Фармацевтически приемлемая соль (е)-n-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2r)-1-метилпирролидин-2-ил]проп-2-енамида, способ ее получения и применение при лечении рака - RU2583056C2
Код документа: RU2583056C2
Описание
Область изобретения
Настоящее изобретение относится к фармацевтически приемлемой соли 6-аминохиназоолина или 3-цианохинолиновым производным, способу ее получения и медицинскому применению. В частности, настоящее изобретение относится к фармацевтически приемлемой соли (E)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамида, способу ее получения и ее применению в качестве терапевтического агента, и в частности, в качестве ингибитора протеинкиназы.
Предшествующий уровень техники
Трансдукция сигнала представляет собой фундаментальный механизм, посредством которого внеклеточные стимулы передаются внутрь клеток, регулируя соответствующие физические ответы, включающие пролиферацию, дифференцировку и апоптоз. Большая часть этих процессов трансдукции сигнала использует процесс обратимого фосфорилирования белков, включающие специфические протеинкиназы и фосфатазы.
Существуют два класса протеинкиназ (PK): белковые тирозинкиназы (PTK) и серин-треониновые киназы (STK). PTK могут фосфорилировать тирозиновый остаток на белке. STK могут фосфорилировать сериновый или/и треониновый остаток. Тирозинкиназы могут быть разделены на тирозинкиназы рецепторного типа (рецепторная тирозинкиназа, RTK) или нерецепторного типа (нерецепторная тирозинкиназа). В настоящее время в человеческом геноме идентифицированы приблизительно 90 тирозинкиназ, из которых приблизительно 60 относятся к рецепторному типу и приблизительно 30 относятся к нерецепторному типу.
Семейство рецепторных тирозинкиназ (RTK) включает множество подсемейств, таких как (1) семейство рецептора EGF (эпидермального фактора роста), включающее EGFR, HER-2, HER-3 и HER-4; (2) семейство инсулинового рецептора, включающее инсулиновый рецептор (IR), рецептор инсулиноподобного фактора роста-I (IGF-IR) и инсулиноподобный рецептор (IRR); (3) семейство класса III, такие как рецептор фактора роста тромбоцитов (PDGFR), рецептор фактора стволовых клеток (SCF) (c-Kit) RTK, рецептор fms-связанной тирозинкиназы 3 (Flt3) и рецептор колониестимулирующего фактора 1 (CSF-1R) и т.п. Иначе, рецептор фактора роста гепатоцитов c-Met, фактор роста эндотелия сосудов (VEGFR) и т.п. также относятся к семейству RTK. Они играют критическую роль в контроле клеточного роста и дифференцировки, и представляют собой ключевые медиаторы клеточных сигналов, приводящих к продукции цитокинов, таких как факторы роста (Schlessinger and Ullrich, Neuron 1992, 9, 383).
Подсемейство EGFR (ErbB, HER) играет критическую роль в контроле клеточной пролиферации и выживания. Эти RTK состоят из внеклеточного гликозилированного связывающего лиганд домена, трансмембранного домена и внутриклеточного цитоплазматического каталитического домена. Ферментативная активность рецепторных тирозинкиназ может быть стимулирована путем опосредованной лигандами гомодимеризации или гетеродимеризации. Димеризация приводит в результате к фосфорилированию тирозиновых остаток на рецепторах в каталитическом домене, приводя впоследствии к сайту связывания. За ним следует активация внутриклеточных сигнальных путей, таких как пути, вовлекающие ассоциирующуюся с микротрубочками протеинкиназу (MAP киназу) и фосфатидилинозитол-3-киназу (PI3 киназу). Активация этих путей связана с регуляцией клеточного цикла и апоптоза. Обнаружили, что такие мутантные и сверхэкспрессированнные формы тирозинкиназ, такие как EGFR, HER-2, представлены в большой доле обычных видов рака человека, таких как рак молочной железы, рак предстательной железы, немелкоклеточный рак легкого, рак пищевода, рак яичников и рак поджелудочной железы и т.п. Преобладание и релевантность тирозинкиназ подтверждается в онкогенезе и раковом росте.
Предполагают синтезировать новые соединения, обладающие активностями против пролиферации опухолевых клеток. Ожидается, что эти соединения ингибируют одну или более чем одну RTK или STK, и полезны для лечения или уменьшения интенсивности физиологических расстройств с сверхпролиферацией клеток, опосредованных RTK или STK, и опосредованных ангиогенезом.
Описание настоящего изобретения относится к сериям фармацевтически приемлемых солей (Е)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамида и их применениям, в частности в качестве ингибиторов протеинкиназ.
Автор изобретения обнаружил, что свободное основание (E)-N-[4-[[3-хлор-4-(2-пиридилметокси) фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид плохо растворим в обычных растворителях и, таким образом, является не благоприятным для того, чтобы его готовили в медицинскую лекарственную форму, ограничивающую их биодоступность in vivo. Таким образом, существует острая потребность в улучшении их растворимости и фармакокинетическом поглощении для того, чтобы соответствовать обычному способу получения лекарственных форм. По сравнению со свободным основанием (Е)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамидом растворимость и фармакокинетика фармацевтически приемлемых солей соединения значительно улучшается, и способ синтеза упрощается.
Настоящее изобретение относится к тому, чтобы предложить фармацевтически приемлемые соли соединений формулы (I), таким образом, улучшая их физические/химические свойства и фармакокинетические характеристики.
Краткое описание изобретения
Настоящее изобретение относится к тому, чтобы предложить фармацевтически приемлемую соль формулы (I) и способ ее получения. Предпочтительно, соль дималеат формулы (I) обладает преимуществами в растворимости, биодоступности и фармакокинетике по сравнению с самим соединением формулы (I) и другими его солями.

где
n равен 1, 2 или 3; и
М представляет собой молекулу кислоты.
В первом своем аспекте настоящее изобретение относится к фармацевтически приемлемой соли (Е)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамиду формулы (I), где указанная соль представляет собой обычную неорганическую соль или органическую соль в области техники. Кроме того, указанная неорганическая соль выбрана из группы, состоящей из гидрохлорида, гидробромида, сульфата, нитрата и фосфата, предпочтительно гидрохлорида, более предпочтительно дигидрохлорида; где указанная органическая соль выбрана из группы, состоящей из пара-толуолсульфоната, метансульфоната, малеата, тартрата, сукцината, ацетата, трифторацетата, фумарата, цитрата, бензолсульфоната, бензоата, нафталинсульфоната, лактата и L-малата, предпочтительно L-яблочной кислоты, метансульфоната, сукцината, пара-толуолсульфоната или малеата, наиболее предпочтительно, малеата. В частности, соль малеат соединения формулы (I) обладает преимуществами в растворимости, биодоступности и фармакокинетике по сравнению с самим соединением формулы (I) и другими его солями.
Во втором своем аспекте настоящее изобретение относится к способу получения фармацевтически приемлемой соли (E)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамида, и указанное соединение может быть получено в соответствии с обычным в области техники способом образования соли. В частности, в указанном способе осуществляют стадию взаимодействия (E)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамида с соответствующей кислотой с образованием соли, где указанная кислота представляет собой неорганическую кислоту/органическую кислоту, выбранную из группы, состоящей из ортофосфорной кислоты, соляной кислоты, серной кислоты, азотной кислоты, бромистоводородной кислоты, пара-толуолсульфоновой кислоты, метансульфоновой кислоты, малеиновой кислоты, винной кислоты, янтарной кислоты, уксусной кислоты, трифторуксусной кислоты, фумаровой кислоты, лимонной кислоты, бензолсульфоновой кислоты, бензойной кислоты, нафталинсульфоновой кислоты, молочной кислоты и L-яблочной кислоты.
