Производное акриланилида, способ его получения и его применения в фармакологии - RU2742372C2
Код документа: RU2742372C2
Описание
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к ингибиторам семейства протеиновых тирозинкиназ (PTK) рецептора эпидермального фактора роста (EGFR) и их фармацевтическим применениям.
УРОВЕНЬ ТЕХНИКИ
Рак, включая лейкемию, является одним из основных заболеваний, вызывающих клиническую смерть человека. Смертность от злокачественных опухолей чрезвычайно высока при раке легкого, раке желудка, раке молочной железы, раке поджелудочной железы, раке печени, раке кишечника и раке пищевода. До сих пор до сих пор нет эффективных терапевтических препаратов или методов, которые могут полностью уничтожить или вылечить рак. Существует настоятельная потребность в высококачественных противоопухолевых препаратах с хорошей специфичностью, высокой активностью, низкой токсичностью и отсутствием лекарственной устойчивости в клинических применениях. Заболеваемость, развитие, метастазирование и ухудшение состояния при раке связаны со многими факторами. Аномальность каскадов трансдукции сигналов в нормальных клетках, особенно в отношении мультифункциональных путей передачи сигналов, опосредованных трансмембранными рецепторами, является одним из основных факторов, приводящих к трансформации и возникновению раковых клеток. Протеиновые тирозинкиназы (ПТК, PTK) представляют собой ферменты, которые катализируют фосфорилирование тирозиновых остатков белков и являются необходимыми для мультифизиологических функций клеток, таких как рост, развитие, дифференцировка, обмен веществ, старение и апоптоз. В общем, ПТК можно разделить на две категории: ПТК мембранных рецепторов и цитоплазматические ПТК. Аномалии ПТК могут непосредственно приводить к различным клиническим заболеваниям, например, раку, воспалению, аутоиммунным заболеваниям, неврологическим или сердечно-сосудистым заболеваниям. После десятилетий непрерывных усилий было идентифицировано множество ПТК, таких как EGFR, HER2/3/4, VEGFR, PDGFR, Met, IGF-1R, FGFR, CSF-1R, Trk-рецептор, эфриновый рецептор, ТАМ-рецептор, Tie-2, FLT-3, RET, ALK, BCR-ABL, JAKs, SRC, FAK, BTK, SYK и BLK, которые могут служить целями для лекарств при различных заболеваниях в клинике. Некоторые из таких ингибиторов ПТК успешно применяются в клинической практике и продемонстрировали хорошие терапевтические эффекты.
EGFR является членом семейства EGFR, которое включает в себя четыре трансмембранных рецепторных протеиновых тирозинкиназ: EGFR (HER1/ErbB1), HER2/ErbB2, HER3/ErbB3 и HER4/ErbB4. Киназы семейства EGFR опосредуют важные множественные сигнальные пути в клетках. Они могут контролировать и регулировать многие физиологические функции клеток. Основные научные исследования, большие геномные и клинические данные показывают, что генетические аномалии EGFR, HER2, HER3 и HER4, такие как точечные мутации, делеции, амплификация и сверхэкспрессия, могут не только непосредственно приводить к клеточной злокачественной трансформации и опухолегенезу, но они также тесно связаны с пролиферацией, инвазией, выживанием, метастазированием, инфильтрацией, ангиогенезом и лекарственной устойчивостью опухолевых клеток.
В клинической практике аномальные генетические вариации EGFR (сверхэкспрессия, точечные мутации, делеции, вставки и т.д.) часто присутствуют у пациентов с различными видами рака, особенно раком легких. Рак легких - это вид злокачественной солидной опухоли с чрезвычайно высокой смертностью. Немелкоклеточная карцинома легкого (НМРЛ), в основном включающая аденокарциному, плоскоклеточную карциному и крупноклеточную карциному, составляет около 80% всего семейства рака легких, а высокая частота мутаций EGFR часто встречается при НМРЛ, что приводит к конститутивной активации опосредуемых им сигнальных путей, и к злокачественности клеток. Аналогичным образом, у пациентов с НМРЛ наблюдаются генетические аномалии HER2/ErbB2 (такие как мутации, амплификация и сверхэкспрессия), особенно у пациентов, демонстрирующих амплификацию и/или сверхэкспрессию HER2/ErbB2. Помимо НМРЛ изменения в HER2/ErbB2 часто встречаются при многих других опухолях, при некоторых из них вплоть до 30%, такие как рак молочной железы (20%), рак желудка (22-25%), рак пищевода (10 ~ 25%), рак поджелудочной железы (2 ~ 30%), карцинома мочевого пузыря (5-15%), карцинома слюнных каналов (15 ~ 37%), рак шейки матки (1 ~ 21%), злокачественная глиома (7 ~ 15%), сопровождающаяся НМРЛ (5%), колоректальный рак (2-3%), рак яичников (6-7%), рак головы и шеи (3%), гепатоцеллюлярная карцинома (2,4%) и меланома (0-5%). Кроме того, степень амплификации HER2 и/или сверхэкспрессии не только положительно коррелируют со степенью злокачественности опухоли, но также связаны с приобретенной лекарственной устойчивостью ко многим химиотерапевтическим средствам, таким как паклитаксел / оксалиплатин.
EGFR и HER2 использовались в качестве целей при разработке противораковых препаратов. До сих пор успешно разработаны или разрабатываются различные новые противораковые лекарственные средства, такие как макромолекулярные моноклональные антитела, включая цетуксимаб, панитумумаб и герцептин, которые нацелены на внеклеточный домен белков EGFR/HER2 и низкомолекулярные ингибиторы, такие как гефитиниб, эрлотиниб и лапатиниб, которые воздействуют на внутриклеточный киназный домен белков EGFR/HER2. Они применяются клинически в течение многих лет и продемонстрировали хорошую терапевтическую эффективность. Однако, как и многие другие противораковые лекарственные средства, препараты EGFR также имеют проблему приобретенной лекарственной устойчивости. Например, клиническая лекарственная устойчивость Гефитиниба и Эрлотиниба или Лапатиниба составляет до 50%. Множество факторов может вызвать приобретенную лекарственную устойчивость. Среди них структурные изменения целевого белка является одной из важных причин. Основной структурой соединений ингибиторов EGFR первого поколения, применяемых в клинической практике, является 4-анилинохиназолин, который может связываться с активным сайтом протеинкиназы EGFR и ингибировать активность протеинкиназы, конкурируя с АТФ. Мутации геномной ДНК часто приводят к изменениям аминокислотной последовательности белка и конформации структуры белка. Например, протеинкиназа EGFR может становиться конститутивно активной в результате изменений структуры белка, вызванных делецией экзона 19 EGFR, точечной мутации L858R экзона 21 или других мутаций, таких как G719S, G719A, G719C, L858R, L861Q и S768I, которые являются PTKi-чувствительными мутантами и сравнительно усиливают ингибирующее действие гефитиниба и эрлотиниба на EGFR. Однако некоторые вариации экзона 20 часто приводят к лекарственной резистентности. Например, когда треонин 790, «страж ворот» в домене протеинкиназы EGFR, мутирует в метионин, он значительно увеличивает аффинность между мутантной протеинкиназой и АТФ. Ингибиторы EGFR первого поколения потеряли способность конкурировать с АТФ, что привело к неэффективности препаратов и лекарственной устойчивости. Из-за такой приобретенной резистентности ингибиторы EGFR первого поколения не оказывают терапевтического воздействия на 40-55% пациентов с НМРЛ в клинической практике. Исследования также показали, что различные вставки аминокислот в экзоне 20 EGFR также могут вызывать лекарственную устойчивость. Несмотря на то, что необратимо действующие ингибиторы второго поколения, такие как афатиниб и нератиниб, ковалентно связывающиеся с цистеином 797 (Cys-797) в белке EGFR, были разработаны на основе структуры ингибиторов EGFR первого поколения и проявляют определенную ингибирующую активность в отношении EGFR Т790М in vitro, они по-прежнему проявляют сильное ингибирующее действие на EGFR дикого типа и клинически проявляют частые побочные эффекты и токсичность. Кроме того, поскольку у них не было очевидного преимущества в лечении пациентов с НМРЛ с экспрессией EGFR Т790М при их одиночном применении, их клиническое применение было значительно ограниченным. Кроме того, ингибиторы EGFR второго поколения также генерировали разные степени приобретенной лекарственной устойчивости, которые связаны с новыми мутантами EGFR и частично другими аномалиями онкогена (такими как амплификация Met/HER3, мутация PIK3CA/ BRAF, потеря NF1 и активация сигнализации FGFR).
Недавние исследования показали, что низкомолекулярное соединение WZ4002 с 2,4-пиримидином в качестве новой основной структуры может высокоэффективно ингибировать мутант EGFR Т790М, тогда как эффекты на EGFR дикого типа относительно слабы. Данные ранних клинических исследований соединений СО-1686 и AZD9291 с каркасом ядра на основе 2,4-пиримидина, последовательно разработанные двумя фармацевтическими компаниями, показывают, что они лучше действуют на пациентов с мутацией EGFR Т790М и обладают относительно низкими побочными эффектами. Они представляют собой новое поколение эффективных ингибиторов мутантов EGFR Т790М.
Ингибирование активности EGFR может эффективно ингибировать рост НМРЛ, однако аномальная экспрессия других генов, такая как амплификация и сверхэкспрессия HER2/ErbB2, амплификация HGFR (МЕТ), а также амплификация и перегруппировка киназы анапластической лимфомы (ALK) также тесно связаны со злокачественным ростом и лекарственной устойчивостью НМРЛ. И во многих других злокачественных опухолях, таких как рак желудка, рак молочной железы, рак пищевода и карцинома слюнных протоков, HER2/ErbB2 является еще одной важной мишенью при раке. Хотя противоопухолевый препарат моноклонального антитела против HER2 герцептин и низкомолекулярное соединение лапатиниб на рынке клинически демонстрируют благоприятные терапевтические эффекты, их проблемы, такие как приобретенная лекарственная резистентность и гематоэнцефалический барьер, ограничивают их широкое клиническое применения.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Объектом настоящего изобретения является получение низкомолекулярного соединения ингибитора семейства EGFR PTK с высокой специфичностью, активностью и низкой токсичностью. Заявителем было обнаружено, что новый тип ингибиторов EGFR / HER2, предложенных в настоящем изобретении, имеет неожиданные технические эффекты, которые могут эффективно ингибировать рост опухолевых клеток со сверхэкспрессией EGFR или HER2/ErbB2 и различные клинически частые мутации, особенно мутации приобретенной лекарственной устойчивости, такие как EGFR Т790М, с минимальными побочными эффектами, тем самым завершив настоящее изобретение.
Настоящее изобретение относится к новым производным акриланилида и их фармацевтически приемлемым солям, которые могут высокоселективно воздействовать на рост in vitro и in vivo различных линий опухолевых клеток человека, которые экспрессируют мутантные гены EGFR (такие как активный мутант EGFR delE746-A750, мутанты с приобретенной лекарственной устойчивостью L858R / Т790М и delE746-A750 / Т790М) и амплифицируют HER2 / ERBB2, необратимым ковалентным связыванием. Они имеют ценность для различных клинических способ лечения.
Семейство EGFR PTK - идеальная цель для противораковой терапии, которая успешно применяется в клинике. Несмотря на то, что ингибиторы протеинкиназы EGFR / HER2, выпущенные на рынок, могут эффективно усиливать клинические терапевтические эффекты у пациентов с раком, приобретенная резистентность или серьезные побочные эффекты, вызванные лекарствами, могут существенно повлиять на клинические терапевтические эффекты таких препаратов, которые не могут удовлетворить клинические требования. Настоящие соединения проявляют сильную противоопухолевую активность как в анализах in vitro, так и в экспериментах in vivo у животных. 50% -ное ингибирование роста (GI50) показывает, что соединение может эффективно ингибировать рост раковой клетки, экспрессирующей различные мутанты EGFR, в частности лекарственно резистентный мутант Т790М, в наномолярных концентрациях. Кроме того, ингибирующая активность в отношении линий раковых клеток, экспрессирующих EGFR дикого типа, является относительно низкой и/или небольшой. Это значительно снизит риски, связанные с кожными и желудочно-кишечными побочными эффектами, вызванными ингибированием EGFR дикого типа, такими как сыпь и диарея.
HER2 / ErbB2 является другим членом семейства EGFR РТК. Его аномальное экспрессия часто происходит при и связано со многими злокачественными опухолями. Авторы настоящего изобретения неожиданно обнаружили, что настоящие соединения могут значительно ингибировать рост различных линий опухолевых клеток с высокой экспрессией гена HER2/ErbB2 (таких как NCl-N87, Calu-3, AU-565, SK-BR-30, NCl- Н2170 и ZR-75-30). Концентрация GI50 находится в наномолярном диапазоне. Кроме того, настоящие соединения также демонстрируют определенные эффекты ингибирования роста на резистентной к лапатинибу клеточной линии НСС1954 (лапатиниб является единственным одобренным FDA селективным обратимым ингибитором HER2 / ERB2, применяемым в клинической практике до настоящего времени). Они могут быть клинически использованы при НМРЛ и различных видах рака, таких как рак желудка, рак молочной железы, рак пищевода, карцинома слюнных протоков с высокой экспрессией HER2 / ErbB2.
Настоящее изобретение включает следующее.
[1] Соединение формулы (I), или его фармацевтически приемлемые соли, сольваты или пролекарства:

где:
X, Y и R1 выбраны из любого а), b) и с) ниже:
a) когда R1 представляет собой -NR5R6, X и Y являются одинаковыми или отличными друг от друга, и каждый независимо выбран из N и CR4;
b) когда R1 выбран из -OR5 и -SR5, X и Y являются одинаковыми или отличными друг от друга, и каждый независимо выбран из N и CR4;
c) когда R1 представляет собой -CR5R6, и когда R5 и R6 вместе с атомами углерода, к которым они присоединены, образуют цикл, X представляет собой CR4 и Y представляет собой N;
R2 выбран из группы, состоящей из алкокси, алкилсульфанила и NR6R6;
R3 выбран из группы, состоящей из водорода, N(Ry)(Rz), -N(Rv)RuN(Ry)(Rz), -ORuOR6, -ORuN(Ry)(Rz), -SR6 и -SRuN(Ry)(Rz);
R4 и R'4 каждый выбран из группы, состоящей из водорода, галогена, алкила, алкокси, галоалкила и циано;
R5 и R6 выбраны из любого а), b) и с) ниже:
a) R5 представляет собой возможно замещенный арил, возможно замещенный гетероарил или возможно замещенный гетероциклил; в случае замещения заместитель выбран из 1~5 групп R7; где каждая группа R7 независимо выбранна из группы, состоящей из водорода, галогена, алкила, алкенила, алкинила, алкокси, гидроксила, амино, галоалкокси, циклоалкила, циклоалкилалкила, гидроксилалкила, галоалкила, арила, арилалкила, гетероциклил а, гетероциклил-алкила, гетероарила или гетероарил-алкила; алкил, алкенил, алкинил, алкокси, амино, галоалкокси, циклоалкил, циклоалкил-алкил, гидроксилалкил, галоалкил, арил, арилалкил, гетероциклил, гетероарил или гетероарил-алкил возможно замещены 1-5 группами, выбранными из группы, состоящей из галогена, алкила, алкенила, алкинила, арила, гидроксила, алкокси, галоалкокси, циклоалкила, эфирной группы и циано;
R6 выбран из водорода и алкила;
b) R5 и R6 образуют гетероциклил, гетероарил или конденсированное ароматическое кольцо вместе с атомом азота, присоединенного к ним, где кольцо содержит 0-4 гетероатома, независимо выбранных из О, S и N; в случае замещения заместитель возможно представляет собой 1-5 групп, выбранных из группы, состоящей из галогена, галоалкила, алкила, алкенила и циано;
c) R5 и R6 образуют конденсированное ароматическое кольцо вместе с атомами углерода, присоединенными к ним, где кольцо содержит 0-4 гетероатома, независимо выбранных из О, S и N; в случае замещения заместитель возможно представляет собой 1-4 групп, выбранных из группы, состоящей из галогена, галоалкила, алкила, алкенила и циано;
Each Ru независимо выбран из алкиленалкилена, алкенилена или алкинилена;
Rv выбран из водорода и алкила;
Ry и Rz каждый независимо выбран из а) и b), приведенных далее:
a) Ry и Rz являются каждый независимо, выбранными из группы, состоящей из водорода, алкила, циклоалкила, алкокси алкила, гидроксилалкила, пирролидила, алкиламино или галоалкила;
b) Ry и Rz образуют гетероциклил или гетероарил вместе с атомами азота, присоединенными к ним, где кольцо содержит 0-4 гетероатома, независимо выбранных из О, S и N; указанное кольцо возможно замещено 1-4 группами, выбранными из группы, состоящей из R5 и R7.
[2] Соединение или его фармацевтически приемлемые соли, сольваты или пролекарства, описанные в [1] выше, которые отличаются тем, что соединение формулы (I) представляет собой соединение формулы (IIa), (IIb) или (IIc),

где, R1 выбран из а) или b) ниже:
a) когда соединение формулы (I) представляет собой соединение формулы IIa или IIb, R1 представляет собой -NR5R6, -OR5 или -SR5;
b) когда соединение формулы (I) представляет собой соединение формулы IIc, R1 представляет собой -NR5R6; R5 и R6 вместе с атомами углерода, к которым они присоединены, образуют цикл;
R2 представляет собой алкокси;
R3 выбран из группы, состоящей из водорода, N(Ry)(Rz), -N(Rv)RuN(Ry)(Rz), -ORuOR6, или -ORuN(Ry)(Rz);
R4 и R'4 являются каждый независимо, выбранными из группы, состоящей из водорода, галогена, алкила и галоалкила;
R5 и R6 выбраны из любого а), b) и с) ниже:
a) R5 представляет собой возможно замещенный арил; в случае замещения заместитель выбран из 1~5 групп R7; каждая группа R7 независимо выбрана из группы, состоящей из водорода, галоген, алкила, алкенила, алкинила, алкокси, гидроксила, амино, галоалкокси, циклоалкила, циклоалкилалкила, гидроксилалкила, галоалкила, арила, арилалкила, гетероциклила, гетероциклил алкила, гетероарила или гетероарилалкила; где алкил, алкенил, алкинил, алкокси, амино, галоалкокси, циклоалкил, циклоалкил-алкил, гидроксилалкил, галоалкил, арил, арилалкил, гетероциклил, гетероарил или гетероарилалкил возможно замещены 1-5 группами, выбранными из группы, состоящей из галогена, алкила, алкенила, алкинила, арила, гидроксила, алкокси, галоалкокси, циклоалкила, эфирной группы и циано;
R6 выбран из водорода и алкила;
b) R5 и R6 образуют конденсированное ароматическое кольцо вместе с атомами азота, присоединенными к ним, где кольцо содержит 0-4 гетероатома, независимо выбранных из О, S и N; в случае замещения заместитель возможно представляет собой 1-4 группы, выбранных из группы, состоящей из галогена, галоалкила, алкила, алкенила и циано;
c) R5 и R6 образуют конденсированное ароматическое кольцо вместе с атомами углерода, присоединенными к ним, где кольцо содержит 0-4 гетероатома, независимо выбранных из О, S и N; в случае замещения заместитель возможно представляет собой 1-4 группы, выбранных из группы, состоящей из галогена, галоалкила, алкила, алкенила и циано;
Каждый Ru независимо выбран из алкилена;
Rv выбран из водорода и алкила;
Ry и Rz каждый независимо выбран из а) или b) ниже:
a) Ry и Rz являются каждый независимо, выбранными из группы, состоящей из водорода, алкила и галоалкила;
b) Ry и Rz образуют гетероциклил вместе с атомами азота, присоединенными к ним, где кольцо содержит 0-4 гетероатома, независимо выбранных из О, S и N; указанное кольцо возможно замещено 1-4 группами, выбранными из группы, состоящей из галогена, галоалкила, алкила, алкокси алкила, алкилгидроксила, NR6R6 или гетероциклила.
[3] Соединение или его фармацевтически приемлемые соли, сольваты или пролекарства, описанные в [2] выше, которые отличаются тем, что соединение формулы (IIa) представляет собой соединение формулы (III):
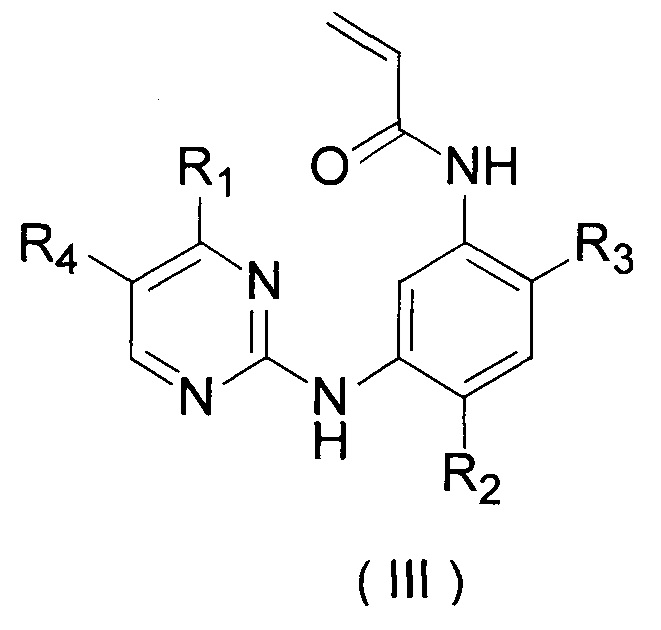
где, R1 выбран из группы, состоящей из

R2 выбран из группы, состоящей из С1-С6 алкокси и С3-С6 циклоалкокси;
R3 выбран из группы, состоящей из

R4 выбран из группы, состоящей из водорода, метила, трифторметила и галогена.
[4] Соединение или его фармацевтически приемлемые соли, сольваты или пролекарства, описанные в [2] выше, которые отличаются тем, что соединение формулы (IIa) представляет собой соединение формулы (IV):

где:
R7a, R7b, R7c, R7d и R7e являются одинаковыми или отличными друг от друга, и каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, алкенила, алкинила, алкокси, гидроксила, амино, галоалкокси, циклоалкила, циклоалкилалкила, гидроксилалкила, галоалкила, арила, арилалкила, гетероциклила, гетероциклилалкила, гетероарила или гетероарилалкила; где алкил, алкенил, алкинил, алкокси, амино, галоалкокси, циклоалкил, циклоалкилалкил, гидроксилалкил, галоалкил, арил, арилалкил, гетероциклил, гетероарил или гетероарилалкил возможно замещены 1-5 группами, выбранными из группы, состоящей из галогена, алкила, алкенила, алкинила, арила, гидроксила, алкокси, галоалкокси, циклоалкила, эфирной группы и циано;
R2 выбран из группы, состоящей из С1-С6 алкокси и С3-С6 циклоалкокси;
R3 выбран из группы, состоящей из

R4 выбран из группы, состоящей из водорода, метила, трифторметила и галогена.
[5] Соединение или его фармацевтически приемлемые соли, сольваты или пролекарства, описанные в [4] выше, где в соединении формулы (IV):
R1 выбран из группы, состоящей из

где, X выбран из группы, состоящей из фтора, хлора и брома;
R2 представляет собой С1-С6 алкокси;
R3 является таким же, как определено в [4];
R4 выбран из водорода, трифторметила и хлора.
[6] Соединение или его фармацевтически приемлемые соли, сольваты или пролекарства, описанные в [2] выше, которые отличаются тем, что соединение формулы (IIb) представляет собой соединение формулы (V):

где, R7a, R7b, R7c, R7d и R7e являются одинаковыми или отличными друг от друга, и каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, алкенила, алкинила, алкокси, гидроксил, амино, галоалкокси, циклоалкил, циклоалкилалкила, гидроксилалкила, галоалкила, арила, арилалкила, гетероциклила, гетероциклилалкила, гетероарила или гетероарилалкила; где, алкил, алкенил, алкинил, алкокси, амино, галоалкокси, циклоалкил, циклоалкилалкил, гидроксилалкил, галоалкил, арил, арилалкил, гетероциклил, гетероарил или гетероарилалкил возможно замещены 1-5 группами, выбранными из группы, состоящей из галогена, алкила, алкенила, алкинила, арила, гидроксила, алкокси, галоалкокси, циклоалкила, эфирной группы и циано; R2 выбран из группы, состоящей из С1-С6 алкокси и С3-С6 циклоалкокси;
R3 выбран из группы, состоящей из
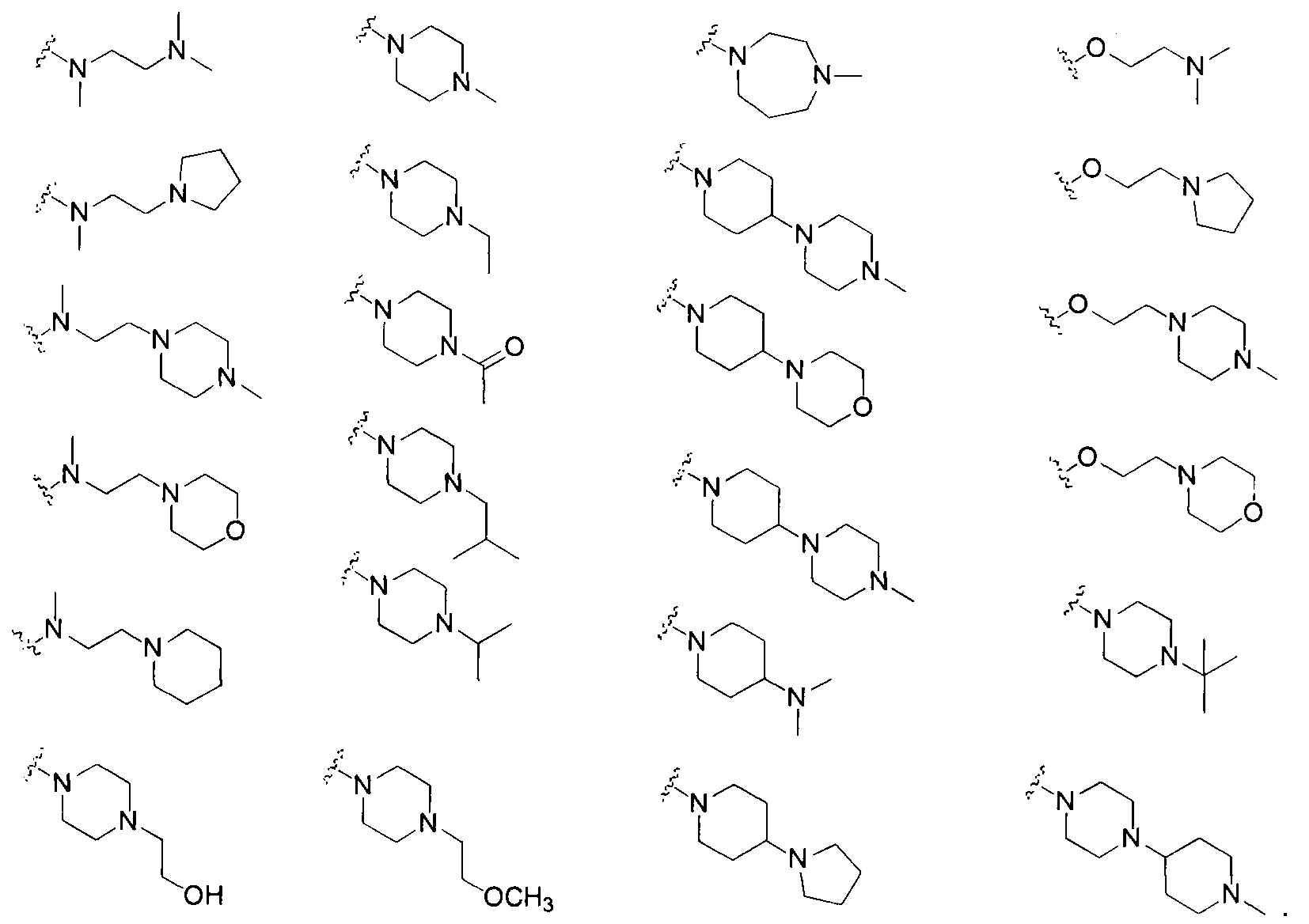
[7] Соединение или его фармацевтически приемлемые соли, сольваты или пролекарства, описанные в [6] выше, где в соединении формулы (V):
R1 выбран из группы, состоящей из
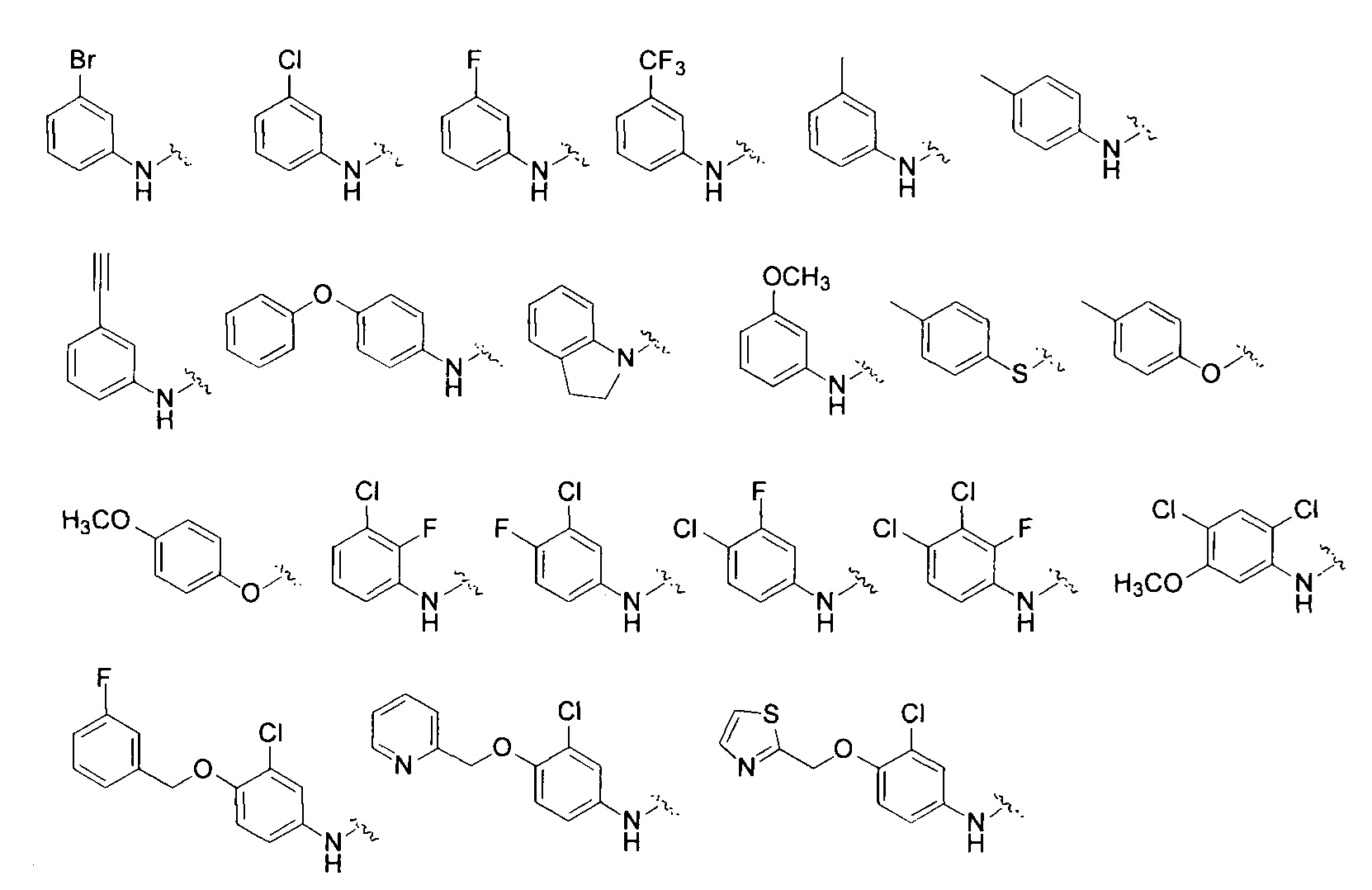
R2 представляет собой С1-С6 алкокси;
R3 является таким же, как определено в [6].
[8] Соединение или его фармацевтически приемлемые соли, сольваты или пролекарства, описанные в [2] выше, где соединение формулы (IIc) представляет собой соединение формулы (VI):

