Производные бензотиазинов, их получение и применение в качестве лекарств - RU2523791C2
Код документа: RU2523791C2
Описание
Целью настоящего изобретения являются производные бензотиазинов, способ получения указанных производных, фармацевтические композиции, содержащие данные соединения, и их применение в качестве лекарств, предназначенных для лечения и/или предупреждения диабета типа 2, ожирения, дислипидемий, артериальной гипертензии и атеросклероза. Данные соединения также могут найти применение в лечении и/или предупреждении гипергликемий, интолерантности к глюкозе, резистентности к инсулину, гипертриглицеридемий, гиперхолестеринемий, рестенозов, панкреатитов, ретинопатий, нефропатий, невропатий (Reichard et al., N. Engl. J. Med. 1993, 329: 304-309), некоторых типов рака (Strickler et al., Diabetes Technology & Therapeutics 2001, 3(2): 263-274) или глауком (Pascale et al., Ophtalmology 2006, 113(7): 1081-86).
Настоящее изобретение также относится к комбинациям описанных соединений и других агентов, используемых в лечении данных патологий. Действительно, лечение таких патологий, как диабет типа 2, часто требует использования одновременно нескольких классов соединений, чтобы добиться рекомендованных показателей гликемии и поддерживать баланс глюкозы (Nathan et al., Diabetes Care 2009 32:193-203). Указанные комбинации также могут быть использованы в комбинированном лечении ожирения и диабетов типа 2 (Grundy et al., Circulation 2005, 112: 2735-2752).
Метаболический синдром является ранней стадией нескольких серьезных сердечно-сосудистых патологий. Он развивается как следствие резистентности к инсулину и характеризуется висцеральным ожирением (Després et al., Nature 2006 444(14): 881-87), ассоциированным с некоторыми факторами риска, такими как интолерантность к глюкозе и некоторые типы дислипидемий, которые могут быть ассоциированы с артериальной гипертензией (Grundy, Nat. Rev. Drug Discov. 2006, 5: 295-309).
Диабет типа 2 является документированной патологией, так как гликемические расстройства могут быть объяснены тремя основными механизмами: недостаточностью функции β-клеток островков Лангерганса поджелудочной железы, уменьшением использования глюкозы в периферических тканях и избыточной продукцией глюкозы в печени (Monnier et al., Diabetes & Metabolism 2008, 34: 207-216). Однако существующие методики лечения не позволяют достигнуть рекомендованных целевых показателей гликемии (в особенности HbA1c) у многих пациенты, страдающие диабетом типа 2. Поэтому всегда существует высокая потребность в методиках лечения данной патологии, основанных на новых механизмах.
Ожирение представляет собой недуг, поражающий все большее и большее количество людей по всему миру. Данное заболевание часто ассоциировано с повышенным риском развития диабета типа 2, сердечно-сосудистых заболеваний, цереброваскулярных инсультов и некоторых типов рака. Поэтому ожирение является основным фактором риска для патологий, ассоциированных с высоким уровнем заболеваемости или смертности.
Глюкокортикоиды (кортизол у человека, кортикостерон у грызунов) представляют собой вездесущие гормоны, которые играют доминирующую роль в регуляции энергетического метаболизма. Они индуцируют глюконеогенез и ингибируют секрецию инсулина бета-клетками поджелудочной железы, а также периферическое поглощение глюкозы (Dallman ef al., Front Neuroendocrinol. 1993, 14: 303-347).
Недавно установлено, что 11β-гидроксистероид-дегидрогеназы (11β-HSD) регулируют уровень глюкокортикоидов в некоторых тканях-мишенях (печени, жировой ткани, почке, головном мозге и т.д.). У человека данный механизм может вызывать локальное повышение уровня кортизола. Повышение уровня кортизола в жировой ткани может приводить к увеличению массы висцеральной жировой ткани вследствие воздействия глюкокортикоидов на дифференцировку преадипоцитов в адипоциты и на липогенез; в некоторых случаях глюкокортикоиды индуцируют липолиз и вредное действие свободных жирных кислот плазмы на печень, поджелудочную железу, скелетные мышцы (липотоксичность). Повышение уровня кортизола в печени может вызывать увеличение гликемии, что может приводить к развитию диабета типа 2.
Известны две изоформы 11β-HSD: тип 1 и тип 2. 11β-HSD2 локализован в основном в почках. Данный фермент катализирует превращение активных глюкокортикоидов в неактивные глюкокортикоиды (у человека превращение кортизола в кортизон) и, следовательно, по существу участвует в защите минералокортикоидных рецепторов (MR) от активации кортизолом (Edwars et al., Lancet, 1988, 2: 986-989). И наоборот, 11β-HSD1 в основном действует подобно 11-кето-редуктазе и превращает неактивные глюкокортикоиды в активные глюкокортикоиды в тканях, где этот фермент имеет высокий уровень экспрессии (в печени и жировой ткани). Поэтому ингибирование данного фермента на уровне печени и адипоцитов должно выражаться в уменьшении описанных выше эффектов. Ряд исследований, в которых использовано моделирование ожирения и/или диабета у животных, подтвердил вовлечение 11β-HSD1 в эти заболевания. Так, уровень экспрессии 11β-HSD1 повышен у диабетических крыс линии Zucker, и данное повышение коррелирует с прогрессированием патологии, моделированной у этих крыс (Duplomb et al., Biochem. Biophys. Res. Commun., 2004, 313: 594-599). Доказано, что мыши с нулевой мутацией в гене, кодирующем 11β-HSD1 (нокаутные мыши), являются устойчивыми к гипергликемии, вызываемой ожирением или стрессом (Kotelevtsev Y. et al. PNAS 1997, 94: 14924-14929). И наоборот, у трансгенных мышей с избирательной сверхэкспрессией 11β-HSD1 в жировой ткани, развивается висцеральное ожирение, инсулин-резистентный диабет и гиперлипидемия (Masuzika et al., Science, 2001, 294: 2166-2170). Приведенные экспериментальные данные указывают на преимущество, которое дает ингибирование 11β-HSD1 в качестве терапевтической мишени (Wamil et al., Drug Discovery Today, 2007, 12: 504-520).
Соединения по настоящему изобретению обладают способностью избирательно ингибировать 11β-HSD1 в сравнении с 11β-HSD2, у человека это должно оказывать благоприятное воздействие на диабет типа 2, ожирение, гиперлипидемии, артериальную гипертензию, атеросклероз и на все патологии, которые с этим ассоциированы, такие как коронарные инсульты, цереброваскулярные инсульты или артериит нижних конечностей (Wilcox et al., Stroke, 2007, 38: 865-873; Wilcox et al., Am. Heart J. 2008, 155:712-7).
Данные соединения отличаются от соединений, известных в данной области техники, своей химической структурой и исключительными биологическими свойствами.
Целью настоящего изобретения являются производные бензотиазинов, обладающие способностью ингибировать 11β-HSD1 не только на ферментном уровне, но также на клеточном уровне.
Соединения по настоящему изобретению представляют собой соединения общей Формулы (I):

где
R1 представляет собой атом водорода; C1-C6 алкил; COR5; SO2R5; CO(CH2)mR6; CO(CH2)mOR6; (CH2)mR6; (CH2)mCONR7R8; (CH2)nNR7R8; (CH2)nOR6; CHR7OR9; (CH2)mR10;
m имеет значение от 1 до 6;
n имеет значение от 2 до 6;
R2 представляет собой фенил, имеющий в качестве заместителей одну или более чем одну группу, выбранную из атома галогена, C1-C6 алкила, CN, ОН, CF3, OCF3, SMe, COMe, CMe(OH)CF3, CH(OH)CF3, COOR7, CONR7R11; нафтил, 1,2,3,4-тетрагидро-нафталин, бифенил, фенил, пиридин или гетероцикл, отличный от индола, в случае, когда R1, R4 и R′4 представляют собой атом водорода, незамещенные или имеющие в качестве заместителей одну или более чем одну группу, выбранную из атома галогена или C1-C6 алкила, CN, OH, CF3, OCF3, OMe, SMe; циклоалкил, незамещенный или имеющий в качестве заместителей OH, CONH2, SO2Me, SO2NH2; C1-C6 алкил арил или циклоалкил арил,
при условии, что группа R2 всегда связана с карбонилом через атом углерода, и, когда R2 представляет собой фенил, заместитель COOR7 никогда не находится в положении 4 относительно карбонила;
R3 представляет собой метил или этил;
R4 и R′4 являются одинаковыми или разными и представляют собой атом водорода; атом галогена; C1-C6 алкил; CN; CF3; OCF3; SMe; OMe; NR7R8; SO2Me;
R5 представляет собой C1-C6 алкил; фенил, незамещенный или имеющий в качестве заместителей одну или более чем одну группу, выбранную из атома галогена, C1-C6 алкила, CN, OH, CF3, OCF3, SMe; нафтил, незамещенный или имеющий в качестве заместителей одну или более чем одну группу, выбранную из атома галогена или C1-C6 алкила, CN, OH, CF3, OCF3, SMe; циклоалкил, незамещенный или имеющий в качестве заместителей CONH2, SO2Me, SO2NH2, гетероарил, незамещенный или имеющий в качестве заместителей одну или более чем одну группу, выбранную из атома галогена, C1-C6 алкила, CN, ОН, CF3, OCF3, SMe;
R6 представляет собой атом водорода; C1-C6 алкил; фенил, незамещенный или имеющий в качестве заместителей одну или более чем одну группу, выбранную из атома галогена, C1-C6 алкила, CN, OH, CF3, OCF3, SMe; нафтил или гетероцикл, незамещенные или имеющие в качестве заместителей одну или более чем одну группу, выбранную из атома галогена или C1-C6 алкила, CN, ОН, CF3, OCF3, SMe; циклоалкил, незамещенный или имеющий в качестве заместителей CONH2, SO2Me, SO2NH2;
R7 представляет собой атом водорода, C1-C6 алкил;
R8 представляет собой атом водорода, C1-C6 алкил, фенил, незамещенный или имеющий в качестве заместителей одну или более чем одну группу, выбранную из атома галогена, C1-C6 алкила, CN, OH, CF3, OCF3, SMe; нафтил или гетероцикл, незамещенные или имеющие в качестве заместителей одну или более чем одну группу, выбранную из атома галогена, C1-C6 алкила, CN, OH, CF3, OCF3, SMe; циклоалкил, незамещенный или имеющий в качестве заместителей CONH2, SO2Me, SO2NH2;
R7 и R8 вместе с атом азота, к которому они присоединены, могут образовывать 4-6-членное кольцо, которое может содержать один или более чем один гетероатом, выбранный из N, S или О, и может быть незамещенным или иметь в качестве заместителей одну или более чем одну группу, выбранную из C1-C6 алкила, C1-C6 алкил арила или арила;
R9 представляет собой COOMe, COOEt;
R10 представляет собой атом галогена, COOH, COOR7;
R11 представляет собой атом водорода, C1-C6 алкил, C1-C6 алкил циклоалкил, циклоалкил, арил, C1-C6 алкил арил;
а также стереоизомеры, соли и сольваты указанных соединений, приемлемые для терапевтического использования.
В вышеупомянутых определениях возможны все комбинации заместителей или переменных при условии, что они дают стабильные соединения; термины, использованные в определениях, имеют следующие значения.
Термин "галоген" относится к атому фтора, хлора, брома или иода.
Термин "алкил" относится к насыщенной или ненасыщенной нормальной или разветвленной алифатической углеводородной цепи, содержащей указанное количество атомов углерода.
Термин "циклоалкил" относится к циклической или полициклической углеводородной цепи, содержащей 3-12 атомов углерода. В качестве примера можно привести адамантил, циклогексил.
Термин "арил" относится к любому моноциклическому или бициклическому углеродному кольцу, которое может содержать до 7 кольцевых атомов, где по меньшей мере одно из колец представляет собой ароматическое кольцо. В качестве примера можно привести фенил, бифенил, нафтил.
Термин "гетероарил" относится к стабильному моноциклу, содержащему 5-7 атомов, или к стабильному бициклу, содержащему 8-11 атомов, ненасыщенным и состоящим из атомов углерода и 1-4 гетероатомов, выбранных из N, О или S. В качестве примера можно привести фуран, тиофен, пиридин, бензотиофен.
Термин "гетероцикл" относится к стабильному моноциклу, содержащему 5-7 атомов, или к стабильному бициклу, содержащему 8-11 атомов, которые могут быть насыщенными или ненасыщенными и состоять из атомов углерода и 1-4 гетероатомов, выбранных из N, O или S. Определение бицикла также включает моноциклические гетероциклы, конденсированные с бензольным кольцом, за исключением индола, когда в Формуле I радикалы R1, R4, и R′4 представляют собой атом водорода. В качестве примера можно привести фуран, пиррол, тиофен, тиазол, изотиазол, оксадиазол, имидазол, оксазол, изоксазол, пиридин, пиримидин, хиназолин, хинолин, хиноксалин, бензофуран, бензотиофен, индолин, индолизин, бензотиазол, бензотиенил, бензопиран, бензоксазол, бензо[1,3]диоксол, бензоизоксазол, бензимидазол, хроман, хромен, дигидробензофуран, дигидробензотиенил, дигидроизоксазол, изохинолин, дигидробензо[1,4]диоксин, имидазо[1,2-а]пиридин, фуро[2,3-с]пиридин, 2,3-дигидро-1H-инден, [1,3]диоксоло[4,5-с]пиридин, пирроло[1,2-с]пиримидин, пирроло[1,2-а]пиримидин, тетрагидронафталин, бензо[b][1,4]оксазин.
OR1 в контексте настоящего изобретения означает сложный или простой эфир, где R1 представляет собой C1-C6 алкил, или COR5, или CO(CH2)mR6, или CO(CH2)mOR6, или (CH2)mR6, или (CH2)mCONR7R8, или (CH2)nNR7R8, или (CH2)nOR6, или CHR7OR9, или (CH2)mR10, такие, как определено выше.
Соли соединений по настоящему изобретению, приемлемые для терапевтического использования, включают стандартные нетоксичные соли соединений по изобретению, например соли, образованные с органическими или неорганическими кислотами или с органическими или неорганическими основаниями. В качестве примера можно привести соли, образованные с неорганическими кислотами, такими как соляная, бромистоводородная, фосфорная, серная кислота, и соли, образованные с органическими кислотами, такими как уксусная, трифторуксусная, пропионовая, янтарная, фумаровая, яблочная, винная, лимонная, аскорбиновая, малеиновая, глутаминовая, бензойная, салициловая, толуолсульфоновая, метансульфоновая, стеариновая, молочная кислота. В качестве примера можно привести соли, образованные с неорганическими основаниями, такими как карбонат натрия, поташ или гидроксид кальция, и соли, образованные с органическими основаниями, такими как лизин или аргинин.
Данные соли могут быть синтезированы из соединений по изобретению, содержащих основную или кислотную группировку, и соответствующих кислот или оснований в соответствии со стандартными химическими методиками.
Сольваты соединений по настоящему изобретению, приемлемые для терапевтического использования, включают стандартные сольваты, например сольваты, образованные на последней стадии получения соединений по изобретению вследствие присутствия растворителей. В качестве примера можно привести сольваты, образованные вследствие присутствия воды или этанола.
В объем настоящего изобретения также включены все стереоизомеры включая все оптические изомеры соединений общей Формулы (I), а также их рацемические смеси.
В соответствии с одним из признаков изобретения соединения общей Формулы (I) представляют собой соединения, у которых
R2 представляет собой фенил, имеющий в качестве заместителей одну или более чем одну группу, выбранную из атома галогена, C1-C6 алкила, CN, OH, CF3, OCF3, SMe; нафтил, 1,2,3,4-тетрагидро-нафталин, бифенил или гетероцикл, отличный от индола, в случае, когда R1, R4 и R′4 представляют собой атом водорода, незамещенные или имеющие в качестве заместителей одну или более чем одну группу, выбранную из атома галогена или C1-C6 алкила, CN, OH, CF3, OCF3, OMe, SMe; циклоалкил, незамещенный или имеющий в качестве заместителей CONH2, SO2Me, SO2NH2,
при условии, что группа R2 всегда связана с карбонилом через атом углерода;
R4 и R′4 являются одинаковыми или разными и представляют собой атом водорода; атом галогена; C1-C6 алкил; CN; CF3; OCF3; SMe; OMe; NR7R8;
R8 представляет собой атом водорода, C1-C6 алкил, фенил, незамещенный или имеющий в качестве заместителей одну или более чем одну группу, выбранную из атома галогена, C1-C6 алкила, CN, OH, CF3, OCF3, SMe; нафтил или гетероцикл, незамещенные или имеющие в качестве заместителей одну или более чем одну группу, выбранную из атома галогена, C1-C6 алкила, CN, OH, CF3, OCF3, SMe; циклоалкил, незамещенный или имеющий в качестве заместителей CONH2, SO2Me, SO2NH2;
R7 и R8 вместе с атом азота, к которому они присоединены, могут образовывать 4-6-членное кольцо, которое может содержать один или более чем один гетероатом, выбранный из N, S или O, и может быть незамещенным или иметь в качестве заместителей одну или более чем одну группу, выбранную из C1-C6 алкила или арила,
R1 является таким, как определено выше, или таким, как определено в данном описании ниже.
Согласно одному из вариантов воплощения изобретения соединения общей Формулы (I) представляют собой соединения, у которых R1 представляет собой атом водорода.
Согласно другому варианту воплощения изобретения соединения общей Формулы (I) представляют собой соединения, у которых OR1 представляет собой сложный или простой эфир, где R1 представляет собой C1-C6 алкил, или COR5, или CO(CH2)mR6, или CO(CH2)mOR6, или (CH2)mR6, или (CH2)mCONR7R8, или (CH2)nNR7R8, или (CH2)nOR6, или CHR7OR9, или (CH2)mR10.
Согласно одному из вариантов воплощения изобретения OR1 представляет собой сложный эфир, где R1 представляет собой COR5, или CO(CH2)mR6, или СО(СН2)mOR6.
Целью настоящего изобретения также являются соединения общей Формулы (I), у которых R2 представляет собой нафтил, или 1,2,3,4-тетрагидро-нафталин, или бифенил, или фенил пиридин, незамещенные или имеющие в качестве заместителей одну или более чем одну группу, выбранную из атома галогена, C1-C6 алкила, CN, OH, CF3, OCF3, OMe, SMe, или фенил, имеющий в качестве заместителей один или более чем один атом галогена, CN, CF3 или C1-C6 алкил.
Согласно одному из вариантов воплощения изобретения соединения общей Формулы (I) представляют собой соединения, у которых R4 и R′4 представляют собой атом водорода.
Среди соединений общей Формулы (I) по настоящему изобретению предпочтительный класс соединений составляют соединения общей Формулы (I), где R1 представляет собой атом водорода и R2 представляет собой нафтил или 1,2,3,4-тетрагидро-нафталин.
Настоящее изобретение также относится к соединениям общей Формулы (I), где OR1 представляет собой сложный или простой эфир и R2 представляет собой нафтил или 1,2,3,4-тетрагидро-нафталин.
Другой предпочтительный класс соединений составляют соединения общей Формулы (I), где R1 представляет собой атом водорода и R2 представляет собой фенил, имеющий в качестве заместителей один или более чем один атом галогена, CN, CF3 или C1-C6 алкил.
Другой предпочтительный класс соединений составляют соединения общей Формулы (I), где R1 представляет собой атом водорода и R2 представляет собой бифенил или фенил пиридин, незамещенные или имеющие заместители, такие, как определено в описании общей Формулы (I).
Настоящее изобретение также относится к соединениям общей Формулы (I), где OR1 представляет собой сложный или простой эфир и R2 представляет собой фенил, имеющий в качестве заместителей один или более чем один атом галогена, CN, CF3 или C1-C6 алкил.
Другой предпочтительный класс соединений составляют соединения общей Формулы (I), где OR1 представляет собой сложный или простой эфир и R2 представляет собой бифенил или фенил пиридин, незамещенные или имеющие заместители, такие как определено в описании общей Формулы (I).
Настоящее изобретение также относится к получению соединений общей Формулы (I) с использованием общих методик, описанных далее на схемах синтеза и при необходимости дополненных другими стандартными процедурами, описанными в литературе или хорошо известными специалисту в данной области техники, или дополнительно проиллюстрированных в экспериментальной части.

На Схеме 1 приведена первая общая методика, которая может быть использована для получения соединений общей Формулы (Ia). На общих формулах, приведенных выше, R2, R3, R4 и R′4, являются такими, как определено в предшествующем описании общей Формулы (I), и R1 представляет собой атом водорода. Х может представлять собой уходящую группу, такую как, например, Cl, Br, I, OSO2CH3, OSO2CF3 или O-тозил. В данном случае реакцию с участием соединения общей Формулы (II) выполняют в присутствии неорганического основания, такого как, например, NaH, в полярном безводном растворителе, таком как ТГФ (тетрагидрофуран) или ДМФА (N,N-диметилформамид), при температуре в диапазоне от -20° до 100°С. Промежуточное соединение общей Формулы (III) превращают в промежуточное соединение общей Формулы (IV) путем перегруппировки в присутствии основания, такого как, например, MeONa, EtONa, в полярном безводном растворителе, таком как МеОН или EtOH (возможно смешанном с неполярным растворителем, таким как толуол), при температуре в диапазоне от 25° до 100°C. Промежуточное соединение общей Формулы (IV) превращают в продукт общей Формулы (Ia) в результате взаимодействия с R3Y, где Y может представлять собой уходящую группу, такую как, например, Cl, Br, I, OSO2CH3, OSO2CF3 или O-тозил, и R3 является таким, как определено выше. В данном случае превращение соединения общей Формулы (IV) выполняют в присутствии неорганического основания, такого как, например, NaH, в полярном безводном растворителе, таком как ТГФ или ДМФА, при температуре в диапазоне от-20° до 100°C.
На Схеме 2 приведена общая методика, которая может быть использована для получения соединений общей Формулы (Ib). На общих формулах, приведенных ниже, R1, R2, R3, R4 и R′4, являются такими, как определено в предшествующем описании общей Формулы (I), за тем исключением, что R1 не является атомом водорода.