Фармацевтически приемлемые соли по настоящему изобретению как правило включают без ограничения:



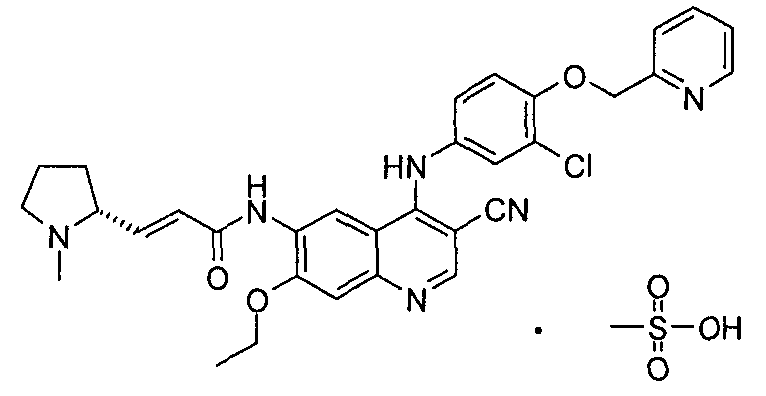




В третьем своем аспекте настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество фармацевтически приемлемой соли (Е)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамида и фармацевтически приемлемые носители. Настоящее изобретение также относится к способу приготовления композиции, при котором осуществляют стадию комбинирования фармацевтически приемлемой соли с фармацевтически приемлемым носителем или разбавителем.
В четвертом своем аспекте настоящее изобретение относится к применению фармацевтически приемлемой соли (E)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамида или его фармацевтической композиции в приготовлении лекарственного средства для лечения заболеваний, связанных с протеинкиназами, где указанные протеинкиназы выбраны из группы, состоящей из рецепторных тирозинкиназ EGFR и рецепторных тирозинкиназ HER-2.
В пятом своем аспекте настоящее изобретение относится к способу лечения заболеваний, связанных с протеинкиназами, при котором осуществляют введение терапевтически эффективного количества фармацевтически приемлемой соли (E)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамида или его фармацевтической композиции субъекту, нуждающемуся в таком введении.
В шестом своем аспекте настоящее изобретение относится к применению фармацевтически приемлемой соли (E)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамида или его фармацевтической композиции при приготовлении лекарственного средства на основе протеинкиназного ингибитора, где указанные протеинкиназы выбраны из группы, состоящей из EGFR и HER-2.
В седьмом своем аспекте настоящее изобретение относится к применению фармацевтически приемлемой соли (E)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамида или фармацевтической композиции в качестве лекарственного средства для лечения заболеваний, связанных с протеинкиназами, где указанные заболевания представляют собой рак, выбранный из группы, состоящей из рака легкого, рака молочной железы, плоскоклеточного рака и рака желудка.
В восьмом своем аспекте настоящее изобретение относится к способу регуляции каталитической активности протеинкиназ, при котором осуществляют приведение в контакт указанной протеинкиназы с фармацевтически приемлемой солью (E)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамида, где указанная протеинкиназа выбрана из группы, состоящей из рецепторной тирозинкиназы EGFR или рецепторной тирозинкиназы HER-2.
Подробное описание изобретения
Настоящее изобретение дополнительно описано при помощи следующих примеров, которые не предназначены для того, чтобы ограничить объем изобретения.
Примеры
Структуры всех соединений идентифицируют при помощи ядерного магнитного резонанса (NMR) и/или масс-спектрометрии (MS). Химический сдвиг NMR (δ) зарегистрирован в виде млн-1 (10-6). NMR осуществляют на спектрометре Bruker AVANCE-400. Растворитель для детекции представляет собой дейтерированный диметилсульфоксид (d-DMSO) с тетраметилсиланом (TMS) в качестве внутреннего стандарта, и химический сдвиг регистрируют в виде млн-1 (10-6).
MS определяют на масс-спектрометре FINNIGAN LCQAd (ESI (ионизация путем распыления электронов)) (Thermo, Model: Finnigan LCQ advantage MAX).
HPLC определяют на спектрометре/жидкостном хроматографе высокого давления Agilent 1200DAD (хроматографическая колонка Sunfire С18 150×4,6 мм) и спектрометре/жидкостном хроматографе высокого давления Waters 2695-2996 (хроматографическая колонка Gimini C18 150×4,6 мм).
В колоночной хроматографии в общем используют в качестве носителя силикагель Yantai Huanghai 200~300 меш.
Исходные вещества по настоящему изобретению известны и могут быть приобретены в ABCR GmbH & Со. KG, Acros Organics, Aldrich Chemical Company и т.п., или они могут быть получены при помощи обычных способов синтеза, известных в области техники.
Если не указано иное, то следующие взаимодействия осуществляют в атмосфере аргона или атмосфере азота.
Термин ″атмосфера аргона″ или ″атмосфера азота″ относится к тому, что реакционный сосуд оборудован баллоном, заполненным приблизительно 1 л аргона или азота.
Термин ″атмосфера водорода″ относится к тому, что реакционный сосуд оборудован баллоном, заполненным приблизительно 1 л водорода.
Если не указано иное, то раствор, используемый в примерах, относится к водному раствору.
Если не указано иное, то температура реакции представляет собой комнатную температуру.
Комнатная температура представляет собой наиболее подходящую температуру реакции, составляющую 20°C-30°C.
Процессы реакции в примерах контролируют при помощи тонкослойной хроматографии (TLC). Система проявляющих растворителей включает систему дихлорметана и метанола, систему н-гексана и этилацетата, систему петролейного эфира и этилацетата и ацетон.
Долю растворителя по объему корректируют в соответствии с полярностью соединений.
Элюирующая система для колоночной хроматографии содержит А:
систему дихлорметана, метанола и ацетона; Б: систему гексана и этилацетата. Долю растворителя по объему корректируют в соответствии с полярностью соединений, и иногда также может быть добавлено небольшое количество аммиака и уксусной кислоты.
Пример 1
(Е)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид малеат
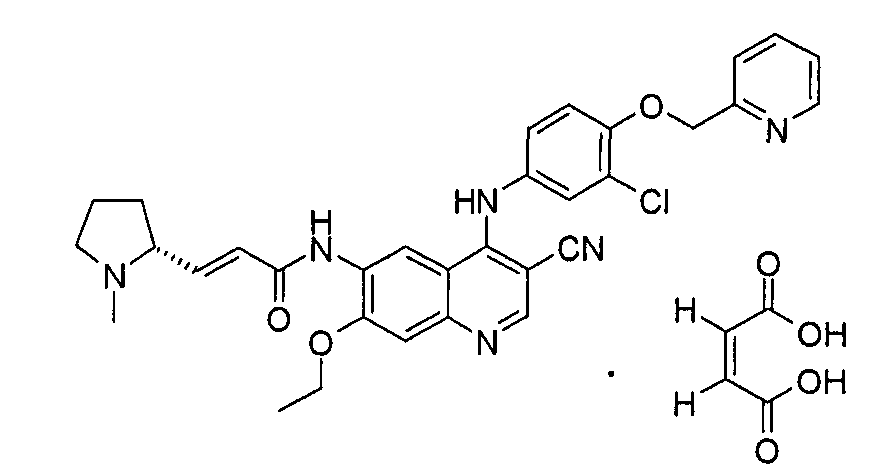


Стадия 1
[(2R)-1-Метилпирролидин-2-ил]метаноллитий алюминий гидрид (230 мг, 6 ммоль) и N-трет-бутоксикарбонил-R-пролинол 1a (400 мг, 2 ммоль) растворяли в 10 мл безводного тетрагидрофурана в бане лед-вода партиями. После обнаружения того, что газ больше не выделяется, реакционную смесь нагревали до температуры дефлегмации в течение 2 часов. В реакционную смесь по каплям добавляли 5 мл метанола в бане лед-вода, а затем добавляли 5 мл воды, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения [(2R)-1-метилпирролидин-2-ил]метанола 1b (221 мг, выход 77,0%) в виде бесцветного масла.
