Конъюгаты и малые молекулы, взаимодействующие с рецептором cd16а - RU2519546C1
Код документа: RU2519546C1
Чертежи














Описание
Изобретение относится к области медицины, в частности к онкологии и иммунологии, к новым соединениям, связывающимся с CD 16а рецептором, и модифицированным ими белкам (конъюгатам), используемым для индукции антителозависимой клеточной цитотоксичности и удаления таким образом из организма определенной целевой группы клеток, например, раковых клеток или аутоиммунных лимфоцитов. Изобретение относится также к способу получения конъюгатов, фармацевтическим композициям и лекарственным средствам, содержащим модифицированные белки (конъюгаты), для лечения онкологических и аутоиммунных заболеваний.
Рецептор FcγIIIa (CD16а) принадлежит к группе рецепторов, отвечающих за связывание Fc-фрагмента антител. CD 16а экспрессируется на поверхности NK-клеток (киллеров) и макрофагов и отвечает за индукцию антителозависимой клеточной цитотоксичности (ADCC), взаимодействуя с Fc-фрагментом связанного с клеткой антитела. ADCC, наряду с комплементзависимой цитотоксичностью (CDC) и апоптозом, является одним из основных механизмов уничтожения раковых клеток из организма. Он же является причиной опосредованных аутоантителами аутоиммунных заболеваний, таких как аутоиммунная полиэндокринопатия первого типа, аутоиммунная гемолитическая анемия, идиопатическая тромбоцитопения, гемолитическая болезнь новорожденных и т.п. То есть антителозависимая клеточная цитотоксичность может играть как положительную, так и отрицательную роль в развитии патологических процессов в организме человека.
Аутоиммунные заболевания - группа заболеваний, развивающихся вследствие выработки иммунного ответа против здоровых тканей организма и приводящих к повреждению этих тканей. В настоящее время для лечения аутоиммунных заболеваний используются иммуносупресанты, подавляющие иммунную систему организма в целом. Избирательное подавление аутоиммунного ответа позволило бы существенно снизить частоту побочных эффектов лечения.
Антитела достаточно давно используются для направленного уничтожения раковых клеток. Примерами могут служить ритуксимаб, трастузумаб, цетуксимаб и многие другие антитела, имеющие мишени на поверхности раковых клеток и действующие через комплиментзависимую цитотоксичность и антитело-зависимую клеточную цитотоксичность. Это позволяет использовать данные неконъюгированные моноклональные антитела в качестве лекарственных средств для лечения рака, например, ритуксимаб - для лечения CD20-позитивной В-клеточной, низкозлокачественной или фолликулярной неходжкинской лимфомы, трастузумаб - для лечения развитого рака молочной железы. Успешное применение этих продуктов обусловлено не только их эффективностью, но и их очень хорошими профилями безопасности (Grillo-Lopez A. - J et al. Semin. Oncol., 26, 1999, pp.66-73).
Все они принесли возможность нового вида терапии. Но, несмотря на ярко выраженную эффективность, примерно половина больных не отвечает на терапию ритуксимабом, а до 60% становятся резистентными при повторном применении. В свете успехов, связанных с этими лекарственными средствами, в настоящее время существует большая потребность в достижении более высокой специфической активности антител по сравнению с той, которая, как правило, обеспечивается при лечении с использованием неконъюгированных антител.
Это послужило причиной для усиления направленного терапевтического эффекта антител к поверхностным антигенам. Первым направлением модификации антител стала разработка модифицированных белков (конъюгатов) антитело - лекарственное средство (ADC) для локальной доставки цитотоксических или цитостатических средств, то есть лекарственных средств, уже используемых для уничтожения или ингибирования опухолевых клеток при лечении злокачественной опухоли [Payne, G. (2003) Cancer Cell 3:207-212; Trail et al. (2003) Cancer Immunol. Immunother. 52:328-337; Syrigos and Epenetos (1999) Anticancer Research 19:605-614; Niculescu-Duvaz and Springer (1997) Adv. Drug Del. Rev. 26:151-172; US 4,975,278]. Такие конъюгаты обеспечивают направленную доставку лекарственного вещества к опухолям и их накопление внутри клеток, добавляя к цитотоксическому действию антител противоопухолевую активность цитотоксических или цитостатических лекарств. Примерами таких конъюгатов являются трастузумаб-DM1, усиленная версия трастузумаба (Герцептина) (WO201169074) и серия конъюгатов с ауристатинами Е (US20120003248).
Альтернативным направлением стало усиление собственной цитотоксичности антител посредством усиления взаимодействия с рецепторами, обуславливающими цитотоксичность. Компания Рош разработала антитело обинутузумаб с усиленным связыванием с рецептором CD16а. Этот эффект достигается за счет инженерии гликозилирования антитела. Обинутузумаб обладает в десятки раз более сильной антителозависимой клеточной цитотоксичностью (WO2005044859, ЕА015009).
Антителозависимая клеточная цитотоксичность - один из основных механизмов цитотоксического действия антител, которые связываются с антигеном на поверхности целевой клетки посредством вариабельных доменов, в то время как константная часть связывается с CD16а рецептором на поверхности клеток киллеров. Этот межклеточный контакт приводит к секреции киллерами перфоринов и гранзимов. Первые образуют поры в клеточной мембране таргетной клетки, а вторые активируют каспазы и другие молекулы апоптоза. CD16а рецептор является членом большого семейства Fc-рецеторов, связывающихся с константным доменом антитела и различающихся локализацией, функцией и сродством к константному домену.
Ниже приведены определения терминов, которые использованы в описании этого изобретения.
«Алкил» означает алифатическую углеводородную линейную или разветвленную группу с 1-12 атомами углерода в цепи. Разветвленная означает, что алкильная цепь имеет один или несколько «низших алкильных» заместителей. Алкил может иметь один или несколько одинаковых или различных заместителей («алкильных заместителей») включая галоген, алкенилокси, циклоалкил, арил, гетероарил, гетероциклил, ароил, циано, гидрокси, алкокси, карбокси, алкинилокси, аралкокси, арилокси, арилоксикарбнил, алкилтио, гетероарилтио, аралкилтио, арилсульфонил, алкилсульфонилгетероаралкилокси, аннелированный гетероарилциклоалкенил, аннелированный гетероарилциклоалкил, аннелированный гетероарилгетероцикленил, аннелированный гетероарилгетероциклил, аннелированный арилциклоалкенил, аннелированный арилциклоалкил, аннелированный арилгетероцикленил, аннелированный арилгетероциклил, алкоксикарбонил, аралкоксикарбонил, гетероаралкилоксикарбонил или RkaRk+1aN-, RkaRk+1aNC(=O)-, RkaRk+1aNC(=S)-, RkaRk+1aNSO2-, где Rka и Rk+1a независимо друг от друга представляют собой «заместители аминогруппы», значение которых определено в данном разделе, например, атом водорода, алкил, арил, аралкил, гетероаралкил, гетероциклил или гетероарил, или Rka и Rk+1a вместе с атомом N, с которым они связаны, образуют через Rka и Rk+1a 4-7- членный гетероциклил или гетероцикленил. Предпочтительными алкильными группами являются метил, трифторметил, циклопропилметил, циклопентилметил, этил, н-пропил, изо-пропил, н-бутил, трет-бутил, н-пентил, 3-пентил, метоксиэтил, карбоксиметил, метоксикарбонилметил, этоксикарбонилметил, бензилоксикарбонилметил метокси-карбонилметил и пиридилметилоксикарбонилметил. Предпочтительными «алкильными заместителями» являются циклоалкил, арил, гетероарил, гетероциклил, гидрокси, алкокси, алкоксикарбонил, аралкокси, арилокси, алкилтио, гетероарилтио, аралкилтио, алкилсульфонил, арилсульфонил, алкоксикарбонил, аралкоксикарбонил, гетероаралкилоксикарбонил или RkaRk+1aN-, RkaRk+1aNC(=0)-, аннелированный арилгетероцикленил, аннелированный арилгетероциклил.
«Аминоалкил» означает CnH2n+1NH- или (CnH2n+1)(CnH2n+1)N- группу, в которой алкил определен в данном разделе. Предпочтительными алкиламино группами являются метиламино, этиламино, н-пропиламино, изо-пропиламино и н-бутиламино. «Антитело» - белок (иммуноглобулин), синтезируемый В-лимфоцитами в организме в ответ на попадание в него чужеродного вещества и обладающий специфическим сродством к этому веществу. Они являются важнейшим фактором специфического гуморального иммунитета. Антитела выполняют две функции: антигенсвязывающую и эффекторную (вызывают тот или иной иммунный ответ).
«Аутоантигены» - свободные молекулы веществ или молекулы в составе клеток, органов и тканей, которые распознаются при определенных условиях иммунной системой как чужеродные и в связи с этим вызывают клеточный или гуморальный иммунный ответ со стороны своего организма. Это, как правило, - нормальные белки или белковые комплексы (а также комплексы белков с ДНК или РНК), которые распознаются иммунной системой у пациентов с аутоиммунными заболеваниями. Такие антигены в норме не должны узнаваться иммунной системой, но, ввиду генетических факторов или условий окружающей среды, иммунологическая толерантность к таким антигенам может быть утеряна.
Свойствами аутоантигенов могут обладать так называемые естественные аутоантигены (секвестированные). К ним относят белки, синтез которых начинается после созревания иммунной системы (сперма, молоко); макромолекулы органов, отделенных от иммунной системы гистогематическим барьером; макромолекулы, входящие в состав ядер и цитоплазмы клеток; макромолекулы с наличием новых чужеродных детерминантных групп вследствие действия эндогенных (иммунные комплексы, некроз, воспаление) или экзогенных (температура, химические вещества, в том числе лекарственные, микробы и их токсины, вирусы и др.) факторов; эмбриональные белки с возобновляющимся при определенных состояниях синтезом (напимер, при опухолях). Они могут индуцировать иммунный ответ, приводящий к образованию аутоантител или сенсибилизированных Т-лимфоцитов и развитию аутоиммунных болезней. В результате начинается развитие аутоиммунной реакции. Они могут приводить к развитию самых разнообразных аутоиммунных заболеваний. К ним, в частности, относятся аутоиммунная полиэндокринопатия первого типа, аутоиммунная гемолитическая анемия, идиопатическая тромбоцитопения, гемолитическая болезнь новорожденных, рассеянный склероз, аутоиммунный тиреоидит и др.
Аутоиммунитет - процесс и связанные с ним заболевания, обусловленные приобретением иммунной системой способности распознавать собственные антигены (аутоантигены) организма и реагировать на них образованием аутоантител или аутоиммунных Т-лимфоцитов. Аутоиммунный процесс - процесс и связанные с ними заболевания, основой которых является поражение тканей, обусловленное последствиями взаимодействия аутоантител или аутоиммунных Т-лимфоцитов с аутоантигенами.
«Конъюгат» - это модифицированный химическими соединениями белок, антиген или антитело. Образование конъюгата - один из важных этапов проведения имунно-ферментного анализа (ИФА). При формировании конъюгата подбирают такой оптимальный метод введения химического соединения, чтобы компонент конъюгата, антиген или антитело, сохранял свою биологическую активность - антигенность и антигенсвязывающую активность, соответственно. Способность чужеродных соединений и метаболитов вступать в реакции конъюгации зависит от наличия в их молекулах определенных функциональных групп.
«Лекарственное средство (препарат)» - вещество (или смесь веществ в виде фармацевтической композиции) в виде таблеток капсул инъекций, мазей и др. готовых форм, предназначенное для восстановления, исправления или изменения физиологических функций у человека и животных, а также для лечения и профилактики болезней, диагностики, анестезии, контрацепции, косметологии и прочего. «Рецепторы» (от латинского recipere - получать, узнавать) представляют собой биологические макромолекулы, расположенные на цитоплазматической мембране клетки или внутриклеточно, способные специфически взаимодействовать с ограниченным набором физиологически активных веществ (лигандов) и трансформировать сигнал об этом взаимодействии в определенный клеточный ответ.
«Сольваты» - продукты присоединения растворителя к растворенным веществам; частный случай сольватов - гидраты (растворитель - вода). Обычно сольваты образуются в растворе, но нередко (при охлаждении раствора, испарении растворителя и др.) могут быть получены в виде кристаллических фаз - кристаллосольватов.
«Фармацевтическая композиция» обозначает композицию, включающую в себя активный компонент (модифицированный белок) и, по крайней мере, один из компонентов, выбранных из группы, состоящей из фармацевтически приемлемых и фармакологически совместимых наполнителей, растворителей, разбавителей, носителей, вспомогательных, распределяющих и воспринимающих средств, средств доставки, таких как консерванты, стабилизаторы, наполнители, измельчители, увлажнители, эмульгаторы, суспендирующие агенты, загустители, подсластители, отдушки, ароматизаторы, антибактериальные агенты, фунгициды, лубриканты, регуляторы пролонгированной доставки, выбор и соотношение которых зависит от природы и способа назначения и дозировки. Примерами суспендирующих агентов являются этоксилированный изостеариловый спирт, полиоксиэтилен, сорбитол и сорбитовый эфир, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и -трагакант, а также смеси этих веществ. Защита от действия микроорганизмов может быть обеспечена с помощью разнообразных антибактериальных и противогрибковых агентов, например, таких как парабены, хлорбутанол, сорбиновая кислота и подобные им соединения. Композиция может включать также изотонические агенты, например, сахара, хлористый натрий и им подобные. Пролонгированное действие композиции может быть обеспечено с помощью агентов, замедляющих абсорбцию активного начала, например, моностеарат алюминия и желатин. Примерами подходящих носителей, растворителей, разбавителей и средств доставки являются вода, этанол, полиспирты, а также их смеси, растительные масла (такие как оливковое масло) и инъекционные органические сложные эфиры (такие как этилолеат). Примерами наполнителей являются лактоза, молочный сахар, цитрат натрия, карбонат кальция, фосфат кальция и им подобные. Примерами измельчителей и распределяющих средств являются крахмал, алгиновая кислота и ее соли, силикаты. Примерами лубрикантов являются стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоль с высоким молекулярным весом. Фармацевтическая композиция для перорального, сублингвального, трансдермального, внутримышечного, внутривенного, подкожного, местного или ректального введения активного начала, одного или в комбинации с другим активным началом, может быть введена животным и людям в стандартной форме введения, в виде смеси с традиционными фармацевтическими носителями. Пригодные стандартные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы, пилюли, порошки, гранулы, жевательные резинки и пероральные растворы или суспензии, сублингвальные и трансбуккальные формы введения, аэрозоли, имплантаты, местные, трансдермальные, подкожные, внутримышечные, внутривенные, интраназальные или внутриглазные формы введения и ректальные формы введения. Фармацевтические композиции, как правило, получают с помощью стандартных процедур, предусматривающих смешение активного соединения с жидким или тонко измельченным твердым носителем.
«Фармацевтически приемлемая соль» означает относительно нетоксичные органические и неорганические соли кислот и оснований, заявленных в настоящем изобретении. Эти соли могут быть получены in situ в процессе синтеза, выделения или очистки соединений или приготовлены специально. В частности, соли оснований могут быть получены специально, исходя из очищенного свободного основания заявленного соединения и подходящей органической или неорганической кислоты. Примерами полученных таким образом солей являются гидрохлориды, гидробромиды, сульфаты, бисульфаты, фосфаты, нитраты, ацетаты, оксалаты, валериаты, олеаты, пальмитаты, стеараты, лаураты, бораты, бензоаты, лактаты, тозилаты, цитраты, малеаты, фумараты, сукцинаты, тартраты, мезилаты, малонаты, салицилаты, пропионаты, этансульфонаты, бензолсульфонаты, сульфаматы и им подобные (Подробное описание свойств таких солей дано в Berge S.M., et al., “Pharmaceutical Salts” J. Pharm. Sci. 1977, 66: 1-19). Соли заявленных кислот также могут быть специально получены реакцией очищенной кислоты с подходящим основанием, при этом могут быть синтезированы соли металлов и аминов. К металлическим относятся соли натрия, калия, кальция, бария, цинка, магния, лития и алюминия, наиболее желательными из которых являются соли натрия и калия. Подходящими неорганическими основаниями, из которых могут быть получены соли металлов, являются гидроксид, карбонат, бикарбонат и гидрид натрия, гидроксид и бикарбонат калия, поташ, гидроксид лития, гидроксид кальция, гидроксид магния, гидроксид цинка. В качестве органических оснований, из которых могут быть получены соли заявленных кислот, выбраны амины и аминокислоты, обладающие достаточной основностью, чтобы образовать устойчивую соль, и пригодные для использования в медицинских целях (в частности, они должны обладать низкой токсичностью). К таким аминам относятся аммиак, метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, бензиламин, дибензиламин, дициклогексиламин, пиперазин, этилпиперидин, трис(гидроксиметил)аминометан и подобные им. Кроме того, для солеобразования могут быть использованы гидроокиси тетраалкиламмония, например, такие как холин, тетраметиламмоний, тетраэтиламмоний и им подобные. В качестве аминокислот могут быть использованы основные аминокислоты - лизин, орнитин и аргинин.
Цель настоящего изобретения заключается в создании новых соединений и модифицированных ими белков (конъюгатов), способных взаимодействовать с рецептором CD16a, используемым для индукции антителозависимой клеточной цитотоксичности и удаления таким образом из организма определенной целевой группы клеток, например, раковых клеток или аутоиммунных лимфоцитов.
Поставленная цель достигается обнаруженными авторами новыми соединениями, обладающими сродством к CD16a рецептору, содержащими активированную группу, способную присоединяться к аминогруппе белка, а именно новыми замещенными 5,5,11-триоксо-10,11-дигидро-5H-дибензо[b,f][1,4]тиазепинами общей формулы 1 или 5,6,7,8,9,10-гексагидро-4H-[1]бензотиено[3,2-f]пирроло[1,2-a][1,4]диазепинами общей формулы 2, или их фармацевтически приемлемыми солями или сольватами,
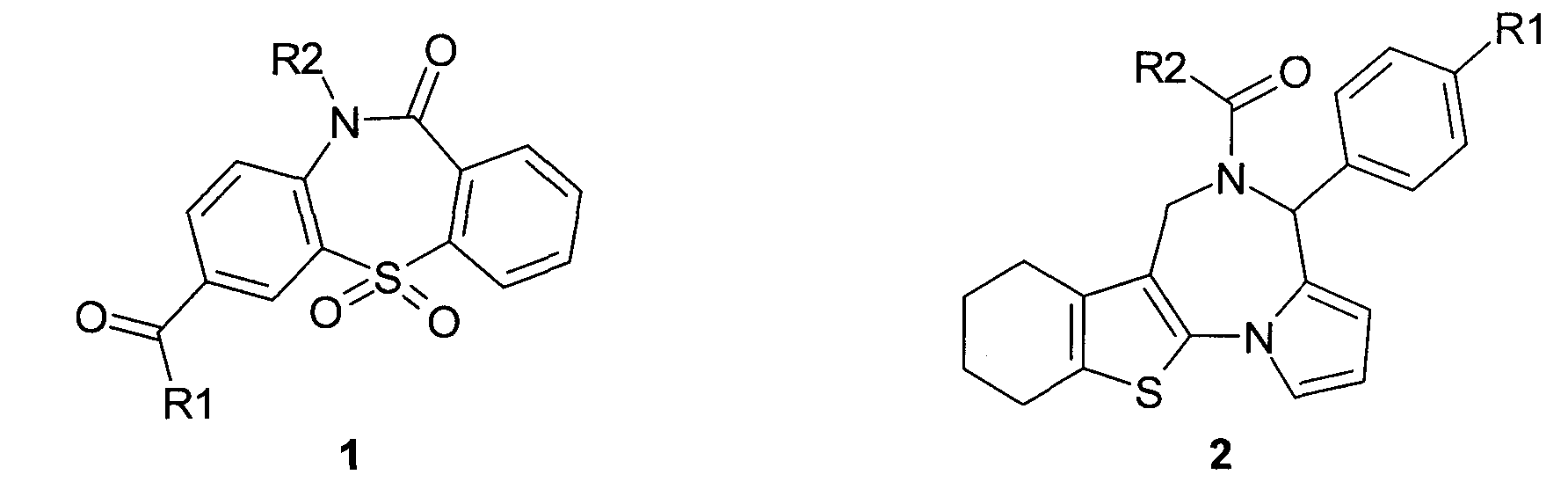
в которых R1 представляет собой (CH3)2N-,

