Новые производные бензодиазепина - RU2683325C2
Код документа: RU2683325C2
Чертежи








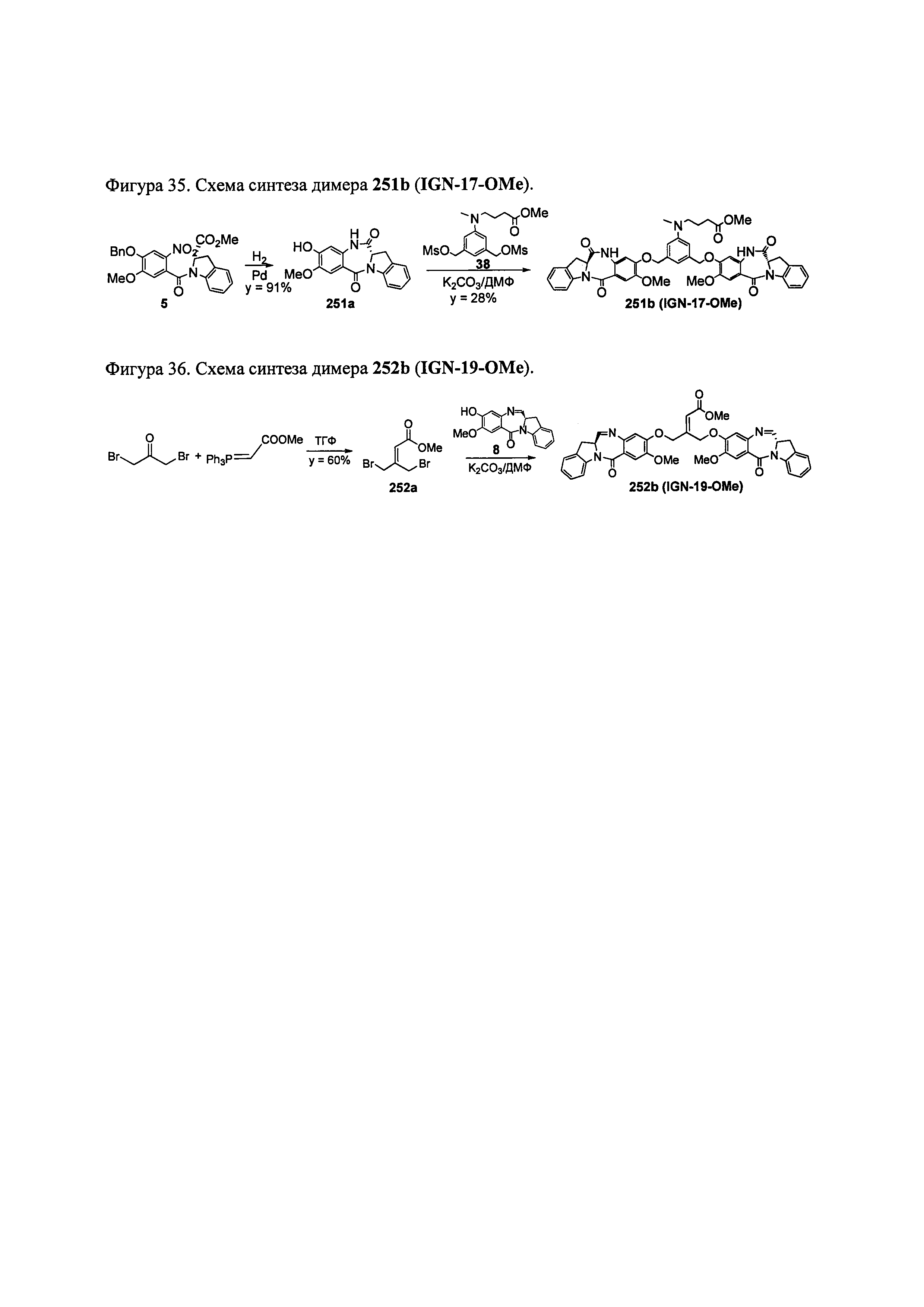






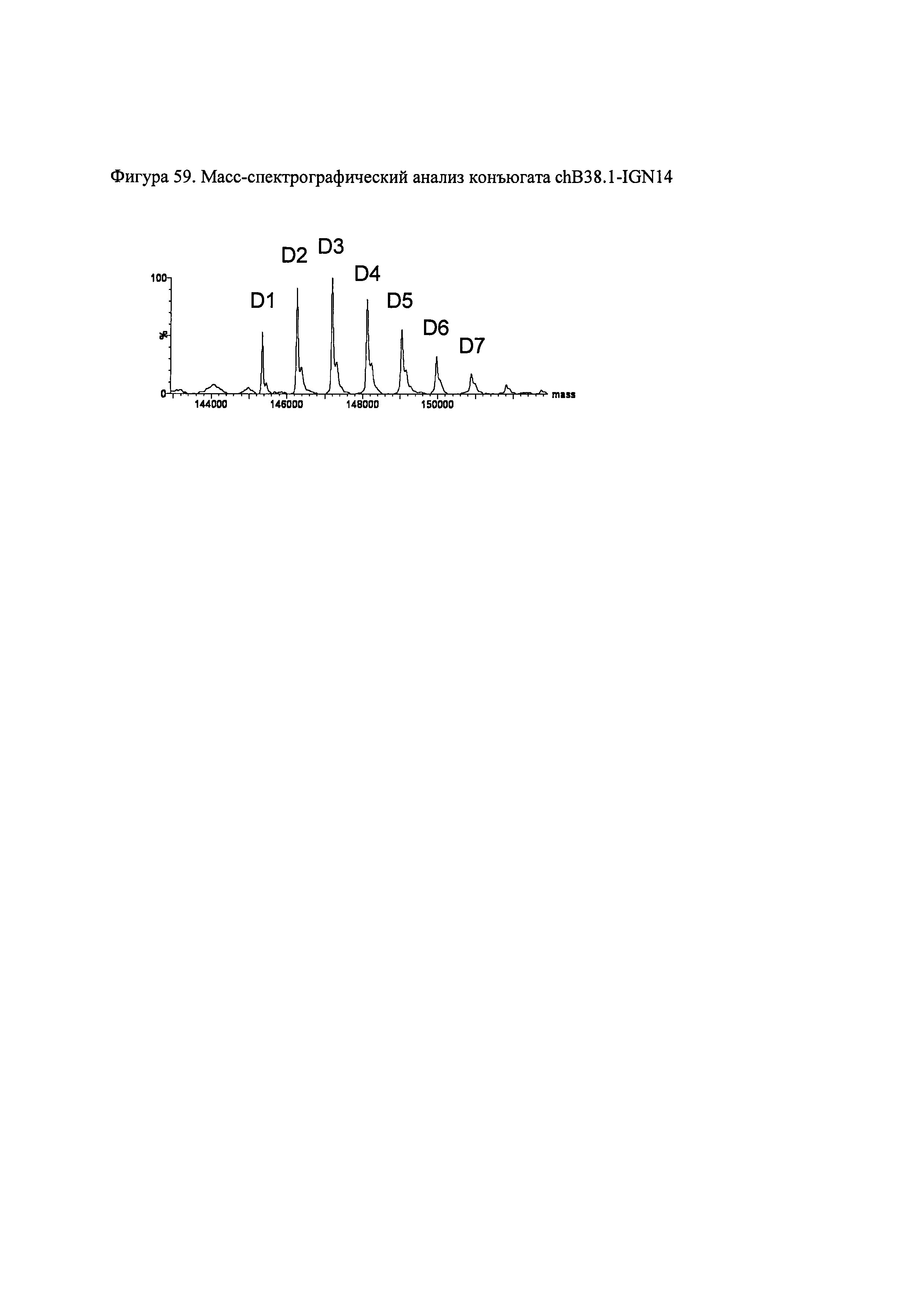
Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к новым цитотоксичным соединениям и цитотоксичным конъюгатам, содержащим эти цитотоксичные соединения и клеточносвязывающие агенты. Более конкретно, настоящее изобретение относится к новым бензодиазепиновым соединениям (напр., индолинобензодиазепинам или оксазолидинобензодиазепинам), их производным, их промежуточным соединениям, их конъюгатам и их фармацевтически приемлемым солям, которые применимы в качестве лекарственных средств, в частности, в качестве антипролиферативных агентов.
Уровень техники
Производные бензодиазепина применяются для лечения различных нарушений и включают такие лекарственные препараты как противоэпилептические средства (имидазо[2,1-b][1,3,5]бензотиадиазепины, патент США №4444688; патент США №4062852), антибактериальные средства (пиримидо[1,2-с][1,3,5]бензотиадиазепины, GB 1476684), диуретические и гипотензивные средства (пирроло(1,2-b)[1,2,5]бензотиадиазепина 5,5 диоксид, патент США №3506646), гиполипидемические средства (WO 03091232), антидепрессанты (патент США №3453266); средства лечения остеопороза (JP 2138272).
Недавно на моделях опухолей у животных было показано, что производные бензодиазепина, такие как пирролобензодиазепины (ПБД), действуют как противоопухолевые агенты (N-2-имидазолил алкил замещенный 1,2,5-бензотиадиазепин-1,1-диоксид, патент США №6156746), бензо-пиридо или дипиридо тиадиазепин (WO 2004/069843), производные пирроло[1,2-b][1,2,5]бензотиадиазепинов и пиррол[1,2-b][1,2,5]бензодиазепина (WO 2007/015280), производные томаймицина (напр., пирроло[1,4]бензодиазепины), такие как описаны в публикациях WO 00/12508, WO 2005/085260, WO 2007/085930 и ЕР 2019104. Известно также, что бензодиазепины влияют на рост клеток и их дифференциацию (Kamal A., et al., Bioorg Med Chem. 2008 Aug 15; 16(16):7804-10 (и приведенные в нем ссылки); Kumar R, Mini Rev Med Chem. 2003 Jun; 3(4):323-39 (и приведенные в нем ссылки); Bednarski J J, et al., 2004; Sutter A. P, et al., 2002; Blatt N B, et al., 2002), Kamal A. et al., Current Med. Chem., 2002; 2; 215-254, Wang J-J., J. Med. Chem., 2206; 49:1442-1449, Alley M.C. et al., Cancer Res. 2004; 64:6700-6706, Pepper C.J., Cancer Res 2004; 74:6750-6755, Thurston D.E. and Bose D.S., Chem Rev 1994; 94:433-465 и Tozuka, Z., et al., Journal of Antibiotics, (1983) 36; 1699-1708. Общая структура ПБД описана в публикации США номер 20070072846. ПБД различаются количеством, типом и положением заместителей как в их ароматических А кольцах, так и в пирроловых С кольцах, а также степенью насыщенности С кольца. Их способность образовывать аддукт в малой бороздке обусловливает возможность их взаимодействия с процессом образования ДНК, что дает возможность их потенциального использования в качестве антипролиферативных агентов.
До сих пор существует необходимость в новых производных бензодиазепина в качестве эффективных и безопасных лекарственных препаратов для лечения различных пролиферативных заболеваний, таких как рак.
Раскрытие изобретения
Одна из целей настоящего изобретения заключается в предоставлении новых бензодиазепинов формулы (I) и (II), в которых диазепиновое кольцо (В) конденсировано с гетероциклическим кольцом (CD), где гетероциклическое кольцо является бициклическим,
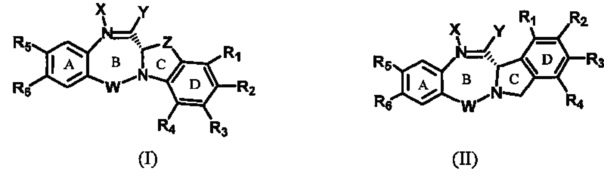
где:
двойная линия

Y выбран из групп: -OR, сложный эфир, представленный группой -OCOR', карбонат, представленный группой -OCOOR', карбамат, представленный группой -OCONR'R'', амин или гидроксиламин, представленный группой NR'R'', амид, представленный группой -NRCOR', пептид, представленный группой NRCOP, где Р является аминокислотой или полипептидом, содержащим от 2 до 20 аминокислотных звеньев, тиоэфир, представленный группой SR', сульфоксид, представленный группой SOR', сульфон, представленный группой -SO2R', сульфит -SO3, бисульфит -OSO3, галоген, циан, азид или тиол, где R, R' и R'' одинаковые или разные и выбраны из групп: Н, замещенный или незамещенный линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 20 углеродных атомов, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000; 5- или 6-членное гетероарильное кольцо, содержащее один или более гетероатомов, независимо выбранных из азота, кислорода и серы, 5-18-членная конденсированная кольцевая система, где по меньшей мере одно кольцо является ароматическим, содержащая один или более гетероатомов, независимо выбранных из азота, кислорода и серы; арил, имеющий от 6 до 18 углеродных атомов, 3-18-членное гетероциклическое кольцо, имеющее от 1 до 6 гетероатомов, выбранных из О, S, N и P, где заместитель выбран из групп: галоген, OR7, NR8R9, NO2, NRCOR', SR10, сульфоксид, представленный группой SOR', сульфон, представленный группой -SO2R', сульфит -SO3, бисульфит -OSO3, сульфонамид, представленный группой SO2NRR', циано, азидо, -COR11, OCOR11 или OCONR11R12, где каждый из R7, R8, R9, R10, R11 и R12 независимо выбран из групп: Н, линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000, 5-или 6-членное гетероарильное кольцо, содержащее один или более гетероатомов, независимо выбранных из азота, кислорода и серы; 5-18-членная конденсированная кольцевая система, где по меньшей мере одно кольцо является ароматическим, содержащая один или более гетероатомов, независимо выбранных из азота, кислорода и серы, арил, имеющий от 6 до 18 углеродных атомов, 3-18-членное гетероциклическое кольцо, имеющее от 1 до 6 гетероатомов, выбранных из О, S, N и Р и R10 является необязательно SR13 или COR13, где R13 выбран из групп: линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000, 5-или 6-членное гетероарильное кольцо, содержащее один или более гетероатомов, независимо выбранных из азота, кислорода и серы, 5-18-членная конденсированная кольцевая система, где по меньшей мере одно кольцо является ароматическим, содержащая один или более гетероатомов, независимо выбранных из азота, кислорода и серы, 3-18-членное гетероциклическое кольцо, имеющее от 1 до 6 гетероатомов, выбранных из О, S, N и Р, и R11 может быть также OR14, где R14 является Н или имеет такое же определение, что и R, необязательно R'' является ОН;
W является С=O, C=S, СН2, ВН (В = бор), SO или SO2;
каждый из R1, R2, R3, R4 независимо выбран из групп: Н, замещенный или незамещенный линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000, или заместитель, выбранный из групп: галоген, OR7, NR8R9, NO2, NRCOR', SR10, сульфоксид, представленный группой SOR', сульфон, представленный группой -SO2R', сульфит -SO3, бисульфит -OSO3, сульфонамид, представленный группой SO2NRR', циано, азидо, гуанидиний [-NH(C=NH)NH2], -COR11, -OCOR11 или -OCONR11R12, где R7, R8, R9, R10, R11 и R12 имеют те же определения, которые представлены выше, необязательно любой из R1, R2, R3, R4 является связывающей группой, обеспечивающей связь с клеточносвязывающим агентом за счет ковалентной связи или выбран из групп: полипирроловое, полииндолиловое, полиимидазолиловое, полипирроло-индолиловое или полиимидазоло-индолиловое звено, необязательно прикрепленное к связывающей группе, обеспечивающей связь с клеточносвязывающим агентом;
R5 выбран из групп: OR15, CRR'OH, SH, CRR'SH, NHR15 или CRR'NHR15, где R15 имеет то же определение, что и R, R и R' имеют то же определение, которое дано выше; необязательно R5 является связывающей группой, обеспечивающей связь с клеточносвязывающим агентом за счет ковалентной связи или выбран из групп: полипирроловое, полииндолиловое, полиимидазолиловое, полипирроло-имидазолиловое, полипирроло-индолиловое или полиимидазоло-индолиловое звено, необязательно прикрепленное к связывающей группе, обеспечивающей связь с клеточносвязывающим агентом;
R6 является OR, SR, NRR', где R и R' имеют то же определение, которое дано выше, или необязательно R6 является связывающей группой;
Z выбран из групп: (СН2)n, гдеn является 1, 2 или 3, CR15R16, NR17, О или S, где каждый из R15, R16 и R17 независимо выбран из групп: Н, линейный, разветвленный или циклический алкил, имеющий от 1 до 10 углеродных атомов, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000; или их фармацевтически приемлемые сольваты, соли, гидраты или гидратированные соли, их оптические изомеры, рацематы, диастереомеры, энантиомеры этих соединений.
при условии, что соединение настоящего изобретения имеет не более одной связывающей группы, обеспечивающей связь с клеточносвязывающим агентом за счет ковалентной связи.
Вторая цель настоящего изобретения заключается в предоставлении новых бензодиазепинов формулы (III), в которой диазепиновое кольцо (В) конденсировано с гетероциклическим кольцом (С), где гетероциклическое кольцо является моноциклическим,

