2-(r-тио)-10-[3-(4-r-пиперазин-1-ил)пропил]-10h-фенотиазины для лечения бета-амилоидопатии или альфа-синуклеопатии, и способ их диагностики или предварительной диагностики - RU2587154C2
Код документа: RU2587154C2
Чертежи
Описание
Накопление белков или фрагментов белков (пептидов) в мозге является существенным признаком возрастных нейродегенеративных заболеваний. При деменции Альцгеймера (болезни Альцгеймера, БА) и церебральной β-амилоидопатии (ЦБА) инициирующим фактором является скопление β-амилоидных пептидов (Аβ), причем основной механизм является неизвестным. При БА и ЦБА нарушена протеостаза Аβ, то есть равновесие между выработкой и разрушением/удалением посредством рецепторов или протеаз. Однако до настоящего времени мало внимания уделялось удалению пептидов Аβ клеточными переносчиками (переносчиками ABC). При болезни Паркинсона накапливается белок α-синуклеин, который, в частности, регулирует выделение допамина в черном веществе. Известно, что при α-синуклеопатии при болезни Паркинсона переносчики ABC играют важную роль в переносе веществ (см. публикацию Kortekaaset и др., Ann Neural 2005, 57, 176-179). Существует несколько подсемейств A-G, которые могут попеременно переносить различные субстраты (метаболиты, медикаменты, пептиды, белки, ионы) и даже способны заменять друг друга в функции переноса веществ (например, АВСВ1 и АВСС1, см. публикацию Тао и др., Cancer Chemotherapy and Pharmacology, 64, 5, 961-969).
Посредством различных моделей на генетически модифицированных мышах было показано, что переносчик ABC (общим структурным элементом переносчика ABC является АТФ-связывающая кассета и пора-переносчик). АВСС1 является важным переносчиком белков/пептидов, в частности переносчиком Аβ, который оказывает необычные функциональные эффекты на накопление белков в мозге. АВСС1 также является важным переносчиком α-синуклеина.
Ниже в качестве примера продемонстрированы результаты исследований действия переносчика по переносу Аβ.
Для определения действия АВСС1 в живом организме трансгенных мышей, экспрессирующих АРР (белок-предшественник амилоида), в каждом случае генетически был удален АВСВ1, ABCG2 или переносчик АВСС1 (мыши с "выключенным" геном).
Было установлено, что:
i) количество Аβ у мышей, лишенных переносчика АВСС1, увеличилось в 12 раз,
ii) потеря переносчика АВСВ1 приводит к увеличению всего лишь в три раза, и
iii) потеря ABCG2 не оказывает какого-либо влияния на накопление Аβ.
Следовательно, задачей настоящего изобретения является создание веществ, надлежащим образом влияющих на переносчик АВСС1, соответственно, для обеспечения возможности лечения нейродегенеративных заболеваний, в частности α-синуклеопатий пациента с β-амилоидопатией. Эта задача была решена посредством 2- (R2-тио)-10-[3-(4-R1-пиперазин-1-ил)пропил]-10H-фенотиазинов по п. 1 формулы изобретения. Дополнительные предпочтительные варианты осуществления изобретения получены из зависимых пунктов формулы изобретения.
Другими словами, эта задача была решена посредством 2-(R2-тио)-10-[3-(4-R1-пиперазин-1-ил)пропил]-10H-фенотиазинов согласно общей формуле I
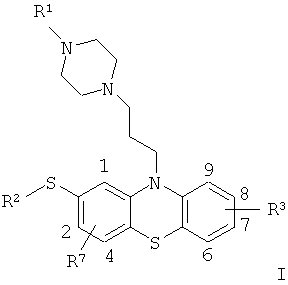
в которых группы
R1 и R2 являются одинаковыми или различными, и каждая из них, независимо друг от друга, представляет собой C1-C6-алкильные группы, которые независимо друг от друга, возможно, но не обязательно, содержат другую замещающую группу, выбранную из алкила, арила, ацила (предпочтительно ацетила), аминогруппы, нитрогруппы, сульфонила, гидроксила, алкоксильной группы, арилоксильной группы, арилтиогруппы, алкилтиогруппы и атомов галогенов, где соответствующие алкильные группы, возможно, но не обязательно, содержат, по меньшей мере, один дополнительный атом галогена, а группа
R3 расположена в одной из позиций 6-9 системы кольца фенотиазина и представляет собой атом водорода или алкил, арил, ацил (предпочтительно ацетил), аминогруппу, нитрогруппу, сульфонил, гидроксил, алкоксильную группу, арилоксильную группу, арилтиогруппу или алкилтиогруппу или атом галогена, где соответствующие алкильные группы, возможно, но не обязательно, содержат, по меньшей мере, один дополнительный атом галогена либо группу NR4R5 или OR6, где группы R4, R5 и R6 являются одинаковыми или различными, и каждая из них независимо друг от друга выбрана из водорода и C1-C3-алкильных групп, а группа
R7 расположена в одной из позиций 1, 2 или 4 системы кольца фенотиазина и представляет собой атом водорода или алкил, арил, ацил (предпочтительно ацетил), аминогруппу, нитрогруппу, сульфонил, гидроксил, алкоксильную группу, арилоксильную группу, арилтиогруппу или алкилтиогруппу или атом галогена, где соответствующие алкильные группы, возможно, но не обязательно, содержат, по меньшей мере, один дополнительный атом галогена либо группу NR8R9 или OR10, где группы R8, R9 и R10 являются одинаковыми или различными, и каждая из них независимо друг от друга выбрана из водорода и C1-C3-алкильной группы,
для лечения β-амилоидопатии или α-синуклеопатии, сопровождаемой отложением белков в мозге.
Кроме того, как в случае α-синуклеопатий, так и в случае β-амилоидопатий существует потребность в идентификации или в диагностике или предварительной диагностике этих болезней.
Задачей настоящего изобретения также является создание способа, посредством которого может быть осуществлена диагностика или предварительная диагностика α-синуклеопатиий, а также β-амилоидопатий. Предпочтительные варианты осуществления изобретения получены из зависимых пунктов формулы изобретения. Другими словами, эта задача решена посредством способа диагностики или предварительной диагностики β-амилоидопатии или α-синуклеопатии или способа определения риска развития такой болезни у обследуемого пациента, где этот обследуемый пациент уже принимает вещества, транспортируемые переносчиком АВСС1 в мозг, состоящего из следующих операций:
a) определяют количество принятого внутрь вещества в пробах жидкости из организма обследуемого пациента в конкретный момент времени;
b) повторяют операцию а) определения, по меньшей мере, в еще один более поздний момент времени;
c) сравнивают количества, определенные при операции а) и при операции b), с количествами, которые были заданы как характеристические величины в эти же самые моменты времени для тех обследуемых пациентов, у которых во время взятия проб не были выявлены клинические симптомы β-амилоидопатии или α-синуклеопатии.
Тот факт, что обследуемый пациент уже принимает, по меньшей мере, одно вещество, транспортируемое посредством переносчика АВСС1 в мозг, означает то, что отсутствует необходимость в приеме этого вещества. Наоборот, оно уже присутствует в теле обследуемого пациента, например, в результате медикаментозного лечения другой болезни. Исследуемыми пробами жидкости из организма обследуемого пациента предпочтительно являются пробы плазмы крови, сыворотки крови и/или спинномозговой жидкости.
β-амилоидопатией предпочтительно является болезнь Альцгеймера, α-синуклеопатией предпочтительно является болезнь Паркинсона. Возможно, но не обязательно, α-синуклеопатией также может являться деменция с тельцами Леви (DLB). Вещества, транспортируемые посредством переносчика АВСС1 в мозг, предпочтительно выбраны из антибиотиков (таких как, например, дифлоксацин, грепафлоксацин), виростатиков/противовирусных медикаментов (таких как, например, саквинавир, ритонавир), противоаллергенных/антигистаминных препаратов (таких как, например, циметидин), сердечно-сосудистых препаратов (таких как, например, верапамил), антидепрессантов (таких как, например, циталопрам), препаратов против гиперурикемии (таких как, например, пробенецид), цитостатиков (таких как, например, метотрексат, этопозит, эдатрексат, ZD1694), витаминов/аналогов витаминов (таких как, например, метотрексат, фолиевая кислота, L-лейковорин), противовоспалительных препаратов (таких как, например, индометацин), противоэпилептических препаратов (таких как, например, вальпроевая кислота), гормонов/производных гормонов (таких как, например, 17β-эстрадиол), лейкотриенов (таких как, например, LTC4), флуоресцентных проб (таких как, например, кальцеин, Fluo-3, BCECF, SNARF), глутатион-(GSH-), сульфат- или глюкуронид-связанных метаболитов природных веществ (произведенных эндогенно), токсинов или медикаментов (таких как, например, 2,4-динитрофенил-SG, биман-SG, N-этилмалеимид-SG, доксорубицин-SG, тиотепа-SG, циклофосфамид-SG, мелфалан-SG, хлорамбуцил-SG, этакриновая кислота-SG, метолахлор-SG, атразин-SG, сульфорафан-SG, афлатоксин В1-эпоксид-SG, 4-нитрохинолин 1-оксид-SG, As(SG)3, этопозид-глюкоза, 4-(метилнитрозамин)-1-(3-пиридил)-1-бутанол (NNAL)-3β-0-глюкоза, SN-38-глюкоза, 4-метилумбеллиферон-β-d-глюкоза, 6-гидрокси-5,7-диметил-2-метиламино-4-(3-пиридилметил)-бензотиазолсульфат (E3040S)-глюкоза, лейкотриен С4, простагландин A2-SG, 15-деокси-Δ12,14 простагландин J2-SG, гидроксиноненал-SG, 17β-эстрадиол-17-β-d-глюкоза, глюкуронозилбилирубин, бис-люкуронозилбилирубин, гиодезоксихолат-6-α-глюкоза, эстрон-3-сульфат, дегидроэпиандростеронсульфат, сульфатолитохолат) (см. также публикацию Deeley RG и др.: Substrate recognition and transport by multi drug resistance protein 1 (ABCC1), FEBS letters 2006, 580 (4), стр.1103-1111).
Этот косвенный анализ транспортного действия переносчиков АВСС1 может использоваться для диагностики/предварительного диагноза соответствующей болезни. У обследуемых пациентов, кто уже принимает переносимые АВСС1 вещества другими способами, может быть исследован профиль концентрации активного вещества в жидкостях организма, предпочтительно, в плазме крови, в сыворотке крови и/или в спинномозговой жидкости. Результаты измерений для тех обследуемых пациентов, имеющих сниженное транспортное действие АВСС1 в зависимости от времени по сравнению со здоровыми обследуемыми пациентами, демонстрируют кривую изменения концентрации вещества с задержкой или со сдвигом (график зависимости концентрации c от времени t), то есть максимум кривой меняется в зависимости от времени.
