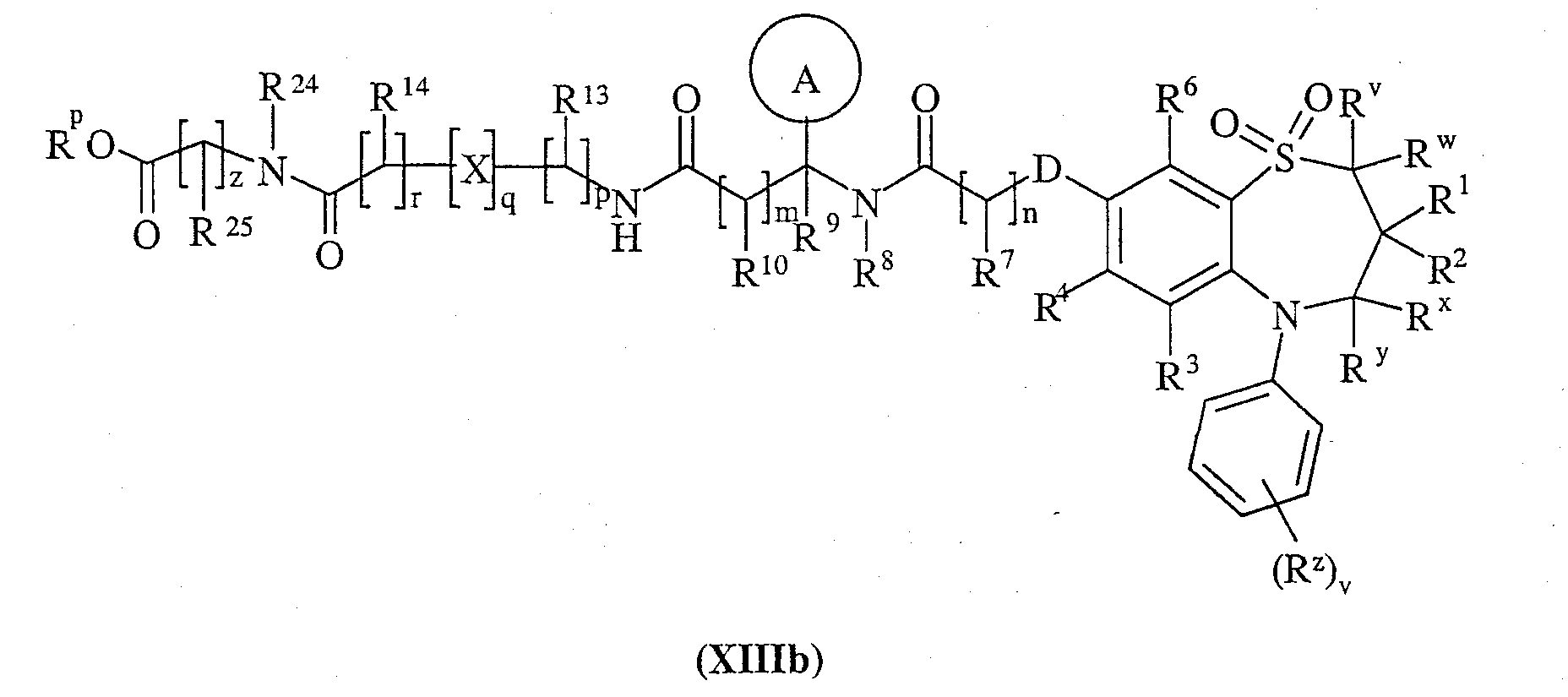
и Rp представляет собой С1-4алкил;
Способ 10) для соединения формулы (I), где Х представляет собой -N(Rq)C(O)-; взаимодействие соединения формулы (XIVa):

или (XIVb):
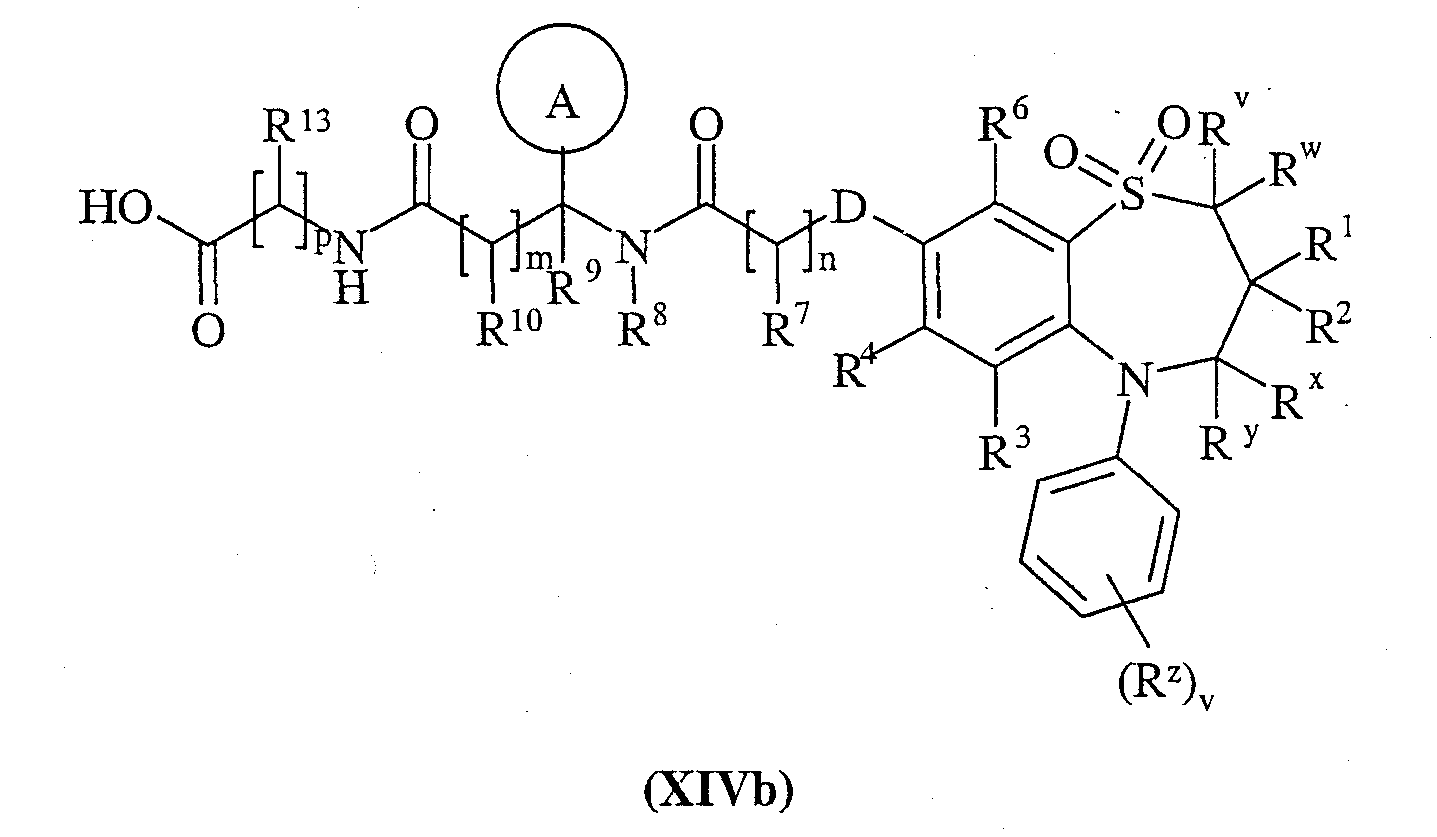
с соединением формулы (XV):
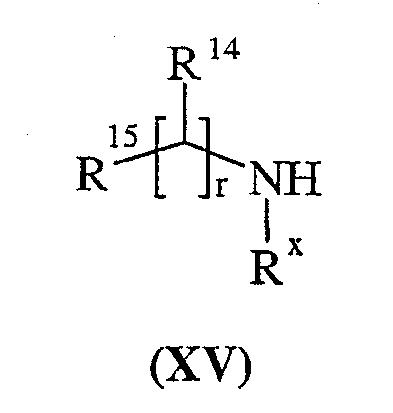
и затем, если это необходимо или желательно:
i) превращение соединения формулы (I) в другое соединение формулы (I);
ii) удаление любых защитных групп;
iii) получение фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства.
Квалифицированному специалисту будет также понятно, что аналогичные способы, соответствующие описанным выше способам, могут использоваться также для получения соединений формулы (I') или соединений формулы (I''), в которых определения групп могут различаться.
L представляет собой замещаемую группу, подходящими значениями для L являются, например, галоген или сульфонилоксигруппа, и в частности, хлор, бром, метансульфонилокси- или толуол-4-сульфонилокси-группа.
Rp представляет собой С1-4алкил. Предпочтительно, Rp представляет собой метил или этил. Более предпочтительно, Rp представляет собой метил.
Специфическими условиями для проведения описанных выше реакций являются следующие условия.
Способ 1): Бензотиазепины формулы (II) могут подвергаться окислению в стандартных условиях окисления серы, например с использованием пероксида водорода и трифторуксусной кислоты при температуры в интервале от 0оС до температуры кипения растворителя, предпочтительно при комнатной температуре или температуре, близкой к комнатной.
Соединения формулы (II) могут быть получены в соответствии со Схемой I для соединений формулы (I), где Rx и Ry представляют собой водород. Квалифицированному специалисту понятно, что когда Rx и Ry не являются водородом, приведенную схему синтеза следует осуществлять в соответствии со стандартными известными методиками.
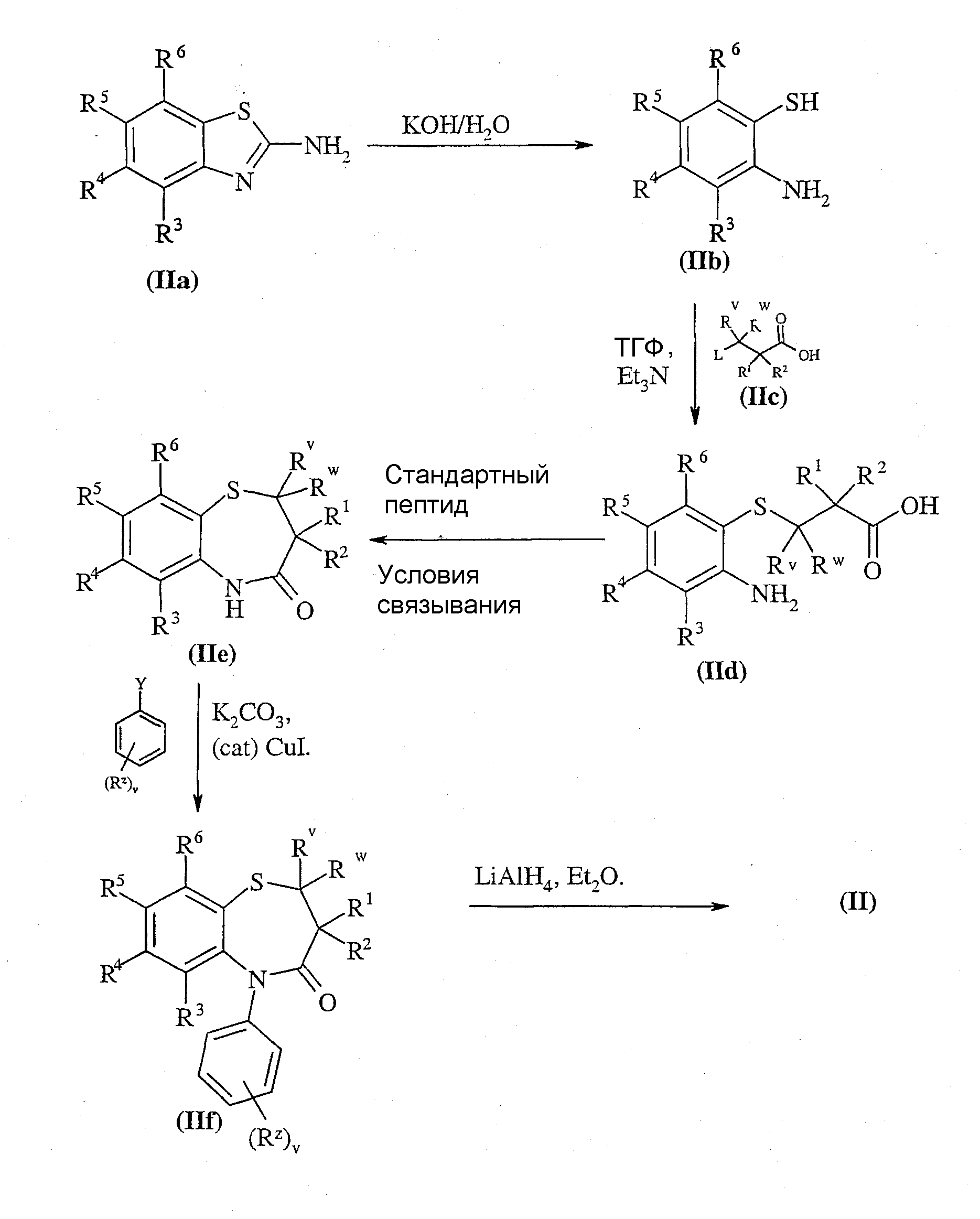
Схема I
где L представляет собой замещаемую группу, которая определена выше, например, галоген.
Соединения формулы (IIa) и (IIc) являются коммерчески доступными соединениями или они описаны в литературе либо могут быть получены известными стандартными способами.
Способ 2): Спирты формулы (IIIa) или (IIIb) могут подвергаться взаимодействию с соединениями формулы (IV) в присутствии основания, например, неорганического основания, такого как карбонат натрия, или органического основания, такого как основание Ханигса (Hunigs base), в присутствии подходящего растворителя, такого как ацетонитрил, дихлорметан или тетрагидрофуран, при температуре в интервале от 0°С до температуры кипения растворителя, предпочтительно при температуре кипения растворителя или близкой к ней.
Соединения формулы (IIIa) или (IIIb) могут быть получены аналогично соединениям формулы (II) (но в которых R4 или R5 представляет собой гидроксильную группу) с последующей стадией окисления (Способ 1).
Соединения формулы (IV) являются коммерчески доступными соединениями, или они известны из литературы либо могут быть получены известными стандартными способами.
Способ 3), Способ 4), Способ 8) и Способ 10): Кислоты и амины могут связываться в присутствии подходящего связывающего реагента. В качестве подходящих связывающих реагентов могут использоваться стандартные пептидные связывающие реагенты, которые известны в данной области техники, или, например, карбонилдиимидазол и дициклогексил-карбодиимид, необязательно, в присутствии катализатора, такого как диметиламинопиридин или 4-пирролидинопиридин, необязательно в присутствии основания, например, триэтиламина, пиридина, или 2,6-диалкилпиридинов, таких как 2,6-лутидина или 2,6-дитретбутилпиридина. Подходящие растворители включают диметилацетамид, дихлорметан, бензол, тетрагидрофуран и диметилформамид. Реакция сочетания может удобно проводиться при температуре в интервале от -40 до 40оС.
Подходящие активированные производные кислот включают галогенангидриды, например, хлорангидриды кислот, и активные сложные эфиры, например, сложные пентафторфениловые эфиры. Взаимодействие соединений таких типов с аминами хорошо известно в данной области, например, они могут взаимодействовать в присутствии основания, такого как описаны выше, и в подходящем растворителе, таком как описаны выше. Реакция может удобно осуществляться при температуре в интервале от -40 до 40оС.
Соединения формулы (Va) или (Vb), где D представляет собой -О-, -NRa- или -S-, могут быть получены в соответствии со схемой 2.

