Трициклические ингибиторы парп - RU2404183C2
Код документа: RU2404183C2
Чертежи



















Описание
Данное изобретение относится к сериям получаемых соединений, которые представляют собой производные трициклических лактам-индолов и трициклических лактам-бензимидазолов и которые ингибируют поли(АДФ-рибозо)полимеразу (ПАРП), и к их применению при лечении рака, в частности, рака молочной железы.
Было показано, что гомологичная рекомбинация (ГР) играет важную роль при репарации повреждений, возникающих в репликативных вилках ДНК в клетках млекопитающих (2). Таким образом, клетки, испытывающие недостаток в ГР, растут медленнее и демонстрируют более высокие уровни генетической нестабильности. Предполагается, что генетическая нестабильность, обусловленная потерей ГР (рекомбинационной) репарации в человеческих клетках, в значительной степени способствует развитию рака в данных клетках (1).
Посттранскрипционная модификация ядерных белков поли(АДФ-рибозилированием) в ответ на разрывы в цепи ДНК играет важную роль при репарации ДНК, регуляции апоптоза и поддержании геномной стабильности.
Поли(АДФ-рибозо)полимераза (ПАРП-1) является главным членом семейства ферментов ПАРП и является распространенным ядерным белком клеток млекопитающих. ПАРП-1 катализирует образование поли(АДФ-рибоза) (ПАР) полимеров, используя NAD+ в качестве субстрата. При повреждении ДНК ПАРП-1 быстро связывается с однонитевым разрывом ДНК (SSB) и катализирует добавление к себе (авторибозилирование) и к другим белкам отрицательно заряженных ПАР цепей [См.(3,4) обзорные статьи]. Полагают, что связывание ПАРП-1 с SSB защищает повреждения ДНК от дальнейшего процессинга, пока ПАРП-1 не диссоциирует с разрывом вследствие накопленного отрицательного заряда, обусловленного полимерами ПАР (5,6).
Хотя ПАРП-1 вовлечена в некоторые процессы в ядре, такие как модуляция структуры хроматина, репликация ДНК, репарация и транскрипция ДНК, мыши, нокаутированные по гену ПАРП-1, развиваются нормально (7). Клетки, выделенные из данных мышей, обладают свойствами гиперрекомбинантности и генетической нестабильности в виде повышенных уровней сестринского хроматинового обмена (СХО), микронуклеуса и тетраплоидии (8, 10). Генетическая нестабильность может также встречаться у данных мышей, нокаутированных по гену ПАРП-1, в виде укорочения теломер, увеличенной частоты слияния хромосом и анэуплоидов (11), хотя все данные результаты не были получены для другого ряда мышей, нокаутированных по гену ПАРП-1 (12). Для первого ряда мышей, нокаутированных по гену, нулевая мутация ПАРП-1 привела к восстановлению нарушенной V (D) J рекомбинации у мышей SCID (13).
Данные результаты подтверждают точку зрения Lindahl и коллег, которая заключается в том, что ПАРП-1 играет защитную роль против рекомбинации (5). Было предположено, что связывание ПАРП-1 с разрывами ssДНК предотвращает их узнавание рекомбинантным комплексом и процессинг повреждений ДНК или, альтернативно, что отрицательные заряды, накопленные последовательным поли(АДФ-рибозил)ированием), отталкивают близлежащие рекомбиногенные последовательности ДНК. Только более поздние модели согласуются с ингибированием собственно ПАРП-1 и экспрессии доминантной отрицательной мутантной формы ПАРП-1, включая СХО, амплификацию гена и гомологичную рекомбинацию (14-18).
Исследования, основанные на обработке клеток ингибиторами ПАРП-1 или клеток, выделенных из мышей, нокаутированных по гену ПАРП-1, показали, что супрессия активности ПАРП-1 повышает чувствительность клеток к агентам, повреждающим ДНК, и ингибирует смыкание разрыва цепи (3, 4, 8-11, 19, 20).
Ингибиторы активности ПАРП-1 использовали в сочетании с традиционными режимами лечения рака, такими как лучевая терапия и химиотерапия (21). Было обнаружено, что при использовании ингибиторов в сочетании с метилирующими агентами, ингибиторами топоизомеразы и ионизирующими излучениями, они увеличивают эффективность данных видов лечения. Однако подобные способы лечения являются неселективными и поэтому приводят к повреждению и гибели нераковых или 'здоровых' клеток. Более того, известно, что подобные способы лечения приводят к возникновению неприятных побочных эффектов.
Таким образом, чрезвычайно желательным является предложение способа лечения, который как эффективен, так и селективен при уничтожении раковых клеток и который не нужно проводить в сочетании с лучевой терапией или химиотерапией.
Неожиданно было обнаружено, что клетки, испытывающие недостаток в гомологичной рекомбинации (ГР), по сравнению с клетками дикого типа являются гиперчувствительными к ингибиторам ПАРП-1.