MS m/z (ESI): 116 [M+1]
Стадия 2
(2R)-1-Метилпирролидин-2-формальдегид
Диметилсульфоксид (820 мкл, 11,46 ммоль) растворяли в 5 мл дихлорметана в бане с сухим льдом, а затем медленно добавляли по каплям оксалилхлорид (968 мг, 7,64 ммоль). После перемешивания в течение 45 минут к раствору добавляли раствор [(2R)-1-метилпирролидин-2-ил]метанола 1b (220 мг, 1,91 ммоль) в 2 мл дихлорметана. Реакционную смесь перемешивали в течение еще 45 минут, и добавляли триэтиламин (1,9 мл, 13,37 ммоль). Реакционную смесь перемешивали в течение 10 минут, затем нагревали до комнатной температуры и перемешивали в течение 1 часа. Реакционную смесь последовательно промывали водой (20 мл) и насыщенным рассолом (10 мл). Объединенные органические экстракты сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, и образующиеся в результате остатки очищали при помощи колоночной хроматографии с щелочным оксидом алюминия с элюирующей системой А с получением указанного в заголовке соединения (2R)-1-метилпирролидин-2-формальдегида 1c (300 мг) в виде желтого твердого вещества, которое непосредственно использовали на следующей стадии без очистки.
Стадия 3
N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-2-диэтоксифосфорил-ацетамид
N,N′-Карбонилдиимидазол (487 мг, 3 ммоль) растворяли в 4 мл тетрагидрофурана. Смесь нагревали до 40°C в масляной бане, по каплям к смеси добавляли раствор диэтилфосфоноуксусной кислоты (588 мг, 3 ммоль) в тетрагидрофуране (4 мл) и перемешивали в течение 30 минут до следующей стадии.
6-Амино-4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-7-этокси-хинолин-3-карбонитрил 1d (446 мг, 1 ммоль, полученный при помощи хорошо известного способа: заявка на патент W02005028443) при 40°C растворяли в 4 мл тетрагидрофурана, а затем по каплям добавляли вышеописанный реакционный раствор. После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении и экстрагировали дихлорметаном (50 мл × 3). Объединенные органические экстракты промывали насыщенным рассолом (30 мл × 2), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, и образующиеся в результате остатки очищали при помощи колоночной хроматографии с силикагелем с элюирующей системой А с получением указанного в заголовке соединения N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-2-диэтоксифосфорил-ацетамида 1e (624 мг, выход 99,9%) в виде светло-желтого твердого вещества.
MS m/z (ESI): 624 [M+1]
Стадия 4
(E)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид
N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-2-диэтоксифосфорил-ацетамид 1е (250 мг, 0,40 ммоль) растворяли в 10 мл безводного тетрагидрофурана в бане с сухим льдом, а затем по каплям добавляли раствор литий бис(триметилсилил)амида (1М) в толуоле (440 мкл, 0,44 ммоль). Реакционную смесь перемешивали в течение 30 минут, по каплям добавляли раствор (2R)-1-метилпирролидин-2-формальдегида 1с (90 мг, 0,80 ммоль) в тетрагидрофуране (5 мл) и перемешивали в течение 30 минут, затем нагревали до комнатной температуры и перемешивали в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении, и образующиеся в результате остатки очищали при помощи колоночной хроматографии с силикагелем с элюирующей системой А с получением указанного в заголовке соединения (Е)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамида 1f (46 мг, выход 19,7%) в виде желтого твердого вещества.
MS m/z (ESI): 583,4 [M+1]
1H NMR (400 МГц, DMSO-d6): δ 9,16 (s, 1H), 8,63 (d, 1H), 8,56 (s, 1H), 8,26 (s, 1H), 7,83-7,80 (dd, 1H), 7,76-7,50 (m, 2H), 7,57-7,56 (m, 1H), 7,40 (s, 1H), 7,38 (s, 1H), 7,19 (d, 1H), 7,06-7,03 (m, 2H), 6,34-6,31 (d, 1H), 5,35 (s, 2H), 4,39 (m, 2H), 4,27-4,26 (m, 1H), 3,32 (m, 1H), 3,10 (m, 1H), 2,73 (s, 3Н), 2,37-2,36 (m, 2H), 2,07-2,01 (m,2H), 1,64 (t, 3H)
Стадия 5
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид малеат
(Е)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид 1f (500 мг, 0,86 ммоль) и малеиновую кислоту (109 мг, 0,94 ммоль) при перемешивании растворяли в 5 мл дихлорметана. После перемешивания в течение 1 часа при к.т. реакционную смесь концентрировали при пониженном давлении с получением (E)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид малеата 1 (609 мг, выход 100%) в виде желтого твердого вещества.