Когда получена сдвинутая кривая по сравнению со случаем здорового пациента, то это является признаком измененного транспортного действия АВСС1. Это означает, что такие вещества, как, например, Аβ или α-синуклеин, переносятся менее эффективно и, следовательно, являются признаком соответствующей болезни.
Как модель на мышах, так и фармакологическое влияние АВСС1 показывают, что он является важным клеточным трансмембранным переносчиком белков Аβ, и подразумевают, что гематоэнцефалический барьер и сосудистое сплетение желудочков мозга занимают ключевую позицию для выведения Аβ из мозга. Может быть показано, что избирательная фармакологическая активизация переносчика АВСС1 значительно уменьшает насыщение мозга Аβ и, следовательно, может использоваться в терапевтических целях для лечения болезней с нарушенной протеостазой мозга. Кроме того, как описано выше, анализ действия переносчика АВСС1 может использоваться для косвенной или прямой диагностики/предварительной диагностики соответствующей болезни. Прямой анализ возможен путем введения в организм веществ, транспортируемых посредством переносчика АВСС1, и их определения. Косвенный анализ уже был описан выше.
Изменения механизмов выведения, которые связаны с переносчиками ABC, могут оказывать существенное влияние на временной профиль агрегирования Aβ и других белков мозговой ткани. Следовательно, влияние функции переносчика АВСС1 оказывает положительный эффект на риск развития нейродегенеративных заболеваний, в частности болезни Альцгеймера. В этом смысле "лечение нейродегенеративных заболеваний" содержит профилактику, а также лечение доклинических стадий болезни.
Роль переносчика ABC в выведении Аβ была первоначально изучена следующим образом: было продемонстрировано, что АВСС1 способен переносить Аβ. Для этого были использованы анализы проб эндотелиальных клеток, полученных "in vitro" с использованием вкладышей "трансвел" (transwell), (анализ проб эндотелиальных клеток, полученных с использованием вкладышей "трансвел", ЕСТА) первичных культур капиллярных эндотелиальных клеток из мозга мыши (подход с использованием культуры клеток).
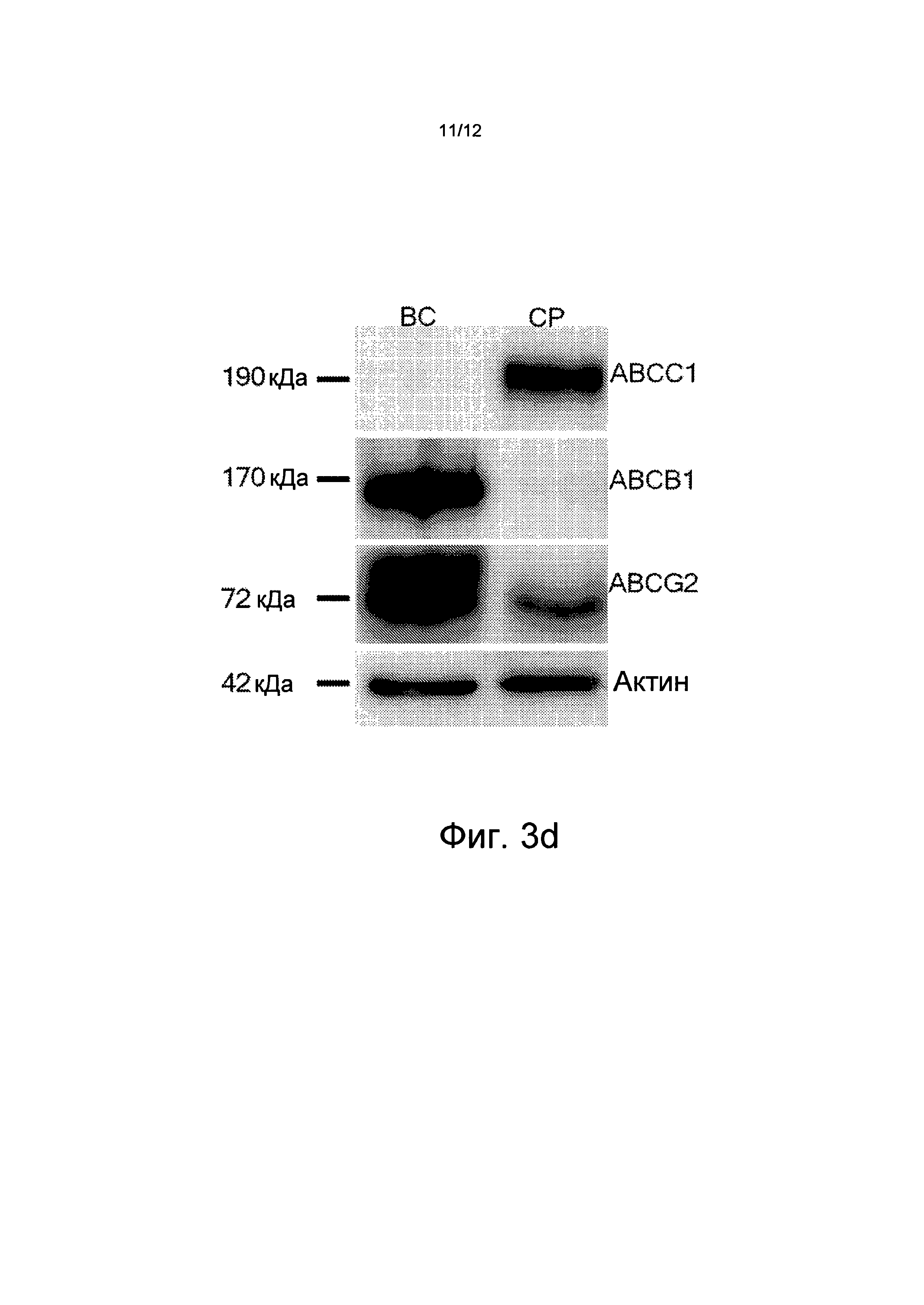
Для исследования Аβ-специфичного транспортного действия использовались первичные культуры эндотелиальных клеток из капилляров мозга мышей (с "выключенным" геном), имеющих дефицит АВСВ1 и дефицит АВСС1, и контрольной группы мышей (C57BI/6, FVB/N). Перенос Аβ из адлюминального пространства (мозг) в люминальное пространство (кровь) ухудшается в эндотелиальных клетках с дефицитом АВСВ1 и с дефицитом АВСС1. Средняя скорость переноса