Таким образом, согласно первому аспекту настоящего изобретения, предлагается соединение для ингибирования активности ПАРП-1, имеющее формулу I:
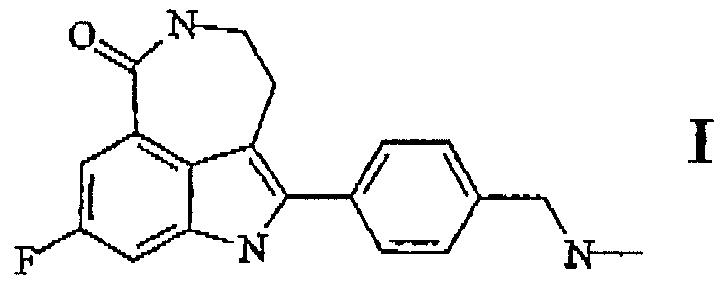
и его фармацевтически приемлимые соли.
Согласно второму аспекту настоящего изобретения предлагается соединение для ингибирования активности ПАРП-1, имеющее формулу II:

и его фармацевтически приемлимые соли.
Согласно третьему аспекту настоящего изобретения предлагается соединение для ингибирования активности ПАРП-1, имеющее формулу III:

и его фармацевтически приемлемые соли.
Соединения, описанные здесь, могут быть получены исходя из синтетических путей, описанных в WO 00/42040 и WO 01/16136.
Должно быть понятно, что ссылки, приведенные в данном описании, на соединения формул с I по III должны быть истолкованы, как охватывающие и другие фармацевтически приемлемые соли и другие фармацевтически приемлимые биопредшественники (формы пролекарств), если уместно. Термин “пролекарство” использованный в настоящем описании для обозначения модифицированных форм или производных фармакологически активного соединения, которые являются биоразлагаемыми или in vivo конвертируются до указанного активного соединения после введения, особенно при пероральном или внутривенном введении, в курсе лечения млекопитающего. Подобные пролекарства обычно выбирают благодаря повышенной растворимости в водном окружении, которая помогает преодолеть проблемы с получением препарата, и также в некоторых случаях обеспечивает относительно медленное или контролируемое высвобождение активного агента.
Как упомянуто в описании, фармацевтически приемлемые соли включают соли металлов, фосфаты и четвертичные амины. Соли металлов могут быть образованы щелочными металлами, такими как литий, натрий или калий.
Предпочтительно, соединение формулы I, указанной выше, вводят в виде фармацевтически приемлемой соли фосфата, имеющей следующую формулу:
Формула I - фосфат

Настоящее изобретение также относится к терапевтической полезности соединений, описанных здесь.
Таким образом, согласно следующему аспекту настоящего изобретения при производстве лекарственного средства предлагается использование терапевтического количества соединения формулы I и его фармацевтически приемлемой соли.
Согласно следующему аспекту настоящего изобретения при производстве лекарственного средства предлагается использование терапевтического количества соединения формулы II и его фармацевтически приемлемой соли.
Согласно следующему аспекту настоящего изобретения предлагается использование терапевтического количества соединения формулы III и его фармацевтически приемлемой соли при производстве лекарственного средства.
Согласно следующему аспекту настоящего изобретения предлагается использование терапевтического количества соединения формулы I, и его фармацевтически приемлемой соли, при производстве лекарственного средства для лечения заболевания или состояния, вызванного генетическим дефектом в гене, который опосредует гомологичную рекомбинацию.
Согласно следующему аспекту настоящего изобретения, предлагается использование терапевтического количества соединения формулы II при производстве лекарственного средства для лечения заболевания или состояния, вызванного генетическим дефектом в гене, который опосредует гомологичную рекомбинацию.
Согласно следующему аспекту настоящего изобретения предлагается использование терапевтического количества соединения формулы III, и его фармацевтически приемлемой соли, при производстве лекарственного средства для лечения заболевания или состояния, вызванного генетическим дефектом в гене, который опосредует гомологичную рекомбинацию.
Заболевания или состояния, вызванные генетическим дефектом в гене, который опосредует гомологичную рекомбинацию, включают, но не ограничиваются, рак, конкретно рак молочной железы.
Упоминаемые в описании “рак” или “опухоль” включают, но не ограничиваются перечисленными, рак легких, толстой кишки, поджелудочной железы, желудка, яичников, шейки матки, молочной железы, предстательной железы, мозга или кожи.
Использование ингибиторов ПАРП является особенно подходящим при лечении рака, который вызван генетическим дефектом в гене, где указанный ген опосредует гомологичную рекомбинацию. Раковые клетки данного типа обычно бывают дефектными в ГР клетках.
Специфическая чувствительность опухолей, обусловленных дефективной ГР, к ингибированию ПАРП означает, что делящиеся нормально “здоровые” клетки пациентов, которые имеют достаточные уровни ГР, не будут сильно затронуты лечением.
Следующим преимуществом лечения с использованием ингибиторов ПАРП является отсутствие необходимости вводить ингибиторы ПАРП в качестве комбинированной терапии вместе с общепринятой лучевой терапией или химиотерапией, избегая, таким образом, побочных эффектов связанных с данными общепринятыми видами лечения.
