Арил- и гетероарилзамещенные тетрагидроизохинолины и их применение для блокирования обратного захвата норэпинефрина, допамина и серотонина - RU2388751C2
Код документа: RU2388751C2
Описание
Настоящая заявка притязает на приоритет по предварительной заявке на выдачу патента США с регистрационным No. 60/588448, поданной 15 июля 2004, которая включена в данное описании в виде ссылки в полном объеме.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новым соединениям 4-замещенных бициклическими карбоциклами и гетероциклами производных тетрагидроизохинолина, фармацевтическим композициям, содержащим такие соединения, способам применения таких соединений для лечения различных неврологических и психических расстройств и для комбинированной терапии.
УРОВЕНЬ ТЕХНИКИ
Хорошо известно, что нейромедиаторы, допамин (DA), норэпинефрин (NE) и серотонин (5-HT), регулируют ряд биологических процессов и что пониженные уровни DA, NE и 5-HT связаны с рядом неврологических расстройств и их физических проявлений. Значительные усилия были затрачены на разработку способов корректировки уровней указанных нейромедиаторов, чтобы получить требуемый фармакологический эффект. Предотвращение обратного захвата указанных нейромедиаторов в любой их комбинации из одного, двух или всех трех, вероятно, должно быть эффективным для лечения указанных расстройств. Доказано, что целенаправленное воздействие на белки переносчики допамина (DAT), переносчики норэпинефрина (NET) и переносчики серотонина (SERT) является эффективным способом повышения уровней соответствующих моноаминов.
Известно, что метилфенидат, который в настоящее время используют для лечения гиперактивного расстройства с дефицитом внимания (ADHD), является избирательным в отношении ингибирования DAT. В патенте США No. 5444070 описаны избирательные ингибиторы обратного захвата допамина в качестве средств лечения болезни Паркинсона и наркомании или злоупотребления лекарственными средствами, включая кокаин и амфетамины.
Также описаны избирательные ингибиторы обратного захвата норэпинефрина (NARI). Например, в патенте США No. 6352986 описаны способы лечения ADHD, расстройств, связанных с наркотической зависимостью, и расстройств, связанных с применением психоактивных веществ, с помощью ребоксетина. Кроме того, в настоящее время продается атомоксетин (Strattera®) в качестве избирательного ингибитора обратного захвата NET для ADHD.
Также показано, что применение избирательных ингибиторов обратного захвата серотонина (SSRI) эффективно для лечения депрессивных расстройств. Сертралин, циталопрам и пароксетин являются хорошо известными примерами SSRI, используемыми для лечения таких расстройств, как депрессия, обсессивно-компульсивное расстройство и острые тревожные состояния с реакцией паники. Существует несколько известных трудностей в случае применения терапевтических средств класса SSRI, включая медленное начало действия, нежелательные побочные эффекты и существование значительной подгруппы популяции, которая не отвечает на терапию SSRI.
Избирательные ингибиторы обратного захвата DAT, NET и SERT также можно вводить совместно друг с другом или с другими лекарственными средствами. В патенте США No. 5532244 описано применение ингибиторов обратного захвата серотонина в комбинации с антагонистом серотонина-1A для лечения обсессивно-компульсивного расстройства, депрессии и ожирения. Применение ингибитора обратного захвата серотонина или норэпинефрина в комбинации с антагонистом рецептора нейрокинина-1 описано в патенте США No. 6121261 для лечения ADHD. В патенте США No. 4843071 описано применение ингибитора обратного захвата норэпинефрина в комбинации с предшественником норэпинефрина для лечения ожирения, наркомании или нарколепсии. В патенте США No. 6596741 описано применение ингибитора NE, DA или 5-HT либо с антагонистом рецептора нейрокинина-1, либо с антагонистом серотонина-1A для лечения широкого круга состояний.
Также полезно применение соединений, которые ингибируют одновременно один или несколько нейромедиаторов. Антидепрессантные качества двойного ингибитора обратного захвата NET и SERT дулоксетина описаны в европейском патенте No. 273658. Венлафаксин описан в патенте США No. 4535186 в качестве ингибитора обратного захвата NE и 5-HT для лечения депрессивных расстройств. В патенте США No. 6635675 описано применение двойного ингибитора обратного захвата NE и 5-HT милнаципрана для лечения синдрома хронической усталости и синдрома фибромиалгии. Кроме того, двойные ингибиторы обратного захвата NE и 5-HT описаны в патенте США No. 6136083 для лечения депрессии. Также известно, что соединения, которые ингибируют обратный захват NE, DA и 5-HT в различных соотношениях, специально не указанные в данном описании, также могут быть полезными.
Лечение заболеваний посредством ингибирования обратного захвата всех трех моноаминов, либо посредством комбинированной терапии, либо «тройными ингибиторами» также может иметь клинические преимущества. В международных публикациях PCT No. WO 03/101453 и WO 97/30997 описан класс соединений, которые являются активными против всех трех переносчиков моноаминов. К основным причинам для включения допамин-усиливающего компонента в антидепрессантную терапию относятся наблюдаемый дефицит допаминергической функции, успех комбинированной терапии агонистами допамина и традиционными антидепрессантами и повышенная чувствительность рецепторов допамина вследствие хронического введения антидепрессантов (Skolnick et al., Life Sciences 73: 3175-3179 (2003)). По существу предполагается, что ингибирующая активность против обратного захвата DA в дополнение к обратному захвату NE и 5-HT обеспечивает более быстрое начало антидепрессантного действия по сравнению с другими смешанными ингибиторами, которые являются избирательными в отношении NET и SERT по сравнению с DAT. Кроме того, в международной публикации PCT No. WO 03/049736 описана серия 4-замещенных пиперидинов, каждый из которых проявляет сходную активность против переносчиков DA, NE и 5-HT. Бицикло[2.2.1]гептаны (Axford et al., Bioorg. Med. Chem. Lett. 13: 3277-3280 (2003)) и азабицикло[3.1.0]гексаны (Skolnick et al., Eur. J. Pharm., 461: 99-104 (2003)) также описаны в качестве тройных ингибиторов трех переносчиков моноаминов.
В патенте США No. 3947456 описаны тетрагидроизохинолины, которые, как указано, применимы в качестве антидепрессантов. В патенте США No. 3666763 описано применение производных фенилтетрагидроизохинолина в качестве антидепрессантов и антигипотензивных средств. В заявке на выдачу патента Канады No. 2015114 описано применение производных фенилтетрагидроизохинолина в качестве антидепрессантов; соединения, описанные в указанной публикации, очевидно, являются неизбирательными в отношении обратного захвата норэпинефрина, серотонина и допамина. В заявке на выдачу патента Великобритании No. 2271566 описано применение производных фенилтетрагидроизохинолина в качестве средств против ВИЧ. В международной публикации PCT No. WO 98/40358 описано применение производных фенилтетрагидроизохинолина как полезных при лечении расстройств путей метаболизма глюкозы. В международной публикации PCT No. WO 97/36876 описано применение производных фенилтетрагидроизохинолина в качестве противоопухолевых средств. В международной публикации PCT No. WO 97/23458 также описаны 4-фенилзамещенные тетрагидроизохинолины в качестве лигандов рецептора NMDA, применимых в случае состояний, связанных с потерей нейронов. Фенилзамещенные тетрагидроизохинолины также описаны в Mondeshka et al., Il Farmaco 49: 475-481 (1994).
В патенте США No. 6579885 описано применение 7-арилзамещенных тетрагидроизохинолинов в качестве полезных средств для лечения расстройств, в которые вовлечена пониженная доступность серотонина, норэпинефрина или допамина. В Tupper et al., J. Heterocyclic Chem. 33: 1123-1129 (1996) описан синтез тетрагидроизохинолинов, замещенных 2- или 3-тиенильной группой в положении 4, в качестве возможных антагонистов допамина D1 и D2. В Prat et al., J. Heterocyclic Chem. 37: 767-771 (2000) описан синтез N-метил-4-пиридил-1,2,3,4-тетрагидроизохинолинов, а также 2-пиридил- и 3-пиридил-аналогов, которые созданы в качестве потенциальных аналогов серотонина, но об их активности не сообщается. В Chandrasekhar et al., Tetrahedron Lett. 43: 1885-1888 (2002) описан синтез транс-4-бензо[1,3]диоксол-5-ил-2-бензил-3-метил-1,2,3,4-тетрагидроизохинолина, однако не сообщается ни о применении, ни об активности. В Lopez et al., Tetrahedron 50: 9097-9106 (1994) описан синтез 2-(1,2,3,4-тетрагидроизохинолин-4-ил)хинолин-3-ола, однако не сообщается ни о применении, ни об активности. В Uno et al., J. Heterocyclic Chem. 38: 341-346 (1991) описан синтез 1-этил-1'-пентафторэтил-1,2,3,4-тетрагидро[4,4']бис(изохинолина), однако не сообщается ни о применение, ни об активности.
Известно, что номифензин®, который является 4-фенилзамещенным производным тетрагидроизохинолина, ингибирует обратный захват нейронами допамина и других катехоламинов, и показана клиническая эффективность в случае ADHD. Однако длительное введение номифензина® приводило к смертельной иммунной гемолитической анемии у очень небольшого количества пациентов, что заставило производителя прекратить поставку указанного лекарственного средства на рынок. Таким образом, остается необходимость в разработке новых соединений, которые лечат ADHD, но не имеют серьезных побочных эффектов, связанных с номифензином® или назначаемыми в настоящее время психостимуляторами.
Настоящее изобретение относится к достижению указанных целей.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Соединения согласно настоящему изобретению представлены химической структурой, определяемой формулой I:

Формула I
где атом углерода, обозначенный *, находится в R- или S-конфигурации;
X означает конденсированный бициклический карбоцикл или гетероцикл, выбранный из группы, состоящей из бензофуранила, бензо[b]тиофенила, бензоизотиазолила, бензоизоксазолила, индазолила, индолила, изоиндолила, индолизинила, бензоимидазолила, бензооксазолила, бензотиазолила, бензотриазолила, имидазо[1,2-a]пиридинила, пиразоло[1,5-a]пиридинила, [1,2,4]триазоло[4,3-a]пиридинила, тиено[2,3-b]пиридинила, тиено[3,2-b]пиридинила, 1H-пирроло[2,3-b]пиридинила, инденила, инданила, дигидробензоциклогептенила, тетрагидробензоциклогептенила, дигидробензотиофенила, дигидробензофуранила, индолинила, нафтила, тетрагидронафтила, хинолинила, изохинолинила, 4H-хинолизинила, 9aH-хинолизинила, хиназолинила, циннолинила, фталазинила, хиноксалинила, бензо[1,2,3]триазинила, бензо[1,2,4]триазинила, 2H-хроменила, 4H-хроменила, и конденсированный бициклический карбоцикл или конденсированный бициклический гетероцикл, необязательно замещенный заместителями (в количестве от 1 до 4), которые определены ниже для R14;
R1 означает H, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, каждый из которых необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, арила, -CN, -OR9 и -NR9R10;
R2 означает H, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил, C4-C7-циклоалкилалкил или C1-C6-галогеналкил, каждый из которых необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, арила, -CN, -OR9и -NR9R10; или
R2 означает гем-диметил;
R3 означает H, галоген, -OR11, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, -NR9R10, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9;
R4 означает H, галоген, -OR11, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, -NR9R10, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9; или
R4 означает фенил, нафтил, инденил, пиридил, пиримидинил, пиридазинил, пиразинил, [1,2,4]триазинил, [1,3,5]триазинил, триазолил, фуранил, тиофенил, пиранил, индазолил, бензимидазолил, хинолинил, хиназолинил, хиноксалинил, фталазинил, циннолинил, изохинолинил, тиенил, имидазолил, тиазолил, бензтиазолил, пуринил, изотиазолил, индолил, пирролил, оксазолил, бензофуранил, бензотиенил, бензтиазолил, изоксазолил, пиразолил, оксадиазолил, тиадиазолил, 3-оксо-[1,2,4]триазоло[4,3-a]пиридинил, имидазо[1,2-a]пиридинил, пиразоло[1,5-a]пиридинил, [1,2,4]триазоло[4,3-a]пиридинил, тиено[2,3-b]пиридинил, тиено[3,2-b]пиридинил, 1H-пирроло[2,3-b]пиридинил или другие гетероциклы, необязательно замещенные заместителями (в количестве от 1 до 4), которые определены ниже для R14;
R5 и R6, каждый независимо, выбраны из группы, состоящей из H, галогена, -OR11, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, -NR9R10, C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила, C4-C7-циклоалкилалкила и фенила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9;
R7 означает H, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9; или
R7 означает гем-диметил;
R8 означает H, галоген, -OR9, -SR9, C1-C6-алкил, -CN или -NR9R10;
R9 и R10, каждый независимо, выбраны из группы, состоящей из H, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксиалкила, C3-C6-циклоалкила, C4-C7-циклоалкилалкила, -C(O)R13, фенила и бензила, где фенил или бензил необязательно замещен 1-3 раза заместителем, выбранным независимо в каждом случае из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила и C1-C4-алкоксигруппы; или
R9 и R10, взятые вместе с атомом азота, с которым они связаны, образуют пиперидин, пирролидин, пиперазин, N-метилпиперазин, морфолин, тиоморфолин, [1,2]оксазинан, изоксазолидин или 2-оксо-2H-пиридин, который необязательно замещен 1-3 раза заместителем, выбранным независимо в каждом случае из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила и C1-C4-алкоксигруппы;
R11 означает H, C1-C4-алкил, C1-C4-галогеналкил, C1-C4-алкоксиалкил, C3-C6-циклоалкил, C4-C7-циклоалкилалкил, -C(O)R13, фенил или бензил, где фенил или бензил необязательно замещен 1-3 раза галогеном, цианогруппой, C1-C4-алкилом, C1-C4-галогеналкилом или C1-C4-алкоксигруппой;
R12 означает H, C1-C4-алкил, C1-C4-галогеналкил, C1-C4-алкоксиалкил, C3-C6-циклоалкил, C4-C7-циклоалкилалкил, фенил или бензил, где фенил или бензил необязательно замещен 1-3 раза галогеном, цианогруппой, C1-C4-алкилом, C1-C4-галогеналкилом или C1-C4-алкоксигруппой; или
R11 и R12, взятые вместе с атомом азота, с которым они связаны, образуют цикл пиперидина, пирролидина, пиперазина, N-метилпиперазина, морфолина или тиоморфолина при условии, что только одна комбинация из R9 и R10 или R11 и R12, взятых вместе с атомом азота, с которым они связаны, образует цикл пиперидина, пирролидина, пиперазина, N-метилпиперазина, морфолина или тиоморфолина;
R13 означает C1-C4-алкил, C1-C4-галогеналкил или фенил;
n равно 0, 1 или 2; и
R14 в каждом случае независимо выбран из заместителя, выбранного из группы, состоящей из галогена, -NO2, -OR11, -NR11R12, -NR11C(O)R12, -NR11C(O)2R12, -NR11C(O)NR12R13, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила, где C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил и C4-C7-циклоалкилалкил необязательно замещены 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, арила, -CN, -OR9 и -NR9R10;
или их оксидом, или их фармацевтически приемлемой солью.
Соединения согласно настоящему изобретению также представлены химической структурой, определяемой формулой I:

Формула I
где атом углерода, обозначенный *, находится в R- или S-конфигурации;
X означает конденсированный ароматический бициклический карбоцикл или гетероцикл, необязательно замещенный заместителями (в количестве от 1 до 4), которые определены ниже для R14, при условии, что X ≠ изохинолинилу, нафтилу или фталимидилу;
R1 означает H, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, каждый из которых необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, арила, -CN, -OR9 и -NR9R10;
R2 означает H, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил, C4-C7-циклоалкилалкил или C1-C6-галогеналкил, каждый из которых необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, арила, -CN, -OR9и -NR9R10; или
R2 означает гем-диметил;
R3 означает H, галоген, -OR11, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, -NR9R10, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9;
R4 означает H, галоген, -OR11, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, -NR9R10, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9; или
R4 означает фенил, нафтил, инденил, пиридил, пиримидинил, пиридазинил, пиразинил, [1,2,4]триазинил, [1,3,5]триазинил, триазолил, фуранил, тиофенил, пиранил, индазолил, бензимидазолил, хинолинил, хиназолинил, хиноксалинил, фталазинил, циннолинил, изохинолинил, тиенил, имидазолил, тиазолил, бензтиазолил, пуринил, изотиазолил, индолил, пирролил, оксазолил, бензофуранил, бензотиенил, бензтиазолил, изоксазолил, пиразолил, оксадиазолил, тиадиазолил, 3-оксо-[1,2,4]триазоло[4,3-a]пиридинил, имидазо[1,2-a]пиридинил, пиразоло[1,5-a]пиридинил, [1,2,4]триазоло[4,3-a]пиридинил, тиено[2,3-b]пиридинил, тиено[3,2-b]пиридинил, 1H-пирроло[2,3-b]пиридинил или другие гетероциклы, необязательно замещенные заместителями (в количестве от 1 до 4), которые определены ниже для R14;
R5 и R6, каждый независимо, выбраны из группы, состоящей из H, галогена, -OR11, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, -NR9R10, C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила, C4-C7-циклоалкилалкила и фенила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9;
R7 означает H, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9; или
R7 означает гем-диметил;
R8 означает H, галоген, -OR9, -SR9, C1-C6-алкил, -CN или -NR9R10;
R9 и R10, каждый независимо, выбраны из группы, состоящей из H, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксиалкила, C3-C6-циклоалкила, C4-C7-циклоалкилалкила, -C(O)R13, фенила и бензила, где фенил или бензил необязательно замещен 1-3 раза заместителем, выбранным независимо в каждом случае из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила и C1-C4-алкоксигруппы; или
R9 и R10, взятые вместе с атомом азота, с которым они связаны, образуют пиперидин, пирролидин, пиперазин, N-метилпиперазин, морфолин, тиоморфолин, [1,2]оксазинан, изоксазолидин или 2-оксо-2H-пиридин, который необязательно замещен 1-3 раза заместителем, выбранным независимо в каждом случае из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила и C1-C4-алкоксигруппы;
R11 означает H, C1-C4-алкил, C1-C4-галогеналкил, C1-C4-алкоксиалкил, C3-C6-циклоалкил, C4-C7-циклоалкилалкил, -C(O)R13, фенил или бензил, где фенил или бензил необязательно замещен 1-3 раза галогеном, цианогруппой, C1-C4-алкилом, C1-C4-галогеналкилом или C1-C4-алкоксигруппой;
R12 означает H, C1-C4-алкил, C1-C4-галогеналкил, C1-C4-алкоксиалкил, C3-C6-циклоалкил, C4-C7-циклоалкилалкил, фенил или бензил, где фенил или бензил необязательно замещен 1-3 раза галогеном, цианогруппой, C1-C4-алкилом, C1-C4-галогеналкилом или C1-C4-алкоксигруппой; или
R11 и R12, взятые вместе с атомом азота, с которым они связаны, образуют цикл пиперидина, пирролидина, пиперазина, N-метилпиперазина, морфолина или тиоморфолина, при условии, что только одна комбинация из R9 и R10 или R11 и R12, взятых вместе с атомом азота, с которым они связаны, образует цикл пиперидина, пирролидина, пиперазина, N-метилпиперазина, морфолина или тиоморфолина;
R13 означает C1-C4-алкил, C1-C4-галогеналкил или фенил;
n равно 0, 1 или 2; и
R14 в каждом случае независимо выбран из заместителя, выбранного из группы, состоящей из галогена, -NO2, -OR11, -NR11R12, -NR11C(O)R12, -NR11C(O)2R12, -NR11C(O)NR12R13, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила, где C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил и C4-C7-циклоалкилалкил необязательно замещены 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, арила, -CN, -OR9 и -NR9R10;
или их оксидом, или их фармацевтически приемлемой солью.
Кроме того, соединения согласно настоящему изобретению представлены химической структурой, определяемой формулой I:
Формула I
где атом углерода, обозначенный *, находится в R- или S-конфигурации;
X означает конденсированный бициклический карбоцикл или гетероцикл, выбранный из группы, состоящей из бензофуранила, бензо[b]тиофенила, бензоизотиазолила, бензоизоксазолила, индазолила, индолила, изоиндолила, индолизинила, бензоимидазолила, бензооксазолила, бензотиазолила, бензотриазолила, имидазо[1,2-a]пиридинила, пиразоло[1,5-a]пиридинила, [1,2,4]триазоло[4,3-a]пиридинила, тиено[2,3-b]пиридинила, тиено[3,2-b]пиридинила, 1H-пирроло[2,3-b]пиридинила, инденила, инданила, дигидробензоциклогептенила, тетрагидробензоциклогептенила, дигидробензотиофенила, дигидробензофуранила, индолинила, нафтила, тетрагидронафтила, хинолинила, изохинолинила, 4H-хинолизинила, 9aH-хинолизинила, хиназолинила, циннолинила, фталазинила, хиноксалинила, бензо[1,2,3]триазинила, бензо[1,2,4]триазинила, 2H-хроменила, 4H-хроменила, и конденсированный бициклический карбоцикл или конденсированный бициклический гетероцикл, необязательно замещенный заместителями (в количестве от 1 до 4), которые определены ниже для R14; при условии, что: (1) когда X = нафтилу и R4 означает NH2 или OR11, R5 не может быть H либо (2) когда X = нафтилу и R5 = OR11, R4 не может быть H;
R1 означает H, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, каждый из которых необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, арила, -CN, -OR9 и -NR9R10;
R2 означает H, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил, C4-C7-циклоалкилалкил или C1-C6-галогеналкил, каждый из которых необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, арила, -CN, -OR9и -NR9R10; или
R2 означает гем-диметил;
R3 означает H, галоген, -OR11, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, -NR9R10, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9;
R4 означает H, галоген, -OR11, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, -NR9R10, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9; или
R4 означает фенил, нафтил, инденил, пиридил, пиримидинил, пиридазинил, пиразинил, [1,2,4]триазинил, [1,3,5]триазинил, триазолил, фуранил, тиофенил, пиранил, индазолил, бензимидазолил, хинолинил, хиназолинил, хиноксалинил, фталазинил, циннолинил, изохинолинил, тиенил, имидазолил, тиазолил, бензтиазолил, пуринил, изотиазолил, индолил, пирролил, оксазолил, бензофуранил, бензотиенил, бензтиазолил, изоксазолил, пиразолил, оксадиазолил, тиадиазолил, 3-оксо-[1,2,4]триазоло[4,3-a]пиридинил, имидазо[1,2-a]пиридинил, пиразоло[1,5-a]пиридинил, [1,2,4]триазоло[4,3-a]пиридинил, тиено[2,3-b]пиридинил, тиено[3,2-b]пиридинил, 1H-пирроло[2,3-b]пиридинил или другие гетероциклы, необязательно замещенные заместителями (в количестве от 1 до 4), которые определены ниже для R14;
R5 и R6, каждый независимо, выбраны из группы, состоящей из H, галогена, -OR11, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, -NR9R10, C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила, C4-C7-циклоалкилалкила и фенила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9;
R7 означает H, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9; или
R7 означает гем-диметил;
R8 означает H, галоген, -OR9, -SR9, C1-C6-алкил, -CN или -NR9R10;
R9 и R10, каждый независимо, выбраны из группы, состоящей из H, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксиалкила, C3-C6-циклоалкила, C4-C7-циклоалкилалкила, -C(O)R13, фенила и бензила, где фенил или бензил необязательно замещен 1-3 раза заместителем, выбранным независимо в каждом случае из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила и C1-C4-алкоксигруппы; или
R9 и R10, взятые вместе с атомом азота, с которым они связаны, образуют пиперидин, пирролидин, пиперазин, N-метилпиперазин, морфолин, тиоморфолин, [1,2]оксазинан, изоксазолидин или 2-оксо-2H-пиридин, который необязательно замещен 1-3 раза заместителем, выбранным независимо в каждом случае из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила и C1-C4-алкоксигруппы;
R11 означает H, C1-C4-алкил, C1-C4-галогеналкил, C1-C4-алкоксиалкил, C3-C6-циклоалкил, C4-C7-циклоалкилалкил, -C(O)R13, фенил или бензил, где фенил или бензил необязательно замещен 1-3 раза галогеном, цианогруппой, C1-C4-алкилом, C1-C4-галогеналкилом или C1-C4-алкоксигруппой;
R12 означает H, C1-C4-алкил, C1-C4-галогеналкил, C1-C4-алкоксиалкил, C3-C6-циклоалкил, C4-C7-циклоалкилалкил, фенил или бензил, где фенил или бензил необязательно замещен 1-3 раза галогеном, цианогруппой, C1-C4-алкилом, C1-C4-галогеналкилом или C1-C4-алкоксигруппой; или
R11 и R12, взятые вместе с атомом азота, с которым они связаны, образуют цикл пиперидина, пирролидина, пиперазина, N-метилпиперазина, морфолина или тиоморфолина, при условии, что только одна комбинация из R9 и R10 или R11 и R12, взятых вместе с атомом азота, с которым они связаны, образует цикл пиперидина, пирролидина, пиперазина, N-метилпиперазина, морфолина или тиоморфолина;
R13 означает C1-C4-алкил, C1-C4-галогеналкил или фенил;
n равно 0, 1 или 2; и
R14 в каждом случае независимо выбран из заместителя, выбранного из группы, состоящей из галогена, -NO2, -OR11, -NR11R12, -NR11C(O)R12, -NR11C(O)2R12, -NR11C(O)NR12R13, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила, где C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил и C4-C7-циклоалкилалкил необязательно замещены 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, арила, -CN, -OR9 и -NR9R10;
или их оксидом, или их фармацевтически приемлемой солью.
Другой аспект настоящего изобретения относится к способу получения соединения формулы I:

Формула I
где атом углерода, обозначенный *, находится в R- или S-конфигурации;
X означает конденсированный бициклический карбоцикл или гетероцикл, выбранный из группы, состоящей из бензофуранила, бензо[b]тиофенила, бензоизотиазолила, бензоизоксазолила, индазолила, индолила, изоиндолила, индолизинила, бензоимидазолила, бензооксазолила, бензотиазолила, бензотриазолила, имидазо[1,2-a]пиридинила, пиразоло[1,5-a]пиридинила, [1,2,4]триазоло[4,3-a]пиридинила, тиено[2,3-b]пиридинила, тиено[3,2-b]пиридинила, 1H-пирроло[2,3-b]пиридинила, инденила, инданила, дигидробензоциклогептенила, тетрагидробензоциклогептенила, дигидробензотиофенила, дигидробензофуранила, индолинила, нафтила, тетрагидронафтила, хинолинила, изохинолинила, 4H-хинолизинила, 9aH-хинолизинила, хиназолинила, циннолинила, фталазинила, хиноксалинила, бензо[1,2,3]триазинила, бензо[1,2,4]триазинила, 2H-хроменила, 4H-хроменила, и конденсированный бициклический карбоцикл или конденсированный бициклический гетероцикл, необязательно замещенный заместителями (в количестве от 1 до 4), которые определены ниже для R14;
R1 означает H, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, каждый из которых необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, арила, -CN, -OR9 и -NR9R10;
R2 означает H, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил, C4-C7-циклоалкилалкил или C1-C6-галогеналкил, каждый из которых необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, арила, -CN, -OR9и -NR9R10; или
R2 означает гем-диметил;
R3 означает H, галоген, -OR11, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, -NR9R10, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9;
R4 означает H, галоген, -OR11, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, -NR9R10, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9; или
R4 означает фенил, нафтил, инденил, пиридил, пиримидинил, пиридазинил, пиразинил, [1,2,4]триазинил, [1,3,5]триазинил, триазолил, фуранил, тиофенил, пиранил, индазолил, бензимидазолил, хинолинил, хиназолинил, хиноксалинил, фталазинил, циннолинил, изохинолинил, тиенил, имидазолил, тиазолил, бензтиазолил, пуринил, изотиазолил, индолил, пирролил, оксазолил, бензофуранил, бензотиенил, бензтиазолил, изоксазолил, пиразолил, оксадиазолил, тиадиазолил, 3-оксо-[1,2,4]триазоло[4,3-a]пиридинил, имидазо[1,2-a]пиридинил, пиразоло[1,5-a]пиридинил, [1,2,4]триазоло[4,3-a]пиридинил, тиено[2,3-b]пиридинил, тиено[3,2-b]пиридинил, 1H-пирроло[2,3-b]пиридинил или другие гетероциклы, необязательно замещенные заместителями (в количестве от 1 до 4), которые определены ниже для R14;
R5 и R6, каждый независимо, выбраны из группы, состоящей из H, галогена, -OR11, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, -NR9R10, C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила, C4-C7-циклоалкилалкила и фенила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9;
R7 означает H;
R8 означает H, галоген, -OR9, -SR9, C1-C6-алкил, -CN или -NR9R10;
R9 и R10, каждый независимо, выбраны из группы, состоящей из H, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксиалкила, C3-C6-циклоалкила, C4-C7-циклоалкилалкила, -C(O)R13, фенила и бензила, где фенил или бензил необязательно замещен 1-3 раза заместителем, выбранным независимо в каждом случае из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила и C1-C4-алкоксигруппы; или
R9 и R10, взятые вместе с атомом азота, с которым они связаны, образуют пиперидин, пирролидин, пиперазин, N-метилпиперазин, морфолин, тиоморфолин, [1,2]оксазинан, изоксазолидин или 2-оксо-2H-пиридин, который необязательно замещен 1-3 раза заместителем, выбранным независимо в каждом случае из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила и C1-C4-алкоксигруппы;
R11 означает H, C1-C4-алкил, C1-C4-галогеналкил, C1-C4-алкоксиалкил, C3-C6-циклоалкил, C4-C7-циклоалкилалкил, -C(O)R13, фенил или бензил, где фенил или бензил необязательно замещен 1-3 раза галогеном, цианогруппой, C1-C4-алкилом, C1-C4-галогеналкилом или C1-C4-алкоксигруппой;
R12 означает H, C1-C4-алкил, C1-C4-галогеналкил, C1-C4-алкоксиалкил, C3-C6-циклоалкил, C4-C7-циклоалкилалкил, фенил или бензил, где фенил или бензил необязательно замещен 1-3 раза галогеном, цианогруппой, C1-C4-алкилом, C1-C4-галогеналкилом или C1-C4-алкоксигруппой; или
R11 и R12, взятые вместе с атомом азота, с которым они связаны, образуют цикл пиперидина, пирролидина, пиперазина, N-метилпиперазина, морфолина или тиоморфолина при условии, что только одна комбинация из R9 и R10 или R11 и R12, взятых вместе с атомом азота, с которым они связаны, образует цикл пиперидина, пирролидина, пиперазина, N-метилпиперазина, морфолина или тиоморфолина;
R13 означает C1-C4-алкил, C1-C4-галогеналкил или фенил;
n равно 0, 1 или 2; и
R14 в каждом случае независимо выбран из заместителя, выбранного из группы, состоящей из галогена, -NO2, -OR11, -NR11R12, -NR11C(O)R12, -NR11C(O)2R12, -NR11C(O)NR12R13, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила, где C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил и C4-C7-циклоалкилалкил необязательно замещены 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, арила, -CN, -OR9 и -NR9R10. Способ заключается в обработке первого промежуточного соединения формулы XVIII:

Формула XVIII
восстановителем в условиях, эффективных для получения соединения продукта.
Результаты последних клинических исследований таких лекарственных средств, как дулоксетин, венлафаксин, атомоксетин и других, которые работают механистически посредством ингибирования обратного захвата переносчиком, представили доказательство того, что активность и избирательность являются важными факторами в продвижении к лекарственным средствам с повышенной эффективностью, улучшенным терапевтическим индексом и применимостью для лечения новых клинических показаний. Дулоксетин, ингибитор переносчиков обратного захвата с двойным действием, является избирательным ингибитором обратного захвата белка-переносчика серотонина («SERT») и белка-переносчика норэпинефрина («NET») (Sorbera et al., Drugs of the Future, 25(9): 907-916 (2000), публикация включена в данное описание в виде ссылки в полном объеме) и находится в стадии клинической разработки для лечения депрессии и недержания мочи при напряжении. В клинических испытаниях исследователи приписывают действие лекарственного средства на широкий спектр симптомов депрессии, которые включают эмоциональные и болевые физические симптомы, а также состояние тревожности, двойному ингибированию обратного захвата как серотонина, так и норэпинефрина. Сообщалось, что венлафаксин, который по сообщениям также является избирательным ингибитором обратного захвата серотонина и норэпинефрина (класс SNRI), имеет более быстрое начало действия. Это было недостатком антидепрессантов первого поколения, т.е. избирательных ингибиторов обратного захвата серотонина одного механизма действия (класс SSRI). В случае Prozac®, прототипа лекарственного средства данного класса, может требоваться четыре недели или больше, чтобы наступило действие полной антидепрессантной активности.
Атомоксетин (Strattera®) недавно одобрен для лечения гиперактивного расстройства с дефицитом внимания (ADHD). Атомоксетин является избирательным ингибитором переносчика обратного захвата норэпинефрина. В отличие от Ritalin®, одного из наиболее часто используемых лекарственных средств для лечения ADHD, атомоксетин обладает небольшой или не обладает активностью по отношению к переносчику допамина. В результате атомоксетин имеет преимущество, состоящее в том, что он не входит в реестр ограниченных в обращении веществ, так как существует минимальная возможность возникновения к нему наркотической зависимости.
Сходным образом с более новыми клиническими средствами, подобными атомоксетину, дулоксетину и венлафаксину, соединения согласно настоящему изобретению могут обладать повышенной эффективностью по отношению к более широкому кругу симптомов депрессии. Соединения согласно настоящему изобретению также могут обладать более быстрым началом действия при лечении заболеваний ЦНС, подобных депрессии. Кроме обеспечения повышенной эффективности соединения согласно настоящему изобретению также могут проявлять меньше нежелательных побочных эффектов. Наконец, вследствие того, что соединения согласно настоящему изобретению имеют разнообразный профиль ингибирования переносчиков обратного захвата, они предположительно применимы для широкого множества расстройств ЦНС.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Соединения согласно настоящему изобретению представлены химической структурой, определяемой формулой I:
Формула I
где атом углерода, обозначенный *, находится в R- или S-конфигурации;
X означает конденсированный бициклический карбоцикл или гетероцикл, выбранный из группы, состоящей из бензофуранила, бензо[b]тиофенила, бензоизотиазолила, бензоизоксазолила, индазолила, индолила, изоиндолила, индолизинила, бензоимидазолила, бензооксазолила, бензотиазолила, бензотриазолила, имидазо[1,2-a]пиридинила, пиразоло[1,5-a]пиридинила, [1,2,4]триазоло[4,3-a]пиридинила, тиено[2,3-b]пиридинила, тиено[3,2-b]пиридинила, 1H-пирроло[2,3-b]пиридинила, инденила, инданила, дигидробензоциклогептенила, тетрагидробензоциклогептенила, дигидробензотиофенила, дигидробензофуранила, индолинила, нафтила, тетрагидронафтила, хинолинила, изохинолинила, 4H-хинолизинила, 9aH-хинолизинила, хиназолинила, циннолинила, фталазинила, хиноксалинила, бензо[1,2,3]триазинила, бензо[1,2,4]триазинила, 2H-хроменила, 4H-хроменила, и конденсированный бициклический карбоцикл или конденсированный бициклический гетероцикл, необязательно замещенный заместителями (в количестве от 1 до 4), которые определены ниже для R14;
R1 означает H, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, каждый из которых необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, арила, -CN, -OR9 и -NR9R10;
R2 означает H, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил, C4-C7-циклоалкилалкил или C1-C6-галогеналкил, каждый из которых необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, арила, -CN, -OR9и -NR9R10; или
R2 означает гем-диметил;
R3 означает H, галоген, -OR11, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, -NR9R10, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9;
R4 означает H, галоген, -OR11, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, -NR9R10, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9; или
R4 означает фенил, нафтил, инденил, пиридил, пиримидинил, пиридазинил, пиразинил, [1,2,4]триазинил, [1,3,5]триазинил, триазолил, фуранил, тиофенил, пиранил, индазолил, бензимидазолил, хинолинил, хиназолинил, хиноксалинил, фталазинил, циннолинил, изохинолинил, тиенил, имидазолил, тиазолил, бензтиазолил, пуринил, изотиазолил, индолил, пирролил, оксазолил, бензофуранил, бензотиенил, бензтиазолил, изоксазолил, пиразолил, оксадиазолил, тиадиазолил, 3-оксо-[1,2,4]триазоло[4,3-a]пиридинил, имидазо[1,2-a]пиридинил, пиразоло[1,5-a]пиридинил, [1,2,4]триазоло[4,3-a]пиридинил, тиено[2,3-b]пиридинил, тиено[3,2-b]пиридинил, 1H-пирроло[2,3-b]пиридинил или другие гетероциклы, необязательно замещенные заместителями (в количестве от 1 до 4), которые определены ниже для R14;
R5 и R6, каждый независимо, выбраны из группы, состоящей из H, галогена, -OR11, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, -NR9R10, C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила, C4-C7-циклоалкилалкила и фенила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9;
R7 означает H, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9; или
R7 означает гем-диметил;
R8 означает H, галоген, -OR9, -SR9, C1-C6-алкил, -CN или -NR9R10;
R9 и R10, каждый независимо, выбраны из группы, состоящей из H, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксиалкила, C3-C6-циклоалкила, C4-C7-циклоалкилалкила, -C(O)R13, фенила и бензила, где фенил или бензил необязательно замещен 1-3 раза заместителем, выбранным независимо в каждом случае из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила и C1-C4-алкоксигруппы; или
R9 и R10, взятые вместе с атомом азота, с которым они связаны, образуют пиперидин, пирролидин, пиперазин, N-метилпиперазин, морфолин, тиоморфолин, [1,2]оксазинан, изоксазолидин или 2-оксо-2H-пиридин, который необязательно замещен 1-3 раза заместителем, выбранным независимо в каждом случае из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила и C1-C4-алкоксигруппы;
R11 означает H, C1-C4-алкил, C1-C4-галогеналкил, C1-C4-алкоксиалкил, C3-C6-циклоалкил, C4-C7-циклоалкилалкил, -C(O)R13, фенил или бензил, где фенил или бензил необязательно замещен 1-3 раза галогеном, цианогруппой, C1-C4-алкилом, C1-C4-галогеналкилом или C1-C4-алкоксигруппой;
R12 означает H, C1-C4-алкил, C1-C4-галогеналкил, C1-C4-алкоксиалкил, C3-C6-циклоалкил, C4-C7-циклоалкилалкил, фенил или бензил, где фенил или бензил необязательно замещен 1-3 раза галогеном, цианогруппой, C1-C4-алкилом, C1-C4-галогеналкилом или C1-C4-алкоксигруппой; или
R11 и R12, взятые вместе с атомом азота, с которым они связаны, образуют цикл пиперидина, пирролидина, пиперазина, N-метилпиперазина, морфолина или тиоморфолина, при условии, что только одна комбинация из R9 и R10 или R11 и R12, взятых вместе с атомом азота, с которым они связаны, образует цикл пиперидина, пирролидина, пиперазина, N-метилпиперазина, морфолина или тиоморфолина;
R13 означает C1-C4-алкил, C1-C4-галогеналкил или фенил;
n равно 0, 1 или 2; и
R14 в каждом случае независимо выбран из заместителя, выбранного из группы, состоящей из галогена, -NO2, -OR11, -NR11R12, -NR11C(O)R12, -NR11C(O)2R12, -NR11C(O)NR12R13, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила, где C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил и C4-C7-циклоалкилалкил необязательно замещены 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, арила, -CN, -OR9 и -NR9R10;
или их оксидом, или их фармацевтически приемлемой солью.
Как использовано выше и на протяжении описания изобретения, следующие термины, если не оговорено особо, следует понимать как имеющие следующие значения.
Термин «конденсированный бициклический карбоцикл» означает бициклическую кольцевую систему, состоящую примерно из 8-11 атомов углерода в цикле, предпочтительно из 9 или 10. Один или оба цикла являются ароматическими. Типичные конденсированные бициклические карбоциклы включают инденил, инданил, нафтил (или нафталенил), дигидронафтил, тетрагидронафтил, бензоциклогептенил, дигидробензоциклогептенил, тетрагидробензоциклогептенил и тому подобные.
Термин «конденсированный бициклический гетероцикл» означает бициклическую кольцевую систему, состоящую примерно из 8-11 атомов в цикле, предпочтительно из 9 или 10, при этом один или несколько атомов в кольцевой системе являются другими элементами, отличными от углерода, например азот, кислород или сера. Приставка аза-, окса- или тиа- перед гетероциклом означает, что по меньшей мере атом азота, кислорода или серы, соответственно, присутствует в качестве атома в цикле. Атом азота гетероарила необязательно окислен до соответствующего N-оксида. Типичные конденсированные бициклические гетероциклы включают бензофуранил, бензо[b]тиофенил, бензоизотиазолил, бензоизоксазолил, индазолил, индолил, изоиндолил, индолизинил, бензоимидазолил, бензооксазолил, бензотиазолил, бензотриазолил, имидазо[1,2-a]пиридинил, пиразоло[1,5-a]пиридинил, [1,2,4]триазоло[4,3-a]пиридинил, тиено[2,3-b]пиридинил, тиено[3,2-b]пиридинил, 1H-пирроло[2,3-b]пиридинил, хроменил, дигидробензотиофенил, дигидробензофуранил, индолинил, хинолинил, изохинолинил, 4H-хинолизинил, 9aH-хинолизинил, хиназолинил, циннолинил, хиноксалинил, бензо[1,2,3]триазинил, бензо[1,2,4]триазинил и тому подобные.
Термин «алкил» означает алифатическую углеводородную группу, которая может быть с прямой или разветвленной цепью, имеющую от примерно 1 до примерно 6 атомов углерода в цепи. «Разветвленная» означает, что одна или несколько низших алкильных групп, таких как метил, этил или пропил, связаны с линейной алкильной цепью. Примеры алкильных групп включают метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, н-пентил и 3-пентил.
Термин «алкенил» означает алифатическую углеводородную группу, содержащую углерод-углеродную двойную связь, которая может быть с прямой или разветвленной цепью, имеющую от примерно 2 до примерно 6 атомов углерода в цепи. Предпочтительные алкенильные группы имеют от 2 до примерно 4 атомов углерода в цепи. «Разветвленная» означает, что одна или несколько низших алкильных групп, таких как метил, этил или пропил, связаны с линейной алкенильной цепью. Примеры алкенильных групп включают этенил, пропенил, н-бутенил и изобутенил.
Термин «алкинил» означает алифатическую углеводородную группу, содержащую углерод-углеродную тройную связь, которая может быть с прямой или разветвленной цепью, имеющую от примерно 2 до примерно 6 атомов углерода в цепи. Предпочтительные алкинильные группы имеют от 2 до примерно 4 атомов углерода в цепи. «Разветвленная» означает, что одна или несколько низших алкильных групп, таких как метил, этил или пропил, связаны с линейной алкинильной цепью. Примеры алкинильных групп включают этинил, пропинил, н-бутинил, 2-бутинил, 3-метилбутинил и н-пентинил.
Термин «арил» означает ароматическую моноциклическую или полициклическую кольцевую систему, имеющую от 6 до примерно 14 атомов углерода, предпочтительно от 6 до примерно 10 атомов углерода. Типичные арильные группы включают фенил и нафтил. Термины «нафтил» и «нафталенил» используют взаимозаменяемо.
Термин «алкоксигруппа» означает алкил-O-группу, где алкильная группа представляет собой определенную в данном описании группу. Примеры алкоксигрупп включают метокси-, этокси-, н-пропокси-, изопропокси-, н-бутокси- и гептоксигруппу.
Подразумевается, что термин «соединения согласно изобретению» и эквивалентные выражения охватывают соединения общей формулы I, которые описаны выше, и указанное выражение включает пролекарства, фармацевтически приемлемые соли и сольваты, например гидраты, там, где контекст это допускает. Подобным образом, ссылка на промежуточные соединения независимо от того, заявлены ли они сами по себе или не заявлены, означает, что они охватывают их соли и сольваты в тех случаях, когда контекст это допускает. Для ясности конкретные случаи, когда контекст это допускает, иногда указаны в тексте, но указанные случаи являются только иллюстративными и не предназначены для исключения других случаев, когда контекст это допускает.
Термин «циклоалкил» означает неароматическую моноциклическую или полициклическую кольцевую систему примерно из 3-7 атомов углерода, предпочтительно примерно из 5-7 атомов углерода. Примеры моноциклических циклоалкильных групп включают циклопентил, циклогексил, циклогептил и тому подобное.
Термин «циклоалкилалкил» означает группу циклоалкил-алкила, в которой циклоалкил и алкил имеют значение, определенное в данном описании. Примеры циклоалкилалкильных групп включают циклопропилметил и циклопентилметил.
Термин «галоген» означает фтор, хлор, бром или йод.
Термин «галогеналкил» означает алкил как с разветвленной, так и с неразветвленной цепью, замещенный одним или несколькими атомами галогена, где алкильная группа имеет значение, определенное в данном описании.
Термин «галогеналкоксигруппа» означает C1-4-алкоксигруппу, замещенную по меньшей мере одним атомом галогена, где алкоксигруппа имеет значение, определенное в данном описании.
Термин «замещенный» или «замещение» атома означает, что один или несколько атомов водорода у указанного атома заменяют на основе выбора из указанной группы при условии, что указанная нормальная валентность атома не превышена. «Незамещенные» атомы несут все атомы водорода, диктуемые их валентностью. Когда заместителем является кетогруппа (т.е. =O), то заменяют два атома водорода на атоме. Комбинации заместителей и/или переменных допустимы, только если такие комбинации приводят к стабильным соединениям; под «стабильным соединением» или «стабильной структурой» подразумевают соединение, которое является достаточно устойчивым, чтобы перенести выделение до пригодной степени чистоты из реакционной смеси и приготовление в виде эффективного терапевтического средства.
Термин «фармацевтически приемлемые соли» означает относительно нетоксичные неорганические и органические кислотно-аддитивные соли и основно-аддитивные соли соединений согласно настоящему изобретению. Указанные соли могут быть получены in situ во время конечного выделения и очистки соединений. В частности, кислотно-аддитивные соли могут быть получены в результате отдельно взаимодействия очищенного соединения в форме его свободного основания с подходящей органической или неорганической кислотой и выделения образованной таким образом соли. Примеры кислотно-аддитивных солей включают гидробромид, гидрохлорид, сульфат, бисульфат, фосфат, нитрат, ацетат, оксалат, валерат, олеат, пальмитат, стеарат, лаурат, борат, бензоат, лактат, фосфат, тозилат, цитрат, малеат, фумарат, сукцинат, тартрат, нафтилат, мезилат, глюкогептонат, лактобионат, сульфаматы, малонаты, салицилаты, пропионаты, метилен-бис-b-гидроксинафтоаты, гентизаты, изотионаты, ди-пара-толуоилтартраты, метансульфонаты, этансульфонаты, бензолсульфонаты, пара-толуолсульфонаты, циклогексилсульфаматы, хинаты, лаурилсульфонат и тому подобные (см., например, Berge et al., «Pharmaceutical Salts», J. Pharm. Sci., 66: 1-sup. 19 (1977) и Remington's Pharmaceutical Sciences, 17th ed, Easton, Pa., Mack Publishing Company, p. 1418 (1985), которые включены в данное описание в виде ссылки в полном объеме). Основно-аддитивные соли также могут быть получены в результате отдельного взаимодействия очищенного соединения в форме его кислоты с подходящим органическим или неорганическим основанием и выделения образованной таким образом соли. Основно-аддитивные соли включают фармацевтически приемлемые соли металлов и аминов. Подходящие соли металлов включают соли натрия, калия, кальция, бария, цинка, магния и алюминия. Предпочтительными являются соли натрия и калия. Подходящие неорганические основно-аддитивные соли получают из оснований металлов, которые включают гидрид натрия, гидроксид натрия, гидроксид калия, гидроксид кальция, гидроксид алюминия, гидроксид лития, гидроксид магния и гидроксид цинка. Подходящие основно-аддитивные аминные соли получают из аминов, которые имеют достаточную основность для образования стабильной соли и предпочтительно включают такие амины, которые часто используют в медицинской химии вследствие их низкой токсичности и приемлемости для медицинского применения. Примеры таких аминов включают аммиак, этилендиамин, N-метилглюкамин, лизин, аргинин, орнитин, холин, N,N'-дибензилэтилендиамин, хлорпрокаин, диэтаноламин, прокаин, N-бензилфенэтиламин, диэтиламин, пиперазин, трис(гидроксиметил)аминометан, гидроксид тетраметиламмония, триэтиламин, дибензиламин, эфенамин, дегидроабиэтиламин, N-этилпиперидин, бензиламин, тетраметиламмоний, тетраэтиламмоний, метиламин, диметиламин, триметиламин, этиламин, основные аминокислоты, такие как лизин и аргинин, дициклогексиламин и тому подобные.
Термин «фармацевтически приемлемые пролекарства» в используемом в данном описании смысле означает такие пролекарства соединений, применимых согласно настоящему изобретению, которые в рамках обоснованного медицинского суждения подходят для применения в контакте с тканями человека и более низкоорганизованных животных, при этом чрезмерная токсичность, раздражение, аллергическая реакция и тому подобное соответствуют рациональному соотношению польза/риск, и они эффективны в случае их планируемого применения, а также в тех случаях, когда это возможно, к формам цвиттерионов соединений согласно изобретению. Термин «пролекарство» означает соединения, которые быстро трансформируются in vivo с образованием исходного соединения указанной выше формулы, например в результате гидролиза в крови. Функциональные группы, которые могут быть быстро трансформированы посредством метаболического расщепления in vivo, образуют класс групп, вступающих в реакцию с карбоксильной группой соединений согласно настоящему изобретению. К ним относятся без ограничения такие группы, как алканоил (такие как ацетил, пропионил, бутирил и тому подобные), незамещенный и замещенный ароил (такой как бензоил и замещенный бензоил), алкоксикарбонил (такой как этоксикарбонил), триалкилсилил (такой как триметил- и триэтилсилил), сложные моноэфиры, образованные с дикарбоновыми кислотами (такие как сукцинил) и тому подобные. Вследствие легкости, с которой метаболически отщепляемые группы соединений, применимых согласно данному изобретению, отщепляются in vivo, соединения, несущие такие группы, действуют как пролекарства. Соединения, несущие метаболически отщепляемые группы, имеют преимущество, состоящее в том, что они могут иметь улучшенную биодоступность в результате повышенной растворимости и/или скорости всасывания, придаваемой исходному соединению вследствие присутствия метаболически отщепляемой группы. Всестороннее обсуждение пролекарств приведено в следующих публикациях: Bundgaard, ed., Design of Prodrugs, Elsevier (1985); Widder et al., Methods in Enzymology, ed., Academic Press, 42: 309-396 (1985); "Design and Applications of Prodrugs", Krogsgaard-Larsen, ed., A Textbook of Drug Design and Development, Chapter 5: 113-191 (1991); Bundgaard, "Advanced Drug Delivery Reviews" 8: 1-38 (1992); Bundgaard et al., Journal of Pharmaceutical Sciences, 77: 285 (1988); Nakeya et al., Chem. Pharm. Bull., 32: 692 (1984); Higuchi, "Pro-drags as Novel Delivery Systems" Roche, ed., A.C.S. Symposium Series, Vol. 14, and "Bioreversible Carriers in Drug Design" American Pharmaceutical Association and Pergamon Press (1987), которые включены в данное описание в виде ссылки в полном объеме. Примеры пролекарств включают без ограничения ацетатные, формиатные и бензоатные производные спирта и функциональных аминных групп соединений согласно изобретению.
Подразумевают, что термин «терапевтически эффективные количества» описывает количество соединения согласно настоящему изобретению, эффективное для повышения уровней серотонина, норэпинефрина или допамина в синапсе и, следовательно, для получения требуемого терапевтического эффекта. Такие количества обычно варьируют в зависимости от ряда факторов, которые специалисты в данной области, имея описание, представленное в данной публикации, способны определить и учесть. Факторы включают без ограничения: конкретного пациента, а также его возраст, массу, рост, общее физической состояние и историю болезни, конкретное используемое соединение, а также носитель, в котором оно приготовлено, и выбранный для него путь введения и природу, и тяжесть состояния, подвергаемого лечению.
Термин «фармацевтическая композиция» означает композицию, содержащую соединение формулы I и по меньшей мере один компонент, выбранный из фармацевтически приемлемых носителей, разбавителей, адъювантов, эксципиентов или наполнителей, таких как консерванты, наполнители, дезинтегрирующие средства, увлажнители, эмульгаторы, суспендирующие средства, подсластители, корригенты, ароматизаторы, антибактериальные средства, противогрибковые средства, скользящие вещества и диспергирующие агенты, в зависимости от природы способа введения и дозированных форм. Примеры суспендирующих средств включают этоксилированные изостеариловые спирты, полиоксиэтиленсорбит и сложные эфиры сорбитана, микрокристаллическую целлюлозу, метагидроксид алюминия, бентонит, агар-агар и трагакант или смеси указанных веществ. Предотвращение действия микроорганизмов можно обеспечить различными антибактериальными и противогрибковыми средствами, например парабенами, хлорбутанолом, фенолом, сорбиновой кислотой и тому подобным. Также может требоваться включение изотонических агентов, например сахаров, хлорида натрия и тому подобного. Пролонгированное всасывание инъекционной фармацевтической формы может быть вызвано использованием средств, замедляющих всасывание, например моностеарата алюминия и желатина. Примеры подходящих носителей, разбавителей, растворителей или наполнителей включают воду, этанол, полиолы, подходящие их смеси, растительные масла (такие как оливковое масло) и инъекционные сложные органические эфиры, такие как этилолеат. Примеры эксципиентов включают лактозу, молочный сахар, цитрат натрия, карбонат кальция, фосфат дикальция. Примеры дезинтегрирующих агентов включают крахмал, альгиновые кислоты и некоторые сложные силикаты. Примеры скользящих веществ включают стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоли с высокой молекулярной массой.
Термин «фармацевтически приемлемый» означает в рамках обоснованного медицинского заключения подходящий для применения в контакте с клетками человека и более низкоорганизованных животных без чрезмерной токсичности, раздражения, аллергической реакции и тому подобного, и соответствующий разумному соотношению польза/риск.
Термин «фармацевтически приемлемые дозированные формы» означает дозированные формы соединения согласно изобретению и включает, например, таблетки, драже, порошки, эликсиры, сиропы, жидкие препараты, включая суспензии, спреи, таблетки для ингаляции, лепешки, эмульсии, растворы, гранулы, капсулы и суппозитории, а также жидкие препараты для инъекций, включая препараты липосом. Способы и препараты, как правило, можно найти в Remington's Pharmaceutical Sciences, 17th ed, Easton, Pa., Mack Publishing Company (1985), который включен в данное описание в виде ссылки в полном объеме.
Один вариант осуществления настоящего изобретения относится к соединению формулы I, где
X выбран из группы, состоящей из бензофуранила, бензо[b]тиофенила, бензоизотиазолила, бензоизоксазолила, индазолила, индолила, изоиндолила, индолизинила, бензоимидазолила, бензооксазолила, бензотиазолила, бензотриазолила, имидазо[1,2-a]пиридинила, пиразоло[1,5-a]пиридинила, [1,2,4]триазоло[4,3-a]пиридинила, тиено[2,3-b]пиридинила, тиено[3,2-b]пиридинила, 1H-пирроло[2,3-b]пиридинила, инденила, инданила, дигидробензоциклогептенила, тетрагидробензоциклогептенила, дигидробензотиофенила, дигидробензофуранила, индолинила, нафтила, тетрагидронафтила, хинолинила, изохинолинила, 4H-хинолизинила, 9aH-хинолизинила, хиназолинила, циннолинила, фталазинила, хиноксалинила, бензо[1,2,3]триазинила, бензо[1,2,4]триазинила, 2H-хроменила, 4H-хроменила, и конденсированного бициклического карбоцикла или конденсированного бициклического гетероцикла, необязательно замещенного заместителями (в количестве от 1 до 4), которые определены ниже для R14;
R1 означает H или C1-C6-алкил;
R2 означает H, C1-C6-алкил или C1-C6-галогеналкил;
R3 означает H, галоген, -OR11, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, -NR9R10, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила или C1-C4-алкоксигруппы, -CN и -OR9;
R4 означает H, галоген, -OR11, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, -NR9R10, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9; или
R4 означает фенил, нафтил, инденил, пиридил, пиримидинил, пиридазинил, пиразинил, [1,2,4]триазинил, [1,3,5]триазинил, триазолил, фуранил, тиофенил, пиранил, индазолил, бензимидазолил, хинолинил, хиназолинил, хиноксалинил, фталазинил, циннолинил, изохинолинил, тиенил, имидазолил, тиазолил, бензтиазолил, пуринил, изотиазолил, индолил, пирролил, оксазолил, бензофуранил, бензотиенил, бензтиазолил, изоксазолил, пиразолил, оксадиазолил, тиадиазолил, 3-оксо-[1,2,4]триазоло[4,3-a]пиридинил, имидазо[1,2-a]пиридинил, пиразоло[1,5-a]пиридинил, [1,2,4]триазоло[4,3-a]пиридинил, тиено[2,3-b]пиридинил, тиено[3,2-b]пиридинил, 1H-пирроло[2,3-b]пиридинил или другие гетероциклы, необязательно замещенные заместителями (в количестве от 1 до 4), которые определены ниже для R14;
R5 и R6, каждый независимо, выбраны из H, галогена, C1-C6-алкила или C1-C4-алкоксиалкила;
R7 означает H или C1-C6-алкил;
R8 означает H, галоген, -OR9, -SR9, C1-C6-алкил, -CN или -NR9R10;
R14 в каждом случае независимо выбран из заместителя, выбранного из группы, состоящей из галогена, -NO2, -OR11, -NR11R12, -NR11C(O)R12, -NR11C(O)2R12, -NR11C(O)NR12R13, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила, где C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил и C4-C7-циклоалкилалкил необязательно замещены 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, арила, -CN, -OR9 и -NR9R10.
Другой вариант осуществления настоящего изобретения относится к соединению формулы I, где
X означает бензофуран-2-ил, 5-хлорбензофуран-2-ил, 4-фторбензофуран-2-ил, 5-фторбензофуран-2-ил, 5-метоксибензофуран-2-ил, 6-фторбензофуран-2-ил, 7-фтор-бензофуран-2-ил, 7-метоксибензофуран-2-ил, бензофуран-3-ил, бензофуран-4-ил, бензофуран-5-ил, бензофуран-6-ил, бензофуран-7-ил, 2,3-дигидробензофуран-5-ил, бензо[b]тиофен-2-ил, 4-хлорбензо[b]тиофен-2-ил, 4-фторбензо[b]тиофен-2-ил, 4-метоксибензо[b]тиофен-2-ил, 5-хлорбензо[b]тиофен-2-ил, 5-фторбензо[b]тиофен-2-ил, 6-хлорбензо[b]тиофен-2-ил, 6-фторбензо[b]тиофен-2-ил, 7-хлорбензо[b]тиофен-2-ил, 7-фторбензо[b]тиофен-2-ил, 1,1-диоксо-1H-1λ6-бензо[b]тиофен-2-ил, бензо[b]тиофен-3-ил, бензо[b]тиофен-4-ил, бензо[b]тиофен-5-ил, 2-метилбензо[b]тиофен-5-ил, 2-хлорбензо[b]тиофен-5-ил, 3-трифторметилбензо[b]тиофен-5-ил, 4-цианобензо[b]тиофен-5-ил, 4-метоксибензо[b]тиофен-5-ил, 4-гидроксибензо[b]тиофен-5-ил, 4-метилбензо[b]тиофен-5-ил, 1,1-диоксо-1H-1λ6-бензо[b]тиофен-5-ил, бензо[b]тиофен-6-ил, 2-хлорбензо[b]тиофен-6-ил, 3-трифторметилбензо[b]тиофен-6-ил, 7-метоксибензо[b]тиофен-6-ил, 7-гидроксибензо[b]тиофен-6-ил, 7-метилбензо[b]тиофен-6-ил, 1,1-диоксо-1H-1λ6-бензо[b]тиофен-6-ил, бензо[b]тиофен-7-ил, 1H-индазол-1-ил, 1H-индазол-3-ил, 1H-индазол-4-ил, 1H-индазол-5-ил, 1-метилиндазол-5-ил, 6-метокси-1H-индазол-5-ил, 7-метокси-1H-индазол-5-ил, 7-фтор-1H-индазол-5-ил, 7-хлор-1H-индазол-5-ил, 7-метокси-1H-индазол-5-ил, 1H-индазол-6-ил, 1-метилиндазол-6-ил, 7-фтор-1H-индазол-6-ил, 1H-индазол-7-ил, индол-1-ил, 1-метилиндол-2-ил, 1H-индол-2-ил, 7-фтор-1H-индол-2-ил, 1H-индол-3-ил, 1H-индол-4-ил, 1H-индол-5-ил, 1-метилиндол-5-ил, 7-фтор-1H-индол-5-ил, 1H-индол-6-ил, 1-метилиндол-6-ил, 7-фтор-1H-индол-6-ил, 2H-изоиндол-1-ил, 2H-изоиндол-2-ил, 2H-изоиндол-4-ил, 2H-изоиндол-5-ил, индолизин-1-ил, индолизин-2-ил, индолизин-3-ил, индолизин-5-ил, индолизин-6-ил, индолизин-7-ил, индолизин-8-ил, бензооксазол-2-ил, бензооксазол-4-ил, бензооксазол-5-ил, 2-метилбензооксазол-5-ил, бензооксазол-6-ил, 2-метилбензооксазол-6-ил, бензооксазол-7-ил, бензотиазол-2-ил, бензотиазол-4-ил, бензотиазол-5-ил, 2-метилбензотиазол-5-ил, бензотиазол-6-ил, 2-метилбензотиазол-6-ил, бензотиазол-7-ил, бензоизотиазол-4-ил, бензоизотиазол-5-ил, бензоизотиазол-6-ил, бензоизотиазол-7-ил, бензоизоксазолил-4-ил, бензоизоксазолил-5-ил, бензоизоксазолил-6-ил, бензоизоксазолил-7-ил, имидазо[1,2-a]пиридин-2-ил, имидазо[1,2-a]пиридин-6-ил, имидазо[1,2-a]пиридин-7-ил, пиразоло[1,5-a]пиридин-2-ил, пиразоло[1,5-a]пиридин-5-ил, пиразоло[1,5-a]пиридин-6-ил, [1,2,4]триазоло[4,3-a]пиридин-6-ил, тиено[2,3-b]пиридин-2-ил, [1,2,4]триазоло[4,3-a]пиридин-7-ил, тиено[2,3-b]пиридин-6-ил, тиено[2,3-b]пиридин-5-ил, тиено[3,2-b]пиридин-2-ил, тиено[3,2-b]пиридин-5-ил, тиено[3,2-b]пиридин-6-ил, 1H-пирроло[2,3-b]пиридин-2-ил, 1H-пирроло[2,3-b]пиридин-5-ил, 1H-пирроло[2,3-b]пиридин-6-ил, 3H-инден-5-ил, индан-5-ил, нафтален-1-ил, 4-метилнафтален-1-ил, нафтален-2-ил, 1-фторнафтален-2-ил, 1-хлорнафтален-2-ил, 1-метоксинафтален-2-ил, 1-метилнафтален-2-ил, 3-фторнафтален-2-ил, 3-хлорнафтален-2-ил, 3-метоксинафтален-2-ил, 3-цианонафтален-2-ил, 4-фторнафтален-2-ил, 4-хлорнафтален-2-ил, 4-метилнафтален-1-ил, 5-фторнафтален-2-ил, 5-хлорнафтален-2-ил, 5-цианонафтален-2-ил, 5-метилнафтален-2-ил, 6-метоксинафтален-2-ил, 6-хлорнафтален-2-ил, 6-фторнафтален-2-ил, 6-цианонафтален-2-ил, 6-метансульфонилнафтален-2-ил, 7-метоксинафтален-2-ил, 7-хлорнафтален-2-ил, 7-фторнафтален-2-ил, 7-цианонафтален-2-ил, 8-метоксинафтален-2-ил, 8-хлорнафтален-2-ил, 8-фторнафтален-2-ил, 8-цианонафтален-2-ил, 5,6,7,8-тетрагидронафтален-2-ил, 2-хинолинил, 3-хинолинил, 6-хинолинил, 7-хинолинил, 1-изохинолинил, 3-изохинолинил, 6-изохинолинил, 7-изохинолинил, 2-хиноксалинил, 6-хиноксалинил, 2-хиназолинил, 2-хиназолинил, 6-хиназолинил, 7-хиназолинил, 3-циннолинил, 6-циннолинил, 7-циннолинил, 6-фталазинил, 2H-хромен-3-ил или 8,9-дигидро-7H-бензоциклогептен-6-ил;
R1 означает H, метил, этил или изопропил;
R2 означает H, метил или гем-диметил;
R3 означает H, метил, гидроксигруппу, метоксигруппу, фтор, хлор или CN;
R4 означает H, C1-C6-алкил, фтор, хлор, -OR11, морфолин-4-ил, 2,6-диметилморфолин-4-ил, пиперазин-1-ил, 4-метилпиперазин-1-ил, пиперидин-1-ил, пирролидин-1-ил, морфолин-4-илметил, 1-метил-1-морфолин-4-илэтил, 1-морфолин-4-илциклопропил, пиперидин-1-илметил, пирролидин-1-илметил, диметиламинометил, 1-диметиламино-1-метилэтил, 1-диметиламиноциклопропанил, метиламинометил, 1-метил-1-метиламиноэтил, 1-метиламиноциклопропил, аминометил, 1-амино-1-метилэтил, 1-аминоциклопропил, метансульфонил или -CN; или
R4 означает фенил, 2-хлорфенил, 3-хлорфенил, 4-хлорфенил, 2-цианофенил, 3-цианофенил, 4-цианофенил, 2-фторфенил, 3-фторфенил, 4-фторфенил, 2,5-дифторфенил, 3,5-дифторфенил, 2,4-дифторфенил, 2,6-дифторфенил, 2-метоксифенил, 3-метоксифенил, 4-метоксифенил, 4-диметиламинофенил, 2-метилсульфонилфенил, 3-метилсульфонилфенил, 4-метилсульфонилфенил, 2-трифторметилфенил, 3-трифторметилфенил, 4-трифторметилфенил, фуран-2-ил, 4-метилфуран-2-ил, 5-метилфуран-2-ил, фуран-3-ил, тиофен-2-ил, тиофен-3-ил, 3,5-диметилизоксазол-4-ил, пиридин-2-ил, 3-метоксипиридин-2-ил, 4-метоксипиридин-2-ил, 3-метилпиридин-2-ил, 4-метилпиридин-2-ил, 6-метоксипиридин-2-ил, пиридин-3-ил, 2-метоксипиридин-3-ил, 6-метоксипиридин-3-ил, пиридин-4-ил, пиримидин-4-ил, пиримидин-2-ил, пиримидин-5-ил, пиразин-2-ил, 3-метилпиразин-2-ил, 5-метилпиразин-2-ил, 6-метилпиразин-2-ил, 3-метоксипиразин-2-ил, 5-метоксипиразин-2-ил, 6-метоксипиразин-2-ил, 6-этилпиразин-2-ил, 6-трифторметилпиразин-2-ил, пиридазин-3-ил, 5-метилпиридазин-3-ил, 6-метилпиридазин-3-ил, 6-диметиламинопиридазин-3-ил, 6-метиламинопиридазин-3-ил, 6-аминопиридазин-3-ил, 6-морфолин-4-илпиридазин-3-ил, 6-трифторметилпиридазин-3-ил, 6-цианопиридазин-3-ил, пиридазин-4-ил, 2-хинолинил, 3-хинолинил, 6-хинолинил, 7-хинолинил, 1-изохинолинил, 3-изохинолинил, 6-изохинолинил, 7-изохинолинил, [1,3,5]триазин-2-ил, [1,2,4]триазин-3-ил, [1,2,4]триазин-5-ил, [1,2,4]триазин-6-ил, циннолин-3-ил, фталазин-1-ил, фталазин-7-ил, хиноксалин-2-ил, хиноксалин-6-ил, хиназолин-2-ил, хиназолин-4-ил, хиназолин-6-ил, хиназолин-7-ил, 3-оксо-[1,2,4]триазоло[4,3-a]пиридин-2-ил или 2-оксо-2H-пиридин-1-ил;
R5 означает H, фтор, хлор, метил, -OH или метоксигруппу;
R6 означает H; фтор, хлор, метил, -OH или метоксигруппу;
R7 означает H; и
R8 означает H, фтор, хлор, -OH, -CN, метил или этил.
Другой вариант осуществления настоящего изобретения относится к соединению формулы I, где атом углерода, обозначенный *, находится в R-конфигурации.
Другой вариант осуществления настоящего изобретения относится к соединению формулы I, где атом углерода, обозначенный *, находится в S-конфигурации.
Другой вариант осуществления настоящего изобретения относится к смеси стереоизомерных соединений формулы I, где атом углерода, обозначенный *, находится в S- или R-конфигурации.
В указанных вариантах выбор конкретного предпочтительного заместителя в любом одном из R1-R8 не влияет на выбор заместителя в любых других R1-R8. То есть конкретные соединения, представленные в данном описании, имеют любые конкретные заместители в любых положениях. Например, как описано выше, R1 предпочтительно означает C1-C6-алкил; выбор R1 в виде любого одного из C1-, C2-, C3-, C4-, C5- или C6-алкила не ограничивает выбор R2, в частности в виде любого одного из H, C1-C6-алкила или C1-C6-галогеналкила. Вернее, в случае R1 в виде любого из C1-, C2-, C3-, C4-, C5- или C6-алкила R2 является любым из H, C1-, C2-, C3-, C4-, C5- или C6-алкила или C1-, C2-, C3-, C4-, C5- или C6-галогеналкила. Подобным образом выбор R2 в виде любого из H, C1-, C2-, C3-, C4-, C5- или C6-алкила или C1-, C2-, C3-, C4-, C5- или C6-галогеналкила не ограничивает выбор R3, в частности, в виде любого одного из H, галогена, -OR11, -S(O)nR12, -CN, -C(O)R12, C1-C6-алкила, C3-C6-циклоалкила, C4-C7-циклоалкилалкила или замещенного C4-C7-циклоалкилалкила.
Другие конкретные соединения согласно изобретению представляют собой соединения со следующими заместителями (Таблица А):

где атом углерода, обозначенный *, находится в R- или S-конфигурации. То есть конкретные соединения в данном случае включают
4-(бензо[b]тиофен-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-2-ил)-1-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-2-ил)-2-этил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-2-ил)-1,2-диметил-1,2,3,4-тетрагидроизохинолин;
4-(4-фторбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(5-фторбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(6-фторбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(7-фторбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(4-хлорбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(5-хлорбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(6-хлорбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(7-хлорбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(4-метоксибензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(5-метоксибензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(6-метоксибензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(7-метоксибензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1,1-диоксо-1H-1λ6-бензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-(бензо[b]тиофен-2-ил)-4-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-2-ил)-2,4-диметил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-карбонитрил;
4-(бензо[b]тиофен-2-ил)-4-фтор-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-2-ил)-4-хлор-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-2-ил)-2,5-диметил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ол;
4-(бензо[b]тиофен-2-ил)-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-2-ил)-2,7-диметил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-2-ил)-7-фтор-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4- тетрагидроизохинолин;
4-(4-фторбензо[b]тиофен-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(5-фторбензо[b]тиофен-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(6-фторбензо[b]тиофен-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(7-фторбензо[b]тиофен-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-2-ил)-2-этил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
[6-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]диметиламин;
[6-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]метиламин;
6-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-иламин;
4-бензо[b]тиофен-2-ил-2-метил-7-(6-морфолин-4-илпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-2-метил-7-(6-трифторметилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
6-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-илкарбонитрил;
4-бензо[b]тиофен-2-ил-8-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-8-метокси-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
4-бензо[b]тиофен-2-ил-2,8-диметил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-2-этил-8-фтор-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-6-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-5-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-бензо[b]тиофен-2-ил-2,4-диметил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-2-ил)-8-фтор-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-2-метил-7-(3-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-7-(3-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-2-метил-7-(6-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-7-(6-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-8-фтор-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-8-фтор-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-8-фтор-2-метил-7-пиримидин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-8-фтор-2-метил-7-пиримидин-5-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-2-ил)-7-(3,5-диметилизоксазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-2-ил)-2-метил-7-(тиазол-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-2-ил)-2-метил-7-(5-метилтиазол-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-2-метил-7-[1,3,5]триазин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-2-метил-7-[1,2,4]триазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-2-метил-7-[1,2,4]триазин-5-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-2-метил-7-[1,2,4]триазин-6-ил-1,2,3,4-тетрагидроизохинолин;
3-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циннолин;
1-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)фталазин;
2-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиноксалин;
2-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
6-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
7-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
2-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-2H-[1,2,4]триазоло[4,3-a]пиридин-3-он;
4-(бензо[b]тиофен-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(4-фторбензо[b]тиофен-2-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(5-фторбензо[b]тиофен-2-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(6-фторбензо[b]тиофен-2-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(7-фторбензо[b]тиофен-2-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-7-(2,6-диметилморфолин-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-2-этил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-8-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-8-метокси-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
4-бензо[b]тиофен-2-ил-2,8-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-2-этил-8-фтор-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-6-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-5-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-бензо[b]тиофен-2-ил-2,4-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-2-метил-7-пиперазин-1-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-2-метил-7-(4-метилпиперазин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-2-ил)-2-метил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-2-метил-7-морфолин-4-илметил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-2-метил-7-(1-метил-1-морфолин-4-илэтил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-2-метил-7-(1-морфолин-4-илциклопропил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-2-метил-7-пиперидин-1-илметил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-2-ил)-2-метил-7-пирролидин-1-илметил-1,2,3,4-тетрагидроизохинолин;
(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)диметиламин;
[1-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]диметиламин;
[1-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]диметиламин;
(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)метиламин;
[1-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]метиламин;
[1-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]метиламин;
C-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)метиламин;
1-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтиламин;
1-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропиламин;
1-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1H-пиридин-2-он;
4-(бензо[b]тиофен-2-ил)-7-метансульфонил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрил;
4-(бензо[b]тиофен-2-ил)-2,8-диметил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-3-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-3-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-3-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-4-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-4-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-1-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-8-фтор-2-метил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-8-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-2-этил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-1,2-диметил-1,2,3,4-тетрагидроизохинолин;
4-(2-метилбензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(2-хлорбензо[b]тиофен-5-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(2-хлорбензо[b]тиофен-5-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(2-хлорбензо[b]тиофен-5-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(2-хлорбензо[b]тиофен-5-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(2-хлорбензо[b]тиофен-5-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(2-хлорбензо[b]тиофен-5-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-7-пиридазин-3-ил-4-(3-трифторметилбензо[b]тиофен-5-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-7-пиримидин-2-ил-4-(3-трифторметилбензо[b]тиофен-5-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-7-пиримидин-4-ил-4-(3-трифторметилбензо[b]тиофен-5-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-7-пиримидин-5-ил-4-(3-трифторметилбензо[b]тиофен-5-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-7-пиразин-2-ил-4-(3-трифторметилбензо[b]тиофен-5-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-7-морфолин-4-ил-4-(3-трифторметилбензо[b]тиофен-5-ил)-1,2,3,4-тетрагидроизохинолин;
5-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)бензо[b]тиофен-4-карбонитрил;
4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
2-этил-4-(4-метоксибензо[b]тиофен-5-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
8-фтор-4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
2-этил-8-фтор-4-(4-метоксибензо[b]тиофен-5-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-5-ол;
4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ол;
5-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)бензо[b]тиофен-4-ол;
5-(2-этил-1,2,3,4-тетрагидроизохинолин-4-ил)бензо[b]тиофен-4-ол;
2-этил-4-(4-метоксибензо[b]тиофен-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1,1-диоксо-1H-1λ6-бензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-бензо[b]тиофен-5-ил-8-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-бензо[b]тиофен-5-ил-7-фтор-2-метил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-(бензо[b]тиофен-5-ил)-4-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-2,4-диметил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-4-карбонитрил;
4-(бензо[b]тиофен-5-ил)-4-фтор-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-4-хлор-2-метил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ол;
4-(бензо[b]тиофен-5-ил)-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-2,7-диметил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-7-фтор-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-этил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
[6-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]диметиламин;
[6-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]метиламин;
6-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-иламин;
4-бензо[b]тиофен-5-ил-2-метил-7-(6-морфолин-4-илпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-метил-7-(6-трифторметилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
6-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-карбонитрил;
4-бензо[b]тиофен-5-ил-8-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-8-метокси-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
4-бензо[b]тиофен-5-ил-2,8-диметил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-этил-8-фтор-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-6-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-5-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-бензо[b]тиофен-5-ил-2,4-диметил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-8-фтор-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-метил-7-(3-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-7-(3-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-метил-7-(6-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-7-(6-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-8-фтор-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-8-метокси-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
4-бензо[b]тиофен-5-ил-2,8-диметил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-8-фтор-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-8-метокси-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
4-бензо[b]тиофен-5-ил-2,8-диметил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-8-фтор-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-8-метокси-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин-8-ол;
4-(бензо[b]тиофен-5-ил)-2,8-диметил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-8-фтор-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-8-метокси-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин-8-ол;
4-(бензо[b]тиофен-5-ил)-2,8-диметил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-7-(3,5-диметилизоксазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(тиазол-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(5-метилтиазол-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-метил-7-[1,3,5]триазин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-метил-7-[1,2,4]триазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-метил-7-[1,2,4]триазин-5-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-метил-7-[1,2,4]триазин-6-ил-1,2,3,4-тетрагидроизохинолин;
3-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циннолин;
1-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)фталазин;
2-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиноксалин;
2-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
6-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
7-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
2-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-2H-[1,2,4]триазоло[4,3-a]пиридин-3-он;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-7-(2,6-диметилморфолин-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-этил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-8-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-8-метокси-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
4-бензо[b]тиофен-5-ил-2,8-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-этил-8-фтор-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-6-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-5-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-бензо[b]тиофен-5-ил-2,4-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-метил-7-пиперазин-1-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-метил-7-(4-метилпиперазин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(морфолин-4-ил)метил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-метил-7-(1-метил-1-морфолин-4-илэтил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-метил-7-(1-морфолин-4-илциклопропил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(пиперидин-1-ил)метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(пирролидин-1-ил)метил-1,2,3,4-тетрагидроизохинолин;
(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)диметиламин;
[1-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]диметиламин;
[1-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]диметиламин;
(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)метиламин;
[1-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]метиламин;
[1-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]метиламин;
C-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)метиламин;
1-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтиламин;
1-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропиламин;
1-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1H-пиридин-2-он;
4-(бензо[b]тиофен-5-ил)-7-метансульфонил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрил;
4-бензо[b]тиофен-5-ил-8-фтор-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрил;
4-(бензо[b]тиофен-5-ил)-2,8-диметил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-1-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-8-фтор-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-2-этил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-1,2-диметил-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(2-метилбензо[b]тиофен-6-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1,1-диоксо-1H-1λ6-бензо[b]тиофен-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-(бензо[b]тиофен-6-ил)-4-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-2,4-диметил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-4-карбонитрил;
4-(бензо[b]тиофен-6-ил)-4-фтор-2-метил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-4-хлор-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(2-хлор-бензо[b]тиофен-6-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(2-хлорбензо[b]тиофен-6-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(2-хлорбензо[b]тиофен-6-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(2-хлорбензо[b]тиофен-6-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(2-хлорбензо[b]тиофен-6-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(2-хлорбензо[b]тиофен-6-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-7-пиридазин-3-ил-4-(3-трифторметилбензо[b]тиофен-6-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-7-пиримидин-2-ил-4-(3-трифторметилбензо[b]тиофен-6-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-7-пиримидин-4-ил-4-(3-трифторметилбензо[b]тиофен-6-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-7-пиримидин-5-ил-4-(3-трифторметилбензо[b]тиофен-6-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-7-пиразин-2-ил-4-(3-трифторметилбензо[b]тиофен-6-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-7-морфолин-4-ил-4-(3-трифторметилбензо[b]тиофен-6-ил)-1,2,3,4-тетрагидроизохинолин;
4-(7-метоксибензо[b]тиофен-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(7-метоксибензо[b]тиофен-6-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
2-этил-4-(7-метоксибензо[b]тиофен-6-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
8-фтор-4-(7-метоксибензо[b]тиофен-6-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
2-этил-8-фтор-4-(7-метоксибензо[b]тиофен-6-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(7-метоксибензо[b]тиофен-6-ил)-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(7-метоксибензо[b]тиофен-6-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(7-метоксибензо[b]тиофен-6-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(7-метоксибензо[b]тиофен-6-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(7-метоксибензо[b]тиофен-6-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(7-метоксибензо[b]тиофен-6-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(7-метоксибензо[b]тиофен-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ол;
4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ол;
(бензо[b]тиофен-6-ил)-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-2,7-диметил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-7-фтор-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-2-этил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
[6-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]диметиламин;
[6-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]метиламин;
6-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-иламин;
4-бензо[b]тиофен-6-ил-2-метил-7-(6-морфолин-4-илпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-2-метил-7-(6-трифторметилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
6-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-карбонитрил;
4-бензо[b]тиофен-6-ил-8-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-8-метокси-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
4-бензо[b]тиофен-6-ил-2,8-диметил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-2-этил-8-фтор-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-6-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-5-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-бензо[b]тиофен-6-ил-2,4-диметил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-8-фтор-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-2-метил-7-(3-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-7-(3-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-2-метил-7-(6-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-7-(6-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-8-фтор-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-8-метокси-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
4-бензо[b]тиофен-6-ил-2,8-диметил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-8-фтор-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-8-метокси-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
4-бензо[b]тиофен-6-ил-2,8-диметил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-8-фтор-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-8-метокси-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин-8-ол;
4-(бензо[b]тиофен-6-ил)-2,8-диметил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-8-фтор-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-8-метокси-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин-8-ол;
4-(бензо[b]тиофен-6-ил)-2,8-диметил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-7-(3,5-диметилизоксазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(тиазол-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(5-метилтиазол-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-2-метил-7-[1,3,5]триазин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-2-метил-7-[1,2,4]триазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-2-метил-7-[1,2,4]триазин-5-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-2-метил-7-[1,2,4]триазин-6-ил-1,2,3,4-тетрагидроизохинолин;
3-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циннолин;
1-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)фталазин;
2-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиноксалин;
2-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
6-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
7-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
2-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-2H-[1,2,4]триазоло[4,3-a]пиридин-3-он;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-7-(2,6-диметилморфолин-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-2-этил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-8-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-8-метокси-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
4-бензо[b]тиофен-6-ил-2,8-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-2-этил-8-фтор-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-6-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-5-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-бензо[b]тиофен-6-ил-2,4-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-2-метил-7-пиперазин-1-ил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-2-метил-7-(4-метилпиперазин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(морфолин-4-ил)метил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-2-метил-7-(1-метил-1-морфолин-4-илэтил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-6-ил-2-метил-7-(1-морфолин-4-илциклопропил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(пиперидин-1-ил)метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(пирролидин-1-ил)метил-1,2,3,4-тетрагидроизохинолин;
(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)диметиламин;
[1-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]диметиламин;
[1-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]диметиламин;
(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)метиламин;
[1-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]метиламин;
[1-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]метиламин;
C-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)метиламин;
1-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтиламин;
1-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропиламин;
1-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1H-пиридин-2-он;
4-(бензо[b]тиофен-6-ил)-7-метансульфонил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрил;
4-бензо[b]тиофен-6-ил-8-фтор-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрил;
4-(бензо[b]тиофен-6-ил)-2,8-диметил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-7-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-7-ил)-2-этил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-7-ил)-8-фтор-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-7-ил)-2-этил-8-фтор-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-7-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[b]тиофен-7-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-циклопропил-1,2,3,4-тетрагидроизохинолин;
(4-бензо[b]тиофен-5-ил-3,4-дигидро-1H-изохинолин-2-ил)ацетонитрил;
[2-(4-бензо[b]тиофен-5-ил-3,4-дигидро-1H-изохинолин-2-ил)этил]диметиламин;
2-(4-бензо[b]тиофен-5-ил-3,4-дигидро-1H-изохинолин-2-ил)этанол;
4-бензо[b]тиофен-5-ил-2-изопропил-1,2,3,4-тетрагидроизохинолин;
4-бензо[b]тиофен-5-ил-2-метил-8-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
или их оксид, их фармацевтически приемлемую соль, их сольват или их пролекарство.
Кроме того, другие конкретные соединения согласно настоящему изобретению представляют собой соединения со следующими заместителями (таблица В):

где атом углерода, обозначенный *, находится в R- или S-конфигурации. То есть конкретные соединения в данном случае включают
4-(бензофуран-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-1-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-этил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-1,2-диметил-1,2,3,4-тетрагидроизохинолин;
4-(5-хлорбензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(5-фторбензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(7-фторбензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(5-метоксибензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(7-метоксибензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(7-метоксибензофуран-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(7-метоксибензофуран-2-ил)-8-фтор-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(7-метоксибензофуран-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(7-метоксибензофуран-2-ил)-8-фтор-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-бензофуран-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-бензофуран-2-ил-4-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2,4-диметил-1,2,3,4-тетрагидроизохинолин;
4-бензофуран-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-4-карбонитрил;
4-(бензофуран-2-ил)-4-фтор-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-4-хлор-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2,5-диметил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-7-фтор-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-7-гидрокси-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2,7-диметил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(4-фторбензофуран-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(5-фторбензофуран-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(6-фторбензофуран-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(7-фторбензофуран-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-этил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
[6-(4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]диметиламин;
[6-(4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]метиламин;
6-(4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-иламин;
4-(бензофуран-2-ил)-2-метил-7-(6-морфолин-4-илпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-(6-трифторметилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
6-(4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-карбонитрил;
4-(бензофуран-2-ил)-8-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-8-метокси-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
4-(бензофуран-2-ил)-2,8-диметил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-этил-8-фтор-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-6-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-5-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-(бензофуран-2-ил)-2,4-диметил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-8-фтор-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-(3-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-7-(3-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-(6-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-7-(6-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-8-фтор-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-8-фтор-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-8-фтор-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-8-фтор-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-7-(3,5-диметилизоксазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-(тиазол-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-(5-метилтиазол-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-[1,3,5]триазин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-[1,2,4]триазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-[1,2,4]триазин-5-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-[1,2,4]триазин-6-ил-1,2,3,4-тетрагидроизохинолин;
3-(4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циннолин;
1-(4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)фталазин;
2-(4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиноксалин;
2-(4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
6-(4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
7-(4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
2-(4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-2H-[1,2,4]триазоло[4,3-a]пиридин-3-он;
4-(бензофуран-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(4-фторбензофуран-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(5-фторбензофуран-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(6-фторбензофуран-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(7-фторбензофуран-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-7-(2,6-диметилморфолин-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-этил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-8-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-8-метокси-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
4-(бензофуран-2-ил)-2,8-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-этил-8-фтор-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-6-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-5-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-(бензофуран-2-ил)-2,4-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-бензофуран-2-ил-2-метил-7-пиперазин-1-ил-1,2,3,4-тетрагидроизохинолин;
4-бензофуран-2-ил-2-метил-7-(4-метилпиперазин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-(морфолин-4-ил)метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-(1-метил-1-морфолин-4-илэтил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-(1-морфолин-4-илциклопропил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-(пиперидин-1-ил)метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-7-(пирролидин-1-ил)метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)диметиламин;
[1-4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]диметиламин;
[1-4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]диметиламин;
(4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)метиламин;
[1-4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]метиламин;
[1-4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]метиламин;
C-4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)метиламин;
1-4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтиламин;
1-4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропиламин;
1-(4-бензофуран-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1H-пиридин-2-он;
4-(бензофуран-2-ил)-7-метансульфонил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрил;
4-(бензофуран-2-ил)-2,8-диметил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-3-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-3-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-3-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-4-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-4-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-1-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-8-фтор-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-этил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-1,2-диметил-1,2,3,4-тетрагидроизохинолин;
4-(3-метилбензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-бензофуран-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-(бензофуран-5-ил)-4-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2,4-диметил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-карбонитрил;
4-(бензофуран-5-ил)-4-фтор-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-4-хлор-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ол;
4-(бензофуран-5-ил)-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2,7-диметил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-7-фтор-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-этил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
[6-4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]диметиламин;
[6-4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]метиламин;
6-4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-иламин;
6-4-(бензофуран-5-ил)-2-метил-7-(6-морфолин-4-илпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
6-4-(бензофуран-5-ил)-2-метил-7-(6-трифторметилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
6-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-карбонитрил;
4-(бензофуран-5-ил)-8-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-8-метокси-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
4-(бензофуран-5-ил)-2,8-диметил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-этил-8-фтор-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-6-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-5-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-(бензофуран-5-ил)-2,4-диметил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-8-фтор-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-(3-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-7-(3-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-(6-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-7-(6-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-8-фтор-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-8-метокси-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
4-(бензофуран-5-ил)-2,8-диметил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-((бензофуран-5-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-8-фтор-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-8-метокси-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
4-(бензофуран-5-ил)-2,8-диметил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-((бензофуран-5-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-8-фтор-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-8-метокси-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин-8-ол;
4-(бензофуран-5-ил)-2,8-диметил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-8-фтор-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-8-метокси-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин-8-ол;
4-(бензофуран-5-ил)-2,8-диметил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-7-(3,5-диметилизоксазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-(тиазол-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-(5-метилтиазол-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-[1,3,5]триазин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-[1,2,4]триазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-[1,2,4]триазин-5-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-[1,2,4]триазин-6-ил-1,2,3,4-тетрагидроизохинолин;
3-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циннолин;
1-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)фталазин;
2-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиноксалин;
2-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
6-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
7-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
2-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-2H-[1,2,4]триазоло[4,3-a]пиридин-3-он;
4-(бензофуран-5-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-7-(2,6-диметилморфолин-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-этил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-8-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-8-метокси-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
4-(бензофуран-5-ил)-2,8-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-этил-8-фтор-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-6-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-5-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-(бензофуран-5-ил)-2,4-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-(морфолин-4-ил)метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-(1-метил-1-морфолин-4-илэтил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-(1-морфолин-4-илциклопропил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-(пиперидин-1-ил)метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-7-(пирролидин-1-ил)метил-1,2,3,4-тетрагидроизохинолин;
(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)диметиламин;
[1-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]диметиламин;
[1-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]диметиламин;
(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)метиламин;
[1-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]метиламин;
[1-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]метиламин;
C-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)метиламин;
1-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтиламин;
1-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропиламин;
1-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1H-пиридин-2-он;
4-(бензофуран-5-ил)-7-метансульфонил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрил;
4-(бензофуран-5-ил)-8-фтор-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрил;
4-(бензофуран-5-ил)-2,8-диметил-1,2,3,4-тетрагидроизохинолин;
4-(2,3-дигидробензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-1-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-этил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-1,2-диметил-1,2,3,4-тетрагидроизохинолин;
4-бензофуран-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-(бензофуран-6-ил)-4-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2,4-диметил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-карбонитрил;
4-(бензофуран-6-ил)-4-фтор-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-4-хлор-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ол;
4-(бензофуран-6-ил)-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2,7-диметил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-7-фтор-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-этил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
[6-(4-бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]диметиламин;
[6-(4-бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]метиламин;
6-(4-бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-иламин;
4-(бензофуран-6-ил)-2-метил-7-(6-морфолин-4-илпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-(6-трифторметилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-карбонитрил;
4-(бензофуран-6-ил)-8-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-8-метокси-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
4-(бензофуран-6-ил)-2,8-диметил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-этил-8-фтор-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-6-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-5-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-8-фтор-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-(3-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-7-(3-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-(6-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-7-(6-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-8-фтор-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-8-метокси-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
4-(бензофуран-6-ил)-2,8-диметил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-8-фтор-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-8-метокси-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
4-(бензофуран-6-ил)-2,8-диметил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-8-фтор-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-8-метокси-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин-8-ол;
4-(бензофуран-6-ил)-2,8-диметил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-[бензофуран-6-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-8-фтор-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-8-метокси-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин-8-ол;
4-(бензофуран-6-ил)-2,8-диметил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-7-(3,5-диметилизоксазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-(тиазол-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-(5-метилтиазол-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-[1,3,5]триазин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-[1,2,4]триазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-[1,2,4]триазин-5-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-[1,2,4]триазин-6-ил-1,2,3,4-тетрагидроизохинолин;
3-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циннолин;
1-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)фталазин;
2-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиноксалин;
2-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
6-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
7-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
2-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-2H-[1,2,4]триазоло[4,3-a]пиридин-3-он;
4-(бензофуран-6-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-7-(2,6-диметилморфолин-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-этил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-8-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-8-метокси-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
4-(бензофуран-6-ил)-2,8-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-этил-8-фтор-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-6-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-5-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-(бензофуран-6-ил)-2,4-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-(морфолин-4-ил)метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-(1-метил-1-морфолин-4-илэтил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-(1-морфолин-4-илциклопропил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-(пиперидин-1-ил)метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-7-(пирролидин-1-ил)метил-1,2,3,4-тетрагидроизохинолин;
(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)диметиламин;
[1-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]диметиламин;
[1-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]диметиламин;
(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)метиламин;
[1-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]метиламин;
[1-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]метиламин;
C-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)метиламин;
1-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтиламин;
1-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропиламин;
1-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1H-пиридин-2-он;
4-(бензофуран-6-ил)-7-метансульфонил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрил;
4-(бензофуран-6-ил)-8-фтор-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрил;
4-(бензофуран-6-ил)-2,8-диметил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-7-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-7-ил)-2-этил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-7-ил)-8-фтор-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-7-ил)-2-этил-8-фтор-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-7-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензофуран-7-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин
или их оксид, их фармацевтически приемлемую соль, их сольват или их пролекарство.
Кроме того, другие конкретные соединения согласно настоящему изобретению представляют собой соединения со следующими заместителями (Таблица С):

где атом углерода, обозначенный *, находится в R- или S-конфигурации. То есть конкретные соединения в данном случае включают
4-(1H-индол-1-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-1-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
2-этил-4-(1H-индол-2-ил)-1,2,3,4-тетрагидроизохинолин;
1,2-диметил-4-(1H-индол-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(5-метокси-1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-(1H-индол-2-ил)-4-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2,4-диметил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-карбонитрил;
4-фтор-4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-хлор-4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ол;
4-(1H-индол-2-ил)-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
2,7-диметил-4-(1H-индол-2-ил)-1,2,3,4-тетрагидроизохинолин;
7-фтор-4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1-метил-1H-индол-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(7-фтор-1H-индол-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
2-этил-4-(1H-индол-2-ил)-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
[6-(4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]диметиламин;
[6-(4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]метиламин;
6-(4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-иламин;
4-(1H-индол-2-ил)-2-метил-7-(6-морфолин-4-илпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-7-(6-трифторметилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
6-(4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-карбонитрил;
8-фтор-4-(1H-индол-2-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
2-этил-8-фтор-4-(1H-индол-2-ил)-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
6-фтор-4-(1H-индол-2-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
5-фтор-4-(1H-индол-2-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-8-метокси-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
2,8-диметил-4-(1H-индол-2-ил)-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ол;
2,4-диметил-4-(1H-индол-2-ил)-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолин;
8-фтор-4-(1H-индол-2-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-7-(3-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-7-(3-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-7-(6-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-7-(6-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-8-фтор-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин;
8-фтор-4-(1H-индол-2-ил)-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
8-фтор-4-(1H-индол-2-ил)-2-метил-7-пиримидин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
8-фтор-4-(1H-индол-2-ил)-2-метил-7-пиримидин-5-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-7-(3,5-диметилизоксазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-7-(тиазол-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-7-(5-метилтиазол-2-ил)-1,2,3,4-тетрагидроизохинолин;
3-(4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циннолин;
1-(4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)фталазин;
2-(4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиноксалин;
2-(4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
6-(4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
7-(4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
2-(4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-2H-[1,2,4]триазоло[4,3-a]пиридин-3-он;
4-(1H-индол-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1-метил-1H-индол-2-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(7-фтор-1H-индол-2-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
7-(2,6-диметилморфолин-4-ил)-4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
2-этил-4-(1H-индол-2-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
8-фтор-4-(1H-индол-2-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
2-этил-8-фтор-4-(1H-индол-2-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
6-фтор-4-(1H-индол-2-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
5-фтор-4-(1H-индол-2-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-8-метокси-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
4-(1H-индол-2-ил)-2,8-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-(1H-индол-2-ил)-2,4-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-7-(морфолин-4-ил)метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-7-(1-метил-1-морфолин-4-илэтил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-7-(1-морфолин-4-илциклопропил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-7-(пиперидин-1-ил)метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-7-(пирролидин-1-ил)метил-1,2,3,4-тетрагидроизохинолин;
(4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)диметиламин;
[1-(4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]диметиламин;
[1-(4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]диметиламин;
(4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)метиламин;
[1-(4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]метиламин;
[1-(4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]метиламин;
C-(4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)метиламин;
1-(4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтиламин;
1-(4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропиламин;
1-(4-(1H-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1H-пиридин-2-он;
4-(1H-индол-2-ил)-7-метансульфонил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-2-ил)-2-метил-l,2,3,4-тетрагидроизохинолин-7-карбонитрил;
2,8-диметил-4-(1H-индол-2-ил)-l,2,3,4-тетрагидроизохинолин;
4-(1H-индол-3-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-1-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
2-этил-4-(1H-индол-5-ил)-1,2,3,4-тетрагидроизохинолин;
1,2-диметил-4-(1H-индол-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-(1H-индол-5-ил)-4-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2,4-диметил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-карбонитрил;
4-фтор-4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-хлор-4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ол;
4-(1H-индол-5-ил)-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
2,7-диметил-4-(1H-индол-5-ил)-1,2,3,4-тетрагидроизохинолин;
7-фтор-4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1-метилиндол-5-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(7-фтор-1H-индол-5-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
2-этил-4-(1H-индол-5-ил)-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
[6-(4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]диметиламин;
[6-(4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]метиламин;
6-(4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-иламин;
4-(1H-индол-5-ил)-2-метил-7-(6-морфолин-4-илпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-7-(6-трифторметилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
6-(4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-карбонитрил;
8-фтор-4-(1H-индол-5-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
2-этил-8-фтор-4-(1H-индол-5-ил)-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
6-фтор-4-(1H-индол-5-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
5-фтор-4-(1H-индол-5-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-8-метокси-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
2,8-диметил-4-(1H-индол-5-ил)-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ол;
2,4-диметил-4-(1H-индол-5-ил)-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолин;
8-фтор-4-(1H-индол-5-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-7-(3-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-7-(3-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-7-(6-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-7-(6-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-8-фтор-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин;
8-фтор-4-(1H-индол-5-ил)-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
8-фтор-4-(1H-индол-5-ил)-2-метил-7-пиримидин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
8-фтор-4-(1H-индол-5-ил)-2-метил-7-пиримидин-5-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-7-(3,5-диметилизоксазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-7-(тиазол-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-7-(5-метилтиазол-2-ил)-1,2,3,4-тетрагидроизохинолин;
3-(4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циннолин;
1-(4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)фталазин;
2-(4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиноксалин;
2-(4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
6-(4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
7-(4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
2-(4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-2H-[1,2,4]триазоло[4,3-a]пиридин-3-он;
4-(1H-индол-5-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1-метил-1H-индол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(7-фтор-1H-индол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
7-(2,6-диметилморфолин-4-ил)-4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
2-этил-4-(1H-индол-5-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
8-фтор-4-(1H-индол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
2-этил-8-фтор-4-(1H-индол-5-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
6-фтор-4-(1H-индол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
5-фтор-4-(1H-индол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-8-метокси-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
2,8-диметил-4-(1H-индол-5-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ол;
2,4-диметил-4-(1H-индол-5-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-7-(морфолин-4-ил)метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-7-(1-метил-1-морфолин-4-илэтил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-7-(1-морфолин-4-илциклопропил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-7-(пиперидин-1-ил)метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-7-(пирролидин-1-ил)метил-1,2,3,4-тетрагидроизохинолин;
(4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)диметиламин;
[1-(4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]диметиламин;
[1-(4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]диметиламин;
(4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)метиламин;
[1-(4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]метиламин;
[1-(4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]метиламин;
C-(4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)метиламин;
1-(4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтиламин;
1-(4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропиламин;
1-(4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1H-пиридин-2-он;
4-(1H-индол-5-ил)-7-метансульфонил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрил;
4-(1H-индол-5-ил)-2,8-диметил-1,2,3,4-тетрагидроизохинолин;
4-(1-бензил-1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-1-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(3-хлор-1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
2-этил-4-(1H-индол-6-ил)-1,2,3,4-тетрагидроизохинолин;
1,2-диметил-4-(1H-индол-6-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-(1H-индол-6-ил)-4-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
2,4-диметил-4-(1H-индол-6-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-карбонитрил;
4-фтор-4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-хлор-4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ол;
4-(1H-индол-6-ил)-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
2,7-диметил-4-(1H-индол-6-ил)-1,2,3,4-тетрагидроизохинолин;
7-фтор-4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1-метилиндол-6-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(7-фтор-1H-индол-6-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
2-этил-4-(1H-индол-6-ил)-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
[6-(4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]диметиламин;
[6-(4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]метиламин;
6-(4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-иламин;
4-(1H-индол-6-ил)-2-метил-7-(6-морфолин-4-илпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-7-(6-трифторметилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
6-(4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-карбонитрил;
8-фтор-4-(1H-индол-6-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
2-этил-8-фтор-4-(1H-индол-6-ил)-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
6-фтор-4-(1H-индол-6-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
5-фтор-4-(1H-индол-6-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-8-метокси-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
2,8-диметил-4-(1H-индол-6-ил)-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ол;
2,4-диметил-4-(1H-индол-6-ил)-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолин;
8-фтор-4-(1H-индол-6-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-7-(3-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-7-(3-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-7-(6-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-7-(6-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
8-фтор-4-(1H-индол-6-ил)-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин;
8-фтор-4-(1H-индол-6-ил)-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
8-фтор-4-(1H-индол-6-ил)-2-метил-7-пиримидин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
8-фтор-4-(1H-индол-6-ил)-2-метил-7-пиримидин-5-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-7-(3,5-диметилизоксазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-7-(тиазол-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-7-(5-метилтиазол-2-ил)-1,2,3,4-тетрагидроизохинолин;
3-(4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циннолин;
1-(4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)фталазин;
2-(4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиноксалин;
2-(4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
6-(4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
7-(4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
2-(4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-2H-[1,2,4]триазоло[4,3-a]пиридин-3-он;
4-(1H-индол-6-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1-метил-1H-индол-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(7-фтор-1H-индол-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
7-(2,6-диметилморфолин-4-ил)-4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
2-этил-4-(1H-индол-6-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
8-фтор-4-(1H-индол-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
2-этил-8-фтор-4-(1H-индол-6-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
6-фтор-4-(1H-индол-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
5-фтор-4-(1H-индол-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-8-метокси-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
2,8-диметил-4-(1H-индол-6-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ол;
2,4-диметил-4-(1H-индол-6-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-7-(морфолин-4-ил)метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-7-(1-метил-1-морфолин-4-илэтил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-7-(1-морфолин-4-илциклопропил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-7-(пиперидин-1-ил)метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-7-(пирролидин-1-ил)метил-1,2,3,4-тетрагидроизохинолин;
(4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)диметиламин;
[1-(4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]диметиламин;
[1-(4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]диметиламин;
(4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)метиламин;
[1-(4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]метиламин;
[1-(4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]метиламин;
C-(4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)метиламин;
1-(4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтиламин;
1-(4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропиламин;
1-(4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1H-пиридин-2-он;
4-(1H-индол-6-ил)-7-метансульфонил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрил;
2,8-диметил-4-(1H-индол-6-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(1-метил-1H-индол-5-ил)-1,2,3,4-тетрагидроизохинолин;
8-фтор-2-метил-4-(1-метил-1H-индол-5-ил)-1,2,3,4-тетрагидроизохинолин;
2-этил-8-фтор-4-(1-метил-1H-индол-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1-бензил-1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
3-[5-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)индол-1-илметил]бензонитрил;
2-[5-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)индол-1-илметил]бензонитрил;
2-метил-4-(1-метил-1H-индол-6-ил)-1,2,3,4-тетрагидроизохинолин;
8-фтор-2-метил-4-(1-метил-1H-индол-6-ил)-1,2,3,4-тетрагидроизохинолин;
2-этил-8-фтор-4-(1-метил-1H-индол-6-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1-бензил-1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
3-[6-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)индол-1-илметил]бензонитрил;
2-[6-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)индол-1-илметил]бензонитрил;
или их оксид, их фармацевтически приемлемая соль, их сольват, или их пролекарство.
Другие конкретные соединения согласно настоящему изобретению представляют собой соединения со следующими заместителями (таблица D):

где атом углерода, обозначенный *, находится в R- или S-конфигурации. То есть конкретные соединения в данном случае включают
4-(индазол-1-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-3-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-1-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(1-метил-1H-индазол-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(6-метоксииндазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(7-метоксииндазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(7-фториндазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(7-хлориндазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(7-метилиндазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
8-фтор-4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
2-этил-4-(1H-индазол-5-ил)-1,2,3,4-тетрагидроизохинолин;
2-этил-8-фтор-4-(1H-индазол-5-ил)-1,2,3,4-тетрагидроизохинолин;
1,2-диметил-4-(1H-индазол-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-(1H-индазол-5-ил)-4-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
2,4-диметил-4-(1H-индазол-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-карбонитрил;
4-фтор-4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-хлор-4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ол;
4-(1H-индазол-5-ил)-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
2,7-диметил-4-(1H-индазол-5-ил)-1,2,3,4-тетрагидроизохинолин;
7-фтор-2-метил-4-(1H-индазол-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1-метил-1H-индазол-5-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(7-фтор-1H-индазол-5-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
2-этил-4-(1H-индазол-5-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
[6-(4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]диметиламин;
[6-(4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]метиламин;
6-(4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-иламин;
4-(1H-индазол-5-ил)-2-метил-7-(6-морфолин-4-илпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-2-метил-7-(6-трифторметилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-7-(3,5-диметилизоксазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-2-метил-7-(тиазол-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-2-метил-7-(5-метилтиазол-2-ил)-1,2,3,4-тетрагидроизохинолин;
3-(4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циннолин;
1-(4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)фталазин;
2-(4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиноксалин;
2-(4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
6-(4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
7-(4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
2-(4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-2H-[1,2,4]триазоло[4,3-a]пиридин-3-он;
4-(1H-индазол-5-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1-метил-1H-индазол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(7-фтор-1H-индазол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
7-(2,6-диметилморфолин-4-ил)-4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
2-этил-4-(1H-индазол-5-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
8-фтор-4-(1H-индазол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
2-этил-8-фтор-4-(1H-индазол-5-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
6-фтор-4-(1H-индазол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
5-фтор-4-(1H-индазол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-8-метокси-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
2,8-диметил-4-(1H-индазол-5-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ол;
2,4-диметил-4-(1H-индазол-5-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-2-метил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-2-метил-7-((морфолин-4-ил)метил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-2-метил-7-(1-метил-1-морфолин-4-илэтил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-2-метил-7-(1-морфолин-4-илциклопропил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-2-метил-7-пиперидин-1-илметил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-2-метил-7-пирролидин-1-илметил-1,2,3,4-тетрагидроизохинолин;
[4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил]диметиламин;
[4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил]метиламин;
1-(4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1H-пиридин-2-он;
4-(1H-индазол-5-ил)-7-метансульфонил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрил;
4-(1H-индазол-5-ил)-2,8-диметил-1,2,3,4-тетрагидроизохинолин;
7-фтор-2-метил-4-(1-метил-1H-индазол-5-ил)-1,2,3,4-тетрагидроизохинолин;
2-этил-7-фтор-4-(1-метил-1H-индазол-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1-бензил-1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
3-[5-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)-индазол-1-илметил]бензонитрил;
2-[5-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)индазол-1-илметил]бензонитрил;
4-(1H-индазол-6-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-1-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(1-метилиндазол-6-ил)-1,2,3,4-тетрагидроизохинолин;
4-(7-фтор-1H-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
2-этил-4-(1H-индазол-6-ил)-1,2,3,4-тетрагидроизохинолин;
1,2-диметил-4-(1H-индазол-6-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-4-метил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-(1H-индазол-6-ил)-4-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
2,4-диметил-4-(1H-индазол-6-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-карбонитрил;
4-фтор-4-(1H-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-хлор-4-(1H-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ол;
4-(1H-индазол-6-ил)-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
2,7-диметил-4-(1H-индазол-6-ил)-1,2,3,4-тетрагидроизохинолин;
7-фтор-2-метил-4-(1H-индазол-6-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(1-метил-1H-индазол-6-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(7-фтор-1H-индазол-6-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
2-этил-4-(1H-индазол-6-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
[6-(4-(1H-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]диметиламин;
[6-(4-(1H-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]метиламин;
6-(4-(1H-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-иламин;
4-(1H-индазол-6-ил)-2-метил-7-(6-морфолин-4-илпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-2-метил-7-(6-трифторметилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-7-(3,5-диметилизоксазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-2-метил-7-(тиазол-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-2-метил-7-(5-метилтиазол-2-ил)-1,2,3,4-тетрагидроизохинолин;
3-(4-(1H-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циннолин;
1-(4-(1H-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)фталазин;
2-(4-(1H-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиноксалин;
2-(4-(1H-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
6-(4-(1H-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
7-(4-(1H-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
2-(4-(1H-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-2H-[1,2,4]триазоло[4,3-a]пиридин-3-он;
4-(1H-индазол-6-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(1-метил-1H-индазол-6-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(7-фтор-1H-индазол-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
7-(2,6-диметилморфолин-4-ил)-4-(1H-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
2-этил-4-(1H-индазол-6-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
8-фтор-4-(1H-индазол-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
2-этил-8-фтор-4-(1H-индазол-6-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
6-фтор-4-(1H-индазол-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
5-фтор-4-(1H-индазол-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-8-метокси-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
2,8-диметил-4-(1H-индазол-6-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ол;
2,4-диметил-4-(1H-индазол-6-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-2-метил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-2-метил-7-морфолин-4-илметил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-2-метил-7-(1-метил-1-морфолин-4-илэтил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-2-метил-7-(1-морфолин-4-илциклопропил)-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-2-метил-7-пиперидин-1-илметил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-2-метил-7-пирролидин-1-илметил-1,2,3,4-тетрагидроизохинолин;
[4-(1H-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил]диметиламин;
[4-(1H-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил]метиламин;
1-(4-(1H-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1H-пиридин-2-он;
4-(1H-индазол-6-ил)-7-метансульфонил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(1H-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрил;
2,8-диметил-4-(1H-индазол-6-ил)-1,2,3,4-тетрагидроизохинолин;
7-фтор-2-метил-4-(1-метил-1H-индазол-6-ил)-1,2,3,4-тетрагидроизохинолин;
2-этил-7-фтор-4-(1-метил-1H-индазол-6-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1-бензил-1H-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
3-[6-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)индазол-1-илметил]бензонитрил;
2-[6-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)индазол-1-илметил]бензонитрил;
4-(1H-индазол-7-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
или их оксид, их фармацевтически приемлемую соль, их сольват, или их пролекарство.
Кроме того, другие конкретные соединения согласно настоящему изобретению представляют собой соединения со следующими заместителями (таблица Е):
где атом углерода, обозначенный *, находится в R- или S-конфигурации. То есть конкретные соединения в данном случае включают
4-(бензооксазол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензооксазол-2-ил)-2-метил(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензооксазол-2-ил)-2-метил(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензооксазол-2-ил)-2-метил(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензооксазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензооксазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензооксазол-5-ил)-2-метил(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензооксазол-5-ил)-2-метил(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензооксазол-5-ил)-2-метил(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(2-метилбензооксазол-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензооксазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензооксазол-6-ил)-2-метил(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензооксазол-6-ил)-2-метил(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензооксазол-6-ил)-2-метил(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(2-метилбензооксазол-6-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензооксазол-7-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензотиазол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензотиазол-2-ил)-2-метил(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензотиазол-2-ил)-2-метил(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензотиазол-2-ил)-2-метил(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензотиазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензотиазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензотиазол-5-ил)-2-метил(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензотиазол-5-ил)-2-метил(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензотиазол-5-ил)-2-метил(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(2-метилбензотиазол-5-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензотиазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензотиазол-6-ил)-2-метил(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензотиазол-6-ил)-2-метил(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензотиазол-6-ил)-2-метил(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(2-метилбензотиазол-6-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензотиазол-7-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[d]изотиазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[d]изотиазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[d]изотиазол-5-ил)-2-метил(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[d]изотиазол-5-ил)-2-метил(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[d]изотиазол-5-ил)-2-метил(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[d]изотиазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[d]изотиазол-6-ил)-2-метил(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[d]изотиазол-6-ил)-2-метил(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[d]изотиазол-6-ил)-2-метил(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(бензо[d]изотиазол-7-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[d]изоксазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[d]изоксазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[d]изоксазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(бензо[d]изоксазол-7-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-имидазо[1,2-a]пиридин-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-имидазо[1,2-a]пиридин-6-ил-2-метил(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-имидазо[1,2-a]пиридин-6-ил-2-метил(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-имидазо[1,2-a]пиридин-6-ил-2-метил(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-имидазо[1,2-a]пиридин-7-ил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-имидазо[1,2-a]пиридин-7-ил-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-имидазо[1,2-a]пиридин-7-ил-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-имидазо[1,2-a]пиридин-7-ил-2-метил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-пиразоло[1,5-a]пиридин-5-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(морфолин-4-ил)-4-пиразоло[1,5-a]пиридин-5-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(пиперидин-1-ил)-4-пиразоло[1,5-a]пиридин-5-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-4-пиразоло[1,5-a]пиридин-5-ил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-пиразоло[1,5-a]пиридин-6-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(морфолин-4-ил)-4-пиразоло[1,5-a]пиридин-6-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(пиперидин-1-ил)-4-пиразоло[1,5-a]пиридин-6-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-4-пиразоло[1,5-a]пиридин-6-ил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-[1,2,4]триазоло[4,3-a]пиридин-6-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(морфолин-4-ил)-4-[1,2,4]триазоло[4,3-a]пиридин-6-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(пиперидин-1-ил)-4-[1,2,4]триазоло[4,3-a]пиридин-6-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(пирролидин-1-ил)-4-[1,2,4]триазоло[4,3-a]пиридин-6-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-4-[1,2,4]триазоло[4,3-a]пиридин-7-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(морфолин-4-ил)-4-[1,2,4]триазоло[4,3-a]пиридин-7-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(пиперидин-1-ил)-4-[1,2,4]триазоло[4,3-a]пиридин-7-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(пирролидин-1-ил)-4-[1,2,4]триазоло[4,3-a]пиридин-7-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-4-тиено[2,3-b]пиридин-2-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(морфолин-4-ил)-4-тиено[2,3-b]пиридин-2-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(пиперидин-1-ил)-4-тиено[2,3-b]пиридин-2-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(пирролидин-1-ил)-4-тиено[2,3-b]пиридин-2-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-4-тиено[2,3-b]пиридин-5-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(морфолин-4-ил)-4-тиено[2,3-b]пиридин-5-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(пиперидин-1-ил)-4-тиено[2,3-b]пиридин-5-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(пирролидин-1-ил)-4-тиено[2,3-b]пиридин-5-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-4-тиено[2,3-b]пиридин-6-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(морфолин-4-ил)-4-тиено[2,3-b]пиридин-6-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(пиперидин-1-ил)-4-тиено[2,3-b]пиридин-6-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(пирролидин-1-ил)-4-тиено[2,3-b]пиридин-6-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-4-тиено[3,2-b]пиридин-2-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(морфолин-4-ил)-4-тиено[3,2-b]пиридин-2-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(пиперидин-1-ил)-4-тиено[3,2-b]пиридин-2-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(пирролидин-1-ил)-4-тиено[3,2-b]пиридин-2-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-4-тиено[3,2-b]пиридин-5-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(морфолин-4-ил)-4-тиено[3,2-b]пиридин-5-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(пиперидин-1-ил)-4-тиено[3,2-b]пиридин-5-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(пирролидин-1-ил)-4-тиено[3,2-b]пиридин-5-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-4-тиено[3,2-b]пиридин-6-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(морфолин-4-ил)-4-тиено[3,2-b]пиридин-6-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(пиперидин-1-ил)-4-тиено[3,2-b]пиридин-6-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(пирролидин-1-ил)-4-тиено[3,2-b]пиридин-6-ил-1,2,3,4-тетрагидроизохинолин;
4-индолизин-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-индолизин-2-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-индолизин-2-ил-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-индолизин-2-ил-2-метил-7-пиримидин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-индолизин-2-ил-2-метил-7-пиримидин-5-ил-1,2,3,4-тетрагидроизохинолин;
4-индолизин-2-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-индолизин-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-индолизин-6-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-индолизин-6-ил-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-индолизин-6-ил-2-метил-7-пиримидин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-индолизин-6-ил-2-метил-7-пиримидин-5-ил-1,2,3,4-тетрагидроизохинолин;
4-индолизин-6-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-индолизин-7-ил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-индолизин-7-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-индолизин-7-ил-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин;
4-индолизин-7-ил-2-метил-7-пиримидин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-индолизин-7-ил-2-метил-7-пиримидин-5-ил-1,2,3,4-тетрагидроизохинолин;
4-индолизин-7-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин;
4-(1H-инден-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(индан-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
или их оксид, их фармацевтически приемлемую соль, их сольват, или их пролекарство.
Кроме того, другие конкретные соединения согласно настоящему изобретению представляют собой соединения со следующими заместителями (таблица F):

где атом углерода, обозначенный *, находится в R- или S-конфигурации. То есть конкретные соединения в данном случае включают
2-метил-4-(нафтален-1-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(4-метилнафтален-1-ил)-1,2,3,4-тетрагидроизохинолин;
4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
1-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
2-этил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
1,2-диметил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
2,5-диметил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин-4-ол;
4-метокси-2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
2,4-диметил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-4-карбонитрил;
4-фтор-2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-хлор-2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(6-метоксинафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(7-метоксинафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(8-метоксинафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(8-метоксинафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-(8-метоксинафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ол;
4-(8-хлорнафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(8-хлорнафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-ол;
4-(8-хлорнафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ол;
4-(8-фторнафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин-7-ол;
7-метокси-4-(нафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
2,7-диметил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
7-фтор-2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(нафтален-2-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1-фторнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1-хлорнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1-метоксинафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1-метилнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(3-фторнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(3-хлорнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(3-метоксинафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
3-(2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ил)нафталин-2-карбонитрил;
4-(4-фторнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(4-хлорнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(5-фторнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(5-хлорнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
6-(2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ил)нафталин-1-карбонитрил;
4-(5-метилнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(6-метоксинафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(6-хлорнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(6-фторнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
6-(2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ил)нафталин-2-карбонитрил;
4-(6-метансульфонилнафтален-2-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин;
4-(7-метоксинафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(7-хлорнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(7-фторнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
7-(2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ил)нафталин-2-карбонитрил;
4-(8-метоксинафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(8-хлорнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
4-(8-фторнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
8-(2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ил)нафталин-1-карбонитрил;
2-этил-4-(нафтален-2-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(6-метилпиридазин-3-ил)-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин;
диметил-[6-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]амин;
метил-[6-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]амин;
6-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-иламин;
2-метил-7-(6-морфолин-4-илпиридазин-3-ил)-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-4-нафтален-2-ил-7-(6-трифторметилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
6-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-илкарбонитрил;
8-фтор-2-метил-4-(нафтален-2-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
8-метокси-2-метил-4-(нафтален-2-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(нафтален-2-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин-8-ол;
2,8-диметил-4-(нафтален-2-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
2-этил-8-фтор-4-(нафтален-2-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
6-фтор-2-метил-4-(нафтален-2-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
5-фтор-2-метил-4-(нафтален-2-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(нафтален-2-ил)-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолин;
8-фтор-2-метил-4-(нафтален-2-ил)-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(нафтален-2-ил)-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(3-метилпиразин-2-ил)-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
7-(3-метоксипиразин-2-ил)-2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(6-метилпиразин-2-ил)-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
7-(6-метоксипиразин-2-ил)-2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
8-фтор-2-метил-4-(нафтален-2-ил)-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
8-метокси-2-метил-4-(нафтален-2-ил)-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(нафтален-2-ил)-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолин-8-ол;
2,8-диметил-4-(нафтален-2-ил)-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(нафтален-2-ил)-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин;
8-фтор-2-метил-4-(нафтален-2-ил)-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин;
8-метокси-2-метил-4-(нафтален-2-ил)-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(нафтален-2-ил)-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин-8-ол;
2,8-диметил-4-(нафтален-2-ил)-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(нафтален-2-ил)-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
8-фтор-2-метил-4-(нафтален-2-ил)-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
8-метокси-2-метил-4-(нафтален-2-ил)-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(нафтален-2-ил)-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин-8-ол;
2,8-диметил-4-(нафтален-2-ил)-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(нафтален-2-ил)-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
8-фтор-2-метил-4-(нафтален-2-ил)-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
8-метокси-2-метил-4-(нафтален-2-ил)-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(нафтален-2-ил)-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин-8-ол;
2,8-диметил-4-(нафтален-2-ил)-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин;
7-(3,5-диметилизоксазол-4-ил)-2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(нафтален-2-ил)-7-(тиазол-2-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(нафтален-2-ил)-7-(5-метилтиазол-2-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-нафтален-2-ил-7-[1,3,5]триазин-2-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-4-нафтален-2-ил-7-[1,2,4]триазин-3-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-4-нафтален-2-ил-7-[1,2,4]триазин-5-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-4-нафтален-2-ил-7-[1,2,4]триазин-6-ил-1,2,3,4-тетрагидроизохинолин;
3-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)циннолин;
1-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)фталазин;
2-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)хиноксалин;
2-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
6-(2-метил-4-нафтален-2-ил-2-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
7-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолин;
2-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)-2H-[1,2,4]триазоло[4,3-a]пиридин-3-он;
2-метил-7-(морфолин-4-ил)-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1-фторнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1-хлорнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1-метоксинафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(1-метилнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(3-фторнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(3-хлорнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(3-метоксинафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
3-(2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинол-4-ил)нафталин-2-карбонитрил;
4-(4-фторнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(4-хлорнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(5-фторнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(5-хлорнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
6-(2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинол-4-ил)нафталин-1-карбонитрил;
2-метил-4-(5-метилнафтален-2-ил)-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(6-метоксинафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(6-хлорнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(6-фторнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
6-(2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинол-4-ил)нафталин-2-карбонитрил;
4-(6-метансульфонилнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(7-метоксинафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(7-хлорнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(7-фторнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
7-(2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинол-4-ил)нафталин-2-карбонитрил;
4-(8-метоксинафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(8-хлорнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
4-(8-фторнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин;
8-(2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинол-4-ил)нафталин-1-карбонитрил;
7-(2,6-диметилморфолин-4-ил)-2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин;
2-этил-7-(морфолин-4-ил)-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин;
8-фтор-2-метил-7-(морфолин-4-ил)-(4-нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
8-метокси-2-метил-7-(морфолин-4-ил)-(4-нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-7-морфолин-4-ил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-8-ол;
2,8-диметил-7-(морфолин-4-ил)-(4-нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
2-этил-8-фтор-7-(морфолин-4-ил)-(4-нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
6-фтор-2-метил-7-(морфолин-4-ил)-(4-нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
5-фтор-2-метил-7-(морфолин-4-ил)-(4-нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(морфолин-4-ил)-(4-нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин-4-ол;
2,4-диметил-7-(морфолин-4-ил)-(4-нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-нафтален-2-ил-7-пиперазин-1-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(4-метилпиперазин-1-ил)-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(нафтален-2-ил)-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(нафтален-2-ил)-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-7-(морфолин-4-ил)метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(нафтален-2-ил)-7-(1-метил-1-морфолин-4-илэтил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(нафтален-2-ил)-7-(1-морфолин-4-илциклопропил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(нафтален-2-ил)-7-(пиперидин-1-ил)метил-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(нафтален-2-ил)-7-(пирролидин-1-ил)метил-1,2,3,4-тетрагидроизохинолин;
диметил-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-илметил)амин;
2-метил-[1-(4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]диметиламин;
2-метил-[1-(4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]диметиламин;
метил-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-илметил)амин;
2-метил-[1-(4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]метиламин;
2-метил-[1-(4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]метиламин;
C-(2-метил-[1-(4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил))]метиламин;
1-(2-метил-[1-(4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил))]-1-метилэтиламин;
1-(2-метил-[1-(4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил))]циклопропиламин;
1-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)-1H-пиридин-2-он;
7-метансульфонил-2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин-7-карбонитрил;
8-фтор-2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин-7-карбонитрил;
2,8-диметил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(5,6,7,8-тетрагидронафтален-2-ил)-1,2,3,4-тетрагидроизохинолин;
или их оксид, их фармацевтически приемлемую соль, их сольват, или их пролекарство.
Кроме того, другие конкретные соединения согласно настоящему изобретению представляют собой соединения со следующими заместителями (таблица G):
где атом углерода, обозначенный *, находится в R- или S-конфигурации. То есть конкретные соединения в данном случае включают
4-(2H-хромен-3-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
2-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)хинолин;
3-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)хинолин;
7-метокси-2-метил-4-хинолин-6-ил-1,2,3,4-тетрагидроизохинолин-4-ол;
6-(7-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)хинолин;
2-метил-4-хинолин-6-ил-1,2,3,4-тетрагидроизохинолин-4-ол;
6-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)хинолин;
6-(2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ил)хинолин;
6-(2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ил)хинолин;
7-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)хинолин;
7-(2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ил)хинолин;
7-(2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ил)хинолин;
2'-метил-1',2',3',4'-тетрагидро-[3,4']биизохинолинил;
2-метил-1,2,3,4-тетрагидро[4,6']биизохинолинил;
2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидро[4,6']биизохинолинил;
2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидро[4,6']биизохинолинил;
2-метил-1,2,3,4-тетрагидро[4,7']биизохинолинил;
2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидро[4,7']биизохинолинил;
2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидро[4,7']биизохинолинил;
2-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)хиноксалин;
6-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)хиноксалин;
6-(2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ил)хиноксалин;
6-(2-метил-7-пиперидин-1-ил-1,2,3,4-тетрагидроизохинолин-4-ил)хиноксалин;
6-(2-метил-7-пирролидин-1-ил-1,2,3,4-тетрагидроизохинолин-4-ил)хиноксалин;
2-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)хиназолин;
6-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)хиназолин;
6-(2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ил)хиназолин;
6-(2-метил-7-пиперидин-1-ил-1,2,3,4-тетрагидроизохинолин-4-ил)хиназолин;
6-(2-метил-7-пирролидин-1-ил-1,2,3,4-тетрагидроизохинолин-4-ил)хиназолин;
7-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)хиназолин;
7-(2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ил)хиназолин;
7-(2-метил-7-пиперидин-1-ил-1,2,3,4-тетрагидроизохинолин-4-ил)хиназолин;
7-(2-метил-7-пирролидин-1-ил-1,2,3,4-тетрагидроизохинолин-4-ил)хиназолин;
6-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)фталазин;
6-(2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ил)фталазин;
6-(2-метил-7-пиперидин-1-ил-1,2,3,4-тетрагидроизохинолин-4-ил)фталазин;
6-(2-метил-7-пирролидин-1-ил-1,2,3,4-тетрагидроизохинолин-4-ил)фталазин;
3-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)циннолин;
6-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)циннолин;
6-(2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ил)циннолин;
6-(2-метил-7-пиперидин-1-ил-1,2,3,4-тетрагидроизохинолин-4-ил)циннолин;
6-(2-метил-7-пирролидин-1-ил-1,2,3,4-тетрагидроизохинолин-4-ил)циннолин;
7-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)циннолин;
7-(2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ил)циннолин;
7-(2-метил-7-пиперидин-1-ил-1,2,3,4-тетрагидроизохинолин-4-ил)циннолин;
7-(2-метил-7-пирролидин-1-ил-1,2,3,4-тетрагидроизохинолин-4-ил)циннолин;
4-(8,9-дигидро-7H-бензоциклогептен-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин;
или их оксид, их фармацевтически приемлемая соль, их сольват, или их пролекарство.
Приведены примеры нескольких оптически чистых соединений, полученных в данном изобретении (Таблица Н).
Другой вариант осуществления настоящего изобретения относится к смеси соединений формулы I, при этом соединение формулы I является радиоактивно меченным, т.е. один или несколько описанных атомов заменены радиоактивным изотопом данного атома (например C, замененный14C, и H, замененный3H). Такие соединения имеют множество возможных применений, например, в качестве стандартов и реагентов для определения способности потенциального фармацевтического средства связываться с белками-переносчиками нейромедиаторов.
Соединения согласно настоящему изобретению также представлены химической структурой, определяемой формулой I:
Формула I
где атом углерода, обозначенный *, находится в R- или S-конфигурации;
X означает конденсированный ароматический бициклический карбоцикл или гетероцикл, необязательно замещенный заместителями (в количестве от 1 до 4), которые определены ниже для R14, при условии, что X ≠ изохинолинилу, нафтилу или фталимидилу;
R1 означает H, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, каждый из которых необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, арила, -CN, -OR9 и -NR9R10;
R2 означает H, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил, C4-C7-циклоалкилалкил или C1-C6-галогеналкил, каждый из которых необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, арила, -CN, -OR9и -NR9R10; или
R2 означает гем-диметил;
R3 означает H, галоген, -OR11, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, -NR9R10, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9;
R4 означает H, галоген, -OR11, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, -NR9R10, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9; или
R4 означает фенил, нафтил, инденил, пиридил, пиримидинил, пиридазинил, пиразинил, [1,2,4]триазинил, [1,3,5]триазинил, триазолил, фуранил, тиофенил, пиранил, индазолил, бензимидазолил, хинолинил, хиназолинил, хиноксалинил, фталазинил, циннолинил, изохинолинил, тиенил, имидазолил, тиазолил, бензтиазолил, пуринил, изотиазолил, индолил, пирролил, оксазолил, бензофуранил, бензотиенил, бензтиазолил, изоксазолил, пиразолил, оксадиазолил, тиадиазолил, 3-оксо-[1,2,4]триазоло[4,3-a]пиридинил, имидазо[1,2-a]пиридинил, пиразоло[1,5-a]пиридинил, [1,2,4]триазоло[4,3-a]пиридинил, тиено[2,3-b]пиридинил, тиено[3,2-b]пиридинил, 1H-пирроло[2,3-b]пиридинил или другие гетероциклы, необязательно замещенные заместителями (в количестве от 1 до 4), которые определены ниже для R14;
R5 и R6, каждый независимо, выбраны из группы, состоящей из H, галогена, -OR11, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, -NR9R10, C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила, C4-C7-циклоалкилалкила и фенила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9;
R7 означает H, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9; или
R7 означает гем-диметил;
R8 означает H, галоген, -OR9, -SR9, C1-C6-алкил, -CN или -NR9R10;
R9 и R10, каждый независимо, выбраны из группы, состоящей из H, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксиалкила, C3-C6-циклоалкила, C4-C7-циклоалкилалкила, -C(O)R13, фенила и бензила, где фенил или бензил необязательно замещен 1-3 раза заместителем, выбранным независимо в каждом случае из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила и C1-C4-алкоксигруппы; или
R9 и R10, взятые вместе с атомом азота, с которым они связаны, образуют пиперидин, пирролидин, пиперазин, N-метилпиперазин, морфолин, тиоморфолин, [1,2]оксазинан, изоксазолидин или 2-оксо-2H-пиридин, который необязательно замещен 1-3 раза заместителем, выбранным независимо в каждом случае из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила и C1-C4-алкоксигруппы;
R11 означает H, C1-C4-алкил, C1-C4-галогеналкил, C1-C4-алкоксиалкил, C3-C6-циклоалкил, C4-C7-циклоалкилалкил, -C(O)R13, фенил или бензил, где фенил или бензил необязательно замещен 1-3 раза галогеном, цианогруппой, C1-C4-алкилом, C1-C4-галогеналкилом или C1-C4-алкоксигруппой;
R12 означает H, C1-C4-алкил, C1-C4-галогеналкил, C1-C4-алкоксиалкил, C3-C6-циклоалкил, C4-C7-циклоалкилалкил, фенил или бензил, где фенил или бензил необязательно замещен 1-3 раза галогеном, цианогруппой, C1-C4-алкилом, C1-C4-галогеналкилом или C1-C4-алкоксигруппой; или
R11 и R12, взятые вместе с атомом азота, с которым они связаны, образуют цикл пиперидина, пирролидина, пиперазина, N-метилпиперазина, морфолина или тиоморфолина при условии, что только одна комбинация из R9 и R10 или R11 и R12, взятых вместе с атомом азота, с которым они связаны, образует цикл пиперидина, пирролидина, пиперазина, N-метилпиперазина, морфолина или тиоморфолина;
R13 означает C1-C4-алкил, C1-C4-галогеналкил или фенил;
n равно 0, 1 или 2; и
R14 в каждом случае независимо выбран из заместителя, выбранного из группы, состоящей из галогена, -NO2, -OR11, -NR11R12, -NR11C(O)R12, -NR11C(O)2R12, -NR11C(O)NR12R13, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила, где C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил и C4-C7-циклоалкилалкил необязательно замещены 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, арила, -CN, -OR9 и -NR9R10;
или их оксидом, или их фармацевтически приемлемой солью.
Кроме того, соединения согласно настоящему изобретению представлены химической структурой, определяемой формулой I:
Формула I
где атом углерода, обозначенный *, находится в R- или S-конфигурации;
X означает конденсированный бициклический карбоцикл или гетероцикл, выбранный из группы, состоящей из бензофуранила, бензо[b]тиофенила, бензоизотиазолила, бензоизоксазолила, индазолила, индолила, изоиндолила, индолизинила, бензоимидазолила, бензооксазолила, бензотиазолила, бензотриазолила, имидазо[1,2-a]пиридинила, пиразоло[1,5-a]пиридинила, [1,2,4]триазоло[4,3-a]пиридинила, тиено[2,3-b]пиридинила, тиено[3,2-b]пиридинила, 1H-пирроло[2,3-b]пиридинила, инденила, инданила, дигидробензоциклогептенила, тетрагидробензоциклогептенила, дигидробензотиофенила, дигидробензофуранила, индолинила, нафтила, тетрагидронафтила, хинолинила, изохинолинила, 4H-хинолизинила, 9aH-хинолизинила, хиназолинила, циннолинила, фталазинила, хиноксалинила, бензо[1,2,3]триазинила, бензо[1,2,4]триазинила, 2H-хроменила, 4H-хроменила, и конденсированный бициклический карбоцикл или конденсированный бициклический гетероцикл, необязательно замещенный заместителями (в количестве от 1 до 4), которые определены ниже для R14; при условии, что (1) когда X = нафтилу и R4 означает NH2 или OR11, R5 не может быть H, и (2) когда X = нафтилу и R5 = OR11, R4 не может быть H;
R1 означает H, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, каждый из которых необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, арила, -CN, -OR9 и -NR9R10;
R2 означает H, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил, C4-C7-циклоалкилалкил или C1-C6-галогеналкил, каждый из которых необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, арила, -CN, -OR9и -NR9R10; или
R2 означает гем-диметил;
R3 означает H, галоген, -OR11, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, -NR9R10, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9;
R4 означает H, галоген, -OR11, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, -NR9R10, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9; или
R4 означает фенил, нафтил, инденил, пиридил, пиримидинил, пиридазинил, пиразинил, [1,2,4]триазинил, [1,3,5]триазинил, триазолил, фуранил, тиофенил, пиранил, индазолил, бензимидазолил, хинолинил, хиназолинил, хиноксалинил, фталазинил, циннолинил, изохинолинил, тиенил, имидазолил, тиазолил, бензтиазолил, пуринил, изотиазолил, индолил, пирролил, оксазолил, бензофуранил, бензотиенил, бензтиазолил, изоксазолил, пиразолил, оксадиазолил, тиадиазолил, 3-оксо-[1,2,4]триазоло[4,3-a]пиридинил, имидазо[1,2-a]пиридинил, пиразоло[1,5-a]пиридинил, [1,2,4]триазоло[4,3-a]пиридинил, тиено[2,3-b]пиридинил, тиено[3,2-b]пиридинил, 1H-пирроло[2,3-b]пиридинил или другие гетероциклы, необязательно замещенные заместителями (в количестве от 1 до 4), которые определены ниже для R14;
R5 и R6, каждый независимо, выбраны из группы, состоящей из H, галогена, -OR11, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, -NR9R10, C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила, C4-C7-циклоалкилалкила и фенила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9;
R7 означает H, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C4-C7-циклоалкилалкил, где каждый из C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, -CN, -OR9, -NR9R10 и фенила, который необязательно замещен 1-3 раза заместителем, выбранным из группы, состоящей из галогена, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксигруппы, -CN и -OR9; или
R7 означает гем-диметил;
R8 означает H, галоген, -OR9, -SR9, C1-C6-алкил, -CN или -NR9R10;
R9 и R10, каждый независимо, выбраны из группы, состоящей из H, C1-C4-алкила, C1-C4-галогеналкила, C1-C4-алкоксиалкила, C3-C6-циклоалкила, C4-C7-циклоалкилалкила, -C(O)R13, фенила и бензила, где фенил или бензил необязательно замещен 1-3 раза заместителем, выбранным независимо в каждом случае из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила и C1-C4-алкоксигруппы; или
R9 и R10, взятые вместе с атомом азота, с которым они связаны, образуют пиперидин, пирролидин, пиперазин, N-метилпиперазин, морфолин, тиоморфолин, [1,2]оксазинан, изоксазолидин или 2-оксо-2H-пиридин, который необязательно замещен 1-3 раза заместителем, выбранным независимо в каждом случае из группы, состоящей из галогена, цианогруппы, C1-C4-алкила, C1-C4-галогеналкила и C1-C4-алкоксигруппы;
R11 означает H, C1-C4-алкил, C1-C4-галогеналкил, C1-C4-алкоксиалкил, C3-C6-циклоалкил, C4-C7-циклоалкилалкил, -C(O)R13, фенил или бензил, где фенил или бензил необязательно замещен 1-3 раза галогеном, цианогруппой, C1-C4-алкилом, C1-C4-галогеналкилом или C1-C4-алкоксигруппой;
R12 означает H, C1-C4-алкил, C1-C4-галогеналкил, C1-C4-алкоксиалкил, C3-C6-циклоалкил, C4-C7-циклоалкилалкил, фенил или бензил, где фенил или бензил необязательно замещен 1-3 раза галогеном, цианогруппой, C1-C4-алкилом, C1-C4-галогеналкилом или C1-C4-алкоксигруппой; или
R11 и R12, взятые вместе с атомом азота, с которым они связаны, образуют цикл пиперидина, пирролидина, пиперазина, N-метилпиперазина, морфолина или тиоморфолина, при условии, что только одна комбинация из R9 и R10 или R11 и R12, взятых вместе с атомом азота, с которым они связаны, образует цикл пиперидина, пирролидина, пиперазина, N-метилпиперазина, морфолина или тиоморфолина;
R13 означает C1-C4-алкил, C1-C4-галогеналкил или фенил;
n равно 0, 1 или 2; и
R14 в каждом случае независимо выбран из заместителя, выбранного из группы, состоящей из галогена, -NO2, -OR11, -NR11R12, -NR11C(O)R12, -NR11C(O)2R12, -NR11C(O)NR12R13, -S(O)nR12, -CN, -C(O)R12, -C(O)NR11R12, C1-C6-алкила, C2-C6-алкенила, C2-C6-алкинила, C3-C6-циклоалкила и C4-C7-циклоалкилалкила, где C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил и C4-C7-циклоалкилалкил необязательно замещены 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-C3-алкила, галогена, арила, -CN, -OR9 и -NR9R10;
или их оксидом или их фармацевтически приемлемой солью.
Другой вариант осуществления настоящего изобретения относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения формулы I и фармацевтически приемлемый носитель.
Другой аспект настоящего изобретения относится к способу лечения расстройства, которое возникает в результате или зависит от пониженной доступности серотонина, норэпинефрина или допамина. Способ заключается во введении пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли.
В другом варианте способ согласно настоящему изобретению, кроме того, включает в себя введение терапевтически эффективного количества антагониста рецептора серотонина 1A или его фармацевтически приемлемой соли.
Антагонистом рецептора серотонина 1A может быть WAY 100135 или спиперон. WAY 100135 (N-(трет-бутил)-3-[α-(2-метоксифенил)пиперазин-1-ил]-2-фенилпропанамид) описан в патенте США No. 4988814 Abou-Gharbia et al., который включен в данное описание в виде ссылки в полном объеме как обладающий аффинностью к рецептору 5-HT1A. Также в публикации Cliffe et al., J. Med. Chem. 36: 1509-10 (1993), которая включена в данное описание в виде ссылки в полном объеме, показано, что соединение является антагонистом 5-HT1A. Спиперон (8-[4-(4-фторфенил)-4-оксобутил]-1-фенил-1,3,8-триазаспиро[4,5]декан-4-он) является хорошо известным соединением и описан в патентах США No. 3155669 и 3155670, которые включены в данное описание в виде ссылки в полном объеме. Активность спиперона как антагониста 5-HT1A показана в публикации Middlemiss et al., Neurosc. and Biobehav. Rev. 16: 75-82 (1992), которая включена в данное описание в виде ссылки в полном объеме.
В другом варианте способ согласно настоящему изобретению дополнительно включает в себя введение терапевтически эффективного количества избирательного антагониста рецептора нейрокинина-1 или его фармацевтически приемлемой соли.
Антагонисты рецептора нейрокинина-1, которые могут быть использованы в комбинации с соединением формулы I в настоящем изобретении, полно описаны, например, в патентах США No. 5373003, 5387595, 5459270, 5494926, 5162339, 5232929, 5242930, 5496833 и 5637699; в международных публикациях патентов PCT No. WO 90/05525, 90/05729, 94/02461, 94/02595, 94/03429, 94/03445, 94/04494, 94/04496, 94/05625, 94/07843, 94/08997, 94/10165, 94/10167, 94/10168, 94/10170, 94/11368, 94/13639, 94/13663, 94/14767, 94/15903, 94/19320, 94/19323, 94/20500, 91/09844, 91/18899, 92/01688, 92/06079, 92/12151, 92/15585, 92/17449, 92/20661, 92/20676, 92/21677, 92/22569, 93/00330, 93/00331, 93/01159, 93/01165, 93/01169, 93/01170, 93/06099, 93/09116, 93/10073, 93/14084, 93/14113, 93/18023, 93/19064, 93/21155, 93/21181, 93/23380, 93/24465, 94/00440, 94/01402, 94/26735, 94/26740, 94/29309, 95/02595, 95/04040, 95/04042, 95/06645, 95/07886, 95/07908, 95/08549, 95/11880, 95/14017, 95/15311, 95/16679, 95/17382, 95/18124, 95/18129, 95/19344, 95/20575, 95/21819, 95/22525, 95/23798, 95/26338, 95/28418, 95/30674, 95/30687, 95/33744, 96/05181, 96/05193, 96/05203, 96/06094, 96/07649, 96/10562, 96/16939, 96/18643, 96/20197, 96/21661, 96/29304, 96/29317, 96/29326, 96/29328, 96/31214, 96/32385, 96/37489, 97/01553, 97/01554, 97/03066, 97/08144, 97/14671, 97/17362, 97/18206, 97/19084, 97/19942, 97/21702 и 97/49710; заявках на выдачу патента Великобритании No. 2266529, 2268931, 2269170, 2269590, 2271774, 2292144, 2293168, 2293169 и 2302689; публикациях европейских патентов No. 0360390, 0517589, 0520555, 0522808, 0528495, 0532456, 0533280, 0536817, 0545478, 0558156, 0577394, 0585913, 0590152, 0599538, 0610793, 0634402, 0686629, 0693489, 0694535, 0699655, 0394989, 0428434, 0429366, 0430771, 0436334, 0443132, 0482539, 0498069, 0499313, 0512901, 0512902, 0514273, 0514274, 0514275, 0514276, 0515681, 0699674, 0707006, 0708101, 0709375, 0709376, 0714891, 0723959, 0733632 и 0776893, которые включены в данное описание в виде ссылки в полном объеме. Получение таких соединений полно описано в указанных выше патентах и публикациях.
В другом варианте способ согласно настоящему изобретению дополнительно включает в себя введение терапевтически эффективного количества предшественника норэпинефрина или его фармацевтически приемлемой соли.
Предшественник норэпинефрина может представлять собой L-тирозин или L-фенилаланин.
Другой аспект настоящего изобретения относится к способу ингибирования синаптического захвата норэпинефрина у нуждающегося в этом пациента, включающему в себя введение терапевтически эффективного ингибирующего количества соединения формулы I.
Другой аспект настоящего изобретения относится к способу ингибирования синаптического захвата серотонина у нуждающегося в этом пациента, включающему в себя введение терапевтически эффективного ингибирующего количества соединения формулы I.
Другой аспект настоящего изобретения относится к способу ингибирования синаптического захвата допамина у нуждающегося в этом пациента, включающему в себя введение терапевтически эффективного ингибирующего количества соединения формулы I.
Другой аспект настоящего изобретения относится к терапевтическому способу, описанному в данной публикации, в котором используют (+)-стереоизомер соединения формулы I.
Другой аспект настоящего изобретения относится к терапевтическому способу, описанному в данной публикации, в котором используют (-)-стереоизомер соединения формулы I.
Другой аспект настоящего изобретения относится к набору, содержащему соединение формулы I и, по меньшей мере, одно соединение, выбранное из группы, состоящей из соединения антагониста рецептора серотонина 1A, соединения избирательного антагониста рецептора нейрокинина-1 и соединения предшественника норэпинефрина.
Другой аспект настоящего изобретения относится к способу лечения расстройства, указанного в описанных выше вариантах, у нуждающегося в этом пациента, включающему в себя ингибирование синаптического захвата серотонина и норэпинефрина посредством введения терапевтически эффективного ингибирующего количества соединения формулы I, которое функционирует как ингибитор захвата и серотонина и норэпинефрина двойного действия.
Другой аспект настоящего изобретения относится к способу лечения расстройства, указанного в описанных выше вариантах, у нуждающегося в этом пациента, включающему в себя ингибирование синаптического захвата серотонина и допамина посредством введения терапевтически эффективного ингибирующего количества соединения формулы I, которое функционирует как ингибитор захвата серотонина и допамина двойного действия.
Другой аспект настоящего изобретения относится к способу лечения расстройства, указанного в описанных выше вариантах, у нуждающегося в этом пациента, включающему в себя ингибирование синаптического захвата допамина и норэпинефрина посредством введения терапевтически эффективного ингибирующего количества соединения формулы I, которое функционирует как ингибитор захвата как допамина, так и норэпинефрина двойного действия.
Другой аспект настоящего изобретения относится к способу лечения расстройства, указанного в описанных выше вариантах, у нуждающегося в этом пациента, включающему в себя ингибирование синаптического захвата норэпинефрина, допамина и серотонина посредством введения терапевтически эффективного ингибирующего количества соединения формулы I, которое функционирует как ингибитор захвата норэпинефрина, допамина и серотонина тройного действия.
Другой аспект настоящего изобретения относится к способу ингибирования захвата серотонина у млекопитающих, который заключается во введении млекопитающему, которому необходима повышенная нейротрансмиссия серотонина, фармацевтически эффективного количества соединения формулы I.
Другой аспект настоящего изобретения относится к способу ингибирования захвата допамина у людей, который заключается во введении человеку, которому требуется повышенная нейротрансмиссия допамина, фармацевтически эффективного количества соединения формулы I.
Другой аспект настоящего изобретения относится к способу ингибирования захвата норэпинефрина у людей, который заключается во введении человеку, которому требуется повышенная нейротрансмиссия норэпинефрина, фармацевтически эффективного количества соединения формулы I.
Другой аспект настоящего изобретения относится к способу подавления у людей желания курить, включающему в себя введение человеку, нуждающемуся в таком подавлении, эффективной для ослабления желания курить дозы соединения формулы I.
Другой аспект настоящего изобретения относится к способу подавления у людей желания потреблять алкоголь, включающему в себя введение человеку, нуждающемуся в таком подавлении, эффективной для ослабления желания потреблять алкоголь дозы соединения формулы I.
Понятно, что некоторые признаки изобретения, которые для ясности описаны в контексте отдельных вариантов осуществления, также могут быть представлены в комбинации в одном варианте осуществления изобретения. Наоборот, различные признаки изобретения, которые для краткости описаны в контексте одного варианта осуществления, также могут быть представлены по отдельности или в любой подходящей подкомбинации.
Соединения согласно изобретению, например исходные вещества, промежуточные соединения или продукты, получают, как описано в данной публикации, или в результате применения, или адаптирования известных способов, под которыми подразумевают способы, применяемые ранее или описанные в литературе.
Соединения, применимые согласно изобретению, могут быть получены в результате применения или адаптирования известных способов, под которыми подразумевают способы, применяемые ранее или описанные в литературе, например способы, описанные Larock, Comprehensive Organic Transformations, VCH publishers (1989), публикация включена в данное описание в виде ссылки в полном объеме.
Соединение формулы I, включающее в себя группу, содержащую один или несколько атомов азота в цикле, может быть превращено в соответствующее соединение, в котором один или несколько входящих в цикл атомов азота группы окисляют до N-оксида, предпочтительно при взаимодействии с перкислотой, например перуксусной кислотой в уксусной кислоте или мета-хлорпероксибензойной кислотой в инертном растворителе, таком как дихлорметан, при температуре от примерно комнатной температуры до температуры кипячения с обратным холодильником, предпочтительно при повышенной температуре.
В реакциях, описанных ниже, может быть необходима защита химически активных функциональных групп, например гидрокси-, амино-, имино-, тио- или карбоксигрупп, в том случае, когда указанные группы требуются в конечном продукте, чтобы избежать их нежелательного участия в реакциях. Можно использовать обычные защитные группы согласно стандартной практике; например, см. Green, Protective Groups in Organic Chemistry, John Wiley and Sons (1991), и McOmie, Protective Groups in Organic Chemistry, Plenum Press (1973), которые включены в данное описание в виде ссылки в полном объеме.
Новые тетрагидроизохинолиновые ингибиторы обратного захвата формулы I согласно данному изобретению могут быть получены согласно общей схеме, изображенной ниже (схема 1). R1-замещенные N-бензиламины формулы III могут быть приобретены из коммерческих источников или, альтернативно, получены на основе простого протокола восстановительного аминирования. Таким образом, карбонилсодержащие соединения формулы II могут быть обработаны H2N-R1 в низших алкиловых спиртовых растворителях (предпочтительно в метаноле или этаноле) при температурах, равных или ниже комнатной температуры. Полученный в результате имин может быть в большинстве случаев восстановлен боргидридами щелочноземельных металлов (предпочтительно боргидридом натрия) с получением требуемых промежуточных аминов.
Схема 1