Аβ в течение первых шести часов после введения в организм пептидов Аβ (Аβ42) составила 2,2 пикограмма в минуту (пг/мин) для клеток контрольной группы. В отличие от этого в клетках с дефицитом АВСС1 была достигнута только половина способности к переносу (1,0 пг/мин). В клетках с дефицитом АВСВ1 перенос Аβ почти не существовал (0,3 пг/мин). Кроме того, исследования капиллярных эндотелиальных клеток и клеток из сосудистого сплетения желудочков мозга выявили, что переносчик АВСВ1 является сильно выраженным в капиллярных эндотелиальных клетках мозга, тогда как эндотелиальная экспрессия АВСС1 в капиллярах мозга является более низкой.
Затем была исследована относительная значимость элементов семейства переносчиков ABC в живом организме с использованием заново созданных моделей на мышах с болезнью Альцгеймера, имеющих дефицит переносчика ABC. Каждая из генетически модифицированных мышей имела дефицит ("выключение") переносчиков конкретных ABC: ABCG2, АВСВ1 или АВСС1.
Аβ-иммуногистохимия участков мозга показала:
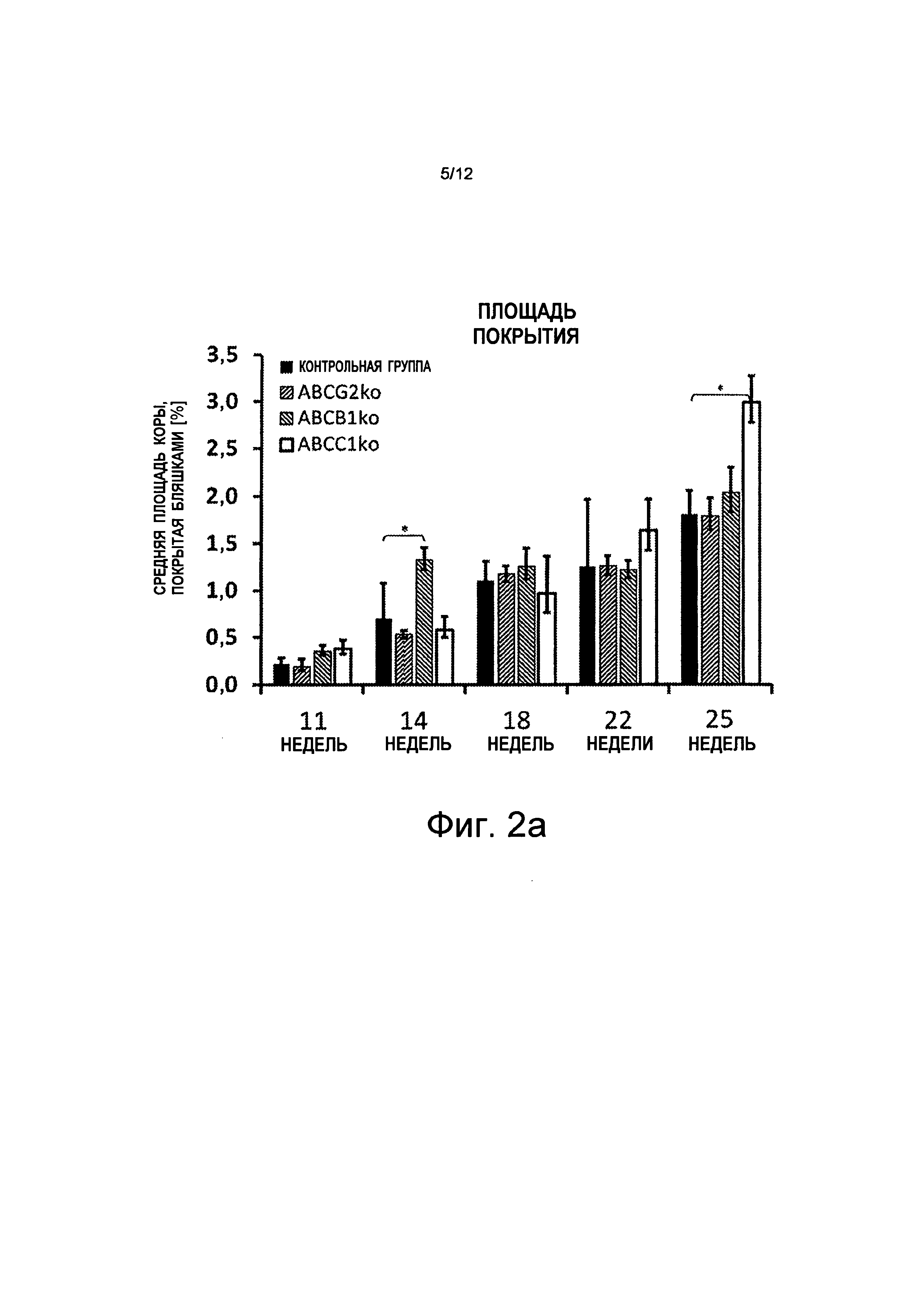
i) значительные увеличения количества и размера кортикальных Аβ-положительных бляшек у мышей с дефицитом АВСС1 по сравнению с мышами из контрольной группы (см. Фиг.1 и Фиг.2а - Фиг.2c);
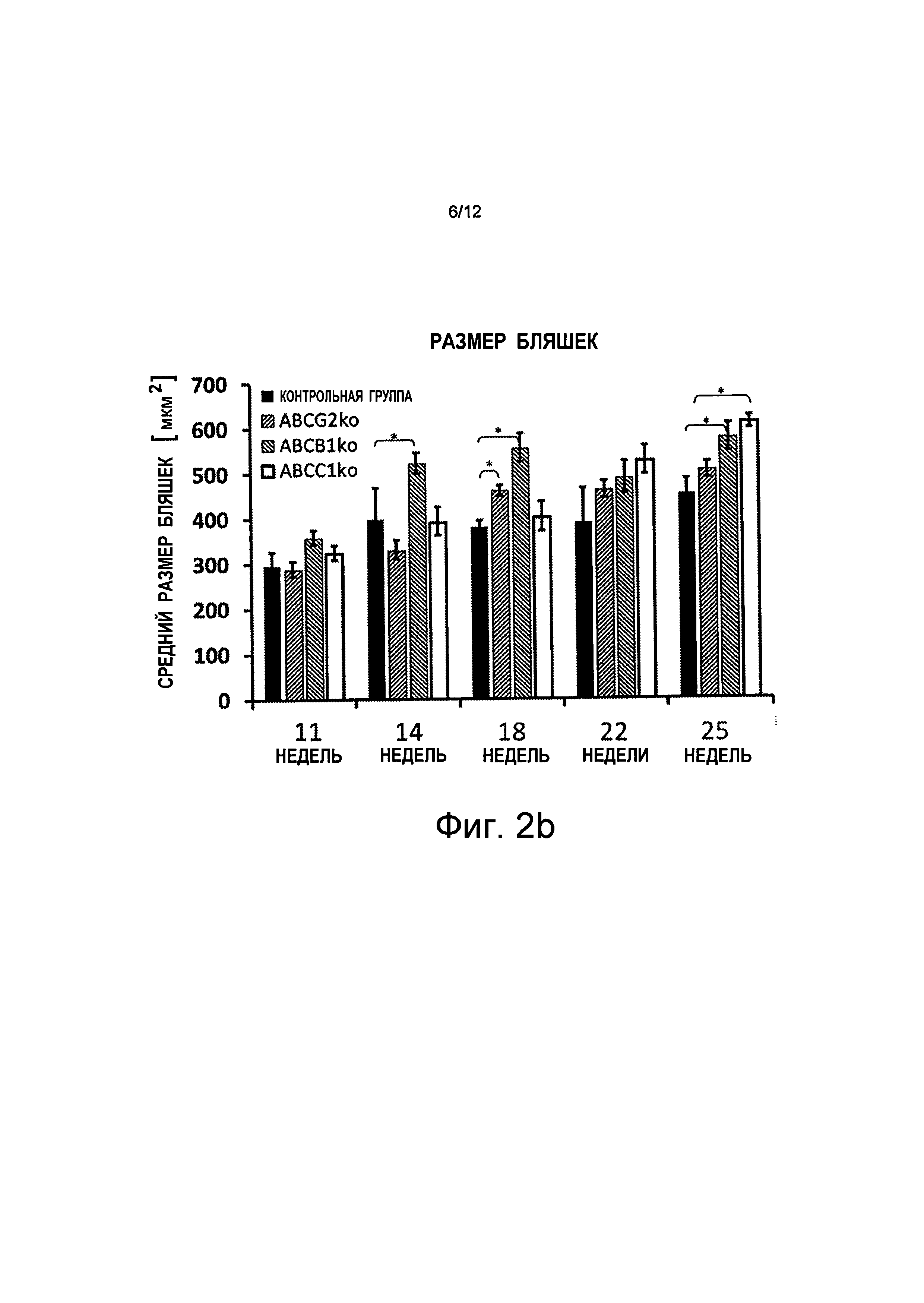
ii) мыши с дефицитом АВСВ1 показали меньшее увеличение количества и размера Аβ-бляшек, чем мыши с дефицитом АВСС1;
iii) не может быть определено какого-либо значительного различия между мышами из контрольной группы и мышами с дефицитом ABCG2 (см. Фиг.2а - Фиг.2c).
Для определения количества Аβ, растворимых в буферном растворе (главным образом, мономеров и меньших олигомеров), и Аβ, растворимых в гуанидине (главным образом, волокнистого или агрегированного вещества), для Аβ использовались иммуноферментные адсорбционные анализы (твердофазные иммуноферментные анализы, ELISA).
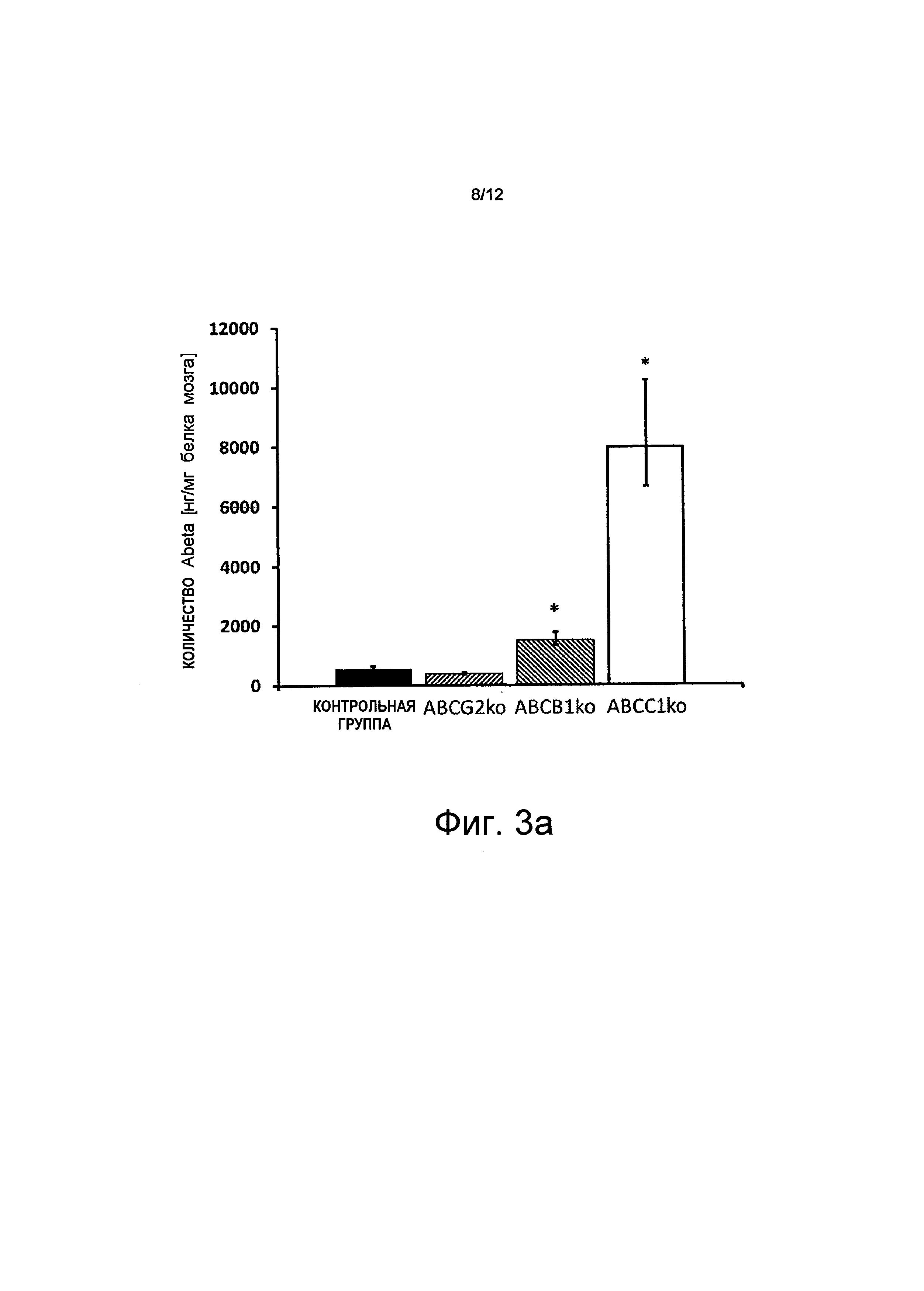
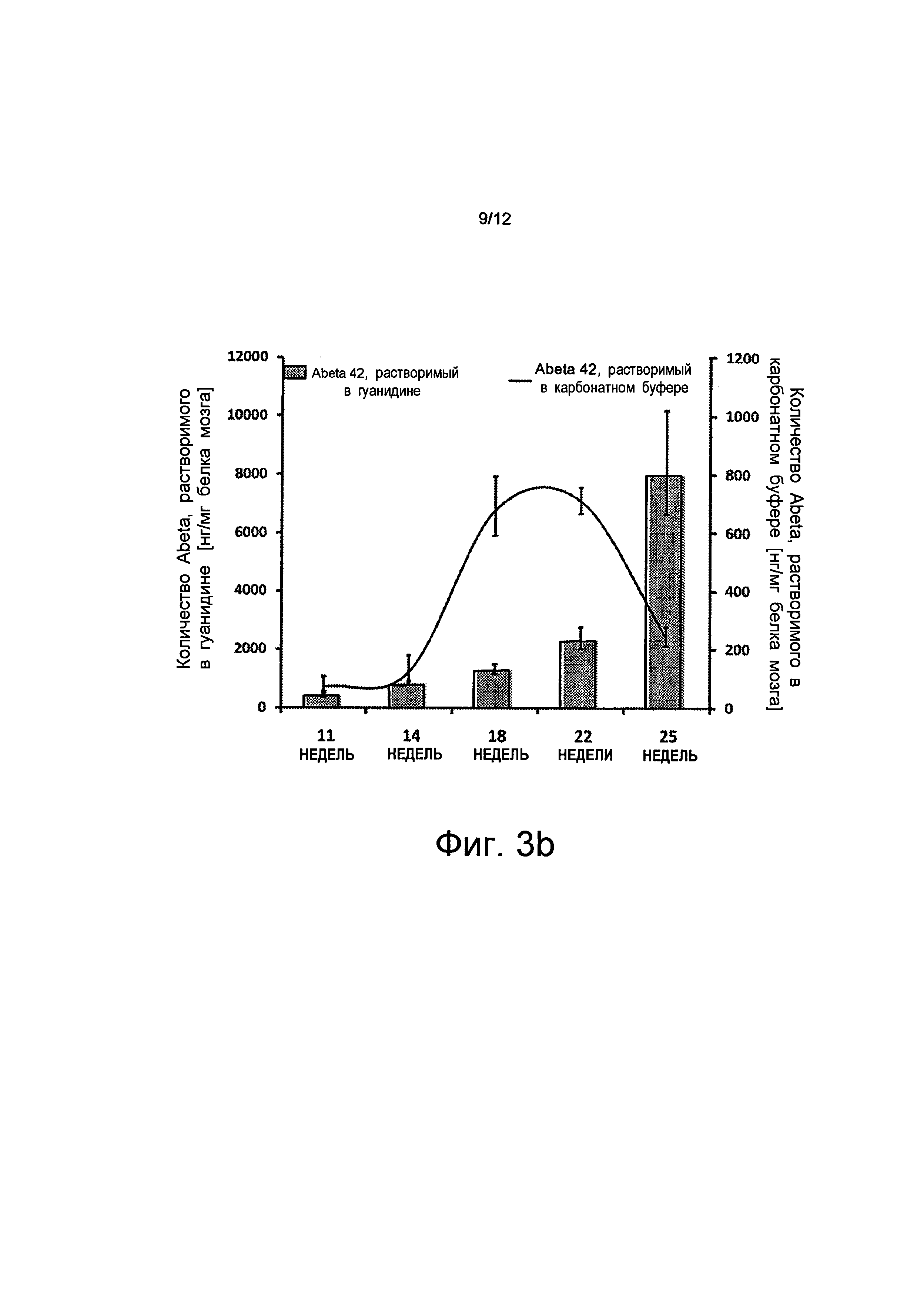
В соответствии с морфологическими результатами иммуногистохимии мыши с дефицитом АВСС1 показали значительное увеличение агрегированных Аβ по сравнению с мышами из контрольной группы во все временных точках измерений. Насыщение мозга Аβ было наиболее сильным в возрасте, равном 25 неделям. В этот момент времени величины Аβ (Аβ42) были в 12 раз более высокими, чем у мышей из контрольной группы. Количество Аβ, растворимых в буферном растворе, также увеличивалось с увеличением возраста, но после 25 недель, в момент времени наивысшего насыщения бляшками, величины растворимых Аβ в группе с дефицитом АВСС1 значительно уменьшились.