Промежуточное соединение общей Формулы (Ia) превращают в соединение общей Формулы (Ib) в результате взаимодействия с R1-Z. Когда R1 представляет собой C1-C6 алкил, (CH2)mR6, (CH2)mCONR7R8, (CH2)nNR7R8, (CH2)nOR6, CHR7OR9 или (CH2)mR10, где R6, R7, R8, R9, R10, тип являются такими, как определено в предшествующем описании общей Формулы (I), за тем исключением, что R10 не являются кислотой, и Z представляет собой уходящую группу, такую как, например, Cl, Br, I, OSO2CH3, OSO2CF3 или O-тозил, превращение енола общей Формулы (Ia) может быть выполнено в присутствии органического или неорганического основания, такого как, например, Et3N, iPr2NEt, NaH, пиридин, Cs2CO3, K2CO3, в полярном безводном растворителе, таком как ТГФ, ДМФА, ДМСО (диметилсульфоксид), ацетон, при температуре в диапазоне от -20° до 140°C с использованием, в качестве катализатора, соли, которая может быть выбрана из KI, Bu4NI, LiI, AgBF4, AgClO4, Ag2CO3, KF, Bu4NF или CsF, или без использования такой соли. Данную реакцию также можно проводить в пробирке, запаянной или закрытой винтовой крышкой, при температуре в диапазоне от 80 до 180°C, используя для нагревания тепловую или микроволновую энергию. Z также может представлять собой спирт. В данном случае превращение промежуточного соединения (Ia) может быть осуществлено с использованием реакции типа реакции Мицунобу, которая может быть выполнена в присутствии диэтилазодикарбоксилата (DEAD) и трифенилфосфина в полярном безводном растворителе, таком как ТГФ, при температуре в диапазоне от 0 до 60°C. Когда R1 представляет собой COR5, SO2R5 или CO(CH2)mR6, где R5, R6 и m являются такими, как определено в предшествующем описании общей Формулы (I), Z может представлять собой атом хлора. В данном случае превращение енола общей Формулы (Ia) сводиться к взаимодействию хлорангидрида и сульфонилхлорида со спиртом. Данное взаимодействие может быть осуществлено в присутствии органического или неорганического основания, такого как, например, Et3N, iPr2NEt, NaH, пиридин, Cs2CO3, K2CO3, в полярном безводном растворителе, таком как ТГФ, ДМФА, ДМСО, дихлорметан, при температуре в диапазоне от -20° до 140°C. Когда R1 представляет собой COR5, CO(CH2)mR6 или СО(CH2)mOR6, где R5, R6 и m являются такими, как определено в предшествующем описании общей Формулы (I), Z также может представлять собой гидроксил. В данном случае превращение енола общей Формулы (Ia) сводиться к взаимодействию кислоты со спиртом. Данное взаимодействие может быть осуществлено с использованием методик и приемов, хорошо известных специалисту в данной области техники. Особенно предпочтительной является методика, которая предполагает выполнение данной реакции конденсации в присутствии 1-(3-диметиламинопропил)-3-этил-карбодиимида (EDC), 3-гидрокси-1,2,3-бензотриазин-4(3Н)-она, третичного амина, такого как диизопропилэтиламин, в полярном апротонном растворителе, таком как дихлорметан, при температуре в диапазоне от -15°C до 40°C.
На Схеме 3 приведена общая методика, которая может быть использована для получения соединений общей Формулы (Ic), где R1 представляет собой (CH2)nNR7R8 или (CH2)nOR6 и R6, R7, R8, n и R2, R3, R4 и R′4 являются такими, как определено в предшествующем описании общей Формулы (I). Промежуточное соединение общей Формулы (Ia) превращают в промежуточное соединение общей Формулы (V) в результате взаимодействия с реагентом общей Формулы Х(СН2)nX′, где Х и X′ представляют собой одинаковые или разные уходящие группы, такие как, например, Cl, Br, I, OSO2CH3, OSO2CF3 или O-тозил, и n имеет такое значение, как определено выше.

Взаимодействие данного реагента и енола общей Формулы (Ia), приводящее к получению промежуточного соединения общей Формулы (V), может быть осуществлено в присутствии органического или неорганического основания, такого как, например, Et3N, iPr2NEt, NaH, пиридин, Cs2CO3, K2CO3, в полярном безводном растворителе, таком как ТГФ, ДМФА, ДМСО, ацетон, при температуре в диапазоне от -20° до 140°C с использованием, в качестве катализатора, соли, которая может быть выбрана из KI, Bu4NI, LiI, AgBF4, AgClO4, Ag2CO3, KF, Bu4NF, или CsF, или без использования такой соли. Данное взаимодействие также может быть осуществлено без растворителя с использованием большого избытка реагента Х(СН2)nX′. Данную реакцию также можно проводить в пробирке, запаянной или закрытой винтовой крышкой, при температуре в диапазоне от 80 до 180°C, используя для нагревания тепловую или микроволновую энергию. Х или X′ также могут представлять собой спирт. В данном случае для получения промежуточного соединения (V) может быть использована реакция типа реакции Мицунобу, которая может быть выполнена в присутствии диэтилазодикарбоксилата (DEAD) и трифенилфосфина в полярном безводном растворителе, таком как ТГФ, при температуре в диапазоне от 0 до 60°C.
Промежуточное соединение общей Формулы (V) превращают в продукт общей Формулы (Ic) в результате взаимодействия с HNR7R8 или HOR6, где R6, R7 и R8 являются такими, как определено в предшествующем описании общей Формулы (I). Данное превращение может быть осуществлено в присутствии органического или неорганического основания, такого как, например, Et3N, iPr2NEt, NaH, пиридин, Cs2CO3, K2CO3, в полярном безводном растворителе, таком как ТГФ, ДМФА, ДМСО, ацетон, при температуре в диапазоне от -20° до 140°C с использованием, в качестве катализатора, растворителя, который может быть выбран из KI, Bu4NI, LiI, AgBF4, AgCl4, Ag2CO3, KF, Bu4NF или CsF, или без его использования. Выбор экспериментальных условий и реагентов, необходимых для проведения данной реакции, конечно, зависит от природы заместителей R6, R7 и R8 и выполняется в соответствии с методиками и приемами, хорошо известными специалисту в данной области техники.
На Схеме 4 приведена общая методика, которая может быть использована для получения соединений общей Формулы (Id), где R1 представляет собой (CH2)mCONR7R8 и R7, R8, m и R2, R3, R4 и R′4, являются такими, как определено в предшествующем описании общей Формулы (I).

Промежуточное соединение общей Формулы (Ia) превращают в промежуточное соединение общей Формулы (VI) в результате взаимодействия с реагентом общей Формулы Y(CH2)mCOOY′, где Y представляет собой уходящую группу, такую как, например, Cl, Br, I, OSO2CH3, OSO2CF3 или O-тозил, m имеет такое значение, как определено выше, и Y′ представляет собой C1-C4 алкильный радикал. Данное превращение может быть осуществлено в присутствии органического или неорганического основания, такого как, например, Et3N, iPr2NEt, NaH, пиридин, Cs2CO3, K2CO3, в полярном безводном растворителе, таком как ТГФ, ДМФА, ДМСО, ацетон, при температуре в диапазоне от -20° до 140°C, с использованием, в качестве катализатора, соли, которая может быть выбрана из KI, Bu4NI, LiI, AgBF4, AgClO4, Ag2CO3, KF, Bu4NF или CsF, или без использования такой соли. Данную реакцию также можно проводить в пробирке, запаянной или закрытой винтовой крышкой, при температуре в диапазоне от 80 до 180°C, используя для нагревания тепловую или микроволновую энергию. Промежуточное соединение общей Формулы (VI) превращают в промежуточное соединение общей Формулы (VII) в результате взаимодействия с неорганическим основанием, таким как, например, NaOH, КОН, LiOH, в полярном растворителе, таком как метанол, этанол, ТГФ и вода, при температуре в диапазоне от 20° до 80°C. Полученная карбоновая кислота (VII) может быть подвергнута взаимодействию с амином с получением соединений общей Формулы (Id). Данное взаимодействие может быть осуществлено с использованием методик и приемов, хорошо известных специалисту в данной области техники. Особенно предпочтительной является методика, согласно которой реакцию конденсации двух указанных соединений выполняют в присутствии 1-(3-диметиламинопропил)-3-этил-карбодиимида (EDC), 3-гидрокси-1,2,3-бензотриазин-4(3Н)-она, третичного амина, такого как, диизопропилэтиламин, в полярном апротонном растворителе, таком как дихлорметан или ДМФА, при температуре в диапазоне от -15°C до 50°C, или, например, с использованием бензотриазол-1-илокси-трис(диметиламино)фосфония гексафторфосфата (ВОР) в присутствии 1-гидроксибензотриазола, третичного амина, такого как диизопропилэтиламин, в полярном растворителе, таком как ДМФА, CH2Cl2 или ДМСО, при температуре в диапазоне от 10° до 50°C. Другой особенно предпочтительной методикой является превращение карбоновой кислоты в хлорангидрид в результате взаимодействия с оксалилхлоридом или тионилхлоридом в присутствии основания, такого как пиридин или триэтиламин, или без использования основания, в растворителе, таком как толуол или дихлорметан, или без использования растворителя, при температуре в диапазоне от 20 до 100°C. Затем полученный хлорангидрид может быть подвергнут взаимодействию с амином HNR7R8 в присутствии основания, такого как пиридин или триэтиламин, в таком растворителе, как дихлорметан, при температуре в диапазоне от 0 до 100°C.
На Схеме 5 приведена общая методика, которая может быть использована для превращения соединений общей Формулы (Ie), где R4 представляет собой атом фтора и R2, R3 и R′4 являются такими, как определено в предшествующем описании общей Формулы (I), в соединения общей Формулы (If), где R4 представляет собой NR7R8 и R7, R8 и R2, R3 и R′4 являются такими, как определено в предшествующем описании общей Формулы (I).

Соединения общей Формулы (Ie) могут быть превращены в соединения общей Формулы (If) в результате взаимодействия с амином общей Формулы HNR7R8 в присутствии органического или неорганического основания, такого как, например, Et3N, iPr2NEt, NaH, Cs2CO3, K2CO3, в полярном безводном растворителе, таком как ДМФА, ДМСО, при температуре в диапазоне от 20° до 140°C.
На Схеме 6 приведена общая методика, которая может быть использована для превращения соединений общей Формулы (Ig), где R3, R4 и R′4 являются такими, как определено в предшествующем описании общей Формулы (I) и где R2 представляет собой фенил, имеющий в качестве заместителя группу X, которая представляет собой атом брома, атом хлора или OTf, в соединения общей Формулы (Ih), где R2 представляет собой бифенил или фенил пиридин, замещенные или незамещенные, и где R3, R4 и R′4 являются такими, как определено в предшествующем описании общей Формулы (I).
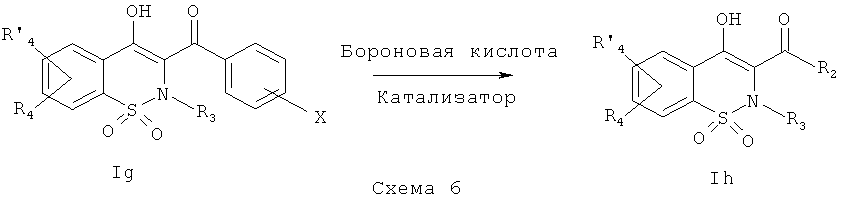
Соединения общей Формулы (Ig) могут быть превращены в соединения общей Формулы (Ih) в результате взаимодействия с бороновой кислотой с использованием реакции типа реакции Сузуки, в присутствии органического или неорганического основания, такого как, например, Et3N, NMP, iPr2NEt, NaH, Cs2CO3 K2CO3, K3PO4, в присутствии катализатора, такого как, например, ацетат палладия, тетракистрифенилфосфин палладия, трис(дибензилиденацетон)дипалладий, в полярном растворителе, таком как, например, ацетон, метилэтилкетон, этанол, ДМЭ (диметоксиэтан), вода, диоксан, и возможно в присутствии фосфина, такого как трифенилфосфин или трициклогексилфосфин, при температуре в диапазоне от 20° до 140°C.
На Схеме 7 приведена общая методика, которая может быть использована для превращения соединений общей Формулы (Ii), где R3, R4 и R′4 являются такими, как определено в предшествующем описании общей Формулы (I) и где Rg представляет собой фенил, имеющий в качестве заместителя группу CN в орто-или мета-положении, в соединения общей Формулы (Ij), где R2 представляет собой фенил, имеющий в качестве заместителя карбоновую кислоту в орто- или мета-положении, и затем в соединения общей Формулы (Ik), где R2 представляет собой фенил, имеющий в качестве заместителя амид Формулы CONR7R11, и где R3, R4, R7, R11 и R′4 являются такими, как определено в предшествующем описании общей Формулы (I).

Соединения общей Формулы (Ii) могут быть превращены в соединения общей Формулы (Ij) путем обработки неорганическим основанием, таким как, например, NaOH, KOH, LiOH, в полярном растворителе, таком как этанол, метанол, ТГФ, вода, при температуре в диапазоне от 20° до 140°C и последующего окисления путем обработки кислотой, такой как HCl, H2SO4, HCOOH. Соединения общей Формулы (Ij) могут быть превращены в соединения общей Формулы (Ik) в результате взаимодействия с амином Формулы HNR7R11. Данное взаимодействие может быть осуществлено с использованием методик и приемов, хорошо известных специалисту в данной области техники. Особенно предпочтительной является методика, согласно которой реакцию конденсации двух указанных соединений выполняют в присутствии 1-(3-диметиламинопропил)-3-этил-карбодиимида (EDC), 3-гидрокси-1,2,3-бензотриазин-4(3Н)-она, третичного амина, такого как диизопропилэтиламин, в полярном апротонном растворителе, таком как дихлорметан или ДМФА, при температуре в диапазоне от -15°C до 50°C, или, например, с использованием бензотриазол-1-илокси-трис(диметиламино)фосфония гексафторфосфата (ВОР) в присутствии 1-гидроксибензотриазола, третичного амина, такого как диизопропилэтиламин, в полярном растворителе, таком как ДМФА, CH2Cl2 или ДМСО, при температуре в диапазоне от 10° до 50°C. Другой особенно предпочтительной методикой является превращение карбоновой кислоты в хлорангидрид в результате взаимодействия с оксалилхлоридом или тионилхлоридом в присутствии основания, такого как пиридин или триэтиламин, или без использования основания, в растворителе, таком как толуол или дихлорметан, или без использования растворителя, при температуре в диапазоне от 20 до 100°C. Затем полученный хлорангидрид может быть подвергнут взаимодействию с амином HNR7R11 в присутствии основания, такого как пиридин или триэтиламин, в таком растворителе, как дихлорметан, при температуре в диапазоне от 0 до 100°C.
Когда желательно выделить соединение общей Формулы (I), содержащее по меньшей мере одну кислотную или основную функциональную группу, в виде соли путем добавления основания или кислоты, желаемая соль может быть получена путем обработки свободного основания или кислоты общей Формулы (I) (которая содержит по меньшей мере одну кислотную или основную функциональную группу) подходящим основанием или кислотой, предпочтительно эквивалентным количеством основания или кислоты.
Следующие далее примеры иллюстрируют данное изобретение, не ограничивая его объем.
Примечание: чистоту всех соединений (если не указано особо) определяли путем ЖХВД с использованием следующих условий:
колонка: Waters XTerra MS C18, 4,6×50 мм, 5 мкм; λ=220 нм; градиент 100% H2O (+0,05% ТФУ) - 100% CH3CN (+0,05% ТФУ) в течение 6 мин и затем 100% CH3CN (+0,05% ТФУ) в течение 1 мин; насос: Waters 600E; скорость потока: 3 мл/мин.
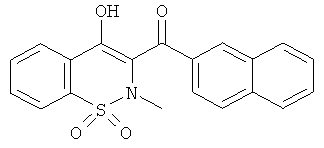
Пример 1
(4-Гидрокси-2-метил-1,1-диоксо-2Н-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанон
Пример 1А - 2-(2-(Нафталин-2-ил)-2-оксоэтил)бензо[d]изотиазол-3(2Н)-он-1,1-диоксид
В трехгорлую колбу, оснащенную термометром и холодильником, вносили сахарин (25 г, 136 ммоль) и ДМФА (350 мл). Данную смесь промывали инертным газом путем последовательного вакуумирования и заполнения азотом (3х). Медленно добавляли гидрид натрия (6 г, 150 ммоль) и затем 2-бром-1-(нафталин-2-ил)этанон (37,4 г, 150 ммоль). Данную реакционную смесь нагревали до 65°C в течение 4 ч и затем охлаждали до комнатной температуры. Образовавшийся осадок отделяли фильтрованием, промывали водой и сушили до постоянной массы с получением 37 г продукта 1А в виде бледно-being твердого вещества (ЖХВД: RT=4,97 мин, 100%). Вторую порцию продукта получали путем добавления в фильтрат воды. Образовавшийся осадок отделяли фильтрованием, промывали водой и затем минимальным количеством этилового спирта и сушили с получением 10 г продукта (ЖХВД: RT=4,97 мин, 93%). Общий выход реакции составлял 96%.
1H ЯМР, ДМСО-d6, δ (м.д.): 5.62 (s, 2H); 7.68 (t, 1H); 7.73 (t, 1H); 8.00-8.25 (m, 7H); 8.39 (d, 1H); 8.92 (s, 1H).
Масс-спектр (ESI+): m/z 352 (MH+, 100%); 369 (MNH4+, 24%).
Пример 1В - (4-Гидрокси-1,1-диоксо-2Н-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанон
В двугорлую колбу, оснащенную холодильником, в атмосфере инертного газа вводили этанол (165 мл) и затем медленно добавляли натрий, нарезанный тонкими слоями и промытый гептаном (8 г, 347 ммоль). После добавления натрия реакционную смесь нагревали до 70°C до тех пор, пока реакция с участием натрия не была завершена. Затем реакционную смесь охлаждали до комнатной температуры и быстро добавляли соединение 1А (47 г, 131 ммоль). Смесь окрашивалась сначала в интенсивный багряно-красный цвет, затем в алый цвет, и выпадал обильный осадок. Реакционную смесь быстро нагревали до 60°C, при данной температуре смесь загустевала. Затем смесь охлаждали до комнатной температуры и разбавляли этилацетат ом (500 мл). Затем добавляли 1 н. водный раствор HCl до тех пор, пока не получали суспензию канареечного цвета. Осадок отделяли фильтрованием, промывали водой и минимальным количеством смеси вода/EtOH (50/50). Затем осадок сушили под вакуумом до постоянной массы с получением продукта 1В в виде твердого вещества канареечного цвета (40,9 г, 88%). ЖХВД: RT=5,15 мин, 100%.
1H ЯМР, ДМСО-d6, δ (м.д.): 7.66 (t, 1H); 7.72 (t, 1H); 7.95 (уширенный s, 3Н); 8.05 (d, 2H); 8.11 (уширенный s, 2H); 8.22 (уширенный s, 1H); 8.64 (s, 1H); 9.99 (s, 1H); 15.59 (s, 1H).
Масс-спектр (ESI+): m/z 352 (MH+, 100%); 369 (MNH4+, 31%).
Пример 1 - (1,1-Диоксо-4-гидрокси-2-метил-2H-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанон
Соединение 1В (40,9 г, 116 ммоль) растворяли в ДМФА (409 мл) в двугорлой колбе в атмосфере инертного газа. Добавляли NaH (6,05 г, 151 ммоль). В ходе реакции выделялось небольшое количество тепла и реакционная смесь приобретала интенсивную алую окраску. Добавляли иодметан (10,8 мл, 174 ммоль) и данную реакционную смесь перемешивали в течение 2 ч при комнатной температуре. Добавляли воду (10 мл) и реакционную смесь концентрировали. Остаток переносили в этилацетат и осадок отделяли фильтрованием, промывали водой и минимальным количеством этилацетата (первая порция твердого вещества). Полученный фильтрат дважды промывали полунасыщенным водным раствором NaCl, затем концентрировали до половины объема и фильтровали. Осадок (вторая порция твердого вещества) промывали минимальным количеством смеси EtOAc/Et2O (50/50). Полученный фильтрат концентрировали. Остаток фильтровали на силикагеле (элюент: гептан/CH2Cl2 (50/50) и затем гептан/CH2Cl2 (25/75)) с получением после выпаривания растворителей желтого порошка (третья порция твердого вещества). Все 3 порции твердого вещества объединяли с получением продукта 1 в виде твердого вещества канареечного цвета (40,1 г, 89%). ЖХВД: RT=5,65 мин, 99%.
1H ЯМР, ДМСО-d6, δ (м.д.): 2.65 (s, 3H); 7.66 (t, 1H); 7.72 (t, 1H); 8.00 (уширенный s, 3H); 8.02 (d, 1H); 8.12 (уширенный s, 3H); 8.22 (уширенный s, 1H); 8.67 (s, 1H).
Масс-спектр (ESI+): m/z 366 (MH+, 100%).
Получение натриевой соли 4-гидрокси-2-метил-1,1-диоксо-2Н-бензо[e][1,2]-тиазин-3-ил)(нафталин-2-ил)метанона
Фракцию, содержащую соединения 1, растворяли в метаноле и обрабатывали при комнатной температуре 1 н. водным раствором карбоната натрия (1,05 экв.). Данную реакционную смесь концентрировали и образовавшееся в остатке твердое вещество промывали смесью дихлорметана и диэтилового эфира. Полученное в результате твердое вещество канареечного цвета сушили под вакуумом в течение нескольких суток.
ЖХВД: RT=11,73 мин, 99,71% (колонка: XBridge C8, 5 мкМ, 4,6×250 мм (Waters); элюент: CH3CN/H2O/KH2PO4 (600/400/6,8 г), рН 4, 25°C; 1 мл/мин; 220 нм).
1H ЯМР, ДМСО-d6, δ (м.д.): 2.61 (s, 3H); 7.50 (уширенный s, 2H); 7.62 (уширенный s, 2H); 7.65-7.72 (m, 2H); 7.80 (d, 1H); 7.89 (уширенный s, 2H); 7.93-7.98 (m, 2H).
Масс-спектр (ESI+): m/z 366 (MH+, 100%).
Примеры 2-12
Соединения 2-12 синтезировали в соответствии с методикой получения производного 1, с использованием на первой стадии сахарина и различных 2-бром-1-(алкил или арил)этанонов и на третьей стадии йодистого метила или йодистого этила. Протокол перегруппировки, используемый на второй стадии, не изменяли.