где:
R1 выбран из группы, состоящей из

R7a, R7b, R7c, R7d и R7e являются одинаковыми или отличными друг от друга, и являются каждый независимо выбранными из группы, состоящей из водорода, галогена, алкила, алкенила, алкинила, алкокси, гидроксила, амино, галоалкокси, циклоалкила, циклоалкилалкила, гидроксилалкила, галоалкила, арила, арилалкила, гетероциклила, гетероциклилалкила, гетероарила и гетероарилалкила; где алкил, алкенил, алкинил, алкокси, амино, галоалкокси, циклоалкил, циклоалкилалкил, гидроксилалкил, галоалкил, арил, арилалкил, гетероциклил, гетероарил или гетероарилалкил возможно замещены 1-5 группами, выбранными из группы, состоящей из галогена, алкила, алкенила, алкинила, арила, гидроксила, алкокси, галоалкокси, циклоалкила, эфирной группы и циано; R2 выбран из группы, состоящей из С1-С6 алкокси и С3-С6 циклоалкокси;
R3 выбран из группы, состоящей из

[9] Соединение или его фармацевтически приемлемые соли, сольваты или пролекарства, описанные в [8] выше, где в соединении формулы (VI):
R1 выбран из группы, состоящей из

R2 представляет собой С1-С6 алкокси;
R3 выбран из группы, состоящей из

[10] Соединение или его фармацевтически приемлемые соли, сольваты или пролекарства, описанные в [2] выше, где соединение формулы (IIc) представляет собой соединение формулы (VII):

где, R7a, R7b, R7c, R7d и R7e являются одинаковыми или отличными друг от друга, и являются каждый независимо, выбранными из группы, состоящей из водорода, галогена, алкила, алкенила, алкинила, алкокси, гидроксила, амино, галоалкокси, циклоалкила, циклоалкилалкила, гидроксилалкила, галоалкила, арила, арилалкила, гетероциклила, гетероциклилалкила, гетероарила или гетероарилалкила; где, алкил, алкенил, алкинил, алкокси, амино, галоалкокси, циклоалкил, циклоалкилалкил, гидроксилалкил, галоалкил, арил, арилалкил, гетероциклил, гетероарил или гетероарилалкил возможно замещены 1-5 группами, выбранными из группы, состоящей из галогена, алкила, алкенила, алкинила, арила, гидроксила, алкокси, галоалкокси, циклоалкила, эфирной группы и циано;
R2 выбран из группы, состоящей из С1-С6 алкокси и С3-С6 циклоалкокси;
R3 является таким же, как определено в [2] выше.
[11] Соединение или его фармацевтически приемлемые соли, сольваты или пролекарства, описанные в [10] выше, где в соединении формулы (VII):

