Производные трициклического лактама в качестве ингибиторов 11-β-гидроксистероидной дегидрогеназы - RU2386617C2
Код документа: RU2386617C2
Описание
Метаболический синдром представляет собой заболевание с возрастающим уровнем распространения не только на Западе, но в Азии и в развивающихся странах. Его отличает ожирение, в частности центральное или висцеральное ожирение, диабет 2 типа, гиперлипидемия, гипертензия, артериосклероз, ишемические болезни сердца и со временем хроническая почечная недостаточность (C.T. Montague et al. (2000), Diabetes, 49, 883-888).
Глюкокортикоиды и 1lβ-HSDl известны как важные факторы дифференцировки жировых стромальных клеток в зрелые адипоциты. В висцеральных стромальных клетках больных, страдающих ожирением, уровень 11β-HSD1 мРНК возрастает в сравнении с подкожной жировой тканью. Кроме того, повышенную экспрессию 11β-HSD1 в жировой ткани у трансгенных мышей связывают с возросшими уровнями кортикостерона в жировой ткани, висцеральным ожирением, инсулинчувствительностью, диабетом 2 типа, гиперлипидемией и гиперфагией (H. Masuzaki et al (2001), Science, 294, 2166-2170). Поэтому наиболее вероятно, что 11β-HSD1 вовлечен в развитие висцерального ожирения и метаболического синдрома.
Ингибирование 11β-HSD1 приводит к уменьшению дифференцировки и возрастанию, пролиферации жировых стромальных клеток. Более того, дефицит глюкокортикоидов (адреналектомия) увеличивает способность инсулина и лептина к стимуляции анорексии и потери массы; введение глюкокортикоидов обращает данный эффект в обратную сторону (P.M. Stewart et al (2002), Trends Endocrin. Metabol, 13, 94-96). Эти данные предполагают, что усиленная реактивация кортизона с помощью 11β-HSD1 может обострить ожирение и это может оказаться полезным для ингибирования этого фермента в жировой ткани больных, страдающих ожирением.
Ожирение также связано с повышенными сердечно-сосудистыми рисками. Существует ощутимая зависимость между скоростью экскреции кортизола и HDL (липопротеиды высокой плотности) холестерином как у мужчин, так и у женщин; означая, что глюкокортикоиды регулируют ключевые компоненты повышенных сердечно-сосудистых рисков. По аналогии потеря аортальной эластичности также связана с висцеральным ожирением у взрослых особей.
Влияние эффекта сниженной активности 11β-HSD1 подчеркивается тем, что мышь воздействием 11β-HSD1 приведенная в шоковое состояние имеет увеличенные плазменные уровни эндогенного активного глюкокортикоида, но несмотря на это сохраняет защищенность от инсулинрезистентности, вызываемой стрессом или ожирением. Дополнительно эти приведенные в стрессовое состояние мыши обнаруживают анти-атерогенный плазмидный липидный профиль и обладают преимуществом сниженного возрастного ослабления познавательной способности.
ГЛЮКОКОРТИКОИДЫ И ГЛАУКОМА
Глюкокортикоиды при экзогенном введении и при некоторых состояниях гиперпродуцирования, таких как синдром Кушинга, повышая внутриглазное давление, увеличивают риск заболевания глаукомой. Рост стимулируемого кортикостероидами внутриглазного давления вызывает повышенная резистентность к оттоку водянистой влаги вследствие вызванных глюкокортикоидами изменений в трабекулярной сети и ее интрацеллюлярном матриксе (Zhou et al. (Int J Mol Med (1998) 1, 339-346). Сообщалось также, что кортикостероиды увеличивают количества фибронектина, так же как и коллагена типа I и типа IV, в трабекулярный сети передних камер глаза культивируемого органа теленка.
11β-HSD1 экспрессируется в базальных клетках корнеального эпителия и непигментированных эпителиальных клетках. Глюкокортикоидную рецепторную мРНК (mRNA) определяли только в трабекулярной сети, тогда как в непигментированных эпителиальных клетках присутствовала глюкокортикоидная, минералокортикоидная рецепторная и 11β-HSD1 мРНК. Введение больным карбеноксолона приводило к значительному снижению внутриглазного давления (S. Rauz et al. (2001), Invest. Ophtalmol. Vis. Science, 42, 2037-2042). Это подчеркивает роль ингибиторов HSD1 при лечении глаукомы.
Соответственно для решения указанной проблемы с помощью настоящего изобретения было необходимо идентифицировать сильные ингибиторы 11β-HSD1, обладающие высокой селективностью по отношению к 11β-HSD1, и использовать их при лечении патологий, связанных с избыточным образованием кортизола, то есть нарушений, при которых необходим сниженный уровень активного глюкокортикоида, таких как метаболический синдром, диабет 2 типа, ослабленная толерантность к глюкозе (IGT), ослабленная утилизация глюкозы (IFG), дислипидемия, гипертензия, ожирение, диабет, сердечно-сосудистые заболевания, связанные с ожирением, артериосклероз, атеросклероз, миопатия, остеопороз, нейродегенеративные и психиатрические расстройства, расстройства, связанные со стрессом, и глаукома. Как показано в дальнейшем, было обнаружено, что 3-замещенные производные 2-пирролидинона формулы (I) оказались пригодными в качестве лекарственного средства, в частности, при получении лекарственного препарата для лечения патологий, связанных с избыточным образованием кортизола.
Blommaert A. et al. (Heterocycles (2001), 55(12), 2273-2278) предлагают получение подложки из производного (R)-фенилглицинола, закрепленной на пиперидин- и пирролидинонподобном полимере и, в частности, описывают, l-[(lR)-2-гидрокси-l-фенилэтил]-3-метил-3-(фенилметил)-2-пирролидинон и, l-[(lR)-2-гидрокси-l-фенилэтил]-3-(фенилметил)-2-пирролидинон, (3R).
Bausanne I. et al. (Tetrahedron: Assymetry (1998), 9(5), 797-804) предлагают получение 3-замещенных пирролидинонов посредством α-алкилирования хирального нерацемического γ-лактона и, в частности, описывают l-(2-гидрокси-l-фенилэтил)-3-бензилпирролидин-2-он.
US 2001/034343; US 6211199; US 6194406; WO 97/22604 и WO 97/19074 представляют собой ряд заявок на патент, поданных Aventis Pharmaceuticals Inc., в которых предлагают пригодные для лечения аллергических заболеваний 4-(lH-бензимидазол-2-ил)[l,4]диазепаны. В этих заявках описывают 3-замещенные пирролидиноны по настоящему изобретению как промежуточное соединение при синтезах указанных 4-(lH-бензимидазол-2-ил)[l,4]-диазепанов. В этих заявках, в частности, описывают 3-[(4-фторфенил)метил]-l-[(lS)-l-фенилэтил]-2-пирролидинон и 3-[(4-фторфенил)метил]-1-[(1R)-1-фенилэтил]-2-пирролидинон.
В PCT публикации WO 03065983 (Merck & Co., Inc.) и WO 2004056744 (Janssen Pharmaceutica N.V.) описывают также соединения подобные адамантилу. Если принять WO 2004056744 за наиболее близкий известный уровень техники, то соединения по настоящей заявке отличает то, что адамантиловый цикл соединен с кольцевым амидным азотом, являющимся частью трициклической кольцевой системы. Вопреки тому, что в WO 03065983 раскрывают, что адамантиловый цикл может быть непосредственно связан с трициклической кольцевой системой, следует заметить, что указанные трициклические кольцевые системы отличаются тем, что имеют 2-адамантилтриазол в качестве структурного элемента остова; и соответственно не следует ожидать, что замена триазола на имидазолидинон или пирролидинон могла бы быть выполнена без потери желаемой активности, то есть способности к сильному ингибированию 11βHSD, при селективности по отношению к 11βHSD1.
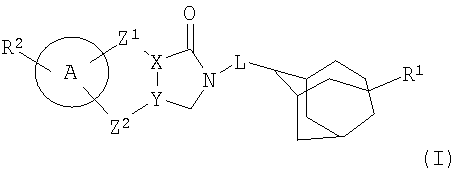
Следовательно, ни в одном из цитируемых документов терапевтическое применение производных трициклического адамантиламида по настоящему изобретению не было не описано, не предложено. Соответственно первый аспект данного изобретения касается соединения формулы (I):

его N-оксидных форм, фармацевтически приемлемых аддитивных солей и их стереохимически изомерных форм, где
X представляет собой С или N;
Y представляет собой С или N;
L представляет собой метил, прямую связь;
Z1 представляет собой направленную связь или C1-2алкил- или двухвалентный радикал формулы
-CH2-CH= (а) или -CH= (b);
Z2 представляет собой прямую связь, C1-2алкил- или двухвалентный радикал формулы
-CH2-CH= (а) или -CH= (b);
R1 представляет собой водород, гало, циано, амино, фенил, гидрокси, C1-4алкилоксикарбонил, -O-(C=O)-C1-4алкил, гидроксикарбонил, NR3R4 или C1-4алкил, где указанный C1-4алкил или -O-(C=O)-C1-4алкил являются необязательно замещенными одним или несколькими заместителями, выбранными из гало, гидроксикарбонила, фенила, C1-4алкилокси или NR5R6, или R1 представляет собой C1-4алкилокси-, необязательно замещенный одним или несколькими заместителями, выбранными из гало, гидроксикарбонила, фенила, C1-4алкилокси или NR7R8;
R2представляет собой водород, гало, C1-4алкил или C1-4алкилокси;
R3 и R4, каждый независимо, представляют собой водород, C1-4алкил или C1-4алкилкарбонил;
R5 и R6, каждый независимо, представляют собой водород, C1-4алкил или C1-4алкилкарбонил;
R7 и R8, каждый независимо, представляют собой водород, C1-4алкил или C1-4алкилкарбонил;
A представляет собой фенил или моноциклический гетероцикл, выбранный из группы, состоящей из тиофенила, фуранила, оксазолила, тиазолила, имидазолила, изоксазолила, изотиазолила, пирридинила, пиридазинила, пиримидинила и пиперазинила.
Используемые в дальнейшем соединения формулы (I) обозначают соединения по настоящему изобретению, включающие в себя соединения формул (Ibis), (Ii), (Iii), (Iiii), (Iiv) и их фармацевтически приемлемые N-оксиды, аддитивные соли, четвертичные амины и стереохимически изомерные формы.
Используемый в предшествующих определениях и в дальнейшем термин «гало» является обобщенным обозначением для фтора, хлора, брома и йода; C1-2алкил обозначает прямые насыщенные углеводородные радикалы, содержащие от 1 до 2 атомов углерода, то есть метил или этил; C1-4алкил обозначает насыщенные углеводородные радикалы с прямой и разветвленной цепью, имеющие от 1 до 4 атомов углерода, такие как, например, метил, этил, пропил, бутил, 1-метилэтил, 2-метилпропил, 2,2-диметилэтил и тому подобное; C1-4алкилокси обозначает прямые или разветвленные насыщенные углеводородные радикалы, имеющие от 1 до 4 атомов углерода, такие как метокси, этокси, пропилокси, бутилокси, 1-метилэтилокси, 2-метилпропилокси и тому подобное.
Гетероциклы, упоминаемые в приведенных выше определениях и используемые в дальнейшем, могут быть соответствующим образом присоединены к остатку молекулы формулы (I) через любой кольцевой углеродный атом или гетероатом. Таким образом, например, если гетероциклом является имидазолил, он может представлять собой 1-имидазолил, 2-имидазолил, 3-имидазолил, 4-имидазолил и 5-имидазолил; если это тиазолил, он может представлять собой 2-тиазолил, 4-тиазолил и 5-тиазолил.
Подразумевают, что фармацевтически приемлемые аддитивные соли, упоминаемые в дальнейшем, содержат терапевтически активные нетоксичные формы аддитивной соли кислоты, которые способны образовывать соединения формулы (I). Последние могут быть получены обработкой основной формы соответствующей кислотой. Соответствующие кислоты включают в себя, например, неорганические кислоты, такие как галоидоводородные кислоты, например, хлористоводородная или бромистоводородная кислота; серная; азотная; фосфорная и подобные кислоты; или органические кислоты, такие, как, например, уксусная, пропановая, оксиуксусная, молочная, пировиноградная, щавелевая, малоновая, янтарная (т.е. бутандиовая кислота), малеиновая, фумаровая, яблочная, винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламовая, салициловая, п-аминосалициловая, памовая и подобные кислоты.
Подразумевают, что упомянутые выше фармацевтически приемлемые аддитивные соли включают в себя терапевтически активные нетоксичные формы основно-аддитивной соли, которые способны образовывать соединения формулы (I). Примеры таких форм основно-аддитивной соли представляют собой, например, соли натрия, калия, кальция, а также соли фармацевтически приемлемых аминов, таких как, например, аммиак, алкиламины, бензатин, N-метил-D-глюкамин, гидрабамин, аминокислоты, например, аргинин, лизин.
И обратно, указанные формы солей могут быть превращены обработкой соответствующим основанием или кислотой в форму свободной кислоты или основания.
Термин «аддитивная соль», используемый выше, также охватывает сольваты, которые соединения формулы (I) способны образовывать, как и соли. Такие сольваты представляют собой, например, гидраты, алкоголяты и тому подобное.
Термин «стереохимически изомерные формы», используемый ранее, обозначает возможные различные изомерные формы, а также конформационные формы, которыми соединения формулы (I) могут обладать. Если не оговорено или не указано иначе, химическое наименование соединений обозначает смесь всех возможных стереохимических и конформационных изомерных форм; при этом указанные смеси включают все диастереомеры, энантиомеры и/или конформеры основной молекулярной структуры. Подразумевают, что все стереохимически изомерные формы соединений формулы (I) и в беспримесном чистом виде и в виде смеси друг с другом охвачены объемом настоящего изобретения.
Подразумевают, что N-оксидные формы соединений формулы (I) включают такие соединения формулы (I), в которых один или несколько атомов азота окислены до так называемого N-оксида.