Схема 2
где L представляет собой замещаемую группу, как определено выше.
Соединения формулы (Va) и (Vb), где D представляет собой -SO- или -SO2-, могут быть получены окислением соединений формулы (Va) и (Vb), синтезированных способом, показанным на Схеме 2, где D представляет собой -S-.
Соединения формулы (Va) или (Vb), где D представляет собой -СН2 -, могут быть получены в соответствии со Схемой 3.
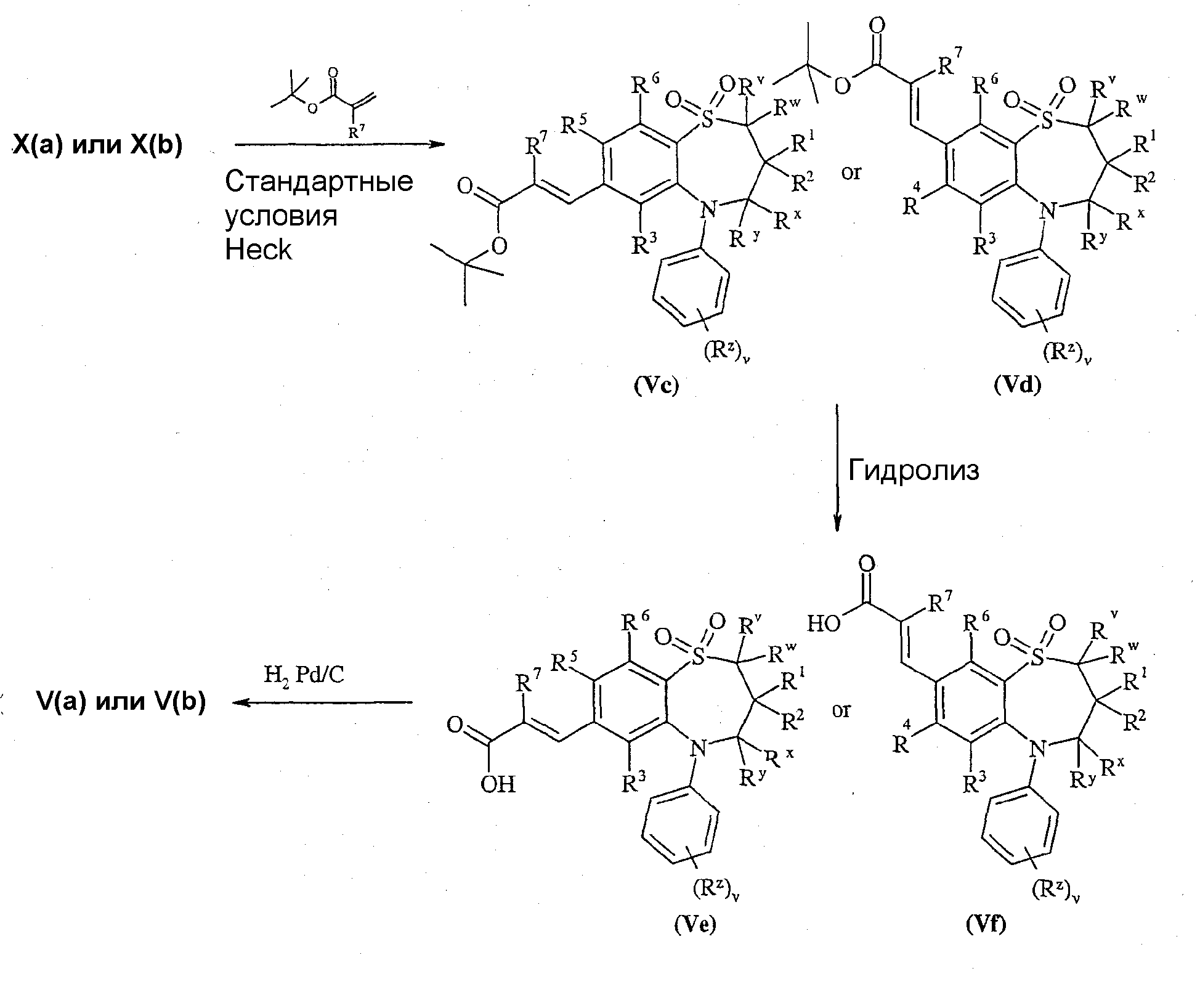
Схема 3
Соединения формулы (XIVa) или (XIVb) могут быть получены любым из способов, описанных в данном изобретении, где R11 представляет собой группу формулы (IB), но когда (IB) представляет собой группу формулы (XVI):
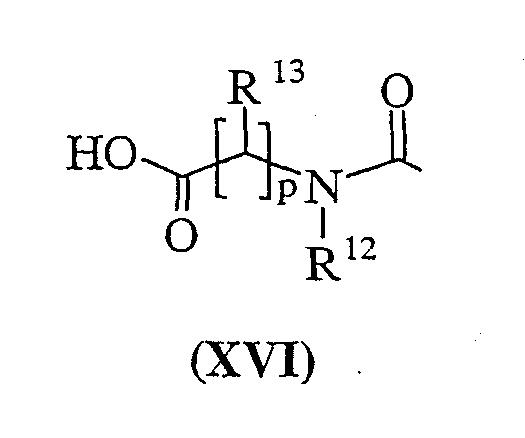
Соединения формулы (Vc), (VI), (VII), (XII), (XV) и (XVI) являются коммерчески доступными соединениями, или они известны в литературе, либо их получают известными стандартными способами.
Способ 5), Способ 6) и Способ 9): в сложных эфирах формулы (VIIIa), (VIIIb), (IXa), (IXb), (XIIIa) и (XIIIb) защитные группы могут быть удалены в стандартных условиях, таких как условия, описанные ниже, например, защитные группы могут удаляться с помощью гидроксида натрия в метаноле при комнатной температуре.
Сложные эфиры формулы (VIIIa), (VIIIb), (IXa), (IXb), (XIIIa) и (XIIIb) могут быть получены в соответствии с любой из методик, описанных выше для получения соединений формулы (I), в которых R11, R15 или R26 представляет собой С1-4алкоксикарбонил.
Способ 7): соединения формулы (Xa) и (Xb) могут подвергаться взаимодействию с тиолами формулы (XI) в присутствии основания, например, неорганического основания, такого как карбонат натрия или органического основания, такого как основание Ханигса, в присутствии подходящего растворителя, такого как ДМФА или ТГФ, при температуре в интервале от 0оС до температуры кипения растворителя.
Соединения формулы (Xa) и (Xb) могут быть получены в соответствии с любой из методик, описанных выше для получения соединений формулы (I), где R4 и R5 представляет собой L.
Соединения формулы (XI) являются коммерчески доступными соединениями, или они известны из литературы, либо могут быть получены известными стандартными способами.
Следует понимать, что некоторые из различных заместителей в кольце в соединениях данного изобретения могут вводиться стандартными реакциями ароматического замещения или вводиться стандартными модификациями функциональных групп либо до, либо сразу после осуществления описанных выше способов, эти реакции сами по себе включены в аспект способа данного изобретения. Такие реакции и модификации включают, например, введение заместителя с помощью реакции ароматического замещения, восстановление заместителей, алкилирование заместителей и окисление заместителей. Реагенты и условия проведения реакций таких методик хорошо известны в области химии. Конкретные примеры реакций ароматического замещения включают введение нитрогруппы с использованием концентрированной азотной кислоты, введение ацильной группы с использованием, например, ацилгалогенида и кислоты Льюиса (такой как трихлорид алюминия) в условиях Фриделя-Крафтса; введение алкильной группы с использованием алкилгалогенида и кислоты Льюиса (такой как трихлорид алюмдиния) в условиях Фриделя Крафтса; и введение группы галогена. Конкретные примеры модификаций функциональных групп включают восстановление нитрогруппы до аминогруппы, например, методом каталитического гидрирования с никелевым катализатором или обработкой железом в присутствии хлористоводородной кислоты при нагревании; окисление алкилтиогруппы до алкилсульфинил или алкилсульфонильной группы.
Следует также понимать, что в некоторых реакциях, описанных в данном изобретении, может быть необходимо/желательно защитить любые чувствительные группы в соединениях. Примеры, когда защита необходима или желательна, и подходящие способы защиты известны квалифицированному в данной области специалисту. Могут использоваться стандартные защитные группы в соответствии со стандартными практическими способами (см., например, T.W. Green, Protective Groups in Organic Synthesis, John Wiley and Sons, 1991). Таким образом, если реагенты включают такие группы, как амино-, карбокси или гидроксильная группы, может быть желательно защитить группу в некоторых описанных реакциях.
Подходящей защитной группой для амино- и алкиламиногруппы является, например, ацильная группа, например алканоильная группа, такая как ацетил, алкоксикарбонильная группа, например, метоксикарбонильная, этоксикарбонильная или трет-бутоксикарбонильная группа, арилметоксикарбонильная группа, например, бензилоксикарбонил, или ароильная группа, например, бензоил. Условия удаления указанных защитных групп необходимо изменять в зависимости от выбора защитной группы. Так, например, ацильная группа, такая как алканоил или алкоксикарбонил, или ароильная группа могут удаляться, например, гидролизом с подходящим основанием, таким как гидроксид щелочного металла, например, гидроксид лития или натрия. Альтернативно, ацильная группа, такая как трет-бутоксикарбонильная группа, может удаляться, например, обработкой подходящей кислотой, такой как хлористоводородная, серная или фосфорная кислота, или трифторуксусной кислотой, и ариметоксикарбонильная группа, такая как бензилоксикарбонильная группа, может удаляться, например, гидрированием над катализатором, таким как палладий на углероде, или обработкой кислотой Льюиса, например, трис(трифторацетатом) бора. Подходящей альтернативной защитной группой для первичной аминогруппы является, например, фталоильная группа, которая может удаляться обработкой алкиламином, например, диметиламинопропиламином или гидразином.
Подходящей защитной группой для гидроксильной группы является, ацильная группа, например, алканоильная группа, такая как ацетил, ароильная группа, такая как бензоил, или арилметильная группа, такая как бензил. Условия удаления указанных защитных групп необходимо менять в зависимости от выбора защитной группы. Так, ацильная группа, такая как алкланоил или ароил, может удаляться, например, гидролизом с подходящим основанием, таким как гидроксид щелочного металла, например, гидроксид лития или натрия. Альтернативно, арилметильная группа, такая как бензильная группа, может удаляться, например, гидрированием над катализатором, таким как палладий на углероде.
Подходящей защитной группой для карбоксильной групп является, например, группа, образующая сложный эфир, такая как метильная или этильная группа, которая может удаляться, например, гидролизом с основанием, таким как гидроксид натрия, или трет-бутильная группа, которая может удаляться, например, обработкой кислотой, например, органической кислотой, такой как трифторуксусная кислота, или бензильная группа, которая может удаляться, например, гидрированием над катализатором, таким как палладий на углероде.
Защитные группы могут удаляться на любой стадии синтеза с использованием стандартных методов, хорошо известных квалифицированному химику.
Как заявлено выше, соединения, определенные в данном изобретении, обладают ингибирующей активностью в отношении IBAT. Эти свойства могут анализироваться количественно, например, in vitro количественным определением влияния на поглощение желчной кислоты в IBAT-трансфектированных клетках (Smith L., Price-Jones M. J., Hugnes K.T., Jones N.R.A.; J. Biomolecular Screening, 3, 227-230) или in vivo изучением влияния на абсорбцию у мышей/крыс желчной кислоты, меченой радиоактивным изотопом (Lewis M.C., Brieaddy L.E., Root C.,J., J. Lip Res., 1995, 36, 108-1105).
В соответствии с дополнительным аспектом данного изобретения, предложена фармацевтическая композиция, которая включает соединение формулы (I) или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство, как определено выше, в сочетании с фармацевтически приемлемым наполнителем или носителем.
Композиция может иметь форму, подходящую для перорального введения, такую как таблетка или капсула, для парентеральной инъекции (включая внутривенное, подкожное, внутримышечное, внутрисосудистое введение или вливание), такую как стерильный раствор, стерильная суспензия или эмульсия, для местного применения, такую как мазь или крем, или для ректального введения, такую как свеча.
Обычно указанные композиции могут быть получены стандартным образом с использованием традиционных наполнителей.
Соединение формулы (I) или его фармацевтически приемлемая соль, сольват, сольват такой соли или пролекарство, обычно будет вводиться теплокровному животному в стандартной дозе в пределах 5 - 5000 мг на кв. м площади тела животного, то есть примерно 0,1 - 100 мг/кг или 0,01-50 мг/кг, и обычно это обеспечивает терапевтически эффективную дозу. Лекарственная форма стандартной дозы, такая как таблетка или капсула, обычно будет содержать, например, 1 - 250 мг активного ингредиента. Предпочтительно применяется суточная доза в интервале 1 - 50 мг/кг. В другом аспекте применяется суточная доза в интервале 0,02 - 20 мг/кг. Однако суточную дозу необходимо подбирать в зависимости от организма, подлежащего лечению, и особенно в зависимости от способа введения и тяжести заболевания, которое подлежит лечению. Таким образом, оптимальная дозировка может определяться врачом, который лечит каждого конкретного пациента.
В соответствии с дополнительным аспектом данного изобретения, предложено соединение формулы (I) или его фармацевтически приемлемая соль, сольват, сольват такой соли или пролекарство, как определено для применения в способе профилактического или терапевтического лечения теплокровного животного, такого как человек.
Заявителями данного изобретения было установлено, что соединения, определенные в данном изобретении, или их фармацевтически приемлемая соль, сольват, сольват такой соли или пролекарство, являются эффективными ингибиторами IBAT и, соответственно представляют интерес в качестве лекарственных средств при лечении болезненных состояний, связанных с состояниями гиперлипемии.
Таким образом, в соответствии с данным аспектом изобретения, предложено соединение формулы (I) или его фармацевтически приемлемая соль, сольват, сольват такой соли или пролекарство, как определено для применения в качестве лекарственного средства.
В соответствии с другим отличительным признаком изобретения, предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства, как определено выше, в производстве лекарственного средства для применения с целью получения IBAT-ингибирующего действия в организме теплокровного животного, такого как человек.
В соответствии с еще одним отличительным признаком изобретения, предложено применение соединения (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства, как определено выше, в производстве лекарственного средства для применения при лечении состояний гиперлипемии у теплокровного животного, такого как человек.
В соответствии с еще одним отличительным признаком данного изобретения, предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства, как определено выше в производстве лекарственного средства для применения при лечении дислипемических состояний и расстройств (dyslipidemic conditions and disorders), таких как гиперлипемия, гиперглицеридемия, гипербеталипопротеинемия (высокое содержание LDL), гиперпребеталипопротеинемия (высокое содержание VLDL), гиперхиломикронемия (hyperchylomicronemia), снижение содержания липопротеинов в крови (hypolipoproteinemia), гиперхолестеринемия, гиперлипопротеинемия и гипоальфалипопротеинемия (снижение содержания HDL) у теплокровного животного, такого как человек.
В соответствии с еще одним отличительным признаком данного изобретения, предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства, как определено выше, в производстве лекарственного средства для применения при лечении различных клинических состояний, таких как атеросклероз, сужение артерий, аритмия, гипертромботические состояния, сосудистая дисфункция, эндотелиальная дисфункция, сердечная недостаточность, коронарные болезни сердца, сердечно-сосудистые болезни, инфаркт миокарда, стенокардия, периферийные сосудистые заболевания, воспаление сердечно-сосудистых тканей, таких как сердце, клапан, сосудистая сеть, артерии и вены, аневризмы, стеноз, рестеноз, сосудистые бляшки, сосудистые жировые прожилки, лейкоцитарная, моноцитарная и/или макрофагоцитарная инфильтрация, уплотнение внутренней оболочки сосудов, медиальное истончение, инфекционная и хирургическая травма и сосудистый тромбоз, удар и временные приступы ишемии, у теплокровного животного, такого как человек.
В соответствии с еще одним отличительным признаком изобретения, предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства, как определено выше, в производстве лекарственного средства с целью применения при лечении атеросклероза, коронарных болезней сердца, инфаркта лиокарда, стенокардии, периферийных сосудистых заболеваний, удара и временных приступов ишемии у теплокровного животного, такого как человек.
В соответствии с еще одним отличительным признаком данного аспекта изобретения, предложен способ получения ингибирующего действия в отношении IBAT у теплокровного животного, такого как человек, при необходимости такого лечения, способ включает введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства.
В соответствии с еще одним отличительным признаком данного аспекта изобретения, предложен способ лечения гиперлипемических состояний у теплокровного животного, такого как человек, при необходимости такого лечения, способ включает введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства.
В соответствии с еще одним отличительным признаком данного аспекта изобретения, предложен способ лечения дислипемических состояний и расстройств, таких как гиперлипемия, гиперглицеридемия, гипербеталипопротеинемия (высокое содержание LDL), гиперпребеталипопротеинемия (высокое содержание VLDL), гиперхиломикронемия, снижение содержания липопротеинов в крови, гиперхолестеринемия, гиперлипопротеинемия и гипоальфалипопротеинемия (снижение содержания HDL) у теплокровного животного, такого как человек, при необходимости такого лечения, способ включает введения указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства.
В соответствии с еще одним отличительным признаком данного аспекта изобретения, предложен способ лечения различных клинических состояний, таких как атеросклероз, сужение артерий, аритмия, гипертромботические состояния, сосудистая дисфункция, эндотелиальная дисфункция, сердечная недостаточность, коронарные болезни сердца, сердечно-сосудистые болезни, инфаркт миокарда, стенокардия, периферийные сосудистые заболевания, воспаление сердечно-сосудистых тканей, таких как сердце, клапаны, сосудистая сеть, артерии и вены, аневризмы, стеноз, рестеноз, сосудистые бляшки, сосудистые жировые прожилки, лейкоцитарная, моноцитарная и/или макрофагоцитарная инфильтрация, уплотнение внутренней оболочки сосудов, медиальное истончение, инфекционная и хирургическая травма и сосудистый тромбоз, удар и временные приступы ишемии, при необходимости такого лечения, способ включает введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства.
В соответствии с еще одним отличительным признаком данного аспекта изобретения, предложен способ лечения атеросклероза, коронарных болезней сердца, инфаркта миокарда, стенокардии, периферийных сосудистых заболеваний, удара и временных приступов ишемии у теплокровного животного, такого как человек, при необходимости такого лечения, способ включает введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства.
Доказано, что ингибитор IBAT потенциально полезен при лечении и/или профилактике желчных конкрементов. В соответствии с дополнительным отличительным признаком данного аспекта изобретения, предложен способ лечения и/или профилактики желчных конкрементов у теплокровного животного, такого как человек, при необходимости такого лечения, способ включает введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства.
Величина дозы, необходимой для терапевтического или профилактического лечения, будет обязательно изменяться в зависимости от организма, подлежащего лечению, способа введения лекарственного средства и тяжести заболевания, подлежащего лечению. Стандартная единичная доза, которая предусмотрена для этой цели, находится в интервале, например, 1 - 100 мг/кг, предпочтительно, 1- 50 мг/кг.
Ингибирующая активность в отношении IBAT, определенная выше, может применяться в качестве единственного терапевтического средства или в дополнение к соединению данного изобретения может включать одно или несколько других веществ и/или курсов лечения. Такое совместное лечение может осуществляться одновременным, последовательным или раздельным введением отдельных компонентов курса лечения. В соответствии с данным аспектом изобретения, предложена фармацевтическая композиция, включающая соединение формулы (I) или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство, как определено выше, и дополнительное соединение, обладающее ингибирующей активностью в отношении IBAT, как определено выше, и дополнительное гиполипемическое лекарственное средство для совместного лечения гиперлипемии.
В другом аспекте данного изобретения соединение формулы (I) или его фармацевтически приемлемая соль, сольват, сольвата такой соли или пролекарство может вводиться в сочетании с ингибитором HMG Co-A редуктазы или его фармацевтически приемлемыми солями, сольватами, сольватами таких солей или пролекарствами. Подходящие ингибиторы HMG Co-A редуктазы, их фармацевтически приемлемые соли, сольваты, сольваты таких солей или пролекарства являются статинами и хорошо известны в данной области. Примерами статинов являются флувастатин, ловастатин, правастатин, симвастатин, атовастатин, церивастатин, бервастатин, далвастатин, мевастатин и (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидрогепт-6-еновая кислота (росувастатин - rosuvastatin), или его фармацевтически приемлемая соль, сольват, сольват такой соли или пролекарство. Специфическим статином является аторвастатин (atorvastatin) или его фармацевтически приемлемая соль, сольват, сольват такой соли или пролекарство. Более специфическим статином является кальциевая соль аторвастатина. Еще одним специфическим статином является (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)амино]пиримидин-5-ил](3R,5S)-3,5-дигидрогепт-6-еновая кислота (росувастатин - rosuvastatin) или его фармацевтически приемлемая соль, сольват, сольват такой соли или пролекарство. Предпочтительным специфическим статином является кальциевая соль росувастатина.
В дополнительном аспекте данного изобретения соединение формулы (I) или его фармацевтически приемлемая соль, сольват, сольват такой соли или пролекарство может вводиться в сочетании с ингибитором HMG Co-A редуктазы или его фармацевтически приемлемой солью, сольватом, сольватом такой соли или пролекарством и/или соединением, связывающим желчную кислоту, посредством чего устраняется возможный риск наличия избытка желчных кислот в кишечнике, вызываемый ингибированием системы транспорта желчной кислоты в подвздошной кишке. Избыток желчных кислот в висцеральном содержимом может вызывать диарею. Таким образом, данное изобретение обеспечивает лечение у пациентов возможного побочного эффекта, такого как диарея, во время курса терапевтического лечения, включающего соединение формулы (I) или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство.
Ингибитор HMG CoA-редуктазы или его фармацевтически приемлемая соль, сольват, сольват такой соли или пролекарство будет посредством своего действия снижать содержание эндогенного холестерина, доступного для синтеза желчных кислот, и оказывать дополнительное действие в сочетании с соединением формулы (I) или его фармацевтически приемлемой солью, сольватом, сольватом такой соли или пролекарством на снижение содержания липидов.
Подходящими веществами, связывающими желчные кислоты, для такой комбинированной терапии являются смолы, такие как холестерамин и холестипол. Одним из преимуществ является то, что доза вещества, связывающего желчные кислоты, может быть снижена по сравнению с терапевтической дозой при лечении холестеринемии с применением только вещества, связывающего желчные кислоты. Снижение дозы вещества, связывающего желчные кислоты, может также приводить к устранению любых возможных побочных эффектов, вызываемых ослабленной толерантностью пациента к терапевтической дозе.
Следовательно, в дополнительном отличительном признаке данного изобретения предложен способ получения ингибирующего действия в отношении IBAT в организме теплокровного животного, такого как человек, нуждающегося в таком лечении, способ включает введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства с одновременным, последовательным или раздельным введением эффективного количества ингибитора HMG Co-A редуктазы, его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства.
Таким образом, в дополнительном отличительном признаке данного изобретения предложен способ получения ингибирующего действия в отношении IBAT у теплокровного животного, например человека, нуждающегося в таком лечении, способ включает введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства с одновременным, последовательным или раздельным введением вещества, связывающего желчные кислоты.
Следовательно, в дополнительном отличительном признаке данного изобретения предложен способ получения ингибирующего действия в отношении IBAT в организме теплокровного животного, такого как человек, нуждающегося в таком лечении, способ включает введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства с одновременным, последовательным или раздельным введением эффективного количества ингибитора HMG Co-A редуктазы, его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства при одновременном, последовательно или раздельном введении вещества, связывающего желчные кислоты.
Следовательно, в дополнительном отличительном признаке данного изобретения предложен способ лечения гиперлипемических состояний у теплокровного животного, например человека, нуждающегося в таком лечении, способ включает введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства при одновременном, последовательном или раздельном введении с эффективным количеством ингибитора HMG Co-A редуктазы или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства.
Таким образом, в дополнительном отличительном признаке данного изобретения предложен способ лечения гиперлипемических состояний у теплокровного животного, например человека, нуждающегося в таком лечении, способ включает введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства при одновременном, последовательном или раздельном введении с эффективным количеством вещества, связывающего желчную кислоту.
Следовательно, в дополнительном отличительном признаке данного изобретения предложен способ лечения гиперлипемических состояний у теплокровного животного, такого как человек, нуждающегося в таком лечении, способ включает введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства при одновременном, последовательном или раздельном введении с эффективным количеством ингибитора HMG Co-A редуктазы или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства и одновременном, последовательном или раздельном введении вещества, связывающего желчные кислоты.
В соответствии с дополнительным аспектом данного изобретения, предложена фармацевтическая композиция, которая включает соединение формулы (I) или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство и ингибитор HMG Co-A редуктазы или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство, в сочетании с фармацевтически приемлемым разбавителем или носителем.
В соответствии с дополнительным аспектом изобретения, предложена фармацевтическая композиция, которая включает соединение формулы (I) или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство и вещество, связывающее желчные кислоты, в сочетании с фармацевтически приемлемым разбавителем или носителем.
В соответствии с дополнительным аспектом данного изобретения, предложена фармацевтическая композиция, которая включает соединение формулы (I) или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство, ингибитор HMG Co-A редуктазы или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство и вещество, связывающее желчную кислоту, в сочетании с фармацевтически приемлемым разбавителем или носителем.
В соответствии с дополнительным аспектом данного изобретения, предложен набор, включающий соединение формулы (I) или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство и ингибитор HMG Co-A редуктазы или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство.
В соответствии с дополнительным аспектом данного изобретения, предложен набор, включающий соединение формулы (I) или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство и вещество, связывающее желчные кислоты.
В соответствии с дополнительным аспектом данного изобретения, предложен набор, включающий соединение формулы (I) или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство, ингибитор HMG Co-A редуктазы или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство и вещество, связывающее желчные кислоты.
В соответствии с дополнительным аспектом данного изобретения, предложен набор, включающий:
а) соединение формулы (I) или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство в форме первой стандартной дозы;
b) ингибитор HMG Co-A редуктазы или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство в форме второй стандартной дозы; и
c) контейнер в качестве тары для указанных первой и второй дозированных лекарственных форм.
В соответствии с дополнительным аспектом данного изобретения, предложен набор, включающий:
a) соединение формулы (I) или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство в форме первой стандартной дозы;
b) вещество, связывающее желчную кислоту, в форме второй стандартной дозы; и
c) контейнер в качестве тары для указанных первой и второй дозированных лекарственных форм.
В соответствии с дополнительным аспектом данного изобретения, предложен набор, включающий:
a) соединение формулы (I) или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство в форме первой стандартной дозы;
b) ингибитор HMG Co-A редуктазы или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство в форме второй стандартной дозы;
c) вещество, связывающее желчную кислоту, в форме третьей стандартной дозы;
d) контейнер в качестве тары для указанных первой, второй и третьей дозированных лекарственных форм.
В соответствии с дополнительным аспектом данного изобретения, предложен набор, включающий:
a) соединение формулы (I) или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство в сочетании с фармацевтически приемлемым разбавителем или носителем в форме первой стандартной дозы;
b) ингибитор HMG Co-A редуктазы или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство в форме второй стандартной дозы; и
c) контейнер в качестве тары для указанных первой и второй дозированных лекарственных форм.
В соответствии с дополнительным аспектом данного изобретения, предложен набор, включающий:
a) соединение формулы (I) или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство в сочетании с фармацевтически приемлемым разбавителем или носителем в форме первой стандартной дозы;
b) вещество, связывающее желчную кислоту, в форме второй стандартной дозы; и
c) контейнер в качестве тары для указанных первой и второй дозированных лекарственных форм.
В соответствии с дополнительным аспектом данного изобретения, предложен набор, включающий:
a) соединение формулы (I) или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство в сочетании с фармацевтически приемлемым разбавителем или носителем в форме первой стандартной дозы;
b) ингибитор HMG Co-A редуктазы или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство в форме второй стандартной дозы;
c) вещество, связывающее желчную кислоту, в форме третьей стандартной дозы; и
d) контейнер в качестве тары для указанных первой, второй и третьей дозированных лекарственных форм.
В соответствии с еще одним отличительным признаком данного изобретения, предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства и ингибитора HMG Co-A редуктазы или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства в производстве лекарственного средства для применения с целью получения ингибирующего действия в отношении IBAT у теплокровного животного, такого как человек.
В соответствии с еще одним отличительным признаком данного изобретения, предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства и вещества, связывающего желчные кислоты, в производстве лекарственного средства для применения с целью получения ингибирующего действия в отношении IBAT у теплокровного животного, такого как человек.
В соответствии с еще одним отличительным признаком данного изобретения, предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства, ингибитора HMG Co-A редуктазы или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства и соединения, связывающего желчные кислоты, при изготовлении лекарственного средства с целью получения ингибирующего действия в отношении IBAT у теплокровного животного, такого как человек.
В соответствии с еще одним отличительным признаком данного изобретения, предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства и ингибитора HMG Co-A редуктазы или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства в производстве лекарственного средства для применения при лечении гиперлипемических состояний у теплокровного животного, такого как человек.
В соответствии с еще одним отличительным признаком данного изобретения, предложен способ применения соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства и акцептора желчных кислот при изготовлении лекарственного средства для применения при лечении гиперлипемических состояний у теплокровного животного, такого как человек.
В соответствии с еще одним отличительным признаком данного изобретения, предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства, ингибитора HMG Co-A редуктазы или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства и соединения, связывающего желчные кислоты, в производстве лекарственного средства для применения при лечении гиперлипемических состояний у теплокровного животного, такого как человек.
В соответствии с еще одним отличительным признаком данного изобретения, предложен способ комбинированного лечения, включающий введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства, необязательно в сочетании с фармацевтически приемлемым разбавителем или носителем, при одновременном, последовательном или раздельном введении ингибитора HMG Co-A редуктазы или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства, необязательно в сочетании с фармацевтически приемлемым разбавителем или носителем, теплокровному животному, например человеку, нуждающемуся в таком терапевтическом лечении.
В соответствии с еще одним отличительным признаком данного изобретения, предложен способ комбинированного лечения, включающий введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства, необязательно в сочетании с фармацевтически приемлемым разбавителем или носителем, при одновременном, последовательном или раздельном введении соединения, связывающего желчные кислоты, необязательно в сочетании с фармацевтически приемлемым разбавителем или носителем, теплокровному животному, например человеку, нуждающемуся в таком терапевтическом лечении.
В соответствии с еще одним аспектом данного изобретения, предложен способ комбинированного лечения, включающий введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства, необязательно в сочетании в фармацевтически приемлемым разбавителем или носителем, при одновременном, последовательном или раздельном введении ингибитора HMG Co-A редуктазы или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства, необязательно в сочетании с фармацевтически приемлемым разбавителем или носителем, и одновременном, последовательном или раздельном введении эффективного количества соединения, связывающего желчные кислоты, необязательно в сочетании с фармацевтически приемлемым разбавителем или носителем, теплокровному животному, например человеку, нуждающемуся в таком терапевтическом лечении.
В соответствии с еще одним дополнительным аспектом данного изобретения, предложен способ комбинированного лечения, включающий введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства, необязательно в сочетании с фармацевтически приемлемым разбавителем или носителем, при одновременном, последовательном или раздельном введении одного или нескольких лекарственных средств, выбранных из:
- ингибитора белка переноса сложных эфиров холестерина (cholesteryl ester transfer protein - СЕТР), например, из ингибиторов, на которые сделана ссылка и которые описаны в WO 00/38725 (стр. 7 строка 22 - страница 10 строка 17), включенной в данное описание в виде ссылки;
- антагониста абсорбции холестерина, например, азетидинонов, таких как SCH 58235, и тех, что описаны в патенте США № 5767115, который включен в данное описание в виде ссылки;
- ингибитора белка переноса микросом, например, описанного в публикации Science, 282, 751-54, 1998, которая включена в данное описание в виде ссылки;
- волокнообразующего производного кислоты; например, клофибрат, гемфиброзил, фенофибрат, ципрофибрат и безафибрат;
- производного никотиновой кислоты, например, никотиновой кислоты (ниацина), аципимокса и ницеритрола;
- фитостеролового соединения, например, станолов;
- пробукола (probucol);
- соединений, использующихся для борьбы с ожирением, таких как орлистат (ЕР 129748) и сибутрамин (Заявка Великобритании № 2184122 и Патент США № 4929629);
- соединения, обладающего гипотензивными свойствами, например, ингибитор ангиотензин-конвертирующего фермента (angiotensin converting enzyme - ACE), антагониста рецептора ангиотензина II, бэта-адренергического блокатора, смешенного альфа/бэта адренергического блокатора, адренергического стимулятора, блокатора кальциевых каналов, диуретика или сосудорасширяющего средства;
- инсулина;
- сульфомочевин, в том числе глибенкламида, толбутамида;
- метформина; и/или
- акаброзы;
или их фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства необязательно в сочетании с фармацевтически приемлемым разбавителем или носителем - теплокровному животному, например человеку, нуждающемуся в таком терапевтическом лечении.
Специфические ингибиторы АСЕ или их фармацевтически приемлемые соли, сольваты, сольваты таких солей или пролекарства, включая активные метаболиты, которые могут использоваться в сочетании с соединением формулы (I), включают, не ограничиваясь только ими, следующие соединения: алацеприл, алатриоприл, алтиоприл, алтоприл кальций, анковенин, беназеприл, беназеприл гидрохлорид, беназеприлат, бензоилкаптоприл, каптоприл, каптоприл-цистеин, каптоприл-глютатион, церанаприл, цераноприл, церонаприл, цилазаприл, цилазаприлат, делаприл, делаприл-дикислота, эналаприл, эналаприлат, энаприл, эпикапроприл, фороксимитин, фосфеноприл, фозеноприл, фозеноприл натрий, фозиноприл, фозиноприл натрий, фозиноприлат, фозиноприловая кислота, гликоприл, геморфин-4, идраприл, имидаприл, индолаприл, индолаприлат, либензаприл, лизиноприл, лициумин А, лициумин В, миксанприл, моексиприл, моексиприлат, мовелтиприл, мурацеин А, мурацеин В, мурацеин С, пентоприл, периндоприл, периндоприлат, пивалоприл, пивоприл, хинаприл, хинаприл гидрохлорид, хинаприлат, рамиприл, рамиприлат, спираприл, спираприл гидрохлорид, спираприлат, спироприл, спироприл гидрохлорид, темокаприл, темокаприл гидрохлорид, тепротид, трандолаприл, трандолаприлат, утибаприл, забициприл, забициприлат, зофеноприл и зофеноприлат. Предпочтительными ингибиторами АСЕ для применения в данном изобретении являются рамиприл, рамиприлат, лизиноприл, эналаприл и эналаприлат. Более предпочтительными ингибиторами АСЕ для применений в данном изобретении являются рамиприл и рамиприлат.
Предпочтительные антагонисты ангиотензина II, их фармацевтически приемлемые соли, сольваты, сольваты таких солей или пролекарства для применения в сочетании с соединением формулы (I) включают, но не ограничиваясь только ими, следующие соединения: кандесартан, кандесартан цилексетил, лозартан, валсартан, ибесартан, тасосартан, телмисартан, эпросартан. Особенно предпочтительными антагонистами ангиотензина II или их фармацевтически приемлемыми производными для применения в данном изобретении являются кандесартан и кандесартан цилексетил.
В другом аспекте данного изобретения соединение формулы (I) или его фармацевтически приемлемая соль, сольват, сольват такой соли или пролекарство может вводиться в сочетании с PPAR альфа и/или гамма агонистом или его фармацевтически приемлемыми солями, сольватами, сольватами таких солей или пролекарствами. Подходящие PPAR альфа и/или гамма агонисты, их фармацевтически приемлемые соли, сольваты, сольваты таких солей или пролекарства хорошо известны в данной области техники. Они включают соединения, описанные в публикациях WO 01/12187, WO 01/12612, WO 99/62870, WO 99/62872, WO 99/62871, WO 98/57941, WO 01/40170 и публикациях J.Med. Chem., 1996, 39, 665, Expert Opinion on Therapeutic Patents, 10 (5), 623-634 (в частности, соединения, описанные в заявках на патент, приведенных на странице 634) и J. Med. Chem., 2000, 43, 527, которые включены в данное описание в виде ссылки. В частности, PPAR альфа и/или гамма агонистом является WY-14643, клофибрат, фенофибрат, бензафибрат, GW 9578, троглитазон, пиоглитазон, розиглитазон, эглитазон, проглитазон, BRL-49634, KRP-297, JTT-501, SB 213068, GW 1929, GW 7845, GW 0207, L-796449, L-165041 и GW 2433. В частности, к PPAR альфа и/или гамма агонистам относится (S)-2-этокси-3-[4-(2-{4-метансульфонилоксифенил}эокси)фенил]пропановая кислота и ее фармацевтически приемлемые соли.
Таким образом, в дополнительном отличительном признаке данного изобретения предложен способ получения ингибирующего действия IBAT у теплокровного животного, такого как человек, нуждающегося в таком лечении, способ включает введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства с одновременным, последовательным или раздельным введением эффективного количества PPAR альфа и/или гамма агониста или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства.
Таким образом, в дополнительном отличительном признаке данного изобретения предложен способ лечения гиперлипидемических состояний у теплокровного животного, например, у человека, нуждающегося в таком лечении, способ включает введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства с одновременным, последовательным или раздельным введением эффективного количества PPAR альфа и/или гамма агониста, его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства.
В соответствии с дополнительным аспектом данного изобретения, предложена фармацевтическая композиция, которая включает соединение формулы (I) или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство и PPAR альфа и/или гамма агонист или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство в сочетании с фармацевтически приемлемым разбавителем или носителем.
В соответствии с дополнительным аспектом данного изобретения, предложен набор, включающий соединение формулы (I) или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство и PPAR альфа и/или гамма агонист или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство.
В соответствии с дополнительным аспектом данного изобретения, предложен набор, включающий:
a)соединение формулы (I) или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство в форме первой стандартной дозы;
b) PPAR альфа и/или гамма агонист или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство в форме второй стандартной дозы; и
c) контейнер в качестве тары для указанных первой и второй дозированных лекарственных форм.
В соответствии с дополнительным аспектом данного изобретения, предложен набор, включающий:
a) соединение формулы (I) или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство в сочетании с фармацевтически приемлемым разбавителем или носителем в форме первой стандартной дозы;
b) PPAR альфа и/или гамма агонист или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство в форме второй стандартной дозы; и
c) контейнер в качестве тары для указанных первой и второй дорированных лекарственных форм.
В соответствии с еще одним отличительным признаком данного изобретения, предложено применение соединения формулы I или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства и PPAR альфа и/или гамма агониста или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства в производстве лекарственного средства для применении с целью получения ингибирующего действия в отношении IBAT у теплокровного животного, такого как человек.
В соответствии с еще одним отличительным признаком изобретения, предложен способ применения соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства и PPAR альфа и/или гамма агониста или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства в производстве лекарственного средства для применении при лечении гиперлипемических состояний у теплокровного животного, такого как человек.
В соответствии с дополнительным аспектом данного изобретения, предложен способ комбинированного лечения, включающий введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства, необязательно в сочетании с фармацевтически приемлемым разбавителем или носителем, с одновременным, последовательным или раздельным введением эффективного количества PPAR альфа и/или гамма агониста или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства, необязательно в сочетании с фармацевтически приемлемым разбавителем или носителем, теплокровному животному, например человеку, нуждающемуся в таком терапевтическом лечении.
Помимо применения в терапии, соединения формулы (I) или их фармацевтически приемлемые соли, сольваты, сольваты таких солей или пролекарства также полезны в качестве фармакологического средства для усовершенствования и стандартизации систем биологических испытаний in vitro и in vivo для оценки действия ингибиторов IBAT в организмах лабораторных животных, таких как кошки, собаки, кролики, обезьяны, крысы и мыши, как часть работы по поиску новых терапевтических средств.
Многие из промежуточных продуктов, описанных в данном изобретении, являются новыми соединениями и, таким образом, обеспечивают дополнительный отличительный признак данного изобретения. Например, соединения формулы (VIIIa), (VIIIb), (IXa), (Xb), (XIIIa) и (XIIIb) проявляют ингибиторную активность в отношении IBAT при испытании в описанном выше опыте in vitro и, следовательно, заявлены в качестве дополнительного объекта данного изобретения.
Таким образом, дополнительный аспект данного изобретения представляет соединение формулы (VIIIa), (VIIIb), (IXa), (Xb), (XIIIa) и (XIIIb) или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство.
Таким образом, в соответствии с дополнительным аспектом данного изобретения, предложена фармацевтическая композиция, которая включает соединение формулы (VIIIa), (VIIIb), (IXa), (Xb), (XIIIa) и (XIIIb) или его фармацевтически приемлемая соль, сольват, сольват такой соли или пролекарство, как определено выше, в сочетании с фармацевтически приемлемым разбавителем или носителем.
Таким образом, в соответствии с дополнительным аспектом данного изобретения, предложено соединение формулы (VIIIa), (VIIIb), (IXa), (Xb), (XIIIa) и (XIIIb) или его фармацевтически приемлемая соль, сольват, сольват такой соли или пролекарство, как определено выше, для применения в способе профилактического или терапевтического лечения теплокровного животного, например человека.
Таким образом, в соответствии с данным аспектом изобретения, предложено соединение формулы (VIIIa), (VIIIb), (IXa), (Xb), (XIIIa) и (XIIIb) или его фармацевтически приемлемая соль, сольват, сольват такой соли или пролекарство, как определено выше, для применения в качестве лекарственного средства.
В соответствии с еще одним отличительным признаком данного изобретения, предложено применение соединения формулы (VIIIa), (VIIIb), (IXa), (Xb), (XIIIa) и (XIIIb) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства, как определено выше, в производстве лекарственного средства для применения с целью получения ингибирующего действия в отношении IBAT у теплокровного животного, такого как человек.
В соответствии с еще одним отличительным признаком данного изобретения, предложено применение соединения формулы (VIIIa), (VIIIb), (IXa), (Xb), (XIIIa) и (XIIIb) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства, как определено выше, в производстве лекарственного средства для применения при лечении гиперлипемических состояний у теплокровного животного, такого как человек.
В соответствии с еще одним отличительным признаком данного изобретения, предложен способ получения ингибирующего действия в отношении IBAT у теплокровного животного, например человека, нуждающегося в таком лечении, способ включает введение указанному животному эффективного количества соединения формулы (VIIIa), (VIIIb), (IXa), (Xb), (XIIIa) и (XIIIb) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства.
В соответствии с еще одним отличительным признаком данного аспекта изобретения, предложен способ лечения гиперлипемических состояний у теплокровного животного, например человека, нуждающегося в таком лечении, способ включает введение указанному животному эффективного количества соединения формулы (VIIIa), (VIIIb), (IXa), (Xb), (XIIIa) и (XIIIb) или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарства.
В описанных выше фармацевтической композиции, процессе, способе, применении и отличительных признаках изготовления лекарственного средства применяются также альтернативные и предпочтительные воплощения соединений данного изобретения.
Примеры
Далее изобретение далее будет описано со ссылкой на приведенные ниже примеры, которые не ограничивают область данного изобретения и в которых, когда это подходит, могут использоваться стандартные методы, известные квалифицированному химику, и методы, аналогичные описанным в этих примерах;
при этом если не указано другого условия, то:
(i) выпаривание проводилось с помощью роторного испарителя в вакууме после удаления фильтрованием твердых остатков, таких как осушители;
(ii) все реакции проводились в инертной атмосфере при комнатной температуре, обычно в интервале 18 - 25°С с использованием растворителей с чистотой, приемлемой для ВЭЖХ, если не указано другого условия;
(iii)колоночная хроматография (флэш-метод) проводилась на силикагеле 40-63 мкм (Merck);
(iv) выходы приведены только для иллюстрации и не являются обязательно максимально возможными;
(v) структуры конечных продуктов формулы (I) были подтверждены методом ядерного (обычно протонного) магнитного резонанса (ЯМР) и масс-спектроскопии; значения химических сдвигов магнитного резонанса определялись в дейтерированном CDCl3 (если не указан другой растворитель) на дельта-шкале (ppm, относительно тетраметилсилана); приведены данные протонного магнитного резонанса, если не указано другого условия; спектры записаны на спектрометрах Varian Mercury-300 MHz, Varian Unity plus-400 MHz, Varian Unity plus-600 MHz или Varian Inova 500 MHz, и если не указано другого условия, данные записывались при 400 МГц; множественность пиков обозначены следующим образом: с - синглет, д - дуплет; дд - двойной дублет; т- триплет; тт - тройной триплет; кв - квартет; ткв - тройной квартет; м - мультиплет; уш. - уширенный сигнал; АВкв - АВ квартет; АВд - АВ дублет; АВдд - АВ дублет дублетов; дАВкв - дублет АВ квартетов; ЖХМС записано на Waters ZMD, ЖХ колонка х Terra MS C8(Waters), обнаружение с помощью HP 1100 MS-детектора, оснащенного диодной антенной; масс-спектр (МС)(петля) записан на VG Platform II (Fisons Instruments) с помощью HP 1100 MS-детектора, оснащенного диодной антенной; если не указано другого условия, приведенный масс-ион представляет собой (MH+); если в тексте не приведены более подробное описание, аналитическая высокоэффективная жидкостная хроматография (ВЭЖХ) проводилась на Prep LC 2000 (Waters), Cromasil C8, 7 μm (Akzo Nobel); MeCN и 10 мМ ацетат аммония в деионизированной воде в качестве подвижной фазы с подходящим составом;
(vii)промежуточные продукты не характеризовались полностью и чистота определялась тонкослойной хроматографией (ТСХ), ВЭЖХ, ИК-, МС и ЯМР анализом.
(viii) при сушке растворов в качестве осушителя использовался сульфат натрия;
(ix) если колонка обозначена как «ISOLUTE», это означает,что колонка содержит 2 г диоксида кремния, причем диоксид кремния помещался в 6 мл шприц и наносился на пористый диск с размером пор 54Å полученный от International Sorbent Technology под названием «ISOLUTE»; «ISOLUTE» - зарегистрированная торговая марка;
(х) могут использоваться следующие аббревиатуры
ДХМ - дихлорметан
ДМФА - N,N-диметилформамид;
TBTU - о-Бензотриазол-1-ил-N,N,N',N'-тетраметилуроний тетрафторборат;
EtOAc - этилацетат;
MeCN - ацетонитрил;
ТФУК - трифторуксусная кислота;
IPA - изопропанол;
DIPEA - диизопропилэтиламин;
ТГФ - тетрагидрофуран.
Пример 1
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-[N-((R)-1'-фенил-1'-карбоксиметил)карбамоилметокси]-2,3,4,5-тетрагидро-1, 5-бензотиазепин
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-[N-((R)-1'-фенил-1'-метоксикарбонилметил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин (метод 29; 300 мг, 0,46 ммоль) растворяют в метаноле (5 мл). К раствору добавляют NaOH (100 мг в 0,2 мл воды) и смесь перемешивают при комнатной температуре в течение 1 часа. Добавляют уксусную кислоту (0,3 мл). Растворитель выпаривают при пониженном давлении и остаток экстрагируют смесью ДХМ/вода. Слой ДХМ отделяют, сушат и выпаривают при пониженном давлении, получая указанное в заголовке соединение: 270 мг (92%). ЯМР, (500 МГц) 0,7-0,8 (м, 6H), 1,0-1,6 (м, 12H), 2,1 (с, 3H) 3,2 (ушир.с, 2H), 3,6-3,8 (м, 2H), 4,6 (с, 2H), 5,6 (д, 1H), 6,6 (с, 1H), 6,9-7,5 (м, 11H), 7,8 (д, 1Н).
Примеры 2-9
Представленные ниже соединения получают в соответствии с методикой примера 1, используя соответствующие исходные вещества.



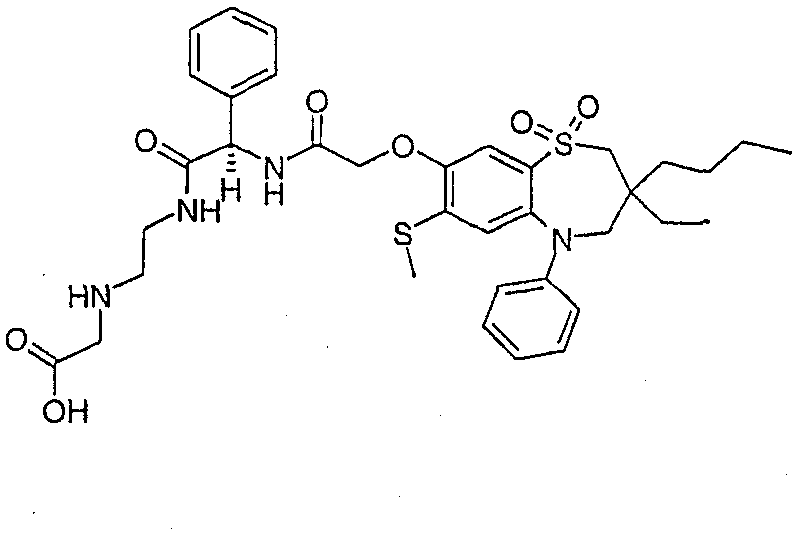

Пример 10
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-бром-8-{1-[N-((R)-1'-фенил-l'-карбоксиметил)карбамоил]этокси}-2,3,4,5-тетрагидро-1, 5-бензотиазепин
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-бром-8-{1-[N-((R)-1'-фенил-1'-метоксикарбонилметил)карбамоил]этокси}2,3,4,5-тетрагидро-1,5-бензотиазепин (метод 33; 103 мг, 0,15 ммоль) растворяют в смеси ТГФ и H2O (2:1, 3 мл). Добавляют LiOH (7 мг, 0,3 ммоль) и смесь перемешивают в течение 7 часов при комнатной температуре. Большую часть растворителя удаляют при пониженном давлении и сырой продукт очищают препаративной ВЭЖХ (элюент: смесь MeCN и ацетат-аммониевого буфера, 45:55), получая указанное в заголовке соединение, 97 мг (96 %). ЯМР (ДМСО-d6) 0,60-0,80 (м, 6H), 0,90-1,60 (м, 11H), 3,15-3,45 (м, 2H), 3,50-3,90 (м, 2H), 4,95-5,25 (м, 2H), 6,85-7,55 (м, 12H), 8,55-8,95 (м, 1H).
Примеры 11-16
Представленные ниже соединения получают в соответствии с методикой примера 10, используя соответствующие исходные вещества.


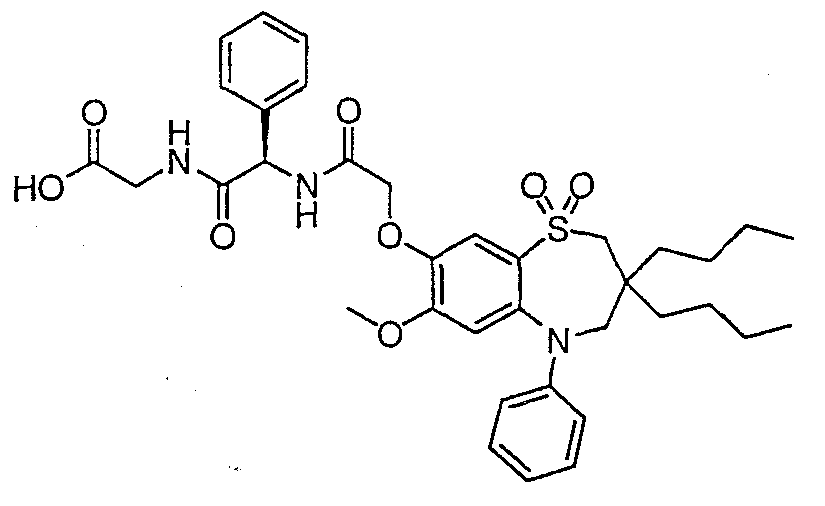
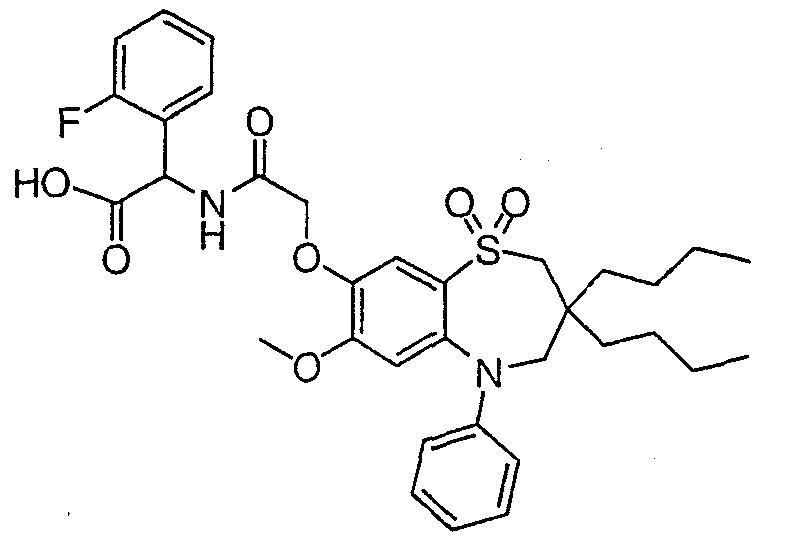


Пример 17
1,1-диоксо-3-бутил-3-этил-5-фенил-7-бром-8-[N -((S)-1'-фенил-1'-карбоксиметил)карбамоилметокси]|-2,3,4,5-тетрагидро-1,5-бензотиазепин
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-бром-8-[N-((S)-1
'-фенил-1'-метоксикарбонилметил)карбамоилметокси]-2,3,4,5-тетрагидро-l,5-бензотиазепин (метод 46; 60 мг, 0,091 ммоль) растворяют в ТГФ (1 мл) и добавляют к раствору гидроксида лития моногидрата (12,6 мг, 0,29 ммоль) в воде (1 мл). Смесь перемешивают несколько раз в течение 30 минут. Добавляют 2M раствор HC1 (0,3 мл) и водный слой экстрагируют ДХМ. Органический слой промывают насыщенным раствором соли, сушат, фильтруют и выпаривают при пониженном давлении, получая указанное в заголовке соединение 48 мг (82%). ЯМР (CD3OD) 0,73-0,84 (м, 6H), 1,0-1,6 (м, 8H), 3,27 (ушир.с, 2H), 3,60-3,, 90 (м, 2H), 4,71 (АВкв, 2H), 5,47-5,55 (м, 1H), 7,02 (ушир.т, 1H), 7,08-7,17 (м, 3H), 7,25-7,46 (м, 7H), 7,52 (с, 1H), 8,43 (д, NH); m/z 643,5.
Примеры 18-21
Представленные ниже соединения получают в соответствии с методикой примера 17, используя соответствующие исходные вещества.

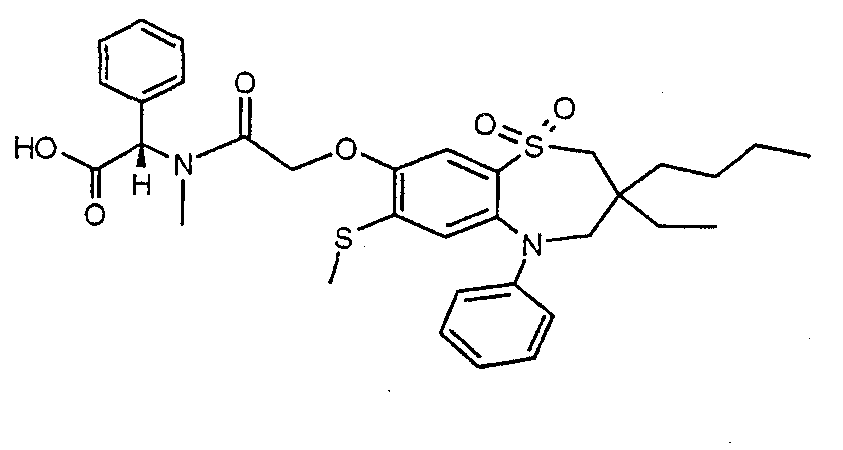


Пример 22
1,1-диоксо-3-бутил-3-этил-5-фенил-7-бром-8-[N-((R)-1'-фенил-1'-карбоксиметил)карбамоилметокси]-2,3,4,5-тетрагидро-l,5-бензотиазепин
Указанное в заголовке соединение синтезируют из 1,1-диоксо-3-бутил-3-этил-5-фенил-7-бром-8-[N-((R)-1'-фенил-1'-метоксикарбонилметил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепина (метод 61) в соответствии с методикой примера 17, с тем отличием, что водный слой экстрагируют EtOAc. Продукт очищают препаративной ВЭЖХ (элюент с градиентом: MeCN/ацетатноаммонивый буфер, от 5:95 до 100/0) ЯМР 0,75-0,83 (м, 6H), 1,0-1,25 (м, 4H), 1,32-1,52 (м, 3H), 1,55-1,70 (м, 1H), 3,20 (АВкв, 2H), 3,65-3,83 (м, 2H), 4,62 (АВкв, 2H), 5,68 (д, 1H), 7,04-7,15 (м, 4H), 7,3-7,5 (м, 8H), 7,87 (ушир.д, 1H); m/z 643,1.
Пример 23
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-бром-8-(N-{(S)-1'-фенил-1'-[N' -(2-сульфоэтил)карбамоил]метил}карбамоилметокси)-2,3,4,5-тетрагидро-l,5-бензотиазепин аммониевая соль
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-бром-8-[N -((S)-1'-фенил-1'-карбоксиметил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин (пример 17; 48 мг, 0,075 ммоль) и 2-аминоэтансульфоновую кислоту (17 мг, 0,14 ммоль) растворяют в ДМФА (2 мл) и DIPEA (0,052 мл, 0,30 ммоль). Смесь перемешивают в течение 15 минут притемпературе 60°C. Добавляют TBTU (31 мг, 0,097 ммоль) и смесь перемешивают в течение 2 часов при температуре 60°C. Растворитель выпаривают при пониженном давлении. Остаток очищают препаративной ВЭЖХ (элюент с градиентом: MeCN/ацетат-аммониевый буфер, от 5/95 до 100/0). Лиофилизация приводит к получению указанного в заголовке соединения, 4 мг (7%). ЯМР (CD3OD) 0,75-0,83 (м, 6H), 0,95-1,65 (м, 8H), 2,85-3,0 (м, 2H), 3,27 (ушир.с, 2H), 3,5-3,9 (м, 4H), 4,72 (АВкв, 2H), 5,48 (с, 1H), 7,02 (ушир.т, 1Н), 7,09-7,15 (м, 3H), 7,25-7,52 (м, 8H); m/z 750,3.
Примеры 24-37
Соединения, представленные ниже, получают с помощью этой же методики. Кислоту (1 экв.) растворяют в ТГФ (1 мл) и добавляют к раствору гидроксида лития моногидрата (12,6 мг, 2,9-6,6 экв.) в воде (1 мл). Смесь время от времени перемешивают и по истечении 1,5-6 часов удаление защитной группы завершается (в соответствии с ЖХ-МС). Добавляют 2M раствор HCl (0,3 мл).
Примеры 24-33
Реакционную смесь отбирают шприцем, в котором содержится hydramatrix®. Продукт элюируют ДХМ. ДХМ сушат, фильтруют и выпаривают при пониженном давлении. Продукт очищают препаративной ВЭЖХ (элюент с градиентом: MeCN/ацетат-аммониевый буфер, от 5:95 до 100:0)
Примеры 34-37
Водный слой дважды экстрагируют ДХМ. Органический слой сушат, фильтруют и выпаривают при пониженном давлении.