выбран из группы, состоящей из

R2 представляет собой С1-С6 алкокси;
R3 выбран из группы, состоящей из

[12] Соединения или их фармацевтически приемлемые соли, сольваты или пролекарства, описанные в любом из [1]~[11] выше, где соединение выбрано из группы, состоящей из
N-(5-(4-(3-бромофениламино)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-хлорфениламино)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-фторфениламино)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-трифторметилфениламино)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-метилфениламино)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(4-метилфениламино)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(4-метилфенилтио)-пиримидин-2-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-ацетенилфениламино)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(4-феноксилфениламино)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(фениламино)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-метоксифенокси)-пиримидин-2-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(3-(4-(3-хлор-4-фтор-фениламино)-пиримидин-2-иламино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-хлор-4-фторфениламино)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-хлор-2-фторфениламино)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3,4-дихлор-2-фторфениламино)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(2,4-дихлор-5-метоксифениламино)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-хлор-4-(пиридин-2-илметокси)-фениламино)-пиримидин-2-иламино)-2-диметиламино-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-хлор-4-(пиридин-2-илметоксил)-фениламино)-пиримидин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксил-фенил)-акриламид;
N-(5-(5-хлор-4-(3-хлор-4-(пиридин-2--илметоксил)-фениламино)-пиримидин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-хлор-4-(пиридин-2-илметокси)-фениламино)-пиримидин-2-иламино)-2-(4-морфолин-4-ил)-пиперидин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-хлор-4-(пиридин-2-илметокси)-фениламино)-пиримидин-2-иламино)-2-(4-(4-метилпиперазин-1-ил)-пиперидин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-хлор-4-(пиридин-2-илметокси)-фениламино)-пиримидин-2-иламино)-2-(4-(1-метилпиперидин-4-ил)-пиперазин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-хлор-4-(пиридин-2-илметокси)-фениламино)-пиримидин-2-иламино)-2-(4-метилпиперазин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-хлор-4-(пиридин-2-илметокси)-фениламино)-пиримидин-2-иламино)-2-(4-(2-гидроксиэтил)пиперазин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-хлор-4-(пиридин-2-илметокси)-фениламино)-пиримидин-2-иламино)-2-(4-метил-[1,4]диазепин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-хлор-4-(пиридин-2-илметокси)-фениламино)-пиримидин-2-иламино)-2-(4-(пиперидин-1-ил)-пиперидин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-хлор-4-(пиридин-2-илметокси)-фениламино)-пиримидин-2-иламино)-2-(4-ацетилпиперазин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-хлор-4-(пиридин-2-илметокси)-фениламино)-пиримидин-2-иламино)-2-(4-(диметиламино)пиперазин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(-4-(3-хлор-4-(бензол-2-метоксил)-фениламино)-пиримидин-2-иламино)-2-(2-(морфолин-4-ил)-этоксил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-хлор-4-(3-фторбензилокси)фениламино)пиримидин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксил-фенил)-акриламид;
N-(2-((2-(диметиламино-этил)-метил-амино)-5-(4-(индолин-1-ил)-пиримидин-2-иламино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(1Н-индол-1-ил)-пиримидин-2-иламино)-4-метоксил-2-(4-ацетил-пиперазин-1-ил)-фенил)-акриламид;
N-(5-(4-(1Н-индол-1-ил)-пиримидин-2-иламино)-4-метоксил-2-(4-(4-метил-пиперазин-1-ил)-пиперидин-1-ил)-фенил)-акриламид;
N-(5-(4-(1Н-индол-1-ил)-пиримидин-2-иламино)-4-метоксил-2-(4-(морфолин-4-ил)-пиперидин-1-ил)-фенил)-акриламид;
N-(5-(4-(1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-(циклогексилметил)-пиперазин-1-ил)-4-метоксифенил)-акриламид;
N-(5-(4-(1Н-индол-1-ил)-пиримидин-2-амино)-4-метоксил-2-(4-метил-1,4-диазепин-1-ил)-фенил)-акриламид;
N-(2-(1,4'-бипиперидин-1'-ил-)-5-(4-(1Н-индол-1-ил)-пиримидин-2-иламино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-диметиламино-пиперидин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-(2-метоксилэтил)-пиперазин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-пирролидил-пиперазин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-(2-цианоэтил)-пиперазин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-трет-бутилпиперазин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(1Н-индол-1-ил)-пиримидин-2-иламино)-4-метоксил-2-(4-(2-(пиридин-4-ил)-этил)-пиперазин-1-ил)-фенил)-акриламид;
N-(5-(4-(1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-бензилпиперазин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-(бензо[d][1,3]диокси-5-метил)-пиперазин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-(2-гидроксипропил-2-ил)-пиперидин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-(гидроксиметил)-пиперидин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-изобутилпиперазин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-(морфолинил)-этоксил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-диметиламино)-этоксил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-пирролидин-1-ил)-этоксил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-4-метил-пиперазин-1-ил)-этоксил)-4-метоксил-фенил)-акриламид;
N-{2-[(2-диметиламино)-этил)-метил-амино]-4-метоксил-5-[4-(6-метоксил-индол-1-ил)-пиримидин-2-иламино]-фенил}-акриламид;
N-{2-[(2-диметиламино)-этил)-метил-амино]-4-метоксил-5-[4-(6-метил-индол-1-ил)-пиримидин-2-иламино]-фенил}-акриламид;
N-{5-[4-(6-циано-индол-1-ил)-пиримидин-2-иламино]-2-[(2-диметиламино-этил)-метил-амино]-4-метоксил-фенил}-акриламид;
N-(5-(4-(6-хлор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-((2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(6-бромо-1Н-индол-1-ил)-пиримидин-2-иламино)-2-((2-(диметиламино)этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(6-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-((2-(диметиламино)этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(6-фтор-1H-индол-1-ил)-пиримидин-2-иламино)-2-(4-(диметиламино)-пиперидинил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(6-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(диметиламино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(6-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-(4-метилпиперазин-1-ил)-пиперидин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(6-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-(1-метилпиперидин-4-ил)-пиперазин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(6-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-(морфолин-4-ил)-пиперидин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(6-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-(2-гидроксил-этил)-пиперазин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(6-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-метил-пиперазин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(6-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-ацетил-пиперазин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(6-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-метил-[1,4]диазепин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(6-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-(пиперидин-1-ил)-пиперидин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(6-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-4-акрилоил-пиперазин-1-ил)-этоксил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(6-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-(морфолин-4-ил)-этоксил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(6-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-пирролидин-1-ил)-этоксил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(5-метил-1Н-индол-1-ил)-пиримидин-2-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(5-метоксил-1Н-индол-1-ил)-пиримидин-2-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
2-((2-акриламидо-5-метоксил-4-(4-(5-метоксил-1Н-индол-1-ил)-пиримидин-2-ил)-амино)-фенил)-метил-N,N-диметил-N-этиламина оксид;
N-(5-(4-(5-хлор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(5-бромо-1Н-индол-1-ил)-пиримидин-2-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(5-трифторметил-1Н-индол-1-ил)-пиримидин-2-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(5-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-((2-(диметиламино-этил-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(5-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-ацетил-пиперазин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(5-фтор-1Н-индол-1-ил-)-пиримидин-2-иламино)-2-(4-(пиперидин-4-ил)-пиперидин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(5-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-(циклогексилметил)-пиперидин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(5-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-метил-[1,4]диазепин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(5-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-(диметиламинометил)-пиперидин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(5-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-(морфолин-4-ил)-пиперидин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(5-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-(4-метилпиперазин-1-ил)-пиперидин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(5-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-метил-пиперазин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(5-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-(1-метилпиперидин-4-ил)-пиперазин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(5-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-(2-гидроксил-этил)-пиперазин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(5-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-(2-(2-гидроксиэтоксил)-этил)-пиперазин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(5-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(диметиламино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(5-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-(морфолин-4-ил)-этоксил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(5-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-пирролидин-1-ил)-этоксил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(4-метил-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(4-метоксил-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(4-циано-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(4-хлор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(4-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(5,6-дифтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(5-фтор-6-хлор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-{5-[4-(6-хлор-5-фтор-индол-1-ил)-пиримидин-2-иламино]-2-[(2-диметиламино-этил)-метил-амино]-4-пропоксил-фенил}-акриламид;
N-(5-(4-бензотриазол-1-ил-пиримидин-2-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-хлор-фениламино)-[1,3,5]триазин-2-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-хлор-4-фтор-фениламино)-[1,3,5]триазин-2-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-хлор-2-фтор-фениламино)-[1,3,5]триазин-2-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-хлор-4-(пиридин-2-илметокси)-фениламино)-[1,3,5]триазин-2-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-хлор-4-(3-фторфенилметокси)-фениламино)-[1,3,5]триазин-2-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-трифторметил-фениламино)-[1,3,5]триазин-2-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-фтор-фениламино)-[1,3,5]триазин-2-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-бромо-фениламино)-[1,3,5]триазин-2-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-[5-[4-(3-бромо-фениламино)-[1,3,5]триазин-2-иламино]-2-(3-диметиламино-пирролидин-1-ил)-4-метоксил-фенил]-акриламид;
N-[5-[4-(3-бромо-фениламино)-[1,3,5]триазин-2-амино]-2-(4-диметиламино-пиперидин-1-ил)-4-метоксил-фенил]-акриламид;
N-[5-[4-(3-бромо-фениламино)-[1,3,5]триазин-2-амино]-4-метоксил-2-(4-(пирролидин-1-ил-пиперидин-1-ил)-фенил-акриламид;
N-{2-[1,4']-пиперидинилпиперидин-1'-ил-5-[4-(3-бромо-фениламино)-[1,3,5]триазин-2-амино]-4-метоксил-фенил}-акриламид;
N-(5-(4-(3-бромо-фениламино)-[1,3,5]триазин-2-иламино)-4-метоксил-2-(4-(4-метил-пиперазин-1-ил)-пиперидин-1-ил)-фенил)-акриламид;
N-(5-(4-(3-бромо-фениламино)-[1,3,5]триазин-2-иламино)-2-(4-этил-пиперазин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-бромо-фениламино)-[1,3,5]триазин-2-иламино)-2-(4-трет-бутил-пиперазин-1-ил)-4-метоксил-фениламино)-акриламид;
N-(5-(4-(3-бромо-фениламино)-[1,3,5]триазин-2-иламино)-2-(4-(3-диметиламино-пропил)-пиперазин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-бромо-фениламино)-[1,3,5]триазин-2-иламино)-4-метоксил-2-(4-(1-метилпиперидин-4-ил)-пиперазин-1-ил)-фенил)-акриламид;
N-{5-[4-(3-бромо-фениламино)-[1,3,5]триазин-2-иламино]-2-[4-(2-циано-этил)-пиперазин-1-ил]-4-метоксил-фенил}-акриламид;
N-{5-[4-(3-бромо-фениламино)-[1,3,5]триазин-2-иламино]-2-[4-(2-гидроксил-этил)-пиперазин-1-ил]-4-метоксил-фенил}-акриламид;
N-{5-[4-(3-бромо-фениламино)-[1,3,5]триазин-2-иламино)-4-метоксил-2-(4-(2-метоксил-этил)-пиперазин-1-ил)-фенил]-акриламид;
N-(5-(4-(3-бромо-фениламино)-[1,3,5]триазин-2-иламино)-2-(4-циклогексил-метил-пиперазин-1-ил)-4-метоксил-фенил)-акриламид;
N-(2-(4-ацетил-пиперазин-1-ил)-5-(4-(3-бромо-фениламино)-[1,3,5]триазин-2-иламино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(3-бромо-фениламино)-[1,3,5]триазин-2-иламино)-4-метоксил-2-(4-метил-[1,4]диазепан-1-ил)-фенил)-акриламид;
N-[5-[4-(3-бромо-фениламино)-[1,3,5]триазин-2-иламино]-4-метоксил-2-(2-морфолин-4-ил-этоксил)-фенил]-акриламид;
N-[5-[4-(3-бромо-фениламино)-[1,3,5]триазин-2-иламино]-4-метоксил-2-(2-метоксил-этоксил)-фенил]-акриламид;
N-(5-(6-(3-бромо-фениламино)-пиримидин-4-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(2-((2-диметиламино)-этил)-метил-амино)-4-метоксил-5-(6-(3-трифторметил-фениламино)-пиримидин-4-иламино)-фенил)-акриламид;
N-(5-(6-(3-алкинил-фениламино)-пиримидин-4-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(2,,4-дихлор-5-метоксил-фениламино)-пиримидин-4-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(2-фтор-3,4-дихлор-фениламино)-пиримидин-4-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(2-фтор-3-хлор-фениламино)-пиримидин-4-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(3-бромо-5-фтор-фениламино)-пиримидин-4-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(3-хлор-4-фтор-фениламино)-пиримидин-4-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(3-хлор-4-фтор-фениламино)-2-метил-пиримидин-4-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(3-хлор-4-фтор-фениламино)-пиримидин-4-иламино)-2-[пиперидин-1-ил]-4-метоксил-фенил)-акриламид;
N-(5-(6-(3-хлор-4-фтор-фениламино)-пиримидин-4-иламино)-4-метоксил-2-(4-морфолин-4-ил-пиперидин-1-ил)-фенил)-акриламид;
N-(5-(6-(3-хлор-4-фтор-фениламино)-пиримидин-4-иламино)-2-(4-метил-[1,4]диазепин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(6-(3-хлор-4-фтор-фениламино)-пиримидин-4-иламино)-4-метоксил-2-(4-метил-пиперазин-1-ил)-фенил)-акриламид;
N-(2-(4-ацетил-пиперазин-1-ил)-5-(6-(3-хлор-4-фтор-фениламино)-пиримидин-4-иламино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(3-хлор-4-фтор-фениламино)-пиримидин-4-иламино)-4-метоксил-2-(4-(1-метилпиперидин-4-ил)-пиперазин-1-ил)-фенил)-акриламид;
N-(5-(6-(3-хлор-4-фтор-фениламино)-пиримидин-4-иламино)-2-(2-диметиламино-этоксил)-4-метоксил-фенил)-акриламид;
N-(5-(6-(3-хлор-4-фтор-фениламино)-пиримидин-4-иламино)-4-метоксил-2-(2-пирролидин-1-ил-этоксил)-фенил)-акриламид;
N-(5-(6-(3-хлор-4-фтор-фениламино)-пиримидин-4-иламино)-2-(2-(4-метил-пиперазин-1-ил)-этоксил)-4-метоксил-фенил)-акриламид;
N-(5-(6-(3-хлор-4-фтор-фениламино)-пиримидин-4-иламино)-4-метоксил-2-(2-морфолин-4-ил-этоксил)-фенил)-акриламид;
N-(5-(6-(3-хлор-4-фтор-фениламино)-пиримидин-4-иламино)-2-(2-метоксилэтоксил)-4-метоксил-фенил)-акриламид;
N-(5-(6-(3-хлор-4-(3-фтор-бензилокси)-фениламино)-пиримидин-4-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(3-хлор-4-(пиридин-2-илметокси)-фениламино)-пиримидин-4-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-бензимидазол-1-ил-пиримидин-4-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-бензотриазол-1-ил-пиримидин-4-иламино)-2-((2-диметиламино-этил-метил-амино)-4-метоксил-фенил)-акриламид;
N-(4-метоксил-5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-2-(4-метилпиперазин-1-ил)-фенил)-акриламид;
N-(2-(4-этил-пиперазин-1-ил)-4-метоксил-5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-фенил)-акриламид;
N-(2-(4-трет-бутил-пиперазин-1-ил)-4-метоксил-5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-фенил)-акриламид;
N-(4-метоксил-5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-2-(4-(1-метилпиперидин-4-ил)-пиперазин-1-ил)-фенил)-акриламид;
N-(2-(4-(2-гидроксил-этил)-пиперазин-1-ил)-4-метоксил-5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-фенил)-акриламид;
N-(4-метоксил-2-(4-(2-метоксил-этил)-пиперазин-1-ил)-5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-фенил)-акриламид;
N-(2-(4-(2-(2-гидроксил-этоксил)-этил)-пиперазин-1-ил)-4-метоксил-5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-фенил)-акриламид;
N-(4-метоксил-5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-2-(4-(2-(пирролидин-1-ил)-этил)-пиперазин-1-ил)-фенил)-акриламид;
N-(2-(4-(2-(диметиламино)-этил)-пиперазин-1-ил)-4-метоксил-5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-фенил)-акриламид;
N-(2-(4-ацетилпиперазин-1-ил)-4-метоксил-5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-фенил)-акриламид;
N-(4-метоксил-5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-2-(4-фенилпиперазин-1-ил)-фенил)-акриламид;
N-(2-(4-бензилпиперазин-1-ил)-4-метоксил-5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-фенил)-акриламид;
N-(4-метоксил-5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-2-(4-(-(пиримидин-3-илметил)-пиперазин-1-ил)-фенил)-акриламид;
N-(4-метоксил-5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-2-(4-метил-2-фенилпиперазин-1-ил)-фенил)-акриламид;
N-(2-(4-(бис-(4-фтор-фенил)-метил)-пиперазин-1-ил)-4-метоксил-5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-фенил)-акриламид;
N-(4-метоксил-2-(4-метил-[1,4]диазепин-1-ил)-5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-фенил)-акриламид;
(S)-N-(2-(3-диметиламино-пирролидин-1-ил)-4-метоксил-5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-фенил)-акриламид;
N-(2-(4-диметиламино-пиперидин-1-ил)-4-метоксил-5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-фенил)-акриламид;
N-(4-метоксил-5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-2-(4-(4-метилпиперазин-1-ил)-пиперидин-1-ил)-фенил)-акриламид;
N-(2-(4-(морфолин-4-ил)пиперидин-1-ил)-4-метоксил-5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-фенил)-акриламид;
N-(4-метоксил-5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-2-(метил-(2-пирролидин-1-ил-этил)-амино)-фенил)-акриламид;
N-(5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-2-иламино)-2-((2-(диметиламино)-этил-метил-амино)-4-метоксил-фенил)-акриламид;
N-{2-[(2-диметиламино-этил)-метил-амино]-5-[6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино]-4-пропил-фенил}-акриламид;
N-{2-((2-диметиламино-этил)-метиламино)-5-[6-(1-этил-1Н-индол-3-ил)-пиримидин-4-иламино]-4-метоксил-фенил}-акриламид;
N-{2-[(2-диметиламино-этил)-метиламино]-5-[6-(1-пропил-1Н-индол-3-ил)-пиримидин-4-иламино]-4-метоксил-фенил}-акриламид;
N-{2-[(2-диметиламино-этил)-метиламино]-5-[6-(1-изопропил-1Н-индол-3-ил)-пиримидин-4-иламино]-4-метоксил-фенил}-акриламид;
N-{2-[(2-диметиламино-этил)-метиламино]-5-[6-(1-изопропил-1Н-индол-3-ил)-пиримидин-4-иламино]-4-пропоксил-фенил}-акриламид;
N-(5-(6-(1-бензил-1Н-индол-3-ил)-пиримидин-4-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(4-метоксил-5-(6-(6-фтор-1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-2-(2-(диметиламино-этил)-метил-амино)-фенил)-акриламид;
N-(2-((2-диметиламино-этил)-метил-амино)-5-(4-(5-фтор-индол-1-ил)-пиримидин-2-иламино)-4-пропил-фенил)-акриламид;
N-(2-((2-диметиламино-этил)-метиламино)-5-(6-(5-фтор-1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-4-метоксил-фенил)-акриламид;
N-(5-[6-(5-фтор-1-изопропил-1Н-индол-3-ил)-пиримидин-4-иламино]-2-{[2-(1-окси-пирролидин-1-ил)-этил]-метил-амино}-4-метоксил-фенил)-акриламид;
N-(5-(6-(5-фтор-1-изопропил-1Н-индол-3-ил)-пиримидин-4-иламино)-4-метокси-2-(4-(4-метил-пиперазин-1-ил)-пиперидин-1-ил)-фенил)-акриламид;
N-(5-(6-(5-фтор-1-изопропил-1Н-индол-3-ил)-пиримидин-4-иламино)-4-метоксил-2-((2-диметиламино-этил)-метил-амино)-фенил)-акриламид;
2-((2-акриламидо-5-метоксил-4-((6-(1-изопропил-5-фтор-1Н-индол-3-ил)-пиримидин-4-ил)-амино)-фенил)-метил)-N,N-диметил-N-этиламина оксид;
N-(5-(6-(5-фтор-1-циклопентил-1Н-индол-3-ил)-пиримидин-4-иламино)-4-метоксил-2-((2-диметиламино-этил)-метиламино)-фенил)-акриламид;
N-(2-((2-(диметиламино)-этил)-метиламино)-4-метоксил-5-(2-метил-6-(1-метил-1Н-индол-3-ил)-пиримидин-4-ил-амино)-фенил)-акриламид;
N-(5-(5-метил-6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-4-метоксил-2-((2-диметиламино-этил)-метил-амино)-фенил)-акриламид;
N-(4-метоксил-2-(2-метоксил-этоксил)-5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-фенил)-акриламид;
N-(5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-4-метокси-2-(2-(4-метилпиперазин-1-ил)-этоксил)-фенил)-акриламид;
N-(5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-4-метокси-2-(2-(морфолин-4-ил)-этоксил)-фенил)-акриламид;
N-(5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-4-метокси-2-(2-(пирролидин-1-ил)-этоксил)-фенил)-акриламид;
N-(5-(4-((3-хлор-4-(пиридин-2-метоксил)фенил)-метил-амино)-пиримидин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксил-фенил)-акриламид;
N-(5-(5-хлор-4-(3-хлор-4-(пиридин-2-метоксил)феноксил)пиримидин-2-илами но)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(4-метоксил-1Н-индол-1-ил)-5-трифторметил-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(4-метоксил-1Н-индол-1-ил)-5-хлор-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(4-гидроксил-1Н-индол-1-ил)-5-хлор-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(4-(2-метоксил-этоксил)-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(4-н-гексилокси-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(5-(2-метоксил-этоксил)-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(4-метоксил-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(4-(4-метил-пиперазин-1-ил)-пиперидин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(4-(6-(2-метоксил-этоксил)-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(4-((тетрагидрофуран-2-ил)-метоксил)-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(4-метоксил-6-бромо-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(4-метоксил-6-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(4-метоксил-6-хлор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(4-(4-(2,5-диметилпиррол-1-ил)-6-бромо-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(3-хлор-4-(2-морфолин-4-ил-этоксил)-фениламино)-пиримидин-4-иламино)-2-((2-диметиламиноэтил)-метил-амино)-4-метоксифенил)-акриламид;
N-(5-(6-(3-хлор-4-(тетрагидропиран-4-ил-метоксил)-фениламино)-пиримидин-4-иламино)-2-((2-диметиламиноэтил)-метил-амино)-4-метоксифенил)-акриламид;
N-(5-(6-(3-(1-(3-метилбутокси)этил)-4-метоксифениламино)-пиримидин-4-иламино)-2-((2-диметиламиноэтил)-метил-амино)-4-метоксифениламино)-акриламид;
N-(5-(6-(3-хлор-4-метоксил-фениламино)-пиримидин-4-иламино)-2-(2-диметиламино-этоксил)-4-метоксил-фенил)-акриламид;
N-(5-(6-(3-хлор-4-(3-метилбутокси)-фениламино)-пиримидин-4-иламино)-2-((2-диметиламиноэтил)-метил-амино)-4-метоксифенил)-акриламид;
5-(6-(5-акриламидо-4-((2-метоксифенил)-метил-амино)-2-метоксифениламино)-пиримидин-4-ил)-2-метоксибензамид;
5-(6-(5-акриламидо-4-((2-метоксифенил)-метил-амино)-2-метоксифениламино)-пиримидин-4-ил)-2-метоксил-N-метилбензамид;
N-(5-(6-(4-метоксил-3-трифторметил-фениламино)-пиримидин-4-ил-амино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(3-хлор-4-(тиазол-2-илметокси)-фениламино)-пиримидин-4-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(4-метоксил-3-бромо-фениламино)-пиримидин-4-ил-амино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(4-метоксил-3-хлор-фениламино)-пиримидин-4-ил-амино)-2-(2-(пирролидин-1-ил)-этоксил)-4-метоксил-фенил)-акриламид;
N-(5-(6-(4-фтор-3-трифторметил-фениламино)-пиримидин-4-ил-амино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(3-хлор-4-метоксил-фениламино)-пиримидин-4-иламино)-2-(4-(1-метилпиперидин-4-ил)-пиперазин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(6-(4-хлор-3-трифторметил-фениламино)-пиримидин-4-ил-амино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(3-циано-фениламино)-пиримидин-4-ил-амино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(5-фтор-3-трифторметил-фениламино)-пиримидин-4-ил-амино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(2-фтор-5-трифторметил-фениламино)-пиримидин-4-ил-амино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(3-метоксил-5-трифторметил-фениламино)-пиримидин-4-ил-амино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(2-метоксил-5-трифторметил-фениламино)-пиримидин-4-ил-амино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(3-хлор-4-трифторметил-фениламино)-пиримидин-4-ил-амино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(3-хлор-4-трифторметокси-фениламино)-пиримидин-4-ил-амино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(3-хлор-5-фтор-4-(2-метоксил-этоксил)-фениламино)-пиримидин-4-ил-амино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(3-метансульфонамидо-4-метоксил-фениламино)-пиримидин-4-ил-амино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(2,4-дихлор-5-метоксил-фениламино)-пиримидин-4-ил-амино)-2-(4-метил-пиперазин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(6-(2,4-дихлор-5-метоксил-фениламино)-пиримидин-4-ил-амино)-2-(4-(морфолин-1-ил)-пиперидин-1-ил)-4-метоксил-фенил)-акриламид;
N-(5-(6-(2,4-дихлор-5-метоксил-фениламино)-пиримидин-4-ил-амино)-2-(метил-(2-(4-метил-пиперазин-1-ил)-2-оксоэтил)-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(2,4-дихлор-5-метоксил-фениламино)-пиримидин-4-иламино)-2-(2-диметиламино-этоксил)-4-метоксил-фенил)-акриламид;
N-(5-(6-(2,4-дихлор-5-метоксил-фениламино)-пиримидин-4-ил-амино)-2-(2-(морфолин-4-ил)-этоксил-4-метоксил-фенил)-акриламид;
N-(5-(б-(2,4-дихлор-5-метоксил-фениламино)-пиримидин-4-ил-амино)-2-(2-(пирролидин-1-ил)-этоксил)-4-метоксил-фенил)-акриламид;
N-(5-(6-(2,4-дихлор-5-метоксил-фениламино)-пиримидин-4-ил-амино)-2-(2-(4-метил-пиперазин-1-ил)-этоксил)-4-метоксил-фенил)-акриламид;
N-(5-(6-(2,4-дихлор-5-метоксил-фениламино)-пиримидин-4-ил-амино)-2-(3-(пирролидин-1-ил)-пропил)-4-метоксил-фенил)-акриламид;
N-(5-(6-(2,4-дихлор-5-метоксил-фениламино)-пиримидин-4-ил-амино)-2-(2-(пиперидин-1-ил)-этоксил-4-метоксил-фенил)-акриламид;
N-(5-(6-(2,4-дихлор-5-метоксил-фениламино)-пиримидин-4-ил-амино)-2-(3-(4-метил-пиперазин-1-ил)-пропил)-4-метоксил-фенил)-акриламид;
N-(5-(6-((2,4-дихлор-5-метоксил-фенил)-метил-амино)-пиримидин-4-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(2,4-дихлор-5-метоксил-фениламино)-пиримидин-4-ил-амино)-2-(3-(морфолин-4-ил)-пропоксил-4-метоксил-фенил)-акриламид;
N-(5-(6-(3-хлор-4-трет-бутокси-фениламино)-пиримидин-4-ил-амино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(3-ацетенил-4-метоксил-фениламино)-пиримидин-4-ил-амино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид;
N-(5-(6-(3-хлор-4-(3-метил-оксетан-3-ил-метоксил)-фениламино)-пиримидин-4-ил-амино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламид
[13] Ингибитор EGFR PTK, содержащий соединение или его фармацевтически приемлемые соли, описанные в любом из [1]~[12] в качестве активного вещества.
[14] Ингибитор HER2/ErbB2 PTK, содержащий соединение или его фармацевтически приемлемые соли, описанные в любом из [1]~[12] в качестве активного вещества.
[15] Применение соединения или его фармацевтически приемлемых солей, описанные в любом из [1]~[12] при получении ингибиторов EGFR и/или HER2/ErbB2 PTK.
[16] Применение соединения или его фармацевтически приемлемых солей, описанные в любом из [1]~[12] при получении лекарственного средства для предотвращения или лечения рака.
Ингибиторы PTF EGFR в соответствии с настоящим изобретением могут эффективно и избирательно воздействовать на мутанты EGFR, включая виды с приобретенной лекарственной устойчивостью и чувствительные (активный тип). Приобретенная резистентная мутация EGFR возникает из мутации EGFR Т790 (например, Т790М), а активирующий мутантный штамм возникает из-за мутации в экзоне 19, экзоне 18 и экзоне 21 EGFR (таких как делеция экзона 19, мутация G719S и мутация L858R) и других мутаций (таких как мутация S761I).
Ингибиторы PTK HER2/ErbB2 в соответствии с настоящим изобретением могут эффективно и избирательно воздействовать на опухолевые клетки с амплификацией или сверхэкспрессией гена HER2/ErbB2 или активирующими мутациями (такими как мутации G776VC или V777M).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Если не указано иного, все технические и научные термины, используемые здесь, должны иметь то же значение, которое обычно понимается специалистом в данной области. Все патенты, заявки, публичные заявки и другие публикации включены сюда путем ссылки. Если существует несколько определений для используемых здесь терминов, если не указано иное, главными являются термины в настоящем описании.
«Галоген» относится к фтору, хлору, брому и йоду.
«Алкил» относится к алкилам с прямой или разветвленной цепью, содержащим от 1 до 18 атомов углерода, предпочтительно от 1 до 10 атомов углерода, более предпочтительно от 1 до 6 атомов углерода, еще более предпочтительно от 1 до 4 атомов углерода, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-амил, изоамил, нео-пентил, гексил, изогексил, н-гептил, изогептил, н-октил, изооктил, н-нонил, н-децил и т.д.. В контексте настоящего изобретения «алкил» также относится к циклоалкилу, содержащему от 3 до 10 атомов углерода, предпочтительно от 3 до 8 атомов углерода и более предпочтительно от 4 до 6 атомов углерода, например циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклононил, циклодецил, декагидронафталинил, норборнан, адамантил и т.д..
«Алкенил» относится к группе с линейной или разветвленной углеводородной цепочкой, состоящей из атомов углерода и водорода, которая содержит по меньшей мере одну двойную связь. Он имеет 2-10 атомов углерода, предпочтительно 2-6 атомов углерода, и соединяется с другими частями молекулы посредством одинарной связи или двойной связи, например, этилен, пропенил, бутенил, пентенил, пентадиенил и гексенил.
«Алкокси» относится к -OR группам, где R означает алкил, как определено выше. Типичные примеры включают, без ограничения, метоксил, этоксил, пропоксил, изопропоксил, бутоксил, изобутокси, втор-бутокси, трет-бутокси, циклопропоксил, циклобутоксил и т.д.
«Алкинил» относится к группам с линейной или разветвленной углеводородной цепочкой, состоящим из атомов углерода и атомов водорода, которые содержат по меньшей мере одну тройную связь. Он имеет 2-10 атомов углерода, предпочтительно 2-6 атомов углерода, и соединяется с другими частями молекулы посредством одинарной связи или тройной связи, например ацетил, пропинил, бутинил, пентинил и гексинил.
«Алкилацил» относится к группам R (С=O), где R обозначает алкил, как определено выше. Типичные примеры включают, без ограничения, ацетил, пропионил, бутирил, валерил, гексаноил и т.д.
«Арил» относится к углеродной кольцевой системе, включая моноциклическую, бициклическую, трициклическую и тетрациклическую кольцевую систему С6-С18, где по меньшей мере одно кольцо является ароматическим. Арил может представлять собой полную ароматическую группу, такую как фенил, нафтил, антрацил и фенантрил. Арил также может быть комбинацией ароматического кольца и неароматического кольца, например инден, флуорен и аценафтен. Предпочтительный арил включает фенил, нафтил и т.д.
«Галоалкил» относится к алкилу, как определено выше, с одним или несколькими атомами водорода, замещенными галогеном. Типичные примеры включают, без ограничения, хлорметил, трифторметил, 1-хлор-2-фторэтил, 2,2-дифторэтил, 2-фторпропил, 2-фторпропан-2-ил, 2,2,2-трифторэтил, 1,1-дифторэтил, 1,3-дифтор-2-метилпропил, 2,2-дифторциклопропил, (трифторметил)циклопропил, 4,4-дифторциклогексил и 2,2,2-трифтор-1,1-диметилэтил.
«Гетероциклил» относится к 3 - 15 членным (например, 3-12 членным или 3-9-членным) гетероциклам с более чем одним, предпочтительно от 1 до 5 кольцевыми атомами, возможно выбранными из гетероатомов О, S и N. Гетероциклил может быть моноциклической, бицикло, трициклической и тетрациклической системами. Он может быть конденсированным кольцом или мостиковым кольцом. N или S атомы в гетероциклиле могут быть возможно окислены. N-атомы могут быть возможно кватернизированы. Гетероциклил может быть частично или полностью насыщенным. Гетероциклическая система может быть связана с основной структурой по любому гетероатому или атому углерода с образованием стабильных соединений. В частности, гетероциклил включает 5-6 членный гетероарил, такой как пирролил, имидазолил, пиразолил, пиридинил, пиридазинил, пиримидинил, пиразинил, триазолил, триазинил, тетразолил, фурил, тиенил, изоксазолил, оксазолил, оксадиазолил, изотиазолил, тиазолил, тиадиазолил; неароматические гетероциклические группы, такие как пиранил, тиазолидинил, пирролидил, пирролинил, имидазолидинил, имидазолинил, пиразолидинил, пиразолинил, пиперидинил, пиперазинил, морфолинил, морфолино, тиоморфолинил, тиоморфолино, дигидропиридинил, тетрагидродигидропиридинил, тетрагидрофуранил, тетрагидропиранил, диазепин и тетрагидродиазепин; бицикло или трициклический конденсированный гетероциклил, такой как индолил, изоиндолил, индазолил, дигидроиндолил, изодиогидроиндолил, хинолил, изохинолил, циннолинил, фталазинил, хиназолинил, нафтиридинил, хиноксалинил, пуринил, птеридил, бензопиранил, бензимидазолил, бензотриазол, бензоизоксазолил, бензоксазолил, бензоксадиазолил, бензоизотиазолил, бензотиазолил, бензофуранил, изобензофуранил, бензотиенил, бензотриазол, тиенопиридинил, имидазотиазолил, бензимидазотиазолил, пиридинопиридазинил, хиназолинил, хинолил, изохинолил и т.д.; предпочтительно пирролил, фурил, имидазолил, изоксазолил, оксазолил, пиримидил, пиридил, тиазолил, тиенил, морфолинил, пиперидинил, пиперазинил, пиранил, пирролидинил, индолил, диазепинтил, бензотиенил, бензотриазол, бензимидазолил и т.д.
Алкильная часть в «арилалкиле», «арилалкилокси», «галогенарилалкилокси», «алкиламино», «алкилациле» и «галогеналкиле» является такой же, как определено выше.
Арильная часть в «арилокси», «ариламино», «арилтио», «арилоксиле», «арилалкиле», «арилалкилоксиле», «галогенарилалкилоксиле» и «галогенариле» является такой же, как определено выше.
Гетероциклильная часть в «гетероциклилалкокси» является такой, как определено выше, и алкоксильная часть в ней также является такой же, как определено выше.
«Возможно замещенный» в настоящем изобретении относится к тому случаю, когда не замещен или замещен одним или несколькими (например, 2, 3 и 4) заместителями. Заместителями являются выбранными из группы, состоящей из галогена, алкила, алкенила, алкинила, галоалкил, алкокси, арила, галоарила, арилокси, арилалкила, арилалкилоксила, гетероциклилалкокси, галоарилалкилоксила, алкиламино, алкилацила, циано и гетероциклила и т.д.. Эти заместители могут быть дополнительно замещены. Например, алкил, выбранный в качестве заместителя, может быть возможно замещен одной или несколькими группами, выбранными из галогена, гидроксила, алкокси, алкиламино, пирролидинила, фенила, пиридинила и галогенфенила. Выбранный в качестве заместителя гетероциклил может быть возможно замещен одной или несколькими группами, выбранными из галогена, алкила и алкокси.
R1 может быть возможно замещенным гетероциклилом, возможно замещенным ариламино, возможно замещенным арилтио или арилоксилом и т.д.
R1 предпочтительно представляет собой возможно замещенный индолил, индолинил, тиенил, индазолил, пирропиридинил, бензотиенил, бензимидазолил или бензотриазолил и т.д., где заместителем предпочтительно является галоген, алкил, циклоалкил, арилалкил и циано. R1 может представлять собой, например, индол-1-ил, индол-3-ил, 1-метил-1Н-индол-3-ил, 1-этил-1Н-индол-3-ил, 1-пропил-1Н-индол-3-ил, 1-изопропил-1Н-индол-3-ил, 1-бензил-1Н-индол-3-ил, 6-фтор-1-метил-1Н-индол-3-ил, 5-фтор-1-метил-1Н-индол-3-ил, 5-фтор-1-циклопентил-1Н-индол-3-ил, бензимидазол-1-ил или бензотриазол-1-ил и т.д.
R1 предпочтительно является возможно замещенным фениламино или нафтиламино и т.д., где заместителем предпочтительно является галоген, алкил, галоалкил, алкокси, алкинил, арилокси, гетероциклилалкокси, арилалкилоксил и галогенарилалкилоксил. R1 может представлять собой, например, феноксилфениламино, метилфениламино, галогенфениламино, метоксифениламино, ацетенилфениламино, трифторметилфениламино, фторбензилоксифениламино или пиридинилметоксифениламино и т.д., предпочтительно галогенфениламино.
R1 предпочтительно является возможно замещенным фенилтио или нафтилтио и т.д., где заместителем предпочтительно является галоген, алкил или алкокси. R1 может представлять собой, например, нафтилтио, метил-фенилтио или метоксил-фенилтио и т.д.
R1 предпочтительно является возможно замещенным фенилоксилом или нафтилоксилом и т.д., где заместителем предпочтительно является галоген, алкил или алкокси. R1 может представлять собой, например, нафтилоксилом, метилфенилоксилом или метоксил-фенилксилом и т.д.
R3 может быть возможно замещенным гетероциклилом, возможно замещенным алкокси или возможно замещенным амино и т.д.
R3 предпочтительно представляет собой возможно замещенный пиперазинил, пиперидинил, пирролидинил, диазепин или пиридинил, где заместителем предпочтительно является галоген, алкил, циано, морфолинил, пиперидинил, алкилпиперазинил, алкиламино, алкилпиперидинил, гидроксиалкил, алкоксиалкил, гидроксилалкоксиалкил, пирролидинилалкил, алкиламил, алкилалкил, арил, пиридинилалкил или галогенарилалкил и т.д. R3 может представлять собой, например, метилпиперазинил, морфолинилпиперидинил, метилпиперазинпиперидинил, диметиламинопиперидинил, трет-бутилпиперазинил, диметиламинопирролидинил, этилпиперазинил, циклогексилметилпиперазинил, ди-пиперидинил, метилдиазепинил, метилпиперидинилпиперазинил, гидроксиметилпиперазинил, метоксилэтилпиперазинил, гидроксиэтоксилэтилпиперазинил, дифторпирролиндиил, пирролидинилпиперазинил, гидроксипропилпиперидинил, пиридилэтилпиперазинил, бензодиазепинпиперазинил, пирролидинилэтилпиперазинил, цианоэтилпиперазинил, диметиламиноэтилпиперазинил, ацетилпиперазинил, бензилпиперазинил, фенилпиперазинил, пиридинилметилпиперазин, 4-метил-2-фенилпиперазинил, бис(фторфенил)метилпиперазинил и т.д.
R3 предпочтительно является возможно замещенным этоксилом, пропоксилом или бутоксилом и т.д., где заместитель предпочтительно представляет собой алкил, алкокси, алкиламино, морфолинил, пирролидинил, алкенилацил или пиперазинил и т.д. R3 может представлять собой, например, метоксилэтоксил, метил пиперазинил этоксил, морфолинилэтоксил, пиррол идинилэтоксил, акрилоилпиперазинилэтоксил или диметиламиноэтоксил и т.д.
Предпочтительно Ry и Rz в R3 представляют собой каждый независимо алкил, галоалкил, гидроксиалкил, пирролидинил или алкиламино и т.д., где атом N может быть окислен. R3 может представлять собой, например, диметиламино, метил(2-пирролидинил)амино, (2-метоксифенил)метиламино, [2-(1-окси-пирролидин-1-ил)этил]метиламино или метил-N,N-диметил-N-окисленный этиламино и т.д.
Фармацевтически приемлемые соли соединения формулы (I) в соответствии с настоящим изобретением могут представлять собой кислотно-аддитивные соли или основно-аддитивные соли, где кислота может быть неорганической кислотой, включая, без ограничения, соляную кислоту, серную кислоту, фосфорную кислоту и бромистоводородную кислоту; или органическую кислоту, включая, без ограничения, лимонную кислоту, малеиновую кислоту, щавелевую кислоту, муравьиную кислоту, уксусную кислоту, пропионовую кислоту, гликолевую кислоту, бензойную кислоту, фумаровую кислоту, трифторуксусную кислоту, янтарную кислоту, винную кислоту, молочную кислоту, глутаминовую кислоту, аспарагиновую кислоту, салициловую кислоту, пировиноградную кислоту, метансульфоновую кислоту, бензолсульфоновую кислоту и п-бензолсульфоновую кислоту; основание может быть неорганическим основанием, включая, без ограничения, гидроксид натрия, гидроксид калия, гидроксид магния и гидроксид кальция; или органическое основание, включая, без ограничения, гидроксид аммония, триэтиламин, аргинин или лизин.
Соединение формулы (I) или его фармацевтически приемлемые соли в соответствии с настоящим изобретением могут присутствовать в форме сольвата или несольвата, такой как гидратная форма.
Пролекарство соединения формулы (I) в соответствии с настоящим изобретением должно соответствовать принципу пролекарственного дизайна. Соединение формулы (I) может быть высвобождено путем ферментации, гидролиза, ацидолиза или метаболической деградации в нормальных физиологических условиях. Пролекарство включает, без ограничения, этерификацию гидроксильных групп соединений (такую как образование фосфата и карбоната), а также защиту аминогрупп и карбоксильных групп. Дизайн пролекарства должен основываться на (1) Karaman R, Prodrugs design based on inter- and intramolecular chemical processes. Chem Biol Drug Des. 82(6):643-68, 2013; (2) Rautio J et al. Prodrugs:design and clinical applications. Nat Rev Drug Discov. 7(3):255-70, 2008; (3) Jampilek J. Prodrugs: pharmaceutical design and current perspectives. Curr Pharm Des. 17(32):3480-1, 2011; (4) Bundgaard H. Design of Progrugs. Elservier, 1985.
С другой стороны, соединение формулы (I) или его фармацевтически приемлемые соли или его предшественники в соответствии с настоящим изобретением могут быть составлены в клинически применимые фармацевтические композиции. Согласно клиническим показаниям, подходам и способам введения лекарственного средства, фармацевтические препараты включают, без ограничения, пероральные препараты, такие как таблетки, гели, мягкие/твердые капсулы, эмульсии, диспергирующий порошок, гранулы, суспензии вода/масло; инъекции, такие как внутривенная инъекция, мышечная инъекция, внутрибрюшинная инъекция, ректальные суппозитории и внутричерепная инъекция, которые могут быть водным раствором и масляным раствором; местные препараты, такие как кремы, мази, гели, водно-масляный раствор и препараты клатратного соединения; ингаляционные препараты, такие как тонкий порошок, жидкие аэрозоли и различные лекарственные формы, пригодные для имплантации in vivo.
Фармацевтическая композиция по настоящему изобретению может быть дополнена обычными фармацевтическими вспомогательными веществами (эксципиентами) в случае необходимости. Такие фармацевтические вспомогательные вещества должны соответствовать правилам подготовки препаратов для фармацевтических препаратов и должны быть совместимы с активными ингредиентами. Вспомогательные вещества для твердых пероральных препаратов включают, без ограничения, маннит, лактозу, крахмал, стеарат магния, целлюлозу, глюкозу, сахарозу, циклодекстрин и молекулярный носитель витамина E-PEG1000, который может способствовать абсорбции в кишечнике. В пероральный препарат может быть добавлено подходящее количество красителя, подсластителя, ароматизатора и консерванта.
Соединение формулы (I) в соответствии с настоящим изобретением можно вводить теплокровным животным в дозе 0,1-100 мг / кг.
Фармацевтические композиции, содержащие соединение формулы (I) или его фармацевтически приемлемые соли, в основном применяются для лечения клинических заболеваний, связанных с EGFR и/или HER2, включая, без ограничения, рак, воспаление, диабет, заболевания иммунной системы, сердечно-сосудистые заболевания, неврологические заболевания и респираторные заболевания.
Среди вышеуказанных клинических заболеваний рак включает, без ограничения, рак легких, рак желудка, рак печени, рак молочной железы, рак носоглотки, рак поджелудочной железы, рак яичников, рак шейки матки, колоректальный рак, глиому, меланому, рак предстательной железы, рак почек, рак пищевода, мезотелиому, рак головы и шеи, карциному мочевого пузыря, рак слюнных желез, анапластическую крупноклеточную лимфому (ALCL), лейкемию, лимфому, включая неходжкинскую лимфому (НХЛ) и множественную миелому.
Фармацевтические композиции настоящего изобретения могут применяться отдельно или в комбинации с одной или несколькими обычными клиническими терапиями, такими как хирургия, лучевая терапия, химиотерапия, иммунотерапия, онколитический вирус, RNAi, адъювантная терапия рака, которые включают, без ограничения, следующие противоопухолевые препараты и терапевтические методы:
1) Алкилирующие агенты, например, цисплатин, карбоплатин, оксалиплатин, хлорамбуцил, циклофосфамид, хлорметамина гидрохлорид, мелфалан, темозоломид, бусульфан и нитрозомочевина.
2) Противоопухолевые антибиотики, например, адриамицин, блеомицин, доксорубицин, даунорубицин, фарморубицин, идарубицин, митомицин С, актиномицин и митрамицин; антимитотические препараты, такие как винкристин, винкалейкобластинус, виндезин, винорелбин, таксол, доцетаксел и ингибитор полокиназы.
3) Антиметаболически и антифолические средства, например, фторпиримидин, метотрексат, цитарабин, ралтитрекс и гидроксимочевина.
4) Ингибиторы топоизомеразы, например, эпиподофиллотоксин и камптотецин.
5) Ингибиторы гормона роста, включая, без ограничения, антиэстрогенные и антиандрогенные лекарственные средства, такие как тамоксифен, фулвестрант, торемифен, ралоксифен, дролоксифен, идоксифен и бикалютамид, флутамид, нилутамид и ацетат ципротерона.
Антагонисты LHRH или агонисты LHRH, например, госерелин, леопрорелин и бусерелин; прогестагены, например, мегестрола ацетат.
Ингибиторы ароматазы, например, анастрозол, летрозол, ворозол, экземестан и ингибиторы 5а-редуктазы, например, финастерид.
6) Средства против инвазии опухоли, включая, без ограничения, ингибиторы семейства c-Src-киназ, ингибиторы металлопротеиназы, ингибиторы активатора урокиназного плазминогена или моноклональные антитела против гепараназы.
7) Ингибиторы клеточного роста, включая, без ограничения, моноклональные антитела против факторов роста или рецепторов факторов роста, такие как антитело против HER2 трастузумаб, антитела против EGFR панитумумаб и цетуксимаб и т.д., и низкомолекулярные ингибиторы протеин тирозин или серин/треонин киназ, таких как FLT3, c-Kit, AbI, FGFR, PDGFR, CSF-1R, IGFR, Aurora, Ras, Raf, MEK, АКТ, PI3K, циклин-зависимые киназы включают ингибиторы CDK2, CDK4 и CDK6.
8) Антиангиогенные агенты, включая, без ограничения, бевацизумаб, моноклональное антитело против сосудистого эндотелиального фактора роста (VEGF) и низкомолекулярные ингибиторы тирозинкиназ рецептора VEGF.
9) Иммунотерапия рака, например, иммунотерапевтические препараты и способы. Включая, без ограничения, улучшающие иммуногенность опухолевых клеток пациентов, такие как цитокины IL-2, IL-4 или GM-CSF; способы снижения анергии Т-клеток, например, моноклональные антитела против ингибиторов иммунных контрольных точек PD-1 / PD-L1; методы использования трансдуцированных иммунных клеток, таких как трансдуцированные цитокинами дендритные клетки; способы снижения функций иммуносупрессивных клеток, включая регуляторные Т-клетки, миелоидные супрессорные клетки и дендритные клетки, экспрессирующие 2,3-дезоксигеназу индоламина; и терапия противораковыми вакцинами с использованием антигенных белков или пептидов, ассоциированных с опухолью.
10) Терапия с помощью химерного антигенного рецептора Т-клеток (CAR-T).
11) Опухолевая генная терапия, такая как CRISPR-Cas 9, RNAi и трансдукция генов.
Следует отметить, что, если количество заместителей не указано (например, галогеналкил), допустимы один или несколько заместителей. Например, «галогеналкил» может содержать один или несколько одинаковых или разных галогенов.
В настоящем изобретении, если химическая структура и химическое название противоречат друг другу, химическая структура является главнее.
Если не указано иное, аббревиатуры любых групп защиты и других соединений, используемых здесь, выражаются в общепринятой форме или в соответствии с комиссией IUPAC-IUB по биохимической номенклатуре (ссылка на Biochem 1972, 77: 942-944).
Примеры
Настоящее изобретение будет подробно проиллюстрировано со ссылкой на следующие примеры. Однако не следует понимать, что настоящее изобретение ограничено этими примерами.
Пример 1
Получение
N-(5-(4-(3-бромофениламино)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламида (Соединение 1)

Стадия 1: Получение (3-бромо-фенил)-(2-хлор-пиримидин-4-ил)-амина
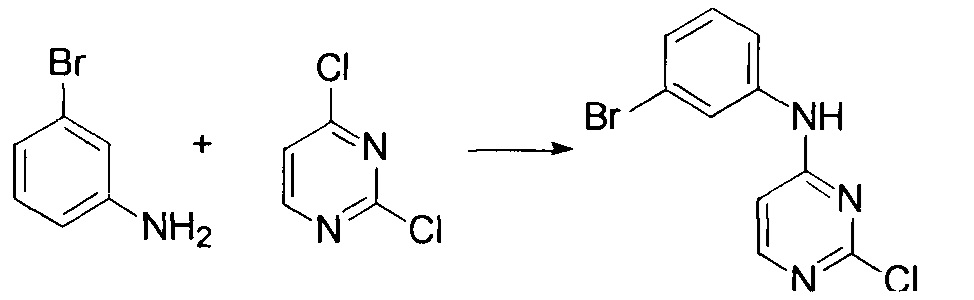
3-Броманилин (2.7 г, 15 ммоль), 2,4-дихлорпиримидин (2.7 г, 18 ммоль) и бикарбонат натрия (2.5 г, 30 ммоль) добавили в 30 мл изопропилового спирта, и затем нагревали при 85°С с перемешиванием в течение 10 ч. Изопропиловый спирт удалили при пониженном давлении, и остаток разбавили водой и этилацетатом. Этилацетатную фазу высушили и сконцентрировали. Остаток очистили с помощью колоночной флеш хроматографии на силикагеле с получением (3-бромо-фенил)-(2-хлор-пиримидин-4-ил)-амина (2.5 г), m/z: ESI МН+ 285.9
Стадия 2: Получение
N4-(3-бромофенил)-N2-(4-((2-диметиламино-этил)-метил-амино)-2-метоксил-5-нитро-фенил)-пиримидин-2,4-диаминтолуолсульфоната.