Первая группа соединений представляет собой такие соединения формулы (I), в которых приняты одно или несколько нижеследующих ограничений;
(i) X представляет собой С или N;
(ii) Y представляет собой С или N;
(iii) L представляет собой метил или прямую связь;
(iv) Z1 представляет собой прямую связь, C1-2алкил или двухвалентный радикал формулы
-CH2-CH= (а) или -CH= (b);
(v) Z2 представляет собой прямую связь, C1-2алкил или двухвалентный радикал формулы
-CH2-CH= (а) или -CH= (b);
(vi) R1 представляет собой водород, гало, циано, амино, фенил, гидрокси, C1-4алкилоксикарбонил, гидроксикарбонил, NR3R4 или C1-4алкил, необязательно замещенный одним или несколькими заместителями, выбранными из гидроксикарбонила, фенила, C1-4алкилокси или NR5R6, или R1 представляет собой C1-4алкилокси, необязательно замещенный одним или несколькими заместителями, выбранными из гидроксикарбонила, фенила, C1-4алкилокси или NR7R8;
(vii) R2 представляет собой водород, гало, C1-4алкил или C1-4алкилокси;
(viii) R3 и R4, каждый независимо, представляют собой водород, C1-4алкил или C1-4алкилкарбонил;
(ix) R5 и R6, каждый независимо, представляют собой водород, C1-4алкил или C1-4алкилкарбонил;
(x) R7 и R8, каждый независимо, представляют собой водород, C1-4алкил или C1-4алкилкарбонил;
(xi) A представляет собой фенил или моноциклический гетероцикл, выбранный из группы, состоящей из тиофенила, фуранила, оксазолила, тиазолила, имидазолила, изоксазолила, изотиазолила, пирридинила, пиридазинила, пиримидинила и пиперазинила.
Интересной группой соединений являются такие соединения формулы (I), к которым применены одно или несколько нижеследующих ограничений:
(i) X представляет собой С или N;
(ii) Y представляет собой С или N;
(iii)L представляет собой метил или прямую связь;
(iv) Z1 представляет собой прямую связь, C1-2алкил или двухвалентный радикал формулы
-CH2-CH= (а) или -CH= (b);
(v) Z2 представляет собой прямую связь, C1-2алкил или двухвалентный радикал формулы
-CH2-CH= (а) или -CH= (b);
(vi) R1 представляет собой водород, гало, циано, амино, фенил, гидрокси, C1-4алкилоксикарбонил, гидроксикарбонил, NR3R4 или C1-4алкил, необязательно замещенный одним или несколькими заместителями, выбранными из гидроксикарбонила, фенила, C1-4алкилокси или NR5R6;
(vii) R2 представляет собой водород, гало, C1-4алкил или C1-4алкилокси;
(viii) R3 и R4, каждый независимо, представляет собой водород, C1-4алкил или C1-4алкилкарбонил;
(ix) R5 и R6, каждый независимо, представляют собой водород, C1-4алкил или C1-4алкилкарбонил;
(x) A представляет собой фенил или моноциклический гетероцикл, выбранный из группы, состоящей из тиофенила, фуранила, оксазолила, тиазолила, имидазолила, изоксазолила, изотиазолила, пирридинила, пиридазинила, пиримидинила и пиперазинила.
Следующая интересная группа соединений состоит из таких соединений формулы (I), к которым применены одно или несколько нижеследующих ограничений:
(i) L представляет собой метил или прямую связь;
(ii) R1 представляет собой водород, гало или гидрокси, в частности гало или гидроксил;
(iii) R2 представляет собой водород, гало или C1-4алкилокси;
(iv) A представляет собой фенил, моноциклический гетероцикл, выбранный из группы, состоящей из пиридинила и тиофенила.
Еще одна группа соединений состоит из таких соединений формулы (I), к которым применены одно или несколько нижеследующих ограничений:
(i) L представляет собой метил или прямую связь;
(ii) R1 представляет собой водород, гало, амино или гидрокси, в частности фтор, амино или гидроксил;
(iii) R2 представляет собой водород, бром или метокси;
(iv) Z1 представляет собой прямую связь, метил, этил или двухвалентный радикал формулы
-CH2-CH= (а);
(v) Z2 представляет собой прямую связь, метил или этил;
(vi) A представляет собой фенил, моноциклический гетероцикл, выбранный из группы, состоящей из пиридинила и тиофенила.
Интерес также представляют такие соединения формулы (I), в которых:
A представляет собой фенил или пиридинил и в которых L представляет собой прямую связь; и/или
R1 представляет собой гало, циано, амино, фенил, гидрокси, C1-4алкилоксикарбонил, гидроксикарбонил, NR3R4 или C1-4алкил, необязательно замещенный одним или несколькими заместителями, выбранными из гидроксикарбонила, фенила, C1-4алкилокси или NR5R6, или R1 представляет собой C1-4алкилокси, необязательно замещенный одним или несколькими заместителями, выбранными из гидроксикарбонила, фенила, C1-4алкилокси или NR7R8; в частности R1 представляет собой гало, циано, амино, фенил, гидрокси, C1-4алкилоксикарбонил, гидроксикарбонил, NR3R4 или C1-4алкил, замещенный одним или несколькими заместителями, выбранными из гидроксикарбонила, фенила, C1-4алкилокси или NR5R6.
В предпочтительном варианте осуществления соединения формулы (I) выбирают из группы, включающей:
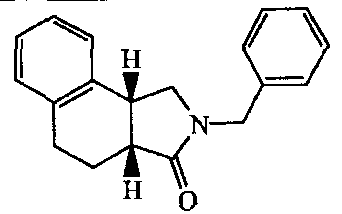
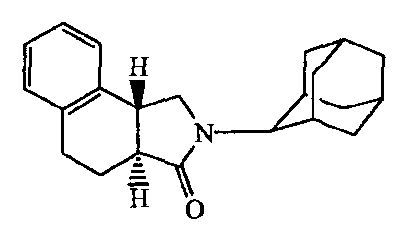
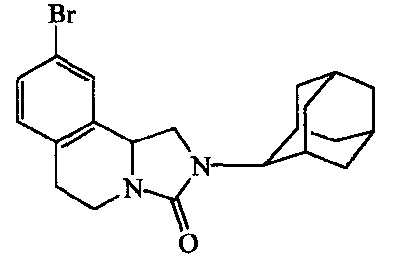
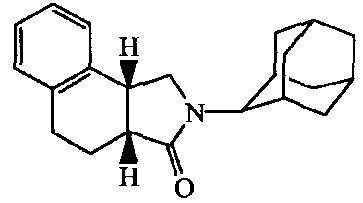
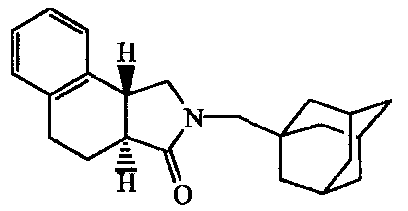
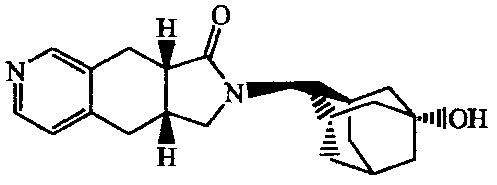
2-Адамантан-2-ил-2,3,3a,4,9,9a-гексагидробензо[f]изоиндол-l-он;
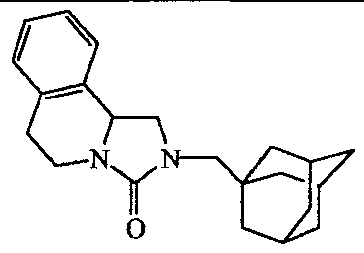
2-Адамантан-2-ил-2,3,10,10a-тетрагидро-5H-имидазо[l,5-b]изохинолин-l-он;
2-Адамантан-2-ил-1,5,10,10a-тетрагидро-2H-имидазо[l,5-b]изохинолин-3-он;
2-Адамантан-1-илметил-1,2,3a,4,5,9b-гексагидробензо[e]изоиндол-3-он;
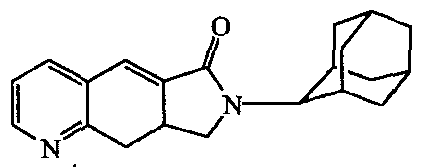
7-Адамантан-2-ил-7,8,8a,9-тетрагидропирроло[3,4-g]хинолин-6-он;
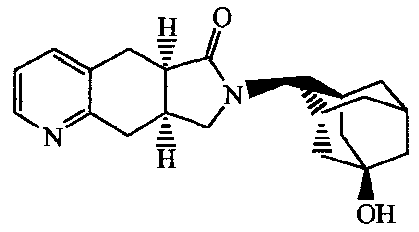
2-(5-Гидроксиадамантан-2-ил)-l,5,6,10b-тетрагидро-2H-имидазо[5,l-a]изохинолин-3-он;
2-(5-Фторадамантан-2-ил)-l,2,3a,4,5,9b-гексагидробензо[e]изоиндол-3-он;
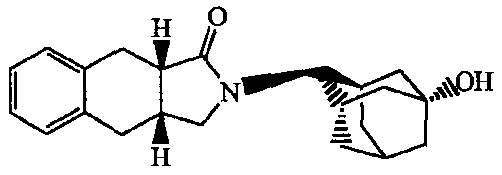
2-(5-Гидроксиадамантан-2-ил)-2,3,3a,4,9,9a-гексагидробензо[f]изоиндол-l-он.
В следующем варианте осуществления в настоящем изобретении предлагают соединения формулы (Ibis)
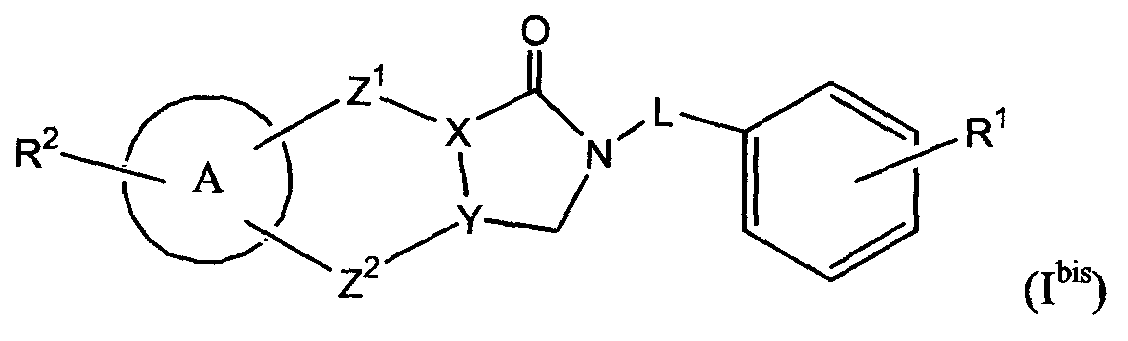
их N-оксидные формы, фармацевтически приемлемые аддитивные соли и стереохимически изомерные формы, в которых:
X представляет собой С или N;
Y представляет собой С или N;
L представляет собой метил или прямую связь;
Z1 представляет собой прямую связь, C1-2алкил или двухвалентный радикал формулы
-CH2-CH= (а) или -CH= (b);
Z2 представляет собой прямую связь, C1-2алкил или двухвалентный радикал формулы
-CH2-CH= (а) или -CH= (b);
R1 представляет собой водород, гало, циано, амино, фенил, гидрокси, C1-4алкилоксикарбонил, -O-(C=O)-C1-4алкил, гидроксикарбонил, NR3R4 или C1-4алкил, в котором указанный C1-4алкил или -O-(C=O)-C1-4алкил являются необязательно замещенными одним или несколькими заместителями, выбранными из гало, гидроксикарбонила, фенила, C1-4алкилокси или NR5R6 или R1 представляет собой C1-4алкилокси, необязательно замещенный одним или несколькими заместителями, выбранными из гало, гидроксикарбонила, фенила, C1-4алкилокси или NR7R8;
R2 представляет собой водород, гало, C1-4алкил или C1-4алкилокси;
R3 и R4, каждый независимо, представляют собой водород, C1-4алкил или C1-4алкилкарбонил;
R5 и R6, каждый независимо, представляют собой водород, C1-4алкил или C1-4алкилкарбонил;
R7 и R8, каждый независимо, представляют собой водород, C1-4алкил или C1-4алкилкарбонил;
A представляет собой фенил или моноциклический гетероцикл, выбранный из группы, состоящей из тиофенила, фуранила, оксазолила, тиазолила, имидазолила, изоксазолила, изотиазолила, пирридинила, пиридазинила, пиримидинила и пиперазинила.
В частности, соединения формулы (Ibis), к которым применены одно или несколько нижеследующих ограничений:
(i) X представляет собой С или N;
(ii) Y представляет собой С или N;
(iii) L представляет собой метил или прямую связь;
(iv) Z1 представляет собой прямую связь, C1-2алкил или двухвалентный радикал формулы
-CH2-CH= (а) или -CH= (b);
(v) Z2 представляет собой прямую связь, C1-2алкил или двухвалентный радикал формулы
-CH2-CH= (а) или -CH= (b);
(vi) R1 представляет собой водород, гало, циано, амино, фенил, гидрокси, C1-4алкилоксикарбонил, гидроксикарбонил, NR3R4 или C1-4алкил, замещенный одним или несколькими заместителями, выбранными из гидроксикарбонила, фенила, C1-4алкилокси или NR5R6, в частности R1 представляет собой водород, гало, амино или гидрокси; даже более конкретно фтор, амино или гидроксил;
(vii) R2 представляет собой водород, гало, C1-4алкил или C1-4алкилокси; в частности R2 представляет собой водород, гало или C1-4алкилокси;
(viii) R3 и R4, каждый независимо, представляет собой водород, C1-4алкил или C1-4алкилкарбонил;
(ix) R5 и R6, каждый независимо, представляют собой водород, C1-4алкил или C1-4алкилкарбонил;
(x) A представляет собой фенил или моноциклический гетероцикл, выбранный из группы, состоящей из тиофенила, фуранила, оксазолила, тиазолила, имидазолила, изоксазолила, изотиазолила, пирридинила, пиридазинила, пиримидинила и пиперазинила; в частности, A представляет собой фенил или моноциклический гетероцикл, выбранный из группы, состоящей из пиридинила и тиофенила.
