Комбинированная терапия антагонистами с-мет и egfr - RU2601892C2
Код документа: RU2601892C2
Чертежи
























Описание
ПЕРЕКРЕСТНЫЕ ССЫЛКИ НА РОДСТВЕННЫЕ ЗАЯВКИ
По настоящей заявке в соответствии с 35 USC § 119(e) испрашивается приоритет предварительной заявки США № 61/034446, поданной 6 марта 2008 года, и предварительной заявки США № 61/044438, поданной 11 апреля 2008 года, содержание которых включено в настоящий документ в качестве ссылки.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ, К КОТОРОЙ ОТНОСИТСЯ ЗАЯВКА
Настоящее изобретение относится в основном к области молекулярной биологии и регуляции факторов роста. Более конкретно, изобретение относится к комбинированной терапии для лечения патологических состояний, таких как рак.
ПРЕДПОСЫЛКИ
HGF является плейотропным фактором мезенхимального происхождения с митогенной, мотогенной и морфогенетической активностью на целый ряд различных типов клеток. Воздействие HGF опосредовано через специфическую тирозинкиназу, c-met, и патологическую экспрессию HGF и c-met часто наблюдают в различных опухолях. Смотри, например, Maulik et al., Cytokine & Growth Factor Reviews (2002), 13:41-59; Danilkovitch-Miagkova & Zbar, J. Clin. Invest. (2002), 109(7):863-867. Регуляция сигнального пути HGF/c-Met вовлечена в развитие и метастазирование опухоли. Смотри, например, Trusolino & Comoglio, Nature Rev. (2002), 2:289-300).
HGF связывается с экстрацеллюлярным доменом рецептора тирозинкиназы c-met (RTK) и регулирует различные биологические процессы, такие как распространение, пролиферация и выживание клеток. Сигнальная система HGF-Met необходима для нормального эмбрионального развития, особенно для миграции мышечных клеток-предшественников и развитии печени и нервной системы (Bladt et al., Nature (1995), 376, 768-771; Hamanoue et al., Faseb J (2000), 14, 399-406; Maina et al., Cell (1996), 87, 531-542; Schmidt et al., Nature (1995), 373, 699-702; Uehara et al., Nature (1995), 373, 702-705). Фенотипы развития нокаутных по Met и HGF мышей очень схожи, что предполагает, что HGF является специфическим лигандом рецептора Met (Schmidt et al., 1995, выше; Uehara et al., 1995, выше). HGF-Met также играет роль в регенерации печени, ангиогенезе и заживлении ран (Bussolino et al., J Cell Biol (1992), 119, 629-641; Matsumoto and Nakamura, Exs (1993), 65, 225-249; Nusrat et al., J Clin Invest (1994) 93, 2056-2065). Предшественник рецептора Met подвергается протеолитическому расщеплению с образованием экстрацеллюлярной субъединицы α и мембранной субъединицы β, соединенных дисульфидными связями (Tempest et al., Br J Cancer (1988), 58, 3-7). Субъединица β содержит цитоплазматический киназный домен и содержит мультисубстратный стыковочный сайт на С-конце, где адаптерные белки связываются и индуцируют сигнальный путь (Bardelli et al., Oncogene (1997), 15, 3103-3111; Nguyen et al., J Biol Chem (1997), 272, 20811-20819; Pelicci et al., Oncogene (1995), 10, 1631-1638; Ponzetto et al., Cell (1994), 77, 261-271; Weidner et al., Nature (1996), 384, 173-176). При связывании HGF активация Met приводит к фосфорилированию тирозина и прохождению сигнала через активацию PI3-киназы и Ras/MAPK, опосредованную Gab1 и Grb2/Sos, соответственно, которые управляют клеточной подвижностью и пролиферацией (Furge et al., Oncogene (2000), 19, 5582-5589; Hartmann et al., J Biol Chem (1994), 269, 21936-21939; Ponzetto et al., J Biol Chem (1996), 271, 14119-14123; Royal and Park, J Biol Chem (1995), 270, 27780-27787).
Было показано, что в обработанной канцерогеном клеточной линии остеосаркомы Met трансформируется (Cooper et al., Nature (1984), 311, 29-33; Park et al., Cell (1986), 45, 895-904). Была отмечена сверхэкспрессия или амплификация гена Met в различных злокачественных опухолях человека. Например, экспрессия белка Met повышается по меньшей мере в 5 раз при колоректальном раке и, как сообщается, происходит амплификация гена в метастазах печени (Di Renzo et al., Clin Cancer Res (1995), 1, 147-154; Liu et al., Oncogene (1992), 7, 181-185). Также было опубликовано, что белок Met сверхэкспрессируется при плоскоклеточной карциноме ротовой полости, гепатоцеллюлярной карциноме, почечноклеточной карциноме, карциноме молочной железы и карциноме легких (Jin et al., Cancer (1997), 79, 749-760; Morello et al., J Cell Physiol (2001), 189, 285-290; Natali et al., Int J Cancer (1996), 69, 212-217; Olivero et al., Br J Cancer (1996), 74, 1862-1868; Suzuki et al., Br J Cancer (1996), 74, 1862-1868). Кроме того, сверхэкспрессия мРНК отмечалась при гепатоцеллюлярной карциноме, карциноме желудка и колоректальной карциноме (Boix et al., Hepatology (1994), 19, 88-91; Kuniyasu et al., Int J Cancer (1993), 55, 72-75; Liu et al., Oncogene (1992), 7, 181-185).
Был обнаружен целый ряд мутаций в киназном домене Met при папиллярной карциноме почек, что приводит к активации конститутивного рецептора (Olivero et al., Int J Cancer (1999), 82, 640-643; Schmidt et al., Nat Genet (1997), 16, 68-73; Schmidt et al., Oncogene (1999), 18, 2343-2350). Эти активирующие мутации обуславливают фосфорилирование тирозина конститутивного Met и приводят к активации MAPK, формированию очага патологического процесса и онкогенезу (Jeffers et al., Proc Natl Acad Sci USA (1997), 94, 11445-11450). Кроме того, эти мутации повышают клеточную подвижность и инвазию (Giordano et al., Faseb J (2000), 14, 399-406; Lorenzato et al., Cancer Res (2002), 62, 7025-7030). HGF-зависимая активация Met в трансформированных клетках опосредует повышенную подвижность, распространение и миграцию, что в конечном счете приводит к инвазивному опухолевому росту и метастазированию (Jeffers et al., Mol Cell Biol (1996), 16, 1115-1125; Meiners et al., Oncogene (1998), 16, 9-20).
Было показано, что Met взаимодействует с другими белками, которые управляют активацией рецептора, трансформацией и инвазией. Было опубликовано, что в опухолевых клетках Met взаимодействует с интегрином α6β4, рецептором компонентов экстрацеллюлярного матрикса, таких как ламинины, для стимуляции HGF-зависимого инвазивного роста (Trusolino et al., Cell (2001), 107, 643-654). Кроме того, было показано, что экстрацеллюлярный домен Met взаимодействует с членом семейства семафоринов, плексином В1, и увеличивает инвазивный рост (Giordano et al., Nat Cell Biol (2002), 4, 720-724). Более того, также было опубликовано, что CD44v6, который вовлечен в онкогенез и метастазирование, образует комплекс с Met и HGF и приводит к активации рецептора Met (Orian-Rousseau et al., Genes Dev (2002), 16, 3074-3086).
Met является членом подсемейства рецепторов тирозинкиназы (RTK), к которым относятся Ron и Sea (Maulik et al., Cytokine Growth Factor Rev (2002), 13, 41-59). Прогнозирование экстрацеллюлярной доменной структуры Met предполагает общую гомологию с семафоринами и плексинами. N-конец Met содержит домен Sema размером приблизительно 500 аминокислот, которые являются консервативными у всех семафоринов и плексинов. Семафорины и плексины относятся к огромному семейству секретируемых и мембраносвязанных белков, впервые описанных в связи с их ролью в развитии нервной системы (Van Vactor and Lorenz, Curr Bio (1999), 19, R201-204). Однако совсем недавно была обнаружена связь сверхэкспрессии семафорина с опухолевой инвазией и метастазированием. Домен PSI, богатый цистеином, (также называемый домен, относящийся к последовательности Met), обнаруженный в плексинах, семафоринах и интегринах, расположен рядом с доменом Sema вслед за четырьмя повторами IPT, которые являются иммуноглобулин-подобными областями, обнаруженными в плексинах и транскрипционных факторах. На основании недавних исследований было сделано предположение, что домен SEMA Met является достаточным для связывания HGF и гепарина (Gherardi et al., Proc Natl Acad Sci USA (2003), 100(21):12039-44).
Как указано выше, рецептор тирозинкиназы Met активируется специфическим лигандом HGF, и фосфорилирование рецептора активирует метаболические пути MAPK, PI3-киназы и PLC-γ (L. Trusolino and P. M. Comoglio, Nat Rev Cancer 2, 289 (2002); C. Birchmeier et al., Nat Rev Mol Cell Biol 4, 915 (2003)). Фосфорилирование Y1234/Y1235 в киназном домене крайне важно для активации киназы Met, в то время как Y1349 и Y1356 в мультисубстратном стыковочном сайте необходимы для связывания с src- гомологичным 2 (SH2), фосфотирозинсвязывающим (PTB) и Met-связывающим доменным (MBD) белками (C. Ponzetto et al., Cell 77, 261 (1994); K. M. Weidner et al., Nature 384, 173 (1996); G. Pelicci et al., Oncogene 10, 1631 (1995)), способствуя активации нисходящих сигнальных путей. Дополнительный околомембранный сайт фосфорилирования, Y1003, был в достаточной степени охарактеризован с точки зрения его связывания с тирозинкиназным связывающим (TKB) доменом Cbl E3-лигазы (P. Peschard et al., Mol Cell 8, 995 (2001); P. Peschard, N. Ishiyama, T. Lin, S. Lipkowitz, M. Park, J Biol Chem 279, 29565 (2004)). Как сообщалось, связывание Cbl запускает эндофилин-опосредованный эндоцитоз рецептора, убиквитинизацию и последующее разрушение рецептора (A. Petrelli et al., Nature 416, 187 (2002)). Этот механизм дезактивации рецептора был уже описан ранее для семейства EGFR, которое также имеет подобный сайт связывания Cbl (K. Shtiegman, Y. Yarden, Semin Cancer Biol 13, 29 (2003); M. D. Marmor, Y. Yarden, Oncogene 23, 2057 (2004); P. Peschard, M. Park, Cancer Cell 3, 519 (2003)). Как уже сообщалось, нарушение регуляции Met и HGF встречается в различных опухолях. Запускаемая лигандом активация Met отмечалась в различных злокачественных опухолях. Было отмечено повышенное содержание сывороточного и внутриопухолевого HGF при раке легкого, молочной железы и множественной миеломе (J. M. Siegfried et al., Ann Thorac Surg 66, 1915 (1998); P. C. Ma et al., Anticancer Res 23, 49 (2003); B. E. Elliott et al. Can J Physiol Pharmacol 80, 91 (2002); C. Seidel, et al, Med Oncol 15, 145 (1998)). Повышенная экспрессия Met и/или HGF, амплификация или мутация Met описана при различных злокачественных опухолях, таких как колоректальный рак, рак легкого, желудка и почки, как предполагается запускает независимую от лиганда активацию рецептора (C. Birchmeier et al, Nat Rev Mol Cell Biol 4, 915 (2003); G. Maulik et al., Cytokine Growth Factor Rev 13, 41 (2002)). Кроме того, индуцибельная сверхэкспрессия Met на мышиной модели печени приводит к возникновению гепатоцеллюлярной карциномы, что указывает на то, что повышенная экспрессия рецептора запускает независимый от лиганда онкогенез (R. Wang, et al, J Cell Biol 153, 1023 (2001)). Наиболее убедительные доказательства того, что Met вовлечен в развитие злокачественных опухолей отмечены у пациентов, имеющих семейную и спорадическую формы папиллярной карциномы почек (RPC). Мутации в киназном домене Met, которые приводят к конститутивной активации рецептора, были идентифицированы как генеративная и соматическая мутации в RPC (L. Schmidt et al., Nat Genet 16, 68 (1997)). Применение этих мутаций в трансгенных мышиных моделях приводит к онкогенезу и метастазированию (M. Jeffers et al., Proc Natl Acad Sci USA 94, 11445 (1997)).
Публикации, касающиеся c-met и антагонистов c-met, включают Martens, T, et al (2006) Clin Cancer Res 12 (20 Pt 1):6144; US 6468529; WO2006/015371; WO2007/063816; WO2006/104912; WO2006/104911; WO2006/113767; US2006-0270594; US2006-US патент № 7481993; WO2009/007427; WO2005/016382; WO2009/002521; WO2007/143098; WO2007/115049; WO2007/126799.
Семейство рецептора эпидермального фактора роста (EGFR) включает четыре близко родственных рецептора (HER1/EGFR, HER2, HER3 и HER4), вовлеченных в клеточные ответы, такие как дифференциация и пролиферация. Повышенная экспрессия киназы EGFR или ее лиганда TGF-альфа как правило связана с большим числом злокачественных опухолей, включая рак молочной железы, легких, колоректальный рак, рак яичника, клеток почечного эпителия, мочевого пузыря и шеи, глиобластомы и астроцитомы, и, по-видимому, участвует в росте этих опухолей. Также было обнаружено, что специфичная делеционная мутация в гене EGFR (EGFRvIII) повышает онкогенность клеток. Активация сигнальных путей стимулированным EGFR запускает большое число процессов, которые являются вероятными стимуляторами развития злокачественной опухоли, например, пролиферацию, ангиогенез, клеточную подвижность и инвазию, сниженный апоптоз и индукцию лекарственной устойчивости. Повышенная экспрессия HER1/EGFR как правило связана с прогрессированием заболевания, метастазированием и плохим прогнозом. Например, было показано, что при NSCLC и раке желудка, повышенная экспрессия HER1/EGFR связана с высокой скоростью метастазирования, низкой степенью дифференциации опухоли и повышенной пролиферацией опухоли.
Мутации, которые активируют внутреннюю белковую активность рецептора тирозинкиназы и/или усиливают нисходящее проведение сигнала, были обнаружены при NSCLC и глиобластоме. Однако роль мутаций как основного механизма придания чувствительности к ингибиторам рецептора EGF, например эрлотинибу (TARCEVA®) или гефитинибу неоднозначна. Было описано, что мутантные формы полноразмерного рецептора EGF рассчитаны на восприимчивость к гефитинибу - ингибитору рецептора тирозинкиназы EGF (Paez, J. G. Et al. (2004) Science 304:1497-1500; Lynch, T. J. et al. (2004) N. Engl. J. Med. 350:2129-2139). Исследования на клеточных культурах показали, что клеточные линии, которые экспрессируют такие мутантные формы рецептора EGF (т.е. H3255), были более чувствительными к ингибированию роста гефитинибом - ингибитором рецептора тирозинкиназы EGF, и что для ингибирования опухолевых клеточных линий, экспрессирующих рецептор EGF дикого типа требуются гораздо более высокие концентрации гефитиниба. Эти наблюдения предполагают, что специфические мутантные формы рецептора EGF могут отражать более высокую чувствительность к ингибиторам рецептора EGF, но не определять полностью невосприимчивый фенотип.
Разработка применения соединений в качестве противоопухолевых средств, которые напрямую ингибируют киназную активность EGFR, а также антител, которые уменьшают киназную активность EGFR путем блокирования активации EGFR, является областью интенсивной научно-исследовательской работы (de Bono J.S. and Rowinsky, E.K. (2002) Trends in Mol. Medicine 8:S19-S26; Dancey, J. and Sausville, E.A. (2003) Nature Rev. Drug Discovery 2:92-313). Различные исследования продемонстрировали, обнаружили или предположили, что некоторые ингибиторы киназы EGFR могут усиливать уничтожение опухолевой клетки или неоплазии при применении в сочетании с некоторыми другими противораковыми или химиотерапевтическими средствами или способами лечения (например, Herbst, R.S. et al. (2001) Expert Opin. Biol. Ther. 1:719-732; Solomon, B. et al (2003) Int. J. Radiat. Oncol. Biol. Phys. 55:713-723; Krishnan, S. et al. (2003) Frontiers in Bioscience 8, e1-13; Grunwald, V. and Hidalgo, M. (2003) J. Nat. Cancer Inst. 95:851-867; Seymour L. (2003) Current Opin. Investig. Drugs 4(6):658-666; Khalil, M.Y. et al. (2003) Expert Rev. Anticancer Ther.3:367-380; Bulgaru, A.M. et al. (2003) Expert Rev. Anticancer Ther.3:269-279; Dancey, J. and Sausville, E.A. (2003) Nature Rev. Drug Discovery 2:92-313; Ciardiello, F. et al. (2000) Clin. Cancer Res. 6:2053-2063; and Patent Publication No: US 2003/0157104).
Эрлотиниб (например, эрлотиниб HCl, также известный как TARCEVA® или OSI-774) является ингибитором киназы EGFR для перорального применения. In vitro эрлотиниб демонстрировал значительную ингибиторную активность в отношении киназы EGFR на многих опухолевых клеточных линиях человека, включая колоректальный рак и рак молочной железы (Moyer J.D. et al. (1997) Cancer Res. 57:4838), и доклинические исследования выявили активность по отношению ко многим EGFR-экспрессирующим опухолевым ксенотрансплантантам человека (Pollack, V.A. et al (1999) J. Pharmacol. Exp. Ther. 291:739). Эрлотиниб продемонстрировал активность в клинических испытаниях при многих показаниях к применению, включая рак головы и шеи (Soulieres, D., et al. (2004) J. Clin. Oncol. 22:77), NSCLC (Perez-Soler R, et al. (2001) Proc. Am. Soc. Clin. Oncol. 20:310a, abstract 1235), CRC (Oza, M., et al. (2003) Proc. Am. Soc. Clin. Oncol. 22:196a, abstract 785) and MBC (Winer, E., et al. (2002) Breast Cancer Res. Treat. 76:5115a, abstract 445; Jones, R.J., et al. (2003) Proc. Am. Soc. Clin. Oncol. 22:45a, abstract 180). В третьей фазе испытаний монотерапия эрлотинибом существенно увеличивала выживаемость, задерживала прогрессирование заболевания и задерживала ухудшение симптомов, связанных с раком легких, у пациентов с прогрессирующим, невосприимчивым к лечению NSCLC (Shepherd, F. et al. (2004) J. Clin. Oncology, 22:14S (July 15 Supplement), Abstract 7022). В ноябре 2004 Управление по контролю за продуктами питания и лекарственными средствами (FDA) США разрешило применение TARCEVA® для лечения пациентов с местно-распространенным или метастатическим немелкоклеточным раком легкого (NSCLC) после безрезультатной по меньшей мере одной предыдущей схемы лечения химиотерапией.
Несмотря на значительный прогресс в лечении рака, все еще необходимы усовершенствованные способы лечения.
Все перечисленные в настоящем документе ссылки, в том числе патентные заявки и публикации, включены в качестве ссылки в полном объеме.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к комбинированной терапии для лечения патологического состояния, такого как рак, где антагонист c-met объединен с антагонистом EGFR, тем самым, обеспечивая значительную противоопухолевую активность.
В одном из аспектов настоящее изобретение относится к способам лечения рака у индивида, включающим введение индивиду терапевтически эффективных количеств антагониста c-met и антагониста EGFR.
Примеры антагонистов c-met включают, но ими не ограничиваются, растворимые рецепторы c-met, растворимые варианты HGF, аптамеры или пептидные антитела, специфичные к c-met или HGF, низкомолекулярные молекулы c-met, антитела анти-c-met и антитела анти-HGF. В некоторых вариантах осуществления изобретения антагонист c-met является анти-c-met антителом.
В одном из вариантов осуществления изобретения антитело анти-c-met содержит вариабельный домен тяжелой цепи, содержащий одну или несколько последовательностей CDR1-HC, CDR2-HC и CDR3-HC, показанных на фигуре 7 (SEQ ID NO:13-15). В некоторых вариантах осуществления изобретения антитело содержит вариабельный домен легкой цепи, содержащий одну или несколько последовательностей CDR1-LC, CDR2-LC и CDR3-LC, показанных на фигуре 7 (SEQ ID NO:5-7). В некоторых вариантах осуществления изобретения вариабельный домен тяжелой цепи содержит последовательности FR1-HC, FR2-HC, FR3-HC и FR4-HC, показанных на фигуре 7 (SEQ ID NO:9-12). В некоторых вариантах осуществления изобретения вариабельный домен легкой цепи содержит последовательности FR1-LC, FR2-LC, FR3-LC и FR4-LC, показанных на фигуре 7 (SEQ ID NO:1-4). В некоторых вариантах осуществления изобретения антитело анти-c-met является моновалентным и содержит участок Fc. В некоторых вариантах осуществления изобретения антитело содержит последовательность Fc, показанную на фигуре 7 (SEQ ID NO:17).
В некоторых вариантах осуществления изобретения антитело является моновалентным и содержит участок Fc, где участок Fc содержит первый и второй полипептид, где первый полипептид содержит последовательность Fc, показанную на фигуре 7 (SEQ ID NO:17), и второй полипептид содержит последовательность Fc, показанную на фигуре 8 (SEQ ID NO:18).
В одном из вариантов осуществления изобретения антитело анти-c-met содержит (а) первый полипептид, содержащий вариабельный домен тяжелой цепи с последовательностью:
QVQLQQSGPELVRPGASVKMSCRASGYTFTSYWLHWVKQRPGQGLEWIGMIDPSNSDTRFNPNFKDKATLNVDRSSNTAYMLLSSLTSADSAVYYCATYGSYVSPLDYWGQGTSVTVSS (SEQ ID NO:19), с последовательностью CH1, показанной на фигуре 7 (SEQ ID NO:16) и с последовательностью Fc, показанной на фигуре 7 (SEQ ID NO:17); и (b) второй полипептид, содержащий вариабельный домен легкой цепи с последовательностью: DIMMSQSPSSLTVSVGEKVTVSCKSSQSLLYTSSQKNYLAWYQQKPGQSPKLLIYWASTRESGVPDRFTGSGSGTDFTLTITSVKADDLAVYYCQQYYAYPWTFGGGTKLEIK(SEQ ID NO:20), и с последовательностью CL1, показанной на фигуре 7 (SEQ ID NO:8); и (с) третий полипептид, содержащий последовательность Fc, показанную на фигуре 8 (SEQ ID NO:18).
В одном из аспектов антитело анти-c-met обладает по меньшей мере одним характерным свойством, которое способствует гетеродимеризации, и в то же время сводя к минимуму гомодимеризацию, последовательностей Fc в пределах фрагмента антитела. Такое характерное свойство(а) улучшает выход и/или степень чистоты и/или гомогенность иммуноглобулиновых популяций. В одном из вариантов осуществления изобретения антитело содержит мутации Fc, формирующие «выпячивания» и «ямки», как описано в WO2005/063816. Например, мутацией, приводящей к образованию «ямки», может быть одна или несколько из мутаций T366A, L368A и/или Y407V в полипептиде Fc, а мутацией, приводящей к образованию «впадины», может быть T366W.
В некоторых вариантах осуществления изобретения антагонистом c-met является SGX-523, PF-02341066, JNJ-38877605, BMS-698769, PHA-665752, SU5416, SU1274, XL-880, MGCD265, ARQ 197, MP-470, AMG 102, антитело 223С4 или гуманизированное антитело 223С4 (WO2009/007427), L2G7, NK4, XL-184, MP-470 или Comp-1.
Антагонисты c-met могут быть использованы для уменьшения или ингибирования одного или нескольких аспектов HGF/c-met-связанных эффектов, включая, но ими не ограничиваясь, активацию c-met, нисходящий молекулярный сигнальный путь (например, фосфорилирование митоген-активируемой протеинкиназы (МАРК), фосфорилирование AKT, фосфорилирование c-met, опосредуемый киназой PI3 сигнальный путь), клеточную пролиферацию, клеточную миграцию, клеточное выживание, клеточный морфогенез и ангиогенез. Эти эффекты могут быть изменены любым биологическим механизмом, включая нарушение связывания лиганда (например, HGF) с c-met, фосфорилирование c-met и/или мультимеризация c-met.
Примеры антагонистов EGFR включают антитела и низкомолекулярные молекулы, которые связываются с EGFR. Антагонисты EGFR также включают низкомолекулярные молекулы, такие как соединения, описанные в US5616582, US5457105, US5475001, US5654307, US5679683, US6084095, US6265410, US6455534, US6521620, US6596726, US6713484, US5770599, US6140332, US5866572, US6399602, US6344459, US6602863, US6391874, WO9814451, WO9850038, WO9909016, WO9924037, WO9935146, WO0132651, US6344455, US5760041, US6002008, US5747498. Специфические низкомолекулярные антагонисты EGFR включают OSI-774 (CP-358774, эрлотиниб, OSI Pharmaceuticals); PD 183805 (CI 1033, 2-пропенамид, N-[4-[(3-хлор-4-фторфенил)амино]-7-[3-(4-морфолинил)пропокси]-6-хиназолинил]-, дигидрохлорид, Pfizer Inc.); Iressa® (ZD1839, гефитиниб, AstraZeneca); ZM 105180 ((6-амино-4-(3-метилфениламино)-хиназолин, Zeneca); BIBX-1382 (N8-(3-хлор-4-фторфенил)-N2-(1-метилпиперидин-4-ил)-пиримидо[5,4-d]пиримидин-2,8-диамин, Boehringer Ingelheim); PKI-166 ((R)-4-[4-[(1-фенилэтил)амино]-1H-пирроло[2,3-d]пиримидин-6-ил]-фенол);(R)-6-(4-гидроксифенил)-4-[(1-фенилэтил)амино]-7H-пирроло[2,3-d]пиримидин); CL-387785 (N-[4-[(3-бромфенил)амино]-6-хиназолинил]-2-бутинамид); EKB-569 (N-[4-[(3-хлор-4-фторфенил)амино]-3-циано-7-этокси-6-хинолинил]-4-(диметиламино)-2-бутенамид); лапатиниб (Tykerb, GlaxoSmithKline); ZD6474 (Zactima, AstraZeneca); CUDC-101 (Curis); канертиниб (CI-1033); AEE788 (6-[4-[(4-этил-1-пиперазинил)метил]фенил]-N-[(1R)-1-фенилэтил]-7H-пирроло[2,3-d]пиримидин-4-амин, WO2003013541, Novartis) и PKI166 4-[4-[[(1R)-1-фенилэтил]амино]-7H-пирроло[2,3-d]пиримидин-6-ил]-фенол, WO9702266 Novartis).
В конкретном варианте осуществления изобретения антагонист EGFR имеет общую формулу I:

в соответствии с патентом США №5757498, приведенным в настоящее описание в качестве ссылки, где:
m равно 1, 2 или 3;
каждый R1 независимо выбран из группы, состоящей из водорода, галогена, гидрокси, гидроксиамино, карбокси, нитро, гуанидино, уреидо, циано, трифторметила и -(С1-С4 алкилен)-W-(фенила), где W представляет собой одинарную связь, О, S или NH;
или каждый R1 независимо выбран из R9 и С1-С4алкила, замещенного циано, где R9выбран из группы, состоящей из R5, -OR6, -NR6R6, -C(O)R7, -NHOR5, -OC(O)R6, циано, А и -YR5; R5 представляет собой С1-С4алкил; R6 независимо представляет собой водород или R5; R7 представляет собой R5, -OR6 или -NR6R6; А выбран из пиперидино, морфолино, пирролидино, 4-R6-пиперазин-1-ил, имидазол-1-ил, 4-пиридон-1-ил, -(С1-С4алкилен)(СО2Н), фенокси, фенил, фенилсульфанил, С2-С4 алкенил и -(С1-С4алкилен)C(O)NR6R6; и Y представляет собой S, SO или SO2; где алкильные фрагменты в R5, -OR6 и -NR6R6 необязательно замещены галозаместителями в количестве от одного до трех, и алкильные фрагменты в R5, -OR6 и -NR6R6 необязательно замещены 1 или 2 R9 группами, и где алкильные фрагменты указанных необзхательных заместителей необязательно замещены галогеном или R9 при условии, что два гетероатома не присоединены к одному и тому же атому углерода;
или каждый R1 независимо выбран из -NHSO2R5, фталимидо-(С1-С4)-алкилсульфониламино, бензамидо, бензенсульфониламино, 3-фенилуреидо, 2-оксопирролидин-1-ил, 2,5-диоксопирролидин-1-ил, и R10-(C2-C4)-алканоиламино, где R10 выбран из галогена, -OR6, С2-С4 алканоилокси, -С(O)R7 и -NR6R6; и где указанные -NHSO2R5, фталимидо-(С1-С4)-алкилсульфониламино, бензамидо, бензенсульфониламино, 3-фенилуреидо, 2-оксопирролидин-1-ил, 2,5-диоксопирролидин-1-ил, и R10-(C2-C4)-алканоиламино R1 группы необязательно замещены 1 или 2 заместителями, независимо выбранными из галогена, С1-С4алкила, циано, метансульфонила и С1-С4алкокси;
или две R1 группы взяты вместе с углеродами, к которым они присоединены для образования 5-8-членной циклической структуры, которая включает 1 или 2 гетероатома, выбранных из O, S и N;
R2 представляет собой водород или С1-С6 алкил, необязательно замещенный заместителями в количестве от 1 до 3, независимо выбранными из галогена, С1-С4 алкокси, -NR6R6 и -SO2R5;
n представляет собой 1 или 2 и каждый R3 независимо выбран из водорода, галогена, гидрокси, С1-С6 алкила, -NR6R6, и С1-С4 алкокси, где алкильные фрагменты указанных R3групп необязательно замещены заместителями в количестве от 1 до 3, независимо выбранными из галогена, С1-С4 алкокси, -NR6R6 и -SO2R; и
R4 представляет собой азидо или -(этинил)-R11, где R11 представляет собой водород или С1-С6 алкил, необязательно замещенный гидрокси, -OR6, или -NR6R6.
В конкретном варианте осуществления изобретения антагонист EGFR является соединением согласно формуле I, выбранным из группы, включающей:
(6,7-диметоксихиназолин-4-ил)-(3-этинилфенил)-амин;
(6,7-диметоксихиназолин-4-ил)-[3-(3'-гидроксипропин-1-ил)фенил]-амин;
[3-(2'-(аминометил)-этинил)фенил]-(6,7-диметоксихиназолин-4-ил)-амин;
(3-этинилфенил)-(6-нитрохиназолин-4-ил)-амин;
(6,7-диметоксихиназолин-4-ил)-(4-этинилфенил)-амин;
(6,7-диметоксихиназолин-4-ил)-(3-этинил-2-метилфенил)-амин;
(6-аминохиназолин-4-ил)-(3-этинилфенил)-амин;
(3-этинилфенил)-(6-метансульфониламинохиназолин-4-ил)-амин;
(3-этинилфенил)-(6,7-метилендиоксихиназолин-4-ил)-амин;
(6,7-диметоксихиназолин-4-ил)-(3-этинил-6-метилфенил)-амин;
(3-этинилфенил)-(7-нитрохиназолин-4-ил)-амин;
(3-этинилфенил)-[6-(4'-толуолсульфониламино)хиназолин-4-ил]-амин;
(3-этинилфенил)-{6-[2'-фталимидо-эфир-1'-ил-сульфониламино]хиназолин-4-ил}-амин;
(3-этинилфенил)-(6-гуанидинохиназолин-4-ил)-амин;
(7-аминохиназолин-4-ил)-(3-этинилфенил)-амин;
(3-этинилфенил)-(7-метоксихиназолин-4-ил)-амин;
(6-карбометоксихиназолин-4-ил)-(3-этинилфенил)-амин;
(7-карбометоксихиназолин-4-ил)-(3-этинилфенил)-амин;
[6,7-бис(2-метоксиэтокси)хиназолин-4-ил]-(3-этинилфенил)-амин;
(3-азидофенил)-(6,7-диметоксихиназолин-4-ил)-амин;
(3-азидо-5-хлорфенил)-(6,7-диметоксихиназолин-4-ил)-амин;
(4-азидофенил)-(6,7-диметоксихиназолин-4-ил)-амин;
(3-этинилфенил)-(6-метансульфонилхиназолин-4-ил)-амин;
(6-этансульфанилхиназолин-4-ил)-(3-этинилфенил)-амин;
(6,7-диметоксихиназолин-4-ил)-(3-этинил-4-фторфенил)-амин;
(6,7-диметоксихиназолин-4-ил)-[3-(пропин-1'-ил)-фенил]-амин;
[6,7-бис-(2-метокси-этокси)-хиназолин-4-ил]-(5-этинил-2-метилфенил)-амин;
[6,7-бис-(2-метоксиэтокси)-хиназолин-4-ил]-(3-этинил-4-фторфенил)-амин;
[6,7-бис-(2-хлорэтокси)-хиназолин-4-ил]-(3-этинилфенил)-амин;
[6-(2-хлорэтокси)-7-(2-метоксиэтокси)-хиназолин-4-ил]-(3-этинилфенил)-амин;
[6,7-бис-(2-ацетоксиэтокси)-хиназолин-4-ил]-(3-этинилфенил)-амин;
2-[4-(3-этинил-фениламино)-7-(2-гидрокси-этокси)-хиназолин-6-илокси]-этанол;
[6-(2-ацетоксиэтокси)-7-(2-метоксиэтокси)-хиназолин-4-ил]-(3-этинилфенил)-амин;
[7-(2-хлорэтокси)-6-(2-метоксиэтокси)-хиназолин-4-ил]-(3-этинилфенил)-амин;
[7-(2-ацетоксиэтокси)-6-(2-метоксиэтокси)-хиназолин-4-ил]-(3-этинилфенил)-амин;
2-[4-(3-этинилфениламино)-6-(2-гидроксиэтокси)-хиназолин-7-илокси]-этанол;
2-[4-(3-этинилфениламино)-7-(2-метоксиэтокси)-хиназолин-6-илокси]-этанол;
2-[4-(3-этинилфениламино)-6-(2-метоксиэтокси)-хиназолин-7-илокси]-этанол;
[6-(2-ацетоксиэтокси)-7-(2-метоксиэтокси)-хиназолин-4-ил]-(3-этинилфенил)-амин;
(3-этинилфенил)-{6-(2-метоксиэтокси)-7-[2-(4-метилпиперазин-1-ил)-этокси]-хиназолин-4-ил}-амин;
(3-этинилфенил)-[7-(2-метоксиэтокси)-6-(2-морфолин-4-ил)-этокси)-хиназолин-4-ил]-амин;
(6,7-диэтоксихиназолин-1-ил)-(3-этинилфенил)-амин;
(6,7-дибутоксихиназолин-1-ил)-(3-этинилфенил)-амин;
(6,7-диизопропоксихиназолин-1-ил)-(3-этинилфенил)-амин;
(6,7-диэтоксихиназолин-1-ил)-(3-этинил-2-метилфенил)-амин;
[6,7-бис-(2-метоксиэтокси)-хиназолин-1-ил]-(3-этинил-2-метилфенил)-амин;
(3-этинилфенил)-[6-(2-гидроксиэтокси)-7-(2-метоксиэтокси)-хиназолин-1-ил]-амин;
[6,7-бис-(2-гидроксиэтокси)-хиназолин-1-ил]-(3-этинилфенил)-амин;
2-[4-(3-этинилфениламино)-6-(2-метоксиэтокси)-хиназолин-7-илокси]-этанол;
(6,7-дипропоксихиназолин-4-ил)-(3-этинилфенил)-амин;
(6,7-диэтоксихиназолин-4-ил)-(3-этинил-5-фторфенил)-амин;
(6,7-диэтоксихиназолин-4-ил)-(3-этинил-4-фторфенил)-амин;
(6,7-диэтоксихиназолин-4-ил)-(5-этинил-2-метилфенил)-амин;
(6,7-диэтоксихиназолин-4-ил)-(3-этинил-4-метилфенил)-амин;
(6-аминометил-7-метоксихиназолин-4-ил)-(3-этинилфенил)-амин;
(6-аминометил-7-метокси-хиназолин-4-ил)-(3-этинилфенил)-амин;
(6-аминокарбонилметил-7-метоксихиназолин-4-ил)-(3-этинилфенил)-амин;
(6-аминокарбонилэтил-7-метоксихиназолин-4-ил)-(3-этинилфенил)-амин;
(6-аминокарбонилметил-7-этокси-хиназолин-4-ил)-(3-этинилфенил)-амин;
(6-аминокарбонилэтил-7-этокси-хиназолин-4-ил)-(3-этинилфенил)-амин;
(6-аминокарбонилметил-7-изопропокси-хиназолин-4-ил)-(3-этинилфенил)-амин;
(6-аминокарбонилметил-7-пропокси-хиназолин-4-ил)-(3-этинилфенил)-амин;
(6-аминокарбонилметил-7-метоксихиназолин-4-ил)-(3-этинилфенил)-амин;
(6-аминокарбонилэтил-7-изопропокси-хиназолин-4-ил)-(3-этинилфенил)-амин; и
(6-аминокарбонилэтил-7-пропоксихиназолин-4-ил)-(3-этинилфенил)-амин;
(6,7-диэтоксихиназолин-1-ил)-(3-этинилфенил)-амин;
(3-этинилфенил)-[6-(2-гидроксиэтокси)-7-(2-метоксиэтокси)-хиназолин-1-ил]-амин;
[6,7-бис-(2-гидроксиэтокси)-хиназолин-1-ил]-(3-этинилфенил)-амин;
[6,7-бис-(2-метокси-этокси)-хиназолин-1-ил]-(3-этинилфенил)-амин;
(6,7-диметоксихиназолин-1-ил)-(3-этинилфенил)-амин;
(3-этинилфенил)-(6-метансульфониламинохиназолин-1-ил)-амин; и
(6-амино-хиназолин-1-ил)-(3-этинилфенил)-амин.
В конкретном варианте осуществления изобретения антагонист EGFR формулы I представляет собой N-(3-этинилфенил)-6,7-бис(2-метоксиэтокси)-4-хиназолинамин. В конкретном варианте осуществления изобретения антагонист EGFR N-(3-этинилфенил)-6,7-бис(2-метоксиэтокси)-4-хиназолинамин находится в форме соли HCl. В другом конкретном варианте осуществления изобретения антагонист EGFR N-(3-этинилфенил)-6,7-бис(2-метоксиэтокси)-4-хиназолинамин находится в форме практически гомогенного кристаллического полиморфа (описан как полиморф В в WO 01/34574), который демонстрирует порошковую рентгенограмму, имеющую характерные пики, выраженные в градусах 2-тета, приблизительно при 6,26, 12,48, 13,39, 16,96, 20,20, 21,10, 22,98, 24,46, 25,14 и 26,91. Такая форма полиморфа N-(3-этинилфенил)-6,7-бис(2-метоксиэтокси)-4-хиназолинамин называется TarcevaTM, а также OSI-774, CP-358774 и эрлотинибом.
Антагонисты EGFR могут быть использованы для уменьшения или ингибирования одного или нескольких лиганд-связанных эффектов EGFR-EGFR, включая, но ими не ограничиваясь, активацию EGFR, нисходящий молекулярный сигнальный путь, клеточную пролиферацию. Эти эффекты могут быть изменены любым встречающимся в природе соответствующим механизмом, включая нарушение связывания лиганда с EGFR и нарушение фосфорилирования EGFR.
Способы настоящего изобретения могут быть использованы для воздействия на любое подходящее патологическое состояние. Например, способы изобретения могут быть применимы для лечения различных злокачественных опухолей, как солидных опухолей, так и опухолей мягких тканей в равной степени. Неограниченное количество примеров злокачественных опухолей, поддающихся лечению по настоящему изобретению, включает рак молочной железы, колоректальный рак, рак прямой кишки, немелкоклеточный рак легкого (NSCLC), неходжкинскую лимфому (NHL), почечноклеточный рак, рак простаты, рак печени, рак поджелудочной железы, саркому мягких тканей, саркому Капоши, карциноидную карциному, рак головы и шеи, глиобластому, меланому, рак яичника, рак желудка, мезотелиому, множественную миелому. В определенных аспектах злокачественные опухоли являются метастатическими. В других аспектах злокачественные опухоли являются не метастатическими.
В одном из вариантов осуществления изобретения антитело анти-c-met и эрлотиниб используются в комбинированной терапии злокачественных опухолей, таких как немелкоклеточная карцинома легкого.
В определенных вариантах осуществления изобретения рак не является устойчивым к антагонисту EGFR (например, эрлотинибу или гефитинибу) раком. В определенных вариантах осуществления изобретения рак не является устойчивым к эрлотинибу или гефитинибу раком.
В определенных вариантах осуществления изобретения рак не является устойчивым к ингибитору тирозинкиназы раком. В определенных вариантах осуществления изобретения рак не является устойчивым к низкомолекулярному ингибитору тирозинкиназы EGFR раком.
В определенных вариантах осуществления изобретения в злокачественной опухоли выявляют экспрессию, амплификацию или активацию c-met и/или EGFR. В определенных вариантах осуществления в злокачественной опухоли не выявляют экспрессию, амплификацию или активацию c-met и/или EGFR. В определенных вариантах осуществления изобретения в злокачественной опухоли выявляют амплификацию c-met. В определенных вариантах осуществления изобретения в злокачественной опухоли выявляют амплификацию c-met и амплификацию EGFR.
В определенных вариантах осуществления изобретения в злокачественной опухоли выявляют ген EGFR дикого типа. В определенных вариантах осуществления изобретения в злокачественной опухоли выявляют ген EGFR дикого типа и амплификацию c-met и/или мутацию c-met.
В определенных вариантах осуществления изобретения в злокачественной опухоли выявляют мутацию EGFR. Мутации могут быть расположены в любой части гена EGFR или регуляторной области, связанной с геном EGFR. Типичные мутации EGFR включают, например, мутации в экзоне 18, 19, 20 или 21, мутации в киназном домене, G719A, L858R, E746K, L747S, E749Q, A750P, A755V, V765M, S768I, L858P, делеция E746-R748, делеция R748-P753, вставка M766-A767 AI, вставка S768-V769 SVA, вставка P772-H773 NS, 2402OC, 2482OA, 2486T>C, 2491 G>C, 2494OC, 251 0OT, 2539OA, 2549OT, 2563OT, 2819T>C, делеция 2482-2490, делеция 2486-2503, 2544-2545 вставка GCCATA, 2554-2555 вставка CCAGCGTGG или 2562-2563 вставка AACTCC. Другие примеры активирующих мутаций EGFR известны в данной области техники (смотри, например, публикацию патента США № 2005/0272083). В определенных вариантах изобретения клетка или клеточная линия не содержит мутацию Т790М в гене EGFR.
В определенных вариантах осуществления изобретения в злокачественной опухоли выявляют активацию c-met и/или EGFR. В определенных вариантах осуществления изобретения в злокачественной опухоли не обнаруживают активацию c-met и/или EGFR.
В определенных вариантах осуществления изобретения в злокачественной опухоли выявляют активацию конститутивных c-met и/или EGFR. В некоторых вариантах осуществления изобретения, конститутивный EGFR содержит мутацию в тирозинкиназном домене. В определенных вариантах осуществления изобретения в злокачественной опухоли не выявляют активацию конститутивных c-met и/или EGFR.
В определенных вариантах осуществления изобретения в злокачественной опухоли выявляют лиганд-независимую активацию c-met и/или EGFR. В определенных вариантах осуществления изобретения в злокачественной опухоли не обнаруживают лиганд-независимую активацию c-met и/или EGFR.
Антагонист c-met может быть введен последовательно или в комбинации с антагонистом EGFR либо в одной и той же композиции или в виде отдельных композиций. Введение антагониста c-met и антагониста EGFR может быть осуществлено одновременно, например, в виде единой композиции или в виде двух или нескольких отдельных композиций, используя одни и те же или различные способы введения препарата. Альтернативно или дополнительно введение может быть осуществлено последовательно в любом порядке. Альтернативно или дополнительно последовательность действий может быть выполнена как комбинация: и последовательно, и одновременно в любом порядке. В определенных вариантах осуществления изобретения интервалы в диапазоне от минут до дней, от недель до месяцев могут присутствовать между применениями двух или большего количества композиций. Например, антагонист EGFR может быть введен первым с последующим приемом антагониста c-met. Однако, одновременное введение или введение антагониста c-met в первую очередь также предусмотрено. Таким образом, в одном аспекте, изобретение относится к способам, включающим введение антагониста c-met (такого как антитело анти-c-met) с последующим введением антагониста EGFR (такого как эрлотиниб (TARCEVA®)). В определенных вариантах осуществления изобретения интервалы в диапазоне от минут до дней, от недель до месяцев могут присутствовать между введениями двух или более композиций.
В одном аспекте изобретение относится к композиции для применения в лечении рака, содержащей эффективное количество антагониста c-met и фармацевтически приемлемого носителя, где указанное применение заключается в одновременном или последовательном введении антагониста EGFR. В некоторых вариантах осуществления изобретения антагонист c-met представляет собой антитело анти-c-met. В некоторых вариантах осуществления изобретения антагонист EGFR представляет собой эрлотиниб (TARCEVA®).