MS m/z (ESI): 584,4 [M+1-116]
1H NMR (400 МГц, CDCl3): δ 9,22 (s, 1H), 8,63 (m, 1H), 8,52 (m, 1H), 8,42 (s, 1H), 7,84 (m, 1H), 7,74 (m, 1H), 7,69 (m, 1H), 7,58 (m, 1H), 7,26 (m, 1H), 7,02 (m, 2H), 6,92 (m, 1H), 6,72 (m, 1H), 6,25 (s, 2H), 5,27 (s, 2H), 4,27 (m, 2H), 3,90 (m, 2H), 3,00 (m, 1H), 2,87 (m, 2H), 2,21 (m, 4H), 2,09 (m, 1H), 1,56 (t, 3H, J=8 Гц)
Пример 2
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид дималеат
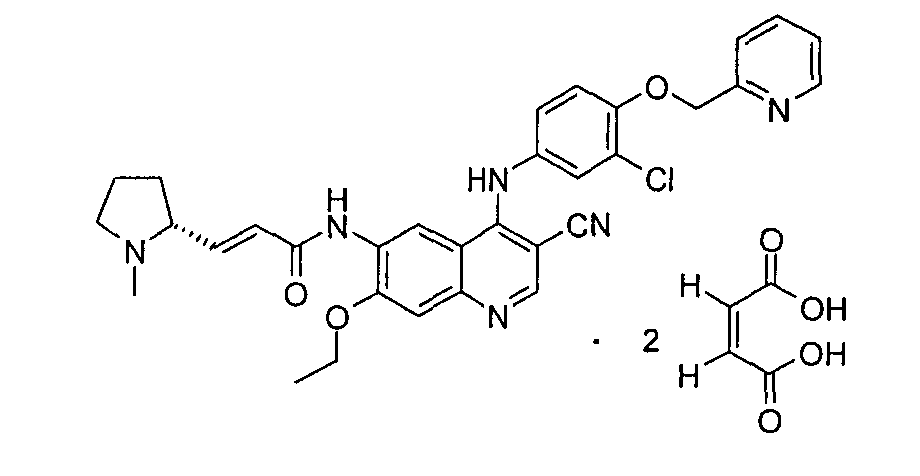
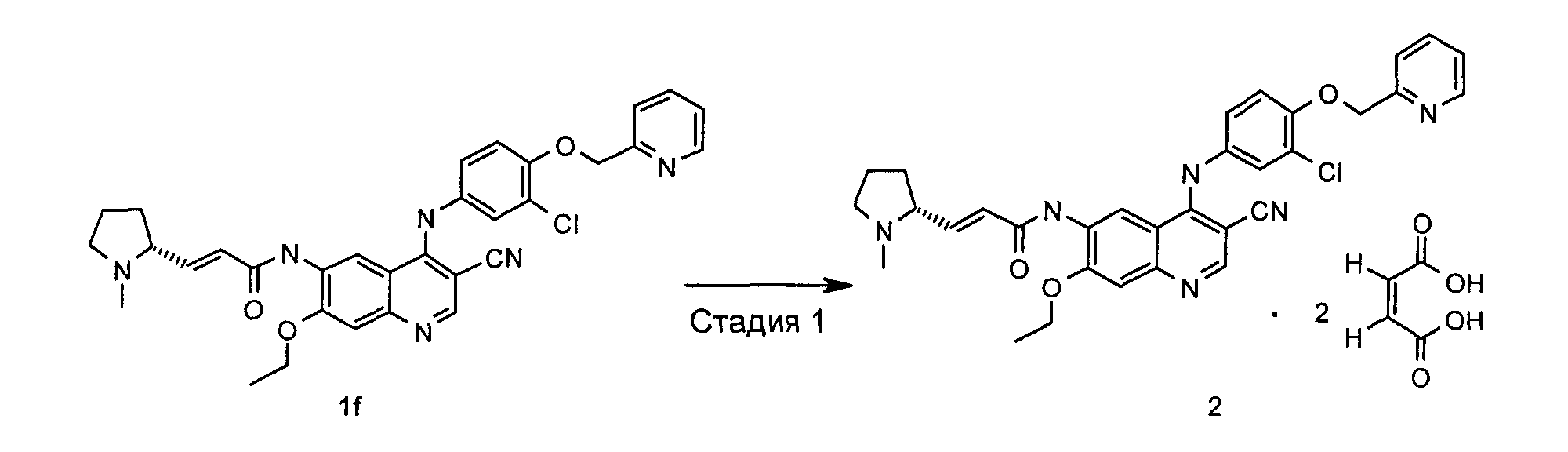
(E)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид дималеат
(E)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид 1f (200 мг, 0,34 ммоль) при перемешивании растворяли в 6 мл этанола, а затем добавляли малеиновую кислоту (80 мг, 0,68 ммоль). После того, как реакционную смесь перемешивали в течение 0,5 часов и оставляли на 12 часов при к.т., в нее добавляли 10 мл диэтилового эфира и фильтровали. Твердое вещество промывали диэтиловым эфиром (20 мл) и сушили в вакууме с получением указанного в заголовке продукта (E)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид дималеата 2 (220 мг, выход 78,6%) в виде желтого твердого вещества.
MS m/z (ESI): 583,4 [M+1-232]
1H NMR (400 МГц, CDCl3): δ 9,12 (s, 1H), 8,57 (m, 1H), 8,51 (m, 1H), 8,37 (s, 1H), 7,79 (m, 1H), 7,71 (m, 1H), 7,63 (m, 1H), 7,52 (m, 1H), 7,21 (m, 1H), 7,00 (m, 2H), 6,90 (m, 1H), 6,57 (m, 1H), 6,25 (s, 4H), 5,23 (s, 2H), 4,21 (m, 2H), 3,91 (m, 2H), 3,11 (m, 1H), 2,85 (m, 2H), 2,22 (m, 4H), 2,01 (m, 1H), 1,54 (t, 3H, J=8 Гц)
Пример 3
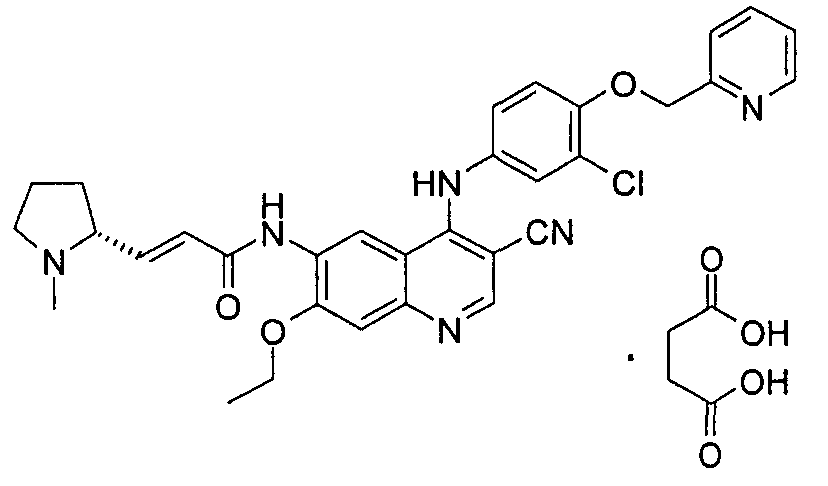
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид L-малат


Стадия 1
(Е)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид L-малат
(Е)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид 1f (300 мг, 0,51 ммоль) при перемешивании растворяли в 5 мл дихлорметана, а затем добавляли L-яблочную кислоту (75,9 мг, 0,56 ммоль). После перемешивания в течение 4 часов при к.т. реакционную смесь концентрировали при пониженном давлении и сушили в вакууме с получением (Е)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид L-малата 3 (375 мг, выход 100%) в виде желтого твердого вещества.
MS m/z (ESI): 583,4 [M+1-134]
1H NMR (400 МГц, MeOD): δ 8,96 (s, 1H), 8,58 (m, 1H), 8,38 (s, 1H), 7,94 (m, 1H), 7,73 (m, 1H), 7,42 (m, 2H), 7,24 (s, 1H), 7,19 (m, 2H), 6,98 (m, 1H), 6,71 (m, 1H), 5,51 (s, 1H), 5,28 (m, 2H), 4,34 (m, 2H), 3,64 (m, 2H), 2,92 (m, 2H), 2,67 (m, 3Н), 2,58 (m, 1H), 2,31 (m, 1H), 2,17 (m, 1H), 2,09 (m, 2H), 1,98 (m, 1H), 1,58 (t, 3Н, J=8 Гц)
Пример 4
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид метансульфонат


Стадия 1
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид метансульфонат
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид 1f (150 мг, 0,26 ммоль) при перемешивании растворяли в 2 мл этанола, а затем добавляли метансульфоновую кислоту (1,67 мл, 0,26 ммоль). После перемешивания в течение 12 часов при к.т. в реакционную смесь добавляли 2 мл диэтилового эфира и фильтровали. Твердое вещество промывали диэтиловым эфиром (10 мл) и сушили в вакууме с получением указанного в заголовке продукта (E)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид метансульфоната 4 (120 мг, выход 69,0%) в виде желтого твердого вещества.