Пример 13
(5-Хлор-4-гидрокси-2-метил-1,1-диоксо-2Н-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанон

Пример 13А - 2-Хлор-6-сульфамоилбензойная кислота
В трехгорлую колбу, оснащенную холодильником, вводили 3-хлор-2-метилбензолсульфонамид (13,27 г, 64,5 ммоль) в присутствии 5% водного раствора карбоната натрия (385 мл). Медленно добавляли перманганат калия (25,5 г, 161 ммоль) и затем реакционную смесь нагревали до 100°C в течение 4 ч. Температуру реакционной смеси доводили до комнатной температуры, смесь фильтровали, подкисляли до рН 1 и 3 раза экстрагировали этилацетатом. Органические фазы объединяли, промывали один раз насыщенным водным раствором NaCl и затем сушили сульфатом магния, фильтровали и концентрировали с получением продукта 13А в виде белого твердого вещества (12,87 г, 83%).
ЖХВД: RT=1,55 мин, 98%.
1H ЯМР, ДМСО-d6, δ (м.д.): 7.48 (s, 2H, обменный); 7.62 (t, 1H); 7.75 (d, 1H); 7.87 (d, 1H); 11-15 (mL, 1H, обменный).
Масс-спектр (ESI-): m/z 234 (M-H-, 55%).
Пример 13В - 4-Хлорсахарин
В колбу вводили соединение 13А (12,87 г, 54,6 ммоль) и затем 38,8 мл концентрированной серной кислоты. Данную реакционную смесь перемешивали в течение 1,5 ч при комнатной температуре и затем вливали в смесь воды со льдом. Образовавшийся осадок отделяли фильтрованием, промывали водой и сушили до постоянной массы с получением соединения 13В в виде белого твердого вещества (9,16 г, 77%).
ЖХВД: RT=2,57 мин, 100%.
1H ЯМР, ДМСО-d6, δ (м.д.): 7.91 (уширенный s, 2H); 8.08 (уширенный s, 1H).
Пример 13С - 4-Хлор-2-(2-(нафталин-2-ил)-2-оксоэтил)бензо[d]изотиазол-3(2Н)-он-1,1-диоксид
Соединение 13С синтезировали из соединения 13В (2,2 г, 10 ммоль) в соответствии с методикой, использованной для получения производного 1А, соединение 13С получали в виде бледно-бежевого твердого вещества (3,3 г, 84%).
ЖХВД: RT=5,11 мин, 99%.
1H ЯМР, ДМСО-d6, δ (м.д.): 5.62 (s, 2H); 7.69 (t, 1H), 7.74 (t, 1H); 7.95-8.20 (m, 6H); 8.38 (d, 1H); 8.92 (s, 1H).
Масс-спектр (ESI+): m/z 386 (MH+, 100%).
Пример 13D - (5-Хлор-1,1-диоксо-4-гидрокси-2Н-бензо[е][1,2]тиазин-3-ил) (нафталин-2-ил)метанон
Соединение 13D синтезировали из соединения 13С (3,3 г, 8,5 ммоль) в соответствии с методикой, использованной для получения производного 1В, соединение 13D получали в виде золотисто-желтого твердого вещества (1,7 г, 51%).
ЖХВД: RT=5,3 мин, 99%.
1H ЯМР, AMCO-d6, δ (м.д.): 7.68 (t, 1H), 7.72 (t, 1H); 7.85-8.15 (m, 8H); 8.59 (s, 1H); 10.11 (s, 1H).
Масс-спектр (ESI+): m/z 386 (MH+, 100%).
Пример 13
Соединение 13 синтезировали из соединения 13D (3 г, 7,7 ммоль) в соответствии с методикой, использованной для получения производного 1. Соединение 13 получали в виде желтого твердого вещества (2,3 г, 70%).
ЖХВД: RT=5,75 мин, 95%.
1H ЯМР, ДМСО-d6, δ (м.д.): 2.69 (s, 3Н); 7.66 (t, 1H); 7.72 (t, 1H); 7.9-8.2 (m, 7H); 8.60 (уширенный s, 1H); 16.15 (уширенный s, 1H, обменный).
Масс-спектр (ESI+): m/z 400 (MH+, 100%).
Пример 14
(5-Хлор-4-гидрокси-2-этил-1,1-диоксо-2Н-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанон

Соединение 14 синтезировали из соединения 13D (1 г, 2,6 ммоль) и 25 иодэтана в соответствии с методикой, использованной для получения производного 1, с выходом 805 мг (60%) желаемого продукта.
ЖХВД: RT=5,77 мин, 81%.
Фракцию, содержащую данный продукт (200 мг), очищали на колонке со сферическим силикагелем (12 г) (скорость потока: 12 мл/мин; 100% гептан (2 мин), 30 градиент 0-50% EtOAc/гептан (30 мин), 50% EtOAc/гептан (5 мин)), с получением 64 мг желаемого продукта в виде желтого твердого вещества.
ЖХВД: RT=5,77 мин, 97%.
1H ЯМР, ДМСО-d6, δ (м.д.): 0.51 (t, 3Н); 3.11 (q, 2Н); 7.66 (t, 1H); 7.72 (t, 1H); 7.85-8.2 (m, 7H); 8.60 (s, 1H); 15.9 (s, 1H, обменный).
Масс-спектр (ESI+): m/z 414 (MH+, 100%).
Пример 15
(6-Фтор-4-гидрокси-2-метил-1,1-диоксо-2Н-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанон

Пример 15А - (6-Фтор-4-гидрокси-1,1-диоксо-2Н-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанон
Соединение 15А синтезировали из 4-фтор-2-метилбензолсульфонамида, используя последовательность стадий, описанную в методике получения соединения 13D. Желаемый продукт получали в виде желтого твердого вещества с общим выходом 79%.
ЖХВД: RT=5,26 мин, 96%.
1H ЯМР, ДМСО-d6, δ (м.д.): 7.66 (t, 1H); 7.72 (t, 1H); 7.80 (t, 1H); 7.94-8.11 (m, 6H); 8.64 (s, 1H); 10.18 (s, 1H, обменный); 15.2 (уширенный s, 1H, обменный).
Масс-спектр (ESI-): m/z 368 (M-H-, 100%).
Пример 15 - (6-Фтор-4-гидрокси-2-метил-1,1-диоксо-2Н-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанон
Соединение 15 синтезировали из соединения 15А (1,5 г, 4 ммоль) в соответствии с методикой, использованной для получения производного 1, с выходом желаемого продукта (1,47 г, 89%) в виде желтого твердого вещества.
ЖХВД: RT=5,6 мин, 93%.
Фракцию, содержащую данный продукт, перекристаллизовывали из этанола с получением 186 мг соединения 15 более высокой чистоты (ЖХВД: RT=5,6 мин, 99,4%).
1H ЯМР, ДМСО-d6, δ (м.д.): 2.68 (s, 3Н); 7.66 (t, 1H); 7.72 (t, 1H); 7.84 (t, 1H); 7.97 (d, 1H); 8.02-8.15 (m, 5H); 8.66 (s, 1H); 15.22 (уширенный s, 1H, обменный).
Масс-спектр (ESI+): m/z 384 (MH+, 100%).
Пример 16
(6-Фтор-4-гидрокси-2-этил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанон
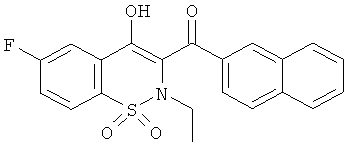
Соединение 16 синтезировали из соединения 15А (1,5 г, 4 ммоль) и иодэтана в соответствии с методикой, использованной для получения производного 1, с выходом желаемого продукта (520 мг, 29%) в виде желтого твердого вещества.
ЖХВД: КТ=5,8 мин, 91%.
Фракцию, содержащую данный продукт, перекристаллизовывали из этанола с получением 71 мг соединения 16 более высокой чистоты.
ЖХВД: RT=5,8 мин, 97%.
1H ЯМР, ДМСО-d6, δ (м.д.): 0.56 (t, 3Н); 3.15 (q, 2Н); 7.66 (t, 1H); 7.72 (t, 1H); 7.82 (t, 1H); 7.97 (d, 1H); 8.00-8.2 (m, 5H); 8.63 (s, 1H); 14.95 (уширенный s, 1H).
Масс-спектр (ESI+): m/z 398 (MH+, 100%).
Пример 17
(7-Фтор-4-гидрокси-2-метил-1,1-диоксо-2Н-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанон

Пример 17А - 5-Фтор-2-метилбензолсульфонамид
К концентрированному раствору аммиака (23 мл) медленно добавляли при 0°C 5-фтор-2-метилбензолсульфонил хлорид (5,00 г, 23,9 ммоль). Затем данную реакционную смесь нагревали до 100°C в течение 1 ч и затем охлаждали до комнатной температуры. Образовавшийся осадок отделяли фильтрованием, промывали водой и сушили до постоянной массы. Соединение 17А получали в виде белого порошка (4,55 г, 100%).
ЖХВД: RT=3,10 мин, 96%.
1H ЯМР, ДМСО-d6, δ (м.д.): 2.54 (s, 3H); 7.35-7.45 (m, 2H); 7.53 (уширенный s, 2Н, обменный); 7.58 (de, 1H).
Масс-спектр (ESI-): m/z 188 (М-Н-, 100%).
Пример 17В - (7-Фтор-4-гидрокси-1,1-диоксо-2Н-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанон
Соединение 17В синтезировали из соединения 17А, используя последовательность стадий, описанную в методике получения соединения 13D. Желаемый продукт получали в виде желтого твердого вещества с общим выходом 73%.
ЖХВД: RT=5,18 мин, 98%.
1H ЯМР, ДМСО-d6, δ (м.д.): 7.66 (t, 1H); 7.72 (t, 1H); 7.81 (t, 1H); 7.90 (d, 1H); 8.04 (d, 2H); 8.11 (уширенный s, 2H); 8.30 (dd, 1H); 8.63 (s, 1H); 10.19 (уширенный s, 1H); 15.63 (уширенный s, 1H).
Масс-спектр (ESI-): m/z 368 (M-H-, 100%).
Масс-спектр (ESI+): m/z 370 (МН+, 100%).
Пример 17 - (7-Фтор-4-гидрокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанон
Соединение 17 синтезировали из соединения 17В (4,00 г, 10,8 ммоль) в соответствии с методикой, использованной для получения производного 1, получали две порции желаемого продукта разной степени очистки.
Первая порция: 3,79 г, бледно-коричневое твердое вещество; ЖХВД: RT=5,65 мин, 94%.
Вторая порция: 320 мг, желтое твердое вещество; ЖХВД: RT=5,65 мин, 99%.
Выход реакции составлял 93%.
1H ЯМР, ДМСО-d6, δ (м.д.): 2.68 (s, 3H); 7.66 (t, 1H); 7.72 (t, 1H); 7.83 (t, 1H); 7.92 (d, 1H); 8.02-8.15 (m, 4H); 8.28 (dd, 1H); 8.62 (s, 1H); 15.62 (уширенный s, 1H, обменный).
Масс-спектр (ESI+): m/z 384 (MH+, 100%).
Пример 18
(7-Фтор-4-гидрокси-2-этил-1,1-диоксо-2Н-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанон

Соединение 18 синтезировали из соединения 17А (1,0 г, 2,7 ммоль) и иодэтана в соответствии с методикой, использованной для получения производного 1, получали две порции желаемого продукта разной степени очистки.
Первая порция: 716 мг, бледно-коричневое твердое вещество; ЖХВД: RT=5,78 мин, 89%.
Вторая порция: 68 мг, желтое твердое вещество; ЖХВД: RT=5,78 мин, 99%.
Выход реакции составлял 65%.
1H ЯМР, ДМСО-d6, δ (м.д.): 0.54 (t, 3Н); 3.14 (q, 2Н); 7.66 (t, 1H); 7.71 (t, 1H); 7.82 (t, 1H); 7.92 (d, 1H); 8.00-8.15 (m, 4H); 8.29 (dd, 1H); 8.60 (s, 1H); 15.45 (уширенный s, 1H, обменный).
Масс-спектр (ESI+): m/z 398 (MH+, 100%).
Пример 19
Бензойной кислоты 2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2Н-бензо[е][1,2]тиазин-4-иловый эфир

Соединение 1 (86 мг, 0,18 ммоль) растворяли в дихлорметане (0,5 мл) и пиридине (0,5 мл) в атмосфере инертного газа. Данную реакционную смесь охлаждали до 0°C и затем добавляли бензоилхлорид (33 мкл, 0,27 ммоль). Охлаждающую баню удаляли и реакционную смесь перемешивали в течение 4 ч при комнатной температуре. Так как реакция была не завершена, добавляли еще одну порцию бензоилхлорида (16 мкл, 0,14 ммоль), реакционную смесь перемешивали при комнатной температуре дополнительно в течение 20 ч и затем концентрировали. Остаток переносили в этилацетат, промывали один раз водой и один раз насыщенным водным раствором NaCl, сушили сульфатом натрия, фильтровали и концентрировали. Полученный остаток упаривали три раза с толуолом с целью удаления оставшегося пиридина. Полученный в результате желтый сироп очищали на колонке со сферическим силикагелем (12 г; скорость потока: 12 мл/мин; градиент: 20-100% CH2Cl2/гептан (30 мин)) с выходом соединения 19 в виде желтой пены (38 мг, 44%).
ЖХВД: RT=5,65 мин, 96%.
1H ЯМР, ДМСО-d6, δ (м.д.): 3.10 (s, 3H); 7.32 (t, 2H); 7.55-7.30 (m, 6H); 7.86 (dd, 2H); 7.90-8.05 (m, 5H); 8.70 (s, 1H).
Пример 20
Циклогексанкарбоновой кислоты 2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2Н-бензо[е][1,2]тиазин-4-иловый эфир

Соединение 1 (86 мг, 0,18 ммоль) растворяли в 0,5 мл пиридина в атмосфере инертного газа. Данную реакционную смесь охлаждали до 0°C и затем добавляли циклогексанкарбонил хлорид (62 мкл, 0,46 ммоль). Охлаждающую баню удаляли и реакционную смесь перемешивали в течение 18 ч при комнатной температуре и затем нагревали до 60°C в течение 8 ч. Затем реакционную смесь концентрировали и три раза упаривали с толуолом. Полученный в результате остаток очищали на колонке со сферическим силикагелем (12 г; скорость потока: 12 мл/мин; градиент: 20-100% CH2Cl2/гептан (20 мин)) с выходом соединения 20 в виде желтой пены (65 мг, 28%).
ЖХВД: RT=5,99 мин, 95%.
1H ЯМР, ДМСО-d6, δ (м.д.): 0.85-1.00 (m, 6Н); 1.38 (de, 2H); 1.49 (de, 2H); 2.28 (tt, 1H); 3.06 (s, 3Н); 7.66 (t, 2H); 7.75 (t, 1H); 7.83 (t, 1H); 7.88 (t, 1H); 7.95-8.15 (m, 5H); 8.66 (s, 1H).
Масс-спектр (ESI+): m/z 493 (MNH4+, 100%).
Пример 21
трет-Бутилкарбоновой кислоты 2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2Н-бензо[е][1,2]тиазин-4-иловый эфир
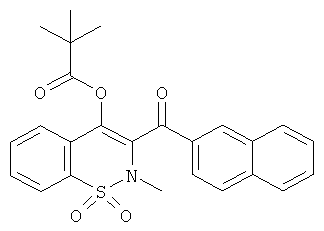
Соединение 1 (86 мг, 0,18 ммоль) растворяли в пиридине (0,5 мл) в атмосфере инертного газа. Данную реакционную смесь охлаждали до 0°C и затем добавляли трет-бутилкарбонил хлорид (57 мкл, 0,46 ммоль). Охлаждающую баню удаляли и реакционную смесь перемешивали в течение 18 ч при комнатной температуре. Затем реакционную смесь концентрировали и три раза упаривали с толуолом. Полученный в результате остаток очищали на колонке со сферическим силикагелем (12 г; скорость потока: 12 мл/мин; градиент: 20-100% CH2Cl2/гептан (20 мин)) с выходом соединения 21, в виде желтой пены (47 мг, 53%).
ЖХВД: RT=5,71 мин, 98%.
1H ЯМР, ДМСО-d6, δ (м.д.): 0.88 (s, 9H); 3.07 (s, 3Н); 7.59 (d, 1H); 7.66 (t, 1H); 7.75 (t, 1H); 7.84 (t, 1H); 7.89 (t, 1H); 8.00-8.09 (m, 4H); 8.12 (d, 1H); 8.69 (s, 1H).
Масс-спектр (ESI+): m/z 467 (MNH4+, 100%).
Пример 22
4-Метилбензойной кислоты 2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2Н-бензо[е][1,2]тиазин-4-иловый эфир
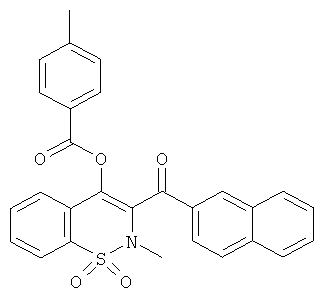
Соединение 22 синтезировали из соединения 1 (86 мг, 0,18 ммоль) и 4-метилбензоил хлорида (62 мкл, 0,46 ммоль) в соответствии с методикой, использованной для получения соединения 21. Желаемый продукт получали в виде желтой пены (27 мг, 31%).
ЖХВД: RT=5,82 мин, 95%.
1H ЯМР, ДМСО-d6, δ (м.д.): 2.28 (s, 3Н); 3.10 (s, 3H); 7.11 (d, 2H); 7.54 (d, 2Н); 7.65 (t, 1Н); 7.73 (te, 2H); 7.86 (dd, 2H); 7.95 (d, 1H); 7.97-8.05 (m, 4H); 8.69 (s, 1H).
Масс-спектр (ESI+): m/z 501 (MNH4+, 100%).
Пример 23
4-Хлорбензойной кислоты 2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2Н-бензо[е][1,2]тиазин-4-иловый эфир

4-Хлорбензойную кислоту (87 мг, 0,55 ммоль) растворяли в толуоле (2 мл) в атмосфере инертного газа. Добавляли при комнатной температуре оксалилхлорид (100 мкл, 1,1 ммоль). Данную реакционную смесь нагревали в течение 2 ч до 80°C и затем концентрировали и три раза упаривали с толуолом. Остаток снова помещали в атмосферу инертного газа и охлаждали до 0°C. Добавляли соединение 1 (86 мг, 0,18 ммоль), которое предварительно растворяли в пиридине (0,5 мл) в атмосфере инертного газа и охлаждали до 0°C. Охлаждающую баню удаляли и реакционную смесь перемешивали в течение 2 ч при комнатной температуре. Затем реакционную смесь концентрировали и три раза упаривали с толуолом. Полученный в результате остаток очищали на колонке со сферическим силикагелем (12 г; скорость потока: 12 мл/мин; градиент: 20-100% CH2Cl2/гептан (20 мин)) с выходом соединения 23 в виде желтой пены (51 мг, 42%).
ЖХВД: RT=5,92 мин, 97%.
1H ЯМР, ДМСО-d6, δ (м.д.): 3.10 (s, 3H); 7.37 (d, 2H); 7.63-7.67 (m, 3H); 7.72 (t, 1H); 7.79 (уширенный s, 1Н); 7.86 (уширенный s, 2H); 7.94 (d, 1H); 7.95-8.07 (m, 4H); 8.66 (s, 1H).
Масс-спектр (ESI+): m/z 521 (MNH4+, 100%); 523 (MNH4+, 37%).
Примеры 24-27
Соединения 24-27 синтезировали из соединения 15 и различных хлорангидридов в соответствии с описанной выше методикой получения соединения 21.

Примеры 28-31
Соединения 28-31 синтезировали из соединения 16 и различных хлорангидридов в соответствии с описанной выше методикой получения соединения 21.