В одном аспекте изобретение относится к композиции для применения в лечении рака, содержащей эффективное количество антагониста c-met и фармацевтически приемлемого носителя, где указанное применение заключается в одновременном или последовательном введении антагониста EGFR. В некоторых вариантах осуществления изобретения антагонист c-met представляет собой антитело анти-c-met. В некоторых вариантах осуществления изобретения антагонист EGFR представляет собой эрлотиниб (TARCEVA®).
В зависимости от специфического признака рака, подлежащего лечению, комбинированная терапия изобретения может сочетаться с дополнительными терапевтическими средствами, такими как химиотерапевтические средства, или дополнительными способами лечения, такими как радиотерапия или хирургическое вмешательство. Многие известные химиотерапевтические средства могут быть использованы в комбинированной терапии согласно изобретению. Предпочтительно будут использованы те химиотерапевтические средства, которые являются стандартными для лечения специфических признаков. Дозирование или частота приема каждого терапевтического средства, подлежащего использованию при комбинировании, предпочтительно те же, или меньше чем, дозирование или частота приема соответствующего средства при использовании без другого средства (средств).
КРАТКОЕ ОПИСАНИЕ ФИГУР
ФИГУРЫ 1А и 1В: Подтверждение совместной экспрессии мРНК EGFR и MET в клеточных линиях и первичных опухолях NSCLC методом кОТ-ПЦР. Экспрессия мРНК EGFR и MET была измерена количественной ОТ-ПЦР в группе клеточных линий NSCLC (1А) или лизатах замороженной первичной опухоли NSCLC (1В). Уровни мРНК EGFR и MET положительно коррелировали в клеточных линиях (ρ=0,59, р<0,0001) и образцах первичной NSCLC (ρ=0,48, р=0,0003).
ФИГУРА 2: Клетки shMet 4.12 EBC1 (shMet 4.12), содержащие тетрациклин-индуцибельную кшРНК, направленную против c-met, или контрольную кшРНК, направленную против GFP (shGFP2) были выращены в контрольной питательной среде (Con) или среде с 0,1 мкг/мл тетрациклинового аналога Доксициклина (Dox) в течение 48 часов. После выращивания клеток в бессывороточной среде в течение 2 часов клетки не были обработаны (-) или были обработаны TGFα (Т, 20нМ) или Херегулином b1 (Hrg, 2нМ) в течение 20 минут. Клеточные лизаты целиком были исследованы на экспрессию суммарного белка и фосфобелков как показано. Бета-актин (β-актин) был измерен, чтобы продемонстрировать эквивалентное нанесение в лунки.
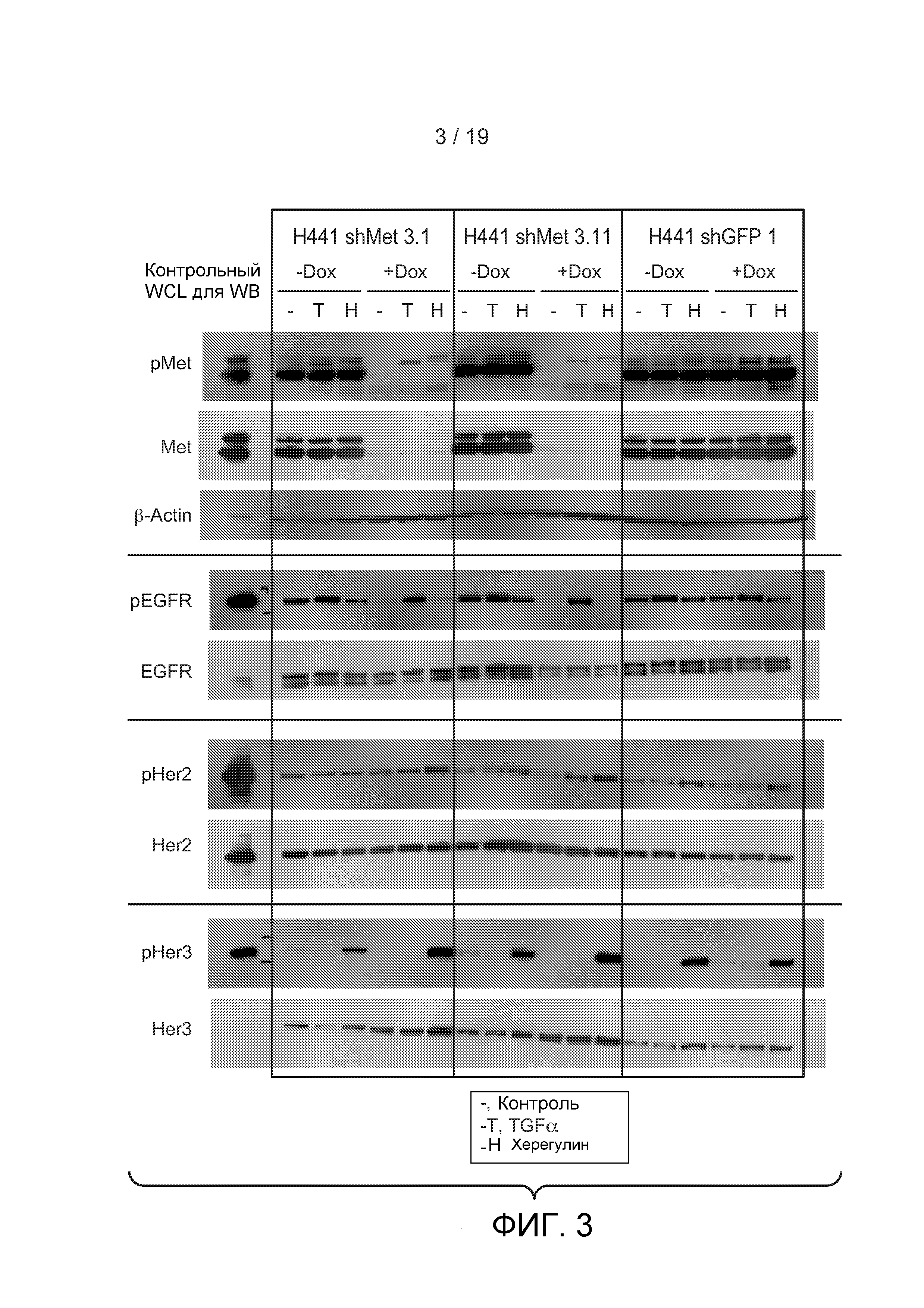
ФИГУРА 3: Клетки NSCLC H441, содержащие индуцибельную кшРНК, направленную против c-met, или контрольную кшРНК, направленную против GFP, были выращены в контрольной питательной среде или среде, содержащей 0,1 мкг/мл доксициклина (Dox) в течение 48 часов. После выращивания клеток в бессывороточной среде в течение 2 часов клетки не были обработаны (-) или были обработаны TGFα (Т) или Херегулином b1 (Н) в течение 20 минут. Бета-актин (β-актин) (4-я панель) был измерен, чтобы продемонстрировать эквивалентное нанесение в лунки.
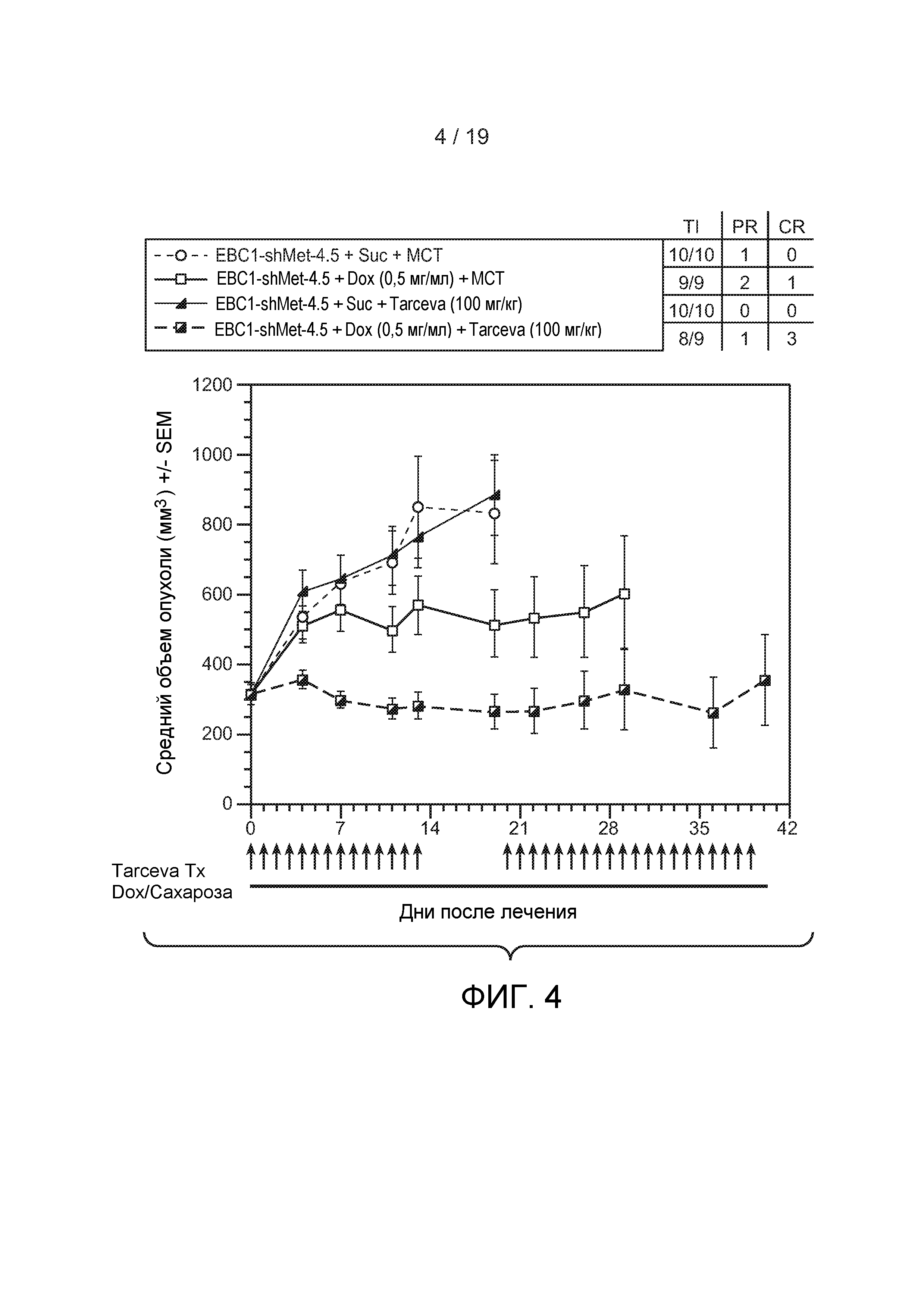
ФИГУРА 4: Эффективность комбинирования эрлотиниба с нокдауном c-met под действием кшРНК в модели ксенотрансплантанта EBC-1 NSCLC. Опухоли EBC-1-shMet-4.5 были детектированы у «голых» (CRL nu/nu) животных и затем обработаны либо разбавителем метилцеллюлозного твина (МСТ) с добавлением питьевой воды, содержащей 5% сахарозу (Suc) (PO, QD где указывают стрелки), MCT с добавлением 1 мг/мл доксициклина (Dox) в питьевой воде, содержащей 5% сахарозу (100 мг/кг; PO, QD где указывают стрелки), эрлотинибом с добавлением питьевой воды, содержащей 5% сахарозу (PO, QD где указывают стрелки) или эрлотинибом с добавлением 1 мг/мл доксициклина в питьевой воде, содержащей 5% сахарозу (PO, QD где указывают стрелки). Пероральное дозирование было выполнено в дни, указанные стрелками. Воду сахарозы или Dox хранили на протяжении исследования в бутылках, взаимозаменяемых каждые 2-3 дня. Объемы опухоли и SEM были вычислены как описано в Примерах.
ФИГУРА 5: Эффективность комбинирования MetMAb с эрлотинибом в модели ксенотрансплантанта NCI-H596 hu-HGF-Tg-C3H-SCID. Опухоли NCI-H596 были выращены в hu-HGF-Tg-C3H-SCID или C3H-SCID однопометных контрольных животных и обработаны либо разбавителем каптисола (PO, QD, х2 недели), эрлотинибом (закрытые кружки, короткая пунктирная линия; 150 мг/кг, PO, QD, х2 недели), MetMAb (30 мг/кг, IP, однократно) или сочетанием MetMAb с эрлотинибом в одинаковых дозах и по одинаковым схемам. Дозирование было как указано в нижней части графика для MetMAb (открытая стрелка) и эрлотиниба или разбавителя (закрытые стрелки). Измерения опухоли были выполнены штангенциркулем два или три раза в неделю в течение 9 недель или до тех пор, пока группы не были отстранены от исследования из-за больших размеров опухоли внутри группы. Объемы опухоли и SEM были вычислены как описано в Примерах.
ФИГУРА 6: Измерения времени удвоения опухоли (TTD), определенного как время, необходимое для удвоения опухоли в размере, были вычислены для каждой группы и использованы для построения кривой выживаемости Каплана-Мейера. Комбинирование MetMAb с эрлотинибом демонстрировало поразительную положительную динамику в развитии опухоли со средним значением TTD равным 49,5 (±2,6) дней по сравнению с 17,8 (±2,2) днями для группы, обработанной MetMAb, 9,5 (±1,2) днями для группы, обработанной эрлотинибом, и 9,5 (±1,2) днями для группы, обработанной разбавителем. Кривые для разбавителя и эрлотиниба полностью наложились друг на друга.
ФИГУРА 7: иллюстрирует аминокислотную последовательность каркасных участков (FR), гипервариабельных областей (HVR), первого константного домена (CL или CH1) и область Fc (Fc) одного варианта осуществления изобретения антитела анти-c-met. Изображенная последовательность Fc содержит мутации T366S, L368A и Y407V как описано в WO 2005/063816.
ФИГУРА 8: иллюстрирует последовательность полипептида Fc, содержащего мутацию T366W как описано в WO 2005/063816. В одном из вариантов осуществления изобретения полипептид Fc, содержащий эту последовательность, образует комплекс с полипептидом Fc, содержащим последовательность Fc Фиг. 7, с образованием области Fc.
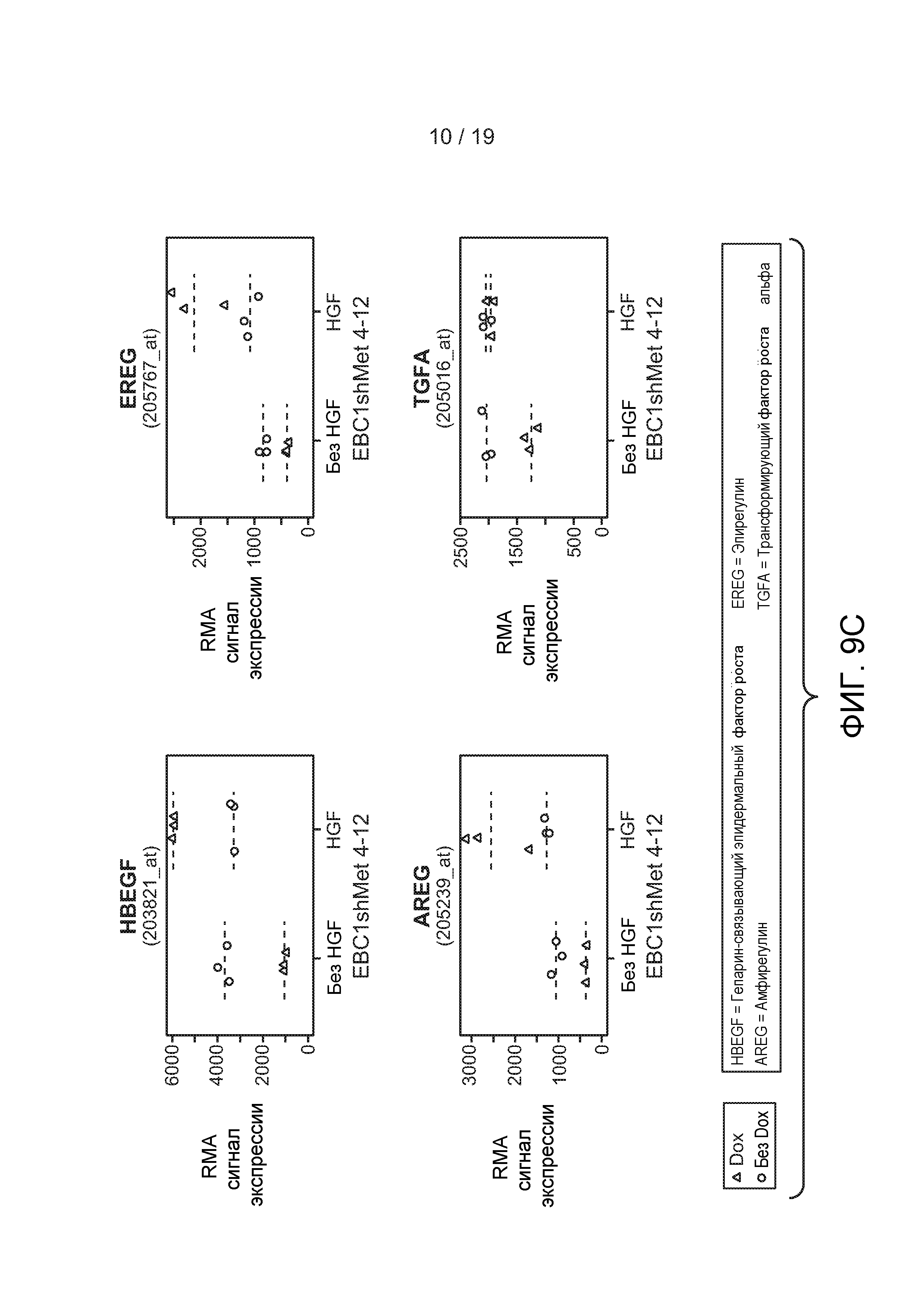
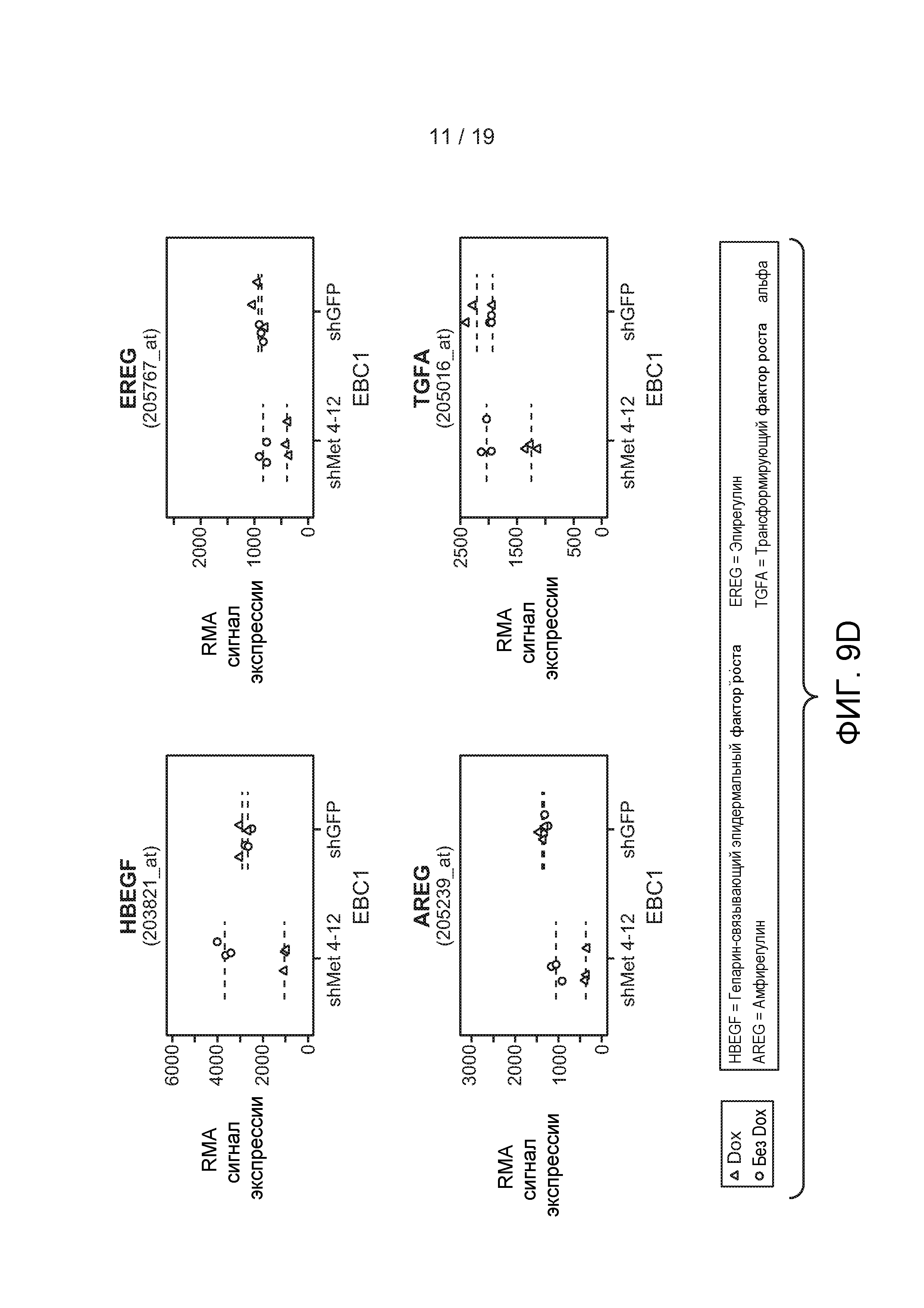
ФИГУРЫ 9А-Е: Активность c-met регулирует экспрессию лигандов EGFR. А) Обработка HGF индуцировала положительную регуляцию лигандов EGFR в клеточной линии NSCLC, чувствительной к HGF. Клетки Hop92 или NCI-H596 растили в бессывороточной среде в течение ночи и затем их не подвергали обработке (без HGF) или обрабатывали HGF (50 нг/мл) в течение 6 часов (HGF). РНК из клеток с -/+ обработкой HGF подвергали микроэррей-анализу как описано в Примерах. RMA=относительный микроэррей. В) Нокдаун c-met снижал экспрессию лигандов EGFR в лиганд-независимой EBC-1 клеточной линии NSCLC. Клоны, стабильно экспрессирующие кшРНК, направленные против c-met, (клоны 3-15 и 4-12) не подвергались обработке (без Dox) или были обработаны Доксициклином (Dox) в течение 24 или 48 часов. РНК из клеток подвергалась микроэррей-анализу, как описано в Примерах. С) Клетки EBC1 shMet4-12, стабильно экспрессирующие кшРНК, направленную против c-met, не подвергались обработке (без Dox) или были обработаны Dox (Dox) в течение 24 часов в отсутствии HGF (без HGF) или в присутствие HGF (100 нг/мл) в течение 2 часов. РНК из клеток были подвергнуты микроэррей-анализам, как описано в Примерах. D) Клетки EBCshMet4-12, стабильно экспрессирующие кшРНК, направленную против c-met, и контрольные клетки, стабильно экспрессирующие shGFP2, не подвергались обработке (без Dox) или были обработаны Dox (Dox) в течение 24 часов. РНК из клеток были подвергнуты микроэррей-анализам, как описано в Примерах. Е) Опухоли из клеток EBC1 shMet-4.12 или EBCshMet-3.15 были детективрованы у «голых» (CRL nu-nu) животных, и мышам была дана питьевая вода, содержащая 1 мг/мл Dox (Dox) в 5% сахарозе, или только 5% сахароза. После 3 дней уровни TGFα в лизатах опухоли были оценены ELISA.
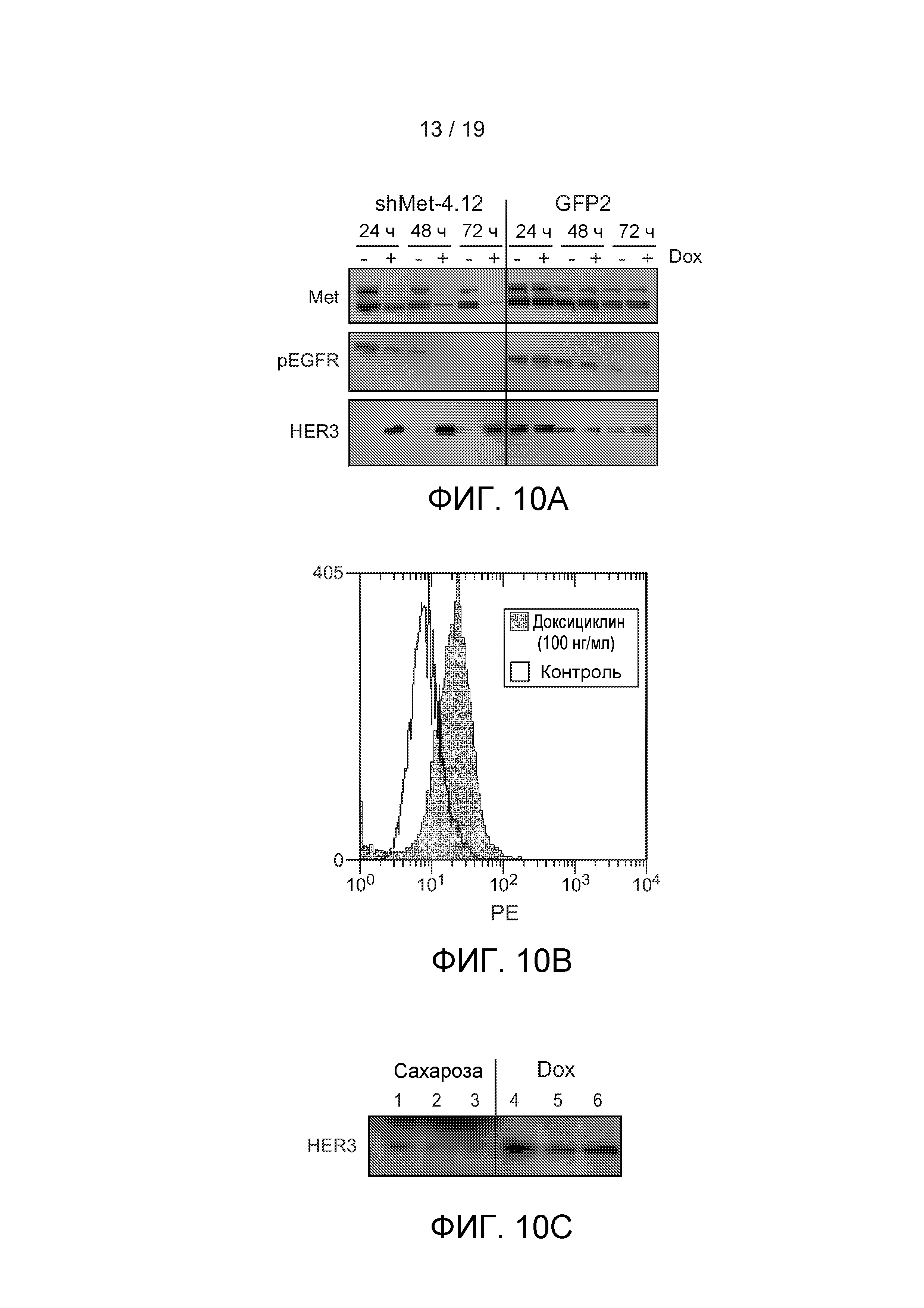
ФИГУРЫ 10А-С: (А) Клетки EBCshMet 4.12 или EBCshGFP2 не были подвергнуты обработке (-) или были обработаны Dox (+) в течение 24, 48 или 72 часов. Белковые лизаты были исследованы на c-met, pEGFR или Her3 вестерн-блоттингом. (В) Клетки EBCshMet 4.12 были обработаны Dox (100 нг/мл) в течение 48 часов и были проанализированы FACS на наличие Her3 на клеточной поверхности. (С) Мышам с опухолями EBCshMet 4.12 была дана питьевая вода с 1 мг/мл Dox в 5% сахарозе (Dox) или только 5% сахароза (Сахароза) в течение 3 дней. Лизаты опухолей были исследованы на белок Her3 вестерн-блоттингом.
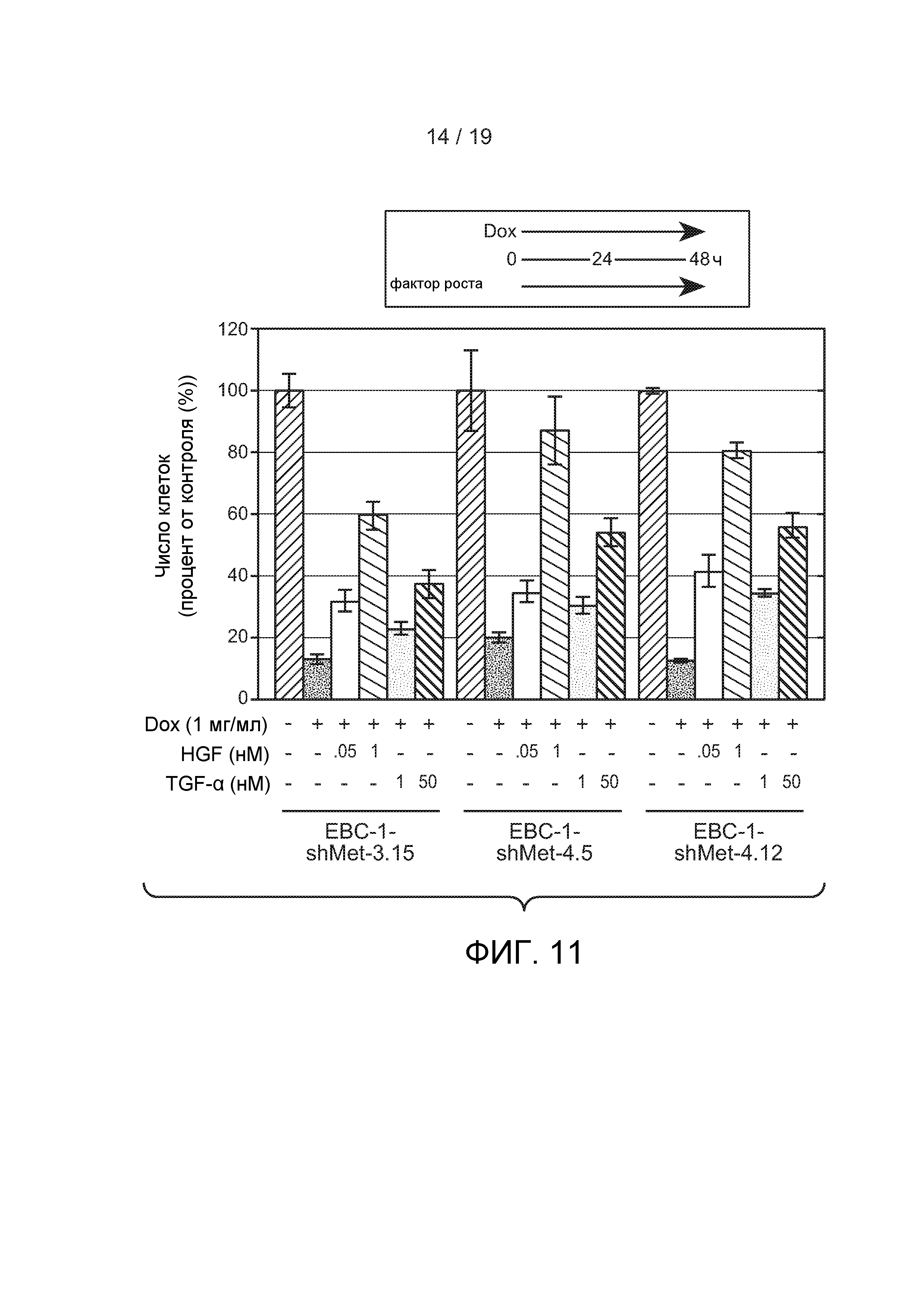
ФИГУРА 11: Клетки EBC-1 shMet (3.15 или 4.5 или 4.12) не подвергались обработке (-) или были обработаны только 100 нг/мл Dox (+) в течение 96 часов или вместе с HGF (5 или 100 нг/мл) или TGFα (1 или 50 нМ), добавленные спустя 48 часов после начала обработки Dox. Число клеток было оценено с использованием Cell Titer Glo.
ФИГУРА 12: Эксперименты по определению временной зависимости были выполнены на клетках NCI-H596 в присутствие (правая панель) или в отсутствии (левая панель) HGF. Клеточные лизаты были приготовлены на 10 минуте (10'), 24 часе, 48 часе или 72 часе (ч) пост-стимуляции и были выполнены вестерн-блот-анализы для измерения суммарного c-met (верхняя панель), фосфо-EGFR (2-я панель) и суммарного EGFR (3-я панель). Бета-актин (β-актин) (4-я панель) был измерен для демонстрации эквивалентного нанесения в лунках.
ФИГУРА 13: Клетки NCI-H596 были высеяны на чашки в отсутствии лигандов, в присутствие только TGF-α, TGF-α + HGF или только HGF. Клеточные лизаты были приготовлены на 10 минуте (10 мин) и 24 часе (ч) пост-стимуляции, и иммунопреципитации (IP) для c-met были выполнены с последующим проведением вестерн-блоттинга для фосфо-тирозина (4G10; верхняя панель), c-met (2-я панель) и EGFR (3-я панель). Фосфо-тирозиновые блоты демонстрируют активацию EGFR (верхняя полоса) и c-met (нижняя полоса) лиганд-зависимым способом, которая ослабевает после 24 часов. Иммунопреципитация c-met осаждает EGFR при всех условиях независимо от состояния активности EGFR или c-met.
ФИГУРА 14: Исследования выживаемости были выполнены на клетках NCI-H596 для оценки ответа клеток на эрлотиниб в присутствие TGF-α и варьирующих концентраций HGF как показано. Было выявлено уменьшение относительной чувствительности к эрлотинибу, так как уровни HGF повышались от 0,5 нг/мл до 50 нг/мл.
ФИГУРА 15: Исследования выживаемости были выполнены на клетках NCI-H596 в присутствие TGF-α и HGF (50 нг/мл) вместе с или в отсутствии MetMAb (1 мкМ) и варьирующих концентраций эрлотиниба. Данные представлены как процент от необработанных контролей. Значения необработанных контролей показаны как индивидуальные точки вверху слева на фигуре.
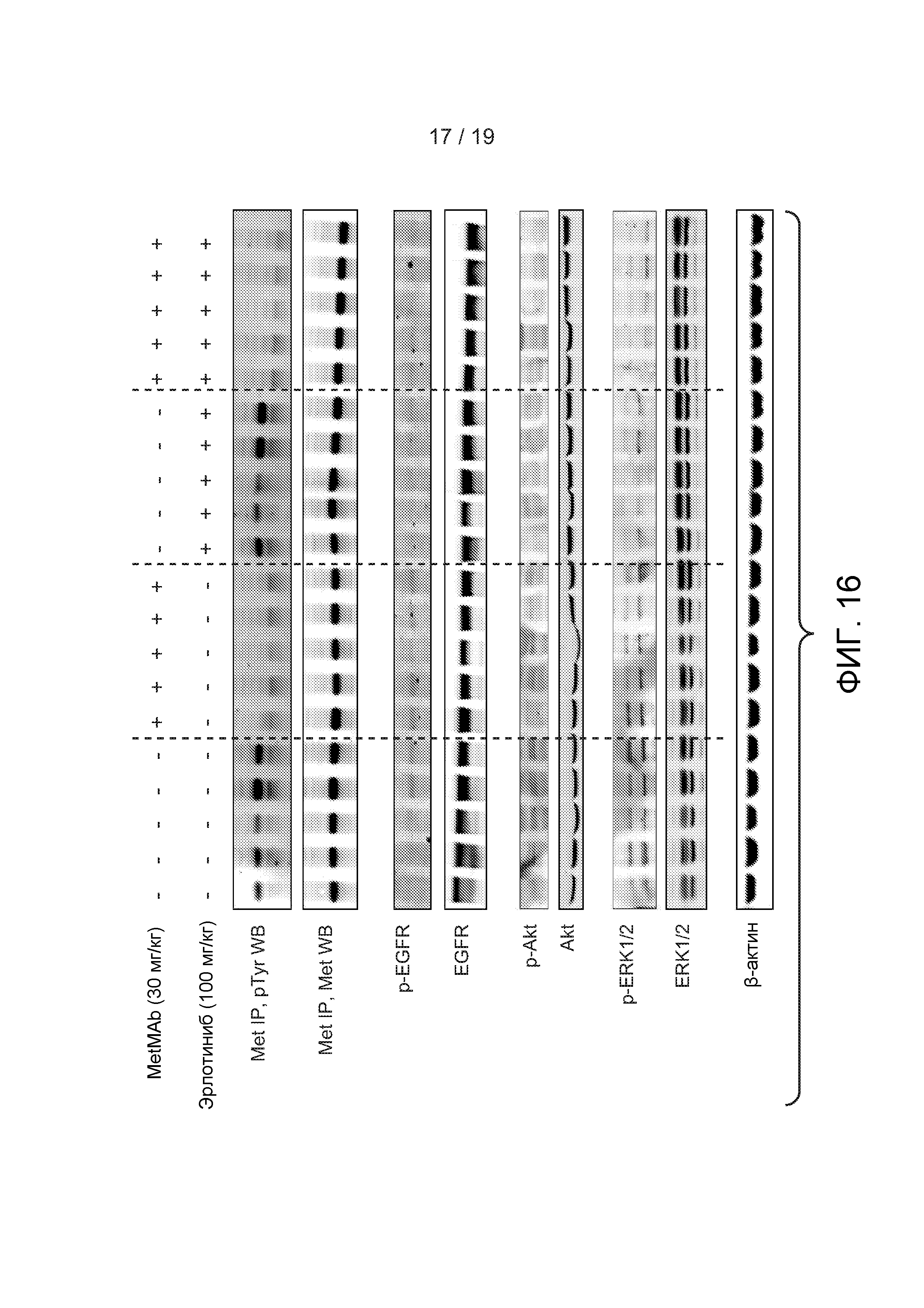
ФИГУРА 16: Комбинированное лечение Эрлотинибом с MetMAb, приводящее к более эффективному ингибированию фосфо-Akt и фосфо-ERK1/2. Человеческий-HGF-трансгенный-SCID (hu-HGF-Tg-SCID) мыши, имеющие опухоли NCI-H596, были подвергнуты лечению разбавителем (буфер MetMAb (100 мкл, IP) и метилцеллюлозный твин (MCT, 100 мкл, РО), MetMAb ((30 мг/кг, IP, однократно) и MCT), эрлотинибом ((100 мг/кг в MCT, 100 мкл, РО) и буфер MetMAb (100 мкл, IP)) или MetMAb и эрлотинибом (те же дозировки, как описано для каждого). Доза MetMAb (или буфер) была дана в нулевое время (t 0 ч), доза эрлотиниба (или MCT) была дана в восемнадцать часов (t 18 ч), мыши были подвергнуты эвтаназии, и опухоли были отобраны в двадцать четыре часа (t 24 ч). Опухолевые лизаты были проанализированы на суммарный белок и фосфо-белки как прямым вестерн-блоттингом, так и иммунопреципитацией с последующим вестерном блотом. Аббревиатура: pTyr = фосфотирозин, EGFR = рецептор эпидермального фактора роста, ERK (регулируемая экстрацеллюлярным сигналом киназа-1 и 2). Бета-актин (β-актин) был измерен для демонстрации эквивалентного нанесения в лунки.
ФИГУРЫ 17 А и 17B: схематически отображают некоторые результаты, описанные в настоящей заявке.
ПОДРОБНОЕ ОПИСАНИЕ
Определения
Термин «фактор роста гепатоцитов» или «HGF» в том смысле, в котором он используется в настоящем описании, относится, если не обозначено иначе, к любому нативному или вариантному (или нативному или синтетическому) полипептиду HGF, способному активировать сигнальный путь HGF/c-met в условиях, подходящих для осуществления такого процесса. Термин «HGF дикого типа» в основном относится к полипептиду, содержащему аминокислотную последовательность встречающегося в природе белка HGF. Термин «HGF последовательность дикого типа» в основном относится к аминокислотной последовательности встречающегося в природе HGF. С-met является известным рецептором HGF, через который внутриклеточная сигнальная система HGF осуществляет свою работу естественным путем.
Термин «вариант HGF» в том смысле, в котором он используется в настоящем описании, относится к полипептиду HGF, который включает одну или несколько мутаций аминокислот в нативной последовательности HGF. Необязательно, одна или несколько мутаций аминокислот включают аминокислотную(ые) замену(ы).
Полипептид «нативной последовательности» содержит полипептид, с той же аминокислотной последовательностью, что и полипептид, встречающийся в природе. Таким образом, полипептид нативной последовательности может иметь аминокислотную последовательность встречающегося в природе полипептида из любого млекопитающего. Такой полипептид нативной последовательности может быть природного происхождения или может быть получен рекомбинантным или синтетическим способом. Термин полипептид «нативной последовательности» главным образом касается встречающихся в природе укороченных или секретируемых форм полипептида (например, последовательность экстрацеллюлярного домена), встречающихся в природе различных форм (например, альтернативно сплайсированные формы) и встречающихся в природе аллельных вариантов полипептида.
«Вариант» полипептида означает биологически активный полипептид, по меньшей мере приблизительно на 80% идентичный аминокислотной последовательности с полипептидом нативной последовательности. Такие варианты включают, например, полипептиды, где один или несколько аминокислотных остатка добавлены или делетированы на N- или С-конце полипептида. Как правило, вариант будет по меньшей мере приблизительно на 80% идентичен аминокислотной последовательности, более предпочтительно по меньшей мере приблизительно на 90% идентичен аминокислотной последовательности и еще более предпочтительно по меньшей мере приблизительно на 95% идентичен аминокислотной последовательности с полипептидом нативной последовательности.
Под «EGFR» (взаимозаменяемо названным «ErbB1», «HER1» и «рецептором эпидермального фактора роста») подразумевают полипептид рецептора тирозинкаиназы, Рецептора Эпидермального Фактора роста, который описан в Ullrich et al, Nature (1984) 309:418425, альтернативно называемый как продукт гена Her-1 и c-erbB, а также их варианты, такие как EGFRvIII. Варианты EGFR также включают делеционные, замещенные и инсерционные варианты, например тех, что описаны в Lynch et al (New England Journal of Medicine 2004, 350:2129), Paez et al (Science 2004, 304:1497), Pao et al (PNAS 2004, 101:13306).
«Биологический образец» (взаимозаменяемо назван «образец» или «образец ткани или клетки») касается различных типов образцов, полученных от индивидуума, и могут быть использованы в диагностическом или мониторирующем анализе. Определение касается крови и других жидких образцов биологической природы, твердых образцов ткани, таких как биоптат или тканевые культуры или полученные из них клетки и их потомство. Определение также включает образцы, с которыми проводили манипуляции любым способом после их получения, такие как обработка реагентами, солюбилизация или обогащение определенных компонентов, таких как белки или полинуклеотиды, или заключение в полутвердый или твердый матрикс с целью изготовления срезов. Термин «биологический образец» касается клинического образца и также включает клетки в культуре, клеточные супернатанты, клеточные лизаты, сыворотку, плазму, биологическую жидкость и тканевые образцы. Источником биологического образца может быть твердая ткань из свежего, замороженного и/или фиксированного органа, или тканевого образца или биопсии или аспирата; кровь или любые компоненты крови; жидкости организма такие как спинномозговая жидкость, амниотическая жидкость, перитонеальная жидкость или интерстициальная жидкость; клетки из любого периода беременности или развития индивида. В некоторый вариантах осуществления изобретения биологический образец получают из первичной или метастатической опухоли. Биологический образец может содержать вещества, которые естественным путем не смешаны с тканью в природе, такие как консерванты, антикоагулянты, буферы, фиксаторы, питательные вещества, антибиотики и т.п.
«Антагонист c-met» (взаимозаменяемо назван «ингибитор c-met» является веществом, которое препятствует активации или функции c-met. Примеры ингибиторов c-met включают анититела c-met; антитела HGF; низкомолекулярные антагонисты c-met; ингибиторы тирозинкиназы c-met; молекулы антисмысловой и ингибиторной РНК (например, кшРНК) (смотри, например, WO2004/87207). Предпочтительно, ингибитором c-met является антитело или низкомолекулярная молекула, которые связываются с c-met. В конкретном варианте осуществления изобретения ингибитор c-met имеет сродство к связыванию (константу диссоциации) с c-met величиной приблизительно 1000 нМ или меньше. В другом варианте осуществления изобретения ингибитор c-met имеет сродство к связыванию с c-met величиной приблизительно 100 нМ или меньше. В другом варианте осуществления изобретения ингибитор c-met имеет сродство к связыванию с c-met величиной приблизительно 50 нМ или меньше. В конкретном варианте осуществления изобретения ингибитор c-met ковалентно связан с c-met. В конкретном варианте осуществления изобретения ингибитор c-met ингибирует сигнальную систему c-met со значением IC50, равным 1000 нМ или меньше. В другом варианте осуществления изобретения ингибитор c-met ингибирует сигнальную систему c-met со значением IC50, равным 500 нМ или меньше. В другом варианте осуществления изобретения ингибитор c-met ингибирует сигнальную систему c-met со значением IC50, равным 50 нМ или меньше.