Были проведены дополнительные исследования, которые предоставили дополнительное доказательство связи между возможно недостаточным выведением посредством АВСС1 и агрегацией Аβ.
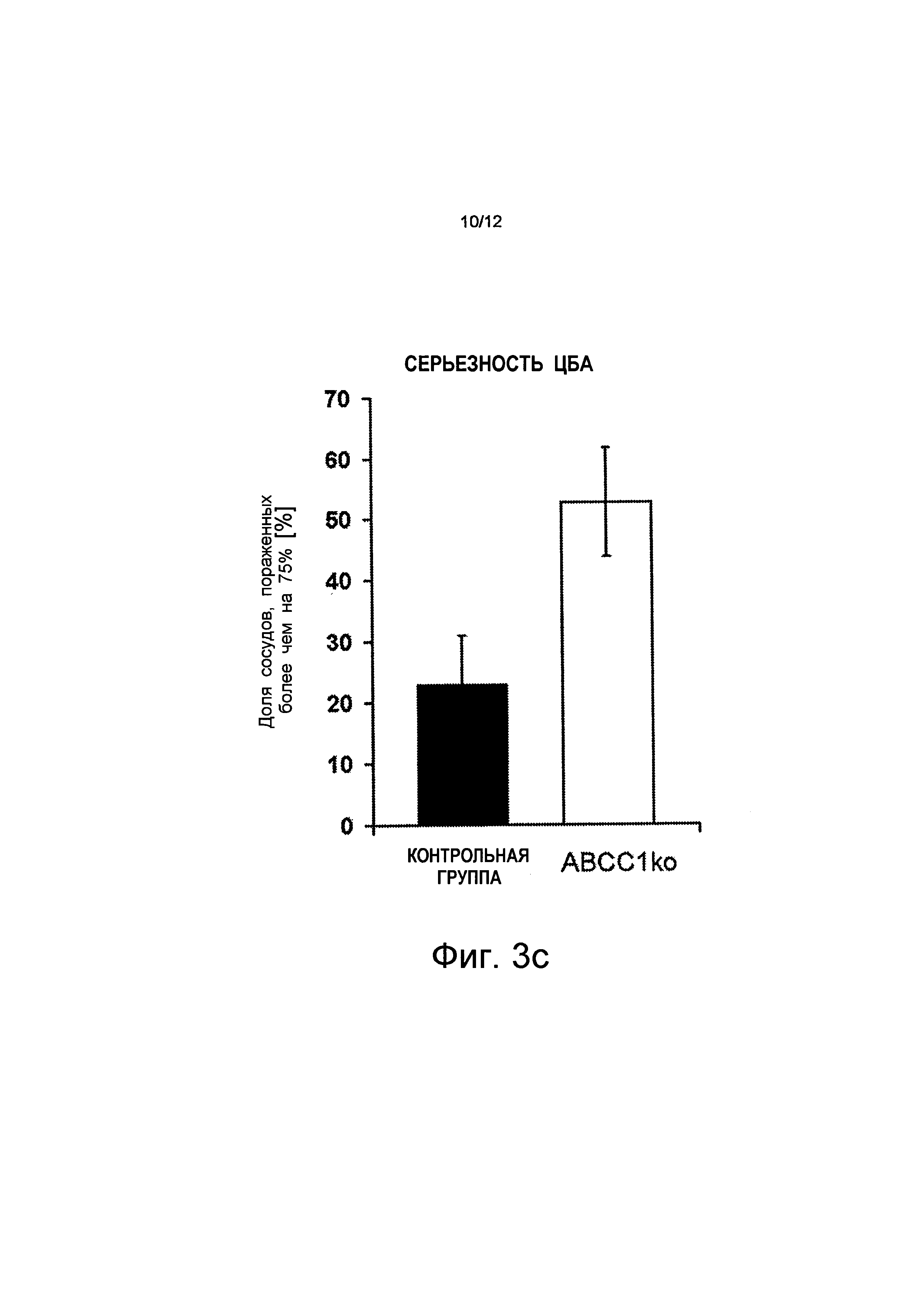
Кинетика переноса, которую имеют переносчики ABC, зависит, в частности, от конкретных характеристик белка/пептида, таких как, например, удельный заряд. Вариант белка-предшественника амилоида голландского типа (голландский мутант, APPdt), который вводит дополнительный отрицательный заряд вблизи границы α-секретазы АРР и, таким образом, приводит к сильному влиянию церебральной амилоидной ангиопатии (ЦБА) на удаление Aβdt через гематоэнцефалический барьер. Анализы капилляров мозга и сосудистых сплетений желудочков мозга (CP) у мышей из контрольной группы методом вестерн-блоттинга показали сильную экспрессию АВСВ1 в капиллярных эндотелиальных клетках мозга (ВС) и АВСС1 в CP (см. чертеж Фиг.3d). Поскольку переносчики ABC играют важную роль в удалении Аβ, то было сделано предположение о том, что APPdt-трансгенные мыши с дефицитом переносчика ABC (в гематоэнцефалическом барьере и в барьере сосудистого сплетения желудочков мозга крови) имеют повышенное накопление Aβdt в менингеальных сосудах. Степень ЦБА у мышей с дефицитом ABC APPdt была определена количественно в возрасте 24 месяцев. В соответствии с этим предположением, у животных с дефицитом АВСС1, по меньшей мере, 51% сосудов были сильно ухудшены (>75% стенок сосудов, насыщенных Аβ по сравнению с 23% у животных из контрольной группы (см. Фиг.3c).
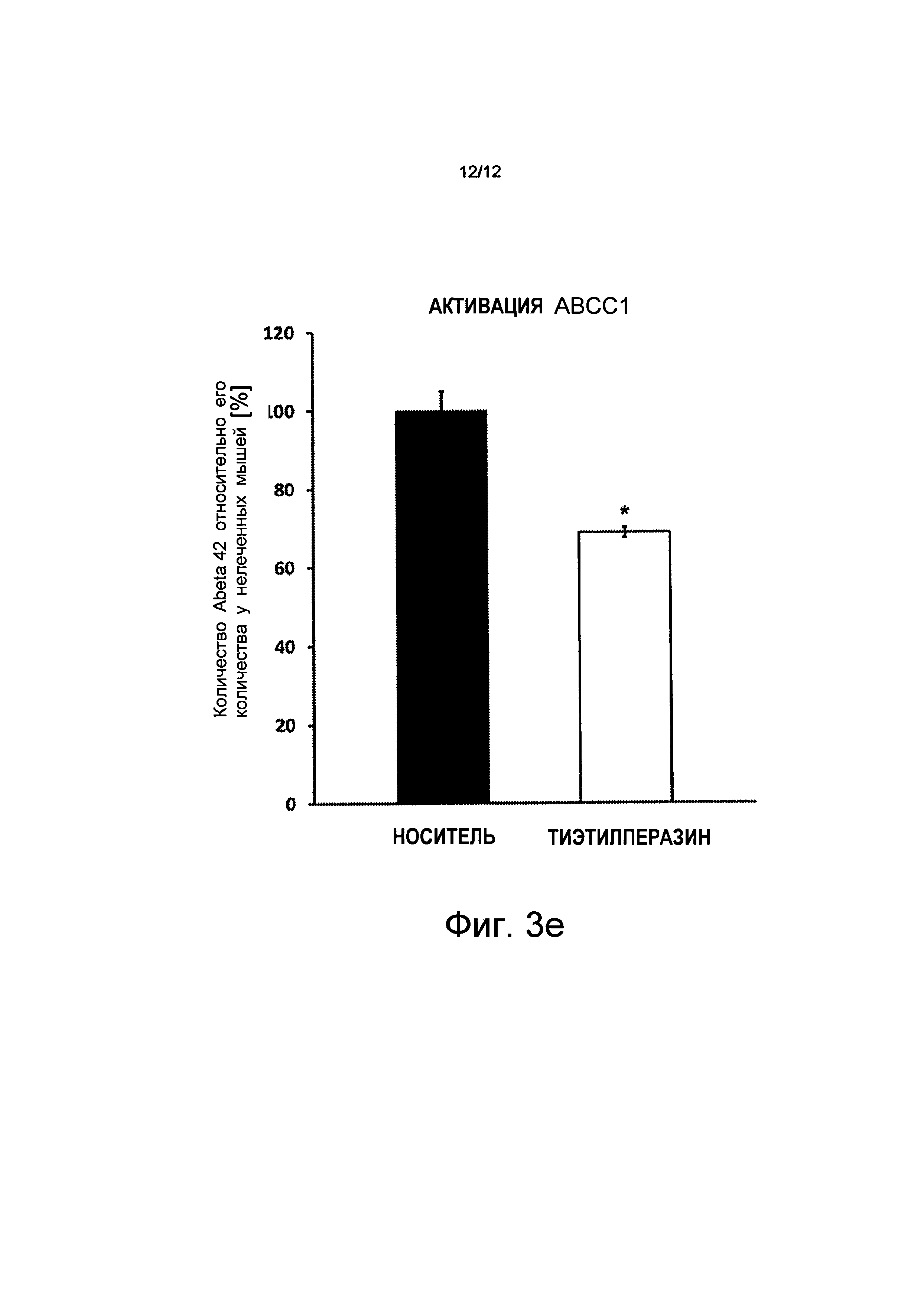
На основании этих результатов были проведены исследования того, насколько содержимое растворимых Аβ в мозге может быть уменьшено за счет активизации переносчиков ABC посредством активного вещества/насколько сильное влияние они оказывают. Мышей с отложениями амилоида лечили в течение 30 дней противорвотным тиэтилперазином (торекан®, 2-(этилтио)-10-[3-(4-метилпиперазин-1-ил)пропил]-10H-фенотиазин). 3 миллиграмма на килограмм веса тела (мг/кг веса тела) вводилось внутримышечно два раза в день. Профилактическое лечение началось до того, как у мышей появились сенильные бляшки. Измерения методом ELISA у животных, подвергнутых лечению, показали снижение количества Аβ, по меньшей мере, на 31% у мышей, подвергнутых лечению, по сравнению с животными, подвергнутыми лечению носителем (носитель = вода) (см. Фиг.3е). Эти результаты воспроизведены графически на Фиг.3.