Примеры 32 и 33
Соединения 32 и 33 синтезировали из 4-хлорбензойной кислоты и соединений 15 и 16, соответственно, с использованием описанной выше методики получения соединения 23.
Пример 34
Нафталин-1-илкарбоновой кислоты 2-метил-3-(4-метилбензоил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-иловый эфир
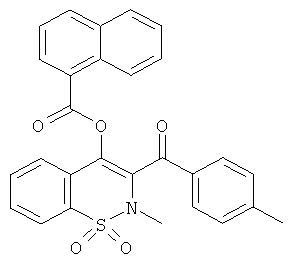
Соединение 3 (150 мг, 0,455 ммоль) растворяли в тетрагидрофуране (3 мл) в атмосфере инертного газа. Добавляли гидрид натрия (27 мг, 0,68 ммоль) и затем, через 30 мин, нафталин-2-илкарбонил хлорид (105 мкл, 0,68 ммоль). Данную реакционную смесь перемешивали в течение 4 часов при комнатной температуре. Затем реакционную смесь нейтрализовали путем добавления воды и водную фазу дважды экстрагировали этилацетатом. Органические фазы объединяли, сушили сульфатом магния, фильтровали и концентрировали. Полученный в результате остаток очищали на колонке со сферическим силикагелем (12 г; скорость потока: 12 мл/мин; градиент 0-45% EtOAc в гептане (20 мин)). Желаемый продукт получали в виде желтого твердого вещества (134 мг, 61%).
ЖХВД: RT=6,59 мин, 98%.
1H ЯМР, ДМСО-d6, δ (м.д.): 2.35 (s, 3Н); 3.08 (s, 3Н); 7.36 (d, 2H); 7.54 (t, 1H); 7.58-7.63 (m, 2H); 7.79-7.87 (m, 3Н); 7.92 (d, 2H); 8.00 (d, 1H); 8.04-8.07 (m, 2H); 8.27 (d, 1H); 8.50-8.55 (m, 1H).
Масс-спектр (ESI+): m/z 501 (MNH4+, 100%).
Примеры 35-45
Соединения 35-45 синтезировали из соединения 3 или из соединения 5 и различных хлорангидридов в соответствии с описанной выше методикой получения соединения 34.
Хлорангидриды, необходимые для синтеза соединений в Примерах 40-45, получали в две стадии из соответствующих ароматических спиртов. Получение (нафталин-2-илокси)ацетил хлорида приведено в качестве примера.
2-Нафтол (3,0 г, 20 ммоль) растворяли в 95 мл метилэтилкетона (МЕК) в присутствии карбоната натрия (40 г, 93 ммоль) в двугорлой колбе, оснащенной холодильником, в атмосфере инертного газа и затем нагревали до 50°C в течение 30 мин. В нагретую смесь добавляли по каплям 2-бромэтановую кислоту (5,76 г, 41 ммоль), растворенную в МЕК (23 мл). Нагревание продолжали еще в течение 4 ч. Затем реакционную смесь охлаждали до комнатной температуры и фильтровали. Твердое вещество собирали путем фильтрования, переносили в смесь этилацетата и 1 н. водного раствора HCl. Две фазы разделяли и водную фазу один раз экстрагировали этилацетатом. Органические фазы собирали, сушили сульфатом магния, фильтровали и концентрировали до появления первых кристаллов. Добавляли гептан (приблизительно 20% от оставшегося объема) и образовавшийся осадок отделяли, промывали гептаном и сушили до постоянной массы с получением (нафталин-2-илокси)уксусной кислоты (3,04 г, 72%) в виде белого твердого вещества.
ЖХВД: RT=4,10 мин, 99%.
1H ЯМР, ДМСО-d6, δ (м.д.): 4.80 (s, 2Н); 7.20 (dd, 1Н); 7.26 (d, 1Н); 7.35 (td, 1Н); 7.45 (td, 1Н); 7.79 (d, 1Н); 7.80-7.86 (m, 2Н); 13.07 (уширенный s, 1Н, обменный).
Macc-спекгр (ESI+): m/z 203 (МН+, 100%).
Масс-спектр (ESI-): m/z 201 (M-H, 100%).
Кислоту, полученную на предыдущей стадии (3,04 г, 15 ммоль), частично растворяли в дихлорметане (34 мл) в атмосфере инертного газа при комнатной температуре. Добавляли оксалилхлорид (1,35 мл, 15,7 ммоль) и затем ДМФА (100 мкл). Внимание: при добавлении ДМФА происходит бурная реакция. Реакционную смесь перемешивали в течение 1 ч, затем концентрировали, дважды упаривали с толуолом и сушили до постоянной массы с получением (нафталин-2-илокси)ацетил хлорида (3,4 г, 100%) в виде оранжевого твердого вещества. Полученный в результате хлорангидрид использовали как есть для синтеза соединений 40 и 41.

Пример 46
Уксусной кислоты 2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2Н-бензо[е][1,2]тиазин-4-иловый эфир
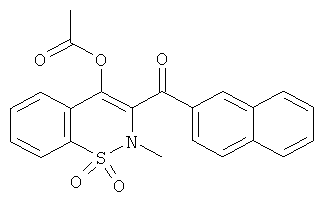
Соединение 1 (100 мг, 0,274 ммоль) растворяли в дихлорметане (2 мл) в атмосфере инертного газа. Добавляли при 0°C триэтиламин (230 мкл, 1,64 ммоль) и затем ацетилхлорид (78 мкл, 1,09 ммоль). Данную реакционную смесь перемешивали в течение 18 ч при комнатной температуре и затем концентрировали. Полученный в результате остаток очищали на колонке со сферическим силикагелем (12 г) с выходом соединения 46 (21 мг, 30%).
ЖХВД: RT=5,34 мин, 97%.
1H ЯМР, ДМСО-d6, δ (м.д.): 1.96 (s, 3Н); 3.03 (s, 3Н); 7.66 (t, 1H); 7.75 (t, 1H); 7.80-7.91 (m, 3Н); 7.99-8.08 (m, 4H); 8.12 (d, 1H); 8.67 (s, 1H).
Масс-спектр (ESI+): m/z 425 (MNH4+, 100%).
Примеры 47-54
Соединения 47-54 синтезировали из соединения 1 и различных хлорангидридов в соответствии с описанной выше методикой получения соединения 46.

Пример 55
(4-Метокси-2-метил-1,1-диоксо-2Н-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанон

Соединение 1 (159 мг, 0,435 ммоль) растворяли в ДМФА (2 мл) в атмосфере инертного газа. Добавляли гидрид натрия (26 мг, 0,65 ммоль) и затем, через 30 мин, иодметан (30 мкл, 0,48 ммоль). Данную реакционную смесь перемешивали в течение 2 ч при комнатной температуре и затем в течение 26 ч при 60°C. На данном этапе реакция была не завершена. Добавляли карбонат цезия (213 мг, 0,65 ммоль) и иодметан (150 мкл, 2,1 ммоль). Данную реакционную смесь перемешивали в течение 24 ч при комнатной температуре и затем нейтрализовали путем добавления воды, водную фазу дважды экстрагировали этилацетатом. Органические фазы объединяли, сушили сульфатом магния, фильтровали и концентрировали. Полученный в результате остаток очищали на колонке со сферическим силикагелем (12 г; скорость потока: 12 мл/мин; градиент 20-60% дихлорметана в гептане) с выходом соединения 55 в виде желтой пены (70 мг, 38%).
ЖХВД: RT=5,27 мин, 90%.
Масс-спектр (ESI+): m/z 380 (MH+, 100%).
Примеры 56-58
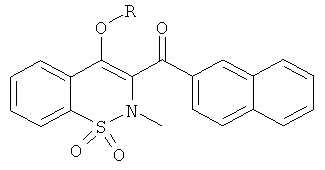
Соединения 56-58 получали в соответствии со следующей методикой. Соединение 1 (150 мг, 0,42 ммоль) растворяли в ДМФА (0,3 мл) в атмосфере инертного газа. Добавляли карбонат цезия (201 мг, 0,61 ммоль) и требуемый алкилиодид (4 ммоль). Данную реакционную смесь перемешивали в течение 18 ч при комнатной температуре, в течение 4 ч при 50°C и затем нейтрализовали путем добавления воды, водную фазу дважды экстрагировали этилацетатом. Органические фазы объединяли, сушили сульфатом магния, фильтровали и концентрировали. Полученный в результате остаток очищали на колонках со сферическим силикагелем (12 г; скорость потока: 12 мл/мин; градиент 20-60% дихлорметана в гептане) с выходом желаемых продуктов.
Пример 59
(4-(2-Хлорэтокси)-2-метил-1,1-диоксо-2Н-бензо[е][1,2]тиазин-3-ил)(пара-толил)метанон

Соединение 3 (100 мг, 0,3 ммоль) растворяли в ТГФ (3 мл) в атмосфере инертного газа в присутствии 2-хлорэтанола (100 мкл, 1,5 ммоль). Данную реакционную смесь охлаждали до 0°C, затем добавляли по каплям последовательно трифенилфосфин (318 мг, 1,2 ммоль) и диэтилдиазен-1,2-дикарбоксилат (DEAD, 211 мг, 1,2 ммоль). Перемешивание продолжали в течение 20 ч при комнатной температуре и затем реакционную смесь нейтрализовали путем добавления насыщенного водного раствора хлорида аммония. Полученную водную фазу дважды экстрагировали этилацетатом. Органические фазы объединяли, сушили сульфатом магния, фильтровали и концентрировали. Полученный в результате остаток очищали на колонке с силикагелем (35 г; скорость потока: 20 мл/мин; градиент 0-100% этилацетата в гептане) с выходом соединения 59 (70 мг, 58%).
ЖХВД: RT=5,28 мин, 99%.
1H ЯМР, ДМСО-d6, δ (м.д.): 2.41 (s, 3Н); 2.91 (s, 3H); 3.57 (t, 2H); 3.92 (t, 2H); 7.40 (d, 2H,); 7.75-7.98 (m, 6H).
Масс-спектр (ESI+): m/z 392 (MH+, 100%); 394 (MH+, 42%).
Пример 60
(4-[2-(Нафталин-2-илокси)этокси]-2-метил-1,1-диоксо-2Н-бензо[е][1,2]тиазин-3-ил)(пара-толил)метанон

Соединение 59 (70 мг, 0,17 ммоль) растворяли в ДМФА (2 мл) в атмосфере инертного газа в присутствии карбоната калия (64 мг, 0,53 ммоль), иодида калия (31 мг, 0,19 ммоль) и 2-нафтола (38 мг, 0,27 ммоль). Данную реакционную смесь нагревали до 65°C в течение 22 ч, затем нейтрализовали путем добавления воды и дважды экстрагировали этилацетатом. Органические фазы объединяли, сушили сульфатом магния, фильтровали и концентрировали. Полученный в результате остаток очищали на колонке Waters Sunfire (19×100 мм, 5 мкм) путем полупрепаративной ЖХВД (скорость потока: 20 мл/мин; градиент 10-100% ацетонитрила в воде (0,1% ТФУ-буфер) в течение 15 мин) с выходом соединения 60 (30 мг, 29%).
ЖХВД: RT=5,95 мин, 99%.
1H ЯМР, ДМСО-d6, δ (м.д.) 2.26 (s, 3Н); 2.90 (s, 3Н); 4.03 (d, 2H); 4.10 (d, 2H); 6.92 (dd, 1H); 7.06 (d, 1H); 7.24 (d, 2H); 7.33 (t, 1H); 7.44 (t, 1H); 7.70-7.90 (m, 7H); 7.94-7.98 (m, 2H).
Масс-спектр (ESI+): m/z 500 (MH+, 100%).
Пример 61
(4-(2-Фенокси-этокси)-2-метил-1,1-диоксо-2Н-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанон

Соединение 1 (100 мг, 0,274 ммоль) растворяли в ДМФА (0,5 мл) в присутствии карбоната калия (90 мг, 0,55 ммоль) и 2-феноксиэтил бромида (110 мг, 0,55 ммоль). Данную реакционную смесь нагревали в герметично закрытой пробирке до 80°C в течение 16 ч. Смесь переносили в этилацетат и затем промывали водой и насыщенным раствором NaCl. Органические фазы объединяли, сушили сульфатом натрия, фильтровали и концентрировали. Полученный в результате остаток очищали на колонке с силикагелем (12 г; скорость потока: 20 мл/мин; градиент 0-10% дихлорметана в гептане) с выходом соединения 61 в виде желтого сиропа (45 мг, 34%).
ЖХВД: RT=5,88 мин, 98%.
1H ЯМР, ДМСО-d6, δ (м.д.) 2.97 (s, 3Н); 3.85-3.87 (m, 2H); 4.01-4.05 (m, 2H); 6.59 (d, 2H); 6.82 (t, 1H); 7.11 (t, 2H); 7.62 (t, 1H); 7.71 (t, 1H); 7.83 (t, 1H); 7.88 (t, 1H); 7.91-8.04 (m, 6H); 8.62 (s, 1H).
Масс-спектр (ESI+): m/z 486 (MH+, 100%).
Пример 62
Метил 2-(2-метил-3-(4-метилбензоил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илокси)ацетат
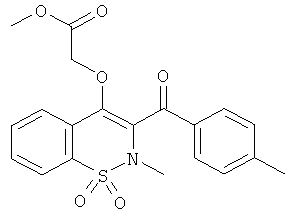
Соединение 62 синтезировали из соединения 3 (300 мг, 0,91 ммоль) и метилгликолята (350 мкл, 4,5 ммоль) в соответствии с методикой, использованной для получения соединения 59, желаемый продукт получали в виде желтого сиропа (300 мг, 79%).
ЖХВД: RT=4,97 мин, 97%.
1H ЯМР, ДМСО-d6, δ (м.д.): 2.42 (s, 3Н); 2.88 (s, 3Н); 3.47 (s, 3H); 4.42 (s, 2H); 7.40 (d, 2H,); 7.75-7.98 (m, 6H).
Масс-спектр (ESI+): m/z 402 (MH+, 100%); 419 (MNH4+, 42%).
Пример 63
(2-Метил-3-(4-метилбензоил)-1,1-диоксо-2Н-бензо[е][1,2]тиазин-4-илокси)уксусная кислота

Соединение 62 (75 мг, 0,18 ммоль) растворяли в ТГФ (1 мл) и добавляли гидроксид лития (1M/H2O, 0,37 ммоль). Данную реакционную смесь перемешивали в течение 2 ч при комнатной температуре и затем разбавляли водой и дважды экстрагировали дихлорметаном. Органические фазы объединяли, сушили сульфатом магния, фильтровали и концентрировали с получением 22 мг желаемого продукта (ЖХВД: RT=4,55 мин, 97%). Выход реакции составлял 30%.
1H ЯМР, ДМСО-d6, δ (м.д.) 2.41 (s, 3Н); 2.88 (s, 3Н); 4.28 (s, 2H); 7.39 (d, 2H); 7.81 (t, 1H); 7.85-7.95 (m, 5H); 12.96 (se, 1H, обменный).
Масс-спектр (ESI+): m/z 388 (MH+, 100%); 405 (MNH4+, 54%.
Примеры 64-66
Соединение 63 (110 мг, 0,28 ммоль) растворяли в ДМФА (3 мл). Добавляли различные амины (0,23 ммоль), DIEA (N,N-диизопропилэтиламин) (82 мкл, 0,472 ммоль), HOOBT (3-гидрокси-1,2,3-бензотриазин-4-он) (35 мг, 0,26 ммоль), EDCI (N-этил-N′-(3-диметиламинопропил)карбодиимида гидрохлорид) (50 мг, 0,26 ммоль). Данную реакционную смесь перемешивали в течение 18 ч при комнатной температуре. Смесь переносили в дихлорметан и затем промывали 1 н. раствором карбоната натрия, водой и насыщенным раствором NaCl. Органические фазы объединяли, сушили сульфатом магния, фильтровали и концентрировали. Полученные в результате остатки очищали на колонках со сферическим силикагелем (12 г; скорость потока: 12 мл/мин; 0-50% AcOEt в гептане) с выходом желаемых продуктов.

Пример 67
Метил 2-(2-метил-3-(4-метилбензоил)-1,1-диоксо-2Н-бензо[е][1,2]тиазин-4-илокси)ацетат

Соединение 1 (1,0 г, 2,74 ммоль) растворяли в ДМФА (2 мл) в присутствии карбоната калия (682 мг, 4,1 ммоль) и бромуксусной кислоты метилового эфира (1,26 мл, 13,68 ммоль). Данную реакционную смесь перемешивали в течение 5 ч при комнатной температуре и затем добавляли еще одну такую же порцию бромуксусной кислоты метилового эфира. Смесь оставляли на ночь при комнатной температуре, затем переносили в этилацетат и затем промывали водой и насыщенным раствором NaCl. Органические фазы объединяли, сушили сульфатом натрия, фильтровали и концентрировали. Полученный в результате остаток очищали на колонке с силикагелем (90 г; скорость потока: 32 мл/мин; градиент 40-100% дихлорметана в гептане) с выходом соединения 67 в виде желтого сиропа (486 мг, 41%).
ЖХВД: RT=5,23 мин, 86%.
1H ЯМР, ДМСО-d6, δ (м.д.) 2.95 (s, 3Н); 3.39 (s, 3Н); 4.45 (s, 2H); 7.64 (t, 1H); 7.72 (t, 1H); 7.81-7.89 (m, 1H); 7.93 (d, 2H); 7.97 (d, 1H); 7.99-8.11 (m, 4H); 8.66 (s, 1H).
Масс-спектр (ESI+): m/z 438 (MH+, 100%).
Пример 68
2-(2-Метил-3-(4-метилбензоил)-1,1-диоксо-2Н-бензо[е][1,2]тиазин-4-илокси)уксусная кислота

Соединение 67 (480 мг, 1,1 ммоль) растворяли в смеси ТГФ/вода (5:1) (6 мл) и затем обрабатывали LiOH (103 мг, 4,39 ммоль) в течение 15 мин при комнатной температуре. Смесь переносили в этилацетат и затем промывали 1 н. HCl, водой и насыщенным раствором NaCl. Органические фазы объединяли, сушили сульфатом натрия, фильтровали и концентрировали. Полученный в результате остаток очищали на колонке с силикагелем (30 г; элюент: дихлорметан/метанол/уксусная кислота (95/4,5/0)) с выходом соединения 68 в виде желтой пены (321 мг, 69%).
ЖХВД: RT=4,86 мин, 99%.
1H ЯМР, ДМСО-d6, δ (м.д.) 2.93 (s, 3Н); 4.29 (s, 2Н); 7.64 (t, 1H); 7.72 (t, 1H); 7.83 (t, 1H); 7.89-8.09 (m, 7H); 8.66 (s, 1H).
Масс-спектр (ESI+): m/z 424 (MH+, 100%).
Примеры 69-71

Соединения 69-71, получали в соответствии со следующей методикой. Соединение 1 (100 мг, 0,23 ммоль) растворяли в дихлорметане (1,5 мл). Добавляли различные амины (0,23 ммоль), DIEA (82 мкл, 0,472 ммоль), HOOBT (35 мг, 0,26 ммоль) и EDCI (50 мг, 0,26 ммоль). Данную реакционную смесь перемешивали в течение 24 ч при комнатной температуре, затем добавляли избыток амина (0,07 ммоль) и полученную смесь перемешивали еще в течение 5 ч. Смесь переносили в дихлорметан и промывали 1 н. раствором карбоната натрия, водой и насыщенным раствором NaCl. Органические фазы объединяли, сушили сульфатом натрия, фильтровали и концентрировали. Полученные в результате остатки очищали на колонках со сферическим силикагелем (12 г; скорость потока: 12 мл/мин; смесь 1% метанол/аммиак (9:1) в дихлорметане) с выходом желаемых продуктов.



Примеры 72-74
Соединения 72-74 синтезировали из соединения 1 и различных хлорангидридов в соответствии с описанной выше методикой получения соединения 46.

Примеры 75 и 76
Соединения 75 и 76 синтезировали из соединения 59 и различных спиртов в соответствии с описанной выше методикой получения соединения 60.

Примеры 77 и 78
Соединения 77 и 78 синтезировали из соединения 3 и из ацетилхлорида и пропаноил хлорида, соответственно, с использованием описанной выше методики получения соединения 34.

Примеры 79 и 80
Соединения 79 и 80 синтезировали из соединения 3 и из метанола и этанола, соответственно, с использованием описанной выше методики получения соединения 59.