где:
двойная линия
Y выбран из групп: -OR, сложный эфир, представленный группой -OCOR', карбонат, представленный группой -OCOOR', карбамат, представленный группой -OCONR'R'', амин или гидроксиламин, представленный группой NR'R'', амид, представленный группой -NRCOR', пептид, представленный группой NRCOP, где Р является аминокислотой или полипептидом, содержащим от 2 до 20 аминокислотных звеньев, тиоэфир, представленный группой SR', сульфоксид, представленный группой SOR', сульфон, представленный группой -SO2R', сульфит -SO3, бисульфит -OSO3, галоген, циано, азидо или тиол, где R, R' и R'' одинаковые или разные и выбраны из групп: Н, замещенный или незамещенный линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000, 5- или 6-членное гетероарильное кольцо, имеющее один или более гетероатомов, независимо выбранных из азота, кислорода и серы, 5-18-членная конденсированная кольцевая система, где по меньшей мере одно кольцо является ароматическим, содержащая один или более гетероатомов, независимо выбранных из азота, кислорода и серы, арил, имеющий от 6 до 18 углеродных атомов, 3-18-членное гетероциклическое кольцо, имеющее 1-6 гетероатомов, выбранных из О, S, N и Р, где заместитель выбран из групп: галоген, OR7, NR8R9, NO2, NRCOR', SR10, сульфоксид, представленный группой SOR', сульфон, представленный группой -SO2R', сульфит -SO3, бисульфит -OSO3, сульфонамид, представленный группой SO2NRR', циано, азидо, -COR11, OCOR11 или OCONR11R12, где каждый из R7, R8, R9, R10, R11 и R12 независимо выбран из групп: Н, линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000, 5- или 6-членное гетероарильное кольцо, содержащее один или более гетероатомов, независимо выбранных из азота, кислорода и серы, 5-18-членная конденсированная кольцевая система, где по меньшей мере одно кольцо является ароматическим, содержащая один или более гетероатомов, независимо выбранных из азота, кислорода и серы, арил, имеющий от 6 до 18 углеродных атомов, 3-18-членное гетероциклическое кольцо, имеющее 1-6 гетероатомов, выбранных из О, S, N и Р и R10 необязательно является SR13 или COR13, где R13 выбран из групп: линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000, 5- или 6-членное гетероарильное кольцо, содержащее один или более гетероатомов, независимо выбранных из азота, кислорода и серы, 5-18-членная конденсированная кольцевая система, где по меньшей мере одно кольцо является ароматическим, содержащая один или более гетероатомов, независимо выбранных из азота, кислорода и серы, арил, имеющий от 6 до 18 углеродных атомов, 3-18-членное гетероциклическое кольцо, имеющее от 1 до 6 гетероатомов, выбранных из О, S, N и Р и R11 может быть также OR14, где R14 является Н или имеет такое же определение, что и R, необязательно R'' является ОН;
W является С=O, C=S, СН2, ВН, SO или SO2;
R5 выбран из групп: OR15, CRR'OH, SH, CRR'SH, NHR15 или CRR'NHR15, где R15 имеет то же определение, что и R, или является связывающей группой, обеспечивающей связь с клеточносвязывающим агентом за счет ковалентной связи или выбран из групп: полипирроловое, полииндолиловое, полиимидазолиловое, полипирроло-имидазолиловое, полипирроло-индолиловое или полиимидазоло-индолиловое звено, необязательно прикрепленное к связывающей группе, обеспечивающей связь с клеточносвязывающим агентом;
R6 является OR, SR или NRR', где R и R' имеют то же определение, которое дано выше, необязательно R6 является связывающей группой;
X' является СН2, NR, СО, ВН, SO или SO2;
Y' является О, СН2, NR или S;
Z' является СН2 или (СН2)n, где n является 2, 3 или 4; или их фармацевтически приемлемые сольваты, соли, гидраты или гидратированные соли, их оптические изомеры, рацематы, диастереомеры, энантиомеры или полиморфные кристаллические структуры этих соединений;
при условии, что соединение настоящего изобретения имеет не более одной связывающей группы, обеспечивающей связь с клеточносвязывающим агентом за счет ковалентной связи.
Третья цель настоящего изобретения заключается в предоставлении цитотоксических димеров (IV), (V) и (VI)
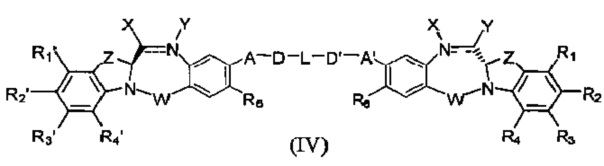
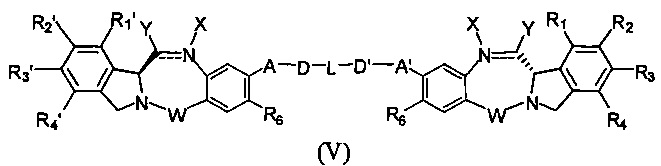
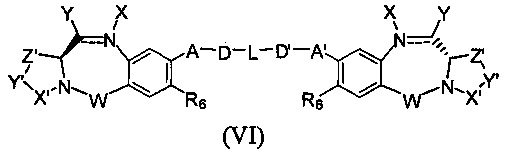
бензодиазепиновых мономеров формул (I), (II) и (III), соответственно, в которых димерные соединения необязательно имеют связывающую группу, обеспечивающую связь с клеточносвязывающими агентами,
где:
двойная линия
Y выбран из групп: -OR, сложный эфир, представленный группой -OCOR', карбонат, представленный группой -OCOOR', карбамат, представленный группой -OCONR'R", амин или гидроксиламин, представленный группой NR'R'', амид, представленный группой -NRCOR', пептид, представленный группой NRCOP, где Р является аминокислотой или полипептидом, содержащим от 2 до 20 аминокислотных звеньев, тиоэфир, представленный группой SR', сульфоксид, представленный группой SOR', сульфон, представленный группой -SO2R', сульфит -SO3, бисульфит -OSO3, галоген, циано, азидо или тиол, где R, R' и R'' одинаковые или разные и выбраны из групп: Н, замещенный или незамещенный линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, полиэтиленгликолевое звено (-ОСН2СН2)n где n является целым числом от 1 до 2000, 5- или 6-членное гетероарильное кольцо, имеющее один или более гетероатомов, независимо выбранных из азота, кислорода и серы, 5-18-членная конденсированная кольцевая система, где по меньшей мере одно кольцо является ароматическим, содержащая один или более гетероатомов, независимо выбранных из азота, кислорода и серы, арил, имеющий от 6 до 18 углеродных атомов, 3-18-членное гетероциклическое кольцо, имеющее 1-6 гетероатомов, выбранных из О, S, N и Р, где заместитель выбран из групп: галоген, OR7, NR8R9, NO2, NRCOR', SR10, сульфоксид, представленный группой SOR', сульфон, представленный группой -SO2R', сульфит -SO3, бисульфит -OSO3, сульфонамид, представленный группой SO2NRR', циано, азидо, -COR11, OCOR11 или OCONR11R12, где каждый из R7, R8, R9, R10, R11 и R12 независимо выбран из групп: Н, линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000, 5- или 6-членное гетероарильное кольцо, содержащее один или более гетероатомов, независимо выбранных из азота, кислорода и серы, 5-18-членная конденсированная кольцевая система, где по меньшей мере одно кольцо является ароматическим, содержащая один или более гетероатомов, независимо выбранных из азота, кислорода и серы, арил, имеющий от 6 до 18 углеродных атомов, 3-18-членное гетероциклическое кольцо, имеющее 1-6 гетероатомов, выбранных из О, S, N и Р и R10 необязательно является SR13 или COR13, где R13 выбран из групп: линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, 5- или 6-членное гетероарильное кольцо, содержащее один или более гетероатомов, независимо выбранных из азота, кислорода и серы, 5-18-членная конденсированная кольцевая система, где по меньшей мере одно кольцо является ароматическим, содержащая один или более гетероатомов, независимо выбранных из азота, кислорода и серы, арил, имеющий от 6 до 18 углеродных атомов, 3-18-членное гетероциклическое кольцо, имеющее от 1 до 6 гетероатомов, выбранных из О, S, N и Р, необязательно R11 является OR14, где R14 имеет такое же определение, что и R, необязательно R'' является ОН;
W является С=O, C=S, СН2, ВН, SO или SO2;
каждый из R1, R2, R3, R4, R1', R2', R3' и R4' независимо выбран из групп: Н, замещенный или незамещенный линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000, или заместитель, выбранный из групп: галоген, гуанидиний [-NH(C=NH)NH2], OR7, NR8R9, NO2, NRCOR', SR10, сульфоксид, представленный группой SOR', сульфон, представленный группой -SO2R', сульфит -SO3, бисульфит -OSO3, сульфонамид, представленный группой SO2NRR', циано, азидо, -COR11, OCOR11 или OCONR11R12, где R7, R8, R9, R10, R11 и R12 имеют те же определения, которые представлены выше, необязательно любой из R1, R2, R3, R4, R1', R2', R3' или R4' является связывающей группой, обеспечивающей связь с клеточносвязывающим агентом за счет ковалентной связи или выбран из групп: полипирроловое, полииндолиловое, полиимидазолиловое, полипирроло-имидазолиловое, полипирроло-индолиловое или полиимидазоло-индолиловое звено, необязательно прикрепленное к связывающей группе, обеспечивающей связь с клеточносвязывающим агентом;
Z выбран из групп: (СН2)n, где n является 1, 2 или 3, CR15R16, NR17, О или S, где каждый из R15, R16 и R17 независимо выбран из групп: Н, линейный, разветвленный или циклический алкил, имеющий от 1 до 10 углеродных атомов, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000;
R6 является OR, SR или NRR', где R и R' имеют то же определение, которое дано выше, необязательно R6 является связывающей группой;
X' выбран из групп: СН2, NR, СО, ВН, SO или SO2, где R имеет то же определение, которое дано выше;
Y' является О, СН2, NR или S, где R имеет то же определение, которое дано выше;
Z' является СН2 или (СН2)n, где n является 2, 3 или 4, при условии, что X', Y' и Z' не являются одновременно СН2;
А и А' одинаковые или различные и выбраны из групп: О, -CRR'O, S, -CRR'S, -NR15 или CRR'NHR15, где R и R' имеют то же определение, которое дано выше и где R15 имеет то же определение, что и R.
D и D' одинаковые или различные и независимо выбраны из групп: линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, необязательно замещенный любой из групп: галоген, OR7, NR8R9, NO2, NRCOR', SR10, сульфоксид, представленный группой SOR', сульфон, представленный группой -SO2R', сульфит -SO3, бисульфит -OSO3, сульфонамид, представленный группой SO2NRR', циано, азидо, -COR11, OCOR11 или OCONR11R12, где определения R7, R8, R9, R10, R11 и R12 такие же, как определено выше, или полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000;
L необязательно является фенильной группой или 3-18-членным гетероциклическим кольцом, имеющим от 1 до 6 гетероатомов, выбранных из О, S, N и Р, необязательно замещенным, где заместитель является связывающей группой, обеспечивающей связь с клеточносвязывающим агентом за счет ковалентной связи, или выбран из групп: линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, необязательно замещенный любой из групп: галоген, OR7, NR8R9, NO2, NRCOR', SR10, сульфоксид, представленный группой SOR', сульфон, представленный группой -SO2R', сульфит -SO3, бисульфит -OSO3, сульфонамид, представленный группой SO2NRR', циано, азидо, -COR11, OCOR11 или OCONR11R12, где R7, R8, R9, R10, R11 и R12 имеют то же определение, которое дано выше, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000; необязательно L является связывающей группой, обеспечивающей связь с клеточносвязывающим агентом за счет ковалентной связи; или их фармацевтически приемлемые сольваты, соли, гидраты или гидратированные соли, их оптические изомеры, рацематы, диастереомеры, энантиомеры или полиморфные кристаллические структуры этих соединений; при условии, что соединение настоящего изобретения имеет не более одной связывающей группы, обеспечивающей связь с клеточносвязывающим агентом за счет ковалентной связи.
Четвертая цель настоящего изобретения заключается в предоставлении конъюгатов клеточносвязывающих агентов с новыми соединениями бензодазепина настоящего изобретения или их производными. Эти конъюгаты могут применяться в качестве лекарственных средств, которые направленно доставляются к клеткам-мишеням и являются цитотоксичными.
Настоящее изобретение включает композиции (напр., фармацевтические композиции), содержащие новые бензодиазепиновые соединения, их производные, их конъюгаты (и/или сольваты, гидраты и/или их соли) и носитель (фармацевтически приемлемый носитель). Настоящее изобретение включает также композиции (напр., фармацевтические композиции), содержащие новые бензодиазепиновые соединения, их производные или их конъюгаты (и/или сольваты, гидраты и/или их соли) и носитель (фармацевтически приемлемый носитель), и дополнительно содержащие второе терапевтическое средство. Настоящие композиции применимы для ингибирования аномального клеточного роста или лечения пролиферативных нарушений у млекопитающих (напр., человека). Настоящие композиции также применимы для лечения депрессии, тревоги, стресса, фобий, паники, дисфории, психиатрических нарушений, боли, воспалительных заболеваний у млекопитающих (напр., человека).
Настоящее изобретение включает способ ингибирования аномального клеточного роста или лечения пролиферативных нарушений у млекопитающих (напр., человека), включая введение оговоренным млекопитающим терапевтически эффективного количества новых бензодиазепиновых соединений, их производных или их конъюгатов (и/или их сольватов и солей) или их композиций, по отдельности или в сочетании со вторым терапевтическим средством.
Настоящее изобретение включает способ получения и использования новых бензодиазепиновых соединений, их производных и их конъюгатов для диагностирования или лечения in vitro, in situ и in vivo клеток и организмов млекопитающих или связанных патологических состояний.
Соединения настоящего изобретения, их производные или их конъюгаты и содержащие их композиции могут применяться для лечения или ослабления степени нарушений, таких как нарушения, характеризующиеся аномальным ростом клеток (напр., рак). Другие применения соединений и конъюгатов настоящего изобретения включают, не ограничиваясь, лечение остеопороза, депрессии, тревоги, стресса, фобий, паники, дисфории, психиатрических нарушений и боли или в качестве противоэпилептических, антибактериальных, диуретических и гипотензивных, гиполипидемических средств и антидепрессантов.
Краткое описание фигур
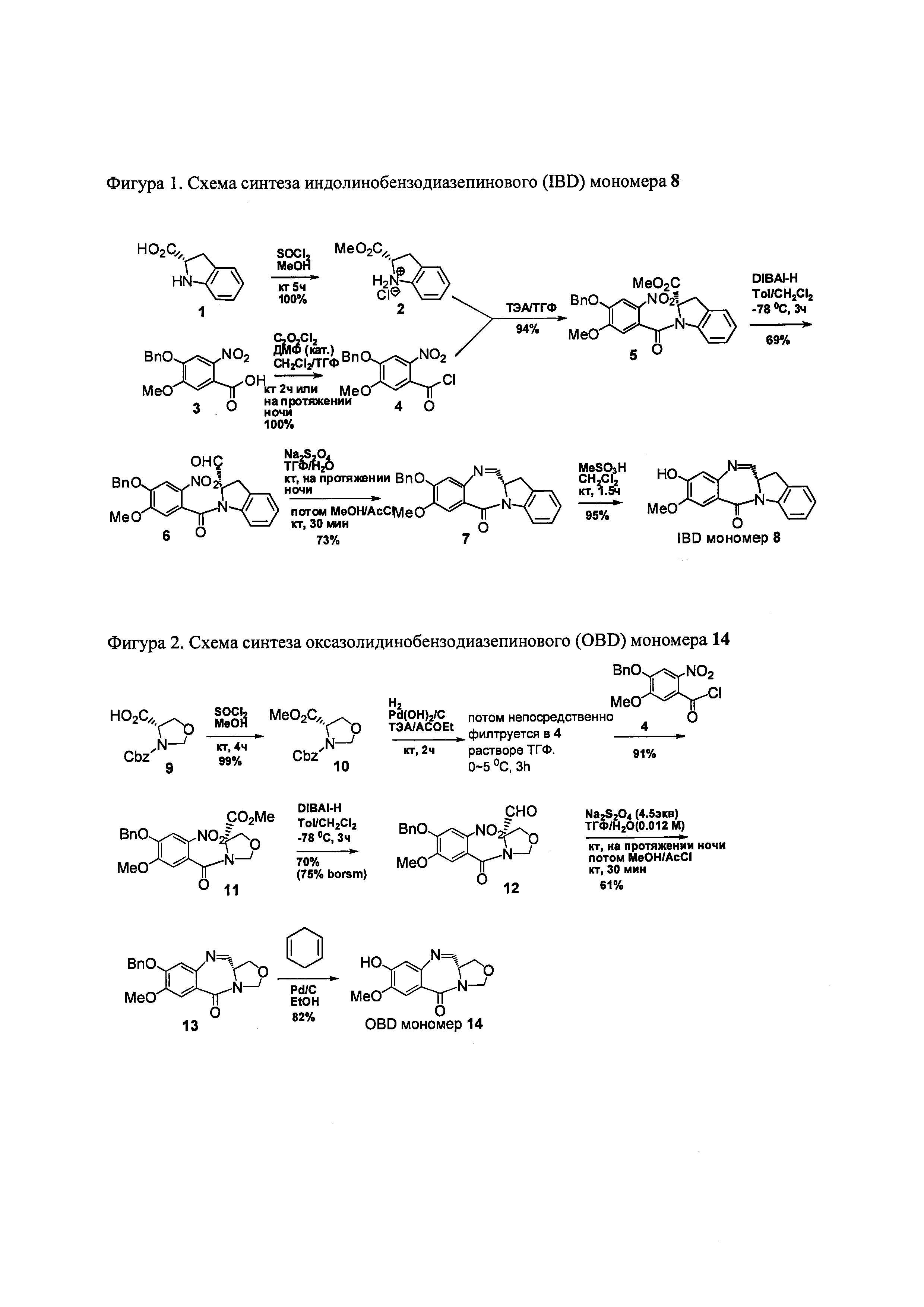
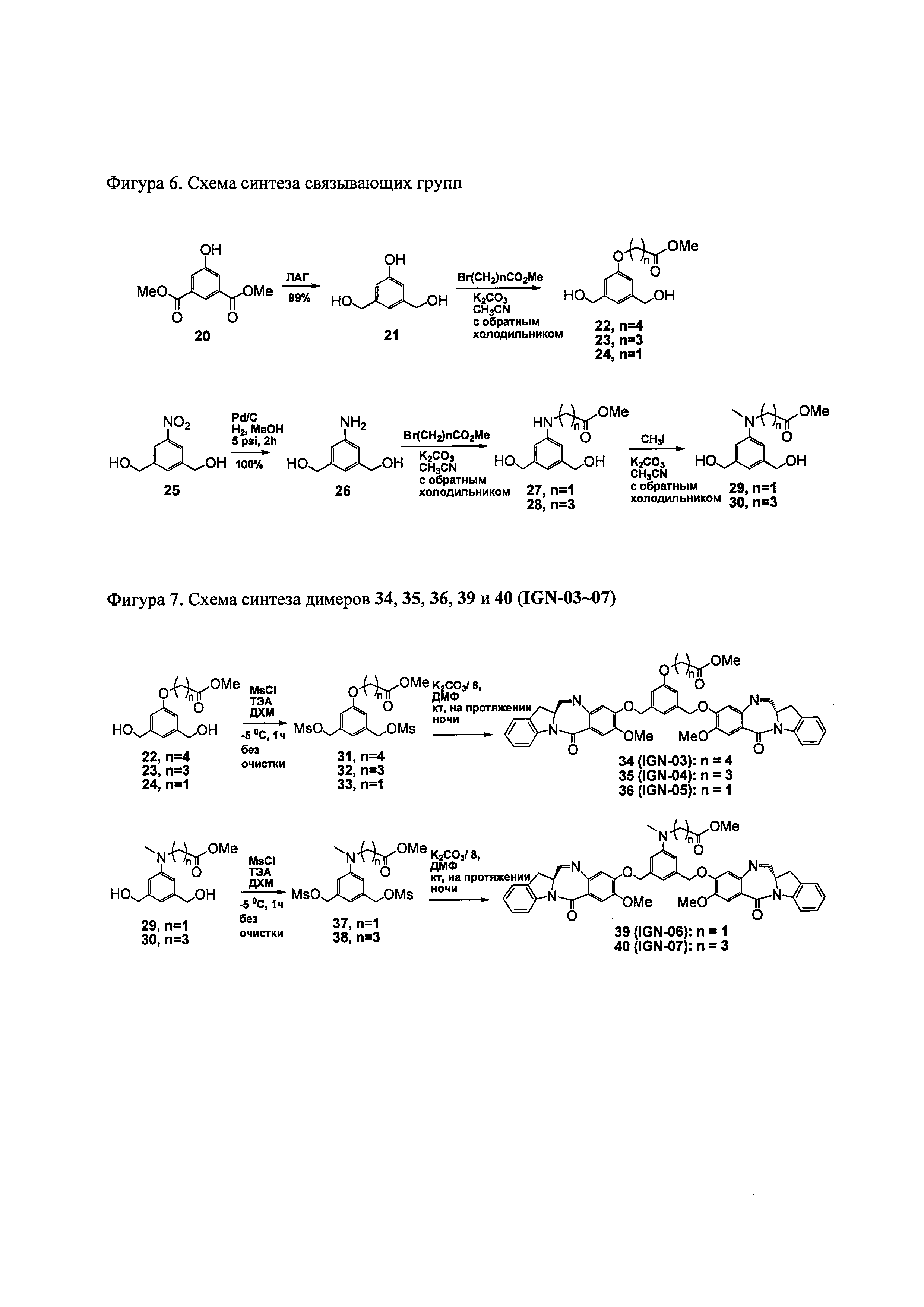
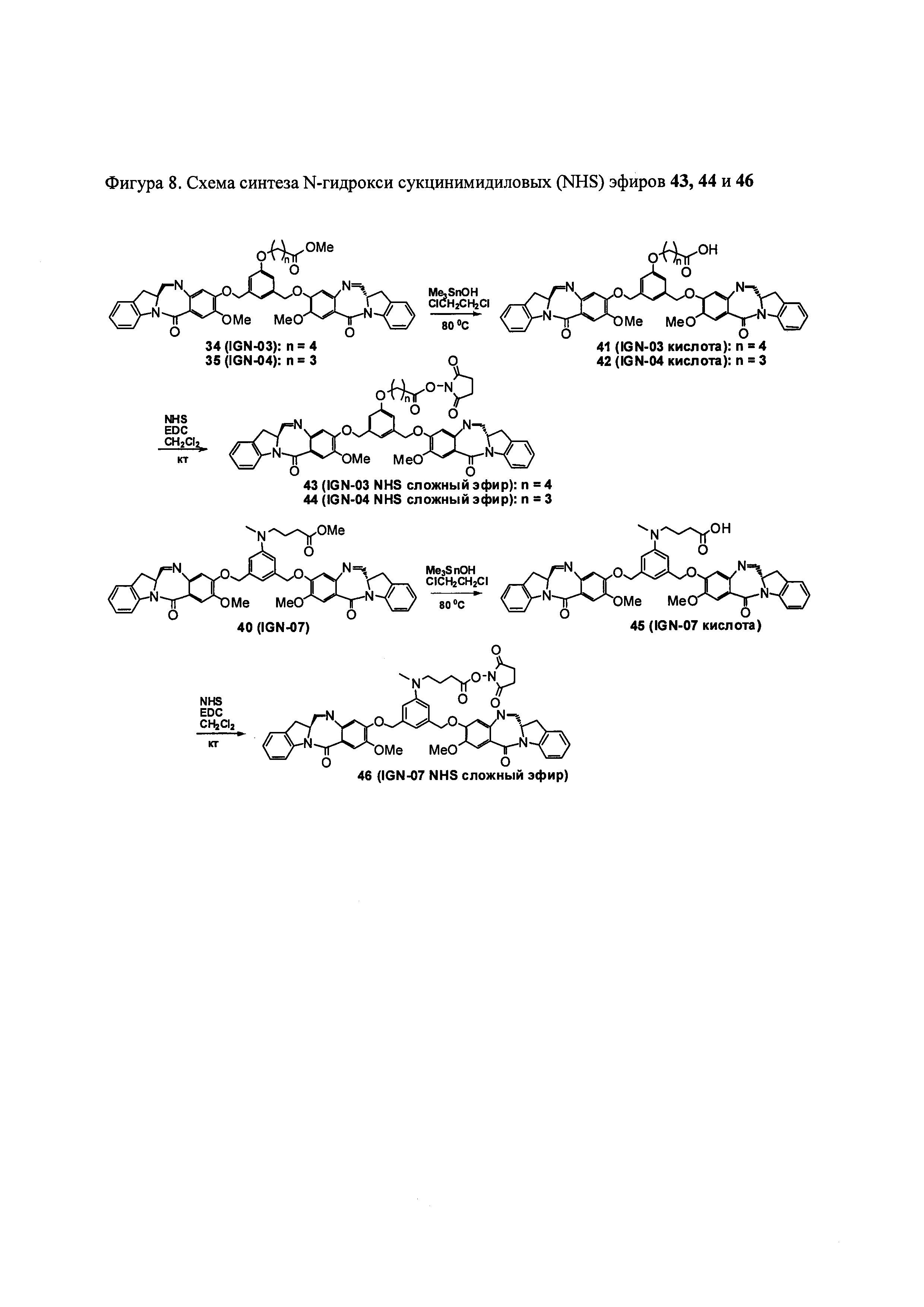
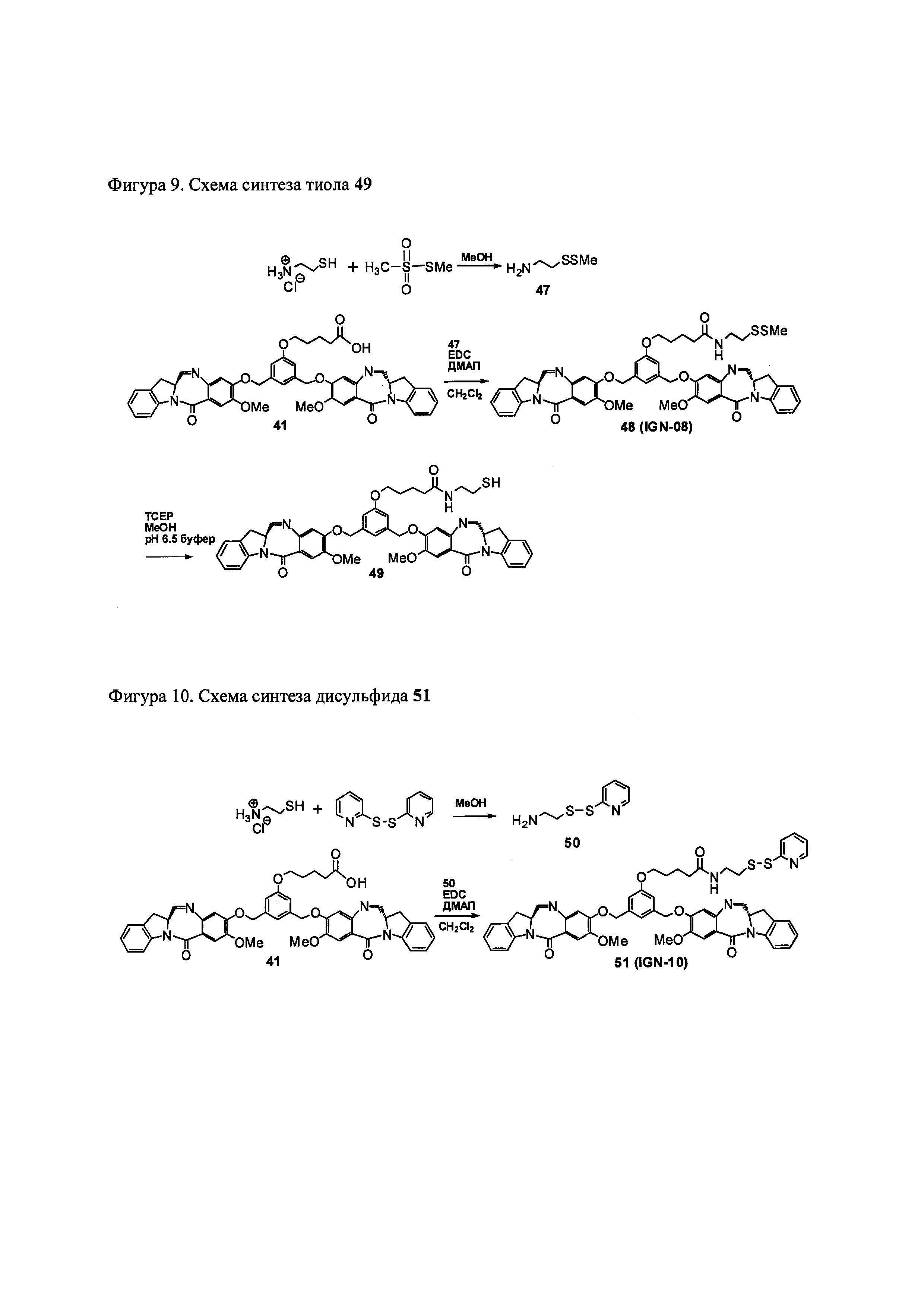
На фиг. 1-10 показаны схемы синтеза индолинобензодиазепиновых и оксзолидинобензодиазепиновых мономеров, представителей сшивающих агентов и димеров настоящего изобретения.
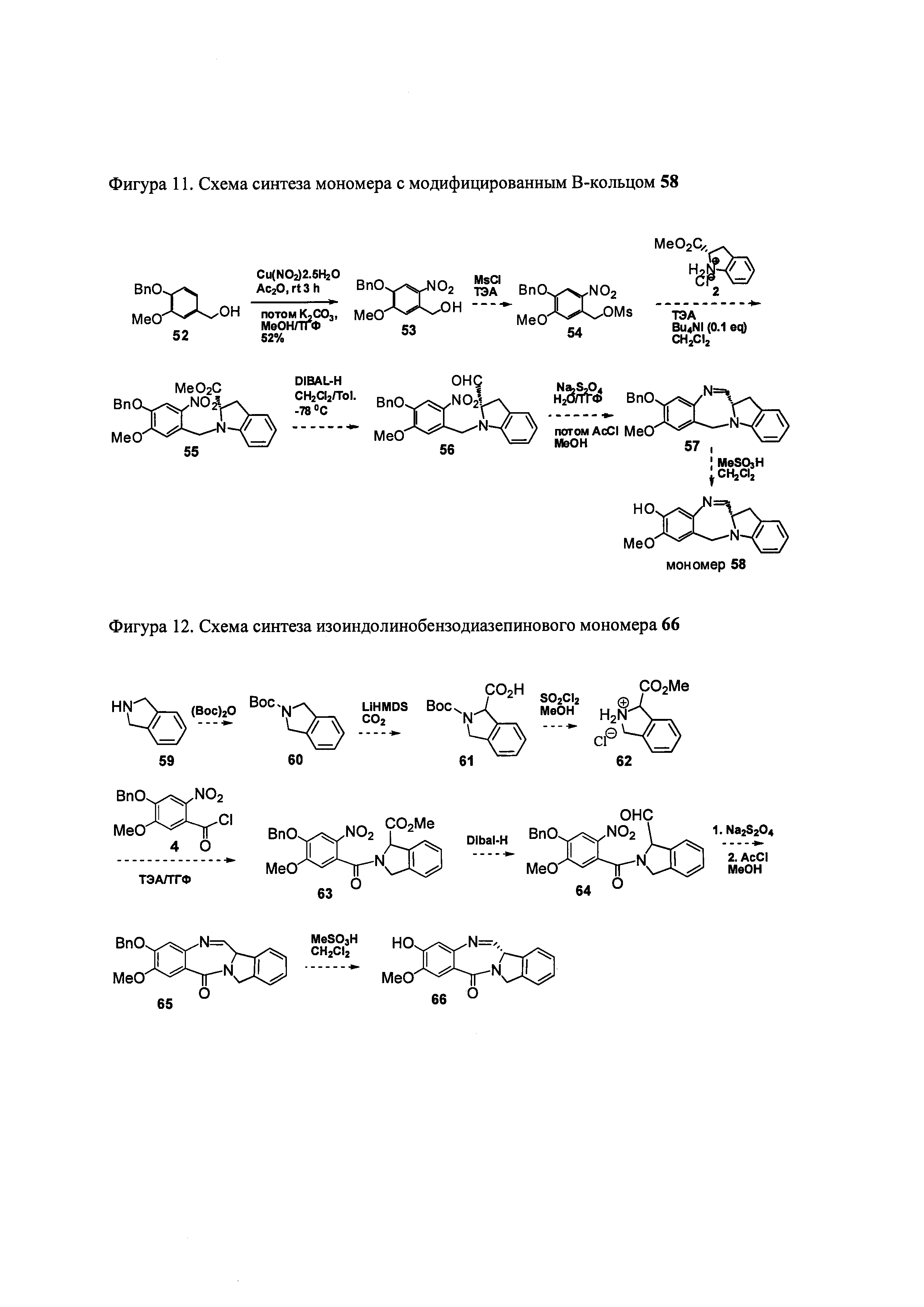
На фиг. 11 показана схема синтеза представителя индолинобензодиазепинового мономера с модифицированным В кольцом.
На фиг. 12 показана схема синтеза представителя изоиндолинобензодиазепинового мономера.
На фиг. 13 показана схема синтеза представителя димера настоящего изобретения со сшивающим агентом, присоединенным непосредственно к индолинобензодиазепиновому фрагменту.
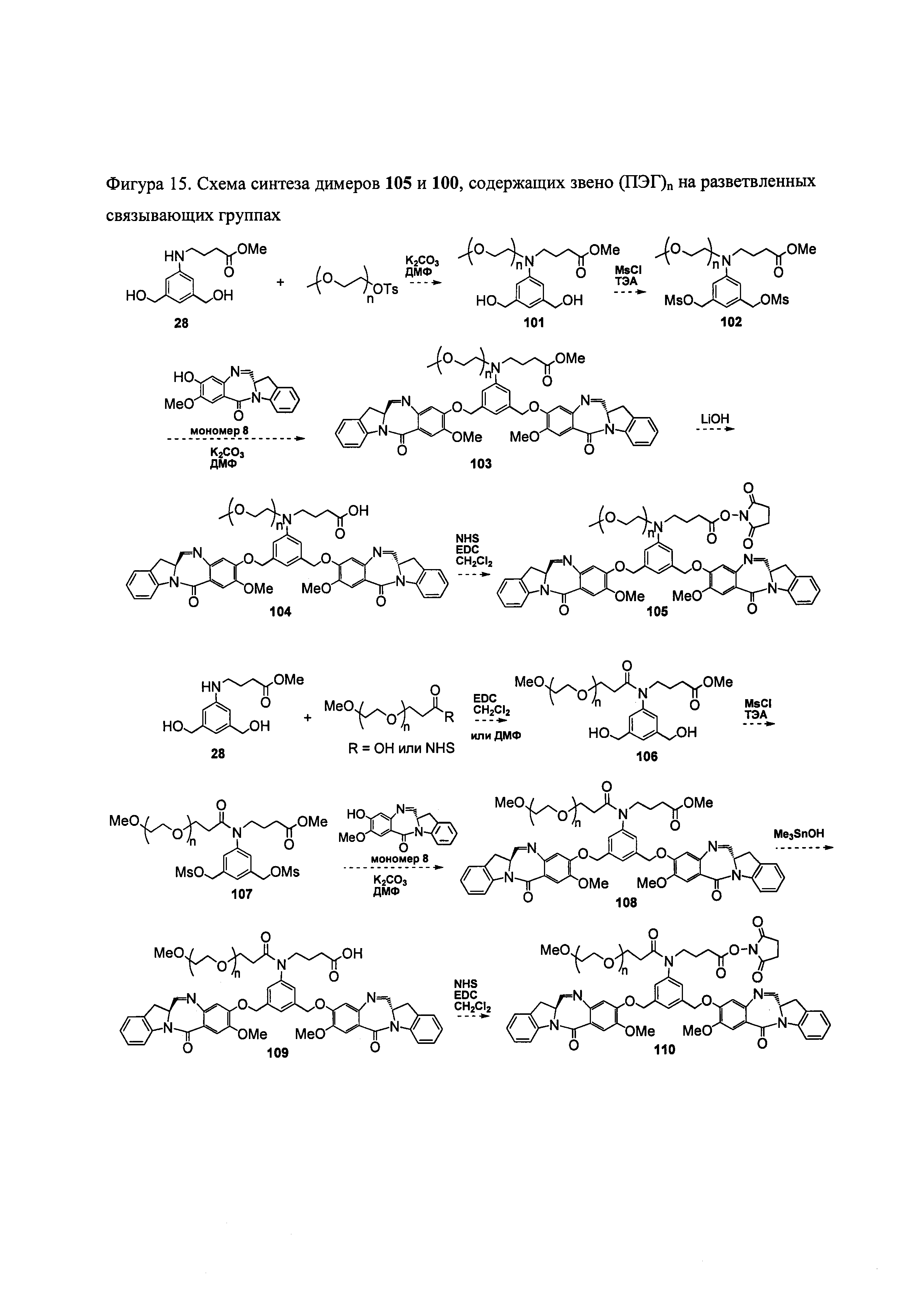
На фиг. 14 и 15 показаны схемы синтеза представителей димеров, содержащих на связывающих группах фрагменты (PEG)n.
На фиг. 16 показаны схемы синтеза представителей смешанных имино-аминных и имино-амидных индолинобензодиазепиновых димеров.
На фиг. 17 показана схема синтеза представителей конъюгатов ИБД-поли(N-метилпиррол-имидазол).
На фиг. 18-19 показана схема синтеза для получения полипирроловых и полипирроло-имидазоловых производных мономеров настоящего изобретения.
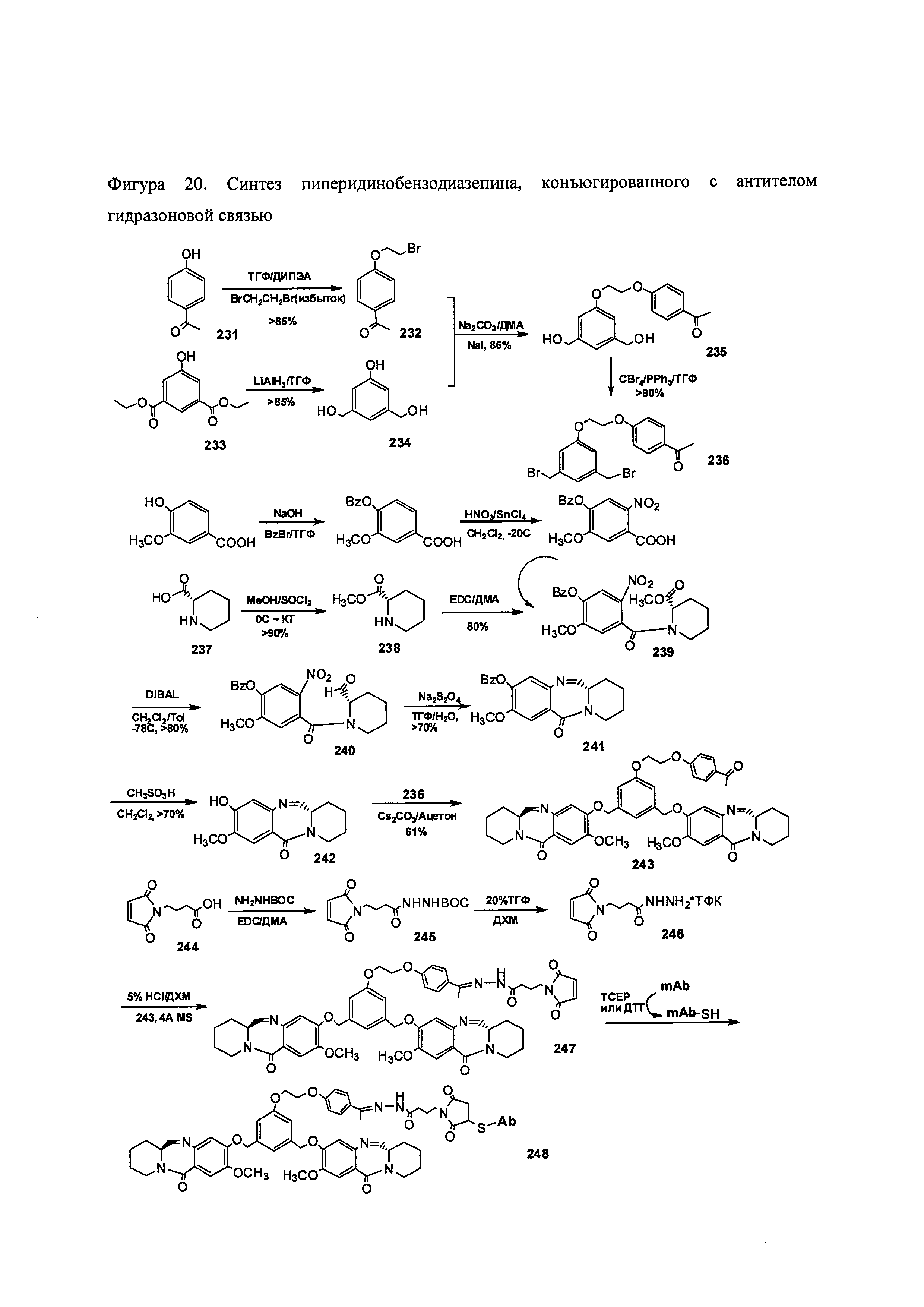
На фиг. 20 показана схема синтеза пиперидинилбензодиазепинов, имеющих связывающую группу.
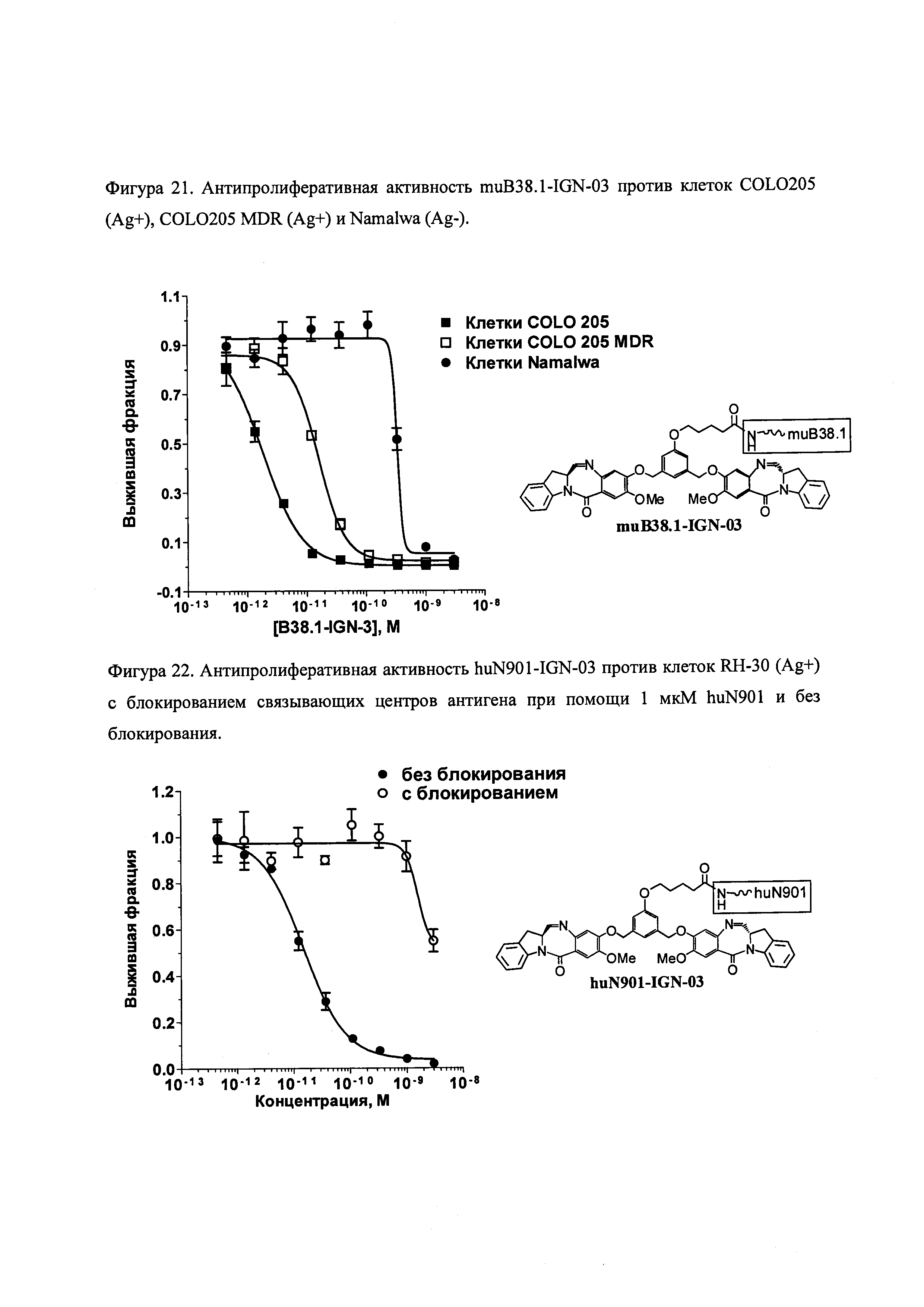
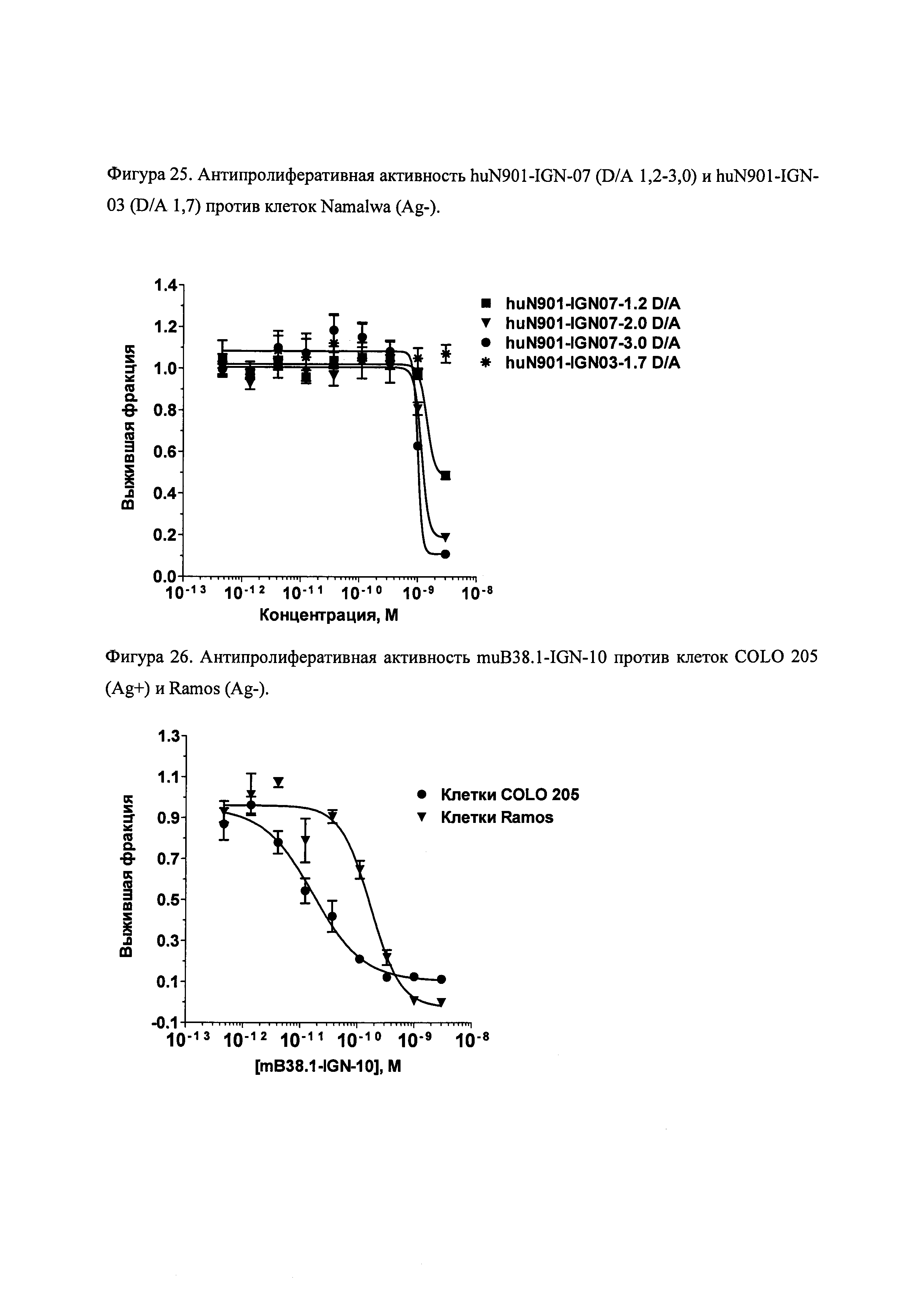
На фиг. 21-26 показана антипролиферативная активность in vitro конъюгатов muB38.1-IGN-03, huN901-IGN-03, huN901-IGN-07 и muB38.1-IGN-10 на антиген-положительных и антиген-отрицательных раковых клеточных линиях, в зависимости от дозировки.
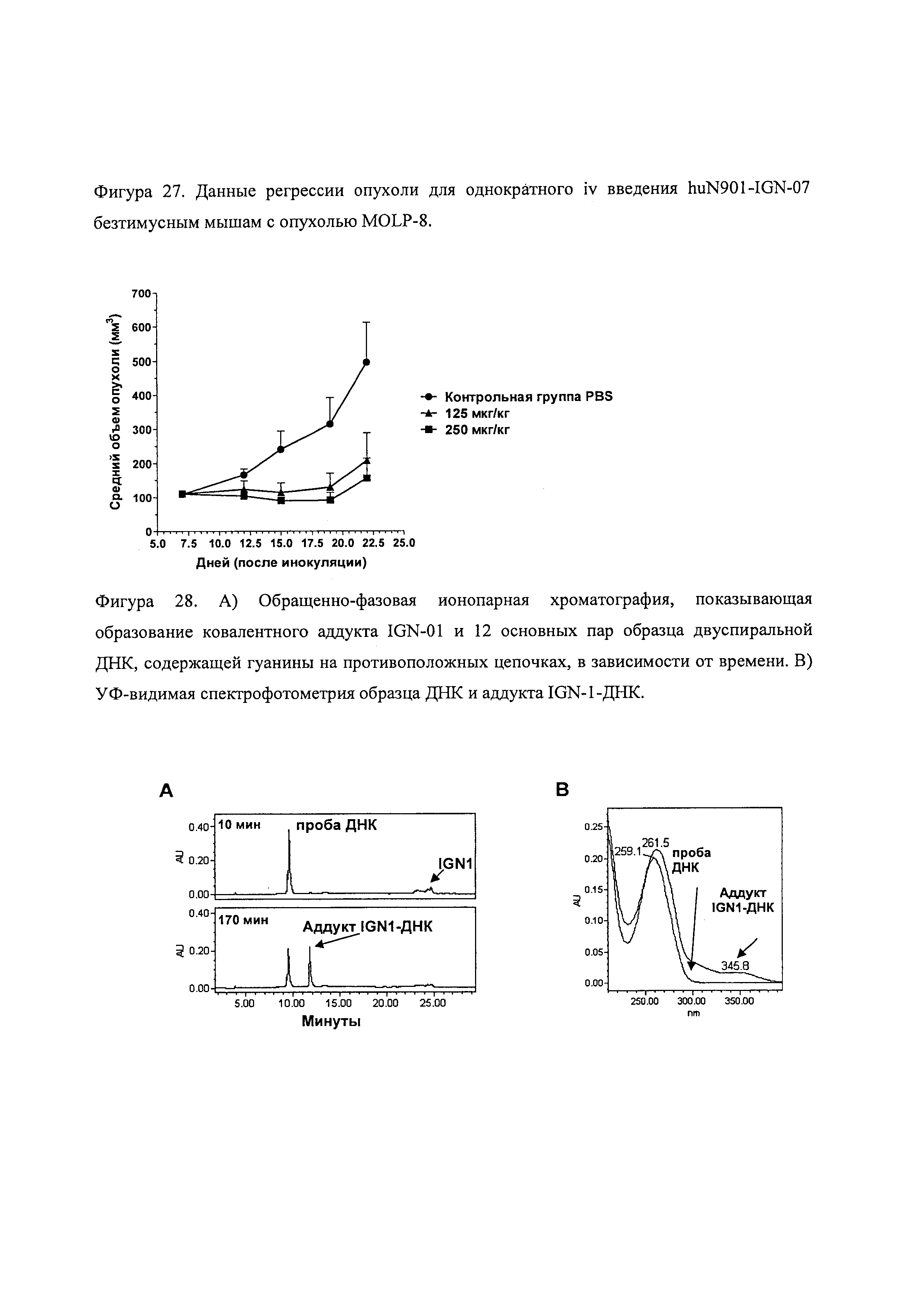
На фиг. 27 показана эффективность in vivo конъюгата huN901-IGN-07 на мышах с опухолью Molp-8.
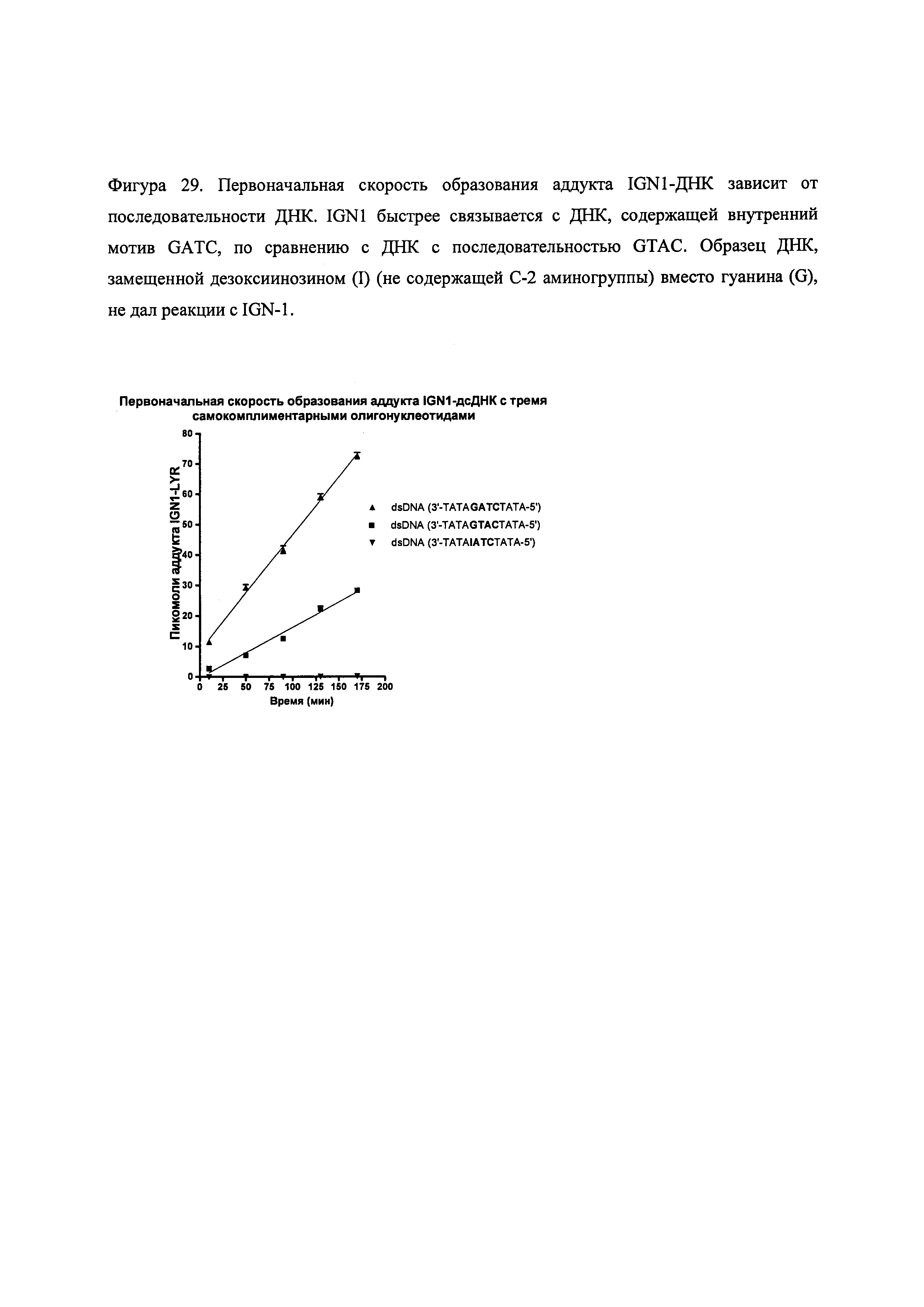
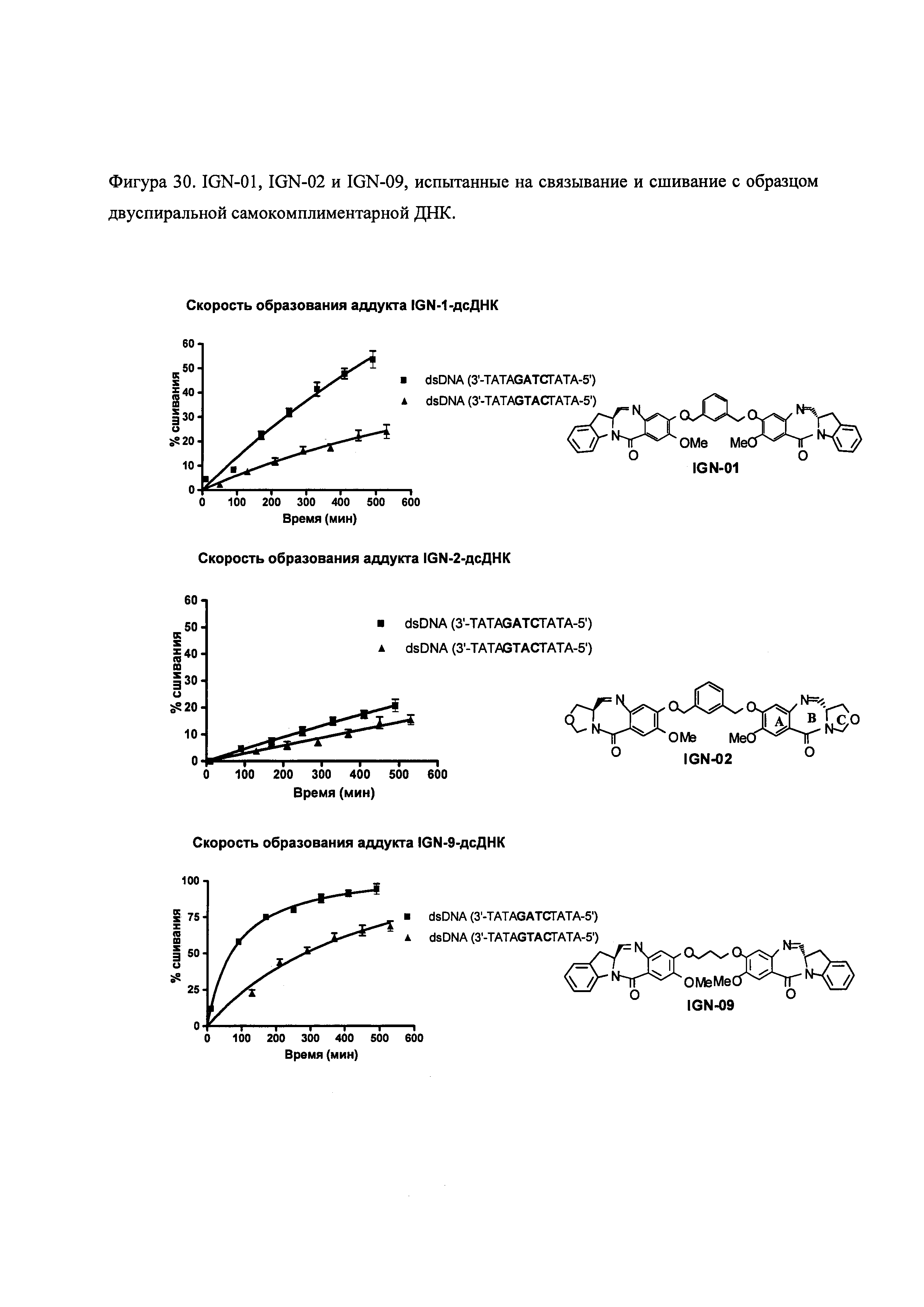
На фиг. 28-30 представлены данные, демонстрирующие, что IGN-01, IGN-02 и IGN-09 связываются с образованием ковалентного аддукта с двуспиральной ДНК, содержащей гуанидиновые остатки на противоположных спиралях.
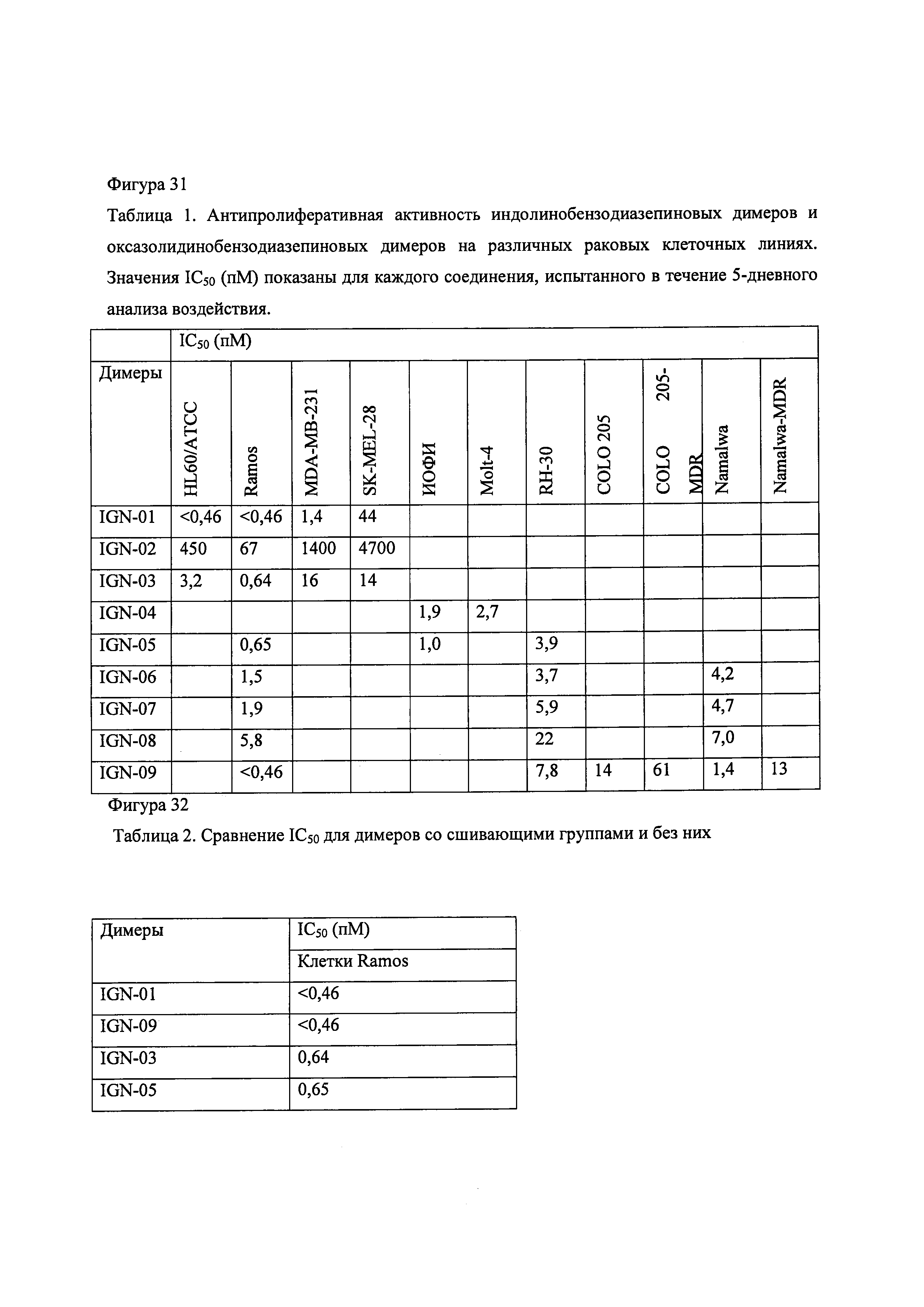
Фиг. 31 содержит Таблицу 1, в которой даны значения IC50 для антипролиферативной активности in vitro индолинобензодиазепиновых димеров и димера оксазолидинобензодиазепина на нескольких раковых клеточных линиях.
Фиг. 32 содержит Таблицу 2, в которой дано сравнение значений IC50 для антипролиферативной активности in vitro индолинобензодиазепиновых димеров со связывающими группами и без них.
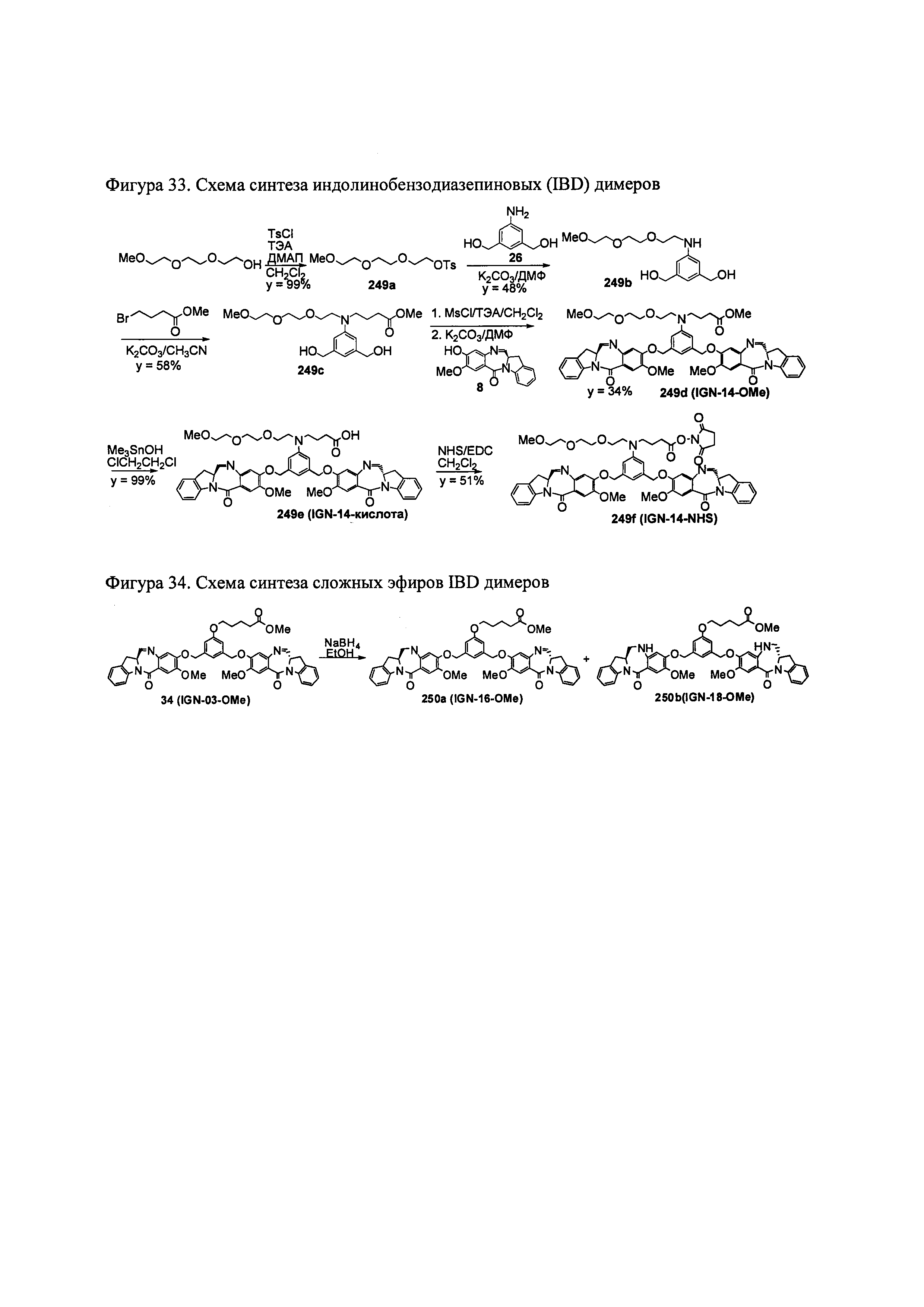
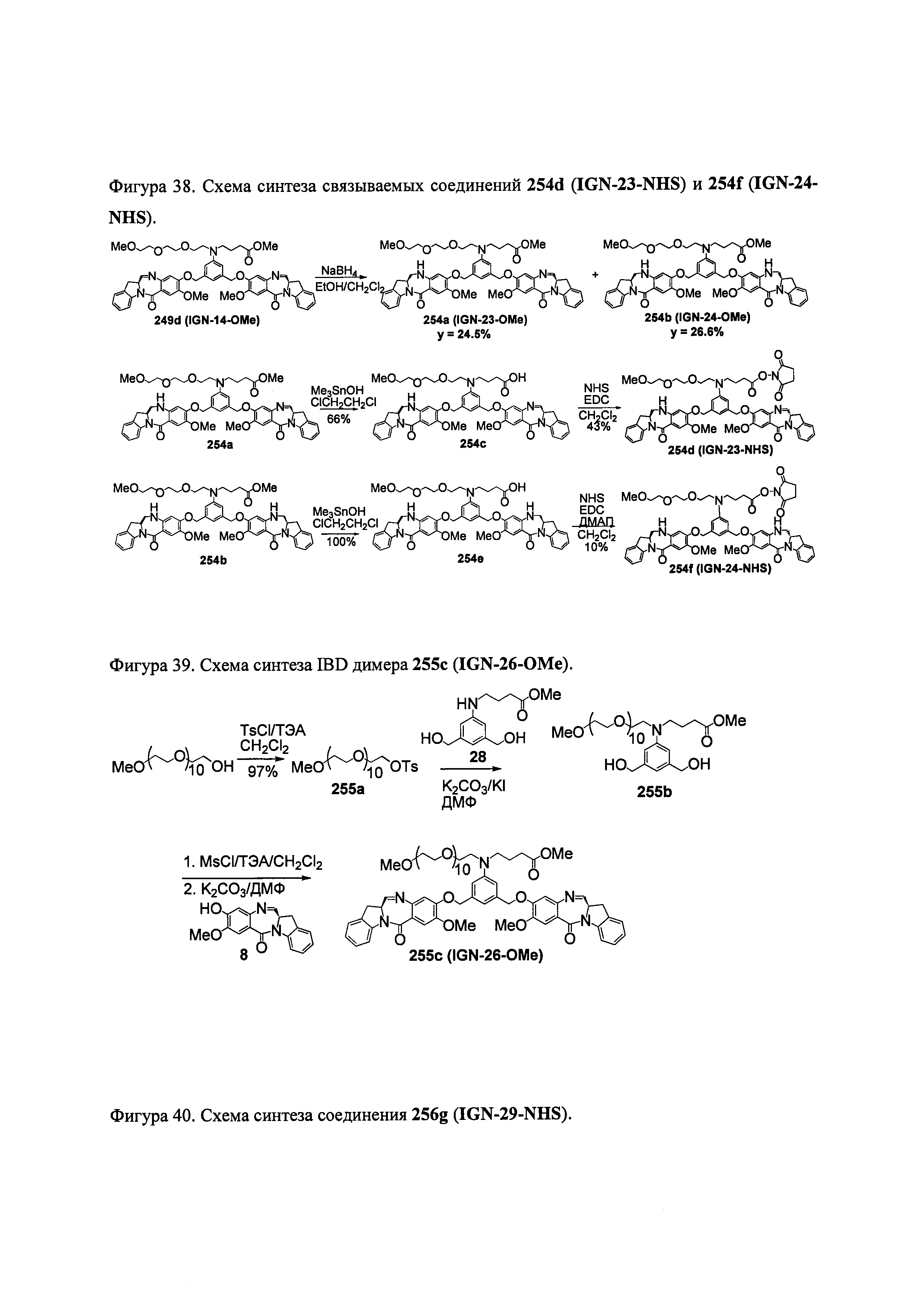
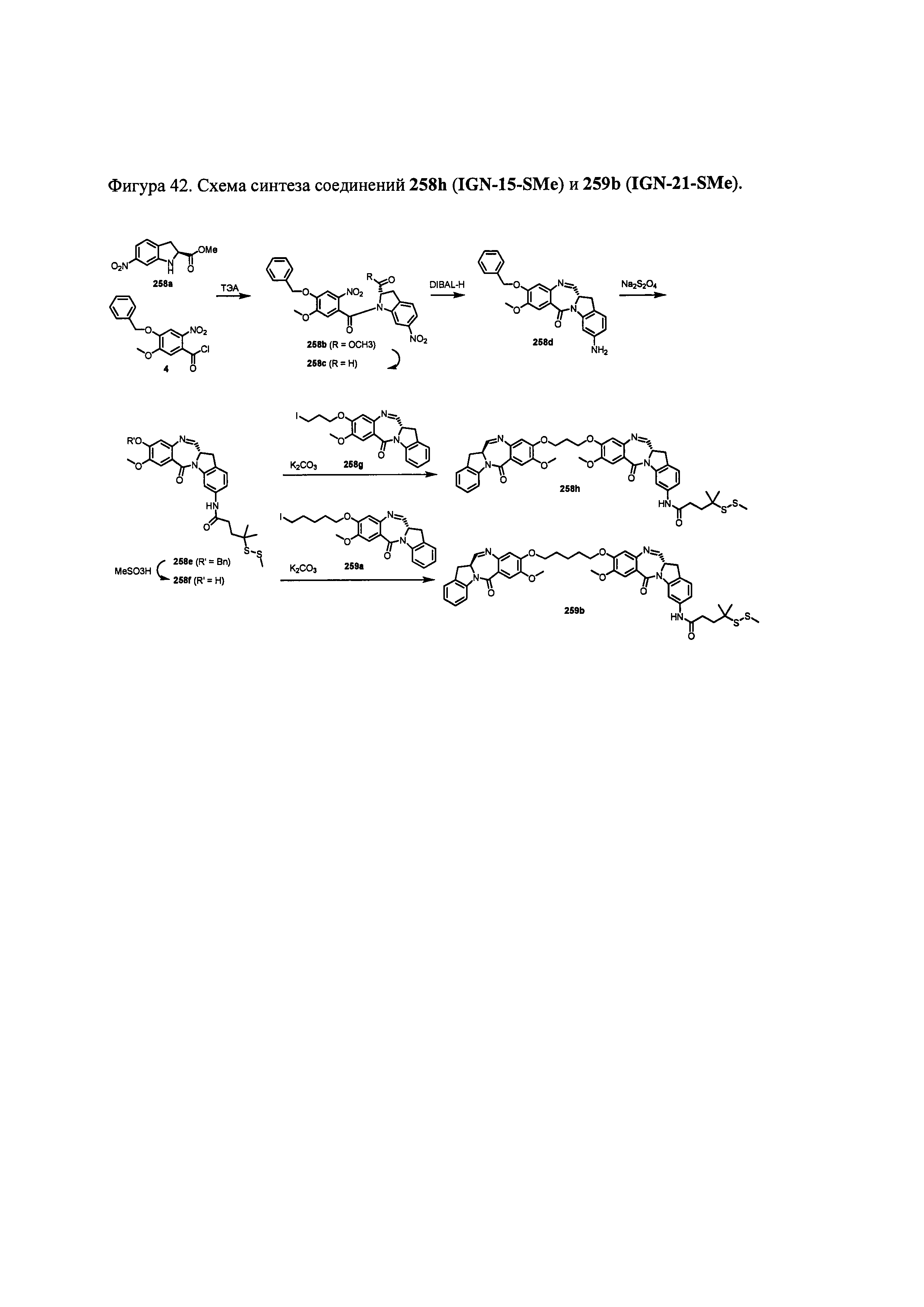
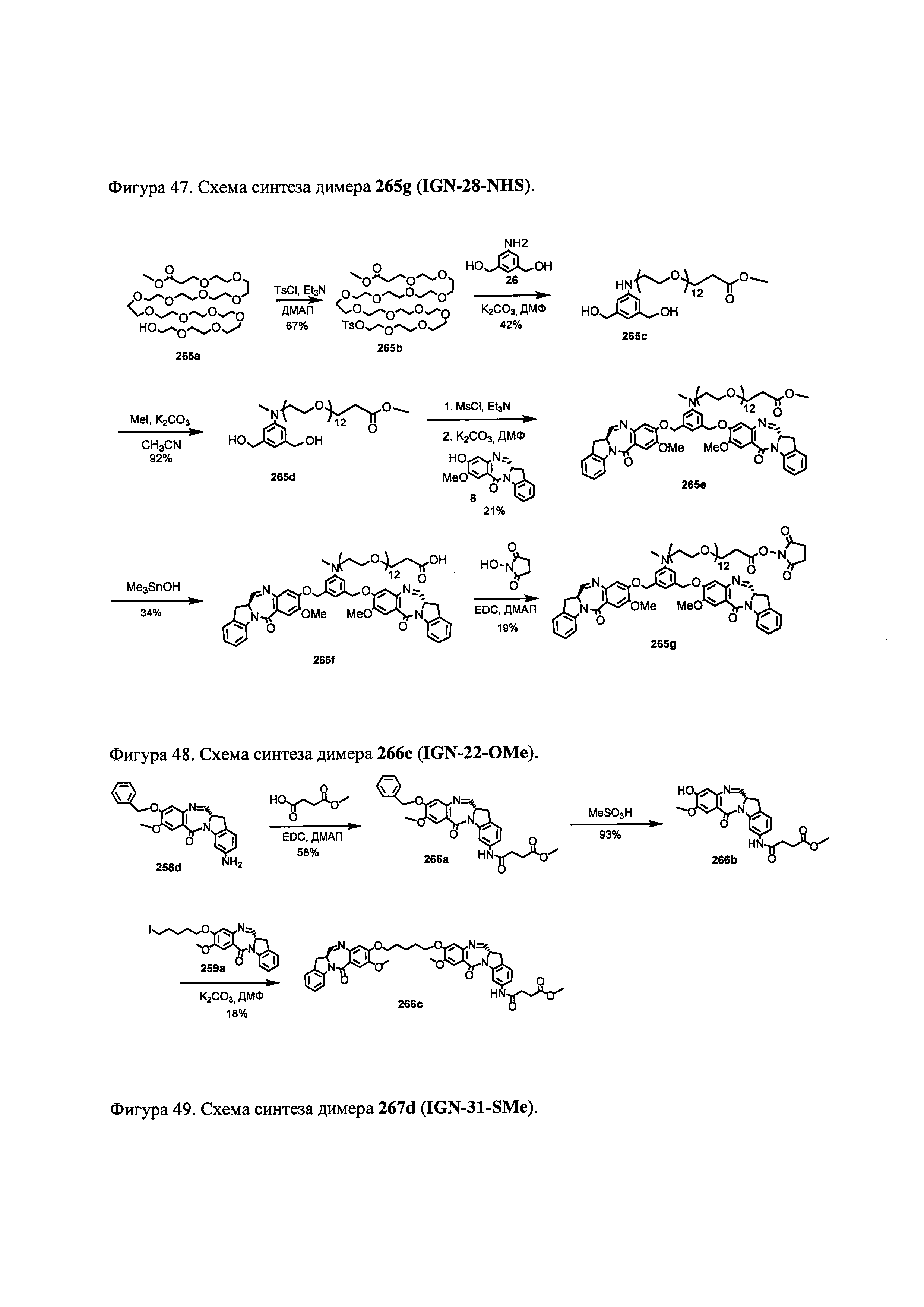
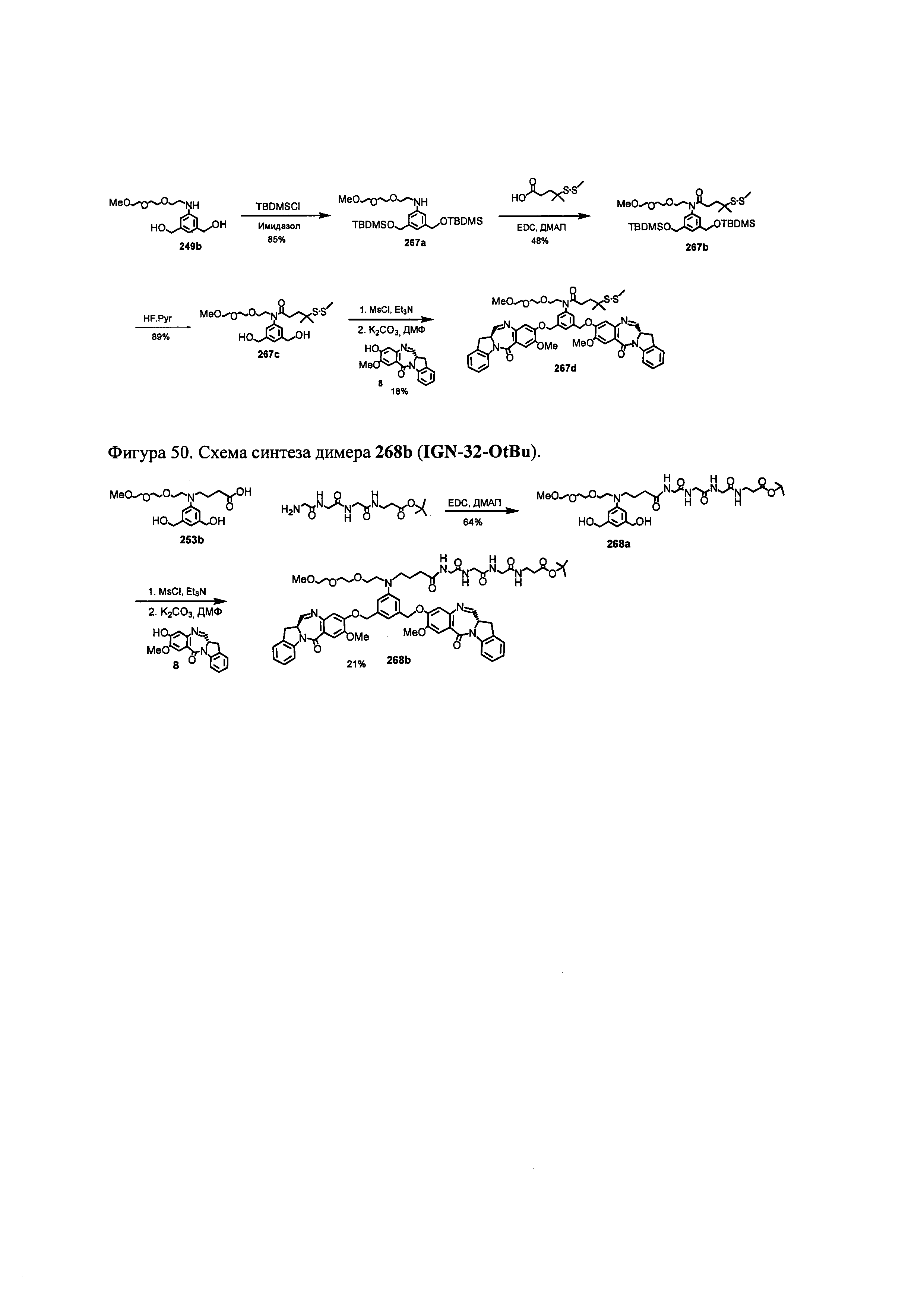
На фиг. 33-36, 39, 42, 43, 44, 48, 49 и 50 показаны схемы синтеза для получения соединений настоящего изобретения.
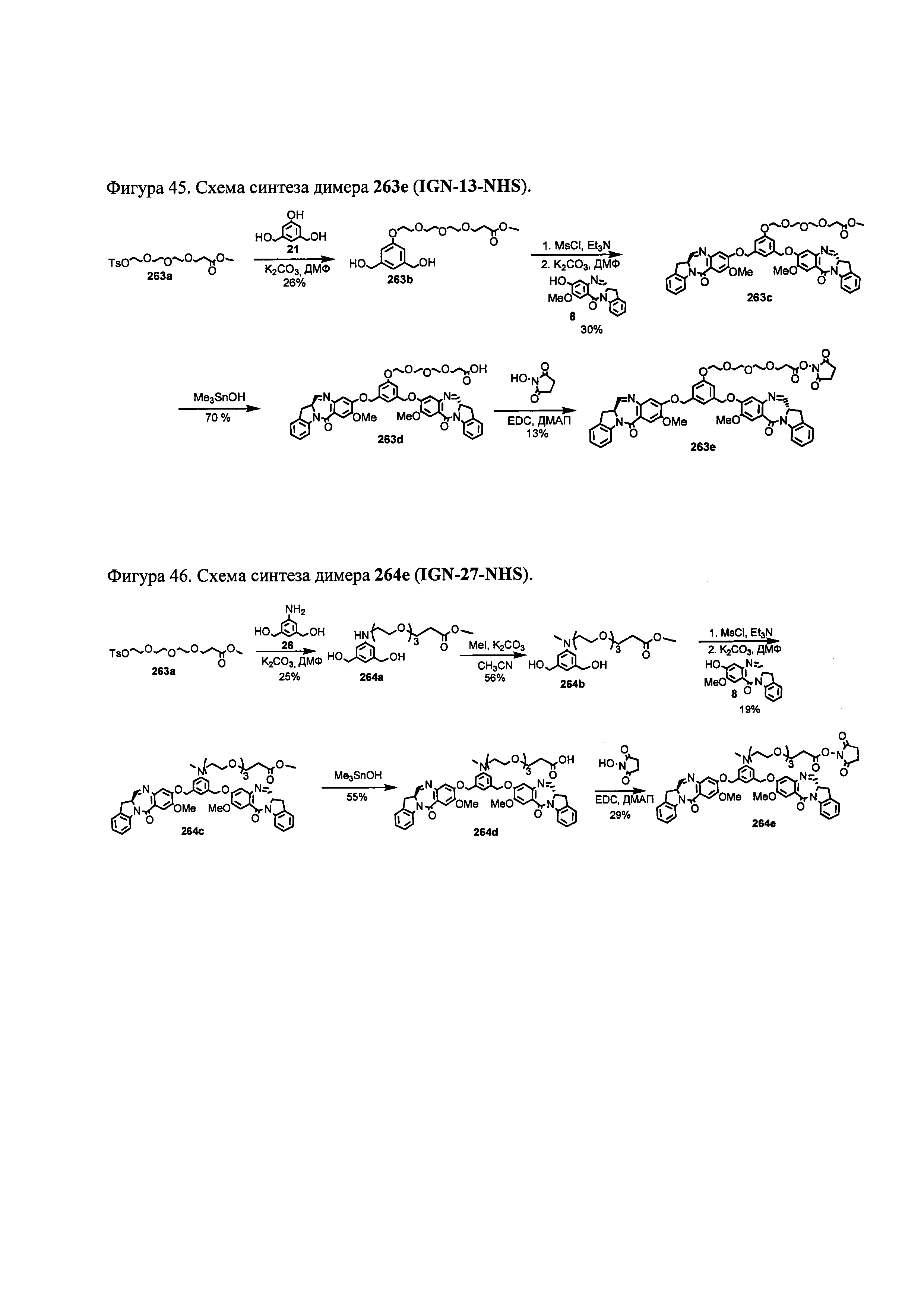
На фиг. 37, 38, 40 и 41, 45, 46, и 47 показаны схемы синтеза для получения связываемых соединений настоящего соединения.
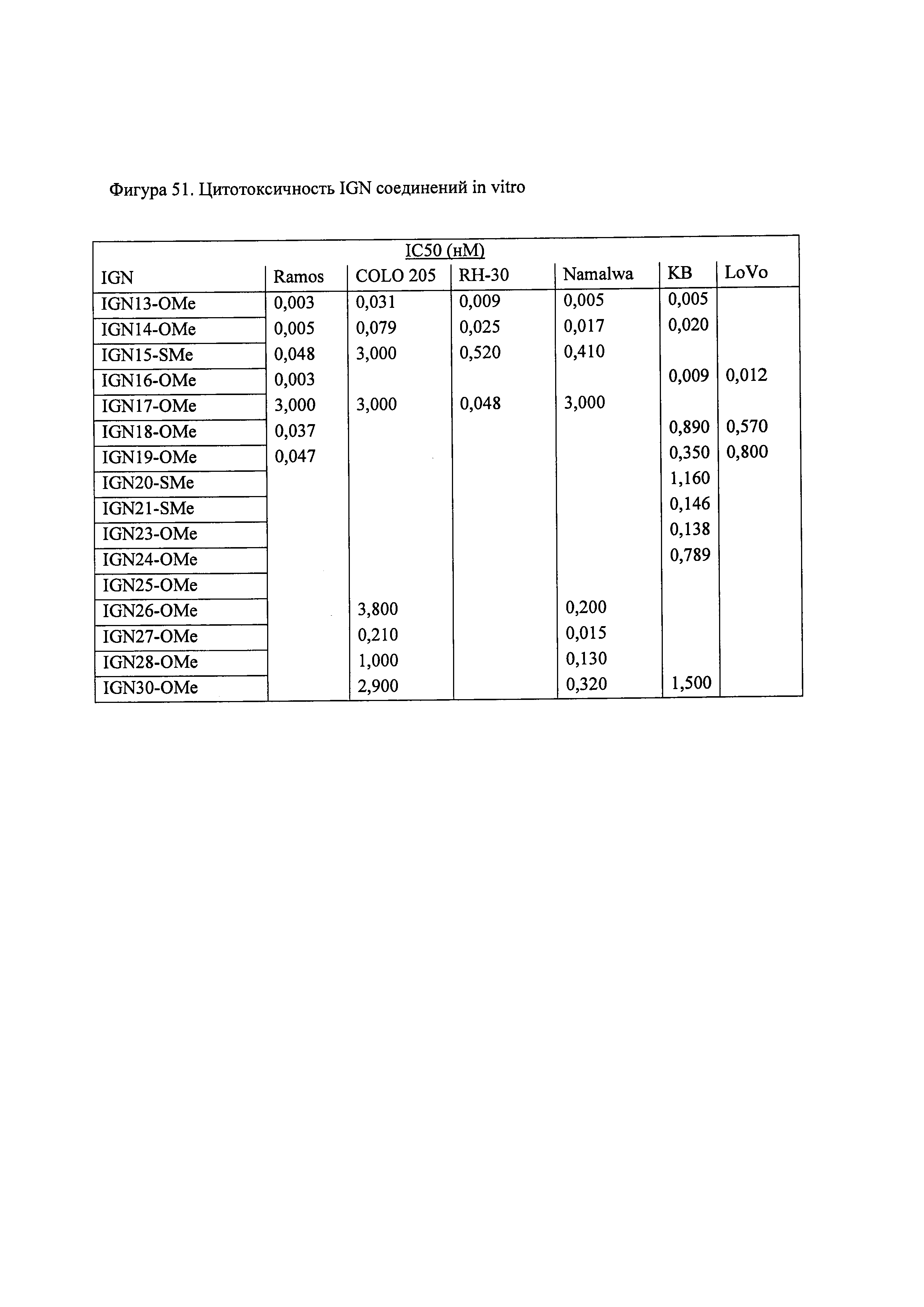
На фиг. 51 показана цитотоксичность соединений настоящего изобретения in vitro.
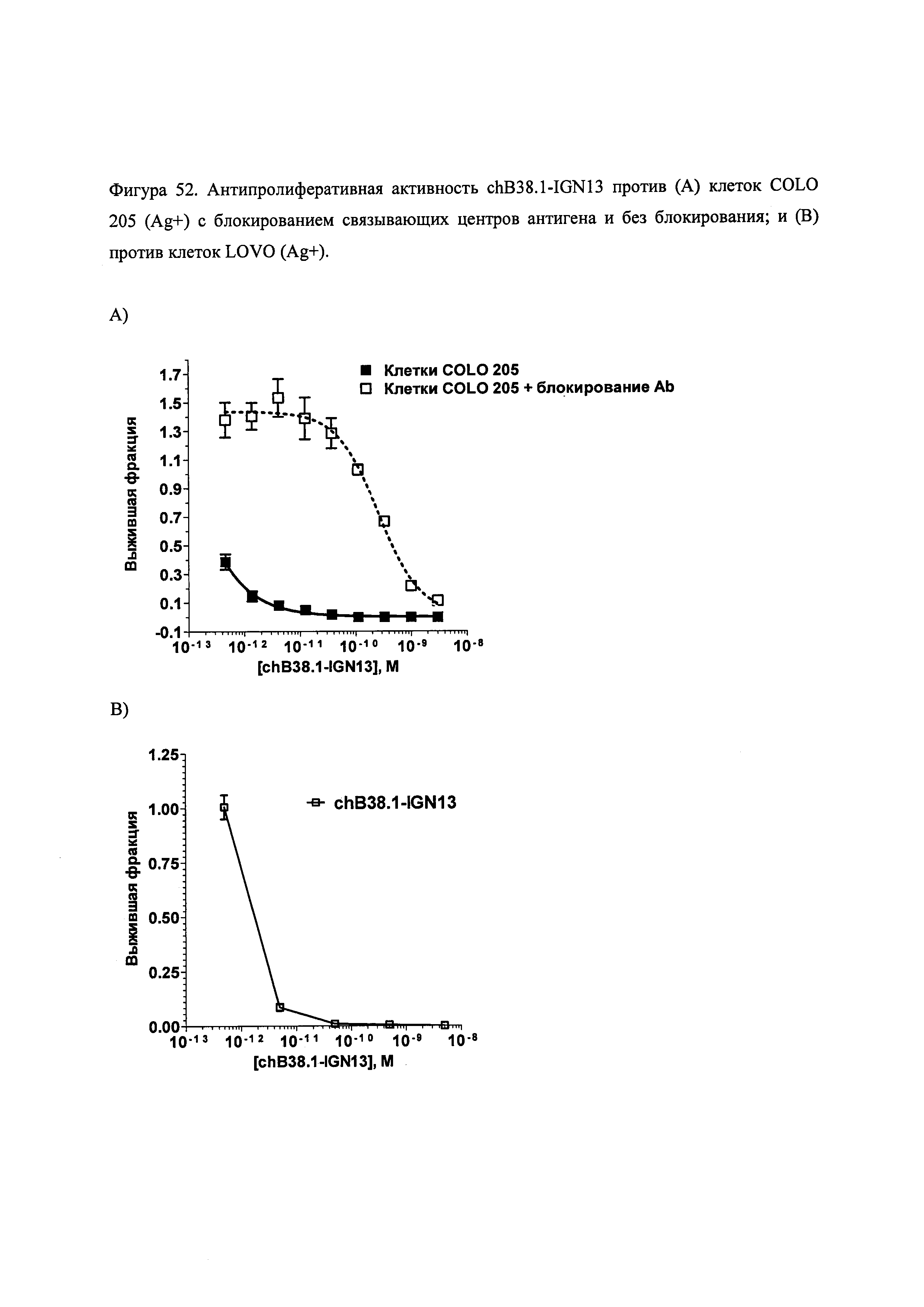
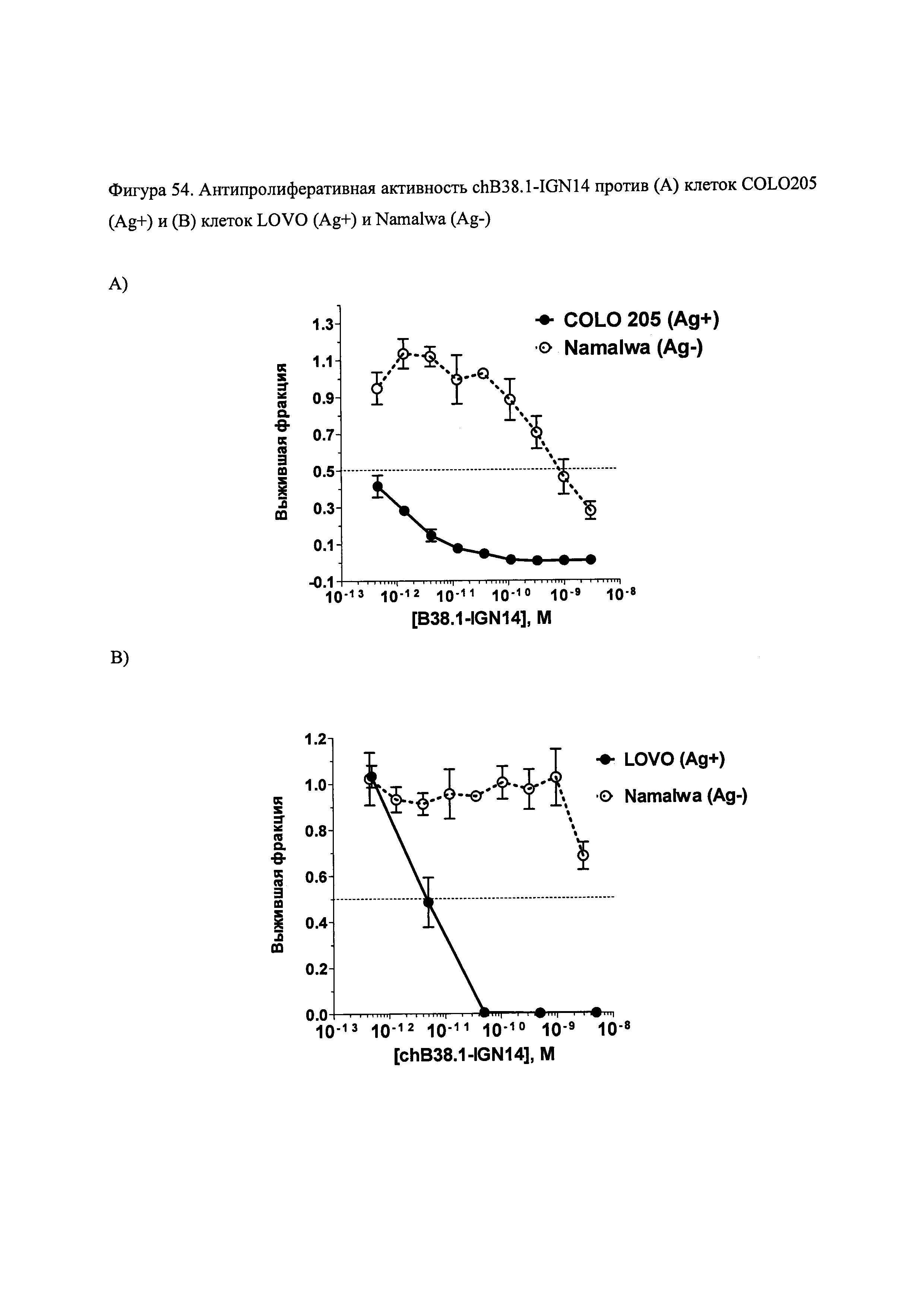
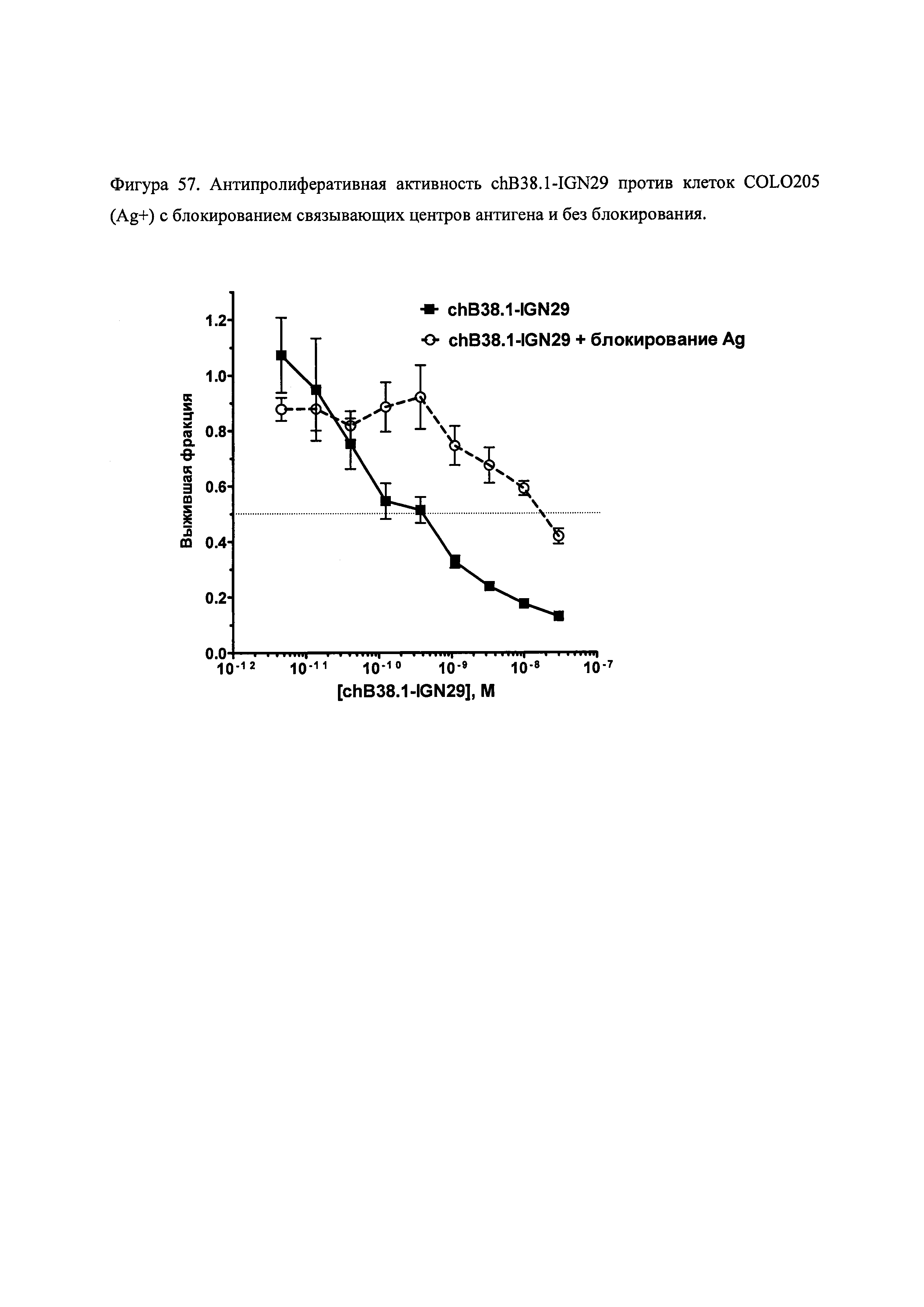
На фиг. 52, 54, 56, 57 и 58 показана цитотоксичность и специфичность конъюгатов chB38.1 in vitro.
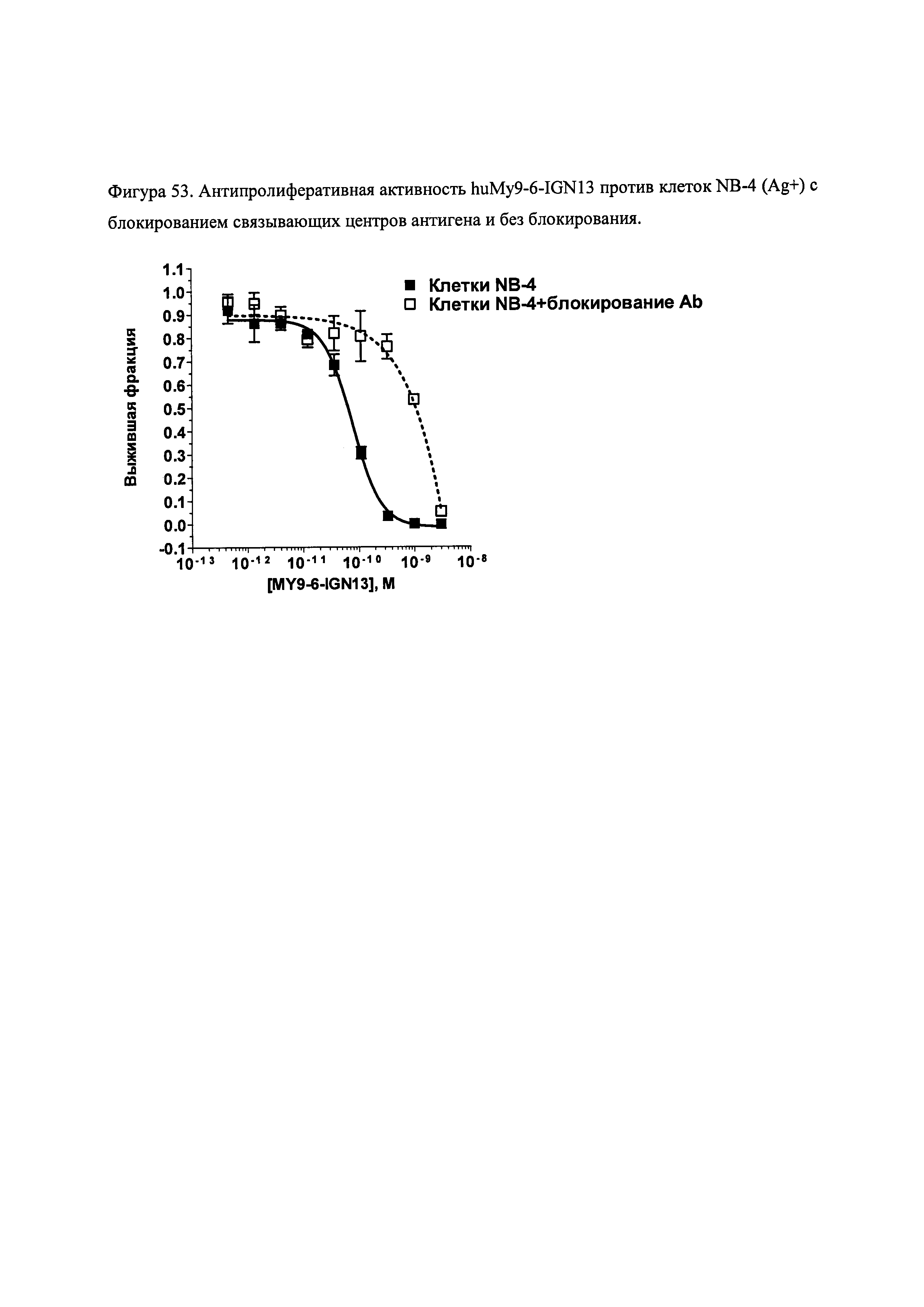
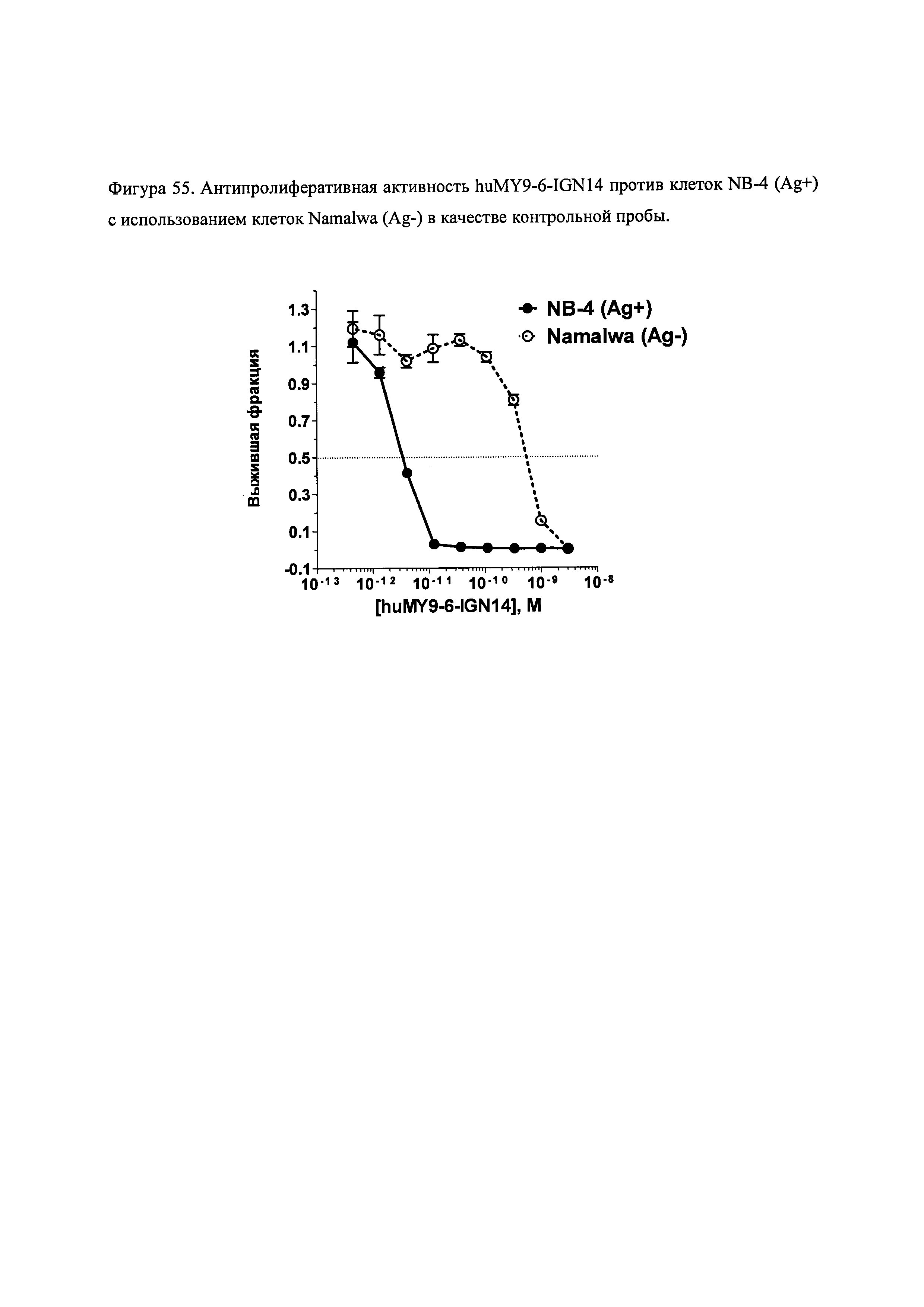
На фиг. 53 и 55 показана цитотоксичность и специфичность конъюгатов huMy9-6 in vitro.
На фиг. 59 показана противоопухолевая активность конъюгата chB38.1.
Осуществление изобретения
Здесь будут подробно рассмотрены некоторые упомянутые варианты настоящего изобретения, примеры которых продемонстрированы сопроводительными структурами и формулами. Несмотря на то, что настоящее изобретение будет описываться в соответствии с пронумерованными вариантами воплощений, следует понимать, что эти варианты не являются ограничивающими для настоящего изобретения. Напротив, в настоящем изобретении сделана попытка охватить все варианты, модификации и эквиваленты, которые могут быть включены в объем настоящего изобретения в соответствии формулой изобретения. Специалист в данной области знает много способов и материалов, подобных или равноценных описанным здесь, которые могут использоваться в ходе осуществления настоящего изобретения.
Определения
Термин «линейный или разветвленный алкил», используемый здесь, относится к насыщенному линейному или разветвленному одновалентному углеводородному радикалу, содержащему от одного до двадцати углеродных атомов. Примеры алкила включают, не ограничиваясь, метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-метил-1-пропил, --СН2СН(СН3)2), 2-бутил, 2-метил-2-пропил, 1-пентил, 2-пентил, 3-пентил, 2-метил-2-бутил, 3-метил-2-бутил, 3-метил-1-бутил, 2-метил-1-бутил, 1-гексил, 2-гексил, 3-гексил, 2-метил-2-пентил, 3-метил-2-пентил, 4-метил-2-пентил, 3-метил-3-пентил, 2-метил-3-пентил, 2,3-диметил-2-бутил, 3,3-диметил-2-бутил, 1-гептил, 1-октил и т.п.
Термин «линейный или разветвленный алкенил» относится к линейному или разветвленному одновалентному углеводородному радикалу, содержащему от двух до двадцати углеродных атомов и по меньшей мере один центр ненасыщенности, т.е. двойную связь углерод-углерод, где алкениловый радикал включает радикалы, имеющие «цис» и «транс» ориентации, или, в качестве альтернативы, «Е» и «Z» ориентации. Примеры включают, не ограничиваясь, этиленил или винил (--СН=СН2), аллил (--СН2СН=СН2) и т.п.
Термин «линейный или разветвленный алкинил» относится к линейному или разветвленному одновалентному углеводородному радикалу, содержащему от двух до двадцати углеродных атомов и по меньшей мере один центр ненасыщенности, т.е. тройную связь углерод-углерод. Примеры включают, не ограничиваясь, этинил, пропинил, 1-бутинил, 2-бутинил, 1-пентинил, 2-пентинил, 3-пентинил, гексинил и т.п.
Термины «циклический алкил», «циклический алкенил», «циклический алкинил», «карбоцикл», «карбоциклил», «карбоциклическое кольцо» и «циклоалкил» относятся к одновалентному неароматическому, насыщенному или частично ненасыщенному кольцу, содержащему от 3 до 12 углеродных атомов в виде моноцикличного кольца или от 7 до 12 углеродных атомов в виде бициклического кольца. Бициклические карбоциклы, содержащие от 7 до 12 атомов, могут быть, например, в виде бицикличных [4,5], [5,5], [5,6] или [6,6] систем, а бицикличные карбоциклы, содержащие 9 или 10 атомов могут быть в виде бицикличных [5,6] или [6,6] систем, или в виде мостиковых систем, таких как бицикло[2.2.1]гептан, бицикло[2.2.2]октан и бицикло[3.2.2]нонан. Примеры моноцикличных карбоциклов включают, не ограничиваясь, циклопропил, циклобутил, циклопентил, 1-циклопент-1-енил, 1-циклопент-2-енил, 1-циклопент-3-енил, циклогексил, 1-циклогекс-1-енил, 1-циклогекс-2-енил, 1-циклогекс-3-енил, циклогексадиенил, циклогептил, циклооктил, циклононил, циклодецил, циклоундецил, циклододецил и т.п.
Термин «арил» означает одновалентный ароматический углеводородный радикал из 6-18 углеродных атомов, полученный отщеплением одного атома водорода от одного атома углерода исходной ароматической кольцевой системы. Некоторые ариловые группы представлены в иллюстративных структурах как «Ar». Арил включает бициклические радикалы, состоящие из ароматического кольца, конденсированного с насыщенным, частично ненасыщенным кольцом или ароматическим карбоциклическим или гетероциклическим кольцом. Типичные ариловые группы включают, не ограничиваясь, радикалы, полученные из бензола (фенил), замещенных бензолов, нафталина, антрацена, инденила, инданила, 1,2-дигидронафталина, 1,2,3,4-тетрагидронафтилаи т.п.
Термины «гетероцикл», «гетероциклил» и «гетероциклическое кольцо» используются здесь взаимозаменяемо и относятся к насыщенному или частично ненасыщенному (напр., имеющему одну или более двойных и/или тройных связей в кольце) карбоциклическому радикалу из 3-18 кольцевых атомов, в которых по меньшей мере один кольцевой атом является гетероатомом, выбранным из азота, кислорода, фосфора и серы, а остальные кольцевые атомы являются С, где один или более кольцевых атомов необязательно замещен независимо одним или более заместителей, описанных ниже. Гетероцикл может быть моноциклом, содержащим от 3 до 7 членов в кольце (2-6 углеродных атомов и 1-4 гетероатомов, выбранных из N, О, Р и S) или бициклом, содержащим от 7 до 10 членов в кольце (4-9 углеродных атомов и 1-6 гетероатомов, выбранных из N, О, Р и S), например: бицикло [4,5], [5,5], [5,6] или [6,6] система. Гетероциклы описаны в публикациях Paquette, Leo А.; «Основы современной химии гетероциклов» (W.A. Benjamin, New York, 1968), в частности, в главах 1, 3, 4, 6, 7 и 9; «Химия гетероциклических соединений, серия монографий» (John Wiley & Sons, New York, 1950 до н.в.), в частности в томах 13, 14, 16, 19 и 28; и публикации J. Am. Chem. Soc. (1960) 82:5566. Термин «гетероциклил» включает также радикалы, где гетероциклические радикалы конденсированы с насыщенным, частично ненасыщенным или ароматическим карбоциклическим или гетероциклическим кольцом. Примеры гетероциклических колец включают, не ограничиваясь, пирролидинил, тетрагидрофуранил, дигидрофуранил, тетрагидротиенил, тетрагидропиранил, дигидропиранил, тетрагидротиопиранил, пиперидино, морфолино, тиоморфолино, тиоксанил, пиперазинил, гомопиперазинил, азетидинил, оксетанил, тиетанил, гомопиперидинил, оксепанил, тиепанил, оксазепинил, диазепинил, тиазепинил, 2-пирролинил, 3-пирролинил, индолинил, 2Н-пиранил, 4Н-пиранил, диоксанил, 1,3-диоксоланил, пиразолинил, дитианил, дитиоланил, дигидропиранил, дигидротиенил, дигидрофуранил, пиразолидинилимидазолинил, имидазолидинил, 3-азабицикло[3.1.0]гексанил, 3-азабицикло[4.1.0]гептанил и азабицикло[2.2.2]гексанил. В это определение включены также спирогруппы. Примерами гетероциклической группы, где кольцевые атомы замещены на оксо (=O) группы являются пиримидинонил и 1,1-диоксо-тиоморфолинил.
Термин «гетероарил» относится к одновалентному ароматическому радикалу из 5- или 6-членных колец и включает конденсированные кольцевые системы (по меньшей мере одно из которых является ароматическим) из 5-18 атомов, содержащие один или более гетероатомов, независимо выбранных из азота, кислорода и серы. Примерами гетероариловых групп являются пиридинил (включая, например, 2-гидроксипиридинил), имидазолил, имидазопиридинил, пиримидинил (включая, например, 4-гидроксипиримидинил), пиразолил, триазолил, пиразинил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксазолил, изотиазолил, пирролил, хинолинил, изохинолинил, индолил, бензимидазолил, бензофуранил, циннолинил, индазолил, идолизинил, фталазинил, пиридазинил, триазинил, изоиндолил, птеридинил, пуринил, оксадиазолил, триазолил, тиадиазолил, фуразанил, бензофуразанил, бензотиофенил, бензотиазолил, бензоксазолил, хиназолинил, хиноксалинил, нафтиридинил и фуропиридинил.
Гетероциклические или гетероариловые группы могут присоединяться, где это возможно, по атому углерода (углерод-связанные) или по атому азота (азот-связанные). В качестве не ограничивающего примера, углерод-связанные гетероциклы или гетероарилы связываются в положении 2, 3, 4, 5 или 6 пиридина, положении 3, 4, 5 или 6 пиридазина, положении 2, 4, 5 или 6 пиримидина, положении 2, 3, 5 или 6 пиразина, положении 2, 3, 4 или 5 фурана, тетрагидрофурана, тиофурана, тиофена, пиррола или тетрагидропиррола, положении 2, 4 или 5 оксазола, имидазола или тиазола, положении 3, 4 или 5 изоксазола, пиразола или изотиазола, положении 2 или 3 азиридина, положении 2, 3 или 4 азетидина, положении 2, 3, 4, 5, 6, 7 или 8 хинолина или положении 1, 3, 4, 5, 6, 7 или 8 изохинолина. В качестве не ограничивающего примера, азот-связанные гетероциклы или гетероарилы связываются в положении 1 азиридина, азетидина, пиррола, пирролидина, 2-пирролина, 3-пирролина, имидазола, имидазолидина, 2-имидазолина, 3-имидазолина, пиразола, пиразолина, 2-пиразолина, 3-пиразолина, пиперидина, пиперазина, индола, индолина, 1Н-индазола, положении 2 изоиндола или изоиндолина, положении 4 морфолина и положении 9 карбазола или О-карболина.
Гетероатомы, присутствующие в гетероариле или гетероцикле, включают окисленные формы, такие как NO, SO и SO2.
Термин «гало» или «галоген» относится к F, Cl, Br или I.
Термин «соединение» или «цитотоксичное соединение» или «цитотоксичный агент», используемый здесь, включает соединения, для которых их структура или формула или любое их производное было описано в настоящем изобретении или соединения, структура или формула которых или любое их производное включено здесь путем ссылки. Термин также включает стереоизомеры, геометрические изомеры, таутомеры, сольваты, метаболиты, соли (напр., фармацевтически приемлемые соли) и пролекарства, пролекарственные соли соединений всех формул, описанных в настоящем изобретении. Термин включает также все сольваты, гидраты и полиморфные формы любого из вышеупомянутых соединений. Конкретное перечисление «стереоизомеров», «геометрических изомеров», «таутомеров», «сольватов», «метаболитов», «солей», «пролекарств», «солей пролекарств», «конъюгатов», «солей конъюгатов», «сольватов», «гидратов» или «полиморфных форм» в одних аспектах настоящего изобретения, описанных в данной заявке, не следует интерпретировать как намеренное упущение этих форм в других аспектах настоящего изобретения, где используется термин «соединение» без перечисления этих других форм.
Термин «конъюгат», используемый здесь, относится к соединению или его производному, которое соединено с клеточносвязьшающим агентом и определяется общей формулой: C-L-CBA, где C = соединение, L = связывающая группа, и СВА = клеточносвязывающий агент.
Термин «связываемый с клеточносвязывающим агентом», используемый здесь, относится к новым бензодиазепиновым соединениям (напр., индолинобензодиазепиновым или оксазолидинобензодиазепиновым), их производным или димерам, содержащим по меньшей мере одну связывающую группу или прекурсор, приемлемый для связывания этих соединений, их производных или димеров с клеточносвязывающим агентом.
Термин «прекурсор» данной группы относится к любой группе, которая может привести к получению этой группы или снятию защиты, химической модификации или реакции связывания.
Термин «связанный с клеточносвязывающим агентом» относится к молекуле конъюгата, включающей по меньшей мере одно из новых бензодиазепиновых соединений (напр., индолинобензодиазепинового или оксазолидинобензодиазепинового), их производных или димеров, связанных с клеточносвязывающим агентом за счет соответствующей связывающей группы или ее прекурсора.
Термин «хиральная» относится к молекулам, обладающим свойством несовмещения с зеркальным отражением, а термин «ахиральная» относится к молекулам, которые полностью совмещаются со своим зеркальным отражением.
Термин «стереоизомер» относится к соединениям, имеющим одинаковую химическую природу и последовательность связей, но различную ориентацию атомов в пространстве, которые не могут быть взаимопревращены вращением вокруг одинарных связей.
Термин «диастереомер» относится к стереоизомеру с двумя или более центрами хиральности, молекулы которого не являются зеркальными отражениями друг друга. Диастереомеры имеют различные физические свойства, напр., температуры плавления и кипения, спектральные свойства и химическую активность. Смеси диастереомеров можно разделить при помощи аналитических методов высокого разрешения, таких как кристаллизация, электрофорез и хроматография.
Термин «энантиомеры» относится к двум стереоизомерам соединения, которые не совмещаются с зеркальным отражением друг друга.
Стереохимические определения и обозначения, используемые здесь, как правило, следуют указаниям Словаря химических терминов, составленного S.P. Parker, Ed., McGraw-Hill (1984) McGraw-Hill Book Company, New York и книги «Стереохимия органических соединений», авторы Eliel, Е. и Wilen, S., John Wiley & Sons, Inc., New York, 1994. Предполагается, что все стереоизомерные формы соединений настоящего изобретения, включая, не ограничиваясь, диастереомеры, энантиомеры и атропоизомеры, а также их смеси, такие как рацемические смеси, являются частью настоящего изобретения. Многие органические соединения существуют в оптически активных формах, т.е. они обладают способностью вращать плоскость плоско-поляризованного света. В описании оптически активного соединения используются приставки D и L, или R и S для обозначения абсолютной конфигурации молекулы относительно ее хирального центра(ов). Приставки d и l или (+) и (-) используются для обозначения знака вращения плоско-поляризованного света соединением настоящего изобретения, где (-) или l обозначает, что соединение является левовращающим. Соединение с приставкой (+) или d является правовращающим. Для данной химической структуры эти стереоизомеры являются идентичными, за исключением того, что они являются зеркальным отражением друг друга. Отдельный стереоизомер может упоминаться также как энантиомер, а смесь таких изомеров часто называется энантиомерной смесью. Смесь энантиомеров 50:50 упоминается как рацемическая смесь или рацемат, что может иметь место, если химическая реакция или метод не является стереоселективным или стереоспецифичным. Термины «рацемическая смесь» и «рацемат» относятся к эквимолярной смеси двух энантиомерных частиц, лишенной оптической активности.
Термин «таутомер» или «таутомерная форма» относится к структурным изомерам различных энергий, которые не могут быть взаимопревращены за счет низкоэнергетичного барьера. Например, протонные таутомеры (также известные как прототропные таутомеры) включают взаимопревращения за счет миграции протона, как например, кето-енольная и имин-енаминная изомеризации. Валентные таутомеры включают взаимопревращения за счет перегруппировки некоторых связывающих электронов.
Заместитель является «замещаемым», если он содержит по меньшей мере один атом углерода, серы, кислорода или азота, связанный с одним или более атомов водорода. Так, например, водород, галоген и циано-группа не входят в это определение.
Если заместитель описан как «замещенный», то неводородный заместитель занимает место водородного заместителя у углерода, кислорода, серы или азота этого заместителя. Так, например, замещенный алкиловый заместитель является алкиловым заместителем, у которого по меньшей мере один неводородный заместитель занимает место водородного заместителя этого алкилового заместителя. Для наглядности, монофторалкил является алкилом, замещенным фтор-заместителем, а дифторалкил является алкилом, замещенный двумя фтор-заместителями. Следует понимать, что если у заместителя имеется более одного замещения, то неводородные заместители могут быть одинаковыми или разными (если не указано иное).
Если заместитель описан как «необязательно замещенный», то заместитель может быть или (1) не замещенным, или (2) замещенным. Если углерод заместителя описан как необязательно замещенный одним или более из списка заместителей, то один или более атомов водорода у углерода (вплоть до всех) могут быть по отдельности и/или одновременно замещены на независимо выбранные возможные заместители. Если азот заместителя описан как необязательно замещенный одним или более из списка заместителей, то один или более атомов водорода у азота (вплоть до всех) могут быть замещены на независимо выбранные возможные заместители. Одним из примеров заместителя является --NR'R'', где R' и R'' вместе с атомом азота, к которому они прикреплены, могут образовывать гетероциклическое кольцо. Гетероциклическое кольцо, образованное R' и R'' вместе с атомом азота, к которому они прикреплены, может быть частично или полностью насыщенным. В одном варианте настоящего изобретения гетероциклическое кольцо состоит из 3-7 атомов. В другом варианте настоящего изобретения гетероциклическое кольцо выбрано из группы, состоящей из групп: пирролил, имидазолил, пиразолил, триазолил, тетразолил, изоксазолил, пиридил и тиазолил.
В этом описании взаимозаменяемо используются термины «заместитель», «радикал» и «группа».
Если группа заместителей совместно описывается как необязательно замещенная одним или более из списка заместителей, то эта группа может включать: (1) незамещаемые заместители, (2) замещаемые заместители, не замещенные возможными заместителями и/или (3) замещаемые заместители, которые замещены одним или более из возможных заместителей.
Если заместитель описан как необязательно замещенный до определенного числа неводородных заместителей, то этот заместитель может быть или (1) незамещенным; или (2) замещенным до определенного числа неводородных заместителей или до максимального числа замещаемых положений заместителя, в зависимости от того, что меньше. Так, например, если заместитель описан как гетероарил, необязательно замещенный не более 3 неводородными заместителями, то любой арил, содержащий менее 3 замещаемых положений, будет необязательно замещаться до такого количества неводородных заместителей, сколько имеется замещаемых положений у гетероарила. Такие заместители, в не ограничивающих примерах, могут быть выбраны из групп: линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, галоген, гуанидиний [-NH(C=NH)NH2], OR7, NR8R9, NO2, NRCOR', SR10, сульфоксид, представленный группой SOR', сульфон, представленный группой -SO2R', сульфит -SO3, бисульфит -OSO3, сульфонамид, представленный группой SO2NRR', циано, азидо, -COR11, OCOR11 или OCONR11R12, где каждый из R7, R8, R9, R10, R11 и R12 независимо выбран из групп: Н, линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000, арил, имеющий от 6 до 10 углеродных атомов, гетероциклическое кольцо, имеющее от 3 до 10 углеродных атомов. Термин «пролекарство», используемый в настоящей заявке, относится к прекурсору или производной форме соединения настоящего изобретения, которое может быть ферментативно или гидролитически активировано или преобразовано в более активную исходную форму. См., напр., «Пролекарства в химиотерапии рака», автор Wilman, Biochemical Society Transactions, 14, с. 375-382, 615th Meeting Belfast (1986) и «Пролекарства: химический подход к целевой доставке лекарства», авторы Stella et al., «Целевая доставка лекарств», авторы Borchardt et al., (ed.), с. 247-267, Humana Press (1985). Пролекарства настоящего изобретения включают, не ограничиваясь, эфир-содержащие пролекарства, фосфат-содержащие пролекарства, тиофосфат-содержащие пролекарства, сульфат-содержащие пролекарства, пептид-содержащие пролекарства, D-аминокислотные модифицированные пролекарства, гликозилатные пролекарства, бета-лактам-содержащие пролекарства, необязательно замещенные феноксиацетамид-содержащие пролекарства, необязательно замещенные фенилацетамид-содержащие пролекарства, 5-фторцитозиновые и другие 5-фторуридиновые пролекарства, которые могут быть превращены в более активные цитотоксичные свободные лекарства. Примеры цитотоксичных лекарств настоящего изобретения, которые могут быть преобразованы в пролекарственную форму применения, включают, не ограничиваясь, соединения настоящего изобретения и химиотерапевтические агенты, описанные выше.
Термин «пролекарство» также включает производное соединения, которое может гидролизоваться, окисляться или другим образом реагировать в биологических условиях (in vitro или in vivo) с образованием соединения настоящего изобретения. Пролекарства могут становиться активными только при протекании таких реакций в биологических условиях, или могут быть активными в непрореагировавшей форме. Примеры пролекарств, рассмотренных в настоящем изобретении, включают, не ограничиваясь, аналоги или производные соединений любой из формул, описанных здесь, которые содержат биогидролизуемые фрагменты, такие как биогидролизуемые амиды, биогидролизуемые сложные эфиры, биогидролизуемые карбаматы, биогидролизуемые карбонаты, биогидролизуемые уреиды и биогидролизуемые фосфатные аналоги. Другие примеры пролекарств включают производные соединений любой из формул, рассмотренных здесь, содержащие группы --NO, --NO2, --ONO или -ONO2. Пролекарства, как правило, можно получить при помощи хорошо известных способов, таких как описаны в книге Медицинская химия и открытие лекарств, автор Burger (1995) 172-178, 949-982 (Manfred Е. Wolff ed., 5-ое изд); см. также Фармакологические основы лекарственных средств, Goodman and Gilman, 8-ое изд., «Биотрансформация лекарств», McGraw-Hill, Int. Ed. 1992.
Если не указано иное, используемые здесь термины «биогидролизуемый амид», «биогидролизуемый сложный эфир», «биогидролизуемый карбамат», «биогидролизуемый карбонат», «биогидролизуемый уреид» и «биогидролизуемый фосфатный аналог» означают амид, сложный эфир, карбамат, карбонат, уреид или фосфатный аналог, соответственно, который или: 1) не нарушает биологическую активность соединения настоящего изобретения и придает этому соединению выгодные свойства in vivo, такие как поглощение, продолжительность действия или начало действия; или 2) является биологически неактивным, но превращается in vivo в биологически активное соединение. Примеры биогидролизуемых амидов включают, не ограничиваясь, низшие алкил амиды, альфа-аминокислотные амиды, алкоксиациламиды и алкиламиноалкилкарбониламиды. Примеры биогидролизуемых сложных эфиров включают, не ограничиваясь, низшие алкилэфиры, алкоксиацилоксиэфиры, алкилациламиноалкилэфиры и холинэфиры. Примеры биогидролизуемых карбаматов включают, не ограничиваясь, низшие алкиламины, замещенные этилендиамины, аминокислоты, гидроксиалкиламины, гетероциклические и гетероароматические амины и полиэфирамины. Наиболее предпочтительными пролекарствами и пролекарственньгми солями являются те, которые увеличивают биодоступность соединений настоящего изобретения при введении таких соединений млекопитающим.
Фраза «фармацевтически приемлемая соль», используемая здесь, относится к фармацевтически приемлемым органическим или неорганическим солям настоящего изобретения. Примеры солей включают, не ограничиваясь, сульфаты, цитраты, ацетаты, оксалаты, хлориды, бромиды, йодиды, нитраты, бисульфаты, фосфаты, кислые фосфаты, изоникотинаты, лактаты, салицилаты, кислые цитраты, тартраты, олеаты, таннаты, пантотенаты, битартраты, аскорбаты, сукцинаты, малеаты, гентизинаты, фумараты, глюконаты, глюкуронаты, сахараты, формиаты, бензоаты, глутаматы, метансульфонаты «мезилаты», этансульфонаты, бензолсульфонаты, п-толуолсульфонаты, памоаты (т.е. 1,1'-метилен-бис-(2-гидрокси-3-нафтоаты)), соли щелочных металлов (напр., натрия и калия), соли щелочноземельных металлов (напр., магния) и соли аммония. Фармацевтически приемлемая соль может включать другую молекулу, такую как ацетат-ион, сукцинат-ион или другой противоион. Противоионом может быть любая органическая или неорганическая группа, стабилизирующая заряд исходного соединения. Кроме того, фармацевтически приемлемая соль может иметь более одного заряженного атома в своей структуре. В случаях, где частью фармацевтически приемлемой соли являются многозарядные атомы, может быть несколько противоионов. Поэтому фармацевтически приемлемая соль может иметь один или более заряженных атомов и/или один или более противоионов.
Если соединение настоящего изобретения является основанием, то желаемая фармацевтически приемлемая соль может быть получена любым подходящим способом, доступным в этой области науки, например, обработкой свободного основания неорганической кислотой, такой как соляная кислота, бромоводородная кислота, серная кислота, азотная кислота, метансульфоновая кислота, фосфорная кислота и т.п., или органической кислотой, такой как уксусная кислота, малеиновая кислота, янтарная кислота, миндальная кислота, фумаровая кислота, малоновая кислота, пировиноградная кислота, щавелевая кислота, гликолевая кислота, салициловая кислота, пиранозидиловая кислота, такая как глюкуроновая кислота или галактуроновая кислота, альфа-гидроксикислота, такая как лимонная кислота или винная кислота, аминокислота, такая как аспарагиновая кислота или глутаминовая кислота, ароматическая кислота, такая как бензойная кислота или коричная кислота, сульфоновая кислота, такая как п-толуолсульфоновая кислота или этансульфоновая кислота и т.п.
Если соединение настоящего изобретения является кислотой, то желаемая фармацевтически приемлемая соль может быть получена любым подходящим способом, например, обработкой свободной кислоты неорганическим или органическим основанием, таким как амин (первичный, вторичный или третичный), гидроксидом щелочного металла или гидроксидом щелочноземельного металла и т.п. Наглядные примеры приемлемых солей включают, не ограничиваясь, органические соли, полученные из аминокислот, таких как глицин и аргинин, аммиак, первичные, вторичные и третичные амины, циклические амины, такие как пиперидин, морфолин и пиперазин и неорганические соли, производные натрия, кальция, калия, магния, марганца, железа, меди, цинка, алюминия и лития.
Используемый здесь термин «сольват» означает соединение, которое дополнительно включает стехиометрическое или нестехиометрическое количество растворителя, такого как вода, изопропанол, ацетон, этанол, метанол, ДМСО, этилацетат, уксусная кислота и этаноламин, дихлорметан, 2-пропанол и т.п., связанного нековалентными межмолекулярными силами. Сольваты или гидраты соединений настоящего изобретения легко получаются добавлением по меньшей мере одного молярного эквивалента гидроксильного растворителя, такого как метанол, этанол, 1-пропанол, 2-пропанол или вода, к соединению настоящего изобретения, в результате чего происходит сольватация или гидратация иминогруппы.
Термины «аномальный клеточный рост» и «пролиферативное нарушение» используются в настоящей заявке взаимозаменяемо. Термин «аномальный клеточный рост», используемый здесь, если не оговорено иное, относится к клеточному росту, который не зависит от нормальных регуляторных механизмов (напр., снижение контактного ингибирования). Это включает, например, аномальный рост: (1) опухолевых клеток (опухолей), которые разрастаются за счет экспрессии мутированной тирозинкиназы или гиперэкспрессии рецептора тирозинкиназы; (2) доброкачественных и злокачественных клеток других пролиферативных нарушений, в которых возникает аберрантная активация тирозинкиназы; (3) любых опухолей, разрастающихся за счет рецептора тирозинкиназы; (4) любых опухолей, которые разрастаются за счет абберантной активации серин/треонинкиназы; и (5) доброкачественных и злокачественных клеток других пролиферативных заболеваний, в которых возникает аберрантная активация серин/треонинкиназы.
Термины «рак» и «раковый» относятся или описывают физиологическое состояние млекопитающего, обычно характеризующееся нерегулируемым клеточным ростом. Термин «опухоль» включает одну или более раковых клеток. Примеры рака включают, не ограничиваясь, карциному, лимфому, бластому, саркому и лейкемию или лимфолейкоз. Более конкретные примеры таких раковых заболеваний включают плоскоклеточный рак (напр., эпителиальный плоскоклеточный рак), рак легких, включая мелкоклеточный рак легкого, немелкоклеточный рак легкого («NSCLC»), аденокарциному легкого и плоскоклеточную карциному легкого, рак брюшины, гепатоцеллюлярный рак, рак желудка, включая гастроинтестинальный рак, рак поджелудочной железы, глиобластому, рак шейки матки, рак яичников, рак печени, рак мочевого пузыря, гепатому, рак груди, рак толстой кишки, рак прямой кишки, колоректальный рак, рак тела матки, карциному слюнной железы, почечный или ренальный рак, рак простаты, рак вульвы, рак щитовидной железы, рак печени, анальный рак, пенильный рак, острый лейкоз, а также рак мозга головы и рак шеи.
Термин «терапевтическое средство» включает как биологический агент, такой как антитело, пептид, белок или фермент, так и химиотерапевтический агент. «Химиотерапевтическое средство» является химическим соединением, применимым для лечения рака. Примеры химиотерапевтических средств включают Эрлотиниб (TARCEVA®, Genentech/OSI Pharm.), Бортезомиб (VELCADE®, Millennium Pharm.), Фулвестрант (FASLODEX®, AstraZeneca), Сутент (SU11248, Pfizer), Летрозол (FEMARA®, Novartis), Иматиниба мезилат (GLEEVEC®, Novartis), PTK 787/ZK 222584 (Novartis), Оксалиплатин (Eloxatin®, Sanofi), 5-FU (5-фторурацил), Лейковорин, Рапамицин (Sirolimus, RAPAMUNE®, Wyeth), Лапатиниб (TYKERB®, GSK572016, Glaxo Smith Kline), Лонафарниб (SCH 66336), Сорафениб (BAY43-9006, Bayer Labs) и Гефитиниб (IRESSA®, AstraZeneca), AG1478, AG1571 (SU 5271; Sugen), алкилирующие средства, такие как тиотепа и циклофосфамид CYTOXAN®; алкилсульфонаты, такие как бусульфан, импросульфан и пипосульфан; азиридины, такие как бензодопа, карбоквион, метуредопа, и уредопа; этиленимины и метиламеламины, включая альтретамин, триэтиленмеламин, триэтиленфосфорамид, триэтилентиофосфорамид и триметиломеламин; ацетогенины (особенно буллатацин и буллатацинон); камптотецин (включая синтетический аналог топотекан); бриостатин; каллистатин; СС-1065 (включая его синтетические аналоги адозелезин, карзелезин и бизелезин); криптофицины (в частности, криптофицин 1 и криптофицин 8); доластатин; дуокармицин (включая синтетические аналоги, KW-2189 и CBI-TMI); элеутеробин; панкратистатин; саркодиктин; спонгистатин; азотистые иприты, такие как хлорамбуцил, хлорнафазин, хлорфосфамид, эстрамустин, ифосфамид, мехлорэтамин, гидрохлорид окиси мехлорэтамина, мелфалан, новембихин, фенестерин, преднимустин, трофосфамид, урацила иприт; нитрозомочевины, такие как кармустин, хлорозотоцин, форемустин, ломустин, нимустин и ранимустин; антибиотики, такие как энедииновые антибиотики (напр., калихеамицин, особенно калихеамицин гаммаll и калихеамицин омегаll, (Agnew, Chem. Intl. Ed. Engl., 33: 183-186 (1994); динемицин, включая динемицин А; бисфосфонаты, такие как клодронат; эсперамицин; а также хромофор неокарциностатина и родственные хромопротеиновые энедииновые антибактериальные хромофоры), аклациномицины, актиномицин, аутрамицин, азасерин, блеомицины, кактиномицин, карабицин, карминомицин, карцинофилин, хромомицины, дактиномицин, даунорубицин, деторубицин, 6-диазо-5-оксо-L-норлейцин, ADRIAMYCIN® (доксорубицин), морфолино-доксорубицин, цианоморфолино-доксорубицин, 2-пирролино-доксорубицин и деоксидоксорубицин, эпирубицин, эзорубицин, идарубицин, марцелломицин, митомицины, такие как митомицин С, микофеноловая кислота, ногаламицин, оливомицины, пепломицин, порфиромицин, пиромицин, квеламицин, родорубицин, стрептонигрин, стрептозоцин, туберцидин, убенимекс, зиностатин, зорубицин; антиметаболиты, такие как метотрексат и 5-фторурацил (5-FU); аналоги фолиевой кислоты, такие как деноптерин, метотрексат, птероптерин, триметрексат; аналоги пуринов, такие как флударабин, 6-меркаптопурин, тиамиприн, тиогуанин; пиримидиновые аналоги, такие как анцитабин, азацитидин, 6-азауридин, кармофур, цитарабин, дидеоксиуридин, доксифлуридин, эноцитабин, флоксуридин; андрогены, такие как калустерон, дромостанолона пропионат, эпитиостанол, мепитиостан, тестолактон; анти-адренальные, такие как аминоглютетимид, митотан, трилостан; пополнитель фолиевой кислоты, такой как фролиновая кислота; ацеглатон; альдофосфамида гликозид; аминолевулиновая кислота; энилурацил; амсакрин; бестрабуцил; бизантрен; эдатраксат; дефофамин; демеколцин; диазиквон; элфорнитин; эллиптиния ацетат; эпотилон; этоглуцид; нитрат галлия; гидроксимочевина; лентинан; лонидамин; майтанзиноиды, такие как майтанзин и ансамитоцины; митогвазон; митоксантрон; мопиданмол; нитраэрин; пентостатин; фенамет; пирарубицин; лозоксантрон; подофиллиновая кислота; 2-этилгидразид; прокарбазин; полисахаридный комплекс PSK® (JHS Natural Products, Eugene, Oreg.); разоксан; ризоксин; сизофуран; спирогерманий; тенуазоновая кислота; триазиквон; 2,2',2''-трихлортриэтиламин; трихотецены (особенно Т-2 токсин, верракурин А, роридин А и ангвидин); уретан; виндезин; дакарбазин; манномустин; митобронитол; митолактол; пипоброман; гацитозин; арабинозид ("Ara-С"); циклофосфамид; тиотепа; таксоиды, напр., TAXOL® (паклитаксел, Bristol Meyers Squibb Oncology, Princeton, N.J.), ABRAXANE® (не содержит Cremophor), композиции из наночастиц паклитаксела на основе альбумина (American Pharmaceutical Partners, Schaumberg, Ill., США) и TAXOTERE® (доцетаксел, Rhone-Poulenc Rorer, Antony, Франция); хлорамбуцил; GEMZAR® (гемцитабин); 6-тиогуанин; меркаптопурин; метотрексат; платиновые аналоги, такие как цисплатин и карбоплатин; винбластин; этопсид (VP-16); ифосфамид; митоксантрон; винкристин; NAVELBINE® (винорелбин); новантрон; тенипозид; эдатрексат; дауномицин; аминоптерин; капецитабин (XELODA®); ибандронат; СРТ-11; ингибитор топоизомеразы RFS 2000; дифторметилорнитин (DMFO); ретиноиды, такие как ретиноевая кислота; и фармацевтически приемлемые соли, кислоты и производные любого вышеуказанного.
Также включенными в определение «химиотерапевтическое средство» являются: (i) антигормональные средства, действие которых направлено на регулирование или ингибирование гормонального эффекта на опухоли, такие как антиэстрогены и селективные модуляторы рецепторов эстрогена (SERM), включая, например, тамоксифен (включая NOLVADEX®; тамоксифена цитрат), ралоксифен, дролоксифен, 4-гидрокситамоксифен, триоксифен, кеоксифен, LY117018, онапристон и FARESTON® (торемифена цитрат); (ii) ингибиторы фермента ароматазы, которые ингибируют фермент ароматазу, регулирующую выработку эстрогенов в надпочечниках, такие как, например, 4(5)-имидазолы, аминоглутетимид, MEGASE® (мегестрола ацетат), AROMASIN® (экземестан; Pfizer), форместан, фадразол, RTVISOR® (ворозол), FEMARA® (летрозол; Novartis) и ARIMIDEX® (анастрозол; AstraZeneca); (iii) антиандрогены, такие как флутамид, нилутамид, бикалутамид, лейпролид и госерилин; а также троксацитабин (аналог 1,3-диоксоланового нуклеозида цитозина); (iv) ингибиторы протеинкиназы; (v) ингибиторы липидкиназы; (vi) антисмысловые олигонуклеотиды, в частности, те, которые ингибируют экспрессию генов в сигнальных путях, включенных в аберрантное разрастание клеток, такие как, например, PKC-альфа, Ralf и H-Ras; (vii) рибозимы, такие как ингибитор экспрессии VEGF (напр., ANGIOZYME®) и ингибиторы экпсрессии HER2; (viii) вакцины, такие как вакцины для генотерапии, например, ALLOVECTIN®, LEUVECTIN® и VAXID®; rIL-2 PROLEUKIN®; ингибитор топоизомеразы 1, такой как LURTOTECAN®; rmRH ABARELIX®; (ix) анти-ангиогенные агенты, такие как бевацизумаб (AVASTIN®, Genentech); и (x) фармацевтически приемлемые соли, кислоты и производные любого из указанных выше. Другие анти-ангиогенные агенты включают ингибиторы ММР-2 (матриксной металлопротеиназы 2), ингибиторы ММР-9 (матриксной металлопротеиназы 9), ингибиторы СОХ-II (циклооксигеназы II) и ингибиторы тирозинкиназы рецептора VEGF. Примеры таких применимых ингибиторов матриксной металлопротеиназы, которые могут использоваться в сочетании с соединениями/композициями настоящего изобретения, описаны в публикациях WO 96/33172, WO 96/27583, ЕР 818442, ЕР 1004578, WO 98/07697, WO 98/03516, WO 98/34918, WO 98/34915, WO 98/33768, WO 98/30566, EP 606,046, EP 931,788, WO 90/05719, WO 99/52910, WO 99/52889, WO 99/29667, WO 99/07675, EP 945864, патенты США NoNo 5863949, 5861510 и EP 780386, все из которых включены здесь в полном объеме путем ссылки. Примеры ингибиторов тирозинкиназы рецептора VEGF включают 4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин (ZD6474; пример 2 в публикации WO 01/32651), 4-(4-фтор-2-метилиндол-5-илокси)-6-метокси-7-(3-пирролидин-1-илпропокси)-хиназолин (AZD2171; пример 240 в публикации WO 00/47212), ваталаниб (PTK787; WO 98/35985) и SU 11248 (сунитиниб; WO 01/60814), а также соединения, описанные в публикациях РСТ NoNo WO 97/22596, WO 97/30035, WO 97/32856 и WO 98/13354).
Другие примеры химиотерапевтических средств, которые могут использоваться в комбинации с соединениями настоящего изобретения, включают ингибиторы PI3K (фосфоинозитид-3 киназы), такие как указаны в журнале Yaguchi et al (2006) Jour. of the Nat. Cancer Inst. 98(8):545-556; патентах США NoNo 7173029; 7037915; 6608056; 6608053; 6838457; 6770641; 6653320; 6403588; публикациях WO 2006/046031; WO 2006/046035; WO 2006/046040; WO 2007/042806; WO 2007/042810; WO 2004/017950; US 2004/092561; WO 2004/007491; WO 2004/006916; WO 2003/037886; US 2003/149074; WO 2003/035618; WO 2003/034997; US 2003/158212; EP 1417976; US 2004/053946; JP 2001247477; JP 08175990; JP 08176070; патенте США No. 6703414 и WO 97/15658, все из которых включены здесь в полном объеме путем ссылки. Конкретные примеры таких ингибиторов PI3K включают SF-1126 (ингибитор PI3K, Semafore Pharmaceuticals), BEZ-235 (ингибитор PI3K, Novartis), XL-147 (ингибитор PI3K Exelixis, Inc.).
«Метаболит» является продуктом, вырабатываемым в процессе метаболизма в организме определенного соединения, его производного, его конъюгата или его соли. Метаболиты соединения, его производного или его конъюгата могут быть идентифицированы при помощи стандартной методики, известной в данной области науки, а их активности определяются при помощи анализов, описанных в настоящем документе. Такие продукты могу образовываться, например, в результате окисления, гидроксилирования, восстановления, гидролиза, амидирования, деамидирования, этерификации, деэтерификации, ферментативного разложения и т.п., введенного соединения. Соответственно, настоящее изобретение включает метаболиты соединений настоящего изобретения, их производных или их конъюгатов, включая соединения, их производные или их конъюгаты, которые получены в процессе, включающем взаимодействие соединения настоящего изобретения, его производного или его конъюгата с организмом млекопитающего в течение времени, достаточного для получения его метаболического продукта.
Фраза «фармацевтически приемлемый» показывает, что вещество или композиция должно быть совместимо химически и/или токсикологически с другими компонентами, входящими в состав композиции, и/или с организмом млекопитающего, подлежащим лечению.
Термин «защитная группа» или «защитная функциональная группа» относится к заместителю, который обычно используется для блокировки или защиты отдельной функциональности, в то время как другие функциональные группы соединения настоящего изобретения, его производного или его конъюгата участвуют в реакции. Например, «защитная группа для аминогруппы» или «защитная функциональная группа для аминогруппы» является заместителем, прикрепленным к аминогруппе, блокирующим или защищающим аминную функциональность соединения настоящего изобретения. Применимые защитные группы для аминогрупп включают ацетил, трифторацетил, трет-бутоксикарбонил (ВОС), бензилоксикарбонил (CBZ) и 9-фторенилметиленоксикарбонил (Fmoc). Аналогично, «защитная группа для гидрокси-группы» относится к заместителю гидрокси-группы, который блокирует или защищает гидроксильную функциональность. Применимые защитные группы включают ацетил и силил. Термин «защитная группа для карбокси-группы» относится к заместителю карбокси-группы, который блокирует или защищает карбоксильную функциональность. Обычные защитные для карбокси-группы включают фенилсульфонилэтил, цианоэтил, 2-(триметилсилил)этил, 2-(триметилсилил)этоксиметил, 2-(п-толуолсульфонил)этил, 2-(п-нитрофенилсульфенил)этил, 2-(дифенилфосфино)-этил, нитроэтил и т.п. Обычные защитные группы тиоловой функциональности включают такие, которые превращают тиол в сложный тиоэфир, например, ацетил, бензоил или трифторацетил, в простой тиоэфир, например, бензил, трет-бутил, трифенилметил, 9-фторенилметил, метоксиметил, 2-тетрагидропиранил или силил, в дисульфид, например, метил, бензил, трет-бутил, пиридил, нитропиридил, фенил, нитрофенил или динитрофенил, в тиокарбонат, например, трет-бутоксикарбонил, в тиокарбамат, например, N-этил. Общее описание защитных групп и их применение описано в книге Защитные группы в органическом синтезе, авторы P. G.M. Wuts & Т.W. Greene, John Wiley & Sons, New York, 2007.
Для новых бензодиазепинов формулы (I) и (II),
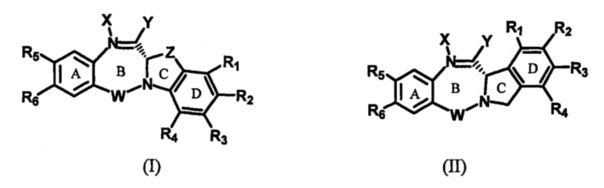
где:
двойная линия