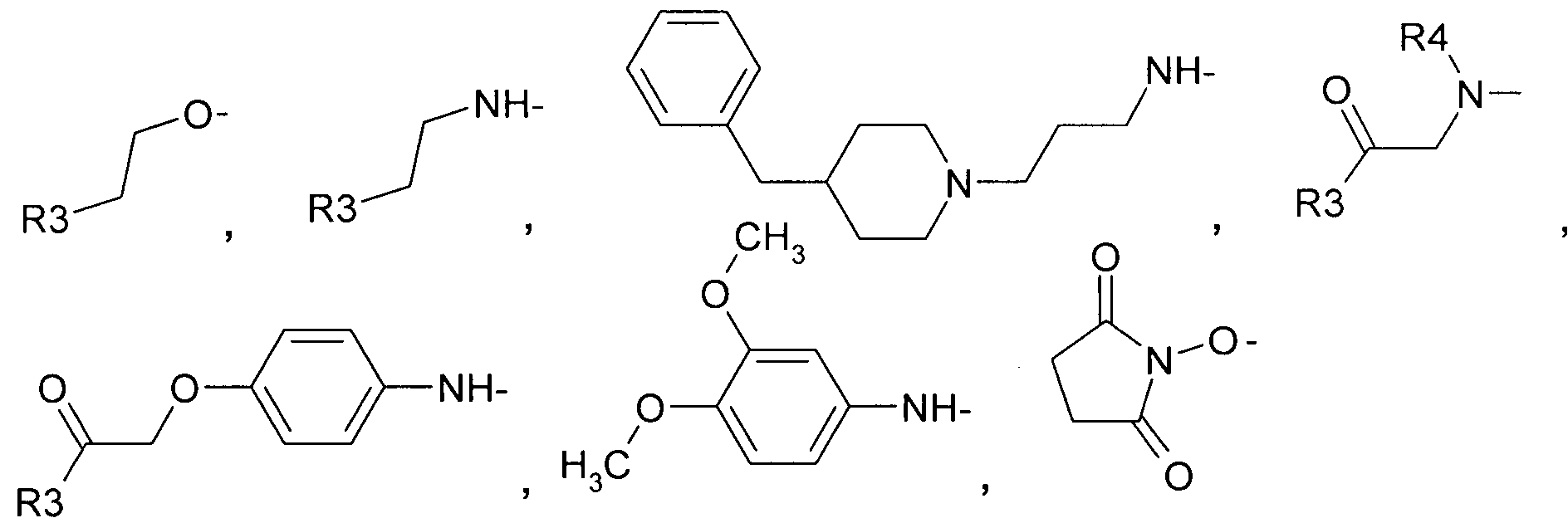
R2 представляет собой
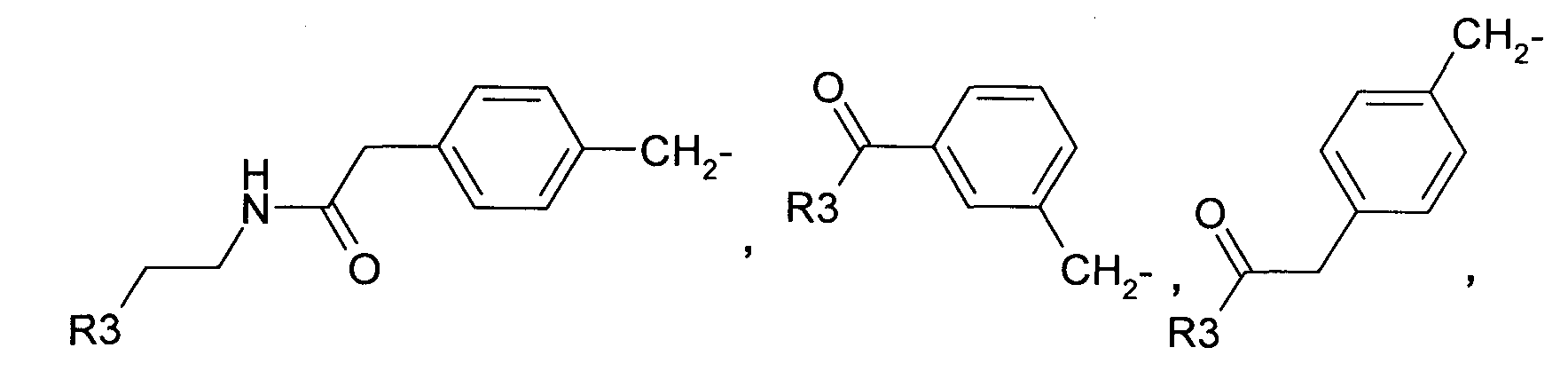
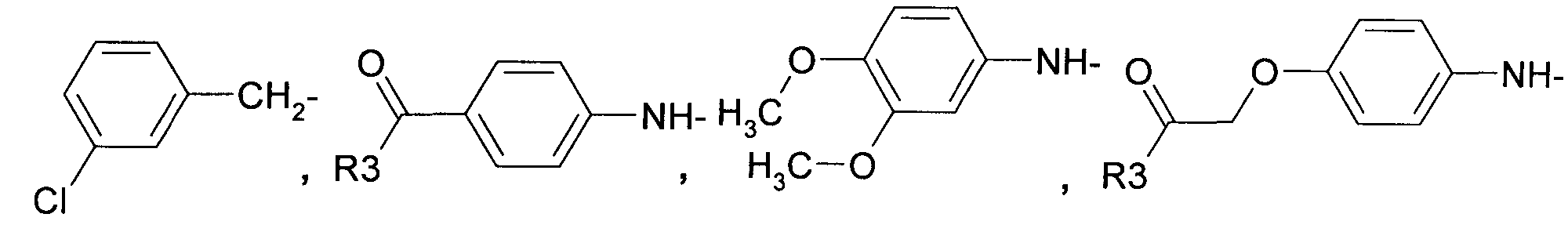
где R3 в качестве концевого заместителя представляет собой незамещенный аминоалкил,
-NH2,
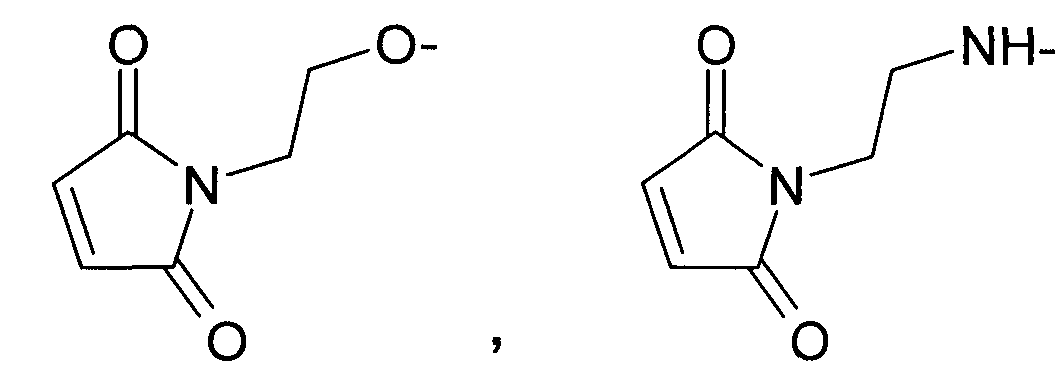
a R4 представляет собой H или C1-C3алкил.
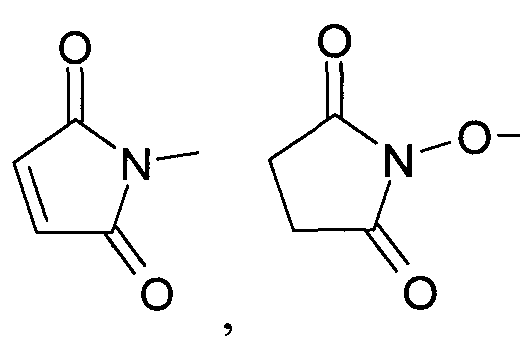
Предпочтительными являются соединения общей формулы 1, в которых R1 представляют собой:

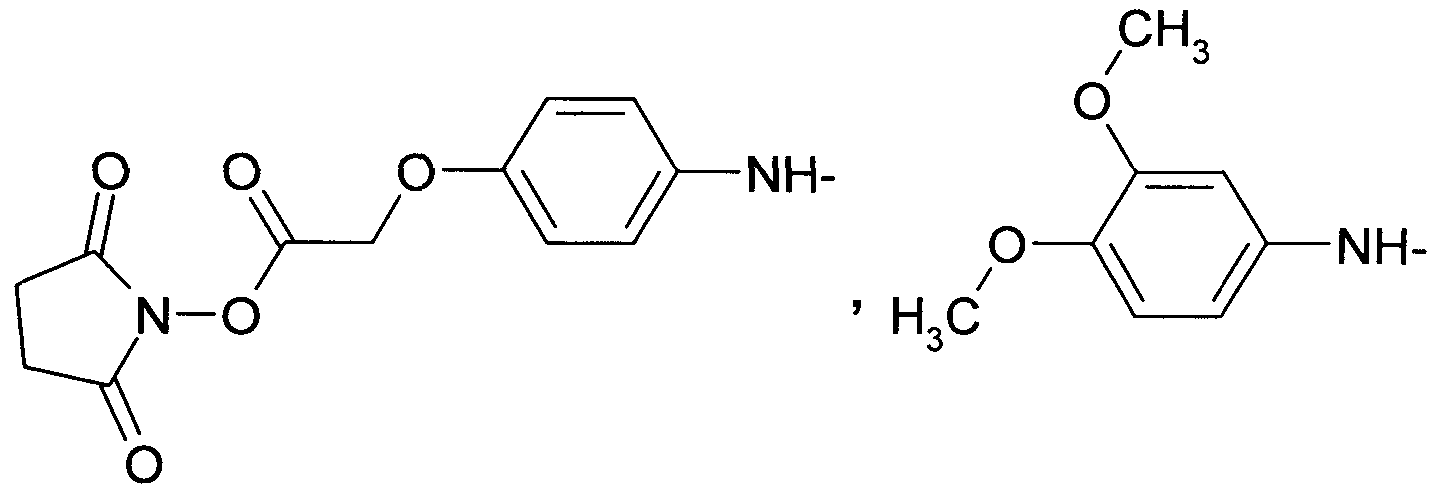

R2 выбран из группы, включающей в себя:

где R4=H или C1-C3алкил.
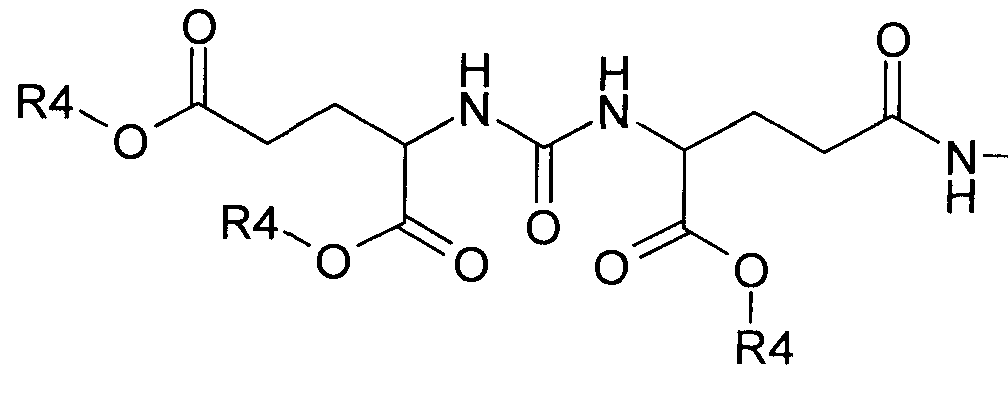
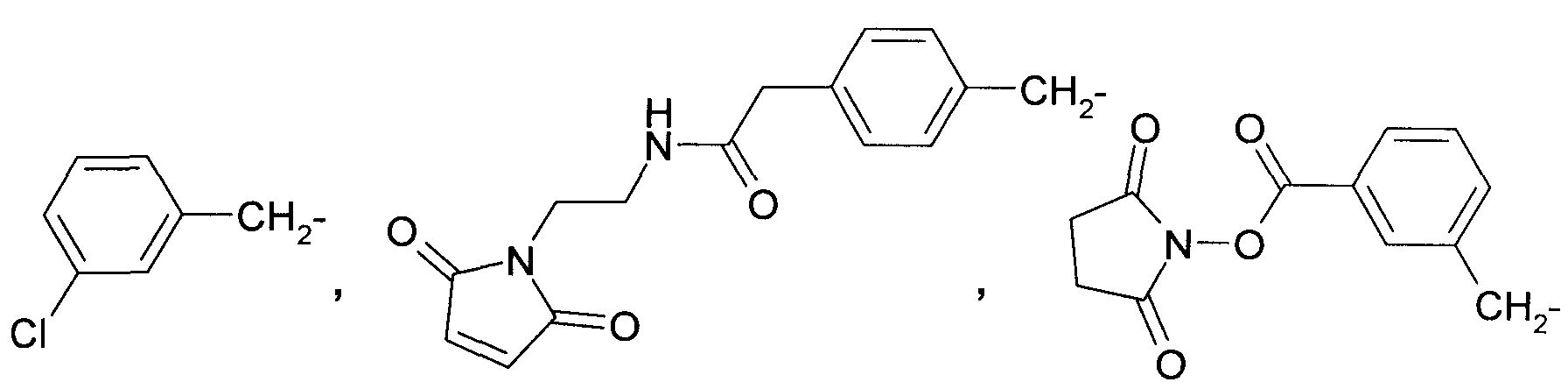
Предпочтительными являются также соединения общей формулы 2, в которых
R1 представляет собой (CH3)2N- или