Было доказано, что способность удаления Аβ является ключевым фактором в регулировании внутримозгового накопления Аβ.
Было доказано, что тиэтилперазин (торекан®) является особо эффективным активатором переносчика АВСС1. Другие производные, начиная с того же самого скелета, также продемонстрировали хорошие результаты по активизации переносчика АВСС1. Соответствующие производные представлены в общей формуле I

в которых группы
R1 и R2 являются одинаковыми или различными, и каждая из них, независимо друг от друга, представляет собой C1-C6-алкильные группы, которые независимо друг от друга, возможно, но не обязательно, содержат другую замещающую группу, выбранную из алкила, арила, ацила (предпочтительно ацетила), аминогруппы, нитрогруппы, сульфонила, гидроксила, алкоксильной группы, арилоксильной группы, арилтиогруппы, алкилтиогруппы и атомов галогенов, где соответствующие алкильные группы, возможно, но не обязательно, содержат, по меньшей мере, один дополнительный атом галогена, а группа
R3 расположена в одной из позиций 6-9 системы кольца фенотиазина и представляет собой атом водорода или алкил, арил, ацил (предпочтительно ацетил), аминогруппу, нитрогруппу, сульфонил, гидроксил, алкоксильную группу, арилоксильную группу, арилтиогруппу или алкилтиогруппу или атом галогена, где соответствующие алкильные группы, возможно, но не обязательно, содержат, по меньшей мере, один дополнительный атом галогена либо группу NR4R5 или OR6, где группы R4, R5 и R6 являются одинаковыми или различными, и каждая из них независимо друг от друга выбрана из водорода и C1-C3-алкильных групп, а группа
R7 расположена в одной из позиций 1, 2 или 4 системы кольца фенотиазина и представляет собой атом водорода или алкил, арил, ацил (предпочтительно ацетил), аминогруппу, нитрогруппу, сульфонил, гидроксил, алкоксильную группу, арилоксильную группу, арилтиогруппу или алкилтиогруппу или атом галогена, где соответствующие алкильные группы, возможно, но не обязательно, содержат, по меньшей мере, один дополнительный атом галогена либо группу NR8R9 или OR10, где группы R8, R9 и R10 являются одинаковыми или различными, и каждая из них независимо друг от друга выбрана из водорода и C1-C3-алкильной группы.
Соответственно, эти производные являются пригодными для лечения нейродегенеративных заболеваний, в частности β-амилоидопатий или α-синуклеопатий, где это лечение, как уже упомянуто выше, содержит как профилактику, так и лечение доклинических стадий болезни. Атом галогена/атомы галогенов предпочтительно выбраны из фтора и хлора. Ацильными группами (-(C=0)-R) групп R1,2,3,7 предпочтительно являются ацетильные группы (-С(=0)CH3). Группы R1 и R2 предпочтительно являются одинаковыми или различными, и каждая из них, независимо друг от друга, представляет собой C1-C6-алкильную группу или C1-C6-алкильную группу (предпочтительно C1-алкил), замещенную ацетильной группой, а группы R3 и R7 представляют собой водород или ацетильную группу. В предпочтительном варианте осуществления изобретения группы R1 и R2 являются одинаковыми или различными, и каждая из них, независимо друг от друга, представляет собой C1-C3-алкильную группу. Кроме того, группами R3 и R7 предпочтительно является водород. Особо предпочтительно, чтобы группой R1 являлась метильная группа, группой R2являлась этильная группа, а группами R3 и R7 являлся водород (тиэтилперазин, торекан®). При использовании для лечения нейродегенеративных заболеваний была доказана целесообразность добавления дополнительных активных веществ, предпочтительно 1-бензогидрилпиперазинов, наиболее предпочтительно 1-бензогидрил-4-циннамилпиперазина (циннаризина).
Согласно настоящему изобретению лечение различных нейродегенеративных заболеваний может производиться производными 2-(R2-тио)-10-[3-(4-R1-пиперазин-1-ил) пропил]-10H-фенотиазина, или они могут быть диагностированы посредством описанного выше косвенного анализа. В наиболее предпочтительном варианте осуществления изобретения нейродегенеративным заболеванием является β-амилоидопатия, в частности болезнь Альцгеймера (БА). Другой вариант осуществления изобретения относится к тому случаю, когда нейродегенеративным заболеванием является α-синуклеопатия, в частности болезнь Паркинсона (БП). Обе болезни, то есть β-амилоидопатия и α-синуклеопатии, характеризуются отложениями белков в мозге, а их лечение может производиться посредством активизации переносчика АВСС1, или они могут быть диагностированы посредством его действия.
Ниже упомянуты другие заболевания, лечение которых также может производиться посредством активизации переносчика АВСС1 или которые могут быть диагностированы посредством действия переносчика АВСС1. Таким образом, другой болезнью, поддающейся лечению, является деменция с тельцами Леви (ДТЛ). Она также характеризуется агрегацией белков в мозге, то есть является α-синуклеопатией, как и болезнь Паркинсона.
Другой вариант осуществления изобретения относится к тому случаю, когда нейродегенеративным заболеванием является болезнь Хантингтона (БХ). Еще один вариант осуществления изобретения относится к тому случаю, когда нейродегенеративным заболеванием является прионная болезнь, в частности болезнь Крейцфельда-Якоба (БКЯ) или фатальная семейная бессонница (ФСБ FFI). Еще один вариант осуществления изобретения относится к тому случаю, когда нейродегенеративным заболеванием является таупатия, в частности кортикобазальная дегенерация (КБД), синдром Стила-Ричардсона-Ольшевского (прогрессирующий супрануклеарный паралич, ПСП) или болезнь Пика (БПк). Еще один вариант осуществления изобретения относится к тому случаю, когда нейродегенеративным заболеванием является лобно-височная лобарная дегенерация (ЛВЛД), в частности убиквитин-положительная дегенерация, TDP43-положительная дегенерация или к случаю убиквитин- и TDP43-отрицательных дегенераций. Еще один вариант осуществления изобретения относится к тому случаю, когда нейродегенеративным заболеванием является боковой амиотрофический склероз (БАС). Еще один вариант осуществления изобретения относится к тому случаю, когда нейродегенеративным заболеванием является спинально-церебеллярная атаксия (СЦА) или спастический парапарез (СПП). Еще один вариант осуществления изобретения относится к тому случаю, когда нейродегенеративным/нейроиммунологическим заболеванием является рассеянный склероз (PC) или синдром, связанный с PC, в частности острый рассеянный энцефаломиелит (ADEM) или синдром Девика.