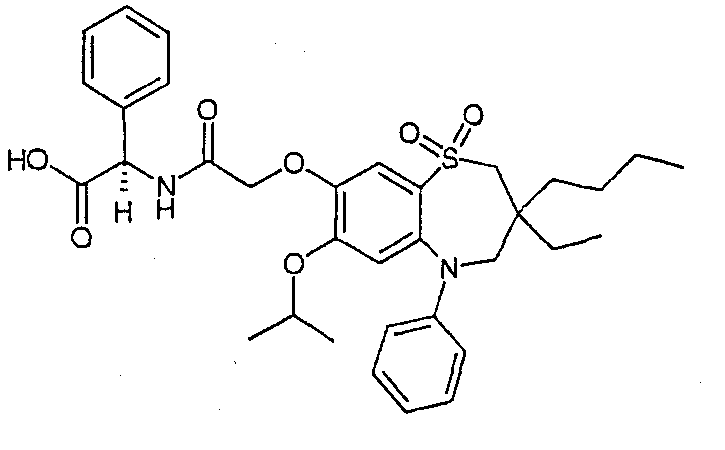
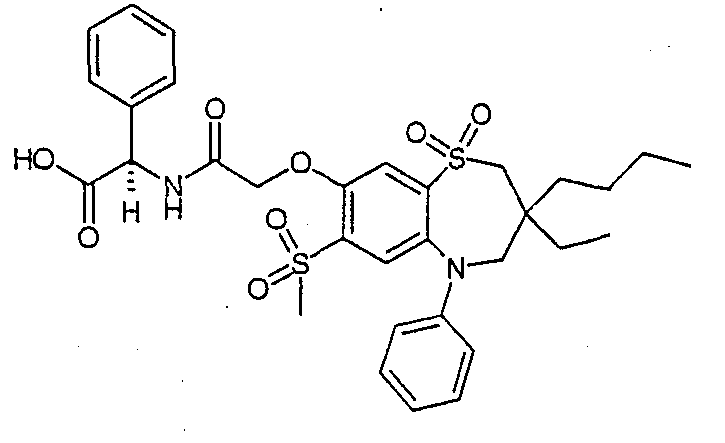
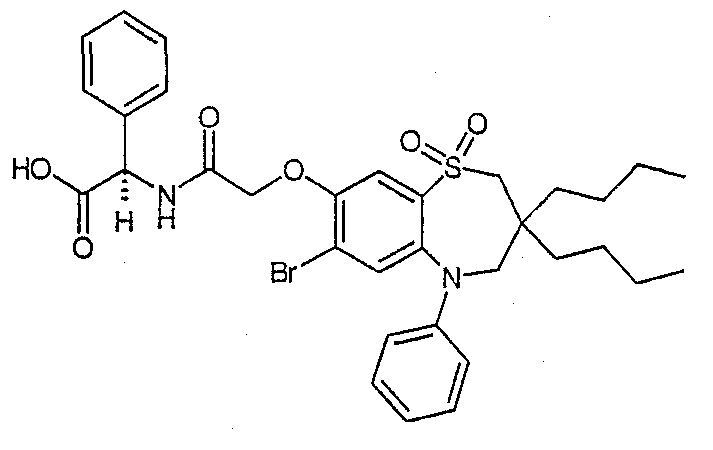
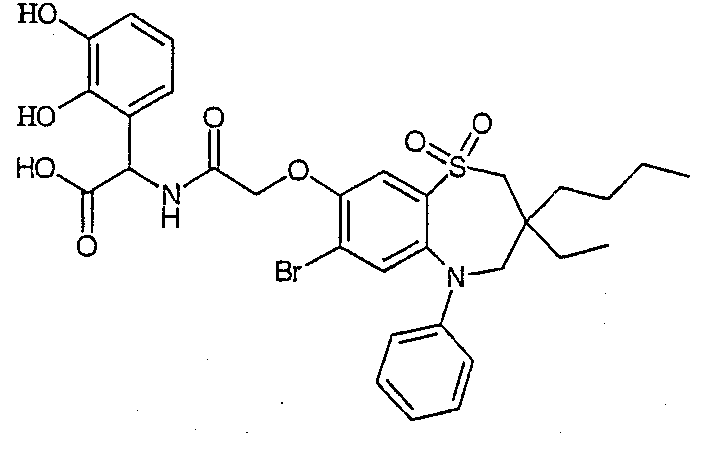
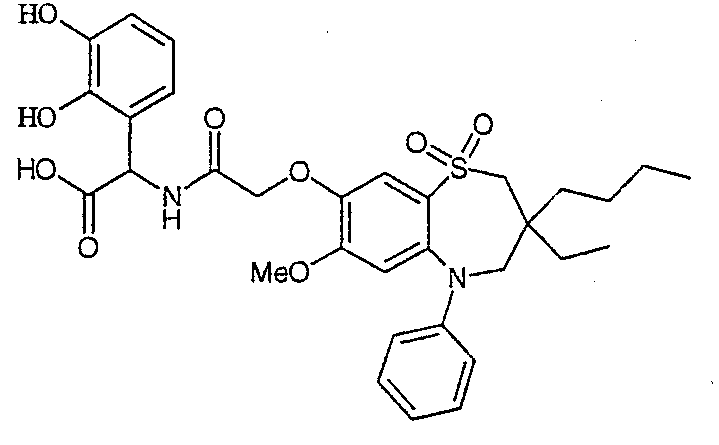

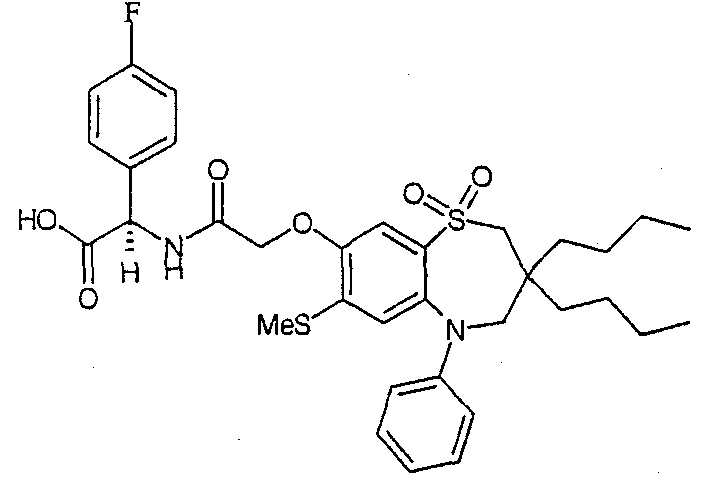

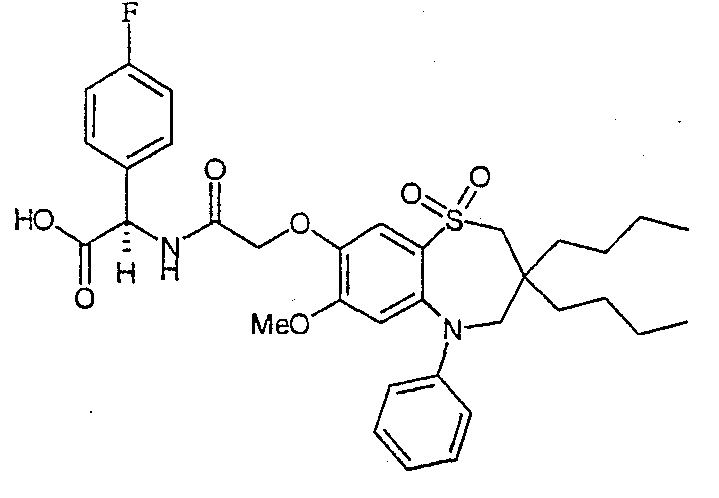
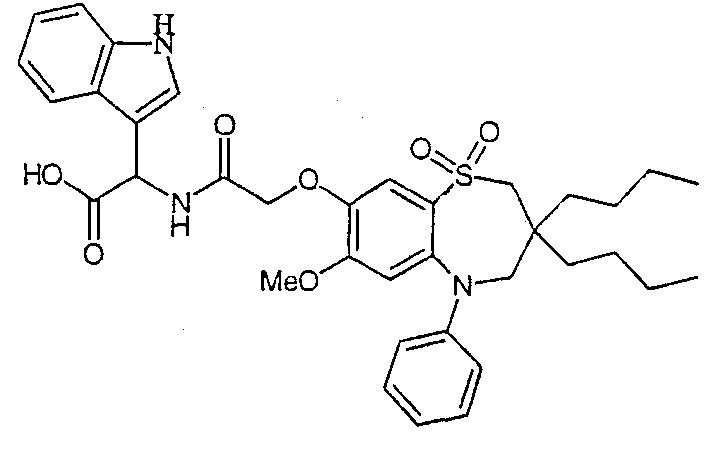
Пример 38
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-[N-((R)-1'-фенил-1'-карбоксиметил)карбамоилметокси]-2,3,4,5-тетрагидро-1, 5-бензотиазепин
1,1-Диоксо-3-бутил-3-этил-3-фенил-7-бром-8-[N-((R)-1'-фенил-1'-карбоксиметил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин (пример 22; 50 мг, 0,078 ммоль) растворяют в ДМФА (1,5 мл). Добавляют метантиолат натрия (20 мг, 0,29 ммоль) и смесь перемешивают в течение 1,5 часов при температуре 50°C. Добавляют уксусную кислоту (40 мг) и растворитель выпаривают при пониженном давлении. Остаток очищают препаративной ВЭЖХ (элюент: MeCN/ацетат-аммониевый буфер, 45:55), получая указанное в заголовке соединение, 29 мг (61%). ЯМР (ДМСО-d6): 0,7-0,8 (м, 6H), 0,9-1,6 (м, 8H), 2,2 (с, 3H), 3,1-3,7 (м, 4H), 4,6-4,8 (м, 3H), 6,7 (с, 1H), 6,8-7,4 (м, 11H), 8,3 (д, 1H).
Пример 39
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-этилтио-8-[N-((R)-1'-фенил-1'-карбоксиметил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин
1, 1-Диоксо-3-бутил-3-этил-5-фенил-7-бром-8-[N-((R)-1'-фенил-1'-карбоксиметил)карбамоилметокси]-2,3,4,5-тетрагидро-l,5-бензотиазепин (пример 22; 50 мг, 0,078 ммоль), этантиол (99 мг, 1,59 ммоль) и карбонат цезия (253 мг, 0,78 ммоль) добавляют в ДМФА (5 мл) и смесь перемешивают в течение 30 часов при температуре 44°C. Раствор фильтруют и выпаривают при пониженном давлении. Остаток очищают препаративной ВЭЖХ (элюент: MeCN/ацетат-аммониевый буфер, 45:55). Остаток очищают колоночной хроматографией (элюент: ДХМ/метанол, 100:15), получая указанное в заголовке соединение, 15 мг (31%). ЯМР (300 МГц, CD3OD) 0,7-0,85 (м, 6H), 1,0-1,6 (м, 11H), 2,65 (кв, 2H), 3,2 (с, 2H), 3,7 (ушир.с, 2H), 4,6 (кв, 2H), 5,3 (с, 1H), 6,75 (с, 1H), 6,9-7,5 (м, 11H).
Пример 40
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-(2-гидроксиэтилтио)-8-[N-((R)-1'-фенил-1'-карбоксиметил)карбамоилметокси]-2,3,4,5-тетрагидро-1, 5-бензотиазепин
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-бром-8-[N-((R)-1'-фенил-1'-карбоксиметил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин (пример 22; 50 мг, 0,078 ммоль), 2-меркаптоэтанол (281 мг, 3,59 ммоль) и карбонат цезия (228 мг, 0,7 ммоль) добавляют в ДМФА (5 мл) и смесь перемешивают в течение 9 часов при температуре 70°C. Растворитель выпаривают при пониженном давлении. Остаток очищают препаративной ВЭЖХ (элюент: MeCN/ацетат-аммониевый буфер, 45:55). Собранные фракции лиофилируют, получая указанное в заголовке соединение, 20 мг (40%). ЯМР (300 МГц, CD3OD) 0,75-0,85 (м, 6H), 1,0-1,6 (м, 8H), 2,9 (т, 2H), 3,2 (с, 2H), 3,55 (т, 2H), 3,7 (ушир.с, 2H), 4,65 (кв, 2H), 5,3 (с, 1Н), 6,9 (с, 1H), 6,95-7,5 (м, 11H).
Пример 41
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-(2-N',N'-диметиламиноэтилтио)-8-[N-((R)-1'-фенил-1'-карбоксиметил)карбамоилметокси]-2,3, 4,5-тетрагидро-1,5-бензотиазепин
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-бром-8-[N-((R)-1'-фенил-1'-карбоксиметил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин (пример 22; 50 мг, 0,078 ммоль), диметиламиноэтантиол гидрохлорид (99 мг, 0,94 ммоль), карбонат калия (129 мг, 0,94 ммоль), DIPEA (100 мг, 0,77 ммоль) и боргидрид натрия (35 мг, 0,93 ммоль) добавляют в ДМФА (10 мл) и смесь перемешивают в течение 24 часов при температуре 85°C. Раствор фильтруют и выпаривают при пониженном давлении. Остаток дважды очищают препаративной ВЭЖХ (элюент: MeCN/ацетат-аммониевый буфер, 45:55). Собранные фракции лиофилируют, получая указанное в заголовке соединение 15 мг (30%). ЯМР (300 МГц, CD3OD) 0,75-0,85 (м, 6H), 1,0-1,65 (м, 8H), 2,65 (с, 6H), 3,05 (т, 2H), 3,2 (т, 2H), 3,3 (с, 2H), 3,75 (ушир.с, 2H), 4,75 (с, 2H), 5,2 (с, 1H), 6,95-7,4 (м, 11), 7,5 (с, 1H).
Пример 42
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-изопропилтио-8-[N-((R)-l'-фенил-1'-карбоксиметил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин
1,1-диоксо-3-бутил-3-этил-5-фенил-7-бром-8-[N-((R)-1'-фенил-1'-карбоксиметил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин (пример 22; 50 мг, 0,078 ммоль), 2-пропантиол (126 мг, 1,65 ммоль), карбонат цезия (152 мг, 0,47 ммоль), боргидрид натрия (25 мг, 0,66 ммоль) добавляют в ДМФА (5 мл) и смесь перемешивают в течение 5 минут при температуре 100°C. Растворитель выпаривают при пониженном давлении. Остаток очищают препаративной ВЭЖХ (элюент: MeCN/ацетат-аммониевый буфер, 45:55). Собранные фракции лиофилируют получая указанное в заголовке соединение, 15 мг (30%). ЯМР (300 МГц, ДМСО-d6) 0,7-0,85 (м, 6H), 0,95-1,65 (м, 14H), 3,3 (с, 2H), 3,7 (ушир.с, 2H), 4,75 (дд, 2H), 5,05 (ушир.с, 1H), 6,75-7,4 (м, 12H), 8,5 (ушир.с, 1H).
Пример 43
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-1'-фенил-1'-[N'-(карбоксиметил)карбамоил]метил}карбамоилметокси)-2,3,4, 5-тетрагидро-1,5-бензотиазепин
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-1'-фенил-1'-[N'-(трет-бутоксикарбонилметил)карбамоил]ме-тил}крбамоилметокси)-2,3,4, 5-тетрагидро-1,5-бензотиазепин (метод 63; 120 мг, 0,17 ммоль) растворяют в ДХМ (2 мл). Добавляют ТФУК (0,7 мл) и смесь перемешивают при комнатной температуре в течение 3 часов. Реакционную смесь выпаривают при пониженном давлении. Остаток очищают препаративной ВЭЖХ (элюент: MeCN/ацетат-аммониевый буфер, 50:50), получая указанное в заголовке соединение, 95 мг (85%). ЯМР (300 МГц, ДМСО-d6) ЯМР (300 МГц, ДМСО-d6) 0,7-0,8 (м, 6H), 0,9-1,6 (м, 12H), 2,2 (с, 3H) 3,2-3,3 (м, 2H), 3,5-3,8 (м, 4H), 4,8 (АВкв, 2H), 5,6 (д;lH), 6,7 (с, 1H), 6,8-7,5 (м, 11H), 8,5-8,7 (м, 2H).
Примеры 44-49
Представленные ниже соединения получают в соответствии с методикой примера 43, используя соответствующие исходные вещества.
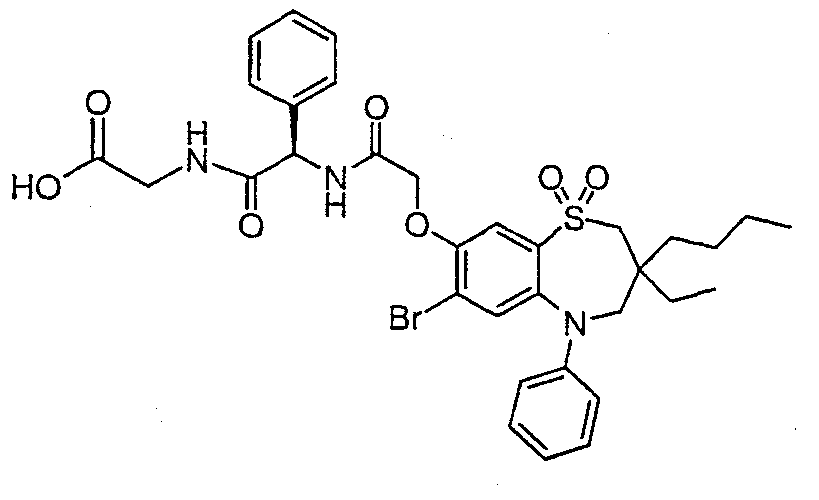
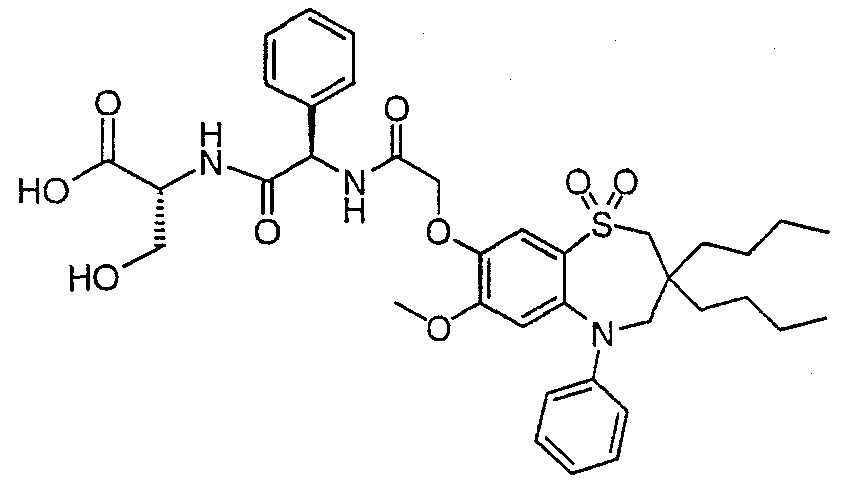


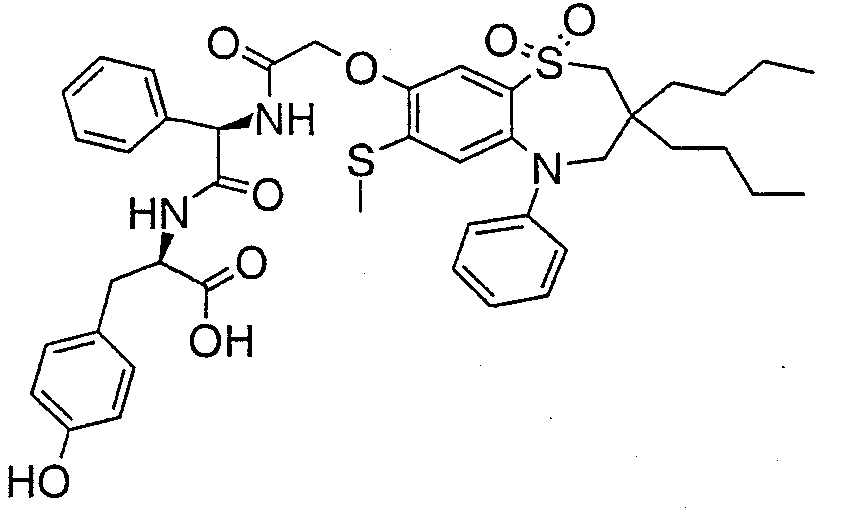

Пример 50
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-1'-фенил-1'-[N'-(2-сульфоэтил)карбамоил]метил}карбамоилметокси)-2,3, 4,5-тетрагидро-1,5-бензотиазепин аммониевая соль
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин (метод 22; 150 мг, 0,30 ммоль) и 2-((2'R)-2'-амино-2'-фенилэтаноиламино)этансульфоновую кислоту (метод 28; содержащую DIPEA гидрохлорид, 150 мг, 0,36 ммоль) растворяют в ДМФА (6 мл). Добавляют DIPEA (0,2 мл, 1,15 ммоль) и TBTU (114 мг, 0,36 ммоль) и смесь перемешивают в течение 2 часов при комнатной температуре. Растворитель выпаривают при пониженном давлении. Остаток очищают препаративной ВЭЖХ (элюент с градиентом: MeCN/ацетат-аммониевый буфер, от 5:95 до 100:0), получая указанное в заголовке соединение, 73 мг (32%). ЯМР (CD3OD) 0,75-0,85 (м, 6H), 1,0-1,3 (м, 8H), 1,3-1,6 (ra, 4H), 2,16 (с, 3H), 2, 85-3,0 (м, 2H), 3,24 (ушир.с, 2H) 3,5-3,85 (м, 4H), 4,70 (АВкв, 2H), 5,47 (с, 1H), 6,71 (с, 1Н), 6,97 (ушир.т, 1H), 7,11 (ушир.д, 2H), 7,23-7,45 (м, 8H); m/z 746,2.
Пример 51
1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-(N-{(R)-1'-фенил-1'-[N'-(2-сульфоэтил)карбамоил]метил}карбамоилметокси)-2,3,4, 5-тетрагидро-1,5-бензотиазепин аммониевая соль
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин (метод 17; 49 мг, 0,10 ммоль) и 2-((2'R)-2'-амино-2'-фенилэтаноиламино)этансульфоновую кислоту (метод 28; содержащую DIPEA гидрохлорид; 52 мг, 0,12 ммоль) растворяют в ДМФА (2 мл). Добавляют DIPEA (0,071 мл, 0,41 ммоль) и TBTU (39 мг, 0,12 ммоль) и смесь перемешивают в течение 2 часов при комнатной температуре. Растворитель выпаривают при пониженном давлении. Остаток очищают препаративной ВЭЖХ (элюент с градиентом: MeCN/ацетат-аммониевый буфер, от 5:95 до 100:0), получая указанное в заголовке соединение, 59 мг (78%). ЯМР (CD3OD) 0,74-0,90 (м, 6H), 0,98-1,3 (м, 4H), 1,35-1,67 (м, 4H), 2,16 (с, 3H), 2, 85-3,02 (м, 2H), 3,23 (ушир.с, 2H) 3,52-3,90 (м, 4H), 4,70 (АВкв, 2H), 5,47 (с, 1H), 6,71 (с, 1H), 6,96 (ушир.т, 1H), 7,09 (ушир.д, 2H), 7,21-7,48 (м, 8H); m/z 718,4.
Пример 52
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-(N-{(R)-1'-фенил-1'-[N-(карбоксиметил)карбамоил]метил}карбамоилметокси)-2,3,4, 5-тетрагидро-1,5-бензотиазепин
1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-(N-{(R)-1'-фенил-1'-[N-(этоксикарбонилметил)карбамоил]метил}карбамоилметокси)-2,3,4, 5-тетрагидро-l,5-бензотиазепин (метод 72; 44 мг, 0,063 ммоль) растворяют в смеси ТГФ:H2O (1:1, 2 мл) и добавляют NaOH (1 M, 0,126 ммоль). Смесь перемешивают при комнатной температуре втечение 1 часа. Реакционную смесь подкисляют HCl (1 M), разбавляют до 10 мл и экстрагируют ДХМ (3x10 мл). Объединенные органические слои сушат(MgSO4) и растворитель выпаривают, получая указанное в заголовке соединение (33 мг, 78%). ЯМР (300 МГц) 0,78-0,85 (м, 6H), 1,02- 1,70 (м, 8H), 2,20 (с, 3Н), 3,15/3,21 (АВкв, 2H), 3,78 (м, 2H), 3,94/4,20 (дАВкв, 2H), 4,64 (кв, 2H), 5,91 (д, 1Н), 6,65 (с, 1Н), 6,98-7,52 (м, 11H), 8,17 (д, 1H).
Пример 53
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-(N -{(R)-1'-фенил-1'-[N-(1"-карбокси-1"-фенилметил)карбамоил]метил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин
Указанное в заголовке соединение синтезируют из 1, 1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-(N-{(R)-1'-фенил-1'- [N-( 1"-метоксикарбонилl-1"-фенилметил)карбамоил]метил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепина (метод 73) в соответствии с методикой примера 52. ЯМР (500 МГц) 0,76-0,84 (м, 6H), 1,05-1,73 (м, 8H), 2,14 (с, 3H), 3,16 (м, 2H), 3,74 (м, 2H), 4,48 (м, 2H), 5,53 (д, 2H), 5,96 (д, 2H), 6,63 (с, 1H), 6,97-7, 48 (м, 13H), 7,86 (м, 1Н), 8,17 (м, 1H).
Пример 54
1,1-Диоксо-3-этил-3-бутил-5-фенил-7-метилтио-8-{1-[N-((R)-α -карбоксибензил)карбамоил]этокси}-2,3,4,5-тетрагидро-l,5-бензотиазепин
К раствору 1,1-диоксо-3-бутил-3-этил-5-фенил-7-бром-8-{1-[N -((R)-1'-фенил-1'-карбоксиметил)карбамоил]этокси}-2,3,4,5-тетрагидро-1,5-бензотиазепина (пример 10, 0,050 г, 7,6x10-5 моль) в ДМФА (4 мл) добавляют тиометилат натрия (0,021 г, 3,0xl0-4моль) и раствор перемешивают в течение 4 часов при комнатной температуре. Смесь концентрируют и остаток распределяют между водой и эфиром. Водную фазу экстрагируют еще два раза эфиром и объединенные органические экстракты сушат (MgSO4), концентрируют и очищают ВЭЖХ. Получают 0,30 г (63%) указанного в заголовке соединения в виде твердого белого вещества. ЯМР (CD3 OD) 0,75-0,90 (м, 6H), 1,00-1,30 (м, 4H), 1,40-1,70 (м, 7H), 2,15 (д, 3H), 3,10-3,30 (м, 2H), 3,55-3,95 (м, 2H), 4,80-4,95 (м, 2H), 5,30 (д, 1H), 6,70-7,50 (м, 12H); m/z 625,3.
Пример 55
1,1-диоксо-3-этил-3-бутил-5-фенил-7-метилтио-8-{α-[N-((R)-α-карбоксибензил)карбамоил]бензоилокси)-2,3,4, 5-тетрагидро-1,5-бензотиазепин
К раствору 1,1-диоксо-3-этил-3-бутил-5-фенил-7-метилтио-8-{α-[N-((R)-α-карбоксибензил)карбамоил]бензоилокси}-2,3,4, 5-тетрагидро-1,5-бензотиазепина (пример 11; 0,018 г, 2,5x10-5моль) в ДМФА (3 мл) добавляют тиометилат натрия (0,007 г, 1,0x10-4моль) и раствор перемешивают в течение 4 часов при комнатной температуре. Смесь концентрируют и остаток распределяют между водой и эфиром. Водную фазу экстрагируют еще два раза эфиром и объединенные органические экстракты сушат (MgSO4), концентрируют и очищают ВЭЖХ. Получают указанное в заголовке соединение (0,015 г, 99 %) в виде твердого белого вещества. ЯМР (CD3OD) 0,70-0,85 (м, 6H), 1,00-1,25 (м, 4H), 1,35-1,65 (м, 4H), 2,20 (д, 3H), 3,10-3,20 (м, 2H), 3,50-3,85 (м, 2H), 5,30 (д, 1Н), 5,80 (д, 1H), 6,70 (с, 1H), 6,90-7,65 (м, 16H).
Пример 56
1, 1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N'-(2-сульфоэтил)карбамоил]-4-гидроксибензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин
2-{[(2R)-2-Амино-2-(4-гидроксифенил)этаноил]амино} этансульфоновую кислоту (метод 80; 32,5 мг, 0,119 ммоль) смешивают с ДМФА (4 мл) и N-метилморфолином (30 μл, 0,272 ммоль). Получают прозрачный раствор и несколькими порциями последовательно добавляют 1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин (метод 22; 50 мг, 0,099 ммоль) и TBTU (38 мг, 0,119 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 30 минут и ДМФА удаляют. Сырой продукт очищают препаративной ВЭЖХ (элюент: MeCN/ацетат-аммониевый буфер, 1:1). Лиофилизация приводит к получению 55 мг указанного в заголовке соединения (71%). ЯМР (500 МГц, MeOD) 0,78-0,86 (м, 6H), 1,0-1,3 (м, 8H), 1,4-1,6 (м, 4H), 2,15 (с, 3H), 2,85-3,00 (м, 2H), 3,25 (с, 2H), 3,55-3,68 (м, 2H), 3,75 (ушир.с, 2Н) 4,65 (АВкв, 2Н) 5,36 (с, 1H), 6,70 (с, 1H), 6,75 (д, 2H), 6,98 (т, 1H), 7,12 (д, 2H), 7,22 (д, 2H) 7,28 (т, 2Н) 7,4 (с, 1H); m/z 762.
Примеры 57-58
Представленные ниже соединения получают в соответствии с методикой примера 56 из соответствующих исходных веществ с тем отличием, что реакцию проводят в течение 64 часов (пример 57) или в течение 2 часов (пример 58) и продукт очищают препаративной ВЭЖХ (элюент с градиентом: MeCN/ацетат-аммониевый буфер, от 45:55 до 60/40).

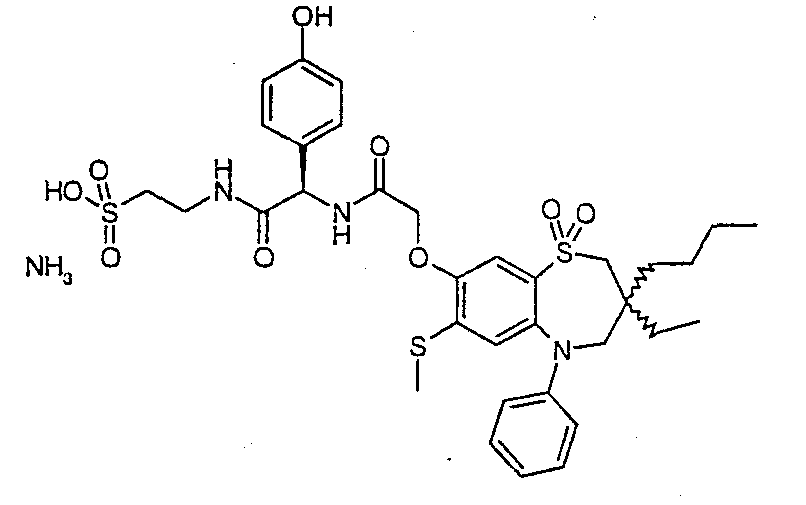
Пример 59
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-(N-{(R)-α-N'-(2-карбоксиэтил)карбамоил]бензил}карбамоилметокси)-2,3,4, 5-тетрагидро-1,5-бензотиазепин
1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-[N-((R)-α-карбоксибензил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин (пример 38; 66,8 мг, 0,109 ммоль) и сложный этиловый эфир β-аланина гидрохлорид (23,0 мг, 0,15 ммоль) растворяют в ДХМ (2,5 мл) и добавляют N-метилморфолин (0,07 мл, 0,64 ммоль). Смесь перемешивают при комнатной температуре в течение 5 минут, затем добавляют TBTU (45,6 мг, 0,142 ммоль) и полученную смесь перемешивают в течение 2 часов. Реакционную смесь фильтруют через небольшую колонку и концентрируют. Сырой сложный эфир растворяют в ТГФ (1,5 мл) и добавляют воду (1,5 мл) и NaOH (1 M, 0,20 ммоль). Смесь перемешивают при комнатной температуре в течение 1 часа, реакцию гасят 1 M HCl. Реакционную смесь разбавляют водой (10 мл) и экстрагируют ДХМ (3 x 5 мл). Органические слои концентрируют и очищают препаративной ВЭЖХ, получая указанное в заголовке соединение (60 мг, 81%). ЯМР (300 МГц) 0,80 (м, 6H), 1,00-1,70 (м, 8H), 2,17 (с, 3H), 2,48 (м, 2H), 3,17 (АВкв, 2H), 3,35 (м, 1H), 3,57 (м, 1Н), 3,70 (м, 2H), 4,62 (АВкв, 2H), 5,77 (д, 1H), 6,64 (с, 1H), 6,98 (т, 1H), 7,06 (д, 2H), 7,28 (м, 4H), 7,42 (м, 3H), 7,56 (м, 1H), 8,10 (м, 1H).
Примеры 60-63
Представленные ниже соединения получают в соответствии с методикой примера 59, используя соответствующие исходные вещества.