Y выбран из групп: -OR, сложный эфир, представленный группой -OCOR', карбонат, представленный группой -OCOOR', карбамат, представленный группой -OCONR'R'', амин или гидроксиламин, представленный группой NR'R'', амид, представленный группой -NRCOR', пептид, представленный группой NRCOP, где Р является аминокислотой или полипептидом, содержащим от 2 до 20 аминокислотных звеньев, тиоэфир, представленный группой SR', сульфоксид, представленный группой SOR', сульфон, представленный группой -SO2R', сульфит -SO3, бисульфит -OSO3, галоген, циано, азидо или тиол, где R, R' и R'' одинаковые или разные и выбраны из групп: Н, замещенный или незамещенный линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 20 углеродных атомов, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000; 5- или 6-членное гетероарильное кольцо, содержащее один или более гетероатомов, независимо выбранных из азота, кислорода и серы, 5-18-членная конденсированная кольцевая система, где по меньшей мере одно кольцо является ароматическим, содержащая один или более гетероатомов, независимо выбранных из азота, кислорода и серы; арил, имеющий от 6 до 18 углеродных атомов, 3-18-членное гетероциклическое кольцо, имеющее от 1 до 6 гетероатомов, выбранных из О, S, N и Р, где заместитель выбран из групп: галоген, OR7, NR8R9, NO2, NRCOR', SR10, сульфоксид, представленный группой SOR', сульфон, представленный группой -SO2R', сульфит -SO3, бисульфит -OSO3, сульфонамид, представленный группой SO2NRR', циано, азидо, -COR11, OCOR11 или OCONR11R12, где каждый из R7, R8, R9, R10, R11 и R12 независимо выбран из групп: Н, линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000, 5- или 6-членное гетероарильное кольцо, содержащее один или более гетероатомов, независимо выбранных из азота, кислорода и серы, 5-18-членная конденсированная кольцевая система, где по меньшей мере одно кольцо является ароматическим, содержащая один или более гетероатомов, независимо выбранных из азота, кислорода и серы, арил, имеющий от 6 до 18 углеродных атомов, 3-18-членное гетероциклическое кольцо, имеющее от 1 до 6 гетероатомов, выбранных из О, S, N и Р, и R10 необязательно является SR13 или COR13, где R13 выбран из групп: линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000, 5- или 6-членное гетероарильное кольцо, содержащее один или более гетероатомов, независимо выбранных из азота, кислорода и серы, 5-18-членная конденсированная кольцевая система, где по меньшей мере одно кольцо является ароматическим, содержащая один или более гетероатомов, независимо выбранных из азота, кислорода и серы; 3-18-членное гетероциклическое кольцо, имеющее от 1 до 6 гетероатомов, выбранных из О, S, N и Р, и R11 может быть также OR14, где R14 является Н или имеет такое же определение, как R, необязательно R'' является ОН;
W является С=O, C=S, СН2, ВН (В = бор), SO или SO2;
каждый из R1, R2, R3, R4 независимо выбран из групп: Н, замещенный или незамещенный линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000, или заместитель, выбранный из групп: галоген, OR7, NR8R9, NO2, NRCOR', SR10, сульфоксид, представленный группой SOR', сульфон, представленный группой -SO2R', сульфит -SO3, бисульфит -OSO3, сульфонамид, представленный группой SO2NRR', циано, азидо, гуанидиний [-NH(C=NH)NH2], -COR11, -OCOR11 или -OCONR11R12, где R7, R8, R9, R10, R11 и R12 имеют те же определения, которые представлены выше, необязательно любой из R1, R2, R3, R4 является связывающей группой, обеспечивающей связь с клеточносвязывающим агентом за счет ковалентной связи или выбран из групп: полипирроловое, полииндолиловое, полиимидазолиловое, полипирроло-имидазолиловое, полипирроло-индолиловое или полиимидазоло-индолиловое звено, необязательно прикрепленное к связывающей группе, обеспечивающей связь с клеточносвязывающим агентом;
R5 выбран из групп: OR15, CRR'OH, SH, CRR'SH, NHR15 или CRR'NHR15, где R15 имеет то же определение, что и R, R и R' имеют то же определение, которое дано выше; необязательно R5 является связывающей группой, обеспечивающей связь с клеточносвязывающим агентом за счет ковалентной связи или выбран из групп: полипирроловое, полииндолиловое, полиимидазолиловое, полипирроло-имидазолиловое, полипирроло-индолиловое или полиимидазоло-индолиловое звено, необязательно прикрепленное к связывающей группе, обеспечивающей связь с клеточносвязывающим агентом;
R6 является OR, SR, NRR', где R и R' имеют то же определение, которое дано выше, или необязательно R6 является связывающей группой;
Z выбран из групп: (СН2)n, где n является 1, 2 или 3, CR15R16, NR17, О или S, где каждый из R15, R16 и R17 независимо выбран из групп: Н, линейный, разветвленный или циклический алкил, имеющий от 1 до 10 углеродных атомов, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000; или их фармацевтически приемлемые сольваты, соли, гидраты или гидратированные соли, их оптические изомеры, рацематы, диастереомеры, энантиомеры этих соединений,
при условии, что соединение настоящего изобретения имеет не более одной связывающей группы, обеспечивающей связь с клеточносвязывающим агентом за счет ковалентной связи.
В одном предпочтительном варианте настоящего изобретения двойная связь
W является С=O, СН2 или SO2;
каждый из R1, R2, R3, R4 является Н; необязательно, независимо, любой из R1, R2, R3 и R4 может быть связывающей группой, обеспечивающей связь с клеточносвязывающим агентом за счет ковалентной связи;
R5 выбран из групп: OR15, CRR'OH, SH, CRR'SH, NHR15 или CRR'NHR15, где R15 является H или имеет то же определение, которое дано выше для R, или выбран из групп: полипирроловое, полииндолиловое, полиимидазолиловое, полипирроло-имидазолиловое, полипирроло-индолиловое или полиимидазоло-индолиловое звено, необязательно прикрепленное к связывающей группе, обеспечивающей связь с клеточносвязывающим агентом, R и R' имеют то же определение, которое дано выше;
R6 является ОСН3;
Z выбран из групп: (СН2)n, где n является 1 или 2, NH, NCH3 или S; или их фармацевтически приемлемые сольваты, соли, гидраты или гидратированные соли, их оптические изомеры, рацематы, диастереомеры, энантиомеры или полиморфные кристаллические структуры этих соединений;
В предпочтительном варианте настоящего изобретения соединения формул (I) и (II) являются соединениями формул (VII), (VIII) или (IX):
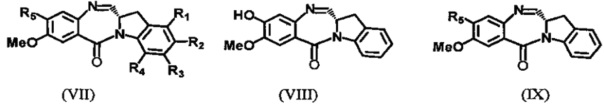
где значения заместителей описаны выше; или их фармацевтически приемлемые сольваты, соли, гидраты или гидратированные соли, их оптические изомеры, рацематы, диастереомеры, энантиомеры или полиморфные кристаллические структуры этих соединений.
Для новых бензодиазепинов формулы (III), в которой диазепиновое кольцо (В) конденсировано с гетероциклическим кольцом (С), где гетероциклическое кольцо является моноциклическим,
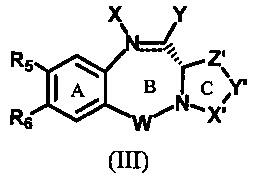
где:
двойная линия
Y выбран из групп: -OR, сложный эфир, представленный группой -OCOR', карбонат, представленный группой -OCOOR', карбамат, представленный группой -OCONR'R'', амин или гидроксиламин, представленный группой NR'R'', амид, представленный группой -NRCOR', пептид, представленный группой NRCOP, где Р является аминокислотой или полипептидом, содержащим от 2 до 20 аминокислотных звеньев, тиоэфир, представленный группой SR', сульфоксид, представленный группой SOR', сульфон, представленный группой -SO2R', сульфит -SO3, бисульфит -OSO3, галоген, циано, азидо или тиол, где R, R' и R'' одинаковые или разные и выбраны из групп: Н, замещенный или незамещенный линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, полиэтиленгликолевое звено (-OCH2CH2)n, где n является целым числом от 1 до 2000, 5- или 6-членное гетероарильное кольцо, имеющее один или более гетероатомов, независимо выбранных из азота, кислорода и серы, 5-18-членная конденсированная кольцевая система, где по меньшей мере одно кольцо является ароматическим, содержащая один или более гетероатомов, независимо выбранных из азота, кислорода и серы, арил, имеющий от 6 до 18 углеродных атомов, 3-18-членное гетероциклическое кольцо, имеющее 1-6 гетероатомов, выбранных из О, S, N и Р, где заместитель выбран из групп: галоген, OR7, NR8R9, NO2, NRCOR', SR10, сульфоксид, представленный группой SOR', сульфон, представленный группой -SO2R', сульфит -SO3, бисульфит -OSO3, сульфонамид, представленный группой SO2NRR', циано, азидо, -COR11, OCOR11 или OCONR11R12, где каждый из R7, R8, R9, R10, R11 и R12 независимо выбран из групп: Н, линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000, 5- или 6-членное гетероарильное кольцо, содержащее один или более гетероатомов, независимо выбранных из азота, кислорода и серы, 5-18-членная конденсированная кольцевая система, где по меньшей мере одно кольцо является ароматическим, содержащая один или более гетероатомов, независимо выбранных из азота, кислорода и серы, арил, имеющий от 6 до 18 углеродных атомов, 3-18-членное гетероциклическое кольцо, имеющее 1-6 гетероатомов, выбранных из О, S, N и Р и R10 является необязательно SR13 или COR13, где R13 выбран из групп: линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000, 5- или 6-членное гетероарильное кольцо, содержащее один или более гетероатомов, независимо выбранных из азота, кислорода и серы, 5-18-членная конденсированная кольцевая система, где по меньшей мере одно кольцо является ароматическим, содержащая один или более гетероатомов, независимо выбранных из азота, кислорода и серы, арил, имеющий от 6 до 18 углеродных атомов, 3-18-членное гетероциклическое кольцо, имеющее от 1 до 6 гетероатомов, выбранных из О, S, N и Р и R11 может быть также OR14, где R14 является Н или имеет такое же определение, что и R, необязательно R'' является ОН;
W является С=O, C=S, СН2, ВН, SO или SO2;
R5 выбран из групп: OR15, CRR'OH, SH, CRR'SH, NHR15 или CRR'NHR15, где R15 является H или имеет то же определение, что и R, или является связывающей группой, обеспечивающей связь с клеточносвязывающим агентом за счет ковалентной связи или выбран из групп: полипирроловое, полииндолиловое, полиимидазолиловое, полипирроло-имидазолиловое, полипирроло-индолиловое или полиимидазоло-индолиловое звено, необязательно прикрепленное к связывающей группе, обеспечивающей связь с клеточносвязывающим агентом; выбран из групп: полипирроловое, полииндолиловое, полиимидазолиловое, полипирроло-имидазолиловое, полипирроло-индолиловое или полиимидазоло-индолиловое звено, необязательно прикрепленное к связывающей группе, обеспечивающей связь с клеточносвязывающим агентом, необязательно R5 является связывающей группой, обеспечивающей связь с клеточносвязывающим агентом за счет ковалентной связи;
R6 является OR, SR или NRR', где R и R' имеют то же определение, которое дано выше, необязательно R6 является связывающей группой;
X' является СН2, NR, СО, ВН, SO или SO2;
Y' является О, СН2, NR или S;
Z' является СН2 или (СН2)n, где n является 2, 3 или 4; или их фармацевтически приемлемые сольваты, соли, гидраты или гидратированные соли, их оптические изомеры, рацематы, диастереомеры, энантиомеры или полиморфные кристаллические структуры этих соединений;
при условии, что соединение настоящего изобретения имеет не более одной связывающей группы, обеспечивающей связь с клеточносвязывающим агентом за счет ковалентной связи.
В одном предпочтительном варианте настоящего изобретения двойная связь
W является С=O, СН2 или SO2;
R5 выбран из групп: OR15, CRR'OH, SH, CRR'SH, NHR15 или CRR'NHR15, где R15 является H или имеет то же определение, которое дано выше для R, или выбран из групп: полипирроловое, полииндолиловое, полиимидазолиловое, полипирроло-имидазолиловое, полипирроло-индолиловое или полиимидазоло-индолиловое звено, необязательно прикрепленное к связывающей группе, обеспечивающей связь с клеточносвязывающим агентом;
R6 является ОСН3;
X' выбран из СН2 или С=O;
Y' является О, СН2, NR или S;
Z' является (СН2)n, где n является 1 или 2, при условии, что X', Y' и Z' не являются одновременно СН2; или их фармацевтически приемлемые сольваты, соли, гидраты или гидратированные соли, их оптические изомеры, рацематы, диастереомеры, энантиомеры или полиморфные кристаллические структуры этих соединений;
В предпочтительном варианте настоящего изобретения соединение формулы III представлено соединением формулы (X) или (XI),

где значения заместителей описаны выше; или их фармацевтически приемлемые сольваты, соли, гидраты или гидратированные соли, их оптические изомеры, рацематы, диастереомеры, энантиомеры или полиморфные кристаллические структуры этих соединений.
Для цитотоксичных димеров, представленных формулами (IV), (V) и (VI)
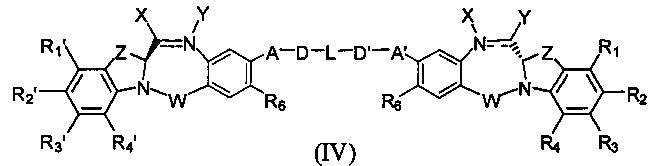
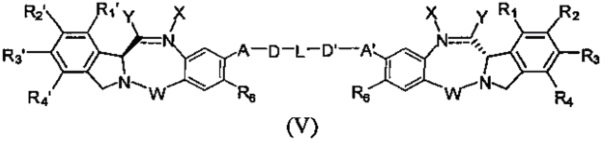
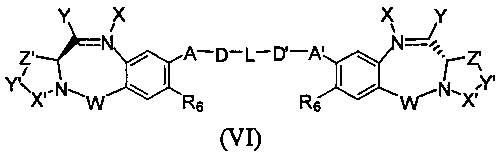
двойная линия
Y выбран из групп: -OR, сложный эфир, представленный группой -OCOR', карбонат, представленный группой -OCOOR', карбамат, представленный группой -OCONR'R'', амин или гидроксиламин, представленный группой NR'R'', амид, представленный группой -NRCOR', пептид, представленный группой NRCOP, где Р является аминокислотой или полипептидом, содержащим от 2 до 20 аминокислотных звеньев, тиоэфир, представлетгый группой SR', сульфоксид, представленный группой SOR', сульфон, представленный группой -SO2R', сульфит -SO3, бисульфит -OSO3, галоген, циано, азидо или тиол, где R, R' и R'' одинаковые или разные и выбраны из групп: Н, замещенный или незамещенный линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000, 5- или 6-членное гетероарильное кольцо, имеющее один или более гетероатомов, независимо выбранных из азота, кислорода и серы, 5-18-членная конденсированная кольцевая система, где по меньшей мере одно кольцо является ароматическим, содержащая один или более гетероатомов, независимо выбранных из азота, кислорода и серы, арил, имеющий от 6 до 18 углеродных атомов, 3-18-членное гетероциклическое кольцо, имеющее 1-6 гетероатомов, выбранных из О, S, N и Р, где заместитель выбран из групп: галоген, OR7, NR8R9, NO2, NRCOR', SR10, сульфоксид, представленный группой SOR', сульфон, представленный группой -SO2R', сульфит -SO3, бисульфит -OSO3, сульфонамид, представленный группой SO2NRR', циано, азидо, -COR11, OCOR11 или OCONR11R12, где каждый из R7, R8, R9, R10, R11 и R12 независимо выбран из групп: Н, линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000, 5- или 6-членное гетероарильное кольцо, содержащее один или более гетероатомов, независимо выбранных из азота, кислорода и серы, 5-18-членная конденсированная кольцевая система, где по меньшей мере одно кольцо является ароматическим, содержащая один или более гетероатомов, независимо выбранных из азота, кислорода и серы, арил, имеющий от 6 до 18 углеродных атомов, 3-18-членное гетероциклическое кольцо, имеющее 1-6 гетероатомов, выбранных из О, S, N и Р и R10 необязательно является SR13 или COR13, где R13 выбран из групп: линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, 5- или 6-членное гетероарильное кольцо, содержащее один или более гетероатомов, независимо выбранных из азота, кислорода и серы, 5-18-членная конденсированная кольцевая система, где по меньшей мере одно кольцо является ароматическим, содержащая один или более гетероатомов, независимо выбранных из азота, кислорода и серы, арил, имеющий от 6 до 18 углеродных атомов, 3-18-членное гетероциклическое кольцо, имеющее от 1 до 6 гетероатомов, выбранных из О, S, N и Р, необязательно R11 является OR14, где R14 имеет такое же определение, что и R, необязательно R'' является ОН;
W является С=O, C=S, СН2, ВН, SO или SO2;
каждый из R1, R2, R3, R4, R1', R2', R3' и R4' независимо выбран из групп: Н, замещенный или незамещенный линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000, или заместитель, выбранный из групп: галоген, гуанидиний [-NH(C=NH)NH2], OR7, NR8R9, NO2, NRCOR', SR10, сульфоксид, представленный группой SOR', сульфон, представленный группой -SO2R', сульфит -SO3, бисульфит -OSO3, сульфонамид, представленный группой SO2NRR', циано, азидо, -COR11, OCOR11 или OCONR11R12, где R7, R8, R9, R10, R11 и R12 - как определено выше, необязательно, любой из R1, R2, R3, R4, R1', R2', R3' или R4' является связывающей группой, обеспечивающей связь с клеточносвязывающим агентом за счет ковалентной связи или выбрана из групп: полипирроловое, полииндолиловое, полиимидазолиловое, полипирроло-имидазолиловое, полипирроло-индолиловое или полиимидазоло-индолиловое звено, необязательно прикрепленное к связывающей группе, обеспечивающей связь с клеточносвязывающим агентом;
Z выбран из групп: (СН2)n, где n является 1, 2 или 3, CR15R16, NR17, О или S, где каждый из R15, R16 и R17 независимо выбран из групп: Н, линейный, разветвленный или циклический алкил, имеющий от 1 до 10 углеродных атомов, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000;
R6 является OR, SR или NRR', где R и R' имеют то же определение, которое дано выше, необязательно R6 является связывающей группой;
X' выбран из групп: СН2, NR, СО, ВН, SO или SO2, где R имеет то же определение, которое дано выше;
Y' является О, СН2, NR или S, где R имеет то же определение, которое дано выше;
Z' является СН2 или (СН2)n, где n является 2, 3 или 4, при условии, что X', Y' и Z' не являются одновременно СН2;
А и А' одинаковые или различные и выбраны из групп: О, -CRR'O, S, -CRR'S, -NR15 или CRR'NHR15, где R и R' имеют то же определение, которое дано выше и где R15 имеет то же определение, что и R.
D и D' одинаковые или различные и независимо выбраны из групп: линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, необязательно замещенный любой из групп: галоген, OR7, NR8R9, NO2, NRCOR', SR10, сульфоксид, представленный группой SOR', сульфон, представленный группой -SO2R', сульфит -SO3, бисульфит -OSO3, сульфонамид, представленный группой SO2NRR', циано, азидо, -COR11, OCOR11 или OCONR11R12, где определения R7, R8, R9, R10, R11 и R12 такие же, как определено выше, или полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000;
L необязательно является фенильной группой или 3-18-членным гетероциклическим кольцом, имеющим от 1 до 6 гетероатомов, выбранных из О, S, N и Р, необязательно замещенным, где заместитель является связывающей группой, обеспечивающей связь с клеточносвязывающим агентом за счет ковалентной связи, или выбран из групп: линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, необязательно замещенный любой из групп: галоген, OR7, NR8R9, NO2, NRCOR', SR10, сульфоксид, представленный группой SOR', сульфон, представленный группой -SO2R', сульфит -SO3, бисульфит -OSO3, сульфонамид, представленный группой SO2NRR', циано, азидо, -COR11, OCOR11 или OCONR11R12, где R7, R8, R9, R10, R11 и R12 имеют то же определение, которое дано выше, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000; необязательно L является связывающей группой, обеспечивающей связь с клеточносвязывающим агентом за счет ковалентной связи; или их фармацевтически приемлемые сольваты, соли, гидраты или гидратированные соли, их оптические изомеры, рацематы, диастереомеры, энантиомеры или полиморфные кристаллические структуры этих соединений; при условии, что это соединение имеет не более одной связывающей группы, обеспечивающей связь с клеточносвязьшаюпгим агентом за счет ковалентной связи.
В одном предпочтительном варианте настоящего изобретения двойная линия
Y выбран из групп: -OR, NR'R'', сульфит -SO3 или бисульфит -OSO3, где R выбран из групп: Н, линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000, арил, имеющий от 6 до 10 углеродных атомов, гетероциклическое кольцо, имеющее от 3 до 10 углеродных атомов;
W является С=O, СН2 или SO2;
каждый из R1, R2, R3, R4, R1', R2', R3' и R4' независимо выбран из групп: Н, NO2 или связывающая группа, обеспечивающая связь с клеточносвязывающим агентом за счет ковалентной связи;
R6 является OR18, где R18 имеет то же определение, что и R;
Z выбран из групп: (СН2)n, где n является 1, 2 или 3, CR15R16, NR17, О или S, где каждый из R15, R16 и R17 независимо выбран из групп: Н, линейный, разветвленный или циклический алкил, имеющий от 1 до 10 углеродных атомов, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000;
X' выбран из СН2 или С=O;
Y' является О, NR или S, где R - как определено выше;
Z' является СН2 или (СН2)2;
оба А и А' являются О;
D и D' одинаковые или разные и независимо выбраны из групп: линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов;
L необязательно является фенильной группой или гетероциклическим кольцом, имеющим от 3 до 10 углеродных атомов, необязательно замещенных, где заместитель является связывающей группой, обеспечивающей связь с клеточносвязывающим агентом за счет ковалентной связи или выбран из групп: линейный, разветвленный или циклический алкил, алкенил или алкинил, имеющий от 1 до 10 углеродных атомов, необязательно замещенный любой из групп: галоген, OR7, NR8R9, NO2, NRCOR', SR10, сульфоксид, представленный группой SOR', сульфон, представленный группой -SO2R', сульфит -SO3, бисульфит -OSO3, сульфонамид, представленный группой SO2NRR', циано, азидо, -COR11, OCOR11 или OCONR11R12, полиэтиленгликолевое звено (-ОСН2СН2)n, где n является целым числом от 1 до 2000; необязательно, L является связывающей группой, обеспечивающей связь с клеточносвязывающим агентом за счет ковалентной связи; или их фармацевтически приемлемые сольваты, соли, гидраты или гидратированные соли, их оптические изомеры, рацематы, диастереомеры, энантиомеры или полиморфные кристаллические структуры этих соединений.
В другом предпочтительном варианте настоящего изобретения соединение формулы
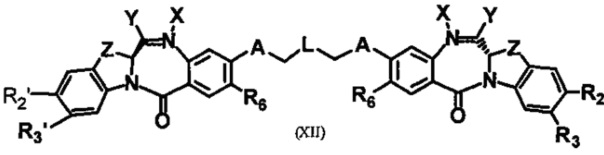
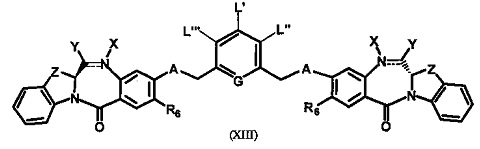
(IV), (V) или (VI) представлено соединениями формул (XII) и (XIII):
где двойная линия
один из R2, R3, R2' и R3' является связывающей группой, обеспечивающей связь с клеточносвязывающим агентом за счет ковалентной связи, а другие является Н, NRCOR' или NO2;
R6 является OR, где R имеет то же определение, что и выше;
Z является СН2 или NR, где R имеет то же определение, что и выше;
А является О или NR15;
L является (СН2)nn, где nn является 0 или целым числом от 1 до 5, или замещенный или незамещенный алкил или алкенил, имеющий от 2 до 4 углеродных атомов, где заместитель выбран из групп: галоген, OR7, NR8R9, NO2, NRCOR', SR10, сульфоксид, представленный группой SOR', сульфон, представленный группой -SO2R', сульфит -SO3, бисульфит -OSO3, сульфонамид, представленный группой SO2NRR', циано, азидо, -COR11, OCOR11 или OCONR11R12, где R7, R8, R9, R10, R11, R12 и R15 имеют то же определение, которое дано выше, необязательно L является связывающей группой, обеспечивающей связь с клеточносвязывающим агентом за счет ковалентной связи;
один из L', L'' или L''' является связывающей группой, обеспечивающей связь с клеточносвязывающим агентом, тогда как остальные являются Н; предпочтительно связывающей группой является L'; и
G является СН или N или их фармацевтически приемлемые сольваты, соли, гидраты или гидратированные соли, их оптические изомеры, рацематы, диастереомеры, энантиомеры или полиморфные кристаллические структуры этих соединений.
Еще в одном предпочтительном варианте настоящего изобретения соединение формулы (IV), (V) или (VI) представлено соединениями формул (XIV) и (XV):
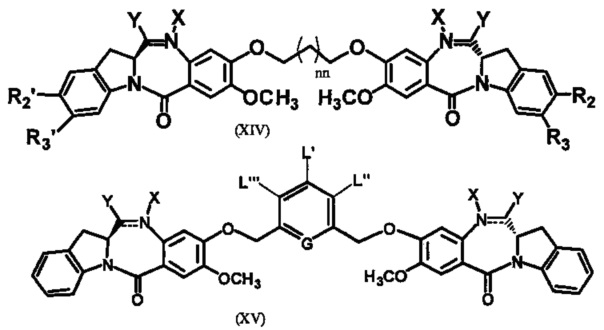
где двойная линия
nn является 0 или целым числом от 1 до 5;
один из R2, R3, R2' и R3' является связывающей группой, обеспечивающей связь с клеточносвязьшающим агентом за счет ковалентной связи, а другие является Н, NRCOR' или NO2;
один из L', L'' или L''' является связывающей группой, обеспечивающей связь с клеточносвязьшающим агентом, при условии, что если один из L', L'' или L''' является связывающей группой, то другие является Н (напр., если L' является связывающей группой, то L'' и L''' являются Н)
G является СН или N или их фармацевтически приемлемые сольваты, соли, гидраты или гидратированные соли, их оптические изомеры, рацематы, диастереомеры, энантиомеры или полиморфные кристаллические структуры этих соединений.
Для связывания цитотоксичных соединений настоящего изобретения (напр., индолинобензодиазепинового или оксазолидинобензодиазепинового), их производных или их димеров с клеточносвязывающим агентом, цитотоксичное соединение имеет связывающую группу. Поскольку связывающая группа, соединяющая две части, является бифункциональной, то сначала один конец связывающей группы может прореагировать с цитотоксичным соединением с получением соединения, имеющего монофункциональную связываюшую группу, которое затем может реагировать с клеточносвязьшающим агентом. В качестве альтернативы, один конец связывающей группы может сначала прореагировать с клеточносвязывающим агентом с получением клеточносвязывающего агента, имеющего монофунциональную связывающую группу, который затем может реагировать с цитотоксичным соединением. Связывающая группа содержит химическую связь, позволяющую высвободить цитотоксичную часть в конкретном месте. Соответствующие химические связи хорошо известны в этой области науки и включают дисульфидные связи, тиоэфирные связи, кислото-неустойчивые связи, фото-неустойчивые связи, пептидаза-неустойчивые связи и эстераза-неустойчивые связи (см., например, патенты США 5208020; 5475092; 6441163; 6716821; 6913748; 7276497; 7276499; 7368565; 7388026 и 7414073). Предпочтительными являются дисульфидные связи, тиоэфирные и пептидаза-неустойчивые связи. Другие связывающие группы, которые могут быть использованы в настоящем изобретении, включают нерасщепляемые связывающие группы, такие как подробно описаны в публикации США номер 20050169933 или заряженные связывающие группы, или гидрофильные связывающие группы, описанные в предварительных заявках на патент 61/049291, поданной 30 апреля 2008 года, 61/147966, поданной 28 января 2009 года и 61/049289, поданной 30 апреля 2008 года, каждая из которых включена здесь в полном объеме путем ссылки.
Соединения формул (I), (II) и (III) (т.е. мономеры) могут связываться через R1, R2, R3, R4 или R5. Из них предпочтительными связывающими группами являются R2, R3 и R5, а наиболее предпочтительной связывающей группой является R5. Примеры соответствующих заместителей у R1, R2, R3, R4 и R5 для соединений формул (I), (II) и (III) включают, не ограничиваясь:
-OH,
-O(CR20R21)m(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-O(CR20R21)m(CR26=CR27)m'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-O(CR20R21)m(алкинил)n'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-O(CR20R21)m(индоло)p'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX''
-O(CR20R21)m(пирроло)q'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-O(CR20R21)m(пирроло)q'(имидазоло)q''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-O(CR20R21)m(имидазоло)q''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-O(CR20R21)m(пирроло)q'(индоло)q''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-O(CR20R21)m(индоло)q'(имидазоло)q''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-O(CR20R21)m(пиперазино)t'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-O(CR20R21)mA''m''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-SH,
-S(CR20R21)m(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-S(CR20R21)m(CR26=CR27)m'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX",
-S(CR20R21)m(алкинил)n'(CR22R23)n(OCH2CH2)p(CH40R41)p''Y''(CR24R25)q(CO)tX'',
-S(CR20R21)m(индоло)p'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-S(CR20R21)m(пирроло)q'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-S(CR20R21)m(имидазоло)q''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-S(CR20R21)m(пирроло)q'(имидазоло)q''(CR22R23)n(OCH2CH2)p(CR40R41)p''X''(CR24R25)q(CO)tX'',
-S(CR20R21)m(пирроло)q'(индоло)q''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-S(CR20R21)m(индоло)q'(имидазоло)q''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-S(CR20R21)m(пиперазино)t'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-S(CR20R21)mA''m''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-NH2,
-NR28(CR20R21)m(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-NR28(CR20R21)m(CR26=CR27)m'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-NR28(CR20R21)m(алкинил)n'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-NR28(CR20R21)m(индоло)p'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-NR28(CR20R21)m(пирроло)q'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-NR28(CR20R21)m(имидазоло)q''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-NR28(CR20R21)m(пирроло)q'(имидазоло)q''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-NR28(CR20R21)m(пирроло)q'(индоло)q''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''-(CR24R25)q(CO)tX'',
-NR28(CR20R21)m(индоло)q'(имидазоло)q''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''-(CR24R25)q(CO)tX'',
-NR28(CR20R21)m(пиперазино)t'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-NR28(CR20R21)mA''m''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-(CR20R21)m(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-(CR20R21)m(CR26=CR27)m'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-(CR20R21)m(алкинил)n'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-(CR20R21)m(индоло)p'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-(CR20R21)m(пирроло)q'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-(CR20R21)m(пиперазино)t'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-(CR20R21)m(пирроло)q'(индоло)q''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-(CR20R21)m(имидазоло)q''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-(CR20R21)m(пирроло)q'(имидазоло)q''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-(CR20R21)m(имидазоло)q''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-(CR20R21)mA''m''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-(CR20R21)m(CR29=N-NR30)n''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-(CR20R21)m(CR29=N-NR30)n''(CR26=CR27)m'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-(CR20R21)m(CR29=N-NR30)n''(алкинил)n'(CR22R23)n(OCH2CH2)p(CH40R41)p''Y''(CR24R25)q(CO)tX'',
-(CR20R21)m(CR29=N-NR30)n''-A''m''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
где:
m, n, p, q, m', n', p', q', q'' являются целым числом от 1 до 10 и могут быть 0;
t, m'', n'' и p'' является 0 или 1;
X'' выбран из групп: OR36, SR37, NR38R39, где R36, R37, R38, R39 является Н, или линейным, разветвленным или циклическим алкилом, алкенилом или алкинилом, имеющим от 1 до 20 углеродных атомов, или полиэтиленгликолевым звеном - (ОСН2СН2)n, необязательно R37 является защитной группой для тиоловой группы, или
если t=1, то СОХ'' образует химически активный сложный эфир, выбранный из следующих эфиров: N-гидроксисукцинимидные эфиры, N-гидроксифталимидные эфиры, N-гидрокси сульфо-сукцинимидные эфиры, эфиры пара-нитрофенила, эфиры динитрофенила, эфиры пентафторфенила и их производные, где указанные производные способствуют образованию амида;
Y'' отсутствует или выбран из О, S, S-S или NR32, где R32 имеет то же определение, которое дано выше для R, или
если Y'' не является S-S и t=0, то X'' выбран из групп: малеимидо-группа, галоацетил-группа или SR37, где R37 имеет то же определение, что и выше;
А'' является аминокислотой, выбранной из кислот: глицин, аланин, лейцин, валин, лизин, цитруллин и глутамат или полипептидом, содержащим от 2 до 20 аминокислотных звеньев;
R20, R21, R22, R23, R24, R25, R26, R27 одинаковые или различные и являются Н или линейным или разветвленным алкилом, имеющим от 1 до 5 углеродных атомов;
R28 является Н или алкилом;
R29 и R30 одинаковые или различные и являются Н или алкилом из 1-5 углеродных атомов;
необязательно, один из R40 и R41 является отрицательно или положительно заряженной функциональной группой, а другой является Н или алкилом, алкенилом, алкинилом, имеющим от 1 до 4 углеродных атомов.
Соединения формул (IV), (V), (VI), (VII), (XII) и (XIII) (т.е. димеры) могут быть связаны через R1, R2, R3, R4, R1', R2', R3', R4', L', L'', L'''. Из них предпочтительными связывающими группами являются R2', R3', R4', L', L'', L''', а наиболее предпочтительными группами являются R2', R3' и L'. Примеры связывающих групп соединений формул (IV), (V), (VI), (VII), (XII) и (XIII) включают, не ограничиваясь:
-O(CR20R21)m(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-O(CR20R21)m(CR26=CR27)m'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-O(CR20R21)m(алкинил)n'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-O(CR20R21)m(пиперазино)t'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-O(CR20R21)m(пирроло)t'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-O(CR20R21)mA''m''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-S(CR20R21)m(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-S(CR20R21)m(CR26=CR27)m'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-S(CR20R21)m(алкинил)n'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-S(CR20R21)m(пиперазино)t'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-S(CR20R21)m(пиролло)t'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-S(CR20R21)mA''m''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-NR33(C=O)p''(CR20R21)m(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-NR33(C=O)р''(CR20R21)m(CR26=CR27)m'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-NR33(C=O)p''(CR20R21)m(алкинил)n'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-NR33(C=O)p''(CR20R21)m(пиперазино)t'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO24R25)q(CO)tX'',
-NR33(C=O)p''(CR20R21)m(пирроло)t'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-NR33(C=O)p''(CR20R21)mA''m''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-(CR20R21)m(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-(CR20R21)m(CR26=CR27)m'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-(CR20R21)m(алкинил)n'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-(CR20R21)m(пиперазино)t'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-(CR20R21)mA''m''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-(CR20R21)m(CR29=N-NR30)n''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-(CR20R21)m(CR29=N-NR30)n''(CR26=CR27)m'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-(CR20R21)m(CR29=N-NR30)n''(алкинил)n'(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
-(CR20R21)m(CR29=N-NR30)n''A''m''(CR22R23)n(OCH2CH2)p(CR40R41)p''Y''(CR24R25)q(CO)tX'',
где:
m, n, p, q, m', n', t' являются целым числом от 1 до 10 или необязательно 0;
t, m'', n'' и р'' является 0 или 1;
X'' выбран из групп: OR36, SR37, NR38R39, где R36, R37, R38, R39 является Н или линейным, разветвленным или циклическим алкилом, алкенилом или алкинилом, имеющим от 1 до 20 углеродных атомов, или полиэтиленгликолевым звеном -(ОСН2СН2)n, R37 необязательно является защитной группой для тиоловой группы
если t=1, то СОХ'' образует химически активный сложный эфир, выбранный из следующих эфиров: N-гидроксисукцинимидные эфиры, N-гидроксифталимидные эфиры, N-гидрокси сульфо-сукцинимидные эфиры, эфиры пара-нитрофенила, эфиры динитрофенила, эфиры пентафторфенила и их производные, где указанные производные способствуют образованию амидной связи;
Y'' отсутствует или выбран из О, S, S-S или NR32, где R32 имеет то же определение, которое дано выше для R, или
если Y'' не является S-S и t=0, то X'' выбран из групп: малеимидо-группа, галоацетил-группа или SR37, где R37 имеет то же определение, что и выше;
А'' является аминокислотой, выбранной из кислот: глицин, аланин, лейцин, валин, лизин, цитруллин и глутамат или полипептидом, содержащим от 2 до 20 аминокислотных звеньев;
R20, R21, R22, R23, R24, R25, R26 и R27 одинаковые или различные и являются Н или линейным или разветвленным алкилом, имеющим от 1 до 5 углеродных атомов;
R29 и R30 одинаковые или различные и являются Н или алкилом из 1-5 углеродных атомов;
R33 является Н или линейным, разветвленным или циклическим алкилом, алкенилом или алкинилом, имеющим от 1 до 12 углеродных атомов, полиэтиленгликолевым звеном -(ОСН2СН2)n или R33 является -COR34, -CSR34, -SOR34 или -SO2R34, где R34 является Н или линейным, разветвленным или циклическим алкилом, алкенилом или алкинилом, имеющим от 1 до 20 углеродных атомов или полиэтиленгликолевым звеном -(ОСН2СН2)n; и
один из R40 и R41 необязательно является отрицательно или положительно заряженной функциональной группой, а другой является Н или алкилом, алкенилом, алкинилом, имеющим от 1 до 4 углеродных атомов.
Далее, несмотря на то, что синтез цитотоксичных соединений (напр., индолинобензодиазепиновых или оксазолидинобензодиазепиновых), их производных или их димеров, имеющих связывающую группу, описан ниже в отношении связывающих групп, содержащих амидную, тиоэфирную или дисульфидную связь в положении L' (в соединении формулы XIII) или R3 (в соединении формулы XII), специалисту в данной области понятно, что связывающие группы в других положениях и с другими химическими связями, описанными выше, также могут использоваться в настоящем изобретении.
Структуры представителей соединений, представителей конъюгатов и заявленных в примерах настоящего изобретения соединений представлены в таблицах 3~9:
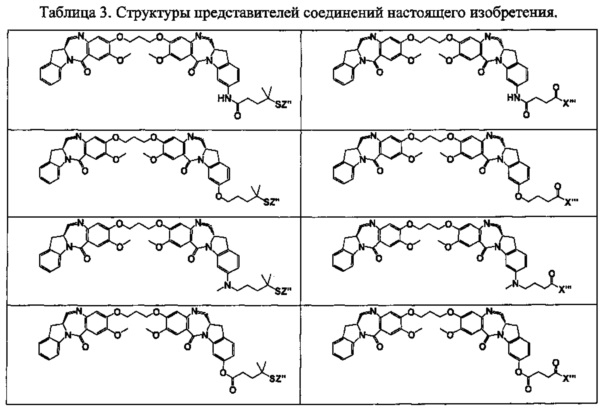
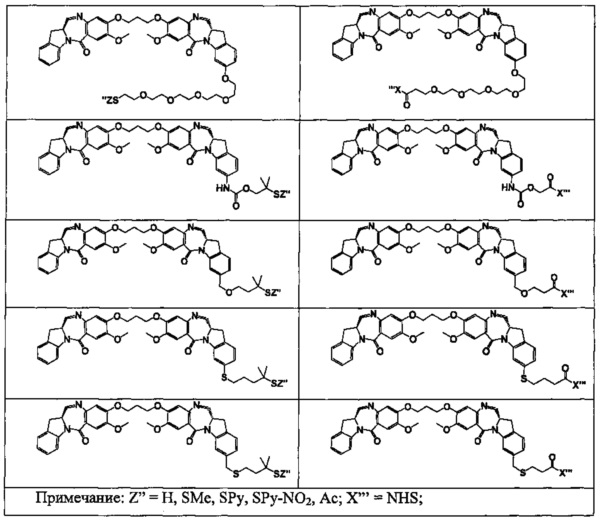