Дефект в гене, который опосредует гомологичную рекомбинацию, может быть обусловлен мутацией в нем, отсутствием или нарушением экспрессии гена, кодирующего белок, вовлеченный в ГР.
Согласно следующему аспекту настоящего изобретения предлагается использование терапевтического количества соединения формулы I при производстве лекарственного средства для индуцирования апоптоза в клетках с дефектом ГР.
Согласно следующему аспекту настоящего изобретения предлагается использование терапевтического количества соединения формулы II при производстве лекарственного средства для индуцирования апоптоза в клетках с дефектом ГР.
Согласно следующему аспекту настоящего изобретения предлагается использование терапевтического количества соединения формулы III при производстве лекарственного средства для индуцирования апоптоза в клетках с дефектом ГР.
Согласно следующему аспекту настоящего изобретения предлагается использование терапевтического количества соединения формулы I при производстве лекарственного средства для лечения рака.
Согласно следующему аспекту настоящего изобретения предлагается использование терапевтического количества соединения формулы II при производстве лекарственного средства для лечения рака.
Согласно следующему аспекту настоящего изобретения предлагается использование терапевтического количества соединения формулы III при производстве лекарственного средства для лечения рака.
Раковые клетки, пригодные для обработки соединениями, описанными здесь, могут быть частично или полностью лишены ГР. Предпочтительно, клетки полностью лишены ГР.
Соединения, описанные здесь, могут быть использованы для лечения наследственных форм рака, при которых пациент, подвергающийся лечению, имеет наследственную предрасположенность к раку. Однако указанные соединения являются особенно предпочтительными для лечения наследственного рака генетической природы, и наиболее предпочтительно, наследственного рака молочной железы, имеющего генетическую природу.
В предпочтительном аспекте, ингибитор ПАРП является полезным при обработке раковых клеток, с нарушенной экспрессией гена, вовлеченного в ГР. Гены, которые как предполагается, функционируют при ГР, включают XRCC1, ADPRT (PARP-1), ADPRTL2, (PARP02) CTPS, RPA, RPA1, RPA2, RPA3, XPD, ERCC1, XPF, MMS19, RAD51, RAD51β, RAD51C, RAD51D, DMC1, XRCCR, XRCC3, BRCA1, BRCA2, RAD52, RAD54, RAD50, MRE11, NB51, WRN, BLM KU70, RU80, ATM, ATR CHK1, CHK2, FANCA, FANCB, FANCC, FANCD1, FANCD2, FANCE, FANCF, FANCG, FANCC, FANCD1, FANCD2, FANCF, FANCG, RAD1, RAD9 [См.(2, 3, 5, 22-28) обзорные статьи].
Ген, вовлеченный в ГР, может быть геном, подавляющим опухоль. Таким образом, данное изобретение относится к обработке раковых клеток с дефектом в экспрессии гена-супрессора опухоли. Предпочтительно, ген-супрессор опухоли представляет собой BRCA1 или BRCA2.
Рак молочной железы является самым распространенным видом рака у женщин в западном мире. Некоторые семьи имеют сильную предрасположенность к раку молочной железы, который часто возникает благодаря наследственной мутации в одном аллеле либо BRCA1 либо BRCA2. Однако один функциональный аллель остается в исходном состоянии. Таким образом, индивиды, несущие указанную мутацию, развиваются нормально и не имеют фенотипических последствий, связанных с данной мутацией. Однако в одной клетке функциональный аллель может быть потерян, это делает данную клетку раковой и в то же время испытывающей недостаток в ГР. Данная стадия является критической при появлении опухоли (1).
Таким образом, согласно еще одному аспекту изобретения предлагается использование терапевтического количества соединения формулы I при производстве лекарственного средства для обработки раковых клеток с дефектом в экспрессии BRCA1 и/или BRCA2.
Согласно следующему аспекту настоящего изобретения предлагается применение терапевтического количества соединения формулы II при производстве лекарственного средства для обработки раковых клеток с дефектом в экспрессии BRCA1 и/или BRCA2.
Согласно следующему аспекту настоящего изобретения предлагается применение терапевтического количества соединения формулы III при производстве лекарственного средства для обработки раковых клеток с дефектом в экспрессии BRCA1 и/или BRCA2.
Раковые клетки, подвергаемые обработке, могут быть частично или полностью лишены экспрессии BRCA1 или BRCA2. Подобные дефекты могут быть идентифицированы с использованием методик множественного анализа ПЦР (29, 30) или с использованием других скринингов, известных специалистам, квалифицированным в данной области. Конкретно полезные методики включают количественную ОТ-ПЦР в реальном времени, Нозерн-блоттинг иммуногистохимию и Вестерн - блоттинг (31, 32).
Соответственно соединения по данному изобретению представляют особенный интерес для лечения ряда указанных раковых опухолей, и изобретение также включает способ лечения пациента, страдающего раковым заболеванием.
Соединения, описанные в данном документе, могут быть введены для эффективного поражения раковых клеток в терапевтически эффективном, нетоксичном количестве любым подходящим способом. Подходящие способы введения включают, без ограничения перечисленными, любой из следующих: пероральный, внутривенный, внутримышечный, подкожный, интраназальный или местный.