Термин «нацеленное на c-met лекарственное средство», в том смысле, в котором он в настоящем описании используется, относится к терапевтическому средству, которое связывается с c-met и ингибирует активацию c-met. Примером нацеленного на c-met лекарственного средства является MetMAb (OA5D5.v2).
«Активация c-met» относится к активации или фосфорилированию рецептора c-met. В основном, активация c-met приводит к проведению сигнала (например, которое обусловлено внутриклеточным киназным доменом рецептора c-met, фосфорилируя тирозиновые остатки в c-met или полипептиде субстрата). Активация c-met может быть опосредована связыванием лиганда c-met (HGF) с мешеневым рецептором c-met. Связывание HGF с c-met может активировать киназный домен c-met и, тем самым приводить к фосфорилированию тирозиновых остатков в c-met и/или фосфорилированию тирозиновых остатков в дополнительном полипептиде(ах) субстрата.
«Антагонист EGFR» (взаимозаменяемо назван «ингибитором EGFR») является веществом, которое препятствует активации или функции EGFR. Примеры ингибиторов EGFR включают антитела EGFR; антитела лиганда EGFR; низкомолекулярные антагонисты EGFR; ингибиторы тирозинкиназы EGFR; молекулы антисмысловой и ингибиторной РНК (например, кшРНК) (смотри, например, WO2004/87207). Предпочтительно, ингибитор EGFR является антителом или низкомолекулярной молекулой, которая связывается с EGFR. В некоторых вариантах осуществления изобретения ингибитор EGFR является нацеленным на EGFR лекарственным средством. В конкретном варианте осуществления изобретения ингибитор EGFR имеет сродство к связыванию (константу диссоциации) с EGFR величиной, равной приблизительно 1000 нМ или меньше. В другом варианте осуществления изобретения ингибитор EGFR имеет сродство к связыванию с EGFR величиной равной приблизительно 100 нМ или меньше. В другом варианте осуществления изобретения ингибитор EGFR имеет сродство к связыванию с EGFR величиной равной приблизительно 50 нМ или меньше. В конкретном варианте осуществления изобретения ингибитор EGFR ковалентно связан с EGFR. В конкретном варианте осуществления изобретения ингибитор EGFR ингибирует сигнальную систему EGFR со значением IC50, равным 1000 нМ или меньше. В другом варианте осуществления изобретения ингибитор EGFR ингибирует сигнальную систему EGFR со значением IC50, равным 500 нМ или меньше. В другом варианте осуществления изобретения ингибитор EGFR ингибирует сигнальную систему EGFR со значением IC50, равным 50 нМ или меньше.
«ErbB2» и «HER2» экспрессии используются взаимозаменяемо в настоящем документе и относятся к белку HER2 человека, описанному, например, в Semba et al., PNAS (USA) 82:6497-6501 (1985) и Yamamoto et al. Nature 319:230-234 (1986) (идентификационный номер в Генбанке Х03363). Термин «erbB2» относится к гену, кодирующего ErbB2 человека и «neu» относится к гену, кодирующему p185neu крысы. Предпочтительный HER2 является человеческий HER2 нативной последовательности.
«ErbB3» и «HER3» относятся к полипептиду рецептора как указано, например, в патентах США №№ 5183884 и 5480968, а также в Kraus et al. PNAS (USA) 86:9193-9197 (1989).
Термины «ErbB4» и «HER4» относятся к полипептиду рецептора как указано, например, в EP Pat Appln № 599274; Plowman et al., Proc. Natl. Acad. Sci. USA, 90:1746-1750 (1993); and Plowman et al., Nature, 366:473-475 (1993), включая их изоформы, например, как указано в WO99/19488, опубликованном 22 апреля 1999 года.
«ErbB» в том смысле, в котором он используется в настоящем описании, относится к полипептидам EGFR, HER2, HER3 и HER4.
«Активация EGFR» относится к активации или фосфорилированию EGFR. В основном активация EGFR приводит к проведению сигнала (например, которое обусловлено внутриклеточным киназным доменом рецетора EGFR, фосфорилируя тирозиновые остатки в EGFR или полипептиде субстрата). Активация EGFR может быть опосредована связыванием лиганда EGFR с димером EGFR, содержащим EGFR. Связывание лиганда EGFR с димером EGFR может активировать киназный домен одного или нескольких EGFR в димере и, тем самым, приводить к фосфорилированию тирозиновых остатков в одном или нескольких EGFR и/или фосфорилированию тирозиновых остатков в дополнительном полипептиде(ах) субстрата.
Термин «нацеленное на EGFR лекарственное средство» в том смысле, в котором он используется в настоящем описании, относится к терапевтическому средству, которое связывается с EGFR и ингибирует активацию EGFR. Примеры таких средств включают антитела и низкомолекулярные молекулы, которые связываются с EGFR. Примеры антител, которые связываются с EGFR, включают MAb 579 (ATCC CRL HB 8506), MAb 455 (ATCC CRL HB8507), MAb 225 (ATCC CRL 8508), MAb 528 (ATCC CRL 8509) (смотри патент США № 4943533, Mendelsohn et al.) и их варианты, такие как химерный 225 (С225 или Cetuximab; ERBUTIX®) и реконструированный человеческий 225 (Н225) (смотри, WO 96/40210, Imclone Systems Inc.); IMC-11F8, полностью человеческое, нацеленное на EGFR антитело (Imclone); антитела, которые связывают мутантный EGFR типа II (Патент США № 5212290); гуманизированные и химерные антитела, которые связывают EGFR, как описано в Патенте США № 5891996; и человеческие антитела, которые связывают EGFR, такие как ABX-EGF (смотри WO98/50433, Abgenix); EMD 55900 (Stragliotto et al. Eur. J. Cancer 32A:636-640 (1996)); EMD7200 (матузумаб) гуманизированное антитело EGFR, направленное против EGFR, которое конкурирует за связывание с EGFR вместе с EGF и TGF-альфа; и mAb 806 или гуманизированное mAb 806 (Johns et al., J. Biol. Chem. 279(29):30375-30384 (2004)). Антитело анти-EGFR может быть конъюгировано с цитотоксическим веществом, таким образом образуя иммуноконъюгат (смотри, например, EP659439A2, Merck Patent GmbH). Примеры низкомолекулярных молекул, которые связываются с EGFR, включают ZD1839 или Gefitinib (IRESSA; Astra Zeneca); CP-358774 или Эрлотиниб (TARCEVATM; Genentech/OSI); и AG1478, AG1571 (SU 5271; Sugen); EMD-7200.
Под «EGFR-устойчивым» раком подразумевают то, что у больного раком происходило развитие заболевания несмотря на получение терапии антагонистом EGFR (т.е., пациент является «EGFR-резистентным») или у пациента прогрессировало заболевание в течение 12 месяцев (например, в пределах одного, двух, трех или шести месяцев) после завершения схемы лечения, основанной на антагонисте EGFR. Например, злокачественные опухоли, которые содержат мутантный EGFR Т790М, устойчивы к терапии эрлотинибом и гефитинибом.
Под «устойчивым к эрлотинибу или гефитинибу» раком подразумевают то, что у больного раком прогрессировало заболевание, несмотря на получение терапии, основанной на эрлотинибе или гефитинибе (т.е. пациент является «эрлотиниб- или гефитиниб резистентными») или у пациента прогрессировало заболевание в течение 12 месяцев (например, в пределах одного, двух, трех или шести месяцев) после завершения схемы лечения, основанной на эрлотинибе или гефитинибе.
Термин «лиганд-независимый» в том смысле, в котором он используется в настоящем описании, например, применительно к рецепторной сигнальной активности, относится к сигнальной активности, которая не зависит от присутствия лиганда. Например, сигнальная система EGFR может быть результатом димеризации с другими членами семейства HER, такими как HER2. Рецептор, имеющий лиганд-независимую киназную активность, не обязательно будет препятствовать связыванию лиганда с данным рецептором для возникновения дополнительной активации киназной активности.
Термин «конститутивный», в том смысле, в котором он используется в настоящем описании, например, применительно к активности рецептора киназы, относится к длительной сигнальной активности рецептора, которая не зависит от присутствия лиганда или других активирующих молекул. Например, в варианте III EGFR (EGFRvIII), который обычно выявляется в мультиформной глиобластоме, делетирована большая часть его экстрацеллюлярного домена. Хотя лиганды не имеют возможности связаться с EGFRvIII, тем не менее, он постоянно активен и связан с аномальной пролиферацией и выживаемостью. В зависимости от природы рецептора вся активность может быть конститутивной или активность рецептора может быть дополнительно активирована путем связывания с другими молекулами (например, лигандами). Процессы в клетке, приводящие к активации рецепторов, хорошо известны специалистам в данной области техники. Например, активация может включать олигомеризацию, например, димеризацию, тримеризацию и т.п., с образованием рецепторных комплексов более высокого порядка. Комплексы могут включать белки одного вида, т.е. гомомерный комплекс. Альтернативно, комплексы могут включать по меньшей мере два различных вида белка, т.е. гетеромерный комплекс. Образование комплекса может быть вызвано, например, повышенной экспрессией нормальных или мутантных форм рецептора на поверхности клетки. Образование комплекса может также быть вызвано специфической мутацией или мутациями в рецепторе.
Фраза «амплификация гена» относится к процессу, с помощью которого многочисленные копии гена или фрагмента гена образуются в определенной клетке или клеточной линии. Дуплицированная область (участок амплифицированной ДНК) обычно именуют как «ампликон». Как правило, образуемое количество матричной РНК (мРНК), т.е. уровень экспрессии гена, также повышается пропорционально числу копий определенного экспрессируемого гена.
«Ингибитор тирозинкиназы» представляет собой молекулу, которая ингибирует до некоторой степени тирозинкиназную активность тирозинкиназы, такой как рецептор c-met.
Образец злокачественной опухоли или биологический образец, который «демонстрирует экспрессию, амплификацию или активацию c-met и/или EGFR», это тот, который в диагностическом тесте экспрессирует (включая сверхэкспрессию) c-met и/или EGFR, имеет амплифицированный ген c-met и/или EGFR, и/или иным способом демонстрирует активацию или фосфорилирование c-met и/или EGFR.
Образец злокачественной опухоли или биологический образец, который «не демонстрирует экспрессию, амплификацию или активацию c-met и/или EGFR», это тот, который в диагностическом тесте не экспрессирует (включая сверхэкспрессию) c-met и/или EGFR, не имеет амплифицированный ген c-met и/или EGFR, и/или иным способом не демонстрирует активацию или фосфорилирование c-met и/или EGFR.
Образец злокачественной опухоли или биологический образец, который «демонстрирует активацию c-met и/или EGFR», это тот, который в диагностическом тесте демонстрирует активацию или фосфорилирование c-met и/или EGFR. Такая активация может быть измерена напрямую (например, путем измерения фосфорилирования c-met и/или EGFR с помощью ELISA) или опосредованно.
Образец злокачественной опухоли или биологический образец, который «не демонстрирует активацию c-met и/или EGFR», это тот, который в диагностическом тесте не демонстрирует активацию или фосфорилирование c-met и/или EGFR. Такая активация может быть измерена напрямую (например, путем измерения фосфорилирования c-met и/или EGFR с помощью ELISA) или опосредованно.
Образец злокачественной опухоли или биологический образец, который «демонстрирует конститутивную активацию c-met и/или EGFR», это тот, который в диагностическом тесте демонстрирует конститутивную активацию или фосфорилирование c-met и/или EGFR. Такая активация может быть измерена напрямую (например, путем измерения фосфорилирования c-met и/или EGFR c помощью ELISA) или опосредованно.
Образец злокачественной опухоли или биологический образец, который «не демонстрирует амплификацию c-met и/или EGFR» это тот, который в диагностическом тесте не имеет амплифицированный ген c-met и/или EGFR.
Образец злокачественной опухоли или биологический образец, который «демонстрирует амплификацию c-met и/или EGFR» это тот, который в диагностическом тесте имеет амплифицированный ген c-met и/или EGFR.
Образец злокачественной опухоли или биологический образец, который «не демонстрирует конститутивную активацию c-met и/или EGFR» это тот, который в диагностическом тесте не демонстрирует конститутивную активацию или фосфорилирование c-met и/или EGFR. Такая активация может быть измерена напрямую (например, путем измерения фосфорилирования c-met и/или EGFR c помощью ELISA) или опосредованно.
Образец злокачественной опухоли или биологический образец, который «демонстрирует лиганд-независимую активацию c-met и/или EGFR» это тот, который в диагностическом тесте демонстрирует лиганд-независимую активацию или фосфорилирование c-met и/или EGFR. Такая активация может быть измерена напрямую (например, путем измерения фосфорилирования c-met и/или EGFR c помощью ELISA) или опосредованно.
Образец злокачественной опухоли или биологический образец, который «не демонстрирует лиганд-независимую активацию c-met и/или EGFR» это тот, который в диагностическом тесте не демонстрирует лиганд-независимую активацию или фосфорилирование c-met и/или EGFR. Такая активация может быть измерена напрямую (например, путем измерения фосфорилирования c-met и/или EGFR c помощью ELISA) или опосредованно.
«Фосфо-ELISA» в настоящем описании является методом, в котором фосфорилирование одного или нескольких c-met и/или EGFR оценивается в ферментном иммуносорбентном анализе (ELISA), используя реагент, обычно антитело, для определения фосфорилированного c-met и/или EGFR, субстрата или молекулу нисходящей сигнальной системы. Предпочтительно, используется антитело, которое выявляет фосфорилированный c-met и/или EGFR. Анализ может быть выполнен на клеточных лизатах, предпочтительно из свежих или замороженных биологических образцов.
Раковая клетка с «сверхэкспрессией или амплификацией c-met и/или EGFR» это та, в которой уровни белка или гена c-met и/или EGFR значительно выше по сравнению с нераковой клеткой того же типа ткани. Такая сверхэкспрессия может быть обусловлена амплификацией гена или увеличенной транскрипцией или трансляцией. Сверхэкспрессия или амплификация c-met и/или EGFR может быть определена в диагностическом или прогностическом анализе путем оценки увеличенных уровней белка c-met и/или EGFR, присутствующего на поверхности клетки (например, с помощью иммуногистохимического анализа; IHC). Альтернативно или дополнительно, каждый может измерить уровни c-met и/или EGFR-кодирующих нуклеиновых кислот в клетке, например, с помощью методов флуоресцентной гибридизации in situ (FISH; смотри WO98/45479, опубликованный в октябре 1998 года), саузерн-блоттинга или полимеразной цепной реакции (ПЦР), такой как количественная ПЦР в реальном времени (кОТ-ПЦР). Помимо вышеперечисленных методик, различные анализы in vivo доступны для квалифицированного практика. Например, клетки в теле пациента можно подвергнуть действию антитела, которое дополнительно помечено детектируемой меткой, например, радиоактивным изотопом, и связывание антитела с клетками в пациенте может быть оценено, например, внешним сканированием радиоактивности или анализом биопсии, взятой у пациента, предварительно подвергнутого действию антитела.
Раковая клетка, которая «не обладает повышенной экспрессией или амплификацией c-met и/или EGFR», это та, которая не имеет более высокий, чем нормальный уровень белка или гена c-met и/или EGFR при сравнении с нераковой клеткой того же типа ткани.
Термин «мутация» в том смысле, в котором он используется в настоящем описании, означает отличие в аминокислотной или нуклеиновой последовательности определенного белка или нуклеиновой кислоты (гена, РНК) относительно белка или нуклеиновой кислоты дикого типа, соответственно. Мутированный белок или нуклеиновая кислота могут быть экспрессированы с или обнаружены на одном аллеле (гетерозигота) или обоих аллелях (гомозигота) гена и могут быть соматической или зародышевой линии. В настоящем изобретении мутации в основном соматические. Мутации включают перестройки последовательности, такие как вставки, делеции и точечные мутации (включая одиночный нуклеотидный/аминокислотный полиморфизмы).
«Ингибировать» означает уменьшить или ослабить активность, функцию и/или количества по сравнению с эталоном.
«Экспрессия» белка относится к преобразованию информации, кодируемой в гене в матричную РНК (мРНК) и затем в белок.
В настоящем изобретении образец или клетка, которые «экспрессируют» исследуемый белок (такой как рецептор HER или лиганд HER), это те, в которых определено наличие мРНК, кодирующей белок или белок, включающий его фрагменты, в образце или клетке.
«Иммуноконъюгат» (взаимозаменяемо называется «конъюгат антитело-лекарственное средство» или «ADC») означает антитело, конъюгированное с одним или несколькими цитотоксическими веществами, такими как химиотерапевтические вещества, лекарственные средства, вещество ингибитора роста, токсин (например. белковый токсин, ферментативно активный токсин бактериального, грибкового, растительного или животного происхождения, или фрагменты вышеперечисленного) или радиоактивный изотоп (т.е. радиоконъюгат).
Термин «участок Fc» в том смысле, в котором он используется в настоящем описании, в основном, относится к димерному комплексу, включающему С-концевые полипептидные последовательности тяжелой цепи иммуноглобулина, где С-концевая полипептидная последовательность это та, которую получают расщеплением папаином интактного антитела. Участок Fc может содержать нативную или вариантную последовательности Fc. Хотя границы последовательности Fc тяжелой цепи иммуноглобулина могут варьировать, как правило, считается, что последовательность Fc тяжелой цепи IgG человека простирается от аминокислотного остатка в позиции приблизительно Cys226 или в позиции приблизительно Pro230 до карбоксильного конца последовательности Fc. Последовательность Fc иммуноглобулина как правило содержит два константных домена, домен СН2 и домен СН3, и дополнительно содержит домен СН4. С-концевой лизин (остаток 447 согласно системе нумерации EU) области Fc может быть удален, например, во время очистки антитела или с помощью рекомбинантного конструирования нуклеиновой кислоты, кодирующей антитело. Соответственно, композиция, содержащая антитело, имеющее область Fc по настоящему изобретению, может содержать антитело с К447, с удаленным К447, или смесь антител, содержащих и не содержащих остаток К447.
Под «полипептидом Fc» в настоящем описании подразумевают один из полипептидов, формирующих участок Fc. Полипептид Fc может быть получен из любого подходящего иммуноглобулина, такого как IgG1,IgG2,IgG3 или IgG4 подтипы, IgA, IgE, IgD или IgM. В некоторых вариантах осуществления изобретения полипептид Fc содержит часть или всю шарнирную последовательность дикого типа (как правило на его N-конце). В некоторых вариантах осуществления изобретения полипептид Fc не содержит функциональную или дикого типа шарнирную последовательность.
«Шарнирная область», «шарнирная последовательность» и их вариации в том смысле, в котором они используются в настоящем описании, включают понятия, известные в данной области техники, проиллюстрированные, например, в Janeway et al., Immuno Biology: the immune system in health and disease, (Elsevier Science Ltd., NY) (4th ed., 1999); Bloom et al., Protein Science (1997), 6:407-415; Humphreys et al., J. Immunol. Methods (1997), 209:193-202.
В настоящем описании изобретения и формуле изобретения нумерация остатков в тяжелой цепи иммуноглобулина находится в соответствии с индексом EU как в Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, Md. (1991), специально включенной в настоящий документ в качестве ссылки. «Индекс EU как в публикации Kabat» относится к нумерации аминокислотных остатков антитела EU IgG1 человека.
Термин «антитело» используется в широком смысле и специфически охватывает моноклональные антитела (включая полноразмерные моноклональные антитела), поликлональные антитела, мультиспецифические антитела (например, биспецифические антитела), моновалентные антитела, мультивалентные антитела и фрагменты антитела такой длины, позволяющей ему проявлять желаемую биологическую активность.
«Фрагменты антитела» содержат только часть интактного антитела, где часть предпочтительно сохраняет по меньшей мере одну, предпочтительно большинство или все функции, обычно ассоциированных с данной частью в случае ее расположения в интактном антителе. В одном из вариантов осуществления изобретения фрагмент антитела содержит сайт связывания антигена интактного антитела и таким образом сохраняет способность связывать антиген. В другом варианте осуществления изобретения фрагмент антитела, например, является таковым, что содержит область Fc, сохраняет по меньшей мере одну из биологических функций, обычно ассоциированных с областью Fc в случае ее расположения в интактном антителе, такую как связывание FcRn, регуляция времени полужизни антитела, функция ADCC и связывание комплемента. В одном из вариантов осуществления изобретения фрагмент антитела является моновалентным антителом, которое имеет время полужизни in vivo, в значительной степени схожее с таковым у интактного антитела. Например, такой фрагмент антитела может содержать антиген-связывающее плечо, соединенное с последовательностью Fc, способной придать стабильность in vivo данному фрагменту. В одном из вариантов осуществления изобретения, антитело изобретения является одноплечевым антителом как описано в WO2005/063816. В одном из вариантов осуществления изобретения одноплечевое антитело содержит мутации Fc, формирующие «выпячивания» и «ямки», как описано в WO2005/063816. Например, мутацией ямки может быть одна или несколько T366A, L368A и/или Y407V в полипептиде Fc, и мутацией впадины может быть T366W.
«Блокирующее» антитело или антительный «антагонист» это то, что ингибирует или снижает биологическую активность антигена, который они связывают. Предпочтительные блокирующие антитела или антительные антагонисты полностью ингибируют биологическую активность антигена.
Если не обозначено иначе, выражение «мультивалентное антитело» используется в настоящем описании изобретения для обозначения антитела, содержащего три или большее количество сайтов связывания антигена. Мультивалентное антитело предпочтительно конструируется так, чтобы иметь три или большее количество сайтов связывания антигена, и в основном не является антителом IgM или IgA с нативной последовательностью.
Фрагмент «Fv» является фрагментом антитела, которое содержит полноразмерный сайт узнавания и связывания антигена. Эта область состоит из димера вариабельного домена одной тяжелой и одной легкой цепи в тесной связи, которая может быть ковалентной, например в scFv. Именно в этой структуре три CDR каждого вариабельного домена взаимодействуют для формирования сайта связывания антигена на поверхности димера VH-VL. В совокупности, шесть CDR или их подтипы придают антителу специфичность связывания антигена. Однако, даже единичный вариабельный домен (или половина Fv, содержащего только три CDR, специфичных для антигена) обладают способностью узнавать и связывать антиген, хотя как правило с более низкой аффинностью нежели весь сайт связывания.
«Вариабельный домен антитела» в том смысле, в котором он используется в настоящем описании, относится к частям легкой и тяжелой цепей молекул антитела, который включает аминокислотные последовательности Участков Определения Комплементарности (CDR; т.е., CDR1, CDR2 и CDR3) и Каркасные Участки (FR). VH относится к вариабельному домену тяжелой цепи. VL относится к вариабельному домену легкой цепи. В соответствии с методами, используемыми в настоящем изобретении, позиции аминокислот, закрепленных за CDR и FR, могут быть определены в соответствии с Kabat (Последовательности Белков для Иммунологических Исследований (Национальные Институты Здоровья, Бетесда, Мэриленд, 1987 и 1991)). Аминокислотная нумерация антител или фрагментов связывания антигена также находится в соответствии с таковой в Kabat.
Термин «Участки Определения Комплементарности» (CDR; т.е., CDR1, CDR2 и CDR3) в том смысле, в котором он используется в настоящем описании, относятся к аминокислотным остаткам вариабельного домена антитела, наличие которых необходимо для связывания антигена. Каждый вариабельный домен как правило имеет три области CDR, идентифицированные как CDR1, CDR2 и CDR3. Каждая область определения комплементарности может содержать аминокислотные остатки из «области определения комплементарности» как определено Kabat (т.е. приблизительно остатки 24-34 (L1), 50-56 (L2) и 89-97 (L3) в вариабельном домене легкой цепи и 31-35 (Н1), 50-65 (Н2) и 95-102 (Н3) в вариабельном домене тяжелой цепи; Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, MD. (1991)) и/или таковые остатки из «гипервариабельной петли» (т.е. приблизительно остатки 26-32 (L1), 50-52 (L2) и 91-96 (L3) в вариабельном домене легкой цепи и 26-32 (H1), 53-55 (H2) и 96-101 (H3) в вариабельном домене тяжелой цепи; Chothia and Lesk J. Mol. Biol. 196:901-917 (1987)). В некоторых примерах область определения комплементарности может включать аминокислоты как области CDR, определенной в соответствии с Kabat, так и гипервариабельной петли. Например, тяжелая цепь CDRH1 антитела 4D5 включает аминокислоты с 26 по 35.
«Каркасные участки» (далее FR) представляют собой остатки вариабельного домена, отличные от остатков CDR. Каждый вариабельный домен как правило имеет четыре FR, идентифицированные как FR1, FR2, FR3 и FR4. При условии, что CDR определены в соответствии с Kabat, остатки легкой цепи FR расположены около остатков 1-23 (LCFR1), 35-49 (LCFR2), 57-88 (LCFR3) и 98-107 (LCFR4), и остатки тяжелой цепи FR расположены около остатков 1-30 (HCFR1), 36-49 (HCFR2), 66-94 (HCFR3) и 103-113 (HCFR4) в остатках тяжелой цепи. Если CDR содержат аминокислотные остатки из гипервариабельной петли, остатки FR легкой цепи расположены около остатков 1-25 (LCFR1), 33-49 (LCFR2), 53-90 (LCFR3) и 97-107 (LCFR4) в легкой цепи, и остатки FR тяжелой цепи расположены около остатков 1-25 (HCFR1), 33-52 (HCFR2), 56-95 (HCFR3) и 102-113 (HCFR4) в остатках тяжелой цепи. В некоторых случаях, когда CDR содержит аминокислоты из CDR, как определено Kabat, и таковые гипервариабельной петли, остатки FR будут добавлены соответствующим образом. Например, когда CDRH1 включает аминокислоты Н26-Н35, остатки FR1 тяжелой цепи находятся в позициях 1-25 и остатки FR2 находятся в позициях 36-49.
Фрагмент «Fab» содержит вариабельный и константный домен легкой цепи и вариабельный домен и первый константный домен (СН1) тяжелой цепи. Фрагменты антитела F(ab')2состоят из пары фрагментов Fab, которые как правило ковалентно связаны вблизи их карбоксильных концов с помощью цистеиновой петли между ними. Другие химические соединения фрагментов антитела также известны в данной области техники.
Фрагменты антитела «одноцепочечного Fv» или «scFv» включают домены VH и VLантитела, где эти домены находятся в одной полипептидной цепи. Обычно полипептид Fv дополнительно содержит полипептидный линкер между доменами VH и VL, который дает возможность scFv образовывать желаемую структуру для связывания антигена. Для обзора scFv смотри Pluckthun in The Pharmacology of Monoclonal Antibodies, Vol 113, Rosenburg and Moore eds. Springer-Verlag, New York, pp. 269-315 (1994).
Термин «диатела» относится к малым фрагментам антитела с двумя антиген-связывающими сайтами, фрагменты которых состоят из вариабельного домена тяжелой цепи (VH), соединенного с вариабельным доменом легкой цепи (VL) в той же полипептидной цепи (VH и VL). С помощью линкера, который слишком короток, чтобы позволить соединение между двумя доменами на той же самой цепи, домены вынуждены соединяться с комплементарными доменами другой цепи и создавать два антиген-связывающих сайтов. Диатела описаны более полно в, например, EP 404097; WO 93/11161; и Hollinger et al., Proc. Natl. Acad. Sci. USA, 90:6444-6448 (1993).
Выражение «линейные антитела» относится к антителам, описанным в Zapata et al., Protein Eng., 8(10):1057-1062 (1995). В нескольких словах, эти антитела содержат пару тандемных сегментов Fd (VH-CH1-VH-CH1), которые вместе с комплементарными полипептидами легкой цепи образуют пару антиген-связывающих областей. Линейные антитела могут быть биспецифичными или моноспецифичными.
Определение «моноклональный» указывает на характерную особенность антитела, получаемого в основном из гомогенной популяции антител, и не ограничивается необходимостью получения антитела определенным методом. Например, моноклональные антитела для применения по настоящему изобретению, могут быть получены любыми методами, включая, например, гибридомный метод (например, Kohler and Milstein, Nature, 256:495-97 (1975); Hongo et al., Hybridoma, 14(3): 253-260 (1995), Harlow et al., Antibodies: A Laboratory Manual, (Cold Spring Harbor Laboratory Press, 2nd ed. 1988); Hammerling et al., in Monoclonal Antibodies and T-Cell Hybridomas 563-681 (Elsevier, N.Y., 1981)), методы рекомбинантной ДНК (смотри, например, Патент США № 4816567), технологии фагового дисплея (смотри, например, Clackson et al., Nature, 352: 624-628 (1991); Marks et al., J. Mol. Biol. 222: 581-597 (1992); Sidhu et al., J. Mol. Biol. 338(2):299-310 (2004); Lee et al., J. Mol. Biol. 340(5): 1073-1093 (2004); Fellouse, Proc. Natl. Acad. Sci. USA 101(34):12467-12472 (2004); и Lee et al., J. Immunol. Methods 284(1-2): 119-132 (2004), и технологии образования человеческих и человекоподобных антител у животных, которые имеют части или все иммуноглобулиновые локусы человека или гены, кодирующие иммуноглобулиновые последовательности человека (смотри, например,WO 1998/24893; WO 1996/34096; WO 1996/33735; WO 1991/10741; Jakobovits et al., Proc. Natl. Acad. Sci. USA 90: 2551 (1993); Jakobovits et al., Nature 362: 255-258 (1993); Bruggemann et al., Year in Immunol. 7:33 (1993); U.S. Patent Nos. 5545807; 5545806; 5569825; 5625126; 5633425; и 5661016; Marks et al., Bio/Technology 10: 779-783 (1992); Lonberg et al., Nature 368: 856-859 (1994); Morrison, Nature 368: 812-813 (1994); Fishwild et al., Nature Biotechnol. 14: 845-851 (1996); Neuberger, Nature Biotechnol. 14: 826 (1996); and Lonberg and Huszar, Intern. Rev. Immunol. 13: 65-93 (1995).
Моноклональные антитела в настоящем описании главным образом включают «химерные» антитела, в которых участок тяжелой и/или легкой цепи идентичен или гомологичен соответствующим последовательностям в антителах, полученных из определенных видов или принадлежащих определенному классу или подклассу антител, в то время как остаток цепи(ей) идентичен или гомологичен соответствующим последовательностям в антителах, полученных из других видов или принадлежащих другому классу или подклассу антител, а также фрагменты таких антител при условии, что они проявляют желаемую биологическую активность (смотри, например, Патент США № 4816567; и Morrison et al., Proc. Natl. Acad. Sci. USA 81:6851-6855 (1984)). Химерные антитела включают антитела PRIMATIZED®, где антиген-связывающий участок антитела получен из антитела, образованного например, иммунизацией обезьян макаки исследуемым антигеном.
«Гуманизированные» формы отличных от человеческих (например, мышиных) антител являются химерными антителами, которые содержат минимальную последовательность, полученную из отличного от человеческого иммуноглобулина. В большинстве своем, гуманизированные антитела являются иммуноглобулинами человека (реципиентное антитело), в которых остатки из гипервариабельного участка реципиента замещены остатками из гипервариабельного участка отличных от человеческих видов (донорское антитело), таких как мышь, крыса, кролик или примат, не относящийся к человеку, имеющих необходимую специфичность, аффинность и способность. В некоторых случаях, остатки каркасного участка (FR) Fv человеческого иммуноглобулина замещены соответствующими, отличными от человеческих остатками. Более того, гуманизированные антитела могут содержать остатки, которые не обнаружены в реципиентном антителе или в донорском антителе. Данные модификации проводят для дополнительного совершенствования функциональной активности антитела. В большинстве случаев, гуманизированное антитело включает большинство (по меньшей мере один, а обычно два) вариабельных доменов, в которых все или большинство гипервариабельных петель соответствуют таковым иммуноглобулина, отличного от человеческого, и все или большинство участков FR представляют собой последовательность иммуноглобулина человека. Гуманизированное антитело дополнительно также включает по меньшей мере часть константного участка (Fc) иммуноглобулина, обычно таковую человеческого иммуноглобулина. Для получения дальнейшей информации смотри Jones et al., Nature 321:522-525 (1986); Riechmann et al., Nature 332:323-329 (1988); и Presta, Curr. Op. Struct. Biol. 2:593-596 (1992).
«Человеческое антитело» это то, которое обладает аминокислотной последовательностью, которая соответствует таковой антитела, вырабатываемого человеком и/или полученного с использованием любой технологии для создания человеческих антител, как рассмотрено в настоящем описании. Это определение человеческого антитела в частности исключает гуманизированное антитело, содержащее антиген-связывающие, отличные от человеческих, остатки. Человеческие антитела могут быть получены, используя различные методики, известные в данной области техники. В одном из вариантов осуществления изобретения человеческое антитело отбирается из фаговой библиотеки, где эта фаговая библиотека экспрессирует человеческие антитела (Vaughan et al. Nature Biotechnology 14:309-314 (1996): Sheets et al. Proc. Natl. Acad.Sci. 95:6157-6162 (1998)); Hoogenboom and Winter, J. Mol. Biol., 227:381 (1991); Marks et al., J. Mol. Biol., 222:581 (1991)). Человеческие антитела могут также быть созданы путем введения локусов иммуноглобулина человека в трансгенные животные, например, мышь, в которой эндогенные иммуноглобулиновые гены были частично или полностью инактивированы. При контрольном заражении наблюдают выработку человеческого антитела, которая точно совпадает с таковой, наблюдаемой у людей во всех отношениях, включая генную перестройку, сборку и спектр антител. Этот метод описан, например, в патентах США №№5545807; 5545806; 5569825; 5625126; 5633425; 5661016 и в следующих научных публикациях:Marks et al., Bio/Technology 10: 779-783 (1992); Lonberg et al., Nature 368: 856-859 (1994); Morrison, Nature 368:812-13 (1994); Fishwild et al., Nature Biotechnology 14: 845-51 (1996); Neuberger, Nature Biotechnology 14: 826 (1996); Lonberg and Huszar, Intern. Rev. Immunol. 13:65-93 (1995).Альтернативно, человеческое антитело может быть получено иммортализацией В-лимфоцитов человека, продуцирующих антитело, направленное против мишеневого антигена (такие В-лимфоциты могут быть выделены из индивидуума или могут быть иммунизированы in vitro). Смотри, например, Cole et al., Monoclonal Antibodies and Cancer Therapy, Alan R. Liss, p. 77 (1985); Boerner et al., J. Immunol., 147 (1):86-95 (1991); и патент США № 5750373.
«Незащищенное антитело» представляет собой антитело, которое не конъюгировано с гетерологичной молекулой, такой как цитотоксическая группа или радиоактивная метка.
Антитело «созревшей аффинности» это то, которое имеет одно или несколько изменений в одной или нескольких его CDR, приводящие к улучшению сродства антитела к антигену по сравнению с исходным антителом, которое не содержит этого(их) изменения(ий). Предпочтительные антитела созревшей аффинности будут иметь наномолярное или даже пикомолярное сродство к мишеневому антигену. Антитела созревшей аффинности синтезируются с помощью методик, известных в данной области техники. Marks et al. Bio/Technology 10:779-783 (1992) описывает созревание аффинности путем перестановки доменов VH и VL. Случайный мутагенез CDR и/или каркасных остатков описан: Barbas et al. Proc Nat. Acad. Sci, USA 91:3809-3813 (1994); Schier et al. Gene 169:147-155 (1995); Yelton et al. J. Immunol. 155:1994-2004 (1995); Jackson et al., J. Immunol. 154(7):3310-9 (1995); and Hawkins et al., J. Mol. Biol. 226:889-896 (1992).
Антитело, имеющее «биологическую характеристику» специализированного антитела, это то, которое обладает одной или несколькими биологическими характеристиками этого антитела, отличающими его от других антител, которые связывают тот же самый антиген.
Чтобы провести отбор антител, которые связываются с эпитопом на антигене, связанного исследуемым антителом, может быть выполнен обычный перекрестный конкурентный анализ, такой, как описано в Antibodies, A Laboratory Manual, Cold Spring Harbor Laboratory, Ed Harlow and David Lane (1988).
Для повышения времени полужизни антител или полипептида, содержащего аминокислотные последовательности настоящего изобртения, можно прикрепить эпитоп связывания рецептора утилизатора к антителу (в частности, фрагменту антитела), как описано, например, в патенте США №5739277. Например, молекула нуклеиновой кислоты, кодирующая эпитоп связывания рецептора утилизатора, может быть соединена в пределах рамки считывания с нуклеиновой кислотой, кодирующей полипептидную последовательность настоящего изобретения с тем, чтобы этот гибридный белок, экспрессируемый сконструированной молекулой нуклеиновой кислоты, содержал эпитоп связывания рецептора утилизатора и полипептидную последовательность настоящего изобретения. Используемый в настоящем описании термин «эпитоп связывания рецептора утилизатора» относится к эпитопу участка Fc молекулы IgG (например, IgG1, IgG2, IgG3 или IgG4), который отвечает за повышение времени полужизни молекулы IgG в сыворотке in vivo (например, Ghetie et al., Ann. Rev. Immunol. 18:739-766 (2000), Таблица 1). Антитела с заменами в этой области Fc и повышенным временем полужизни в сыворотке также описаны в WO00/42072, WO 02/060919; Shields et al., J. Biol. Chem. 276:6591-6604 (2001); Hinton, J. Biol. Chem. 279:6213-6216 (2004)). В другом варианте осуществления изобретения, время полужизни в сыворотке также может быть повышено, например, путем прикрепления других полипептидных последовательностей. Например, антитела или другие полипептиды, используемые в способах настоящего изобретения, могут быть прикреплены к сывороточному альбумину или части сывороточного альбумина, который связывается с рецептором FcRn или пептидом связывания сывороточного альбумина таким образом, что сывороточный альбумин связывается с антителом или полипептидом, например, такие полипептидные последовательности рассмотрены в WO01/45746. В предпочтительном варианте осуществления изобретения, пептид сывороточного альбумина, подлежащий сцеплению, содержит аминокислотную последовательность DICLPRWGCLW (SEQ ID NO:21). В другом варианте осуществления изобретения, время полужизни Fab повышается этими методами. Смотри также Dennis et al. J. Biol. Chem. 277:35035-35043 (2002) для пептидных последовательностей связывания сывороточного альбумина.
«Выделенный» полипептид или «выделенное» антитело представляют собой то, что было идентифицировано и отделено и/или извлечено из составляющего его естественного окружения. Примесные компоненты его естественного окружения являются веществами, которые будут препятствовать диагностическому или терапевтическому использованию полипептида или антитела, и могут включать ферменты, гормоны и другие белковые или небелковые растворенные вещества. В предпочтительных вариантах осуществления изобретения полипептид или антитело подлежат очистке (1) более чем на 95% по весу полипептида или антитела, как определяется методом Лоури, и наиболее предпочтительно более чем на 99% по весу, (2) до степени, достаточной для получения по меньшей мере 15 остатков N-концевой или внутрилежащей аминокислотной последовательности путем использования секвенатора с вращающимся стаканом, или (3) до гомогенности с помощью SDS-PAGE в восстановительных и невосстановительных условиях, используя Кумасси бриллиантовый голубой или, предпочтительно, окрашивание серебром. Выделенный полипептид или антитело включает полипептид или антитело in situ в рекомбинантных клетках, поскольку по меньшей мере одна составляющая естественного окружения полипептида не должна присутствовать. Как правило, однако, выделенный полипептид или антитело получают в ходе по меньшей мере одного этапа очистки.
Под «фрагментом» подразумевают часть полипептида или молекулы нуклеиновой кислоты, которая содержит, предпочтительно, по меньшей мере 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95% или более от полной длины эталонной молекулы нуклеиновой кислоты или полипептида. Фрагмент может содержать 10, 20, 30, 40, 50, 60, 70, 80, 90 или 100, 200, 300, 400, 500, 600 или более нуклеотидов или 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 120, 140, 160, 180, 190, 200 аминокислот или более.
«Лечение» относится как к терапевтическому лечению, так и к профилактическим или предупредительным мерам. Те, кто нуждается в лечении, включают тех, кто уже имеет доброкачественную, предраковую или неметастатическую опухоль, так же как и тех, у которых возникновение или рецидив рака должны быть предотвращены.
Термин «терапевтически эффективное количество» относится к количеству терапевтического средства для лечения или предотвращения заболевания или нарушения у млекопитающих. В случае злокачественных опухолей терапевтически эффективное количество терапевтического средства может уменьшить число раковых клеток; уменьшить размер первичной опухоли; ингибировать (т.е. замедлить до некоторой степени и предпочтительно остановить) инфильтрацию раковыми клетками периферических органов; ингибировать (т.е. замедлить до некоторой степени и предпочтительно остановить) метастазирование опухоли; ингибировать, до некоторой степени, опухолевый рост; и/или снять до некоторой степени один или несколько симптомов, ассоциированных с заболеванием. В зависимости от степени, до которой лекарственное средство может предотвратить рост и/или убить существующие раковые клетки, оно может быть цитостатическим и/или цитотоксическим. Для противораковой терапии, эффективность воздействия in vivo может, например, быть измерена путем оценки продолжительности выживаемости, времени прогрессирования заболевания (ТТР), скорости ответа (RR), продолжительности ответа и/или качества жизни.
Термины «рак» и «злокачественный» относятся к описанию физиологического состояния у млекопитающих, которое как правило характеризуется нерегулируемым клеточным ростом. К настоящему определению относятся доброкачественная опухоль и злокачественная опухоль. «Ранняя стадия рака» или «ранняя стадия опухоли» означает рак, не являющийся инвазивным или метастатическим или классифицирующийся как Стадия 0, I или II рака. Примеры рака включают, но ими не ограничиваются, карциному, лимфому, бластому (включая медуллобластому и ретинобластому), саркому (включая липосаркому и синовиальноклеточную саркому), нейроэндокринные опухоли (включая карциноидные опухоли, гастриному и рак островковых клеток), мезотелиома, шваннома (включая акустическую невриному), менингиому, аденокарциному, меланому и лейкемию или лимфолейкозы. Более конкретные примеры таких раков включают плоскоклеточный рак (например, эпителиальный плоскоклеточный рак), рак легкого, включая мелкоклеточный рак легкого (SCLC), немелкоклеточный рак легкого (NSCLC), аденокарциному легкого и плоскоклеточную карциному легкого, рак брюшины, гепатоцеллюлярный рак, гастральный рак или рак желудка, в том числе рак желудочно-кишечного тракта, рак поджелудочной железы, глиобластому, рак шейки матки, рак яичников, рак печени, рак мочевого пузыря, гепатому, рак молочной железы (включая метастатический рак молочной железы), рак толстой кишки, рак прямой кишки, колоректальный рак, карциному эндометрии или матки, карциному слюнных желез, рак почки или почечный рак, рак простаты, рак вульвы, рак щитовидной железы, печеночную карциному, карциному анального канала, пенильную карциному, рак яичка, рак пищевода, опухоли желчевыводящих протоков, а также рак головы и шеи.
Термин «предраковый» относится к заболеванию или новообразованию, которое, как правило, предшествует или трансформируется в рак. «Предраковое» новообразование содержит клетки, которые характеризуются аномальной регуляцией клеточного цикла, пролиферацией или дифференцировкой, которые могут быть определены с помощью маркеров регуляции клеточного цикла, клеточной пролиферации или дифференцировки.
Под «дисплазией» подразумевают любой аномальный рост или развитие ткани, органа или клеток. Предпочтительно, дисплазия имеет высокую степень дедифференцировки или является предраковой.
Под «метастазированием» подразумевают распространение рака из его первичной локализации в другие части тела. Раковые клетки могут отделяться от первичной опухоли, проникать внутрь лимфатических и кровеносных сосудов, циркулировать в кровотоке и расти в отдаленных очагах (метастазировать) в нормальных тканях в другом месте тела. Метастазы могут быть локальными или отдаленными. Метастазирование является последовательным процессом, зависящим от отделения опухолевых клеток от первичной опухоли, их циркуляции в кровотоке и остановки в удаленном участке. В новом участке клетки обеспечивают кровоснабжение и могут расти с образованием опасной для жизни опухоли.
Как стимулирующий, так и ингибирующий молекулярный каскад реакций в опухолевой клетке регулирует это поведение, и взаимодействия между опухолевой клеткой и клетками хозяина в удаленном участке также очень многозначительны.
Под «не метастатической» подразумевают опухоль, которая является доброкачественной и остается в месте первичной локализации и не проникает в лимфатические или кровеносную систему или в ткани, отличные от первичной локализации. В основном, не метастатический рак является любым раком, имеющим Стадию 0, I или II, и изредка Стадию III.