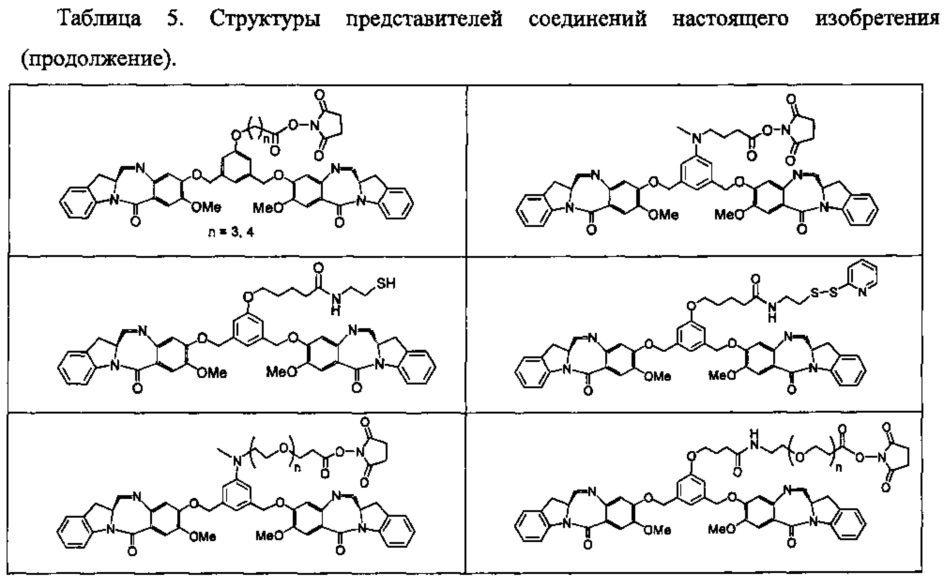

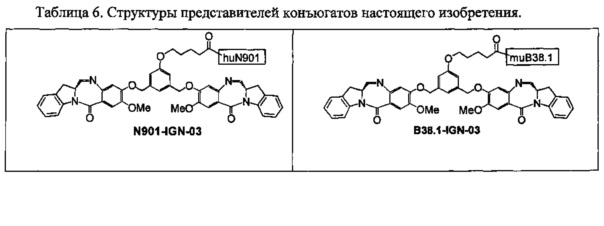

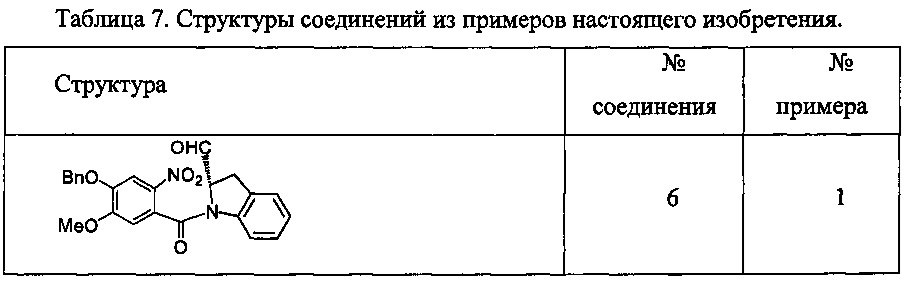
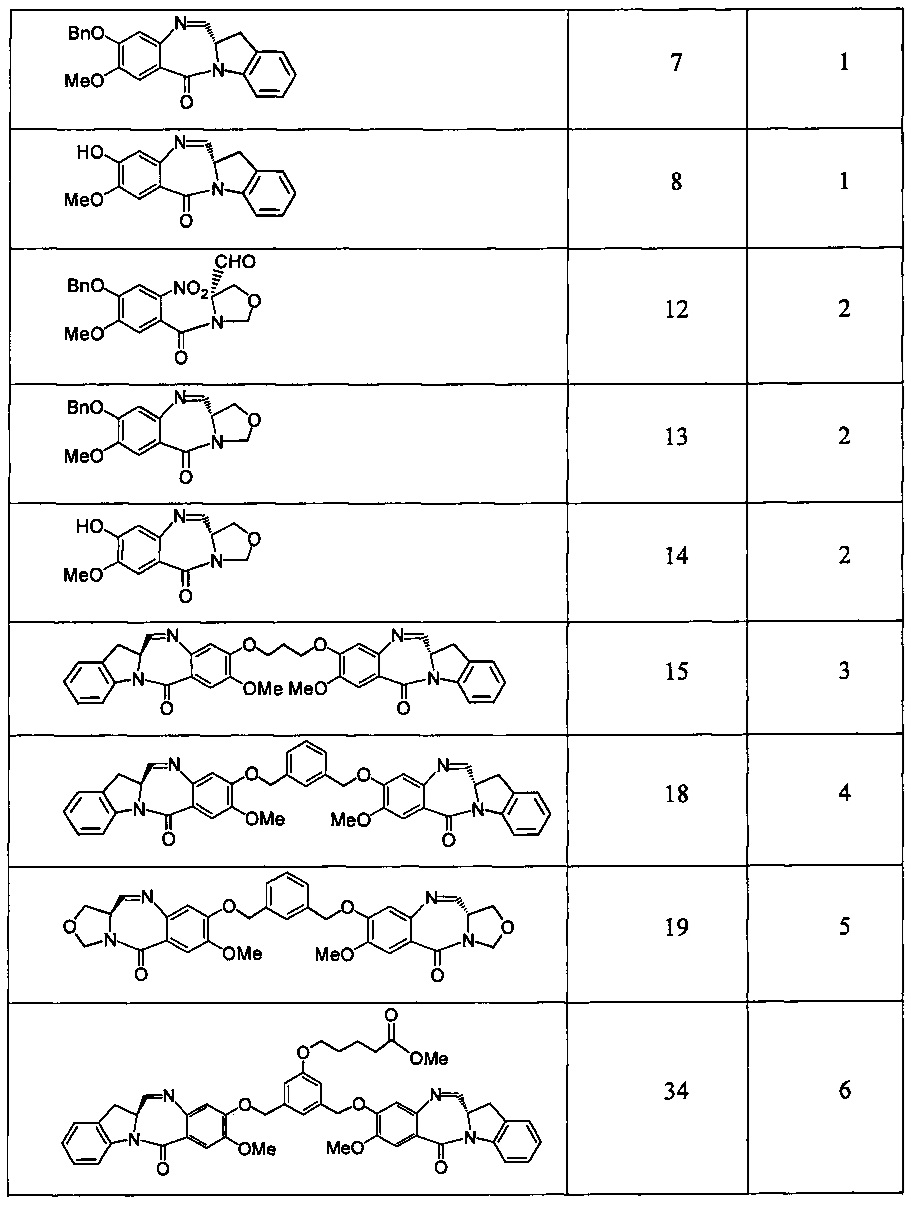
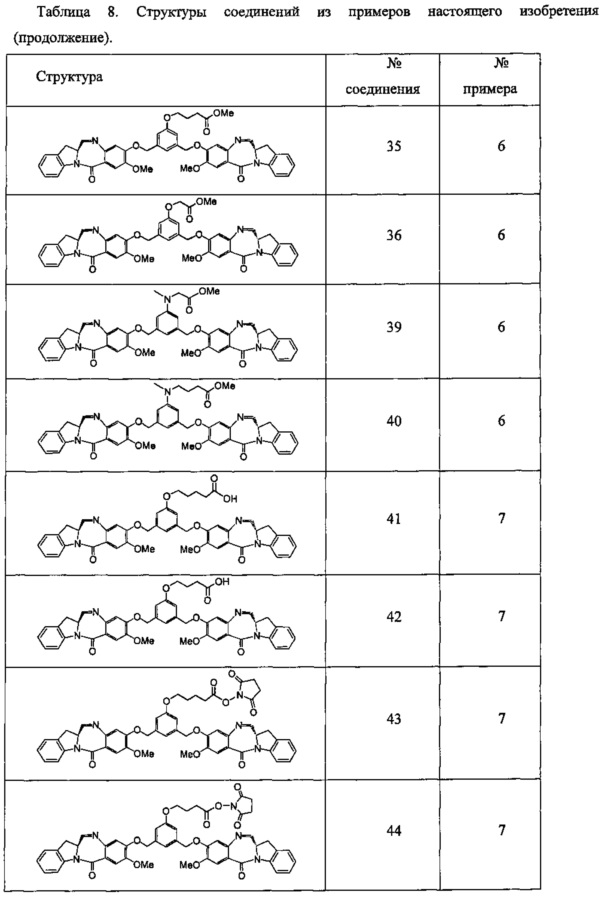
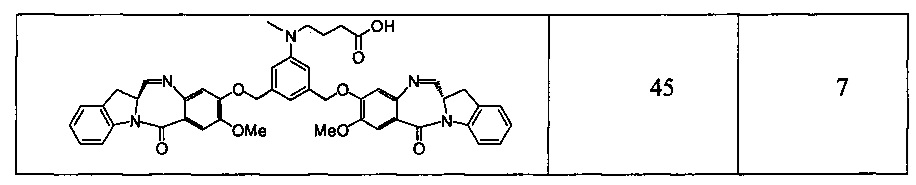
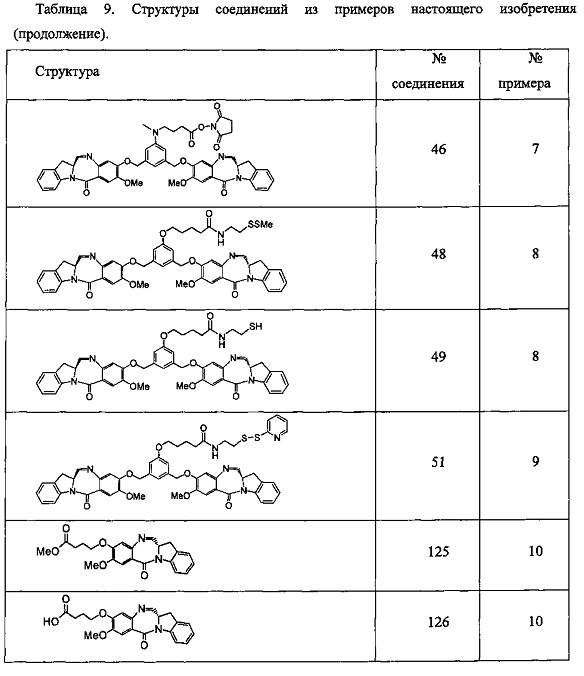
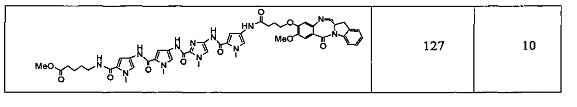
Синтез цитотоксичных соединений
Методика получения представителя мономерного соединения настоящего изобретения на примере индолинобензодиазепинового соединения 8 представлена на фиг. 1. Из имеющейся в продаже индолин-2-карбоновой кислоты 1, был количественно получен ее метиловый эфир 2 по реакции с тионилхлоридом в метаноле. Метил индолин-2-карбоксилат 2 связали с хлорангидридом 4 или напрямую с кислотой 3 для получения амида 5, который затем восстановили гидридом диизобутилалюминия (DIBAL) до альдегида 6. Хотя можно использовать много способов восстановления функциональной нитро-группы формулы 5 до соответствующей аминогруппы, в этом примере использовали дитионит натрия для простого превращения альдегида 6 в соединение 7 с закрытым циклом, после дополнительной обработки метанолом в кислой среде. Защитную группу бензила удалили для получения мономера 8.
Методика получения мономерного оксазолидинобензодиазепинового соединения формулы 14 настоящего изобретения представлена на фиг. 2. Из имеющегося в продаже соединения 9, был количественно получен его сложный метиловый эфир 10 обработкой тионилхлоридом в метаноле. С соединения 10 сняли защиту, а затем связали с ацетилхлоридом 4 или непосредственно с кислотой 3 для получения амида 11, который затем превратили в альдегид 12. Восстановление нитро-группы выполнили обработкой дитионитом натрия с последующим эффективным превращением в соединение с закрытым кольцом 13 после дополнительной обработки метанолом в кислой среде. Защитную группу бензила удалили для получения мономера 14.
Методика получения представителей димерных соединений настоящего изобретения представлена на фиг. 3-5 и 7. Димеры были получены по реакции мономеров формулы 8 или формулы 14 с соединениями, имеющими две уходящие группы, такие как Br, I, трифлат, мезилат или тозилат.
Димеры, имеющие связывающие группы, которые могут реагировать с антителами, получили превращением сложных метиловых эфиров в соответствующие химически активные сложные эфиры уходящих групп, таких как, не ограничиваясь, сложные эфиры N-гидроксисукцинимида, сложные эфиры N-гидроксифталимида, сложные эфиры N-гидрокси сульфо-сукцинимида, сложные эфиры пара-нитрофенила, сложные эфиры динитрофенила, сложные эфиры пентафторфенила. Типичные примеры синтеза связываемых димеров показаны на фиг. 8. Синтез димеров, имеющих тиоловую или дисульфидную группу для обеспечения связи с клеточносвязывающими агентами за счет восстанавливаемых или невосстанавливаемых связей, показан на фиг. 9 и 10. Мономер 58 с модифицированным кольцом В, лишенный карбонильной группы, получен из соединения бензилового спирта 52 по этапам, показанным на фиг. 11. Изоиндолиновый мономер 66 может быть получен из изоиндола 59, как показано на фиг. 12. Связывающая группа может также присоединяться непосредственно к индолиновой группе. Метил индолино-2-карбоксилат можно превратить в связываемый димер 82 по этапам синтеза, показанным на фиг. 13. Синтез связываемых димеров, имеющих ПЭГ звено, показан на фиг. 14 и 15.
Так, в одном аспекте настоящего изобретения предложена методика получения индолинобензодиазепинового мономера (ИБД) (I) (фиг. 1), методика включает следующие этапы:
a) связывание соединения формулы (1) и соединения формулы (2) с получением соединения формулы (3);
b) превращение соединения формулы (3) в альдегид формулы (4); и
c) превращение соединения формулы (4) в соединение формулы (I),
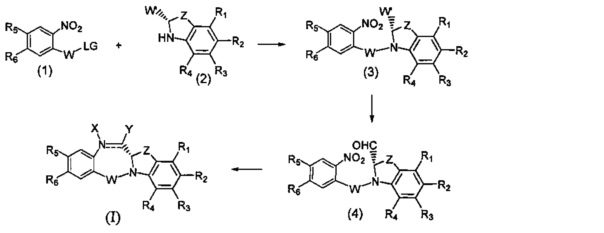
где LG является уходящей группой, W' является COOR или CH2OW'', где R имеет такое же определение, как и выше, и W'' является защитной группой; R1, R2, R3, R4, R5, R6, W, Z, X, Y и
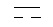
В другом аспекте настоящего изобретения предложена методика получения соединения формулы (II), включающая следующие этапы:
а) связывание соединения формулы (1) и соединения формулы (5) с получением соединения формулы (6);
b) превращение соединения формулы (6) в альдегид формулы (7); и
c) превращение соединения формулы (7) в соединение формулы (II),

где LG является уходящей группой, W' является COOR или CH2OW'', где R имеет такое же определение, как и выше, и W'' является защитной группой; R1, R2, R3, R4, R5, R6, W, X, Y и
В другом аспекте настоящего изобретения предложена методика получения соединения формулы (III), включающая следующие этапы:
a) связывание соединения формулы (1) и соединения формулы (8) с получением соединения формулы (9);
b) превращение соединения формулы (9) в альдегид формулы (10); и
c) превращение соединения формулы (10) в соединение формулы (II),
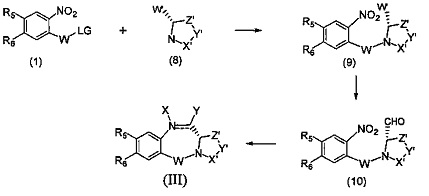
где LG является уходящей группой, W' является COOR или CH2OW'', где R имеет такое же определение, как и выше, и W'' является защитной группой; R5, R6, W, X, Y, X', Y', Z' и
В другом аспекте настоящего изобретения предложена методика получения соединения формулы (IV), включающая следующие этапы:
связывание соединения формулы (11), соединения формулы (11)' и соединения формулы (12) с образованием соединения формулы (IV),
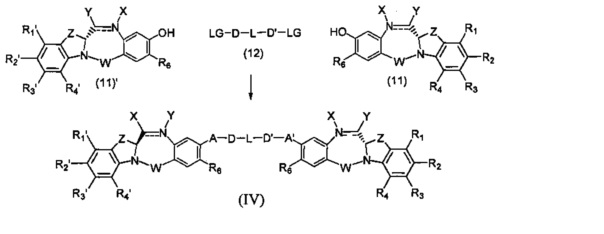
где LG является уходящей группой; R1, R2, R3, R4, R1', R2', R3', R4', R6, W, X, Y, Z, A, A', D, D', L и
В другом аспекте настоящего изобретения предложена альтернативная методика получения соединения формулы (IV) настоящего изобретения, включающая следующие этапы:
a) превращение соединения формулы (15) в альдегид формулы (16); и
b) превращение соединения формулы (16) в соединение формулы (IV),
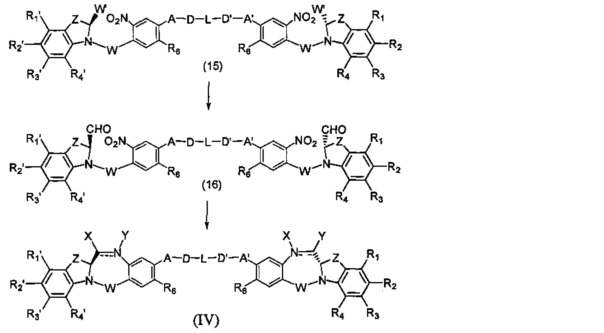
где W' является COOR или CH2OW'', где R имеет то же определение, что и выше и W'' является защитной группой; R1, R2, R3, R4, R1', R2', R3', R4', R6, W, X, Y, Z, A, A', D, D', L и
В другом аспекте настоящего изобретения представлена методика получения соединения формулы (V), включающая этап связывания соединения формулы (13), соединения формулы (13)' и соединения формулы (12) с получением соединения формулы (V).
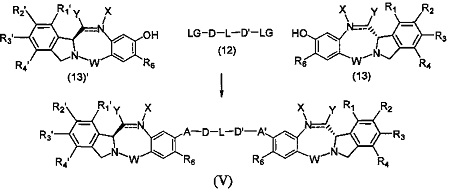
где LG является уходящей группой; R1, R2, R3, R4, R1', R2', R3', R4', R6, W, X, Y, A, A', D, D', L и
В другом аспекте настоящего изобретения предложена альтернативная методика получения соединения формулы (V) настоящего изобретения, включающая следующие этапы:
a) превращение соединения формулы (17) в альдегид формулы (18); и
b) превращение соединения формулы (18) в соединение формулы (V),
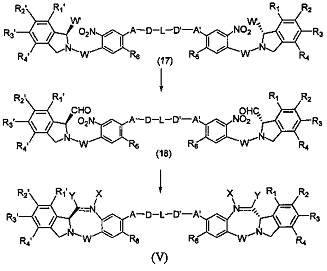
где W' является COOR или CH2OW'', где R имеет то же определение, что и выше и W'' является защитной группой; R1, R2, R3, R4, R1', R2', R3', R4', R6, W, X, Y, A, A', D, D', L и
В другом аспекте настоящего изобретения представлена методика получения соединения формулы (VI), включающая этап связывания соединения формулы (14), соединения формулы (14)' и соединения формулы (12) с получением соединения формулы (VI),
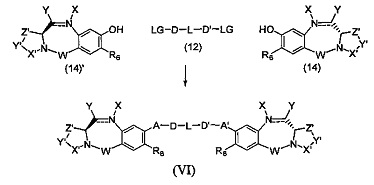
где LG является уходящей группой; R6, W, X, Y, X', Y', Z' A, A', D, D', L и
В другом аспекте настоящего изобретения предложена методика получения соединения формулы (VI) настоящего изобретения, включающая следующие этапы:
a) превращение соединения формулы (19) в альдегид формулы (20); и
b) превращение соединения формулы (20) в соединение формулы (VI),

где W' является COOR или CH2OW'', где R имеет то же определение, что и выше и W'' является защитной группой; R6, W, X, Y, X', Y', Z' A, A', D, D', L и
Цитотоксичность соединений in vitro
Цитотоксичность in vitro цитотоксичных соединений настоящего изобретения (напр., индолинобензодиазепиновых или оксазолидинобензодазепиновых), их производных, их димеров или их конъюгатов может быть оценена по их способности подавлять разрастание различных раковых клеточных линий in vitro (таблицы 1, 2 на фиг. 31, 32). Например, такие клеточные линии как линия рака молочной железы человека SK-Br-3 или клеточная линия эпидермоидного рака человека KB, могут использоваться для оценки цитотоксичности этих новых соединений. Клетки, подлежащие оценке, подвергаются обработке соединениями настоящего изобретения в течение 72 часов, а выжившие фракции клеток измеряются прямыми анализами известными способами. Затем по результатам анализов рассчитывается значение IC50.
Примеры цитотоксичности in vitro соединений настоящего изобретения, испытанные на панели раковых клеточных линий, и их данные представлены в таблице 1. Все испытанные димерные соединения индолинобензодиазепина обладают сильным действием со значениями IC50 в нижнем пикомолярном диапазоне. IGN-09 по большей части сохранил свою эффективность на клеточных линиях, устойчивых к действию различных лекарств, таких как COLO205-MDR (IC50 лишь в 4 раза выше, чем COLO205). Соединения настоящего изобретения в 1000-10000 раз более цитотоксичны, чем другие лекарства, взаимодействующие с ДНК, используемые при лечении рака, такие как доксорубицин, мелфалан и цис-платин. При прямом сравнении, активность соединений IGN1, не имеющих связывающей группы (соединение 18) и IGN09 (соединение 15), была сравнена с активностью соединений, имеющих связывающую группу, IGN03 (соединение 34) и IGN05 (соединение 36), испытание провели по отношению к репрезентативной клеточной линии Ramos. Как показано в таблице 2, все четыре соединения высоко эффективны и имеют значения IC50 менее 1 пикомоль, показывая, что внедрение связывающих групп не ухудшает эффективности.
Клеточносвязывающие агенты
Эффективность соединений настоящего изобретения (напр., индолинобензодазепиновых или оксазолидинобензодиазепиновых), их производных, их димеров или их конъюгатов в качестве терапевтических средств зависит от тщательного выбора соответствующего клеточносвязывающего агента. Клеточносвязывающие агенты могут быть любого известного в настоящее время типа, или становящегося известным типа, включая пептиды и непептидные соединения. Как правило, это могут быть антитела (особенно моноклональные антитела), лимфокины, гормоны, факторы роста, витамины, молекулы, транспортирующие питательные вещества (такие как трансферрин) или любые другие клеточносвязывающие молекулы или вещества.
Более конкретные примеры клеточносвязывающих агентов, которые могут быть использованы, включают:
поликлональные антитела;
моноклональные антитела;
фрагменты антител, такие как Fab, Fab' и F(ab')2, Fv (Parham, J. Immunol. 131:2895-2902 (1983); Spring et al. J. Immunol. 113:470-478 (1974); Nisonoff et al. Arch Biochem. Biophys. 89:230-244 (1960));
интерфероны (напр., альфа, бета, гамма);
лимфокины, такие как IL-2, IL-3, IL-4, IL-6;
гормоны, такие как инсулин, TRH (рилизинг-гормон тиротропина), MSH (меланоцит-стимулирующий гормон), стероидные гормоны, такие как андрогены и эстрогены;
факторы роста и колониестимулирующие факторы, такие как EGF, TGF-альфа, FGF, VEGF, G-CSF, M-CSF и GM-CSF (Burgess, Immunology Today 5:155-158 (1984));
трансферрин (O'Keefe et al. J. Biol. Chem. 260:932-937 (1985)); и
витамины, такие как фолат.
Методики моноклональных антител позволяют создавать чрезвычайно специфичные клеточносвязывающие агенты в форме специфичных моноклональных антител. Особенно хорошо известны в данной области науки методики создания моноклональных антител, получаемых за счет иммунизации мышей, крыс, хомяков или любых других млекопитающих целевым антигеном, таким как здоровые клетки-мишени, антигены, выделенные из клеток-мишеней, цельный вирус, аттенуированный цельный вирус и вирусные белки, такие как белки вирусной оболочки. Можно использовать также сенсибилизированные клетки человека. Другой способ создания моноклональных антител заключается в использовании фаговой библиотеки scFv (одноцепочечная V-область), в частности, scFv человека (см., напр., Griffiths et al., патенты США NoNo 5885793 и 5969108; McCafferty et al., WO 92/01047; Liming et al., WO 99/06587). Кроме того, могут использоваться так же антитела с измененной поверхностью, описанные в патенте США No 5639641, химерные антитела и гуманизированные антитела. Выбор соответствующего клеточносвязыващего агента является предметом выбора, зависящим от конкретной клеточной популяции, являющейся мишенью, но в целом предпочтительны моноклональные антитела человека, если соответствующие антитела доступны.
Например, моноклональное антитело MY9 является мышечным антителом IgG1, специфически связьшающимся с антигеном CD33 {J.D. Griffin et al 8 Leukemia Res., 521 (1984)} и может использоваться, если экспрессия клеток-мишеней {J.D. Griffin et al 8 Leukemia Res., 521 (1984)} такая же, как в случае заболевания острым миелогенным лейкозом (AML). Аналогично, моноклональное антитело анти-В4 является мышечным антителом IgG1, связывающимся с антигеном CD19 на В клетках {Nadler et al, 131 J. Immunol. 244-250 (1983)} и может использоваться, если клетками-мишенями являются В клетки или поврежденные клетки, экспрессирующие этот антиген, такие как лимфома Неходжкина или хронический лимфобластный лейкоз HuB4 является антителом с измененной поверхностью, полученным из мышечного антитела анти-В4 (Roguska et al., 1994, Proc. Natl. Acad. Sci., 91, c. 969-973). HuN901 является гуманизированным антителом, связывающимся с антигеном CD56, экспрессированном на небольших клетках легочного рака, множественной миеломы, рака яичников и других твердых опухолей, включая нейроэндокринные раковые опухоли (Roguska et al., 1994, Proc. Natl. Acad. Sci., 91, c. 969-973). B38.1 является химерным антителом против ЕрСАМ. Цельные антитела человека, такие как панитумумаб, направленный против рецептора EGF, экспрессированного на нескольких твердых опухолях, также могут использоваться (Van Cutsem et al., J Clin Oncol. 2007; 25(13):1658-1664). Клеточносвязьшающий агент, включающий конъюгаты и модифицированные клеточносвязывающие агенты настоящего изобретения, могут быть любого известного в настоящее время типа или становящегося известным типа, и включает пептиды и непептидные соединения. Клеточносвязывающий агент может быть любым соединением, которое может связываться с клеткой, как специфическим, так и неспецифическим образом. Как правило, это могут быть антитела (особенно моноклональные антитела и фрагменты антител), интерфероны, лимфокины, гормоны, факторы роста, витамины, молекулы, транспортирующие питательные вещества (такие как трансферрин) или любые другие клеточносвязывающие молекулы или вещества.
Если клеточносвязывающий агент является антителом, то он связывается с антигеном, который является полипептидом, и может быть трансмембранной молекулой (напр., рецептор) или лигандом, таким как фактор роста. Примеры антигенов включают такие молекулы как ренин; гормон роста, включая гормон роста человека и бычий гормон роста; рилизинг-фактор гормона роста; гормон паратироид; тироид-стимулирующий гормон; липопротеины; альфа-1-антитрипсин; А-цепь инсулина; В-цепь инсулина; проинсулин, фолликул-стимулирующий гормон; кальцитонин; лютеинизирующий гормон; глюкагон, коагулирующие факторы, такие как фактор vmc, фактор IX, тканевый фактор (TF) и фактор фон Виллебранда; анти-коагулирующие факторы, такие как протеин С; атриальный натрийуретический фактор; легочный сурфактант; активатор плазминогена, такой как урокиназа или активатор плазминогена из человеческой мочи или тканевого типа (t-PA); бомбезин; тромбин; гемопоэтический фактор роста; альфа- и бета-фактор некроза опухоли; энкефалиназа; RANTES (регулируемый при активации нормально экспрессированных и секретированных Т-клеток); макрофагальный белок воспаления человека (MIP-1-альфа); сывороточный альбумин, такой как сывороточный альбумин человека; ингибирующее вещество Мюллера; А-цепь релаксина; В-цепь релаксина; прорелаксин; гонадотропин-ассоциированный белок мышей; белок микробов, такой как бета-лактамаза; DNase; IgE; цитотоксичный Т-лимфоцит-ассоциированный антиген (CTLA), такой как CTLA-4; ингибин, активин; васкулярный эндотелиальный фактор роста (VEGF); рецепторы для гормонов или факторов роста; белок А или D; ревматоиные факторы; нейротрофный фактор, такой как костный нейтротрофный фактор (BDNF), нейротрофин-3, -4, -5 или -6 (NT-3, NT4, NT-5 или NT-6) или фактор роста нервов, такой как NGF-β; тромбоцитарный фактор роста (PDGF); фактор роста фибробластов, такой как aFGF и bFGF, эпидермальньгй фактор роста (EGF); трансформирующий фактор роста (TGF), такой как TGF-альфа и TGF-бета, включая TGF-β1, TGF-β2, TGF-β3, TGF-β4 или TGF-β5; инсулин-подобный фактор роста-I и -II (IGF-I и IGF-II); дез(1-3)-IGF-I (мозговой IGF-I), инсулин-подобный фактор роста, связывающий белки, ЕрСАМ, GD3, FLT3, PSMA, PSCA, MUC1, MUC16, STEAP, СЕА, TENB2, рецепторы EphA, рецепторы EphB, рецепторы фолатов, FOLR1, мезотелин, крипто, alphavbeta6, интегрины, VEGF, VEGFR, рецептор трансферрина, IRTA1, IRTA2, IRTA3, IRTA4, IRTA5; CD белки, такие как CD2, CD3, CD4, CD5, CD6, CD8, CD11, CD14, CD19, CD20, CD21, CD22, CD25, CD26, CD28, CD30, CD33, CD36, CD37, CD38, CD40, CD44, CD52, CD55, CD56, CD59, CD70, CD79, CD80. CD81, CD103, CD105, CD134, CD137, CD138, CD152 или антитело, связывающееся с одним или более опухолевых антигенов или рецепторы поверхности клетки, описанными в публикации США No 20080171040 или Публикации США No 20080305044 и включены в полном объеме путем ссылки; эритропоиэтин; остеоиндуктивные факторы; иммунотоксины; костный морфогенетический белок (BMP); интерферон, такой как интерферон-альфа, -бета и -гамма; колониестимулирующие факторы (CSF), напр., M-CSF, GM-CSF и G-CSF; интерлейкины (IL), напр., от IL-1 до IL-10; супероксидная дисмутаза; Т-клеточные рецепторы; белки поверхностной мембраны; фактор ускорения распада; вирусный антиген, такой как, например, часть развития HIV; транспортные белки, «хоминг»-рецепторы; аддрессины; регулирующие белки; интегрины, такие как CD11a, CD11b, CD11c, CD18, ICAM, VLA-4 и VCAM; опухолеассоциированный антиген, такой как рецептор HER2, HER3 или HER4; и фрагменты любого из вышеперечисленных полипептидов.
Кроме того, GM-CSF, связьшающийся с миелоидными клетками, может использоваться в качестве клеточносвязывающего агента для клеток, пораженных острым миелогенным лейкозом. IL-2, который связывается с активированными Т-клетками, может использоваться для предотвращения отторжения трансплантата, для лечения или предупреждения реакции "трансплантат против хозяина" и для лечения острого лейкоза Т-клеток. MSH, связывающийся с меланоцитами, может использоваться для лечения меланомы. Фолиевая кислота может использоваться для таргетирования фолат-рецептора, экспрессированного на опухоли яичника и других опухолях. Эпидермальный фактор роста может использоваться для таргетирования плосколеточного рак, такие как рак легкого, головы и шеи. Соматостатин может использоваться для таргетирования нейробластом и других типов опухолей.
На рак груди и яичек можно успешно целенаправленно воздействовать эстрогенами (или аналогами эстрогенов) или андрогенами (или аналогами андрогенов), соответственно, в качестве клеточносвязывающих агентов.
Получение цитотоксичных конъюгатов
Настоящее изобретение также представляет цитотоксичные конъюгаты соединение-клетка-связывающий агент, содержащие клеточносвязьшающий агент, связанный с одним или более цитотоксичных соединений за счет различных связывающих групп, включая, не ограничиваясь, дисульфидные связывающие группы, тиоэфирные связывающие группы, амид-связанные связывающие группы, пептидаза-неустойчивые связывающие группы, кислото-неустойчивые связывающие группы, эстераза-неустойчивые связывающие группы. Представителями цитотоксичных конъюгатов настоящего изобретения являются: антитело/цитотоксичное соединение, фрагмент антитела/цитотоксичное соединение, эпидермальный фактор роста (EGF)/цитотоксичное соединение, меланоцит-стимулирующий гормон (MSH)/цитотоксичное соединение, тироид-стимулирующий гормон (TSH)/цитотоксичное соединение, соматостатин/цитотоксичное соединение, фолат/цитотоксичное соединение, эстроген/цитотоксичное соединение, аналог эстрогена/цитотоксичное соединение, андроген/цитотоксичное соединение и аналог андрогена/цитотоксичное соединение.
В предпочтительном варианте настоящего изобретения предложен конъюгат индолинобензодиазепиновый димер-клетка-связывающий агент, содержащий цитотоксичный агент и клеточносвязьшающий агент, связанный за счет ковалентной связи. Связывающий агент может расщепляться на месте опухоли/нежелательно разрастающихся клеток, обеспечивая доставку цитотоксичного агента к его мишени несколькими способами. Связывающий агент может расщепляться, например, при низком pH (гидразон), в восстановительной среде (дисульфид), за счет протеолиза (амидная/пептидная связь) или за счет ферментативной реакции (эстераза/гликозидаза).
В предпочтительном варианте представителями цитотоксичных конъюгатов настоящего изобретения являются: антитело/индолинобензодиазепиновый димер, фрагмент антитела/индолинобензодиазепиновый димер, эпидермальный фактор роста (EGF)/индолинобензодиазепиновый димер, меланоцит-стимулирующий гормон (MSH)/индолинобензодиазепиновый димер, тироид-стимулирующий гормон (TSH)/индолинобензодиазепиновый димер, соматостатин/индолинобензодиазепиновый димер, фолат/индолинобензодиазепиновый димер, эстроген/индолинобензодиазепиновый димер, аналог эстрогена/индолинобензодиазепиновый димер, ингибитор простата-специфичного мембранного антигена (PSMA)/индолинобензодиазепиновый димер, ингибитор матриптазы/индолинобензодиазепиновый димер, белки с заданным анкириновым повтором (DARP)/индолинобензодиазепиновый димер, андроген/индолинобензодиазепиновый димер и аналог андрогена/индолинобензодиазепиновый димер.
Дисульфид-содержащие цитотоксичные конъюгаты могут быть получены по реакции тиол-содержащего цитотоксичного агента, такого как 49, с соответствующим образом модифицированным клеточносвязывающим агентом. Эти конъюгаты могут быть очищены для удаления несвязанного цитотоксичного агента при помощи гель-фильтрации, ионообменной хроматографии, хроматографии на керамическом гидроксиапатите (СНТ), хроматографии гидрофобных взаимодействий (СНТ), фильтрования тангенциальным потоком (TFF) или ВЭЖХ.
Раствор антитела в водном буферном растворе инкубируется с молярным избытком агента, модифицирующего антитело, такого как N-сукцинимидил-3-(2-пиридилдитио)пропионат (SPDP) или с N-сукцинимидил-4-(2-пиридилдитио)бутаноат (SPDB) для получения дитиопиридиловых групп. Затем модифицированное антитело реагирует с тиол-содержащим цитотоксичным агентом, таким как соединение 49 для получения дисульфид-связанного конъюгата антитело-индолинобензодиазепиновый димер. Цитотоксичный клеточносвязывающий конъюгат может быть очищен при помощи любого из выше упомянутых способов.
В качестве альтернативы, антитело инкубируется с молярным избытком агента, модифицирующего антитело, таким как 2-иминотиолан, L-гомоцистеина тиолактон (или производные) или N-сукцинимидил-S-ацетилтиоацетат (SATA) для получения сульфгидрильных групп. Затем модифицированное антитело реагирует с соответствующим дисульфид-содержащим цитотоксичным агентом, таким как соединение 51 для получения дисульфид-связанного конъюгата антитело-цитотоксичный агент. Конъюгат антитело-цитотоксичный агент затем может быть очищен гель-фильтрацией или другими выше упомянутыми способами.
Количество связей цитотоксичных молекул на одну молекулу антитела можно определить спектрофотометрически путем измерения степени поглощения при 280 нм и 330 нм. По этому способу может связываться в среднем 1-10 цитотоксичных молекул на одну молекулу антитела. Предпочтительное среднее количество связанных цитотоксичных молекул на одну молекулу антитела составляет 2-5, и наиболее предпочтительно 3-4,5.
В качестве альтернативы раствор антитела в водном буферном растворе инкубируется с молярным избытком агента, модифицирующего антитело, таким как N-сукцинимидил-4-(N-малеимидометил)-циклогексан-1-карбоксилат для получения малеимидо-групп или с N-сукцинимидил-4-(йодацетил)-аминобензоатом (SIAB) для получения йодоацетильных групп. Затем модифицированное антитело реагирует с тиол-содержащим цитотоксичным агентом для получения тиоэфир-связанного конъюгата антитело-цитотоксичный агент. Конъюгат антитело-цитотоксичный агент может быть затем очищен гель-фильтрацией или другими способами, упомянутыми выше, или способами, известными специалисту в данной области.
Цитотоксичные агенты, содержащие связывающие группы, заканчивающиеся N-гидрокси сукцинимидил (NHS) эфиром, такие как соединения 43, 44 и 46, могут реагировать с антителом с прямым получением амид-связанных конъюгатов, таких как huN901-IGN-03 и huN901-IGN-07. Конъюгат антитело-цитотоксичный агент может быть затем очищен гель-фильтрацией или другими выше упомянутыми способами.
При помощи соответствующих связывающих групп могут быть получены следующие конъюгаты клеточносвязьшающий агент/цитотоксичный агент. Димеры 1 и 2 с пептид-расщепляемыми связывающими группами могут быть получены из соответствующих NHS эфиров, димер 3 может быть получен по реакции соответствующего тиол-содержащего цитотоксичного агента с модифицированным SMCC клеточносвязывающим агентом, а кислото-неустойчивый гидразонный димер 4 может быть получен путем конденсации цитотоксичного агента, содержащего алкил, арилкетон, с гадразид-модифицированным клеточносвязывающим агентом.
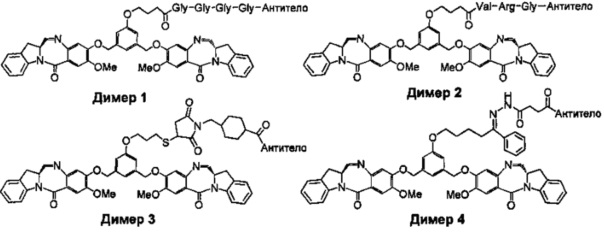
Асимметричные конъюгаты индолинобензодиазепиновых димеров, такие как димеры 5-8, также могут быть получены описанными выше способами.
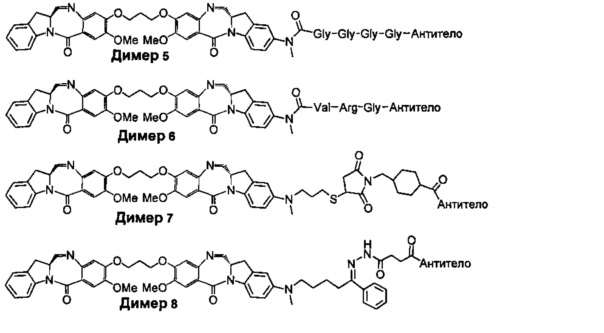
Конъюгаты клеточносвязьтающих агентов с цитотоксичными агентами настоящего изобретения можно оценить по их способности подавлять разрастание различных нежелательных клеточных линий in vitro. Например, для оценки цитотоксичности этих конъюгатов могут использоваться такие клеточные линии, как линия рака толстой кишки человека COLO 205, клеточная линия рабдомиосаркомы RH-30 и клеточная линия множественной миеломы MOLP-8. Клетки, подлежащие оценке, подвергаются обработке соединениями настоящего изобретения в течение 1-5 дней, а выжившие фракции клеток измеряются прямыми анализами известными способами. Затем по результатам анализов рассчитываются значения IC50.
Примеры эффективности in vitro и целевой специфичности конъюгатов антитело-цитотоксичный агент настоящего изобретения представлены на фиг. 21-26. Все конъюгаты с соотношением цитотоксичный агент/антитело 1-3 обладают чрезвычайной цитотоксичностью по отношению к антиген-положительным клеточным клеткам со значениями IC50 в нижнем пикомолярном диапазоне. Антиген-отрицательные клеточные линии остаются жизнеспособными при воздействии на них тех же конъюгатов. Целевая специфичность конъюгатов индолинобензодиазепиновых димеров >1000 с антителами huN901 (анти-CD56) и muB38.1 (анти-ЕрСАМ). Например, конъюгат B38.1-IGN-3 убивает антиген-положительные клетки COLO 205 при значении IC50 1,86 пМ, тогда как антиген-отрицательная клеточная линия Namalwa примерно в 200 раз менее чувствительна, значение IC50 составило 336,3 пМ, что демонстрирует антиген-специфичность. Кроме того, этот конъюгат также высоко эффективен по отношению к клеточной линии COLO 205 MDR, устойчивой к различным лекарствам, значение IC50 составляет 16 пМ. Аналогично, конъюгат huN901-IGN3 показал высокую эффективность, значение IC50 составило 15 пМ для антиген-положительных клеток RH30 (фиг. 22). Добавление избытка неконъюгированного антитела huN901 уничтожило этот цитотоксический эффект (IC50>3 нМ), что демонстрирует антиген-специфичность. Другой конъюгат huN901-IGN (huN901-IGN-07) также показал высокую эффективность по отношению к антиген-экспрессирующим клеткам RH-30, с цитотоксичностью, зависящей от дозировки лекарства, и значениями IC50 16 пМ, 3 пМ и 2 пМ, соответственно, для конъюгатов, имеющих 1,2, 2,0 и 3,0 связанных частиц лекарства на одну молекулу антитела (фиг. 23). Аналогичные результаты были получены для huN901-IGN07 и huN901-IGN03 по отношению к антиген-положительным клеткам Molp-8. Hu901-IGN07 дал значения IC50 5 пМ, 3 пМ и 2 пМ, соответственно, для содержания IGN07 1,2, 2,0 и 3,0 (фиг. 24). Конъюгаты huN901-IGN07 и IGN03 были значительно менее эффективными по отношению к антиген-отрицательным клеткам Namalwa со значениями IC50 от 1000 пМ до >3000 пМ (фиг. 25). Конъюгат B38.1-IGN10 также показал специфическую эффективность, убивая антиген-положительные клетки COLO 205, со значением IC50 17 пМ, и меньшую эффективность (170 пМ) для антиген-отрицательных клеток Ramos (фиг. 26).
В одном примере была измерена эффективность конъюгата клеточносвязывающий агент/цитотоксичный агент in vivo. Безтимусные мыши с опухолью MOLP-8 человека, подвергались лечению конъюгатом huN901-IGN-07, и у них наблюдалась значительная регрессия опухоли по сравнению с быстрым ростом опухоли у мышей, не подвергавшихся лечению (фиг. 27).
Индолинобензодиазепиновые димеры настоящего изобретения связывают и алкилируют двуспиральную ДНК (дсДНК), содержащую гуанидиновые остатки на противоположных цепочках, отстоящих на 4 пары оснований. На фиг. 28-30 представлены данные анализов обращенно-фазовой ионопарной хроматографии, показывающие степень связывания IGN-01, IGN-02 и IGN-09 и сшивания с дсДНК. Индолино-группа (IGN-01) является предпочтительней оксазоловой группы (IGN-02) для быстрого связывания ДНК и внутриспирального сшивания (ICL). Исходная скорость образования аддукта IGN1-ДНК зависит от последовательности ДНК. IGN1 быстрее связывается с ДНК, содержащей внутренний мотив GATC, по сравнению с ДНК с последовательностью GTAC. Образец ДНК, замещенный дезоксиинозином (I) (не содержащий С-2 аминогруппы) вместо гуанина (G) не дал реакции с IGN-1 (фиг. 29).
Значения IC50 различных соединений настоящего изобретения по отношению к панелям клеточных линий перечислены на фиг. 31. Сравнение эффективности in vitro связываемых и несвязываемых соединений настоящего изобретения показано на фиг. 32. Внедрение связывающего агента существенно не влияет на эффективность исходных соединений.
Композиции и способы применения
Настоящее изобретение включает композицию (напр., фармацевтическую композицию), содержащую новые бензодиазепиновые соединения (напр., индолинобензодиазепиновые или оказолидинобензодиазепиновые), их производные, их конъюгаты (и/или сольваты, гидраты и/или их соли) и носитель (фармацевтически приемлемый носитель). Настоящее изобретение включает также композицию (напр., фармацевтическую композицию), содержащую новые бензодиазепиновые соединения, их производные или их конъюгаты (и/или сольваты, гидраты и/или их соли) и носитель (фармацевтически приемлемый носитель), и дополнительно содержащую второе терапевтическое средство. Настоящие композиции применимы для ингибирования аномального клеточного роста или лечения пролиферативных нарушений у млекопитающих (напр., человека). Настоящие композиции также применимы для лечения депрессии, тревоги, стресса, фобий, паники, дисфории, психиатрических нарушений, боли, воспалительных заболеваний у млекопитающих (напр., человека).
Настоящее изобретение включает способ ингибирования аномального клеточного роста или лечения пролиферативных нарушений у млекопитающих (напр., человека), включая введение оговоренным млекопитающим терапевтически эффективного количества новых бензодиазепиновых соединений (напр., индолинобензодиазепиновых или оксазолидинобензодиазепиновых), их производных или их конъюгатов (и/или их сольватов и солей) или их композиций, по отдельности или в сочетании со вторым терапевтическим средством.
В настоящем изобретении предложены также способы лечения, включающие введение пациенту, нуждающемуся в лечении, эффективного количества любого из выше описанных конъюгатов.
Аналогично, в настоящем изобретении предложен способ инициации гибели клеток в выбранных клеточных популяциях, включающий контакт клеток или тканей-мишеней, содержащих клетки-мишени, с эффективным количеством цитотоксичного агента, включая любой из конъюгатов цитотоксичного соединения с клеточносвязывающим агентом (напр., индолинобензодиазепиновый или оксазолидинобензодиазепиновый димер, связанный с клеточносвязывающим агентом) настоящего изобретения, его соль или сольват. Клетки-мишени является клетками, с которыми может связываться клеточносвязьшающий агент.
При желании вместе с конъюгатом настоящего изобретения можно вводить другие активные агенты, такие как другие противоопухолевые агенты.
Соответствующие фармацевтически приемлемые носители, разбавители и наполнители хорошо известны и могут быть определены специалистом в данной области в соответствии с клинической ситуацией.
Примеры применимых носителей, разбавителей и/или наполнителей включают: (1) фосфатный солевой буферный раствор Dulbecco, рН около 7,4, содержащий или не содержащий от около 1 мг/мл до 25 мг/мл альбумина сыворотки человека, (2) 0,9% раствор соли (0,9% масс. NaCl) и (3) 5% (масс.) декстрозы; и может также содержать антиоксидант, такой как триптамин и стабилизирующий агент, такой как Твин 20.
Способ инициирования гибели клеток в выбранных клеточных популяциях может быть осуществлен in vitro, in vivo или ex vivo.
Примеры использования in vitro включают лечение аутологического костного мозга перед его трансплантацией тому же пациенту для уничтожения пораженных или злокачественных клеток: лечение костного мозга перед его трансплантацией для уничтожения конкурирующих Т-клеток и предупреждения реакции "трансплантат против хозяина" (GVHD); обработка клеточных культур для уничтожения всех клеток, за исключением нужных вариантов, не экспрессирующих целевой антиген; или для уничтожения вариантов, экспрессирующих нежелательный антиген.
Условия неклинического использования in vitro легко определяются специалистом в данной области.
Примеры клинического использования ex vivo заключаются в удалении опухолевых клеток или лимфоидных клеток из клеточного мозга перед аутологической трансплантацией при лечении рака или при лечении аутоиммунного заболевания, или для удаления Т-клеток и других лимфоидных клеток из аутологического или аллогенного костного мозга или ткани перед пересадкой для предотвращения GVHD. Лечение может быть выполнено следующим образом. Получают костный мозг пациента или другого организма и инкубируют в среде, содержащей сыворотку, в которую добавлен цитотоксичный агент настоящего изобретения, диапазон концентраций от около 10 мкМ до 1 пМ, в течение от около 30 минут до около 48 часов при температуре около 37°С. Точная концентрация и время инкубации, т.е., доза, легко определяются специалистом в данной области. После инкубации клетки костного мозга промывают средой, содержащей сыворотку, и возвращают пациенту внутривенно в соответствии с известными способами. Если пациент получает другое лечение, такое как курс аблативной химиотерапии или тотальное облучение всего организма между отбором костного мозга и обратного введения обработанных клеток, то обработанные клетки костного мозга хранятся замороженными в жидком азоте при помощи стандартного медицинского оборудования.
Для клинического использования in vivo, цитотоксичный агент настоящего изобретения вводится в виде раствора или лиофилизированного порошка, протестированного на стерильность и уровень эндотоксинов. Примеры соответствующих протоколов введения конъюгатов представлены ниже. Конъюгаты вводятся еженедельно в течение 4 недель в виде внутривенного болюса раз в неделю. Дозы болюса даются в изотоническом растворе объемом 50-1000 мл, в который может быть добавлено 5-10 мл альбумина сыворотки человека. Дозы составляют от 10 мкг до 2000 мг за одно введение, внутривенно (диапазон от 1000 нг до 20 мг/кг в день). Через четыре недели лечения пациент может продолжать получать лечение на еженедельной основе. Конкретные клинические протоколы с учетом способа введения, наполнителей, разбавителей, дозировок, интервалов и т.п., могут быть определены специалистом в данной области в зависимости от клинической ситуации.
Примеры медицинских состояний, которые могут лечиться в соответствии с in vivo или ex vivo способами индуцирования гибели клеток в выбранных клеточных популяциях включают злокачественность любого типа, включая, например, рак легкого, груди, прямой кишки, простаты, почек, поджелудочной железы, яичников и лимфатический органов; аутоиммунные заболевания, такие как системная волчанка, ревматоидный артрит и множественный склероз; отторжение трансплантата, такое как отторжение почечного трансплантата, отторжение печеночного трансплантата, отторжение легочного трансплантата, отторжение сердечного трансплантата и отторжение трансплантата костного мозга; реакция "трансплантат против хозяина"; вирусные инфекции, такие как инфекция CMV, инфекция HIV, AIDS и т.п.; и паразитные инфекции, такие как лямблиоз, амебиаз, шистосомоз и другие, определенные специалистом в данной области.
Лечение рака и дозировки, способы введения и рекомендуемое использование известно в данной области и описано в такой литературе как Настольный справочник врача (Physician's Desk Reference (PDR)). PDR описывает дозировки агентов, используемых для лечения различных видов рака. Режим дозирования и терапевтически эффективные дозы этих вышеупомянутых химиотерапевтических лекарств зависят от конкретного вида рака, подлежащего лечению, масштаба заболевания и других факторов, известных специалисту в данной области, и определяются врачом. Содержание PDR в прямой форме включено здесь в полном объеме путем ссылки. Специалист в данной области может обратиться к PDR, используя один или более из следующих параметров, для определения режима дозирования и доз химиотерапевтических агентов и конъюгатов, которые могут использоваться в соответствии с указаниями настоящего изобретения. Эти параметры включают:
Общий индекс
По производителю
Продукты (по компаниям или торговым названиям лекарств)
Индекс категории
Общий/химический индекс (общепринятые названия лекарств, не торговые марки)
Цветовые обозначения лекарственных средств
Информация о продукте, совместимая с маркировкой FDA
Химическая информация
Назначение/действие
Показания и противопоказания
Пробные испытания, побочные эффекты, предостережения
Аналоги и производные
Специалист в области цитотоксичных агентов понимает, что каждый из цитотоксичных агентов, описанных здесь, может быть модифицирован таким способом, чтобы полученное соединение все еще сохраняло специфичность и/или активность исходного соединения. Опытный специалист также понимает, что многие из этих соединений могут использоваться вместо цитотоксичных соединений, описанных здесь. Так, цитотоксичные агенты настоящего изобретения включают аналоги и производные описанных здесь соединений.
Все приведенные здесь и в последующих примерах ссылки включены здесь в полном объеме путем ссылки.
ПРИМЕРЫ
Теперь настоящее изобретение будет проиллюстрировано ссылками на неограничивающие примеры. Если не оговорено иное, все проценты, отношения, доли и т.п. представлены в массовом выражении. Все реагенты были закуплены у компании Aldrich Chemical Co., Нью-Джерси, или из других доступных источников. Спектры ядерного магнитного резонанса (1Н ЯМР) были получены на приборе Bruker 400 МГц, а масс-спектры были получены на приборе Bruker Daltonics Esquire 3000 с использованием электроспрей ионизации.
Пример 1
метиловый эфир (2S)-1-[5-метокси-2-нитро-4-(фенилметокси)-бензоил]-2-индолинкарбоновой кислоты 5:

К перемешанному раствору 4-бензилокси-5-метокси-2-нитробензойной кислоты 3 (7,01 г, 23,1 ммоль) в безводном дихлорметане (100 мл) и ТГФ (10 мл) добавили оксалилхлорид (4,1 мл, 46,2 ммоль) и ДМФ (30 мкл, 0,38 ммоль) при комнатной температуре. После добавления ДМФ образовалось большое количество пузырьков. Смесь перемешивали в течение ночи (реакция обычно заканчивается за 3 часа), а затем растворители удалили ротационным выпариванием in vacuo. Осадок упарили еще раз при добавлении безводного дихлорметана и отогнали под высоким вакуумом для получения ацетилхлорида 4 в виде твердого желтого вещества, которое напрямую использовали для следующих этапов.
К перемешанному раствору (s)-(-)-индолин-2-карбоновой кислоты 1 (3,43 г, 21,0 ммоль) в безводном метаноле (42 мл) по каплям добавили тионилхлорид (3,1 мл, 42,0 ммоль) при 0°С. Ледяную баню убрали через 30 минут, а смесь продолжали перемешивать при комнатной температуре в течение 5 часов. Растворитель удалили при пониженном давлении и дополнительно высушили осадок под высоким вакуумом для получения сложного метилового эфира 2, который растворили в безводном ТГФ (70 мл) в 500 мл круглодонной колбе. Раствор охладили до 0°С и добавили триэтиламин (9,7 мл, 69,3 ммоль), а затем быстро прибавили свежеприготовленный ацетилхлорид 4 в безводном ТГФ (70 мл) через канюлю при 0°С. Смесь перемешивали при 0~5°С еще в течение 1,5 часов, а затем при комнатной температуре в течение 30 минут. Реакцию погасили добавлением холодной 5% HCl, а затем разбавили этилацетатом и водой. Водный слой экстрагировали этилацетатом три раза. Затем промыли объединенные органические слои насыщенным солевым раствором, насыщенным раствором бикарбоната натрия и солевым раствором, высушили над безводным сульфатом натрия и отфильтровали. Растворители выпарили при пониженном давлении и очистили осадок силикагелевой хроматографией (гексаны/этилацетат, 2:1, 1,5:1) для получения метилового эфира (2S)-1-[5-метокси-2-нитро-4-(фенилметокси)-бензоил]-2-индолинкарбоновой кислоты 5 в виде твердого желтого вещества (9,1 г, выход = 94%).1Н ЯМР (400 Гц, CDCl3): соединение получено в виде трех отдельных ротамеров. δ 8.27 (d, J=8.4 Hz, 0.3Н), 7.90 (s, 0.1Н), 7.82 (s, 0.6H), 7.79 (s, 0.3H), 7.50-7.28 (m, 5.4H), 7.20-7.09 (m, 1.3H), 7.05 (s, 0.6H), 6.97-6.81 (m, 1.6H), 6.76 (s, 0.1H), 5.85 (d, J=8.0 Hz, 0.1H), 5.70 (d, J=8.0 Hz, 0.6H), 5.45-5.41 (m, 0.6H), 5.33-5.21 (m, 2.1H), 4.55 (dd, J1=10.8 Hz, J2=2.8 Hz, 0.3H), 3.98 (s, 1.8H), 3.94 (s, 0.9H), 3.83-3.81 (m, 2.4H), 3.62 (dd, J1=16.4 Hz, J2=11.4 Hz, 1H), 3.56 (s, 0.9H), 3.27-3.13 (m, 1H);13C NMR (400 Hz, CDCl3): 171.5, 164.7, 155.2, 154.4, 148.6, 148.3, 140.3, 137.4, 135.11, 135.05, 130.5, 129.2, 128.7, 128.4, 127.9, 127.6, 127.5, 126.7, 125.5, 124.8, 124.3, 123.9, 117.6, 112.4, 110.1, 109.2, 108.8, 71.3, 71.2, 61.5, 60.2, 60.1, 56.7, 56.5, 52.5, 52.4, 33.6, 31.4; HRMS (ESI, m/z): расч. 463.1505 (M+H)+, найдено 463,1516.
(2S)-1-[5-метокси-2-нитро-4-(фенилметокси)-бензоил]-2-индолинальдегид 6:
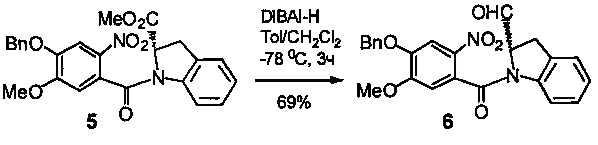
К перемешанному раствору метилового эфира 5 (4,4 г, 9,5 ммоль) в безводном дихлорметане (11 мл) и толуоле (33 мл) по каплям добавили диизобутилалюминия гидрид (19 мл, 1,0 М в толуоле) через шприцевой насос в течение 30 минут при -78°С. Смесь продолжали перемешивать при -78°С в течение 3 часов, и тонкослойная хроматография (гексаны/AcOEt, 1:1,5) показала, что исходный материал полностью израсходован. Реакцию погасили метанолом (0,4 мл) и 5% HCl (30 мл) при -78°С. Добавили этилацетат (100 мл) и убрали баню из сухого льда/ацетона. Смесь перемешивали при комнатной температуре в течение 30 минут, а затем переместили в делительную воронку. Водный слой дважды экстрагировали AcOEt и промыли объединенные органические слои насыщенным солевым раствором, насыщенным раствором бикарбоната натрия и солевым раствором и высушили над безводным сульфатом натрия. Профильтровали через целит и удалили растворители при пониженном давлении (температура <35°С). Осадок очистили флэш-хроматографией (гексаны/AcOEt, 1,5:1, 1:1, 1:1,5) для получения альдегида 6 в виде твердого желтого вещества (2,85 г, выход = 69%).1Н ЯМР (400 Гц, CDCl3): соединение получено в виде трех отдельных ротамеров. δ 10.02 (s, 0.3Н), 9.85 (s, 0.5Н), 9.45 (s, 0.2Н), 8.32-8.31 (m, 0.2Н), 7.93 (s, 0.3H), 7.83 (s, 0.5H), 7.79 (s, 0.2H), 7.53-7.34 (m, 5.2H), 7.26-7.14 (m, 1.3H), 7.08 (s, 0.5H), 7.01-6.94 (m, 1H), 6.91-6.82 (m, 1H), 5.78 (d, J=8.4 Hz, 0.3H), 5.71 (d, J=8.4 Hz, 0.5H), 5.52-5.48 (m, 0.5H), 5.35-5.21 (m, 2.3H), 4.53-4.50 (m, 0.2H), 4.06 (s, 1.5H), 3.98 (s, 0.6H), 3.94 (s, 0.9H), 3.63-3.17 (m, 2H); HRMS (ESI, m/z): расч. 433.1400 (M+H)+, найдено 433,1387.
Соединение 7:
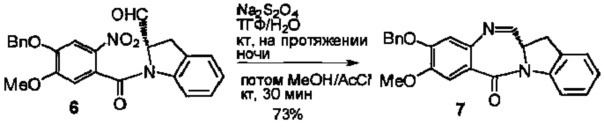
К перемешанному раствору альдегида 6 (2,16 г, 5 ммоль) в ТГФ (230 мл) добавили деионизированную воду (150 мл) и дитионит натрия (85%, 4,61 г, 22,5 ммоль). Полученный слегка мутный раствор стал прозрачным после добавления еще 5 мл деионизированной воды. Прозрачную смесь перемешивали при комнатной температуре в течение 16 часов и добавили 30 мл МеОН. После перемешивания в течение еще 2 часов растворители удалили при пониженном давлении (температура бани ниже 35°С). Осадок суспендировали в ацетонитриле и выпарили для облегчения удаления остатков воды. Полученное твердое белое вещество дополнительно полностью высушили, оставив под высоким вакуумом на несколько часов. Осадок суспендировали в смеси дихлорметан/метанол (1:1) и отфильтровали через целит. Колбу и твердое вещество тщательно промыли смесью дихлорметан/метанол (1:1). Фильтрат десорбировали при пониженном давлении. Осадок растворили в метаноле (50 мл), а затем по каплям добавили ацетилхлорид (1,86 мл, 25 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут и концентрировали при пониженном давлении (температура бани ниже 35°С) для удаления половины метанола. Остаток погасили насыщенным раствором бикарбоната натрия, а затем добавили дихлорметан (150 мл) и воду (150 мл). Водный слой экстрагировали дихлорметаном (2×100 мл) и промыли объединенные органические слои насыщенным солевым раствором, высушили над безводным сульфатом натрия и отфильтровали. Растворители удалили при пониженном давлении и очистили осадок силикагелевой хроматографией (гексаны/AcOEt, 1:1, 1:1,3, 1:1,5) для получения соединения 7 в виде твердого желтого вещества (1.41 g, y=73%).1Н NMR (400 Hz, CDCl3): δ 8.26 (d, J=8.0 Hz, 1H), 7.83 (d, J=4.4 Hz, 1H), 7.57 (s, 1H), 7.46-7.23 (m, 7H), 7.11-7.08 (m, 1H), 6.86 (s, 1H), 5.23 (d, J=12 Hz, 1H), 5.18 (d, J=12 Hz, 1H), 4.44 (ddd, J1=11.2 Hz, J2=4.4 Hz, J3=4.0 Hz, 1H), 3.97 (s, 3H), 3.67 (dd, J1=16.4 Hz, J2=11.2 Hz, 1H), 3.46 (dd, J1=16.4 Hz, J2=4.0 Hz, 1H);13C NMR (400 Hz, CDCl3): δ 163.8, 163.0, 150.9, 148.3, 141.96, 139.97, 136.0, 129.4, 128.6, 128.1, 128.08, 127.3, 124.7, 124.69, 120.7, 116.8, 111.9, 111.3, 70.8, 56.2, 54.9, 32.5; HRMS (ESI, m/z): расч. 385.1552 (M+H)+, найдено 385.1592.
Индолинобензодиазепиновый (ИБД) мономер 8:
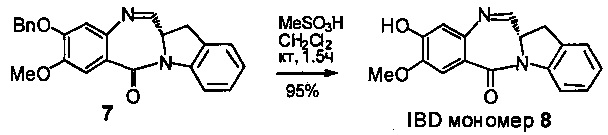
К перемешанному раствору исходного материала 7 (1,41 г, 3,67 ммоль) в дихлорметане (26 мл) добавили свежеперемешанный раствор метансульфоновой кислоты (26 мл) в дихлорметане (52 мл) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 1,5 часов и разбавили дихлорметаном (100 мл). Смесь вылили в смесь льда (~200 г) и МеОН (10 мл). pH полученного раствора довели до 7 насыщенным раствором NaHCO3, твердым NaHCO3 и водой. Смесь разделили и промыли дихлорметановый слой насыщенным солевым раствором. Объединенные водные слои экстрагировали этилацетатом (3×80 мл). Этил ацетатные слои объединили и промыли насыщенным солевым раствором. Дихлорметан объединили с этилацетатом, высушили над безводным сульфатом натрия и отфильтровали. Растворители удалили, а осадок (1,26 г) очистили силикагелевой хроматографией (CH2Cl2/МеОН, 20:1, 15:1) для получения ИБД мономера 8 в виде твердого желтого вещества (1.02 g, y=95%).1Н NMR (400 Hz, CDCl3): δ 8.29 (d, J=8.0Hz, 1H), 7.91 (d, J=4.8 Hz, 1H), 7.59 (s, 1H), 7.32-7.28 (m, 2H), 7.13 (t, J=7.2 Hz, 1H), 6.94 (s, 1H), 6.02 (s, -OH), 4.50 (dt, J1=10.8 Hz, J2=4.4 Hz, 1H), 4.02 (s, 3H), 3.73 (dd, J1=16.8 Hz, J2=10.8 Hz, 1H), 3.52 (dd, J1=16.8 Hz, J2=3.6 Hz, 1H); HRMS (ESI, m/z): расч. 295.1083 (M+H)+, найдено 295,1076.
Пример 2
Метиловый эфир (s)-(-)-3-(бензилоксикарбонил)-4-оксазолидинкарбоновой кислоты 10:

К перемешанному раствору (s)-(-)-3-(бензилоксикарбонил)-4-оксазолидинкарбоновой кислоты 9 (1,75 г, 6,96 ммоль) в безводном метаноле (15 мл) добавили тионилхлорид (1,02 мл, 13,9 ммоль) при 0°С. Через 30 минут убрали водно-ледяную баню, а реакционную смесь продолжали перемешивать при комнатной температуре в течение 3,5 часов. Реакцию погасили добавлением насыщенного раствора бикарбоната натрия и разбавили дихлорметаном (100 мл) и водой (50 мл). Смесь разделили и экстрагировали водный слой дихлорметаном (2×50 мл). Объединенные органические слои промыли насыщенным солевым раствором, высушили над безводным сульфатом натрия и отфильтровали. Растворители удалили при пониженном давлении, а осадок очистили силикагелевой хроматографией (гексаны/AcOEt, 1,5:1) для получения метилового эфира (s)-(-)-3-(бензилоксикарбонил)-4-оксазолидинкарбоновой кислоты 10 в виде бесцветной маслянистой жидкости (1,84 г, выход = 99%).1Н ЯМР (400 Гц, CDCl3): соединение получено в виде двух отдельных ротамеров. δ 7.35 (bs, 5Н), 5.22-4.99 (m, 4Н), 4.53-4.45 (m, 1Н), 4.22-4.09 (m, 2Н), 3.76 (s, 1.5Н), 3.65 (s, 1.5Н); MS (m/z): найдено 288,0 (M+Na)+.
Соединение 11:

К перемешанному раствору метилового эфира (s)-(-)-3-(бензилоксикарбонил)-4-оксазолидинкарбоновой кислоты 10 (1,04 г, 3,92 ммоль) в этилацетате (16 мл) добавили триэтиламин (1,4 мл, 10 ммоль) и гидроксид палладия на углероде (20%, 267 мг, 0,337 ммоль). Воздух из реакционной колбы удалили под вакуумом, затем добавили колбу водорода и перемешивали смесь в атмосфере водорода при комнатной температуре в течение 2 часов. К раствору ацетилхлорида 4 (приготовленному из 1,3 г, 4,3 ммоль 4-бензилокси-5-метокси-2-нитробензойной кислоты 2 по вышеописанной методике) в безводном ТГФ (15 мл) добавили триэтиламин (1,1 мл, 7,9 ммоль) при 0°С, а затем добавили реакционную смесь вышеуказанной реакции гидрогенизации путем фильтрации через целит. Палладиевый катализатор/целит промыли безводным ТГФ (15 мл). Полученную смесь перемешивали при 0°С в течение 3 часов. Разбавили этилацетатом и насыщенным раствором хлорида аммония. рН смеси довели до 6~7 добавлением 5% соляной кислоты. Смесь разделили и экстрагировали водный слой этилацетатом (2×80 мл). Объединенные органические слои промыли насыщенным солевым раствором, высушили над безводным сульфатом натрия и отфильтровали. Растворители удалили при пониженном давлении и очистили осадок силикагелевой хроматографией (гексаны/AcOEt, 1:2, 1:3) для получения соединения 11 в виде твердого бледно-желтого вещества (1,49 г, выход = 91%).1Н ЯМР (400 Гц, CDCl3): соединение получено в виде двух отдельных ротамеров. δ 7.78 (s, 0.5Н), 7.75 (s, 0.5Н), 7.48-7.37 (m, 5Н), 6.97 (s, 0.5Н), 6.91 (s, 0.5H), 5.39 (d, J=4.8 Hz, 0.5H), 5.26-5.23 (m, 2.5H), 4.95 (dd, J1=7.2 Hz, J2=4.4 Hz, 0.5H), 4.81 (d, J=3.6 Hz, 0.5H), 4.67 (d, J=3.6 Hz, 0.5H), 4.37-4.30 (m, 1H), 4.25-4.11 (m, 1.5H), 4.02 (s, 1.5H), 3.97 (s, 1.5H), 3.87 (s, 1.5H), 3.67 (s, 1.5H); HRMS (ESI, m/z): расч. 417.1298 (M+H)+, найдено 417,1305.
Альдегид 12:

К перемешанному раствору метилового эфира 11 (1,49 г, 3,6 ммоль) в безводном дихлорметане (4 мл) и толуоле (12 мл) по каплям добавили диизобутилалюминия гидрид (6,5 мл, 1,0 М в толуоле) через шприцевой насос в течение 30 минут при -78°С. Смесь продолжали перемешивать при -78°С в течение 2 часов. Реакцию погасили метанолом (146 мкл, 3,6 ммоль) и 5% HCl (30 мл) при -78°С. Добавили этилацетат (100 мл) и убрали баню из сухого льда/ацетона. Смесь перемешивали при комнатной температуре в течение 30 минут, а затем переместили в делительную воронку. Водный слой дважды экстрагировали AcOEt. Все органические слои объединили, промыли насыщенным солевым раствором, насыщенным раствором бикарбоната натрия и солевым раствором. Высушили над безводным сульфатом натрия и отфильтровали через целит. Фильтрат выпарили при пониженном давлении и очистили осадок силикагелевой хроматографией (гексаны/AcOEt, 1:5, 1:10) для получения соединения 12 в виде твердого бледно-желтого вещества (980 мг, выход = 70%).1Н ЯМР (400 Гц, CDCl3): соединение получено в виде двух отдельных ротамеров. δ 9.83 (s, 0.67Н), 9.45 (s, 0.33Н), 7.77 (s, 0.67Н), 7.72 (s, 0.33Н), 7.45-7.37 (m, 5Н), 6.90 (s, 1Н), 5.31-5.19 (m, 3H), 4.77 (bs, 1H), 4.67-4.56 (m, 1H), 4.36-3.94 (m, 5H); HRMS (ESI, m/z): расч. 387.1192 (M+H)+, найдено 387,1184.
Соединение 13:

К перемешанному раствору альдегида 12 (154 мг, 0,4 ммоль) в ТГФ (21 мл) добавили деионизированную воду (14 мл) и дитионит натрия (85%, 369 мг, 1,8 ммоль). Прозрачную смесь перемешивали при комнатной температуре в течение 16 часов и добавили 5 мл МеОН. После перемешивания в течение еще 2 часов растворители удалили при пониженном давлении (температура бани ниже 35°С). Осадок суспендировали в ацетонитриле и выпарили для облегчения удаления остатков воды. Полученное твердое белое вещество дополнительно полностью высушили, оставив под высоким вакуумом на несколько часов. Осадок суспендировали в смеси дихлорметан/метанол (2:1) и отфильтровали через целит. Колбу и твердое вещество тщательно промыли смесью дихлорметан/метанол (1:1). Фильтрат десорбировали при пониженном давлении. Осадок растворили в метаноле (5 мл) и быстро добавили свежеприготовленный раствор ацетилхлорида (0,15 мл)/МеОН (5 мл). Смесь перемешивали при комнатной температуре в течение 30 минут и погасили добавлением насыщенного раствора бикарбоната натрия. Разбавили дихлорметаном и водой. Два слоя разделили и экстрагировали водный слой дихлорметаном. Объединенные дихлорметановые слои промыли насыщенным солевым раствором и высушили над безводным сульфатом натрия. Растворители удалили при пониженном давлении для получения 127 мг неочищенного продукта. Водный слой и промывочный раствор объединили и подкислили до рН 2~3 при помощи KHSO4. Концентрировали раствор до половины объема при пониженном давлении (температура <40°С) и экстрагировали дихлорметаном. Объединенный дихлорметан промыли насыщенным раствором бикарбоната натрия и насыщенным солевым раствором, высушили над безводным сульфатом натрия. Смесь отфильтровали, а фильтрат выпарили при пониженном давлении. Осадок объединили с полученными ранее 127 мг неочищенного продукта и очистили силикагелевой хроматографией (гексаны/AcOEt, 1:3, 1:5, 1:8) для получения соединения 13 в виде бесцветной пены (80 мг, выход = 61%).1Н NMR (400 Hz, CDCl3): δ 7.77 (d, J=4.0 Hz, 1H), 7.52 (s, 1H), 7.46-7.28 (m, 5H), 6.88 (s, 1H), 5.28 (d, J=5.2 Hz, 1H), 5.23 (d, J=12 Hz, 1H), 5.17 (d, J=12 Hz, 1H), 5.05 (d, J=5.2 Hz, 1H), 4.49 (dd, J1=9.6 Hz, J2=3.2 Hz, 1H), 4.33 (dd, J1=9.6 Hz, J2=6.4 Hz, 1H), 3.96 (s, 3H), 3.83 (dd, J1=6.4 Hz, J2=3.2 Hz, 1H); MS (m/z): найдено 361.1 (M+Na)+, 379.1 (M+H2O+Na)+, 339.1 (M+H)+.
Оксазолидинобензодиазеттиновый (ОВР) мономер 14:
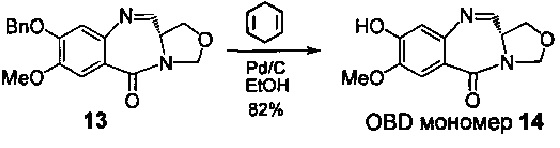
Раствор соединения 13 (90 мг, 0,27 ммоль) и Pd/C (10%, 90 мг) в абсолютном этаноле (1,5 мл) продували аргоном. Добавили 1,4-циклогексадиен (496 мкл, 5,3 ммоль) и продолжали продувать аргон в течение 3 часов до исчезновения исходного материала (тонкослойная хроматография, дихлорметан/метанол 10:1). Затем смесь отфильтровали через целит и промыли целит метанолом. Фильтрат выпарили при пониженном давлении для получения 63 мг неочищенного продукта в виде бесцветной пены, который очистили силикагелевой хроматографией (дихлорметан/метанол, 20:1) для получения OBD мономера 14 (55 мг, выход = 82%) в виде твердого белого вещества.1Н ЯМР (400 Гц, CDCl3): соединение получено в виде смеси имина и его простых метиловых эфиров, C11(R) и C11(S) (2:3:1). δ 7.71 (bs, 1Н), 7.43 (s, 0.5H), 7.41 (s, 1H), 7.18 (s, 1.5H), 6.83 (s, 1H), 6.36 (s, 1.5H), 6.13 (s, 0.5H), 5.25 (d, J=4.8 Hz, 0.5H), 5.22-5.20 (m, 1H), 5.14 (d, J=5.2 Hz, 1.5H), 5.10 (d, J=4.8 Hz, 0.5H), 5.05 (d, J=5.2 Hz, 1.5H), 5.00-4.97 (m, 1H), 4.47 (d, J=8.8 Hz, 1.5H), 4.44-4.41 (m, 1H), 4.32 (apt, J=8.0 Hz, 0.5H), 4.28-4.25 (m, 1H), 4.18-4.00 (m, 2x1.5H+2x0.5H=4H), 3.84 (bs, 3x1H+0.5H=3.5H), 3.76 (bs, 3x1.5H+1H=5.5H), 3.73 (s, 3x0.5H=1.5H), 3.56 (dt, J1=8.8 Hz, J2=2.8 Hz, 1.5H), 3.34 (s, 3x1.5H=4.5H), 3.22 (s, 3x0.5H=1.5H); MS (m/z): найдено 303,1 (M+MeOH+Na)+, 271,1 (M+Na)+.
Пример 3
Димер 15 (IGN-09):

К раствору ИБД мономера 8 (147 мг, 0,5 ммоль) и 1,3-дийодпропана (23 мкл, 0,2 ммоль) в безводном ДМФ (1,0 мл) добавили карбонат калия (111 мг, 0,8 ммоль). Смесь перемешивали при комнатной температуре в течение ночи (16 часов) и разбавили дихлорметаном. Промыли насыщенным раствором хлорида аммония и насыщенным солевым раствором, высушили над безводным сульфатом натрия и отфильтровали. Фильтрат выпарили при пониженном давлении и очистили осадок препаративной обращенно-фазовой ВЭЖХ (колонка С18, ацетонитрил/вода) для получения димера 15 (IGN-09) (18,9 мг, выход = 15%) в виде твердого белого вещества.1H NMR (400 Hz, CDCl3): δ 8.26 (d, J=8.0 Hz, 2H), 7.87 (d, J=4.4 Hz, 2H), 7.55 (s, 2H), 7.26 (s, 4H), 7.12-7.08 (m, 2H), 6.88 (s, 2H), 4.45 (ddd, J1=10.8 Hz, J2=4.4 Hz, J3=4.0 Hz, 2H), 4.36-4.26 (m, 4H), 3.94 (s, 6H), 3.70 (dd, J1=16.8 Hz, J2=10.8 Hz, 2H), 3.50 (dd, J1=16.8 Hz, J2=4.0 Hz, 2H), 2.45 (p, J=6.0 Hz, 2H); HRMS (ESI, m/z): расч. 629.2400 (M+H)+, найдено 629,2400.
Пример 4
Димер 18 (IGN-01):
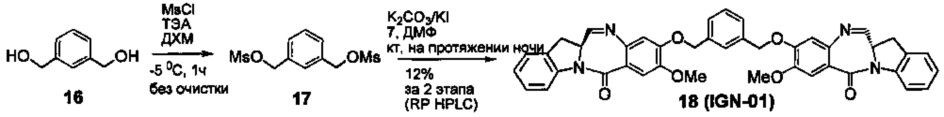
К перемешанному раствору 1,3-бензолдиметанола 16 (11 мг, 0,08 ммоль) в безводном дихлорметане (0,8 мл) добавили триэтиламин (33 мкл, 0,24 ммоль), а затем по каплям в течение 15 минут добавили метансульфонилхлорид (16 мкл, 0,21 ммоль) при -5~-10°С. Раствор перемешивали при -5~-10°С в течение еще 60 минут и погасили смесью лед/вода, разбавили холодным этилацетатом. Смесь разделили и промыли органический слой холодной водой, высушили над безводным сульфатом натрия. Отфильтровали и выпарили фильтрат ротационным выпариванием in vacuo (температура <35°С). Осадок 17 выдержали под высоким вакуумом в течение нескольких часов, а затем растворили в безводном ДМФ (1,5 мл). Затем добавили ИБД мономер 7 (94 мг, 0,32 ммоль), безводный карбонат калия (50 мг, 0,36 ммоль) и йодид калия (27 мг, 0,16 ммоль). Смесь перемешивали при комнатной температуре в течение 17 часов (проверяли по масс-спектру) и разбавили дихлорметаном. Промыли насыщенным солевым раствором, высушили над безводным сульфатом натрия и отфильтровали. Фильтрат выпарили при пониженном давлении, а осадок очистили обращенно-фазовой ВЭЖХ (колонка С18, CH3CN/H2O, загруженная колонка с CH3CN/H2O, 3:1, перемешивали в течение 30 минут и центрифугировали перед вводом) для получения димера 18 (IGN-01, 6,6 мг) в виде твердого белого вещества.1H NMR (400 Hz, CDCl3): δ 8.21 (d, J=8.0 Hz, 2H), 7.79 (d, J=4.4 Hz, 2H), 7.51 (s, 2H), 7.46 (s, 1H), 7.36 (bs, 3H), 7.23-7.18 (m, 4H), 7.06-7.03 (m, 2H), 6.79 (s, 2H), 5.20 (d, J=12.4 Hz, 2H), 5.14 (d, J=12.4 Hz, 2H), 4.41 (ddd, J1=10.8 Hz, J2=4.4 Hz, J3=4.0 Hz, 2H), 3.92 (s, 6H), 3.64 (dd, J1=17.2 Hz, J2=11.2 Hz, 2H), 3.42 (dd, J1=16.8 Hz, J2=4.0 Hz, 2H); HRMS (ESI, m/z): расч. 691.2557 (M+H)+, найдено 691,2570.
Пример 5
Димер 19 (IGN-02):
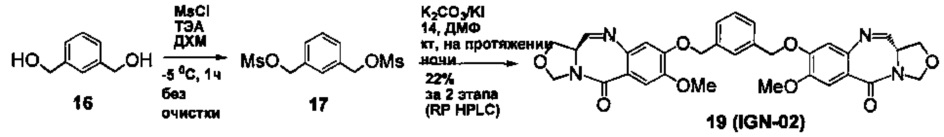
К перемешанному раствору 1,3-бензолдиметанола 16 (10 мг, 0,074 ммоль) в безводном дихлорметане (0,8 мл) добавили триэтиламин (31 мкл, 0,22 ммоль), а затем по каплям в течение 15 минут добавили метансульфонилхлорид (15 мкл, 0,19 ммоль) при -5~-10°С. Раствор перемешивали при -5~-10°С в течение еще 60 минут и погасили смесью лед/вода, разбавили холодным этилацетатом. Смесь разделили и промыли органический слой холодной водой, высушили над безводным сульфатом натрия. Отфильтровали и выпарили фильтрат ротационным выпариванием in vacuo (температура <35°С). Осадок 17 выпарили под высоким вакуумом, а затем растворили в безводном ДМФ (1,5 мл). Затем добавили OBD мономер 14 (70 мг, 0,28 ммоль), безводный карбонат калия (51 мг, 0,37 ммоль) и йодид калия (25 мг, 0,15 ммоль). Смесь перемешивали при комнатной температуре в течение 17 часов (проверяли по масс-спектру) и разбавили дихлорметаном. Промыли насыщенным солевым раствором, высушили над безводным сульфатом натрия и отфильтровали. Фильтрат выпарили при пониженном давлении и очистили осадок обращенно-фазовой ВЭЖХ (колонка С18, CH3CN/H2O, загруженная колонка с CH3CN/H2O, 3:1, перемешивали в течение 30 минут и центрифугировали перед вводом) для получения димера 19 (IGN-02, 10,0 мг) в виде твердого белого вещества.1Н NMR (400 Hz, CDCl3): δ 7.75 (d, J=4.0 Hz, 2H), 7.50-7.48 (bs, 3Н), 7.38 (bs, 3Н), 6.83 (s, 2H), 5.26 (d, J=5.2 Hz, 2H), 5.21 (d, J=14.4 Hz, 2H), 5.15 (d, J=14.0 Hz, 2H), 5.03 (d, J=5.6 Hz, 2H), 4.34-4.30 (m, 2H), 3.94 (s, 6H), 3.86-3.76 (m, 2H); HRMS (ESI, m/z): расч. 599.2142 (M+H)+, найдено 599,2184.
Пример 6
Триол 21:
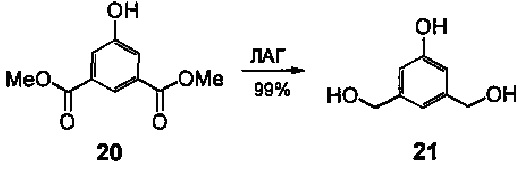
К перемешанному раствору диметил 5-гидроксиизофталата 20 (2,1 г, 10 ммоль) в безводном ТГФ (50 мл) добавили гидрид лития-алюминия (2,0 М в ТГФ, 10 мл, 20 ммоль) при -20~-30°С через шприцевой насос в течение 30 минут. Охлаждающую баню убрали через 30 минут, а смесь продолжали перемешивать при комнатной температуре в течение 4 часов. Смесь охладили до 0~-10°С и погасили насыщенным раствором сульфата натрия. Смесь разбавили ацетонитрилом и добавили 5% соляную кислоту (20 мл). Смесь перемешивали в течение 30 минут и высушили над безводным сульфатом натрия. Смесь отфильтровали через целит, а фильтрат выпарили при пониженном давлении. Осадок очистили силикагелевой хроматографией (дихлорметан/метанол, 10:1, 8:1, 5:1) для получения триола 21 (1,5 г, выход = 99%) в виде бесцветной маслянистой жидкости, которая при стоянии превратилась в твердое белое вещество.1Н ЯМР (400 Гц, MeOD): δ 6,78, (с, 1Н), 6,69 (с, 2Н), 4,50 (с, 4Н).13С ЯМР (400 Гц, MeOD): δ 158,7, 144,4, 117,8, 113,8, 65,2; MS (m/z): найдено 153,0 [М-Н]-.
Соединение 22:
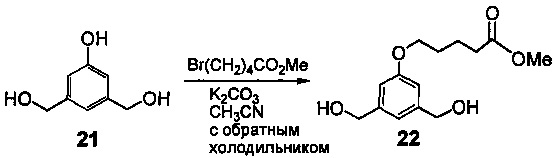
К раствору триола 21 (827 мг, 5,37 ммоль) и метил 5-бромвалерата (998 мг, 5,12 ммоль) в ацетонитриле (40 мл) добавили карбонат калия (3,71 г, 26,9 ммоль). Смесь поместили на масляную баню при 86°С и дефлегмировали в течение 6 часов. Реакционную смесь сняли с масляной бани, охладили до комнатной температуры и выпарили растворители при пониженном давлении (температура <35°С). Осадок разбавили дихлорметаном и отфильтровали. Фильтрат промыли насыщенным солевым раствором, высушили над безводным сульфатом натрия и отфильтровали. Фильтрат десорбировали при пониженном давлении и очистили осадок силикагелевой хроматографией (гексаны/этилацетат, 1:2, 1:3) для получения соединения 22 (1,15 г, выход = 84%) в виде твердого белого вещества.1Н NMR (400 Hz, CDCl3): δ 6.89 (s, 1Н), 6.80 (s, 2H), 4.62 (s, 4H), 3.98-3.95 (m, 2H), 3.67 (s, 3Н), 2.41-2.37 (m, 2H), 2.23 (bs, -OHx2), 1.84-1.78 (m, 4H); MS (m/z): найдено 291,1 (M+Na)+.
Соединение 23:

По методике получения соединения 22 синтезировали соединение 23 (1,43 г, выход = 75%) в виде твердого белого вещества из триола 21 (1,16 г, 7,53 ммоль), метил 4-бромбутирата (1,52 г, 8,39 ммоль) и карбоната калия (5,2 г, 37,6 ммоль).1Н NMR (400 Hz, CDCl3): δ 6.90 (s, 1Н), 6.80 (s, 2H), 4.62 (s, 4H), 4.00 (t, J=6.0 Hz, 2H), 3.68 (s, 3H), 2.51 (t, J=7.2 Hz, 2H), 2.19 (s, -OHx2), 2.13-2.06 (m, 2H); MS (m/z): найдено 277,1 (M+Na)+.
Соединение 24:
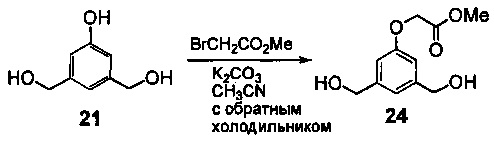
По методике получения соединения 22 синтезировали соединение 24 (515 мг, выход = 37%) в виде вязкого белого вещества из триола 21 (953 мг, 6,19 ммоль), метил бромацетата (587 мкл, 6,19 ммоль) и карбоната калия (4,3 г, 31 ммоль).1Н NMR (400 Hz, CDCl3): δ 6.95 (s, 1Н), 6.81 (s, 2H), 4.64 (s, -OHx2), 4.61 (s, 4H), 3.81 (s, 3Н), 2.41 (s, 2H);13C NMR (400 Hz, CDCl3): δ 169.4, 158.1, 143.0, 118.5, 112.1, 65.2, 64.8, 52.3; MS (m/z): найдено 249,0 (M+Na)+.
Соединение 27:

К раствору 5-нитро-м-ксилол-α,α'-диола 25 (1,07 г, 5,84 ммоль) в метаноле (50 мл) добавили Pd/C (10%, 311 мг, 0,29 ммоль). Для вытеснения воздуха ввели водород, затем смесь гидрировали (Н2, 5 фунтов/кв.дюйм) в течение 2 часов при комнатной температуре. Раствор отфильтровали через целит и выпарили фильтрат ротационным выпариванием in vacuo для получения соединения 26 в виде твердого белого вещества (900 мг, выход = 100%).1H NMR (400 Hz, MeOD): δ 6.71 (s, 1Н), 6.66 (s, 2H), 4.51 (s, 4H);13C NMR (400 Hz, MeOD): δ 148.9, 143.8, 116.7, 114.3, 65.5; растворили в безводном ацетонитриле (30 мл) и этилбромацетате (443 мкл, 4,67 ммоль), добавили карбонат калия (807 мг, 5,84 ммоль). Смесь поместили на масляную баню при 86°С и дефлегмировали в течение 17 часов. Реакционную смесь сняли с масляной бани, охладили до комнатной температуры и разбавили дихлорметаном. Смесь отфильтровали через целит и промыли твердое вещество дихлорметаном. В фильтрате появился белый осадок. Осадок собрали фильтрацией для получения соединения 27 (414 мг, выход = 39%) в виде твердого белого вещества.1H NMR (400 Hz, MeOD): δ 6.67 (s, 1Н), 6.53 (s, 2H), 4.51 (s, 4H), 3.94 (s, 2H), 3.73 (s, 3H);13C NMR (400 Hz, MeOD): δ 174.0, 149.7, 143.9, 116.2, 111.6, 65.6, 52.6, 46.5; MS (m/z): найдено 248,0 (M+Na)+.
Соединение 28:

К раствору 5-нитро-м-ксилол-α,α'-диола 25 (564 мг, 3,08 ммоль) в метаноле (35 мл) добавили Pd/C (10%, 164 мг, 0,154 ммоль). Для вытеснения воздуха ввели водород, затем смесь гидрировали (Н2, 5 фунтов/кв.дюйм) в течение 2 часов при комнатной температуре. Раствор отфильтровали через целит и выпарили фильтрат ротационным выпариванием in vacuo для получения соединения 26, которое растворили в безводном ацетонитриле (15 мл) и добавили метил 4-бромбутират (557 мг, 3,08 ммоль) и карбонат калия (426 мг, 3,08 ммоль). Смесь поместили на масляную баню при 86°С и дефлегмировали в течение 18 часов. Реакционную смесь сняли с масляной бани, охладили до комнатной температуры и разбавили дихлорметаном. Отфильтровали через целит и промыли твердое вещество смесью дихлорметан/ацетонитрил (1:1). Фильтрат выпарили при пониженном давлении и очистили осадок силикагелевой хроматографией (Combiflash, дихлорметан/метанол) для получения соединения 28 (292 мг, выход = 37%) в виде твердого белого вещества.1Н NMR (400 Hz, MeOD): δ 6.62 (s, 1Н), 6.55 (s, 2H), 4.50 (s, 4H), 3.65 (s, 3H), 3.13 (d, J=7.2 Hz, 2H), 2.43 (d, J=7.2 Hz, 2H), 1.89 (p, J=7.2 Hz, 2H);13C NMR (400 Hz, MeOD): δ 175.9, 150.5, 143.7, 115.5, 111.7, 65.7, 52.2, 44.3, 32.5, 25.8; MS (m/z): найдено 276,0 (M+Na)+.
Соединение 29:

К раствору соединения 27 (230 мг, 1,02 ммоль) в безводном ацетонитриле (7 мл) добавили метилйодид (70 мкл, 1,12 ммоль) и карбонат калия (155 мг, 1,12 ммоль). Смесь поместили на масляную баню при 86°С и дефлегмировали в течение 17 часов. Реакционную смесь сняли с масляной бани, охладили до комнатной температуры и разбавили дихлорметаном. Отфильтровали через целит и промыли твердое вещество смесью дихлорметан/метанол (10:1). Фильтрат выпарили при пониженном давлении и очистили осадок силикагелевой хроматографией (Combiflash, дихлорметан/метанол) для получения соединения 29 (98 мг, выход = 40%) в виде твердого белого вещества.1Н NMR (400 Hz, MeOD): δ 6.70 (s, 1Н), 6.63 (s, 2H), 4.84 (s, 2x-OH), 4.54 (s, 4H), 4.16 (s, 2H), 3.69 (s, 3H), 3.05 (s, 3H);13C NMR(400 Hz, MeOD): δ 173.6, 150.9, 143.8, 115.6, 111.0, 65.7, 54.9, 52.4, 39.8; MS (m/z): найдено 262,0 (M+Na)+.
Соединение 30:
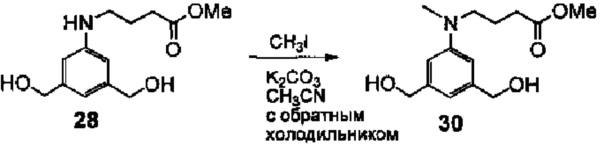
К раствору соединения 28 (151 мг, 0,597 ммоль) в безводном ацетонитриле (4 мл) добавили метилйодид (74 мкл, 1,19 ммоль) и карбонат калия (99 мг, 0,716 ммоль). Смесь поместили на масляную баню при 86°С и дефлегмировали в течение 17 часов. Реакционную смесь сняли с масляной бани, охладили до комнатной температуры и разбавили дихлорметаном. Отфильтровали через целит и промыли твердое вещество смесью дихлорметан/метанол (10:1). Фильтрат выпарили при пониженном давлении и очистили осадок силикагелевой хроматографией (Combiflash, дихлорметан/метанол) для получения соединения 30 (63 мг, выход = 39%) в виде бесцветной маслянистой жидкости.1Н NMR (400 Hz, MeOD): δ 6.67 (s, 2Н), 6.65 (s, 1Н), 4.54 (s, 4H), 3.65 (s, 3H), 3.36 (t, J=7.2 Hz, 2H), 2.92 (s, 3H), 2.36 (t, J=7.2 Hz, 1H), 1.87 (p, J=7.2 Hz, 2H);13C NMR (400 Hz, MeOD): δ 175.7, 151.3, 143.7, 115.0, 111.4, 65.9, 53.0, 52.2, 38.9, 32.2, 23.3; MS (m/z): найдено 290,0 (M+Na)+.
Соединение 34 (IGN-03):
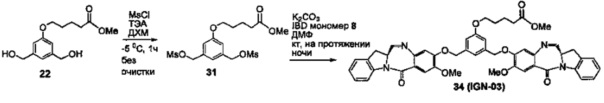
К перемешанному раствору соединения 22 (80,4 мг, 0,3 ммоль) в безводном дихлорметане (2 мл) добавили триэтиламин (125 мкл, 0,9 ммоль), а затем по каплям за 15 минут добавили метансульфонилхлорид (60 мкл, 0,78 ммоль) при -5 ~ -10°С. Раствор перемешивали при -5 ~ 10°С в течение еще 60 минут и погасили смесью лед/вода, разбавили холодным этилацетатом. Смесь разделили и промыли органический слой холодной водой, высушили над безводным сульфатом натрия. Отфильтровали и выпарили фильтрат ротационным выпариванием in vacuo (температура <35°С). Осадок 31 выпарили под высоким вакуумом, а затем растворили в безводном ДМФ (3 мл). Добавили ИБД мономер 7 (221 мг, 0,75 ммоль) и безводный карбонат калия (207 мг, 1,5 ммоль). Смесь перемешивали при комнатной температуре в течение 20 часов (проверяли по масс-спектру) и разбавили дихлорметаном. Смесь промыли водой и насыщенным солевым раствором, высушили над безводным сульфатом натрия и отфильтровали. Фильтрат выпарили при пониженном давлении и очистили осадок силикагелевой хроматографией (гексаны/этилацетат, 1:3, 1:4, 1:6, 1:10, затем этилацетат/метанол, 10:1) для получения соединения 34 (169 мг, выход = 68%, чистота 86% по данным аналитической обращенно-фазовой ВЭЖХ) в виде твердого желтоватого вещества. Фракции, содержащие примеси и соединение 34, также собрали и выпарили растворители для получения 70 мг твердого желтоватого вещества. Два твердых желтоватых вещества объединили и дополнительно очистили обращенно-фазовой ВЭЖХ (колонка С18, CH3CN/H2O, загруженная колонка с CH3CN/H2O, 3:1, перемешивали в течение 30 минут и центрифугировали перед вводом) для получения димера 34 (IGN-03, 103 мг, выход = 41%) в виде твердого белого вещества.1Н NMR (400 Hz, CDCl3): δ 8.27 (d, J=8.0 Hz, 2H), 7.85 (d, J=3.2 Hz, 2H), 7.58 (s, 2H), 7.29-7.24 (m, 4H), 7.12-7.07 (m, 3H), 6.94 (s, 2H), 6.83 (s, 2H), 5.22 (d, J=12.8 Hz, 2H), 5.16 (d, J=12.8 Hz, 2H), 4.47 (dt, J1=11.2 Hz, J2=4.4 Hz, 2H), 3.98 (bs, 8H), 3.73-3.64 (m, 2H), 3.68 (s, 3H), 3.48 (dd, J1=16.8 Hz, J2=3.6 Hz, 2H), 2.42-2.38 (m, 2H), 1.83-1.80 (m, 4H); HRMS (ESI, m/z): расч. 821.3187 (M+H)+, найдено 821,3188.
Соединение 35 (IGN-04):
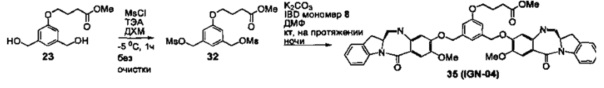
По методике получения соединения 34 синтезировали соединение 35 (IGN-04) (151 мг, выход = 62%, чистота 88% по данным аналитической обращенно-фазовой ВЭЖХ) в виде твердого желтоватого вещества. Часть его дополнительно очистили обращенно-фазовой ВЭЖХ для анализа1Н ЯМР.1Н NMR (400 Hz, CDCl3): δ 8.17 (d, J=8.0 Hz, 2H), 7.74 (d, J=5.2 Hz, 2H), 7.48 (s, 2H), 7.20-7.15 (m, 4H), 7.03-6.99 (m, 3H), 6.85 (s, 2H), 6.75 (s, 2H), 5.12 (d, J=12.8 Hz, 2H), 5.06 (d, J=12.8 Hz, 2H), 4.37 (dt, J1=11.2 Hz, J2=4.4 Hz, 2H), 3.93 (t, J=6.0 Hz, 2H), 3.86 (s, 6H), 3.64-3.57 (m, 2H), 3.60 (s, 3H), 3.39 (dd, J1=16.8 Hz, J2=3.6 Hz, 2H), 2.44 (t, J=7.2 Hz, 2H), 2.02 (p, J=6.4 Hz, 2H); HRMS (ESI, m/z): расч. 807.3030 (M+H)+, найдено 807,3008.
Соединение 36 (IGN-05):
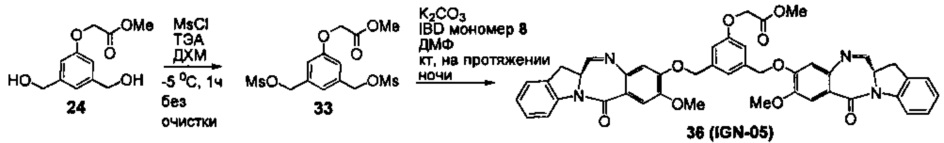
По методике получения соединения 34 синтезировали соединение 36 (IGN-05) (84,5 мг, выход = 18%) в виде твердого белого вещества после препаративной обращенно-фазовой ВЭЖХ.1Н NMR (400 Hz, CDCl3): δ 8.24 (d, J=8.0 Hz, 2H), 7.79 (d, J=4.4 Hz, 2H), 7.55 (s, 2H), 7.26-7.22 (m, 4H), 7.12-7.07 (m, 3H), 6.96 (s, 2H), 6.81 (s, 2H), 5.18 (d, J=12.8 Hz, 2H), 5.12 (d, J=12.8 Hz, 2H), 4.64 (s, 2H), 4.44 (dt, J1=10.8 Hz, J2=4.4 Hz, 2H), 3.95 (s, 6H), 3.77 (s, 3H), 3.73-3.62 (m, 2H), 3.44 (dd, J1=16.8 Hz, J2=3.6 Hz, 2H); HRMS (ESI, m/z): расч. 779.2717 (M+H)+, найдено 779,2703.
Соединение 39 (IGN-06):

По методике получения соединения 34 синтезировали соединение 39 (IGN-06) с выходом 6% в виде твердого белого вещества после препаративной обращенно-фазовой ВЭЖХ.1Н NMR (400 Hz, CDCl3): δ 8.28 (d, J=8.0 Hz, 2H), 7.86 (d, J=4.0 Hz, 2H), 7.58 (s, 2H), 7.31-7.26 (m, 4H), 7.12 (t, J=7.2 Hz, 2H), 6.90-6.86 (m, 3H), 6.72 (s, 2H), 5.22 (d, J=12.4 Hz, 2H), 5.13 (d, J=12.4 Hz, 2H), 4.51-4.46 (m, 2H), 3.99 (s, 6H), 3.74-3.68 (m, 2H), 3.71 (s, 3H), 3.49 (dd, J1=16.8 Hz, J2=3.6 Hz, 2H), 3.09 (s, 3H); HRMS (ESI, m/z): расч. 792.3033 (M+H)+, найдено 792,3013.
Соединение 40 (IGN-07):
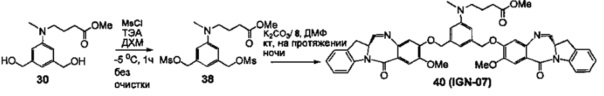
По методике получения соединения 34 синтезировали соединение 40 (IGN-07) с выходом 21% в виде твердого белого вещества после препаративной обращенно-фазовой ВЭЖХ.1Н NMR (400 Hz, CDCl3): δ 8.27 (d, J=8.0 Hz, 2H), 7.84 (d, J=4.4 Hz, 2H), 7.58 (s, 2H), 7.30-7.23 (m, 4H), 7.21-7.02 (m, 3H), 6.88 (s, 2H), 6.74 (s, 2H), 5.23-5.13 (m, 4H), 4.50-4.42 (m, 2H), 3.99 (s, 6H), 3.74-3.70 (m, 2H), 3.67 (s, 3H), 3.51-3.33 (m, 4H), 2.92 (s, 3H), 2.36-2.30 (m, 2H), 1.93-1.84 (m, 2H); HRMS (ESI, m/z): расч. 820.3346 (M+H)+, найдено 820,3329.
Пример 7
Соединение 41:
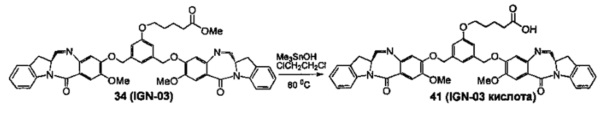
К раствору соединения 34 (42 мг, 0,051 ммоль) в безводном 1,2-дихлорэтане (1 мл) добавили гидроксид триметилолова (139 мг, 0,77 ммоль). Смесь нагревали при 78~82°С (масляная баня 80°С) и перемешивали в течение ночи. Тонкослойная хроматография (CH2Cl2/МеОН, 10:1) показала исчезновение исходного материала. Реакционную смесь охладили до комнатной температуры и разбавили дихлорметаном. Смесь промыли каплями 5% соляной кислоты в насыщенном солевом растворе, насыщенным раствором хлорида аммония и солевым раствором, высушили над безводным сульфатом натрия, отфильтровали и выпарили. Осадок очистили силикагелевой хроматографией (Combiflash, CH2Cl2/МеОН, от 1:0 до 5:1) для получения IGN-03 кислоты 41 (33,8 мг, выход = 82%) в виде твердого желтоватого вещества. Осадок может также использоваться для следующих этапов без очистки. MS (m/z): найдено 805,1 (М-Н)-, 823,0 (М+H2O-Н)-, 829,2 (М+Na)+, 847,2 (М+H2O+Na)+.
Соединение 42:
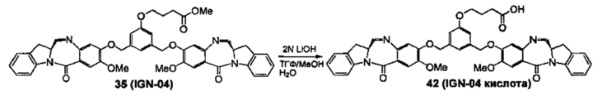
К перемешанному раствору соединения 35 (32 мг, 0,040 ммоль) в смеси ТГФ (0,4 мл), метанола (0,1 мл) и деионизированной воды (0,1 мл) добавили свежеприготовленный 2н раствор LiOH (24 мкл, 0,048 ммоль) при 0°С. Охлаждающую баню убрали и перемешивали смесь при комнатной температуре в течение 8 часов. Реакционную смесь разбавили этилацетатом и водой. рН смеси довели до 4~5 добавлением 5% соляной кислоты. Смесь промыли насыщенным солевым раствором, высушили над безводным сульфатом натрия и отфильтровали. Фильтрат выпарили при пониженном давлении и очистили осадок препаративной обращено-фазовой ВЭЖХ (колонка С18, ацетонитрил/вода) для получения IGN-04 кислоты 42 (4,2 мг, выход = 13%) в виде твердого белого вещества. MS (m/z): найдено 791,0 (М-Н)-, 809,0 (М+H2O-Н)-, 815,2 (М+Na)+, 833,1 (М+H2O+Na)+.
Соединение 43:

К перемешанному раствору IGN-03 кислоты 41 (8,9 мг, 0,011 ммоль) в безводном дихлорметане (0,2 мл) добавили N-гидроксисукцинимид (2,6 мг, 0,022 ммоль), N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорид (4,2 мг, 0,022 ммоль) и маленький кусочек димегиламинопиридина. Смесь перемешивали при комнатной температуре в течение ночи и разбавили дихлорметаном. Смесь промыли насыщенным раствором хлорида аммония и насыщенным солевым раствором, высушили над безводным сульфатом натрия и отфильтровали. Фильтрат выпарили при пониженном давлении. Осадок очистили силикагелевой хроматографией (Combiflash, CH2Cl2/МеОН, от 1:0 до 10:1) для получения NHS эфира IGN-03 43 (7,9 мг, выход = 79%) в виде твердого желтоватого вещества. В результате очистки обращенно-фазовой препаративной ВЭЖХ (колонка С18, CH3CN/H2O, фракции продукта экстрагировали дихлорметаном) получили 3,2 мг твердого белого вещества для анализа1Н ЯМР.1Н NMR (400 Hz, CDCl3): δ 8.28 (d, J=8.0 Hz, 2H), 7.87 (d, J=4.0 Hz, 2H), 7.59 (s, 2H), 7.31-7.27 (m, 4H), 7.15-7.10 (m, 3H), 6.97 (s, 2H), 6.86 (s, 2H), 5.25 (d, J=12.4 Hz, 2H), 5.18 (d, J=12.4 Hz, 2H), 4.49 (dt, J1=10.8 Hz, J2=4.0 Hz, 2H), 4.04 (t, J=5.6 Hz, 2H), 4.01 (s, 6H), 3.72 (dd, J1=16.8 Hz, J2=10.8 Hz, 2H), 3.51 (dd, J1=16.8 Hz, J2=4.0 Hz, 2H), 2.85 (bs, 4H), 2.72 (t, J=6.8 Hz, 2H), 1.99-1.91 (m, 4H); HRMS (ESI, m/z): расч. 904.3194 (M+H)+, найдено 904,3182.
Соединение 44:

По методике получения соединения 43 синтезировали соединение 44 с выходом 86% в виде твердого желоватого вещества. MS (m/z): найдено 944,2 (М+МеОН+Na)+, 976,2 (М+2MeOH+Na)+.
Соединение 45 (IGN-07 кислота):
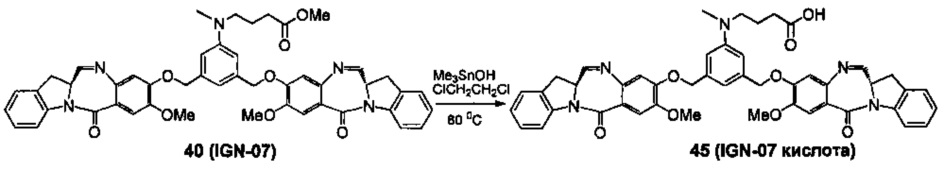
К раствору соединения 40 (14 мг, 0,017 ммоль) в безводном 1,2-дихлорэтане (0,5 мл) добавили гидроксид триметилолова (62 мг, 0,34 ммоль). Смесь нагревали при 78~82°С (масляная баня 80°С) и перемешивали в течение ночи. Тонкослойная хроматография (CH2Cl2/МеОН, 10:1) показала исчезновение исходного материала. Реакционную смесь охладили до комнатной температуры и разбавили дихлорметаном. Смесь промыли насыщенным раствором хлорида аммония и солевым раствором, высушили над безводным сульфатом натрия, отфильтровали и выпарили для получения IGN-07 кислоты 45 в виде бледно-желтого твердого вещества (29,2 мг, с примесями гидроксида триметилолова). MS (m/z): найдено 804,1 (М-Н)-, 822,1 (М+H2O-Н)-, 828,2 (М+Na)+, 846,2 (М+H2O+Na)+. Вещество использовали для следующих этапов без очистки.
Соединение 46:

К перемешанному раствору IGN-07 кислоты 45 из выше приведенной реакции (0,017 ммоль) в безводном дихлорметане (0,5 мл) добавили N-гидроксисукцинимид (6,1 мг, 0,051 ммоль), N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорид (9,8 мг, 0,051 ммоль) и маленький кусочек диметиламинопиридина. Смесь перемешивали при комнатной температуре в течение ночи и разбавили дихлорметаном. Смесь промыли насыщенным раствором хлорида аммония и насыщенным солевым раствором, высушили над безводным сульфатом натрия и отфильтровали. Фильтрат выпарили при пониженном давлении. Осадок очистили силикагелевой хроматографией (Combiflash, CH2Cl2/МеОН, от 1:0 до 10:1) для получения NHS эфира IGN-07 46 (9,1 мг, выход = 59% для двух этапов из IGN-07) в виде твердого желтоватого вещества.1Н ЯМР (400 Гц, CDCl3): δ 8.25 (d, J=7.6 Hz, 2Н), 7.82 (d, J=4.4 Hz, 2H), 7.55 (s, 2H), 7.26-7.18 (m, 5H), 7.09 (t, J=7.6 Hz, 2H), 6.84 (s, 2H), 6.74 (s, 2H), 5.21 (d, J=12.4 Hz, 2H), 5.15 (d, J=12.4 Hz, 2H), 4.46-4.42 (m, 2H), 3.98 (s, 6H), 3.72-3.64 (m, 2H), 3.44-3.37 (m, 4H), 2.95 (s, 3H), 2.74 (bs, 4H), 2.57 (t, J=7.2 Hz, 2H), 1.95 (t, J=7.2 Hz, 2H); HRMS (ESI, m/z): расч. 903.3354 (M+H)+, найдено 903,3347.
Пример 8
Соединение 47:
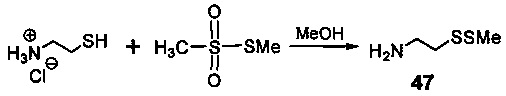
К перемешанному раствору цистеамина гидрохлорида (568 мг, 5 ммоль) в безводном метаноле (15 мл) добавили S-метил метантиосульфонат (519 мкл, 5,5 ммоль) при 0°С. Смесь перемешивали при комнатной температуре в течение ночи. Добавили триэтиламин (1,4 мл, 10 ммоль) и удалили растворители при пониженном давлении. Осадок растворили в 50 мл безводного дихлорметана и получили 0,1 М раствор соединения 47 в дихлорметане (принимая выход за 100%). Аликвоту раствора (0,2 мл) использовали для реакции на следующем этапе. Остаток раствора разбавили дихлорметаном, промыли насыщенным раствором бикарбоната натрия и солевым раствором, высушили над безводным сульфатом натрия и отфильтровали. Фильтрат выпарили при пониженном давлении и очистили осадок силикагелевой хроматографией (дихлорметан/метанол, 10:1 с 1% триэтиламина) для получения соединения 47 (82 мг, выход = 13%, потери продукта при операциях с водой из-за его хорошей растворимости в воде) в виде бесцветной маслянистой жидкости.1Н ЯМР (400 Гц, CDCl3): δ 3,02 (т, J=6,4 Гц, 2Н), 2,77 (т, J=6,4 Гц, 2Н), 2,41 (с, 3Н), 1,34 (шс, 2Н).
Соединение 48 (IGN-08):
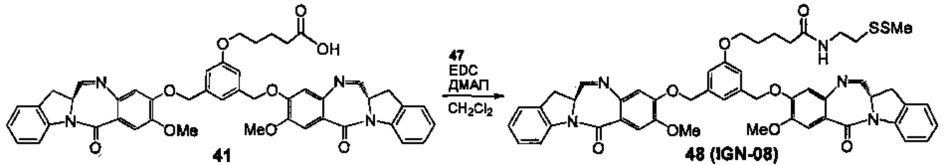
В колбу, содержащую IGN-03 кислоту 41 (8,1 мг, 0,01 ммоль) добавили выше указанный 0,1 М раствор соединения 47 в безводном дихлорметане (0,2 мл). Добавили N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорид (3,8 мг, 0,02 ммоль), триэтиламин (1,4 мкл, 0,01 ммоль) и маленький кусочек диметиламинопиридина. Смесь перемешивали при комнатной температуре в течение ночи и разбавили дихлорметаном. Смесь промыли насыщенным раствором хлорида аммония и насыщенным солевым раствором, высушили над безводным сульфатом натрия и отфильтровали. Фильтрат выпарили при пониженном давлении и очистили осадок препаративной обращенно-фазовой ВЭЖХ (колонка С18, ацетонитрил/H2O) для получения соединения 48 (4,0 мг, выход = 44%) в виде твердого белого вещества.1Н NMR (400 Hz, CDCl3): δ 8.25 (d, J=8.0 Hz, 2H), 7.84 (d, J=4.4 Hz, 2H), 7.57 (s, 2H), 7.29-7.24 (m, 4H), 7.10 (t, J=7.6 Hz, 2H), 7.06 (s, 1H), 6.92 (s, 2H), 6.82 (s, 2H), 5.22 (d, J=12.8 Hz, 2H), 5.17 (d, J=12.4 Hz, 2H), 4.46 (dt, J1=11.2 Hz, J2=4.4 Hz, 2H), 3.98 (bs, 8H), 3.69 (dd, J1=16.8 Hz, J2=10.8 Hz, 2H), 3.62 (d, J=6.4 Hz, 1H), 3.58 (d, J=6.0 Hz, 1H), 3.48 (dd, J1=17.2 Hz, J2=3.6 Hz, 2H), 2.82 (t, J=6.4 Hz, 2H), 2.39 (s, 3H), 2.23 (t, J=6.8 Hz, 2H), 1.80-1.78 (m, 4H); HRMS (ESI, m/z): расч. 912.3101 (M+H)+, найдено 912,3118.
Соединение 49:

К суспензии трис(2-карбоксиэтил)фосфина гидрохлорида (ТСЕР⋅HCl, 3,8 мг, 0,013 ммоль) в капле деионизированной воды (~50 мкл) по каплям добавили насыщенный раствор бикарбоната натрия (~25 мкл), чтобы довести pH примерно до 6~7, затем добавили буферный раствор с pH 6,5 (0,1 М фосфатный буферный раствор, 0,3 мл). Полученную смесь добавили к раствору соединения 48 (IGN-08, 4,0 мг, 0,0044 ммоль) в метаноле (1,0 мл) и ацетонитриле (1,0 мл). Раствор перемешивали при комнатной температуре в течение 1,5 часов и разбавили буферным раствором с pH 6,5 и дихлорметаном (реакцию проверили по масс-спектру, который показал только сигналы продукта). Смесь разделили и промыли органический слой насыщенным солевым раствором, высушили над безводным сульфатом натрия и отфильтровали. Фильтрат выпарили при пониженном давлении и очистили осадок силикагелевой хроматографией (Combiflash, дихлорметан/МеОН) для получения продукта 49 в виде твердого бледно-желтого вещества (2,7 мг, выход = 71%). MS (m/z): найдено 864,0 (М-Н)-, 932,0 (М+МеОН+2H2O-Н)-, 888,1 (М+Na)+, 920,2 (М+МеОН+Na)+, 952,2 (М+2МеОН+Na)+.
Пример 9
Соединение 50:
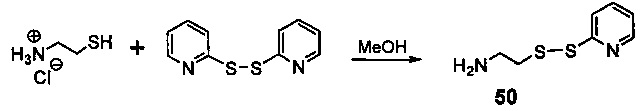
К перемешанному раствору цистеамина гидрохлорида (227 мг, 2 ммоль) в безводном метаноле (10 мл) добавили альдритиол (661 мг, 3 ммоль) Реакционный раствор из прозрачного бесцветного стал прозрачным желтым после добавления альдритиола. Реакционную смесь перемешивали при комнатной температуре в течение 21 часа. Добавили триэтиламин (279 мкл, 2 ммоль) и удалили растворители при пониженном давлении. Осадок очистили силикагелевой хроматографией (Combiflash, дихлорметан/метанол, от 1:0 до 15:1 с 0,1% триэтиламина) для получения соединения 50 (301 мг, выход = 81%) в виде бесцветной маслянистой жидкости.1Н ЯМР (400 Гц, CDCl3): δ 8,52-8,49 (м, 1Н), 7,69-7,60 (м, 2Н), 7,15-7,10 (м, 1Н), 3,04 (т, J=6,0 Гц, 2Н), 2,92 (т, J=6,0 Гц), 1,92 (шс, 2Н).
Соединение 51 (IGN-10):
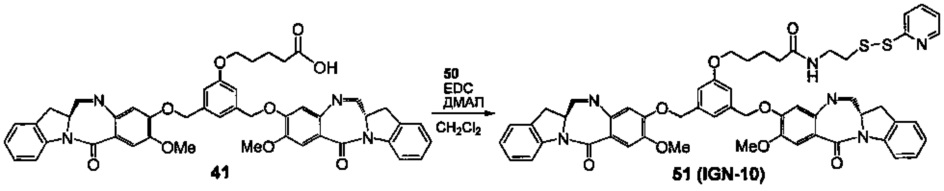
К раствору IGN-03 кислоты 41 (из 0,05 ммоль IGN-03 без очистки) в безводном дихлорметане (1 мл) добавили соединение 50 (37 мг, 0,2 ммоль), N-(3-диметиламинопропил)-N'-этилкарбодаимида гидрохлорид (38 мг, 0,2 ммоль) и маленький кусочек диметиламинопиридина. Смесь перемешивали при комнатной температуре в течение ночи и разбавили дихлорметаном. Смесь промыли насыщенным раствором хлорида аммония и насыщенным солевым раствором, высушили над безводным сульфатом натрия и отфильтровали. Фильтрат выпарили при пониженном давлении. Осадок очистили силикагелевой хроматографией (Combiflash, дихлорметан/метанол, от 1:0 до 5:1) для получения 51 мг желтой пены, которую дополнительно очистили препаративной обращенно-фазовой ВЭЖХ (колонка С18, ацетонитрил/Н2О) для получения соединения 51 (7,4 мг, выход = 15%) в виде твердого желтоватого вещества.1Н NMR (400 Hz, CDCl3): δ 8.50 (d, J=4.4 Hz, 1H), 8.28 (d, J=8.0 Hz, 2H), 7.87 (d, J=4.4 Hz, 2H), 7.63-7.59 (m, 3H), 7.52 (d, J=8.0 Hz, 1H), 7.31-7.21 (m, 4H), 7.14-7.09 (m, 4H), 6.96 (s, 2H), 6.85 (s, 2H), 5.23 (d, J=12.8 Hz, 2H), 5.18 (d, J=12.4 Hz, 2H), 4.49 (dt, J1=11.2 Hz, J2=4.4 Hz, 2H), 4.03-4.00 (m, 8H), 3.72 (dd, J1=16.8 Hz, J2=11.2 Hz, 2H), 3.60 (d, J=5.6 Hz, 1H), 3.57 (d, J=5.6 Hz, 1H), 3.50 (dd, J1=16.8 Hz, J2=3.6 Hz, 2H), 2.95 (t, J=5.6 Hz, 2H), 2.30 (t, J=6.4 Hz, 2H), 1.85-1.84 (m, 4H); HRMS (ESI, m/z): расч. 975.3210 (M+H)+, найдено 975,3190.
Соединение 53:

К перемешанному раствору 4-бензилокси-3-метоксибензилового спирта 52 (2,5 г, 10 ммоль) в уксусном ангидриде (30 мл) медленно частями добавили гидрат нитрата меди (II) (2,7 г, 11 ммоль) при 0°С. Полученную суспензию продолжали перемешивать при 0°С в течение 1 часа и при комнатной температуре в течение 3 часов. Реакционную смесь вылили в смесь льда и воды и перемешивали в течение 1 часа. Смесь отфильтровали, чтобы собрать твердое желтое вещество, которое затем растворили в смеси МеОН/ТГФ (1:1, об./об., 30 мл). Добавили карбонат калия (2,1 г, 15 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 3 часов. Смесь концентрировали при пониженном давлении и разбавили осадок дихлорметаном, промыли водой и насыщенным солевым раствором, высушили над безводным сульфатом натрия и отфильтровали. Фильтрат выпарили при пониженном давлении, и очистили осадок силикагелевой хроматографией (CH2Cl2/МеОН, 20:1, 18:1, 15:1) для получения соединения 53 (1,50 г, выход = 52%) в виде твердого желтого вещества.1Н ЯМР (400 Гц, CDCl3): δ 7,78 (с, 1Н), 7,48-7,33 (м, 5Н), 7,20 (с, 1Н), 5,18 (с, 2Н), 4,96 (с, 2Н), 4,01 (с, 3Н).
Пример 10
Соединение 123:
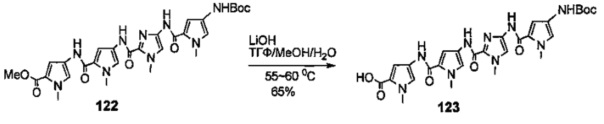
К перемешанному раствору соединения 122 (137 мг, 0,22 ммоль) в ТГФ (1,5 мл) и МеОН (0,5 мл) добавили раствор моногидрата гидроксида лития (46 мг, 1,1 ммоль) в деионизированной воде (0,5 мл). Смесь перемешивали на масляной бане при 60°С в течение 6 часов. Смесь охладили до комнатной температуры и разбавили этилацетатом и водой. рН смеси довели до 4~5 добавлением 5% соляной кислоты. Водный слой экстрагировали этилацетатом. Объединенные органические слои промыли насыщенным раствором бикарбоната натрия и солевым раствором, высушили над безводным сульфатом натрия и отфильтровали. Фильтрат десорбировали для получения соединения 123 (87,5 мг, выход = 65%). MS (m/z): найдено 606,1 [М-Н]-.
Соединение 124:

К раствору кислоты 123 (87,5 мг, 0,14 ммоль) в безводном ДМФ (1 мл) добавили диметиламинопиридин (ДМАП) (21 мг, 0,17 ммоль) метил 5-аминовалерата гидрохлорид (26 мг, 0,15 ммоль) и EDC (40 мг, 0,21 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и разбавили этилацетатом. Смесь промыли насыщенным раствором хлорида аммония, насыщенным солевым раствором, насыщенным раствором бикарбоната натрия и солевым раствором, высушили над безводным сульфатом натрия и отфильтровали. Фильтрат десорбировали и очистили осадок силикагелевой хроматографией (Combiflash, дихлорметан/МеОН) для получения соединения 124 (71 мг, выход = 70%) в виде желтой пены.1Н NMR (400 Hz, CDCl3): δ 9.07 (s, 1Н), 8.62 (s, 1H), 8.40 (s, 1H), 7.19 (s, 1H), 7.17 (s, 1H), 7.09 (s, 1H), 7.00 (s, 1H), 6.74 (s, 1H), 6.62 (s, 3H), 6.46 (s, 1H), 3.94 (s, 3H), 3.85 (bs, 12H), 3.34-3.31 (m, 2H), 2.32 (t, J=7.2 Hz, 2H), 1.68-1.55 (m, 4H), 1.48 (s, 9H); MS (ESI, m/z): найдено 721,0 (M+H)+.
Соединение 125:
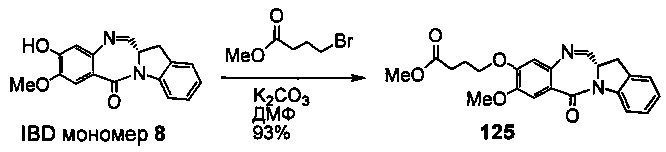
К раствору ИБД мономера 8 (118 мг, 0,4 ммоль) и метил 4-бромбутирата (109 мг, 0,6 ммоль) в безводном ДМФ (1,5 мл) добавили карбонат калия (111 мг, 0,8 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и разбавили этилацетатом, промыли насыщенным раствором хлорида аммония и насыщенным солевым раствором. Высушили над безводным сульфатом натрия и отфильтровали. Фильтрат десорбировали при пониженном давлении для получения соединения 125 (146 мг, выход = 93%) в виде желтой пены.1Н NMR (400 Hz, CDCl3): δ 8.25 (d, J=8.0 Hz, 1H), 7.84 (d, J=4.4 Hz, 1H), 7.52 (s, 1H), 7.26-7.22 (m, 2H), 7.10-7.06 (m, 1H), 6.81 (s, 1H), 4.44 (dt, J1=10.8 Hz, J2=4.0 Hz, 1H), 4.15-4.07 (m, 2H), 3.92 (s, 3H), 3.68 (s, 3H), 3.67-3.64 (m, 1H), 3.46-3.43 (m, 1H), 2.55 (t, J=7.2 Hz, 2H), 2.22-2.15 (m, 2H); MS (ESI, m/z): найдено 465,2 (M+МеОН+K)+.
Соединение 126:

Смесь соединения 125 (146 мг, 0,37 ммоль) и гидроксида триметилолова (669 мг, 3,7 ммоль) в безводном 1,2-дихлорэтане (2 мл) нагрели до 80°С (температура масляной бани) и перемешивали при этой температуре в течение 18 часов. Убрали масляную баню и разбавили смесь дихлорметаном, промыли смесью насыщенного солевого раствора и 5% HCl (0,5 мл), насыщенным раствором бикарбоната натрия и солевым раствором, высушили над безводным сульфатом натрия и отфильтровали. Фильтрат десорбировали и очистили осадок силикагелевой хроматографией (Combiflash, дихлорметан/МеОН) для получения соединения 126 (90 мг, выход = 64%) в виде твердого желтого вещества.1Н NMR (400 Hz, CDCl3): δ 8.26 (d, J=8.0 Hz, 1H), 7.83 (bs, 1H), 7.54 (s, 1H), 7.30-7.25 (m, 2H), 7.11 (t, J=7.6 Hz, 1H), 6.88 (s, 1H), 4.48 (dt, J1=11.2 Hz, J2=4.0 Hz, 1H), 4.16-4.13 (m, 2H), 3.94 (s, 3H), 3.71 (dd, J1=16 Hz, J2=11.2 Hz, 1H), 3.47 (d, J=16 Hz, 1H), 2.60 (t, J=6.4 Hz, 2H), 2.22-2.18 (m, 2H).
Соединение 127 (IGN-11):
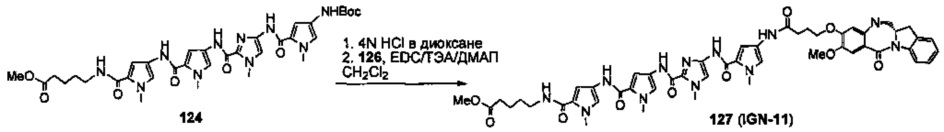
В колбу, содержащую соединение 124 (71 мг, 0,099 ммоль) добавили 4н раствор HCl в диоксане (4 мл). Смесь перемешивали при комнатной температуре в течение 2 часов и десорбировали при пониженном давлении. Осадок растворили в безводном дихлорметане (1,5 мл). Затем добавили соединение 126 (42 мг, 0,11 ммоль), триэтиламин (14 мкл, 0,1 ммоль), EDC (38 мг, 0,2 ммоль) и ДМАП (1 мг, 0,0099 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 22 часов и разбавили дихлорметаном, промыли насыщенным раствором хлорида аммония и насьпценным солевым раствором. Высушили над безводным сульфатом натрия и отфильтровали. Фильтрат десорбировали при пониженном давлении и очистили осадок силикагелевой хроматографией (Combiflash, дихлорметан/МеОН) для получения соединения 127 (14 мг, выход = 49%) в виде твердого желтого вещества. HRMS (ESI, m/z): расч. 983,4164 (М+Н)+, найдено 983,4167.
Пример 11
Получение исходного раствора NHS эфира IGN-03 (соединение 43) и NHS эфира IGN-07 (соединение 46):
Приготовили свежие растворы NHS эфира IGN-03 и NHS эфира IGN-07 концентрацией 0,006 М в диметилацетамиде (ДМА) на основании молекулярного веса 903,93 г/моль (NHS эфир IGN-03) или 902,95 (NHS эфир IGN-07). Исходный раствор проанализировали спектрофотометрически при помощи справочного коэффициента экстинкции, определенного при 330 нм (ε330 нм=15231 М-1 см-1).
Пример 12
4-(трет-бутоксикарбониламино)-1-метил-1Н-пиррол-2-карбоновая кислота
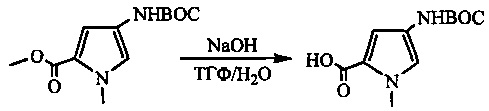
К метил 4-(трет-бутоксикарбониламино)-1-метил-1Н-пиррол-2-карбоксилату (Eldon Е. Baird and Peter В. Dervan, J. Am. Chem. Soc. 1996, 118, 6141-6146) (5,0 r, 19,67 ммоль) в 120 мл смеси THF/H2O 1:1 добавили 8 г NaOH в 30 мл воды. Смесь перемешивали в течение ночи, концентрировали, разбавили водой, экстрагировали смесью EtAc/гексаны (1:1). Водный раствор довели до pH 4,0 20%-ной H3PO4 и экстрагировали EtAc (4×60 мл). Органические растворы объединили, высушили над MgSO4, отфильтровали, выпарили и кристаллизовали со смесью этанол/EtAc/гексан для получения 3,81 г (81%) указанного в заголовке продукта.1H NMR (CD3OD) 12.79 (s, 1Н), 10.48 (br, 1Н), 7.51 (s, 1H), 6.99 (s, 1Н), 3.78 (s, 3Н), 1.49 (s, 9Н);13С NMR 158.47, 153.82, 123.64, 121.56, 109.58, 79.52, 37.06, 28.42; MS m/z- 239.2 (М-Н).
4-(трет-бутоксикарбониламино)-1-метил-1Н-имидазол-2-карбоновая кислота
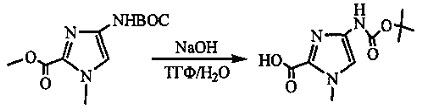
К метил 4-(трет-бутоксикарбониламино)-1-метил-1H-имидазол-2-карбоксилату (5,0 г, 19,59 ммоль) в 120 мл смеси ТГФ/H2O 1:1 добавили 8 г NaOH в 30 мл воды. Смесь перемешивали в течение ночи, концентрировали, разбавили водой, экстрагировали смесью EtAc/гексаны (1:1). Водный раствор довели до pH 4,0 20%-ной Н3РО4 и экстрагировали EtAc (4×60 мл). Органические растворы объединили, высушили над MgSO4, отфильтровали, выпарили и кристаллизовали со смесью этанол/EtAc/гексан для получения 3,85 г (81%) указанного в заголовке продукта.1Н ЯМР (ДМСО) 9,32 (с, 1Н), 7,29 (с, 1Н), 3,57 (с, 3Н), 1,42 (с, 9Н);13С ЯМР 172,45, 159,78, 136,93, 135,44, 132,85, 79,50, 35,57, 28,07; MS m/z- 240,8 (M-H).
1-Метил-4-нитро-1Н-пиррол-2-карбоновая кислота
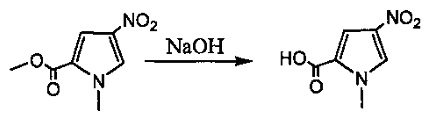
К метил 1 -метил-4-нитро-1Н-пиррол-2-карбоксилату (5,0 г, 27,17 ммоль) в 120 мл смеси ТГФ/Н2О 1:1 добавили 8 г NaOH в 30 мл воды. Смесь перемептивали в течение ночи, концентрировали, разбавили водой, экстрагировали смесью EtAc/гексаны (1:1). Водный раствор довели до рН 3~4 20%-ной Н3РО4 и экстрагировали EtAc (4×60 мл). Органические растворы объединили, высушили над MgSO4 отфильтровали, выпарили и кристаллизовали со смесью этанол/EtAc/гексан для получения 4,06 г (88%) указанного в заголовке продукта.1Н ЯМР (ДМСО) 13,12 (с, 1Н), 8,21 (с, 1Н), 7,25 (с, 3Н), 3,91 (с, 9Н);13С ЯМР 160,97, 134,01, 129,16, 123,81, 111,38, 37,47; MS m/z- 169,1 (M-H).
Метил 1-метил-4-(1-метил-4-нитро-1Н-пиррол-2-карбоксамидо)-1Н-пиррол-2-карбоксилат
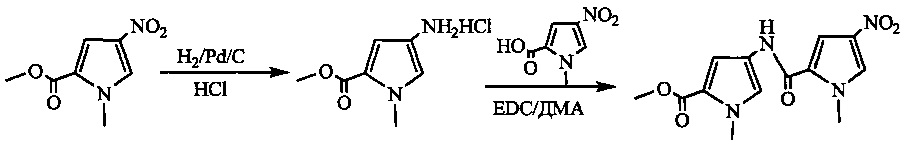
В колбу для гидрогенизации добавили метил 1-метил-4-нитро-1Н-пиррол-2-карбоксилат (3,0 г, 16,30 ммоль), 80 мл ТГФ, 405 мг 10% Pd/C и 1,3 мл HCl (конц.). После вакуумирования в колбу закачали водород под давлением 30 фунтов/кв.дюйм и встряхивали в течение 5 часов. Смесь отфильтровали через целиты, выпарили до сухости без дальнейшей очистки. К сухой смеси добавили 1-метил-4-нитро-1Н-пиррол-2-карбоновую кислоту (2,75 г, 16,18 ммоль), 80 мл ДМА, EDC (8,51 г, 44,27 ммоль) и диизопропилэтиламин (ДИПЭА) (2,80 мл, 16,10 ммоль). Смесь перемешивали под Ar в течение ночи, концентрировали, разбавили смесью ТГФ/EtAc (1:2, 150 мл) и промыли смесью 1 М раствора NaH2PO4/NaCl (конц.) и отдельно NaHCO3 (конц.). Органический слой отделили и высушили над MgSO4, отфильтровали, концентрировали и кристаллизовали со смесью ТГФ/H2O для получения 3,74 г (75%) указанного в заголовке продукта.1Н NMR (DMSO) 10.25 (s, 1Н), 8.17 (s, 1Н), 7.25 (s, 1Н), 6.52 (s, 1Н), 6.08 (s, 1H), 3.90 (s, 3Н), 3.78 (s, 3Н), 3.56 (s, 3Н);13С NMR 157.87, 156.84, 133.76, 128.16, 123.39, 119.13, 118.18, 111.83, 107.50, 104.17, 51.55, 37.41, 36.03; MS m/z+ 329.1 (M+Na).
Метил 4-(4-(трет-бутоксикарбониламино)-1-метил-1Н-пиррол-2-карбоксамидо)-1-метил-1Н-имидазол-2-карбоксилат
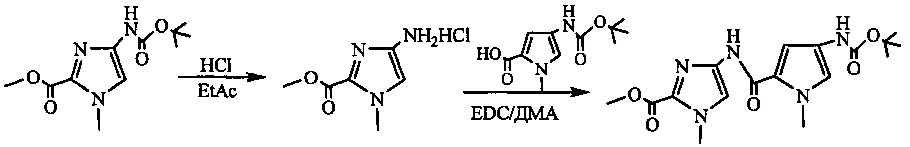
К метил 4-(трет-бутоксикарбониламино)-1-метил-1Н-имидазол-2-карбоксилату (2,50 г, 9,80 ммоль) в 30 мл EtAc добавили 6 мл HCl (конц.). После перемешивания в течение 45 минут, смесь разбавили этанолом и толуолом, концентрировали и упарили со смесью этанол/толуол (1:1, 3×50 мл) до сухости без дополнительной очистки. К сухому соединению добавили 4-(трет-бутоксикарбониламино)-1-метил-1Н-пиррол-2-карбоновую кислоту (2,35 г, 9,8 ммоль), EDC (5,60 г, 29,1 ммоль) и ДИПЭА (1,70 мл, 9,8 ммоль) и 80 мл ДМА. Смесь перемешивали под Ar в течение ночи, концентрировали, разбавили смесью ТГФ/EtAc (1:2, 150 мл) и промыли смесью 1 М раствора NaH2PO4/NaCl (конц.) и отдельно NaHCO3 (конц.). Слой органического растворителя отделили и высушили над MgSO4, отфильтровали, концентрировали и очистили SiO2 хроматографией, используя элюирующую смесь EtAc/ДХМ (от 1:25 до 1:15) для получения 2,72 г (73%) указанного в заголовке продукта.1Н NMR (DMF-d7) 10.27 (s, 1H), 9.08 (s, 1H), 7.41 (s, 1H), 7.32 (s, 1H), 7.07 (s, 1H), 4.10 (s, 3H), 3.93 (s, 3H), 3.84 (s, 3H), 1.47 (s, 9H);13C NMR 162.62, 161.20, 153.82, 145.32, 144.12, 132.56, 128.46, 124.39, 119.83, 79.51, 52.75, 36.06, 35.83, 28.88; MS m/z+ 400.2 (M+Na).
Метил 4-(4-(трет-бутоксикарбониламино)-1-метил-1Н-имидазол-2-карбоксамидо)-1-метил-1Н-имидазол-2-карбоксилат
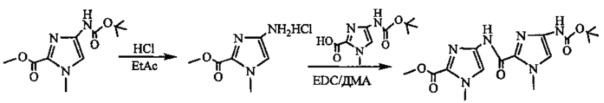
К метил 4-(трет-бутоксикарбониламино)-1-метил-1Н-имидазол-2-карбоксилату (2,50 г, 9,80 ммоль) в 30 мл EtAc добавили 6 мл HCl (конц.). После перемешивания в течение 30 минут, смесь разбавили этанолом и толуолом, концентрировали и упарили со смесью этанол/толуол (1:1, 3×50 мл) до сухости соединения без дополнительной очистки. К сухому соединению добавили 4-(трет-бутоксикарбониламино)-1-метил-1Н-имидазол-2-карбоновую кислоту (2,36 г, 9,8 ммоль), EDC (5,90 г, 30,7 ммоль), ДИПЭА (1,70 мл, 9,8 ммоль) и 80 мл ДМА. Смесь перемешивали под Ar в течение ночи, концентрировали, разбавили смесью ТГФ/EtAc (1:2, 150 мл) и промыли смесью 1 М раствора NaH2PO4/NaCl (конц.) и отдельно NaHCO3 (конц.). Слой органического растворителя отделили и высушили над MgSO4, отфильтровали, концентрировали и очистили SiO2 хроматографией, используя элюирующую смесь EtAc/ДХМ (от 1:25 до 1:15) для получения 2,65 г (71,5%) указанного в заголовке продукта.1Н NMR (DMSO) 11.17 (s, 1H), 10.48 (s, 1H), 7.58 (s, 1H), 7.32 (s, 1H), 4.01 (s, 3H), 3.94 (s, 3H), 3.92 (s, 3H), 1.45 (s, 9H);13C NMR 160.60, 157.30, 135.92, 135.45, 132.86, 126.12, 114.83, 79.50, 52.70, 35.58, 34.92, 28.08; MS m/z+ 401.8 (M+Na).
1-Метил-4-(1-метил-4-нитро-1Н-пиррол-2-карбоксамидо)-1Н-пиррол-2-карбоновая кислота
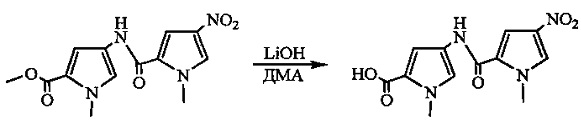
К метил 1-метил-4-(1-метил-4-нитро-1Н-пиррол-2-карбоксамидо)-1Н-пиррол-2-карбоксилату (2,0 г, 6,53 ммоль) в 50 мл ДМА добавили 2 г LiOH в 30 мл воды. Смесь перемешивали в течение ночи, концентрировали, разбавили водой, экстрагировали смесью EtAc/гексан (1:1). Водный раствор довели до pH 4,0 20%-ной H3PO4 для образования осадков. Осадки отфильтровали, промыли водой и высушили над P2O5 с вакуумом для получения 1,4 г (73%) указанного в заголовке продукта.1Н NMR (DMF-d7) 10.34 (br, 1H), 8.17 (s, 1H), 7.62 (s, 1H), 7.51 (s, 1H), 7.00 (s, 1H), 4.09 (s, 1H), 3.91 (s, 1H);13C NMR 158.47, 135.61, 129.11, 127.77, 123.65, 121.57, 121.50, 109.48, 108.52, 38.38, 37.05; MS m/z- 291.0 (M-H).
4-(4-(трет-Бутоксикарбониламино)-1-метил-1Н-пиррол-2-карбоксамидо)-1-метил-1H-имидазол-2-карбоновая кислота
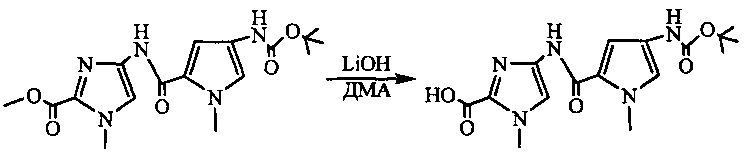
К метил 4-(4-(трет-бутоксикарбониламино)-1-метил-1Н-пиррол-2-карбоксамидо)-1-метил-1Н-имидазол-2-карбоксилату (2,0 г, 5,30 ммоль) в 50 мл ДМА добавили 2 г LiOH в 30 мл воды. Смесь перемешивали в течение ночи, концентрировали, разбавили водой, экстрагировали смесью EtAc/гексан (1:1). Водный раствор довели до pH 4,0 20%-ной H3PO4 для образования осадков. Осадки отфильтровали, промыли водой и высушили над P2O5 с вакуумом для получения 1,44 г (75%) указанного в заголовке продукта.1H NMR (DMSO) 10.41 (br, 1H), 9.07 (s, 1H), 7.48 (s, 1H), 6.97 (s, 1H), 6.88 (s, 1H), 3.92 (s, 1H), 3.81 (s, 1H), 1.47 (s, 9H);13C NMR 160.46, 158.42, 152.85, 145.21, 135.81, 129.11, 127.77, 122.39, 121.57, 113.58, 79.81, 36.06, 35.25,28.17; MS m/z- 362.1 (M-H).
Метил 4-(4-(4-(4-(трет-бутоксикарбониламино)-1-метил-1Н-пиррол-2-карбоксамидо)-1-метил-1Н-имидазол-2-карбоксамидо)-1-метил-1Н-пиррол-2-карбоксамидо)-1-метил-1Н-пиррол-2-карбоксилат
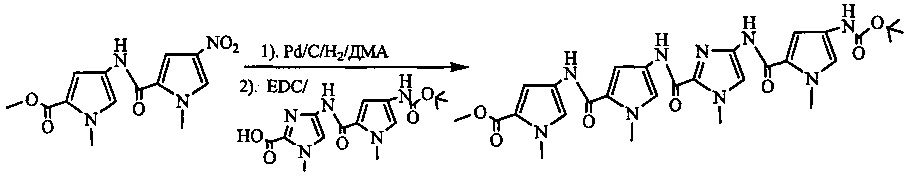
В колбу для гидрогенизации добавили метил 1-метил-4-(1-метил-4-нитро-1Н-пиррол-2-карбоксамидо)-1Н-пиррол-2-карбоксилат (1,0 г, 3,27 ммоль), 20 мл ТГФ, 305 мг 10% Pd/C (50% влажности) и 0,25 мл HCl (конц.). После вакуумирования в колбу закачали водород под давлением 50 фунтов/кв.дюйм и встряхивали в течение 4 часов. Смесь отфильтровали через целит, выпарили до сухости без дополнительной очистки. К высушенной смеси добавили 4-(4-(трет-бутоксикарбониламино)-1-метил-1Н-пиррол-2-карбоксамидо)-1-метил-1Н-имидазол-2-карбоновую кислоту (1,15 г, 3,16 ммоль), 10 мл ДМА, EDC (2,0 г, 10,4 ммоль) и ДИПЭА (0,70 мл, 4,02 ммоль). Смесь перемешивали под Ar в течение ночи, концентрировали, разбавили смесью гексан/EtAc (1:1, 10 мл) и 10 мл воды для образования осадков. Осадки отфильтровали, промыли 1 М NaH2PO4, 1 М NaHCO3 и водой, высушили над Р2О5 с вакуумом для получения 1,61 г (82%) указанного в заголовке продукта.1Н NMR (DMF-d7) 10.29 (s, 1Н), 10.20 (s, 1H), 10.12 (s, 1H), 9.08 (s, 1H), 7.58 (s, 1H), 7.47 (d, 1H, J=1.7 Hz), 7.26 (d, 1H, J=1.5 Hz), 7.15 (d, 1H, J=1.5 Hz), 6.98 (s, 1H), 6.91 (d, 1H, J=1.8 Hz), 6.86 (s, 1H), 3.97 (s, 3H), 3.82 (s, 3H), 3.73 (s, 3H), 3.56 (s, 3H), 1.45 (s, 9H);13C NMR 162.16, 160.05, 159.90, 157.20, 154.31, 137.88, 135.35, 124.56, 124.39, 124.24, 123.09, 120.09, 119.82, 115.32, 105.58, 102.27, 79.31, 51.51, 38.13, 36.01, 35.80, 35.08, 28.79; MS m/z+ 644,2 (M+Na).
Метил 1-метил-4-(1-метил-4-(1-метил-4-(1-метил-4-нитро-1Н-пиррол-2-карбоксамидо)-1Н-пиррол-2-карбоксамидо)-1Н-пиррол-2-карбоксамидо)-1Н-пиррол-2-карбоксилат
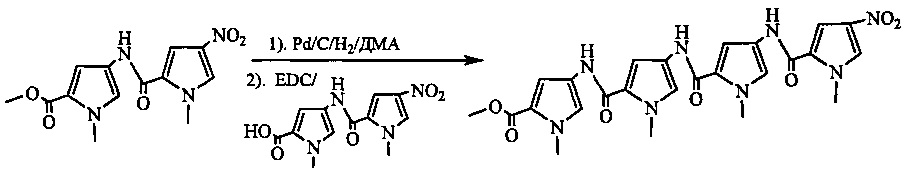
В колбу для гидрогенизации добавили метил 1-метил-4-(1-метил-4-нитро-1Н-пиррол-2-карбоксамидо)-1Н-пиррол-2-карбоксилат (2,0 г, 6,53 ммоль), 80 мл ДМА, 500 мг 10% Pd/C (50% влажности) и 0,4 мл HCl (конц.). После вакуумирования в колбу закачали водород под давлением 50 фунтов/кв.дюйм и встряхивали в течение 4 часов. Смесь отфильтровали через целит, выпарили до сухости без дополнительной очистки. К сухой смеси добавили 1-метил-4-(1-метил-4-нитро-1Н-пиррол-2-карбоксамидо)-1Н-пиррол-2-карбоновую кислоту (1,49 г, 5,10 ммоль), 30 мл ДМА, EDC (4,0 г, 20,8 ммоль) и ДИПЭА (1,0 мл, 5,75 ммоль). Смесь перемешивали под Ar в течение ночи, концентрировали, разбавили смесью гексан/EtAc (1:1, 10 мл) и 10 мл воды для образования осадков. Осадки отфильтровали, промыли 1 М NaH2PO4, 1 М NaHCO3 и водой, высушили над Р2О5 с вакуумом для получения 2,13 г (76%) указанного в заголовке продукта.1Н NMR (DMSO) 10.28 (s, 1Н), 10.25 (s, 1Н), 9.78 (s, 1Н), 8.18 (s, 1Н), 7.86 (s, 1Н), 7.52 (s, 1Н), 7.31 (d, 1H, J=1.7 Hz), 7.25 (s, 1Н), 7.23 (s, 1H), 7.17 (d, 1H, J=1.5 Hz), 6.98 (s, 1H), 6.71 (s, 1H), 4.02 (s, 3H), 3.94 (s, 3H), 3.83 (s, 3H), 3.73 (s, 3H), 3.56 (s, 3H), 1.47 (s, 9H);13C NMR 160.78, 158.93, 158.06, 157.81, 135.25, 127.28, 126.36, 123.78, 122.57, 121.91, 121.40, 120.94, 119.65, 110.73, 108.39, 107.34, 103.75, 80.81, 51.57, 39.74, 38.52, 38.22, 37.08, 28.63; MS m/z+ 573.2 (M+Na).
4-(4-(4-(4-(трет-бутоксикарбониламино)-1-метил-1Н-пиррол-2-карбоксамидо)-1-метил-1Н-имидазол-2-карбоксамидо)-1-метил-1Н-пиррол-2-карбоксамидо)-1-метил-1Н-пиррол-2-карбоновая кислота
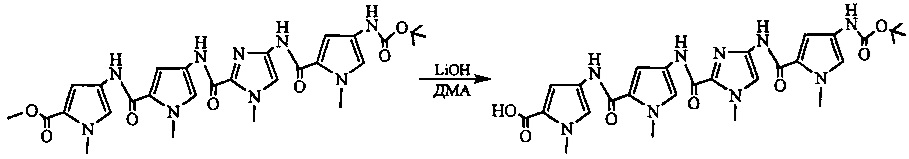
К метил 4-(4-(4-(4-(трет-бутоксикарбониламино)-1-метил-1Н-пиррол-2-карбоксамидо)-1-метил)-1Н-имидазол-2-карбоксамидо)-1-метил-1Н-пиррол-2-карбоксамидо)-1-метил-1Н-пиррол-2-карбоксилату (510 мг, 0,82 ммоль) в 10 мл ДМА добавили 0,8 г LiOH в 10 мл воды. Смесь перемешивали в течение ночи, концентрировали, разбавили водой, экстрагировали смесью EtAc/гексан (1:1). Водный раствор довели до рН 4,0 20%-ной Н3РО4 для образования осадков. Осадки отфильтровали, промыли водой и высушили над P2O5 с вакуумом для получения 363 мг (73%) указанного в заголовке продукта.1Н NMR (DMF-d7) 10.31 (s, 1Н), 10.18 (s, 1H), 10.11 (s, 1H), 9.10 (s, 1H), 7.58 (s, 1H), 7.54 (s, 1H), 7.41 (s, 1H), 7.33 (s, 1H), 7.21 (s, 1H), 7.10 (s, 1H), 7.06 (s, 1H), 4.10 (s, 1H), 3.98 (s, 1H), 3.95 (s, 1H), 3.93 (s, 1H), 1.47 (s, 9H);13C NMR 162.16, 160.05, 159.90, 157.20, 154.31, 137.88, 135.35, 124.56, 124.39, 123.51, 123.09, 121.76, 120.09, 119.83, 118.96, 115.32, 109.53, 105.58, 102.27, 79.32, 38.13, 36.02, 35.81, 34.88, 28.79; MS m/z- 606.2 (M-H).
S-3-(трет-бутоксикарбониламино)пропил этантиоат