MS m/z (ESI): 583,4 [M+1-96]
1H NMR (400 МГц, DMSO-d6): δ 11,10 (m, 1H), 10,00 (s, 1H), 9,91 (m, 1H), 9,11 (s, 1H), 9,01 (s, 1H), 8,60 (d, 1H, J=4 Гц), 8,03 (m, 1H), 7,57 (m, 2H), 7,47 (m, 4H), 6,92 (m, 2H), 5,31 (s, 2H), 4,43 (m, 2H), 4,01 (m, 2H), 3,73 (m, 3H), 3,14 (s, 2H), 2,80 (m, 2H), 2,32 (m, 1H), 2,27 (m, 3H), 1,51 (t, 3H, J=8 Гц)
Пример 5
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамиддиметансульфонат
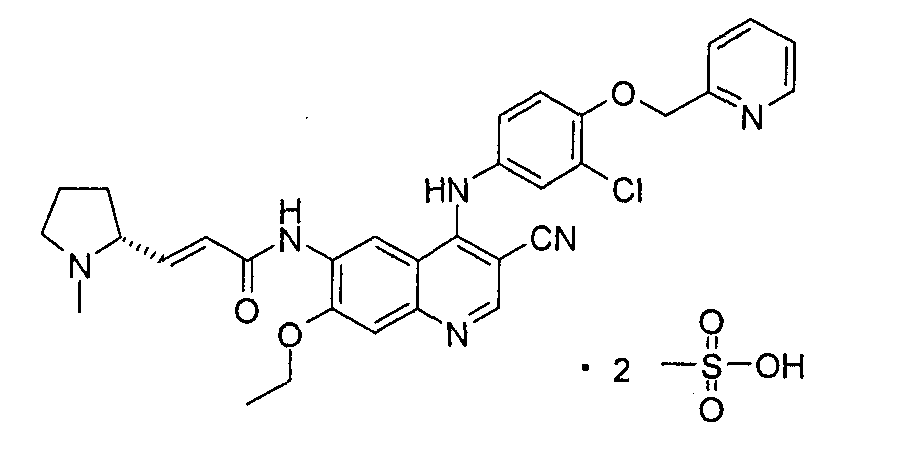

Стадия 1
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамиддиметансульфонат
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид 1f (200 мг, 0,34 ммоль) при перемешивании растворяли в 2 мл этанола, а затем добавляли метансульфоновую кислоту (4,45 мл, 0,68 ммоль). После перемешивания в течение 12 часов при к.т. в реакционную смесь добавляли 2 мл диэтилового эфира и фильтровали, твердое вещество промывали диэтиловым эфиром (10 мл) и сушили в вакууме с получением указанного в заголовке продукта (Е)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид диметансульфоната 5 (180 мг, выход 67,7%) в виде желтого твердого вещества.
MS m/z (ESI): 583,4 [M+1-192]
1H NMR (400 МГц, DMSO-d6): δ 11,17 (m, 1H), 10,04 (s, 1H), 9,98 (m, 1H), 9,18 (s, 1H), 9,05 (s, 1H), 8,67 (d, 1H, J=4 Гц), 8,01 (m, 1H), 7,68 (m, 2H), 7,50 (m, 4H), 6,91 (m, 2H), 5,39 (s, 2H), 4,40 (m, 2H), 4,14 (m, 2H), 3,71 (m, 6H), 3,22 (s, 2H), 2,82 (m, 2H), 2,36 (m, 1H), 2,32 (m, 3Н), 1,52 (t, 3H, J=8 Гц)
Пример 6
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамидтриметансульФонат


Стадия 1
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамидтриметансульфонат
(E)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид 1f (150 мг, 0,26 ммоль) при перемешивании растворяли в 1 мл этанола, а затем добавляли метансульфоновую кислоту (5,00 мл, 0,78 ммоль). После перемешивания в течение 12 часов в реакционную смесь добавляли 2 мл диэтилового эфира и фильтровали, твердое вещество промывали диэтиловым эфиром (10 мл) и сушили в вакууме с получением указанного в заголовке продукта (E)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид триметансульфоната 6 (140 мг, выход 62,5%) в виде желтого твердого вещества.
MS m/z (ESI): 583,4 [M+1-288]
1H NMR (400 МГц, DMSO-d6): δ 10,97 (m, 1H), 9,99 (s, 1H), 9,89 (m, 1H), 9,27 (s, 1H), 9,08 (s, 1H), 8,71 (d, 1H, J=4 Гц), 7,93m, 1H), 7,62 (m, 2H), 7,51 (m, 4H), 6,94 (m, 2H), 5,27 (s, 2H), 4,37 (m, 2H), 4,09 (m, 2H), 3,67 (m, 9H), 3,19 (s, 2H), 2,78 (m, 2H), 2,31 (m, 1H), 2,27 (m, 3H), 1,52 (t, 3H, J=8 Гц)
Пример 7
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид пара-толуолсульфонат

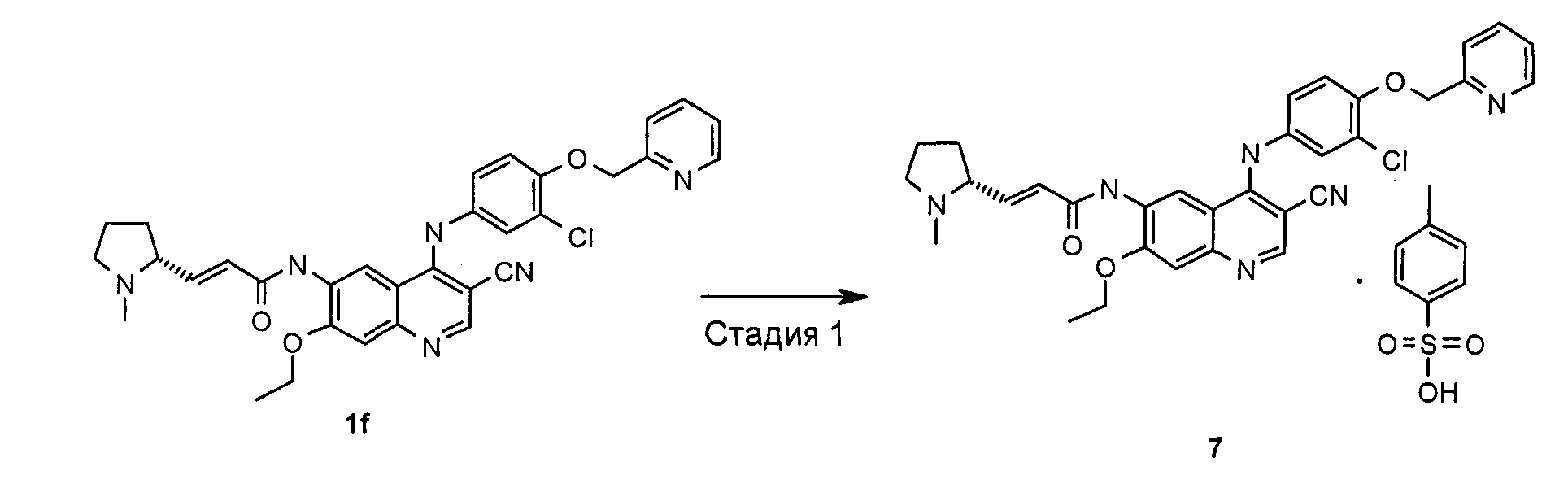
Стадия 1
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид пара-толуолсульфонат
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид 1f (300 мг, 0,514 ммоль) при перемешивании растворяли в 3 мл смешанного растворителя н-пропанол/вода (3/1), а затем добавляли пара-толуолсульфоновой кислоты моногидрат (117 мг, 0,617 ммоль). После перемешивания в течение 0,5 часов при к.т. реакционную смесь концентрировали при пониженном давлении и сушили в вакууме с получением указанного в заголовке продукта (E)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид пара-толуолсульфоната 7 (410 мг, выход 98,0%) в виде желтого твердого вещества.