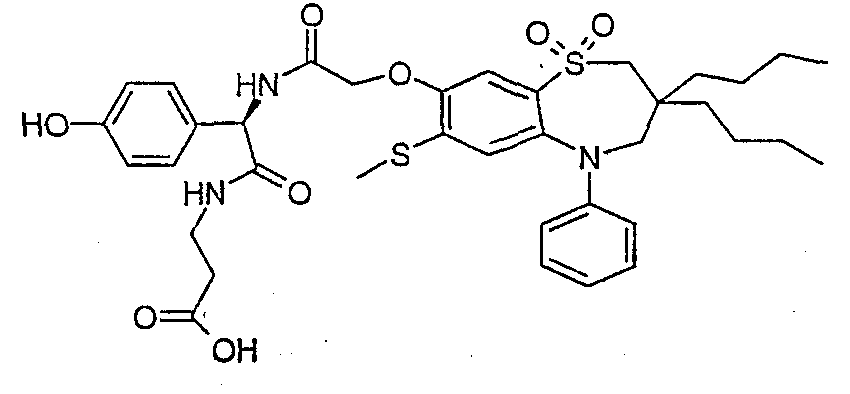
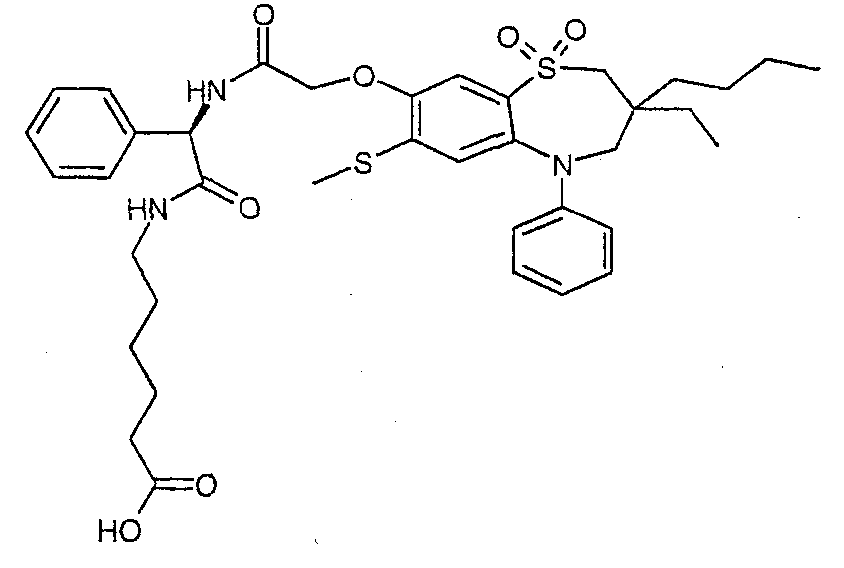
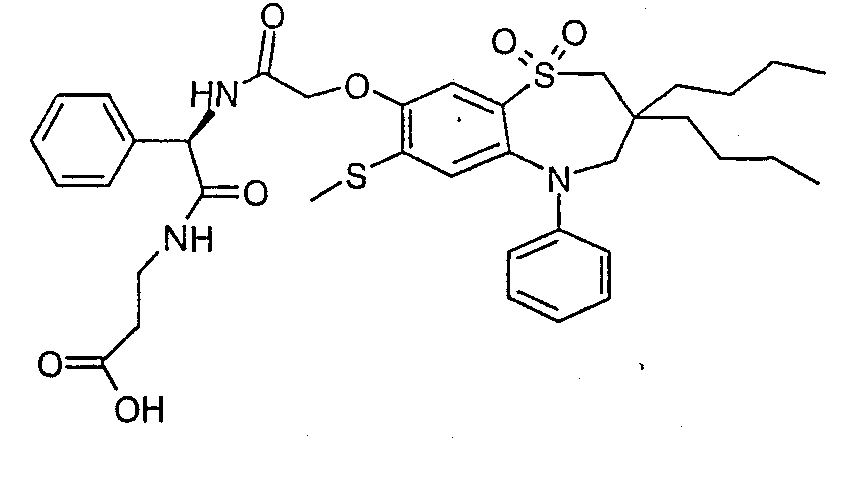
Пример 64
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-[N-((R)-α -карбокси-4-метоксибензил)карбамоилметокси]-2,3,4,5-тетрагидро-l,5-бензотиазепин
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-{N-[(R)-α -(трет-бутоксикарбонил)-4-гидроксибензил]карбамоилметокси}-2,3,4,5-тетрагидро-1,5-бензотиазепин (метод 78; 48 мг, 0,070 ммоль), бромэтил(триметиламмонийбромид) (57 мг, 0,230 ммоль), тетрабутиламмоний бромид (3 мг, 0,009 ммоль) и Cs2CO3 (71 мг, 0,22 ммоль) добавляют к CH3CN (1,0 мл) и реакционную смесь кипятят с обратным холодильником в течение ночи. Растворитель выпаривают и остаток переносят в воду (10 мл), экстрагируют ДХМ (3 x 5 мл) и сушат (MgSO4). Сырой сложный эфир растворяют в ДХМ (2,5 мл), добавляют ТФУК (0,3 мл) и реакционную смесь перемешивают при комнатной температуре в течение ночи. Растворители выпаривают и сырой продукт очищают препаративной ВЭЖХ, получая указанное в заголовке соединение (23 мг, 51 %). ЯМР (ДМСО-d6) 0,74 (м, 6H), 0,94-1,60 (м, 8H), 2,17 (с, 3H), 3,25 (м, 2H), 3,69 (с, 3H), 4,70 (АВкв, 2H), 4,95 (ушир.с, 1Н) 6,71 (с, 1H), 6,83 (м, 3H), 6,97 (д, 2H), 7,20 (м, 4H), 7,27 (с, 1H), 8,37 (ушир.с, 1H).
Пример 65
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(α-[N' -(2-сульфоэтил)карбамоил]-α-метилбензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин аммониевая соль
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-[N-(α -карбокси-α-метилбензил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин (пример 18; 27 мг, 0,041 ммоль) растворяют в ДХМ (2 мл). Последовательно добавляют таурин (таурин) тетрабутиламмониевую соль (45 мг, 0,123 ммоль) и TBTU (16 мг, 0,050 ммоль) и смесь перемешивают в течение 5 часов при комнатной температуре. Растворитель выпаривают и продукт очищают препаративной ВЭЖХ (элюент: MeCN/ацетат-аммониевый буфер, 50:50). Лиофилизация приводит к получению указанного в заголовке соединения (20 мг, 62%). ЯМР показывает, что 16% продукта сохраняется в виде тетрубутиламмониевой соли. ЯМР (500 МГц) 0,75-0,9 (м, 6H), 1,0-1,3 (м, 8H), 1,3-1,6 (м, 4H), 1,95 (с, 3H), 2,1 (с, 3H), 2,9 (ушир.с, 2H), 3,05 (ушир.с, 2H), 3,55 (АВд, 2H), 3,75 (ушир.с, 2H), 4,55 (АВкв, 2H), 6,6 (с, 1H), 6,9-7,6 (м, 12H), 8,2-8,3 (ушир.с, 1H); m/z 777 (M+NH4+).
Примеры 66-67
Представленные ниже соединения получают в соответствии с методикой примера 65, используя соответствующие исходные вещества.


Пример 68
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{α-[N'-(карбоксиметил)карбамоил]-α-метилбензил}карбамоилметокси)-2,3,4,5-тетрагидро-1, 5-бензотиазепин
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{α-[N'-(метоксикарбонилметил)карбамоил]-α-метилбензил}карбамоилметокси)-2,3,4, 5-тетрагидро-1,5-бензотиазепин (метод 44; 20 мг, 0,028 ммоль) растворяют в 2,5 мл смеси ТГФ/вода (4/1). Добавляют LiOH (2 мг, 0,084 ммоль) и смесь перемешивают в течение 1 часа при комнатной температуре. Указанное в заголовке соединение очищают препаративной ВЭЖХ (элюент: MeCN/ацетат-аммониевый буфер, 50:50). MeCN выпаривают и оставшийся буфер подкисляют уксусной кислотой. Лиофилизация приводит к получению 10 мг продукта (51%). ЯМР 0,7-0,9 (м, 6H), 1,0-1,35 (м, 8H), 1,35-1,6 (м, 4H), 2,0 (с, 3H), 2,2 (с, 3H), 3,2 (ушир.с, 2H), 3,65-3,85 (ушир.с, 2H), 3,9-4,1 (д, 2H), 4,5-4,7 (АВкв, 2H), 6,6 (с, 1Н), 6,8 (ушир.с, 1Н), 6,9-7,5 (м, 11H), 8,1 (с, 1Н) ; m/z 727 (M +NH4+).
Пример 69
1, 1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{α-[N'-( 2-сульфоэтил)карбамоил]-2-фторбензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин
1,1-Диоксо-3, 3-дибутил-5-фенил-7-метилтио-8-{N-[α-карбокси-2-фторбензил]карбамоилметокси}-2,3,4,5-тетрагидро-1,5-бензотиазепин (пример 15; 20 мг, 0,030 ммоль), таурин тетрабутиламмониевую соль (20 мг, 0,054 ммоль) и DIPEA (25 мг, 0,19 ммоль) растворяют в ДМФА (0,4 мл). Добавляют TBTU (15 мг, 0,047 ммоль) и смесь перемешивают в течение 30 минут при комнатной температуре. Продукт выделяют из реакционной смеси препаративной ВЭЖХ (элюент: MeCN/ацетат-аммониевый буфер, 50:50). Получают 7 мг (29%) указанного в заголовке соединения. M/z = 764,5.
Пример 70
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-(R)-{α-[N'-(R)-{α-[N" -(карбоксиметил)карбамоил]бензил}крбамоил)метилкарбамоил]бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N -{(R)- 1'-фенил-1'-[N'-(карбоксиметил)карбамоил]метил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин (пример 43; 35 мг, 0,050 ммоль) и (R)-α -[N-(трет-бутоксикарбонилметил)карбамоил]бензиламин (метод 86; 20 мг, 0,076 ммоль) растворяют в ДХМ (2 мл) и добавляют 2,6-лутидин (0,03 мл, 0,26 ммоль). Смесь перемешивают при комнатной температуре в течение 5 минут, затем добавляют TBTU (20 мг, 0,062 ммоль) и перемешивание продолжают в течение 3 часов. Реакционную смесь фильтруют через колонку (элюент: ДХМ/EtOAc, 3:1). Трет-Бутиловый эфир после этого растворяют в ДХМ (6 мл) и добавляют ТФУК (1 мл). Смесь перемешивают при комнатной температуре в течение ночи и растворители выпаривают. Добавляют толуол и выпаривают его, и повторяют эту процедуру. Получают указанное в заголовке соединение (40 мг, 93 %), которому не требуется дополнительная очистка. ЯМР (500 МГц, ДМСО-d6) 0,75 (м, 6H), 0,95-1,50 (м, 12H), 2,16 (с, 3H), 3,25 (м, 2H), 3,75 (м, 2H), 3,90 (дд, 1Н), 4,73/4,84 (АВкв, 2H), 5,54 (м, 2H), 5,58 (д, 1H), 6,68 (с, 1H), 6,85 (т, 1H), 6,99 (д, 2H), 7,18-7,46 (м, 13H), 8,51-8,73 (м, 4H).
Пример 71
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-(S)-{α-карбокси-4-гидроксибензил)карбамоилметокси]-2,3,4,5-тетрагидро-1, 5-бензотиазепин
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин (метод 22; 61 мг, 0,12 ммоль) и метил (2S)-амино(4-гидроксифенил)ацетат гидрохлорид (31 мг, 0,14 ммоль) растворяют в ДХМ (4 мл) и добавляют 2,6-лутидин (0,04 мл, 0,34 ммоль). Смесь перемешивают при комнатной температуре в течение 5 минут, затем добавляют TBTU (53 мг, 0,17 ммоль) добавляют и перемешивание продолжают в течение 2 часов. Реакционную смесь фильтруют через небольшую колонку. Сырой сложный метиловый эфир растворяют в ТГФ (1,5 мл) и добавляют воду (1,0 мл) и NaOH (водн., 1 M, 0,39 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 8 часов, реакцию гасят HCl (1 M) и экстрагируют ДХМ (3 x 5 мл). Собранные органические слои концентрируют и очищают препаративной ВЭЖХ (элюент: MeCN/ацетат-аммониевый буфер, 50:50), получая указанное в заголовке соединение (57 мг, 72 %). ЯМР (500 МГц, CD3OD) 0,81 (м, 6H), 1,05-1,26 (м, 8H), 1,40-1,55 (м, 4H), 2,17 (с, 3H), 3,24 (ушир.с, 2H), 3,74 (ушир.с, 1Н), 4,66 (АВкв, 2H), 6,70-6,75 (м, 3H), 6,99 (т, 1Н), 7,11 (д, 2H), 7,22-7,30 (м, 4H), 7, 40 (с, 1H).
Пример 72
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-(S)-{α-[N' -(2-сульфоэтил)карбамоил]-4-гидроксибензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин аммониевая соль
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-[N-(S)-(α -карбокси-4-гидроксибензил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин (пример 71; 31 мг, 0,047 ммоль) и тетрабутиламмоний таурин (57 мг, 0,155 ммоль) растворяют в ДХМ (2 мл). Смесь перемешивают при комнатной температуре в течение 5 минут, затем добавляют TBTU (24 мг, 0,075 ммоль) и перемешивание продолжают в течение 6 часов. Растворитель выпаривают и остаток очищают препаративной ВЭЖХ (дважды, для удалений всей тетрабутиламмониевой соли) (элюент: MeCN/ацетат-аммониевый буфер, 45:55), получая указанное в заголовке соединение (6 мг, 16%). M/z 762,2.
Примеры 73-74
1,1-Диоксо-3-(R/S)-3-бутил-3-этил-5-фенил-7-метилтио-8-(N-(R)-{α-[N' -(R)-(2-имидазол-5-ил-1-карбоксиэтил)карбамоил]бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-[N -((R)-1'-фенил-1'-карбоксиметил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин (пример 38; 56,4 мг, 0,092 ммоль) и метил D-гистидинат дигидрохлорид (25,2 мг, 0,104 ммоль) добавляют к ДХМ (3 мл). Затем добавляют N-метилморфолин (0,05 мл, 0,41 ммоль) и TBTU (40 мг, 0,12 ммоль). Реакционную смесь перемешивают при температуре 4°C в течение 1,5 часов и при комнатной температуре в течение 3 часов. Добавляют дополнительное количество TBTU (15 мг, 0,047 ммоль) и DIPEA (0,025 мл, 0,14 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение дополнительных 30 минут. Растворитель выпаривают и остаток фильтруют через небольшую колонку (элюент: MeOH). Сырой сложный метиловый эфир растворяют в ТГФ (1,0 мл) и добавляют воду (1,0 мл) и NaOH (водн., 1 M, 0,15 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов и реакцию гасят HCl (1 M). Растворители выпаривают, остаток очищают препаративной ВЭЖХ (элюент: MeCN/ацетат-аммониевый буфер). Соединение элюируют в виде двух пиков, которые объясняют наличием двух диастереомеров. Первый пик (10 мг, 14%). Второй пик (16,8 мг, 24%).
Первый пик: ЯМР (ДМСО-d6) 0,74 (м, 6H), 0,95-1,60 (м, 8H), 2,17 (с, 3H), 2,82 (м, 2H), 3,23 (м, 2H), 4,27 (м, 1H), 4,80 (АВкв, 2H), 5,60 (д, 1H), 6,55 (ушир.с, 1H), 6,70 (с, 1H), 6,84 (т, 1H), 6,96 (д, 2H), 7,14-7,28 (м, 6H), 7,33 (с, 1H), 7,44 (ушир.с, 1Н), 8,54 (д, 1H), 8,60 (ушир.с, 1H); m/z 748,4.
Второй пик: ЯМР (ДМСО-d6) 0,74 (м, 6H), 0,95-1,60 (м, 8H), 2,17 (с, 3H), 2,92 (дАВкв, 2H), 3,23 (м, 2H), 4,41 (м, 1Н), 4,79 (АВкв, 2H), 5,60 (д, 1H), 6,70 (с, 1H), 6,78 (с, 1H), 6,84 (т, 1Н), 6,96 (д, 2H), 7,16-7,34 (м, 6H), 7,40 (м, 2H), 7,55 (с, 1Н), 8,55 (д, 1H), 8,71 (д, 1H); m/z 748,4.
Пример 75
1,1-Диоксо-3,3-дибутил-5-(4-трет-бутилфенил)-7-метилтио-8-(N-{(R)-α-[N' -(карбоксиметил)карбамоил]бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин
Указанное в заголовке соединение выделяют как побочный продукт в синтезе 1,1-диоксо-3, 3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-1'-фенил-1'-[N'-(карбоксиметил)карбамоил]метил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепина (пример 43). Примерно 1 г данного соединения очищают препаративной ВЭЖХ (элюент: MeCN/ацетат-аммониевый буфер, 50:50), получая указанное в заголовке соединение (32 мг). ЯМР (500 МГц, ДМСО-d6) 0,73 (м, 6H), 0,90-Г,40 (м, 12H), 1,24 (с, 9H), 2,16 (с, 3H), 3,23 (м, 2H), 3,65/3,75 (дАВкв, 2H), 4,72/4,82 (АВкв, 2H), 5,60 (д, 1H), 6,65 (с, 1H), 6,97 (д, 2H), 7,23-7,35 (м, 6H), 7,45 (д, 2H), 8,58 (д, 1H), 8,62 (т, 1H).
Пример 76
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-(N-((R)-α-карбоксибензил)карбамоилметилтио)-8-метокси-2,3,4,5-тетрагидро-1, 5-бензотиазепин
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-карбоксиметилтио-8-метокси-2,3,4,5-тетрагидро-1,5-бензотиазепин (метод 81; 38 мг, 0,080 ммоль) и метиловый эфир D-фенилглицина гидрохлорид (24 мг, 0,12 ммоль) растворяют в ДХМ (2 мл) и добавляют N-метилморфолин (0,05 мл, 0,42 ммоль). Смесь перемешивают при комнатной температуре в течение 5 минут, затем добавляют TBTU (44 мг, 0,14 ммоль) и перемешивание продолжают в течение 2 часов. Реакционную смесь фильтруют через небольшую колонку. Полученный продукт растворяют в ТГФ (1 мл), добавляют воду (1 мл) и NaOH (водн., 0,2 мл, 1 M) и реакционную смесь перемешивают при комнатной температуре в течение 2 часов. Реакцию гасят добавлением HCl (1 M), разбавляют смесь водой (10 мл) и экстрагируют ДХМ (3 x 3 мл). Очистка препаративной ВЭЖХ приводит к получению указанного в заголовке соединения (40 мг, 82 %). ЯМР (ДМСО-d6) 0,75 (м, 6H), 0,96-1,60 (м, 8H), 3,22 (м, 2H), 3,56 (АВкв, 2H), 3,89 (с, 3H), 4,81 (д, 1Н), 6,78 (т, 1H), 6,83 (д, 2H), 6,89 (с, 1H), 7,11-7,23 (м, 7H), 7,31 (с, 1H), 8,37 (м, 1H).
Пример 77
1, 1-диоксо-3-бутил-3-этил-5-фенил-7-карбоксиметилтио-8-[N-(α-карбоксибензил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин
1, 1-Диоксо-3-бутил-3-этил-5-фенил-7-этоксикарбонилметилтио-8-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин (метод 82; 21 мг, 0,038 ммоль) и метиловый эфир фенилглицина гидрохлорид (12 мг, 0,061 ммоль) растворяют в ДХМ (1,5 мл) и добавляют N-метилморфолин (0,02 мл, 0,19 ммоль). Смесь перемешивают при комнатной температуре в течение 5 минут, затем добавляют TBTU (18 мг, 0,056 ммоль) и перемешивание продолжают в течение 2 часов. Реакционную смесь фильтруют через небольшую колонку. Сырой диэфир растворяют в ТГФ (1 мл) и добавляют воду (1 мл) и NaOH (водн., 0,1 мл, 1 M). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов, гасят HC1 (1 M), разбавляют водой (10 мл) и экстрагируют ДХМ (3x3 мл). Собранные органические слои концентрируют и очищают препаративной ВЭЖХ (элюент с градиентом: MeCN/ацетат-аммониевый буфер, от 30:70 до 40:60), получая указанное в заголовке соединение (20 мг, 80%). ЯМР (CD3OD) 0,80 (м, 6H), 1,03-1,26 (м, 4H), 1,38-1,65 (м, 4H), 1,96 (с, 3H), 3,20 (с, 2H), 3,44 (с, 2H), 3,67 (ушир.с, 1H), 3,76 (ушир.с, 1H), 4,67 (АВкв, 2H), 5,29 (с, 1H), 6,89 (с, 1H), 6,92 (т, 1Н), 7,05 (д, 2H), 7,19-7,32 (м, 5H), 7,41 (с, 1Н), 7,45 (д, 2H).
Пример 78
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-[N-(R)-(α-{N'-[(R)- N''-(2-гидрокси-1-карбоксиэтил)карбамоилметил]карбамоил}бензил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N -{(R)-1'-фенил-1'-[N'-(карбоксиметил)карбамоил]метил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин (пример 43; 50 мг, 0,072 ммоль), трет-бутил o-(трет -бутил)-D-серинат гидрохлорид (22 мг, 0,087 ммоль) и N-метилморфолин (40 мг, 0,40 ммоль) растворяют в ДХМ (1 мл). Добавляют TBTU (29 мг, 0,090 ммоль) и смесь перемешивают в течение 1 часа при комнатной температуре. Реакционную смесь выпаривают и остаток фильтруют через небольшую колонку (элюент: ДХМ/EtOAc, 1:4). Полученное вещество (˜60 мг) растворяют в ДХМ (1 мл). Добавляют ТФУК (0, 59 г, 5,2 ммоль) и смесь перемешивают в течение 2 часов при комнатной температуре. Растворитель выпаривают и остаток очищают препаративной ВЭЖХ (элюент: MeCN/ацетат-аммониевый буфер, 50:50). Получают 38 мг (72%) указанного в заголовке соединения. ЯМР (300 МГц, ДМСО-d6) 0,7-0,8 (м, 6H), 0,9-1,5 (м, 12H), 2,2 (с, 3H), 3,2-3,9 (м, 10H), 4,2 (ушир.с, 1H), 4,8 (АВкв, 2H), 5,6 (д, 1H), 6,7 (с, 1H), 6,8-7,5 (м, 11H), 8,0 (д, 1Н), 8,6 (д, 1H), 8,7 (т, 1Н).
Пример 79
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-[N -(R)-(α-{N'-[(S)-N"-(2-гидрокси-1-карбоксиэтил)карбамоилметил]карбамоил}бензил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин
1,1-Диоксо-3, 3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-l'-фенил-l'-[N'-(карбоксиметил)карбамоил]метил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин (пример 43; 50 мг, 0,072 ммоль), трет- бутил O-(трет-бутил)-L-серинат гидрохлорид (22 мг, 0,087 ммоль) и N-метилморфолин (40 мг, 0,40 ммоль) растворяют в ДХМ (1 мл). Добавляют TBTU (29 мг, 0,090 ммоль) и смесь перемешивают в течение 1 часа при комнатной температуре. Реакционную смесь выпаривают и остаток фильтруют через небольшую колонку (элюент: ДХМ/EtOAc, 1:4). Полученное вещество (˜ 60 мг) растворяют в ДХМ (1 мл). Добавляют ТФУК (0,44 г, 3,9 ммоль) и смесь перемешивают в течение 18 часов при комнатной температуре. Растворитель выпаривают и остаток очищают препаративной ВЭЖХ (элюент: MeCN/ацетат-аммониевый буфер, 50:50). Получают 33 мг (63%) указанного в заголовке соединения. ЯМР (300 МГц, ДМСО-d6) 0,7-0,8 (м, 6H), 0,9-1,5 (м, 12H), 2,2 (с, 3H), 3,2-3,9 (м, 10H), 4,2 (м, 1Н), 4,8 (АВкв, 2H), 5,6 (д, 1H), 6,7 (с, 1H), 6,8-7,5 (м, 11H), 7,9 (д, 1H), 8,6 (д, 1Н), 8,7 (т, 1Н).
Пример 80
1, 1-Диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-(N-{(R)-α-[N'-(1,1-дикарбоксиметил)карбамоил]бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин
1, 1-Диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-[N-((R)-α-карбоксибензил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин (пример 38; 50 мг, 0,082 ммоль), диметиламиномалонат (60 мг, 0,13 ммоль) и N-метилморфолин (55 мкл,0,5 ммоль) растворяют в ДХМ (3 мл), добавляют TBTU (42 мг, 0,13 ммоль) и смесь перемешивают в течение 15 минут. Растворитель выпаривают при пониженном давлении. Остаток растворяют в этаноле (95%) (2 мл) и добавляют раствор NaOH (80 мг, 2 ммоль) в воде (80 мкл). Реакционную смесь перемешивают в течение 4 часов. Смесь нейтрализуют уксусной кислотой. Растворитель выпаривают при пониженном давлении и остаток очищают препаративной ВЭЖХ (элюент: MeCN/ацетат-аммониевый буфер, 50:50). Собранные фракции лиофилируют, получая 4 мг (7%) указанного в заголовке соединения. ЯМР (300 МГц, CD3OD) 0,75-0,9 (м, 6H), 1,0-1,3 (м, 4H), 1,4-1,65 (м, 4H), 2,15 (с, 3Н) 3,25 (с, 2Н) 3,7 (ушир.с, 2H), 4,65-4,8 (м, 2H), 5,75 (с, 1H), 6,75 (с, 1Н), 6, 9-7,55 (м, 11H).
Примеры 81-87
Представленные ниже соединения получают в соответствии с методикой примера 80 из соответствующих исходных веществ, но с тем отличием, что вместо N-метилморфолина используют 2,6-лутидин и изменяется соотношение компонентов элюента MeCN/аценатноаммониевый буфер (45:55). Незначительно изменяют продолжительность каждой стадии.
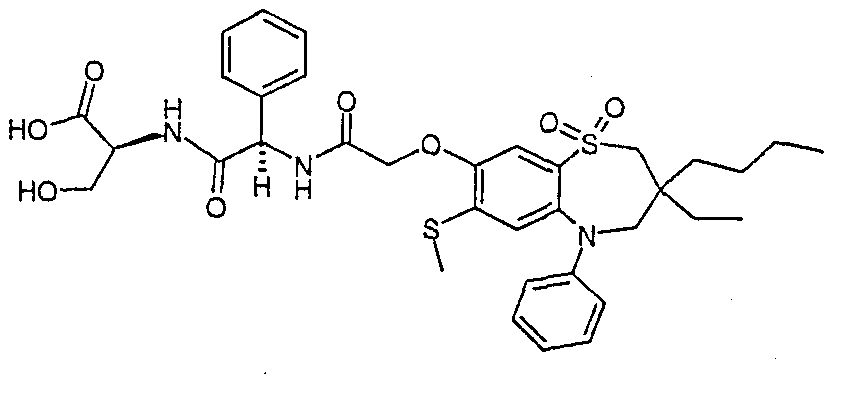
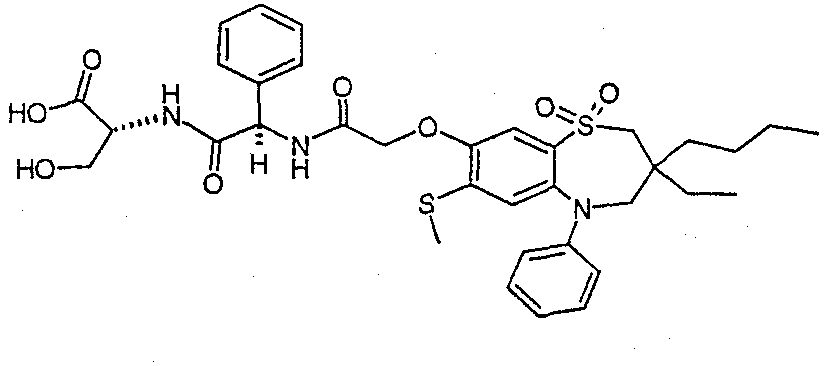

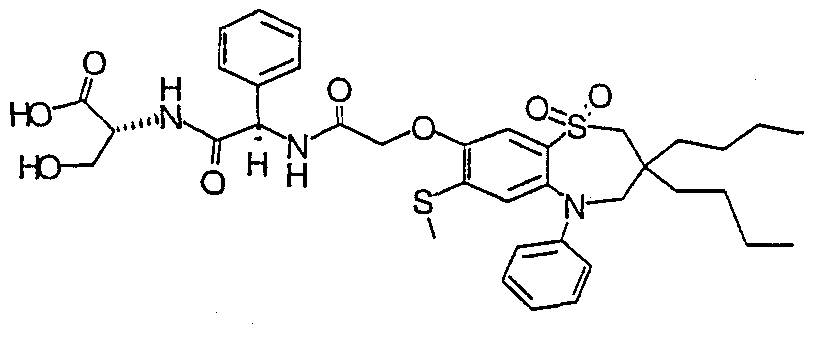

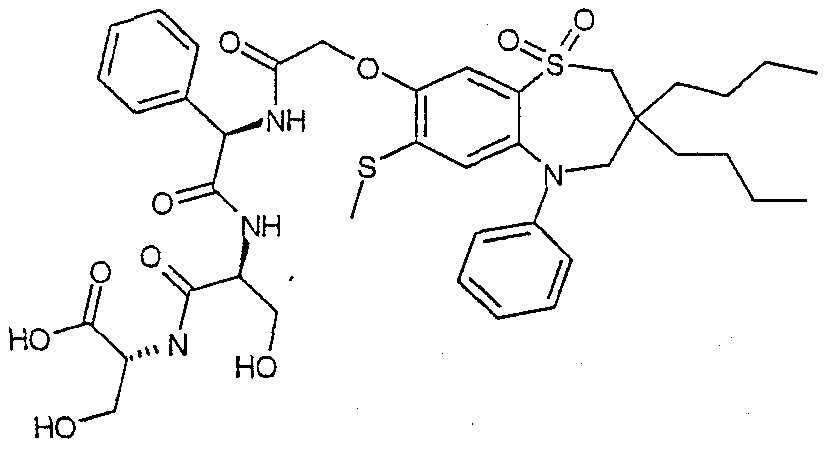
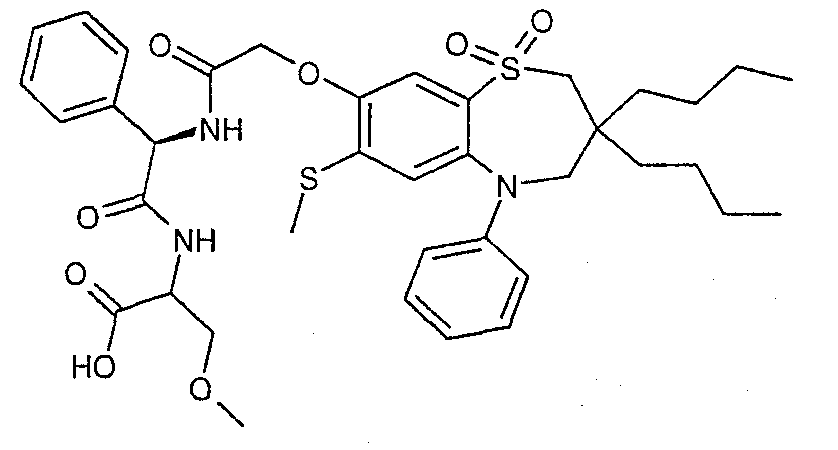
Пример 88
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-[N-(S)-(α -карбоксибензил)карбамоилметоксиl-2,3,4,5-тетрагидро-1,5-бензотиазепин
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-[N-(S)-(α-метоксикарбонилбензил)карбамоилметокси]-2, 3,4,5-тетрагидро-1,5-бензотиазепин (метод 87; 55,2 мг, 0,064 ммоль) растворяют в ТГФ (2 мл) и 0,5 мл воды. Добавляют LiOH (3,1 мг, 0,127 ммоль) и смесь перемешивают в течение 1 часа. Добавляют воду (1 мл), смесь подкисляют 0,1M HCl и экстрагируют ДХМ (3 x 2 мл). Фазу ДХМ сушат и концентрируют. Твердый продукт выпаривают в диэтиловом эфире и растворяют в воде (сорта: чистая для ВЭЖХ). Лиофилизация приводит к получению указанного в заголовке соединения в виде твердого белого вещества (28 мг, 68%). ЯМР 0,77-0,85 (м, 6H), 1,03-1,25 (м, 8H), 1,34-1,57 (м, 4Н) 2,16 (с, 3H), 3,18 (ушир.с, 2H), 3,75 (ушир.с, 2H), 4,65 (АВкв, 2H), 5,7 (д, 1H), 6,63 (с, 1Н), 7,0 (т, 1H), 7,1 (д, 2H), 7,26-7,48 (м, 8H), 7,85 (д, 1H); m/z 639.
Пример 89
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-[N-{(S)-α-[N'-(карбоксиметил)карбамоил]бензил}карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-[N-{(S)-α-[N'-(метоксикарбонилметил)карбамоил]бензил}карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин (метод 88; 19 мг, 0,027 ммоль) гидролизуют с помощью LiOH (1,3 мг, 0,054 ммоль) в ТГФ (1 мл) и воду (0,3 мл). Спустя 1 час добавляют воду (3 мл), смесь подкисляют, используя 0,1M HC1, и экстрагируют ДХМ (3 x 3 мл). Органический слой сушат и выпаривают, получая 16 мг (82%) указанного в заголовке соединения. ЯМР 0,77-0,85 (м, 6H), 1,0-1,3 (м, 8H), 1,34-1,57 (м, 4H), 2,17 (с, 3H), 3,18 (с, 2H), 3,75 (ушир.с, 2H), 3, 90-4,20 (м, 2H), 4,65 (АВкв, 2H), 5,87 (м, 1H), 6,63 (с, 1H), 6,98-7,50 (м, 12H), 8,12-8,20 (м, 1H); m/z 696.
Пример 90
1, 1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-[N-{(S)-α-[N'-(2-сульфоэтил)карбамоил]бензил}карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин натриевая соль
1, 1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-[N-(S)-(α-карбоксибензил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин (пример 88; 41 мг, 0,064 ммоль) растворяют в 3 мл ДХМ. Добавляют тетрабутиламмониевую соль таурина (70 мг, 0,191 ммоль) и TBTU (25 мг, 0,078 ммоль) и смесь перемешивают в течение ночи при комнатной температуре. Растворитель выпаривают и продукт очищают препаративной ВЭЖХ (элюент с градиентом: MeCN/ацетат-аммониевый буфер, от 45:55 до 55:45). Лиофилизация собранных фракций и затем ионно-обменная хроматография над 4 г Amberlite CG 120 (форма Na+) приводит к получению указанного в заголовке соединения (42 мг, 85%). ЯМР 0,7-0,8 (м, 6H), 0,9-1,2 (м, 8H), 1,3-1,5 (м, 4H), 2,0 (с, 3H), 2,9-3,2 (м, 2H+2H), 3,3-3,8 (м, 2H+2H), 4,4-4,7 (м, 2H), 5,6 (м, 1H), 6,57 (с, 1H), 6,9-7,5 (м, 11H), 7,8-8,1 (м, 2H); m/z 746.
Пример 91
Представленное ниже соединение получают в соответствии с методикой примера 90 из соответствующих исходных веществ, но с тем отличием, что продукт очищают с использованием буферного элюента с градиентом от 40/60 до 70/30, и последующая лиофилизация приводит к получению аммониевой соли.

Пример 92
1, 1-Диоксо-3-бутил-3-этил-5-фенил-7-[N-{(R)-α-[N'-(карбоксиметил)карбамоил]бензил}карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин натриевая соль
1, 1-Диоксо-3-бутил-3-этил-5-фенил-7-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин, натриевую соль (WO 01/66533; 120 мг, 0,278 ммоль), растворенную в ДХМ (4 мл), добавляют к раствору α-[ N-(трет-бутоксикарбонилметил)карбамоил]бензиламина (метод 86; 90%, 150 мг, 0,511 ммоль) в ДХМ (3 мл). Добавляют 2,6-диметилпиридин (65 мл, 0,559 ммоль) и TBTU (137 мг, 0,427 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение ночи. Раствор фильтруют через колонку (элюент: ДХМ/EtOAc, 8:2). Растворитель выпаривают. Добавляют ДХМ (4 мл) и ТФУК (0,6 мл) и смесь перемешивают в течение ночи. Растворитель выпаривают и сырой продукт очищают препаративной ВЭЖХ на колонке с наполнителем Chromasil C18, в качестве подвижной фазы использют смесь MeCN/ацетат-аммониевый буфер с градиентом (от 50:50 до 100:0). MeCN выпаривают, и лиофилизация приводит к получению указанного в заголовке соединения (62 мг, 36%). ЯМР 0,73-0,82 (м, 6H), 1,00-1,23 (м, 4H), 1,30-1,65 (м, 4H), 3,05-3,18 (м, 2B), 3,65 (ушир.с, 2H), 3,75 (АВдд, 2H), 4,46 (АВкв, 2H), 5,70 (д, 1Н), 6,79-7,24 (м, 10H), 7,36 (д, 2H), 7,46 (д, 1Н), 7,83 (д, 1H), 8,00 (ушир.с, IB); m/z 622.
Пример 93-94
Представленные ниже соединения получают в соответствии с методикой примера 92 из соответствующих исходных веществ, но с тем отличием, что ВЭЖХ осуществляют на колонке с наполнителем Chromasil C8(элиюрование с градиентом: от 45/55 до 60/40).