ОПИСАНИЕ ЧЕРТЕЖЕЙ
На чертежах изображено следующее:
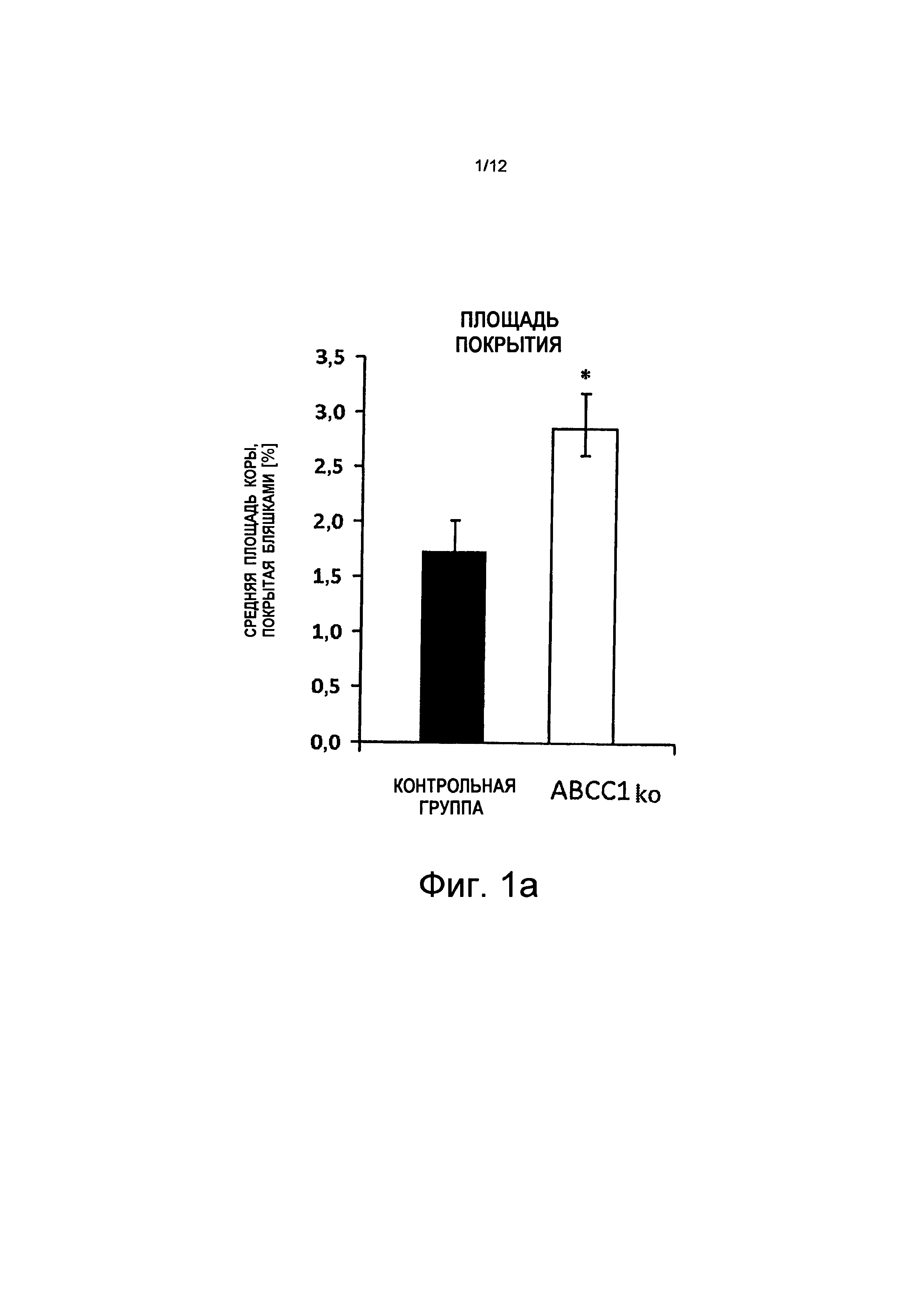
на Фиг.1а показано, что кортикальная плотность нейритических бляшек у мышей с дефицитом АВСС1 (ABCC1ko) увеличилась на ~75%;
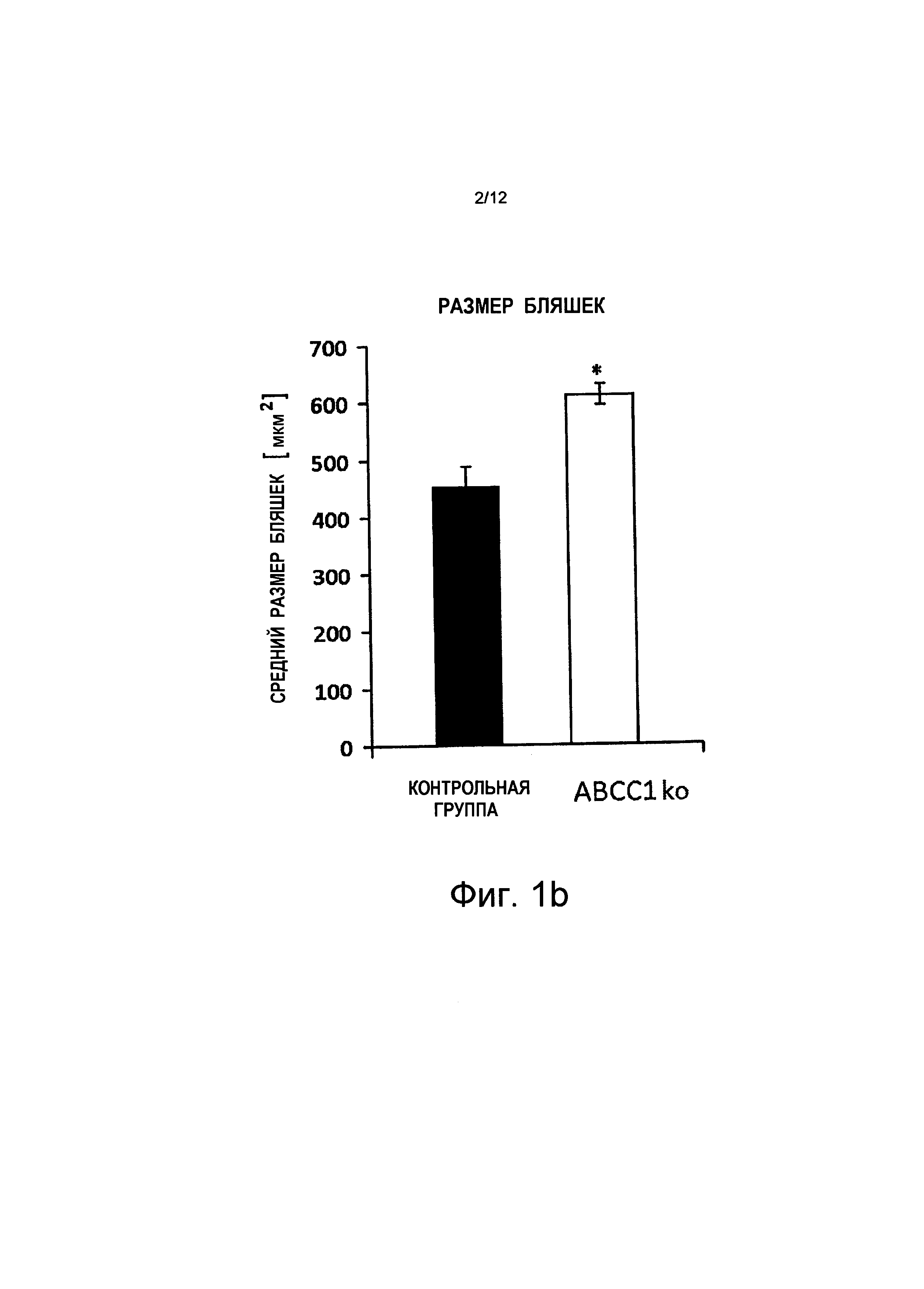
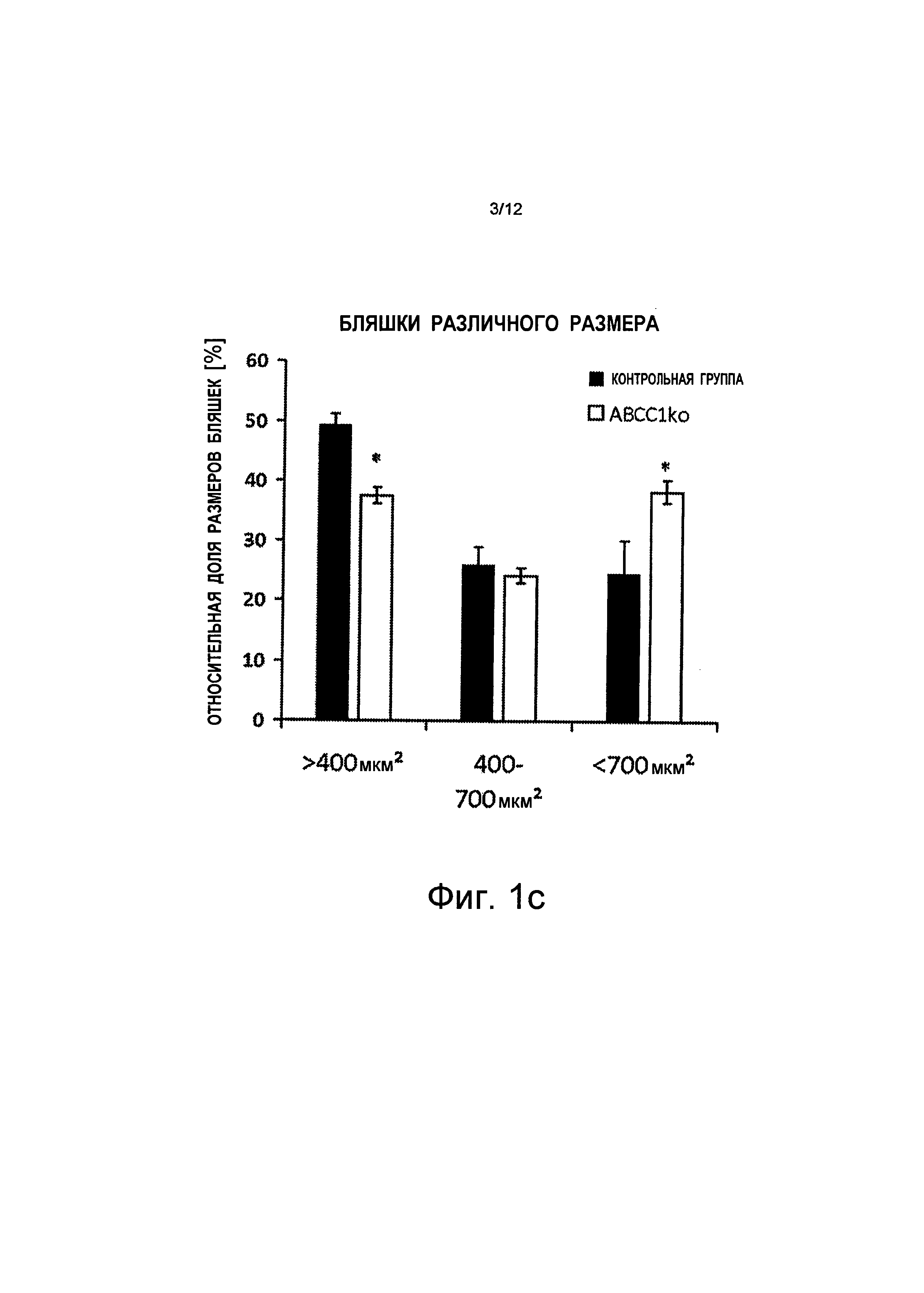
на Фиг.1b и Фиг.1c показано, что средний размер бляшки увеличился (+34%) в результате того, что большее количество бляшек (+63%) имеют размер свыше 700 мкм2 (квадратных микрометров), и более низкой частоты встречаемости бляшек меньшего размера (-24%). Показаны планки погрешностей, среднеквадратичная погрешность (n≥3);
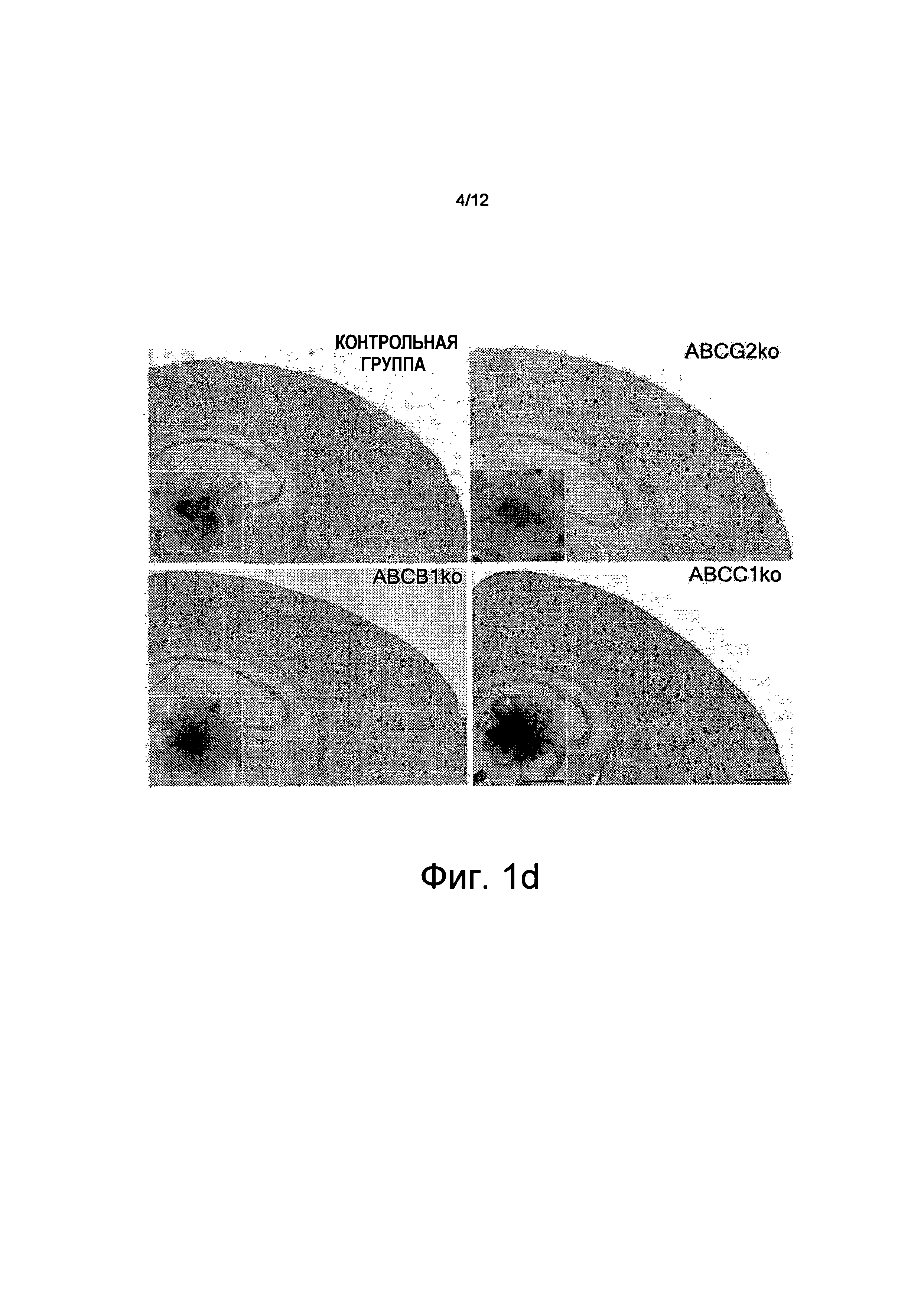
на Фиг.1d показано, что иммуногистохимическое (IHC) окрашивание у мышей с дефицитом ABCG2 (ABCG2ko), с дефицитом АВСВ1 (ABCB1ko), с дефицитом АВСС1 (ABCC1ko) и у мышей из контрольной группы показало более высокую поверхностную плотность Аβ у животных с дефицитом АВСС1. Типичные бляшки одинакового размера показаны в разрезе, масштабные полоски отображают 500 мкм (общий вид) и 50 мкм (разрез) (*p<0,05);
на Фиг.2а показано, что плотность бляшек в коре (площадь покрытия) у мышей с "выключенным" переносчиком конкретного ABC увеличена. В частности, мыши с дефицитом АВСС1 (ABCC1ko) показывают повышенное насыщение Аβ-амилоидом (светло-серые полосы, в каждом случае снаружи справа в отдельных группах), w = неделя на абсциссе;
на Фиг.2b показано, что общий размер бляшек у мышей с дефицитом АВСС1 (ABCC1ko) и с дефицитом ABCB1 (ABCB1ko) в возрасте 25 недель увеличен, w = неделя на абсциссе;
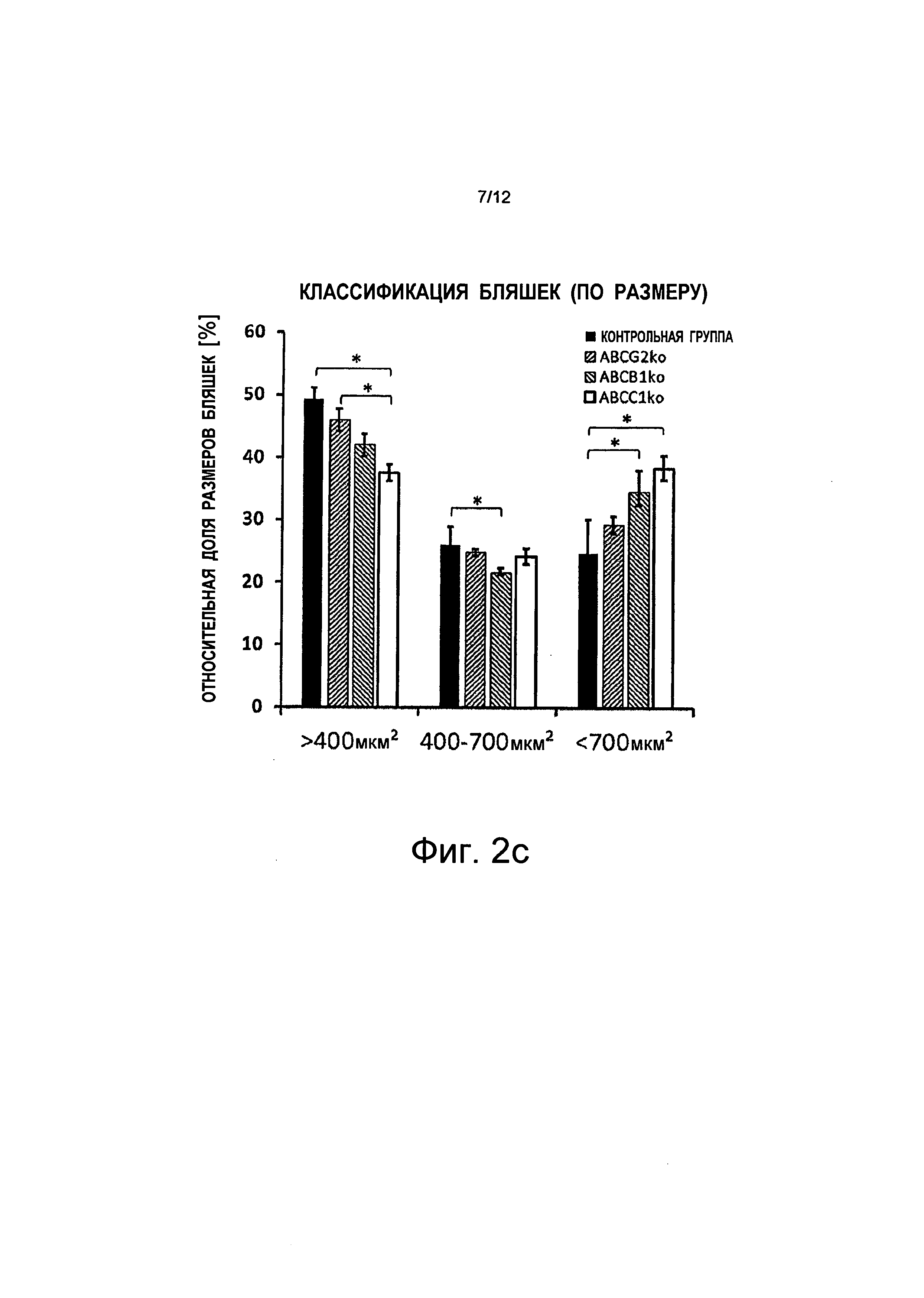
на Фиг.2c показано, что общее увеличение размера бляшек связано с меньшим количеством бляшек меньшего размера и с более значительным количеством бляшек большего размера (>700 мкм2), тогда как количество бляшек среднего размера остается равным тому же самому значению, показаны планки погрешностей, среднеквадратичная погрешность (n≥5), *p<0,05;