MS m/z (ESI): 584,2 [M+1-172]
1H NMR (400 МГц, MeOD): δ 8,56 (s, 1H), 8,46 (s, 1H), 8,26 (s, 1H), 7,85 (m, 1H), 7,65 (m, 4H), 7,40 (m, 5H), 7,12 (m, 1H), 7,00 (m, 2H), 6,81 (m, 2H), 5,12 (s, 2H), 4,14 (m, 4H), 3,54 (m, 7H), 2,83 (m, 3H), 2,33 (m, 6H), 1,56 (t, 3H, J=7,2 Гц)
Пример 8
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид сукцинат

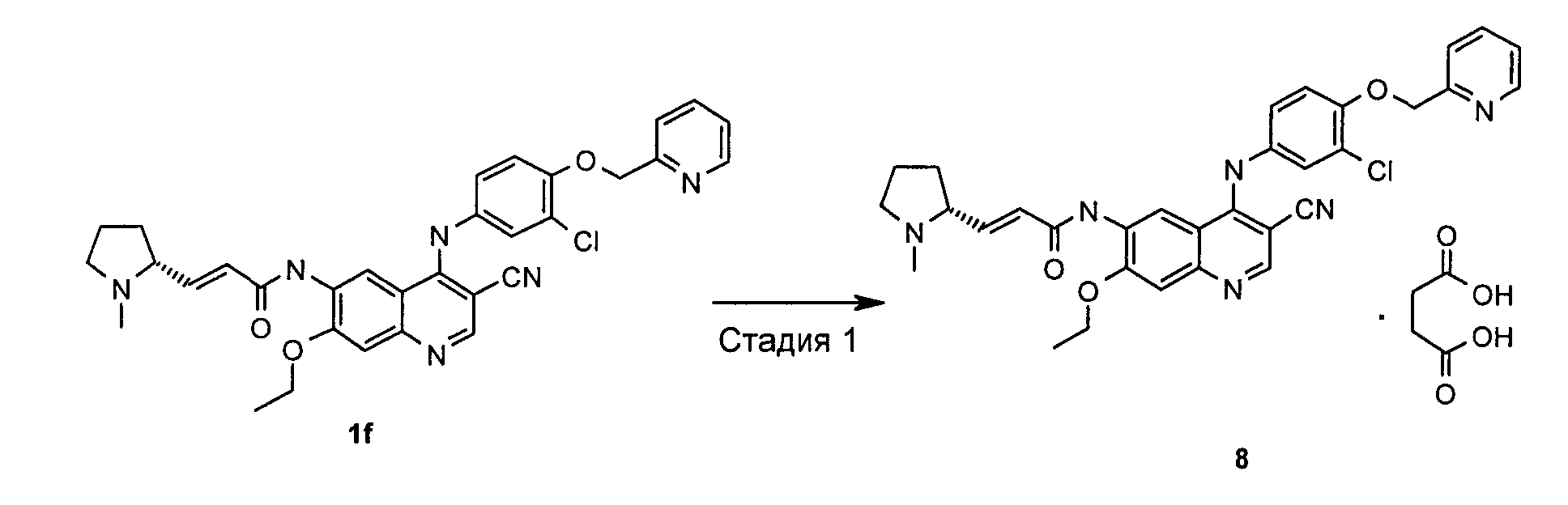
Стадия 1
(E)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид сукцинат
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид 1f (400 мг, 0,69 ммоль) при перемешивании растворяли в 5 мл дихлорметана, а затем добавляли янтарную кислоту (89,0 мг, 0,75 ммоль). После перемешивания в течение 0,5 часов при к.т. реакционную смесь концентрировали при пониженном давлении и сушили в вакууме с получением указанного в заголовке продукта (E)-N-[4-[[3-хлор-4-(2-пиридилметокси)сренил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид сукцината 8 (442 мг, выход 90,4%) в виде желтого твердого вещества.
MS m/z (ESI): 583,4 [M+1-118]
1H NMR (400 МГц, MeOD): δ 12,21 (s, 2H), 9,62 (s, 1H), 8,96 (s, 1H), 8,61 (s, 1H), 8,47 (m, 1H), 7,87 (m, 1H), 7,59 (m, 1H0,7,39 (m, 2H), 7,25 (m, 2H), 6,66 (m, 1H), 5,76 (m, 1H), 5,29 (s, 2H), 4,31 (m, 2H), 4,03 (m, 1H), 3,32 (m, 2H), 2,50 (m, 2H), 2,24 (m, 1H), 1,48 (t, 3H, J=7,2 Гц)
Пример 9
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид дигидрохлорид

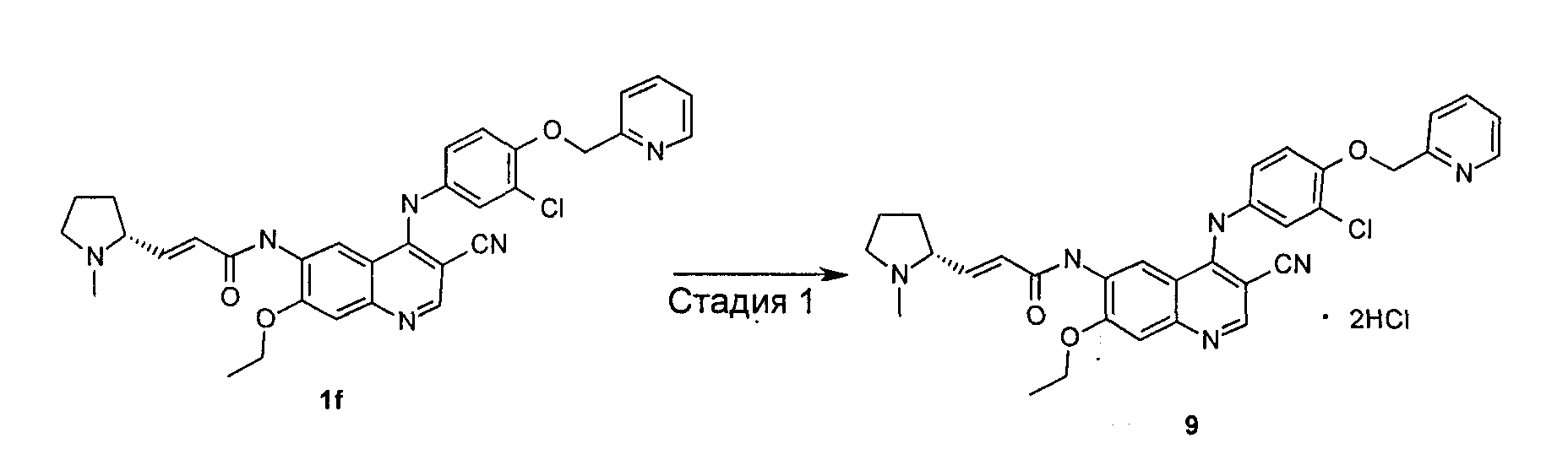
Стадия 1
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид дигидрохлорид
(E)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид 1f (1000 мг, 1,71 ммоль) при перемешивании растворяли в 5 мл дихлорметана, а затем добавляли раствор соляной кислоты в диэтиловом эфире (1М, 3,42 мл, 3,42 ммоль). После перемешивания в течение 0,5 часов в ледяной бане реакционную смесь концентрировали при пониженном давлении и сушили в вакууме с получением указанного в заголовке продукта (E)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид дигидрохлорида 9 (1500 мг, выход 100%) в виде желтого твердого вещества.