Следующий аспект настоящего изобретения касается применения любого из соединений упомянутой выше группы соединений в качестве лекарственного средства. В частности, для лечения или предупреждении патологий, связанных с избыточным образованием кортизола, таких как ожирение, диабет, сердечно-сосудистые заболевания, связанные с ожирением, стресс и глаукома.
PCT Международная заявка на патент WO 2004/089416 показывает преимущества комбинированной терапии, включающей введение ингибитора 11β-HSD1 и гипотензивного средства при лечении, например, инсулинрезистентности, дислипидемии, ожирения и гипертензии, в частности для лечения гипертензии. Соответственно целью настоящего изобретения является обеспечение любым соединением из упомянутой выше группы соединений для комбинированной терапии с гипотензивным средством, таким, как например, алпренолол, атенолол, тимолол, пиндолол, пропранолол, метопролол, бисопрололфумерат, эсмолол, ацебутелол, ацебутолол, бетаксолол, селипролол, небиволол, тертатолол, окспренолол, амузолалул, карведилол, лабеталол, S-атенолол, OPC-1085, квинаприл, лизиноприл, эналаприл, каптоприл, беназеприл, периндоприл, трандолаприл, фозиноприл, рамиприл, цилазаприл, делаприл, имидаприл, моэксиприл, спираприл, темокаприл, зофеноприл, S-5590, фазидотрил, Hoechst-Marion Roussel: 100240 (EP00481522), омапатрилат, гемопатрилат и GW-660511, нифедипин, фелодипин, никардипин, израдипин, нимодипин, дилтиазем, амлодипин, нитрендипин, верапамил, ласидипин, леракандипин, аранидипин, цилиндипин, клевидипин, азелнидипин, барнидипин, эфонодипин, иазидипин, иэмилдипин, иэркандипин, манидипин, нилвадипин, пранидипин, фурнидипин, доксазосин, урапидил, празосин, теразосин, буназосин и OPC-28326, бендрофлуметазид, хлорталидон, гидрохлортиазид и клопамид, буметанид, фуросемид, тораземид, амилорид, спиронолактон, ABT-546, амбризетан, антразентан, SB-234551, CI-1034, S-0139, YM-598, босентан, J-104133, алискирен, OPC-21268, толваптан, SR-121463, OPC-31260, Незиритид, ирбезартан, кандесартансилексетил, лозартан, валсартан, телмисартан, эпросартан, кандесартан, CL-329167, иосартан, олмесартан, пратосартан, TA-606, YM-358, фенолдопам, кетансерин, нафтопидил, N-0861, FK-352, KT2-962, экадотрил, LP-805, MYD-37, ноломирол, омакор, трепростинил, берапрост, экрапрост, PST-2238, KR-30450, PMD-3117, Индапамиды, CGRP-униген, стимуляторы гуанилатциклазы, гидралазины, метиидопа, докарпамин, моксонидин, Ко-Апровел и Мондо-Биотех-811. Указанное изобретение предоставляет собой фармацевтическую композицию, которая содержит комбинацию ингибитора 11β-HSD1 по настоящему изобретению и гипотензивного средства.
PCT Международная заявка на патент WO 2004/089415 обеспечивает преимущество комбинированной терапии, которая включает введение ингибитора 11β-HSD1 и агониста глюкокортикоидного рецептора для снижения нежелательных побочных эффектов, происходящих в процессе проведения терапии, использующей агонист глюкокортикоидного рецептора, и для лечения некоторых форм рака, заболеваний и расстройств, включающих воспалительный компонент. В частности, при снижении неблагоприятных воздействий терапии, использующей агонист глюкокортикоидного рецептора, при симптомах заболевания Кушинга, синдрома Кушинга, аллергических воспалительных заболеваний, неблагоприятных воздействий при лечении расстройств респираторной системы с помощью агониста глюкокортикоидного рецептора, неблагоприятных воздействий при лечении с помощью агониста глюкокортикоидного рецептора воспалительного заболевания пищеварительного тракта; неблагоприятных воздействий при лечении расстройства иммунной системы, соединительной ткани и суставов, используя агонист глюкокортикоидного рецептора; неблагоприятных воздействий от лечения эндокринологических заболеваний с помощью агониста глюкокортикоидного рецептора; неблагоприятных воздействий от лечения гематологических заболеваний с помощью агониста глюкокортикоидного рецептора; неблагоприятных воздействий от лечения рака с помощью агониста глюкокортикоидного рецептора, тошноты, вызванной применением химиотерапии, неблагоприятных воздействий, вызванных лечением заболеваний мышц и места нейромышечного соединения агонистом глюкокортикоидного рецептора; неблагоприятных воздействий лечения агонистом глюкокортикоидного рецептора при хирургическом вмешательстве, трансплантации; неблагоприятных воздействий, вызванных лечением агонистом глюкокортикоидного рецептора абсцесса головного мозга, тошноты/рвоты, инфекций, гиперкальцемии, гиперплазии надпочечника, аутоиммунного гепатита, заболевания спинного мозга, мешковидных аневризм.
Примерами симптомов, при которых комбинация соединения 11β-HSD1 по настоящему изобретению с агонистами глюкокортикоидного рецептора может оказаться полезной, являются нижеследующие симптомы: заболевание Кушинга, синдром Кушинга, астма, атопический дерматит, муковисцидоз, эмфизема, бронхит, гиперчувствительность, пневмонит, эозинофильная пневмония, пневмофиброз, болезнь Крона, неспецифический язвенный колит, рeактивный артрит, ревматоидный артрит, синдром Шегрена, системная красная волчанка, волчаночный нефрит, пурпура Геноха-Шенлейна, грануломатоз Вегенера, височный артерит, системный склероз, васкулит, саркоидоз, дерматомиозит-полимиозит, пузырчатка обыкновенная, гипертиреоз, гипоальдостеронизм, гипопитуитаризм, гемолитическая анемия, тромбоцитопения, пароксизмальная ночная гемоглобинурия, неопластическое сдавливание спинного мозга, опухоли головного мозга, острый лимфобластный лейкоз, болезнь Ходжинкина, тошнота, вызванная применением химиотерапии, тяжелая псевдопаралитическая миастения, наследственные миопатии, мышечная дистрофия Дюшенна, травма, послеоперационный стресс, операционный стресс, трансплантация почек, трансплантация печени, трансплантация легких, трансплантация панкреатического островка, трансплантация стволовых клеток крови, трансплантация костного мозга, трансплантация сердца, трансплантация надпочечника, трансплантация трахеи, трансплантация кишечника, трансплантация роговицы, пересадка кожи, кератопластика, имплантация хрусталика, абсцесс головного мозга, тошнота/рвота, инфекции, гиперкальцемия, гиперплазия надпочечников, аутоиммунный гепатит, заболевания спинного мозга и мешковидные аневризмы. Соответственно объектом настоящего изобретения является обеспечение любого из упомянутой выше группы соединений для комбинированной терапии с агонистом глюкокортикоидного рецептора, так же как фармацевтические препараты, содержащие указанную комбинацию соединения по настоящему изобретению с агонистом глюкокортикоидного рецептора. Агонист глюкокортикоидного рецептора, например, выбирают из группы, состоящей из: бетаметазона, дексаметазона, гидрокортизона, метилпреднизолона, преднизолона, преднизона, белкометазона, бутиксикорта, клобетазола, флунисолид, флукатизона (и аналогов), мометазона, триамцинолонацетонида, триамцинолонгексацетонида GW-685698, NXC-1015, NXC-1020, NXC-1021, NS-126, P-4112, P-4114, RU-24858 and T-25 series.
Чтобы упростить представление соединений формулы (I), группу
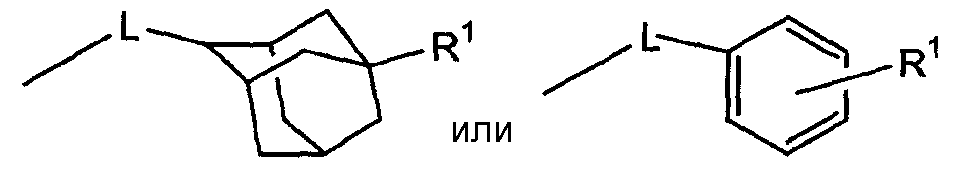
в дальнейшем будут обозначать как -D.
Производные "изогнутого" трициклического адамантиламида по настоящему изобретению, в дальнейшем обозначаемые как соединения формулы (Ii), обычно получают путем конденсирования на первой стадии коммерчески доступной бензоциклобутанкарбоновой кислоты (II) соответствующим амином в условиях, известных в данной области техники (схема 1). Затем полученный таким образом амид (III) восстанавливают, используя, например, алюмогидрид лития или борандиметилсульфидный комплекс, чтобы получить амин формулы (IV). В результате указанный амин ацилируют акроилхлоридом с последующей реакцией циклизации, следуя известным процедурам в данной области техники, таким, как например, нагревание амида (V) в толуоле при 190°C, для получения смеси производных цис- и транс-изомеров "изогнутого" трициклического адамантиламида по настоящему изобретению:
СХЕМА 1
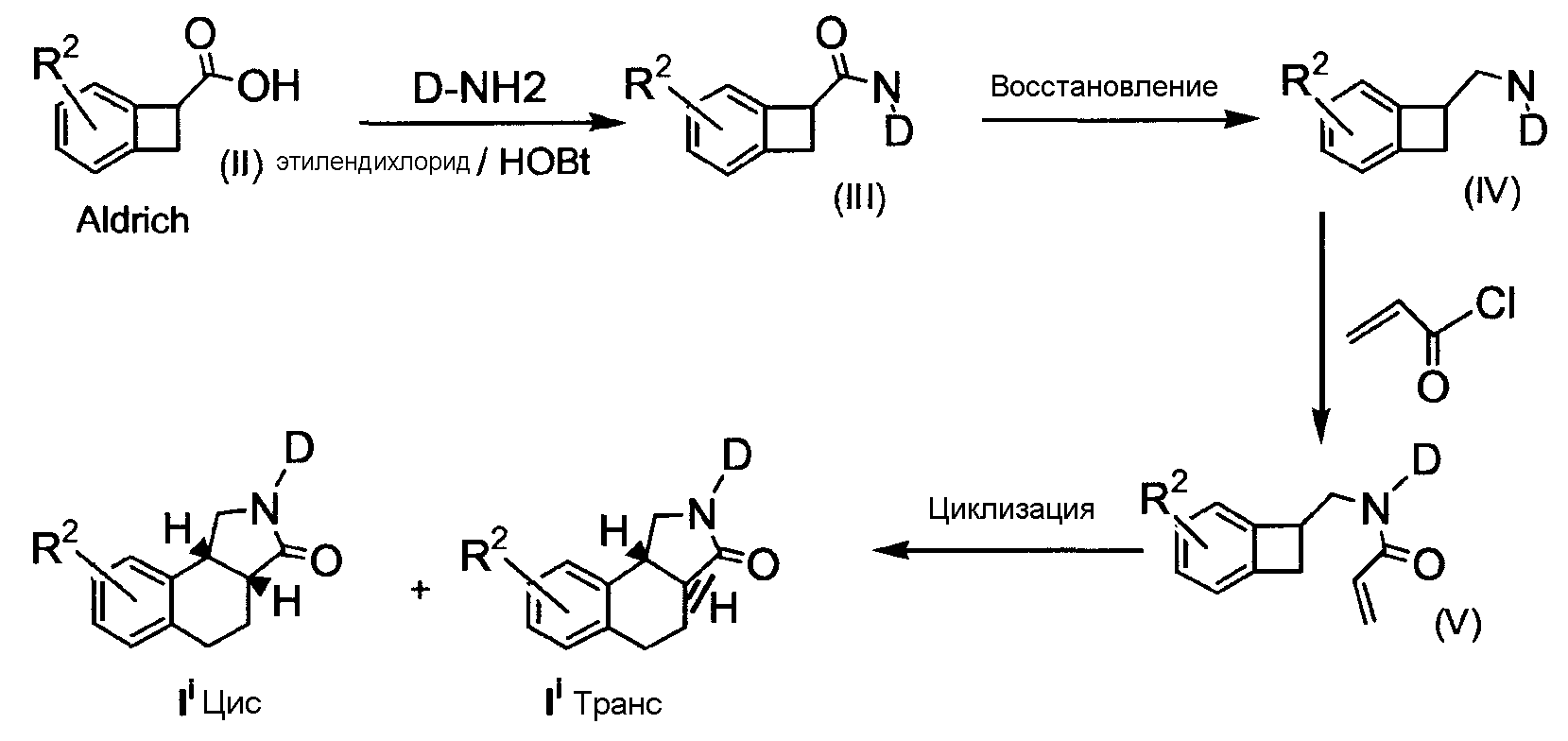
где R2 обозначает то же, что и для соединений формулы (I), описанных ранее.