Под «первичной опухолью» или «первичным раком» подразумевают первоначальный рак и не метастатический очаг, расположенный в другой ткани, органе или локализации в теле индивида.
Под «доброкачественной опухолью» или «доброкачественным раком» подразумевают то, что опухоль остается локализованной в месте происхождения и не обладает способностью к инфильтрации, инвазии или метастазированию в удаленные участки.
Под «общей массой опухоли» подразумевают число раковых клеток, размер опухоли или объем рака в теле. Общая масса опухоли также упоминается как опухолевая масса.
Под «количеством опухоли» подразумевают число опухолей.
Под «индивидом» подразумевают млекопитающее, включая, но ими не ограничиваясь, человека или млекопитающего, отличного от человека, такого как бык, лошадь, собака, овца или кошка. Предпочтительно, индивидом является человек.
Термин «противораковая терапия» означает терапию, пригодную для лечения рака. Примеры противораковых терапевтических средств включают, но ими не ограничиваются, например, химиотерапевтические средства, ингибирующие рост вещества, цитотоксические средства, вещества, используемые в радиотерапии, вещества против ангиогенеза, апоптотические вещества, антитубулиновые вещества и другие вещества для лечения рака, антитела анти-CD20, ингибиторы тромбоцитарного фактора роста (например, GleevecTM (Imatinib Mesylate)), ингибитор COX-2 (например, целекоксиб), интерфероны, цитокины, антагонисты (например, нейтрализующие антитела), которые связывают одну или несколько следующих мишеней ErbB2, ErbB3, ErbB4, PDGFR-бета, BlyS, APRIL, BCMA или рецептор(ы) VEGF, TRAIL/Apo2 и другие биоактивные и органические химические вещества. Комбинации вышеперечисленного также включены в настоящее изобретение.
Термин «цитотоксическое средство» в том смысле, в котором он используется в настоящем описании, относится к веществу, которое ингибирует или предотвращает функцию клеток и/или вызывает разрушение клеток. Термин предназначен, чтобы включать в себя радиоактивные изотопы (например. I131, I125, Y90и Re186), химиотерапевтические вещества и токсины, такие как ферментативно активные токсины бактериального, грибкового, растительного или животного происхождения или их фрагменты.
«Химиотерапевтическое средство» является химическим соединением, пригодным для лечения рака. Например, химиотерапевтическое средство включает химическое(ие) соединение(ия) подходящее(ие) для лечения рака. Примеры химиотерапевтических средств включают алкилирующий агент, такой как тиотепа, циклофосфамид CYTOXAN®; алкилсульфонаты, такие как бисульфан, импросульфан и пипосульфан; азиридины, такие как бензодопа, карбокуон, метуредопа и уредопа; этиленимины и метиламеламины, включая алтретамин, триэтиленмеламин, триэтиленфосфорамид, триэтилентиофосфорамид и триметилоломеламин; ацетогенины (в частности, буллатацин и буллатацинон); камптотецин (в том числе синтетический аналог топотекан); бриостатин; каллистатин; СС-1065 (в том числе его синтетические аналоги адозелезин, карзелезин и бизелезин); криптофицины (в частности, криптофицин 1 и криптофицин 8); доластатин; дуокармицин (в том числе синтетические аналоги, KW-2189 и CB1-TM1); элеутеробин; панкратистатин; саркодиктиин; спонгистатин; азотистые иприты, такие как хлорамбуцил, хлорнафазин, холофосфамид, эстрамустин, ифосфамид, мехлоретамин, гидрохлорид оксида мехлоретамина, мелфалан, новембихин, фенестерин, преднимустин, трофосфамид, урамустин; нитрозомочевины, такие как кармустин, хлорозотоцин, фотемустин, ломустин, нимустин и ранимнустин; антибиотики, такие как энедииновые антибиотики (например, калихимицин, в частности, калихимицин-гамма1I и калихимицин-омегаI1 (смотри, например, Agnew, Chem Intl. Ed. Engl., 33: 183-186 (1994)); динемицин, в том числе динемицин A; бисфосфонаты, такие как клодронат; эсперамицин; а также хромофор неокарциностатина и родственные ему хромофоры хромопротеинового энедиинового антибиотика), аклациномизины, актиномицин, аутрамицин, азасерин, блеомицины, кактиномицин, карабицин, карминомицин, карцинофилин, хромомицин, дактиномицин, даунорубицин, деторубицин, 6-диазо-5-оксо-L-норлейцин, доксорубицин ADRIAMYCIN® (в том числе морфолинодоксорубицин, цианморфолинодоксорубицин, 2-пирролиндоксорубицин и дезоксидоксорубицин), эпирубицин, эзорубицин, идарубицин, марцелломицин, митомицины, такие как митомицин C, микофеноловая кислота, ногаламицин, оливомицины, пепломицин, потфиромицин, пуромицин, квеламицин, родорубицин, стрептонигрин, стрептозоцин, туберцидин, убенимекс, зиностатин, зорубицин; антиметаболиты, такие как метотрексат и 5-фторурацил (5-FU); аналоги фолиевой кислоты, такие как деноптерин, метотрексат, птероптерин, триметрексат; аналоги пурина, такие как флударабин, 6-меркаптопурин, тиамиприн, тиогуанин; аналоги пиримидина, такие как анцитабин, азацитидин, 6-азауридин, кармофур, цитарабин, дидезоксиуридин, доксифлуридин, эноцитабин, флоксуридин; андрогены, такие как калустерон, пропионат дромостанолона, эпитиостанол, мепитиостан, тестолактон; противоадреналовые средства, такие как аминоглутетимид, митотан, трилостан; компенсатор фолиевой кислоты, такой как фолиновая кислота; ацеглатон; гликозид альдофосфамида; аминолевулиновую кислоту; энилурацил; амсакрин; бестрабуцил; бисантрен; эдатрексат; дефофамин; демеколцин; диазиквон; элфорнитин; ацетат эллиптиния; эпотилон; этоглуцид; нитрат галлия; гидроксимочевину; лентинан; лонидаинин; майтанзиноиды, такие как майтанзин и ансамитоцины; митогуазон; митоксантрон; мопиданмол; нитраэрин; пентостатин; фенамет; пирарубицин; лозоксантрон; подофиллиновую кислоту; 2-этилгидразид; прокарбазин; полисахаридный комплекс PSK® (JHS Natural Products, Eugene, OR); разоксан; ризоксин; сизофиран; спирогерманий; тенуазоновую кислоту; триазиквон; 2,2',2''-трихлортриэтиламин; трихотецены (в частности, токсин T-2, верракурин A, роридин A и ангуидин); уретан; виндезин; дакарбазин; манномустин; митобронитол; митолактол; пипоброман; гацитозин; арабинозид ("Ara-C"); циклофосфамид; тиотепу; таксоиды, например, паклитаксел TAXOL® (Bristol-Myers Squibb Oncology, Princeton, N.J.), не содержащий кремофор ABRAXANETM, сконструированный с альбумином препарат наночастиц паклитаксела (American Pharmaceutical Partners, Schaumberg, Illinois) и доксетаксел TAXOTERE® (Rhône-Poulenc Rorer, Antony, France); хлоранбуцил; гемцитабин GEMZAR®; 6-тиогуанин; меркаптопурин; метотрексат; аналоги платины, такие как цисплатин и карбоплатин; винбластин; платину; этопозид (VP-16); ифосфамид; митоксантрон; винкристин; винорелбин NAVELBINE®; новантрон; тенипозид; эдатрексат; дауномицин; аминоптерин; кселоду; ибандронат; иринотекан (Камптосар, CPT-11) (включая схему лечения иринотеканом с 5-FU и лейковорином); ингибитор топоизомеразы RFS 2000; дифторметилорнитин (DMFO); ретиноиды, такие как ретиноевая кислота; капецитабин; комбретастатин; лейковорин (LV); оксалиплатин, включая схему лечения оксалиплатином (FOLFOX); ингибиторы PKC-альфа, Raf, H-Ras, EGFR (например, эрлотиниб (TarcevaTM)) и VEGF-A, который уменьшает клеточную пролиферацию, и фармацевтически приемлемые соли, кислоты или производные любого из указанного выше.
Также в это определение включены антигормональные средства, которые действуют, регулируя или ингибируя воздействие гормона на опухоли, такие как антиэстрогенные средства и избирательные регуляторы рецептора эстрогена (SERM), включая, например, тамоксифен (в том числе тамоксифен NOLVADEX®), ралоксифен, дролоксифен, 4-гидрокситамоксифен, триоксифен, кеоксифен, LY117018, онапристон и торемифен FARESTON; ингибиторы ароматазы, которые ингибируют фермент ароматазу, регулирующую продукцию эстрогена в надпочечниках, такие как, например, 4(5)-имидазолы, аминоглутетимид, ацетат мегестрола MEGASE®, экземестан AROMASIN®, форместан, фадрозол, ворозол RIVISOR®, летрозол FEMARA® и анастрозол ARIMIDEX®; и антиандрогенные средства, такие как флутамид, нилутамид, бикалутамид, леупролид и гозерелин; а также троксацитабин (аналог 1,3-диоксолана нуклеозида цитозина); антисмысловые олигонуклеотиды, в частности те, которые ингибируют экспрессию генов в сигнальных путях, вовлеченных в аберрантную клеточную пролиферацию, таких как, например, PKC-альфа, Raf и H-Ras; рибозимы, такие как ингибитор экпсрессии VEGF (например, рибозим ANGIOZYME®) и ингибитор экпсрессии HER2; вакцины, такие как вакцины для генотерапии, например, вакцина ALLOVECTIN®, вакцина LEUVECTIN® и вакцина VAXID®; rIL-2 PROLEUKIN®; ингибитор топоизомеразы 1 LURTOTECAN®; rmRH ABARELIX®; винорелбин и эсперамицины (смотри патент США № 4675187) и фармацевтически приемлемые соли, кислоты или производные любого из указанного выше.
Термин «пролекарство» в том смысле, в котором он используется в настоящем описании, относится к предшественнику или производному фармацевтически активного вещества, которое является менее цитотоксическим в отношении опухолевых клеток по сравнению с исходным препаратом, и которое способно ферментативно активироваться или превращаться в более активную исходную форму. Смотри, например, Wilman, "Prodrugs in Cancer Chemotherapy", Biochemical Society Transactions, 14, pp. 375-382, 615th Meeting Belfast (1986) и Stella et al., "Prodrugs: A Chemical Approach to Targeted Drug Delivery", Directed Drug Delivery, Borchardt et al., (ed.), pp.247-267, Humana Press (1985). Пролекарства настоящего изобретения включают, но ими не ограничиваются, фосфатсодержащие пролекарства, тиофосфатсодержащие пролекарства, сульфатсодержащие пролекарства, пептидсодержащие пролекарства, модифицированные D-аминокислотные пролекарства, гликозилированные пролекарства, β-лактамсодержащие пролекарства, необязательно замещенные феноксиацетамидсодержащие пролекарства или необязательно замещенные фенилацетамидсодержащие пролекарства, 5-фторцитозин и другие 5-фторуридиновые пролекарства, которые могут быть превращены в более активные цитотоксические лекарственные препараты. Примеры цитотоксических препаратов, которые могут быть превращены в пролекарство для применения по настоящему изобретению, включают, но не ограничиваются химиотерапевтическими средствами, описанными выше.
Под «радиационной терапией» подразумевают использование направленных гамма-лучей или бета-лучей с целью индукции достаточного повреждения клетки в той степени, чтобы ограничить ее способность нормально функционировать, или полного разрушения клетки. Следует понимать, что существует много способов, известных в данной области техники, для определения дозы и продолжительности лечения. Стандартное лечение происходит в виде однократного назначения, и стандартные дозы находятся в диапазоне от 10 до 200 единиц (грей) в день.
Терапевтические средства
Настоящее изобретение описывает использование антагонистов c-met и антагонистов EGFR в комбинированной терапии для лечения патологического состояния, такого как опухоль, у индивида.
Антагонисты c-met
Антагонисты c-met, используемые в методах настоящего изобретения, включают полипептиды, которые специфически связываются с c-met, антителами анти-c-met, низкомолекулярными молекулами c-met, молекулами рецептора и производными, которые специфически связываются с c-met, и гибридные белки. Антагонисты c-met также включают антагонистические варианты полипептидов c-met, РНК-аптамеры и пептидные антитела против c-met и HGF. Также рассматриваемыми в качестве антагонистов c-met, используемых в способах настоящего изобретения, являются антитела анти-HGF, полипептиды анти-HGF, молекулы рецептора c-met и производные, которые специфически связываются с HGF. Примеры каждого из них описаны ниже.
Антитела анти-c-met, используемые в способах по настоящему изобретению, включают любое антитело, которое связывается с достаточной аффинностью и специфичностью с c-met и может снизить или ингибировать активность c-met. Отобранное антитело будет как правило иметь достаточно сильное сродство к связыванию c-met, например, антитело может связать c-met человека со значением Kd в диапазоне 100 нМ-1 пМ. Аффинность антитела может быть определена с помощью анализа, основанного на поверхностном плазмонном резонансе, (таком как анализ BIAcore, как описано в публикации заявки PCT № WO2005/012359); ферментного иммуносорбентного анализа (ELISA); и конкурентным анализом (например, RIA'), например. Предпочтительно, антитела анти-c-met по изобретению могут быть использованы в качестве терапевтических средств с целью направленного действия на заболевание или препятствия развития заболевания, где вовлечена активность c-met/HGF. Также, антитело может быть использовано в других анализах биологической активности, например, чтобы оценить его эффективность в качестве терапевтического средства. Такие анализы известны в данной области техники и зависят от мишеневого антигена и предназначения антитела.
Антитела анти-c-met известны в данной области техники (смотри, например, Martens, T, et al (2006) Clin Cancer Res 12 (20 Pt 1):6144; US 6468529; WO2006/015371; WO2007/063816; US7408043; WO2009/007427; WO2005/016382; WO2007/126799. В одном из вариантов осуществления изобретения антитело анти-c-met содержат вариабельный домен тяжелой цепи, содержащий один или несколько последовательностей CDR1-HC, CDR2-HC и CDR3-HC, представленных на фигуре 7 (SEQ ID NO:13-15). В некоторых вариантах осуществления изобретения антитело содержит вариабельный домен легкой цепи, содержащий один или несколько последовательностей CDR1-LC, CDR2-LC и CDR3-LC, представленных на фигуре 7 (SEQ ID NO:5-7). В некоторых вариантах осуществления изобретения вариабельный домен тяжелой цепи содержит последовательности FR1-HC, FR2-HC, FR3-HC и FR4-HC, представленные на фигуре 7 (SEQ ID NO:9-12). В некоторых вариантах осуществления изобретения вариабельный домен легкой цепи содержит последовательности FR1-LC, FR2-LC, FR3-LC и FR4-LC, представленные на фигуре 7 (SEQ ID NO:1-4). В некоторых вариантах осуществления изобретения антитело анти-c-met является моновалентным и содержит участок Fc. В некоторых вариантах осуществления изобретения антитело содержит последовательность Fc, представленную на фигуре 7 (SEQ ID NO:17).
В некоторых вариантах осуществления изобретения антитело является моновалентным и содержит участок Fc, где участок Fc содержит первый и второй полипептид, где первый полипептид включает последовательность Fc, представленную на фигуре 7 (SEQ ID NO:17) и второй полипептид включает последовательность Fc, представленную на фигуре 8 (SEQ ID NO:18).
В одном из вариантов осуществления изобретения антитело анти-c-met содержит (а) первый полипептид, содержащий вариабельный домен тяжелой цепи с последовательностью: QVQLQQSGPELVRPGASVKMSCRASGYTFTSYWLHWVKQRPGQGLEWIGMIDPSNSDTRFNPNFKDKATLNVDRSSNTAYMLLSSLTSADSAVYYCATYGSYVSPLDYWGQGTSVTVSS (SEQ ID NO:19), с последовательностью СН1, представленной на фигуре 7 (SEQ ID NO:16) и с последовательностью Fc, представленной на фигуре 7 (SEQ ID NO:17); и (b) второй полипептид, включающий вариабельный домен легкой цепи, с последовательностью: DIMMSQSPSSLTVSVGEKVTVSCKSSQSLLYTSSQKNYLAWYQQKPGQSPKLLIYWASTRESGVPDRFTGSGSGTDFTLTITSVKADDLAVYYCQQYYAYPWTFGGGTKLEIK (SEQ ID NO:20), и с последовательностью CL1, представленной на фигуре 7 (SEQ ID NO:8); и (с) третий полипептид, содержащий последовательность Fc, представленную на фигуре 8 (SEQ ID NO:18).
В других вариантах осуществления изобретения антитело анти-c-met является моноклональным антителом, продуцируемым клеточной линией гибридомы, зарегистрированной под Идентификационным Номером Американской Коллекции Типовых Культур ATCC HB-11894 (гибридома 1А3.3.13) или HB-11895 (гибридома 5D5.11.6). В других вариантах осуществления изобретения антитело содержит одну или несколько последовательностей CDR моноклонального антитела, продуцируемого клеточной линией гибридомы, зарегистрированной под Идентификационным Номером Американской Коллекции Типовых Культур ATCC HB-11894 (гибридома 1А3.3.13) или HB-11895 (гибридома 5D5.11.6).
В других вариантах осуществления, антитело c-met по изобретению специфически связывается по меньшей мере с частью домена Sema с-met или его варианта. В одном примере антитело-антагонист по изобретению специфически связывается по меньшей мере с одной из последовательностей, выбранной из группы, состоящей из LDAQT (SEQ ID NO:22) (например, остатки 269-273 c-met), LTEKRKKRS (SEQ ID NO:23) (например, остатки 300-308 c-met), KPDSAEPM (SEQ ID NO:24) (например, остатки 350-357 c-met) и NVRCLQHF (SEQ ID NO:25) (например, остатки 381-388 c-met). В одном из вариантов осуществления, антитело-антагонист по изобретению специфически связывается с конформационным эпитопом, сформированным частью или всей, по меньшей мере, одной из последовательностей, выбранной из группы, состоящей из LDAQT (SEQ ID NO:22) (например, остатки 269-273 c-met), LTEKRKKRS (SEQ ID NO:23) (например, остатки 300-308 c-met), KPDSAEPM (SEQ ID NO:24) (например, остатки 350-357 c-met) и NVRCLQHF (SEQ ID NO:25) (например, остатки 381-388 c-met). В одном из вариантов осуществления, антитело-антагонист по изобретению специфически связывается с аминокислотной последовательностью, имеющей по меньшей мере 50%, 60%, 70%, 80%, 90%, 95%, 98% идентичность или схожесть с последовательностью LDAQT (SEQ ID NO:22), LTEKRKKRS (SEQ ID NO:23), KPDSAEPM (SEQ ID NO:24) и/или NVRCLQHF (SEQ ID NO:25).
Антитела анти-HGF хорошо известны в данной области техники. Смотри, например, Kim KJ., et al. Clin Cancer Res. (2006) 12(4):1292-8; WO2007/115049; WO2009/002521; WO2007/143098; WO2007/017107; WO2005/017107; L2G7; AMG-102.
Молекулы рецептора c-met или его фрагменты, которые специфически связываются с HGF, могут быть использованы в методах настоящего изобретения, например, для связывания и блокировать белок HGF, таким образом, предотвращая его от участия в сигнальной системе. Предпочтительно, молекула рецептора c-met или его фрагменты связывания HGF представляют собой растворимую форму. В некоторых вариантах осуществления изобретения, растворимая форма рецептора оказывает ингибирующее воздействие на биологическую активность белка c-met путем связывания с HGF, таким образом, предотвращая его связывание с его природными рецепторами, находящимися на поверхности клеток-мишеней. Также включенными для рассмотрения являются гибридные белки рецептора c-met, примеры которых описаны ниже.
Растворимый белок рецептора c-met или химерные белки рецептора c-met настоящего изобретения включают белки рецептора c-met, которые не прикреплены к поверхности клеток посредством трансмембранного домена. В этой связи, растворимые формы рецептора c-met, включая белки химерного рецептора, несмотря на способность связывания и инактивации HGF, не содержат трансмембранный домен и таким образом в основном не становятся ассоциированными с клеточной мембраной клеток, в которых экспрессируется данная молекула. Смотри, например, Kong-Beltran, M et al Cancer Cell (2004) 6(1): 75-84.
Молекулы HGF или его фрагменты, которые специфически связываются с c-met и блокируют или уменьшают активацию c-met, тем самым предотвращая его участие в сигнальной системе, могут быть использованы в способах по изобретению.
Аптамеры представляют собой молекулы нуклеиновой кислоты, которые образуют третичные структуры, которые специфически связываются с молекулой-мишенью, такой как полипептид HGF. Получение и терапевтическое использование аптамеров хорошо разработаны в данной области техники. Смотри, например, патент США № 5475096. Аптамер HGF представляет собой пегилированный модифицированный олигонуклеотид, который принимает трехмерную конформацию, позволяющую ему связываться с экстрацеллюлярным HGF. Дополнительную информацию по аптамерам можно найти в публикации патентной заявки США № 20060148748.
Пептидное антитело представляет собой последовательность, присоединенную к аминокислотной последовательности, кодирующей фрагмент или часть молекулы иммуноглобулина. Полипептиды могут быть получены из случайных последовательностей, отобранных любым методом для специфического связывания, включая, но ими не ограничиваясь, технологию фагового дисплея. В предпочтительном варианте осуществления изобретения, отобранный полипептид может быть соединен с аминокислотной последовательностью, кодирующей часть Fc иммуноглобулина. Пептидные антитела, которые специфически связываются с и противодействуют HGF или c-met, также пригодны в способах по изобретению.
Антагонисты c-met включают низкомолекулярные молекулы, такие как соединения, описанные в US 5792783; US 5834504; US 5880141; US 6297238; US 6599902; US 6790852; US 2003/0125370; US 2004/0242603; US 2004/0198750; US 2004/0110758; US 2005/0009845; US 2005/0009840; US 2005/0245547; US 2005/0148574; US 2005/0101650; US 2005/0075340; US 2006/0009453; US 2006/0009493; WO 98/007695; WO 2003/000660; WO 2003/087026; WO 2003/097641; WO 2004/076412; WO 2005/004808; WO 2005/121 125; WO 2005/030140; WO 2005/070891; WO 2005/080393; WO 2006/014325; WO 2006/021886; WO 2006/021881, WO 2007/103308). PHA-665752 представляет собой низкомолекулярную молекулу, конкурирующую с АТФ, ингибитор активного сайта каталитической активности c-Met, а также клеточного роста, клеточной подвижности, инвазии и морфологии различных опухолевых клеток (Ma et al (2005) Clin. Cancer Res. 11:2312-2319; Christensen et al (2003) Cancer Res. 63:7345-7355).
Антагонисты EGFR
Антагонисты EGFR включают антитела, такие как гуманизированные моноклональные антитела, известные как нимотузумаб (YM Biosciences), полностью человеческое ABX-EGF (панитумумаб, Abgenix Inc.), а также полностью человеческие антитела, известные как E1.1, E2.4, E2.5, E6.2, E6.4, E2.11, E6.3 и E7.6.3 и описанные в US 6235883; MDX-447 (Medarex Inc.). Пертузумаб (2С4) является гуманизированным антителом, которое связывается непосредственно с HER2, но препятствует димеризации HER2-EGFR, тем самым ингибируя сигнальную систему EGFR. Другие примеры антител, которые связываются с EGFR, включают MAb 579 (ATCC CRL HB 8506), MAb 455 (ATCC CRL HB8507), MAb 225 (ATCC CRL 8508), MAb 528 (ATCC CRL 8509) (смотри патент США № 4943533, Mendelsohn et al.) и их варианты, такие как химерное 225 (С225 или цетуксимаб; ERBUTIX®) и реконструированное человеческое 225 (Н225) (смотри, WO 96/40210, Imclone Systems Inc.); IMC-11F8, полностью человеческое, EGFR-ориентированное антитело (Imclone); антитела, которые связывают мутантный EGFR типа II (патент США № 5212290); гуманизированные и химерные антитела, которые связывают EGFR как описано в патенте США № 5891996; и человеческие антитела, которые связывают EGFR, такие как ABX-EGF (смотри WO98/50433, Abgenix); EMD 55900 (Stragliotto et al. Eur. J. Cancer 32A:636-640 (1996)); гуманизированное EGFR-антитело EMD7200 (матузумаб), направленное против EGFR, которое конкурирует как с EGF, так и TGF-альфа за связывание с EGFR; и mAb 806 или гуманизированное mAb 806 (Johns et al., J. Biol. Chem. 279(29):30375-30384 (2004)). Антитело анти-EGFR может быть конъюгировано с цитотоксическим веществом, тем самым образуя иммуноконъюгат (смотри, например, EP659439 A2, Merck Patent GmbH).
Антитела анти-EGFR, пригодные в способах по изобретению, включают любое антитело, которое связывается с достаточной аффинностью и специфичностью с EGFR и может снизить или ингибировать активность EGFR. Отобранное антитело будет, как правило, иметь достаточно сильное сродство к связыванию EGFR, например, антитело может связать c-met человека со значением Kd в диапазоне 100 нМ-1 пМ. Аффинность антитела может быть определена с помощью анализа, основанного на поверхностном плазмонном резонансе, (такого как анализ BIAcore, как описано в публикации заявки PCT № WO2005/012359); ферментного иммуносорбентного анализа (ELISA); и с помощью конкурентного анализа (например, RIA'), например. Предпочтительно, антитела анти-c-met по изобретению могут быть использованы в качестве терапевтических средств с целью направленного воздействия на заболевание или препятствия развития заболеваний или состояний, где вовлечена активность лиганда EGFR/EGFR. Также, антитело может быть использовано в других анализах биологической активности, например, чтобы оценить его эффективность в качестве терапевтического средства. Такие анализы известны в данной области техники и зависят от мишеневого антигена и предназначения антитела.
Биспецифические антитела представляют собой антитела, имеющие специфичность связывания по меньшей мере к двум различным эпитопам. Типичные биспецифические антитела могут связываться с EGFR и c-met. В другом примере, типичное специфическое антитело может связаться с двумя различными эпитопами одного и того же белка, например, белка c-met. Альтернативно, спейсер c-met или EGFR может быть соединен со спейсером, который связывается с триггерными молекулами на лейкоците, такими как молекула Т-клеточного рецептора (например, CD2 или CD3) или рецепторы Fc для IgG (FcγR), такие как FcγRI (CD64), FcγRII (CD32) и FcγRIII (CD16), с тем, чтобы сконцентрироваться на клеточных защитных механизмах в клетке, экспрессирующей c-met или EGFR. Биспецифические антитела могут также быть использованы для локализации цитотоксических веществ в клетках, экспрессирующих EGFR или c-met. Эти антитела содержат EGFR- или c-met-связывающий спейсер и спейсер, который связывает цитотоксическое вещество (например, сапорин, анти-интерферон-α, алкалоид барвинка, цепь рицина А, метотрексат или радиоактивный изотоп гаптен). Биспецифические антитела могут быть получены в виде полноразмерных антител или фрагментов антитела (например, биспецифические антитела F(ab')2).
Антагонисты EGFR также включают низкомолекулярные молекулы, такие как соединения, описанные в US5616582, US5457105, US5475001, US5654307, US5679683, US6084095, US6265410, US6455534, US6521620, US6596726, US6713484, US5770599, US6140332, US5866572, US6399602, US6344459, US6602863, US6391874, WO9814451, WO9850038, WO9909016, WO9924037, WO9935146, WO0132651, US6344455, US5760041, US6002008, US5747498. Специфические низкомолекулярные антагонисты EGFR включают OSI-774 (CP-358774, эрлотиниб, OSI Pharmaceuticals); PD 183805 (CI 1033, 2-пропенамид, N-[4-[(3-хлор-4-фторфенил)амино]-7-[3-(4-морфолинил)пропокси]-6-хиназолинил]-, дигидрохлорид, Pfizer Inc.); Iressa® (ZD1839, гефитиниб, AstraZeneca); ZM 105180 ((6-амино-4-(3-метилфениламино)-хиназолин, Zeneca); BIBX-1382 (N8-(3-хлор-4-фтор-фенил)-N2-(1-метилпиперидин-4-ил)-пиримидо[5,4-d]пиримидин-2,8-диамин, Boehringer Ingelheim); PKI-166 ((R)-4-[4-[(1-фенилэтил)амино]-1H-пирроло[2,3-d]пиримидин-6-ил]-фенол);(R)-6-(4-гидроксифенил)-4-[(1-фенилэтил)амино]-7H-пирроло[2,3-d]пиримидин); CL-387785 (N-[4-[(3-бромфенил)амино]-6-хиназолинил]-2-бутинамид); EKB-569 (N-[4-[(3-хлор-4-фторфенил)амино]-3-циано-7-этокси-6-хинолинил]-4-(диметиламино)-2-бутенамид); лапатиниб (Tykerb, GlaxoSmithKline); ZD6474 (Zactima, AstraZeneca); CUDC-101 (Curis); канертиниб (CI-1033); AEE788 (6-[4-[(4-этил-1-пиперазинил)метил]фенил]-N-[(1R)-1-фенилэтил]-7H-пирроло[2,3-d]пиримидин-4-амин, WO2003013541, Novartis) и PKI166 4-[4-[[(1R)-1-фенилэтил]амино]-7H-пирроло[2,3-d]пиримидин-6-ил]-фенол, WO9702266 Novartis).
В одном из вариантов осуществления изобретения антагонист EGFR имеет общую формулу I:
в соответствии с патентом США №5757498, приведенным в настоящее описание в качестве ссылки, где:
m равно 1, 2 или 3;
каждый R1 независимо выбран из группы, состоящей из водорода, галогена, гидрокси, гидроксиамино, карбокси, нитро, гуанидино, уреидо, циано, трифторметила и -(С1-С4 алкилен)-W-(фенила), где W представляет собой одинарную связь, О, S или NH;
или каждый R1 независимо выбран из R9 и С1-С4алкила, замещенного циано, где R9выбран из группы, состоящей из R5, -OR6, -NR6R6, -C(O)R7, -NHOR5, -OC(O)R6, циано, А и -YR5; R5 представляет собой С1-С4алкил; R6 независимо представляет собой водород или R5; R7 представляет собой R5, -OR6 или -NR6R6; А выбран из пиперидино, морфолино, пирролидино, 4-R6-пиперазин-1-ил, имидазол-1-ил, 4-пиридон-1-ил, -(С1-С4алкилен)(СО2Н), фенокси, фенил, фенилсульфанил, С2-С4 алкенил и -(С1-С4алкилен)C(O)NR6R6; и Y представляет собой S, SO или SO2; где алкильные фрагменты в R5, -OR6 и -NR6R6 необязательно замещены галозаместителями в количестве от одного до трех, и алкильные фрагменты в R5, -OR6 и -NR6R6 необязательно замещены 1 или 2 R9 группой(ами), и где алкильные фрагменты указанных необязательных заместителей необязательно замещены галогеном или R9 при условии, что два гетероатома не присоединены к одному и тому же атому углерода;
или каждый R1 независимо выбран из -NHSO2R5, фталимидо-(С1-С4)-алкилсульфониламино, бензамидо, бензенсульфониламино, 3-фенилуреидо, 2-оксопирролидин-1-ил, 2,5-диоксопирролидин-1-ил, и R10-(C2-C4)-алканоиламино, где R10 выбран из галогена, -OR6, С2-С4 алканоилокси, -С(O)R7 и -NR6R6; и где указанные -NHSO2R5, фталимидо-(С1-С4)-алкилсульфониламино, бензамидо, бензенсульфониламино, 3-фенилуреидо, 2-оксопирролидин-1-ил, 2,5-диоксопирролидин-1-ил, и R10-(C2-C4)-алканоиламино R1 группы необязательно замещены 1 или 2 заместителями, независимо выбранными из галогена, С1-С4алкила, циано, метансульфонила и С1-С4алкокси;
или две R1 группы взяты вместе с углеродами, к которым они присоединены для образования 5-8-членной циклической структуры, которая включает 1 или 2 гетероатома, выбранных из O, S и N;
R2 представляет собой водород или С1-С6 алкил, необязательно замещенный заместителями в количестве от 1 до 3, независимо выбранными из галогена, С1-С4 алкокси, -NR6R6 и -SO2R5;
n представляет собой 1 или 2 и каждый R3 независимо выбран из водорода, галогена, гидрокси, С1-С6 алкил, -NR6R6, и С1-С4 алкокси, где алкильные фрагменты указанных R3групп необязательно замещены заместителями в количестве от 1 до 3, независимо выбранными из галогена, С1-С4 алкокси, -NR6R6 и -SO2R; и
R4 представляет собой азидо или -(этинил)-R11, где R11 представляет собой водород или С1-С6 алкил, необязательно замещенный гидрокси, -OR6, или -NR6R6.
В конкретном варианте осуществления изобретения антагонист EGFR является соединением согласно формуле I, выбранным из группы, включающей:
(6,7-диметоксихиназолин-4-ил)-(3-этинилфенил)-амин;
(6,7-диметоксихиназолин-4-ил)-[3-(3'-гидроксипропин-1-ил)фенил]-амин;
[3-(2'-(аминометил)-этинил)фенил]-(6,7-диметоксихиназолин-4-ил)-амин;
(3-этинилфенил)-(6-нитрохиназолин-4-ил)-амин;
(6,7-диметоксихиназолин-4-ил)-(4-этинилфенил)-амин;
(6,7-диметоксихиназолин-4-ил)-(3-этинил-2-метилфенил)-амин;
(6-аминохиназолин-4-ил)-(3-этинилфенил)-амин;
(3-этинилфенил)-(6-метансульфониламинохиназолин-4-ил)-амин;
(3-этинилфенил)-(6,7-метилендиоксихиназолин-4-ил)-амин;
(6,7-диметоксихиназолин-4-ил)-(3-этинил-6-метилфенил)-амин;
(3-этинилфенил)-(7-нитрохиназолин-4-ил)-амин;
(3-этинилфенил)-[6-(4'-толуолсульфониламино)хиназолин-4-ил]-амин;
(3-этинилфенил)-{6-[2'-фталимидо-эфир-1'-ил-сульфониламино]хиназолин-4-ил}-амин;
(3-этинилфенил)-(6-гуанидинохиназолин-4-ил)-амин;
(7-аминохиназолин-4-ил)-(3-этинилфенил)-амин;
(3-этинилфенил)-(7-метоксихиназолин-4-ил)-амин;
(6-карбометоксихиназолин-4-ил)-(3-этинилфенил)-амин;
(7-карбометоксихиназолин-4-ил)-(3-этинилфенил)-амин;
[6,7-бис(2-метоксиэтокси)хиназолин-4-ил]-(3-этинилфенил)-амин;
(3-азидофенил)-(6,7-диметоксихиназолин-4-ил)-амин; (3-азидо-5-хлорфенил)-(6,7-диметоксихиназолин-4-ил)-амин;
(4-азидофенил)-(6,7-диметоксихиназолин-4-ил)-амин;
(3-этинилфенил)-(6-метансульфонилхиназолин-4-ил)-амин;
(6-этансульфанил-хиназолин-4-ил)-(3-этинилфенил)-амин;
(6,7-диметоксихиназолин-4-ил)-(3-этинил-4-фторфенил)-амин;
(6,7-диметоксихиназолин-4-ил)-[3-(пропин-1'-ил)-фенил]-амин;
[6,7-бис-(2-метоксиэтокси)-хиназолин-4-ил]-(5-этинил-2-метилфенил)-амин;
[6,7-бис-(2-метоксиэтокси)-хиназолин-4-ил]-(3-этинил-4-фторфенил)-амин;
[6,7-бис-(2-хлорэтокси)-хиназолин-4-ил]-(3-этинилфенил)-амин;
[6-(2-хлорэтокси)-7-(2-метоксиэтокси)-хиназолин-4-ил]-(3-этинилфенил)-амин;
[6,7-бис-(2-ацетокси-этокси)-хиназолин-4-ил]-(3-этинилфенил)-амин;
2-[4-(3-этинилфениламино)-7-(2-гидроксиэтокси)-хиназолин-6-илокси]-этанол;
[6-(2-ацетоксиэтокси)-7-(2-метоксиэтокси)-хиназолин-4-ил]-(3-этинилфенил)-амин;
[7-(2-хлорэтокси)-6-(2-метоксиэтокси)-хиназолин-4-ил]-(3-этинилфенил)-амин;
[7-(2-ацетоксиэтокси)-6-(2-метоксиэтокси)-хиназолин-4-ил]-(3-этинил-фенил)-амин;
2-[4-(3-этинилфениламино)-6-(2-гидроксиэтокси)-хиназолин-7-илокси]-этанол;
2-[4-(3-этинилфениламино)-7-(2-метоксиэтокси)-хиназолин-6-илокси]-этанол;
2-[4-(3-этинилфениламино)-6-(2-метоксиэтокси)-хиназолин-7-илокси]-этанол;
[6-(2-ацетоксиэтокси)-7-(2-метокси-этокси)-хиназолин-4-ил]-(3-этинилфенил)-амин;
(3-этинилфенил)-{6-(2-метоксиэтокси)-7-[2-(4-метилпиперазин-1-ил)-этокси]-хиназолин-4-ил}-амин;
(3-этинилфенил)-[7-(2-метоксиэтокси)-6-(2-морфолин-4-ил)-этокси)-хиназолин-4-ил]-амин;
(6,7-диэтоксихиназолин-1-ил)-(3-этинилфенил)-амин;
(6,7-дибутоксихиназолин-1-ил)-(3-этинилфенил)-амин;
(6,7-диизопропоксихиназолин-1-ил)-(3-этинилфенил)-амин;
(6,7-диэтоксихиназолин-1-ил)-(3-этинил-2-метилфенил)-амин;
[6,7-бис-(2-метоксиэтокси)-хиназолин-1-ил]-(3-этинил-2-метил-фенил)-амин;
(3-этинилфенил)-[6-(2-гидроксиэтокси)-7-(2-метоксиэтокси)-хиназолин-1-ил]-амин;
[6,7-бис-(2-гидроксиэтокси)-хиназолин-1-ил]-(3-этинилфенил)-амин;
2-[4-(3-этинилфениламино)-6-(2-метоксиэтокси)-хиназолин-7-илокси]-этанол;
(6,7-дипропоксихиназолин-4-ил)-(3-этинилфенил)-амин;
(6,7-диэтоксихиназолин-4-ил)-(3-этинил-5-фторфенил)-амин;
(6,7-диэтоксихиназолин-4-ил)-(3-этинил-4-фторфенил)-амин;
(6,7-диэтоксихиназолин-4-ил)-(5-этинил-2-метилфенил)-амин;
(6,7-диэтоксихиназолин-4-ил)-(3-этинил-4-метилфенил)-амин;
(6-аминометил-7-метоксихиназолин-4-ил)-(3-этинилфенил)-амин;
(6-аминометил-7-метоксихиназолин-4-ил)-(3-этинилфенил)-амин;
(6-аминокарбонилметил-7-метоксихиназолин-4-ил)-(3-этинилфенил)-амин;
(6-аминокарбонилэтил-7-метоксихиназолин-4-ил)-(3-этинилфенил)-амин;
(6-аминокарбонилметил-7-этоксихиназолин-4-ил)-(3-этинилфенил)-амин;
(6-аминокарбонилэтил-7-этоксихиназолин-4-ил)-(3-этинилфенил)-амин;
(6-аминокарбонилметил-7-изопропоксихиназолин-4-ил)-(3-этинилфенил)-амин;
(6-аминокарбонилметил-7-пропоксихиназолин-4-ил)-(3-этинилфенил)-амин;
(6-аминокарбонилметил-7-метоксихиназолин-4-ил)-(3-этинилфенил)-амин;
(6-аминокарбонилэтил-7-изопропоксихиназолин-4-ил)-(3-этинилфенил)-амин; и
(6-аминокарбонилэтил-7-пропоксихиназолин-4-ил)-(3-этинилфенил)-амин;
(6,7-диэтоксихиназолин-1-ил)-(3-этинилфенил)-амин;
(3-этинилфенил)-[6-(2-гидроксиэтокси)-7-(2-метоксиэтокси)-хиназолин-1-ил]-амин;
[6,7-бис-(2-гидроксиэтокси)-хиназолин-1-ил]-(3-этинилфенил)-амин;
[6,7-бис-(2-метоксиэтокси)-хиназолин-1-ил]-(3-этинилфенил)-амин;
(6,7-диметоксихиназолин-1-ил)-(3-этинилфенил)-амин;
(3-этинилфенил)-(6-метансульфониламинохиназолин-1-ил)-амин; и
(6-аминохиназолин-1-ил)-(3-этинилфенил)-амин.
В конкретном варианте осуществления изобретения антагонист EGFR формулы I представляет собой N-(3-этинилфенил)-6,7-бис(2-метоксиэтокси)-4-хиназолинамин. В конкретном варианте осуществления изобретения антагонист EGFR, N-(3-этинилфенил)-6,7-бис(2-метоксиэтокси)-4-хиназолинамин, находится в форме соли HCl. В другом конкретном варианте осуществления изобретения антагонист EGFR, N-(3-этинилфенил)-6,7-бис(2-метоксиэтокси)-4-хиназолинамин, находится в основном в форме гомогенного кристаллического полиморфа (описан как полиморф В в WO 01/34574), который демонстрирует на порошковой рентгенограмме характерные пики, выраженные в градусах 2-тета, приблизительно при 6,26, 12,48, 13,39, 16,96, 20,20, 21,10, 22,98, 24,46, 25,14 и 26,91. Такая форма полиморфа N-(3-этинилфенил)-6,7-бис(2-метоксиэтокси)-4-хиназолинамин именуется TarcevaTM, а также OSI-774, CP-358774 и эрлотинибом.
Соединения формулы I, фармацевтически приемлемые соли и их пролекарства (далее называемые активными соединениями) могут быть получены в ходе любого процесса, известного для применения в получении химически-связанных соединений. В большинстве случаев, активные соединения могут быть получены из соответствующим образом замещенного хиназолина, используя соответствующим образом замещенный амин, как показано на общей схеме I, представленной в патенте США 5747498:
Схема I

Как показано на Схеме I соответствующий 4-замещенный хиназолин 2, где Х представляет собой подходящую замещаемую уходящую группу, такую как галогено, арилокси, алкилсульфинил, алкилсульфонил как, например, трифторметансульфонилокси, арилсульфинил, арилсульфонил, силокси, циано, пиразоло, триазоло или тетразоло, предпочтительно 4-хлорхиназолин, вступает в реакцию с соответствующим амином или гидрохлоридамином 4 или 5, где R4 представляет собой как описано выше, и Y представляет собой Br, I или трифторметан-сульфонилокси в растворителе, таком как спирт (С1-С6), диметилформамид (DMF), N-метилпирролидин-2-он, хлороформ, ацетонитрил, тетрагидрофуран (THF), 1-4 диоксан, пиридин или другие апротонные растворители. Реакция может осуществляться в присутствии основания, предпочтительно карбоната или гидроксида щелочного металла или щелочноземельного металла или третичного аммониевого основания, такого как пиридин, 2,6-лутидин, коллидин, N-метил-морфолин, триэтиламин, 4-диметиламино-пиридин или N,N-диметиланилин. Эти основания далее относятся к используемым основаниям. Реакционную смесь выдерживают при температуре, находящейся в диапазоне приблизительно от температуры окружающей среды до приблизительно температуры флегмы растворителя, предпочтительно приблизительно от 35°С до приблизительно температуры флегмы, до тех пор, пока не будут определяться остатки 4-галогенхиназолина, обычно приблизительно от 2 до приблизительно 24 часов. Предпочтительно, реакцию проводят в атмосфере инертного газа, такого как сухой азот.
Обычно реагенты вступают в реакцию стехиометрически. Когда основание амина используется для тех соединений, где используется соль (обычно соль HCl) амина 4 или 5, предпочтительным является использование избытка основания амина, как правило, избыточный эквивалент основания амина. (Альтернативно, если основание амина не используется, избыток амина 4 или 5 может быть использован).
Для тех соединений, где используется стерически затрудненный амин 4 (такой как 2-алкил-3-этиниланилин) или очень реакционно-способный 4-галогенхиназолин, предпочтительно использовать t-бутиловый спирт или полярный апротонный растворитель, такой как DMF или N-метилпирролидин-2-он в качестве растворителя.