При обработке промежуточных продуктов формулы III промежуточными продуктами формулы V точно образуются продукты алкилирования формулы VI. Реакции алкилирования могут быть осуществлены во множестве разнообразных условий, известных специалисту в области органического синтеза. Типичные растворители включают ацетонитрил, толуол, диэтиловый эфир, тетрагидрофуран, диметилсульфоксид, диметилформамид, метиленхлорид и низшие алкиловые спирты, включая этанол. Реакции могут быть успешно осуществлены при температурах в диапазоне от 0°C вплоть до точки кипения используемого растворителя. Ход реакции обычно контролируют стандартными хроматографическими и спектроскопическими способами. Реакцию алкилирования необязательно осуществляют с добавлением ненуклеофильного органического основания, такого как, но без ограничения, пиридин, триэтиламин и диизопропилэтиламин.
Указанный выше промежуточный продукт формулы V может быть приобретен из коммерческих источников или получен обработкой необязательно замещенного кетона формулы IV общепринятыми бромирующими агентами, такими как, но без ограничения, бром, NBS или трибромид тетрабутиламмония, который легко дает требуемые бромацетофеноны формулы V. Указанные реакции оптимально проводят в уксусной кислоте или метиленхлориде с метанолом, используемым в качестве сорастворителя для реагента трибромида, при температурах реакции, равных или ниже комнатной температуры. Другой вариант осуществления указанной методики может включать в себя применение соединений хлорацетофенона формулы V.
Кетоны формулы IV также доступны из коммерческих источников или их легко получают несколькими хорошо известными способами, включая обработку соответствующих промежуточных ароматических или гетероароматических карбоновых кислот двумя стехиометрическими эквивалентами метиллития (см., например, Jorgenson, Organic Reactions, 18: 1 (1970), публикация включена в данное описание в виде ссылки в полном объеме). Альтернативно можно обработать соответствующие ароматические или гетероароматические альдегиды нуклеофильным алкилмагнийгалогенидом Гриньяра (например, MeMgBr) или алкиллитием (например, MeLi) с последующим обычным окислением до кетона (см., например, Larock, Comprehensive Organic Transformations, VCH Publishers, New York, p. 604 (1989), публикация включена в данное описание в виде ссылки в полном объеме).
Восстановление соединений формулы VI до вторичных спиртов формулы VII происходит со многими восстановителями, включая, например, боргидрид натрия, боргидрид лития, боран, гидрид диизобутилалюминия и алюмогидрид лития. Восстановление осуществляют в течение периода времени от 1 часа до 3 суток при комнатной температуре или повышенной температуре вплоть до температуры кипячения с обратным холодильником используемого растворителя. Если используют боран, то он может быть использован в виде комплекса, например, но без ограничения, комплекса боран-метилсульфид, комплекса боран-пиперидин или комплекса боран-тетрагидрофуран. Специалисту в данной области будет понятна необходимая оптимальная комбинация восстановителей и условий реакции или он может найти инструкции в тексте Larock, R. C, Comprehensive Organic Transformations, VCH Publishers, New York, p. 604 (1989), который включен в данное описание в виде ссылки в полном объеме.
Соединения формулы VII могут быть циклизованы до соединений тетрагидроизохинолина формулы VIII согласно данному изобретению кратковременной обработкой сильной кислотой. Подходящие кислоты включают, но не ограничены указанным, концентрированную серную кислоту, полифосфорную кислоту, метансульфоновую кислоту и трифторуксусную кислоту. Реакции проводят в чистом виде или необязательно в присутствии сорастворителя, такого как, например, метиленхлорид или 1,2-дихлорэтан. Циклизация может быть проведена при температурах в диапазоне от 0°C вплоть до температуры кипячения с обратным холодильником используемого растворителя. Специалисту в области химии гетероциклов будут легко понятны такие условия или можно получить справку в руководствах Mondeshka, Il Farmaco, 49: 475-480 (1994) и Venkov, Synthesis, 253-255 (1990), которые включены в данное описание в виде ссылки в полном объеме. Циклизацию также можно осуществить обработкой соединений формулы VII сильными кислотами Льюиса, такими как трихлорид алюминия, обычно в галогенированных растворителях, таких как метиленхлорид. Специалисту в данной области будет известен прецедент, описанный Kaiser, J. Med. Chem. 27: 28-35 (1984) и Wyrick, J. Med. Chem. 24: 1013-1015 (1981), публикации включены в данное описание в виде ссылки в полном объеме.
Наконец, целевые соединения формулы I согласно данному изобретению могут быть получены обработкой соединений формулы (VIII; Y = Br, I, OSO2CF3) арил- или гетероарилбороновыми кислотами или сложными эфирами арил- или гетероарилбороновых кислот, при этом Z эквивалентно B(OH)2 или B(ORa)(ORb) (где Ra и Rb представляют собой низший алкил, т.е. C1-C6, или вместе взятые Ra и Rb представляют собой низший алкилен, т.е. C2-C12) в присутствии металлического катализатора с основанием или без основания в инертном растворителе с получением соединений изохинолина формулы XIII. Металлические катализаторы включают без ограничения соли или фосфиновые комплексы Cu, Pd или Ni (например, Cu(OAc)2, PdCl2 (PPh3)2 и NiCl2 (PPh3)2). Основания могут включать без ограничения карбонаты щелочноземельных металлов, бикарбонаты щелочноземельных металлов, гидроксиды щелочноземельных металлов, карбонаты щелочных металлов, бикарбонаты щелочных металлов, гидроксиды щелочных металлов, гидриды щелочных металлов (предпочтительно гидрид натрия), алкоксиды щелочных металлов (предпочтительно метоксид натрия или этоксид натрия), гидриды щелочноземельных металлов, диалкиламиды щелочных металлов (предпочтительно диизопропиламид лития), бис(триалкилсилил)амиды щелочных металлов (предпочтительно бис(триметилсилил)амид натрия), триалкиламины (предпочтительно диизопропилэтиламин или триэтиламин) или ароматические амины (предпочтительно пиридин). Инертные растворители могут включать, но без ограничения, ацетонитрил, простые диалкиловые эфиры (предпочтительно диэтиловый эфир), циклические эфиры (предпочтительно тетрагидрофуран или 1,4-диоксан), N,N-диалкилацетамиды (предпочтительно диметилацетамид), N,N-диалкилформамиды (предпочтительно диметилформамид), диалкилсульфоксиды (предпочтительно диметилсульфоксид), ароматические углеводороды (предпочтительно бензол или толуол) или галогеналканы (предпочтительно метиленхлорид). Предпочтительные температуры реакции находятся в диапазоне от комнатной температуры до точки кипения используемого растворителя. Реакции могут быть осуществлены в обычной стеклянной посуде или в одной из многих коммерчески доступных установок для параллельного синтеза. Недоступные из коммерческих источников бороновые кислоты или сложные эфиры бороновых кислот могут быть получены из соответствующего необязательно замещенного арилгалогенида, как описано Gao, Tetrahedron, 50: 979-988 (1994), публикация включена в данное описание в виде ссылки в полном объеме. Также специалисту в данной области будет понятно, что соединения формулы VIII могут быть превращены в бороновую кислоту или сложный эфир бороновой кислоты и затем обработаны требуемым необязательно замещенным арил- или гетероарилгалогенидом ступенчато или последовательно, как описано в Baudoin, J. Org. Chem. 67: 1199-1207 (2002), публикация включена в данное описании в полном объеме.
Соединения формулы VI могут быть обработаны реагентом C1-C4-алкиллитием или C1-C4-алкиловым реагентом Гриньяра. Полученные в результате третичные спирты затем могут быть превращены в соединения формулы VIII, где R8 означает соответствующий C1-C4-алкил, затем в соединения формулы I, где R8 означает соответствующий C1-C4-алкил, используя указанные выше способы.
Соединения формулы I также могут быть получены согласно схеме, изображенной ниже (схема 2). 4-замещенные изохинолины формулы IX могут быть приобретены из коммерческих источников. Обработка соединений формулы IX (обычно Y = Br) бициклической карбоциклической или бициклической гетероциклической бороновой кислотой или бициклическим карбоциклическим или бициклическим гетероциклическим сложным эфиром бороновой кислоты, где Z эквивалентно B(OH)2 или B(ORa)(ORb) (где Ra и Rb означают низший алкил, т.е. C1-C6, или вместе взятые Ra и Rb означают низший алкилен, т.е. C2-C12) в присутствии металлического катализатора с основанием или без основания в инертном растворителе дает соединения изохинолина соединения формулы X. Металлические катализаторы включают, но без ограничения, соли или фосфиновые комплексы Cu, Pd или Ni (например, Cu(OAc)2, PdCl2(PPh3)2 и NiCl2(PPh3)2). Основания могут включать, но без ограничения, карбонаты щелочноземельных металлов, бикарбонаты щелочноземельных металлов, гидроксиды щелочноземельных металлов, карбонаты щелочных металлов, бикарбонаты щелочных металлов, гидроксиды щелочных металлов, гидриды щелочных металлов (предпочтительно гидрид натрия), алкоксиды щелочных металлов (предпочтительно метоксид натрия или этоксид натрия), гидриды щелочноземельных металлов, диалкиламиды щелочных металлов (предпочтительно диизопропиламид лития), бис(триалкилсилил)амиды щелочных металлов (предпочтительно бис(триметилсилил)амид натрия), триалкиламины (предпочтительно диизопропилэтиламин или триэтиламин) или ароматические амины (предпочтительно пиридин). Инертные растворители могут включать, но без ограничения, ацетонитрил, простые диалкиловые эфиры (предпочтительно диэтиловый эфир), циклические простые эфиры (предпочтительно тетрагидрофуран или 1,4-диоксан), N,N-диалкилацетамиды (предпочтительно диметилацетамид), N,N-диалкилформамиды (предпочтительно диметилформамид), диалкилсульфоксиды (предпочтительно диметилсульфоксид), ароматические углеводороды (предпочтительно бензол или толуол) или галогеналканы (предпочтительно метиленхлорид). Предпочтительные температуры реакции находятся в диапазоне от комнатной температуры до точки кипения используемого растворителя. Реакции могут быть осуществлены в обычной стеклянной посуде или в одной из многих коммерчески доступных установок для параллельного синтеза. Недоступные из коммерческих источников бороновые кислоты или сложные эфиры бороновых кислот могут быть получены из соответствующего необязательно замещенного арилгалогенида, как описано Gao, Tetrahedron, 50: 979-988 (1994), публикация включена в данное описание в виде ссылки в полном объеме. Также специалисту в данной области будет понятно, что соединения формулы IX могут быть превращены в соответствующую бороновую кислоту или сложный эфир бороновой кислоты и затем обработаны требуемым необязательно замещенным арил- или гетероарилгалогенидом ступенчато или последовательно, как описано в Baudoin, J. Org. Chem. 67: 1199-1207 (2002), публикация включена в данное описание в полном объеме с получением требуемых изохинолинов формулы X.
Обработка промежуточных продуктов формулы X подходящим алкилирующим реагентом R1-W, где W может быть эквивалентным, но без ограничения, I, Br, -OSO2CF3 (предпочтительно -OSO2CF3) дает изохинолиний формулы XI. Подходящие растворители включают, но без ограничения, метиленхлорид, дихлорэтан, толуол, диэтиловый эфир и тетрагидрофуран. Реакции могут быть успешно осуществлены при температурах, равных или ниже комнатной температуры. Реакции могут быть осуществлены в обычной стеклянной посуде или в одной из многих коммерчески доступных установок для параллельного синтеза.
Схема 2

Соединения формулы XI могут быть восстановлены до тетрагидроизохинолина в большинстве случаев цианоборгидридом натрия с получением соединения формулы I. Подходящие растворители для данной реакции включают, но без ограничения, низшие спирты, включая метанол и этанол. Реакции могут быть осуществлены при температурах в диапазоне от 0°C до точки кипения используемого растворителя.
Соединения формулы I, где R8 = OH, согласно данному изобретению могут быть получены в соответствии с общей схемой, изображенной ниже (схема 3). R1-замещенный кетон формулы XIV может быть получен с использованием способа, описанного Hanna, J. Med. Chem. 17(9): 1020-1023 (1974), публикация включена в данное описание в виде ссылки в полном объеме. Таким образом, сложные бис-эфиры формулы XII легко получают из коммерчески доступных, необязательно замещенных орто-метилбензойных кислот или орто-метил/этилбензоата, используя несколько хорошо известных способов, таких как без ограничения этерификация, бромирование, N-алкилирование, конденсация типа конденсации Клайзена и декарбоксилирование. Соединения формулы XIV легко могут быть превращены специалистом в данной области в соединения формулы I, где R8 = OH, обработкой реагентом Гриньяра с формулой X-MgBr или литиевым реагентом с формулой X-Li.
Схема 3

Кроме того, соединения формулы I, в которых R8 = OH, могут быть легко алкилированы (см. выше) с получением соединений формулы I, где R8 = OR11. Обработка соединений формулы I, в которых R8 = OH, фторирующим реагентом, таким как, но без ограничения, диэтиламиносератрифторид (DAST), легко дает соединения формулы I, в которых R8 = F.
Обработка соединений формулы I, в которых R8 = OH, хлорирующим реагентом, таким как, но без ограничения, тионилхлорид или трихлорид фосфора, может давать соединения формулы I, в которых R8 = Cl. Дополнительные ссылки можно найти в обзоре Hudlicky, Organic Reactions 35: 513-637 (1985), который включен в данное описание в виде ссылки в полном объеме. Обработка соединений формулы I, в которых R8 = OH, реагентов цианирования, таким как, но без ограничения, цианид натрия или цианид триметилсилила, может давать соединения формулы I, в которых R8 = CN.
Соединения формулы I, в которых R8 = OH, согласно данному изобретению также могут быть получены в соответствии с руководствами Kihara, Tetrahedron 48: 67-78 (1992) и Blomberg, Synthesis, p. 18-30, (1977), которые включены в данное описание в виде ссылки в полном объеме. Таким образом кетоновые соединения формулы VI, которые имеют орто-йодид, могут быть обработаны сильными основаниями, такими как, но без ограничения, основания низший алкил(C1-C6)-лития (предпочтительно t-BuLi или n-BuLi), чтобы получить ожидаемый обмен галоген-металл с последующей внутримолекулярной циклизацией Барбье с образованием соединений формулы I, где R8 = OH. Необходимы инертные растворители, такие как диалкиловые эфиры (предпочтительно диэтиловый эфир), циклические эфиры (предпочтительно тетрагидрофуран или 1,4-диоксан), и поддерживают низкие температуры реакции (от -78 до -25°C), чтобы избежать побочных продуктов. Альтернативно обмен галоген-металл также может быть осуществлен в присутствии никеля с нулевой валентностью, и в данном случае N,N-диалкилформамиды (предпочтительно диметилформамид) служат в качестве идеальных растворителей.
Соединения формулы I, в которых R8 = OH, обычно превращают в соединения формулы I, в которых R8 = H, с помощью дегидратации с последующим восстановлением таким восстановителем, как, но без ограничения, цианоборгидрид натрия.
В другом варианте осуществления настоящего изобретения соединения формулы I, в которых R7 = H, могут быть получены из соединений формулы XV, как изображено ниже (схема 4). Соединения формулы XV получают из коммерческих источников или они могут быть легко получены хорошо известными способами. Превращение фенилуксусных кислот формулы XV в соответствующие ацилхлориды может быть достигнуто с использованием обычных реагентов, таких как, но без ограничения, тионилхлорид или оксалилхлорид, в таких растворителях, как, но без ограничения, толуол. Образованный таким образом ацилхлорид может быть легко трансформирован без отделения от реакционной смеси в амиды формулы XVI при обработке H2N-R1.
Схема 4

Превращение амидов формулы XVI в дигидроизохинолиноны формулы XVII происходит с альдегидами R2CHO в присутствии кислой среды, такой как, но без ограничения, полифосфорная кислота, пирофосфорная кислота или реагент Итона (пентоксид фосфора, раствор 7,7% мас. в метансульфоновой кислоте).
Обработка дигидроизохинолинонов формулы XVII соединением формулы X-галогенид (предпочтительно бромид) или соединением формулы X-OSO2CF3 в присутствии металлического катализатора с использованием основания в инертном растворителе дает дигидроизохинолиноны формулы XVIII. Металлические катализаторы могут включать без ограничения соли или фосфиновые комплексы Cu, Pd или Ni [например, Pd(OAc)2, CuI, PdCl2(PPh3)2, PdCl2dppf, NiCl2(PPh3)2]. Предпочтительные металлические катализаторы могут быть созданы in situ смешиванием солей металлов (предпочтительно Pd(OAc)2) с фосфиновыми лигандами (предпочтительно 2,8,9-три-изобутил-2,5,8,9-тетрааза-1-фосфабицикло[3.3.3]ундекан, 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил или 2,2'-бис(дифенилфосфино)-1,1'-бинафтил). Основания могут включать, но без ограничения, карбонаты щелочноземельных металлов, бикарбонаты щелочноземельных металлов, гидроксиды щелочноземельных металлов, карбонаты щелочных металлов, бикарбонаты щелочных металлов, фосфат щелочного металла, гидроксиды щелочных металлов, гидриды щелочных металлов, алкоксиды щелочных металлов (предпочтительно трет-бутоксид натрия), гидриды щелочноземельных металлов, диалкиламиды щелочных металлов, бис(триалкилсилил)амиды щелочных металлов (предпочтительно бис(триметилсилил)амид натрия), алкиламины (предпочтительно диизопропилэтиламин или триэтиламин) или ароматические основания (предпочтительно пиридин). Инертные растворители могут включать, но без ограничения, ацетонитрил, циклические эфиры (предпочтительно тетрагидрофуран или 1,4-диоксан), N,N-диалкилацетамиды (предпочтительно диметилацетамид), N,N-диалкилформамиды (предпочтительно диметилформамид), диалкилсульфоксиды (предпочтительно диметилсульфоксид), ароматические углеводороды (предпочтительно толуол). Предпочтительные температуры реакции находятся в диапазоне от комнатной температуры до точки кипения используемого растворителя. Реакции могут быть осуществлены в обычной стеклянной посуде или в одной из многих коммерчески доступных установок для параллельного синтеза.
Восстановление дигидроизохинолинонов формулы XVIII до соединений формулы I происходит с восстановителями, включая, например, но без ограничения, боргидрид натрия, боргидрид лития, боран, гидрид диизобутилалюминия и алюмогидрид лития. Восстановление осуществляют при температуре от 0°C до повышенной температуры вплоть до точки кипения используемого растворителя. Если используют боран, то он может быть использован в виде комплекса, например, но без ограничения, комплекса боран-метилсульфид, комплекса боран-пиперидин или комплекса боран-тетрагидрофуран. Специалистам в данной области будет понятна оптимальная комбинация восстановителей и необходимых условий реакции или они могут найти инструкции в тексте Larock, R. C, Comprehensive Organic Transformations, VCH Publishers, New York, p. 604 (1989), который включен в данное описание в виде ссылки в полном объеме.
Соединения формулы I, где R4 означает арил, гетероарил или NR9R10 и R7 = H, могут быть получены с использованием сходной методики, описанной на схеме 4, или как изображено на схеме 5.
Схема 5