(3-бромо-фенил)-(2-хлор-пиримидин-4-ил)-амин (1.4 г, 5 ммоль), 2-метоксил-4-фтор-5-нитроанилин (0.9 г, 5 ммоль) и n-толуолсульфоновую кислоту (1.03 г, 6 ммоль) добавили в 15 мл 2-амилового спирта и затем нагревали в течение 3 ч при 115°С. Реакционную смесь охладили до комнатной температуры и отфильтровали. Остаток на фильтре промыли дважды метилтрет-бутиловым эфиром и затем высушили с получением N4-(3-бромофенил)-N2-(4-((2-диметиламино-этил)-метил-амино)-2-метоксил-5-нитро-фенил)-пиримидин-2,4-диаминтолуолсульфоната. m/z: ESI МН+ 434.0
Стадия 3: Получение N4-(4-(3-бромофениламино)-пиримидин-2-ил)-N1-(2-диметиламино-этил)-5-метоксил-N1-метил-фенил-1,2,4-триамина.

N4-(3-бромофенил)-N2-(4-((2-диметиламино-этил)-метил-амино)-2-метоксил-5-нитро-фенил)-пиримидин-2,4-диаминтолуолсульфонат (0.6 г, 1 ммоль), N,N,N'-триметилэтилендиамин (0.15 г, 1.5 ммоль) и безводный карбонат калия (0.42 г, 3 ммоль) добавили в 2 мл ДМФ, и затем нагревали в течение 3 ч при 90°С. Реакционную смесь охладили до комнатной температуры, и затем разбавили водой и этилацетатом. Слой этилацетата сконцентрировали, и к остатоку добавили порошок железа (0.28 г, 5 ммоль), хлорид аммония (0.27 г, 5 ммоль), воду (5 мл) и этанол (15 мл). Смесь нагревали в течение 5 ч при 80°С. Осадок железа отфильтровали горячим, и фильтрат разбавили водой и дихлорметаном после концентрирования. Дихлорметановую фазу высушили и сконцентрировали. Остаток очистили с помощью колоночной хроматографии с получением N4-(4-(3-бромофениламино)-пиримидин-2-ил)-N1-(2-диметиламино-этил)-5-метоксил-N1-метил-фенил-1,2,4-триамина (0.24 г). m/z: ESI МН+ 486.1
Стадия 4: Получение N-(5-(4-(3-бромофениламино)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламида

N4-(4-(3-бромофениламино)-пиримидин-2-ил)-N1-(2-диметиламино-этил)-5-метоксил-N1-метил-фенил-1,2,4-триамин (0.24 г, 0.5 ммоль) и N,N-ди(изопропил)этиламин (0.2 г, 1.5 ммоль) добавили в тетрагидрофуран. Смесь охладили на бане с водой/льдом и разбавили акрилоилхлоридом (0.05 г, 0.55 ммоль) по каплям. Реакционную смесь перемешивали в течение 1 часа после добавления и затем перемешивали в течение 2 часов при комнатной температуре. Смесь разбавили водой и дихлорметаном. Дихлорметановую фазу высушили над безводным Na2SO4 и затем сконцентрировали. Остаток очистили посредством колоночной хроматографии с получением соединения, указанного в заголовке (70 мг).
Пример 2
Получение N-(5-(4-(3-метоксифенокси)-пиримидин-2-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламида (Соединение 11)

Стадия 1: 2-хлор-4-(3-метоксил-фенил)пиримидин

3-метоксил-фенол (500 мг, 4 ммоль), 2,4-дихлорпиримидин (900 мг, 6 ммоль) и карбонат калия (1.1 г, 8 ммоль) добавили в 5 мл ДМФ, и нагревали в течение ночи при 60°С. Реакционную смесь влили в воду, и затем экстрагировали этилацетатом. Слой этилацетата высушили и затем сконцентрировали. Остаток очистили посредством колоночной хроматографии с получением 2-хлор-4-(3-метоксифенил)пиримидина (0.63 г). m/z: ESI МН+ 237.1.
Стадии 2-4 примера 1 повторили с получением соединения, указанного в заголовке.
Пример 3
Получение
N-(5-(4-(3-хлор-4-(пиридин-2-метоксил)фениламино)пиримидин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксил-фенил)-акриламида (Соединение 18)

Стадия 1: Получение (3-хлор-4-(пиридин-2-ил-метоксил)фенил)-(2-хлорпиримидин-4-ил)-амина

3-хлор-4-(пиридин-2-ил-метоксил)анилин (58.7 г, 250 ммоль), 2,4-дихлорпиримидин (44.7 г, 300 ммоль) и бикарбонат натрия (31.5 г, 375 ммоль) добавили в 400 мл изопропилового спирта и затем нагревали на масляной бане при 85°С в течение 24 ч. Реакционную смесь охладили до комнатной температуры и затем разбавили 500 мл воды. Смесь отфильтровали и осадок промыли 500 мл воды, и затем высушили с получением промежуточного соединения (84 г). m/z: ESI МН+ 347.0.
Стадия 2: Синтез N4-(3-хлор-4-(пиридин-2-ил-метоксил)-фенил)-N2-(4-фтор-2-метоксил-5-нитро-фенил)-пиримидин-2,4-диамина мезилата

(3-хлор-4-(пиридин-2-ил-метоксил)фенил)-(2-хлорпиримидин-4-ил)-амин (1.75 г, 5 ммоль), 2-метоксил-4-фтор-5-нитроанилин (0.98 г, 5.25 ммоль) и метансульфоновую кислоту (0.96 г, 10 ммоль) добавили в 20 мл изопропилового спирта и затем нагревали при 75°С в течение 12 ч. Реакционную смесь охладили до комнатной температуры и высушили с получением N4-(3-фтор-4-(пиридин-2-ил-метоксил)-фенил)-N2-(4-фтор-2-метоксил-5-нитро-фенил)-пиримидин-2,4-диамина мезилата (2.1 г). m/z: ESI МН+ 497.1.
Стадия 3: Синтез N4-(4-(3-хлор-4-(пиридин-2-ил-метоксил)фениламино)-пиримидин-2-ил)-N1-(2-диметиламино-этил)-5-метоксил-N1-метил-фенил-1,2,4-триамина

N4-(3-хлор-4-(пиридин-2-ил-метоксил)-фенил)-N2-(4-фтор-2-метоксил-5-нитро-фенил)-пиримидин-2,4-диамина мезилата (11.8 г, 20 ммоль), N,N,N'-триметилэтилендиамин (3.06 г, 30 ммоль) и безводный карбонат калия (8.3 г, 60 ммоль) добавили в 40 мл ДМФ и затем нагревали при 90°С в течение 4 ч. Реакционную смесь охладили до комнатной температуры, и затем разбавили водой и этилацетатом. После концентрирования этилацетата порошок железа (5.6 г, 100 ммоль), хлорид аммония (5.6 г, 105 ммоль), воду (80 мл) и этанол (80 мл) добавили и затем нагревали при 80°С в течение 5 ч. Осадок железа отфильтровали горячим. Фильтрат сконцентрировали и затем разбавили водой и дихлорметаном. Дихлорметановый слой высушили и затем сконцентрировали. Остаток очистили с помощью хроматографии на колонке с получением N4-(4-(3-хлор-4-(пиридин-2-ил-метоксил)фениламино)-пиримидин-2-ил)-N1-(2-диметиламино-этил)-5-метоксил-N1-метил-фенил-1,2,4-триаммония (3.1 г), m/z: ESI МН+ 549.3.
Стадия 4: N-(5-(4-(3-хлор-4-(пиридин-2-илметокси)фениламино)-пиримидин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксил-фенил)-акриламид
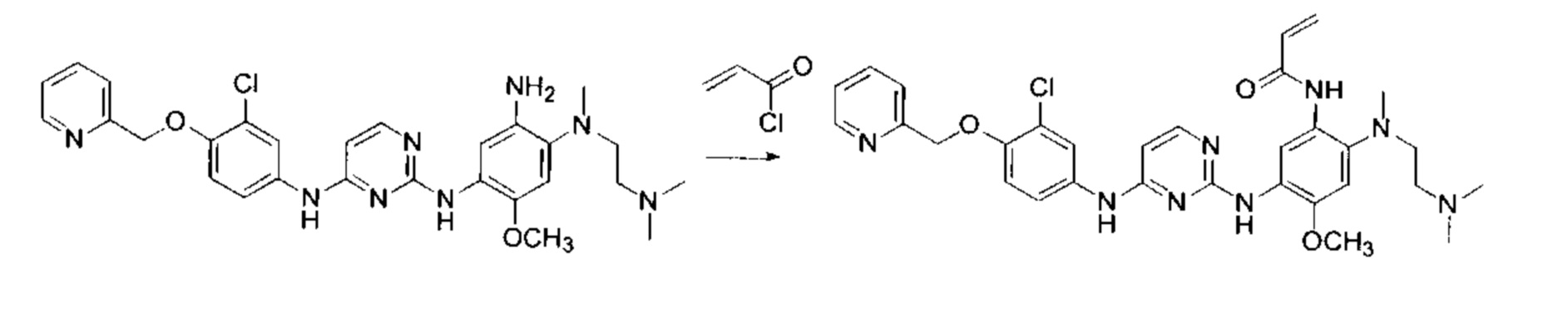
N4-(4-(3-хлор-4-(пиридин-2-ил-метоксил)фениламино)-пиримидин-2-ил)-N1-(2-диметиламино-этил)-5-метоксил-N1-метил-фенил-1,2,4-триамин (0.55 г, 1 ммоль) и N,N-ди(изопропил)этиламин (0.38 г, 3 ммоль) добавили в тетрагидрофуран. Смесь охладили на ледяной бане и затем разбавили акрилоилхлоридом (0.11 г, 1.2 ммоль). После добавления смесь перемешивали в течение 1 часа, и затем довели до комнатной температуры и продолжили перемешивание в течение 2 часов. Реакционную смесь разбавили водой и дихлорметаном. Дихлорметановый слой высушили над безводным Na2SO4 и затем сконцентрировали. Остаток очистили посредством колоночной хроматографии с получением соединения, указанного в заголовке (110 мг).
Пример 4
Получение
N-(5-(-4-(3-хлор-4-(бензол-2-метоксил)-фениламино)-пиримидин-2-иламино)-2-(2-(морфолин-4-ил)-этоксил)-4-метоксил-фенил)-акриламида (Соединение 29)

Стадии 1 и 2 являются такими же, как описано в примере 3.
Стадия 3: Синтез N2-(5-амино-2-метоксил-4-(2-морфолин-4-ил-этоксил)-фенил)-N4-(3-хлор-4-(пиридин-2-метоксил)-фенил)-пиримидил-2,4-диамин

N4-(3-хлор-4-(пиридин-2-ил-метоксил)-фенил)-N2-(4-фтор-2-метоксил-5-нитро-фенил)-пиримидин-2,4-диамина мезилат (0.59 г, 1 ммоль), 4-(2-гидроксиэтил-морфолин) (0.2 г, 1.5 ммоль) и гидроксид натрия (0.12 г, 3 ммоль) добавили в 3 мл ДМФ и затем нагревали при 60°С в течение 3 ч. Реакционную смесь охладили до комнатной температуры и затем разбавили водой и этилацетатом. После концентрирования этилацетатного слоя порошок железа (0.28 г, 5 ммоль), хлорид аммония (0.27 г, 5 ммоль), воду (5 мл) и этанол (15 мл) добавили и затем нагревали при 80°С в течение 5 ч. Осадок железа отфильтровали горячим. Фильтрат сконцентрировали и затем разбавили водой и дихлорметаном. Дихлорметановый слой высушили, и затем сконцентрировали. Остаток очистили посредством колоночной хроматографии с получением N2-(5-амино-2-метоксил-4-(2-морфолин-4-ил-этоксил)-фенил)-N4-(3-хлор-4-(пиридин-2-метоксил)-фенил)-пиримидил-2,4-диамина (0.21 г), m/z: ESI МН+ 578.3.
Стадию 4 примера 3 повторили с получением соединения, указанного в заголовке.
Пример 5
Получение
N-(5-(4-(3,4-дихлор-2-фторфениламино)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламида (Соединение 15)

Стадия 1: Получение (3,4-дихлор-2-фторфенил)-(2-хлорпиримидин-4-ил)-амина

3,4-дихлор-2-фторанилин (1.8 г, 10 ммоль), 2,4-дихлорпиримидин (2.25 г, 15 ммоль) и соляную кислоту (0.5 мл, 12 М) добавили в 5 мл изопропилового спирта и затем подвергли кипению с обратным холодильником в течение 2 ч. Реакционную смесь охладили до комнатной температуры и затем отфильтровали с получением промежуточного соединения, указанного в заголовке (1.2 г). m/z: ESI МН+ 292.0.
Стадии 2~4 примера 1 повторили с получением соединения, указанного в заголовке.
Соединения 1-6, 8-10, 12-14 и 30-31 в Таблице 1 получили в соответствии с такой же методикой, как описано в Примере 1. Соединения 7 и 11 в Таблице 1 получили в соответствии с такой же методикой, как описано в Примере 2. Соединения 17-28 в Таблице 1 получили в соответствии с такой же методикой, как описано в Примере 3. Соединение 29 в Таблице 1 получили в соответствии с такой же методикой, как описано в Примере 4. Соединения 15-16 в Таблице 1 получили в соответствии с такой же методикой, как описано в Примере 5.
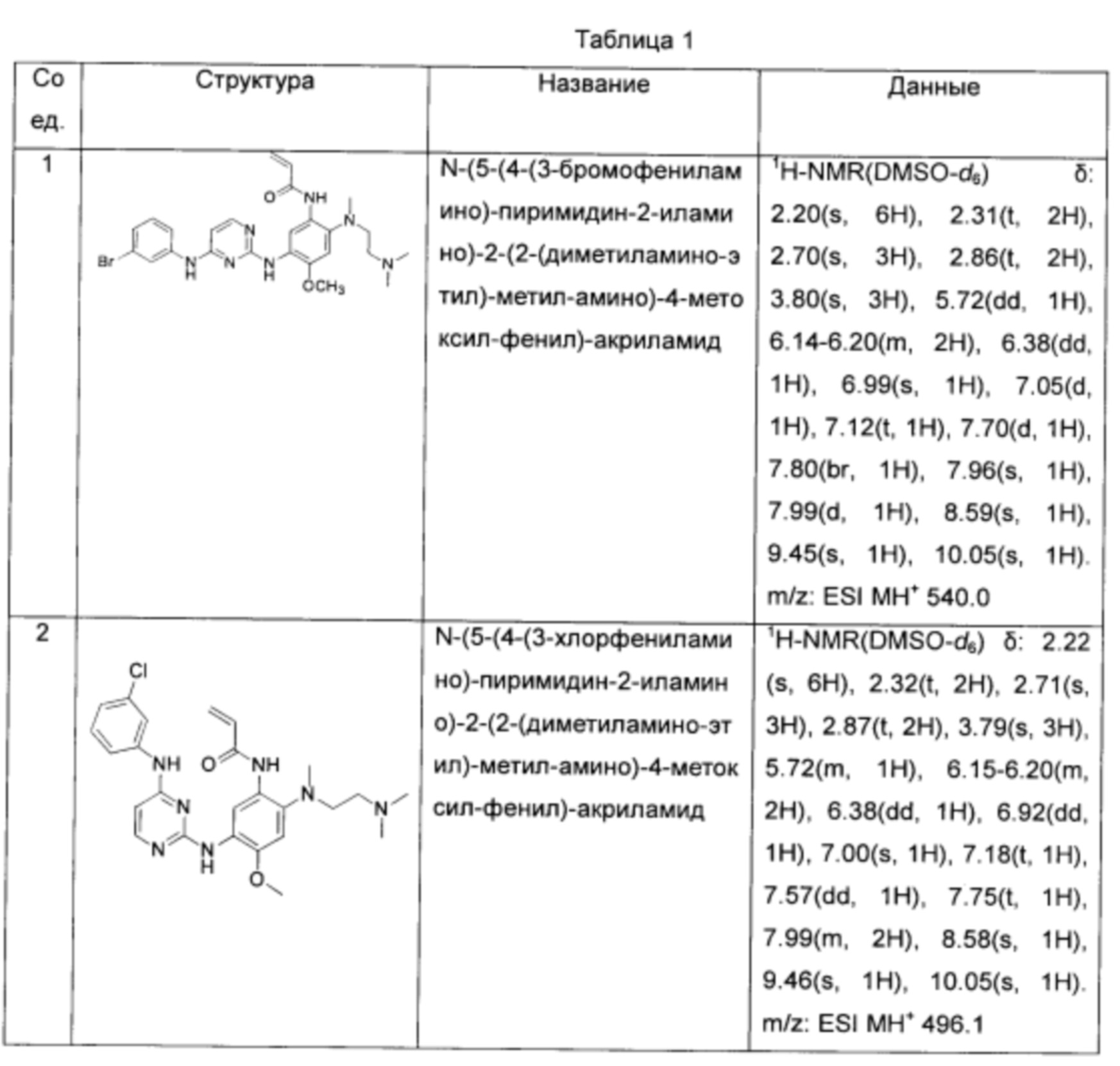













Пример 6
Получение
N-(5-(4-(4-метоксил-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламида (Соединение 99)
Стадия 1: Получение 1-(2-хлорпиримидин-4-ил)-6-метоксил-1Н-индола

4-Метоксил-индол (4.5 г, 30 ммоль), 2,4-дихлорпиримидин (6.8 г, 45 ммоль), HOBT (0.8 г, 6 ммоль), безводный карбонат калия (8.4 г, 60 ммоль) добавили в 25 мл ДМФ и затем перемешивали при 75°С в течение 15 ч. Реакционную смесь разбавили водой и затем отфильтровали. Осадок добавили в 50 мл изопропилового спирта и перемешивали при кипении с обратным холодильником. Реакционную смесь охладили до комнатной температуры и затем отфильтровали и высушили с получением неочищенного материала промежуточного соединения (9.6 г). MS m/zES+MH+ 260.1.
Стадия 2: Получение (4-фтор-2-метоксил-5-нитро-фенил)-[4-(4-метоксил-индол-1-ил)-пиримидин-2-ил]-амина толуолсульфоната

1-(2-хлорпиримидин-4-ил)-6-метоксил-1Н-индол (9.6 г,30 ммоль), 2-метоксил-4-фтор-5-нитроанилин (6.5 г, 35 ммоль), п-толуосульфоновой кислоты гидрат (7.7 г, 40.7 ммоль) добавили в 100 мл 2-амилового спирта и затем нагревали при 100°С в течение 2 ч. Реакционную смесь охладили до комнатной температуры, и затем отфильтровали и высушили с получением неочищенного продукта, указанного в заголовке (11 г). MS m/z:ES+MH+ 410.1.
Стадия 3: Получение N4-(2-диметиламиноэтил)-2-метоксил-N1-(4-(4-метоксил-индол-1-ил)-пиримидин-2-ил)-N4-метил-5-нитро-1,4-фенилендиамина

(4-фтор-2-метоксил-5-нитро-фенил)-(4-(4-метоксил-индол-1-ил)-пиримидин-2-ил)-амина толуолсульфонат (1.1 г, 1.89 ммоль), триметилэтилендиамин (0.27 г, 2.64 ммоль) и карбонат калия (0.65 г, 4.7 ммоль) добавили в 5 мл ДМФ и затем нагревали при 70°С в течение 4 ч. Реакционную смесь охладили до комнатной температуры, и затем разбавили водой и этилацетатом. Слой этилацетата высушили и сконцентрировали. Остаток очистили посредством колоночной хроматографии с получением промежуточного соединения, указанного в заголовке (0.4 г). MS m/z:ES+MH+ 492.2.
Стадия 4: Получение N1-(2-диметиламиноэтил)-5-метоксил-N4-(4-(4-метоксил-индол-1-ил)-пиримидин-2-ил)-N1-метил-1,2,4-бензолтриамина

N4-(2-диметиламиноэтил)-2-метоксил-N1-(4-(4-метоксил-индол-1-ил)-пиримидин-2-ил)-N4-метил-5-нитро-1,4-фенилендиамин (0.4 г, 0.8 ммоль), порошок железа (0.34 г, 6 ммоль) и хлорид аммония (0.33 г, 6 ммоль) добавили в 10 мл этанола и затем нагревали при 70°С в течение 2 ч. Осадок железа отфильтровали и фильтрат сконцентрировали. Остаток очистили посредством колоночной хроматографии с получением промежуточного соединения, указанного в заголовке (0.15 г). MS m/z:ES+MH+ 462.3.
Стадия 5: Получение N-(5-(4-(4-метоксил-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламида

AddN1-(2-диметиламиноэтил)-5-метоксил-N4-[4-(4-метоксил-индол-1-ил)-пиримидин-2-ил]-N1-метил-1,2,4-бензолтриамин (0.15 г, 0.32 ммоль) и N,N-диизопропилэтиламин (0.13 г, 1 ммоль) добавили в 3 мл тетрагидрофурана, и затем добавили акрилоилхлорид при 0°С (45 мг, 0.5 ммоль). Смесь перемешивали в течение 2 ч, и затем разбавили водой и этилацетатом. Колоночная хроматография дала соединение, указанного в заголовке (45 мг).
Пример 7
Получение
N-(5-(4-(6-фтор-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-(морфолин-4-ил)-этоксил)-4-метоксил-фенил)-акриламида (Соединение 75)

Стадия 1 и 2: Получение N-(4-(6-фтор-1Н-индол-1-ил)пиримидин-2-ил)-4-фтор-2-метоксил-5-нитроанилина
Стадии 1 и 2 являются такими же, как в примере 6, за исключением замены 4-метоксилиндола на 6-фториндол.
Стадия 3: Синтез N-(4-(6-фтор-1Н-индол-1-ил)пиримидин-2-ил)-4-(2-морфолин-4-ил-этоксил)-2-метоксил-5-нитроанилина

N-(4-(6-фтор-1Н-индол-1-ил)пиримидин-2-ил)-4-фтор-2-метоксил-5-нитроанилин (1.1 г, 2 ммоль), 2-(морфолин-4-ил)этанол (0.39 г, 3 ммоль) и гидроксид натрия (0.2 г, 5 ммоль) добавили в 5 мл ДМФ, и затем нагревали на масляной бане при 60°С в течение 3 ч. Реакционную смесь разбавили водой и этилацетатом. Органический слой высушили и затем сконцентрировали. Остаток очистили посредством колоночной хроматографии с получением промежуточного соединения, указанного в заголовке (0.45 г). m/z: ESI МН+ 508.2,
Стадии 4 и 5 примера 6 повторили с получением соединения, указанного в заголовке.
Соединения 32-52, 57-73, 77-95, 98-105 в Таблице 2 получили в соответствии с методикой в примере 6.
Соединения 53-56, 74-76, 96-97 в Таблице 2 получили в соответствии с методикой в примере 7.