Пример 95
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-{N-[(R)-α -(N'-{2-[(этокси)(метил)фосфорил]этил}карбамоил)бензил]карбамоилметокси}-2,3,4,5-тетрагидро-1,5-бензотиазепин
К раствору 2-[(метил)(этил)фосфорил]этиламина (Helv.Chim.Acta; GE; 75; 8; 1992; 2545-2552; 16 мг, 0,106 ммоль) в ДХМ (2 мл) при температуре 0°C в атмосфе аргона добавляют 1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-[N -((R)-1'-фенил-1'-карбоксиметил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин (пример 38; 50 мг, 0,082 ммоль), DIPEA (42 мг, 0,328 ммоль) и TBTU (34 мг, 0,106 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 110 минут, затем добавляют ДХМ и раствор промывают NaHCO3 (водн., насыщ.) и насыщенным раствором соли. Органический слой сушат и растворитель выпаривают при пониженном давлении. Остаток очищают хроматографией, элюируя продукт смесью ДХМ/метанол (100:5). Получают 43 мг (71%) указанного в заголовке соединения. ЯМР (500 МГц) 0, 78-0,85 (м, 6 H), 1,02-1,54 (м, 12H), 1,6-1,75 (ушир., 1H), 1,8-2,10 (м, 3H), 2,21 (с, 3H), 3,10-3,25 (м, 2Н), 3,51-3,84 (м, 4 Н), 3,9-3,99 (м, 1Н), 4,01-4,09 (м, 1Н), 4,54-4,69 (дд, 2H), 5,51 (д, 1H), 6,68 (с,1 H), 6,96-7,02 (м, 1H), 7,03-7,18 (м, 3H), 7,25-7,42 (м, 6H), 7,43-7,48 (м, 2H), 8,05-8,15 (м, 1H).
Примеры 96-97
Представленные ниже соединения получают в соответствии с методикой примера 95, используя соответствующие исходные вещества.
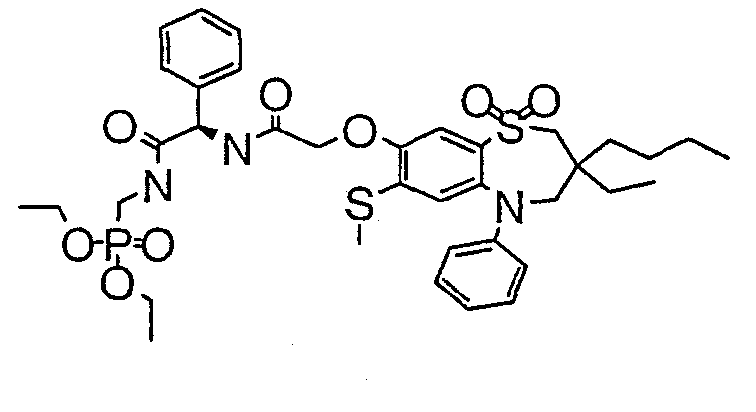

Пример 98
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-{N-[(R)-α-(N' -{2-[{гдрокси)(метил)фосфорил]этил}карбамоил)бензил]карбамоилметокси}-2,3,4,5-тетрагидро-1,5-бензотиазепин
К раствору 1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-{N-[(R)-α -(N'-{2-[(этокси)(метил)фосфорил]этил}карбамоил)бензил]карбамоилметокси}-2,3,4,5-тетрагидро-1,5-бензотиазепин (пример 95; 27 мг, 0,036 ммоль) в этаноле (1,5 мл) при температуре 0°C добавляют 2 M водный раствор NaOH (0,22 мл, 0,44 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 24 часов. Добавляют уксусную кислоту (0,2 мл). Растворитель выпаривают при пониженном давлении и остаток экстрагируют смесью ДХМ/вода. Слой ДХМ отделяют, промывают насыщенным раствором соли, сушат и выпаривают при пониженном давлении. Перекристаллизация остатка из смеси ДХМ/эфир/петролейный эфир приводит к получению указанного в заголовке соединения (23 мг, 89%). (600 МГц) 0,74-0,82 (м, 6H), 1,0-1,70 (м, 11H), 1,90-2,09 (м, 2H), 2,16 (с, 3H), 3,05-3,24 (м, 2H), 3, 40-3,85 (м, 4H), 4,50-4,65 (дд, 2H), 5,55 (д, 1H), 6,63 (с, 1H), 6,93-7,07 (м, 3H), 7,20-7,50 (м, 9H), 8,10 (д, 1H); m/z 716,3.
Пример 99
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-[N-((R)-α-{N'-[(гидрокси)(этокси)фосфорилметил]карбамоил}бензил)карбамоилметокси]-2,3,4,5-тетрагидро-1, 5-бензотиазепин
К раствору 1,1-Диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-[N-((R)-α-{N'-[(диэтокси)фосфорилметил]карбамоил}бензил)карбамоилметокси]-2,3,4, 5-тетрагидро-l,5-бензотиазепина (пример 96; 13 мг, 0,017 ммоль) в MeCN (0,5 мл) по каплям добавляют 1 M водный раствор LiOH (0,171 мл, 0,171 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 3 дней. Добавляют уксусную кислоту и растворитель выпаривают при пониженном давлении. Сырой продукт очищают препаративной ВЭЖХ (элюент: MeCN/ацетат-аммониевый буфер, 45:55), получая указанное в заголовке соединение 11 мг (88%). ЯМР (600 МГц, CD3OD) 0,77-0,84 (м, 6H), 1,00-1,30 (м, 7H), 1,40-1,65 (м, 4H), 2,17 (с, 3H), 3,23 (ушир.с, 2H), 3,51 (д, 2H), 3,6-3,85 (м, 4H), 4,70 (дд, 2H), 5,57 (с, 1H), 6,72 (с, 1H), 6,96 (т, 1Н), 7,09 (д, 2H), 7,25-7,31 (м, 3H), 7,32-7,36 (м, 2H), 7,40 (с, 1H), 7,45 (д, 2H); m/z 732,4.
Пример 100
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-[N-((R)-α-{N'-[(гидрокси)(метил)фосфорилметил]карбамоил}бензил)карбамоилметокси]-2,3,4, 5-тетрагидро-1,5-бензотиазепин
К раствору 1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-[N-((R)-α-{N' -[(этокси)(метил)фосфорилметил]карбамоил}бензил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепина (пример 97; 85 мг, 0,12 ммоль) в MeCN (2,4 мл) при температуре 0°C по каплям добавляют 1 M водный раствор LiOH (1,17 мл, 1,17 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 20 часов. Добавляют уксусную кислоту и растворитель выпаривают при пониженном давлении. Сырой продукт очищают колоночной хроматографией (элюент: ДХМ/MeOH/Et3N; 100:15:0,2 и 100:30:0,2), получая указанное в заголовке соединение (62 мг, 76%). ЯМР (CD3OD) 0,75-0,84 (м, 6H), 1,0-1,70 (м, 11H), 2,15 (с, 3H), 3,22 (ушир.с, 2H), 3,35 (д, 2H), 3,60-3,90 (м, 2H), 4,70 (дд, 2H), 3,55 (с, 1H), 6,71 (с, 1H), 6,96 (т, 1 H), 7,09 (д, 2H), 7,23-7,38 (м, 5H), 7,40 (с, 1H), 7,46 (д, 2H); m/z 702,3
Пример 101
Представленные ниже соединения получают в соответствии с методикой примера 100, используя соответствующие исходные вещества.
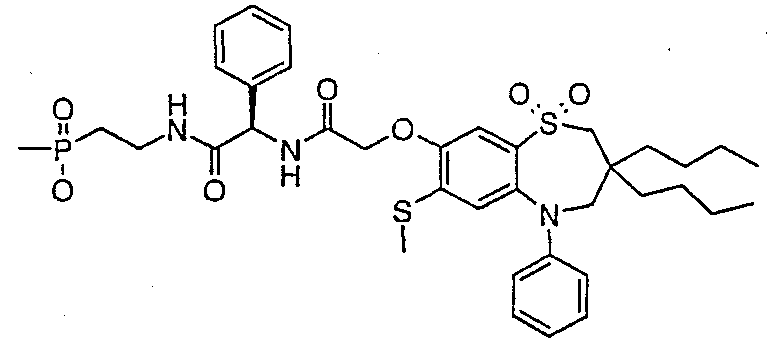
Пример 102
1, 1-Диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-[N-((R)-α-{N'-[ди-(трет-бутокси)фосфорилметилlкарбамоил}бензил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин
К раствору 1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-[N-((R)-1'-фенил-1'-карбоксиметил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепина (пример 38; 80 мг, 0,131 ммоль) и ди(трет-бутокси)фосфорилметиламина (Tet. Lett.; EN; 33; 1; 1992; 77-80; 37 мг, 0,164 ммоль) в ДХМ (5 мл) добавляют 2,6-лутидин (28 мг, 0,262 ммоль) и TBTU (53 мг, 0,164 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов 50 минут. Растворитель выпаривают при пониженном давлении и сырой продукт очищают колоночной хроматографией (элюент: ДХМ/MeOH, 100:4), получая указанное в заголовке соединение (92 мг, 86%). ЯМР (500 МГц) 0,77-0,86 (м, 6H), 1,03-1,75 (м, 26H), 2,22 (с, 3H), 2,10-2,25 (м, 2H), 3,45-3,90 (м, 4H), 4,61 (дд, 2H), 5,52 (д, 1H), 5,94 (ушир.с, 1H), 6,67 (с, 1H), 7,0 (т, 1H), 7,07 (д, 2H), 7,26-7,48 (м, 8H), 8,12 (д, 1H); m/z 704,22 [M-2(трет-бутил)+2H].
Пример 103
1, 1-Диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-[N-((R)-α-{N'-[ди(гидрокси)фосфорилметил]карбамоил}бензил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин
К раствору 1, 1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-[N-((R)-α-{N'-[ди-(трет-бутокси)фосфорилметил]карбамоил}бензил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепина (пример 102; 72 мг, 0,088 ммоль) в ДХМ (4 мл) при температуре 0°C добавляют ТФУК (1 мл). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов. Растворитель выпаривают при пониженном давлении и остаток экстрагируют смесью ДХМ/вода. Органический слой отделяют, промывают насыщенным раствором соли, сушат и выпаривают при пониженном давлении. Остаток суспендируют в эфире и кристаллы отфильтровывают, получая указанное в заголовке соединение (60 мг, 97%). ЯМР (500 МГц, ДМСО-d6) 0,70-0,80 (м, 6H), 0,99-1,61 (м, 8H), 2,18 (с, 3H), 2,80-4,0 (м, 6H), 4,80 (дд, 2H), 5,65 (д, 1Н), 6,71 (с, 1H), 6,80-7,02 (м, 3H), 7,15-7,35 (м, 6H), 7,48 (д, 2H), 8,50-9,20 (м, 2H); m/z 704,3
Пример 104
1,1-Диоксо-3, 3-дибутил-5-фенил-7-метилтио-8-{N-[(R)-α-(N'-{2-[(метил)(этил)фосфорил]этил}карбамоил)бензил]карбамоилметокси}-2,3,4,5-тетрагидро-1,5-бензотиазепин
К раствору 1, 1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-[N-((R)-l'-фенил-l'-карбоксиметил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин (пример 1; 60 мг, 0,094 ммоль) и 2-[(метил)(этил)фосфорил]этиламин (Helv. Chim. Acta; GE; 75; 8; 1992; 2545-2552; 20 мг, 0,132 ммоль) при температуре 0°C в атмосфере аргона добавляют 2,6-лутидин (20 мг, 0,19 ммоль) и TBTU (39 мг, 0,121 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 70 минут, затем добавляют ДХМ и раствор промывают водой и насыщенным раствором соли. Органический слой сушат и растворитель выпаривают при пониженном давлении. Остаток очищают колоночной хроматографией (элюент: ДХМ/MeOH, 100:5), получая указанное в заголовке соединение (67 мг, 92%). ЯМР (300 МГц) 0,74-86 (м, 6H), 1,0-1,60 (м, 18H), 1,80-2,05 (м, 2H), 2,20 (с, 3H), 2,17 (с, 2H), 3,47-3,80 (м, 4H), 3,8S-4,10 (дд, 2H), 5,52 (д, 1H), 6,65 (с, 1H), 6,95-7,12 (м, 3H), 7,13-7,42 (м, 7H), 7,43-7,49 (м, 2H), 8, 05-8,16 (м, 1H); m/z 772,4.
Пример 105
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N' -(2-меркапто-1-карбоксиэтил)карбамоил]бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин
К раствору 1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-[N-((R)-α -{N'-[2-(трифенилметилсульфанил)-1-(трет-бутоксикарбонил)этил]карбамоил}бнзил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепина (метод 91; 37 мг, 0,036 ммоль) в ДХМ (1 мл) в атмосфере аргона добавляют при температуре 0°C добавляют ТФУК (1 мл). Ледяную баню удаляют и добавляют триэтилсилан (42 мг, 0,36 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов и после этого растворитель выпаривают при пониженном давлении. Сырой продукт очищают препаративной ВЭЖХ (элюент с градиентом: MeCN/ацетат-аммониевый буфер, от 40:60 до 60:40), получая указанное в заголовке соединение (16 мг, 59%). ЯМР (500 МГц, CD3OD) 0,76-0,85 (м, 6H), 1,05-1,60 (м, 12H), 2,17 (с, 3H), 2,77-2,92 (м, 2H), 3,24 (ушир.с, 2H), 3,61-3,88 (м, 2H), 4,56 (т, 1H), 4,70 (дд, 2H), 5,65 (с, 1H), 6,71 (с, 1H), 6,98 (т, 1H), 7,12 (д, 2H), 7,25-7,43 (м, 6H), 7,50 (д, 2H); m/z 742,4.
Пример 106
Соединение, представленное ниже, получают в соответствии с методикой примера 105, используя соответствующие исходные вещества.
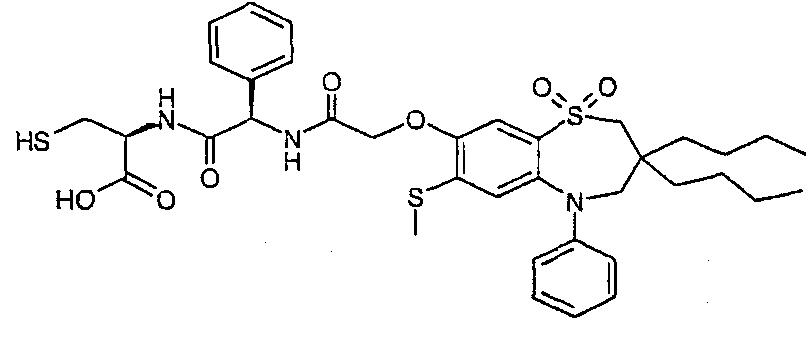
Пример 107
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-бром-8-(2-{N-[(R)-α-(карбокси)бензил]карбамоил}этокси)-2,3,4, 5-тетрагидро-1,5-бензотиазепин
К раствору 1,1-диоксо-3-бутил-3-этил-5-фенил-7-бром-8-(2-{N-[(R)-α-(трет-бутоксикарбонил)бензил]карбамоил}эокси)-2,3,4,5-тетрагидро-1, 5-бензотиазепина (метод 90; 77 мг, 0,108 ммоль) в ДХМ (3 мл) при температуре 0°C добавляют ТФУК (0,75 мл). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов 45 минут. Растворитель выпаривают при пониженном давлении и сырой продукт очищают препаративной ВЭЖХ (элюент с градиентом: MeCN/ацетат-аммониевый буфер, от 40:60 до 50:50), получая указанное в заголовке соединение (60 мг, 82%). ЯМР (500МГц, CD3OD) 0,75-0,85 (м, 6H), 1,0-1,25 (м, 4H), 1,40-1,64 (м, 4H), 2,75-2,90 (м, 2H), 3,26 (с, 2H), 3,50-3,90 (м, 2H), 4,30-4,41 (м, 2H), 5,43 (с, 1H), 6, 99 (т, 1Н), 7,05-7,13 (м, 3H), 7,23-7,34 (м, 5H), 7,45 (д, 2H), 7,52 (с, 1H); m/z 658.
Пример 108
Соединение, представленное ниже, получают в соответствии с методикой примера 107, используя соответствующие исходные вещества.
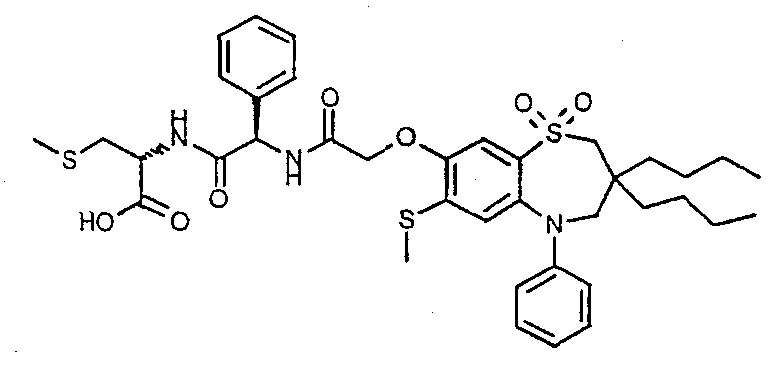
Пример 109
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-{N-[(R)-α-( N'-{2-[(метил)(этил)фосфорил]этил}карбамоил)-4-гидроксибензил]карбамоилметокси}-2,3,4,5-тетрагидро-1,5-бензотиазепин
К раствору 1,1-диоксо-3, 3-дибутил-5-фенил-7-метилтио-8-[N-((R)-α-карбокси-4-гидроксибензил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепина (пример 2; 80 мг, 0,122 ммоль) и 2-[(метил)(этил)фосфорил]этиламина (Helv. Chim. Acta; GE; 75; 8; 1992; 2545-2552; 24 мг, 0,159 ммоль) в ДХМ (2 мл) под атмосферой аргона добавляют 2,6-лутидин (26 мг, 0,244 ммоль) и TBTU (51 мг, 0,159 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 60 минут, затем разбавляют ДХМ. Раствор промывают водой, насыщенным раствором соли, сушат и растворитель выпаривают при пониженном давлении. Остаток очищают колоночной хроматографией (элюент: ДХМ/MeOH; 100:7), получая указанное в заголовке соединение (67 мг, 92%). ЯМР (600 МГц), 0,74-0,80 (м, 6H), 1,0-1,55 (м, 18H), 1,82-1-98 (м, 2H), 2,15 (с, 3H), 3,14 (ушир.с, 2H), 3,40-3,56 (м, 2H), 3,70 (ушир.с, 2H), 3,89-4,02 (м, 2H), 4,51 (дд, 2H), 5,33 (т, 1Н), 6,61 (с, 1Н), 6,65-6,72 (м, 2H), 6,95 (т, 1H), 7,03 (д, 2H), 7,12-7,19 (м, 3H), 7,22-7,26 (м, 2H), 7,32 (с, 1H), 8,11 (т, 1H); m/z 788,56.
Пример 110
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-{N-[(R)-α-( N'-{2-[(метил)(гидрокси)фосфорил]этил}карбамоил)-4-гидрокси-бензил]карбамоилметокси}-2,3,4,5-тетрагидро-1,5-бензотиазепин
К раствору 1,1-диоксо-3, 3-дибутил-5-фенил-7-метилтио-8-{N-[(R)-α-(N'-{2-[(метил)(этил)фосфорил]этил}карбамоил)-4-гидроксибензил]карбамоилметокси}-2,3,4,5-тетрагидро-1,5-бензотиазепина (пример 104; 37 мг, 0,047 ммоль) в смеси MeCN/MeOH (4 мл, 1:1) добавляют 1 M водного раствора LiOH (0,8 мл, 0,8 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 40 минут. Добавляют уксусную кислоту и растворитель выпаривают при пониженном давлении. Сырой продукт очищают препаративной ВЭЖХ (элюент: MeCN/ацетат-аммониевый буфер, 40:60 и 45:55), получая указанное в заголовке соединение (35 мг, 96%). ЯМР (500 МГц, CD3OD) 0,78-0,85 (м, 6H), 1,06-1,28 (м, 11H), 1,39-1,57 (м, 4H), 1,72-1,85 (м, 2H), 2,16 (с, 3H), 2,24 (с, 2H), 3,40-3,50 (м, 2H), 3,65-3,84 (м, 2H), 4,69 (дд, 2H), 5,36 (с, 1H), 6,71 (с, 1Н), 6,76 (д, 2H), 6,99 (т, 1H), 7,13 (д, 2H), 7,22-7,33 (м, 4H), 7,39 (с, 1H); m/z 760,27
Пример 111
1, 1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[(R)-N'-(2-метилсульфинил-1-карбоксиэтил)карбамоил]бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин
Указанное в заголовке соединение выделяют как побочный продукт синтеза примера 108. ЯМР (500 МГц, CD3OD) 0,78-0,85 (м, 6H), 1,02-1,60 (м, 12H), 2,16 (д, 3H), 2,53 (д, 3H), 3,08-3, 18 (м, 1H), 3,24 (с, 2H), 3,35 (v ушир., 1H), 3,75 (v ушир., 2H), 4,62 (v ушир., 1Н), 4,71 (дд, 2H), 5,60 (д, 1H), 7,71 (с, 1H), 6,98 (т, 1Н), 7,12 (д, 2H), 7,25-7,42 (м, 6H), 7,47 (д, 2H); m/z 772, 25.
Пример 112
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[(S)-N' -(3-метилтио-2-карбоксипропил)карбамоил]бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин
К раствору 1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α -[(S)-N'-(3-метилтио-2-метоксикарбонилпропил)карбамоил]бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепина (метод 94; 68 мг, 0,087 ммоль) в этаноле (5 мл) добавляют NaOH (9 мг в 0,4 мл воды) при температуре 0°C. Реакционную смесь перемешивают при комнатной температуре в течение 2,5 часов. Добавляют уксусную кислоту и растворитель выпаривают при пониженном давлении. Сырой продукт очищают препаративной ВЭЖХ (элюент с градиентом: MeCN/ацетат-аммониевый буфер, от 40:60 до 60:40), получая указанное в заголовке соединение (52 мг, 76%). ЯМР (500 МГц, CD3OD) 0,79-0, 86 (м, 6H), 1,05-1,29 (м, 8H), 1,40-1,58 (м, 4H), 1,84-1,93 (м, 4H), 2,01-2,21 (м, 5H), 2,26-2,34 (м, 1H), 3,26 (с, 2H), 3,76 (ушир.с, 2H), 4,52-4,58 (м, 1H), 4,70 (дд, 2H), 5,61 (с, 1Н) 6,73 (с, 1Н), 7,0 (т, 1H), 7,14 (д, 2H), 7,27-7,43 (м, 6H), 7,49 (д, 2H); m/z 770,16.
Пример 113
1,1-Диоксо-3, 3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[(S)-N'-(2-метилтио-1-карбоксиэтил)карбамоил]бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин
К раствору 1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[(S)-N'-(2-меркапто-1-карбоксиэтил)карбамоил]бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепина (пример 106; 15 мг, 0,02 ммоль) в метаноле (1,5 мл) в атмосфере азота добавляют метоксид натрия (0,104 ммоль в 0,14 мл метанола) и метилйодид (0,16 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 50 минут. Добавляют уксусную кислоту. Растворитель выпаривают при пониженном давлении и остаток экстрагируют смесью ДХМ/вода. Органический слой отделяют, промывают насыщенным раствором соли, сушат и выпаривают при пониженном давлении, получая указанное в заголовке соединение (4 мг, 26%). ЯМР (500 МГц, CD3OD) 0,75-8,30 (м, 6H), 1,03-1,57 (м, 12H), 2,10 (с, 3H), 2,17 (с, 3H), 2, 83-2,30 (м, 1Н), 3,0- 3,25 (м, 1H), 3,26 (с, 2H), 3,77 (ушир.с, 2H), 4,58-4,63 (м, 1Н), 4,72 (дд, 2H), 5,64 (с, 1H), 6,72 (с, 1H), 7,0 (т, 1H), 7,12 (д, 2H), 7,28-7,52 (м, 8H); m/z 756,25.
Пример 114
1,1-Диоксо-3,3-дибутил-5-(4-хлорфенил)-7-метилтио-8-[N-{(R)-α-[N' -(карбоксиметил)карбамоил]бензил}карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин
К раствору 1,1-диоксо-3,3-дибутил-5-(4-хлорфенил)-7-метилтио-8-[N-{(R)-α-[ N'-(трет-бутоксикарбонилметил)карбамоил]бензил}карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепина (метод 102; 129 мг, 0,164 ммоль) в ДХМ (5 мл) в атмосфере азота при температуре 0°C добавляют ТФУК (1,5 мл). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов. Растворитель выпаривают при пониженном давлении и сырой продукт очищают препаративной ВЭЖХ (элюент с градиентом: MeCN/ацетат-аммониевый буфер, от 40:60 до 50:50), получая указанное в заголовке соединение (77 мг, 63%). ЯМР (500 МГц, CD3OD) 0,84 (т, 6H), 1,10-1,22 (м, 8H), 1,35-1,45 (м, 4H), 2,34 (с, 3H), 3,19-3,27 (м, 2H), 3,55 (с, 2H), 3,87 (дд, 2H), 4,67 (дд, 2H), 5,61 (с, 1Н), 7,09-7,15 (м, 3H), 7,27-7,37 (м, 6H), 7,47 (д, 2H); m/z 748,03 (M+NH3).
Пример 115
1,1-Диоксо-3,3-дипропил-5-фенил-7-метилтио-8-[N-{(R)-α-[N' -(2-сульфоэтил)карбамоил]-4-гидроксибензил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин
К раствору 1,1-диоксо-3,3-дипропил-5-фенил-7-метилтио-8-карбоксиметокси-2,3,4, 5-тетрагидро-1,5-бензотиазепина (метод 118; 0,050 г, 0,105 ммоль) в ДМФА (4 мл) добавляют 2-{[(2R)-2-амино-2-(4-гидроксифенил)этаноил]амино}этансульфоновую кислоту (метод 80; 0,037 г, 0,135 ммоль) и N-метилморфолин (0,040 мл, 0,363 ммоль). Смесь перемешивают в течение 10 минут и затем добавляют TBTU (0,044 г, 0,137 ммоль). Реакционную смесь перемешивают в течение двух дней, после чего растворитель удаляют при пониженном давлении. Остаток очищают препаративной ВЭЖХ (элюент: MeCN/ацетат-аммониевый буфер), получая указанное в заголовке соединение (0,042 г, 55 %) в виде твердого белого вещества. ЯМР (ДМСО-d6) 0,60-0,80 (м, 6H), 1,05-1,50 (м, SH), 2,15 (с, 3H), 2,45-2,55 (м, 2H), 3,05-3,80 (м, 6H), 4,70 (АВд, 1Н), 4,80 (АВд, 1H), 5,25 (д, 1Н), 6,65-6,75 (м, 3H), 6,80-7,05 (м, 3H), 7,10-7,25 (м, 4H), 7,30 (с, 1H), 8,20-8,30 (м, 1H), 8,45 (д, 1H).
Пример 116
1,1-Диоксо-3, 3-дипропил-5-фенил-7-метилтио-8-[N-{(R)-α-[N'-(карбоксиметил)карбамоил]-4-гидроксибензил}карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин
К раствору 1, 1-диоксо-3,3-дипропил-5-фенил-7-метилтио-8-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепина (метод 118; 0,050 г, 0,105 ммоль) в ДХМ (4 мл) добавляют (R)-α-[N -(трет-бутоксикарбонилметил)карбамоил]бензиламин (метод 86; 0,036 г, 0.136 ммоль) и N-метилморфолин (0,040 мл, 0,363 ммоль). Смесь перемешивают в течение 5 минут и затем добавляют TBTU (0,044 г, 0,137 ммоль). Реакционную смесь перемешивают в течение двух дней, затем добавляют ТФУК (1,5 мл). Спустя 1,5 часа, раствор разбавляют толуолом, затем растворитель удаляют при пониженном давлении. Остаток очищают препаративной ВЭЖХ (элюент: MeCN/ацетат-аммониевый буфер), получая указанное в заголовке соединение (0,020 г, 29 %) в виде твердого белого вещества. ЯМР (ДМСО-d6) 0,60-0,80 (м, 6H), 1,05-1,50 (м, 8H), 2,15 (с, 3H), 3,10-3,80 (м, 6H), 4,70 (АВд, 1H), 4,85 (АВд, 1H), 5,60 (д, 1Н), 6,70 (с, 1H), 6,80-7,05 (м, 3H), 7,15-7,50 (м, 8H), 8,35 (ушир.с, 1H), 8,55 (д, 1H).
Пример 117
1,1-Диоксо-3,3-дибутил-5-фенил-7-метокси-8-[N-{(R)-α-[N' -(2-сульфоэтил)карбамоил]-4-гидроксибензил}карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин
К раствору 1,1-диоксо-3,3-дибутил-5-фенил-7-метокси-8-карбоксиметокси-2,3,4, 5-тетрагидро-1,5-бензотиазепина (метод 6; 0,020 г, 4,09х10-5моль) в ДМФА (4 мл) добавляют 2-{[(2R)-2-амино-2-(4-гидроксифенил)этаноил]амино}эансульфоновую кислоту (метод 80; 0,014 г, 5, 10х10-5моль) и N-метилморфолин (0,020 мл, 1,81х10-4моль). Смесь перемешивают в течение 10 минут и затем добавляют TBTU (0,016 г, 4,98х10-5моль). Реакционную смесь перемешивают в течение 3 часов и затем растворитель удаляют при пониженном давлении. Остаток очищают препаративной ВЭЖХ (элюент: MeCN/ацетат-аммониевый буфер), получая указанное в заголовке соединение (0,023 г, 75 %) в виде твердого белого вещества. ЯМР (500 МГц, ДМСО-d6) 0,65-0,80 (м, 6H), 0,80-1,50 (м, 12H), 2,40-2,60 (м, 2H), 3,15-3,45 (м, 4H), 3,60 (с, 3H), 3,65 (ушир.с, 2H), 4,60 (АВд, 1H), 4,70 (АВд, 1H), 5,25 (д, 1H), 6,50 (с, 1Н), 6,70-7,25 (м, 10H), 7,35 (с, 1H), 8,20-8,30 (м, 1H), 8,50 (д, 1H), 9,40 (ушир.с, 1H).
Пример 118
1,1-диоксо-3-бутил-3-этил-5-фенил-8-[N-{(R)-α-[N'-сульфоэтил)карбамоил]-4-гидроксибензил}карбамоилметокси]-2,3,4,5-тетрагидро-1, 5-бензотиазепин
К раствору 1,1-диоксо-3-бутил-3-этил-5-фенил-8-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепина (метод 115; 0,020 г, 4,63х10-5моль) в ДМФА (4 мл) добавляют 2-{[(2R)-2-амино-2-(4-гидроксифенил)этаноил]амино}этансульфоновую кислоту (метод 80; 0,017 г, 6,20х10-5моль) и N-метилморфолин (0,016 мл, 1,46х10-4моль). Смесь перемешивают в течение 10 минут и затем добавляют TBTU (0,019 г, 5,92х10-5моль). Реакционную смесь перемешивают в течение ночи и затем растворитель удаляют при пониженном давлении. Остаток очищают препаративной ВЭЖХ (элюент: смесь MeCN/ацетат-аммониевый буфер), получая указанное в заголовке соединение (0,008 г, 24 %) в виде твердого белого вещества. ЯМР (500 МГц, ДМСО-d6) 0, 65-0,80 (м, 6H), 0,80-1,60 (м, 8H), 2,40-2,55 (м, 2H), 3,20-3,40 (м, 4H), 3,65 (ушир.с, 2H), 4,65 (АВд, 1H), 4,70 (АВд, 1H), 5,25 (д, 1H), 6,65-7,45 (м, 13H), 8,20-8,30 (м, 1H), 8,60 (д, 1H), 9,40 (ушир.с, 1H).
Пример 119
1,1-Диоксо-3,3-дибутил-5-(4-трет-бутоксикарбониламинофенил)-8-[N-(α -(R)-карбоксибензил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин
Указанное в заголовке соединение синтезируют из 1,1-диоксо-3, 3-дибутил-5-(4-трет-бутоксикарбониламинофенил)-8-[N-(α-(R)-метоксикарбонилlбензил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепина (метод 45) в соответствии с методикой метода 109. ЯМР (CD3OD) 0,81 (ушир.т, 6H), 1,03-1,3 (м, 8H), 1,32-1,59 (м, 13H), 3,24 (ушир.с, 2H), 3,57-3,77 (м, 2H), 4,61 (ушир.с, 2H), 5,51 (с, 1H), 6,83 (д, 1H), 7,0-7,1 (м, 3H), 7,26-7,43 (м, 7H), 7,49 (д, 1H); m/z 708,5.
Пример 120
1,1-Диоксо-3,3-дибутил-5-[4-(N'-трет-бутилуреидо)фенил]-8-[N-(α -(R)-карбоксибензил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин
1,1-Диоксо-3,3-дибутил-5-(4-(N'-трет-бутилуреидо)фенил)-8-[N-(α -(R)-метоксикарбонилlбензил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин (метод 111; 30 мг, 0,042 ммоль) растворяют в ТГФ (1,5 мл), H2O (0,5 мл) и добавляют LiOH (42 мг, 0,064 ммоль, моногидрат) добавляют. Смесь перемешивают в течение 2 часов. Продукт очищают препаративной ВЭЖХ (элюент с градиентом: MeCN/ацетат-аммониевый буфер, от 5:95 до 100:0), получая указанное в заголовке соединение (24 мг, 82%). ЯМР (CD3OD) 0,81 (ушир.т, 6H), 1,05-1,26 (м, 8H), 1,35 (с, 9H), 1,38-1,57 (м, 4H), 3,25 (ушир.с, 2H), 3,6-3,77 (м, 2H), 4,61 (АВкв, 2H), 5,45 (с, 1H), 6, 84 (д, 1H), 7,01-7,11 (м, 3H), 7,24 (д, 2H), 7,26-7,37 (м, 3H), 7,37-7,42 (м, 2H), 7,50 (д, 1H); m/z 707,5.
Получение исходных веществ
Исходные вещества для примеров, описанных выше, являются либо коммерчески доступными, либо могут быть легко получены стандартными методами из известных материалов. Приведенные далее реакции являются иллюстрацией, но не ограничением, получения некоторых исходных веществ, использованных в описанных выше реакциях.
Метод 1
1, 1-диоксо-3-бутил-3-этил-5-фенил-7-бром-8-[1'-(этоксикарбонил)этокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин
К раствору 1,1-диоксо-3-бутил-3-этил-5-фенил-7-бром-8-гидрокси-2,3,4, 5-тетрагидро-1,5-бензотиазепина (WO 96/16051; 0,300 г, 0,663 ммоль) в MeCN (10 мл) добавляют карбонат натрия (0,30 г, 2,83 ммоль), этиловый эфир 2-бромпропановой кислоты (0,145 г, 0,796 ммоль) и тетрабутиламмоний бромид (0,022 г, 0,07 ммоль). Суспензию кипятят с обратным холодильником в течение ночи. Растворитель выпаривают и неочищенную смесь экстрагируют (ДХМ/H20), сушат (MgSO4), выпаривают и очищают флэш-хроматографией (гексан:EtOAc - 5:1), получая указанное в заголовке соединение (0,346 г, 95 %) в виде твердого белого вещества. ЯМР 0,70-0,85 (м, 6H), 1, 00-1,75 (м, 8H), 1,35 (т, 3H), 1,70 (д, 3H), 3,05-3,25 (м, 2H), 3,55-3,90 (м, 2H), 4,20-4,35 (м, 2H), 4,80 (кв, 1H), 7,00-7,10 (м, 3H), 7,15 (с, 1Н), 7,25-7,35 (м, 2H), 7,45 (с, 1H).
Метод 2
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-бром-8-[1'-карбоксиэтокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин
К раствору 1, 1-диоксо-3-бутил-3-этил-5-фенил-7-бром-8-[1-(этоксикарбонил)этокси]-2,3,4,5-тетрагидро-1,5-бензотиазепина (метод 1; 0,050 г, 0,090 ммоль) в EtOH (4 мл, 95 %) добавляют гидроксид натрия (0,045 г, 1,13 ммоль) и полученную смесь кипятят с обратным холодильником в течение 1,5 часа. Добавляют AcOH (0,2 мл) и большую часть растворителя удаляют при пониженном давлении. Сырой продукт экстрагируют (ДХМ/H20), сушат (MgSO4) и выпаривают, получая указанное в заголовке соединение (0,031 г, 65 %) в виде твердого белого вещества. ЯМР (500 МГц, CD3OD) 0,70-0,85 (м, 6H), 0,95-1,25 (м, 4H), 1,35-1,70 (м, 4H), 2,65 (д, 3H), 3,10-3,35 (м, 2H), 3,45-3,95 (м, 2H), 4,70 (кв, 1H), 6,90-7,35 (м, 6H), 7,45 (с, 1H).
Метод 3
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-бром-8-[1'-фенил-1'-этоксикарбонилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин
К раствору 1, 1-диоксо-3-бутил-3-этил-5-фенил-7-бром-8-гидрокси-2,3,4,5-тетрагидро-1,5-бензотиазепина (WO 96/16051; 0,200 г, 0,442 ммоль) в MeCN (6 мл) добавляют этил α-бромфенилацетат (0,139 г), Na2CO3 (0,200 г) и тетрабутиламмоний бромид (0,034 г). Суспензию кипятят с обратным холодильником в течение ночи, затем растворитель удаляют при пониженном давлении. Сырой продукт экстрагируют (ДХМ/H20) и очищают флэш-хроматографией (гексан:EtOAc-5:l) получая указанное в заголовке соединение (0,256 г, 94 %) в виде твердого белого вещества. ЯМР 0,65-0,85 (м, 6H), 0, 95-1,65 (м, 8H), 3,00-3,15 (м, 2H), 3,50-3,80 (м, 2H), 3,70-3,80 (2s, 3H), 5,60 (с, 1H), 5,65 (д, 1H) 7,00-7,60 (м, 17H), 8,05-8,20 (2д, 1H).
Метод 4
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-бром-8-[1'-фенил-1'-карбоксиметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин
К раствору 1, 1-диоксо-3-бутил-3-этил-5-фенил-7-бром-8-[1'-фенил-1'-этоксикарбонилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепина (метод 3; 0,244 г, 0,397 ммоль) в смеси ТГФ/H2O (2/1, 3 мл) добавляют гидроксид лития (0,019 г). Спустя 2 дня растворитель удаляют при пониженном давлении и неочищенную смесь очищают ВЭЖХ, получая указанное в заголовке соединение (0,215 г,92 %) в виде твердого белого вещества. ЯМР (CD3OD) 0,60-0,80 (м, 6H), 0,90-1,25 (м, 4H), 1,30-1,60 (м, 4H), 3,05-3,30 (м, 2H), 3,40-3,90 (м, 2H), 5,55 (с, 1Н), 6,85-7,70 (м, 12H).
Метод 5
1,1-Диоксо-3,3-дибутил-5-фенил-7-метокси-8-этоксикарбонилметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
К раствору 1,1-диоксо-3, 3-дибутил-5-фенил-7-метокси-8-гидрокси-2,3,4,5-тетрагидро-1,5-бензотиазепина (синтезирован в соответствии с методикой публикации W09616051 для соответствующего 3-бутил-3-этилового аналога; 0,400 г, 0, 927 ммоль) в MeCN (10 мл) добавляют этилбромацетат (0,13 мл), Na2CO3 (0,40 г) и тетрабутиламмоний бромид (0,030 г). Суспензию кипятят с обратным холодильником в течение ночи, затем большую часть растворителя удаляют при пониженном давлении. Сырой продукт экстрагируют (ДХМ/H2O) и фильтруют через колонку небольшой высоты с диоксидом кремния (ДХМ:EtOAc-1:4), получая указанное в заголовке соединение (0,476 г, 99 %). ЯМР 0,65-0,85 (м, 6H), 0,95-1,65 (м, 8H), 3,00-3,15 (м, 2H), 3,50-3,80 (м, 2H), 3,70-3,80 (с, 3H), 5,60 (с, 1H), 5,65 (д, 1Н) 7,00-7,60 (м, 17H), 8,05-8,20 (д, 1H).
Метод 6
1,1-Диоксо-3,3-дибутил-5-фенил-7-метокси-8-карбоксиметокси-2,3,4,5-тетрагидро-1, 5-бензотиазепин
К раствору 1,1-диоксо-3,3-дибутил-5-фенил-7-метокси-8-этоксикарбонилметокси-2,3,4,5-тетрагидро-1,5-бензотиазепина (метод 5; 0,448 г, 0,865 ммоль) в смеси ТГФ/H2O (2/1, 6 мл) добавляют гидроксид лития (0,062 г). Спустя 1 час добавляют AcOH (0,5 мл), добавляют и большую часть растворителя удаляют при пониженном давлении. Сырой продукт очищают ВЭЖХ (MeCN), получая указанное в заголовке соединение (0,408 г, 96 %) в виде твердого белого вещества. ЯМР (CD3OD) 0,75-0,85 (м, 6H), 1,00-1,30 (м, 8H), 1,35-1,55 (м, 4H), 3,20 (с, 2H), 3,65 (с, 3H), 3,70 (ушир.с, 2H), 4,50 (с, 2H), 6,50 (с, 1H), 6,90-7,30 (м, 5H), 7,40 (с, 1H).
Метод 7
1, 1-Диоксо-3-бутил-3-этил-5-фенил-7-метокси-8-этоксикарбонилметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-метокси-8-гидрокси-2,3,4,5-тетрагидро-1, 5-бензотиазепин (WO 9616051; 1,0 г), этилбромацетат (0,50 г), карбонат натрия (1,2 г) и тетрабутиламмоний бромид (60 мг) в MeCN (15 мл) кипятят с обратным холодильником в течение ночи. Растворитель удаляют при пониженном давлении и остаток экстрагируют (ДХМ/H2O). Органический слой отделяют и растворитель удаляют при пониженном давлении. Остаток очищают хроматографией (ДХМ/EtOAc, 90:10), получая указанное в заголовке соединение (1,2 г, 98%). ЯМР (CD3OD) 0,75-0,85 (м, 6H), 1,00-1,30 (м, 8H), 1,35-1,55 (м, 4H), 3,20 (с, 2H), 3,65 (с, 3H), 3,70 (ушир.с, 2Н) 4,50 (с, 2H), 6,50 (с, 1Н), 6,90-7,30 (м, 5H), 7,40 (с, 1H).
Метод 8
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-бром-8-этоксикарбонилметокси-2,3, 4,5-тетрагидро-1,5-бензотиазепин
Смесь 1,1-диоксо-3-бутил-3-этил-5-фенил-7-бром-8-гидрокси-2,3,4,5-тетрагидро-1,5-бензотиазепина (WO 96/16051; 0,3 г), этилбромацетата (0,14 г), карбоната натрия (0,3 г), тетрабутиламмония бромида (0,02 г) в MeCN (10 мл) кипятят с обратным холодильником в течение 4 часов. Растворитель удаляют при пониженном давлении. Остаток распределяют между ДХМ/H2O и органический слой отделяют. Растворитель выпаривают и остаток очищают хроматографией (ДХМ/EtOAc, 90:10), получая указанное в заголовке соединение (0,34 г, 95%). ЯМР (500 МГц) 0, 7-0,9 (м, 6H), 1,0-1,8 (м, 11H), 3,2 (м, 2H), 3,6-3,8 (ушир.с, 2H), 4,3 (кв, 2H), 4,7 (с, 2H), 7,0-7,1 (м, 3H), 7,15 (с, 1Н), 7,3 (м, 2H), 7,4 (с, 1Н).
Метод 9
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-бром-8-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
1, 1-Диоксо-3-бутил-3-этил-5-фенил-7-бром-8-этоксикарбонилметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин (метод 8; 0,34 г) и гидроксид натрия (0,3 г) растворяют в этаноле и смесь кипятят с обратным холодильником в течение 1 часа. Добавляют уксусную кислоту (1 мл) и растворитель удаляют при пониженном давлении. Остаток распределяют между ДХМ/H2O и органический слой отделяют и сушат. Растирание остатка с н-гексаном приводит к получению указанного в заголовке соединения (0,29 г, 90%) в виде твердого вещества. ЯМР (500 МГц) 0,7-0,8 (м, 6H), 1,0-1,7 (м, 8H), 3,1-3,2 (м, 2H), 3, 6 (ушир.с, 2H), 4,6 (с, 2H), 6,9-7,1 (м, 4H), 7,2 (м, 2H), 7,5 (с, 1Н).
Метод 10
1, 1-Диоксо-3-бутил-3-этил-5-фенил-7-метокси-8-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-метокси-8-этоксикарбонилметокси-2,3,4, 5-тетрагидро-1,5-бензотиазепин (метод 7; 1,2 г) растворяют в этаноле (20 мл). К полученному раствору добавляют гидроксид натрия (0,5 г), растворенный в H2O (1 мл), реакционную смесь нагревают до 40°C и выдерживают при этой температуре в течение 30 минут. Добавляют уксусную кислоту (1 мл) и растворитель удаляют при пониженном давлении. Остаток распределяют между ДХМ/H2O и органический слой отделяют и сушат. Растирание остатка с н-гексаном приводит к получению указанного в заголовке соединения (1,1 г, 97%) в виде твердого вещества. ЯМР 0,75-0,85 (м, 3H), 0,9 (т, 3H), 1,0-1,7 (м, 8H), 3,2 (кв, 2H), 3,65 (с, 3H), 3,65-3,85 (м, 2H), 4,7 (с, 2H), 6,4 (с, 1Н), 7,0 (т, 1H), 7,1 (д, 2H), 7,3 (т, 2H), 7,5 (с, 1H).
Метод 11
1,1-Диоксо-3,3-дибутил-5-фенил-7-бром-8-этоксикарбонилметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
1,1-Диоксо-3,3-дибутил-5-фенил-7-бром-8-гидрокси-2, 3,4,5-тетрагидро-1,5-бензотиазепин (синтезированный в соответствии с методикой публикации W09616051 для соответствующего 3-бутил-3-этилового аналога; 2,0 г, 4,16 ммоль), этилбромацетат (0,84 г, 5,03 ммоль), карбонат натрия (2,0 г, 189 ммоль) и тетрабутиламмоний бромид (80 мг, 0,25 ммоль) добавляют к MeCN (20 мл). Смесь кипятят с обратным холодильником в течение 2 часов и затем выпаривают при пониженном давлении. Остаток экстрагируют смесью ДХМ/вода. Слой ДХМ отделяют и выпаривают при пониженном давлении. Остаток очищают колоночной хроматографией. Продукт элюируют смесью ДХМ/EtOAc (90:10), получая указанное в заголовке соединение (2,2 г, 93%). ЯМР 0,7-0,8 (м, 6H), 1,0-1,6 (м, 15H), 3,2 (ушир.с, 2H), 3,7 (ушир.с, 2H), 4,3 (кв, 2H), 4,7 (с, 2H), 7,0-7,3 (м, 6H), 7,4 (с, 1H).
Методы 12-13
Соединения, представленные ниже, синтезируют в соответствии с методикой метода 11, используя соответствующие кислоту и амин (когда вещество является коммерчески доступным, источник не указывается).