Соединения формулы (XIX; V = Br, Cl или OSO2CF3) могут быть получены из коммерческих источников и легко превращены в соединения формулы XXIII через химические стадии, которые описаны для превращения соединений формулы XV в соединения формулы I на схеме 4. Целевые соединения формулы I согласно данному изобретению могут быть получены обработкой соединений формулы XXIII арил- или гетероарилбороновыми кислотами, или сложными эфирами арил-, гетероарилбороновых кислот, или HNR9R10 в присутствии металлического катализатора с основанием или без основания в инертном растворителе. Металлические катализаторы могут включать, но без ограничения, соли или фосфиновые комплексы Cu, Pd или Ni [например, Pd(OAc)2, CuI, PdCl2(PPh3)2, PdCl2dppf и NiCl2(PPh3)2]. Предпочтительные металлические катализаторы могут быть созданы in situ смешиванием солей металлов (предпочтительно Pd(OAc)2) с фосфиновыми лигандами. Основания могут включать, но без ограничения, карбонаты щелочноземельных металлов, бикарбонаты щелочноземельных металлов, гидроксиды щелочноземельных металлов, карбонаты щелочных металлов, бикарбонаты щелочных металлов, фосфат щелочного металла, гидроксиды щелочных металлов, гидриды щелочных металлов, алкоксиды щелочных металлов, гидриды щелочноземельных металлов, диалкиламиды щелочных металлов, бис(триалкилсилил)амиды щелочных металлов (предпочтительно бис(триметилсилил)амид натрия), триалкиламины (предпочтительно диизопропилэтиламин или триэтиамин) или ароматические амины (предпочтительно пиридин). Инертные растворители могут включать, но без ограничения, ацетонитрил, циклические эфиры (предпочтительно тетрагидрофуран или 1,4-диоксан), N,N-диалкилацетамиды (предпочтительно диметилацетамид), N,N-диалкилформамиды (предпочтительно диметилформамид), диалкилсульфоксиды (предпочтительно диметилсульфоксид), ароматические углеводороды (предпочтительно толуол). Предпочтительные температуры реакции находятся в диапазоне от комнатной температуры до точки кипения используемого растворителя. Реакции могут быть осуществлены в обычной стеклянной посуде или в одной из многих коммерчески доступных установок для параллельного синтеза.
Альтернативно соединения формулы XXIII могут быть превращены в соответствующие бороновые кислоты или сложные эфиры бороновых кислот, при этом V = B(OH)2 или B(ORa)(ORb) (где Ra и Rb означают низший алкил, т.е. C1-C6, или вместе взятые Ra и Rb представляют собой низший алкилен, т.е. C2-C12) с использованием хорошо известных способов. Обработка указанных образованных таким образом бороновых кислот или сложных эфиров бороновых кислот арил- или гетероарилгалогенидом или арил- или гетероарилтрифлатом в присутствии металлического катализатора с основанием или без основания в инертном растворителе может давать соединения формулы I. Металлические катализаторы могут включать, но без ограничения, соли или фосфиновые комплексы Cu, Pd или Ni [например, Pd(OAc)2, CuI, PdCl2(PPh3)2, PdCl2dppf и NiCl2(PPh3)2]. Основания могут включать, но без ограничения, карбонаты щелочноземельных металлов, бикарбонаты щелочноземельных металлов, гидроксиды щелочноземельных металлов, карбонаты щелочных металлов, бикарбонаты щелочных металлов, фосфат щелочного металла, гидроксиды щелочных металлов, гидриды щелочных металлов, алкоксиды щелочных металлов (предпочтительно трет-бутоксид натрия), гидриды щелочноземельных металлов, диалкиламиды щелочных металлов, бис(триалкилсилил)амиды щелочных металлов (предпочтительно бис(триметилсилил)амид натрия), триалкиламины (предпочтительно диизопропилэтиламин или триэтиамин) или ароматические амины (предпочтительно пиридин). Инертные растворители могут включать, но без ограничения, ацетонитрил, циклические эфиры (предпочтительно тетрагидрофуран или 1,4-диоксан), N,N-диалкилацетамиды (предпочтительно диметилацетамид), N,N-диалкилформамиды (предпочтительно диметилформамид), диалкилсульфоксиды (предпочтительно диметилсульфоксид), ароматические углеводороды (предпочтительно толуол). Предпочтительные температуры реакции находятся в диапазоне от комнатной температуры до точки кипения используемого растворителя. Реакции могут быть осуществлены в обычной стеклянной посуде или в одной из многих коммерчески доступных установок для параллельного синтеза.
Обработка дигидроизохинолинонов формулы XXI арил- или гетероарилбороновыми кислотами, или сложными эфирами арил- или гетероарилбороновых кислот, или HNR9R10 в присутствии металлического катализатора с основанием или без основания в инертном растворителе создает соединения формулы XXIV. Металлические катализаторы могут включать, но без ограничения, соли или фосфиновые комплексы Cu, Pd или Ni [например, Pd(OAc)2, CuI, PdCl2(PPh3)2, PdCl2dppf и NiCl2(PPh3)2]. Предпочтительные металлические катализаторы могут быть созданы in situ смешиванием солей металлов (предпочтительно Pd(OAc)2) с фосфиновыми лигандами (предпочтительно 2,8,9-три-изобутил-2,5,8,9-тетрааза-1-фосфабицикло[3.3.3]ундекан, 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил или 2,2'-бис(дифенилфосфино)-1,1'-бинафтил) или CuI с L-пролином. Основания могут включать, но без ограничения, карбонаты щелочноземельных металлов, бикарбонаты щелочноземельных металлов, гидроксиды щелочноземельных металлов, карбонаты щелочных металлов, бикарбонаты щелочных металлов, фосфат щелочного металла (предпочтительно K3PO4), гидроксиды щелочных металлов, гидриды щелочных металлов, алкоксиды щелочных металлов (предпочтительно трет-бутоксид натрия), гидриды щелочноземельных металлов, диалкиламиды щелочных металлов, бис(триалкилсилил)амиды щелочных металлов (предпочтительно бис(триметилсилил)амид натрия), триалкиламины (предпочтительно диизопропилэтиламин или триэтиламин) или ароматические амины (предпочтительно пиридин). Инертные растворители могут включать, но без ограничения, ацетонитрил, циклические эфиры (предпочтительно тетрагидрофуран или 1,4-диоксан), N,N-диалкилацетамиды (предпочтительно диметилацетамид), N,N-диалкилформамиды (предпочтительно диметилформамид), диалкилсульфоксиды (предпочтительно диметилсульфоксид), ароматические углеводороды (предпочтительно толуол). Предпочтительные температуры реакции находятся в диапазоне от комнатной температуры до точки кипения используемого растворителя. Реакции могут быть осуществлены в обычной стеклянной посуде или в одной из многих коммерчески доступных установок для параллельного синтеза.
Наконец, превращение соединений формулы XXIV в соединения формулы XVIII, затем в соединения формулы I можно осуществить подобно способам, описанным для превращения соединений формулы XVII в соединения формулы I (схема 4).
Энантиомерно обогащенные соединения формулы XVIII и соединения формулы XXII могут быть получены с использованием хиральных лигандов, таких как, например, но без ограничения, (+)- или (-)-2,2'-бис(дифенилфосфино)-1,1'-бинафтил.
Способы, которые описаны для путей синтеза, используемых на схеме 4 и 5, также применимы для получения соединений формулы I, в которых X является моноциклическим арилом или моноциклическим гетероарилом.
Соединения формулы I могут быть получены в форме чистых (R)- и (S)-энантиомеров кристаллизацией с хиральными солями, такими как, но без ограничения, соль (+)-ди-пара-толуоил-D-винной кислоты, соль (-)-ди-пара-толуоил-L-винной кислоты, соль (1S)-(+)-10-камфорсульфоновой кислоты, соль (+)-дибензоил-D-винной кислоты, соль (-)-дибензоил-L-винной кислоты, соль L-(+)-винной кислоты, соль D-(-)-винной кислоты, соль D-(+)-яблочной кислоты, соль L-(-)-яблочной кислоты, соль S-(+)-миндальной кислоты, соль R-(-)-миндальной кислоты, соль S-(-)-кислоты Мошера [α-метокси-α-(трифторметил)фенилуксусной кислоты], соль R-(+)-кислоты Мошера [α-метокси-α-(трифторметил)фенилуксусной кислоты]. Дополнительные хиральные соли могут быть описаны в книге Eliel et al., Stereochemistry of Organic Compounds, Wiley-Interscience Publication: New York, p. 334 (1994), которая включена в данное описание в виде ссылки в полном объеме.
Способ разделения может быть осуществлен в широком множестве условий, известных специалисту в области органического синтеза. Обычные растворители включают ацетонитрил, тетрагидрофуран, метилэтилкетон, ацетон, метанол, этанол, изопропиловый спирт и смесь двух или более указанных растворителей. Способ разделения может на отдельных стадиях иметь температуры от -50°C до точки кипения используемого растворителя.
Альтернативно соединения формулы I могут быть получены в форме чистых (R)- и (S)-энантиомеров из соответствующей рацемической смеси с помощью хиральной ВЭЖХ, используя коммерчески доступные хиральные колонки.
Соединения формулы I в форме чистых (R)- или (S)-энантиомеров могут быть превращены в соответствующую рацемическую смесь соединений обработкой основанием в присутствии или в отсутствии краун-эфирного катализатора в инертном растворителе. Основания могут включать, но без ограничения, гидроксид щелочного металла, гидрид щелочного металла, алкоксиды щелочных металлов (предпочтительно трет-бутоксид натрия), алканы щелочных металлов, диалкиламиды щелочных металлов, бис(триалкилсилил)амиды щелочных металлов (предпочтительно бис(триметилсилил)амид натрия). Инертные растворители могут включать, но без ограничения, ацетонитрил, простые эфиры (предпочтительно тетрагидрофуран или 1,4-диоксан), N,N-диалкилформамиды (предпочтительно диметилформамид), диалкилсульфоксиды (предпочтительно диметилсульфоксид), ароматические углеводороды (предпочтительно толуол). Когда основанием является алкоксид щелочного металла, такой спирт как, но без ограничения, этанол, изопропанол, н-бутанол, трет-бутанол, изобутанол или этиленгликоль, является предпочтительным в качестве сорастворителя или единственного растворителя. Типичным краун-эфиром является 18-краун-6. Предпочтительные температуры реакции находятся в диапазоне от 0°C до точки кипения используемого растворителя. Указанная реакция также может быть осуществлена в сосуде под давлением или микроволновом реакторе.
Методики синтеза, описанные на схеме 4 и 5, поддаются осуществлению при крупномасштабном (масштаб большого количества килограммов) синтетическом процессе.
Будет понятно, что соединения, применимые согласно настоящему изобретению, могут содержать асимметричные центры. Такие асимметричные центры независимо могут быть либо в R-, либо в S-конфигурации, и такие соединения способны вращать плоскость поляризованного света в поляриметре. Если соединение заставляет указанную плоскость поляризованного света вращаться в направлении против часовой стрелки, то говорят, что соединение является (-)-стереоизомером соединения. Если соединение заставляет указанную плоскость поляризованного света вращаться в направлении по часовой стрелке, то говорят, что соединение является (+)-стереоизомером соединения. Специалистам в данной области будет понятно, что некоторые соединения, применимые согласно изобретению, также могут проявлять геометрическую изомерию. Следует понимать, что настоящее изобретение включает отдельные геометрические изомеры и стереоизомеры и их смеси, включая рацемические смеси соединений формулы I, описанных выше. Такие изомеры могут быть выделены из их смесей в результате применения или адаптации известных способов, например хроматографических способов и способов перекристаллизации, или их отдельно получают из соответствующих изомеров их промежуточных продуктов.
Радиоактивно меченные соединения согласно изобретению синтезируют рядом способов, хорошо известных специалистам в данной области, например с использованием исходных веществ, включающих в себя один или несколько радиоизотопов. Соединения согласно настоящему изобретению, в которые стабильные радиоизотопы, такие как углерод-14, тритий, йод-121 или другой радиоизотоп, были введены при синтезе, являются применимыми диагностическим средствами для идентификации областей головного мозга или центральной нервной системы, которые могут быть поражены расстройствами, в которые вовлечены переносчики норэпинефрина, допамина или серотонина и механизм их захвата.
Настоящее изобретение относится к композициям, содержащим соединения, описанные в данной публикации, включая, в частности, фармацевтические композиции, содержащие терапевтически эффективные количества соединений и фармацевтически приемлемые носители.
Следующим объектом настоящего изобретения являются наборы, имеющие множество активных ингредиентов (с носителем или без носителя), которые вместе могут быть эффективно использованы для осуществления новой комбинированной терапии согласно изобретению.
Другим объектом изобретения является новая фармацевтическая композиция, которая эффективна сама по себе для применения в целебной комбинированной терапии, так как она содержит множество активных ингредиентов, которые могут быть использованы согласно изобретению.
Настоящее изобретение также относится к наборам или отдельным упаковкам, в которых объединены два или более активных ингредиента, применимых для лечения заболевания. В наборе могут быть предоставлены (отдельно или в сочетании с фармацевтически приемлемым разбавителем или носителем) соединение формулы I и дополнительный активный ингредиент (отдельно или в сочетании с разбавителем или носителем), выбранный из антагониста рецептора серотонина 1A, избирательного антагониста рецептора нейрокинина-1 и предшественника норэпинефрина.
На практике соединения согласно настоящему изобретению, как правило, можно вводить парентерально, внутривенно, подкожно, внутримышечно, в толстую кишку, назально, внутрибрюшинно, ректально или перорально.
Продукты согласно изобретению могут быть представлены в формах, допускающих введение наиболее подходящим путем, и изобретение также относится к фармацевтическим композициям, содержащим по меньшей мере один продукт согласно изобретению, который подходит для применения на человеке или в ветеринарии. Указанные композиции могут быть получены согласно обычным способам с использованием одного или нескольких фармацевтически приемлемых адъювантов или эксципиентов. Адъюванты наряду с прочим включают разбавители, стерильные водные среды и различные нетоксичные органические растворители. Композиции могут быть представлены в форме таблеток, пилюль, гранул, порошков, водных растворов или суспензий, инъекционных растворов, эликсиров или сиропов и могут содержать один или несколько агентов, выбранных из группы, состоящей из подсластителей, корригентов, красителей или стабилизаторов, чтобы получить фармацевтически приемлемые препараты.
Выбор наполнителя и содержание активного вещества в наполнителе обычно определяют в соответствии с растворимостью и химическими свойствами продукта, конкретным способом введения и условиями, которые необходимо соблюдать в фармацевтической практике. Например, эксципиенты, такие как лактоза, цитрат натрия, карбонат кальция, фосфат дикальция, и дезинтегрирующие средства, такие как крахмал, альгиновые кислоты и некоторые сложные силикаты, в сочетании со скользящими веществами, такими как стеарат магния, лаурилсульфат натрия и тальк, могут быть использованы для получения таблеток. Для получения капсулы предпочтительно использовать лактозу и полиэтиленгликоли с высокой молекулярной массой. Когда используют водные суспензии, они могут содержать эмульгаторы или агенты, которые облегчают суспендирование. Также можно использовать разбавители, такие как сахароза, этанол, полиэтиленгликоль, пропиленгликоль, глицерин и хлороформ или их смеси.
Для парентерального введения используют эмульсии, суспензии или растворы продуктов согласно изобретению в растительном масле, например кунжутном масле, арахисовом масле или оливковом масле, или водно-органические растворы, такие как вода и пропиленгликоль, инъекционные сложные органические эфиры, такие как этилолеат, а также стерильные водные растворы фармацевтически приемлемых солей. Растворы солей продуктов согласно изобретению особенно применимы для введения посредством внутримышечной или подкожной инъекции. Водные растворы, также содержащие растворы солей в чистой дистиллированной воде, могут быть использованы для внутривенного введения, при условии, что их pH скорректирован соответствующим образом, что они нужным образом забуферены и сделаны изотоничными с помощью достаточного количества глюкозы или хлорида натрия, и что они стерилизованы нагреванием, облучением или микрофильтрацией.
Подходящие композиции, содержащие соединения согласно изобретению, могут быть получены обычными способами. Например, соединения согласно изобретению могут быть растворены или суспендированы в носителе, подходящем для применения в распылителе, или в суспензии, или растворе для аэрозоля либо могут быть абсорбированы или адсорбированы на подходящем твердом носителе для применения в ингаляторе сухого порошка.
Твердые композиции для ректального введения включают суппозитории, приготовленные согласно известным способам и содержащие по меньшей мере одно соединение формулы I.
Процентное содержание активного ингредиента в композициях согласно изобретению можно варьировать, при этом необходимо, чтобы оно составляло такую часть, чтобы получить подходящую дозу. Очевидно, что несколько дозированных лекарственных форм можно вводить примерно в одно и то же время. Применяемую дозу будет определять лечащий врач, и она зависит от требуемого терапевтического эффекта, пути введения и продолжительности лечения, и состояния пациента. Для взрослого человека дозы, как правило, составляют от примерно 0,01 до примерно 100, предпочтительно от примерно 0,01 до примерно 10 мг/кг массы тела в сутки при ингаляции, от примерно 0,01 до примерно 100, предпочтительно от 0,1 до 70, более конкретно от 0,5 до 10 мг/кг массы тела в сутки при пероральном введении, и от примерно 0,01 до примерно 50, предпочтительно от 0,01 до 10 мг/кг массы тела в сутки при внутривенном введении. В каждом конкретном случае дозы будут определены в соответствии с факторами, отличительными для субъекта, подвергаемого лечению, такими как возраст, масса, общее состояние здоровья и другие характеристики, которые могут влиять на эффективность лекарственного продукта.
Продукты согласно изобретению можно вводить так часто, как это необходимо, чтобы получить требуемый терапевтический эффект. Некоторые пациенты могут быстро отвечать на более высокую или более низкую дозу и могут считать адекватными намного более слабые поддерживающие дозы. Для других пациентов может быть необходимо длительное лечение с частотой 1-4 дозы в сутки в соответствии с физиологическими потребностями каждого конкретного пациента. Как правило, активный продукт можно вводить перорально от 1 до 4 раз в сутки. Само собой разумеется, что для других пациентов будет необходимо назначение не более одной или двух доз в сутки.
Настоящее изобретение относится к соединениям, которые ингибируют синаптический захват норэпинефрина, допамина и серотонина и поэтому считаются применимыми для лечения расстройства, которое вызвано или зависит от пониженной доступности серотонина, норэпинефрина или допамина. Хотя соединения формулы I ингибируют синаптический захват норэпинефрина, допамина и серотонина, в случае любого отдельного соединения указанные ингибирующие эффекты могут проявляться при одних и тех же или сильно отличающихся концентрациях или дозах. В результате некоторые соединения формулы I применимы для лечения такого расстройства в дозах, при которых синаптический захват норэпинефрина может быть в значительной степени ингибирован, но при которых синаптический захват серотонина или допамина существенно не ингибируется, или наоборот. Также некоторые соединения формулы I применимы для лечения такого расстройства в дозах, при которых синаптический захват допамина может быть в значительной степени ингибирован, но при которых синаптический захват норэпинефрина или серотонина существенно не ингибируется, или наоборот. И наоборот, некоторые соединения формулы I применимы для лечения такого расстройства в дозах, при которых синаптический захват серотонина может быть в значительной степени ингибирован, но при которых синаптический захват норэпинефрина или допамина существенно не ингибируется, или наоборот. Другие соединения формулы I применимы для лечения такого расстройства в дозах, при которых синаптический захват норэпинефрина, допамина и серотонина в значительной степени ингибируется.
Настоящее изобретение относится к соединениям, в случае которых ингибирующее действие на захват серотонина и норэпинефрина происходит при сходных или даже одинаковых концентрациях указанных соединений, тогда как действие на ингибирование захвата допамина происходит при сильно отличающихся концентрациях или дозах. В результате некоторые соединения формулы I применимы для лечения такого расстройства в дозах, при которых синаптический захват серотонина и норэпинефрина может быть в значительной степени ингибирован, но при которых синаптический захват допамина существенно не ингибируется, или наоборот.
Настоящее изобретение относится к соединениям, в случае которых ингибирующее действие на захват серотонина и допамина происходит при сходных или даже одинаковых концентрациях указанных соединений, тогда как действие на ингибирование захвата норэпинефрина происходит при сильно отличающихся концентрациях или дозах. В результате некоторые соединения формулы I применимы для лечения такого расстройства в дозах, при которых синаптический захват серотонина и допамина может быть в значительной степени ингибирован, но при которых синаптический захват норэпинефрина существенно не ингибируется, или наоборот.
Настоящее изобретение относится к соединениям, в случае которых ингибирующее действие на захват норэпинефрина и допамина происходит при сходных или даже одинаковых концентрациях указанных соединений, тогда как действие на ингибирование захвата допамина происходит при сильно отличающихся концентрациях или дозах. В результате некоторые соединения формулы I применимы для лечения такого расстройства в дозах, при которых синаптический захват норэпинефрина и допамина может быть в значительной степени ингибирован, но при которых синаптический захват серотонина существенно не ингибируется, или наоборот.
Настоящее изобретение относится к соединениям, в случае которых ингибирующее действие на захват норэпинефрина, допамина и серотонина возникает при сходных или даже при одной и той же концентрации. В результате некоторые соединения формулы I применимы для лечения такого расстройства в дозах, при которых синаптические захваты норэпинефрина, допамина и серотонина могут быть в значительной степени ингибированы.
Концентрации или дозы, при которых тестируемое соединение ингибирует синаптический захват норэпинефрина, допамина и серотонина, легко определяют с использованием стандартного анализа и способов, хорошо известных и понятных специалисту в данной области. Например, степень ингибирования при конкретной дозе у крыс может быть определена способом Dudley, J. Pharmacol. Exp. Ther. 217: 834-840 (1981), публикация включена в данное описание в виде ссылки в полном объеме.
Терапевтически эффективной ингибирующей дозой является доза, которая эффективна для ингибирования синаптического захвата норэпинефрина, синаптического захвата допамина или синаптического захвата серотонина или для ингибирования синаптического захвата двух или более веществ: норэпинефрина, допамина и серотонина. Терапевтически эффективная ингибирующая доза может быть легко определена специалистами в данной области с использованием обычных способов нахождения диапазона и аналогичных результатов, полученных в системах тестирования, описанных выше.
Соединения согласно данному изобретению обеспечивают чрезвычайно благоприятный терапевтический индекс по сравнению с другими соединениями, имеющимися для лечения сходных расстройств. Не намереваясь ограничиваться теорией, предполагают, что это по меньшей мере частично является следствием того, что некоторые соединения обладают более высокими аффинностями связывания по отношению к одному или двум переносчикам нейромедиаторов, например избирательно по отношению к белку-переносчику норэпинефрина («NET») по сравнению с переносчиками других нейрохимических агентов, например белком-переносчиком допамина («DAT») и белком-переносчиком серотонина («SERT»).
Другие соединения согласно данному изобретению могут проявлять избирательность по отношению к SERT по сравнению с переносчиками других нейрохимических агентов, например DAT и NET.
Другие соединения согласно данному изобретению могут проявлять избирательность по отношению к DAT по сравнению с переносчиками других нейрохимических агентов, например SERT и NET.
Другие соединения согласно данному изобретению могут проявлять избирательность по отношению к SERT и NET по сравнению с переносчиком другого нейрохимического агента, например DAT.
Другие соединения согласно данному изобретению могут проявлять избирательность по отношению к SERT и DAT по сравнению с переносчиком другого нейрохимического агента, например NET.
Другие соединения согласно данному изобретению могут проявлять избирательность по отношению к NET и DAT по сравнению с переносчиком другого нейрохимического агента, например SERT.
Наконец, другие соединения обладают почти идентичной аффинностью по отношению к NET, DAT и SERT.
Аффинности связывания показывают рядом способов, хорошо известных специалистам в данной области, включая, но без ограничения, способы, описанные в разделе примеров ниже. Коротко, например, содержащие белок экстракты из клеток, например клеток HEK293E, экспрессирующих белки-переносчики, инкубируют с радиоактивно меченными лигандами белков. Связывание радиоактивных лигандов с белками обратимо в присутствии других лигандов белков, например соединений согласно данному изобретению; указанная обратимость, как описано ниже, обеспечивает средства для измерения аффинностей связывания соединений с белками (Ki). Более высокое значение Ki для соединения является показателем того, что соединение имеет меньшую аффинность связывания по отношению к белку, чем соединение с меньшей Ki; наоборот, меньшие значения Ki являются показателем более высоких аффинностей связывания.
Соответственно различие в избирательности соединений по отношению к белкам показано более низким значением Ki по отношению к белку, к которому соединение является более избирательным, и более высоким значением Ki по отношению к белку, к которому соединение менее избирательно. Таким образом, чем больше отношение значений Ki соединения по отношению к белку A по сравнению с белком B, тем выше избирательность соединений по отношению к последнему по сравнению с первым (первый имеет более высокое значение Ki, а последний более низкое значение Ki по отношению к данному соединению). Соединения, предлагаемые в данном изобретении, обладают широким диапазоном профилей избирательности по отношению к переносчикам норэпинефрина, допамина и серотонина, которые отражаются в отношениях экспериментально определенных значений Ki.
Выбранные соединения («ингибиторы обратного захвата переносчиков одного механизма действия») согласно настоящему изобретению обладают высокой аффинностью связывания по отношению к каждому из переносчиков биогенных аминов NET, DAT или SERT. Например, выбранные соединения согласно данному изобретению обладают высокой (Ki NET <100 нМ) и избирательной аффинностью связывания NET, при этом отношение Ki DAT/NET и SERT/NET выше 10:1. Другие выбранные соединения согласно настоящему изобретению обладают высокой (Ki SERT <100 нМ) и избирательной аффинностью связывания SERT, при этом отношение Ki NET/SERT и DAT/SERT выше 10:1. Другие выбранные соединения согласно настоящему изобретению обладают высокой (Ki DAT <100 нМ) и избирательной аффинностью связывания DAT, при этом отношение Ki NET/DAT и SERT/DAT выше 10:1.
Выбранные соединения («ингибиторы обратного захвата переносчиков двойного действия») согласно настоящему изобретению обладают высокой аффинностью связывания по отношению к двум из переносчиков биогенных аминов NET, DAT или SERT. Например, выбранные соединения согласно данному изобретению обладают высокой (значения Ki NET и SERT <100 нМ) и избирательной аффинностью связывания NET и SERT, при этом отношение Ki DAT/NET и DAT/SERT выше 10:1, тогда как отношение Ki SERT/NET или NET/SERT меньше 10:1. Другие выбранные соединения согласно данному изобретению обладают высокой (значения Ki NET и DAT <100 нМ) и избирательной аффинностью связывания NET и DAT, при этом отношение Ki SERT/NET и SERT/DAT выше 10:1, тогда как отношение Ki DAT/NET или NET/DAT меньше 10:1. Другие выбранные соединения согласно данному изобретению обладают высокой (значения Ki DAT и SERT <100 нМ) и избирательной аффинностью связывания DAT и SERT, при этом отношение Ki NET/DAT и SERT/DAT выше 10:1, тогда как отношение Ki SERT/NET или NET/SERT меньше 10:1.
Выбранные соединения («ингибиторы обратного захвата переносчиков тройного действия») согласно настоящему изобретению обладают высокой аффинностью связывания одновременно по отношению ко всем трем переносчикам биогенных аминов NET, DAT или SERT. Например, выбранные соединения согласно данному изобретению обладают высокой аффинностью связывания (значения Ki NET, DAT и SERT <100 нМ), при этом все отношения Ki NET/DAT, NET/SERT, DAT/NET, DAT/SERT, SERT/NET и SERT/DAT меньше 10:1.
Выбранные соединения согласно настоящему изобретению обладают высокой аффинностью связывания (значения Ki <100 нМ) по отношению к одному, двум или трем из переносчиков биогенных аминов, NET, DAT и SERT, при этом отношения Ki для любых NET/SERT, NET/DAT, DAT/NET, DAT/SERT, SERT/NET и SERT/DAT выпадают из границ, определенных для «ингибиторов обратного захвата переносчиков одинарного, двойного или тройного действия», определенных выше.
Выбранные соединения согласно настоящему изобретению обладают менее высокой аффинностью связывания (значения Ki от 100 нМ до 1000 нМ) по отношению к одному, двум или трем переносчикам биогенных аминов, NET, DAT и SERT, при этом отношения Ki для любых NET/SERT, NET/DAT, DAT/NET, DAT/SERT, SERT/NET и SERT/DAT попадают в границы, определенные для «ингибиторов обратного захвата переносчиков одинарного, двойного или тройного действия», определенных выше.
Наконец, выбранные соединения согласно настоящему изобретению обладают менее высокой аффинностью связывания (значения Ki от 100 нМ до 1000 нМ) по отношению к одному, двум или трем переносчикам биогенных аминов, NET, DAT и SERT, при этом отношения Ki для любых NET/SERT, NET/DAT, DAT/NET, DAT/SERT, SERT/NET и SERT/DAT выпадают из границ, определенных для «ингибиторов обратного захвата переносчиков одинарного, двойного или тройного действия», определенных выше.
Настоящее изобретение относится к способам лечения субъектов, пораженных различными неврологическими и психиатрическими расстройствами, посредством введения указанным субъектам дозы фармацевтической композиции, предлагаемой в данном изобретении. Указанные расстройства включают, без ограничения, гиперактивное расстройство с дефицитом внимания (ADHD), когнитивное расстройство, тревожные расстройства, особенно генерализованное тревожное расстройство (GAD), расстройство панического типа, биполярное расстройство, также известное как маниакальная депрессия или маниакально-депрессивное расстройство, обсессивно-компульсивное расстройство (OCD), посттравматическое стрессовое расстройство (PTSD), острое стрессовое расстройство, социофобию, простую фобию, предменструальное дисфорическое расстройство (PMDD), социальное тревожное расстройство (SAD), большое депрессивное расстройство (MDD), супрануклеарный паралич, расстройства пищевого поведения, особенно ожирение, нервную анорексию, нейрогенную булимию и расстройство неумеренного потребления пищи, анальгезию (включая невропатическую боль, особенно диабетическую невропатию), расстройства, связанные со злоупотреблением психоактивными веществами (включая химические зависимости), подобные привыканию к никотину, привыканию к кокаину, привыканию к алкоголю и амфетамину, синдром Леша-Найхана, нейродегенеративные заболевания, подобные болезни Паркинсона, синдрому поздней фазы лютеинизации или нарколепсии, психиатрические симптомы гнева, такие как чувствительность к отказу, двигательные расстройства, подобные экстрапирамидальному синдрому, тикозные расстройства и синдром усталых ног (RLS), позднюю дискинезию, супрануклеарный паралич, расстройство пищевого поведения, связанное со сном (SRED), синдром ночной еды (NES), недержание мочи (включая недержание мочи при напряжении (SUI) и смешанное недержание мочи), мигрень, синдром фибромиалгии (FS), синдром хронической усталости (CFS), половую дисфункцию, особенно преждевременную эякуляцию и импотенцию у мужчин, расстройства терморегуляции (например, приливы, которые могут быть связаны с менопаузой) и боль в нижнем отделе спины.
Соединения, предлагаемые в данном изобретении, в частности, применимы для лечения указанных и других расстройств по меньшей мере отчасти вследствие их способности избирательно связываться с белками-переносчиками некоторых нейрохимических агентов с более высокой аффинностью, чем с белками-переносчиками других нейрохимических агентов.
Соединения согласно изобретению, способы их получения и их биологическая активность, по-видимому, будут более понятны на основе изучения следующих примеров, которые представлены только в качестве иллюстрации и которые не следует рассматривать как ограничивающие объем изобретения.
ПРИМЕРЫ
Следующие примеры предлагаются для иллюстрации вариантов осуществления настоящего изобретения, но никоим образом не предназначены для ограничения его объема.
Если не оговорено особо, использовали реагенты и растворители, которые получали от коммерческих поставщиков. Все реакции осуществляли в атмосфере азота, если не указано другое. Протекание реакций контролировали с помощью ТСХ, ГХ или ВЭЖХ. Тонкослойную хроматографию (ТСХ) осуществляли, используя пластины силикагеля Analtech или EMD и визуализировали в ультрафиолетовом (УФ) свете (254 нм). ГХ осуществляли в следующих условиях: колонка Hewlett Packard 5-MS, 30 м × 0,25 мм × 0,25 мкм, температура впрыскивания 150°C, начальная температура 100°C, начальное время 2,0 минуты, конечная температура 320°C, скорость 20°C/минуту. ВЭЖХ осуществляли в следующих условиях: Phenomenex Synergi polar-RP, 4 мк, 150×4,6 мм; или Phenomenex Luna C18(2), 4 мк, 150×4,6 мм; ацетонитрил:вода (содержащая TFA или уксусную кислоту), элюент 1 мл/минуту, регистрация при 220, 230 или 254 нм. Спектры ядерного магнитного резонанса протонов получали на спектрометре Bruker AV 500 или Bruker AV-300. Спектры приведены в м.д. (δ) и константы взаимодействия, J, даны в Герцах. Тетраметилсилан использовали в качестве внутреннего стандарта для протонных спектров и пик растворителя использовали в качестве эталонного пика для углеродных спектров. Масс-спектры получали на масс-спектрометре с ионизацией при атмосферном давлении (APCI) Perkin Elmer Sciex 100, масс-спектрометре на основе ионной ловушки с ионизацией в электроспрее (ESI) для ЖХМС Finnigan LCQ Duo или Thermofinnigan AQA. Очистку с использованием препаративной ВЭЖХ осуществляли, используя колонку Phenomenex Luna 10 мк C18(2) (250×21,20 мм) на Varian Prostar с УФ-регистрацией при 220, 230 или 254 нм. Хиральное разделение осуществляли на колонке CHIRALCEL OD, CHIRALPAK AD, CHIRACEL OJ или CHIRALPAK ADH. Хроматография относится к флэш-хроматографии, которую осуществляли на силикагеле (силикагель EMD 60 Е) или жидкостной хроматографии среднего давления, осуществляемой с использованием системы CombiFlash Companion или Biotage Horizon. Оптические вращения измеряли на поляриметре Perkin Elmer (модель 341 или модель 343) для свободных оснований. Элементные анализы осуществляли в Robertson Microlit Laboratories, Inc. или Quantitative Technologies Inc. Указанные выходы не оптимизированы.
Пример 1 - Получение 4-(бензо[b]тиофен-2-ил)-1,2,3,4-тетрагидроизохинолина
Стадия A: Продукт со стадии A примера 3 (0,34 г, 1,3 ммоль) в CH3OH (10 мл) перемешивали в течение 10 минут при комнатной температуре в атмосфере N2. Добавляли цианоборгидрид натрия (0,49 г, 7,8 ммоль) и перемешивали в течение 2 часов. Добавление 3 М HCl (4 мл по каплям) изменяло реакционную смесь от молочно-белого раствора до желтого/зеленого и обратно до молочно-белого раствора. Смесь подщелачивали 2N раствором Na2CO3 и затем концентрировали в вакууме до получения белого твердого вещества. Остаток распределяли между H2O (30 мл) и EtOAc (30 мл). Смесь экстрагировали EtOAc (3×30 мл). Объединенный органический слой промывали насыщенным раствором соли (50 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Хроматография (SiO2, 250 г, 1:49 метанол:EtOAc) давала чистый продукт в виде масла (0,29 г); ВЭЖХ: чистота 94,2%;1H ЯМР (500 МГц, CDCl3) δ 7,70 (д, J=7,84 Гц, 1H), 7,61 (д, J=7,76 Гц, 1H), 7,20 (м, 3H), 7,11 (д, J=3,85 Гц, 2H), 7,05 (д, J=7,47 Гц, 1H), 6,89 (с, 1H), 4,32 (т, J=5,16 Гц, 1H), 4,06 (дд, J=24,6, 16,6 Гц, 2H), 3,39 (дд, J=13,0, 4,64 Гц, 1H), 3,30 (дд, J=13,0, 4,64 Гц, 1H) 1,88-2,10 (ушир.с, 1H); ESI МС m/z 266 [М+1].
Стадия B: Продукт со стадии A (0,18 г, 6,8 ммоль) перемешивали в EtOH (3,0 мл) и добавляли фумаровую кислоту (0,08 г, 6,8 ммоль), растворенную в CH3OH (0,5 мл). Часть реакционной смеси преципитировалась, но перемешивание продолжали в течение 1 часа, затем добавляли диэтиловый эфир (2 мл) и реакционную смесь фильтровали, получая белое твердое веществе (0,16 г); ВЭЖХ: чистота 98,6%;1H ЯМР (500 МГц, ДМСО-d6) δ 7,83 (д, J=7,90 Гц, 1H), 7,74 (д, J=7,76 Гц, 1H), 7,32 (м, 1H), 7,26 (м, 2H), 7,16 (т, J=4,92 Гц, 1Н), 7,10 (м, 3H), 6,55 (с, 1H), 4,50 (т, J=7,20 Гц, 1H), 4,06 (д, J=16,1 Гц, 1H), 3,99 (д, J=16,1 Гц, 1H), 3,36 (дд, J=12,4, 4,9 Гц, 1H), 3,16 (дд, J=12,4, 6,15 Гц, 1Н); ESI МС m/z 266 [М+1].
Пример 2 - Получение гидрохлоридной соли (+)-4-(бензо[b]фуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина и гидрохлоридной соли и (-)-4-(бензо[b]фуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Рацемический 4-(бензо[b]фуран-2-ил)-2-метил-1,2,3,4- тетрагидроизохинолин получали из 2-бензо[b]фуранбороновой кислоты и 4-бромизохинолина, как описано в примере 36 (стадии D-F). Указанное рацемическое соединение (120 мг) разделяли с помощью полупрепаративной хиральной ВЭЖХ (chiralcel OD-H, 1×25 см, элюент: 3% изопропанол в гептане, расход: 3,1 мл/минуту, впрыскивания 500 мкл, 20 мг/впрыскивание). Полученные в результате свободные основания растворяли в этилацетате и обрабатывали 2 М раствором HCl в диэтиловом эфире (2 экв.), получая гидрохлоридную соль (+)-4-(бензо[b]фуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (13 мг, 99,6% AUC ВЭЖХ, 100% AUC хиральная ВЭЖХ):1H ЯМР (300 МГц, ацетон-d6) δ 7,71-7,80 (м, 1H), 7,50-7,58 (м, 1H), 7,31-7,45 (м, 2H), 7,01-7,23 (м, 5H); 5,35-5,50 (м, 1H), 4,47-4,70 (м, 2H), 3,70-4,00 (м, 2H), 3,11 (с, 3H), 2,43 (с, 3H); EI МС m/z=263 [C18H17NO]+; [α]25D +37° (c 0,5, MeOH, свободное основание) и гидрохлоридную соль (-)-4-(бензо[b]фуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (49 мг, 99,7% AUC ВЭЖХ, 100% хиральная ВЭЖХ):1H ЯМР (300 МГц, ацетон-d6) δ 7,64-7,71 (м, 1H), 7,42-7,49 (м, 1H), 7,25-7,36 (м, 2H), 6,93-7,15 (м, 4H), 5,22-4,40 (м, 1H), 3,40-4,60 (м, 2H), 3,60-3,90 (м, 2H), 3,02 (с, 3H), 2,35 (с, 3H); EI МС m/z=263 [C18H17NO]+; [α]25D -40° (c 0,5, MeOH, свободное основание).
Пример 3 - Получение фумаратной соли 4-(бензо[b]тиофен-2-ил)-2-этил-1,2,3,4-тетрагидроизохинолина
Стадия A: Смесь 4-бромизохинолина (0,64 г, 3,0 ммоль) в диметиловом эфире этиленгликоля (4 мл), бензотиофен-2-бороновой кислоты (0,69 г, 4,0 ммоль) и 2 N Na2CO3 (3 мл) дегазировали в течение 5 минут, затем дважды в течение 5 минут продували в атмосфере N2. Добавляли каталитическое количество Pd(PPh3)4 (0,36 г, 0,3 ммоль) и реакционную смесь дегазировали и продували N2. Реакционную смесь нагревали до 80°C при перемешивании в течение 12 часов, в течение которых раствор становился темно-коричневого цвета. Реакционную смесь охлаждали до комнатной температуры, разбавляли H2O (20 мл) и экстрагировали EtOAc (3×50 мл). Объединенный органический слой промывали насыщенным раствором соли (30 мл), сушили Na2SO4, фильтровали и концентрировали в вакууме. Хроматография (SiO2, 200 г, 1:3 этилацетат/гексан) давала чистый продукт в виде масла (0,65 г); ВЭЖХ: чистота 99,5%;1H ЯМР (500 МГц, CDCl3) δ 9,27 (с, 1H), 8,70 (с, 1H), 8,29 (д, J=8,48 Гц, 1H), 8,05 (д, J=8,1 Гц, 1H), 7,86-7,91 (м, 2H), 7,72-7,75 (м, 1H), 7,64-7,67 (м, 1H), 7,5 (с, 1Н), 7,37-7,44 (м, 2H); ESI МС m/z 262 [М+1].
Стадия B: Продукт со стадии A (0,35 г, 1,3 ммоль) перемешивали в метиленхлориде (10 мл) и охлаждали на ледяной бане в атмосфере N2. Добавляли этилтрифторметансульфонат (0,2 мл, 1,6 ммоль) и перемешивали при комнатной температуре в течение 2 часов (ESI МС показала M+l = 290), тонкослойная хроматография показала отсутствие исходного вещества. Реакционную смесь концентрировали в вакууме и добавляли CH3OH (30 мл) при перемешивании в атмосфере N2. Добавляли цианоборгидрид натрия (0,2 г, 3,2 ммоль) и реакционную смесь перемешивали в течение ночи. Реакционную смесь концентрировали в вакууме и распределяли в H2O (30 мл) и EtOAc (30 мл). Смесь экстрагировали EtOAc (3×30 мл). Объединенный органический слой промывали насыщенным раствором соли (50 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Хроматография (SiO2, 150 г, 1:5 этилацетат/гексан) давала чистый продукт в виде масла (0,26 г); ВЭЖХ: чистота 97,3%;1H ЯМР (500 МГц, CDCl3) δ 7,7 (д, J=7,92 Гц, 1H), 7,66 (д, J=7,81 Гц, 1H) 7,25-7,28 (м, 1Н), 7,18-7,22 (м, 1Н), 7,13 (т, J=10,7 Гц, 3H), 7,07 (т, J=9,04 Гц, 2H), 4,53 (т, J=7,33 Гц, 1H), 3,77 (д, J=14,9 Гц, 1H), 3,68 (д, J=14,9 Гц, 1H), 3,02 (дд, J=11,4, 4,86 Гц, 1H), 2,91 (дд, J=11,4, 6,27 Гц, 1H), 2,56-2,67 (м, 2H), 1,17 (т, J=9,57 Гц, 3H); ESI МС m/z 294 [М+1].
Стадия C: Продукт со стадии B (0,25 г, 0,85 ммоль) перемешивали в EtOH (2,5 мл) и добавляли фумаровую кислоту (0,09 г, 0,85 ммоль). Реакционную смесь осторожно нагревали вплоть до образования осадка. Смесь оставляли в морозильной камере в течение 1 часа, затем растирали в диэтиловом эфире и фильтровали, получая белое твердое вещество (0,16 г), ВЭЖХ: чистота 98,5%;1H ЯМР (500 МГц, CD3OD) δ 7,78 (д, J=7,95 Гц, 1H), 7,75 (д, J=8,07 Гц, 1H), 7,28-7,35 (м, 4H), 7,24 (т, J=10,6 Гц, 2H), 7,17 (д, J=7,78 Гц, 1H), 6,67 (с, 2H), 4,94 (дд, J=9,79, 5,79 Гц, 1H), 4,39 (д, J=15,2 Гц, 1H), 4,28 (д, J=15,2 Гц, 1H), 3,73-3,77 (м, 1H), 3,42 (дд, J=12,1, 10,0 Гц, 1H), 3,15-3,20 (м, 2H), 1,37 (т, J=9,70 Гц, 3H); ESI МС m/z 294 [М+1].
Пример 4 - Получение гидрохлоридной соли (+)-4-(бензофуран-2-ил)-2,7-диметил-1,2,3,4-тетрагидроизохинолина и гидрохлоридной соли (-)-4-(бензофуран-2-ил)-2,7-диметил-1,2,3,4-тетрагидроизохинолина
Рацемический 4-бензофуран-2-ил-2,7-диметил-1,2,3,4- тетрагидроизохинолин получали из 2-ацетилбензофурана и 3-толилметиламина, как описано в примере 26. Указанное рацемическое соединение (105 мг) разделяли с помощью полупрепаративной хиральной ВЭЖХ (chiralcel OD-H, 1×25 см, элюент: 3% этанол в гептане, расход: 4 мл/минуту, впрыскивание 500 мкл, 5 мг/впрыскивание). Полученные в результате свободные основания растворяли в этилацетате и обрабатывали 2 М раствором HCl в диэтиловом эфире (2 экв.), получая гидрохлоридную соль (+)-4-бензофуран-2-ил-2,7-диметил-1,2,3,4-тетрагидроизохинолина (39 мг, 100% AUC ВЭЖХ, 99,4% AUC хиральная ВЭЖХ):1H ЯМР (300 МГц, ацетон-d6) δ 7,63-7,70 (м, 1H), 7,20-7,47 (м, 6H), 7,00-7,08 (м, 2H), 5,31-5,43 (м, 1H), 4,43-4,62 (м, 2H), 3,70-3,88 (м, 2H), 3,01 (с, 3H); EI МС m/z=277 [C19H19NO]+; [α]25D+20° (c 1,0, MeOH, свободное основание) и гидрохлоридную соль (-)-4-бензофуран-2-ил-2,7-диметил-1,2,3,4-тетрагидроизохинолина (40 мг, 100% AUC ВЭЖХ, 100% хиральная ВЭЖХ):1H ЯМР (300 МГц, ацетон-d6) δ 7,63-7,69 (м, 1H), 7,21-7,47 (м, 6H), 7,00-7,10 (м, 2H), 5,32-5,45 (м, 1H), 4,43-4,66 (м, 2H), 3,67-3,90 (м, 2H), 3,02 (с, 3H); EI МС m/z=277 [C19H19NO]+; [α]25D-17° (c 1,0, MeOH, свободное основание).
Пример 5 - Получение малеата 4-(бензофуран-2-ил)-2,5-диметил-1,2,3,4-тетрагидроизохинолина
Малеат 4-бензофуран-2-ил-2,5-диметил-1,2,3,4-тетрагидроизохинолина (99,3% AUC ВЭЖХ) получали, как описано в примере 26, из 2-ацетилбензофурана и 3-толилметиламина:1H ЯМР (300 МГц, MeOD) δ 7,17-7,49 (м, 7H), 6,20 (с, 1H), 6,14 (с, 2H), 4,87 (с, 1H), 4,62 (д, J=15,3 Гц, 1H), 4,41 (д, J=15,6 Гц, 1H), 4,11 (д, J=12,6 Гц, 2H), 3,81 (дд, J=12,6, 5,1 Гц, 1H), 3,05 (с, 3H), 2,22 (с, 3H), EI МС m/z=278 [C19H19NO+H]+. Элементный анализ, вычислено для C23H23NO5: С, 70,07; Н, 5,85; N, 3,55 с 0,11% Н2O. Найдено: C, 66,80; Н, 5,65; N, 3,29.
Пример 6 - Получение малеата 4-(бензофуран-2-ил)-2,8-диметил-1,2,3,4-тетрагидроизохинолина
Малеат 4-бензофуран-2-ил-2,8-диметил-1,2,3,4-тетрагидроизохинолина (99,0% AUC ВЭЖХ) получали, как описано в примере 26, из 2-ацетилбензофурана и 2-толилметиламина:1H ЯМР (300 МГц, MeOD) δ 7,57 (д, J=6,9 Гц, 1H), 7,43 (д, J=8,1 Гц, 1Н), 7,10-7,31 (м, 5H), 6,67 (с, 1H), 6,21 (с, 2H), 4,87-4,89 (м, 1H), 4,51 (с, 2H), 3,89-3,93 (м, 2H), 3,15 (с, 3H), 2,35 (с, 3H), EI МС m/z=278 [C19H19NO+H]+. Элементный анализ, вычислено для C23H23NO5: С, 69,39; Н, 6,00; N, 3,45 с 0,46% Н2O и 2,5% EtOH. Найдено: C, 68,38; Н, 6,30; N, 3,30.
Пример 7 - Получение гидрохлоридной соли 4-(5-хлор-1-бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Гидрохлоридную соль 4-(5-хлор-1-бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (16 мг, 95,8% AUC ВЭЖХ) получали из 5-хлорсалицилальдегида, как описано в примере 8:1H ЯМР (400 МГц, ДМСО-d6) δ 7,50-7,63 (м, 1H), 7,34-7,49 (м, 2H), 7,06-7,25 (м, 4H), 6,80-6,93 (м, 1Н), 4,74-4,96 (м, 1H), 4,26-4,53 (м, 2H), 3,47-3,83 (м, 2H), 2,81 (ушир.с, 3H); DI МС m/z 299 [C18H16ClNO+H]+.
Пример 8 - Получение гидрохлоридной соли 4-(5-фторбензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: Хлорметилтриметилсилан (5,0 г, 41 ммоль) и йодид натрия (6,1 г, 41 ммоль) добавляли к смеси 5-фторсалицилальдегида (5,2 г, 37 ммоль) и карбоната калия (15,2 г, 110 ммоль) в ДМФА (100 мл). Полученную в результате смесь нагревали при 65°C в течение 15 часов, затем охлаждали до комнатной температуры, гасили водой и дважды экстрагировали диэтиловым эфиром. Объединенные органические экстракты промывали водой и сушили над безводным сульфатом магния, получая требуемый промежуточный продукт в виде не совсем белого твердого вещества (7,7 г, 93%, 97,2% AUC ВЭЖХ):1H ЯМР (500 МГц, ДМСО-d6) δ 10,30-10,10 (м, 1H), 7,50-7,30 (м, 1H), 7,30-7,10 (м, 2H), 3,65 (ушир.с, 2H), 0,20 (ушир.с, 9H).
Стадия B: Смесь продукта со стадии A (7,7 г, 34 ммоль) и фторида цезия (15,6 г, 103 ммоль) в ДМФА (100 мл) нагревали при 95°C в течение 3 дней. Полученную в результате смесь разбавляли насыщенным водным раствором бикарбоната натрия и экстрагировали диэтиловым эфиром и дихлорметаном. Объединенные органические экстракты промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, получая сырой остаток. Полученный остаток разбавляли MTBE, промывали водой и насыщенным раствором соли, сушили, получая требуемый промежуточный гидроксидигидробензофуран в виде коричневой жидкости (3,9 г, 76,2% AUC ГХ):1H ЯМР (500 МГц, CDCl3) δ 6,91 (дд, J=7,6, 2,9 Гц, 1H), 6,78 (тд, J=8,9, 2,9 Гц, 1H), 6,60 (дд, J=8,9, 4,1 Гц, 1H), 5,12 (дд, J=6,6, 2,5 Гц, 1H), 4,36 (дд, J=10,7, 6,7 Гц, 1H), 4,23 (дд, J=10,7, 2,6 Гц, 1H), 2,57 (ушир.с, 1H).
Стадия C: Продукт со стадии B (3,7 г, 24,0 ммоль) дегидратировали тионилхлоридом (10 экв.) в пиридине (75 мл), получая промежуточный продукт бензофуран (1,97 г, 60%, 89,1% AUC ВЭЖХ), как описано в примере 9 (стадия B).
Стадия D: Гидрохлоридную соль 4-(5-фтор-l-бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (99,1% AUC ВЭЖХ) получали из продукта со стадии C и 4-бромизохинолина, как описано в примере 10, используя альтернативный способ получения гидрохлоридной соли. Свободное основание (53 мг, 188 ммоль) растворяли в этилацетате (2 мл) и обрабатывали HCl в диэтиловом эфире (92 мкл, 2 М) при комнатной температуре. Твердые вещества начинали выпадать в осадок. Добавляли MTBE и полученную в результате смесь фильтровали, получая требуемую гидрохлоридную соль в виде желтого твердого вещества (36 мг, 77%, 95,2% AUC ВЭЖХ):1H ЯМР (400 МГц, D2O) δ 7,05-7,49 (м, 7Н), 6,95-7,00 (тд, J=9,6, 2,3 Гц, 1H), 4,59-4,81 (м, 3H), 3,65-4,03 (м, 2H), 3,00 (ушир.с, 3H); DI МС m/z 282 [C18H16FNO+H]+.
Пример 9 - Получение гидрохлоридной соли 4-(7-фторбензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: Йодметилтриметилсилан (6,0 мл, 40 ммоль) добавляли к смеси 3-фторсалицилальдегида (5,0 г, 36 ммоль) и карбоната калия (15,2 г, 110 ммоль) в диметилформамиде (100 мл). Полученную в результате смесь нагревали при 60°C в течение 6 часов, после этого добавляли дополнительное количество йодметилтриметилсилана. После перемешивания при 60°C в течение 15 часов смесь охлаждали до комнатной температуры и твердые вещества удаляли при фильтровании. К фильтрату добавляли фторид цезия (16,4 г, 108 ммоль) и полученную в результате смесь нагревали при 105°C в течение 36 часов. Смесь экстрагировали метиленхлоридом, промывали водой и сушили над безводным сульфатом магния, получая красное/коричневое масло (3,6 г), которое отверждалось при отстаивании. Полученные неочищенные твердые вещества растирали в гексанах, получая светло-коричневые игольчатые кристаллы (2,3 г, 42%):1H ЯМР (500 МГц, ДМСО-d6) δ 7,06 (д, J=7,6 Гц, 1Н), 7,00 (дд, J=11,4, 8,5 Гц, 1Н), 6,75 (ддд, J=12,0, 7,9, 4,4 Гц, 1H), 5,61 (д, J=5,7 Гц, 1H), 5,18 (ушир.с, 1H), 4,46 (дд, J=10,1, 6,9 Гц, 1H), 4,19 (дд, J=10,1, 2,8 Гц, 1H).
Стадия B: Тионилхлорид (11,0 мл, 150 ммоль) в течение периода времени, составляющего 5 минут, добавляли к раствору продукта со стадии A (2,3 г, 15 ммоль) в пиридине (12 мл) при 0°C и полученную в результате смесь перемешивали при 0°C в течение 90 минут. Добавляли дихлорметан (100 мл) и реакцию осторожно гасили 10% водным раствором бикарбоната натрия (200 мл), достигая pH 5. Затем добавляли твердый бикарбонат натрия (30 г), затем воду (100 мл). Два слоя разделяли и водную фазу экстрагировали дихлорметаном (100 мл). Объединенные органические слои промывали 5% водным раствором бикарбоната натрия (100 мл) и водой (100 мл) и сушили над безводным сульфатом магния. Сырой продукт растворяли в дихлорметане и промывали дважды 1N HCl и водой, чтобы удалить остаточный пиридин. Органический слой сушили над безводным сульфатом магния, получая требуемый бензофуран (1,5 г, 73%, 98,3% AUC ВЭЖХ):1H ЯМР (500 МГц, ДМСО-d6) δ 7,95 (д, J=1,9 Гц, 1H), 7,36 (д, J=7,6 Гц, 1H), 7,17-7,05 (м, 2H), 6,94 (т, J=2,5 Гц, 1H).
Стадия C: Гидрохлоридную соль 4-(7-фтор-1-бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (99,1% AUC ВЭЖХ) получали из продукта со стадии B и 4-бромизохинолина, как описано в примере 10:1H ЯМР (500 МГц, CDCl3) δ 7,07-7,38 (м, 5H), 6,92-7,04 (м, 2H), 6,81-6,87 (м, 1H), 4,57 (д, J=16,1 Гц, 1H), 4,29-4,50 (м, 1H), 4,11-4,28 (м, 1H), 3,73-3,83 (м, 1H), 3,51-3,66 (м, 1H), 3,00 (ушир.с, 3H); DI МС m/z 282 [C18H16FNO+H]+.
Пример 10 - Получение гидрохлоридной соли 4-(5-метоксибензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: 5-Метоксибензофуран (1 г, 6,8 ммоль) растворяли в тетрагидрофуране (10 мл) и охлаждали до -30°C. Раствор обрабатывали n-BuLi (3,5 мл, 8,9 ммоль, 2,5 М в гексанах) в течение 30 минут, поддерживая во время добавления внутреннюю температуру -30°C, получая красный раствор. Спустя 1 час при -30°C добавляли триметилборат (1 мл, 8,8 ммоль) в течение 10 минут, и раствор становился бледно-коричневым. Полученному в результате раствору давали возможность медленно нагреться до 10°C в течение 2 часов, после этого гасили 6 М HCl (10 мл) и экстрагировали этилацетатом (30 мл). Органическую часть промывали водой (30 мл) и насыщенным раствором соли (30 мл) и сушили над безводным сульфатом магния. После фильтрования органический раствор концентрировали и бороновую кислоту преципитировали добавлением гексанов. Кристаллы отфильтровывали и промывали гексанами, получая не совсем белое твердое вещество (983 мг, 76%):1H ЯМР (500 МГц, ДМСО-d6) δ 8,53 (ушир.с, 2H), 4,47 (ушир.д, J=8,8 Гц, 1H), 7,20 (ушир.с, 1H), 6,94 (ушир.дд, J=8,8, 2,5 Гц, 1H), 3,79 (ушир.с, 3H).
Стадия B: Раствор продукта со стадии A (1,0 г, 5,1 ммоль) в 1,2-диметоксиэтане (10 мл) обрабатывали водным раствором карбоната натрия (6,0 мл, 2 М раствор), 4-бромизохинолином (1,0 г, 4,8 ммоль) и каталитическим ацетатом палладия и трифенилфосфином. Смесь кипятили с обратным холодильником в течение 2 часов. Дополнительно добавляли ацетат палладия и трифенилфосфин и реакционную смесь кипятили с обратным холодильником в течение ночи. Реакционную смесь охлаждали до комнатной температуры и разбавляли водой. Продукт дважды экстрагировали метиленхлоридом, промывали водой и сушили над безводным сульфатом магния, получая коричневое масло (1,5 г, 83,1% AUC ВЭЖХ).
Стадия C: Раствор продукта со стадии B (1,5 г, 5,5 ммоль) в хлороформе (20 мл) обрабатывали йодметаном (1,0 мл, 16,4 ммоль) и нагревали при 60°C в течение 3,5 часов. Дополнительно добавляли йодметан и смесь нагревали при 60°C в течение ночи. Полученную в результате взвесь охлаждали до температуры окружающей среды. Твердые вещества отфильтровывали и промывали хлороформом, получая требуемый метилированный изохинолин в виде желтого твердого вещества (1,7 г, 84% из 4-бромизохинолина, 90,8% AUC ВЭЖХ):1H ЯМР (500 МГц, ДМСО-d6) δ 10,03 (с, 1Н), 9,22 (с, 1H), 8,85 (д, J=8,5 Гц, 1H), 8,62-8,54 (м, 1H), 8,45-8,36 (м, 1H), 8,22-8,14 (м, 1H), 7,81 (с, 1H), 7,72 (д, J=9,2 Гц, 1H), 7,36 (ушир.д, J=1,9 Гц, 1H), 7,11 (ушир.дд, J=8,8, 2,2 Гц, 1H), 4,55 (с, 3H), 3,87 (с, 3H).
Стадия D: Продукт со стадии C (0,50 г, 1,2 ммоль) суспендировали в метаноле (25 мл) и обрабатывали цианоборгидридом натрия (0,17 мг, 1,7 ммоль) и несколькими каплями раствора бромкрезолового зеленого в метаноле. Полученную в результате зеленую смесь обрабатывали 2,0 М раствором хлористого водорода в диэтиловом эфире вплоть до того, как окраска раствора становилась желтой. Смесь перемешивали в течение 2 часов при комнатной температуре, поддерживая желтую окраску добавлением дополнительного количества хлористого водорода в эфире. Добавляли водный раствор гидроксида натрия (10 мл, 3,0 M), чтобы погасить реакцию, и смесь экстрагировали этилацетатом. Органические экстракты промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, получая неочищенное желтое масло (0,28 г). Соответствующую гидрохлоридную соль получали, пропуская поток газа хлористого водорода через раствор неочищенного продукта в этилацетате. Объем органической части уменьшали и обрабатывали гексанами, чтобы преципитировать требуемую соль. Взвесь фильтровалии, промывали гексанами, получая бледно-желтое твердое вещество (0,24 г, 60%, 100% AUC ВЭЖХ):1H ЯМР (500 МГц, ДМСО-d6) δ 7,60-6,80 (м, 8H), 5,15-4,82 (м, 1H), 4,70-4,30 (м, 2H), 4,20-3,55 (м, 5H), 3,00 (с, 3H); DI МС m/z=294 [C19H19NO2+H]+.
Пример 11 - Получение гидрохлоридной соли (+)-4-(7-метоксибензо[b]фуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина и гидрохлоридной соли (-)-4-(7-метоксибензо[b]фуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Рацемический 4-(7-метоксибензо[b]фуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин получали из 2-(7-метоксибензо[b]фуран)бороновой кислоты и 4-бромизохинолина, как описано в примере 36 (стадии D-F). Полученное рацемическое соединение (260 мг) разделяли с помощью полупрепаративной хиральной ВЭЖХ (chiralcel OD-H, 1×25 см, элюент: 2% этанол в гептане, расход: 3,7 мл/мин, впрыскивание 500 мкл, 20 мг/впрыскивание). Полученные в результате свободные основания растворяли в этилацетате и обрабатывали 2 М раствором HCl в диэтиловом эфире (2 экв.), получая гидрохлоридную соль (+)-4-(7-метоксибензо[b]фуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (48 мг, 95,0% AUC ВЭЖХ, 100% AUC хиральная ВЭЖХ):1H ЯМР (300 МГц, MeOD) δ 7,25-7,40 (м, 4H), 7,13-7,19 (м, 2H), 6,90 (дд, J=6,0, 3,0 Гц, 1H), 6,70 (ушир.с, 1Н), 4,75-4,92 (м, 1H), 4,59 (д, J=5,1 Гц, 2H), 3,95 (с, 3H), 3,90-3,98 (м, 1H), 3,22-3,33 (м, 1Н), 3,10 (с, 3H); E1 МС m/z=294 [C19H19NO2+H]+, [α]25D +5,3° (c 1,0, MeOH, свободное основание) и гидрохлоридную соль (-)-4-(7-метоксибензо[b]фуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (48 мг, 97,3% AUC ВЭЖХ, 100% хиральная ВЭЖХ):1H ЯМР (300 МГц, MeOD) δ 7,25-7,40 (м, 4H), 7,13-7,19 (м, 2H), 6,85-6,92 (м, 1H), 6,70 (ушир.с, 1Н), 4,78-4,92 (м, 1H), 4,50-4,56 (м, 2H), 3,95 (с, 3H), 3,85-3,95 (м, 2H), 3,09 (с, 3H); EI МС m/z=294 [C19H19NO2+H]+; [α]25D -10,4° (c 1,0, MeOH, свободное основание).
Пример 12 - Получение гидрохлоридной соли 4-(бензо[b]фуран-3-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: Раствор 2-метоксифенилацетона (5 г, 30,5 ммоль) в диметилацетале N,N-диметилформамида (8,7 мл, 73,1 ммоль) перемешивали при 80°C в течение 4 часов. Смесь концентрировали в вакууме. Полученный в результате остаток растворяли в дихлорметане (40 мл), охлаждали до 0°C и обрабатывали раствором трибромида бора (5 мл, 52,9 ммоль) в дихлорметане (10 мл). После перемешивания при 0°C в течение 1 часа добавляли дополнительное количество трибромида бора (5 мл, 52,9 ммоль). Смесь перемешивали при 0°C еще в течение 10 минут и вливали в смесь льда и насыщенного водного раствора бикарбоната натрия. Полученную в результате смесь три раза экстрагировали дихлорметаном. Объединенные органические экстракты один раз промывали водой и сушили над безводным сульфатом натрия, получая 3-ацетилбензофуран (4,7 г, 96%, 94,5% AUC ВЭЖХ):1H ЯМР (300 МГц, CDCl3) δ 8,16-8,19 (м, 2H), 7,44-7,48 (м, 1H), 7,29-7,32 (м, 2Н), 2,50 (с, 3H).
Стадия B: Раствор трибромида тетрабутиламмония (12,3 г, 25,4 ммоль) в дихлорметане (60 мл) по каплям добавляли к раствору продукта со стадии A (3,7 г, 23,1 ммоль) в дихлорметане (15 мл) и метаноле (15 мл) при комнатной температуре. После завершения добавления полученный в результате красно-оранжевый раствор перемешивали при комнатной температуре в течение 15 часов. Смесь концентрировали в вакууме и остаток помещали в этилацетат и воду. Слои разделяли и водную фазу экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли и сушили над безводным сульфатом натрия, получая требуемое бромсодержащее соединение (5,5 г, 99%):1H ЯМР (300 МГц, CDCl3) δ 8,41 (с, 1Н), 8,22-8,26 (м, 1H), 7,56-7,60 (м, 1H), 7,41-7,44 (м, 2Н), 4,36 (с, 2H).
Стадия C: Бензилметиламин (2,8 г, 23,0 ммоль) по каплям добавляли к раствору продукта со стадии B (5,5 г, 23,0 ммоль) в дихлорметане (45 мл) при 0°C. После завершения добавления полученную в результате смесь перемешивали при указанной температуре в течение 15 минут и по каплям добавляли диизопропилэтиламин (4,4 мл, 25,3 ммоль). Смесь перемешивали при комнатной температуре в течение 15 часов, после этого гасили насыщенным водным раствором бикарбоната натрия и дважды экстрагировали дихлорметаном. Объединенные органические экстракты промывали насыщенным водным раствором бикарбоната натрия и сушили над безводным сульфатом натрия. Концентрирование фильтрата и очистка хроматографией (9:1 гептан/этилацетат) давали требуемый промежуточный продукт (3,2 г, 50%):1H ЯМР (300 МГц, CDCl3) δ 8,69 (с, 1H); 8,26-8,29 (м, 1H), 7,53-7,56 (м, 1H), 7,28-7,41 (м, 7H), 3,68 (с, 2H), 3,61 (с, 2H), 2,39 (с, 3H).
Стадия D: Боргидрид натрия (0,43 г, 11,3 ммоль) добавляли к раствору продукта со стадии C (3,2 г, 11,4 ммоль) в метаноле (35 мл) при 0°C. Полученную в результате смесь перемешивали при комнатной температуре в течение 15 часов. Метанол удаляли в вакууме. Остаток помещали в воду и дважды экстрагировали дихлорметаном. Объединенные органические экстракты промывали водой и насыщенным раствором соли и сушили над безводным сульфатом натрия, получая после хроматографии требуемый промежуточный спирт (2,4 г, 75%):1H ЯМР (300 МГц, CDCl3) δ 7,60 (с, 1H), 7,46-7,49 (м, 2H), 7,17-7,39 (м, 7H), 5,03 (дд, J=11,1, 0,6 Гц, 1H), 3,82 (д, J=12,9 Гц, 1H), 3,58 (д, J=13,2 Гц, 1H), 2,94 (дд, J=12,3, 10,8 Гц, 1H), 2,68 (дд, J=12,3, 3,3 Гц, 1H), 2,41 (с, 3H).
Стадия E: Хлорид алюминия (2,0 г, 15,3 ммоль) порциями добавляли к раствору продукта со стадии D (2,4 г, 8,5 ммоль) в дихлорметане (90 мл) при 0°C. После окончания добавления смесь перемешивали в течение 1,5 часов при 0°C, вливали в лед и дихлорметан и перемешивали еще в течение 15 минут. Смесь промывали водой и насыщенным водным раствором бикарбоната натрия, экстрагировали дихлорметаном и сушили над сульфатом натрия. Полученный в результате сырой продукт очищали хроматографией (4:1 гептан/этилацетат) и препаративной ТСХ (1:1 гептан/этилацетат), получая 4-бензофуран-3-ил-2-метил-1,2,3,4-тетрагидроизохинолин (0,13 г, 6%, 99,6% AUC ВЭЖХ):1H ЯМР (300 МГц, CDCl3) δ 7,46-7,51 (м, 2H), 7,26-7,32 (м, 2H), 7,08-7,18 (м, 5H), 4,53 (т, J=3,6 Гц, 1H), 4,14 (дд, J=14,4, 7,2 Гц, 1H), 3,81 (д, J=15,0 Гц, 1Н), 3,69 (д, J=14,7 Гц, 1H), 3,05 (ддд, J=11,4, 5,7, 1,2 Гц, 1H), 2,82 (дд, J=11,1, 8,1 Гц, 1H), 2,48 (с, 3H).
Стадия F: Продукт со стадии E (0,12 г, 0,48 ммоль) растворяли в этилацетате (2 мл) и обрабатывали 2 М HCl в диэтиловом эфире (0,48 мл, 0,95 ммоль), получая гидрохлоридную соль 4-бензофуран-3-ил-2-метил-1,2,3,4-тетрагидроизохинолина (0,12 г, 88%, 99,3% AUC ВЭЖХ) после фильтрования:1H ЯМР (300 МГц, ДМСО-d6) δ 8,18 (ушир.с, 1H), 7,62 (д, J=8,4 Гц, 1H), 6,88-7,36 (м, 7H), 4,90-5,02 (м, 1H), 4,52-4,70 (м, 2H), 3,61-3,82 (м, 2H), 2,95 (с, 3H); EI МС m/z=264 [C18H17NO+H]+. Элементный анализ, вычислено для C18H18ClNO: C, 72,11; Н, 6,05; N, 4,67. Найдено: C, 71,97; Н, 6,31; N,4,52.
Пример 13 - Получение малеата 4-(бензо[b]фуран-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
4-Бензо[b]фуран-4-ил-2-метил-1,2,3,4-тетрагидроизохинолин получали из 3-бромфенола, как описано в примере 14 (стадии A и B), и превращали в его соль малеат (0,39 г, 100% AUC ВЭЖХ), как описано в примере 69:1H ЯМР (300 МГц, MeOD) δ 7,74 (д, J=2,4 Гц, 1H), 7,52-7,55 (м, 1H), 7,15-7,38 (м, 5H), 6,90 (д, J=7,8 Гц, 1H), 6,50-6,51 (м, 1Н), 6,26 (с, 2H), 4,92-4,99 (м, 1H), 4,62-4,68 (м, 2H), 3,86-3,94 (м, 1H), 3,58-3,68 (м, 1Н), 3,11 (с, 3H); E1 МС m/z=264 [C18H17NO+H]+. Элементный анализ, вычислено для C22H21NO5: C, 69,64; Н, 5,58; N, 3,69. Найдено: C, 69,36; Н, 5,80; N, 3,34.
Пример 14 - Получение гидрохлоридной соли (+)-4-(бензо[b]фуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина и гидрохлоридной соли (-)-4-(бензо[b]фуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: Смесь 4-бромфенола (5,0 г, 29 ммоль), диэтилацеталя бромацетальдегида (4,5 мл, 30 ммоль), карбоната калия (4,1 г, 30 ммоль) и йодида калия (0,2 г, 1 ммоль) в бутаноне (30 мл) перемешивали при 80°C в течение 8 дней. Смесь охлаждали до комнатной температуры, разбавляли водой и три раза экстрагировали этилацетатом. Объединенные органические экстракты промывали водным раствором гидроксида натрия (1 М), водой и насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали в вакууме, получая требуемый продукт (3,8 г, 46%, 100% AUC ГХ) после хроматографии (9:1 гептан/этилацетат):1H ЯМР (300 МГц, CDCl3) δ 7,38 (дт, J=9,0, 3,3 Гц, 2H), 6,83 (дт, J=9,0, 3,3 Гц, 2H), 4,83 (т, J=5,1 Гц, 1H), 3,99 (д, J=5,1 Гц, 2H), 3,78 (кв.д, J=9,3, 7,2 Гц, 2H), 3,65 (кв.д, J=9,3, 7,2 Гц, 2H), 1,26 (т, J=7,2 Гц, 6H).
Стадия B: 4-бензо[b]фуран-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин (0,5 г, 98,1%) получали из продукта со стадии A, как описано в примере 40 (стадии B-E):1H ЯМР (300 МГц, CDCl3) δ 7,62 (д, J=2,1 Гц, 1H), 7,42-7,45 (м, 2H), 7,05-7,19 (м, 4H), 7,00 (д, J=7,5 Гц, 1H), 6,72 (дд, J=2,4, 0,9 Гц, 1H), 4,40 (т, J=7,2 Гц, 1Н), 3,80 (д, J=15,0 Гц, 1H), 3,67 (д, J=14,7 Гц, 1H), 3,09 (ддд, J=11,4, 5,7, 1,2 Гц, 1H), 2,64 (дд, J=11,4, 8,7 Гц, 1Н), 2,46 (с, 3H).
Стадия C: Продукт со стадии B разделяли с помощью полупрепаративной хиральной ВЭЖХ (chiralcel OD-H, 1×25 см, элюент: 2% этанол в гептане (0,1% Et3N), расход: 4 мл/минуту, впрыскивание 500 мкл, 5 мг/впрыскивание). Полученные в результате свободные основания растворяли в этилацетате и обрабатывали 2 М раствором HCl в диэтиловом эфире (2 экв.), получая гидрохлоридную соль (+)-4-бензо[b]фуран-5-ил-2-метил-1,2,3,4-тетрагидроизохинолина (33 мг, 99% AUC ВЭЖХ, 100% AUC хиральная ВЭЖХ):1H ЯМР (300 МГц, MeOD, свободное основание) δ 7,81 (д, J=2,1 Гц, 1Н), 7,57 (ушир.с, 1Н), 7,53 (д, J=8,7 Гц, 1H), 7,16-7,40 (м, 4H), 6,92 (д, J=7,8 Гц, 1H), 6,84 (ушир.с, 1H), 4,60-4,85 (м, 3H), 3,82-3,94 (м, 1Н), 3,59 (т, J=12,3 Гц, 1H), 3,11 (с, 3H); EI МС m/z=264 [C18H17NO+H]+; [α]25D+37° (c 1,0, MeOH, свободное основание), и гидрохлоридную соль (-)-4-бензо[b]фуран-5-ил-2-метил-1,2,3,4-тетрагидроизохинолина (28 мг, 99,5% AUC ВЭЖХ, 100% хиральная ВЭЖХ):1H ЯМР (300 МГц, MeOD) δ 7,81 (д, J=2,1 Гц, 1H), 7,57 (ушир.с, 1H), 7,54 (д, J=8,7 Гц, 1H), 7,25-7,38 (м, 3H), 7,18 (дд, J=8,7, 1,8 Гц, 1H), 6,92 (д, J=7,8 Гц, 1H), 6,84 (ушир.с, 1H), 4,55-4,88 (м, 3H), 3,80-3,96 (м, 1H), 3,59 (т, J=11,7 Гц, 1H), 3,11 (с, 3H); EI МС m/z=264 [C18H17NO+H]+, [α]25D -37° (c 1,0, MeOH, свободное основание).
Пример 15 - Получение малеата 4-(бензо[b]фуран-5-ил)-2,7-диметил-1,2,3,4-тетрагидроизохинолина
4-Бензо[b]фуран-5-ил-2,7-диметил-1,2,3,4-тетрагидроизохинолин получали из 7-метилциннамовой кислоты и 5-бромфурана, как описано в примере 41 (стадии A-H), и превращали в его соответствующую соль малеат (0,29 г, 98,3% AUC ВЭЖХ), как описано в примере 69:1H ЯМР (300 МГц, MeOD) δ 7,79 (д, J=1,8 Гц, 1H), 7,50-7,53 (м, 2H), 7,07-7,18 (м, 3H), 6,80-6,83 (м, 2H), 6,24 (с, 2H), 4,67 (дд, J=10,8, 6,0 Гц, 1H), 4,51-4,58 (м, 2H), 3,87 (дд, J=12,0, 5,7 Гц, 1H), 3,54-3,63 (м, 1H), 3,09 (с, 3H), 2,34 (с, 3H); EI МС m/z=278 [C19H19NO+H]+. Элементный анализ, вычислено для С23Н23NO5: С, 69,97; Н, 5,91; N, 3,52 с 0,08 экв. EtOH. Найдено: C, 69,31; Н, 5,88; N, 3,39.
Пример 16 - Получение гидрохлоридной соли 4-(бензо[b]фуран-5-ил)-7-фтор-2-метил-1,2,3,4-тетрагидроизохинолина
Гидрохлоридную соль 4-бензо[b]фуран-5-ил-7-фтор-2-метил-1,2,3,4-тетрагидроизохинолина (0,26 г, 96,5% AUC ВЭЖХ) получали из 7-фторциннамовой кислоты и 5-бромфурана, как описано в примере 41:1H ЯМР (300 МГц, ДМСО-d6) δ 11,65 (ушир.с, 1H), 8,03 (д, J=2,1 Гц, 1H), 7,62 (д, J=8,4 Гц, 1H), 7,55 (с, 1H), 6,96-7,50 (м, 4H), 6,78 (ушир.с, 1H), 4,66-4,74 (м, 1H), 4,53 (ушир.с, 1H), 3,45-3,80 (м, 2H), 2,94 (с, 3H), 2,51 (с, 3H); EI МС m/z=282 [C18H16FNO+H]+. Элементный анализ, вычислено для C18H17ClFNO: C, 67,21; Н, 5,51; N, 4,36 с 1,1 экв. НC1. Найдено: C, 67,32; Н, 5,41; N, 4,19.
Пример 17 - Получение малеатной соли 4-(бензо[b]фуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
4-Бензо[b]фуран-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин получали из 3-бромфенола, как описано в примере 14 (стадии A и B), и превращали в его соль малеат (0,28 г, 98,2% AUC ВЭЖХ), как описано в примере 69:1H ЯМР (300 МГц, MeOD) δ 7,79 (д, J=2,1 Гц, 1H), 7,65 (д, J=8,1 Гц, 1H), 7,25-7,42 (м, 4H), 7,14 (дд, J=8,1, 1,5 Гц, 1H), 6,96 (д, J=7,8 Гц, 1H), 6,86-6,87 (м, 1H), 6,26 (с, 2H), 4,60-4,76 (м, 3H), 3,86-3,92 (м, 1H), 3,62 (т, J=12,0 Гц, 1H), 3,09 (с, 3H); EI МС m/z=264 [C18H17NO+H]+. Элементный анализ, вычислено для C22H21NO5: C, 69,64; Н, 5,58; N, 3,69. Найдено: C, 69,67; Н, 5,65; N, 3,39.
Пример 18 - Получение гидрохлоридной соли 4-(бензо[b]фуран-6-ил)-7-фтор-2-метил-1,2,3,4-тетрагидроизохинолина
Гидрохлоридную соль 4-бензо[b]фуран-6-ил-7-фтор-2-метил-1,2,3,4-тетрагидроизохинолина (0,21 г, 97,3% AUC ВЭЖХ) получали из 7-фторциннамовой кислоты и 6-бромфурана, как описано в примере 41:1H ЯМР (300 МГц, ДМСО-d6) δ 11,53 (ушир.с, 1H), 8,02 (д, J=2,4 Гц, 1H), 7,68 (д, J=8,1 Гц, 1H), 7,53 (ушир.с, 1H), 6,97-7,20 (м, 4H), 6,79 (ушир.с, 1H), 4,68-4,74 (м, 1Н), 4,53 (ушир.с, 21H), 3,39-3,82 (м, 2H), 2,92 (с, 3H), 2,50 (с, 3H); EI МС m/z=282 [C18H16FNO+H]+. Элементный анализ, вычислено для C18H17ClFNO: C, 67,20; Н, 5,51; N, 4,36 с 1,1 экв.HCl. Найдено: C, 67,30; Н, 5,66; N, 4,30.
Пример 19 - Получение малеатной соли 4-(бензо[b]фуран-7-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
4-Бензо[b]фуран-7-ил-2-метил-1,2,3,4-тетрагидроизохинолин получали из 2-бромфенола, как описано в примере 14 (стадии A и B), и превращали в его соль малеат (0,64 г, 100% AUC ВЭЖХ), как описано в примере 69:1H ЯМР (300 МГц, MeOD) δ 7,74 (д, J=2,1 Гц, 1H), 7,64 (дд, J=7,8, 1,2 Гц, 1H), 7,15-7,33 (м, 5H), 6,90 (д, J=2,1 Гц, 2H), 6,26 (с, 2H), 5,06-5,12 (м, 1H), 4,64 (с, 2H), 3,79-3,96 (м, 2H), 3,11 (с, 3H); EI МС m/z=264 [C18H17NO+H]+. Элементный анализ, вычислено для C22H21NO5: C, 69,64; Н, 5,58; N, 3,69. Найдено: C, 69,67; Н, 5,82; N, 3,49.
Пример 20 - Получение фумаратной соли (+) и (-)-4-(бензо[b]тиофен-5-ил)-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору изохинолин-4-бороновой кислоты (378 мг, 2,013 ммоль) и 5-бромбензотиофена (286 мг, 1,342 ммоль) в дихлорэтане (8 мл) добавляли карбонат натрия (2 М водный раствор, 2 мл, 4 ммоль) и раствор дегазировали, поочередно три раза откачивая газы и выпуская аргон. К полученной гетерогенной смеси добавляли тетракистрифенилфосфин палладия (76 мг, 0,6711 ммоль), реакционную смесь 3 раза дегазировали и нагревали до 90°C при встряхивании в течение 12 часов. К полученной смеси добавляли EtOAc (100 мл), смесь промывали водой (3×100 мл) и сушили над безводным сульфатом натрия. Очистка хроматографией на колонке (SiO2, 30 г, 50% этилацетат/гексан) давала продукт в виде белого твердого вещества (316 мг):1H ЯМР (300 МГц, CDCl3) δ 9,29 (с, 1H), 8,55 (с, 1H), 8,06-7,97 (м, 4H), 7,68-7,65 (м, 2H), 7,56-7,42 (м, 3H). ESI-МС вычислено для C17H11NS [M+H]+ 262. Найдено 262.
Стадия B: К раствору продукта со стадии A (400 мг, 1,530 ммоль) в безводном ТГФ (15 мл) при 0°C добавляли боргидрид лития (1 М раствор в ТГФ, 18 мл, 18 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Добавляли метанол (50 мл), смесь перемешивали при комнатной температуре в течение 15 минут и концентрировали досуха. Остаток растворяли в EtOAc (250 мл), органический слой промывали водой (50 мл) и насыщенным раствором соли (50 мл) и сушили над безводным сульфатом натрия. Очистка хроматографией на колонке (SiO2, 30 г, 33%-100% этилацетат/гексан, затем 10% MeOH/хлороформ) давала продукт в виде бесцветного масла (228 мг):1H ЯМР (300 МГц, CDCl3) δ 7,80 (д, J=8,3 Гц, 1H), 7,49 (с, 1H), 7,41 (д, J=5,4 Гц, 1Н), 7,25-7,07 (м, 4H), 6,92 (д, J=7,5 Гц, 1H), 4,25-4,07 (м, 5H), 3,43 (дд, J=5,2, 10,0 Гц, 1Н), 3,15 (дд, J=6,5, 13,0 Гц, 1H), ESI-МС вычислено для C17H15NS [M+H]+ 266. Найдено 266.
Стадия C: Свободное основание превращали в его ацетат, концентрируя раствор продукта со стадии B (65 мг, 0,245 ммоль) в уксусной кислоте (5 мл) досуха. Лиофилизация из ацетонитрила (2 мл) и воды (2 мл) давала рацемический продукт в виде белого твердого вещества (45 мг): т.пл. 118-120°C.1H ЯМР (CD3OD, 300 МГц) δ 7,85 (д, J=9,0 Гц, 1H), 7,72 (д, J=7,4 Гц, 1H), 7,8 (д, J=5,5 Гц, 1H), 7,33 (д, J=5,1 Гц, 1H), 7,23-7,11 (м, 4Н), 6,86 (д, J=7,6 Гц, 1H), 4,47-4,22 (м, 3H), 3,62-3,57 (м, 1Н), 3,32-3,30 (м, 1Н), 1,92 (с, 3H), ESI-МС вычислено для C17H15NS [M+H]+266. Найдено 266.
Стадия D: Отдельные энантиомеры получали посредством хиральной хроматографии (Chiralpak AD, 10% изопропанол/гептан с 0,1% диэтиламина) продукта со стадии B. (+)-энантиомер (80 мг): [α]D +6,0 (0,15, метанол), (-)-энантиомер (80 мг): [α]D -30,7 (0,15, метанол).
Стадия E: К раствору (-)-энантиомера, полученного на стадии D (73 мг), в этаноле (5 мл) добавляли раствор фумаровой кислоты (32 мг) в метаноле (1 мл) при 0°C. Раствор перемешивали при 0°C в течение 2 часов, затем концентрировали и остаток промывали этанолом. Соль фумарат выделяли в виде белого порошка (73 мг): т.пл. 192-197°C; ВЭЖХ >99% ee (колонка Chiralpak AD);1H ЯМР (300 МГц, CD3OD) δ 7,89 (д, J=8,3 Гц, 1H), 7,69 (с, 1H), 7,59 (д, J=5,5 Гц, 1H), 7,34-7,16 (м, 5H), 6,90 (д, J=6,7 Гц, 1H), 6,67 (с, 2H), 4,58-4,32 (м, 3H), 3,73-3,67 (м, 1H), 3,44-3,40 (м, 1H), ESI МС вычислено для C17H15NS [M+H]+ 266. Найдено 266.
Фумарат (+)-энантиомера получали сходным образом. (+)-энантиомер; т.пл. 165-172°C. ВЭЖХ 95,7% ee (колонка Chiralpak AD).
Пример 21 - Получение 4-(бензотиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
4-(бензотиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин получали из 2-бензотиофенбороновой кислоты и 4-бромизохинолина, как описано в примере 36 (стадии D-F): т.пл. 89-91°C;1H ЯМР (500 МГц, CDCl3) δ 7,72 (д, J=7,9 Гц, 1H), 7,67 (д, J=7,5 Гц, 1H), 7,32-7,23 (м, 3H), 7,07-7,17 (м, 4H), 4,55 (т, J=5,0 Гц, 1H), 3,77 (д, J=15,0 Гц, 1H), 3,62 (д, J=15,0 Гц, 1H), 3,00 (дд, J=12,0, 5,0 Гц, 1H), 2,89 (дд, J=12,0, 6,0 Гц, 1H), 2,48 (с, 3H); ИК (КВr) 2945, 2783, 1493, 1458 см-1; CI МС m/z=280 [C18H17NS +Н]+; ВЭЖХ 99,1%, tr = 16,07 мин. Элементный анализ, вычислено для C18H17NS: C, 77,38; Н, 6,13; N, 5,01. Найдено; C, 77,23; Н, 6,16; N, 4,97.
Пример 22 - Получение малеатной соли 4-(бензотиофен-2-ил)-2,8-диметил-1,2,3,4-тетрагидроизохинолина
Малеатную соль 4-бензотиофен-2-ил-2,8-диметил-1,2,3,4-тетрагидроизохинолина (98,7% AUC ВЭЖХ) получали, как описано в примере 26, из 2-ацетилбензотиофена и 2-толилметиламина:1H ЯМР (300 МГц, MeOD) δ 7,75-7,83 (м, 2H), 7,19-7,39 (м, 5H), 7,08 (т, J=4,5 Гц, 1H), 6,21 (с, 2H), 5,03 (дд, J=9,6, 5,7 Гц, 1H), 4,57 (д, J=15,6 Гц, 1Н), 4,45 (д, J=15,6 Гц, 1H), 3,93 (дд, J=12,0, 5,4 Гц, 1H), 3,72 (дд, J=12,0, 9,9 Гц, 1H), 3,14 (с, 3H), 2,35 (с, 3H), EI МС m/z=294 [C19H19NS+H]+. Элементный анализ, вычислено для C23H23NO4S: С, 66,83; Н, 5,74; N, 3,33; S, 7,61 с 0,36% Н2O и 2,00% EtOH. Найдено: C, 65,68; Н, 5,60; N, 3,20; S, 7,70.
Пример 23 - Получение малеатной соли 4-(4-фторбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Малеатную соль 4-(4-фторбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (45 мг, 95,6% AUC ВЭЖХ) получали из 3-фторбензолтиола и диэтилацеталя бромацетальдегида, как описано в примере 36:1H ЯМР (300 МГц, ДМСО-d6) δ 7,56 (д, J=7,8 Гц, 1H), 7,31 (с, 1H), 6,95-7,19 (м, 6H), 5,89 (с, 2H), 4,74 (т, J=6,3 Гц, 1H), 3,98-4,21 (м, 2H), 3,32-3,59 (м, 2H), 2,64 (с, 3H); EI МС m/z=298 [C18H16FNS+H]+.
Пример 24 - Получение 4-(бензо[b]тиофен-5-ил)-2-этил-1,2,3,4-тетрагидроизохинолина
К раствору продукта со стадии A примера 20 (100 мг, 0,383 ммоль) в безводном дихлорметане (2,5 мл) при 0°C добавляли этилтрифторметансульфонат (60 мкл, 0,459 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов, концентрировали досуха и перерастворяли в смеси метанол (5 мл)/дихлорметан (5 мл). К полученному раствору добавляли цианоборгидрид натрия (240 мг, 2,00 ммоль), смесь перемешивали при комнатной температуре в течение 12 часов и концентрировали досуха. Остаток растворяли в этилацетате (25 мл) и органический слой промывали насыщенным раствором бикарбоната натрия (25 мл), насыщенным раствором соли (25 мл) и сушили над безводным сульфатом натрия. Очистка полупрепаративной ВЭЖХ давала продукт в виде бесцветного масла (81 мг): ESI МС, рассчитано для C19H19NS [M+H]+ 394. Найдено 394.
Пример 25 - Получение малеатной соли 4-(бензотиофен-2-ил)-2,5-диметил-1,2,3,4-тетрагидроизохинолина
Малеатную соль 4-бензотиофен-2-ил-2,5-диметил-1,2,3,4-тетрагидроизохинолина (97,3% AUC ВЭЖХ) получали, как описано в примере 26, из 2-ацетилбензотиофена и 3-толилметиламина:1H ЯМР (300 МГц, MeOD) δ 7,82 (т, J=5,1 Гц, 1H), 7,67 (т, J=4,2 Гц, 1H), 7,28-7,41 (м, 4H), 7,20 (д, J=7,5 Гц, 3H), 6,23 (с, 3H), 5,00 (т, J=4,2 Гц, 1H), 4,64 (д, J=15,6 Гц, 1H), 4,45 (д, J=15,6 Гц, 1H), 3,94 (д, J=3,6 Гц, 2H), 3,05 (с, 3H), 2,18 (с, 3H), EI МС m/z=294 [C19H19NS+H]+. Элементный анализ, вычислено для C25H25NO6S: C, 63,34; Н, 5,28; N, 2,96; S, 6,76 с 1,28% Н2O. Найдено: C, 63,76; Н, 5,45; N, 2,76; S, 6,53.
Пример 26 - Получение малеатной соли 4-(бензотиофен-2-ил)-2,7-диметил-1,2,3,4-тетрагидроизохинолина
Стадия A: Раствор трибромида тетрабутиламмония (15,0 г, 312 ммоль) в дихлорметане (80 мл) по каплям добавляли к раствору 2-ацетилбензотиофена (5,0 г, 28 ммоль) в дихлорметане (20 мл) и метаноле (20 мл) при комнатной температуре. После завершения добавления полученный в результате красно-оранжевый раствор перемешивали при комнатной температуре в течение 15 часов. Смесь концентрировали в вакууме и остаток помещали в этилацетат и воду. Слои разделяли и водную фазу экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли и сушили над безводным сульфатом натрия, получая требуемое бромсодержащее соединение (7,3 г, 100%):1H ЯМР (300 МГц, CDCl3) δ 8,07 (с, 1H), 7,91 (дд, J=12,0, 8,1 Гц, 2Н), 7,42-7,53 (м, 2H), 4,47 (с, 2H).
Стадия B: 3-Толилметиламин (1,6 г, 11,7 ммоль) по каплям добавляли к раствору продукта со стадии A (3,0 г, 11,7 ммоль) в дихлорметане (25 мл) при 0°C. После завершения добавления полученную в результате смесь перемешивали при 0°C в течение 15 минут и по каплям добавляли диизопропилэтиламин (2,3 мл, 12,9 ммоль). Смесь перемешивали при комнатной температуре в течение 15 часов, после этого гасили насыщенным водным раствором бикарбоната натрия и дважды экстрагировали дихлорметаном. Объединенные органические экстракты промывали насыщенным водным раствором бикарбоната натрия и сушили над сульфатом натрия, получая требуемый промежуточный продукт (1,8 г, 49%) после хроматографии (9:1 гептан/этилацетат):1H ЯМР (300 МГц, CDCl3) δ 7,99 (с, 1H), 7,78 (д, J=7,8 Гц, 2H), 7,29-7,41 (м, 2H), 6,98-7,18 (м, 4H), 3,67 (с, 2H), 3,60 (с, 2H), 2,34 (с, 3H), 2,27 (с, 3H).
Стадия C: Боргидрид натрия (0,2 г, 5,8 ммоль) добавляли к раствору продукта со стадии B (1,8 г, 5,8 ммоль) в метаноле (20 мл) при 0°C. Полученную в результате смесь перемешивали при комнатной температуре в течение 15 часов. Метанол удаляли в вакууме. Остаток помещали в воду и три раза экстрагировали дихлорметаном. Объединенные органические экстракты промывали водой и насыщенным раствором соли и сушили над сульфатом натрия, получая требуемый спирт (1,6 г, 86%) после хроматографии (от 9:1 до 2:1 гептан/этилацетат):1H ЯМР (300 МГц, CDCl3) δ 7,73 (дд, 7,81-7,85 (м, 1H), J=6,6, 1,5 Гц, 1H), 7,23-7,38 (м, 4H), 7,12-7,15 (м, 3H), 5,10 (дд, J=10,8, 3,9 Гц, 1H), 3,73 (д, J=12,9 Гц, 1H), 3,57 (д, J=13,2 Гц, 1Н), 2,85 (дд, J=12,6, 10,2 Гц, 1H), 2,74 (дд, J=12,6, 3,9 Гц, 1Н), 2,38 (с, 3H), 2,35 (с, 3H).
Стадия D: Хлорид алюминия (1,20 г, 8,7 ммоль) порциями добавляли к раствору продукта со стадии C (1,50 г, 4,8 ммоль) в дихлорметане (60 мл) при 0°C. После окончания добавления смесь перемешивали при 0°C в течение 1,5 часов, вливали в лед и дихлорметан и перемешивали еще в течение 15 минут. Смесь промывали водой и насыщенным водным раствором бикарбоната натрия, экстрагировали дихлорметаном и сушили над сульфатом натрия, получая неочищенную смесь, содержащую региоизомеры в соотношении 2 к 1. Очистка хроматографией (от 4:1 до 2:1 гептан/этилацетат) давала 4-бензотиофен-2-ил-2,7-диметил-1,2,3,4-тетрагидроизохинолин (0,57 г, 40%, 99,1% AUC ВЭЖХ):1H ЯМР (300 МГц, CDCl3) δ 7,58-7,66 (м), 7,13-7,24 (м, 2H), 7,07 (с, 1H), 6,96 (с, 2H), J=7,5 Гц, 1H), 6,84 (д, J=11,4 Гц, 1H), 4,44 (т, J=5,4 Гц, 1H), 3,65 (д, J=15,0 Гц, 1Н), 3,51 (д, J=15,0 Гц, 1H), 2,91 (дд, J=11,7, 5,1 Гц, 1Н), 2,80 (дд, J=11,4, 6,3 Гц, 1H), 2,40 (с, 3H), 2,22 (с, 3H) и 4-бензотиофен-2-ил-2,5-диметил-1,2,3,4-тетрагидроизохинолин (0,21 г, 15%, 96,0% AUC ВЭЖХ):1H ЯМР (300 МГц, CDCl3) δ 7,74 (д, J=7,63 (дд = 7,8 Гц, 1H), J=6,9, 1,2 Гц, 1H), 7,16-7,32 (м, 3H), 7,03 (т, J=14,7 Гц, 1H), 6,89 (с, 1H), 4,45 (ушир.с, 1H), 4,02 (д, J=15,0 Гц, 1H), 3,40 (д, J=15,0 Гц, 1H), 3,13 (дт, J=11,4, 1,8 Гц, 1H), 2,79 (дд, J=11,4, 4,2 Гц, 1Н), 2,42 (с, 3H), 2,19 (с, 3H).
Стадия E: 4-Бензотиофен-2-ил-2,7-диметил-1,2,3,4-тетрагидроизохинолин (0,57 г) кристаллизовали с малеиновой кислотой (1 экв.) в этаноле, получая малеатную соль 4-бензотиофен-2-ил-2,7-диметил-1,2,3,4-тетрагидроизохинолина (0,20 г, 26%, 99,5% AUC ВЭЖХ):1H ЯМР (300 МГц, MeOD) δ 7,76-7,84 (м, 2H), 7,31-7,40 (м, 3H), 7,12-7,18 (м, 3H), 6,25 (с, 2,7H, малеиновая кислота), 5,00 (дд, J=9,6, 6,0 Гц, 1H), 4,58 (д, J=15,3 Гц, 1H), 4,52 (д, J=15,9 Гц, 1H), 3,98 (дд, J=12,3, 5,7 Гц, 1H), 3,74 (дд, J=12,0, 10,2 Гц, 1H), 3,13 (с, 3H), 2,37 (с, 3H), EI МС m/z=294 [C19H19NS+H]+. Элементный анализ, вычислено для С23Н23НО4S: С, 64,70; Н, 5,43; N, 3,08; S, 7,05 с 0,39% Н2O и 1,35 экв. малеиновой кислоты. Найдено: C, 62,86; Н, 5,25; N, 2,90; S, 7,20.
Пример 27 - Получение фумаратной соли 4-(бензо[b]тиофен-5-ил)-4-гидрокси-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору 5-бром-1-бензотиофена (511 мг, 2,4 ммоль) при -75°C по каплям добавляли трет-бутиллитий (1,7 М в пентане, 1,6 мл, 2,6 ммоль). Реакционную смесь перемешивали при -75°C в течение 1 часа. К полученной в результате темно-коричневой смеси добавляли 2-метил-2,3-дигидро-1H-изохинолин-4-он (323 мг, 2,0 ммоль), который получали, используя способ, описанный в Hanna et al., J. Med. Chem. 17(9): 1020-1023 (1974), публикация включена в данное описание в виде ссылки в полном объеме. Реакционную смесь перемешивали в течение 15 часов с постепенным нагреванием. Смесь гасили насыщенным раствором хлорида аммония и экстрагировали этилацетатом (3×50 мл). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией среднего давления на силикагеле (10-40% этилацетат/гексаны) с последующей препаративной ВЭЖХ давала продукт (65 мг, 11%):1H ЯМР (500 МГц, CDCl3) δ 7,81 (д, 1H, J=8,5 Гц), 7,69 (д, 1H, J=8,5 Гц), 7,34-7,23 (м, 4H), 7,20 (с, 1H), 7,18 (д, 1H, J=7,3 Гц), 7,10 (д, 1H, J=7,6 Гц), 3,91 (с, 1H), 3,84 (д, 1H, J=15,0 Гц), 3,53 (д, 1H, J=15,0 Гц), 3,11 (дд, 1H, J=1,4, 11,6 Гц), 2,87 (д, 1H, J=11,6 Гц), 2,50 (с, 3H), ESI МС m/z=296 [M+Н]+.
Стадия B: Продукт со стадии A (59,1 мг, 0,2 ммоль) растворяли в этаноле (1 мл) и добавляли раствор фумаровой кислоты (24 мг, 0,2 ммоль) в метаноле (0,5 мл). Растворитель удаляли при пониженном давлении. Остаток растирали в этилацетате. Полученный в результате преципитат собирали фильтрованием, промывали этилацетатом и сушили при 50°C в вакууме, получая продукт в виде белого твердого вещества (60 мг, 73%):1H ЯМР (CD3OD, 500 МГц) δ 7,81 (д, 1Н, J=7,2 Гц), 7,71 (д, 1H, J=7,2 Гц), 7,40 (д, 1H, J=7,8 Гц), 7,36-7,23 (м, 4H), 7,25 (д, 1H, J=7,6 Гц), 7,20 (с, 1H), 6,70 (с, 2H), 4,25 (д, 1Н, J=15,4 Гц), 4,20 (д, 1H, J=15,4 Гц), 3,54 (д, 1H, J=12,1 Гц), 3,48 (д, 1H, J=12,1 Гц), 2,83 (с, 3H), ESI МС m/z=296 [M+H]+.
Пример 28 - Получение малеатной соли 4-(5-фторбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Малеатную соль 4-(5-фторбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (638 мг, 97,6% AUC ВЭЖХ) получали из 4-фторбензолтиола и диэтилацеталя бромацетальдегида, как описано в примере 36:1H ЯМР (300 МГц, ДМСО-d6) δ 7,98 (дд, J=5,1, 3,6 Гц, 1H), 7,69 (дд, J=2,4, 9,9 Гц, 1Н), 7,44 (с, 1Н), 7,18-7,38 (м, 5H), 6,13 (с, 2H), 4,86-4,99 (м, 1H), 4,40 (д, J=11,4 Гц, 1H), 4,27 (д, J=15,3 Гц, 1H), 3,41-3,79 (м, 2H), 2,87 (с, 3H); EI МС m/z=298 [C18H16FNS+H]+. Элементный анализ, вычислено для C22H20FNO4S: C, 63,32; Н, 4,89; N, 3,36 с 0,83% Н2O. Найдено: C, 63,07; Н, 4,60; N, 3,46.
Пример 29 - Получение малеатной соли 4-(6-фторбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Малеатную соль 4-(6-Фторбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (252 мг, 96,5% AUC ВЭЖХ) получали из 3-фторбензолтиола и диэтилацеталя бромацетальдегида, как описано в примере 36:1H ЯМР (300 МГц, ДМСО-d6) δ 7,82-7,86 (м, 2H), 7,41 (с, 1H), 7,21-7,34 (м, 4H), 7,15 (д, J=7,2 Гц, 1H), 6,09 (с, 2H), 4,89 (т, J=7,8 Гц, 1H), 4,40 (д, J=15,3 Гц, 1H), 4,26 (д, J=15,3 Гц, 1H), 3,42-3,73 (м, 2H), 2,86 (с, 3H); EI МС m/z=298 [C18H16FNS+H]+. Элементный анализ, вычислено для C22H20FNO4S: C, 63,40; Н, 4,90; N, 3,36; S, 7,68 с 0,70% Н2O. Найдено: C, 62,97; Н, 4,70; N, 3,01; S, 7,60.
Пример 30 - Получение малеатной соли 4-(7-фторбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Малеатную соль 4-(7-фторбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (617 мг, 99,7% AUC ВЭЖХ) получали из 2-фторбензолтиола и диэтилацеталя бромацетальдегида, как описано в примере 36:1H ЯМР (300 МГц, ДМСО-d6) δ 7,68 (д, J=7,8 Гц, 1H), 7,53 (с, 1H), 7,41 (дт, J=7,8, 5,4 Гц, 1H), 7,16-7,34 (м, 5H), 6,10 (с, 2H), 4,86-4,98 (м, 1H), 4,37 (д, J=15,6 Гц, 1H), 4,20 (д, J=13,8 Гц, 1H), 3,39-3,71 (м, 2H), 2,83 (с, 3H); EI МС m/z=298 [C18H16FNS+H]+. Элементный анализ, вычислено для C22H20FNO4S: C, 63,51; Н, 4,86; N, 3,37; S, 7,70 с 0,54% Н2O. Найдено: C, 63,22; Н, 4,49; N, 3,29; S, 7,94.
Пример 31 - Получение малеатной соли 4-(4-хлорбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Малеатную соль 4-(4-хлорбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (290 мг, 96,1% AUC ВЭЖХ) получали из 3-хлорбензолтиола и диэтилацеталя бромацетальдегида, как описано в примере 36:1H ЯМР (300 МГц, ДМСО-d6) δ 7,97 (д, J=7,9 Гц, 1H), 7,23-7,57 (м, 7H), 6,15 (с, 2H), 4,96-5,13 (м, 1H), 4,47 (д, J=15,5 Гц, 1H), 4,32 (д, J=15,4 Гц, 1H), 3,52-3,85 (м, 2H), 2,91 (с, 3H); EI МС m/z=314 [C18H16ClNS+H]+. Элементный анализ, вычислено для C22H20ClNO4S: C, 60,65; Н, 4,73; N, 3,22; S, 7,35 с 1,24% Н2O. Найдено: C, 60,31; Н, 4,49; N, 3,14; S, 7,64.
Пример 32 - Получение 4-(5-хлорбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
4-(5-Хлорбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин получали из 4-хлорбензолтиола и диэтилацеталя бромацетальдегида, как описано в примере 36 (стадии A-F):1H ЯМР (300 МГц, CDCl3) δ 7,57 (д, J=1,8 Гц, 1H), 7,54 (д, J=8,4 Гц, 1H), 7,01-7,14 (м, 6H), 4,45 (т, J=5,1 Гц, 1H), 3,73 (д, J=15,0 Гц, 1H), 3,52 (д, J=15,0 Гц, 1H), 2,87 (ддд, J=18,3, 11,4, 4,8 Гц, 2H), 2,41 (с, 3H); EI МС m/z=314 [C18H16ClNS+H]+.
Пример 33 - Получение малеатной соли 4-(6-хлорбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Малеатную соль 4-(6-хлорбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (364 мг, 100% AUC ВЭЖХ) получали из 3-хлорбензолтиола и диэтилацеталя бромацетальдегида, как описано в примере 36:1H ЯМР (300 МГц, ДМСО-d6) δ 8,08 (с, 1H), 7,83 (д, J=8,4 Гц, 1H), 7,24-7,43 (м, 5H), 7,15 (д, J=7,5 Гц, 1H), 6,09 (с, 2H), 4,89 (т, J=6,6 Гц, 1H), 4,37 (д, J=14,7 Гц, 1Н), 4,23 (д, J=14,4 Гц, 1H), 3,45-3,75 (м, 2H), 2,84 (с, 3H); EI МС m/z=314 [C18H16ClNS+H]+. Элементный анализ, вычислено для C22H20ClNO4S: C, 61,46; Н, 4,69; N, 3,26; S, 7,46. Найдено: C, 61,24; Н, 4,51; N, 3,36; S, 7,53.
Пример 34 - Получение малеатной соли 4-(7-хлорбензо[b]тиофен -2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Малеатную соль 4-(7-хлорбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (373 мг, 98,4% AUC ВЭЖХ) получали из 2-хлорбензолтиола и диэтилацеталя бромацетальдегида, как описано в примере 36:1H ЯМР (300 МГц, ДМСО-d6) δ 7,82 (д, J=6,6 Гц, 1H), 7,54 (с, 1H), 7,21-7,49 (м, 5H), 7,17 (д, J=7,2 Гц, 1Н), 6,10 (с, 2H), 4,85-5,03 (м, 1H), 4,39 (д, J=14,4 Гц, 1H), 4,23 (д, J=13,5 Гц, 1H), 3,38-3,75 (м, 2H), 2,84 (с, 3H); EI МС m/z=314 [C18H16ClNS+H]+. Элементный анализ, вычислено для C22H20NO4S: C, 60,14; Н, 4,78; N, 3,19; с 2,06% Н2O. Найдено: C, 58,57; Н, 4,84; N, 2,92.
Пример 35 - Получение малеатной соли 4-(4-метоксибензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Малеатную соль 4-(4-метоксибензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (73 мг, 99,1% AUC ВЭЖХ) получали из 3-метоксибензолтиола и диэтилацеталя бромацетальдегида, как описано в примере 36:1H ЯМР (300 МГц, MeOD) δ 7,25-7,41 (м, 7H), 6,87 (д, J=7,8 Гц, 1H), 6,25 (с, 2H), 4,99-5,04 (м, 1H), 4,50-4,61 (м, 2H), 3,93-3,98 (м, 4H), 3,69-3,76 (м, 1H), 3,09 (с, 3H); EI МС m/z=310 [C19H19NOS+H]+.
Пример 36 - Получение малеатной соли 4-(5-метоксибензотиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: Диэтилацеталь бромацетальдегида (10,2 мл, 68 ммоль) по каплям добавляли к суспензии 4-метоксибензолтиола (10,0 г, 71 ммоль) и карбоната калия (9,8 г, 71 ммоль) в ацетоне (120 мл) при комнатной температуре. После перемешивания в указанных условиях в течение 15 часов реакционную смесь фильтровали через диатомовую землю, промывали ацетоном и фильтрат концентрировали в вакууме. Полученный в результате остаток помещали в воду и этилацетат. Два слоя разделяли и водную фазу дважды экстрагировали этилацетатом. Объединенные органические экстракты промывали водным раствором гидроксида натрия (1 М) и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали в вакууме. Сырой продукт очищали хроматографией (19:1 гептан/этилацетат), получая требуемый продукт (16,2 г, 89%, 94,1% AUC ГХ):1H ЯМР (300 МГц, CDCl3) δ 7,38-7,43 (м, 2H), 6,83-6,87 (м, 2H), 4,61 (т, J=5,7 Гц, 1H), 3,80 (с, 3H), 3,49-3,71 (м, 4H), 3,02 (д, J=6,0 Гц, 2H), 1,19 (т, J=7,5 Гц, 6H).
Стадия B: Раствор продукта со стадии A (15,0 г, 58,5 ммоль) в хлорбензоле (65 мл) по каплям добавляли к раствору полифосфорной кислоты (125 г) в хлорбензоле (375 мл), нагретой до 135°C. После перемешивания при 135°C в течение 1,5 часов смесь охлаждали ниже 50°C. Хлорбензольный слой выливали из реакционной колбы и концентрировали в вакууме. В это время в реакционную колбу добавляли воду, чтобы разложить полифосфорную кислоту. Полученную в результате водную фазу добавляли к остатку хлорбензольного слоя. Добавляли дополнительно воду и дихлорметан и две фазы разделяли. Водный слой дважды экстрагировали дихлорметаном. Объединенные органические экстракты сушили над сульфатом натрия и концентрировали в вакууме. Сырой продукт очищали хроматографией (15:1 гептан/этилацетат), получая циклизованный продукт (4,5 г, 47%, 100% AUC ГХ): H ЯМР (300 МГц, CDCl3) δ 7,87 (д, J=9,0 Гц, 1H), 7,58 (д, J=5,4 Гц, 1H), 7,39-7,42 (м, 2H), 7,14 (дд, J=9,0, 2,4 Гц, 1H), 4,01 (с, 3H).
Стадия C: N,N,N',N'-тетраметилэтилендиамин (2,0 мл, 13,4 ммоль) по каплям добавляли к раствору продукта со стадии B (2,0 г, 12,2 ммоль) в тетрагидрофуране (50 мл) при -40°C. После завершения добавления смесь перемешивали в течение 15 минут в указанных условиях. По каплям добавляли н-бутиллитий (9,0 мл, 1,6 M) и полученную в результате смесь перемешивали в течение 1 часа, давая возможность температуре достичь -30°C. По каплям добавляли триметилборат (1,5 мл, 13,4 ммоль) и смеси давали возможность нагреться до 20°C в течение ночи. Реакцию гасили 1 М хлористоводородной кислотой, три раза экстрагировали этилацетатом и сушили над безводным сульфатом натрия, получая требуемую неочищенную бороновую кислоту (2,6 г, количественный выход). Полученный продукт использовали на следующей стадии без очистки.
Стадия D: Неочищенный продукт со стадии C (2,5 г, 12,0 ммоль) добавляли к раствору 4-бромизохинолина (1,7 г, 8,0 ммоль) и трифенилфосфина (0,4 г, 1,6 ммоль) в 1,2-диметоксиэтане (40 мл). Смесь три раза дегазировали, используя аргон. Добавляли ацетат палладия (0,2 г, 0,8 ммоль) и суспензию перемешивали при комнатной температуре в течение 20 минут. Добавляли водный раствор карбоната натрия (9,6 мл, 2 М раствор) и суспензию три раза дегазировали, используя аргон. После перемешивания при 85°C в течение 15 часов охлажденную смесь разбавляли водой и дважды экстрагировали дихлорметаном. Объединенные экстракты сушили над сульфатом натрия, концентрировали в вакууме и очищали хроматографией (5:1 гептан/этилацетат), получая требуемый изохинолин (1,8 г, 76%, 97,9% AUC ВЭЖХ) после очистки хроматографией:1H ЯМР (300 МГц, CDCl3) δ 9,21 (с, 1H), 8,62 (с, 1H), 8,24 (д, J=8,1 Гц, 1H), 7,99 (д, J=7,8 Гц, 1H), 7,58-7,71 (м, 3H), 7,38 (с, 1Н), 7,26 (д, J=2,1 Гц, 1H), 7,19 (с, 1H), 6,99 (дд, J=9,0, 2,4 Гц, 1H), 3,84 (с, 3H).
Стадия E: Раствор продукта со стадии D (1,8 г, 6,2 ммоль) и йодметана (1,2 мл, 18,5 ммоль) в хлороформе (40 мл) перемешивали при 60°C в течение 15 часов. Охлажденную смесь концентрировали в вакууме, получая метилированный неочищенный продукт (2,6 г, 97%, 79% AUC ВЭЖХ):1H ЯМР (300 МГц, CDCl3) δ 10,83 (с, 1H), 8,71 (д, J=8,1 Гц, 1H), 8,46 (д, J=8,4 Гц, 1H), 8,34 (с, 1H), 8,10 (дт, J=7,2, 1,2 Гц, 1H), 7,96 (т, J=7,2 Гц, 1H), 7,68-7,74 (м, 2H), 7,32 (д, J=2,4 Гц), 7,07 (дд, J=9,0, 2,7 Гц, 1H), 4,75 (с, 3H), 3,85 (с, 3H).
Стадия F: Цианоборгидрид натрия (0,85 г, 13,5 ммоль), затем две капли метанольного раствора бромкрезолового зеленого добавляли к раствору неочищенного продукта со стадии E (2,6 г, 6,0 ммоль) в метаноле (10 мл). Наблюдали голубую окраску. Добавляли HCl в метаноле вплоть до того, как окраска реакционной смеси становилась желтой. Полученную в результате смесь перемешивали при комнатной температуре в течение 1 часа с периодическим добавлением HCl в метаноле для поддержания желтой окраски. Смесь гасили добавлением водного раствора гидроксида натрия (10 мл, 3 М) и дважды экстрагировали этилацетатом. Органические экстракты промывали насыщенным раствором соли, сушили над сульфатом магния и концентрировали, получая требуемый тетрагидроизохинолин (1,6 г, 86%, 99,3% AUC ВЭЖХ) после очистки хроматографией (5:1 гептан/этилацетат):1H ЯМР (300 МГц, CDCl3) δ 7,60 (д, J=8,7 Гц, 1H), 7,09-7,21 (м, 6H), 6,92 (дд, J=8,7, 2,4 Гц, 1H), 4,55 (т, J=10,8 Гц, 1H), 3,86 (с, 3H), 3,79 (д, J=15,0 Гц, 1H), 3,64 (д, J=15,0 Гц, 1H), 3,02 (дд, J=11,4, 4,5 Гц, 1H), 2,91 (дд, J=11,4, 6,0 Гц, 1H), 2,50 (с, 3H).
Стадия G: Продукт со стадии F (1,6 г) кристаллизовали с малеиновой кислотой в дихлорметане. Полученные в результате твердые вещества растирали в MTBE и сушили в условиях высокого вакуума, получая малеатную соль 4-(5-метоксибензотиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (2,1 г, 95%, 99,3% AUC ВЭЖХ):1H ЯМР (300 МГц, МеОD) δ 7,68 (д, J=9,0 Гц, 1H), 7,25-7,37 (м, 6H), 6,99 (дд, J=9,0, 2,7 Гц, 1H), 6,24 (с, 2H), 5,04 (дд, J=9,9, 6,0 Гц, 1H), 4,59 (с, 2H), 3,99 (дд, J=12,6, 6,0 Гц, 1H), 3,85 (с, 3H), 3,75 (дд, J=12,3, 10,2 Гц, 1H), 3,11 (с, 3H), EI МС m/z=310 [C19H19NOS+H]+. Элементный анализ, вычислено для C23H23NO5S: C, 63,17; Н, 5,44; N, 3,15; S, 7,20 с 1,7% Н2O. Найдено: C, 62,94; Н, 5,42; N, 2,75; S, 6,80.
Пример 37 - Получение 4-(6-метоксибензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
4-(6-Метоксибензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин (29 мг, 96,9% AUC ВЭЖХ) получали из 3-метоксибензолтиола и диэтилацеталя бромацетальдегида, как описано в примере 36 (стадии A-F):1H ЯМР (300 МГц, CDCl3) δ 7,57 (д, J=8,7 Гц, 1H), 7,24 (д, J=2,4 Гц, 1H), 7,09-7,22 (м, 4H), 7,07 (с, 1H), 6,95 (дд, J=2,4, 8,7 Гц, 1H), 4,54 (т, J=5,4 Гц, 1H), 3,85 (с, 3H), 3,78 (д, J=15,0 Гц, 1H), 3,65 (д, J=15,0 Гц, 1H), 3,02 (дд, J=4,8, 11,4 Гц, 1H), 2,90 (дд, J=6,3, 11,4 Гц, 1H), 2,51 (с, 3H); EI МС m/z=310 [C19H19NOS+H]+.
Пример 38 - Получение 4-(бензо[b]тиофен-3-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
4-(Бензо[b]тиофен-3-ил)-2-метил-1,2,3,4-тетрагидроизохинолин получали из 3-бензо[b]тиофенбороновой кислоты и 4-бромизохинолина, как описано в примере 36 (стадии D-F):1H ЯМР (300 МГц, CDCl3) δ 7,55-7,58 (м, 7,88-7,82 (м, 1H), 1H), 7,22-7,27 (м, 2H), 6,90-7,10 (м, 5H), 4,69 (дд, J=6,9, 6,6 Гц, 1H), 3,72 (д, J=15,0 Гц, 1H), 3,63 (д, J=15,0 Гц, 1H), 3,00 (дд, J=11,4, 5,4 Гц, 1Н), 2,73 (дд, J=11,4, 8,1 Гц, 1H), 2,38 (с, 3H); EI МС m/z=280 [C18H17NS+H]+.
Пример 39 - Получение малеатной соли 4-(бензо[b]тиофен-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Малеатную соль 4-(бензо[b]тиофен-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (0,13 г, 99,6% AUC ВЭЖХ) получали из 3-бромбензолтиола, как описано в примере 69:1H ЯМР (300 МГц, ДМСО-d6) δ 7,87 (д, J=7,9 Гц, 1H), 7,68 (д, J=5,5 Гц, 1H), 7,01-7,34 (м, 6H), 6,62 (д, J=7,7 Гц, 1H), 4,87-4,92 (м, 1H), 4,39 (кв., J=15,5 Гц, 2H), 3,60-3,64 (м, 1H), 3,4 (т, J=11,4 Гц, 1H), 2,78 (с, 3H); EI МС m/z=280 [C18H17NS+H]+. Элементный анализ, вычислено для C22H21NO4S: C, 64,60; Н, 5,48; N, 3,42; S, 7,83. Найдено: C, 66,55; Н, 5,52; N, 3,46; S, 7,87.
Пример 40 - Получение гидрохлоридной соли (+)-4-(бензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина и гидрохлоридной соли (-)-4-(бензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: Диэтилацеталь бромацетальдегида (19 мл, 132 ммоль) по каплям добавляли к суспензии 4-бромбензолтиола (25 г, 126 ммоль) и карбоната калия (18 г, 132 ммоль) в ацетоне (200 мл) при комнатной температуре. После перемешивания в течение 15 часов в указанных условиях реакционную смесь фильтровали через диатомовую землю, промывали ацетоном и фильтрат концентрировали в вакууме. Полученный в результате остаток помещали в воду и этилацетат. Два слоя разделяли и водную фазу дважды экстрагировали этилацетатом. Объединенные органические экстракты промывали водным раствором гидроксида натрия (1 М) и насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали в вакууме, получая требуемый продукт (35 г, 91%, 100% AUC ГХ) после хроматографии (9:1 гептан/этилацетат):1H ЯМР (300 МГц, CDCl3) δ 7,24-7,45 (м, 4H), 4,64 (т, J=5,5 Гц, 1H), 3,49-3,74 (м, 4H), 1,71 (д, J=5,5 Гц, 2H), 1,18-1,27 (м, 6H).
Стадия B: Раствор продукта со стадии A (35 г, 92 ммоль) в хлорбензоле (50 мл) по каплям добавляли к раствору полифосфорной кислоты (100 г) в хлорбензоле (250 мл) при 135°C. После перемешивания при 135°C в течение 1 часа смесь охлаждали ниже 50°C. Хлорбензольный слой выливали из реакционной колбы и концентрировали в вакууме. В это время в реакционную колбу добавляли воду при 0°C, чтобы разложить полифосфорную кислоту. Полученную в результате водную фазу добавляли к остатку хлорбензольного слоя. Добавляли дополнительное количество воды и дихлорметана и две фазы разделяли. Водный слой дважды экстрагировали дихлорметаном. Объединенные органические экстракты сушили над безводным сульфатом натрия и концентрировали в вакууме, получая циклизованный продукт (18 г, 72%) после хроматографии (100% гептан):1H ЯМР (300 МГц, CDCl3) δ 7,98 (д, J=1,8 Гц, 1H), 7,76 (д, J=8,6 Гц, 1H), 7,44-7,50 (м, 2H), 7,29 (д, J=5,8 Гц, 1H).
Стадия C: Изохинолин-4-илбороновую кислоту (2,4 г, 14,0 ммоль) добавляли к раствору продукта со стадии B (2,0 г, 9,4 ммоль) и трифенилфосфина (0,5 г, 1,9 ммоль) в 1,2-диметоксиэтане (50 мл). Суспензию дегазировали, используя азот. Добавляли ацетат палладия (0,2 г, 0,9 ммоль) и порцию перемешивали при комнатной температуре в течение 20 минут. Добавляли водный раствор карбоната натрия (11,3 мл, 2 М раствор) и суспензию снова дегазировали, используя азот. Смесь перемешивали при 85°C в течение 3,5 часов, охлаждали, разбавляли водой и дважды экстрагировали этилацетатом. Объединенные органические экстракты сушили над безводным сульфатом натрия, концентрировали в вакууме и очищали хроматографией (5:1 гептан/этилацетат), получая требуемый изохинолин (1,8 г, 74%, 98% AUC ВЭЖХ):1H ЯМР (300 МГц, CDCl3) δ 9,30 (с, 1Н), 8,59 (с, 1H), 7,95-8,07 (м, 4H), 7,43-7,72 (µ, 5Н).
Стадия D: Раствор продукта со стадии C (1,8 г, 7,0 ммоль) и йодметана (1,3 мл, 21,0 ммоль) в хлороформе (30 мл) перемешивали при 60°C в течение 15 часов. Охлажденную смесь концентрировали в вакууме, получая метилированный неочищенный продукт (3,1 г, 81,9% AUC ВЭЖХ):1H ЯМР (300 МГц, ДМСО-d6) δ 10,99 (с, 1H), 8,81 (д, J=8,1 Гц, 1H), 8,27 (с, 1H), 8,02-8,17 (м, 4H), 7,49-7,55 (м, 4H), 4,86 (с, 3H).
Стадия E: Цианоборгидрид натрия (1,1 г, 17,3 ммоль), а затем две капли метанольного раствора бромкрезолового зеленого добавляли к раствору неочищенного продукта со стадии D (3,1 г, 7,7 ммоль) в метаноле (50 мл). Наблюдали голубую окраску. Добавляли HCl в метаноле вплоть до того, как реакционная смесь становилась желтой. Полученную в результате смесь перемешивали при комнатной температуре в течение 5 часов с периодическим добавлением HCl в метаноле, чтобы поддержать желтую окраску. Смесь гасили добавлением водного раствора гидроксида натрия (3 М) и дважды экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным раствором соли, сушили над безводным сульфатом магния и концентрировали, получая требуемый тетрагидроизохинолин (1,5 г, 78% для 2 стадий, 97,4% AUC ВЭЖХ) после очистки хроматографией (3:1 гептан/этилацетат):1H ЯМР (300 МГц, CDCl3) δ 7,81 (д, J=8,3 Гц, 1H), 7,68 (с, 1H), 7,44 (д, J=5,5 Гц, 1H), 7,3 (с, 1H), 7,10-7,22 (м, 4H), 6,91 (д, J=7,6 Гц, 1H), 4,42 (т, J=7,5 Гц, 1H), 3,81 (д, J=15,0 Гц, 1H), 3,68 (д, J=14,7 Гц, 1H), 3,10 (ддд, J=11,4, 5,7, 1,2 Гц, 1H), 2,63-2,69 (м, 1H), 2,46 (с, 3H).
Стадия F: Рацемическое соединение со стадии E (105 мг) разделяли с помощью полупрепаративной хиральной ВЭЖХ (chiralcel OD-H, 1×25 см, элюент: 3% изопропанол в гептане, расход: 4 мл/минуту, впрыскивание 500 мкл, 5 мг/впрыскивание). Полученные в результате свободные основания растворяли в этилацетате и обрабатывали 2 М раствором HCl в диэтиловом эфире (2 экв.), получая гидрохлоридную соль (+)-4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолина (47 мг, 100% AUC ВЭЖХ, 100% AUC хиральная ВЭЖХ):1H ЯМР (300 МГц, MeOD) δ 7,94 (д, J=8,4 Гц, 1H), 7,82 (с, 1Н), 7,65 (д, J=5,4 Гц, 1H), 7,19-7,37 (м, 5H), 6,93 (д, J=7,5 Гц, 1H), 4,64-4,81 (м, 3H), 3,90-3,96 (м, 1H), 3,62 (т, J=12,3 Гц, 1H), 3,12 (с, 3H); EI МС m/z=280 [C18H17NS+H]+; [α]25D +60° (c 1,0, MeOH, свободное основание), и гидрохлоридную соль (-)-4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолина (46 мг, 99,7% AUC ВЭЖХ, 100% хиральная ВЭЖХ):1H ЯМР (300 МГц, MeOD) δ 7,94 (дд, J=8,4, 2,4 Гц, 1H), 7,82 (с, 1H), 7,65 (дд, J=5,4, 2,4 Гц, 1H), 7,19-7,39 (м, 5H), 6,93 (д, J=7,8 Гц, 1H), 4,64-4,82 (м, 3H), 3,86-3,96 (м, 1H), 3,61 (т, J=12,6 Гц, 1H), 3,12 (с, 3H); EI МС m/z=280 [C18H17NS+H]+, [α]25D -60° (c 1,0, MeOH, свободное основание).
Пример 41 - Получение гидрохлоридной соли 4-(бензо[b]тиофен-5-ил)-2,7-диметил-1,2,3,4-тетрагидроизохинолина
Стадия A: Этилхлорформиат (28 мл, 293 ммоль) по каплям добавляли к раствору 4-метилциннамовой кислоты (40 г, 247 ммоль) и триэтиламина (69 мл, 492 ммоль) в ацетоне (300 мл) при 0°C и полученную в результате смесь перемешивали при 0°C в течение 1 часа. По каплям добавляли раствор азида натрия (26 г, 400 ммоль) в воде (100 мл), поддерживая температуру смеси ниже 5°C. После завершения добавления смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли воду (400 мл) и ацетон удаляли в вакууме (примечание: температуру бани роторного испарителя поддерживали при 40°C и перед роторным испарителем помещали взрывозащитный экран). Полученную в результате взвесь экстрагировали толуолом (3×100 мл). Объединенные органические экстракты сушили над безводным сульфатом магния и фильтровали. Фильтрат по каплям добавляли к раствору трибутиламина (120 мл, 503 ммоль) в дифениловом эфире (200 мл) при 190°C. Толуол отгоняли из реакционной смеси по мере осуществления добавления. После окончания добавления смесь перемешивали при 210°C в течение 2 часов и охлаждали до комнатной температуры. Полученную в результате взвесь фильтровали и промывали гептаном, получая требуемый изохинолон (24 г, 62%) в виде желтого твердого вещества:1H ЯМР (300 МГц, CDCl3) δ 8,25 (с, 1Н), 7,47-7,51 (м, 2H), 7,18 (д, J=6,9 Гц, 1H), 6,57 (д, J=6,9 Гц, 1H), 2,52 (с, 3H).
Стадия B: Раствор брома (4,8 мл, 94 ммоль) в уксусной кислоте (50 мл) по каплям добавляли к раствору продукта со стадии A (15,0 г, 94 ммоль) в уксусной кислоте (300 мл) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 4 часов, вливали в ледяную воду, три раза экстрагировали дихлорметаном и сушили над сульфатом магния, получая требуемый продукт (24,2 г):1H ЯМР (300 МГц, CDCl3) δ 8,24 (с, 1H), 7,80 (д, J=8,4 Гц, 1Н), 7,63 (дд, J=8,4, 1,8 Гц, 1H), 7,39 (с, 1H), 2,55 (с, 3H).
Стадия C: Раствор продукта со стадии B (24,2 г, 102 ммоль) в оксихлориде фосфора (250 мл) перемешивали при 110°C в течение 4 часов. Смесь охлаждали до комнатной температуры и оксихлорид фосфора выпаривали в вакууме. Остаток гасили насыщенным раствором бикарбоната натрия при 0°C и три раза экстрагировали дихлорметаном. Объединенные органические экстракты промывали насыщенным раствором бикарбоната натрия и сушили над сульфатом магния, получая требуемое вещество (22,0 г, 91% для 2 стадий):1H ЯМР (300 МГц, CDCl3) δ 8,41 (с, 1H), 8,10 (д, J=0,6 Гц, 1H), 8,06 (д, J=8,7 Гц, 1H), 7,69 (дд, J=8,7, 1,5 Гц, 1H), 2,63 (с, 3H).
Стадия D: Красный фосфор (3,9 г, 125 ммоль) добавляли к раствору продукта со стадии C (9,5 г, 27 ммоль) в йодистоводородной кислоте (22,7 мл, 57% мас. в воде) и смесь перемешивали при 140°C в течение 5 часов. После охлаждения до комнатной температуры смесь вливали в насыщенный раствор бикарбоната натрия (500 мл, содержащий 10 г сульфита натрия). Добавляли дихлорметан (250 мл) и смесь фильтровали через диатомовую землю, чтобы удалить фосфор. Фильтрат три раза экстрагировали дихлорметаном и сушили над сульфатом магния, получая 4-бром-7-метилизохинолин (4,6 г, 76%, 98,3% AUC ГХ) после очистки хроматографией (5:1 гептан/этилацетат):1H ЯМР (300 МГц, CDCl3) δ 9,01 (с, 1H), 8,61 (с, 1H), 7,96 (д, J=8,7 Гц, 1H), 7,64 (с, 1H), 7,56 (д, J=8,7 Гц, 1H), 2,53 (с, 3H).
Стадия E: Бис(пинаколато)дибор (1,3 г, 5,12 ммоль), дихлор[1,1'-бис(дифенилфосфино)ферроцен]палладия(II) дихлорметан (0,1 г, 0,14 ммоль) и ацетат калия (1,4 г, 14,26 ммоль) загружали в 3-горлую круглодонную колбу объемом 100 мл, которая была высушена с помощью струйной сушилки в вакууме и охлаждена в азоте перед использованием. Смесь дегазировали три раза с использованием азота. Добавляли раствор 5-бромтиофена (1,0 г, 4,69 ммоль) в диметилсульфоксиде (20 мл), смесь снова три раза дегазировали и перемешивали при 85°C в течение 1,5 часов. После охлаждения до комнатной температуры смесь фильтровали через диатомовую землю и промывали водой и этилацетатом. Дополнительно добавляли воду к фильтрату, который три раза экстрагировали этилацетатом. Объединенные органические экстракты один раз промывали насыщенным раствором соли и сушили над сульфатом натрия, получая требуемое вещество (0,8 г, 64%, 100% AUC ГХ) после хроматографии (от 9:1 до 5:1 гептан/этилацетат):1H ЯМР (300 МГц, CDCl3) δ 8,24 (с, 1H), 7,81 (д, J=8,1 Гц, 1H), 7,68 (дд, J=8,1, 0,6 Гц, 1H), 7,34 (д, J=5,4 Гц, 1H), 7,28 (дд, J=5,4, 0,6 Гц, 1H), 1,30 (с, 12H).
Стадия F: Водный раствор бикарбоната натрия (4,30 мл, 2 М) добавляли к раствору бромизохинолина со стадии D (0,64 г, 2,9 ммоль), продукта со стадии E (0,75 г, 2,9 ммоль) и трифенилфосфина (0,30 г, 1,2 ммоль) в ДМФА (20 мл) при комнатной температуре. Полученную в результате смесь три раза дегазировали, используя азот. Добавляли ацетат палладия (II) (0,07 г, 0,3 ммоль), смесь три раза дегазировали, используя азот, и перемешивали при 80°C в течение 15 часов. После охлаждения до комнатной температуры смесь фильтровали через диатомовую землю и промывали водой и этилацетатом. Фильтрат промывали два раза водой и насыщенным раствором соли и сушили над сульфатом магния, получая требуемый связанный изохинолин (0,70 г, 88%) после хроматографии (от 5:1 до 3:1 гептан/этилацетат):1H ЯМР (300 МГц, CDCl3) δ 7,94 (д, J=8,4 Гц, 1H), 7,88 (д, J=1,2 Гц, 1H), 7,75-7,78 (м, 2H), 7,47 (д, J=5,4 Гц, 1H), 7,40-7,44 (м, 2H), 7,34 (д, J=5,4 Гц, 1H), 2,50 (с, 3H).
Стадия G: Продукт со стадии F (0,7 г, 2,5 ммоль) метилировали согласно способу, описанному в примере 40 (стадия D), получая требуемый метилированный изохинолин (1,0 г, 95%):1H ЯМР (300 МГц, ДМСО-d6) δ 9,93 (с, 1H), 8,79 (д, J=0,9 Гц, 1H), 8,30-8,33 (м, 2H), 8,07-8,16 (м, 3H), 7,96 (д, J=5,7 Гц, 1H), 7,58-7,63 (м, 2H), 4,53 (с, 3H), 2,65 (с, 3H).
Стадия H: Продукт со стадии G (1,01 г, 2,4 ммоль) восстанавливали, следуя способу, описанному в примере 40 (стадия E), получая требуемый тетрагидроизохинолин (0,38 г, 53%, 97,3% AUC ВЭЖХ):1H ЯМР (300 МГц, CDCl3) δ 7,80 (д, J=8,4 Гц, 1H), 7,68 (д, J=1,5 Гц, 1H), 7,43 (д, J=5,4 Гц, 1H), 7,28-7,29 (м, 1H), 7,20 (дд, J=8,4, 1,8 Гц, 1H), 6,89-6,94 (м, 2H), 6,79 (д, J=7,8 Гц, 1H), 4,38 (т, J=7,8 Гц, 1H), 3,76 (д, J=14,7 Гц, 1H), 3,63 (д, J=15,0 Гц, 1H), 3,08 (ддд, J=11,4, 5,7, 1,2 Гц, 1Н), 2,63 (дд, J=11,4, 8,4 Гц, 1H), 2,45 (с, 3H), 2,32 (с, 3H).
Стадия I: Гидрохлоридную соль 4-бензо[b]тиофен-5-ил-2,7-диметил-1,2,3,4-тетрагидроизохинолина (0,24 г, 64%) получали растворением продукта со стадии H (0,33 г, 1,1 ммоль) в этилацетате и обрабатывая полученный в результате раствор 2 М хлористым водородом в диэтиловом эфире (2 экв.) при комнатной температуре:1H ЯМР (300 МГц, ДМСО-d6) δ 11,52 (ушир.с, 1Н), 8,01 (д, J=8,1 Гц, 1H), 7,79-7,82 (м, 2H), 7,45 (д, J=4,2 Гц, 1H), 7,18 (д, J=8,4 Гц, 1H), 7,07 (с, 1H), 7,05 (д, J=6,3 Гц, 1H), 6,64 (д, J=7,8 Гц, 1H), 4,64-4,75 (м, 1H), 4,49 (с, 2H), 3,45-3,82 (м, 2H), 2,92 (с, 3H), 2,28 (с, 3H); EI МС m/z=294 [C19H19NS+H]+. Элементный анализ, вычислено для C19H20ClNS: C, 68,44; Н, 6,03; N, 4,20 с 1,1 экв. НCl. Найдено: C, 68,49; Н, 5,76; N,4,05.
Пример 42 - Получение гидрохлоридной соли 4-(2-метилбензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: Смесь 4-бромтиофенола (5,0 г, 26,4 ммоль), 2,3-дихлор-1-пропена (2,6 г, 23,8 ммоль) и карбоната калия (4,4 г, 31,7 ммоль) в ацетоне (20 мл) перемешивали при 55°C в течение 6 часов. После охлаждения до комнатной температуры ацетон удаляли в вакууме и остаток помещали в воду и дважды экстрагировали этилацетатом. Объединенные органические слои промывали 1 М гидроксидом натрия, водой и насыщенным раствором соли, сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. К остатку добавляли диэтиланилин (30 мл) и полученную в результате смесь перемешивали при 185°C в течение 15 часов. К охлажденной смеси добавляли этилацетат и диэтиланилин удаляли промывкой 1 М HCl четыре раза. Органический слой сушили над карбонатом натрия, получая 5-бром-2-метилбензотиофен (5,3 г, 88%, 95,1% AUC ГХ):1H ЯМР (300 МГц, CDCl3) δ 7,70 (с, 1H), 7,52 (д, J=8,7 Гц, 1H), 7,26 (дд, J=8,7, 1,8 Гц, 1H), 6,83 (с, 1H), 2,51 (с, 3H).
Стадия B: 4-(2-метилбензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин получали из 5-бром-2-метилбензотиофена и изохинолин-4-илбороновой кислоты, как описано в примере 40 (стадии C-E). Свободное основание растворяли в этилацетате и обрабатывали 2 М хлористым водородом в диэтиловом эфире (2 экв.), получая соответствующую гидрохлоридную соль (0,41 г, 98,8% AUC ВЭЖХ):1H ЯМР (300 МГц, ДМСО-d6) δ 11,09 (ушир.с, 1H), 7,88 (д, J=8,4 Гц, 1H), 7,63 (с, 1H), 7,09-7,27 (м, 5H), 6,76 (д, J=7,5 Гц, 1H), 4,69 (дд, J=10,8, 6,3 Гц, 1H), 4,55 (ушир.с, 2H), 3,54-3,83 (м, 2H), 2,94 (с, 1H), 2,57 (с, 3H); EI МС m/z=293 [C19H19NS]+. Элементный анализ, вычислено для C19H20ClNS: C, 68,61; Н, 6,10; N, 4,21 с 0,73% Н2O. Найдено: C, 68,35; Н, 6,55; N, 4,00.
Пример 43 - Получение гидрохлоридной соли 4-(бензо[b]тиофен-5-ил)-7-фтор-2-метил-1,2,3,4-тетрагидроизохинолина
Гидрохлоридную соль 4-(бензо[b]тиофен-5-ил)-7-фтор-2-метил-1,2,3,4-тетрагидроизохинолина (0,31 г, 99,5% AUC ВЭЖХ) получали из 7-фторциннамовой кислоты и 5-бромтиофена, как описано в примере 41:1H ЯМР (300 МГц, ДМСО-d6) δ 11,48 (ушир.с, 1H), 8,03 (д, J=8,1 Гц, 1H), 7,81-7,82 (м, 2H), 7,46 (д, J=5,1 Гц, 1H), 7,20 (д, J=8,7 Гц, 1H), 7,00-7,08 (м, 1H), 6,79 (ушир.с, 1H), 4,68-4,76 (м, 1H), 4,54 (с, 2H), 3,54-3,82 (м, 3H), 2,93 (с, 3H), 2,51 (с, 3H); EI МС m/z=298 [C18H16FNS+H]+. Элементный анализ, вычислено для C18H17ClFNS: C, 64,01; Н, 5,07; N, 4,15 с 1,1 экв. НCl. Найдено: C, 63,91; Н, 5,50; N, 3,86.
Пример 44 - Получение малеатной соли 4-(бензо[b]тиофен-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Малеатную соль 4-(бензо[b]тиофен-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (0,09 г, 99,7% AUC ВЭЖХ) получали из 3-бромбензолтиола, как описано в примере 69:1H ЯМР (300 МГц, ДМСО-d6) δ 7,80-7,86 (м, 2H), 7,71 (д, J=5,4 Гц, 1H), 7,39 (д, J=5,4 Гц, 1H), 7,11-7,22 (м, 4H), 6,76 (д, J=7,6 Гц, 1H), 6,00 (с, 2H), 4,53-4,59 (м, 1H), 4,34-4,46 (м, 2H), 3,65-3,81 (м, 1H), 3,46 (т, J=11,3 Гц, 1H), 3,26 (с, 3H); EI МС m/z=280 [C18H17NS+H]+. Элементный анализ, вычислено для C22H21NO4S: C, 66,82; Н, 5,35; N, 3,54; S, 8,11. Найдено: C, 66,76; Н, 5,26; N, 3,42; S, 7,97.
Пример 45 - Получение гидрохлоридной соли 4-индол-1-ил-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: 4-Индол-1-илизохинолин (0,88 г, 3,6 ммоль) и йодметан (0,67 мл, 10,8 ммоль) в хлороформе (18 мл) перемешивали при 60°C в течение 15 часов. Охлажденную смесь концентрировали в вакууме, неочищенное метилированное вещество (1,5 г, 76,4% AUC ВЭЖХ):1H ЯМР (300 МГц, CDCl3) δ 11,05 (с, 1H), 8,86-8,89 (м, 1H), 8,39-8,42 (м, 1H), 8,08-8,13 (м, 2H), 7,89-7,92 (м, 1H), 7,77-7,80 (м, 1H), 7,58 (д, J=3,3 Гц, 1H), 7,24-7,30 (м, 2H), 7,09-7,12 (м, 1H), 6,93 (д, J=3,3 Гц, 1H), 4,86 (с, 3H).
Стадия B: Цианоборгидрид натрия (0,55 г, 8,7 ммоль), а затем две капли раствора бромкрезолового зеленого в метаноле добавляли к раствору неочищенного продукта со стадии A (1,5 г, 3,9 ммоль) в метаноле (20 мл). Наблюдали голубую окраску. Добавляли HCl в метаноле до тех пор, пока реакционная смесь не становилась желтой. Полученную в результате смесь перемешивали при комнатной температуре в течение 5 часов с периодическим добавлением HCl в метаноле, чтобы поддержать желтую окраску. Смесь гасили добавлением водного раствора гидроксида натрия (3 М) и дважды экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным раствором соли, сушили над безводным сульфатом магния и концентрировали, получая требуемый тетрагидроизохинолин (0,65 г, 64% для двух стадий, 94,9% AUC ВЭЖХ) после двух раз очистки хроматографией (5:1 гептан/этилацетат):1H ЯМР (300 МГц, CDCl3) δ 7,57 (д, J=7,8 Гц, 1H), 7,29 (д, J=8,1 Гц, 1H), 7,01-7,19 (м, 3H), 6,94 (д, J=3,0 Гц, 1H), 6,87 (д, J=7,8 Гц, 1H), 6,39-6,40 (м, 1H), 5,70-5,74 (м, 1H), 3,66 (с, 2H), 3,00 (дд, J=11,4, 5,1 Гц, 1H), 2,82 (дд, J=11,4, 6,9 Гц, 1H), 2,37 (с, 3H).
Стадия C: Продукт со стадии B (37 мг, 0,14 ммоль) растворяли в этилацетате (1 мл) и обрабатывали 2 М раствором HCl в диэтиловом эфире (0,14 мл), получая гидрохлоридную соль 4-индол-1-ил-2-метил-1,2,3,4-тетрагидроизохинолина (28 мг, 64%, 98% AUC ВЭЖХ):1H ЯМР (300 МГц, MeOD) δ 7,62 (д, J=7,5 Гц, 1H), 7,09-7,40 (м, 8H), 6,59 (с, 1H), 6,29 (м, 1H), 4,59-4,78 (м, 2H), 3,81-4,08 (м, 2H), 3,15 (с, 3H); EI МС m/z=263 [C18H18N2+H]+; элементный анализ, вычислено для C18H18N2 с 1,2 НCl: C, 70,59; Н, 6,27; N, 9,15. Найдено: C, 70,74; Н, 6,41; N, 8,96.
Пример 46 - Получение 4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: Индол (3,00 г, 25,6 ммоль) и ди-трет-бутилдикарбонат (6,15 г, 28,2 ммоль) объединяли, как описано для синтеза в примере 48, стадия A. Неочищенный продукт переносили на стадию B после концентрирования в вакууме.
Стадия B: Неочищенный продукт, полученный на стадии A, и триизопропилборат (7,22 г, 38,4 ммоль) объединяли, как описано для синтеза в примере 48, стадия B, получая при перекристаллизации продукт в виде белого твердого вещества (3,41 г, 51% для стадий A и B):1H ЯМР (300 МГц, CD3OD) δ 8,10 (д, J=7,7 Гц, 1H), 7,54 (д, J=6,3 Гц, 1H), 7,28-7,17 (м, 2H), 6,63 (с, 1H), 1,68 (с, 9H).
Стадия C: Бороновую кислоту, полученную на стадии B (1,50 г, 5,74 ммоль), и 4-бромизохинолин (956 мг, 4,60 ммоль) объединяли, как описано для синтеза в примере 48, стадия C, получая после хроматографии продукт в виде белого твердого вещества (1,05 г, 66%).1H ЯМР (300 МГц, CDCl3) δ 9,29 (с, 1H), 8,57 (с, 1H), 8,37 (д, J=8,3 Гц, 1H), 8,07-8,01 (м, 1Н), 7,68-7,62 (м, 4H), 7,42-7,32 (м, 2H), 6,72 (с, 1H), 0,87 (с, 9H).
Стадия D: Продукт, полученный на стадии C (1,05 г, 3,05 ммоль), и метилтрифторметансульфонат (551 мг, 3,36 ммоль) объединяли, как описано для синтеза в примере 48, стадия D, получая желтую соль, которую переносили на стадию E.
Стадия E: Неочищенный продукт, полученный на стадии D, и цианоборгидрид натрия (767 мг, 12,2 ммоль) объединяли, как описано для синтеза в примере 48, стадия E, получая продукт (698 мг, 87% для стадий D и E) в виде светло-коричневого твердого вещества.
Стадия F: Продукт, полученный на стадии E, и малеиновую кислоту (340 мг, 2,93 ммоль) объединяли, как описано для синтеза в примере 48, стадия F, получая после перекристаллизации продукт (530 мг, 48%) в виде белого твердого вещества, т.пл. 167-169°C;1H ЯМР (300 МГц, CD3OD) δ 7,48 (д, J=7,8 Гц, 1H), 7,35-7,27 (м, 4H), 7,15-6,97 (м, 3H), 6,34 (с, 1H), 6,24 (с, 2H), 4,84-4,80 (м, 1H), 4,53 (д, J=4,6 Гц, 2H), 3,91-3,87 (м, 1H), 3,73-3,69 (м, 1H), 3,07 (с, 3H); ESI-МС m/z 263 [C18H18N2+H]+; элементный анализ, вычислено для C18H18N2-C4H4O4: C, 69,83; Н, 5,86; N, 7,40. Найдено: C, 69,52; Н, 5,86; N, 7,18.
Пример 47 - Получение 4-(1H-индазол-5-ил)-2,7-диметил-1,2,3,4-тетрагидроизохинолина
Стадия A: К смеси 1H-индол-2-карбоновой кислоты (2,50 г, 15,5 ммоль) и ацетилхлорида (31 мл, 0,43 моль) в Et2O (31,5 мл) порциями добавляли пентахлорид фосфора (3,55 г, 17,1 ммоль) и затем смесь кипятили с обратным холодильником в течение 1,5 часов. Охлажденную реакционную смесь концентрировали при пониженном давлении и полученный таким образом сырой продукт перекристаллизовывали из гептанов, получая 1H-индол-2-карбонилхлорид (2,04 г, 73%) в виде желтых игольчатых кристаллов.
Стадия B: К ледяной двухфазной смеси 40% водного раствора KOH (7,88 г KOH в 19,7 г раствора) и Et2O (67 мл) добавляли 1-метил-3-нитро-1-нитрозогуанидин (4,35 г, 29,5 ммоль) в течение 15 минут. Через 30 минут ярко-желтый Et2O-раствор диазометана декантировали в охлажденную на льду колбу Эрленмейера, содержащую таблетки KOH. Через 2 часа ледяной раствор диазометана декантировали во вторую охлажденную на льду колбу Эрленмейера. К указанному выше раствору добавляли хлорангидрид (2,04 г, 11,4 ммоль) в течение 15 минут. Полученную в результате смесь перемешивали при 0°C в течение 1 часа и затем хранили при -30°C в течение ночи. В течение 30 минут барботировали азот через холодную реакционную смесь, которую затем разбавляли EtOAc (60 мл). Реакционную смесь промывали водой и насыщенным раствором соли, сушили (MgSO4), фильтровали и концентрировали, получая сырой продукт, который как таковой использовали на стадии C.
Стадия C: К ледяной суспензии неочищенного продукта со стадии B в Et2O (50 мл) по каплям добавляли 48% HBr (3 мл). Через 30 минут реакционную смесь разбавляли Et2O (25 мл) и промывали водой. Водный слой повторно экстрагировали Et2O (25 мл). Органические экстракты объединяли, промывали насыщенным раствором соли, сушили (MgSO4), фильтровали и концентрировали при пониженном давлении, получая сырой продукт (грубый выход 1,88 г), который использовали на стадии D без дополнительной очистки.
Синтез м-толиламина: К раствору м-толуальдегида (6,4 мл, 54,0 ммоль) в MeOH (50 мл) добавляли 40% водный метиламин (4,3 мл, 55,1 ммоль). Через 20 минут смесь охлаждали на ледяной бане и небольшими порциями добавляли боргидрид натрия (3,07 г, 81,1 ммоль) в течение 20 минут. Затем смесь нагревали до комнатной температуры и перемешивали в течение ночи. Затем реакционную смесь концентрировали при пониженном давлении и распределяли между водой и CH2Cl2 (100 мл каждого). Водный слой повторно экстрагировали CH2Cl2 (2×50 мл) и органические экстракты объединяли и промывали 2 N HCl (3×30 мл). Водный слой промывали CH2Cl2 (50 мл), обрабатывали концентрированным NH4OH (до pH 12) и экстрагировали CH2Cl2 (3×100 мл). Органические экстракты объединяли, промывали насыщенным раствором соли, сушили (MgSO4), фильтровали и концентрировали при пониженном давлении, получая м-толиламин (5,46 г, 75%), который использовали на стадии D без дополнительной очистки.1H ЯМР (300 МГц, CDCl3) δ 7,26-7,05 (м, 4H), 3,72 (с, 2H), 2,46 (с, 3H), 2,35 (с, 3H).
Стадия D: К ледяному раствору продукта со стадии C (1,88 г, неочищенный продукт) в CH2Cl2 (15,8 мл) добавляли метил-м-толиламин (1,1 г, 7,9 ммоль), затем диизопропилэтиламин (1,8 мл, 10,6 ммоль). Реакционную смесь сохраняли холодной в течение 30 минут и затем перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли водой и продукт экстрагировали в CH2Cl2 (3×40 мл). Органические экстракты объединяли, промывали насыщенным раствором соли и концентрировали при пониженном давлении. Очистка флэш-хроматографией на колонке (90:10 CH2Cl2/гексаны, затем CH2Cl2, 99:1 CH2Cl2/MeOH и 98:2 CH2Cl2/MeOH) давала частично очищенный продукт (1,86 г), который как таковой использовали на стадии E.1H ЯМР (300 МГц, CDCl3) δ 9,84 (ушир.с, 1H), 7,69 (д, J=8,0 Гц, 1H), 7,41-7,09 (м, 8H), 3,75 (с, 2Н), 3,67 (с, 2H), 2,42 (с, 3H), 2,35 (с, 3H).
Стадия E: К смеси продукта со стадии D (1,86 г) и DMAP (40 мг, 0,31 ммоль) в CH3CN (25 мл) добавляли Boc2O (1,46 г, 6,68 ммоль) и смесь перемешивали при комнатной температуре в течение 45 минут. Затем смесь разбавляли водой и CH2Cl2 (50 мл каждого). Органический слой отделяли, промывали насыщенным раствором соли и концентрировали при пониженном давлении. Очистка флэш-хроматографией на колонке (градиент от 95:5 гексаны/EtOAc до 75:25 гексаны/EtOAc) давала продукт (1,02 г, 17% для 5 стадий) в виде бледно-желтого масла:1H ЯМР (300 МГц, CDCl3) δ 8,04 (д, J=8,4 Гц, 1H), 7,59 (д, J=7,8 Гц, 1H), 7,42-7,39 (м, 1H), 7,27-7,25 (м, 2H), 7,16 (д, J=7,3 Гц, 1H), 7,08-7,03 (м, 4H), 3,64 (с, 2H), 3,60 (с, 2H), 2,37 (с, 3H), 2,28 (с, 3H), 1,59 (с, 9H).
Стадия F: К ледяному раствору продукта со стадии E (1,02 г, 2,60 ммоль) в MeOH (6,7 мл) небольшими порциями добавляли NaBH4 (0,11 г, 2,86 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи и в этот момент к смеси дополнительно добавляли NaBH4 (30 мг, 0,79 ммоль) и перемешивание продолжали еще в течение 2 часов. Затем реакционную смесь концентрировали досуха. Очистка флэш-хроматографией (градиент от 95:5 гексаны/EtOAc до 60:40 EtOAc/гексаны) давала требуемый продукт (0,29 г, 28%) в виде желтого масла:1H ЯМР 7,99 (д, J=8,3 Гц, 1H), 7,49 (д, J=7,2 Гц, 1H), 7,26-7,06 (м, 6H), 6,70 (с, 1H), 5,35-5,25 (м, 1H), 3,67 (д, J=13,0 Гц, 1H), 3,51 (д, J=13,0 Гц, 1H), 3,00-2,90 (м, 1H), 2,70-2,60 (м, 1H), 2,32 (с, 3H), 2,31 (с, 3H), 1,67 (с, 9H).
Стадия G: Раствор продукта со стадии F, описанной выше, (0,29 г, 0,74 ммоль) в 1,2-дихлорэтане (2,4 мл) по каплям через капельную воронку добавляли к метансульфоновой кислоте (2,7 мл) при 40°C. Через 30 минут реакционную смесь охлаждали до комнатной температуры и выливали на лед. pH полученной в результате смеси доводили до pH 9 концентрированным NH4OH и продукт экстрагировали в CH2Cl2 (4×15 мл). Органические экстракты объединяли, промывали насыщенным раствором соли, сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении. Очистка флэш-хроматографией на колонке (градиент от 90:10 гексаны/EtOAc, содержащий 0,05% TEA, до 55:45 гексаны/EtOAc, содержащий 0,05% TEA) давала требуемый продукт (59 мг, 29%) в виде коричневого масла.
Стадия H: К раствору продукта со стадии G, описанной выше, (59 мг, 0,21 ммоль) в EtOH (0,5 мл) при -30°C добавляли малеиновую кислоту (24 мг, 0,21 ммоль). Описанный выше раствор разбавляли EtOH (1 мл) и по каплям добавляли к Et2O (50 мл) при -30°C. Образованный преципитат отфильтровывали, промывали Et2O и сушили. Полученное не совсем белое твердое вещество затем растирали в смеси EtOAc/гексаны, фильтровали и сушили при пониженном давлении, получая продукт (47 мг, 51%) в виде не совсем белого твердого вещества:1H ЯМР (300 МГц, CD3OD) δ 7,47 (д, J=8,1 Гц, 1H), 7,28 (д, J=8,0 Гц, 1H), 7,11-6,96 (м, 5H), 6,31 (ушир.с, 1H), 6,25 (с, 2H), 4,76-4,72 (м, 1H), 4,42 (ушир.с, 2H), 3,83-3,79 (м, 1H), 3,60-3,50 (м, 1H), 3,01 (с, 3H), 2,00 (с, 3H); ESI МС m/z 277 [C19H20N2 + Н]+; элементный анализ, вычислено для C19H20N2-1,25C4H4O4-0,5H2O: C, 66,82; Н, 6,10; N, 6,49. Найдено: C, 67,16; Н, 6,25; N, 6,29.
Пример 48 - Получение малеатной соли 4-(5-метокси-1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору 5-метоксииндола (3,00 г, 20,4 ммоль) в CH3CN (15 мл) добавляли ди-трет-бутилдикарбонат (4,90 г, 22,4 ммоль) и каталитическое количество DMAP. Раствор перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли холодной 1 N HCl (30 мл) и экстрагировали EtOAc (3×30 мл). Органическую фазу сушили (Na2CO3) и концентрировали при пониженном давлении. Очистка флэш-хроматографией на колонке (CH2Cl2) давала N-трет-бутилкарбоксилат-5-метоксииндол (4,89 г, 97%) в виде белого твердого вещества.1H ЯМР (300 МГц, CDCl3) δ 8,01 (д, J=8,3 Гц, 1H), 7,56 (д, J=3,3 Гц, 1H), 7,02 (с, 1H), 6,922 (дд, J=9,0 Гц, 2,5 Гц, 1H), 6,49 (д, J=3,6 Гц, 1H), 3,85 (с, 3H), 1,66 (с, 9H).
Стадия B: К ледяному раствору N-трет-бутилкарбоксилат-5-метоксииндола (2,45 г, 9,90 ммоль) и триизопропилбората (2,83 г, 15,1 ммоль) в ТГФ (12,5 мл) добавляли 2,0 М LDA (6,3 мл в ТГФ, 12,5 мл). Реакционную смесь перемешивали при 0°C в течение 1 часа, после этого гасили 2 N HCl (водный раствор, 30 мл) и затем экстрагировали CH2Cl2 (40 мл). Органическую фазу сушили (Na2SO4) и концентрировали при пониженном давлении. Перекристаллизация концентрата в смеси 1:1 CH3CN/H2O (40 мл) давала N-трет-бутилкарбоксилат-5-метокси-2-индолбороновую кислоту (2,70 г, 94%) в виде белого твердого вещества.1H ЯМР (300 МГц, ДМСО-d6) δ 8,18 (с, 2H), 7,95 (д, J=8,9 Гц, 1H), 7,07 (д, J=2,5 Гц, 1H), 6,87 (дд, J=9,0 Гц, 2,6 Гц, 1H), 6,55 (с, 1H), 3,77 (с, 3H), 1,59 (с, 9H).
Стадия C: Смесь 4-бромизохинолина (1,05 г, 5,04 ммоль), продукта, полученного на стадии B (2,20 г, 7,56 ммоль), DME (13 мл) и 2 М Na2CO3 (6,3 мл, 12,6 ммоль) дегазировали (пять раз, вакуум/аргон). К полученной смеси добавляли Pd(PPh3)4 (291 мг, 0,252 ммоль). Полученную в результате смесь дегазировали (пять раз, вакуум/аргон) и затем нагревали при кипячении с обратным холодильником в течение ночи. Охлажденную реакционную смесь фильтровали и осадок на фильтре промывали CH2Cl2. Фильтрат обрабатывали 1 N NaOH (водный раствор, 20 мл) и экстрагировали CH2Cl2 (2×20 мл). Объединенную органическую фазу сушили (Na2SO4) и концентрировали при пониженном давлении. Очистка флэш-хроматографией на колонке (1:1 EtOAc/гексаны, смесь загружали на колонку в виде раствора в CH2Cl2) давала продукт (1,12 г, 59%) в виде прозрачного масла.1H ЯМР (300 МГц, CDCl3) δ 9,29 (с, 1H), 8,55 (с, 1H), 8,26 (д, J=9,0 Гц, 1H), 8,05-8,02 (м, 1H), 7,68-7,63 (м, 3H), 7,09 (д, J=2,5 Гц, 1H), 7,03 (дд, J=9,0 Гц, 2,6 Гц, 1H), 6,64 (с, 1H), 3,90 (с, 3H), 0,87 (с, 9H).
Стадия D: Метилтрифторметансульфонат (120 мг, 0,732 ммоль) добавляли по каплям к ледяному раствору продукта со стадии C (250 мг, 0,668 ммоль) в CH2Cl2 (2,3 мл). Полученную в результате взвесь перемешивали в течение 30 минут при комнатной температуре. Избыток метилтрифторметансульфоната гасили MeOH и полученный в результате раствор концентрировали в вакууме, получая соль пиридиния в виде желтого твердого вещества, который переносили на стадию E без дополнительной очистки.
Стадия E: Цианоборгидрид натрия (105 мг, 1,67 ммоль) добавляли к раствору продукта со стадии C в MeOH (5 мл). Реакционную смесь перемешивали в течение ночи при комнатной температуре. MeOH удаляли в вакууме и остаток разбавляли CH2Cl2. Раствор промывали 1 N NaOH (водный), сушили (Na2SO4) и концентрировали в вакууме. Неочищенное масло использовали на стадии F.
Стадия F: Раствор продукта со стадии E и малеиновую кислоту (83 мг, 0,711 ммоль) в EtOH (5 мл) перемешивали при -30°C в течение 1 часа. Образованную взвесь фильтровали при пониженном давлении и промывали EtOAc, получая продукт (120 мг, 36% для стадий E и F) в виде белого твердого вещества: т.пл. 117-120°C;1H ЯМР (300 МГц, CD3OD) δ 7,35-7,26 (м, 3H), 7,19-7,13 (м, 2H), 7,00 (с, 1H), 6,75 (дд, J=8,8 Гц, 2,4 Гц, 1H), 6,27-6,24 (м, 3H), 4,82-4,77 (м, 1H), 4,54 (д, J=3,7 Гц, 2H), 3,92-3,87 (м, 1H), 3,79 (с, 3H), 3,74-3,69 (м, 1H), 3,08 (с, 3H); ESI-МС m/z 293 [C19H20N2O+H]+; элементный анализ, вычислено для C19H20N2O-l,5C4H4O4: C, 64,37; Н, 5,62; N, 6,01. Найдено: C, 64,00; Н, 5,87; N, 6,17.
Пример 49 - Получение 4-(1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: 5-Бром-N-трет-бутилиндолкарбоксилат (1,28 мг, 4,34 ммоль) и 4-изохинолинбороновую кислоту (900 мг, 5,20 ммоль) объединяли, как описано для синтеза в примере 48, стадия C, получая после хроматографии продукт (314 мг, 21%) в виде коричневого масла.1H ЯМР (300 МГц, CDCl3) δ 9,26 (с, 1H), 8,54 (с, 1Н), 8,28 (д, J=8,4 Гц, 1H), 8,07-8,04 (м, 1H), 7,97-7,94 (м, 1H), 7,70-7,60 (м, 4H), 7,46 (дд, J=8,5 Гц, 1,7 Гц), 6,66 (д, J=3,6 Гц, 1H), 1,72 (с, 9H).
Стадия B: Продукт, полученный на стадии A (314 мг, 0,918 ммоль), и метилтрифторметансульфонат (164 мг, 1,00 ммоль) подвергали взаимодействию, как описано для синтеза в примере 48, стадия D, получая желтую соль, которую использовали на стадии C.
Стадия C: Неочищенный продукт, полученный на стадии B, и цианоборгидрид натрия (231 мг, 3,67 ммоль) объединяли, как описано для синтеза в примере 48, стадия E. Затем реакционную смесь концентрировали в вакууме, разбавляли CH2Cl2 и промывали 1 N NaOH (водным). Органическую фазу сушили (Na2SO4) и концентрировали при пониженном давлении. Очистка флэш-хроматографией на колонке (2:5 EtOAc/гексаны, смесь загружали на колонку в виде раствора в CH2Cl2) давала продукт (217 мг, 65% для стадий B и C) в виде прозрачного масла.1H ЯМР (300 МГц, CDCl3) δ 8,04 (д, J=8,7 Гц, 1H), 7,58 (д, J=3,6 Гц, 1H), 7,39 (д, J=1,5 Гц, 1H), 7,16-7,02 (м, 4H), 6,87 (д, J=7,7 Гц, 1H), 6,50 (д, J=3,7 Гц, 1H), 4,38 (т, J=7,0 Гц, 1Н), 3,82-3,61 (м, 2H), 3,11-3,05 (м, 1H), 2,65-2,58 (м, 1H), 2,44 (с,3H), 1,66 (с,9H).
Стадия D: К раствору продукта, полученного на стадии C (217 мг, 0,599 ммоль), в CH2Cl2 (10 мл) добавляли TFA (5 мл). Раствор перемешивали при комнатной температуре в течение 3 часа, после этого раствор концентрировали в вакууме. Затем концентрат растворяли в EtOH (10 мл), к которому добавляли концентрированный NH4OH (6 мл). Раствор перемешивали при комнатной температуре в течение 30 минут и концентрировали в вакууме, после этого концентрат растворяли в CH2Cl2 (30 мл) и промывали 2 N NaOH (водным). Органическую фазу сушили (Na2SO4) и концентрировали при пониженном давлении. Очистка флэш-хроматографией на колонке (98/1,5/0,5 CH2Cl2/MeOH/NH4OH, смесь загружали на колонку в виде раствора в CH2Cl2) давала продукт (40 мг, 25%) в виде светло-коричневого твердого вещества.1H ЯМР (500 МГц, CDCl3) δ 8,24 (с, 1H), 7,48 (с, 1H), 7,29 (д, J=8,4 Гц, 1H), 7,16-7,08 (м, 3H), 7,03 (т, J=7,4 Гц, 1H), 6,99 (д, J=7,4 Гц, 1H), 6,91 (д, J=7,7 Гц, 1H), 6,48 (с, 1H), 4,39 (т, J=7,5 Гц, 1H), 3,82 (д, J=14,8 Гц, 1Н), 3,62 (д, J=14,8 Гц, 1H), 3,13-3,01 (м, 1H), 2,62 (т, J=10,4 Гц, 1H), 2,45 (3H).
Стадия E: Продукт, полученный на стадии D, и малеиновую кислоту (19 мг, 0,167 ммоль) объединяли, как описано для синтеза в примере 48, стадия F, получая после перекристаллизации продукт (38 мг, 55%) в виде не совсем белого твердого вещества: т.пл. 185-189°C;1H ЯМР (500 МГц, CD3OD) δ 7,44 (м, 1H), 7,38 (д, J=8,4 Гц, 1H), 7,31-7,21 (м, 4H), 6,97 (д, J=7,7 Гц, 1H), 6,92 (дд, J=8,4 Гц, 1,6 Гц, 1H), 6,41 (д, J=2,5 Гц, 1H), 6,24 (с, 2H), 4,64-4,55 (м, 3H), 3,87-3,83 (м, 1H), 3,65-3,58 (м, 1H), 3,07 (с, 3H); ESI-МС m/z 263 [C18H18N2+H]+; элементный анализ, вычислено для Cl8H18N2-l,3C4H4O4: C, 67,50; Н, 5,66; N, 6,80. Найдено: C, 67,43; Н, 5,66; N, 6,78.
Пример 50 - Получение гидрохлоридной соли 2-метил-4-(2-метилбензо[b]тивзол-5-ил)-1,2,3,4-тетрагидроизохинолина
Стадия A: Нитрит натрия (0,9 г, 13 ммоль) порциями добавляли к суспензии дигидрохлорида 2-метил-5-аминобензотиазола (2,0 г, 8 ммоль) в бромистоводородной кислоте (24 мл) при 0°C. Полученную в результате смесь добавляли по каплям к раствору бромида меди (I) (4,0 г, 14 ммоль) в бромистоводородной кислоте (50 мл) при 0°C. После перемешивания при 0°C в течение 2 часов добавляли воду. Реакционную смесь подщелачивали до pH 9, используя водный раствор гидроксида аммония, и экстрагировали этилацетатом. Объединенные органические слои сушили над сульфатом магния, получая 2-метил-5-бромбензотиазол (1,0 г, 52%, 82% AUC ГХ) после хроматографии (9:1 гептан/этилацетат):1H ЯМР (300 МГц, ДМСО-d6) δ 8,12 (д, J=1,8 Гц, 8,02 (дд, J=8,4, 1,5 Гц, 1H), 7,56 (дд, J=8,7, 2,1 Гц, 1H), 2,81 (с, 3H).
Стадия B: Гидрохлоридную соль 2-метил-4-(2-метилбензо[b]тиазол-5-ил)-1,2,3,4-тетрагидроизохинолина (0,06 г, 97,4% AUC ВЭЖХ) получали из продукта со стадии A и 4-бромизохинолина, как описано в примере 41 (стадии E-I):1H ЯМР (300 МГц, MeOD) δ 7,98 (д, J=8,4 Гц, 1H), 7,85 (с, 1H), 7,24-7,36 (м, 4H), 6,92 (д, J=7,8 Гц, 1H), 4,75-4,85 (м, 1H), 4,65 (ушир.с, 2H), 3,85-4,00 (м, 1H), 3,55-3,68 (м, 1H), 3,07 (с, 3H), 2,87 (с, 3H); EI МС m/z=295 [C18H18N2S+H]+. Элементный анализ, вычислено для C18H19ClN2S: C, 61,91; Н, 5,45; N, 8,02 с 1,5 экв. НCl. Найдено: C, 61,61; Н, 5,79; N, 7,90.
Пример 51 - Получение малеатной соли 4-(1H-инден-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: 1H-инден-2-иловый сложный эфир трифторметансульфоновой кислоты синтезировали с выходом 87%, следуя способу, опубликованному в работе Huffman et al., J. Med. Chem. 39: 3875-3877 (1996), которая включена в данное описание в виде ссылки в полном объеме, используя 2-инданон:1H ЯМР (300 МГц, CDCl3) δ 8,05 (с, 1Н), 7,43-7,20 (м, 4H), 6,68 (с, 1H), 3,66 (с, 2H).
Стадия B: Раствор 1H-инден-2-илового сложного эфира трифторметансульфоновой кислоты (2,58 г, 10,39 ммоль), бис(пинаколато)дибора (5,28 г, 20,79 ммоль) и KOAc (3,06 г, 31,18 ммоль) в ДМСО (54 мл) продували аргоном. К полученной смеси добавляли PdCl2dppf-CH2Cl2 (0,679 г, 0,83 ммоль). Полученную в результате смесь продували аргоном и нагревали до 80°C в течение 5 часов. Охлажденную реакционную смесь разбавляли водой (50 мл) и экстрагировали CH2Cl2 (1×200 мл, 2×100 мл). Объединенные органические слои концентрировали до меньшего объема при пониженном давлении. Остаток разбавляли EtOAc (200 мл), промывали водой (1×50 мл, 2×25 мл), насыщенным раствором соли (2×25 мл), сушили (Na2SO4) и концентрировали при пониженном давлении, получая 2-(1H-инден-2-ил)-4,4,5,5-тетраметил-[1,3,2]диоксаборолан в виде черного масла, которое использовали на стадии C без дополнительной очистки.
Стадия C: Раствор продукта, полученного на стадии B (~10 ммоль), 4-бромизохинолина (1,39 г, 6,66 ммоль) и Pd(PPh3)4 (0,46 г, 0,40 ммоль) в ДМФА (33 мл) продували аргоном. К полученной смеси добавляли раствор Cs2CO (8,68 г, 26,64 ммоль) в воде (13 мл). Полученную в результате смесь продували аргоном и нагревали до 88°C в течение ночи. Охлажденную реакционную смесь разбавляли водой (25 мл) и экстрагировали EtOAc (3×150 мл). Объединенный органический слой концентрировали при пониженном давлении. Очистка флэш-хроматографией (1:1:1 CH2Cl2/гексаны/EtOAc) давала продукт (0,649 г, 40%) в виде коричневого твердого вещества:1H ЯМР (300 МГц, CDCl3) δ 9,18 (с, 1H), 8,57 (с, 1H), 8,28 (д, 1H, J=8,5 Гц), 8,02 (д, 1H, J=8,2 Гц), 7,77-7,45 (м, 4H), 7,40-7,18 (м, 2H), 3,94 (с, 2H).
Стадия D: Метилтрифторметансульфонат (0,153 мл, 1,35 ммоль) добавляли по каплям к ледяному раствору продукта со стадии C (0,30 г, 1,23 ммоль) в CH2Cl2 (5 мл). Полученный в результате раствор перемешивали при 0°C в течение 1,5 часов и затем при комнатной температуре в течение 0,5 часа. Смесь концентрировали досуха при пониженном давлении, получая соль пиридиния в виде желтого твердого вещества, которое использовали на стадии E без дополнительной очистки.
Стадия E: Цианоборгидрид натрия (0,144 г, 2,26 ммоль) добавляли к раствору продукта со стадии C в MeOH (6 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь разбавляли водой (10 мл) и 2 N NaOH (10 мл) и экстрагировали CH2Cl2 (3×100 мл). Объединенный органический раствор сушили (Na2SO4) и концентрировали при пониженном давлении. Очистка флэш-хроматографией (элюент: от 98 до 96:4 до 92:8 CH2Cl2/MeOH) давала продукт (0,24 г, 75% для двух стадий) в виде масла.
Стадия F: Раствор продукта со стадии E (0,236 г, 0,90 ммоль) и малеиновой кислоты (0,107 г, 0,92 ммоль) в EtOH (2 мл) охлаждали до -20°C. Образованную взвесь фильтровали при пониженном давлении и промывали EtOH, получая продукт в виде не совсем белого твердого вещества (0,135 г, 39%): т.пл. 184-187°C;1H ЯМР (300 МГц, CD3OD) δ 7,43-7,19 (м, 7H), 7,14 (тд, 1H, J=7,3, 1,2 Гц), 6,75 (с, 1H), 6,22 (с, 2H), 4,66-4,55 (м, 1H), 4,50 (с, 2H), 3,83-3,69 (м, 1H), 3,68-3,57 (м, 1H), 3,46-3,32 (м, 2H), 3,13 (с, 3H); ESI m/z 261 [C19H19N+Н]+. Элементный анализ, вычислено для C19H19N-1,05C4H4O4: C, 72,71; Н, 6,10; N, 3,65. Найдено: C, 72,78; Н, 6,17; N, 3,65.
Пример 52 - Получение гидрохлоридной соли 4-(индан-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
4-Индан-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин получали из 5-ацетилиндана и бензиламина, как описано в примере 58 (стадии A-D). Его растворяли в этилацетате и обрабатывали 2 М хлористым водородом в диэтиловом эфире (2 экв.), получая соответствующую гидрохлоридную соль (2,15 г, 99,4% AUC ВЭЖХ):1H ЯМР (300 МГц, CDCl3) δ1H ЯМР (300 МГц, MeOD) δ 7,22-7,34 (м, 4H), 7,11 (с, 1H), 7,02 (д, J=6,9 Гц, 1H), 6,91 (д, J=6,9 Гц, 3H), 4,54-4,66 (м, 3H), 3,81-3,87 (м, 1H), 3,51 (т, J=12,3 Гц, 1H), 3,10 (с, 3Н), 2,87-2,94 (м, 4H), 2,03-2,14 (м, 2H), EI МС m/z=264 [C19H21N+H]+. Элементный анализ, вычислено для C19H22ClN: C, 76,11; Н, 7,40; N, 4,67. Найдено: C, 75,87; Н, 7,57; N, 4,52.
Пример 53 - Получение 2-метил-4-(нафтален-1-ил)-1,2,3,4-тетрагидроизохинолина
2-Метил-4-нафтален-1-ил-1,2,3,4-тетрагидроизохинолин (0,16 г, 99,5% AUC ВЭЖХ) получали из 1-нафталинбороновой кислоты и 4-бромизохинолина, как описано в примере 56:1H ЯМР (300 МГц, CDCl3) δ 7,92 (дд, J=4,5, 4,8 Гц, 1H), 7,77 (д, J=8,4 Гц, 1H), 7,47-7,57 (м, 2H), 7,40 (т, J=7,7 Гц, 1H), 7,15-7,22 (м, 3H), 7,05-7,10 (м, 1H), 6,89 (д, J=7,8 Гц, 1H), 5,17 (ушир.с, 1H), 3,72-3,88 (м, 2H), 3,16 (дд, J=11,4, 5,7 Гц, 1Н), 2,72-2,88 (м, 1H), 2,45 (с, 3H); EI МС m/z=274 [C20H19N+H]+.
Пример 54 - Получение 2-метил-4-(4-метилнафтален-1-ил)-1,2,3,4-тетрагидроизохинолина
2-Метил-4-(4-метилнафтален-1-ил)-1,2,3,4-тетрагидроизохинолин (0,17 г, 98,9% AUC ВЭЖХ) получали из 4-метил-1-нафталинбороновой кислоты и 4-бромизохинолина, как описано в примере 56:1H ЯМР (300 МГц, CDCl3) δ 8,07-8,10 (м, 1H), 7,50-7,60 (м, 2H), 7,30-7,26 (м, 6H), 6,90 (д, J=7,5 Гц, 1H), 5,14 (ушир.с, 1H), 3,70-3,82 (м, 2H), 3,16 (дд, J=11,4, 5,7 Гц, 1Н), 2,70-2,88 (м, 1H), 2,70 (с, 3H), 2,44 (с, 3H); EI МС m/z=288 [C21H21N+H]+.
Пример 55 - Получение 2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина
Стадия A: 2-Нафталинбороновую кислоту (1,2 г, 7,2 ммоль) добавляли к раствору 4-бромизохинолина (1,0 г, 4,8 ммоль) и трифенилфосфина (0,3 г, 0,9 ммоль) в 1,2-диметоксиэтане (10 мл). Смесь дегазировали аргоном. Добавляли ацетат палладия (II) (0,1 г, 0,5 ммоль) и суспензию перемешивали при комнатной температуре в течение 20 минут. Добавляли водный раствор карбоната натрия (5,8 мл, 2 М) и суспензию дегазировали, используя аргон. После перемешивания при 85°C в течение 3 часов охлажденную смесь разбавляли водой и один раз экстрагировали дихлорметаном. Объединенные экстракты сушили сульфатом натрия, концентрировали в вакууме и очищали хроматографией (5:1 гептан/этилацетат), получая требуемый продукт (1,1 г, 90%, 95% AUC ГХ):1H ЯМР (300 МГц, CDCl3) δ 9,32 (с, 1H), 8,62 (с, 1H), 7,92-8,12 (м, 6H), 7,56-7,72 (м, 5Н).
Стадия B: Раствор продукта со стадии A (1.0 г, 4 ммоль) и йодметана (0,8 мл, 12 ммоль) в хлороформе (15 мл) перемешивали при 60°C в течение 2 часов. Смесь концентрировали в вакууме, получая требуемый промежуточный продукт (1,0 г, 61%, 86% AUC ВЭЖХ):1H ЯМР (300 МГц, CDCl3) δ 10,79 (с, 1H), 8,58 (д, J=8,1 Гц, 1H), 8,09 (д, J=1,2 Гц, 1H), 7,74-7,93 (м, 7H), 7,37-7,43 (м, 3H), 4,63 (с, 3H).
Стадия C: Цианоборгидрид натрия (0,34 г, 5,4 ммоль), а затем две капли раствора бромкрезолового зеленого в метаноле добавляли к раствору продукта со стадии C (0,97 г, 2,4 ммоль) в метаноле (40 мл). Наблюдали голубую окраску. Раствор HCl в метаноле (3 М) добавляли до получения желтой окраски. Полученную в результате смесь перемешивали при комнатной температуре в течение 1 часа с периодическим добавлением HCl в метаноле, чтобы поддерживать желтую окраску. Смесь гасили добавлением водного раствора гидроксида натрия (20 мл, 3 М) и дважды экстрагировали этилацетатом. Органические экстракты промывали насыщенным раствором соли, сушили над сульфатом магния и концентрировали, получая 2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин (0,61 г, 93%, 96% AUC ВЭЖХ) в виде белого твердого вещества:1H ЯМР (300 МГц, CDCl3) δ 7,62-7,69 (м, 3H), 7,57 (с, 1H), 7,28-7,36 (м, 2H), 7,14 (дд, J=8,4, 1,5 Гц, 1H), 6,90-7,06 (м, 3H), 6,76 (д, J=7,8 Гц, 1H), 4,36 (дд, J=8,4, 6,3 Гц, 1H), 3,72 (д, J=15,0 Гц, 1H), 3,58 (д, J=14,7 Гц, 1H), 3,02 (ддд, J=11,7, 6,0, 1,5 Гц, 1Н), 2,59 (дд, J=11,4, 9,0 Гц, 1H), 2,35 (с, 3H); EI МС m/z=274 [C20H19N+H]+.
Пример 56 - Получение малеатной соли 2,5-диметил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина
Малеатную соль 2,5-диметил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолина (0,16 г, 98,5% AUC ВЭЖХ) получали, как описано в примере 58:1H ЯМР (300 МГц, MeOD) δ 7,84-7,92 (м, 2H), 7,69-7,73 (м, 1H), 7,44-7,51 (м, 2H), 7,34-7,39 (м, 3H), 7,22-7,24 (м, 2H), 6,17 (с, 2H), 4,86 (т, J=5,7 Гц, 1H), 4,66 (д, J=15,3 Гц, 1Н), 4,47 (д, J=15,3 Гц, 1H), 3,97 (дд, J=12,6, 6,0 Гц, 1H), 3,72-3,85 (м, 1H), 3,01 (с, 3H), 1,94 (с, 3H), EI МС m/z=288 [C21H21N+H]+. Элементный анализ, вычислено для C25H25NO4: C, 72,59; Н, 6,38; N, 3,35 с 2,00% Н2O и 1,37% EtOH. Найдено: C, 72,24; Н, 6,38; N, 3,05.
Пример 57 - Получение малеатной соли 2,7-диметил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина
Стадия A: Раствор трибромида тетрабутиламмония (15,6 г, 32 ммоль) в дихлорметане (80 мл) добавляли по каплям к раствору 2-ацетилнафталина (5,0 г, 29 ммоль) в дихлорметане (20 мл) и метаноле (20 мл) при комнатной температуре. После завершения добавления полученный в результате красно-оранжевый раствор перемешивали при комнатной температуре в течение 15 часов. Смесь концентрировали в вакууме и остаток помещали в этилацетат и воду. Слои разделяли и водную фазу экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли и сушили над безводным сульфатом натрия, получая требуемое бромсодержащее соединение (7,6 г, 100%):1H ЯМР (300 МГц, CDCl3) δ 8,05 (с, 1H), 7,04 (дд, J=4,2, 1,8 Гц, 1Н), 8,02 (д, J=1,8 Гц, 1H), 7,56-7,67 (м, 2H), 4,59 (с, 2H).
Стадия B: 3-Толилметиламин (2,7 г, 20 ммоль) добавляли по каплям к раствору продукта со стадии A (5,0 г, 20 ммоль) в дихлорметане (25 мл) при 0°C. После завершения добавления полученную в результате смесь перемешивали при 0°C в течение 15 минут и по каплям добавляли диизопропилэтиламин (3,8 мл, 22 ммоль). Смесь перемешивали при комнатной температуре в течение 15 часов, после этого смесь гасили насыщенным водным раствором бикарбоната натрия и дважды экстрагировали дихлорметаном. Объединенные органические экстракты промывали насыщенным водным раствором бикарбоната натрия и сушили над сульфатом натрия, получая требуемый промежуточный продукт (2,7 г, 45%) после хроматографии (от 19:1 до 9:1 гептан/этилацетат):1H ЯМР (300 МГц, CDCl3) δ 8,50 (с, 1H), 8,03 (дд, J=8,7, 1,8 Гц, 1H), 7,87-7,95 (м, 3H), 7,56-7,64 (м, 2H), 7,11-7,28 (м, 4H), 3,92 (с, 2H), 3,71 (с, 2H), 2,44 (с, 3H), 2,36 (с, 3H).
Стадия C: Боргидрид натрия (0,34 г, 8,9 ммоль) добавляли к раствору продукта со стадии B (2,70 г, 8,9 ммоль) в метаноле (40 мл) при 0°C. Полученную в результате смесь перемешивали при комнатной температуре в течение 15 часов. Метанол удаляли в вакууме. Остаток помещали в воду и три раза экстрагировали дихлорметаном. Объединенные органические экстракты промывали водой и насыщенным раствором соли и сушили над сульфатом натрия, получая требуемый спирт (1,5 г, 55%) после хроматографии (от 19:1 до 3:1 гептан/этилацетат):1H ЯМР (300 МГц, CDCl3) δ 7,84-7,88 (м, 4H), 7,47-7,52 (м, 3H), 7,27 (т, J=7,5 Гц, 1H), 7,12-7,17 (м, 3H), 6,46 (дд, J=9,6, 3,9 Гц, 1H), 3,77 (д, J=12,9 Гц, 1H), 3,55 (д, J=12,9 Гц, 1H), 2,62-2,75 (м, 2H), 2,39 (с, 3H), 2,38 (с, 3H).
Стадия D: Хлорид алюминия (1,20 г, 8,8 ммоль) порциями добавляли к раствору продукта со стадии C (1,50 г, 4,9 ммоль) в дихлорметане (40 мл) при 0°C. После окончания добавления смесь перемешивали при 0°C в течение 1,5 часов, вливали в лед и дихлорметан и перемешивали еще в течение 15 минут. Смесь промывали водой и насыщенным водным раствором бикарбоната натрия, экстрагировали дихлорметаном и сушили над сульфатом натрия, получая неочищенную смесь, содержащую региоизомеры в отношении от 1,4 до 1. Очитка хроматографией (от 5:1 до 1:1 гептан/этилацетат) давала 4-нафтален-2-ил-2,7-диметил-1,2,3,4-тетрагидроизохинолин (0,23 г, 16%, 98,7% AUC ВЭЖХ):1H ЯМР (300 МГц, CDCl3) δ 7,59-7,73 (м, 4H), 7,31-7,39 (м, 2H), 7,19 (дд, J=8,4, 1,5 Гц, 1H), 6,84 (с, 1H), 6,78 (д, J=7,8 Гц, 1H), 6,67 (д, J=7,8 Гц, 1H), 4,32 (т, J=7,8 Гц, 1H), 3,67 (д, J=15,0 Гц, 1Н), 3,53 (д, J=15,0 Гц, 1H), 2,99 (ддд, J=11,4, 5,4, 1,2 Гц, 1H), 2,56 (дд, J=11,4, 9,0 Гц, 1H), 2,35 (с, 3H), 2,21 (с, 3H) и 4-нафтален-2-ил-2,5-диметил-1,2,3,4-тетрагидроизохинолин (0,44 г, 31%, 99,1% AUC ВЭЖХ)1H ЯМР (300 МГц, CDCl3) δ 7,63-7,72 (м, 3H), 7,41 (с, 1H), 7,29-7,34 (м, 2H), 7,18-7,21 (м, 1H), 7,08 (т, J=7,5 Гц, 1H), 6,95 (д, J=7,5 Гц, 1H), 6,92 (д, J=7,2 Гц, 1H), 4,22 (т, J=3,9 Гц, 1H), 3,87 (д, J=15,0 Гц, 1H), 3,35 (д, J=14,7 Гц, 1H), 2,75-2,83 (м, 2H), 2,79 (дд, J=11,4, 4,2 Гц, 1H), 2,23 (с, 3H), 1,83 (с, 3H).
Стадия E: 4-Нафтален-2-ил-2,7-диметил-1,2,3,4-тетрагидроизохинолин (0,18 г) кристаллизовали с малеиновой кислотой (1 экв.) в спиртовом реагенте, получая малеатную соль 4-нафтален-2-ил-2,7-диметил-1,2,3,4-тетрагидроизохинолина (0,07 г, 27%, 99,4% АUC ВЭЖХ):1H ЯМР (300 МГц, MeOD) δ 7,80-7,91 (м, 4H), 7,51-7,54 (м, 2H), 7,29 (д, J=8,4, 1,5 Гц, 1H), 7,08-7,149 (м, 2H), 6,84 (д, J=7,8 Гц, 1H), 6,26 (с, 2H), 6,74 (дд, J=10,5, 6,6 Гц, 1H), 4,62 (д, J=15,3 Гц, 1H), 4,56 (д, J=15,6 Гц, 1H), 3,91 (дд, J=12,0, 6,0 Гц, 1H), 3,66 (т, J=11,7 Гц, 1H), 3,10 (с, 3H), 2,36 (с, 3H), EI МС m/z=288 [C21H21N+H]+.
Пример 58 - Получение гидрохлоридной соли 2,8-диметил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолина
Стадия A: о-толуальдегид превращали в метил-(2-метилбензил)амин посредством восстановительного аминирования, используя водный раствор метиламина и NaBH4. Затем метил-(2-метилбензил)амин алкилировали 2-бром-2'-ацетонафтоном с последующим восстановлением NaBH4, получая 2-[метил-(2-метилбензил)амино]-1-нафтален-2-илэтанол в виде прозрачного светло-желтого масла:1H ЯМР (300 МГц, CDCl3) δ 7,78-7,87 (м, 4Н), 7,40-7,51 (м, 3Н), 7,14-7,30 (м, 4Н), 4,89 (дд, J=10,1, 3,9 Гц, 1Н), 3,71 (д, J=12,9 Гц, 1Н), 3,54 (д, J=12,9 Гц, 1Н), 2,70 (дд, J=12,3, 10,3 Гц, 1Н), 2,62 (дд, J=12,4, 3,9 Гц, 1Н), 2,41 (с, 3Н), 2,35 (с, 3Н); CI МС m/z 306 [C21H23NO+H]+.
Стадия B: Продукт со стадии A (1,25 г, 4,1 ммоль) растворяли в трифторуксусной кислоте (TFA, 7,5 мл), затем добавляли трифторуксусный ангидрид (TFAA, 7,5 мл). После перемешивания в течение 24 часов растворители удаляли в вакууме, остаток растворяли в MeOH и растворитель удаляли в вакууме. Остаток растворяли в водном NH4OH, затем дважды экстрагировали CH2Cl2. Экстракт сушили над Na2SO4, фильтровали, растворитель удаляли в вакууме и остаток очищали хроматографией на колонке с силикагелем (40 г), элюируя смесью от 5% до 30% EtOAc/гексаны с 1% Et3N, получая 2,8-диметил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин (0,73 г, 61%) в виде прозрачного темно-желтого масла:1H ЯМР (300 МГц, CDCl3) δ 7,66-7,88 (м, 4Н), 7,38-7,48 (м, 2Н), 7,26 (дд, J=8,3, 1,6 Гц, 1Н), 6,93-7,04 (м, 2Н), 6,74 (д, J=7,2 Гц, 1Н), 4,45 (т, J=6,9 Гц, 1Н), 3,75 (д, J=15,4 Гц, 1Н), 3,51 (д, J=15,4 Гц, 1Н), 3,06 (ддд, J=11,2, 5,6, 0,9 Гц, 1Н), 2,67 (ддд, J=11,4, 8,5 Гц, 1Н), 2,48 (с, 3Н), 2,27 (с, 3H).
Стадия C: Используя HCl в эфире и метанол, продукт со стадии B (0,73 г, 2,5 ммоль) превращали в его гидрохлоридную соль (136 мг, 17%, 98,4% AUC ВЭЖХ) в виде белого аморфного твердого вещества: т.пл. 210-214°C;1H ЯМР (300 МГц, CD3OD) δ 7,76-7,90 (м, 4H), 7,47-7,55 (м, 2Н), 7,26 (дд, J=8,6, 1,6 Гц, 1Н), 7,10-7,21 (м, 2Н), 6,77 (д, J=7,3 Гц, 1Н), 4,78 (дд, J=11,2, 6,0 Гц, 1H), 4,65 (д, J=15,7 Гц, 1Н), 4,48 (д, J=15,6 Гц, 1Н), 3,87 (дд, J=12,1, 6,0 Гц, 1H), 3,66 (т, J=11,8 Гц, 1Н), 3,13 (с, 3Н), 2,36 (с, 3Н), ИК (КВr) 3434, 2926, 2546, 1459, 754, 479 см-1; Cl МС m/z 288 [C21H21N+H]+; элементный анализ, вычислено для C21H21N·HCl·0,5 Н2O: C, 75,77; Н, 6,96; N, 4,21. Найдено: C, 76,04; Н, 6,85; N, 4,14.
Пример 59 - Получение малеатной соли 4-(8-хлорнафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: 7-Бром-3,4-дигидронафтален-1-он получали из бромбензола и янтарного ангидрида, как описано в примере 64 (стадии A-C).
Стадия B: Ацетат натрия (12,4 г, 150 ммоль) добавляли к раствору гидрохлорида гидроксиламина (10,6 г, 150 ммоль) в метаноле (230 мл) при комнатной температуре и полученную в результате смесь перемешивали при комнатной температуре в течение 30 минут. Продукт со стадии A (30,9 г, 140 ммоль) добавляли порциями в течение 1 часа и смесь перемешивали при комнатной температуре в течение 2,5 часов. Добавляли воду (300 мл), смесь сначала становилась прозрачной, а затем твердые вещества начинали выпадать в осадок. Суспензию перемешивали при комнатной температуре в течение 1 часа и затем фильтровали. Полученные в результате твердые вещества три раза подвергали азеотропной перегонке с этилацетатом, получая требуемый оксим (28,3 г, 86%):1H ЯМР (300 МГц, ДМСО-d6) δ 7,94 (д, J=0,9 Гц, 1H), 7,40 (дд, J=8,4, 1,2 Гц, 1H), 7,15 (д, J=8,1 Гц, 1H), 2,61-2,68 (м, 4H), 1,68-1,78 (м, 2H).
Стадия C: Концентрированную серную кислоту (33,9 мл, 636 ммоль) добавляли к смеси продукта со стадии B (28,3 г, 118 ммоль) и уксусного альдегида (33,9 мл, 359 ммоль) в уксусной кислоте (170 мл) при комнатной температуре и полученную в результате смесь перемешивали при 95°C в течение 1 часа. После охлаждения до комнатной температуры добавляли воду (170 мл). Смесь подщелачивали до pH 13 6 М раствором гидроксида натрия и экстрагировали метил-трет-бутиловым эфиром. Объединенные органические экстракты сушили над сульфатом магния, получая 2-амино-7-бромнафталин (14 г, 53%) после очистки хроматографией (19:1 гексаны/этилацетат).
Стадия D: Смесь продукта со стадии C (6,0 г, 30 ммоль) и 6 М HCl (13,4 мл) перемешивали при комнатной температуре до образования твердого вещества. Смесь охлаждали до 0°C и медленно добавляли раствор нитрита натрия (1,9 г, 30 ммоль) в воде (5,6 мл), поддерживая температуру ниже 5°C. Полученную в результате коричневую взвесь перемешивали при 0°C в течение 15 минут и затем добавляли раствор хлорида меди (I) (3,3 г, 30 ммоль) в 6 М HCl (15,4 мл). Смесь перемешивали при 0°C в течение 10 минут, затем нагревали до комнатной температуры и перемешивали при 60°C в течение 30 минут. После охлаждения до комнатной температуры добавляли дихлорметан. Смесь фильтровали через диатомовую землю и фильтрат три раза экстрагировали дихлорметаном. Объединенные органические слои промывали водой и сушили над сульфатом магния, получая 7-бром-1-хлорнафталин (2,8 г, 43%) после хроматографии (100% гексаны):1H ЯМР (300 МГц, ДМСО-d6) δ 8,30 (д, J=1,8 Гц, 1H), 7,96-8,02 (м, 2H), 7,74-7,78 (м, 2H), 7,55 (т, J=7,8 Гц, 1H).
Стадия E: Малеатную соль 4-(8-хлорнафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (0,29 г, 99,5% AUC ВЭЖХ) получали из продукта со стадии D, как описано в примере 63 (стадии C и D):1H ЯМР (300 МГц, ДМСО-d6) δ 8,12 (с, 1H), 8,04 (д, J=8,4 Гц, 1H), 7,96 (д, J=8,4 Гц, 1H), 7,74 (дд, J=7,2, 0,9 Гц, 1H), 7,53 (д, J=8,4 Гц, 1H), 7,42 (дд, J=8,4, 1,5 Гц, 1H), 7,29-7,32 (м, 2H), 7,18-7,25 (м, 1H), 6,83 (д, J=7,5 Гц, 1H), 6,05 (с, 2H), 4,79 (дд, J=10,5, 6,3 Гц, 1H), 4,54 (д, J=15,3 Гц, 1H), 4,44 (д, J=15,3 Гц, 1H), 3,80 (дд, J=12,0, 6,3 Гц, 1H), 3,56 (т, J=11,4 Гц, 1Н), 2,90 (с, 3H); EI МС m/z=308 [C20H18ClN+H]+. Элементный анализ, вычислено для C24H22ClNO4: C, 67,35; Н, 5,23; Cl, 8,28; N, 3,27 с 0,95% Н2O. Найдено: C, 67,55; Н, 5,53; Cl, 8,45; N, 3,15.
Пример 60 - Получение малеатной соли 4-(8-фторнафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: 2-Амино-7-бромнафталин (14 г) получали, как описано в примере 60 (стадии A-C).
Стадия B: Раствор продукта со стадии A (5 г, 21 ммоль) в метил-трет-бутиловом эфире (20 мл) обрабатывали раствором хлористого водорода в диэтиловом эфире (16,9 мл, 2 М) и полученную в результате смесь перемешивали при комнатной температуре. Через 30 минут твердые вещества выпадали в осадок и их отфильтровывали и промывали метил-трет-бутиловым эфиром. Полученные твердые вещества добавляли к 6 М HCl (57,7 мл) и полученную в результате суспензию охлаждали до 0°C. Добавляли нитрит натрия (4,7 г, 70 ммоль), а затем тетрафторборат натрия (7,4 г, 70 ммоль) порциями, поддерживая температуру 0°C. Смесь перемешивали при 0°C в течение 30 минут, затем фильтровали. Фильтрат промывали ледяной водой и экстрагировали ледяным метил-трет-бутиловым эфиром, получая 7-бром-1-фторнафталин (4 г, 85%):1H ЯМР (300 МГц, ДМСО-d6) δ 8,18 (д, J=1,8 Гц, 1H), 7,97 (дд, J=9,0, 1,8 Гц, 1H), 7,80 (д, J=8,1 Гц, 1H), 7,72 (дд, J=8,7, 2,1 Гц, 1H), 7,55 (тд, J=7,8, 5,4 Гц, 1H), 7,39 (ддд, J=10,8, 7,8, 0,9 Гц, 1H).
Стадия C: Малеатную соль 4-(8-фторнафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (0,23 г, 965% AUC ВЭЖХ) получали из продукта со стадии B, как описано в примере 63 (стадии C и D):1H ЯМР (300 МГц, ДМСО-d6) δ 7,99-8,04 (м, 2H), 7,79 (д, J=8,1 Гц, 1H), 7,19-7,56 (м, 6H), 6,84 (д, J=7,5 Гц, 1Н), 6,05 (с, 2H), 4,77 (дд, J=10,5, 6,6 Гц, 1H), 4,54 (д, J=15,3 Гц, 1Н), 4,44 (д, J=15,6 Гц, 1H), 3,78 (дд, J=12,0, 6,3 Гц, 1H), 3,57 (т, J=11,1 Гц, 1H), 2,91 (с, 3H); EI МС m/z=292 [C20H18FN+H]+. Элементный анализ, вычислено для C24H22FNO4: C, 70,47; Н, 5,47; F, 4,64; N, 3,43 с 0,38% Н2O. Найдено: C, 70,33; Н, 5,78; F, 4,38; N, 3,30.
Пример 61 - Получение 4-(6-метоксинафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
4-(6-Метоксинафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин получали из 6-метокси-2-нафталинбороновой кислоты и 4-бромизохинолина, как описано в примере 56:1H ЯМР (300 МГц, CDCl3) δ 7,55-7,63 (м, 3H), 6,96-7,16 (м, 6H), 6,82 (д, J=7,8 Гц, 1H), 4,35 (дд, J=8,4, 6,0 Гц, 1H), 3,84 (с, 3H), 3,74 (д, J=14,7 Гц, 1H), 3,59 (д, J=15,0 Гц, 1H), 3,03 (ддд, J=11,4, 5,7, 1,2 Гц, 1Н), 2,59 (дд, J=11,4, 9,0 Гц, 1H), 2,39 (с, 3H); EI МС m/z=304 [C21H21NO+H]+.
Пример 62 - Получение малеатной соли 4-(7-метоксинафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: Гидроксид натрия (20 мл, 1 М) и диметилсульфат (15,0 мл, 158 ммоль) по каплям добавляли к раствору 2,7-дигидроксинафталина (6,6 г, 40 ммоль) в дихлорметане (100 мл) и воде (60 мл). Добавляли дополнительное количестве гидроксида натрия (20 мл, 1 М) и диметилсульфата (15,0 мл, 158 ммоль) и полученную в результате смесь перемешивали при комнатной температуре в течение 2 часов. Две фазы разделяли и водный слой дважды экстрагировали дихлорметаном. Объединенные органические экстракты промывали 1 М HCl, сушили над сульфатом магния и концентрировали в вакууме. Сырой продукт очищали хроматографией на колонке (5:1 гептан/этилацетат), получая 2-гидрокси-7-метоксинафталин (2,1 г, 30%, 100% AUC ВЭЖХ):1H ЯМР (300 МГц, CDCl3) δ 7,66-7,70 (м, 2H), 6,94-7,08 (м, 4H), 4,90 (с, 1H), 3,92 (с, 3H).
Стадия B: Ангидрид трифторметансульфоновой кислоты (6,7 мл, 6,6 ммоль) добавляли к раствору продукта со стадии A (1,0 г, 5,7 ммоль) и триэтиламина (1,9 мл, 13,3 ммоль) в дихлорметане при -23°C. После завершения добавления смеси давали возможность нагреться до комнатной температуры и перемешивали в течение 3 часов. Реакционную смесь разбавляли водой и дважды экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным раствором соли и сушили над сульфатом натрия, получая продукт (1,9 г, 100% AUC ГХ):1H ЯМР (300 МГц, CDCl3) δ 7,66-7,86 (м, 3H), 7,15-7,25 (м, 3H), 3,95 (с, 3H).
Стадия C: Изохинолин-4-илбороновую кислоту (0,76 г, 4,4 ммоль) добавляли к раствору продукта со стадии B (0,9 г, 2,9 ммоль) и трифенилфосфина (0,15 г, 0,6 ммоль) в 1,2-диметоксиэтане (15 мл). Суспензию дегазировали, используя азот. Добавляли ацетат палладия (0,06 г, 0,3 ммоль) и порцию перемешивали при комнатной температуре в течение 20 минут. Добавляли водный раствор карбоната натрия (3,5 мл, 2 М раствор) и суспензию дегазировали, снова используя азот. Смесь перемешивали при 85°C в течение 3,5 часов, охлаждали, разбавляли водой и дважды экстрагировали этилацетатом. Объединенные органические экстракты сушили над безводным сульфатом натрия, концентрировали в вакууме и очищали хроматографией (5:1 гептан/этилацетат), получая требуемый изохинолин (0,46 г, 55%, 98,7% AUC ВЭЖХ):1H ЯМР (300 МГц, CDCl3) δ 9,30 (с, 1H), 8,59 (с, 1H), 8,06-8,10 (м, 1H), 7,83-8,00 (м, 4H), 7,64-7,69 (м, 2H), 7,50 (дд, J=8,1, 1,5 Гц, 1H), 7,24-7,27 (м, 2H), 3,96 (с, 3H).
Стадия D: 4-(7-Метоксинафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин получали из продукта со стадии C, как описано в примере 56 (стадии B и C), и превращали в соответствующую малеатную соль (0,15 г, 99,0% AUC ВЭЖХ) посредством кристаллизации с малеиновой кислотой (1 эквивалент) в этаноле:1H ЯМР (300 МГц, ДМСО-d6) δ 7,83 (дд, J=8,4, 4,3 Гц, 2H), 7,73 (с, 1H), 7,16-7,30 (м, 6H), 6,84 (д, J=7,6 Гц, 1H), 6,05 (с, 2H), 4,61-4,67 (м, 1H), 4,43-4,55 (м, 2H), 3,87 (с, 3H), 3,78 (дд, J=6,0, 11,4 Гц, 1H), 3,55 (т, J=11,2 Гц, 1H), 2,93 (с, 3H); EI МС m/z=304 [C21H21NO+H]+. Элементный анализ, вычислено для C25H25NO5: C, 71,58; Н, 6,01; N,3,34. Найдено: C, 71,55; Н, 6,22; N, 3,28.
Пример 63 - Получение малеатной соли 4-(8-метоксинафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: Бромбензол (150 мл, 1,42 моль) одной порцией добавляли к смеси хлорида алюминия (109 г, 0,82 ммоль) и янтарного ангидрида (41 г, 0,41 моль) в циклогексане (200 мл) при 80°C и полученную в результате смесь перемешивали при 80°C в течение 2 часов. После охлаждения ниже 40°C смесь медленно вливали в 6 М HCl (200 мл) и лед, и желтые твердые вещества выпадали в осадок. Добавляли метил-трет-бутиловый эфир, чтобы растворить твердые вещества, и фазы разделяли. Органический слой три раза промывали водой и экстрагировали в 2 М NaOH. Водные экстракты подкисляли до pH 1, используя 6 М HCl, экстрагировали метил-трет-бутиловым эфиром, сушили над сульфатом магния и концентрировали до получения остатка. Полученное вещество перекристаллизовывали, растворяя остаток в смеси 50:35:15 циклогексан/толуол/изопропанол при 70°C с последующим охлаждением до комнатной температуры, фильтрованием и азеотропной перегонкой полученных в результате твердых веществ с гексанами, получая требуемое соединение (57 г, 55%) в виде белого твердого вещества:1H ЯМР (300 МГц, ДМСО-d6) δ 7,89-7,93 (м, 2H), 7,72-7,77 (м, 2H), 3,23 (т, J=6,0 Гц, 2H), 2,58 (т, J=6,6 Гц, 2H).
Стадия B: Смесь продукта со стадии A (57 г, 223 ммоль), гидроксида калия (43 г, 760 ммоль) и гидрата гидразина (26 мл, 830 ммоль) в диэтиленгликоле (2865 мл) перемешивали при 195°C в течение 3 часов. После охлаждения ниже 40°C смесь разбавляли водой (300 мл), вливали в 3 М NaOH и три раза промывали дихлорметаном. Добавляли насыщенный раствор соли, чтобы разрушить эмульсию. Водный слой подкисляли до pH 1, используя 6 М HCl, и три раза экстрагировали метил-трет-бутиловым эфиром. Объединенные органические экстракты сушили над сульфатом магния, получая 4-(4-бромфенил)масляную кислоту (41 г, 75%):1H ЯМР (300 МГц, ДМСО-d6) δ 7,44-7,48 (м, 2H), 7,14-7,20 (м, 2H), 2,56 (т, J=7,2 Гц, 2H), 2,20 (т, J=7,5 Гц, 2H), 1,72-1,82 (м, 2H).
Стадия C: Продукт со стадии B (97 г, 397 ммоль) добавляли к полифосфорной кислоте (580 г) и полученную в результате смесь перемешивали при 90°C в течение 10 минут. После охлаждения до 0°C добавляли 6 М NaOH (2 л) и смесь экстрагировали метил-трет-бутиловым эфиром. Органические экстракты сушили над сульфатом магния, получая 7-бром-3,4-дигидронафтален-1-он (49 г, 55%) после хроматографии (от 6:1 до 4:1 гептан/этилацетат) и перекристаллизации из циклогексана:1H ЯМР (300 МГц, ДМСО-d6) δ 7,91 (д, J=2,1 Гц, 1H), 7,71 (дд, J=8,1, 2,1 Гц, 1H), 7,34 (д, J=8,1 Гц, 1H), 2,90 (т, J=6,0 Гц, 2H), 2,61 (т, J=6,3 Гц, 2H), 1,99-2,07 (м, 2H).
Стадия D: Раствор трибромида тетрабутиламмония (11,8 г, 24,4 ммоль) в дихлорметане (80 мл) добавляли по каплям к раствору продукта со стадии C (5,0 г, 22,2 ммоль) в дихлорметане (20 мл) и метаноле (20 мл) при комнатной температуре в течение 1 часа. После завершения добавления смесь перемешивали при комнатной температуре в течение 15 часов и затем концентрировали. Остаток помещали в дихлорметан и три раза промывали насыщенным раствором бикарбоната натрия. Органический слой концентрировали и остаток растворяли в диметилформамиде (100 мл). Добавляли карбонат лития (5,3 г, 71,1 ммоль) и бромид лития (4,1 г, 46,6 ммоль) и полученную в результате смесь перемешивали при 140°C в течение 1,5 часов. После охлаждения до комнатной температуры твердые вещества отфильтровывали и промывали этилацетатом. Фильтрат четыре раза промывали водой и сушили над сульфатом натрия, получая 7-бромнафтален-1-ол (2,7 г, 56%):1H ЯМР (300 МГц, CDCl3) δ 8,41 (д, J=1,8 Гц, 1H), 7,68 (д, J=8,7 Гц, 1H), 7,57 (дд, J=8,7, 1,8 Гц, 1H), 7,41 (д, J=8,4 Гц, 1H), 7,28-7,35 (м, 1H), 6,62 (д, J=7,2 Гц, 1H), 5,80 (ушир.с, 1Н).
Стадия E: Карбонат калия (3,3 г, 23,9 ммоль) и метилйодид (1,5 мл, 23,9 ммоль) добавляли к раствору продукта со стадии D (2,7 г, 11,9 ммоль) в ацетоне (40 мл) при комнатной температуре и смесь перемешивали при 65°C в течение 2 часов. Твердые вещества отфильтровывали, промывали этилацетатом и фильтрат упаривали. Остаток растворяли в этилацетате и дважды промывали водой. Водные слои экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным раствором соли и сушили над сульфатом натрия, получая 7-бром-1-метоксинафталин (2,5 г, 88%) после хроматографии (19:1 гептан/этилацетат):1H ЯМР (300 МГц, CDCl3) δ 8,49 (д, J=2,1 Гц, 1H), 7,68 (д, J=8,7 Гц, 1H), 7,59 (дд, J=8,7, 2,1 Гц, 1H), 7,39-7,43 (м, 2H), 6,85 (дд, J=5,4, 3,0 Гц, 1H), 3,15 (с, 3H).
Стадия F: Малеатную соль 4-(8-метоксинафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (0,29 г, 96,2% AUC ВЭЖХ) получали из продукта со стадии E, как описано в примере 63 (стадии C и D):1H ЯМР (300 МГц, MeOD) δ 8,18 (с, 1H), 7,84 (д, J=8,4 Гц, 1H), 7,23-7,47 (м, 6H), 6,93-7,00 (м, 2H), 6,23 (с, 2H), 4,76-4,82 (м, 1H), 4,62 (ушир.с, 2H), 3,88-3,96 (м, 1H), 3,59-3,70 (м, 1H), 3,10 (с, 3H); EI МС m/z=304 [C21H21NO+H]+. Элементный анализ, вычислено для C25H25NO5: C, 71,05; Н, 6,10; N, 3,27 с 0,2% Н2O и 0,17 EtOH. Найдено: C, 70,53; Н, 6,04; N, 3,13.
Пример 64 - Получение гидрохлоридной соли 2-метил-4-(5,6,7,8-тетрагидронафтален-2-ил)-1,2,3,4-тетрагидроизохинолина
2-Метил-4-(5,6,7,8-тетрагидронафтален-2-ил)-1,2,3,4-тетрагидроизохинолин получали из 6-ацетилтетралина и бензиламина, как описано в примере 58 (стадии A-D). Его растворяли в этилацетате и обрабатывали 2 М хлористым водородом в диэтиловом эфире (2 эквивалента), получая соответствующую гидрохлоридную соль (0,43 г, 98,6% AUC ВЭЖХ):1H ЯМР (300 МГц, MeOD) δ 7,22-7,34 (м, 3H), 7,08 (д, J=7,8 Гц, 1H), 6,94 (ушир.с, 3H), 4,49-4,62 (м, 3H), 3,81 (дд, J=12,0, 6,0 Гц, 1H), 3,53 (т, J=11,7 Гц, 1H), 3,08 (с, 3Н), 2,76 (ушир.с, 4H), 1,82 (ушир.с, 4H), EI МС m/z=278 [C20H23N+H]+. Элементный анализ, вычислено для C20H24ClN: C, 75,60; Н, 7,59; N, 4,41 с 1,1 экв. НCl. Найдено: C, 75,44; Н, 8,11; N, 4,26.
Пример 65 - Получение малеатной соли 4-(2H-хромен-3-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: К ледяной суспензии NaH (0,49 г, 12,4 ммоль, 60% дисперсия в масле) в ТГФ (34,3 мл) добавляли салицилальдегид (1,1 мл, 10,3 ммоль) в течение 25 минут. Через 2,5 часа при 0°C к реакционной смеси добавляли триметил-2-фосфоноакрилат (1,6 мл, 10,3 ммоль) в течение 10 минут. Баню со льдом убирали и реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем при 70°C в течение 2 часов. Охлажденную реакционную смесь гасили водой и продукт экстрагировали в Et2O (3×75 мл). Et2O-экстракт промывали насыщенным раствором соли, сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении. Полученный остаток перерастворяли в Et2O (100 мл), промывали 10% NaHSO3 и насыщенным раствором соли, сушили (MgSO4), фильтровали и концентрировали, получая продукт (1,96 г, количественный выход, неочищенный) в виде белого твердого вещества:1H ЯМР (300 МГц, CDCl3) δ 7,44 (с, 1H), 7,23-7,20 (м, 1Н), 7,13 (дд, J=7,2, 1,6 Гц, 1H), 6,92 (тд, J=7,4, 0,9 Гц, 1H), 6,84 (д, J=8,1 Гц, 1Н), 5,00 (д, J=1,3 Гц, 2H), 3,82 (с, 3H).
Стадия B: К ледяному раствору сложного эфира (1,96 г, 10,3 ммоль) со стадии A в смеси 2:1:1 ТГФ/H2O/MeOH (120 мл) добавляли LiOH-H2O (0,87 г, 20,6 ммоль). Баню со льдом убирали и реакционную смесь кипятили с обратным холодильником в течение 30 минут. Охлажденную реакционную смесь концентрировали при пониженном давлении и подкисляли до pH 3-4 концентрированной HCl. Образовавшийся белый осадок отфильтровывали, перерастворяли в CH2Cl2/MeOH, сушили (MgSO4), фильтровали и концентрировали, получая 2H-хромен-3-карбоновую кислоту (1,43 г, 79% для 2 стадий):1H ЯМР (300 МГц, ДМСО-d6) δ 7,45 (с, 1H), 7,34-7,31 (м, 1H), 7,25 (дд, J=7,9, 1,5 Гц, 1H), 6,95 (тд, J=7,4, 0,9 Гц, 1H), 6,85 (д, J=8,1 Гц, 1H), 4,91 (д, J=1,3 Гц, 2H).
Стадия C: К раствору LiOAc-2H2O (0,16 г, 1,62 ммоль) в смеси 97:3 CH3CN/H2O (30 мл) добавляли 6-хлор-2H-1-бензопиран-3-карбоновую кислоту (1,43 г, 8,12 ммоль) с последующим добавлением NBS (1,52 г, 8,53 ммоль) и смесь перемешивали при комнатной температуре в течение 5 часов. Затем реакционную смесь концентрировали досуха при пониженном давлении. Очистка флэш-хроматографией на колонке (градиент, гексаны, затем от 98:2 до 90:10 гексаны/Et2O) давала 3-бром-6-хлор-2H-хромен (0,47 г, 27%) в виде бледно-желтого масла:1H ЯМР (300 МГц, CDCl3) δ 7,14-7,10 (м, 1H), 6,94-6,88 (м, 2H), 6,79 (д, J=8,1 Гц, 1H), 6,75 (с, 1H), 4,88 (д, J=1,5 Гц, 2H).
Стадия D: К смеси пинаколдиборана (0,62 г, 2,45 ммоль), KOAc (0,65 г, 6,68 ммоль) и бромида (0,47 г, 2,23 ммоль) со стадии C, описанной выше, добавляли ДМСО (21,8 мл). Раствор дегазировали (три раза, вакуум/аргон) и к нему добавляли PdCl2dppf-CH2Cl2 (110 мг, 0,13 ммоль). Реакционную смесь снова дегазировали (три раза, вакуум/аргон) и нагревали при 80°C в течение 3,5 часов. Охлажденную реакционную смесь разбавляли водой, экстрагировали CH2Cl2 (3×50 мл), промывали насыщенным раствором соли (50 мл), сушили (Na2SO4), фильтровали и концентрировали, получая неочищенный сложный эфир бороновой кислоты, который используют на следующей стадии без дополнительной очистки.
Стадия E: К смеси неочищенного сложного эфира бороновой кислоты со стадии D, описанной выше, и 4-бромхинолина (0,42 г, 2,03 ммоль) в ДМФА (8,4 мл) добавляли раствор Cs2CO3 (2,64 г, 8,11 ммоль) в воде (3,8 мл). Смесь дегазировали (три раза, вакуум/аргон) и к ней добавляли Pd(PPh3)4 (0,16 г, 0,14 ммоль). Реакционную смесь снова дегазировали (три раза, вакуум/аргон) и нагревали при 80°C в течение ночи. Охлажденную реакционную смесь разбавляли водой, экстрагировали CH2Cl2 (3×50 мл), промывали насыщенным раствором соли (50 мл), сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении. Очистка флэш-хроматографией на колонке (градиент, от 95:5 до 70:30 гексаны/EtOAc) давала частично очищенный продукт (0,36 г), который использовали на стадии F без дополнительной очистки.
Стадия F: К ледяному раствору продукта со стадии E, описанной выше (0,36 г), в CH2Cl2 (4,68 мл) по каплям добавляли метилтрифторметансульфонат (0,17 мл, 1,53 ммоль). Баню со льдом убирали и смесь перемешивали при комнатной температуре в течение 40 минут. Реакцию гасили MeOH (1 мл) и реакционную смесь концентрировали при пониженном давлении и использовали как таковую на стадии G.
Стадия G: К раствору неочищенного продукта со стадии F, описанной выше, в MeOH (48 мл) добавляли цианоборгидрид натрия (0,19 г, 3,06 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали, разбавляли водой и экстрагировали CH2Cl2 (5×25 мл). Органические экстракты объединяли, промывали насыщенным раствором соли, сушили (Na2SO4) и концентрировали при пониженном давлении. Очистка флэш-хроматографией на колонке (градиент, от 90:10 до 70:30 гексаны/EtOAc) давала частично очищенный продукт (0,20 г), который подвергали флэш-хроматографии (элюент: 90:10 EtOAc/гексаны и 80:20 EtOAc/гексаны) с последующей обращенно-фазовой ВЭЖХ (полученную соль TFA превращали в соответствующее свободное основание, используя концентрированный NH4OH), получая требуемый продукт (54 мг, 8% для 4 стадий) в виде бледно-желтого масла:1H ЯМР (300 МГц, CDCl3) δ 7,26-6,86 (м, 7H), 6,76 (д, J=7,6 Гц, 1Н), 6,33 (с, 1H), 4,74 (дд, J=14,5, 1,3 Гц, 1H), 4,50 (дд, J=14,5, 1,3 Гц, 1H), 3,90-3,80 (м, 1H), 3,58 (д, J=3,0 Гц, 1H), 2,85-2,75 (м, 1H), 2,61 (дд, J=11,5, 6,8 Гц, 1H), 2,42 (с, 3H).
Стадия H: К раствору продукта со стадии G (54 мг, 0,18 ммоль) в EtOH (0,5 мл) добавляли малеиновую кислоту (21 мг, 0,18 ммоль) и раствор охлаждали до -30°C. Образованную густую суспензию разбавляли EtOH (0,5 мл) и нагревали до комнатной температуры. Полученный прозрачный желтый раствор концентрировали при пониженном давлении, получая пену, которую растирали в EtOAc, затем в смеси EtOAc/гексаны, получая требуемый продукт (26 мг, 36%) в виде не совсем белого твердого вещества:1H ЯМР (500 МГц, CD3OD) δ 7,37-7,35 (м, 3H), 7,27 (ушир.с, 1H), 7,14-7,11 (м, 1H), 7,03 (ушир.с, 1H), 6,89 (ушир.с, 1H), 6,77-6,76 (м, 1H), 6,50-6,40 (ушир.с, 1H), 6,25 (с, 2H), 4,65-4,62 (м, 1H), 4,52-4,50 (м, 1H), 4,45 (с, 2H), 4,23 (ушир.с, 1H), 3,70 (ушир.с, 1H), 3,50-3,40 (м, 1H), 3,04 (с, 3H); ESI МС m/z 278 [C19H19NO+Н]+; элементный анализ, вычислено для C19H19NO-C4H4O4-0,25H2O: C, 69,42; Н, 5,95; N, 3,52. Найдено: C, 69,28; Н, 5,58; N, 3,47.
Пример 66 - Малеатная соль 4-(8,9-дигидро-7H-бензоциклогептен-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: 8-Бром-6,7-дигидро-5H-бензоциклогептен синтезировали, следуя способу, опубликованному в работе Paquette et al., J. Org. Chem. 56: 6199-6205 (1991), которая включена в данное описание в виде ссылки в полном объеме.1H ЯМР (300 МГц, CDCl3) δ 7,17-7,03 (м, 4H), 6,94 (с, 1H), 2,95-2,79 (м, 4H), 2,00-2,89 (м, 2H).
Стадия B: 8-Бром-6,7-дигидро-5H-бензоциклогептен (1,0 г, 4,48 ммоль) и бис(пинаколато)дибор (2,28 г, 8,98 ммоль) подвергали взаимодействию, как описано для синтеза в примере 67, стадия D. Сырой продукт использовали без дополнительной очистки на стадии C.
Стадия C: Продукт со стадии B (~0,48 ммоль) и 4-бромизохинолин (0,865 г, 4,07 ммоль) подвергали взаимодействию, как описано для синтеза в примере 67, стадия E, получая после хроматографии продукт в виде не совсем белого твердого вещества (1,08 г, 82% для 2 стадий):1H ЯМР (300 МГц, CDCl3) δ 9,19 (с, 1H), 8,47 (с, 1H), 8,06-7,97 (м, 2H), 7,74-7,50 (м, 2H), 7,24-7,14 (м, 4H), 6,62 (с, 1H), 3,05-3,00 (м, 2H), 2,74-2,67 (м, 2H), 2,28-2,17 (м, 2H).
Стадия D: Продукт со стадии C (1,0 г, 3,68 ммоль) и метилтрифторметансульфонат (0,46 мл, 4,06 ммоль) подвергали взаимодействию, как описано для синтеза в примере 67, стадия F, получая желтое твердое вещество, которое переносили на стадию E без дополнительной очистки.
Стадия E: Продукт со стадии D (~3,68 ммоль) и цианоборгидрид натрия (0,578 г, 9,20 ммоль) объединяли, как описано для синтеза в примере 67, стадия G, получая после хроматографии продукт в виде не совсем белого твердого вещества (1,08 г, количественный выход 2 стадий).1H ЯМР (300 МГц, ДМСО-d6) δ 7,25-7,05 (м, 8H), 6,50 (с, 1H), 3,85 (т, 1H, J=6,7 Гц), 3,57 (д, 1/2 AB, 1H, J=15,0 Гц), 3,43 (д, 1/2 AB, 1H, J=15,0 Гц), 2,91-2,79 (м, 1Н), 2,69-2,60 (м, 2H), 2,56-2,46 (м, 2H), 2,33 (с, 3H), 2,12-1,81 (м, 3H).
Стадия F: Продукт со стадии E (1,0 г, 3,45 ммоль) и малеиновую кислоту (0,43 г, 0,74 ммоль) в EtOH (3 мл) при комнатной температуре перемешивали, используя ультразвуковой генератор. Взвесь фильтровали при пониженном давлении, получая требуемый продукт (0,926 г, 93%) в виде белого твердого вещества: т.пл. 178-180°C;1H ЯМР (300 МГц, CD3OD) δ 7,43-7,30 (м, 3H), 7,28-7,22 (м, 1H), 7,20-7,12 (м, 4H), 6,68 (с, 1H), 4,48 (с, 2H), 4,26 (дд, 1H, J=11,4, 6,5 Гц), 3,79 (дд, 1H, J=12,2, 6,4 Гц), 3,49 (т, 1Н, J=11,6 Гц), 3,07 (с, 3H), 2,89-2,71 (м, 2H), 2,11-1,89 (м, 4H) ESI m/z 289 [C21H23N + Н]+. Элементный анализ, вычислено для C21H23N-C4H4O4: С, 74,05; Н, 6,71; N, 3,45. Найдено: С, 73,91; Н, 6,73; N, 3,43.
Пример 67 - Получение малеатной соли 4-(бензо[b]тиофен-7-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
4-Бензо[b]тиофен-7-ил-2-метил-1,2,3,4-тетрагидроизохинолин получали из 2-бромбензолтиола, как описано в примере 40 (стадии A-E), и превращали в соответствующую малеатную соль (0,14 г, 99,2% AUC ВЭЖХ) кристаллизацией с малеиновой кислотой (1 эквивалент) в этаноле (2 мл):1H ЯМР (300 МГц, ДМСО-d6) δ 7,91 (д, J=7,9 Гц, 1H), 7,79 (д, J=5,4 Гц, 1H), 7,56 (д, J=5,4 Гц, 1H), 7,47 (т, J=7,5 Гц, 1H), 7,35 (д, J=3,8 Гц, 2H), 7,20-7,28 (м, 2H), 6,82 (д, J=6,2 Гц, 1H), 6,09 (с, 2H), 4,89-4,95 (м, 1H), 4,54 (с, 2H), 3,76-3,87 (м, 1H), 3,57-3,65 (м, 1H), 2,96 (с, 3H); EI МС m/z=280 [C18H17NS+H]+. Элементный анализ, вычислено для C22H21NO4S: C, 64,60; Н, 5,48; N,3,42; S, 7,83 с 3,2% Н2O. Найдено: C, 64,11; Н, 5,56; N, 2,98; S, 7,87.
Пример 68 - Получение фумарата 4-гидрокси-7-метокси-2-метил-4-(хинолин-6-ил)-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору 6-бромхинолина (624 мг, 3,0 ммоль) при -75°C по каплям добавляли трет-бутиллитий (1,7 М в пентане, 1,95 мл, 3,3 ммоль). Реакционную смесь перемешивали при -75°C в течение 1 часа. К полученной в результате темно-коричневой смеси добавляли 7-метокси-2-метил-2,3-дигидро-1H-изохинолин-4-он (383 мг, 2,0 ммоль), который получали, используя способ, описанный в публикации Hanna et al., J. Med. Chem., 17(9): 1020-1023 (1974), которая включена в данное описание в виде ссылки в полном объеме. Реакционную смесь перемешивали в течение 15 часов с постепенный нагреванием. Смесь гасили насыщенным раствором хлорида аммония и экстрагировали этилацетатом (3×50 мл). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией среднего давления на силикагеле (0-5% метанол/дихлорметан) давала продукт (225 мг, 35%):1H ЯМР (300 МГц, CDCl3) δ 8,90 (дд, 1H, J=1,7, 4,3 Гц), 8,22-8,18 (м, 2H), 8,00 (д, 1H, J=8,9 Гц), 7,50 (дд, 1H, J=2,0, 8,9 Гц), 7,41 (дд, 1H, J=4,3, 8,3 Гц), 6,85 (д, 1H, J=8,6 Гц), 6,68 (дд, 1H, J=2,6, 8,6 Гц), 6,60 (д, 1Н, J=2,5 Гц), 4,50 (ушир.с, 1H), 3,80 (с, 3H), 3,69 (д, 1Н, J=15,0 Гц), 3,43 (д, 1H, J=15,0 Гц), 2,98 (д, 1H, J=11,7 Гц), 2,75 (д, 1H, J=11,7 Гц), 2,44 (с, 3H), ESI МС m/z=321 [M + Н]+.
Стадия B: Продукт со стадии A (45 мг, 0,14 ммоль) растворяли в этаноле (1 мл) и добавляли раствор фумаровой кислоты (17 мг, 0,14 ммоль) в метаноле (0,5 мл). Растворитель удаляли при пониженном давлении. Остаток растирали с этилацетатом. Полученный в результате преципитат собирали фильтрованием, промывали этилацетатом и сушили при 50°C в вакууме, получая продукт в виде не совсем белого твердого вещества (35 мг, 57%):1H ЯМР (CD3OD, 500 МГц) δ 8,87 (дд, 1H, J=1,6, 4,3 Гц), 8,39 (д, 1H, J=7,7 Гц), 8,20 (д, 1H, J=1,8 Гц), 7,98 (д, 1H, J=8,9 Гц), 7,64 (дд, 1H, J=2,0, 8,9 Гц), 7,58 (дд, 1H, J=4,3, 8,3 Гц), 6,89-6,81 (м, 3H), 6,70 (с, 2H), 4,38 (д, 1H, J=15,5 Гц), 4,27 (д, 1H, J=15,5 Гц), 3,81 (с, 3H), 3,53 (д, 1H, J=12,3 Гц), 3,43 (д, 1H, J=12,3 Гц), 2,88 (с, 3H), ESI МС m/z=321 [M + Н]+.
Пример 69 - Получение фумаратной соли 7-метокси-2-метил-4-(хинолин-6-ил)-1,2,3,4-тетрагидроизохинолина
Стадия A: Продукт со стадии A примера 70 (250 мг, 0,78 ммоль) нагревали при кипячении с обратным холодильником в смеси HCl/этанол (полученной смешиванием 1,5 мл ацетилхлорида с 25 мл этанола) в течение 2 часов. Растворитель удаляли при пониженном давлении. Остаток перерастворяли в метаноле (25 мл) и к раствору добавляли боргидрид натрия (300 мг, 7,8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Анализ ESI-МС показал, что реакция не была завершена. К указанной смеси дополнительно добавляли цианоборгидрид натрия (250 мг, 4 ммоль). Полученную в результате реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Затем смесь гасили водой и экстрагировали этилацетатом (3×50 мл). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией среднего давления на силикагеле (0-5% метанол/дихлорметан) давала продукт (105 мг, 44%):1H ЯМР (CD3OD, 500 МГц) δ 8,80 (дд, 1H, J=1,6, 4,3 Гц), 8,30 (д, 1H, J=8,4 Гц), 7,95 (д, 1H, J=8,7 Гц), 7,77 (д, 1H, J=1,8 Гц), 7,56 (дд, 1H, J=1,9, 8,7 Гц), 7,51 (дд, 1H, J=4,3, 8,3 Гц), 6,74-6,66 (м, 3H), 4,48 (дд, 1H, J=6,2, 8,9 Гц), 3,81 (д, 1H, J=15,0 Гц), 3,76 (с, 3H), 3,66 (д, 1Н, J=15,0 Гц), 3,17-3,13 (м, 1H), 2,67 (дд, 1Н, J=9,4, 11,6 Гц), 2,75 (д, 1H, J=11,7 Гц), 2,44 (с, 3H), ESI МС m/z=305 [M+H]+.
Стадия B: Продукт со стадии A (51 мг, 0,16 ммоль) растворяли в этаноле (1 мл) и добавляли раствор фумаровой кислоты (19 мг, 0,16 ммоль) в метаноле (0,5 мл). Растворитель удаляли при пониженном давлении. Остаток растирали в этилацетате. Полученный в результате преципитат собирали фильтрованием, промывали этилацетатом и сушили при 50°C в вакууме, получая продукт в виде не совсем белого твердого вещества (55 мг, 82%):1H ЯМР (CD3OD, 500 МГц) δ 8,85 (дд, 1H, J=1,6, 4,3 Гц), 8,34 (д, 1H, J=8,2 Гц), 8,01 (д, 1H, J=8,8 Гц), 7,87 (д, 1H, J=1,6 Гц), 7,60-7,54 (м, 2H), 6,83 (с, 1H), 6,79 (д, 2H, J=1,4 Гц), 4,72 (дд, 1H, J=6,1, 10,6 Гц), 4,37 (с, 2H), 3,79 (с, 3H), 3,71 (дд, 1H, J=6,1, 12,1 Гц), 3,39 (д, 1H, J=11,1 Гц), 2,91 (с, 3H), ESI МС m/z=305 [M+H]+.
Пример 70 - Получение малеатной соли (+)-4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина и малеатной соли (-)-4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору 2-ацетилбензо[b]тиофен (5,0 г, 28 ммоль) в хлороформе (120 мл) добавляли пербромид бромида пиридиния (9,6 г, 28 ммоль) двумя порциями. Реакционный раствор перемешивали при комнатной температуре в течение 2 часов и затем промывали водой, водным раствором HCl (2 М), водой и насыщенным раствором соли. Полученный в результате раствор сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый бромкетон (грубый выход 7,4 г) в виде коричневого твердого вещества, который использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 8,07 (с, 1H), 7,94-7,87 (м, 2H), 7,52-7,25 (м, 2H), 4,46 (с, 2H).
Стадия B: К раствору 3-метоксибензилметиламина (4,3 г, 29 ммоль) в дихлорметане (150 мл) добавляли триэтиламин (10 мл) и бромкетон (7,3 г, 29 ммоль) со стадии A, описанной выше. Реакционный раствор перемешивали при комнатной температуре в течение 2 часов и затем гасили водным насыщенным раствором бикарбоната натрия. Органический экстракт отделяли, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от 90:10 до 85:15 гексаны/этилацетат), получая требуемый продукт (7,1 г, 76% для 2 стадий) в виде красноватого масла:1H ЯМР (CDCl3, 500 МГц) δ 8,09 (с, 1H), 7,86 (д, J=8,0 Гц, 2H), 7,45 (дд, J=8,0, 1,1 Гц, 1H), 7,40 (дд, J=8,0, 1,0 Гц, 1Н), 7,24 (д, J=8,1 Гц, 1H), 6,97-6,95 (м, 2H), 6,83 (д, J=8,5 Гц, 1Н), 3,78 (с, 3H), 3,76 (с, 2H), 3,70 (с, 2H), 2,43 (с, 3H); ESI-МС m/z 326 [M+H]+.
Стадия C: К раствору кетона (7,1 г, 22 ммоль) со стадии B, описанной выше, в метаноле (100 мл) при 0°C добавляли боргидрид натрия (0,89 г, 24 ммоль) небольшими порциями. Реакционный раствор перемешивали при 0°C в течение 1 часа. Затем растворитель удаляли при пониженном давлении и полученный остаток растворяли в дихлорметане. Полученный в результате раствор промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый спирт (6,9 г, грубый выход 96%) в виде желтого масла. Сырой продукт использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,81 (д, J=7,9 Гц, 1H), 7,70 (д, J=8,1 Гц, 1Н), 7,33-7,23 (м, 3H), 7,21 (с, 1H), 6,91-6,87 (м, 3H), 5,07 (дд, J=10,1, 3,5 Гц, 1H), 4,40-4,03 (м, 1H), 3,81 (с, 3H), 3,72 (д, J=13,0 Гц, 1Н), 3,56 (д, J=13,0 Гц, 1H), 2,83 (дд, J=12,4, 10,2 Гц, 1H), 2,72 (дд, J=12,4, 3,7 Гц, 1H), 2,34 (с, 3H); ESI-МС m/z 328 [M+H]+.
Стадия D: К раствору спирта (6,9 г, 21 ммоль) со стадии C, описанной выше, в дихлорметане (150 мл) по каплям добавляли метансульфоновую кислоту (14 мл, 210 ммоль). После добавления реакционный раствор перемешивали при комнатной температуре в течение 20 минут и затем медленно добавляли к ледяному водному раствору гидроксида натрия (120 мл, 2 М) и перемешивали в течение 20 минут. Органический экстракт отделяли и водный экстракт дважды промывали дихлорметаном. Объединенный органический экстракт промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от 95:5 до 50:50 гексаны/этилацетат), получая требуемый циклизованный продукт (3,4 г, 52%) в виде красноватого масла:1H ЯМР (CDCl3, 500 МГц) δ 7,72 (д, J=8,0 Гц, 1H), 7,67 (д, J=8,0 Гц, 1H), 7,29-7,23 (м, 2H), 7,13 (с, 1H), 7,06 (д, J=8,5 Гц, 1Н), 6,69 (д, J=2,7 Гц, 1H), 6,61 (д, J=2,6 Гц, 1Н), 4,50 (т, J=5,3 Гц, 1H), 3,77 (с, 3H), 3,73 (д, J=14,5 Гц, 1H), 3,59 (д, J=14,5 Гц, 1H), 2,99 (дд, J=11,5, 4,9 Гц, 1H), 2,87 (дд, J=11,4, 6,2 Гц, 1H), 2,47 (с, 3H); ESI-МС m/z 310 [M+H]+.
Стадия E: К раствору 7-метокситетрагидроизохинолина (1,6 г, 5,1 ммоль) со стадии D, описанной выше, в уксусной кислоте (25 мл) добавляли водный раствор бромистого водорода (25 мл, 48%). Реакционный раствор нагревали при 100°C в течение 20 часов и затем охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток помещали в смесь дихлорметана, воды и небольшого количества метанола. Полученный в результате раствор осторожно гасили насыщенным водным раствором бикарбоната натрия плоть до pH >8. Органический экстракт отделяли, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый фенол (грубый выход 1,6 г) в виде коричневого твердого вещества, который использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,72 (д, J=7,7 Гц, 1H), 7,67 (д, J=7,7 Гц, 1H), 7,29 (тд, J=7,3, 1,2 Гц, 1H), 7,23 (тд, J=7,3, 1,3 Гц, 1H), 7,13 (с, 1H), 7,01 (д, J=8,4 Гц, 1H), 6,59 (дд, J=8,4, 2,5 Гц, 1H), 6,53 (д, J=2,3 Гц, 1H), 4,49 (т, J=5,7 Гц, 1H), 3,68 (д, J=15,1 Гц, 1H), 3,57 (д, J=15,0 Гц, 1H), 3,00 (дд, J=11,6, 5,4 Гц, 1H), 2,85 (дд, J=11,5, 6,4 Гц, 1H), 2,47 (с, 3H), 1,90-1,45 (м, 1Н); ESI-МС m/z 296 [M+H]+.
Стадия F: К раствору полученного фенола (1,6 г, 5,1 ммоль) со стадии E, описанной выше, в дихлорметане (50 мл) при 0°C последовательно добавляли пиридин (0,80 мл, 10 ммоль) и ангидрид трифторметансульфоновой кислоты (1,1 мл, 6,6 ммоль) по каплям. Полученный в результате реакционный раствор перемешивали при 0°C в течение 1 часа и затем гасили насыщенным водным раствором бикарбоната натрия. Органический экстракт отделяли и водный слой дважды экстрагировали дихлорметаном. Объединенный органический экстракт промывали смесью 1:1 вода/насыщенный раствор соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая сырой продукт. Очистка флэш-хроматографией на колонке (от дихлорметана до смеси 98:2 дихлорметан/метанол) давала требуемый трифторметансульфонат (1,6 г, 73% за две стадии) в виде желтого масла:1H ЯМР (CDCl3, 500 МГц) δ 7,74 (д, J=8,0 Гц, 1H), 7,70 (д, J=7,8 Гц, 1H), 7,32-7,24 (м, 3H), 7,16 (с, 1H), 7,02-7,00 (м, 2Н), 4,54 (т, J=5,4 Гц, 1H), 3,78 (д, J=15,4 Гц, 1H), 3,65 (д, J=15,4 Гц, 1H), 3,01 (дд, J=11,6, 5,0 Гц, 1H), 2,91 (дд, J=11,6, 6,0 Гц, 1H), 2,50 (с, 3H); ESI-МС m/z 428 [M+H]+.
Стадия G: К смеси трифторметансульфоната (1,0 г, 2,3 ммоль) со стадии F, описанной выше, бис(пинаколато)дибора (0,65 г, 2,6 ммоль) и ацетата калия (0,69 г, 7,0 ммоль) добавляли диметилсульфоксид (15 мл). Полученный в результате раствор продували аргоном в течение 10 минут и затем к нему добавляли 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий (57 мг, 0,07 ммоль). Реакционный раствор снова дегазировали, используя аргон, в течение 5 минут и нагревали при 80°C в течение 1 часа. Затем реакционный раствор охлаждали до комнатной температуры, разбавляли этилацетатом, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от дихлорметана до смеси 95:5 дихлорметан/метанол), получая требуемый продукт (грубый выход 1,3 г) в виде красноватого масла, который использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,72 (д, J=7,9 Гц, 1H), 7,67 (д, J=7,9 Гц, 1H), 7,56-7,54 (м, 2H), 7,29 (тд, J=7,3, 1,1 Гц, 1H), 7,23 (тд, J=8,2, 2,6 Гц, 1H), 7,16 (д, J=7,5 Гц, 1H), 7,13 (с, 1Н), 4,57 (т, J=5,3 Гц, 1H), 3,77 (д, J=14,9 Гц, 1H), 3,63 (д, J=14,9 Гц, 1H), 3,00 (дд, J=11,3, 4,8 Гц, 1H), 2,89 (дд, J=11,4, 6,1 Гц, 1H), 2,48 (с, 3H), 1,33 (с, 12H); ESI-МС m/z 406 [M+H]+.
Стадия H: К смеси сложного эфира бороновой кислоты (1,3 г, неочищенный продукт) со стадии G, описанной выше, 3,6-дихлорпиридазина (0,69 г, 4,6 ммоль) и карбоната натрия (0,75 г, 7,1 ммоль) добавляли N,N-диметилформамид (20 мл) и воду (5,1 мл). Реакционный раствор продували аргоном в течение 10 минут и затем к нему добавляли 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий (94 мг, 0,12 ммоль). Реакционный раствор снова дегазировали, используя аргон, в течение 5 минут и нагревали при 80°C в течение 2 часов. Затем реакционный раствор охлаждали до комнатной температуры, разбавляли этилацетатом, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали, используя систему MPLC Biotage (от дихлорметана до смеси 95:5 дихлорметан/метанол), получая частично очищенный продукт (0,83 г, 91%) в виде светло-коричневого твердого вещества, который использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,87 (с, 1H), 7,80-7,69 (м, 4H), 7,54 (д, J=9,0 Гц, 1H), 7,53-7,24 (м, 3H), 7,20 (с, 1H), 4,62 (ушир.с, 1Н), 3,86 (д, J=15,3 Гц, 1H), 3,74 (д, J=15,0 Гц, 1H), 3,07-3,04 (м, 1H), 2,96-2,92 (м, 1H), 2,53 (с, 3H); ESI-МС m/z 392 [M+H]+.
Стадия I: К частично растворенному хлорпиридазину (0,40 г, 1,0 ммоль) со стадии H, описанной выше, в смеси этанола (55 мл) и метанола (20 мл) добавляли моногидрат гидразина (2,0 мл, 42 ммоль) и палладий-на-угле (150 мг). Реакционный раствор нагревали при кипячении с обратным холодильником в течение 16 часов и затем охлаждали до комнатной температуры, фильтровали через слой целита и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от дихлорметана до смеси 97:3 дихлорметан/метанол), получая требуемый продукт (0,21 г, 59%) в виде светло-желтой пены:1H ЯМР (CDCl3, 500 МГц) δ 9,14 (дд, J=4,9, 1,5 Гц, 1H), 7,93 (д, J=1,5 Гц, 1H), 7,82 (дд, J=8,6, 1,6 Гц, 1H), 7,77-7,73 (м, 2H), 7,70 (д, J=7,8 Гц, 1H), 7,51 (дд, J=9,2, 5,4 Гц, 1H), 7,33-7,23 (м, 3H), 7,20 (с, 1H), 4,63 (т, J=5,4 Гц, 1H), 3,87 (д, J=15,1 Гц, 1H), 3,75 (д, J=15,0 Гц, 1H), 3,06 (дд, J=11,6, 4,9 Гц, 1H), 2,94 (дд, J=11,6, 6,1 Гц, 1H), 2,53 (с, 3H).
Стадия J: К раствору 7-пиридазинилтетрагидроизохинолина (0,21 г, 0,60 ммоль) со стадии I, описанной выше, в смеси метанола (10 мл) и дихлорметана (3 мл) добавляли малеиновую кислоту (69 мг, 0,60 ммоль). Реакционный раствор концентрировали при пониженном давлении до ~3 мл и разбавляли водой (10 мл). Полученный в результате раствор лиофилизировали в течение ночи, получая малеатную соль 4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина (280 мг, 99%) в виде светло-желтого твердого вещества: т.пл. 113-115°C;1H ЯМР (CD3OD, 500 МГц) δ 9,18 (дд, J=4,9, 1,5 Гц, 1H), 8,20 (дд, J=8,7, 1,5 Гц, 1H), 8,09 (с, 1H), 8,02 (дд, J=7,7, 1,2 Гц, 1H), 7,84-7,79 (м, 3H), 7,44 (д, J=8,3 Гц, 1H), 7,40-7,32 (м, 3H), 6,24 (с, 2H), 5,09 (дд, J=9,8, 6,0 Гц, 1H), 4,65 (ушир.с, 2H), 3,98 (дд, J=12,4, 6,0 Гц, 1Н), 3,75 (т, J=11,6 Гц, 1H), 3,08 (с, 3H); ESI-МС m/z 358 [M+H]+.
Стадия K: Свободное основание малеатной соли 4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина (0,28 г) разделяли препаративной хиральной ВЭЖХ (колонка CHIRALPAK OD, используя смесь 80:20:0,1 гептан/2-пропанол/диэтиламин в качестве элюента), получая (+)-энантиомер [[α]25D +122,7° (c 0,11, хлороформ)] и (-)-энантиомер [[α]25D -39,1° (c 0,11, хлороформ)]. (+)-Энантиомер (0,11 г, 0,31 ммоль) растворяли в смеси метанола (3 мл) и дихлорметана (3 мл) и к раствору добавляли один эквивалент малеиновой кислоты (35 мг, 0,31 ммоль). Полученный в результате раствор концентрировали при пониженном давлении и полученный остаток растворяли в смеси метанола (5 мл) и воды (25 мл) и лиофилизировали в течение ночи, получая малеатную соль (+)-4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина (0,14 г, 98%, >99% AUC ВЭЖХ) в виде белого твердого вещества: т.пл. 99-102°C;1H ЯМР (CD3OD, 500 МГц) δ 9,18 (дд, J=4,9, 1,5 Гц, 1H), 8,20 (дд, J=8,7, 1,5 Гц, 1H), 8,08 (с, 1H), 8,02 (дд, J=7,7, 1,2 Гц, 1H), 7,84-7,77 (м, 3H), 7,44 (д, J=8,3 Гц, 1H), 7,40-7,32 (м, 3H), 6,25 (с, 2H), 5,09 (дд, J=9,8, 6,0 Гц, 1H), 4,63 (ушир.с, 2H), 3,97 (дд, J=12,4, 6,0 Гц, 1H), 3,73 (т, J=11,6 Гц, 1Н), 3,09 (с, 3H); ESI-МС m/z 358 [M+H]+; элементный анализ, вычислено для C22H19N3S-C4H4O4-H2O: C, 63,53; Н, 5,13; N, 8,55. Найдено: C, 63,66; Н 4,90; N, 8,43.
(-)-Энантиомер (0,11 г, 0,31 ммоль) растворяли в смеси метанола (3 мл) и дихлорметана (3 мл) и к раствору добавляли один эквивалент малеиновой кислоты (35 мг, 0,31 ммоль). Полученный в результате раствор концентрировали при пониженном давлении и полученный остаток растворяли в смеси метанола (5 мл) и воды (25 мл) и лиофилизировали в течение ночи, получая малеатную соль (-)-4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина (0,14 г, 98%, >99% AUC ВЭЖХ) в виде белого твердого вещества: т.пл. 100-104°C;1H ЯМР (CD3OD, 500 МГц) δ 9,18 (дд, J=4,9, 1,5 Гц, 1H), 8,20 (дд, J=8,7, 1,5 Гц, 1H), 8,08 (с, 1H), 8,02 (дд, J=7,7, 1,2 Гц, 1H), 7,84-7,77 (м, 3H), 7,44 (д, J=8,3 Гц, 1H), 7,40-7,32 (м, 3H), 6,25 (с, 2H), 5,09 (дд, J=9,8, 6,0 Гц, 1H), 4,63 (ушир.с, 2H), 3,97 (дд, J=12,4, 6,0 Гц, 1H), 3,73 (т, J=11,6 Гц, 1H), 3,09 (с, 3H); ESI-МС m/z 358 [M+H]+; элементный анализ, вычислено для C22H19N3S-C4H4O4-H2O: C, 63,53; Н, 5,13; N, 8,55. Найдено: C, 63,46; Н 5,07; N, 8,43.
Пример 71 - Получение малеатной соли (+/-)-4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору 2-ацетилбензо[b]тиофена (5,0 г, 28 ммоль) в хлороформе (120 мл) добавляли пербромид бромида пиридиния (9,6 г, 28 ммоль) двумя порциями. Реакционный раствор перемешивали при комнатной температуре в течение 2 часов и затем промывали водой, водным раствором HCl (2 М), водой и насыщенным раствором соли. Полученный в результате раствор сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый бромкетон (грубый выход 7,4 г) в виде коричневого твердого вещества, которое использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 8,07 (с, 1H), 7,94-7,87 (м, 2H), 7,52-7,25 (м, 2H), 4,46 (с, 2H).
Стадия B: К раствору 3-метоксибензилметиламина (4,3 г, 29 ммоль) в дихлорметане (150 мл) добавляли триэтиламин (10 мл) и бромкетон (7,3 г, 29 ммоль) со стадии A, описанной выше. Реакционный раствор перемешивали при комнатной температуре в течение 2 часов и затем гасили насыщенным водным раствором бикарбоната натрия. Органический экстракт отделяли, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от 90:10 до 85:15 гексаны/этилацетат), получая требуемый продукт (7,1 г, 76% для 2 стадий) в виде красноватого масла:1H ЯМР (CDCl3, 500 МГц) δ 8,09 (с, 1H), 7,86 (д, J=8,0 Гц, 2H), 7,45 (дд, J=8,0, 1,1 Гц, 1H), 7,40 (дд, J=8,0, 1,0 Гц, 1Н), 7,24 (д, J=8,1 Гц, 1H), 6,97-6,95 (м, 2H), 6,83 (д, J=8,5 Гц, 1H), 3,78 (с, 3H), 3,76 (с, 2H), 3,70 (с, 2H), 2,43 (с, 3H); ESI-МС m/z 326 [M+H]+.
Стадия C: К раствору (7,1 г, 22 ммоль) со стадии B, описанной выше, в метаноле (100 мл) при 0°C небольшими порциями добавляли боргидрид натрия (0,89 г, 24 ммоль). Реакционный раствор перемешивали при 0°C в течение 1 часа. Затем растворитель удаляли при пониженном давлении и полученный остаток растворяли в дихлорметане. Полученный в результате раствор промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый спирт (6,9 г, грубый выход 96%) в виде желтого масла. Неочищенный продукт использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,81 (д, J=7,9 Гц, 1H), 7,70 (д, J=8,1 Гц, 1H), 7,33-7,23 (м, 3H), 7,21 (с, 1H), 6,91-6,87 (м, 3H), 5,07 (дд, J=10,1, 3,5 Гц, 1H), 4,40-4,03 (м, 1H), 3,81 (с, 3H), 3,72 (д, J=13,0 Гц, 1H), 3,56 (д, J=13,0 Гц, 1H), 2,83 (дд, J=12,4, 10,2 Гц, 1H), 2,72 (дд, J=12,4, 3,7 Гц, 1H), 2,34 (с, 3H); ESI-МС m/z 328 [M+H]+.
Стадия D: К раствору спирта (6,9 г, 21 ммоль) со стадии C, описанной выше, в дихлорметане (150 мл) по каплям добавляли метансульфоновую кислоту (14 мл, 210 ммоль). После добавления реакционный раствор перемешивали при комнатной температуре в течение 20 минут и затем медленно добавляли к ледяному водному раствору гидроксида натрия (120 мл, 2 М) и перемешивали в течение 20 минут. Органический слой отделяли и водный слой дважды экстрагировали дихлорметаном. Объединенный органический экстракт промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от 95:5 до 50:50 гексаны/этилацетат), получая требуемый продукт (3,4 г, 52%) в виде красноватого масла:1H ЯМР (CDCl3, 500 МГц) δ 7,72 (д, J=8,0 Гц, 1H), 7,67 (д, J=8,0 Гц, 1H), 7,29-7,23 (м, 2H), 7,13 (с, 1H), 7,06 (д, J=8,5 Гц, 1H), 6,69 (д, J=2,7 Гц, 1H), 6,61 (д, J=2,6 Гц, 1Н), 4,50 (т, J=5,3 Гц, 1H), 3,77 (с, 3H), 3,73 (д, J=14,5 Гц, 1H), 3,59 (д, J=14,5 Гц, 1H), 2,99 (дд, J=11,5, 4,9 Гц, 1H), 2,87 (дд, J=11,4, 6,2 Гц, 1H), 2,47 (с, 3H); ESI-МС m/z 310 [M+H]+.
Стадия E: К раствору 7-метокситетрагидроизохинолина (1,6 г, 5,1 ммоль) со стадии D, описанной выше, в уксусной кислоте (25 мл) добавляли водный раствор бромистого водорода (25 мл, 48%). Реакционный раствор нагревали при 100°C в течение 20 часов и затем охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток помещали в смесь дихлорметана, воды и небольшого количества метанола. Полученный в результате раствор осторожно гасили насыщенным водным раствором бикарбоната натрия вплоть до pH >8. Органический экстракт отделяли, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый фенол (грубый выход 1,6 г) в виде коричневого твердого вещества, которое использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,72 (д, J=7,7 Гц, 1H), 7,67 (д, J=7,7 Гц, 1H), 7,29 (тд, J=7,3, 1,2 Гц, 1H), 7,23 (тд, J=7,3, 1,3 Гц, 1H), 7,13 (с, 1H), 7,01 (д, J=8,4 Гц, 1H), 6,59 (дд, J=8,4, 2,5 Гц, 1H), 6,53 (д, J=2,3 Гц, 1H), 4,49 (т, J=5,7 Гц, 1H), 3,68 (д, J=15,1 Гц, 1H), 3,57 (д, J=15,0 Гц, 1H), 3,00 (дд, J=11,6, 5,4 Гц, 1H), 2,85 (дд, J=11,5, 6,4 Гц, 1H), 2,47 (с, 3H), 1,90-1,45 (м, 1H); ESI-МС m/z 296 [M+H]+.
Стадия F: К раствору полученного фенола (1,6 г, 5,1 ммоль) со стадии E, описанной выше, в дихлорметане (50 мл) при 0°C последовательно по каплям добавляли пиридин (0,80 мл, 10 ммоль) и ангидрид трифторметансульфоновой кислоты (1,1 мл, 6,6 ммоль). Полученный в результате реакционный раствор перемешивали при 0°C в течение 1 часа и затем гасили насыщенным водным раствором бикарбоната натрия. Органический экстракт отделяли и водный слой дважды экстрагировали дихлорметаном. Объединенный органический экстракт промывали смесью 1:1 вода/насыщенный раствор соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая сырой продукт. Очистка флэш-хроматографией на колонке (от дихлорметана до смеси 98:2 дихлорметан/метанол) давала требуемый трифторметансульфонат (1,6 г, 73% для двух стадий) в виде желтого масла:1H ЯМР (CDCl3, 500 МГц) δ 7,74 (д, J=8,0 Гц, 1H), 7,70 (д, J=7,8 Гц, 1H), 7,32-7,24 (м, 3H), 7,16 (с, 1H), 7,02-7,00 (м, 2H), 4,54 (т, J=5,4 Гц, 1H), 3,78 (д, J=15,4 Гц, 1H), 3,65 (д, J=15,4 Гц, 1H), 3,01 (дд, J=11,6, 5,0 Гц, 1H), 2,91 (дд, J=11,6, 6,0 Гц, 1H), 2,50 (с, 3H); ESI-МС m/z 428 [M+H]+.
Стадия G: К смеси трифторметансульфоната (1,0 г, 2,3 ммоль) со стадии F, описанной выше, бис(пинаколато)дибора (0,65 г, 2,6 ммоль) и ацетата калия (0,69 г, 7,0 ммоль) добавляли диметилсульфоксид (15 мл). Полученный в результате раствор продували аргоном в течение 10 минут и затем к нему добавляли 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий (57 мг, 0,07 ммоль). Реакционный раствор снова дегазировали, используя аргон, в течение 5 минут и нагревали при 80°C в течение 1 часа. Затем реакционный раствор охлаждали до комнатной температуры, разбавляли этилацетатом, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от дихлорметана до смеси 95:5 дихлорметан/метанол), получая требуемый продукт (грубый выход 1,3 г) в виде красноватого масла, которое использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,72 (д, J=7,9 Гц, 1H), 7,67 (д, J=7,9 Гц, 1H), 7,56-7,54 (м, 2H), 7,29 (тд, J=7,3, 1,1 Гц, 1H), 7,23 (тд, J=8,2, 2,6 Гц, 1Н), 7,16 (д, J=7,5 Гц, 1H), 7,13 (с, 1H), 4,57 (т, J=5,3 Гц, 1H), 3,77 (д, J=14,9 Гц, 1H), 3,63 (д, J=14,9 Гц, 1H), 3,00 (дд, J=11,3, 4,8 Гц, 1H), 2,89 (дд, J=11,4, 6,1 Гц, 1H), 2,48 (с, 3H), 1,33 (с, 12H); ESI-МС m/z 406 [M+H]+.
Стадия H: К смеси сложного эфира бороновой кислоты (0,12 г, 0,3 ммоль) со стадии G, описанной выше, 2-хлорпиразина (0,053 мл, 0,59 ммоль) и карбоната натрия (95 мг, 0,90 ммоль) добавляли диметилформамид (3 мл) и воду (0,75 мл). Реакционный раствор продували аргоном в течение 10 минут и затем к нему добавляли 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий (12 мг, 0,015 ммоль). Реакционный раствор снова дегазировали, используя аргон, в течение 5 минут и нагревали при 80°C в течение 3 часов. Затем реакционный раствор охлаждали до комнатной температуры, разбавляли этилацетатом, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали препаративной тонкослойной хроматографией (95:5 дихлорметан/метанол), получая требуемый продукт (67 мг, 62%) в виде светло-желтого твердого вещества:1H ЯМР (CDCl3, 500 МГц) δ 8,99 (д, J=1,5 Гц, 1Н), 8,61 (дд, J=2,5, 1,6 Гц, 1H), 8,49 (д, J=2,5 Гц, 1H), 7,79 (с, 1H), 7,75-7,68 (м, 3H), 7,32-7,25 (м, 3H), 7,19 (с, 1H), 4,63 (т, J=5,6 Гц, 1H), 3,87 (д, J=15,0 Гц, 1H), 3,74 (д, J=15,0 Гц, 1H), 3,07-3,04 (м, 1H), 2,96-2,92 (м, 1H), 2,53 (с, 3Н). К раствору вновь полученного 7-пиразинилтетрагидроизохинолина (67 мг, 0,19 ммоль) в метаноле (2 мл) добавляли малеиновую кислоту (22 мг, 0,9 ммоль) с последующим медленным добавлением воды (10 мл). Полученный в результате реакционный раствор лиофилизировали в течение ночи, получая малеатную соль (+/-)-4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолина (87 мг) в виде не совсем белого твердого вещества: т.пл. 102-105°C;1H ЯМР (CD3OD, 500 МГц) δ 9,14 (д, J=1,5 Гц, 1H), 8,69 (дд, J=2,5, 1,6 Гц, 1H), 8,56 (д, J=2,5 Гц, 1H), 8,06-8,03 (м, 2H), 7,83 (д, J=7,3 Гц, 1H), 7,78 (д, J=7,5 Гц, 1H), 7,42-7,32 (м, 4H), 6,25 (с, 2H), 5,08-5,05 (м, 1H), 4,60 (кажущийся с, 2H), 3,94 (дд, J=11,6, 5,3 Гц, 1H), 3,74-3,68 (м, 1H), 3,08 (с, 3H); ESI-МС m/z 358 [M+H]+.
Пример 72 - Получение малеатной соли (+/-)-4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору 2-ацетилбензо[b]тиофена (5,0 г, 28 ммоль) в хлороформе (120 мл) добавляли пербромид бромида пиридиния (9,6 г, 28 ммоль) двумя порциями. Реакционный раствор перемешивали при комнатной температуре в течение 2 часов и затем промывали водой, водным раствором HCl (2 М), водой и насыщенным раствором соли. Полученный в результате раствор сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый бромкетон (грубый выход 7,4 г) в виде коричневого твердого вещества, которое использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 8,07 (с, 1H), 7,94-7,87 (м, 2H), 7,52-7,25 (м, 2H), 4,46 (с, 2H).
Стадия B: К раствору 3-метоксибензилметиламина (4,3 г, 29 ммоль) в дихлорметане (150 мл) добавляли триэтиламин (10 мл) и бромкетон (7,3 г, 29 ммоль) со стадии A, описанной выше. Реакционный раствор перемешивали при комнатной температуре в течение 2 часов и затем гасили насыщенным водным раствором бикарбоната натрия. Органический экстракт отделяли, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от 90:10 до 85:15 гексаны/этилацетат), получая требуемый продукт (7,1 г, 76% для 2 стадий) в виде красноватого масла:1H ЯМР (CDCl3, 500 МГц) δ 8,09 (с, 1H), 7,86 (д, J=8,0 Гц, 2H), 7,45 (дд, J=8,0, 1,1 Гц, 1H), 7,40 (дд, J=8,0, 1,0 Гц, 1H), 7,24 (д, J=8,1 Гц, 1H), 6,97-6,95 (м, 2H), 6,83 (д, J=8,5 Гц, 1H), 3,78 (с, 3H), 3,76 (с, 2H), 3,70 (с, 2H), 2,43 (с, 3H); ESI-МС m/z 326 [M+H]+.
Стадия C: К раствору кетона (7,1 г, 22 ммоль) со стадии B, описанной выше, в метаноле (100 мл) при 0°C небольшими порциями добавляли боргидрид натрия (0,89 г, 24 ммоль). Реакционный раствор перемешивали при 0°C в течение 1 часа. Затем растворитель удаляли при пониженном давлении и полученный остаток растворяли в дихлорметане. Полученный в результате раствор промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый спирт (6,9 г, грубый выход 96%) в виде желтого масла. Неочищенный продукт использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,81 (д, J=7,9 Гц, 1H), 7,70 (д, J=8,1 Гц, 1H), 7,33-7,23 (м, 3H), 7,21 (с, 1H), 6,91-6,87 (м, 3H), 5,07 (дд, J=10,1, 3,5 Гц, 1H), 4,40-4,03 (м, 1H), 3,81 (с, 3H), 3,72 (д, J=13,0 Гц, 1H), 3,56 (д, J=13,0 Гц, 1H), 2,83 (дд, J=12,4, 10,2 Гц, 1H), 2,72 (дд, J=12,4, 3,7 Гц, 1H), 2,34 (с, 3H); ESI-МС m/z 328 [M+H]+.
Стадия D: К раствору спирта (6,9 г, 21 ммоль) со стадии C, описанной выше, в дихлорметане (150 мл) по каплям добавляли метансульфоновую кислоту (14 мл, 210 ммоль). После добавления реакционный раствор перемешивали при комнатной температуре в течение 20 минут и затем медленно добавляли к ледяному водному раствору гидроксида натрия (120 мл, 2 М) и перемешивали в течение 20 минут. Органический слой отделяли и водный слой дважды экстрагировали дихлорметаном. Объединенный органический экстракт промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от 95:5 до 50:50 гексаны/этилацетат), получая требуемый продукт (3,4 г, 52%) в виде красноватого масла:1H ЯМР (CDCl3, 500 МГц) δ 7,72 (д, J=8,0 Гц, 1H), 7,67 (д, J=8,0 Гц, 1H), 7,29-7,23 (м, 2H), 7,13 (с, 1H), 7,06 (д, J=8,5 Гц, 1H), 6,69 (д, J=2,7 Гц, 1H), 6,61 (д, J=2,6 Гц, 1H), 4,50 (т, J=5,3 Гц, 1H), 3,77 (с, 3H), 3,73 (д, J=14,5 Гц, 1H), 3,59 (д, J=14,5 Гц, 1H), 2,99 (дд, J=11,5, 4,9 Гц, 1H), 2,87 (дд, J=11,4, 6,2 Гц, 1H), 2,47 (с, 3H); ESI-МС m/z 310 [M+H]+.
Стадия E: К раствору 7-метокситетрагидроизохинолина (1,6 г, 5,1 ммоль) со стадии D, описанной выше, в уксусной кислоте (25 мл) добавляли водный раствор бромистого водорода (25 мл, 48%). Реакционный раствор нагревали при 100°C в течение 20 часов и затем охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток помещали в смесь дихлорметана, воды и небольшого количества метанола. Полученный в результате раствор осторожно гасили насыщенным водным раствором бикарбоната натрия до pH >8. Органический экстракт отделяли, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый фенол (грубый выход 1,6 г) в виде коричневого твердого вещества, которое использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,72 (д, J=7,7 Гц, 1H), 7,67 (д, J=7,7 Гц, 1H), 7,29 (тд, J=7,3, 1,2 Гц, 1H), 7,23 (тд, J=7,3, 1,3 Гц, 1H), 7,13 (с, 1H), 7,01 (д, J=8,4 Гц, 1H), 6,59 (дд, J=8,4, 2,5 Гц, 1H), 6,53 (д, J=2,3 Гц, 1H), 4,49 (т, J=5,7 Гц, 1H), 3,68 (д, J=15,1 Гц, 1H), 3,57 (д, J=15,0 Гц, 1H), 3,00 (дд, J=11,6, 5,4 Гц, 1H), 2,85 (дд, J=11,5, 6,4 Гц, 1H), 2,47 (с, 3H), 1,90-1,45 (м, 1H); ESI-МС m/z 296 [M+H]+.
Стадия F: К раствору полученного фенола (1,6 г, 5,1 ммоль) со стадии E, описанной выше, в дихлорметане (50 мл) при 0°C последовательно по каплям добавляли пиридин (0,80 мл, 10 ммоль) и ангидрид трифторметансульфоновой кислоты (1,1 мл, 6,6 ммоль). Полученный в результате реакционный раствор перемешивали при 0°C в течение 1 часа и затем гасили насыщенным водным раствором бикарбоната натрия. Органический экстракт отделяли и водный слой дважды экстрагировали дихлорметаном. Объединенный органический экстракт промывали смесью 1:1 вода/насыщенный раствор соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая сырой продукт. Очистка флэш-хроматографией на колонке (от дихлорметана до смеси 98:2 дихлорметан/метанол) давала требуемый трифторметансульфонат (1,6 г, 73% для двух стадий) в виде желтого масла:1H ЯМР (CDCl3, 500 МГц) δ 7,74 (д, J=8,0 Гц, 1H), 7,70 (д, J=7,8 Гц, 1H), 7,32-7,24 (м, 3H), 7,16 (с, 1H), 7,02-7,00 (м, 2H), 4,54 (т, J=5,4 Гц, 1H), 3,78 (д, J=15,4 Гц, 1H), 3,65 (д, J=15,4 Гц, 1H), 3,01 (дд, J=11,6, 5,0 Гц, 1H), 2,91 (дд, J=11,6, 6,0 Гц, 1H), 2,50 (с, 3H); ESI-МС m/z 428 [M+H]+.
Стадия G: К смеси трифторметансульфоната (1,0 г, 2,3 ммоль) со стадии F, описанной выше, бис(пинаколато)дибора (0,65 г, 2,6 ммоль) и ацетата калия (0,69 г, 7,0 ммоль) добавляли диметилсульфоксид (15 мл). Полученный в результате раствор продували аргоном в течение 10 минут и затем к нему добавляли 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий (57 мг, 0,07 ммоль). Реакционный раствор снова дегазировали, используя аргон, в течение 5 минут и нагревали при 80°C в течение 1 часа. Затем реакционный раствор охлаждали до комнатной температуры, разбавляли этилацетатом, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от дихлорметана до смеси 95:5 дихлорметан/метанол), получая требуемый продукт (грубый выход 1,3 г) в виде красноватого масла, которое использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,72 (д, J=7,9 Гц, 1H), 7,67 (д, J=7,9 Гц, 1H), 7,56-7,54 (м, 2H), 7,29 (тд, J=7,3, 1,1 Гц, 1H), 7,23 (тд, J=8,2, 2,6 Гц, 1H), 7,16 (д, J=7,5 Гц, 1Н), 7,13 (с, 1H), 4,57 (т, J=5,3 Гц, 1H), 3,77 (д, J=14,9 Гц, 1H), 3,63 (д, J=14,9 Гц, 1H), 3,00 (дд, J=11,3, 4,8 Гц, 1H), 2,89 (дд, J=11,4, 6,1 Гц, 1H), 2,48 (с, 3H), 1,33 (с, 12H); ESI-МС m/z 406 [M+H]+.
Стадия H: К смеси сложного эфира бороновой кислоты (0,12 г, 0,3 ммоль) со стадии G, описанной выше, 2-хлорпиримидина (68 мг, 0,59 ммоль) и карбоната натрия (95 мг, 0,90 ммоль) добавляли N,N-диметилформамид (3 мл) и воду (0,75 мл). Реакционный раствор продували аргоном в течение 10 минут и затем к раствору добавляли 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий (12 мг, 0,015 ммоль). Реакционный раствор снова дегазировали, используя аргон, в течение 5 минут и нагревали при 80°C в течение 3 часов. Затем реакционный раствор охлаждали до комнатной температуры, разбавляли этилацетатом, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали препаративной тонкослойной хроматографией (95:5 дихлорметан/метанол), получая требуемый продукт (34 мг, 31%) в виде светло-желтого масла:1H ЯМР (CDCl3, 500 МГц) δ 8,79 (д, J=4,8 Гц, 2H), 8,20-8,18 (м, 2H), 7,74 (д, J=8,0 Гц, 1H), 7,68 (д, J=7,8 Гц, 1H), 7,31-7,25 (м, 3H), 7,18 (с, 1H), 7,17 (т, J=4,7 Гц, 1H), 4,64-4,62 (м, 1H), 3,87 (д, J=15,0 Гц, 1H), 3,73 (д, J=15,0 Гц, 1H), 3,10-2,93 (м, 2H), 2,52 (с, 3H).
К раствору 7-пиримидинилтетрагидроизохинолина (34 мг, 0,095 ммоль) в метаноле (2 мл) добавляли малеиновую кислоту (11 мг, 0,095 ммоль) с последующим медленным добавлением воды (10 мл). Полученный в результате реакционный раствор лиофилизировали в течение ночи, получая малеатную соль (+/-)-4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолина (42 мг) в виде светло-желтого твердого вещества: т.пл. 103-106°C;1H ЯМР (CD3OD, 500 МГц) δ 8,86 (д, J=4,9 Гц, 2Н), 8,36-8,34 (м, 2Н), 7,83 (д, J=7,8 Гц, 1H), 7,78 (д, J=7,4 Гц, 1H), 7,40-7,32 (м, 5H), 6,25 (с, 2H), 5,07 (дд, J=9,1, 5,8 Гц, 1H), 4,62 (ушир.с, 2H), 3,96 (дд, J=12,3, 5,6 Гц, 1H), 3,77-3,62 (м, 1H), 3,09 (с, 3H); ESI-МС m/z 358 [M+H]+.
Пример 73 - Получение малеатной соли (+/-)-4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору 2-ацетилбензо[b]тиофена (5,0 г, 28 ммоль) в хлороформе (120 мл) добавляли пербромид бромида пиридиния (9,6 г, 28 ммоль) двумя порциями. Реакционный раствор перемешивали при комнатной температуре в течение 2 часов и затем промывали водой, водным раствором HCl (2 М), водой и насыщенным раствором соли. Полученный в результате раствор сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый бромкетон (грубый выход 7,4 г) в виде коричневого твердого вещества, которое использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 8,07 (с, 1H), 7,94-7,87 (м, 2H), 7,52-7,25 (м, 2H), 4,46 (с, 2H).
Стадия B: К раствору 3-метоксибензилметиламина (4,3 г, 29 ммоль) в дихлорметане (150 мл) добавляли триэтиламин (10 мл) и бромкетон (7,3 г, 29 ммоль) со стадии A, описанной выше. Реакционный раствор перемешивали при комнатной температуре в течение 2 часов и затем гасили насыщенным водным раствором бикарбоната натрия. Органический экстракт отделяли, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от 90:10 до 85:15 гексаны/этилацетат), получая требуемый продукт (7,1 г, 76% для 2 стадий) в виде красноватого масла:1H ЯМР (CDCl3, 500 МГц) δ 8,09 (с, 1H), 7,86 (д, J=8,0 Гц, 2H), 7,45 (дд, J=8,0, 1,1 Гц, 1H), 7,40 (дд, J=8,0, 1,0 Гц, 1H), 7,24 (д, J=8,1 Гц, 1H), 6,97-6,95 (м, 2H), 6,83 (д, J=8,5 Гц, 1H), 3,78 (с, 3H), 3,76 (с, 2H), 3,70 (с, 2H), 2,43 (с, 3H); ESI-МС m/z 326 [M+H]+.
Стадия C: К раствору кетона (7,1 г, 22 ммоль) со стадии B, описанной выше, в метаноле (100 мл) при 0°C небольшими порциями добавляли боргидрид натрия (0,89 г, 24 ммоль). Реакционный раствор перемешивали при 0°C в течение 1 часа. Затем растворитель удаляли при пониженном давлении и полученный остаток растворяли в дихлорметане. Полученный в результате раствор промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый спирт (6,9 г, грубый выход 96%) в виде желтого масла. Неочищенный продукт использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,81 (д, J=7,9 Гц, 1H), 7,70 (д, J=8,1 Гц, 1H), 7,33-7,23 (м, 3H), 7,21 (с, 1H), 6,91-6,87 (м, 3H), 5,07 (дд, J=10,1, 3,5 Гц, 1H), 4,40-4,03 (м, 1H), 3,81 (с, 3H), 3,72 (д, J=13,0 Гц, 1H), 3,56 (д, J=13,0 Гц, 1H), 2,83 (дд, J=12,4, 10,2 Гц, 1Н), 2,72 (дд, J=12,4, 3,7 Гц, 1H), 2,34 (с, 3H); ESI-МС m/z 328 [M+H]+.
Стадия D: К раствору спирта (6,9 г, 21 ммоль) со стадии C, описанной выше, в дихлорметане (150 мл) по каплям добавляли метансульфоновую кислоту (14 мл, 210 ммоль). После добавления реакционный раствор перемешивали при комнатной температуре в течение 20 минут и затем медленно добавляли к ледяному водному раствору гидроксида натрия (120 мл, 2 М) и перемешивали в течение 20 минут. Органический слой отделяли и водный слой дважды экстрагировали дихлорметаном. Объединенный органический экстракт промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от 95:5 до 50:50 гексаны/этилацетат), получая требуемый продукт (3,4 г, 52%) в виде красноватого масла:1H ЯМР (CDCl3, 500 МГц) δ 7,72 (д, J=8,0 Гц, 1H), 7,67 (д, J=8,0 Гц, 1H), 7,29-7,23 (м, 2H), 7,13 (с, 1H), 7,06 (д, J=8,5 Гц, 1Н), 6,69 (д, J=2,7 Гц, 1H), 6,61 (д, J=2,6 Гц, 1H), 4,50 (т, J=5,3 Гц, 1Н), 3,77 (с, 3H), 3,73 (д, J=14,5 Гц, 1H), 3,59 (д, J=14,5 Гц, 1H), 2,99 (дд, J=11,5, 4,9 Гц, 1H), 2,87 (дд, J=11,4, 6,2 Гц, 1H), 2,47 (с, 3H); ESI-МС m/z 310 [M+H]+.
Стадия E: К раствору 7-метокситетрагидроизохинолина (1,6 г, 5,1 ммоль) со стадии D, описанной выше, в уксусной кислоте (25 мл) добавляли водный раствор бромистого водорода (25 мл, 48%). Реакционный раствор нагревали при 100°C в течение 20 часов и затем охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток помещали в смесь дихлорметана, воды и небольшого количества метанола. Полученный в результате раствор осторожно гасили насыщенным водным раствором бикарбоната натрия до pH >8. Органический экстракт отделяли, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый фенол (грубый выход 1,6 г) в виде коричневого твердого вещества, которое использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,72 (д, J=7,7 Гц, 1H), 7,67 (д, J=7,7 Гц, 1H), 7,29 (тд, J=7,3, 1,2 Гц, 1H), 7,23 (тд, J=7,3, 1,3 Гц, 1H), 7,13 (с, 1H), 7,01 (д, J=8,4 Гц, 1H), 6,59 (дд, J=8,4, 2,5 Гц, 1Н), 6,53 (д, J=2,3 Гц, 1H), 4,49 (т, J=5,7 Гц, 1H), 3,68 (д, J=15,1 Гц, 1H), 3,57 (д, J=15,0 Гц, 1H), 3,00 (дд, J=11,6, 5,4 Гц, 1H), 2,85 (дд, J=11,5, 6,4 Гц, 1Н), 2,47 (с, 3H), 1,90-1,45 (м, 1H); ESI-МС m/z 296 [M+H]+.
Стадия F: К раствору полученного фенола (1,6 г, 5,1 ммоль) со стадии E, описанной выше, в дихлорметане (50 мл) при 0°C последовательно по каплям добавляли пиридин (0,80 мл, 10 ммоль) и ангидрид трифторметансульфоновой кислоты (1,1 мл, 6,6 ммоль). Полученный в результате реакционный раствор перемешивали при 0°C в течение 1 часа и затем гасили насыщенным водным раствором бикарбоната натрия. Органический экстракт отделяли и водный слой дважды экстрагировали дихлорметаном. Объединенный органический экстракт промывали смесью 1:1 вода/насыщенный раствор соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая сырой продукт. Очистка флэш-хроматографией на колонке (от дихлорметана до смеси 98:2 дихлорметан/метанол) давала требуемый трифторметансульфонат (1,6 г, 73% для двух стадий) в виде желтого масла:1H ЯМР (CDCl3, 500 МГц) δ 7,74 (д, J=8,0 Гц, 1H), 7,70 (д, J=7,8 Гц, 1H), 7,32-7,24 (м, 3H), 7,16 (с, 1H), 7,02-7,00 (м, 2H), 4,54 (т, J=5,4 Гц, 1H), 3,78 (д, J=15,4 Гц, 1H), 3,65 (д, J=15,4 Гц, 1H), 3,01 (дд, J=11,6, 5,0 Гц, 1H), 2,91 (дд, J=11,6, 6,0 Гц, 1H), 2,50 (с, 3H); ESI-МС m/z 428 [M+H]+.
Стадия G: К смеси трифторметансульфоната (1,0 г, 2,3 ммоль) со стадии F, описанной выше, бис(пинаколато)дибора (0,65 г, 2,6 ммоль) и ацетата калия (0,69 г, 7,0 ммоль) добавляли диметилсульфоксид (15 мл). Полученный в результате раствор продували аргоном в течение 10 минут и затем к нему добавляли 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий (57 мг, 0,07 ммоль). Реакционный раствор снова дегазировали, используя аргон, в течение 5 минут и нагревали при 80°C в течение 1 часа. Затем реакционный раствор охлаждали до комнатной температуры, разбавляли этилацетатом, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от дихлорметана до смеси 95:5 дихлорметан/метанол), получая требуемый продукт (грубый выход 1,3 г) в виде красноватого масла, которое использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,72 (д, J=7,9 Гц, 1H), 7,67 (д, J=7,9 Гц, 1H), 7,56-7,54 (м, 2H), 7,29 (тд, J=7,3, 1,1 Гц, 1H), 7,23 (тд, J=8,2, 2,6 Гц, 1H), 7,16 (д, J=7,5 Гц, 1H), 7,13 (с, 1H), 4,57 (т, J=5,3 Гц, 1H), 3,77 (д, J=14,9 Гц, 1H), 3,63 (д, J=14,9 Гц, 1H), 3,00 (дд, J=11,3, 4,8 Гц, 1H), 2,89 (дд, J=11,4, 6,1 Гц, 1H), 2,48 (с, 3H), 1,33 (с, 12H); ESI-МС m/z 406 [M+H]+.
Стадия H: К смеси сложного эфира бороновой кислоты (0,12 г, 0,3 ммоль) со стадии G, описанной выше, 5-бромпиримидина (94 мг, 0,59 ммоль) и карбоната натрия (95 мг, 0,90 ммоль) добавляли N,N-диметилформамид (3 мл) и воду (0,75 мл). Реакционный раствор продували аргоном в течение 10 минут и затем к раствору добавляли 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий (12 мг, 0,015 ммоль). Реакционный раствор снова дегазировали, используя аргон, в течение 5 минут и нагревали при 80°C в течение 3 часов. Затем реакционный раствор охлаждали до комнатной температуры, разбавляли этилацетатом, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали препаративной тонкослойной хроматографией (95:5 дихлорметан/метанол), получая требуемый продукт (49 мг, 45%) в виде светло-желтого твердого вещества:1H ЯМР (CDCl3, 500 МГц) δ 9,19 (с, 1H), 8,92 (с, 2H), 7,74 (д, J=8,0 Гц, 1H), 7,70 (д, J=7,8 Гц, 1H), 7,32-7,25 (м, 5H), 7,21 (с, 1H), 4,63-4,61 (м, 1H), 3,85 (д, J=15,1 Гц, 1H), 3,72 (д, J=15,1 Гц, 1H), 3,08-3,04 (м, 1H), 2,97-2,93 (м, 1H), 2,53 (с, 3H). К раствору 7-пиримидинилтетрагидроизохинолина (49 мг, 0,14 ммоль) в метаноле (2 мл) добавляли малеиновую кислоту (16 мг, 0,14 ммоль) с последующим медленным добавлением воды (10 мл). Полученный в результате реакционный раствор лиофилизировали в течение ночи, получая малеатную соль (+/-)-4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина (64 мг) в виде не совсем белого твердого вещества: т.пл. 103-107°C;1H ЯМР (CD3OD, 500 МГц) δ 9,16 (с, 1H), 9,09 (с, 2H), 7,82 (дд, J=7,9, 5,7 Гц, 1H), 7,78 (д, J=7,5 Гц, 1H), 7,70-7,68 (м, 2H), 7,42-7,32 (м, 4H), 6,25 (с, 2H), 5,06 (дд, J=9,9, 5,9 Гц, 1H), 4,58 (кажущийся с, 2H), 3,93 (дд, J=12,2, 5,9 Гц, 1H), 3,70 (дд, J=9,8, 6,5 Гц, 1H), 3,07 (с, 3H); ESI-МС m/z 358 [M+H]+.
Пример 74 - Получение малеатной соли (+/-)-4-(бензо[b]тиофен-2-ил)-2-метил-7-(3,5-диметилизоксазол-4-ил)-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору 2-ацетилбензо[b]тиофена (5,0 г, 28 ммоль) в хлороформе (120 мл) добавляли пербромид бромида пиридиния (9,6 г, 28 ммоль) двумя порциями. Реакционный раствор перемешивали при комнатной температуре в течение 2 часов и затем промывали водой, водным раствором HCl (2 М), водой и насыщенным раствором соли. Полученный в результате раствор сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый бромкетон (грубый выход 7,4 г) в виде коричневого твердого вещества, которое использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 8,07 (с, 1H), 7,94-7,87 (м, 2H), 7,52-7,25 (м, 2H), 4,46 (с, 2H).
Стадия B: К раствору 3-метоксибензилметиламина (4,3 г, 29 ммоль) в дихлорметане (150 мл) добавляли триэтиламин (10 мл) и бромкетон (7,3 г, 29 ммоль) со стадии A, описанной выше. Реакционный раствор перемешивали при комнатной температуре в течение 2 часов и затем гасили насыщенным водным раствором бикарбоната натрия. Органический экстракт отделяли, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от 90:10 до 85:15 гексаны/этилацетат), получая требуемый продукт (7,1 г, 76% для 2 стадий) в виде красноватого масла:1H ЯМР (CDCl3, 500 МГц) δ 8,09 (с, 1Н), 7,86 (д, J=8,0 Гц, 2H), 7,45 (дд, J=8,0, 1,1 Гц, 1H), 7,40 (дд, J=8,0, 1,0 Гц, 1H), 7,24 (д, J=8,1 Гц, 1H), 6,97-6,95 (м, 2H), 6,83 (д, J=8,5 Гц, 1H), 3,78 (с, 3H), 3,76 (с, 2H), 3,70 (с, 2H), 2,43 (с, 3H); ESI-МС m/z 326 [M+H]+.
Стадия C: К раствору кетона (7,1 г, 22 ммоль) со стадии B, описанной выше, в метаноле (100 мл) при 0°C небольшими порциями добавляли боргидрид натрия (0,89 г, 24 ммоль). Реакционный раствор перемешивали при 0°C в течение 1 часа. Затем растворитель удаляли при пониженном давлении и полученный остаток растворяли в дихлорметане. Полученный в результате раствор промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый спирт (6,9 г, грубый выход 96%) в виде желтого масла. Неочищенный продукт использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,81 (д, J=7,9 Гц, 1H), 7,70 (д, J=8,1 Гц, 1H), 7,33-7,23 (м, 3H), 7,21 (с, 1H), 6,91-6,87 (м, 3H), 5,07 (дд, J=10,1, 3,5 Гц, 1H), 4,40-4,03 (м, 1H), 3,81 (с, 3H), 3,72 (д, J=13,0 Гц, 1H), 3,56 (д, J=13,0 Гц, 1H), 2,83 (дд, J=12,4, 10,2 Гц, 1H), 2,72 (дд, J=12,4, 3,7 Гц, 1H), 2,34 (с, 3H); ESI-МС m/z 328 [M+H]+.
Стадия D: К раствору спирта (6,9 г, 21 ммоль) со стадии C, описанной выше, в дихлорметане (150 мл) по каплям добавляли метансульфоновую кислоту (14 мл, 210 ммоль). После добавления реакционный раствор перемешивали при комнатной температуре в течение 20 минут и затем медленно добавляли к ледяному водному раствору гидроксида натрия (120 мл, 2 М) и перемешивали в течение 20 минут. Органический слой отделяли и водный слой дважды экстрагировали дихлорметаном. Объединенный органический экстракт промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от 95:5 до 50:50 гексаны/этилацетат), получая требуемый продукт (3,4 г, 52%) в виде красноватого масла:1H ЯМР (CDCl3, 500 МГц) δ 7,72 (д, J=8,0 Гц, 1H), 7,67 (д, J=8,0 Гц, 1H), 7,29-7,23 (м, 2H), 7,13 (с, 1H), 7,06 (д, J=8,5 Гц, 1H), 6,69 (д, J=2,7 Гц, 1Н), 6,61 (д, J=2,6 Гц, 1H), 4,50 (т, J=5,3 Гц, 1H), 3,77 (с, 3H), 3,73 (д, J=14,5 Гц, 1H), 3,59 (д, J=14,5 Гц, 1H), 2,99 (дд, J=11,5, 4,9 Гц, 1H), 2,87 (дд, J=11,4, 6,2 Гц, 1H), 2,47 (с, 3H); ESI-МС m/z 310 [M+H]+.
Стадия E: К раствору 7-метокситетрагидроизохинолина (1,6 г, 5,1 ммоль) со стадии D, описанной выше, в уксусной кислоте (25 мл) добавляли водный раствор бромистого водорода (25 мл, 48%). Реакционный раствор нагревали при 100°C в течение 20 часов и затем охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток помещали в смесь дихлорметана, воды и небольшого количества метанола. Полученный в результате раствор осторожно гасили насыщенным водным раствором бикарбоната натрия до pH >8. Органический экстракт отделяли, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый фенол (грубый выход 1,6 г) в виде коричневого твердого вещества, которое использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,72 (д, J=7,7 Гц, 1H), 7,67 (д, J=7,7 Гц, 1Н), 7,29 (тд, J=7,3, 1,2 Гц, 1Н), 7,23 (тд, J=7,3, 1,3 Гц, 1H), 7,13 (с, 1H), 7,01 (д, J=8,4 Гц, 1H), 6,59 (дд, J=8,4, 2,5 Гц, 1H), 6,53 (д, J=2,3 Гц, 1H), 4,49 (т, J=5,7 Гц, 1Н), 3,68 (д, J=15,1 Гц, 1H), 3,57 (д, J=15,0 Гц, 1H), 3,00 (дд, J=11,6, 5,4 Гц, 1Н), 2,85 (дд, J=11,5, 6,4 Гц, 1H), 2,47 (с, 3H), 1,90-1,45 (м, 1H); ESI-МС m/z 296 [M+H]+.
Стадия F: К раствору полученного фенола (1,6 г, 5,1 ммоль) со стадии E, описанной выше, в дихлорметане (50 мл) при 0°C последовательно по каплям добавляли пиридин (0,80 мл, 10 ммоль) и ангидрид трифторметансульфоновой кислоты (1,1 мл, 6,6 ммоль). Полученный в результате реакционный раствор перемешивали при 0°C в течение 1 часа и затем гасили насыщенным водным раствором бикарбоната натрия. Органический экстракт отделяли и водный слой дважды экстрагировали дихлорметаном. Объединенный органический экстракт промывали смесью 1:1 вода/насыщенный раствор соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая сырой продукт. Очистка флэш-хроматографией на колонке (от дихлорметана до смеси 98:2 дихлорметан/метанол) давала требуемый трифторметансульфонат (1,6 г, 73% для двух стадий) в виде желтого масла:1H ЯМР (CDCl3, 500 МГц) δ 7,74 (д, J=8,0 Гц, 1H), 7,70 (д, J=7,8 Гц, 1H), 7,32-7,24 (м, 3H), 7,16 (с, 1H), 7,02-7,00 (м, 2H), 4,54 (т, J=5,4 Гц, 1H), 3,78 (д, J=15,4 Гц, 1H), 3,65 (д, J=15,4 Гц, 1H), 3,01 (дд, J=11,6, 5,0 Гц, 1H), 2,91 (дд, J=11,6, 6,0 Гц, 1H), 2,50 (с, 3H); ESI-МС m/z 428 [M+H]+.
Стадия G: К смеси трифторметансульфоната (1,0 г, 2,3 ммоль) со стадии F, описанной выше, бис(пинаколато)дибора (0,65 г, 2,6 ммоль) и ацетата калия (0,69 г, 7,0 ммоль) добавляли диметилсульфоксид (15 мл). Полученный в результате раствор продували аргоном в течение 10 минут и затем к нему добавляли 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий (57 мг, 0,07 ммоль). Реакционный раствор снова дегазировали, используя аргон, в течение 5 минут и нагревали при 80°C в течение 1 часа. Затем реакционный раствор охлаждали до комнатной температуры, разбавляли этилацетатом, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от дихлорметана до смеси 95:5 дихлорметан/метанол), получая требуемый продукт (грубый выход 1,3 г) в виде красноватого масла, которое использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,72 (д, J=7,9 Гц, 1H), 7,67 (д, J=7,9 Гц, 1H), 7,56-7,54 (м, 2H), 7,29 (тд, J=7,3, 1,1 Гц, 1H), 7,23 (тд, J=8,2, 2,6 Гц, 1H), 7,16 (д, J=7,5 Гц, 1H), 7,13 (с, 1H), 4,57 (т, J=5,3 Гц, 1H), 3,77 (д, J=14,9 Гц, 1H), 3,63 (д, J=14,9 Гц, 1H), 3,00 (дд, J=11,3, 4,8 Гц, 1H), 2,89 (дд, J=11,4, 6,1 Гц, 1H), 2,48 (с, 3H), 1,33 (с, 12H); ESI-МС m/z 406 [M+H]+.
Стадия H: К смеси сложного эфира бороновой кислоты (0,12 г, 0,3 ммоль) с описанной выше стадии G, 4-йод-3,5-диметилизоксазола (0,13 г, 0,59 ммоль) и карбоната натрия (95 мг, 0,90 ммоль) добавляли N,N-диметилформамид (3 мл) и воду (0,75 мл). Реакционный раствор продували аргоном в течение 10 минут и затем к раствору добавляли 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий (12 мг, 0,015 ммоль). Реакционный раствор снова дегазировали, используя аргон, в течение 5 минут и нагревали при 80°C в течение 3 часов. Затем реакционный раствор охлаждали до комнатной температуры, разбавляли этилацетатом, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали препаративной тонкослойной хроматографией (95:5 дихлорметан/метанол), получая требуемый продукт (28 мг, 25%):1H ЯМР (CDCl3, 500 МГц) δ 7,74 (д, J=8,0 Гц, 1Н), 7,70 (д, J=7,9 Гц, 1H), 7,32 (тд, J=7,3, 1,1 Гц, 1H), 7,28-7,21 (м, 3H), 7,00 (дд, J=7,9, 1,6 Гц, 1H), 6,97 (с, 1H), 4,59 (кажущийся с, 1H), 3,80 (д, J=15,0 Гц, 1H), 3,67 (д, J=15,2 Гц, 1Н), 3,06-3,04 (м, 1H), 2,94 (дд, J=11,3, 5,9 Гц, 1H), 2,52 (с, 3H), 2,38 (с, 3H), 2,25 (с, 3H); ESI-МС m/z 375 [M+H]+. К раствору 7-изоксазолтетрагидроизохинолина (28 мг, 0,075 ммоль) в метаноле (2 мл) добавляли малеиновую кислоту (8,7 мг, 0,075 ммоль) с последующим медленным добавлением воды (10 мл). Полученный в результате реакционный раствор лиофилизировали в течение ночи, получая малеатную соль (+/-)-4-(бензо[b]тиофен-2-ил)-2-метил-7-(3,5-диметилизоксазол-4-ил)-1,2,3,4-тетрагидроизохинолина (35 мг) в виде светло-желтого твердого вещества: т.пл. 92-95°C;1H ЯМР (CD3OD, 500 МГц) δ 7,83 (д, J=7,8 Гц, 1H), 7,78 (д, J=7,3 Гц, 1H), 7,39-7,29 (м, 6H), 6,25 (с, 2H), 5,05 (дд, J=9,9, 5,6 Гц, 1H), 4,59 (кажущийся с, 2H), 3,97 (дд, J=12,2, 5,6 Гц, 1H), 3,77-3,72 (м, 1H), 3,09 (с, 3H), 2,42 (с, 3H), 2,26 (с, 3H); ESI-МС m/z 375 [M+H]+.
Пример 75 - Получение малеатной соли (+)-4-(бензо[b]тиофен-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина и малеатной соли (-)-4-(бензо[b]тиофен-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору 2-ацетилбензо[b]тиофена (5,0 г, 28 ммоль) в хлороформе (120 мл) добавляли пербромид бромида пиридиния (9,6 г, 28 ммоль) двумя порциями. Реакционный раствор перемешивали при комнатной температуре в течение 2 часов и затем промывали водой, водным раствором HCl (2 М), водой и насыщенным раствором соли. Полученный в результате раствор сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый бромкетон (грубый выход 7,4 г) в виде коричневого твердого вещества, которое использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 8,07 (с, 1H), 7,94-7,87 (м, 2H), 7,52-7,25 (м, 2H), 4,46 (с, 2H).
Стадия B: К раствору 3-метоксибензилметиламина (4,3 г, 29 ммоль) в дихлорметане (150 мл) добавляли триэтиламин (10 мл) и бромкетон (7,3 г, 29 ммоль) со стадии A, описанной выше. Реакционный раствор перемешивали при комнатной температуре в течение 2 часов и затем гасили насыщенным водным раствором бикарбоната натрия. Органический экстракт отделяли, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от 90:10 до 85:15 гексаны/этилацетат), получая требуемый продукт (7,1 г, 76% для 2 стадий) в виде красноватого масла:1H ЯМР (CDCl3, 500 МГц) δ 8,09 (с, 1H), 7,86 (д, J=8,0 Гц, 2H), 7,45 (дд, J=8,0, 1,1 Гц, 1H), 7,40 (дд, J=8,0, 1,0 Гц, 1H), 7,24 (д, J=8,1 Гц, 1H), 6,97-6,95 (м, 2H), 6,83 (д, J=8,5 Гц, 1Н), 3,78 (с, 3H), 3,76 (с, 2H), 3,70 (с, 2H), 2,43 (с, 3H); ESI-МС m/z 326 [M+H]+.
Стадия C: К раствору кетона (7,1 г, 22 ммоль) со стадии B, описанной выше, в метаноле (100 мл) при 0°C небольшими порциями добавляли боргидрид натрия (0,89 г, 24 ммоль). Реакционный раствор перемешивали при 0°C в течение 1 часа. Затем растворитель удаляли при пониженном давлении и полученный остаток растворяли в дихлорметане. Полученный в результате раствор промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый спирт (6,9 г, грубый выход 96%) в виде желтого масла. Неочищенный продукт использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,81 (д, J=7,9 Гц, 1H), 7,70 (д, J=8,1 Гц, 1H), 7,33-7,23 (м, 3H), 7,21 (с, 1H), 6,91-6,87 (м, 3H), 5,07 (дд, J=10,1, 3,5 Гц, 1H), 4,40-4,03 (м, 1H), 3,81 (с, 3H), 3,72 (д, J=13,0 Гц, 1H), 3,56 (д, J=13,0 Гц, 1H), 2,83 (дд, J=12,4, 10,2 Гц, 1H), 2,72 (дд, J=12,4, 3,7 Гц, 1H), 2,34 (с, 3H); ESI-МС m/z 328 [M+H]+.
Стадия D: К раствору спирта (6,9 г, 21 ммоль) со стадии C, описанной выше, в дихлорметане (150 мл) по каплям добавляли метансульфоновую кислоту (14 мл, 210 ммоль). После добавления реакционный раствор перемешивали при комнатной температуре в течение 20 минут и затем медленно добавляли к ледяному водному раствору гидроксида натрия (120 мл, 2 М) и перемешивали в течение 20 минут. Органический слой отделяли и водный слой дважды промывали дихлорметаном. Объединенный органический экстракт промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от 95:5 до 50:50 гексаны/этилацетат), получая требуемый циклизованный продукт (3,4 г, 52%) в виде красноватого масла:1H ЯМР (CDCl3, 500 МГц) δ 7,72 (д, J=8,0 Гц, 1H), 7,67 (д, J=8,0 Гц, 1H), 7,29-7,23 (м, 2H), 7,13 (с, 1H), 7,06 (д, J=8,5 Гц, 1H), 6,69 (д, J=2,7 Гц, 1H), 6,61 (д, J=2,6 Гц, 1H), 4,50 (т, J=5,3 Гц, 1Н), 3,77 (с, 3H), 3,73 (д, J=14,5 Гц, 1H), 3,59 (д, J=14,5 Гц, 1Н), 2,99 (дд, J=11,5, 4,9 Гц, 1Н), 2,87 (дд, J=11,4, 6,2 Гц, 1H), 2,47 (с, 3H); ESI-МС m/z 310 [M+H]+.
Стадия E: К раствору 7-метокситетрагидроизохинолина (1,6 г, 5,1 ммоль) со стадии D, описанной выше, в уксусной кислоте (25 мл) добавляли водный раствор бромистого водорода (25 мл, 48%). Реакционный раствор нагревали при 100°C в течение 20 часов и затем охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток помещали в смесь дихлорметана, воды и небольшого количества метанола. Полученный в результате раствор осторожно гасили насыщенным водным раствором бикарбоната натрия до pH >8. Органический экстракт отделяли, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый фенол (грубый выход 1,6 г) в виде коричневого твердого вещества, которое использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,72 (д, J=7,7 Гц, 1H), 7,67 (д, J=7,7 Гц, 1H), 7,29 (тд, J=7,3, 1,2 Гц, 1H), 7,23 (тд, J=7,3, 1,3 Гц, 1H), 7,13 (с, 1H), 7,01 (д, J=8,4 Гц, 1H), 6,59 (дд, J=8,4, 2,5 Гц, 1H), 6,53 (д, J=2,3 Гц, 1H), 4,49 (т, J=5,7 Гц, 1Н), 3,68 (д, J=15,1 Гц, 1H), 3,57 (д, J=15,0 Гц, 1H), 3,00 (дд, J=11,6, 5,4 Гц, 1H), 2,85 (дд, J=11,5, 6,4 Гц, 1H), 2,47 (с, 3H), 1,90-1,45 (м, 1H); ESI-МС m/z 296 [M+H]+.
Стадия F: К раствору полученного фенола (1,6 г, 5,1 ммоль) со стадии E, описанной выше, в дихлорметане (50 мл) при 0°C последовательно по каплям добавляли пиридин (0,80 мл, 10 ммоль) и ангидрид трифторметансульфоновой кислоты (1,1 мл, 6,6 ммоль). Полученный в результате реакционный раствор перемешивали при 0°C в течение 1 часа и затем гасили насыщенным водным раствором бикарбоната натрия. Органический экстракт отделяли и водный слой дважды экстрагировали дихлорметаном. Объединенный органический экстракт промывали смесью 1:1 вода/насыщенный раствор соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая сырой продукт. Очистка флэш-хроматографией на колонке (от дихлорметана до смеси 98:2 дихлорметан/метанол) давала требуемый трифторметансульфонат (1,6 г, 73% для двух стадий) в виде желтого масла:1H ЯМР (CDCl3, 500 МГц) δ 7,74 (д, J=8,0 Гц, 1H), 7,70 (д, J=7,8 Гц, 1H), 7,32-7,24 (м, 3H), 7,16 (с, 1H), 7,02-7,00 (м, 2H), 4,54 (т, J=5,4 Гц, 1H), 3,78 (д, J=15,4 Гц, 1H), 3,65 (д, J=15,4 Гц, 1H), 3,01 (дд, J=11,6, 5,0 Гц, 1H), 2,91 (дд, J=11,6, 6,0 Гц, 1H), 2,50 (с, 3H); ESI-МС m/z 428 [M+H]+.
Стадия G: К раствору трифторметансульфоната (0,10 г, 0,23 ммоль) со стадии F, описанной выше, в толуоле (2,5 мл) добавляли карбонат цезия (67 мг, 0,14 ммоль), 2-(дициклогексилфосфино)-2',4',6'-триизопропил-1,1'-бифенил (0,19 г, 0,58 ммоль) и морфолин (41 мкл, 0,47 ммоль). Реакционную смесь продували аргоном в течение 5 минут и затем добавляли ацетат палладия (8 мг, 0,04 ммоль). Реакционную колбу закрывали и нагревали в микроволновой печи (160°C) в течение 2 часов. Затем реакционный раствор охлаждали до комнатной температуры, фильтровали через слой целита и концентрировали при пониженном давлении. Полученный сырой продукт очищали, используя флэш-хроматографию на колонке (от дихлорметана до смеси 95:5 дихлорметан/MeOH), получая требуемый продукт (57 мг, 68%) в виде желтой пены:1H ЯМР (CDCl3, 500 МГц) δ 7,72 (д, J=7,9 Гц, 1H), 7,67 (д, J=7,9 Гц, 1H), 7,31-7,22 (м, 2H), 7,13 (с, 1H), 7,05 (д, J=8,6 Гц, 1H), 6,71 (дд, J=8,6, 2,7 Гц, 1H), 6,60 (д, J=2,5 Гц, 1H), 4,49 (т, J=5,4 Гц, 1Н), 3,85-3,83 (м, 4H), 3,72 (д, J=14,9 Гц, 1H), 3,58 (д, J=15,0 Гц, 1H), 3,13-3,11 (м, 4H), 2,99 (дд, J=11,3, 5,0 Гц, 1H), 2,86 (дд, J=11,4, 6,2 Гц, 1H), 2,47 (с, 3H).
Стадия H: К раствору 7-морфолинилтетрагидроизохинолина (56 мг, 0,15 ммоль) со стадии G, описанной выше, в метаноле (5 мл) добавляли малеиновую кислоту (18 мг, 0,15 ммоль). Полученный в результате раствор концентрировали при пониженном давлении до ~2 мл и разбавляли водой (5 мл). Полученный в результате раствор лиофилизировали в течение ночи, получая соответствующую малеатную соль (73 мг, 99%) в виде светло-желтого твердого вещества: т.пл. 110-113°C;1H ЯМР (CD3OD, 500 МГц) δ 7,80 (дд, J=7,8, 0,6 Гц, 1H), 7,75 (д, J=7,4 Гц, 1H), 7,38-7,30 (м, 3H), 7,10 (д, J=8,7 Гц, 1H), 6,95 (дд, J=8,7, 2,4 Гц, 1Н), 6,80 (д, J=2,5 Гц, 1H), 6,25 (с, 2H), 4,91 (дд, J=9,7, 6,1 Гц, 1H), 4,50 (д, J=15,2 Гц, 1H), 4,45 (д, J=15,4 Гц, 1H), 3,91 (дд, J=12,2, 5,6 Гц, 1H), 3,83-3,81 (м, 4H), 3,71-3,60 (м, 1Н), 3,17-3,15 (м, 4H), 3,06 (с, 3H); ESI-МС m/z 365 [M+H]+.
Стадия I: Свободное основание продукта со стадии H (0,27 г) разделяли препаративной хиральной ВЭЖХ (колонка CHIRALPAK AD, используя смесь 80:20:0,1 гептан/изопропанол/диэтиламин в качестве элюента), получая (+)-энантиомер [[α]25D +44,8° (c 0,11, метанол)] и (-)-энантиомер [[α]25D -45,5° (c 0,13, метанол)]. К раствору (+)-энантиомера (0,13 г, 0,36 ммоль) в метаноле (5 мл) добавляли малеиновую кислоту (41 мг, 0,35 ммоль) с последующим медленным добавлением воды (25 мл). Полученный в результате раствор лиофилизировали в течение ночи, получая малеатную соль (+)-4-(бензо[b]тиофен-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина (0,17 г, 97%, AUC ВЭЖХ >99%) в виде не совсем белого твердого вещества: т.пл. 142-146°C;1H ЯМР (CD3OD, 500 МГц) δ 7,80 (дд, J=7,8, 0,6 Гц, 1H), 7,75 (д, J=7,5 Гц, 1H), 7,74-7,29 (м, 3H), 7,10 (д, J=8,7 Гц, 1H), 6,95 (дд, J=8,7, 2,2 Гц, 1H), 6,80 (д, J=2,3 Гц, 1H), 6,25 (с, 2H), 4,92 (дд, J=9,6, 5,9 Гц, 1H), 4,50 (д, J=15,2 Гц, 1H), 4,45 (д, J=15,3 Гц, 1H), 3,91 (дд, J=12,4, 5,9 Гц, 1H), 3,83-3,80 (м, 4H), 3,70-3,59 (м, 1H), 3,18-3,15 (м, 4H), 3,06 (с, 3H); ESI-МС m/z 365 [M+H]+; элементный анализ, вычислено для C22H24N2OS-1,125C4H4O4-0,75H2O: C, 62,56; Н, 5,95; N, 5,51. Найдено: C, 62,45; Н 5,95; N, 5,14. К раствору (-)-энантиомера (0,13 г, 0,36 ммоль) в метаноле (5 мл) добавляли малеиновую кислоту (40 мг, 0,35 ммоль) с последующим медленным добавлением воды (25 мл). Полученный в результате раствор лиофилизировали в течение ночи, получая малеатную соль (-)-4-(бензо[b]тиофен-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина (0,16 г, 98%, AUC ВЭЖХ >99%) в виде не совсем белого твердого вещества: т.пл. 142-145°C;1H ЯМР (CD3OD, 500 МГц) δ 7,80 (дд, J=7,8, 0,6 Гц, 1H), 7,75 (д, J=7,5 Гц, 1H), 7,74-7,29 (м, 3H), 7,10 (д, J=8,7 Гц, 1H), 6,95 (дд, J=8,7, 2,2 Гц, 1H), 6,80 (д, J=2,3 Гц, 1H), 6,25 (с, 2H), 4,92 (дд, J=9,6, 5,9 Гц, 1H), 4,50 (д, J=15,2 Гц, 1H), 4,45 (д, J=15,3 Гц, 1H), 3,91 (дд, J=12,4, 5,9 Гц, 1H), 3,83-3,80 (м, 4H), 3,70-3,59 (м, 1H), 3,18-3,15 (м, 4H), 3,06 (с, 3H); ESI-МС m/z 365 [M+Н]+. Элементный анализ, вычислено для C22H24N2OS-C4H4O4-H2O: C, 62,63; Н, 6,06; N, 5,62. Найдено: C, 62,45; Н 5,89; N, 5,32.
Пример 76 - Получение малеатной соли (+)-4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолина и малеатной соли (-)-4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору 2-ацетилбензо[b]тиофена (5,0 г, 28 ммоль) в хлороформе (120 мл) добавляли пербромид бромида пиридиния (9,6 г, 28 ммоль) двумя порциями. Реакционный раствор перемешивали при комнатной температуре в течение 2 часов и затем промывали водой, водным раствором HCl (2 М), водой и насыщенным раствором соли. Полученный в результате раствор сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый бромкетон (грубый выход 7,4 г) в виде коричневого твердого вещества, которое использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 8,07 (с, 1H), 7,94-7,87 (м, 2H), 7,52-7,25 (м, 2H), 4,46 (с, 2H).
Стадия B: К раствору 3-метоксибензилметиламина (4,3 г, 29 ммоль) в дихлорметане (150 мл) добавляли триэтиламин (10 мл) и бромкетон (7,3 г, 29 ммоль) со стадии A, описанной выше. Реакционный раствор перемешивали при комнатной температуре в течение 2 часов и затем гасили насыщенным водным раствором бикарбоната натрия. Органический экстракт отделяли, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от 90:10 до 85:15 гексаны/этилацетат), получая требуемый продукт (7,1 г, 76% для 2 стадий) в виде красноватого масла:1H ЯМР (CDCl3, 500 МГц) δ 8,09 (с, 1H), 7,86 (д, J=8,0 Гц, 2H), 7,45 (дд, J=8,0, 1,1 Гц, 1H), 7,40 (дд, J=8,0, 1,0 Гц, 1H), 7,24 (д, J=8,1 Гц, 1H), 6,97-6,95 (м, 2H), 6,83 (д, J=8,5 Гц, 1H), 3,78 (с, 3H), 3,76 (с, 2H), 3,70 (с, 2H), 2,43 (с, 3H); ESI-МС m/z 326 [M+Н]+.
Стадия C: К раствору кетона (7,1 г, 22 ммоль) со стадии B, описанной выше, в метаноле (100 мл) при 0°C небольшими порциями добавляли боргидрид натрия (0,89 г, 24 ммоль). Реакционный раствор перемешивали при 0°C в течение 1 часа. Затем растворитель удаляли при пониженном давлении и полученный остаток растворяли в дихлорметане. Полученный в результате раствор промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый спирт (6,9 г, 96% грубый выход) в виде желтого масла. Неочищенный продукт использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,81 (д, J=7,9 Гц, 1H), 7,70 (д, J=8,1 Гц, 1H), 7,33-7,23 (м, 3H), 7,21 (с, 1H), 6,91-6,87 (м, 3H), 5,07 (дд, J=10,1, 3,5 Гц, 1H), 4,40-4,03 (м, 1Н), 3,81 (с, 3H), 3,72 (д, J=13,0 Гц, 1H), 3,56 (д, J=13,0 Гц, 1H), 2,83 (дд, J=12,4, 10,2 Гц, 1Н), 2,72 (дд, J=12,4, 3,7 Гц, 1H), 2,34 (с, 3H); ESI-МС m/z 328 [M+H]+.
Стадия D: К раствору спирта (6,9 г, 21 ммоль) со стадии C, описанной выше, в дихлорметане (150 мл) по каплям добавляли метансульфоновую кислоту (14 мл, 210 ммоль). После добавления реакционный раствор перемешивали при комнатной температуре в течение 20 минут и затем медленно добавляли к ледяному водному раствору гидроксида натрия (2 М) и перемешивали в течение 20 минут. Органический слой отделяли и водный слой дважды промывали дихлорметаном. Объединенный органический экстракт промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от 95:5 до 50:50 гексаны/этилацетат), получая требуемый циклизованный продукт (3,4 г, 52%) в виде красноватого масла:1H ЯМР (CDCl3, 500 МГц) δ 7,72 (д, J=8,0 Гц, 1H), 7,67 (д, J=8,0 Гц, 1Н), 7,29-7,23 (м, 2H), 7,13 (с, 1H), 7,06 (д, J=8,5 Гц, 1H), 6,69 (д, J=2,7 Гц, 1H), 6,61 (д, J=2,6 Гц, 1H), 4,50 (т, J=5,3 Гц, 1H), 3,77 (с, 3H), 3,73 (д, J=14,5 Гц, 1H), 3,59 (д, J=14,5 Гц, 1H), 2,99 (дд, J=11,5, 4,9 Гц, 1H), 2,87 (дд, J=11,4, 6,2 Гц, 1H), 2,47 (с, 3H); ESI-МС m/z 310 [M+H]+.
Стадия E: К раствору 7-метокситетрагидроизохинолина (1,6 г, 5,1 ммоль) со стадии D, описанной выше, в уксусной кислоте (25 мл) добавляли водный раствор бромистого водорода (25 мл, 48%). Реакционный раствор нагревали при 100°C в течение 20 часов и затем охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток помещали в смесь дихлорметана, воды и небольшого количества метанола. Полученный в результате раствор осторожно гасили насыщенным водным раствором бикарбоната натрия до pH >8. Органический экстракт отделяли, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый фенол (грубый выход 1,6 г) в виде коричневого твердого вещества, которое использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,72 (д, J=7,7 Гц, 1H), 7,67 (д, J=7,7 Гц, 1H), 7,29 (тд, J=7,3, 1,2 Гц, 1H), 7,23 (тд, J=7,3, 1,3 Гц, 1H), 7,13 (с, 1H), 7,01 (д, J=8,4 Гц, 1H), 6,59 (дд, J=8,4, 2,5 Гц, 1H), 6,53 (д, J=2,3 Гц, 1H), 4,49 (т, J=5,7 Гц, 1H), 3,68 (д, J=15,1 Гц, 1H), 3,57 (д, J=15,0 Гц, 1Н), 3,00 (дд, J=11,6, 5,4 Гц, 1H), 2,85 (дд, J=11,5, 6,4 Гц, 1H), 2,47 (с, 3H), 1,90-1,45 (м, 1H); ESI-МС m/z 296 [M+H]+.
Стадия F: К раствору полученного фенола (1,6 г, 5,1 ммоль) со стадии E, описанной выше, в дихлорметане (50 мл) при 0°C последовательно по каплям добавляли пиридин (0,80 мл, 10 ммоль) и ангидрид трифторметансульфоновой кислоты (1,1 мл, 6,6 ммоль). Полученный в результате реакционный раствор перемешивали при 0°C в течение 1 часа и затем гасили насыщенным водным раствором бикарбоната натрия. Органический экстракт отделяли и водный слой дважды экстрагировали дихлорметаном. Объединенный органический экстракт промывали смесью 1:1 вода/насыщенный раствор соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая сырой продукт. Очистка флэш-хроматографией на колонке (от дихлорметана до смеси 98:2 дихлорметан/метанол) давала требуемый трифторметансульфонат (1,6 г, 73% для двух стадий) в виде желтого масла:1H ЯМР (CDCl3, 500 МГц) δ 7,74 (д, J=8,0 Гц, 1H), 7,70 (д, J=7,8 Гц, 1H), 7,32-7,24 (м, 3H), 7,16 (с, 1H), 7,02-7,00 (м, 2H), 4,54 (т, J=5,4 Гц, 1H), 3,78 (д, J=15,4 Гц, 1H), 3,65 (д, J=15,4 Гц, 1H), 3,01 (дд, J=11,6, 5,0 Гц, 1H), 2,91 (дд, J=11,6, 6,0 Гц, 1H), 2,50 (с, 3H); ESI-МС m/z 428 [M+H]+.
Стадия G: К раствору трифторметансульфоната (0,60 г, 1,4 ммоль) со стадии F, описанной выше, в толуоле (18 мл) добавляли карбонат цезия (1,15 г, 3,5 ммоль) и 2-(дициклогексилфосфино)-2',4',6'-триизопропил-1,1'-бифенил (0,40 г, 0,85 ммоль). Реакционную смесь продували аргоном в течение 10 минут и затем к ней добавляли ацетат палладия (II) (47 мг, 0,21 ммоль). Полученный в результате раствор продували аргоном в течение 5 минут и затем через шприц добавляли пиперидин (0,29 мл, 2,8 ммоль). Реакционную колбу закрывали и нагревали при 100°C в течение 20 часов и затем охлаждали до комнатной температуры, фильтровали через слой целита и концентрировали при пониженном давлении. Полученный сырой продукт очищали флэш-хроматографией на колонке (от дихлорметана до смеси 96:4 дихлорметан/метанол), получая требуемый продукт (0,38 г, 74%) в виде желтой пены:1H ЯМР (CD3OD, 500 МГц) δ 7,73 (д, J=7,8 Гц, 1H), 7,69 (д, J=7,5 Гц, 1H), 7,31-7,23 (м, 2H), 7,19 (с, 1H), 6,97 (д, J=8,5 Гц, 1H), 6,79 (т, J=6,5 Гц, 1H), 6,72 (д, J=1,5 Гц, 1H), 4,59 (ушир.с, 1H), 3,72 (д, J=15,0 Гц, 1H), 3,60 (д, J=15,1 Гц, 1H), 3,15-3,10 (м, 5H), 2,76 (дд, J=11,2, 8,7 Гц, 1H), 2,45 (с, 3H), 1,72-1,57 (м, 6H); ESI-МС m/z 363 [M+H]+.
Стадия H: 7-пиперидинилтетрагидроизохинолин (0,42 г, 1,2 ммоль) со стадии G, описанной выше, разделяли препаративной хиральной ВЭЖХ (колонка CHIRALCEL OJ, используя смесь 80:20:0,1 гептан/этанол/диэтиламин в качестве элюента), получая (+)-энантиомер [[α]25D +52,7° (c 0,15, метанол)] и (-)-энантиомер [[α]25D -55,6° (c 0,14, метанол)]. (+)-Энантиомер (0,20 г, 0,55 ммоль) растворяли в метаноле (5 мл) и к нему добавляли малеиновую кислоту (64 мг, 0,55 ммоль). К полученному раствору добавляли воду (25 мл). Полученный в результате раствор лиофилизировали в течение ночи, получая малеатную соль (+)-4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолина (0,26 г, 98%, >99% AUC ВЭЖХ) в виде не совсем белого твердого вещества: т.пл. 114-116°C;1H ЯМР (CD3OD, 500 МГц) δ 7,80 (дд, J=8,3, 1,0 Гц, 1H), 7,75 (д, J=7,3 Гц, 1H), 7,37-7,28 (м, 3H), 7,07 (д, J=8,7 Гц, 1H), 6,94 (дд, J=8,7, 2,5 Гц, 1Н), 6,79 (д, J=2,5 Гц, 1H), 6,24 (с, 2H), 4,90 (дд, J=9,6, 6,0 Гц, 1H), 4,48 (д, J=15,3 Гц, 1H), 4,43 (д, J=15,3 Гц, 1H), 3,89 (дд, J=12,4, 6,0 Гц, 1H), 3,63 (т, J=10,4 Гц, 1H), 3,20-3,18 (м, 4H), 3,05 (с, 3H), 1,72-1,58 (м, 6H); ESI-МС m/z 363 [M+H]+; элементный анализ, вычислено для C23H25N2S-C4H4O4-1,5H2O: C, 64,14; Н, 6,58; N, 5,54. Найдено: C, 63,94; Н 6,20; N, 5,40.
(-)-Энантиомер (0,20 г, 0,55 ммоль) растворяли в метаноле (5 мл) и добавляли малеиновую кислоту (63 мг, 0,55 ммоль). К полученному раствору добавляли воду (25 мл) и полученный в результате раствор лиофилизировали в течение ночи, получая малеатную соль (-)-4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолина (0,25 г, 98%, >99% AUC ВЭЖХ) в виде не совсем белого твердого вещества: т.пл. 113-115°C;1H ЯМР (CD3OD, 500 МГц) δ 7,80 (дд, J=8,3, 1,0 Гц, 1H), 7,75 (д, J=7,3 Гц, 1H), 7,37-7,28 (м, 3H), 7,07 (д, J=8,7 Гц, 1Н), 6,94 (дд, J=8,7, 2,5 Гц, 1H), 6,79 (д, J=2,5 Гц, 1H), 6,24 (с, 2H), 4,90 (дд, J=9,6, 6,0 Гц, 1H), 4,48 (д, J=15,3 Гц, 1H), 4,43 (д, J=15,3 Гц, 1H), 3,89 (дд, J=12,4, 6,0 Гц, 1H), 3,63 (т, J=10,4 Гц, 1H), 3,20-3,18 (м, 4H), 3,05 (с, 3H), 1,72-1,58 (м, 6H); ESI-МС m/z 363 [M+H]+; элементный анализ, вычислено для C23H25N2S-C4H4O4-0,75Н2О: С, 65,90; Н, 6,45; N, 5,69. Найдено: C, 65,74; Н 6,27; N, 5,51.
Пример 77 - Получение малеатной соли (+/-)-4-(бензо[b]тиофен-2-ил)-2-метил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору 2-ацетилбензо[b]тиофена (5,0 г, 28 ммоль) в хлороформе (120 мл) добавляли пербромид бромида пиридиния (9,6 г, 28 ммоль) двумя порциями. Реакционный раствор перемешивали при комнатной температуре в течение 2 часов и затем промывали водой, водным раствором HCl (2 М), водой и насыщенным раствором соли. Полученный в результате раствор сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый бромкетон (грубый выход 7,4 г) в виде коричневого твердого вещества, которое использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 8,07 (с, 1H), 7,94-7,87 (м, 2H), 7,52-7,25 (м, 2H), 4,46 (с, 2H).
Стадия B: К раствору 3-метоксибензилметиламина (4,3 г, 29 ммоль) в дихлорметане (150 мл) добавляли триэтиламин (10 мл) и бромкетон (7,3 г, 29 ммоль) со стадии A, описанной выше. Реакционный раствор перемешивали при комнатной температуре в течение 2 часов и затем гасили насыщенным водным раствором бикарбоната натрия. Органический экстракт отделяли, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от 90:10 до 85:15 гексаны/этилацетат), получая требуемый продукт (7,1 г, 76% для 2 стадий) в виде красноватого масла:1H ЯМР (CDCl3, 500 МГц) δ 8,09 (с, 1H), 7,86 (д, J=8,0 Гц, 2H), 7,45 (дд, J=8,0, 1,1 Гц, 1H), 7,40 (дд, J=8,0, 1,0 Гц, 1H), 7,24 (д, J=8,1 Гц, 1H), 6,97-6,95 (м, 2H), 6,83 (д, J=8,5 Гц, 1H), 3,78 (с, 3H), 3,76 (с, 2H), 3,70 (с, 2H), 2,43 (с, 3H); ESI-МС m/z 326 [M+H]+.
Стадия C: К раствору кетона (7,1 г, 22 ммоль) со стадии B, описанной выше, в метаноле (100 мл) при 0°C небольшими порциями добавляли боргидрид натрия (0,89 г, 24 ммоль). Реакционный раствор перемешивали при 0°C в течение 1 часа. Затем растворитель удаляли при пониженном давлении и полученный остаток растворяли в дихлорметане. Полученный в результате раствор промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый спирт (6,9 г, грубый выход 96%) в виде желтого масла. Неочищенный продукт использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,81 (д, J=7,9 Гц, 1H), 7,70 (д, J=8,1 Гц, 1H), 7,33-7,23 (м, 3H), 7,21 (с, 1H), 6,91-6,87 (м, 3H), 5,07 (дд, J=10,1, 3,5 Гц, 1H), 4,40-4,03 (м, 1H), 3,81 (с, 3H), 3,72 (д, J=13,0 Гц, 1H), 3,56 (д, J=13,0 Гц, 1H), 2,83 (дд, J=12,4, 10,2 Гц, 1H), 2,72 (дд, J=12,4, 3,7 Гц, 1H), 2,34 (с, 3H); ESI-МС m/z 328 [M+H]+.
Стадия D: К раствору спирта (6,9 г, 21 ммоль) со стадии C, описанной выше, в дихлорметане (150 мл) по каплям добавляли метансульфоновую кислоту (14 мл, 210 ммоль). После добавления реакционный раствор перемешивали при комнатной температуре в течение 20 минут и затем медленно добавляли к ледяному водному раствору гидроксида натрия (120 мл, 2 М) и перемешивали в течение 20 минут. Органический слой отделяли и водный слой дважды экстрагировали дихлорметаном. Объединенный органический экстракт промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от 95:5 до 50:50 гексаны/этилацетат), получая требуемый продукт (3,4 г, 52%) в виде красноватого масла:1H ЯМР (CDCl3, 500 МГц) δ 7,72 (д, J=8,0 Гц, 1H), 7,67 (д, J=8,0 Гц, 1H), 7,29-7,23 (м, 2H), 7,13 (с, 1H), 7,06 (д, J=8,5 Гц, 1H), 6,69 (д, J=2,7 Гц, 1H), 6,61 (д, J=2,6 Гц, 1H), 4,50 (т, J=5,3 Гц, 1H), 3,77 (с, 3H), 3,73 (д, J=14,5 Гц, 1H), 3,59 (д, J=14,5 Гц, 1H), 2,99 (дд, J=11,5, 4,9 Гц, 1H), 2,87 (дд, J=11,4, 6,2 Гц, 1H), 2,47 (с, 3H); ESI-МС m/z 310 [M+H]+.
Стадия E: К раствору 7-метокситетрагидроизохинолина (1,6 г, 5,1 ммоль) со стадии D, описанной выше, в уксусной кислоте (25 мл) добавляли водный раствор бромистого водорода (25 мл, 48%). Реакционный раствор нагревали при 100°C в течение 20 часов и затем охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток помещали в смесь дихлорметана, воды и небольшого количества метанола. Полученный в результате раствор осторожно гасили насыщенным водным раствором бикарбоната натрия до pH >8. Органический экстракт отделяли, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый фенол (грубый выход 1,6 г) в виде коричневого твердого вещества, которое использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,72 (д, J=7,7 Гц, 1H), 7,67 (д, J=7,7 Гц, 1Н), 7,29 (тд, J=7,3, 1,2 Гц, 1H), 7,23 (тд, J=7,3, 1,3 Гц, 1H), 7,13 (с, 1H), 7,01 (д, J=8,4 Гц, 1H), 6,59 (дд, J=8,4, 2,5 Гц, 1H), 6,53 (д, J=2,3 Гц, 1H), 4,49 (т, J=5,7 Гц, 1H), 3,68 (д, J=15,1 Гц, 1H), 3,57 (д, J=15,0 Гц, 1Н), 3,00 (дд, J=11,6, 5,4 Гц, 1H), 2,85 (дд, J=11,5, 6,4 Гц, 1H), 2,47 (с, 3H), 1,90-1,45 (м, 1H); ESI-МС m/z 296 [M+H]+.
Стадия F: К раствору полученного фенола (1,6 г, 5,1 ммоль) со стадии E, описанной выше, в дихлорметане (50 мл) при 0°C последовательно по каплям добавляли пиридин (0,80 мл, 10 ммоль) и ангидрид трифторметансульфоновой кислоты (1,1 мл, 6,6 ммоль). Полученный в результате реакционный раствор перемешивали при 0°C в течение 1 часа и затем гасили насыщенным водным раствором бикарбоната натрия. Органический экстракт отделяли и водный слой дважды экстрагировали дихлорметаном. Объединенный органический экстракт промывали смесью 1:1 вода/насыщенный раствор соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая сырой продукт. Очистка флэш-хроматографией на колонке (от дихлорметана до смеси 98:2 дихлорметан/метанол) давала требуемый трифторметансульфонат (1,6 г, 73% для двух стадий) в виде желтого масла:1H ЯМР (CDCl3, 500 МГц) δ 7,74 (д, J=8,0 Гц, 1H), 7,70 (д, J=7,8 Гц, 1H), 7,32-7,24 (м, 3H), 7,16 (с, 1H), 7,02-7,00 (м, 2H), 4,54 (т, J=5,4 Гц, 1Н), 3,78 (д, J=15,4 Гц, 1H), 3,65 (д, J=15,4 Гц, 1H), 3,01 (дд, J=11,6, 5,0 Гц, 1H), 2,91 (дд, J=11,6, 6,0 Гц, 1Н), 2,50 (с, 3H); ESI-МС m/z 428 [M+H]+.
Стадия G: К раствору трифторметансульфоната (0,11 г, 0,26 ммоль) со стадии F, описанной выше, в толуоле (3 мл) добавляли карбонат цезия (0,21 г, 0,66 ммоль) и 2-(дициклогексилфосфино)-2',4',6'-триизопропил-1,1'-бифенил (37 мг, 0,08 ммоль) и реакционную смесь продували аргоном в течение 10 минут. К полученному раствору добавляли ацетат палладия (II) (4,7 мг, 0,02 ммоль). Реакционный раствор снова продували аргоном в течение 5 минут и затем через шприц добавляли пирролидин (44 мкл, 0,52 ммоль). Реакционную колбу закрывали и нагревали при 100°C в течение 20 часов. Затем реакционный раствор охлаждали до комнатной температуры, фильтровали через слой целита и концентрировали при пониженном давлении. Полученный сырой продукт очищали флэш-хроматографией на колонке (99:1 дихлорметан/метанол) с последующей препаративной тонкослойной хроматографией (66:34 гексаны/этилацетат), получая требуемый продукт (60 мг, 66%) в виде белой пены:1H ЯМР (CD3OD, 500 МГц) δ 7,71 (д, J=8,1 Гц, 1H), 7,65 (д, J=7,8 Гц, 1H), 7,28-7,25 (м, 1H), 7,22 (дд, J=8,1, 1,2 Гц, 1H), 7,13 (с, 1H), 6,98 (д, J=8,5 Гц, 1H), 6,38 (дд, J=8,0, 2,6 Гц, 1H), 6,25 (д, J=2,5 Гц, 1H), 4,48 (т, J=5,6 Гц, 1H), 3,72 (д, J=14,7 Гц, 1H), 3,58 (д, J=14,7 Гц, 1H), 3,26-3,23 (м, 4H), 2,99-2,96 (м, 1H), 2,85 (дд, J=14,4, 6,3 Гц, 1H), 2,46 (с, 3H), 1,99-1,95 (м, 4H); ESI-МС m/z 349 [M+H]+.
Стадия H: К раствору 7-пирролидинтетрагидроизохинолина (60 мг, 0,17 ммоль) со стадии G, описанной выше, в метаноле (2 мл) добавляли малеиновую кислоту (20 мг, 0,17 ммоль) с последующим медленным добавлением воды (10 мл). Полученный в результате раствор лиофилизировали в течение ночи, получая малеатную соль (+/-)-4-(бензо[b]тиофен-2-ил)-2-метил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолина (79 г, 98%) в виде белого твердого вещества: т.пл. 105-108°C;1H ЯМР (CD3OD, 500 МГц) δ 7,80 (д, J=7,9 Гц, 1H), 7,74 (д, J=7,6 Гц, 1H), 7,37-7,27 (м, 3H), 7,02 (д, J=8,5 Гц, 1H), 6,57 (дд, J=8,4, 2,1, Гц, 1H), 6,38 (д, J=2,2 Гц, 1H), 6,24 (с, 2H), 4,90-4,84 (м, 1H), 4,50 (д, J=15,0 Гц, 1H), 4,44 (д, J=15,0 Гц, 1H), 3,90 (дд, J=12,5, 6,1 Гц, 1H), 3,69-3,56 (м, 1H), 3,31-3,26 (м, 4H), 3,06 (с, 3H), 2,05-2,01 (м, 4H); ESI-МС m/z 349 [M+H]+.
Пример 78 - Получение малеатной соли (+)-4-(бензо[b]тиофен-2-ил)-2-метил-7-(морфолин-4-илметил)-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору (+)-энантиомера, свободного основания со стадии H примера 79 (0,25 г, 0,82 ммоль) в толуоле (10 мл) при - 78°C добавляли гидрид диизобутилалюминия (0,90 мл, 0,90 ммоль, 1 М в толуоле). После добавления реакционный раствор медленно нагревали до комнатной температуры и перемешивали в течение ночи. Затем к полученной в результате реакционной смеси медленно добавляли насыщенный раствор сегнетовой соли и полученную взвесь энергично перемешивали в течение 2 часов. Органическую фазу отделяли и водный раствор дважды экстрагировали этилацетатом. Объединенный органический экстракт промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученный сырой продукт очищали препаративной тонкослойной хроматографией (95:5 дихлорметан/метанол), получая требуемый продукт (78 мг, 30%) в виде бесцветного масла:1H ЯМР (CDCl3, 500 МГц) δ 9,95 (с, 1H), 7,73 (д, J=8,1 Гц, 1Н), 7,70 (д, J=7,9 Гц, 1H), 7,63-7,61 (м, 2H), 7,33-7,25 (м, 3H), 7,18 (с, 1Н), 4,61 (т, J=5,4 Гц, 1H), 3,84 (д, J=15,2 Гц, 1H), 3,70 (д, J=15,2 Гц, 1H), 3,04 (дд, J=11,6, 5,0 Гц, 1Н), 2,93 (дд, J=11,6, 5,9 Гц, 1H), 2,52 (с, 3H).
Стадия B: К раствору альдегида (78 мг, 0,25 ммоль) со стадии A, описанной выше, в дихлорметане (8 мл) при комнатной температуре добавляли морфолин (28 мкл, 0,32 ммоль), триацетоксиборгидрид натрия (80 мг, 0,38 ммоль), уксусную кислоту (5 мкл) и молекулярные сита 4Е (0,5 г). Реакционный раствор перемешивали при комнатной температуре в течение ночи и затем разбавляли дихлорметаном, промывали водным раствором карбоната натрия (2 М), сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученный сырой продукт очищали препаративной тонкослойной хроматографией (94:6 дихлорметан/метанол), получая требуемый продукт (68 мг, 72%) в виде белой пены: [α]24D +37,2° (c 0,11, метанол);1H ЯМР (CDCl3, 500 МГц) δ 7,72 (д, J=8,0 Гц, 1H), 7,68 (д, J=7,9 Гц, 1H), 7,30-7,23 (м, 2H), 7,16 (с, 1H), 7,11-7,05 (м, 3H), 4,54 (т, J=5,0 Гц, 1H), 3,75 (д, J=15,0 Гц, 1H), 3,73-3,69 (м, 4H), 3,62 (д, J=14,9 Гц, 1H), 3,44 (с, 2H), 3,00 (дд, J=11,5, 4,9 Гц, 1H), 2,89 (дд, J=11,4, 6,1 Гц, 1H), 2,49 (с, 3H), 2,45-2,41 (м, 4H); ESI-МС m/z 379 [M+H]+.
К раствору тетрагидроизохинолина (62 мг, 0,16 ммоль) в метаноле (3 мл) добавляли малеиновую кислоту (38 мг, 0,33 ммоль), затем воду (15 мл). Полученный в результате раствор лиофилизировали в течение ночи, получая малеатную соль (+/-)-4-(бензо[b]тиофен-2-ил)-2-метил-7-(морфолин-4-илметил)-1,2,3,4-тетрагидроизохинолина (94 мг, 91%, AUC ВЭЖХ >99%): т.пл. 80-82°C;1H ЯМР (CD3OD, 500 МГц) δ 7,80 (д, J=7,9 Гц, 1H), 7,76 (д, J=7,5 Гц, 1H), 7,40-7,31 (м, 6H), 6,26 (с, 4H), 5,01 (дд, J=9,6, 5,8 Гц, 1H), 4,50-4,47 (м, 2H), 4,19 (с, 2H), 3,90-3,85 (м, 5H), 3,64 (дд, J=12,3, 10,2 Гц, 1H), 3,13-3,09 (м, 4H), 3,03 (с, 3H); ESI-МС m/z 379 [M+H]+; элементный анализ, вычислено для C22H24N2OS-2C4H4O4-1,875H2O: C, 57,77; Н, 5,90; N, 4,35. Найдено: C, 58,09; Н 5,58; N, 3,95.
Пример 79 - Получение малеатной соли (+)-4-(бензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрила и малеатной соли (-)-4-(бензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрила
Стадия A: К раствору 2-ацетилбензо[b]тиофена (5,0 г, 28 ммоль) в хлороформе (120 мл) добавляли пербромид бромида пиридиния (9,6 г, 28 ммоль) двумя порциями. Реакционный раствор перемешивали при комнатной температуре в течение 2 часов и затем промывали водой, водным раствором HCl (2 М), водой и насыщенным раствором соли. Полученный в результате раствор сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый бромкетон (грубый выход 7,4 г) в виде коричневого твердого вещества, которое использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 8,07 (с, 1H), 7,94-7,87 (м, 2H), 7,52-7,25 (м, 2H), 4,46 (с, 2H).
Стадия B: К раствору 3-метоксибензилметиламина (4,3 г, 29 ммоль) в дихлорметане (150 мл) добавляли триэтиламин (10 мл) и бромкетон (7,3 г, 29 ммоль) со стадии A, описанной выше. Реакционный раствор перемешивали при комнатной температуре в течение 2 часов и затем гасили насыщенным водным раствором бикарбоната натрия. Органический экстракт отделяли, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от 90:10 до 85:15 гексаны/этилацетат), получая требуемый продукт (7,1 г, 76% для 2 стадий) в виде красноватого масла:1H ЯМР (CDCl3, 500 МГц) δ 8,09 (с, 1H), 7,86 (д, J=8,0 Гц, 2H), 7,45 (дд, J=8,0, 1,1 Гц, 1H), 7,40 (дд, J=8,0, 1,0 Гц, 1H), 7,24 (д, J=8,1 Гц, 1Н), 6,97-6,95 (м, 2H), 6,83 (д, J=8,5 Гц, 1H), 3,78 (с, 3H), 3,76 (с, 2H), 3,70 (с, 2H), 2,43 (с, 3H); ESI-МС m/z 326 [M+H]+.
Стадия C: К раствору кетона (7,1 г, 22 ммоль) со стадии B, описанной выше, в метаноле (100 мл) при 0°C небольшими порциями добавляли боргидрид натрия (0,89 г, 24 ммоль). Реакционный раствор перемешивали при 0°C в течение 1 часа. Затем растворитель удаляли при пониженном давлении и полученный остаток растворяли в дихлорметане. Полученный в результате раствор промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый спирт (6,9 г, 96% грубый выход) в виде желтого масла. Неочищенный продукт использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,81 (д, J=7,9 Гц, 1H), 7,70 (д, J=8,1 Гц, 1H), 7,33-7,23 (м, 3H), 7,21 (с, 1H), 6,91-6,87 (м, 3H), 5,07 (дд, J=10,1, 3,5 Гц, 1H), 4,40-4,03 (м, 1H), 3,81 (с, 3H), 3,72 (д, J=13,0 Гц, 1H), 3,56 (д, J=13,0 Гц, 1H), 2,83 (дд, J=12,4, 10,2 Гц, 1H), 2,72 (дд, J=12,4, 3,7 Гц, 1H), 2,34 (с, 3H); ESI-МС m/z 328 [M+H]+.
Стадия D: К раствору спирта (6,9 г, 21 ммоль) со стадии C, описанной выше, в дихлорметане (150 мл) по каплям добавляли метансульфоновую кислоту (14 мл, 210 ммоль). После добавления реакционный раствор перемешивали при комнатной температуре в течение 20 минут и затем медленно добавляли к ледяному водному раствору гидроксида натрия (120 мл, 2 М) и перемешивали в течение 20 минут. Органический слой отделяли и водный слой дважды экстрагировали дихлорметаном. Объединенный органический экстракт промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от 95:5 до 50:50 гексаны/этилацетат), получая требуемый продукт (3,4 г, 52%) в виде красноватого масла: H ЯМР (CDCl3, 500 МГц) δ 7,72 (д, J=8,0 Гц, 1H), 7,67 (д, J=8,0 Гц, 1H), 7,29-7,23 (м, 2H), 7,13 (с, 1H), 7,06 (д, J=8,5 Гц, 1H), 6,69 (д, J=2,7 Гц, 1H), 6,61 (д, J=2,6 Гц, 1H), 4,50 (т, J=5,3 Гц, 1H), 3,77 (с, 3H), 3,73 (д, J=14,5 Гц, 1H), 3,59 (д, J=14,5 Гц, 1H), 2,99 (дд, J=11,5, 4,9 Гц, 1H), 2,87 (дд, J=11,4, 6,2 Гц, 1H), 2,47 (с, 3H); ESI-МС m/z 310 [M+H]+.
Стадия E: К раствору 7-метокситетрагидроизохинолина (1,6 г, 5,1 ммоль) со стадии D, описанной выше, в уксусной кислоте (25 мл) добавляли водный раствор бромистого водорода (25 мл, 48%). Реакционный раствор нагревали при 100°C в течение 20 часов и затем охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток помещали в смесь дихлорметана, воды и небольшого количества метанола. Полученный в результате раствор осторожно гасили насыщенным водным раствором бикарбоната натрия до pH >8. Органический экстракт отделяли, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый фенол (грубый выход 1,6 г) в виде коричневого твердого вещества, которое использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,72 (д, J=7,7 Гц, 1H), 7,67 (д, J=7,7 Гц, 1H), 7,29 (тд, J=7,3, 1,2 Гц, 1H), 7,23 (тд, J=7,3, 1,3 Гц, 1H), 7,13 (с, 1H), 7,01 (д, J=8,4 Гц, 1H), 6,59 (дд, J=8,4, 2,5 Гц, 1H), 6,53 (д, J=2,3 Гц, 1Н), 4,49 (т, J=5,7 Гц, 1H), 3,68 (д, J=15,1 Гц, 1H), 3,57 (д, J=15,0 Гц, 1H), 3,00 (дд, J=11,6, 5,4 Гц, 1H), 2,85 (дд, J=11,5, 6,4 Гц, 1H), 2,47 (с, 3H), 1,90-1,45 (м, 1Н); ESI-МС m/z 296 [M+H]+.
Стадия F: К раствору полученного фенола (1,6 г, 5,1 ммоль) с описанной выше стадии E в дихлорметане (50 мл) при 0°C последовательно по каплям добавляли пиридин (0,80 мл, 10 ммоль) и ангидрид трифторметансульфоновой кислоты (1,1 мл, 6,6 ммоль). Полученный в результате реакционный раствор перемешивали при 0°C в течение 1 часа и затем гасили насыщенным водным раствором бикарбоната натрия. Органический экстракт отделяли и водный слой дважды экстрагировали дихлорметаном. Объединенный органический экстракт промывали смесью 1:1 вода/насыщенный раствор соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая сырой продукт. Очистка флэш-хроматографией на колонке (от дихлорметана до смеси 98:2 дихлорметан/метанол) давала требуемый трифторметансульфонат (1,6 г, 73% для двух стадий) в виде желтого масла:1H ЯМР (CDCl3, 500 МГц) δ 7,74 (д, J=8,0 Гц, 1H), 7,70 (д, J=7,8 Гц, 1H), 7,32-7,24 (м, 3H), 7,16 (с, 1H), 7,02-7,00 (м, 2H), 4,54 (т, J=5,4 Гц, 1H), 3,78 (д, J=15,4 Гц, 1H), 3,65 (д, J=15,4 Гц, 1H), 3,01 (дд, J=11,6, 5,0 Гц, 1H),2,91 (дд, J=11,6, 6,0 Гц, 1H), 2,50 (с, 3H); ESI-МС m/z 428 [M+H]+.
Стадия G: К раствору трифторметансульфоната (1,8 г, 4,3 ммоль) с описанной выше стадии F в N,N-диметилформамиде (15 мл) добавляли цианид цинка (1,0 г, 8,6 ммоль). Реакционный раствор продували аргоном в течение 10 минут и затем к нему добавляли тетракис(трифенилфосфин)палладий (0) (0,50 г, 0,43 ммоль). Полученный в результате раствор дегазировали, используя аргон, в течение 5 минут и нагревали при 120°C в течение 4 часов. Затем реакционную смесь охлаждали до комнатной температуры, разбавляли водой, экстрагировали этилацетатом. Органический экстракт промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (элюент от 83:17 до 66:34 гексаны/этилацетат), получая требуемый продукт (0,50 г, 38%) в виде желтой пены:1H ЯМР (CDCl3, 500 МГц) δ 7,74 (д, J=8,0 Гц, 1H), 7,69 (д, J=7,8 Гц, 1H), 7,40-7,26 (м, 5H), 7,17 (с, 1H), 4,57 (т, J=5,3 Гц, 1H), 3,77 (д, J=15,3 Гц, 1H), 3,64 (д, J=15,4 Гц, 1Н), 3,02 (дд, J=11,6, 5,0 Гц, 1H), 2,91 (дд, J=11,6, 5,2 Гц, 1H), 2,51 (с, 3H).
Стадия H: 7-карбонитрилтетрагидроизохинолин (0,67 г) со стадии G, описанной выше, разделяли препаративной хиральной ВЭЖХ (колонка CHIRALPAK AD, используя смесь 85:15:0,1 гептан/этанол/диэтиламин в качестве элюента), получая (+)-энантиомер [[α]25D +129,3° (c 0,16, метанол)] и (-)-энантиомер [[α]25D -133,6° (c 0,11, метанол)]. (+)-Энантиомер (83 мг, 0,27 ммоль) растворяли в метаноле (3 мл) и обрабатывали малеиновой кислотой (32 мг, 0,27 ммоль) с последующим медленным добавлением воды (10 мл). Полученный в результате раствор лиофилизировали в течение ночи, получая малеатную соль (+)-4-(бензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрила (105 мг, 91%, AUC ВЭЖХ >99%) в виде желтого твердого вещества: т.пл. 91-94°C;1H ЯМР (CD3OD, 500 МГц) δ 7,82 (д, J=7,8 Гц, 1Н), 7,78 (д, J=7,5 Гц, 1H), 7,71 (с, 1H), 7,64 (д, J=8,0 Гц, 1H), 7,41-7,32 (м, 4H), 6,26 (с, 3H), 5,05 (дд, J=9,6, 6,0 Гц, 1H), 4,51 (кажущийся с, 2H), 3,89 (дд, J=12,4, 5,9 Гц, 1H), 3,66 (дд, J=12,0, 10,3 Гц, 1H), 3,03 (с, 3H); ESI-МС m/z 305 [M+H]+; элементный анализ, вычислено для C19H16N2S-1,5C4H4О4-1,5H2О: C, 59,40; Н, 4,98; N, 5,54. Найдено: C, 59,36; Н, 4,60; N, 5,64.
Стадия I: К раствору (-)-энантиомера (0,18 г, 0,59 ммоль), который получен на стадии H, в метаноле (3 мл) добавляли малеиновую кислоту (68 мг, 0,59 ммоль) с последующим медленным добавлением воды (15 мл). Полученный в результате раствор лиофилизировали в течение ночи, получая малеатную соль (-)-4-(бензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрила (0,24 г, 96%, 97,1% AUC ВЭЖХ) в виде светло-желтого твердого вещества: т.пл. 80-84°C;1H ЯМР (CD3OD, 500 МГц) δ 7,82 (д, J=7,8 Гц, 1H), 7,78 (д, J=7,5 Гц, 1H), 7,71 (с, 1H), 7,64 (д, J=8,0 Гц, 1H), 7,41-7,32 (м, 4H), 6,26 (с, 3H), 5,01 (дд, J=9,6, 6,0 Гц, 1H), 4,46 (кажущийся с, 2H), 3,84 (дд, J=12,4, 5,6 Гц, 1H), 3,62 (дд, J=12,0, 10,3 Гц, 1H), 3,00 (с, 3H); ESI-МС m/z 305 [M+H]+.
Пример 80 - Получение малеатной соли (+/-)-4-(бензо[b]тиофен-4-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору 3-бромтиофенола (14,8 г, 78 ммоль) в N,N-диметилформамиде (270 мл) при 0°C порциями добавляли гидрид натрия (3,6 г, 90 ммоль). После добавления реакционный раствор перемешивали при комнатной температуре в течение 30 минут и к нему добавляли диметилацеталь бромацетальдегида (9,7 мл, 82 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Полученный в результате раствор разбавляли этилацетатом, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от гексанов до смеси 95:5 гексаны/этилацетат), получая требуемый продукт (20,6 г, 95%) в виде светло-желтого масла:1H ЯМР (CDCl3, 500 МГц) δ 7,51 (т, J=1,8 Гц, 1H), 7,32-7,28 (м, 2H), 7,14 (т, J=7,9 Гц, 1Н), 4,53 (т, J=5,5 Гц, 1H), 3,38 (с, 6H), 3,11 (д, J=5,5 Гц, 2H).
Стадия B: К раствору полифосфорной кислоты (90 г) в хлорбензоле (250 мл) при кипячении с обратным холодильником по каплям добавляли раствор тиофенолацеталя (27,6 г, 99,6 ммоль) с описанной выше стадии A в хлорбензоле (120 мл). После добавления реакционный раствор нагревали при кипячении с обратным холодильником в течение 2 часов и затем охлаждали до комнатной температуры. Верхний слой отделяли; нижний слой разбавляли водой и затем дважды экстрагировали дихлорметаном. Объединенный органический экстракт промывали водой и насыщенным водным раствором бикарбоната натрия и сушили и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (гексаны), получая смесь 1:1 4-бромбензо[b]тиофена и 6-бромбензо[b]тиофена (15,5 г, 73%), которую использовали на следующей стадии без дополнительной очистки или характеристики.
Стадия C: К раствору изомерного бромбензо[b]тиофена (14,2 г, 66,5 ммоль) со стадии B, описанной выше, в N,N-диметилформамиде (130 мл) добавляли пиридин (7,5 мл) и цианид меди (I) (7,75 г). Реакционную смесь нагревали при кипячении с обратным холодильником в течение 20 часов. Затем реакционный раствор охлаждали до 60°C и вливали в раствор этилендиамина в воде. Полученную в результате смесь перемешивали в течение 30 минут и экстрагировали диэтиловым эфиром и этилацетатом. Объединенный органический экстракт промывали смесью 1:1 насыщенный раствор соли/вода, сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученный сырой продукт очищали флэш-хроматографией на колонке (от 95:5 до 91:9 гексаны/этилацетат), получая 4-цианобензо[b]тиофен (4,15 г, 39%) в виде светло-желтого твердого вещества и 6-цианобензо[b]тиофен (4,34 г, 41%) в виде красноватого масла:1H ЯМР для 4-цианобензо[b]тиофена (CDC13, 500 МГц) δ 8,10 (д, J=8,2 Гц, 1H), 7,74-7,70 (м, 2H), 7,62 (дд, J=5,5, 0,6 Гц, 1H), 7,43 (д, J=7,8 Гц, 1Н);1H ЯМР для 6-цианобензо[b]тиофена (CDC13, 500 МГц) δ 8,22 (кажущийся с, 1H), 7,90 (д, J=8,3 Гц, 1H), 7,72 (д, J=5,4 Гц, 1H), 7,59 (д, J=8,3 Гц, 1H), 7,42 (дд, J=5,4, 0,5 Гц, 1H).
Стадия D: В реакционную колбу, оборудованную механической мешалкой, в атмосфере аргона добавляли бромид меди (I) (0,19 г, 1,3 ммоль) и метилмагнийбромид (26 мл, 78 ммоль, 3 М в диэтиловом эфире) при -78°C. Затем к полученной в результате смеси по каплям добавляли раствор 4-цианобензо[b]тиофен (4,15 г, 26,1 ммоль) со стадии C, описанной выше, в тетрагидрофуране (50 мл) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Затем реакционный раствор медленно при перемешивании добавляли к насыщенному водному раствору хлорида аммония. Затем полученную в результате смесь дважды экстрагировали этилацетатом. Объединенный органический экстракт промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученный сырой продукт очищали флэш-хроматографией на колонке (94:6 гексаны/этилацетат), получая частично очищенный продукт (грубый выход 5,9 г), который использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 8,37 (д, J=5,5 Гц, 1Н), 8,08 (д, J=8,1 Гц, 1H), 7,96 (д, J=7,5 Гц, 1H), 7,65 (д, J=5,6 Гц, 1H), 7,43 (т, J=7,7 Гц, 1H), 2,73 (с, 3H).
Стадия E: К раствору метилкетона (5,9 г, 24 ммоль) со стадии D, описанной выше, в хлороформе (100 мл) добавляли пербромид бромида пиридиния (8,0 г, 24 ммоль) двумя порциями. Реакционный раствор перемешивали при комнатной температуре в течение 2 часов и затем промывали водой, водным раствором HCl (2 М), водой и насыщенным раствором соли. Полученный в результате раствор сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый бромкетон (грубый выход 6,4 г), который использовали на следующей стадии без очистки:1H ЯМР (CDCl3, 500 МГц) δ 8,34 (дд, J=5,5, 0,6 Гц, 1H), 8,13 (д, J=8,0 Гц, 1H), 7,97 (дд, J=7,6, 0,8 Гц, 1H), 7,70 (д, J=5,6 Гц, 1H), 7,45 (т, J=7,8 Гц, 1H), 4,59 (с, 2H).
Стадия F: К раствору 3-метоксибензилметиламина (4,4 г, 29 ммоль) в дихлорметане (150 мл) добавляли триэтиламин (10 мл) и бромкетон (6,4 г, 24 ммоль) со стадии E, описанной выше. Реакционный раствор перемешивали при комнатной температуре в течение 18 часов и затем гасили насыщенным водным раствором бикарбоната натрия (100 мл). Органический экстракт отделяли, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученный сырой продукт очищали флэш-хроматографией на колонке (86:14 гексаны/этилацетат), получая требуемый продукт (3,9 г, 46% для 3 стадий) в виде светло-желтого масла:1H ЯМР (CDCl3, 500 МГц) δ 8,29 (д, J=5,6 Гц, 1Н), 8,06 (д, J=8,1 Гц, 1H), 7,95 (д, J=7,5 Гц, 1H), 7,63 (д, J=5,6 Гц, 1H), 7,37 (т, J=7,8 Гц, 1Н), 7,22 (т, J=7,7 Гц, 1Н), 6,93-6,90 (м, 2H), 6,80 (дд, J=7,7, 2,5 Гц, 1H), 3,88 (с, 2H), 3,77 (с, 3H), 3,69 (с, 2H), 2,42 (с, 3H).
Стадия G: К раствору кетона (3,9 г, 12 ммоль) со стадии F, описанной выше, в метаноле (80 мл) при 0°C небольшими порциями добавляли боргидрид натрия (0,5 г, 13 ммоль). Реакционный раствор перемешивали при 0°C в течение 2 часов и затем растворитель удаляли при пониженном давлении. Полученный остаток растворяли в дихлорметане, промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли. Органический экстракт сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый спирт (6,9 г, грубый выход 96%) в виде желтого масла, которое использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,80 (д, J=8,1 Гц, 1H), 7,46 (д, J=7,2 Гц, 1H), 7,41 (д, J=5,6 Гц, 1H), 7,37 (д, J=5,6 Гц, 1H), 7,33 (т, J=7,7 Гц, 1H), 7,27 (т, J=7,8 Гц, 1H), 6,93 (д, J=7,6 Гц, 1H), 6,90 (ушир.с, 1H), 6,84 (дд, J=8,2, 2,5 Гц, 1H), 5,20 (дд, J=10,6, 3,3 Гц, 1H), 4,44-4,03 (м, 1H), 3,82 (с, 3H), 3,76 (д, J=13,0 Гц, 1H), 3,52 (д, J=13,0 Гц, 1H), 2,77 (дд, J=12,6, 11,2 Гц, 1H), 2,64 (дд, J=12,6, 3,4 Гц, 1H), 2,42 (с, 3H); ESI-МС m/z 328 [M+H]+.
Стадия H: К раствору спирта (3,9 г, 12 ммоль) со стадии G, описанной выше, в дихлорметане (150 мл) по каплям при комнатной температуре добавляли пентоксид фосфора (2 г) и метансульфоновую кислоту (1,6 мл, 24 ммоль). После перемешивания в течение 2 часов к раствору добавляли дополнительное количество метансульфоновой кислоты (1,6 мл, 24 ммоль) в дихлорметане (3 мл). После добавления реакционный раствор перемешивали при комнатной температуре в течение 1 часа и затем органический слой отделяли и промывали насыщенным водным раствором бикарбоната натрия, водой и насыщенным раствором соли. Полученный в результате органический экстракт сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученный сырой продукт очищали флэш-хроматографией на колонке (от 95:5 до 50:50 гексаны/этилацетат), получая требуемый продукт (1,4 г, 38%) в виде желтого масла:1H ЯМР (CDCl3, 500 МГц) δ 7,77 (д, J=8,1 Гц, 1H), 7,39 (д, J=5,5 Гц, 1H), 7,28-7,25 (м, 2H), 7,10 (д, J=7,2 Гц, 1H), 6,76 (д, J=8,5 Гц, 1H), 6,66 (д, J=2,5 Гц, 1H), 6,62 (дд, J=8,5, 2,6 Гц, 1H), 4,79-4,76 (м, 1H), 3,81 (д, J=15,3 Гц, 1H), 3,78 (с, 3H), 3,64 (д, J=15,0 Гц, 1H), 3,13-3,08 (м, 1Н), 2,68 (дд, J=11,5, 8,2 Гц, 1H), 2,43 (с, 3H); ESI-МС m/z 310 [M+H]+.
Стадия I: К раствору 7-метокситетрагидроизохинолина (1,4 г, 4,5 ммоль) с описанной выше стадии H в уксусной кислоте (25 мл) добавляли 48% водный раствор бромистого водорода (25 мл). Полученную в результате смесь нагревали при 100°C в течение 20 часов и затем охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток растворяли в дихлорметане и осторожно гасили насыщенным водным раствором бикарбоната натрия до pH >8. Органический экстракт отделяли, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый фенол (грубый выход 1,4 г) в виде серой пены, который использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,77 (д, J=8,1 Гц, 1Н), 7,38 (д, J=5,5 Гц, 1H), 7,28-7,24 (м, 2H), 7,10 (д, J=7,3 Гц, 1H), 6,70 (д, J=8,3 Гц, 1H), 6,56-6,50 (м, 2H), 4,78-4,75 (м, 1H), 3,77 (д, J=15,0 Гц, 1Н), 3,60 (д, J=15,0 Гц, 1H), 3,15-3,11 (м, 1H), 2,68 (дд, J=11,5, 9,5 Гц, 1H), 2,44 (с, 3H), 1,95-1,56 (м, 1H).
Стадия J: К раствору фенола (1,4 г, 4,5 ммоль) со стадии I, описанной выше, в дихлорметане (50 мл) при 0°C по каплям добавляли пиридин (0,73 мл, 9,0 ммоль), затем ангидрид трифторметансульфоновой кислоты (1,0 мл, 5,9 ммоль). Реакционный раствор перемешивали при 0°C в течение 1 часа и затем гасили насыщенным водным раствором бикарбоната натрия. Органический экстракт отделяли, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от дихлорметана до смеси 98:2 дихлорметан/метанол), получая требуемый трифторметансульфонат (1,6 г, 85% для двух стадий) в виде красноватого масла:1H ЯМР (CDCl3, 500 МГц) δ 7,81 (д, J=8,1 Гц, 1H), 7,44 (д, J=5,5 Гц, 1H), 7,28-7,22 (м, 2H), 7,07-7,04 (м, 2Н), 6,94 (кажущийся с, 2H), 4,83-4,80 (м, 1H), 3,86 (д, J=15,3 Гц, 1H), 3,68 (д, J=15,3 Гц, 1H), 3,16-3,11 (м, 1H), 2,72 (дд, J=11,6, 9,1 Гц, 1H), 2,46 (с, 3H); ESI-МС m/z 428 [M+H]+.
Стадия K: К раствору трифторметансульфоната (0,10 г, 0,23 ммоль) со стадии J, описанной выше, в толуоле (2,5 мл) добавляли карбонат цезия (67 мг, 0,14 ммоль), 2-(дициклогексилфосфино)-2',4',6'-триизопропил-1,1'-бифенил (0,19 г, 0,58 ммоль) и морфолин (41 мкл, 0,47 ммоль). Реакционную смесь продували аргоном в течение 5 минут и затем добавляли ацетат палладия (II) (8 мг, 0,04 ммоль). Реакционную колбу закрывали, нагревали в микроволновой печи (160°C) в течение 45 минут и затем охлаждали до комнатной температуры. Реакционный раствор фильтровали через слой целита и концентрировали. Полученный сырой продукт очищали флэш-хроматографией на колонке (от дихлорметана до смеси 97:3 дихлорметан/метанол), получая требуемый продукт (11 мг, 13%) в виде желтого масла:1H ЯМР (CDCl3, 500 МГц) δ 7,77 (д, J=8,0 Гц, 1H), 7,39 (д, J=5,4 Гц, 1H), 7,28-7,24 (м, 2H), 7,10 (д, J=7,4 Гц, 1H), 6,75 (д, J=8,1 Гц, 1H), 6,65-6,63 (м, 2H), 4,77 (ушир.с, 1H), 3,86-3,84 (м, 4H), 3,79 (д, J=14,5 Гц, 1H), 3,64 (д, J=14,5 Гц, 1H), 3,14-3,11 (м, 5H), 2,71-2,68 (м, 1H), 2,44 (с, 3H); ESI-МС m/z 365 [M+H]+.
Стадия L: К раствору 7-морфолинилтетрагидроизохинолина (11 мг, 0,03 ммоль) со стадии K, описанной выше, в метаноле (1 мл) добавляли малеиновую кислоту (3,5 мг, 0,03 ммоль), затем воду (5 мл). Полученный в результате раствор лиофилизировали в течение ночи, получая малеатную соль (+/-)-4-(бензо[b]тиофен-4-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина (15 мг, 99%) в виде светло-желтого твердого вещества: т.пл. 103-107°C;1H ЯМР (CD3OD, 500 МГц) δ 7,92 (д, J=8,1 Гц, 1Н), 7,61 (д, J=4,8 Гц, 1H), 7,36 (т, J=7,6 Гц, 1H), 7,34-7,10 (м, 2H), 6,87-6,84 (м, 2H), 6,75 (ушир.с, 1H), 6,25 (с, 2H), 5,02-4,98 (м, 1H), 4,67-4,50 (м, 2H), 3,87 (дд, J=12,1, 6,1 Гц, 1H), 3,86-3,82 (м, 4H), 3,72-3,54 (м, 1H), 3,16-3,13 (м, 4H), 3,08 (с, 3H); ESI-МС m/z 365 [M+H]+.
Пример 81 - Получение малеатной соли (+/-)-4-(бензо[b]тиофен-4-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору 3-бромтиофенола (14,8 г, 78 ммоль) в N,N-диметилформамиде (270 мл) при 0°C порциями добавляли гидрид натрия (3,6 г, 90 ммоль). После добавления реакционный раствор перемешивали при комнатной температуре в течение 30 минут и к нему добавляли диметилацеталь бромацетальдегида (9,7 мл, 82 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Полученный в результате раствор разбавляли этилацетатом, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от гексанов до смеси 95:5 гексаны/этилацетат), получая требуемый продукт (20,6 г, 95%) в виде светло-желтого масла:1H ЯМР (CDCl3, 500 МГц) δ 7,51 (т, J=1,8 Гц, 1H), 7,32-7,28 (м, 2H), 7,14 (т, J=7,9 Гц, 1H), 4,53 (т, J=5,5 Гц, 1H), 3,38 (с, 6H), 3,11 (д, J=5,5 Гц, 2H).
Стадия B: К раствору полифосфорной кислоты (90 г) в хлорбензоле (250 мл) при кипячении с обратным холодильником по каплям добавляли раствор тиофенолацеталя (27,6 г, 99,6 ммоль) со стадии A, описанной выше, в хлорбензоле (120 мл). После добавления реакционный раствор кипятили с обратным холодильником в течение 2 часов и затем охлаждали до комнатной температуры. Верхний слой отделяли; нижний слой разбавляли водой и затем дважды экстрагировали дихлорметаном. Объединенный органический экстракт промывали водой и насыщенным водным раствором бикарбоната натрия и сушили и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (гексаны), получая смесь 1:1 4-бромбензо[b]тиофена и 6-бромбензо[b]тиофена (15,5 г, 73%), которую использовали на следующей стадии без дополнительной очистки или характеристики.
Стадия C: К раствору изомерного бромбензо[b]тиофена (14,2 г, 66,5 ммоль) со стадии B, описанной выше, в N,N-диметилформамиде (130 мл) добавляли пиридин (7,5 мл) и цианид меди (I) (7,75 г). Реакционную смесь кипятили с обратным холодильником в течение 20 часов. Затем реакционный раствор охлаждали до 60°C и вливали в раствор этилендиамина в воде. Полученную в результате смесь перемешивали в течение 30 минут и экстрагировали диэтиловым эфиром и этилацетатом. Объединенный органический экстракт промывали смесью 1:1 насыщенный раствор соли/вода, сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученный сырой продукт очищали флэш-хроматографией на колонке (от 95:5 до 91:9 гексаны/этилацетат), получая 4-цианобензо[b]тиофен (4,15 г, 39%) в виде светло-желтого твердого вещества и 6-цианобензо[b]тиофен (4,34 г, 41%) в виде красноватого масла:1H-ЯМР для 4-цианобензо[b]тиофена (CDCl3, 500 МГц) δ 8,10 (д, J=8,2 Гц, 1H), 7,74-7,70 (м, 2H), 7,62 (дд, J=5,5, 0,6 Гц, 1H), 7,43 (д, J=7,8 Гц, 1H);1H-ЯМР для 6-цианобензо[b]тиофена (CDC13, 500 МГц) δ 8,22 (кажущийся с, 1H), 7,90 (д, J=8,3 Гц, 1H), 7,72 (д, J=5,4 Гц, 1H), 7,59 (д, J=8,3 Гц, 1H), 7,42 (дд, J=5,4, 0,5 Гц, 1H).
Стадия D: В реакционную колбу, оборудованную механической мешалкой, в атмосфере аргона добавляли бромид меди (I) (0,19 г, 1,3 ммоль) и метилмагнийбромид (26 мл, 78 ммоль, 3 М в диэтиловом эфире) при -78°C. Затем к полученной в результате взвеси по каплям добавляли раствор 4-цианобензо[b]тиофена (4,15 г, 26,1 ммоль) со стадии C, описанной выше, в тетрагидрофуране (50 мл) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Затем реакционный раствор медленно при перемешивании добавляли к насыщенному водному раствору хлорида аммония. Полученную в результате смесь затем дважды экстрагировали этилацетатом. Объединенный органический экстракт промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученный сырой продукт очищали флэш-хроматографией на колонке (94:6 гексаны/этилацетат), получая частично очищенный продукт (грубый выход 5,9 г), который использовали на следующей стадии без дополнительной очистки1H ЯМР (CDCl3, 500 МГц) δ 8,37 (д, J=5,5 Гц, 1Н), 8,08 (д, J=8,1 Гц, 1H), 7,96 (д, J=7,5 Гц, 1H), 7,65 (д, J=5,6 Гц, 1H), 7,43 (т, J=7,7 Гц, 1H), 2,73 (с, 3H).
Стадия E: К раствору метилкетона (5,9 г, 24 ммоль) со стадии D, описанной выше, в хлороформе (100 мл) добавляли пербромид бромида пиридиния (8,0 г, 24 ммоль) двумя порциями. Реакционный раствор перемешивали при комнатной температуре в течение 2 часов и затем промывали водой, водным раствором HCl (2 М), водой и насыщенным раствором соли. Полученный в результате раствор сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый бромкетон (грубый выход 6,4 г), который использовали на следующей стадии без очистки:1H ЯМР (CDCl3, 500 МГц) δ 8,34 (дд, J=5,5, 0,6 Гц, 1H), 8,13 (д, J=8,0 Гц, 1H), 7,97 (дд, J=7,6, 0,8 Гц, 1H), 7,70 (д, J=5,6 Гц, 1H), 7,45 (т, J=7,8 Гц, 1H), 4,59 (с, 2H).
Стадия F: К раствору 3-метоксибензилметиламина (4,4 г, 29 ммоль) в дихлорметане (150 мл) добавляли триэтиламин (10 мл) и бромкетон (6,4 г, 24 ммоль) со стадии E, описанной выше. Реакционный раствор перемешивали при комнатной температуре в течение 18 часов и затем гасили насыщенным водным раствором бикарбоната натрия (100 мл). Органический экстракт отделяли, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученный сырой продукт очищали флэш-хроматографией на колонке (86:14 гексаны/этилацетат), получая требуемый продукт (3,9 г, 46% для 3 стадий) в виде светло-желтого масла:1H ЯМР (CDCl3, 500 МГц) δ 8,29 (д, J=5,6 Гц, 1H), 8,06 (д, J=8,1 Гц, 1H), 7,95 (д, J=7,5 Гц, 1H), 7,63 (д, J=5,6 Гц, 1H), 7,37 (т, J=7,8 Гц, 1H), 7,22 (т, J=7,7 Гц, 1H), 6,93-6,90 (м, 2H), 6,80 (дд, J=7,7, 2,5 Гц, 1H), 3,88 (с, 2H), 3,77 (с, 3H), 3,69 (с, 2H), 2,42 (с, 3H).
Стадия G: К раствору кетона (3,9 г, 12 ммоль) со стадии F, описанной выше, в метаноле (80 мл) при 0°C небольшими порциями добавляли боргидрид натрия (0,5 г, 13 ммоль). Реакционный раствор перемешивали при 0°C в течение 2 часов и затем растворитель удаляли при пониженном давлении. Полученный остаток растворяли в дихлорметане, промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли. Органический экстракт сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый спирт (6,9 г, грубый выход 96%) в виде желтого масла, которое использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,80 (д, J=8,1 Гц, 1H), 7,46 (д, J=7,2 Гц, 1H), 7,41 (д, J=5,6 Гц, 1Н), 7,37 (д, J=5,6 Гц, 1H), 7,33 (т, J=7,7 Гц, 1H), 7,27 (т, J=7,8 Гц, 1H), 6,93 (д, J=7,6 Гц, 1H), 6,90 (ушир.с, 1H), 6,84 (дд, J=8,2, 2,5 Гц, 1H), 5,20 (дд, J=10,6, 3,3 Гц, 1H), 4,44-4,03 (м, 1H), 3,82 (с, 3H), 3,76 (д, J=13,0 Гц, 1H), 3,52 (д, J=13,0 Гц, 1H), 2,77 (дд, J=12,6, 11,2 Гц, 1Н), 2,64 (дд, J=12,6, 3,4 Гц, 1H), 2,42 (с, 3H); ESI-МС m/z 328 [M+H]+.
Стадия H: К раствору спирта (3,9 г, 12 ммоль) со стадии G, описанной выше, в дихлорметане (150 мл) по каплям при комнатной температуре добавляли пентоксид фосфора (2 г) и метансульфоновую кислоту (1,6 мл, 24 ммоль). После перемешивания в течение 2 часов добавляли раствор дополнительного количества метансульфоновой кислоты (1,6 мл, 24 ммоль) в дихлорметане (3 мл). После добавления реакционный раствор перемешивали при комнатной температуре в течение 1 часа и затем органический слой отделяли и промывали насыщенным водным раствором бикарбоната натрия, водой и насыщенным раствором соли. Полученный в результате органический экстракт сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученный сырой продукт очищали флэш-хроматографией на колонке (от 95:5 до 50:50 гексаны/этилацетат), получая требуемый продукт (1,4 г, 38%) в виде желтого масла:1H ЯМР (CDCl3, 500 МГц) δ 7,77 (д, J=8,1 Гц, 1H), 7,39 (д, J=5,5 Гц, 1H), 7,28-7,25 (м, 2H), 7,10 (д, J=7,2 Гц, 1H), 6,76 (д, J=8,5 Гц, 1H), 6,66 (д, J=2,5 Гц, 1Н), 6,62 (дд, J=8,5, 2,6 Гц, 1H), 4,79-4,76 (м, 1H), 3,81 (д, J=15,3 Гц, 1H), 3,78 (с, 3H), 3,64 (д, J=15,0 Гц, 1H), 3,13-3,08 (м, 1H), 2,68 (дд, J=11,5, 8,2 Гц, 1H), 2,43 (с, 3H); ESI-МС m/z 310 [M+H]+.
Стадия I: К раствору 7-метокситетрагидроизохинолина (1,4 г, 4,5 ммоль) со стадии H, описанной выше, в уксусной кислоте (25 мл) добавляли 48% водный раствор бромистого водорода (25 мл). Полученную в результате смесь нагревали при 100°C в течение 20 часов и затем охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток растворяли в дихлорметане и осторожно гасили насыщенным водным раствором бикарбоната натрия до pH >8. Органический экстракт отделяли, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая требуемый фенол (грубый выход 1,4 г) в виде серой пены, который использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,77 (д, J=8,1 Гц, 1H), 7,38 (д, J=5,5 Гц, 1H), 7,28-7,24 (м, 2H), 7,10 (д, J=7,3 Гц, 1H), 6,70 (д, J=8,3 Гц, 1H), 6,56-6,50 (м, 2H), 4,78-4,75 (м, 1H), 3,77 (д, J=15,0 Гц, 1H), 3,60 (д, J=15,0 Гц, 1H), 3,15-3,11 (м, 1Н), 2,68 (дд, J=11,5, 9,5 Гц, 1H), 2,44 (с, 3H), 1,95-1,56 (м, 1Н).
Стадия J: К раствору фенола (1,4 г, 4,5 ммоль) со стадии I, описанной выше, в дихлорметане (50 мл) при 0°C по каплям добавляли пиридин (0,73 мл, 9,0 ммоль), затем ангидрид трифторметансульфоновой кислоты (1,0 мл, 5,9 ммоль). Реакционный раствор перемешивали при 0°C в течение 1 часа и затем гасили насыщенным водным раствором бикарбоната натрия. Органический экстракт отделяли, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от дихлорметана до смеси 98:2 дихлорметан/метанол), получая требуемый трифторметансульфонат (1,6 г, 85% для двух стадий) в виде красноватого масла:1H ЯМР (CDCl3, 500 МГц) δ 7,81 (д, J=8,1 Гц, 1Н), 7,44 (д, J=5,5 Гц, 1H), 7,28-7,22 (м, 2H), 7,07-7,04 (м, 2Н), 6,94 (кажущийся с, 2H), 4,83-4,80 (м, 1H), 3,86 (д, J=15,3 Гц, 1H), 3,68 (д, J=15,3 Гц, 1H), 3,16-3,11 (м, 1H), 2,72 (дд, J=11,6, 9,1 Гц, 1H), 2,46 (с, 3H); ESI-МС m/z 428 [M+H]+.
Стадия K: К раствору трифторметансульфоната (1,4 г, 3,3 ммоль) со стадии J, описанной выше, в диметилсульфоксиде (22 мл) добавляли бис(пинаколато)дибор (0,91 г, 3,6 ммоль) и ацетат калия (0,98 г, 10 ммоль). Реакционный раствор продували аргоном в течение 10 минут и затем к нему добавляли 1,1'- бис(дифенилфосфино)ферроцендихлорпалладий (81 мг, 0,10 ммоль). Реакционный раствор дегазировали, используя аргон, в течение 5 минут, нагревали при 80°C в течение 1 часа и затем охлаждали до комнатной температуры. Полученный в результате реакционный раствор разбавляли этилацетатом, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученный сырой продукт очищали флэш-хроматографией на колонке (от дихлорметана до смеси 95:5 дихлорметан/метанол), получая требуемый продукт (грубый выход 1,4 г), который использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,78 (д, J=8,1 Гц, 1Н), 7,59 (с, 1H), 7,47 (д, J=7,7 Гц, 1H), 7,38 (д, J=5,4 Гц, 1H), 7,27-7,24 (м, 2H), 7,08 (д, J=7,2 Гц, 1H), 6,87 (д, J=7,7 Гц, 1H), 4,85 (т, J=7,1 Гц, 1H), 3,88 (д, J=14,7 Гц, 1H), 3,67 (д, J=14,9 Гц, 1H), 3,14 (дд, J=11,7, 6,1 Гц, 1H), 2,70 (дд, J=11,4, 9,4 Гц, 1H), 2,44 (с, 3H), 1,34 (с, 12H).
Стадия L: В реакционную колбу со сложным эфиром бороновой кислоты (грубый выход 1,4 г) со стадии K, описанной выше, 3,6-дихлорпиридазином (0,98 г, 6,6 ммоль) и карбонатом натрия (1,08 г, 10 ммоль) добавляли N,N-диметилформамид (30 мл) и воду (7,6 мл). Полученный в результате раствор продували аргоном в течение 10 минут и затем добавляли 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий (0,14 г, 0,17 ммоль). Реакционный раствор снова дегазировали, используя аргон, в течение 5 минут, нагревали при 80°C в течение 2 часов и затем охлаждали до комнатной температуры. Полученный в результате раствор разбавляли этилацетатом, промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали, используя систему MPLC Biotage (от 99:1 до 95:5 дихлорметан/метанол), получая частично очищенный продукт (0,97 г, 75%) в виде розовой пены, который использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,94 (д, J=1,4 Гц, 1H), 7,80 (т, J=8,2 Гц, 2H), 7,64 (дд, J=8,1, 1,7 Гц, 1Н), 7,54 (д, J=9,0 Гц, 1H), 7,41 (д, J=5,3 Гц, 1H), 7,31-7,26 (м, 2H), 7,14 (д, J=7,1 Гц, 1H), 7,01 (д, J=8,1 Гц, 1H), 4,89 (т, J=7,8 Гц, 1H), 3,97 (д, J=15,0 Гц, 1H), 3,75 (д, J=15,0 Гц, 1H), 3,20-3,16 (м, 1H), 2,76 (дд, J=11,6, 8,2 Гц, 1H), 2,49 (с, 3H); ESI-МС m/z 392 [M+H]+.
Стадия M: К раствору хлорпиридазина (0,55 г, 1,4 ммоль) со стадии L, описанной выше, в смеси этанола (30 мл) и метанола (30 мл) добавляли гидразин (1,4 мл, 28 ммоль) и палладий-на-угле (0,11 г). Реакционный раствор нагревали при кипячении с обратным холодильником в течение 16 часов, затем охлаждали до комнатной температуры, фильтровали через слой целита и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от дихлорметана до смеси 97:3 дихлорметан/метанол), получая требуемый продукт (0,28 г, 54%) в виде светло-желтой пены:1H ЯМР (CDCl3, 500 МГц) δ 9,14 (дд, J=4,9, 1,6 Гц, 1H), 7,99 (д, J=1,4 Гц, 1Н), 7,82 (тд, J=8,6, 1,6 Гц, 2H), 7,67 (дд, J=8,1, 1,7 Гц, 1H), 7,51 (дд, J=8,1, 4,8 Гц, 1H), 7,41 (д, J=5,4 Гц, 1H), 7,29 (т, J=7,8 Гц, 2H), 7,15 (д, J=7,0 Гц, 1Н), 7,02 (д, J=8,1 Гц, 1Н), 4,90 (т, J=6,9 Гц, 1H), 3,98 (д, J=15,0 Гц, 1H), 3,76 (д, J=14,9 Гц, 1H), 3,18 (дд, J=11,6, 6,0 Гц, 1H), 2,76 (дд, J=11,5, 9,2 Гц, 1H), 2,49 (с, 3H); ESI-МС m/z 358 [M+H]+.
Стадия N: К раствору 7-пиридазинилтетрагидроизохинолина (0,28 г, 0,71 ммоль) со стадии M, описанной выше, в метаноле (3 мл) добавляли малеиновую кислоту (84 мг, 0,71 ммоль) с последующим медленным добавлением воды (15 мл). Полученный в результате раствор лиофилизировали в течение ночи, получая малеатную соль (+/-)-4-(бензо[b]тиофен-4-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина (0,32 г, 95%) в виде светло-желтого твердого вещества: т.пл. 94-97°C;1H ЯМР (CD3OD, 500 МГц) δ 9,17 (дд, J=4,9, 1,5 Гц, 1H), 8,18 (дд, J=8,7, 1,5 Гц, 1H), 8,14 (д, J=1,2 Гц, 1H), 7,97 (д, J=8,1 Гц, 1H), 7,91 (дд, J=8,2, 1,4 Гц, 1H), 7,81 (дд, J=8,7, 5,0 Гц, 1H), 7,65 (д, J=5,6 Гц, 1H), 7,40 (т, J=7,7 Гц, 1H), 7,31-7,23 (м, 2H), 7,08 (д, J=8,3 Гц, 1H), 6,25 (с, 2H), 5,19 (дд, J=11,3, 6,6 Гц, 1H), 4,79 (кажущийся с, 2H), 3,97 (дд, J=12,4, 6,3 Гц, 1H), 3,76 (т, J=12,0 Гц, 1H), 3,14 (с, 3H); ESI-МС m/z 358 [M+H]+.
Пример 82 - Получение фумаратной соли (+)-4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина и фумаратной соли (-)-4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: 5-Бром-4-метоксибензотиофен получали, следуя способам, описанным в литературе (Chenard et al., J. Org. Chem., 48: 4312-4317 (1983), публикация включена в данное описание в виде ссылки в полном объеме). Смесь 5-бром-4-метоксибензотиофена (2,43 г, 10 ммоль), изохинолин-4-бороновой кислоты (2,08 г, 12 ммоль) и карбоната цезия (9,77 г, 30 ммоль) в 1,2-диметоксиэтане (50 мл) и воде (10 мл) дегазировали, используя аргон. Добавляли Pd(PPh3)4 (693 мг, 0,6 ммоль) и реакционную смесь нагревали при кипячении с обратным холодильником в течение 15 часов. Охлажденную реакционную смесь разбавляли водой и дважды экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали. Очистка хроматографией на колонке (силикагель, от 10% до 50% этилацетат/гексаны) давала требуемый продукт (2,02 г, 69%):1H ЯМР (CDCl3, 500 МГц) δ 9,31 (с, 1Н), 9,27 (с, 1H), 8,07-8,05 (м, 1H), 7,75 (д, J=8,2 Гц, 1Н), 7,70-7,62 (м, 3H), 7,56 (д, J=5,4 Гц, 1H), 7,50 (д, J=5,5 Гц, 1H), 7,31 (д, J=8,2 Гц, 1H), 3,52 (с, 3H); ESI-МС m/z=292 [M+H]+.
Стадия B: К ледяному раствору продукта со стадии A (1,0 г, 3,43 ммоль) в дихлорметане (15 мл) добавляли метилтрифторметансульфонат (676 мг, 4,12 ммоль). Реакционную смесь перемешивали при 0°C в течение 0,5 часа. Затем растворитель удаляли при пониженном давлении, получая требуемый продукт (1,6 г, грубый выход >99%): ESI МС m/z=306 [M]+. Полученный сырой продукт использовали на следующей стадии без дополнительной очистки.
Стадия C: К раствору неочищенного продукта со стадии B (1,6 г, 3,43 ммоль) в метаноле (50 мл) добавляли цианоборгидрид натрия (260 мг, 4,12 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 0,5 часа. Растворитель удаляли при пониженном давлении. Остаток разбавляли водой и дважды экстрагировали этилацетатом. Объединенные экстракты промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали. Очистка хроматографией на колонке (силикагель, от 10% до 50% этилацетат/гексаны) давала требуемый продукт (700 мг, 66% для 2 стадий):1H ЯМР (CDCl3, 500 МГц) δ 7,51 (д, J=8,6 Гц, 1H), 7,47 (дд, J=5,6, 0,6 Гц, 1H), 7,42 (д, J=5,6 Гц, 1H), 7,13-7,09 (м, 2H), 7,06-7,04 (м, 1H), 6,99 (д, J=8,4 Гц, 1H), 6,84 (д, J=7,7 Гц, 1H), 4,90-4,89 (м, 1H), 3,95 (с, 3H), 3,81 (д, J=14,8 Гц, 1H), 3,64 (д, J=14,8 Гц, 1H), 3,09-3,06 (м, 1H), 2,60 (дд, J=11,4, 9,0 Гц, 1H), 2,45 (с, 3H); ESI МС m/z=310 [M+H]+. Полученное соединение разделяли препаративной хиральной ВЭЖХ (колонка CHIRALPAK AD, используя смесь 99:1:0,1 гептан/IPA/диэтиламин в качестве элюента), получая (+)-энантиомер [[α]25D +66,4° (c 0,1, метанол)] (323 мг, 98,6% AUC ВЭЖХ) и (-)-энантиомер [[α]25D -36,0° (c 0,1, метанол)] (346 мг, >99% AUC ВЭЖХ).
Стадия D: К раствору (+)-энантиомера со стадии C (103 мг, 0,333 ммоль) в метаноле (2 мл) добавляли фумаровую кислоту (39 мг, 0,333 ммоль). Растворитель удаляли при пониженном давлении. Остаток растирали в этилацетате и диэтиловом эфире. Полученный в результате преципитат собирали фильтрованием, промывали диэтиловым эфиром и сушили при 50°C в вакууме получая фумаратную соль (+)-4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина в виде белого твердого вещества (85 мг, 60%, >99% AUC ВЭЖХ): т.пл. 97-99°C;1H ЯМР (CD3OD, 500 МГц) δ 7,70-7,57 (м, 2H), 7,50 (д, J=5,5 Гц, 1H), 7,29-7,26 (м, 2H), 7,22-7,15 (м, 1H), 7,08 (д, J=8,3 Гц, 1H), 6,89 (д, J=7,7 Гц, 1H), 6,69 (с, 2H), 5,02-4,99 (м, 1H), 4,48 (д, J=15,0 Гц, 1H), 4,42 (д, J=15,0 Гц, 1H), 3,77 (с, 3H), 3,72-3,65 (м, 1H), 3,52-3,46 (м, 1H), 2,98 (с, 3H); ESI МС m/z = 310 [M+H]+.
Стадия E: К раствору (-)-энантиомер со стадии C (106 мг, 0,342 ммоль) в метаноле (2 мл) добавляли фумаровую кислоту (40 мг, 0,342 ммоль). Растворитель удаляли при пониженном давлении. Остаток растирали этилацетатом и диэтиловым эфиром. Полученный в результате преципитат собирали фильтрованием, промывали диэтиловым эфиром и сушили при 50°C в вакууме, получая фумаратную соль (-)-4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина в виде белого твердого вещества (70 мг, 48%, >99% AUC ВЭЖХ): т.пл. 94-96°C;1H ЯМР (CD3OD, 500 МГц) δ 7,66-7,61 (м, 2H), 7,50 (д, J=5,5 Гц, 1H), 7,26 (д, J=4,0 Гц, 2H), 7,22-7,18 (м, 1H), 7,08 (д, J=8,3 Гц, 1H), 6,89 (д, J=7,6 Гц, 1Н), 6,68 (с, 2H), 4,67 (дд, J=9,9, 6,1 Гц, 1H), 4,49 (д, J=15,1 Гц, 1H), 4,43 (д, J=15,1 Гц, 1H), 3,76 (с, 3H), 3,72-3,68 (м, 1Н), 3,53-3,46 (м, 1H), 2,98 (с, 3H); ESI МС m/z=310 [M+H]+.
Пример 83 - Получение малеатной соли (+)-4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина
Стадия A: Бис(пинаколато)дибор (217 мг, 0,854 ммоль) добавляли к смеси неочищенного продукта со стадии A примера 85 (390 мг, 0,776 ммоль) и ацетата калия (229 мг, 2,33 ммоль) в диметилсульфоксиде (5 мл). Реакционную смесь дегазировали, используя аргон. Добавляли PdCl2(dppf) (34 мг, 0,047 ммоль) и реакционную смесь перемешивали при 80°C в течение 2 часов, охлаждали, разбавляли водой и дважды экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали, получая требуемый боронат (546 мг, грубый выход >99%). ESI МС m/z=436 [M+H]+. Полученный сырой продукт использовали на следующей стадии без дополнительной очистки.
Стадия B: 3,6-Дихлорпиридазин (180 мг, 1,17 ммоль) добавляли к смеси неочищенного продукта со стадии A (546 мг, 0,776 ммоль) и карбоната натрия (2 М, 1,2 мл, 2,4 ммоль) в диметилформамиде (6 мл). Реакционную смесь дегазировали, используя аргон. Добавляли PdCl2(dppf) (34 мг, 0,047 ммоль) и реакционную смесь перемешивали при 100°C в течение 1,5 часа, охлаждали, разбавляли водой и дважды экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали. Очистка хроматографией на колонке (силикагель, от 2% до 8% MeOH/CH2Cl2) давала требуемый продукт (177 мг, 54% для 3 стадий):1H ЯМР (CDCl3, 500 МГц) δ 7,88 (д, J=1,2 Гц, 1H), 7,78 (д, J=9,0 Гц, 1H), 7,66 (дд, J=8,1, 1,7 Гц, 1H), 7,55-7,52 (м, 2H), 7,49 (д, J=5,5 Гц, 1H), 7,44 (д, J=5,5 Гц, 1H), 7,01 (д, J=8,3 Гц, 2H), 4,96- 4,92 (м, 1H), 3,97 (с, 3H), 3,92 (д, J=15,0 Гц, 1H), 3,73 (д, J=15,0 Гц, 1H), 3,14- 3,10 (м, 1H), 2,65 (дд, J=11,4, 9,0 Гц, 1H), 2,49 (с, 3H); ESI МС m/z=422 [M+H]+.
Стадия C: К раствору продукта со стадии B (177 мг, 0,419 ммоль) в этаноле (10 мл) добавляли гидразин (210 мг, 4,19 ммоль) и 10% Pd/C (80 мг). Реакционную смесь нагревали при кипячении с обратным холодильником в течение 4 часов. Затем смесь фильтровали через целит и слой целита промывали метанолом. Фильтрат концентрировали и очищали хроматографией на колонке (силикагель, от 3% до 5% MeOH/CH2Cl2), получая требуемый продукт (100 мг, 62%):1H ЯМР (CDCl3, 300 МГц) δ 9,13 (дд, J=4,9, 1,5 Гц, 1H), 7,94 (д, J=1,5 Гц, 1H), 7,82 (дд, J=8,6, 1,6 Гц, 1H), 7,70 (дд, J=8,1, 1,8 Гц, 1H), 7,55-7,48 (м, 3H), 7,44 (д, J=5,5 Гц, 1H), 7,03-7,00 (м, 2H), 4,95 (дд, J=8,1, 6,5 Гц, 1H), 3,97 (с, 3Н), 3,94 (д, J=15,1 Гц, 1Н), 3,73 (д, J=15,1 Гц, 1H), 3,16-3,09 (м, 1H), 2,65 (дд, J=11,4, 9,0 Гц, 1Н), 2,49 (с, 3H); ESI-МС m/z=388 [M+Н]+.
Стадия D: К раствору продукта со стадии C (92 мг, 0,237 ммоль) в метаноле (3 мл) добавляли малеиновую кислоту (27 мг, 0,237 ммоль). Растворитель удаляли при пониженном давлении, получая малеатную соль (+)-4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина в виде не совсем белого твердого вещества (119 мг, 96%, >99% AUC ВЭЖХ):1H ЯМР (CD3OD, 500 МГц) δ 9,19 (дд, J=4,8, 1,3 Гц, 1H), 8,17 (дд, J=8,7, 1,4 Гц, 1H), 8,09 (с, 1H), 7,94 (дд, J=8,2, 1,4 Гц, 1H), 7,80 (дд, J=8,7, 4,9 Гц, 1H), 7,70 (д, J=8,4 Гц, 1H), 7,65 (д, J=5,6 Гц, 1H), 7,53 (д, J=5,6 Гц, 1H), 7,16-7,11 (м, 2H), 6,23 (с, 2H), 5,11-5,07 (м, 1Н), 4,72 (д, J=15,2 Гц, 1H), 4,67 (д, J=15,2 Гц, 1H), 3,87-3,82 (м, 1Н), 3,81 (с, 3Н), 3,73-3,68 (м, 1H), 3,12 (с, 3H); ESI МС m/z=388 [M+H]+; элементный анализ, вычислено для C23H21N3OS·C4H4O4·H2O: C, 62,17; Н, 5,22; N, 8,06. Найдено: C, 62,43; Н, 5,09; N, 7,71.
Пример 84 - Получение малеатной соли (+)-4-(4-метоксибензотиофен-5-ил)-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина
Стадия A: 3-Хлор-6-метилпиридазин (230 мг, 1,78 ммоль) добавляли к смеси неочищенного продукта со стадии A примера 83 (545 г, 1,19 ммоль) и карбоната натрия (2 М, 1,8 мл, 3,6 ммоль) в диметилформамиде (10 мл). Реакционную смесь дегазировали, используя аргон. Добавляли PdCl2(dppf) (52 мг, 0,071 ммоль) и реакционную смесь перемешивали при 100°C в течение 3 часов, охлаждали, разбавляли водой и дважды экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали. Очистка хроматографией на колонке (силикагель, от 3% до 6% MeOH/CH2Cl2) давала требуемый продукт (300 мг, 63% для 3 стадий):1H ЯМР (CDCl3, 500 МГц) δ 7,90 (д, J=1,3 Гц, 1H), 7,72-7,67 (м, 2H), 7,53 (д, J=8,4 Гц, 1H), 7,48 (д, J=5,5 Гц, 1H), 7,44 (д, J=5,5 Гц, 1Н), 7,35 (д, J=8,7 Гц, 1Н), 7,03-6,98 (м, 2H), 4,96-4,93 (м, 1Н), 3,97 (с, 3H), 3,92 (д, J=14,9 Гц, 1H), 3,73 (д, J=14,9 Гц, 1H), 3,14-3,10 (м, 1Н), 2,74 (с, 3H), 2,65 (дд, J=11,4, 9,0 Гц, 1H), 2,49 (с, 3H); ESI МС m/z=402 [M+Н]+.
Стадия B: К раствору продукта со стадии A (102 мг, 0,254 ммоль) в метаноле (3 мл) добавляли малеиновую кислоту (29,5 мг, 0,254 ммоль). Растворитель удаляли при пониженном давлении, получая малеатную соль (+)-4-(4-метоксибензотиофен-5-ил)-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина в виде светло-коричневого твердого вещества (122 мг, 93%, 95,3% AUC ВЭЖХ):1H ЯМР (CD3OD, 500 МГц) δ 8,07 (с, 1H), 8,06 (д, J=8,8 Гц, 1H), 7,91 (дд, J=8,2, 1,6 Гц, 1H), 7,71-7,64 (м, 3H), 7,53 (д, J=5,5 Гц, 1H), 7,16 (д, J=8,3 Гц, 1H), 7,11 (д, J=8,2 Гц, 1H), 6,24 (с, 2H), 5,09-5,08 (м, 1H), 4,75-4,72 (м, 2H), 3,91-3,87 (м, 1H), 3,80 (с, 3H), 3,79-3,75 (м, 1H), 3,15 (с, 3H), 2,72 (с, 3H); ESI МС m/z=402 [M+Н]+.
Пример 85 - Получение малеатной соли (+)-4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина
Стадия A: Ангидрид трифторметансульфоновой кислоты (416 мг, 1,74 ммоль) добавляли к раствору (+)-энантиомера со стадии F примера 87 (434 мг, 1,33 ммоль) и триэтиламина (202 мг, 2,0 ммоль) в дихлорметане (15 мл) при 0°C. Реакционную смесь перемешивали при 0°C в течение 0,5 часа и затем разбавляли водой и дважды экстрагировали дихлорметаном. Объединенные органические экстракты промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали, получая требуемый трифторметансульфонат (670 мг, грубый выход >99%): ESI-МС m/z=458 [M+H]+. Полученный неочищенный продукт использовали на следующей стадии без дополнительной очистки.
Стадия B: К раствору неочищенного продукта со стадии A (280 мг, 0,557 ммоль) в толуоле (20 мл) добавляли морфолин (97 мг, 1,11 ммоль), карбонат цезия (544 мг, 1,67 ммоль), X-Phos (159 мг, 0,334 ммоль), и Pd(OAc)2 (19 мг, 0,084 ммоль). Реакционную смесь дегазировали, используя аргон, нагревали при кипячении с обратным холодильником в течение 15 часов и затем давали возможность остыть до комнатной температуры. Смесь разбавляли этилацетатом и фильтровали через целит. Фильтрат промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали. Очистка хроматографией на колонке (силикагель, от 2% до 4% MeOH/CH2Cl2) давала требуемый продукт (96 мг, 44% для 2 стадий): [[α]25D +55,0° (c 0,1, метанол);1H ЯМР (CDCl3, 500 МГц) δ 7,51 (д, J=8,4 Гц, 1H), 7,47 (д, J=5,5 Гц, 1H), 7,42 (д, J=5,5 Гц, 1H), 7,00 (д, J=8,4 Гц, 1H), 6,74 (д, J=8,4 Гц, 1H), 6,66-6,63 (м, 2H), 4,83-4,80 (м, 1H), 3,96 (с, 3H), 3,84 (т, J=4,8 Гц, 4H), 3,76 (д, J=14,8 Гц, 1H), 3,61 (д, J=14,8 Гц, 1H), 3,11 (т, J=4,8 Гц, 4H), 3,06 (дд, J=11,2, 5,6 Гц, 1H), 2,57 (дд, J=11,2, 9,0 Гц, 1Н), 2,44 (с, 3H); ESI-МС m/z=395 [M+H]+.
Стадия C: К раствору продукта со стадии B (86 мг, 0,218 ммоль) в метаноле (2 мл) добавляли малеиновую кислоту (25 мг, 0,218 ммоль). Растворитель удаляли при пониженном давлении, получая малеатную соль (+)-4-(4-метоксибензотиофен-5-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолин в виде не совсем белого твердого вещества (111 мг, 97%, 98,7% AUC ВЭЖХ):1H ЯМР (CD3OD, 500 МГц) δ 7,67-7,62 (м, 2H), 7,51-7,50 (м, 1H), 7,08 (д, J=8,2 Гц, 1H), 6,89-6,87 (м, 1H), 6,83-6,78 (м, 2H), 6,25 (с, 2H), 4,91-4,89 (м, 1H), 4,56-4,53 (м, 2H), 3,82-3,79 (м, 8H), 3,58-3,56 (м, 1H), 3,14-3,12 (м, 4H), 3,09 (с, 3H); ESI МС m/z=395 [M+H]+; элементный анализ, вычислено для C23H26N2O2S·C4H4O4·0,75H2O: C, 61,87; Н, 6,06; N, 5,34. Найдено: C, 62,01;H, 5,83; N, 4,94.
Пример 86 - Получение малеатной соли (+/-)-4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-5-ола
Стадия A: К раствору 5-гидроксирегиоизомера со стадии E примера 87 (96 мг, 0,295 ммоль) в метаноле (3 мл) добавляли малеиновую кислоту (34,6 мг, 0,295 ммоль). Растворитель удаляли при пониженном давлении, получая малеатную соль (+/-)-4-(4-метоксибензотиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-5-ола соли в виде не совсем белого твердого вещества (130 мг, 99%, >99% AUC ВЭЖХ):1H ЯМР (CD3OD, 500 МГц) δ 7,57-7,47 (м, 3H), 7,22-7,19 (м, 1H), 6,85-6,74 (м, 3H), 6,21 (с, 2H), 5,01 (т, J=6,8 Гц, 1Н), 4,57 (д, J=14,8 Гц, 1H), 4,43 (д, J=14,8 Гц, 1H), 3,87 (с, 3H), 3,86 (ушир.с, 1H), 3,53 (ушир.с, 1Н), 3,01 (с, 3H); ESI МС m/z=326 [M+H]+.
Пример 87 - Получение малеатной соли (-)-4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ола и малеатной соли (+)-4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ола
Стадия A: 5-Бром-4-метоксибензотиофен получали, следуя способам, описанным в литературе (Chenard et al., J. Org. Chem., 48: 4312-4317 (1983), публикация включена в данное описание в виде ссылки в полном объеме). Смесь 5-бром-4-метоксибензотиофена (9,39 г, 38,6 ммоль), н-бутилвинилового эфира (19,3 г, 193 ммоль), карбоната калия (6,4 г, 46,3 ммоль), dppp (1,05 г, 2,55 ммоль) и Pd(OAc)2 (260 мг, 1,16 ммоль) в ДМФА (100 мл) и воде (10 мл) нагревали при кипячении с обратным холодильником в течение 6 часов. Смеси давали возможность остыть до комнатной температуры и затем добавляли 2 N HCl (150 мл). Полученную в результате смесь перемешивали при комнатной температуре в течение 0,5 часа и три раза экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным раствором бикарбоната натрия и насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали. Очистка хроматографией на колонке (силикагель, от 5% до 15% этилацетат/гексаны) давала требуемый продукт (7,06 г, 88%):1H ЯМР (CDCl3, 500 МГц) δ 7,71 (д, J=8,5 Гц, 1H), 7,64 (дд, J=8,5, 0,5 Гц, 1H), 7,54 (дд, J=5,5, 0,5 Гц, 1H), 7,47 (д, J=5,5 Гц, 1H), 4,04 (с, 3H), 2,73 (с, 3H).