К трет-бутил 3-гидроксипропилкарбамату (3,22 г, 18,37 ммоль) в 100 мл ДХМ при 0°С добавили тиолуксусную кислоту (2,0 мл, 26,73 ммоль) и трифенилфосфин (7,0 г, 26,73 ммоль) под Ar. После перемешивания при 0°С в течение 15 мин. добавили DIAD (6,0 мл, 28,93). Смесь перемешивали при 0°С в течение 2 часов, а затем при комнатной температуре в течение ночи. Смесь концентрировали, разбавили 120 мл смеси EtAc/гексан (1:2), отфильтровали через целит.Раствор промыли смесью NaHCO3 (конц.)/NaCl (конц.) и 1 М NaH2PO4 соответственно, высушили над MgSO4, отфильтровали, выпарили и очистили SiO2 хроматографией, используя элюирующую смесь EtAc/гексан (от 1:7 до 1:6) для получения 3,22 г (75%) указанного в заголовке соединения.1Н NMR (CDCl3) 3.09 (t, 2Н, J=6.5 Hz), 2.84 (t, 2Н, J=6.9 Hz), 2.27 (s, 3Н), 1.69 (dt, 2H, J=6.8, 13.5 Hz), 1.38 (s, 9H);13C NMR196.35, 156.16, 79.50, 39.26, 30.79, 30.24, 28.61, 26.44; MS m/z+ 256.0 (M+Na).
S-3-(трет-бутоксикарбонил(метил)амино)пропил этантиоат

К раствору NaH (0,57 г, 60%, 14,25 ммоль) в 20 мл ТГФ при 0°С добавили S-3-(трет-бутоксикарбониламино)пропил этантиоат (1,25 г, 5,36 ммоль) под Ar. После перемешивания при 0°С в течение 30 минут к смеси добавили MeI (1,0 мл, 16,06 ммоль). Перемешивание продолжали при 0°С в течение 2 часов, а затем при комнатной температуре в течение ночи. Смесь концентрировали, повторно растворили в 120 мл смеси EtAc/гексан (1:2), промыли 1 М NaH2PO4, NaCl (конц.), высушили над MgSO4, отфильтровали, вьшарили и очистили SiO2 хроматографией, используя элюирующую смесь EtAc/гексан (1:7) для получения 1,121 г (85%) указанного в заголовке соединения.1Н NMR (CDCl3) 3.69 (t, 2Н, J=7.3 Hz), 2.41 (t, 2Н, J=7.3 Hz), 2.39 (s, 3Н), 2.03 (s, 3Н), 1.76 (m, 2H), 1.47 (s, 9H);13C NMR 173.21, 153.39, 83.28, 43.67, 31.84, 28.26, 28.19, 27.11, 15.65; MS m/z+ 270.0 (M+Na).
S-3-(Метиламино)пропил этантиоата солянокислая соль
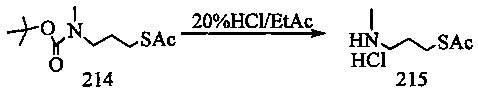
К S-3-(трет-бутоксикарбонил(метил)амино)пропил этантиоату (206 мг, 0,834 ммоль) в 4 мл EtAc добавили 1,0 мл HCl (конц.) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 1 часа, разбавили смесью этанол/толуол (6 мл, 1:1), вьпарили и упарили со смесью этанол/толуол (3×10 мл), кристаллизовали со смесью этанол/EtAc/гексан, отфильтровали и высушили под вакуумом для получения 135 мг (88%) указанного в заголовке продукта.1Н NMR (CDCl3) 9.70 (br, 1Н), 8.56 (br, 1Н), 3.42 (m, 2Н), 2.52 (m, 2Н), 2.35 (s, 3Н), 2.05 (s, 3Н), 1.88 (m, 2Н);13С NMR 174.64, 40.57, 31.57, 27.69, 20.94,15.62; MS m/z+ 170.0 (M+Na), 148.10 (М+Н).
трет-Бутил 2-(пиридин-2-илдисульфанил)этилкарбамат (217)

К раствору 2,2'-дитиолпиридина (3,97 г, 18,02 ммоль) в 100 мл метанола и 80 мл 1 М NaH2PO4, pH 6,8 добавили трет-бутил 2-меркаптоэтилкарбамат (1,00 г, 5,65 ммоль) в 50 мл метанола. Смесь перемешивали под Ar в течение ночи, концентрировали, экстрагировали дихлорметаном, высушили над MgSO4, отфильтровали, выпарили и очистили SiO2 хроматографией, используя элюирующую смесь EtAc/гексан (от 1:10 до 1:6) для получения 1,341 г (83%) указанного в заголовке соединения.1Н NMR (CDCl3) 8.39 (m, 1Н), 7.56 (m, 1H), 7.49 (m, 1H), 7.03 (m, 1H), 7.00 (m, 1H), 3.34 (m, 2H), 2.84 (m, 2H),1.37 (s, 9H);13C NMR 160.05, 159.39, 159.07, 149.87, 137.21, 120.78, 79.48, 39.58, 38.96, 28.57; MS m/z+ 309.2 (M+Na).
2-(пиридин-2-илдисульфанил)этанамин
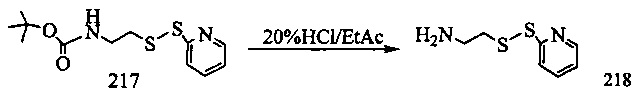
К трет-бутил 2-(пиридин-2-илсульфанил)этилкарбамату (1,06 г, 3,70 ммоль) в 16 мл EtAc добавили 4,0 мл HCl (конц.) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 0,5 часа, разбавили смесью этанол/толуол (6 мл, 1:1), выпарили и упарили со смесью этанол/толуол (3×10 мл), кристаллизовали со смесью этанол/EtAc/гексан, отфильтровали и высушили под вакуумом для получения 135 мг (88%) указанного в заголовке продукта.1Н NMR (CD3OD) 7.58 (m, 1Н), 7.47 (m, 1H), 7.06 (m, 1H), 6.83 (m, 1H), 3.34 (m, 2H), 3.02 (m, 2H);13C NMR 158.69, 149.07, 137.81, 122.48, 120.98, 39.52, 36.94; MS m/z+ 187.10 (M+H).
Метил 4-бромбутаноат (223)
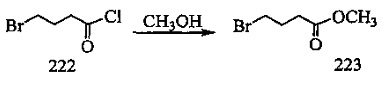
4-Бромбутаноил хлорид (3,1 мл, 25,28 ммоль) добавили к 15 мл сухого метанола при 0°С. Перемешивание продолжали при 0°С под Ar в течение 2 часов, а затем при комнатной температуре в течение ночи. Смесь выпарили, разбавили смесью EtAc/гексан (1:5), отфильтровали через гель SiO2 и выпарили для получения 4,50 г (99%) указанного в заголовке соединения.1Н NMR (CDCl3) 3.65 (s, 3Н), 3.43 (t, 2Н, J=6.5 Hz), 2.47 (t, 2Н, J=7.1 Hz), 2.13 (dt, 2H, J=6.7, 13.6 Hz);13C NMR 173.08, 51.84, 32.82, 32.34, 27.89; MS m/z+ 203.0 (M+Na).
(Z)-метил 4-(7-метокси-2',3'-бензо[e]-5-оксо-5,11а-дигидро-1Н-бензо[е]пирроло[1,2-а][1,4]диазепин-8-илокси)бутаноат
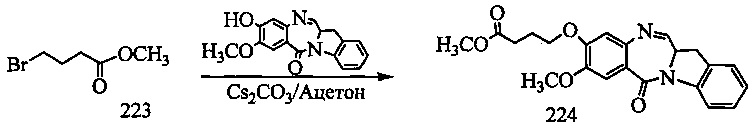
К (Z)-2,3-бензо-8-гидрокси-7-метокси-1Н-бензо[е]пирроло[1,2-а][1,4]диазепин-5(11аН)-ону (60 мг, 0,20 ммоль) в 4 мл ацетона добавили Cs2CO3 (90 мг, 0,28 ммоль), а затем добавили метил 4-бромбутаноат (50 мг, 0,27 ммоль). Смесь перемешивали под Ar в течение ночи, выпарили и очистили SiO2 хроматографией, используя элюирующую смесь EtAc/ДХМ (от 1:5 до 1:3) для получения 50,1 мг (63%) указанного в заголовке соединения.1Н NMR (CDCl3) 8.19 (d, 1Н, J=7.9 Hz), 7.80 (d, 1H, J=4.2 Hz), 7.48 (s, 1H), 7.19 (m, 2H), 7.03 (d, 1H, J=7.4 Hz), 6.77 (s, 1H), 4.41 (m, 1H), 3.88 (s, 3H), 3.64 (m, 2H), 3.62 (s, 3H), 3.42 (dd, 1H, J=3.4, 13.7 Hz), 2.50 (t, 2H, J=7.2 Hz), 2.12 (t, 2H, J=6.8 Hz);13C NMR, 173.64, 164.12, 163.24, 152.25, 148.41, 142.28, 140.34, 129.69, 128.39, 124.97, 120.85, 117.15, 112.15, 110.68, 68.08, 56.40, 55.18, 51.90, 32.84, 30.64, 24.50; MS m/z+ 187.10 (M+H). MS m/z+ 417.2 (M+Na), 435.2 (M+Na+H2O).
4-(7-метокси-2,3-бензо[e]-5-оксо-5,11а-дигидро-1Н-бензо[е]пирроло[1,2-а][1,4]диазепин-8-илокси)бутановая кислота
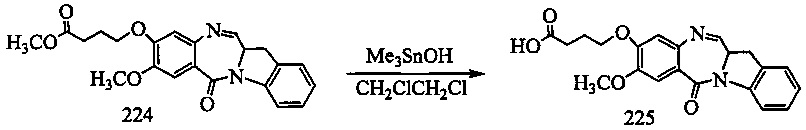
(Z)-метил 4-(7-метокси-2',3'-бензо[е]-5-оксо-5,11а-дигидро-1Н-бензо[е]пирроло[1,2-а][1,4]диазепин-8-илокси)бутаноат (41 мг, 0,104 ммоль) и гидроксид триметилолова (302 мг, 1,67 ммоль) в 15 мл дихлорэтана дефлегмировали при 85°С под Ar в течение ночи. Смесь промыли 1 М NaH2PO4, pH 3,5, высушили над MgSO4, отфильтровали, выпарили и очистили SiO2 хроматографией, используя элюирующую смесь EtAc/ДХМ/HCl (1:25:0,01%) для получения 30 мг (76%) указанного в заголовке соединения.1Н NMR (CDCl3) 8.18 (d, 1Н, J=7.9 Hz), 7.85 (m, 1Н), 7.46 (s, 1H), 7.20 (m, 2H), 7.04 (d, 1H, J=7.4 Hz), 6.81 (s, 1H), 4.40 (m, 1H), 3.86 (s, 3H), 3.63 (m, 2H), 3.23 (dd, 1H, J=10.2, 16.3 Hz), 2.52 (t, 2H, J=7.2 Hz), 2.12 (t, 2H, J=6.8 Hz);13C NMR, 173.64, 164.12, 163.24, 152.25, 148.41, 142.28, 140.34, 129.69, 128.39, 125.10, 120.85, 117.19, 112.15, 110.68, 67.94, 56.43, 55.18, 31.81, 30.64,24.21; MS m/z- 397.0 (M+H2O-H).
Метиловый эфир 4-{[4-({4-[4-(4-(7-метокси-2',3'-бензо[е]-5-оксо-5,11а-дигидро-1H-бензо[е]пирроло[1,2-а][1,4]диазепин-8-илокси)бутириламино]-1-метил-1H-пиррол-2-карбонил}амино)-1-метил-1H-имидазол-2-карбонил]амино}-1-метил-1H-пиррол-2-карбонил]-амино}-1-метил-1H-пиррол-2-карбоновой кислоты (226)
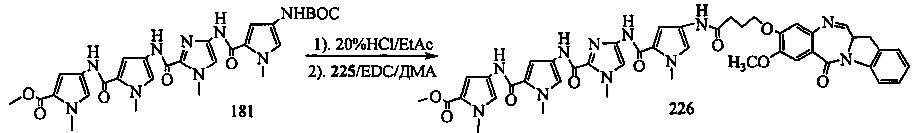
К метил 4-(4-(4-(4-(трет-бутоксикарбониламино)-1-метил-1Н-пиррол-2-карбоксамидо)-1-метил-1Н-имидазол-2-карбоксамидо)-1-метил-1Н-пиррол-2-карбоксамидо)-1-метил-1Н-пиррол-2-карбоксилату (15 мг, 0,024 ммоль) в 4 мл EtAc добавили 1,0 мл HCl (конц.). Смесь перемешивали при комнатной температуре в течение 0,5 часа, разбавили смесью этанол/толуол (6 мл, 1:1), вьшарили и упарили со смесью этанол/толуол (3×10 мл) и высушили под вакуумом. Твердое соединение использовали напрямую, без дополнительной очистки. К твердому веществу добавили 4-(7-метокси-2',3'-бензо[е]-5-оксо-5,11а-дигидро-1Н-бензо[е]пирроло[1,2-а][1,4]диазепин-8-илокси)бутановую кислоту (6 мг, 0,0155 ммоль), EDC (40 мг, 0,21 ммоль), ДИПЭА (4 мкл, 0,023 ммоль) и 1 мл ДМА. Смесь перемешивали под Ar в течение ночи, выпарили и очистили препаративной ВЭЖХ, колонка С18 (колонка Ф10 мм × 200 мм, скорость потока 9 мл/мин., градиентная система растворителей от соотношения растворителей А:В 75:25 на период 0-5 минут до 40:60 А:В через 15 минут, затем 20:80 А:В через 25 минут до 10:90 А:В через 30 минут. Растворитель А - вода, растворитель В - ацетонитрил/диоксан (1:2)) и лиофилизировали для получения твердого белого вещества (4,2 мг (30%) указанного в заголовке соединения). MS m/z- 900,3 (М+H2O-Н).
S-3-(4-(4-(4-(4-(трет-бутоксикарбониламино)-1-метил-1Н-пиррол-2-карбоксамидо)-1-метил-1Н-имидазол-2-карбоксамидо)-1-метил-1Н-пиррол-2-карбоксамидо)-N,1-диметил-1Н-пиррол-2-карбоксамидо)пропил этантиоат (227).
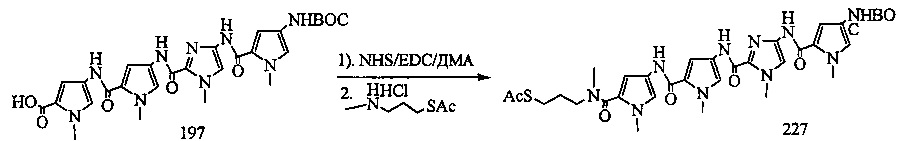
4-(4-(4-(4-(трет-бутоксикарбониламино)-1-метил-1Н-пиррол-2-карбоксамидо)-1-метил-1Н-имидазол-2-карбоксамидо)-1-метил-1Н-пиррол-2-карбоксамидо)-1-метил-1Н-пиррол-2-карбоновую кислоту (256 мг, 0,42 ммоль), NHS (60 мг, 0,52 ммоль) и EDC (500 мг, 2,60 ммоль) в 4 мл ДМА перемешивали под Ar в течение 2 часов, затем добавили S-3-(метиламино)пропил этантиоата солянокислую соль (76,5 мг, 0,42 ммоль) и продолжали перемешивать смесь в течение 24 часов, выпарили и очистили SiO2 хроматографией, используя элюирующую смесь ТГФ/ДХМ (от 1:5 до 1:4) для получения 198 мг (64%) указанного в заголовке соединения.1Н NMR (DMSO) 10.21 (s, 1Н), 10.09 (s, 1Н), 10.06 (s, 1Н), 9.08 (s, 1Н), 7.76 (d, 1Н, J=1.7 Hz), 7.52 (s, 1H), 7.28 (s, 1Н), 7.21 (d, 1H, J=1.7 Hz), 6.97 (s, 1H), 6.87 (s, 1H), 3.98 (s, 1H), 3.86 (s, 3H), 3.75 (s, 3H), 3.73 (s, 3H), 3.66 (m, 2H), 2.85 (s, 3H), 2.60 (m, 2H), 2.01 (s, 3H), 1.45 (s, 9H);13C NMR 173.31, 162.16, 160.05, 159.90, 157.20, 154.31, 137.88, 135.35, 124.56, 124.39, 123.51, 123.09, 121.76, 120.09, 119.83, 118.96, 115.32, 109.53, 105.58, 102.27, 79.32, 43.67, 38.13, 36.02, 35.81, 34.88, 31.84, 28.79, 28.26, 28.21, 27.01; MS m/z+ 759.2 (M+Na).
(Z)-S-3-(4-(4-(4-(4-(4-(7-метокси-2,3-бензо[е]-5-оксо-5,11а-дигидро-1Н-бензо[е]пирроло[1,2-а][1,4]диазепин-8-илокси)бутанамидо)-1-метил-1Н-пиррол-2-карбоксамидо)-1-метил-1Н-имидазол-2-карбоксамидо)-1-метил-1Н-пиррол-2-карбоксамидо)-N,1-диметил-1Н-пиррол-2-карбоксамидо)пропил этантиоат
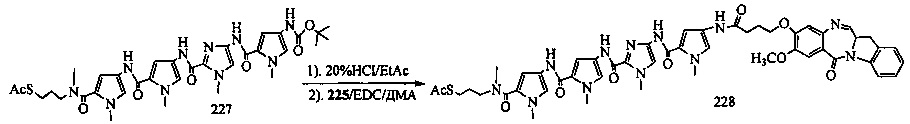
S-3-(4-(4-(4-(4-(трет-бутоксикарбониламино)-1-метил-1Н-пиррол-2-карбоксамидо)-1-метил-1Н-имидазол-2-карбоксамидо)-1-метил-1Н-пиррол-2-карбоксамидо)-N,1-диметил-1Н-пиррол-2-карбоксамидо)пропил этантиоат (227) (27 мг, 0,037 ммоль) перемешивали в 2 мл диоксана и 0,5 мл HCl (конц.) в течение 15 минут, разбавили смесью этанол/толуол (6 мл, 1:1), выпарили и упарили со смесью этанол/толуол (4×10 мл), кристаллизовали со смесью EtOH/ДХМ/гексан и высушили под вакуумом для получения 21 мг твердого вещества. Твердое соединение использовали напрямую, без дополнительной очистки. К твердому веществу добавили 4-(7-метокси-2,3-бензо[е]-5-оксо-5,11а-дигидро-1Н-бензо[е]пирроло[1,2-а][1,4]диазепин-8-илокси)бутановую кислоту (10 мг, 0,026 ммоль), EDC (101 мг, 0,52 ммоль), ДИПЭА (5 мкл, 0,028 ммоль) и 2 мл ДМА. Смесь перемешивали в течение ночи, выпарили, разбавили ДХМ, промыли смесью 1 М NaH2PO4/NaCl (конц.), pH 4,0, высушили над MgSO4, отфильтровали, вьшарили и очистили на препаративной колонке С18 ВЭЖХ (колонка Ф10 мм × 200 мм, скорость потока 9 мл/мин., градиентная система растворителей, начиная от соотношения растворителей А:В 75:25 на период 0-5 минут до 40:60 А:В через 15 минут, затем 20:80 А:В через 25 минут до 10:90 А:В через 30 минут. Растворитель А - вода, растворитель В - ацетонитрил/диоксан (1:2)) и лиофилизировали для получения твердого белого вещества 8,2 мг (32%) указанного в заголовке соединения. MS m/z- 1015,1 (М+H2O-Н), УФ ε(1=305 нм)=32800 M-1 см-1.
трет-Бутил 1-метил-5-(1-метил-2-(1-метил-5-(1-метил-5-(2-(пиридин-2-илдисульфанил)этилкарбамоил)-1Н-пиррол-3-илкарбамоил)-1Н-пиррол-3-илкарбамоил)-1Н-имидазол-4-илкарбамоил)-1Н-пиррол-3-илкарбамат (229)
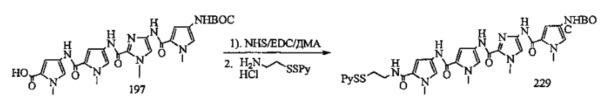
4-(4-(4-(4-(трет-бутоксикарбониламино)-1-метил-1Н-пиррол-2-карбоксамидо)-1-метил-1Н-имидазол-2-карбоксамидо)-1-метил-1Н-пиррол-2-карбоксамидо)-1-метил-1Н-пиррол-2-карбоновую кислоту (102 мг, 0,17 ммоль), 2-(пиридин-2-илдисульфанил)этанамина солянокислую соль (40 мг, 0,18 ммоль), ДИПЭА (30 мкл, 0,17 ммоль) и EDC (200 мг, 1,04 ммоль) в 2 мл ДМА перемешивали под Ar в течение 24 часов, выпарили и очистили SiO2 хроматографией, используя элюирующую смесь ТГФ/ДХМ (от 1:5 до 1:4) для получения 90 мг (68%) указанного в заголовке соединения.1Н NMR (DMSO) 10.93 (s, 1Н), 10.19 (s, 1H), 10.06 (s, 1Н), 9.03 (s, 1Н), 8.81 (m 1Н), 8.29 (m, 1Н), 8.03 (m, 1Н), 7.68 (s, 1H), 7.47 (s, 1Н), 7.28 (s, 1Н), 7.24 (s, 1H), 7.18 (m, 1Н), 6.87 (s, 1Н), 3.96 (s, 1Н), 3.86 (s, 3Н), 3.75 (s, 3Н), 3.73 (s, 3Н), 3.58 (m, 2Н), 2.48 (m, 2Н), 1.45 (s, 9Н); MS m/z+ 798.0 (М+Na), 776.0 (М+Н).
4-(4-(4-(7-метокси-2,3-бензо[е]-1-5-оксо-5,11а-дигидро-1Н-бензо[е]пирроло[1,2-а][1,4]диазепин-8-илокси)бутанамидо)-1-метил-1Н-пиррол-2-карбоксамидо)-1-метил-N-(1-метил-5-(1-метил-5-(метил(2-(пиридин-2-иддисульфанил)этил)карбамоил)-1Н-пиррол-3-илкарбамоил)-1Н-пиррол-3-ил)-1Н-имидазол-2-карбоксамид
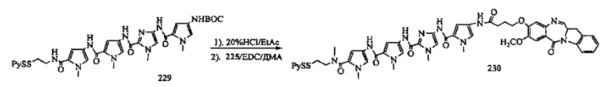
трет-Бутил 1-метил-5-(1-метил-2-(1-метил-5-(1-метил-5-(2-(пиридин-2-илдисульфанил)этилкарбамоил)-1Н-пиррол-3-илкарбамоил)-1Н-пиррол-3-илкарбамоил)-1Н-имидазол-4-илкарбамоил)-1Н-пиррол-3-илкарбамат (229) (30 мг, 0,038 моль) перемешивали в 2 мл диоксана и 0,5 мл HCl (конц.) в течение 15 минут, разбавили смесью этанол/толуол (6 мл, 1:1), выпарили и упарили со смесью этанол/толуол (4×10 мл), кристаллизовали со смесью EtOH/ДХМ/гексан и высушили под вакуумом для получения 19,5 мг твердого вещества. Твердое соединение использовали напрямую, без дополнительной очистки. К этому твердому веществу добавили 4-(7-метокси-2,3-бензо[е]-5-оксо-5,11а-дигидро-1Н-бензо[е]пирроло[1,2-а][1,4]диазепин-8-илокси)бутановую кислоту (10 мг, 0,026 ммоль), EDC (102 мг, 0,52 ммоль), ДИПЭА (5 мкл, 0,028 ммоль) и 2 мл ДМА. Смесь перемешивали в течение ночи, выпарили, разбавили ДХМ, промыли смесью 1 М NaH2PO4/NaCl (конц.), pH 4,0, высушили над MgSO4, отфильтровали, выпарили и очистили на препаративной колонке С18 ВЭЖХ (колонка Ф10 мм × 200 мм, скорость потока 9 мл/мин., градиентная система растворителей, начиная от соотношения растворителей А:В 75:25 на период 0-5 минут до 40:60 А:В через 15 минут, затем 20:80 А:В через 25 минут до 10:90 А:В через 30 минут. Растворитель А - вода, растворитель В - ацетонитрил/диоксан (1:2)) и лиофилизировали для получения твердого белого вещества, 7,5 мг (27%) указанного в заголовке соединения. MS m/z- 1050,0 (М+Н2О-Н), УФ ε(1=305 нм)=32855 M-1 см-1.
1-(4-(2-бромэтокси)фенил)этанон
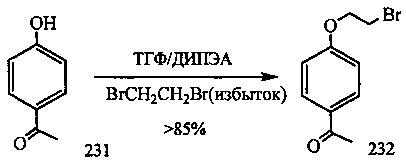
1-(4-гидроксифенил)этанон (8,2 г, 60,2 ммоль), карбонат калия (15,2 г, 110,1 ммоль) и KI (1,0 г, 6,0 ммоль) в 100 ДМФ перемешивали в течение 5 минут, затем добавили 1,2-дибромэтан (60 мл, 696,2 ммоль). Смесь перемешивали в течение ночи, вьшарили, разбавили смесью EtAc/гексан (1:1), промыли 0,1 М HCl/NaCl (конц.), высушили над MgSO4, отфильтровали, выпарили и очистили SiO2 хроматографией, используя элюирующую смесь EtAc/гексан (от 1:3 до 2:3) для получения 12,41 г (85,2%) указанного в заголовке соединения.1Н NMR (CDCl3) 7.87 (ddd, 2Н, J=2.8, 4.9, 9.7 Hz), 6.88 (ddd, 2Н, J=2.8, 4.9, 9.6 Hz), 4.29 (t, 2H, J=6.2 Hz), 3.59 (t, 2H, J=6.2 Hz);13C NMR 196.88, 162.11, 131.15, 130.54, 113.80, 68.06, 29.50, 26.62; MS m/z+ 264.80 (M+Na), 266.80 (M+2+Na).
(5-гидрокси-1,3-фенилен)диметанол
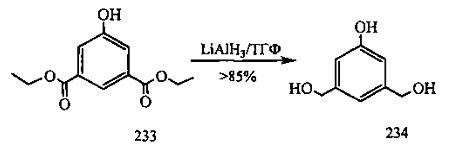
К 100 мл 2,0 M раствора LiAlH4 в ТГФ при 0°С в течение 15 минут добавили диметил 5-гидрокси изофталат (12,3 г, 58,5 ммоль) в 120 мл ТГФ под Ar. Смесь перемешивали при 0°С в течение 30 минут, а затем при комнатной температуре в течение ночи. Смесь погасили 20 мл метанола при 0°С и довели рН смеси до 5,0 добавлением H3PO4, отфильтровали через целит, выпарили и кристаллизовали со смесью эфир/гексан для получения 76,6 (85%) указанного в заголовке соединения.1Н ЯМР (ДМСО) 6,68 (с, 1Н), 6,61 (с, 2Н), 4,69 (с, 4Н); MS m/z+ 177,0 (М+Na).
1-(4-(2-(3,5-бис(гидроксиметил)фенокси)этокси)фенил)этанон (235)
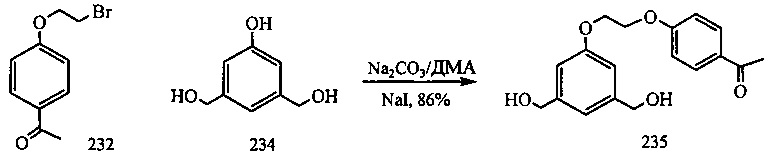
К перемешанному раствору (5-гидрокси-1,3-фенилен)диметанола (3,0, 20,0 ммоль), карбоната натрия (2,5 г, 29,0 ммоль) и йодида натрия (0,45 г, 2,9 ммоль) в 60 мл ДМА добавили 1-(4-(2-броэтокси)фенил)этанон (5,0, 20,57 ммоль). Смесь перемешивали в течение ночи, выпарили и очистили SiO2 хроматографией, используя элюирующую смесь EtAc/ДХМ (от 4:1 до 5:1) для получения 5,41 г (86%) указанного в заголовке соединения.1Н NMR (CD3OD) 7.99 (ddd, 2Н, J=2.8, 4.8, 9.8 Hz), 7.07 (ddd, 2Н, J=2.8, 4.7, 9.8 Hz), 6.94 (s, 1H), 6.89 (s, 2H), 4.58 (s, 4H), 4.42 (dd, 2H, J=2.2, 6.1 Hz), 4.37 (m, 2H), 2.55 (s, 3H);13C NMR 199.55, 164.66, 160.59, 144.72, 132.03, 131.74, 119.16, 115.64, 113.11, 68.36, 67.87, 65.20, 26.53; MS m/z+ 339.2 (M+Na).
1-(4-(2-(3,5-бис(бромметил)фенокси)этокси)фенил)этанон (236)
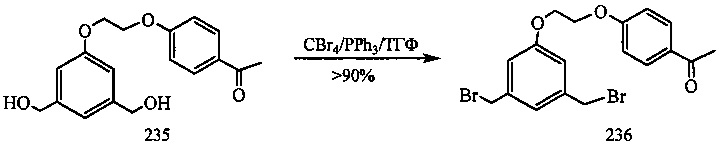
1-(4-(2-(3,5-бис(гидроксиметил)фенокси)этокси)фенил)этанон (0,216 г, 0,68 ммоль), тетрабромуглерод (0,50 г, 1,50 ммоль и PPh3 (0,40 г, 1,52 ммоль) перемешивали в 18 мл ТГФ под Ar в течение ночи и отфильтровали. Раствор концентрировали, очистили SiO2 хроматографией, используя элюирующую смесь EtAc/гексан (1:4) и кристаллизовали со смесью эфир/гексан для получения 277 мг (92%) указанного в заголовке соединения.1Н NMR (CDCl3) 7.94 (ddd, 2Н, J=2.7, 4.6, 9.6 Hz), 7.02 (s, 1Н), 6.98 (ddd, 2H, J=2.7, 4.6, 9.6 Hz), 6.91 (d, 2H, J=1.2 Hz), 4.62 (s, 4H), 4.35 (m, 4H), 2.55 (s, 3H);13C NMR 197.05, 162.63, 159.14, 139.98, 130.96, 130.85, 122.57, 155.60, 114.52, 66.78, 66.73, 32.88, 26.57; MS m/z+ 462.9 (M+Na), 464.9 (M+2+Na)
(R)-Метил пиперидин-2-карбоксилат (238)
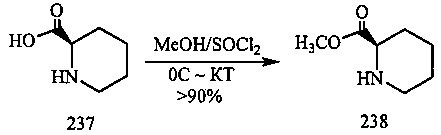
К (R)-пиперидин-2-карбоновой кислоте (5,00 г, 38,73 ммоль) в 150 мл сухого метанола при 0°С добавили тионилхлорид (5,2 мл, 71,28 ммоль) под Ar. Смесь перемешивали при 0°С в течение 30 минут, а затем при комнатной температуре в течение ночи, выпарили и кристаллизовали со смесью EtOH/гексан для получения 4,96 г (92%) указанного в заголовке продукта.1Н NMR (CD3OD) 3.67 (s, 3Н), 3.57 (m, 1H), 2.79 (m, 1H), 2.69 (m, 1H), 2.01 (m, 1H), 1.98 (m, 1H), 1.73 (m, 1H), 1.55-1.45 (m, 4H);13C NMR 171.22, 62.50, 51.35, 45.35, 29.52, 28.41, 23.82; MS m/z+ 144.0 (M+H).
(R)-Метил 1-(4-(бензилокси)-5-метокси-2-нитробензоил)пиперидин-2-карбоксилат (239)
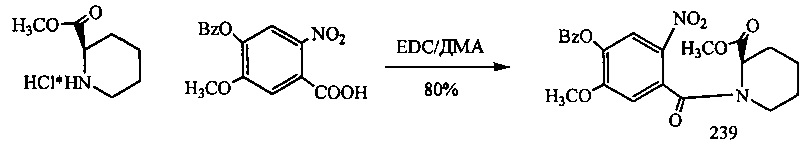
4-(Бензоилокси)-5-метокси-2-нитробензойную кислоту (1,70 г, 5,61 ммоль), (R)-метил пиперидин-2-карбоксилат (1,05 г, 5,84 ммоль), EDC (3,90 г, 20,31 ммоль) и ДИПЭА (1,0 мл, 5,75 ммоль) перемешивали в 20 мл ДМА в течение ночи. Смесь выпарили, разбавили ДХМ, промыли отдельно 1 М NaH2PO4/NaCl (конц.) и 0,1 М NaHCO3/NaCl (конц.). Слой органического растворителя отделили и высушили над MgSO4, отфильтровали, концентрировали и очистили хроматографией на SiO2, используя элюирующую смесь EtAc/ДХМ (1:15) для получения 1,772 г (74%) указанного в заголовке продукта.1Н NMR (CDCl3) 7.69 (s, 1Н), 7.40-7.38 (m, 2H), 7.35-7.27 (m, 3Н), 6.76 (d, 1H), 5.15 (s, 2H), 3.91 (s, 3H), 3.83 (s, 1H), 3.73 (s, 3H), 3.18 (m, 2H), 1.70 (m 2H), 1.47 (m, 4H);13C NMR 171.89, 171.33, 155.10, 154.78, 148.32, 135.59, 129.05, 128.74, 127.80, 109.66, 109.58, 109.41, 71.63, 56.92, 52.70, 52.19, 45.70, 39.92, 27.29, 26.35, 21.63; MS m/z+ 451.2 (M+Na).
(R)-1-(4-(бензилокси)-5-метокси-2-нитробензоил)пиперидин-2-карбальдегид

К (R)-Метил 1-(4-(бензоилокси)-5-метокси-2-нитробензоил)пиперидин-2-карбоксилату (1,50 г, 3,50 ммоль) в 30 мл смеси ДХМ/бензол 1:1 при -78°С добавили 7,5 мл 1,0 М гидрида диизобутилалюминия в толуоле под Ar в течение 10 минут. Смесь перемешивали при -78°С в течение 1 часа и погасили реакцию 0,5 мл метанола. Смесь разбавили 150 мл EtAc и 100 мл 0,2 М HCl. Слой органического растворителя отделили, а водный слой экстрагировали EtOAc (3×80 мл). Органические слои объединили, высушили над MgSO4, отфильтровали, концентрировали и очистили хроматографией на SiO2, используя элюирующую смесь EtAc/гексан (3:2) для получения 1,52 г (90%) указанного в заголовке продукта.1Н NMR (CDCl3), 9.60 (s, 1Н), 7.70 (s, 1H), 7.65-7.28 (m, 5Н), 6.78 (m, 1Н), 5.16 (s, 2Н), 3.92 (s, 3Н), 3.22, (m, 1H), 3.01 (m, 1Н), 2.20 (m, 1H), 1.84 (m, 1H), 1.65-1.40 (m, 4Н);13С NMR 200.24, 171.31, 155.13, 154.78, 148.41, 146.20, 137.57, 135.47, 129.03, 128.73, 127.31, 109.83, 109.41, 71.61, 64.50, 56.96, 45.98, 25.25, 23.42, 18.70; MS m/z+ 421.1 (М+Na).
(R,Z)-3-(бензилокси)-2-метокси-7,8,9,10-тетрагидробензо[е]пиридо[1,2-а][1,4]диазепин-12(6аН)-он
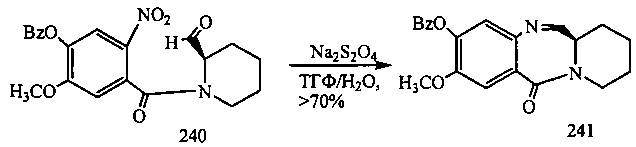
К раствору (R)-1-(4-(бензилокси)-5-метокси-2-нитробензоил)пиперидин-2-карбальдегиду (1,0 г, 2,51 ммоль) в смеси 25 мл ТГФ и 15 мл воды добавили Na2S2O4 (3,0 г, 17,25 ммоль). Смесь перемешивали в течение 4 часов, разбавили метанолом и диоксаном, вьшарили и упарили с диоксаном (3×60 мл) до сухости. Твердое вещество диспергировали смесью CH3OH/CH2Cl2 (1:1, 80 мл), отфильтровали и выпарили до получения твердого вещества. Полученное твердое вещество растворили в СН3ОН (100 мл) и добавили 0,4 мл HCl (конц.). Смесь перемешивали в течение 1 часа, нейтрализовали до рН 3,0 0,1 М NaHCO3, концентрировали и экстрагировали CH2Cl2 (4×60 мл). Органические слои объединили, промыли 1 М NaHCO3/NaCl (конц.), высушили над Na2SO4, отфильтровали, выпарили и очистили хроматографией на SiO2, используя элюирующую смесь EtAc/CH2Cl2 (1:3) для получения 615 мг (70%) указанного в заголовке продукта.1Н NMR (CDCl3), 7.81 (d, 1H, J=5.7 Hz), 7.38~7.23 (m, 6Н), 6.74 (s, 1Н), 5.12 (dd, 2H, J=2.3, 21.8 Hz), 4.18 (m, 1H), 3.88 (d, 3H), 3.69 (m, 1H), 3.15 (m, 1H), 1.99 (m, 1H), 1.87 (m, 1H), 1.79~1.65 (m, 4H);13C NMR 167.76, 163.31, 150.72, 148.48, 140.09, 136.46, 128.87, 128.28, 127.53, 121.77, 111.01, 71.02, 56.41, 49.84, 39.93, 24.76, 23.21, 18.62; MS m/z+ 373.2 (M+Na), 391.2 (M+Na+H2O), 405.3 (M+Na+CH3OH).
(R,Z)-3-Гидрокси-2-метокси-7,8,9,10-тетрагидробензо[е]пиридо[1,2-а][1,4]диазепин-12(6аН)-он (242)
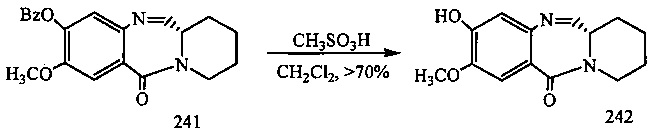
К (R,Z)-3-(бензилокси)-2-метокси-7,8,9,10-тетрагидробензо[е]пиридо[1,2-а][1,4]диазегган-12(6аН)-ону (241) (215 мг, 0,614 ммоль) в 25 мл CH2Cl2 при 0°С добавили 25 мл CH2SO3H. Смесь перемешивали при 0°С в течение 10 минут, а затем при комнатной температуре в течение 2 часов, разбавили CH2Cl2, нейтрализовали холодным 1,0 М NaHCO3, экстрагировали CH2Cl2, высушили над Na2SO4, отфильтровали, выпарили и очистили хроматографией на SiO2, используя элюирующую смесь СН3ОН/CH2Cl2 (1:15) для получения 122 мг (70%) указанного в заголовке продукта.1Н NMR (CDCl3), 7.75 (d, 1Н, J=5.7 Hz), 7.28 (s, 1Н), 6.70 (s, 1H), 4.08 (m, 1H), 3.83 (d, 3H), 3.61 (m, 1H), 3.08 (m, 1H), 1.91 (m, 1H), 1.81 (m, 1H), 1.71~1.55 (m, 4H);13C NMR 167.81, 163.46, 148.53, 145.71, 140.84, 121.23, 111.89, 111.39, 56.45, 49.83, 39.96, 24.71, 23.22, 18.60; MS m/z+ 283.7 (M+Na).
(5Z,5'Z,6aR,6а'R)-3,3'-(5-(2-(4-Ацетилфенокси)этокси)-1,3-фенилен)бис(метилен)бис(окси)бис(2-метокси-7,8,9,10-тетрагидробензо[е]пиридо[1,2-a][1,4]диазепин-12(6аН)-он) (243)
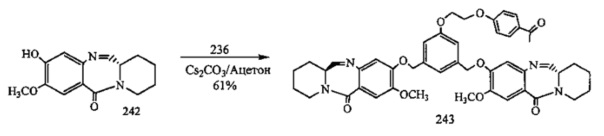
К перемешиваемому раствору (R,Z)-3-гидрокси-2-метокси-7,8,9,10-тетрагидробензо[е]пиридо[1,2-а][1,4]диазепин-12(6аН)-она (242) (42 мг, 0,16 ммоль), Cs2CO3 (100 мг, 0,307 ммоль), KI (3,2 мг, 0,018 ммоль) в 5 мл ацетона добавили 1-(4-(2-(3,5-бис(бромметил)фенокси)этокси)фенил)этанон (236) (36 мг, 0,081 ммоль). Смесь перемешивали в течение ночи, выпарили и очистили на колонке С18 препаративной ВЭЖХ (колонка Ф10 мм × 200 мм, скорость потока 9 мл/мин., градиентная система растворителей от соотношения растворителей А:В 80:20 на период 0-5 минут до 50:50 А:В через 15 минут, затем 30:70 А:В через 25 минут до 10:90 А:В через 30 минут. Растворитель А - вода, растворитель В - диоксан) и лиофилизировали для получения твердого белого вещества, 39,1 мг (61%) указанного в заголовке соединения.1Н NMR (DMF-d7), 8.30 (m, 2Н), 7.75 (d, 2Н, J=5.7 Hz), 7.30 (s, 2H), 7.01 (m, 2H), 6.71 (s, 2H), 6.68 (s, 1H), 6.63 (s, 2H), 5.21 (s, 4H), 4.43 (m, 2H), 4.32 (m, 2H), 4.08 (m, 2H), 3.83 (s, 6H), 3.61 (m, 2H), 3.08 (m, 2H), 2.56 (s, 3Н), 1.91 (m, 2H), 1.81 (m, 2H), 1.71~1.55 (m, 8H); MS m/z+ 823.2 (M+Na), 839.3 (M+K), 857.3 (M+K+H2O); MS m/z- 799.2 (M-H).
трет-Бутил 2-(4-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)бутаноил)гидразинкарбоксилат (245)
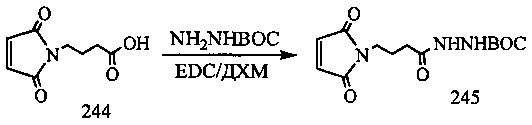
4-Малеимидомасляную кислоту (245 мг, 1,33 ммоль), трет-бутил гидразинкарбоксилат (201 мг, 1,52 ммоль) и EDC (400 мг, 2,08 ммоль) в 5 мл CH2Cl2 перемешивали в течение ночи под Ar, промыли 1 М NaH2PO4,/NaCl (конц.), высушили над MgSO4, отфильтровали, выпарили и очистили хроматографией на SiO2, используя элюирующую смесь МеОН/ДХМ (1:25) для получения 335 мг (85%) указанного в заголовке соединения.1Н NMR (CDCl3), 7.83 (br, 1Н), 6.65 (s, 2Н), 6.50 (br, 1Н), 3.58 (t, 2Н, J=6.3 Hz), 2.15 (t, 2Н, J=7.0 Hz), 1.90 (dt, 2H, J=6.8, 13.4 Hz), 1.40 (s, 9H);13C NMR 171.30, 155.61, 134.41, 82.00, 37.13, 31.38, 28.36, 24.95; MS m/z+ 320.2 (M+Na).
4-(2,5-Диоксо-2,5-дигидро-1Н-пиррол-1-ил)бутангидразида трифторуксуснокислая соль(246)
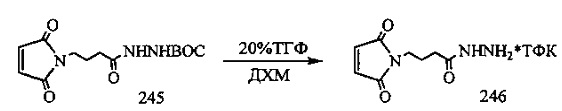
К трет-бутил 2-(4-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)бутаноил)-гидразинкарбоксилату (245) (200 мг, 0,673 ммоль) в 8 мл ДХМ добавили 2 мл трифторуксусной кислоты (ТФК). Смесь перемешивали в течение 45 минут, разбавили смесью этанол/толуол (8 мл, 1:1), выпарили и упарили со смесью этанол/толуол (3×10 мл), кристаллизовали со смесью этанол/EtAc/reKcaH, отфильтровали и высушили под вакуумом для получения 188 мг (90%) указанного в заголовке продукта.1Н NMR (CD3OD) 6.72 (s, 2Н), 5.39 (s, 0.6Н), 3.47 (t, 2Н, J=6.6 Hz), 2.20 (m, 2Н), 1.85 (m, 2Н);13С NMR 172.72, 135.56, 54.93, 39.20, 37.99, 25.20; MS m/z+ 197.9 (M+H).
(E)-N'-(1-(4-(2-(3,5-бис(((S,Z)-2-метокси-12-оксо-6a,7,8,9,10,12-гексагидробензо[e]пиридо[1,2-a][1,4]диазепин-3-илокси)метил)фенокси)этокси)фенил)этилиден)-4-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)бутангидразид (247)
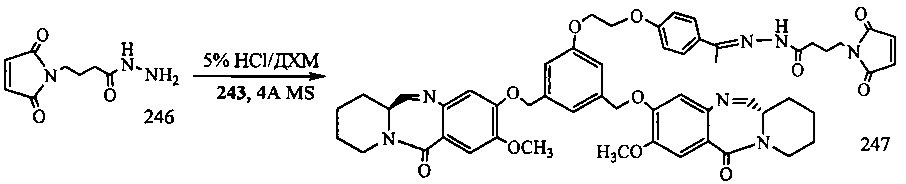
4-(2,5-Диоксо-2,5-дигидро-1Н-пиррол-1-ил)бутангидразида трифторуксуснокислую соль (246) (3 мг, 0,0096 ммоль), (5Z,5'Z,6aR,6а'R)-3,3'-(5-(2-(4-ацетилфенокси)этокси)-1,3-фенилен)бис(метилен)бис(окси)бис(2-метокси-7,8,9,10-тетрагидробензо[е]пиридо[1,2-а][1,4]диазепин-12(6аН)-он) (243) (7,5 мг, 0,0093 ммоль) и 50 мг 4 Å молекулярных сит перемешивали в 2 мл сухой 5% НАс в ДХМ (за день до этого высушили на 4 Å молекулярных ситах) в течение 2 часов, нейтрализовали 0,5 мл ДИПЭА, выпарили и очистили на колонке С18 препаративной ВЭЖХ (колонка Ф10 мм × 200 мм, скорость потока 9 мл/мин., градиентная система растворителей, начиная от соотношения растворителей А:В 80:20 на период 0-5 минут до 50:50 А:В через 15 минут, затем 30:70 А:В через 25 минут до 10:90 А:В через 30 минут. Растворитель А - вода, растворитель В - метанол/диоксан (2:1)) и лиофилизировали для получения твердого белого вещества, 5,6 мг (61%) указанного в заголовке соединения. MS m/z+ 1066,3 (М+2СН3ОН+Na).
Пример 13
Получение конъюгата huN901-IGN-07:
Для конъюгирования производных IGN выбрали антитело huN901, связывающееся с антигеном CD56. Раствор антитела huN901 концентрацией 3 мг/мл в водном буферном растворе, содержащем 0,05 М N-(2-гидроксиэтил)-пиперазин-N'-2-этансульфоновой кислоты (HEPES) и 2 мМ этилендиаминтетрауксусной кислоты (ЭДТА), pH 8, обработали 20-кратным избытком раствора NHS эфира IGN-07 в диметилацетамиде (ДМА), так что конечная концентрация ДМА в буферном растворе составила 10% об./об. Реакционную смесь перемешивали при комнатной температуре в течение 120 минут, а затем загрузили в гель-фильтрационную колонку Sephadex G25 (обессоливающая колонка HiPrep™ 26/10 GE# 17-5087-01), предварительно уравновешенную водным буферным раствором, содержащим 0,01 М цитрата натрия, 0,135 М хлорида натрия, pH 5,5. Фракции, содержащие конъюгированное антитело, собрали и объединили для получения продукта. Объединенную выборку подвергали диализу в течение ночи против такого же элюирующего буферного раствора (0,01 М цитрата натрия, 0,135 М хлорида натрия, рН 5,5) для дополнительной очистки продукта.
Полученный конъюгат анализировали методом спектрофотометрии, используя коэффициенты экстинкции, определенные для IGN-07 (ε330 нм=15231 М-1 см-1 и ε280 нм=26864 М-1 см-1). В среднем с одной молекулой антитела связалось 3,1 молекул IGN.

Получение исходного раствора IGN-10 ("соединение 51):
Приготовили свежий 0,004 М раствор IGN-10 в диметилацетамиде (ДМА) на основании молекулярного веса 975,14 г/моль. Исходный раствор проанализировали методом спектрофотометрии, используя справочный коэффициент экстинкции, определенный при 330 нм (ε330 нм=15500 М-1 см-1).
Пример 14
Получение конъюгата muB38.1-IGN-10:
Для конъюгирования производных IGN за счет дисульфидной связи выбрали антитело muB38.1, связывающееся с антигеном ЕрСАМ. Раствор антитела muB38.1 концентрацией 2,5 мг/мл в водном буферном растворе, содержащем фосфатную буферную соль (PBS), pH 7,4, обработали 120-кратным молярным избытком 1-гомоцистеин тиолактона в течение 12 часов при 37°С. Реакционную смесь загрузили в гель-фильтрационную колонку Sephadex G25 (обессоливающая колонка HiPrep™ 26/10 GE# 17-5087-01), предварительно уравновешенную раствором PBS pH 7,4. Фракции, содержащие антитело, собрали, объединили и проанализировали на содержание активного тиола, используя реактив Эльмана. Затем модифицированное антитело обработали 4-кратным молярным избытком IGN-10 (в ДМА) в пересчете на свободный тиол, и оставили реакционную смесь при комнатной температуре на 8 часов. Реакционную смесь загрузили в гель-фильтрационную колонку Sephadex G25 (обессоливающая колонка HiPrep™ 26/10 GE# 17-5087-01), предварительно уравновешенную водным буферным раствором, содержащим 0,01 М цитрата натрия, 0,135 М хлорида натрия, pH 5,5. Фракции, содержащие конъюгированное антитело, собрали и объединили для получения продукта. Объединенную выборку подвергали диализу в течение ночи против такого же элюирующего буферного раствора (0,01 М цитрата натрия, 0,135 М хлорида натрия, pH 5,5) для дополнительной очистки продукта.
Полученный конъюгат анализировали спектрофотометрически, используя коэффициенты экстинкции, определенные для IGN-10 (ε330 нм=15500 М-1 см-1 и ε280 нм=26864 М-1 см-1) и антитела muB38.1 (ε280 нм=215525 М-1 см-1). В среднем с одной молекулой антитела связалось 0,7 молекул IGN.
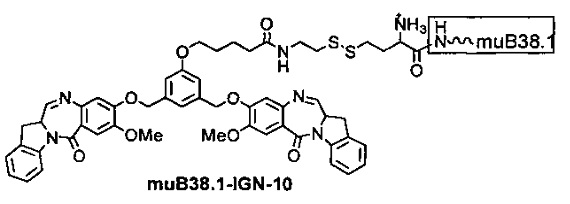
Пример 15
Анализ пробы ДНК для измерения связывания и алкилирования димера IGN с двуспиральной ДНК (дсДНК):
Условия реакции: дсДНК (конечная концентрация 25 мкМ) в 100 мМ TRIS, 1 мМ ЭДТА, pH 8, смешали с 3,7 молярных эквивалентов IGN-01 (соединение 18), IGN-02 (соединение 19) или IGN-09 (соединение 15), растворенных в ацетонитриле (конечная концентрация в ацетонитриле <2% по объему). Реакцию инкубировали при 15°С (ниже температуры плавления дсДНК) и в различные временные точки после смешивания ввели 10 мкл аликвоты на обращенно-фазовой ВЭЖХ.
Условия ВЭЖХ: колонка Waters Xbridge С8, 2,1×50 мм, буфер А: 100 мМ гексафторизопропанола, 16,3 мМ триэтиламина, в воде, буфер В: Метанол; 98% А → 100% В более, чем за 32 минуты, поток 0,25 мл/мин., нагрев колонки 60°С, обнаружение 260 нм. Для определения % сшивания в каждой временной точке инкубации использовали площади под кривой (AUC) для пика образца ДНК и пика полученного аддукта IGN/ДНК.
Ренатурация ДНК: односпиральную ДНК (Invitrogen) ренатурировали в дсДНК, используя амплификатор Peltier thermal cycler (РТС-200, MJ Research). 1 мМ ДНК в 100 мМ TRIS, 1 мМ ЭДТА, pH 8, нагрели до 80°С и постепенно охладили до 4°С в течение 90 минут, 15-градусными этапами. Полученную дсДНК хранили при 4°С до использования в анализах. В контрольных экспериментах IGN-01, IGN-02 и IGN-09 не образовали ковалентных аддуктов с односпиральной ДНК (осДНК).
Пример 16
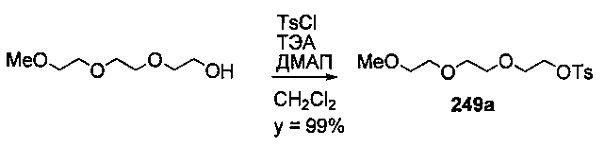
2-(2-(2-метоксиэтокси)этокси)этил 4-метилбензолсульфонат:
К перемешанному раствору 2-(2-(2-метоксиэтокси)этокси)этанола (1,64 г, 10 ммоль) в безводном дихлорметане (30 мл) добавили триэтиламин (2,53 г, 25 ммоль), тозилхлорид (3,81 г, 20 ммоль), а затем диметиламинопиридин (ДМАП) (0,061 г, 0,5 ммоль) при комнатной температуре. Смесь продолжали перемешивать в течение ночи, разбавили этилацетатом и отфильтровали для удаления твердого гидрохлорида триэтиламина. Твердое вещество промыли этилацетатом и выпарили фильтрат. Осадок разбавили этилацетатом и отфильтровали для удаления дополнительного осадка. Фильтрат выпарили для получения жидкого неочищенного продукта. Его очистили силикагелевой хроматографией (дихлорметан/метанол) для получения соединения 249а в виде маслянистой жидкости (3,16 г, выход = 99%).1Н NMR (400 Hz, CDCl3): δ 7.81 (d, J=8.0 Hz, 2H), 7.35 (d, J=8.0 Hz, 2H), 4.17 (t, J=3.2 Hz, 2H), 3.70 (t, J=4.8 Hz, 2H), 3.64-3.60 (m, 6H), 3.54 (t, J=4.8 Hz, 2H), 3.38 (s, 3H), 2.46 (s, 3H);13C NMR (400 Hz, CDCl3): δ 144.7, 133.0, 129.8, 127.9, 71.9, 70.7, 70.52, 70.50, 69.2, 68.6, 59.0, 21.6; MS (m/z): найдено 341,1 (M+Na)+.
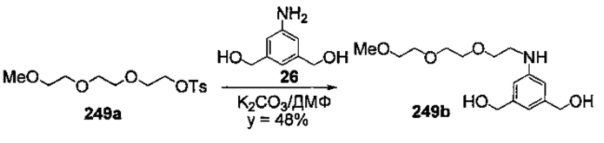
(5-(2-(2-(2-метоксиэтокси)токси)этиламино)-1,3-фенилен)диметанол:
К смеси тозилата 249а (1,85 г, 5,81 ммоль) и анилинового соединения 26 (1,78 г, 11,6 ммоль) в безводном ДМФ (9 мл) добавили безводный карбонат калия (1,61 г, 11,6 ммоль). Смесь нагрели до 85°C и перемешивали при этой температуре в течение ночи. Раствор охладили до комнатной температуры и разбавили дихлорметаном. Смесь отфильтровали через целит и промыли твердое вещество дихлорметаном. Фильтрат выпарили и разбавили осадок дихлорметаном, снова отфильтровали для удаления дополнительного твердого вещества. Фильтрат выпарили и очистили осадок силикагелевой хроматографией (дихлорметан/метанол) для получения соединения 249b в виде светло-желтой маслянистой жидкости (835 мг, выход = 48%).1H NMR (400 Hz, CDCl3): δ 6.60 (s, 1H), 6.47 (s, 2H), 4.48 (s, 4H), 4.31 (bs, 1H), 3.66-3.59 (m, 8H), 3.55-3.52 (m, 2H), 3.36 (s, 3H), 3.24 (t, J=4.8 Hz, 2H);13C NMR (400 Hz, CDCl3): δ 148.5, 142.4, 114.6, 110.7, 71.8, 70.4, 70.3, 70.1, 69.4, 64.9, 58.9, 43.5; MS (m/z): найдено 322.2 (M+Na)+.
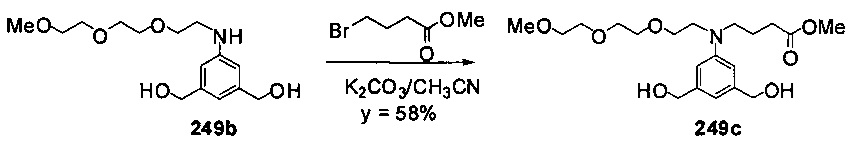
Соединение 249c (связыващая группа IGN-14):
К раствору соединения 249b (319 мг, 1,07 ммоль) и метил 4-бромбутирата (248 мг, 1,37 ммоль) в безводном ацетонитриле (5 мл) добавили безводный карбонат калия (177 мг, 1,28 ммоль). Смесь перемешивали и нагревали с дефлегмацией (масляная баня 86°С) в течение ночи. Смесь охладили до комнатной температуры и разбавили дихлорметаном. Смесь отфильтровали через целит и выпарили фильтрат. Осадок очистили силикагелевой хроматографией (дихлорметан/метанол) для получения соединения 249с (связывающая группа IGN-14) в виде бесцветной маслянистой жидкости (246 мг, выход = 58%).1Н NMR (400 Hz, CDCl3): δ 6.69 (s, 2Н), 6.66 (s, 1H), 4.64 (s, 4H), 3.71 (s, 3Н), 3.64-3.62 (m, 8H), 3.57-3.54 (m, 4H), 3.40-3.38 (m, 5H), 2.38 (t, J=7.2 Hz, 2H), 1.93 (p, J=7.2 Hz, 2H); MS (m/z): найдено 422.3 (M+Na)+.