a R2 представляет собой

Более предпочтительными соединениями являются:
2,5-диоксопирролидин-1-иловый эфир (3-хлорбензил)-5,5,11-триоксо-10,11-дигидро-5H-дибензо[b,f][1,4]тиазепин-7-карбоновой кислоты 1(1),
2,5-диоксопирролидин-1-иловый эфир (4-{[10-(3-хлорбензил)-5,5,11-триоксо-10,11-дигидро-5H-дибензо[b,f][1,4]тиазепин-7-карбонил]-амино}-фенокси)-уксусной кислоты 1(2),
2,5-диоксопирролидин-1-иловый эфир 4-{[10-(3-хлорбензил)-5,5,11-триоксо-10,11-дигидро-5H-дибензо[b,f][1,4]тиазепин-7-карбонил]-амино}-фенилкарбоновой кислоты 1(3),
2,5-диоксо-пирролидин-1-иловый эфир 3-[8-(3,4-диметоксифенилкарбамоил)-5,5,11-триоксо-5,11-дигидро-дибензо[b,f][1,4]тиазепин-10-илметил]-бензойной кислоты 1(4),
2,5-диоксо-пирролидин-1-иловый эфир (4-{8-[3-(4-бензилпипермдин-1-ил)-пропилкарбамоил]-5,5,11-триоксо-5,11-дигидро-дибензо[b,f][1,4]тиазепин-10-илметил}-фенил)-уксусной кислоты 1(5),
10-(3-хлорбензил)-5,5,11-триоксо-10,11-дигидро-5H-дибензо[b,f][1,4]тиазепин-8-карбоновой кислоты (2-аминоэтил)-амид 1(6),
10-{4-[(2-амино-этилкарбамоил)-метил]-бензил}-5,5,11-триоксо-10,11-дигидро-5H-дибензо[b,f][1,4]тиазепин-8-карбоновой кислоты [3-(4-бензил-пиперидин-1-ил)-пропил]-амид 1(7),
2-(2,5-диоксо-2,5-дигидро-пиррол-1-ил)-этиловый эфир 10-(3-хлорбензил)-5,5,11-триоксо-10,11-дигидро-5H-дибензо[b,f|[1,4]тиазепин-8-карбоновой кислоты 1(8),
10-(4-{[2-(2,5-диоксо-2,5-дигидро-пиррол-1-ил)-этилкарбамоил]-метил}-бензил)-5,5,11-триоксо-10,11-дигидро-5H-дибензо[b,f][1,4]тиазепин-8-карбоновой кислоты [3-(4-бензил-пиперидин-1-ил)-пропил]-амид 1(9),
N-[2-({N-(метоксикарбонил)-N-[(1,5-диметокси-1,5-диоксопентан-2-ил)карбамоил]-β-аланил}амино)этил]-10-(3-хлорбензил)-5,5,11-триоксо-10,11-дигидродибензо[b,f][1,4]тиазепин-7-карбоксамид 1(10),
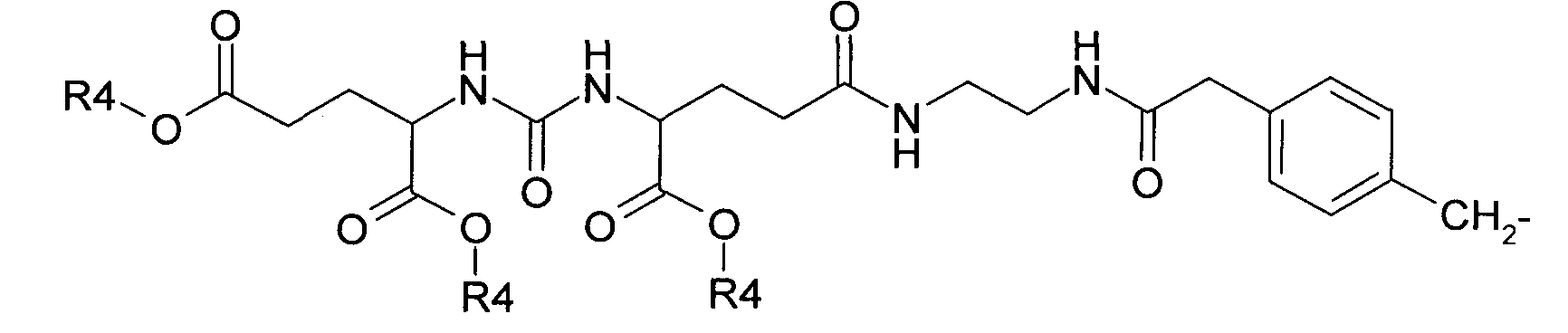
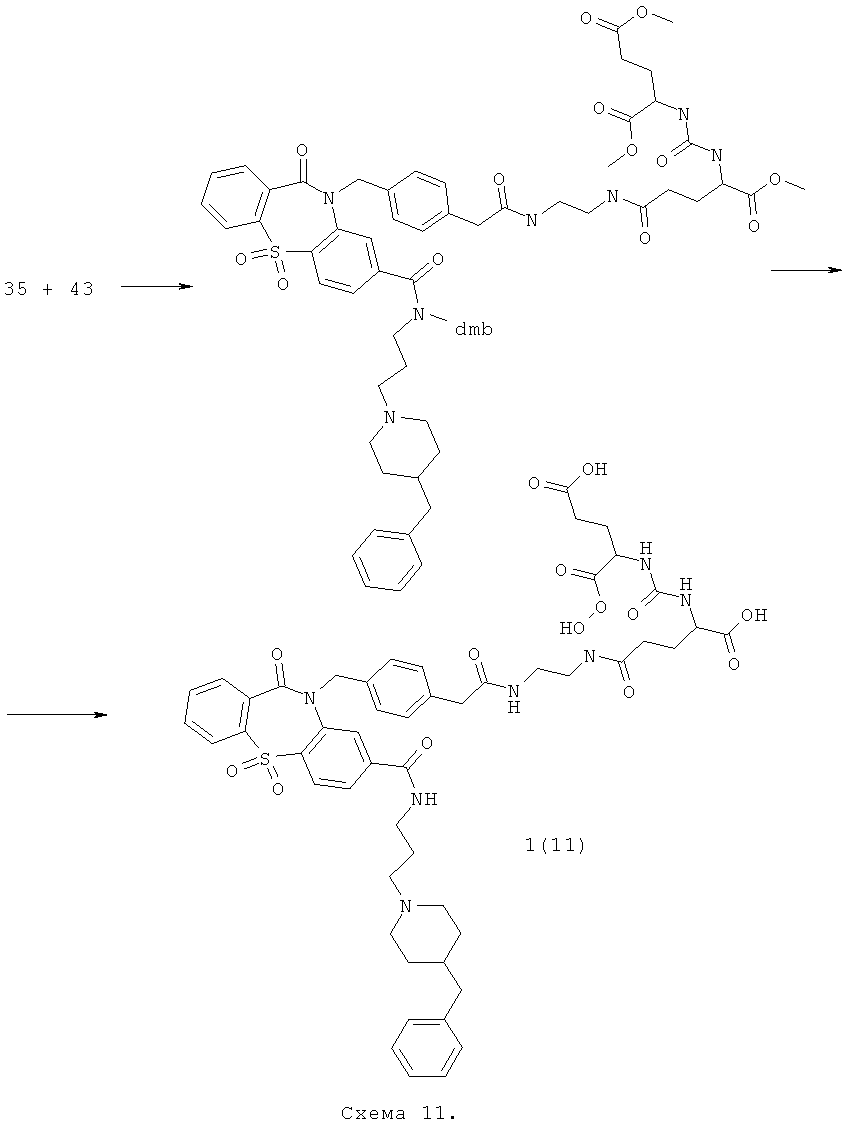
N5-(2-{[(4-{[7-{[[3-(4-бензилпиперидин-1-ил)пропил](фенил)амино]-карбонил}-5,5,11-триоксо-дибензо[b,f][1,4]тиазепин-10(11H)-ил]метил}фенил)ацетил]-амино}этил)-N2-{[(1,3-дикарбоксипропил)амино]карбонил}глутамин 1(11),
4-[4-(диметиламино)фенил]-N-(4-{[(2,5-диоксопирролидин-1-ил)окси]карбонил}фенил)-7,8,9,10-тетрагидро-4H-[1]бензотиено[3,2-,f]пирроло[1,2-α][1,4]диазепин-5(6H)-карбоксамид 2(1),
4-[4-(диметиламино)фенил]-N-(4-{2-[(2,5-диоксопирролидин-1-ил)окси]-2-оксоэтокси}фенил)-7,8,9,10-тетрагидро-4H-[1]бензотиено [3,2-f]пирроло[1,2-α][1,4] диазепин-5(6H)-карбоксамид 2(2),
2,5-диоксопирролидин-1-ил N-[4-(5-{[(3,4-диметоксифенил)амино] -карбонил}-5,6,7,8,9,10-гексагидро-4H-[1]бензотиено[3,2-f]пирроло[1,2-α][1,4]диазепин-4-ил)фенил]-N-метилглицинат 2(3).
Авторы впервые обнаружили модифицированный белок (конъюгат), активный по отношению к CD16а рецептору. Поэтому предметом данного изобретения является модифицированный белок (конъюгат), активный по отношению к CD16а рецептору, полученный взаимодействием белка с модифицирующим соединением, обладающим сродством к CD16а рецептору.
Более предпочтительным является модифицированный белок (конъюгат), активный по отношению к CD16а рецептору, полученный взаимодействием белка с модифицирующим соединением общей формулы 1 или 2.
Более предпочтительным является модифицированный белок (конъюгат, полученный из антитела и соединения общей формулы 1 или 2, в котором антитело представляет собой ритуксимаб, трастузумаб или цетуксимаб.
Более предпочтительным является также модифицированный белок (конъюгат), представляющий собой ритуксимаб, модифицированный соединением общей формулы 1 или 2.
Более предпочтительным является также модифицированный белок (конъюгат), представляющий собой трастузумаб, модифицированный соединением общей формулы 1 или 2.
Более предпочтительным является также модифицированный белок (конъюгат), представляющий собой цетуксимаб, модифицированный соединением общей формулы 1 или 2.
Более предпочтительным является также модифицированный белок (конъюгат), полученный из аутоантигена и соединения общей формулы 1 или 2, в котором аутоантиген представляет собой интерферон альфа или главный белок миелина, или белок комплимента C1q.
Более предпочтительным является также модифицированный белок (конъюгат), представляющий собой интерферон альфа, модифицированный соединением общей формулы 1 или 2.
Более предпочтительным является также модифицированный белок (конъюгат), представляющий собой главный белок миелина, модифицированный соединением общей формулы 1 или 2.
Более предпочтительным является также модифицированный белок (конъюгат), представляющий собой белок комплимента C1q, модифицированный соединением общей формулы 1 или 2.
Исследование сравнительной эффективности белков и их конъюгатов по отношению к CD16а рецептору показало, что конъюгаты на 1-3 порядка активнее своих немодифицированных белков.
Предметом данного изобретения является также способ получения модифицированного белка (конъюгата), согласно которому подвергают взаимодействию белок с соединением общей формулы 1 или 2, растворенным в среде органического растворителя, например, диметилсульфоксиде, в интервале молярного соотношения от 1:3 до 1:100 в среде фосфатного солевого буферного раствора (рН 7.4) при комнатной температуре при постоянном перемешивании.
Предметом данного изобретения является также фармацевтическая композиция, активная по отношению к CD16а рецептору, содержащая модифицированного белок (конъюгат) в терапевтически эффективном количестве и фармацевтически приемлемые разбавитель, носитель или эксципиент.
Фармацевтические композиции могут включать фармацевтически приемлемые эксципиенты. Под фармацевтически приемлемыми эксципиентами подразумеваются применяемые в сфере фармацевтики разбавители, вспомогательные агенты и/или носители. Фармацевтическая композиция наряду с модифицированным белком (конъюгатом), полученным взаимодействием белка с модифицирующим соединением общей формулы 1 или 2, или его фармацевтически приемлемой солью или сольватом, по настоящему изобретению может включать и другие активные субстанции, в том числе обладающие противогриппозной активностью, при условии, что они не вызывают нежелательных эффектов.
При необходимости использования фармацевтической композиции по настоящему изобретению в клинической практике она может смешиваться с традиционными фармацевтическими носителями.
Носители, используемые в фармацевтических композиций по настоящему изобретению, представляют собой носители, которые применяются в сфере фармацевтики для получения распространенных форм, в том числе: пероральных, форм для инъекций, местных форм.
Предметом данного изобретения является также лекарственное средство, активное по отношению к CD16а рецептору, в форме таблеток, капсул или инъекций, помещенных в фармацевтически приемлемую упаковку, предназначенное для лечения заболеваний, вызванных патологическими клетками, включающее в свой состав новый модифицированного белок (конъюгат) или новую фармацевтическую композицию в терапевтически эффективном количестве.