Пример 81
[4-(2-Бром-этокси)-2-метил-1,1-диоксо-2Н-бензо[е][1,2]тиазин-3-ил]-нафталин-2-ил-метанон

Соединение 1 (150 мг, 0,41 ммоль) растворяли в метилэтилкетоне (3 мл) и затем обрабатывали дибромэтаном (71 мкл, 0,82 ммоль) в присутствии K2CO3 (170 мг, 1,02 ммоль). Данную реакционную смесь нагревали в микроволновой печи в герметично закрытой пробирке в течение 4 ч 30 мин при 130°C. Смесь переносили в этилацетат и затем промывали водой и насыщенным раствором NaCl. Органические фазы объединяли, сушили сульфатом натрия, фильтровали и концентрировали. Полученный в результате остаток (коричневый сироп, 197 мг) как есть использовали в следующей реакции.
Пример 82
{4-[2-(4-Хлор-фенокси)-этокси]-2-метил-1,1-диоксо-2Н-бензо[е][1,2]тиазин-3-ил}-нафталин-2-ил-метанон

Соединение 81 (197 мг, 0,41 ммоль) растворяли в метилэтилкетоне (1,5 мл) и затем обрабатывали 4-хлорфенолом (107 мкл, 0,82 ммоль) в присутствии K2CO3 (173 мг, 1,04 ммоль). Данную реакционную смесь нагревали в микроволновой печи в герметично закрытой пробирке в течение 2 ч при 130°C. Смесь переносили в этилацетат и затем промывали водой и насыщенным раствором NaCl. Органические фазы объединяли, сушили сульфатом натрия, фильтровали и концентрировали. Полученный в результате остаток очищали на колонке с силикагелем (12 г; скорость потока: 12 мл/мин; градиент 10-100% дихлорметана в гептане) с выходом соединения 82 в виде желтого сиропа (21 мг, 14%).
ЖХВД: RT=6,10 мин, 89%.
1H ЯМР, ДМСО-d6, δ (м.д.) 2.96 (s, 3Н); 3.85 (m, 2Н); 3.97 (m, 2H); 6.57 (d, 2Н); 7.08 (d, 2H); 7.61 (t, 1H); 7.70 (t, 1H); 7.83 (t, 1H); 7.89-8.02 (m, 7H); 8.60 (s, 1H).
Масс-спектр (ESI+): m/z 520 (MH+, 66%).
Пример 83
Угольной кислоты этил 1-[2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2Н-бензо[е][1,2]тиазин-4-илокси]-этиловый эфир
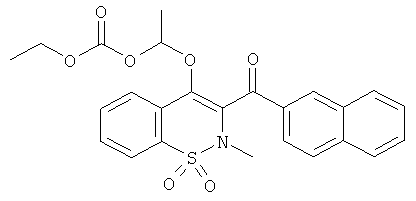
Соединение 1 (100 мг, 0,27 ммоль) растворяли в ДМФА (1 мл) и затем обрабатывали этил 2-хлорпропаноатом (110 мкл, 0,82 ммоль) в присутствии K2CO3 (91 мг, 0,55 ммоль). Данную реакционную смесь нагревали в герметически закрытом сосуде до 60°C в течение ночи и затем добавляли еще одну такую же порцию этил 2-хлорпропаноата и реакционную смесь перемешивали дополнительно течение 24 ч. Смесь переносили в этилацетат и затем промывали водой и насыщенным раствором NaCl. Органические фазы объединяли, сушили сульфатом натрия, фильтровали и концентрировали. Полученный в результате остаток очищали на колонке с силикагелем (12 г; скорость потока: 12 мл/мин; градиент 25-80% дихлорметана в гептане) с выходом соединения 83 в виде желтого сиропа (100 мг, 76%).
ЖХВД: RT=5,55 мин, 97%.
1H ЯМР, ДМСО-d6, δ (м.д.) 0.94 (t, 3Н); 1.25 (а, 3Н); 2.94 (s, 3H); 3.80-3.93 (m, 2Н); 5.99 (q, 1H); 7.65 (t, 1H); 7.73 (t, 1H); 7.85 (m, 1H); 7.90 (d, 2Н); 7.97 (d, 1H); 8.01-8.11(m,4H); 8.69 (s, 1H).
Масс-спектр (ESI+): m/z 504 (MNa+, 100%).
Пример 84
[2-Метил-1,1-диоксо-4-(2-пиперидин-1-ил-этокси)-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-ил]-нафталин-2-ил-метанон

Соединение 1 (100 мг, 0,27 ммоль) растворяли в метилэтилкетоне (0,5 мл) и затем обрабатывали 1-(2-хлорэтил)пиперидином (252 мг, 1,37 ммоль) в присутствии K2CO3 (159 мг, 0,96 ммоль). Данную реакционную смесь нагревали до 80°C в течение ночи. Затем смесь обрабатывали этилацетатом и затем промывали водой и насыщенным раствором NaCl. Органические фазы объединяли, сушили сульфатом натрия, фильтровали и концентрировали. Полученный в результате остаток очищали на колонке с силикагелем (12 г; скорость потока: 12 мл/мин; дихлорметан/MeOH/NH4OH (99/09/01)) с выходом соединения 84 в виде желтого сиропа (50 мг, 43%).
ЖХВД: RT=4,17 мин, 92%.
1H ЯМР, ДМСО-d6, δ (м.д.): 1.21 (уширенный s, 6H); 1.99 (уширенный s, 4H); 2.19 (t, 2H); 2.98 (s, 3Н); 3.72 (t, 2H); 7.64 (t, 1H); 7.72 (t, 1H); 7.83 (t, 1H); 7.90 (t, 1H); 7.97 (d, 1H); 8.01-8.11 (m, 5H); 8.66 (s, 1H).
Масс-спектр (ESI+): m/z 477 (MH+, 100%).
Примеры 85-96
Соединения 85-96 синтезировали в соответствии со следующей методикой. Соединение 13, 15 или 17 (100 мг) растворяли в дихлорметане (2 мл) в атмосфере инертного газа в присутствии триэтиламина (6 экв.) и затем обрабатывали различными хлорангидридами (4 экв.) при 0°C. Реакционные смеси перемешивали в течение 2 ч при 0°C и затем в течение 20 ч при комнатной температуре. Смеси переносили в этилацетат и затем промывали водой и насыщенным раствором NaCl. Органические фазы объединяли, сушили сульфатом натрия, фильтровали и концентрировали. Полученные в результате остатки очищали на колонке с силикагелем (12 г; скорость потока: 12 мл/мин; градиент 0-20% этилацетата в гептане) с выходом желаемых соединений.

Примеры 97-104
Соединения 97-104 синтезировали из соединения 13, 15 или 17 и из соответствующих алкил иодидов или сульфатов в соответствии с методикой, использованной для получения соединений в Примерах 56-58.

Пример 105
[7-Фтор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2Н-бензо[е][1,2]тиазин-4-илокси]-уксусной кислоты метиловый эфир

Соединение 105 синтезировали из соединения 17 (1 г, 2,61 ммоль) в соответствии с методикой, использованной для получения соединения в Примере 67. Желаемый продукт получали в виде желтого сиропа (810 мг, 68%).
ЖХВД: RT=5,37 мин, 95%.
1H ЯМР, ДМСО-d6, δ (м.д.): 2.95 (s, 3Н); 3.40 (s, 3Н); 4.45 (s, 2H); 7.64 (t, 1H); 7.70 (t, 1H); 7.80 (dt, 1H); 7.91 (dd, 1H); 7.99-8.10 (m, 5H); 8.65 (s, 1H).
Масс-спектр (ESI+): m/z 456 (MH+, 100%).
Пример 106
[7-Фтор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илокси]-уксусной кислоты метиловый эфир

Соединение 106 синтезировали из соединения 105 (598 мг, 1,31 ммоль) в соответствии с методикой, использованной для получения соединения в Примере 68. Желаемый продукт получали в виде бежевого порошка (262 мг, 45%).
ЖХВД: RT=4,92 мин, 97%.
Масс-спектр (ESI+): m/z 442 (МН+, 100%).
Примеры 107-109
Соединения 107-109 синтезировали из соединения 106 и соответствующих аминов в соответствии с методикой, использованной для получения соединений в Примерах 69-71.

Пример 110
Бензолсульфоновой кислоты 7-фтор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2Н-бензо[е][1,2]тиазин-4-иловый эфир
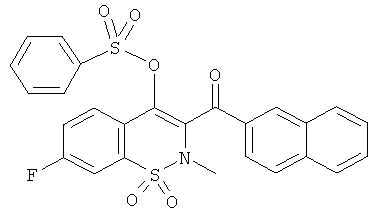
Соединение 17 (200 мг, 0,52 ммоль) растворяли в дихлорметане (2 мл) и затем обрабатывали при 0°C бензолсульфоновой кислоты хлорангидридом (67 мкл, 0,52 ммоль) в присутствии Et3N (145 мкл, 1,04 ммоль). Данную реакционную смесь перемешивали в течение 4 ч (пока температура реакционной смеси не повышалась от 0°C до комнатной температуры) и затем смесь переносили в дихлорметан и промывали водой и насыщенным раствором NaCl. Органические фазы объединяли, сушили сульфатом магния, фильтровали и концентрировали. Полученный в результате остаток очищали на колонке с силикагелем (12 г; скорость потока: 12 мл/мин; градиент 20-50% дихлорметана в гептане) с выходом соединения 110 в виде порошка кремового цвета (177 мг, 65%).
ЖХВД: RT=5,72 мин, 94%.
1H ЯМР, ДМСО-d6, δ (м.д.): 2.98 (s, 3H); 7.40 (t, 2H); 7.54-7.61 (m, 4H); 7.66 (t, 1H); 7.75 (m, 2H); 7.84-7.90 (m, 2H); 8.04-8.09 (m, 3H); 8.57 (s, 1H).
Масс-спектр (ESI+): m/z 541 (MNH4+, 100%).
Примеры 111-117
Соединения 111-117 синтезировали из соединения 1 или 17 и соответствующих сульфонилхлоридов в соответствии с методикой, использованной для получения соединения в Примере 110.

Пример 118
(4-Гидрокси-2-метил-7-пиперидин-1-ил-1,1-диоксо-2Н-бензо[е][1,2]тиазин-3-ил)-нафталин-2-ил-метанон

Соединение 17 (200 мг, 0,52 ммоль) растворяли в ДМСО (2 мл) в присутствии K2CO3 (144 мг, 1,04 ммоль) и затем обрабатывали при комнатной температуре пиперидином (154 мкл, 1,56 ммоль). Данную реакционную смесь перемешивали в течение 20 ч при 100°C и затем смесь переносили в этилацетат и промывали водой и насыщенным раствором NaCl. Органические фазы объединяли, сушили сульфатом магния, фильтровали и концентрировали. Полученный в результате остаток очищали на колонке с силикагелем (12 г; скорость потока: 12 мл/мин; градиент 0-50% этилацетата в гептане) с выходом соединения 118 в виде желтого порошка (22 мг, 9,5%).
ЖХВД: RT=6,39 мин, 96%.
1H ЯМР, ДМСО-d6, δ (м.д.): 1.64 (уширенный s, 6H); 2.63 (s, 3H); 3.57 (уширенный s, 4Н); 7.22 (а, 1Н); 7.34 (dd, 1H); 7.65-7.70 (m, 2H); 7.96 (d, 1H); 8.03 (d, 1H); 8.09-8.11 (m, 3H); 8.62 (s, 1H).
Масс-спектр (ESI+): m/z 449 (MH+, 100%).
Примеры 119-121
Соединения 119-121 синтезировали из соединения 17 и соответствующих аминов в соответствии с методикой, использованной для получения соединения в Примере 118.


Пример 122
(7-трет-Бутил-4-гидрокси-2-метил-1,1-диоксо-2Н-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанон

Соединение 122 синтезировали из 2-метил-5-трет-бутилбензолсульфонил хлорида, используя последовательность стадий, описанную в методике получения соединения 17. Соединение получали в виде желтого твердого вещества с общим выходом 10%.
ЖХВД: RT=6,33 мин, 95%.
1H ЯМР, ДМСО-d6, δ (м.д.): 1.38 (s, 9Н); 2.65 (s, 3Н); 7.66 (t, 1H); 7.72 (t, 1H); 7.88 (s, 1H); 8.05 (d, 2H); 8.12-8.17 (m, 4H); 8.65 (s, 1H); 15.69 (s, 1H, обменный).
Масс-спектр (ESI+): m/z 422 (МН+, 100%).
Примеры 123-130

Соединения 123-130 синтезировали из сахарина и соответствующих 2-бром-1-арилэтанонов, используя последовательность стадий, описанную в методике получения соединения 1 (для 123, 125, 127 и 128), соединения 8 (для 124 и 126) и соединения 17 (для 129 и 130).
Примеры 131-143

Соединения 131-143 синтезировали из сахарина и соответствующих 2-бром-1-арилэтанонов, используя последовательность стадий, описанную в методике получения соединения 1 (R2=Me) или соединения 8 (R2=Et).










Примеры 144-146

Соединения 144-146 синтезировали из сахарина и соответствующих 2-бром-1-арилэтанонов, используя последовательность стадий, описанную в методике получения соединения 1.
2-Бром-1-арилэтаноны получали путем бромирования соответствующих арилэтанонов в соответствии с описанной выше методикой получения соединения 144А. 1-(3,4-Диметилфенил)этанон (2,5 г, 16,9 ммоль) растворяли в ТГФ (42 мл) при комнатной температуре в атмосфере азота. Добавляли трифторуксусную кислоту (1,5 мл, 16,9 ммоль) и затем трибромид пиридиния (6,5 г, 20,2 ммоль). Раствор становился багряно-красным, и постепенно формировался белый осадок. Реакционную смесь перемешивали в течение трех часов при комнатной температуре, затем нейтрализовали путем добавления воды (50 мл) и затем экстрагировали этилацетатом (100 мл). Органическую фазу промывали насыщенным раствором CuSO4 (40 мл), насыщенным раствором NaCl (40 мл) и затем сушили сульфатом магния, фильтровали и концентрировали при пониженном давлении. Остаток очищали на колонке силикагеля (130) градиентом 0%-5% этилацетата в гептане с получением двух порций 2-бром-1-(3,4-диметил-фенил)-этанона (144А. 57%).
Порция 1: 1,25 г, ЖХВД: RT=4,90 мин, 90%.
Порция 2: 1,90 г, ЖХВД: RT=4,90 мин, 70%.


Пример 147
Адамантан-2-ил-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-ил)-метанон

Пример 147А - Адамантан-2-карбонитрил
Адамантанон (2,5 г, 16,6 ммоль) растворяли в 1,2-диметоксиэтане (ДМЭ) (58 мл) в присутствии этанола (1,7 мл) и TosMIC (4,22 г, 21,6 ммоль) в атмосфере азота. Данную реакционную смесь охлаждали с помощью ледяной бани. Медленно добавляли трет-бутилат калия (5,72 г, 51 ммоль), поддерживая температуру реакционной смеси в диапазоне от 2 до 11°C. Реакционную смесь перемешивали в течение 30 мин, поддерживая температуру реакционной смеси в диапазоне от 5 до 12°C, затем нагревали до комнатной температуры и продолжали перемешивание в течение 2 ч. Затем реакционную смесь фильтровали, отфильтрованный белый осадок промывали ДМЭ. Фильтрат концентрировали. Полученный в результате остаток очищали на силикагеле (5% этилацетат в гептане) с выходом соединения 147А в виде белого твердого вещества (2,38 г, 88%).
1H ЯМР, ДМСО-d6, δ (м.д.): 1.65-1.95 (m, 12 Н); 2.07 (уширенный s, 2H); 3.14 (уширенный s, 1H).
Масс-спектр (ESI+): m/z 162 (MH+, 20%); 194 (MH+. МеОН, 100%).
Пример 147В - 1-Адамантан-2-ил-этанон
Соединение 147А (4,63 г, 28,7 ммоль) растворяли в диэтиловом эфире (61 мл) в атмосфере азота и затем охлаждали с помощью ледяной бани. Добавляли по каплям метиллитий (27 мл, 1,6 M/Et2O, 43 ммоль), поддерживая температуру реакционной смеси в диапазоне от 5°C до 12°C. Сразу после завершения добавления холодную баню удаляли и перемешивание продолжали в течение 30 мин при комнатной температуре. Затем реакционную смесь нейтрализовали путем добавления воды (46 мл). Отбирали органическую фазу, которую сушили сульфатом магния, сушили и концентрировали при пониженном давлении. Остаток переносили в смесь ацетона (28 мл) и 6 н. HCl (28 мл) и затем нагревали при температуре дефлегмации в течение 80 мин. Затем выпаривали ацетон и оставшуюся водную фазу дважды экстрагировали этилацетатом. Органические фазы объединяли, сушили сульфатом магния, фильтровали и концентрировали. Полученный в результате остаток очищали на колонке с силикагелем (90 г; 32 мл/мин; 6% этилацетат в гептане) с выходом соединения 147В в виде желтого твердого вещества (3,66 г, 71%).
1H ЯМР, ДМСО-d6, δ (м.д.): 1.45-1.55 (m, 2H); 1.65-1.90 (m, 10Н); 2.09 (s, 3H); 2.29 (уширенный s, 2H); 2.54 (уширенный s, 1Н).
Масс-спектр (ESI+): m/z 179 (МН+, 100%).
Пример 147С - 1-Адамантан-2-ил-2-бром-этанон
Соединение 147В (500 мг, 2,8 ммоль) растворяли в метаноле (8,6 мл) в атмосфере азота, и затем охлаждали до 0°C. Медленно добавляли бром (151 мкл, 2,94 ммоль). Данную реакционную смесь перемешивали в течение 1 ч 40 мин при 0°C и затем нейтрализовали путем добавления воды и дважды экстрагировали этилацетатом. Органические фазы объединяли, сушили сульфатом магния, фильтровали и концентрировали. Полученный в результате остаток очищали на колонке с силикагелем (35 г; 20 мл/мин; градиент 0%-15% этилацетата в гептане в течение 25 мин) с выходом соединения 147С (1,37 г, 85%).
1H ЯМР, ДМСО-d6, δ (м.д.): 1.5-1.6 (m, 2H); 1.65-1.90 (m, 10Н); 2.38 (уширенный s, 2H); 2.86 (уширенный s, 1Н); 4.45 (s, 2H).
Пример 147 - Адамантан-2-ил-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанон
Соединение 147 синтезировали из сахарина и соединения 147С, используя последовательность стадий, описанную в методике получения соединения 1 (выход после трех стадий: 11%).
Белое твердое вещество.
ЖХВД: RT=6,28 мин, 97%.
1H ЯМР, ДМСО-d6, δ (м.д.): 1.54-1.61 (m, 2H); 1.68-1.93 (m, 8H); 2.05-2.25 (m, 2H); 2.36 (уширенный s, 2H); 2.89 (s, 3H); 3.27 (s, 1Н); 7.93-7.96 (m, 3H); 8.08-8.11 (m, 1Н); 15.22 (s, 1Н, обменный).
Масс-спектр (ESI-): m/z 372 (М-Н-, 100%).
Пример 148
Хроман-6-ил-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-ил)-метанон

Пример 148А - Хроман
4-Хроманон (5,0 г, 33,7 ммоль) растворяли в ТГФ (102 мл) в атмосфере азота. Добавляли при комнатной температуре BF3.OEt2 (12,8 мл, 101 ммоль) и медленно добавляли цианоборгидрид натрия (4,33 г, 67,4 ммоль) (бурная реакция). Полученную в результате белую суспензию нагревали до 65°C в течение 18 ч и затем нейтрализовали водой. Реакционную смесь дважды экстрагировали этилацетатом. Органические фазы объединяли, промывали последовательно насыщенным раствором NaHCO3 и насыщенным раствором NaCl и затем сушили сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный в результате остаток очищали на силикагеле (градиент 0%-50% дихлорметана в гептане и затем 10% этилацетат в гептане) с выходом частично очищенного соединения 148A (3,34 г, 61%).
ЖХВД: RT=4,56 мин, 83%.
1H ЯМР, ДМСО-d6, δ (м.д.): 1.91 (q, 2Н); 2.72 (t, 2Н); 4.11 (t, 2H); 6.70 (d, 1H); 6.80 (t, 1H); 7.0-7.05 (m, 2H).
Пример 148В - 1-Хроман-6-ил-этанон
Хроман (4,64 г, 29,4 ммоль) растворяли в 30 мл безводного дихлорметана (ДХМ) в атмосфере азота. Данную реакционную смесь охлаждали до -30°C и затем в течение 5 мин добавляли холодный (-10°C) раствор этаноил хлорида (4,75 мл, 67 ммоль) в безводном ДХМ (20 мл). Данную смесь перемешивали в течение 45 мин при -15°C и затем выливали на смесь льда (100 г) и концентрированной HCl (50 мл) и три раза экстрагировали ДХМ. Органические фазы объединяли, сушили сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный в результате остаток очищали на колонке с силикагелем (120 г; градиент 0%-20% EtOAc в гептане в течение 60 мин) с получением двух порций соединения 148В (70%).
Порция 1: 2,25 г, ЖХВД: RT=4,09 мин, 96,6%.
1H ЯМР, ДМСО-d6, δ (м.д.): 1.93 (q, 2H); 2.49 (s, 3H); 2.79 (t, 2H); 4.21 (t, 2H); 6.81 (d, 1H); 7.65-7.75 (m, 2H).
Масс-спектр (ESI+): m/z 177 (MH+, 100%).
Порция 2: 1,79 г, ЖХВД: RT=4,09 мин, 81%.
Пример 148С - 2-Бром-1-хроман-6-ил-этанон
Соединение 148С синтезировали из соединения 148В в соответствии с методикой получения соединения 144А (выход: 65%).
ЖХВД: RT=4,59 мин, 74%.
1H ЯМР, ДМСО-d6, δ (м.д.): 1.95 (q, 2H); 2.80 (t, 2H); 4.20 (t, 2H); 4.80 (s, 2H); 6.85 (d, 1Н); 7.70-7.85 (m, 2H).
Пример 148 - Хроман-6-ил-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанон
Соединение 148 синтезировали из сахарина и соединения 148С, используя последовательность стадий, описанную в методике получения соединения 1, с общим выходом 36%.
ЖХВД: RT=5,45 мин, 98%.
1H ЯМР, ДМСО-d6, δ (м.д.): 1.98 (t, 2H); 2.69 (s, 3H); 2.84 (t, 2H); 4.27 (t, 2H); 6.96 (d, 1Н); 7.89 (s, 1Н); 7.97-8.02 (m, 4H); 8.17-8.18 (m, 1Н); 16.02 (s, 1Н, обменный).
Масс-спектр (ESI+): m/z 372 (МН+, 100%).
Пример 149
(4-Хлор-3-трифторметил-фенил)-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-ил)-метанон