Соединение 249d (IGN-14-OMe):
К перемешанному раствору соединения 249 с (120 мг, 0,3 ммоль) в безводном дихлорметане (3 мл) добавили триэтиламин (146 мкл, 1,05 ммоль). Смесь охладили до -10°С и медленно, в течение 15 минут, добавили метансульфонилхлорид (70 мкл, 0,9 ммоль). Раствор продолжали перемешивать при температуре между -10°С и -5°С в течение 60 минут и погасили добавлением водно-ледяной смеси. Смесь разбавили этилацетатом и промыли холодной водой. Органический слой высушили над безводным сульфатом натрия, отфильтровали, вьшарили и отогнали под высоким вакуумом для получения мезилата в виде бесцветной маслянистой жидкости. Мезилат перенесли в 10 мл круглодонную колбу с этилацетатом, вьшарили и отогнали под высоким вакуумом. Добавили соединение 8 (221 мг, 0,75 ммоль), а затем безводный ДМФ (2 мл) и безводный карбонат калия (207 мг, 1,5 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Разбавили дихлорметаном и промыли насыщенным солевым раствором. Органический слой высушили над безводным сульфатом натрия, отфильтровали и вьшарили. Осадок очистили препаративной обращенно-фазовой ВЭЖХ (колонка С18, элюированная смесью CH3CN/H2O) для получения соединения 249d (IGN-14-OMe) в виде твердого светло-желтого вещества (98 мг, выход = 34%).1Н NMR (400 Hz, CDCl3): δ 8.29 (d, J=8.0 Hz, 2H), 7.86 (d, J=4.4 Hz, 2H), 7.58 (s, 2H), 7.31-7.26 (m, 4H), 7.12 (t, J=8.0 Hz, 2H), 6.88 (s, 2H), 6.83 (s, 1H), 6.76 (s, 2H), 5.18 (dd, J1=23.2 Hz, J2=12.4 Hz, 4H), 4.49 (dt, J1=10.8 Hz, J2=4.4 Hz, 2H), 3.99 (s, 6H), 3.73-3.52 (m, 19H), 3.40-3.37 (m, 5H), 2.35 (t, J=7.2 Hz, 2H), 1.90 (p, J=7.2 Hz, 2H);13C NMR (400 Hz, CDCl3): δ 173.7, 164.9, 163.2, 151.1, 148.5, 148.4, 142.1, 140.2, 137.8, 129.7, 128.2, 124.9, 120.7, 117.0, 113.8, 112.0, 111.4, 110.4, 72.0, 71.3, 70.7, 70.6, 68.6, 59.1, 56.3, 55.0, 51.7, 50.7, 32.7, 31.3, 22.4; MS (m/z): найдено 974.6 (M+Na)+, 992.7 (M+H2O+Na)+, 1010.7 (M+2H2O+Na)+, 950.3 (M-H)-, 1022.3 (M+4H2O-H)-.
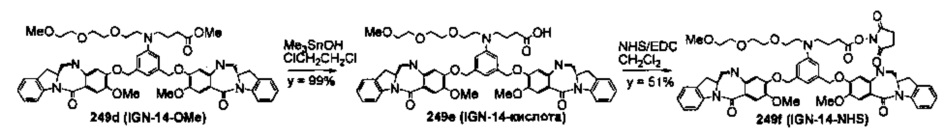
Соединение 249f (IGN-14-NHS):
К раствору соединения 249d (105 мг, 0,11 ммоль) в безводном 1,2-дихлорэтане (2 мл) добавили гидроксид триметилолова (299 мг, 1,65 ммоль). Смесь нагрели до 80°С и перемешивали в течение ночи. Смесь охладили до комнатной температуры, разбавили дихлорметаном и промыли смесью насыщенного раствора хлорида натрия и 5% соляной кислоты (~1 мл), затем насыщенным солевым раствором. Органический слой высушили над безводным сульфатом натрия, отфильтровали и выпарили. Осадок пропустили через короткую силикагелевую колонку и элюировали смесью дихлорметан/метанол для удаления избытка гидроксида триметилолова. Фракции продукта выпарили и отогнали под высоким вакуумом для получения кислоты 249е в виде твердого желтоватого вещества (102 мг, выход = 99%). MS (m/z): найдено 936,1 (М-Н)-, 960,3 (М+Na)+. Затем соединение 249е растворили в безводном дихлорметане (1 мл). Затем добавили N-гидроксисукцинимид (NHS), 37,5 мг, 0,326 ммоль) и N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорид (EDC, 62,5 мг, 0,326 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и разбавили дихлорметаном, промыли насыщенным солевым раствором и высушили над безводным сульфатом натрия. Отфильтровали, выпарили и очистили осадок препаративной обращенно-фазовой ВЭЖХ (колонка С18, элюированная смесью ацетонитрил/вода). Фракции продукта объединили и экстрагировали дихлорметаном. Органические слои высушили над безводным сульфатом натрия, отфильтровали, выпарили и отогнали под высоким вакуумом для получения соединения 249f (IGN-14-NHS) в виде твердого светло-желтоватого вещества (57,8 мг, выход = 51%).1H NMR (400 Hz, CDCl3): δ 8.28 (d, J=7.6 Hz, 2H), 7.86 (d, J=4.4 Hz, 2H), 7.58 (s, 2H), 7.31-7.27 (m, 4H), 7.12 (t, J=7.6 Hz, 2H), 6.87 (s, 2H), 6.81 (s, 1H), 6.74 (s, 2H), 5.23 (dd, J1=26.4 Hz, J2=12.4 Hz, 4H), 4.49 (dt, J1=10.8 Hz, J2=4.4 Hz, 2H), 4.00 (s, 6H), 3.73-3.47 (m, 18H), 3.37 (s, 3H), 2.79-2.74 (m, 4H), 2.59 (t, J=7.2 Hz, 2H), 1.97 (p, J=7.2 Hz, 2H); MS (m/z): найдено 1057.4 (M+Na)+.
Пример 17
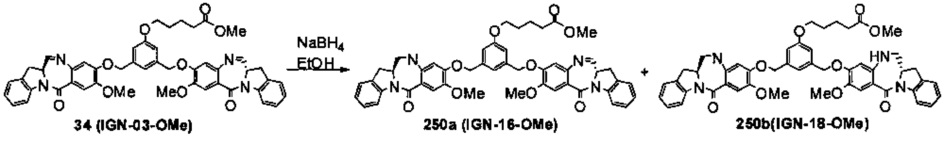
Соединения 250a (IGN-16-OMe) и 250b QGN-18-OMe):
К перемешанному раствору соединения 34 (111 мг, 0,135 ммоль) в абсолютном этаноле (1,0 мл) и безводном дихлорметане (0,5 мл) добавили боргидрид натрия (1,0 мг, 0,027 ммоль) при 0°С. Водно-ледяную баню убрали через 30 минут, а смесь продолжали перемешивать при комнатной температуре в течение 3 часов. Реакцию погасили добавлением насыщенного раствора хлорида аммония и разбавили дихлорметаном. Смесь разделили и промыли органический слой насыщенным солевым раствором, высушили над безводным сульфатом натрия и отфильтровали. Растворители удалили при пониженном давлении и очистили осадок препаративной обращенно-фазовой ВЭЖХ (колонка С18, элюированная смесью ацетонитрил/вода) для получения соединений 250а (IGN-16-OMe, 6,6 мг) и 250b (8,0 мг) в виде твердых белых веществ. 250а: MS (m/z), найдено 845,3 (М+Na)+, 863,3 (М+H2O+Na)+. 250b:1Н NMR (400 Hz, CDCl3), δ 8.34 (d, J=8.0 Hz, 2H), 7.49 (s, 2H), 7.27-7.03 (m, 6H), 6.89-6.87 (m, 3H), 6.05 (s, 2H), 4.96 (dd, J1=20.8 Hz, J2=12.8 Hz, 4H), 4.40-4.34 (m, 2H), 3.94-3.91 (m, 2H), 3.87 (s, 6H), 3.67 (s, 3H), 3.53-3.42 (m, 6H), 2.78 (dd, J1=16.8 Hz, J2=4.0 Hz, 2H), 2.38-2.37 (m, 2H), 1.79-1.77 (m, 4H); MS (m/z), найдено 847,3 (M+Na)+.
Пример 18
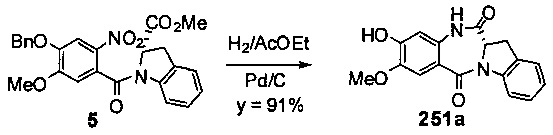
Соединение 251а:
К перемешанному раствору соединения 5 (840 мг, 1,82 ммоль) в этилацетате (10 мл) добавили палладий на древесном угле (10%, 193 мг, 0,182 ммоль). Колбу быстро вакуумировали и заместили колбой Н2. Смесь продолжали гидрогенировать в течение ночи и отфильтровали через целит. Твердое вещество промыли метанолом и обработали фильтрат 5% соляной кислотой (0,1 мл). Раствор десорбировали при пониженном давлении и очистили осадок силикагелевой хроматографией (дихлорметан/метанол) для получения соединения 251а в виде твердого белого вещества (512 мг, выход = 91%).1Н NMR (400 Hz, CDCl3), δ 8.21 (d, J=8.0 Hz, 1H), 8.09 (bs, NH), 7.53 (s, 1H), 7.31-7.25 (m, 2H), 7.12 (t, J=7.6 Hz, 1H), 6.61 (s, 1H), 6.22 (bs, 1H), 4.73 (dd, J1=10.4 Hz, J2=2.8 Hz, 1H), 4.07 (dd, J1=16.4 Hz, J2=2.4 Hz, 1H), 3.98 (s, 3H), 3.34 (dd, J1=16.4 Hz, J2=10.4 Hz, 1H); MS (m/z), найдено 333,1 (M+Na)+, 308,9 (M-H)-.

Соединение 251b (IGN-17-OMe):
К раствору соединения 38 (0,165 ммоль, полученного из 44 мг соединения 30 по методике, описанной в примере 6) и 251а (128 мг, 0,413 ммоль) в безводном ДМФ (1,5 мл) добавили безводный карбонат калия (114 мг, 0,825 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и разбавили дихлорметаном, промыли насыщенным солевым раствором и высушили над безводным сульфатом натрия и сульфатом магния. Смесь отфильтровали, выпарили и очистили часть осадка препаративной обращенно-фазовой ВЭЖХ (колонка С18, элюированная смесью ацетонитрил/вода) для получения 1,9 мг соединения 251b в виде твердого белого вещества. Остаток осадка очистили препаративной тонкослойной хроматографией (дихлорметан/метанол, 12:1) для получения 36,8 мг продукта в виде твердого белого вещества. В целом было выделено 38,7 мг соединения 251b (IGN-17-OMe) (выход = 28%).1Н,1Н NMR (400 Hz, CDCl3): δ 8.61 (s, 2H), 8.15 (d, J=8.0 Hz, 2H), 7.48 (s, 2H), 7.25 (d, J=7.6 Hz, 2H), 7.20 (t, J=7.6 Hz, 2H), 7.07 (t, J=7.6 Hz, 2H), 6.73 (s, 1H), 6.69 (s, 2H), 6.58 (s, 2H), 5.02 (dd, J1=17.6 Hz, J2=13.2 Hz, 4H), 4.66 (dd, J1=10.4 Hz, J2=2.8 Hz, 2H), 4.00 (dd, J1=16.4 Hz, J2=2.4 Hz, 2H), 3.90 (s, 6H), 3.68 (s, 3H), 3.39-3.23 (m, 4H), 2.89 (s, 3H), 2.44-2.30 (m, 2H), 1.91-1.92 (m, 2H);13C NMR (400 Hz, CDCl3): δ 174.5, 169.1, 164.2, 151.6, 149.6, 146.9, 141.2, 137.3, 130.6, 129.4, 127.5, 124.9, 124.8, 119.6, 117.1, 114.2, 112.5, 110.9, 106.0, 71.4, 58.0, 56.2, 51.9, 51.7, 38.3, 31.1, 28.2, 21.8; MS (m/z), найдено 874,3 (M+Na)+, 850,2 (M-H)-.
Пример 19
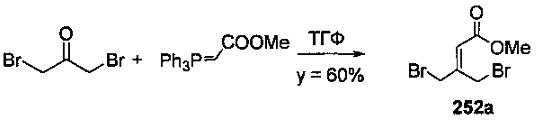
Соединение 252a:
Смесь 1,3-дибромацетона (0,863 г, чистота 75%, 3,0 ммоль) и метил (трифенилфосфоранилиден)ацетата (1,505 г, 4,5 ммоль) в безводном ТГФ (15 мл) нагревали с дефлегматором в течение 4,5 часов. Раствор охладили до комнатной температуры и выпарили. Осадок очистили силикагелевой хроматографией (гексаны/этилацетат) для получения соединения 252а в виде бесцветной жидкости (485 мг, выход = 60%).1Н NMR (400 Hz, CDCl3): δ 6.06 (s, 1Н), 4.76 (s, 2H), 4.19 (s, 2H), 3.79 (s, 3Н);13C NMR (400 Hz, CDCl3): δ 165.1, 150.4, 121.3, 51.8, 33.6, 25.5.
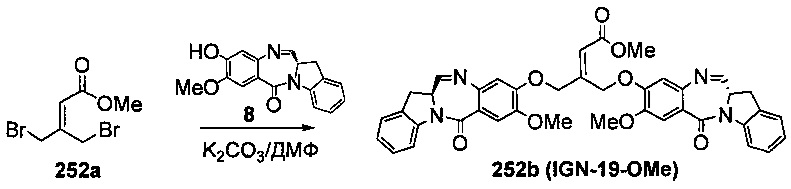
Соединение 252b (IGN-19-OMe):
Смесь соединения 252a (32 мг, 0,118 ммоль), мономера 8 (87 мг, 0,294 ммоль) и безводного карбоната калия (49 мг, 0,353 ммоль) в безводном ДМФ (1 мл) перемешивали при комнатной температуре в течение ночи. Смесь разбавили дихлорметаном, промыли насыщенным солевым раствором и высушили над безводным сульфатом натрия. Раствор отфильтровали, выпарили и очистили силикагелевой хроматографией (дихлорметан/метанол) для получения 105 мг соединения 252b, смешанного с побочными продуктами, в виде желтоватой пены. Часть продуктов дополнительно очистили препаративной обращенно-фазовой ВЭЖХ (колонка С18, элюированная смесью ацетонитрил/вода) для получения 10 мг соединения 252b (IGN-19-OMe) в виде твердого белого вещества. MS (m/z): найдено 721.2 (М+Na)+, 739.2 (М+H2O+Na)+, 757.2 (М+2H2O+Na)+, 697.1 (М-Н)-, 769.1 (М+4H2O-Н)-.
Пример 20
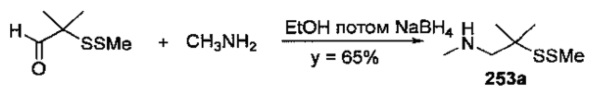
Соединение 253а:
К раствору 2-(метилдитио)-изобутиральдегида (690 мг, 4,59 ммоль) в абсолютном спирте (15 мл) добавили метиламин (629 мкл, 33% вес, 5,05 ммоль). Смесь перемешивали при комнатной температуре в течение четырех часов и охладили до 0°С, а затем добавили боргидрид натрия (174 мг, 4,59 ммоль). Через один час реакцию погасили несколькими каплями холодной 5% соляной кислоты, а затем подщелочили насыщенным раствором бикарбоната натрия. Смесь разбавили дихлорметаном и промыли насыщенным солевым раствором. Органический слой высушили над безводным сульфатом натрия, отфильтровали и выпарили при пониженном давлении. Осадок очистили силикагелевой хроматографией (0,2% триэтиламина в смеси дихлорметан/метанол) для получения летучего соединения 253а в виде светло-желтой жидкости (491 мг, выход = 65%).1Н NMR (400 Hz, CDCl3): δ 2.61 (s, 2Н), 2.45 (s, 3Н), 2.39 (s, 3Н), 1.32 (s, 6Н), 1.20 (s, NH);13C NMR (400 Hz, CDCl3): δ 61.2, 51.7, 37.2, 26.5, 25.3; MS (m/z): найдено 166,0 (M+H)+.

Соединение 253b:
Смесь соединения 249c (117 мг, 0,293 ммоль) и гидроксида триметилолова (794 мг, 4,39 ммоль) в безводном 1,2-дихлорэтане (1,5 мл) нагрели до 80°С и перемешивали в течение ночи. Смесь охладили до комнатной температуры, разбавили дихлорметаном и промыли смесью насыщенного раствора хлорида натрия и 5% соляной кислоты (~1 мл), затем насыщенным солевым раствором. Органический слой высушили над безводным сульфатом натрия, отфильтровали и выпарили. Осадок очистили силикагелевой хроматографией (дихлорметан/метанол) для получения кислоты 253b в виде маслянистой жидкости (93,9 мг, выход = 99%).1Н NMR (400 Hz, CDCl3): δ 6.62 (s, 2Н), 6.57 (s, 1Н), 4.50 (s, 4H), 3.63-3.54 (m, 8H), 3.53-3.46 (m, 4H), 3.36-3.31 (m, 5H), 2.29 (t, J=6.8 Hz, 2H), 1.83 (p, J=6.8 Hz, 2H);13C NMR (400 Hz, CDCl3): δ 177.0, 148.2, 142.4, 113.8, 110.1, 71.9, 70.7, 70.6, 70.4, 68.8, 65.2, 59.0, 50.8, 50.7, 31.4, 22.3; MS (m/z): найдено 384,2 (M-H)-, 408,4 (M+Na)+.
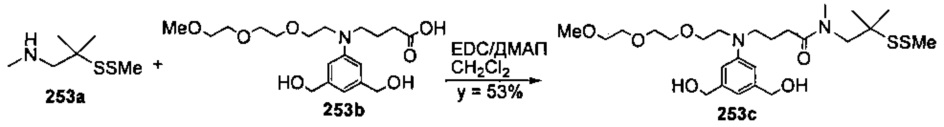
Соединение 253c:
К раствору амина 253a (44,3 мг, 0,268 ммоль) и карбоновой кислоты 253b (93,3 мг, 0,244 ммоль) в безводном дихлорметане (1,5 мл) добавили N-гидроксисукцинимид (NHS, 70,1 мг, 0,365 ммоль) и диметиламинопиридин (ДМАП) (5,95 мг, 0,049 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и разбавили дихлорметаном, промыли насыщенным раствором хлорида аммония, высушили над безводным сульфатом натрия и выпарили. Осадок очистили силикагелевой хроматографией (дихлорметан/метанол) для получения соединения 253с в виде бесцветной маслянистой жидкости (69,1 мг, выход = 53%).1H NMR (400 Hz, CDCl3): δ 6.71 (s, 2Н), 6.64 (s, 1Н), 4.57 (s, 4H), 3.63-3.59 (m, 8H + 2OH), 3.54-3.51 (m, 4H), 3.38-3.34 (m, 5H), 3.08 (s, 2.35H), 3.00 (s, 0.65H), 2.86 (bs, 2H), 2.43 (s, 3H), 2.34 (t, J=6.8 Hz, 2H), 1.95-1.88 (m, 2H), 1.36 (s, 1.3H), 1.31 (s, 4.7H);13C NMR (400 Hz, CDCl3): δ 173.7, 148.5, 142.7, 113.2, 109.8, 72.0, 70.8, 70.7, 70.6, 68.9, 65.6, 59.1, 56.5, 53.0, 52.2, 51.0, 50.8, 38.8, 30.6, 26.6, 25.6, 22.3; MS (m/z): найдено 555,5 (M+Na)+.
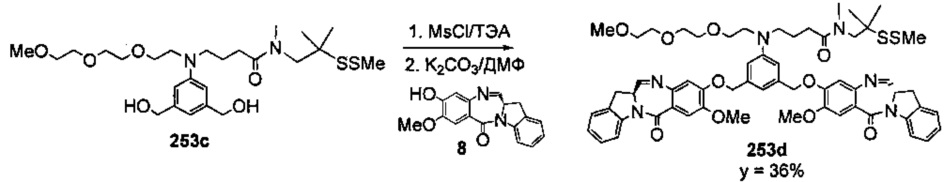
Соединение 253d:
К перемешанному раствору соединения 253с (69,1 мг, 0,13 ммоль) в безводном дихлорметане (1,5 мл) добавили триэтиламин (63 мкл, 0,454 ммоль). Смесь охладили до -10°С и медленно, в течение 15 минут, добавили метансульфонилхлорид (30 мкл, 0,389 ммоль). Раствор продолжали перемешивать при температуре между -10°С и -5°С в течение 60 минут и погасили добавлением водно-ледяной смеси. Смесь разбавили этилацетатом и промыли холодной водой. Органический слой высушили над безводным сульфатом натрия, отфильтровали, выпарили и отогнали под высоким вакуумом для получения мезилата в виде бесцветной маслянистой жидкости. Мезилат перенесли в 10 мл круглодонную колбу с этилацетатом, выпарили и отогнали под высоким вакуумом. Добавили соединение 8 (99 мг, 0,338 ммоль), а затем добавили безводный ДМФ (1 мл) и безводный карбонат калия (90 мг, 0,65 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Разбавили дихлорметаном и промыли насыщенным солевым раствором. Органический слой высушили над безводным сульфатом натрия, отфильтровали и выпарили. Осадок очистили силикагелевой хроматографией (дихлорметан/метанол) для получения 150 мг желтоватой пены, которую дополнительно очистили препаративной обращенно-фазовой ВЭЖХ (колонка С18, элюированная смесью CH3CN/H2O) для получения соединения 253d в виде твердого желтоватого вещества (50,7 мг, выход = 36%). MS (m/z): найдено 1107,7- (М+Na)+, 1125.7 (М+H2O+Na)+, 1143.7 (М+2H2O+Na)+, 1083.4 (М-Н)-, 1155.5 (М+4H2O-Н)-.

Соединение 253е:
В небольшую пробирку, содержающую трис(2-карбоксиэтил)фосфина гидрохлорид (ТСЕР, 19,1 мг, 0,067 ммоль) добавили несколько капель деионизированной воды. По каплям добавили насыщенный раствор бикарбоната натрия до рН около 7 по индикаторной рН бумаге. Затем смесь разбавили фосфатным буферным раствором с рН 6,5 (0,4 мл) для получения свежего раствора ТСЕР. К перемешанному раствору соединения 253d (24,1 мг, 0,022 ммоль) в метаноле (3 мл) и ацетонитриле (1 мл) добавили раствор ТСЕР и перемешивали при комнатной температуре в течение 1,5 часов. Смесь разбавили дихлорметаном и промыли насыщенным солевым раствором, высушили над безводным сульфатом натрия, отфильтровали и выпарили для получения тиола в виде твердого желтоватого вещества (21,9 мг), который использовали напрямую на следующих этапах (тиол нельзя очистить из-за агрегации). К раствору тиола (21,9 мг, 0,021 ммоль) в безводном дихлорметане (0,1 мл) и метаноле (0,4 мл) добавили 4-(2-пиридилдитио)бутановую кислоту (24,2 мг, 0,105 ммоль) и триэтиламин (29 мкл, 0,211 ммоль). Смесь перемешивали при комнатной температуре в течение пяти часов и погасили насыщенным раствором хлорида аммония. Смесь разбавили дихлорметаном, разделили и промыли органический слой насыщенным солевым раствором, высушили над безводным сульфатом натрия, отфильтровали и выпарили. Осадок очистили препаративной обращенно-фазовой ВЭЖХ (колонка С18, элюированная смесью ацетонитрил/вода) для получения соединения 253е в виде твердого белого вещества (7,3 мг, выход = 30%).1H NMR (400 Hz, CDCl3): δ 8.28 (d, J=7.6 Hz, 2H), 7.89 (bs, 2H), 7.60 (bs, 2H), 7.31-7.26 (m, 4H), 7.12 (t, J=7.6 Hz, 2H), 6.91-6.78 (m, 5H), 5.22-5.13 (m, 4H), 4.54-4.49 (m, 2H), 3.99 (s, 6H), 3.68-3.41 (m, 20H), 3.38 (s, 3H), 3.07 (s, 3H), 2.78-2.77 (m, 2H), 2.47 (bs, 2H), 2.35 (bs, 2H), 2.01-1.95 (m, 4H), 1.31 (s, 6H); MS (m/z): найдено 1179.7 (M+Na)+, 1197.7 (M+H2O+Na)+, 1073.6 (M+H2O-H)-, 1191.5 (M+2H2O-H)-.
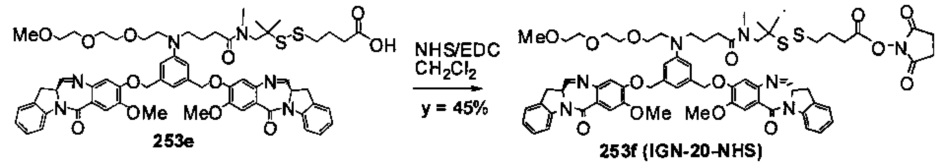
Соединение 253f:
К раствору соединения 253e (9,0 мг, 0,00778 ммоль) в безводном дихлорметане (0,5 мл) добавили N-гидроксисукцинимид (NHS, 2,68 мг, 0,023 ммоль), а затем N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорид (EDC, 4,47 мг, 0,023 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и разбавили дихлорметаном, промыли насыщенным солевым раствором и высушили над безводным сульфатом натрия. Отфильтровали, выпарили и очистили осадок препаративной обращенно-фазовой ВЭЖХ (колонка С18, элюированная смесью ацетонитрил/вода). Фракции продукта объединили и экстрагировали дихлорметаном. Органические слои высушили над безводным сульфатом натрия, отфильтровали, выпарили и отогнали под высоким вакуумом для получения соединения 253f (IGN-20-NHS) в виде желтоватой пены (4,4 мг, выход = 45%). MS (m/z): найдено 1276,7 (М+Na)+.
Пример 21

Соединение 254а (IGN-23-OMe) и 254b (IGN-24-OMe):
К перемешанному раствору соединения 249d (91,8 мг, 0,103 ммоль) в абсолютном этаноле (1,0 мл) и безводном дихлорметане (0,4 мл) добавили боргидрид натрия (0,4 мг, 0,0106 ммоль) при 0°С. Водно-ледяную баню убрали через 30 минут, а смесь продолжали перемешивать при комнатной температуре в течение 3 часов. Реакцию погасили добавлением насыщенного раствора хлорида аммония и разбавили дихлорметаном. Смесь разделили и промыли органический слой насыщенным солевым раствором, высушили над безводным сульфатом натрия и отфильтровали. Растворители удалили при пониженном давлении и очистили осадок препаративной обращенно-фазовой ВЭЖХ (колонка С18, элюированная смесью ацетонитрил/вода) для получения соединений 254а (IGN-23-OMe, 24,2 мг, выход = 24,5%) и 254b (IGN-24-OMe, 26,3 мг выход = 26,6%) в виде твердых желтоватых веществ. 254а:1Н NMR (400 Hz, CDCl3): δ 8.34 (d, J=8.0 Hz, 1H), 8.27 (d, J=7.6 Hz, 1H), 7.83 (d, J=4.4 Hz, 1H), 7.57 (s, 1H), 7.46 (s, 1H), 7.29-7.02 (m, 6H), 6.87 (s, 1H), 6.75 (s, 1H), 6.70-6.66 (m, 2H), 6.10 (s, 1H), 5.21-5.02 (m, 4H), 4.49-4.39 (m, 2H), 3.99 (s, 3H), 3.89 (s, 3H), 3.66 (s, 3H), 3.64-3.41 (m, 19H), 3.39-3.34 (m, 4H), 2.78 (dd, J1=16.4 Hz, J2=3.6 Hz, 1H), 2.33 (t, J=7.2 Hz, 2H), 1.90-1.84 (m, 2H);13C NMR (400 Hz, CDCl3): δ 173.8, 166.8, 164.0, 163.5, 152.3, 151.2, 148.7, 148.5, 143.0, 142.1, 140.7, 140.2, 138.5, 137.8, 129.8, 129.7, 128.3, 127.9, 125.0, 124.7, 123.9, 120.9, 117.5, 117.0, 114.6, 113.4, 113.2, 112.1, 111.6, 110.2, 110.1, 104.2, 72.1, 71.4, 71.2, 70.80, 70:76, 70.70, 68.7, 59.2, 57.3, 56.5, 56.4, 55.1, 54.8, 51.8, 50.9, 50.7, 33.3, 32.7, 31.3, 22.4; MS (m/z), найдено 976,7 (M+Na)+, 994,6 (M+H2O+Na)+; 254b: MS (m/z), найдено 978,7 (M+Na)+.

Соединение 254c и 254d (IGN-23-NHS):
К раствору соединения 254a (31,8 мг, 0,033 ммоль) в безводном 1,2-дихлорэтане (1 мл) добавили гидроксид триметилолова (90 мг, 0,5 ммоль). Смесь нагрели до 80°С и перемешивали в течение ночи. Смесь охладили до комнатной температуры, разбавили дихлорметаном и промыли смесью насыщенного раствора хлорида натрия и 5% соляной кислоты (~1 мл), затем насыщенным солевым раствором. Органический слой высушили над безводным сульфатом натрия, отфильтровали и выпарили. Осадок пропустили через короткую силикагелевую колонку и элюировали смесью дихлорметан/метанол для удаления избытка гидроксида триметилолова. Фракции продукта выпарили и отогнали под высоким вакуумом для получения кислоты 254с в виде твердого желтоватого вещества (20,8 мг, выход = 66%). MS (m/z): найдено 938,2 (М-Н)-, 962,3 (М+Na)+. Затем соединение 254с (20,8 мг, 0,022 ммоль) растворили в безводном дихлорметане (1 мл). Реакционную колбу быстро вакуумировали и заместили аргоном. Затем добавили N-гидроксисукцинимид (NHS, 5,09 мг, 0,044 ммоль) и N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорид (EDC, 8,48 мг, 0,044 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и разбавили дихлорметаном, промыли насыщенным солевым раствором и высушили над безводным сульфатом натрия. Отфильтровали, выпарили и очистили осадок препаративной обращенно-фазовой ВЭЖХ (колонка С18, элюированная смесью ацетонитрил/вода). Фракции продукта объединили и экстрагировали дихлорметаном. Органические слои высушили над безводным сульфатом натрия, отфильтровали, выпарили и отогнали под высоким вакуумом для получения соединения 254d (IGN-23-NHS) в виде твердого светло-желтоватого вещества (9,8 мг, выход = 43%). MS (m/z): найдено 1059,6 (М+Na)+, 1077,6 (М+H2O+Na)+.
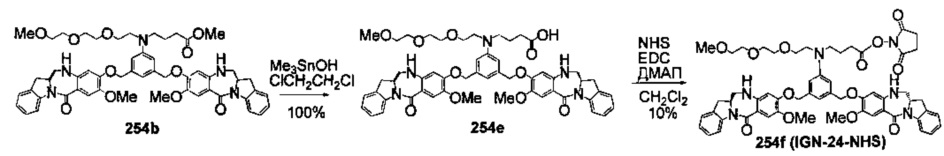
Соединение 254е и 254f (IGN-24-NHS):
К раствору соединения 254b (26,3 мг, 0,028 ммоль) в безводном 1,2-дихлорэтане (1 мл) добавили гидроксид триметилолова (74,6 мг, 0,413 ммоль). Смесь нагрели до 80°С и перемешивали в течение ночи. Смесь охладили до комнатной температуры, разбавили дихлорметаном и промыли смесью насыщенного раствора хлорида натрия и 5% соляной кислоты (~1 мл), затем насыщенным солевым раствором. Органический слой высушили над безводным сульфатом натрия, отфильтровали и выпарили. Осадок пропустили через короткую силикагелевую колонку и элюировали смесью дихлорметан/метанол для удаления избытка гидроксида триметилолова. Фракции продукта выпарили и отогнали под высоким вакуумом для получения кислоты 254е в виде твердого желтоватого вещества (26 мг, выход = 100%). MS (m/z): найдено 940,5 (М-Н)-, 964,6 (М+Na)+. Затем соединение 2542 (26 мг, 0,028 ммоль) растворили в безводном дихлорметане (1 мл). Затем добавили N-гидроксисукцинимид (NHS, 9,57 мг, 0,083 ммоль) и N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорид (EDC, 15,9 мг, 0,083 ммоль) и диметиламинопиридин (ДМАП) (0,34 мг, 0,0028 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и разбавили дихлорметаном, промыли насыщенным раствором хлорида аммония и насыщенным солевым раствором, высушили над безводным сульфатом натрия. Отфильтровали, выпарили и очистили осадок препаративной обращенно-фазовой ВЭЖХ (колонка С18, элюированная смесью ацетонитрил/вода). Фракции продукта объединили и экстрагировали дихлорметаном. Органические слои высушили над безводным сульфатом натрия, отфильтровали, выпарили и отогнали под высоким вакуумом для получения соединения 254f (IGN-24-NHS) в виде твердого светло-желтоватого вещества (3,0 мг, выход = 10%). MS (m/z): найдено 1061,7 (М+Na)+. Примечание: Не следует добавлять ДМАП, это может быть причиной низкого выхода.
Пример 22
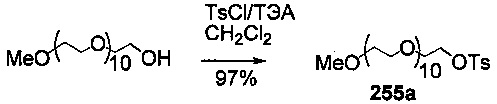
Соединение 255а:
К перемешанному раствору О-метил-ундекаэтиленгликоля (500 г, 0,968 ммоль) в безводном дихлорметане (3 мл) добавили триэтиламин (270 мкл, 1,94 ммоль), тозилхлорид (277 мг, 1,45 ммоль), а затем диметиламинопиридин (ДМАП) (5,91 г, 0,048 ммоль) при комнатной температуре. Смесь продолжали перемешивать в течение ночи, разбавили этилацетатом и отфильтровали для удаления твердого гидрохлорида триэтиламина. Твердое вещество промыли этилацетатом и выпарили фильтрат. Осадок разбавили этилацетатом и отфильтровали для удаления дополнительного осадка. Фильтрат выпарили для получения жидкого неочищенного продукта. Его очистили силикагелевой хроматографией (дихлорметан/метанол) для получения соединения 255а в виде светло-желтоватой маслянистой жидкости (630 мг, выход = 97%).1Н NMR (400 Hz, CDCl3): δ 7.81 (d, J=8.0 Hz, 2H), 7.35 (d, J=8.0 Hz, 2H), 4.17 (t, J=4.8 Hz, 2H), 3.72-3.54 (m, 42H), 3.39 (s, 3H), 2.46 (s, 3H); MS (m/z): найдено 693,6 (M+Na)+.

Соединение 255b:
К смеси тозилата 255a (630 мг, 0,939 ммоль) и анилина 28 (238 мг, 0,939 ммоль) в безводном ДМФ (3 мл) добавили безводный карбонат калия (195 мг, 1,409 ммоль) и йодид калия (31,2 мг, 0,188 ммоль). Смесь нагрели до 85°С и перемешивали при этой температуре в течение ночи. Раствор охладили до комнатной температуры и разбавили дихлорметаном. Смесь отфильтровали через целит и промыли твердое вещество дихлорметаном. Фильтрат выпарили и разбавили осадок дихлорметаном, снова отфильтровали для удаления дополнительного твердого вещества. Фильтрат выпарили и очистили осадок силикагелевой хроматографией (гексаны/10% метанол в ТГФ) для получения соединения 255b в виде бесцветной маслянистой жидкости (41,8 мг, выход = 5,9%).1Н NMR (400 Hz, CDCl3): δ 6.66 (s, 2Н), 6.65 (s, 1H), 4.60 (s, 4Н), 3.69 (s, 3Н), 3.66-3.58 (m, 42Н), 3.56-3.53 (m, 2H), 3.39-3.36 (m, 5H), 2.52 (broad s, 2OH), 2.36 (t, J=7.2 Hz, 2H), 1.91 (p, J=7.2 Hz, 2H);13C NMR (400 Hz, CDCl3): δ 173.9, 148.5, 142.8, 113.4, 109.9, 72.1, 70.8, 70.7, 68.9, 65.7, 59.2, 51.8, 50.9, 50.7, 31.3, 22.4; MS (m/z): найдено 774,3 (M+Na)+.
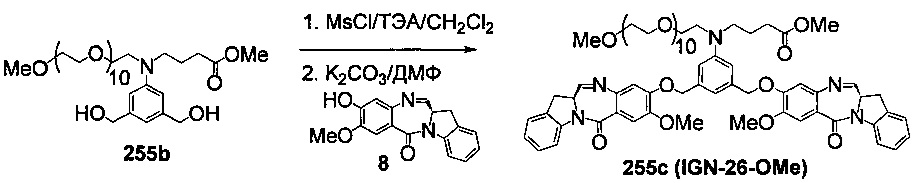
Соединение 255c (IGN-26-OMe):
К перемешанному раствору соединения 255b (41,8 мг, 0,056 ммоль) в безводном дихлорметане (1 мл) добавили триэтиламин (27 мкл, 0,196 ммоль). Смесь охладили до -10°С и медленно, в течение 15 минут, добавили метансульфонилхлорид (12,9 мкл, 0,167 ммоль). Раствор продолжали перемешивать при температуре между -10°С и -5°С в течение 60 минут и погасили добавлением водно-ледяной смеси. Смесь разбавили этилацетатом и промыли холодной водой. Органический слой высушили над безводным сульфатом натрия, отфильтровали, выпарили и отогнали под высоким вакуумом для получения мезилатов в виде бесцветной маслянистой жидкости. MS (m/z): найдено 930,3 (М+Na)+. Мезилаты (30 мг, 0,033 ммоль) перенесли в 5 мл круглодонную колбу с этилацетатом, выпарили и отогнали под высоким вакуумом. Добавили соединение 8 (29,2 мг, 0,099 ммоль), а затем добавили безводный ДМФ (0,5 мл), безводный карбонат калия (22,8 мг, 0,165 ммоль) и йодид калия (5,5 мг, 0,033 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Разбавили дихлорметаном и промыли насыщенным солевым раствором. Органический слой высушили над безводным сульфатом натрия, отфильтровали и выпарили. Осадок очистили силикагелевой хроматографией (гексаны/10% метанола в ТГФ) для получения 20,5 мг смеси, содержащей соединение 255с. Смесь растворили в безводном дихлорметане (0,3 мл). Затем добавили триэтиламин (4 мкл, 0,03 ммоль), тозилхлорид (3,8 мг, 0,02 ммоль) и ДМАП (0,2 мг, 0,002 ммоль) при комнатной температуре. Смесь продолжали перемешивать при комнатной температуре в течение ночи, а затем выпарили. Осадок очистили силикагелевой хроматографией (дихлорметан/метанол) для получения 11 мг светло-желтоватой пены. Ее дополнительно очистили препаративной обращенно-фазовой ВЭЖХ (колонка С18, элюированная смесью CH3CN/H2O) для получения соединения 255с (IGN-26-OMe) в виде бесцветной пены (1,6 мг, выход = 2,2%). MS (m/z): найдено 1326,5 (М+2H2O+Na)+, 1344.6 (М+H2O+Na)+, 1362.5 (М+2H2O+Na)+.
Пример 23
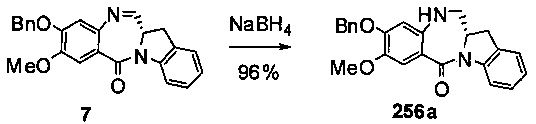
Соединение 256а:
К перемешанному раствору соединения 7 (384 мг, 1,0 ммоль) в абсолютном этаноле (6 мл) и безводном дихлорметане (2 мл) добавили боргидрид натрия (37,8 мг, 1,0 ммоль) при 0°С. Водно-ледяную баню убрали через 30 минут, а смесь продолжали перемешивать при комнатной температуре в течение ночи. Реакцию погасили добавлением насыщенного раствора хлорида аммония и разбавили дихлорметаном. Смесь разделили и промыли органический слой насыщенным солевым раствором, высушили над безводным сульфатом натрия и отфильтровали. Растворители удалили при пониженном давлении для получения соединения 256а в виде твердого белого вещества (369 мг, выход = 96%).1Н NMR (400 Hz, CDCl3): δ 8.37 (d, J=8.0 Hz, 1H), 7.50 (s, 1H), 7.40-7.24 (m, 6H), 7.18 (d, J=7.2 Hz, 1H), 7.05 (t, J=7.2 Hz, 1H), 6.12 (s, 1H), 5.06 (s, 2H), 4.40 (tt, J1=10.0 Hz, J2=3.6 Hz, 1H), 3.87 (s, 3H), 3.52-3.41 (m, 3H), 2.78 (dd, J1=16.8 Hz, J2=3.6 Hz, 1H);13C NMR (400 Hz, CDCl3): δ 166.5, 152.1, 142.73, 142.70, 140.4, 136.3, 129.5, 128.5, 127.9, 127.7, 127.1, 124.5, 123.8, 117.2, 114.5, 112.7, 103.4, 70.5, 57.1, 56.2, 54.5, 33.1; MS (m/z), найдено 409.2 (M+Na)+.

Соединение 256b:
К раствору соединения 256a (369 мг, 0,955 ммоль) в безводном ацетонитриле (9 мл) добавили йодметан (65 мкл, 1,05 ммоль) и карбонат калия (158 мг, 1,15 ммоль). Смесь перемешивали, нагрели до 82°С и дефлегмировали в течение ночи. Реакционную смесь сняли с масляной бани, охладили до комнатной температуры и разбавили дихлорметаном. Смесь отфильтровали через целит и выпарили фильтрат при пониженном давлении. Осадок очистили силикагелевой хроматографией (гексаны/этилацетат) для получения соединения 256b в виде бесцветной пены (195 мг, выход = 51%). Также было получено 123 мг исходного материала 256а.1Н NMR (400 Hz, CDCl3): δ 8.29 (d, J=8.0 Hz, 1H), 7.46 (s, 1H), 7.44 (s, 1H), 7.39-7.24 (m, 5H), 7.16 (d, J=7.2 Hz, 1H), 7.04 (t, J=7.6 Hz, 1H), 6.46 (s, 1H), 5.19 (dd, J1=15.2 Hz, J2=12.4 Hz, 2H), 4.36-4.29 (m, 1H), 3.89 (s, 3H), 3.38-3.31 (m, 2H), 3.02 (dd, J1=10.8 Hz, J2=4.0 Hz, 1H), 2.70 (dd, J1=16.8 Hz, J2=2.8 Hz, 1H), 2.65 (s, 3H);13C NMR (400 Hz, CDCl3): δ 166.9, 151.2, 144.2, 142.1, 141.9, 136.7, 129.8, 128.6, 128.0, 127.8, 127.3, 125.1, 123.9, 121.7, 117.1, 113.5, 104.7, 71.1, 64.2, 57.2, 56.3, 40.2, 32.0; MS (m/z): найдено 423.2 (M+Na)+.
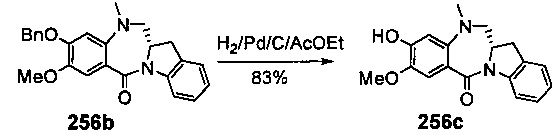
Соединение 256c:
К перемешанному раствору соединения 256b (195 мг, 0,487 ммоль) в этилацетате (2,5 мл) добавили палладий на древесном угле (10%, 25,9 мг, 0,024 ммоль). Колбу быстро вакуумировали и заместили колбой Н2. Смесь продолжали гидрогенировать в течение ночи и отфильтровали через целит. Фильтрат десорбировали при пониженном давлении и очистили осадок силикагелевой хроматографией (дихлорметан/метанол) для получения соединения 256с в виде твердого белого вещества (126 мг, выход = 83%).1Н NMR (400 Hz, MeOD): δ 8.14 (d, J=8.0 Hz, 1H), 7.30-7.23 (m, 2H), 7.22 (s, 1H), 7.10 (t, J=7.6 Hz, 1H), 6.56 (s, 1H), 4.46-4.38 (m, 1H), 3.88 (s, 3H), 3.48-3.37 (m, 2H), 3.12 (dd, J1=10.8 Hz, J2=4.4 Hz, 1H), 2.84 (dd, J1=16.8 Hz, J2=2.8 Hz, 1H), 2.80 (s, 3H).
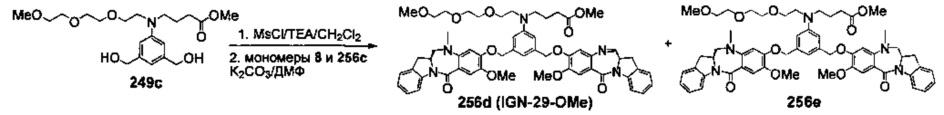
Соединение 256d (IGN-29-OMe):
К перемешанному раствору соединения 249 с (136 мг, 0,34 ммоль) в безводном дихлорметане (2 мл) добавили триэтиламин (142 мкл, 1,02 ммоль). Смесь охладили до -10°С и медленно, в течение 15 минут, добавили метансульфонилхлорид (66 мкл, 0,85 ммоль). Раствор продолжали перемешивать при температуре между -10°С и -5°С в течение 60 минут и погасили добавлением водно-ледяной смеси. Смесь разбавили этилацетатом и промыли холодной водой. Органический слой высушили над безводным сульфатом натрия, отфильтровали, выпарили и отогнали под высоким вакуумом для получения мезилата в виде бесцветной маслянистой жидкости. Мезилат перенесли в 10 мл круглодонную колбу с этилацетатом, вьшарили и отогнали под высоким вакуумом. К нему добавили соединение 8 (120 мг, 0,41 ммоль) и соединение 256 с (106 мг, 0,34 ммоль), а затем добавили безводный ДМФ (1,5 мл) и безводный карбонат калия (235 мг, 1,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Разбавили дихлорметаном и промыли насыщенным солевым раствором. Органический слой высушили над безводным сульфатом натрия, отфильтровали и выпарили. Осадок очистили препаративной обращенно-фазовой ВЭЖХ (колонка С18, элюированная смесью CH3CN/H2O) для получения соединения 256d (IGN-29-OMe) в виде твердого светло-желтоватого вещества (46 мг, выход = 14%) и соединения 256е. 256d:1Н NMR (400 Hz, CDCl3), δ 8.27 (d, J=8.0 Hz, 2H), 7.84 (d, J=4.8 Hz, 1H), 7.57 (s, 1H), 7.32-7.04 (m, 7H), 6.87 (s, 1H), 6.82 (s, 1H), 6.76-6.70 (m, 2H), 6.50 (s, 1H) 5.18-5.12) m, 4H), 4.49-4.43 (m, 1H), 4.40-4.35 (m, 1H), 3.98 (s, 3H), 3.89 (s, 3H), 3.68-3.52 (m, 18H), 3.41-3.36 (m, 6H), 3.08 (dd, J1=10.8 Hz, J2=4.4 Hz, 1H), 2.56 (dd, J1=16.8 Hz, J2=2.8 Hz, 1H), 2.70 (s, 3H), 2.34 (t, J=7.2 Hz, 2H), 1.92-1.85 (m, 2H); MS (m/z): найдено 990.6 (M+Na)+, 1008.6 (M+H2O+Na)+. 256e: MS (m/z): найдено 1006,6 (M+Na)+.
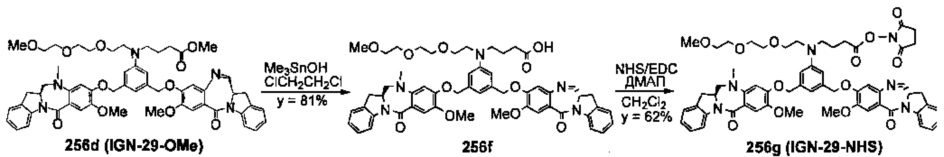
Соединение 256f и соединение 256g (IGN-29-NHS):
К раствору соединения 256d (46 мг, 0,048 ммоль) в безводном 1,2-дихлорэтане (1,5 мл) добавили гидроксид триметилолова (129 мг, 0,71 ммоль). Смесь нагрели до 80°С и перемешивали в течение ночи. Смесь охладили до комнатной температуры, разбавили дихлорметаном и промыли смесью насыщенного раствора хлорида натрия и 5% соляной кислоты (~1 мл), затем насыщенным солевым раствором. Органический слой высушили над безводным сульфатом натрия, отфильтровали и выпарили. Осадок пропустили через короткую силикагелевую колонку и элюировали смесью дихлорметан/метанол для удаления избытка гидроксида триметилолова. Фракции продукта выпарили и отогнали под высоким вакуумом для получения кислоты 256f в виде твердого желтоватого вещества (36,9 мг, выход = 81%). MS (m/z): найдено 952,8 [М-Н]-. Затем соединение 256f (36,9 мг, 0,039 ммоль) растворили в безводном дихлорметане (0,8 мл). Затем добавили N-гидроксисукцинимид (NHS, 13,4 мг, 0,12 ммоль) и N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорид (EDC, 22,2 мг, 0,12 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и разбавили дихлорметаном, промыли насыщенным солевым раствором и высушили над безводным сульфатом натрия. Отфильтровали, вьшарили и очистили осадок препаративной обращенно-фазовой ВЭЖХ (колонка С18, элюированная смесью ацетонитрил/вода). Фракции, содержащие продукт, объединили и экстрагировали дихлорметаном. Органические слои высушили над безводным сульфатом натрия, отфильтровали, выпарили и отогнали под высоким вакуумом для получения соединения 256g (IGN-29-NHS) в виде твердого светло-желтоватого вещества (25,4 мг, выход = 62%). MS (m/z): найдено 1073,4 (М+Na)+, 1091,4 (М+H2O+Na)+, 1103,3 (М+3H2O-H)-.
Пример 24
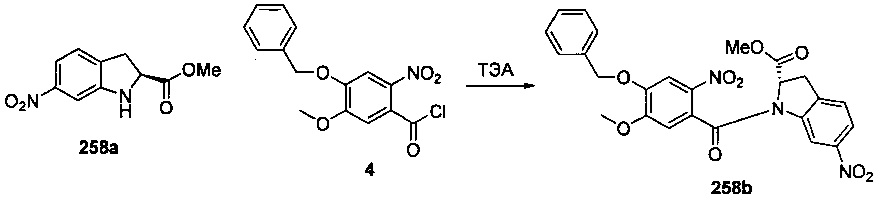
метил 1-(4-(бензилокси)-5-метокси-2-нитробензоил)-6-нитроиндолин-2-карбоксилат (258b):
Метил 6-нитроиндолин-2-карбоксилат (258а) (0,233 г, 1,048 ммоль) растворили в безводном тетрагидрофуране (4 мл) в отдельной колбе и охладили до 0°С на ледяной бане. В другой колбе растворили 4-(бензилокси)-5-метокси-2-нитробензоил хлорид (4) (0,371 г, 1,153 ммоль) в безводном тетрагидрофуране (4 мл) и охладили до 0°С на ледяной бане. В колбу, содержащую индолин, добавили триэтиламин (0,438 мл, 3,15 ммоль) через шприц и быстро добавили в реакционную смесь ацетилхлорид 4 через канюлю при 0°С. Реакционную смесь перемешивали в течение 90 минут при 0°С, а затем при комнатной температуре еще в течение 1 часа. Затем реакцию погасили 5% HCl и экстрагировали этилацетатом (3х). Объединенные органические слои промыли насыщенным солевым раствором, высушили над сульфатом натрия и концентрировали in vacuo. Осадок очистили силикагелевой хроматографией, используя 30% смесь ацетона в гексане для получения 1-(4-(бензилокси)-5-метокси-2-нитробензоил)-6-нитроиндолин-2-карбоксилата (258b) (0,220 г, 0,434 ммоль, выход 41,4%) в виде желтоватой пены.1Н NMR (400 Hz, CDCl3): δ 3.30 (m, 1Н), 3.60 (s, 3Н), 3.69 (m, 1H), 3.86 (s, 3H), 4.64 (dd, 1H, J=2.4 Hz, 10.8), 5.23 (s, 2H), 7.31 (m, 1H), 7.46 (m, 6H), 7.99 (dd, 1H, J=2.0, 8.0 Hz), 9.04 (d, 1H, J=2.0 Hz). MS (m/z), найдено 530,1 ([M]++Na).

1-(4-(бензилокси)-5-метокси-2-нитробензоил)-6-нитроиндолин-2-карбальдегид (258с):
Метил 1-(4-(бензилокси)-5-метокси-2-нитробензоил)-6-нитроиндолин-2-карбоксилат (258b) (1,023 г, 2,016 ммоль) растворили в смеси безводного дихлорметана (2,5 мл) и толуола (7,5 мл) и охладили до -78°С на бане из сухого льда и ацетона. Через 15 минут добавили диизобутилалюминия гидрид (1,0 М в ТГФ) (4,03 мл, 4,03 ммоль) через шприцевой насос в течение около 20 минут. Полученный раствор перемешивали в течение 2 часов при -78°С, после чего по каплям добавили метанол (1 мл) для погашения реакции при -78°С. Затем реакционную смесь разбавили 5% HCl и этилацетатом и нагрели до комнатной температуры. Водный слой промыли дополнительным количеством этилацетата, а объединенные органические слои промыли насыщенным солевым раствором и высушили над безводным сульфатом натрия. Реакционную смесь пропустили через слой целита и концентрировали in vacuo. Неочищенный осадок очистили силикагелевой хроматографией, используя 40% смесь ацетона в гексане для получения 1-(4-(бензилокси)-5-метокси-2-нитробензоил)-6-нитроиндолин-2-карбальдегида (258с) (621 мг, 1,301 ммоль, выход 64,5%).1Н NMR (400 Hz, CDCl3): δ 3.15-3.60 (m, 2Н), 3.90 (s, 0.6Н), 3.92 (s, 1.2H), 3.97 (s, 1.2H), 4.57 (d, 0.2H, J=4.8 Hz), 5.21 (m, 2.4H), 5.5 (m, 0.4H), 6.39 (s, 0.4H), 6.46 (s, 0.2H), 6.76 (s, 0.2H), 6.89 (s, 0.4H), 7.01 (s, 0.4H), 7.19-7.41 (m, 5.6H), 7.60-7.77 (m, 1.6H), 7.86-7.91 (m, 0.8H), 8.94 (s, 0.4H), 9.34 (s, 0.4H), 9.74 (s, 0.4H), 9.90 (s, 0.2H). MS (m/z), найдено 500,1 ([M]++Na).
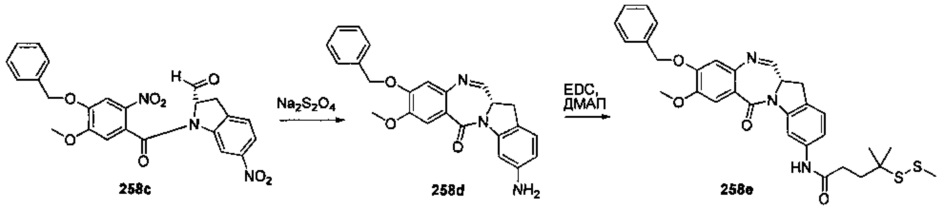
Соединение 258e:
1-(4-(бензилокси)-5-метокси-2-нитробензоил)-6-нитроиндолин-2-карбальдегид (258c) (0,125 r, 0,262 ммоль) растворили в тетрагидрофуране (8 мл) и воде (5,33 мл). К этому раствору добавили гидросульфит натрия (0,456 г, 2,62 ммоль), накрыли реакционную смесь мембраной и вентилировали через иглу (не требуется использовать азот/аргон) и перемешивали в течение ночи. К реакционной смеси добавили метанол и перемешивали еще 30 минут, затем реакционную смесь концентрировали in vacuo для удаления всех растворителей. Осадок растворили в смеси метанола и дихлорметана 1:1 (20 мл), в результате чего остался нерастворенный осадок. Смесь пропустили через короткую подушку диоксида кремния, расположенную в верхней части короткой подушки целита, и тщательно промыли смесью метанола и дихлорметана 1:1. Фильтрат снова отфильтровали через целит, а затем при перемешивании добавили раствор HCl в диоксане (4 М) до достижения рН ~3-4. Затем реакционную смесь перемешивали еще 30 минут, а затем добавили водный раствор бикарбоната натрия до щелочной реакции (~рН 8-9), добавили дополнительное количество дихлорметана и удалили органический слой. Водный слой промыли дополнительным количеством дихлорметана и объединили полученные органические слои и промыли насыщенным солевым раствором, высушили над безводным сульфатом натрия и концентрировали in vacuo. Осадок, содержащий соединение 258d (0,105 г, 0,263 ммоль, выход 100%) использовали на следующих этапах без дополнительной очистки. MS (m/z), найдено 454,2 ([M]++Na+CH3OH).
В небольшой пробирке растворили 4-метил-4-(метилдисульфанил)пентановую кислоту (0,061 г, 0,313 ммоль), EDC (0,060 г, 0,313 ммоль) и ДМАП (0,038 г, 0,313 ммоль) в дихлорметане (1 мл) при перемешивании. К этой смеси добавили соединение 258d (0,125 г, 0,313 ммоль), растворенное в дихлорметане (1,5 мл) и перемешивали смесь при комнатной температуре в течение ночи. Добавили воду и разделили слои. Органический слой промыли насыщенным солевым раствором, высушили над безводным сульфатом натрия и концентрировали in vacuo. Осадок очистили на силикагелевой колонке, используя 50% раствор этилацетата в гексане для получения соединения 258е (0,037 г, 0,064 ммоль, выход 20,54%).1Н NMR (400 Hz, CDCl3): δ 1.27 (s, 6Н), 1.97 (t, 2Н, J=8.0 Hz), 2.06 (t, 2H, J=8.0 Hz), 2.45 (s, 3H), 3.48 (m, 1H), 3.67 (m, 1H), 3.99 (s, 3H), 4.49 (m, 1H), 5.24 (q, 2H, J=8.4 Hz), 6.90 (s, 1H), 7.22 (d, 1H, J=8.0 Hz), 7.39 (m, 5H), 7.55 (s, 1H), 7.82 (d, 1H, J=8.0 Hz), 7.87 (d, 1H, J=4.0 Hz), 8.07 (s, 1H). MS (m/z), найдено 630,3 ([M]++Na+MeOH).
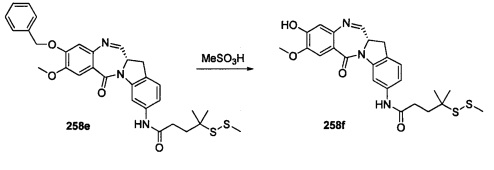
Соединение 258f:
Соединение 258е (0,0185 г, 0,032 ммоль) растворили в безводном дихлорметане (0,5 мл) и к этому раствору добавили метансульфоновую кислоту (0,021 мл, 0,321 ммоль), растворенную в безводном дихлорметане (0,500 мл), и полученную смесь перемешивали при комнатной температуре в течение трех часов. Реакционную смесь перелили в смесь льда и метанола и нейтрализовали до рН 7 водным раствором бикарбоната натрия. Затем реакционную смесь разбавили дихлорметаном и разделили слои. Органический слой промыли насыщенным солевым раствором, высушили над безводным сульфатом натрия и концентрировали in vacuo. Осадок очистили препаративной тонкослойной хроматографией на диоксиде кремния, используя 3% смесь метанола в дихлорметане для получения IGN мономера NH(4-метил-4-метилдитио-пентаноат)-индол (0,007 г, 0,014 ммоль, выход 44,9%). MS (m/z), найдено 484,0 ([М]+-1).
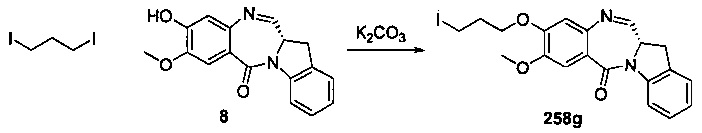
Соединение 258g:
В небольшой пробирке растворили соединение 8 (0,033 г, 0,112 ммоль) в ДМФ (1,5 мл) при перемешивании при комнатной температуре. Добавили 1,3-дийодпропан (0,065 мл, 0,561 ммоль), а затем добавили карбонат калия (0,023 г, 0,168 ммоль). Реакционную смесь накрыли фольгой и перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавили дихлорметаном и промыли водным раствором хлорида аммония и насыщенным солевым раствором. Органический слой высушили над безводным сульфатом натрия, отфильтровали и концентрировали in vacuo. Осадок очистили препаративной тонкослойной хроматографией на диоксиде кремния, используя 50% смесь этилацетата в гексане для получения соединения 258g (0,018 г, 0,039 ммоль, выход 34,7%). MS (m/z), найдено 533,0 ([М]++K).
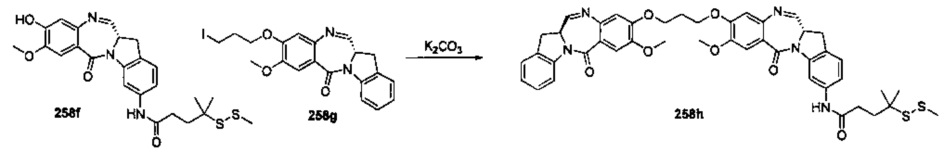
Соединение 258h (IGN-15-SMe):
В небольшой пробирке растворили соединение 258f (0,007 г, 0,014 ммоль) в диметилформамиде (1 мл) при перемешивании при комнатной температуре. Добавили соединение 8 (6,66 мг, 0,014 ммоль), а затем добавили карбонат калия (1,992 г, 0,014 ммоль). Реакционную смесь накрыли фольгой и перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавили дихлорметаном и промыли водным раствором хлорида аммония и насыщенным солевым раствором. Органический слой высушили над безводным сульфатом натрия, отфильтровали и концентрировали in vacuo. Осадок очистили препаративной тонкослойной хроматографией на пластине диоксида кремния в 5% смеси метанола в дихлорметане для получения соединения 258h (IGN-15-SMe) (0,005 г, 7,32 мкмоль, выход 50,8%). MS (m/z), найдено 906,3 ([M]++Na+2CH3OH).
Пример 25
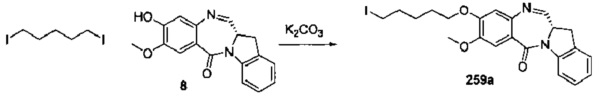
Соединение 259а:
В небольшой пробирке растворили соединение 8 (0,100 г, 0,340 ммоль) в ДМФ (5 мл) при перемешивании при комнатной температуре. Добавили 1,5-дийодпентан (0,506 мл, 3,40 ммоль), а затем добавили карбонат калия (0,070 г, 0,510 ммоль). Реакционную смесь накрыли фольгой и перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавили дихлорметаном и промыли водным раствором хлорида аммония и насыщенным солевым раствором. Органический слой высушили над безводным сульфатом натрия, отфильтровали и концентрировали in vacuo. Осадок очистили препаративной тонкослойной хроматографией на диоксиде кремния, используя 50% смесь этилацетата в гексане для получения соединения 259а (0,045 г, 7,32 мкмоль, выход 27%).1Н NMR (400 Hz, CDCl3): δ 1.64 (m, 2Н), 1.94 (М, 4Н), 3.24 (t, 2Н, J=6.5 Hz), 3.52 (dd, 1Н, J=4.0, 16.6 Hz), 3.73 (dd, 1H, J=10.5, 16.6 Hz), 3.98 (s, 3H), 4.12 (m, 2H), 4.50 (dt, 1H, J=4.0, 11.2 Hz), 6.84 (s, 1H), 7.13 (t, 1H, J=6.0 Hz), 7.29 (m, 2H), 7.57 (s, 1H), 7.90 (d, 1H, J=4.4 Hz), 8.29 (d, 1H, J=8.0 Hz). MS (m/z), найдено 533,3 ([M]++K).
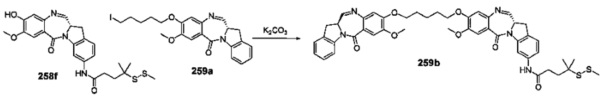
Соединение 259b (IGN-21-SMe):
В небольшой пробирке растворили соединение 258f (15 мг, 0,031 ммоль) в диметилформамиде (1 мл) при перемешивании при комнатной температуре. Добавили соединение 259а (17,42 мг, 0,036 ммоль), а затем добавили карбонат калия (4,27 г, 0,031 ммоль). Реакционную смесь накрыли фольгой и перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавили дихлорметаном и промыли водным раствором хлорида аммония и насыщенным солевым раствором. Органический слой высушили над безводным сульфатом натрия, отфильтровали и концентрировали in vacuo. Осадок очистили препаративной тонкослойной хроматографией на пластине диоксида кремния в 5% смеси метанола в дихлорметане для получения соединения 259а (IGN-15-SMe) (0,006 г, 7,32 мкмоль, выход 22%). MS (m/z), найдено 934,1 ([M]++Na+2CH3OH).
Пример 26

Соединение 260а:
Соединение 256а (55 мг, 0,142 ммоль) растворили в безводном дихлорметане, а затем добавили 4-метокси-4-оксобутановую кислоту (76 мг, 0,575 ммоль), EDC (70 мг, 0,365 ммоль) и ДМАП (8,69 мг, 0,071 ммоль). Смесь перемешивали в течение ночи при комнатной температуре и проверили по ВЭЖХ отсутствие остатков исходного материала. Затем реакционную смесь разбавили водой и этилацетатом. После дополнительной экстракции этилацетатом, органический слой промыли насыщенным солевым раствором, высушили над сульфатом натрия, отфильтровали и концентрировали in vacuo. Неочищенный осадок очистили силикагелевой хроматографией, используя 50% смесь этилацетата в гексане для получения соединения 260а (54 мг, выход 76%).1Н NMR (400 Hz, CDCl3): δ 8.21 (d, J=8.0 Hz, 1H), 7.45-7.25 (m, 7H), 7.20 (d, J=7.2 Hz, 1H), 7.08 (t, J=7.4 Hz, 1H), 6.825 (s, 1H), 5.27 (q, J=15.1 Hz, 2H), 4.56 (t, J=12.6 Hz, 1H), 4.35-4.29 (m, 1H), 3.99 (s, 3H), 3.65 (s, 3H), 3.44-3.38 (m, 2H), 2.88 (dd, J1=16.4 Hz, J2=2 Hz, 1H), 2.58-2.50 (m, 1H), 2.40-2.33 (m, 1H), 2.26-2.18 (m, 1H), 1.99-1.92 (m, 1H); MS (m/z), найдено 523,1 ([M]++Na)+.