Поскольку при формировании конъюгата его компонент, антиген или антитело, сохраняют свою биологическую активность - антигенность и антигенсвязывающую активность, соответственно, то конъюгаты по настоящему изобретению могут использоваться для лечения тех же заболеваний, для которых используются неконъюгированные моноклональные антитела в качестве лекарственных средств, таких как низкозлокачественная или фолликулярная неходжкинская лимфома, рак молочной железы. Также известно, что активность антител по отношению к CD16а рецептору используется для лечения аутоиммунных или онкологических заболеваний (R.L.Ferris, et al. J. Clinical Oncology, 2010, Oct 1, Vol.28, No 28: 4390-4399), в том числе таких как лимфома (К.-Н.Heider, et al. Blood, 2011, 118: 4159-4168) или лимфатическая лейкемия (J.A.Bowels, et al. Blood, 2006 108: 2648-2654).
Предметом данного изобретения является способ лечения заболевания, вызванного патологическими клетками, которое можно лечить путем опосредованного воздействия на CD16а рецептор, согласно которому пациенту, нуждающемуся в лечении, вводят терапевтически эффективное количество модифицированного белка (конъюгата), или фармацевтической композиции, или лекарственного средства, активных по отношению к CD16а рецептору.
Предпочтительным является способ лечения аутоиммунных или онкологических заболеваний, определенных выше в данном разделе, в том числе таких как лимфома, лимфатическая лейкемия или рак молочной железы.
Предпочтительным является также способ лечения аутоиммунной полиэндокринопатии первого типа.
Лекарственные средства могут вводиться перорально или парентерально (например, внутривенно, подкожно, внутрибрюшинно или местно). Клиническая дозировка модифицированного белка (конъюгата), или фармацевтической композиции, или лекарственного средства, активных по отношению к CD16а рецептору, у пациентов может корректироваться в зависимости от терапевтической эффективности и биодоступности активных ингредиентов в организме, скорости их обмена и выведения из организма, а также в зависимости от возраста, пола и стадии заболевания пациента, при этом суточная доза у взрослых обычно составляет 300~1200 мг, предпочтительно - 500~1000 мг в случае, когда белок в конъюгате представляет собой антитело, и 0,01~100 мг, предпочтительно - 0,1~10 мг в случае, когда белок в конъюгате представляет собой аутоантиген. Поэтому во время приготовления фармацевтических композиций по настоящему изобретению в виде единиц дозировки необходимо учитывать вышеназванную эффективную дозировку препарата. В соответствии с указаниями врача или фармацевта данные препараты могут приниматься несколько раз в течение определенных промежутков времени (предпочтительно - от одного до шести раз).
Изобретение поясняется следующими чертежами:
Фиг.1. Спектр протонного магнитного резонанса (ПМР спектр) 2,5-диоксопирролидин-1-илового эфира (3-хлорбензил)-5,5,11-триоксо-10,11-дигидро-5H-дибензо[b,f|[1,4]тиазепин-7-карбоновой кислоты 1(1).
Фиг.2. ПМР-спектр 2,5-диоксопирролидин-1-илового эфира (4-{[10-(3-хлорбензил)-5,5,11-триоксо-10,11-дигидро-5H-дибензо[b,f][1,4]тиазепин-7-карбонил]-амино}-фенокси)-уксусной кислоты 1(2).
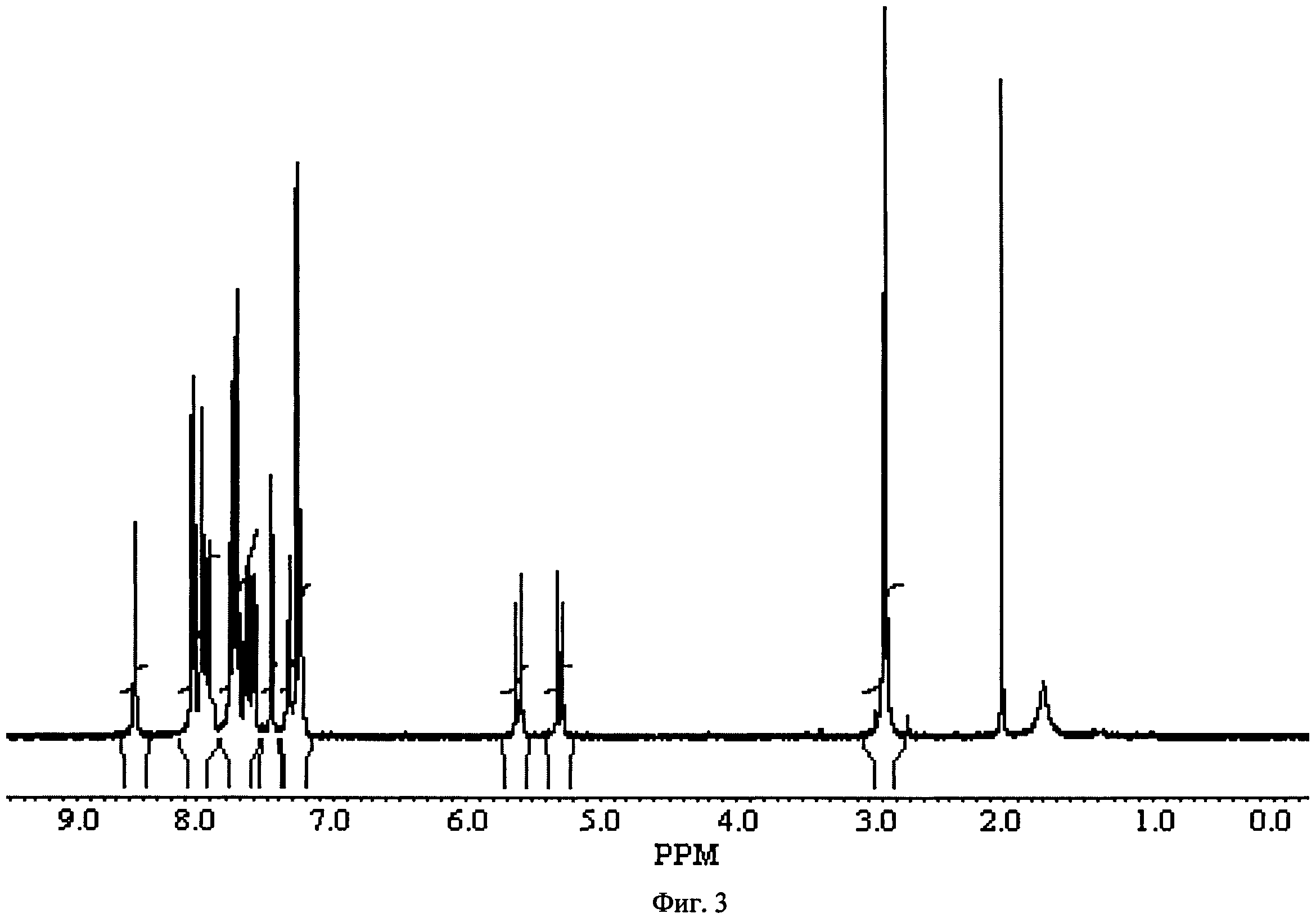
Фиг.3. ПМР-спектр 2,5-диоксопирролидин-1-илового эфира 4-{[10-(3-хлорбензил)-5,5,11-триоксо-10,11-дигидро-5H-дибензо[b,f|[1,4]тиазепин-7-карбонил]-амино}-фенилкарбоновой кислоты 1(3).
Фиг.4. LCMS-спектр 2,5-диоксо-пирролидин-1-илового эфира 3-[8-(3,4-диметоксифенилкарбамоил)-5,5,11-триоксо-5,11-дигидро-5H-дибензо[b,f][1,4]тиазепин-10-илметил]-бензойной кислоты 1(4).
Фиг 5. ПМР-спектр 2,5-диоксо-пирролидин-1-илового эфира (4-{8-[3-(4-бензилпипермдин-1-ил)-пропилкарбамоил]-5,5,11-триоксо-5,11-дигидро-дибензо[b,f][1,4]тиазепин-10-илметил}-фенил)-уксусной кислоты 1(5).
Фиг.6. ПМР-спектр 10-(3-хлорбензил)-5,5,11-триоксо-10,11-дигидро-5H-дибензо[b,f][1,4]тиазепин-8-карбоновой кислоты (2-аминоэтил)-амида 1(6).
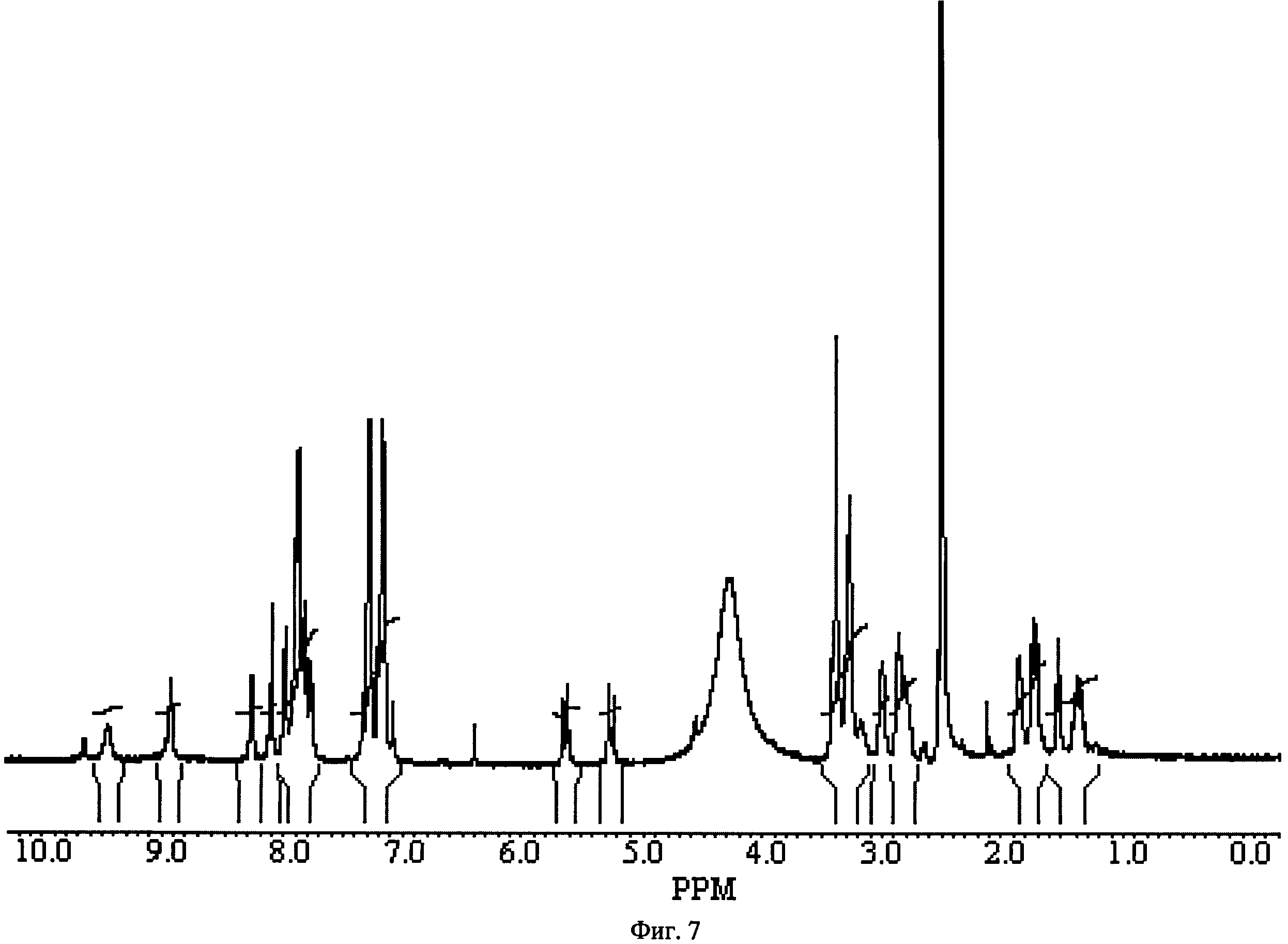
Фиг.7. ПМР-спектр 10-{4-[(2-амино-этилкарбамоил)-метил]-бензил}-5,5,11-триоксо-10,11-дигидро-5H-дибензо[b,f][1,4]тиазепин-8-карбоновой кислоты [3-(4-бензил-пиперидин-1-ил)-пропил]-амида 1(7).
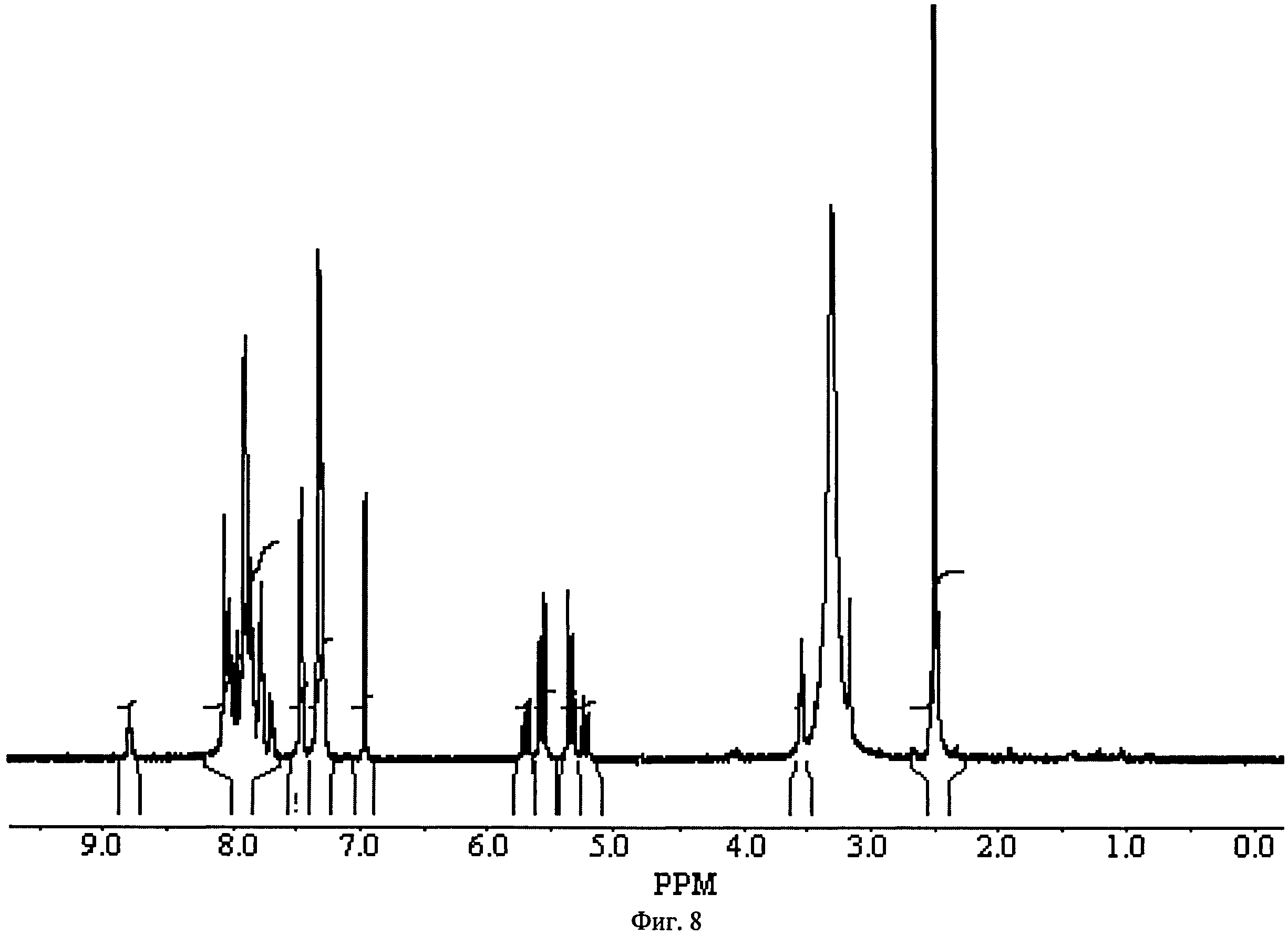
Фиг.8. ПМР-спектр 2-(2,5-диоксо-2,5-дигидро-пиррол-1-ил)-этилового эфира 10-(3-хлорбензил)-5,5,11-триоксо-10,11-дигидро-5Я-дибензо[b,f][1,4]тиазепин-8-карбоновой кислоты 1(8).
Фиг.9. ПМР-спектр 10-(4-{[2-(2,5-диоксо-2,5-дигидро-пиррол-1-ил)-этилкарбамоил]-метил}-бензил)-5,5,11-триоксо-10,11-дигидро-5H-дибензо[b,f][1,4]тиазепин-8-карбоновой кислоты [3-(4-бензил-пиперидин-1-ил)-пропил]-амида 1(9).
Фиг.10. ПМР-спектр соединения N-[2-({N-(Метоксикарбонил)-N-[(1,5-диметокси-1,5-диоксопентан-2-ил)карбамоил]-β-аланил}амино)этил]-10-(3-хлорбензил)-5,5,11-триоксо-10,11-дигидродибензо [b,f][1,4]тиазепин-7-карбоксамида 1(10).
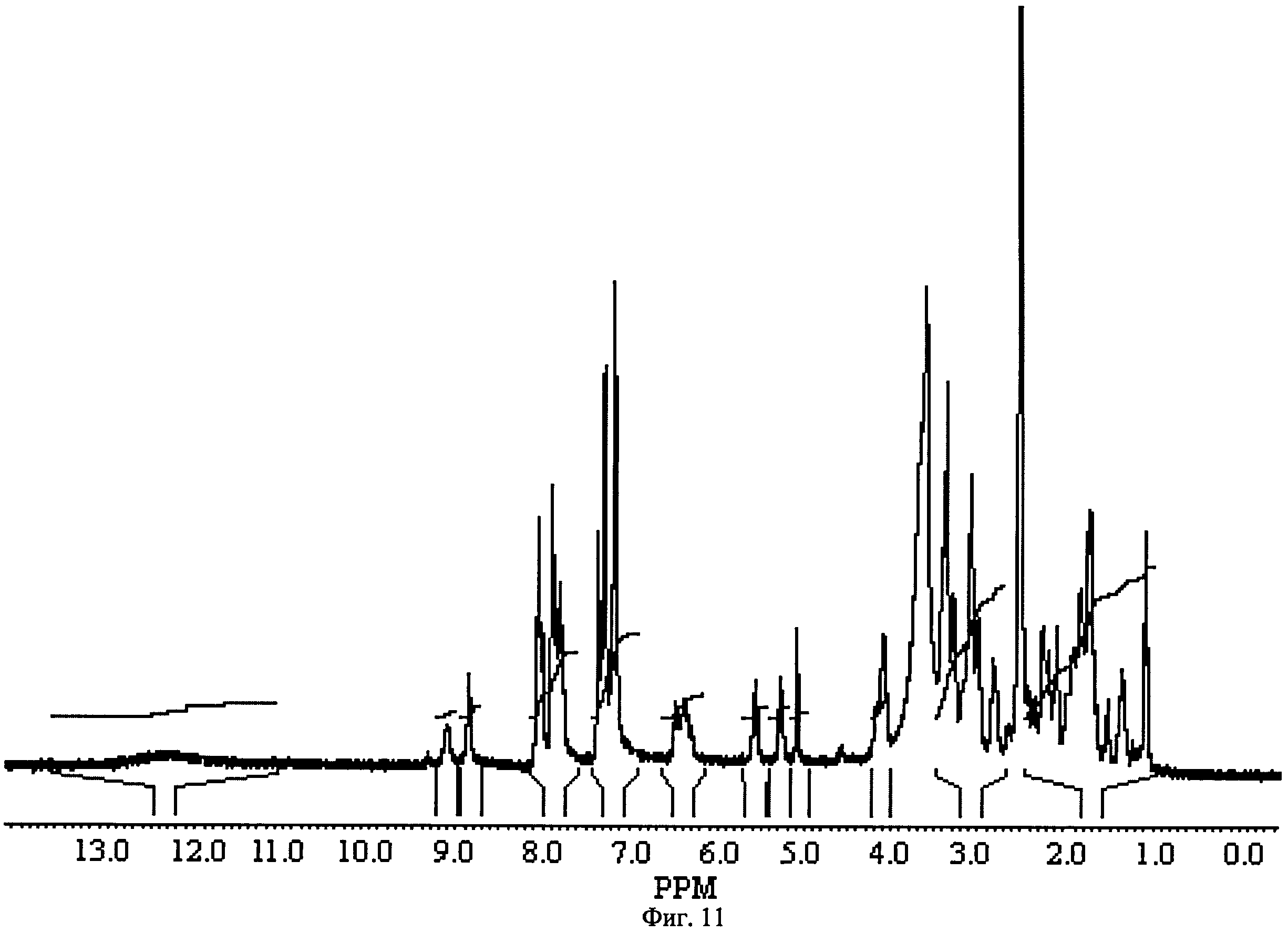
Фиг.11. ПМР-спектр N5-(2-{[(4-{[7-{[[3-(4-Бензилпиперидин-1-ил)пропил](фенил)амино]карбонил}-5,5,11-триоксо-дибензо[b,f][1,4]тиазепин-10(11H)-ил]метил}фенил)ацетил]амино}этил)-N2-{[(1,3-дикарбоксипропил)амино]-карбонил}глутамина 1(11).
Фиг 12. LCMS-спектр 4-[4-(диметиламино)фенил]-N-(4-{[(2,5-диоксопирролидин-1-ил)окси]карбонил}фенил)-7,8,9,10-тетрагидро-4H-[1]бензотиено[3,2-f]пирроло[1,2-α][1,4]диазепин-5(6H)-карбоксамида 2(1).
Фиг.13 ПМР-спектр 4-[4-(диметиламино)фенил]-N-(4-{2-[(2,5-диоксопирролидин-1-ил)окси]-2-оксоэтокси}фенил)-7,8,9,10-тетрагидро-4H-[1]бензотиено[3,2-/]пирроло[1,2-α][1,4]диазепин-5(6H)-карбоксамида 2(2).
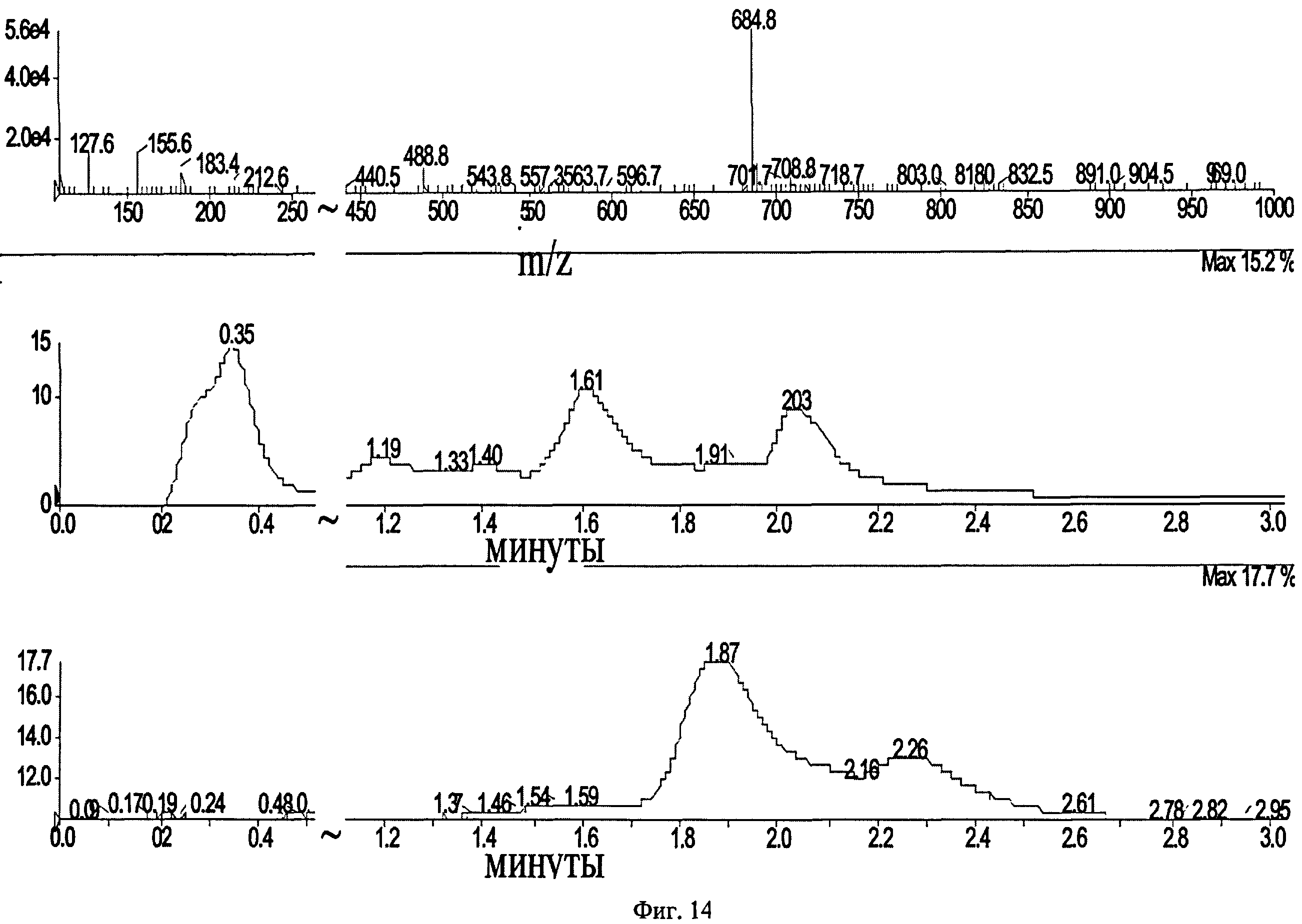
Фиг.14. LCMS-спектр 2,5-диоксопирролидин-1-ил N-[4-(5-{[(3,4-диметоксифенил)-амино]карбонил}-5,6,7,8,9,10-гексагидро-4H-[1]бензотиено[3,2-f)пирроло[1,2-α][1,4]диазепин-4-ил)фенил]-N-метилглицината 2(3).
Фиг.15. Хроматограмма конъюгата КР1(1), на колонке TSK GEL SUPER SW3000.
Фиг 16. Имунно-ферментный анализ (ИФА) связывания конъюгатов ритуксимаба с CD16a рецептором. Зависимость оптической плотности на длине волны 450 нм от концентрации введенного в ИФА конъюгата.
Фиг 17. Имунно-ферментный анализ (ИФА) связывания интерферона (И) и конъюгата КИ1(1) с CD16а рецептором. Зависимость оптической плотности на длине волны 450 нм от концентрации введенного в ИФА конъюгата.
Фиг.18. Сравнение эффективности ритуксимаба (Р) и конъюгата КР1(1) в тесте антителозависимой цитотоксичности.
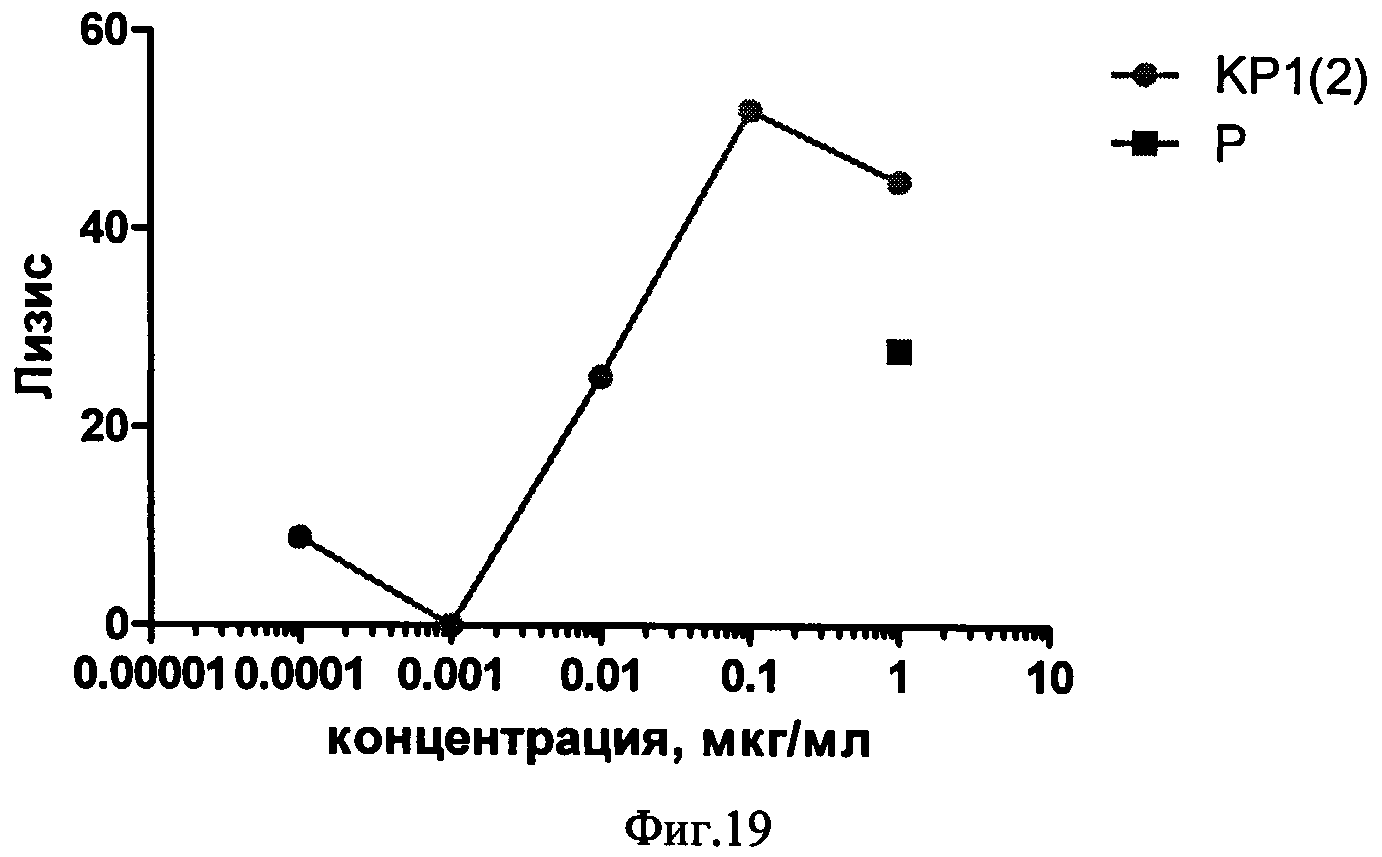
Фиг.19. Сравнение эффективности ритуксимаба (Р) и конъюгата КР1(2) в тесте антителозависимой цитотоксичности.
Фиг 20. Имунно-ферментный анализ (ИФА) связывания ритуксимаба (Р), трастузумаба (Т) и их конъюгатов КР1(7), КТ1(7) по отношению к CD16a рецептору.
Представленные ниже примеры иллюстрируют, но не ограничивают данное изобретение.
Пример 1. 2,5-Диоксопирролидин-1-иловый эфир (3-хлорбензил)-5,5,11-триоксо-10,11-дигидро-5H-дибензо[b,f][1,4]тиазепин-7-карбоновой кислоты 1(1) получают по нижеследующей Схеме 1.