Стадия B: К смеси продукта со стадии A (7,06 г, 34,2 ммоль) в этилацетате (75 мл) и хлороформе (60 мл) добавляли бромид меди (II) (15,45 г, 68,4 ммоль). Реакционную смесь нагревали при кипячении с обратным холодильником в течение 4 часов. Смеси давали возможность остыть до комнатной температуры и затем фильтровали через целит. Фильтрат концентрировали и очищали хроматографией на колонке (силикагель, от 5% до 20% этилацетат/гексаны), получая требуемый продукт (6,37 г, 65%):1H ЯМР (CDCl3, 500 МГц) δ 7,74 (д, J=8,5 Гц, 1H), 7,67 (дд, J=8,5, 0,5 Гц, 1H), 7,56 (дд, J=5,5, 0,5 Гц, 1H), 7,51 (д, J=5,5 Гц, 1Н), 4,68 (с, 2H), 4,11 (с, 3H).
Стадия C: К раствору продукта со стадии B (4,37 г, 15,3 ммоль) в дихлорметане (60 мл) добавляли диизопропилэтиламин (2,97 г, 23,0 ммоль) и N-(3-гидроксибензил)метиламин (2,52 г, 18,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов и затем разбавляли дихлорметаном (100 мл). Смесь промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали. Очистка хроматографией на колонке (силикагель, от 1% до 4% метанол/дихлорметан) давала требуемый продукт (3,96 г, 81%):1H ЯМР (CDCl3, 300 МГц) δ 7,62 (с, 2H), 7,48 (дд, J=10,0, 5,6 Гц, 2H), 7,15 (т, J=7,7 Гц, 1H), 6,89-6,84 (м, 2H), 6,75-6,72 (м, 1H), 3,93 (с, 2H), 3,92 (с, 3H), 3,68 (с, 2H), 2,43 (с, 3H), ESl-МС m/z=342 [M+H]+.
Стадия D: К охлажденному на льду раствору продукта со стадии C (3,96 г, 11,6 ммоль) в метаноле (60 мл) добавляли боргидрид натрия (910 мг, 24 ммоль). Реакционную смесь перемешивали при 0°C в течение 1 часа. Растворитель удаляли при пониженном давлении. Остаток распределяли между дихлорметаном и водой. Органические экстракты промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали, получая требуемый продукт (3,90 г, грубый выход 98%): ESI-МС m/z=344 [M+H]+. Полученный неочищенный продукт использовали на следующей стадии без дополнительной очистки.
Стадия E: К раствору продукта со стадии D (3,60 г, 10,5 ммоль) в дихлорметане (300 мл) по каплям добавляли метансульфоновую кислоту (7,41 г, 77,1 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 20 минут. Затем смесь охлаждали на бане со льдом и добавляли насыщенный раствор бикарбоната натрия (250 мл). Органический слой отделяли и водный слой экстрагировали дихлорметаном. Объединенные экстракты промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали. Очистка хроматографией на колонке (силикагель, от 1% до 6% метанол/дихлорметан) давала 5-гидроксирегиоизомер (1,56 г, 46%) и 7-гидроксирегиоизомер (1,09 г, 32%). 5-Гидроксирегиоизомер:1H ЯМР (CDCl3, 500 МГц) δ 7,50 (д, J=8,4 Гц, 1H), 7,46 (д, J=5,5 Гц, 1H), 7,42-7,40 (м, 2H), 7,05 (т, J=7,8 Гц, 1H), 6,70 (д, J=7,6 Гц, 1H), 6,58 (д, J=8,0 Гц, 1H), 5,60 (ушир.с, 1H), 4,63 (т, J=4,8 Гц, 1H), 4,05 (с, 3H), 3,84 (д, J=14,9 Гц, 1H), 3,46 (д, J=14,9 Гц, 1H), 2,91 (дд, J=11,5, 5,5 Гц, 1H), 2,78 (дд, J=11,5, 4,3 Гц, 1H), 2,38 (с, 3H); ESI МС m/z=326 [M+H]+. 7-Гидроксирегиоизомер:1H ЯМР (CDCl3, 500 МГц) δ 7,52 (д, J=8,4 Гц, 1H), 7,45 (д, J=5,5 Гц, 1H), 7,41 (д, J=5,5 Гц, 1H), 6,99 (д, J=8,4 Гц, 1H), 6,66 (д, J=8,4 Гц, 1H), 6,52 (дд, J=8,4, 2,5 Гц, 1H), 6,43 (ушир.с, 1H), 4,81 (дд, J=9,1, 8,1 Гц, 1H), 3,89 (с, 3H), 3,68 (д, J=14,9 Гц, 1H), 3,53 (д, J=14,9 Гц, 1H), 3,09 (дд, J=11,4, 5,9 Гц, 1H), 2,59 (дд, J=11,3, 9,7 Гц, 1H), 2,44 (с, 3H); ESI-МС m/z=326 [M+H]+.
Стадия F: 7-гидроксирегиоизомер со стадии E (1,03 г) разделяли препаративной хиральной ВЭЖХ (колонка CHIRALCEL OD, с использованием смеси 80:20:0,1 гептан/IPA/диэтиламин в качестве элюента), получая (-)-энантиомер [[α]25D -48,1° (c 0,104, метанол)] (493 мг, >99% AUC ВЭЖХ) и (+)-энантиомер [[α]25D +58,3° (c 0,108, метанол)] (485 мг, >99% AUC ВЭЖХ).
Стадия G: К раствору (-)-энантиомера со стадии F (50 мг, 0,154 ммоль) в метаноле (2 мл) добавляли малеиновую кислоту (18 мг, 0,154 ммоль). Растворитель удаляли при пониженном давлении, получая малеатную соль (-)- 4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ола в виде белого твердого вещества (65 мг, 95%, 97,5% AUC ВЭЖХ):1H ЯМР (CD3OD, 500 МГц) δ 7,67-7,61 (м, 2H), 7,50 (д, J=5,5 Гц, 1H), 7,08 (д, J=8,1 Гц, 1H), 6,73-6,68 (м, 3H), 6,25 (с, 2H), 4,92-4,90 (м, 1H), 4,53-4,51 (м, 2H), 3,80-3,78 (м, 1H), 3,79 (с, 3H), 3,63-3,61 (м, 1H), 3,08 (с, 3H); ESI-МС m/z=326 [M+H]+.
Стадия H: К раствору (+)-энантиомера со стадии F (50 мг, 0,154 ммоль) в метаноле (2 мл) добавляли малеиновую кислоту (18 мг, 0,154 ммоль). Растворитель удаляли при пониженном давлении, получая малеатную соль (+)-4-(4-метоксибензотиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ола в виде белого твердого вещества (65 мг, 95%, >99% AUC ВЭЖХ):1H ЯМР (CD3OD, 500 МГц) δ 7,67-7,61 (м, 2H), 7,50 (д, J=5,5 Гц, 1H), 7,08 (д, J=8,2 Гц, 1H), 6,74-6,68 (м, 3H), 6,24 (с, 2H), 4,90-4,86 (м, 1H), 4,53-4,51 (м, 2H), 3,79 (с, 3H), 3,79-3,76 (м, 1H), 3,65-3,62 (м, 1H), 3,08 (с, 3H); ESI МС m/z=326 [M+H]+.
Пример 88 - Получение фумаратной соли (+)-5-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)бензо[b]тиофен-4-ола и фумаратной соли (-)-5-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)бензо[b]тиофен-4-ола
Стадия A: Смесь (+)-энантиомера со стадии C примера 82 (220 мг, 0,711 ммоль) в 48% HBr (5 мл) и уксусной кислоте (1 мл) нагревали при кипячении с обратным холодильником в течение 2 часов. Растворитель и избыток HBr удаляли при пониженном давлении. Остаток распределяли между этилацетатом и насыщенным раствором бикарбоната натрия. Водный слой экстрагировали этилацетатом. Объединенные экстракты промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали. Очистка хроматографией на колонке (силикагель, от 20% до 50% этилацетат/гексаны) давала требуемый фенол (100 мг, 48%, >99% AUC ВЭЖХ): [[α]25D +220,0° (c 0,12, метанол)];1H ЯМР (CDCl3, 500 МГц) δ 13,84 (с, 1H), 7,40 (д, J=5,5 Гц, 1H), 7,29 (д, J=8,1 Гц, 1H), 7,23 (д, J=8,1 Гц, 1H), 7,19 (д, J=5,5 Гц, 1H), 7,13-7,04 (м, 4H), 4,16 (д, J=5,1 Гц, 1H), 4,11 (дд, J=14,7, 1,2 Гц, 1H), 3,59 (д, J=14,7 Гц, 1H), 3,31 (д, J=11,9 Гц, 1H), 2,97 (дд, J=11,9, 5,3 Гц, 1H), 2,61 (с, 3H); ESI-МС m/z=296[M+Н]+.
Стадия B: К раствору продукта со стадии A (98 мг, 0,33 ммоль) в метаноле (3 мл) добавляли фумаровую кислоту (39 мг, 0,33 ммоль). Растворитель удаляли при пониженном давлении. Остаток растирали в этилацетате и диэтиловом эфире. Полученный в результате преципитат собирали фильтрованием, промывали диэтиловым эфиром и сушили при 50°C в вакууме, получая фумаратную соль (+)-5-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)бензо[b]тиофен-4-ола в виде белого твердого вещества (80 мг, 71%, >99% AUC ВЭЖХ): т.пл. 213-215°C;1H ЯМР (CD3OD, 500 МГц) δ 7,39 (д, J=5,3 Гц, 1H), 7,34-7,30 (м, 2H), 7,17-7,08 (м, 4H), 7,03 (д, J=7,7 Гц, 1H), 6,70 (с, 0,8H), 4,52-4,50 (м, 1H), 4,23 (д, J=15,0 Гц, 1H), 3,90 (д, J=11,3 Гц, 1H), 3,38-3,26 (м, 2H), 2,73 (с, 3H), ESI-МС m/z=296 [M+Н]+.
Стадия C: Смесь (-)-энантиомера со стадии D примера 82 (240 мг, 0,775 ммоль) в 48% HBr (5 мл) и уксусной кислоте (1 мл) нагревали при кипячении с обратным холодильником в течение 2 часов. Растворитель и избыток HBr удаляли при пониженном давлении. Остаток распределяли между этилацетатом и насыщенным раствором бикарбоната натрия. Водный слой экстрагировали этилацетатом. Объединенные экстракты промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали. Очистка хроматографией на колонке (силикагель, от 10% до 50% этилацетат/гексаны) давала требуемый фенольный продукт (110 мг, 48%, >99% AUC ВЭЖХ): [[α]25D -211,3° (c 0,12, метанол)];1H ЯМР (CDCl3, 500 МГц) δ 13,84 (с, 1H), 7,40 (д, J=5,4 Гц, 1H), 7,30 (д, J=8,1 Гц, 1H), 7,23 (д, J=8,1 Гц, 1H), 7,19 (д, J=5,4 Гц, 1H), 7,13-7,04 (м, 4H), 4,16 (д, J=4,8 Гц, 1H), 4,11 (д, J=14,7 Гц, 1H), 3,59 (д, J=14,7 Гц, 1H), 3,31 (д, J=11,9 Гц, 1H), 2,97 (дд, J=11,9, 5,3 Гц, 1H), 2,61 (с, 3H), ESI МС m/z=296 [M+Н]+.
Стадия D: К раствору продукта со стадии C (106 мг, 0,359 ммоль) в метаноле (3 мл) добавляли фумаровую кислоту (42 мг, 0,359 ммоль). Растворитель удаляли при пониженном давлении. Остаток растирали в этилацетате и диэтиловом эфире. Полученный в результате преципитат собирали фильтрованием, промывали диэтиловым эфиром и сушили при 50°C в вакууме, получая фумаратную соль (-)-5-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)бензо[b]тиофен-4-ола в виде белого твердого вещества (95 мг, 75%, >99% AUC ВЭЖХ): т.пл. 214-216°C;1H ЯМР (CD3OD, 500 МГц) δ 7,40 (д, J=5,4 Гц, 1H), 7,35-7,31 (м, 2H), 7,16-7,09 (м, 4H), 7,03 (д, J=7,7 Гц, 1H), 6,70 (с, 1Н), 4,55-4,54 (м, 1H), 4,25 (д, J=14,9 Гц, 1H), 3,95-3,94 (м, 1H), 3,41-3,26 (м, 2H), 2,76 (с, 3H), ESI-МС m/z=296 [M+H]+.
Пример 89 - Получение фумаратной соли (+)-5-(2-этил-1,2,3,4-тетрагидроизохинолин-4-ил)бензо[b]тиофен-4-ола и фумаратной соли (-)-5-(2-этил-1,2,3,4-тетрагидроизохинолин-4-ил)бензо[b]тиофен-4-ола
Стадия A: Смесь (+)-энантиомера со стадии B примера 90 (225 мг, 0,695 ммоль) в 48% HBr (5 мл) и уксусной кислоте (1 мл) нагревали при кипячении с обратным холодильником в течение 2 часов. Растворитель и избыток HBr удаляли при пониженном давлении. Остаток распределяли между этилацетатом и насыщенным раствором бикарбоната натрия. Водный слой экстрагировали этилацетатом. Объединенные экстракты промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали. Очистка хроматографией на колонке (силикагель, от 0% до 2% метанол/дихлорметан) давала требуемый фенол (130 мг, 60%, >99% AUC ВЭЖХ): [[α]25D +250,8° (c 0,13, метанол)];1H ЯМР (CDCl3, 300 МГц) δ 13,84 (с, 1H), 7,41 (д, J=5,5 Гц, 1H), 7,29 (д, J=8,1 Гц, 1H), 7,25-7,17 (м, 2H), 7,11-7,06 (м, 4H), 4,22-4,16 (м, 2H), 3,57 (д, J=14,6 Гц, 1Н), 3,40 (д, J=11,7 Гц, 1H), 2,93 (дд, J=11,9, 5,2 Гц, 1H), 2,79 (кв., J=7,3 Гц, 2H), 1,29 (т, J=7,3 Гц, 3H); ESI МС m/z=310 [M+Н]+.
Стадия B: К раствору продукта со стадии A (124 мг, 0,40 ммоль) в метаноле (3 мл) добавляли фумаровую кислоту (47 мг, 0,40 ммоль). Растворитель удаляли при пониженном давлении. Остаток растирали в этилацетате и диэтиловом эфире. Полученный в результате преципитат собирали фильтрованием, промывали диэтиловым эфиром и сушили при 50°C в вакууме, получая фумаратную соль (+)-5-(2-этил-1,2,3,4-тетрагидроизохинолин-4-ил)бензо[b]тиофен-4-ола в виде белого твердого вещества (120 мг, 70%, >99% AUC ВЭЖХ): т.пл. 158-159°C;1H ЯМР (CD3OD, 500 МГц) δ 7,45 (д, J=5,4 Гц, 1H), 7,39-7,34 (м, 2H), 7,21-7,09 (м, 4H), 7,00-6,97 (м, 1Н), 6,70 (с, 2H), 4,74-4,73 (м, 1H), 4,39 (д, J=15,0 Гц, 1H), 4,19-4,17 (м, 1H), 3,53-3,46 (м, 2H), 3,14-3,12 (м, 2H), 1,37 (т, J=7,2 Гц, 3H); ESI-МС m/z=310 [M+H]+.
Стадия C: Смесь (-)-энантиомера со стадии B примера 90 (230 мг, 0,711 ммоль) в 48% HBr (5 мл) и уксусной кислоте (1 мл) нагревали при кипячении с обратным холодильником в течение 2 часов. Растворитель и избыток HBr удаляли при пониженном давлении. Остаток распределяли между этилацетатом и насыщенным раствором бикарбоната натрия. Водный слой экстрагировали этилацетатом. Объединенные экстракты промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали. Очистка хроматографией на колонке (силикагель, от 0% до 4% метанол/дихлорметан) давала требуемый фенол (115 мг, 52%, >99% AUC ВЭЖХ): [[α]25D -266,4° (c 0,13, метанол)],1H ЯМР (CDCl3, 300 МГц) δ 13,84 (с, 1H), 7,41 (д, J=5,5 Гц, 1H), 7,29 (д, J=8,1 Гц, 1H), 7,25-7,17 (м, 2H), 7,11-7,06 (м, 4H), 4,22-4,16 (м, 2H), 3,57 (д, J=14,6 Гц, 1H), 3,40 (д, J=11,7 Гц, 1H), 2,93 (дд, J=11,9, 5,2 Гц, 1H), 2,79 (кв., J=7,3 Гц, 2H), 1,29 (т, J=7,3 Гц, 3H); ESI-МС m/z=310 [M+H]+.
Стадия D: К раствору продукта со стадии C (109 мг, 0,352 ммоль) в метаноле (3 мл) добавляли фумаровую кислоту (41 мг, 0,352 ммоль). Растворитель удаляли при пониженном давлении. Остаток растирали в этилацетате и диэтиловом эфире. Полученный в результате преципитат собирали фильтрованием, промывали диэтиловым эфиром и сушили при 50°C в вакууме, получая фумаратную соль (-)-5-(2-этил-1,2,3,4-тетрагидроизохинолин-4-ил)бензо[b]тиофен-4-ола в виде белого твердого вещества (60 мг, 41%, >99% AUC ВЭЖХ): т.пл. 155-157°C;1H ЯМР (CD3OD, 500 МГц) δ 7,46 (д, J=5,5 Гц, 1H), 7,40-7,35 (м, 2H), 7,21-7,10 (м, 4H), 6,99-6,98 (м, 1H), 6,70 (с, 1,8H), 4,76-4,75 (м, 1H), 4,39 (д, J=14,9 Гц, 1H), 4,21-4,20 (м, 1H), 3,53-3,44 (м, 2H), 3,15-3,13 (м, 2H), 1,37 (т, J=7,1 Гц, 3H); ESI МС m/z=310 [M+H]+.
Пример 90 - Получение фумаратной соли (+)-4-(4-метоксибензо[b]тиофен-5-ил)-2-этил-1,2,3,4-тетрагидроизохинолина и фумаратной соли (-)-4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: К охлажденному на льду раствору продукта со стадии A примера 82 (1,0 г, 3,43 ммоль) в дихлорметане (15 мл) добавляли этилтрифторметансульфонат (672 мг, 3,77 ммоль). Реакционную смесь перемешивали при 0°C в течение 0,5 часа. Затем растворитель удаляли при пониженном давлении, получая требуемый продукт (1,7 г, грубый выход >99%): ESI-МС m/z=320 [M]+. Полученный неочищенный продукт использовали на следующей стадии без дополнительной очистки.
Стадия B: К раствору неочищенного продукта со стадии A (1,7 г, 3,43 ммоль) в метаноле (50 мл) добавляли цианоборгидрид натрия (260 мг, 4,12 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель удаляли при пониженном давлении. Остаток разбавляли водой и дважды экстрагировали этилацетатом. Объединенные экстракты промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали. Очистка хроматографией на колонке (силикагель, 30% этилацетат/гексаны) давала требуемый продукт (710 мг, 64% для 2 стадий):1H ЯМР (CDCl3, 500 МГц) δ 7,52 (д, J=8,4 Гц, 1H), 7,47 (д, J=5,5 Гц, 1H), 7,42 (д, J=5,5 Гц, 1H), 7,13-7,10 (м, 2H), 7,05-7,01 (м, 2H), 6,83 (д, J=7,7 Гц, 1H), 4,91-4,88 (м, 1H), 3,94 (с, 3H), 3,93 (д, J=14,7 Гц, 1H), 3,64 (д, J=14,7 Гц, 1H), 3,20-3,16 (м, 1H), 2,64-2,56 (м, 3H), 1,17 (т, J=7,2 Гц, 3H), ESI МС m/z=324 [M+H]+.
Полученное вещество разделяли препаративной хиральной ВЭЖХ (колонка CHIRALPAK AD, с использованием смеси 98:2:0,1 гептан/IPA/диэтиламин в качестве элюента), получая (+)-энантиомер [[α]25D +51,2° (c 0,1, метанол)] (331 мг, >99% AUC ВЭЖХ) и (-)-энантиомер [[α]25D -88,5° (c 0,1, метанол)] (342 мг, >99% AUC ВЭЖХ).
Стадия C: К раствору (+)-энантиомера со стадии B (106 мг, 0,328 ммоль) в метаноле (2 мл) добавляли фумаровую кислоту (38 мг, 0,328 ммоль). Растворитель удаляли при пониженном давлении. Остаток растирали в этилацетате и диэтиловом эфире. Полученный в результате преципитат собирали фильтрованием, промывали диэтиловым эфиром и сушили при 50°C в вакууме, получая фумаратную соль (+)-4-(4-метоксибензо[b]тиофен-5-ил)-2-этил-1,2,3,4-тетрагидроизохинолина в виде не совсем белого твердого вещества (94 мг, 65%, 98,6% AUC ВЭЖХ): т.пл. 91-92°C;1H ЯМР (CD3OD, 500 МГц) δ 7,66 (дд, J=8,2, 0,5 Гц, 1H), 7,62 (д, J=5,6 Гц, 1Н), 7,51 (дд, J=5,6, 0,5 Гц, 1Н), 7,31-7,26 (м, 2H), 7,23-7,19 (м, 1H), 7,10 (д, J=8,3 Гц, 1H), 6,90 (д, J=7,7 Гц, 1H), 6,68 (с, 2H), 5,00 (дд, J=11,2, 6,3 Гц, 1H), 4,57 (д, J=15,1 Гц, 1H), 4,43 (д, J=15,1 Гц, 1H), 3,77 (с, 3H), 3,77-3,74 (м, 1H), 3,51-3,46 (м, 1H), 3,32-3,28 (м, 2H), 1,42 (т, J=7,3 Гц, 3H); ESI МС m/z=324 [M+H]+.
Стадия D: К раствору (-)-энантиомера со стадии B (112 мг, 0,346 ммоль) в метаноле (2 мл) добавляли фумаровую кислоту (40 мг, 0,346 ммоль). Растворитель удаляли при пониженном давлении. Остаток растирали в этилацетате и диэтиловом эфире. Полученный в результате преципитат собирали фильтрованием, промывали диэтиловым эфиром и сушили при 50°C в вакууме, получая фумаратную соль (-)-4-4-(метоксибензо[b]тиофен-5-ил)-2-этил-1,2,3,4-тетрагидроизохинолина в виде не совсем белого твердого вещества (70 мг, 46%, 98,8% AUC ВЭЖХ): т.пл. 88-90°C;1H ЯМР (CD3OD, 500 МГц) δ 7,66 (д, J=8,4 Гц, 1H), 7,62 (д, J=5,5 Гц, 1H), 7,51 (д, J=5,5 Гц, 1Н), 7,31-7,26 (м, 2H), 7,23-7,19 (м, 1H), 7,10 (д, J=8,3 Гц, 1H), 6,90 (д, J=7,7 Гц, 1Н), 6,68 (с, 2H), 5,01 (дд, J=11,2, 6,3 Гц, 1H), 4,59 (д, J=15,2 Гц, 1H), 4,45 (д, J=15,5 Гц, 1Н), 3,79-3,76 (м, 1H), 3,76 (с, 3H), 3,53-3,48 (м, 1H), 3,34-3,30 (м, 2H), 1,43 (т, J=7,3 Гц, 3H); ESI-МС m/z=324 [M+H]+.
Пример 91 - Получение фумаратной соли (+)-5-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)бензо[b]тиофен-4-карбонитрила и фумаратной соли (-)-5-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)бензо[b]тиофен-4-карбонитрила
Стадия A: Рацемический продукт со стадии C примера 82 (0,063 г, 2,0 ммоль) растворяли в бромистоводородной кислоте (25 мл) и кипятили с обратным холодильником в течение 3,5 часов. Раствор концентрировали в вакууме и перерастворяли в метаноле и концентрировали. Твердое вещество обрабатывали насыщенным раствором бикарбоната натрия и экстрагировали метиленхлоридом. Экстракт промывали насыщенным раствором соли, затем сушили над сульфатом натрия, фильтровали и упаривали до не совсем белого твердого вещества (0,58 г, 96%). ESI-МС m/z 296 [M+H]+.
Стадия B: Продукт со стадии A (0,57 г, 0,002 ммоль) растворяли в метиленхлориде. Смесь охлаждали на бане со льдом и добавляли триэтиламин (0,41 мл) и ангидрид трифторметансульфоновой кислоты (0,40 мл) и перемешивали в течение 3 часов. К смеси добавляли насыщенный раствор хлорида натрия и дважды экстрагировали метиленхлоридом. Объединенный органический экстракт сушили над сульфатом натрия, фильтровали и концентрировали до получения желтого твердого вещества (0,84 г, 99%); ESI-МС m/z 428 [M+H]+.
Стадия C: Продукт со стадии B (0,84 г, 2,0 ммоль) и цианид цинка (0,46 г, 40 ммоль) в N,N-диметилформамиде дегазировали, используя аргон. Добавляли тетракис(трифенилфосфин)палладий(0) (0,23 г, 0,2 ммоль), трис(дибензилиденацетон)дипалладий(0) (0,72 г, 0,79 ммоль), и 1,1'-бис(дифенилфосфино)ферроцен (0,17 г, 0,31 ммоль) и затем реакционную смесь нагревали до 100°C в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом.
Смесь сначала промывали насыщенным раствором бикарбоната натрия, затем насыщенным раствором соли и экстрагировали этилацетатом. Органический экстракт сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Масло очищали хроматографией на колонке (силикагель, 20% этилацетат/гексаны), получая требуемый продукт в виде желтого масла (0,09 г, 15%):1H ЯМР (500 МГц, CDCl3) δ 7,90 (д, J=8,5 Гц, 1Н), 7,68 (д, J=5,5 Гц, 1Н), 7,63-7,62 (м, 1Н), 7,20-7,17 (м, 2H), 7,14 (д, J=7,0 Гц, 1H), 7,11-7,08 (м, 1H), 6,87 (д, J=7,7 Гц, 1H), 4,86 (т, J=5,7 Гц, 1H), 3,80 (д, J=14,9 Гц, 1H), 3,64 (д, J=15,0 Гц, 1H), 3,06 (дд, J=11,6, 5,4 Гц, 1H), 2,78 (дд, J=11,6, 6,2 Гц, 1H), 2,42 (с, 3H).
Полученное соединение разделяли препаративной хиральной ВЭЖХ (колонка CHIRALPAK AD, с использованием смеси 90% гептан/10% изопропиловый спирт/0,1% диэтиламин), получая (+)-энантиомер [[α]25D +68,6° (c = 0,08, метанол) и (-)-энантиомер [[α]25D -75,0° (c = 0,05, метанол). (+)-Энантиомер (30 мг, 99 ммоль) превращали в соль фумаровой кислоты растворением свободного основания в минимальном количестве этанола, добавлением одного эквивалента фумаровой кислоты в достаточном количестве метанола, чтобы полностью растворить кислоту, затем объединяя два раствора и перемешивания в течение 2 часов. Раствор концентрировали до минимального объема, затем охлаждали до -30°C до образования кристаллов. Фильтрование давало фумаратную соль 5-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)бензо[b]тиофен-4-карбонитрила (14,8 мг, >99%, 93,8% AUC ВЭЖХ) в виде не совсем белого твердого вещества:1H ЯМР (500 МГц, CD3OD) δ 8,17 (д, J=8,5 Гц, 1H), 7,98 (д, J=5,5 Гц, 1H), 7,60 (д, J=5,5 Гц, 1H), 7,30-7,17 (м, 4H), 6,80 (д, J=5,5 Гц, 1H), 6,71 (д, J=2,0 Гц, 2H), 5,05 (д, J=6,2 Гц, 1H), 4,26-4,19 (м, 2H), 3,61 (д, J=5,1 Гц, 1H), 3,27-3,23 (м, 1H), 2,82 (с, 3H).
Такой же способ использовали для превращения (-)-энантиомера (37,0 мг, 115,0 ммоль), получая фумаратную соль 5-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)бензо[b]тиофен-4-карбонитрила (10,7 мг, >99%) в виде не совсем белого твердого вещества:1H ЯМР (500 МГц, CD3OD) δ 8,17 (д, J=8,5 Гц, 1H), 7,98 (д, J=5,5 Гц, 1H), 7,60 (д, J=5,5 Гц, 1Н), 7,30-7,18 (м, 4H), 6,81 (д, J=7,8 Гц, 1H), 6,7 (с, 2Н), 5,08-5,05 (м, 1Н), 4,30-4,21 (м, 2H), 3,63 (т, J=5,9 Гц, 1H), 3,26 (д, J=10,6 Гц, 1H), 2,82 (д, J=5,4 Гц, 3H).
Пример 92 - Получение фумаратной соли (+)-4-(бензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-ола и (-)-4-(бензотиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-ола
Стадия A: К раствору 5-бромбензотиофена (511 мг, 2,4 ммоль) при -75°C по каплям добавляли трет-бутиллитий (1,7 М в пентане, 1,6 мл, 2,6 ммоль). Реакционную смесь перемешивали при -75°C в течение 1 часа. К полученной в результате темно-коричневой смеси добавляли 2-метил-2,3-дигидро-1H-изохинолин-4-он (323 мг, 2,0 ммоль), который получали, используя способ, описанный в работе Hanna et al., J. Med. Chem., 17(9): 1020-1023 (1974), которая включена в данное описание в виде ссылки в полном объеме. Реакционную смесь перемешивали в течение 15 часов с постепенным нагреванием. Смесь гасили насыщенным раствором хлорида аммония и экстрагировали этилацетатом (3×50 мл). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией среднего давления на силикагеле (10-40% этилацетат/гексаны) с последующей препаративной ВЭЖХ давала 4-(бензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-ол (65 мг, 11%) и 4-(5-бромбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-ол (58 мг, 8%). 4-(Бензотиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-ол:1H ЯМР (CDCl3, 500 МГц) δ 7,81 (д, J=8,5 Гц, 1H), 7,69 (д, J=8,5 Гц, 1H), 7,34-7,23 (м, 4H), 7,20 (с, 1H), 7,18 (д, J=7,3 Гц, 1H), 7,10 (д, J=7,6 Гц, 1H), 3,91 (с, 1H), 3,84 (д, J=15,0 Гц, 1H), 3,53 (д, J=15,0 Гц, 1H), 3,11 (дд, J=11,6, 1,4 Гц, 1H), 2,87 (д, J=11,6 Гц, 1H), 2,50 (с, 3H); ESI МС m/z=296 [M+H]+. 4-(5-Бромбензотиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-ол:1H ЯМР (CDCl3, 300 МГц) δ 7,81 (д, J=1,8 Гц, 1H), 7,66 (д, J=8,5 Гц, 1H), 7,38 (дд, J=8,5, 1,9 Гц, 1H), 7,30-7,23 (м, 2H), 7,18 (д, J=7,3 Гц, 1H), 7,11 (с, 1H), 7,07 (д, J=7,6 Гц, 1H), 3,69 (д, J=15,0 Гц, 1H), 3,48 (д, J=15,0 Гц, 1H), 3,10 (дд, J=11,6, 1,3 Гц, 1H), 2,83 (д, J=11,6 Гц, 1H), 2,47 (с, 3H); ESI МС m/z=374 [M+H]+.
Стадия B: 4-(бензотиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-ол со стадии A (59,1 мг, 0,2 ммоль) растворяли в этаноле (1 мл) и добавляли к раствору фумаровой кислоты (24 мг, 0,2 ммоль) в метаноле (0,5 мл). Растворитель удаляли при пониженном давлении. Остаток растирали в этилацетате. Полученный в результате преципитат собирали фильтрованием, промывали этилацетатом и сушили при 50°C в вакууме, получая фумаратную соль 4-(бензотиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-ола в виде белого твердого вещества (60 мг, 73%, >99% AUC ВЭЖХ): т.пл. 172-174°C;1H ЯМР (CD3OD, 500 МГц) δ 7,81 (д, J=7,2 Гц, 1H), 7,71 (д, J=7,2 Гц, 1H), 7,40 (д, J=7,8 Гц, 1Н), 7,36-7,23 (м, 4H), 7,25 (д, J=7,6 Гц, 1H), 7,20 (с, 1H), 6,70 (с, 2H), 4,25 (д, J=15,4 Гц, 1H), 4,20 (д, J=15,4 Гц, 1Н), 3,54 (д, J=12,1 Гц, 1H), 3,48 (д, J=12,1 Гц, 1H), 2,83 (с, 3H); ESI МС m/z=296 [M+H]+.
Стадия C: Полученное соединение со стадии B разделяли препаративной хиральной ВЭЖХ (колонка CHIRALPAK AD), получая (+)-энантиомер [α]25D +69,1° (c = 0,06, метанол) и (-)-энантиомер [α]25D -72,7° (c = 0,06, метанол).
Стадия D: (+)- Энантиомер (0,14 г, 0,47 ммоль) превращали в соль фумаровой кислоты растворением свободного основания в минимальном количестве этанола, добавляли один эквивалент фумаровой кислоты в достаточном количестве метанола, чтобы полностью растворить кислоту, затем объединяя два раствора и перемешивая в течение 2 часов. Раствор концентрировали до минимального объема, затем охлаждали до -30°C до образования кристаллов. Фильтрованием давало малеатную соль 4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-4-ола (124 мг, 64%, >99% AUC ВЭЖХ) в виде не совсем белого твердого вещества: т.пл. 95-97°C;1H ЯМР (500 МГц, CD3OD) δ 8,03 (д, J=1,2 Гц, 1H), 7,88 (д, J=8,5 Гц, 1H), 7,61 (д, J=5,4 Гц, 1H), 7,38-7,34 (м, 2H), 7,30-7,23 (м, 3H), 7,00 (д, J=7,5 Гц, 1H), 6,69 (с, 2H), 4,46 (д, J=15,4 Гц, 1H), 4,34 (д, J=15,3 Гц, 1H), 3,58 (д, J=12,3 Гц, 1H), 3,48 (д, J=12,4 Гц, 1H) 2,92 (с, 3H).
Пример 93 - Получение соли (+)-4-бензо[b]тиофен-5-ил-2,4-диметил-1,2,3,4-тетрагидроизохинолина и фумаровой кислоты и соли (-)-4-бензо[b]тиофен-5-ил-2,4-диметил-1,2,3,4-тетрагидроизохинолина и фумаровой кислоты
Стадия A: N-метилбензиламин (0,26 мл, 2,0 ммоль) добавляли к перемешиваемому раствору 1-бензотиофен-5-ил-2-бромэтанона (0,5 г, 1,99 ммоль) со стадии C примера 96 в тетрагидрофуране (10 мл) и N,N-диизопропилэтиламине (0,45 мл, 2,6 ммоль) в атмосфере N2 в течение 3,0 часов. Растворитель удаляли и к остатку добавляли воду и три раза экстрагировали этилацетатом. Органический экстракт сушили над сульфатом натрия, фильтровали и концентрировали до желтого масла. Масло очищали хроматографией на колонке (силикагель, 25% этилацетат/гексаны), получая требуемый продукт (0,36 г, 62%) в виде желтого масла:1H ЯМР (500 МГц, CDCl3) δ 8,43 (д, J=0,9 Гц, 1H), 7,95-7,89 (м, 2H), 7,51 (д, J=5,4 Гц, 1H), 7,41-7,27 (м, 6H), 3,85 (с, 2H), 3,71 (с, 2H), 2,40 (с, 3H).
Стадия B: Продукт со стадии A (0,35 г, 1,2 ммоль) растворяли в безводном диэтиловом эфире (2,0 мл) и раствор по каплям добавляли к раствору метилмагнийиодида (0,8 мл, 2,4 ммоль) в диэтиловом эфире (8 мл), охлажденном при -65°C. Реакционной смеси давали возможность нагреться до комнатной температуры в течение 3 часов, затем концентрировали (0,29 г, грубый выход 80%) до получения желтого масла. Полученный неочищенный продукт использовали на следующей стадии без дополнительной очистки. ESI-МС m/z 312 [M+H]+.
Стадия C: Метансульфоновую кислоту (10 мл) нагревали до 40°C в атмосфере азота. Неочищенный продукт со стадии B (0,29 г, 0,9 ммоль) растворяли в дихлорэтане (6 мл) и добавляли к теплой кислоте, затем смесь нагревали до 80°C. После перемешивания в течение 30 минут при 80°C смесь вливали в лед и доводили до pH 9-10 добавлением концентрированного гидроксида аммония. Раствор дважды экстрагировали метиленхлоридом. Органические слои объединяли и сушили над сульфатом натрия, фильтровали и концентрировали до получения коричневого масла. Масло очищали хроматографией на колонке (силикагель, 25%-100% этилацетат/гексаны), получая требуемый продукт (99 мг, 37%) в виде бесцветного масла:1H ЯМР (500 МГц, CDCl3) δ 7,73-7,69 (м, 2H), 7,38-7,35 (м, 1H), 7,27-7,24 (м, 1H), 7,21-7,06 (м, 4H), 6,91 (д, J=7,9 Гц, 1H), 3,73 (д, J=14,7 Гц, 1H), 3,60 (д, J=14,7 Гц, 1H), 2,73 (д, J=11,5 Гц, 1H), 2,66 (д, J=11,5 Гц, 1H), 2,35 (с, 3H), 1,82 (с, 3H).
Стадия D: Продукт со стадии C (0,10 г, 0,34 ммоль) превращали в соль фумаровой кислоты растворением свободного основания в минимальном количестве этанола, добавлением одного эквивалента фумаровой кислоты в достаточном количестве метанола, чтобы полностью растворить кислоту, затем объединяя два раствора и перемешивая в течение 2 часов. Раствор концентрировали до минимального объема, затем охлаждали при -30°C до образования кристаллов. Фильтрование давало фумаратную соль (+/-)-4-бензо[b]тиофен-5-ил-2,4-диметил-1,2,3,4-тетрагидроизохинолина (98,5 мг, 86%) в виде не совсем белого твердого вещества:1H ЯМР (500 МГц, CDCl3) δ 7,84 (д, J=8,5 Гц, 1H), 7,72 (с, 1Н), 7,57 (д, J=5,4 Гц, 1H), 7,32-7,20 (м, 5H), 7,08 (д, J=7,7 Гц, 1H), 6,69 (с, 2H), 4,29 (м, 2H), 3,56 (д, J=12,4 Гц, 1H), 3,40 (д, J=12,4 Гц, 1H), 2,83 (с, 3H), 1,92 (с, 3H).
Стадия E: Свободное основание со стадии D (58,8 мг, 212 ммоль) разделяли препаративной хиральной ВЭЖХ (колонка CHIRALPAK AD с использованием смеси 90% гептан/10% изопропиловый спирт/0,1% диэтиламин), получая (+)-энантиомер [α]25D +39,6° (c 0,06, метанол) и (-)-энантиомер [α]25D -31,6° (c 0,06, метанол). (+)-Энантиомер (8,5 мг, 0,03 ммоль) превращали в соль фумаровой кислоты растворением свободного основания в минимальном количестве этанола, добавлением одного эквивалента фумаровой кислоты в достаточном количестве метанола, чтобы полностью растворить кислоту, затем объединяя два раствора и перемешивая в течение 2 часов. Раствор концентрировали до минимального объема, затем охлаждали при -30°C до образования кристаллов. Фильтрование давало фумаратную соль 4-бензо[b]тиофен-5-ил-2,4-диметил-1,2,3,4-тетрагидроизохинолина (12,0 мг, >99%, 97,3% AUC ВЭЖХ) в виде не совсем белого твердого вещества: т.пл. 95-97°C;1H ЯМР (500 МГц, CD3OD) δ 7,81 (д, J=8,6 Гц, 1H), 7,71 (д, J=1,4 Гц, 1H), 7,55 (д, J=5,4 Гц, 1H), 7,30 (д, J=5,4 Гц, 1H), 7,26-7,18 (м, 4H), 7,03 (д, J=6,9 Гц, 1H), 6,69 (с, 2H), 4,12-4,07 (м, 2H), 3,35-3,34 (м, 1H), 3,19-3,20 (м, 1H), 2,67 (с, 3H), 1,89 (с, 3H).
Такой же способ использовали для превращения (-)-энантиомера (12 мг, 0,04 ммоль) в его фумаратную соль, получая фумаратную соль 4-бензо[b]тиофен-5-ил-2,4-диметил-1,2,3,4-тетрагидроизохинолина (16,5 мг, >99%, 98,5% AUC ВЭЖХ) в виде не совсем белого твердого вещества: т.пл. 95-97°C;1H ЯМР (500 МГц, CD3OD) δ 7,81 (д, J=8,6 Гц, 1Н), 7,71 (с, 1H), 7,54 (д, J=5,4 Гц, 1H), 7,30-7,29 (м, 1H), 7,24-7,18 (м, 4H), 7,02 (д, J=7,5 Гц, 1H), 6,69 (с, 2H), 4,07-4,02 (м, 2H), 3,27-3,26 (м, 1H), 3,16-3,17 (м, 1H), 2,65 (с, 3H), 1,88 (с, 3H).
Пример 94 - Получение фумаратной соли (+/-)4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-4-карбонитрила
Стадия A: Хлорид олова IV в дихлорметане (0,51 мл, 1,0 М) добавляли по каплям к суспензии 4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-4-ола (300 мг, 1,02 ммоль) из примера 92 и триметилсилилцианида (0,68 мл, 5,1 ммоль) в дихлорметане (4,5 мл) при 0°C. Полученному в результате раствору давали возможность перемешиваться при комнатной температуре в течение ночи, затем добавляли дополнительное количество хлорида олова IV в дихлорметане (1,02 мл, 1,0 М) и раствор перемешивали при комнатной температуре еще в течение 24 часов. Добавляли карбонат калия (636 мг, 4,60 ммоль), гидрат фторида калия (435 мг, 7,49 ммоль) и затем воду (0,135 мл, 7,50 ммоль) и смесь перемешивали в течение ночи. Добавляли силикагель (2,5 г) и достаточное количество воды, чтобы смесь можно было перемешать, и смесь фильтровали. Фильтровальную подушку промывали этилацетатом. Органический слой промывали насыщенным раствором бикарбоната натрия, сушили над сульфатом натрия и концентрировали в вакууме. Сырое вещество фильтровали через слой диоксида кремния (1:1 гексан/этилацетат). Фильтрат концентрировали в вакууме. Остаток очищали хроматографией на колонке (95:5, затем 90:10 гексан/этилацетат), получая 4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-4-карбонитрил (17 мг, 6%). Перекристаллизация из этанола давала 10 мг белого твердого вещества. Карбонитрил (10 мг, 0,033 ммоль) превращали в фумаратную соль растворением в минимальном количестве метанола и обработкой раствором фумаровой кислоты (3,0 мг, 0,026 ммоль) в метаноле. Раствор концентрировали досуха и остаток растворяли в смеси 1:1 ацетонитрил/вода и лиофилизировали, получая фумаратную соль 4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-4-карбонитрила (13 мг, 94%, >99,0% AUC ВЭЖХ): т.пл. 170-172°C;1H ЯМР (500 МГц, CDCl3) δ 7,92 (д, J=1,8 Гц, 1H), 7,89 (д, J=11,5 Гц, 1H), 7,63 (д, J=5,5 Гц, 1H), 7,38 (д, J=5,5 Гц, 1H), 7,35-7,32 (м, 1H), 7,28-7,20 (м, 3H), 7,04 (д, J=8,00 Гц, 1H), 6,75 (с, 1,4H), 3,90 (д, J=15,2 Гц, 1H), 3,70 (д, J=15,2 Гц, 1H), 3,34 (д, J=12,0 Гц, 1H), 2,95 (д, J=12,0 Гц, 1H), 2,48 (с, 3H).
Пример 95 - Получение фумаратной соли (+)-4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ола
К раствору (+)-энантиомера, полученного со стадии F примера 96 (свободное основание, 0,5 г, 1,6 ммоль) в AcOH (20 мл), добавляли HBr (20 мл, 48%). Реакционную смесь кипятили с обратным холодильником в течение 3,5 часа. После охлаждали до комнатной температуры растворитель удаляли в вакууме. Остаток нейтрализовали насыщенным водным раствором NaHCO3. Продукт экстрагировали EtOAc (2×100 мл). Органические слои объединяли, промывали насыщенным раствором соли (100 мл), сушили (Na2SO4) и концентрировали, получая не совсем белое твердое вещество, которое очищали, используя хроматографию среднего давления (элюент: MeOH/дихлорметан от 1:99 до 5:95), получая (+)-4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ол в виде белого твердого вещества (460 мг, 1,5 ммоль, 96%): [α]25D +85° (c 0,02, метанол). Твердое вещество (28 мг, 0,09 ммоль) растворяли в MeOH (5 мл) и к полученному раствору добавляли фумаровую кислоту (11 мг, 0,10 ммоль). Раствор концентрировали до объема менее 1 мл. К полученному раствору добавляли воду (5 мл). Затем полученную в результате суспензию лиофилизировали в сублимационной сушилке, получая фумаратную соль (+)-4-бензо[b]тиофен-5-ил-7-гидрокси-2-метил-1,2,3,4-тетрагидроизохинолина (33 мг, 85%, >99% AUC ВЭЖХ) в виде белого твердого вещества: т.пл. 155-157°C;1H ЯМР (500 МГц, CD3OD) δ 7,88 (д, J=8,3 Гц, 1H), 7,71 (с, 1Н), 7,59 (д, J=5,5 Гц, 1H), 7,34 (д, J=5,5 Гц, 1Н), 7,17 (дд, J=8,3, 1,6 Гц, 1Н), 6,73-6,71 (м, 1H), 6,69 (с, 2H), 6,66-6,64 (м, 2H), 4,54 (дд, J=11,2, 6,2 Гц, 1H), 4,32 (с, 2H), 3,68 (дд, J=12,2, 6,0 Гц, 1Н), 3,32-3,31 (м, 1H), 2,91 (с, 3H); ESI-МС m/z 296 [M+H]+.
Пример 96 - Получение фумаратной соли (+)-4-бензо[b]тиофен-5-ил-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: Смесь CuCN (86 г, 960 ммоль), 5-бромбензо[b]тиофена (157 г, 723 ммоль), пиридина (80 мл) и ДМФА (1400 мл) нагревали при кипячении с обратным холодильником в течение 14 часов. После охлаждения до 80°C реакционную смесь вливали в холодный водный раствор этилендиамина (400 мл в 2 л воды), охлажденный на бане со льдом. Продукт экстрагировали эфиром (2×1,5 л). Эфирный слой промывали насыщенным раствором соли (1 л), сушили (Na2SO4) и концентрировали. Остаток перекристаллизовывали из смеси CHCl3/гексаны (50 мл/2000 мл), получая бензо[b]тиофен-5-карбонитрил (106 г, 90%) в виде белого твердого вещества:1H ЯМР (300 МГц, CDCl3) δ 8,15 (с, 1H), 7,97 (д, J=8,3 Гц, 1H), 7,61 (д, J=5,4 Гц, 1Н), 7,56 (д, J=8,3 Гц, 1H), 7,41 (д, J=5,4 Гц, 1Н).
Стадия B: К холодной (-78°C) смеси метилмагнийбромида в ТГФ (3 М, 693 мл, 2,08 моль) и CuBr (5,5 г, 38 ммоль) добавляли раствор бензо[b]тиофен-5-карбонитрила (106 г, 667 ммоль) и TBSCl (199 г, 1,32 моль) в ТГФ (500 мл). Охлаждающую баню убирали и реакционной смеси давали возможность нагреться до комнатной температуры. Смесь перемешивали при комнатной температуре в течение 40 минут. Реакционную смесь медленно вливали в ледяную воду (100 мл). Продукт экстрагировали дихлорметаном (2×100 мл). Органический слой промывали насыщенным водным раствором NH4Cl (100 мл), насыщенным раствором соли (100 мл), сушили (Na2SO4) и концентрировали, получая требуемый продукт (860 мг, 78%) в виде не совсем белого твердого вещества:1H ЯМР (300 МГц, CDCl3) δ 8,43 (с, 1H), 7,95-7,94 (м, 2H), 7,53 (д, J=5,4 Гц, 1Н), 7,44 (д, J=5,4 Гц, 1H), 2,69 (с, 3H).
Стадия C: К раствору продукта, полученного на стадии B (1- бензо[b]тиофен-5-илэтанона) (60 г, 340 ммоль) в CHCl3 (1 л), добавляли трибромид пиридиния (110 г, 343 ммоль) при комнатной температуре. Смесь перемешивали механической мешалкой при комнатной температуре в течение 6 часов. Смесь промывали 1 N HCl (2×1 л), чтобы удалить пиридин. Затем органический слой промывали насыщенным раствором соли, сушили (Na2SO4) и концентрировали. Остаток очищали хроматографией среднего давления (элюент: CH2Cl2/гексан 30:70), получая требуемый продукт (50 г, 57%) в виде белого твердого вещества:1H ЯМР (300 МГц, CDCl3) δ 8,48 (с, 1H), 7,97-7,96 (м, 2H), 7,56 (д, J=5,4 Гц, 1H), 7,47 (д, J=5,4 Гц, 1H), 4,54 (с, 2H).
Стадия D: К холодному (0°C) и перемешиваемому раствору 3-метокси-N-бензиламина (46 г, 305 ммоль) и диизопропилэтиламина (40 мл, 276 ммоль) в CH2Cl2 (500 мл) добавляли продукт, полученный на стадии C (71 г, 276 ммоль) в CH2Cl2 (500 мл). Затем реакционную смесь перемешивали при 0°C в течение 2 часов. Смесь промывали водой (500 мл), насыщенным водным раствором NaHCO3 (500 мл), насыщенным раствором соли, сушили и концентрировали, получая требуемый продукт в виде светло-желтой жидкости (107 г, количественный выход сырого продукта):1H ЯМР (300 МГц, CDCl3) δ 8,43 (с, 1H), 7,94-7,92 (м, 2H), 7,51 (д, J=5,4 Гц, 1H), 7,40 (д, J=5,4 Гц, 1H), 7,25-7,21 (м, 1H), 6,93-6,80 (м, 3H), 3,83 (с, 2H), 3,76 (с, 3H), 3,67 (с, 2H), 2,40 (с, 3H); ESI-МС m/z 326 [M+H]+.
Стадия E: К холодному (0°C) и перемешиваемому раствору продукта, полученного на стадии D (107 г, неочищенный продукт), в метаноле (1000 мл) медленно добавляли NaBH4 (11 г, 290 ммоль). Полученный в результате раствор перемешивали при 0°C в течение 3 часов. Растворитель удаляли и остаток собирали в воду (500 мл). Продукт экстрагировали дихлорметаном (2×1000 мл). Органические слои объединяли, промывали насыщенным раствором соли, сушили и концентрировали, получая требуемый продукт (107 г, количественный выход сырого продукта) в виде густой жидкости:1H ЯМР (300 МГц, CDCl3) δ 7,85-7,82 (м, 2H), 7,43 (д, J=5,4 Гц, 1H), 7,32-7,30 (м, 2H), 6,95-6,80 (м, 4H), 4,88 (дд, J=6,9, 3,6 Гц, 1Н), 3,81-3,81 (м, 4H), 3,74 (д, J=12,9 Гц, 1H), 3,52 (д, J=12,9 Гц, 1H), 2,65-2,58 (м, 2H), 2,35 (с, 3H); ESI-МС m/z 328 [M+H]+.
Стадия F: К раствору продукта, полученного на стадии E (25,0 г, 76,5 ммоль), в CH2Cl2 (500 мл) медленно при комнатной температуре добавляли MsOH (74 г, 770 ммоль). Раствор перемешивали при комнатной температуре в течение 15 минут. Смесь медленно добавляли к охлажденному на льду раствору NaOH (2 N, 500 мл). Органический слой отделяли и водный слой еще раз экстрагировали дихлорметаном (200 мл). Органические слои объединяли, промывали водой (500 мл) и насыщенным раствором соли (300 мл), сушили (Na2SO4) и концентрировали. Остаток очищали флэш-хроматографией на колонке (элюент: MeOH/EtOAc/гексан 1:9:15), получая 4-бензо[b]тиофен-5-ил-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолин (12,5 г, 53%) в виде пенообразного твердого вещества. Затем его разделяли на колонке CHRIALPAK AD (элюент: 10 IPA/90 гептан/0,1 DEA). Небольшое количество (+)-энантиомера [α]25D +74,3° (c 0,07, метанол)] (65 мг, 0,21 ммоль) растворяли в метаноле (5 мл) и к полученному раствору добавляли фумаровую кислоту (24 мг, 0,21 ммоль). Раствор концентрировали до объема менее 1 мл. К полученному раствору добавляли воду (5 мл). затем полученную в результате суспензию лиофилизировали в сублимационной сушилке, получая фумарат (+)-4-бензо[b]тиофен-5-ил-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолина (55 мг, 62%, >99% AUC ВЭЖХ) в виде белого твердого вещества: т.пл. 103-105°C;1H ЯМР (500 МГц, CD3OD) δ 7,89 (д, J=8,3 Гц, 1H), 7,72 (с, 1Н), 7,60 (д, J=5,4 Гц, 1H), 7,34 (д, J=5,5 Гц, 1H), 7,17 (дд, J=8,3, 1,6 Гц, 1Н), 6,84-6,80 (м, 3H), 6,72 (с, 4H), 4,60 (дд, J=10,5, 6,0 Гц, 1H), 4,48-4,42 (м, 2H), 3,37-3,74 (м, 4H), 3,44 (т, J=10,5 Гц, 1H), 2,98 (с, 3H); ESI-МС m/z 310 [M+H]+.
Пример 97 - Получение фумаратной соли (+)-4-бензо[b]тиофен-5-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору продукта, полученного согласно примеру 95 (свободное основание, 6,0 г, 20,3 ммоль) в дихлорметане (200 мл), охлажденному до 0°C, добавляли ангидрид трифторметансульфоновой кислоты (11,2 г, 40,7 ммоль). После перемешивания при комнатной температуре в течение 1 часа реакционную смесь вливали в насыщенный водный раствор NaHCO3 (200 мл). Отделенный органический слой промывали насыщенным раствором соли (300 мл), сушили (Na2SO4) и концентрировали, получая требуемый трифторметансульфонат (9,0 г, количественный выход сырого продукта) в виде светло-желтого масла:1H ЯМР (300 МГц, CDCl3) δ 7,80 (д, J=8,3 Гц, 1H), 7,61 (с, 1H), 7,44 (д, J=5,5 Гц, 1H), 7,28-7,26 (м, 1H), 7,15 (д, J=8,3 Гц, 1H), 7,05-7,03 (м, 1H), 6,97-6,94 (м, 2H), 4,39 (т, J=12,0 Гц, 1H), 3,81 (д, J=15,0 Гц, 1H), 3,67 (д, J=15,0 Гц, 1H), 3,09 (дд, J=10,5, 6,0 Гц, 1Н), 2,64 (дд, J=11,6, 8,7 Гц, 1Н), 2,45 (с, 3H); ESI-MS 428 [M+H]+.
Стадия B: Смесь трифторметансульфоната (2,5 г, неочищенный, 5,9 ммоль), полученного на стадии A, бис(пинаколато)дибора (1,49 г, 5,9 ммоль), KOAc (1,74 г, 17,7 ммоль) и ДМСО (20 мл) продували аргоном. К смеси добавляли PdCl2dppf (722 мг, 0,77 ммоль) и систему снова продували аргоном. Смесь нагревали при 100°C в течение 3 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли дихлорметаном (100 мл) и фильтровали через целит. Фильтрат промывали H2O (100 мл) и насыщенным раствором соли (100 мл), сушили (Na2SO4) и концентрировали, получая требуемый сложный эфир бороновой кислоты (5,2 г, неочищенный продукт) в виде темного масла:1H ЯМР (300 МГц, CDCl3) δ 7,78 (д, J=8,3 Гц, 1Н), 7,64 (с, 1H), 7,57 (с, 1H), 7,49 (д, J=7,8 Гц, 1H), 7,41 (д, J=5,4 Гц, 1H), 7,26 (с, 1H), 7,15 (д, J=8,2 Гц, 1H), 6,90 (д, J=7,9 Гц, 1H), 4,42-4,40 (м, 1Н), 3,80 (д, J=15,9 Гц, 1H), 3,66 (д, J=15,9 Гц, 1H), 3,10-3,08 (м, 1H), 2,62 (т, J=11,4 Гц, 1H), 2,43 (с, 3H), 1,33 (с, 12H); ESI-MS 406 [M+H]+.
Стадия C: Смесь сложного эфира (5,2 г, неочищенный продукт, 5,9 ммоль), полученного на стадии B, 3-пиридазинилхлорида (1,0 г, 8,8 ммоль), Na2CO3 (1,93 г, 17,9 ммоль), H2O (5 мл) и ДМФА (25 мл) продували аргоном. Добавляли PdCl2dppf (722 мг, 0,88 ммоль). Смесь нагревали при 100°C в течение 5 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли дихлорметаном (100 мл) и фильтровали через целит. Фильтрат промывали водой (200 мл) и насыщенным раствором соли (200 мл), сушили (Na2SO4) и концентрировали. Остаток очищали хроматографией среднего давления (элюент: MeOH/EtOAc 1:9), получая (+)-4-бензо[b]тиофен-5-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин (390 мг, 19% для трех стадий) в виде светло-коричневого масла. Продукт растворяли в MeOH (5 мл) при комнатной температуре и к полученному раствору добавляли фумаровую кислоту (125 мг, 1,07 ммоль). Раствор концентрировали примерно до 2 мл. К раствору добавляли воду (20 мл). Полученную в результате суспензию лиофилизировали в сублимационной сушилке в течение ночи, получая фумаратную соль (+)-4-бензо[b]тиофен-5-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина (355 мг, 59%, >99% AUC ВЭЖХ) в виде светло-коричневого твердого вещества: [α]25D +7,3° (c 0,07, метанол); т.пл. 122-124°C;1H ЯМР (500 МГц, CD3OD) δ 9,15 (д, J=4,9 Гц, 1H), 8,17 (д, J=8,7 Гц, 1H), 8,04 (с, 1H), 7,93-7,88 (м, 2H), 7,80-7,77 (м, 2H), 7,61 (д, J=5,5 Гц, 1H), 7,35 (д, J=5,5 Гц, 1H), 7,22 (д, J=8,3 Гц, 1H), 7,11 (д, J=8,2 Гц, 1H), 6,71 (с, 4H), 4,71 (дд, J=10,5, 6,5 Гц, 1H), 4,52 (д, J=15,1 Гц, 1H), 4,44 (д, J=15,1 Гц, 1H), 3,73 (дд, J=12,0, 6,0 Гц, 1H), 3,42-3,38 (м, 1H), 2,91 (с, 3H); ESI-MS 358 [M+H]+; элементный анализ, вычислено для C22H19N3S·1,75C4H4O4: C, 62,13; Н, 4,67; N, 7,50. Найдено: C, 61,80; Н, 4,81; N, 7,17.
Пример 98 - Получение фумаратной соли (+)-[6-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]диметиламина
Стадия A: Смесь продукта, полученного на стадии B примера 97 (2,0 г неочищенного продукта), 3,6-дихлорпиридазин (1,0 г), Na2CO3 (1,56 г, 14,7 ммоль), H2O (4 мл) и ДМФА (20 мл) продували аргоном. Добавляли PdCl2dppf (600 мг, 0,74 ммоль). Смесь нагревали при 80°C в течение 4 часов и при 85°C в течение 1 часа. После охлаждения до комнатной температуры реакционную смесь разбавляли дихлорметаном (200 мл) и фильтровали через целит. Фильтрат промывали водой (200 мл) и насыщенным раствором соли (200 мл), сушили (Na2SO4) и концентрировали. Остаток очищали хроматографией среднего давления (элюент: MeOH/EtOAc/гексаны 1:9:10), получая требуемый продукт (410 мг, 87%) в виде не совсем белого твердого вещества:1H ЯМР (300 МГц, CDCl3) δ 7,89 (ушир., 1Н), 7,82 (д, J=8,3 Гц, 1H), 7,81 (д, J=8,9 Гц, 1H), 7,68-7,65 (м, 2H), 7,54 (д, J=8,9 Гц, 1H), 7,44 (д, J=5,4 Гц, 1Н), 7,29-7,27 (м, 1H), 7,19 (д, J=5,4 Гц, 1H), 7,06 (д, J=8,1 Гц, 1Н), 4,48-4,43 (м, 1Н), 3,90 (д, J=15,1 Гц, 1H), 3,75 (д, J=15,1 Гц, 1H), 3,13 (дд, J=11,8, 5,6 Гц, 1H), 2,71-2,64 (м, 1H), 2,48 (с, 3H); ESI-MS 392 [M+H]+.
Стадия B: Смесь продукта, полученного на стадии A (200 мг, 0,51 ммоль), Me2NH (40% водный раствор, 4 мл, 35 ммоль) и ДМФА (10 мл) помещали в герметичную пробирку. Затем пробирку нагревали при 110°C в течение 14 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли дихлорметаном (50 мл) и промывали водой, насыщенным раствором соли, сушили и концентрировали. Остаток очищали хроматографией среднего давления (элюент: MeOH/EtOAc 1:9), получая [6-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]диметиламин (200 мг, 98%) в виде желтого полутвердого вещества. Соединение растворяли в метаноле (2 мл) и к полученному раствору добавляли фумаровую кислоту (60 мг, 0,52 ммоль). К полученному раствору добавляли воду (15 мл). Полученную в результате суспензию лиофилизировали в сублимационной сушилке в течение ночи, получая фумаратную соль (+)-[6-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]диметиламина (230 мг, 88%, 98,0% AUC ВЭЖХ) в виде не совсем белого твердого вещества: [α]25D +32,5° (c 0,08, метанол); т.пл. 116-119°C;1H ЯМР (500 МГц, CD3OD) δ 7,92 (д, J=8,3 Гц, 1H), 7,87 (ушир.с, 1Н), 7,81 (д, J=10,0 Гц, 1H), 7,77 (ушир.с, 1H), 7,75 (д, J=8,3 Гц, 1Н), 7,61 (д, J=5,4 Гц, 1H), 7,35 (д, J=5,4 Гц, 1H), 7,22 (д, J=8,3 Гц, 1H), 7,19 (д, J=9,6 Гц, 1H), 7,04 (д, J=8,3 Гц, 1H), 6,71 (с, 4H), 4,73-4,67 (м, 1Н), 4,60-4,49 (м, 2H), 3,81-3,77 (м, 1H), 3,50-3,43 (м, 1H), 3,21 (с, 6H), 2,99 (с, 3H); ESI-МС m/z 401 [M+H]+; элементный анализ, вычислено для C22H24N4S·2,0C4H4О4·0,25H2O: C, 60,32; Н, 5,14; N, 8,79. Найдено: C, 60,03; Н, 4,94; N, 8,87.
Пример 99 - Получение (+)-4-бензо[b]тиофен-5-ил-2-метил-7-пиридазин-4-ил-1,2,3,4-тетрагидроизохинолина и (-)-4-бензо[b]тиофен-5-ил-2-метил-7-пиридазин-4-ил-1,2,3,4-тетрагидроизохинолина
Стадия A: Смесь (-)-4-бензо[b]тиофен-5-ил-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолина (10,00 г, 32,3 ммоль), который получали согласно стадия F получения примера 98, и этантиолата натрия (11,3 г, 134,3 ммоль) в ДМФА (250 мл) нагревали при 140°C в атмосфере N2 в течение 14 часов. Реакционную смесь охлаждали на ледяной бане перед добавлением насыщенного раствора NH4Cl (250 мл). Водную фазу экстрагировали ДХМ (3×750 мл) и объединенные органические экстракты промывали водой (3×500 мл), насыщенным раствором соли, сушили над Na2SO4 и концентрировали в вакууме. Сырой продукт очищали хроматографией на колонке с силикагелем (элюент: 5% MeOH/CH2Cl2), получая (-)-4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ол (6,64 г, 70%) в виде оранжево-коричневого твердого вещества:1H ЯМР (300 МГц, CDCl3) δ 7,76 (д, J=8,3 Гц, 1Н), 7,62 (с, 1H), 7,40 (д, J=5,4 Гц, 1Н), 7,26-7,20 (м, 2H), 7,13 (д, J=8,4 Гц, 1H), 6,66 (д, J=8,3 Гц, 1H), 6,50-6,40 (м, 2H), 4,34 (дд, J=9,0, 6,0 Гц, 1H), 3,68 (д, J=15,0 Гц, 1H), 3,54 (д, J=15,0 Гц, 1H), 3,11 (дд, J=11,5, 5,7 Гц, 1H), 2,58 (дд, J=11,3, 9,7 Гц, 1H), 2,43 (с, 3H); ESI-МС m/z=296 [M+H]+.
Стадия B: К раствору (-)-4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ола (6,64 г, 22,5 ммоль) в дихлорметане (210 мл), охлажденному до 0°C, добавляли ангидрид трифторметансульфоновой кислоты (4,92 мл, 29,2 ммоль). После перемешивания в течение 2 часов реакционной смеси давали возможность нагреться до комнатной температуры в течение 30 минут, затем вливали в насыщенный водный раствор NaHCO3 раствор (200 мл). Отделенный органический слой промывали водой (200 мл) и насыщенным раствором соли, сушили (Na2SO4) и летучие вещества удаляли в вакууме. Сырой продукт очищали хроматографией на колонке с силикагелем (элюент: Et2O), получая сложный 4-бензо[b]тиофен-ил-2-метил-1,2,3,4-тетрагидроизохино-7-иловый эфир (-)-трифторметансульфоновой кислоты (7,88 г, 82%) в виде желтого масла:1H ЯМР (300 МГц, CDCl3) δ 7,80 (д, J=8,3 Гц, 1Н), 7,61 (с, 1Н), 7,44 (д, J=5,5 Гц, 1Н), 7,28-7,26 (м, 1Н), 7,15 (д, J=8,3 Гц, 1Н), 7,05-7,03 (м, 1Н), 6,97-6,94 (м, 2H), 4,39 (т, J=12,0 Гц, 1H), 3,81 (д, J=15,0 Гц, 1H), 3,67 (д, J=15,0 Гц, 1H), 3,09 (дд, J=10,5, 6,0 Гц, 1H), 2,64 (дд, J=11,6, 8,7 Гц, 1H), 2,45 (с, 3H); ESI-МС m/z=428 [M+H]+.
Стадия C: К раствору 2,2,6,6-тетраметилпиперидина (4,75 мл, 28,1 ммоль) в ТГФ (120 мл) при -30°C в атмосфере N2 добавляли n-BuLi (11,28 мл 2,5 М раствора в гексанах, 28,1 ммоль). Раствору давали возможность нагреться до 0°C в течение 30 минут, затем охлаждали до -78°C. По каплям добавляли раствор пиридазина (2,04 мл, 28,1 ммоль) в ТГФ (9 мл). Темно-красный раствор перемешивали при -78°C в течение 30 минут перед добавлением ZnCl2 (112,30 мл 0,5 М раствора в ТГФ, 56,3 ммоль). Полученной в результате коричневой суспензии давали возможность нагреться до комнатной температуры и добавляли раствор сложного 4-бензо[b]тиофен-ил-2-метил-1,2,3,4-тетрагидроизохино-7-илового эфира (-)-трифторметансульфоновой кислоты (6,00 г, 14,1 ммоль) в ТГФ (36 мл) с последующим добавлением тетракис(трифенилфосфин)палладия(0) (1,62 г, 1,41 ммоль). Систему продували аргоном и смесь нагревали при 70°C в течение 14 часов. После охлаждения до комнатной температуры добавляли насыщенный раствор NH4Cl (200 мл) и смесь распределяли между EtOAc (500 мл) и водой (300 мл). Слои фильтровали, чтобы обеспечить отделение органической фазы. Органическую фазу промывали насыщенным раствором соли, сушили над Na2SO4 и концентрировали в вакууме. Полученный в результате остаток частично очищали хроматографией на колонке (элюент: 5% MeOH/CH2Cl2), получая смесь 3- и 4-пиридазинилрегиоизомеров. Полученное вещество растворяли в EtOAc (15 мл) и раствор оставляли стоять в течение ночи. Преципитированное твердое вещество отфильтровывали, получая (-)-4-бензо[b]тиофен-5-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин (0,92 г, 18%) в виде светло-коричневого твердого вещества. Оставшийся маточный раствор концентрировали в вакууме и остаток элюировали через флэш-колонку ISCO combi 120 г (элюент: 5% MeOH/EtOAc), получая дополнительное количество соединения примера 99 [(-)-энантиомер] (1,01 г, 20%) плюс дополнительные смешанные фракции, содержащие, главным образом, региоизомерный продукт. Смешанные фракции концентрировали в вакууме и очищали, используя флэш-колонку ISCO combi 40 г (элюент: от 5 до 10% MeOH/EtOAc), получая (-)-4-бензо[b]тиофен-5-ил-2-метил-7-пиридазин-4-ил-1,2,3,4-тетрагидроизохинолин (210 мг, 4%) в виде не совсем белого твердого вещества:1H ЯМР (CDCl3, 500 МГц) δ 9,45 (м, 1Н), 9,20 (дд, J=5,4, 1,0 Гц, 1Н), 7,82 (д, J=8,3 Гц, 1Н), 7,69 (с, 1H), 7,62 (дд, J=5,4, 2,5 Гц, 1H), 7,46 (д, J=9,7 Гц, 1Н), 7,42 (с, 1H), 7,37 (дд, J=8,1, 1,8 Гц, 1Н), 7,29 (д, J=5,4 Гц, 1Н), 7,18 (дд, J=8,3, 1,5 Гц, 1H), 7,08 (д, J=8,1 Гц, 1H), 4,45 (т, J=6,5 Гц, 1H), 3,88 (д, J=15,0 Гц, 1H), 3,74 (д, J=15,0 Гц, 1H), 3,18 (дд, J=11,5, 5,6 Гц, 1H), 2,69 (дд, J=11,5, 8,7 Гц, 1Н), 2,48 (с, 3H), ESI-МС m/z=358 [M+H]+.
Стадия D: К раствору (-)-4-бензо[b]тиофен-5-ил-2-метил-7- пиридазин-4-ил-1,2,3,4-тетрагидроизохинолина (210 мг, 0,59 ммоль) в трет-бутаноле (6 мл) добавляли трет-бутоксид калия (396 мг, 3,53 ммоль). Смесь нагревали в атмосфере N2 при 95°C в течение 20 часов. После охлаждения до комнатной температуры добавляли насыщенный раствор NH4Cl (3 мл) и смесь экстрагировали EtOAc (30 мл). Органический экстракт промывали насыщенным раствором соли, сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали хроматографией, используя флэш-колонку ISCO combi 12 г (элюент: 10% MeOH/EtOAc), получая (+/-)-4-бензо[b]тиофен-5-ил-2-метил-7-пиридазин-4-ил-1,2,3,4-тетрагидроизохинолин в виде не совсем белого твердого вещества (200 мг, 95%). Рацемат разделяли, используя колонку Chiralpak AD (элюент: 20 IPA/80 гептан/0,1 DEA), получая (+)-4-бензо[b]тиофен-5-ил-2-метил-7-пиридазин-4-ил-1,2,3,4-тетрагидроизохинолин (77 мг, 77%) в виде не совсем белого твердого вещества: [[α]25D +77,4° (c 0,10, метанол)] и (-)-4-бензо[b]тиофен-5-ил-2-метил-7-пиридазин-4-ил-1,2,3,4-тетрагидроизохинолин (75 мг, 75%) в виде не совсем белого твердого вещества: [[α]25D -77,7° (c 0,08, метанол)].
Пример 100 - Получение фумаратной соли (+)-4-бензо[b]тиофен-5-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина
Смесь продукта, полученного на стадии A примера 97 (2,5 г, неочищенный продукт), морфолина (1,03 г, 11,8 ммоль), Pd(OAc)2 (198 мг, 0,88 ммоль), X-phos (1,67 г, 3,51 ммоль), Cs2CO3 (5,58 г, 17,6 ммоль) и толуола (20 мл) помещали в реакционный сосуд для микроволновой печи. Сосуд нагревали в микроволновом реакторе (программа: 10 мин ramp, 160°C, 30 минут). После охлаждения до комнатной температуры реакционную смесь разбавляли дихлорметаном (100 мл) и фильтровали через целит. Фильтрат промывали насыщенным водным раствором хлорида аммония (100 мл) и насыщенным раствором соли (100 мл), сушили (Na2SO4) и концентрировали. Остаток очищали хроматографией среднего давления (элюент: MeOH/EtOAc/гексаны 1:19:20) с последующей другой хроматографией среднего давления (элюент: MeOH/CH2Cl2 от 1:39 до 1:19), получая 4-бензо[b]тиофен-5-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин (320 мг, 14% для трех стадий) в виде бесцветного масла. Затем продукт растворяли в MeOH (5 мл) и к полученному раствору добавляли фумаровую кислоту (100 мг, 0,86 ммоль). Раствор концентрировали примерно до 2 мл. К полученному в результате раствору добавляли воду (20 мл), затем раствор лиофилизировали в сублимационной сушилке в течение 48 часов, получая фумаратную соль (+)-4-бензо[b]тиофен-5-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина (284 мг, 56%, 98.6% AUC ВЭЖХ) в виде не совсем белого твердого вещества: [α]25D +12,7° (c 0,06, метанол); т.пл. 118-120°C;1H ЯМР (500 МГц, CD3OD) δ 7,89 (д, J=8,5 Гц, 1H), 7,72 (с, 1H), 7,60 (д, J=5,4 Гц, 1H), 7,33 (д, J=5,4 Гц, 1H), 7,17 (д, J=8,3 Гц, 1H), 6,88-6,85 (м, 1H), 6,80-6,78 (м, 2H), 6,71 (с, 4H), 4,58 (дд, J=10,9, 6,0 Гц, 1H), 4,44-4,37 (м, 2H), 3,82-3,80 (м, 4H), 3,74 (дд, J=12,0, 6,1 Гц, 1H), 3,43-3,38 (м, 1Н), 3,14-3,12 (м, 4H), 2,96 (с, 3H); ESI-МС m/z 365 [M+Н]+; элементный анализ, вычислено для C22H24N2OS·1,75C4H4O4·0,5H2O: C, 60,3; Н, 5,60; N, 4,85. Найдено: C, 60,61; Н, 5,57; N, 4,62.
Пример 101 - Получение фумаратной соли (+/-)-(4-бензо[b]тиофен-5-ил-2-метил-7-морфолин-4-илметил-1,2,3,4-тетрагидроизохинолина
Смесь продукта, полученного в примере 103 (свободное основание, 69 мг, 0,22 ммоль), ди(этиленгликоль)ди-пара-тозилата (92,5 мг, 0,22 ммоль), Na2CO3 (71 мг, 0,67 ммоль) и ацетонитрила кипятили с обратным холодильником в течение ночи. После охлаждения до комнатной температуры твердое вещество фильтровали. Фильтрат разбавляли дихлорметаном (50 мл), промывали насыщенным раствором соли (100 мл), сушили (Na2SO4) и концентрировали. Остаток очищали хроматографией среднего давления (элюент: NH4OH/MeOH/CH2Cl2 3:27:970) с последующей препаративной ВЭЖХ, получая (+/-)-(4-бензо[b]тиофен-5-ил-2-метил-7-морфолин-4-илметил-1,2,3,4-тетрагидроизохинолин (26 мг, 31%) в виде полутвердого вещества. Твердое вещество растворяли в метаноле (2 мл) и к полученному раствору добавляли фумаровую кислоту (8,0 мг, 0,069 ммоль). Раствор концентрировали примерно до 1 мл и к полученному раствору добавляли воду (5 мл). Полученную в результате суспензию лиофилизировали в сублимационной сушилке в течение ночи, получая фумаратную соль (+/-)-(4-бензо[b]тиофен-5-ил-2-метил-7-морфолин-4-илметил-1,2,3,4-тетрагидроизохинолина (30 мг, 87%) в виде белого твердого вещества: т.пл. 115-118°C;1H ЯМР (500 МГц, CD3OD) δ 7,89 (д, J=8,4 Гц, 1H), 7,73 (с, 1H), 7,60 (д, J=5,4 Гц, 1Н), 7,34 (д, J=5,4 Гц, 1H), 7,26 (с, 1Н), 7,21 (д, J=8,0 Гц, 1H), 7,16 (д, J=8,4 Гц, 1H), 6,90 (д, J=8,0 Гц, 1H), 6,70 (с, 3H), 4,62-4,61 (м, 1H), 4,40-4,30 (м, 2H), 3,71-3,63 (м, 7H), 3,34-3,32 (м, 1H), 2,89 (с, 3H), 2,59 (м, 4H); ESI-МС m/z 379 [M+H]+.
Пример 102 - Получение фумаратной соли (+/-)-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)диметиламина
Стадия A: К холодному (0°C) раствору (+/-)-4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрила (180 мг, 0,59 ммоль), который получали таким же способом, как способ получения продукта примера 104, в ТГФ (5 мл) добавляли LAH в ТГФ (1,8 мл, 1,0 М, 1,8 ммоль). Реакционной смеси давали возможность нагреться до комнатной температуры и перемешивали при комнатной температуре в течение 5 часов. Реакцию гасили ледяной водой. Продукт экстрагировали дихлорметаном (2×100 мл). Органические слои объединяли, промывали насыщенным раствором соли (100 мл), сушили (Na2SO4) и концентрировали. Остаток очищали хроматографией среднего давления (элюент: NH4OH/MeOH/ДХМ 1:9:190), получая 4-бензо[b]тиофенил-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбальдегид (35 мг, 19%) в виде бесцветного масла: ESI-МС m/z 308 [M+H]+.
Стадия B: К раствору продукта, полученного на стадии A (35 мг, 0,11 ммоль) в ТГФ (2 мл), добавляли диметиламин в ТГФ (0,12 мл, 2,0 М) при комнатной температуре. После перемешивания при комнатной температуре в течение 2 часов к реакционной смеси при комнатной температуре добавляли NaBH4 (13 мг, 0,34 ммоль). Реакционную смесь дополнительно перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь вливали в насыщенный водный раствор хлорида аммония (50 мл) и продукт экстрагировали дихлорметаном (2×50 мл). Органические слои объединяли, промывали насыщенным раствором соли (50 мл), сушили (Na2SO4) и концентрировали. Остаток очищали на колонке Biotage (элюент: NH4OH/MeOH/ДХМ 3:27:970) с последующей ВЭЖХ, получая (+/-)-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)диметиламин (10 мг, 26%) в виде бесцветного густого масла. Соединение растворяли в метаноле (2 мл) и к полученному раствору добавляли фумаровую кислоту (5,0 мг, 0,043 ммоль). Раствор концентрировали примерно до 1 мл и к полученному раствору добавляли воду (5 мл). Полученную в результате суспензию лиофилизировали в сублимационной сушилке в течение ночи, получая фумаратную соль (+/-)-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)диметиламина (14 мг, 85%) в виде белого твердого вещества: т.пл. 123-125°C;1H ЯМР (500 МГц, CD3OD) δ 7,85 (д, J=8,3 Гц, 1H), 7,70 (с, 1Н), 7,58 (д, J=5,4 Гц, 1H), 7,32-7,31 (м, 2H), 7,22 (д, J=8,3 Гц, 1Н), 7,15 (д, J=8,3 Гц, 1H), 6,99 (д, J=8,0 Гц, 1H), 6,70 (с, 5H), 4,58-4,52 (м, 1H), 4,19 (с, 2H), 4,02-3,94 (м, 2H), 3,43-3,38 (м, 1H), 2,95-2,94 (м, 1Н), 2,80 (с, 6H), 2,65 (с, 3H); ESI-МС m/z 337 [M+H]+.
Пример 103 - Получение фумаратной соли (+)-C-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)метиламина
К холодному (0°C) и перемешиваемому раствору продукта, полученного согласно примеру 104 (свободное основание, 120 мг, 0,40 ммоль) в ТГФ (5 мл), добавляли LAH в ТГФ (1,2 мл, 1,2 ммоль). Реакционной смеси давали возможность нагреться до комнатной температуры и перемешивали в течение 24 часов при комнатной температуре. Реакционную смесь медленно добавляли к ледяной воде и продукт экстрагировали дихлорметаном (2×100 мл). Органические слои объединяли, промывали насыщенным раствором соли (100 мл), сушили (Na2SO4) и концентрировали. Остаток очищали хроматографией среднего давления (элюент: NH4OH/MeOH/CH2Cl2 1:9:190), получая C-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)метиламин (86 мг, 70%) [[α]25D +71,1° (c 0,045, метанол)]. Полученное соединение (16 мг, 0,052 ммоль) растворяли в метаноле (2 мл) и к раствору добавляли фумаровую кислоту (6,0 мг, 0,052 ммоль). Раствор концентрировали примерно до 1 мл и к полученному раствору добавляли воду (5 мл). Полученную в результате суспензию лиофилизировали в сублимационной сушилке в течение ночи, получая фумаратную соль C-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)метиламина (18 мг, 56%, 95,3% AUC ВЭЖХ): т.пл. 143-145°C;1H ЯМР (500 МГц, CD3OD) δ 7,86 (д, J=8,4 Гц, 1H), 7,71 (с, 1H), 7,59 (д, J=5,4 Гц, 1Н), 7,31 (д, J=5,4 Гц, 1H), 7,29 (с, 1H), 7,22 (д, J=8,1 Гц, 1H), 7,14 (д, J=8,4 Гц, 1H), 6,95 (д, J=8,1 Гц, 1H), 6,67 (с, 5H), 4,62-4,60 (м, 1H), 4,29-4,15 (м, 2H), 4,08 (с, 2H), 3,59-3,55 (м, 1H), 3,20-3,15 (м, 1H), 2,79 (с, 3H); ESI-МС m/z 309 [M+H]+; элементный анализ, вычислено для C19H20N2S·2,5C4H4O4·2,25H2O: C, 54,50; C, 5,44; N, 4,38. Найдено: C, 54,72; Н, 5,37; N, 4,04.
Пример 104 - Получение фумаратной соли (+)-4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрила
Смесь продукта, полученного на стадии A примера 97 (300 мг, 0,702 ммоль), цианида цинка (165 мг, 1,40 ммоль) и ДМФА (6 мл) продували аргоном. К смеси добавляли Pd(PPh3)4 (123 мг, 0,105 ммоль) и смесь дополнительно продували аргоном. Смесь нагревали при 120°C в течение 4 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли дихлорметаном (100 мл) и фильтровали через целит. Фильтрат промывали насыщенным раствором соли (100 мл), сушили (Na2SO4) и концентрировали. Остаток очищали хроматографией среднего давления (элюент: MeOH/EtOAc/гексаны 3:57:140), получая (+)-4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрил (68 мг, 32%) [[α]25D +97,8° (c 0,05, метанол)] в виде бесцветного полутвердого вещества. Соединение (68 мг, 0,22 ммоль) растворяли в метаноле (2 мл) и к полученному раствору добавляли фумаровую кислоту (26 мг, 0,07 ммоль). Раствор концентрировали примерно до 1 мл и к полученному раствору добавляли воду (5 мл). Полученную в результате суспензию лиофилизировали в сублимационной сушилке в течение ночи, получая фумаратную соль (+)-4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрила (50 мг, 53%, 96% AUC ВЭЖХ) в виде белого твердого вещества: т.пл. 112-114°C;1H ЯМР (500 МГц, CD3OD) δ 7,88 (д, J=8,3 Гц, 1H), 7,71 (с, 1H), 7,63 (с, 1Н), 7,60 (д, J=5,4 Гц, 1H), 7,46 (д, J=7,8 Гц, 1H), 7,33 (д, J=5,4 Гц, 1H), 7,16 (д, J=8,4 Гц, 1H), 7,06 (д, J=8,1 Гц, 1H), 6,72 (с, 3H), 4,59 (дд, J=10,0, 6,3 Гц, 1H), 4,22 (д, J=15,5 Гц, 1H), 4,09 (д, J=15,5 Гц, 1H), 3,49 (дд, J=11,9, 6,0 Гц, 1H), 3,10-3,08 (м, 1H), 2,73 (с, 3H); ESI-МС m/z 305 [M+H]+; элементный анализ, вычислено для C19H16N2S·1,75C4H4O4·H2O: C, 59,42; Н, 4,79; N, 5,33. Найдено: C, 59,27; Н, 4,43; N, 5,33.
Пример 105 - Получение фумаратной соли (+/-)-4-(бензо[b]тиофен-5-ил)-1,2-диметил-1,2,3,4-тетрагидроизохинолина
Стадия A: Смесь 5-бромбензо[b]тиофена (1,28 г, 6,0 ммоль), изохинолин-4-бороновой кислоты (1,04 г, 6 ммоль) и карбоната цезия (1,95 г, 6,0 ммоль) в 1,2-диметоксиэтане (50 мл) и 2 М карбонате натрия (6 мл) дегазировали, используя аргон. Добавляли Pd(PPh3)4 (416 мг, 0,36 ммоль) и реакционную смесь нагревали при кипячении с обратным холодильником в течение 15 часов. Охлажденную реакционную смесь разбавляли водой и дважды экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали. Очистка хроматографией на колонке (силикагель, от 10% до 30% этилацетат/гексаны) давала требуемый продукт (510 мг, 32%):1H ЯМР (CDCl3, 500 МГц) δ 9,28 (с, 1H), 8,55 (с, 1H), 8,07-8,02 (м, 2H), 7,96-7,93 (м, 2H), 7,69-7,62 (м, 2H), 7,55 (д, J=5,4 Гц, 1H), 7,49 (дд, J=8,2, 1,6 Гц, 1H), 7,42 (дд, J=5,4, 0,4 Гц, 1H); ESI-МС m/z = 262 [M+H]+.
Стадия B: К охлажденному на льду раствору продукта со стадии A (240 мг, 0,918 ммоль) в дихлорметане (4 мл) добавляли метилтрифторметансульфонат (181 мг, 1,102 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель удаляли при пониженном давлении и остаток растворяли в ТГФ, охлаждали на бане со льдом и обрабатывали метилмагнийиодидом (3M в эфире, 0,62 мл, 1,86 ммоль). Реакционную смесь перемешивали при 0°C в течение 2 часов, гасили насыщенным раствором хлорида аммония и три раза экстрагировали этилацетатом. Объединенные экстракты промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали, получая требуемый продукт (300 мг, выход сырого продукта >99%): ESI МС m/z=292 [M+H]+. Полученный сырой продукт использовали на следующей стадии без дополнительной очистки.
Стадия C: К раствору неочищенного продукта со стадии B (300 мг, 0,918 ммоль) в метаноле (25 мл) добавляли цианоборгидрид натрия (300 мг, 4,77 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли еще две порции цианоборгидрида (300 мг, 4,77 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи до завершения реакции. Затем реакционную смесь гасили водой и три раза экстрагировали этилацетатом. Объединенные экстракты промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали. Очистка хроматографией на колонке (силикагель, от 15% до 40% этилацетат/гексаны) давала требуемый продукт (70 мг, 26% для 3 стадий):1H ЯМР (CDCl3, 300 МГц) δ 7,79 (д, J=8,3 Гц, 1H), 7,65 (д, J=1,4 Гц, 1H), 7,42 (д, J=5,5 Гц, 1H), 7,28-7,12 (м, 4H), 7,03 (т, J=8,0 Гц, 1H), 6,81 (д, J=7,7 Гц, 1Н), 4,41 (дд, J=9,4, 5,1 Гц, 1H), 3,71 (кв., J=6,4 Гц, 1H), 3,18 (дд, J=11,6, 5,1 Гц, 1Н), 2,74 (дд, J=11,6, 9,6 Гц, 1H), 2,48 (с, 3H), 1,50 (д, J=6,4 Гц, 3H); ESI МС m/z=294 [M+H]+.
Стадия D: К раствору продукта со стадии C (67 мг, 0,228 ммоль) в метаноле (2 мл) добавляли фумаровую кислоту (27 мг, 0,228 ммоль). Растворитель удаляли при пониженном давлении. Остаток растирали в этилацетате и диэтиловом эфире. Полученный в результате преципитат собирали фильтрованием, промывали диэтиловым эфиром и сушили при 50°C в вакууме, получая фумаратную соль (+/-)-4-(бензотиофен-5-ил)-1,2-диметил-1,2,3,4-тетрагидроизохинолина в виде белого твердого вещества (70 мг, 75%, 98,0% AUC ВЭЖХ): т.пл. 179-181°C;1H ЯМР (CD3OD, 500 МГц) δ 7,89 (д, J=8,3 Гц, 1H), 7,73 (д, J=1,4 Гц, 1Н), 7,60 (д, J=5,5 Гц, 1H), 7,40 (д, J=8,0 Гц, 1H), 7,35-7,31 (м, 2H), 7,21-7,14 (м, 2H), 6,87 (д, J=7,8 Гц, 1H), 6,69 (с, 2H), 4,70-4,63 (м, 2H), 3,74 (дд, J=12,4, 5,8 Гц, 1H), 3,56-3,51 (м, 1H), 2,96 (с, 3H), 1,78 (д, J=6,7 Гц, 3H); ESI МС m/z = 294 [M+H]+.
Пример 106 - Получение фумаратной соли (+/-)-4-(бензо[b]тиофен-5-ил)-8-метокси-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору 2-анисальдегид (5,56 г, 40 ммоль) в метаноле (40 мл) добавляли метиламин (40% мас. раствор в воде, 6,9 мл, 80 моль) и уксусную кислоту (240 мг, 4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов и затем охлаждали на бане со льдом. К полученной охлажденной на льду смеси порциями добавляли боргидрид натрия (1,51 г, 40 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Большую часть растворителя удаляли при пониженном давлении. Остаток разбавляли водой (100 мл) и этилацетатом (100 мл). Органический слой отделяли и водный слой дважды экстрагировали этилацетатом. Объединенные экстракты промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали, получая требуемый 2-метоксибензилметиламин (4,7 г, 79%):1H ЯМР (CDCl3, 500 МГц) δ 7,23 (д, J=7,4 Гц, 2H), 6,91 (т, J=7,4 Гц, 1Н), 6,86 (д, J=7,9 Гц, 1H), 3,83 (с, 3H), 3,75 (с, 2H), 2,42 (с, 3H); ESI МС m/z=152 [M+H]+.
Полученный сырой продукт использовали на следующей стадии без дополнительной очистки.
Стадия B: К охлажденной на льду смеси продукта со стадии A (348 мг, 2,3 ммоль) и диизопропилэтиламина (394 мг, 3,0 ммоль) в дихлорметане (10 мл) добавляли 1-(бензотиофен-5-ил)-2-бромэтанон (см. получение в примере 96) (600 мг, 2,03 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов и затем разбавляли дихлорметаном (100 мл). Смесь промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали, получая требуемый продукт (800 мг, выход сырого продукта >99%): ESI-МС m/z=326 [M+H]+. Полученный сырой продукт использовали на следующей стадии без дополнительной очистки.
Стадия C: К охлажденному на льду раствору продукта со стадии B (800 мг, 2,0 ммоль) в метаноле (10 мл) добавляли боргидрид натрия (80 мг, 2,1 ммоль). Реакционную смесь перемешивали при 0°C в течение 1 часа. Растворитель удаляли при пониженном давлении. Остаток распределяли между дихлорметаном и водой. Водный слой дважды экстрагировали дихлорметаном. Объединенные экстракты промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали, получая требуемый продукт (700 мг, выход сырого продукта 99%): ESI-МС m/z=328 [M+H]+. Полученный сырой продукт использовали на следующей стадии без дополнительной очистки.
Стадия D: К охлажденному на льду раствору неочищенного продукта со стадии C (500 мг, 1,43 ммоль) в дихлорметане (300 мл) по каплям добавляли метансульфоновую кислоту (1 мл, 15,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли еще 1 мл метансульфоновой кислоты и реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь доводили до pH >8 добавлением насыщенного раствора бикарбоната натрия. Органический слой отделяли и водный слой дважды экстрагировали дихлорметаном. Объединенные экстракты промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали. Очистка хроматографией на колонке (силикагель, от 10 до 30% этилацетат/гексаны) давала требуемый продукт (37 мг, 8% для 4 стадий):1H ЯМР (CDCl3, 500 МГц) δ 7,78 (д, J=8,3 Гц, 1H), 7,65 (д, J=1,4 Гц, 1H), 7,41 (д, J=5,4 Гц, 1H), 7,26 (д, J=6,0 Гц, 1H), 7,17 (дд, J=8,3, 1,6 Гц, 1H), 7,03 (т, J=7,9 Гц, 1H), 6,69 (д, J=8,1 Гц, 1Н), 6,50 (д, J=7,8 Гц, 1H), 4,39-4,37 (м, 1H), 3,86 (с, 3H), 3,84 (д, J=15,8 Гц, 1H), 3,49 (д, J=15,8 Гц, 1H), 3,05-3,01 (м, 1H), 2,61 (дд, J=11,4, 8,5 Гц, 1H), 2,46 (с, 3H); ESI-МС m/z=310 [M+H]+.
Стадия E: К раствору продукта со стадии D (37 мг, 0,119 ммоль) в метаноле (1 мл) добавляли фумаровую кислоту (14 мг, 0,119 ммоль). Растворитель удаляли при пониженном давлении. Остаток растирали в этилацетате и диэтиловом эфире. Полученный в результате преципитат собирали фильтрованием, промывали диэтиловым эфиром и сушили при 50°C в вакууме, получая фумаратную соль (+/-)-4-(бензо[b]тиофен-5-ил)-8-метокси-2-метил-1,2,3,4-тетрагидроизохинолина в виде белого твердого вещества (40 мг, 91%, >99% AUC ВЭЖХ): т.пл. 197-199°C;1H ЯМР (CD3OD, 500 МГц) δ 7,85 (д, J=8,4 Гц, 1H), 7,69 (д, J=1,3 Гц, 1Н), 7,58 (д, J=5,4 Гц, 1H), 7,32 (д, J=5,6 Гц, 1H), 7,15-7,12 (м, 2H), 6,86 (д, J=8,2 Гц, 1Н), 6,66 (с, 1H), 6,46 (д, J=7,6 Гц, 1H), 4,60-4,56 (м, 1Н), 4,35 (д, J=15,8 Гц, 1Н), 3,90 (д, J=15,8 Гц, 1H), 3,89 (с, 3H), 3,52-3,48 (м, 1H), 3,15-3,11 (м, 1H), 2,80 (с, 3H); ESI-МС m/z=310 [M+H]+.
Пример 107 - Получение (+/-)-4-бензо[b]тиофен-5-ил-8-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-4-ола
Стадия A: К смеси (2-йод-6-метоксифенил)метанола (2,00 г, 7,65 ммоль) в хлороформе (35 мл) добавляли оксид марганца IV (17,96 г, 206,5 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 24 часов. Охлажденную реакционную смесь фильтровали через небольшой слой диатомовой земли. Фильтрат концентрировали. Очистка флэш-хроматографией на колонке (силикагель; 10:90 этилацетат/гексаны) давала требуемый продукт (0,43 г, 22%) в виде желтого масла;1H ЯМР (CDCl3, 300 МГц) δ 10,25 (с, 1H), 7,59 (д, J=7,7 Гц, 1H), 7,13 (т, J=8,0 Гц, 1H), 6,99 (д, J=8,4 Гц, 1H), 3,91 (с, 3H).
Стадия B: Продукт со стадии A (0,43 г, 1,64 ммоль) и метиламин (40% водный раствор в воде, 0,14 мл, 1,67 ммоль) перемешивали в метаноле (2,5 мл) при комнатной температуре в течение 30 минут. Затем реакционную смесь охлаждали до 0°C и обрабатывали боргидридом натрия (90 мг, 2,46 ммоль). Реакционную смесь перемешивали при 0°C в течение 1 часа, нагревали до комнатной температуры и перемешивали еще в течение 3,5 часов. Реакционную смесь разбавляли водой, три раза экстрагировали метиленхлоридом, сушили над сульфатом натрия, фильтровали и растворитель выпаривали, получая требуемый продукт (0,42 г, 93%) в виде желтого масла:1H ЯМР (CDCl3, 500 МГц) δ 7,42 (д, J=7,9 Гц, 1H), 6,92 (т, J=8,1 Гц, 1H), 6,84 (д, J=7,8 Гц, 1H), 3,85 (с, 2H), 3,80 (с, 3H), 2,43 (с, 3H).
Стадия C: Продукт со стадии B (0,43 г, 1,55 ммоль) и N,N-диизопропилэтиламин (0,26 г, 2,02 ммоль) в метиленхлориде (4 мл) охлаждали до 0°C и обрабатывали 1-бензо[b]тиофен-5-ил-2-бромэтаноном, который получали согласно стадии C примера 98 (0,475 г, 1,86 ммоль), порциями в течение 10-минутного периода времени. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь три раза промывали водой, сушили над сульфатом натрия, фильтровали и растворитель выпаривали. Очистка хроматографией на колонке (40 г силикагеля; 70:30 гексаны/этилацетат) давала продукт (0,39 г, 56%) в виде светло-желтого твердого вещества:1H ЯМР (CDCl3, 500 МГц) δ 8,49 (с, 1H), 7,94 (д, J=8,5 Гц, 1H), 7,86 (д, J=8,5 Гц, 1H), 7,54 (д, J=8,0 Гц, 1H), 7,48 (д, J=5,5 Гц, 1H), 7,40 (д, J=5,4 Гц, 1H), 6,97 (т, J=8,1 Гц, 1H), 6,86 (д, J=8,2 Гц, 1H), 3,86 (с, 2H), 3,79 (с, 2H), 3,73 (с, 3H), 2,45 (с, 3H).
Стадия D: К продукту со стадии C (0,39 г, 0,864 ммоль) в ТГФ (4 мл) по каплям добавляли н-бутиллитий (1,6 М в гексане, 0,7 мл, 1,12 ммоль) при -70°C. Реакционную смесь перемешивали еще в течение 10 минут. Реакционную смесь гасили водой и водным раствором хлорида аммония (5 мл). Раствор четыре раза экстрагировали этилацетатом, три раза промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на колонке (40 г силикагеля; 60:40 этилацетат/гексан) давала 4-бензо[b]тиофен-5-ил-8-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-4-ол (0,15 г, 53%, 99% AUC ВЭЖХ) в виде не совсем белого твердого вещества:1H ЯМР (CDCl3, 500 МГц) δ 8,06 (с, 1Н), 7,79 (д, J=8,5 Гц, 1H), 7,43 (д, J=5,4 Гц, 1Н), 7,32 (д, J=5,4 Гц, 1Н), 7,29-7,26 (м, 1H), 7,08 (т, J=8,0 Гц, 1H), 6,75 (д, J=7,5 Гц, 1H), 6,56 (д, J=7,8 Гц, 1H), 4,10 (д, J=8,7 Гц, 1H), 3,86 (с, 3H), 3,26 (д, J=15,9 Гц, 1H), 2,90 (д, J=11,5 Гц, 1H), 2,75 (д, J=11,5 Гц, 1H), 2,52 (с, 3H); ESI МС m/z 326 [M+H]+.
Пример 108 - Получение соли фумаровой кислоты и (+)-4-бензо[b]тиофен-5-ил-7-фтор-2-метил-1,2,3,4-тетрагидроизохинолин-4-ола и соли фумаровой кислоты и (-)-4-бензо[b]тиофен-5-ил-7-фтор-2-метил-1,2,3,4-тетрагидроизохинолин-4-ола
Стадия A: Раствор нитрита натрия (4,5 г, 65 ммоль) в воде
(75 мл) охлаждали и добавляли к 2-амино-5-фторбензойной кислоте (10,0 г, 65 ммоль) в 2 N хлористоводородной кислоте (150 мл) при -5°C и смесь перемешивали в течение 30 минут.
В отдельном сосуде йодид калия (21,5 г, 130 ммоль) и йодид меди (I) (6,2 г, 32,5 ммоль) растворяли в воде (75 мл) и охлаждали до -5°C. К полученному раствору по каплям добавляли указанный выше раствор диазония. Образовавшемуся в результате красно-коричневому преципитату давали возможность нагреться до комнатной температуры в течение 4-часового периода времени. Преципитат выделяли фильтрованием и твердое вещество промывали водой и сушили в вакууме в течение 24 часов. Коричневое твердое вещество суспендировали в трет-бутилметиловом эфире и нагревали до 56°C; неорганическую соль отфильтровывали. Фильтрат концентрировали до получения взвеси и добавляли гексаны, вызывая большее образование преципитата. Смеси давали возможность перемешиваться еще в течение 1 часа, затем фильтровали и промывали гексанами и сушили дополнительно в вакуумной печи, получая 5-фтор-2-йодбензойную кислоту (9,0 г, 52%):1H ЯМР (500 МГц, CD3OD) δ 8,01 (дд, J=8,7, 5,4 Гц, 1H), 7,55 (дд, J=9,2, 3,0 Гц, 1Н), 7,05-7,01 (м, 1Н).
Стадия B: Продукт со стадии A (9,0 г, 34 ммоль) растворяли в тетрагидрофуране (50 мл) и охлаждали на бане со льдом в атмосфере азота. По каплям добавляли 2,0 М раствор боран-метилсульфидного комплекса (42 мл, 8,5 ммоль) в тетрагидрофуране и смесь перемешивали в течение 30 минут. Баню со льдом убирали и реакционную смесь кипятили с обратным холодильником в течение 2 часов при 70°C. Растворитель концентрировали в вакууме и остаток растворяли в насыщенном растворе хлорида аммония и дважды экстрагировали метиленхлоридом. Органический экстракт промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая требуемый продукт (6,2 г, 73%) в виде не совсем белого твердого вещества:1H ЯМР (500 МГц, CDCl3) δ 7,78-7,69 (м, 1H), 7,31-7,22 (м, 1H), 6,83-6,75 (м, 1H), 4,64 (с, 2H), 2,04 (с, 1H).
Стадия C: Продукт со стадии B (6,2 г, 25,0 ммоль) растворяли в хлороформе (170 мл) и раствор добавляли к суспензии оксида марганца IV (43,0 г, 675 ммоль) в хлороформе (150 мл) и смесь перемешивали в течение ночи при 75°C. Реакционную смесь фильтровали через слой диатомовой земли и концентрировали в вакууме, получая требуемый продукт (3,7 г, выход сырого продукта 60%) в виде желтого твердого вещества:1H ЯМР (500 МГц, CDCl3) δ 10,01 (д, J=3,0 Гц, 1Н), 7,92 (дд, J=8,7, 5,0 Гц, 1H), 7,60 (дд, J=8,6, 3,1 Гц, 1H), 7,10-7,06 (м, 1Н).
Стадия D: Неочищенный продукт со стадии C (3,7 г, 14,8 ммоль) растворяли в метаноле (20 мл) и 40% водном растворе N-метиламина (1,3 мл, 15,0 ммоль) и смесь перемешивали в течение 1,5 часа. Затем реакционную смесь охлаждали на бане со льдом и порциями добавляли боргидрид натрия (0,8 г, 22,0 ммоль) и смесь перемешивали при комнатной температуре еще в течение 2 часов. Смесь концентрировали в вакууме и остаток растворяли в воде (20 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические экстракты промывали насыщенным раствором соли (20 мл), сушили над сульфатом натрия и концентрировали в вакууме получая желтое масло (3,3 г, выход сырого продукта 83%):1H ЯМР (500 МГц, CDCl3) δ 7,74-7,67 (м, 1H), 7,12-7,02 (м, 1Н), 6,68-6,64 (м, 1H), 3,66 (с, 2H), 2,39 (с, 3H), 1,58 (ушир.с, 1H).
Стадия E: К раствору 1-бензо[b]тиофен-5-ил-2-бромэтанона (см. пример 98, стадия C) добавляли N,N-диизопропилэтиламин (0,51 мл, 2,9 ммоль) и смесь перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь гасили водой (5 мл) и органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, затем очищали хроматографией на колонке (5-40% этилацетат/гексаны), получая требуемый продукт (0,97 г, 92%) в виде желтого масла:1H ЯМР (500 МГц, CDCl3) δ 8,47 (д, J=1,0 Гц, 1H), 7,96-7,91 (м, 2H), 7,77 (дд, J=8,6, 5,6 Гц, 1H), 7,52 (д, J=5,4 Гц, 1H), 7,42 (д, J=5,4 Гц, 1Н), 7,30 (дд, J=9,7, 3,1 Гц, 1H), 6,76-6,72 (м, 1H), 3,98 (с, 2H), 3,74 (с, 2H), 2,45 (с, 3H).
Стадия F: 1,6 М раствор н-бутиллития в гексанах (3,3 мл, 5,3 ммоль) добавляли к охлажденному (-60°C) раствору продукта, полученного на стадии E (1,7 г, 4,1 ммоль) в безводном тетрагидрофуране в течение 10 минут. Реакционную смесь гасили водой (10 мл) и насыщенным раствором хлорида аммония (5 мл). Органический слой сушили над сульфатом натрия, концентрировали в вакууме и очищали хроматографией на колонке (5-50% этилацетат/гексаны), получая требуемый продукт (0,72 г, 60%):1H ЯМР (500 МГц, CDCl3) δ 8,08 (с, 1Н), 7,89 (д, J=1,5 Гц, 1H), 7,79 (д, J=8,5 Гц, 1H), 7,54 (д, J=5,4 Гц, 1H), 7,32 (д, J=5,4 Гц, 1Н), 7,26 (дд, J=8,5, 1,7 Гц, 1H), 7,05 (дд, J=8,6, 5,7 Гц, 1H), 6,94-6,86 (м, 2H), 3,84 (д, J=15,2 Гц, 1Н), 3,58 (д, J=15,2 Гц, 1H), 2,96 (дд, J=11,9, 1,1 Гц, 1H), 2,81 (д, J=11,9 Гц, 1Н), 2,42 (с, 3H).
Стадия G: Полученное соединение со стадии F разделяли препаративной хиральной ВЭЖХ (колонка CHIRALCEL OJ, с использованием смеси 90% гептан/10% изопропиловый спирт/0,1% диэтиламин), получая (+)-энантиомер [α]25D +73,8° (0,07, метанол) и (-)-энантиомер [α]25D -60,0° (0,06, метанол). (+)-Энантиомер (14 мг, 0,05 ммоль) превращали в соль фумаровой кислоты растворением свободного основания в минимальном количестве этанола, добавлением одного эквивалента фумаровой кислоты в достаточном количестве метанола, чтобы полностью растворить кислоту, затем объединяя два раствора и перемешивая в течение 2 часов. Раствор концентрировали до минимального объема, затем охлаждали до -30°C до образования кристаллов. Фильтрование давало фумаратную соль 4-бензо[b]тиофен-5-ил-7-фтор-2-метил-1,2,3,4-тетрагидроизохинолин-4-ола (12,0 мг, >99%, 97,3% AUC ВЭЖХ) в виде не совсем белого твердого вещества: т.пл. 95-97,0°C;1H ЯМР (500 МГц, CD3OD) δ 8,00 (д, J=1,3 Гц, 1H), 7,87 (д, J=8,5 Гц, 1H), 7,60 (д, J=5,4 Гц, 1H), 7,37 (д, J=5,4 Гц, 1H), 7,28 (дд, J=1,5, 8,5 Гц, 1H), 7,06-6,97 (м, 3H), 6,71 (с, 2H), 4,32 (д, J=15,5 Гц, 1H), 4,18 (д, J=15,6 Гц, 1H), 3,43-3,35 (м, 2H), 2,81 (с, 3H).
Такой же способ использовали для превращения (-)-энантиомера (15 мг, 0,05 ммоль) в фумаратную соль 4-бензо[b]тиофен-5-ил-7-фтор-2-метил-1,2,3,4-тетрагидроизохинолин-4-ола (16,5 мг, >99%, 98,5% AUC ВЭЖХ) в виде не совсем белого твердого вещества: т.пл. 95-97°C;1H ЯМР (500 МГц, CD3OD) δ 8,00 (д, J=1,2 Гц, 1H), 7,87 (д, J=8,5 Гц, 1H), 7,60 (д, J=5,4 Гц, 1Н), 7,37 (д, J=5,5 Гц, 1H), 7,28 (дд, J=1,4 Гц, 8,5 Гц, 1H), 7,06-6,97 (м, 3H), 6,71 (с, 2H), 4,32 (д, J=15,5 Гц, 1H), 4,18 (д, J=15,4 Гц, 1H), 3,42-3,34 (м, 2H), 2,81 (с, 3H).
Пример 109 - Получение фумаратной соли (+/-)-4-бензо[b]тиофен-5-ил-2-циклопропил-1,2,3,4-тетрагидроизохинолина
Смесь 4-бензо[b]тиофен-5-ил-1,2,3,4-тетрагидроизохинолина (250 мг, 0,94 ммоль, получение которого описано в примере 20), (1-этоксициклопропокси)триметилсилана (987 мг, 5,66 ммоль), уксусной кислоты (600 мг, 9,42 ммоль), цианоборгидрида натрия (296 мг, 4,72 ммоль) и молекулярных сит (4Е) нагревали при кипячении с обратным холодильником в течение 16 часов, затем охлаждали до комнатной температуры. Смесь обрабатывали водой, концентрировали в вакууме, остаток растворяли в этилацетате и промывали 1N раствором NaOH, затем насыщенным раствором соли. Органический слой сушили над сульфатом магния, концентрировали и остаток очищали хроматографией на колонке (SiO2, 12 г, от 85% до 0% гексаны/этилацетат), получая требуемый тетрагидроизохинолин (87 мг, 30%). Полученный продукт растворяли в метаноле и обрабатывали раствором фумаровой кислоты в метаноле (5 мл) при 0°C. Раствор перемешивали при комнатной температуре в течение 12 часов, затем концентрировали и твердое вещество промывали диэтиловым эфиром, получая фумаратную соль 4-бензо[b]тиофен-5-ил-2-циклопропил-1,2,3,4-тетрагидроизохинолина (100 мг, 95,5% AUC ВЭЖХ):1H ЯМР (CD3OD, 300 МГц) δ 7,84 (д, J=8,3 Гц, 1Н), 7,70 (д, J=1,4 Гц, 1H), 7,56 (д, J=5,5 Гц, 1H), 7,32 (д, J=5,1 Гц, 1H), 7,20-7,07 (м, 4H), 6,84 (д, J=7,5 Гц, 1Н), 6,73 (с, 2H), 4,48-4,43 (м, 1H), 4,23-4,10 (м, 2H), 3,57-3,47 (м, 2H), 3,13-3,06 (м, 1H), 2,21-2,18 (м, 1H), 0,71-0,64 (м, 4H); ESI-МС m/z=306 [+H]+.
Пример 110 - Получение фумаратной соли (+/-)-(4-бензо[b]тиофен-5-ил-3,4-дигидро-1H-изохинолин-2-ил)ацетонитрила
Смесь 4-бензо[b]тиофен-5-ил-1,2,3,4-тетрагидроизохинолина (250 мг, 0,94 ммоль, получение которого описано в примере 20), хлорацетонитрила (285 мг, 3,77 ммоль), и карбоната цезия (614 мг, 1,88 ммоль) в ДМФА (2 мл) перемешивали при комнатной температуре в течение 3 часов. Смесь разбавляли этилацетатом, промывали 5% раствором LiOH, затем насыщенным раствором соли и органический слой сушили над сульфатом магния и концентрировали. Остаток очищали хроматографией на колонке (SiO2, 12 г, от 85% до 0% гексан/этилацетат), получая требуемый тетрагидроизохинолин (90 мг, 31%). Полученный продукт растворяли в метаноле и обрабатывали раствором фумаровой кислоты в метаноле (5 мл) при 0°C. Раствор перемешивали при комнатной температуре в течение 12 часов, затем концентрировали и твердое вещество промывали диэтиловым эфиром, получая фумаратную соль (4-бензо[b]тиофен-5-ил-3,4-дигидро-1H-изохинолин-2-ил)ацетонитрила (91 мг, 73%, 97,6% AUC ВЭЖХ):1H ЯМР (ДМСО-d6, 300 МГц) δ 7,91 (д, J=8,3 Гц, 1H), 7,74-7,68 (м, 2H), 7,39 (д, J=5,4 Гц, 1Н), 7,24-7,06 (м, 4H), 6,83 (д, J=7,6 Гц, 1H), 6,63 (с, 2H), 4,43-4,39 (м, 1H), 3,99-3,76 (м, 4H), 3,12-3,07 (м, 1H), 2,83-2,77 (м, 1H); ESI-МС m/z=306 [M+H].
Пример 111 - Получение фумаратной соли (+/-)-[2-(4-бензо[b]тиофен-5-ил-3,4-дигидро-1H-изохинолин-2-ил)этил]диметиламина
Стадия A: Смесь 4-бензо[b]тиофен-5-ил-1,2,3,4-тетрагидроизохинолина (250 мг, 0,94 ммоль, получение которого описано в примере 20), гидрохлоридной соли 2-диметиламиноэтилхлорида (271 мг, 1,89 ммоль) и карбоната цезия (1,23 г, 3,77 ммоль) в ДМФА (4 мл) перемешивали при комнатной температуре в течение 3 суток. Смесь разбавляли этилацетатом, промывали 5% раствором LiOH, затем насыщенным раствором соли и органический слой сушили над сульфатом магния и концентрировали. Остаток очищали хроматографией на колонке (SiO2, 12 г, от 85% до 0% гексан/этилацетат), получая требуемый тетрагидроизохинолин (7,0 мг, 2%). Полученный продукт растворяли в метаноле и обрабатывали раствором фумаровой кислоты в метаноле (5 мл) при 0°C. Раствор перемешивали при комнатной температуре в течение 12 часов, затем концентрировали и твердое вещество промывали диэтиловым эфиром, получая фумаратную соль [2-(4-бензо[b]тиофен-5-ил-3,4-дигидро-1H-изохинолин-2-ил)этил]диметиламина (9,2 мг, 98%, 97,2% AUC ВЭЖХ):1H ЯМР (CD3OD, 500 МГц) δ 7,82 (д, J=8,3 Гц, 1H), 7,63 (д, J=1,4 Гц, 1H), 7,54 (д, J=5,5 Гц, 1H), 7,29-7,28 (м, 1H), 7,20-7,10 (м, 4H), 6,90-6,88 (м, 1H), 6,75 (с, 2H), 4,46-4,44 (м, 1H), 4,04-3,94 (м, 2H), 3,16-2,98 (м, 6H), 2,64 (с, 3H), 2,60 (с, 3H); ESI-МС m/z=337 [M+H]+.
Пример 112 - Получение фумаратной соли (+/-)-2-(4-бензо[b]тиофен-5-ил-3,4-дигидро-1H-изохинолин-2-ил)этанола
Стадия A: Смесь 4-бензо[b]тиофен-5-ил-1,2,3,4-тетрагидроизохинолина (40 мг, 0,15 ммоль, получение которого описано в примере 20), 2-бромэтанола (23 мг, 0,18 ммоль) и карбоната цезия (98 мг, 0,30 ммоль) в ДМФА (4 мл) перемешивали при комнатной температуре в течение 12 часов. Смесь разбавляли этилацетатом, промывали 5% раствором LiOH, затем насыщенным раствором соли и органический слой сушили над сульфатом магния и концентрировали. Остаток очищали хроматографией на колонке (SiO2, 12 г, от 85% до 0% гексан/этилацетат), получая требуемый тетрагидроизохинолин (13 мг, 28%). Полученный продукт растворяли в метаноле и обрабатывали раствором фумаровой кислоты в метаноле (5 мл) при 0°C. Раствор перемешивали при комнатной температуре в течение 12 часов, затем концентрировали и твердое вещество промывали диэтиловым эфиром, получая фумаратную соль 2-(4-бензо[b]тиофен-5-ил-3,4-дигидро-1H-изохинолин-2-ил)этанола (18 мг, 99%, 96,0% AUC ВЭЖХ):1H ЯМР (CD3OD, 500 МГц) δ 7,92 (д, J=8,4 Гц, 1H), 7,79 (д, J=0,9 Гц, 1Н), 7,62 (д, J=5,4 Гц, 1H), 7,36-7,30 (м, 3H), 7,25-7,20 (м, 2H), 6,92 (д, J=7,8 Гц, 1H), 6,74 (с, 2H), 4,77-4,74 (м, 1H), 4,69 (с, 2H), 4,01-3,97 (м, 3H), 3,65-3,58 (м, 1H), 3,48-3,46 (м, 2H), ESI-МС m/z=310 [M+H]+.
Пример 113 - Получение фумаратной соли (+)-4-(7-метоксибензотиофен-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина и фумаратной соли (-)-4-(7-метоксибензотиофен-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: 2-(3-Тиенил)этанол (4,26 г, 33,0 ммоль) растворяли в ацетонитриле (10 мл) и добавляли к охлажденному раствору дибромтрифенилфосфорана (14,01 г, 33,0 ммоль) в ацетонитриле (20 мл) и пиридине (2,68 мл, 33,0 ммоль) и перемешивали в течение 2 часов. Растворители концентрировали и остаток растворяли в эфире (50 мл) и промывали насыщенным раствором бикарбоната натрия, водой и насыщенным раствором соли. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали до получения полутвердого вещества. Сырой продукт очищали хроматографией на колонке (силикагель, 10% этилацетат/гексаны), получая 3-(2- бромэтил)тиофен (3,96 г, 62%) в виде бледно-желтого масла:1H ЯМР (500 МГц, CDCl3) δ 7,28 (дд, J=4,9, 2,9 Гц, 1H), 7,06 (т, J=1,0 Гц, 1Н), 6,97 (дд, J=4,9, 1,1 Гц, 1H), 3,56 (т, J=7,5 Гц, 2H), 3,20 (т, J=7,5 Гц, 2H).
Стадия B: Продукт со стадии A (3,96 г, 21,0 ммоль) растворяли в тетрагидрофуране и медленно по каплям добавляли к охлажденной смеси гидрида натрия (0,83 г, 40,0 ммоль) в тетрагидрофуране (60 мл) и диэтилмалонате (3,32 г, 21,0 ммоль). Смесь перемешивали еще в течение 10 минут до того, как убирали баню со льдом, затем перемешивали в течение часа при комнатной температуре. К реакционной смеси добавляли каталитическое количество йодида натрия и перемешивали в течение 48 часов. Смесь концентрировали до получения твердого вещества и перерастворяли в этилацетате, затем промывали водой и дважды экстрагировали этилацетатом. Органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали до получения желтого масла. Масло очищали хроматографией на колонке (силикагель, 10% этилацетат/гексаны), получая требуемый малонат (3,0 г, 54%) в виде бесцветного масла:1H ЯМР (500 МГц, CDCl3) δ 7,26-7,25 (м, 1H), 6,98-6,94 (м, 2H), 4,22-4,17 (м, 4H), 3,34 (т, J=7,5 Гц, 1H), 2,71-2,68 (м, 2H), 2,25-2,20 (м, 2H), 1,28-1,26 (м, 6H).
Стадия C: Продукт со стадии B (3,0 г, 11,0 ммоль) растворяли в 20% растворе гидроксида калия (20 мл) и нагревали до 60°C в течение 2,5 часов. Температуру реакционной смеси возвращали к комнатной температуре, затем смесь охлаждали на бане со льдом и добавляли холодный 6 N раствор хлористого водорода (25 мл) и диэтиловый эфир (25 мл). Баню со льдом убирали и реакционную смесь нагревали до 40°C в течение ночи. Температуру реакционной смеси возвращали к комнатной температуре и водный слой насыщали хлоридом натрия. Органический слой дважды экстрагировали диэтиловым эфиром, сушили над сульфатом натрия, фильтровали и концентрировали, получая 2-(2-тиофен-3-илэтил)малоновую кислоту (2,4 г, 99%) в виде масла:1H ЯМР (500 МГц, CDCl3) δ 7,24 (дд, J=4,6, 2,9 Гц, 1H), 6,99-6,95 (м, 2H), 3,34 (т, J=7,3 Гц, 1H), 2,76-2,73 (м, 2H), 2,26-2,12 (м, 2H).
Стадия D: Продукт со стадии C (2,4 г, 11,0 ммоль) растворяли в 2-метоксиэтиловом эфире (20 мл) и кипятили с обратным холодильником в течение 24 часов. Реакционную смесь охлаждали до комнатной температуры и подщелачивали до pH 9 концентрированным гидроксидом аммония. Смесь один раз экстрагировали диэтиловым эфиром и отставляли в сторону. Водный слой подкисляли концентрированной хлористоводородной кислотой и четыре раза экстрагировали диэтиловым эфиром. Экстракт объединяли и сушили над сульфатом натрия, фильтровали и концентрировали, получая 4- тиофен-3-илмасляную кислоту (1,8 г, 95%) в виде масла.
Стадия E: Продукт со стадии D (1,8 г, 11,0 ммоль) растворяли в 2-метоксиэтиловом эфире (2 мл) и добавляли тионилхлорид (0,89 мл, 12,2 ммоль) и пиридин (1 каплю) и нагревали до 70°C в течение 24 часов. Смесь очищали хроматографией на колонке (силикагель, 0-15% этилацетат/гексаны в качестве элюента), получая 5,6-дигидро-4H-бензотиофен-7-он (1,16 г, 72%):1H ЯМР (500 МГц, CDCl3) δ 7,61 (д, J=4,9 Гц, 1H), 6,97 (д, J=4,9 Гц, 1Н), 2,89-2,87 (м, 2H), 2,63-2,60 (м, 2H), 2,21-2,16 (м, 2H).
Стадия F: Продукт со стадии E (1,16 г, 7,6 ммоль) растворяли в хлороформе (20 мл) и добавляли к горячей суспензии (70°C) бромида меди (6,9 г, 30,0 ммоль) в этилацетате (15 мл) и перемешивали в течение 48 часов. Температуру реакционной смеси возвращали к комнатной температуре и фильтровали через слой диатомовой земли. Фильтрат концентрировали и перерастворяли в диэтиловом эфире. Активированный оксид алюминия, нейтральный для хроматографии, 50-200 микрон, использовали, чтобы избавиться от очень темной окраски. Суспензию фильтровали через слой диатомовой земли и сульфата натрия. Смесь концентрировали, получая 6,6-дибром-5,6-дигидро-4H-бензотиофен-7-он (1,95 г, 87% сырого продукта) в виде коричневого твердого вещества:1H ЯМР (500 МГц, CDCl3) δ 7,75 (д, J=4,9 Гц, 1H), 6,99 (д, J=4,9 Гц, 1H), 3,13-3,11 (м, 2H), 3,01-2,99 (м, 2H).
Стадия G: Продукт со стадии F (1,90 г, 6,0 ммоль) растворяли в N,N-диметилформамиде (15 мл) и добавляли карбонат натрия (3,25 г, 30,0 ммоль) и нагревали до 100°C в течение 1,5 часа. Затем суспензию перемешивали в течение 24 часов при комнатной температуре. Реакционную смесь фильтровали через сульфат натрия и промывали диэтиловым эфиром (50 мл). Фильтрат подкисляли до pH 1 концентрированной хлористоводородной кислотой. Органический слой промывали (2×50 мл) водой, затем экстрагировали 5% водным раствором гидроксида калия (3×50 мл). Щелочной водный раствор подкисляли концентрированной хлористоводородной кислотой и экстрагировали (3×50 мл) диэтиловым эфиром. Объединенные эфирные экстракты сушили над сульфатом натрия, фильтровали и концентрировали, получая 6-бромбензотиофен-7-ол (1,09 г, выход сырого продукта 77%) в виде красно-коричневого твердого вещества:1H ЯМР (500 МГц, CDCl3) δ 7,45-7,42 (м, 2H), 7,31-7,29 (м, 2H), 5,93 (с, 1H).
Стадия H: Продукт со стадии G (1,08 г, 4,7 ммоль) растворяли в ацетоне (10 мл) и добавляли карбонат калия (1,30 г, 9,4 ммоль) и диметилсульфат (0,9 мл, 9,4 ммоль) и смесь перемешивали при 50°C в течение 24 часов. Смесь охлаждали до комнатной температуры и фильтровали через сульфат натрия. Затем реакционную смесь концентрировали до получения масла и перерастворяли в метаноле (10 мл) и добавляли гидроксид натрия (1,5 г), чтобы разложить избыток диметилсульфата. Смесь перемешивали в течение 1 часа. Метанол удаляли и остаток растворяли в воде (5 мл) и экстрагировали (2×30 мл) диэтиловым эфиром. Объединенный органический экстракт сушили над сульфатом натрия, фильтровали и концентрировали до получения масла. Масло очищали хроматографией на колонке (силикагель, 0-100% этилацетат/гексаны в качестве элюента), получая 6-бром-7-метоксибензотиофен (0,96 г, 84%):1H ЯМР (500 МГц, CDCl3) δ 7,50 (д, J=8,4 Гц, 1H), 7,44-7,41 (м, 2H), 7,32 (д, J=5,4 Гц, 1H), 4,05 (с, 3H).
Стадия I: Продукт со стадии H (0,59 г, 2,4 ммоль), 4-изохинолинбороновую кислоту (0,63 г, 3,6 ммоль), трифенилфосфин (0,13 г, 0,5 ммоль), 2 N карбонат натрия (1,5 мл) и диметиловый эфир этиленгликоля (10 мл) добавляли в реакционный сосуд и создавали вакуум, затем продували в атмосфере аргона в течение 15 минут. Затем к реакционной смеси добавляли ацетат палладия и смесь нагревали до 80°C в течение 6 часов. Реакционную смесь разбавляли водой (5 мл) и три раза экстрагировали этилацетатом. Экстракты объединяли и промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали до получения коричневого масла. Масло очищали хроматографией на колонке (силикагель, 5-90% этилацетат/гексаны в качестве элюента), получая 4-(7-метоксибензотен-6-ил)изохинолин (0,50 г, 70%) в виде желтого масла:1H ЯМР (300 МГц, CDCl3) δ 9,33 (с, 1Н), 8,60 (с, 1Н), 8,10-8,07 (м, 1H), 7,73-7,65 (м, 4H), 7,56 (д, J=5,4 Гц, 1H), 7,47 (д, J=5,4 Гц, 1Н), 7,36 (д, J=8,1 Гц, 1H), 3,57 (с, 3H).
Стадия J: Продукт со стадии I (0,99 г, 3,4 ммоль) растворяли в метиленхлориде (10 мл) и добавляли метилтрифторметансульфонат (0,46 мл, 0,41 ммоль) и перемешивали в течение 30 минут. Растворитель концентрировали до получения желтого твердого вещества. Твердое вещество перерастворяли в метаноле (10 мл) и охлаждали на бане со льдом. К реакционной смеси добавляли цианоборгидрид натрия (1,29 г, 21 ммоль) и перемешивали в течение 15 минут, затем реакционной смеси давали возможность нагреться до комнатной температуры в течение 2 часов. Реакционную смесь концентрировали и распределяли между водой (20 мл) и этилацетатом (40 мл). Смесь экстрагировали этилацетатом (2×30 мл). Объединенный экстракт промывали насыщенным раствором соли (50 мл) и органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Масло очищали хроматографией на колонке (силикагель, 10-70% этилацетат/гексаны в качестве элюента), получая 4-(7-метоксибензотиофен-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (0,61 г, 58%) в виде желтого масла:1H ЯМР (300 МГц, CDCl3) δ 7,47 (д, J=8,2 Гц, 1H), 7,41 (д, J=5,4 Гц, 1Н), 7,30 (д, J=5,4 Гц, 1Н), 7,13-7,00 (м, 4H), 6,84 (д, J=7,6 Гц, 1H), 4,91-4,86 (м, 1H), 3,98 (с, 3H), 3,82 (д, J=14,8 Гц, 1H), 3,64 (д, J=14,8 Гц, 1H), 3,11-3,05 (м, 1H), 2,62 (дд, J=11,3, 9,0 Гц, 1H), 2,45 (с, 3H); ESI-MS m/z 310 [M+H]+.
Полученное соединение разделяли препаративной хиральной ВЭЖХ (колонка CHIRALPAK AD, с использованием смеси 95% гептан/5% изопропиловый спирт/0,1% диэтиламин), получая (+)-энантиомер [[α]25D +74,7° (c=0,19, метанол)] и (-)-энантиомер [[α]25D -84,0° (c=0,1, метанол)]. (+)-Энантиомер (58 мг, 0,2 ммоль) превращали в соль фумаровой кислоты растворением масла в минимальном количестве этанола, добавлением одного эквивалента фумаровой кислоты в достаточном количестве метанола, чтобы полностью растворить кислоту, затем объединяя два раствора и перемешивая в течение 2 часов. Раствор концентрировали до минимального объема, затем охлаждали при -30°C вплоть до образования кристаллов. Фильтрование давало фумаратную соль (+)-4-(7-метоксибензо[b]тиофен-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (14,8 мг, >99%, 96,2% AUC ВЭЖХ) в виде не совсем белого твердого вещества: т.пл. 98-99°C;1H ЯМР (500 МГц, CD3OD) δ 7,61-7,58 (м, 2H), 7,39 (д, J=5,3 Гц, 1Н), 7,26 (д, J=3,2 Гц, 2H), 7,21-7,18 (м, 1H), 7,10 (д, J=8,2 Гц, 1Н), 6,89 (д, J=7,7 Гц, 1H), 6,70 (с, 2H), 4,98 (дд, J=10,8, 6,3 Гц, 1H), 4,45-4,34 (м, 2H), 3,80 (с, 3H), 3,67-3,64 (м, 1H), 3,45-3,41 (м, 1H), 2,94 (с, 3H).
Такой же способ использовали для превращения (-)-энантиомера (63,6 мг, 0,21 ммоль) в его соль, получая фумаратную соль (-)- 4-(7-метоксибензотиофен-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (10,7 мг, >99%, 97,4% AUC ВЭЖХ) в виде не совсем белого твердого вещества: т.пл. 98-99°C;1H ЯМР (500 МГц, CD3OD) δ 7,61-7,58 (м, 2H), 7,39 (д, J=5,3 Гц, 1H), 7,26 (д, J=3,2 Гц, 2H), 7,21-7,18 (м, 1H), 7,10 (д, J=8,2 Гц, 1H), 6,89 (д, J=7,7 Гц, 1H), 6,70 (с, 2H), 4,98 (дд, J=10,8, 6,3 Гц, 1H), 4,45-4,34 (м, 2H), 3,80 (с, 3H), 3,67-3,64 (м, 1H), 3,45-3,41 (м, 1H), 2,94 (с, 3H).
Пример 114 - Получение малеатной соли (+/-)-4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ола
Стадия A: Смесь 3-метокситиофенола (25,0 г, 175,0 ммоль), карбоната калия (26,6 г, 192,0 ммоль) и бромацетальдегид-диметилацеталя (32,3 мл, 175,0 ммоль) добавляли в ацетон (250,0 мл). Реакционную смесь перемешивали в течение 24 часов. К реакционной смеси добавляли воду и смесь три раза экстрагировали этилацетатом. Объединенный органический слой промывали насыщенным раствором бикарбоната натрия, насыщенным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали, получая требуемый продукт (48,0 г, 99% выход сырого продукта) в виде масла:1H ЯМР (500 МГц, CDCl3) δ 7,20-7,17 (м, 1H), 6,96-6,92 (м, 2H), 6,73-6,71 (м, 1H), 4,65 (т, J=5,5 Гц, 1H), 3,79 (с, 3H), 3,68 (дд, J=9,3, 7,0 Гц, 2H), 3,55 (дд, J=9,3, 7,0 Гц, 2H), 3,14 (д, J=5,6 Гц, 2H), 1,21 (т, J=7,1 Гц, 6H).
Стадия B: Продукт со стадии A (15,0 г, 58,5 ммоль) растворяли в метиленхлориде (125 мл) и раствор добавляли к раствору комплекса диэтилового эфира и трифторида бора (7,86 мл, 62 ммоль) в метиленхлориде (900 мл) при комнатной температуре в атмосфере азота. Реакционную смесь перемешивали в течение 30 минут. К смеси добавляли насыщенный раствор бикарбоната натрия до тех, пока обе фазы не становились прозрачными. Органический слой дважды экстрагировали метиленхлоридом. Объединенные органические экстракты сушили над сульфатом натрия и концентрировали до получения масла. Масло очищали хроматографией на колонке (100% гексаны), получая требуемый 6-метоксибензотиофен (5,0 г, 52%) в виде бесцветного масла:1H ЯМР (500 МГц, CDCl3) δ 7,50 (д, J=5,6 Гц, 1H), 7,46 (д, J=8,1 Гц, 1H), 7,3 (д, J=5,6 Гц, 1Н), 7,29-7,25 (м, 1H), 6,75 (д, J=7,8 Гц, 1H), 3,96 (с, 3H).
Стадия C: Продукт со стадии B (9,1 г, 55,0 ммоль) добавляли неразбавленным к гидрохлориду пиридина (25,6 г, 22 ммоль) при 200°C в течение 2,5 часов. Смеси давали возможность остыть, затем добавляли ледяную воду и дважды экстрагировали метиленхлоридом. Объединенный экстракт сушили над сульфатом натрия, фильтровали и концентрировали до получения масла. Масло отверждалось при отстаивании, затем его растирали в гексанах, получая требуемый продукт (4,42 г, 53,4%):1H ЯМР (500 МГц, CDCl3) δ 7,67 (д, J=8,6 Гц, 1Н), 7,31 (д, J=2,2 Гц, 1Н), 7,26-7,22 (м, 2H), 6,91 (дд, J=8,6, 2,3 Гц, 1H), 4,81 (с, 1H).
Стадия D: Продукт со стадии C (2,9 г, 19,0 ммоль) растворяли в метиленхлориде (40 мл) и триэтиламине (4,0 мл, 29,0 ммоль). Реакционную смесь охлаждали на бане со льдом и добавляли ангидрид трифторметансульфоновой кислоты (3,75 мл, 21,0 ммоль) и перемешивали в течение 30 минут. К смеси добавляли насыщенный раствор хлорида натрия и дважды экстрагировали метиленхлоридом. Органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали до получения желтого твердого вещества (5,4 г, 99%):1H ЯМР (500 МГц, CDCl3) δ 7,86 (д, J=8,8 Гц, 1H), 7,81 (д, J=2,2 Гц, 1H), 7,56 (д, J=5,4 Гц, 1H), 7,37 (д, J=5,5 Гц, 1Н), 7,28 (дд, J=8,7, 2,3 Гц, 1H).
Стадия E: Смесь продукта со стадии D (5,4 г, 19 ммоль), н-бутилвинилового эфира (9,84 мл), триэтиламина (5,33 мл, 38 ммоль) и 1,3-бис(дифенилфосфино)пропана (4,73 г, 11 ммоль) в N,N-диметилформамиде дегазировали, используя аргон, и перемешивали. Затем к реакционной смеси добавляли ацетат палладия (II), затем смесь нагревали до 100°C в течение 4 часов. Охлажденную реакционную смесь фильтровали через слой диатомовой земли и концентрировали до получения желтого твердого вещества. Твердое вещество растворяли в 1 N хлористоводородной кислоте (50,0 мл) и перемешивали в течение 1 часа. Затем смесь концентрировали и очищали хроматографией на колонке (5-25% этилацетат/гексаны в качестве элюента), получая требуемый кетон (2,95 г, 87,5%) в виде желтого твердого вещества:1H ЯМР (500 МГц, CDCl3) δ 8,52-8,51 (м, 1H), 7,97 (дд, J=8,4, 1,6 Гц, 1H), 7,88 (д, J=8,4 Гц, 1H), 7,67 (д, J=5,5 Гц, 1H), 7,41-7,39 (м, 1H), 2,68 (с, 3H).
Стадия F: Продукт со стадии E (2,9 г, 16 ммоль) растворяли в этилацетате (20 мл) и добавляли к суспензии бромида меди (II) (7,35 г, 33,0 ммоль) в хлороформе (40 мл). Реакционную смесь кипятили с обратным холодильником в течение 3 часов, затем давали возможность остыть до комнатной температуры. Смесь фильтровали через слой диатомовой земли и концентрировали до получения коричневого твердого вещества, которое очищали хроматографией на колонке (5-10% этилацетат/гексаны), получая требуемый продукт (3,71 г, 88,5%):1H ЯМР (500 МГц, CDCl3) δ 8,56-8,55 (м, 1H), 7,98 (дд, J=8,4, 1,6 Гц, 1H), 7,90 (д, J=8,4 Гц, 1H), 7,72 (д, J=5,5 Гц, 1H), 7,41 (дд, J=5,4, 0,5 Гц, 1H), 4,53 (с, 2H).
Стадия G: Продукт со стадии F (3,70 г, 14,5 ммоль), 4-гидрокси-N-метилбензиламин (2,37 г, 17,5 ммоль) и N,N-диизопропилэтиламин (2,8 мл, 16,0 ммоль) суспендировали в метиленхлориде и перемешивали в течение 24 часов. К реакционной смеси добавляли воду и экстрагировали метиленхлоридом. Затем органический слой промывали 1N HCl, затем насыщенным раствором хлорида натрия. Раствор сушили над сульфатом натрия, фильтровали, концентрировали в вакууме и очищали хроматографией на колонке (2-10% метанол/метиленхлорид в качестве элюента), получая требуемый продукт (2,94 г, 65%):1H ЯМР (500 МГц, CDCl3) δ 8,52 (д, J=0,5 Гц, 1Н), 7,95 (дд, J=8,4, 1,5 Гц, 1Н), 7,84 (д, J=8,4 Гц, 1H), 7,66 (д, J=5,4 Гц, 1H), 7,38 (д, J=5,4 Гц, 1H), 7,20 (д, J=7,8 Гц, 1H), 6,92-6,90 (м, 2H), 6,77-6,75 (м, 1H), 3,86 (с, 2H), 3,65 (с, 2H), 2,40 (с, 3H).
Стадия H: Продукт со стадии G (2,94 г, 9,0 ммоль) растворяли в метаноле и охлаждали на бане со льдом. К реакционной смеси добавляли боргидрид натрия (0,43 г, 11,0 ммоль) и перемешивали при комнатной температуре в течение 1 часа. Метанол концентрировали в вакууме и твердое вещество перерастворяли в метиленхлориде, дважды промывали водой и дважды экстрагировали метиленхлоридом. Объединенный органический экстракт промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали до получения желтого твердого вещества (2,75 г, 93%):1H ЯМР (500 МГц, CDCl3) δ 7,91 (с, 1Н), 7,77 (д, J=8,2 Гц, 1Н), 7,41 (д, J=5,4 Гц, 1H), 7,32-7,30 (м, 2H), 7,20 (т, J=7,7 Гц, 1Н), 6,87 (д, J=7,6 Гц, 1H), 6,81 (с, 1H), 6,75 (д, J=8,1 Гц, 1H), 4,88 (дд, J=3,6 Гц, 10,3 Гц, 1Н), 3,71 (д, J=13,1 Гц, 1Н), 3,49 (д, J=12,1 Гц, 1H), 2,67-2,56 (м, 2H), 2,34 (с, 3H); ESI-МС m/z 314 [M+Н]+.
Стадия I: Продукт со стадии H (2,75 г, 8,8 ммоль) растворяли в метиленхлориде (80 мл) и добавляли к раствору метансульфоновой кислоты (7,0 мл, 105 ммоль) в метиленхлориде (400 мл) и перемешивали в течение 10 минут. Реакционную смесь охлаждали на бане со льдом, затем гасили насыщенным раствором бикарбоната натрия и перемешивали в течение 1 часа. Органический слой промывали насыщенным раствором хлорида натрия, сушили над сульфатом натрия. Смесь фильтровали, концентрировали и очищали хроматографией на колонке (10% метанол/метиленхлорид), получая требуемый 4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ол (0,98 г, 38%):1H ЯМР (500 МГц, CDCl3) δ 7,72 (т, J=8,2 Гц, 2H), 7,38 (д, J=5,4 Гц, 1H), 7,29 (д, J=5,3 Гц, 1H), 7,18 (д, J=8,1 Гц, 1H), 6,73 (д, J=8,3 Гц, 1H), 6,54 (дд, J=2,6 Гц, 8,3 Гц, 1H), 6,49 (с, 1H), 4,33 (т, J=5,9 Гц, 1H), 3,67 (д, J=14,9 Гц, 1Н), 3,56 (д, J=7,4 Гц, 1H), 3,09-3,05 (м, 1H), 2,59 (т, J=8,9 Гц, 1H), 2,42 (с, 3H).
Стадия J: Продукт со стадии I (0,10 г, 0,35 ммоль) превращали в соль малеиновой кислоты растворением свободного основания в минимальном количестве этанола, добавлением одного эквивалента малеиновой кислоты в достаточном количестве метанола, чтобы полностью растворить кислоту, затем объединяя два раствора и перемешивая в течение 1 часа. Раствор концентрировали до минимального объема, затем охлаждали при -30°C до образования кристаллов, получая малеатную соль 4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ола (0,13 г, 90%, 98,7% AUC ВЭЖХ):1H ЯМР (500 МГц, CD3OD) δ 7,84 (д, J=8,2 Гц, 1H), 7,79 (с, 1H), 7,59 (д, J=5,4 Гц, 1H), 7,37 (д, J=5,4 Гц, 1H), 7,20 (д, J=8,2 Гц, 1H), 6,74-6,6,8 (м, 3H), 6,23 (с, 2H), 4,61-4,59 (м, 1Н), 4,52-4,48 (м, 2H), 3,84-3,82 (м, 1H), 3,49-3,47 (м, 1H), 3,05 (с, 3H).
Пример 115 - Получение малеатной соли (+)-4-бензо[b]тиофен-6-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина и малеатной соли (-)-4-бензо[b]тиофен-6-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина
Стадия A: Продукт со стадии J (1,54 г, 3,6 ммоль) со стадии K получения согласно примеру 148, бис(пинаколато)диборан (1,4 г, 5,4 ммоль), ацетат калия (1,06 г, 10,8 ммоль) в диметилсульфоксиде (50,0 мл) и дегазировали с использованием аргона. Добавляли комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (0,24 г, 0,3 ммоль) и смесь нагревали до 100°C в течение 4 часов. Реакционную смесь фильтровали через слой диатомовой земли и промывали этилацетатом. Смесь дважды промывали водой и один раз насыщенным раствором соли, затем три раза экстрагировали этилацетатом. Объединенный органический экстракт сушили над сульфатом натрия и концентрировали до получения темно-коричневого масла (1,4 г, 99%):1H ЯМР (500 МГц, CDCl3) δ 7,65 (д, J=8,1 Гц, 1Н), 7,62 (с, 1H), 7,50 (с, 1Н), 7,43 (д, J=7,4 Гц, 1H), 7,33-7,31 (м, 2H), 7,22 (д, J=5,4 Гц, 1Н), 7,09 (д, J=6,7 Гц, 1H), 4,39-4,36 (м, 1H), 3,76 (д, J=14,9 Гц, 1H), 3,62 (д, J=14,7 Гц, 1H), 3,06-3,03 (м, 1H), 2,61-2,56 (м, 1H), 2,39 (с, 3H); ESI-МС m/z 406 [M+Н]+.
Стадия B: Продукт со стадии A (1,0 г, 2,6 ммоль), 3,6-дихлорпиридазин (0,59 г, 3,9 ммоль), 2 М карбонат натрия (4,0 мл), и комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II) с дихлорметаном (0,15 г, 0,18 ммоль) в N,N-диметилформамиде нагревали до 100°C в течение 4 часов. К реакционной смеси добавляли воду и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором хлорида натрия и дважды экстрагировали этилацетатом. Объединенный органический слой сушили над сульфатом натрия, концентрировали в вакууме и очищали хроматографией на колонке (2-10% метанол/метиленхлорид), получая требуемый продукт (0,39 г, 45%):1H ЯМР (500 МГц, CDCl3) δ 7,88 (с, 1H), 7,80-7,78 (м, 1Н), 7,74 (с, 1Н), 7,67 (д, J=8,1 Гц, 1H), 7,53 (д, J=8,9 Гц, 1H), 7,40 (д, J=5,4 Гц, 1H), 7,31 (д, J=5,8 Гц, 1H), 7,21 (дд, J=1,5 Гц, 8,1 Гц, 1H), 7,06 (д, J=8,1 Гц, 1H), 4,45 (т, J=6,5 Гц, 1H), 3,88 (д, J=15,0 Гц, 1H), 3,74 (д, J=15,0 Гц, 1H), 3,14-3,10 (м, 1H), 2,71-2,67 (м, 1H), 2,47 (с, 3H).
Стадия C: Продукт со стадии B (0,38 г, 0,98 ммоль) растворяли в этаноле (50 мл) и моногидрате гидразина (0,47 мл, 9,8 ммоль). К реакционной смеси добавляли палладий-на-угле (10% мас., 0,20 г) и нагревали до 100°C в течение 4 часов. Смесь охлаждали до комнатной температуры и фильтровали через слой диатомовой земли, затем промывали метиленхлоридом. Смесь концентрировали в вакууме и очищали хроматографией на колонке (2-10% метанол/метиленхлорид), получая требуемый продукт (0,21 г, 60%):1H ЯМР (500 МГц, CDCl3) δ 9,14 (д, J=4,8 Гц, 1H), 7,94 (с, 1H), 7,82 (д, J=8,6 Гц, 1H), 7,76 (с, 1Н), 7,74 (с, 1H), 7,71 (д, J=8,1 Гц, 1H), 7,52-7,50 (м, 1Н), 7,40 (д, J=5,4 Гц, 1H), 7,31 (д, J=5,4 Гц, 1Н), 7,22 (д, J=8,2 Гц, 1Н), 7,06 (д, J=8,1 Гц, 1Н), 4,46 (т, J=6,4 Гц, 1H), 3,88 (д, J=14,9 Гц, 1H), 3,75 (д, J=14,9 Гц, 1H), 3,14-3,11 (м, 1H), 2,71-2,67 (м, 1H), 2,48 (с, 3H).
Полученное соединение разделяли препаративной хиральной ВЭЖХ (колонка CHIRALPAK AD, с использованием смеси 80% гептан/20% этанол/0,1% диэтиламин), получая (+)-энантиомер [α]25D +53,3° (c = 0,105, метанол) и (-)-энантиомер [α]25D -67,9° (c = 0,109, метанол).
Стадия D: (+)-Энантиомер (0,09 г, 0,25 ммоль) со стадии C превращали в соль малеиновой кислоты растворением свободного основания в минимальном количестве метанола, добавлением одного эквивалента малеиновой кислоты в достаточном количестве метанола, чтобы полностью растворить кислоту, затем объединяя два раствора и перемешивая в течение 1 часа. Раствор концентрировали до минимального объема, затем охлаждали при -30°C до образования кристаллов. Фильтрование давало малеатную соль (+)-4-бензо[b]тиофен-6-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина (86,0 мг, 99%, >99% AUC ВЭЖХ) в виде коричневого твердого вещества:1H ЯМР (500 МГц, CD3OD) δ 9,17 (д, J=4,8 Гц, 1H), 8,18 (д, J=8,7 Гц, 1H), 8,09 (с, 1Н), 7,94 (дд, J=1,6 Гц, 8,1 Гц, 1H), 7,89 (с, 1H), 7,87 (с, 1H), 7,81-7,79 (м, 1H), 7,62 (д, J=5,4 Гц, 1H), 7,39 (д, J=5,4 Гц, 1Н), 7,27 (д, J=8,2 Гц, 1H), 7,14 (д, J=8,2 Гц, 1H), 6,24 (с, 2H), 4,81-4,77 (м, 2H), 4,70 (с, 2H), 3,94-3,90 (м, 1H), 3,69-3,66 (м, 1Н), 3,10 (с, 3H).
Такой же способ использовали для превращения (-)-энантиомера (0,09 мг, 0,25 ммоль) в его малеатную соль, получая 4-бензо[b]тиофен-6-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина (16,5 мг, 98,9%, >99% AUC ВЭЖХ) в виде не совсем белого твердого вещества:1H ЯМР (500 МГц, CD3OD) δ 9,17 (д, J=4,9 Гц, 1H), 8,17 (д, J=8,6 Гц, 1H), 8,08 (с, 1H), 7,94 (д, J=6,6 Гц, 1H), 7,89 (с, 1Н), 7,87 (с, 1H), 7,81-7,79 (м, 1Н), 7,62 (д, J=5,4 Гц, 1H), 7,39 (д, J=5,4 Гц, 1H), 7,26 (д, J=8,2 Гц, 1Н), 7,14 (д, J=8,2 Гц, 1H), 6,24 (с, 2H), 4,79-4,77 (м, 1H), 4,68 (с, 2H), 3,93-3,89 (м, 1Н), 3,67-3,65 (м, 1H), 3,09 (с, 3H).
Пример 116 -Получение гидрохлоридной соли (+/-)-4-бензо[b]тиофен-7-ил-2-метил-1,2,3,4-тетрагидроизохинолина и гидрохлоридной соли (-)-4-бензо[b]тиофен-7-ил-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: В герметично закрытой пробирке раствор 7-бромбензотиофена (330 мг, 1,55 ммоль), 4-трибутилстаннанилизохинолина (648 мг, 1,55 ммоль) и трифенилфосфина (41 мг, 0,155 ммоль) в ДМФА (3 мл) дегазировали замораживанием/оттаиванием. Добавляли триэтиламин (108 мкл, 0,775 ммоль), ацетат палладия (II) (17 мг, 0,078 ммоль) и йодида меди (59 мг, 0,31 ммоль) и реакционную смесь нагревали до 100°C в течение 3 суток. Сырой продукт получали посредством обработки водой, используя CH2Cl2 в качестве экстрагирующего растворителя.
Стадия B: Метилтрифторметансульфонат (179 мкл, 1,58 ммоль) добавляли к раствору неочищенного продукта со стадии A (375 мг, 1,44 ммоль) и метиленхлорида (3 мл) и перемешивали при комнатной температуре в течение 3 часов. Растворитель выпаривали. Остаток разбавляли метанолом (3 мл) и обрабатывали цианоборгидридом натрия (226 мг, 3,6 ммоль) и перемешивали при комнатной температуре в течение ночи. Растворитель выпаривали и остаток разбавляли метиленхлоридом, промывали водным раствором гидроксида натрия, сушили над сульфатом натрия и концентрировали досуха.
Продукт разделяли хиральной ВЭЖХ (Chiralpak AD, 97:3 гептан/EPA с 0,1% диэтиламина). Получали (+)-энантиомер (127 мг, 35%) [α]24D +42,6° (c 0,6, метанол).
Стадия C: Раствор продукта со стадии B (127 мг, 0,455 ммоль) и 2N раствора HCl (455 мкл, 0,909 ммоль) в метиленхлориде (1 мл) перемешивали при комнатной температуре. Растворитель выпаривали. Остаток разбавляли диэтиловым эфиром и фильтровали, получая (+/-)4-бензо[b]тиофен-7-ил-2-метил-1,2,3,4-тетрагидроизохинолин (97 мг, 68%) в виде бледно-зеленого твердого вещества: т.пл. 228-230°C;1H ЯМР (CDCl3, 500 МГц) δ 13,63 (ушир., 1Н), 7,82 (д, J=7,7 Гц, 1H), 7,46-7,14 (м, 7H), 6,93-6,92 (м, 1H), 5,47-5,43 (м, 1H), 4,91 (д, J=14,9 Гц, 1H), 4,26 (д, J=13,6 Гц, 1H), 3,80-3,76 (м, 1H), 3,48-3,46 (м, 1H), 2,98 (с, 3H); ESI-МС m/z 280 [M+H]+. Элементный анализ, вычислено для C18H17NS-HCl-0,25H2O: C, 67,48; Н, 5,82; N, 4,37; Cl 11,07. Найдено: C, 67,78; Н 5,45; N, 4,24; Cl, 11,00.
Пример 117 - Получение малеатной соли (+/-)-4-бензофуран-2-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору бензофуран-2-илметилкетона (10 г, 62,4 ммоль) в сероуглероде (80 мл) по каплям в течение 3 часов добавляли раствор брома (3,2 мл, 62,4 ммоль) в сероуглероде (80 мл). Реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении. Полученный сырой продукт очищали перекристаллизацией из горячего этанола, получая требуемый α-бромкетон (10,14 г, 68%, две порции) в виде зеленовато-желтых кристаллов:1H ЯМР (500 МГц, CDCl3) δ 7,74 (д, J=7,9 Гц, 1Н), 7,66 (д, J=0,8 Гц, 1H), 7,60 (дд, J=8,5, 0,6 Гц, 1H), 7,52 (тд, J=7,2, 1,2 Гц, 1H), 7,35 (тд, J=7,5, 0,8 Гц, 1H), 4,45 (с, 2H).
Стадия B: К смеси (3-метоксибензил)метиламина (4,7 г, 40 ммоль) и триэтиламина (8,6 мл, 62 ммоль) в дихлорметане (62 мл) небольшими порциями добавляли α-бромкетон со стадии A (7,4 г, 31 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем вливали в воду. Органический слой отделяли, дважды промывали водой и насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Сырой продукт очищали хроматографией на колонке (85:15 гексаны/этилацетат, затем 80:20 и 75:25 гексаны/этилацетат), получая требуемый третичный амин (6,78 г, 70%) в виде коричневато-оранжевого масла:1H ЯМР (500 МГц, CDCl3) δ 7,69 (д, J=7,8 Гц, 1H), 7,60 (д, J=0,9 Гц, 1H), 7,56 (дд, J=8,7, 0,8 Гц, 1H), 7,49-7,45 (м, 1H), 7,32-7,29 (м, 1H), 7,23 (д, J=8,0 Гц, 1Н), 6,98-6,93 (м, 2H), 6,83-6,81 (м, 1H), 3,79 (с, 5H), 3,71 (с, 2H), 2,45 (с, 3H).
Стадия C: К ледяному раствору продукта со стадии B (6,78 г, 21,9 ммоль) в метаноле (56,5 мл) небольшими порциями добавляли боргидрид натрия (0,91 г, 24,1 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем концентрировали при пониженном давлении. Полученный остаток распределяли между дихлорметаном и водой. Органический слой отделяли и водный слой снова дважды экстрагировали дихлорметаном.
Объединенный органический экстракт промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали, получая требуемый спирт (6,1 г, 89%) в виде коричневато-оранжевого масла:1H ЯМР (300 МГц, CDCl3) δ 7,60-7,50 (м, 1H), 7,45 (д, J=8,2 Гц, 1Н), 7,40-7,10 (м, 3H), 6,95-6,68 (м, 4H), 4,91 (дд, J=9,8 Гц, 3,7 Гц, 1Н), 3,78 (с, 3H), 3,70 (д, J=14,9 Гц, 1H), 3,59-3,51 (м, 1H), 3,00 (дд, J=12,4, 9,9 Гц, 1H), 2,77 (дд, J=12,4, 3,7 Гц, 1H), 2,32 (с, 3H).
Стадия D: К раствору неочищенного продукта со стадии C (6,1 г, 19,6 ммоль) в дихлорметане (128 мл) по каплям через капельную воронку добавляли метансульфоновую кислоту (12,7 мл, 196 ммоль). Темно-коричневую смесь перемешивали при комнатной температуре в течение 40 минут и затем добавляли по каплям к ледяному раствору гидроксида натрия (128 мл, 2 М). Полученную смесь разбавляли водой и органический слой отделяли. Водный слой снова дважды экстрагировали дихлорметаном и объединенный органический экстракт промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Сырой продукт очищали хроматографией на колонке (градиент 95:5-70:30 гексаны/этилацетат) и полученный частично очищенный продукт растворяли в дихлорметане и промывали 2 М HCl (Примечание: гидрохлоридная соль требуемого продукта не может быть экстрагирована в водный слой). Органический слой концентрировали и снова очищали хроматографией на колонке (90:10 дихлорметан/метанол), получая гидрохлоридную соль требуемого продукта. Раствор гидрохлоридной соли в дихлорметане промывали насыщенным раствором бикарбоната натрия, сушили над сульфатом натрия, фильтровали и концентрировали, получая требуемый продукт (1,3 г, 23%) в виде бледно-желтого масла:1H ЯМР (500 МГц, CDCl3) δ 7,44-7,00 (м, 2H), 7,21 (тд, J=8,0, 1,5 Гц, 1Н), 7,16 (тд, J=7,4, 1,1 Гц, 1H), 7,10 (д, J=8,5 Гц, 1Н), 6,73 (дд, J=8,5, 2,7 Гц, 1Н), 6,63 (д, J=2,6 Гц, 1H), 6,34 (д, J=0,7 Гц, 1H), 4,38 (т, J=5,8 Гц, 1Н), 3,79 (с, 3H), 3,71 (д, J=15,0 Гц, 1H), 3,58 (д, J=15,0 Гц, 1H), 3,06 (дд, J=11,4, 6,5 Гц, 1H), 2,95 (дд, J=11,4, 5,2 Гц, 1H), 2,45 (с, 3H).
Также выделяли нежелательный 5-метоксирегиоизомер (0,99 г, 17%) в виде желтого твердого вещества.
Стадия E: К раствору продукта со стадии D (1,3 г, 4,4 ммоль) в уксусной кислоте (20 мл) добавляли раствор бромистого водорода в уксусной кислоте (20 мл, 33% раствор). Смесь нагревали при 100°C в течение ночи и охлажденную реакционную смесь концентрировали при пониженном давлении. Полученный остаток разбавляли водой и дихлорметаном и подщелачивали насыщенным раствором бикарбоната натрия. Органический слой отделяли и водный слой снова дважды экстрагировали дихлорметаном. Объединенный органический экстракт сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая требуемый фенол (1,18 г, 96%) в виде коричневой пены:1H ЯМР (300 МГц, CDCl3) δ 7,44-7,39 (м, 2H), 7,26-7,15 (м, 2H), 7,03 (д, J=8,3 Гц, 1H), 6,62 (дд, J=8,3, 2,6 Гц, 1H), 6,51 (д, J=2,1 Гц, 1Н), 6,36 (с, 1H), 4,39 (т, J=5,9 Гц, 1Н), 3,64 (д, J=15,1 Гц, 1H), 3,54 (д, J=15,1 Гц, 1H), 3,11-2,95 (м, 2H), 2,46 (с, 3H).
К ледяному раствору фенола (1,18 г, 4,23 ммоль) в дихлорметане (42 мл) добавляли пиридин (0,7 мл, 8,5 ммоль) и ангидрид трифторметансульфоновой кислоты (0,9 мл, 5,5 ммоль). Реакционную смесь перемешивали при 0°C в течение 45 минут и затем гасили насыщенным раствором бикарбоната натрия. Органический слой отделяли и водный слой снова дважды экстрагировали дихлорметаном. Объединенный органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Сырой продукт очищали хроматографией на колонке (90:10 дихлорметан/гексаны, затем дихлорметан), получая требуемый трифторметансульфонат (1,34 г, 74%) в виде желтого масла:1H ЯМР (300 МГц, CDCl3) δ 7,49-7,47 (м, 1H), 7,42 (д, J=8,1 Гц, 1H), 7,29-7,19 (м, 3H), 7,08-7,00 (м, 2H), 6,39 (с, 1H), 4,44 (т, J=5,8 Гц, 1H), 3,74 (д, J=15,5 Гц, 1H), 3,65 (д, J=15,3 Гц, 1Н), 3,08 (дд, J=11,6, 6,7 Гц, 1H), 2,99 (дд, J=11,6, 5,2 Гц, 1H), 2,47 (с, 3H).
Стадия F: К дегазированной смеси (Ar, 10 минут) трифторметансульфоната со стадии E (1,02 г, 2,48 ммоль), ацетата калия (0,73 г, 7,44 ммоль) и бис(пинаколато)дибора (0,69 г, 2,72 ммоль) в диметилсульфоксиде (16 мл) добавляли дихлорбис(фосфиноферроцен)палладий(II) (0,06 г, 0,07 ммоль) и смесь нагревали при 80°C в течение 3 часов. Охлажденную реакционную смесь распределяли между этилацетатом и водой и водный слой снова экстрагировали дихлорметаном. Объединенный органический экстракт сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Сырой продукт очищали хроматографией на колонке (дихлорметан, затем 99:1 и 98:2 дихлорметан/метанол), получая частично очищенный сложный эфир бороновой кислоты (0,99 г) в виде бледно-желтого масла:1H ЯМР (300 МГц, CDCl3) δ 7,65-7,55 (м, 2H), 7,45-7,30 (м, 2H), 7,26-7,10 (м, 3H), 6,34 (с, 1Н), 4,45 (т, J=5,6 Гц, 1H), 3,74 (д, J=15,2 Гц, 1H), 3,62 (д, J=14,9 Гц, 1H), 3,07 (дд, J=11,5, 6,5 Гц, 1H), 3,04-2,90 (м, 1Н), 2,45 (с, 3H), 1,34 (с, 12H).
К смеси сложного эфира бороновой кислоты (0,99 г, 2,56 ммоль) и 3,6-дихлорпиридазина (0,76 г, 5,11 ммоль) в диметилформамиде (22 мл) добавляли раствор карбоната натрия (0,83 г, 7,87 ммоль) в воде (5,7 мл). Раствор дегазировали (Ar, 10 минут) и к нему добавляли дихлорбис(фосфиноферроцен)палладий(II) (0,17 г, 0,20 ммоль). Реакционную смесь нагревали при 80°C в течение 4 часов, охлаждали и распределяли между дихлорметаном и водой. Затем водный слой снова дважды экстрагировали дихлорметаном и объединенный органический экстракт сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Сырой продукт очищали хроматографией на колонке (дихлорметан, затем 99:1, 98:2, 97:3 и 96:4 дихлорметан/метанол), получая требуемый продукт (0,41 г, 43%) в виде коричневого твердого вещества:1H ЯМР (300 МГц, CDCl3) δ 7,88 (д, J=1,3 Гц, 1H), 7,82-7,70 (м, 2H), 7,55 (д, J=9,0 Гц, 1H), 7,50-7,40 (м, 2H), 7,35 (д, J=8,2 Гц, 1H), 7,26-7,15 (м, 2H), 6,43 (с, 1H), 4,52 (т, J=5,9 Гц, 1H), 3,83 (д, J=15,2 Гц, 1Н), 3,74 (д, J=15,2 Гц, 1H), 3,11 (дд, J=11,5, 6,8 Гц, 1H), 3,03 (дд, J=11,4, 5,4 Гц, 1H), 2,48 (с, 3H).
Стадия G: К раствору продукта со стадии F (0,2 г, 0,5 ммоль) в этаноле (10 мл) и метаноле (7 мл) добавляли моногидрат гидразина (0,5 г, 10 ммоль), затем 10% палладий-на-угле (50 мг). Смесь нагревали при 70°C в течение ночи и охлаждали до комнатной температуры. Добавляли дополнительное количество моногидрата гидразина (0,25 г, 5,0 ммоль) и 10% палладия-на-угле (50 мг) к реакционной смеси, которую затем нагревали при кипячении с обратным холодильником в течение 90 минут. Охлажденную реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (90:10 дихлорметан/гексаны, затем дихлорметан и, наконец, 99:1, 98:2, 97:3, 96:4 и 95:5 дихлорметан/метанол), получая требуемое производное пиридазина (0,11 г, 61%) в виде не совсем белой пены:1H ЯМР (500 МГц, CDCl3) δ 9,16 (дд, J=4,9, 1,6 Гц, 1H), 7,94 (д, J=1,4 Гц, 1H), 7,84 (дд, J=8,6, 1,6 Гц, 1H), 7,81 (дд, J=8,1, 1,9 Гц, 1H), 7,52 (дд, J=8,6, 4,9 Гц, 1H), 7,48 (дд, J=7,8, 1,1 Гц, 1H), 7,43 (дд, J=8,3, 0,6 Гц, 1H), 7,35 (д, J=8,1 Гц, 1H), 7,26-7,17 (м, 2H), 6,43 (с, 1H), 4,52 (т, J=5,9 Гц, 1H), 3,83 (д, J=15,1 Гц, 1H), 3,75 (д, J=15,1 Гц, 1H), 3,11 (дд, J=11,4, 6,7 Гц, 1H), 3,04 (дд, J=11,5, 5,2 Гц, 1H), 2,51 (с, 3H).
Стадия H: К раствору продукта со стадии G (0,11 г, 0,31 ммоль) в метаноле (2 мл) добавляли малеиновую кислоту (36 мг, 0,31 ммоль). Через 1 час раствор разбавляли водой (12 мл) и затем лиофилизировали, получая малеатную соль (+/-)-4-бензофуран-2-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина (142 мг, теоретический выход, 98,6% AUC ВЭЖХ) в виде бледно-желтого твердого вещества: т.пл. 96-98°C;1H ЯМР (500 МГц, CD3OD) δ 9,18 (дд, J=4,9, 1,5 Гц, 1H), 8,20 (дд, J=8,7, 1,5 Гц, 1H), 8,10 (с, 1H), 8,03 (д, J=8,2 Гц, 1Н), 7,81 (дд, J=8,7, 4,9 Гц, 1Н), 7,59 (д, J=7,9 Гц, 1H), 7,47-7,37 (м, 2H), 7,32-7,21 (м, 2H), 6,78 (ушир.с, 1Н), 6,24 (с, 2H), 4,97 (т, J=7,1 Гц, 1H), 4,63 (кажущийся с, 2H), 3,94 (кажущийся д, J=6,6 Гц, 2H), 3,11 (с, 3H); ESI-МС m/z 342 [M+H]+.
Пример 118 - Получение малеатной соли (+/-)-4-бензофуран-2-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору бензофуран-2-илметилкетона (10 г, 62,4 ммоль) в сероуглероде (80 мл) по каплям в течение 3 часов добавляли раствор брома (3,2 мл, 62,4 ммоль) в сероуглероде (80 мл). Реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении. Полученный сырой продукт очищали перекристаллизацией из горячего этанола, получая требуемый α-бромкетон (10,14 г, 68%, две порции) в виде зеленовато-желтых кристаллов:1H ЯМР (500 МГц, CDCl3) δ 7,74 (д, J=7,9 Гц, 1H), 7,66 (д, J=0,8 Гц, 1H), 7,60 (дд, J=8,5, 0,6 Гц, 1H), 7,52 (тд, J=7,2, 1,2 Гц, 1H), 7,35 (тд, J=7,5, 0,8 Гц, 1H), 4,45 (с, 2H).
Стадия B: К смеси (3-метоксибензил)метиламина (4,7 г, 40 ммоль) и триэтиламина (8,6 мл, 62 ммоль) в дихлорметане (62 мл) небольшими порциями добавляли α-бромкетон со стадии A (7,4 г, 31 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем вливали в воду. Органический слой отделяли, промывали два раза водой и насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Сырой продукт очищали хроматографией на колонке (85:15 гексаны/этилацетат, затем 80:20 и 75:25 гексаны/этилацетат), получая требуемый третичный амин (6,78 г, 70%) в виде коричневато-оранжевого масла:1H ЯМР (500 МГц, CDCl3) δ 7,69 (д, J=7,8 Гц, 1H), 7,60 (д, J=0,9 Гц, 1H), 7,56 (дд, J=8,7, 0,8 Гц, 1Н), 7,49-7,45 (м, 1H), 7,32-7,29 (м, 1Н), 7,23 (д, J=8,0 Гц, 1H), 6,98-6,93 (м, 2H), 6,83-6,81 (м, 1H), 3,79 (с, 5H), 3,71 (с, 2H), 2,45 (с, 3H).
Стадия C: К ледяному раствору продукта со стадии B (6,78 г, 21,9 ммоль) в метаноле (56,5 мл) небольшими порциями добавляли боргидрид натрия (0,91 г, 24,1 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем концентрировали при пониженном давлении. Полученный остаток распределяли между дихлорметаном и водой. Органический слой отделяли и водный слой снова дважды экстрагировали дихлорметаном.
Объединенный органический экстракт промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали, получая требуемый спирт (6,1 г, 89%) в виде коричневато-оранжевого масла:1H ЯМР (300 МГц, CDCl3) δ 7,60-7,50 (м, 1H), 7,45 (д, J=8,2 Гц, 1H), 7,40-7,10 (м, 3H), 6,95-6,68 (м, 4H), 4,91 (дд, J=9,8 Гц, 3,7 Гц, 1H), 3,78 (с, 3H), 3,70 (д, J=14,9 Гц, 1H), 3,59-3,51 (м, 1H), 3,00 (дд, J=12,4, 9,9 Гц, 1H), 2,77 (дд, J=12,4, 3,7 Гц, 1H), 2,32 (с, 3H).
Стадия D: К раствору неочищенного продукта со стадии C (6,1 г, 19,6 ммоль) в дихлорметане (128 мл) по каплям через капельную воронку добавляли метансульфоновую кислоту (12,7 мл, 196 ммоль). Темно-коричневую смесь перемешивали при комнатной температуре в течение 40 минут и затем добавляли по каплям к ледяному раствору гидроксида натрия (128 мл, 2 М). Полученную смесь разбавляли водой и органический слой отделяли. Водный слой снова дважды экстрагировали дихлорметаном и объединенный органический экстракт промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Сырой продукт очищали хроматографией на колонке (градиент 95:5-70:30 гексан/этилацетат) и полученный частично очищенный продукт растворяли в дихлорметане и промывали 2 М HCl (Примечание: гидрохлоридная соль требуемого продукта не может быть экстрагирована в водный слой). Органический слой концентрировали и снова очищали хроматографией на колонке (90:10 дихлорметан/метанол), получая гидрохлоридную соль требуемого продукта. Раствор гидрохлоридной соли в дихлорметане промывали насыщенным раствором бикарбоната натрия, сушили над сульфатом натрия, фильтровали и концентрировали, получая требуемый продукт (1,3 г, 23%) в виде бледно-желтого масла:1H ЯМР (500 МГц, CDCl3) δ 7,44-7,00 (м, 2H), 7,21 (тд, J=8,0, 1,5 Гц, 1H), 7,16 (тд, J=7,4, 1,1 Гц, 1H), 7,10 (д, J=8,5 Гц, 1H), 6,73 (дд, J=8,5, 2,7 Гц, 1H), 6,63 (д, J=2,6 Гц, 1H), 6,34 (д, J=0,7 Гц, 1H), 4,38 (т, J=5,8 Гц, 1Н), 3,79 (с, 3H), 3,71 (д, J=15,0 Гц, 1H), 3,58 (д, J=15,0 Гц, 1H), 3,06 (дд, J=11,4, 6,5 Гц, 1H), 2,95 (дд, J=11,4, 5,2 Гц, 1Н), 2,45 (с, 3H). Также выделяли нежелательный 5-метоксирегиоизомер (0,99 г, 17%) в виде желтого твердого вещества.
Стадия E: К раствору продукта со стадии D (1,3 г, 4,4 ммоль) в уксусной кислоте (20 мл) добавляли раствор бромистого водорода в уксусной кислоте (20 мл, 33% раствор). Смесь нагревали при 100°C в течение ночи и охлажденную реакционную смесь концентрировали при пониженном давлении. Полученный остаток разбавляли водой и дихлорметаном и подщелачивали насыщенным раствором бикарбоната натрия. Органический слой отделяли и водный слой снова дважды экстрагировали дихлорметаном. Объединенный органический экстракт сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая требуемый фенол (1,18 г, 96%) в виде коричневой пены:1H ЯМР (300 МГц, CDCl3) δ 7,44-7,39 (м, 2H), 7,26-7,15 (м, 2H), 7,03 (д, J=8,3 Гц, 1H), 6,62 (дд, J=8,3, 2,6 Гц, 1H), 6,51 (д, J=2,1 Гц, 1H), 6,36 (с, 1H), 4,39 (т, J=5,9 Гц, 1H), 3,64 (д, J=15,1 Гц, 1H), 3,54 (д, J=15,1 Гц, 1H), 3,11-2,95 (м, 2H), 2,46 (с, 3H).
К ледяному раствору фенола (1,18 г, 4,23 ммоль) в дихлорметане (42 мл) добавляли пиридин (0,7 мл, 8,5 ммоль) и ангидрид трифторметансульфоновой кислоты (0,9 мл, 5,5 ммоль). Реакционную смесь перемешивали при 0°C в течение 45 минут и затем гасили насыщенным раствором бикарбоната натрия. Органический слой отделяли и водный слой снова дважды экстрагировали дихлорметаном. Объединенный органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Сырой продукт очищали хроматографией на колонке (90:10 дихлорметан/гексаны, затем дихлорметан), получая требуемый трифторметансульфонат (1,34 г, 74%) в виде желтого масла:1H ЯМР (300 МГц, CDCl3) δ 7,49-7,47 (м, 1H), 7,42 (д, J=8,1 Гц, 1Н), 7,29-7,19 (м, 3H), 7,08-7,00 (м, 2H), 6,39 (с, 1Н), 4,44 (т, J=5,8 Гц, 1Н), 3,74 (д, J=15,5 Гц, 1Н), 3,65 (д, J=15,3 Гц, 1H), 3,08 (дд, J=11,6, 6,7 Гц, 1H), 2,99 (дд, J=11,6, 5,2 Гц, 1H), 2,47 (с, 3H).
Стадия F: К смеси трифторметансульфоната со стадии E (0,1 г, 0,24 ммоль), 2-(дициклогексилфосфино)-2',4',6'-триизопропилбифенила (69 мг, 0,14 ммоль) и карбоната цезия (0,20 г, 0,61 ммоль) в толуоле (2,6 мл) добавляли морфолин (42 мкл, 0,49 ммоль) и смесь дегазировали (Ar, 10 минут). К смеси добавляли ацетат палладия (II) (8 мг, 0,04 ммоль) и затем смесь нагревали при кипячении с обратным холодильником в течение ночи. Охлажденную реакционную смесь разбавляли метанолом, фильтровали через целит и фильтрат концентрировали при пониженном давлении.
Указанную реакцию повторяли еще два раза (0,24 ммоль трифторметансульфоната на реакцию) и объединенный сырой продукт частично очищали хроматографией на колонке (дихлорметан, затем 98:2, 97:3 и 96:4 дихлорметан/метанол), получая требуемый продукт (103 мг) в виде коричневого масла. Полученный продукт дополнительно очищали препаративной тонкослойной хроматографией (1-мм пластины Analtech; элюент 95:5 дихлорметан/метанол), получая требуемый продукт (23 мг, 9%) в виде бледно-желтого масла:1H ЯМР (500 МГц, CDCl3) δ 7,45-7,43 (м, 1H), 7,41 (д, J=8,0 Гц, 1H), 7,22-7,18 (м, 1H), 7,16 (тд, J=7,5, 1,1 Гц, 1Н), 7,09 (д, J=8,5 Гц, 1H), 6,75 (дд, J=8,5, 2,6 Гц, 1H), 6,62 (д, J=2,5 Гц, 1H), 6,35 (с, 1H), 4,37 (т, J=5,7 Гц, 1H), 3,85 (т, J=4,8 Гц, 4H), 3,69 (д, J=15,2 Гц, 1H), 3,57 (д, J=14,9 Гц, 1H), 3,13 (т, J=4,8 Гц, 4H), 3,05 (дд, J=11,4, 6,5 Гц, 1H), 2,94 (дд, J=11,4, 5,2 Гц, 1H), 2,45 (с, 3H).
К раствору производного морфолина (23 мг, 0,06 ммоль) в метаноле (2 мл) добавляли малеиновую кислоту (7,5 мг, 0,06 ммоль) и смесь перемешивали при комнатной температуре в течение 1 часа. Раствор концентрировали досуха и остаток дважды растирали в диэтиловом эфире (2 мл) и метаноле (2-3 капли). Надосадок удаляли пипеткой и остаток растворяли в метаноле (2 мл) и воде (10 мл) и раствор лиофилизировали, получая малеатную соль (+/-)-4-бензофуран-2-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина (26 мг, 86%, 98,4% AUC ВЭЖХ) в виде желто-коричневого твердого вещества: т.пл. 95-97°C;1H ЯМР (500 МГц, CD3OD) δ 7,55 (д, J=7,6 Гц, 1H), 7,42 (д, J=8,0 Гц, 1H), 7,29-7,25 (м, 1H), 7,22 (тд, J=7,4, 0,9 Гц, 1Н), 7,15 (ушир.с, 1H), 7,00-6,95 (м, 1H), 6,82 (д, J=2,4 Гц, 1H), 6,75-6,50 (ушир.с, 1H), 6,25 (с, 2H), 4,80-4,75 (м, 2H), 4,60-4,40 (м, 2H), 3,88 (ушир.с, 1H), 3,82 (кажущийся т, J=4,8 Гц, 4H), 3,16 (кажущийся т, J=4,8 Гц, 4H), 3,08 (с, 3H); ESI-МС m/z 349 [M+H]+.
Пример 119 - Получение фумаратной соли (+)-4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: Рацемический 4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин получали из 6-бром-1H-индола способом, сходным со способом, описанным в примере 122 (стадии A-D). Полученное рацемическое соединение (450 мг) разделяли полупрепаративной хиральной ВЭЖХ (Chiralcel OD, 90% гептан/изопропанол с 0,1% диэтиламином). Каждый из полученных в результате энантиомеров растворяли в метаноле и обрабатывали раствором малеиновой кислоты в метаноле (5 мл) при 0°C. Раствор перемешивали при комнатной температуре в течение ночи, затем концентрировали и твердое вещество суспендировали с диэтиловом эфире, фильтрование давало фумаратную соль (+)-4-(1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина [160 мг, 42%, >99% AUC ВЭЖХ, 100% AUC хиральная ВЭЖХ (свободное основание)]:1H ЯМР (CD3OD, 300 МГц) δ 7,55 (д, J=8,2 Гц, 1H), 7,31-7,21 (м, 5H), 6,99 (д, J=7,7 Гц, 1H), 6,85 (дд, J=8,2, 1,5 Гц, 1H), 6,74 (с, 4H), 6,46-6,44 (м, 1H), 4,67-4,58 (м, 3H), 3,89-3,83 (м, 1H), 3,64-3,60 (м, 1H), 3,07 (с, 3H); ESI-МС m/z=263 [M+H]+, [α]D25 +108,0° (c 0,08, MeOH, свободное основание). Элементный анализ, вычислено для C18H18N2•2C4H4O4 0,375 Н2O: C, 62,29; Н, 5,38; N, 5,59. Найдено: C, 61,94; Н, 5,19; N, 6,18.
Пример 120 - Получение малеатной соли (+/-)-4-(3-хлор-1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору 4-бром-2-нитротолуола (7,9 г, 36,6 ммоль) в диметилформамиде (73 мл) добавляли диметилацеталь N,N-диметилформамида (14,5 мл, 110 ммоль) и пирролидин (4,7 мл) и смесь нагревали при 110°C в течение 90 минут. Охлажденную реакционную смесь разбавляли диэтиловым эфиром и промывали водой. Водный слой снова дважды экстрагировали диэтиловым эфиром и объединенный органический экстракт сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток растворяли в водном растворе уксусной кислоты (245 мл, 80%) и нагревали до 75°C. Цинковую пыль (20,8 г, 318 ммоль) добавляли к горячему раствору небольшими порциями в течение 2 часов. Затем реакционную смесь нагревали при 85°C в течение 3 часов 30 минут, охлаждали до комнатной температуры и затем до 0°C. Образованный преципитат удаляли фильтрованием и фильтрат разбавляли этилацетатом и дважды промывали водой. Органический экстракт сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая коричневое масло. Сырой продукт очищали флэш-хроматографией на колонке (95:5 гексаны/этилацетат, затем 90:10 гексаны/этилацетат), получая 6-броминдол в виде серого твердого вещества (2,61 г, 36%):1H ЯМР (500 МГц, CDCl3) δ 8,14 (ушир.с, 1H), 7,53 (с, 1H), 7,49 (д, J=8,4 Гц, 1H), 7,21 (дд, J=8,4, 1,7 Гц, 1H), 7,17-7,15 (м, 1H), 6,53-6,51 (м, 1Н).
Стадия B: К ледяному раствору 6-броминдола (0,5 г, 2,55 ммоль) в метаноле (10 мл) небольшими порциями добавляли N-хлорсукцинимид (0,34 г, 2,55 ммоль). Реакционную смесь перемешивали при 0°C в течение 15 минут и при комнатной температуре в течение 1 часа и затем вливали в ледяную воду. Водную смесь четыре раза экстрагировали диэтиловым эфиром и объединенный органический экстракт промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток растворяли в дихлорметане, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. К полученному остатку добавляли ди-трет-бутилдикарбонат (0,56 г, 2,55 ммоль), затем N,N-диметиламинопиридин (25 мг) и ацетонитрил (7 мл) и смесь перемешивали в течение 30 минут. Реакционную смесь вливали в воду и продукт дважды экстрагировали этилацетатом. Объединенный органический экстракт промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Сырой продукт очищали хроматографией на колонке (95:5 гексаны/этилацетат), получая требуемое соединение (0,61 г, 72%) в виде коричневого твердого вещества:1H ЯМР (500 МГц, CDCl3) δ 8,38 (ушир.с, 1H), 7,53 (с, 1H), 7,46-7,41 (м, 2H), 1,66 (с, 9H).
Стадия C: К смеси продукта со стадии B (0,15 г, 0,45 ммоль) и изохинолин-4-бороновой кислоты (0,10 г, 0,54 ммоль) в диметиловом эфире этиленгликоля (2,1 мл) добавляли раствор карбоната цезия (0,45 мл, 2 М) и смесь дегазировали (Ar, 10 минут). К смеси добавляли тетракис(трифенилфосфин)палладий (0) (26 мг, 0,02 ммоль) и затем смесь нагревали при кипячении с обратным холодильником в течение 5 часов. Охлажденную реакционную смесь распределяли между этилацетатом и водой и водный слой снова дважды экстрагировали этилацетатом. Объединенный органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (98:2 дихлорметан/метанол), получая требуемый продукт (82 мг, 48%) в виде розовой пены:1H ЯМР (500 МГц, CDCl3) δ 9,28 (с, 1H), 8,55 (с, 1H), 8,32 (ушир.с, 1H), 8,06 (д, J=7,6 Гц, 1Н), 7,95 (д, J=8,2 Гц, 1Н), 7,74 (д, J=8,0 Гц, 1H), 7,68-7,64 (м, 3H), 7,47 (дд, J=8,0, 1,4 Гц, 1Н), 1,60 (с, 9H).
Стадия D: К ледяному раствору продукта со стадии C (80 мг, 0,21 ммоль) в дихлорметане по каплям добавляли метилтрифторметансульфонат (26 мкл, 0,23 ммоль). Реакционную смесь перемешивали при 0°C в течение 15 минут и затем концентрировали досуха. Остаток собирали в метаноле (2,5 мл) и охлаждали до 0°C. Небольшими порциями добавляли цианоборгидрид натрия (0,13 г, 2,11 ммоль) и реакционную смесь перемешивали при 0°C в течение 1 часа 15 минут. Реакционную смесь концентрировали и распределяли между водой и дихлорметаном. Водный слой снова экстрагировали дихлорметаном и объединенный органический экстракт сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Сырой продукт очищали препаративной тонкослойной хроматографией (1-мм пластины Analtech; элюент: 95:5 дихлорметан/метанол), получая требуемый продукт (55 мг, 65%) в виде белого твердого вещества:1H ЯМР (500 МГц, CDCl3) δ 8,00 (ушир.с, 1H), 7,54 (с, 1H), 7,48 (д, J=8,1 Гц, 1H), 7,15-7,04 (м, 4H), 6,87 (д, J=7,7 Гц, 1H), 4,42 (т, J=6,7 Гц, 1H), 3,76 (д, J=15,1 Гц, 1H), 3,65 (д, J=14,8 Гц, 1H), 3,07 (дд, J=11,5, 5,4 Гц, 1H), 2,64 (дд, J=11,3, 8,5 Гц, 1H), 2,44 (с, 3H), 1,59 (с, 9H).
К раствору полученного, как указано выше, производного хлориндола (55 мг, 0,14 ммоль) в диоксанах (3 мл) добавляли 2 М HCl (5 мл, раствор в эфире) и смесь перемешивали при комнатной температуре в течение 5 суток. Реакционную смесь концентрировали, растворяли в 4 М HCl (5 мл, раствор в диоксанах) и перемешивали в течение ночи. Реакционную смесь концентрировали и сырой продукт очищали препаративной тонкослойной хроматографией (1-мм пластины Analtech; элюент: 95:4,5:0,5 дихлорметан/метанол/концентрированный гидроксид аммония), получая требуемый продукт (25 мг, 60%) в виде темно-желтого твердого вещества:1H ЯМР (500 МГц, CDCl3) δ 8,00 (ушир.с, 1H), 7,54 (с, 1H), 7,48 (д, J=8,1 Гц, 1H), 7,15-7,04 (м, 5H), 6,87 (д, J=7,7 Гц, 1H), 4,42 (т, J=6,7 Гц, 1H), 3,76 (д, J=15,1 Гц, 1Н), 3,65 (д, J=14,8 Гц, 1Н), 3,07 (дд, J=11,5, 5,4 Гц, 1H), 2,64 (дд, J=11,3, 8,5 Гц, 1H), 2,44 (с, 3H).
К смеси свободного основания требуемого продукта (25 мг, 0,08 ммоль) и малеиновой кислоты (9,6 мг, 0,08 ммоль) добавляли метанол (2 мл) и дихлорметан (2 мл) и смесь перемешивали в течение 1 часа. Раствор концентрировали и остаток растворяли в метаноле и воде и лиофилизировали, получая малеатную соль (+/-)-4-(3-хлор-1H-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (34 мг, количественный выход, 93,9% AUC ВЭЖХ при 254 нм, 98,2% AUC ВЭЖХ при 220 нм) в виде не совсем белого твердого вещества: т.пл. 94-98°C;1H ЯМР (500 МГц, CD3OD) δ 7,53 (д, J=8,2 Гц, 1H), 7,40-7,20 (м, 5H), 6,96 (д, J=7,7 Гц, 2H), 6,25 (с, 2H), 4,71-4,40 (м, 3H), 3,90-3,85 (м, 1Н), 3,79 (ушир.с, 1H), 3,07 (с, 3H); ESI МС m/z 297 [M+Н]+.
Пример 121 - Получение малеатной соли (+/-)-4-(1-бензил-1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: К ледяному раствору 5-броминдола (2 г, 10,2 ммоль) в ДМФА (17,6 мл) добавляли трет-бутоксид калия (2,40 г, 21,4 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут и снова охлаждали до 0°C. К реакционной смеси по каплям добавляли бензилбромид (2,4 мл, 1,96 ммоль), затем смесь перемешивали при комнатной температуре в течение 1 часа 45 минут. Реакционную смесь вливали в ледяную воду (400 мл) и перемешивали в течение 15 минут. Образовавшийся не совсем белый преципитат отфильтровывали, промывали водой и сушили при пониженном давлении при 45°C в течение ночи, получая требуемое бензилированное соединение (2,70 г, 92%) в виде не совсем белого твердого вещества:1H ЯМР (500 МГц, ДМСО-d6) δ 7,74 (д, J=1,8 Гц, 1H), 7,56 (д, J=3,0 Гц, 1H), 7,42 (д, J=8,8 Гц, 1H), 7,34-7,15 (м, 6H), 6,48 (д, J=2,8 Гц, 1H), 5,42 (с, 2H).
Стадия B: К смеси продукта со стадии A (0,50 г, 1,77 ммоль) и изохинолин-4-бороновой кислоты (0,4 г, 2,13 ммоль) в диметиловом эфире этиленгликоля (8,1 мл) добавляли раствор карбоната цезия (1,77 мл, 2 М) и смесь дегазировали (Ar, 10 минут). К смеси добавляли тетракис(трифенилфосфин)палладий (0) (0,10 г, 0,89 ммоль), затем смесь нагревали при кипячении с обратным холодильником в течение 5 часов. Охлажденную реакционную смесь распределяли между этилацетатом и водой и водный слой снова дважды экстрагировали этилацетатом. Объединенный органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (80:20 гексаны/этилацетат, затем 70:30-60:40 гексаны/этилацетат), получая требуемый продукт (0,33 г, 56%) в виде розового твердого вещества:1H ЯМР (500 МГц, CDCl3) δ 9,24 (с, 1H), 8,54 (с, 1Н), 8,06-7,95 (м, 2H), 7,78 (д, J=1,3 Гц, 1H), 7,67-7,57 (м, 2H), 7,42 (д, J=8,4 Гц, 1H), 7,36-7,29 (м, 4H), 7,24 (д, J=3,1 Гц, 1Н), 7,21-7,18 (м, 2H), 6,64 (д, J=3,1 Гц, 1Н), 5,40 (с, 2H).
Стадия C: К ледяному раствору продукта со стадии B (0,06 г, 0,17 ммоль) в дихлорметане по каплям добавляли метилтрифторметансульфонат (21 мкл, 0,19 ммоль). Реакционную смесь перемешивали при 0°C в течение 10 минут и концентрировали досуха. Остаток собирали в метаноле (1,9 мл), и охлаждали до 0°C. Небольшими порциями добавляли цианоборгидрид натрия (53 мг, 0,85 ммоль) и реакционную смесь перемешивали при 0°C в течение 20 минут. К реакционной смеси добавляли метанол (12 мл) и дихлорметан (6 мл), затем цианоборгидрид натрия (53 мг, 0,85 ммоль). Смесь нагревали до комнатной температуры и перемешивали в течение ночи. Смесь вливали в воду и экстрагировали этилацетатом. Органический экстракт сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Сырой продукт частично очищали препаративной тонкослойной хроматографией (1-мм пластины Analtech; элюент: 93:7 дихлорметан/метанол), получая требуемый продукт (40 мг).
Указанную реакцию повторяли еще раз с продуктом со стадии B (0,25 г, 0,75 ммоль). Сырой продукт реакции частично очищали хроматографией на колонке (дихлорметан, затем 98:2 и 97:3 дихлорметан/метанол), получая требуемый продукт (225 мг, частично очищенный).
Две порции объединяли и очищали препаративной обращенно-фазовой ВЭЖХ (колонка Phenomenex Luna C18 (2)). Полученный раствор соли трифторуксусной кислоты и требуемого продукта концентрировали (чтобы удалить ацетонитрил) и затем подщелачивали насыщенным раствором бикарбоната натрия. Основной раствор четыре раза экстрагировали дихлорметаном и объединенный органический экстракт сушили над сульфатом натрия, фильтровали и концентрировали, получая требуемый продукт (58 мг, 18%):1H ЯМР (500 МГц, CDCl3) δ 7,48 (с, 1H), 7,30-7,26 (м, 3H), 7,20 (д, J=8,4 Гц, 1Н), 7,15-6,97 (м, 8H), 6,48 (д, J=3,1 Гц, 1H), 5,29 (с, 2H), 4,38 (т, J=7,3 Гц, 1H), 3,80 (д, J=14,8 Гц, 1H), 3,61 (д, J=14,7 Гц, 1H), 3,09 (дд, J=11,4, 5,7 Гц, 1H), 2,61 (дд, J=11,4, 9,3 Гц, 1H), 2,43 (с, 3H).
К раствору полученного, как описано выше, продукта (57 мг, 0,16 ммоль) в метаноле (2 мл) добавляли малеиновую кислоту (19 мг, 0,16 ммоль) и смесь перемешивали в течение 40 минут. К полученному раствору добавляли метанол (6 мл) и воду (18 мл) и смесь лиофилизировали, получая малеатную соль (+/-)-4-(1-бензил-1H-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (75 мг, 99%, 97,6% AUC ВЭЖХ) в виде не совсем белого твердого вещества: т.пл. 81-85°C;1H ЯМР (500 МГц, CD3OD) δ 7,45 (ушир.с, 1H), 7,35-7,20 (м, 8H), 7,13 (д, J=7,1 Гц, 2H), 7,05-6,84 (м, 2H), 6,48 (д, J=3,1 Гц, 1H), 5,38 (с, 2H), 6,24 (с, 2H), 4,68-4,42 (м, 3H), 3,85-3,78 (м, 1H), 3,68-3,56 (ушир.м, 1H), 3,05 (с, 3H); ESI МС m/z 353 [M+Н]+.
Пример 122 - Получение фумаратной соли (+)-4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина и фумаратной соли (-)-4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору изохинолин-4-бороновой кислоты (1,90 г, 10,1 ммоль) и трет-бутилового эфира 5-броминдазол-1-карбоновой кислоты (2,00 г, 6,74 ммоль) в диметоксиэтане (40 мл) добавляли водный раствор карбоната цезия (2 М, 6,7 мл, 13,47 ммоль) и раствор дегазировали, три раза чередуя откачивание газов и продувку аргоном. К полученной гетерогенной смеси добавляли тетракис(трифенилфосфин)палладий (389 мг, 0,337 ммоль), реакционную смесь дегазировали три раза и нагревали до 85°C при встряхивании в течение 2 часов. Смесь охлаждали, добавляли EtOAc (150 мл), органический слой промывали водой (3×50 мл) и сушили над безводным сульфатом натрия, затем концентрировали. Остаток очищали хроматографией на колонке (SiO2, 40 г, 100%-50% гексан/этилацетат), получая продукт в виде желтого твердого вещества (910 мг, 45%):1H ЯМР (CDCl3, 300 МГц) δ 9,29 (с, 1Н), 8,53 (с, 1Н), 8,34 (д, J=8,5 Гц, 1Н), 8,27 (д, J=0,7 Гц, 1H), 8,09-8,07 (м, 1H), 7,88-7,85 (м, 2H), 7,71-7,65 (м, 3H), 1,77 (с, 9H); ESI МС m/z=346 [M+H]+.
Стадия B: К раствору продукта со стадии A (910 мг, 2,64 ммоль) в безводном дихлорметане (40 мл) при 0°C добавляли метилтрифторметансульфонат (328 мкл, 0,289 ммоль). Смесь перемешивали при 0°C в течение 1 часа, концентрировали досуха и перерастворяли в метаноле (100 мл). К полученному раствору добавляли цианоборгидрид натрия (1,66 г, 26,3 ммоль), смесь перемешивали при комнатной температуре в течение 12 часов и концентрировали досуха. Остаток растворяли в этилацетате (250 мл) и органический слой промывали раствором гидроксида натрия (0,05 М, 50 мл), насыщенным раствором соли (2×100 мл), сушили над безводным сульфатом натрия и концентрировали. Остаток очищали хроматографией на колонке (SiO2, 40 г, от 100% до 50% гексан/этилацетат), получая требуемый тетрагидроизохинолин в виде бесцветного масла (540 мг, 74%): ESI МС m/z=364 [M+H]+.
Стадия C: К раствору продукта со стадии B (540 мг) в дихлорметане (20 мл) при 0°C добавляли TFA (10 мл). Смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали. Остаток очищали хроматографией на колонке (SiO2, 40 г, от 100% до 50% гексан/этилацетат, затем 90% этилацетат/метанол + 1% гидроксид аммония), получая продукт в виде белого твердого вещества (530 мг, 95%).1H ЯМР (CD3OD, 300 МГц) δ 7,97 (с, 1H), 7,60 (с, 1Н), 7,46 (д, J=8,7 Гц, 1H), 7,16-7,02 (м, 4H), 6,80 (д, J=7,6 Гц, 1Н), 4,46-4,41 (м, 1Н), 3,87 (д, J=4,9 Гц, 1H), 3,62 (д, J=4,7 Гц, 1H), 3,18-3,06 (м, 1Н), 2,65-2,61 (м, 1H), 2,24 (с, 3H). ESI-МС m/z=264 [M+H]+.
Стадия D: Отдельные энантиомеры получали с помощью хиральной ВЭЖХ (Chiralcel OD, 99% гептан/1% изопропанол с 0,1% диэтиламина) продукта со стадии C. (+)-энантиомер: ВЭЖХ >99% ee (колонка Chiralpak AD. [α]D25 +102,9° (c 0,07, метанол). (-)-энантиомер: ВЭЖХ >99% ee (колонка Chiralpak AD. [α]D25 -54,0° (c 0,1, метанол).
Стадия E: К раствору каждого из отдельных энантиомеров, полученных на стадии D (320 мг, 1,22 ммоль), в метаноле (25 мл) добавляли раствор фумаровой кислоты (141 мг, 1,22 ммоль) в метаноле (15 мл) при 0°C. Раствор перемешивали при комнатной температуре в течение 5 часов, концентрировали и остаток суспендировали в диэтиловом эфире. Фумаратную соль (+)-4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина и фумаратную соль (-)-4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина получали фильтрованием в виде белого порошка (399 мг, 87%):1H ЯМР (CD3OD, 300 МГц) δ 8,03 (д, J=0,9 Гц, 1H), 7,71 (с, 1H), 7,55 (д, J=8,7 Гц, 1H), 7,33-7,19 (м, 4H), 6,91 (д, J=8,7 Гц, 1H), 6,71 (с, 2H), 4,76-4,70 (м, 1H), 4,57 (с, 3H), 3,84 (дд, J=12,2, 6,1 Гц, 1H), 3,05 (м, 3H), ESI МС m/z=264 [M+H]+, (+)-энантиомер; т.пл. 105-114°C, ВЭЖХ >99% AUG. Элементный анализ, вычислено для C17H17N3•2,25C4H4O4•Н2O: C, 57,56; Н, 5,20; N, 7,75. Найдено: C, 57,57; Н, 4,99; N, 7,57, (-)-энантиомер: т.пл. >200°C, ВЭЖХ 96,9% AUG.
Стадия F: К ледяному раствору (+)-энантиомера, полученного на стадии D (38 мг, 0,125 ммоль), в ДМФА (2 мл) добавляли NaH (60% в минеральном масле, 9 мг, 0,190 ммоль) и суспензию перемешивали при 0°C в течение 1 часа. К полученной смеси добавляли раствор α-бром-м-толунитрила (24,4 мг, 0,124 ммоль) в ДМФА (2 мл) при 0°C и раствор нагревали до комнатной температуры и перемешивали в течение 3 часов. Смесь гасили водой (2 мл), экстрагировали EtOAc (2×50 мл) и концентрировали. Остаток очищали хроматографией на колонке (SiO2, 12 г, от 50% до 100% этилацетат/гексан), получая целевое соединение в виде бесцветного масла (15,4 мг, 33%):1H ЯМР (CD3OD, 300 МГц) δ 8,06 (с, 1H), 7,64-7,48 (м, 6H), 7,23-7,16 (м, 3H), 7,09-7,05 (м, 1H), 6,84-6,81 (м, 1Н), 5,69 (с, 2H), 4,49-4,44 (м, 1Н), 3,90-3,85 (д, J=4,9 Гц, 1Н), 3,67-3,63 (д, J=4,9 Гц, 1Н), 3,18-3,13 (м, 1Н), 2,66-2,59 (м, 1Н), 2,47 (с, 3H), ESI МС m/z=379 [M+H]+, [a]D25 +26,2° (c 0,07, метанол), ВЭЖХ >99% ее (колонка Chiralpak AD).
Стадия G: К раствору продукта, полученного на стадии D (14 мг, 0,037 ммоль) в метаноле (3 мл), добавляли раствор малеиновой кислоты (4 мг, 0,037 ммоль) в метаноле (1 мл) при 0°C. Раствор перемешивали при комнатной температуре в течение ночи, затем концентрировали и твердое вещество промывали этанолом и диэтиловым эфиром, получая целевое соединение в виде белого порошка (13,4 мг, 73%): т.пл. 83-88°C. ВЭЖХ >99% AUC.1H ЯМР (CD3OD, 500 МГц) δ 8,08 (с, 1H), 7,70-7,46 (м, 6H), 7,31-7,19 (м, 4H), 6,92-6,90 (м, 1H), 6,24 (с, 2H), 5,70 (с, 2H), 4,68-4,65 (м, 1H), 4,42 (с, 2H), 3,74-3,70 (м, 1H), 3,42-3,38 (м, 1H), 2,94 (с, 3H); ESI-МС m/z=379 [M+H]+.
Пример 123 - Получение малеатной соли (+)-4-(6-метокси-1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: Рацемический 4-(6-метокси-1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин получали из 5-бром-6-метокси-1H-индазола, как описано в примере 1 (стадии A-D). Указанное рацемическое соединение (500 мг) разделяли с помощью полупрепаративной хиральной ВЭЖХ (Chiralcel OJ, 80% гептан/20% этанол с 0,1% диэтиламина). Каждый полученный в результате энантиомер растворяли в метаноле и обрабатывали раствором малеиновой кислоты в метаноле (5 мл) при 0°C. Раствор перемешивали при комнатной температуре в течение ночи, затем концентрировали и твердое вещество промывали диэтиловым эфиром, получая малеатную соль 4-(6-метокси-1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина (отдельный энантиомер) [130 мг, 54%, >99% AUC ВЭЖХ, 100% AUC хиральная ВЭЖХ (свободное основание)]:1H ЯМР (CD3OD, 500 МГц) δ 7,88 (с, 1Н), 7,30-7,25 (м, 4Н), 7,07 (с, 1Н), 7,00-6,94 (м, 1Н), 6,25 (с, 2H), 4,94-4,93 (м, 1Н), 4,60-4,50 (м, 2Н), 3,90-3,70 (м, 5Н), 3,07 (м, 3Н); ESI-МС m/z=294 [M+H], [α]D25+28,0° (с 0,05, MeOH, свободное основание); элементный анализ, вычислено для C18H19N3•1,25C4H4O4•0,75H2O: C, 61,12; Н, 5,69; N, 9,30. Найдено: C, 61,11; Н, 5,33; N, 9,19 и малеатную соль (+)-4-(6-метокси-1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина [48 мг, 54%, 98,0% AUC ВЭЖХ, 100% AUC хиральная ВЭЖХ (свободное основание)]:1H ЯМР (CD3OD, 500 МГц) δ 7,88 (с, 1H), 7,30-7,25 (м, 4H), 7,07 (с, 1Н), 7,00-6,94 (м, 1H), 6,25 (с, 6H), 4,94-4,93 (м, 1Н), 4,60-450 (м, 2H), 3,90-3,70 (м, 5H), 3,07 (м, 3H), ESI МС m/z=294 [M+H], [α]D25+4,0° (c 0,08, MeOH, свободное основание).
Пример 124 - Получение малеатной соли (+)-4-(7-хлор-1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: Смесь изохинолин-4-илбороновой кислоты (1,00 г, 5,78 ммоль), 5-бром-7-хлор-1H-индазола (892 мг, 3,85 ммоль), 2,0 М карбоната натрия (3,85 мл, 7,71 ммоль) и диметоксиэтана дегазировали, используя аргон. Добавляли тетракис(трифенилфосфин)палладий(0) (233 мг, 0,193 ммоль), смесь снова дегазировали, используя аргон, затем нагревали до 85°C в течение 24 часов. Реакционную смесь охлаждали и распределяли между этилацетатом и водой, органический слой промывали и сушили сульфатом натрия. Растворитель удаляли в вакууме и остаток очищали хроматографией на колонке (градиент 9/1-6/4 гексаны/этилацетат), получая 4-(7-хлор-1H-индазол-5-ил)изохинолин (777 мг, 72%):1H ЯМР (300 МГц, CDCl3) δ 10,52 (с, 1H) 9,31 (с, 1H), 8,53 (с, 1H), 8,22 (с, 1H), 8,09 (дд, J=7,4, 1,62 Гц, 1H), 7,28 (дд, J=7,4, 1,2 Гц, 1H), 7,87 (д, J=9,0 Гц, 1H), 7,81 (д, J=1,2 Гц, 1H), 7,72-7,64 (м, 2H), 7,57 (д, J=1,3 Гц, 1H).
Стадия B: К суспензии продукта, полученного на стадии A (776 мг, 2,77 ммоль), и ди-трет-бутилдикарбоната (909 мг, 4,16 ммоль) в ацетонитриле (18 мл) добавляли 4-диметиламинопиридин. Полученный в результате раствор перемешивали в течение ночи, растворитель удаляли в вакууме и остаток очищали хроматографией на колонке (3:1 гексаны/этилацетат), получая требуемый BOC-защищенный продукт (778 мг, 74%):1H ЯМР (500 МГц, CDCl3) δ 9,26 (с, 1H), 8,76 (с, 1H), 8,53 (с, 1H), 8,07 (д, J=7,9 Гц, 1H), 7,88 (д, J=8,3, Гц, 1Н), 7,73-7,67 (м, 3H), 7,54 (д, J=1,3 Гц, 1Н), 1,75 (с, 9H).
Стадия C: К раствору продукта, полученного на стадии B (758 мг, 2,00 ммоль), в дихлорметане с температурой 0°C добавляли анизол (1 мл) и метилтрифторметансульфонат (344 мг, 2,09 ммоль). Раствору давали возможность нагреться 10°C в течение 1,5 часа и концентрировали досуха в вакууме. В колбу загружали анизол (1 мл) и смесь растворяли в метаноле. Затем добавляли цианоборгидрид натрия (503 мг, 8,00 ммоль) и реакционную смесь перемешивали в течение 2,5 часов. Реакционную смесь вливали в смесь насыщенного раствора соли (196 мл) и воды (30 мл) и три раза экстрагировали этилацетатом и объединенные органические слои промывали насыщенным раствором соли, сушили над сульфатом натрия, концентрировали в вакууме и остаток очищали хроматографией на колонке (градиент от 75:25 до 60:40 гексаны/этилацетат), получая требуемый тетрагидроизохинолин (655 мг, 82%):1H ЯМР (500 МГц, CDCl3) δ 8,56 (с, 1H), 7,34 (с, 1H), 7,26 (с, 1H), 7,21 (д, J=1,2 Гц, 1H), 7,18-6,89 (м, 3H), 4,27 (т, J=6,50 Гц, 1H), 3,71 (д, J=15,0 Гц, 1H), 3,67 (д, J=15,0 Гц, 1H), 2,99 (дд, J=11,4, 5,4 Гц, 1H), 2,66 (дд, J=11,6, 7,70 Гц, 1H), 2,44 (с, 3H), 1,71 (с, 9H).
Стадия D: К раствору продукта, полученного на стадии C (655 мг, 1,65 ммоль), в метаноле (18,3 мл) добавляли 1N HCl в эфире (17,9 мл, 17,9 ммоль). Раствор перемешивали в течение ночи, концентрировали в вакууме и остаток очищали хроматографией на колонке (градиент от 95:5:0,2 до 90:10:0,2 дихлорметан/метанол/концентрированный гидроксид аммония), получая требуемый индазол с удаленной защитой (427 мг, 84%):1H ЯМР (500 МГц, CDCl3) δ 10,18 (с, 1Н), 8,04 (с, 1H), 7,49 (д, J=1,0 Гц, 1H), 7,26-7,06 (м, 3H), 6,87 (д, J=7,8 Гц, 1Н), 4,36 (т, J=6,50 Гц, 1H), 3,72 (д, J=14,9 Гц, 1H), 3,69 (д, J=15,0 Гц, 1H), 3,03 (дд, J=11,5, 5,5 Гц, 1H), 2,65 (дд, J=11,5, 7,80 Гц, 1H), 2,44 (с, 3H).
Полученное соединение разделяли препаративной хиральной ВЭЖХ (колонка CHIRALPAK AD, с использованием смеси 8:2:0,01 гептан/IPA/триэтиламин в качестве элюента), получая (+)-энантиомер [[α]25D +45,1° (c 0,04, метанол)] и (-)-энантиомер [[α]25D -81,3° (c 0,03, метанол)]. (+)-Энантиомер (105 мг, 0,353 ммоль) превращали в его малеатную соль растворением в абсолютном этаноле (1 мл), добавлением одного эквивалента малеиновой кислоты в этаноле и концентрированием досуха в вакууме. Остаток растворяли в смеси 1:1 ацетонитрил/вода и лиофилизировали, получая малеатную соль (+)-4-(7-хлор-1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина в виде не совсем белого твердого вещества (143 мг, 98%, >99% AUC ВЭЖХ): т.пл. 107,4-110,9°C;1H ЯМР (500 МГц, CD3OD) δ 8,12 (с, 1H), 7,66 (с, 1H), 7,35-7,25 (м, 4H), 6,94 (д, J=7,7 Гц, 1H), 6,24 (с, 2H) 4,71 (дд, J=10,4, 6,2 Гц, 1H), 4,58 (д, J=15,3 Гц, 1H), 4,54 (д, J=15,3 Гц, 1H), 3,85 (дд, J=12,4, 6,4 Гц, 1H), 3,58 (т, J=11,8 Гц, 1H), 3,05 (с, 3H); ESI-МС m/z 298 [M+H]+.
Пример 125 - Получение малеатной соли (-)-4-(7-хлор-1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Свободное основание (-)-энантиомера (112 мг, 0,376 ммоль) со стадии D примера 126 превращали в его малеатную соль растворением в абсолютном этаноле (1 мл) добавлением одного эквивалента малеиновой кислоты в этаноле и концентрированием досуха в вакууме. Остаток растворяли в смеси 1:1 ацетонитрил/вода и лиофилизировали, получая малеатную соль (-)-4-(7-хлор-1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина в виде белого твердого вещества (143 мг, 92%, >99% AUC ВЭЖХ): т.пл. 104-110°C;1H ЯМР (500 МГц, CD3OD) δ 8,12 (с, 1H), 7,67 (с, 1H), 7,35-7,25 (м, 4H), 6,95 (д, J=7,8 Гц, 1H), 6,24 (с, 2Н), 4,73 (дд, J=11,0, 6,3 Гц, 1H), 4,63 (д, J=15,3 Гц, 1H), 4,57 (д, J=15,3 Гц, 1H), 3,85 (дд, J=12,3, 6,2 Гц, 1H), 3,63 (т, J=11,8 Гц, 1H), 3,08 (с, 3H); ESI-МС m/z 298 [M+H]+.
Пример 126 - Получение фумаратной соли (+)-4-(1H-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина и фумаратной соли (-)-4-(1H-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Рацемический 4-(7-метил-1H-индазол-5-ил)-2-метил-1,2,3,4- тетрагидроизохинолин получали из 5-бром-7-метил-1H-индазола способом, сходным со способом, описанным в примере 131 (стадии A-D). Полученное рацемическое соединение (180 мг) разделяли с помощью полупрепаративной хиральной ВЭЖХ (Chiralpak AD, 80% гептан/20% изопропанол с 0,1% диэтиламина). Полученные в результате свободные основания растворяли в метаноле и обрабатывали раствором малеиновой кислоты в метаноле (5 мл) при 0°C. Раствор перемешивали при комнатной температуре в течение ночи, затем концентрировали и твердое вещество промывали диэтиловым эфиром, получая малеатную соль (+)-4-(7-метил-1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина [23 мг, 9%, >99% AUC ВЭЖХ, 96,6% AUC хиральная ВЭЖХ (свободное основание)]:1H ЯМР (500 МГц, CD3OD) δ 8,01 (с, 1H), 7,52 (с, 1Н), 7,31-7,23 (м, 3H), 6,98-6,93 (м, 2H), 6,23 (с, 2Н), 4,70-4,56 (м, 3Н), 3,88-3,84 (м, 1Н), 3,64-3,60 (м, 1H), 3,08 (с, 3H), 2,52 (с, 3H), ESI МС m/z=278 [C18H19N3+H], [α]D25 +51,7° (c 0,07, MeOH, свободное основание) и малеатную соль (-)-4-(7-метил-1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина [71 мг, 71%, 92,7% AUC ВЭЖХ, 91,3% AUC хиральная ВЭЖХ (свободное основание)]:1H ЯМР (CD3OD, 500 МГц) δ 8,01 (с, 1H), 7,52 (с, 1Н), 7,31-7,23 (м, 3H), 6,98-6,93 (м, 2H), 6,23 (с, 2H), 4,70-4,56 (м, 3H), 3,88-3,84 (м, 1H), 3,64-3,60 (м, 1H), 3,08 (с, 3H), 2,52 (с, 3H), ESI-МС m/z=278 [M+H], [α]D25-43,5° (c 0,06, MeOH, свободное основание).
Пример 127 - Получение малеатной соли (+/-)-4-(1H-индазол-5-ил-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору Boc-защищенного 5-броминдазола (2,0 г, 6,7 ммоль) и н-бутилвинилового эфира (4,4 мл, 33,6 ммоль) в диоксане (20 мл) добавляли три-трет-бутилфосфин (0,41 г, 2,0 ммоль) и N-метилдициклогексиламин (1,6 мл, 7,4 ммоль). Реакционный раствор продували аргоном в течение 5 минут и затем к нему добавляли трис(дибензилиденацетон)дипалладий (0,37 г, 0,4 ммоль). Реакционную колбу продували аргоном в течение 5 минут и затем закрывали и нагревали при 40°C в течение 16 часов. Затем реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом. Органический раствор промывали водой, насыщенным водным раствором хлорида аммония и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток растворяли в смеси тетрагидрофурана (60 мл) и воды (30 мл) и обрабатывали уксусной кислотой (5 мл). Полученный в результате раствор перемешивали при комнатной температуре в течение 2 часов и затем растворитель удаляли при пониженном давлении. Полученный остаток разбавляли этилацетатом, дважды промывали водой, дважды насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученный сырой продукт очищали, используя систему MPLC Biotage (от 95:5 до 65:35 гексаны/этилацетат), получая требуемый продукт (0,91 г, 52%) в виде красноватого масла:1H ЯМР (CDCl3, 500 МГц) δ 8,38 (т, J=0,7 Гц, 1H), 8,28 (с, 1Н), 8,25 (д, J=8,9 Гц, 1H), 8,16 (дд, J=8,9, 1,5 Гц, 1H), 2,69 (с, 3H), 1,74 (с, 9H).
Стадия B: К раствору метилкетона (0,85 г, 3,2 ммоль) с описанной выше стадии A в тетрагидрофуране (12 мл) добавляли гидротрибромид 2-пирролидинона (1,75 г, 3,4 ммоль) и 2-пирролидинон (0,26 мл). Реакционный раствор нагревали при кипячении с обратным холодильником в течение 1 часа и затем охлаждали до комнатной температуры и фильтровали, чтобы удалить образованный преципитат. Полученный фильтрат концентрировали при пониженном давлении, получая сырой продукт, который очищали, используя систему MPLC Biotage (от 95:5 до 63:37 гексаны/этилацетат), получая требуемый продукт (0,91 г, 70%) в виде желтого твердого вещества:1H ЯМР (CDCl3, 500 МГц) δ 8,44 (д, J=1,2 Гц, 1H), 8,30 (с, 1H), 8,29 (д, J=9,0 Гц, 1H), 8,18 (дд, J=8,8 Гц, 1H), 4,51 (с, 2H), 1,74 (с, 9H).
Стадия C: К раствору метил-(3-морфолин-4-илбензил)амина (0,11 г, 0,1 ммоль) в дихлорметане (5 мл) добавляли α-бромметилкетон (0,16 г, 0,47 ммоль) с описанной выше стадии B и N,N-диизопропилэтиламин (0,13 мл, 0,77 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение 3 часов и затем разбавляли дихлорметаном. Полученный раствор промывали водой, сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученный сырой продукт частично очищали флэш-хроматографией на колонке (98:2 дихлорметан/метанол), получая требуемый продукт (0,22 г), который использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 8,38 (с, 1H), 8,23 (д, J=0,5 Гц, 1H), 8,21 (д, J=8,8 Гц, 1H), 8,16 (дд, J=8,8, 1,5 Гц, 1H), 7,23 (т, J=7,8 Гц, 1H), 6,91 (с, 1H), 6,85-6,81 (м, 2H), 3,87-3,82 (м, 4H), 3,79 (с, 2H), 3,62 (с, 2H), 3,17-3,12 (м, 4H), 2,39 (с, 3H), 1,74 (с, 9H); ESI-МС m/z 465 [M+H]+.
Стадия D: К раствору кетона (0,22 г, 0,47 ммоль) с описанной выше стадии C в метаноле (15 мл) при 0°C добавляли боргидрид натрия (19 мг, 0,52 ммоль). Реакционную смесь перемешивали в течение 45 минут и затем растворитель удаляли при пониженном давлении. Полученный остаток растворяли в дихлорметане, промывали водой и насыщенным водным раствором бикарбоната натрия, сушили над сульфатом натрия и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (от 98:2 до 95:5 дихлорметан/метанол), получая требуемый продукт (0,13 г, 58% для 2 стадий) в виде светло-желтого масла:1H ЯМР (CDCl3, 500 МГц) δ 8,15-8,13 (м, 2H), 7,75 (д, J=0,6 Гц, 1H), 7,49 (дд, J=8,8, 1,5 Гц, 1H), 7,27-7,23 (м, 1H), 6,89 (с, 1H), 6,86-6,83 (м, 2H), 4,87 (дд, J=10,0, 4,0 Гц, 1H), 4,26-4,06 (м, 1Н), 3,88-3,86 (м, 4H), 3,73 (д, J=13,0 Гц, 1H), 3,50 (д, J=13,0 Гц, 1H), 3,18-3,16 (м, 4H), 2,61-2,57 (м, 2H), 2,36 (с, 3H), 1,72 (с, 9H).
Стадия E: К раствору спирта (0,12 г, 0,25 ммоль) с описанной выше стадии D в дихлорметане (6 мл) добавляли N,N-диизопропилэтиламин (70 мкл, 0,50 ммоль), затем метансульфонилхлорид (23 мкл, 0,30 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение 90 минут и затем к нему двумя порциями добавляли метансульфоновую кислоту (0,16 мл, 2,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем гасили насыщенным водным раствором бикарбоната натрия и дважды экстрагировали дихлорметаном. Объединенный органический экстракт сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученный сырой продукт очищали препаративной тонкослойной хроматографией (95:4,5:0,5 этилацетат/метанол/концентрированный гидроксид аммония), получая требуемый продукт (10 мг, 11%) в виде белой пены:1H ЯМР (CDCl3, 500 МГц) δ 10,15-9,73 (ушир.с, 1H), 8,00 (д, J=0,5 Гц, 1H), 7,59 (с, 1H), 7,39 (д, J=8,9 Гц, 1H), 7,23 (дд, J=8,7, 1,5 Гц, 1H), 6,78 (д, J=8,5 Гц, 1H), 6,68-6,62 (м, 2H), 4,34-4,31 (м, 1H), 3,86-3,83 (м, 4H), 3,72 (д, J=14,9 Гц, 1H), 3,63 (д, J=14,9 Гц, 1H), 3,14-3,11 (м, 4H), 3,06-3,00 (м, 1H), 2,61 (дд, J=11,2, 8,5 Гц, 1H), 2,43 (с, 3H); ESI-МС m/z 349 [M+H]+.
Стадия F: К раствору 7-морфолинилтетрагидроизохинолина (10 мг, 0,028 ммоль) с описанной выше стадии E в метаноле (2 мл) добавляли малеиновую кислоту (6,6 мг, 0,057 ммоль), затем воду (10 мл). Полученный в результате раствор лиофилизировали в течение ночи, получая малеатную соль (+/-)-4-(1H-индазол-5-ил-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина (16 мг, 96%) в виде светло-желтого твердого вещества: т.пл. 71-75°C;1H ЯМР (CD3OD, 500 МГц) δ 8,02 (с, 1H), 7,78-7,57 (м, 1H), 7,55 (д, J=8,6 Гц, 1H), 7,21 (д, J=8,5 Гц, 1H), 6,89 (д, J=7,9 Гц, 1H), 6,82-6,81 (м, 2H), 6,28 (с, 5H), 4,65-4,43 (м, 3H), 3,86-3,80 (м, 5H), 3,63-3,44 (м, 1H), 3,15-3,13 (м, 4H), 3,07 (с, 3H); ESI-МС m/z 349 [M+H]+.
Пример 128 - Получение малеатной соли (+)-3-[5-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)индазол-1-илметил]бензонитрила
Стадия A: К раствору изохинолин-4-бороновой кислоты (1,90 г,
10,1 ммоль) и трет-бутилового эфира 5-броминдазол-1-карбоновой кислоты (2,00 г, 6,74 ммоль) в диметоксиэтане (40 мл) добавляли водный раствор карбоната цезия (2 М, 6,7 мл, 13,47 ммоль) и раствор дегазировали, три раза чередуя откачку газов и продувку аргоном. К полученной гетерогенной смеси добавляли тетракис(трифенилфосфин)палладий (389 мг, 0,337 ммоль), реакционную смесь три раза дегазировали и нагревали до 85°C при встряхивании в течение 2 часов. Смесь охлаждали, добавляли EtOAc (150 мл), органический слой промывали водой (3×50 мл) и сушили над безводным сульфатом натрия, затем концентрировали. Остаток очищали хроматографией на колонке (SiO2, 40 г, от 100% до 50% гексан/этилацетат), получая продукт в виде желтого твердого вещества (910 мг, 45%):1H ЯМР (CDCl3, 300 МГц) δ 9,29 (с, 1H), 8,53 (с, 1H), 8,34 (д, J=8,5 Гц, 1H), 8,27 (д, J=0,7 Гц, 1H), 8,09-8,07 (м, 1Н), 7,88-7,85 (м, 2H), 7,71-7,65 (м, 3H), 1,77 (с, 9H); ESI-МС m/z=346 [M+H]+.
Стадия B: К раствору продукта со стадии A (910 мг, 2,64 ммоль) в безводном дихлорметане (40 мл) при 0°C добавляли метилтрифторметансульфонат (328 мкл, 0,289 ммоль). Смесь перемешивали при 0°C в течение 1 часа, концентрировали досуха и перерастворяли в метаноле (100 мл). К полученному раствору добавляли цианоборгидрид натрия (1,66 г, 26,3 ммоль), смесь перемешивали при комнатной температуре в течение 12 часов и концентрировали досуха. Остаток растворяли в этилацетате (250 мл) и органический слой промывали раствором гидроксида натрия (0,05 М, 50 мл), насыщенным раствором соли (2×100 мл), сушили над безводным сульфатом натрия и концентрировали. Остаток очищали хроматографией на колонке (SiO2, 40 г, от 100% до 50% гексан/этилацетат), получая требуемый тетрагидроизохинолин в виде бесцветного масла (540 мг, 74%): ESI-МС m/z=364 [M+H]+.
Стадия C: К раствору продукта со стадии B (540 мг) в дихлорметане (20 мл) при 0°C добавляли TFA (10 мл). Смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали. Остаток очищали хроматографией на колонке (SiO2, 40 г, от 100% до 50% гексан/этилацетат, затем 90% этилацетат/метанол + 1% концентрированного гидроксида аммония), получая 4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин в виде белого твердого вещества (530 мг, 95%).1H ЯМР (CD3OD, 300 МГц) δ 7,97 (с, 1H), 7,60 (с, 1Н), 7,46 (д, J=8,7 Гц, 1H), 7,16-7,02 (м, 4H), 6,80 (д, J=7,6 Гц, 1H), 4,46-4,41 (м, 1Н), 3,87 (д, J=4,9 Гц, 1H), 3,62 (д, J=4,7 Гц, 1H), 3,18-3,06 (м, 1H), 2,65-2,61 (м, 1H), 2,24 (с, 3H), ESI МС m/z=264 [M+H]+.
Стадия D: Отдельные энантиомеры получали с помощью хиральной хроматографии (Chiralcel OD, 99% гептан/изопропанол с 0,1% диэтиламина) продукта со стадии C. (+)-энантиомер: ВЭЖХ >99% ee (колонка Chiralpak AD. [α]D25 +102,9° (c 0,07, метанол). (-)-энантиомер: ВЭЖХ >99% ee (колонка Chiralpak AD. [α]D25 -54,0° (c 0,1, метанол).
Стадия E: К ледяному раствору (+)-энантиомера, полученного на стадии D (38 мг, 0,125 ммоль) в ДМФА (2 мл), добавляли NaH (60% в минеральном масле, 9 мг, 0,190 ммоль) и суспензию перемешивали при 0°C в течение 1 часа. К полученной смеси добавляли раствор α-бром-м-толунитрила (24,4 мг, 0,124 ммоль) в ДМФА (2 мл) 0°C и раствор нагревали до комнатной температуры и перемешивали в течение 3 часов. Смесь гасили водой (2 мл), экстрагировали EtOAc (2×50 мл) и концентрировали. Остаток очищали хроматографией на колонке (SiO2, 12 г, от 50% до 100% этилацетат/гексан), получая (+)-3-[5-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)индазол-1-илметил]бензонитрил в виде бесцветного масла (15,4 мг, 33%):1H ЯМР (CD3OD, 300 МГц) δ 8,06 (с, 1H), 7,64-7,48 (м, 6H), 7,23-7,16 (м, 3H), 7,09-7,05 (м, 1H), 6,84-6,81 (м, 1H), 5,69 (с, 2H), 4,49-4,44 (м, 1H), 3,90-3,85 (д, J=4,9 Гц,1Н), 3,67-3,63 (д, J=4,9 Гц, 1H), 3,18-3,13 (м, 1H), 2,66-2,59 (м, 1H), 2,47 (с, 3H), ESI-МС m/z=379 [M+H]+, [α]D25 +26,2° (c 0,07, метанол), ВЭЖХ >99% ее (колонка Chiralpak AD).
Стадия F: К раствору продукта, полученного на стадии D (14 мг, 0,037 ммоль) в метаноле (3 мл), добавляли раствор малеиновой кислоты (4 мг, 0,037 ммоль) в метаноле (1 мл) при 0°C. Раствор перемешивали при комнатной температуре в течение ночи, затем концентрировали и твердое вещество промывали этанолом и диэтиловым эфиром, получая малеатную соль (+)-3-[5-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)индазол-1-илметил]бензонитрила в виде белого порошка (13,4 мг, 73%): т.пл. 83-88°C. ВЭЖХ >99% AUC.1H ЯМР (CD3OD, 500 МГц) δ 8,08 (с, 1H), 7,70-7,46 (м, 6H), 7,31-7,19 (м, 4H), 6,92-6,90 (м, 1H), 6,24 (с, 2H), 5,70 (с, 2H), 4,68-4,65 (м, 1Н), 4,42 (с, 2H), 3,74-3,70 (м, 1H), 3,42-3,38 (м, 1Н), 2,94 (с, 3H), ESI-МС m/z=379[M+H]+.
Пример 129 - Получение фумаратной соли (+)-4-(1H-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина и фумаратной соли (-)-4-(1H-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору 6-броминдазола (2,57 г, 13,0 ммоль) в ацетонитриле (50 мл) добавляли ди-трет-бутилдикарбонат (4,3 мг, 19,6 ммоль) и DMAP (80 мг, 6,52 ммоль). Смесь перемешивали при комнатной температуре в течение 26 часов и концентрировали досуха. Остаток очищали хроматографией на колонке, получая очищенный продукт (2,09 г, 64%) в виде белого твердого вещества:1H ЯМР (CDCl3, 300 МГц) δ 8,43 (с, 1H), 8,13 (д, J=0,7 Гц, 1H), 7,60 (д, J=8,4 Гц, 1H), 7,44 (дд, J=8,4 Гц, 1,6 Гц, 1H), 1,73 (с, 9H).
Стадия B: К раствору изохинолин-4-бороновой кислоты (4,36 г, 23,1 ммоль) и продукта со стадии A (4,58 г, 15,4 ммоль) в диметоксиэтане (40 мл) добавляли водный раствор карбоната цезия (2 М, 15 мл, 30,8 ммоль) и раствор дегазировали, три раза чередуя откачивание газов и продувку аргоном. К полученной гетерогенной смеси добавляли тетракис(трифенилфосфин)палладий (890 мг, 0,77 ммоль). Реакционную смесь три раза дегазировали и нагревали до 85°C при встряхивании в течение 4 часов. Охлажденную реакционную смесь разбавляли EtOAc (250 мл), промывали водой (3×150 мл), сушили (сульфат натрия) и концентрировали. Остаток очищали хроматографией на колонке (SiO2, 40 г, от 100% до 0% гексан/этилацетат), получая продукт в виде желтого твердого вещества (4,00 г, 70%):1H ЯМР (CDCl3, 300 МГц) δ 9,31 (с, 1H), 8,56 (с, 1H), 8,39 (с, 1H), 8,28 (с, 1H), 8,10-8,07 (м, 1H), 7,93-7,87 (м, 2H), 7,71-7,66 (м, 2H), 7,50-7,47 (м, 1H), 1,70 (с, 9H); ESI-МС m/z=345 [M+H]+.
Стадия C: К раствору продукта со стадии B (2,00 г, 5,79 ммоль) в безводном дихлорметане (60 мл) при 0°C добавляли метилтрифторметансульфонат (720 мкл, 6,37 ммоль). Смесь перемешивали при 0°C в течение 1 часа, концентрировали досуха и растворяли в метаноле (60 мл). К полученному раствору добавляли цианоборгидрид натрия (1,40 г, 23,2 ммоль), смесь перемешивали при комнатной температуре в течение 12 часов и концентрировали досуха. Остаток растворяли в этилацетате (250 мл) и органический слой промывали раствором гидроксида натрия (0,05 М, 50 мл), насыщенным раствором соли (2×100 мл), сушили над безводным сульфатом натрия и концентрировали. Остаток очищали хроматографией на колонке (SiO2, 40 г, от 85% до 0% гексан/этилацетат), получая тетрагидроизохинолин в виде бесцветного масла (1,56 г, 63%): ESI МС m/z=364 [M+H]+.
Стадия D: К раствору продукта со стадии C (1,56 г) в дихлорметане (20 мл) при 0°C добавляли TFA (10 мл). Смесь перемешивали при комнатной температуре в течение 2,5 часов и концентрировали. Остаток очищали хроматографией на колонке (SiO2, 40 г, от 1:0 до 1:1 гексан/этилацетат, затем 90% этилацетат/10% метанол + 1% гидроксида аммония), получая продукт в виде белого твердого вещества (970 мг, 95%):1H ЯМР (CD3OD, 300 МГц) δ 7,99 (д, J=0,6 Гц, 1Н), 7,69 (д, J=8,2 Гц, 1H), 7,36 (с, 1Н), 7,21-7,15 (м, 2H), 7,10-7,04 (м, 1Н), 6,95-6,92 (м, 1H), 6,84-6,82 (д, J=7,6 Гц, 1Н), 4,49-4,44 (м, 1H), 3,87 (д, J=15,0 Гц, 1Н), 3,64 (д, J=14,9 Гц, 1H), 3,20-3,14 (м, 1H), 2,64 (дд, J=11,6, 9,9 Гц, 1H), 2,45 (с, 3H); ESI-МС m/z=264 [M+H]+.
Стадия E: Отдельные энантиомеры получали с помощью хиральной хроматографии (Chiralcel OD5 95% гептан/5% изопропанол с 0,1% диэтиламина) продукта со стадии D. (+)-энантиомер: [α]D25 +72,0° (c 0,05, метанол); ВЭЖХ >99% ee (колонка Chiralpak AD). (-)-энантиомер: [α]D25 -89,5° (c 0,16, метанол); ВЭЖХ >99% ee (колонка Chiralpak AD).
Стадия F: К раствору отдельного энантиомера, полученного на стадии E (400 мг, 1,52 ммоль) в метаноле (40 мл) добавляли раствор фумаровой кислоты (352 мг, 3,04 ммоль) в метаноле (15 мл) при 0°C. Раствор перемешивали при комнатной температуре в течение 12 часов, затем концентрировали и остаток суспендировали в диэтиловом эфире. Фумаратную соль выделяли фильтрованием в виде белого порошка (650 мг, 87%):1H ЯМР (CD3OD, 300 МГц) δ 8,04 (с, 1Н), 7,79-7,76 (д, J=8,4 Гц, 1H), 7,46 (с, 1H), 7,30-7,20 (м, 3H), 6,99-6,92 (м, 2H), 6,71 (с, 4H), 4,73-4,67 (м, 1H), 4,46 (с, 1H), 3,80-3,74 (м, 1H), 3,53-3,43 (м, 1H), 2,97 (с, 3H); ESI-MS = 264 [M+H]+, (+)-энантиомер: т.пл. 114-125°C, ВЭЖХ >99% AUG. Элементный анализ, вычислено для C17H17N3•2C4H4O4•0,5H2O: C, 59,52; Н, 5,19; N, 8,33. Найдено: C, 59,41; Н, 5,02; N, 8,34, (-)-энантиомер: т.пл. 110-117°C, ВЭЖХ >99% AUG.
Пример 130 - Получение малеатной соли (+)-4-(1H-индазол-6-ил)-2-этил-1,2,3,4-тетрагидроизохинолина и малеатной соли (-)-4-(1H-индазол-6-ил]-2-этил-1,2,3,4-тетрагидроизохинолина
Рацемический 4-(1H-индазол-6-ил)-2-этил-1,2,3,4-тетрагидроизохинолин получали из трет-бутилового эфира 6-изохинолин-4-илиндазол-1-карбоновой кислоты и этилтрифторметансульфоната способом, сходным со способом, описанным в примере 131. Полученное рацемическое соединение (500 мг, 74%) разделяли с помощью полупрепаративной хиральной ВЭЖХ (Chiralcel OD, 95% гептан/5% изопропанол с 0,1% диэтиламина). Полученные в результате свободные основания растворяли в метаноле и обрабатывали раствором малеиновой кислоты в метаноле (5 мл) при 0°C. Раствор перемешивали при комнатной температуре в течение 12 часов, затем концентрировали и твердое вещество промывали диэтиловым эфиром, получая малеатную соль (+)-4-(1H-индазол-6-ил)-2-этил-1,2,3,4-тетрагидроизохинолина [250 мг, 87%, >99% AUC ВЭЖХ, 99,6% AUC, хиральная ВЭЖХ (свободное основание)]:1H ЯМР (500 МГц, CD3OD) δ 8,03 (д, J=0,86 Гц, 1H), 7,71 (с, 1H), 7,55 (д, J=8,7 Гц, 1H), 7,33-7,19 (м, 4H), 6,91 (д, J=8,7 Гц, 1H), 6,24 (с, 2H), 4,76-4,70 (м, 1H), 4,70-4,50 (м, 2H), 4,57 (с, 3H), 3,84 (дд, J=12,2, 6,1 Гц, 1H), 1,47-1,44 (м, 3H); ESI-МС m/z=278 [M+H]+; [α]25D +70,0° (c 0,11, MeOH, свободное основание); элементный анализ, вычислено для C18H19N3•C4H4O4•0,5Н2O: C, 65,66; Н, 6,01; N, 10,44. Найдено: C, 65,35; Н, 5,80; N, 10,27, и малеатную соль (-)-4-(1H-индазол-6-ил)-2-этил-1,2,3,4-тетрагидроизохинолина [280 мг, 97,0% AUC ВЭЖХ, 100% AUC, хиральная ВЭЖХ (свободное основание)]:1H ЯМР (CD3OD, 500 МГц) δ 8,03 (д, J=0,86 Гц, 1H), 7,71 (с, 1H), 7,55 (д, J=8,7 Гц, 1Н), 7,33-7,19 (м, 4H), 6,91 (д, J=8,7 Гц, 1H), 6,71 (с, 2H), 4,76-4,70 (м, 1H), 4,70-4,50 (м, 2H), 4,57 (с, 3H), 3,84 (дд, J=12,2, 6,1 Гц, 1Н), 1,47-1,44 (м, 3H), ESI-МС m/z=278 [M+H]+; [α]25D -84,7,0° (c 0,08, MeOH, свободное основание).
Пример 131 - Получение малеатной соли (+/-)-4-бензооксазол-2-ил-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: Рацемический 4-бензооксазол-2-ил-2-метил-1,2,3,4- тетрагидроизохинолин получали способом, сходным со способом, описанным в примере 132, начиная с бензооксазола (стадии A-B). Свободное основание (20 мг, 0,076 ммоль) растворяли в метаноле (5 мл), добавляли раствор малеиновой кислоты (8 мг, 0,076 ммоль) в метаноле (2 мл) при 0°C. Раствор перемешивали при комнатной температуре в течение 12 часов, концентрировали и остаток суспендировали в диэтиловом эфире. Фильтрование давало малеатную соль 4-бензооксазол-2-ил-2-метил-1,2,3,4-тетрагидроизохинолина в виде белого порошка (28 мг, 98%):1H ЯМР (CD3OD, 500 МГц) δ 7,69 (дд, J=8,3, 6,8 Гц, 2H), 7,62 (дд, J=8,3, 1,3 Гц, 2H), 7,48-7,36 (м, 5H), 7,28 (д, J=2,9 Гц, 1Н), 5,42 (с, 2H), 4,98-4,97 (м, 1H), 4,43-4,33 (м, 2H), 4,03 (с, 1Н), 3,81-3,77 (м, 1Н), 3,04 (с, 3H); ESI-МС m/z=265 [M+H]+; т.пл. 135-140°C; ВЭЖХ 97,5% AUC.
Пример 132 - Получение малеатной соли (+/-)4-бензотиазол-2-ил-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору 4-бромизохинолина (2,00 г, 9,61 ммоль), бензотиазола (1,08 г, 8,01 ммоль) и бромида меди (374 мг, 1,60 ммоль) в ДМФА (50 мл) добавляли карбонат цезия (3,16 г, 9,70 ммоль) и раствор дегазировали, три раза чередуя откачивание газов и продувку аргоном. К полученной гетерогенной смеси добавляли ацетат палладия (II) (540 мг, 0,801 ммоль), реакционную смесь три раза дегазировали и нагревали до 150°C при встряхивании в течение 3 часов. Смесь охлаждали, добавляли диэтиловый эфир (150 мл), органический слой промывали водой (3×50 мл) и сушили над безводным сульфатом натрия, затем концентрировали. Остаток очищали хроматографией на колонке (SiO2, 40 г, от 100% до 0% гексан/этилацетат), получая продукт в виде желтого твердого вещества (646 мг, 31%): ESI-МС m/z=263 [M+H]+.
Стадия B: К раствору продукта со стадии A (100 мг, 0,381 ммоль) в безводном дихлорметане (5 мл) при 0°C по каплям добавляли метилтрифторметансульфонат (43 мкл, 0,381 ммоль) при -40°C. Смесь перемешивали при -40°C в течение 1 часа, концентрировали досуха и перерастворяли в метаноле (100 мл). К полученному раствору добавляли цианоборгидрид натрия (1,66 г, 26,3 ммоль) при 0°C, смесь перемешивали при комнатной температуре в течение 12 часов и концентрировали досуха. Остаток очищали полупрепаративной обращенно-фазовой ВЭЖХ, получая требуемый тетрагидроизохинолин в виде белого твердого вещества (18 мг, 17%). ESI-МС m/z=281 [M+H]+.
Стадия C: К раствору продукта со стадии B (18 мг, 0,064 ммоль) в метаноле (5 мл) добавляли раствор малеиновой кислоты (7 мг, 0,064 ммоль) в метаноле (2 мл) при 0°C. Раствор перемешивали при комнатной температуре в течение 12 часов, концентрировали и остаток суспендировали в диэтиловом эфире. Фильтрование давало малеатную соль (+/-)-4-бензотиазол-2-ил-2-метил-1,2,3,4-тетрагидроизохинолина в виде белого порошка (18 мг, 71%):1H ЯМР (CD3OD, 500 МГц) δ 8,00-7,92 (м, 2H), 7,70-7,29 (м, 6H), 6,26 (с, 4H), 5,11-5,04 (м, 1H), 4,64-4,44 (м, 2H), 4,32-4,19 (ушир.с, 1H), 3,96-3,84 (м, 1H), 3,19-3,10 (м, 3H). ESI МС m/z=281 [M+H]+, т.пл 165-170°C, ВЭЖХ 95,7% AUC.
Пример 133 - Получение малеатной соли (+/-)4-бензотиазол-6-ил-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: Рацемический 4-бензотиазол-6-ил-2-метил-1,2,3,4- тетрагидроизохинолин получали из 6-бромбензотиазола способом, сходным со способом, описанным в примере 129 (стадии B-C). Требуемый тетрагидроизохинолин (20,5 мг) растворяли в метаноле и обрабатывали раствором малеиновой кислоты в метаноле (1 мл) при 0°C. Раствор перемешивали при комнатной температуре в течение ночи, затем концентрировали и твердое вещество промывали диэтиловым эфиром, получая малеатную соль (+/-)4-бензотиазол-6-ил-2-метил-1,2,3,4-тетрагидроизохинолина (30 мг, 98%, >99% AUC ВЭЖХ):1H ЯМР (CD3OD, 500 МГц) δ 9,28 (д, J=3,9 Гц, 1H), 8,09-8,08 (м, 1H), 8,00-7,99 (м, 1H), 7,44-7,42 (м, 1H), 7,36-7,25 (м, 3H), 6,93-6,92 (м, 1H), 6,26-6,25 (с, 2H), 4,80-4,78 (м, 1H), 4,64-4,56 (м, 2H), 3,93-3,89 (м, 1H), 3,63-3,61 (м, 1H), 3,08 (с, 3H); ESI-МС m/z=281 [M+H]+.
Пример 134 - Получение фумаратной соли (-)-4-бензо[d]изотиазол-6-ил-2-метил-1,2,3,4-тетрагидроизохинолина и фумаратной соли (+)-4-бензо[d]изотиазол-6-ил-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: Изохинолин-4-бороновую кислоту (1,01 г, 6,0 ммоль) и 2 М раствор карбоната натрия (5 мл) добавляли к раствору 6-бромбензо[d]изотиазол (1,12 г, 5,0 ммоль) в N,N-диметилформамиде. Смесь дегазировали, используя аргон, и энергично перемешивали. Затем к реакционной смеси добавляли тетракис(трифенилфосфин)палладий (0) (0,6 г, 0,5 ммоль) и смесь нагревали до 85°C в течение 4 часов. Температуру реакционной смеси возвращали к комнатной температуре и к смеси добавляли воду (10 мл) и три раза экстрагировали этилацетатом. Органический экстракт сушили над сульфатом натрия, фильтровали и концентрировали до получения коричневого масла. Масло очищали хроматографией на колонке (силикагель, от 5:95 до 40:60 этилацетат/гексаны в качестве элюента), получая требуемый продукт (0,76 г, 55%) в виде желтого твердого вещества:1H ЯМР (300 МГц, CDCl3) δ 9,33 (с, 1H), 9,03 (с, 1H), 8,56 (с, 1H), 8,22 (д, J=8,2 Гц, 1H), 8,11-8,09 (м, 2H), 7,89-7,87 (м, 1H), 7,72-7,60 (м, 3H).
Стадия B: Продукт со стадии A (0,74 г, 2,8 ммоль) растворяли в безводном метиленхлориде (10 мл). Раствор охлаждали на ледяной бане, затем по каплям добавляли метилтрифторметансульфонат (0,38 мл) и перемешивали в течение 5 минут. Раствор концентрировали, получая (0,78 г, >99%) в виде желтого твердого вещества. Полученный сырой продукт использовали на следующей стадии без дополнительной очистки.
Стадия C: Сырой продукт со стадии B (0,78 г, 2,8 ммоль) растворяли в метаноле (20 мл) и раствор охлаждали на ледяной бане. Цианоборгидрид натрия (0,35 г, 5,6 ммоль) порциями добавляли к реакционной смеси и перемешивали в течение 2 минут. Ледяную баню убирали и реакционную смесь перемешивали еще в течение 15 минут. Этанол удаляли и остаток растворяли в метиленхлориде и воде. Смесь три раза экстрагировали метиленхлоридом. Органические экстракты объединяли, сушили над сульфатом натрия, фильтровали и концентрировали. Смесь очищали хроматографией на колонке (силикагель, 2,5% метанол/метиленхлорид), получая требуемый продукт (0,55 г, 79%) в виде желтого масла:1H ЯМР (300 МГц, CDCl3) δ 8,86 (д, J=0,6 Гц, 1H), 7,97 (д, J=8,3 Гц, 1H), 7,80 (с, 1H), 7,30 (дд, J=8,3, 1,3 Гц, 1H), 7,18 (т, J=7,3 Гц, 1H), 7,13 (д, J=7,0 Гц, 1H), 7,09 (т, J=7,4 Гц, 1H), 6,87 (д, J=7,7 Гц, 1H), 4,44 (т, J=6,4 Гц, 1Н), 3,72 (с, 2H), 3,05 (дд, J=11,5, 5,5 Гц, 1H), 2,71-2,69 (м, 1H), 2,43 (с, 3H). Полученное соединение разделяли препаративной хиральной ВЭЖХ (колонка CHIRALPAK AD, с использованием смеси 90:10 гептан/изопропанол с 0,1% диэтиламина в качестве элюента), получая (+)-энантиомер [α]25D +67,9° (c 0,36, метанол) и (-)-энантиомер [α]25D-72,8° (c 0,36, метанол).
Стадия D: (+)-Энантиомер со стадии C превращали в соль фумаровой кислоты растворением свободного основания в минимальном количестве этанола, добавлением одного эквивалента фумаровой кислоты в достаточном количестве метанола, чтобы полностью растворить кислоту, затем смешивая два раствора и перемешивая в течение 2 часов. Раствор концентрировали до минимального объема, затем охлаждали при -30°C до образования кристаллов. Фильтрование давало (+)-энантиомер, фумаратную соль 4-бензо[d]изотиазол-6-ил-2-метил-1,2,3,4-тетрагидроизохинолина (326 мг, 88%, 96,6% AUC ВЭЖХ) в виде не совсем белого твердого вещества:1H ЯМР (500 МГц, CD3OD) δ 8,96 (д, J=0,8 Гц, 1H), 8,15 (д, J=8,3 Гц, 1Н), 7,98 (с, 1H), 7,33 (дд, J=8,4 Гц, 1H), 7,28-7,25 (м, 2H), 7,21-7,19 (м, 1H), 6,88 (д, J=7,8 Гц, 1H), 6,70 (с, 2H), 4,74-4,71 (м, 1H), 4,30 (д, J=6,9 Гц, 2H), 3,67-3,63 (м, 1H), 3,32-3,27 (м, 1H), 2,85 (с, 3H).
Такой же способ использовали для превращения (-)-энантиомера в фумаратную соль 4-бензо[d]изотиазол-6-ил-2-метил-1,2,3,4-тетрагидроизохинолина (302 мг, 93%, 97,9% AUC ВЭЖХ) в виде не совсем белого твердого вещества:1H ЯМР (500 МГц, CD3OD) δ 8,96 (с, 1Н), 8,14 (д, J=8,3 Гц, 1H), 7,97 (с, 1H), 7,34-7,26 (м, 3H), 7,20-7,19 (м, 1H), 6,88 (д, J=7,8 Гц, 1H), 6,70 (с, 2H), 4,72-4,69 (м, 1H), 4,26 (д, J=8,9 Гц, 2H), 3,62-3,59 (м, 1H), 3,30-3,23 (1Н), 2,82 (с, 3H).
Пример 135 - Получение малеатной соли (-)-4-бензо[d]изотиазол-5-ил-2-метил-1,2,3,4-тетрагидроизохинолина и малеатной соли (+)-4-бензо[d]изотиазол-5-ил-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: Раствор 2-бром-5-фторбензальдегида (8,5 г, 41,9 ммоль) добавляли к смеси бензилмеркаптана (5,2 г, 41,9 ммоль) и карбоната калия (7,5 г, 54,4 ммоль) в ДМФА (55 мл). Реакционную смесь нагревали до 80°C в течение 4 часов. После охлаждения до комнатной температуры растворитель упаривали. Остаток разбавляли водой и три раза экстрагировали эфиром, промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на колонке (120 г диоксида кремния, 90:10 гексан/этилацетат) давала требуемый продукт (6,1 г, 48%) в виде светло-желтого твердого вещества:1H ЯМР (CDCl3, 500 МГц) δ 10,17 (с, 1H), 7,92 (с, 1H), 7,58 (д, J=8,4 Гц, 1H), 7,32-7,21 (м, 6H), 4,09 (с, 2H).
Стадия B: Сульфурилхлорид (1,6 мл, 20,3 ммоль) по каплям добавляли к суспензии продукта со стадии A (6,1 г, 19,9 ммоль) в 1,2-дихлорэтане (30 мл) при комнатной температуре. Растворитель упаривали. Остаток суспендировали в ТГФ (30 мл) и обрабатывали аммиаком (7 N в метаноле, 30 мл) и перемешивали при комнатной температуре в течение 2 часов. После завершения перемешивания реакционную смесь разбавляли водой и органический слой отделяли. Водный слой три раза экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая 5-бромбензо[d]изотиазол (3,63 г, 84%) в виде желтого твердого вещества:1H ЯМР (CDCl3, 500 МГц) δ 8,85 (с, 1H), 8,21 (с, 1H), 7,84 (д, J=7,9 Гц, 1H), 7,62 (д, J=8,5 Гц, 1H).
Стадия C: Смесь 5-бромбензо[d]изотиазола, полученного на стадии B (1,33 г, 6,21 ммоль), изохинолин-4-бороновой кислоты (1,61 г, 9,32 ммоль) и 2 N Na2CO3 (9 мл) в диметиловом эфире этиленгликоля (19 мл) дегазировали, используя аргон. К полученной смеси добавляли Pd(Ph3P)4 (0,72 г, 0,62 ммоль). Полученную в результате смесь дегазировали, используя аргон, и затем кипятили с обратным холодильником в течение ночи. После охлаждения до комнатной температуры реакционную смесь разбавляли водой и органический слой отделяли. Водный слой три раза экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на колонке (80 г силикагеля; 60:40 гексаны/этилацетат) давала продукт (1,04 г, 64%) в виде не совсем белого твердого вещества:1H ЯМР (CDCl3, 500 МГц) δ 9,32 (с, 1H), 9,01 (с, 1H), 8,55 (с, 1H), 8,21 (с, 1H), 8,13-8,08 (м, 2H), 7,70 (д, J=8,5 Гц, 1H), 7,68-7,50 (м, 3H).
Стадия D: Метилтрифторметансульфонат (0,25 мл, 2,29 ммоль) по каплям добавляли к ледяному раствору продукта со стадии C (0,500 г, 3,81 ммоль) в метиленхлориде (2,5 мл). Полученную в результате взвесь перемешивали в течение 10 минут при комнатной температуре. Реакционную смесь концентрировали в вакууме. Остаток (1,05 г, 3,8 ммоль) разбавляли метанолом (12 мл) и обрабатывали цианоборгидридом натрия (600 мг, 9,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 20 минут. Растворитель удаляли в вакууме и остаток разбавляли этилацетатом, три раза промывали водой, сушили над сульфатом натрия и концентрировали в вакууме. Очистка хроматографией на колонке (80 г диоксида кремния; 95:5 метиленхлорид/метанол) давала продукт (280 мг, 26%). Полученный продукт разделяли хиральной ВЭЖХ (Chiralpak AD, 95:5 гептан/изопропанол с 0,1% диэтиламина). (-)-Энантиомер (230 мг, 43%) получали в виде желтого масла: [α]25D -58,6° (c 0,10, метанол);1H ЯМР (500 МГц, CDCl3) δ 8,84 (с, 1Н), 7,89-7,86 (м, 2H), 7,40 (д, J=8,4 Гц, 1Н), 7,19-7,06 (м, 3H), 6,87 (д, J=7,7 Гц, 1H), 4,43 (т, J=6,5 Гц, 1H), 3,72 (с, 2H), 3,06-3,02 (м, 1H), 2,69-2,66 (м, 1Н), 2,43 (с, 3H). (+)-Энантиомер (230 мг, 43%) получали в виде желтого масла: [α]25D +55,0° (c 0,10, метанол);1H ЯМР (500 МГц, CDCl3) δ 8,84 (с, 1H), 7,89-7,86 (м, 2H), 7,40 (д, J=8,4 Гц, 1H), 7,19-7,06 (м, 3H), 6,87 (д, J=7,7 Гц, 1H), 4,43 (т, J=6,5 Гц, 1H), 3,72 (с, 2H), 3,06-3,02 (м, 1H), 2,69-2,66 (м, 1H), 2,43 (с, 3H).
Стадия E: Раствор (-)-энантиомера со стадии D (230 мг, 0,820 ммоль) и малеиновой кислоты (95 мг, 0,820 ммоль) в метаноле (1 мл) перемешивали при комнатной температуре. Растворитель упаривали. Лиофилизация из смеси ацетонитрил (0,5 мл)/вода (0,5 мл) давала малеатную соль (-)-4-бензо[d]изотиазол-5-ил-2-метил-1,2,3,4-тетрагидроизохинолина (306 мг, 91%, >99,0% AUC ВЭЖХ) в виде белого твердого вещества: т.пл. 127-129°C;1H ЯМР (500 МГц, CD3OD) δ 8,95 (с, 1H), 8,11-8,08 (м, 2H), 7,44 (д, J=8,5 Гц, 1H), 7,35-7,24 (м, 3H), 6,92 (д, J=7,7 Гц, 1H), 6,23 (с, 2H), 4,62-4,55 (м, 2H), 3,91-3,87 (м, 1H), 3,61 (т, J=11,7 Гц, 1H), 3,33-3,30 (м, 1H), 3,06 (с, 3H); ESI МС m/z 281 [M+H]+.
Стадия F: Раствор (+)-энантиомера со стадии D (230 мг, 0,820 ммоль) и малеиновой кислоты (95 мг, 0,230 ммоль) в метаноле (1 мл) перемешивали при комнатной температуре. Растворитель упаривали. Лиофилизация из смеси ацетонитрил (0,5 мл)/вода (0,5 мл) давала малеатную соль (+)-4-бензо[d]изотиазол-5-ил-2-метил-1,2,3,4-тетрагидроизохинолина (303 мг, 88%, 97,9% AUC ВЭЖХ) в виде белого твердого вещества: т.пл. 145-147°C;1H ЯМР (500 МГц, CD3OD) δ 8,95 (с, 1H), 8,11-8,08 (м, 2H), 7,44 (д, J=8,5 Гц, 1H), 7,35-7,24 (м, 3H), 6,92 (д, J=7,7 Гц, 1H), 6,23 (с, 2H), 4,62-4,55 (м, 2H), 3,91-3,87 (м, 1H), 3,61 (т, J=11,7 Гц, 1H), 3,33-3,30 (м, 1H), 3,06 (с, 3H); ESI МС m/z 281 [M+H]+.
Пример 136 - Получение малеатной соли (+)-8-метокси-2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ола и малеатной соли (-)-8-метокси-2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ола
Стадия A: Карбонат калия (29,2 г, 0,275 моль) добавляли к раствору 2,3-дигидроксибензальдегида (38,0 г, 0,275 моль) в ДМФА (500 мл) при 25°C в атмосфере азота. Смесь перемешивали в течение 30 минут и по каплям добавляли метилйодид. Смесь перемешивали в течение ночи, распределяли между EtOAc (3×200 мл) и водой (200 мл). Объединенные органические экстракты промывали 5% раствором LiCl (2×200 мл), сушили (Na2SO4) и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (элюент гексаны/EtOAc 10:1), получая метоксибензальдегид (24,8 г, 59%) в виде желтого твердого вещества:1H ЯМР (CDCl3, 300 МГц) δ 10,27 (с, 1H), 7,38-7,40 (м, 1H), 7,12-7,36 (м, 2H), 5,75 (с, 1H), 3,98 (с, 3H).
Стадия B: Раствор метоксибензальдегида (7,10 г, 4,67 ммоль) с описанной выше стадии A и метиламина (1,73 г, 56,0 ммоль, 40% мас. в воде) в метаноле (150 мл) перемешивали в течение 15 минут при 25°C в атмосфере азота. Смесь охлаждали до 0°C и порциями добавляли боргидрид натрия (880 мг, 23,3 ммоль). Реакционную смесь перемешивали в течение 2 часов и концентрировали при пониженном давлении. Остаток обрабатывали EtOAc (3×200 мл) и водой (200 мл), сушили (Na2SO4) и концентрировали при пониженном давлении, получая метиламиносоединение (5,0 г, 64%) в виде желто-коричневого твердого вещества, которое использовали на следующей стадии без какой-либо дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 6,88-6,90 (м, 1H), 6,75-6,78 (м, 2H), 4,30 (ушир.с, 1H), 3,81 (с, 3H), 3,77 (с, 1H), 3,74 (с, 2H), 2,48 (с, 3H).
Стадия C: Раствор метиламиносоединения (5,5 г, 32,9 ммоль) с описанной выше стадии B и α-бром-2'-ацетонафтона (8,2 г, 32,9 ммоль) и N,N-диизопропилэтиламина (4,25 г, 32,9 ммоль) в метиленхлориде (150 мл) перемешивали в течение 3 часов при 25°C в атмосфере азота. Реакционную смесь распределяли между EtOAc (3×150 мл) и водой (200 мл), сушили (Na2SO4) и концентрировали при пониженном давлении, получая требуемый кетон (10,4 г, 95% сырого продукта) в виде желто-коричневого твердого вещества, которое использовали на следующей стадии без какой-либо дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 8,44 (с, 1Н), 7,98 (д, J=8,7 Гц, 1H), 7,85-7,91 (м, 3H), 7,54-7,60 (м, 3H), 6,92-7,01 (м, 3H), 3,96 (с, 2H), 3,83 (с, 2H), 3,79 (с, 3H), 2,45 (с, 3H).
Стадия D: К раствору кетона (10,4 г, 31,3 ммоль) с описанной выше стадии C в метаноле (150 мл) при 0°C небольшими порциями добавляли боргидрид натрия (1,40 г, 37,6 ммоль). Реакционный раствор перемешивали при 0°C в течение 2 часов и растворитель удаляли при пониженном давлении. Полученный остаток распределяли между EtOAc (2×250 мл) и водой (200 мл). Органические экстракты сушили (Na2SO4) и концентрировали при пониженном давлении, получая требуемый спирт, который использовали на следующей стадии без дополнительной очистки и характеристики. Полученный остаток собирали в метиленхлориде (200 мл) и по каплям добавляли концентрированную серную кислоту (10 мл). Реакционную смесь перемешивали 1 час, вливали в ледяную воду и подщелачивали с использованием концентрированного гидроксида аммония до pH 10 и экстрагировали EtOAc (3×500 мл), сушили (Na2SO4) и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке (элюент гексаны/EtOAc 7:3), получая требуемый тетрагидроизохинолин (3,90 г, 39%) в виде желто-коричневой пены:1H ЯМР (CDCl3, 500 МГц) δ 7,74-7,81 (м, 3H), 7,67 (с, 1H), 7,42-7,51 (м, 2H), 7,27-7,28 (м, 1H), 6,70 (д, J=8,4 Гц, 1H), 6,56 (д, J=8,4 Гц, 1H), 5,30 (с, 1H), 4,38 (дд, J=8,5 Гц, 6,0 Гц, 1H), 3,91 (д, J=15,3 Гц, 1H), 3,87 (с, 3H), 3,58 (д, J=15,4 Гц, 1H), 3,01 (дд, J=8,5 Гц, 6,0 Гц, 1H), 2,60-2,64 (м, 1H), 2,46 (с, 3H); ESI-МС m/z 320 [M+H]+.
Свободное основание кетона (300 мг) разделяли препаративной хиральной ВЭЖХ (колонка CHIRALPAK AD, с использованием смеси 85:15:0,1 гептан/EtOH/диэтиламин в качестве элюента), получая (+)-энантиомер [[α]23D +80,0° (c 0,08, метанол)] и (-)-энантиомер [[α]23D -87,4° (c 0,135, метанол)]. (+)-Энантиомер (56,7 мг, 0,177 ммоль) растворяли в метаноле (3 мл) и добавляли один эквивалент малеиновой кислоты (20,6 мг, 0,177 ммоль). Полученный в результате раствор концентрировали при пониженном давлении, получая малеатную соль (+)-8-метокси-2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ола (69,0 мг, 89%, 98,7% AUC ВЭЖХ) в виде белого твердого вещества: т.пл. 85-90°C;1H ЯМР (CD3OD, 500 МГц) δ 7,82-7,87 (м, 3H), 7,77 (с, 1H), 7,47-7,52 (м, 2H), 7,26 (дд, J=8,5 Гц, 1,4 Гц, 1H), 6,76 (д, J=8,5 Гц, 1H), 6,50 (д, J=8,5 Гц, 1H), 6,24 (с, 2H), 4,63-4,71 (м, 2H), 4,44 (д, J=15,1 Гц, 1H), 3,93 (с, 3H), 3,80-3,85 (м, 1H), 3,54-3,59 (м, 1H), 3,10 (с, 3H); ESI-МС m/z 320 [M+Н]+; элементный анализ, вычислено для C21H21NO2-C4H4O4-0,90H2O: C, 66,37; Н, 5,77; N, 3,10. Найдено: C, 66,49; Н 5,54; N, 3,08.
(-)-Энантиомер (100 мг, 0,312 ммоль) растворяли в метаноле (3 мл) и добавляли один эквивалент малеиновой кислоты (35,1 мг, 0,312 ммоль). Полученный в результате раствор концентрировали при пониженном давлении и растирали в диэтиловом эфире, получая малеатную соль (-)-8-метокси-2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ола (101,1 мг, 69%, 93,8% AUC ВЭЖХ) в виде белого твердого вещества: т.пл. 85-90°C;1H ЯМР (CD3OD, 500 МГц) δ 7,82-7,87 (м, 3H), 7,77 (с, 1H), 7,47-7,52 (м, 2H), 7,26 (дд, J=8,5 Гц, 1,4 Гц, 1H), 6,76 (д, J=8,5 Гц, 1H), 6,50 (д, J=8,5 Гц, 1H), 6,24 (с, 2H), 4,63-4,71 (м, 2H), 4,44 (д, J=15,1 Гц, 1H), 3,93 (с, 3H), 3,80-3,85 (м, 1H), 3,54-3,59 (м, 1H), 3,10 (с, 3H); ESI-МС m/z 320 [M+Н]+.
Пример 137 - Получение малеатной соли (-)-2-метил-4-нафтален-2-ил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина и малеатной соли (+)-2-метил-4-нафтален-2-ил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору (3-бромбензил)метиламина (2,0 г, 9,99 ммоль) в метиленхлориде (40 мл) добавляли диизопропилэтиламин (3,5 мл, 20,0 ммоль). Реакционную смесь охлаждали до 0°C и порциями обрабатывали 2-бром-2'-ацетонафтоном (2,49 г, 9,99 ммоль) в течение 10-минутного периода времени. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 3 часов. Реакционную смесь три раза промывали водой, сушили над сульфатом натрия, фильтровали и растворитель выпаривали, получая продукт (3,57 г, 98%) в виде вязкого оранжевого масла:1H ЯМР (CDCl3, 500 МГц) δ 8,49 (с, 1H), 8,00 (д, J=8,6 Гц, 1H), 7,95 (д, J=8,1 Гц, 1H), 7,87 (т, J=8,2 Гц, 2H), 7,61-7,54 (м, 3H), 7,40 (д, J=7,0 Гц, 1H), 7,31 (д, J=7,6 Гц, 1H), 7,19 (т, J=7,7 Гц, 1H), 3,91 (с, 2H), 3,71 (с, 2H), 2,40 (с, 3H).
Стадия B: Боргидрид натрия (440 мг, 11,6 ммоль) добавляли в течение 10-минутного периода к охлажденному на льду раствору продукта со стадии A (3,57 г, 9,69 ммоль) в метаноле (45 мл). Реакционной смеси давали возможность нагреться до комнатной температуры и продолжали перемешивать еще в течение 5 часов. Растворитель удаляли при пониженном давлении и остаток разбавляли водой и три раза экстрагировали метиленхлоридом. Объединенные экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая продукт (2,74 г, 77%) в виде оранжевого масла:1H ЯМР (500 МГц, CDCl3) δ 7,84-7,81 (м, 4H), 7,48-7,40 (м, 5Н), 7,26 (с, 1H), 7,20 (т, J=7,7 Гц, 1H), 4,92 (дд, J=3,8 Гц, 10,0 Гц, 1H), 3,99 (с, 1H), 3,71 (д, J=13,2 Гц, 1H), 3,52 (д, J=13,2 Гц, 1H), 2,69-2,61 (м, 2H), 2,34 (с, 3H).
Стадия C: Продукт со стадии B (2,74 г, 7,4 ммоль) в 1,2-дихлорэтане (45 мл) добавляли по каплям через капельную воронку к метансульфоновой кислоте (27 мл, 414 ммоль) при 40°C и перемешивали еще в течение 3,5 часов. Охлажденную реакционную смесь вливали в лед и делали основной до pH 9, используя концентрированный гидроксид аммония. Реакционную смесь четыре раза экстрагировали этилацетатом. Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на колонке (80 г диоксида кремния, 99:1 метиленхлорид/метанол) давала продукт (490 мг, 19%) в виде желтого твердого вещества:1H ЯМР (500 МГц, CDCl3) δ 7,81-7,75 (м, 3H), 7,66 (с, 1H), 7,46-7,44 (с, 1H), 7,27 (с, 1Н), 7,24-7,23 (м, 2H), 7,16 (д, J=8,3 Гц, 1H), 6,75 (д, J=8,3 Гц, 1H), 4,35 (т, J=6,29 Гц, 1H), 3,75 (д, J=15,1 Гц, 1H), 3,62 (д, J=15,1 Гц, 1Н), 3,10-3,06 (м, 1H), 2,66-2,61 (м, 1H), 2,43 (с, 3H).
Стадия D: Смесь продукта со стадии C (490 мг, 1,39 ммоль), бис(пинаколато)дибора (389 мг, 1,53 ммоль), KOAc (409 мг, 4,17 ммоль) дегазировали, используя аргон. К полученной смеси добавляли PdCl2(dppf) (70 мг, 0,083 ммоль). Полученную в результате смесь дегазировали, используя аргон и затем нагревали при 80°C в течение 6 часов. Охлажденную смесь разбавляли водой и органический слой отделяли. Водный слой три раза экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая коричневое масло, которое использовали на стадии E без дополнительной очистки.
Стадия E: Смесь сырого продукта со стадии D (555 мг, 1,39 ммоль), 3,6-дихлорпиридазина (260 мг, 1,74 ммоль) и карбоната цезия (1,36 г, 4,17 ммоль) в ДМФА (25 мл) и воде (5 мл) дегазировали, используя аргон. К полученной смеси добавляли PdCl2(dppf) (68 мг, 0,08 ммоль). Полученную в результате смесь дегазировали, используя аргон, и затем нагревали при 80°C в течение 4,5 часов. Охлажденную смесь фильтровали через слой диатомовой земли и три раза промывали водой. Органические слои отделяли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на колонке (40 г, 95:5 метиленхлорид/метанол) давала продукт (470 мг, 88%) в виде светло-коричневого твердого вещества;1H ЯМР (500 МГц, CDCl3) δ 7,90 (с, 1H), 7,81-7,77 (м, 4H), 7,71 (с, 1H), 7,67-7,64 (м, 1H), 7,53 (д, J=9,0 Гц, 1H), 7,47-7,45 (м, 3H), 7,05 (д, J=8,0 Гц, 1H), 4,51 (т, J=6,2 Гц, 1H), 3,91 (д, J=15,0 Гц, 1H), 3,75 (д, J=15,0 Гц, 1H), 3,17-3,13 (м, 1H), 2,73-2,69 (м, 1H), 2,47 (с, 3H).
Стадия F: Гидразин (2,49 мл, 51,2 ммоль) и 10% палладий-на-угле (250 мг) добавляли к раствору продукта со стадии E (470 мг, 1,22 ммоль) в этаноле (40 мл) и нагревали при кипячении с обратным холодильником в течение 6,5 часов. Охлажденную смесь фильтровали через слой диатомовой земли и промывали метанолом. Растворитель выпаривали. Очистка хроматографией на колонке (40 г диоксида кремния, 95:5 метиленхлорид/метанол) давала требуемый продукт (150 мг, 35%). Полученный продукт разделяли хиральной ВЭЖХ (Chiralpak AD, 70:30 гептан/изопропанол с 0,1% диэтиламина). (-)-Энантиомер (60 мг, 40%) получали в виде светло-оранжевого твердого вещества [α]25D -37,0° (0,1, метанол);1H ЯМР (500 МГц, CDCl3) δ 9,14 (д, J=4,9 Гц, 1H), 7,96 (с, 1H), 7,95-7,77 (м, 4H), 7,71-7,68 (м, 2H), 7,52-7,46 (м, 3H), 7,30 (д, J=8,4 Гц, 1H), 7,05 (д, J=8,2 Гц, 1H), 4,52 (т, J=6,7 Гц, 1H), 3,92 (д, J=15,0 Гц, 1H), 3,76 (д, J=14,9 Гц, 1H), 3,17-3,14 (м, 1H), 2,73 (м, 1H), 2,49 (с, 3H).
Стадия G: Раствор (-)-энантиомера со стадии F (60 мг, 0,171 ммоль) и малеиновой кислоты (20 мг, 0,171 ммоль) в метаноле (1 мл) перемешивали при комнатной температуре. Растворитель выпаривали. Лиофилизация из смеси ацетонитрил (0,5 мл)/вода (0,5 мл) давала малеатную соль (-)-2-метил-4-нафтален-2-ил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина (76 мг, 99%) в виде не совсем белого твердого вещества: т.пл. 91-93°C;1H ЯМР (CD3OD, 500 МГц) δ 9,16 (д, J=4,8 Гц, 1H), 8,17 (д, J=8,6 Гц, 1H), 8,07 (с, 1H), 7,90-7,78 (м, 6H), 7,52-7,50 (м, 2H), 7,32 (д, J=8,5 Гц, 1H), 7,12 (д, J=8,2 Гц, 1H), 6,24 (с, 0,8Н), 4,81-4,77 (м, 1H), 4,58-4,55 (м, 2H), 3,82-3,78 (м, 1H), 3,54-3,52 (м, 1H), 3,00 (с, 3H); ESI-МС m/z 352 [M+Н]+.
Стадия H: Раствор (+)-энантиомера со стадии F (80 мг, 0,228 ммоль) и малеиновой кислоты (26 мг, 0,228 ммоль) в метаноле (1 мл) перемешивали при комнатной температуре. Растворитель выпаривали. Лиофилизация из смеси ацетонитрил (0,5 мл)/вода (0,5 мл) давала малеатную соль (+)-2-метил-4-нафтален-2-ил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина (91,6 мг, 78%) в виде не совсем белого твердого вещества: т.пл.86-88°C;1H ЯМР (CD3OD, 500 МГц) δ 9,16 (д, J=4,8 Гц, 1H), 8,17 (д, J=8,6 Гц, 1H), 8,07 (с, 1Н), 7,90-7,78 (м, 6H), 7,52-7,50 (м, 2H), 7,32 (д, J=8,5 Гц, 1H), 7,12 (д, J=8,2 Гц, 1H), 6,24 (с, 0,8Н), 4,81-4,77 (м, 1H), 4,58-4,55 (м, 2H), 3,82-3,78 (м, 1H), 3,54-3,52 (м, 1H), 3,00 (с, 3H); ESI-МС m/z 352 [M+Н]+.
Пример 138 - Получение малеатной соли (+)-4-(1-метоксинафтален-2-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина и малеатной соли (-)-4-(1-метоксинафтален-2-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина
Стадия A: Карбонат калия (5,2 г, 37,6 ммоль) и диметилсульфат (2,7 мл, 28,2 ммоль) добавляли к раствору 1'-гидрокси-2'-ацетонафтона (3,5 г, 18,8 ммоль) в ацетоне (50 мл) и нагревали при кипячении с обратным холодильником в течение 22,5 часов. Растворитель из охлажденной реакционной смеси выпаривали. Остаток растворяли в метаноле (40 мл), обрабатывали гидроксидом натрия (5,6 г) и перемешивали при комнатной температуре в течение 1,5 часа. Растворитель выпаривали и остаток разбавляли водой и три раза экстрагировали эфиром. Объединенные органические экстракты промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка флэш-хроматографией на колонке (80 г силикагеля, 100% гексаны) давала требуемый продукт (3,33 г, 89%) в виде не совсем белого твердого вещества:1H ЯМР (CDCl3, 300 МГц) δ 8,24-8,21 (м, 1H), 7,87-7,84 (м, 1H), 7,74 (д, J=8,6 Гц, 1Н), 7,64-7,55 (м, 3H), 4,01 (с, 3H), 2,78 (с, 3H).
Стадия B: Бромид меди (7,28 г, 32,6 ммоль) в этилацетате (50 мл) нагревали до 78°C в течение 15 минут. Продукт со стадии A (3,33 г, 16,6 ммоль) в хлороформе (50 мл) добавляли к смеси через капельную воронку. После завершения добавления реакционную смесь продолжали перемешивать при 78°C в течение 23 часов. Охлажденную реакционную смесь фильтровали через слой диатомовой земли и промывали избытком этилацетата. Растворитель выпаривали. Очистка хроматографией на колонке (120 г силикагеля, 100% гексаны) давала требуемый продукт (1,73 г, 37%) в виде не совсем белого твердого вещества: ЯМР (CDCl3, 500 МГц) δ 8,21 (д, J=7,6 Гц, 1H), 7,87 (д, J=7,3 Гц, 1H), 7,74 (д, J=8,6 Гц, 1Н), 7,67-7,58 (м, 3H), 4,74 (с, 2H), 4,04 (с, 3H).
Стадия C: К раствору (3-бромбензил)метиламина (1,24 г, 6,20 ммоль) в метиленхлориде (25 мл) добавляли диизопропилэтиламин (2,16 мл, 12,4 ммоль). Реакционную смесь охлаждали до 0°C и обрабатывали порциями 2-бром-1-(3-метоксинафтален-2-ил)этанона, полученного на стадии B (1,73 г, 6,20 ммоль), в течение 10-минутного периода. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 3 часов. Реакционную смесь три раза промывали водой, сушили над сульфатом натрия, фильтровали и растворитель выпаривали, получая требуемый продукт (количественный выход) в виде вязкого оранжевого масла:1H ЯМР (CDCl3, 500 МГц) δ 8,19-8,17 (м, 1H), 7,87-7,84 (м, 1H), 7,64 (с, 2H), 7,60-7,54 (м, 2H), 7,51 (с, 1Н), 7,37 (д, J=8,9 Гц, 1H), 7,28-7,25 (м, 1H), 7,15 (т, J=7,7 Гц, 1H), 3,97 (с, 2H), 3,90 (с, 3H), 3,70 (с, 2H), 2,42 (с, 3H).
Стадия D: Боргидрид натрия (281 мг, 7,44 ммоль) добавляли в течение 10-минутного периода к охлажденному на льду раствору продукта со стадии C (2,47 г, 6,20 ммоль) в метаноле (25 мл). Реакционной смеси давали возможность нагреться до комнатной температуры и продолжали перемешивать еще в течение 5 часов. Растворитель удаляли при пониженном давлении и остаток разбавляли водой и три раза экстрагировали метиленхлоридом. Объединенные экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая требуемый продукт (2,37 г, 96%) в виде оранжевого масла:1H ЯМР (500 МГц, CDCl3) δ 8,07 (д, J=8,2 Гц, 1H), 7,83 (д, J=7,7 Гц, 1H), 7,64-7,60 (м, 3H), 7,51-7,47 (м, 4H), 7,27-7,21 (м, 2H), 5,33-5,29 (м, 1H), 3,95 (с, 3H), 3,90 (с, 1H), 3,74 (д, J=13,3 Гц, 1H), 2,66-2,64 (м, 1H), 2,42 (с, 1H), 2,37 (с, 3H).
Стадия E: Продукт со стадии D (2,37 г, 5,92 ммоль) в 1,2-дихлорэтане (35 мл) по каплям через капельную воронку добавляли к метансульфоновой кислоте (22 мл, 331 ммоль) при 40°C и перемешивали еще в течение 19 часов. Охлажденную реакционную смесь вливали в лед и подщелачивали до pH 9 концентрированным гидроксидом аммония. Реакционную смесь четыре раза экстрагировали этилацетатом. Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на колонке (80 г силикагеля, 99:1 метиленхлорид/метанол) давала 7-бром-4-(1-метоксинафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин (690 мг, 31%) в виде не совсем белого твердого вещества;1H ЯМР (500 МГц, CDCl3) δ 8,13 (д, J=8,4 Гц, 1H), 7,80 (д, J=8,0 Гц, 1Н), 7,55-7,46 (м, 3H), 7,13-7,10 (м, 2H), 7,04 (д, J=8,5 Гц, 1H), 6,80 (д, J=8,2 Гц, 1H), 4,92 (т, J=8,5 Гц, 1H), 3,98 (с, 3H), 3,80 (д, J=15,0 Гц, 1H), 3,61 (д, J=15,0 Гц, 1H), 3,11-3,08 (м, 1H), 2,62-2,58 (м, 1H), 2,46 (с, 3H).
Стадия F: Смесь продукта со стадии E (690 мг, 1,80 ммоль), бис(пинаколато)дибора (504 мг, 1,99 ммоль) и KOAc (530 мг, 5,40 ммоль) в ДМСО (12 мл) дегазировали, используя аргон. К полученной смеси добавляли PdCl2(dppf) (88 мг, 0,108 ммоль). Полученную в результате смесь дегазировали, используя аргон, и затем нагревали при 80°C в течение 3 часов. После завершения реакции (на основе анализа тонкослойной хроматографией) охлажденный продукт разбавляли водой и органический слой отделяли. Водный слой три раза экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая коричневое масло, которое использовали на стадии G без дополнительной очистки.
Стадия G: Смесь сырого продукта со стадии F (773 мг, 1,80 ммоль), 3,6-дихлорпиридазина (335 мг, 2,25 ммоль) и карбоната цезия (1,76 г, 5,4 ммоль) в ДМФА (30 мл) и воде (6 мл) дегазировали, используя аргон. К полученной смеси добавляли PdCl2(dppf) (88 мг, 0,108 ммоль). Полученную в результате смесь дегазировали, используя аргон, и затем нагревали при 80°C в течение 4 часов. Охлажденную смесь фильтровали через слой диатомовой земли и три раза промывали водой. Органические слои отделяли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на колонке (80 г силикагеля, 98:2 метиленхлорид/метанол) давала требуемый продукт (570 мг, 76%) в виде светло-коричневого твердого вещества;1H ЯМР (500 МГц, CDCl3) δ 8,15 (д, J=8,2 Гц, 1H), 7,90 (с, 1Н), 7,82-7,77 (м, 2H), 7,64 (д, J=7,3 Гц, 1H), 7,55-7,47 (м, 4H), 7,09 (д, J=8,5 Гц, 1Н), 6,99 (д, J=8,0 Гц, 1H), 5,07-5,04 (м, 1H), 4,03 (с, 3H), 3,95 (д, J=14,8 Гц, 1H), 3,73 (д, J=15,0 Гц, 1H), 3,18-3,15 (м, 1Н), 2,69-2,65 (м, 1H), 2,50 (с, 3H).
Стадия H: Гидразин (2,8 мл, 57,5 ммоль) и 10% палладий-на-угле (280 мг) добавляли к раствору продукта со стадии G (570 мг, 1,37 ммоль) в этаноле (45 мл) и реакционную смесь нагревали при кипячении с обратным холодильником в течение 5,5 часов. После завершения реакции (по данным анализа тонкослойной хроматографией) охлажденный продукт фильтровали через слой диатомовой земли и промывали метанолом. Растворитель выпаривали. Очистка хроматографией на колонке (80 г силикагеля, 98:2 метиленхлорид/метанол) давала требуемый продукт (320 мг, 61%). Полученный продукт разделяли хиральной ВЭЖХ (Chiralpak AD, 60:40 гептан/изопропанол с 0,1% диэтиламина). (-)-Энантиомер (140 мг, 44%) получали в виде светло-оранжевого твердого вещества: [α]25D -191,3° (c 0,11, метанол);1H ЯМР (500 МГц, CDCl3) δ 9,14 (д, J=6,4 Гц, 1H), 8,16 (д, J=8,1 Гц, 1H), 7,95 (с, 1H), 7,82 (д, J=8,6 Гц, 2H), 7,68 (д, J=8,0 Гц, 1H), 7,56-7,48 (м, 4H), 7,10 (д, J=8,5 Гц, 1Н), 6,99 (д, J=8,1 Гц, 1H), 5,07 (т, J=6,9 Гц, 1H), 4,03 (с, 3H), 3,97 (д, J=14,9 Гц, 1H), 3,74 (д, J=15,0 Гц, 1H), 3,19-3,15 (м, 1H), 3,67 (т, J=9,2 Гц, 1H), 2,51 (с, 3H). (+)-Энантиомер (110 мг, 34%) получали в виде светло-желтого твердого вещества: [α]25D +195,3° (c 0,11, метанол);1H ЯМР (500 МГц, CDCl3) δ 9,14 (д, J=6,4 Гц, 1H), 8,16 (д, J=8,1 Гц, 1H), 7,95 (с, 1H), 7,82 (д, J=8,6 Гц, 2H), 7,68 (д, J=8,0 Гц, 1H), 7,56-7,48 (м, 4H), 7,10 (д, J=8,5 Гц, 1H), 6,99 (д, J=8,1 Гц, 1H), 5,07 (т, J=6,9 Гц, 1H), 4,03 (с, 3H), 3,97 (д, J=14,9 Гц, 1H), 3,74 (д, J=15,0 Гц, 1H), 3,19-3,15 (м, 1H), 3,67 (т, J=9,2 Гц, 1H), 2,51 (с, 3H).
Стадия I: Раствор (-)-энантиомера со стадии H (140 мг, 0,367 ммоль) и малеиновой кислоты (42 мг, 0,367 ммоль) в метаноле (1 мл) перемешивали при комнатной температуре. Растворитель выпаривали. Лиофилизация из смеси ацетонитрил (0,5 мл)/вода (0,5 мл) давала малеатную соль (-)-4-(1-метоксинафтален-2-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина (152,10 мг, 81%, 99% AUC ВЭЖХ) в виде не совсем белого твердого вещества: т.пл. 88-90°C;1H ЯМР (CD3OD, 500 МГц) δ 9,16 (с, 1H), 8,18-8,12 (м, 3H), 7,96-7,88 (м, 2H), 7,81-7,79 (м, 1H), 7,69 (д, J=8,5 Гц, 1H), 7,60-7,55 (м, 2H), 7,19 (д, J=8,6 Гц, 1H), 7,11 (д, J=8,2 Гц, 1H), 6,25 (с, 1H), 5,27-5,24 (м, 1H), 4,81-4,66 (м, 2H), 3,94-3,90 (м, 4H), 3,78-3,76 (м, 1H), 3,16 (с, 3H); ESI МС m/z 382 [M+Н]+.
Стадия J: Раствор (+)-энантиомера со стадии G (110 мг, 0,228 ммоль) и малеиновой кислоты (33,5 мг, 0,228 ммоль) в метаноле (1 мл) перемешивали при комнатной температуре. Растворитель выпаривали. Лиофилизация из смеси ацетонитрил (0,5 мл)/вода (0,5 мл) давала малеатную соль (+)-4-(1-метоксинафтален-2-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина (138,20 мг, 97%, >99,0% AUC ВЭЖХ) в виде светло-желтого твердого вещества: т.пл.90-92°C;1H ЯМР (CD3OD, 500 МГц) δ 9,16 (д, J=4,8 Гц, 1H), 8,16 (т, J=9,9 Гц, 2H), 8,11 (ушир.с, 1H), 7,92-7,88 (м, 2H), 7,81-7,78 (м, 1H), 7,68 (д, J=8,6 Гц, 1Н), 7,61-7,50 (м, 2H), 7,18 (д, J=8,5 Гц, 1H), 7,11 (д, J=8,0 Гц, 1H), 6,24 (с, 1H), 5,27-5,24 (м, 1H), 4,77-4,68 (м, 2H), 3,94 (с, 3H), 3,91-3,87 (м, 1H), 3,72-3,69 (м, 1H), 3,13 (с, 3H); ESI МС m/z 382 [M+Н]+.
Пример 139 - Получение малеатной соли (+/-)-2-метил-4-нафтален-2-ил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору (3-бромбензил)метиламина (7,01 г, 35,0 ммоль) в метиленхлориде (120 мл) добавляли диизопропилэтиламин (12 мл, 70,0 ммоль). Реакционную смесь охлаждали до 0°C и порциями обрабатывали 2-бром-2'-ацетонафтоном (8,73 г, 35,0 ммоль) в течение 30-минутного периода. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 3 часов. Реакционную смесь три раза промывали водой, сушили над сульфатом натрия, фильтровали и растворитель выпаривали, получая требуемый продукт (12,54 г, 97%) в виде вязкого оранжевого масла:1H ЯМР (CDCl3, 500 МГц) δ 8,49 (с, 1Н), 8,00 (д, J=8,6 Гц, 1Н), 7,95 (д, J=8,1 Гц, 1H), 7,87 (т, J=8,2 Гц, 2H), 7,61-7,54 (м, 3H), 7,40 (д, J=7,0 Гц, 1Н), 7,31 (д, J=7,6 Гц, 1H), 7,19 (т, J=7,7 Гц, 1H), 3,91 (с, 2H), 3,71 (с, 2H), 2,40 (с, 3H).
Стадия B: Боргидрид натрия (1,55 г, 40,9 ммоль) добавляли в течение 30-минутного периода к охлажденному на льду раствору продукта со стадии A (12,54 г, 34,0 ммоль) в метаноле (140 мл). Реакционной смеси давали возможность нагреться до комнатной температуры и продолжали перемешивать еще в течение 4 часов. Растворитель удаляли при пониженном давлении и остаток разбавляли водой и три раза экстрагировали метиленхлоридом. Объединенные экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая требуемый продукт (11,67 г, 93%) в виде оранжевого масла:1H ЯМР (500 МГц, CDCl3) δ 7,84-7,81 (м, 4H), 7,48-7,40 (м, 5H), 7,24-7,20 (м, 2H), 4,92 (т, J=6,1 Гц, 1H), 3,71 (д, J=13,3 Гц, 1H), 3,52 (д, J=13,3 Гц, 1H), 2,69-2,60 (м, 2H), 2,33 (с, 3H).
Стадия C: Продукт со стадии B (11,6 г, 31,3 ммоль) в 1,2-дихлорэтане (170 мл) добавляли по каплям через капельную воронку к метансульфоновой кислоте (110 мл, 1754 ммоль) при 40°C и перемешивали еще в течение 6,5 часов. Затем охлажденную реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь вливали в лед и подщелачивали до pH 9 концентрированным гидроксидом аммония. Реакционную смесь четыре раза экстрагировали этилацетатом. Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на колонке (330 г силикагеля, 90:10 гексан/этилацетат) давала 7-бром-2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин (4,24 г, 38%) в виде не совсем белого твердого вещества;1H ЯМР (500 МГц, CDCl3) δ 7,81-7,75 (м, 3H), 7,66 (с, 1Н), 7,46-7,44 (с, 1Н), 7,27 (с, 1H), 7,24-7,23 (м, 2H), 7,16 (д, J=8,3 Гц, 1Н), 6,75 (д, J=8,3 Гц, 1Н), 4,35 (т, J=6,29 Гц, 1H), 3,75 (д, J=15,1 Гц, 1H), 3,62 (д, J=15,1 Гц, 1H), 3,10-3,06 (м, 1H), 2,66-2,61 (м, 1H), 2,43 (с, 3H).
Стадия D: Смесь продукта со стадии C (1,00 мг, 2,80 ммоль), бис(пинаколато)дибора (793 мг, 3,12 ммоль), KOAc (830 мг, 8,50 ммоль) дегазировали, используя аргон. К полученной смеси добавляли PdCl2(dppf) (99 мг, 0,122 ммоль). Полученную в результате смесь дегазировали, используя аргон, и затем нагревали при 80°C в течение 3 часов. После завершения реакции по данным анализа тонкослойной хроматографией охлажденный продукт разбавляли водой и органический слой отделяли. Водный слой три раза экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая коричневое масло, которое использовали на стадии E без дополнительной очистки.
Стадия E: Смесь сырого продукта со стадии D (565 мг, 1,42 ммоль), 2-хлорпиразина (200 мг, 1,77 ммоль) и карбоната цезия (1,5 г, 4,26 ммоль) в ДМФА (21 мл) и воде (3 мл) дегазировали, используя аргон. К полученной смеси добавляли PdCl2(dppf) (70 мг, 0,085 ммоль). Полученную в результате смесь дегазировали, используя аргон, и затем нагревали при 80°C в течение 4 часов. Охлажденную смесь фильтровали через слой диатомовой земли и три раза промывали водой. Органические слои отделяли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на колонке (80 г силикагеля, 100% метиленхлорид) давала продукт (50 мг, 10%) в виде светло-красного твердого вещества;1H ЯМР (500 МГц, CDCl3) δ 9,00 (с, 1H), 8,61 (с, 1H), 8,48 (с, 1H), 7,82-7,76 (м, 4H), 7,71-7,68 (м, 2H), 7,47-7,44 (м, 2H), 7,29 (д, J=8,4 Гц, 1H), 7,04 (д, J=8,1 Гц, 1H), 4,51 (т, J=6,5 Гц, 1H), 3,91 (д, J=14,9 Гц, 1H), 3,75 (д, J=14,9 Гц, 1H), 3,16-3,13 (м, 1H), 2,73-2,69 (м, 1H), 2,48 (с, 3H).
Стадия F: Раствор продукта со стадии E (40 мг, 0,114 ммоль) и малеиновой кислоты (13 мг, 0,114 ммоль) в метаноле (1 мл) перемешивали при комнатной температуре. Растворитель выпаривали. Лиофилизация из смеси ацетонитрила (0,5 мл)/вода (0,5 мл) давала малеатную соль 2-метил-4-нафтален-2-ил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолина (52,2 мг, 98%, >99% AUC ВЭЖХ) в виде коричневого твердого вещества: т.пл. 89-91°C;1H ЯМР (CD3OD, 500 МГц) δ 9,12 (с, 1H), 8,69-8,68 (м, 1H), 8,56-8,55 (м, 1H), 8,08 (с, 1H), 7,98 (д, J=8,1 Гц, 1H), 7,91-7,85 (м, 4H), 7,54-7,50 (м, 2H), 7,33 (д, J=8,5 Гц, 1H), 7,12 (д, J=8,2 Гц, 1H), 6,24 (с, 1H), 4,72 (ушир.с, 2H), 3,98-3,94 (м, 1H), 3,72 (т, J=11,8 Гц, 1H), 3,30-3,29 (м, 1H), 3,09 (с, 3H); ESI-МС m/z 352 [M+Н]+.
Пример 140 - Получение малеатной соли (+)-2-метил-4-нафтален-2-ил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолина и малеатной соли (-)-2-метил-4-нафтален-2-ил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору (3-бромбензил)метиламина (29,4 г, 146,9 ммоль) в метиленхлориде (300 мл) добавляли диизопропилэтиламин (51 мл, 293,8 ммоль). Реакционную смесь охлаждали до 0°C и порциями обрабатывали 2-бром-2'-ацетонафтоном (36,6 г, 146,9 ммоль) в течение 40-минутного периода. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 5,5 часов. Реакционную смесь три раза промывали водой, сушили над сульфатом натрия, фильтровали и растворитель выпаривали, получая требуемый продукт (количественный выход) в виде вязкого оранжевого масла:1H ЯМР (CDCl3, 500 МГц) δ 8,49 (с, 1Н), 8,00 (д, J=8,6 Гц, 1Н), 7,95 (д, J=8,1 Гц, 1H), 7,87 (т, J=8,2 Гц, 2H), 7,61-7,54 (м, 3H), 7,40 (д, J=7,0 Гц, 1H), 7,31 (д, J=7,6 Гц, 1H), 7,19 (т, J=7,7 Гц, 1Н), 3,91 (с, 2H), 3,71 (с, 2H), 2,40 (с, 3H).
Стадия B: Боргидрид натрия (54,11 г, 155,4 ммоль) добавляли в течение 45-минутного периода к охлажденному на льду раствору продукта со стадии A (7,05 г, 186,4 ммоль) в метаноле (400 мл). Реакционной смеси давали возможность нагреться до комнатной температуры и продолжали перемешивать еще в течение 21,5 часа. Растворитель удаляли при пониженном давлении и остаток разбавляли водой и три раза экстрагировали метиленхлоридом. Объединенные экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая требуемый продукт (47,78 г, 83%) в виде оранжевого масла:1H ЯМР (500 МГц, CDCl3) δ 7,84-7,81 (м, 4H), 7,48-7,40 (м, 5H), 7,24-7,20 (м, 2H), 4,92 (т, J=6,1 Гц, 1Н), 3,71 (д, J=13,3 Гц, 1H), 3,52 (д, J=13,3 Гц, 1Н), 2,69-2,60 (м, 2H), 2,33 (с, 3H).
Стадия C: Продукт со стадии B (47,78 г, 129,0 ммоль) в 1,2- дихлорэтане (500 мл) добавляли по каплям через капельную воронку к метансульфоновой кислоте (469 мл, 7226 ммоль) при комнатной температуре и перемешивали еще в течение 6,5 часов. Затем охлажденную реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь вливали в лед и подщелачивали до pH 9 концентрированным гидроксидом аммония. Реакционную смесь четыре раза экстрагировали этилацетатом. Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на колонке (силикагель, 98:2 метиленхлорид/метанол) давала 7-бром-2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин (7,12 г, 16%) в виде светло-коричневого твердого вещества;1H ЯМР (500 МГц, CDCl3) δ 7,81-7,75 (м, 3H), 7,66 (с, 1H), 7,46-7,44 (с, 1H), 7,27 (с, 1H), 7,24-7,23 (м, 2H), 7,16 (д, J=8,3 Гц, 1H), 6,75 (д, J=8,3 Гц, 1H), 4,35 (т, J=6,29 Гц, 1Н), 3,75 (д, J=15,1 Гц, 1Н), 3,62 (д, J=15,1 Гц, 1H), 3,10-3,06 (м, 1H), 2,66-2,61 (м, 1H), 2,43 (с, 3H).
Стадия D: Смесь продукта со стадии C (0,72 г, 2,0 ммоль), бис(пинаколато)дибора (571 мг, 2,24 ммоль), KOAc (600 мг, 6,12 ммоль) дегазировали, используя аргон. К полученной смеси добавляли PdCl2(dppf) (99 мг, 0,122 ммоль). Полученную в результате смесь дегазировали, используя аргон, и затем нагревали при 80°C в течение 3 часов. Охлажденную смесь разбавляли водой и органический слой отделяли. Водный слой три раза экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая коричневое масло, которое использовали на стадии E без дополнительной очистки.
Стадия E: Смесь сырого продукта со стадии D (565 мг, 1,42 ммоль), 2-хлорпиримидина (395 мг, 1,77 ммоль) и карбоната цезия (1,5 г, 4,26 ммоль) в ДМФА (21 мл) и воде (3 мл) дегазировали, используя аргон. К полученной смеси добавляли PdCl2(dppf) (70 мг, 0,085 ммоль). Полученную в результате смесь дегазировали, используя аргон, и затем нагревали при 80°C в течение 4 часов. Охлажденную смесь фильтровали через слой диатомовой земли и три раза промывали водой. Органические слои отделяли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на колонке (80 г силикагеля, 98:2 метиленхлорид/метанол) давала 2-метил-4-нафтален-2-ил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин (430 мг, 60%). Полученный продукт разделяли хиральной ВЭЖХ (Chiralpak OD, 90:10 гептан/этанол с 0,1% диэтиламина). (+)-Энантиомер (130 мг, 30%) получали в виде светло-желтого твердого вещества: [α]24D +209,47° (c 0,095, метанол);1H ЯМР (500 МГц, CDCl3) δ 8,78 (с, 2H), 8,22 (с, 1H), 8,12 (д, J=8,1 Гц, 1H), 7,82-7,75 (м, 3H), 7,71 (с, 1H), 7,48-7,42 (м, 2H), 7,29 (д, J=8,4 Гц, 1H), 7,16 (т, J=4,8 Гц, 1H), 7,03 (д, J=8,1 Гц, 1H), 4,51 (т, J=6,4 Гц, 1H), 3,91 (д, J=14,9 Гц, 1H), 3,75 (д, J=14,9 Гц, 1H), 3,75 (д, J=14,9 Гц, 1Н), 3,16-3,12 (м, 1H), 2,72-2,66 (м, 1H), 2,48 (с, 3H). (-)-Энантиомер (130 мг, 30%) получали в виде светло-желтого твердого вещества [α]24D -71,66° (c = 0,10, метанол);1H ЯМР (500 МГц, CDCl3) δ 8,78 (с, 2H), 8,22 (с, 1H), 8,12 (д, J=8,1 Гц, 1H), 7,82-7,75 (м, 3H), 7,71 (с, 1H), 7,48-7,42 (м, 2H), 7,29 (д, J=8,4 Гц, 1H), 7,16 (т, J=4,8 Гц, 1H), 7,03 (д, J=8,1 Гц, 1H), 4,51 (т, J=6,4 Гц, 1H), 3,91 (д, J=14,9 Гц, 1H), 3,75 (д, J=14,9 Гц, 1H), 3,16-3,12 (м, 1H), 2,72-2,66 (м, 1H), 2,48 (с, 3H).
Стадия F: Раствор (+)-энантиомера со стадии E (127,7 мг, 0,363 ммоль) и малеиновой кислоты (42,2 мг, 0,363 ммоль) в метаноле (1 мл) перемешивали при комнатной температуре. Растворитель выпаривали. Лиофилизация из смеси ацетонитрил (0,5 мл)/воду (0,5 мл) давала малеатную соль (+)-2-метил-4-нафтален-2-ил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолина (144,40 мг, 79%, 95,2% AUC ВЭЖХ) в виде не совсем белого твердого вещества: т.пл. 84-85°C;1H ЯМР (500 МГц, CD3OD) δ 8,85 (с, 2H), 8,38 (с, 1H), 8,29 (д, J=8,4 Гц, 1H), 7,92-7,85 (м, 4H), 7,53 (д, J=4,1 Гц, 2H), 7,38 (д, J=9,7 Гц, 1H), 7,33 (д, J=8,5 Гц, 1Н), 7,10 (д, J=8,1 Гц, 1H), 6,25 (с, 2,5H), 4,73 (ушир.с, 2H), 3,98-3,94 (м, 1H), 3,75-3,73 (м, 1Н), 3,13 (с, 3H); ESI МС m/z 352 [M+Н]+; элементный анализ, вычислено для C24H21N3-1,25C4H4O4-0,25H2O: C, 69,52; Н, 5,33; N, 8,39. Найдено: C, 69,61; Н 5,24; N, 8,41.
Стадия G: Раствор (-)-энантиомера со стадии E (130 мг, 0,369 ммоль) и малеиновой кислоты (42,9 мг, 0,369 ммоль) в метаноле (1 мл) перемешивали при комнатной температуре. Растворитель выпаривали. Лиофилизация из смеси ацетонитрил (0,5 мл)/вода (0,5 мл) давала малеатную соль (-)-2-метил-4-нафтален-2-ил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолина (140,6 мг, 82%, 95,6% AUC ВЭЖХ) в виде не совсем белого твердого вещества: т.пл. 86-88°C;1H ЯМР (CD3OD, 500 МГц) δ 8,85 (с, 2H), 8,38 (с, 1H), 8,29 (д, J=8,2 Гц, 1H), 7,91-7,81 (м, 4H), 7,52 (д, J=9,8 Гц, 1H), 7,38 (д, J=9,7 Гц, 1H), 7,33 (д, J=8,5 Гц, 1H), 7,09 (д, J=8,2 Гц, 1H), 6,24 (с, 2H), 4,73 (м, 2H), 3,98-3,94 (м, 1H), 3,75-3,71 (м, 1Н), 3,13 (с, 3H); ESI-МС m/z 352 [M+Н]+.
Пример 141 - Получение малеатной соли (+)-2-метил-4-нафтален-2-ил-7-пиримидин-5-ил-1,2,3,4-тетрагидроизохинолина и малеатной соли (-)-2-метил-4-нафтален-2-ил-7-пиримидин-5-ил-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору (3-бромбензил)метиламина (29,4 г, 146,9 ммоль) в метиленхлориде (300 мл) добавляли диизопропилэтиламин (51 мл, 293,8 ммоль). Реакционную смесь охлаждали до 0°C и обрабатывали порциями 2-бром-2'-ацетонафтона (36,6 г, 146,9 ммоль) в течение 40-минутного периода. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 5,5 часов. Реакционную смесь три раза промывали водой, сушили над сульфатом натрия, фильтровали и растворитель выпаривали, получая требуемый продукт (количественный выход) в виде вязкого оранжевого масла:1H ЯМР (CDCl3, 300 МГц) δ 8,49 (с, 1H), 8,00 (д, J=8,6 Гц, 1H), 7,95 (д, J=8,1 Гц, 1Н), 7,87 (т, J=8,2 Гц, 2H), 7,61-7,54 (м, 3H), 7,40 (д, J=7,0 Гц, 1H), 7,31 (д, J=7,6 Гц, 1H), 7,19 (т, J=7,7 Гц, 1H), 3,91 (с, 2H), 3,71 (с, 2H), 2,40 (с, 3H).
Стадия B: Боргидрид натрия (54,11 г, 155,4 ммоль) добавляли в течение 45-минутного периода к охлажденному на льду раствору продукта со стадии A (7,05 г, 186,4 ммоль) в метаноле (400 мл). Реакционной смеси давали возможность нагреться до комнатной температуры и продолжали перемешивать еще в течение 21,5 часа. Растворитель удаляли при пониженном давлении и остаток разбавляли водой и три раза экстрагировали метиленхлоридом. Объединенные экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая требуемый продукт (47,78 г, 83%) в виде оранжевого масла:1H ЯМР (500 МГц, CDCl3) δ 7,84-7,81 (м, 4H), 7,48-7,40 (м, 5H), 7,24-7,20 (м, 2H), 4,92 (т, J=6,1 Гц, 1H), 3,71 (д, J=13,3 Гц, 1Н), 3,52 (д, J=13,3 Гц, 1H), 2,69-2,60 (м, 2H), 2,33 (с, 3H).
Стадия C: Продукт со стадии B (47,78 г, 129,0 ммоль) в 1,2- дихлорэтане (500 мл) добавляли по каплям через капельную воронку к метансульфоновой кислоте (469 мл, 7226 ммоль) при комнатной температуре и перемешивали еще в течение 6,5 часов. Затем охлажденную реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь вливали в лед и подщелачивали до pH 9 концентрированным гидроксидом аммония. Реакционную смесь четыре раза экстрагировали этилацетатом. Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на колонке (силикагель; 98:2 метиленхлорид/метанол) давала 7-бром-2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин (7,12 г, 16%) в виде светло-коричневого твердого вещества;1H ЯМР (500 МГц, CDCl3) δ 7,76-7,75 (м, 3H), 7,66 (с, 1H), 7,46-7,44 (с, 1H), 7,27 (с, 1Н), 7,24-7,23 (м, 2H), 7,16 (д, J=8,3 Гц, 1H), 6,75 (д, J=8,3 Гц, 1H), 4,35 (т, J=6,29 Гц, 1H), 3,75 (д, J=15,1 Гц, 1H), 3,62 (д, J=15,1 Гц, 1H), 3,10-3,06 (м, 1H), 2,66-2,61 (м, 1H), 2,43 (с, 3H).
Стадия D: Смесь продукта со стадии C (680 мг, 1,93 ммоль), бис(пинаколато)дибора (538 мг, 2,12 ммоль), KOAc (568 мг, 5,79 ммоль) дегазировали, используя аргон. К полученной смеси добавляли PdCl2(dppf) (95 мг, 0,116 ммоль). Полученную в результате смесь дегазировали, используя аргон, и затем нагревали при 80°C в течение 4,5 часов. После завершения реакции (по данным анализа тонкослойной хроматографией), охлажденный продукт разбавляли водой и органический слой отделяли. Водный слой три раза экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая коричневое масло, которое использовали на стадии E без дополнительной очистки.
Стадия E: Смесь сырого продукта со стадии D (771 мг, 1,93 ммоль), 5-бромпиримидина (429 мг, 2,70 ммоль) и карбоната цезия (2,04 г, 5,79 ммоль) в ДМФА (21 мл) и воде (3 мл) дегазировали, используя аргон. К полученной смеси добавляли PdCl2(dppf) (95 мг, 0,116 ммоль). Полученную в результате смесь дегазировали, используя аргон, и затем нагревали при 80°C в течение 5 часов. Охлажденную смесь фильтровали через слой диатомовой земли и три раза промывали водой. Органические слои отделяли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на колонке (80 г силикагеля, 98:2 метиленхлорид/метанол) давала 2-метил-4-нафтален-2-ил-7-пиримидин-5-ил-1,2,3,4-тетрагидроизохинолин (370 мг, 55%). Полученный продукт разделяли хиральной ВЭЖХ (Chiralpak AD, 70:30 гептан/IPA с 0,1% диэтиламина). (-)-Энантиомер (160 мг, 43%) получали в виде не совсем белого твердого вещества: [α]24D -73,0° (c 0,14, метанол);1H ЯМР (500 МГц, CDCl3) δ 9,18 (с, 1Н), 8,93 (с, 2H), 7,82-7,77 (м, 3H), 7,72 (с, 1H), 7,49-7,45 (м, 2H), 7,33-7,26 (м, 3H), 7,05 (д, J=7,9 Гц, 1Н), 4,51 (т, J=7,6 Гц, 1H), 3,90 (д, J=15,0 Гц, 1H), 3,75 (д, J=14,7 Гц, 1H), 3,19-3,13 (м, 1H), 2,76-2,69 (м, 1H), 2,50 (с, 3H). (+)-Энантиомер (150 мг, 41%) получали в виде светло-красного твердого вещества: [α]24D +76,3° (c 0,14, метанол);1H ЯМР (500 МГц, CDCl3) δ 9,18 (с, 1H), 8,93 (с, 2H), 7,82-7,77 (м, 3H), 7,72 (с, 1H), 7,49-7,45 (м, 2H), 7,33-7,26 (м, 3H), 7,05 (д, J=7,9 Гц, 1H), 4,51 (т, J=7,6 Гц, 1H), 3,90 (д, J=15,0 Гц, 1H), 3,75 (д, J=14,7 Гц, 1H), 3,19-3,13 (м, 1H), 2,76-2,69 (м, 1H), 2,50 (с, 3H).
Стадия F: (+)-Энантиомер со стадии E (160 мг, 0,455 ммоль) в метилэтилкетоне (6 мл) нагревали до 60°C. Добавляли раствор малеиновой кислоты в диоксане (1 М, 48 мкл) и перемешивали при 60°C в течение 1 часа. Реакционную смесь медленно охлаждали до комнатной температуры и перемешивали в течение ночи. Растворитель выпаривали, получая малеатную соль (+)-2-метил-4-нафтален-2-ил-7-пиримидин-5-ил-1,2,3,4-тетрагидроизохинолина (168,3 мг, 70%, 98% AUC ВЭЖХ) в виде светло-коричневого твердого вещества: т.пл. 62-64°C;1H ЯМР (500 МГц, CD3OD) δ 9,16 (с, 1H), 9,08 (с, 2Н), 7,91-7,85 (м, 4Н), 7,71 (с, 1Н), 7,64 (д, J=7,9 Гц, 1H), 7,53-7,51 (м, 2H), 7,32 (д, J=8,4 Гц, 1H), 7,13 (д, J=8,1 Гц, 1Н), 6,26 (с, 3,0H), 4,73 (с, 2H), 3,99-3,92 (м, 1H), 3,82-3,68 (м, 1Н), 3,39-3,30 (м, 1H), 3,11 (с, 3H); ESI МС m/z 352 [M+Н]+.
Стадия G: (-)-Энантиомер со стадии E (140 мг, 0,398 ммоль) в метилэтилкетоне (5 мл) нагревали до 60°C. Добавляли раствор малеиновой кислоты в диоксане (1 М, 42 мкл) и перемешивали при 60°C в течение 1 часа. Реакционную смесь медленно охлаждали до комнатной температуры и перемешивали в течение ночи. Растворитель выпаривали, получая малеатную соль (-)-2-метил-4-нафтален-2-ил-7-пиримидин-5-ил-1,2,3,4-тетрагидроизохинолина (195,0 мг, 94%, 97% AUC ВЭЖХ) в виде светло-коричневого твердого вещества: т.пл.62-64°C;1H ЯМР (500 МГц, CD3OD) δ 9,16 (с, 1H), 9,08 (с, 2H), 7,91-7,85 (м, 4H), 7,71 (с, 1H), 7,64 (д, J=7,9 Гц, 1H), 7,53-7,51 (м, 2H), 7,32 (д, J=8,4 Гц, 1H), 7,13 (д, J=8,1 Гц, 1Н), 6,26 (с, 2,4H), 4,73 (с, 2H), 3,99-3,92 (м, 1H), 3,82-3,68 (м, 1Н), 3,39-3,30 (м, 1H), 3,11 (с, 3H); ESI МС m/z 352 [M+Н]+. Элементный анализ, вычислено для C24H21N3-1,25C4H4O4-1,5H2O: C, 66,53; Н, 5,58; N, 8,03. Найдено: C, 66,28; Н 5,39; N, 7,66.
Пример 142 - Получение малеатной соли (+/-)-7-(3,5-диметилизоксазол-4-ил)-2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору (3-бромбензил)метиламина (7,01 г, 35,0 ммоль) в метиленхлориде (120 мл) добавляли диизопропилэтиламин (12 мл, 20,0 ммоль). Реакционную смесь охлаждали до 0°C и обрабатывали порциями 2-бром-2'-ацетонафтона (8,73 г, 35,0 ммоль) в течение 30-минутного периода. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 3 часов. Реакционную смесь три раза промывали водой, сушили над сульфатом натрия, фильтровали и растворитель выпаривали, получая требуемый продукт (12,54 г, 97%) в виде вязкого оранжевого масла:1H ЯМР (CDCl3, 500 МГц) δ 8,49 (с, 1H), 8,00 (д, J=8,6 Гц, 1H), 7,95 (д, J=8,1 Гц, 1H), 7,87 (т, J=8,2 Гц, 2H), 7,61-7,54 (м, 3H), 7,40 (д, J=7,0 Гц, 1H), 7,31 (д, J=7,6 Гц, 1H), 7,19 (т, J=7,7 Гц, 1H), 3,91 (с, 2H), 3,71 (с, 2H), 2,40 (с, 3H).
Стадия B: Боргидрид натрия (1,55 г, 40,9 ммоль) добавляли в течение 30-минутного периода к охлажденному на льду раствору продукта со стадии A (12,54 г, 34,0 ммоль) в метаноле (140 мл). Реакционной смеси давали возможность нагреться до комнатной температуры и продолжали перемешивать еще в течение 4 часов. Растворитель удаляли при пониженном давлении и остаток разбавляли водой и три раза экстрагировали метиленхлоридом. Объединенные экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая требуемый продукт (11,67 г, 93%) в виде оранжевого масла:1H ЯМР (500 МГц, CDCl3) δ 7,84-7,81 (м, 4H), 7,48-7,40 (м, 5H), 7,24-7,20 (м, 2H), 4,92 (т, J=6,1 Гц, 1H), 3,71 (д, J=13,3 Гц, 1H), 3,52 (д, J=13,3 Гц, 1H), 2,69-2,60 (м, 2H), 2,33 (с, 3H).
Стадия C: Продукт со стадии B (11,6 г, 31,3 ммоль) в 1,2- дихлорэтане (170 мл) добавляли по каплям через капельную воронку к метансульфоновой кислоте (110 мл, 1754 ммоль) при 40°C и перемешивали еще в течение 6,5 часов. Затем охлажденную реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь вливали в лед и подщелачивали до pH 9 концентрированным гидроксидом аммония. Реакционную смесь четыре раза экстрагировали этилацетатом. Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на колонке (330 г силикагеля, 90:10 гексан/этилацетат) давала 7-бром-2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин (4,24 г, 38%) в виде не совсем белого твердого вещества;1H ЯМР (500 МГц, CDCl3) δ 7,81-7,75 (м, 3H), 7,66 (с, 1H), 7,46-7,44 (с, 1H), 7,27 (с, 1H), 7,24-7,23 (м, 2H), 7,16 (д, J=8,3 Гц, 1H), 6,75 (д, J=8,3 Гц, 1H), 4,35 (т, J=6,29 Гц, 1H), 3,75 (д, J=15,1 Гц, 1H), 3,62 (д, J=15,1 Гц, 1Н), 3,10-3,06 (м, 1H), 2,66-2,61 (м, 1H), 2,43 (с, 3H).
Стадия D: Смесь продукта со стадии C (0,50 г, 1,4 ммоль), бис(пинаколато)дибора (397 мг, 1,56 ммоль), KOAc (415 мг, 4,25 ммоль) дегазировали, используя аргон. К полученной смеси добавляли PdCl2(dppf) (70 мг, 0,085 ммоль). Полученную в результате смесь дегазировали, используя аргон, и затем нагревали при кипячении с обратным холодильником в течение 3 часов. После завершения реакции по данным анализа тонкослойной хроматографией охлажденный продукт разбавляли водой и органический слой отделяли. Водный слой три раза экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая коричневое масло, которое использовали на стадии E без дополнительной очистки.
Стадия E: Смесь сырого продукта со стадии D (565 мг, 1,42 ммоль), 3,5-диметил-4-йодизоксазола (395 мг, 1,77 ммоль) и карбоната цезия (1,5 г, 4,26 ммоль) в ДМФА (21 мл) и воде (3 мл) дегазировали, используя аргон. К полученной смеси добавляли PdCl2(dppf) (70 мг, 0,085 ммоль). Полученную в результате смесь дегазировали, используя аргон, и затем нагревали при 80°C в течение 4 часов. Охлажденную смесь фильтровали через слой диатомовой земли и три раза промывали водой. Органические слои отделяли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на колонке (80 г силикагеля, 98:2 метиленхлорид/метанол) давала требуемый продукт (100 мг, 19%) в виде коричневого твердого вещества;1H ЯМР (500 МГц, CDCl3) δ 7,81-7,79 (м, 3H), 7,77 (с, 1H), 7,48-7,45 (м, 2H), 7,32 (д, J=8,4 Гц, 1H), 6,99 (с, 1H), 6,94-6,93 (м, 2H), 4,47 (т, J=1,95 Гц, 1H), 3,83 (д, J=14,9 Гц, 1H), 3,69 (д, J=14,8 Гц, 1H), 3,16-3,12 (м, 1H), 2,73-2,69 (м, 1H), 2,47 (с, 3H), 2,39 (с, 3H), 2,26 (с, 3H).
Стадия F: Раствор продукта со стадии E (98 мг, 0,266 ммоль) и малеиновой кислоты (31 мг, 0,266 ммоль) в метаноле (1 мл) перемешивали при комнатной температуре. Растворитель выпаривали. Лиофилизация из смеси ацетонитрил (0,5 мл)/вода (0,5 мл) давала малеатную соль 7-(3,5-диметилизоксазол-4-ил)-2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолина (128,89 мг, 99%, >99% AUC ВЭЖХ) в виде коричневого твердого вещества: т.пл. 88-90°C;1H ЯМР (CD3OD, 500 МГц) δ 7,91-7,85 (м, 4H), 7,53-7,50 (м, 2H), 7,33-7,31 (м, 2H), 7,23 (д, J=8,0 Гц, 1H), 7,04 (д, J=8,0 Гц, 1Н), 6,23 (с, 1H), 4,81-4,78 (м, 1H), 4,64 (с, 2H), 3,93-3,91 (м, 1H), 3,70-3,67 (м, 1H), 3,09 (с, 3H), 2,40 (с, 3H), 2,24 (с, 3H); ESI МС m/z 369 [M+Н]+.
Пример 143 - Получение малеатной соли (+)-2-метил-7-морфолин-4-ил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолина и малеатной соли (-)-2-метил-7-морфолин-4-ил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору (3-бромбензил)метиламина (2,0 г, 10,0 ммоль) в метиленхлориде (40 мл) добавляли диизопропилэтиламин (3,5 мл, 20,0 ммоль). Реакционную смесь охлаждали до 0°C и обрабатывали порциями 2-бром-2'-ацетонафтона (2,49 г, 10,0 ммоль) в течение 10-минутного периода. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 5,5 часов. Реакционную смесь три раза промывали водой, сушили над сульфатом натрия, фильтровали и растворитель выпаривали, получая требуемый продукт (3,67 г, 99%) в виде вязкого оранжевого масла:1H ЯМР (CDCl3, 500 МГц) δ 8,49 (с, 1H), 8,00 (д, J=8,6 Гц, 1H), 7,95 (д, J=8,1 Гц, 1H), 7,87 (т, J=8,2 Гц, 2H), 7,61-7,54 (м, 3H), 7,40 (д, J=7,0 Гц, 1Н), 7,31 (д, J=7,6 Гц, 1H), 7,19 (т, J=7,7 Гц, 1H), 3,91 (с, 2H), 3,71 (с, 2H), 2,40 (с, 3H).
Стадия B: Боргидрид натрия (453 мг, 12,0 ммоль) добавляли в течение 10-минутного периода к охлажденному на льду раствору продукта со стадии A (3,68 г, 10,0 ммоль) в метаноле (45 мл). Реакционной смеси давали возможность нагреться до комнатной температуры и продолжали перемешивать еще в течение 2,5 часов. Растворитель удаляли при пониженном давлении и остаток разбавляли водой и три раза экстрагировали метиленхлоридом. Объединенные экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая продукт (2,99 г, 81%) в виде оранжевого масла:1H ЯМР (500 МГц, CDCl3) δ 7,84-7,81 (м, 4H), 7,48-7,40 (м, 5H), 7,24-7,20 (м, 2H), 4,92 (т, J=6,1 Гц, 1H), 3,71 (д, J=13,3 Гц, 1H), 3,52 (д, J=13,3 Гц, 1H), 2,69-2,60 (м, 2H), 2,33 (с, 3H).
Стадия C: Продукт со стадии B (2,99 г, 8,07 ммоль) в 1,2- дихлорэтане (50 мл) добавляли по каплям через капельную воронку к метансульфоновой кислоте (30 мл, 452 ммоль) при 40°C и перемешивали еще в течение 4 часов. Охлажденную реакционную смесь вливали в лед и подщелачивали до pH 9 концентрированным гидроксидом аммония. Реакционную смесь четыре раза экстрагировали этилацетатом. Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на колонке (80 г диоксида кремния, 95:5 гексан/этилацетат) давала 7-бром-2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин (790 мг, 28%) в виде не совсем белого твердого вещества;1H ЯМР (500 МГц, CDCl3) δ 7,81-7,75 (м, 3H), 7,66 (с, 1H), 7,46-7,44 (с, 1H), 7,27 (с, 1H), 7,24-7,23 (м, 2H), 7,16 (д, J=8,3 Гц, 1H), 6,75 (д, J=8,3 Гц, 1H), 4,35 (т, J=6,29 Гц, 1H), 3,75 (д, J=15,1 Гц, 1H), 3,62 (д, J=15,1 Гц, 1H), 3,10-3,06 (м, 1H), 2,66-2,61 (м, 1H), 2,43 (с, 3H).
Стадия D: Смесь продукта со стадии C (790 мг, 2,24 ммоль), морфолина (390 мг, 4,48 ммоль), карбоната цезия (1,82 г, 5,6 ммоль), Xphos (641 мг, 1,34 ммоль) в толуоле (25 мл) дегазировали, используя аргон. К полученной смеси добавляли ацетат палладия (II) (75 мг, 0,336 ммоль). Полученную в результате смесь дегазировали, используя аргон, и затем нагревали при кипячении с обратным холодильником в течение 16 часов. После завершения реакции (по данным анализа тонкослойной хроматографией) охлажденный продукт фильтровали через слой диатомовой земли и растворитель выпаривали. Очистка хроматографией на колонке (80 г силикагеля, 95:5 метиленхлорид/метанол) давала продукт (570 мг, 57%) в виде желтого твердого вещества. Полученный продукт разделяли хиральной ВЭЖХ (Chiralpak AD, 80:20 гептан/изопропанол с 0,1% диэтиламина). (-)Энантиомер (150 мг, 35%) получали в виде не совсем белого твердого вещества [α]25D -65,1° (c 0,13, метанол);1H ЯМР (500 МГц, CDCl3) δ 7,79-7,74 (м, 3H), 7,68 (с, 1H), 7,45-7,42 (м, 2H), 7,29-7,25 (м, 1H), 6,78 (д, J=8,3 Гц, 1H), 6,65 (д, J=9,5 Гц, 2H), 4,38-4,36 (м, 1Н), 3,84 (т, J=4,7 Гц, 4H), 3,75 (д, J=14,8 Гц, 1H), 3,62 (д, J=14,7 Гц, 1H), 3,12 (т, J=4,8 Гц, 4H), 3,07-3,06 (м, 1H), 2,65-2,61 (м, 1H), 2,43 (с, 3H). (+)-Энантиомер (191 мг, 42%) получали в виде не совсем белого твердого вещества [α]25D +65,1° (c 0,11, метанол);1H ЯМР (500 МГц, CDCl3) δ 7,79-7,74 (м, 3H), 7,68 (с, 1H), 7,45-7,42 (м, 2H), 7,29-7,25 (м, 1H), 6,78 (д, J=8,3 Гц, 1H), 6,65 (д, J=9,5 Гц, 2H), 4,38-4,36 (м, 1H), 3,84 (т, J=4,7 Гц, 4H), 3,75 (д, J=14,8 Гц, 1H), 3,62 (д, J=14,7 Гц, 1H), 3,12 (т, J=4,8 Гц, 4H), 3,07-3,06 (м, 1H), 2,65-2,61 (м, 1H), 2,43 (с, 3H).
Стадия E: Раствор (-)-энантиомера со стадии D (150 мг, 0,419 ммоль) и малеиновой кислоты (48,5 мг, 0,419 ммоль) в метаноле (1 мл) перемешивали при комнатной температуре. Растворитель выпаривали. Лиофилизация из смеси ацетонитрил (0,5 мл)/вода (0,5 мл) давала малеатную соль (-)-2-метил-7-морфолин-4-ил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолина (197,80 мг, 99%, >99% AUC ВЭЖХ) в виде не совсем белого твердого вещества: т.пл.98-99°C;1H ЯМР (CD3OD, 500 МГц) δ 7,87-7,81 (м, 3H), 7,77 (с, 1H), 7,50-7,48 (м, 2H), 7,27 (д, J=8,5 Гц, 1H), 6,87-6,85 (м, 1H), 6,82-6,80 (м, 2H), 6,24 (с, 1H), 4,67-4,63 (м, 1H), 4,53-4,45 (м, 2H), 3,84-3,80 (м, 5H), 3,30-3,26 (м, 1H), 3,16-3,13 (м, 4H), 3,30 (с, 3H); ESI МС m/z 358 [M+Н]+.
Стадия F: Раствор (+)-энантиомера со стадии D (184 мг, 0,513 ммоль) и малеиновой кислоты (59,6 мг, 0,513 ммоль) в метаноле (1 мл) перемешивали при комнатной температуре. Растворитель выпаривали. Лиофилизация из смеси ацетонитрил (0,5 мл)/вода (0,5 мл) давала малеатную соль (+)-2-метил-7-морфолин-4-ил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолина (190,9 мг, 77%, >99% AUC ВЭЖХ) в виде не совсем белого твердого вещества: т.пл. 94-96°C;1H ЯМР (CD3OD, 500 МГц) δ 7,87-7,82 (м, 3H), 7,77 (с, 1H), 7,52-7,48 (м, 2H), 7,27 (д, J=8,5 Гц, 1H), 6,88 (д, J=8,6 Гц, 1H), 6,82-6,80 (м, 2H), 6,24 (с, 1H), 4,68-4,65 (м, 1H), 4,57-4,48 (м, 2H), 3,87-3,80 (м, 5H), 3,60-3,58 (м, 1Н), 3,16-3,13 (м, 4H), 3,06 (с, 3H); ESI МС m/z 358 [M+Н].
Пример 144 - Получение малеатной соли (+/-)-2-метил-4-нафтален-2-ил-7-пиперидин-1-ил-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору (3-бромбензил)метиламина (7,01 г, 35,0 ммоль) в метиленхлориде (120 мл) добавляли диизопропилэтиламин (12 мл, 20,0 ммоль). Реакционную смесь охлаждали до 0°C и обрабатывали порциями 2-бром-2'-ацетонафтоном (8,73 г, 35,0 ммоль) в течение 30-минутного периода. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 3 часов. Реакционную смесь три раза промывали водой, сушили над сульфатом натрия, фильтровали и растворитель выпаривали, получая требуемый продукт (12,54 г, 97%) в виде вязкого оранжевого масла:1H ЯМР (CDCl3, 500 МГц) δ 8,49 (с, 1H), 8,00 (д, J=8,6 Гц, 1H), 7,95 (д, J=8,1 Гц, 1H), 7,87 (т, J=8,2 Гц, 2H), 7,61-7,54 (м, 3H), 7,40 (д, J=7,0 Гц, 1H), 7,31 (д, J=7,6 Гц, 1H), 7,19 (т, J=7,7 Гц, 1H), 3,91 (с, 2H), 3,71 (с, 2H), 2,40 (с, 3H).
Стадия B: Боргидрид натрия (1,55 г, 40,9 ммоль) добавляли в течение 30-минутного периода к охлажденному на льду раствору продукта со стадии A (12,54 г, 34,0 ммоль) в метаноле (140 мл). Реакционной смеси давали возможность нагреться до комнатной температуры и продолжали перемешивать еще в течение 4 часов. Растворитель удаляли при пониженном давлении и остаток разбавляли водой и три раза экстрагировали метиленхлоридом. Объединенные экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая требуемый продукт (11,67 г, 93%) в виде оранжевого масла:1H ЯМР (500 МГц, CDCl3) δ 7,84-7,81 (м, 4H), 7,48-7,40 (м, 5H), 7,24-7,20 (м, 2H), 4,92 (т, J=6,1 Гц, 1H), 3,71 (д, J=13,3 Гц, 1H), 3,52 (д, J=13,3 Гц, 1H), 2,69-2,60 (м, 2H), 2,33 (с, 3H).
Стадия C: Продукт со стадии B (11,6 г, 31,3 ммоль) в 1,2-дихлорэтане (170 мл) добавляли по каплям через капельную воронку к метансульфоновой кислоте (110 мл, 1754 ммоль) при 40°C и перемешивали еще в течение 6,5 часов. Затем охлажденную реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь вливали в лед и подщелачивали до pH 9 концентрированным гидроксидом аммония. Реакционную смесь четыре раза экстрагировали этилацетатом. Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на колонке (330 г силикагеля, 90:10 гексаны/этилацетат) давала 7-бром-2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин (4,24 г, 38%) в виде не совсем белого твердого вещества;1H ЯМР (500 МГц, CDCl3) δ 7,81-7,75 (м, 3H), 7,66 (с, 1H), 7,46-7,44 (с, 1H), 7,27 (с, 1H), 7,24-7,23 (м, 2H), 7,16 (д, J=8,3 Гц, 1Н), 6,75 (д, J=8,3 Гц, 1H), 4,35 (т, J=6,29 Гц, 1Н), 3,75 (д, J=15,1 Гц, 1Н), 3,62 (д, J=15,1 Гц, 1Н), 3,10-3,06 (м, 1H), 2,66-2,61 (м, 1H), 2,43 (с, 3H).
Стадия D: Смесь продукта со стадии C (260 мг, 0,738 ммоль), пиперидина (126 мг, 1,48 ммоль), карбоната цезия (0,601 г, 1,85 ммоль), Xphos (0,211 мг, 0,443 ммоль) в толуоле (12 мл) дегазировали, используя аргон. К полученной смеси добавляли ацетат палладия(II) (25 мг, 0,111 ммоль). Полученную в результате смесь дегазировали, используя аргон, и затем нагревали при кипячении с обратным холодильником в течение 17 часов. Охлажденную смесь фильтровали через слой диатомовой земли и растворитель выпаривали. Очистка хроматографией на колонке (40 г силикагеля, 95:5 метиленхлорид/метанол) давала требуемый продукт (110 мг, 38%) в виде светло-желтого твердого вещества;1H ЯМР (500 МГц, CDCl3) δ 7,80-7,73 (м, 3H), 7,67 (с, 1H), 7,46-7,40 (м, 2H), 7,29 (д, J=8,4 Гц, 1H), 6,74 (д, J=11,0 Гц, 1H), 6,68-6,66 (м, 2H), 4,36 (т, J=5,9 Гц, 1H), 3,73 (д, J=14,7 Гц, 1H), 3,61 (д, J=14,7 Гц, 1H), 3,11 (т, J=5,4 Гц, 4H), 3,08-3,04 (м, 1H), 2,67-2,55 (м, 1H), 2,42 (с, 3H), 1,71-1,67 (м, 4H), 1,58-1,54 (м, 2H).
Стадия E: Раствор продукта со стадии D (110 мг, 0,28 ммоль) и малеиновой кислоты (32 мг, 0,28 ммоль) в метаноле (1 мл) перемешивали при комнатной температуре. Растворитель выпаривали. Лиофилизация из смеси ацетонитрил (0,5 мл)/вода (0,5 мл) давала малеатную соль (+/-)-2-метил-4-нафтален-2-ил-7-пиперидин-1-ил-1,2,3,4-тетрагидроизохинолина (131,45 мг, 99%, 94% AUC ВЭЖХ) в виде светло-желтого твердого вещества: т.пл. 88-90°C;1H ЯМР (CD3OD, 500 МГц) δ 7,87-7,81 (м, 3H), 7,77 (с, 1Н), 7,51-7,47 (м, 2H), 7,27 (д, J=8,4 Гц, 1Н), 6,87 (д, J=8,6 Гц, 1H), 6,81 (с, 1H), 6,77 (д, J=8,7 Гц, 1Н), 6,23 (с, 1H), 4,66-4,63 (м, 1H), 4,56 (кв., J=15,2 Гц, 2H), 3,84-3,81 (м, 1Н), 3,54 (т, J=11,9 Гц, 1H), 3,26-3,15 (м, 4H), 3,04 (с, 3H), 1,69-1,67 (м, 4H), 1,61-1,58 (м, 2H); ESI-МС m/z 357 [M+Н]+.
Пример 145 - Получение малеатной соли (+/-)-2-метил-4-нафтален-2-ил-7-пирролидин-1-ил-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору (3-бромбензил)метиламина (29,40 г, 146,92 ммоль) в метиленхлориде (300 мл) добавляли диизопропилэтиламин (37,9 мл, 293,85 ммоль). Реакционную смесь охлаждали до 0°C и обрабатывали порциями 2-бром-2'-ацетонафтона (36,6 г, 146,92 ммоль) в течение 40-минутного периода. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 5,5 часов. Реакционную смесь три раза промывали водой, сушили над сульфатом натрия, фильтровали и растворитель выпаривали, получая требуемый продукт (количественный выход) в виде вязкого оранжевого масла:1H ЯМР (CDCl3, 500 МГц) δ 8,49 (с, 1Н), 8,00 (д, J=8,6 Гц, 1Н), 7,95 (д, J=8,1 Гц, 1H), 7,87 (т, J=8,2 Гц, 2H), 7,61-7,54 (м, 3H), 7,40 (д, J=7,0 Гц, 1H), 7,31 (д, J=7,6 Гц, 1Н), 7,19 (т, J=7,7 Гц, 1H), 3,91 (с, 2H), 3,71 (с, 2H), 2,40 (с, 3H).
Стадия B: Боргидрид натрия (7,05 г, 186,44 ммоль) добавляли в течение 45-минутного периода к охлажденному на льду раствору продукта со стадии A (54,11 г, 155,36 ммоль) в метаноле (400 мл). Реакционной смеси давали возможность нагреться до комнатной температуры и продолжали перемешивать еще в течение 21,5 часов. Растворитель удаляли при пониженном давлении и остаток разбавляли водой и три раза экстрагировали метиленхлоридом. Объединенные экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая требуемый продукт (47,78 г, 83% для двух стадий) в виде оранжевого масла:1H ЯМР (500 МГц, CDCl3) δ 7,84-7,81 (м, 4H), 7,48-7,40 (м, 5H), 7,24-7,20 (м, 2H), 4,92 (т, J=6,1 Гц, 1H), 3,71 (д, J=13,3 Гц, 1H), 3,52 (д, J=13,3 Гц, 1H), 2,69-2,60 (м, 2H), 2,33 (с, 3H).
Стадия C: Продукт со стадии B (47,78 г, 129,04 ммоль) в 1,2-дихлорэтане (500 мл) добавляли по каплям через капельную воронку к метансульфоновой кислоте (469 мл, 7226 ммоль) при комнатной температуре и перемешивали еще в течение 1 часа. Реакционную смесь вливали в лед и подщелачивали до pH 9 концентрированным гидроксидом аммония (600 мл). Реакционную смесь четыре раза экстрагировали этилацетатом. Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на колонке (силикагель, 50:50 гексан/этилацетат) давала 7-бром-2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин (4,72 г, 10%) в виде коричневого твердого вещества;1H ЯМР (500 МГц, CDCl3) δ 7,81-7,75 (м, 3H), 7,66 (с, 1Н), 7,46-7,44 (с, 1H), 7,27 (с, 1H), 7,24-7,23 (м, 2H), 7,16 (д, J=8,3 Гц, 1H), 6,75 (д, J=8,3 Гц, 1H), 4,35 (т, J=6,29 Гц, 1H), 3,75 (д, J=15,1 Гц, 1H), 3,62 (д, J=15,1 Гц, 1H), 3,10-3,06 (м, 1H), 2,66-2,61 (м, 1H), 2,43 (с, 3H).
Стадия D: Смесь продукта со стадии C (470 мг, 1,33 ммоль), пирролидина (0,22 мл, 1,48 ммоль), карбоната цезия (1,08 г, 3,32 ммоль), Xphos (0,38 г, 0,798 ммоль) в толуоле (20 мл) дегазировали, используя аргон. К полученной смеси добавляли ацетат палладия(II) (112 мг, 0,498 ммоль). Полученную в результате смесь дегазировали, используя аргон, и затем кипятили с обратным холодильником в течение 22,5 часов. Охлажденную смесь фильтровали через слой диатомовой земли и растворитель выпаривали. Очистка хроматографией на колонке (40 г силикагеля, 98:2 метиленхлорид/метанол) давала требуемый продукт (220 мг, 48%) в виде светло-желтого твердого вещества;1H ЯМР (500 МГц, CDCl3) δ 7,78-7,72 (м, 3H), 7,69 (с, 1H), 7,44-7,42 (м, 2H), 7,30 (д, J=8,4 Гц, 1H), 6,71 (д, J=8,4 Гц, 1H), 6,33-6,29 (м, 2H), 4,36 (т, J=5,9 Гц, 1H), 3,75 (д, J=14,6 Гц, 1H), 3,63 (д, J=14,7 Гц, 1H), 3,25 (т, J=6,5 Гц, 4H), 3,08-3,04 (м, 1H), 2,63-2,59 (м, 1H), 2,43 (с, 3H), 1,99-1,96 (м, 4H).
Стадия E: Раствор продукта со стадии D (220 мг, 0,584 ммоль) и малеиновой кислоты (67,8 мг, 0,584 ммоль) в метаноле (1 мл) перемешивали при комнатной температуре. Растворитель выпаривали. Лиофилизация из смеси ацетонитрил (0,5 мл)/вода (0,5 мл) давала малеатную соль (+/-)-2-метил-4-нафтален-2-ил-7-пирролидин-1-ил-1,2,3,4-тетрагидроизохинолина (236,10 мг, 88%, 98,7% AUC ВЭЖХ) в виде светло-желтого твердого вещества: т.пл.97-100°C;1H ЯМР (CD3OD, 500 МГц) δ 7,86-7,81 (м, 3H), 7,76 (с, 1H), 7,51-7,47 (м, 2H), 7,28 (д, J=8,2 Гц, 1H), 6,72 (д, J=7,5 Гц, 1H), 6,50 (д, J=8,6 Гц, 1H), 6,40 (с, 1H), 6,24 (с, 1H), 4,66-4,63 (м, 1H), 4,57-4,50 (м, 2H), 3,86-3,83 (м, 1H), 3,44-3,43 (м, 1H), 3,30-3,25 (м, 4H), 3,06 (с, 3H), 2,03-2,00 (м, 4H); ESI МС m/z 343 [M+Н]+.
Пример 146 - Получение фумаратной соли (+/-)-8-метокси-2-метил-4-нафтален-2-ил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору рацемического фенола (2,0 г, 6,26 ммоль) со стадии D примера 138 в дихлорметане (30 мл) при 0°C добавляли триэтиламин (1,30 мл, 9,39 ммоль), затем ангидрид трифторметансульфоновой кислоты (1,26 мл, 7,51 ммоль) по каплям. Реакционный раствор перемешивали при 0°C в течение 2 часов. Реакционную смесь обрабатывали CH2Cl2 (2×100 мл) и водой (100 мл), сушили (Na2SO4) и концентрировали при пониженном давлении, получая требуемый трифторметансульфонат (3,10 г, 100% выход сырого продукта) в виде рыжевато-коричневой пены, который использовали на следующей стадии без какой-либо дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,76-7,83 (м, 3H), 7,68 (с, 1H), 7,46-7,52 (м, 2H), 7,24-7,26 (м, 1H), 6,95 (д, J=8,6 Гц, 1Н), 6,70 (д, J=8,9 Гц, 1H), 4,54-4,57 (м, 1H), 3,99 (д, J=15,9 Гц, 1H), 3,94 (с, 3H), 3,69 (д, J=15,9 Гц, 1H), 3,10-3,14 (м, 1H), 2,67-2,70 (м, 1H), 2,53 (с, 3H); ESI-МС m/z 452 [M+H]+.
Стадия B: К раствору трифторметансульфоната (2,10 г, 3,3 ммоль) с описанной выше стадии A в ДМСО (25 мл) добавляли бис(пинаколато)дибор (1,30 г, 5,11 ммоль) и ацетат калия (1,37 г, 13,9 ммоль). Раствор продували азотом в течение 10 минут и затем добавляли 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий(II) (190 мг, 0,233 ммоль). Реакционный раствор нагревали при 80°C в течение 4 часов и затем охлаждали до комнатной температуры. Полученный в результате реакционный раствор разбавляли EtOAc (120 мл), промывали водой (2×50 мл), сушили (Na2SO4) и концентрировали при пониженном давлении, получая требуемый продукт (2,1 г, 100% выход сырого продукта), который использовали на следующей стадии без дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,74-7,81 (м, 3H), 7,68 (с, 1H), 7,41-7,51 (м, 2H), 7,26-7,28 (м, 1H), 6,70 (д, J=8,4 Гц, 1H), 6,54 (д, J=8,4 Гц, 1H), 4,43 (дд, J=8,5 Гц, 6,0 Гц, 1H), 3,90 (д, J=15,3 Гц, 1H), 3,87 (с, 3H), 3,60 (д, J=15,4 Гц, 1H), 3,01-3,15 (м, 1H), 2,60-2,64 (м, 1H), 2,46 (с, 3H), 1,35 (с, 12H).
Стадия C: В реакционную колбу со сложным эфиром бороновой кислоты (1,43 г сырого продукта) с описанной выше стадии B, хлорпиридазина (460 мг, 4,01 ммоль) и карбоната натрия (1,06 г, 10,0 ммоль) добавляли ДМФА (15 мл) и воду (4.0 мл). Полученный в результате раствор продували азотом в течение 10 минут и затем добавляли 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий (137 мг, 0,167 ммоль). Реакционный раствор нагревали при 100°C в течение 5 часов и обрабатывали EtOAc (3×150 мл) и водой (150 мл), сушили (Na2SO4) и концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на колонке с силикагелем (элюент CHCl3/EtOH 95:5, затем CH2Cl2/i-PrOH 90:10), получая требуемый продукт (40 мг, 4%) в виде желто-коричневого твердого вещества:1H ЯМР (CDCl3, 500 МГц) δ 9,14 (дд, J=3,2 Гц, 1,7 Гц, 1H), 8,06 (дд, J=6,9 Гц, 1,7 Гц, 1H), 7,77-7,83 (м, 3H), 7,72 (с, 1Н), 7,62 (д, J=8,1 Гц, 1H), 7,43-7,52 (м, 3H), 7,29 (дд, J=6,7 Гц, 1,8 Гц, 1H), 6,85 (д, J=8,2 Гц, 1H), 4,50-4,52 (м, 1H), 3,99-4,03 (м, 1H), 3,63 (д, J=15,6 Гц, 1H), 3,54 (с, 3H), 3,11-3,15 (м, 1H), 2,68 (дд, J=8,8 Гц, 2,7 Гц, 1H), 2,52 (с, 3H); ESI-МС m/z 382 [M+H]+.
Стадия D: К раствору 7-пиридазинилтетрагидроизохинолина (38,7 мг, 0,101 ммоль) с описанной выше стадии C в метаноле (3 мл) добавляли фумаровую кислоту (12,0 мг, 0,101 ммоль). Полученный в результате раствор концентрировали при пониженном давлении, получая фумаратную соль (+/-)-8-метокси-2-метил-4-нафтален-2-ил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина (41,0 мг, 82%, AUC ВЭЖХ = 97,4%) в виде желто-коричневого твердого вещества: т.пл. 182-186°C;1H ЯМР (CD3OD, 500 МГц) δ 9,17 (дд, J=4,9 Гц, 1,6 Гц, 1H), 8,14 (дд, J=8,6 Гц, 1,6 Гц, 1H), 7,79-7,88 (м, 5H), 7,57 (д, J=8,2 Гц, 1H), 7,48-7,52 (м, 2H), 7,31 (дд, J=8,5 Гц, 1,8 Гц, 1H), 7,31 (д, J=8,5 Гц, 1H), 6,71 (с, 2H), 4,72-4,76 (м, 1H), 4,56 (д, J=15,8 Гц, 1H), 4,22 (д, J=15,6 Гц, 1H), 3,63-3,67 (м, 1H), 3,56 (с, 3H), 3,32-3,35 (м, 1H), 2,91 (с, 3H); ESI-МС m/z 382 [M+H]+.
Пример 147 - Получение малеатной соли (+/-)-4-(хиноксалин-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия A: Смесь 6-бромхиноксалина (1,09 г, 5,06 ммоль), н-бутилвинилового эфира (2,53 г, 25,3 ммоль), карбоната калия (839 мг, 6,07 ммоль), dppp (138 мг, 0,334 ммоль) и Pd(OAc)2 (34 мг, 0,152 ммоль) в ДМФА (13 мл) и воде (1,5 мл) нагревали при кипячении с обратным холодильником в течение 6 часов. Смеси давали возможность остыть до комнатной температуры и затем добавляли 2 N HCl (20 мл). Полученную в результате смесь перемешивали при комнатной температуре в течение 0,5 часа и три раза экстрагировали этилацетатом. Объединенные органические экстракты промывали насыщенным раствором бикарбоната натрия и насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали. Очистка хроматографией на колонке (силикагель, от 20% до 40% этилацетат/гексаны) давала требуемый продукт (324 мг, 37%):1H ЯМР (CDCl3, 500 МГц) δ 8,95 (с, 2H), 8,69 (д, J=1,9 Гц, 1H), 8,36 (дд, J=8,8, 1,9 Гц, 1H), 8,19 (д, J=8,8 Гц, 1H), 2,79 (с, 3H); ESI-МС m/z=229 [M+H]+.
Стадия B: К суспензии бромида меди(II) (840 мг, 3,76 ммоль) в этилацетате (6 мл) добавляли раствор продукта со стадии A (324 мг, 1,88 ммоль) в хлороформе (5 мл). Реакционную смесь кипятили с обратным холодильником в течение 4 часов. Смеси давали возможность остыть до комнатной температуры. Смесь разбавляли этилацетатом (20 мл) и насыщенным раствором хлорида аммония (20 мл) и затем фильтровали через целит. Слой целита промывали этилацетатом. Органический слой отделяли и промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали. Очистка хроматографией на колонке (силикагель, от 20% до 40% этилацетат/гексаны) давала требуемый продукт (174 мг, 43%):1H ЯМР (CDCl3, 500 МГц) δ 8,97 (с, 2H), 8,74 (д, J=1,9 Гц, 1H), 8,36 (дд, J=8,8, 1,9 Гц, 1H), 8,22 (д, J=8,8 Гц, 1H), 4,61 (с, 2H).
Стадия C: К раствору продукта со стадии B (174 мг, 0,693 ммоль) в дихлорметане (5 мл) добавляли диизопропилэтиламин (134 мг, 1,04 ммоль) и N-бензилметиламин (130 мг, 1,04 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов и затем разбавляли дихлорметаном (20 мл). Смесь промывали водой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали. Очистка хроматографией на колонке (силикагель, от 20% до 60% этилацетат/гексаны) давала требуемый продукт (103 мг, 51%):1H ЯМР (CDCl3, 300 МГц) δ 8,94 (д, J=1,7 Гц, 1H), 8,93 (д, J=1,7 Гц, 1H), 8,77 (д, J=1,9 Гц, 1H), 8,32 (дд, J=8,8, 1,9 Гц, 1H), 8,16 (д, J=8,8 Гц, 1H), 7,38-7,26 (м, 5Н), 3,94 (с, 2H), 3,74 (с, 2H), 2,42 (с, 3H); ESI-МС m/z=292 [M+H]+.
Стадия D: К охлажденному на льду раствору продукта со стадии C (103 мг, 0,353 ммоль) в метаноле (5 мл) добавляли боргидрид натрия (15 мг, 0,4 ммоль). Реакционную смесь перемешивали при 0°C в течение 1 часа. Растворитель удаляли при пониженном давлении. Остаток распределяли между дихлорметаном и водой. Органические экстракты промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали, получая требуемый продукт (100 мг, 97% выход сырого продукта): ESI МС m/z=294 [M+H]+. Полученный сырой продукт использовали на следующей стадии без дополнительной очистки.
Стадия E: К раствору продукта со стадии D (100 мг, 0,34 ммоль) в 1,2-дихлорэтане (10 мл) добавляли реагент Итона (0,9 мл). Реакционную смесь перемешивали при 90°C в течение 24 часов. Затем смесь охлаждали на бане со льдом и добавляли насыщенный раствор бикарбоната натрия. Органический слой отделяли и водный слой экстрагировали дихлорметаном. Объединенные экстракты промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали. Очистка хроматографией на колонке (силикагель, от 4% до 5% метанол/дихлорметан) давала требуемый продукт (37 мг, 39%):1H ЯМР (CDCl3, 500 МГц) δ 8,81 (д, J=1,8 Гц, 1H), 8,80 (д, J=1,8 Гц, 1Н), 8,01 (д, J=8,7 Гц, 1H), 7,97 (д, J=1,9 Гц, 1Н), 7,64 (дд, J=8,7, 1,9 Гц, 1H), 7,19-7,13 (м, 2Н), 7,09-7,06 (м, 1Н), 6,90 (д, J=7,7 Гц, 1H), 4,52 (т, J=6,3 Гц, 1H), 3,76 (д, J=15,0 Гц, 1H), 3,71 (д, J=15,0 Гц, 1H), 3,08 (дд, J=11,5, 5,5 Гц, 1H), 2,78 (дд, J=11,5, 7,2 Гц, 1H), 2,44 (с, 3H); ESI-МС m/z = 276 [M+H]+.
Стадия F: К раствору продукта со стадии E (37 мг, 0,134 ммоль) в метаноле (1 мл) добавляли малеиновую кислоту (15,6 мг, 0,134 ммоль). Растворитель удаляли при пониженном давлении, получая малеатную соль (+/-)-4-(хиноксалин-6-ил)-2-метил- 1,2,3,4-тетрагидроизохинолина в виде светло-коричневого твердого вещества (52 мг, 99%, 96,7% AUC ВЭЖХ):1H ЯМР (CD3OD, 500 МГц) δ 8,91 (с, 2H), 8,13 (д, J=8,7 Гц, 1Н), 8,04 (с, 1H), 7,70 (дд, J=8,7, 2,0 Гц, 1H), 7,39-7,33 (м, 2Н), 7,30-7,26 (м, 1Н), 6,96 (д, J=7,8 Гц, 1Н), 6,24 (с, 1H), 4,95-4,92 (м, 1Н), 4,67 (д, J=15,3 Гц, 1H), 4,61 (д, J=15,3 Гц, 1H), 3,99-3,95 (м, 1Н), 3,74-3,69 (м, 1Н), 3,10 (с, 3H); ESI-МС m/z = 276 [M+H]+.
Пример 148 - Получение малеатной соли (+)-4-бензо[b]тиофен-6-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина и малеатной соли (-)-4-бензо[b]тиофен-6-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина
Стадия A: Смесь 3-метокситиофенола (25,0 г, 175,0 ммоль), карбоната калия (26,6 г, 192,0 ммоль) и бромацетальдегид-диметилацеталя (32,3 мл, 175,0 мл) добавляли в ацетон (250,0 мл). Реакционную смесь перемешивали в течение 24 часов. К реакционной смеси добавляли воду и смесь три раза экстрагировали этилацетатом. Объединенный органический слой промывали насыщенным раствором бикарбоната натрия, затем насыщенным раствором хлорида натрия и сушили над сульфатом натрия. Затем смесь фильтровали и концентрировали, получая требуемый продукт (48,0 г, выход сырого продукта 99%) в виде масла:1H ЯМР (500 МГц, CDCl3) δ 7,20-7,17 (м, 1H), 6,96-6,92 (м, 2H), 6,73-6,71 (м, 1H), 4,65 (т, J=5,5 Гц, 1H), 3,79 (с, 3H), 3,68 (дд, J=9,3, 7,0 Гц, 2H), 3,55 (дд, J=9,3, 7,0 Гц, 2H), 3,14 (д, J=5,6 Гц, 2H), 1,21 (т, J=7,1 Гц, 6H).
Стадия B: Продукт со стадии A (15,0 г, 58,5 ммоль) растворяли в метиленхлориде (125 мл) и раствор добавляли к раствору комплекса диэтилового эфира и трифторида бора (7,86 мл, 62 ммоль) в метиленхлориде (900 мл) при комнатной температуре в атмосфере азота. Реакционную смесь перемешивали в течение 30 минут. Насыщенный раствор бикарбоната натрия добавляли к смеси до тех пор, пока обе фазы становились прозрачными. Органический слой дважды экстрагировали метиленхлоридом. Объединенный органический слой сушили над сульфатом натрия и концентрировали до получения масла. Масло очищали хроматографией на колонке (100% гексаны), получая требуемый 6-метоксибензотиофен (5,0 г, 52%), в виде бесцветного масла:1H ЯМР (500 МГц, CDCl3) δ 7,50 (д, J=5,6 Гц, 1H), 7,46 (д, J=8,1 Гц, 1H), 7,3 (д, J=5,6 Гц, 1H), 7,29-7,25 (м, 1H), 6,75 (д, J=7,8 Гц, 1Н), 3,96 (с, 3H).
Стадия C: Продукт со стадии B (9,1 г, 55,0 ммоль) добавляли неразбавленным к гидрохлориду пиридина (25,6 г, 22 ммоль) при 200°C в течение 2,5 часов. Смеси давали возможность остыть, затем добавляли ледяную воду и дважды экстрагировали метиленхлоридом. Объединенный экстракт сушили над сульфатом натрия, фильтровали и концентрировали до получения масла. Масло отверждалось при отстаивании, затем его растирали в гексанах, получая требуемый продукт (4,42 г, 53,4%):1H ЯМР (500 МГц, CDCl3) δ 7,67 (д, J=8,6 Гц, 1H), 7,31 (д, J=2,2 Гц, 1H), 7,26-7,22 (м, 2H), 6,91 (дд, J=8,6, 2,3 Гц, 1Н), 4,81 (с, 1H).
Стадия D: Продукт со стадии C (2,9 г, 19,0 ммоль) растворяли в метиленхлориде (40 мл) и добавляли триэтиламин (4,0 мл, 29,0 ммоль). Реакционную смесь охлаждали на ледяной бане и добавляли ангидрид трифторметансульфоновой кислоты (3,75 мл, 21,0 ммоль) и перемешивали в течение 30 минут. К смеси добавляли насыщенный раствор хлорида натрия и дважды экстрагировали метиленхлоридом. Объединенный органический экстракт сушили над сульфатом натрия, фильтровали и концентрировали до получения желтого твердого вещества (5,4 г, 99%):1H ЯМР (500 МГц, CDCl3) δ 7,86 (д, J=8,8 Гц, 1H), 7,81 (д, J=2,2 Гц, 1H), 7,56 (д, J=5,4 Гц, 1Н), 7,37 (д, J=5,5 Гц, 1H), 7,28 (дд, J=8,7, 2,3 Гц, 1H).
Стадия E: Продукт со стадии D (5,4 г, 19 ммоль), н-бутилвиниловый эфир (9,84 мл), триэтиламин (5,33 мл, 38 ммоль) и 1,3-бис(дифенилфосфино)пропан (4,73 г, 11 ммоль) в N,N-диметилформамиде дегазировали, используя аргон, и перемешивали. Затем к реакционной смеси добавляли ацетат палладия(II), затем смесь нагревали до 100°C в течение 4 часов. Охлажденную реакционную смесь фильтровали через слой диатомовой земли и концентрировали до получения желтого твердого вещества. Твердое вещество растворяли в 1 N хлористоводородной кислоте (50,0 мл) и перемешивали в течение 1 часа. Затем смесь концентрировали и очищали хроматографией на колонке (от 5:95 до 25:75 этилацетат/гексаны в качестве элюента), получая требуемый кетон (2,95 г, 87,5%), в виде желтого твердого вещества:1H ЯМР (500 МГц, CDCl3) δ 8,52-8,51 (м, 1H), 7,97 (дд, J=8,4, 1,6 Гц, 1H), 7,88 (д, J=8,4 Гц, 1Н), 7,67 (д, J=5,5 Гц, 1H), 7,41-7,39 (м, 1H), 2,68 (с, 3H).
Стадия F: Продукт со стадии E (2,9 г, 16 ммоль) растворяли в этилацетате (20 мл) и добавляли к суспензии бромида меди(II) (7,35 г, 33,0 ммоль) в хлороформе (40 мл). Реакционную смесь кипятили с обратным холодильником в течение 3 часов, затем давали возможность остыть до комнатной температуры. Смесь фильтровали через слой диатомовой земли и концентрировали до получения коричневого твердого вещества, затем очищали хроматографией на колонке (5-10% этилацетат/гексаны), получая требуемый продукт (3,71 г, 88,5%):1H ЯМР (500 МГц, CDCl3) δ 8,56-8,55 (м, 1H), 7,98 (дд, J=8,4, 1,6 Гц, 1H), 7,90 (д, J=8,4 Гц, 1H), 7,72 (д, J=5,5 Гц, 1H), 7,41 (дд, J=5,4, 0,5 Гц, 1H), 4,53 (с, 2H).
Стадия G: Продукт со стадии F (3,70 г, 14,5 ммоль), 4-гидрокси-N-метилбензиламин (2,37 г, 17,5 ммоль) и N,N-диизопропилэтиламин (2,8 мл, 16,0 ммоль) суспендировали в метиленхлориде и перемешивали в течение 24 часов. К реакционной смеси добавляли воду и экстрагировали метиленхлоридом. Органический слой промывали 1N HCl, затем насыщенным раствором хлорида натрия. Раствор сушили над сульфатом натрия, фильтровали, концентрировали в вакууме и очищали хроматографией на колонке (2-10% метанол/метиленхлорид в качестве элюента), получая требуемый продукт (2,94 г, 65%):1H ЯМР (500 МГц, CDCl3) δ 8,52 (д, J=0,5 Гц, 1H), 7,95 (дд, J=8,4, 1,5 Гц, 1H), 7,84 (д, J=8,4 Гц, 1H), 7,66 (д, J=5,4 Гц, 1H), 7,38 (д, J=5,4 Гц, 1Н), 7,20 (д, J=7,8 Гц, 1H), 6,92-6,90 (м, 2H), 6,77-6,75 (м, 1H), 3,86 (с, 2H), 3,65 (с, 2H), 2,40 (с, 3H).
Стадия H: Продукт со стадии G (2,94 г, 9,0 ммоль) растворяли в метаноле и охлаждали на бане со льдом. К реакционной смеси добавляли боргидрид натрия (0,43 г, 11,0 ммоль) и перемешивали при комнатной температуре в течение 1 часа. Метанол концентрировали в вакууме и твердое вещество перерастворяли в метиленхлориде, дважды промывали водой и дважды экстрагировали метиленхлоридом. Объединенный органический экстракт промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали до получения желтого твердого вещества (2,75 г, 93%):1H ЯМР (500 МГц, CDCl3) δ 7,91 (с, 1H), 7,77 (д, J=8,2 Гц, 1Н), 7,41 (д, J=5,4 Гц, 1Н), 7,32-7,30 (м, 2H), 7,20 (т, J=7,7 Гц, 1H), 6,87 (д, J=7,6 Гц, 1H), 6,81 (с, 1H), 6,75 (д, J=8,1 Гц, 1H), 4,88 (дд, J=3,6 Гц, 10,3 Гц, 1H), 3,71 (д, J=13,1 Гц, 1H), 3,49 (д, J=12,1 Гц, 1H), 2,67-2,56 (м, 2H), 2,34 (с, 3H); ESI МС m/z 314 [M+Н]+ .
Стадия I: Продукт со стадии H (2,75 г, 8,8 ммоль) растворяли в метиленхлориде (80 мл) и добавляли к раствору метансульфоновой кислоты (7,0 мл, 105 ммоль) в метиленхлориде (400 мл) и перемешивали в течение 10 минут. Реакционную смесь охлаждали на бане со льдом, затем гасили насыщенным раствором бикарбоната натрия и перемешивали в течение 1 часа. Органический слой промывали насыщенным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали, концентрировали и очищали хроматографией на колонке (10:90 метанол/метиленхлорид), получая требуемый 4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ол (0,98 г, 38%):1H ЯМР (500 МГц, CDCl3) δ 7,72 (т, J=8,2 Гц, 2H), 7,38 (д, J=5,4 Гц, 1H), 7,29 (д, J=5,3 Гц, 1H), 7,18 (д, J=8,1 Гц, 1H), 6,73 (д, J=8,3 Гц, 1H), 6,54 (дд, J=2,6 Гц, 8,3 Гц, 1H), 6,49 (с, 1H), 4,33 (т, J=5,9 Гц, 1H), 3,67 (д, J=14,9 Гц, 1H), 3,56 (д, J=7,4 Гц, 1H), 3,09-3,05 (м, 1H), 2,59 (т, J=8,9 Гц, 1H), 2,42 (с, 3H).
Стадия J: Продукт со стадии I (0,10 г, 0,35 ммоль) превращали в соль малеиновой кислоты растворением свободного основания в минимальном количестве этанола, добавлением одного эквивалента малеиновой кислоты в достаточном количестве метанола, чтобы полностью растворить кислоту, затем объединяя два раствора и перемешивая в течение 1 часа. Раствор концентрировали до минимального объема, затем охлаждали при -30°C до образования кристаллов, получая малеатную соль 4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ола (0,13 г, 90%, 98,7% AUC ВЭЖХ):1H ЯМР (500 МГц, CD3OD) δ 7,84 (д, J=8,2 Гц, 1H), 7,79 (с, 1H), 7,59 (д, J=5,4 Гц, 1H), 7,37 (д, J=5,4 Гц, 1H), 7,20 (д, J=8,2 Гц, 1H) 6,74-6,6,8 (м, 3H), 6,23 (с, 2H), 4,61-4,59 (м, 1Н), 4,52-4,48 (м, 2H), 3,84-3,82 (м, 1H), 3,49-3,47 (м, 1H), 3,05 (с, 3H).
Стадия K: Продукт со стадии I (0,97 г, 3,3 ммоль) растворяли в метиленхлориде (40 мл) и триэтиламине (0,69 мл, 4,9 ммоль). Реакционную смесь охлаждали на бане со льдом, добавляли ангидрид трифторметансульфоновой кислоты (0,64 мл, 3,6 ммоль) и перемешивали в течение 30 минут. К смеси добавляли насыщенный раствор хлорида натрия и дважды экстрагировали метиленхлоридом. Объединенный органический экстракт сушили над сульфатом натрия, фильтровали и концентрировали до получения желтого твердого вещества.1H ЯМР (500 МГц, CDCl3) δ 7,82 (д, J=8,2 Гц, 1Н), 7,76 (с, 1H), 7,51 (д, J=5,4 Гц, 1H), 7,35 (д, J=5,4 Гц, 1H), 7,14-7,11 (м, 3H), 7,04 (д, J=9,2 Гц, 1H), 5,30-5,02 (м, 1H), 4,70-4,65 (м, 1H), 4,30-4,27 (м, 1H), 3,91-3,79 (м, 1H), 3,26-3,25 (м, 1H), 3,03 (с, 3H).
Стадия L: Продукт со стадии K (0,54 г, 1,3 ммоль), 2- (дициклогексилфосфино)-2',4',6'-триизопропил-1,1'-бифенил (0,36 г, 0,76 ммоль), морфолин (0,22 мл, 2,5 ммоль), карбонат цезия (1,03 г, 3,2 ммоль) в толуоле перемешивали и дегазировали с использованием аргона. К реакционной смеси добавляли ацетат палладия(II) (0,04 г, 1,9 ммоль) и нагревали до 100°C в течение 24 часов. Охлажденную реакционную смесь фильтровали через слой диатомовой земли и концентрировали до получения желтого твердого вещества. Твердое вещество очищали хроматографией на колонке (5:95 метанол/метиленхлорид), получая требуемый продукт (0,23 г, 50%):1H ЯМР (500 МГц, CDCl3) δ 7,73-7,70 (м, 2H), 7,37 (д, J=5,4 Гц, 1H), 7,29 (д, J=5,4 Гц, 1H), 7,19 (дд, J=1,4 Гц, 8,2 Гц, 1H), 6,79 (д, J=8,5 Гц, 1H), 6,67-6,62 (м, 2H), 4,32 (с, 1H), 3,84 (т, J=4,7 Гц, 4H), 3,71 (д, J=17,4 Гц, 1H), 3,61 (д, J=14,8 Гц, 1H), 3,12 (т, J=4,8 Гц, 4H), 3,04 (дд, J=5,6 Гц, 11,3 Гц, 1H), 2,60 (т, J=8,5 Гц, 1H), 2,42 (с, 3H).
Полученное соединение разделяли препаративной хиральной ВЭЖХ (колонка CHIRALPAK AD, с использованием смеси 80% гептан/20% изопропанол/0,1% диэтиламин), получая (+)-энантиомер [α]25D +47,8° (c 0,069, метанол) и (-)-энантиомер [α]25D -38,8° (c 0,17, метанол).
Стадия M: (+)-Энантиомер со стадии L (0,10 г, 0,34 ммоль) превращали в соль малеиновой кислоты растворением свободного основания в минимальном количестве метанола, добавлением одного эквивалента малеиновой кислоты в достаточном количестве метанола, чтобы полностью растворить кислоту, затем объединяя два раствора и перемешивая в течение 1 часа. Раствор концентрировали до минимального объема, затем охлаждали при -30°C до образования кристаллов. Фильтрование давало малеатную соль (+)-4-бензо[b]тиофен-6-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина (86,0 мг, 64%, >99% AUC ВЭЖХ) в виде коричневого твердого вещества:1H ЯМР (500 МГц, CD3OD) δ 7,84 (д, J=8,0 Гц, 1H), 7,79 (с, 1Н), 7,59 (дд, J=1,4 Гц, 5,4 Гц, 1H), 7,37 (д, J=4,4 Гц, 1H), 7,20 (д, J=8,2 Гц, 1H), 6,90-6,88 (м, 1H), 6,81 (с, 2H), 6,25 (с, 2H), 4,63-4,52 (м, 3H), 3,87-3,80 (м, 5H), 3,59-3,57 (м, 1Н), 3,14 (с, 3H), 3,06 (с, 1H).
Такой же способ использовали для превращения (-)-энантиомера (12 мг, 0,34 ммоль) в его соль малеиновой кислоты, получая малеатную соль (-)-4-бензо[b]тиофен-6-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина (16,5 мг, >99%, >99% AUC ВЭЖХ) в виде не совсем белого твердого вещества:1H ЯМР (500 МГц, CD3OD) δ 7,84 (д, J=8,0 Гц, 1H), 7,79 (с, 1H), 7,59 (дд, J=1,4 Гц, 5,4 Гц, 1H), 7,37 (д, J=4,4 Гц, 1Н), 7,20 (д, J=8,2 Гц, 1H), 6,90-6,88 (м, 1H), 6,81 (с, 2H), 6,25 (с, 2H), 4,63-4,52 (м, 3H), 3,87-3,80 (м, 5H), 3,59-3,57 (м, 1H), 3,14 (с, 3H), 3,06 (с, 1H).
Пример 149 - Получение фумаратной соли (+/-)-4-бензо[b]тиофен-5-ил-2-метил-8-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору 1,3-дибромбензола (29,2 г, 0,123 моль) в ТГФ (300 мл) при -78°C по каплям в атмосфере азота добавляли диизопропиламид лития (83 мл, 0,148 моль, 1,8 М в ТГФ). Через 30 минут при -78°C к оранжевой суспензии по каплям добавляли N,N- диметилформамид (10,9 г, 0,148 моль). Реакционную смесь перемешивали в течение 30 минут и по каплям при 0°C добавляли разбавленную серную кислоту (~500 мл). Водный слой экстрагировали EtOAc (3×350 мл), сушили (Na2SO4) и концентрировали при пониженном давлении, получая дибромбензальдегид (32,3 г, 99%) в виде оранжевого твердого вещества, которое использовали непосредственно на следующей стадии без дополнительной очистки.1H ЯМР (CDCl3, 500 МГц) δ 10,26 (с, 1H), 7,65 (д, J=8,0 Гц, 2H), 7,21-7,24 (м, 1H).
Стадия B: Раствор дибромбензальдегида (10,00 г, 4,67 ммоль) с описанной выше стадии A и метиламина (1,40 г, 45,4 ммоль, 40% мас. в воде) в метаноле (200 мл) перемешивали в течение 15 минут при 25°C в атмосфере азота. Смесь охлаждали до 0°C и порциями добавляли боргидрид натрия (720 мг, 18,9 ммоль). Реакционную смесь перемешивали в течение 3 часов и концентрировали при пониженном давлении. Остаток обрабатывали EtOAc (3×200 мл) и водой (200 мл), сушили (Na2SO4) и концентрировали при пониженном давлении, получая метиламиносоединение (8,9 г, 84%) в виде оранжевого масла, которое использовали на следующей стадии без какой-либо дополнительной очистки:1H ЯМР (CDCl3, 500 МГц) δ 7,52-7,54 (м, 2H), 6,95-6,98 (м, 1H), 4,09 (с, 2H), 2,46 (с, 3H).
Стадия C: Суспензию 5-бромбензо[c]тиофена (4,6 г, 21,6 ммоль), 1,1'-бис(дифенилфосфино)ферроцена (2,40 г, 4,31 ммоль), ацетата таллия(I) (8,50 г, 32,4 ммоль) и 1-(трет-бутилдиметилсилилокси)метоксиэтена (4,90 г, 28,1 ммоль) в ТГФ (150 мл) продували азотом в течение 15 минут и затем добавляли ацетат палладия(II) (485 мг, 2,16 ммоль). Полученную в результате смесь кипятили с обратным холодильником в течение ночи, обрабатывали EtOAc (3×200 мл) и водой (200 мл), сушили (Na2SO4) и концентрировали при пониженном давлении, получая маслянистый остаток. Остаток очищали хроматографией на колонке с силикагелем (элюент гексаны/этилацетат 10:1), получая требуемый сложный метиловый эфир (2,63 г, 59%) в виде белого восковидного твердого вещества:1H ЯМР (500 МГц, CDCl3) δ 7,73 (д, J=8,3 Гц, 1H), 7,64 (с, 1H), 7,34 (д, J=5,4 Гц, 1H), 7,15-7,24 (м, 2H), 3,64 (с, 2H), 3,60 (с, 3H).
Стадия D: Раствор сложного метилового эфира (2,63 г, 12,8 ммоль) с описанной выше стадии C и гидроксида лития (763 мг, 31,9 ммоль) в ТГФ (60 мл) и воде (10 мл) перемешивали при температуре окружающей среды в течение ночи. Реакционную смесь концентрировали при пониженном давлении, обрабатывали EtOAc (100 мл) и водой (100 мл). Водный слой подкисляли 1N HCl (120 мл) и экстрагировали EtOAc (3×200 мл). Объединенные органические экстракты сушили (Na2SO4) и концентрировали при пониженном давлении, получая требуемую кислоту (1,90 г, 78%) в виде белого твердого вещества:1H ЯМР (CD3OD, 500 МГц) δ 7,83 (д, J=8,3 Гц, 1H), 7,77 (с, 1H), 7,54 (д, J=5,4 Гц, 1H), 7,32 (д, J=5,4 Гц, 1H), 7,27 (д, J=8,3 Гц, 1H), 3,72 (с, 2H).
Стадия E: Раствор кислоты (1,40 г, 7,29 ммоль) с описанной выше стадии D, метиламиносоединения (2,13 г, 7,65 ммоль) с описанной выше стадии B, EDC (1,82 г, 9,47 ммоль), 1-гидроксибензотриазола (1,28 г, 9,47 ммоль) и N,N-диизопропилэтиламина (2,82 г, 21,8 ммоль) в метиленхлориде (50 мл) перемешивали в течение ночи при температуре окружающей среды в атмосфере азота. Смесь обрабатывали CH2Cl2 (3×150 мл) и водой (150 мл), сушили (Na2SO4) и концентрировали при пониженном давлении, получая маслянистый остаток. Остаток очищали хроматографией на колонке с силикагелем (элюент гексаны/этилацетат 5:1), получая требуемый амид (1,68 г, 48%) в виде желтого твердого вещества:1H ЯМР (CDCl3, 500 МГц) δ 7,82 (д, J=8,2 Гц, 1H), 7,76 (д, J=7,3 Гц, 1H), 7,54-7,59 (м, 2H), 7,42-7,44 (м, 1H), 7,29-7,31 (м, 2H), 7,00-7,03 (м, 1H), 5,05 (с, 1,5H, ротамер), 4,85 (с, 0,5H, ротамер), 4,09 (с, 0,5H, ротамер), 3,88 (с, 1,5H, ротамер), 2,75 (с, 2,2H, ротамер), 2,69 (с, 0,8H, ротамер).
Стадия F: Раствор амида (1,68 г, 3,71 ммоль) с описанной выше стадии E в диоксане (8,0 мл) в атмосфере азота по каплям добавляли к дегазированному с использованием азота раствору трет-бутоксида калия (624 мг, 5,56 ммоль), бис(дибензилиденацетон)палладия(0) (213 мг, 0,371 ммоль) и 1,3-бис(дифенилфосфино)этана (221 мг, 0,556 ммоль) в диоксане (10 мл). Реакционную смесь нагревали при кипячении с обратным холодильником в течение 1 часа, обрабатывали EtOAc (3×100 мл) и водой (100 мл), сушили (Na2SO4) и концентрировали при пониженном давлении, получая маслянистый остаток. Остаток очищали хроматографией на колонке с силикагелем (элюент гексаны/этилацетат 5:1), получая требуемый лактам (365 мг, 26%) в виде желтой пены:1H ЯМР (CDCl3, 500 МГц) δ 7,79 (д, J=8,3 Гц, 1H), 7,54 (д, J=7,7 Гц, 1H), 7,49-7,51 (м, 1H), 7,41-7,43 (м, 1H), 7,24-7,26 (м, 1H), 7,11-7,17 (м, 3H), 4,98 (с, 1H), 4,58 (с, 2H), 3,15 (с, 3H); ESI-МС m/z 372, 374 [M+H]+.
Стадия G: Комплекс боран-диметилсульфид (5,80 мл, 5,80 ммоль, 1,0 М в ТГФ) добавляли по каплям к раствору лактама (362 мг, 0,972 ммоль) с описанной выше стадии F в ТГФ (8,0 мл). Реакционную смесь нагревали при кипячении с обратным холодильником в течение 30 минут в атмосфере азота. Смесь концентрировали при пониженном давлении и к остатку добавляли карбонат калия (1,30 г, 9,72 ммоль), диоксан (8,0 мл) и воду (4,0 мл). Смесь кипятили с обратным холодильником в течение 4 часов, обрабатывали EtOAc (3×40 мл) и водой (50 мл), сушили (Na2SO4) и концентрировали при пониженном давлении, получая требуемый тетрагидроизохинолин (346 мг, 100% выход сырого продукта) в виде масла:1H ЯМР (CDCl3, 500 МГц) δ 7,78 (д, J=8,3 Гц, 1H), 7,63 (с, 1H), 7,43 (д, J=5,2 Гц, 1H), 7,39 (д, J=7,8 Гц, 1H), 7,26-7,28 (м, 1H), 7,14 (д, J=8,3 Гц, 1H), 6,92-6,95 (м, 1H), 6,85 (д, J=7,7 Гц, 1H), 4,38-4,43 (м, 1H), 3,83 (д, J=15,8 Гц, 1H), 3,56 (д, J=15,9 Гц, 1H), 3,04 (дд, J=14,1 Гц, 6,6 Гц, 1H), 2,64 (дд, J=11,4 Гц, 8,4 Гц, 1H), 2,49 (с, 3H); ESI-МС m/z 358, 360 [M+H]+.
Стадия H: Смесь тетрагидроизохинолина (260 мг, 0,725 ммоль) с описанной выше стадии G, морфолина (126 мг, 1,45 ммоль), карбоната цезия (710 мг, 2,18 ммоль) и 2-(дициклогексилфосфино)-2',4',6'-триизопропил-1,1'-бифенила (104 мг, 0,217 ммоль) в м-ксилене (10 мл) дегазировали, используя поток азота, в течение 10 минут и добавляли ацетат палладия(II) (16 мг, 0,0725 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 6 часов и смесь фильтровали через небольшой слой целита, промывая метанолом. Фильтрат концентрировали при пониженном давлении, получая коричневый остаток, который очищали хроматографией на колонке с силикагелем (элюент CH2Cl2/MeOH 98:2), получая целевой морфолин (30 мг, 11%) в виде желтого твердого вещества: ESI-МС m/z 365 [M+H]+.
Стадия I: К раствору целевого морфолина (52 мг, 0,143 ммоль) с описанной выше стадии H в метаноле (3 мл) добавляли фумаровую кислоту (16,5 мг, 0,143 ммоль). Раствор концентрировали и сушили в вакууме в течение ночи, получая фумаратную соль (+/-)-4-бензо[b]тиофен-5-ил-2-метил-8-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина (AUC ВЭЖХ = 97,4%) в виде желтого твердого вещества: т.пл. 148-153°C;1H ЯМР (CD3OD, 500 МГц) δ 7,88 (д, J=8,4 Гц, 1Н), 7,72 (с, 1H), 7,59 (д, J=5,4 Гц, 1H), 7,33 (д, J=5,4 Гц, 1H), 7,12-7,23 (м, 3H), 6,71 (д, J=7,8 Гц, 1H), 6,95 (с, 2H), 4,66-4,72 (м, 2H), 4,24 (д, J=15,3 Гц, 1H), 3,83-3,93 (м, 4H), 3,70-3,74 (м, 1H), 3,32-3,34 (м, 1H), 3,01-3,06 (м, 2H), 2,97 (с, 3H), 2,81-2,85 (м, 2H); ESI-МС m/z 365 [M+H]+.
Пример 150 - Получение фумаратной соли (+/-)-2-(4-бензо[b]тиофен-5-ил-3,4-дигидро-1H-изохинолин-2-ил)этанола
Стадия A: Смесь 4-бензо[b]тиофен-5-ил-1,2,3,4-тетрагидроизохинолина (40 мг, 0,15 ммоль, получение которого описано в примере 20), 2-бромэтанола (23 мг, 0,18 ммоль) и карбоната цезия (98 мг, 0,30 ммоль) в ДМФА (4 мл) перемешивали при комнатной температуре в течение 12 часов. Смесь разбавляли этилацетатом, промывали 5% раствором LiOH, затем насыщенным раствором соли и органический слой сушили над сульфатом магния и концентрировали. Остаток очищали хроматографией на колонке (SiO2, 12 г, от 85% до 0% гексан/этилацетат), получая требуемый тетрагидроизохинолин (13 мг, 28%). Полученный продукт растворяли в метаноле и обрабатывали раствором фумаровой кислоты в метаноле (5 мл) при 0°C. Раствор перемешивали при комнатной температуре в течение 12 часов, затем концентрировали и твердое вещество промывали диэтиловым эфиром, получая фумаратную соль (+/-)-2-(4-бензо[b]тиофен-5-ил-3,4-дигидро-1H-изохинолин-2-ил)этанола (18 мг, 99%, 96,0% AUC ВЭЖХ):1H ЯМР (CD3OD, 500 МГц) δ 7,92 (д, J=8,4 Гц, 1H), 7,79 (д, J=0,9 Гц, 1H), 7,62 (д, J=5,4 Гц, 1H), 7,36-7,30 (м, 3H), 7,25-7,20 (м, 2H), 6,92 (д, J=7,8 Гц, 1H), 6,74 (с, 2H), 4,77-4,74 (м, 1H), 4,69 (с, 2H), 4,01-3,97 (м, 3H), 3,65-3,58 (м, 1H), 3,48-3,46 (м, 2H), ESI МС m/z=310 [M+H].
Пример 151 - Получение (+)-4-бензо[b]тиофен-5-ил-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолина
Смесь неочищенного (+)-энантиомера сложного эфира бороновой кислоты (200 мг, 0,89 ммоль), полученного согласно стадии B примера 97, 2-хлорпиримидина (100 мг, 0,90 ммоль), Cs2CO3 (869 мг, 2,67 ммоль), ДМФА (10 мл) и воды (1,5 мл) продували аргоном из баллона. Добавляли PdCl2(dppf) (44 мг, 0,05 ммоль) и смесь нагревали при 90°C в течение 5 часов. После охлаждения реакционную смесь распределяли между EtOAc (150 мл) и водой (150 мл). Органический слой промывали водой (2×100 мл), насыщенным раствором соли, сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали хроматографией на колонке с силикагелем, используя картридж ISCO 40 г (элюент: от 100:0 до 95:5 EtOAc/MeOH), получая (+)-4-бензо[b]тиофен-5-ил-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин (52 мг, 16%) в виде не совсем белого твердого вещества:1H ЯМР (300 МГц, CDCl3) δ 8,77 (д, J=4,8 Гц, 2H), 8,22 (с, 1H), 8,13 (дд, J=8,2, 1,2 Гц, 1Н), 7,80 (д, J=8,3 Гц, 1H), 7,67 (с, 1H), 7,42 (д, J=5,4 Гц, 1H), 7,27 (д, J=5,3 Гц, 1H), 7,13-7,22 (м, 2H), 7,03 (д, J=8,2 Гц, 1H), 4,47 (м, 1H), 3,91 (д, J=15,0 Гц, 1H), 3,74 (д, J=15,0 Гц, 1H), 3,13 (дд, J=11,5, 5,7 Гц, 1H), 2,67 (дд, J=11,4, 8,7 Гц, 1H), 2,47 (с, 3H); ESI-МС m/z 358 [M+H]+.
Пример 152 - Получение (+/-)-4-бензо[b]тиофен-5-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору 4-бромфенилуксусной кислоты (50 г, 233 ммоль) в SOCl2 (170 мл, 2,32 моль) добавляли ДМФА (2 мл, 26 ммоль). После перемешивания при указанной температуре в течение 1 часа избыток SOCl2 удаляли при пониженном давлении. Остаток растворяли в CH2Cl2 (300 мл) и полученный в результате раствор по каплям добавляли к охлажденному (0°C) раствору CH3NH2 (90 мл, 40% мас. в H2O, 1,03 моль) в ТГФ (200 мл). После завершения добавления раствор дополнительно перемешивали при 0°C в течение 15 мин. К реакционному раствору добавляли воду (400 мл) и органический слой отделяли. Водный слой экстрагировали CH2Cl2 (200 мл). Органические слои объединяли, промывали насыщенным раствором соли (500 мл), сушили и концентрировали, получая 4-бромфенил-N-метилацетамид (54 г, количественный выход) в виде белого твердого вещества:1H ЯМР (500 МГц, CDCl3) δ 7,48 (д, J=8,9 Гц, 2H), 7,15 (д, J=8,9 Гц, 2H), 3,51 (с, 2H), 2,77 (с, 3H); ESI-МС m/z 228 [M+H]+.
Стадия B: 4-Бромфенил-N-метилацетамид, полученный на стадии A (3,8 г, 16,7 ммоль), параформальдегид (550 мг, 18,4 ммоль) и H4P2O7 (30 г) смешивали и нагревали при 150-160°C в течение 1 часа. Реакционную смесь вливали в ледяную воду (200 мл). Продукт экстрагировали CH2Cl2 (2×100 мл). Органический слой промывали NaHCO3 (100 мл), насыщенным раствором соли (100 мл), сушили и концентрировали, получая требуемый продукт (3,4 г, 85%) в виде желтого твердого вещества:1H ЯМР (500 МГц, CDCl3) δ 7,38 (д, J=8,1 Гц, 1H), 7,32 (с, 1H), 7,03 (д, J=8,1 Гц, 1Н), 4,46 (с, 2H), 3,56 (с, 2H), 3,10 (с, 3H); ESI-МС m/z 240 [M+H]+.
Стадия C: К раствору 5-бромбензотиофена (11,6 г, 55,5 ммоль) в толуоле (150 мл) добавляли Pd(OAc)2. Смесь продували аргоном; к смеси добавляли 2,8,9-триизобутил-2,5,8,9-тетрааза-1-фосфабицикло[3,3,3]ундекан (2,6 г, 7,6 ммоль). После перемешивания при комнатной температуре в течение 2 мин к смеси добавляли NaO-t-Bu (5,4 г, 56 ммоль), затем продукт, полученный на стадии B (9,0 г, 37,5 ммоль). Полученную в результате смесь нагревали при 70°C в течение 30 мин. После охлаждения до комнатной температуры реакционную смесь разбавляли CH2Cl2 (200 мл), промывали насыщенным водным раствором хлорида аммония (200 мл) и насыщенным раствором соли (200 мл), сушили и концентрировали. Остаток очищали хроматографией среднего давления (элюент: EtOAc/CH2Cl2 1:4), получая требуемый продукт (8,84 г, 63%) в виде желтого твердого вещества:1H ЯМР (300 МГц, CDCl3) δ 7,78 (д, J=8,4 Гц, 1H), 7,47-7,41 (м, 4H), 7,23 (д, J=5,6 Гц, 1H), 4,93 (с, 1H), 4,64 (д, J=16,2 Гц, 1Н), 4,31 (д, J=16,2 Гц, 1H), 3,09 (с, 3H); ESI-МС m/z 372 [M+H]+.
Стадия D: К охлажденному (0°C) раствору продукта, полученного на стадии C (3,13 г, 8,4 ммоль) в ТГФ (50 мл), добавляли BH3-Me2S в ТГФ (7,7 мл, 2,0 М, 15,4 ммоль) в течение 15-минутного периода. Полученный в результате раствор перемешивали при температуре от 0°C до комнатной температуры в течение 20 мин. Затем раствор нагревали при 50°C в течение 20 мин. После охлаждения до комнатной температуры растворитель удаляли. К остатку добавляли диоксан (45 мл) и HCl (6 N, 15 мл). Полученный в результате раствор кипятили с обратным холодильником в течение 30 мин. После охлаждения до комнатной температуры раствор нейтрализовали 2 N NaOH. Продукт экстрагировали CH2Cl2 (2×100 мл). Органический слой промывали насыщенным раствором соли (100 мл), сушили и концентрировали. Остаток очищали хроматографией среднего давления (элюент: MeOH/EtOAc/гексан 1:9:40), получая требуемый продукт (2,48 г, 82%) в виде белой пены:1H ЯМР (500 МГц, ДМСО-d6) δ 7,89 (д, J=8,3 Гц, 1H), 7,72 (д, J=5,4 Гц, 1H), 7,67 (с, 1H), 7,40-7,38 (м, 2H), 7,24-7,19 (м, 2H), 6,76 (д, J=8,3 Гц, 1H), 4,30 (т, J=6,3 Гц, 1H), 3,62 (д, J=15,0 Гц, 1H), 3,58 (д, J=15,0 Гц, 1Н), 2,92 (м, 1H), 2,62 (м, 1H), 2,31 (с, 3H); ESI-МС m/z 358 [M+H]+.
Стадия E: Продукт, полученный на стадии D (8,0 г, 22,4 ммоль), BINAP (1,4 г, 2,24 ммоль), KO-t-Bu (5,5 г, 49 ммоль), морфолин (7,8 г, 90 ммоль) и толуол смешивали и смесь продували аргоном. К смеси добавляли Pd(OAc)2 (502 мг, 2,24 ммоль). Полученную в результате смесь нагревали при кипячении с обратным холодильником в течение 0,5 часа. После охлаждения до комнатной температуры смесь разбавляли CH2Cl2 (200 мл), промывали насыщенным водным раствором хлорида аммония (200 мл), насыщенным раствором соли (200 мл), сушили и концентрировали. Остаток очищали хроматографией среднего давления (элюент: MeOH/EtOAc 1:9), получая требуемый продукт (5,2 г, 64%) в виде бледной пены:1H ЯМР (300 МГц, CDCl3) δ 7,78 (д, J=8,3 Гц, 1H), 7,65 (с, 1H), 7,41 (д, J=5,3 Гц, 1H), 7,18 (д, J=8,3 Гц, 1H), 6,79 (д, J=8,4 Гц, 1Н), 6,68-6,63 (м, 2H), 4,34-4,30 (м, 1H), 3,86-3,83 (м, 4H), 3,73 (д, J=14,6 Гц, 1Н), 3,61 (д, J=14,6 Гц, 1Н), 3,14-3,02 (м, 5H), 2,62-2,56 (м, 1H), 2,42 (с, 3H); ESI-MS 365 [M+H]+.
Также получали (+/-)-4-бензо[b]тиофен-5-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин, используя альтернативную последовательность реакций, которая описана для стадий F-H ниже:
Стадия F: К раствору продукта, полученного на стадии B (10 г, 41,7 ммоль), добавляли L-пролин (1,9 г, 16,7 ммоль), морфолин (28,6 г, 336 ммоль), CuI (1,57 г, 83,4 ммоль) и K3PO4 (17,5 г, 83,4 ммоль). Смесь нагревали при 100°C в течение 40 часов. После охлаждения до комнатной температуры смесь промывали насыщенным водным раствором хлорида аммония (300 мл), насыщенным раствором соли (300 мл), сушили и концентрировали. Остаток очищали на флэш-колонке (элюент: MeOH/EtOAc 1:19), получая требуемый продукт (6,5 г, 63%) в виде желтого твердого вещества:1H ЯМР (300 МГц, CDCl3) δ 7,06 (д, J=8,4 Гц, 1H), 6,84 (д, J=8,4 Гц, 1H), 6,68 (с, 1H), 4,46 (с, 2H), 3,88-3,85 (м, 4H), 3,55 (с, 2H), 3,14-3,10 (м, 7H); ESI-МС m/z 247 [M+H]+.
Стадия G: Продукт, полученный на стадии F (5,5 г, 22 ммоль), 5-бромбензотиофен (4,76 г, 22 ммоль) и BINAP (684 мг, 2,2 ммоль) смешивали с толуолом (50 мл). Смесь продували аргоном. К смеси добавляли Pd(OAc)2 (264 мг, 1,1 ммоль), затем NaO-t-Bu (3,17 г, 33 ммоль). Затем смесь нагревали при 70°C в течение 1,5 часа. После охлаждения до комнатной температуры смесь разбавляли CH2Cl2 (200 мл) и промывали насыщенным водным раствором хлорида аммония (200 мл), насыщенным раствором соли (100 мл), сушили и концентрировали. Остаток очищали хроматографией среднего давления (элюент: EtOAc), получая требуемый продукт (6,5 г, 79%) в виде желтого твердого вещества:1H ЯМР (300 МГц, CDCl3) δ 7,77 (д, J=8,4 Гц, 1H), 7,50 (с, 1H), 7,39 (д, J=5,4 Гц, 1H), 7,23-7,19 (м, 2H), 7,05 (д, J=8,4 Гц, 1H), 6,90-6,85 (м, 1H), 6,77 (с, 1H), 4,91 (с, 1H), 4,64 (д, J=15,9 Гц, 1H), 4,28 (д, J=15,9, 1H), 3,90-3,87 (м, 4H), 3,20-3,17 (м, 4H), 3,08 (с, 3H); ESI-МС m/z 379 [M+H]+.
Стадия H: К раствору продукта, полученного на стадии B (11,5 г, 30,4 ммоль), в ТГФ (200 мл), нагретому до температуры кипячения с обратным холодильником, по каплям добавляли BH3-Me2S в CH2Cl2 (61 мл, 1,0 М, 61 ммоль). Раствор кипятили с обратным холодильником в течение 1 часа. После охлаждения до комнатной температуры растворитель удаляли. К остатку добавляли MeOH (250 мл) и тетраметилэтилендиамин (35 мл). Смесь кипятили с обратным холодильником в течение 4,5 часа. После охлаждения до комнатной температуры растворитель удаляли. Остаток очищали хроматографией среднего давления (элюент: MeOH/EtOAc 1:9), получая требуемый продукт (7,0 г, 62%) в виде белого твердого вещества:1H ЯМР (500 МГц, CDCl3) δ 7,78 (д, J=8,3 Гц, 1H), 7,65 (с, 1H), 7,41 (д, J=5,3 Гц, 1H), 7,18 (д, J=8,3 Гц, 1H), 6,79 (д, J=8,4 Гц, 1H), 6,68-6,63 (м, 2H), 4,34-4,30 (м, 1H), 3,86-3,83 (м, 4H), 3,73 (д, J=14,6 Гц, 1Н), 3,61 (д, J=14,6 Гц, 1H), 3,14-3,02 (м, 5H), 2,62-2,56 (м, 1H), 2,42 (с, 3H); ESI-MS 365 [M+H]+.
Также получали 4-бензо[b]тиофен-5-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин, используя альтернативную последовательность реакций, которая описана для стадий I-M ниже:
Стадии I и J: 7-Хлор-2-метил-l,4-дигидро-2H-изохинолин-3-он получали, используя 4-хлорфенилуксусную кислоту (69,22 г, 406,5 ммоль), в две стадии с общим выходом 71% в виде желтого твердого вещества, следуя способу, сходному со способом, описанным для стадий A и B:1H ЯМР (500 МГц, CDCl3) δ 7,28-7,22 (м, 1H), 7,16 (с, 1H), 7,08 (д, J=8,2 Гц, 1Н), 4,46 (с, 2H), 3,57 (с, 2H), 3,10 (с, 3H).
Стадия K: 4-Бензо[b]тиофен-5-ил-7-хлор-2-метил-1,4-дигидро-2H-изохинолин-3-он получали, используя 7-хлор-2-метил-1,4-дигидро-2H-изохинолин-3-он, полученный на стадии J, с выходом 63% в виде желтой пены, следуя способу, сходному со способом, описанным на стадии C:1H ЯМР (500 МГц, CDCl3) δ 7,78 (д, J=8,4 Гц, 1H), 7,48-7,45 (м, 1H), 7,42 (д, J=5,4 Гц, 1H), 7,30-7,28 (м, 2H), 7,23 (дд, J=0,5, 5,4 Гц, 1Н), 7,17 (дд, J=1,7, 8,4 Гц, 1H), 7,09 (д, J=8,8 Гц, 1H), 4,94 (с, 1Н), 4,63 (д, J=19,6 Гц, 1H), 4,30 (д, J=16,0 Гц, 1H), 3,09 (с, 3H).
Стадия L: 4-Бензо[b]тиофен-5-ил-7-хлор-2-метил-1,2,3,4-тетрагидроизохинолин получали, используя 4-бензо[b]тиофен-5-ил-7-хлор-2-метил-1,4-дигидро-2H-изохинолин-3-он, полученный на стадии K, с выходом 97% в виде прозрачного масла, следуя способу, сходному со способом, который описан для стадии D:1H ЯМР (500 МГц, CDCl3) δ 7,78 (д, J=8,4 Гц, 1H), 7,63 (д, J=1,2 Гц, 1H), 7,43 (д, J=5,5 Гц, 1H), 7,27 (д, J=5,4 Гц, 1H), 7,14 (дд, J=8,3, 1,5 Гц, 1H), 7,10 (д, J=1,9 Гц, 1H), 7,02 (дд, J=8,3, 2,1 Гц, 1H), 6,82 (д, J=8,3 Гц, 1H), 4,36-4,31 (м, 1H), 3,73 (д, J=15,1 Гц, 1H), 3,61 (д, J=15,1 Гц, 1H), 3,09-3,02 (м, 1H), 2,61 (дд, J=11,5, 8,5 Гц, 1H), 2,43 (с, 3H).
Стадия M: (+/-)-4-бензо[b]тиофен-5-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин получали, используя 4-бензо[b]тиофен-5-ил-7-хлор-2-метил-1,2,3,4-тетрагидроизохинолин, полученный на стадии L, с выходом 72% в виде прозрачного масла, следуя способу, сходному со способом, описанным для стадии E:1H ЯМР (300 МГц, CDCl3) δ 7,78 (д, J=8,3 Гц, 1H), 7,65 (с, 1H), 7,41 (д, J=5,3 Гц, 1H), 7,27-7,25 (м, 1H), 7,18 (д, J=8,3 Гц, 1H), 6,79 (д, J=8,4 Гц, 1H), 6,68-6,63 (м, 2H), 4,34-4,30 (м, 1H), 3,86-3,83 (м, 4H), 3,73 (д, J=14,6 Гц, 1Н), 3,61 (д, J=14,6 Гц, 1Н), 3,14-3,02 (м, 5H), 2,62-2,56 (м, 1H), 2,42 (с, 3H); ESI-МС m/z 365 [M+Н]+.
Пример 153 - Получение соли (+)-ди-пара-толуоил-D-винной кислоты и (+)-4-бензо[b]тиофен-5-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина
(+-)-4-бензо[b]тиофен-5-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин, полученный в примере 152 (0,302 г, 8,30 ммоль) растворяли в ацетоне (5,0 мл, 60 мг/мл) при 50°C с перемешиванием. После растворения добавляли (+)-ди-пара-толуоил-D-винную кислоту (0,320 г, 1 эквивалент) в виде твердого вещества. Температуру поддерживали на уровне 50°C в течение 10 минут, за это время начинал образовываться преципитат. Реакционную смесь охлаждали до комнатной температуры со скоростью 20°C/ч. Реакционной смеси давали возможность перемешиваться в течение ночи. Твердые вещества отфильтровывали и анализировали хиральной ВЭЖХ. Хиральная ВЭЖХ для твердых веществ показала выход 90,52%. Реакция давала 0,247 г (39,7%) после сушки в вакууме при 55°C. Полученный продукт перекристаллизовывали из ТГФ (11 мл) при 60°C. Твердые вещества отфильтровывали, сушили в вакууме при 55°C и анализировали хиральной ВЭЖХ. Кристаллизация давала 0,158 г вещества (общий выход 25,40% из (+-)-4-бензо[b]тиофен-5-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина. Выход в % для данного продукта повышали до 98,97%.
Пример 154 - Получение (+/-)-4-бензо[b]тиофен-5-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина
Стадия A: К раствору продукта, полученного на стадии D примера 152 (4,31 г, 12,07 ммоль), бис-пинаколатодибора (4,68 г, 18,42 ммоль) и ацетата калия (4,93 г, 50,23 ммоль) в ДМСО (87 мл) добавляли PdCl2dppf (1,1 г, 1,34 ммоль). Реакционную смесь нагревали до 70°C в течение ночи, затем охлаждали до комнатной температуры. Затем реакционную смесь разбавляли EtOAc (500 мл), полученную в результате смесь промывали водой (3×20 мл). Органическую фазу сушили (Na2SO4) и концентрировали досуха при пониженном давлении. Остаток очищали с использованием хроматографии среднего давления (элюент: EtOAc/гексаны от 95:5 до EtOAc 100 до EtOAc/MeOH 99:1), получая требуемый сырой продукт в виде черного масла (~11 г):1H ЯМР (300 МГц, CDCl3) δ 7,78 (д, J=8,3 Гц, 1Н), 7,64 (с, 1H), 7,57 (с, 1H), 7,49 (д, J=7,8 Гц, 1H), 7,41 (д, J=5,4 Гц, 1H), 7,26 (с, 1H), 7,15 (д, J=8,2 Гц, 1H), 6,90 (д, J=7,9 Гц, 1H), 4,42-4,40 (м, 1Н), 3,80 (д, J=15,9 Гц, 1H), 3,66 (д, J=15,9 Гц, 1H), 3,10-3,08 (м, 1H), 2,62 (т, J=11,4 Гц, 1H), 2,43 (с, 3H), 1,33 (с, 12H); ESI-МС m/z 406 [M+H]+.
Стадия B: К раствору неочищенного продукта, полученного на стадии A, Na2CO3 (4,45 г, 41,99 ммоль), 3,6-дихлорпиридазина (3,75 г, 25,17 ммоль) в смеси ДМФА (210 мл) и H2O (42 мл) добавляли PdCl2dppf (1,1 г, 1,5 ммоль). Реакционную смесь нагревали до 65°C в течение ночи. Охлажденную до комнатной температуры реакционную смесь разбавляли H2O (100 мл). Полученную в результате смесь экстрагировали CH2Cl2 (3×300 мл). Органические слои объединяли, затем концентрировали досуха при пониженном давлении, получая коричневое твердое вещество, которое растирали в смеси EtOAc/MeOH (9:1). Твердое вещество выделяли фильтрованием, затем последовательно промывали EtOAc, EtOAc/CH2Cl2 (9:1) и диэтиловым эфиром, сушили, получая 4-бензо[b]тиофен-5-ил-7-(6-хлорпиридазин-3-ил)-2-метил-1,2,3,4-тетрагидроизохинолин (2,82 г, 60% для 2 стадий) в виде не совсем белого твердого вещества. Фильтрат концентрировали досуха при пониженном давлении и очищали, используя хроматографию среднего давления (элюент: от EtOAc 100 до EtOAc/MeOH 98:2), получая вторую порцию требуемого продукта (1,20 г, 25% для 2 стадий) в виде коричневого твердого вещества:1H ЯМР (500 МГц, ДМСО-d6) δ 8,28 (д, J=9,1 Гц, 1H), 7,99 (д, J=9,1 Гц, 1H), 7,96 (с, 1H), 7,91 (д, J=8,3 Гц, 1H), (дд, J=8,8, 1,4 Гц, 1H), 7,74-7,71 (м, 2H), 7,40 (д, J=5,4 Гц, 1H), 7,27 (дд, J=8,4, 1,1 Гц, 1H), 7,00 (д, J=8,2 Гц, 1H), 4,42 (т, J=5,9 Гц, 1H), 3,77 (д, J=15,2 Гц, 1H), 3,72 (д, J=15,2 Гц, 1H), 2,98 (дд, J=11,4, 5,5 Гц, 1Н), 2,71-2,65 (м, 1H), 2,36 (с, 3H).
Стадия C: К раствору продукта, полученного на стадии B (2,02 г, 5,15 ммоль), и гидрата гидразина (8 мл) в MeOH (100 мл) добавляли 10% Pd/C (0,25 г). Реакционную смесь нагревали при кипячении с обратным холодильником 8 часов, затем реакционную смесь фильтровали через небольшой слой целита. Фильтрат концентрировали досуха при пониженном давлении. Остаток разбавляли CH2Cl2 (60 мл), затем промывали H2O (1×40 мл). Водную фазу экстрагировали дополнительным количеством CH2Cl2 (3×20 мл). Объединенные органические фазы сушили (Na2SO4) и концентрировали досуха при пониженном давлении. Остаток очищали, используя хроматографию среднего давления (элюент: EtOAc/MeOH 9:1), получая (+/-)-4-бензо[b]тиофен-5-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин (1,3 г, 71%, >99% AUC ВЭЖХ) в виде белого твердого вещества:1H ЯМР (500 МГц, CDCl3) δ 9,14 (дд, J=1,6, 4,9 Гц, 1H), 7,94 (д, J=1,5 Гц, 1H), 7,84-7,79 (м, 2H), 7,71-7,67 (м, 2H), 7,51 (дд, J=8,6, 4,9 Гц, 1H), 7,44 (д, J=5,4 Гц, 1H), 7,28-7,26 (м, 1H), 7,20 (дд, J=8,3, 1,6 Гц, 1H), 7,06 (д, J=8,1 Гц, 1H), 4,46 (м, 1H), 3,90 (д, J=14,6 Гц, 1H), 3,76 (д, J=14,6 Гц, 1H), 3,15-3,10 (м, 1H), 2,71-2,65 (м, 1H), 2,48 (с, 3H); ESI-МС m/z 358 [M+H]+.
Пример 155 - Первичный анализ связывания
Чтобы оценить относительную аффинность различных соединений по отношению к переносчикам NE, DA и 5HT, создавали линии клеток HEK293E, экспрессирующих каждый из трех переносчиков человека. кДНК, содержащие полные кодирующие области каждого переносчика, амплифицировали с помощью ПЦР из библиотек головного мозга человека. кДНК, находящиеся в векторах pCRII, секвенировали, чтобы подтвердить их идентичность, и затем субклонировали в основанной на вирусе Эпштейна-Барра экспрессирующей плазмиде (Shen et al., Gene 156: 235-239 (1995), публикация включена в данное описание в виде ссылки в полном объеме). Указанную плазмиду, содержащую кодирующую последовательность для одного из человеческих переносчиков, трансфицировали в клетки HEK93E. Успешную трансфекцию подтверждали по способности известных блокаторов обратного захвата ингибировать захват меченных тритием NE, DA или 5HT.
Для анализа связывания клетки гомогенизировали, центрифугировали и затем ресуспендировали в буфере для инкубации (5 мМ трис, 120 мМ NaCl, 5 мМ KCl, pH 7,4). Затем добавляли соответствующий радиолиганд. Для связывания NET добавляли [3H]-низоксетин (86,0 кюри/ммоль, NEN/DuPont) до конечной концентрации примерно 5 нМ. Для связывания DAT добавляли [3H] WIN 35428 (84,5 кюри/ммоль) в концентрации 15 нМ. Для связывания 5HTT добавляли [3H]-цитолапрам (85,0 кюри/ммоль) в концентрации 1 нМ. Затем различные концентрации (от 10-5 до 10-11 М) представляющего интерес соединения инкубировали в течение 1 часа в 96-луночном планшете. После инкубации планшеты помещали в устройство для сбора и быстро 4 раза промывали (50 мМ трис, 0,9% NaCl, pH 7,4), при этом клеточные мембраны, содержащие связанную радиоактивную метку, улавливали на фильтрах Whatman GF/B. К фильтрам добавляли смесь для сцинтилляции и затем считали в счетчике Packard TopCount. Аффинности связывания представляющих интерес соединений определяли по кривой нелинейной регрессии, используя компьютерную программу GraphPad Prism 2.01. Неспецифичное связывание определяли с помощью замены 10 микромолями мазиндола.
Пример 156 - Функциональное ингибирование in vitro захвата нейромедиаторов в синаптосомах крыс
Функциональную способность соединений ингибировать захват нейромедиаторов оценивали, измеряя ингибирование захвата [3H]-норадреналина, [3H]-серотонина и [3H]-допамина синаптосомами головного мозга крыс. Описание применяемых способов приведено ниже:
Захват [3H]-серотонина в синаптосомы головного мозга крыс
Приготовление синаптосом
Кору лобных долей гомогенизировали в ледяной 0,32 М сахарозе (1:20 мас./об.), используя свободно притертый (зазор: 0,5 мм), изготовленный из стекла/тефлона гомогенизатор с электроприводом (12 ударов, 800 об/мин). Ядра и обломки клеток удаляли центрифугированием при 1500×g в течение 10 минут. Полученный в результате надосадок центрифугировали при 18000×g в течение 10 минут. Полученный неочищенный осадок синаптосом затем ресуспендировали в ледяном буфере Кребса-Хензелайта, pH 7,4 при 25°C (эквивалент 8,3 мг сырой массы ткани/мл). Все центрифугирования осуществляли при 4°C.
Анализ захвата
Неочищенные синаптосомы коры лобных долей инкубировали на водяной бане на качалке в течение 15 минут при 37°C. Затем аликвоты (150 мкл; эквивалент 1,25 мг сырой массы ткани/пробирку) добавляли в пробирки, содержащие 275 мкл буфера Кребса-Хензелайта и 50 мкл буфера (общий захват) или 50 мкл раствора лекарственного средства в 10 концентрациях в диапазоне от 10-11 до 10-4 М или 50 мкл зимелдина (10-5 М, неспецифичный захват). Захват инициировали добавлением 25 мкл свежеприготовленного [3H]5-HT (2 нМ) с последующим встряхиванием и продолжали в течение 5 минут при 37°C на водяной бане на качалке.
Захват останавливали фильтрованием в вакууме через фильтры Skatron 11734, используя устройство для сбора клеток Skatron. Фильтры быстро промывали ледяным физиологическим раствором (установки для промывки 9,9,0). Диски из фильтровальной бумаги для счета помещали в пластиковые флаконы для сцинтилляции и в четыре флакона пипеткой вносили по 25 мкл [3H]5-HT для точного определения концентрации, добавляемой в каждую пробирку. Радиоактивность (распады в минуту) определяли счетом жидкостной сцинтилляции (1 мл сцинтиллятора Packard MV Gold).
Захват [3H]-норадреналина в синаптосомы головного мозга крыс
Приготовление синаптосом
Кору лобных долей гомогенизировали в ледяной 0,32 М сахарозе (1:10 мас./об.), используя свободно притертый (зазор: 0,5 мм), изготовленный из стекла/тефлона гомогенизатор с электроприводом (12 ударов, 800 об/мин). Ядра и обломки клеток удаляли центрифугированием при 1500×g в течение 10 минут. Затем надосадок центрифугировали при 18000×g в течение 10 минут. Полученный неочищенный осадок синаптосом затем ресуспендировали в ледяном физиологическом буфере Кребса, обработанном смесью газов 95% O2/5% CO2 (эквивалент 16,7 мг сырой массы ткани/мл). Все центрифугирования осуществляли при 4°C.
Анализ захвата
Неочищенные синаптосомы коры лобных долей инкубировали на водяной бане на качалке в течение 15 минут при 37°C. Затем аликвоты (150 мкл; эквивалент 2,5 мг сырой массы ткани/пробирку) добавляли в пробирки, содержащие 275 мкл физиологического буфера Кребса и 50 мкл буфера (общий захват) или 50 мкл раствора лекарственного средства в 10 концентрациях в диапазоне от 10-11 до 10-4 М или 50 мкл дезипрамина (10-5 М, неспецифичный захват). Захват инициировали добавлением 25 мкл свежеприготовленного [3H]-норадреналина (10 нМ) с последующим встряхиванием и продолжали в течение 5 минут при 37°C на водяной бане на качалке.
Захват останавливали фильтрованием в вакууме через фильтры Skatron 11734, используя устройство для сбора клеток Skatron. Фильтры быстро промывали ледяным физиологическим раствором (установки для промывки 9,9,0). Диски из фильтровальной бумаги для счета помещали в пластиковые флаконы для сцинтилляции и в четыре флакона пипеткой вносили по 25 мкл [3H]-норадреналина для точного определения концентрации, добавляемой в каждую пробирку. Радиоактивность (распады в минуту) определяли счетом жидкостной сцинтилляции (1 мл сцинтиллятора Packard MV Gold).
Захват [3H]-допамина в синаптосомы головного мозга крыс
Приготовление синаптосом
Полосатые тела гомогенизировали в ледяной 0,32 М сахарозе (1:40 мас./об.), используя свободно притертый (зазор: 0,5 мм), изготовленный из стекла/тефлона гомогенизатор с электроприводом (12 ударов, 800 об/мин). Ядра и обломки клеток удаляли центрифугированием при 1500×g в течение 10 минут. Затем полученный в результате надосадок центрифугировали при 18000×g в течение 10 минут. Полученный неочищенный осадок синаптосом затем ресуспендировали в ледяном физиологическом буфере Кребса-Хензелайта, pH 7,4 при 25°C (эквивалент 4,17 мг сырой массы ткани/мл). Все центрифугирования осуществляли при 4°C.
Анализ захвата
Неочищенные синаптосомы полосатого тела инкубировали на водяной бане на качалке в течение 15 минут при 37°C. Затем аликвоты (150 мкл; эквивалент 0,625 мг сырой массы ткани/пробирку) добавляли в пробирки, содержащие 275 мкл буфера Кребса-Хензелайта и 50 мкл буфера (общий захват) или 50 мкл раствора лекарственного средства в 10 концентрациях в диапазоне от 10-11 до 10-4 М или 50 мкл GBR 12909 (10-5 М, неспецифичный захват). Захват инициировали добавлением 25 мкл свежеприготовленного [3H]-допамина (2,5 нМ) с последующим встряхиванием и продолжали в течение 5 минут при 37°C на водяной бане на качалке.
Захват останавливали фильтрованием в вакууме через фильтры Skatron 11734, используя устройство для сбора клеток Skatron. Фильтры быстро промывали ледяным физиологическим раствором (установки для промывки 9,9,0). Диски из фильтровальной бумаги для счета помещали в пластиковые флаконы для сцинтилляции и в четыре флакона пипеткой вносили по 25 мкл [3H]-допамина для точного определения концентрации, добавляемой в каждую пробирку. Радиоактивность (распады в минуту) определяли счетом жидкостной сцинтилляции (1 мл сцинтиллятора Packard MV Gold).
Анализ данных
Константы ингибирования (значения Ki)
Концентрацию соединения, необходимую для ингибирования 50% специфичного захвата (IC50), рассчитывали, используя программу Prism, в которую данные счета (распады в минуту) поступали непосредственно из анализатора жидкостной сцинтилляции. Указанная программа рассчитывает специфичный захват в отсутствие и в присутствии соединения в диапазоне концентраций и затем превращает значения специфичного захвата в присутствии и в отсутствие соединения в каждой концентрации в проценты от специфичного захвата в отсутствие соединения, как описано для одной концентрации.
Специфичный захват в процентах при каждой концентрации соединения затем откладывали против десятичного логарифма концентрации соединения. IC50 рассчитывали, используя следующую формулу:

где 100 = максимальное связывание (т.е. связывание в отсутствие соединения),
P = коэффициент наклона, который аналогичен коэффициенту наклона Хилла,
D = концентрация соединения (M).
Наклон Хилла рассчитывали, чтобы найти отклонения от простых взаимодействий в одном сайте. Коэффициент наклона Хилла, приближающийся к единице, показывает отклонение от одного сайта, значительно меньший единицы свидетельствует об отклонении от множества сайтов, и значительно больший единицы свидетельствует о позитивной кооперативности.
Затем будет рассчитана константа аффинности (Ki) соединения в отношении сайта захвата с использованием уравнения Ченга-Прусова:

где [L] = концентрация радиолиганда (M),
Kd = аффинность сайта захвата для радиолиганда.
Концентрация радиолиганда [L] нМ
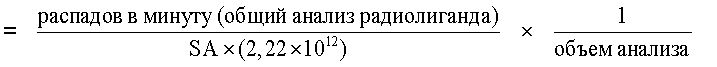
где SA = удельная активность радиолиганда (кюри/ммоль).
Пример 157 - Определение занятости переносчиков in vivo с помощью связывания ex vivo у крыс
Модель связывания ex vivo у крысы использовали для оценки способности тестируемых соединений достигать переносчиков-мишеней в головном мозге крыс после перорального дозирования. Протокол для связывания ex vivo у крыс был сходен с опубликованными экспериментальными способами (Bymaster et al. Neuropsychopharmacology, 25: 871-880 (2001), публикация включена в данное описание в виде ссылки в полном объеме) оценки занятости переносчика дулоксетина (норадреналина и серотонина) в ткани головного мозга. Дулоксетин является известным SNRI, продаваемым в настоящее время как Cymbalta® для лечения депрессии. Исследования связывания ex vivo у крыс осуществляли, используя [3H]-низоксетин и [3H]-циталопрам в качестве радиолигандов в анализе. Чтобы измерить занятость переносчика допамина разработали протокол связывания ex vivo у крыс с радиолигандом [3H]-WIN 35428. Кроме предоставления подтверждения того, что тестируемые соединения достигли требуемых мишеней в головном мозге крыс, такой эксперимент обеспечивает заменитель измерений фармакокинетических свойств соединений, т.е. пероральной биодоступности и проникновения в головной мозг.
Описание используемых способов приведено ниже:
Животные
Самцов крыс Sprague-Dawley (250-300 г) получали от Charles River (Margate, Kent) и содержали группами при 21±4°C и влажности 55±20% в условиях нормального цикла по 12 часов освещенность/темнота (свет включали в 07.00 часов) со свободным доступом к стандартному корму для грызунов и водопроводной воде по меньшей мере в течение одной недели до использования.
Обработка лекарственным средством
В день тестирования животным (n=5) перорально вводили дозу наполнителя, соединения AMRI или дулоксетина (позитивный контроль). Крыс забивали через 1 час после обработки. Извлекали целый головной мозг и вырезали кору лобных долей и полосатое тело и замораживали сухим льдом. Ткань хранили при -80°C до дня анализа. Кору лобной доли из каждой полусферы замораживали отдельно. Одну использовали для определения занятости сайтов NA-переносчиков, а другую - для определения занятости сайтов переносчиков 5-HT. Полосатое тело использовали для определения занятости сайтов переносчиков DA. Остальную ткань (за исключением мозжечка) вырезали и анализировали для определения концентрации лекарственного средства в головном мозге.
Препарат мембран
Кору лобной доли из каждой полусферы каждого животного или полосатое тело гомогенизировали отдельно в ледяном буфере для анализа, используя плотно притертый, изготовленный из стекла/тефлона гомогенизатор, и сразу же использовали в анализе связывания.
Связывание [3H]-циталопрама с сайтами переносчика 5-HT в головном мозге крыс
Мембраны коры лобной доли (400 мкл; эквивалент 1,25 мг сырой массы ткани/пробирку) инкубировали с 50 мкл [3H]-циталопрама в одной концентрации 1,3 нМ и либо 50 мкл буфера (общее связывание), либо 50 мкл пароксетина (0,5 мкМ; неспецифичное связывание) в течение 1 часа при 27°C в трех повторах.
Связанную с мембранами радиоактивность извлекали фильтрованием в вакууме через фильтры Skatron 11734, предварительно замоченные в 0,5% полиэтиленимине (PEI), используя устройство для сбора клеток Skatron. Фильтры быстро промывали ледяным 50 мМ трис-буфером (установки для промывки 9,9,0) и радиоактивность определяли счетом жидкостной сцинтилляции (1 мл сцинтиллятора Packard MV Gold).
Связывание [3H]-низоксетина с сайтами переносчика норадреналина в головном мозге крыс
Мембраны коры лобной доли (400 мкл; эквивалент 6,0 мг сырой массы ткани/пробирку) инкубировали с 50 мкл [3H]-низоксетина в одной концентрации 0,6 нМ и либо 50 мкл буфера (общее связывание), либо 50 мкл мазиндола (1 мкМ; неспецифичное связывание) в течение 4 часов при 4°C в трех повторах.
Связанную с мембранами радиоактивность извлекали фильтрованием в вакууме через фильтры Skatron 11734, используя устройство для сбора клеток Skatron. Фильтры быстро промывали ледяным 50 мМ трис-буфером (установки для промывки 9,9,0) и радиоактивность определяли счетом жидкостной сцинтилляции (1 мл сцинтиллятора Packard MV Gold).
Связывание [3H]-WIN 35428 с сайтами переносчика DA в головном мозге крыс
Мембраны полосатого тела (200 мкл; эквивалент 2 мг сырой массы ткани/пробирку) инкубировали с 25 мкл [3H]-WIN 35428 в одной концентрации 24 нМ и либо 25 мкл буфера (общее связывание), либо 25 мкл GBR12935 (1 мкМ; неспецифичное связывание) в течение 2 часов при 4°C в трех повторах.
Связанную с мембранами радиоактивность извлекали фильтрованием в вакууме через фильтры Skatron 11734, предварительно замоченные в 0,5% PEI, используя устройство для сбора клеток Skatron. Фильтры быстро промывали ледяным фосфатным буфером (установки для промывки 9,9,0) и радиоактивность (распады в минуту) определяли счетом жидкостной сцинтилляции (1 мл сцинтиллятора Packard MV Gold).
Анализ данных
Значение специфичного связывания (распады в минуту) получали вычитанием среднего неспецифичного связывания (распады в минуту) из среднего общего связывания (распады в минуту) для каждого животного. Данные представляли в виде среднего специфичного связывания в процентах от обработанного наполнителем контроля и применяли соответствующий статистический анализ.
Пример 158 - Определение функциональной активности у мыши с помощью экспериментов по исследованию обмена биогенных аминов
Предпосылка и цель исследования - Ингибирование метаболизма 5-HT в головном мозге мышей
5-Гидрокситриптамин (5-HT) образуется после действия фермента декарбоксилазы ароматических аминокислот на 5-гидрокситриптофан (5-HTP). Ранее обнаружено, что измерение 5-HTP является применимым показателем метаболизма 5-HT (Fuller et al., Federation Proc, 36: 2154-2158 (1977), публикация включена в данное описание в виде ссылки в полном объеме). В указанных исследованиях ингибитор декарбоксилазы ароматических аминокислот, м-гидроксибензилгидразин (NSD-1015), используют для предотвращения декарбоксилирования 5-HTP в 5-HT. Накопление 5-HTP затем можно измерить, используя высокоэффективную жидкостную хроматографию с электрохимической детекцией (ВЭЖХ-ECD). Целью данного исследования является измерение влияния новых лекарственных средств на уровни 5-HTP в головном мозге мышей. Соединения, ингибирующие обратный захват 5-HT ex vivo, уменьшают уровни 5-HTP. Избирательный ингибитор обратного захвата 5-HT, пароксетин, использовали в качестве позитивного контроля для сравнения.
Животные
Эксперименты проводили на 40 самцах мышей CD1 (20-30 г), полученных от Charles River (Margate, Kent). Животных содержали группами при 21±4°C и влажности 55±20% в условиях нормального цикла по 12 часов освещенность/темнота (свет включали в 07.00 часов) со свободным доступом к стандартному корму для грызунов и водопроводной воде. Животным давали возможность акклиматизироваться к помещению для животных по меньшей мере в течение одной недели до эксперимента.
Экспериментальные способы
В каждый эксперимент включали 5 разных групп обработки (наполнитель, пароксетин и 3 группы обработки соединением (например, одна доза 3 разных лекарственных средств или три дозы одного лекарственного средства, n=8). В день тестирования мышам перорально вводили дозы наполнителя, пароксетина или тестируемого соединения. Через 30 минут всем мышам давали NSD 1015 (100 мг/кг в/б), чтобы предотвратить метаболизм 5-HTP в 5-HT. Еще через 30 минут (т.е. через 60 минут после введения лекарственного средства) животных забивали и извлекали целый головной мозг без мозжечка и замораживали, используя жидкий азот. Ткани готовили для анализа ВЭЖХ-ECD, используя хорошо разработанные способы. Концентрации 5-HTP в головном мозге определяли, используя высоту пика по отношению к стандартному раствору 5-HTP, впрыскиваемому отдельно.
Лекарственные средства и реагенты
Все тестируемые лекарственные средства предоставлены AMRI. NSD-1015 приобретали от Sigma-Aldrich (Poole). Все лекарственные средства готовили свежими каждый день. Лекарственные средства растворяли в подходящем наполнителе и давали дозу в объеме в диапазоне 1-10 мл/кг. Все дозы лекарственных средств выражены в виде свободного основания, если не оговорено особо. Все реагенты для анализа ВЭЖХ-ECD и стандарт 5-HTP получали от Sigma-Aldrich или сходного коммерческого поставщика.
Данные и статистический анализ
Результаты выражали в виде средних ± SEM. Статистический анализ выполняли собственными силами. Точные используемые статистические критерии зависели от полученных данных, однако статистические сравнения между уровнями 5-HTP у разных групп мышей обычно осуществляли анализом дисперсии с последующими подходящими T-критериями или критерием множественного сравнения (двухсторонним), чтобы сравнить каждую группу обработки с обработанными наполнителем контролями. P<0,05 считается статистической значимой.
Предпосылка и цель исследования - ингибирование метаболизма норадреналина в головном мозге мышей
Норадреналин является важным нейромедиатором в центральной нервной системе. Один путь инактивации норадреналина заключается в превращении сначала в 3,4-дигидроксифенилгликольальдегид моноаминоксидазой (MAO) и затем в 3,4-дигидроксифенилгликоль (DHPG) альдегидредуктазой. DHPG затем метилируется с образованием 3-метокси-4-гидроксифенилгликоля (MHPG) катехол-O-метилтрансферазой (COMT). Так как COMT расположена вне нейронов, она идеально подходит для обеспечения маркера высвобождения и функциональной утилизации норадреналина. Целью данного исследования было изучение влияния новых соединений на метаболизм норадреналина посредством измерения уровней MHPG в головном мозге мышей с использованием высокоэффективной жидкостной хроматографии с электрохимической детекцией (ВЭЖХ-ECD). Действие ингибиторов обратного захвата норадреналина в виде увеличения уровней норадреналина как в теле клетки, так и концевых областях, приводит к активации соматодендритических и терминальных α2-адренергических ауторецепторов, которые ослабляют импульсацию нейронов и высвобождение нейромедиаторов соответственно. Соединения, ингибирующие обратный захват норадреналина ex vivo, могут снижать уровни MHPG в головном мозге мышей. Дезипрамин использовали в качестве позитивного контроля для сравнения.
Животные
Эксперименты проводили на 40 самцах мышей CD1 (20-30 г), полученных от Charles River (Margate, Kent). Животных содержали группами при 21±4°C и влажности 55±20% в условиях нормального цикла по 12 часов освещенность/темнота (свет включали в 07.00 часов) со свободным доступом к стандартному корму для грызунов и водопроводной воде. Животным давали возможность акклиматизироваться к помещению для животных по меньшей мере в течение одной недели до эксперимента.
Экспериментальные способы
В каждый эксперимент включали 5 разных групп обработки (наполнитель, дезипрамин и 3 группы обработки лекарственным средством, например, одна доза 3 разных лекарственных средств или три дозы одного лекарственного средства, n=8). В день тестирования мышам перорально вводили дозы наполнителя, дезипрамина или тестируемого соединения. Через 60 минут животных забивали и извлекали целый головной мозг без мозжечка и замораживали, используя жидкий азот. Ткани готовили для анализа ВЭЖХ-ECD, используя хорошо разработанные способы. Концентрации MHPG в головном мозге определяли, используя высоту пика по сравнению с отношением внутреннего стандарта (изо-MHPG) к MHPG.
Лекарственные средства и реагенты
Все тестируемые лекарственные средства предоставлены AMRI. Гидрохлорид дезипрамина приобретали от Sigma-Aldrich. Все лекарственные средства готовили свежими каждый день. Лекарственные средства растворяли в подходящем наполнителе и вводили дозу в объеме в диапазоне 1-10 мл/кг. Все дозы лекарственных средств выражены в виде свободного основания, если не оговорено особо. Все реагенты для анализа ВЭЖХ-ECD и MHPG получали от Sigma-Aldrich или сходного коммерческого поставщика.
Данные и статистический анализ
Результаты выражали в виде средних ± SEM. Статистический анализ выполняли собственными силами. Точные используемые статистические критерии зависели от полученных данных, однако статистические сравнения между уровнями MHPG у разных групп мышей обычно осуществляли анализом дисперсии с последующими подходящими T-критериями или критерием множественного сравнения (двухсторонним), чтобы сравнить каждую группу обработки с обработанными наполнителем контролями. P<0,05 считается статистической значимой.
Пример 159 - Микродиализное исследование
Целью данного исследования является применение микродиализа с двойными зондами для определения влияния перорального введения соединений на внеклеточные концентрации норадреналина (NA) и 5-HT в предлобной коре и допамина (DA) в полосатом теле свободно двигающихся крыс Sprague-Dawley.
Крыс анестезировали изофлураном (5% для индукции, 2% для поддержания) в смеси O2/N2O (1 литр · мин-1 каждый), доставляемой посредством аппарата для анестезии (St Bernard Medical Services, UK). Концентрические зонды для микродиализа с открытым 2- и 4-мм мембранным концом Hospal (CMA) стереотаксически имплантировали в предлобную кору и полосатое тело, соответственно, используя координаты, взятые из стереотаксического атласа Paxinos and Watson (Paxinos et al., «The Rat Brain in Stereotaxic Coordinates», 2nd Edition, London: Academic Press (1986), который включен в данное описание в виде ссылки в полном объеме). Верхнюю пластину режущего инструмента устанавливали на 3,3 мм ниже интерауральной линии, так чтобы поверхность черепа между брегмой и ламбдой была горизонтальной. Дополнительные трепанационные отверстия делали для черепных винтов (стальных) и зонды закрепляли, используя зубной цемент. После хирургической операции животных содержали отдельно в круглых камерах (размеры 450 мм внутренний диаметр, 320 мм высота стенок), при этом микродиализные зонды соединяли с вращающимся промывочным устройством и кронштейном с противовесом, чтобы обеспечить возможность неограниченного движения. Крысам давали время на восстановление по меньшей мере 16 часов с доступом к корму и воде по желанию. В течение указанного периода зонды непрерывно перфузировали искусственной спинномозговой жидкостью (aCSF; Harvard Apparatus, UK) со скоростью потока 1,2 мкл·мин-1.
Эксперимент проводили на следующий день после операции. Образцы диализата собирали каждый 30 минут в пробирки Эппендорф, содержащие перхлорную кислоту, чтобы предотвратить окисление. Собрали всего 11 образцов для каждой области головного мозга (3 до введения лекарственного средства и 8 после введения лекарственного средства). Для определения и количественной оценки 5-HT и NA в коре лобной доли микродиализные образцы делили и анализировали на отдельных системах ВЭЖХ с помощью обращенно-фазовой ионпарной ВЭЖХ в сочетании с электрохимической детекцией. В случае DA в полосатом теле микродиализные образцы анализировали на отдельной системе ВЭЖХ. Анализ ВЭЖХ проводили остальную часть недели после эксперимента.
Пример 160 - Тест поведенческого отчаяния у крыс
Способ, также называемый «тестом принудительного плавания» или «тест Порсолта», который выявляет антидепрессантную активность, осуществляли, следуя способу, описанному Porsolt et al. (Eur. J. Pharmacol., 47: 379-391 (1978), публикация включена в данное описание в виде ссылки в полном объеме). Крысы, которых вынуждали плавать в ситуации, из которой они не могут спастись, быстро становились неподвижными. Антидепрессанты снижают продолжительность неподвижности. Данное исследование осуществляли согласно Porsolt and Partners Pharmacology (Dr. Vincent Castagne, France).
Крыс по отдельности помещали в цилиндр (высота = 40 см; диаметр = 20 см), содержащий 13 см воды (25°C) на 15 минут в первый день эксперимента (сеанс 1) и затем возвращали в воду через 24 часа для 5-минутного теста (сеанс 2). Измеряли продолжительность неподвижности в течение 5-минутного теста.
Исследовали по 6 крыс на группу. Тест осуществляли вслепую.
Тестируемые вещества оценивали при пероральном введении (п/о) 3 раза: за 24 часа, 4 часа и 60 минут до теста (сеанс 2) и сравнивали с контрольной группой, обработанной наполнителем.
Имипрамин (64 мг/кг п/о), вводимый в таких же экспериментальных условиях, использовали в качестве эталонного вещества.
Пример 161 - Анализ с использованием тетрабеназина
Активность in vivo соединений согласно данному изобретению у животных оценивали на основе определения способности соединений предотвращать седативное действие тетрабеназина (TBZ) (см., например, G. Stille, Arzn. Forsch. 14: 534-537 (1964), публикация включена в данное описание в виде ссылки в полном объеме). Рандомизированные и закодированные дозы тестируемых соединений перорально вводили мышам, также затем вводили дозу тетрабеназина. Затем животных оценивали в отношении антагонизма индуцированной тетрабеназином потери исследовательской активности и птоза через конкретные временные интервалы после введения лекарственного средства. Исследовательскую активность, например, оценивают, помещая животного в центр круга и затем оценивая количество времени, которое требуется животному, чтобы пересечь периметр круга - в общем, чем больше времени требуется животному, чтобы осуществить такое пересечение, тем у него больше потеря исследовательской активности. Кроме того, считали, что у животного имеется птоз, если его веки закрыты по меньшей мере на 50%. Предполагается, что более 95% контрольных (обработанных наполнителем) мышей имеют потерю исследовательской активности и птоз; связанную с соединениями активность затем рассчитывают в виде процента мышей, не способных отвечать на контрольную дозу тетрабеназина, при этом предполагается, что терапевтически более эффективные соединения лучше уменьшают потерю исследовательского поведения и птоза. Эффективность в анализе мышей с использованием тетрабеназина, осуществляемым, как описано выше, является показателем фармакокинетических свойств (пероральной биодоступности) соединений согласно настоящему изобретению и также способности соединений проникать в центральную нервную систему и достигать требуемой мишени, переносчиков биогенных аминов, в головном мозге. Номифензин, агент ЦНС, который проявлял хорошие антидепрессантные свойства в клинических испытаниях на человеке, проявлял высокую биологическую активность в данном анализе in vivo. Ritalin®, широко используемое лекарственное средство для лечения гиперактивного расстройства с дефицитом внимания, также проявлял высокую активность в данном анализе.
Самцов мышей CFI (Charles River Breeding Laboratories) массой при тестировании 18-25 г содержали минимум 6 дней в тщательно контролируемых условиях среды (22,2±1,1°C; средняя влажность 50%, 12-часовой цикл освещенности/24 часа). Мыши голодали в течение ночи (16-22 часов) перед тестированием. Мышей помещали в прозрачные поликарбонатные «башмаки»-коробки (17 см × 28,5 см × 12 см). Рандомизированные и кодированные дозы тестируемых соединений вводили п/о. Дозу 45 мг/кг тетрабеназина вводили в/б за 30 минут до момента счета. Все соединения вводили в объеме 0,1 мл/10 г массы тела. Животных оценивали в отношении антагонизма индуцированной тетрабеназином потери исследовательской активности и птоза через конкретные интервалы времени после введения лекарственного средства. В указанный временной интервал мышей исследовали в отношении симптомов исследовательской активности и птоза. Исследовательскую активность оценивали, помещая животное в центр круга размером 5 дюймов. Животному позволяли 15 минут двигаться и пересечь периметр. Это рассматривали как антагонизм тетрабеназина и присваивали оценку 0. Неспособность покинуть круг расценивали как потерю исследовательской активности и давали оценку 4. Считали, что у животного имеется птоз, если его веки были закрыты по меньшей мере на 50%, и давали оценку 4, если они были полностью закрыты; при отсутствии закрытия давали оценку 0. Предполагали, что более 95% контрольных (обработанных наполнителем) мышей имели потерю исследовательской активности и птоз. Активность лекарственного средства рассчитывали в виде процента мышей, неспособных отвечать на контрольную дозу тетрабеназина.
Пример 162 - Статистическая оценка
Средние эффективные дозы (ED50) и 95% доверительные интервалы определяли в числовом выражении способом Litchfield and Wilcoxon, Journal of Pharmacology и Experimental Therapeutics, 96: 99-113 (1949), публикация включена в данное описание в виде ссылки в полном объеме.
В разделе «Уровень техники» даны ссылки на известные основанные на тетрагидроизохинолине соединения, которые имеют гетероароматический цикл, введенный в качестве заместителя в 4-положении цикла тетрагидроизохинолина. В таблице 1, показанной ниже, приведено сравнение аффинностей связывания (значения Ki определяли, как описано в экспериментальном разделе выше) для соединений с «моноциклическими гетероциклическими циклами, введенными в качестве заместителя в 4-положении» 1,2,3,4-тетрагидроизохинолина, с аффинностями связывания для соединений согласно настоящему изобретению, которые имеют «бициклические гетероароматические циклы, введенные в качестве заместителя в 4-положении».