MS m/z (ESI): 583,2 [M+1-72]
1H NMR (400 МГц, CDCl3): δ 9,16 (s, 1H), 8,60 (s, 1H), 8,51 (s, 1H), 8,13 (s, 1H), 7,79 (m, 1H), 7,68 (m, 1H), 7,45 (m, 1H), 7,35 (s, 1H), 7,33 (d, 1H, J=4 Гц), 7,25 (m, 1H), 7,13 (m, 1H), 7,01 (m, 1H), 6,98 (m, 1H), 6,19 (m, 1H), 5,31 (s, 2H), 4,35 (dd, 2H, J=8 Гц, J=16 Гц), 4,24 (m, 1H), 3,19 (m, 1H), 2,85 (m, 1H), 2,33 (m, 4H), 2,09 (m, 1H), 1,60 (t, 3H, J=8 Гц)
Примеры тестов
Биологическая оценка
Пример 1: анализ ингибирования пролиферации клеток egfr
Следующий анализ in vitro предназначен для определения активности соединений по настоящему изобретению в отношении ингибирования пролиферации клеток человеческой эпидермоидной карциномы А431, имеющих высокий уровень экспрессии EGFR.
Следующий анализ in vitro предназначен для определения активности тестируемых соединений в отношении ингибирования пролиферации раковых клеток, обладающих высоким уровнем экспрессии EGFR. Активность представлена значением IC50 (средней ингибирующей концентрации). Общие процедуры анализа являются следующими: выбрали раковые клетки А431, имеющие высокий уровень экспрессии EGFR, и засевали в 96-луночный культуральный планшет с подходящей плотностью (например 5000 клеток/мл среды). Клетки затем инкубировали в инкубаторе с диоксидом углерода до достижения ими 85% конфлюэнтности. Затем клеточную культуральную среду заменяли на свежую среду, содержащую тестируемые соединения с сериями разведении (как правило, 6-7 концентраций). Клетки вновь помещали в инкубатор и непрерывно выращивали в течение 72 часов. Активность тестируемых соединений в отношении ингибирования клеточной пролиферации определяли с использованием способа с сульфородамином В (SRB). Значения IC50 рассчитывали в соответствии с уровнем ингибирования при различных концентрациях тестируемых соединений.
Активность соединений по настоящему изобретению:
Биологическую активность соединений по настоящему изобретению тестировали путем использования вышеописанного анализа. Значения IC50 рассчитывали и представили в следующей таблице:
Заключение: малеат и другие соли, и свободное основание соединений формулы (I) обладают очевидной активностью в отношении ингибирования пролиферации клеток А431, обладающих высоким уровнем экспрессии EGFR.
Пример 2: Анализ киназной активности EGFR
Киназную активность EGFR in vitro тестировали при помощи следующего анализа.
Следующий анализ может быть использован для определения активности соединений по настоящему изобретению в отношении ингибирования киназной активности EGFR. Среднюю ингибирующую концентрацию IC50 (концентрацию тестируемого соединения, демонстрирующую 50% ингибирование ферментативной активности) для каждого соединения определяли путем инкубации нескольких различных концентраций тестируемых соединений со специфическим ферментом и субстратом. Киназа EGFR, используемая в этом анализе, представляет собой человеческий рекомбинантный белок, который инкубировали с пептидным субстратом при различных концентрациях тестируемых соединений в буферном растворе, содержащем 60 мМ HEPES (рН 7,5), 5 мМ MgCl2, 5 мМ MnCl2, 3 мкМ Na3VO4, 1,25 М DTT (дитиотреитол) (1000×) и 20 мкМ АТР (аденозинтрифосфат) при 25°C в течение 45 минут. Киназную активность EGFR количественно определяли путем использования способа флуоресценции с разрешением по времени.
Активность соединений по настоящему изобретению:
Биологическую активность соединений по настоящему изобретению тестировали путем использования вышеописанного анализа. Значения IC50 рассчитывали и представили в следующей таблице:
Заключение: малеат и другие соли, и свободное основание соединений формулы (I) обладают очевидной активностью в отношении ингибирования киназной активности EGFR.
Пример 3: анализ активности клеток Пег-2
Следующий анализ in vitro предназначен для определения активности соединений по настоящему изобретению в отношении ингибирования пролиферации клеток человеческой карциномы SK-BR-3, имеющих высокий уровень экспрессии HER-2
Следующий анализ in vitro предназначен для определения активности тестируемых соединений в отношении ингибирования ангиогенеза и пролиферации раковых клеток, обладающих высоким уровнем экспрессии HER-2. Активность представлена значением IC50. Общие процедуры анализа являются следующими: выбрали линию раковых клеток SK-BR-3, имеющих высокий уровень экспрессии HER-2, и засевали в 96-луночный культуральный планшет с подходящей плотностью. Клетки затем инкубировали в инкубаторе с диоксидом углерода до достижения ими 60% конфлюэнтности. Затем клеточную культуральную среду заменяли на свежую среду, содержащую тестируемые соединения в различных концентрациях (как правило, 6-7 концентраций). Клетки вновь помещали в инкубатор и непрерывно выращивали в течение 96 часов. Активность тестируемых соединений в отношении ингибирования клеточной пролиферации определяли с использованием способа с сульфородамином В (SRB). Значения IC50 рассчитывали в соответствии с уровнем ингибирования при различных концентрациях тестируемых соединений.
Активность соединений по настоящему изобретению:
Биологическую активность соединений по настоящему изобретению тестировали путем использования вышеописанного анализа. Значения IC50 рассчитывали и представили в следующей таблице:
Заключение: малеат и другие соли, и свободное основание соединений формулы (I) обладают очевидной активностью в отношении ингибирования пролиферации клеток SK-BR-3, обладающих высоким уровнем экспрессии Her-2.
Пример 4: Анализ киназной активности Her-2
Киназную активность Her-2 in vitro тестировали при помощи следующего анализа.
Следующий анализ может быть использован для определения активности соединений по настоящему изобретению в отношении ингибирования киназы Her-2. Среднюю ингибирующую концентрацию IC50 (концентрацию тестируемого соединения, демонстрирующую 50% ингибирование ферментативной активности) для каждого соединения определяли путем инкубации нескольких различных концентраций тестируемых соединений со специфическим ферментом и субстратом. Киназа Her-2, используемая в этом анализе, представляет собой человеческий рекомбинантный белок, который инкубировали с пептидным субстратом при различных концентрациях тестируемых соединений в буферном растворе, содержащем 60 мМ HEPES (pH 7,5), 5 мМ MgCl2, 5 мМ MnCl2, 3 мкМ Na3VO4, 1,25 М DTT (дитиотреитол) (1000×) и 20 мкМ АТР (аденозинтрифосфат) при 25°C в течение 45 минут. Киназную активность Her-2 определяли путем использования способа флуоресценции с разрешением по времени.