на Фиг.3 показано, что дефицит АВСС1 способствует накопление Аβ и Aβdt и что активизация АВСС1 (путем введения торекана в организм) уменьшает величины Аβ; где
на Фиг.3а показано, что в возрасте 25 недель дефицит АВСС1 приводит к заметному росту (~12 раз) нерастворимых Аβ; и
на Фиг.3b показано, что количество Аβ42, растворимых в буферном растворе, в возрасте 25 недель заметно уменьшено по сравнению с возрастом, равным 22 неделям (-56%). Это, вероятно, обусловлено отложением нерастворимых отложений. В том же самом возрасте область, покрытая отложениями Аβ, которая измерена методом иммуногистохимии, увеличена на 83% (планки погрешностей, среднеквадратичная погрешность n≥5, p<0,05);
на Фиг.3с показано, что 53% кровеносных сосудов сильно ухудшены вследствие ЦБА (>75% стенок сосудов демонстрируют Аβ). Это относится к мышам с дефицитом АВСС1 (ABCC1ko) по сравнению с 23% в контрольной группе (n=3);
на Фиг.3d показано, что экспрессия АВСС1 может быть замечена преимущественно в сосудистом сплетении желудочков мозга (CP), тогда как ABCD1 преимущественно выражен в капиллярах мозга (BP);
на Фиг.3е показано, что активизация АВСС1 тиэтилперазином (тореканом) снижает величины Аβ у мышей (-28%), показаны планки погрешностей, среднеквадратичная погрешность (n=4, *p<0,05).
ПРИМЕРЫ
Животные
АРР-трансгенные мыши (АРР, APPdt) были получены из Лаборатории Джексона (г.Бар-Харбор, США) и университета Тюбингена (г.Тюбинген, Германия). - ДЕФИЦИТНЫЕ НЭПОМ Мыши с деаицитом NEP были получены из Исследовательского института мозга Riken (г.Сайтама, Япония). Мыши с дефицитом ABCG2, АВСВ1 и АВСС1 были получены из фирмы "Таконик-фармз" (Дания). Все линии трансгенных мышей и мышей с "выключенным" геном были гибридизированы, по меньшей мере, в течение 9 поколений на генетическом FVB-фоне. Мыши содержались в условиях цикла 12 часов светлого времени/12 часов темноты при 23°C со свободным доступом к еде и воде.