Исследование аффинности связывания (значения Ki) для моноциклических гетероароматических циклов, например тиофен-2-ила, 4-метилтиофен-2-ила, 5-метилфуран-2-ила, 4-(3,5-диметилизоксазол-4-ила или 6-метоксипиридин-3-ила, показывает, что указанные соединения намного менее эффективны по отношению ко всем трем переносчикам биогенных аминов, чем соединения согласно настоящему изобретению, которые имеют бициклические гетероароматические циклы, введенные в качестве заместителя в 4-положении 1,2,3,4-тетрагидроизохинолина. Бициклические гетероароматические циклы, введенные в качестве заместителя в 4-положении 1,2,3,4-тетрагидроизохинолина, представляют собой важную структурную особенность, которая приводит к неожиданным улучшениям эффективности и избирательности соединений согласно настоящему изобретению.
Соединения согласно настоящему изобретению являются сильными ингибиторами обратного захвата моноаминов переносчиками допамина, норэпинефрина и серотонина. Соединения согласно настоящему изобретению могут быть сгруппированы согласно профилю ингибирования обратного захвата переносчиков, в соответствии с которым конкретное соединение влияет на переносчики. Предполагается, что благодаря различным профилям избирательности соединения, заявленные в настоящем изобретении, могут иметь широкое применение для лечения множества патологических состояний, в которые вовлечены уровни нейромедиаторов, допамина, норэпинефрина и серотонина.
Хотя изобретение описано подробно в целях иллюстрации, предполагается, что такие подробности приведены только для такой цели, и специалистами в данной области могут быть осуществлены изменения, не отходя от сути и не выходя за рамки объема изобретения, который определен следующей далее формулой изобретения.
Реферат
Изобретение относится к новым соединениям формулы I: ! ! где атом углерода, обозначенный *, находится в R- или S-конфигурации; Х означает конденсированный бициклический карбоцикл или гетероцикл, выбранный из группы, состоящей из бензофуранила, бензо[b]тиофенила, бензоизотиазолила, индазолила, индолила, бензооксазолила, бензотиазолила, инденила, инданила, дигидробензоциклогептенила, нафтила, тетрагидронафтила, хинолинила, изохинолинила, хиноксалинила, 2Н-хроменила, имидазо[1,2-а]пиридинила, пиразоло[1,5-а]пиридинила, и конденсированный бициклический карбоцикл или конденсированный бициклический гетероцикл, необязательно замещен заместителями (в количестве от 1 до 4), которые определены ниже для R14; R1 означает Н, C1-С6-алкил, С3-С6-циклоалкил, C1-С3-алкил, замещенный OR11, -NR9R10 или -CN; R2 означает Н, C1-С6-алкил, или гем-диметил; R3 означает Н, -OR11, C1-С6-алкил или галоген; R4 означает Н, галоген, -OR11, -CN, C1-С6-алкил, C1-С6-алкил, замещенный -NR9R10, С3-С6-циклоалкил, замещенный -NR9R10, C(O)R12; или R4 означает морфолинил, пиперидинил, пиримидинил, пиридазинил, пиразинил, пирролил, изоксазолил, пирролидинил, пиперазинил, 2-оксо-2Н-пиридинил, [1,2,4]триазоло[4,3-а]пиридинил, 3-оксо-[1,2,4]триазоло[4,3-а]пиридинил, хиноксалинил, которые необязательно замещены заместителями (в количестве от 1 до 4), которые определены ниже для R14; R5 означает Н или C1-С6-алкил; R6 означает Н, C1-С6-алкил или -OR11; R7 означает Н; R8 означает Н, -OR9, C1-С6-алкил, -CN; R9 означает Н или C1-C4-алкил; R10 означает Н или С1-С4-алкил; или R9 и R10, взятые вместе с атомом азота, с которым они связаны, образуют морфолин; R11 означает Н, С1-С4-алкил; R12 означает С1-С4-алкил; R14 в каждом случае независимо выбирают из заместителя, выбранного из гр
Формула