Для получения стереоизомеров производных "изогнутого" трициклического адамантиламида формулы (Ii), указанную ранее, коммерчески доступную бензоциклобутанкарбоновую кислоту (II) конденсируют аллил-2-адамантиламином (VI) для получения амида общей формулы (VII), который при электроциклическом замыкании цикла дает производные "изогнутого" трициклического адамантиламида формулы (Ii) (схема 2):
СХЕМА 2
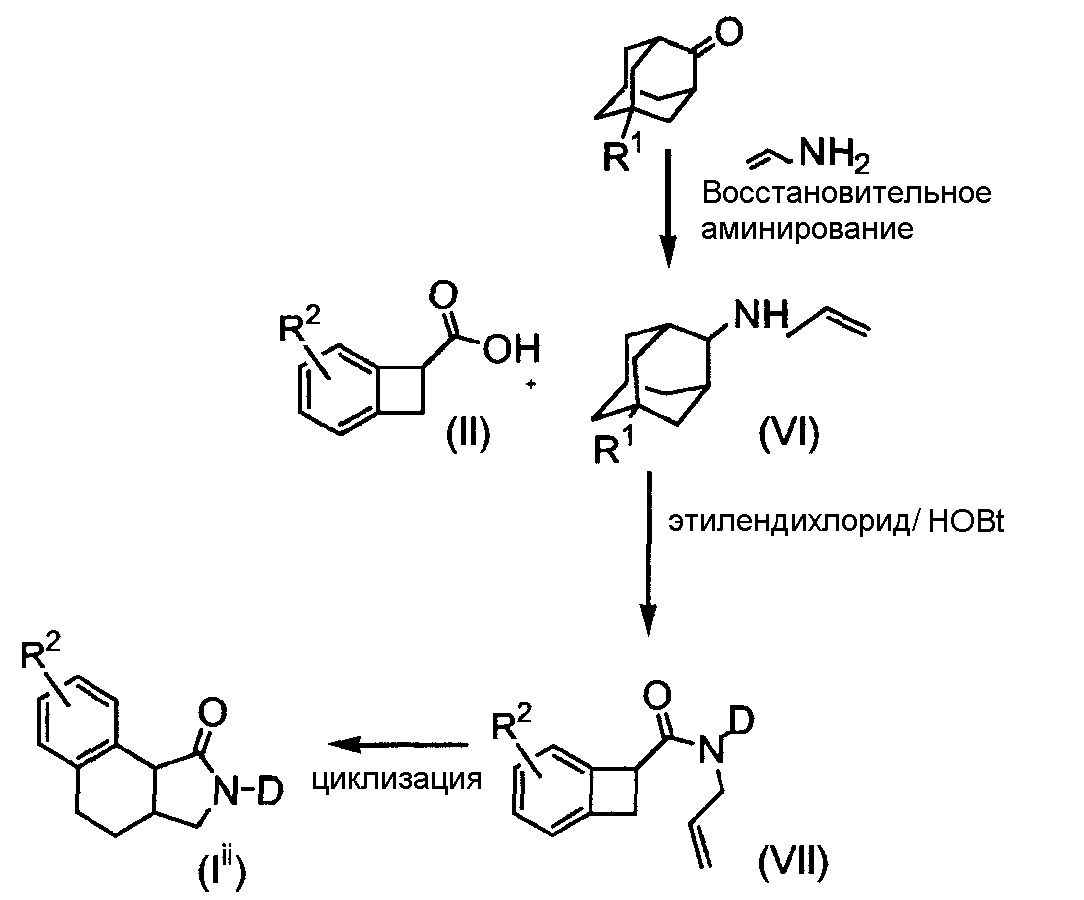
где R1 и R2 обозначают то же, что и для соединений формулы (I), описанной ранее. Эти соединения формулы (I), в которых X представляет собой N, в дальнейшем обозначаемые как мочевины формулы (Iii), обычно получают по реакционным схемам 3 и 4, приведенным далее. В первом альтернативном варианте мочевины получают, исходя из коммерчески доступной BОК (бензилоксикарбонил)-защищенной тетрагидрохинолин-3-карбоновой кислоты (оба энантиомера); реакция с аминоадамантаном и восстановление амида давали диамин формулы (VIII). Последующая циклизация согласно процедурам, известным в данной области техники, дала циклические мочевины формулы (Iiii):
СХЕМА 3
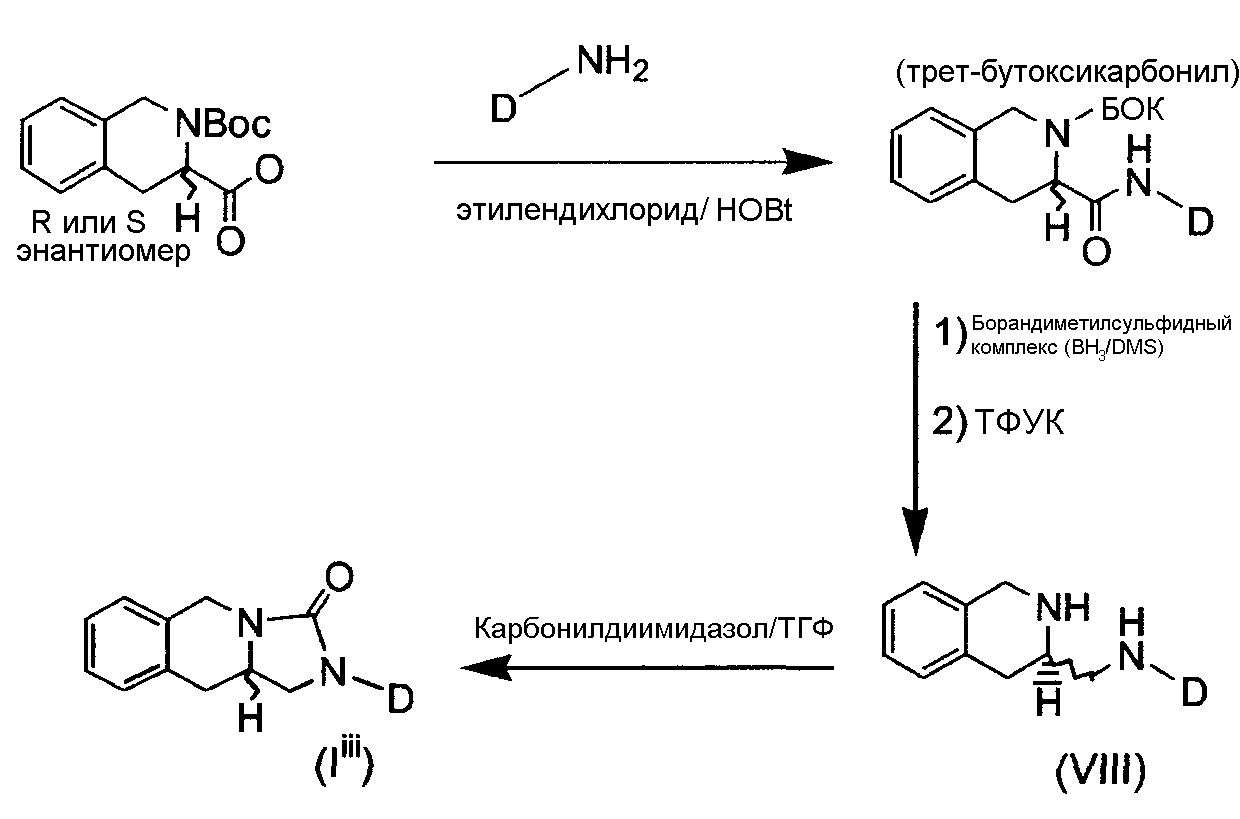
Во втором альтернативном варианте производные мочевины получают по известным в данной области техники методикам, путем сочетания коммерчески доступных хинолин-2-карбоновых кислот или изохинолин-1-карбоновых кислот с соответствующим амином для получения соответствующего амида формулы (IX). Селективное гидрирование пиридинового цикла дало тетрагидро(изо)хинолинацетамиды (X), которые восстанавливали, используя, например, BH3.DMS (борандиметилсульфидный комплекс) в толуоле для получения диаминов общей формулы (XI). Последующая циклизация, использующая, например, карбонилдиимидазол (CDI), дала циклические мочевины формулы (Iiii):
СХЕМА 4

где R2 обозначает то же, что и для соединений формулы (I), описанной ранее,
-A-A- представляет собой -N-СН2- или -CH2-N- и -A=A- представляет собой -N=CH- или -CH=N-.
В случаях, когда замещенные изохинолин-1-карбоновые кислоты не были коммерчески доступны, замещенные трициклические производные получали, исходя из фенетиламинов (XII) и этилхлорформата (схема 5). Полученный карбамат подвергали циклизации, используя способы, известные в данной области техники, такие как, например, модифицированная реакция Бишлера-Напиральского (Larsen, Robert D., et al., A модифицированный Bischler-Napieralski procedure for the synthesis of 3-aryl-3,4-dihydroisoquinolines., Journal of Organic Chemistry (1991), 56(21), 6034-8.), с получением защищенной по аминогруппе тетрагидроизохинолин-l-карбоновой кислоты формулы (X'). Следующий синтез замещенных трициклических производных осуществляют, как показано на реакционной схеме 4, приведенной выше:
СХЕМА 5

Производные "линейного" трициклического адамантиламида формулы (IiV) могут быть получены по реакционным схемам 6 и 7, приведенным далее. По первому альтернативному варианту производные "линейного" трициклического адамантиламида получают исходя из арил- или гетероарилзамещенной акриловой кислоты или хлорида кислоты (ацилхлорида) (XIII). Реакция с соответствующим амином дает амид формулы (XIV), который при электроциклическом замыкании цикла в условиях, известных в данной области техники, например в толуоле при 220°C, дает трициклическую систему формулы (IiV):
СХЕМА 6
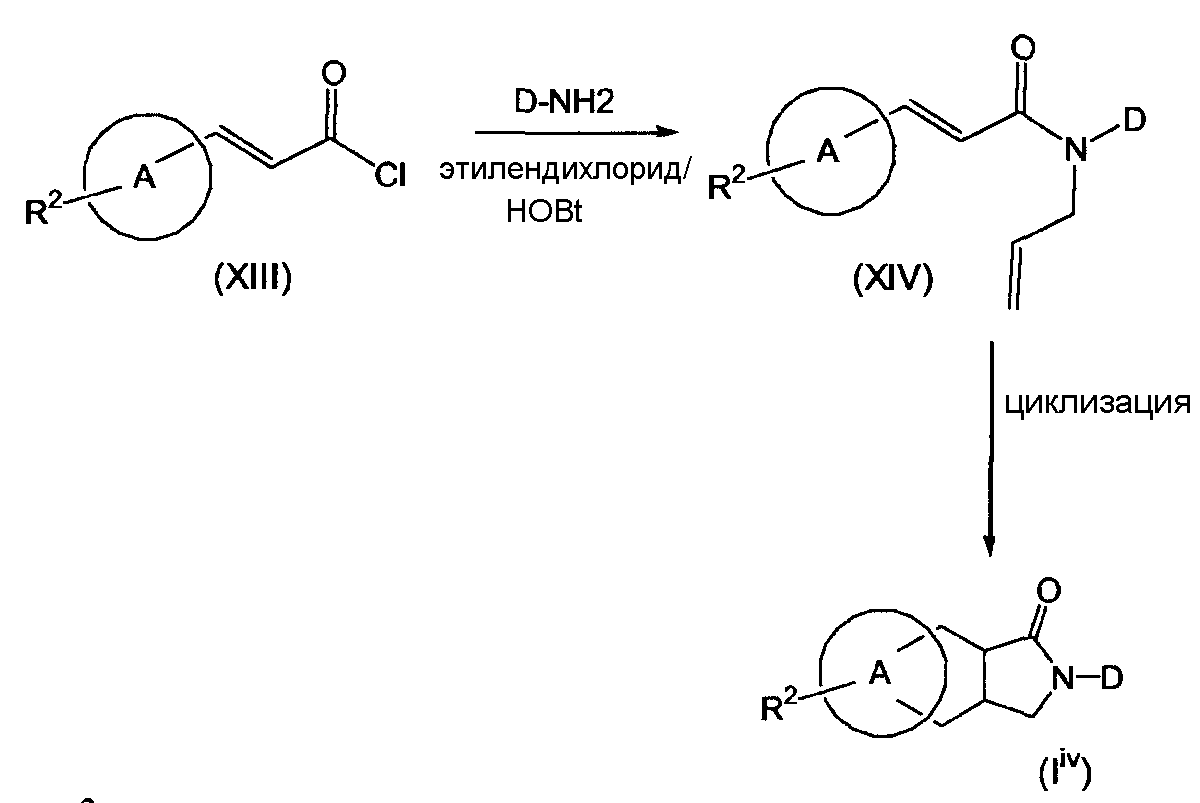
где A и R2 обозначают то же, что и в соединениях формулы (I), приведенных ранее.