Терапевтически эффективное количество соединений, описанных в данном документе, обычно представляет собой количество, достаточное для достижения желаемого эффекта и может варьироваться согласно природе и степени тяжести болезненного состояния, и эффективности соединения. Следует понимать, что концентрации, применяемые для профилактики, отличаются от концентраций, используемых для лечения активного заболевания.
Для введения млекопитающим, и конкретно человеку, ожидается, что ежедневный уровень дозирования активного агента будет составлять от 0,01 до 50 мг/кг для мышей и от 0,01 мг/м2 до 50 мг/м2 поверхности тела для человека. В конечном счете, однако, количество вводимого активного ингредиента и частота введения будет выбрана по усмотрению врача.
Предпочтительно, уже очень низкие дозы соединений, ингибирующих ПАРП, необходимы для достижения терапевтического эффекта при лечении рака, уменьшая таким образом системное накопление соединений и минимизируя токсичные эффекты.
Хотя соединения, описанные в данном документе, можно вводить отдельно, в качестве 'сырого' соединения, предпочтительно представить соединение в фармацевтической композиции.
Все способы приготовления лекарственного средства при приготовлении подобных фармацевтических композиций в основном включают стадию приведения одного из соединений, описанных здесь, в ассоциацию с носителем, который состоит из одного или более вспомогательного ингредиентов. Обычно препараты получают однородным и плотным приведением соединения формулы I в ассоциацию с жидким носителем или с тонко измельченным твердым носителем, или обоими и затем, при необходимости, приданием продукту формы желаемых препаратов.
Препараты по настоящему изобретению, пригодные для перорального введения, могут быть представлены в виде дискретных единиц, таких как капсулы, саше, таблетки или пастилки, каждая содержащая заданное количество одного из описанных здесь соединений; порошок или гранулы; или суспензия в водной жидкости или неводной жидкости, такая как сироп, эликсир, эмульсия или аэрозоль. Любое из описанных здесь соединений также может быть представлено в виде шарика, лекарственной каши или массы. Таблетки могут быть получены прессованием или формованием, при желании с одним или более вспомогательным ингредиентом. Прессованные таблетки могут быть получены прессованием в подходящей машине сыпучей формы любого из описанных здесь соединений, такой как порошок или гранулы, при желании перемешанной со связующим веществом, смазывающим веществом, инертным разбавителем, поверхностно-активным или диспергирующим агентом. Формованные таблетки могут быть изготовлены формованием в подходящей машине смеси любого из описанного здесь соединения в виде порошка с любым пригодным носителем.
Сироп может быть получен добавлением любого из описанных здесь соединений, к концентрированному водному раствору сахара, например сахарозы, к которому может быть добавлен любой желаемый дополнительный ингредиент. Подобный дополнительный ингредиент(ы) может включать ароматизаторы, агент для замедления кристаллизации сахара или агент для увеличения растворимости любого другого ингредиента, такого как многоатомный спирт, например, глицерин или сорбит.
Препараты для ректального введения могут быть представлены в виде суппозитория с обычным носителем, таким как масло какао.
Препараты, подходящие для парентерального введения, обычно включают стерильные водные препараты любого из описанных здесь соединений, которые предпочтительно являются изотоническими по отношению к крови реципиента.
В дополнение к вышеупомянутым ингредиентам, препараты по данному изобретению, например мази, кремы и подобные, могут включать один или более дополнительных ингредиентов, например, разбавитель, буфер, ароматизирующий агент, связующее вещество, поверхностно-активный агент, загуститель, смазывающее вещество и/или консервант (включая антиоксидант) или другой фармацевтически приемлемый инертный наполнитель.
Соединения по данному изобретению для введения могут также быть включены в липосомальные препараты, которые могут быть получены способами, хорошо известными в данной области.
Таким образом, согласно следующему аспекту настоящего изобретения предложена фармацевтическая композиция, содержащая в качестве активного агента соединение формулы I или его фармацевтически приемлемую соль.
Согласно следующему аспекту настоящего изобретения предложена фармацевтическая композиция, содержащая в качестве активного агента соединение формулы II или его фармацевтически приемлемую соль.
Согласно следующему аспекту настоящего изобретения предложена фармацевтическая композиция, содержащая в качестве активного агента соединение формулы III или его фармацевтически приемлемую соль.
Фармацевтическая композиция может содержать по меньшей мере один дополнительный ингредиент, обеспечивающий совместимую фармацевтически приемлемую добавку, носитель, разбавитель-носитель или наполнитель, и может быть представлена в единой дозированной форме.
Носитель(ли) должен быть фармацевтически приемлемым в отношении совместимости с другими ингредиентами препарата и не должен быть опасным для реципиента.
Возможные препараты включают таковые, пригодные для перорального, ректального, местного и парентерального (включая подкожно, внутримышечно и внутривенно) введения или для введения в легкое или другой абсорбирующий участок, такой как носовые ходы.
Соединения, упоминаемые в описании, могут быть введены в сочетании с другими противоопухолевыми соединениями.