где атом углерода, обозначенный *, находится в R-или S-конфигурации;
Х означает конденсированный бициклический карбоцикл или гетероцикл, выбранный из группы, состоящей из бензофуранила, бензо[b]тиофенила, бензоизотиазолила, индазолила, индолила, бензооксазолила, бензотиазолила, инденила, инданила, дигидробензоциклогептенила, нафтила, тетрагидронафтила, хинолинила, изохинолинила, хиноксалинила, 2Н-хроменила, имидазо[1,2-а]пиридинила, пиразоло[1,5-а]пиридинила, и конденсированный бициклический карбоцикл или конденсированный бициклический гетероцикл необязательно замещен заместителями (в количестве от 1 до 4), которые определены ниже для R14;
R1 означает Н, C1-С6-алкил, С3-С6-циклоалкил, C1-С3-алкил, замещенный OR11,-NR9R10 или-CN;
R2 означает Н, C1-С6-алкил, или гем-диметил;
R3 означает Н, -OR11, C1-С6-алкил или галоген;
R4 означает Н, галоген, -OR11, -CN, C1-С6-алкил, C1-С6-алкил, замещенный -NR9R10,
С3-С6-циклоалкил, замещенный -NR9R10, C(O)R12; или
R4 означает морфолинил, пиперидинил, пиримидинил, пиридазинил, пиразинил, пирролил, изоксазолил, пирролидинил, пиперазинил, 2-оксо-2Н-пиридинил, [1,2,4]триазоло[4,3-а]пиридинил, 3-оксо-[1,2,4]триазоло[4,3-а]пиридинил, хиноксалинил, которые необязательно замещены заместителями (в количестве от 1 до 4), которые определены ниже для R14;
R5 означает Н или C1-С6-алкил;
R6 означает Н, C1-С6-алкил или-OR11;
R7 означает Н;
R8 означает Н, -OR9, C1-С6-алкил, -CN;
R9 означает Н или С1-С4-алкил;
R10 означает Н или С1-С4-алкил;
или R9 и R10, взятые вместе с атомом азота, с которым они связаны, образуют морфолин;
R11 означает Н, С1-С4-алкил;
R12 означает С1-С4-алкил;
R14 в каждом случае независимо выбирают из заместителя, выбранного из группы, состоящей из галогена, -OR11, -NR11R12, C1-С6-алкила, который необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-С3-алкила, арила;
или его фармацевтически приемлемая соль.
R2 означает Н, СН3 или гем-диметил;
R3 означает Н, СН3, ОН, ОСН3 или F;
R4 означает Н, СН3, ОН, ОСН3, -CN или F;
R5 означает Н;
R означает Н или метоксигруппу;
R7 означает Н;
R8 означает Н, ОН, ОСН3, -CN или СН3; и
Х выбирают из группы, состоящей из бензофуранила, бензо[b]тиофенила, бензоизотиазолила, индазолила, индолила, бензооксазолила, бензотиазолила, имидазо[1,2-а]пиридинила, пиразоло[1,5-а]пиридинила, хроменила, инденила, инданила, и конденсированный бициклический карбоцикл или конденсированный бициклический гетероцикл, необязательно замещен 1-4 заместителями R14.
Х выбирают из группы, состоящей из бензофуран-2-ила, 5-хлорбензофуран-2-ила, 4-фторбензофуран-2-ила, 5-фторбензофуран-2-ила, 5-метоксибензофуран-2-ила, 6-фторбензофуран-2-ила, 7-фторбензофуран-2-ила, 7-метоксибензофуран-2-ила, бензофуран-3-ила, бензофуран-4-ила, бензофуран-5-ила, бензофуран-6-ила, бензофуран-7-ила, 2,3-дигидробензофуран-5-ила, бензо[b]тиофен-2-ила, 4-хлорбензо[b]тиофен-2-ила, 4-фторбензо[b]тиофен-2-ила, 4-метоксибензо[b]тиофен-2-ила, 5-хлорбензо[b]тиофен-2-ила, 5-фторбензо[b]тиофен-2-ила, 6-хлорбензо[b]тиофен-2-ила, 6-фторбензо[b]тиофен-2-ила, 7-хлорбензо[b]тиофен-2-ила, 7-фторбензо[b]тиофен-2-ила, бензо[b]тиофен-3-ила, бензо[b]тиофен-4-ила, бензо[b]тиофен-5-ила, 2-метилбензо[b]тиофен-5-ила, 2-хлорбензо[b]тиофен-5-ила, 4-метоксибензо[b]тиофен-5-ила, 4-гидроксибензо[b]тиофен-5-ила, 4-метилбензо[b]тиофен-5-ила, бензо[b]тиофен-6-ила, 2-хлорбензо[b]тиофен-6-ила, 7-метоксибензо[b]тиофен-6-ила, 7-гидроксибензо[b]тиофен-6-ила, 7-метилбензо[b]тиофен-6-ила, бензо[b]тиофен-7-ила, индазол-1-ила, 1H-индазол-3-ила, 1Н-индазол-4-ила, 1H-индазол-5-ила, 1-метилиндазол-5-ила, 6-метокси-1H-индазол-5-ила, 7-метокси-1Н-индазол-5-ила, 7-фтор-1Н-индазол-5-ила, 7-хлор-1Н-индазол-5-ила, 7-метокси-1Н-индазол-5-ила, 1H-индазол-6-ила, 1-метилиндазол-6-ила, 7-фтор-1Н-индазол-6-ила, 1Н-индазол-7-ила, индол-1-ила, 1-метилиндол-2-ила, 1Н-индол-2-ила, 7-фтор-1Н-индол-2-ила, 1Н-индол-3-ила, 1H-индол-4-ила, 1Н-индол-5-ила, 1-метилиндол-5-ила, 7-фтор-1H-индол-5-ила, 1H-индол-6-ила, 1-метилиндол-6-ила, 7-фтор-1Н-индол-6-ила, бензооксазол-2-ила, бензооксазол-4-ила, бензооксазол-5-ила, 2-метилбензооксазол-5-ила, бензооксазол-6-ила, 2-метилбензооксазол-6-ила, бензооксазол-7-ила, бензотиазол-2-ила, бензотиазол-4-ила, бензотиазол-5-ила, 2-метилбензотиазол-5-ила, бензотиазол-6-ила, 2-метилбензотиазол-6-ила, бензотиазол-7-ила, бензоизотиазол-4-ила, бензоизотиазол-5-ила, бензоизотиазол-6-ила, бензоизотиазол-7-ила, имидазо[1,2-а]пиридин-2-ила, имидазо[1,2-а]пиридин-6-ила, имидазо[1,2-а]пиридин-7-ила, пиразоло[1,5-а]пиридин-2-ила, пиразоло[1,5-а]пиридин-5-ила, пиразоло[1,5-а]пиридин-6-ила, 3Н-инден-5-ила и индан-5-ила.
R1 означает метил, этил или изопропил;
R2 означает Н, СН3 или гем-диметил;
R3 означает Н, СН3, ОН, ОСН3 или F;
R4 означает пиримидинил, пиридазинил, пиразинил, хиноксалинил, изоксазолил, 3-оксо-[1,2,4]триазоло[4,3-а]пиридинил, [1,2,4]триазоло[4,3-а]пиридинил, которые необязательно замещены 1-4 заместителями R14;
R5 означает Н;
R6 означает Н или метоксигруппу;
R7 означает Н;
R8 означает Н, ОН, ОСН3, -CN или СН3; и
Х выбирают из группы, состоящей из бензофуранила, бензо[b]тиофенила, бензоизотиазолила, индазолила, индолила, бензооксазолила, бензотиазолила, имидазо[1,2-а]пиридинила, пиразоло[1,5-а]пиридинила, хроменила, инденила, инданила, дигидробензоциклогептенила, и конденсированный бициклический карбоцикл или конденсированный бициклический гетероцикл необязательно замещен 1-4 заместителями R14.
R4 выбирают из группы, состоящей из 3,5-диметилизоксазол-4-ила, пиримидин-4-ила, пиримидин-2-ила, пиримидин-5-ила, пиразин-2-ила, 3-метилпиразин-2-ила, 5-метилпиразин-2-ила, 6-метилпиразин-2-ила, 3-метоксипиразин-2-ила, 5-метоксипиразин-2-ила, 6-метоксипиразин-2-ила, 6-этилпиразин-2-ила, пиридазин-3-ил, 5-метилпиридазин-3-ила, 6-метилпиридазин-3-ила, 6-диметиламинопиридазин-3-ила, 6-метиламинопиридазин-3-ила, пиридазин-4-ила, хиноксалин-2-ила, хиноксалин-6-ила, 3-оксо-[1,2,4]триазоло[4,3-а]пиридин-2-ила и 2-оксо-2Н-пиридин-1-ила; и
Х выбирают из группы, состоящей из бензофуран-2-ила, 5-хлорбензофуран-2-ила, 4-фторбензофуран-2-ила, 5-фторбензофуран-2-ила, 5-метоксибензофуран-2-ила, 6-фторбензофуран-2-ила, 7-фторбензофуран-2-ила, 7-метоксибензофуран-2-ила, бензофуран-3-ила, бензофуран-4-ила, бензофуран-5-ила, бензофуран-6-ила, бензофуран-7-ила, 2,3-дигидробензофуран-5-ила, бензо[b]тиофен-2-ила, 4-хлорбензо[b]тиофен-2-ила, 4-фторбензо[b]тиофен-2-ила, 4-метоксибензо[b]тиофен-2-ила, 5-хлорбензо[b]тиофен-2-ила, 5-фторбензо[b]тиофен-2-ила, 6-хлорбензо[b]тиофен-2-ила, 6-фторбензо[b]тиофен-2-ила, 7-хлорбензо[b]тиофен-2-ила, 7-фторбензо[b]тиофен-2-ила, бензо[b]тиофен-3-ила, бензо[b]тиофен-4-ила, бензо[b]тиофен-5-ила, 2-метилбензо[b]тиофен-5-ила, 2-хлорбензо[b]тиофен-5-ила, 3-трифторметилбензо[b]тиофен-5-ила, 4-метоксибензо[b]тиофен-5-ила, 4-гидроксибензо[b]тиофен-5-ила, 4-метилбензо[b]тиофен-5-ила, бензо[b]тиофен-6-ила, 2-хлорбензо[b]тиофен-6-ила, 7-метоксибензо[b]тиофен-6-ила, 7-гидроксибензо[b]тиофен-6-ила, 7-метилбензо[b]тиофен-6-ила, бензо[b]тиофен-7-ила, 1Н-индазол-1-ила, 1Н-индазол-3-ила, 1Н-индазол-4-ила, 1Н-индазол-5-ила, 1-метилиндазол-5-ила, 6-метокси-1Н-индазол-5-ила, 7-метокси-1Н-индазол-5-ила, 7-фтор-1H-индазол-5-ила, 7-хлор-1Н-индазол-5-ила, 7-метокси-1Н-индазол-5-ила, 1Н-индазол-6-ила, 1-метилиндазол-6-ила, 7-фтор-1Н-индазол-6-ила, 1Н-индазол-7-ила, индол-1-ила, 1-метилиндол-2-ила, 1Н-индол-2-ила, 7-фтор-1Н-индол-2-ила, 1H-индол-3-ила, 1Н-индол-4-ила, 1Н-индол-5-ила, 1-метилиндол-5-ила, 7-фтор-1Н-индол-5-ила, 1H-индол-6-ила, 1-метилиндол-6-ила, 7-фтор-1H-индол-6-ила, бензооксазол-2-ила, бензооксазол-4-ила, бензооксазол-5-ила, 2-метилбензооксазол-5-ила, бензооксазол-6-ила, 2-метилбензооксазол-6-ила, бензооксазол-7-ила, бензотиазол-2-ила, бензотиазол-4-ила, бензотиазол-5-ила, 2-метилбензотиазол-5-ила, бензотиазол-6-ила, 2-метилбензотиазол-6-ила, бензотиазол-7-ила, бензоизотиазол-4-ила, бензоизотиазол-5-ила, бензоизотиазол-6-ила, бензоизотиазол-7-ила, имидазо[1,2-а]пиридин-2-ила, имидазо[1,2-а]пиридин-6-ила, имидазо[1,2-а]пиридин-7-ила, пиразоло[1,5-а]пиридин-2-ила, пиразоло[1,5-а]пиридин-5-ила, пиразоло[1,5-а]пиридин-6-ила, 3Н-инден-5-ила и индан-5-ила.
R4 выбирают из группы, состоящей из морфолин-4-ила, 2,6-диметилморфолин-4-ила, пиперазин-1-ила, 4-метилпиперазин-1-ила, пиперидин-1-ила, пирролидин-1-ила, диметиламинометила, 1-аминоциклопропила; и
Х выбирают из группы, состоящей из бензофуран-2-ила, 5-хлорбензофуран-2-ила, 4-фторбензофуран-2-ила, 5-фторбензофуран-2-ила, 5-метоксибензофуран-2-ила, 6-фторбензофуран-2-ила, 7-фторбензофуран-2-ила, 7-метоксибензофуран-2-ила, бензофуран-3-ила, бензофуран-4-ила, бензофуран-5-ила, бензофуран-6-ила, бензофуран-7-ила, 2,3-дигидробензофуран-5-ила, бензо[b]тиофен-2-ила, 4-хлорбензо[b]тиофен-2-ила, 4-фторбензо[b]тиофен-2-ила, 4-метоксибензо[b]тиофен-2-ила, 5-хлорбензо[b]тиофен-2-ила, 5-фторбензо[b]тиофен-2-ила, 6-хлорбензо[b]тиофен-2-ила, 6-фторбензо[b]тиофен-2-ила, 7-хлорбензо[b]тиофен-2-ила, 7-фторбензо[b]тиофен-2-ила, бензо[b]тиофен-3-ила, бензо[b]тиофен-4-ила, бензо[b]тиофен-5-ила, 2-метилбензо[b]тиофен-5-ила, 2-хлорбензо[b]тиофен-5-ила, 4-метоксибензо[b]тиофен-5-ила, 4-гидроксибензо[b]тиофен-5-ила, 4-метилбензо[b]тиофен-5-ила, бензо[b]тиофен-6-ила, 2-хлорбензо[b]тиофен-6-ила, 7-метоксибензо[b]тиофен-6-ила, 7-гидроксибензо[b]тиофен-6-ила, 7-метилбензо[b]тиофен-6-ила, бензо[b]тиофен-7-ила, 1H-индазол-1-ила, 1Н-индазол-3-ила, 1Н-индазол-4-ила, 1Н-индазол-5-ила, 1-метилиндазол-5-ила, 6-метокси-1Н-индазол-5-ила, 7-метокси-1Н-индазол-5-ила, 7-фтор-1Н-индазол-5-ила, 7-хлор-1Н-индазол-5-ила, 7-метокси-1Н-индазол-5-ила, 1Н-индазол-6-ила, 1-метилиндазол-6-ила, 7-фтор-1Н-индазол-6-ила, 1Н-индазол-7-ила, индол-1-ила, 1-метилиндол-2-ила, 1H-индол-2-ила, 7-фтор-1Н-индол-2-ила, 1H-индол-3-ила, 1Н-индол-4-ила, 1Н-индол-5-ила, 1-метилиндол-5-ила, 7-фтор-1Н-индол-5-ила, 1H-индол-6-ила, 1-метилиндол-6-ила, 7-фтор-1H-индол-6-ила, бензооксазол-2-ила, бензооксазол-4-ила, бензооксазол-5-ила, 2-метилбензооксазол-5-ила, бензооксазол-6-ила, 2-метилбензооксазол-6-ила, бензооксазол-7-ила, бензотиазол-2-ила, бензотиазол-4-ила, бензотиазол-5-ила, 2-метилбензотиазол-5-ила, бензотиазол-6-ила, 2-метилбензотиазол-6-ила, бензотиазол-7-ила, бензоизотиазол-4-ила, бензоизотиазол-5-ила, бензоизотиазол-6-ила, бензоизотиазол-7-ила, имидазо[1,2-а] пиридин-2-ила, имидазо[1,2-а]пиридин-6-ила, имидазо[1,2-а]пиридин-7-ила, пиразоло[1,5-а]пиридин-2-ила, пиразоло[1,5-а]пиридин-5-ила, пиразоло[1,5-а] пиридин-6-ила, 3Н-инден-5-ила и индан-5-ила.
4-(бензо[b]тиофен-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-2-ил)-1-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-2-ил)-2-этил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-2-ил)-1,2-диметил-1,2,3,4-тетрагидроизохинолина;
4-(4-фторбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(5-фторбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(6-фторбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(7-фторбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(4-хлорбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(5-хлорбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(6-хлорбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(7-хлорбензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(4-метоксибензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(5-метоксибензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(6-метоксибензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(7-метоксибензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1,1-диоксо-1Н-1λ6-бензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-ола;
4-(бензо[b]тиофен-2-ил)-4-метокси-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-2-ил)-2,4-диметил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-карбонитрила;
4-(бензо[b]тиофен-2-ил)-4-фтор-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-2-ил)-4-хлор-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-2-ил)-2,5-диметил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ола;
4-(бензо[b]тиофен-2-ил)-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-2-ил)-2,7-диметил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-2-ил)-7-фтор-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(4-фторбензо[b]тиофен-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(5-фторбензо[b]тиофен-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(6-фторбензо[b]тиофен-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(7-фторбензо[b]тиофен-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-2-ил)-2-этил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
[6-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]ди-метиламина;
[6-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]ме-тиламина;
6-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-илами-на;
4-бензо[b]тиофен-2-ил-2-метил-7-(6-морфолин-4-илпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-2-метил-7-(6-трифторметилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
6-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-илкарб-онитрила;
4-бензо[b]тиофен-2-ил-8-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохино-лина;
4-бензо[b]тиофен-2-ил-8-метокси-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохино-лина;
4-бензо[b]тиофен-2-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-8-
ола;
4-бензо[b]тиофен-2-ил-2,8-диметил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-2-этил-8-фтор-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохиноли-на;
4-бензо[b]тиофен-2-ил-6-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохино-лина;
4-бензо[b]тиофен-2-ил-5-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохино-лина;
4-бензо[b]тиофен-2-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-
ола;
4-бензо[b]тиофен-2-ил-2,4-диметил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-2-ил)-8-фтор-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-2-метил-7-(3-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-7-(3-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-2-метил-7-(6-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-7-(6-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-8-фтор-2-метил-7-пиразин-2-ил-1,2,3,4-
тетрагидроизохинолина;
4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-8-фтор-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохино-лина;
4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-8-фтор-2-метил-7-пиримидин-4-ил-1,2,3,4-тетрагидроизохино-лина;
4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-8-фтор-2-метил-7-пиримидин-5-ил-1,2,3,4-тетрагидроизохино-лина;
4-(бензо[b]тиофен-2-ил)-7-(3,5-диметилизоксазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-2-ил)-2-метил-7-(тиазол-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-2-ил)-2-метил-7-(5-метилтиазол-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-2-метил-7-[1,3,5]триазин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-2~метил-7-[1,2,4]триазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-2-метил-7-[1,2,4]триазин-5-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-2-метил-7-[1,2,4]триазин-6-ил-1,2,3,4-тетрагидроизохинолина;
3-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циннолина;
1-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)фталазина;
2-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиноксалина;
2-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
6-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
7-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
2-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-2Н-[1,2,4]триазоло[4,3-а]пиридин-3-она;
4-(бензо[b]тиофен-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(4-фторбензо[b]тиофен-2-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(5-фторбензо[b]тиофен-2-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(6-фторбензо[b]тиофен-2-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(7-фторбензо[b]тиофен-2-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-7-(2,6-диметилморфолин-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-2-этил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-8-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохино-лина;
4-бензо[b]тиофен-2-ил-8-метокси-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохино-лина;
4-бензо[b]тиофен-2-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-8-
ола;
4-бензо[b]тиофен-2-ил-2,8-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-2-этил-8-фтор-7-морфолин-4-ил-1,2,3,4-
тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-6-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохино-лина;
4-бензо[b]тиофен-2-ил-5-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохино-лина;
4-бензо[b]тиофен-2-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-
ола;
4-бензо[b]тиофен-2-ил-2,4-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-2-метил-7-пиперазин-1-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-2-метил-7-(4-метилпиперазин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-2-ил)-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-2-ил)-2-метил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-2-метил-7-морфолин-4-илметил-1,2,3,4-
тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-2-метил-7-(1-метил-1-морфолин-4-илэтил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-2-метил-7-(1-морфолин-4-илциклопропил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-2-метил-7-пиперидин-1-илметил-1,2,3,4-
тетрагидроизохинолина;
4-(бензо[b]тиофен-2-ил)-2-метил-7-пирролидин-1-илметил-1,2,3,4-тетрагидроизохинолина;
(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)диметила-мина;
[1-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]диметиламина;
[1-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]диме-тиламина;
(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)
метиламина;
[1-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]метиламина;
[1-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]метиламина;
С-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)метиламина;
1-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтиламина;
1-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропиламина;
1-(4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1Н-пиридин-2-она;
4-(бензо[b]тиофен-2-ил)-7-метансульфонил-2-метил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрила;
4-(бензо[b]тиофен-2-ил)-2,8-диметил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-3-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-3-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-3-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-4-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-4-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-1-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-8-фтор-2-метил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-8-метокси-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-2-этил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-1,2-диметил-1,2,3,4-тетрагидроизохинолина;
4-(2-метилбензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(2-хлорбензо[b]тиофен-5-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(2-хлорбензо[b]тиофен-5-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(2-хлорбензо[b]тиофен-5-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(2-хлорбензо[b]тиофен-5-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(2-хлорбензо[b]тиофен-5-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(2-хлорбензо[b]тиофен-5-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-7-пиридазин-3-ил-4-(3-трифторметилбензо[b]тиофен-5-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-7-пиримидин-2-ил-4-(3-трифторметилбензо[b]тиофен-5-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-7-пиримидин-4-ил-4-(3-трифторметилбензо[b]тиофен-5-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-7-пиримидин-5-ил-4-(3-трифторметилбензо[b]тиофен-5-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-7-пиразин-2-ил-4-(3-трифторметилбензо[b]тиофен-5-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-7-морфолин-4-ил-4-(3-трифторметилбензо[b]тиофен-5-ил)-1,2,3,4-тетрагидроизохинолина;
5-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)бензо[b]тиофен-4-карбонитрила;
4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
2-этил-4-(4-метоксибензо[b]тиофен-5-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
8-фтор-4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
2-этил-8-фтор-4-(4-метоксибензо[b]тиофен-5-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-5-ола;
4-(4-метоксибензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ола;
5-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)бензо[b]тиофен-4-ола;
5-(2-этил-1,2,3,4-тетрагидроизохинолин-4-ил)бензо[b]тиофен-4-ола;
2-этил-4-(4-метоксибензо[b]тиофен-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1,1-диоксо-1Н-λ6-бензо[b]тиофен-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-4-ола;
4-бензо[b]тиофен-5-ил-8-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-4-ола;
4-бензо[b]тиофен-5-ил-7-фтор-2-метил-1,2,3,4-тетрагидроизохинолин-4-ола;
4-(бензо[b]тиофен-5-ил)-4-метокси-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-2,4-диметил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-4-карбонитрила;
4-(бензо[b]тиофен-5-ил)-4-фтор-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-4-хлор-2-метил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ола;
4-(бензо[b]тиофен-5-ил)-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-2,7-диметил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-7-фтор-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-2-этил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
[6-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]ди-метиламина;
[6-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]ме-тиламина;
6-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-илами-на;
4-бензо[b]тиофен-5-ил-2-метил-7-(6-морфолин-4-илпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-2-метил-7-(6-трифторметилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
6-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-карбо-нитрила;
4-бензо[b]тиофен-5-ил-8-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохино-лина;
4-бензо[b]тиофен-5-ил-8-метокси-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохино-лина;
4-бензо[b]тиофен-5-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-8-
ола;
4-бензо[b]тиофен-5-ил-2,8-диметил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-2-этил-8-фтор-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохиноли-на;
4-бензо[b]тиофен-5-ил-6-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохино-лина;
4-бензо[b]тиофен-5-ил-5-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохино-лина;
4-бензо[b]тиофен-5-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-
ола;
4-бензо[b]тиофен-5-ил-2,4-диметил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-8-фтор-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-2-метил-7-(3-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-7-(3-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-2-метил-7-(6-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-7-(6-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-8-фтор-2-метил-7-пиразин-2-ил-1,2,3,4-
тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-8-метокси-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохино-лина;
4-бензо[b]тиофен-5-ил-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолин-8-ола;
4-бензо[b]тиофен-5-ил-2,8-диметил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-8-фтор-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохино-лина;
4-бензо[b]тиофен-5-ил-8-метокси-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохи-нолина;
4-бензо[b]тиофен-5-ил-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин-8-
ола;
4-бензо[b]тиофен-5-ил-2,8-диметил-7-пиримидин-2-ил-1,2,3,4-
тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-8-фтор-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-8-метокси-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин-8-ола;
4-(бензо[b]тиофен-5-ил)-2,8-диметил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-8-фтор-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-8-метокси-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин-8-ола;
4-(бензо[b]тиофен-5-ил)-2,8-диметил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-7-(3,5-диметилизоксазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(тиазол-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(5-метилтиазол-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-2-метил-7-[1,3,5]триазин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-2-метил-7-[1,2,4]триазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-2-метил-7-[1,2,4]триазин-5-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-2-метил-7-[1,2,4]триазин-6-ил-1,2,3,4-тетрагидроизохинолина;
3-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циннолина;
1-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)фталазина;
2-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиноксалина;
2-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
6-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
7-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
2-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-2Н-[1,2,4]триазоло[4,3-а]пиридин-3-она;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-7-(2,6-диметилморфолин-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-2-этил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-8-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохино-лина;
4-бензо[b]тиофен-5-ил-8-метокси-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохи-нолина;
4-бензо[b]тиофен-5-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-8-
ола;
4-бензо[b]тиофен-5-ил-2,8-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-2-этил-8-фтор-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-6-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохино-лина;
4-бензо[b]тиофен-5-ил-5-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохино-лина;
4-бензо[b]тиофен-5-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-
ола;
4-бензо[b]тиофен-5-ил-2,4-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-2-метил-7-пиперазин-1-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-2-метил-7-(4-метилпиперазин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(морфолин-4-ил)метил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-2-метил-7-(1-метил-1-морфолин-4-илэтил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-2-метил-7-(1-морфолин-4-илциклопропил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(пиперидин-1-ил)метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-5-ил)-2-метил-7-(пирролидин-1-ил)метил-1,2,3,4-тетрагидроизохинолина;
(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)диметил-амина;
[1-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]диметиламина;
[1-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]диме-тиламина;
(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)
метиламина;
[1-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил] метиламина;
[1-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]метил-амина;
С-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)метиламина;
1-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтиламина;
1-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропиламина;
1-(4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1Н-пиридин-2-она;
4-(бензо[b]тиофен-5-ил)-7-метансульфонил-2-метил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрила;
4-бензо[b]тиофен-5-ил-8-фтор-2-метил-1,2,3,4-тетрагидроизохинолин-7-
карбонитрила;
4-(бензо[b]тиофен-5-ил)-2,8-диметил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-1-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-8-фтор-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-2-этил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-1,2-диметил-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(2-метилбензо[b]тиофен-6-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1,1-диоксо-1Н-1λ6-бензо[b]тиофен-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-4-ола;
4-(бензо[b]тиофен-6-ил)-4-метокси-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-2,4-диметил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-4-карбонитрила;
4-(бензо[b]тиофен-6-ил)-4-фтор-2-метил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-4-хлор-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(2-хлорбензо[b]тиофен-6-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(2-хлорбензо[b]тиофен-6-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(2-хлорбензо[b]тиофен-6-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(2-хлорбензо[b]тиофен-6-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(2-хлорбензо[b]тиофен-6-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(2-хлорбензо[b]тиофен-6-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-7-пиридазин-3-ил-4-(3-трифторметилбензо[b]тиофен-6-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-7-пиримидин-2-ил-4-(3-трифторметилбензо[b]тиофен-6-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-7-пиримидин-4-ил-4-(3-трифторметилбензо[b]тиофен-6-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-7-пиримидин-5-ил-4-(3-трифторметилбензо[b]тиофен-6-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-7-пиразин-2-ил-4-(3-трифторметилбензо[b]тиофен-6-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-7-морфолин-4-ил-4-(3-трифторметилбензо[b]тиофен-6-ил)-1,2,3,4-тетрагидроизохинолина;
4-(7-метоксибензо[b]тиофен-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(7-метоксибензо[b]тиофен-6-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
2-этил-4-(7-метоксибензо[b]тиофен-6-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
8-фтор-4-(7-метоксибензо[b]тиофен-6-ил)-2-метил-7-(пиридазин-3-ил)1,2,3,4-тетрагидроизохинолина;
2-этил-8-фтор-4-(7-метоксибензо[b]тиофен-6-ил)-7-(пиридазин-3-ил)1,2,3,4-тетрагидроизохинолина;
4-(7-метоксибензо[b]тиофен-6-ил)-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(7-метоксибензо[b]тиофен-6-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(7-метоксибензо[b]тиофен-6-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(7-метоксибензо[b]тиофен-6-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(7-метоксибензо[b]тиофен-6-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(7-метоксибензо[b]тиофен-6-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(7-метоксибензо[b]тиофен-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ола;
4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ола;
4-(бензо[b]тиофен-6-ил)-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-2,7-диметил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-7-фтор-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-2-этил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
[6-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]ди-метиламина;
[6-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]ме-тиламина;
6-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-иламина;
4-бензо[b]тиофен-6-ил-2-метил-7-(6-морфолин-4-илпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-2-метил-7-(6-трифторметилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
6-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-карбо-нитрила;
4-бензо[b]тиофен-6-ил-8-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохино-лина;
4-бензо[b]тиофен-6-ил-8-метокси-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохи-нолина;
4-бензо[b]тиофен-6-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-8-
ола;
4-бензо[b]тиофен-6-ил-2,8-диметил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-2-этил-8-фтор-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохиноли-на;
4-бензо[b]тиофен-6-ил-6-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохино-лина;
4-бензо[b]тиофен-6-ил-5-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохино-лина;
4-бензо[b]тиофен-6-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-
ола;
4-бензо[b]тиофен-6-ил-2,4-диметил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-8-фтор-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-2-метил-7-(3-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-7-(3-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-2-метил-7-(6-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-7-(6-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-8-фтор-2-метил-7-пиразин-2-ил-1,2,3,4-
тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-8-метокси-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохино-лина;
4-бензо[b]тиофен-6-ил-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолин-8-ола;
4-бензо[b]тиофен-6-ил-2,8-диметил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-8-фтор-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохино-лина;
4-бензо[b]тиофен-6-ил-8-метокси-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин-8-
ола;
4-бензо[b]тиофен-6-ил-2,8-диметил-7-пиримидин-2-ил-1,2,3,4-
тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-8-фтор-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-8-метокси-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин-8-ола;
4-(бензо[b]тиофен-6-ил)-2,8-диметил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-8-фтор-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-8-метокси-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин-8-ола;
4-(бензо[b]тиофен-6-ил)-2,8-диметил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-7-(3,5-диметилизоксазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(тиазол-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(5-метилтиазол-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-2-метил-7-[1,3,5]триазин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-2-метил-7-[1,2,4]триазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-2-метил-7-[1,2,4]триазин-5-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-2-метил-7-[1,2,4]триазин-6-ил-1,2,3,4-тетрагидроизохинолина;
3-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циннолина;
1-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)фталазина;
2-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиноксалина;
2-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
6-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
7-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
2-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-2Н-[1,2,4]триазоло[4,3-а]пиридин-3-она;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-7-(2,6-диметилморфолин-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-2-этил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-8-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохино-лина;
4-бензо[b]тиофен-6-ил-8-метокси-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохи-нолина;
4-бензо[b]тиофен-6-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-8-
ола;
4-бензо[b]тиофен-6-ил-2,8-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-2-этил-8-фтор-7-морфолин-4-ил-1,2,3,4-
тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-6-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохино-лина;
4-бензо[b]тиофен-6-ил-5-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохино-лина;
4-бензо[b]тиофен-6-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-
ола;
4-бензо[b]тиофен-6-ил-2,4-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-2-метил-7-пиперазин-1-ил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-2-метил-7-(4-метилпиперазин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(морфолин-4-ил)метил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-2-метил-7-(1-метил-1-морфолин-4-илэтил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-6-ил-2-метил-7-(1-морфолин-4-илциклопропил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(пиперидин-1-ил)метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-2-метил-7-(пирролидин-1-ил)метил-1,2,3,4-тетрагидроизохинолина;
(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)диметил-амина;
[1-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]диметиламина;
[1-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]диме-тиламина;
(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)
метиламина;
[1-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]метиламина;
[1-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]метил-амина;
С-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)метиламина;
1-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтиламина;
1-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропиламина;
1-(4-бензо[b]тиофен-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1Н-пиридин-2-она;
4-(бензо[b]тиофен-6-ил)-7-метансульфонил-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрила;
4-бензо[b]тиофен-6-ил-8-фтор-2-метил-1,2,3,4-тетрагидроизохинолин-7-
карбонитрила;
4-(бензо[b]тиофен-6-ил)-2,8-диметил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-7-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-7-ил)-2-этил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-7-ил)-8-фтор-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-7-ил)-2-этил-8-фтор-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-7-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[b]тиофен-7-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-2-циклопропил-1,2,3,4-тетрагидроизохинолина;
(4-бензо[b]тиофен-5-ил-3,4-дигидро-1Н-изохинолин-2-ил)ацетонитрила;
[2-(4-бензо[b]тиофен-5-ил-3,4-дигидро-1Н-изохинолин-2-ил)этил]диметиламина;
2-(4-бензо[b]тиофен-5-ил-3,4-дигидро-1Н-изохинолин-2-ил)этанола;
4-бензо[b]тиофен-5-ил-2-изопропил-1,2,3,4-тетрагидроизохинолина;
4-бензо[b]тиофен-5-ил-2-метил-8-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина.
4-(бензофуран-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-1-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-этил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-1,2-диметил-1,2,3,4-тетрагидроизохинолина;
4-(5-хлорбензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(5-фторбензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(7-фторбензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(5-метоксибензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(7-метоксибензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(7-метоксибензофуран-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(7-метоксибензофуран-2-ил)-8-фтор-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(7-метоксибензофуран-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(7-метоксибензофуран-2-ил)-8-фтор-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-бензофуран-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-4-ола;
4-бензофуран-2-ил-4-метокси-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2,4-диметил-1,2,3,4-тетрагидроизохинолина;
4-бензофуран-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-4-карбонитрила;
4-(бензофуран-2-ил)-4-фтор-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-4-хлор-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2,5-диметил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-7-фтор-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-7-гидрокси-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2,7-диметил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(4-фторбензофуран-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(5-фторбензофуран-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(6-фторбензофуран-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(7-фторбензофуран-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-этил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
[6-(4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]диметиламина;
[6-(4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]метиламина;
6-(4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-иламина;
4-(бензофуран-2-ил)-2-метил-7-(6-морфолин-4-илпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-7-(6-трифторметилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
6-(4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-карбонитрила;
4-(бензофуран-2-ил)-8-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-8-метокси-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-8-ола;
4-(бензофуран-2-ил)-2,8-диметил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-этил-8-фтор-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-6-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-5-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-мегил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ола;
4-(бензофуран-2-ил)-2,4-диметил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-8-фтор-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-7-(3-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-7-(3-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-7-(6-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-7-(6-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-8-фтор-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-8-фтор-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-8-фтор-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-8-фтор-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-7-(3,5-диметилизоксазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-7-(тиазол-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-7-(5-метилтиазол-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-7-[1,3,5]триазин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-7-[1,2,4]триазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-7-[1,2,4]триазин-5-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-7-[1,2,4]триазин-6-ил-1,2,3,4-тетрагидроизохинолина;
3-(4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циннолина;
1-(4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)фталазина;
2-(4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиноксалина;
2-(4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
6-(4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
7-(4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
2-(4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-2Н-[1,2,4]триазоло[4,3-а]пиридин-3-она;
4-(бензофуран-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(4-фторбензофуран-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(5-фторбензофуран-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(6-фторбензофуран-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(7-фторбензофуран-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-7-(2,6-диметилморфолин-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-этил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-8-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-8-метокси-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-8-ола;
4-(бензофуран-2-ил)-2,8-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-этил-8-фтор-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-6-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-5-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ола;
4-(бензофуран-2-ил)-2,4-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-бензофуран-2-ил-2-метил-7-пиперазин-1-ил-1,2,3,4-тетрагидроизохинолина;
4-бензофуран-2-ил-2-метил-7-(4-метилпиперазин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-7-(морфолин-4-ил)метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-7-(1-метил-1-морфолин-4-илэтил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-7-(1-морфолин-4-илциклопропил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-7-(пиперидин-1-ил)метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-7-(пирролидин-1-ил)метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)диметиламина;
[1-4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]диметиламина;
[1-4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]диметиламина;
(4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)метиламина;
[1-4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил] метиламина;
[1-4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]метиламина;
С-4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)метиламина;
1-4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтиламина;
1-4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропиламина;
1-(4-бензофуран-2-ил-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1Н-пиридин-2-она;
4-(бензофуран-2-ил)-7-метансульфонил-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрила;
4-(бензофуран-2-ил)-2,8-диметил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-3-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-3-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-3-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-4-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-4-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-1-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-8-фтор-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-этил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-1,2-диметил-1,2,3,4-тетрагидроизохинолина;
4-(3-метилбензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-бензофуран-5-ил-2-метил-1,2,3,4-тетрагидроизохинолин-4-ола;
4-(бензофуран-5-ил)-4-метокси-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2,4-диметил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-карбонитрила;
4-(бензофуран-5-ил)-4-фтор-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-4-хлор-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ола;
4-(бензофуран-5-ил)-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2,7-диметил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-7-фтор-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-этил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
[6-4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]диметиламина;
[6-4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]метиламина;
6-4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-иламина;
6-4-(бензофуран-5-ил)-2-метил-7-(6-морфолин-4-илпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
6-4-(бензофуран-5-ил)-2-метил-7-(6-трифторметилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
6-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-карбонитрила;
4-(бензофуран-5-ил)-8-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-8-метокси-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-8-ола;
4-(бензофуран-5-ил)-2,8-диметил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-этил-8-фтор-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-6-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-5-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ола;
4-(бензофуран-5-ил)-2,4-диметил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-8-фтор-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-(3-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-7-(3-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-(6-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-7-(6-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-8-фтор-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-8-метокси-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолин-8-ола;
4-(бензофуран-5-ил)-2,8-диметил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-8-фтор-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-8-метокси-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин-8-ола;
4-(бензофуран-5-ил)-2,8-диметил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-8-фтор-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-8-метокси-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин-8-ола;
4-(бензофуран-5-ил)-2,8-диметил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-8-фтор-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-8-метокси-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин-8-ола;
4-(бензофуран-5-ил)-2,8-диметил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-7-(3,5-диметилизоксазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-(тиазол-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-(5-метилтиазол-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-[1,3,5]триазин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-[1,2,4]триазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-[1,2,4]триазин-5-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-[1,2,4]триазин-6-ил-1,2,3,4-тетрагидроизохинолина;
3-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циннолина;
1-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)фталазина;
2-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиноксалина;
2-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
6-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
7-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
2-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-2Н-[1,2,4]триазоло[4,3-а]пиридин-3-она;
4-(бензофуран-5-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-7-(2,6-диметилморфолин-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-этил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-8-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-8-метокси-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-8-ола;
4-(бензофуран-5-ил)-2,8-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-этил-8-фтор-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-6-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-5-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ола;
4-(бензофуран-5-ил)-2,4-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-(морфолин-4-ил)метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-(1-метил-1-морфолин-4-илэтил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-(1-морфолин-4-илциклопропил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-(пиперидин-1-ил)метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-7-(пирролидин-1-ил)метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)диметиламина;
[1-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]диметиламина;
[1-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]диметиламина;
(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)метиламина;
[1-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил] метиламина;
[1-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]метиламина;
С-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)метиламина;
1-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтиламина;
1-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропиламина;
1-(4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1Н-пиридин-2-она;
4-(бензофуран-5-ил)-7-метансульфонил-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрила;
4-(бензофуран-5-ил)-8-фтор-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрила;
4-(бензофуран-5-ил)-2,8-диметил-1,2,3,4-тетрагидроизохинолина;
4-(2,3-дигидробензофуран-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-1-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-этил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-1,2-диметил-1,2,3,4-тетрагидроизохинолина;
4-бензофуран-6-ил-2-метил-1,2,3,4-тетрагидроизохинолин-4-ола;
4-(бензофуран-6-ил)-4-метокси-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2,4-диметил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-карбонитрила;
4-(бензофуран-6-ил)-4-фтор-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-4-хлор-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ола;
4-(бензофуран-6-ил)-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2,7-диметил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-7-фтор-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-этил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
[6-(4-бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]диметиламина;
[6-(4-бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]метиламина;
6-(4-бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-иламина;
4-(бензофуран-6-ил)-2-метил-7-(6-морфолин-4-илпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-(6-трифторметилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-карбонитрила;
4-(бензофуран-6-ил)-8-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-8-метокси-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-8-ола;
4-(бензофуран-6-ил)-2,8-диметил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-этил-8-фтор-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-6-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-5-фтор-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-8-фтор-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-(3-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-7-(3-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-(6-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-7-(6-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-8-фтор-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-8-метокси-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолин-8-ола;
4-(бензофуран-6-ил)-2,8-диметил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-8-фтор-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-8-метокси-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолин-8-ола;
4-(бензофуран-6-ил)-2,8-диметил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-8-фтор-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-8-метокси-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин-8-ола;
4-(бензофуран-6-ил)-2,8-диметил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-8-фтор-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-8-метокси-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин-8-ола;
4-(бензофуран-6-ил)-2,8-диметил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-7-(3,5-диметилизоксазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-(тиазол-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-(5-метилтиазол-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-[1,3,5]триазин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-[1,2,4]триазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-[1,2,4]триазин-5-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-[1,2,4]триазин-6-ил-1,2,3,4-тетрагидроизохинолина;
3-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циннолина;
1-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)фталазина;
2-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиноксалина;
2-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
6-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
7-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
2-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-2Н-[1,2,4]триазоло[4,3-а]пиридин-3-она;
4-(бензофуран-6-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-7-(2,6-диметилморфолин-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-этил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-8-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-8-метокси-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-8-ола;
4-(бензофуран-6-ил)-2,8-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-этил-8-фтор-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-6-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-5-фтор-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ола;
4-(бензофуран-6-ил)-2,4-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-(морфолин-4-ил)метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-(1-метил-1-морфолин-4-илэтил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-(1-морфолин-4-илциклопропил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-(пиперидин-1-ил)метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-7-(пирролидин-1-ил)метил-1,2,3,4-тетрагидроизохинолина;
(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)диметиламина;
[1-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]диметиламина;
[1-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]диметиламина;
(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)метиламина;
[1-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]метиламина;
[1-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил] метиламина;
С-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)метиламина;
1-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтиламина;
1-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропиламина;
1-(4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1Н-пиридин-2-она;
4-(бензофуран-6-ил)-7-метансульфонил-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрила;
4-(бензофуран-6-ил)-8-фтор-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрила;
4-(бензофуран-6-ил)-2,8-диметил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-7-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-7-ил)-2-этил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-7-ил)-8-фтор-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-7-ил)-2-этил-8-фтор-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-7-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензофуран-7-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина.
4-(1Н-индол-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-1-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
2-этил-4-(1Н-индол-2-ил)-1,2,3,4-тетрагидроизохинолина;
1,2-диметил-4-(1Н-индол-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(5-метокси-1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-ола;
4-(1Н-индол-2-ил)-4-метокси-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2,4-диметил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-карбонитрила;
4-фтор-4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-хлор-4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ола;
4-(1Н-индол-2-ил)-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолина;
2,7-диметил-4-(1Н-индол-2-ил)-1,2,3,4-тетрагидроизохинолина;
7-фтор-4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1-метил-1Н-индол-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(7-фтор-1Н-индол-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
2-этил-4-(1Н-индол-2-ил)-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
[6-(4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]диметиламина;
[6-(4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]метиламина;
6-(4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-иламина;
4-(1Н-индол-2-ил)-2-метил-7-(6-морфолин-4-илпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-7-(6-трифторметилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
6-(4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-карбонитрила;
8-фтор-4-(1Н-индол-2-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
2-этил-8-фтор-4-(1Н-индол-2-ил)-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
6-фтор-4-(1Н-индол-2-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
5-фтор-4-(1Н-индол-2-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-8-метокси-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-8-ола;
2,8-диметил-4-(1Н-индол-2-ил)-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ола;
2,4-диметил-4-(1Н-индол-2-ил)-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолина;
8-фтор-4-(1Н-индол-2-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-7-(3-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-7-(3-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-7-(6-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-7-(6-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-8-фтор-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолина;
8-фтор-4-(1Н-индол-2-ил)-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
8-фтор-4-(1Н-индол-2-ил)-2-метил-7-пиримидин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
8-фтор-4-(1Н-индол-2-ил)-2-метил-7-пиримидин-5-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-7-(3,5-диметилизоксазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-7-(тиазол-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-7-(5-метилтиазол-2-ил)-1,2,3,4-тетрагидроизохинолина;
3-(4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циннолина;
1-(4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)фталазина;
2-(4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиноксалина;
2-(4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
6-(4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
7-(4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
2-(4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-2Н-[1,2,4]триазоло[4,3-а]пиридин-3-она;
4-(1Н-индол-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1-метил-1Н-индол-2-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(7-фтор-1Н-индол-2-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
7-(2,6-диметилморфолин-4-ил)-4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
2-этил-4-(1Н-индол-2-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
8-фтор-4-(1Н-индол-2-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
2-этил-8-фтор-4-(1Н-индол-2-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
6-фтор-4-(1Н-индол-2-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
5-фтор-4-(1Н-индол-2-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-8-метокси-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-8-ола;
4-(1Н-индол-2-ил)-2,8-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ола;
4-(1Н-индол-2-ил)-2,4-диметил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-7-(морфолин-4-ил)метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-7-(1-метил-1-морфолин-4-илэтил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-7-(1-морфолин-4-илциклопропил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-7-(пиперидин-1-ил)метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-7-(пирролидин-1-ил)метил-1,2,3,4-тетрагидроизохинолина;
(4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)диметиламина;
[1-(4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]диметиламина;
[1-(4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]диметиламина;
(4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)метиламина;
[1-(4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]метиламина;
[1-(4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]метиламина;
С-(4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)метиламина;
1-(4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтиламина;
1-(4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропиламина;
1-(4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1Н-пиридин-2-она;
4-(1Н-индол-2-ил)-7-метансульфонил-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрила;
2,8-диметил-4-(1Н-индол-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-3-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-1-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
2-этил-4-(1Н-индол-5-ил)-1,2,3,4-тетрагидроизохинолина;
1,2-диметил-4-(1Н-индол-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-ола;
4-(1Н-индол-5-ил)-4-метокси-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2,4-диметил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-карбонитрила;
4-фтор-4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-хлор-4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ола;
4-(1Н-индол-5-ил)-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолина;
2,7-диметил-4-(1Н-индол-5-ил)-1,2,3,4-тетрагидроизохинолина;
7-фтор-4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1-метилиндол-5-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(7-фтор-1Н-индол-5-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
2-этил-4-(1Н-индол-5-ил)-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
[6-(4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]диметиламина;
[6-(4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил] метиламина;
6-(4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-иламина;
4-(1Н-индол-5-ил)-2-метил-7-(6-морфолин-4-илпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-7-(6-трифторметилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
6-(4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-карбонитрила;
8-фтор-4-(1Н-индол-5-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
2-этил-8-фтор-4-(1Н-индол-5-ил)-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
6-фтор-4-(1Н-индол-5-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
5-фтор-4-(1Н-индол-5-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-8-метокси-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-8-ола;
2,8-диметил-4-(1Н-индол-5-ил)-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ола;
2,4-диметил-4-(1Н-индол-5-ил)-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолина;
8-фтор-4-(1Н-индол-5-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-7-(3-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-7-(3-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-7-(6-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-7-(6-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-8-фтор-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолина;
8-фтор-4-(1Н-индол-5-ил)-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
8-фтор-4-(1Н-индол-5-ил)-2-метил-7-пиримидин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
8-фтор-4-(1Н-индол-5-ил)-2-метил-7-пиримидин-5-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-7-(3,5-диметилизоксазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-7-(тиазол-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-7-(5-метилтиазол-2-ил)-1,2,3,4-тетрагидроизохинолина;
3-(4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циннолина;
1-(4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)фталазина;
2-(4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиноксалина;
2-(4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
6-(4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
7-(4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
2-(4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-2Н-[1,2,4]триазоло[4,3-а]пиридин-3-она;
4-(1Н-индол-5-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1-метил-1Н-индол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(7-фтор-1Н-индол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
7-(2,6-диметилморфолин-4-ил)-4-(1Н-индол-5-ил)-2-метил-1,2,3,4 тетрагидроизохинолина;
2-этил-4-(1Н-индол-5-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
8-фтор-4-(1Н-индол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
2-этил-8-фтор-4-(1Н-индол-5-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
6-фтор-4-(1Н-индол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
5-фтор-4-(1Н-индол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-8-метокси-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-8-ола;
2,8-диметил-4-(1Н-индол-5-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ола;
2,4-диметил-4-(1Н-индол-5-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-7-(морфолин-4-ил)метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-7-(1-метил-1-морфолин-4-илэтил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-7-(1-морфолин-4-илциклопропил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-7-(пиперидин-1-ил)метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-7-(пирролидин-1-ил)метил-1,2,3,4-тетрагидроизохинолина;
(4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)диметиламина;
[1-(4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]диметиламина;
[1-(4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]диметиламина;
(4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)метиламина;
[1-(4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]метиламина;
[1-(4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]метиламина;
С-(4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)метиламина;
1-(4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтиламина;
1-(4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропиламина;
1-(4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1Н-пиридин-2-она;
4-(1Н-индол-5-ил)-7-метансульфонил-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрила;
4-(1Н-индол-5-ил)-2,8-диметил-1,2,3,4-тетрагидроизохинолина;
4-(1-бензил-1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-1-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(3-хлор-1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
2-этил-4-(1Н-индол-6-ил)-1,2,3,4-тетрагидроизохинолина;
1,2-диметил-4-(1Н-индол-6-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-ола;
4-(1Н-индол-6-ил)-4-метокси-2-метил-1,2,3,4-тетрагидроизохинолина;
2,4-диметил-4-(1Н-индол-6-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-карбонитрила;
4-фтор-4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-хлор-4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ола;
4-(1Н-индол-6-ил)-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолина;
2,7-диметил-4-(1Н-индол-6-ил)-1,2,3,4-тетрагидроизохинолина;
7-фтор-4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1-метилиндол-6-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(7-фтор-1Н-индол-6-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
2-этил-4-(1Н-индол-6-ил)-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
[6-(4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]диметиламина;
[6-(4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил] метиламина;
6-(4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-иламина;
4-(1Н-индол-6-ил)-2-метил-7-(6-морфолин-4-илпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-7-(6-трифторметилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
6-(4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-карбонитрила;
8-фтор-4-(1Н-индол-6-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
2-этил-8-фтор-4-(1Н-индол-6-ил)-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
6-фтор-4-(1Н-индол-6-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
5-фтор-4-(1Н-индол-6-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-8-метокси-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-8-ола;
2,8-диметил-4-(1Н-индол-6-ил)-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ола;
2,4-диметил-4-(1H-индол-6-ил)-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолина;
8-фтор-4-(1Н-индол-6-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-7-(3-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-7-(3-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-7-(6-метилпиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-7-(6-метоксипиразин-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
8-фтор-4-(1Н-индол-6-ил)-2-метил-7-пиразин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолина;
8-фтор-4-(1Н-индол-6-ил)-2-метил-7-пиримидин-2-мл-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
8-фтор-4-(1Н-индол-6-ил)-2-метил-7-пиримидин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
8-фтор-4-(1Н-индол-6-ил)-2-метил-7-пиримидин-5-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-7-(3,5-диметилизоксазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-7-(тиазол-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-7-(5-метилтиазол-2-ил)-1,2,3,4-тетрагидроизохинолина;
3-(4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циннолина;
1-(4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)фталазина;
2-(4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиноксалина;
2-(4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
6-(4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
7-(4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
2-(4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-2Н-[1,2,4]триазоло[4,3-а]пиридин-3-она;
4-(1Н-индол-6-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1-метил-1Н-индол-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(7-фтор-1Н-индол-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
7-(2,6-диметилморфолин-4-ил)-4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
2-этил-4-(1Н-индол-6-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
8-фтор-4-(1Н-индол-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
2-этил-8-фтор-4-(1Н-индол-6-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
6-фтор-4-(1Н-индол-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
5-фтор-4-(1Н-индол-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-8-метокси-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-8-ола;
2,8-диметил-4-(1Н-индол-6-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ола;
2,4-диметил-4-(1Н-индол-6-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-7-(морфолин-4-ил)метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-7-(1-метил-1-морфолин-4-илэтил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-7-(1-морфолин-4-илциклопропил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-7-(пиперидин-1-ил)метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-7-(пирролидин-1-ил)метил-1,2,3,4-тетрагидроизохинолина;
(4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)диметиламина;
[1-(4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]диметиламина;
[1-(4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]диметиламина;
(4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил)метиламина;
[1-(4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]метиламина;
[1-(4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]метиламина;
С-(4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)метиламина;
1-(4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтиламина;
1-(4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропиламина;
1-(4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1Н-пиридин-2-она;
4-(1Н-индол-6-ил)-7-метансульфонил-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрила;
2,8-диметил-4-(1Н-индол-6-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(1-метил-1Н-индол-5-ил)-1,2,3,4-тетрагидроизохинолина;
8-фтор-2-метил-4-(1-метил-1Н-индол-5-ил)-1,2,3,4-тетрагидроизохинолина;
2-этил-8-фтор-4-(1-метил-1Н-индол-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1-бензил-1Н-индол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
3-[5-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)индол-1-илметил] бензонитрила;
2-[5-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)индол-1-илметил] бензонитрила;
2-метил-4-(1-метил-1Н-индол-6-ил)-1,2,3,4-тетрагидроизохинолина;
8-фтор-2-метил-4-(1-метил-1Н-индол-6-ил)-1,2,3,4-тетрагидроизохинолина;
2-этил-8-фтор-4-(1-метил-1Н-индол-6-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1-бензил-1Н-индол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
3-[6-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)индол-1-илметил]бензонитрила и
2-[6-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)индол-1-илметил]бензонитрила.
4-(индазол-1-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-3-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-1-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(1-метил-1Н-индазол-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(6-метоксииндазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(7-метоксииндазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(7-фториндазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(7-хлориндазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(7-метилиндазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
8-фтор-4-(1Н-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
2-этил-4-(1Н-индазол-5-ил)-1,2,3,4-тетрагидроизохинолина;
2-этил-8-фтор-4-(1Н-индазол-5-ил)-1,2,3,4-тетрагидроизохинолина;
1,2-диметил-4-(1Н-индазол-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-ола;
4-(1Н-индазол-5-ил)-4-метокси-2-метил-1,2,3,4-тетрагидроизохинолина;
2,4-диметил-4-(1Н-индазол-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-карбонитрила;
4-фтор-4-(1Н-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-хлор-4-(1H-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ола;
4-(1Н-индазол-5-ил)-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолина;
2,7-диметил-4-(1Н-индазол-5-ил)-1,2,3,4-тетрагидроизохинолина;
7-фтор-2-метил-4-(1Н-индазол-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1-метил-1Н-индазол-5-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(7-фтор-1Н-индазол-5-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
2-этил-4-(1Н-индазол-5-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
[6-(4-(1Н-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]диметиламина;
[6-(4-(1Н-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]метиламина;
6-(4-(1Н-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-иламина;
4-(1Н-индазол-5-ил)-2-метил-7-(6-морфолин-4-илпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-2-метил-7-(6-трифторметилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-7-(3,5-диметилизоксазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-2-метил-7-(тиазол-2-ил)-1,2,3,4-тетрагидроизохинолина;
3-(4-(1Н-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циннолина;
1-(4-(1Н-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)фталазина;
2-(4-(1Н-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиноксалина;
2-(4-(1Н-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
6-(4-(1Н-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
7-(4-(1Н-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
2-(4-(1Н-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-2Н-[1,2,4]триазоло[4,3-а]пиридин-3-она;
4-(1Н-индазол-5-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1-метил-1Н-индазол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(7-фтор-1Н-индазол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
7-(2,6-диметилморфолин-4-ил)-4-(1Н-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
2-этил-4-(1Н-индазол-5-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
8-фтор-4-(1Н-индазол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
2-этил-8-фтор-4-(1Н-индазол-5-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
6-фтор-4-(1Н-индазол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
5-фтор-4-(1Н-индазол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-8-метокси-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-8-ола;
2,8-диметил-4-(1Н-индазол-5-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ола;
2,4-диметил-4-(1Н-индазол-5-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-2-метил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-2-метил-7-((морфолин-4-ил)метил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-2-метил-7-(1-метил-1-морфолин-4-илэтил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-2-метил-7-(1-морфолин-4-илциклопропил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-2-метил-7-пиперидин-1-илметил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-2-метил-7-пирролидин-1-илметил-1,2,3,4-тетрагидроизохинолина;
[4-(1Н-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил]диметиламина;
[4-(1Н-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил]метиламина;
1-(4-(1Н-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1Н-пиридин-2-она;
4-(1Н-индазол-5-ил)-7-метансульфонил-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрила;
4-(1Н-индазол-5-ил)-2,8-диметил-1,2,3,4-тетрагидроизохинолина;
7-фтор-2-метил-4-(1-метил-1Н-индазол-5-ил)-1,2,3,4-тетрагидроизохинолина;
2-этил-7-фтор-4-(1-метил-1Н-индазол-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1-бензил-1Н-индазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
3-[5-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)индазол-1-илметил]бензонитрила;
2-[5-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)индазол-1-илметил]бензонитрила;
4-(1Н-индазол-6-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-6-ил)-1-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(1-метилиндазол-6-ил)-1,2,3,4-тетрагидроизохинолина;
4-(7-фтор-1Н-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
2-этил-4-(1Н-индазол-6-ил)-1,2,3,4-тетрагидроизохинолина;
1,2-диметил-4-(1Н-индазол-6-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-6-ил)-4-метил-1,2,3,4-тетрагидроизохинолин-4-ола;
4-(1Н-индазол-6-ил)-4-метокси-2-метил-1,2,3,4-тетрагидроизохинолина;
2,4-диметил-4-(1Н-индазол-6-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-карбонитрила;
4-фтор-4-(1Н-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-хлор-4-(1Н-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ола;
4-(1Н-индазол-6-ил)-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолина;
2,7-диметил-4-(1Н-индазол-6-ил)-1,2,3,4-тетрагидроизохинолина;
7-фтор-2-метил-4-(1Н-индазол-6-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-6-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(1-метил-1Н-индазол-6-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(7-фтор-1Н-индазол-6-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
2-этил-4-(1Н-индазол-6-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-6-ил)-2-метил-7-(6-метилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
[6-(4-(1Н-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]диметиламина;
[6-(4-(1Н-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]метиламина;
6-(4-(1Н-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-иламина;
4-(1Н-индазол-6-ил)-2-метил-7-(6-морфолин-4-илпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-6-ил)-2-метил-7-(6-трифторметилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-6-ил)-2-метил-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-6-ил)-2-метил-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-6-ил)-2-метил-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-6-ил)-2-метил-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-6-ил)-2-метил-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-6-ил)-7-(3,5-диметилизоксазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-6-ил)-2-метил-7-(тиазол-2-ил)-1,2,3,4-тетрагидроизохинолина;
3-(4-(1Н-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)циннолина;
1-(4-(1Н-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)фталазина;
2-(4-(1Н-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиноксалина;
2-(4-(1Н-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
6-(4-(1Н-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
7-(4-(1Н-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
2-(4-(1Н-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-2Н-[1,2,4]триазоло[4,3-а]пиридин-3-она;
4-(1Н-индазол-6-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(1-метил-1Н-индазол-6-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(7-фтор-1Н-индазол-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
7-(2,6-диметилморфолин-4-ил)-4-(1Н-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
2-этил-4-(1Н-индазол-6-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
8-фтор-4-(1Н-индазол-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
2-этил-8-фтор-4-(1Н-индазол-6-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
6-фтор-4-(1Н-индазол-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
5-фтор-4-(1Н-индазол-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-6-ил)-8-метокси-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-8-ола;
2,8-диметил-4-(1Н-индазол-6-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-6-ил)-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ола;
2,4-диметил-4-(1Н-индазол-6-ил)-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-6-ил)-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-6-ил)-2-метил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-6-ил)-2-метил-7-морфолин-4-илметил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-6-ил)-2-метил-7-(1-метил-1-морфолин-4-илэтил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-6-ил)-2-метил-7-(1-морфолин-4-илциклопропил)-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-6-ил)-2-метил-7-пиперидин-1-илметил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-6-ил)-2-метил-7-пирролидин-1-илметил-1,2,3,4-тетрагидроизохинолина;
[4-(1Н-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил]диметиламина;
[4-(1Н-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-илметил]метиламина;
1-(4-(1Н-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1Н-пиридин-2-она;
4-(1Н-индазол-6-ил)-7-метансульфонил-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(1Н-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-карбонитрила;
2,8-диметил-4-(1Н-индазол-6-ил)-1,2,3,4-тетрагидроизохинолина;
7-фтор-2-метил-4-(1-метил-1Н-индазол-6-ил)-1,2,3,4-тетрагидроизохинолина;
2-этил-7-фтор-4-(1-метил-1Н-индазол-6-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1-бензил-1Н-индазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
3-[6-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)индазол-1-илметил]бензонитрила;
2-[6-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)индазол-1-илметил]бензонитрила и
4-(1Н-индазол-7-ил)-2-метил-1,2,3,4-тетрагидроизохинолина.
4-(бензооксазол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензооксазол-2-ил)-2-метил(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензооксазол-2-ил)-2-метил(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензооксазол-2-ил)-2-метил(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензооксазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензооксазол-5-ил)-2-мегил-1,2,3,4-тетрагидроизохинолина;
4-(бензооксазол-5-ил)-2-метил(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензооксазол-5-ил)-2-метил(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензооксазол-5-ил)-2-метил(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(2-метилбензооксазол-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензооксазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензооксазол-6-ил)-2-метил(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензооксазол-6-ил)-2-метил(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензооксазол-6-ил)-2-метил(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(2-метилбензооксазол-6-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензооксазол-7-ил)-2-метил-1,2,3,4-тетрагидроизохинолина.
4-(бензотиазол-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензотиазол-2-ил)-2-метил(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензотиазол-2-ил)-2-метил(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензотиазол-2-ил)-2-метил(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензотиазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензотиазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензотиазол-5-ил)-2-метил(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензотиазол-5-ил)-2-метил(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензотиазол-5-ил)-2-метил(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(2-метилбензотиазол-5-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензотиазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензотиазол-6-ил)-2-метил(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензотиазол-6-ил)-2-метил(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензотиазол-6-ил)-2-метил(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(2-метилбензотиазол-6-ил)-1,2,3,4-тетрагидроизохинолина и
4-(бензотиазол-7-ил)-2-метил-1,2,3,4-тетрагидроизохинолина.
4-(бензо[d]изотиазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[d]изотиазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[d]изотиазол-5-ил)-2-метил(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[d]изотиазол-5-ил)-2-метил(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо(d]изотиазол-5-ил)-2-метил(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[d]изотиазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[d]изотиазол-6-ил)-2-метил(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[d]изотиазол-6-ил)-2-метил(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[d]изотиазол-6-ил)-2-метил(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(бензо[d]изотиазол-7-ил)-2-метил-1,2,3,4-тетрагидроизохинолина.
4-(бензо[d]изоксазол-4-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[d]изоксазол-5-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(бензо[d]изоксазол-6-ил)-2-метил-1,2,3,4-тетрагидроизохинолина и
4-(бензо[d]изоксазол-7-ил)-2-метил-1,2,3,4-тетрагидроизохинолина.
4-имидазо[1,2-а]пиридин-6-ил-2-метил-1,2,3,4-тетрагидроизохинолина;
4-имидазо[1,2-а]пиридин-6-ил-2-метил(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-имидазо[1,2-а] пиридин-6-ил-2-метил(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-имидазо[1,2-а]пиридин-6-ил-2-метил(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-имидазо[1,2-а]пиридин-7-ил-2-метил-1,2,3,4-тетрагидроизохинолина;
4-имидазо[1,2-а]пиридин-7-ил-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-имидазо[1,2-а]пиридин-7-ил-2-метил-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-имидазо[1,2-а]пиридин-7-ил-2-метил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-пиразоло[1,5-а]пиридин-5-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(морфолин-4-ил)-4-пиразоло[1,5-а]пиридин-5-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(пиперидин-1-ил)-4-пиразоло[1,5-а]пиридин-5-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-4-пиразоло[1,5-а]пиридин-5-ил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-пиразоло[1,5-а]пиридин-6-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(морфолин-4-ил)-4-пиразоло[1,5-а]пиридин-6-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(пиперидин-1-ил)-4-пиразоло[1,5-а] пиридин-6-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-4-пиразоло[1,5-а]пиридин-6-ил-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-[1,2,4]триазоло[4,3-а]пиридин-6-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(морфолин-4-ил)-4-[1,2,4]триазоло[4,3-а]пиридин-6-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(пиперидин-1-ил)-4-[1,2,4]триазоло[4,3-а]пиридин-6-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(пирролидин-1-ил)-4-[1,2,4]триазоло[4,3-а]пиридин-6-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-4-[1,2,4]триазоло[4,3-а]пиридин-7-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(морфолин-4-ил)-4-[1,2,4]триазоло[4,3-а]пиридин-7-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(пиперидин-1-ил)-4-[1,2,4]триазоло[4,3-а]пиридин-7-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(пирролидин-1-ил)-4-[1,2,4]триазоло[4,3-а]пиридин-7-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-4-тиено[2,3-b]пиридин-2-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(морфолин-4-ил)-4-тиено[2,3-b]пиридин-2-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(пиперидин-1-ил)-4-тиено[2,3-b]пиридин-2-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(пирролидин-1-ил)-4-тиено[2,3-b]пиридин-2-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-4-тиено[2,3-b]пиридин-5-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(морфолин-4-ил)-4-тиено[2,3-b]пиридин-5-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(пиперидин-1-ил)-4-тиено[2,3-b]пиридин-5-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(пирролидин-1-ил)-4-тиено[2,3-b]пиридин-5-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-4-тиено[2,3-b]пиридин-6-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(морфолин-4-ил)-4-тиено[2,3-b]пиридин-6-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(пиперидин-1-ил)-4-тиено[2,3-b]пиридин-6-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(пирролидин-1-ил)-4-тиено[2,3-b]пиридин-6-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-4-тиено[3,2-b]пиридин-2-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(морфолин-4-ил)-4-тиено[3,2-b]пиридин-2-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(пиперидин-1-ил)-4-тиено[3,2-b]пиридин-2-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(пирролидин-1-ил)-4-тиено[3,2-b]пиридин-2-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-4-тиено[3,2-b]пиридин-5-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(морфолин-4-ил)-4-тиено[3,2-b]пиридин-5-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(пиперидин-1-ил)-4-тиено[3,2-b]пиридин-5-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(пирролидин-1-ил)-4-тиено[3,2-b]пиридин-5-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-4-тиено[3,2-b]пиридин-6-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(морфолин-4-ил)-4-тиено[3,2-b]пиридин-6-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(пиперидин-1-ил)-4-тиено[3,2-b]пиридин-6-ил-1,2,3,4-
тетрагидроизохинолина и
2-метил-7-(пирролидин-1-ил)-4-тиено[3,2-b]пиридин-6-ил-1,2,3,4-
тетрагидроизохинолина.
4-индолизин-2-ил-2-метил-1,2,3,4-тетрагидроизохинолина;
4-индолизин-2-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-индолизин-2-ил-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-индолизин-2-ил-2-метил-7-пиримидин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-индолизин-2-ил-2-метил-7-пиримидин-5-ил-1,2,3,4-тетрагидроизохинолина;
4-индолизин-2-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-индолизин-6-ил-2-метил-1,2,3,4-тетрагидроизохинолина;
4-индолизин-6-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-индолизин-6-ил-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-индолизин-6-ил-2-метил-7-пиримидин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-индолизин-6-ил-2-метил-7-пиримидин-5-ил-1,2,3,4-тетрагидроизохинолина;
4-индолизин-6-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-индолизин-7-ил-2-метил-1,2,3,4-тетрагидроизохинолина;
4-индолизин-7-ил-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-индолизин-7-ил-2-метил-7-пиримидин-2-ил-1,2,3,4-тетрагидроизохинолина;
4-индолизин-7-ил-2-метил-7-пиримидин-4-ил-1,2,3,4-тетрагидроизохинолина;
4-индолизин-7-ил-2-метил-7-пиримидин-5-ил-1,2,3,4-тетрагидроизохинолина;
4-индолизин-7-ил-2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолина.
R1 означает метил, этил или изопропил;
R2 означает Н, СН3 или гем-диметил;
R3 означает Н, СН3, ОН, ОСН3 или F;
R4 означает Н, СН3, ОН, ОСН3, -CN или F;
R5 означает Н;
R6 означает Н или метоксигруппу;
R7 означает Н;
R8 означает Н, ОН, ОСН3, CN или СН3; и
Х выбирают из группы, состоящей из нафтила, тетрагидронафтила, хинолинила, изохинолинила, хиноксалинила, 2Н-хроменила, дигидробензоциклогептенила, и конденсированный бициклический карбоцикл или конденсированный бициклический гетероцикл необязательно замещен 1-4 заместителями R14.
R1 означает метил, этил или изопропил;
R2 означает Н, СН3 или гем-диметил;
R3 означает Н, СН3, ОН, ОСН3 или F;
R4 выбирают из группы, состоящей из пиримидинила, пиридазинила, пиразинила, пирролила, изоксазолила, 3-оксо-[1,2,4]триазоло[4,3-а]пиридинила, необязательно замещенных 1-4 заместителями R14;
R5 означает Н;
R6 означает Н или метоксигруппу;
R7 означает Н;
R8 означает Н, ОН, ОСН3, -CN или СН3; и
Х выбирают из группы, состоящей из нафтила, тетрагидронафтила, хинолинила, изохинолинила, хиноксалинила, 2Н-хроменила, дигидробензоциклогептенила, и конденсированный бициклический карбоцикл или гетероцикл необязательно замещен 1-4 заместителями R14.
R4 выбирают из группы, состоящей из 3,5-диметилизоксазол-4-ила, пиримидин-4-ила, пиримидин-2-ила, пиримидин-5-ила, пиразин-2-ила, 3-метилпиразин-2-ила, 5-метилпиразин-2-ила, 6-метилпиразин-2-ила, 3-метоксипиразин-2-ила, 5-метоксипиразин-2-ила, 6-метоксипиразин-2-ила, 6-этилпиразин-2-ила, пиридазин-3-ила, 5-метилпиридазин-3-ила, 6-метилпиридазин-3-ила, 6-диметиламинопиридазин-3-ила, 6-метиламинопиридазин-3-ила, 6-аминопиридазин-3-ила, пиридазин-4-ила, хиноксалин-2-ила, хиноксалин-6-ила, хиназолин-2-ила, хиназолин-4-ила, хиназолин-6-ила, хиназолин-7-ила и 3-оксо-[1,2,4]триазоло[4,3-а]пиридин-2-ила и 2-оксо-2Н-пиридин-1-ила; и
Х означает нафтален-1-ил, 4-метилнафтален-1-ил, нафтален-2-ил, 1-фторнафтален-2-ил, 1-хлорнафтален-2-ил, 1-метоксинафтален-2-ил, 1-метилнафтален-2-ил, 3-фторнафтален-2-ил, 3-хлорнафтален-2-ил, 3-метоксинафтален-2-ил, 4-фторнафтален-2-ил, 4-хлорнафтален-2-ил, 4-метилнафтален-1-ил, 5-фторнафтален-2-ил, 5-хлорнафтален-2-ил, 5-метилнафтален-2-ил, 6-метоксинафтален-2-ил, 6-хлорнафтален-2-ил, 6-фторнафтален-2-ил, 6-цианонафтален-2-ил, 6-метансульфонилнафтален-2-ил, 7-метоксинафтален-2-ил, 7-хлорнафтален-2-ил, 7-фторнафтален-2-ил, 8-метоксинафтален-2-ил, 8-хлорнафтален-2-ил, 8-фторнафтален-2-ил, 5,6,7,8-тетрагидронафтален-2-ил, 2-хинолинил, 3-хинолинил, 6-хинолинил, 7-хинолинил, 1-изохинолинил, 3-изохинолинил, 6-изохинолинил, 7-изохинолинил, 2-хиноксалинил, 6-хиноксалинил, 2Н-хромен-3-ил или 8,9-дигидро-7Н-бензоциклогептен-6-ил.
R4 выбирают из группы, состоящей из морфолин-4-ила, 2,6-диметилморфолин-4-ила, пиперазин-1-ила, 4-метилпиперазин-1-ила, пиперидин-1-ила, пирролидин-1-ила, диметиламинометила, аминометила, 1-аминоциклопропила; и
Х означает нафтален-1-ил, 4-метилнафтален-1-ил, нафтален-2-ил, 1-фторнафтален-2-ил, 1-хлорнафтален-2-ил, 1-метоксинафтален-2-ил, 1-метилнафтален-2-ил, 3-фторнафтален-2-ил, 3-хлорнафтален-2-ил, 3-метоксинафтален-2-ил, 4-фторнафтален-2-ил, 4-хлорнафтален-2-ил, 4-метилнафтален-1-ил, 5-фторнафтален-2-ил, 5-хлорнафтален-2-ил, 5-метилнафтален-2-ил, 6-метоксинафтален-2-ил, 6-хлорнафтален-2-ил, 6-фторнафтален-2-ил, 7-метоксинафтален-2-ил, 7-хлорнафтален-2-ил, 7-фторнафтален-2-ил, 8-метоксинафтален-2-ил, 8-фторнафтален-2-ил, 8-метоксинафтален-2-ил, 5,6,7,8-тетрагидронафтален-2-ил, 2-хинолинил, 3-хинолинил, 6-хинолинил, 7-хинолинил, 1-изохинолинил, 3-изохинолинил, 6-изохинолинил, 7-изохинолинил, 2-хиноксалинил, 6-хиноксалинил, 2Н-хромен-3-ил или 8,9-дигидро-7Н-бензоциклогептен-6-ил.
2-метил-4-(4-метилнафтален-1-ил)-1,2,3,4-тетрагидроизохинолина;
4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
1-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
2-этил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
1,2-диметил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
2,5-диметил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин-4-ола;
4-метокси-2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
2,4-диметил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-4-карбонитрила;
4-фтор-2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-хлор-2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(6-метоксинафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(7-метоксинафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(8-метоксинафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(8-метоксинафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-ола;
4-(8-метоксинафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ола;
4-(8-хлорнафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
4-(8-хлорнафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-4-ола;
4-(8-хлорнафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолин-7-ола;
4-(8-фторнафтален-2-ил)" 2-метил-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин-7-ола;
7-метокси-4-(нафтален-2-ил)-2-метил-1,2,3,4-тетрагидроизохинолина;
2,7-диметил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
7-фтор-2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(нафтален-2-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1-фторнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1-хлорнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1-метоксинафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1-метилнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(3-фторнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(3-хлорнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(3-метоксинафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
3-(2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ил)нафталин-2-карбонитри-ла;
4-(4-фторнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(4-хлорнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(5-фторнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(5-хлорнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
6-(2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ил)нафталин-1-карбонитри-ла;
4-(5-метилнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(6-метоксинафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(6-хлорнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(6-фторнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
6-(2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ил)нафталин-2-карбонитри-ла;
4-(6-метансульфонилнафтален-2-ил)-2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолина;
4-(7-метоксинафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(7-хлорнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(7-фторнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
7-(2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ил)нафталин-2-карбонитри-ла;
4-(8-метоксинафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(8-хлорнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
4-(8-фторнафтален-2-ил)-2-метил-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
8-(2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ил)нафталин-1-карбонитри-ла;
2-этил-4-(нафтален-2-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(6-метилпиридазин-3-ил)-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолина;
диметил-[6-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]амина;
метил-[6-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил]амина;
6-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-иламина;
2-метил-7-(6-морфолин-4-илпиридазин-3-ил)-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-4-нафтален-2-ил-7-(6-трифторметилпиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
6-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)пиридазин-3-ил-карбонит-рила;
8-фтор-2-метил-4-(нафтален-2-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
8-метокси-2-метил-4-(нафтален-2-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(нафтален-2-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолин-8-ола;
2,8-диметил-4-(нафтален-2-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
2-этил-8-фтор-4-(нафтален-2-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
6-фтор-2-метил-4-(нафтален-2-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
5-фтор-2-метил-4-(нафтален-2-ил)-7-(пиридазин-3-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(нафтален-2-ил)-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолина;
8-фтор-2-метил-4-(нафтален-2-ил)-7-(пиридазин-4-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(нафтален-2-ил)-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(3-метилпиразин-2-ил)-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
7-(3-метоксипиразин-2-ил)-2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(6-метилпиразин-2-ил)-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
7-(6-метоксипиразин-2-ил)-2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
8-фтор-2-метил-4-(нафтален-2-ил)-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
8-метокси-2-метил-4-(нафтален-2-ил)-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(нафтален-2-ил)-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолин-8-ола;
2,8-диметил-4-(нафтален-2-ил)-7-(пиразин-2-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(нафтален-2-ил)-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолина;
8-фтор-2-метил-4-(нафтален-2-ил)-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолина;
8-метокси-2-метил-4-(нафтален-2-ил)-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(нафтален-2-ил)-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолин-8-ола;
2,8-диметил-4-(нафтален-2-ил)-7-(пиримидин-2-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(нафтален-2-ил)-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
8-фтор-2-метил-4-(нафтален-2-ил)-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
8-метокси-2-метил-4-(нафтален-2-ил)-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(нафтален-2-ил)-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолин-8-ола;
2,8-диметил-4-(нафтален-2-ил)-7-(пиримидин-4-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(нафтален-2-ил)-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
8-фтор-2-метил-4-(нафтален-2-ил)-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
8-метокси-2-метил-4-(нафтален-2-ил)-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(нафтален-2-ил)-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолин-8-ола;
2,8-диметил-4-(нафтален-2-ил)-7-(пиримидин-5-ил)-1,2,3,4-тетрагидроизохинолина;
7-(3,5-диметилизоксазол-4-ил)-2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(нафтален-2-ил)-7-(тиазол-2-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(нафтален-2-ил)-7-(5-метилтиазол-2-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-нафтален-2-ил-7-[1,3,5]триазин-2-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-4-нафтален-2-ил-7-[1,2,4]триазин-3-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-4-нафтален-2-ил-7-[1,2,4]триазин-5-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-4-нафтален-2-ил-7-[1,2,4]триазин-6-ил-1,2,3,4-тетрагидроизохинолина;
3-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)циннолина;
1-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)фталазина;
2-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)хиноксалина;
2-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
6-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
7-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)хиназолина;
2-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)-2Н-[1,2,4]триазоло[4,3-а]пиридин-3-она;
2-метил-7-(морфолин-4-ил)-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1-фторнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1-хлорнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1-метоксинафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(1-метилнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(3-фторнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(3-хлорнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(3-метоксинафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
3-(2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинол-4-ил)нафталин-2-карбонитрила;
4-(4-фторнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(4-хлорнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(5-фторнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(5-хлорнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
6-(2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинол-4-ил)нафталин-1-карбонитрила;
2-метил-4-(5-метилнафтален-2-ил)-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(6-метоксинафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(6-хлорнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(6-фторнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
6-(2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинол-4-ил)нафталин-2-карбонитрила;
4-(6-метансульфонилнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(7-метоксинафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(7-хлорнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(7-фторнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
7-(2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинол-4-ил)нафталин-2-карбонитрила;
4-(8-метоксинафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(8-хлорнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
4-(8-фторнафтален-2-ил)-2-метил-7-(морфолин-4-ил)-1,2,3,4-тетрагидроизохинолина;
8-(2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинол-4-ил)нафталин-1-карбонитрила;
7-(2,6-диметилморфолин-4-ил)-2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолина;
2-этил-7-(морфолин-4-ил)-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолина;
8-фтор-2-метил-7-(морфолин-4-ил)-(4-нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
8-метокси-2-метил-7-(морфолин-4-ил)-(4-нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-7-морфолин-4-ил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-8-ола;
2,8-диметил-7-(морфолин-4-ил)-(4-нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
2-этил-8-фтор-7-(морфолин-4-ил)-(4-нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
6-фтор-2-метил-7-(морфолин-4-ил)-(4-нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
5-фтор-2-метил-7-(морфолин-4-ил)-(4-нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(морфолин-4-ил)-(4-нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин-4-ола;
2,4-диметил-7-(морфолин-4-ил)-(4-нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-нафтален-2-ил-7-пиперазин-1-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(4-метилпиперазин-1-ил)-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(нафтален-2-ил)-7-(пиперидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(нафтален-2-ил)-7-(пирролидин-1-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-7-(морфолин-4-ил)метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(нафтален-2-ил)-7-(1-метил-1-морфолин-4-илэтил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(нафтален-2-ил)-7-(1-морфолин-4-илциклопропил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(нафтален-2-ил)-7-(пиперидин-1-ил)метил-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(нафтален-2-ил)-7-(пирролидин-1-ил)метил-1,2,3,4-тетрагидроизохинолина;
диметил-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-илметил)амина;
2-метил-[1-(4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]диметиламина;
2-метил-[1-(4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил]диметиламина;
метил-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-илметил)амина;
[2-метил-[1-(4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтил]метиламина;
[2-метил-[1-(4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропил] метиламина;
С-(2-метил-[1-(4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)метиламина;
1-(2-метил-[1-(4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)-1-метилэтиламина;
1-(2-метил-[1-(4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)циклопропиламина;
1-(2-метил-4-нафтален-2-ил-1,2,3,4-тетрагидроизохинолин-7-ил)-1Н-пиридин-2-она;
7-метансульфонил-2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина;
2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин-7-карбонитрила;
8-фтор-2-метил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолин-7-карбонитрила;
2,8-диметил-4-(нафтален-2-ил)-1,2,3,4-тетрагидроизохинолина и 2-метил-4-(5,6,7,8-тетрагидронафтален-2-ил)-1,2,3,4-тетрагидроизохинолина.
2-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)хинолина;
3-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)хинолина;
7-метокси-2-метил-4-хинолин-6-ил-1,2,3,4-тетрагидроизохинолин-4-ола;
6-(7-метокси-2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)хинолина;
2-метил-4-хинолин-6-ил-1,2,3,4-тетрагидроизохинолин-4-ола;
6-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)хинолина;
6-(2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ил)хинолина;
6-(2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ил)хинолина;
7-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)хинолина;
7-(2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидроизохинолин-4-ил)хинолина;
7-(2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ил)хинолина;
2'-метил-1',2',3',4'-тетрагидро-[3,4']биизохинолинила;
2-метил-1,2,3,4-тетрагидро[4,6']биизохинолинила;
2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидро[4,6']биизохинолинила;
2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидро[4,6']биизохинолинила;
2-метил-1,2,3,4-тетрагидро[4,7']биизохинолинила;
2-метил-7-пиридазин-3-ил-1,2,3,4-тетрагидро[4,7']биизохинолинила;
2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидро[4,7']биизохинолинила.
2-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)хиноксалина;
6-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)хиноксалина;
6-(2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ил)хиноксалина;
6-(2-метил-7-пиперидин-1-ил-1,2,3,4-тетрагидроизохинолин-4-ил)хиноксалина;
6-(2-метил-7-пирролидин-1-ил-1,2,3,4-тетрагидроизохинолин-4-ил)хиноксалина.
2-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)хиназолина;
6-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)хиназолина;
6-(2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ил)хиназолина;
6-(2-метил-7-пиперидин-1-ил-1,2,3,4-тетрагидроизохинолин-4-ил)хиназолина;
6-(2-метил-7-пирролидин-1-ил-1,2,3,4-тетрагидроизохинолин-4-ил)хиназолина;
7-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)хиназолина;
7-(2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ил)хиназолина;
7-(2-метил-7-пиперидин-1-ил-1,2,3,4-тетрагидроизохинолин-4-ил)хиназолина;
7-(2-метил-7-пирролидин-1-ил-1,2,3,4-тетрагидроизохинолин-4-ил)хиназолина.
6-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)фталазина;
6-(2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ил)фталазина;
6-(2-метил-7-пиперидин-1-ил-1,2,3,4-тетрагидроизохинолин-4-ил)фталазина;
6-(2-метил-7-пирролидин-1-ил-1,2,3,4-тетрагидроизохинолин-4-ил)фталазина.
6-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)циннолина;
6-(2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ил)циннолина;
6-(2-метил-7-пиперидин-1-ил-1,2,3,4-тетрагидроизохинолин-4-ил)циннолина;
6-(2-метил-7-пирролидин-1-ил-1,2,3,4-тетрагидроизохинолин-4-ил)циннолина;
7-(2-метил-1,2,3,4-тетрагидроизохинолин-4-ил)циннолина;
7-(2-метил-7-морфолин-4-ил-1,2,3,4-тетрагидроизохинолин-4-ил)циннолина;
7-(2-метил-7-пиперидин-1-ил-1,2,3,4-тетрагидроизохинолин-4-ил)циннолина;
7-(2-метил-7-пирролидин-1-ил-1,2,3,4-тетрагидроизохинолин-4-ил)циннолина.
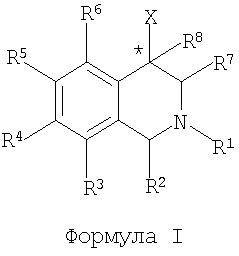
где атом углерода, обозначенный *, находится в R- или S-конфигурации;
Х означает конденсированный бициклический карбоцикл или гетероцикл, выбранный из группы, состоящей из бензофуранила, бензо[b]тиофенила, бензоизотиазолила, индазолила, индолила, бензооксазолила, бензотиазолила, инденила, инданила, дигидробензоциклогептенила, нафтила, тетрагидронафтила, хинолинила, изохинолинила, хиноксалинила, 2Н-хроменила, имидазо[1,2-а] пиридинила, пиразоло[1,5-а] пиридинила, и конденсированный бициклический карбоцикл или конденсированный бициклический гетероцикл необязательно замещен заместителями (в количестве от 1 до 4), которые определены ниже для R14; при условии, что (1) когда Х=нафтил и R4 означает OR11, R5 не может быть Н;
R1 означает Н, C1-С6-алкил, С3-С6-циклоалкил, C1-С6-алкил, замещенный -OR11, -NR9R10 или -CN;
R2 означает Н, C1-С6-алкил, или гем-диметил;
R3 означает Н, -OR11, C1-С6-алкил или галоген;
R4 означает Н, галоген, -OR11, -CN, C1-С6-алкил, C1-С6-алкил, замещенный -NR9R10,
С3-С6-циклоалкил, замещенный -NR9R10, C(O)R12;
R4 означает морфолинил, пиперидинил, пиримидинил, пиридазинил, пиразинил, пирролил, изоксазолил, пирролидинил, пиперазинил, 2-оксо-2Н-пиридинил, [1,2,4]триазоло[4,3-а]пиридинил, 3-оксо-[1,2,4]триазоло[4,3-а]пиридинил, хиноксалинил, которые необязательно замещены заместителями (в количестве от 1 до 4), которые определены ниже для R14;
R5 означает Н или C1-С6-алкил;
R6 означает Н, С1-С6-алкил или -OR11;
R7 означает Н;
R8 означает Н, -OR9, C1-С6-алкил, -CN;
R9 означает Н или С1-С4-алкил;
R10 означает Н или С1-С4-алкил;
или R9 и R10, взятые вместе с атомом азота, с которым они связаны, образуют морфолин;
R11 означает Н, С1-С4-алкил;
R12 означает С1-С4-алкил;
R14 в каждом случае независимо выбран из заместителя, выбранного из группы, состоящей из галогена, -OR11, -NR11R12, C1-С6-алкила, необязательно замещенного 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-С3-алкила, арила;
или его фармацевтически приемлемая соль.
где атом углерода, обозначенный *, находится в R- или S-конфигурации;
Х означает конденсированный бициклический карбоцикл или гетероцикл, выбранный из группы, состоящей из бензофуранила, бензо[b]тиофенила, бензоизотиазолила, индазолила, индолила, бензооксазолила, бензотиазолила, инденила, инданила, дигидробензоциклогептенила, нафтила, тетрагидронафтила, хинолинила, изохинолинила, хиноксалинила, 2Н-хроменила, имидазо[1,2-а] пиридинила, пиразоло[1,5-а] пиридинила, и конденсированный бициклический карбоцикл или конденсированный бициклический гетероцикл необязательно замещен заместителями (в количестве от 1 до 4), которые определены ниже для R14;
R1 означает Н, C1-С6-алкил, С3-С6-циклоалкил, C1-С3-алкил, замещенный OR11, -NR9R10 или -CN;
R2 означает Н, C1-С6-алкил или гем-диметил;
R3 означает Н, -OR11, C1-С6-алкил или галоген;
R4 означает Н, галоген, -OR11, -CN, C1-С6-алкил, C1-С6-алкил, замещенный -NR9R10, С3-С6-циклоалкил, замещенный -NR9R10, C(O)R12; или
R4 означает морфолинил, пиперидинил, пиримидинил, пиридазинил, пиразинил, пирролил, изоксазолил, пирролидинил, пиперазинил, 2-оксо-2Н-пиридинил, [1,2,4]триазоло[4,3-а]пиридинил, 3-оксо-[1,2,4]триазоло[4,3-а]пиридинил, хиноксалинил, которые необязательно замещены заместителями (в количестве от 1 до 4), которые определены ниже для R14;
R5 означает Н или C1-С6-алкил;
R6 означает Н, C1-С6-алкил или-OR11;
R7 означает Н;
R8 означает Н, -OR9, C1-С6-алкил, -CN;
R9 означает Н или С1-С4-алкил;
R10 означает Н или С1-С4-алкил;
или R9 и R10, взятые вместе с атомом азота, с которым они связаны, образуют морфолин;
R11 означает Н, С1-С4-алкил;
R12 означает С1-С4-алкил;
R14 в каждом случае независимо выбирают из заместителя, выбранного из группы, состоящей из галогена, -OR11, -NR11R12, C1-С6-алкила, который необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из С1-С3-алкила, арила;
включающий
обработку первого промежуточного соединения формулы (XVIII)