К раствору 31.8 г КОН в 170 мл воды присыпают порциями 25 г соединения 4, после его растворения добавляют 30 г соединения 3 и перемешивают при 60°С в течение 15 ч. Затем реакционную смесь охлаждают, подкисляют 10% соляной кислотой до рН 3, фильтруют, промывают водой, сушат. Получают соединение 5 с выходом 70%. Растворяют 50 г соединения 5 в 500 мл водного аммиака растворяют и порциями присыпают 80 г дитионита, после чего реакционную смесь кипятят с обратным холодильником 1 ч. Затем реакционную смесь охлаждают, упаривают на роторном испарителе аммиак, а водный раствор подкисляют конц. соляной кислотой до рН 1, перемешивают 1 ч, фильтруют осадок, промывают водой, сушат. Получают соединение 6 с выходом 60%. Добавляют порциями 45 г соединения 6 к 200 мл полифосфорной кислоты при 50°С, после чего перемешивают 10 ч при 90°С. Выливают на 500 мл льда, фильтруют, промывают водой, сушат. Получают соединение 7 с выходом 60%. Растворяют 30 г соединения 7 в 500 мл уксусной кислоты, добавляют 70 мл 33% перекиси водорода, перемешивают ночь при 70°С. Затем охлаждают, уксусную кислоту упаривают на роторном испарителе, к остатку добавляют 600 мл воды. Выпавший осадок фильтруют, промывают водой, сушат. Получают соединение 8 с выходом 70%. К раствору 22 г соединения 8 в 300 мл этанола прикапывают 8 мл хлористого тионила при 10°С, после чего смесь кипятят 5 часов. Охлаждают, упаривают метанол на роторном испарителе, добавляют 300 мл воды. Выпавший осадок фильтруют, промывают водой, сушат. Получают соединение 9 с выходом 90%. Растворяют 8.2 г соединения 9 в 80 мл ДМФА, добавляют 7 г поташа и после этого 5.2 г м-хлорбензилхлорида. Смесь перемешивают при 50°С ночь, упаривают ДМФА на роторном испарителе, к остатку добавляют 200 мл воды. Выпавший осадок фильтруют, промывают водой, сушат. Получают соединение 10 с выходом 90%. Растворяют 11 г соединения 10 в 150 мл 50% водном этаноле, добавляют 2.7 г КОН и перемешивают ночь при комнатной температуре. После этого этанол упаривают на роторном испарителе, водный раствор подкисляют 10% соляной кислотой до рН 3. Выпавший осадок фильтруют, промывают водой, сушат. Получают соединение 11 с выходом 80%. Растворяют 1.8 г соединения 11 в 50 мл ТГФ, в атмосфере аргона при перемешивании добавляют 0.48 г N-гидроксисукцинимида и 0.87 г дициклогексилкарбодиимида. Смесь перемешивают ночь при комнатной температуре ночь, после этого выпавший осадок фильтруют, маточный раствор упаривают, чистят флэш-хроматографией на силикагеле (элюент этилацетат). Получают соединение 1(1) с выходом 50%. Спектр протонного магнитного резонанса (ПМР спектр) соединения 1(1) представлен на Фиг.1.
Пример 2. 2,5-Диоксопирролидин-1-иловый эфир (4-{[10-(3-хлорбензил)-5,5,11-триоксо-10,11-дигидро-5H-дибензо[b,f][1,4]тиазепин-7-карбонил]-амино}-фенокси)-уксусной кислоты 1(2) получают по нижеследующей Схеме 2.
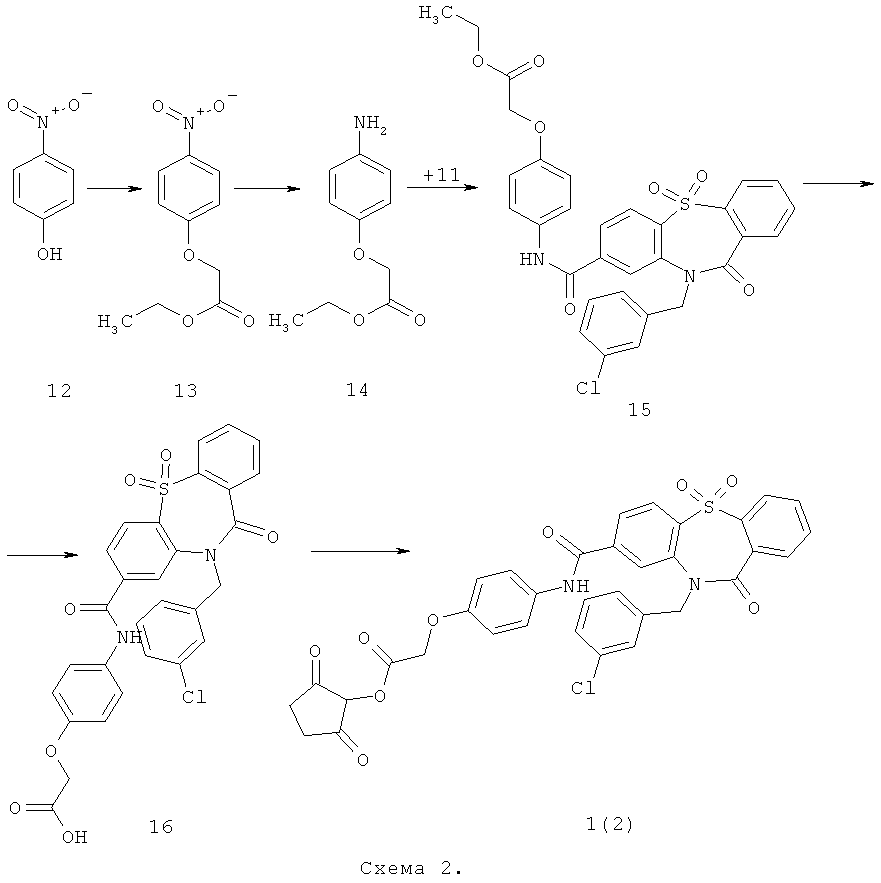
К суспензии 30 г поташа в 200 мл ацетонитрила добавляют 15 г п-нитрофенола 12, перемешивают 1 ч, после чего прикапывают 19,8 г этилового эфира бромуксусной кислоты. Перемешивают ночь при 60°С, фильтруют, упаривают на роторном испарителе. Полученный продукт 13 используют дальше без очистки. Растворяют 14.3 г соединения 13 в 200 мл 50% водной уксусной кислоты, нагревают до 70°С, после чего присыпают небольшими порциями 10 г железа так, чтобы смесь кипела. После этого перемешивают еще 15 мин, охлаждают, добавляют 500 мл воды. Экстрагируют 3x150 мл этилацетата, объединенные экстракты промывают концентрированным раствором NaHCO3, сушат, растворитель упаривают на роторном испарителе. Получают продукт 14 с выходом 75%. Смешивают в 50 мл диоксана 1.9 г соединения 11 и 0.87 г соединения 14. Перемешивают 1 ч, после чего добавляют 1.2 мл триэтиламина и 0.89 г хлорокиси фосфора. Перемешивают 3 ч при 50°С, добавляют 150 мл воды, выпавший осадок фильтруют, промывают водой, сушат. Получают продукт 15 с выходом 60%. Растворяют 2.7 г соединения 15 в 20 мл 50% водного этанола, добавляют 0.93 г LiOH и перемешивают ночь при комнатной температуре. После этого этанол упаривают на роторном испарителе, водный раствор подкисляют 10% соляной кислотой до рН 3. Выпавший осадок фильтруют, промывают водой, сушат. Продукт очищают колоночной хроматографией в системе хлороформ-метанол-триэтиламин 10-1-1. Получают продукт 16 с выходом 11%. Растворяют 290 мг соединения 16 в 50 мл ТГФ, в атмосфере аргона при перемешивании добавляют 58 г N-гидроксисукцинимида и 104 мг дициклогексилкарбо-диимида. Смесь перемешивают при комнатной температуре ночь, после этого выпавший осадок фильтруют, маточный раствор упаривают, чистят флэш-хроматографией на силикагеле (элюент этилацетат). Получают продукт 1(2) с выходом 50%. ПМР-спектр соединения 1(2) представлен на Фиг.2.
Пример 3. 2,5-Диоксопирролидин-1-иловый эфир 4-{[10-(3-хлорбензил)-5,5,11-триоксо-10,11-дигидро-5N-дибензо[b,f][1,4]тиазепин-7-карбонил]-амино}-фенилкарбоновой кислоты 1(3) получают по нижеследующей Схеме 3.