Активность соединений по настоящему изобретению:
Биологическую активность соединений по настоящему изобретению тестировали путем использования вышеописанного анализа. Значения IC50 рассчитывали и представили в следующей таблице:
Заключение: малеат и другие соли, и свободное основание соединений формулы (I) обладали очевидной активностью в отношении ингибирования киназной активности Her-2.
Анализ растворимости
В соответствии с обычным измерением растворимости, растворимость соединения формулы (I) и его солей определяли в трех различных системах: вода, физиологический раствор и метанол. Результаты представлены в таблице 1:
Заключение: По сравнению со свободным основанием и другими солями соединения формулы (I) растворимость малеата соединения формулы (I) была значимо лучше.
Фармакокинетический анализ
Пример теста 1
Фармакокинетический анализ соединений по настоящему изобретению
1. Задача теста
В качестве тестируемых животных использовали крыс. Соединение формулы (I) и другие его соли вводили внутрижелудочно, малеат соединения формулы (I) инъецировали в хвостовую вену для определения концентрации лекарства в плазме крови в различные моменты времени при помощи способа LC/MS/MS (жидкостная хроматография/тандемная масс-спектрометрия). Фармакокинетическое поведение, характеристики и абсолютная биодоступность при пероральном введении соединений по настоящему изобретению исследовали и оценивали у крыс.
2. Протокол
2.1 Тестируемые образцы
Соединение 1f в качестве примера, соединения 1, 2, 4, 5, 6, 7 и 9 в качестве примера.
2.2 Тестируемые животные
28 здоровых взрослых крыс SD, самцы и самки поровну, приобретены в SINO-BRITSH SIPPR/BK LAB. ANIMAL LTD., CO, Номер лицензии: SCXK (Shanghai) 2008-0016 и разделены на 7 групп (по 4 крысы в каждой группе).
2.3 Оборудование
Тройной квадрупольный масс-спектрометр TSQ Quantum Ultra AM triple, Thermo Finnigan (US);
Система высокоэффективной жидкостной хроматографии Agilent 1200, Agilent (US).
2.4 Приготовление тестируемых соединений
Группа для внутривенной инъекции: подходящее количество соединений взвешивали, растворяли в DMSO (диметилсульфооксид) и разбавляли нормальным физиологическим раствором до конечного объема. Концентрация образца составляла 2,5 мг/мл.
Группа для внутрижелудочного введения: подходящее количество соединений взвешивали и добавляли в 0,5% CMC-Na с получением суспензии 2,5 мг/мл.
2.5 Введение
32 здоровые взрослые крысы SD, самцы и самки поровну, разделили на 7 групп (по 4 крысы в каждой группе). После ночного поста соединение 1f в качестве примера, соединение 1, 2, 4, 5, 6, 7 и 9 в качестве примера в дозе 25 мг/кг (рассчитанной по основной части) и объеме 10 мл/кг вводили внутрижелудочно или инъецировали в хвостовую вену.
2.6 Отбор образцов
В группе с внутривенной инъекцией образцы крови (0,2 мл) отбирали из орбитального синуса перед введением и через 2 мин, 15 мин, 30 мин, 1,0 ч, 2,0 ч, 4,0 ч, 6,0 ч, 8,0 ч, 12,0 ч, 24,0 ч и 36,0 ч после введения, хранили в гепаринизированных пробирках и центрифугировали в течение 10 минут при 3500 об./мин для отделения плазмы крови. Образцы плазмы крови хранили при -20°C.
В группе с внутрижелудочным введением образцы крови отбирали перед введением и через 0,5, 1,0, 2,0, 3,0, 4,0, 6,0, 8,0, 12,0, 24,0 и 36,0 часов после введения. Способ обработки образцов был таким же, как для группы с внутривенной инъекцией. Крыс кормили через 2 часа после введения.
3. Способ
50 мкл плазмы крови крысы добавляли в 50 мкл серии стандартных растворов соответственно с получением концентрации в плазме крови 50,0, 100, 200, 500, 1000, 2000 и 5000 нг/мл, затем образцы плазмы крови в различных концентрациях смешивали с 50 мкл метанола в течение 3 мин путем использования вортекса, и смесь центрифугировали в течение 10 минут (13500 об./мин). 10 мкл супернатанта анализировали при помощи LC-MS/MS (жидкостной хроматографии/тандемной масс-спектрометрии).
2.9 Расчет фармакокинетических параметров
4. Результаты фармакокинетических параметров
Фармакокинетические параметры соединений по настоящему изобретению представлены на таблице 2.
Заключение: По сравнению со свободным основанием и другими солями соединений формулы (I) малеат соединения формулы (I) значимо улучшает фармакокинетические характеристики и биодоступность и обладает очевидным фармакокинетическим преимуществом.
Для того чтобы продемонстрировать лучшую активность соединений по изобретению по сравнению с известными из уровня техники, Заявитель дополнительно осуществил сравнительные испытания соединений по изобретению и соединений, раскрытых в документе уровня техники CN 101824029. Приведенный ниже сравнительный эксперимент показывает, что хиральное соединение согласно настоящему изобретению (Пример 1f) и его соли имеют более сильный эффект, нежели рацемическое вещество, раскрытое в CN 101824029.
Для соединения согласно CN 101824029 и соединений согласно настоящему изобретению был проведен сравнительный анализ активности в отношении киназы EGFR in vitro и активности в отношении киназы HER-2 in vitro. Полученные результаты показаны ниже.


Как показано в таблице выше:
EGFR(BIO): Значения IC50 для солей согласно изобретению находятся в интервале 0,009-0,035 мкМ; для соединения 1f (R-изомер) это значение составляет 0,013 мкМ; в то время как для рацемата в CN 101824029 оно составляет 0,22 мкМ; то есть, активность указанных солей в 7-25 раз выше, чем активность соединения согласно CN 101824029, и активность соединения 1f в 17 раз выше активности соединения согласно CN 101824029;
HER2(BIO): Значения IC50 указанных солей находятся в интервале 0,045-0,078 мкМ; для соединения 1f (R-изомер) это значение составляет 0,097 мкМ; для рацемата согласно CN 101824029 оно составляет 0,19 мкМ; то есть, активность указанных солей в 2,4-4 раза выше, чем соединения согласно CN 101824029, и активность соединения 1f в 2 раза выше активности соединения согласно CN 101824029.
Таким образом, активность стереоизомерного соединения 1f и его солей согласно настоящему изобретению значительно выше по сравнению с активностью рацемического соединения согласно CN 101824029.
Реферат
Изобретение относится к области органической химии, а именно к фармацевтически приемлемой соли (E)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамида (формулы (I)), где n равен 1, 2 или 3; и М представляет собой молекулу кислоты, где указанная соль выбрана из группы, состоящей из гидрохлорида, пара-толуолсульфоната, метансульфоната, малеата, сукцината и L-малата. Также изобретение относится к способу получения соли соединения формулы (I), фармацевтической композиции на основе соли соединения формулы (I) и к применению соли соединения формулы (I). Технический результат: получены новые соли соединения формулы (I), полезные при лечении рака. 6 н. и 4 з.п. ф-лы, 7 табл., 13 пр.
Формула

где
n равен 1, 2 или 3; и
М представляет собой молекулу кислоты, где указанная соль выбрана из группы, состоящей из гидрохлорида, пара-толуолсульфоната, метансульфоната, малеата, сукцината и L-малата.
Документы, цитированные в отчёте о поиске
3-цианохинолины в качестве ингибиторов egf-r и her2 киназ
Комментарии