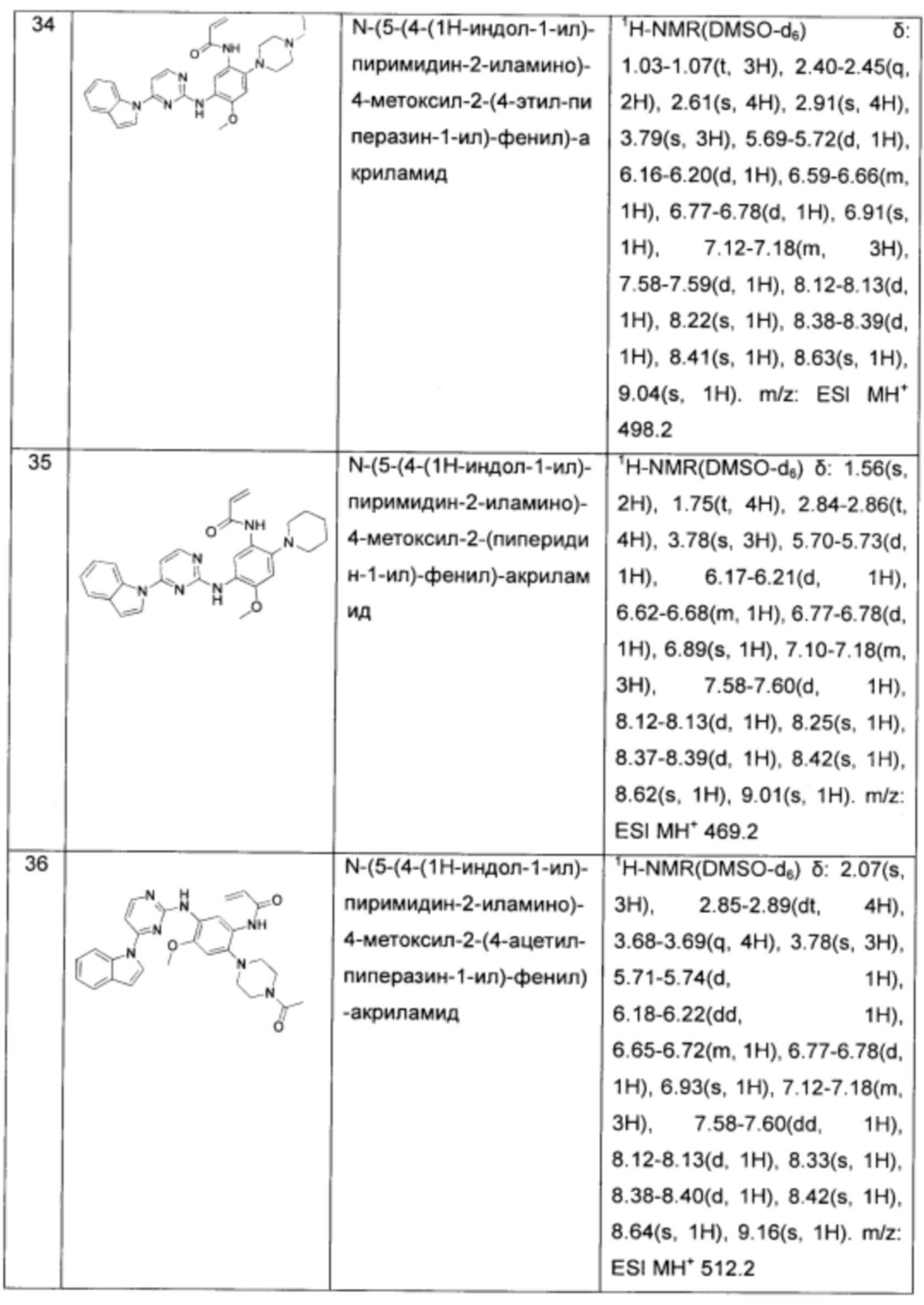













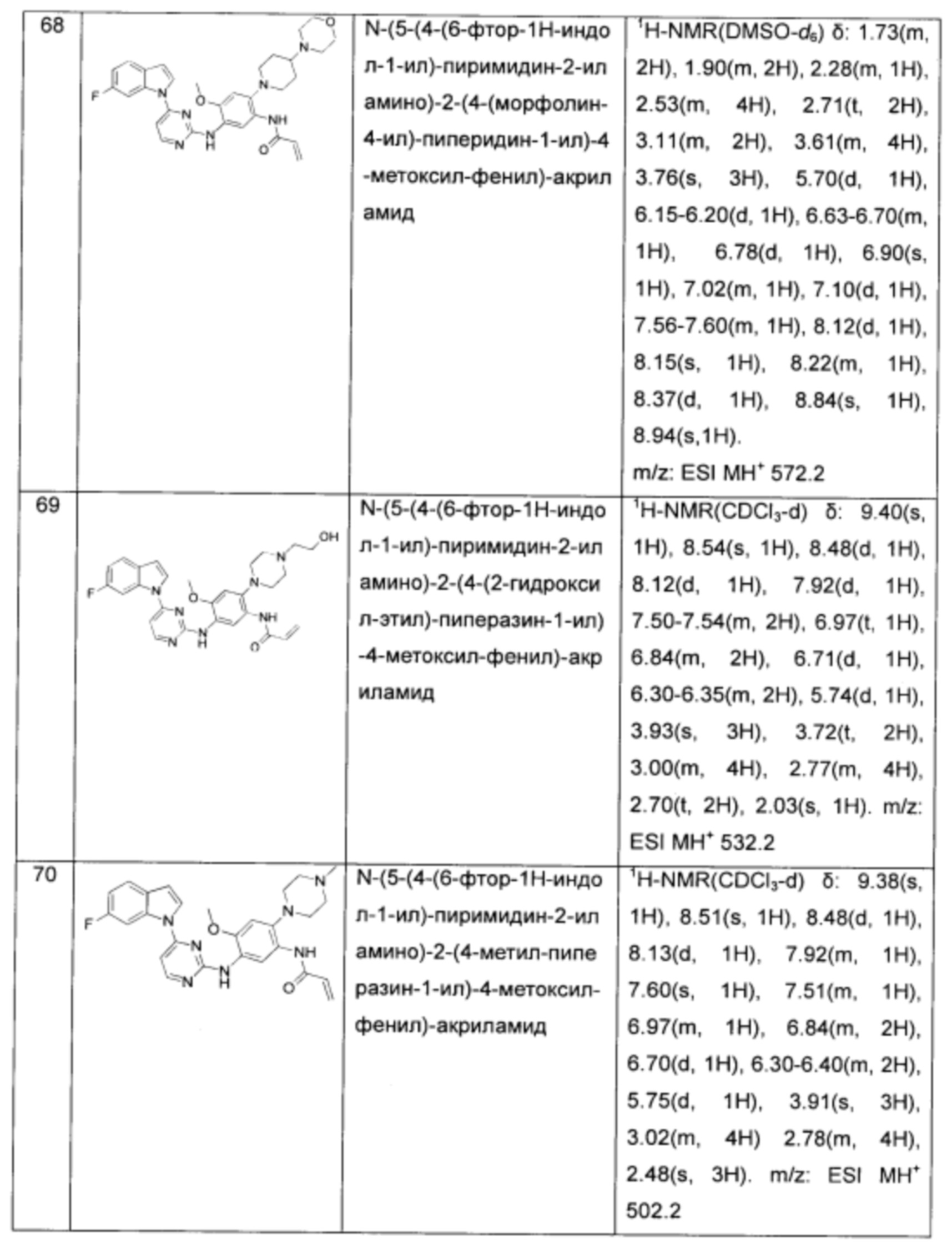
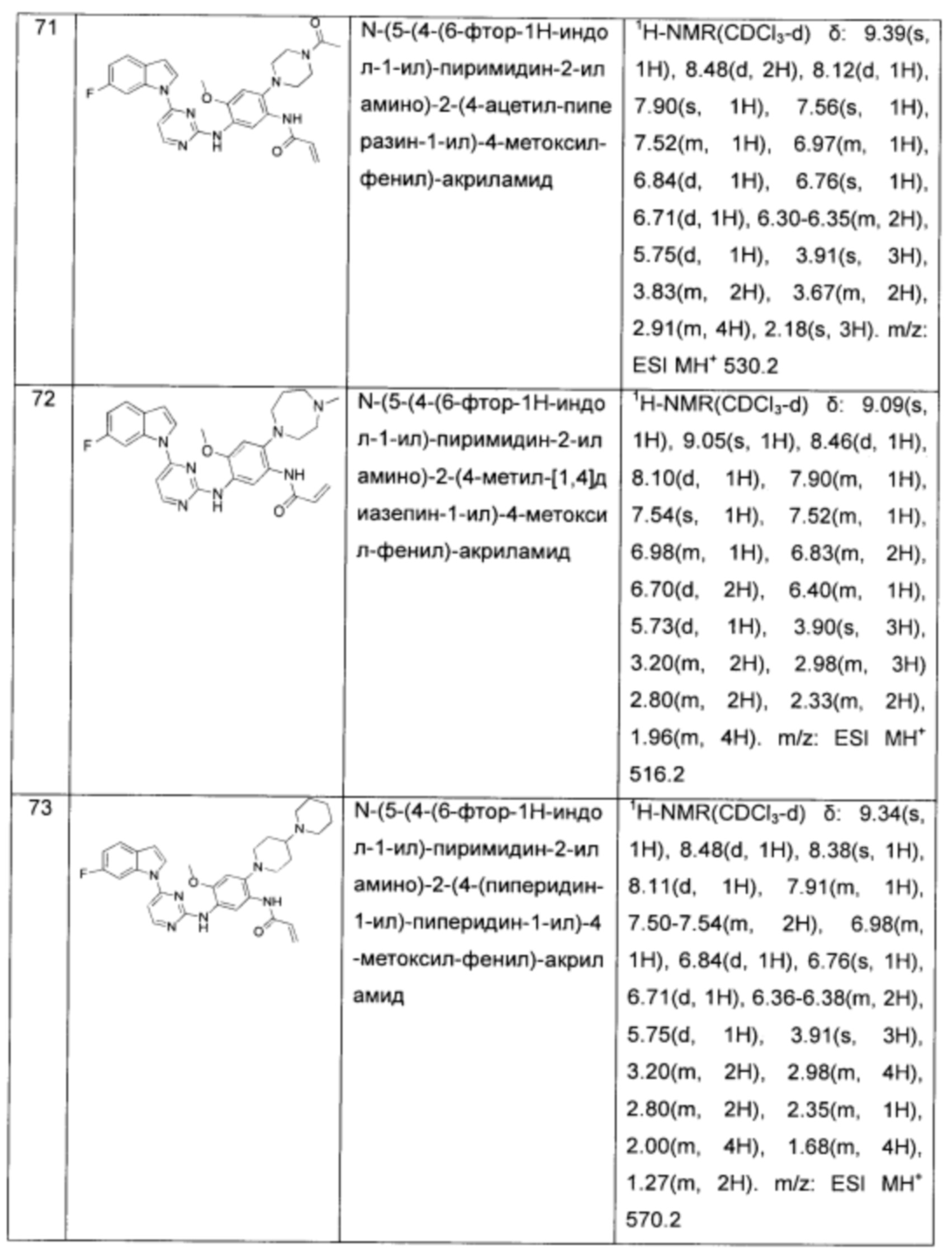
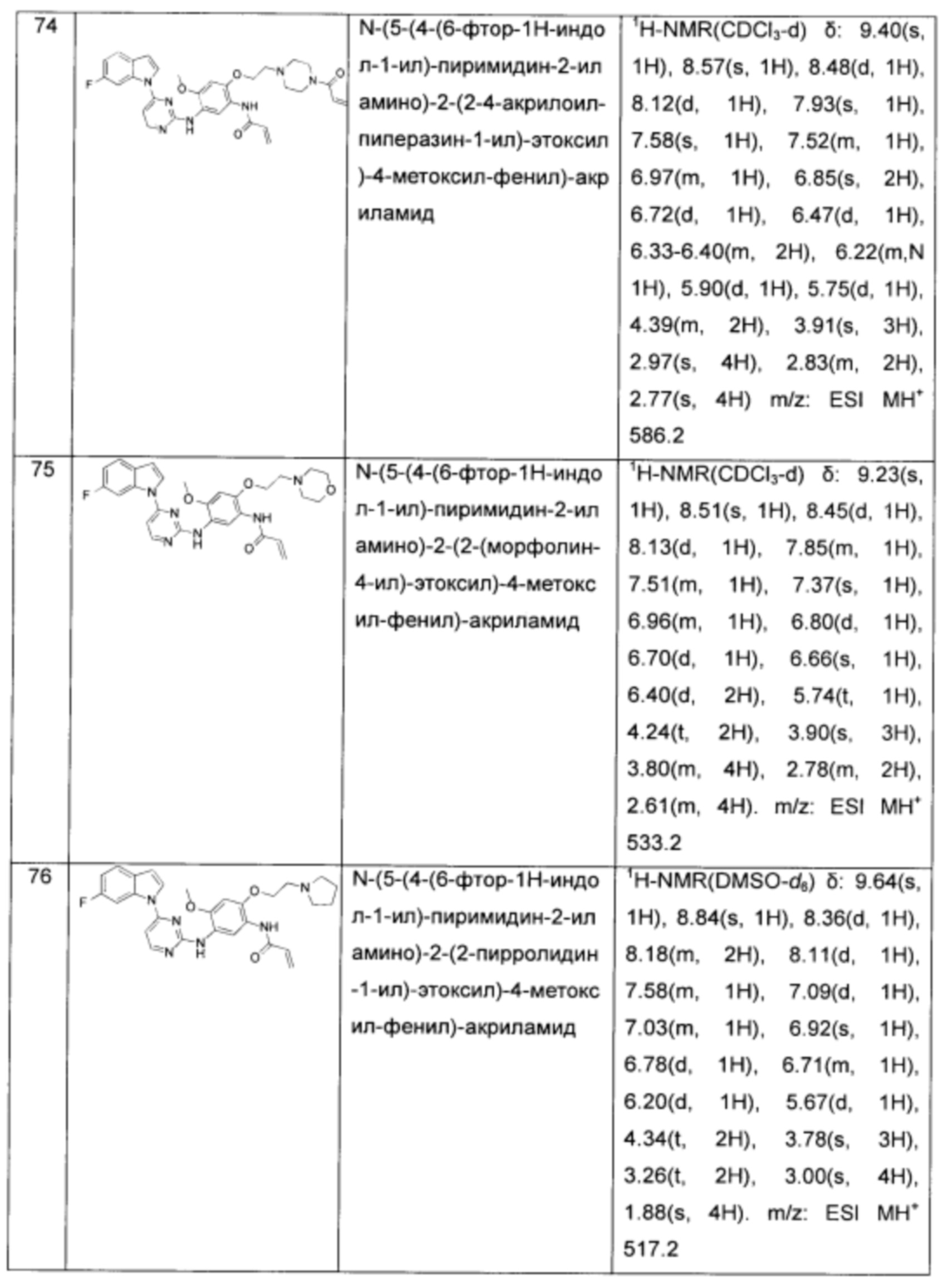

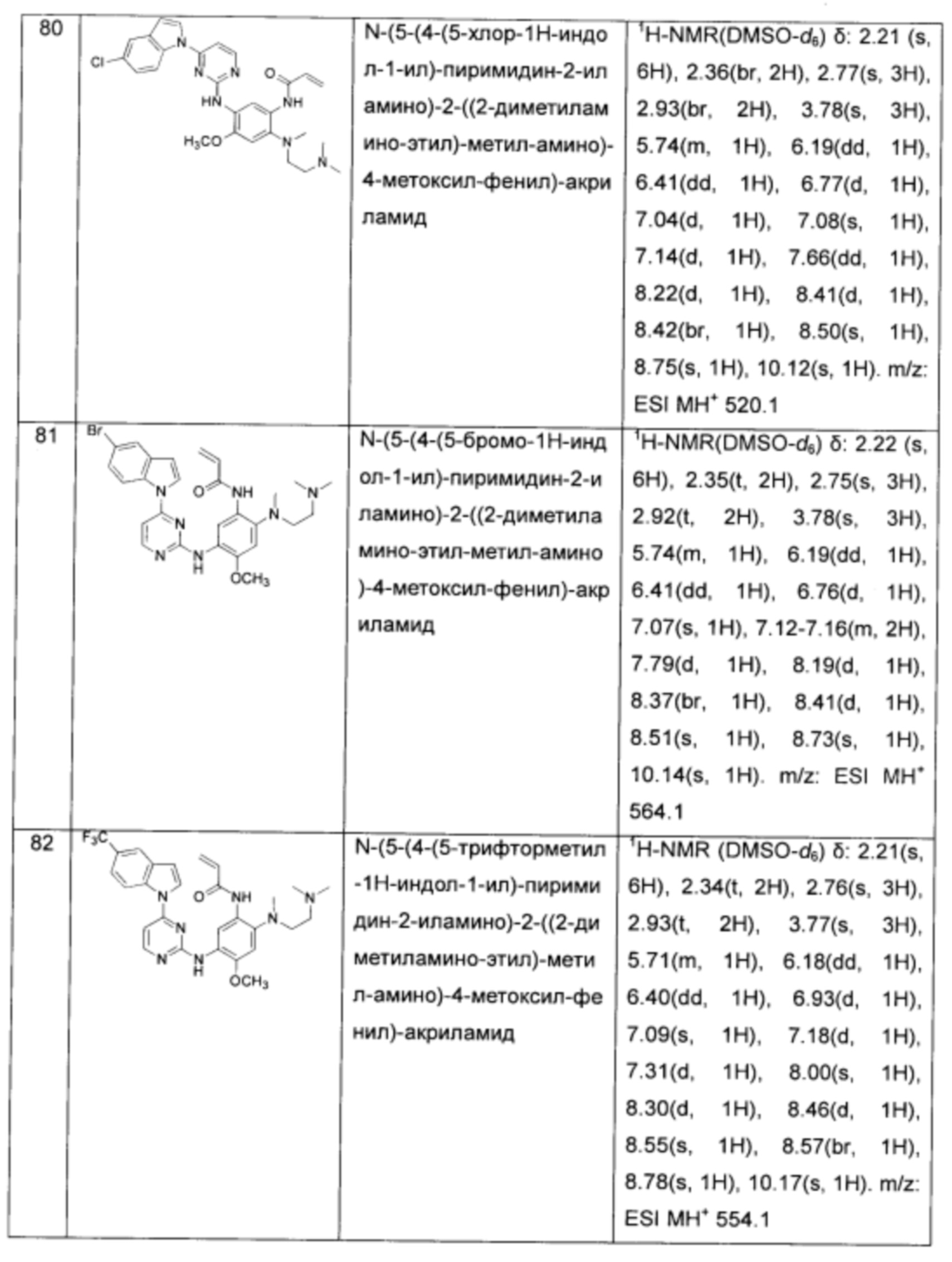









Пример 8
Получение N-(5-(4-(-1H-бензо[d]имидазол-1-ил)пиримидин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксил)фенил)акриламид (соединение 106)

Стадия 1: Синтез 1-(2-хлорпиримидин-4-ил)-1Н-бензимидазола
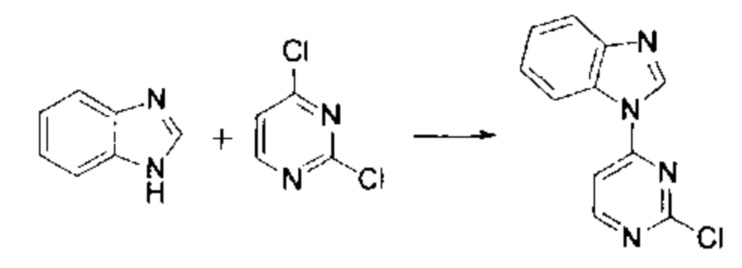
Бензимидазол (2.4 г, 20 ммоль), карбонат калия (5.6 г, 40 ммоль) и 2,4-дихлорпиримидин (4.5 г, 30 ммоль) добавили в 5 мл ДМФ, и затем перемешивали в течение 1 ч при комнатной температуре. Реакционную смесь влили в воду и затем экстрагировали этилацетатом. Слой этилацетата высушили и затем сконцентрировали. Остаток очистили посредством колоночной хроматографии с получением промежуточного соединения, указанного в заголовке (2.1 г), m/z: ESI МН+ 231.0.
Стадии 2~5 примера 6 повторили с получением целевых соединений. Соединения 106-107 в Таблице 3 получили в соответствии с методикой в примере 1.

Пример 9
Получение
N-(5-(4-(3-хлор-фениламино)-[1,3,5]триазин-2-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламида (Соединение 108)

Стадия 1: Получение (3-хлорфенил)-(4-хлор-[1,3,5]триазин-2-ил)-амина

3-Хлоранилин (2.56 г, 20 ммоль) и DIEA (3.08 г, 24 ммоль) добавили в 20 мл ДМФ, и затем охладили на бане с ледяной солью. Раствор 2,4-дихлор-1,3,5-триазина (3.29 г, 22 ммоль) в ДМФ добавили по каплям, и смесь перемешивали в течение 2.5 ч на бане с ледяной солью. Реакционную смесь влили в воду, и затем добавили этилацетат. Слой этилацетата высушили и сконцентрировали. Остаток очистили посредством колоночной хроматографии с получением промежуточного соединения, указанного в заголовке (3.65 г), m/z: ESI МН+ 241.0.
Стадии 2~4 примера 1 повторили с получением целевого соединения.
Соединения 108-130 в Таблице 4 получили в соответствии с методикой в примере 9.
Соединения 131-132 в Таблице 4 получили в соответствии со стадиями 1 и 2 Примера 9 и стадиями 3 и 4 примера 4.










Пример 10
Получение N-(5-(6-(3-бромофениламино)-пиримидин-4-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламида (Соединение 133)

Стадия 1: Получение N-(6-хлорпиримидин-4-ил)-3-броманилин

3-Броманилин (1.72 г, 10 ммоль), 4,6-дихлорпиримидин (2.25 г, 15 ммоль) и триэтиламин (3.03 г, 30 ммоль) добавили в 20 мл этанола и перемешивали при кипении с обратным холодильником в течение 20 ч. Изопропиловый спирт удалили при пониженном давлении. Остаток очистили посредством колоночной хроматографии с получением промежуточного соединения, указанного в заголовке (1.8 г). m/z: ESI МН+ 286.0.
Стадия 2: Получение N-(3-бромофенил)-N'-(4-фтор-2-метоксил-5-нитрилфенил)-пиримидин-4,6-диаминтолуолсульфоната

N-(6-хлорпиримидин-4-ил)-3-броманилин (1.43 г, 5 ммоль), 4-фтор-2-метоксил-5-нитроанилин (0.93 г, 5 ммоль) и п-толуолсульфоновую кислоту (1.05 г, 6 ммоль) добавили в 20 мл 2-амилового спирта и затем перемешивали в течение 15 ч при 110°С. Реакционную смесь охладили до комнатной температуры и затем отфильтровали с получением промежуточного соединения, указанного в заголовке (0.8 г). m/z: ESI МН+ 434.1.
Стадия 3: Получение N-(3-бромофенил)-N'-(4-((2-диметиламиноэтил)-метиламино)-2-метоксил-5-нитрофенил)-пиримидин-4,6-диамина

N-(3-Бромофенил)-N'-(4-фтор-2-метоксил-5-нитрофенил)-пиримидин-4,6-диаминтолуолсульфонат (0.61 г, 1 ммоль), триметилэтилендиамин (0.15 г, 1.5 ммоль) и карбонат калия (0.56 г, 4 ммоль) добавили в 4 мл ДМФ и затем нагревали на масляной бане при 70°С в течение 2 ч. Реакционную смесь разбавили водой и этилацетатом. Слой этилацетата высушили и затем сконцентрировали. Остаток очистили посредством колоночной хроматографии с получением промежуточного соединения, указанного в заголовке (0.22 г). m/z: ESI МН+ 518.2.
Стадия 4: Получение N-(3-бромофенил)-N'-(4-((2-диметиламиноэтил)-метиламино)-2-метоксил-5-аминофенил)-пиримидин-4,6-диамин
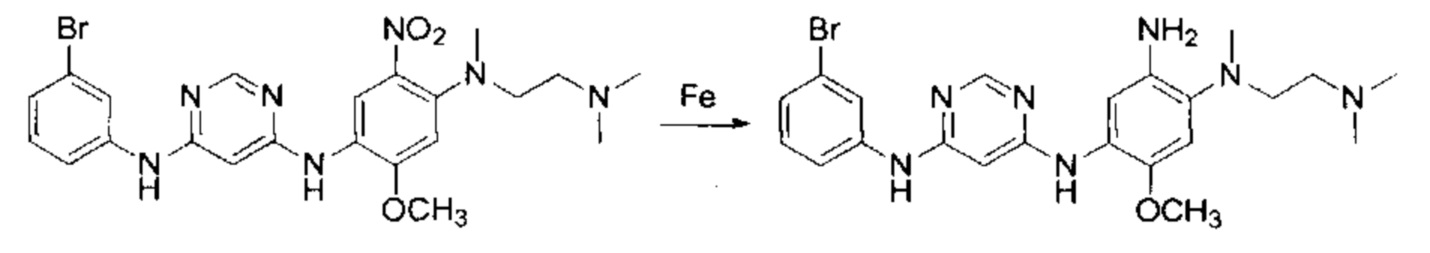
N-(3-бромофенил)-N'-(4-((2-диметиламиноэтил)-метиламино)-2-метоксил-5-нитрофенил)-пиримидин-4,6-диамин (0.22 г, 0.42 ммоль), порошок железа (0.17 г, 3 ммоль) и хлорид аммония (0.16 г, 3 ммоль) добавили в 10 мл этанола и 5 мл воды, и затем нагревали на масляной бане при 60°С в течение 2 ч. Осадок железа отфильтровали и фильтрат сконцентрировали. Остаток очистили посредством колоночной хроматографии с получением промежуточного соединения, указанного в заголовке (0.13 г). m/z: ESI МН+ 486.2.
Стадия 5: Получение N-(5-(6-(3-бромо-фениламино)-пиримидин-4-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламида

N-(3-Бромофенил)-N'-(4-((2-диметиламиноэтил)-метиламино)-2-метоксил-5-аминофенил)-пиримидин-4,6-диамин (0.13 г, 0.29 ммоль) и DIEA (0.13 г, 1 ммоль) добавили в 5 мл тетрагидрофурана, и затем охладили на бане с ледяной солью. Акрилоилхлорид (45 мг, 0.5 ммоль) добавили и затем перемешивали в течение 1 ч. Реакционную смесь разбавили водой и этилацетатом. Слой этилацетата высушили и затем сконцентрировали. Остаток очистили посредством колоночной хроматографии с получением промежуточного соединения, указанного в заголовке (35 мг).
Пример 11
Получение
N-(5-(6-(3-хлор-4-фтор-фениламино)-пиримидин-4-иламино)-2-(2-диметиламино-этоксил)-4-метоксил-фенил)-акриламида
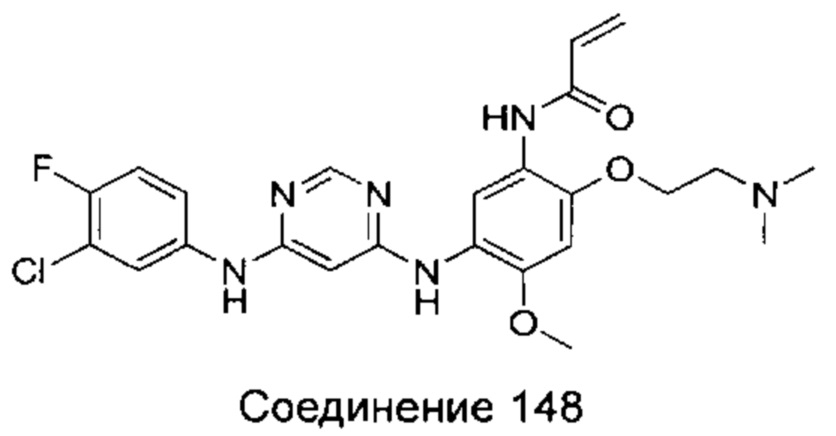
Стадии 1 и 2: Получение N-(4-фтор-3-хлорфенил)-N'-(4-фтор-2-метоксил-5-нитрофенил)-пиримидин-4,6-диаминтолуолсульфоната
Стадии 1 и 2 являются такими же, как в примере 10 за исключением замены 3-броманилина на 4-фтор-3-хлоранилин.
Стадия 3: Получение N-(4-фтор-3-хлорфенил)-N'-(4-(2-диметиламино-этоксил)-2-метоксил-5-нитрофенил)-пиримидин-4,6-диамина

N-(4-фтор-3-хлорфенил)-N'-(4-фтор-2-метоксил-5-нитрофенил)-пиримидин-4,6-диаминтолуолсульфонат (0.58 г, 1 ммоль), диметиламиноэтанол (0.18 г, 2 ммоль) и гидроксид натрия (0.16 г, 4 ммоль) добавили в 4 мл ДМФ и затем нагревали на масляной бане при 60°С в течение 4 ч. Реакционную смесь разбавили водой и этилацетатом. Слой этилацетата высушили и сконцентрировали. Остаток очистили посредством колоночной хроматографии с получением промежуточного соединения, указанного в заголовке (0.15 г). m/z: ESI МН+ 477.1.
Стадии 4 и 5 примера 10 повторили с получением соединения, указанного в заголовке.
Пример 12
Получение N-(5-(6-(2,4-дихлор-5-метоксил-фениламино)-пиримидин-4-иламино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламида (Соединение 136)