Соединение 260b:
К раствору соединения 260а (50 мг, 0,100 ммоль) в безводном дихлорметане (11,5 мл) по каплям добавили метансульфоновую кислоту (0,389 мл, 5,99 ммоль), получили желтый раствор. Реакционную смесь перемешивали при комнатной температуре и контролировали по ВЭЖХ до завершения реакции через 30 минут.Смесь разбавили водой и метанолом, затем нейтрализовали до рН 7 насыщенным раствором бикарбоната натрия. Водный слой экстрагировали дихлорметаном и высушили органический слой над сульфатом натрия. Неочищенный продукт очистили силикагелевой хроматографией, используя 6% смесь метанола в дихлорметане для получения соединения 260b (40 мг, выход = 98%).1Н NMR (400 Hz, CDCl3): δ 8.22 (d, J=8.0 Hz, 1H), 7.35 (s, 1H), 7.28 (t, J=7.8 Hz, 1H), 7.22 (d, J=7.2 Hz, 1H), 7.09 (t, J=7.4 Hz, 1H), 6.90 (s, 1H), 6.06 (s, 1H), 4.63 (t, J=12.6 Hz, 1H), 4.38-4.30 (m, 1H), 4.00 (s, 3H), 3.66 (s, 3H), 3.47-3.39 (m, 2H), 2.90 (dd, J1=16.2 Hz, J2=2.2 Hz, 1H), 2.69-2.59 (m, 2H), 2.52-2.45 (m, 1H), 2.22-2.14 (m, 1H); MS (m/z), найдено 433 (M+Na)+.
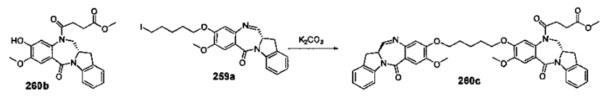
Соединение 260c:
Соединение 260b (20 мг, 0,049 ммоль) и соединение 259а (30 мг, 0,061 ммоль) растворили в безводном N,N-диметилформамиде (1 мл). Добавили карбонат калия (20,20 мг, 0,146 ммоль) и перемешивали реакционную смесь в течение ночи при комнатной температуре. Реакцию погасили водой и экстрагировали дихлорметаном. Органический слой промыли насыщенным солевым раствором и высушили над сульфатом натрия. Неочищенный продукт очистили силикагелевой хроматографией, используя 5% смесь метанола в дихлорметане для получения соединения 260с (25 мг, выход = 66%). MS (m/z), найдено 813,5 (М+Na+H2O)+.
Пример 27
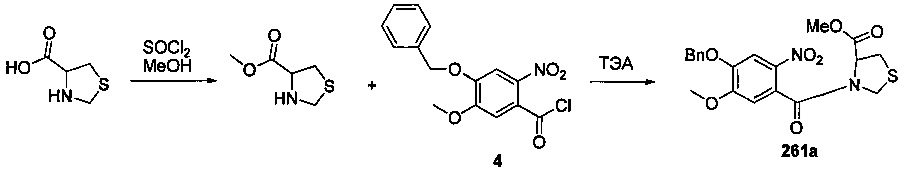
Соединение 261а:
Доступный в продаже исходный материал, тиазолидин-4-карбоновую кислоту (1,3 г, 9,59 ммоль) растворили в безводном метаноле (19,18 мл) и охладили до 0°С на ледяной бане. По каплям добавили тионилхлорид (1,40 мл, 19,18 ммоль) и перемешивали реакционную смесь в течение 30 минут. Убрали ледяную баню и продолжали перемешивание в течение 4-5 часов или в течение ночи. Растворитель десорбировали и поместили продукт под высокий вакуум для получения 4-(метоксикарбонил)тиазолидин-3-иум хлорид. Без дополнительной очистки, принимая выход за 100%, 4-(метоксикарбонил)тиазолидин-3-иум хлорид (1,761 г, 9,59 ммоль) и соединение 4 (3,39 г, 10,55 моль) растворили по отдельности в тетрагидрофуране (32,0 мл) и охладили до 0°С. Добавили триэтиламин (4,41 мл, 31,6 ммоль) в раствор с 4-(метоксикарбонил)тиазолидин-3-иум хлоридом, а затем через канюлю быстро добавили соединение 4. Через 20 минут проверили pH раствора, чтобы убедиться, что среда щелочная. Реакционную смесь перемешивали при 0°С в течение 1,5 часов, а затем при комнатной температуре в течение 30 минут, проверили по масс-спектру. Погасили холодной 5% соляной кислотой и разбавили холодным этилацетатом и водой. Раствор экстрагировали этилацетатом три раза и промыли объединенные органические слои насыщенным солевым раствором, насыщенным раствором бикарбоната натрия и снова солевым раствором. Высушили над безводным сульфатом натрия, отфильтровали и десорбировали. Неочищенный материал очистили силикагелевой хроматографией, используя градиент от 50% до 75% этилацетата в гексанах для получения соединения 261а (4,1 г, выход = 99%).1Н ЯМР (400 Гц, CDCl3): соединение получено в виде двух отдельных ротамеров. δ 7.78 (s, 0.6Н), 7.74 (s, 0.4Н), 7.48-7.35 (m, 5Н), 6.96 (s, 0.4Н), 6.92 (s, 0.6H), 5.40 (dd, J1=7.0Hz, J2=3.4 Hz, 0.6H), 5.31-5.22 (m, 2H), 5.13 (d, 9.6Hz, 0.4H), 4.60 (d, J=9.6 Hz, 0.4H), 4.46 (dd, J1=4.4Hz, J2=3.2 Hz, 0.4 H), 4.36 (d, J=8.4 Hz, 0.6 H), 4.26 (d, J=8.4 Hz, 0.6H), 4.02 (s, 1.8H), 3.96 (s, 1.2H), 3.86 (s, 1.8H), 3.71 (s, 1.2H), 3.48-3.43 (m, 0.6H), 3.36-3.29 (m, 1.4H); MS (m/z), найдено 455,3 (M+Na)+.
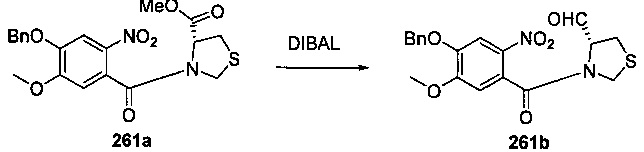
Соединение 261b:
Соединение 261а (4,1 г, 9,48 ммоль) растворили в дихлорметане (11 мл) и толуоле (33 мл), затем охладили до -78°С на бане из ацетона и сухого льда. Очень медленно, в течение не менее 30 минут, добавили диизобутилалюминия гидрид (18,96 мл, 18,96 ммоль), при помощи шприцевого насоса. Реакционную смесь перемешивали при -78°С в течение 3 часов и погасили метанолом (0,4 мл), а затем 5% соляной кислотой (30 мл). Добавили этилацетат (100 мл) и убрали ледяную баню. Смесь продолжали перемешивать при комнатной температуре в течение 30 минут. Смесь экстрагировали этилацетатом и промыли объединенные органические слои насыщенным солевым раствором, насыщенным раствором бикарбоната натрия и снова солевым раствором. Высушили над безводным сульфатом натрия и отфильтровали через целит. Неочищенный продукт очистили силикагелевой хроматографией, используя 75% смесь этилацетата в гексанах для получения соединения 261b (2,3 г, выход = 60%).1Н ЯМР (400 Гц, CDCl3): соединение получено в виде двух ротамеров. δ 9.80 (s, 0.8Н), 9.41 (s, 0.2Н), 7.80 (s, 0.8Н), 7.73 (s, 0.2H), 7.49-7.36 (m, 5H), 6.91 (s, 0.2H), 6.84 (s, 0.8H), 5.25-5.22 (m, 2H), 4.85-4.73 (m, 1H), 4.35-4.30 (m, 1H), 4.22-4.17 (m, 1H), 4.04-3.97 (m, 3H), 3.40-3.26 (m, 2H); MS (m/z), найдено 425,0 (M+Na)+.
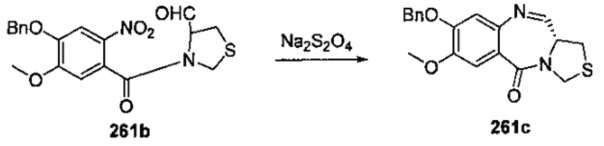
Соединение 261c:
Соединение 261b растворили в тетрагидрофуране (230 мл), а затем в воде (150 мл). Медленно, маленькими порциями добавили гидросульфит натрия (5,27 г, 25,7 ммоль). Если раствор остался мутным, по каплям добавляли дополнительное количество воды до прозрачности раствора. Реакционную смесь накрыли мембраной, оставили иглу для отвода SO2 газа и перемешивали в течение ночи. Раствор из желтого стал очень светлым, почти бесцветным. На следующее утро добавили воду до прозрачности раствора, а затем добавили метанол (30 мл). Перемешивали раствор еще 2 часа, затем выпарили растворители и повторно упарили осадок с ацетонитрилом не мене двух раз. Белый осадок поместили под высокий вакуум на несколько часов. Осадок повторно растворили в смеси метанол: дихлорметан [1:1], отфильтровали через целит и десорбировали. Этап фильтрации повторяли до тех пор, пока при разбавлении в метаноле не образовался прозрачный раствор без твердых частиц. Промежуточное соединение поместили под высокий вакуум до полного высушивания, затем растворили в безводном метаноле (50 мл). По каплям добавили ацетилхлорид (1,9 мл, 26,7 ммоль) при комнатной температуре, что вызвало образование желтого осадка. Смесь перемешивали при комнатной температуре в течение 30 минут и погасили реакцию насыщенным раствором бикарбоната натрия. Смесь разбавили дихлорметаном и водой (130 мл/85 мл) и экстрагировали дихлорметаном. Водный слой подкислили гидросульфатом натрия, концентрировали для уменьшения объема и повторно экстрагировали. Объединенный органический слой промыли насыщенным раствором бикарбоната натрия и высушили над сульфатом натрия. Десорбированный осадок очистили силикагелевой хроматографией, используя 60% смесь этилацетата в гексанах для получения соединения 261с (1,2 г, выход 59%).1Н NMR (400 Hz, CDCl3): δ 7.69 (d, J=4.4Hz, 1H), 7.52-7.28 (m, 6H), 6.87 (s, 1H), 5.22 (q, J=12.3 Hz, 2H), 4.85, (d, J=10.4 Hz, 1H), 4.58 (d, J=10.4 Hz, 1H), 4.03-4.02 (m, 1H), 3.98 (s, 3H), 3.51-3.47 (m, 1H), 3.45-3.23 (m, 1H); MS (m/z), найдено 377,3 (M+Na)+.
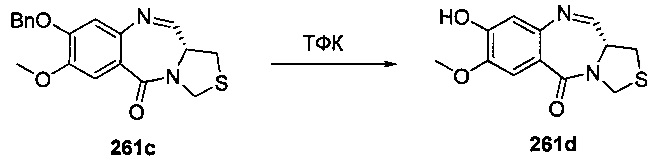
Соединение 261d:
Соединение 261c (75 мг, 0,212 ммоль) растворили в чистой трифторуксусной кислоте (0,4 мл, 5,19 ммоль). Дефлегмировали смесь в течение около 1 часа при 50°С, затем увеличили температуру до 80°С. Через 3 часа от начала реакции выпарили растворитель. Осадок напрямую очистили препаративной тонкослойной хроматографией, используя 5% смесь метанола в дихлорметане для получения соединения 261d (19,4 мг, 35%).1Н NMR (400 Hz, CDCl3): δ 7.72 (d, J=4.4 Hz, 1H), 7.51 (s, 1H), 6.91 (s, 1H), 6.18 (s, 1H), 4.85 (d, J=10.4 Hz, 1H), 4.58 (J=10.4 Hz, 1H), 4.05-4.02 (m, 1H), 3.99 (s, 3H), 3.50 (dd, J1=12.4 Hz, J2=6 Hz, 1H), 3.32, (dd, J1=12.4 H, J2=2 Hz, 1H); MS (m/z), найдено 319,0 (M+Na+MeOH)+.
Пример 28
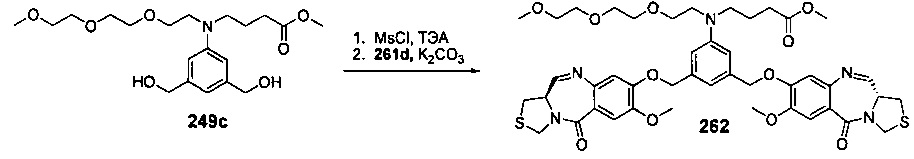
Соединение 262:
Соединение 249с (18 мг, 0,045 ммоль) растворили в безводном дихлорметане (0,45 мл) и затем охладили на бане изо льда и насыщенного солевого раствора. Сначала добавили триэтиламин (0,022 мл, 0,158 ммоль), а затем метансульфонилхлорид (10,46 мкл, 0,135 ммоль); второе вещество - очень медленно. Смесь продолжали перемешивать на бане в течение 1 часа. Реакцию погасили смесью льда и воды и разбавили холодным этилацетатом. После разделения снова промыли органический слой холодной водой и высушили над сульфатом натрия. Отфильтровали и выпарили при пониженном давлении, поддерживая температуру ниже 20°С, а затем поместили под высокий вакуум для прямого использования. После полного высушивания продукт и соединение 261d (28,5 мг, 0,108 ммоль) растворили в безводном N,N-диметилформамиде (350 мкл). Добавили карбонат калия (29,8 мг, 0,216 ммоль). После перемешивания в течение ночи при комнатной температуре реакционную смесь разбавили дихлорметаном, промыли насыщенным солевым раствором, высушили над сульфатом натрия, отфильтровали и десорбировали.
Неочищенный продукт сначала очистили силикагелевой хроматографией, используя 4% смесь метанола в дихлорметане для удаления основных примесей. Полученный материал дополнительно очистили обращенно-фазовой ВЭЖХ (колонка С18, CH3CN/H2O, загруженная колонка со смесью 3:1, центрифугировали перед вводом) для получения соединения 262 в виде твердого вещества.1Н NMR (400 Hz, CDCl3): δ 7.68 (dd, J1=4.4 Hz, J2=1.6 Hz, 2H), 7.51 (s, 2H), 6.86 (s, 2H), 6.78 (s, 1H), 6.71 (s, 2H), 5.16 (dq, J1=8.4 Hz, J2=2.2, 4H), 4.85 (d, J=10.4 Hz, 2H), 4.58 (J=10.4 Hz, 2H), 4.04-3.97 (m, 7H), 3.68-3.38 (m, 18 H), 3.40-3.29 (m. 7H), 2.33 (t, 7.2 Hz, 2H), 1.89-1.35 (m, 2H) MS (m/z), найдено 914,1 (M+Na)+.
Пример 29 (IGN-13)
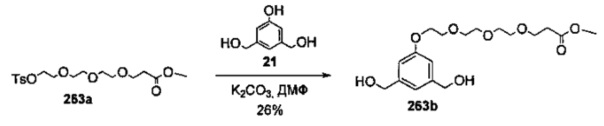
метил 3-(2-(2-(2-(3,5-бис(гидроксиметил)фенокси)этокси)этокси)этокси)пропаноат (263b):
К перемешанной смеси метил 3-(2-(2-(2-(тозилокси)этокси)этокси)этокси)пропаноата (263а) (1,504 г, 3,85 ммоль) и (5-гидрокси-1,3-фенилен)диметанола (21) (0,54 г, 3,50 ммоль) в безводном ДМФ (7,8 мл) добавили карбонат калия (0,726 г, 5,25 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов при 75°С. Смесь оставили остывать до комнатной температуры, погасили водой и экстрагировали этилацетатом. Органические экстракты промыли насыщенным солевым раствором, высушили над безводным сульфатом магния, отфильтровали и концентрировали. В результате очистки силикагелевой хроматографией (5% МеОН/CH2Cl2) получили метил 3-(2-(2-(2-(3,5-бис(гидроксиметил)фенокси)этокси)этокси)этокси)пропаноат (263b) (340 мг, 26%).1Н NMR (400 Hz, CDCl3): δ 6.83 (s, 1Н), 6.75 (s, 2H), 4.52 (s, 4H), 4.05 (t, J=4.8 Hz, 2H), 3.79 (t, J=4.8 Hz, 2H), 3.70 (t, J=6.4 Hz, 2H), 3.65 (s, 3H), 3.70-3.56 (m, 8H), 3.26 (s, 2H), 2.55 (t, J=6.4 Hz, 2H);13C NMR (400 Hz, CDCl3): δ 172.31, 159.1, 143.0, 117.7, 112.1, 70.8, 70.7, 70.5, 70.4, 69.8, 67.5, 66.6, 64.7, 51.8, 34.9: MS (m/z), найдено 395,2 (M+Na)+.

Соединение 263c:
К перемешанному раствору метил 3-(2-(2-(2-(3,5-бис(гидроксиметил)фенокси)этокси) этокси)этокси)пропаноата (263b) (145 мг, 0,389 ммоль) в безводном дихлорметане (5,5 мл) добавили триэтиламин (0,163 мл, 1,168 ммоль). Смесь охладили до -5°С и медленно добавили метансульфонилхлорид (0,076 мл, 0,973 ммоль). После перемешивания в течение одного часа при -5°С реакцию погасили холодной водой и экстрагировали холодным этилацетатом. Органические экстракты промыли холодной водой, высушили над безводным сульфатом натрия, отфильтровали и концентрировали при пониженном давлении для получения метил 3-(2-(2-(2-(3,5-бис((метилсульфонилокси)метил)фенокси)этокси)этокси)этокси) пропаноата. MS (m/z), найдено 551,1 (М+Na)+. К перемешанной смеси метил 3-(2-(2-(2-(3,5-бис((метилсульфонилокси)метил)фенокси)этокси)этокси)этокси)пропаноата (206 мг, 0,390 ммоль) и соединения 8 (287 мг, 0,974 ммоль) в безводном ДМФ (3,9 мл) добавили карбонат калия 269 мг, 1,949 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Смесь погасили водой и экстрагировали три раза дихлорметаном. Органические экстракты промыли водой и насыщенным солевым раствором, высушили над безводным сульфатом натрия и концентрировали in vacuo. В результате очистки силикагелевой флэш-хроматографией (5%, МеОН/CH2Cl2) с последующей препаративной обращенно-фазовой ВЭЖХ (колонка С18, элюированная смесью CH3CN/H2O) получили соединение 263с (110 мг, 30%) в виде твердого белого вещества.1Н NMR (400 Hz, CDCl3): δ 8.18 (d, J=8.0 Hz, 2H), 7.77 (m, 2H), 7.49 (s, 2H), 7.19 (m, 4H), 7.02 (m, 2H), 6.89 (s, 2H), 6.87 (s, 1H), 6.75 (s, 2H), 5.10 (m, 4H), 4.39 (m, 2H), 4.05 (m, 2H), 3.90 (s, 6H), 3.77 (m, 2H), 3.67 (t, J=6.4 Hz, 2H), 3.64 (m, 2H), 3.59 (s, 3H), 3.70-3.54 (m, 8H), 3.40 (m, 2H), 2.51 (t, J=6.4 Hz, 2H); MS (m/z), найдено 965,3 (M+H2O+Na)+, 983,3 (M+2H2O+Na)+.
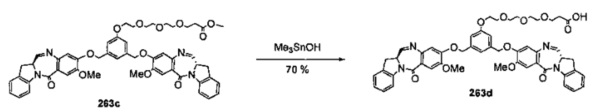
Соединение 263d:
К раствору соединения 263c (51 мг, 0,055 ммоль) в 1,2-дихлорэтане (2,2 мл) добавили гидроксид триметилолова (199 мг, 1,103 ммоль). Реакционную смесь перемешивали в течение 18 часов при 80°С, затем охладили до комнатной температуры и погасили насыщенным раствором хлорида аммония. Смесь экстрагировали дихлорметаном. Органический слой промыли насыщенным солевым раствором, высушили над безводным сульфатом натрия, отфильтровали и концентрировали. В результате очистки силикагелевой хроматографией (10% МеОН/CH2Cl2) получили соединение 263d (35 мг, 70%).1Н NMR (400 Hz, CDCl3): δ 8.26 (d, J=8.0 Hz, 2H), 7.88 (m, 2H), 7.58 (s, 2H), 7.28 (m, 4H), 7.11 (m, 3Н), 7.00 (s, 2H), 6.88 (s, 2H), 5.21 (m, 4H), 4.49 (m, 2H), 4.18 (m, 2H), 4.00 (s, 6H), 3.89 (m, 2H), 3.79 (m, 2H), 3.70 (m, 10H), 3.51 (m, 2H), 2.62 (m, 2H); MS (m/z), найдено 909,2 (M-1)-, 927,2 (M-1+H2O)-, 945,2 (M-1+2H2O)-.
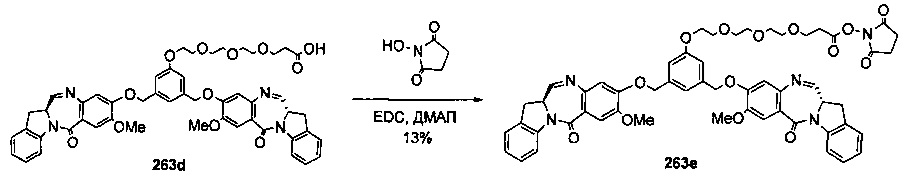
Соединение 263е:
К раствору соединения 263d (30 мг, 0,033 ммоль) в безводном дихлорметане (2,5 мл) добавили N-гидроксисукцинимид (9,77 мг, 0,082 ммоль), EDC (15,78 мг, 0,082 ммоль) и ДМАП (0,406 мг, 3,29 мкмоль). Реакционную смесь перемешивали в течение 18 часов при комнатной температуре, затем разбавили дихлорметаном. Смесь промыли насыщенным раствором хлорида аммония и насыщенным солевым раствором. Органический слой высушили над безводным сульфатом натрия, отфильтровали и концентрировали in vacuo. Неочищенный материал очистили препаративной обращенно-фазовой ВЭЖХ (колонка С18, элюированная смесью CH3CN/H2O). Фракции, содержащие продукт, экстрагировали дихлорметаном, высушили над безводным сульфатом натрия, отфильтровали и упарили с ацетонитрилом при пониженном давлении для получения соединения 263е (4,5 мг, 13%) в виде твердого белого вещества; MS (m/z), найдено 1030,4 (М+Na)+, 1046,3 (М+K)+.
Пример 30 (IGN-27)
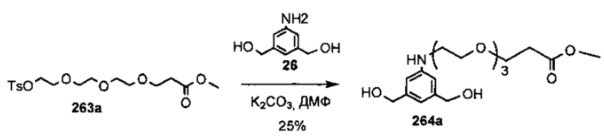
метил 3-(2-(2-(2-(3,5-бис(гидроксиметил)фениламино)этокси)этокси)этокси)пропаноат (264а):
К смеси метил 3-(2-(2-(2-(тозилокси)этокси)этокси)этокси)пропаноата (263а) (250 мг, 0,640 ммоль) и (5-амино-1,3-фенилен)диметанола (26) (108 мг, 0,704 ммоль) в безводном ДМФ (1,4 мл) добавили карбонат калия (133 г, 0,960 ммоль). Реакционную смесь перемешивали в течение 18 часов при 80°С, затем оставили остывать до комнатной температуры. Смесь погасили водой и дважды экстрагировали этилацетатом. Органические экстракты промыли насыщенным солевым раствором, высушили над безводным сульфатом магния, отфильтровали и концентрировали. В результате очистки силикагелевой хроматографией (5% метанол/метиленхлорид) получили метил 3-(2-(2-(2-(3,5-бис(гидроксиметил)фениламино)этокси)этокси)этокси)пропаноат (264а) (61 мг, 25%);1Н NMR (400 Hz, CDCl3): δ 6.58 (s, 1Н), 6.47 (s, 2H), 4.49 (s, 4H), 3.67 (t, J=6.4 Hz, 2H), 3.62 (s, 3Н), 3.64-3.54 (m, 10Н), 3.21 (t, J=5.2 Hz, 2H), 2.51 (t, J=6.4 Hz, 2H); MS (m/z), найдено 394,3 (M+Na)+.

Соединение 264b:
К раствору метил 3-(2-(2-(2-(3,5-бис(гидроксиметил)фениламино)этокси) этокси)этокси)пропаноата (264а) (60 мг, 0,162 ммоль) в ацетонитриле (1,6 мл) добавили йодметан (0,013 мл, 0,210 ммоль) и карбонат калия (26,8 мг, 0,194 ммоль). Реакционную смесь перемешивали при 82°С в течение 18 часов. Смесь охладили до комнатной температуры, затем удалили растворитель при пониженном давлении. Неочищенный материал разбавили смесью CH2Cl2/МеОН 3:1 и отфильтровали через целит.Фильтрат концентрировали и очистили силикагелевой хроматографией, элюируя 5% смесью метанол/дихлорметан для получения соединения 264b (35 мг, 56%).1Н NMR (400 Hz, CDCl3): δ 6.58 (s, 3Н), 4.52 (s, 4Н), 3.64 (t, J=6.4 Hz, 2H), 3.60 (s, 3Н), 3.53 (m, 12H), 2.91 (s, 3Н), 2.51 (t, J=6.4 Hz, 2H), 2.28 (s, 2H);13C NMR (400 Hz, CDCl3): δ 172.1, 149.8, 142.4, 113.4, 109.9, 70.7, 70.6, 70.4, 70.3, 68.6, 66.5, 65.6, 52.3, 51.7, 38.9, 34.8; MS (m/z), найдено 408,4 (M+Na)+.
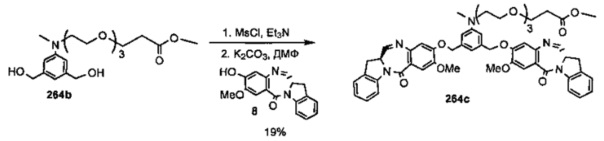
Соединение 264c:
К перемешанному раствору соединения 246b (60 мг, 0,156 ммоль) в безводном дихлорметане (2,8 мл) добавили триэтиламин (0,065 мл, 0,467 ммоль). Смесь охладили до -5°С и медленно добавили метансульфонилхлорид (0,030 мл, 0,389 ммоль). После перемешивания в течение одного часа при -5°С реакцию погасили холодной водой и экстрагировали холодным этилацетатом. Органический слой промыли холодной водой, высушили над безводным сульфатом натрия, отфильтровали и концентрировали при пониженном давлении для получения димезилатного промежуточного соединения. MS (m/z), найдено 564,0 (M+Na)+. К смеси связующего димезилата (49 мг, 0,090 ммоль) и соединения 8 (66,6 мг, 0,226 ммоль) в безводном ДМФ (0,9 мл) добавили карбонат калия (62,5 мг, 0,452 ммоль). Реакционную смесь перемешивали в течение 18 часов при комнатной температуре, погасили реакцию водой и экстрагировали три раза дихлорметаном. Органические экстракты промыли водой и насыщенным солевым раствором, высушили над безводным сульфатом натрия и концентрировали in vacuo. В результате очистки силикагелевой флэш-хроматографией (5%, МеОН/CH2Cl2) с последующей препаративной обращенно-фазовой ВЭЖХ (колонка С18, элюированная смесью CH3CN/H2O) получили соединение 264с (16 мг, 19%) в виде твердого белого вещества.1H NMR (400 Hz, CDCl3): δ 8.18 (d, J=8.0 Hz, 2H), 7.76 (m, 2H), 7.48 (s, 2H), 7.18 (m, 4H), 7.02 (t, J=7.2 Hz, 2H), 6.79 (m, 2H), 6.74 (s, 1H), 6.65 (s, 2H), 5.08 (m, 4H), 4.39 (m, 2H), 3.89 (s, 6H), 3.66 (t, J=6.4 Hz, 2H), 3.62 (m, 2H), 3.60 (s, 3H), 3.53 (m, 12H), 3.40 (m, 2H), 2.91 (s, 3H), 2.51 (t, J=6.4 Hz, 2H); MS (m/z), найдено 978,3 (M+H2O+Na)+, 996,3 (M+2H2O+Na)+.
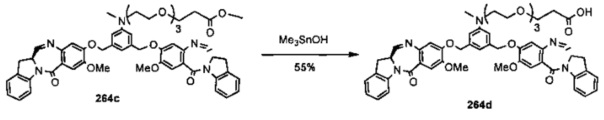
Соединение 264d:
К раствору соединения 264c (26 мг, 0,028 ммоль) в безводном 1,2-дихлорэтане (1,1 мл) добавили гидроксид триметилолова (100 мг, 0,554 ммоль). Реакционную смесь перемешивали в течение 18 часов при 80°С. Смесь оставили остывать до комнатной температуры и экстрагировали дихлорметаном и насыщенным раствором хлорида аммония. Органические экстракты промыли насыщенным солевым раствором, высушили над безводным сульфатом натрия, отфильтровали и концентрировали in vacuo. В результате очистки препаративной тонкослойной хроматографией в 5% смеси метанол/метиленхлорид получили соединение 264d (14 мг, 55%). MS (m/z), найдено 922,1 (М-1)-, 940,0 (М-1+H2O)-, 958,1 (М-1+2H2O)-.
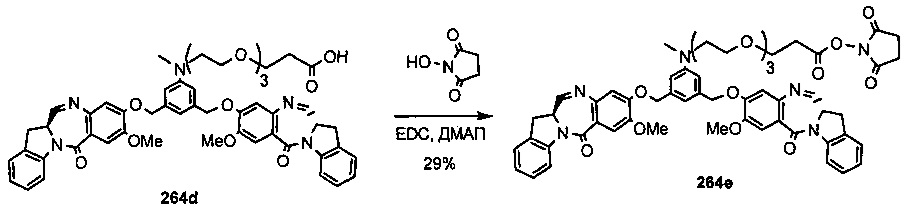
Соединение 264е:
К перемешанному раствору соединения 264d (13 мг, 0,014 ммоль) в безводном дихлорметане (1,0 мл) добавили N-гидроксисукцинимид (5,01 мг, 0,042 ммоль), EDC (8,09 мг, 0,042 ммоль) и ДМАП (0,172 мг, 1,407 мкмоль). Реакционную смесь перемешивали в течение 18 часов при комнатной температуре. Смесь экстрагировали дихлорметаном и насыщенным раствором хлорида аммония. Органический слой промыли насыщенным солевым раствором, высушили над безводным сульфатом натрия, отфильтровали и концентрировали in vacuo. Неочищенный материал очистили препаративной обращенно-фазовой ВЭЖХ (колонка С18, элюированная смесью CH3CN/H2O). Фракции, содержащие продукт, объединили и экстрагировали дихлорметаном, высушили над безводным сульфатом натрия, отфильтровали и упарили с ацетонитрилом при пониженном давлении для получения соединения 264е (4,1 мг, 29%). MS (m/z), найдено 1021,3 (М+Н)+, 1043,2 (М+Na)+, 1061,2 (М+H2O+Na)+, 1079,2 (М+2H2O+Na)+.
Пример 31 (IGN-28)
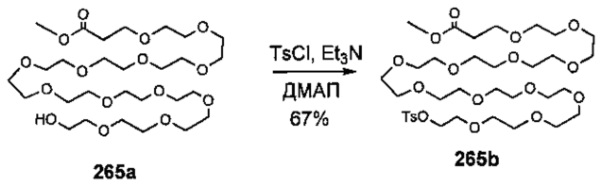
метил 1-(тозилокси)-3,6,9,12,15,18,21,24,27,30,33,36-додекаоксанонатриаконтан-39-оат (265b):
К перемешанному раствору метил 1-гидрокси-3,6,9,12,15,18,21,24,27,30,33,36-додекаоксанонатриаконтан-39-оата (265а) (1,2 г, 1,897 ммоль) в дихлорметане (9,48 мл) при 0°С добавили триэтиламин (0,529 мл, 3,79 ммоль), толуол сульфонилхлорид (0,542 г, 2,84 ммоль) и ДМАП (0,023 г, 0,190 ммоль). Смесь перемешивали в течение одного часа при 0°С, а затем три часа при комнатной температуре, после чего ее погасили водой и дважды экстрагировали дихлорметаном. Органические экстракты промыли насыщенным солевым раствором, высушили над безводным сульфатом магния, отфильтровали и концентрировали in vacuo. В результате очистки силикагелевой хроматографией (5% МеОН/CH2Cl2) получили метил 1-(тозилокси)-3,6,9,12,15,18,21,24,27,30,33,36-додекаоксанонатриаконтан-39-оат (265b) (1,0 г, 67%) в виде светло-желтой маслянистой жидкости.1Н NMR (400 Hz, CDCl3): δ 7.80 (d, J=8.4Hz, 2H), 7.35 (d, J=8.0Hz, 2H), 4.16 (t, J=4.8Hz, 2H), 3.75 (t, J=6.4Hz, 2H), 3.69 (s, 3H), 3.64 (m, 46H), 2.60 (t, J=6.4 Hz, 2H), 2.45 (s, 3H).

метил 1-(3,5-бис(гидроксиметил)фениламино)-3,6,9,12,15,18,21,24,27,30,33,36-додекаоксанонатриаконтан-39-оат (265с):
К перемешанной смеси метил 1-(тозилокси)-3,6,9,12,15,18,21,24,27,30,33,36-додекаоксанонатриаконтан-39-оата (265b) (700 мг, 0,890 ммоль) и (5-амино-1,3-фенилен)диметанола (26) (150 мг, 0,978 ммоль) в безводном ДМФ (2,0 мл) добавили карбонат калия (184 мг, 1,334 ммоль). Реакционную смесь перемешивали при 80°С в течение ночи. Смесь охладили до комнатной температуры, погасили водой и экстрагировали 10% смесью метанол/метиленхлорид. Органический слой промыли насыщенным солевым раствором, высушили над безводным сульфатом магния, отфильтровали и концентрировали in vacuo. Неочищенный продукт очистили силикагелевой хроматографией (элюированной 5→15% смесью МеОН/CH2Cl2) для получения метил 1-(3,5-бис(гидроксиметил)фениламино)-3,6,9,12,15,18,21,24,27,30,33,36-додекаоксанонатриаконтан-39-оата (265с) (285 мг, 42%).1Н NMR (400 Hz, CDCl3): δ 6.62 (s, 1Н), 6.51 (s, 2H), 4.52 (s, 4H), 3.72 (t, J=6.4 Hz, 2H), 3.65 (s, 3H), 3.61 (m, 48H), 2.94 (s, 2H), 2.63 (s, 1H), 2.57 (t, J=6.4 Hz, 2H); MS (m/z), найдено 790,4 (M+Na)+.

метил 2-(3,5-бис(гидроксиметил)фенил)-5,8,11,14,17,20,23,26,29,32,35,38-додекаокса-2-азагентетраконтан-41-оат (265d):
К перемешанному раствору метил 1-(3,5-бис(гидроксиметил)фениламино)-3,6,9,12,15,18,21,24,27,30,33,36-додекаоксанонатриаконтан-39-оата (265с) (67 мг, 0,087 моль) в безводном ДМФ (1,0 мл) добавили йодметан (7,06 мкл, 0,113 ммоль) и карбонат калия (14,47 мг, 0,105 ммоль). Реакционную смесь перемешивали при 82°С в течение 18 часов. Смесь охладили до комнатной температуры, разбавили водой и экстрагировали дихлорметаном. Органический слой высушили над безводным сульфатом натрия, отфильтровали и концентрировали in vacuo. В результате очистки препаративной тонкослойной хроматографией (10% МеОН/CH2Cl2) получили метил 2-(3,5-бис(гидроксиметил)фенил)-5,8,11,14,17,20,23,26,29,32,35,38-додекаокса-1-азагентетраконтан-41-оат (265d) (62 мг, 92%).1Н NMR (400 Hz, CDCl3): δ 6.65 (s, 3Н), 4.59 (d, J=5.6 Hz, 4H), 3.74 (t, J=6.4 Hz, 2H), 3.67 (s, 3H), 3.61 (m, 46H), 3.54 (t, J=6.0 Hz, 2H) 2.98 (s, 3H), 2.59 (t, J=6.4 Hz, 2H), 2.55 (m, 2H); MS (m/z), найдено 820,5 (M+K)+.
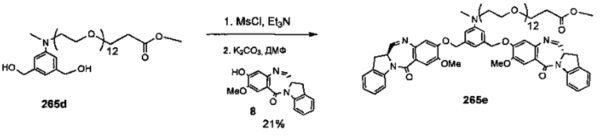
Соединение 265e:
К перемешанному раствору метил 2-(3,5-бис(гидроксиметил)фенил)-5,8,11,14,17,20,23,26,29,32,35,38-додекаокса-2-азагентетраконтан-41-оата (265d) (71 мг, 0,091 ммоль) в безводном дихлорметане (1,4 мл) добавили триэтиламин (0,038 мл, 0,272 ммоль). Смесь охладили до -5°С и медленно добавили метансульфонилхлорид (0,018 мл, 0,227 ммоль). После перемешивания в течение одного часа при -5°С реакцию погасили холодной водой и экстрагировали холодным этилацетатом. Органические экстракты промыли холодной водой, высушили над безводным сульфатом натрия, отфильтровали и концентрировали при пониженном давлении для получения метил 2-(3,5-бис((метилсульфонилокси)метил)фенил)-5,8,11,14,17,20,23,26,29,32,35,38-додекаокса-2-азагентетраконтан-41-оата. MS (m/z), найдено 960,2 (М+Na)+. К смеси метил 2-(3,5-бис((метилсульфонилокси)метил)фенил)-5,8,11,14,17,20,23,26,29,32,35,38-додекаокса-2-азагентетраконтан-41-оата (69 мл, 0,074 ммоль) и соединения 8 (54,1 мг, 0,184 ммоль) в безводном ДМФ (0,8 мл) добавили карбонат калия (50,8 мг, 0,368 ммоль). Реакционную смесь перемешивали в течение 18 часов при комнатной температуре. Реакцию погасили водой и дважды экстрагировали дихлорметаном. Оставшийся водный слой дважды экстрагировали 50% смесью МеОН/CH2Cl2. Объединенные органические экстракты промыли насыщенным солевым раствором, высушили над безводным сульфатом натрия, отфильтровали и концентрировали in vacuo. В результате очистки силикагелевой флэш-хроматографией (5% МеОН/CH2Cl2) с последующей препаративной обращенно-фазовой ВЭЖХ (колонка С18, элюированная смесью CH3CN/H2O) получили соединение 265е (23 мг, 23%). MS (m/z), найдено 1375,4 (М+Na+H2O)+, 1393,4 (М+Na+2H2O)+.
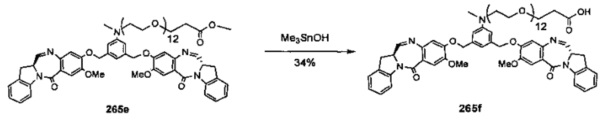
Соединение 265f:
К перемешанному раствору соединения 265е (22 мг, 0,016 ммоль) в безводном 1,2-дихлорэтане (300 мкл) добавили гидроксид триметилолова (44,7 мг, 0,247 ммоль). Реакционную смесь перемешивали при 90°С в течение 18 часов. Реакционную смесь охладили до комнатной температуры, а затем разбавили дихлорметаном. Органический слой промыли насыщенным солевым раствором, содержащим несколько капель 5%-ной соляной кислоты, а затем только насыщенным солевым раствором, высушили над безводным сульфатом натрия, отфильтровали и концентрировали in vacuo. В результате очистки препаративной тонкослойной хроматографией (2х 5% МеОН/CH2Cl2) получили соединение 265f (7,5 мг, 34%). MS (m/z), найдено 1318,4 (М-1)-, 1336,4 (М-1+H2O)-, 1354,4 (М-1+2H2O)-.
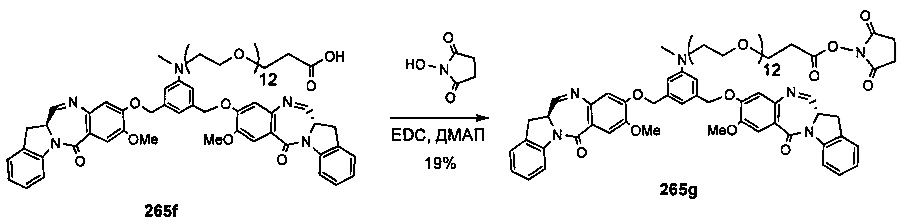
Соединение 265g:
К перемешанному раствору соединения 265f (7,5 мг, 5,68 мкмоль) в безводном дихлорметане (400 мкл) добавили N-гидроксисукцинимид (1,961 мг, 0,017 ммоль), EDC (3,27 мг, 0,017 ммоль) и ДМАП (0,069 мг, 0,568 мкмоль). Реакционную смесь перемешивали в течение 18 часов при комнатной температуре. Смесь экстрагировали дихлорметаном и насыщенным раствором хлорида аммония. Органический слой промыли насыщценным солевым раствором, высушили над безводным сульфатом натрия, отфильтровали и удалили растворитель при пониженном давлении. Неочищенный материал очистили препаративной обращенно-фазовой ВЭЖХ (колонка С18, элюированная смесью CH3CN/H2O). Фракции, содержащие продукт, объединили и экстрагировали дихлорметаном, высушили над безводным сульфатом натрия, отфильтровали и упарили с ацетонитрилом для получения соединения 265g (1,5 мг, 19%). MS (m/z), найдено 1439,9 (M+Na)+, 1457,9 (M+H2O+Na)+.
Пример 32 (IGN-22)
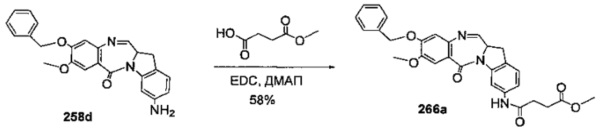
Соединение 266а:
К раствору соединения 258d (20 мг, 0,050 ммоль) в дихлорметане (1,0 мл) добавили моно-метилсукцинат (13,23 мг, 0,100 ммоль), EDC (19,20 мг, 0,100 ммоль) и ДМАП (3,06 мг, 0,025 ммоль). Реакционную смесь перемешивали в течение 18 часов при комнатной температуре. Смесь разбавили водой и экстрагировали этилацетатом. Органические экстракты промыли насыщенным солевым раствором, отфильтровали и концентрировали при пониженном давлении. В результате очистки силикагелевой хроматографией (3% МеОН/CH2Cl2) получили соединение 266а (15 мг, 58%). MS (m/z), найдено 568,4 (М+Na+МеОН)+.
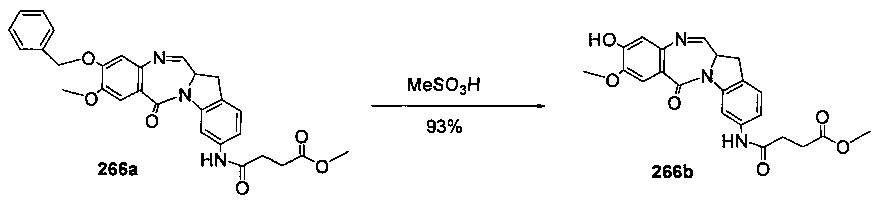
Соединение 266b:
К раствору соединения 266а (15 мг, 0,029 ммоль) в дихлорметане (3,5 мл) добавили метансульфоновую кислоту (0,114 мл, 1,753 ммоль). Реакционную смесь перемешивали в течение одного часа при комнатной температуре, затем разбавили метанолом и водой. Смесь нейтрализовали насыщенным раствором бикарбоната натрия до рН=7 и три раза экстрагировали дихлорметаном. Органический слой высушили над безводным сульфатом натрия, отфильтровали и концентрировали in vacuo. В результате очистки препаративной тонкослойной хроматографией (2х 5% МеОН/CH2Cl2) получили соединение 266b (11,5 мг, 93%). MS (m/z), найдено 446,4 (М+Na)+, 478,4 (М+Na+MeOH)+.
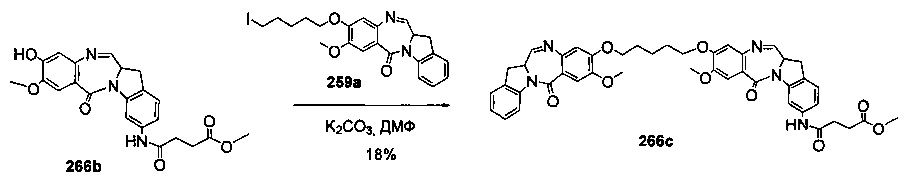
Соединение 266с:
К смеси соединения 266b (11,5 мг, 0,027 ммоль) и соединения 259а (19,98 мг, 0,041 ммоль) в безводном ДМФ (0,5 мл) добавили карбонат калия (11,26 мг, 0,081 ммоль). Реакционную смесь перемешивали в течение 18 часов при комнатной температуре. Смесь погасили водой и экстрагировали три раза дихлорметаном. Органический слой промыли насыщенным солевым раствором, высушили над безводным сульфатом натрия, отфильтровали и концентрировали при пониженном давлении. В результате очистки препаративной тонкослойной хроматографией (5% МеОН/CH2Cl2) с последующей препаративной обращенно-фазовой ВЭЖХ (колонка С18, элюированная смесью CH3CN, H2O) получили соединение 266с (4 мг, 18%).1Н NMR (400 Hz, CDCl3): δ 8.27 (d, J=8.0Hz, 1H), 8.06 (s, 1H), 7.87 (m, 2H), 7.74 (m, 1H), 7.55 (s, 1H), 7.52 (s, 1H), 7.49 (m, 1H), 7.26 (m, 1H) 7.19 (d, J=8.8Hz, 1H), 7.10 (m, 1H), 6.82 (m, 2H), 4.49 (m, 2H), 4.12 (m, 4H), 3.95 (s, 6H), 3.71 (s, 3H), 3.48 (m, 4H), 2.75 (m, 2H), 2.66 (m, 2H), 1.98 (m, 4H), 1.70 (m, 2H); MS (m/z), найдено 824,1 (M+K)+.
Пример 33 (IGN-31)

3,5-бис((трет-бутилдиметилсилилокси)метил)-N-(2-(2-(2-метоксиэтокси)этокси)этил)анилин (267a):
К раствору (5-(2-(2-(2-метоксиэтокси)этокси)этиламино)-1,3-фенилен)диметанола (249b) (0,4 г, 1,336 ммоль) в дихлорметане (6,68 мл) добавили трет-бутилдиметилсилил хлорид (0,604 г, 4,01 ммоль) и имидазол (0,318 г, 4,68 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 90 минут. Смесь разбавили дихлорметаном и отфильтровали через целит. Фильтрат концентрировали и очистили силикагелевой хроматографией, элюируя 20% смесью этилацетат/гексаны для получения 3,5-бис((трет-бутилдиметилсилилокси)метил)-N-(2-(2-(2-метоксиэтокси)этокси)этил)анилина (267а) (600 мг, 85%). MS (m/z), найдено 550,3 (М+Na)+.
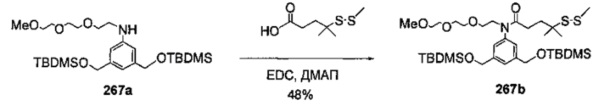
N-(3,5-бис((трет-бутилдиметилсилилокси)метил)фенил)-N-(2-(2-(2-метоксиэтокси)этокси)этил)-4-метил-4-(метилдисульфанил)пентанамид (267b):
К смеси 3,5-бис((трет-бутилдиметилсилилокси)метил)-N-(2-(2-(2-метоксиэтокси)-этокси)этил)анилина (267а) (525 мг, 0,995 ммоль) и 4-метил-4-(метилдисульфанил)пентановой кислоты (232 мг, 1,193 ммоль) в безводном дихлорметане (9,0 мл) добавили EDC (229 мг, 1,193 ммоль) и ДМАП (12,15 мг, 0,099 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение пяти часов. Смесь разбавили дихлорметаном и водой. Органический слой промыли насыщенным солевым раствором, высушили над безводным сульфатом натрия, отфильтровали и концентрировали при пониженном давлении. В результате очистки силикагелевой хроматографией (30% смесь этилацетат/гексаны) получили N-(3,5-бис((трет-бутилдиметилсилилокси)метил)фенил)-N-(2-(2-(2-метоксиэтокси)этокси)этил)-4-метил-4-(метилдисульфанил)пентанамид (267b) (335 мг, 48%).
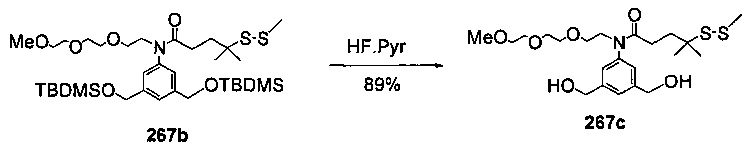
N-(3,5-бис(гидроксиметил)фенил)-N-(2-(2-(2-метоксиэтокси)этокси)этил)-4-метил-4-(метилдисульфанил)пентанамид (267с):
К перемешанному раствору N-(3,5-бис((трет-бутилдиметилсилилокси)метил)фенил)-N-(2-(2-(2-метоксиэтокси)этокси)этил)-4-метил-4-(метилдисульфанил)пентанамида (267b) (315 мг, 0,447 ммоль) в безводном ацетонитриле (7,0 мл) при 0°С добавили безводный пиридин (7,00 мл), а затем по каплям добавили HF.пиридин (3,1 мл, 1 мл/100 мг). Реакционную смесь перемешивали при 0°С в течение двух часов. Смесь разбавили этилацетатом и медленно погасили насьпценным раствором бикарбоната натрия. Смесь экстрагировали этилацетатом три раза. Органический слой промыли насыщенным солевым раствором, высушили над сульфатом натрия, отфильтровали и концентрировали. В результате очистки силикагелевой хроматографией, элюируя 5% смесью МеОН/CH2Cl2, получили N-(3,5-бис((гидроксиметил)фенил)-N-(2-(2-(2-метоксиэтокси)этокси)этил)-4-метил-4-(метилдисульфанил)пентанамид (267с) (190 мг, 89%).1Н NMR (400 Hz, CDCl3): δ 7.21 (s, 1H), 7.16 (s, 2H), 4.63 (s, 4H), 3.79 (t, J=5.2,5.6 Hz, 2H), 3.53 (m, 6H), 3.48 (m, 4H), 3.29 (s, 3H), 2.53 (s, 2H), 2.27 (s, 3H), 2.07 (m, 2H), 1.84 (m, 2H), 1.08 (s, 6H); MS (m/z), найдено 498,2 (M+Na)+.
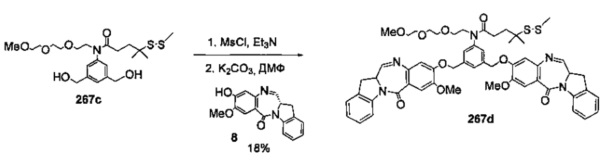
Соединение 267d:
К перемешанному раствору N-(3,5-бис(гидроксиметил)фенил)-N-(2-(2-(2-метоксиэтокси)этокси)этил)-4-метил-4-(метиддисульфанил)пентанамида (267с) (72 мг, 0,151 ммоль) в безводном дихлорметане (3,0 мл) добавили триэтиламин (0,063 мл, 0,454 ммоль). Смесь охладили до -5°С и медленно добавили метансульфонилхлорид (0,029 мл, 0,378 ммоль). После перемешивания в течение одного часа при -5°С реакцию погасили холодной водой и экстрагировали холодным этилацетатом. Органический слой промыли холодной водой, высушили над безводным сульфатом натрия, отфильтровали и концентрировали при пониженном давлении для получения (5-(N-(2-(2-(2-метоксиэтокси)этокси)этил)-4-метил-4-(метилдисульфанил)пентанамидо)-1,3-фенилен)бис(метилен)диметансульфоната. MS (m/z), найдено 654,1 (М+Na)+. К смеси (5-(N-(2-(2-(2-метоксиэтоси)этокси)этил)-4-метил-4-(метилдисульфанил)пентанамидо)-1,3-фенилен)бис(метилен)диметансульфоната (89 мг, 0,141 ммоль) и соединения 8 (83 мг, 0,282 ммоль) в безводном ДМФ (1,5 мл) добавили карбонат калия (97 мг, 0,704 ммоль). Реакционную смесь перемешивали в течение 18 часов при комнатной температуре. Смесь погасили водой и дважды экстрагировали дихлорметаном. Органический слой промыли насыщенным солевым раствором, высушили над безводным сульфатом натрия, отфильтровали и концентрировали in vacuo. В результате очистки силикагелевой хроматографией (5% МеОН/CH2Cl2) и препаративной обращенно-фазовой ВЭЖХ (колонка С18, элюированная смесью CH3CN/H2O) получили соединение 267d (27 мг, 18%).1Н NMR (400 Hz, CDCl3): δ 8.28 (d, J=4.8 Hz, 2H), 7.87 (m, 2H), 7.61 (s, 2H), 7.37-7.27 (m, 7H), 7.13 (t, J=7.2, 7.6 Hz, 2H), 6.88 (s, 2H), 5.25 (m, 4H), 4.50 (m, 2H), 4.00 (s, 6H), 3.90 (m, 2H), 3.73 (m, 2H), 3.60 (m, 6H), 3.51 (m, 6H), 3.30 (s, 3H), 2.32 (s, 3H), 2.15 (m, 2H), 1.90 (m, 2H), 1.13 (s, 6H); MS (m/z), найдено 1050.3 (M+Na)+, 1068.3 (M+H2O+Na)+, 1086.3 (M+2H2O+Na)+.
Пример 34 (IGN-32)

Соединение 268а:
К смеси соединения 253b (150 мг, 0,389 ммоль) и трет-бутил 3-(2-(2-(2-аминоацетамидо)ацетамидо)ацетамидо)пропаноата (148 мг, 0,467 ммоль) в безводном ДМФ (1,5 мл) добавили EDC (90 мг, 0,467 ммоль) и ДМАП (4,75 мг, 0,039 ммоль). Реакционную смесь перемешивали в течение 18 часов при комнатной температуре. Смесь напрямую очистили препаративной обращенно-фазовой ВЭЖХ (колонка С18, элюированная смесью CH3CN/H2O + 0,1% муравьиной кислоты). В результате дополнительной очистки препаративной тонкослойной хроматографией (15% МеОН/CH2Cl2) получили соединение 268а (170 мг, 64%).1Н NMR (400 Hz, CDCl3): δ 7.62 (m, 1Н), 7.56 (m, 1H), 7.38 (m, 1H), 7.11 (m, 1H), 6.55 (s, 2H), 6.52 (s, 1H), 4.45 (s, 4H), 4.17 (s, 2H), 3.63 (m, 6H), 3.55-3.40 (m, 12H), 3.28 (m, 7H), 2.33 (t, J=6.4 Hz, 2H), 2.16 (m, 2H), 1.79 (m, 2H), 1.36 (s, 9H); MS (m/z), найдено 706,3 (M+Na)+.
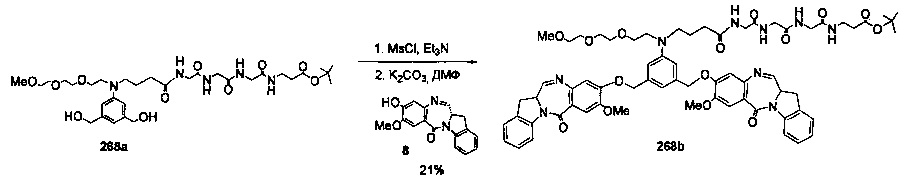
Соединение 268b:
К перемешанному раствору соединения 268а (59 мг, 0,086 ммоль) в безводном дихлорметане (1,75 мл) добавили триэтиламин (0,036 мл, 0,259 ммоль). Смесь охладили до -5°С и медленно добавили метансульфонилхлорид (0,017 мл, 0,216 ммоль). После перемешивания в течение одного часа при -5°С реакцию погасили холодной водой и экстрагировали холодным этилацетатом. Органические экстракты промыли холодной водой, высушили над безводным сульфатом натрия, отфильтровали и концентрировали при пониженном давлении для получения желаемого димезилатного промежуточного соединения. MS (m/z), найдено 862,3 (М+Na)+.
К раствору промежуточного димезилатного соединения (65 мг, 0,077 ммоль) и соединения 8 (114 мг, 0,387 ммоль) в безводном ДМФ (1,0 мл) добавили карбонат калия (86 мг, 0,619 ммоль). Реакционную смесь перемешивали в течение 18 часов при комнатной температуре, погасили водой и экстрагировали три раза дихлорметаном. Органический слой промыли насыщенным солевым раствором, высушили над безводным сульфатом натрия, отфильтровали и концентрировали при пониженном давлении. В результате очистки силикагелевой хроматографией (2%→10% МеОН/CH2Cl2) получили соединение 268b (22 мг, 21%).1H NMR (400 Hz, CDCl3): δ 8.26 (d, J=8.0 Hz, 2H), 7.88 (m, 2H), 7.58 (s, 2H), 7.28 (m, 4H), 7.13 (t, J=7.2 Hz, 2H), 6.89 (s, 2H), 6.81 (s, 1H), 6.73 (s, 2H), 5.19 (m, 4H), 4.48 m, 2H), 3.99 (s, 6H), 3.7-3.4 (m, 26H), 3.34 (s, 3H), 2.45 (t, J=6.4 Hz, 2H), 2.30 (m, 2H), 1.81 (m, 2H), 1.44 (s, 9H).
Пример 35
Получение конъюгата chB38.1-IGN14:
Раствор антитела chB38.1 концентрацией 2 мг/мл в водном буферном растворе, содержащем 0,05 М N-(2-гидроксиэтил)-пиперазин-N'-2-этансульфоновой кислоты (HEPES) и 2 мМ этилендиаминтетрауксусной кислоты (ЭДТА), рН 8, обработали 10-кратным молярным избытком раствора IGN14-NHS в диметилацетамиде (ДМА), так что конечная концентрация ДМА в буферном растворе составила 10% об./об. Реакционную смесь перемешивали при комнатной температуре в течение 120 минут, а затем загрузили в гель-фильтрационную колонку Sephadex G25 (обессоливающая колонка HiPrep™ 26/10 GE# 17-5087-01), предварительно уравновешенную водным буферным раствором, содержащим 10 мМ гистидина, 250 мМ глицина, 1% сахарозы, рН 5,5. Фракции, содержащие конъюгированное антитело, собрали и объединили для получения продукта. Объединенную выборку подвергали диализу в течение ночи против такого же элюирующего буферного раствора для дополнительной очистки продукта. Полученный конъюгат анализировали спектрофотометрически, используя коэффициенты экстинкции, определенные для IGN-14 (ε330 нм=15231 М-1 см-1 и ε280 нм=26864 М-1 см-1) и антитела chB38.1 (ε280 нм=204000 М-1 см-1). В среднем с одной молекулой антитела связалось 3,3 молекул IGN14.
Пример 36
Получение конъюгата huMy9-6-IGN23
Раствор антитела huMy9-6 концентрацией 2 мг/мл в водном буферном растворе, содержащем 0,05 М N-(2-гидроксиэтил)-пиперазин-N'-2-этансульфоновой кислоты (HEPES) и 2 мМ этилендиаминтетрауксусной кислоты (ЭДТА), рН 8,5, обработали 12,5-кратным молярным избытком раствора IGN23-NHS в диметилацетамиде (ДМА), глицерине и сахарозе. Окончательная концентрация ДМА, глицерина и сахарозы в буферном растворе составила 15%, 5% и 5% (об./об.), соответственно. Реакционную смесь перемешивали при комнатной температуре в течение 120 минут, а затем загрузили в гель-фильтрационную колонку Sephadex G25 (обессоливающая колонка HiPrep™ 26/10 GE# 17-5087-01), предварительно уравновешенную водным буферным раствором, содержащим 10 мМ гистидина, 250 мМ глицина, 1% сахарозы, рН 5,5. Фракции, содержащие конъюгированное антитело, собрали и объединили для получения продукта. Объединенную выборку концентрировали на фильтрующей центрифуге Millipore, а затем подвергали диализу в течение ночи против того же элюирующего буферного раствора для дополнительной очистки продукта.
Полученный конъюгат анализировали спектрофотометрически, используя коэффициенты экстинкции, определенные для IGN-23 (ε330 нм=15231 М-1 см-1 и ε280 нм=26864 М-1 см-1) и huMy9-6 (ε280 нм=206460 М-1 см-1). В среднем с одной молекулой антитела связалось 2,2 молекул IGN23.
Пример 37
Получение конъюгата chB38.1-IGN27
Раствор антитела chB38.1 концентрацией 2 мг/мл в водном буферном растворе, содержащем 0,05 М N-(2-гидроксиэтил)-пиперазин-N'-2-этансульфоновой кислоты (HEPES) и 2 мМ этилендиаминтетрауксусной кислоты (ЭДТА), рН 8,5, обработали 12-кратным молярным избытком раствора IGN27-NHS в диметилацетамиде (ДМА, исходный раствор 5 мМ), так что конечная концентрация ДМА в буферном растворе составила 15% об./об. Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, а затем загрузили в гель-фильтрационную колонку Sephadex G25 (обессоливающая колонка HiPrep™ 26/10 GE# 17-5087-01), предварительно уравновешенную водным буферным раствором, содержащим PBS, рН 7,4. Фракции, содержащие конъюгированное антитело, собрали и объединили для получения продукта. Объединенную выборку подвергали диализу в течение ночи против такого же элюирующего буферного раствора для дополнительной очистки продукта.
Полученный конъюгат анализировали спектрофотометрически, используя коэффициенты экстинкции, определенные для IGN-27 (ε330 нм=15231 М-1 см-1 и ε280 нм=26864 М-1 см-1) и антитела chB38.1 (ε280 нм=204000 М-1 см-1). В среднем с одной молекулой антитела связалось 2,9 молекул IGN27.
Пример 38
Эффективность In Vitro свободных лекарств и IGN конъюгатов:
Общая использованная методика: Образцы лекарств, не содержащих IGN или IGN конъюгаты, добавили в 96-луночные плоскодонные планшеты тканевых культур и титровали серийными растворами, охватывающими нужный молярный диапазон. Антиген-положительные (Ag+) или антиген-отрицательные (Ag-) клетки добавили в лунки с определенными плотностями клеток, так что получили тройные образцы для каждой концентрации лекарства для каждой соответствующей клеточной линии. Затем планшеты инкубировали при 37°С в атмосфере 5% CO2 в течение 4-5 дней в зависимости от клеточной линии. COLO 205 (1000 клеток в лунке), Namalwa (3000 клеток в лунке) - 4 дня; RH30 (1000 клеток в лунке), Ramos (10000 клеток в лунке), KB (1000 клеток в лунке) - 5 дней.
В конце инкубационного периода проанализировали цитотоксические активности, используя WST-анализ жизнеспособности клеток, а выжившие клетки измерили выращиванием с WST (2-7 часов). Измерили поглощение в каждой лунке и отметили на графике выжившую фракцию клеток при каждой концентрации для выявления цитотоксичности и антиген-специфичности (конъюгатов).
Измерили цитотоксичность лекарств, не содержащих IGN, и активность и специфичность IGN конъюгатов против панели клеточных линий рака человека, выбранных из COLO 205, NB-4, LOVO, Namalwa, RH30, Ramos, KB, и/или LOVO. Результаты представлены на фиг. 51-58.
Фиг. 51: Таблица, показывающая высокую эффективность (в нМ) лекарств, не содержащих IGN, против различных клеточных линий. В целом найдено, что лекарства, не содержащие IGN, эффективны против этой панели клеточных линий в нижнем пикомолярном диапазоне.
Фиг. 52: (А) Найдено, что конъюгат chB38.1-IGN13 (3,8 IGN/Ab) эффективен в суб-пикомолярных дозировках против клеток COLO 205 (Ag+), и что его эффективность существенно уменьшилась (0,26 нМ) при блокировании связывающих центров антигена 1 мкМ неконъюгированным антителом chB38.1, что показывает высокую специфичность этого конъюгата (>260 раз). (В) Найдено, что конъюгат chB38.1-IGN13 (3.8 IGN/Ab) эффективен в пикомолярных дозировках (0,002 пМ) против клеток LOVO (Ag+) в клоногенном анализе.
Фиг. 53: (А) Найдено, что конъюгат huMy9-6-IGN13 (3,4 IGN/Ab) эффективен в пикомолярных дозировках против клеток NB-4 (Ag+) (0,077 нМ), и что его эффективность существенно уменьшилась (1,0 нМ) при блокировании связывающих центров антигена 1 мкМ неконъюгированным антителом huMy9-6, что показывает высокую специфичность этого конъюгата.
Фиг. 54: (А) Найдено, что конъюгат chB38.1-IGN14 (3,1 IGN/Ab) эффективен в суб-пикомолярных дозировках против клеток COLO 205 (Ag+), и что его эффективность значительно ниже против клеток Namalwa (Ag-) (0,9 нМ), что показывает высокую специфичность этого конъюгата (>900 раз). (В) Найдено, что конъюгат chB38.1-IGN14 (2,6 IGN/Ab) высоко эффективен против клеток LOVO (Ag+), и что его эффективность значительно ниже против клеток Namalwa (Ag-) (>3,0 нМ), что показывает высокую специфичность этого конъюгата (>250 раз).
Фиг. 55: Найдено, что конъюгат huMy9-6-IGN14 (3,3 IGN/Ab) высоко эффективен против клеток NB-4 (Ag+), и что его эффективность значительно ниже против клеток Namalwa (Ag-) (0,6 нМ), что показывает высокую специфичность этого конъюгата.
Фиг. 56: (А) Найдено, что конъюгат chB38.1-IGN23 (2,5 IGN/Ab) эффективен в пикомолярных дозировках против клеток LOVO (Ag+) (0,063 нМ), и что его эффективность значительно ниже против клеток Namalwa (Ag-) (>3,0 нМ), что показывает высокую специфичность этого конъюгата. (В) Найдено, что конъюгат chB38.1-IGN23 (3,4 IGN/Ab) эффективен в пикомолярных дозировках против клеток COLO 205 (Ag+) (0,006 нМ), и что его эффективность существенно уменьшилась (2,5 нМ) при блокировании связывающих центров антигена 1 мкМ chB38.1, что показывает специфичность этого конъюгата.
Фиг. 57: Найдено, что конъюгат chB38.1-IGN29 (2,8 IGN/Ab) эффективен в суб-наномолярных дозировках против клеток COLO 205 (Ag+) (0,410 нМ), и что его эффективность существенно уменьшилась (18 нМ) при блокировании связывающих центров антигена 1 мкМ chB38.1, что показывает специфичность этого конъюгата.
Пример 39
Эффективность конъюгата chB38.1-IGN14 In vivo на безтимусных мышах с опухолью COLO 205:
В этом исследовании была изучена противоопухолевая активность chB38.1-IGN14 на безтимусных самках мышей с опухолью COLO 205, модели рака толстой кишки человека. Опухолевые клетки COLO 205, 2×106 клеток на одну мышь, были инокулированы подкожно в объеме 0,1 мл/мышь в области около правого плеча безтимусной самки мыши в возрасте 5 недель. Через восемь дней после инокуляции опухолевых клеток мышей случайным образом разделили на группы (n=6 в каждой группе) по объему опухоли. Лечение начали в день разделения на группы, среди групп была также контрольная группа, которой ввели PBS (200 мкл/инъекция), оголенное антитело chB38.1 (2,8 мг/кг), ненаправленный конъюгат chKTI-IGN14 (50 мкг/кг) и chB38.1-IGN14 (дозировка IGN14 50 мкг/кг; дозировка антитела 2,5 мг/кг). Лечение проводили дважды, раз в неделю (на 8 и 15 день после клеточной инокуляции). Стрелки указывают время введения дозировок после инокуляции. Во всех случаях лечения отмечена хорошая переносимость со средней потерей массы тела, сравнимой с потерей массы у контрольных мышей PBS. Средний объем опухоли в зависимости от времени представлен (фиг. 58) с данными, демонстрирующими противоопухолевую активность конъюгата chB38.1-IGN14. Ненаправленные и оголенные антитела не проявили активности выше, чем контрольный носитель, указывая на то, что противоопухолевая активность, наблюдаемая с конъюгатом chB38.1-IGN-14, является антиген-специфичной.
Пример 40
На фиг. 59 представлен масс-спектр chB38.1-IGN14 (дегликозилированное антитело). Пики помечены D1-D7 для обозначения количества молекул IGN14, прикрепленных к одному антителу. Рассчитано, что среднее количество молекул IGN14 на одно антитело составляет 3,5 (согласуется с дозировкой лекарственного средства, рассчитанного по УФ-видимой спектрофотометрии).
Реферат
Изобретение относится к соединениям формулы (III) и формулы (VI):или их фармацевтически приемлемым солям, цитотоксичным димерам, конъюгатам мономеров и димеров этих соединений, где: двойная линиямежду N и C представляет собой двойную связь, Х отсутствует, а Y является H; W является C=O; Rявляется OR, SH или NHR; необязательно Rявляется связывающей группой или выбран из полипирролового, полииндолилового, полиимидазолилового, полипирроло-имидазолилового, полипирроло-индолилового или полиимидазоло-индолилового звена, необязательно несущего связывающую группу; Rявляется OR; R является H или линейным или разветвленным алкилом, имеющим от 1 до 3 углеродных атомов; X’ является CH; Y’ является O; и Z’ является CH; каждый A и A’ является O; каждый D и D’ является линейным алкилом, имеющим от 1 до 10 углеродных атомов; L отсутствует или является фенильной группой, которая необязательно является замещенной, где заместитель является связывающей группой, обеспечивающей связь с клеточносвязывающим агентом за счет ковалентной связи, или выбран из OR, NRRи OCOR; R является H или линейным или разветвленным алкилом, имеющим от 1 до 3 углеродных атомов; каждый R, R, R, и Rнезависимо является H, линейным или разветвленным алкилом, имеющими от 1 до 10 углеродных атомов, или полиэтиленгликолевым фрагментом –(OCHCH), где n является целым числом от 1 до 10. Изобретение также относится к способам применения соединений или конъюгатов для in vitro, in situ и in vivo диагностирования или лечения клеток млекопитающих или связанных патологических состояний. 8 н. и 10 з.п. ф-лы, 59 ил., 7 табл., 40 пр.
Формула
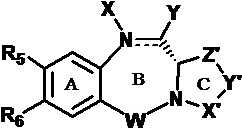


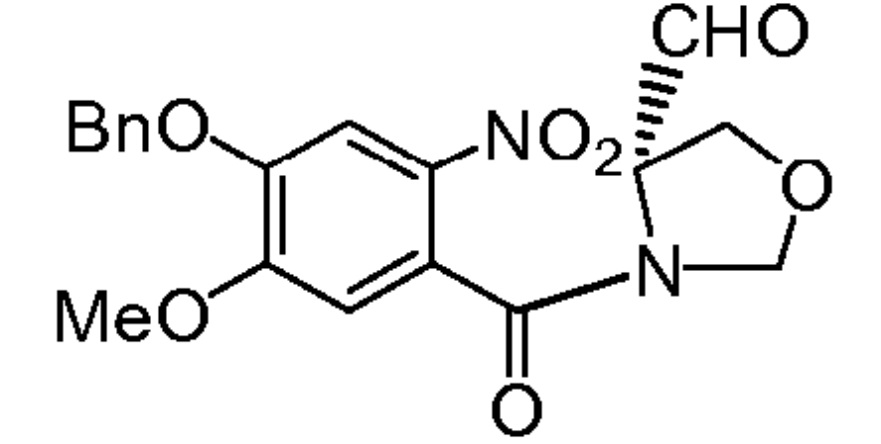

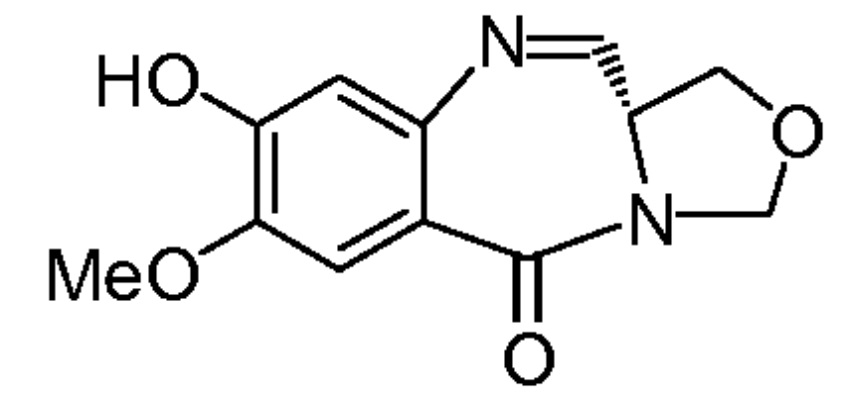
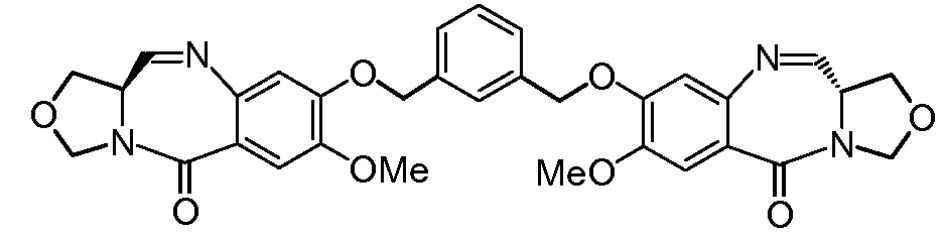


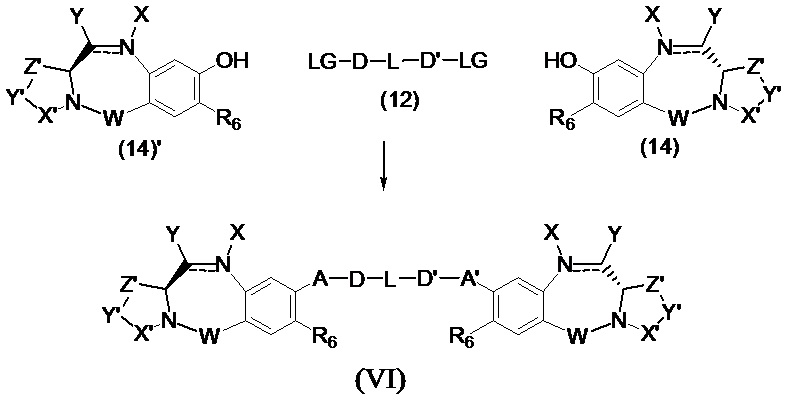
Комментарии