СПОСОБЫ
Приготовление препаратов тканей
Для приготовления препаратов тканей мыши были умерщвлены путем смещения шейных позвонков и была произведена их транскардиальная перфузия PBS (фосфат-буферизованным физиологическим раствором). Мозг был удален, и одно полушарие было сохранено в забуференном 4%-ном параформальдегиде для заливки в парафин и для иммуногистохимии. Другое полушарие было подвергнуто резкой заморозке в жидком азоте и сохранено при -80°C для биохимических анализов.
Твердофазный иммуноферментный анализ (ELISA)
Для количественного анализа Аβ использовались наборы (TH40HS, TK42HS) для твердофазного иммуноферментного анализа (ELISA), производимые фирмой "Дженетикс Компани" (The Genetics Company (TGC г.Шлирен, Швейцария. Полушария мозга были гомогенизированы с использованием гомогенизатора PreCellys24 (12 с, 6500 об/мин). После добавления карбонатного буфера (pH 8,0) гомогенизаты были смешаны с использованием гомогенизатора PreCellys (5 с, 5000 об/мин) и подвергнуты центрифугированию в течение 90 минут при температуре 4°C и с ускорением 24000 g для отделения нерастворимых компонентов Аβ от растворимых компонентов Аβ. Оставшийся супернатант (фракция, растворимая в буферном растворе) был смешан с 8М гидрохлорида гуанидина в отношении 1:1,6. Для извлечения агрегированных компонентов Аβ сгусток после центрифугирования был растворен в 8 объемах 5М гидрохлорида гуанидина, подвергнут перемешиванию при комнатной температуре в течение 3 часов и подвергнут центрифугированию с ускорением 24000 g в течение 20 минут при 4°C. Оставшийся супернатант образовал растворимую в гуанидине фракцию (GuaHCI). Содержание белков во всех пробах было измерено три раза с использованием спектрофотометра Nanodrop1000 (выпущенного фирмой "Термо Фишер Саентифик" (Thermo Fisher Scientific), г.Уилмингтон, США). Твердофазные иммуноферментные анализы (ELISA) выполнялись согласно инструкциям изготовителя с использованием соответствующих разведений.
Вестерн-блоттинги
Гомогенизаты тканей были подготовлены для вестерн-блоттингов. Общие концентрации белков в экстрактах были определены путем анализа с использованием бицинхониновой кислоты (ВСА) (фирма "Пирс" (Pierce), являющаяся частью фирмы "Термо Фишер Саентифик" (Thermo Fisher Scientific), г.Рокфорд, США). После электрофореза всего 10 мкг (микрограмм) белков для каждого следа был произведен блоттинг белков на поливинилиденфторидных (PVDF) мембранах. После блокирования в 5%-ном сухом молоке в TBST-буфере (50 миллимолей трис-буфера pH 7,4, 150 миллимолей NaCl, 0,1% Tween20) в течение 1 часа при комнатной температуре блоты были исследованы на содержание любого из следующих веществ: АВСВ1 (1:500, D-11, Santa Cruz), АВСС1 (1:200, Alexis Bio) или β-актина (1:20.000, Sigma), за ночь при температуре 4°C. В качестве идентифицирующих антител использовались антитела мышей, меченные пероксидазой хрена (anti-mouse-HRP), антитела крыс, меченные пероксидазой хрена (anti-rat-HRP), или антитела зайцев, меченные пероксидазой хрена (anti-hare-HRP). Для визуализации использовался набор для обнаружения "Amersham ECL Plus Detection kit" и камера "Roper CoolSnap HQ2".
Иммуногистохимия (IHC)
Зафиксированные в формалине мозги были залиты парафином и нарезаны на срезы толщиной 4 мкм. После удаления парафина срезы были дополнительно обработаны красителем BondMax(TM) Autostainer (фирма "Менарини/Лейка" (Menarini/Leica), Германия). Иммунное окрашивание было инициировано после блокирования эндогенной пероксидазы (5 минут) и демаскировки антигена в течение 5 минут с использованием 95%-ной муравьиной кислоты (для антител 6F3D, произведенной фирмой "Дако" (Dako), Германия) и 70%-ной муравьиной кислоты (для антител 4G8, произведенной фирмой "Миллипор" (Millipore), Германия). Первичные антитела обычно инкубировались при комнатной температуре в течение 30 минут с приведенными ниже степенями разведения: 6F3D (1:100), 4G8 (1:500). Первичные антитела были обнаружены при помощи набора для обнаружения "BondMax (ТМ) Bond Polymer Refine detection kit" и согласно стандартному протоколу DAB R30. Срезы были полностью переведены в цифровую форму с разрешающей способностью 230 нм с использованием сканера "MiraxDesk/MiraxMidi", а затем автоматически проанализированы с использованием пакета программ "AxioVision" (фирмы "Цейсе" (Zeiss), Германия).
Оценка серьезности ЦБА
Срезы мозга с APPdt были окрашены антителом 4G8. По меньшей мере, два непоследовательных среза были исследованы на ЦБА менингеальных сосудов способом с маскированием. Все менингеальные сосуды были подсчитаны вручную, и серьезность ЦБА была классифицирована следующим образом:
Категория I: без негативного влияния
Категория II: положительно окрашено ≤25% периферии
Категория III: положительно окрашено ≤50% периферии
Категория IV: положительно окрашено ≤75% периферии
Категория V: положительно окрашено ≤100% периферии
Для каждой категории было вычислено среднее количество сосудов относительно общего количества идентифицированных сосудов.
Анализ эндотелиальных клеток с использованием вкладышей "трансвел" (ЕСТА)
Эндотелиальные клетки капилляров мозга мыши были подготовлены так, как описано в публикации Койсна (Coisne) и др. (Coisne С. и др. Mouse syngenic in vitro blood-brain barrier model: a new tool to examine inflammatory events in cerebral endothelium. Laboratory Investigation; 85, 734-746 (2005)). Мыши в возрасте, по меньшей мере, 3-4 недели были обезглавлены, и их мозг был извлечен. После рассечения ствола мозга, белого вещества и мягких мозговых оболочек ткань была гомогенизирована в двух объемах промывочного буфера В (WBB) (буферного солевого раствора Хенкса (HBBS), 10 миллимолей N-2-гидроксиэтилпиперазин-N'-2-этансульфоновой кислоты (HEPES), 0,1% бычьего сывороточного альбумина (BSA)) с использованием 15-миллилитрового стеклянного гомогенизатора Даунса (фирмы "Уитон Индастриз" (Wheaton Industries), г.Миллвилль, штат Нью-Джерси; США). К гомогенизату был добавлен один объем 30%-ного раствора декстрана. Этот состав был дважды повергнут центрифугированию с ускорением 3000 g и при температуре 4°C. Сгусток после центрифугирования, содержащий сосуды, был повторно суспендирован в WBB, и большие сосуды были разрушены вручную путем жесткого прокапывания раствора из пипетки. Для отделения больших сосудов от капилляров использовалась вакуумная фильтрация через 60-микронные мембраны (фирмы SEFAR, Швейцария). После комбинированной обработки коллагеназой/диспазой (HBSS, 10 миллимолей N-2-гидроксиэтилпиперазин-N'-2-этансульфоновой кислоты (HEPES), 0,15 г/мл TCLK, 10 мкг/мл дезоксирибонуклеазы-l, 1 мг/мл коллагеназы/диспазы (фирмы "Рош" (Roche)) была получена суспензия отдельных клеток путем дальнейшего жесткого прокапывания раствора из пипетки. Эндотелиальные клетки были введены в покрытые матригелем вкладыши "трансвел" (с порами 0,4 мкм, фирмы "Грейнер Био-Ван" (Greiner Bio-One), Германия) с плотностью 120000 клеток на каждый вкладыш, и была обеспечена возможность их роста на поддерживающей глиальной культуре.
Для определения трансклеточного переноса во время анализа использовалась желтая сера. Питательная среда адлюминального отсека была замещена раствором, содержащим 10 нанограммов Ass42 (конечная концентрация 1,6 наномолей). Затем через 2 часа, 6 часов или 24 часа были взяты пробы из люминального отсека и было определено содержание Аβ методом твердофазного иммуноферментного анализа (ELISA). (высокая чувствительность к TK42-high, TGC, Швейцария). Скорость переноса была описана в публикации Койсна и др. (Coisne С. и др. Mouse syngenic in vitro blood-brain barrier model: a new tool to examine inflammatory events in cerebral endothelium. Laboratory Investigation; 85, 734-746 (2005)).
Статистические данные твердофазного иммуноферментного анализа (ELISA)
К данным, полученным методом твердофазного иммуноферментного анализа (ELISA), и к данным, полученным методом твердофазного иммуноферментного анализа (ELISA), которые подвернуты логарифмическому преобразованию, был применен критерий согласия Лиллифорса (Lilliefors) (альфа=0,05) для проведения различий между предположением о том, что выборочные данные имеют нормальное распределение, и предположением о том, что выборочные данные имеют логарифмически нормальное распределение. Несмотря на небольшой размер выборки, для обоих наборов данных нулевая гипотеза (H0) была отклонена для 5 из 44 проб. В соответствии с наблюдением преимущественно положительных (с отклонением) и строго положительных выборочных данных было отклонено предположение о том, что данные имеют нормальное распределение. Были вычислены средние доверительные интервалы в предположении основного логарифмически нормального распределения. Был применен критерий суммы рангов Уилкоксона (Wilcoxon) для сравнения данных о различных штаммах мышей, полученных методом твердофазного иммуноферментного анализа (ELISA), для каждого момента времени.
Реферат
Изобретение относится к применению 2-(R-тио)-10-[3-(4-R-пиперазин-1-ил)пропил]-10H-фенотиазина общей формулы I для лечения β-амилоидопатии или α-синуклеопатии, сопровождаемой отложением белков в головном мозге и сниженной активностью АВСС1-переносчика в головном мозге, в том числе деменции Альцгеймера, болезни Паркинсона или деменции с тельцами Леви. В общей формуле I R- метильная группа, R- этильная группа, R- водород, и R- водород. 2 з.п. ф-лы, 12 ил.
Формула
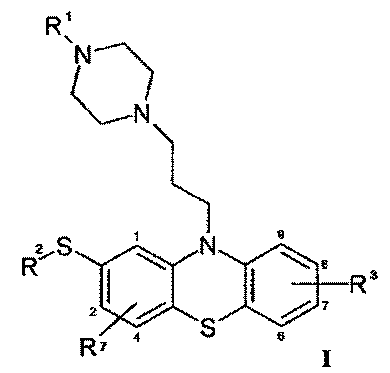
где
R1 - метильная группа,
R2 - этильная группа,
R3 - водород и
R7 - водород,
для лечения β-амилоидопатии или α-синуклеопатии, сопровождаемой отложением белков в головном мозге и сниженной активностью АВСС1-переносчика в головном мозге.
Комментарии