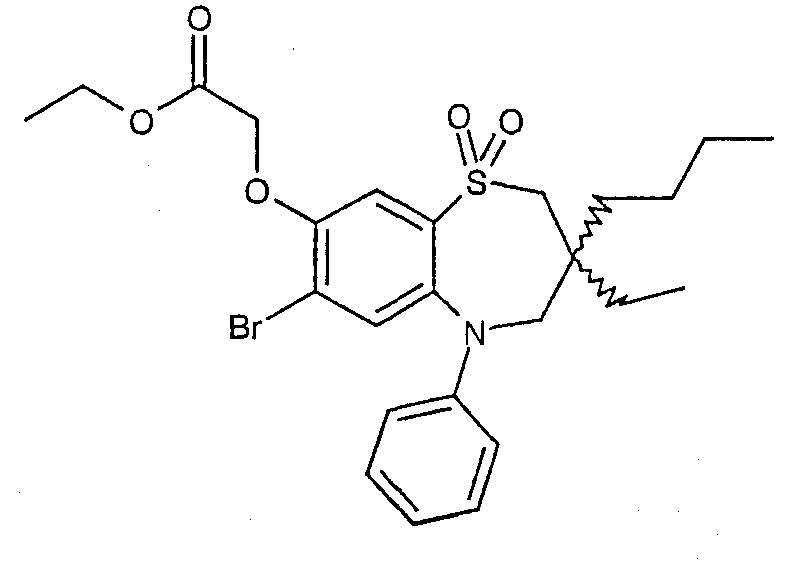
Метод 14
1,1-Диоксо-3,3-дибутил-5-фенил-7-бром-8-карбоксиметокси-2,3,4,5-тетрагидро-1, 5-бензотиазепин
1,1-Диоксо-3,3-дибутил-5-фенил-7-бром-8-этоксикарбонилметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин (метод 11; 2,2 г, 3,88 ммоль) растворяют в этаноле (15 мл). К полученному раствору добавляют NaOH (0,8 г в 1,5 мл воды) и смесь перемешивают в течение 30 минут при комнатной температуре. Добавляют уксусную кислоту (2 мл). Растворитель выпаривают при пониженном давлении и остаток экстрагируют смесью EtOAc/вода. Слой EtOAc отделяют, сушат и выпаривают при пониженном давлении, получая указанное в заголовке соединение (2,0 г, 95%). ЯМР (500 МГц) 0,7-0,8 (м, 6Н), 1,0-1,5 (м, 12H), 3,2 (ушир.с, 2H), 3,7 (ушир.с, 2H), 4,7 (с, 2H), 7,0-7,3 (м, 6H), 7,4 (с, 1H).
Метод 15
1, 1-Диоксо-3-бутил-3-этил-5-фенил-7-изопропокси-8-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
В изопропиловый спирт (12 мл) добавляют натрий (115 мг, 5 ммоль), после этого температура возрастает до 80°C и образуется солевая форма спирта. После того как весь натрий растворится, одной порцией добавляют 1,1-диоксо-3-бутил-3-этил-5-фенил-7-бром-8-карбоксиметокси-2,3, 4,5-тетрагидро-1,5-бензотиазепин (метод 9; 100 мг, 0,2 ммоль). После этого реакционную смесь кипятят с обратным холодильником в течение ночи, затем охлаждают до комнатной температуры и гасят уксусной кислотой. Растворитель затем удаляют при пониженном давлении и остаток растворяют в смеси воды и MeCN (70/30) и частично очищают ВЭЖХ. Остаток растворяют в этиленгликоле и добавляют NaOH (500 мг). Полученную реакционную смесь нагревают до 125°C и выдерживают при этой температуре в течение ночи, затем охлаждают до комнатной температуры, гасят уксусной кислотой и добавляют EtOAc (100 мл). Этиленгликоль удаляют промывкой органического слоя подкисленной водой три раза. Органический слой затем концентрируют и остаток снова очищают, как описано выше, получая указанное в заголовке соединение (40 мг, 41 %). ЯМР (300 МГц) 0,7-1,0 (м, 6H), 1,0-1,8 (м, 15H), 3,2 (кв, 2H), 3,75 (м, 2H), 4,3 (м, 1H), 4,6 (с, 2H), 6,35 (с, 1H), 6,95-7,2 (м, 3H), 7,2-7,4 (м, 2H), 7,55 (с, 1H).
Метод 16
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-этоксикарбонилметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
К 1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-гидрокси-2,3,4,5-тетрагидро-1,5-бензотиазепину (метод 25; 500 мг, 1,2 ммоль) добавляют MeCN (30 мл), тетрабутиламмоний бромид (30 мг, 0,08 ммоль), безводный карбонат натрия (500 мг, 4,7 ммоль), этилбромацетат (0,14 мл, 1,26 ммоль) и карбонат цезия (20 мг, 0,06 ммоль). Полученную реакционную смесь затем перемешивают в течение ночи при температуре 80°C. Затем растворитель удаляют при пониженном давлении, добавляют воду и ДХМ и водную фазу три раза экстрагируют ДХМ. Объединенные органические фазы затем сушат, концентрируют и очищают флэш-хроматографией [ДХМ/EtOAc; 1:0, 9:1], получая указанное в заголовке соединение (600 мг, 99%). ЯМР (300 МГц) 0,8-1,0 (м, 6H), 1,0-1,8 (м, 11H), 2,2 (с, 3H), 3,2 (кв, 2H) 3,75 (ушир.кв, 2H), 4,3 (кв, 2H), 4,75 (с, 1H), 6,7 (с, 1H), 6,95 (т, 1H), 7,05 (д, 2H), 7,25 (т, 2H), 7,3 (с, 1H).
Метод 17
1, 1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
К 1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-этоксикарбонилметокси-2,3,4, 5-тетрагидро-1,5-бензотиазепину (метод 16; 478 мг, 0,95 ммоль) добавляют ТГФ (15 мл), воду (3 мл) и LiOH (34 мг, 1,4 ммоль). После этого реакционную смесь перемешивают в течение 1 часа, затем добавляют уксусную кислоту (0,2 мл), воду (10 мл) и ДХМ (10 мл). Водный слой три раза экстрагируют ДХМ. Объединенные органические фазы сушат и концентрируют, получая указанное в заголовке соединение (450 мг, 99%). ЯМР 0,7-0,9 (м, 6H), 1,0-1,7 (м, 8H), 2,2 (с, 3H), 3,2 (кв, 2H), 3,7 (м, 2H), 4,8 (с, 2H), 6,65 (с, 1H), 6,95 (т, 1H), 7,05 (д, 2H), 7,25 (т, 2H), 7,35 (с, 1H), 8,4 (ушир.с, 1H).
Метод 18-19
Соединения, представленные ниже, синтезируют в соответствии с методикой Метода 17, используя соответствующую кислоту и амин (когда соединения коммерчески доступны, источник не указывается), с тем отличием, что используют два эквивалента LiOH и экстракцию выполняют с использованием EtOH по истечение 2 часов реакции.
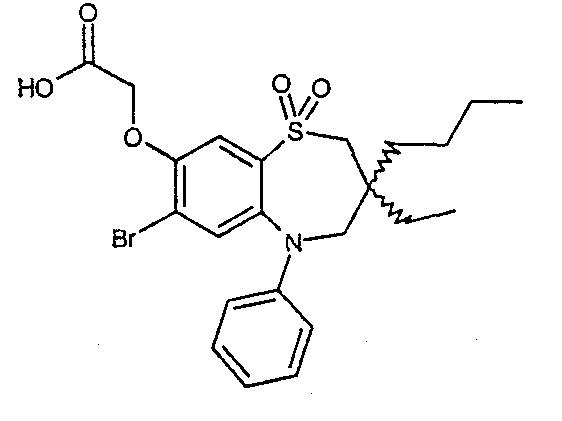

Метод 20
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-мезил-8-этоксикарбонилметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
К 1, 1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-этоксикарбонилметокси-2,3,4,5-тетрагидро-1,5-бензотиазепину (метод 16; 122 мг, 0,24 ммоль) добавляют ДХМ (3 мл), воду (3 мл) и карбонат калия (120 мг, 0,87 ммоль). Реакционную смесь охлаждают до 0°C и одной порцией добавляют м-хлорпероксибензойную кислоту (160 мг, 0,51 ммоль). Через 5 часов реакцию гасят добавлением ДХМ и насыщенным раствором гидрокарбоната натрия. Водную фазу три раза экстрагируют ДХМ. Объединенные органические фазы сушат, концентрируют и очищают флэш-хроматографией [ДХМ/EtOAc, 9:1], получая указанное в заголовке соединение (46 мг, 35%). ЯМР 0,7-0,8 (м, 6H), 1,0-1,65 (м, 11Н), 3,2 (кв, 2H), 3,3 (с, 3H), 3,7 (ушир.с, 1H), 4,25 (кв, 2H), 4,8 (с, 2H), 7,0-7,1 (м, 3H), 7,2-7,3 (м, 2H), 7,5 (с, 2H).
Метод 21
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-мезил-8-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
К 1, 1-диоксо-3-бутил-3-этил-5-фенил-7-мезил-8-этоксикарбонилметокси-2,3,4,5-тетрагидро-1,5-бензотиазепину (метод 20; 46 мг, 0,085 ммоль) добавляют ТГФ (5 мл), воду (1 мл) и LiOH (10 мг, 0,4 ммоль). Реакционную смесь перемешивают в течение 1 часа и затем реакцию гасят избытком уксусной кислоты. Добавляют воду и ДХМ, и водную фазу три раза экстрагируют ДХМ. Объединенные органические фазы сушат и концентрируют, получая указанное в заголовке соединение (40 мг, 91%). ЯМР 0,7-0,85 (м, 6H), 1,0-1,7 (м, 8H), 3,2 (м, 2H), 3,3 (с, 3H), 3,8 (с, 2H), 4,9 (с, 2H), 5,0 (ушир.с, 1Н), 7,05-7,15 (м, 3H), 7, 3-7,4 (т, 2H), 7,5 (с, 1H), 7,6 (с, 1Н).
Метод 22 (Способ получения 1)
1,1-Диоксо-3, 3-дибутил-5-фенил-7-метилтио-8-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
1,1-Диоксо-3,3-дибутил-5-фенил-7-бром-8-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин (метод 14; 500 мг, 0,93 ммоль) растворяют в ДМФА (10 мл). Добавляют метантиолат натрия (200 мг, 2,85 ммоль) и смесь перемешивают в течение 2 часов при температуре 50°C. Добавляют уксусную кислоту (0,4 мл) и растворитель выпаривают при пониженном давлении. Остаток экстрагируют смесью EtOAc/вода. Этилацетатный слой отделяют, сушат и выпаривают при пониженном давлении, получая указанное в заголовке соединение (450 мг, 96%). ЯМР (300 МГц) 0,7-0,8 (м, 6H), 1,0-1,6 (м, 12H), 2,2 (с, 2H), 3,2 (ушир.с, 2H), 3,7 (ушир.с, 2H), 4,8 (с, 2H), 6,6 (с, 1H), 6,9-7,1 (м, 3H), 7,2-7,4 (м, 3H).
Метод 22 (Способ получения 2)
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
К раствору 1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-этоксикарбонил-2,3,4,5-тетрагидро-1,5-бензотиазепина (метод 114; 15,45 г, 28,71 ммоль) в EtOH (160 мл) добавляют раствор NaOH (4,67 г, 116 ммоль) в воде (10 мл). Раствор перемешивают в течение 30 минут при комнатной температуре. Растворитель удаляют при пониженном давлении и остаток распределяют между EtOAc и 1,0 M HCl. Водный слой дополнительно экстрагируют два раза EtOAc и объединенные органические экстракты промывают насыщенным раствором соли и концентрируют, получая указанное в заголовке соединение (14,28 г, 98 %) в виде белого порошка. ЯМР (500 МГц, ДМСО-d6) ЯМР (500 МГц, ДМСО-d6) 0,65-0,80 (м, 6H), 0,90-1,50 (м, 12H), 2,20 (с, 3Н) 3,25 (с, 2H), 3,65 (ушир.с, 2H), 4,80 (с, 2H), 6,70 (с, 1H), 6,80-7,30 (м, 6H), 13,20 (с, 1H).
Метод 23-24
Соединения, представленные ниже, синтезируют в соответствии с методикой метода 22 (способ получения 1), используя соответствующую кислоту и амин (когда соединения коммерчески доступны, источник не указывается), с тем отличием, что реакции проводят при комнатной температуре и в Методе 24 в течение более длительного периода времени.


Метод 25
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-гидрокси-2,3,4, 5-тетрагидро-1.5-бензотиазепин
К 1,1-диоксо-3-бутил-3-этил-5-фенил-7-бром-8-метокси-2,3,4,5-тетрагидро-1,5-бензотиазепину (WО9616051; 600 мг, 1,29 ммоль) добавляют ДМФА (5 мл) и метантиолат натрия (450 мг, 6,42 ммоль). Реакционную смесь нагревают до 60°C и выдерживают при этой температуре в течение 1 часа. После этого масляную баню нагревают 120°C и выдерживают смесь при этой температуре в течение 4 часов. Для остановки реакции температуру снижают до комнатной температуры и быстро добавляют избыток уксусной кислоты. Реакционную смесь выдерживают под потоком азота, проходящим над гипохлоридом натрия, в течение 2 часов. Добавляют воду и EtOAc и водную фазу три раза экстрагируют EtOAc. Объединенные органические фазы промывают водой, сушат и концентрируют при пониженном давлении. Остаток очищают флэш-хроматографией [ДХМ/EtOAc, 9:1], получая указанное в заголовке соединение (0,5 г, 92%). ЯМР 0,65-0,8 (м, 6H), 0,95-1,6 (м, 8Н), 3,1 (кв, 2H), 3,6 (ушир.кв, 2H), 6,75 (с, 1H), 6,8 (т, 1H), 6,9 (д, 2H), 7,15 (т, 2H), 7,55 (с, 1Н).
Метод 26
1,1-Диоксо-3, 3-дибутил-5-фенил-7-метилтио-8-гидрокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
К 1,1-диоксо-3,3-дибутил-5-фенил-7-бром-8-метокси-2,3,4,5-тетрагидро-1,5-бензотиазепину (синтезированному в соответствии с методикой публикации WО9616051 для соответствующего 3-бутил-3-этилового аналога; 40 мг, 0,08 ммоль) добавляют ДМФА (2 мл), метантиолат натрия (60 мг, 0,85 ммоль) и боргидрид натрия (60 мг, 1,6 ммоль). Реакционную смесь выдерживают в течение ночи при температуре 60°C. Добавляют дополнительное количество боргидрида натрия (60 мг, 1,6 ммоль) и метантиолата натрия(60 мг, 0,85 ммоль) и температуру повышают до 120°C. Реакционную смесь выдерживают при этой температуре в течение в течение 4 часов и затем охлаждают до комнатной температуры. Добавляют уксусную кислоту и выдерживают под потоком азота, проходящим через гипохлорид натрия, в течение ночи. Добавляют воду и EtOAc и водную фазу три раза экстрагируют EtOAc. Объединенные органические фазы промывают HCl (1M), сушат и концентрируют при пониженном давлении. Остаток очищают флэш-хроматографией [EtOAc/гептан, 1:4], получая указанное в заголовке соединение (0,34 г, 93%). ЯМР 0,7-0,9 (м, 6H), 1,0-1,6 (м, 12H), 2,2 (с, 3H), 3,1 (с, 2H), 3,4 (с, 2H), 3,7 (ушир.с, 2H), 6,7 (с, 1H), 6,85-7,05 (м, 2H), 7,2-7,4 (м, 2H).
Метод 27
Аммониевая соль 2-[(2'R)-2'-(трет-бутоксикарбониламино)-2'-фенилэтаноиламино]этансульфоновой кислоты
2-Аминоэтансульфоновую кислоту (740 мг, 5,91 ммоль) и (2R)-2-(трет-бутоксикарбониламино)-2-фенилуксусную кислоту (1,09 г, 4,34 ммоль) растворяют в ДМФА (20 мл). Добавляют DIPEA (2,8 мл, 16,1 ммоль) и TBTU (1,53 г, 4,78 ммоль) и смесь перемешивают в течение 2 часов при температуре 60°C. Растворитель выпаривают при пониженном давлении. Остаток очищают препаративной ВЭЖХ (элюент с градиентом: MeCN/ацетат-аммониевый буфер, от 5:95 до 100:0), получая указанное в заголовке соединение (589 мг, 32%). ЯМР (CD3OD) 1,43 (с, 9H), 2,85-3,0 (м, 2H), 3,53-3,68 (м, 2H), 5,1 (ушир.с, 1H), 7,25-7,45 (м, 5Н).
Метод 28
Аммониевая соль 2-((2'R)-2'-амино-2'-фенилэтаноилами-но)этансульфоновой кислоты
Аммониевую соль 2-[(2'R)-2'-(трет-бутоксикарбониламино)-2'-фенилэтаноиламино]этансульфоновой кислоты (метод 27; 589 мг, 1,57 ммоль) растворяют в EtOAc (20 мл) и смесь охлажадют на ледяной бане. Газообразный хлористый водород барботируют через реакционную смесь, ледяную баню удаляют и реакционную смесь оставляют на 30 минут при комнатной температуре. Растворитель выпаривают при пониженном давлении. Остаток повторно растворяют в EtOAc (20 мл) и охлажадют на ледяной бане. Газообразный хлористый водород снова барботируют через реакционную смесь, ледяную баню удаляют и реакционную смесь оставляют на 30 минут при комнатной температуре. Растворитель выпаривают при пониженном давлении. Добавляют DIPEA в ДХМ и смесь выпаривают при пониженном давлении. Ту методику повторяют дважды. Смесь лиофилируют, получая указанное в заголовке соединение (563 мг, 85%), содержащее 1 эквивалент диизопропиламмонийхлорида. ЯМР (D2O) 1,35-1,38 (м, 15H), 2,96-3,12 (м, 2H), 3,21 (кв, 2H), 3,50-3,80 (м, 4H), 5,11 (ушир.с, 1Н), 7,45-7,55 (м, 5H).
Метод 29
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-[N -((R)-1'-фенил-1'-метоксикарбонилметил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-карбоксиметокси-2,3,4,5-тетрагидро-1, 5-бензотиазепин (метод 22; 250 мг, 0,49 ммоль), метиловый эфир (R)-2-фенилглицина гидрохлорид (120 мг, 0,60 ммоль) и DIPEA (300 мг, 2,3 ммоль) растворяют в ДХМ (10 мл) и смесь перемешивают в течение 5 минут при комнатной температуре. Добавляют TBTU (210 мг, 0,65 ммоль) и смесь перемешивают в течение 30 минут при комнатной температуре. Растворитель выпаривают при пониженном давлении и остаток переносят в колонку. Продукт элюируют ДХМ/EtOAc (90:10), получая указанное в заголовке соединение (306 мг, 95%). ЯМР (500 МГц) 0,7-0,8 (м, 6H), 1,0-1,6 (м, 12H), 2,1 (с, 3H) 3,2 (ушир.с, 2H), 3,6-3,8 (м, 5H), 4,6 (АВкв, 2H), 5,6 (д, 1H), 6,6 (с, 1H), 6,9-7,5 (м, 11Н), 7,9 (д, 1H).
Методы 30-45
Соединения, представленные ниже, синтезируют в соответствии с методикой метода 29, из соответствующей кислоты и амина (когда вещества коммерчески доступны, источник не указан), но с тем отличием, что в некоторых методах время реакции увеличивают до 2 часов.
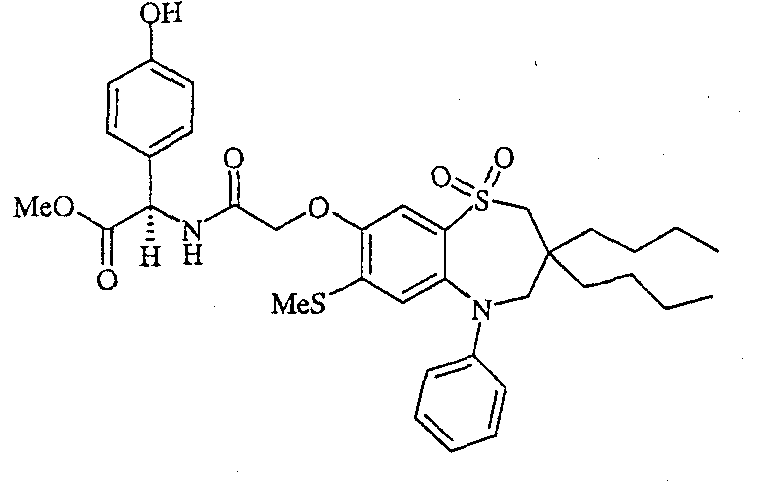


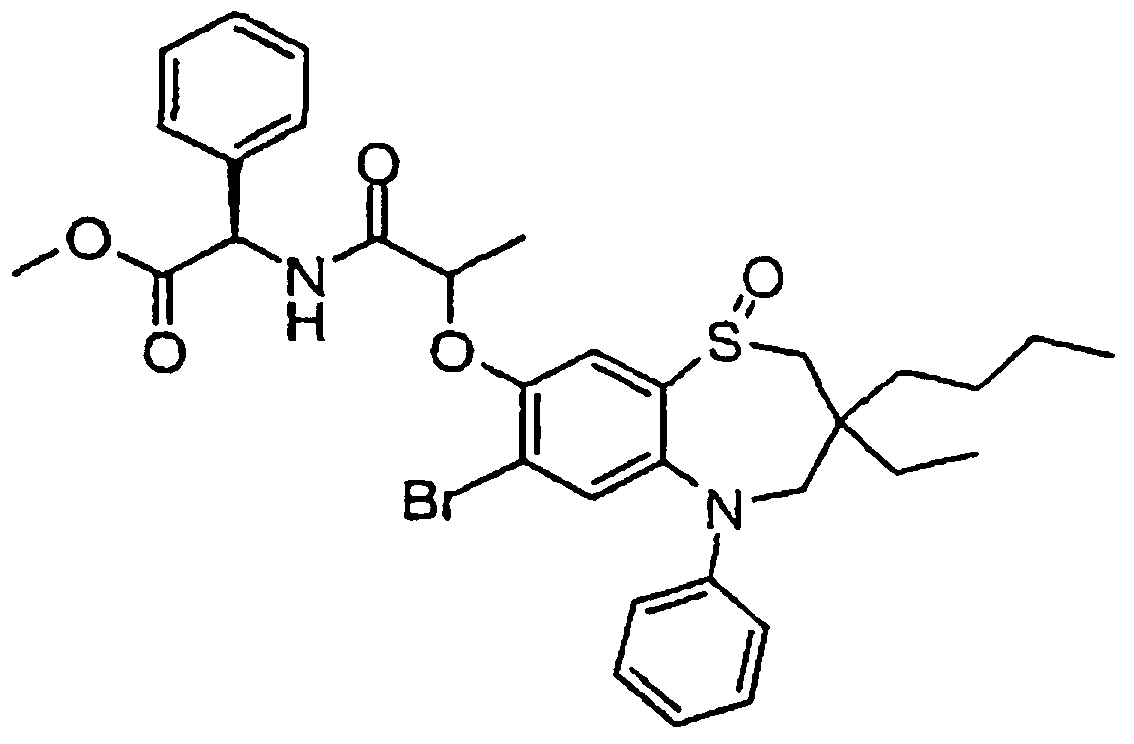


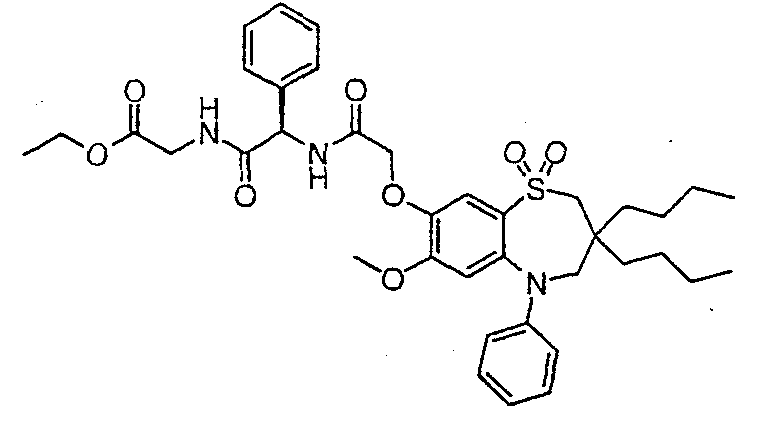




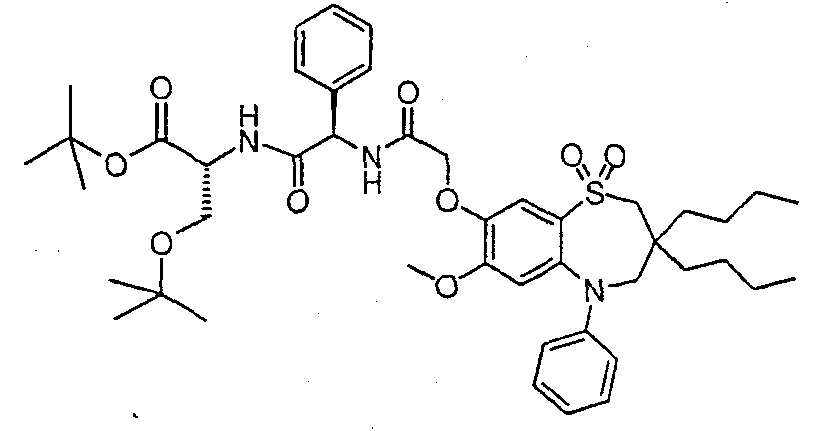

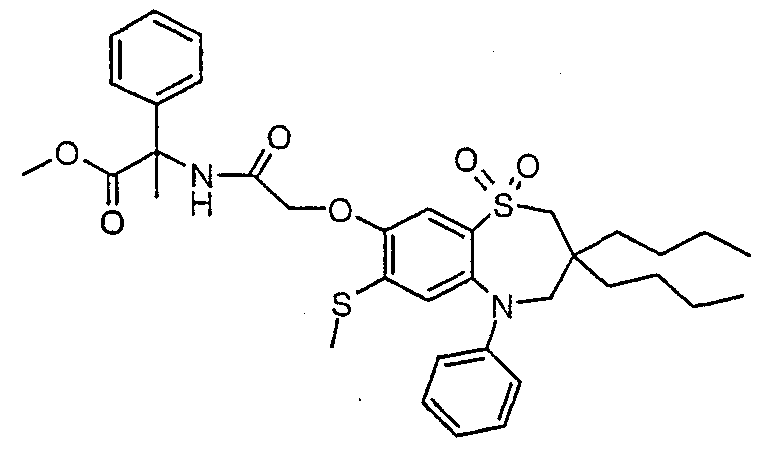
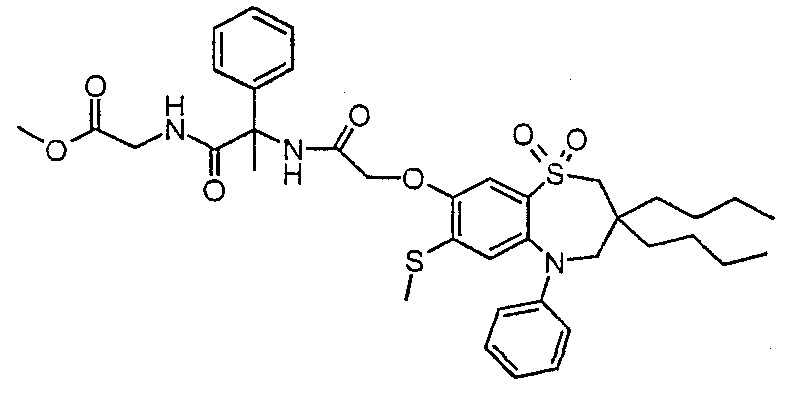
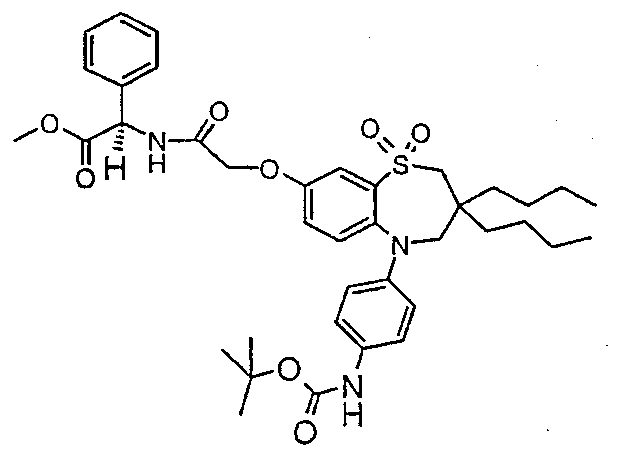
Метод 46
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-бром-8-[N -((S)-1'-фенил-l'-метоксикарбонилметил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-бром-8-карбоксиметокси-2,3,4,5-тетрагидро-1, 5-бензотиазепин (метод 9; 50 мг, 0,098 ммоль) растворяют в ДХМ (2 мл). К полученному раствору добавляют метил (2S)-амино(фенил)ацетат (19 мг, 0,12 ммоль) и DIPEA (0,068 мл, 0,39 ммоль) и реакционную смесь перемешивают в течение 2 минут. Добавляют TBTU (42 мг, 0,13 ммоль) и смесь перемешивают в течение 1,5 часа при комнатной температуре. Смесь переносят в колонку с наполнителем ISOLUTE и элюируют 10 мл смеси ДХМ/EtOAc (8:2), получая указанное в заголовке соединение (60 мг,93%). M/z 657,5.
Методы 47-62
Соединения, представленные ниже, синтезируют в соответствии с методикой метода 46 (но реакции проводят в течение ночи) из соответствующей кислоты и амина (когда вещества коммерчески доступны, источник не указан).