Альтернативно, 4-замещенный хиназолин 2, где Х представляет собой гидроксил или оксо (и 2-азот гидрогенизован) вступает в реакцию с четыреххлористым углеродом и необязательно замещается триарилфосфином, который необязательно снабжен инертным полимером (например, трифенилфосфином на полимерной подложке, Aldrich Cat. No. 36645-5, которая представляет собой 2% дивинилбензоловый сшитый полистирол, содержащий 3 ммоль фосфора на грамм полимера) в растворителе, таком как тетрахлорид углерода, хлороформ, дихлорэтан, тетрагидрофуран, ацетонитрил или другие апротонные растворители или их смеси. Реакционную смесь выдерживают при температуре, находящейся в диапазоне приблизительно от температуры окружающей среды до температуры флегмы, предпочтительно приблизительно от 35°С до температуры флегмы, от 2 до 24 часов. Эта смесь вступает в реакцию с соответствующим амином или гидрохлоридом амина 4 или 5 либо непосредственно, либо после удаления растворителя, например, путем выпаривания в вакууме, и добавления подходящего альтернативного растворителя, такого как спирт (C1-C6), DMF, N-метилпирролидин-2-он, пиридин или 1-4 диоксан. Затем реакционную смесь выдерживают при температуре, находящейся в диапазоне приблизительно от температуры окружающей среды до температуры флегмы растворителя, предпочтительно приблизительно от 35°С до приблизительно температуры флегмы до тех пор, пока не будет достигнуто полное образование продукта, как правило приблизительно от 2 до приблизительно 24 часов. Предпочтительно, реакцию проводят в атмосфере инертного газа, такого как сухой азот.
Когда соединение 4, где Y представляет собой Br, I или трифторметансульфонилокси, используется в качестве исходного материала в реакции с хиназолином 2, образуется соединение формулы 3, где R1, R2, R3 и Y описаны выше. Соединение 3 превращается в соединения формулы 1, где R4 представляет собой этинил R11, и R11 охарактеризован выше, в реакции с соответствующим реагентом палладия, таким как дихлорид тетракис(трифенилфосфин)палладия или бис(трифенилфосфин)палладия в присутствии соответствующей кислотой Льюиса, такой как хлористая медь, и соответствующего алкина, такого как триметилсилилацетилен, пропаргиловый спирт или 3-(N,N-диметиламин)-пропин в растворителе, таком как диэтиламин или триэтиламин. Соединения 3, где Y представляет собой NH2, могут превращаться в соединения 1, где R4 представляет собой азид путем обработки соединения 3 диазотирующимся веществом, таким как кислота и нитрит (например, уксусная кислота и NaNO2) c последующей обработкой образующегося продукта азидом, таким как NaN3.
Для получения этих соединений Формулы I, где R1 представляет собой амино- или гидроксиаминогруппу, применяется восстановление соответствующего Формуле I соединения, где R1 представляет собой нитро.
Восстановление обычно выполняется любым из многочисленных способов, известных для таких преобразований. Восстановление может быть выполнено, например, гидрогенизацией нитросоединения в реакционно-инертном растворителе в присутствии соответствующего металлического катализатора, такого как палладий, платина или никель. Дополнительным подходящим восстановителем является, например, активированный металл, такой как активированное железо (получаемое путем промывки железного порошка слабым раствором кислоты, такой как соляная кислота). Таким образом, например, восстановление может быть выполнено путем нагревания смеси нитросоединения и активированного металла с концентрированной соляной кислотой в растворителе, таком как смесь воды и спирта, например, метанола или этанола, до температуры в диапазоне, например, от 50°С до 150°С, обычно при или около 70°С. Другим подходящим классом восстановителей являются дитиониты щелочных металлов, такие как дитионит натрия, который может быть использован в (C1-C4)алкановых кислотах, (C1-C6)алканолах, воде или их смесях.
Для получения этих соединений Формулы I, где R2 или R3 включен в первичную или вторичную аминогруппу (другие нежели аминогруппы, предусмотренные для взаимодействия с хиназолином), такие свободные аминогруппы предпочтительно защищены до прохождения вышеописанной реакции с последующим снятием защиты после прохождения вышеописанной реакции с 4-(замещенным)хиназолином 2.
Несколько хорошо известных защитных азотных групп могут быть использованы. Такие группы включают (C1-C6)алкоксикарбонил, необязательно замещенный бензилоксикарбонил, арилоксикарбонил, тритил, винилоксикарбонил, О-нитрофенилсульфонил, дифенилфосфинил, п-толуолсульфонил и бензил. Добавление защитной азотной группы может быть выполнено в хлорированном углеводородном растворителе, таком как метиленхлорид или 1,2-дихлорэтан, или эфирном растворителе, таком как глим, диглим или THF в присутствии или в отсутствие третичного амина, такого как триэтиламин, диизопропилэтиламин или пиридин, предпочтительно триэтиламин, при температуре приблизительно от 0°С до приблизительно 50°С, предпочтительно приблизительно при комнатной температуре. Альтернативно, защитные группы обычно присоединяют, используя условия реакции Шоттена-Баумана.
После описанной выше реакции связывания соединений 2 и 5 защитные группы могут быть удалены путям снятия защиты методами, известными специалистам в данной области техники, такими как обработка трифторуксусной кислотой в хлористом метилене для защищенных трет- бутоксикарбонилом продуктов.
Для описания защитных групп и их использования смотри T. W. Greene and P. G. M. Wuts, «Prorective Groups in Organic Synthesis» Second Ed., John Wiley & Sons, New York, 1991.
Для получения соединений Формулы I, где R1 или R2 представляют собой гидрокси, предпочтительным является расщепление соединения Формулы I, где R1 или R2 представляют собой (C1-C4)алкокси.
Реакция расщепления обычно выполняется любым из многочисленных методов, известных для таких преобразований. Обработка защищенного производного формулы I расплавленным пиридингидрохлоридом (20-30 экв.) при температуре от 150° до 170°С может быть выполнена для О-деалкилирований. Альтернативно, реакция расщепления может быть выполнена, например, путем обработки защищенного производного хиназолина (C1-C4)алкилульфидом щелочного металла, таким как этантиолат натрия или путем обработки диарилфосфидом щелочного металла, таким как дифенилфосфид лития. Реакция расщепления также обычно выполняется путем обработки защищенного производного хиназолина трехгалоидным соединением бора или алюминия, таким как трибромид бора. Такие реакции предпочтительно выполняют в присутствии реакционно-инертного растворителя при подходящей температуре.
Соединения Формулы I, где R1 или R2представляют собой (C1-C4)алкилсульфинильную или (C1-C4)алкилсульфонильную группу предпочтительно получают путем оксисления соединения формулы I, где R1 или R2 представляет собой (C1-C4)алкилсульфанильную группу. Пригодные окислители известны в данной области техники для окисления сульфанила до сульфинила и/или сульфонила, например, перекись водорода, перкислота (такая как 3-хлорпероксибензойная или пероксиуксусная кислота), пероксисульфат щелочного металла (такой как пероксимоносульфат калия), триоксид хрома или газообразный кислород в присутствии платины. Окисление в основном выполняют при, насколько это возможно, мягких условиях, используя стехиометрическое количество окислителя, чтобы уменьшить риск избыточного окисления и повреждения других функциональных групп. Обычно реакцию выполняют в подходящем растворителе, таком как метиленхлорид, хлороформ, ацетон, тетрагидрофуран или трет-бутилметиловый эфир и при температуре в диапазоне приблизительно от -25°С до 50°С, предпочтительно при комнатной температуре или температуре, близкой к комнатной, т.е. в диапазоне от 15° до 35°С. Когда требуется соединение, несущее сульфинильную группу, следует использовать мягкий окислитель, такой как метапериодат натрия или калия, обычно в полярном растворителе, таком как уксусная кислота или этанол. Соединения формулы I, содержащие (C1-C4)алкилсульфонильную группу, могут быть получены путем окисления соответствующего (C1-C4)алкилсульфинильного соединения также как и соответствующего (C1-C4)алкилсульфанильного соединения.
Соединения формулы I, где R1 представляет собой необязательно замещенный (C2-C4)алканоиламино, уреидо, 3-фенилуреидо, бензамидо или сульфонамидо, могут быть получены путем ацилирования или сульфонилирования соответствующего соединения, где R1 представляет собой амино. Пригодные ацилирующие реагенты представляют собой любые вещества, известные в данной области техники для ацилирования амина с образованием ациламина, например, ацилгалогениды, например, (C2-C4)алканоилхлорид или бромид или бензоилхлорид или бромид, ангидриды алкановой кислоты или смешанные ангидриды (например, уксусный ангидрид или смешанный ангидрид, образованный в ходе реакции алкановой кислоты и (C1-C4)алкоксикарбонилгалогенида, например, (C1-C4)алкоксикарбонилхлорида, в присутствии подходящего основания. Для образования этих соединений Формулы I, где R1 представляет собой уреидо или 3-фенилуреидо, подходящим ацилирующим реагентом является, например, цианат, например, цианат щелочного металла, такой как цианат натрия, или изоцианат, такой как фенилизоцианат. N-сульфонилирование может быть выполнено с подходящими сульфонилгалогенидами или сульфонилангидридами в присутствии третичного аммониевого основания. В основном, ацилирование или сульфонилирование выполняют в реакционно-инертном растворителе и при температуре в диапазоне приблизительно от -30°С до 120°С, обычно при комнатной температуре или температуре, близкой к комнатной.
Соединения Формулы I, где R1 представляет собой (C1-C4)алкокси или замещенный (C1-C4)алкокси или R1 представляет собой (C1-C4)алкиламино или замещенный моно-N- или ди-N,N-(C1-C4)алкиламино, получают путем алкилирования, предпочтительно в присутствии подходящего основания, соответствующего соединения, где R1 представляет собой гидрокси или амино, соответственно. Используемый алкилирующий реагент включает алкил или замещенный алкилгалогенид, например, дополнительно замещенный (C1-C4)алкилхлорид, бромид или иодид, в присутствии подходящего основания в реакционно-инертном растворителе и при температуре в диапазоне приблизительно от 10° до 140°С, обычно при комнатной температуре или температуре, близкой к комнатной.
Для образования этих соединений Формулы I, где R1 представляет собой амино-, окси- или циано-замещенный (C1-C4)алкильный заместитель, соответствующее соединение, где R1 представляет собой (C1-C4)алкильный заместитель, несущий группу, которая замещается амино-, алкокси- или цианогруппой, вступает в реакцию с соответствующим амином, спиртом или цианидом, предпочтительно в присутствии подходящего основания. Реакция предпочтительно выполняется в реакционно-инертном растворителе или разбавителе и при температуре в диапазоне приблизительно от 10° до 100°С, предпочтительно при комнатной температуре или температуре, близкой к комнатной.
Соединения Формулы I, где R1 представляет собой карбокси-заместитель или заместитель, который содержит карбоксигруппу, получают путем гидролиза соответствующего соединения, где R1 представляет собой (C1-C4)алкоксикарбонильный заместитель или заместитель, который содержит(C1-C4)алкоксикарбонильную группу. Гидролиз может быть удобно выполнен, например, в щелочных условиях, например, в присутствии гидроксида щелочного металла.
Соединения Формулы I, где R1 представляет собой амино, (C1-C4)алкиламино, ди-[(C1-C4)алкил]амино, пирролидин-1-ил, пиперидино, морфолино, пиперазин-1-ил, 4-(C1-C4)алкилпиперазин-1-ил или (C1-C4)алкилсульфанил, могут быть образованы в ходе реакции, в присутствии подходящего основания, соответствующего соединения, где R1 представляет собой аминовую или тиоловую замещаемую группу с соответствующим амином или тиолом. Реакцию предпочтительно выполняют в реакционно-инертном растворителе или разбавителе и при температуре в диапазоне приблизительно от 10°С до 180°С, обычно в диапазоне от 100° до 150°С.
Соединения Формулы I, где R1 представляет собой 2-оксопирролидин-1-ил или 2-оксопиперидин-1-ил, получают путем кристаллизации, в присутствии подходящего основания, соответствующего соединения, где R1 представляет собой галоген-(C2-C4)алканоиламиногруппу. Реакцию предпочтительно проводят в реакционно-инертном растворителе или разбавителе и при температуре в диапазоне приблизительно от 10°С до 100°С, обычно при комнатной температуре или температуре, близкой к комнатной.
Для получения соединений Формулы I, в которой R1 представляет собой карбамоил, замещенный карбамоил, алканоилокси или замещенный алканоилокси, целесообразно провести карбамоилирование или ацилирование соответствующего соединения, где R1представляет собой гидрокси.
Подходящие ацилирующие реагенты, известные в данной области техники для ацилирования гидроксиарильных групп с образованием алканоилоксиарильных групп, включают, например, (C2-C4)алканоилгалогениды, (C2-C4)алканоилангидриды и смешанные ангидриды, как описано выше, и подходящие замещенные их производные могут быть использованы, обычно в присутствии подходящего основания. Альтернативно, (C2-C4)алкановые кислоты или соответствующим образом замещенные их производные могут быть объединены с соединением Формулы I, где R1 представляет собой гидрокси, с помощью конденсирующего средства, такого как карбодиимид. Для образования этих соединений Формулы I, в которой R1 представляет собой карбамоил или замещенный карбамоил, подходящими карбамоилирующими средством являются, например, цианаты или алкил или арилизоцианаты, обычно в присутствии подходящего основания.
Альтернативно, соответствующие интермедиаты, такие как хлороформат- или карбонилимидазолил-производное соединения Формулы I, в котором R1 представляет собой гидрокси, могут быть образованы, например, путем обработки указанного производного фосгеном (или эквивалентом фосгена) или карбонилдиимидазол. Получаемый промежуточный продукт может затем вступать в реакцию с соответствующим амином или замещенным амином с целью образования необходимых карбамоиловых производных.
Соединения формулы I, где R1 представляет собой аминокарбонил или замещенный аминокарбонил, могут быть получены путем аминолиза соответствующего интермедиата, в котором R1 представляет собой карбокси.
Активация и объединение соединений формулы I, где R1 представляет собой карбокси, могут быть выполнены различными методами, известными специалистам в данной области техники. Подходящие методы включают активацию карбоксила в виде галоидангидрида, азида, симметричного или смешанного ангидрида или активного эфира соответствующей реакционной способности для соединения с нужным амином. Примеры интермедиатов такого типа и их получение и использование при присоединении аминов могут быть найдены с подробным описанием в литературе; например, M. Bodansky and A. Bodansky, "The Practice of Peptide Synthesis", Springer-Verlag, New York, 1984. Образующиеся соединения формулы I могут быть выделены и очищены стандартными методами, такими как удаление растворителя и рекристаллизация или хроматография.
Исходные реагенты для описанной схемы I реакции (например, амины, хиназолины и защитные группы амина) легко доступны или легко могут быть синтезированы специалистами в данной области техники, используя стандартные методы органического синтеза. Например, получение 2,3-дигидро-1,4-бензоксазин-производных описано в R.C. Elderfield, W.H. Todd, S. Gerber, Ch. 12 in "Heterocyclic Compounds", Vol. 6, R. C. Elderfield ed., John Wiley and Sons, Inc., N.Y., 1957. Замещенные соединения 2,3-дигидробензотиазинила описаны R. C. Elderfield и E. E. Harris в главе 13 тома 6 книги Elderfield "Heterocyclic Compounds".
В другом конкретном варианте осуществления изобретения, антагонист EGFR имеет общую формулу II, как описано в патенте США № 5457105, включенном в настоящее описание в качестве ссылки:

где:
m равно 1, 2 или 3 и
каждый R1 независимо представляет собой 6-гидрокси, 7-гидрокси, амино, карбокси, карбамоил, уреидо, (1-4С)алкоксикарбонил, N-(1-4C)алкилкарбамоил, N,N-ди-[(1-4C)алкил]карбамоил, гидроксиамино, (1-4С)алкоксиамино, (2-4С)алканоилоксиамино, трифторметокси, (1-4С)алкил, 6-(1-4С)алкокси, 7-(1-4С)алкокси, (1-3С)алкилендиокси, (1-4С)алкиламино, ди-1[(1-4C)алкил]амино, пирролидин-1-ил, пиперидино, морфолино, пиперазин-1-ил, 4-(1-4С)алкилпиперазин-1-ил, (1-4С)алкилтио, (1-4С)алкилсульфинил, (1-4С)алкилсульфонил, бромметил, дибромметил, гидрокси-(1-4С)алкил, (2-4С)алканоилокси-(1-4С)алкил, (1-4С)алкокси-(1-4С)алкил, карбокси-(1-4С)алкил, (1-4С)алкоксикарбонил-(1-4С)алкил, карбамоил-(1-4С)алкил, N-(1-4C)алкилкарбамоил-(1-4С)алкил, N, N,-ди-[(1-4С)алкил]карбамоил-(1-4С)алкил, амино-(1-4С)алкил, (1-4С)алкиламино-(1-4С)алкил, ди-[(1-4С)алкил]амино-(1-4С)алкил, пиперидино-(1-4С)алкил, морфолино-(1-4С)алкил, пиперазин-1-ил-(1-4С)алкил, 4-(1-4С)алкилпиперазин-1-ил-(1-4С)алкил, гидрокси-(2-4С)алкокси-(1-4С)алкил, (1-4С)алкокси-(2-4С)алкокси-(1-4С)алкил, гидрокси-(2-4С)алкиламино-(1-4С)алкил, (1-4С)алкокси-(2-4С)алкиламино-(1-4С)алкил, (1-4С)алкилтио-(1-4С)алкил, гидрокси-(2-4С)алкилтио-(1-4С)алкил, (1-4С)алкокси-(2-4С)алкилтио-(1-4С)алкил, фенокси-(1-4С)алкил, анилино-(1-4С)алкил, фенилтио-(1-4С)алкил, циано-(1-4С)алкил, галоген-(2-4С)алкокси, гидрокси-(2-4С)алкокси, (2-4С)алканоилокси-(2-4С)алкокси, (1-4С)алкокси-(2-4С)алкокси, карбокси-(1-4С)алкокси, (1-4С)алкоксикарбонил-(1-4С)алкокси, карбамоил-(1-4С)алкокси, N-(1-4С)алкилкарбамоил-(1-4С)алкокси, N,N-ди[(1-4С)алкил]карбамоил-(1-4С)алкокси, амино-(2-4С)алкокси, (1-4С)алкиламино-(2-4С)алкокси, ди-[(1-4С)алкил]амино-(2-4С)алкокси, (2-4С)алканоилокси, гидрокси-(2-4С)алканоилокси, (1-4С)алкокси-(2-4С)алканоилокси, фенил-(1-4С)алкокси, фенокси-(2-4С)алкокси, анилино-(2-4С)алкокси, фенилтио-(2-4С)алкокси, пиперидино-(2-4С)алкокси, морфолино-(2-4С)алкокси, пиперазин-1-ил-(2-4С)алкокси, 4-(1-4С)алкилпиперазин-1-ил-(2-4С)алкокси, галоген-(2-4С)алкиламино, гидрокси-(2-4С)алкиламино, (2-4С)алканоилокси-(2-4С)алкиламино, (1-4С)алкокси-(2-4С)алкиламино, карбокси-(1-4С)алкиламино, (1-4С)алкоксикарбонил-(1-4С)алкиламино, карбамоил-(1-4С)алкиламино, N-(1-4С)алкилкарбамоил-(1-4С)алкиламино, N,N-ди-[(1-4С)алкил]карбамоил-(1-4С)алкиламино, амино-(2-4С)алкиламино, (1-4С)алкиламино-(2-4С)алкиламино, ди-1(1-4С)алкил]амино-(2-4С)алкиламино, фенил-(1-4С)алкиламино, фенокси-(2-4С)алкиламино, анилино-(2-4С)алкиламино, фенилтио-(2-4С)алкиламино, (2-4С)алканоиламино, (1-4С)алкоксикарбониламино, (1-4С)алкилсульфониламино, бензамидо, бензенсульфонамидо, 3-фенилуреидо, 2-оксопирролидин-1-ил, 2,5-диоксопирролидин-1-ил, галоген-(2-4С)алканоиламино, гидрокси-(2-4С)алканоиламино, (1-4С)алкокси-(2-4С)алканоиламино, карбокси-(2-4С)алканоиламино, (1-4С)алкоксикарбонил-(2-4С)алканоиламино, карбамоил-(2-4С)алканоиламино, N-(1-4С)алкилкарбамоил-(2-4С)алканоиламино, N,N-ди-[(1-4C)алкил]карбамоил-(2-4С)алканоиламино, амино-(2-4С)алканоиламино, (1-4С)алкиламино-(2-4С)алканоиламино или ди-[(1-4C)алкил]амино-(2-4С)алканоиламино, и где указанный бензамидо или бензенсульфонамидо-заместитель или любая анилино, фенокси или фенил-группа в заместителе R1 может необязательно иметь один или два галоген, (1-4С)алкил или (1-4С)алкокси заместителя;
n равно 1 или 2 и
каждый R2 независимо представляет собой водород, гидрокси, галоген, трифторметил, амино, нитро, циано, (1-4С)алкил, (1-4С)алкокси, (1-4С)алкиламино, ди-[(1-4C)алкил]амино, (1-4С)алкилтио, (1-4С)алкилсульфинил или (1-4С)алкилсульфонил; или их фармацевтически приемлемые соли; причем 4-(4'-гидроксианилино)-6-метоксихиназолин, 4-(4,-гидроксианилино)-6,7-метилендиоксихиназолин, 6-амино-4-(4'-аминоанилино)хиназолин, 4-анилино-6-метилхиназолин или их хлористоводородные соли и 4-анилино-6,7-диметоксихиназолин или его хлористоводородные соли исключены.
В конкретном варианте осуществления изобретения, антагонист EGFR является соединением в соответствии с формулой II, выбранной из группы, состоящей из:
4-(3'-хлор-4'-фторанилино)-6,7-диметоксихиназолин;
4-(3',4'-дихлоранилино)-6,7-диметоксихиназолин;
6,7-диметокси-4-(3'-нитроанилино)-хиназолин;
6,7-диэтокси-4-(3'-метиланилино)-хиназолин;
6-метокси-4-(3'-метиланилино)-хиназолин;
4-(3'-хлоранилино)-6-метоксихиназолин;
6,7-этилендиокси-4-(3'-метиланилино)-хиназолин;
6-амино-7-метокси-4-(3'-метиланилино)-хиназолин;
4-(3'-метиланилино)-6-уреидохиназолин;
6-(2-метоксиэтоксиметил)-4-(3'-метиланилино)-хиназолин;
6,7-ди-(2-метоксиэтокси)-4-(3'-метиланилино)-хиназолин;
6-диметиламино-4-(3'-метиланилино)хиназолин;
6-бензамидо-4-(3'-метиланилино)хиназолин;
6,7-диметокси-4-(3'-трифторметиланилино)-хиназолин;
6-гидрокси-7-метокси-4-(3'-метиланилино)-хиназолин;
7-гидрокси-6-метокси-4-(3'-метиланилино)-хиназолин;
7-амино-4-(3'-метиланилино)-хиназолин;
6-амино-4-(3'-метиланилино)хиназолин;
6-амино-4-(3'-хлоранилино)-хиназолин;
6-ацетамидо-4-(3'-метиланилино)-хиназолин;
6-(2-метоксиэтиламино)-4-(3'-метиланилино)-хиназолин;
7-(2-метоксиацетамидо)-4-(3'-метиланилино)-хиназолин;
7-(2-гидроксиэтокси)-6-метокси-4-(3'-метиланилино)-хиназолин;
7-(2-метоксиэтокси)-6-метокси-4-(3'-метиланилино)-хиназолин;
6-амино-4-(3'-метиланилино)-хиназолин.
Производное хиназолина формулы II или его фармацевтически приемлемые соли могут быть получены любым способом, известным для применения с целью получения химически-связанных соединений. Подходящий способ, например, проиллюстрирован в патенте США № 4322420. Необходимые исходные реагенты могут быть коммерчески доступны или получены стандартными методами органической химии.
(а) Реакция, обычно в присутствии подходящего основания, хиназолина (i), где Z представляет собой замещаемую группу, с анилином (ii).

Используемая замещаемая группа Z представляет собой, например, галоген, алкокси, арилокси или сульфонилокси группу, например, хлор, бром, метокси, фенокси, метансульфонилокси или толуол-п-сульфонилокси группу.
Пригодным основанием является, например, органическое основание амина, такое как, например, пиридин, 2,6-лутидин, коллидин, 4-диметиламинопиридин, триэтиламин, морфолин, N-метилморфолин или диазабицикло[5.4.0]ундец-7-ен, или, например, карбонат или гидроксид щелочного или щелочно-земельного металла, например, карбонат натрия, карбонат калия, карбонат кальция, гидроксид натрия или гидроксид калия.
Реакцию предпочтительно проводят в присутствии подходящего инертного растворителя или разбавителя, например алканола или сложного эфира, такого как метанол, этанол, изопропанол или этилацетат, галогенизированный растворитель, такой как метиленхлорид, хлороформ или тетрахлорид углерода, простой эфир, такой как тетрагидрофуран или 1,4-диоксан, ароматический растворитель, такой как толуол, или биполярный апротонный растворитель, такой как N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидин-2-он или диметилульфоксид. Реакцию обычно проводят при температуре в диапазоне, например, от 10° до 150°С, предпочтительно в диапазоне от 20° до 80°С.
Производное хиназолина формулы II может быть получено в ходе этого процесса в форме свободного основания или альтернативно может быть получено в форме соли с кислотой, имеющей формулу H-Z, где Z имеет смысловое содержание, определенное выше. Когда необходимо получить свободное основание из соли, соль может быть обработана подходящим основанием, как определено выше, используя стандартную процедуру.
(b) Для получения тех соединений формулы II, где R1 или R2 представляет собой гидрокси, расщепление производного хиназолина формулы II, где R1 или R2 представляет собой (1-4С)алкокси.
Реакция расщепления может удобно быть проведена любым из многочисленных способов, известных для таких преобразований. Реакция может быть выполнена, например, путем обработки производного хиназолина (1-4С)алкилсульфидом щелочного металла, таким как этантиолат натрия или, например, путем обработки диарилфосфидом щелочного металла, таким как дифенилфосфид лития. Альтернативно, реакция расщепления может удобно быть проведена, например, путем обработки производного хиназолина трехгалоидным бором или алюминием, таким как трибромид бора. Такие реакции предпочтительно проводят в присутствии подходящего инертного растворителя или разбавителя, как определено выше, и при подходящей температуре.
(с) Для получения тех соединений формулы II, где R1 или R2 представляет собой (1-4С)алкилсульфинильную или (1-4С)алкилсульфонильную группу, окисление производного хиназолина формулы II, где R1 или R2 представляет собой (1-4С)алкилтиогруппу.
Подходящим окислителем является, например, любое вещество, известное в данной области техники для окисления тио до сульфинила и/или сульфонила, например, перекись водорода, перкислота (такая как 3-хлорпероксибензойная или пероксиуксусная кислота), пероксисульфат щелочного металла (такой как пероксимоносульфат калия), триоксид хрома или газообразный кислород в присутствии платины. Окисление в основном выполняют при, насколько это возможно, мягких условиях, используя стехиометрическое количество окислителя, чтобы уменьшить риск избыточного окисления и повреждения других функциональных групп. В основном реакцию выполняют в подходящем растворителе или разбавителе, таком как метиленхлорид, хлороформ, ацетон, тетрагидрофуран или трет-бутилметиловый эфир и при температуре, например, в диапазоне от -25°С до 50°С, обычно при комнатной температуре или температуре, близкой к комнатной, то есть в диапазоне от 15° до 35°С. Когда требуется соединение, несущее сульфинильную группу, мягкий окислитель может быть также использован, например, метапериодат натрия или калия, обычно в полярном растворителе, таком как уксусная кислота или этанол. Следует понимать, что когда требуется соединение формулы II, содержащее (1-4С)алкилсульфонильную группу, оно может быть получено путем окисления соответствующего (1-4С)алкилсульфинильного соединения, а также соответствующего(1-4С)алкилтиосоединения.
(d) Для получения тех соединений формулы II, где R1 представляет собой амино, восстановление производного хиназолина формулы I, где R1 представляет собой нитро.
Восстановление может быть удобно выполнено любым из многочисленных способов, известных для таких преобразований. Восстановление может быть выполнено, например, гидрогенизацией раствора нитросоединения в инертном растворителе или разбавителе, как определено выше, в присутствии металлического катализатора, такого как палладий или платина. Дополнительным подходящим восстановителем, например, является активированный металл, такой как активированное железо (получаемое путем промывки железного порошка слабым раствором кислоты, такой как соляная кислота). Таким образом, например, восстановление может быть выполнено путем нагревания смеси нитросоединения и активированного металла в подходящем растворителе или разбавителе, таком как смесь воды и спирта, например, метанола или этанола, до температуры в диапазоне, например, от 50°С до 150°С, обычно при или около 70°С.
(е) Для получения тех соединений формулы II, где R1 представляет собой (2-4С)алканоиламино или замещенный (2-4С)алканоиламино, уреидо, 3-фенилуреидо или бензамидо, или R2 представляет собой ацетамидо или бензамидо, ацилирование производного хиназолина формулы II, где R1 или R2представляет собой амино.
Пригодным ацилирующим реагентом является, например, любой известный реагент, известный в данной области техники для ацилирования амино с образованием ациламино, например, ацилгалогенида, например, (2-4С)алканоилхлорида или бромида или бензоилхлорида или бромида, обычно в присутствии подходящего основания, как определено выше, ангидрида алкановой кислоты или смешанного ангидрида, например, ангидрида (2-4С)алкановой кислоты, такого как уксусный ангидрид или смешанный ангидрид, образованный в ходе реакции алкановой кислоты и (1-4С)алкоксикарбонилгалогенида, например, (1-4С)алкоксикарбонилхлорида, в присутствии подходящего основания, как определено выше. Для получения этих компонентов формулы II, где R1 представляет собой уреидо или 3-фенилуреидо, подходящим ацилирующим реагентом является, например, цианат, например, цианат щелочного металла, такой как цианат натрия или, например, изоцианат, такой как фенилизоцианат. В основном, ацилирование выполняют в подходящем инертном растворителе или разбавителе, как определено выше, и при температуре в диапазоне, например, от -30° до 120°С, обычно при комнатной температуре или температуре, близкой к комнатной.
(f) Для получения тех соединений формулы II, где R1представляет собой (1-4С)алкокси или замещенный (1-4С)алкокси или R1представляет собой (1-4С)алкиламино или замещенный (1-4С)алкиламино, алкилирование, предпочтительно в присутствии подходящего основания, как определено выше, производного хиназолина формулы II, где R1представляет собой гидрокси или амино в соответствующих случаях.
Подходящим алкилирующим реагентом является, например, любое вещество, известное в данной области техники для алкилирования гидрокси с образованием алкокси или замещенного алкокси, или для алкилирования амино с образованием алкиламино или замещенного алкиламино, например, алкила или замещенного алкилгалогенида, например, (1-4С)алкилхлорида, бромида или иодида или замещенного (1-4С)алкилхлорида, бромида или иодида, в присутствии подходящего основания, как определено выше, в подходящем инертном растворителе или разбавителе, как определено выше, и при температуре в диапазоне, например, от 10° до 140°С, обычно при комнатной температуре или температуре, близкой к комнатной.
(g) Для получения тех соединений формулы II, где R1представляет собой карбокси-заместитель или заместитель, который содержит карбоксигруппу, гидролиз производного хиназолина формулы II, где R1представляет собой (1-4С)алкоксикарбонил-заместитель или заместитель, который содержит (1-4С)алкоксикарбоксильную группу.
Гидролиз может быть удобно выполнен, например, в щелочных условиях.
(h) Для получения тех соединений формулы II, где R1представляет собой амино-, окси-, тио- или циано-замещенный (1-4С)алкилзаместитель, реакция, предпочтительно в присутствии подходящего основания, как определено выше, производного хиназолина формулы II, где R1представляет собой (1-4С)алкил-заместитель, несущий замещаемую группу, как определено выше, с соответствующим амином, спиртом, тиолом или цианидом.
Реакция предпочтительно выполняется в подходящем инертном растворителе или разбавителе, как определено выше, и при температуре в диапазоне, например, от 10° до 100°С, обычно при комнатной температуре или температуре, близкой к комнатной.
Когда необходима фармацевтически приемлемая соль производного хиназолина формулы II, она может быть получена, например, в ходе реакции указанного соединения с, например, подходящей кислотой, используя стандартную методику.
В конкретном варианте осуществления изобретения, антагонист EGFR представляет собой соединение в соответствии с формулой II', как рассмотрено в патенте США № 5770599, приведенном в настоящем описании в качестве ссылки,:

где:
n равно 1, 2 или 3;
каждый R2 независимо представляет собой галоген или трифторметил
R3 представляет собой (1-4С)алкокси; и
R1 представляет собой ди-[(1-4С)алкил]амино-(2-4С)алкокси, пирролидин-1-ил-(2-4С)алкокси, пиперидино-(2-4С)алкокси, морфолино-(2-4С)алкокси, пиперазин-1-ил-(2-4С)алкокси, 4-(1-4С)алкилпиперазин-1-ил-(2-4С)алкокси, имидазол-1-ил-(2-4С)алкокси, ди-[(1-4С)алкокси-(2-4С)алкил]амино-(2-4С)алкокси, тиаморфолино-(2-4С)алкокси, 1-оксотиаморфолино-(2-4С)алкокси или 1,1-диоксотиаморфолино-(2-4С)алкокси, и где любой из вышеупомянутых R1-заместителей, включающий СН2 (метиленовую) группу, которая не присоединена к атому N или О, необязательно несет на указанной CH2-группе гидрокси-заместитель;
или их фармацевтически приемлемые соли.
В определенном варианте осуществления изобретения, антагонист EGFR представляет собой соединение в соответствии с формулой II', выбранной из группы, состоящей из:
4-(3'-хлор-4'-фторанилино)-7-метокси-6-(2-пирролидин-1-илэтокси)-хиназолин;
4-(3'-хлор-4'-фторанилино)-7-метокси-6-(2-морфолинэтокси)-хиназолин;
4-(3'-хлор-4'-фторанилино)-6-(3-диэтиламинопропокси)-7-метоксихиназолин;
4-(3'-хлор-4'-фторанилино)-7-метокси-6-(3-пирролидин-1-илпропокси)-хиназолин;
4-(3'-хлор-4'-фторанилино)-6-(3-диметиламинопропокси)-7-метоксихиназолин;
4-(3',4'-дифторанилино)-7-метокси-6-(3-морфолинопропокси)-хиназолин;
4-(3'-хлор-4'-фторанилино)-7-метокси-6-(3-пиперидинпропокси)-хиназолин;
4-(3'-хлор-4'-фторанилино)-7-метокси-6-(3-морфолинопропокси)-хиназолин;
4-(3'-хлор-4'-фторанилино)-6-(2-диметиламиноэтокси)-7-метоксихиназолин;
4-(2',4'-дифторанилино)-6-(3-диметиламинопропокси)-7-метоксихиназолин;
4-(2',4'-дифторанилино)-7-метокси-6-(3-морфолинопропокси)-хиназолин;
4-(3'-хлор-4'-фторанилино)-6-(2-имидазол-1-илэтокси)-7-метоксихиназолин;
4-(3'-хлор-4'-фторанилино)-6-(3-имидазол-1-илпропокси)-7-метоксихиназолин;
4-(3'-хлор-4'-фторанилино)-6-(2-диметиламиноэтокси)-7-метоксихиназолин;
4-(2',4'-дифторанилино)-6-(3-диметиламинопропокси)-7-метоксихиназолин;
4-(2',4'-дифторанилино)-7-метокси-6-(3-морфолинопропокси)-хиназолин;
4-(3'-хлор-4'-фторанилино)-6-(2-имидазол-1-илэтокси)-7-метоксихиназолин; и
4-(3'-хлор-4'-фторанилино)-6-(3-имидазол-1-илпропокси)-7-метоксихиназолин.
В конкретном варианте осуществления изобретения антагонист EGFR представляет собой соединение в соответствии с формулой II', являющееся 4-(3'-хлор-4'-фторанилино)-7-метокси-6-(3-морфолинопропокси)-хиназолином, альтернативно называемый как ZD 1839, гефитиниб и Iressa®.
Производное хиназолина формулы II', или его фармацевтически приемлемые соли, могут быть получены в ходе любого процесса, известного для применения в получении химически-связанных соединений. Соответствующие процессы включают, например, таковые, проиллюстрированные в US5616582, US5580870, US5475001 и US5569658. Если не утверждается иное, n, R2, R3 и R1 несут в себе любое смысловое содержание, определенное выше для производного хиназолина формулы II'. Необходимые исходные реагенты могут быть коммерчески доступны или получены стандартными методами органической химии.
(а) Реакция, обычно в присутствии подходящего основания, хиназолина (iii), где Z представляет собой замещаемую группу, с анилином (iv)

Подходящая замещаемая группа Z представляет собой, например, галоген, алкокси, арилокси или сульфонилокси группу, например, хлор, бром, метокси, фенокси, метансульфонилокси или толуол-4-сульфонильную группу.
Пригодным основанием является, например, органическое основание амина, такое как, например, пиридин, 2,6-лутидин, коллидин, 4-диметиламинопиридин, триэтиламин, морфолин, N-метилморфолин или диазабицикло[5.4.0]ундец-7-ен, или, например, карбонат или гидроксид щелочного или щелочно-земельного металла, например, карбонат натрия, карбонат калия, карбонат кальция, гидроксид натрия или гидроксид калия. Альтернативно, пригодным основанием является, например, амид щелочного металла или щелочно-земельного металла, например, амид натрия или бис(триметилсилил)амид натрия.
Реакцию предпочтительно проводят в присутствии подходящего инертного растворителя или разбавителя, например, алканола или эфира, такого как метанол, этанол, изопропанол или этилацетат, галогенизированного растворителя, такого как метиленхлорид, хлороформ или тетрахлорид углерода, эфира, такого как тетрагидрофуран или 1,4-диоксан, ароматического растворителя, такого как толуол, или биполярного апротонного растворителя, такого как N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидин-2-он или диметилсульфоксид. Реакцию обычно проводят при температуре в диапазоне, например, от 10° до 150°С, предпочтительно в диапазоне от 20° до 80°С.
Производное хиназолина формулы II' может быть получено в ходе этого процесса в форме свободного основания или альтернативно оно может быть получено в форме соли с кислотой формулы H-Z, где Z имеет смысловое содержание, определенное выше. Когда требуется получить свободное основание из соли, соль может быть обработана подходящим основанием, как определено выше, используя стандартную методику.
(b) Для получения тех соединений формулы II', где R1 представляет собой аминозамещенную (2-4С)алкокси группу, алкилирование, обычно в присутствии подходящего основания, как определено выше, производного хиназолина формулы II', где R1 представляет собой гидроксигруппу.
Подходящим алкилирующим реагентом является, например, любое вещество, известное в данной области техники для алкилирования гидрокси с образованием аминозамещенного алкокси, например, аминозамещенного алкилгалогенида, например, аминозамещенного (2-4С)алкилхлорида, бромида или иодида, в присутствии подходящего основания, как определено выше, в пригодном инертном растворителе или разбавителе, как определено выше, и при температуре в диапазоне, например, от 10° до 140°С, обычно при или около 80°С.
(с) Для получения тех соединений формулы II', где R1 представляет собой аминозамещенную (2-4С)алкоксигруппу, реакция, обычно в присутствии подходящего основания, как определено выше, соединения формулы II', где R1 представляет собой гидрокси-(2-4С)алкоксигруппу, или его реакционно-способное производное, с соответствующим амином.
Подходящим реакционно-способным производным соединения формулы II', где R1 представляет собой гидрокси-(2-4С)алкоксигруппу, является, например, галоген- или сульфонилокси-(2-4С)алкокси группа, такая как бром- или метансульфонилокси-(2-4С)алкокси группа.
Реакция предпочтительно выполняется в присутствии подходящего инертного растворителя или разбавителя, как определено выше, и при температуре в диапазоне, например, от 10° до 150°С, обычно при или около 50°С.
(d) Для получения тех соединений формулы II', где R1 представляет собой гидрокси-амино-(2-4С)алкокси группу, реакция соединения формулы II', где R1 представляет собой 2,3-эпоксипропокси или 3,4-эпоксибутокси группу с соответствующим амином.
Реакцию предпочтительно выполняют в присутствии подходящего инертного растворителя или разбавителя, как определено выше, и при температуре в диапазоне, например, от 10° до 150°С, обычно при или около 70°С.
Когда необходима фармацевтически приемлемая соль производного хиназолина формулы II', например, моно- или ди-кислотно-аддитивная соль производного хиназолина формулы II', она может быть получена, например, в ходе реакции указанного соединения с, например, подходящей кислотой, используя стандартную методику.
В конкретном варианте осуществления изобретения, антагонист EGFR является соединением в соответствии с формулой III, как рассмотрено в WO9935146, включенного в настоящее описание в качестве ссылки:

или соль или сольват вышеперечисленного; где
X представляет собой N или CH;
Y представляет собой CR1 и V представляет собой N;
или Y представляет собой N и V представляет собой CR1;
или Y представляет собой CR1 и V представляет собой CR2;
или Y представляет собой CR2 и V представляет собой CR1;
R1 представляет собой CH3SO2CH2CH2NHCH2-Ar-, где Ar выбран из фенила, фурана, тиофена, пиррола и тиазола, каждый из которых может дополнительно быть замещенным одной или двумя галоген, С1-4алкил или С1-4алкокси группами;
R2 выбран из группы, включающей водород, галоген, гидрокси, С1-4алкил, С1-4алкокси, С1-4алкиламино и ди[С1-4алкил]амино;
U представляет собой фенил, пиридил, 3Н-имидазолил, индолил, изоиндолил, индолинил, изоиндолинил, 1Н-индазолил, 2,3-дигидро-1Н-индазолил, 1Н-бензимидазолил, 2,3-дигидро-1Н-бензимидазолил или 1Н-бензотриазолил группу, замещенную R3 группой и дополнительно замещенную по меньшей мере одной независимо выбранной R4 группой;
R3 выбран из группы, включающей бензил, галоген-, дигалоген- и тригалогенбензил, бензоил, пиридилметил, пиридилметокси, фенокси, бензилокси, галоген-, дигалоген- и тригалогенбензилокси и бензенсульфонил; или R3 представляет собой тригалогенметилбензил или тригалогенметилбензилокси;
или R3 представляет собой группу формулы

где каждый R5 независимо выбран из галогена, С1-4алкил и С1-4алкокси; и n принимает значения от 0 до 3; и
каждый R4 независимо представляет собой гидрокси, галоген, С1-4алкил, С2-4алкенил, С2-4алкинил, С1-4алкокси, амино, С1-4алкиламино, ди[С1-4алкил]амино, С1-4алкилтио, С1-4алкилсульфинил, С1-4алкилсульфонил, С1-4алкилкарбонил, карбокси, карбамоил, С1-4алкоксикарбонил, С1-4алканоиламино, N-(С1-4алкил)карбамоил, N,N-ди(С1-4алкил)карбамоил, циано, нитро и трифторметил.
В конкретном варианте осуществления изобретения, антагонисты EGFR формулы III исключают:
(1-бензил-1Н-индазол-5-ил)-(6-(5-((2-метансульфонил-этиламино)-метил)-фуран-2-ил)-пиридо[3,4-d]пиримидин-4-ил-амин;
(4-бензилоксифенил)-(6-(5-((2-метансульфонилэтиламино)-метил)-фуран-2-ил)-пиридо[3,4-d]пиримидин-4-ил-амин;
(1-бензил-1Н-индазол-5-ил)-(6-(5-((2-метансульфонилэтиламино)-метил)-фуран-2-ил)-хиназолин-4-ил-амин;
(1-бензил-1Н-индазол-5-ил)-(7-(5-((2-метансульфонил-этиламино)-метил)-фуран-2-ил)-хиназолин-4-ил-амин; и
(1-бензил-1Н-индазол-5-ил)-(6-(5-((2-метансульфонилэтиламино)-метил)-1-метилпиррол-2-ил)-хиназолин-4-ил-амин.