Настоящее изобретение также включает способ лечения рака у млекопитающих введением описанных здесь соединений и их фармацевтически приемлемых солей.
Таким образом, согласно следующему аспекту настоящего изобретения, предложен способ лечения рака у млекопитающих, включающий введение соединения формулы I или его фармацевтически приемлемой соли.
Таким образом, согласно следующему аспекту настоящего изобретения, предложен способ лечения рака у млекопитающих, включающий введение соединения формулы II или его фармацевтически приемлемой соли.
Таким образом, согласно следующему аспекту настоящего изобретения предложен способ лечения рака у млекопитающих, включающий введение соединения формулы III или его фармацевтически приемлемой соли.
Настоящее изобретение далее описано посредством примера, со ссылкой на прилагаемые чертежи, где:
Фиг.1 представляет собой график, показывающий выживаемость клеток в присутствии ингибитора ПАРП формулы III для клеточной линии АА8, клеточной линии IsrISF и клеточной линии CxR3.
Фиг.2 представляет собой график, показывающий выживаемость клеток в присутствии ингибитора ПАРП формулы III для клеточной линии V79, клеточной линии VC8 и клеточной линии VC8B2.
Фиг.3 представляет собой график, показывающий выживаемость клеток в присутствии ингибитора ПАРП формулы I для клеточной линии V79, клеточной линии VC8 и клеточной линии VC8B2.
Фиг.4 представляет собой гистограмму, показывающую активность ПАРП в клеточных линиях VC8, V79, VC8#13 и VC8, VC8#13 и VC8+B2 в присутствии ингибитора ПАРП формулы III.
Фиг.5 представляет собой пару графиков, показывающих ингибирование клеточной активности ПАРП в присутствии ингибитора ПАРП формулы I и III в клетках L1210 с нарушенной проницаемостью мембраны (верхний график) и интактных (нижний график).
Фиг.6 представляет собой пару гистограмм, отражаающих фармакокинетику и фармакодинамику в крови и в опухоли фосфата соединения формулы I в дозе 1 мг/кг (верхний) и 10 мг/кг (нижний) у мышей, несущих ксенотрансплантаты SW620.
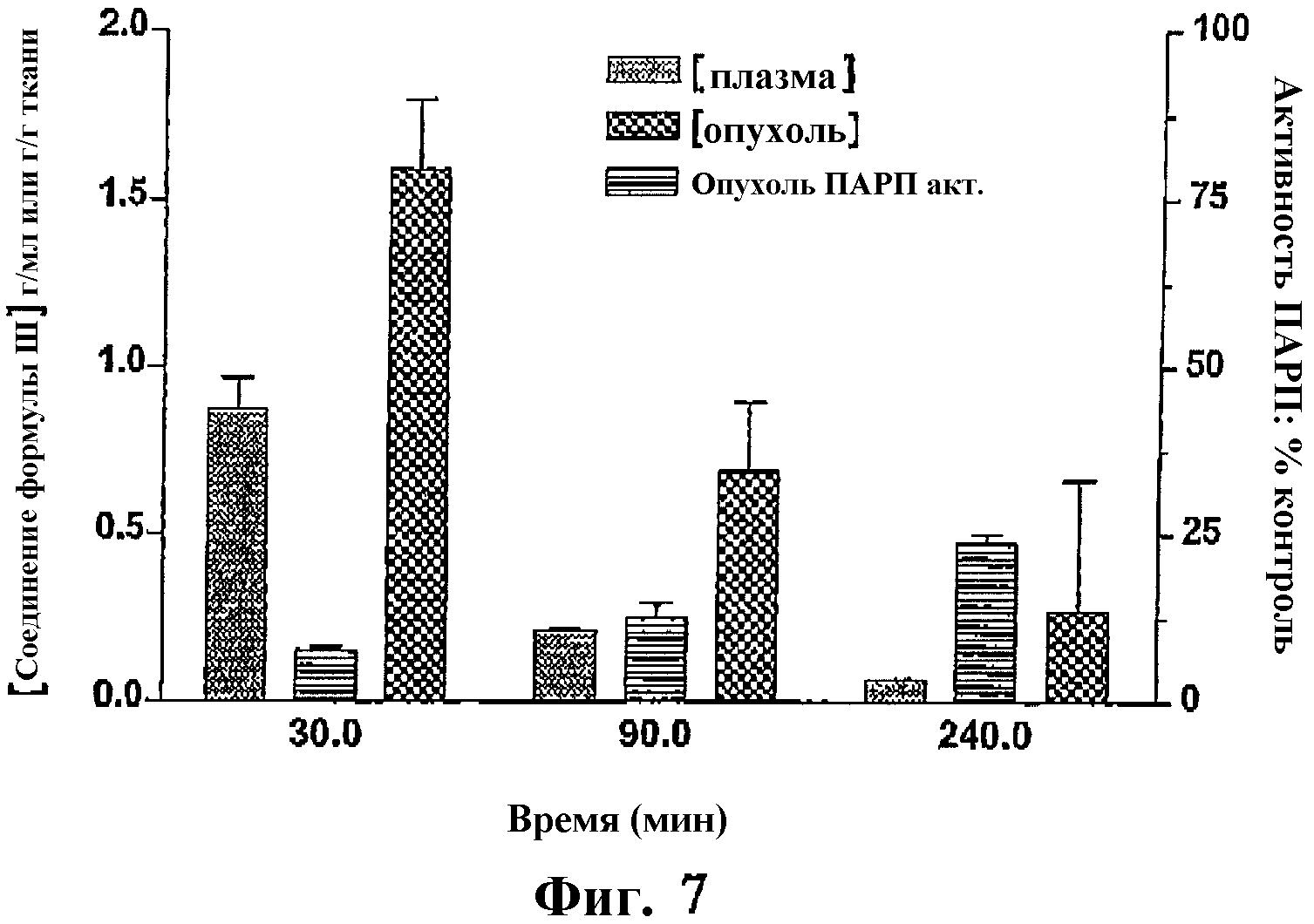
Фиг.7 представляет собой гистограмму, показывающую фармакокинетику и фармакодинамику соединения формулы III у мышей, несущих ксенотрансплантаты SW620.