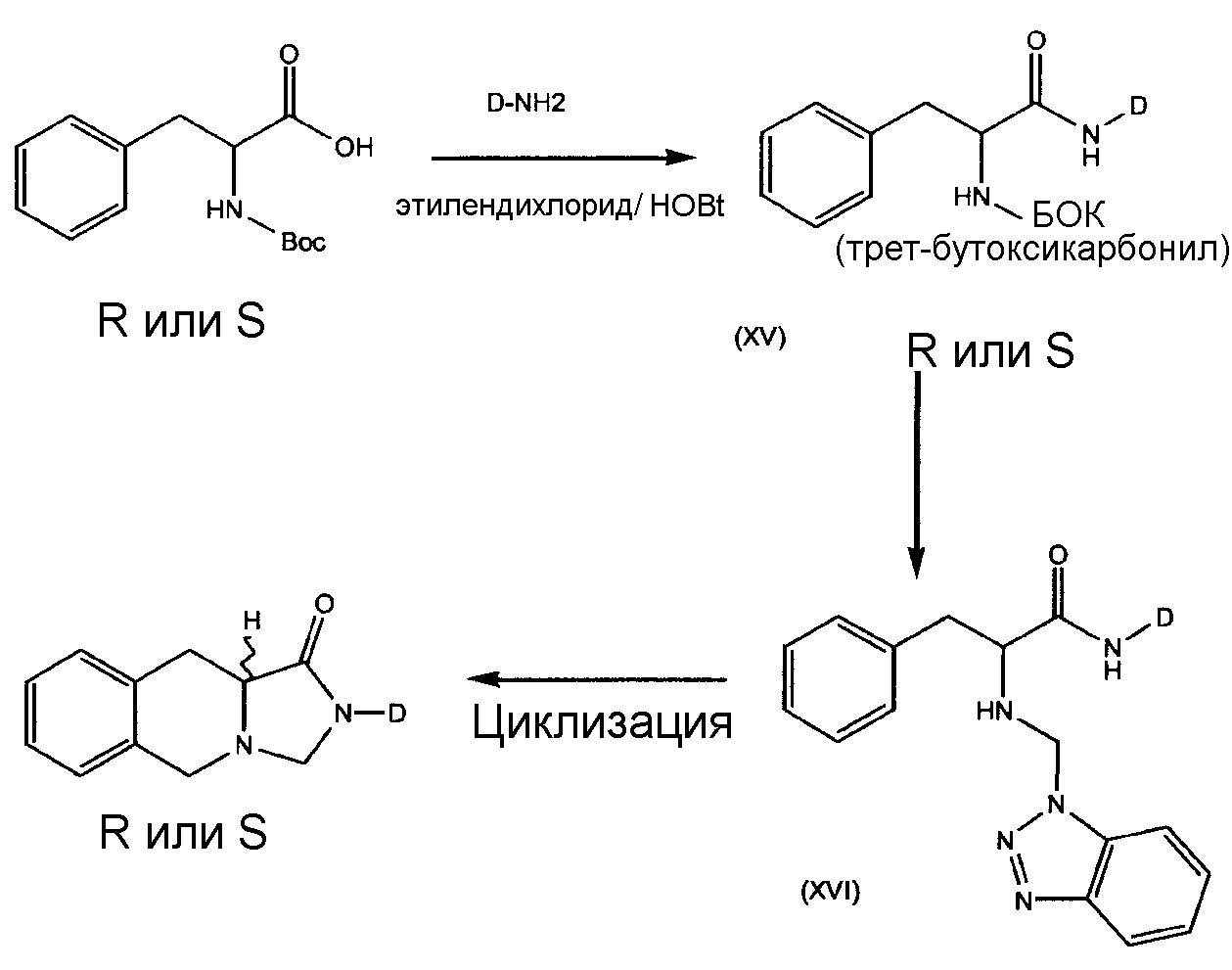
Во втором альтернативном варианте производные "линейного" трициклического адамантиламида формулы (IiV), где A представляет собой фенил, Y представляет собой N, могут быть получены путем сочетания группы D, защищенной по аминогруппе, или L-фенилаланина с соответствующим амином с получением α-аминоамида формулы (XV); см., например, реакционные условия, описанные в J.Org.Chem. 2002, 67, 8224. Снятие защиты с последующей конденсацией по Mannich бензотриазолом и формальдегидом дает промежуточное соединение формулы (XVI). Электроциклическое замыкание цикла дает производные "линейного" трициклического адамантиламида формулы (IiV):
СХЕМА 7
Следующие примеры синтеза соединений формулы (I), использующие любой из упомянутых выше методов синтеза, приведены в экспериментальной части, приведенной далее.
Если необходимо или желательно, в любом порядке могут быть осуществлены любые одна или несколько нижеследующих стадий:
(i) удаление любой остающейся защитной группы(групп);
(ii) превращение соединения формулы (I) или его защищенной формы в следующее соединение формулы (I) или его защищенную форму;
(iii) превращение соединения формулы (I) или его защищенной формы в N-оксид, соль, четвертичный амин или сольват соединения формулы (I) или его защищенной формы;
(iv) превращение N-оксида, соли, четвертичного амина или сольвата соединения формулы (I) или его защищенной формы в соединение формула (I) или его защищенную форму;
(v) превращение N-оксида, соли, четвертичного амина или сольвата соединения формулы (I) или его защищенной формы в еще один N-оксид, фармацевтически приемлемую аддитивную соль, четвертичный амин или сольват соединения формулы (I) или его защищенной формы;
(vi) в том случае, когда соединение формулы (I) получают в виде смеси (R) и (S) энантиомеров, разделение смеси с получением желательного энантиомера.
Как будет понятно специалистам в данной области техники, в способах, описанных выше, может оказаться необходимой блокировка защитными группами функциональных групп промежуточного соединения.
Функциональные группы, которые нуждаются в защите, включают в себя гидрокси, амино и карбоксильную группы. Пригодные защитные группы для гидрокси включают в себя триалкилсилильные группы (например, трет-бутилдиметилсилил, трет-бутилдифенилсилил или триметилсилил), бензил и тетрагидропиранил. Пригодные защитные группы для амино включают в себя трет-бутилоксикарбонил или бензилоксикарбонил. Пригодные защитные группы для карбоновой кислоты включают в себя C(1-6)алкиловые или бензиловые сложные эфиры.
Защита функциональных групп и удаление защиты могут иметь место до или после соответствующей стадии реакции.
Использование защитных групп подробно описано в “Protective Groups in Оrganic Synthesis” 2-nd edition, TW Greene & P G M Wutz, Wiley Interscience (1991).
Дополнительно N-атомы в соединениях формулы (I) могут быть метилированы известными способами в данной области техники, используя СН3-I в соответствующем растворителе, таком как, например, 2-пропанон, тетрагидрофуран или диметилформамид.
Соединения формулы (I) также могут быть превращены друг в друга, используя способы превращения функциональных групп, известные в данной области техники, некоторые примеры которых были приведены выше.
Соединения формулы (I) также могут быть превращены в соответствующие N-оксидные формы известными в данной области техники способами превращения трехвалентного азота в N-оксидную форму. Указанная реакция N-окисления обычно может быть осуществлена взаимодействием исходного вещества формулы (I) с 3-фенил-2-(фенилсульфонил)оксазиридином или с соответствующим органическим или неорганическим пероксидом. Соответствующие неорганические пероксиды включают, например, перекись водорода, пероксиды щелочных или щелочно-земельных металлов, например пероксид натрия, пероксид калия; соответствующие органические пероксиды могут включать надкислоты, такие как, например бензолкарбопероксокислота или галогензамещенная бензолкарбопероксокислота, например 3-хлор бензолкарбопероксокислота, пероксоалкановые кислоты, например пероксоуксусная кислота, алкилгидропероксиды, например т-бутилгидропероксид. Пригодными растворителями являются, например, вода, низшие спирты, например этанол и тому подобное; углеводороды, например толуол; кетоны, например 2-бутанон; галогенированные углеводороды, например дихлорметан и смеси таких растворителей.
Беспримесные стереохимически изомерные формы соединений формулы (I) могут быть получены путем использования методик, известных в данной области техники. Диастереомеры можно разделить физическими методами, такими как избирательная кристаллизация и хроматография, например противоточное распределение, жидкостная хроматография и тому подобное.
Некоторые из соединений формулы (I) и некоторые из промежуточных соединений по настоящему изобретению могут содержать асимметричный углеродный атом. Чистые стереохимически изомерные формы указанных соединений и указанных промежуточных соединений могут быть получены при использовании методик, известных в данной области техники. Например, диaстереоизомеры могут быть разделены с помощью физических методов, таких как избирательная кристаллизация и хроматография, например противоточное распределение, жидкостная хроматография и подобные методы. Энантиомеры можно получить из рацемических смесей, сначала превращая указанные рацемические смеси с помощью пригодных растворяющих агентов, таких как, например, хиральные кислоты, в смеси диастереомерных солей или соединений, с последующим физическим разделением указанных смесей диастереомерных солей или соединений посредством, например, избирательной кристаллизации или хроматографии, например жидкостной хроматографии и подобных методов; и в конечном счете превращением указанных разделенных диастереомерных солей или соединений в соответствующие энантиомеры. Чистые стереохимически изомерные формы можно также получить из чистых стереохимически изомерных форм соответствующих промежуточных соединений и исходных веществ при условии, что происходящие реакции протекают стереоспецифично.
Альтернативный способ разделения энантиомерных форм соединений формулы (I) и промежуточных соединений включает жидкостную хроматографию, в частности жидкостную хроматографию, использующую хиральную стационарную фазу.
Некоторые из промежуточных соединений и исходных веществ, используемых в реакционных методиках, упомянутых выше, являются известными соединениями и могут быть коммерчески доступными или могут быть получены по известным в данной области методикам.
Соединения по настоящему изобретению применимы, потому что они обладают фармакологическими характеристиками. Поэтому они могут быть использованы как лекарственные средства, в частности, для лечения патологий, связанных с избыточным образованием кортизола, то есть расстройств, при которых желателен сниженный уровень активного глюкокортикоида, таких как метаболический синдром, диабет 2 типа, ослабленная толерантность к глюкозе (IGT), нарушенная утилизация глюкозы (IFG), дислипидемия, гипертензия, ожирение, диабет, сердечно-сосудистые заболевания, связанные с ожирением, артериосклероз, атеросклероз, миопатия, остеопороз, нейродегенеративные и психиатрические расстройства, расстройства, вызванные стрессом, и глаукома. В частности, для лечения патологий, таких как, например, ожирение, диабет, диабет 2 типа, сердечно-сосудистые заболевания, связанные с ожирением, стресс и глаукома.
Как описано в приведенной далее экспериментальной части, ингибирующее действие настоящих соединений на активность 1lβ-HSDl-редуктазы (превращение кортизона в кортизол) показали in vitro при ферментативном анализе, использующем рекомбинантный фермент 1lβ-HSD1, посредством измерения степени превращения кортизона в кортизол, применяя для очистки и количественных определений методы (HPLC) ВЭЖХ. Ингибирование 11β-HSD1-редуктазы также демонстрировали in vitro при клеточном анализе, который включал контакт экспрессирующих 11β-HSD1 клеток с соединениями, подлежащими тестированию, и оценку влияния указанных соединений на образование кортизола в целлюлярной среде этих клеток. Клетки, предпочтительно используемые в опыте по настоящему изобретению, выбирают из группы, состоящей из 3T3-L1 клеток мышиных фибробластов, HepG2 клеток, клеток свиной почки, в частности LCC-PK1 клеток, и гепатоцитов крысы.
Соответственно в настоящем изобретении предлагают для терапевтического использования соединения формула (I) и их фармацевтически приемлемые N-оксиды, аддитивные соли, четвертичные амины стереохимически изомерные формы. В частности, для лечения патологий, связанных с избыточным образованием кортизола, то есть нарушений, при которых желателен сниженный уровень активного глюкокортикоида, таких как метаболический синдром, диабет 2 типа, ослабленная толерантность к глюкозе (IGT), нарушенная утилизация глюкозы (IFG), дислипидемия, гипертензия, ожирение, диабет, сердечно-сосудистые заболевания, связанные с ожирением, артериосклероз, атеросклероз, миопатия, остеопороз, нейродегенеративные и психиатрические расстройства, расстройства, вызванные стрессом, и глаукома. Более конкретно для лечения патологий, таких как, например, ожирение, диабет 2 типа, диабет, сердечно-сосудистые заболевания, связанные с ожирением, стресс и глаукома. Еще более конкретно при лечении или предупреждении патологий, связанных с избыточным образованием кортизола, таких как ожирение, диабет, сердечно-сосудистые заболевания, связанные с ожирением и глаукома.
Ввиду эффективности соединений по изобретению предложен метод лечения животных, например млекопитающих, включая людей, страдающих от патологии, связанной с избыточным образованием кортизола, который включает в себя введение эффективного количества соединения по настоящему изобретению.
Указанный способ, включающий системное или местное введение теплокровным животным, включая людей, эффективного количества соединения по изобретению.
Таким образом, целью настоящего изобретения является получение соединения по настоящему изобретению для использования в качестве лекарственного средства. В частности, для использования соединения по настоящему изобретению при получении лекарственных препаратов для лечения патологий, связанных с избыточным образованием кортизола, таких как, например, метаболический синдром, диабет 2 типа, ослабленная толерантность к глюкозе (IGT), нарушенная утилизация глюкозы (IFG), дислипидемия, гипертензия, ожирение, диабет, сердечно-сосудистые заболевания, связанные с ожирением, артериосклероз, атеросклероз, миопатия, остеопороз, нейродегенеративные и психиатрические расстройства, расстройства, вызванные стрессом, и глаукома; в частности, ожирение, диабет, сердечно-сосудистые заболевания, связанные с ожирением, стресс и глаукома.
Количество соединения по настоящему изобретению, обозначаемого здесь как активный ингредиент, которое необходимо для достижения терапевтического эффекта, будет, безусловно, меняться вместе с конкретным соединением, способом введения, возрастом и состоянием реципиента и конкретным расстройством или заболеванием, подлежащим лечению. Подходящая ежедневная доза могла бы составлять от 0,001 мг/кг до 500 мг/кг массы тела, в частности от 0,005 мг/кг до 100 мг/кг массы тела. Способ лечения может также включать в себя введение активного ингредиента в режиме от одного до четырех приемов в день.
Хотя возможно введение активного ингредиента самого по себе, предпочтительно представлять его в виде фармацевтической композиции. Соответственно настоящее изобретение, кроме того, предлагает фармацевтические композиции, содержащие соединение по настоящему изобретению вместе с фармацевтически приемлемым носителем или разбавителем. Носитель или разбавитель должен быть "приемлемым", т.е. совместимым с другими ингредиентами композиции, и не причинять вреда реципиентам.