Растворяют 1 экв n-аминобензойной кислоты в трет-бутиловом спирте и добавляют 1.1 экв EDC. Кипятят реакционную смесь 18 ч. Охлаждают до 0°С, добавляют воду и экстрагируют диэтиловым эфиром. Упаренный органический слой пускают на следующую стадию без очистки. Получают продукт 17 с выходом 60%. Смешивают в 50 мл диоксана 2.2 г соединения 11 и 0.85 г трет-бутилового эфира т-аминобензойной кислоты 17. Перемешивают 1 ч, после чего добавляют 1.4 мл триэтиламина и 1.0 г хлорокиси фосфора. Перемешивают 3 ч при 50°С, добавляют 150 мл воды, выпавший осадок фильтруют, промывают водой, сушат. Получают продукт 18 с выходом 60%. Соединение 18 растворяют в трифторуксусной кислоте и перемешивают ночь при 50°С. Реакционную массу упаривают досуха. Получают продукт 19 с выходом 88%, который используют в следующую стадию без очистки. Растворяют 760 мг соединения 19 в 50 мл ТГФ, в атмосфере аргона при перемешивании добавляют 160 мг N-гидроксисукцинимида и 287 мг дициклогексилкарбодиимида. Смесь перемешивают ночь при комнатной температуре, выпавший осадок фильтруют, маточный раствор упаривают, чистят флэш-хроматографией на силикагеле (элюент-этилацетат). Получают продукт 1(4) с выходом 50%. ПМР-спектр соединения 1(4) представлен на Фиг.3.
Пример 4. 2,5-Диоксо-пирролидин-1-иловый эфир 3-[8-(3,4-диметоксифенилкарбамоил)-5,5,11-триоксо-5,11-дигидро-дибензо[b,f][1,4]тиазепин-10-илметил]-бензойной кислоты 1(4) получают по нижеследующей Схеме 4.
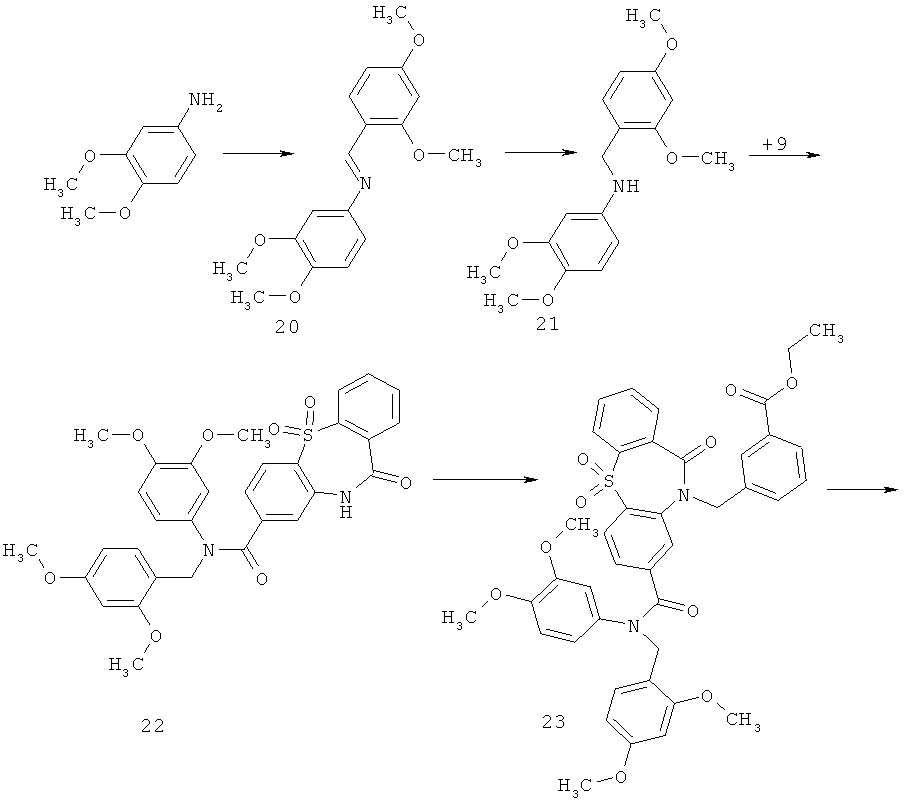

Кипятят 10 г 2,4-диметоксибензальдегида и 9.2 г 3,4-диметоксианилина в 150 мл толуола с насадкой дина-старка до прекращения выделения воды. После этого смесь охлаждают, упаривают толуол на роторном испарителе, полученный продукт 20 используют дальше без очистки. Растворяют соединение 20 в 100 мл метанола и при перемешивании присыпают 2.8 г боргидрида натрия, перемешивают при комнатной температуре еще 1 ч, упаривают метанол на роторном испарителе, добавляют 100 мл воды, подщелачивают 10% раствором КОН до рН 9, экстрагируют хлористым метиленом, сушат, упаривают. Получают продукт 21 с выходом 80%. Смешивают в 50 мл диоксана 2.2 г соединения 9 и 3.0 г соединения 21. Перемешивают 1 ч, после чего добавляют 5.5 мл триэтиламина и 2.0 г хлорокиси фосфора. Перемешивают 3 ч при 50°С, добавляют 150 мл воды, выпавший осадок фильтруют, промывают водой, сушат. Получают продукт 22 с выходом 60%. Растворяют 3.0 г соединения 22 в 30 мл ДМФА, добавляют 1.7 г поташа и 1.3 г этил 3-бромметилбензоата. Смесь перемешивают при 50°С ночь, упаривают ДМФА в вакууме, к остатку добавляют 70 мл воды. Выпавший осадок фильтруют, промывают водой, сушат. Получают продукт 23 с выходом 90%. Растворяют 3.0 г соединения 23 в 40 мл хлористого метилена, добавляют 1.4 г трифторуксусной кислоты и кипятят с обратным холодильником ночь. После этого смесь промывают конц. раствором NaНСО3, сушат, упаривают хлористый метилен. Чистят продукт колоночной хроматографией, элюент хлороформ / метанол 40/1. Выход продукта 24 составляет 30%. Растворяют 0.9 г соединения 24 в 20 мл 50% водного этанола, добавляют 0.25 г LiOH и перемешивают ночь при комнатной температуре. После этого этанол упаривают на роторном испарителе, водный раствор подкисляют 10% соляной кислотой до рН 3. Выпавший осадок фильтруют, промывают водой, сушат. Продукт очищают колоночной хроматографией в системе хророформ-метанол-триэтиламин 10-1-1. Получают продукт 25 с выходом 20%. Растворяют 140 мг соединения 25 в 20 мл ТГФ, в атмосфере аргона при перемешивании добавляют 42 мг N-гидроксисукцинимида и 76 мг дициклогексилкарбодиимида. Смесь перемешивают ночь при комнатной температуре ночь, после этого выпавший осадок фильтруют, маточный раствор упаривают, чистят флэш-хроматографией на силикагеле (элюент этилацетат). Получают продукт 1(4) с выходом 50%. LCMS-спектр соединения 1(4): (М+1=670) представлен на Фиг.4.
Пример 5. 2,5-Диоксо-пирролидин-1-иловый эфир (4-{8-[3-(4-бензилпиперидин-1-ил)-пропилкарбамоил]-5,5,11-триоксо-5,11-дигидро-дибензо[b,f][1,4]тиазепин-10-илметил}-фенил)-уксусной кислоты 1(5) получают по нижеследующей Схеме 5.
Кипятят 3.8 г 2,4-диметоксибензальдегида и 5.3 г соединения 26 в 100 мл толуола с насадкой дина-старка до прекращения выделения воды. После этого смесь охлаждают, упаривают толуол на роторном испарителе. Полученный продукт 27 используют дальше без очистки. Растворяют соединение 27 в 50 мл метанола и при перемешивании присыпают 1.3 г боргидрида натрия, перемешивают при комнатной температуре еще 1 ч, упаривают метанол на роторном испарителе, добавляют 50 мл воды, подщелачивают 10% раствором КОН до рН 9, экстрагируют хлористым метиленом, сушат, упаривают. Получают соединение 28 с выходом 80%. Растворяют 2.4 г соединения 9 в 100 мл ТГФ, в атмосфере аргона при перемешивании добавляют 1.0 г N-гидроксисукцинимида и 1.1 г дициклогексилкарбодиимида. Смесь перемешивают ночь при комнатной температуре ночь, после этого добавляют 3 г соединения 28 и перемешивают 6 ч при 60°С. Выпавший осадок отфильтровывают, маточный раствор упаривают на роторном испарителе. Остаток чистят колоночной хроматографией, элюент хлороформ:метанол 19:1. Получают соединение 29 с выходом 70%. Смесь 10.0 г п-толилуксусной кислоты, 13.0 г N-бромсукцинимида и 0.1 г 2,2'-азабисизобутиронитрила в 60 мл четыреххлористого углерода кипятят с обратным холодильником 4 ч. Смесь охлаждают до комнатной температуры, выливают на 100 мл воды, выпавший осадок отфильтровывают, сушат. Растворяют в 50 мл этанола, добавляют при 0°С 3.7 мл тионилхлорида, перемешивают ночь при комнатной температуре, упаривают растворитель.


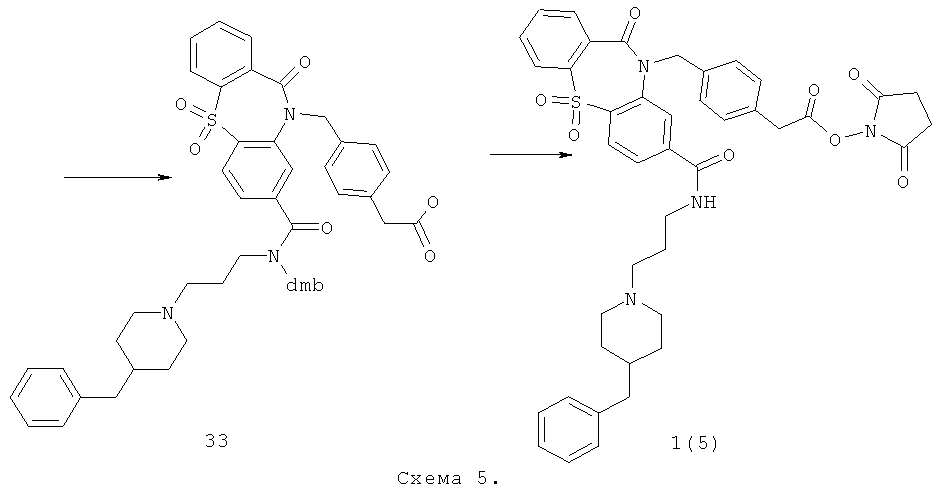
Получают соединение 30 с выходом 50%. Полученный продукт используют дальше без очистки. Растворяют 4.5 г соединения 29 в 50 мл ДМФА, добавляют 2.2 г поташа и после этого 2.0 г этилового эфира п-бромметилфенилуксусной кислоты 30. Смесь перемешивают при 50°С ночь, упаривают ДМФА на роторном испарителе, к остатку добавляют 100 мл воды. Выпавший осадок фильтруют, промывают водой, сушат. Получают соединение 31 с выходом 80%. Растворяют 3.5 г соединения 31 в 40 мл хлористого метилена, добавляют 2.0 г трифторуксусной кислоты и кипятят с обратным холодильником ночь. После этого смесь встряхивают несколько раз с конц. раствором NaHCO3, сушат, упаривают хлористый метилен на роторном испарителе. Чистят продукт колоночной хроматографией, элюент хлороформ / метанол 40/1. Получают соединение 32 с выходом 55%. Растворяют 1.6 г соединения 32 в 20 мл 50% водного этанола, добавляют 0.29 г LiOH и перемешивают ночь при комнатной температуре. После этого этанол упаривают на роторном испарителе, водный раствор подкисляют 10% соляной кислотой до рН 3. Выпавший осадок фильтруют, промывают водой, сушат. Продукт очищают колоночной хроматографией в системе хророформ-метанол-триэтиламин 10-1-1. Получают соединение 33 с выходом 15%. Растворяют 180 мг соединения 33 в 20 мл ТГФ, в атмосфере аргона при перемешивании добавляют 47 мг N-гидроксисукцинимида и 84 мг дициклогексилкарбодиимида. Смесь перемешивают ночь при комнатной температуре ночь, после этого выпавший осадок фильтруют, маточный раствор упаривают, чистят флэш-хроматографией на силикагеле (элюент этилацетат). Получают соединение 1(5) с выходом 50%. ПМР-спектр соединения 1(5) представлен на Фиг.5.
Пример 6. 10-(3-Хлорбензил)-5,5,11-триоксо-10,11-дигидро-5H-дибензо[b,f][1,4]тиазепин-8-карбоновой кислоты (2-аминоэтил)-амид 1(6) получают по нижеследующей Схеме 6.

Соединение 11 растворяют в хлороформе, добавляют 1.1 экв КДИ и перемешивают при комнатной температуре час, затем прибавляют бок-этилендиамин и перемешивают ночь при комнатной температуре. Промывают водой и органический слой упаривают. Чистим на колонке в элюенте хлороформ-метанол 20:1. Получают соединение 34 с выходом 70%.
Соединение 34 перемешивают в диоксане, насыщенном HCl, при комнатной температуре 1 ч, осадок отфильтровывают. Получают соединение 1(6) с выходом 78%. ПМР-спектр соединения 1(6) представлен на Фиг.6.
Пример 7. 10-{4-[(2-Амино-этилкарбамоил)-метил]-бензил}-5,5,11-триоксо-10,11-дигидро-5H-дибензо[b,f][1,4]тиазепин-8-карбоновой кислоты [3-(4-бензил-пиперидин-1-ил)-пропил]-амид 1(7) получают по нижеследующей Схеме 7.

Соединение 35 растворяют в хлороформе, добавляют 1.1 экв КДИ и перемешивают при комнатной температуре 1 ч, затем прибавляют бок-этилендиамин и перемешивают ночь при комнатной температуре. Промывают водой и органический слой упариваем. Хроматографируют на силикагеле смесью хлороформа с метанолом (20:1). Получают соединение 36 с выходом 70%. Соединение 36 перемешивают в диоксане насыщенном НСl при комнатной температуре 1 ч, осадок отфильтровываем. Получают соединение 1(7) с выходом 78%. ПМР-спектр соединения 1(7) представлен на Фиг.7.
Пример 8. 2-(2,5-Диоксо-2,5-дигидро-пиррол-1-ил)-этиловый эфир 10-(3-хлорбензил)-5,5,11-триоксо-10,11-дигидро-5H-дибензо[b,f][1,4]тиазепин-8-карбоновой кислоты 1(8) получают по нижеследующей Схеме 8.
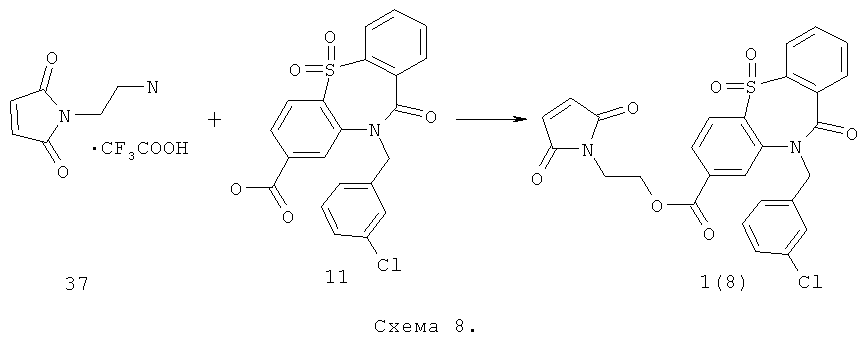
Соединения 37 и 11 растворяют в ТГФ в соотношении 1:1 и при перемешивании добавляют 1 экв дициклогексилкарбодиимида и 1 экв Et3N. Перемешивают ночь при 50°С. Охлаждают, выпавший осадок фильтруют, а маточник упаривают и чистят на HPLC. Получают соединение 1(8) с выходом 10%. ПМР-спектр соединения 1(8) представлен на Фиг.8.
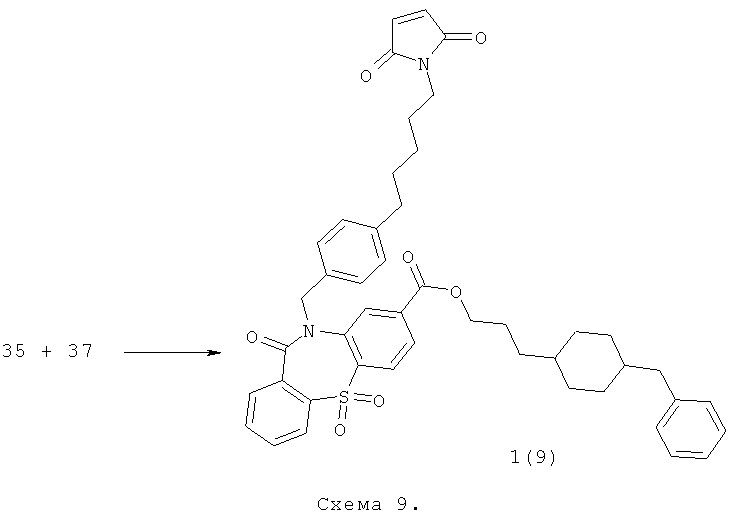
Пример 9. 10-(4-{[2-(2,5-диоксо-2,5-дигидро-пиррол-1-ил)-этилкарбамоил]-метил}-бензил)-5,5,11-триоксо-10,11-дигидро-5H-дибензо[b,f][1,4]тиазепин-8-карбоновой кислоты [3-(4-бензил-пиперидин-1-ил)-пропил]-амид 1(9) получают по нижеследующей Схеме 9.
Соединения 35 и 37 растворяют в ТГФ в соотношении 1:1 и при перемешивании добавляют 1 экв дициклогексилкарбодиимида и 1 экв Et3N. Перемешивают ночь при 50°С. Охлаждают, выпавший осадок фильтруют, а маточник упаривают и чистят на HPLC. Получают соединение 1(9) с выходом 10%. ПМР-спектр соединения 1(9) представлен на Фиг.9.
Пример 10. N-[2-({N-(Метоксикарбонил)-N-[(1,5-диметокси-1,5-диоксопентан-2-ил)карбамоил]-β-аланил}амино)этил]-10-(3-хлорбензил)-5,5,11-триоксо-10,11-дигидродибензо[b,f][1,4]тиазепин-7-карбоксамид 1(10) получают по нижеследующей Схеме 10.