Фиг.8 представляет собой график, показывающий рост опухоли (относительный средний объем опухоли) у мышей, несущих ксенотрансплантаты SW620, после обработки соединением формулы III в сочетании с темозоломидом (ТМЗ) и ТМЗ отдельно.
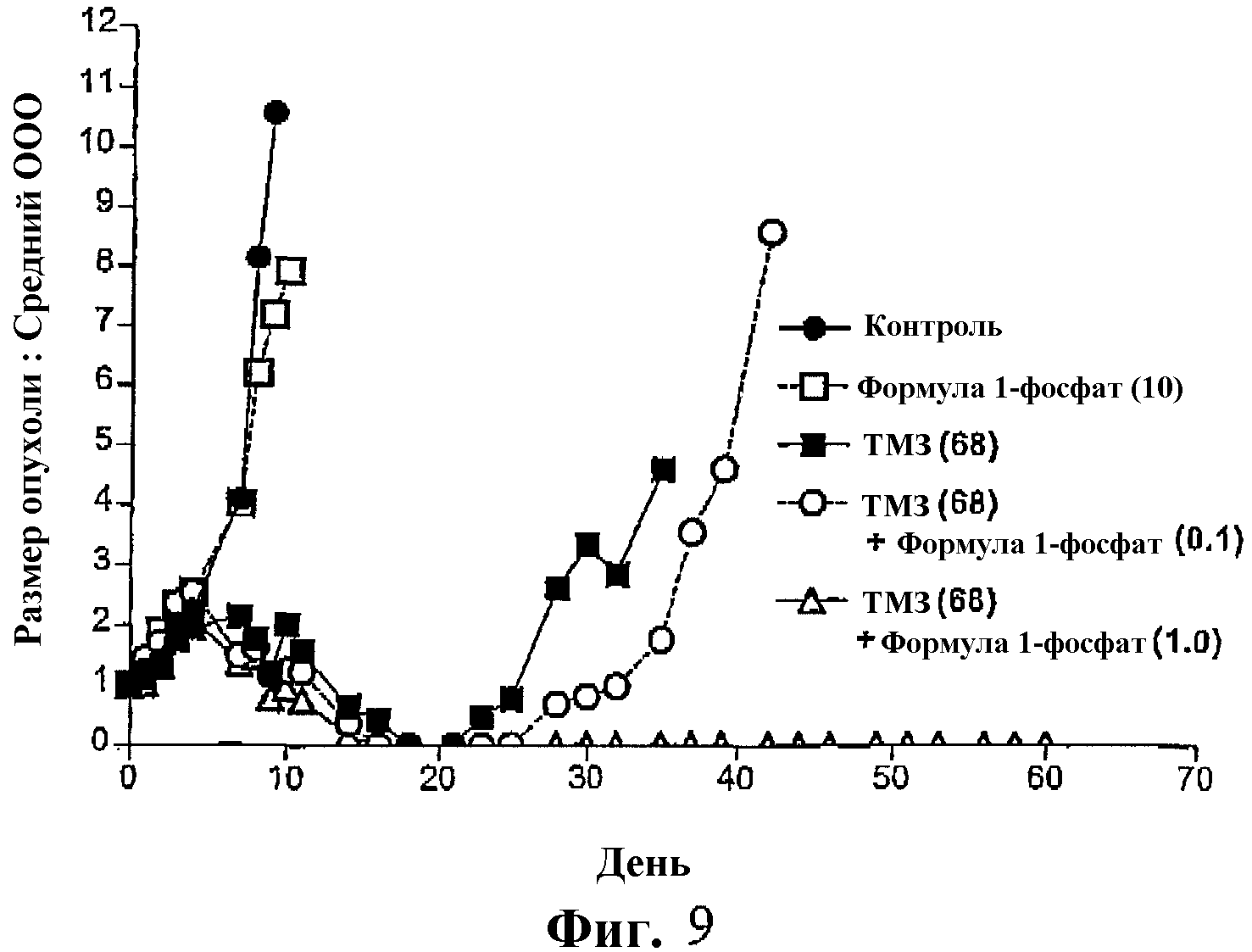
Фиг.9 представляет собой график, показывающий рост опухоли (относительный средний объем опухоли) у мышей, несущих ксенотрансплантаты SW620, после обработки фосфатом соединения формулы I в сочетании с темозоломидом (ТМЗ) и ТМЗ отдельно.