где R1, R2, R3, R4, R5, R6, R8 и Х имеют значения, указанные в п.1, восстановителем в условиях, эффективных для получения соединения.
введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (I)
где атом углерода, обозначенный *, находится в R- или S-конфигурации;
Х означает конденсированный бициклический карбоцикл или гетероцикл, выбранный из группы, состоящей из бензофуранила, бензо[b]тиофенила, бензоизотиазолила, индазолила, индолила, бензооксазолила, бензотиазолила, инденила, инданила, дигидробензоциклогептенила, нафтила, тетрагидронафтила, хинолинила, изохинолинила, хиноксалинила, 2Н-хроменила, имидазо[1,2-а]пиридинила, пиразоло[1,5-а]пиридинила, и конденсированный бициклический карбоцикл или конденсированный бициклический гетероцикл необязательно замещен заместителями (в количестве от 1 до 4), которые определены ниже для R14;
R1 означает Н, C1-С6-алкил, С3-С6-циклоалкил, С1-С3-алкил, замещенный OR11, -NR9R10 или -CN;
R2 означает Н, C1-С6-алкил или гем-диметил;
R3 означает Н,-OR11, C1-С6-алкил или галоген;
R4 означает Н, галоген, -OR11, -CN, C1-С6-алкил, C1-С6-алкил, замещенный -NR9R10,
С3-С6-циклоалкил, замещенный -NR9R10, C(O)R12; или
R4 означает морфолинил, пиперидинил, пиримидинил, пиридазинил, пиразинил, пирролил, изоксазолил, пирролидинил, пиперазинил, 2-оксо-2Н-пиридинил, [1,2,4]триазоло[4,3-а]пиридинил, 3-оксо-[1,2,4]триазоло[4,3-а]пиридинил, хиноксалинил, которые необязательно замещены заместителями (в количестве от 1 до 4), которые определены ниже для R14;
R5 означает Н или C1-С6-алкил;
R6 означает Н, C1-С6-алкил или -OR11;
R7 означает Н;
R8 означает Н, -OR9, C1-С6-алкил, -CN;
R9 означает Н или С1-С4-алкил;
R10 означает Н или С1-С4-алкил;
или R9 и R10, взятые вместе с атомом азота, с которым они связаны, образуют морфолин;
R11 означает Н, С1-С4-алкил;
R12 означает С1-С4-алкил;
R14 в каждом случае независимо выбирают из заместителя, выбранного из группы, состоящей из галогена, -OR11, -NR11R12, C1-С6-алкила, который необязательно замещен 1-3 заместителями, в каждом случае независимо выбранными из группы, состоящей из C1-С3-алкила, арила;
или его фармацевтически приемлемой соли.
Комментарии