К суспензии трифосгена (4.2 г, 0014 моль) в CH2Cl2 при 0°С медленно прикапывают раствор, состоящий из 11 г (0.038 моль) соединения 38 и 12.6 г (0.098 моль) DIPEA в CH2Cl2. Перемешивают смесь 30 минут при 0°С, затем добавляют одной порцией раствор 8.1 г (0.038 моль) соединения 39 и 10.8 г (0.084 моль) DIPEA в CH2Cl2. Затем перемешивают еще 15 мин, органический слой отделили, промыли водой и хроматографируют на силикагеле СНСl3/МеОН (98:2). Получают соединение 40 с выходом, 53%. Растворяют 6 г соединения 40 в этилацетате, добавляют 0.6 г Pd/VC (10%). Смесь гидрируют в автоклаве при 2 атм 17 ч. Катализатор отфильтровывают, фильтрат упарили. Получают 5 г соединения 41, которое используют на следующей стадии без дополнительной очистки. К раствору 4.8 г (0.013 моль) соединения 41 в CH2Cl2 при комнатной температуре присыпали 2.6 г КДИ (0.016 моль). Через 2 часа добавляют 2.2 г (0.013 моль) бок-этилендиамина. Смесь перемешивают около 18 часов, разбавляют водой, продукт экстрагируют CH2Cl2 и хроматографируют на силикагеле смесью СНСl3/МеОН (99:1). Получают 4.4 г (66%) соединения 42. К раствору 4.4 г соединения 42 в диоксане добавляют 5 экв 3N НСl в диоксане. Смесь перемешивают ночь при комнатной температуре, затем упаривают досуха. Получают соединение 43 с выходом 90%. Смешивают 0.8 г (0.002 моль) соединения 43, 0.85 г (0.002 моль) соединения 11, 0.4 г NEt3 (0.004 моль) и 0.4 г (0.002 моль) N-(диметиламинопропил)-N'-этилкарбодиимида гидрохлорид в THF. Реакционную массу перемешивают при 60°С два дня. Затем добавляют 0.5 г (0.012 моль) LiOH·H2O и 2 мл воды. Через 24 часа добавляют 0.72 г (0.012 моль) уксусной кислоты. Реакционную массу упаривают и очистили HPLC. Получают соединение 44 с выходом 7%. ПМР-спектр соединения 1(10) представлен на Фиг.10. Гидрохлорид 1(10)*НСl получают добавлением к соединению 1(10) растворением в этилацетате, насыщенном НСl. Полученный в осадке гидрохлорид фильтруют, сушат, очищают перекристаллизацией. Аналогичным образом получают гидрохлориды соединений, полученных в примерах 1-9 и 11-14.
Пример 11. N5-(2-{[(4-{[7-{[[3-(4-Бензилпиперидин-1-ил)пропил](фенил)амино]-карбонил}-5,5,11-триоксо-дибензо[b,f][1,4]тиазепин-10(11H)-ил]метил}фенил)ацетил]-амино}этил)-N2-{[(1,3-дикарбоксипропил)амино]карбонил}глутамин 1(11) получают по нижеследующей Схеме 11.
Смешивают 1.6 г (0.0024 моль) кислоты 35, 1 г (0.0024 моль) эфира 43, 0.5 г NEt3 (0.0048 моль) и 0.45 г(0.0024 моль) N-(диметиламинопропил)]-N'этилкарбодиимида гидрохлорид в THF. Реакционную массу перемешивают при 60°С два дня. Затем, не выделяя продукт 45, добавляют 0.0144 моль, 0.6 г LiOH·Н2O и 2 мл воды. Через 24 ч добавляют 0.86 г (0.014 моль) уксусной кислоты. Реакционную массу упаривают и очистили HPLC. Получают соединение 1(11) с выходом 10%. ПМР-спектр соединения 1(11) представлен на Фиг.11.
Пример 12. 4-[4-(Диметиламино)фенил]-N-(4-{[(2,5-диоксопирролидин-1 -ил)окси]карбонил}фенил)-7,8,9,10-тетрагидро-4H-[1]бензотиено[3,2-f]пирроло[1,2-α][1,4]диазепин-5(6H)-карбоксамид 2(1) получают по нижеследующей Схеме 12.

Смесь 3.0 г соединения 46 и 1.7 г 4-диметиламинобензальдегида в 40 мл этанола кипятят с обратным холодильником 10 ч, охлаждают до комнатной температуры, фильтруют выпавший осадок. Осадок растворяют в 50 мл воды и подщелачивают 10% КОН до рН 10. Выпавший осадок фильтруют, промывают водой, сушат. Получают соединение 47 с выходом 35%. Растворяют 0.6 г соединения 47 и 0.32 г этил 4-изоцианатобензоата в 30 мл диоксана и перемешивают ночь при комнатной температуре. Выпавший осадок фильтруют. Получают соединение 48 с выходом 30%. Растворяют 140 мг соединения 48 в 5 мл 50% водного этанола, добавляют 32 мг LiOH и перемешивают ночь при комнатной температуре. Этанол упаривают на роторном испарителе, водный раствор подкисляют 10% соляной кислотой до рН 3. Выпавший осадок фильтруют, промывают водой, сушат. Получают соединение 49 с выходом 60%. Растворяют 80 мг соединения 49 в 10 мл ТГФ, в атмосфере аргона при перемешивании добавляют 22 мг N-гидроксисукцинимида и 39 мг дициклогексилкарбодиимида. Смесь перемешивают ночь при комнатной температуре ночь, после этого выпавший осадок фильтруют, маточный раствор упаривают, чистят флэш- хроматографией на силикагеле (элюент этилацетат). Получают соединение 2(1) с выходом 50%. LCMS-спектр соединения 1(12) представлен на Фиг.12 (М+1)=624.
Пример 13. 4-[4-(Диметиламино)фенил]-N-(4-{2-[(2,5-диоксопирролидин-1-ил)окси]-2-оксоэтокси}фенил)-7,8,9,10-тетрагидро-4H-[1]бензотиено[3,2-f]пирроло[1,2-α][1,4]диазепин-5(6H)-карбоксамид 2(2) получают по нижеследующей Схеме 13.

Растворяют в 30 мл диоксана 0.8 г соединения 50 и 0.5 г соединения 47 и перемешивают ночь при комнатной температуре. Растворитель упаривают, остаток чистят колоночной хроматографией. Элюент хлороформ - метанол 39:1. Получают соединение 51 с выходом 5%. Растворяют 6 мг соединения 51 в 5 мл 50% водного этанола, добавляют 14 мг LiOH и перемешивают ночь при комнатной температуре. После этого этанол упаривают на роторном испарителе, водный раствор подкисляют 10% соляной кислотой до рН 3. Выпавший осадок фильтруют, промывают водой, сушат. Получают соединение 52 с выходом 60%. Растворяют 50 мг соединения 52 в 10 мл ТГФ, в атмосфере аргона при перемешивании добавляют 10 мг N-гидроксисукцинимида и 19 мг дициклогексилкарбодиимида. Смесь перемешивают при комнатной температуре ночь, после этого выпавший осадок фильтруют, маточный раствор упаривают, чистят флэш-хроматографией на силикагеле (элюент этилацетат). Получают соединение 1(13) с выходом 50%. ПМР-спектр соединения соединения 2(2) представлен на Фиг.13.
Пример 14. 2,5-Диоксопирролидин-1-ил N-[4-(5-{[(3,4-диметоксифенил)амино]-карбонил}-5,6,7,8,9,10-гексагидро-4H-[1] бензотиено[3,2-f]пирроло[1,2-α][1,4]диазепин-4-ил)фенил]-N-метилглицинат 2(3) получают по нижеследующей Схеме 14.

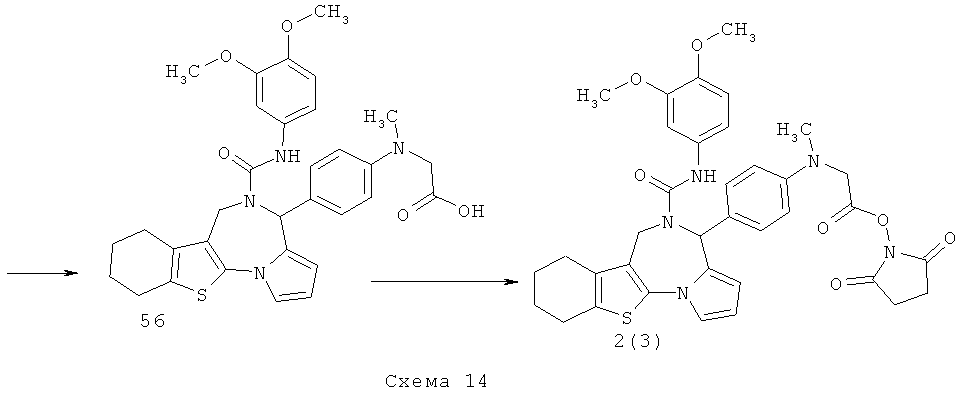
Смесь 1.68 г соединения 46 и 1.45 г соединения 53 в 25 мл этанола кипятят с обратным холодильником 10 ч, охлаждают до комнатной температуры, фильтруют, выпавший осадок растворяют в 50 мл воды и подщелачивают 10% КОН до рН 10. Выпавший осадок фильтруют, промывают водой, сушат. Получают соединение 54 с выходом 19%. Растворяют в 10 мл диоксана 0.54 г соединения 54 и 0.29 г диметоксианилина и перемешивают ночь при комнатной температуре. Растворитель упаривают, остаток чистят колоночной хроматографией. Элюент - хлороформ: метанол 39:1. Получают соединение 55 с выходом 65%. Растворяют 500 мг соединения 55 в 5 мл 50% водного этанола, добавляют 32 мг LiOH и перемешивают ночь при комнатной температуре. Этанол упаривают на роторном испарителе, водный раствор подкисляют 10% соляной кислотой до рН 3. Выпавший осадок фильтруют, промывают водой, сушат. Получают соединение 56 с выходом 60%. Растворяют 360 мг соединения 56 в 10 мл ТГФ, в атмосфере аргона при перемешивании добавляют 71 мг N-гидроксисукцинимида и 127 мг дициклогексилкарбодиимида. Смесь перемешивают ночь при комнатной температуре ночь, после этого выпавший осадок фильтруют, маточный раствор упаривают, чистят флэш-хроматографией на силикагеле (элюент этилацетат). Получают соединение 2(3) с выходом 50%. LCMS-спектр соединения 2(3) представлен на Фиг.14 (М+1)=684.
Пример 15. Общий способ получение модифицированных белков (конъюгатов). Белок модифицируют веществом общей формулы 1 или 2 в молярном соотношениии от 1:1 до 1:100. Навеску вещества общей формулы 1 или 2 растворяют в 50 мкл ДМСО, затем медленно добавляют фосфатный солевой буферный раствор (ФСБ, рН 7,4) и добавляют раствор белка в ФСБ. Конечная концентрация белка в реакционных смесях составляла 1 мг/мл. Реакцию ведут при комнатной температуре в течение 20 ч при постоянном перемешивании. Реакционную массу центрифугируют, фильтрат наносят на гель Superdex 75 (GE, США); высота столба носителя - 23 см; подвижная фаза - ФСБ; скорость потока - 57,3 см/час; объем инжекции - 2,2% от объема носителя. Чистоту конъюгата определяют с использованием ВЭЖХ на аффинной колонке Bio-Monolith Protein A (Agilent, США).
По данному способу получены, в частности, конъюгаты:
КР1(1) - ритуксимаба (Р) с соединением 1(1), в соотношении 1:10,
КР1(2) - ритуксимаба (Р) с соединением 1(2), в соотношении 1:100,
КР1(3) - ритуксимаба (Р) с соединением 1(3), в соотношении 1:100,
КР1(4) - ритуксимаба (Р) с соединением 1(4), в соотношении 1:100,
КР1(5) - ритуксимаба (Р) с соединением 1(5), в соотношении 1:10,
КР2(1) - ритуксимаба (Р) с соединением 2(1), в соотношении 1:100,
КЦ1(1) - цетуксимаба (Ц) с соединением 1(1), в соотношении 1:10,
КИ1(1) - интерферона альфа (И) с соединением 1(1), в соотношении 1:10,
КМ1(1) - белка миелина (Р) с соединением 1(1), в соотношении 1:10,
КС1(1) - белка комплимента C1q (С) с соединением 1(1), в соотношении 1:10.
На Фиг.15 приведена для примера хроматограмма конъюгата КР1(1), свидетельствующая о содержании основного вещества более 98%.
Пример 16. Определение активности конъюгатов по отношению к CD 16а рецептору. В эксперименте используют стандартные материалы и оборудование для иммуно-ферментного анализа. В лунки 96-луночного планшета сорбируют CD16а из раствора 3,3 мкг/мл в фосфатно-солевом буфере в объеме 75 мкл/лунку. Сорбцию вели при 4°С в течение 16 часов. Раствор CD16а удаляют из планшета, лунки заполняли 2%-ным раствором БСА в ФСБ-П (150 мкл/лунку). Планшеты инкубируют при 37°С в течение 2 часов и трижды отмывали буфером ФСБ-П (каждый раз по 300 мкл/лунку). Затем лунки планшета заполняли растворами конъюгатами в ФСБ-П по 50 мкл/лунку, в разведениях от 0,25 до 256 мкг/мл. Каждому разведению одного конъюгата в планшете соответствовало три лунки. Для контроля над неспецифической сорбцией образцов часть лунок заполняли 2%-ным раствором БСА. Планшет инкубируют один час при 37°С в шейкере для ИФА (скорость вращения 180 оборотов/мин). Планшет отмывали пять раз по 300 мкл/лунку ФСБ-П и заполняли раствором белка, модифицированного пероксидазой хрена (по 75 мкл/лунку, в рабочем разведении, которое указано в описании к иммуноконъюгату). Планшет инкубируют 30 минут, отмывали пять раз по 300 мкл/лунку ФСБ-П и заполняли раствором ТМБ (100 мкл/лунку). Визуально оценивали глубину протекания ферментативной реакции и останавливали последнюю добавкой 50 мкл/лунку 500 мМ раствора серной кислоты. Измеряют оптическую плотность в лунках планшета при комнатной температуре на длине волны 450 нм. Для обработки результатов эксперимента используют программу GraphPad Prism 5.0.
На Фиг.16 представлены зависимости связывания некоторых конъюгатов по отношению к CD 16а рецептору, из которых рассчитаны (Таблица 1) константы диссоциации комплексов конъюгатов с CD16а рецептором (активность конъюгатов по отношению к CD16а рецептору - Kd). Как видно из таблицы, активность конъюгатов ритуксимаба в 10-100 раз выше активности неконъюгированного ритуксимаба.
На Фиг.17 представлены зависимости связывания интерферона альфа и конъюгата интерферона альфа с соединением 1(1) с CD16а рецептором. Зависимость оптической плотности на длине волны 450 нм от концентрации введенного в ИФА конъюгата. Как видно из таблицы 2, активность конъюгата интерферона выше активности неконъюгированного интерферона альфа.
Пример 17. Эффективность конъюгатов в тесте на антителозависимую (ритуксимаб-зависимую) клеточную цитотоксичность. Эффективность конъюгатов ритуксимаба в сравнении с неконъюгированным ритуксимаба была оценены в тесте на антителозависимую клеточную токсичность. В качестве клеток-мишеней были использованы СD20-положительные клетки линии Daudi (В-клеточная лимфома Беркита). В качестве эффекторных клеток используют пулированные периферические мононуклеарные клетки крови из 5 здоровых доноров, выделенные по стандартному протоколу.
Разведения исследуемых конъюгатов и ритуксимаба были приготовлены в среде RPMI 1640. 50 мкл растворов исследуемых конъюгатов и ритуксимаба были помещены в лунки 96-луночного круглодонного планшета. Клетки линии Daudi ресуспендируют в среде RPMI 1640 до концентрации 8×105 клеток/мл. 75 мкл полученной клеточной суспензии добавляют к приготовленным растворам ритуксимаба в планшете. Планшет инкубируют в термостате при температуре 37°С в течение 50 минут. Добавляют 75 мкл суспензии клеток РВМС (размороженных непосредственно перед экспериментом) в лунки к клеткам Daudi в соотношении 50:1 (РВМС: клетки Daudi). В качестве контрольных растворов добавляют среду RPMI 1640 (негативный контроль, содержавший только эффекторные клетки), а также 4% раствор Тритона Х-100 (положительный контроль, содержавший только клетки-мишени). Планшеты центрифугируют 3 минуты со скоростью 500 об/мин. Инкубируют планшеты в термостате в течение 8 часов при 37°С.
После инкубации полученные образцы были осаждены центрифугированием при 1200 об/мин в течение 10 минут. 50 мкл супернатанта были перенесены в новый планшет, где были смешаны с 50 мкл реакционной смеси (набор для детекции лактат дегидрогеназы, Promega (США), каталожный номер G1780). Полученную смесь инкубируют при комнатной температуре в течение 25 минут, после чего реакцию останавливали. Измеряют поглощение на плашечном спектрофотометре при длине волны 490 нм.
Цитотоксичность рассчитывалась по формуле (все величины в единицах оптической плотности):
цитотоксичность, %=100·(А-(SE-ST))/(HE-ST),
где А - уровень сигнала в смеси эффекторных клеток и клеток-мишеней;
SE - уровень сигнала эффекторных клеток;
ST - уровень сигнала клеток-мишеней;
HE - сигнал, соответствующий максимальному лизису клеток-мишеней.
Результат измерения эффективности полученных конъюгатов показан на примере конъюгата КР1(1) (Фиг.18) CON 4564. Полученный конъюгат обладал усиленным цитотоксическим действием на клетки-мишени (Daudi). В частности, ~35%-ный лизис клеток достигается при концентрации ритуксимаба ~1 мкг/мл, в то же время сходный цитотоксический эффект конъюгата КР1(1) CON 4564 достигается при концентрации ~0,001 мкг/мл. Аналогичная зависимость наблюдается и в случае конъюгата КР1(2) (Фиг.19). Таким образом, активность у конъюгатов ритуксимаба на 2-3 порядка выше, чем у ритуксимаба.
Пример 18. Получение конъюгата ритуксимаба КР1(7) и трастузумаба КТ1(7). К 1,5 г сахарной части антитела в растворе 100 мМ ацетата натрия, 200 мМ хлорида натрия, рН 5,5 добавляют 2,0 г периодата натрия, инкубируют 30 мин при комнатной температуре в темноте при перемешивании. Растворяют 1200 мг соединения 1(7) в 4 мл DMSO. К раствору антитела добавляют 8,4 г гидрокарбоната натрия, затем по каплям добавляют 1230 мкл растворенного в DMSO соединения 1(7) и инкубируют 2 часа.
Очистку проводили на SP-Sepharose (колонка ХК26 GE) ступенчатым градиентом NaCl (буфер 1: 25 мМ цитрат натрия, рН 4,5. Буфер 2: 25 мМ цитрат натрия, рН ~ 10,270 мМ). Конъюгат антитела концентрируют в ячейке Amicon через мембрану Millipore (NMWL 30000, cat. # PLTK07610) до конечной концентрации конъюгата 10 мг/мл. После фильтрации в конъюгат добавляют полисорбат-80 до конечной концентрации конъюгата КР1(7) или КТ1(7) 0,01%.
Пример 19. Сравнительная активность ритуксимаба (P) и трастузумаба (Т) и их конъюгатов КР1(7), КТ1(7) по отношению к CD16a рецептору. Изучение связывания полученных конъюгатов трастузумаба и ритуксимаба с рецептором CD16а проводили идентично примеру 17.
Сравненительная эффективность и активность (Kd) по отношению к CD16а рецептору ритуксимаба (Р), трастузумаба (Т) и их конъюгатов КР1(7), КТ1(7) в тесте антителозависимой цитотоксичности представлена на Фиг.20.
Как видно из полученных данных, активность конъюгатов КР1(7), КТ1(7) по отношению к CD16а рецептору на 1-2 порядка выше активности немодифицированных ритуксимаба (Р), трастузумаба (Т) (Таблица 3).
Реферат
Изобретение относится к новым соединениям общей формулыилиили их фармацевтически приемлемым солям, обладающим сродством к CD16а рецептору. Изобретение также относится к модифицированным этими соединениями белкам (конъюгатам), усиливающим и направляющим антителозависимую клеточную цитотоксичность. Соединения могут найти применение для лечения онкологических и аутоиммунных заболеваний. Изобретение также характеризует способ получения конъюгатов, фармацевтическую композицию и лекарственное средство, которые содержат модифицированные белки. В общих формулах 1 или 2R1 выбран из группы, представляющей собой (CH)N-,R2 выбран из группы, представляющей собойгде R3 в качестве концевого заместителя представляет собой -NH,,, илиa R4 представляет собой H или C-Cалкил. 6 н. и 12 з.п. ф-лы,20 ил., 3 табл.,19 пр.
Формула