Фармацевтические композиции по данному изобретению могут быть получены любыми способами, хорошо известными в данной области фармации, например использование таких способов, как описанные Gennaro et al. Remington's Pharmaceutical Sciences (18th ed., Mack Publishing Company, 1990, в особенности смотреть part 8: Pharmaceutical preparations and their Manufacture). Терапевтически эффективное количество конкретного соединения в основной форме или в форме аддитивной соли в качестве активного ингредиента объединяют в однородную смесь с фармацевтически приемлемым носителем, который может принимать самые разнообразные формы в зависимости от формы препарата, требуемой для введения. Эти фармацевтические композиции желательно иметь в однократной лекарственной форме, предпочтительно пригодной для системного введения, такого как введение через кожу, оральное или парентеральное введение, или для местного введения, такого как ингаляция, назальное опрыскивание, закапывание в глаза или введение через крем, гель, шампунь и тому подробное. Например, при получении композиций в оральной лекарственной форме может быть использована любая из обычных фармацевтических сред, таких как, например, вода, гликоли, масла, спирты и тому подобное, для случая жидких оральных препаратов, таких как суспензии, сиропы, эликсиры и растворы; или твердые носители, такие как крахмалы, сахара, каолин, смазки, связующие вещества, расщепляющие агенты и тому подобное, для случая порошков, пилюль, капсул и таблеток. Из-за легкости их введения таблетки и капсулы представляют собой наиболее преимущественные оральные стандартные лекарственные формы, очевидно, в этом случае используют твердые фармацевтические носители. Для парентеральных композиций носитель обычно будет содержать стерильную воду, по крайней мере, в значительной части, хотя могут быть включены другие ингредиенты, например, для обеспечения растворимости. Например, могут быть получены инъецируемые растворы, в которых носитель содержит солевой раствор, раствор глюкозы или смесь солевого раствора и раствора глюкозы. Также могут быть получены инъецируемые суспензии, в этом случае могут быть использованы соответствующие жидкие носители, суспендирующие агенты и тому подобное. В композициях, пригодных для введения через кожу, носитель необязательно содержит агент, повышающий проникновение и/или пригодный смачивающий агент, необязательно объединенный с подходящими добавками любой природы в малых пропорциях, эти добавки не оказывают на кожу каких-либо существенных вредных воздействий. Указанные добавки могут облегчать введение через кожу и/или могут способствовать получению желательных композиций. Эти композиции могут быть введены различными способами, например, в виде трансдермального пластыря, в виде пятна или в виде мази. В качестве подходящих композиций для местного применения могут быть перечислены все композиции, обычно используемые как лекарственные средства для местного введения, например кремы, гели, повязки, шампуни, настойки, пластыри, мази, целебные мази, порошки и тому подобное. Указанные композиции можно использовать в виде аэрозоля, например, с такими пропеллентами, как азот, двуокись углерода, фреон, или без использования пропеллента, с помощью пульверизатора, или как капли, лосьоны, или полутвердые вещества, такие как загущенные композиции, которые можно наносить с помощью аппликаторов. В частности, удобно будет использовать полутвердые композиции, такие как мази, кремы, гели, мази и тому подробное.
Особенно большое преимущество для облегчения введения и однородности дозировки дает формирование упомянутых выше фармацевтических композиций в стандартной лекарственной форме. Термин «стандартная лекарственная форма», используемый в описании и формуле изобретения, обозначает здесь физически дискретные единицы, пригодные в качестве однократных доз; при этом каждая единица содержит предварительно определенное, рассчитанное для получения требуемого терапевтического эффекта количество активного ингредиента в сочетании с необходимым фармацевтическим носителем. Примерами таких стандартных лекарственных форм являются таблетки (включая размеченные рисками таблетки или таблетки с покрытием), капсулы, пилюли, порошки в пакетиках, облатки, растворы или суспензии для инъекцирования, отмеренные количества, соответствующие чайной ложке, столовой ложке и тому подобное, и отдельные многочисленные их варианты.
Чтобы увеличить растворимость и/или стабильность соединений формулы (I) в фармацевтических композициях, может оказаться полезным использовать α-, β- или γ-циклодекстрины, или их производные. Такие со-растворители, как спирты, также могут улучшить растворимость и/или стабильность соединений формулы (I) в фармацевтических композициях. При получении водных композиций аддитивные соли обсуждаемых соединений, очевидно, являются более подходящими вследствие их увеличенной растворимости в воде.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В методиках, описанных далее, были использованы нижеследующие сокращения: "THF" обозначает тетрагидрофуран; "DIPE" обозначает диизопропиловый эфир; "EtOAc" обозначает этилацетат; "DMF" обозначает N,N-диметилформамид, "BMS" обозначает тригидро[тиобис[метан]]бор [13292-87-0].
«ExtrelutTM» является продуктом фирмы Merck KgaA (Darmstadt, Germany) и представляет собой короткую колонку, содержащую диатомовую землю. «Supelco» является колонкой для жидкостной хроматографии, предварительно заполненной силикагелем.
Для некоторых химических веществ были использованы химические формулы, например для дихлорметана использовали обозначение CH2Cl2, CH3OH - для метанола, HC1 - для хлористоводородной кислоты, KOH - для гидроксида калия, NaOH - для гидроксида натрия, Na2CO3 - для карбоната натрия, NaHCO3 - для гидрокарбоната натрия, MgSO4 - для сульфата магния, N2 - для газообразного азота, CF3COOH - для трифторуксусной кислоты.
A. ПОЛУЧЕНИЕ ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ
Пример Al
Получение
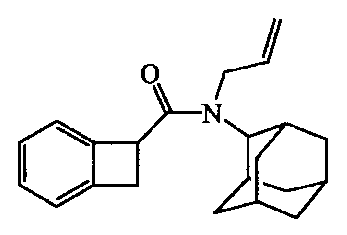
Тионилхлорид (0,5 мл) добавляли к раствору бицикло[4.2.0]окта-1,3,5-триен-7-карбоновой кислоты [14381-41-0] (0,001 моль) в дихлорметане. Реакционную смесь кипятили с обратным холодильником в течение 1 часа. Затем перемешивали в течение ночи при комнатной температуре. Растворители 2 раза выпаривали совместно с бензолом с получением бицикло[4.2.0]окта-1,3,5-триен-7-карбонилхлорида [1473-47-8], который растворяли в DIPE. Полученный раствор добавляли по каплям к охлажденной смеси (0°C) N-аллил-2-адамантанамина [24161-63-5] и карбоната натрия в DIPE. Реакционную смесь перемешивали 30 минут на льду и затем в течение 2 часов при комнатной температуре. Смесь выливали в воду и экстрагировали дихлорметаном. Органический слой фильтровали через Extrelut™ и фильтрат упаривали. Остаток очищали флеш колоночной хроматографией на TRIKONEX FlashTube™ (элюент: CH2Cl2/EtOAc 90/10). Полученные фракции собирали, и растворители выпаривали, получая 0,13 г промежуточного соединения 1.
Пример A2
a) Получение
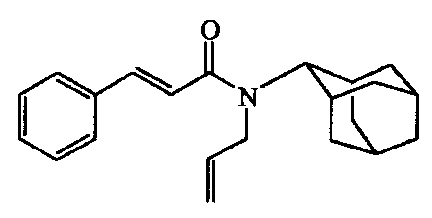
Смесь 3-фенил-2-пропеновой кислоты [140-10-3] (0,01 моль) и тионилхлорида (30 мл) кипятили с обратным холодильником в течение 2 часов. Растворитель выпаривали совместно с метилбензолом. Остаток растворяли в DIPE (20 мл), и полученный раствор добавляли по каплям к смеси N-аллил-2-адамантанамина [24161-63-5] (0,01 моль) и карбоната натрия (2 г) в DIPE (50 мл) на льду. Реакционную смесь перемешивали в течение ночи, выливали в дихлорметан и промывали водой. Органически слой отделяли, сушили (MgSO4), фильтровали, и растворитель выпаривали. Остаток очищали колоночной хроматографией на силикагеле (элюент: CH2CI2). Полученные фракции собирали, и растворитель выпаривали. Остаток растирали в DIPE, и требуемый продукт собирали, получая 1,68 г (56%) промежуточного соединения 2.
Пример A3
a) Получение
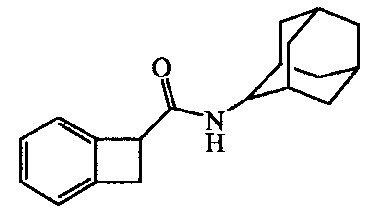
Раствор бицикло[4.2.0]окта-1,3,5-триен-7-карбоновой кислоты [14381-41-0] (0,0033 моль) в дихлорметане (25 мл) и N,N-диэтилэтанамин (5 мл) перемешивали и добавляли 1-гидрокси-lH-бензотриазол (0,0035 моль). Затем добавляли N-(этилкарбонимидоил)-N,N-диметил-l,3-пропандиамина моногидрохлорид (0,0035 моль), и смесь перемешивали в течение 10 минут. Добавляли трицикло[3.3.1.13,7]декан-2-амина гидрохлорид (1:1) [10523-68-9] (0,0035 моль), и реакционную смесь перемешивали в течение 2 дней. Смесь промывали 15% раствором лимонной кислоты и раствором карбоната натрия. Органический слой отделяли, сушили, фильтровали и растворитель выпаривали. Остаток растирали в DIPE и требуемый продукт собирали, получая 0,6 г промежуточного соединения 3.
b) Получение
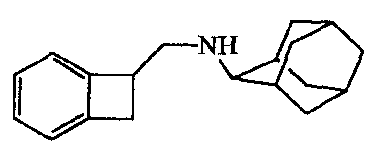
Алюмогидрид лития (0,0042 моль) перемешивали в диэтиловом эфире (10 мл) (на льду) и добавляли хлорид алюминия (0,0042 моль); смесь перемешивали в течение 15 минут и по частям добавляли промежуточное соединение 3 (0,0021 моль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем гасили разбавленным раствором HC1. Добавляли разбавленный раствор KOH до pH 10; и полученную смесь экстрагировали дихлорметаном. Органический слой отделяли и сушили, затем фильтровали через Extrelut™, и фильтрат упаривали, получая выход 0,489 г промежуточного соединения 4.
c) Получение
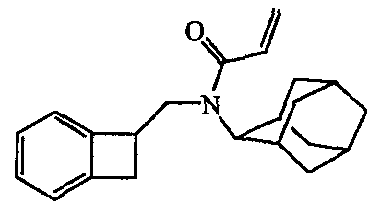
Смесь промежуточного соединения 4 (0,0018 моль) и карбоната натрия (0,3 г) в дихлорметане (10 мл) перемешивали на льду. По каплям добавляли 2-пропеноилхлорид [814-68-6] (0,002 моль); и реакционную смесь перемешивали в течение ночи при комнатной температуре. Смесь промывали водой (4 мл) и фильтровали через Extrelut; фильтрат упаривали, получая выход 0,497 г промежуточного соединения 5.
Пример A4
a) Получение
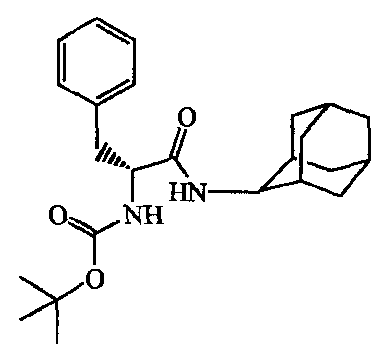
К смеси N-[(1,1-диметилэтокси)карбонил]-D-фенилаланина [18942-49-9] (0,0075 моль) и N,N-диэтилэтанамина (5 мл) в дихлорметане (100 мл) добавляли l-гидрокси-lH-бензотриазол (0,02 моль). После 5-минутного перемешивания добавляли N-(этилкарбонимидоил)-N,N-диметил-1,3-пропандиамина моногидрохлорид [25952-53-8] (0,02 моль). После перемешивания в течение 10 минут добавляли трицикло[3.3.1.13,7]декан-2-амина гидрохлорид [10523-68-9] (0,015 моль); и реакционную смесь перемешивали в течение ночи при комнатной температуре. Смесь выливали в воду и экстрагировали дихлорметаном. Органический слой сушили, фильтровали, и растворитель выпаривали, получая 2,5 г промежуточного соединения 6.
b) Получение
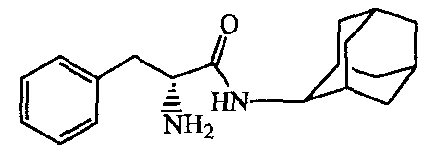
Смесь промежуточного соединения 6 (0,0075 моль) в дихлорметане (50 мл) и трифторуксусной кислоты (10 мл) перемешивали в течение ночи, и растворители выпаривали. Остаток растворяли в дихлорметане и промывали раствором карбоната натрия. Органический слой сушили, фильтровали и растворитель выпаривали. Остаток растирали в DIPE и требуемый продукт собирали, получая 1,4 г промежуточного соединения 7.
c) Получение
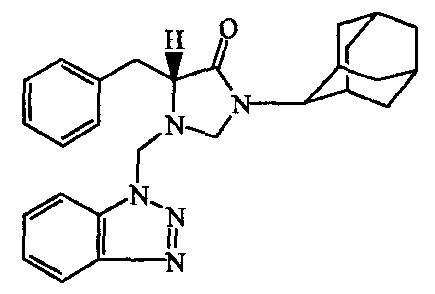
Смесь промежуточного соединения 7 (0,0046 моль), lH-бензотриазола [95-14-7] (0,0092 моль), параформальдегида (0,0138 моль) и 4-метилбензолсульфоновой кислоты [104-15-4] (0,18 г) в бензоле (60 мл) кипятили с обратным холодильником над установкой Dean-Starck в течение 3 часов. Затем перемешивали в течение ночи при комнатной температуре. Растворитель выпаривали, добавляли толуол (60 мл); смесь кипятили с обратным холодильником над установкой Dean-Starck в течение следующих 2 часов. Смесь охлаждали и промывали раствором NaOH (2M). Органический слой сушили над MgS04, фильтровали и растворитель выпаривали, получая 2,3 г промежуточного соединения 8.