Стадия 1: Получение
(6-хлор-пиримидин-4-ил)-4-фтор-2-метоксил-5-нитроанилина

2,4-дихлор-5-метоксиланилин (11.5 г, 60 ммоль), 4,6-дихлорпиримидин (13.4 г, 90 ммоль) и метансульфоновую кислоту (6.9 г, 72 ммоль) добавили в 100 мл изопропилового спирта и затем перемешивали при кипении с обратным холодильником в течение 7 ч. Реакционную смесь охладили до комнатной температуры и отфильтровали с получением промежуточного соединения, указанного в заголовке (18 г). m/z: ESI МН+ 304.0.
Стадии 2~5 примера 10 повторили с получением соединения, указанного в заголовке.
Соединения 133-135, 138-147, 153-154 в Таблице 5 получили в соответствии с методикой в примере 10.
Соединения 148-152 в Таблице 5 получили в соответствии с методикой в примере 11.
Соединения 136-137 в Таблице 5 получили в соответствии с методикой в примере 12.
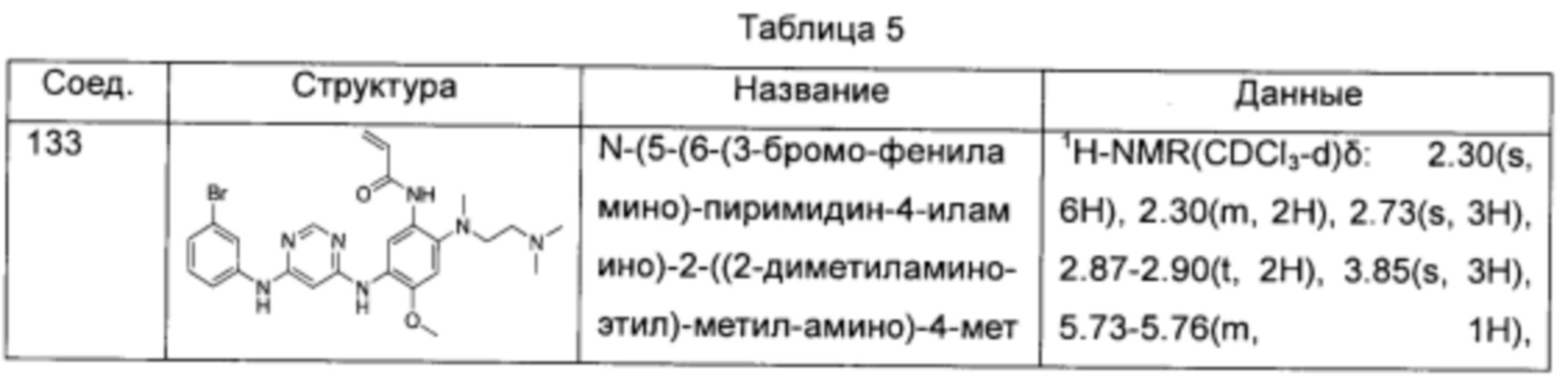



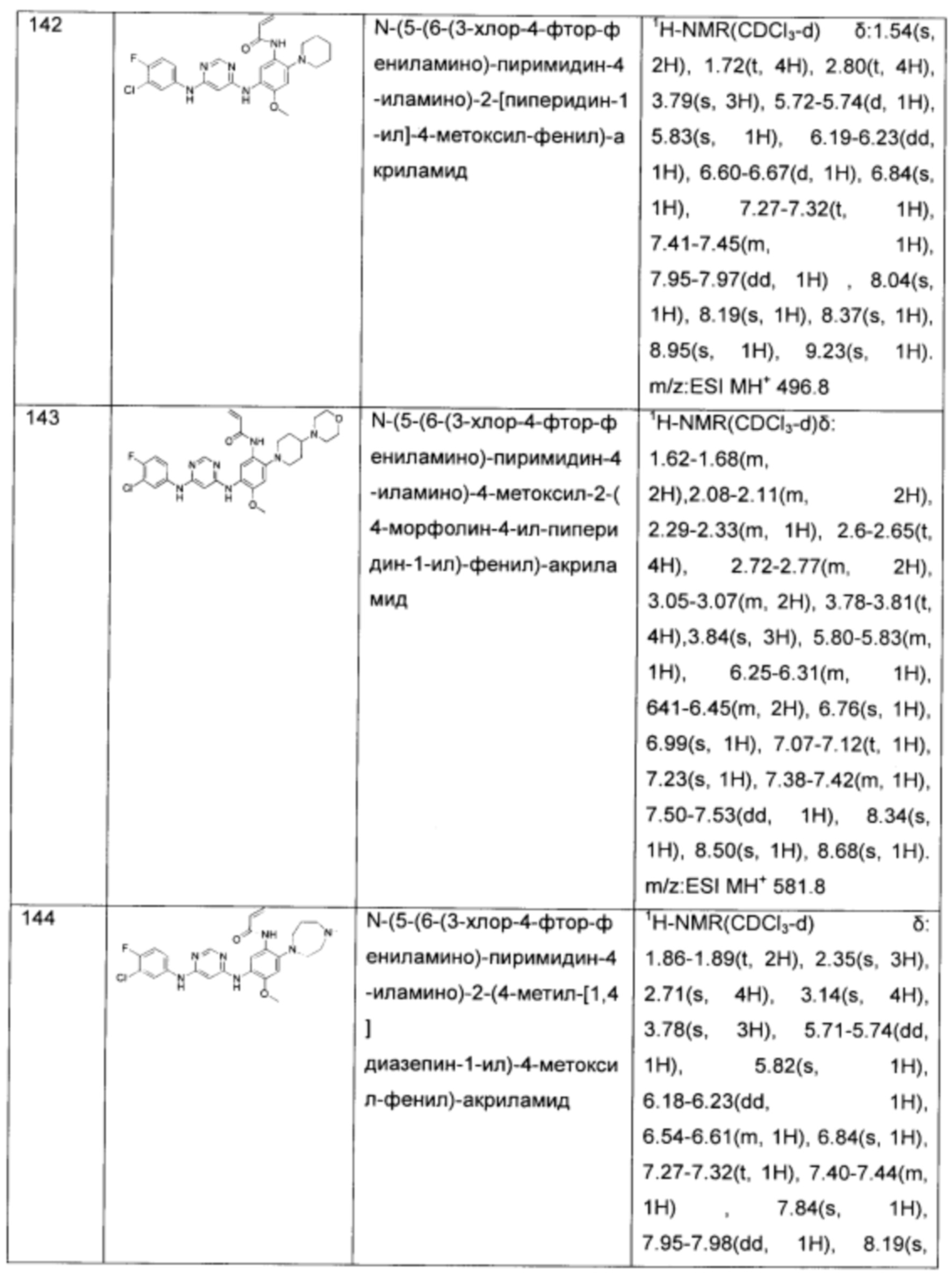




Соединения 155-157 в Таблице 6 получили в соответствии с методикой в примере 8.


Пример 13
Получение
N-(4-метоксил-5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-иламино)-2-(4-метилпиперазин-1-ил)-фенил)-акриламида (Соединение 158)


Стадия 1: Синтез 3-(6-хлорпиримидин-4-ил)-1-метил-1Н-индола

1-Метилиндол (2.5 мл, 23 ммоль), 4,6-дихлорпиримидин (3 г, 23 ммоль) и безводный трихлорид алюминия (3 г, 23 ммоль) добавили в 30 мл 1,2-дихлоржтана и затем нагревали при 45°С в течение 4 ч. Реакционную смесь охладили до комнатной температуры, и добавили 1М разбавленной соляной кислоты и дихлорметан. Дихлорметановый слой высушили и затем сконцентрировали. Остаток очистили посредством колоночной хроматографии с получением промежуточного соединения, указанного в заголовке (3.5 г), m/z: ESI МН+ 244.1.
Стадия 2: Синтез (4-фтор-2-метоксил-5-нитро-фенил)-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-ил)-амина

3-(6-хлорпиримидин-4-ил)-1-метил-1Н-индол (1.2 г, 5 ммоль), 2-метоксил-4-фтор-5-нитроанилин (0.9 г, 5 ммоль) и п-толуолсульфоновую кислоту (1.03 г, 6 ммоль) добавили в 15 мл 2-амилового спирта и затем нагревали при 115°С в течение 3 ч. Реакционную смесь охладили до комнатной температуры и затем отфильтровали. Остаток на фильтре промыли дважды метилтрет-бутиловым эфиром и высушили с получением промежуточного соединения, указанного в заголовке (1.4 г), m/z: ESI МН+ 394.1.
Стадии 3-5 являются такими же, как в примере 6, за исключением замены триметилэтилендиамина на N-метилпиперазин.
Пример 14
Получение N-(4-метоксил-2-(2-метоксил-этоксил)-5-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-ил амино)-фенил)-акриламида (Соединение 196)

Стадии 1 и 2: Синтез (4-фтор-2-метоксил-5-нитро-фенил)-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-ил)-амина
Способ получения был таким же, как в Примере 13.
Стадия 3: Получение (2-метоксил-4-(2-метоксил-этоксил)-5-нитро-фенил)-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-ил)-амина
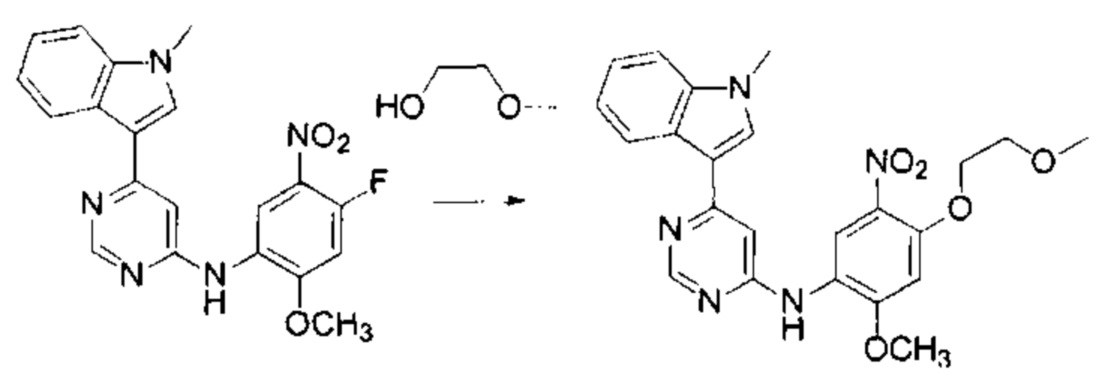
4-фтор-2-метоксил-5-нитро-фенил)-(6-(1-метил-1Н-индол-3-ил)-пиримидин-4-ил)-амин (0.4 г, 1 ммоль), монометиловый эфир этиленгликоля (0.15 г, 2 ммоль) и гидроксид натрия (0.16 г, 4 ммоль) добавили в 2 мл ДМФ и затем нагревали при 60°С в течение 5 ч. Реакционную смесь охладили до комнатной температуры и затем разбавили водой и этилацетатом. После концентрирования этилацетатного слоя порошок железа (0.28 г, 5 ммоль), хлорид аммония (0.27 г, 5 ммоль), воду (5 мл) и этанол (15 мл) добавили и затем нагревали при 80°С в течение 5 ч. Осадок железа отфильтровали горячим. Фильтрат сконцентрировали и затем разбавили водой и дихлорметаном. Дихлорметановый слой высушили и затем сконцентрировали. Остаток очистили посредством колоночной хроматографии с получением промежуточного соединения, указанного в заголовке (0.18 г), m/z: ESI МН+ 450.2.
Стадии 4 и 5 примера 13 повторили с получением соединения, указанного в заголовке.
Соединения 158-186 и 188-195 в Таблице 7 получили в соответствии с методикой в примере 13.
Соединение 187 в Таблице 7 получили в соответствии с методикой в примере 6.
Соединения 196-199 в Таблице 7 получили в соответствии с методикой в примере 14.


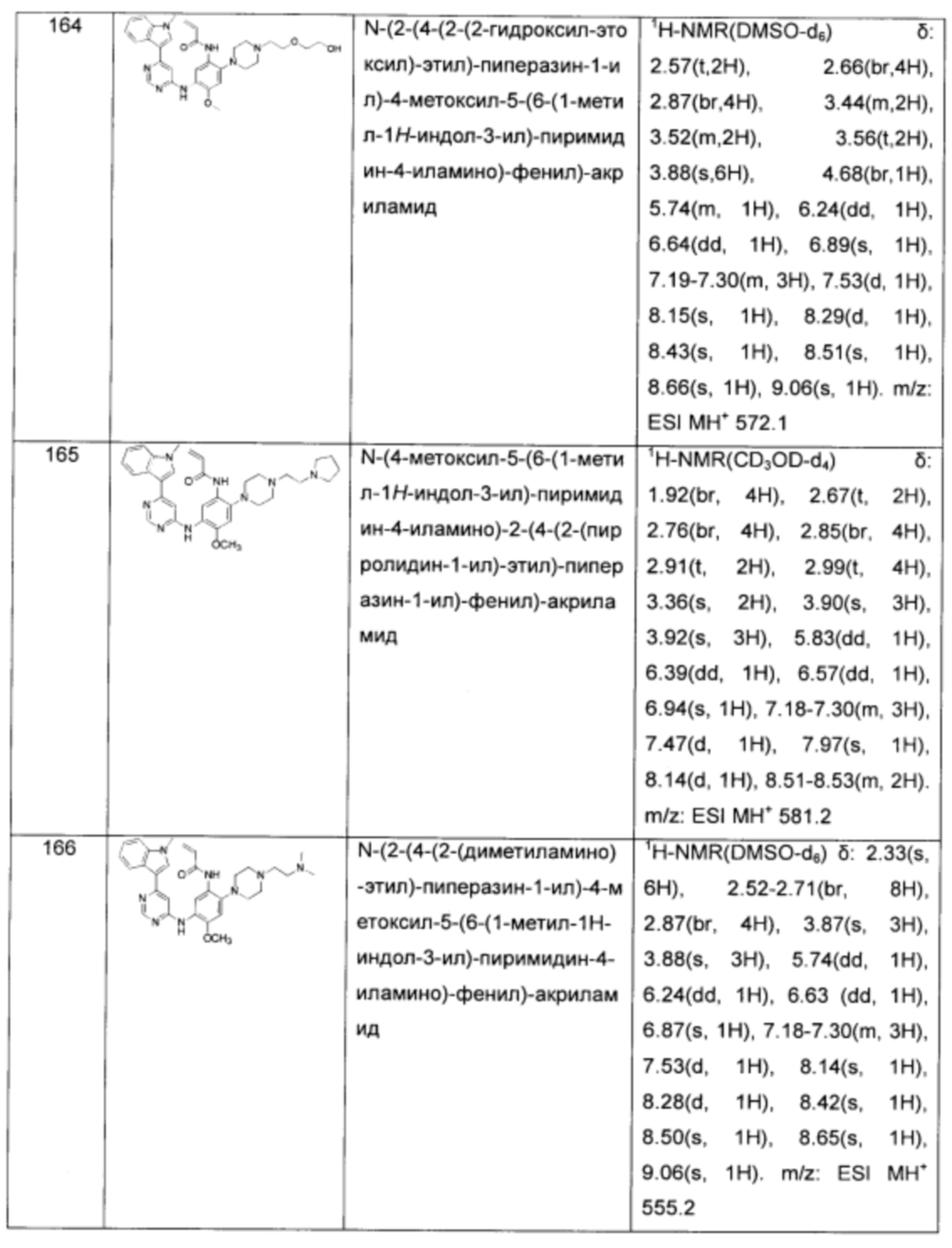







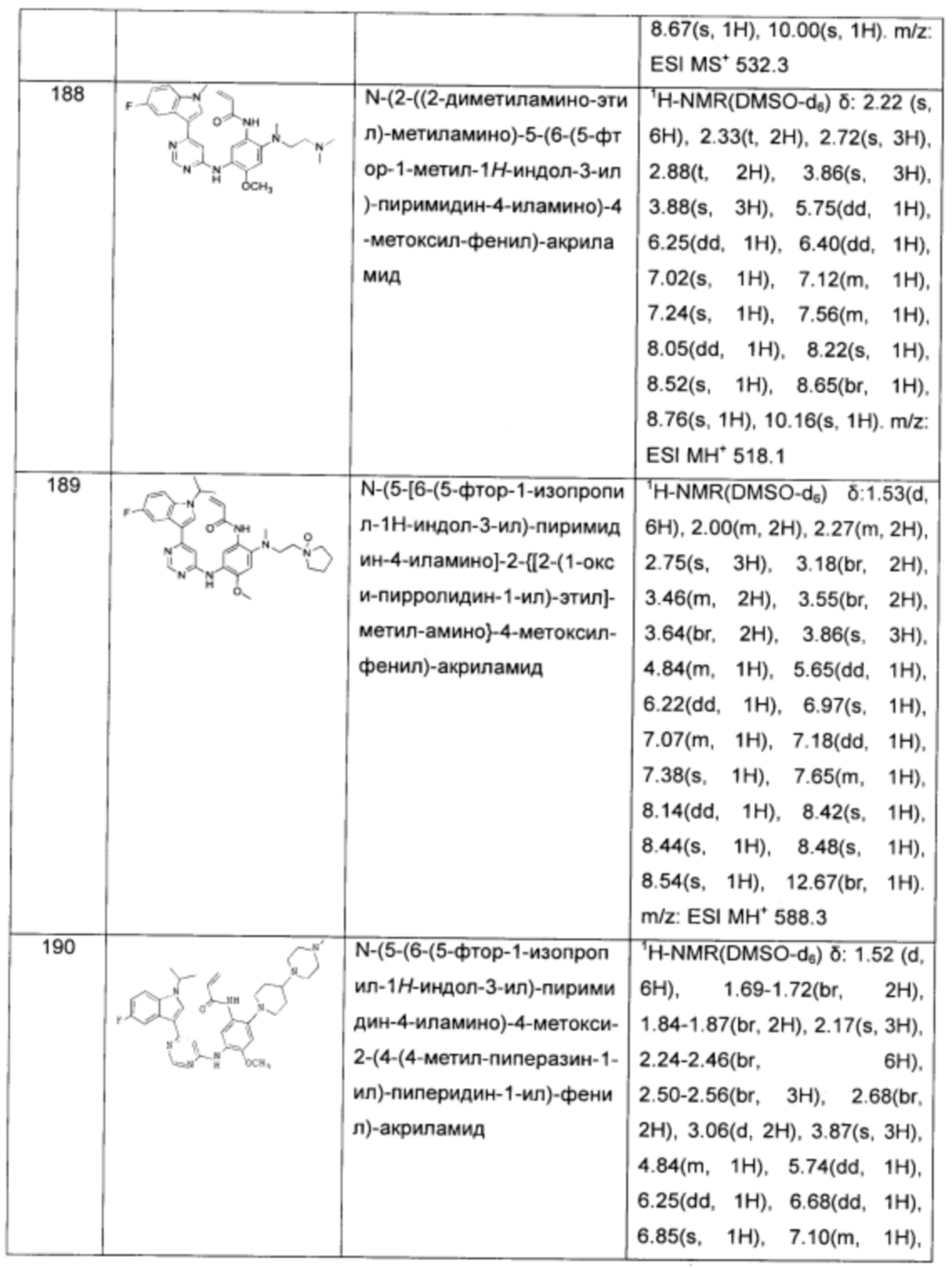




Пример 15
Получение N-(5-(4-((3-хлор-4-(пиридин-2-илметоксил)фенил)-метил-амино)-пиримидин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксил-фенил)-акриламида (Соединение 200)

Соединение 200
Стадия 1: Синтез 4-(3-хлор-4-(пиридин-2--илметоксил)фениламино)-2-хлорпиридина
Стадия 1 является такой же, как в примере 1.
Стадия 2: Синтез 4-((3-хлор-4-(пиридин-2-метоксил)фенил)-метил-амино)-2-хлор-пиримидина
4-(3-хлор-4-(пиридин-2--илметоксил) фениламино)-2-хлорпиримидин (0.7 г, 2 ммоль), карбонат калия (0.41 г, 3 ммоль) и метилйодид (0.34 г, 2.4 ммоль) добавили в 4 мл ДМФ и затем перемешивали в течение 36 ч. Реакционную смесь разбавили водой и этилацетатом. Слой этилацетата высушили и затем сконцентрировали. Остаток очистили посредством колоночной хроматографии с получением 4-((3-хлор-4-(пиридин-2-метоксил)фенил) -метил-амино)-2-хлорпиримидина (0.5 г). m/z: ESI МН+ 361.1.
Стадии 3~5 примера 3 повторили с получением соединения, указанного в заголовке.
Соединение 200 в Таблице 8 получили в соответствии с методикой в примере 15.
Соединение 201 в Таблице 8 получили в соответствии с методикой в примере 2.

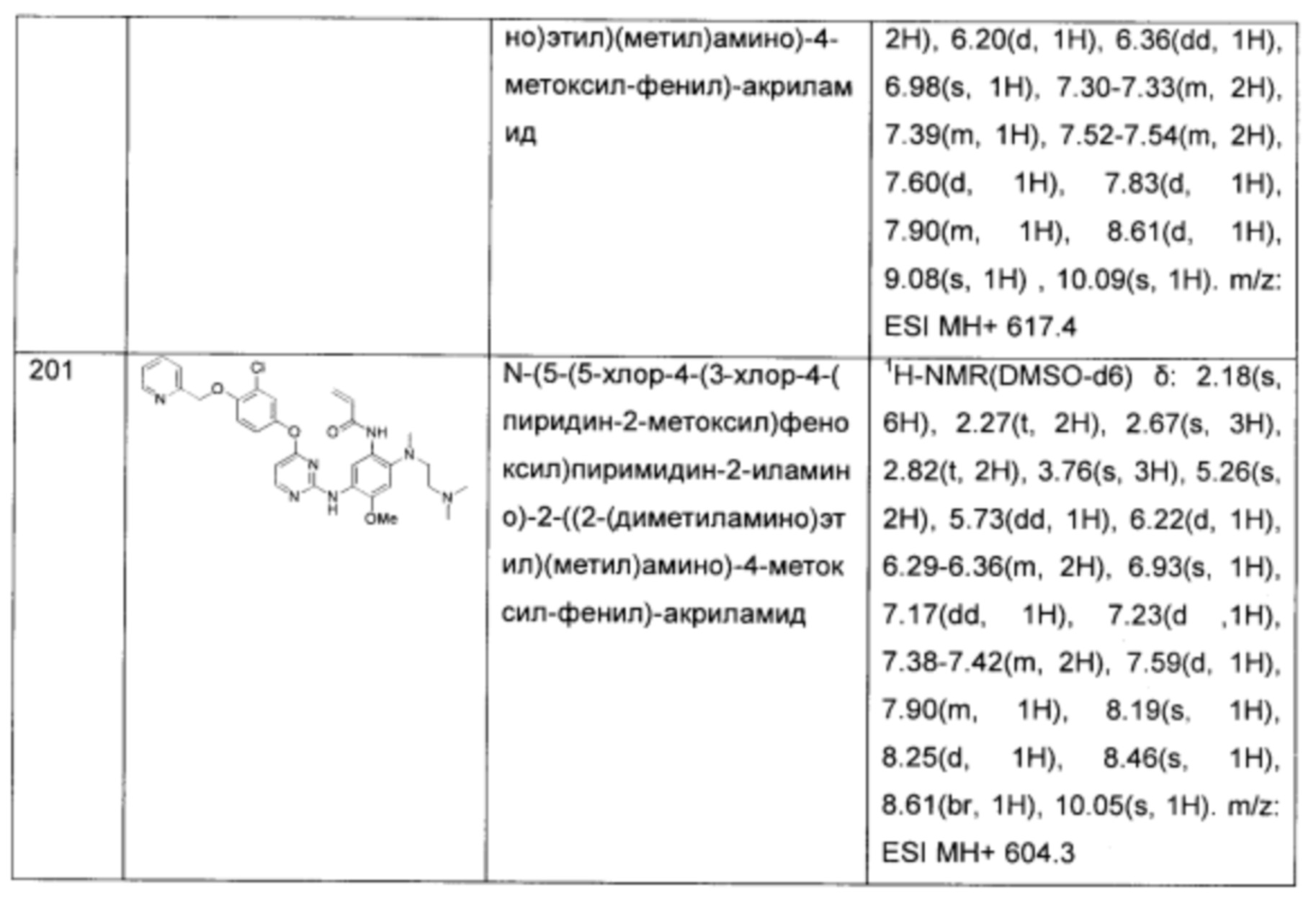
Пример 16
Получение N-(5-(4-(4-метоксил-1Н-индол-1-ил)-5-трифторметилпиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламида (Соединение 202)

Соединение 202
Стадия 1: Синтез 4-хлор-2-(4-нитрофеноксил)-5-трифторметил пиримидин
4-Нитрофенол (1.39 г, 10 ммоль) и N-метилморфолин (1.0 г, 10 ммоль) растворили в 15 мл изопропилового спирта и затем охладили на бане с ледяной солью и разбавили 2,4-дихлор-5-трифторметилпиримидином (2.17 г, 10 ммоль). После перемешивания в течение 1 ч реакционную смесь разбавили 35 мл воды и затем отфильтровали с получением неочищенного продукта (3 г).
Стадия 2: Синтез 4-(4-метоксил-1Н-индол-1-ил)-2-(4-нитро феноксил)-5-трифторметил пиримидина

4-хлор-2-(4-нитрофеноксил)-5-трифторметил пиримидин (2.0 г, 6.25 ммоль), 4-метоксил-1Н-индол (0.92 г, 6.25 ммоль) и карбонат цезия (4.0 г, 12.5 ммоль) добавили в 8 мл ДМФ и затем перемешивали в течение 6 ч. Реакционную смесь разбавили водой и этилацетатом. Слой этилацетата высушили и сконцентрировали. Остаток очистили посредством колоночной хроматографии с получением промежуточного соединения, указанного в заголовке (1.5 г), m/z: ESI МН+ 431.1.
Стадия 3: N4-(2-диметиламиноэтил)-2-метоксил-N1-(4-(4-метоксил-индол-1-ил)-5-трифторметил-пиримидин-2-ил)-N4-метил-5-нитро-фенил-1,4-диамин

4-(4-метоксил-1Н-индол-1-ил)-2-(4-нитро феноксил-5-трифторметил пиримидин (0.86 г, 2 ммоль) и N4-(2-диметиламиноэтил)-2-метоксил-N4-метил-5-нитро-фенил-1,4-диамин (0.54 г, 2 ммоль) добавили в 5 мл ДМФ и затем разбавили гидридом натрия (240 мг, 6 ммоль). Смесь перемешивали в течение 3 ч и затем разбавили водой и этилацетатом. Органический слой высушили и затем сконцентрировали. Остаток очистили посредством колоночной хроматографии с получением промежуточного соединения, указанного в заголовке (0.32 г), m/z: ESI МН+ 560.3.
Стадии 4 и 5 являются такими же, как в примере 6 с получением соединения, указанного в заголовке.
Пример 17
Получение N-(5-(4-(4-метоксил-1Н-индол-1-ил)-5-хлорпиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламида (Соединение 203)

Соединение 203
Стадия 1: Получение 1-(2,5-дихлорпиримидин-4-ил)-6-метоксил-1Н-индола

Стадия 1 является такой же, как в примере 6 за исключением замены 2,4-дихлорпиримидина на 2,4,5-трихлорпиримидин.
Стадия 2: Получение (4-фтор-2-метоксил-5-нитро-фенил)-(4-(4-метоксил-индол-1-ил)-5-хлорпиримидин-2-ил)-амина бензолсульфоната

1-(2, 5-дихлорпиримидин-4-ил)-6-метоксил-1Н-индол (1 г, 3.4 ммоль), 2-метоксил-4-фтор-5-нитроанилин (0.64 г, 3.4 ммоль) и бензолсульфоновую кислоту (0.65 г, 4.1 ммоль) добавили в 15 мл хлорбензола и затем нагревали при 130°C в течение 20 ч. Реакционную смесь охладили до комнатной температуры и затем разбавили 15 мл петролейного эфира и отфильтровали. Осадок высушили с получением неочищенного промежуточного соединения (1.5 г). m/z:ESI МН+ 444.2.
Стадии 3-5 примера 6 повторили с получением соединения, указанного в заголовке.
Пример 18
Получение
N-(5-(4-(4-гидроксил-1Н-индол-1-ил)-пиримидин-2-иламино)-2-(2-(диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламида (Соединение 204)

Соединение 204
Стадии 1~3: Получение N4-(2-диметиламиноэтил)-2-метоксил-N1-(4-(4-бензилокси-индол-1-ил)-пиримидин-2-ил)-N4-метил-5-нитро-1,4-фенилендиамина

Стадии 1~3 являются такими же, как в примере 6 за исключением замены 4-метоксилиндола на 4-бензилоксииндол.
Стадия 4: Синтез 1-(2-(5-амино-4-((2-диметиламино-этил)-метил-амино)-2-метоксил-фениламино)-пиримидин-4-ил)-1Н-4-гидроксииндола
1-(2-(5-амино-4-((2-диметиламино-этил)-метил-амино)-2-метоксил-фениламино)-пиримидин-4-ил)-1Н-4-бензилоксииндол (200 мг, 0.35 ммоль) и гидроксид палладия (30 мг) добавили в 5 мл метанола и затем перемешивали в атмосфере водорода в течение ночи. Гидроксид палладия отфильтровали и фильтрат сконцентрировали с получением неочищенного продукта, который сразу использовали на следующей стадии.
Стадия 5 примера 6 повторили с получением соединения, указанного в заголовке.
Соединение 202 в Таблице 9 получили в соответствии с методикой в примере 16.
Соединение 203 в Таблице 9 получили в соответствии с методикой в примере 17.
Соединение 204 в Таблице 9 получили в соответствии с методикой в примере 18.
Соединения 205-214 в Таблице 9 получили в соответствии с методикой в примере 6.