В конкретном варианте осуществления изобретения, антагонист EGFR формулы III выбран из группы, состоящей из:
4-(4-фторбензилокси)-фенил)-(6-(5-((2-метансульфонилэтиламино)метил)-фуран-2-ил)-пиридо[3,4-d]пиримидин-4-ил)-амин;
(4-(3-фторбензилокси)-фенил)-(6-(5-((2-метанульфонилэтиламино)метил)фуран-2-ил)-пиридо[3,4-d]пиримидин-4-ил)-амин;
(4-бензенсульфонил-фенил)-(6-(5-((2-метансульфонилэтиламино)-метил)-фуран-2-ил)-пиридо[3,4d]пиримидин-4-ил)-амин;
(4-бензилоксифенил)-(6-(3-((2-метансульфонилэтиламино)-метил)-фенил)-пиридо[3,4-d]пиримидин-4-ил)-амин;
(4-бензилокси-фенил)-(6-(5-((2-метансульфонилэтиламино)-метил)-фуран-2-ил)хиназолин-4-ил)-амин;
(4-(3-фторбензилоксифенил)-(6-(4-((2-метансульфонилэтиламино)-метил)-фуран-2-ил)-пиридо[3,4-d]пиримидин-4-ил)-амин;
(4-бензилокси-фенил)-(6-(2-((2-метансульфонилэтиламино)-метил)-тиазол-4-ил)хиназолин-4-ил)-амин;
N-{4-[(3-фторбензил)окси]фенил}-6-[5-({[2-(метансульфонил)этил]амино}метил)-2-фурил]-4-хиназолинамин;
N-{4-[(3-фторбензил)окси]-3-метоксифенил}-6-[5-({[2-(метансульфонил)этил]амино}метил)-2-фурил]-4-хиназолинамин;
N-[4-(бензилокси)фенил]-7-метокси-6-[5-({[2-(метансульфонил)этил]амино}метил)-2-фурил]-4-хиназолинамин;
N-[4-бензилокси)фенил]-6-[4-({[2-(метансульфонил)этил]амино}метил)-2-фурил]-4-хиназолинамин;
N-{4-[(3-фторбензил)окси]-3-метоксифенил}-6-[2-({[2-метансульфонил)этил]амино}метил)-1,3-тиазол-4-ил]-4-хиназолинамин;
N-{4-[(3-бромбензил)окси]фенил}-6-[2-({[2-(метансульфонил)этил]амино}метил)-1,3-тиазол-4-ил]-4-хиназолинамин;
N-{4-[(3-фторбензил)окси]фенил)-6-[2-({[2-(метансульфонил)этил]амино}метил)1,3-тиазол-4-ил]-4-хиназолинамин;
N-[4-(бензилокси)-3-фторфенил]-6-[2-({[2-(метансульфонил)этил]амино)метил)-1,3-тиазол-4-ил]-4-хиназолинамин;
N-(1-бензил-1Н-индазол-5-ил)-7-метокси-6-[5-({[2-(метансульфонил)этил]амино)метил)-2-фурил]-4-хиназолинамин;
6-[5-({[2-(метансульфонил)этил]амино)метил)-2-фурил]-N-(4-{[3-(трифторметил)бензил]окси)фенил)-4-хиназолинамин;
N-{3-фтор-4-[(3-фторбензил)окси]фенил}-6-[5-({[2-(метансульфонил)этил]амино)метил)-2-фурил]-4-хиназолинамин;
N-{4-[(3-бромбензил)окси]фенил)-6-[5-({[2-(метансульфонил)этил]амино)метил)-2-фурил]-4-хиназолинамин;
N-[4-(бензилокси)фенил]-6-[3-({[2-(метансульфонил)этил]амино}метил)-2-фурил]-4-хиназолинамин;
N-[1-(3-фторбензил)-1Н-индазол-5-ил]-6-[2-({[2-(метансульфонил)этил]амино}метил)-1,3-тиазол-4-ил]-4-хиназолинамин;
6-[5-({[2-(Метансульфонил)этил]амино)метил)-2-фурил]-N-[4-(бензенсульфонил)фенил]-4-хиназолинамин;
6-[2-({[2-(метансульфонил)этил]амино)метил)-1,3-тиазол-4-ил]-N-[4-(бензенсульфонил)фенил]-4-хиназолинамин;
6-[2-({[2-(метансульфонил)этил]амино}метил)-1,3-тиазол-4-ил]-N-(4-{[3-(трифторметил)бензил]окси)фенил)-4-хиназолинамин;
N-{3-фтор-4-[{3-фторбензил)окси]фенил)-6-[2-({[2-(метансульфонил)этил]амино}метил)-1,3-тиазол-4-ил]-4-хиназолинамин;
N-(1-бензил-1Н-индазол-5-ил)-6-[2-({[2-(метансульфонил)этил]амино)метил)-1,3-тиазол-4-ил]-4-хиназолинамин;
N-(3-фтор-4-бензилоксифенил)-6-[2-({[2-(метансульфонил)этил]амино)метил)-1,3-тиазол-4-ил]-4-хиназолинамин;
N-(3-хлор-4-бензилоксифенил)-6-[2-({[2-(метансульфонил)этил)амино)метил)-1,3-тиазол-4-ил]-4-хиназолинамин;
N-{3-хлор-4-[(3-фторбензил)окси]фенил}-6-[5-({[2-(метансульфонил)этил]амино)метил)-2-фурил]-4-хиназолинамин;
6-[5-({[2-(метансульфонил)этил]амино)метил)-2-фурил]-7-метокси-N-(4-бензенсульфонил)фенил-4-хиназолинамин;
N-[4-(бензилокси)фенил]-7-фтор-6-[5-({[2-(метансульфонил)этил]амино)метил)-2-фурил]-4-хиназолинамин;
N-(1-бензил-1Н-индазол-5-ил)-7-фтор-6-[5-({[2-(метансульфонил)этил]амино}метил)-2-фурил]-4-хиназолинамин;
N-[4-(бензенсульфонил)фенил]-7-фтор-6-[5-({[2-(метансульфонил)этил]амино}метил)-2-фурил]-4-хиназолинамин;
N-(3-трифторметил-4-бензилоксифенил)-6-[5-({[2-(метансульфонил)этил]амино)метил)-4-фурил]-4-хиназолинамин; и
соли и сольваты вышеперечисленного.
В конкретном варианте осуществления изобретения, антагонист EGFR представляет собой: соль дитозилата N-[3-хлор-4-[(3-фторфенил)метокси]фенил]-6-[5-[[[2-(метилсульфонил)этил]амино]метил]-2-фуранил]-4-хиназолинамина (лапатиниб).
В конкретном варианте осуществления изобретения, антагонист EGFR является соединением в соответствии с формулой IV, как рассмотрено в WO0132651, включенном в настоящий документ в качестве ссылки:

где:
m принимает целочисленное значение от 1 до 3;
R1 представляет собой галоген или C1-3алкил;
X1 представляет собой -О-;
R2 выбирают из одной из следующих трех групп:
1) C1-5алкилR3 (где R3 представляет собой пиперидин-4-ил, который может нести один или два заместителя, выбранных из гидрокси, галогена, C1-4алкила, C1-4гидроксиалкила и C1-4алкокси;
2) C2-5алкенилR3 (где R3 представляет собой соединение, как определено в настоящем документе);
3) C2-5алкинилR3 (где R3 представляет собой соединение, как определено в настоящем документе);
и где любая алкильная, алкенильная или алкинильная группа может нести одну или несколько заместителей, выбранных из гидрокси, галогена или амино; или соли вышеперечисленного.
В конкретном варианте осуществления изобретения антагониста EGFR выбирают из группы, состоящей из:
4-(4-хлор-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин;
4-(2-фтор-4-метиланилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин;
4-(4-бром-2-фторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин;
4-(4-хлор-2,6-дифторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин;
4-(4-бром-2,6-дифторанилино)-6-метокси-7-(1-метилпиперидин-4-илметокси)хиназолин;
4-(4-хлор-2-фторанилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолин;
4-(2-фтор-4-метиланилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолин;
4-(4-бром-2-фторанилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолин;
4-(4-хлор-2,6-дифторанилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолин;
4-(4-бром-2,6-дифторанилино)-6-метокси-7-(пиперидин-4-илметокси)хиназолин; и
их фармацевтически приемлемые соли и сольваты.
В конкретном варианте осуществления изобретения, антагонистом EGFR является 4-(4-бром-2-фторанилино)-6-метокси-7-(I-метилпиперидин-4-илметокси)хиназолин (Zactima) и его соль.
Комбинированные способы лечения
Настоящее изобретение описывает комбинированное использование антагониста c-met и антагониста EGFR как часть специфической схемы лечения, предназначенной для обеспечения положительного эффекта комбинированной активности этих терапевтических средств. Положительный эффект комбинирования включает, в числе прочего, фармакокинетическое и фармакодинамическое совместное действие, возникающее вследствие комбинирования терапевтических средств. Настоящее изобретение особенно подходит в лечении рака различных типов на различных стадиях.
Термин рак охватывает ряд пролиферативных нарушений, включая, но ими не ограничиваясь, предраковые новообразования, доброкачественные опухоли и злокачественные опухоли. Доброкачественные опухоли остаются локализованными в участке происхождения и не обладают способностью к инфильтрации, инвазии или метастазированию в удаленные участки. Злокачественные опухоли будут внедряться в и разрушать другие ткани, их окружающие. Они также приобретают способность отделяться от участка возникновения и распространяться в другие части тела (метастазировать), обычно через кровоток или через лимфатическую систему, где расположены лимфатические узлы. Первичные опухоли классифицируют по типу ткани, из которой они возникают; метастатические опухоли классифицируют по типу ткани, из которой происходят раковые клетки. Со временем, клетки злокачественной опухоли становятся более аномальными и менее похожими на нормальные клетки. Это изменение внешнего вида раковых клеток названо степенью злокачественности, и раковые клетки описывают как высокодифференцированные (низкая степень), среднедифференцированные, низкодифференцированные или недифференцированные (высокая степень). Высокодифференцированные клетки имеют вполне нормальный внешний вид и напоминают нормальные клетки, из которых они произошли. Недифференцированные клетки - это те клетки, которые стали настолько аномальными, что невозможно определить происхождение этих клеток.
Система определения стадии рака описывает насколько далеко распространен рак анатомически и пытается распределить пациентов со схожим прогнозом и лечением в одну и ту же группу стадирования. Можно провести ряд исследований, чтобы разобраться в стадии рака, включая биопсию и определенные процедуры получения изображения, такие как рентгенография органов грудной клетки, маммография, остеосцинтиграфия, изображение КТ и изображение ЯМР. Исследование крови и клиническое обследование также используются для оценки общего состояния здоровья пациента и определения, распространился ли рак на определенные органы.
Для определения стадии рака Американский Объединенный Онкологический Комитет первыми ввели буквенные обозначения рака, в частности солидных опухолей, используя TNM систему классификации. Злокачественные опухоли обозначены буквой Т (размер опухоли), N (пальпируемые лимфатические узлы) и/или М (метастазы). Т1, Т2, Т3 и Т4 описывают увеличение размера первичной опухоли; N0, N1, N2, N3 обозначает прогрессивно развивающееся вовлечение лимфатических узлов; и М0 и М1 отражает отсутствие или присутствие отдаленных метастазов.
Во втором методе стадирования, также известном как Общая Группировка по Стадиям или Стадирование Римскими Цифрами, раки подразделяют на стадии от 0 до IV, включая размер первичных опухолей, а также наличие распространения на лимфоузлы и отдаленных метастазов. В этой системе, пациентов группируют по четырем стадиям, обозначенных римскими цифрами от I до IV, или классифицируют как «рецидивирующий». Для некоторых раков, стадия 0 относится к «in situ» или «Tis», таких как протоковая карцинома in situ или лобулярная карцинома in situ для раков молочной железы. Аденомы высокой степени дифференцировки также можно классифицировать как стадия 0. В основном, рак I стадии представляет собой небольшую, локализованную злокачественную опухоль, которая обычно излечима, в то время как IV стадия обычно представляет собой неоперабельный или метастатический рак. Стадия II и III рак обычно является местно-распространенным и/или характеризуется вовлечением локальных лимфатических узлов. Как правило, более высокий номер стадии указывает на более распространенный патологический процесс, включая больший размер опухоли и/или большее распространение рака на близлежащие лимфатические узлы и/или органы, расположенные рядом с первичной опухолью. Эти стадии четко определены, но это определение различно для каждого вида рака и известно специалистам в данной области техники.
Многие раковые регистры, такие как Программа Национального Института Рака по контролю, эпидемиологии и конечным результатам (NCI's Surveillance, Epidemiology, and End Results Program (SEER)), используют общее стадирование. Эта система используется для всех типов рака. Она группирует больных раков по пяти основным категориям:
In situ является начальной стадией рака, который расположен только в тех слоях клеток, из которых он начал свое развитие.
Локализованный представляет собой рак, который не выходит за пределы органа, в котором он начал свое развитие, без признаков распространения.
Местно-распространенный представляет собой рак, который распространяется за пределы исходного (первичного) сайта локализации на близлежащие лимфатические узлы или органы и ткани.
Метастатический представляет собой рак, который распространяется из первичного сайта локализации в отдаленные органы или отдаленные лимфатические узлы.
Неизвестный используется для описания больных раком, для которых нет достаточной информации, чтобы указать стадию.
При этом, раку свойственно рецидивировать в течение месяцев или нескольких лет после того, как была удалена первичная опухоль. Рак, который снова возникает после устранения всей видимой опухоли, называется рецидивирующим заболеванием. Заболевание, которое вновь возникает в области первичной опухоли, называется локально рецидивирующим, а заболевание, которое вновь возникает в виде метастазов, относят к метастатическому рецидиву.
Опухоль может быть солидной опухолью или несолидной или опухолью мягких тканей. Примеры опухолей мягких тканей включают лейкемию (например, хронический миелолейкоз, острый миелобластный лейкоз, острый лимфобластный лейкоз взрослых, острый лимфобластный лейкоз из зрелых В-клеток, хронический лимфоцитарный лейкоз, пролимфоцитарный лейкоз, или волосатоклеточный лейкоз) или лимфома (например, неходжкинская лимфома, Т-клеточная лимфома кожи, или болезнь Ходжкина). Солидная опухоль включает любой рак тканей тела за исключением крови, костного мозга или лимфатической системы. Солидные опухоли могут быть дополнительно подразделены на таковые эпителиально-клеточного происхождения и таковые не эпителиально-клеточного происхождения. Примеры эпителиально-клеточных солидных опухолей включают опухоли желудочно-кишечного тракта, толстой кишки, молочной железы, простаты, легкого, почки, печени, поджелудочной железы, яичника, головы и шеи, ротовой полости, желудка, двенадцатиперстной кишки, тонкой кишки, толстой кишки, анального отверстия, желчного пузыря, губы, носоглотки, кожи, матки, мужских половых органов, органы мочевой системы, мочевого пузыряи и кожи. Солидные опухоли не эпителиального происхождения включают саркомы, опухоли мозга и опухоли костей.
В некоторых вариантах осуществления изобретения, пациент подвергается диагностическому исследованию, например, до и/или во время и/или после терапии. В основном, если выполнено диагностическое исследование, образец может быть получен от пациента, нуждающегося в терапии. В зависимости от того, где индивид имеет рак, образец может быть опухолевым образцом или другим биологическим образцом, таким как биологическая жидкость, включая без ограничений кровь, мочу, слюну, жидкость перитонеального выпота или производные, такие как сыворотка крови или плазма крови и т.п.
Биологический образец настоящего изобретения может быть фиксированным образцом, например, фиксированным в формалине, заключенным в парафин (FFPE) образцом или замороженным образцом.
Различные методы определения экспрессии мРНК или белка включают, но ими не ограничиваются, анализ профиля экспрессии гена, полимеразная цепная реакция (ПЦР), включая количественную ПЦР в реальном времени (кОТ-ПЦР), анализ с применением микроматриц, серийный анализ экспрессии гена (SAGE), MassARRAY, Анализ Экспрессии Гена Массивно-Параллельным Опознавательным Секвенированием (MPSS), протеомику, иммуногистохимию (IHC) и др. Предпочтительно количественному анализу подвергают мРНК. Такой анализ мРНК предпочтительно выполняют, используя технологию полимеразной цепной реакции (ПЦР) или анализ на микроматрицах. Там где применяется ПЦР, предпочтительной формой ПЦР является количественная ПЦР в реальном времени (кОТ-ПЦР). В одном из вариантов осуществления изобретения экспрессия одного или нескольких указанных выше генов считается положительной экспрессией, если она находится на уровне медианы или выше ее, например, по сравнению с другими образцами опухоли того же типа. Срединное значение уровня экспрессии может быть определено фактически в одно и то же время с измерением экспрессии гена или может быть заранее определено.
Этапы репрезентативного протокола для определения профиля экспрессии гена, используя фиксированные, заключенные в парафин ткани в качестве источника РНК, включая выделение мРНК, очистку, удлинение праймера и амплификацию, представлены в различных опубликованных статьях в журналах (например, Godfrey et al. J. Molec. Diagnostics 2: 84-91 (2000); Specht et al., Am. J. Pathol. 158: 419-29 (2001)). В нескольких словах, репрезентативный процесс начинается с нарезания срезов толщиной приблизительно 10 микрограммов образцов ткани опухоли, заключенных в парафин. Затем выделяют РНК, а белок и ДНК удаляют. После анализа концентрации РНК, этапы восстановления и/или амплификации РНК могут быть включены, по мере необходимости, и проводят обратную транскрипцию РНК, используя ген-специфичные промоторы, с последующей ПЦР. В конечном итоге, данные анализируют для идентификации наилучших(его) параметров(а) лечения, доступного для пациента на основе присущего паттерна генной экспрессии, идентифицированного в исследуемом образце опухоли.
Определение экспрессии гена или белка может быть измерено напрямую или опосредованно.
Можно оценить экспрессию или амплификацию c-met и/или EGFR в злокачественной опухоли (напрямую или опосредованно). Для этого имеются в распоряжении различные диагностические/прогностические методы. В одном из вариантов осуществления изобретения, сверхэкспрессия c-met и/или EGFR может быть проанализирована IHC. Срезы заключенной в парафин ткани из биопсии опухоли могут быть подвергнуты IHC-анализу и окрашиванию белка c-met и/или EGFR со следующим критерием интенсивности:
При 0 баллов не наблюдается никакого окрашивания или наблюдается окрашивание мембраны у менее, чем 10% опухолевых клеток.
При 1+ балле едва различимое окрашивание мембраны определяется у более, чем 10% опухолевых клеток. Окрашивается только часть мембран клеток.
При 2+ баллах наблюдается полное окрашивание мембран (от слабой до умеренной интенсивности) у более, чем 10% опухолевых клеток.
При 3+ баллах наблюдается полное окрашивание мембран (от умеренной до сильной интенсивности) у более, чем 10% опухолевых клеток.
В некоторых вариантах осуществления изобретения, те опухоли, которые имеют от 0 до 1+ балла при оценке сверхэкспрессии c-met и/или EGFR, могут быть охарактеризованы как не обладающие сверхэкспрессией c-met и/или EGFR, в то время как те опухоли, которые имеют от 2+ до 3+ баллов могут быть охарактеризованы как сверхэкспрессирующие c-met и/или EGFR.
В некоторых вариантах осуществления изобретения, опухоли, обладающие сверхэкспрессией c-met и/или EGFR, могут быть оценены по иммуногистохимическим баллам, соответствующим числу копий молекул c-met и/или EGFR, экспрессируемых в одной клетке, и могут быть определены биохимически:
0 = 0-10000 копий/клетка,
1+ = по меньшей мере приблизительно 200000 копий/клетка,
2+ = по меньшей мере приблизительно 500000 копий/клетка,
3+ = по меньшей мере приблизительно 2000000 копий/клетка.
Альтернативно или дополнительно FISH-анализ может быть выполнен на фиксированной в формалине, заключенной в парафин опухолевой ткани для определения степени (если имеется) амплификации c-met и/или EGFR в опухоли.
Активация c-met или EGFR может быть определена напрямую (например, путем оценки фосфо-ELISA или другими способами детектирования фосфорилированного рецептора) или опосредованно (например, путем выявления компонентов активированного нисходящего сигнального пути, выявления димеров рецептора (например, гомодимеров, гетеродимеров), определения профилей генной экспрессии и т.п.
Подобным образом, конститутивная активация c-met или EGFR или наличие лиганд-независимого EGFR или c-met может быть выявлена напрямую или опосредованно (например, путем выявления рецепторных мутаций, связанных с конститутивной активностью, путем оценки амплификации рецептора, связанной с конститутивной активностью и т.п.).
Методы определения мутаций нуклеиновой кислоты хорошо известны в данной области техники. Как правило, хотя не обязательно, мишеневую нуклеиновую кислоту в образце амплифицируют, чтобы обеспеченить необходимым количеством вещества для определения того, присутствует ли мутация. Методики амплификации хорошо известны в данной области техники. Например, амплифицируемый продукт может заключать или не заключать в себе всю последовательность нуклеиновой кислоты, кодирующей интересующий белок, поскольку амплифицируемый продукт содержит определенный участок аминокислотной/нуклеиново-кислотной последовательности, где предполагается наличие мутации.
В одном примере, присутствие мутации может быть определено путем взаимодействия нуклеиновой кислоты из образца с нуклеиновой кислотой зонда, способного специфически гибридизоваться с нуклеиновой кислотой, кодирующей мутированную нуклеиновую кислоту, и путем обнаружения указанной гибридизации. В одном из вариантов осуществления изобретения, зонд маркируют для детектирования, например, радиоизотопом (3H,32P,33P и др.), флуоресцентным веществом (родамином, флуоресцеин и др.) или хромогенным веществом. В некоторых вариантах осуществления изобретения, зондом является антисмысловой олигомер, например, PNA, морфолино-фосфорамидат, LNA или 2'-алкоксиалкокси. Зонд может быть размером приблизительно от 8 нуклеотидов до приблизительно 100 нуклеотидов, или приблизительно от 10 до приблизительно 75, или приблизительно от 15 до приблизительно 50, или приблизительно от 20 до приблизительно 30. В другом аспекте, нуклеиново-кислотные зонды настоящего изобретения предоставляются в наборе реагентов для идентификации мутаций c-met в образце, указанный набор, включающий олигонуклеотид, который специфически гибридизуется с сайтом мутации в нуклеиновой кислоте, кодирующей c-met, или расположенным рядом с ним участком. Набор реагентов может дополнительно содержать инструкции по лечению пациентов, имеющих опухоли, содержащие мутации c-met, с помощью антагонистов c-met, основываясь на результатах гибридизационного анализа с использованием набора реагентов.
Мутации также могут быть выявлены путем сравнения электрофоретической подвижности амплифицированной нуклеиновой кислоты с электрофоретической подвижностью соответствующей нуклеиновой кислоты, кодирующей c-met дикого типа. Различие в подвижности указывает на наличие мутации в амплифицированной последовательности нуклеиновой кислоты. Электрофоретическая подвижность может быть определена любым подходящим способом разделения молекул, например, в полиакриламидном геле.
Нуклеиновые кислоты могут также быть проанализированы для определения мутаций, используя Ферментативное Определение Мутаций (EMD) (Del Tito et al, Clinical Chemistry 44:731-739, 1998). EMD использует эндонуклеазу VII резольвазы бактериофага Т4, которая перемещается вдоль двухцепочечной ДНК до тех пор, пока она не выявляет и не расщепляет структурное искажение, вызванное некомплементарностью пары оснований из-за изменений в нуклеиновой кислоте, таких как точечные мутации, вставки и делеции. Выявление двух коротких фрагментов, образованных расщеплением резольвазой, например, с помощью гель-электрофореза, указывает на наличие мутации. Преимуществами метода EMD являются один протокол для идентификации точечных мутаций, делеций или вставок, анализируемых непосредственно после реакций амплификации, устраняя необходимость в очистке образца, укорачивая время гибридизации и повышая отношение сигнала к шуму. Могут быть проанализированы смешанные образцы, содержащие вплоть до 20-кратного избытка нормальных нуклеиновых кислот и фрагменты длиной до 4 тысяч оснований. Однако EMD сканирование не идентифицирует конкретные изменения оснований, происходящие в содержащих мутацию образцах, тем самым, как правило, требуя дополнительных процедур секвенирования для идентификации специфических мутаций, если это необходимо. Фермент CEL I может быть использован подобно эндонуклеазе VII резольвазы Т4, как продемонстрировано в патенте США № 5869245.
Другим простым набором для определения мутаций является тестовая пластина для обратной гибридизации, подобная Haemochromatosis StripAssayTM (Viennalabs http://www.bamburghmarrsh.com/pdf/4220.pdf) для выявления множественных мутаций в генах HFE, TFR2 и FPN1, вызывающих гемохроматоз. Такой анализ основан на сайт-специфической гибридизации вслед за амплификацией с помощью ПЦР. Для анализа единичных мутаций может быть использована система детектирования основанная на микроплашках, в то время как для анализа множественных мутаций, тестовые пластины могут быть использованы в качестве «макро-матриц». Наборы могут включать готовые к использованию реактивы для получения образца, амплификации и определения мутации. Протоколы по мультиплексной амплификации удобны и позволяют тестировать образцы с очень ограниченным объемом. Используя эффективный формат StripAssay, анализ двадцати и большего количества мутаций может быть завершен менее чем за пять часов без дорогостоящего оборудования. ДНК выделяют из образца и мишеневую нуклеиновую кислоту амплифицируют in vitro (например, с помощью ПЦР) и помечают биотином, обычно в одной («мультиплексной») реакции амплификации. Продукты амплификации затем селективно гибридизуются с олигонуклеотидными зондами (дикого типа и мутант-специфичного), иммобилизованными на твердой подложке, такой как тестовая пластина, в которой зонды иммобилизованы в виде параллельных линий или полос. Связанные биотинилированные ампликоны выявляют, используя стрептавидин-щелочную фосфатазу и цветные субстраты. Такой анализ может выявить все или любой подкласс мутаций настоящего изобретения. С учетом полосы определенного мутантного зонда возможен один из трех сигнальных паттерна: (i) полоса только для зонда дикого типа, что означает нормальную последовательность нуклеиновой кислоты, (ii) полосы как для зонда дикого типа, так и для мутантного зонда, что означает гетерозиготный генотип, и (iii) полоса только для мутантного зонда, что означает гомозиготный мутантный генотип. Соответственно, в одном аспекте, изобретение относится к способу определения мутаций изобретения, включающему выделение и/или амплификацию мишеневой последовательности нуклеиновой кислоты c-met из образца, такого, при котором продукт амплификации включает лиганд, взаимодействие продукта амплификации с зондом, который включает детектируемый партнер связывания с лигандом и зонд, способный специфически гибридизоваться с мутацией изобретения, и последующее выявление гибридизации указанного зонда с указанным продуктом амплификации. В одном из вариантов осуществления изобретения, лигандом является биотин, и партнер связывания включает авидин или стрептавидин. В одном из вариантов осуществления изобретения, партнер связывания включает стрептавидин-щелочную фосфатазу, детектируемая цветными субстратами. В одном из вариантов осуществления изобретения, зонды иммобилизованы, например, на тестовой пластине, где зонды, комплементарные различным мутациям, отделены друг от друга. Альтернативно, амплифицированную нуклеиновую кислоту маркируют изотопом, при этом нет необходимости включать в зонд детектируемую метку.
Изменения гена дикого типа включает все формы мутаций, такие как вставки, инверсии, делеции и/или точечные мутации. В одном из вариантов осуществления изобретения, мутации являются соматическими. Соматические мутации это те, которые происходят только в определенных тканях, например, в опухолевой ткани, и не унаследованы в зародышевой линии. Мутации зародышевой линии могут быть обнаружены в любой ткани организма.
Образец, включающий мишеневую нуклеиновую кислоту, может быть получен методами, хорошо известными в данной области техники и которые подходят для определенного типа и определенной локализации опухоли. Тканевая биопсия обычно используется для получения репрезентативного кусочка ткани опухоли. Альтернативно, клетки опухоли могут быть получены опосредовано в виде тканей/жидкостей, в которых известно или предполагается наличие представляющих интерес опухолевых клеток. Например, образцы участка повреждения раком легкого могут быть получены в ходе резекции, бронхоскопии, тонкоигольной аспирационной биопсии, бронхиальной браш-биопсии или из мокроты, плевральной жидкости или крови. Мутантные гены или продукты гена могут быть определены в опухоли или других образцах организма, таких как моча, мокрота или сыворотка. Те же самые методики, обсуждаемые выше, для определения мутантных мишеневых генов или генных продуктов в опухолевых образцах могут быть применены к другим образцам организма. Раковые клетки отторгаются от опухолей и появляются в таких образцах организма. Путем исследования таких образцов организма, несложная ранняя диагностика может быть проведена для заболеваний, таких как рак. При этом эффективность терапии может быть отслежена проще путем тестирования таких образцов организма на мутантные мишеневые гены или генные продукты.
Способы обогащения тканевых препаратов опухолевыми клетками известны в данной области техники. Например, ткань может быть выделена из парафиновых или замороженных срезов. Раковые клетки могут быть также отделены от нормальных клеток проточной цитометрией или лазерной захватывающей микродиссекцией. Эти, а также другие методы отделения опухолевых от нормальных клеток хорошо известны в данной области техники. Если опухолевая ткань сильно загрязнена нормальными клетками, выявление мутаций может быть более затруднительной, хотя технологии минимизации загрязнения и/или ложноположительных/ложноотрицательных результатов известны, некоторые из которых описаны ниже. Например, образец может быть также проанализирован на присутствие биомаркера (включая мутацию), как известно, ассоциированного с исследуемой опухолевой клеткой, а не с соответствующей нормальной клеткой и наоборот.
Определение точечных мутаций в мишеневых нуклеиновых кислотах может быть выполнено с помощью молекулярного клонирования мишеневых нуклеиновых кислот и секвенирования нуклеиновых кислот, используя методики, хорошо известные в данной области техники. Альтернативно, техника амплификации, такая как полимеразная цепная реакция (ПЦР), может быть использована для амплификации мишеневых последовательностей нуклеиновых кислот непосредственно в препаратах геномной ДНК из опухолевой ткани. Затем может быть определена последовательность нуклеиновой кислоты амплифицированной последовательности и в ней идентифицирована мутация. Техника амплификации хорошо известна в данной области техники, например, полимеразная цепная реакция, как описано в Saiki et al., Science 239:487, 1988; патентах США №№ 4683203 и 4683195.
Следует отметить, что создание и подбор подходящих праймеров являются общепризнанными методиками в данной области техники.
Лигазная цепная реакция, которая известна в данной области техники, также может быть использована для амплификации мишеневых последовательностей нуклеиновой кислоты. Смотри, например, Wu et al., Genomics, Vol. 4, pp. 560-569 (1989). Кроме того, методика, известная как аллель-специфичная ПЦР, также может быть использована. Смотри, например, Ruano and Kidd, Nucleic Acids Research, Vol. 17, p. 8392, 1989. Согласно этой методики, используются праймеры, которые гибридизуются своими 3'-концами с определенной мутацией мишеневой нуклеиновой кислоты. Если отсутствует определенная мутация, продукта амплификации не наблюдают. Амплификационная система для идентификации мутаций (ARMS) также может быть использована, как рассмотрено в Европейской публикации патентной заявки № 0332435 и в Newton et al., Nucleic Acids Research, Vol. 17, p. 7, 1989. Вставки и делеции генов также могут быть выявлены с помощью клонирования, секвенирования и амплификации. Кроме того, зонды полиморфизма длин рестрикционных фрагментов (RFLP) для гена или окружающих маркерных генов могут быть использованы, чтобы подсчитать количество изменений аллеля или вставки в полиморфном фрагменте. Анализ одноцепочечного конформационного полиморфизма (SSCP) также может быть использован для выявления вариантов нуклеотидной замены в аллеле. Смотри, например. Orita et al., Proc. Natl. Acad. Sci. USA Vol. 86, pp. 2766-2770, 1989 и Genomics, Vol. 5, pp. 874-879, 1989. Другие методики для определения вставок и делеций, известные в данной области техники, также могут быть использованы.
Изменение генов дикого типа также могут быть выявлены на основе изменения экспрессии продукта гена дикого типа. Такие продукты экспрессии включают мРНК, а также белковый продукт. Точечные мутации могут быть выявлены путем амплификации и секвенирования мРНК или путем молекулярного клонирования кДНК, синтезированной на мРНК. Последовательность клонированной кДНК может быть определена, используя методики секвенирования ДНК, хорошо известные в данной области техники. кДНК также может быть секвенирована путем полимеразной цепной реакции (ПЦР).
Ошибочно гибридизированные основания нуклеотидов представляют собой дуплексы нуклеиновой кислоты, которые не комплементарны на 100%. Отсутствие общей комплементарности может быть результатом делеций, вставок, инверсий, замен или мутаций со сдвигом рамки считывания. Выявление ошибочно спаренных оснований нуклеотидов может быть использовано для определения точечных мутаций в мишеневой нуклеиновой кислоте. Хотя эти методики могут быть менее чувствительными, чем секвенирование, они проще при анализе большого количества тканевых образцов. Примером методики расщепления ошибочно спаренных оснований нуклеотидов является метод защиты от РНКазы, который подробно описан в Winter et al., Proc. Natl. Acad. Sci. USA, Vol. 82, p. 7575, 1985 и Meyers et al., Science, Vol. 230, p. 1242, 1985. Например, метод по изобретению может включать использование меченого рибозонда, комплементарного человеческой мишеневой нуклеиновой кислоте дикого типа. Рибозонд и мишеневая нуклеиновая кислота, полученная из тканевого образца, подвергаются отжигу (гибридизуются) друг с другом и затем расщепляются ферментом РНКазой А, который способен выявить некоторые ошибочно спаренные основания нуклеотидов в дуплексной структуре РНК. Если ошибочно спаренное основание нуклеотида выявляется РНКазой А, она расщепляет в сайте ошибочно спаренного основания нуклеотида. Таким образом, при разделении отожженного препарата РНК в электрофоретическом гелевом матриксе, если ошибочно спаренное основание нуклеотида было обнаружено и расщеплено РНКазой А, будет визуализирован продукт РНК меньшего размера, чем полноразмерный дуплекс РНК для рибозонда и мРНК или ДНК. Рибозонд не обязательно должен быть полноразмерным как мРНК или ген мишеневой нуклеиновой кислоты, а может быть частью мишеневой нуклеиновой кислоты при условии, что она содержит предположительно мутированный нуклеотид. Если рибозонд содержит только часть мРНК или гена мишеневой нуклеиновой кислоты, желательно использовать несколько таких зондов для проверки всей последовательности мишеневой нуклеиновой кислоты на предмет наличия ошибочно спаренных оснований нуклеотидов.
Подобным образом, ДНК-зонды могут быть использованы для определения ошибочно спаренных оснований нуклеотидов, например, посредством ферментативного или химического расщепления. Смотри, например, Cotton et al., Proc. Natl. Acad. Sci. USA, Vol. 85, 4397, 1988; и Shenk et al., Proc. Natl. Acad. Sci. USA, Vol. 72, p.989, 1975. Альтернативно, ошибочно спаренные основания нуклеотидов могут быть выявлены по сдвигу в электрофоретической подвижности ошибочно спаренных дуплексов относительно комплементарных дуплексов. Смотри, например, Cariello, Human Genetics, Vol. 42, p. 726, 1988. мРНК или ДНК мишеневой нуклеиновой кислоты, которая может содержать мутацию, может быть амплифицирована либо с рибозондами, либо с ДНК-зондами перед гибридизацией. Изменения в ДНК мишеневой нуклеиновой кислоты могут также быть выявлены, используя Саузерн-гибридизацию, особенно, если эти изменения представляют собой значительные перестройки, такие как делеции и вставки.
ДНК-последовательности мишеневой нуклеиновой кислоты, которые были амплифицированы, могут быть также подвергнуты скринингу, используя аллель-специфичные зонды. Эти зонды представляют собой олигомеры нуклеиновой кислоты, каждый из которых содержит участок гена мишеневой нуклеиновой кислоты, несущий известную мутацию. Например, один олигомер может быть длиной приблизительно в 30 нуклеотидов, соответствуя части мишеневой последовательности гена. Путем использования набора таких аллель-специфичных зондов продукты амплификации мишеневой нуклеиновой кислоты могут быть подвергнуты скринингу с целью определения наличия ранее идентифицированной мутации в мишеневом гене. Гибридизация аллель-специфичных зондов с амплифицированными последовательностями мишеневой нуклеиновой кислоты может быть выполнена, например, на нейлоновом фильтре. Гибридизация со специфическим зондом в жестких условиях гибридизации означает наличие той же мутации в опухолевой ткани, как и в аллель-специфичном зонде.
Изменение мишеневых генов дикого типа может также быть выявлено путем проверки изменений соответствующего белка дикого типа. Например, моноклональные антитела, иммунореактивные к продукту мишеневого гена, могут быть использованы для исследования ткани, например, антитело, которое, как известно, связывается с определенным мутированным сайтом продукта гена (белком). Например, используемое антитело может связывать делетированный экзон (например, экзон 14) или связывать конформационный эпитоп, включающий делетированную часть мишеневого белка. Отсутствие узнаваемого антигена будет означать мутацию. Антитела, специфичные к продуктам мутантных аллелей могли бы быть также использованы для определения продукта мутантного гена. Антитела могут быть идентифицированы из библиотек фагового дисплея. Такие иммунологические анализы могут быть выполнены в любом удобном формате, известном в данной области техники. Они включают Вестерн-блоты, иммуногистохимические анализы и анализ ELISA. Любые способы определения измененного белка могут быть использованы для выявления изменения мишеневых генов дикого типа.
Пары праймеров используются для определения нуклеотидной последовательности мишеневой нуклеиновой кислоты, используя методики амплификации нуклеиновой кислоты, такие как полимеразная цепная реакция. Пары одноцепочечных ДНК-праймеров могут подвергаться отжигу с последовательностями внутри последовательности мишеневой нуклеиновой кислоты или рядом с ней, чтобы служить затравкой для амплификации мишеневой последовательности. Аллель-специфичные праймеры также могут быть использованы. Такие праймеры отжигаются только к специфичной мутантной мишеневой последовательности, и, таким образом, будут только амплифицировать продукт в присутствии мутантной мишеневой последовательности в качестве матрицы. Для облегчения последующего клонирования амплифицированных последовательностей праймеры могут иметь последовательности сайта ферментативного расщепления, добавленных к их концам. Такие ферменты и сайты хорошо известны в данной области техники. Сами праймеры могут быть синтезированы, используя методики, хорошо известные в данной области техники. В основном, праймеры могут быть получены, используя олигонуклеотидные синтезаторы, которые коммерчески доступны. Создание специфичных праймеров хорошо известно специалистам в данной области техники.
Зонды нуклеиновых кислот пригодны для многих целей. Они могут быть использованы в Саузерн-гибридизации с геномной ДНК и в методе защиты от РНКазы для определения точечных мутаций, как уже обсуждалось выше. Зонды могут быть использованы для выявления продуктов амплификации мишеневой нуклеиновой кислоты. Они могут также быть использованы для выявления ошибочного спаривания оснований нуклеотидов с геном дикого типа или мРНК, используя другие методики. Ошибочное спаривание оснований нуклеотидов может быть выявлено, используя либо ферменты (например, S1 нуклеазу), химические вещества (например, гидроксиламин или четырехокись осмия и пиперидин), либо изменения в электрофоретической подвижности ошибочно спаренных гибридов при сравнении с полностью комплементарными гибридами. Эти методы хорошо известны в данной области техники. Смотри Novack et al., Proc. Natl. Acad. Sci. USA, Vol. 83, p. 586, 1986. Как правило, зонды комплементарны последовательностям вне киназного домена. Целый набор зондов нуклеиновой кислоты может быть использован для создания набора реагентов по выявлению мутаций в мишеневых нуклеиновых кислотах. Набор реагентов позволяет проводить гибридизацию с протяженным участком исследуемой мишеневой последовательности. Зонды могут перекрываться друг с другом или быть смежными.
Если рибозонд используется для выявления ошибочного спаривания нуклеотидов с мРНК, он, в большинстве случаев, комплементарен мРНК мишеневого гена. Рибозонд, таким образом, представляет собой антисмысловой зонд в том, что он не кодирует соответствующий продукт гена, поскольку он комплементарен смысловой цепи. Рибозонд, как правило, маркируют радиоактивным, колориметрическим или флуорометрическим веществом, что может быть выполнено любым способом, известным в данной области техники. Если рибозонд используется для выявления ошибочного спаривания нуклеотидов с ДНК, он может быть любой полярности, смысловым или анти-смысловым. Подобным образом, ДНК-зонды также могут быть использованы для определения ошибочного спаривания оснований нуклеотидов.
В некоторых случаях, в злокачественной опухоли сверхэкспрессируется или не сверхэкспрессируется рецептор c-met и/или EGFR. Сверхэкспрессия рецептора может быть выявлена в ходе диагностического или прогностического анализа путем оценки повышенных уровней белка рецептора, находящегося на поверхности клетки (например, с помощью иммуногистохимического анализа; IHC). Альтернативно или дополнительно, можно измерить уровень нуклеиновой кислоты, кодирующей рецептор, в клетке, например, с помощью методов флуоресцентной гибридизации in situ (FISH; смотри WO98/45479, опубликованный в октябре 1998 года), саузерн-блоттинга или полимеразной цепной реакции (ПЦР), такой как количественная ПЦР в реальном времени (кОТ-ПЦР). Помимо упомянутых выше анализов, различные анализы in vivo доступны для специалистов в данной области техники. Например, клетки в теле пациента можно подвергнуть действию антитела, которое дополнительно помечено детектируемой меткой, например, радиоактивным изотопом, и связывание антитела с клетками в пациенте может быть оценено, например, внешним сканированием радиоактивности или анализом биопсии, взятой у пациента, предварительно подвергнутого действию антитела.
Химиотерапевтические средства
Комбинированная терапия по настоящему изобретению может дополнительно включать один или несколько химиотерапевтических средств. Комбинированное назначение включает совместное назначение или одновременное назначение, используя отдельные композиции или единую фармацевтическую композицию, и последовательное назначение в любом порядке, где предпочтительным является период времени, когда оба (или все) активных вещества одновременно проявляют свои биологические активности.
Химиотерапевтическое средство в случае назначения обычно назначают в дозах, известных для него, или при желании дозу снижают из-за комбинированного действия лекарственных средств или отрицательных побочных эффектов, присущих назначению антиметаболитного химиотерапевтического средства. Приготовление и режим дозирования для таких химиотерапевтических средств могут быть использованы в соответствии с инструкциями производителя или эмпирическим подбором специалистами в данной области техники.
Различные химиотерапевтические средства, которые могут быть скомбинированы, рассмотрены выше. Предпочтительные химиотерапевтические средства, подлежащие комбинированию, выбраны из группы, состоящей из таксана (включая доцетаксел и паклитаксел), барвинка (такого как винорелбин или винбластин), соединения платины (такого как карбоплатин или цисплатин), ингибитора ароматазы (такого как летрозол, анастразол или экземестан), анти-эстрогена (например, фулвестрант или тамоксифен), этопозид, тиотепа, циклофосфамид, метотрексат, липосомальный доксорубицин, пегилированный липосомальный доксорубицин, капецитабин, гемцитабин, ингибитор COX-2 (например, целекоксиб) или ингибитор протеосомы (например, PS342).
Композиции, дозировки и назначения
Терапевтические средства, используемые в изобретении, будут составлять основу композиции, дозированы и назначены в соответствии с надлежащей медицинской практикой. В этой связи рассматриваемые факторы включают специфические нарушения, подлежащие лечению, конкретный индивид, подлежащий лечению, клиническое состояние индивидуального пациента, причина нарушения, место доставки лекарственного средства, метод назначения, график назначения, взаимодействие лекарственных средств, подлежащих комбинированию, друг с другом, и другие факторы, известные специалистам медицикам.
Терапевтические композиции получаютют, используя стандартные методы, известные в данной области техники, путем смешивания активного ингредиента, имеющего желаемую степень чистоты, с дополнительными физиологически приемлемыми носителями, эксципиентами или стабилизаторами (Remington's Pharmaceutical Sciences (20th edition), ed. A. Gennaro, 2000, Lippincott, Williams & Wilkins, Philadelphia, PA). Приемлемые носители включают физиологический раствор или буферы, такие как фосфатный, лимоннокислый и другие органические кислоты; антиоксиданты, включая аскорбиновую кислоту; низкомолекулярные (менее чем приблизительно 10 аминокислотных остатков) полипептиды; белки, такие как сывороточный альбумин, желатин или иммуноглобулины; гидрофильные полимеры, такие как поливинилпирролидон, аминокислоты, такие как глицин, глутамин, аспарагин, аргинин или лизин; моносахариды, дисахариды и другие карбогидраты, включая глюкозу, маннозу или декстрины; хелатирующие агенты, такие как EDTA; сахарные спирты, такие как маннитол или сорбитол; солеобразующие противоионы, такие как натрий; и/или неионные поверхностно-активные вещества, такие как TWEENTM, PLURONICSTMили PEG.
Необязательно, но предпочтительно, композиция содержит фармацевтически приемлемую соль, предпочтительно хлорид натрия и предпочтительно приблизительно в физиологических концентрациях. Необязательно, композиции изобретения могут содержать фармацевтически приемлемые консерванты. В некоторых вариантах осуществления изобретения концентрация консерванта находится в диапазоне от 0,1 до 2,0%, обычно объем/объем. Пригодные консерванты включают таковые, известные в фармацевтической области техники. Бензиловый спирт, фенол, м-крезол, метилпарабен и пропилпарабен являются предпочтительными консервантами. Необязательно, композиции изобретения могут включать фармацевтически приемлемые поверхностно-активные вещества в концентрации от 0,005 до 0,02%.
Композиция по настоящему изобретению может также содержать более чем одно активное соединение в соответствии с необходимостью лечения определенного симптома, предпочтительно таковые с дополняющими друг друга активностями, которые не оказывают противоположного воздействия друг на друга. Такие молекулы соответственно присутствуют в комбинации в количествах, эффективных для поставленной цели.
Активные ингредиенты могут быть также заключены в микрокапсулу, приготовленную, например, методом коацервации или путем межфазной полимеризации, например, гидроксиметилцеллюлозная или желатиновая микрокапсула и поли-(метилметацилат)-микрокапсула, соответственно, в коллоидных системах доставки лекарственного средства (например, липосомах, альбуминовых микросферах, микроэмульсиях, наночастицах и нанокапсулах) или в макроэмульсиях. Такие методики рассмотрены в Remington's Pharmaceutical Sciences, смотри ранее.