Пример A5
a) Получение
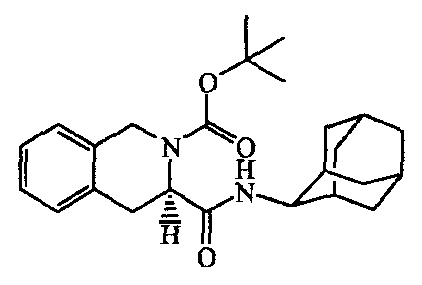
1-Гидрокси-lH-бензотриазол (0,0012 моль) и N-(этилкарбонимидоил)-N,N-диметил-1,3-пропандиамина моногидрохлорид [25952-53-8] (0,0012 моль) добавляли к смеси 2-(l,l-диметилэтилового)эфира (3R)-3,4-дигидро-2,3(lH)-изохинолиндикарбоновой кислоты, [115962-35-1] (0,001 моль) в DMF (10 мл) и N,N-диэтилэтанамина (0,2 мл). Смесь перемешивали в течение 20 минут при комнатной температуре. Добавляли трицикло[3.3.1.13,7]декан-2-амина гидрохлорид [10523-68-9] (0,0012 моль), и реакционную смесь перемешивали в течение ночи. Смесь выливали в воду и перемешивали 10 минут, затем полученный осадок отфильтровывали и растворяли в дихлорметане. Полученный раствор промывали водой, сушили над MgSO4, фильтровали и растворитель выпаривали, получая 0,38 г промежуточного соединения 9.
b) Получение
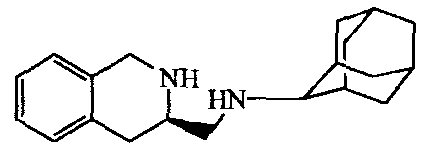
Смесь промежуточного соединения 9 (0,00087 моль) в толуоле (10 мл) перемешивали на льду (в атмосфере N2). По каплям добавляли BMS (0,001 моль), затем реакционную смесь 30 минут перемешивали на льду. Смесь кипятили с обратным холодильником в течение ночи. Смесь охлаждали и промывали раствором Nа2СО3. Органический растворитель выпаривали. Остаток растворяли в смеси CH2CI2/CF3COOH (20%) и 20 часов перемешивали при комнатной температуре. Растворители выпаривали. Остаток растворяли в CH2Cl2 и промывали раствором Nа2СО3. Органический слой концентрировали, и остаток очищали на колонке «Supelco», заполненной силикагелем (элюент: CH2Cl2/CH3OH, градиентное элюирование). Полученные фракции собирали, растворители выпаривали, получая 0,120 г промежуточного соединения 10.
Пример A6
a) Получение
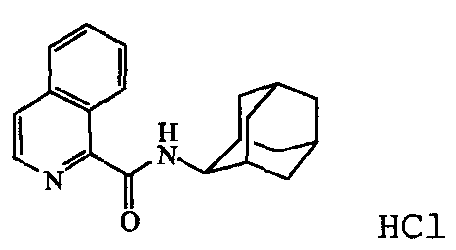
К перемешенному раствору 1-изохинолинкарбоновой кислоты (0,0056 моль) и N,N-диэтилэтанамина (0,7 г) в DMF (50 мл) добавляли l-гидрокси-lH-бензотриазол (0,0067 моль) и N'-(этилкарбонимидоил)-N,N-диметил-l,3-пропандиамина моногидрохлорид [25952-53-8] (0,0067 моль). Смесь 20 минут перемешивали при комнатной температуре. Добавляли трицикло[3.3.1.13,7]декан-2-амина гидрохлорид [10523-68-9] (0,0067 моль), и реакционную смесь перемешивали в течение ночи. Смесь выливали в воду, 10 минут перемешивали и экстрагировали дихлорметаном. Органический слой отделяли, сушили над MgSO4, фильтровали и растворитель выпаривали. Остаток растворяли в 2-пропаноле и под действием смеси HCl/2-пропанол превращали в соль хлористоводородной кислоты (1:1). Требуемый продукт фильтровали, получая 1,2 г промежуточного соединения 11.
b) Получение

Раствор промежуточного соединения 11 (0,0035 моль) в HC1, 2-пропанол (1 мл) и метанол (50 мл) в течение ночи подвергали гидрированию с платиновым катализатором, нанесенным на активированный уголь (0,5 г). После поглощения водорода (2 экв.) катализатор отфильтровывали и фильтрат упаривали. Остаток растворяли в дихлорметане и промывали раствором Na2CO3. Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали. Остаток очищали на колонке «Supelco», заполненной силикагелем (элюент: смесь CH2Cl2/CH3OH 99/1). Две полученные фракции собирали, и растворитель выпаривали, получая 0,370 г промежуточного соединения 12.
c) Получение

Раствор промежуточного соединения 12 (0,0012 моль) в толуоле (10 мл) перемешивали на льду (N2). Добавляли по каплям BMS (0,002 моль), затем реакционную смесь 30 минут перемешивали на льду и при 100°C перемешивали в течение ночи. Смесь промывали раствором NaHCO3 и экстрагировали с помощью CH2Cl2. Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали, получая остаток с массой 0,29 г. Остаток растирали в DIPE, и осадок фильтровали. Фильтрат упаривали, получая 0,22 г промежуточного соединения 13.
Пример А7
a) Получение
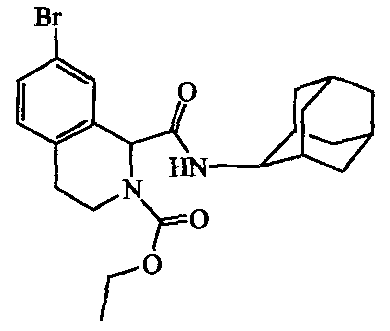
Смесь 2-этилового эфира 7-бром-3,4-дигидро-l,2(lH)-изохинолиндикарбоновой кислоты, [135335-12-5] (0,006 моль) и N,N-диэтилэтанамина (5 мл) в DMF (40 мл) перемешивали и добавляли l-гидрокси-lH-бензотриазол (0,0067 моль). Затем добавляли N'-(этилкарбонимидоил)-N,N-диметил-1,3-пропандиамина моногидрохлорид [25952-53-8] (0,0067 моль), и смесь 20 минут перемешивали. Добавляли трицикло[3.3.1.13,7]декан-2-амина гидрохлорид [10523-68-9] (0,0067 моль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь выливали в воду, перемешивали 10 минут. Полученный осадок фильтровали, растворяли в CH2Cl2, сушили над MgSO4, фильтровали и растворитель выпаривали. Остаток растирали в DIPE, требуемый продукт собирали, получая 1,6 г промежуточного соединения 14.
b) Получение
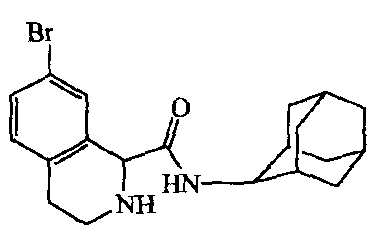
Раствор промежуточного соединения 14 (0,0034 моль) в смеси HBr/CH3COOH (50 мл) перемешивали при комнатной температуре в течение 1 недели. Смесь выливали в воду и перемешивали 15 минут. Осадок отфильтровывали и растворяли в CH2CI2. Раствор промывали раствором NaHCO3, сушили (MgSO4), фильтровали и растворитель выпаривали. Остаток растирали в DIPE, требуемую фракцию собирали (получая 0,7 г). Эту фракцию растворяли в разбавленном растворе HC1, и полученный раствор промывали CH2CI2. Водный слой подщелачивали раствором Nа2СО3 и экстрагировали CH2C12. Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали, получая 0,35 г промежуточного соединения 15.
c) Получение
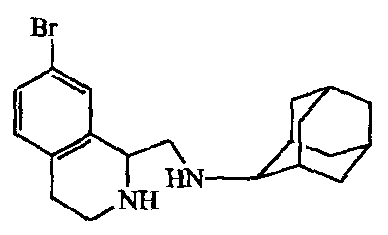
Смесь промежуточного соединения 15 (0,00089 моль) в толуоле (50 мл) и THF (20 мл) перемешивали в атмосфере N2 до полного растворения, и затем раствор перемешивали на льду в атмосфере N2. По каплям добавляли BMS (0,002 моль), и реакционную смесь 30 минут перемешивали на льду в атмосфере N2. Далее смесь перемешивали при 100°C в течение ночи и затем охлаждали. Добавляли 1н. раствор HC1 (50 мл). Смесь перемешивали и 2 часа кипятили с обратным холодильником. Полученную смесь охлаждали, нейтрализовали раствором Nа2СО3и экстрагировали CH2CI2. Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали, получая 0,3 г промежуточного соединения 16.
B. ПОЛУЧЕНИЕ СОЕДИНЕНИЙ
Пример Bl
Получение
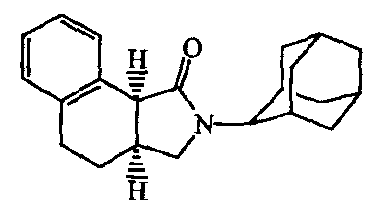
Смесь промежуточного соединения 1 (0,00093 моль) в безводном метилбензоле (10 мл) перемешивали 6 часов при 190°C и затем при комнатной температуре перемешивали в течение ночи. Растворитель выпаривали, и остаток очищали колоночной хроматографией над силикагелем (элюент: CH2CI2). Полученные фракции собирали, и растворитель выпаривали, получая 0,19 г (63%) соединения 1.
Пример B2
Получение
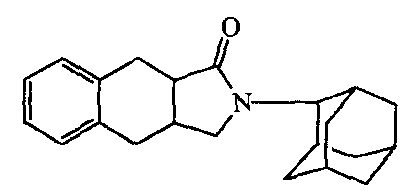
Смесь промежуточного соединения 2 (0,00031 моль) и 4-метоксифенола (каталитическое количество) в метилбензоле (10 мл) один час перемешивали при 220°C. Растворитель выпаривали, остаток очищали (2 раза) флеш-колоночной хроматографией на TRIKONEX FlashTube™ (элюент: смесь CH2Cl2/EtOAc 90/10). Полученные фракции собирали с получением 0,008 г соединения 2.
Пример B3
Получение
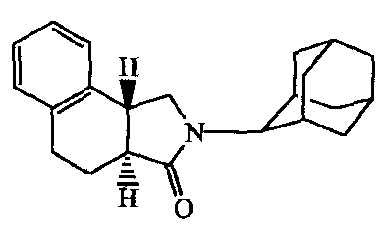
Раствор промежуточного соединения 5 (0,0015 моль) в метилбензоле (15 мл) перемешивали в автоклаве при 190°C 6 часов. Затем при комнатной температуре реакционную смесь перемешивали в течение ночи. Растворитель выпаривали, и остаток очищали на колонке фирмы Supelco, заполненной силикагелем (элюент: CH2Cl2). Фракции собирали, и растворитель и выпаривали, получая 0,1 г соединения 3.
Пример B4
Получение

Промежуточное соединение 8 (0,006 моль) в дихлорметане (250 мл) перемешивали и добавляли хлорид алюминия (0,018 моль). Реакционную смесь кипятили с обратным холодильником 3 часа. Смесь охлаждали и промывали КОН (1М). Органический слой промывали, сушили, фильтровали и растворитель выпаривали, получая 0,7 г остатка. Часть (0,3 г) остатка очищали над силикагелем (элюент: смесь CH2Cl2/EtOAc 90/10). Полученные фракции собирали, растворитель выпаривали, получая 0,133 г соединения 4.
Пример В5
Получение

Раствор промежуточного соединения 10 (0,00040 моль) в тетрагидрофуране (10 мл) перемешивали и добавляли l,l'-карбонилбис-lH-имидазол [530-62-1] (0,00045 моль). Смесь кипятили с обратным холодильником в течение ночи. После охлаждения добавляли воду (2 мл). Смесь экстрагировали дихлорметаном, и органический слой фильтровали через Extrelut™. Полученный остаток очищали колоночной хроматографией на силикагеле (Supelco) (элюент: CH2Cl2). Полученные фракции собирали, растворитель выпаривали, получая 0,063 г соединения 5.
Пример В6
Получение
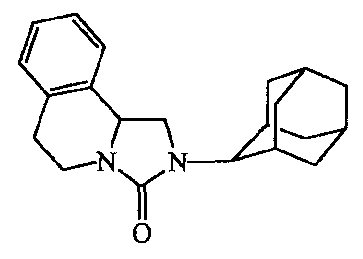
1,1'-Карбонилбис-1H-имидазол [530-62-1] (0,00185 моль) добавляли к перемешенному раствору промежуточного соединения 13 (0,00048 моль) в тетрагидрофуране (15 мл). Реакционную смесь перемешивали 48 часов при 60°C и охлаждали. Добавляли воду (4 мл). Смесь перемешивали 10 минут и экстрагировали дихлорметаном (10 мл). Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали. Остаток (0,337 г) очищали 2 раза на колонке фирмы Supelco, заполненной силикагелем (элюент: CH2Cl2) Полученные фракции собирали, растворитель выпаривали, получая 0,051 г соединения 6.
Пример B7
Получение
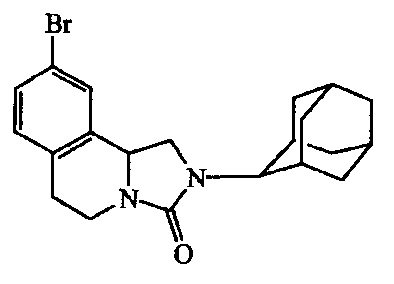
Смесь промежуточного соединения 16 (0,0008 моль) в тетрагидрофуране (5 мл) перемешивали и добавляли l,l'-карбонилбис-1H-имидазол (0,5 г). Реакционную смесь перемешивали в течение ночи при комнатной температуре, и растворитель выпаривали. Остаток очищали колоночной хроматографией (Supelco) над силикагелем (элюент: смесь CH2Cl2/EtOAc 90/10). Полученные фракции собирали, растворители выпаривали, получая 0,068 г соединения 7.
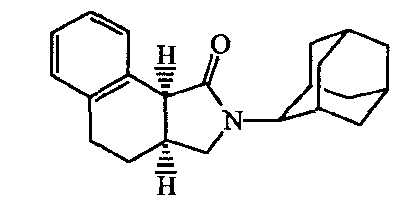
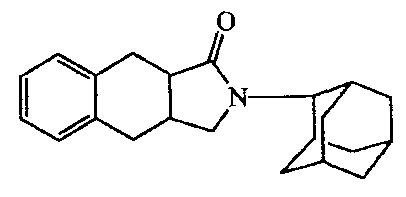
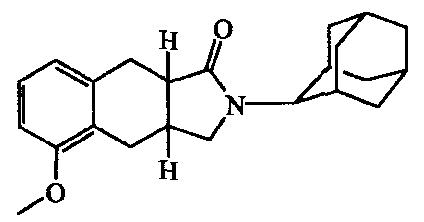
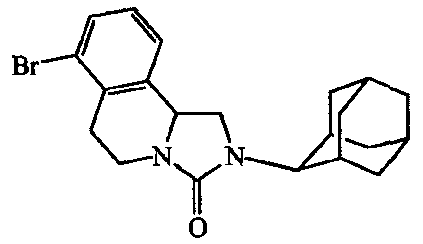
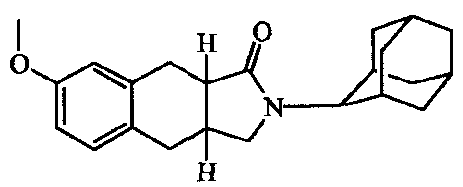
В таблице F-l перечислены соединения, которые были получены согласно одному из приведенных выше примеров.