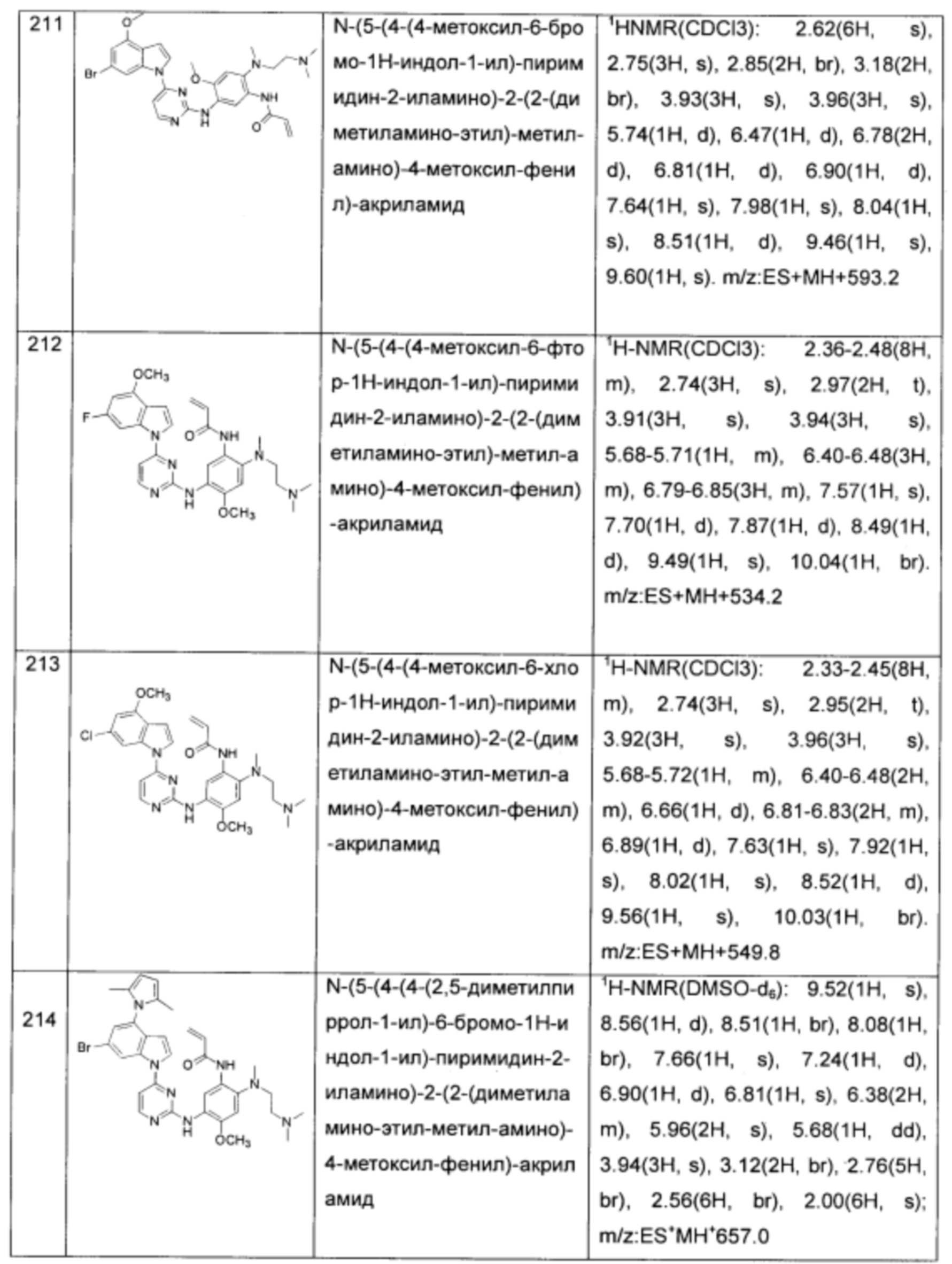
Соединения 215-238 в Таблице 10 получили в соответствии с методикой в примере 10.
Соединения 239-241 в Таблице 10 получили в соответствии с методикой в примере 12.
Соединения 242-248 в Таблице 10 получили повторением первых двух стадий примера 12 за исключением замены диметиламиноэтанола на соответствующий спирт.
Соединение 249 в Таблице 10 получили как в Примерах 12 и 15.




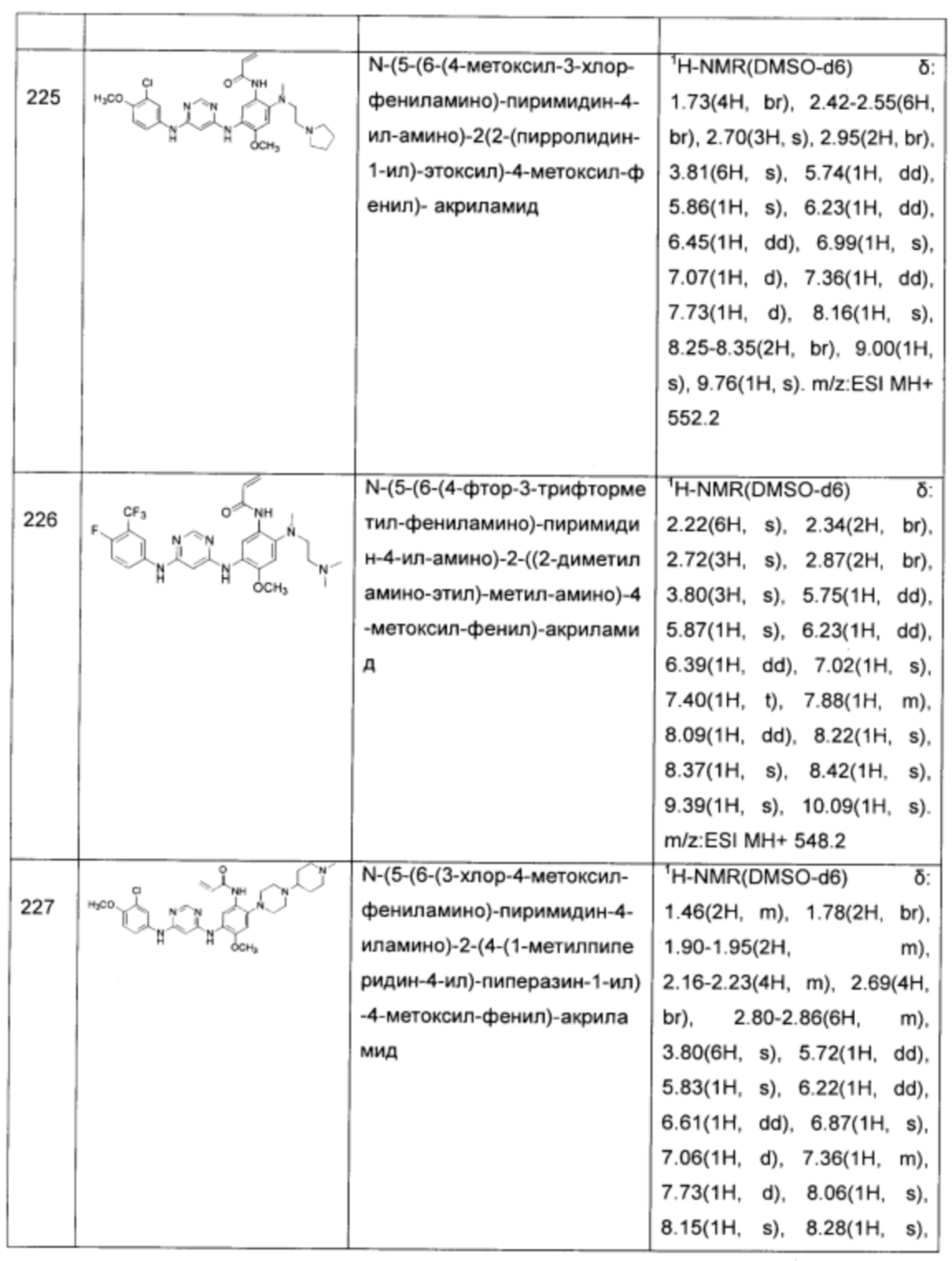
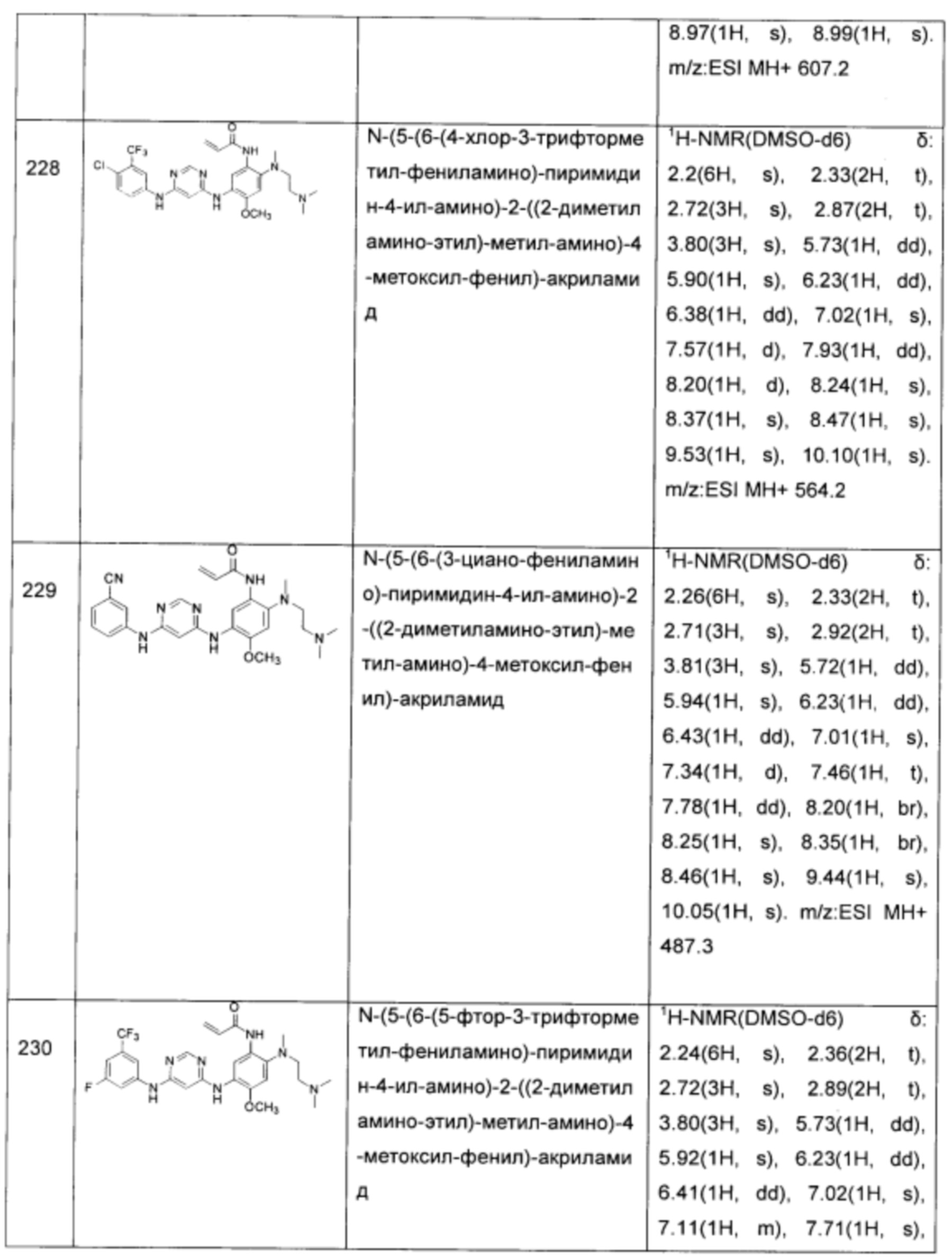



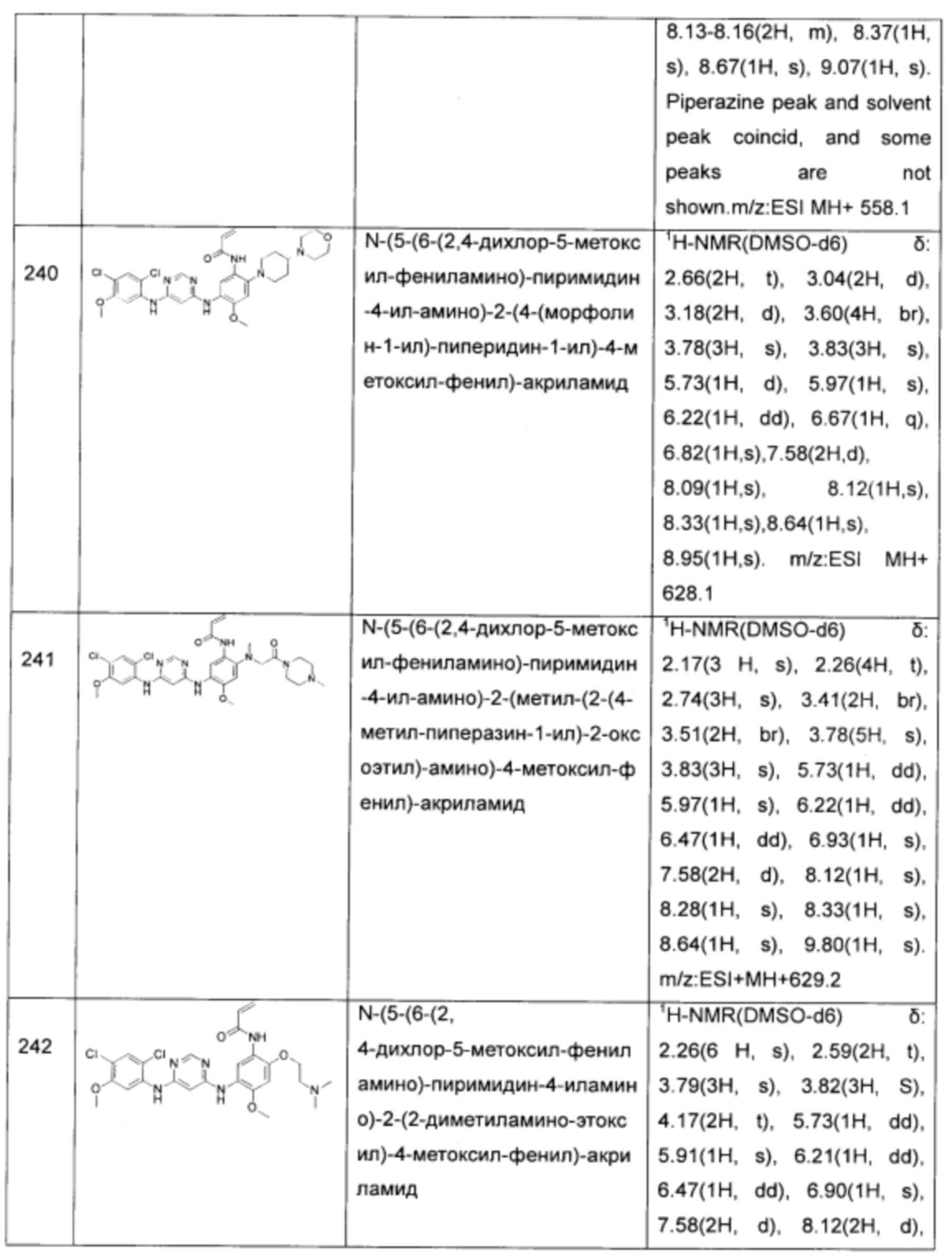



Пример 19
Получение
N-(5-(6-(3-хлор-4-трет-бутокси-фениламино)-пиримидин-4-иламино)-2-((2-диметиламиноэтил)-метил-амино)-4-метоксил-фенил)-акриламида (Соединение 250)

Соединение 250
Стадия 1: Получение (6-хлор-пиримидин-4-ил)-4-фтор-2-метоксил-5-нитроанилина

2-метоксил-4-фтор-5-нитроанилин (9.3 г, 50 ммоль), 4,6-дихлорпиримидин (11.3 г, 75 ммоль) и метансульфоновую кислоту (5.3 г, 55 ммоль) добавили в 150 мл изопропилового спирта и затем перемешивали при кипении с обратным холодильником в течение 6 ч. Реакционную смесь отфильтровали с получением промежуточного соединения, указанного в заголовке (11.5 г). m/z:ESI МН+ 299.0.
Стадии 2 и 3: Получение N-(4-трет-бутокси-3-хлорфенил)-N'-(4-((2-диметиламиноэтил)-метил-амино)-2-метоксил-5-нитрофенил)-пиримидин-4,6-диамина

3-хлор-4-трет-бутоксифениамин (1.95 г, 10 ммоль), (6-хлор-пиримидин-4-ил)-4-фтор-2-метоксил-5-нитроанилин (0.75 г, 2.5 ммоль) смешали и затем нагревали при 100°C в течение 2 ч в атмосфере азота. Реакционную смесь охладили до комнатной температуры и затем добавили триметилэтилендиамин (0.51 г, 5 ммоль), карбонат калия (1.38 г, 10 ммоль) и 6 мл ДМА. Смесь нагревали на масляной бане при 85°C в течение 1 ч, и затем разбавили водой и этилацетатом. Органический слой высушили и затем сконцентрировали. Остаток очистили посредством колоночной хроматографии с получением промежуточного соединения, указанного в заголовке (0.45 г). m/z:ESI МН+ 544.2.
Стадии 4 и 5 являются такими же, как в примере 1 с получением соединения, указанного в заголовке.
Пример 20
Получение
N-(5-(6-(3-хлор-4-(3-метил-оксетан-3-ил-метоксил)-фениламино)-пиримидин-4-ил-амино)-2-((2-диметиламино-этил)-метил-амино)-4-метоксил-фенил)-акриламида (Соединение 252)

Соединение 252
Стадии 1~3: Получение 2-хлор-4-(6-(4-((2-диметиламино-этил)-метил-амино)-2-метоксил-5-нитро-фениламино)-пиримидин-4-иламино)-фенола
Стадии 1-3 примера 19 повторили за исключением замены 3-хлор-4-трет-бутоксифениламина на 2-хлор-4-аминофенол.
Стадия 4: Получение N-(4-(3-метил-оксетан-3-ил-метоксил)-3-хлорфенил)-N'-(4-((2-диметиламиноэтил)-метил-амино)-2-метоксил-5-нитрофенил)-пиримидин-4,6-диамина

N-(4-гидроксил-3-хлорфенил)-N'-(4-((2-диметиламиноэтил)-метил-амино)-2-метоксил-5-нитрофенил)-пиримидин-4,6-диамин (0.49 г, 1 ммоль, способ получения такой же, как в примере 19), 3-метил-3-хлорометил-оксетан (0.15 г, 1.2 ммоль) и гидроксид натрия (0.08 г, 2 ммоль) добавили к 3 мл ДМА и затем нагревали на масляной бане при 55°C в течение 20 ч. Реакционную смесь разбавили водой и этилацетатом. Органический слой высушили и затем сконцентрировали. Остаток очистили посредством колоночной хроматографии с получением продукта (0.23 г). m/z:ESI МН+ 572.2.
Стадии 5 и 6 являются такими же как стадии 4 и 5 в примере 19 с получением соединения, указанного в заголовке.
Соединения 250-251 в Таблице 11 получили в соответствии с методикой в примере 19.
Соединение 252 в Таблице 11 получили в соответствии с методикой в примере 20.
Соединения 253-256 в Таблице 11 получили в соответствии с методикой в примере 10.



ПРИМЕРЫ ТЕСТИРОВАНИЯ Определение активности соединения
Тестовый пример 1
Определение 50% ингибирования роста (GI50) in vitro заявленным соединением в линиях EGFR дикого типа и мутантных клеточных линиях или HER2/ErbB2 положительных линиях опухолевых клеток.
Экспериментальные материалы и методы
1. Линии опухолевых клеток и клеточная культура
Линии опухолевых клеток являются эффективными клеточными моделями для изучения ингибирования роста опухолевых клеток или пролиферации in vitro. Некоторые типичные линии опухолевых клеток были выбраны для определения активности настоящего соединения. Все клеточные линии, использованные в эксперименте, были приобретены в АТСС и Клеточном Банке Китайской академии наук, соответственно. Условия и способы культивирования клеток проводили в соответствии с требованиями для каждой клеточной линии. Каждая клеточная линия культуры in vitro не должна превышать трех пассажей. Выделение и идентификация моноклональных клеток могут проводиться для клеточных линий при необходимости.
В качестве среды для культивирования клеток были выбраны RPMI1640 (Gibco), MEM (Gibco), McCOY'S5A (Gibco) и IMDM (Gibco). Полную среду получали с добавлением 5-20% фетальной телячьей сыворотки (Gibco), 1% двойных антибиотиков (10000 единиц/мл пеницилтина и 10000 единиц/мл), 2 мМ глутамина или 1 мМ пирувата натрия, соответственно.
(1) EGFR дикого типа (WT) или мутантные опухолевые клеточные линии
Клеточные линии с EGFR дикого типа: клеточная линия эпидермального рака человека А431 (EGFR WT, амплификация и сверхэкспрессия, из Шанхайского клеточного банка Китайской академии наук и АТСС), клеточные линии НМРЛ NCI-H460 (EGFR WT, Kras G61H и PI3KCA E545K мутанты, АТСС) и NCI-H1299 (EGFR WT, АТСС), клеточная линия меланомы А375 (мутант EGFR WT и BRAF V600E из Шанхайского клеточного банка Китайской академии наук, АТСС), клеточная линия рака легких человека NCI-H292 (EGFR WT, из человеческой мукоэпидермоидной карциномы легкого/лимфатических метастаз, приобретенная у Шанхайского клеточного банка Китайской академии наук), культивировали в полной среде 1×RPMI1640, с подпиткой 10% FBS. Клеточную линию НМРЛ А549 (EGFR WT и мутант Kras G12S из Шанхайского клеточного банка Китайской академии наук) культивировали в полной среде 1×Ham F12K, с подпиткой 10% FBS, 1% двойных антибиотиков и 2 мМ глутамина.
Клеточные линии с мутантным EGFR: обе клеточные линии НМРЛ РС-9 (АТСС) и НСС827 (Шанхайский клеточный банк Китайской академии наук) имеют делецию экзона 19 EGFR (Е746-А750). Две клеточные линии являются чувствительными к терапии ингибитором ПТК EGFR первого поколения. NLCLC-клеточная линия NCI-H1975 экспрессирует EGFR L858R/T790M (АТСС) и является резистентной к ингибиторам EGFR первого поколения. PC-9ER является клеточной линией РС-9, разработанной инвестором, с приобретенной лекарственной резистентностью к эрлотинибу. В этой клеточной линии EGFR представляет собой мутантный тип deIE746-A750 / Т790М и присутствует устойчивость к ингибиторам EGFR первого поколения. Четыре вышеуказанные клеточные линии культивировали в полной среде 1×RPMI1640 (добавка 10% FBS). Опухолевую клеточную линию рака толстой кишки SW48 (EGFR G719S позитивная, АТСС) культивировали в полной среде L15 (10% FBS).
(2) клеточные линии с амплификацией и высокой экспрессией или мутацией HER2
Клеточную линию рака желудка человека NCI-N87 (АТСС), клеточную линию рака молочной железы человека ZR-75-30 (АТСС), клеточную линию аденокарциномы человека AU565 (АТСС), клеточную линию карциномы плоскоклеточного рака легкого человека NCI-H2170 (АТСС) и линия рака молочной железы человека НСС1954 (клеточная линия, резистентная как в отношении селективного обратимого ингибитора HER2/ERB2 лапатиниба, так и в отношении моноклонального антитела к HER2 герцептина) культивировали в полной среде 1×RPMI1640 (10% FBS) соответственно. Линию клеток аденокарциномы легкого человека Calu-3 (АТСС) культивировали в полной среде 1×MEM (10% FBS, 1% двойных антибиотиков, 1% NEAA(Gibco), 2 мМ глутамина и 1 мМ пирувата натрия). Линию клеток рака молочной железы человека SK-BR-3 культивировали в полной среде McCOY'S5A (10% FBS). Клеточную линию рака толстой кишки SNU1040 (HER2 V777M позитивная) (АТСС) и клеточную линию бронхоальвеолярной аденокарциномы человека NCI-H1781 (экспрессирующая HER2 с мутацией в экзоне 20 G776VC) (АТСС) культивировали в полной среде 1×RPMI1640 (10% FBS) соответственно.
(3) Линии опухолевых клеток с амплификацией гена МЕТ
Линию клеток рака желудка человека MKN-45 (АТСС) и клеточную линию НМРЛ NCI-H1993 (АТСС) культивировали в полной среде 1×RPMI1640, которая была дополнена 10% FBS, соответственно.
(4) Линии опухолевых клеток, экспрессирующие слитые и мутантные гены ALK
Клеточную линию НМРЛ человека NCI-H2228 (АТСС) (EML4-ALK-позитивная) и клеточную линию ALCL человека Karcas-299 (АТСС) (NPM-ALK позитивную) культивировали в полной среде 1×RPMI1640 (добавка 10% FBS) соответственно. Клеточные линии нейробластомы SH-SY5Y (ALK F1174L позитивную) (Шанхайский клеточный банк Китайской академии наук) культивировали в полной среде MEM (добавка 10% FBS, 1% NEAA, 1% двойных антибиотиков и 1 мМ пирувата натрия).
(5) BCR-ABL позитивная клеточная линия
Линию клеток лейкемии человека K562 (экспрессирующая слитый белок BCR-ABL) (АТСС) культивировали в полной среде 1×RPMI1640 (добавка 10% FBS).
(6) FLT3-ITD позитивная клеточная линия
FLT3 является разновидностью ПТК и около 20-30% пациентов с острой миелоцитарной лейкемией (АМЛ) в клинике экспрессируют FLT3-ITD мутированный белок. FLT3-ITD позитивную клеточную линию MV4-11 культивировали в полной среде 1×RPMI1640 (добавка с 10% FBS).
(7) Jak2 V617F позитивная клеточная линия
Jak2 является нерецепторной PTK (Янус киназа 2), имеющей большое значение в росте клеток, дифференцировке и трансформации. Мутация гена Jak2 часто приводит к гиперплазии костного мозга и трансформации. Особенно характерна мутация Jak2 V167F для клинических пациентов. Линию клеток эритролейкемии человека HEL, экспрессирующую Jak2 V167F, культивировали в полной среде 1×RPMI1640 (добавка 10% FBS).
(8) Клеточная линия, позитивная в отношении протеин тирозинкиназы Брутона (BTK)
RAMOS - линия клеток лимфоцитарной лейкемии человека, положительно экспрессирующая BTK. Ее культивировали в полной среде 1×RPMI1640 (добавка 10% FBS).
(9) c-Kit позитивная клеточная линия
Kasumi-1 представляет собой линию клеток лейкемии, положительно экспрессирующую мутант c-kit N822K. Аномальные изменения c-Kit часто проявляются у больных раком. Линию клеток Kasumi-1 культивировали в полной среде 1×RPMI1640 (добавка 10% FBS).
(10) Клеточные линии с амплификацией FGFR1 или FGFR2
Клеточная линия НМРЛ человека NCI-H1581 (АТСС) содержит амплификацию гена FGFR1 и высокую экспрессию. Линия клеток рака желудка человека SNU-16 (АТСС) содержит амплификацию гена FGFR2 и высокую экспрессию. Их культивировали в 1×RPMI1640 полной среде (10% FBS), соответственно.
2. Терапия лекарственными средствами
Адгезивные клетки отщепляли 0,25% панкреатин-ЭДТА (Gibco). Суспензию клеток собрали центрифугированием. Супернатант отбрасывали и осадок клеток ресуспендировали и подсчитывали. Различные концентрации клеток (5~10 × 104 клеток/мл) получали в соответствии с циклом роста каждой клеточной линии и затем высевали на 96-луночный планшет (Corning), 100 мкл/лунку. Клетки инкубировали в течение ночи при 37°C, 5% CO2. Соединения добавили в клетки на второй день с 2 лунками в параллели. Конечная концентрация органического растворителя не должна превышать