Могут быть получены препараты замедленного высвобождения. Подходящие примеры препаратов замедленного действия включают полупроницаемые матрицы твердых гидрофобных полимеров, содержащих антитело, матрицы которых представлены в форме профилированных изделий, например, пленки или микрокапсулы. Примеры матриц замедленного высвобождения включают полиэфиры, гидрогели (например, поли(2-гидроксэтил-метакрилат) или поли(виниловый спирт)), полиактиды (патент США № 3773919), сополимеры L-глутаминовой кислоты и γ-этил-L-глутамата, неразлагающийся этиленвинилацетат, разлагающиеся кополимеры молочной кислоты-гликолевой кислоты, такие как LUPRON DEPOTTM (инъецируемые микросферы, состоящие из кополимера молочной кислоты-гликолевой кислоты и лейпролид ацетата) и поли-D-(-)-3-гидроксимасляная кислота. Хотя полимеры, такие как этиленвинилацетат и молочная кислота-гликолевая кислота способствуют высбождению молекул в течение 100 дней, определенные гидрогели высвобождают белки в более короткий период времени. Когда инкапсулированные антитела остаются в теле в течение длительного времени, они могут денатурировать или агрегировать в результате воздействия влажности при 37°С, что приводит к потере биологической активности и возможным изменениям иммуногенности. Рациональные стратегии могут быть разработаны для стабилизации в зависимости от вовлеченного механизма. Например, если обнаружено, что механизм агрегации заключается в образовании межмолекулярной S-S связи путем тио-дисульфидной реакции обмена, стабилизация может быть достигнута путем модификации сульфгидрильных остатков, лиофилизации из кислых растворов, контролирования содержания влаги, используя подходящие добавки, и развития композиций специфического полимерного матрикса.
Терапевтические средства изобретения назначают пациенту-человеку в соответствии с известными методами, такими как внутривенное введение в виде болюса или путем непрерывной инфузии в течение периода времени, внутримышечным, интраперитонеальным, интрацереброспинальным, подкожным, внурисуставным, интрасиновиальным, интратекальным, пероральным, местным или ингаляционным способом применения. В случае антагонистов VEGF местное применение особенно желательно, если значительные побочные эффекты или токсичность ассоциирована с VEGF-антагонизмом. Метод ex vivo также может быть использован для терапевтического применения. Методы ex vivo включают трансфецирование или трансдуцирование клеток, полученных от индивида, полинуклеотидом, кодирующим антагонист c-met или EGFR. Трансфецированные или трансдуцированные клетки затем возвращают в тело индивида. Клетки могут быть любыми из разнообразных типов, включая, без ограничения, гемопоэтические клетки (например, клетки костного мозга, макрофаги, моноциты, дендритные клетки, Т-клетки или В-клетки), фибробласты, эпителиальные клетки, эндотелиальные клетки, кератиноциты или мышечные клетки.
Например, если антагонист c-met или EGFR является антителом, антитело назначают любым подходящим способом, включая парентеральное, подкожное, интраперитонеальное, внутрилегочное и интраназальное и, если необходимо для локального иммуносупрессивного лечения, вводимое внутрь пораженной ткани применение. Парентеральные инфузии включают внутримышечное, внутривенное, внутриартериальное, интраперитонеальное или подкожное применение. При этом, антитело удобно применяют путем импульсной инфузии, в частности уменьшающимися дозами антитела. Предпочтительно, дозирование осуществляется с помощью инъекций, наиболее предпочтительно внутривенными или подкожными инъекциями, частично в зависимости от того, является ли применение кратковременным или длительным.
В другом примере, соединение антагониста c-met или EGFR назначается местно, например, в виде прямых инъекций, когда нарушение или локализация опухоли это позволяет, и инъекции могут повторяться периодически. Антагонист c-met или EGFR также может доставляться систематически индивиду или напрямую к опухолевым клеткам, например, к опухоли или ложе опухоли после хирургической экстирпации опухоли, чтобы предотвратить или уменьшить местный рецидив или метастазирование.
Введение терапевтических средств в виде комбинации обычно выполняется в течение определенного периода времени (обычно минут, часов, дней или недель в зависимости от выбранной комбинации). Комбинированная терапия предназначена включать введение этих терапевтических средств последовательно, то есть, где каждое терапевтическое средство вводится в различное время, а также введение этих терапевтических средств или, по меньшей мере, двух терапевтических средств осуществляется практически одновременно.
Терапевтическое средство может вводится одним и тем же способом или разными способами. Например, антагонист c-met или EGFR в комбинации могут вводиться внутривенной инъекцией, в то время как ингибитор протеинкиназы в комбинации может вводиться только перорально. Альтернативно, например, оба терапевтических средства могут вводиться перорально, или оба терапевтических средства могут вводиться путем внутривенной инъекции в зависимости от специфических терапевтических средств. Последовательность, в которой терапевтические средства вводятся, также варьирует в зависимости от специфических терапевтических средств.
В зависимости от типа и тяжести заболевания приблизительно, от 1 мкг/кг до 100 мг/кг (например, 0,1-20 мг/кг) каждого терапевтического средства является начальной возможной дозировкой для введения пациенту, будь то, например, одно или несколько отдельных введений или непрерывная инфузия. Типичная дневная дозировка может варьировать приблизительно от 1 мкг/кг до приблизительно 100 мг/кг или более в зависимости от факторов, упомянутых выше. Для повторных введений в течение нескольких дней или дольше, в зависимости от патологического состояния, лечение продолжают до тех пор, пока рак не будет вылечен, что оценивается методами, описанными выше. Однако может быть пригоден другой режим дозирования. В одном примере, если антагонист c-met или EGFR является антителом, антитело изобретения вводится каждые две-три недели в дозе в диапазоне приблизительно от 5 мг/кг до приблизительно 15 мг/кг. Если антагонист c-met или EGFR является пероральным низкомолекулярным соединением, лекарственное средство вводится ежедневно в дозировке в диапазоне приблизительно от 25 мг/кг до приблизительно 50 мг/кг. Более того, пероральное соединение изобретения может быть введено либо по традиционной высокодозированной прерывистой схеме приема лекарственного препарата, либо используя более низкое и более частое дозирование без регулярных перерывов (называется «периодической терапией»). Когда используется прерывистая схема лечения, например, лекарственное средство может вводиться ежедневно в течение двух-трех недель с последующим недельным перерывом; или ежедневно в течение четырех недель с последующим двухнедельным перерывом в зависимости от ежедневной дозы и особых указаний. Эффективность терапии изобретения легко контролируется стандартными методами и анализами.
Настоящая заявка изобретения рассматривает назначение антагониста c-met и/или EGFR путем генной терапии. Смотри, например, WO96/07321, опубликованный 14 марта 1996 года, касающийся использования генной терапии с целью выработки внутриклеточных антител.
Существует два основных способа доставки нуклеиновой кислоты (дополнительно содержащейся в векторе) в клетки пациента; in vivo и ex vivo. При доставке in vivo нуклеиновую кислоту инъецируют непосредственно в пациента, обычно в участке, где необходимо антитело. При лечении ex vivo клетки пациента извлекаются, нуклеиновую кислоту вводят в эти выделенные клетки, и видоизмененные клетки вводят пациенту либо напрямую, либо, например, инкапсулированными в пористые мембраны, которые имплантируют в пациента (смотри, например, патент США №№ 4892538 и 5283187). Существуют различные методики, предназначенные для введения нуклеиновых кислот в живые клетки. Методики различаются в зависимости от того, переносят ли нуклеиновую кислоту в культивируемые клетки in vitro или in vivo в клетки предполагаемого хозяина. Методики, подходящие для переноса нуклеиновой кислоты в клетки млекопитающих in vitro, включают использование липосом, электропорации, микроинъекции, слияние клеток, DEAE-декстран, метод преципитации фосфатом кальция и др. Традиционно используемым вектором для доставки гена ex vivo является ретровирус.
В настоящее время предпочитаемые методы переноса нуклеиновой кислоты in vivo включают трансфекцию с вирусными векторами (такими как аденовирус, вирус простого герпеса I или адено-ассоциированый вирус) и основанные на липидах системы (пригодными липидами для липид-опосредованного переноса гена являются DOTMA, DOPE и DC-Chol, например). В некоторых ситуациях желательно обеспечить источнику нуклеиновой кислоты вещество, которое ориентировано на клетки-мишени, такое как антитело, специфичное для мембранного белка клеточной поверхности или для клетки-мишени, лиганд для рецептора на клетки-мишени и др. В случае применения липосом, белки, которые связываются с мембранным белком клеточной поверхности, ассоциированным с эндоцитозом, могут быть использованы для мишеневого воздействия и/или для облегчения захвата, например, капсидные белки или их фрагменты, тропные к определенному клеточному типу, антитела к белкам, которые подвергаются циклической интернализации, и белки, которые целенаправленно воздействуют на внутриклеточную локализацию и повышают внутриклеточное время полужизни. Метод рецептор-опосредованного эндоцитоза описан, например, в Wu et al., J. Biol. Chem. 262:4429-4432 (1987); и Wagner et al., Proc. Natl. Acad. Sci. USA 87:3410-3414 (1990). Для обзора по известным на данный момент протоколам маркирования гена и генной терапии смотри Anderson et al., Science 256:808-813 (1992). Смотри также WO 93/25673 и указанные здесь ссылки.
Далее представлены примеры способов и композиций изобретения. Понятно, что могут применяться различные другие варианты осуществления изобретения при условии, что основное описание предоставлено выше.
ПРИМЕРЫ
Пример 1: Анализ экспрессии c-met и EGFR в клеточных линиях NSCLC и опухолевых образцах.
Материалы и способы
Исследования с помощью микроматриц. Анализ базальной экспрессии гена в клеточных линиях NSCLC и первичных опухолях был выполнен, используя РНК, выделенную из субконфлюэнтных клеточных культур или лизатов замороженной опухоли, на Affymetrix (Santa Clara, CA) микроматрице (HGU133Plus_2.0 chips). Получение комплементарной РНК, гибридизация с микроматрицей и дальнейший анализ данных были выполнены, используя протоколы производителя, в частности, как описано в Hoffman EP et al., Nat Rev Genet 5:229-37 (2004).
Для оценки связи экспрессии c-met с экспрессией других рецепторов тирозинкиназы (RTK), экспрессируемых в образцах NSCLC, был использован вариационный фильтр, чтобы исключить гены с минимальными вариациями для всей совокупности анализируемых образцов. Гены с минимальной экспрессией (те, для которых абсолютная вариация (максимум-минимум) для всей совокупности образцов была < 1000) были исключены для дальнейшего анализа. При этом один зонд был выбран для представления одного гена. Коэффициенты ранговой корреляции Спирмена (ρ) были посчитаны для каждого гена против мРНК MET (зонд ID, 203510_at) или белка c-met (IHC).
Количественная ПЦР. Уровни экспрессии мРНК EGFR и MET были проанализированы количественной ОТ-ПЦР, используя стандартную методику Taqman. Уровни транскрипта были нормированы на рибосомный белок L19 (RPL19) гена домашнего хозяйства и результаты были представлены либо как нормированные значения экспрессии (=2-∆Ct), либо как нормированная экспрессия относительно пулированного тканевого источника (=2-∆∆Ct). Следующие наборы праймер/зонд были использованы:
RPL19: прямой праймер, 5'-ACCCCAATGAGACCAATGAAATC-3' (SEQ ID NO:26), обратный праймер, 5'-ATCTTTGATGAGCTTCCGGATCT-3' (SEQ ID NO:27),
зонд, 5'(VIC)-AATGCCAACTCCCGTCAG-(MGBNFQ)-3'(SEQ ID NO:28);
MET: прямой праймер, 5'-CATTAAAGGAGACCTCACCATAGCTAAT-3' (SEQ ID NO:29),
обратный праймер, 5'-CCTGATCGAGAAACCACAACCT-3'(SEQ ID NO:30),
зонд, 5'(FAM)-CATGAAGCGACCCTCTGATGTCCCA-(BHQ-1)-3' (SEQ ID NO:31).
Наборы праймер/зонд для EGFR были приобретены у Applied Biosystems (cat#4331182, Hs00193306; Foster City, CA).
Иммуногистохимия (IHC). Фиксированные в формалине и заключенные в парафин образцы были нарезаны толщиной в 5 микрон на предметные стекла. После депарафинизации и регидрации срезы были подвергнуты IHC анализу c-met и EGFR. IHC EGFR была выполнена с помощью набора реагентов EGFR pharmDxTM Kit (Dako, Glostrup, Denmark) согласно инструкциям производителя. Для иммуногистохимии (IHC) c-met извлечение антигена было выполнено, используя предварительно нагретый при 99°С в течение 40 минут буфер Target Retrieval (Dako, Glostrup, Denmark) для IHC c-met. Эндогенная пероксидазная активность была блокирована раствором KPL Blocking Solution (KPL, Gaithersburg, MD) при комнатной температуре в течение 4 минут. Эндогенный авидин/биотин был блокирован набором реагентов Vector Avidin Biotin Blocking Kit (Vector Laboratories, Burlingame, CA). Впоследствии срезы были инкубированы в присутствии 10 мкг/мл мышиного анти-c-met (клон DL-21) моноклонального антитела (Upstate Biotechnology Inc. Lake Placid, NY) в блокирующей сыворотке в течение 60 минут при комнатной температуре с последующей инкубацией в присутствии биотинилированных вторичных антимышиных антител лошади в течение 30 минут. Реагент Vectastain ABC Elite (Vector Laboratory, Burlingame, CA) вместе с Metal Enhanced DAB (Pierce Biotechnology, Inc. Rockford, IL) был использован для проявления покровных стекол со срезами. Уровни экспрессии были определены как отрицательные (-), слабые (+), средние (++) или высокие (+++). Клеточные линии или опухолевые образцы, содержащие более чем 10% опухолевых клеток со слабой, средней или высокой степенью окрашивания, рассматривались как положительные.
Клеточная культура и опухолевые образцы.
Клеточные линии были получены из Американской Коллекции Типовых Культур (American Type Culture Collection), Подразделения Национального института рака по Лечению и Диагностике рака (NCI Division of Cancer Treatment and Diagnosis) и Японского хранилища ресурсов медицинских наук (Japanese Health Sciences Resources depositories), как показано в Таблице 1. Все клеточные линии поддерживали в RPMI 1640 с добавлением 10% FBS (Sigma, St. Louis, MO) и 2 мМ L-глутамина. Опухолевые образцы были получены из Объединенной сети тканей человека и Интегрированной лаборатории Университета штата Мичиган (University of Michigan, Cybrdio, Cooperative Human Tissue Network and Integrated Laboratory services).
Результаты
Экспрессия мРНК MET связана с экспрессией мРНК EGFR в клеточных линиях NSCLC.
Чтобы оценить, связана ли экспрессия c-met с экспрессией EGFR и других рецепторов тирозинкиназы (RTK) в клеточных линиях NSCLC, коэффициенты ранговой корреляции Спирмена были посчитаны по данным экспрессии генов, полученным на микроматрицах, в 50 клеточных линиях NSCLC, показанных в Таблице 1. Уровни мРНК EGFR и MET положительно коррелировали в клеточных линиях (ρ=0,54, р<0,0001), и экспрессия EGFR была высоко связана с экспрессией MET (Таблица 2).
Экспрессия белка cMET связана с экспрессией мРНК EGFR в клеточных линиях NSCLC.
Чтобы оценить связана ли экспрессия белка c-met, выявленная иммуногистохимией (IHC), с экспрессией EGFR и других рецепторнов тирозинкиназы (RTK) в клеточных линиях NSCLC, коэффициенты ранговой корреляции Спирмена были посчитаны по данным экспрессии генов, полученным на микроматрицах, в 50 клеточных линиях NSCLC, показанных в Таблице 1. Уровни мРНК EGFR и белка c-met положительно коррелировали в клеточных линиях (ρ=0,50, р=0,002), и экспрессия EGFR была высоко связана с экспрессией белка c-met (Таблица 3).
Экспрессия мРНК MET связана с экспрессией мРНК EGFR в опухолевых образцах NSCLC.
Чтобы оценить связана ли экспрессия мРНК c-met с экспрессией EGFR и других рецепторов тирозинкиназы (RTK) в клеточных линиях NSCLC, показанных в Таблице 1, коэффициенты ранговой корреляции Спирмена были посчитаны по данным экспрессии генов, полученным на микроматрицах, в 78 опухолях NSCLC. Экспрессии мРНК EGFR и мРНК MET были положительно связаны в опухолях NSCLC (ρ=0,26, р=0,02) (Таблица 4).
Коэкспрессия EGFR и MET в клеточных линиях NSCLC и первичных опухолях.
Чтобы оценить происходит ли коэкспрессия c-met и EGFR в образцах клеточных линий NSCLC и первичных опухолей, экспрессия мРНК EGFR и MET была измерена количественной ОТ-ПЦР в выборке клеточных линий NSCLC (как указано в Таблице 1) или лизатов замороженных первичных опухолей NSCLC. Уровни мРНК EGFR и MET были положительно связаны в клеточных линиях (ρ=0,59, р<0,0001) (Фигура 1, левая панель) и в образцах первичных опухолей NSCLC (ρ=0,48, р=0,0003) (Фигура 1, правая панель). Эти данные демонстрируют, что существует перекрывание в экспрессии MET и EGFR в клеточных линиях NSCLC и образцах первичных опухолей.
Подтверждение коэкспрессии EGFR и MET методом IHC в клеточных линиях NSCLC и первичных опухолях.
Сорок семь клеточных линий немелкоклеточного рака легкого (NSCLC) (как указано в Таблице 1) и сто тридцать восемь образцов первичной NSCLC (коллекция Genentech) были исследованы на предмет экспрессии в них c-met и EGFR методом IHC. Уровни экспрессии были расценены как отрицательные (-), слабые (+), средние (++) или высокие (+++), и клеточная линия или опухолевый образец, содержащие более чем 10% опухолевых клеток со слабой, средней или высокой степенью окрашивания, рассматривались как положительные.
79% (37/47) клеточных линий и 68% (94/138) опухолей NSCLC окрашивались положительно по EGFR (Таблица 5). Положительные по EGFR образцы (79% клеточных линий и 68% первичных опухолей) в дальнейшем были распределены по группам, основываясь на уровнях экспрессии в них c-met (Таблица 5). Положительные по EGFR клеточные линии демонстрировали слабую (22%), среднюю (57%) и высокую (19%) экспрессию c-met, и EGFR-положительные образцы первичных опухолей были только слабо или средне положительными. Подтип аденокарциномы был чаще положительным по c-met-окрашиванию, нежели подтип плоских клеток (70% против 40%), с большим количеством случаев среднего окрашивания (30% против 7%). Эти данные демонстрируют значительное перекрывание между экспрессией c-met и EGFR в клеточных линиях NSCLC и опухолевых образцах, особенно в опухолевом подтипе аденокарциномы.
Пример 2: Снижение экспрессии белка c-met в клетках NSCLC увеличивает лиганд-индуцируемую активацию EGFR, Her2 и Her3.
Материалы и способы
Ретровирусные конструкты кшРНК. Олигонуклеотиды, кодирующие последовательности кшРНК против c-met (5'-GATCCCCGAACAGAATCACTGACATATTCAAGAGATATGTCAGTGATTCTGTTCTTTTTTGGAAA-3' (SEQ ID NO:32) (shMet 3) и
5'GATCCCCGAAACTGTATGCTGGATGATTCAAGAGATCATCCAGCATACAGTTTCTTTTTTGGAAA (SEQ ID NO:33) (shMet 4)) были клонированы в участки BglII/HindIII вектора pShuttle-H1 ниже промотора Н1 (David Davis, GNE). Текст, выделенный жирным шрифтом, означает последовательность гибридизации мишени. Эти конструкты были рекомбинированы с ретровирусным вектором pHUSH-GW (Gray D et al BMC Biotechnology. 2007; 7:61), используя фермент Clonase II (Invitrogen), образуя конструкт, в котором экспрессия кшРНК находится под контролем индуцибельного промотора. Обработка тетрациклиновым аналогом доксициклина приводит к экспрессии кшРНК. Контрольный ретровирусный конструкт shGFP2, содержащий кшРНК, направленный против GFP (Hoeflich et al. Cancer Res. (2006) 66(2):999-1006), был предоставлен David Davis, Genentech, Inc. shGFP2 содержит следующий олигонуклеотид:
(EGFP)кшРНК
(смысловой)5'-GATCCCCAGATCCGCCACAACATCGATTCAAGAGATCGATGTTGTGGCGGATCTTGTTTTTTGGAAA-3' (SEQ ID NO:34).
Клеточная культура. Упаковывающие клетки GP-293 (Clontech) поддерживали в среде HGDMEM (GNE) с добавлением 10% Tet-Free FBS (Clontech), 2 мМ L-глутамина (GNE) и 100 ед/мл пенициллина и 100 ед/мл стрептомицина (Gibco). Клетки H441 (ATCC № HTB-174) поддерживали в среде 50:50 (DMEM:F12, MediaTech) с добавлением 10% Tet-Free FBS (Clontech), 2 мМ L-глутамина (GNE) и 100 ед/мл пенициллина и 100 ед/мл стрептомицина (Gibco). Клетки EBC-1 (Japanese Health Sciences Resources; смотри Cancer Res. (2005) 65(16):7276-82) поддерживали в RPMI 1640 (GNE) с добавлением 10% Tet-Free FBS (Clontech), 2 мМ L-глутамина (GNE) и 100 ед/мл пенициллина и 100 ед/мл стрептомицина (Gibco). Клетки поддерживали при 37°С в 5% CO2.
Получение рекомбинантного ретровируса и стабильных клеточных линий. Упаковывающие клетки GP-293 были котрансфецированы, используя набор реагентов FuGene 6 (Roche) и CalPhos Mammalian Transfection kit (Clontech), pVSV-G (Clontech) и вышеупомянутыми рекомбинантными ретровирусными конструктами. Среда, содержащая рекомбинантный вирус, была затем добавлена к клеткам EBC-1 и H441, и клетки были отобраны в пуромицине (Clontech). Клетки, стабильно экспрессирующие ретровирусные конструкты, были затем аутоклонированы через FACS в 96-луночных плашках.
Вестерн блот. Чтобы разделить белки, 20 мкг клеточного лизата было нанесено в гель NuPAGE 4-12% Bis-Tris в буфере MOPS (Invitrogen). Гели были уравновешены в буфере для блоттинга 2Х NUPAGE вместе с антиоксидантным буфером, затем были перенесены на 0,2 мкм мембрану PVDF с помощью iBlot. Мембраны затем были блокированы в TBST (10мМ TRIS, pH 8,0, 150 мМ NaCl, 0,1% Tween 20), содержащем 5% BSA, в течение 1 часа при комнатной температуре, затем инкубированы в течение ночи с первичными антителами, разбавленными в блокирующем буфере, при 4°С. Мембраны были промыты TBST, затем инкубированы с HRP-конъюгированными вторичными антителами (GE Healthcare) в TBST с 5% обезжиренным молоком в течение часа при комнатной температуре. Антитела выявляли с помощью хемилюминесценции (GE Healthcare, ECL Plus).
Скрининг стабильных клеточных линий. Клоны, стабильно трансдуцированные ретровирусными конструктами, были выращены в оптимальной среде +/- 1 мкг/мл доксициклин (Clontech) для индукции экспрессии кшРНК и проверены посредством вестерн-блотов на предмет нокдауна c-met, используя антитела анти-c-met C-12 (Santa Cruz Biotech). Фосфо-с-met был блотирован для использования антител анти-Фосфо-c-met Y1003 (Biosource) и анти-Фосфо-c-met Y1234/1234 (Cell Signaling). В качестве контроля, актин был блотирован для использования антитела анти-Актин I-19 (Santa Cruz Biotech). Клон 3.15 EBC и клон 4.12 EBC демонстрировали значительное снижение экспрессии с-met и уровней фосфо-c-met, клон 3.11 Н441 и клон 3.1 Н441 демонстрировали промежуточное снижение экспрессии с-met и экспрессии фосфо-met, и клон 4.5 EBC демонстрировал меньшее снижение экспрессии c-met и фосфо-c-met.
Клон 4.5 клеточных линий EBC, клон 4.12 EBC содержали конструкт shMet4, и клон 3.1 клеточных линий Н441, клон 3.11 Н441 и клон 3.15 EBC содержали конструкт shMet3.
Эксперименты по ответу на действие лиганда. Клетки, пересеваемые с/без доксициклина в течение 48 часов (EBC shMet) или 6 дней (Н441 shMet) были помещены в количестве 1х106 клеток/лунка в 6-луночный планшет с/без Dox (0,1 мкг/мл) в 10% FBS-RPMI, затем инкубированы в течение ночи при 37°С. Клетки ополаскивали в PBS, и среда была заменена на 0,5% BSA-RPMI (с/без доксициклина) у бессывороточных клеток на 2 часа при 37°С. Среда, содержащая лиганд (20 нМ TGFα или 2нМ HRG), была добавлена в лунки и инкубирована в течение 20 минут при 37°С. Лунки ополаскивали холодным TBS, затем осуществляли лизис в TBS, 1% NP-40, полном коктейле ингибиторов протеаз (Roche) и коктейли 1 и 2 ингибиторов фосфотаз (Sigma). Монослой и супернатант были собраны из лунки и перенесены в микроцентрифужную пробирку, где лизат был инкубирован на льду в течение 10-30 минут. Клеточные обломки были осаждены микрофугированием, и супернатант был перенесен в новую пробирку. Концентрация белка была количественно определена BCA-анализом (Pierce), и лизаты хранили при -20°С до размораживания для электрофореза. 20 мкг (EBC1) или 15 мкг (Н441) клеточного лизата были нанесены на гель и блотированы для выявления фосфо-c-met (YY1234/35, 3126 из Cell Signaling Technology), суммарного c-met (C12, sc-10 из San Cruz Biotechnology), β-актина (I-19, sc-1616 из San Cruz Biotechnology), фосфо-EGFR (Y1173, 04-341 из Upstate), суммарного EGFR (MI-12-1, из MBL), фосфо-Her2 (YY1121/22, 2243 из Cell Signaling Technology), суммарного Her3 (С18, sc-284, из San Cruz Biotechnology), фосфо-Her3 (Y1289, 4791 из Cell Signaling Technology) или суммарного Her3 (C17, sc-285, из San Cruz Biotechnology) как описано выше.
Результаты
Ретровирусы, несущие тетрациклин-индуцибельную короткую шпилечную РНК (кшРНК), которая целенаправленно воздействует на c-met, были использованы для образования стабильных клонов клеточной линии NSCLC, у которых может быть индуцирована экспрессия кшРНК для нокдауна экспрессии с-met. Для изучения воздействия нокдауна c-met на экспрессию и фосфорилирование членов семейства EGFR в EBC1 клеточной линии NSCLC, клетки shMet 4.12 EBC1, содержащие индуцибельную кшРНК, направленную против met, или контрольную кшРНК, направленную против GFP, были выращены в контрольной среде или среде, содержащей 0,1 мкг/мл Dox, в течение 48 часов. После выращивания в бессывороточной среде в течение двух часов, клетки были обработаны или не обработаны TGFα или херегулином b1 в течение 20 минут. Клеточные лизаты были проанализированы на предмет экспрессии суммарных и фосфо-белков, как показано.
Dox-обработанные клетки EBC1, в которых экспрессия белка с-met была подавлена, используя кшРНК (Фигура 2; EBCshMet 4.12, Dox, левая панель), но не Dox-обработанные контрольные клетки EBC1 (Фигура 2; shGFP2, правая панель), демонстрировали увеличенный pEGFR и pHer2 в ответ на обработку TGFα и увеличенный pHer3 в ответ на обработку херегулином, а также увеличенный pAKT либо при обработке TGFα, либо херегулином. Dox-обработанные shMet 4.12 клетки EBC (без стимуляции лигандом) демонстрировали увеличенный суммарный Her2 и суммарный Her3 и сниженный pEGFR и pHer3. Клетки EBC1 не демонстрировали значительную индукцию pEGFR, pHer2, pHer3 или pAKT в ответ на обработку TGFα или херегулином в отсутствие воздействия “накдауна” c-met.
С целью исследования воздействия накдауна c-met на экспрессию и фосфорилирование членов семейства EGFR в других клеточных линиях NSCLC, клетки NSCLC H441, содержащие индуцибельную кшРНК, направленную против met, или контрольную кшРНК, направленную против GFP, были выращены в контрольной среде или среде, содержащей 0,1 мкг/мл Dox, в течение 48 часов. После выращивания в бессывороточной среде в течение двух часов, клетки не были обработаны или были обработаны TGFα или херегулином b1 в течение 20 минут. Клеточные лизаты были проанализированы на предмет экспрессии суммарных и фосфо-белков, как показано.
Клетки Н441, в которых был подавлен (накдаун) c-met, используя кшРНК (Фигура 3; Dox-обработанные shMet 3.1, левая панель и Dox-обработанные shMet 3.11 средняя панель), но не Dox-обработаннные контрольные клетки Н441 (Фигура 3; shGFP1, правая панель), демонстрировали увеличенные pHer2 и pHer3 в ответ на обработку херегулином. Dox-обработанные клетки shMet 3.1 и shMet 3.11 также демонстрировали увеличенный суммарный Her3 и пониженный pEGFR. В отличие от клеток EBC1, клетки Н441 имели слабый ответ на TGFα (pEGFR) и херегулин (pHer2 и pHer3) в отсутствие накдауна c-met. Клетки EBC1 имели более высокие уровни c-met нежели клетки Н441.
Эти эксперименты продемонстрировали, что снижение экспрессии c-met в клеточных линиях NSCLC приводит к сниженной базальной активации EGFR (pEGFR) и повышенной лиганд-индуцированной активации Her 2 и Her 3, предполагая, что ингибирование Met увеличивает чувствительность к лигандам семейства EGF.
Пример 3: Комбинирование накдауна met и обработки эрлотинибом, ингибитором EGFR, значительно ингибирует опухолевый рост в модели ксенотрансплантанта
Чтобы исследовать, играет ли роль EGFR в поддержании опухолевой жизнеспособности в клетке, в которой функция с-met частично ингибирована, животные, несущие опухоль EBC-1 shMet-4.5 были лечены комбинацией эрлотиниба (TarcevaTM) и Dox.
Материалы и способы
Материал для исследования. Эрлотиниб (TarcevaTM) был предоставлен OSI Pharmaceuticals в Отдел приготовления лекарственных форм в Genentech и взвешен наряду с достаточным количеством наполнителя (метилцеллюлозным твином (MCT)). Вещества хранились в холодильнике, установленном на поддержание температуры в диапазоне от 4°С до 8°С. Моновалентное моноклональное анти-c-met антитело MetMAb (rhuOA5D5v5)(WO2007/063816) было предоставлено Подразделением Создания Антител в компании Genetech (Antibody Engineering Department at Genetech, Inc.) в очищенной жидкой форме. Клеточная линия EBC-1 была получена из Японской Коллекции Исследовательских Биоресурсов (JCRB).
Вид организмов. Сорок «голых» мышей (nu/nu) были получены из компании Charles River Laboratories (CRL), и была проведена их акклиматизация, по меньшей мере, в течение одной недели до их использования в исследовании. Животных содержали в вентилируемых клеточных приспособлениях в комнатах с фильтрами, обеспечивающими высокоэффективный распыленный воздух (HEPA). Только те животные, которые оказались здоровыми и были без очевидных аномалий, были взяты для исследования.
План исследования. Клетки EBC-1 были культивированы в питательной среде, которая состояла из среды RPMI 1640 (Invitrogen), 2 мМ L-глутамина и 10% фетальной бычьей сыворотки. Чтобы приготовить клетки для инокуляции мышам, клетки были трипсинизированы, промыты десятью миллилитрами стерильного 1Х фосфатно-солевого буфера (PBS). Субпопуляция клеток была подсчитана методом исключения трипанового синего, и оставшиеся клетки были ресуспендированы в 100 мкл стерильного 1Х PBS в конечной концентрации, равной 5 × 107 клеток в миллилитре. Мышам инокулировали подкожно в правую подлопаточную область клетками EBC-1 в количестве 5 × 106. За опухолями осуществлялся мониторинг до достижения ими среднего объема величиной 300 мм3.
Мыши, которым имплантировали опухолевые клетки, были разделены на четыре группы по 10 мышей в каждой до начала каждого вида лечения (суммировано в Таблице 6). Мышам в Группе 1 (контрольная группа) проводили лечение 100 мкл наполнителя, метилцеллюлозным твином (МСТ), каждый день (QD) через ротовой зонд (РО), и их переключали на питьевую воду, содержащую 5% сахарозу. Мышам в Группе 2 (группе с нокдауном c-met) проводили лечение 100 мкл MCT, QD, PO, но их переключали на питьевую воду, содержащую 0,5 мг/мл доксициклина (Dox) в 5% сахарозе. Мышей в Группе 3 (леченной эрлотинибом группе) проводили лечение 100 мг/кг эрлотиниба в объеме 100 мкл, приготовленным в MCT, QD, PO, и их переключали на питьевую воду, содержащую 5% сахарозу. Мышам в Группе 4 (леченной эрлотинибом группа с нокдауном c-met) проводили лечение 100 мг/кг эрлотиниба в объеме 100 мкл, приготовленным в MCT, QD, PO, и их переключали на питьевую воду, содержащую 1 мг/мл доксициклина (Dox) в 5% сахарозе. Dox и воду с сахарозой меняли каждые 2-3 дня. Эрлотиниб и МСТ дозировались в течение 14 дней с перерывом в 6 дней и затем возобновляли лечение ими в течение оставшегося периода исследования (20 дней). Животных исключали из исследования, если опухоли достигали размеров более чем 1000 мм3 или если в опухолях обнаруживали признаки некротических поражений. Если более чем 3 животных должны были исключить из исследования из любой из этих групп, лечение в этой группе было прекращено и все животные были исключены из исследования. Все исследования и обращение с мышами соответствовали основным требованиям комитета по уходу за животными и использованию (IACUC).
Измерение опухоли и массы тела. Объемы опухолей были измерены в двух измерениях (длина и ширина), используя калипер UltraCal-IV (модель 54-10-111, Fred V. Fowler Company, Inc.; Newton, MA). Следующая формула была использована в Excel v11.2 (Microsoft Corporation; Redmond, WA) для вычисления объема опухоли:
Объем опухоли (мм3)=(длина•ширина2)•0,5
Анализ данных эффективности. Ингибирование опухоли было представлено в графическом изображении, используя KaleidaGraph 3.6 (Synergy Software; Reading, PA). Процент ингибирования роста (%Inh) на 17 день был вычислен следующим образом:
%Inh=100 X [Размер опухоли (наполнитель) - {Размер опухоли (MetMAb)/Размер опухоли (наполнитель)}]
Частота опухолей (TI) была определена по числу поддающихся измерению опухолей в каждой группе на 17 день. Частичная регрессия (PR) определена как регрессия опухоли >50%, но <100% от исходного объема опухоли на любой день во время исследования. Полная регрессия (CR) определена как регрессия опухоли на 100% от исходного объема опухоли на любой день во время исследования.
Средний объем опухоли и стандартная ошибка среднего (SEM) были вычислены, используя программное обеспечение JMP, версия 5.1.2 (SAS Institute; Cary, NC). Анализ данных и вычисление р-значений, используя либо критерий Стьюдента, либо критерий Дуннетта, был также выполнен, используя программное обеспечение JMP, версия 5.1.2.
Результаты
Комбинирование нокдауна c-met и лечения эрлотинибом значительно ингибирует опухолевый рост в модели ксенотрансплантанта.
Чтобы исследовать роль c-met в регуляции опухолевого роста на модели EBC-1, стабильные клоны EBC-1, которые могут быть индуцированы с целью экспрессии кшРНК, чтобы подавить экспрессию c-met, были получены, используя ретровирусы, несущие тетрациклин-индуцибельную короткошпилечную РНК (кшРНК), целенаправленно воздействующую на c-met. Клеточная линия EBC-1 немелкоклеточного рака легкого (NSCLC) является высоко амплифицированной по с-met и экспрессирует значительные количества рецептора c-met, который работает лиганд-независимым способом при регуляции клеточного и опухолевого роста. Ген EGFR относится к дикому типу в клеточной линии EBC-1.
Вслед за индукцией экспрессии кшРНК тетрациклиновым аналогом доксициклина клон shMet-3.15 EBC-1 демонстрировал эффективное, преимущественно полное подавление экспрессии c-met. Индукция кшРНК также блокировала пролиферацию этих клеток, как это было проанализировано методами по оценке жизнеспособности клеток Cell Titer Glo и Alamar Blue. Остановка роста с последующим апоптозом отмечалась в клетках shMet3.15 EBC1 спустя 24-72 часа после индукции кшРНК. Тот же самый клон клеточной линии был имплантирован в животную модель, в основном, как описано выше (за исключением того, что животных не подвергали лечению эрлотинибом) и дали возможность образоваться опухолям. Индукция экспрессии кшРНК после образования опухоли в этих животных привела к регрессии опухоли in vivo. Эти результаты показывают, что экспрессия c-met существенна для роста и выживаемости клеток EBC-1 in vitro и in vivo.
Клон shMet-4.5 EBC-1 демонстрировал частичное подавление экспрессии с-met вслед за индукцией экспрессии кшРНК с использованием Dox. Снижение экспрессии c-met также оказало воздействие на клеточный рост и выживаемость в этом клоне: индукция экспрессии кшРНК уменьшала число клеток, как показал анализ in vitro по оценке выживаемости клеток, и индукция экспрессии кшРНК после образования опухоли в модели ксенотрансплантанта ингибировала опухолевый рост, но не вызывала регрессию опухоли.
Клон shMet-4.5 был выбран для использования в экспериментах по оценке воздействия комбинирования подавления экспрессии c-met с обработкой эрлотинибом, как описано ниже.
Клеточная линия EBC-1 shMet-4.5 NSCLC была инокулирована «голым» мышам, и затем за животными осуществлялся мониторинг на предмет опухолевого роста до тех пор, пока привитые клетки не образовывали опухоли размером приблизительно 300 мм3. Мыши затем были распределены по четырем группам, получающим лечебное воздействие; Группа 1: наполнитель, Группа 2: доксициклин (Dox), Группа 3: эрлотиниб (100 мг/кг) и Группа 4: эрлотиниб + Dox (смотри Таблицу 6).
Лечение мышей эрлотинибом не оказало никакого воздействия на опухолевый рост (-6% ингибирования опухоли; Фигура 4), в то время как лечение Dox (ингибирующее экспрессию met) приводило к 38% снижению опухолевого роста при сравнении с контролем наполнителем на 19 день (Фигура 4; критерий Стьюдента, р = 0,084), снижаясь статистически незначимо. Однако, снижение опухолевого роста было статистически значимо при сравнении только с группой воздействия эрлотинибом (критерий Стьюдента, р = 0,004; Фигура 4). Комбинирование эрлотиниба и Dox приводило к значительному улучшению эффективности, приводя к 68% снижению опухолевого роста при сравнении с контролем наполнителем на 19 день (критерий Стьюдента, р = 0,001; Фигура 4). Комбинированное лечение эрлотинибом и Dox также приводило к статистически значимому снижению опухолевого роста при сравнении с лечением одним только Dox (критерий Стьюдента, р = 0,03) или лечением одним только эрлотинибом (критерий Стьюдента, р < 0,0001).
Лечение Dox и эрлотинибом приводило к более высокому числу частичных ответов (PR; определены как регрессия опухоли >50%, но <100% от исходного объема опухоли на любой день во время исследования) и полных ответов (CR; определены как регрессия опухоли на 100% от исходного объема опухоли на любой день во время исследования). В частности, комбинирование эрлотиниба с Dox приводило к 1 PR и 3 CR, в то время как лечение эрлотинибом не приводило ни к PR, ни к CR, и лечение Dox (нокдаун c-Met) приводило к 2 PR и 1 CR. Эти данные демонстрируют, что комбинирование ингибирования met (лечение Dox) и ингибирование EGFR (лечение эрлотинибом) более вероятно вызовет полную регрессию опухоли, нежели ингибирование c-met или EGFR по отдельности, даже хотя анализ опухолевых данных по индивидуальным животным выявил, что не все опухоли четко отвечали на комбинирование ингибирования c-met и эрлотиниба.
Эти результаты демонстрируют, что ингибирование c-met и EGFR в модели ксенотрансплантанта shMet-4.5 EBC-1 приводило к значительному снижению опухолевого роста. Таким образом, опухоли, в которых экспрессия и активность c-met частично ингибированы, используют сигнальный путь EGFR для обеспечения роста и выживаемости опухоли. Это означает, что EGFR играет роль в опухолевом росте и выживаемости в опухолях, в которых ингибирован c-met.
Пример 4: Лечение антителом анти-c-met и эрлотинибом, ингибитором EGFR, демонстрировало значительное улучшение эффективности воздействия по сравнению с лечением антителом анти-c-met или эрлотинибом по отдельности.
Материалы и методы
Материал для исследования. Моновалентное моноклональное анти-c-met антитело MetMAb (rhuOA5D5v5) было предоставлено Подразделением Создания Антител в компании Genetech (Antibody Engineering Department at Genetech, Inc.) в очищенной жидкой форме в концентрации 10,6 мг/мл. Наполнителем был 10 мМ гистидин сукцинат, 4% дигидрат трегалозы, 0,02% полисорбат 20, рН 5,7. Эрлотиниб (TARCEVATM) был предоставлен OSI Pharmaceuticals в Отдел приготовления лекарственных форм в Genentech и взвешен наряду с достаточным количеством наполнителя (метилцеллюлозным твином (MCT)). Весь материал был доставлен из компании Genentech в Научно-исследовательский институт Van Andel (VARI; Grand Rapids, MI) и была составлена его рецептура до начала лечения животных. Материалы хранили в холодильнике, установленном на поддержание температуры в диапазоне от 4°С до 8°С. Клеточная линия NCI-H596 была получена из Американской Коллекции Типовых Культур (Manassas, VA).
Вид организмов. Сорок трансгенных по человеческому HGF мышей C3H-SCID (hu-HGF-Tg-C3H-SCID) были получены из собственной колонии мышей в Научно-исследовательском институте Van Andel (VARI; Grand Rapids, MI). Пять мышей C3H-SCID были получены из Лаборатории Jackson. Животные были в возрасте 4-6 недель и весили 21-22 грамма каждый. Мыши были акклиматизированы к условиям исследования в течение, по меньшей мере, трех дней до инокуляции клетками опухоли. Мышей содержали в душевом огражденном сооружении. Животных содержали в вентилируемых клеточных приспособлениях в комнатах с фильтрами, обеспечивающими высокоэффективный распыленный воздух (HEPA). Только те животные, которые оказались здоровыми и были без очевидных аномалий, были взяты для исследования.
План исследования. Поскольку большинство HGF-чувствительных опухолей регулируется паракринным способом, была выбрана модель ксенотрансплантанта, моделирующая паракринную регуляцию роста. Мышиный HGF является слабым лигандом для человеческого c-met, приводя к слабому биологическому ответу клеточных линий, экспрессирующих c-met человека, на действие HGF мыши (Bhargava, M., et al., 1992; Rong, S., et al., 1992). Таким образом, чтобы смоделировать паракринные HGF-регулируемые опухоли человека, были получены трансгенные мыши (hu-HGF-Tg-SCID), которые экспрессируют HGF человека в убиквитарном виде с металлотионеинового промотора (Zhang, Y., et al, 2005). Уровни сывороточного HGF в мыши hu-HGF-Tg-SCID в ~5-10 раз выше, чем физиологические уровни (1-5 нг/мл против 0,2-0,5 нг/мл), и клеточные линии, которые отвечают на HGF пролиферацией in vitro, демонстрируют потенциальное увеличение опухолевого роста, когда выращены в виде ксенотрансплантантных опухолей в мышах hu-HGF-Tg-SCID.
Клеточная линия NCI-H596 немелкоклеточного рака легкого (NSCLC) была выбрана для исследований эффективности воздействий in vivo в мыши hu-HGF-Tg-SCID, поскольку данная клеточная линия высокочувствительна к HGF, и антитело анти-c-met, MetMAb, блокирует HGF-регулируемую пролиферацию этой клеточной линии in vitro (Kong-Beltran, M., et al., 2006). Клеточная линия NCI-H596 несет мутированную форму гена c-met c отсутствующим экзоном 14, который кодирует сайт связывания Е3 убиквитин-лигазы Cbl (Kong-Beltran, M., 2006). Cbl-связывающий сайт фосфорилируется по тирозину 1003 (Y1003) вслед за связыванием HGF, позволяя Cbl связаться и убиквитинировать c-met, таким образом нацеливая его для протеосомальной деградации (Peschard, P., et al., 2001). Восприимчивость NCI-H596 также может быть прослежена in vivo, поскольку клеточная линия быстро образует опухоли в мышах HGF-Tg-SCID (экспрессирующих HGF человека, как отмечено выше), но не будет образовывать опухоли в мышах с нарушенной иммунологической реактивностью, у которых отсутствует HGF человека («голые» мыши nu/nu или мыши SCID). Клетки NCI-H596, как полагают, образуют c-met-регулируемые опухоли. Клетки NCI-H596 обладают геном EGFR дикого типа и чувствительны к ингибитору EGFR, эрлотинибу (TARCEVATM), когда они растут в присутствии TGFα, как было продемонстрировано сниженной клеточной жизнеспособностью при их выращивании в присутствии эрлотиниба и TGFα.