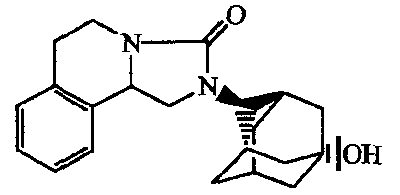
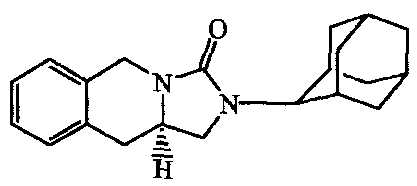


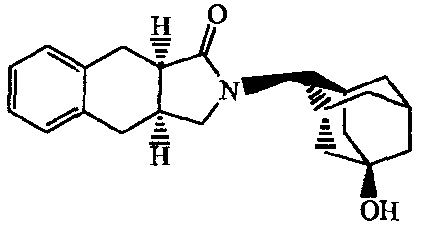
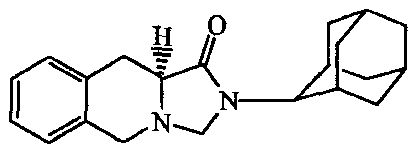




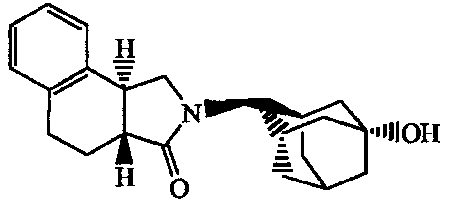
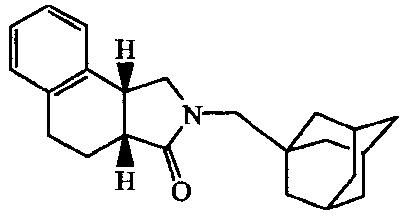


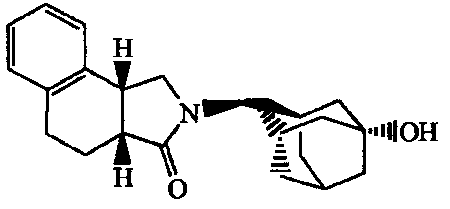
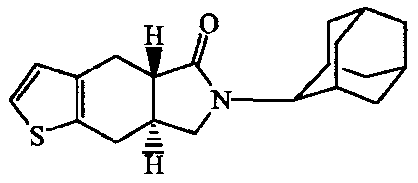
В таблице F-2 приведены данные по химическим сдвигам сигналов1Н ЯМР и13С ЯМР для образцов соединений по настоящему изобретению, при использовании в качестве растворителя CDCl3.
C. ФАРМАКОЛОГИЧЕСКИЕ ПРИМЕРЫ
Пример C1:
Ферментативный анализ, исследующий действие соединений на 11β-гидроксистероидную дегидрогеназу типа 1 и типа 2
Воздействия соединений на 11β-HSDl - зависимое превращение кортизона в кортизол (активность редуктазы), изучали на реакционной смеси, содержащей 30 мМ Tris-HC1 буфера с pH 7,2, 180 мкМ NADPH (НАДФ), 1мМ EDTA (ЭДТА), 2 мкМ кортизона, 1 мкл лекарственного средства и/или растворителя и 11 мкг рекомбинантного белка в конечном объеме, составляющем 100 мкл.
Воздействие на активность 11β-HSDl-дегидрогеназы (превращение кортизола в кортизон) измеряли в реакционной смеси, содержащей 0,1M натрийфосфатного буфера с pH 9,0, 300 мкМ NADP(НАДФ), 25 мкМ кортизола, 1 мкл лекарственного средства и/или растворителя и 3,5 мкг рекомбинантного белка в конечном объеме, составляющем 100 мкл.
Воздействия на 11β-HSD2 - зависимую активность дегидрогеназы изучали на реакционной смеси, содержащей 0,1M натрийфосфатного буфера с pH 7,5, 300 мкМ NAD, 100 нM кортизола (из которого 2 нM помечены радиоактивным тритием,3H), 1 мкл лекарственного средства и/или растворителя и 2,5 мкг рекомбинантного белка в конечном объеме, составляющем 100 мкл.
Все инкубации осуществляли на водяной бане при 37°C в течение 45 мин. Реакцию останавливали добавлением 100 мкл ацетонитрила, содержащего в качестве внутреннего стандарта 20 мкг кортикостерона. После центрифугирования тестирование образования продукта проводили в надосадочной жидкости посредством ВЭЖХ (HPLC) на колонке Hypersyl BDS-C18, используя в качестве растворителя 0,05 мкМ смеси ацетат аммония/метанол (50/50). Во всех упомянутых выше опытах лекарственные средства, подлежащие тестированию, брали из основного раствора и тестировали при конечной концентрации в диапазоне от 10-5М до 3,10-9М. Из полученных таким образом кривых ответной реакции на дозу вычисляли величины pIC50и оценивали в баллах следующим образом: балл 1=величина pIC50<5, балл 2=величина pIC50 находится в интервале от 5 до 6, балл 3=величина pIC50>6. Некоторые из полученных таким образом результатов суммированы в таблице, приведенной ниже.
Пример C2: Клеточный опыт по тестированию действия соединений на 11β-гидроксистероидную дегидрогеназу типа 1 и типа 2
Воздействие на активность 11β-HSDl измеряли на дифференцированных 3T3-L1 клетках и гепатоцитах крысы.
Клетки 3T3-L1 мышиных фибробластов (ATCC-CL-173) высеивали с плотностью 16500 клеток/мл в 12-луночные планшеты и выращивали в течение 7 дней в среде DMEM (с добавлением 10% инактивированной нагреванием околоплодной сыворотки теленка, 2мМ глутамина и 25 мг гентамицина) при 37°C во влажной 5% CO2 атмосфере. Среду регенерировали дважды в неделю. Дифференцировка фибробластов в адипоциты происходила при 37°C во влажной 5% CO2 атмосфере в среде для выращивания, содержащей 2 мкг/мл инсулина, 55 мкг/мл IBMX и 39,2 мкг/мл дексаметазона.
Первичные гепатоциты самцов крысы высевали в обычные 12-луночные планшеты фирмы Falcon с плотностью 250000 клеток на лунку и инкубировали в течение 16 часов при 37°C во влажной 5% CO2 атмосфере в среде DMEM-HAM's F12, содержащей 5% Nu(нейтрализующей) сыворотки, 100 Е(U)/мл пенициллина, 100 мкг/мл стрептомицина, 0,25 мкг/мл амфотерицина B, 50 мкг/мл гентамицинсульфата, 5 мкг/мл инсулина 392 нг/мл дексаметазона. После 4 часов преинкубации с тестируемым соединением к 3T3-L1 культурам добавляли 0,5 мкКи3H-кортизона или дегидрокортикостерона. Через час среду экстрагировали на Extrelut3колонках 15 мл диэтилового эфира, и экстракт анализировали с помощью ВЭЖХ (HPLC), как было описано выше. Воздействие JNJ-соединений на HSD1 активность крысиного гепатоцита измеряли после 90-минутного инкубационного периода с помощью 0,5 мкКи3H-дегидрокортикостерона. Анализ на образование кортикостерона проводили с помощью ВЭЖХ.
Влияние на активность 11β-HSD2 изучали на HepG2- и LCC-PK1-клетках. HepG2-клетки (ATCC HB-8065) высаживали в 12-луночные планшеты с плотностью 100000 клеток/мл и выращивали при 37°C в увлажненной 5% CO2 атмосфере в MEM-Rega-3 среде с добавлением 10% инактивированной нагреванием околоплодной сыворотки теленка, 2мМ L-глутамина и бикарбоната натрия. Среду регенерировали дважды в неделю.
Клетки почек свиньи (LCC-PK1, ATCC CRL-1392) высаживали в 12-луночные планшеты с плотностью 150000 клеток/мл и выращивали при 37°C в увлажненной 5% CO2 атмосфере в среде 199 с добавлением модифицированного солевого раствора Эрла, 100 Е/мл пенициллина, 100 мкг/мл стрептомицина и 10% околоплодной сыворотки теленка. Среду регенерировали дважды в неделю. За двадцать четыре часа до начала эксперимента среду заменяли средой, содержащей 10% околоплодную сыворотку теленка, очищенную от легких элементов активированным углем.
После 4 часов преинкубации с тестируемым соединением к культурам добавляли 0,5 мкКи3H-кортизола или кортикостерона. Через час среду экстрагировали на Extrelut3колонках 15 мл диэтилового эфира, и экстракт анализировали с помощью ВЭЖХ (HPLC), как было описано выше.
Что касается ферментативных анализов, то соединения, подлежащие тестированию, брали из основного раствора и тестировали при конечной концентрации в диапазоне от 10-5М до 3,10-9М. Из полученных таким образом кривых ответной реакции на дозу вычисляли величины pIC50и оценивали в баллах следующим образом: балл 1 - величина pIC50<5, балл 2 - величина pIC50 находится в интервале от 5 до 6, балл 3 - величина pIC50>6. Некоторые из полученных таким образом результатов суммированы в таблице, приведенной ниже.
D. Примеры композиций
Нижеследующие препараты служат примерами типичных фармацевтических композиций по настоящему изобретению, пригодных для системного или местного введения пациенту, животному и человеку.
Термин "активный ингредиент" (A.I.), используемый на всем протяжении описания данных примеров, обозначает соединение формулы (I) или его фармацевтически приемлемую аддитивную соль.
Пример Dl: Таблетки с пленочным покрытием.
Получение центральной части таблетки.
Смесь A.I. (100 г), лактозы (570 г) и крахмала (200 г) тщательно перемешивали и после этого увлажняли раствором додецилсульфата натрия (5 г) и поливинилпирролидона (10 г) в приблизительно 200 мл воды. Смоченную смесь порошков просеивали, сушили и просеивали снова. Затем добавляли микрокристаллическую целлюлозу (100 г) и гидрогенизированное растительное масло (15 г). Все это тщательно перемешивали и прессовали в таблетки, получая 10000 таблеток, каждая из которых содержала 10 мг активного ингредиента.
Покрытие
К раствору метилцеллюлозы (10 г) в денатурированном этаноле (75 мл) добавляли раствор этилцеллюлозы (5 г) в CH2C12 (150 мл). Затем добавляли CH2C12 (75 мл) и 1,2,3-пропантриол (2,5 мл). Полиэтиленгликоль (10 г) плавили и растворяли в дихлорметане (75 мл). Последний раствор добавляли к предыдущему и затем добавляли октадеканоат магния (2,5 г), поливинилпирролидон (5 г) и концентрированную цветную суспензию (30 мл); и все в целом гомогенизировали. Центральную часть таблетки покрывали смесью, полученной таким образом в специальных аппаратах для покрытия.
Реферат
Изобретение относится к соединениям общей формулы (I), их N-оксидным формам, фармацевтически приемлемым аддитивным солям и стереохимически изомерным формам в качестве ингибитора 11-HSD1, к их применению, фармацевтической композиции на их основе и способу ее получения. Соединения могут найти применение для лечения и профилактики заболеваний, опосредованных 11-HSD1. В общей формуле (I) ! ! X представляет собой С или N; Y представляет собой С или N; L представляет собой метил или прямую связь; Z1 представляет собой прямую связь, С1-2алкил или радикал формулы -СН=; Z2 представляет собой прямую связь, С1-2алкил; R1 представляет собой водород, гало, гидрокси; R2 представляет собой водород, гало или С1-4алкилокси; А представляет собой фенил или моноциклический гетероцикл, выбранный из группы, состоящей из тиофенила или пирридинила. 6 н. и 3 з.п. ф-лы, 3 табл.
Формула
его N-оксидные формы, фармацевтически приемлемые аддитивные соли и стереохимически изомерные формы,
где Х представляет собой С или N;
Y представляет собой С или N;
L представляет собой метил или прямую связь;
Z1 представляет собой прямую связь, С1-2алкил или радикал формулы -СН=;
Z2 представляет собой прямую связь, C1-2алкил;
R1 представляет собой водород, гало, гидрокси;
R2 представляет собой водород, гало или С1-4алкилокси;
А представляет собой фенил или моноциклический гетероцикл, выбранный из группы, состоящей из тиофенила или пирридинила.
А представляет собой фенил или пиридинил, и где L представляет собой прямую связь; и/или
R1 представляет собой гало, гидрокси.
2-адамантан-2-ил-2,3,3а,4,9,9а-гексагидробензо[f]изоиндол-1-он;
2-адамантан-2-ил-2,3,10,10а-тетрагидро-5Н-имидазо[1,5-b]изохинолин-1-он;
2-адамантан-2-ил-1,5,10,10а-тетрагидро-2Н-имидазо[1,5-b]изохинолин-3-он;
2-адамантан-1-илметил-1,2,3а,4,5,9b-гексагидробензо[e]изоиндол-3-он;
7-адамантан-2-ил-7,8,8а,9-тетрагидропирроло [3,4-g]хинолин-6-он;
2-(5-гидроксиадамантан-2-ил)-1,5,6,10b-тетрагидро-2Н-имидазо[5,1-а]изохинолин-3-он;
2-(5-фторадамантан-2-ил)-1,2,3а,4,5,9b-гексагидробензо[е]изоиндол-3-он и
2-(5-гидроксиадамантан-2-ил)-2,3,3а,4,9,9а-гексагидробензо[f]изоиндол-1-он.
Комментарии