Пример 149А - 1-(4-Хлор-3-трифторметил-фенил)-этанол
4-Хлор-3-трифторметил-бензальдегид (6,19 г, 29,7 ммоль) растворяли в ТГФ (124 мл) в атмосфере азота. Данную реакционную смесь охлаждали до -78°C, затем добавляли по каплям MeMgBr (13 мл, 3M/Et2O, 38,6 ммоль) и затем перемешивали в течение 2 ч при низкой температуре (-78°C), в заключение нейтрализовали путем добавления насыщенного раствора NH4Cl (60 мл). Затем реакционную смесь дважды экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором NaCl и затем сушили сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный в результате остаток очищали на колонке с силикагелем (120 г; 92 мл/мин; градиент 0%-35% этилацетата в гептане в течение 40 мин) с выходом соединения 149А (5,71 г, 62%).
ЖХВД: RT=5,71 мин, 98% (колонка XBridge).
1H ЯМР, ДМСО-d6, δ (м.д.): 1.33 (а, 3Н); 4.81 (quintet, 1H); 5.46 (d, 1H, обменный); 7.60-7.69 (m, 2H); 7.81 (s, 1H).
Пример 149В - 1-(4-Хлор-3-трифторметил-фенил)-этанон
Соединение 149А (2,62 г, 11,7 ммоль) растворяли в ДХМ (53 мл) в присутствии целита (3,8 г). Добавляли при комнатной температуре РСС (хлорхромат пиридиния) (3,77 г, 17,5 ммоль) и данную реакционную смесь перемешивали в течение ночи и затем фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали на колонке с силикагелем (80 г; 32 мл/мин; градиент 0%-30% EtOAc в гептане в течение 26 мин) с выходом соединения 149В (2,27 г, 87%).
ЖХВД: RT=5,84 мин, 97% (колонка XBridge).
1H ЯМР, ДМСО-d6, δ (м.д.): 2.65 (s, 3Н); 7.92 (d, 1H); 8.22-8.27 (m, 2H).
Пример 149С - 2-Бром-1-(4-хлор-3-трифторметил-фенил)-этанон
Соединение 149С синтезировали из соединения 149В в соответствии с методикой получения соединения 144А (выход: 66%).
ЖХВД: RT=6,18 мин, 89% (колонка XBridge).
1H ЯМР, ДМСО-d6, δ (м.д.): 5.05 (s, 2H); 7.96 (d, 1H); 8.28 (d, 1H); 8.34 (s, 1H).
Пример 149 - (4-Хлор-3-трифторметил-фенил)-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанон
Соединение 149 синтезировали из сахарина и соединения 149С, используя последовательность стадий, описанную в методике получения соединения 1, с общим выходом 20%.
ЖХВД: RT=6,71 мин, 99% (колонка XBridge).
1H ЯМР, ДМСО-d6, δ (м.д.): 2.68 (s, 3H); 7.98-8.04 (m, 4H); 8.18-8.21 (m, 1H); 8.29 (d, 1H); 8.40 (s, 1H); 14.82 (s, 1H, обменный).
Масс-спектр (ESI-): m/z 416 (M-H-, 100%); 418 (M-H-, 25%).
Пример 150
(7-Бром-4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-ил)-нафталин-2-ил-метанон

Пример 150А - 4-Бром-2-сульфамоил-бензойная кислота
5-Бром-2-метил-бензолсульфонамид (2,5 г, 9,99 ммоль) растворяли в 5% водном растворе карбоната натрия (62 мл). Данную реакционную смесь нагревали до 100°C, и постепенно, в течение 15 мин, добавляли КМnO4 (6,43 г, 25 ммоль). Нагревание продолжали в течение 140 мин. Реакционную смесь охлаждали до комнатной температуры и затем фильтровали. рН фильтрата доводили до 1,2 концентрированным раствором HCl и затем фильтровали. Осадок промывали этилацетатом. Две фазы фильтрата разделяли и полученную водную фазу один раз экстрагировали этилацетатом. Органические фазы собирали, сушили сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением соединения 150А (1,64 г, 58%).
ЖХВД: RT=0,29 мин, 99,5%.
1H ЯМР, ДМСО-d6, δ (м.д.): 7.35 (s, 2Н, обменный); 7.67 (d, 1H); 7.91 (d, 1H); 8.08 (s, 1H); 13.83 (уширенный s, 1H, обменный).
Масс-спектр (ESI-): m/z 278 (M-H-, 100%); 280 (M-H-, 87%).
Пример 150В - 6-Бром-1,1-диоксо-1,2-дигидро-2H-бензо[d]изотиазол-3-он
Соединение 150А (1,59 г, 5,56 ммоль) растворяли в концентрированной серной кислоте (6 мл) при комнатной температуре и данную реакционную смесь перемешивали в течение 3 ч, затем выливали на лед. Полученную суспензию фильтровали. Осадок промывали три раза водой и затем сушили в течение 24 ч при 50°C под вакуумом. Соединение 150В (1,33 г, 89%) получали в виде белого твердого вещества.
ЖХВД: RT=3,16 мин, 96%.
1H ЯМР, ДМСО-d6, δ (м.д.): 7.85 (d, 1Н); 8.08 (d, 1H); 8.48 (s, 1H).
Масс-спектр (ESI-): m/z 260 (M-H-, 100%); 262 (M-H-, 94%).
Пример 150 - (7-Бром-4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-ил)-нафталин-2-ил-метанон
Соединение 150 синтезировали из соединения 150В и 2-бром-1-(нафталин-2-ил)этанона, используя последовательность стадий, описанную в методике получения соединения 1, с общим выходом 55%.
ЖХВД: RT=6,14 мин, 97%.
1H ЯМР, ДМСО-d6, δ (м.д.): 2.68 (s, 3Н); 7.66 (t, 1H); 7.73 (t, 1H); 8.04-8.15 (m, 5H); 8.19-8.22 (m, 2H); 8.65 (s, 1H); 15.38 (s, 1H, обменный).
Масс-спектр (ESI+): m/z 444 (MH+, 100%); 446 (MH+, 99%).
Пример 151
(7-Хлор-4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-ил)-нафталин-2-ил-метанон

Пример 151А - 6-Хлор-1,1-диоксо-1,2-дигидро-2Н-бензо[d]изотиазол-3-он
Метил 2-амино-4-хлорбензоат (5 г, 26,9 ммоль) нагревали в 20% водном растворе HCl (18 мл) до полного растворения и затем охлаждали до 0°C. Добавляли по каплям раствор NaNO2 (1,85 г, 26,9 ммоль) в воде (4,5 мл), поддерживая температуру в диапазоне от 2°C до 6°C. Затем реакционную смесь перемешивали в течение 1 ч при комнатной температуре. В другой колбе смесь уксусной кислоты (22 мл) и воды (2,3 мл) при 0°C барботировали газообразным SO2 (приблизительно 15 г). Затем добавляли CuCl (666 мг, 6,7 ммоль). Затем к полученному сине-зеленому раствору при температуре в диапазоне от 1°C до 3°C добавляли реакционную смесь из первой колбы. Наблюдалось выделение газа; перемешивание при низкой температуре продолжали в течение 45 мин, затем охлаждающую баню удаляли и реакционную смесь выливали на лед (100 г) и три раза экстрагировали этилацетатом. Органические фазы собирали, промывали насыщенным раствором NaHCO3, сушили сульфатом магния, фильтровали и концентрировали при пониженном давлении. Остаток переносили в ТГФ (5 мл) при 0°C и медленно добавляли концентрированный раствор аммиака (2,8 мл). Охлаждающую баню удаляли и перемешивание продолжали в течение 1 ч. Затем реакционную смесь концентрировали, обрабатывали насыщенным раствором NaHCO3, один раз промывали диэтиловым эфиром и затем рН доводили снова до 1 концентрированным раствором HCl. Образовавшийся осадок отделяли фильтрованием, промывали водой и сушили при пониженном давлении при 50°C с получением соединения 151А (896 мг, 15%).
ЖХВД: RT=3,07 мин, 99%.
1H ЯМР, ДМСО-d6, δ (м.д.): 7.94 (уширенный d, 2Н); 8.38 (s, 1H).
Масс-спектр (ESI-): m/z 216 (M-H-, 100%); 218 (М-Н-, 32%).
Пример 151 - (7-Хлор-4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-ил)-нафталин-2-ил-метанон
Соединение 151 синтезировали из соединения 151А и 2-бром-1-(нафталин-2-ил)этанона, используя последовательность стадий, описанную в методике получения соединения 1, с общим выходом 39%.
ЖХВД: RT=6,13 мин, 95%.
1H ЯМР, ДМСО-d6, δ (м.д.): 2.68 (s, 3Н); 7.66 (t, 1H); 7.74 (t, 1H); 8.03-8.16 (m, 6H); 8.21 (d, 1H); 8.65 (s, 1H); 15.41 (se, 1H, обменный).
Масс-спектр (ESI-): m/z 398 (M-H-, 100%); 400 (M-H-, 32%).
Пример 152
(4-Гидрокси-2,7-диметил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-ил)-нафталин-2-ил-метанон

Пример 152А - 2-Циано-5-метил-бензолсульфонамид
2-Амино-4-метил-бензонитрил (2,5 г, 18,9 ммоль) нагревали в 20% водном растворе HCl (12 мл) до полного растворения и затем охлаждали до 0°C. Добавляли по каплям раствор NaNO2 (1,3 г, 18,9 ммоль) в воде (3,2 мл), поддерживая температуру в диапазоне от 2°C до 6°C. Затем реакционную смесь перемешивали в течение 1 ч при комнатной температуре. В другой колбе смесь уксусной кислоты (15 мл) и воды (1,6 мл) при 0°C барботировали газообразным SO2 (приблизительно 15 г). Затем добавляли CuCl (468 мг, 4,7 ммоль). Затем к полученному сине-зеленому раствору при температуре в диапазоне от 1°C до 3°C добавляли реакционную смесь из первой колбы. Перемешивание при низкой температуре продолжали в течение 45 мин, затем удаляли охлаждающую баню и затем реакционную смесь выливали на лед (70 г) и три раза экстрагировали смесью 20% метанол/ДХМ. Органические фазы собирали, промывали насыщенным раствором NaHCO3, сушили сульфатом магния, фильтровали и концентрировали при пониженном давлении. Остаток переносили в ТГФ (5 мл) при 0°C и медленно добавляли концентрированный раствор аммиака (2,8 мл). Охлаждающую баню удаляли и перемешивание продолжали в течение 1 ч. Затем реакционную смесь концентрировали, обрабатывали насыщенным раствором NaHCO3, один раз промывали диэтиловым эфиром и затем рН доводили снова до 1 концентрированным раствором HCl. Образовавшийся осадок отделяли фильтрованием, промывали водой и сушили при пониженном давлении при 50°C с получением соединения 152А (800 мг, 21%).
ЖХВД: RT=3,67 мин, 99% (колонка XBridge).
1H ЯМР, ДМСО-d6, δ (м.д.): 7.61 (d, 1Н); 7.77 (s, 1H); 7.99 (d, 1H); 8.77 (уширенный s, 2H).
Macc-спекгр (ESI+): m/z 197 (MH+, 100%).
Пример 152В - 4-Метил-2-сульфамоил-бензойная кислота
Соединение 152А (620 мг, 3,15 ммоль) растворяли в 30% водном растворе KOH (7,5 мл), содержащем перекись водорода (530 мкл 30% водного раствора). Данную реакционную смесь нагревали при температуре дефлегмации в течение 4 ч и затем охлаждали до комнатной температуры, рН доводили снова до 1 концентрированным раствором HCl и три раза экстрагировали смесью 20% метанол/ДХМ. Органические фазы собирали, сушили сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением соединения 152В (428 г, 60%).
ЖХВД: RT=4,11 мин, 95% (колонка XBridge).
1H ЯМР, ДМСО-d6, δ (м.д.): 2.41 (s, 3Н); 7.19 (уширенный s, 2H, обменный); 7.48 (d, 1H); 7.65 (d, 1H); 7.77 (s, 1H); 12.5-14.5 (уширенный s, 1H, обменный).
Масс-спектр (ESI-): m/z 214 (M-H-, 100%).
Пример 152С - 6-Метил-1,1-диоксо-1,2-дигидро-2Н-бензо[d]изотиазол-3-он
Соединение 152В (428 мг, 1,94 ммоль) растворяли в концентрированной серной кислоте (3 мл) при комнатной температуре. Данную реакционную смесь перемешивали в течение 2 ч и затем выливали на лед и фильтровали. Осадок промывали большим количеством воды и затем сушили с получением соединения 152С (359 мг, 93%) в виде розового твердого вещества.
ЖХВД: RT=4,00 мин, 96% (колонка XBridge).
1H ЯМР, ДМСО-d6, δ (м.д.): 7.74 (d, 1Н); 7.89 (d, 1Н); 8.00 (s, 1H).
Масс-спектр (ESI-): m/z 196 (М-Н-, 100%).
Пример 152 - (4-Гидрокси-2,7-диметил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-ил)-нафталин-2-ил-метанон.
Соединение 152 синтезировали из соединения 152С и 2-бром-1-(нафталин-2-ил)этанона, используя последовательность стадий, описанную в методике получения соединения J., с общим выходом 21%.
ЖХВД: RT=6,87 мин, 98% (колонка XBridge).
1H ЯМР, ДМСО-d6, δ (м.д.): 2.55 (s, 3Н); 2.64 (s, 3Н); 7.66 (t, 1Н); 7.72 (t, 1Н); 7.80 (d, 1Н); 7.84 (s, 1Н); 8.05 (d, 1Н); 8.11-8.11 (m, 4H); 8.67 (s, 1Н); 15.75 (s, 1Н, обменный).
Масс-спектр (APCI+ (химическая ионизация при атмосферном давлении)): m/z 380(MH+, 24%).
Пример 153
Бифенил-3-ил-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-ил)-метанон

Пример 153 - Бифенил-3-ил-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-ил)-метанон
Соединение 142 (200 мг, 0,5 ммоль) растворяли в ацетоне (1,1 мл) и воде (1,2 мл) в присутствии фенилбороновой кислоты (68 мг, 0,55 ммоль), карбоната калия (175 мг, 1,27 ммоль) и ацетата палладия (5 мг, 0,02 ммоль) в атмосфере инертного газа. Данную реакционную смесь нагревали до 85°C в течение 1 ч и 30 мин и затем охлаждали до комнатной температуры, разбавляли водой и три раза экстрагировали ДХМ. Органические фазы объединяли, сушили сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный в результате остаток очищали на колонке с силикагелем (12 г), используя для элюирования ДХМ, с выходом соединения 153 (141 мг, 68%).
ЖХВД: RT=7,07 мин, 96% (колонка XBridge).
1H ЯМР, ДМСО-d6, δ (м.д.): 2.68 (s, 3H); 7.44 (t, 1H); 7.53 (t, 2H); 7.69-7.76 (m, 3Н); 7.97-8.04 (m, 5H); 8.21 (уширенный s, 1H); 8.31 (s, 1H); 15.36 (уширенный s, 1H, обменный).
Масс-спектр (ESI-): m/z 390 (M-H-, 100%).
Примеры 154-169

Соединения 154-169 синтезировали из соединения 142 и различных бороновых кислот в соответствии с описанной выше методикой получения соединения 153.












Пример 170
(4-Гидрокси-7-метансульфонил-2-метил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-ил)-нафталин-2-ил-метанон

Пример 170 - (4-Гидрокси-7-метансульфонил-2-метил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-ил)-нафталин-2-ил-метанон
Соединение 170 синтезировали из 5-метансульфонил-2-метил-бензолсульфонил хлорида, используя последовательность стадий, описанную в методике получения соединения 17, с общим выходом 7%.
ЖХВД: RT=5,43 мин, 97%.
1H ЯМР, ДМСО-d6, δ (м.д.): 2.71 (s, 3Н); 3.47 (s, 3Н); 7.67 (t, 1H); 7.73 (t, 1H); 8.05-8.15 (m, 4H); 7.42-8.49 (m, 3Н); 8.67 (s, 1H); 14.76 (se, 1H, обменный).
Масс-спектр (ESI+): m/z 444 (MH+, 100%).
Пример 171
(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2Я-бензо[е][1,2]тиазин-3-ил)-(1-фенил-циклопропил)-метанон

Пример 171А - 1-(1-Фенил-циклопропил)-этанон
Соединение 171А синтезировали из 1-фенил-циклопропанкарбонитрила в соответствии с описанной выше методикой получения соединения 147В (выход: 40%).
ЖХВД: RT=4,30 мин, 96% (колонка XBridge).
1H ЯМР, ДМСО-d6, δ (м.д.): 1.15 (dd, 2H); 1.47 (dd, 1H); 1.92 (s, 3H); 7.23-7.40 (m, 5H).
Масс-спектр (ESI+): m/z 161 (MH+, 100%).
Пример 171 - (4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-ил)-(1-фенил-циклопропил)-метанон
Соединение 171 синтезировали из соединения 171А, используя последовательность стадий, описанную в методике получения соединения 144, с общим выходом 6%.
ЖХВД: RT=6,40 мин, 96% (колонка XBridge).
1H ЯМР, ДМСО-d6, δ (м.д.): 1.38-1.41 (m, 2H); 1.65-1.68 (m, 2H); 2.46 (s, 3H); 7.25-7.37 (m, 5H); 7.78-7.83 (m, 1H); 7.86-7.91 (m, 2H); 8.06-8.08 (m, 1H), 15.31 (s, 1H, обменный).
Масс-спектр (ESI+): m/z 356 (МН+, 100%).
Пример 172
1-[3-(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-карбонил)-фенил]-этанон

Пример 172 - 1-[3-(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-карбонил)-фенил]-этанон
Соединение 172 синтезировали из 1-(3-ацетил-фенил)-этанона, используя последовательность стадий, описанную в методике получения соединения 144, с общим выходом 21%.
ЖХВД: RT=6,25 мин, 97% (колонка XBridge).
1H ЯМР, ДМСО-d6, δ (м.д.): 2.64 (s, 3H); 2.66 (s, 3H); 7.79 (t, 1H); 8.00 (уширенный s, 3H); 8.15-8.35 (m, 3H); 8.58 (s, 1H); 15.21 (уширенный s, 1H, обменный).
Масс-спектр (ESI-): m/z 356 (M-H-, 100%).
Пример 173
(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-[3-(2,2,2-трифтор-1-гидрокси-1-метил-этил)-фенил]-метанон

Пример 173А - 1-[3-(2,2,2-Трифтор-1-гидрокси-1-метил-этил)-фенил]-этанон
1-(3-Ацетил-фенил)-этанон (2,5 г, 15,4 ммоль) растворяли в ТГФ (120 мл) в атмосфере азота при 0°C в присутствии TMS-CF3 (2,7 мл, 18,4 ммоль). В течение 20 мин с помощью поршневого насоса добавляли TBAF (тетрабутиламмония фторид) (1МЛТФ, 18,4 мл, 18,4 ммоль). Охлаждающую баню удаляли и реакционную смесь перемешивали дополнительно течение 18 ч, затем нейтрализовали путем добавления насыщенного раствора NaHCO3 и в заключение 3 раза экстрагировали этилацетатом. Органические фазы объединяли, промывали водой и затем сушили сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный в результате остаток очищали на силикагеле (градиент 0%-50% этилацетата в гептане в течение 20 мин) с выходом частично очищенного соединения 173А (2,87 г), которое использовали как есть на следующей стадии.
Пример 173В - 2-Бром-1-[3-(2,2,2-трифтор-1-гидрокси-1-метил-этил)-фенил]-этанон
Соединение 173В синтезировали из соединения 173А в соответствии с методикой получения соединения 144А (выход: 57%).
ЖХВД: RT=5,50 мин, 74% (колонка XBridge).
1H ЯМР, ДМСО-d6, δ (м.д.): 1.73 (s, 3H); 4.96 (s, 2H); 6.81 (уширенный s, 1H, обменный); 7.60 (t, 1H); 7.89 (d, 1H); 8.03 (d, 1H); 8.17 (s, 1H).
Пример 173 - (4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-ил)-[3-(2,2,2-трифтор-1-гидрокси-1-метил-этил)-фенил]-метанон
Соединение 173 синтезировали из сахарина и соединения 173В, используя последовательность стадий, описанную в методике получения соединения 1, с общим выходом 52%.
ЖХВД: RT=6,05 мин, 95% (колонка XBridge).
1H ЯМР, ДМСО-d6, δ (м.д.): 1.76 (s, 3Н); 2.61 (s, 3Н); 6.84 (s, 1H, обменный); 7.67 (t, 1H); 7.90-8.05 (m, 5H); 8.17-8.23 (m, 1H); 8.36 (s, 1H); 15.47 (уширенный s, 1H, обменный).
Масс-спектр (ESI+): m/z 445 (MNH4+, 100%).
Пример 174
(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-ил)-[3-(2,2,2-трифтор-1-гидрокси-этил)-фенил]-метанон