в котором R1 выбран из группы, представляющей собой (CH3)2N-,
R2 выбран из группы, представляющей собой

где R3 в качестве концевого заместителя представляет собой -NH2,
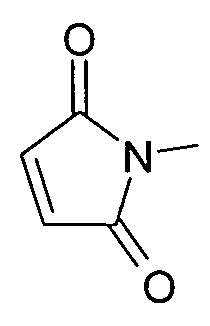
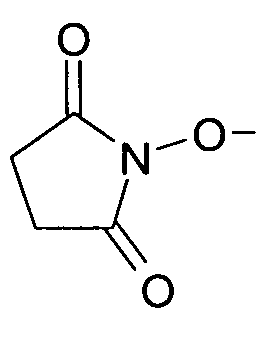
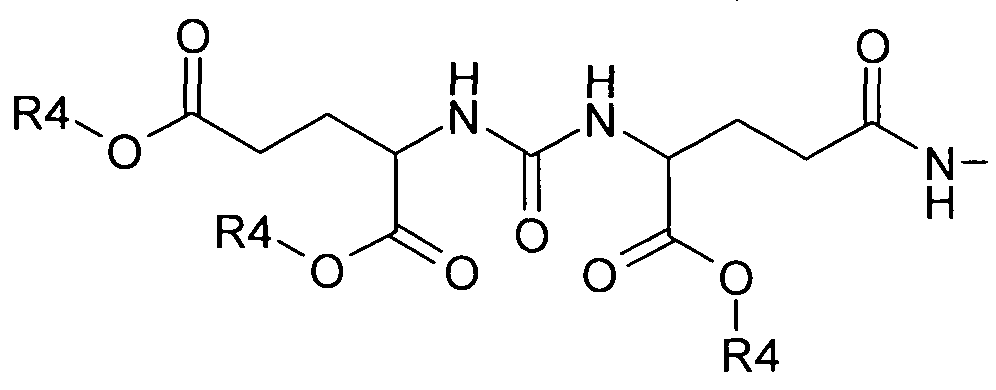
a R4 представляет собой H или C1-C3алкил.

R2 выбран из группы, включающей в себя:

где R4=H или C1-C3алкил.
R1 представляет собой (CH3)2N- или

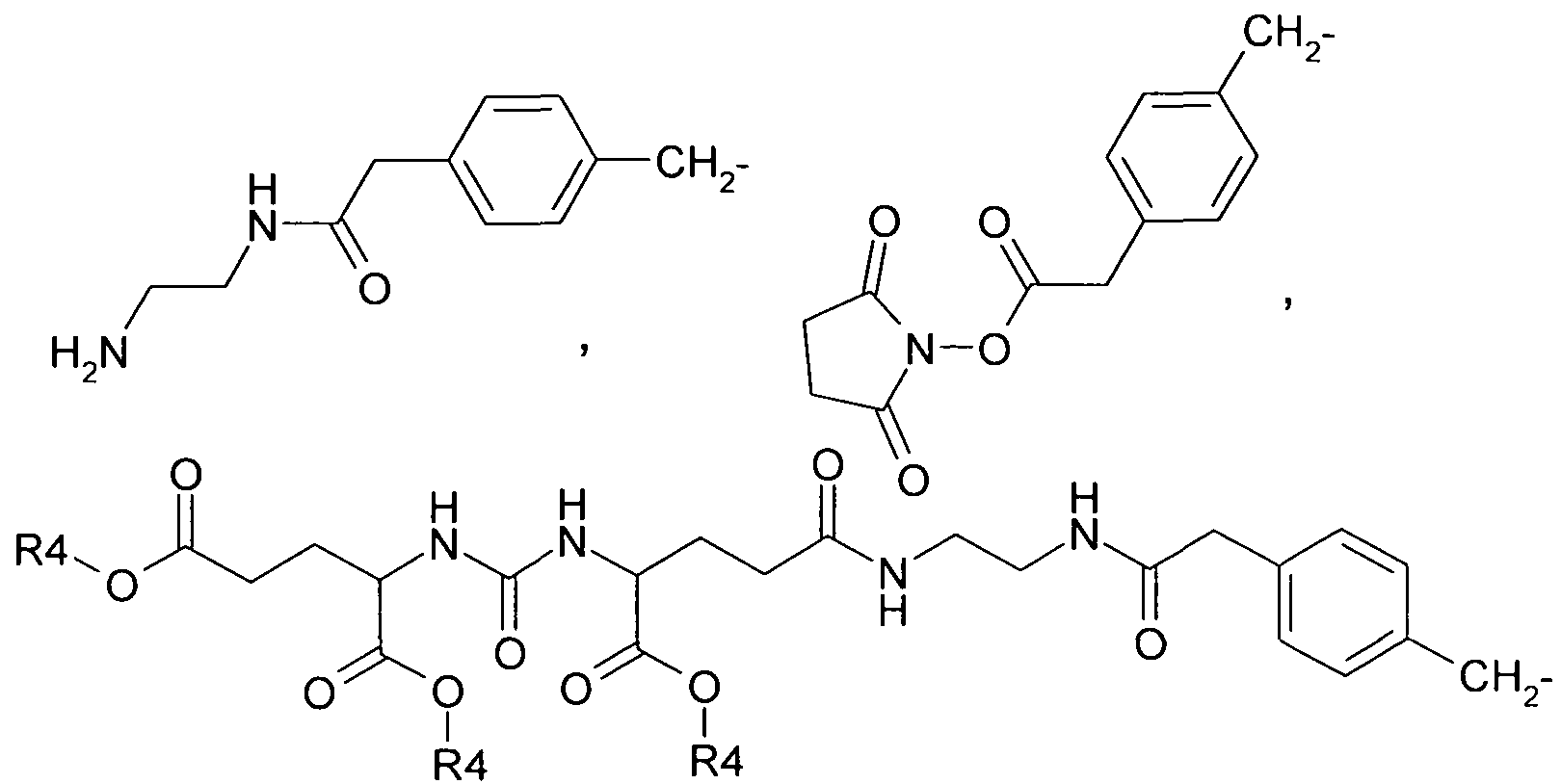
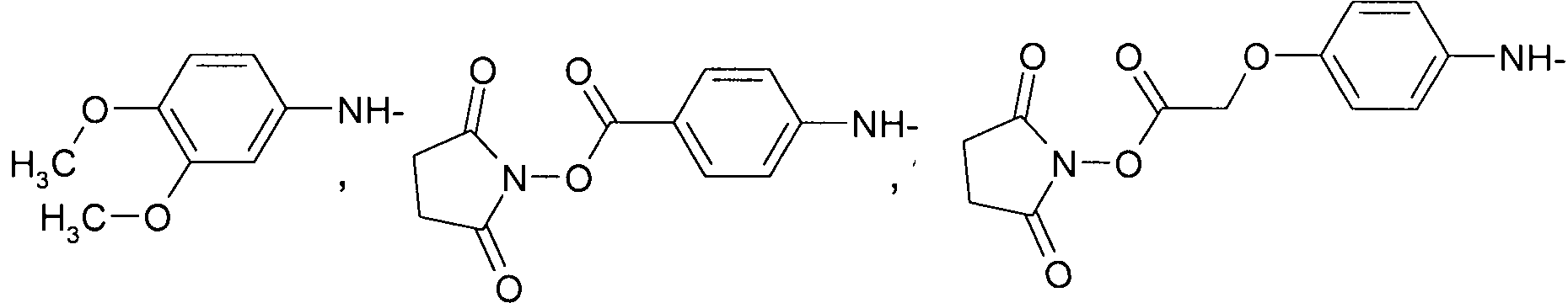
R2 выбран из группы, включающей в себя:
2,5-диоксопирролидин-1-иловый эфир (4-{[10-(3-хлорбензил)-5,5,11-триоксо-10,11-дигидро-5H-дибензо[b,f][1,4]тиазепин-7-карбонил]-амино}-фенокси)-уксусной кислоты,
2,5-диоксопирролидин-1-иловый эфир 4-{[10-(3-хлорбензил)-5,5,11-триоксо-10,11-дигидро-5H-дибензо[b,f][1,4]тиазепин-7-карбонил]-амино}-фенилкарбоновой кислоты,
2,5-диоксо-пирролидин-1-иловый эфир 3-[8-(3,4-диметоксифенилкарбамоил)-5,5,11-триоксо-5,11-дигидро-дибензо[b,f][1,4]тиазепин-10-илметил]-бензойной кислоты,
2,5-диоксо-пирролидин-1-иловый эфир (4-{8-[3-(4-бензилпиперидин-1-ил)-пропилкарбамоил]-5,5,11-триоксо-5,11-дигидро-дибензо[b,f][1,4]тиазепин-10-илметил}-фенил)-уксусной кислоты,
10-(3-хлорбензил)-5,5,11-триоксо-10,11-дигидро-5H-дибензо[b,f|[1,4]тиазепин-8-карбоновой кислоты (2-аминоэтил)-амид,
10-{4-[(2-амино-этилкарбамоил)-метил]-бензил}-5,5,11-триоксо-10,11-дигидро-5H-дибензо[b,f][1,4]тиазепин-8-карбоновой кислоты [3-(4-бензил-пиперидин-1-ил)-пропил]-амид,
2-(2,5-диоксо-2,5-дигидро-пиррол-1-ил)-этиловый эфир 10-(3-хлорбензил)-5,5,11-триоксо-10,11-дигидро-5H-дибензо[b,f] [1,4]тиазепин-8-карбоновой кислоты,
10-(4-{[2-(2,5-диоксо-2,5-дигидро-пиррол-1-ил)-этилкарбамоил]-метил}-бензил)-5,5,11-триоксо-10,11-дигидро-5H-дибензо[b,f][1,4]тиазепин-8-карбоновой кислоты [3-(4-бензил-пиперидин-1-ил)-пропил]-амид,
N-[2-({N-(метоксикарбонил)-N-[(1,5-диметокси-1,5-диоксопентан-2-ил)карбамоил]-β-аланил}амино)этил]-10-(3-хлорбензил)-5,5,11-триоксо-10,11-дигидродибензо[b,f][1,4]тиазепин-7-карбоксамид,
N5-(2-{[(4-{[7-{[[3-(4-бензилпиперидин-1-ил)пропил](фенил)амино]-карбонил}-5,5,11-триоксо-дибензо[b,f] [1,4]тиазепин-10(11H)-ил]метил}фенил)ацетил]-амино}этил)-N2-{[(1,3-дикарбоксипропил)амино]карбонил}глутамин,
4-[4-(диметиламино)фенил]-N-(4-{[(2,5-диоксопирролидин-1-ил)окси]карбонил}фенил)-7,8,9,10-тетрагидро-4H-[1]бензотиено[3,2-f]пирроло[1,2-а][1,4]диазепин-5(6H)-карбоксамид,
4-[4-(диметиламино)фенил]-N-(4-{2-[(2,5-диоксопирролидин-1-ил)окси]-2-оксоэтокси}фенил)-7,8,9,10-тетрагидро-4H-[1]бензотиено [3,2-f]пирроло[1,2-а][1,4]диазепин-5(6H)-карбоксамид, и
2,5-диоксопирролидин-1-ил N-[4-(5-{[(3,4-диметоксифенил)амино] -карбонил}-5,6,7,8,9,10-гексагидро-4H-[1]бензотиено[3,2-f]пирроло[1,2-а][1,4]диазепин-4-ил)фенил]-N-метилглицинат.
Комментарии