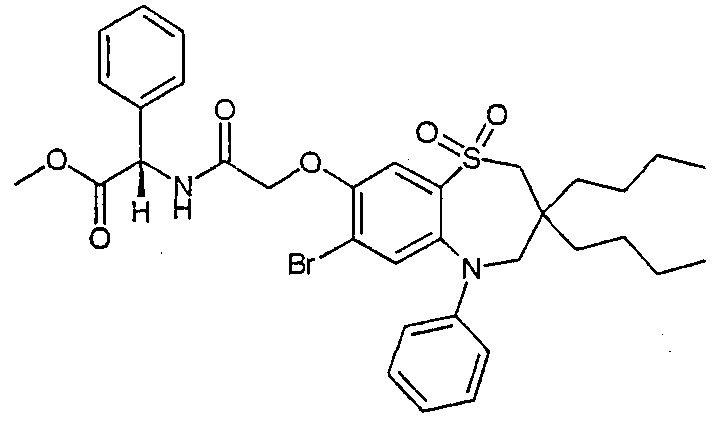


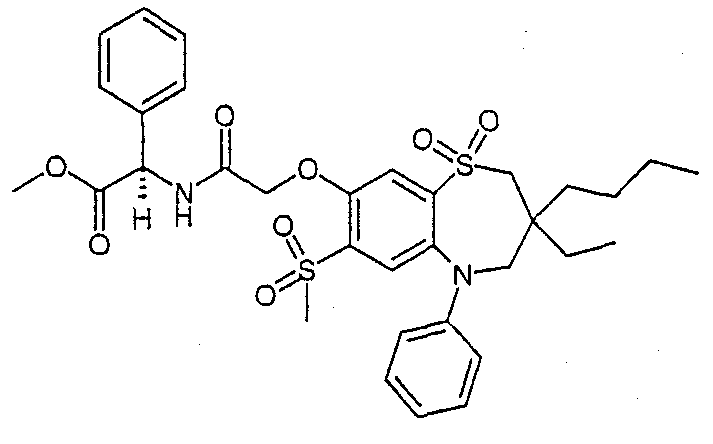
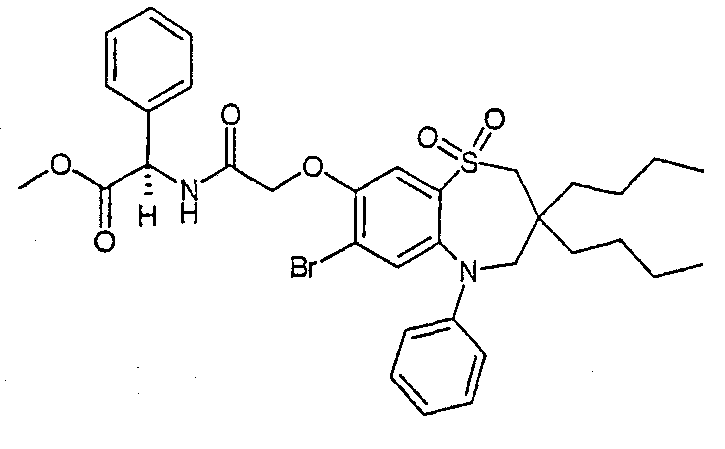





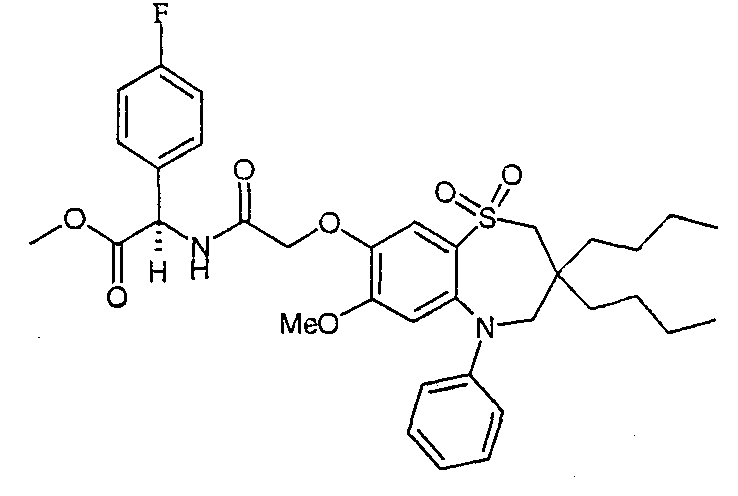
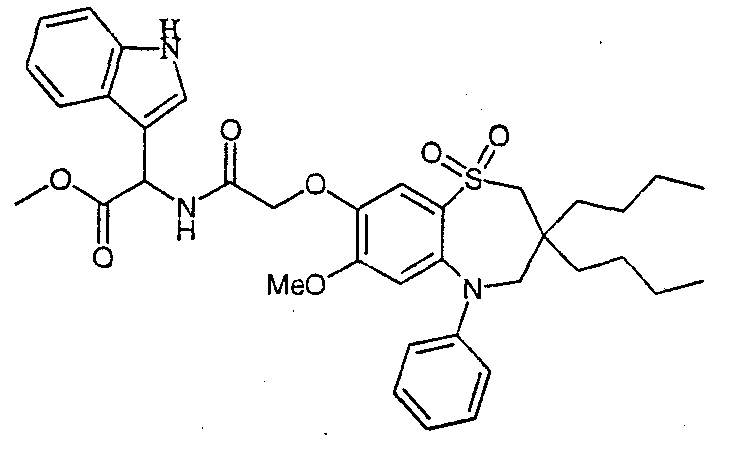


Метод 63
1, 1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N- {(R)-1'-фенил-1'-[N'-(трет-бутоксикарбонилметил)карбамоил]метил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин
1, 1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-[N-((R)-1'-фенил-1'-карбоксиметил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин (пример 1; 110 мг, 0,17 ммоль), трет-бутиловый эфир глицина (30 мг, 0,23 ммоль) и DIPEA (120 мг, 0,93 ммоль) растворяют в ДХМ (2 мл). Смесь перемешивают в течение 5 минут при комнатной температуре. Добавляют TBTU (72 мг, 0,22 ммоль) и смесь перемешивают в течение 1 часа при комнатной температуре. Растворитель выпаривают при пониженном давлении, остаток переносят в колонку и продукт элюируют смесью ДХМ/EtOAc (90:10), получая указанное в заголовке соединение (122 мг, 94%). ЯМР (300 МГц) 0,7-0,8 (м, 6H), 1,0-1,6 (м, 21H), 2,2 (с, 3H) 3,2 (с, 2H), 3,7-4,0 (м, 4H), 4,6 (АВкв, 2H), 5,6 (д, 1H), 6,4 (т, 1H), 6,6 (с, 1Н), 6,9-7,5 (м, 11H), 8,1 (д, 1Н).
Методы 64-69
Соединения, представленные ниже, синтезируют в соответствии с методикой метода 63 из соответствующей кислоты и амина (когда вещества коммерчески доступны, источник не указан).


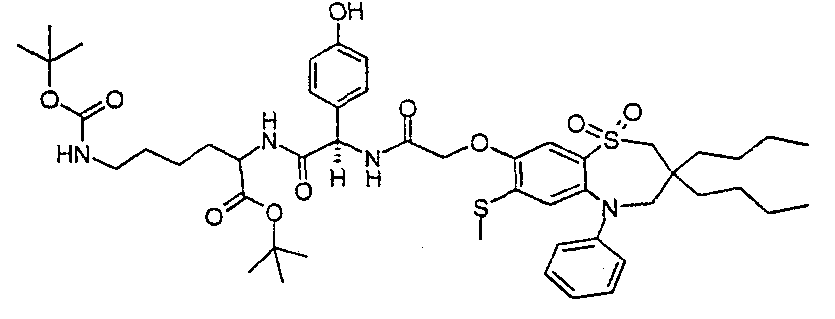



Метод 70
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-бром-8-{N -[(S)-1'-фенил-1'-{дэтоксифосфорилметил]карбамоилметокси}-2,3,4,5-тетрагидро-1,5-бензотиазепин
Указанное в заголовке соединение синтезируют из 1, 1-диоксо-3-бутил-3-этил-5-фенил-7-бром-8-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин (метод 9) и диэтил (S)-амино(фенил)метилфосфоната в соответствии с методикой примера 33. ЯМР (600 МГц) 7,77-7,72 (Н), м), 7,47-7,42 (3H, м), 7,36-7,27 (5H, м), 7,14 (1H, с), 7,10-7,03 (2H, м), 5,55-5,48 (1H, м), 4,63-4,51 (2H, м), 4,14-4,02 (2H, м), 3,99-3,92 (1H, м), 3,81-3,60 (3H, м), 3,22-3,10 (2H, м), 1,65-1,25 (8H, м), 1,19-0,95 (6H, м), 0,78-0,73 (6H, м).
Метод 71
4-Трифторметил-α-метоксикарбонилбензиламин
4-Трифторметил-α-карбоксибензиламин (1,4 г, 1,83 ммоль) и тионилхлорид добавляют к метанолу (8 мл) и смесь кипятят с обратным холодильником в течение 2 часов. Растворитель выпаривают при пониженном давлении. Остаток суспендируют в диэтиловом эфире, продукт отфильтровывают, промывают эфиром и сушат, получая указанное в заголовке соединение (0,34 г, 69%). ЯМР (300 МГц, ДМСО-d6) 3,3 (с, 1Н), 5,45 (с, 1H), 7,7-7,9 (м, 4H), 9,25 (ушир.с, 3H).
Метод 72
1, 1-Диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-(N-{(R)-1'-фенил-1'-N-(этоксикарбонилметил)карбамоил]метил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин
1, 1-Диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-[N-((R)-1'-фенил-1'-карбоксиметил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин (пример 38; 52 мг, 0,082 ммоль) и этиловый эфир глицина гидрохлорид (18 мг, 0,129 ммоль) растворяют в ДХМ (2 мл) и добавляют DIPEA (0,70 мл, 0,42 ммоль). Смесь перемешивают при комнатной температуре в течение 5 минут, добавляют TBTU (34 мг, 0,11 ммоль) и снова перемешивают в течение 2 часов. Растворитель выпаривают и остаток очищают флэш-хроматографией (элюирование: ДХМ/EtOAc, 10:3), получая указанное в заголовке соединение 50 мг (88%). ЯМР (500 МГц) 0,86 (м, 6H), 1,10-1,75 (м, 8H), 1,28 (м, 3H), 2,23 (с, 3H), 3,19 (кв, 2H), 3,75 (м, 2H), 3,99-4,25 (м, 4H), 4,64 (кв, 2H), 5,64 (м, 1H), 6,35 (ушир.с, 1H), 6,69 (с, 1H), 7,03 (т, 1H), 7,09 (д, 1H), 7, 29-7,52 (м,7H), 8,10(д, 1H).
Метод 73
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-[N-{(R)-1'-фенил-1'-[N -(1"-метоксикарбонил-1"-фенилметилкарбамоил]метил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин
Указанное в заголовке соединение синтезируют в соответствии с методикой метода 72 из 1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-[N-((R)-1'-фенил-1'-карбоксиметил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепина (пример 38) и метил (2R)-амино(фенил)ацетата гидрохлорида.
Метод 74
1-(1'-Метоксикарбонил-1'-аминометил)-2,3-дигидроксифенил гидрохлорид
1-(1'-Карбокси-1'-аминометил)-2,3-дигидроксифенил (40 г, 0,218 ммоль) смешивают с метанолом (230 мл). Газообразный HCl барботируют через раствор. Смесь кипятят с обратным холодильником в течение 2 часов. Растворитель выпаривают при пониженном давлении. Продукт кристаллизуют из смеси метанол/EtOAc/диэтиловый эфир с получением 35,5 г (70 %) указанного в заголовке соединения. ЯМР (600 МГц, CD3OD) 3,76 (с, 3H), 5,19 (с, 1H), 6,68-6-75 (м, 2H), 6,85 (дд, 1H).
Метод 75
(R)-1-(1'-Метоксикарбонил-1'-аминометил)-4-фторфенил гидрохлорид
(2R)-Амино(4-фторфенил)уксусную кислоту (570 мг, 2,77 ммоль) растворяют в метаноле (5 мл) и полученный раствор охлаждают на ледяной бане. По каплям добавляют тионилхлорид (2 мл) и смеси дают нагреться до комнатной температуры. Через 5 часов смесь выпаривают при пониженном давлении. Эту обработку повторяют и после этого реакционную смесь перемешивают в течение ночи. Смесь выпаривают при пониженном давлении, получая указанное в заголовке соединение с количественным выходом. ЯМР (500 МГц, CD3OD) 3,84 (с, 3H), 5,26 (с, 1H), 7,26 (т, 2H), 7,53 (дд, 2H).
Методы 76-77
Соединения, представленные ниже, синтезируют в соответствии с методикой метода 75 из соответствующей кислоты и амина (когда вещества коммерчески доступны, источник не указан).
Метод 78
1, 1-Диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-{N-[(R)-α-трет-бутоксикарбонил)-4-гидроксибензил]карбамоилметокси}-2,3,4,5-тетрагидро-1,5-бензотиазепин
трет-Бутил (2R)-амино(4-гидроксифенил)ацетат (104 мг, 0,47 ммоль) и 1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин (метод 17; 185 мг, 0,39 ммоль) растворяют в ДХМ (5 мл) и полученному раствору добавляют лутидин (0,09 мл, 0,77 ммоль). Смесь перемешивают при комнатной температуре в течение 5 минут, затем добавляют o-(7-азабензотриазол-1-ил)-N,N, N',N'-тетраметилуроний гексафторфосфат (208 мг, 0,55 ммоль) и полученную смесь перемешивают при комнатной температуре в течение 2 часов. Очистка флэш-хроматографией (элюирование с градиентом: ДХМ:EtOAc; 10:1→5:1) приводит к получению указанного в заголовке соединения (175 мг, 66 %). ЯМР (300 МГц) 0,81 (м, 6H), 1,05-1,65 (м, 8H), 1,42 (с, 9H), 2,21 (с, 3H), 3,17 (АВкв, 2H), 3,74 (м, 2H), 4,60 (АВкв, 2H), 5,22 (ушир.с, 1H), 5,49 (д, 1H), 6,67 (с, 1H), 6,79 (м, 2H), 7,00 (т, 1H), 7,07 (д, 2H), 7,23-7,30 (м, 3H), 7,40 (с, 1H), 7,82 (ушир.д, 1H).
Метод 79
Соединения, представленные ниже, синтезируют в соответствии с методикой метода 78 из подходящих исходных веществ.

Метод 80
2-{[(2R)-2-Амино-2-(4-гидроксифенил)этаноил]амино}этансульфоновая кислота
N-Boc-4-гидроксифенилглицин (1,00 г, 3,21 ммоль) растворяют в ДМФА (5 мл) и к полученному раствору добавляют тетрабутиламмоний таурин (2,36 г, 6,42 ммоль) с дополнительным количеством ДМФА (5 мл). Полученную суспензию охлаждают на ледяной бане и добавляют TBTU (1,24 г, 3,85 ммоль) добавляют. Ледяную баню удаляют через 30 минут и смесь перемешивают в течение 2 часов, затем фильтруют и концентрируют. В реакционную смесь добавляют ТФУК в ДХМ (20%, 20 мл) и полученную смесь перемешивают в течение ночи. Добавляют этанол (20 мл) и растворители выпаривают. Сырой продукт кипятят с обратным холодильником в этаноле (100 мл) в течение 1 часа. После фильтрования получают чистое соединение, указанное в заголовке, в виде твердого белого вещества (626 мг, 71%). ЯМР (ДМСО-d6) 2,4-2,6 (м, 2H), 3,2-3,4 (м, 2H), 4,79 (с, 1H), 6,78 (д, 2H), 7,23 (д, 2H), 8,22 (т, 1H), 8,4 (ушир.с, 3H), 9,7 (с, 1H).
Метод 81
1, 1-Диоксо-3-бутил-3-этил-5-фенил-7-карбоксиметилтио-8-метокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-бром-8-метокси-2,3,4,5-тетрагидро-1, 5-бензотиазепин (113 мг, 0,24 ммоль), Cs2CO3 (170 мг, 0,52 ммоль) и этилтиогликолят (0,060 мл, 0,54 ммоль) в ДМФА (4,0 мл) подвергают микроволновому облучению в аппарате Смита при температуре 80°C в течение 3 минут и затем при температуре 90°C в течение 8 минут. Реакционную смесь рабавляют водой (250 мл), экстрагируют ДХМ (5 x 10 мл) и объединенные органические фракции сушат (MgSO4), концентрируют и очищают но колонке небольшой высоты (элюирование с градиентом: петролейный эфир:EtOAc 4:1→2:1). Полученный продукт растворяют в ТГФ (2 мл), добавляют воду (2 мл) и NaOH (водн., 0,5 мл, 1 M) и реакционную смесь перемешивают при комнатной температуре в течение 2 часов. Реакцию гасят добавлением HCl (1 M), и реакционную смесь разбавляют водой (10 мл) и экстрагируют ДХМ (3 x 3 мл). Очистка препаративной ВЭЖХ приводит к получению указанного в заголовке соединения (58 мг, 59 %). ЯМР (300 МГц, CD3OD) 0,81 (м, 6H), 1, 00-1,70 (м, 8H), 3,21 (м, 2H), 3,42 (м, 2H), 3,71 (м, 2H), 3,92 (с, 3H), 6,88 (м, 2H), 7,02 (м, 2H), 7,23 (т, 2H), 7,40 (с, 1H).
Метод 82
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-этоксикарбонилметил-тио-8-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
1, 1-Диоксо-3-бутил-3-этил-5-фенил-7-бром-8-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин (Метод 9; 50 мг, 0,098 ммоль) и CS2CO3 (51 мг, 0,15 ммоль) добавляют в ДМФА (2,0 мл) и к полученному раствору добавляют этилтиогликолят (0,02 мл, 0,15 ммоль). Реакционную смесь подвергают микроволновому облучению в аппарате Смита при температуре 150°C в течение 5 минут. Реакционную смесь разбавляют водой (100 мл), подкисляют HCl (1 M), экстрагируют ДХМ (3 x 10 мл) и объединенные органические слои сушат (MgSO4) с получением указанного в заголовке соединения в виде сырого продукта (54 мг). M/z 550,2.
Метод 83 и Метод 84
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-бром-8-гидрокси-2,3,4, 5-тетрагидро-1,5-бензотиазепин (энантиомер 1): и
1,1-диоксо-3-бутил-3-этил-5-фенил-7-бром-8-гидрокси-2,3,4,5-тетрагидро-1,5-бензотиазепин (энантиомер 2)
Два энантиомера 1,1-диоксо-3-бутил-3-этил-5-фенил-7-бром-8-гидрокси-2,3,4,5-тетрагидро-1,5-бензотиазепина (WO 96/16051) получают разеделением соответствующего рацемической смеси с использованием препаративной ВЭЖХ (колонка: Chiralpak AD (20 x 250 мм, 10 мкм); подвижная фаза: гептан/IPA, 90/10). Впрыскиваемый рацемат (17,3 мг в IPA (1 мл)) элюируют с объемной скоростью потока 10 мл/мин, хроматограмма проявляется в УФ свете при 285 нм. Всего разделяют 260 мг рацемата с получением 121 мг энантиомера первого элюирования (энантиомер 1) и 115 мг энантиомера второго элюирования (энантиомер 2). Общий выход 91%. Каждый из двух энантиомеров получают с выходом 99,4%.
Метод 85
(R)-N -Бензилоксикарбонил-α-[N'-(трет-бутоксикарбонил-метил)карбамоил]бензиламин
(R)-N-Бензоилоксикарбонил-α-карбоксибензиламин (10 г, 35,0 ммоль) и трет-бутилглицин гидрохлорид (6,3 г, 37,4 ммоль) растворяют в ДХМ (200 мл) и добавляют 2,6-лутидин (8,2 мл, 70,4 ммоль). Полученную смесь перемешивают 5 минут при температуре 0°C, добавляют TBTU (12,4 г, 38,6 ммоль) и перемешивание продолжают 1 час 30 минут при температуре 0°C и 3 часа 45 минут при комнатной температуре. Реакционную смесь промывают водой (2 x 100 мл), сушат (MgSO4) и очищают флэш-хроматографией (элюирование с градиентом: ДХМ:EtOAc 7:1→5:1), получая указанное в заголовке соединение (13 г, 94 %). ЯМР (500 МГц) 1,45 (с, 9H), 3,84 (д, 1H), 4,00 (дд, 1H), 5,10 (м, 2H), 5,28 (ушир.с, 1H), 6,13 (ушир.с, 1H), 6,23 (ушир.с, 1H), 7,30-7,44 (м, 10H).
Метод 86
(R)-α-[N-(трет-Бутоксикарбонилметил)карбамоил]бензиламин
(R)-(N-Бензоилоксикарбонил-α-[N'-(трет-бутоксикарбонил-метил)карбамоил]бензиламин (метод 85; 12,8 г, 32,2 ммоль) растворяют в EtOH (99%, 200 мл) и толуолее (50 мл). Добавляют Pd/C (10%, 0,65 г) и проводят гидрирование при атмосферной давлении в течение 5 часов 30 минут при комнатной температуре. Реакционную смесь фильтруют через диатомовую землю, затем растворители выпаривают, получая указанное в заголовке соединение (8,4 г, 99 %). ЯМР (600 МГц) 1,45 (с, 9H), 3,93 (м, 2H), 4,54 (с, 1H), 7,31-7,42 (м, 5H), 7,51 (ушир.с, 1Н).
Метод 87
1,1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-[N-(S)-(α -метоксикарбонилбензил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин (метод 22; 50 мг, 0,099 ммоль) растворяют в ДХМ (2 мл). Добавляют метиловый эфир (S)-фенилглицина гидрохлорид (24,8 мг, 0,123 ммоль) и диизопропилэтиламин (70 мкл, 0,401 ммоль). Смесь перемешивают в течение 15 минут, затем добавляют TBTU (38 мг, 0,118 ммоль). Реакция завершается через 1,5 часа (ЖХ/МС). Сырой продукт очищают флэш-хроматографией (элюирование: хлороформ/EtOAc, 8/2), получая указанное в заголовке соединение (88,6%; 55,2 мг, 0,064 ммоль). M/z 653.
Метод 88
1,1-Диоксо-3, 3-дибутил-5-фенил-7-метилтио-8-[N-{(S)-α-[N'-(метоксикарбонилметил)карбамоил]бензил}карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин
1,1-Диоксо-3, 3-дибутил-5-фенил-7-метилтио-8-[N-(S)-(α-карбоксибензил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин (пример 88; 25 мг, 0,039 ммоль) и метиловый эфир глицина (7,5 мг, 0,059 ммоль) растворяют в ДХМ (2 мл). К полученной смеси последовательно добавляют диизопропилэтиламин (27 мкл, 0,158 ммоль) и TBTU (15 мг, 0,047 ммоль) и смесь перемешивают в течение 2 часов при комнатной температуре. Сырой продукт очищают флэш-хроматографией (элюирование: ДХМ/EtOAc, 8/2), получая указанное в заголовке соединение (22 мг, 79%). M/z 710.
Метод 89
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-бром-8-(2-карбоксиэтокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин
Гидроксид натрия (38 мг, 0,95 ммоль) растворяют в этаноле (2,5 мл) и добавляют 1,1-диоксо-3-бутил-3-этил-5-фенил-7-бром-8-гидрокси-2,3,4,5-тетрагидро-1,5-бензотиазепин (WO 96/16051; 200 мг, 0,443 ммоль). Смесь перемешивают при комнатной температуре в течение 5 минут, добавляют 3-бромпропионовую кислоту (68 мг, 0,443 ммоль) и реакционную смесь кипятят с обратным холодильником в течение 20 часов. Добавляют уксусную кислоту. Растворитель выпаривают при пониженном давлении и остаток экстрагируют EtOAc/водой. Органический слой отделяют, промывают водой, сушат и выпаривают при пониженном давлении. Сырой продукт очищают колоночной хроматографией (элюирование: ДХМ/MeOH, 100:5), получая указанное в заголовке соединение (89 мг, 38%). ЯМР (CD3OD) 0,75-0,83 (м, 6H), 1,0-1,25 (м, 4H), 1,38-1,65 (м, 4H), 2,82 (м, 2H), 3,26 (с, 2H), 3,50-3, 90 (м, 2H), 4,33 (т, 2H), 6,99 (т, 1H), 7,07-7,13 (м, 3H), 7,28 (т, 2H), 7,53 (с, 1H).
Метод 90
1, 1-диоксо-3-бутил-3-этил-5-фенил-7-бром-8-(2-{N-[(R)-α-(трет-бутоксикарбонил)бензил]карбамоил}этокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин
К раствору 1, 1-диоксо-3-бутил-3-этил-5-фенил-7-бром-8-(2-карбоксиэтокси)-2,3,4,5-тетрагидро-1,5-бензотиазепина (метод 89; 70 мг, 0,134 ммоль) и (R)-α-(трет-бутоксикарбонил)бензиламин (J. Amer. Chem. Soc.; EN; 117; 44; 1995; 10879-10888; 35 мг, 0,169 ммоль) в ДХМ (2,5 мл) добавляют 2,6-лутидин (29 мг, 0,268 ммоль) и TBTU (56 мг, 0,174 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 2,5 часов, зате разбавляют ДХМ. Раствор промывают NaHCO3 (водн., насыщ.), водой, сушат и растворитель выпаривают при пониженном давлении. Остаток суспензируют в смеси эфир/петролейный эфир и кристаллы отфильтровывают, получая указанное в заголовке соединение (85 мг, 89%). (500 МГц) 0,79-0,86 (м, 6H), 1,04-1,28 (м, 4H), 1,35-1,56 (м, 11H), 1,60-1,77 (м, 2H), 2,82 (т, 2H), 3,13-3,25 (м, 2H), 3,72 (ушир.с, 2H), 4,35-4,44 (м, 2H), 5,54 (д, 1H), 6,95 (д, 1H), 7,04 (т, 1Н), 7,08 (д, 2H), 7,15 (с, 1H), 7,29-7,43 (м, 6H), 7,52 (с, 1H).
Методы 91-94
Соединения, представленные ниже, синтезируют в соответствии с методикой примера 104 из соответствующих исходных веществ (указан исходный амин, когда он не является коммерчески доступным).