Пример 174А - 1-[3-(2,2,2-Трифтор-1-триметилсиланилокси-этил)-фенил]-этанон
3-Ацетилбензальдегид (1,27 г, 8,57 ммоль) растворяли в ДМФА (30 мл) в атмосфере азота в присутствии карбоната калия (59 мг, 0,42 ммоль) и ТМS-CF3 (1,52 мл, 10,3 ммоль). Данную реакционную смесь перемешивали в течение 30 мин при комнатной температуре, затем нейтрализовали путем добавления насыщенного раствора NH4Cl (1 мл) и концентрировали при пониженном давлении. Остаток переносили в этилацетат и затем один раз промывали 1 н. раствором HCl в воде, сушили сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением частично очищенного соединения 174А (2,55 г), которое использовали как есть на следующей стадии.
ЖХВД: RT=5,15 мин, 42% (ОН) в течение 6,91 мин, 40% (OTMS) (колонка XBridge, частичное снятие защитных групп на колонке).
1H ЯМР, ДМСО-d6, δ (м.д.): 0.09 (s, 9Н); 2.60 (s, 3Н); 5.60 (q, 1H); 7.60 (t. 1H); 7.76 (d, 1H); 8.02 (d, 1H); 8.08 (s, 1H).
Масс-спектр (ESI+): m/z 291 (MH+, 100%).
Пример 174В - 2-Бром-1-[3-(2,2,2-трифтор-1-гидрокси-этил)-фенил]-этанон
Соединение 174В синтезировали из соединения 174А в соответствии с методикой получения соединения 144А (выход: 60%).
ЖХВД: RT=5,23 мин, 83% (колонка XBridge).
1H ЯМР, ДМСО-d6, δ (м.д.): 4.93 (s, 2H); 5.32 (q, 1H); 7.03 (уширенный s, 1H, обменный); 7.61 (t, 1H); 7.81 (d, 1H); 8.05 (d, 1H); 8.11 (s, 1H).
Пример 174 - (4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-ил)-[3-(2,2,2-трифтор-1-гидрокси-этил)-фенил]-метанон
Соединение 174 синтезировали из сахарина и соединения 174В, используя последовательность стадий, описанную в методике получения соединения 1, с общим выходом 33%.
ЖХВД: RT=5,92 мин, 92% (колонка XBridge).
1H ЯМР, ДМСО-d6, δ (м.д.): 2.60 (s, 3Н); 5.32 (уширенный s, 1H); 7.05 (d, 1H, обменный); 7.67 (t, 1H); 7.80 (d, 1H); 7.98 (уширенный s, 3Н); 8.08 (d, 1H); 8.20 (уширенный s, 2H); 15.45 (уширенный s, 1H, обменный).
Масс-спектр (ESI+): m/z 431 (MNH4+, 100%).
Пример 175
3-(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-карбонил)-бензойная кислота

Пример 175 - 3-(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-карбонил)-бензойная кислота
Соединение 143 (100 мг, 0,29 ммоль) растворяли в 30% растворе КОН в воде (1 мл) в присутствии этанола (330 мкл) и затем нагревали до 70°C в течение 18 ч. Данную реакционную смесь разбавляли водой (10 мл), дважды промывали диэтиловым эфиром, рН доводили снова до 26 н. водным раствором HCl и в заключение смесь три раза экстрагировали 20% раствором метанола в ДХМ. Органические фазы объединяли, сушили сульфатом магния, фильтровали и концентрировали с получением соединения 175 в виде желтого твердого вещества (99 мг, 93%).
ЖХВД: RT=5,49 мин, 98% (колонка XBridge).
1H ЯМР, ДМСО-d6, δ (м.д.): 2.64 (s, 3H); 7.77 (t, 1H); 7.99-8.00 (m, 3H); 8.20-8.24 (m, 2H); 8.28 (d, 1H); 8.61 (s, 1H); 13.33 (s, обменный, 1Н); 15.32 (s, обменный, 1H).
Масс-спектр (ESI-): m/z 358 (M-H-, 100%).
Пример 176
3-(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-карбонил)-N-метил-бензамид

Пример 176 - 3-(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-карбонил)-N-метил-бензамид
Соединение 175 (150 мг, 0,41 ммоль) растворяли в ДМФА (3 мл) в присутствии (3-диметиламино-пропил)-этил-карбодиимида гидрохлорида (120 мг, 0,62 ммоль), 3-гидрокси-3Н-бензо[d][1,2,3]триазин-4-она (102 мг, 0,62 ммоль) и iPr2NEt (162 мг, 1,25 ммоль) в атмосфере инертного газа и затем перемешивали в течение 72 ч при комнатной температуре. Затем реакционную смесь концентрировали при пониженном давлении, разбавляли ДХМ (20 мл) и дважды промывали 1 н. водным раствором HCl. Водные фазы собирали и экстрагировали ДХМ. Органические фазы объединяли, сушили сульфатом магния, фильтровали и концентрировали. Полученный в результате остаток очищали на колонке с силикагелем (12 г; 12 мл/мин; градиент 0%-25% ацетона в ДХМ в течение 20 мин) с выходом соединения 176 в виде желтого твердого вещества (101 мг, 64%).
ЖХВД: RT=5,21 мин, 97% (колонка XBridge).
1H ЯМР, ДМСО-d6, δ (м.д.): 2.63 (s, 3H); 2.82 (d, 3H); 7.72 (t, 1H), 7.99 (d, 3H); 8.10 (d, 1H); 8.20-8.22 (m, 2H); 8.42 (s, 1H); 8.66 (d, 1H); 15.35 (уширенный s, 1H, обменный).
Масс-спектр (ESI+): m/z 373 (MH+, 100%).
Примеры 177-183

Соединения 177-183 синтезировали из соединения 175 и различных аминов в соответствии с описанной выше методикой получения соединения 176.







Пример 184
3-(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-карбонил)-бензамид

Пример 184 - 3-(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-карбонил)-бензамид
Соединение 175 (150 мг, 0,42 ммоль) растворяли в ТГФ (3 мл) в присутствии РуВОР (239 мг, 0,46 ммоль), аммиака (152 мкл, 1,25 ммоль) и DIEA (80 мкл, 0,46 ммоль) и перемешивали в течение 4 ч при комнатной температуре. Затем реакционную смесь разбавляли ДХМ (20 мл) и промывали 1 н. водным раствором HCl. Водную фазу четыре раза экстрагировали ДХМ. Органические фазы объединяли, сушили сульфатом магния, фильтровали и концентрировали с получением соединения 184 в виде желтого твердого вещества (71 мг, 46%).
ЖХВД: RT=5,06 мин, 97,4% (колонка XBridge).
1H ЯМР, ДМСО-d6, δ (м.д.): 2.63 (s, 3Н); 7.57 (s, 1Н); 7.71 (t, 1H); 7.99 (d, 3H); 8.14-8.23 (m, 4H); 8.46 (s, 1H); 15.36 (уширенный s, обменный, 1H).
Масс-спектр (ESI+): m/z 359 (MH+, 100%).
Пример 185
3-(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-карбонил)-бензойной кислоты этиловый эфир

Пример 185 - 3-(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-карбонил)-бензойной кислоты этиловый эфир
Соединение 175 (150 мг, 0,42 ммоль) растворяли в этаноле (6 мл) в присутствии pTsOH (8 мг, 0,04 ммоль) и нагревали при температуре дефлегмации в течение 18 ч при перемешивании. Затем реакционную смесь концентрировали при пониженном давлении. Полученный в результате остаток очищали на колонке с силикагелем (12 г; 12 мл/мин; ДХМ) с выходом соединения 185 в виде желтого твердого вещества (138 мг, 84%).
ЖХВД: RT=6,28 мин, 95,8% (колонка XBridge).
1H ЯМР, ДМСО-d6, δ (м.д.): 1.36 (t, 3Н); 2.64 (s, 3Н); 4.37 (q, 2Н); 7.79 (t, 1H); 8.00 (d, 3Н); 8.20-8.30 (m, 3Н); 8.65 (s, 1H), 15.23 (уширенный s, обменный, 1H).
Масс-спектр (ESI+): m/z 405 (МН+, 100%).
Пример 186
(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-ил)-(3-пиридин-3-ил-фенил)-метанон
Пример 186 - (4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2Н-бензо[е][1,2]тиазин-3-ил)-(3-пиридин-3-ил-фенил)-метанон
Соединение 142 (200 мг, 0,5 ммоль) растворяли в 1,4-диоксане (1,5 мл) в присутствии пиридин-3-илбороновой кислоты (104 мг, 0,76 ммоль), ортофосфата калия (1,27 моль/л, 679 мкл, 0,86 ммоль), трис(дибензилиденацетон)дипалладия (23 мг, 0,025 ммоль) и трицикло-гексилфосфина (21 мг, 0,076 ммоль) в атмосфере инертного газа. Данную реакционную смесь нагревали до 100°C в течение 18 ч и затем охлаждали до комнатной температуры, разбавляли ДХМ и промывали насыщенным раствором NH4Cl. Органическую фазу сушили сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный в результате остаток очищали на колонке с силикагелем (12 г; 16 мл/мин; градиент 0%-5% метанола в течение 7 мин) с выходом соединения 186 (128 мг, 62%).
ЖХВД: RT=5,07 мин, 97,5% (колонка XBridge).
1H ЯМР, ДМСО-d6, δ (м.д.): 2.67 (s, 3Н); 7.77-7.82 (m, 3Н); 7.98-8.01 (m, 3H); 8.11-8.15 (m, 2H); 8.22 (dd, 1H); 8.39 (s, 1H); 8.71 (d, 2H); 15.20 (уширенный s, 1H, обменный).
Масс-спектр (ESI+): m/z 393 (MH+, 100%).
Примеры 187-195

Соединения 187-195 синтезировали из соединения 144 и различных бороновых кислот в соответствии с описанной выше методикой получения соединения 186.









Примеры 196 и 197

Соединения 196 и 197 синтезировали из сахарина и из 2-бром-1-(3-хлорфенил)этанона и 2-бром-1-(3-фторфенил)этанона, соответственно, с использованием описанной выше методики получения соединения 1.
В модельных экспериментах, результаты которых приведены ниже, показано, что соединения по настоящему изобретению избирательно ингибируют 11β-HSD1 в сравнении с 11β-HSD2.
1) Ферментативная активность 11β-HSD1 из макросом печени человека после обработки ингибирующими соединениями (% ингибирования)
Данный ферментативный тест основан на превращении кортизона в кортизол с помощью 11β-HSD1. Ферментативную реакцию запускали путем добавления в лунки 96-луночных планшетов с половинным объемом лунок, содержащие 160 нмоль кортизона в 20 мМ Трис-буфере (рН 7,4), содержащем 5 мМ ЭДТА, 200 мкМ НАДФ(Н) (никотинамидадениндинуклеотидфосфат, восстановленный) и ингибирующее соединение или носитель (1% ДМСО), по 1 мкг препарата микросом печени человека (Xenotech) (объем реакционной смеси 50 мкл). Данные для построения калибровочной кривой по известным концентрациям кортизола получали одновременно с экспериментальными данными с использованием одних и тех же условий. Планшеты инкубировали в течение 2 ч при 37°C (ферментативная фаза). Ферментативная реакция может быть остановлена путем добавления 25 мкл конъюгата кортизол-d2 и 25 мкл меченого Eu3+-криптатом антитела против кортизола на лунку; после инкубации в течение 2 ч при комнатной температуре количество образовавшегося кортизола (фаза регистрации) может быть определено с использованием HTRF® (CISBIO international, кит 62CO2PEC). Измерение флюоресценции проводили с помощью ридера Fusion™ α (Perkin Elmer). Флюоресценцию каждой лунки измеряли при 620 нм и при 665 нм. Вычисляли соотношение (λ665 нм/λ620 нм) и специфический FRET-сигнал, по которому может быть определен процент ингибирования для каждой концентрации анализируемого ингибирующего соединения.
Ссылки (постеры):
IC50 determination of Carbenoxolone and Glycyrrhetinic acid on 11-beta hydroxysteroid dehydrogenase type 1 activity by HTRF®: C.Tokuda et al., Screentech, March 2004, San Diego (USA)
New Cortisol assay for 11-beta hydroxysteroid dehydrogenase type 1 activity using a new HTRF® acceptor, d2: M.Amoravain et al., SBS, 12th Annual Conference, September 2006, Seatle (USA)
2) Ферментативная активность 11β-HSD1 из микросом печени человека после обработки ингибирующими соединениями (% ингибирования или EC50)
Данный ферментативный тест основан на превращении [3H]-кортизона в [3H]-кортизол с помощью 11β-HSD1. Ферментативную реакцию запускали путем добавления в лунки 96-луночных планшетов Optiplate™; содержащие 20 нмоль [1,2-3H]-кортизона (удельная активность 40-50 Ки/ммоль, Amersham-GE Healthcare) в 50 мМ HEPES-буфере (рН 7,4), содержащем 100 мМ KCl, 5 мМ NaCl, 2 мМ MgCl2, 1 мМ НАДФ(Н) и ингибирующее соединение или носитель (1% ДМСО), по 1 мкг препарата микросом печени человека (Xenotech) (стандартизованное количество препарата, обеспечивающее превращение максимум 80% субстрата в используемых экспериментальных условиях) (объем реакционной смеси 50 мкл). Герметично закрытые планшеты центрифугировали на низкой скорости для перемешивания компонентов и затем инкубировали в течение 2 ч при 37°C (ферментативная фаза). Ферментативную реакцию останавливали путем добавления в каждую лунку 70 мкл комплекса (10 мг/мл), полученного путем предварительной инкубации иттрий-силикатных SPA-шариков, покрытых А-белком (GE Healthcare), с моноклональным антителом против кортизола (East Coast Biologies, ME), содержащего 10 мкмоль 18β-глицирритиновой кислоты. Планшеты герметично закрывали и затем инкубировали при слабом орбитальном перемешивании в течение 2 ч при комнатной температуре (фаза регистрации). После центрифугирования проводили измерения с использованием сцинтилляционного счетчика TopCount NXT (Perkin Elmer). Для каждой концентрации анализируемого соединения вычисляли процент ингибирования ферментативной активности относительно стандартной ферментативной активности (носитель: 1% ДМСО); затем на основе этих данных определяли активность каждого соединения (значение ЕС50 (концентрация, при которой достигается 50% эффект), вычисляемое с помощью программы SigmaPlot v.11 с использованием 4-х параметрического логистического уравнения).
Ссылки:
Development and application of a scintillation proximity assay (SPA) for identification of selective inhibitors of 11β-hydroxysteroid dehydrogenase type 1: S.Mundt et al., ASSAY and Drug Development Technologies, volume 3, number 4, 367-375, 2005 High-throughput screening of 11β-hydroxysteroid dehydrogenase type 1 in scintillation proximity assay format: K.Solly et al., ASSAY and Drug Development Technologies, volume 3, number 4, 377-384, 2005
3) Ферментативная активность 11β-HSD2 из макросом почек человека после обработки ингибирующими соединениями (% ингибирования)
Данный ферментативный тест основан на превращении [3H]-кортизола в [3Н]-кортизон с помощью 11β-HSD2. Ферментативную реакцию запускали путем добавления в лунки 96-луночных планшетов Optiplate™, содержащие 8 нМ [1,2,6,7-3H] кортизол (удельная активность 70-75 Ки/ммоль, Amersham-GE Healthcare) в 50 мМ HEPES-буфере (рН 7,4), содержащем 100 мМ KCl, 5 мМ NaCl, 2 мМ MgCl2, 1 мМ NAD+ и ингибирующее соединение или носитель (1% ДМСО), по 0,75 мкг микросом почек человека (Xenotech) (стандартизованное количество препарата, обеспечивающее превращение максимум 80% субстрата в используемых экспериментальных условиях) (объем реакционной смеси 50 мкл). Герметично закрытые планшеты центрифугировали на низкой скорости для перемешивания компонентов и затем инкубировали в течение 2 ч при 37°C (ферментативная фаза). Ферментативную реакцию останавливали путем добавления в каждую лунку 70 мкл комплекса (10 мг/мл), полученного путем предварительной инкубации иттрий-силикатных SPA-шариков, покрытых А-белком (GE Healthcare), с моноклональным антителом против кортизола (East Coast Biologies, ME), содержащего 10 мкмоль 18β-глицирритиновой кислоты. Планшеты герметично закрывали и затем инкубировали при слабом орбитальном перемешивании в течение 2 ч при комнатной температуре (фаза регистрации). После центрифугирования проводили измерения с использованием сцинтилляционного счетчика TopCount NXT (Perkin Elmer). Для каждой концентрации анализируемого соединения вычисляли процент ингибирования ферментативной активности относительно стандартной ферментативной активности (носитель: 1% ДМСО).
Ссылки:
Development and application of a scintillation proximity assay (SPA) for identification of selective inhibitors of 11β-hydroxysteroid dehydrogenase type 1: S.Mundt et al., ASSAY and Drug Development Technologies, volume 3, number 4, 367-375, 2005 High-throughput screening of 11β-hydroxysteroid dehydrogenase type 1 in scintillation proximity assay format: K.Solly et al., ASSAY and Drug Development Technologies, volume 3, number 4, 377-384, 2005
Результаты
Несколько приведенных ниже примеров, выбранных из соединений по настоящему изобретению, иллюстрируют совершенно неожиданную способность данных соединений избирательно ингибировать 11β-HSD1 в сравнении с 11β-HSD2.
[01874]
Целью настоящего изобретения являются соединения общей Формулы (I), или один из их стереоизомеров, или одна из их солей, приемлемых для фармацевтического использования, предназначенные для применения в качестве лекарств.
Целью настоящего изобретения также являются фармацевтические композиции, содержащие в качестве активного ингредиента соединение общей Формулы (I), или один из его стереоизомеров, или одну из его солей, приемлемых для фармацевтического использования, в сочетании с фармацевтически приемлемым носителем, в качестве лекарств. Данные композиции могут находиться в форме, например, твердых и жидких композиций, эмульсий, лосьонов или кремов.
Фармацевтические композиции, содержащие в качестве активного ингредиента соединение общей Формулы (I), или один из его стереоизомеров, или одну из его солей, приемлемых для фармацевтического использования, можно применять для ингибирования 11β-гидроксистероид-дегидрогеназы типа 1 (11βHSD1).
Фармацевтические композиции, содержащие в качестве активного ингредиента соединение общей Формулы (I), или один из его стереоизомеров, или одну из его солей, приемлемых для фармацевтического использования, можно применять в лечении и профилактике диабета типа 2.
Фармацевтические композиции, содержащие в качестве активного ингредиента соединение общей Формулы (I), или один из его стереоизомеров, или одну из его солей, приемлемых для фармацевтического использования, можно применять в лечении и профилактике расстройств, связанных с 11β-гидроксистероид-дегидрогеназой типа 1 (11βHSD1); или ожирения; или дислипидемий; или артериальной гипертензии; или атеросклероза и клинических патологий, которые являются следствием данного заболевания, таких как коронарные инсульты, или цереброваскулярные инсульты, или артериит нижних конечностей; или гипергликемий; или интолерантности к глюкозе; или резистентности к инсулину; или гипертриглицеридемий; или гиперхолестеринемий; или рестенозов, или панкреатитов; или ретинопатий; или нефропатий; или невропатий; или некоторых типов рака или глауком.
Данные композиции могут быть введены в сочетании с противодиабетическим средством, таким как бигуаниды (например, метформин), различные формы инсулина, сульфонилмочевины (например карбутамид, глиборнурид, глипизид, гликлазид, глибенкламид, глимепирид), меглитиниды (например, натеглинид, репаглинид, митиглинид), модуляторы PPAR (рецептора активатора пролиферации пероксисом) (например, пиоглитазон), ингибиторы альфа-глюкозидазы (например, акарбоза, миглитол, воглибоза), аналоги GLP-1 (глюкагоноподобного пептида) (например, эксенатид, лираглутид), ингибиторы DPP-4 (дипептидилпептидазы 4) (например, ситаглиптин, вилдаглиптин), аналоги амилина (например, прамлинтид).
Данные композиции также могут быть введены в сочетании с агентом, противодействующим ожирению, таким как, например, орлистат или сибутрамин.
В качестве твердых композиций для перорального введения могут быть использованы таблетки, пилюли, порошки (желатиновые капсулы, таблетки) или гранулы. В данных композициях активный ингредиент по изобретению смешивают в потоке аргона с одним или более чем с одним инертным разбавителем, таким как крахмал, целлюлоза, сахароза, лактоза или диоксид кремния. Данные композиции кроме разбавителей также могут содержать другие вещества, например одно или более чем одно смазывающее вещество, такое как стеарат магния или тальк, окрашивающий агент, покрытие (драже) или лак.
В качестве жидких композиций для перорального введения могут быть использованы фармацевтически приемлемые растворы, суспензии, эмульсии, сиропы и эликсиры, содержащие инертные разбавители, такие как вода, этанол, глицерин, растительные масла или парафиновое масло. Данные композиции кроме разбавителей могут содержать другие вещества, например смачивающие агенты, подсластители, загустители, корригенты или стабилизаторы.
Стерильные композиции для парентерального введения предпочтительно готовят в форме водных или неводных растворов, суспензий или эмульсий. В качестве растворителя или носителя могут быть использованы вода, пропиленгликоль, полиэтиленгликоль, растительные масла, в частности оливковое масло, органические сложные эфиры для инъекций, например этилолеат, или другие подходящие органические растворители. Данные композиции также могут содержать адъюванты, в частности смачивающие, изотонические, эмульгирующие, диспергирующие и стабилизирующие агенты. Стерилизация может быть выполнена несколькими способами, например путем антисептического фильтрования, путем добавления к композиции стерилизующих агентов, путем облучения или нагревания. Данные композиции также могут быть приготовлены в виде стерильных твердых композиций, которые могут быть растворены непосредственно перед использованием в стерильной воде или любой другой стерильной среде для инъекций.
Композиции для ректального введения могут быть приготовлены в форме суппозиториев или ректальных капсул, которые кроме активного продукта содержат эксципиенты, такие как масло какао, полусинтетические глицериды или полиэтиленгликоли.
Композиции для местного введения могут быть приготовлены в форме, например, кремов, лосьонов, глазных капель, растворов для полоскания рта, назальных капель или аэрозолей.
Величина дозы зависит от желаемого эффекта, продолжительности лечения и используемого пути введения, доза для взрослого субъекта, предпочтительно пероральная, обычно составляет от 0,001 г до 1 г (предпочтительно от 0,005 г до 0,75 г) в сутки, стандартная доза находится в диапазоне от 0,1 мг до 500 мг активного вещества. Обычно лечащий врач определяет подходящую дозу в зависимости от возраста, массы тела и всех других факторов, специфичных для конкретного субъекта, нуждающегося в лечении.
Реферат
Изобретение относится к производным бензотиазинов, которые представлены общей Формулой (I):,где Rпредставляет собой атом водорода; C-Cалкил; COR; SOR; CO(CH)OR; (CH)R; (CH)CONRR; (CH)NRR; (CH)OR; CHROR; (CH)R; m имеет значение от 1 до 6; n имеет значение от 2 до 6; Rпредставляет собой фенил; нафтил, 1,2,3,4-тетрагидро-нафталин, бифенил, фенилпиридин или бензоловое кольцо, конденсированное с насыщенным или ненасыщенным моноциклическим гетероциклом, содержащим 5-7 атомов и состоящим из атомов углерода и 1-4 гетероатомов, выбранных из N, O или S, отличным от индола, Rпредставляет собой метил или этил; Rи R′являются одинаковыми или разными и представляют собой атом водорода; атом галогена; C-Cалкил; NRR; SOMe; а также их стереоизомерам, солям и сольватам, приемлемым для терапевтического использования, и которые обладают способностью ингибировать 11β-HSD1 на ферментативном и на клеточном уровне. 9 н. и 8 з.п. ф-лы, 197 пр.
Формула

