Соединения и композиции для подавления активности shp2 - RU2744988C2
Код документа: RU2744988C2
Описание
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЯ
[0001] Настоящее изобретение относится к соединениям, способным подавлять активность SHP2. В изобретении дополнительно предложены способ получения соединений данного изобретения, фармацевтические препараты, содержащие такие соединения, и методы использования таких соединений и композиций в лечении заболеваний или расстройств, связанных с отличающейся от нормальной активностью SHP2.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
[0002] Фосфатаза с Src-гомологичным доменом 2 (SHP2) представляет собой нерецепторную тирозинфосфатазу белка, кодируемую геном PTPN11, который участвует во многих клеточных функциях, включая пролиферацию, дифференциацию, поддержание клеточного цикла и миграцию. SHP2 участвует в сигнальном процессе через каскадные пути Ras-митоген-активированной протеинкиназы, JAK-STAT или фосфоинозитол-3-киназы-AKT.
[0003] SHP2 имеет два N-концевых Src-гомологичных домена 2 (N-SH2 и C-SH2), каталитический домен (PTP) и C-концевой хвост. Два SH2 домена контролируют субклеточную локализацию и функциональную регуляцию SHP2. Молекула существует в неактивной, самоингибированной конформации, стабилизированной связывающей сетью, в которой участвуют остатки из обоих доменов N-SH2 и PTP. Стимулирование, например, цитокинов или факторов роста, приводит к обнажению каталитических сайтов, приводящему к ферментативной активации SHP2.
[0004] Мутации в гене PTPN11, а впоследствии и в SHP2, были обнаружены в нескольких болезнях человека, таких как синдром Нунан, синдром Leopard, ювенильные миеломоноцитарные лейкозы, нейробластома, меланома, острый миелоидный лейкоз и раковые заболевания молочной железы, легкого и толстой кишки. SHP2, таким образом, представляет собой очень привлекательную цель для разработки новых способов терапевтического лечения различных заболеваний. Соединения настоящего изобретения направлены на удовлетворение потребности в малых молекулах, которые подавляют активность SHP2.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
[0005] В одном аспекте настоящего изобретения предложены соединения Формулы I:

[0006] в которых:
[0007] R1 выбирают из:

[0008] R2 и R3 вместе с атомом азота, к которому присоединены оба R2 и R3, образуют кольцо, выбранное из пиперидинила, пиперазинила, 2-окса-8-азаспиро[4.5]декан-8-ила, 8-азаспиро[4.5]декан-8-ила и пирролидинила; где указанные пирролидинил, пиперазинил, 2-окса-8-азаспиро[4.5]декан-8-ил, 8-азаспиро[4.5]декан-8-ил или пиперидинил незамещены или замещены 1-3 группами, независимо выбранными из аминогруппы, метила, этила, амино-метила, метил-аминогруппы, гидроксила, цианогруппы, фтор-метила, фтора и ((((5-метил-2-оксо-1,3-диоксол-4-ил)метокси)карбонил)амино)метила; R4 выбран из гидроксила, C1-3алкоксигруппы и OC(O)C1-3алкила; R5 выбран из H и метила; R6 выбран из водорода, метила и фенила; R7 выбран из водорода, метила, этила, фенила и бензила; R8 выбран из водорода и метила; Y1 выбран из N и CH; Y2 выбран из N и CH; Y3 выбран из NH и CH2; Y4 выбран из N и CH; Y5 выбран из N и CH; или их фармацевтически приемлемые соли.
[0009] Во втором аспекте настоящего изобретения предложена фармацевтическая композиция, которая содержит соединение Формулы I или его N-оксидное производное, таутомер, отдельные изомеры или смесь изомеров; или его фармацевтически приемлемую соль с добавлением одного или нескольких подходящих формообразующих.
[0010] В третьем аспекте настоящего изобретения предложен способ лечения заболевания у животного, в котором изменение активности SHP2 может предотвратить, подавить или облегчить течение патологического процесса и/или симптомы заболевания, причем способ включает введение животному терапевтически эффективного количества соединения Формулы I или его N-оксидного производного, его отдельных изомеров или смеси изомеров, или его фармацевтически приемлемой соли.
[0011] В четвертом аспекте настоящего изобретения предложен способ лечения заболевания у животного, в котором изменение активности SHP2 может предотвратить, подавить или облегчить течение патологического процесса и/или симптомы заболевания, причем способ включает введение животному терапевтически эффективного количества соединения Формулы I или его N-оксидного производного, его отдельных изомеров или смеси изомеров, или его фармацевтически приемлемой соли одновременно или последовательно в комбинации с противораковым терапевтическим средством.
[0012] В пятом аспекте настоящего изобретения предложено применение соединения Формулы I в производстве лекарственного средства для лечения заболевания у животного, при котором активность SHP2 содействует течению патологического процесса и/или проявлению симптомов заболевания.
[0013] В шестом аспекте настоящего изобретения предложен способ получения соединений Формулы I и их N-оксидных производных, производных пролекарств, защищенных производных, отдельных изомеров и смеси изомеров, и их фармацевтически приемлемых солей.
Определения
[0014] Общие термины, используемые ранее и далее, предпочтительно имеют в контексте настоящего раскрытия следующие значения, если не указано иное, где более общие термины, которые могут использоваться независимо друг от друга, могут заменяться более конкретными определениями или не изменяться, таким образом определяя более подробные варианты осуществления изобретения:
[0015] «Алкил» относится к полностью насыщенному разветвленному или неразветвленному углеводородному фрагменту, содержащему вплоть до 20 атомов углерода. Если не указано иное, алкил относится к углеводородным остаткам, имеющим от 1 до 7 атомов углерода (C1-7алкил) или от 1 до 4 атомов углерода (C1-4алкил). Типичные примеры алкила включают, но не ограничиваются ими, метил, этил, н-пропил, изо-пропил, н-бутил, втор-бутил, изо-бутил, трет-бутил, н-пентил, изопентил, неопентил, н-гексил, 3-метилгексил, 2,2-диметилпентил, 2,3-диметилпентил, н-гептил, н-октил, н-нонил, н-децил и т.п. Замещенный алкил представляет собой алкильную группу, содержащую один или несколько, например, один, два или три заместителя, выбранных из галогена, гидрокси- или алкоксигрупп. Галогензамещенный алкил и галогензамещенная алкоксигруппа могут быть либо линейными, либо разветвленными и включают в себя метоксигруппу, этоксигруппу, дифторметил, трифторметил, пентафторэтил, дифторметоксигруппу, трифторметоксигруппу и т.п.
[0016] «Арил» означает моноциклическую или конденсированную бициклическую ароматическую циклическую систему, содержащую от шести до десяти кольцевых атомов углерода. Например, арил может представлять собой фенил или нафтил, предпочтительно фенил. «Арилен» означает двухвалентный радикал, полученный из арильной группы.
[0017] «Гетероарил» такой же, как определено выше для арила, где один или несколько членов кольца представляют собой гетероатом. Например, C5-10гетероарил состоит из минимум 5 членов, на что указывают атомы углерода, но эти атомы углерода могут быть заменены гетероатомом. Соответственно, C5-10гетероарил включает пиридил, индолил, индазолил, хиноксалинил, хинолинил, бензофуранил, бензопиранил, бензотиопиранил, бензо[1,3]диоксол, имидазолил, бензо-имидазолил, пиримидинил, фуранил, оксазолил, изоксазолил, триазолил, тетразолил, пиразолил, тиенил и т.п.
[0018] «Циклоалкил» означает насыщенную или частично ненасыщенную моноциклическую, конденсированную бициклическую или мостиковую полициклическую кольцевую систему, содержащую указанное количество атомов кольца. Например, C3-10циклоалкил включает циклопропил, циклобутил, циклопентил, циклогексил, циклогексенил и т.п.