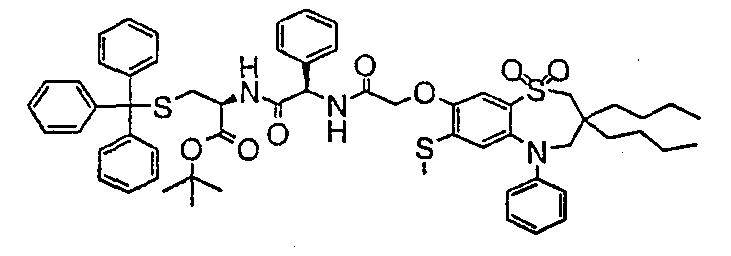

Метод 95
3,3-Дибутил-4-оксо-5-(4-хлорфенил)-7-бром-8-метокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
Смесь 3,3-дибутил-4-оксо-7-бром-8-метокси-2,3,4,5-тетрагидро-1,5-бензотиазепина (WO 95/04534; 1,0 г, 2,5 ммоль), 4-бромхлорбензола (4,78 г, 24,98 ммоль), бромида меди (36 мг, 0,25 ммоль) и карбоната калия (0,35 г, 2,5 ммоль) кипятят с обратным холодильником в течение 20 часов. Реакционную смесь загружают в колонку и продукт элюируют смесью 5% EtOAc/петролейный эфир (0,8 г, 63%). ЯМР (500 МГц) 0,86-0,92 (м, 6H), 1,16-1,35 (м, 8H), 1,45-1,65 (м, 4Н) 3,16 (с, 2H), 3,96 (с, 3H), 7,06-7,10 (м, 2H), 7,19 (с, 1H), 7,29 (с, 1H), 7,33-7,38 (м, 2H). M/z 511.
Метод 96
1,1-Диоксо-3,3-дибутил-4-оксо-5-(4-хлорфенил)-7-бром-8-метокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
К смеси 3, 3-дибутил-4-оксо-5-(4-хлорфенил)-7-бром-8-метокси-2,3,4,5-тетрагидро-1,5-бензотиазепина (метод 95; 0,67 г, 1,304 ммоль), ДХМ (34 мл), воды (34 мл) и карбоната калия (0,554 г, 4,0 ммоль) при температуре 0°C одной порцией добавляют м-хлорпероксибензойную кислоту (0,78 г, 3,2 ммоль). Реакционную смесь перемешивают при температуре 0 °C в течение 10 часов и затем при комнатной температуре в течение 14 часов. Добавляют ДХМ (100 мл) и NaHCO3 (водн., насыщ.; 150 мл). Органический слой отделяют, промывают насыщенным раствором соли, сушат и выпаривают при пониженном давлении, получая указанное в заголовке соединение (0,68 г, 96%). ЯМР (600 МГц) 0,7-0,92 (м, 6H), 1,0-1,60 (м, 10Н), 1,70-1,92 (м, 2H), 2,30-3,7 (м, 2H), 3,99 (с, 3Н), 7,16-7,20 (м, 2H), 7,24 (с, 1Н), 7,34-7,37 (м, 2H), 7,44 (с, 1H); m/z 543.
Метод 97
1,1-Диоксо-3,3-дибутил-4-оксо-5-(4-хлорфенил)-7-метилтио-8-метокси-2,3,4, 5-тетрагидро-1,5-бензотиазепин
К раствору 1,1-диоксо-3,3-дибутил-4-оксо-5-(4-хлорфенил)-7-бром-8-метокси-2,3,4,5-тетрагидро-1,5-бензотиазепина (метод 96; 0,66 г, 1,22 ммоль) в безводном ДМФА (11 мл) в атмосфере азота добавляют метантиолат натрия (0,43 г, 6,08 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 72 часов. Растворитель выпаривают при пониженном давлении и остаток экстрагируют трихлорметаном и водой. Органический слой отделяют, промывают насыщенным раствором соли, сушат и выпаривают при пониженном давлении. Сырой продукт очищают колоночной хроматографией (элюирование: ДХМ), получая указанное в заголовке соединение 0,6 г (96%). ЯМР (500 МГц) 0,80-1,0 (м, 6H), 1,10-1,6 (м, 10H), 1,70-2,0 (м, 2H), 2,28 (с, 3H), 3,37-3,70 (м, 2H), 4,04 (с, 3H), 6,65 (с, 1H), 7,25-7,30 (м, 2Н) 7,35-7,42 (м, 3H); m/z 510,4.
Метод 98
1,1-Диоксо-3, 3-дибутил-5-(4-хлорфенил)-7-метилтио-8-метокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
К раствору 1,1-диоксо-3,3-дибутил-4-оксо-5-(4-хлорфенил)-7-метилтио-8-метокси-2,3,4, 5-тетрагидро-1,5-бензотиазепина (метод 97; 0,41 г, 0,79 ммоль) в безводном эфире (15 мл) в атмосфере азота добавляют LiAlH4 (0,15 г, 3,97 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 2,5 часов. Реакционую колбу охлаждают до 0°C и избыток LiAlH4 гасят добавлением воды (0,3 мл) и 2 M водного раствора NaOH (0,3 мл). Смесь фильтруют и фильтрат сушат и выпаривают при пониженном давлении. Сырой продукт очищают колоночной хроматографией (элюирование: ДХМ), получая указанное в заголовке соединение (0,265 г, 68%). ЯМР (300 МГц) 0,8-0,90 (м, 6H), 1,0-1,47 (м, 12H), 2,33 (с, 3H), 3,17 (с, 2H), 3,70 (с, 2H), 3,93 (с, 3H), 7,03-7,08 (м, 3H), 7,23-7,32 (м, 3H); m/z 496.
Метод 99
1,1-Диоксо-3,3-дибутил-5-(4-хлорфенил)-7-метилтио-8-гидрокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
К раствору 1,1-диоксо-3, 3-дибутил-5-(4-хлорфенил)-7-метилтио-8-метокси-2,3,4,5-тетрагидро-1,5-бензотиазепина (метод 98; 0,26 г 0,52 ммоль) в безводном ДХМ (10 мл) в атмосфере азота добавляют трибромид бора (2,63 г, 10,48 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 2,5 часов. Реакционную колбу охлаждают до 0°C, добавляют воду (20 мл) и гидразин моногидрат (0,5 мл). Органический слой отделяют, сушат и выпаривают при пониженном давлении. Сырой продукт очищают колоночной хроматографией (элюирование: ДХМ/EtOAc, 100:5 и 100:10), получая указанное в заголовке соединение (0,20 г, 80%). ЯМР (500 МГц) 0,85 (т, 6H), 1,03-1,28 (м, 8H), 1,35-1,46 (м, 4H), 2,39 (с, 3H), 3,21 (с, 2H), 3,73 (с, 2H), 7,04 (д, 2H), 7,29-7,34 (м, 3H), 7,44 (с, 1H); m/z 482.
Метод 100
1,1-Диоксо-3,3-дибутил-5-(4-хлорфенил)-7-метилтио-8-этоксикарбонилметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
К смеси 1,1-диоксо-3,3-дибутил-5-(4-хлорфенил)-7-метилтио-8-гидрокси-2,3,4,5-тетрагидро-1,5-бензотиазепина (метод 99; 0,194 г, 0,402 ммоль), безводного Na2CO3 (0,192 г, 1,81 ммоль) и бромида тетрабутиламмония в MeCN (5 мл) добавляют этилбромацетат (0,101 г, 0,604 ммоль). Реакционную смесь кипятят с обратным холодильником в течение 3,5 часов. Растворитель выпаривают при пониженном давлении и остаток экстрагируют смесью ДХМ/вода. Органический слой отделяют, сушат и выпаривают при пониженном давлении. Сырой продукт очищают колоночной хроматографией (элюирование: ДХМ/EtOAc, 100/5 и 100/10), получая указанное в заголовке соединение (0,197 г, 86%). ЯМР (300 МГц) 0,80-0,89 (м, 6H), 1,0-1,45 (м, 15H), 2,34 (с, 3H), 3,16 (с, 2H), 3,68 (с, 2H), 4,30 (кв, 2H), 4,71 (с, 2H), 7,05-7,11 (м, 3H), 7,19 (с, 1H), 7,29-7,35 (м, 2H).
Метод 101
1,1-Диоксо-3, 3-дибутил-5-(4-хлорфенил)-7-метилтио-8-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
К раствору 1,1-диоксо-3,3-дибутил-5-(4-хлорфенил)-7-метилтио-8-этоксикарбонилметокси-2, 3,4,5-тетрагидро-1,5-бензотиазепина (метод 100; 0,195 г, 0,343 ммоль) в этаноле (8 мл) добавляют NaOH (1,03 ммоль в 0,5 мл воды). Реакционную смесь перемешивают при комнатной температуре в течение 70 минут и затем гасят добавлением уксусной кислоты (0,3 мл). Растворитель выпаривают при пониженном давлении и остаток экстрагируют смесью ДХМ/вода. Органический слой отделяют, промывают насыщенным раствором соли, сушат и выпаривают при пониженном давлении, получая указанное в заголовке соединение (0,169 г, 91%). ЯМР (500 МГц, CD3OD) 0,86 (т, 6H), 1,11-1,28 (м, 8H), 1,37-1,44 (м, 4H), 2,33 (с, 3H), 3,25 (с, 2H), 3,55 (с, 2H), 4,73 (с, 2H), 7,10-7,15 (м, 3H), 7,26 (с, 1H), 7,28-7,32 (м, 2H).
Метод 102
1, 1-Диоксо-3,3-дибутил-5-(4-хлорфенил-7-метилтио-8-[N-{(R)-α-[N'-(трет-бутоксикарбонилметил)карбамоил]бензил}карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин
К раствору 1,1-диоксо-3,3-дибутил-5-(4-хлорфенил)-7-метилтио-8-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепина (метод 101; 100 мг, 0,185 ммоль) и (R)-α-[N -(трет-бутоксикарбонилметил)карбамоил]бензиламина (метод 86; 56 мг, 0,213 ммоль) в ДХМ (4 мл) добавляют 2,6-лутидин (40 мг, 0,37 ммоль) и TBTU (89 мг, 0,28 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов, затем добавляют EtOAc и раствор промывают водой. Органический слой отделяют, сушат и выпаривают при пониженном давлении. Сырой продукт очищают колоночной хроматографией (элюирование: ДХМ/MeOH, 100:3), получая указанное в заголовке соединение (0,129 г, 89%). ЯМР (600 МГц) 0,78-82 (м, 6H), 1,01-1,23 (м, 8H), 1,30-1,42 (м, 13H), 2,32 (с, 3H), 3,10-3,16 (м, 2H), 3,62-3,68 (м, 2H), 3,81-3,87 (м, 1H), 3,95-4,03 (м, 1H), 4,52 (дд, 2H), 5,57 (д, 1H), 6,27 (т, 1H), 7,01-7,07 (м, 3H), 7,20-7,43 (м, 8H), 8,02 (д, 1H).
Метод 103
3,3-Дибутил-4-оксо-5-(4-нитрофенил)-8-метокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
К 3,3-дибутил-4-оксо-8-метокси-2,3,4,5-тетрагидро-l, 5-бензотиазепину (синтезированному в соответствии с методикой публикации WO9616051 для соответствующего 3-бутил-3-этилового аналога; 2,9 г, 9,0 ммоль) добавляют п-нитрофенилбромид (24 г, 119 ммоль), K2CO3 (1,6 г, 12 ммоль) и CuI (180 мг, 0,95 ммоль). Реакционную смесь нагревают до 200°C и выдерживают при этой температуре в течение ночи. Затем смеси дают охладиться до комнатной температуры и образующееся твердое вещество очищают хроматографией (элиюрование: ДХМ). Фракции, содержащие продукт, концентрируют при пониженном давлении и добавляют EtOH (95%) и нерастворимый п-нитрофенилбромид отфильтровывают. Остаток снова очищают флэш-хроматографией (элюирование: ДХМ). Продукт еще не является чистым, поэтому остаток затем очищают флэш-хроматографией (элюирование: EtOAc/гептан, 1:9), получая указанное в заголовке соединение (2,57 г, 64%). ЯМР (600 МГц) 0,77-0,87 (м, 6H), 1,12-1,31 (м, 8H), 1,4-1,6 (м, 4H), 3,09 (ушир.с, 2Н) 3,79 (с, 3H), 6,72-6,83 (м, 2H), 7,18-7,27 (м, 3H), 8,3 (д, 2H).
Метод 104
1,1-Диоксо-3,3-дибутил-4-оксо-5-(4-нитрофенил)-8-метокси-2,3,4, 5-тетрагидро-1,5-бензотиазепин
К 3,3-дибутил-4-оксо-5-(4-нитрофенил)-8-метокси-2,3,4,5-тетрагидро-1,5-бензотиазепину (метод 103; 2,57 г, 5,8 ммоль) добавляют ДХМ (130 мл), воду (130 мл) и Ka2CO3 (2,44 г, 17,6 ммоль). Реакционную смесь охлаждают до 0°C и одной порцией добавляют м-хлорпероксибензойную кислоту (3,42 г, 13,9 ммоль). Реакционную смесь оставляют на ночь, давая ей возможность медленно нагреться до комнатной температуры. После этого добавляют водн. NaHCO3 (насыщ.) и разделяют два слоя. Водный слой три раза экстрагируют ДХМ. Объединенные органические слои сушат, фильтруют и выпаривают при пониженном давлении. Продукт очищают флэш-хроматографией (элюирование: ДХМ), получая указанное в заголовке соединение (2,4 г, 87%). M/z 475,4.
Метод 105
1,1-Диоксо-3,3-дибутил-5-(4-аминофенил)-8-метокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
К LiAlH4 (5,76 г, 151 ммоль) добавляют ТГФ (200 мл). Реакционную смесь охлаждают до 0°C и медленно с помощью шприца добавляют H2SO4 (4,06 мл, 76 ммоль). После завершения добавления реакционную смесь перемешивают в течение 10 минут. Затем при температуре 0°C добавляют 1,1-диоксо-3,3-дибутил-4-оксо-5-(4-нитрофенил)-8-метокси-2,3,4,5-тетрагидро-1, 5-бензотиазепин (метод 104; 2,57 г, 5,06 ммоль), растворенный в ТГФ (50 мл). После энергичного перемешивания в течение 1 часа охлаждающую баню удаляют, реакционную смесь нагревают до 40°C и выдерживают при этой температуре в течение ночи. Посли этого в указанном порядке добавляют Na2SO4·10H2O (3-4 чайные ложки), воду (8 мл), NaOH (15%, водн.) (8 мл), воду (25 мл) и MeOH (30 мл) добавляют. Осадок удаляют фильтрованием и продывают ДХМ/MeOH. Растворитель сушат, фильтруют и концентрируют при пониженном давлении. Остаток очищают флэш-хроматографией (элюирование: ДХМ/EtOAc, 9:1 затем 3:1), получая указанное в заголовке соединение (0,6 г, 27%). M/z 431,3.
Метод 106
1,1-Диоксо-3,3-дибутил-5-(4-аминофенил)-8-гидрокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
1,1-Диоксо-3,3-дибутил-5-(4-аминофенил)-8-метокси-2,3,4,5-тетрагидро-1, 5-бензотиазепин (метод 105; 918 мг, 2,13 ммоль) растворяют в ДМФА (сухой, 20 мл). К полученному раствору добавляют тиометоксид натрия (810 мг, 11,6 ммоль). Реакционную смесь обрабатывают при температуре 100-120°C в течение четырех дней, затем при комнатной температуре в течение ночи. К полученной смеси добавляют уксусную кислоту (3 мл), смесь продувают азотом (газообр.) и газ пропускают через колбу с гипохлоритом натрия для разложения образующегося метилмеркаптана. Добавляют воду и водный слой дважды экстрагируют EtOAc. Объединенные органические слои промывают насыщенным раствором соли, сушат, фильтруют и выпаривают при пониженном давлении. Добавляют смесь, содержащую ДМФА, толуол и насыщенный раствор соли (не растворяют). Водный слой дважды экстрагируют толуолом. Объединенные органические экстракты промывают насыщенным раствором соли. Длительную воронку промывают EtOAc для растворения любых остатков. Толуоловый и этилацетатный растворы объединяют, сушат, фильтруют и выпаривают при пониженном давлении. Остаток очищают флэш-хроматографией (элюирование: ДХМ/EtOAc, 7:3), получая указанное в заголовке соединение (0,6 г,27%). M/z 417,4.
Метод 107
1,1-Диоксо-3,3-дибутил-5-(4-трет-бутоксикарбониламинофенил)-8-гидрокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
1, 1-Диоксо-3,3-дибутил-5-(4-аминофенил)-8-гидрокси-2,3,4,5-тетрагидро-1,5-бензотиазепин (метод 106; 600 мг, 1,44 ммоль растворяют в ТГФ (10 мл). Добавляют ди-трет-бутилдикарбонат (314 мг, 1,44 ммоль) и смесь перемешивают при температуре 60°C в течение двух часов и при комнатной температуре в течение 3 дней. Растворитель выпаривают при пониженном давлении. Добавляют EtOAc и органический слой промывают раствором KHSO4 (0,3M, водн.) и насыщенным раствором соли, сушат, фильтруют и выпаривают при пониженном давлении. Остаток очищают флэш-хроматографией (элюирование: ДХМ/EtOAc, 9:1), получая указанное в заголовке соединение (0,597 г, 80%). M/z 517,3.
Метод 108
1,1-Диоксо-3, 3-дибутил-5-(4-трет-бутоксикарбониламинофенил)-8-этоксикарбонилметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
1,1-Диоксо-3, 3-дибутил-5-(4-трет-бутоксикарбониламинофенил)-8-гидрокси-2,3,4,5-тетрагидро-1,5-бензотиазепин (метод 107; 597 мг, 1,16 ммоль) растворяют в MeCN (20 мл), добавляют K2CO3 (480 мг, 3,5 ммоль), бромид тетрабутиламмония (54 мг, 0,17 ммоль) и этилбромацетат (167 мкл, 1,5 ммоль). Смесь нагревают до 60°C и выдерживают при этой температуре в течение ночи. Растворитель выпаривают при пониженном давлении. Добавляют EtOAc и воду и водный слой дважды экстрагируют EtOAc. Объединенные органические экстракты промывают насыщенным раствором соли, сушат, фильтруют и выпаривают при пониженном давлении, получая указанное в заголовке соединение (0,617 г, 89%). M/z 603,3.
Метод 109
1,1-Диоксо-3, 3-дибутил-5-(4-трет-бутоксикарбониламинофенил)-8-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
1,1-Диоксо-3, 3-дибутил-5-(4-трет-бутоксикарбониламинофенил)-8-этоксикарбонилметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин (метод 108; 607 мг, 1,0 ммоль) растворяют в ТГФ (6 мл), добавляют H2O (6 мл) и LiOH (127 мг, 3,02 ммоль, моногидрат). Смесь перемешивают в течение 1 часа. После этого смесь выливают в воду и раствор подкисляют, используя водный раствор HCl (1M). Водный слой дважды экстрагируют EtOAc. Объединенные органические экстракты промывают насыщенным раствором соли, сушат, фильтруют и выпаривают при пониженном давлении, получая указанное в заголовке соединение (0,571 г, 99%). M/z 575, 4.
Метод 110
1,1-Диоксо-3,3-дибутил-5-(4-аминофенил)-8-[N-(α-(R)-метоксикарбонилбензил)карбамоилметокси]-2,3,4, 5-тетрагидро-1,5-бензотиазепин
1,1-Диоксо-3,3-дибутил-5-(4-трет-бутоксикарбониламинофенил)-8-[N-(α-(R)-метоксикарбонилбензил)карбамоилметокси]-2,3,4,5-тетрагидро-1, 5-бензотиазепин (метод 45; 562 мг, 0,78 ммоль) растворяют в ДХМ (18 мл). Добавляют ТФУК (4 мл) и реакционную смесь перемешивают в течение 3 часов. Растворитель выпаривают при пониженном давлении. Остаток распределяют между EtOAc и водным раствором NaOH (1M). Водную фазу экстрагируют еще раз EtOAc. Объединенные органические экстракты промывают насыщенным раствором соли, сушат, фильтруют и выпаривают при пониженном давлении, получая указанное в заголовке соединение (440 мг, 91%). M/z 622,5.
Метод 111
1,1-Диоксо-3, 3-дибутил-5-[4-(N'-трет-бутилуреидо)фенил]-8-[N-(α-(R)-метоксикарбонилlбензил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин
1,1-Диоксо-3, 3-дибутил-5-(4-аминофенил)-8-[N-(α-(R)-метоксикарбонилбензил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин (метод 110; 40 мг, 0,064 ммоль) растворяют в ДМФА (1 мл). Добавляют трет-бутилизоцианат (8,3 мкл, 0,071 ммоль). Реакционную смесь перемешивают при температуре 60-80°C в течение ночи. Добавляют трет-бутилизоцианат (20 мкл, 0,171 ммоль). Реакционную смесь перемешивают при температуре 60-80°C в течение 2 дней и затем при комнатной температуре в течение нескольких дней. Растворитель выпаривают при пониженном давлении. Продукт очищают препаративной ВЭЖХ (элюирование с градиентом: MeCN/ацетат-аммониевый буфер, от 5/95 до 100/0), получая указанное в заголовке соединение (30 мг, 65%). M/z 721,6.
Метод 112
1,1-Диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-[N-(α-метоксикарбонилметил-бензил)карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин
Указанное в заголовке соединение синтезируют из 1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепина (метод 17) и метил 3-амино-3-фенилпропионата (Helv. Chim. Acta; EN; 83; 6; 2000; 1256 - 1267) в соответствии с методикой примера 56. M/z 639,4.
Метод 113
трет-Бутил D-(S-тритил)цистеината гидрохлорида
К суспензии S-тритил-D-цистеина (2,0 г, 5,5 ммоль) в трет-бутилацетате (35 мл) при энергичном перемешивании по каплям добавляют 70% HClO4 (1,6 мл). Реакционную смесь перемешивают при комнатной температуре в течение 70 минут, затем добавляют EtOAc (50 мл) и NaHCO3 (водн., насыщ.) до pH 8,0. Осадок, непрореагировавший S-тритил-D-цистеин, удаляют фильтрованием. Органический слой отделяют, промывают 0,5 M HCl (2 x 75 мл) и насыщенным раствором соли, сушат и выпаривают, получая указанное в заголовке соединение (2,02 г, 81%). ЯМР (500 МГц): 1,43 (с, 9H), 2,83-2,95 (м, 2H), 3,41-3,48 (м, 1H), 7,21-7,37 (м, 9H), 7,46 (д, 6H).
Метод 114
1, 1-Диоксо-3,3-дибутил-5-фенил-7-метилтио-8-этоксикарбонил-2,3,4,5-тетрагидро-1,5-бензотиазепин
К суспензии 1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-гидрокси-2,3,4,5-тетрагидро-1, 5-бензотиазепина (метод 26; 12,85 г, 28,71 ммоль) в MeCN (150 мл) добавляют этилбромацетат (3,85 мл, 34,6 ммоль), бромид тетрабутиламмония (0,925 г, 2,869 ммоль) и карбонат натрия (12,85 г, 121,2 ммоль). Смесь кипятят с обратным холодильником в течение 5 часов. Растворитель удаляют при пониженном давлении и остаток распределяют между ДХМ и 0,5 M HCl. Органический слой промывают насыщенным раствором соли, сушат (MgSO4) и концентрируют. Хроматография (элюирование: ДХМ/EtOAc, 9:1) приводит к получению желаемого продукта (15,45 г) в виде темного масла. ЯМР 0,70-0,85 (м, 6H), 1, 00-1,55 (м, 15H), 2,15 (с, 3H), 3,10 (с, 2H), 3,70 (ушир.с, 2H), 4,25 (кв, 2H), 4,70 (с, 2H), 6,65 (с, 1H), 6,90-7,30 (м, 6H).
Метод 115
1,1-Диоксо-3-бутил-3-этил-5-фенил-8-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
1,1-Диоксо-3-бутил-3-этил-5-фенил-8-этоксикарбонилметокси-2,3,4, 5-тетрагидро-1,5-бензотиазепин (метод 116; 0,48 г, 1,04 ммоль) растворяют в этаноле (10 мл). Добавляют NaOH (0,30 г, 7,5 ммоль) и смесь кипятят с обратным холодильником в течение 30 минут. Добавляют уксусную кислоту (1 мл). Растворитель выпаривают при пониженном давлении и остаток экстрагируют смесью ДХМ/вода. Слой ДХМ отделяют, сушат и выпаривают. Получают 0,44 г (97%) указанного в заголовке соединения. ЯМР (300 МГц) 0,7-0,8 (м, 6H), 1,0-1,6 (м, 8H), 3,1-3,3 (м, 2H), 3,5-3,8 (м, 2H), 4,6 (с, 3H), 6,8-7,3 (м, 7H), 7,5 (с, 1H).
Метод 116
1,1-Диоксо-3-бутил-3-этил-5-фенил-8-этоксикарбонилметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
К MeCN (10 мл) добавляют 1, 1-диоксо-3-бутил-3-этил-5-фенил-8-гидрокси-2,3,4,5-тетрагидро-1,5-бензотиазепин (WO9616051; 0,40 г, 1,07 ммоль), этилбромацетат (0,23 г, 1,38 ммоль), карбонат натрия (0,50 г, 4,7 ммоль) и бромид тетрабутиламмония (30 мг, 0,093 ммоль). Смесь кипятят с обратным холодильником в течение 18 часов и затем выпаривают при пониженном давлении. Остаток экстрагируют смесью ДХМ/вода. Слой ДХМ отделяют и выпаривают при пониженном давлении. Остаток очищают колоночной хроматографией (элюирование: ДХМ/EtOAc, 90:10). Получают 0,480 г (97%) указанного в заголовке соединения. ЯМР (300 МГц) 0,7-0,85 (м, 6H), 1,0-1,7 (м, 11H), 3,1-3,3 (м, 2H), 3,6-3,8 (м, 2H), 4,3 (кв, 2H), 4,6 (с, 2H), 6,9-7,3 (м, 7H), 7,5 (д, 1H).
Метод 117
1, 1-Диоксо-3,3-дипропил-5-фенил-7-метилтио-8-гидрокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
К суспензии 1,1-диоксо-3,3-дипропил-5-фенил-7-бром-8-метокси-2,3,4,5-тетрагидро-1, 5-бензотиазепина (получен в соответствии с методикой публикации WO 96/16051 при использовании идентичных стадий синтеза, но с тем отличием, что исходное вещество выбрано с целью получения дипропилового, а не бутил/этилового производного; 0,756 г, 1,62 ммоль) в ДМФА (40 мл) добавляют NaSMe (0,605 г, 8,20 ммоль, 95 %), и смесь перемешивают в течение ночи при температуре 120°C. Растворитель удаляют при пониженном давлении и остаток распределяют между EtOAc и 0,5 M HCl. Водный слой экстрагируют еще два раза EtOAc и объединенные органические экстракты сушат (MgSO4) и концентрируют. Получают указанное в заголовке соединение (0,665 г, 98 %). ЯМР (500 МГц, ДМСО-d6) ЯМР (500 МГц, ДМСО-d6) 0,60-0,80 (м, 6H), 1,05-1,50 (м, 8H), 2,15 (с, 3H), 3,20 (с, 2Н) 3,65 (ушир.с, 2H), 6,65 (с, 1H), 6,75-6,95 (м, 3H), 7,10-7,25 (м, 2H), 7,30 (с, 1H), 10,5 (с, 1H).
Метод 118
1, 1-Диоксо-3,3-дипропил-5-фенил-7-метилтио-8-карбоксиметокси-2,3,4,5-тетрагидро-1,5-бензотиазепин
К суспензии 1,1-диоксо-3,3-дипропил-5-фенил-7-метилтио-8-гидрокси-2,3,4, 5-тетрагидро-1,5-бензотиазепина (Метод 117; 0,665 г, 1,58 ммоль) в MeCN (10 мл) добавляют этилбромацетат (0,262 мл, 2,35 ммоль), бромид тетрабутиламмония (0,051 г, 0,158 ммоль) и карбонат натрия (0, 870 г, 8,21 ммоль). Смесь перемешивают в течение ночи при температуре 80°C. Растворитель удаляют при пониженном давлении и остаток распределяют между EtOAc и 0,5 M HCl. Органический слой промывают насыщенным раствором соли, сушат (MgSO4) и концентрируют. Остаток фильтруют через колонку небольшой высоты, наполненную диоксидом кремния (элюирование: ДХМ/EtOAc, 9:1), концентрируют и растворяют в EtOH (10 мл). Добавляют раствор NaOH (0,25 г, 6,25 ммоль) в воде (1 мл) и полученную смесь перемешивают в течение ночи при комнатной температуре. Растворитель удаляют при пониженном давлении и остаток распределяют между EtOAc и 0,5 M HCl. Водный слой экстрагируют еще два раза EtOAc, объединенные органические экстракты промывают насыщенным раствором соли и концентрируют. Сырой продукт очищают препаративной ВЭЖХ (элюент: MeCN/ацетат-аммониевый буфер), получая указанное в заголовке соединение (0,441 г, 58%) в виде твердого белого вещества. ЯМР (ДМСО-d6) 0,55-0,75 (м, 6H), 1,05-1,50 (м, 8H), 2,15 (с, 3H), 3,20 (с, 2H), 3,65 (ушир.с, 2H), 4,50 (с, 2H), 6,65 (с, 1H), 6,80-7,00 (м, 3H), 7,15 (с, 1H), 7,15-7,25 (м,2H).
Пример 121
Приведенные далее композиции являются типичными примерами фармацевтических дозированных форм, содержащих соединение формулы (I) или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарство (соединение Х) для терапевтического или профилактического применения у людей:
Примечание
Представленные препараты могут быть получены стандартными способами, хорошо известными в фармацевтической области. Таблетки (а) - (с) стандартными способами могут быть покрыты энтеросолюбильным покрытием, например покрытием из целлюлозоацетатфталата.
Реферат
Данное изобретение относится к 1,5-бензотиазепинам формулы (I), где RV и RW независимо выбраны из водорода или С1-6алкила; R1 и R2 независимо выбраны из С1-6алкила; Rx и Ry независимо выбраны из водорода или С1-6алкила, или один из Rx и Ry представляет собой водород или C1-6алкил, а другой представляет собой гидроксил или C1-6алкокси; Rz выбран из галогена, нитро, циано, гидроксила, амино, карбокси, карбамоила, меркапто, сульфамоила, С1-6алкила и др. значений указанных в п.1 формулы изобретения; v принимает значение от 0 до 5; один из R4 и R5 представляет собой группу формулы (IA): R3 и R6 и второй из R4 и R5 независимо выбраны из водорода, галогена, нитро, циано, гидроксила, амино, карбокси, карбамоила, меркапто, сульфамоила, С1-4алкила и др. значений указанных в п.1 формулы изобретения; причем R3 и R6 и второй из R4 и R5 могут быть необязательно замещенными по углероду одним или несколькими R16; D представляет собой -О-, -N(Ra)-, -S(O)b- или -CH(Ra)-; где, Ra представляет собой водород или C1-6алкил, и b принимает значение от 0 до 2; кольцо А представляет собой арил или гетероарил, причем кольцо А является необязательно замещенным одним или несколькими заместителями, выбранными из R17; R7 представляет собой водород, С1-4алкил, карбоциклил или гетероциклил, причем R7 является необязательно замещенным одним или несколькими заместителями, выбранными из R18; R8 представляет собой водород или С1-4алкил; R9представляет собой водород или С1-4алкил; R10 представляет собой водород, C1-4алкил, карбоциклил или гетероциклил, причем R10 является необязательно замещенным одним или несколькими заместителями, выбранными из R19; R11 представляет собой карбокси, сульфо, сульфино, фосфоно, тетразолил, -P(O)(ORc)(ORd), -P(O)(OH)(ORc), -Р(O)(ОН)(Рd) или -P(O)(ORc)(Rd), где Rc и Rd независимо выбраны из C1-6алкила; либо R11 представляет собой группу формулы (IB): где: Х представляет собой -N(Rq)-, -N(Rq)C(O)-, -О- и -S(O)a; где а принимает значение от 0 до 2, и Rqпредставляет собой водород или С1-4алкил; R12 представляет собой водород или C1-4алкил; R13 и R14независимо выбраны из водорода, С1-4алкила, карбоциклила, гетероциклила или R23, причем указанные С1-4алкил, карбоциклил или гетероциклил могут быть независимо необязательно замещенными одним или несколькими заместителями, выбранными из R20; R15 представляет собой карбокси, сульфо, сульфино, фосфоно, тетразолил, -P(O)(ORe)(ORf), -P(O)(OH)(ORe), -P(O)(OH)(Re) или -Р(O)(ORe)(Rf), где Re и Rf независимо выбраны из C1-6алкила; или R15 представляет собой группу формулы (IC): где R24 выбран из водорода или C1-4алкила; R25 выбран из водорода, С1-4алкила, карбоциклила, гетероциклила или R27, причем указанные С1-4алкил, карбоциклил или гетероциклил могут быть независимо необязательно замещенными одним или несколькими заместителями, выбранными из R28; R26 выбран из карбокси, сульфо, сульфино, фосфоно, тетразолила, -P(O)(ORg)(ORh), -Р(O)(ОН)(ORg), -Р(O)(ОН)(Rg) или -Р(O)(ORg)(Rh), где Rg и Rh независимо выбраны из С1-6алкила; р принимает значение от 1 до 3; где значения R13 могут быть одинаковыми или разными; q принимает значение от 0 до 1; r принимает значение от 0 до 3; где значения R14 могут быть одинаковыми или разными; m принимает значение от 0 до 2; где значения R10 могут быть одинаковыми или разными; n принимает значение от 1 до 3; где значения R7 могут быть одинаковыми или разными; z принимает значение от 0 до 3; где значения R25 могут быть одинаковыми или разными; R16, R17 и R18 независимо выбраны из галогена, нитро, циано, гидроксила, амино, карбокси, карбамоила, меркапто, сульфамоила, С1-4 алкила, С2-4алкенила, С2-4алкинила, С1-4алкокси, С1-4алканоила, С1-4алканоилокси, N-(С1-4алкил)2амино, N,N-(С1-4 алкил)2амино, С1-4алканоиламино, N-(С1-4алкил)карбамоила, N,N-(С1-4алкил)2карбамоила, С1-4алкилS(O)а, где а принимает значение от 0 до 2, С1-4алкоксикарбонила, N-(С1-4алкил)сульфамоила и N,N-(C1-4алкил)2сульфамоила; где R16, R17 и R18 могут быть независимо необязательно замещенными по углероду одним или несколькими R21; R19, R20, R23, R27 и R28 независимо выбраны из галогена, нитро, циано, гидроксила, амино, карбокси, карбамоила, меркапто, сульфамоила, С1-4алкила, С2-4алкенила, С2-4алкинила, С1-4алкокси, С1-4 алканоила, С1-4алканоилокси, N-(С1-4алкил)амино, N,N-(C1-4алкил)2амино, С1-4алканоиламино, N-(С1-4алкил)карбамоила, N,N-(С1-4алкил)2карбамоила, C1-4алкилS(O)а, где а принимает значение от 0 до 2, С1-4алкоксикарбонила, N-(С1-4алкил)сульфамоила, N,N-(С1-4алкил)2сульфамоила, карбоциклила, гетероциклила, сульфо, сульфино, амидино, фосфоно, P(O)(ORa)(ORb), -P(O)(OH)(ORa), -P(O)(OH)(Ra) или -P(О)(ORa)(Rb), где Ra и Rb независимо выбраны из C1-6алкила; и где R19, R20, R23, R27 и R28 могут быть независимо необязательно замещенными по углероду одним или несколькими R22; R21 и R22 независимо выбраны из галогена, гидроксила, циано, карбамоила, уреидо, амино, нитро, карбокси, карбамоила, меркапто, сульфамоила, трифторметила, трифторметокси, метила, этила, метокси, этокси, винила, аллила, этинила, метоксикарбонила, формила, ацетила, формамидо, ацетиламино, ацетокси, метиламино, диметиламино, N-метилкарбамоила, N,N-диметилкарбамоила, метилтио, метилсульфинила, мезила, N-метилсульфамоила и N,N-диметилсульфамоила; или его фармацевтически приемлемая соль, сольват или сольват такой соли. Также описываются способы получения соединений формулы (I), фармацевтические композиции на основе соединений формулы (I), способ получения ингибирующего действия в отношении IBAT и промежуточные соединения. 14 н. и 22 з.п. ф-лы.

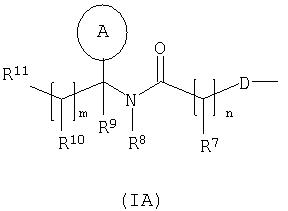
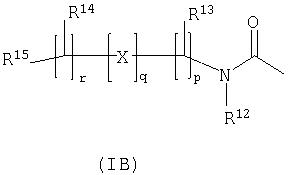
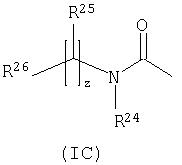
Формула
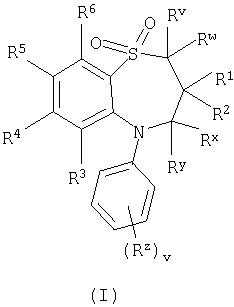
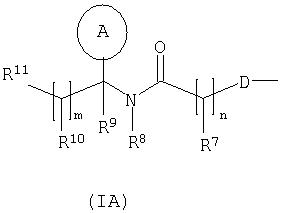
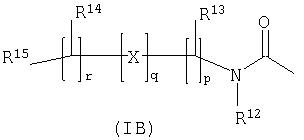
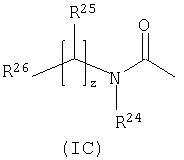
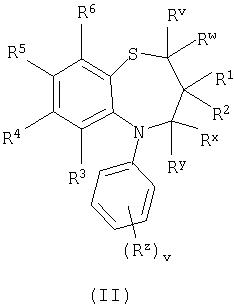
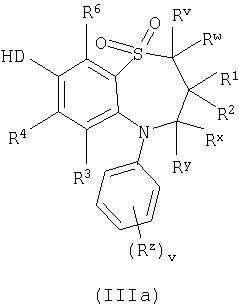
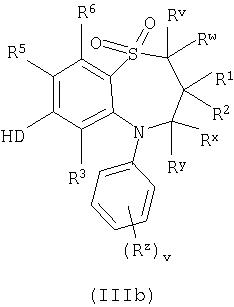
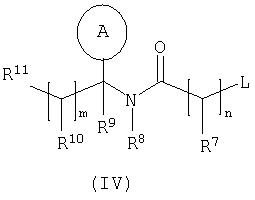
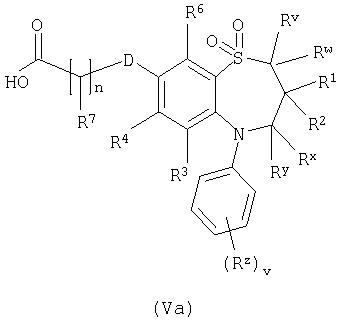


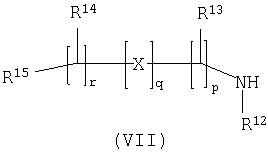



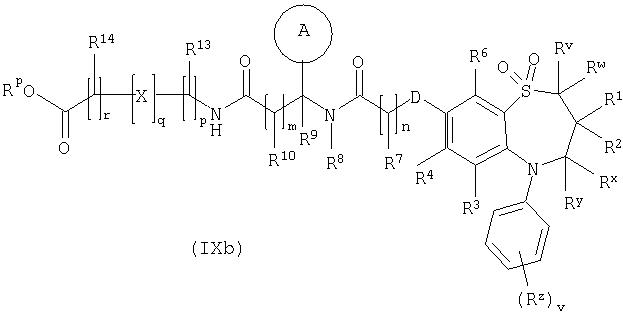
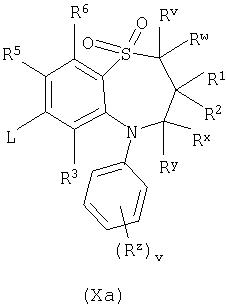

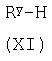
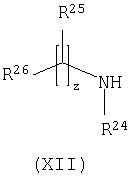

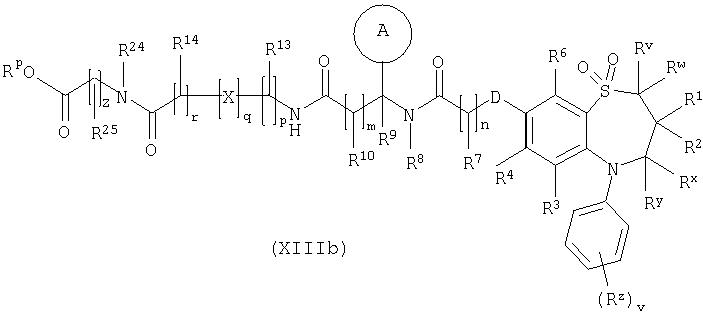
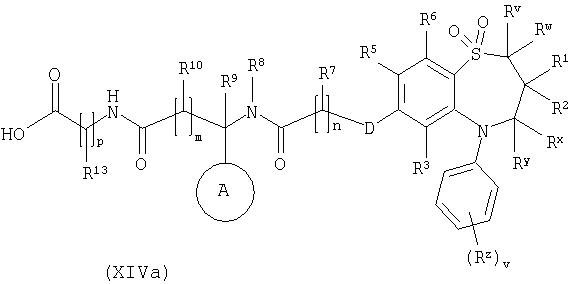


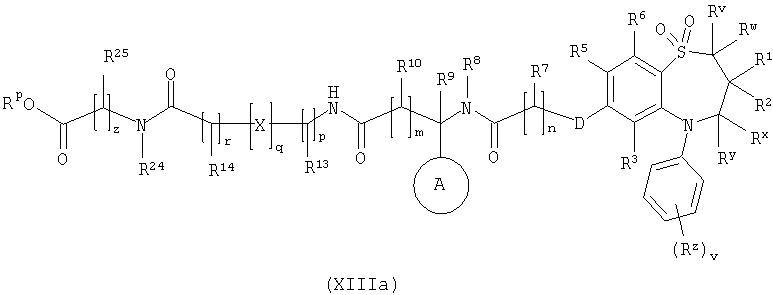

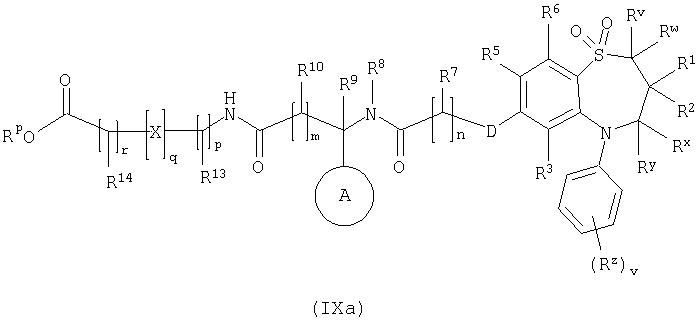
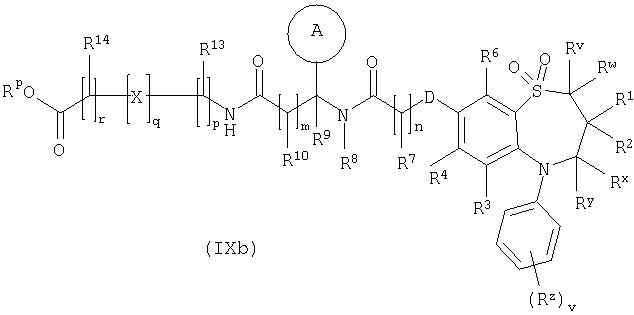

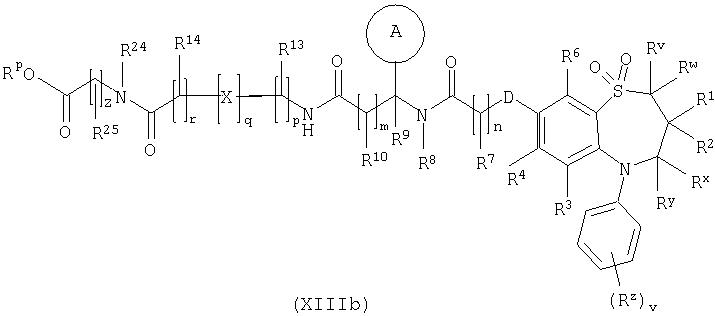
Комментарии