где
R1 представляет собой атом водорода; C1-C6 алкил; COR5; SO2R5; CO(CH2)mOR6;
(CH2)mR6; (CH2)mCONR7R8; (CH2)nNR7R8; (CH2)nOR6; CHR7OR9; (CH2)mR10;
m имеет значение от 1 до 6;
n имеет значение от 2 до 6;
R2 представляет собой фенил, имеющий в качестве заместителей одну или более чем одну группу, выбранную из атома галогена, C1-C6 алкила, CN, CF3, СОМе, CMe(OH)CF3, CH(OH)CF3, COOR7, CONR7R11; нафтил, 1,2,3,4-тетрагидро-нафталин, бифенил, фенилпиридин или бензоловое кольцо, конденсированное с насыщенным или ненасыщенным моноциклическим гетероциклом, содержащим 5-7 атомов и состоящим из атомов углерода и 1-4 гетероатомов, выбранных из N, O или S, отличным от индола, в случае, когда R1, R4 и R′4 представляют собой атом водорода, незамещенные или имеющие в качестве заместителей одну или более чем одну группу, выбранную из атома галогена или C1-C6 алкила, CN, CF3, ОМе; циклическое или полициклическое углеводородное кольцо, содержащее 3-12 атомов углерода; C1-C6 алкил арил или циклоалкил арил,
при условии, что группа R2 всегда связана с карбонилом через атом углерода,
и, когда R2 представляет собой фенил, заместитель COOR7 никогда не находится в положении 4 относительно карбонила;
R3 представляет собой метил или этил;
R4 и R′4 являются одинаковыми или разными и представляют собой атом водорода; атом галогена; C1-C6 алкил; NR7R8; SO2Me;
R5 представляет собой C1-C6 алкил; фенил, незамещенный или имеющий в качестве заместителей одну или более чем одну группу, выбранную из атома галогена, C1-C6 алкила, CN; нафтил, незамещенный или имеющий в качестве заместителей одну или более чем одну группу, выбранную из атома галогена или C1-C6 алкила, CN; циклическое или полициклическое углеводородное кольцо, содержащее 3-12 атомов углерода, незамещенный моноциклический гетероарил, содержащий 5-7 атомов и состоящий из атомов углерода и 1-4 гетероатомов, выбранных из N, О или S;
R6 представляет собой атом водорода; C1-C6 алкил; фенил, незамещенный или имеющий в качестве заместителей одну или более чем одну группу, выбранную из атома галогена; нафтил;
R7 представляет собой атом водорода, C1-C6 алкил;
R8 представляет собой атом водорода; нафтил или, циклическое или полициклическое углеводородное кольцо, содержащее 3-12 атомов углерода;
R7 и R8 вместе с атом азота, к которому они присоединены, могут образовывать 4-6-членное кольцо, которое может содержать один или более чем один гетероатом, выбранный из N, S или О, и может быть незамещенным или иметь в качестве заместителей одну или более чем одну группу, выбранную из C1-C6 алкила, C1-C6 алкил арила или арила;
R9 представляет собой СООМе, COOEt;
R10 представляет собой атом галогена, СООН, COOR7;
R11 представляет собой атом водорода, C1-C6 алкил, C1-C6 алкил циклоалкил,
циклоалкил, арил, C1-C6 алкил арил;
а также их стереоизомеры, соли и сольваты, приемлемые для терапевтического использования.
(4-Гидрокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанона,
(4-Гидрокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(адамантан-1-ил)метанона,
(4-Гидрокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(4-метилфенил)метанона,
(4-Гидрокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(4-хлорфенил)метанона,
(4-Гидрокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(4-цианофенил)метанона,
Бифенил-4-ил-(4-гидрокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)метанона,
(4-Гидрокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(2,4-дихлорфенил)метанона,
(4-Гидрокси-2-этил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(адамантан-1-ил)метанона,
(4-Гидрокси-2-этил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанона,
(4-Гидрокси-2-этил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(4-метилфенил)метанона,
(4-Гидрокси-2-этил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(4-хлорфенил)метанона,
Бифенил-4-ил-(4-гидрокси-2-этил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)метанона,
(5-Хлор-4-гидрокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанона,
(5-Хлор-4-гидрокси-2-этил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанона,
(6-Фтор-4-гидрокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанона,
(6-Фтор-4-гидрокси-2-этил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанона,
(7-Фтор-4-гидрокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанона,
(7-Фтор-4-гидрокси-2-этил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанона,
Бензойной кислоты 2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
Циклогексанкарбоновой кислоты 2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
трет-Бутилкарбоновой кислоты 2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
4-Метилбензойной кислоты 2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
4-Хлорбензойной кислоты 2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
тpeт-Бутилкарбоновой кислоты 16-фтор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
Циклогексанкарбоновой кислоты 6-фтор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
Бензойной кислоты 6-фтор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
4-Метилбензойной кислоты 6-фтор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
тpeт-Бутилкарбоновой кислоты 6-фтор-2-этил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
Циклогексанкарбоновой кислоты 6-фтор-2-этил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
Бензойной кислоты 6-фтор-2-этил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
4-Метилбензойной кислоты 6-фтор-2-этил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
4-Хлорбензойной кислоты 6-фтор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
4-Хлорбензойной кислоты 6-фтор-2-этил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
Нафталин-1-илкарбоновой кислоты 2-метил-3-(4-метилбензоил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
Нафталин-2-илкарбоновой кислоты 2-метил-3-(4-метилбензоил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
Нафталин-1-илкарбоновой кислоты 2-метил-3-(4-цианобензоил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
Нафталин-2-илкарбоновой кислоты 2-метил-3-(4-цианобензоил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
4-Хлорбензойной кислоты 2-метил-3-(4-метилбензоил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
4-Хлорбензойной кислоты 2-метил-3-(4-цианобензоил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
(Нафталин-2-илокси)уксусной кислоты 2-метил-3-(4-метилбензоил)-1,1-диоксо-2H-бензо [е][1,2]тиазин-4-илового эфира,
(Нафталин-2-илокси)уксусной кислоты 2-метил-3-(4-цианобензоил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
(Нафталин-1-илокси)уксусной кислоты 2-метил-3-(4-метилбензоил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
(Нафталин-1-илокси)уксусной кислоты 2-метил-3-(4-цианобензоил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
(4-Хлорфенокси)уксусной кислоты 2-метил-3-(4-метилбензоил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
(4-Хлорфенокси)уксусной кислоты 2-метил-3-(4-цианобензоил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
Уксусной кислоты 2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
2,4-Дихлорбензойной кислоты 2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
4-Фторбензойной кислоты 2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
Циклопентановой кислоты 2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
2-Фурановой кислоты 2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
Тиофен-2-карбоновой кислоты 2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
3-Хлорбензойной кислоты 2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
2-Хлорбензойной кислоты 2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
Феноксиуксусной кислоты 2-метил-3-(нафталин-2-илкарбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
(4-Метокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанона,
(4-Этокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанона,
(4-Пропилокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанона,
(4-Бутилокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанона,
(4-(2-Хлорэтокси)-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(пара-толил)метанона,
(4-[2-(Нафталин-2-илокси)этокси]-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил) (пара-толил)метанона,
(4-(2-Фенокси-этокси)-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанона,
Метил 2-(2-метил-3-(4-метилбензоил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илокси)ацетата,
2-(2-Метил-3-(4-метилбензоил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илокси)уксусной кислоты,
2-(2-Метил-3-(4-метилбензоил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илокси)-N-(нафталин-1-ил)ацетамида,
2-(2-Метил-3-(4-метилбензоил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илокси)-N-(адамантан-1-ил)ацетамида,
2-(2-Метил-3-(4-метилбензоил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илокси)-N-(адамантан-2-ил)ацетамида,
Метил 2-(1,1-диоксо-2-метил-3-(4-метилбензоил)-2H-бензо[е][1,2]тиазин-4-илокси)ацетата,
(1,1-Диоксо-2-метил-3-(4-метилбензоил)-2H-бензо[е][1,2]тиазин-4-илокси)уксусной кислоты,
2-[2-Метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илокси]-1-пиперидин-1-ил-этанона,
2-[2-Метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илокси]-1-(4-метил-пиперазин-1-ил)-этанона,
1-(4-Бензил-пиперазин-1-ил)-2-[2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илокси]-этанона,
(4-Хлор-фенокси)-уксусной кислоты 2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
(Нафталин-1-илокси)-уксусной кислоты 2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
(Нафталин-2-илокси)-уксусной кислоты 2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
(4-[2-(Нафталин-1-илокси)этокси]-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(пара-толил)метанона,
(4-[2-(4-Хлорфенилокси)этокси]-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(пара-толил)метанона,
Уксусной кислоты 2-метил-3-(4-метилбензоил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
Пропановой кислоты 2-метил-3-(4-метилбензоил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
(4-Метилокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(пара-толил)метанона,
(4-Этилокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(пара-толил)метанона, [4-(2-Бром-этокси)-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил]-нафталин-2-ил-метанона,
{4-[2-(4-Хлор-фенокси)-этокси]-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил}-нафталин-2-ил-метанона,
Угольной кислоты этил 1-[2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илокси]-этилового эфира,
[2-Метил-4-(2-пиперидин-1-ил-этокси)-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил]-нафталин-2-ил-метанона,
4-Хлор-бензойной кислоты 5-хлор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
Циклогексанкарбоновой кислоты 5-хлор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
Бензойной кислоты 5-хлор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
4-Хлор-бензойной кислоты 6-фтор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
Циклогексанкарбоновой кислоты 6-фтор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
Бензойной кислоты 6-фтор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
4-Хлор-бензойной кислоты 7-фтор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
Циклогексанкарбоновой кислоты 7-фтор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-иловый эфир,
Бензойной кислоты 7-фтор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
Уксусной кислоты 7-фтор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
Фенокси-уксусной кислоты 7-фтор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
(4-Хлор-фенокси)-уксусной кислоты 7-фтор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
(5-Хлор-4-этокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)-нафталин-2-ил-метанона,
(5-Хлор-4-пропокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)-нафталин-2-ил-метанона,
(6-Фтор-4-метокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)-нафталин-2-ил-метанона,
(6-Фтор-4-этокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)-нафталин-2-ил-метанона,
(6-Фтор-4-пропокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)-нафталин-2-ил-метанона,
(7-Фтор-4-метокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)-нафталин-2-ил-метанона,
(7-Фтор-4-этокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)-нафталин-2-ил-метанона,
(7-Фтор-4-пропокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)-нафталин-2-ил-метанона,
[7-Фтор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илокси]-уксусной кислоты метилового эфира,
[7-Фтор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илокси]-уксусной кислоты метилового эфира,
2-[7-Фтор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илокси]-1-пиперидин-1-ил-этанона,
2-[7-Фтор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илокси]-1-(4-метил-пиперазин-1-ил)-этанона,
1-(4-Бензил-пиперазин-1-ил)-2-[7-фтор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илокси]-этанона,
Бензолсульфоновой кислоты 7-фтор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
Бензолсульфоновой кислоты 2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
4-Хлор-бензолсульфоновой кислоты 2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
4-Метил-бензолсульфоновой кислоты 2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
4-Циано-бензолсульфоновой кислоты 2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
4-Хлор-бензолсульфоновой кислоты 7-фтор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
4-Метил-бензолсульфоновой кислоты 7-фтор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
4-Циано-бензолсульфоновой кислоты 7-фтор-2-метил-3-(нафталин-2-карбонил)-1,1-диоксо-2H-бензо[е][1,2]тиазин-4-илового эфира,
(4-Гидрокси-2-метил-1,1-диоксо-7-пиперидин-1-ил-2H-бензо[е][1,2]тиазин-3-ил)-нафталин-2-ил-метанона,
(7-Диметиламино-4-гидрокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)-нафталин-2-ил-метанона,
(4-Гидрокси-2-метил-1,1-диоксо-7-пиррол идин-1-ил-2H-бензо[е][1,2]тиазин-3-ил)-нафталин-2-ил-метанона,
[4-Гидрокси-2-метил-1,1-диоксо-7-(4-фенил-пиперазин-1-ил)-2H-бензо[е][1,2]тиазин-3-ил]-нафталин-2-ил-метанона,
(7-трет-Бутил-4-гидрокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(нафталин-2-ил)метанона,
(4-Гидрокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(3,4-дихлорфенил)метанона,
(4-Гидрокси-2-этил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(3,4-дихлорфенил)метанона,
(4-Гидрокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(бензофуран-2-ил)метанона,
(4-Гидрокси-2-этил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)(бензофуран-2-ил)метанона,
(4-Гидрокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)-(5,6,7,8-тетрагидро-нафталин-2-ил)-метанона,
(4-Гидрокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)-(5,5,8,8-тетраметил-5,6,7,8-тетрагидро-нафталин-2-ил)-метанона,
(7-Фтор-4-гидрокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)-(5,6,7,8-тетрагидро-нафталин-2-ил)-метанона,
(7-Фтор-4-гидрокси-2-метил-1,1-диоксо-2H-бензо[е][1,2]тиазин-3-ил)-(5,5,8,8-тетраметил-5,6,7,8-тетрагидро-нафталин-2-ил)-метанона,
(2,3-Дигидро-бензофуран-5-ил)-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
(2,3-Дигидро-бензофуран-5-ил)-(4-гидрокси-2-этил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
Бензо[1,3]диоксол-5-ил-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
Бензо[1,3]диоксол-5-ил-(4-гидрокси-2-этил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
(2,3-Дигидро-бензо[1,4]диоксин-6-ил)-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
(2,3-Дигидро-бензо[1,4]диоксин-6-ил)-(4-гидрокси-2-этил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
Бензо[b]тиофен-5-ил-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
Бензофуран-5-ил-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-(1-метил-1Н-бензоимидазол-5-ил)-метанона,
Бензо[b]тиофен-2-ил-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
(4-трет-Бутил-фенил)-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
(3-Бром-фенил)-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
3-(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-карбонил)-бензонитрила,
(3,4-Диметил-фенил)-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-(3-трифторметил-фенил)-метанона,
(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-(4-трифторметил-фенил)-метанона,
Адамантан-2-ил-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
Хроман-6-ил-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
(4-Хлор-3-трифторметил-фенил)-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
(7-Бром-4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-нафталин-2-ил-метанона,
(7-Хлор-4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-нафталин-2-ил-метанона,
(4-Гидрокси-2,7-диметил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-нафталин-2-ил-метанона,
Бифенил-3-ил-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
(2′-Фтор-бифенил-3-ил)-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
(3′-Фтор-бифенил-3-ил)-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
(4′-Фтор-бифенил-3-ил)-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
(2′-Хлор-бифенил-3-ил)-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
(3′-Хлор-бифенил-3-ил)-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
(4′-Хлор-бифенил-3-ил)-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
(2′-Метил-бифенил-3-ил)-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
(3′-Метил-бифенил-3-ил)-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
(4′-Метил-бифенил-3-ил)-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
(2′-Метокси-бифенил-3-ил)-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
(3′-Метокси-бифенил-3-ил)-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
(4′-Метокси-бифенил-3-ил)-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-(3′-трифторметил-бифенил-3-ил)-метанона,
(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-(4′-трифторметил-бифенил-3-ил)-метанона,
3′-(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-карбонил)-бифенил-3-карбонитрила,
3′-(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-карбонил)-бифенил-4-карбонитрила,
(4-Гидрокси-7-метансульфонил-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-нафталин-2-ил-метанона,
(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-(1-фенил-циклопропил)-метанона,
1-[3-(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-карбонил)-фенил]-этанона,
(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-[3-(2,2,2-трифтор-1-гидрокси-1-метил-этил)-фенил]-метанона,
(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-[3-(2,2,2-трифтор-1-гидрокси-этил)-фенил]-метанона,
3-(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-карбонил)-бензойной кислоты,
3-(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-карбонил)-N-метил-бензамида,
3-(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-карбонил)-N,N-диметил-бензамида,
N,N-Этил-3-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-карбонил)-бензамида,
3-(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-карбонил)-N,N-диэтил-бензамида,
N-Циклопропил-3-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-карбонил)-бензамида,
N-Циклопропилметил-3-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-карбонил)-бензамида,
3-(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-карбонил)-N-фенил-бензамида,
N-Бензил-3-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-карбонил)-бензамида,
3-(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-карбонил)-бензамида,
3-(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-карбонил)-бензойной кислоты этилового эфира,
(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-(3-пиридин-3-ил-фенил)-метанона,
(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-(3-пиридин-4-ил-фенил)-метанона,
(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-[3-(6-метил-пиридин-3-ил)-фенил]-метанона,
(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-[3-(5-метил-пиридин-3-ил)-фенил]-метанона,
(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-[3-(4-метил-пиридин-3-ил)-фенил]-метанона,
(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-[3-(2-метил-пиридин-3-ил)-фенил]-метанона,
(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-[3-(4-метокси-пиридин-3-ил)-фенил]-метанона,
(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-[3-(6-фтор-пиридин-3-ил)-фенил]-метанона,
(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-[3-(2-метокси-пиридин-3-ил)-фенил]-метанона,
(4-Гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-[3-(6-метокси-пиридин-3-ил)-фенил]-метанона,
(3-Хлор-фенил)-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона,
(3-Фтор-фенил)-(4-гидрокси-2-метил-1,1-диоксо-1,2-дигидро-2H-бензо[е][1,2]тиазин-3-ил)-метанона.

где R1 представляет собой атом водорода, отличающийся тем, что промежуточное соединение общей Формулы (IV)

где R2, R4 и R′4 являются такими, как определено в п.1, конденсируют с промежуточным соединением общей Формулы R3-Y, где R3 является таким, как определено в п.1, и Y представляет собой уходящую группу.

где R1 не является атомом водорода, отличающийся тем, что промежуточное соединение общей Формулы (Ia)

где R2, R3, R4 и R′4 являются такими, как определено в п.1, конденсируют с промежуточным соединением общей Формулы R1-Z, где R1 является таким, как определено выше, и Z представляет собой уходящую группу.

где R1 представляет собой (CH2)nNR7R8 или (CH2)nOR6, отличающийся тем, что промежуточное соединение общей Формулы (V)

где R2, R3, R4, R′4, n являются такими, как определено в п.1, и X′ представляет собой уходящую группу, конденсируют с промежуточным соединением общей Формулы R7R8NH или R8OH, где R7 R8 и R6 являются такими, как определено в п.1.

где R1 представляет собой (CH2)nCONR7R8, отличающийся тем, что промежуточное соединение общей Формулы (VII)

где R2, R3, R4, R′4 и m являются такими, как определено в п.1, конденсируют с промежуточным соединением общей Формулы R7R8NH, где R7 и R8 являются такими, как определено в п.1.

где R4 представляет собой NR7R8, отличающийся тем, что промежуточное соединение общей Формулы (Ie)
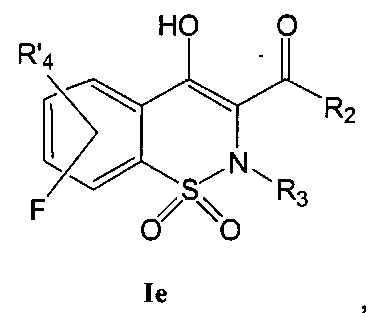
где R2 R3 и R′4 являются такими, как определено в п.1, конденсируют с промежуточным соединением общей Формулы R7R8NH, где R7 и R8 являются такими, как определено в п.1.

где R2 представляет собой бифенил или фенил пиридин, замещенные или незамещенные, отличающийся тем, что промежуточное соединение общей Формулы (Ig)

где R3, R4, R′4 являются такими, как определено в п.1, и X представляет собой уходящую группу, конденсируют с бороновой кислотой.

где R2 представляет собой фенил, имеющий в качестве заместителя амид в орто- или мета-положении, отличающийся тем, что промежуточное соединение общей Формулы (Ij)
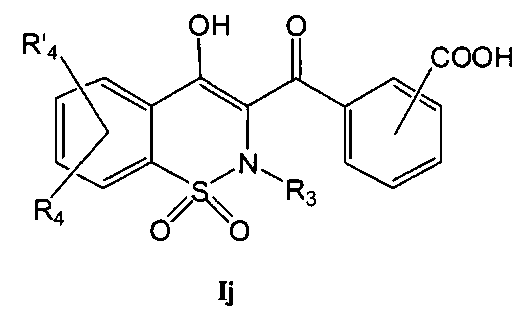
где R3, R4, R′4 являются такими, как определено в п.1, конденсируют с амином общей Формулы R7R11NH, где R7 и R11 являются такими, как определено в п.1.
Комментарии