На фиг.1 показана доля в процентах выживаемости клеточных линий AA8, IrS ISF и CxR3 при обработке соединением формулы III в различных концентрациях. Было обнаружено, что формула III является наиболее активной для IrS ISF, которая лишена XRCC3, имея ЛК50 (концентрация активного компонента, при которой гибнет 50% клеток) 100 нМ.
На фиг.2 показана доля в процентах выживаемости клеточных линий V79-Z, VC8 и VC8B2 при обработке соединением формулы III в различных концентрациях. Было обнаружено, что формула III является самым эффективным для клеточной линии VC8, которая лишена BRCA2, имея величину ЛК50 43 нМ и ЛК90 (концентрация активного компонента, при которой гибнет 90% клеток) составляла 1200 нМ.
На фиг.3 показана доля в процентах выживаемости клеточных линий V79-Z, VC8 и VC8B2 при обработке соединением формулы I в различных концентрациях. Было обнаружено, что соединения формулы III являются самыми эффективными для клеточной линии VC8, которая лишена BRCA2, имея величину ЛК50 12 нМ, ЛК90 была 27 нМ.
На фиг.4 показана активность ПАРП различных клеточных линий при обработке соединением формулы III в различных концентрациях. На графике фиг.4 представлены четыре серии результатов для каждой соответствующей клеточной линии. Первый столбец каждой серии показывает фоновую активность ПАРП (олиго отсутствуют, следовательно, активность ПАРП зависит от разрывов в цепи ДНК), второй столбец отражает полностью стимулированную (олиго) активность ПАРП и третий и четвертый столбцы показывают активность ПАРП в присутствии соединения формулы III.
На фиг.5 показано действие соединений формулы I и III на активность ПАРП.
У клеток, использованных для получения результатов, показанных на фиг.5, либо была нарушена проницаемостью мембраны с помощью дигитонина и затем было проведено исследование на общую стимулированную (олиго) активность ПАРП в присутствии или отсутствии ингибитора ПАРП формулы I и формулы III либо они были подвергнуты воздействию одного из указанных ингибиторов ПАРП в течение 20 минут перед нарушением проницаемости мембраны и подвергнуты анализу на общую стимулированную активность ПАРП.
Также не было обнаружено различий в ингибиторной активности ПАРП соединений формулы I и формулы III при нарушении проницаемости клеточной мембраны перед добавлением соединения, но соединение формулы I было более эффективным в интактных клетках, возможно, благодаря большей степени его накопления внутри клеток.
На фиг.6 показаны концентрации соединения формулы I в плазме и опухоли и его фармакокинетический эффект для лимфоцитов из периферической крови мыши (лпк парп) и ксенотрансплантатов SW620 (опухоль ПАРП) в различные сроки после внутрибрюшинного введения фосфата соединения формулы I. Фосфатная соль соединения формулы I обеспечивает повышение растворимости соединения формулы I. Однако при введении в плазму животного (включая человека) фосфатазы разлагают фосфатные соли формулы I (формула I - фосфат) до исходного соединения, т.е. формулы I.
Из фиг.6 видно, что через тридцать минут после введения формулы I - фосфат 10 мг/кг, самые высокие уровни исходного соединения определялись как в плазме, так и в опухоли. Концентрация формулы I повышалась с течением времени в плазме быстрее, чем в опухоли и через 24 часа после введения определялись значительные уровни в опухоли, но ничего не определялось в плазме. Происходило полное и замедленное ингибирование активности ПАРП как в лпк, так и в опухоли: <50% контроль через 24 ч.
После введения фосфата соединения формулы I в дозе 1 мг/кг более низкие уровни соединения формулы I могут быть обнаружены как в плазме, так и в опухоли и, следовательно, влияние на активность ПАРП менее выражено.
На фиг.7 показаны концентрации соединения формулы III в плазме и опухоли, и его фармакокинетический эффект для ксенотрансплантатов SW620 (опухоль ПАРП активна) в различные сроки после внутрибрюшинного введения соединения формулы III в дозе 10 мг/кг. Также данное соединение хорошо распространяется в опухоли и селективно накапливается с течением времени и таким образом ингибирует активность ПАРП в опухоли.
Из фиг.8 видно, что в течение 20 дней после введения темолозомида (68 мг/кг ежедневно х5) ксенотрансплантат опухоли постепенно уменьшался в размерах. Однако вскоре после данного срока размер опухоли начал увеличиваться. При введении соединения формулы III (5 мг/кг ежедневно х5) вместе с темозоломидом опухоль значительно уменьшается за приблизительно 15 дней до недетектируемого размера, размер опухоли остается недетектируемым в течение следующих 50 дней. Затем она начинает увеличиваться в размере. При введении более высокой дозы соединения формулы III (15 мг/кг ежедневно х5) размер опухоли остается недетектируемым в течение следующих 80 дней до конца эксперимента, при вскрытии опухоль не была обнаружена, т.е. произошла полная регрессия опухоли.