Соединения по изобретению и референсные соединения растворили в ДМСО (Sigma), соответственно. Чистота соединения была более 98%. Концентрация для хранения соединений составляла 10 мМ и поддерживалась при -20°C. Они были разбавлены в 2 раза или 10 раз серийным разбавлением перед использованием.
3. Анализ МТТ и расчет GI50
Набор реагентов Dojindo CCK8 использовался в качестве теста МТТ. В качестве микропланшетного ридера использовался измеритель THERMO MULTISKAN FC.
Среду для алгезионного культивирования клеток отбрасывали и немедленно заменяли свежеприготовленной средой, которая содержала 10% реагента CCK8 (100 мкл/лунка). Для суспензионных клеток CCK8 был непосредственно добавлен до 10% от конечной концентрации. Клетки непрерывно инкубировали в течение 1-4 ч. Когда цвет контрольных лунок стал темно-желтым, значение поглощения измерялось при OD450 нм. Скорость роста клеток рассчитывали по следующей формуле: скорость роста клеток (%) = 100* (T-T0)/(C-T0), Т = оптическая плотность лунки, обработанной лекарственным средством - оптическая плотность пустой контрольной лунки; Т0 = оптическая плотность лунки с клетками до обработки лекарственным средством - оптическая плотность пустой контрольной лунки; С = оптическая плотность лунки с клетками контрольной группы растворителя - оптическая плотность пустой контрольной лунки. Рассчитали значение концентрации 50% ингибирования роста клеток (GI50). Эксперимент был независимо повторен 1~3 раза и подвергался биологическому статистическому анализу.
Результаты эксперимента
Результаты этого исследования суммированы в Таблице 12 и Таблице 13, показывающие, что 50% ингибирование роста (или апоптоз клеток) настоящего соединения тестировалось в выбранных клеточных линиях с диким типом или мутантным EGFR, а также в клеточных линиях, позитивных в отношении HER2/ErbB2. Активность ингибирования роста настоящим соединением будет сильнее, когда значение GI50 будет меньше. Если настоящее соединение имеет небольшое значение GI50 в клетках с мутантным EGFR (например, Н1975, РС9, PC9ER и НСС827) и большое значение GI50 в клетках с WT EGFR, таких как А431, А549, Н460 и Н1299, или большое отношение WT EGFR GI50 к мутанту EGFR GI50, это указывает на то, что соединение является более избирательным в отношении мутантного EGFR, чем WT EGFR.


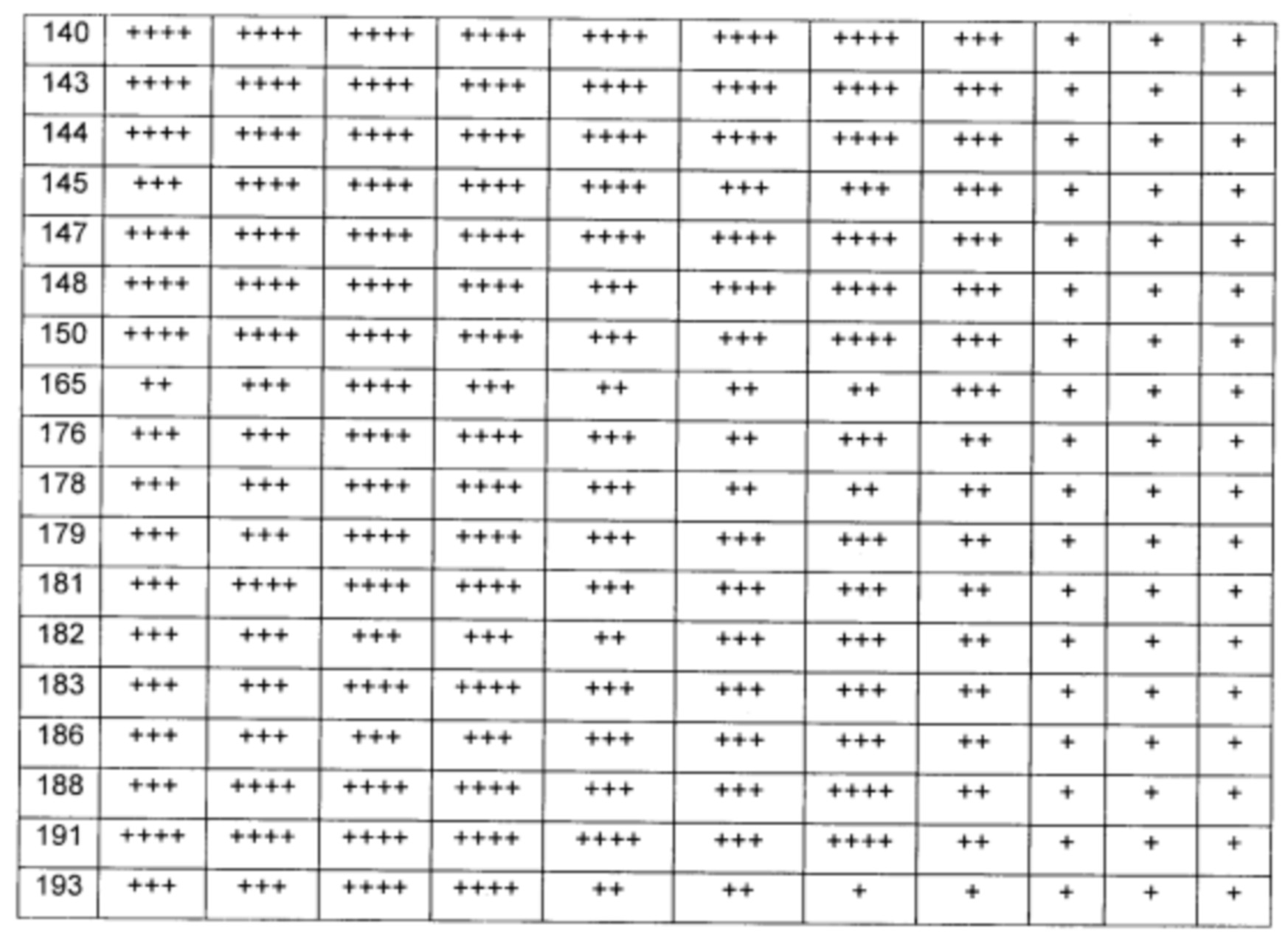



Результаты Таблицы 12 и Таблицы 13 показывают, что соединения по настоящему изобретению обладают значительной активностью ингибирования роста в клеточных линиях с мутантным EGFR Н1975, PC9ER, РС-9 и НСС827 и HER2/ErbB2, N87, AU565 и SK-BR-3. Значение GI50 может быть меньше 10 нМ. Однако для большинства клеточных линий с EGFR дикого типа А539, Н460 и Н1299 значение GI50 составляет более 1000 нм. Даже в клетках А431 эпидермоидной карциномы человека, линии с амплификацией/сверхэкспрессией EGFR дикого типа, настоящие соединения имеют относительно слабую активность ингибирования роста по сравнению с активирующими или мутантными по EGFR Т790М клетками.
Тестовый пример 2
Определение значений GI50 настоящего соединения в разных опухолевых клеточных линиях, экспрессирующих онкогены
Сначала были выбраны девять соединений настоящего изобретения (2, 18, 19, 106, 114, 118, 140, 147 и 183), а в качестве референса выбрали AZD9291, ингибитор EGFR третьего поколения. Анализ ингибирования роста проводили в линиях клеток, показанных в таблице 14, с различными онкогенными мутациями. Каждое соединение начинали при 1000 нм в качестве начальной концентрации и разбавляли в 2 раза серийным разведением до 0,03 нМ. Значение GI50 каждого соединения рассчитывали в соответствии с тестовым примером 1.3. Результаты показаны в Таблице 14.
Исходное соединение AZD9291 синтезировали в соответствии со способом согласно WO 212/014448 А1.



wt - дикий тип
amp - амплификация
Результаты показывают, что соединение по настоящему изобретению обладает избирательно высокой ингибирующей активностью роста клеточных линиях с мутантным EGFR Н1975, РС-9 и НСС827, а также клеточных линиях с амплификации HER2 / ErbB2 (N87, AU565, SK-BR-3, Н2178, ZR-75 -30, Calu-3 и НСС1954). GI50 составляет менее 20 нМ. Относительно высокий уровень GI50 необходим для проявления явного эффекта ингибирования EGFR WT сверхэкспрессирующей клеточной линии А431. Однако в большинстве нормально экспрессирующих EGFR клеточных линиях они проявляют небольшой, если он вообще присутствует, эффект ингибирования. Активность настоящих соединений против ErbB2 позитивных клеток намного выше, чем активность референсного соединения AZD9291. За исключением того, что соединения 114, 118, 140, 147 и 183 оказывают умеренное ингибирующее действие на клетки FLT3-ITD+ MV4-11 и BTK+ Ramos, соединения по настоящему изобретению проявляют небольшой, если он вообще присутствует, эффект ингибирования на клеточные линии, позитивные по другим онкогенам.
Тестовый пример 3
Сравнение ингибирующей активности (GI50) настоящего соединения в клетках с EGFR дикого типа и мутантных опухолевых клеток
Кроме того, настоящие соединения (99, 136, 205, 211, 212, 213, 216, 229, 230, 231, 232, 233 и 236) были выбрано для проведения теста ингибирования роста для некоторых линий опухолевых клеток, которые экспрессируют дикий тип и мутантный EGFR. AZD9291, ингибитор EGFR третьего поколения, использовали в качестве референса (AZD9291 синтезировали в соответствии со способом А1 из WO 212/014448).
Каждое соединение начинали при 1000 нм в качестве начальной концентрации и разбавляли в 2 раза серийным разведением до 0,03 нМ. Значение GI50 каждого соединения рассчитывали в соответствии с тестовым примером 1.3. Результаты показаны в Таблице 15.

Результаты показывают, что соединения настоящего изобретения обладают высокой ингибирующей активностью по отношению к клеточным линиям Н1975, РС-9 и SW48 (GI50 < 20 нм), при небольшом, если оно имеется, ингибированием клеточных линий А548 и Н292, которые экспрессируют EGFR дикого типа, даже при 1000 нм.
Тестовый пример 4
Сравнение активности ингибирования (GI50) настоящими соединениями роста опухолевых клеток с амплификацией/высокой экспрессией или мутациями в HER2
Кроме того, настоящие соединения (99, 136, 205, 211, 212, 213, 216, 229, 230, 231, 232, 233 и 236) были выбраны для проведения теста ингибирования роста опухолевых клеток с амплификацией или высокой экспрессией или мутациями в HER2/ERBB2. Афатиниб и AZD9291 были использованы в качестве референсных как ингибиторы EGFR второго и третьего поколений.
Афатиниб синтезировали согласно способу, раскрытому в патенте US6251912.
Каждое соединение начинали при 1000 нм в качестве начальной концентрации и разбавляли в 2 раза серийным разведением до 0,03 нМ. Значение GI50 каждого соединения рассчитывали в соответствии с тестовым примером 1.3. Результаты показаны в Таблице 16.

Результаты показывают, что настоящее соединение, используемое в эксперименте, обладает значительной ингибирующей активностью в опухолевых клетках с амплификацией / сверхэкспрессией или мутантных HER2. Их ингибирующая активность находится в том же диапазоне, что и у афатиниба, но намного сильнее, чем у AZD9291.
Тестовый пример 5
Эксперимент по ингибированию роста опухоли in vivo
Модели опухолевых ксенотрансплантатов иммунодефицитных мышей являются эффективным инструментом для проверки противоопухолевой активности соединения in vivo. Как правило, эффективность в модели ксенотрансплантата опухолевых клеток человека положительно коррелирует с клиническим лечением опухоли человека. Bab/c иммунодефицитные мыши являются одним из наиболее часто используемых животных с ксенотрансплантатом опухолевых клеток человека. Чтобы проверить, могут ли соединения настоящего изобретения ингибировать рост опухолевых клеток in vivo, экспрессирующих мутантный EGFR или сверхэкспрессирующих HER2/ErbB2, в экспериментах использовали линии клеток Н1975, РС-9 и NCI-N87. Когда клетки Н1975, РС-9 и NCI-N87 находились в логарифмической фазе роста, они были отщеплены панкреатином. Соответствующее количество культуральной среды 1×RPMI1640 (без сыворотки) добавили для прекращения отщепления. Клетки собрали в 50 мл центрифужную пробирку (Corning) и центрифугировали при 1700 об/мин в течение 3 мин. Супернатант отбросили, затем клетки суспендировали в культуральной среде 1×RPMI1640 и подсчитали их количество. Получили 5 ~ 10 × 107 клеток / мл, а затем поместили на лед. 5 ~ 10 × 106 (0,2 мкл) клеток подкожно инокулировали с правой стороны спины 6-8-недельным самкам Bab/c «голых» мышей (около 20 г в весе). Когда средний объем опухоли увеличился до 100~200 мм3 мышей случайным образом поделили на группы (3-6 мышей на группу) и промаркировали метками с помощью прокалывания ушей и взвесили. Тестируемые соединения и референсное соединение (с чистотой более 99% и индивидуальными примесями не более 0,2%) были сформированы в эмульсионную суспензию с СМ (30% полиэтиленгликоля 400, 0,5% Tween-80 и 2,5% пропиленгликоля) соответственно. Внутрижелудочное введение продолжали один раз в день при дозе 25 мг / кг (0,1 мл / 10 г мыши). Опухоль измеряли 3 раза в неделю. Когда средний объем опухоли в контрольной группе СМ составлял до 1500 мм3 или после введения в течение 30 дней, эксперимент был завершен.
Объем опухоли (V) рассчитывали как V=1/2*а*b2 (а - длина опухоли, b - ширина опухоли). Ингибирование роста опухоли (TGI) рассчитывали как: TGI (%) = 100 - (VT - VT0) / (VC - VC0) * 100%; где VT0 и VT - объемы опухолей в первый и последний дни дозированных групп; и VC0 и VC - объемы опухолей в первый и последний день для контрольной группы, соответственно.
Результаты ингибирования роста опухоли in vivo (TGI) настоящими соединениями на опухолевых клетках РС-9 и Н1975 (19-й день) показаны в Таблице 17.

Результаты in vivo ингибирования роста опухоли (TGI) соединениями по настоящему изобретению в клеточной линии N78 с HER2/ ErbB2 амплификацией (день 19) показаны в Таблице 18.
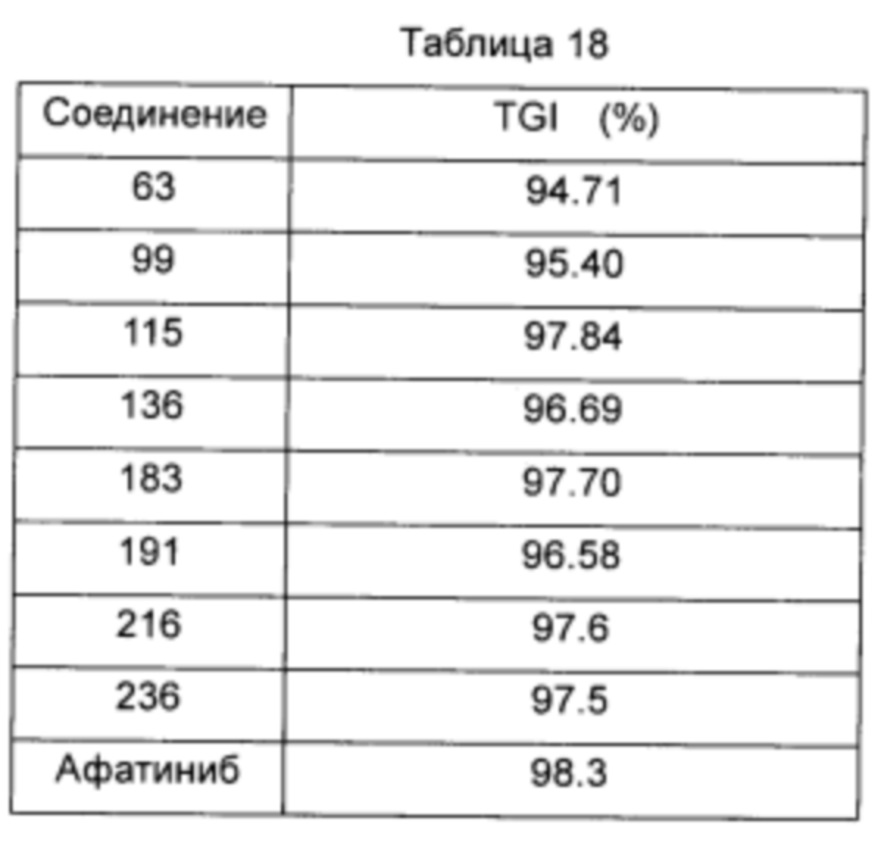
Результаты показывают, что соединения в тестируемой дозе могут эффективно ингибировать рост мутантных клеточных линий EGFR РС-9 и Н1975, а также клеточную линию N87? позитивную по HER2/ErbB2 у «голых» мышей с ксенотрансплантатом без снижения массы тела животных.
Тестовый пример 6
Изучение фармакокинетики соединения настоящего изобретения на крысах
Крысы Спрег-Доули (SD) (мужчины) были приобретены у компании Shanghai SIPPR BK Laboratory Animals Со. и содержались в особых условиях без патогенов в течение 7 дней в учреждении по уходу за животными в Шанхае в Университете традиционной китайской медицины (все исследования на животных проводились с утверждением Комитета по этике университета). Крыс случайным образом разделяли на 2 группы (3 крысы/группа) для каждого соединения. Одной группе назначали инъекцию в хвостовую вену (iv), а другой группе вводили внутрижелудочно (ро). Соединение готовили в прозрачном водном растворе с метансульфоновой кислотой, РН> 3,5. Крысы голодали в течение 12 часов до введения лекарственного средства, и образцы крови собирались из орбитального сплетения через 0 мин (до введения препарата), 2 мин, 5 мин, 15 мин, 30 мин, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч и 24 ч соответственно. Каждый образец крови собирали в 1,5 мл центрифужную пробирку с гепарином натрия и затем центрифугировали при 8000 об/мин при 4°C в течение 3 мин. Супернатант собрали. Концентрацию соединения в плазме определяли с помощью ЖХ-МС/МС. Параметры фармакокинетики рассчитывали с помощью профессионального программного обеспечения для фармакокинетики WinNonlin. Эксперимент повторили один раз. Результаты показаны в Таблице 19.

Результаты показывают, что соединения настоящего изобретения обладают хорошей пероральной биодоступностью у крыс.
Реферат
Изобретение относится к соединениям общей формулы (I) и их фармацевтически приемлемым солям, где в формуле (I) R1представляет собой -NR5R6, -OR5или -SR5; X представляет собой CR4, Y представляет собой N; R2выбран из группы, состоящей из C1-C6алкокси или C1-C6алкилсульфанила; R3выбран из -N(Rv)RuN(Ry)(Rz), -ORuOR6, -ORuN(Ry)(Rz) и -SRuN(Ry)(Rz); R4и R'4каждый выбран из группы, состоящей из водорода, галогена, C1-C6алкила и C1-C6галоалкила; R5, R6, Ry, Rzявляются такими, как указано в п.1 формулы изобретения; Ruпредставляет собой C1-C6алкилен; Rvвыбран из водорода и C1-C6алкила. Также изобретение относится к конкретным соединениям, указанным в п.7 формулы, к фармацевтической композиции и к применению для изготовления лекарственного средства. Технический результат – соединения формулы (I) и конкретные соединения по п.7 формулы в качестве ингибиторов EGFR и/или HER2/ErbB2 PTK. 6 н. и 5 з.п. ф-лы, 19 табл., 26 пр.
Формула



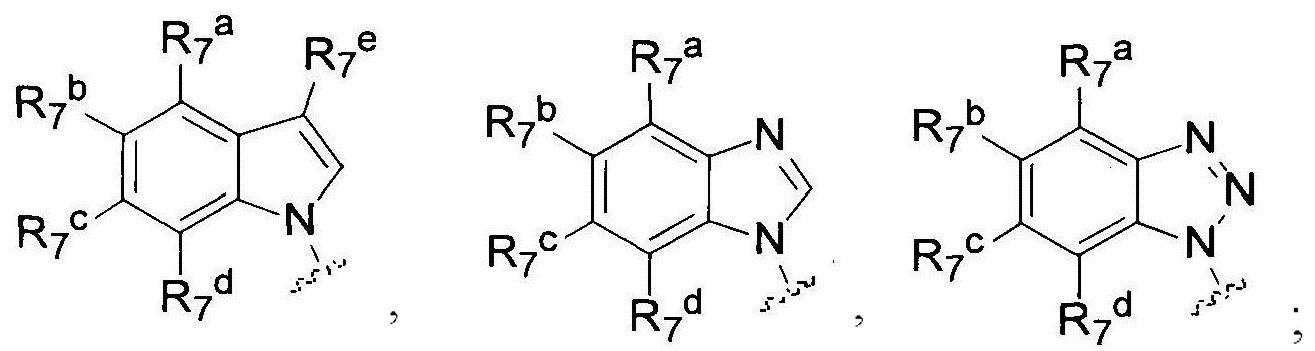


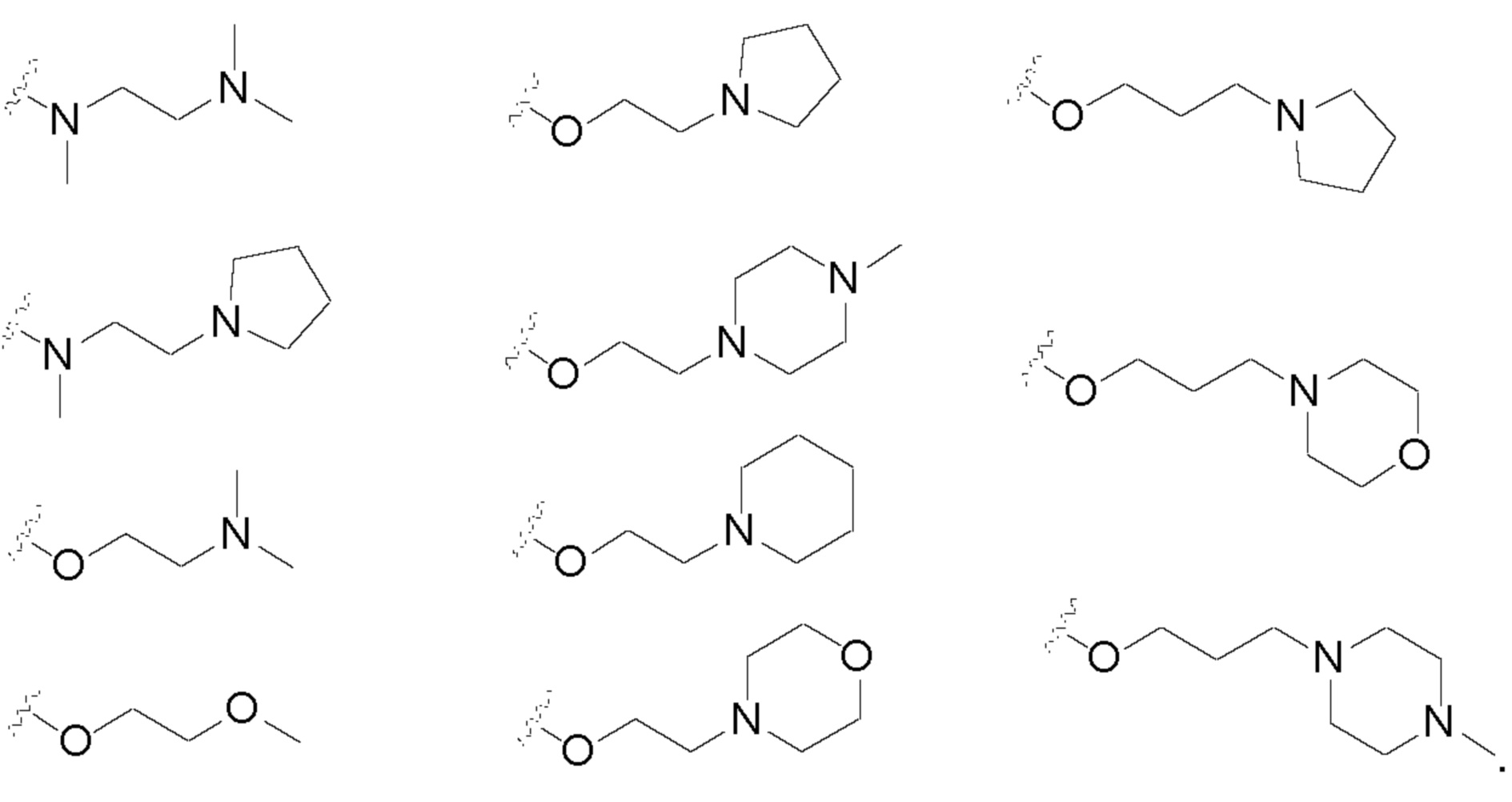
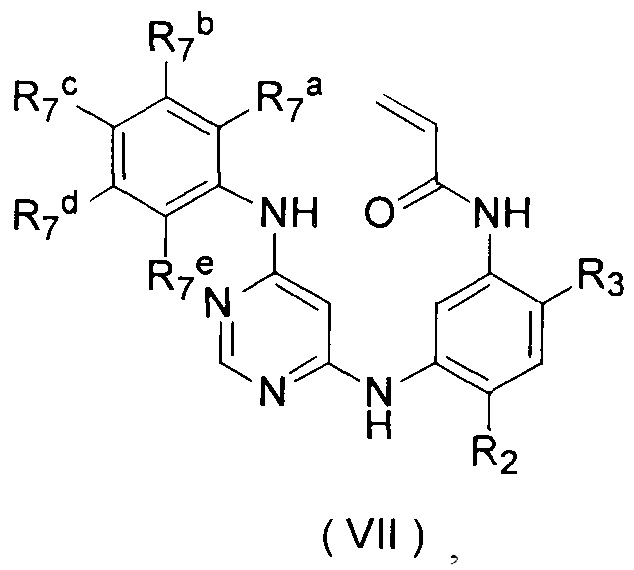




Документы, цитированные в отчёте о поиске
Гетероарильные соединения и их применение
Комментарии