Клетки NCI-H596 культивировали в питательной среде, состоящей из среды RPMI 1640 (Invitrogen), 2 мМ L-глутамина и 10% фетальной бычьей сыворотки. Чтобы приготовить клетки для инокуляции мышам, клетки были трипсинизированы, промыты десятью миллилитрами стерильного 1Х фосфатно-солевого буфера (PBS). Субпопуляция клеток была подсчитана методом исключения трипанового синего, и оставшиеся клетки были ресуспендированы в 100 мкл стерильного 1Х PBS в конечной концентрации, равной 5 × 106 клеток в миллилитре.
Мыши были подготовлены для инокуляции сбриванием дорсальной области ножницами. На следующий день каждой мыши инокулировали подкожно в правую подлопаточную область клетками NCI-H596 в количестве 5 × 105. За опухолями осуществлялся мониторинг до достижения ими среднего объема величиной 100 мм3.
Мыши HGF-Tg-C3H-SCID были разделены на две группы по одиннадцать мышей в каждой, и им была введена интраперитонеальная инъекция исследуемого материала два раза в неделю в течение четырех недель. Животным в Группе 1 вводили 100 мкл наполнителя и животным в Группе 2 вводили 30 мг/кг моновалентного моноклонального анти-c-met антитела MetMAb. План исследования представлен в Таблице 7. Опухоли измеряли три раза в неделю в течение пяти недель, начиная со дня лечения. К мышам была применена эвтаназия спустя пять недель, хотя к некоторым животным эвтаназия была применена раньше вследствие больших объемов опухолей (> 1500 мм3). Контрольные мыши C3H-SCID были также инокулированы, чтобы служить отрицательным контролем опухолевого роста, и за ними осуществляли мониторинг на предмент опухолевого роста в течение пяти недель.
Все исследования и обращение с мышами соответствовали основным требованиям комитета по уходу за животными и использованию (IACUC).
Измерение опухоли и массы тела. Объемы опухолей были измерены в двух измерениях (длина и ширина), используя калипер UltraCal-IV (модель 54-10-111, Fred V. Fowler Company, Inc.; Newton, MA). Следующая формула была использована в Excel v11.2 (Microsoft Corporation; Redmond, WA) для вычисления объема опухоли:
Объем опухоли (мм3)=(длина•ширина2)•0,5
Анализ данных эффективности. Ингибирование опухоли было представлено в графическом изображении, используя KaleidaGraph 3.6 (Synergy Software; Reading, PA). Процент ингибирования роста (%Inh) на 17 день был вычислен следующим образом:
%Inh=100 X [Размер опухоли (наполнитель) - {Размер опухоли (MetMAb)/Размер опухоли (наполнитель)}]
Частота опухолей (TI) была определена по числу поддающихся измерению опухолей в каждой группе на 17 день. Частичная регрессия (PR) определена как регрессия опухоли >50%, но <100% от исходного объема опухоли на любой день во время исследования. Полная регрессия (CR) определена как регрессия опухоли на 100% от исходного объема опухоли на любой день во время исследования.
Средний объем опухоли и стандартная ошибка среднего (SEM) были вычислены, используя программное обеспечение JMP, версия 5.1.2 (SAS Institute; Cary, NC). Анализ данных и вычисление р-значений, используя либо критерий Стьюдента, либо критерий Дуннетта, был также выполнен, используя программное обеспечение JMP, версия 5.1.2.
Оценочные показатели кривой выживаемости Каплана Мейера были графически представлены для периода времени удвоения опухоли для каждой группы. Были выполнены попарные сравнения между группами. Статистические сравнения были выполнены, используя логарифмический ранговый критерий. Анализ данных был выполнен, используя программное обеспечение JMP.
Результаты
Клеточная линия NCI-H596 NSCLC была инокулирована животным hu-HGF-Tg-C3H-SCID, и за животными осуществлялся мониторинг на предмет опухолевого роста до тех пор, пока привитые клетки не образовывали опухоли размером приблизительно 100 мм3. Мыши затем были распределены по четырем группам, получающим лечебное воздействие; группа 1: наполнитель, Группа 2: эрлотиниб, Группа 3: MetMAb и Группа 4: эрлотиниб + MetMAb (смотри Таблицу 7). Группы, леченные MetMAb, получали только однократную дозу, в то время как группы, леченные эрлотинибом, получали дозированный препарат каждый день в течение двух недель, затем лечение было прекращено, и за ростом опухоли наблюдали два-три раза в неделю. Контрольных мышей C3H-SCID также инокулировали, и у них осуществлялся мониторинг роста опухолей NCI-H596, не подвергавшихся воздействию HGF человека.
Рост опухолей NCI-H596 был значительно улучшен у мышей hu-HGF-Tg-C3H-SCID при сравнении с контрольными мышами C3H-SCID (Фигура 5; сравните контрольную группу, леченную наполнителем, с C3H-SCID). Лечение мышей моновалентным моноклональным анти-c-met антителом MetMAb привело к 67% снижению опухолевого роста при сравнении с контролем наполнителем на 20 день (Фигура 5; критерий Стьюдента, р = 0,0044), что согласуется с предыдущими исследованиями MetMAb на моделях NCI-H596. Лечение несущих опухоль мышей NCI-H596 эрлотинибом привело к статистически незначимому снижению опухолевого роста при сравнении с контролем наполнителем на 20 день (Фигура 5; критерий Стьюдента, р = 0,165). Комбинированное лечение MetMAb и эрлотинибом продемонстрировало значительное улучшение эффективности воздействия, приведя к 89% снижению опухолевого роста при сравнении с контролем наполнителем на 20 день (Фигура 5; критерий Стьюдента, р = 0,0035).
Лечение мышей MetMAb привело к 67% снижению опухолевого роста при сравнении с контролем наполнителем на 20 день (Фигура 5; критерий Стьюдента, р = 0,0044), что согласуется с предыдущими исследованиями MetMAb на моделях NCI-H596. Лечение несущих опухоль мышей NCI-H596 эрлотинибом привело к статистически незначимому снижению опухолевого роста при сравнении с контролем наполнителем на 20 день (Фигура 5; критерий Стьюдента, р = 0,165). Комбинированное лечение MetMAb и эрлотинибом продемонстрировало значительное улучшение эффективности воздействия относительно любого из веществ в отдельности, приведя к 89% снижению опухолевого роста при сравнении с контролем наполнителем на 20 день (Фигура 5; критерий Стьюдента; MetMAb+ эрлотиниб в сравнении с наполнителем, день 20 - р = 0,0035; MetMAb+ эрлотиниб в сравнении с эрлотинибом в отдельности, день 26 - р = 0,0009; MetMAb+ эрлотиниб в сравнении с MetMAb в отдельности, день 48 - р = 0,0149).
Данные по опухолевому объему были собраны в течение девяти недель после окончания дозирования, чтобы узнать приводит ли комбинирование MetMAb и эрлотиниба к улучшениям во время развития опухоли. Чтобы ответить на данный вопрос, измерения времени удвоения опухоли (TTD), определенное как время, необходимое для удвоения размера опухоли, были вычислены для каждой группы и использованы для построения кривых выживаемости Каплана-Мейера. Комбинирование MetMAb и эрлотиниба продемонстрировало значительное улучшение во время развития опухоли со средним значением TTD, равным 49,5 (± 2,6) дней, по сравнению с 17,8 (± 2,2) днями для леченной MetMAb группы, 9,5 (± 1,2) днями для леченной эрлотинибом группы и 9,5 (± 1,2) днями для леченной наполнителем контрольной группы (Фигура 6). Эти данные показывают, что комбинирование MetMAb и эрлотиниба значительно улучшает время развития опухоли по сравнению с любым из веществ в отдельности (логарифмический ранговый критерий; наполнитель в сравнении с MetMAb - р < 0,0001; наполнитель в сравнении с MetMAb + эрлотиниб - р < 0,0001; эрлотиниб в сравнении с MetMAb + эрлотиниб - р < 0,0001; и MetMAb в сравнении с MetMAb + эрлотиниб - р = 0,0009).
Эти данные демонстрируют, что комбинированное лечение MetMAb и эрлотинибом приводит к очень значительным улучшениям в плане ингибирования опухолевого роста и развития опухоли относительно лечения MetMAb или эрлотинибом по отдельности.
Пример 5: Сигнальная система c-met регулирует сигнальную систему EGFR
Материалы и способы
Исследования с помощью микроматриц: Три эксперимента на микроматрицах были выполнены, используя матрицы Affymetrix HGU133 Plus 2.0. В каждом случае, приготовление комплементарной РНК, гибридизация с матрицей и последующий анализ данных были выполнены, используя протоколы производителя, в частности, как описано в Hoffman EP et al., Nat Rev Genet 5:229-37 (2004). Исходные данные по экспрессии, в виде файлов Affymetrix CEL, были нормированы как группа, чтобы избавиться от небиологических источников вариации между данными по индивидуальным образцам, используя метод нормирования RMA (Irizarry, Biostatistics, 2003, PubMed ID 12925520), как предоставлено программным обеспечением Partek GS 6.3b (Partek, Saint Louis, MO). Полученные в результате нормированные значения экспрессии, представленные в log2 масштабе, были проанализированы, как указано далее, и были трансформированы в линейные величины для построения графических изображений.
В первом эксперименте лиганд-чувствительные клетки NSCLC HOP92 и H596 не были обработаны или были стимулированы 50 нг/мл HGF в течение 6 часов до анализа профиля экспрессии мРНК. Кратко, клетки поместили в 6-луночные плашки в концентрации приблизительно 5х105клеток/лунка. Спустя день, клетки были промыты, затем перенесены в среду RPMI + 0,1% BSA. На 3 день клетки простимулировали в течение 6 часов HGF в концентрации 50 нг/мл в среде RPMI + 0,1% BSA. Клетки были однократно промыты холодным PBS, лизированы лизирующим буфером RNAeasy, и РНК была получена согласно протоколу производителя. Образцы HOP92 и Н596 были проанализированы отдельно, используя критерий для определения значимости (Р-значение) различия в уровнях экспрессии для каждого гена в условиях + HGF и - HGF. Эти Р-значения были преобразованы в Q-значения путем внесения поправки при многочисленных анализах, используя метод Бенджамина и Хочберга (Benjamini and Hochberg, 1995). Затем гены были ранжированы по статистической значимости (Q-значениям) различия в уровне экспрессии в каждой клеточной линии.
Во втором эксперименте, уровни экспрессии мРНК клонов EBCshMet3-15 и EBCshMet4-12 были проанализированы спустя 24 и 48 часов инкубации в присутствии или отсутствие 50 нг/мл доксициклина. Паттерн экспрессии на каждой Affymetrix-панели зондов (генов) был проанализирован, используя линейную статистическую модель (ANOVA), с помощью которой оценивали влияние клона (3-15 или 4-12), лечения (контроль или доксициклин) и временные точки (24 или 48 часов), а также взаимозависимость временных точек и лечебных эффектов. Метод ANOVA выдал величины значимости (Р-значения) для каждого из этих четырех воздействий. Эти Р-значения были преобразованы в Q-значения путем внесения поправки при многочисленных анализах, используя метод Бенджамина и Хочберга. Затем гены были ранжированы по статистической значимости (Q-значениям) различия в уровне экспрессии между доксициклиновыми и контрольными образцами.
В третьем эксперименте клетки EBCMet кшРНК 4-12 или контрольные клетки EBCGFP кшРНК инкубировали только в среде или в среде с 50 нг/мл доксициклина в течение 24 часов. После дополнительной обработки (+/-HGF 100 нг/мл в течение 2 часов), был проведен анализ экспрессии мРНК с помощью микроматриц. Паттерн экспрессии на каждой Affymetrix-панели зондов (генов) был проанализирован, используя линейную статистическую модель (ANOVA), с помощью которой оценивали влияние мишени кшРНК (Met или GFP), индукции кшРНК (доксициклин или контроль) и HGF-обработки, а также взаимодействие этих трех переменных. Эти Р-значения были затем преобразованы в Q-значения различия в уровне экспрессии между условиями плюс-HGF и минус-HGF в обработанных доксициклином образцах EBCMetshRNA4-12. Принимая во внимание пороговый уровень Q-значения, равное 0,05 (5% False Discovery Rate), и двукратное изменение экспрессии при сравнении групп +/- HGF, было отобрано 188 панельных зондов.
ELISA TGFα: Опухоли ксенотрансплантанта EBC-1-shMet были образованы, и был дозирован Dox, в частности как описано в Примере 3, за исключением того, что Dox был использован в концентрации 1 мг/мл в 5% сахарозе и размер опухоли мог достигать 300-400 мм3 до начала лечения. Животным вводили дозы препарата 3 дня, затем их умерщвляли. Быстрозамороженные образцы опухоли ксенотрансплантанта EBC-1-shMet-4.12 помещали в 2 мл холодного лизирующего буфера (PBS + 1%TritonX-100 + Фосфотазный коктейль 2 (Sigma cat#P5726)) и ингибитор протеазы Complete Mini EDTA-Free (Roche#11836170001)(1 таблетка на 10 мл раствора). Опухоли были гомогенизированы ручным гомогенизатором, и лизаты были инкубированы на льду в течение 1 часа с периодическим покачиванием. Лизаты были центрифугированы при 10000xG в течение 10 минут при 4°С, перенесены в новую пробирку и была проведена количественная оценка белка Her3, используя метод BCA (Pierce cat#23225).
Поликлональное антитело анти-TGF-альфа (R&D Systems, Minneapolis, MN) было разведено до концентрации 1 мкг/мл в фосфатно-солевом буфере (PBS) и нанесено на плашки ELISA (25 мкл/лунка, 384-луночные плашки с поверхностью MaxiSorp, Nunc, Neptune, NJ) во время ночной инкубации при 4°С. После шестикратного промывания в промывочном буфере (PBS / 0,05% Tween-20) плашки были блокированы с помощью PBS / 0,5% бычий сывороточный альбумин (BSA) в течение 1-2 часов. Эта и все последующие инкубации были проведены при комнатной температуре в орбитальном встряхивателе. Образцы были разведены, используя буфер для образцов (PBS / 0,5% BSA / 0,5% Tween-20 / 0,2% бычий гамма-глобулин / 0,25% CHAPS / 5 мМ EDTA / 10 частей на миллион Proclin). Используя этот же самый буфер, были приготовлены серийные разведения рекомбинантного TGF-альфа человека (R&D Systems) с диапазоном калибровочной кривой, равным 400 - 12,5 пг/мл. Замороженные контрольные образцы, предварительно разведенные для количественного анализа в высокой, средней и низкой области калибровочной кривой, были разморожены. Плашки были промыты шесть раз, и образцы, стандарты и контроли были добавлены (25 мкл/лунка) и инкубированы в течение 2 часов. После промывания плашек двенадцать раз, биотинилированное поликлональное козье антитело анти-TGF-альфа (R&D Systems), разбавленное до концентрации 1 мкг/мл в буфере для образцов, было добавлено (25 мкл/лунка). После часовой инкубации плашки были промыты двенадцать раз. Конъюгат стрептавидина с пероксидазой хрена (GE Healthcare, Piscataway, NJ), разведенный 1/4000 в буфере для образцов, был затем добавлен (25 мкл/лунка). После финальной 30-минутной инкубации плашки были промыты двенадцать раз, и был добавлен тетраметилбензидин (TMB, Kirkegaard & Perry Laboratories, Gaithersburg, MD). На проявление цветной реакции было отведено от 6 до 8 минут при комнатной температуре, и реакция была остановлена путем добавления 1 М фосфорной кислоты. Значения оптической плотности были получены, используя планшет-ридер (450 нм, 620 стандарт), и концентрации образца были вычислены из 4-параметрических стандартных кривых.
Результаты
Активация с-met обработкой HGF увеличивала экспрессию мРНК лигандов EGFR (HB-EGF, эпирегулина, амфирегулина, TGFα) в лиганд-чувствительных клеточных линиях NSCLC Hop-92 и NCI-H596 (Фигура 9А). Напротив, ингибирование экспрессии c-met, используя кшРНК в лиганд-независимых клетках EBC1 клеточной линии NSCLC, снижало экспрессию мРНК этих лигандов EGFR (Фигура 9В). Обработка HGF dox-обработанной клеточной линии 4-12 EBC1 shMet восстанавливала экспрессию лигандов EGFR (Фигура 9С). Снижения экспрессии лигандов EGFR не происходило в контрольных клетках EBC-1, которые экспрессировали миРНК, направленные против GFP (Фигура 9D). Снижение экспрессии c-met в опухолях ксенотрансплантанта EBC-1-sh привело к уменьшению в опухоли уровней белка TGFα на третий день после обработки (Фигура 9Е).
Эти данные демонстрируют, что активность c-met может регулировать сигнальную систему EGFR в с-met-амплифицированных HGF-независимых клетках (EBC1), а также HGF-зависимых клеточных линиях (Hop92 и NCI-H596). Более конкретно, сигнальная система c-met увеличивала и поддерживала экспрессию семейства EGFR лигандов, которые могли затем стимулировать их собственное семейство EGFR рецепторов аутокринным способом. Напротив, ингибирование сигнальной системы c-met приводило к снижению экспрессии лигандов EGFR. Препятствие механизму аутокринной петли, по-видимому, является причиной сниженного pEGFR, что наблюдается в клетках EBC1 вслед за подавлением с-met (Фигура 10), и повышенной чувствительности к лиганд-индуцированной активации EGFR вслед за подавлением c-met, описанной в Примере 2. Эти результаты предполагают, что активность EGFR может компенсировать потерю активности сигнальной системы c-met в HGF-зависимых и HGF-независимых опухолях, и согласуются со значительно повышенной эффективностью воздействия на опухоль ксенотрансплантанта, наблюдаемую при обработке опухоли комбинированием ингибиторов EGFR и c-met (Пример 4).
Пример 6: активность c-met регулирует экспрессию Her3.
Материалы и способы
Вестерн-блот анализ pEGFR и белка Her3: Клетки были посеяны плотностью 1×106 и инкубированы 18 часов при 37°С в 10% Tet-approved FBS в RPMI 1640. На следующий день среда была удалена и заменена на свежую стандартную среду с добавлением или без добавления 0,1 мкг/мл Dox. Через 24, 48 и 72 часа после смены среды белки были выделены с помощью 1% NP-40/TBS/полного коктейля ингибитора протеазы Roche/коктейлей 1 и 2 ингибитора фосфатазы Sigma после ополаскивания холодным TBS. 15 мкг суммарного белка были нанесены в гель NuPAGE 4-12% Bis-Tris (Invitrogen) в буфере MOPS и перенесены на PVDF с помощью Invitrogen's iBlot. Мембраны были подвергнуты иммуноблоттингу для выявления фосфорилированных белков (pEGFR (Y1173) Upstate 04-341 в разведении 1:1000 в 5% BSA/TBST), десорбированы восстанавливающим десорбирующим буфером Pierce, затем повторно зондированы для выявления суммарных белков (c-met: SCBT sc-10 в разведении 1:10000; Her3: SCBT sc-285 в разведении 1:2000 в 5% нежирном сухом молоке и TBST). Белки определяли с помощью HRP-конъюгированных вторичных антител Amersham (Amersham антикроличьи-HRP, #NA934V; Amersham антимышиные-HRP), используя набор реагентов Amersham's ECL Plus chemiluminescent kit согласно инструкциям производителя.
FACS Her3: Клетки EBC-1 shMet 4-12 были высеяны в количестве 106 на 10 см чашку в RPMI 1640 (как указано выше), и чашки инкубировали в течение ночи. Dox был добавлен в чашки в конечной концентрации 100 нг/мл. Плашки были инкубированы 48 часов. Вслед за инкубацией клетки были трипсинизированы, центрифугированы, затем ресуспендированы в холодном 200 мкл PBS + 2% FBS (буфер FACS) и перенесены в 96-луночные плашки. Клетки были центрифугированы и ресуспендированы в буфере FACS с 10 мкг/мл антителом Her3:1638 (3E9.2G6) из Genetech. Клетки инкубировали в течение 1 часа на льду, затем промыты холодным буфером FACS и ресуспендированы в буфере FACS + 1:200 RPE, конъюгированных с F(ab')2 козьим антимышиным IgG + IgM (H+L) (Jackson Immuno cat# 115-116-068). Клетки были инкубированы на льду в течение 30 минут, затем однократно промыты холодным буфером FACS и ресуспендированы в буфере FACS с 7AAD (BD Pharmingen cat#559925). Анализ FACS был выполнен в соответствии с инструкциями производителя.
Лизаты опухолей: Опухоли ксенотрансплантанта EBC-1-shMet были образованы, и Dox был дозирован, в частности, как описано в Примере 3, за исключением того, что Dox был использован в концентрации 1 мг/мл в 5% сахарозе, и размеры опухоли могли достигать 300-400 мм3 до начала лечения. Животным вводили дозы препарата в течение трех дней, затем их умерщвляли. Быстрозамороженные образцы опухоли ксенотрансплантанта EBC-1-shMet-4.12 помещали в 2 мл холодного лизирующего буфера (PBS + 1%TritonX-100 + (3Х) Фосфотазный коктейль 2 (Sigma cat#P5726)) и ингибитор протеазы Complete Mini EDTA-Free (Roche#11 836 170 001). Опухоли были гомогенизированы ручным гомогенизатором, и лизаты были инкубированы на льду в течение 1 часа с периодическим покачиванием. Лизаты были центрифугированы при 10000xG в течение 10 минут при 4°С, перенесены в новую пробирку, и была проведена количественная оценка белка Her3, используя метод BCA (Pierce cat#23225).
Результаты
кшРНК-опосредованное подавление экспрессии c-met снижало уровни pEGFR и значительно увлеличивало уровни белка HER3 (Фигура 10А). Анализ FACS выявил повышенные уровни поверхностного HER3 после подавления c-met (Фигура 10В). Подавление c-met в опухолях ксенотрансплантанта EBC-1-shMet-4.12 привело к повышению уровней белка HER3 (Фигура 10С).
Эти данные демонстрируют, что активность c-met может регулировать уровень экспрессии HER3. В частности, ингибирование c-met приводило к повышению уровней белка HER3 и сниженным уровням pEGFR. Снижение уровня pEGFR после ингибирования c-met, по-видимому, происходит из-за сниженной работы аутокринной сигнальной системы лигандами EGFR (смотри Фигура 9), и увеличенные уровни HER3 могут повышать чувствительность к эрлотинибу, как было продемонстрировано другими исследователями (например, Yauch et al. Clin Cancer Res (2005) 11:8686-98). Эти результаты предполагают, что активность HER3 (например, сигнальная система, опосредованная действием HER2) может усиливать последующее ингибирование сигнальной системы c-met, и дополнительно поддерживают использование комбинированной терапии ингибиторами c-met и HER3 для лечения рака.
Пример 7: Активация сигнального пути EGFR может восстанавливать клеточную пролиферацию и жизнеспособность клетки, в которой активность с-met ингибирована
Материалы и способы
Клетки EBC-1 shMet были высеяны в количестве 5000/лунка в среду RPMI 1640 (содержащей 10% Tet-Free FBS from Clontech cat#631107) в 96-луночную плашку с темными стенками, и плашки были инкубированы в течение ночи. Среда была заменена на свежую среду +/- 100 нг/мл dox, и плашки инкубировали 48 часов. Лиганды EGFR были затем добавлены до конечной концентрации, как описано ниже, и плашки инкубировали дополнительные 48 часов, затем было определено число клеток, используя Cell TiterGlo (Promega #G7570) как описано в настоящем документе: Dox + 100 нг/мл HGF; Dox + 50 нМ TGFα; Dox + 5 нг/мл HGF; и Dox + 1 нМ TGFα.
Результаты
Подавление экспрессии c-met с помощью кшРНК привело к занчительному падению числа клеток, предполагая снижение клеточной выживаемости и пролиферации. EGFR лиганды HGF и TGFα обладали способностью сохранять число клеток доза-зависимым способом, хотя HGF, как оказалось, сохранял число клеток несколько лучше, нежели TGFα. Эти результаты демонстрируют, что активация сигнальной системы EGFR может восстанавливать клеточную пролиферацию и клеточную жизнеспособность в клетках, в которых активность сигнальной системы c-met ингибирована. Таким образом, сигнальная система EGFR (и/или других членов семейства HER) компенсирует потерю активности сигнальной системы c-met. Эти результаты поддерживают использование комбинированной терапии с использованием ингибиторов c-met и EGFR и согласуются со значительно увеличенной эффективностью воздействия на опухоль ксенотрансплантанта, наблюдаемой при комбинированном лечении опухолей ингибиторами EGFR и c-met (Пример 4).
Пример 8: Активация c-met приводит к активации EGFR, c-met взаимодействует с EGFR независимо от активации сигнального пути c-met или EGFR, и активация c-met ослабляет ответ на ингибитор EGFR.
Материалы и способы
Клетки: Клетки NCI-H596 были получены из Американской Коллекции Типовых Культур (ATCC) и поддерживались в RPMI 1640 с добавлением 10% фетальной бычьей сыворотки (FBS; Sigma, St. Louis, MO) и 2 мМ L-глутамина. Среду для анализа клеток меняли, как описано ниже в зависимости от эксперимента.
Лекарственные средства и факторы роста: Эрлотиниб и MetMAb были предоставлены компанией Genentech, Inc., как описано выше. HGF и TGFα были получены в Genentech.
Иммунопреципитация и иммуноблоттинг: Клетки голодали ночь в 0,1% BSA/RPMI до стимуляции лигандом и/или обработки дозой вещества, как описано в тексте. Лиганды HGF и TGF-α были собственного производства. В момент сбора клеток они были мгновенно однократно промыты в ледяном PBS с последующим лизированием в лизирующем буфере (CST#9803) с добавлением 1 мМ каждого из следующих реагентов: Ингибиторов протеаз (Sigma Cat#P3840), Ингибиторов фосфатаз (Sigma Cat#P2850 и Р3726), NaF, Na3VO4 и PMSF. Образцы были помещены в 180° ротор при 4°С, с последующей очисткой при 14000 об/мин, 20 мин 4°С. Концентрация белка была определена, используя метод Брэдфорда.
Клеточные лизаты были либо непосредственно нанесены на гели (Фигура 12, эквивалентная концентрация лизата 40 мкг/лунка) или иммунопреципитированы (Фигура 13, эквивалентная концентрация лизата 1,6 мг/образец). Иммунопреципитация была выполнена с каждым из следующих антител; конъюгированные с агарозой cMET:(Santa Cruz Biotechnology Cat#SC-161AC), EGFR (Neomarkers MS-609-P) + протеин А-сефарозные шарики быстрого потока. Образцы были помещены в 180° ротор на ночь при 4°С с последующими тремя промываниями лизисным буфером и последующей денатурацией в SDS-буфере для образцов, содержащем бета-меркаптоэтанол. Образцы были нагреты в течение 5 минут при 95°С с последующим нанесением на 4-12% градиентные гели и переносом на нитроцеллюлозные мембраны, используя стандартные методики вестерн-блоттинга. Мембраны были блокированы в 5% молоке/TBST в течение 1 часа, RT и затем было проведено зондирование со следующими фосфоантителами в течение ночи при 4°С, как указано в тексте:p-c-met: pTyr 4G10 (Upstate Cat#05-777); pEGFR (Cell Signaling Technologies Cat#2264). Мембраны были десорбированы восстанавливающим десорбирующим буфером (Pierce Cat#21059) и затем повторно зондированы с антителами к суммарному белку: cMET DL-21 (Upstate Biotech Cat #05-238); EGFR (mbl Cat#MI-12-1); бета-актин (Santa Cruz Biotechnologies Cat#SC-1616). Вторичные антитела были получены из Jackson Laboratories. Проводили оценку иммуноблотам, используя метод ECL в соответствии с рекомендациями производителя.
Анализ клеточной жизнеспособности: Для анализа клеточной жизнеспособности клетки были высеяны в четырех экземплярах в количестве 1×103 клеток на лунку в 384-луночные плашки в среду RPMI, содержащую 0,5% FBS (среда для исследования), инкубировали в течение ночи до стимуляции средой для исследования, содержащей 3 нМ TGF-α +/- HGF. Эрлотиниб был добавлен в различных концентрациях, и через 72 часа была измерена клеточная жизнеспособность, используя Люминесцентный анализ клеточной жизнеспособности Celltiter-Glo (Promega, Madison, WI).
Результаты
Активация c-met под действием HGF приводит к активации EGFR
Поскольку аткивация c-met приводила к положительной регуляции разнообразных лигандов EGFR в клетках NCI-H596, мы предположили, что активация c-met приводит к трансактивации сигнального пути EGFR. Чтобы проверить данную гипотезу, клетки NCI-H596 NSCLC были обработаны в присутствии и в отсутствие HGF in vitro, и клеточные лизаты были проанализированы на десятой минуте, 24, 48 и 72 часах для исследования активации сигнального пути EGFR.
Активация сигнальной системы c-met приводит к активации сигнальной системы EGFR (Фигура 12). Индукция уровня pEGFR наблюдалась уже на десятой минуте после стимуляции HGF, предполагая, что активация c-met напрямую трансактивирует сигнальную систему EGFR (Фигура 12). Увеличенные уровни pEGFR наблюдались в более поздних временных точках (24, 48 и 72 часа после стимуляции HGF) (Фигура 12). Замедленная кинетика активации pEGFR согласуется с данными, показывающими, что активность c-met приводит к повышенной экспрессии лигандов EGFR, которые могут отвечать за отсроченную (>24 часов) активацию сигнальной системы EGFR. На этой модели активация EGFR прогнозируется стать увеличенной в более поздние временные точки и оставаться относительно высокой, что согласуется с данными, показанными в настоящем документе.
C-met взаимодействует с EGFR независимо от состояния активации сигнального пути c-met или EGFR.
Эксперименты по коиммунопреципитации (co-IP) были выполнены для выяснения, может ли активность c-met и/или EGFR приводить к физической ассоциации c-met c EGFR. Клетки NCI-H596 были обработаны без лиганда, одним TGF-α, одним HGF или TGF-α плюс HGF в течение 10 минут или 24 часов. После этой обработки была проведена иммунопреципитация c-met с последующим вестерн-блоттингом по выявлению любого фосфотирозина (4G10), EGFR или с-met.
Иммунопреципитация с-met осаждала EGFR в отсутствие любого лиганда и в поздние временные точки, когда уровни pc-met и pEGFR падали, указывая на то, что c-met взаимодействовал с EGFR независимо от состояния активации сигнального пути c-met или EGFR (Фигура 13). C-met IP с проведением блоттинга для выявления фосфотирозина выявили, что активация EGFR и c-met была лиганд-зависимой и ослабевала после 24 часов. Активация c-met под действием HGF приводила к коиммунопреципитации pEGFR; однако уровни pEGFR были намного ниже, чем уровни pEGFR, наблюдаемые при стимуляции клеток одним TGF-α или в комбинации с HGF. Активация c-met или EGFR их соответствующими лигандами показала, что каждый сигнальный путь может быть активирован независимо друг от друга.
Активация c-met ослабляла ответ клеток NCI-H596 на действие ингибитора EGFR, и обработка MetMAb антителом анти-c-met сохраняла ответ на действие ингибитора EGFR.
Клетки NCI-H596 чувствительны к эрлотинибу (TARCEVATM), ингибитору EGFR, когда растут в присутствии TGF-α, как продемонстрировано сниженной клеточной жизнеспособностью при выращивании в присутствии эрлотиниба и TGF-α. Чтобы выяснить, может ли активация сигнального пути c-met изменить ответ клеток NCI-H596 на действие эрлотиниба, клетки были стимулированы TGF-α, обработаны эрлотинибом и/или HGF, затем был проведен анализ клеточной жизнеспособности.
Низкие уровни HGF демонстрировали среднее воздействие на клеточную чувствительность к эрлотинибу; однако чувствительность к эрлотинибу значительно снижалась доза-зависимым способом по мере повышения концентраций HGF (Фигура 14), как показано увеличенной жизнеспособностью клеток в этих условиях. Эти данные указывают на то, что HGF-активация сигнального пути Met достаточна для ослабления ответа клеток NCI-H596 на действие эрлотиниба.
Чтобы выяснить, снижает ли комбинация ингибиторов c-met и ингибиторов EGFR клеточную жизнеспособность клеточных линий, которые коактивированы HGF и TGFα, оценка жизнеспособности клеток NCI-H596 была проведена в присутствии HGF, TGFα и различных доз эрлотиниба и/или MetMAb антитела антагониста c-met (1 мкМ).
Присутствие HGF ослабляет ответ клеток NCI-H596 на действие эрлотиниба (Фигура 15). Ингибирование сигнального пути c-met действием MetMAb в значительной степени восстанавливало чувствительность к эрлотинибу (Фигура 15), тем самым, предполагая, что обработка ингибиторами c-met и EGFR может оказывать комбинированное воздействие, влияющее на клеточную жизнеспособность в клеточной линии NCI-H596.
Вместе взятые эти исследования подтверждают гипотезу о том, что активация сигнального пути c-met напрямую активировала сигнальный путь EGFR как посредством индукции экспрессии лиганда EGFR, так же как и через непосредственное взаимодействие c-met с EGFR. Эти результаты согласуются со значительно повышенной эффективностью воздействия на опухоль ксенотрансплантанта, наблюдаемой при лечении опухолей комбинированием ингибиторов EGFR и c-met (Пример 4).
Пример 9: Комбинированное лечение антагонистом c-met и антагонистом EGFR приводило к лучшему ингибированию пролиферации и сигнального пути, отвечающего за выживаемость, в опухолях ксенотрансплантанта NCI-H596.
Материалы и способы
Опухоли ксенотрансплантанта NCI-H596 hu-HGF-Tg-SCID: Ксенотрансплантанты NCI-H596 были обнаружены у мышей hu-HGF-Tg-SCID, как описано в Примере 4. Размеры опухолей могли достигать 200-300 мм3 до начала лечения. Дозирование было выполнено, как описано в Таблице 8. В нескольких словах, доза MetMAb (30 мг/кг) или буфера MetMAb была введена в нулевой временной точке (0 часов), и доза наполнителя метилцеллюлозного твина (MCT) или эрлотиниба (150 мг/кг) была введена в 18-часовой временной точке (18 часов). Мышей подвергли эвтаназии, и опухоли и плазма были собраны в 24-часовой временной точке (24 часа). Опухоли были быстро заморожены в жидком азоте и затем хранились при -70°С до использования в иммунопреципитации и иммуноблоттинге.
Иммунопреципитации и иммуноблоттинг: Чтобы провести анализ белка в опухолях, опухоли были сначала гомогенизированы, используя стеклянный гомогенизатор Даунса, в лизирующем буфере (Cell Signaling Technology, Inc., Danvers, MA) с добавлением 1 мМ PMSF, дополнительный коктейл ингибитора протеазы и коктейли I и II ингибитора фосфатазы (Sigma, Inc., St. Louis, MO). Лизаты были инкубированы на льду в течение часа и затем центрифугированы при 14000 × g в течение пяти минут, и были собраны супернатанты. Концентрации белка были определены, используя набор реагентов BCATMProtein Assay Kit (Pierce, Inc., Rockford, IL), и образцы были подвергнуты иммуноблоттингу. Чтобы выполнить иммунопреципитации, 1,5 мг лизатов опухолей были использованы для осаждения Met, используя С-28 античеловеческое c-Met поликлональное антитело (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), конъюгированное с агарозными шариками, или EGFR, используя антитело MI-12-1 (MBL, Inc., Woburn, MA), при 4°С ночь с перемешиванием. Шарики были промыты три раза лизирующим буфером при 4°С с последующим ресуспендированием в буфере 1X Novex Tris-Glycine SDS Running Buffer (Invitrogen, Inc., Carlsbad, CA), содержащем 2,5% (вес/объем) бета-меркаптоэтанолом. Для прямого вестерн-блота 50 мкг лизата опухолей было нанесено в лунку. Образцы были затем проанализированы с помощью SDS-PAGE и иммуноблоттингом. Использованные антитела включают мышиные античеловеческие c-Met DL-21, мышиные антифосфотирозиновые mAb 4G10 (оба из Upstate Biotechnology/Millepore, Inc., Charlottesville, VA), анти-Akt, анти-p44/42 MAP киназа (ERK-1/2), анти-фосфо-Akt (Ser473), анти-фосфо-р44/42 MAP киназа (ERK-1/2) (Thr202/Tyr204), все используемые в соответствии с рекомендациями производителя (все из Cell Signaling Technology, Inc. (Danvers, MA)). Козьи антимышиные-IRdye800 (Rockland Immunochemicals, Inc., Gilbertsville, PA) и козьи антикроличьи-AlexaFluor680 (Molecular Probes, Inc., Eugene, OR) были использованы в качестве вторичных антител. Были получены изображения иммуноблотов, и уровни фосфо-белков были количественно определены и нормированы к суммарным уровням белка (например, pEGFR относительно суммарного EGFR), используя устройство для визуализации Odyssey (LI-COR Biosciences, Lincoln, NE).
Результаты
Активация сигнальных путей c-met и EGFR была исследована в опухолях ксенотрансплантанта, образованных в мышиной модели ксенотрансплантанта NCI-H596 hu-HGF-Tg-SCID и леченных ингибитором EGFR, ингибитором c-met и комбинацией ингибиторов c-met и EGFR. Двадцать мышей hu-HGF-Tg-SCID были инокулированы клетками NCI-H596, и образование опухолей было установлено, как описано ранее. Как только опухоли достигали размера в диапазоне 200-300 мм3, мыши были поровну разделены на четыре группы, основываясь на объеме опухоли, и было начато введение доз препаратов (Таблица 8). Доза MetMAb была введена за 24 часа до забора опухоли, в то время как доза эрлотиниба была введена за 6 часов до забора опухоли. Времена дозирования были выбраны, исходя из относительного времени полужизни каждого терапевтического средства. В 24-часовой временной точке мыши были подвергнуты эвтаназии, опухоли были собраны и были подвергнуты анализу с помощью иммунопреципитаций (IP) и/или иммуноблотов на выявление фосфорилированного и суммарного Met, EGFR, Akt и ERK1/2.
Лечение одним MetMAb привело к ингибированию фосфорилирования c-met до 12% (+/- 3,6%) от контроля наполнителем (Фигура 16), и комбинированное лечение с помощью MetMAb и эрлотиниба привело к ингибированию фосфорилирования c-met до 6% (+/- 3,5%) от контроля наполнителем (Фигура 16) (р=0,039). Лечение одним эрлотинибом (Фигура 16) не снижал фосфорилирования c-met. Лечение одним эрлотинибом ингибировало фосфорилирование EGFR до 16% (+/- 7,9%) от контроля наполнителем, и комбинированная терапия эрлотинибом и MetMAb ингибировала фосфорилирование EGFR до 19% (+/- 15%) от контроля наполнителем (Фигура 16). Лечение одним MetMAb также умеренно ингибировало pEGFR до 62% (+/- 21,6%) от контроля наполнителем (р=0,006).
Эти результаты демонстрируют, что MetMAb и эрлотиниб каждый эффективно ингибируют активацию их соответствующих мишеней, и что блокирование c-met может ингибировать ответ pEGFR в NCI-H596 hu-HGF-Tg-SCID модели.
Комбинированное лечение MetMAb и эрлотинибом также приводило к более эффективному ингибированию сигнальных путей PI-3K/Akt и Ras-RAF-MEK-ERK1/2, которые активируются в нисходящем направлении от активации Met и EGFR, где сигнальные пути действуют для активации выживаемости и пролиферации опухолевых клеток, соответственно, и помогают регулировать онкогенез. Фосфо-Akt и фосфо-ERK-1/2 были исследованы в опухолях ксенотрансплантанта из животных, леченных MetMAb, эрлотинибом или MetMAb с эрлотинибом.
Лечение одним MetMAb привело к ингибированию pAkt до 72% (+/- 27,9%) от контроля наполнителем и ингибированию pERK-1/2 до 72% (+/- 40,3%) от контроля наполнителем (Фигура 15, Таблица 9). Лечение эрлотинибом привело к более сильному ингибированию pAkt до 45% (+/- 25,7%) и ERK-1/2 на 39% (+/- 8,9%) от контроля наполнителем, соответственно (Фигура 16, Таблица 9). Комбинированное лечение MetMAb и эрлотинибом продемонстрировало сильное ингибирование pAkt и pERK-1/2 до 24% (+/- 13,8%) от контроля наполнителем и 29% (+/- 2,9%) от контроля наполнителем, соответственно (Фигура 15, Таблица 9). Эти результаты показали, что комбинированное лечение MetMAb и эрлотинибом ингибировало нисходящие сигнальные пути более эффективно, чем лечение MetMAb или эрлотинибом по отдельности.
Таблица 9. Сводная информация о количественной оценке уровней фосфо-белков в виде процента от контроля наполнителем после лечения мышей, несущих опухоль NCI-H596, с помощью MetMAb, эрлотиниба или комбинации MetMAb и эрлотиниба. Уровни фосфо-белков были определены путем количественной оценки интенсивности сигнала полос с помощью Li-Cor, нормализации к уровням суммарного белка (за вычетом фона). Данные представлены как процент от контроля наполнителем (значения представляют собой среднее по опухолям от 5 леченных различных животных, как показано на фигуре 16).
Фигуры 17А и 17В в виде диаграммы суммируют некоторые данные, раскрытые в настоящем описании следующим образом:
(1) c-met и EGFR были коэкспрессированы в клеточных линиях и опухолях NSCLC;
(2) активность c-met осуществляла положительную регуляцию экспрессии лигандов EGFR и pEGFR;
(3) активность c-met осуществляла отрицательный контроль экспрессии HER3;
(4) лечение TGFα избавляло лиганд-независимые c-met-активируемые клетки от потери жизнеспособности, опосредованной ингибитором c-met; и
(5)активация c-met уменьшала ответ на действие эрлотиниба in vitro и in vivo.
Часть списка литературных источников
Bhargava, M, Joseph, A, Knesel, J, Halaban, R, Li, Y, Pang, S, Goldberg, I, Setter, E, Donovan, MA, Zarnegar, R, Michalopoulos, GA, Nakamura, T, Faletto, D. and Rosen, E. (1992). Scatter factor and hepatocyte growth factor: activities, properties, and mechanism. Cell Growth Differ., 3(1); 11-20.
Kong-Beltran, M., Seshagiri, S., Zha, J., Zhu, W., Bhawe, K., Mendoza, N., Holcomb, T., Pujara, K., Stinson, J., Fu, L., Severin, C., Rangell, L., Schwall, R., Amler, L., Wickramasinghe, D., Yauch, R. (2006). Somatic Mutations Lead to an Oncogenic Deletion of Met in Lung Cancer. Cancer Res, 66 (1); 283-289.
Peschard, P.,.Fournier, T.M., Lamorte, L, Naujokas, M.A., Band, H., Langton, W.Y., Park, M. (2001). Mutation of the c-Cbl TKB domain binding site on the Met receptor tyrosine kinase converts it into a transforming protein. Mol Cell., 8(5); 995-1004.
Ridgeway, J.B.B., Presta, L.G., Carter, P. (1996). 'Knobs-into-holes'engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Engin. 9 (7): 617-621.
Rong, S., Bodescot, M., Blair, D., Dunn, J., Nakamura, T., Mizuno, K., Park, M., Chan, A., Aaronson, S., Vande Woude, G.F. (1992). Tumorigenicity of the met proto-oncogene and the gene for the hepatocyte growth factor. Mol Cell Biol., 12(11); 5152-5158.
Zhang, Y-W., Su, Y., Lanning, N., Gustafson, M., Shinomiya, N., Zhao, P., Cao, B., Tsarfaty, G., Wang, L-M, Hay, R., Vande Woude, G.F. (2005). Enhanced growth of human met-expressing xenografts in a new strain of immunocompromised mice transgenic for human hepatocyte growth factor/scatter factor. Oncogene, 24; 101-106.
Хотя вышеуказанное изобретение в некоторых вариантах осуществления было описано в качестве иллюстрации и примеров для ясности, описание и примеры не следует рассматривать как ограничивающие рамки изобретения.
Реферат
Настоящее изобретение относится в основном к областям молекулярной биологии и медицины. В частности, изобретение относится к комбинированной терапии патологических состояний, таких как рак. Способ лечения рака включает введение терапевтически эффективного количества антагониста c-met и антагониста EGFR. 4 з.п. ф-лы, 9 табл., 9 пр., 25 ил.
Комментарии