На фиг.9 показан образец, подобный таковому на фиг.8 после введения фосфата соединения формулы I (при 0,1 мг/кг и 1,0 мг/кг) в сочетании с темолозомидом.
Таблица 1
Генотип и происхождение клеточных линий, использованных в данном исследовании

Материалы и методы
Цитотоксичность ингибиторов ПАРП для клеток, испытывающих недостаток в ГР (XRCC3 или BRCA2)
Клеточная культура
Клеточные линии АА8, irs1SF и CXR3 были предоставлены Larry Thompson [41].
VC-8 VC-8+B2 VC-8#13 были подарены Malgorzata Zdienicka [42]. Все клеточные линии для данного исследования выращивались в модифицированной среде Игла по способу Дульбекко (МДСИ) с 10% сывороткой крови эмбрионов крупного рогатого скота и пенициллином (100 мкМ/мл) и стрептомицина сульфатом (100 мкг/мл) при 37єС в атмосфере, содержащей 5% СО2.
Анализ токсичности - анализ клоногенной выживаемости
Клетки в стадии экспоненциального роста в 6-луночных планшетах подвергали воздействию соединения формулы III при концентрациях, показанных на фиг.2 в 1% ДМСО или 1% ДМСО отдельно в среде в течение 24 часов.
Клетки собирали трипсинизацией, считали и высевали при различных плотностях в 10 см чашки в свежую среду в отсутствие лекарственного средства для выращивания колоний.
Через 7-10 дней чашки фиксировали в растворе метанол:уксусная кислота в соотношении 3:1 и прокрашивали 0,4% кристаллическим фиолетовым.
Колонии подсчитывали и доля выживаемости была вычислена по отношению к контролю - клеткам, обработанным 1% ДМСО.
Анализ активности ПАРП
Клетки в экспоненциальной фазе роста с нарушенной дигитонином проницаемостью клеточной мембраны, или интактные в течение 20 минут перед промыванием и нарушением клеточной мембраны дигитонином были подвергнуты действию 1% ДМСО в культуральной среде (контроль) или действию соединения формул I или III в 1% ДМСО при концентрациях, указанных на фиг.4. Активность ПАРП измеряли включением [32Р]меченого NAD+субстрата в полимеры, осаждаемые ТХУ после стимулирования добавлением олигонуклеотида с «тупыми» концами и сравнением с ненуклеотидным стимулированием клеток. Активность ПАРП в гомогенатах опухоли (1 в 40 в изотоническом буфере) мыши, подвергнутой действию формулы III, была измерена подобным образом. Активность ПАРП в гомогенатах лпк и опухоли мыши, подвергнутой действию фосфата соединения формулы I, была измерена иммуннологическим определением полимера с использованием антитела 10Н. Кратко, гомогенаты опухоли, разведенные до более чем 1:1000 в изотоническом буфере, инкубировали с 350 мкМ NAD в течение 6 мин и переносили на нитроцеллюлозную мембрану. Образование полимера поли(АДФ-рибоза) (ПАР) количественно определяли детектированием хемилюминисценции с использованием УФ-иллюминатора Fuji LAS3000, исходя из последовательных разведений стандартов PAR после инкубирования с антителом 10Н с PAR и вторичным антимышиным антителом. Результаты были стандартизированы, исходя из измеренного содержания белка в гомогенате.
Конечно, должно быть понятно, что изобретение не ограничено деталями вышеуказанных воплощений, описанных только в качестве примера.
Ссылки:





Реферат
Настоящее изобретение относится к применению ингибиторов поли(АДФ-рибозо)полимеразы формул I, II и III для лечения раковых опухолей, в клетках которых имеет место генетический дефект гена, опосредующего гомологичную рекомбинацию. При применении соединений формул I, II и III предварительно выбирают млекопитающее с генетическим дефектом гена, опосредующего гомологичную рекомбинацию, после чего соединения формул I, II, III используют для индуцирования апоптоза в клетках с дефектом гена, опосредующего гомологичную рекомбинацию ! ! ! Технический результат - разработка способа лечения рака у млекопитающих с генетическим дефектом гена, опосредующего гомологичную рекомбинацию. 3 н. и 20 з.п. ф-лы, 9 ил., 1 табл.
Формула



или его фармацевтически приемлемой соли при производстве цитотоксического лекарственного средства для лечения рака, где рак вызван генетическим дефектом гена, который опосредует гомологичную рекомбинацию.
или его фармацевтически приемлемой соли, при производстве цитотоксического лекарственного средства для индуцирования апоптоза в клетках с дефектом гена, который опосредует гомологичную рекомбинацию.
вводят млекопитающему соединение формулы I, II или III
или его фармацевтически приемлемую соль.
Комментарии