[0019] «Гетероциклоалкил» означает циклоалкил, как определено в этой заявке, при условии, что один или несколько из указанных кольцевых атомов углерода заменены на фрагмент, выбранный из -O-, -N=, -NR-, -C(O)-, -S-, -S(O)- или -S(O)2-, где R представляет собой водород, C1-4алкил или защитную группу азота. Например, C3-8гетероциклоалкил, используемый в контексте данной заявки для обозначения соединений данного изобретения, включает морфолино, пирролидинил, пирролидинил-2-он, пиперазинил, пиперидинил, пиперидинилон, 1,4-диокса-8-аза-спиро[4.5]дец-8-ил, тиоморфолино, сульфаноморфолино, сульфономорфолино и т.п.
[0020] «Галоген» (или галоген-) предпочтительно обозначает атом хлора или фтора, то также может быть атомом брома или иода.
[0021] «SHP2» означает «фосфатаза с Src-гомологичным доменом 2» и она также известна как SH-PTP2, SH-PTP3, Syp, PTP1D, PTP2C, SAP-2 или PTPN11.
[0022] Онкологические заболевания, при которых встречаются «мутации PTPN11», включают, но не ограничиваются ими: N58Y; D61Y, V; E69K; A72V, T, D; E76G, Q, K (ALL); G60A; D61Y; E69V; F71K; A72V; T73I; E76G, K; R289G; G503V (AML); G60R, D61Y, V, N; Y62D; E69K; A72T, V; T73I; E76K, V, G, A, Q; E139D; G503A, R; Q506P (JMML); G60V; D61V; E69K; F71L; A72V; E76A (MDS); Y63C (CMML); Y62C; E69K; T507K (нейробластома); V46L; N58S; E76V (рак легкого); R138Q (меланома); E76G (рак толстой кишки).
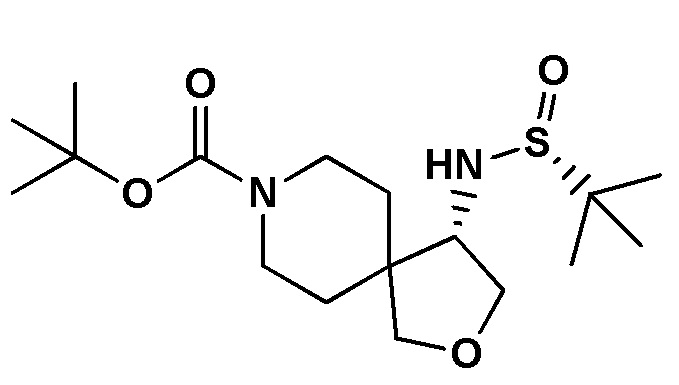
[0023] В данной заявке указанная выше структура представлена (S)-трет-бутил-4-((R)-1,1-диметилэтилсульфинамидо)-2-окса-8-азаспиро[4.5]декан-8-карбоксилатом (название, созданное с использованием chemdraw) и (S)-трет-бутил-4-((R)-1,1-диметилэтилсульфинамино)-2-окса-8-азаспиро[4.5]декан-8-карбоксилатом (название, созданное с использованием chemdraw).
[0024] Соединения формулы I могут иметь разные изомерные формы. Например, любой асимметричный атом углерода может находиться в (R)-, (S)- или (R,S)-конфигурации, предпочтительно в (R)- или (S)-конфигурации. Заместители при двойной связи или, особенно, в кольце могут находиться в цис- (= Z-) или транс- (= E-) форме. Таким образом, соединения могут присутствовать в виде смесей изомеров или предпочтительно в виде чистых изомеров, предпочтительно в виде чистых диастереомеров или чистых энантиомеров.
[0025] Когда используется форма множественного числа (например, соединения, соли), это включает форму единственного числа (например, одно соединение, одну соль). Употребление слова «соединение» не исключает того, что (например, в фармацевтическом составе) присутствует более одного соединения формулы I (или его соли) и представляет неопределенную форму единственного числа. Существительное в неопределенной форме единственного числа можно понимать как «один или несколько», и менее предпочтительно как «один».
[0026] При любом употреблении слов соединение или соединения формулы I также означает, что подразумеваются N-оксиды таких соединений и/или их таутомеры.
[0027] Выражение «и/или его N-оксид, его таутомер и/или его (предпочтительно фармацевтически приемлемая) соль» особенно означает, что соединение формулы I может быть само по себе или в смеси с его N-оксидом, в виде таутомера (например, вследствие кето-енольной, лактам-лактимовой, амид-имидокислотной или енамин-иминной таутомерии) или в (например, в результате равновесной реакции) смеси с его таутомером, или в виде соли соединения формулы I и/или любой из этих форм или смесей двух или более таких форм.
[0028] Настоящее изобретение также включает все подходящие изотопные разновидности соединений данного изобретения или их фармацевтически приемлемых солей. Изотопная разновидность соединения данного изобретения или его фармацевтически приемлемой соли определены как такая, в которой по меньшей мере один атом замещен атомом, имеющим такой же атомный номер, но его атомная масса отличается от атомной массы, обычно встречающейся в природе. Примеры изотопов, которые могут быть включены в соединения данного изобретения и их фармацевтически приемлемые соли, включают, но не ограничиваются ими, изотопы водорода, углерода, азота и кислорода, такие как2H,3H,11C,13C,14C,15N,17O,18O,35S,18F,36Cl и123I. Некоторые изотопные разновидности соединений данного изобретения и их фармацевтически приемлемых солей, например те, в которые включен радиоактивный изотоп, такой как3H или14C, полезны для исследований распределения в тканях лекарств и/или субстратов. В конкретных примерах изотопы3H и14C могут использоваться благодаря легкости их получения и распознавания. В других примерах замещение изотопами, такими как2H, может предоставлять определенные терапевтические преимущества, основанные на большей метаболической стабильности, такой как повышенное время полувыведения in vivo или потребность в более низком дозировании. Изотопные разновидности соединений данного изобретения или их фармацевтически приемлемых солей можно обычно получать традиционными методиками, используя подходящие изтопные разновидности подходящих реагентов.
Описание предпочтительных вариантов осуществления
[0029] Настоящее изобретение относится к соединениям, способным подавлять активность SHP2. В одном аспекте данного изобретения, где он касается соединений формул I и II, предложены соединения формулы II:

[0030] в которых: R1 выбирают из:

[0031] R2 и R3 вместе с атомом азота, к которому присоединены оба R2 и R3, образуют кольцо, выбранное из пиперидинила, пиперазинила, 2-окса-8-азаспиро[4.5]декан-8-ила, 8-азаспиро[4.5]декан-8-ила и пирролидинила; где указанные пирролидинил, пиперазинил, 2-окса-8-азаспиро[4.5]декан-8-ил, 8-азаспиро[4.5]декан-8-ил или пиперидинил незамещены или замещены 1-3 группами, независимо выбранными из аминогруппы, метила, этила, амино-метила, метил-аминогруппы, гидроксила, цианогруппы, фтор-метила, фтора и ((((5-метил-2-оксо-1,3-диоксол-4-ил)метокси)карбонил)амино)метила; R4 выбран из гидроксила; R5 выбран из H и метила; R6 выбран из водорода, метила и фенила; R7 выбран из водорода, метила, этила, фенила и бензила; R8 выбран из водорода и метила; Y1 выбран из N и CH; Y2 выбран из N и CH; Y3 выбран из NH и CH2; Y4 выбран из N и CH; Y5 выбран из N и CH; или их фармацевтически приемлемая соль.
[0032] В следующем варианте осуществления предложены соединения или их фармацевтически приемлемая соль, в которых: R1 выбирают из:

[0033] R4 выбран из гидроксила; R5 выбран из H и метила; R6 выбран из водорода, метила и фенила; R7 выбран из водорода, метила, этила, фенила и бензила; R8 выбран из водорода и метила; Y4 выбран из N и CH; и Y5 выбран из N и CH.
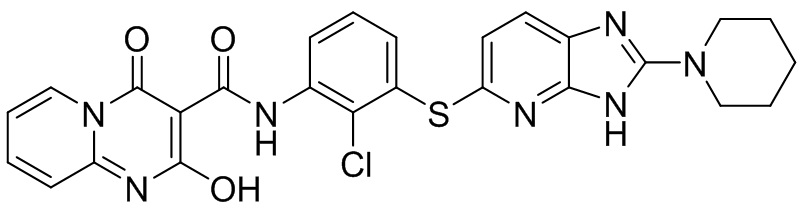
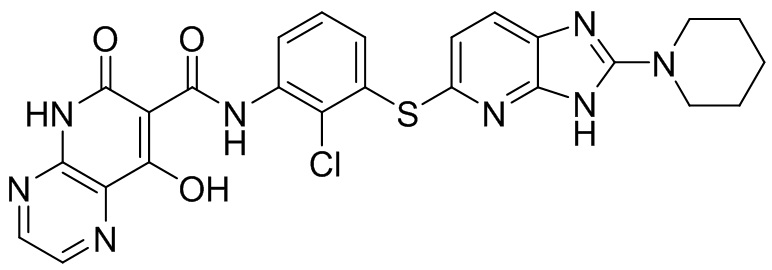
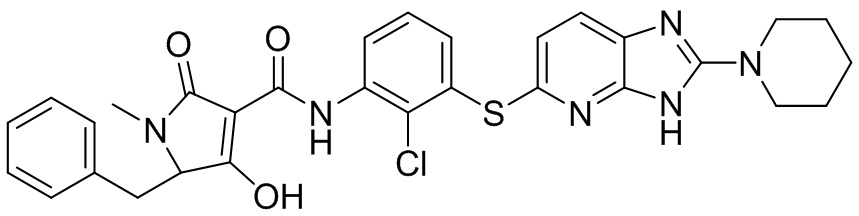
[0034] В следующем варианте осуществления предложены соединения или их фармацевтически приемлемая соль, выбранные из:



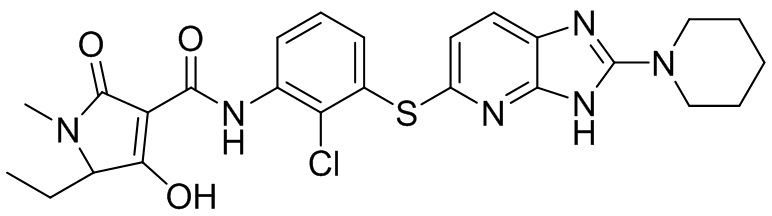
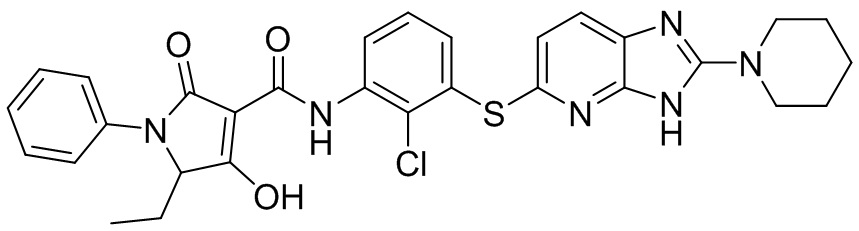
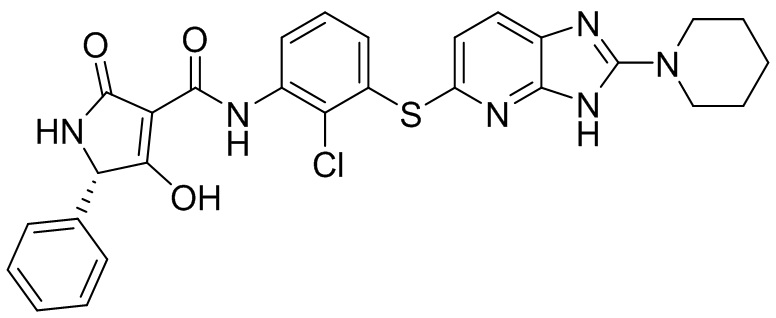

[0035] В другом варианте осуществления предложены соединения формулы I:

[0036] в которых R1 выбирают из:

[0037] R2 и R3 вместе с атомом азота, к которому присоединены оба R2 и R3, образуют кольцо, выбранное из пиперидинила, пиперазинила, 2-окса-8-азаспиро[4.5]декан-8-ила, 8-азаспиро[4.5]декан-8-ила и пирролидинила; где указанные пирролидинил, пиперазинил, 2-окса-8-азаспиро[4.5]декан-8-ил, 8-азаспиро[4.5]декан-8-ил или пиперидинил незамещены или замещены 1-3 группами, независимо выбранными из аминогруппы, метила, этила, амино-метила, метил-аминогруппы, гидроксила, цианогруппы, фтор-метила, фтора и ((((5-метил-2-оксо-1,3-диоксол-4-ил)метокси)карбонил)амино)метила; R4 выбран из гидроксила; R5 выбран из H и метила; R6 выбран из водорода, метила и фенила; R7 выбран из водорода, метила, этила, фенила и бензила; R8 выбран из водорода и метила; Y1 выбран из N и CH; Y2 выбран из N и CH; Y3 выбран из NH и CH2; Y4 выбран из N и CH; Y5 выбран из N и CH; или их фармацевтически приемлемая соль.
[0038] В следующем варианте осуществления предложены соединения или их фармацевтически приемлемая соль, выбранные из:

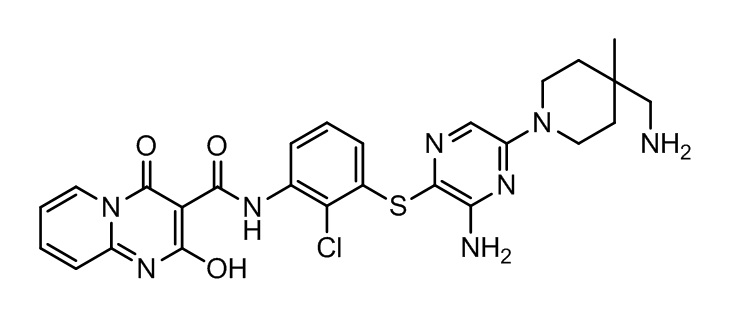
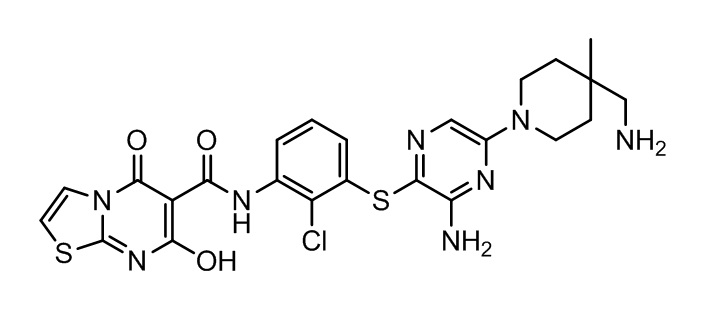

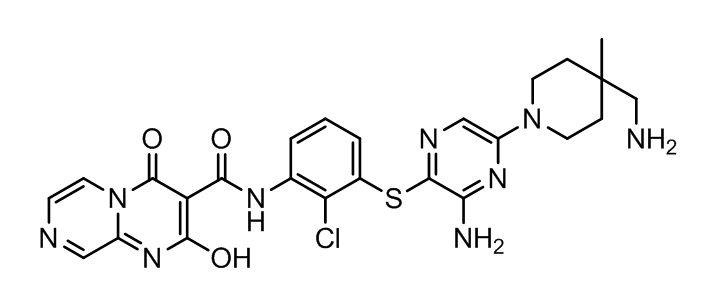


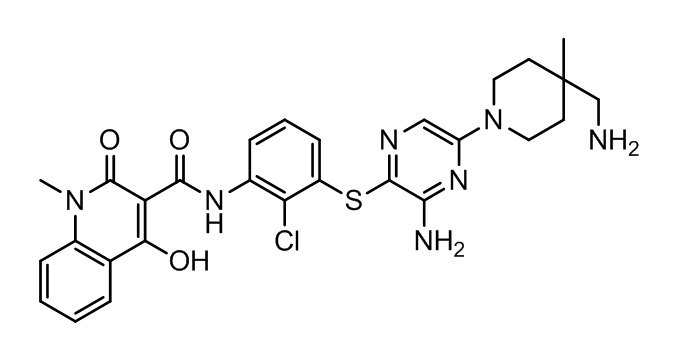
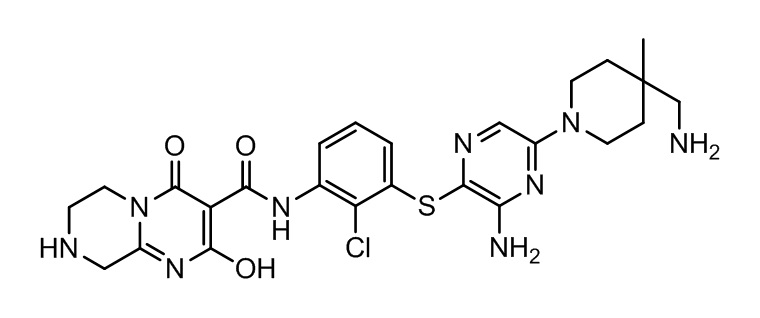


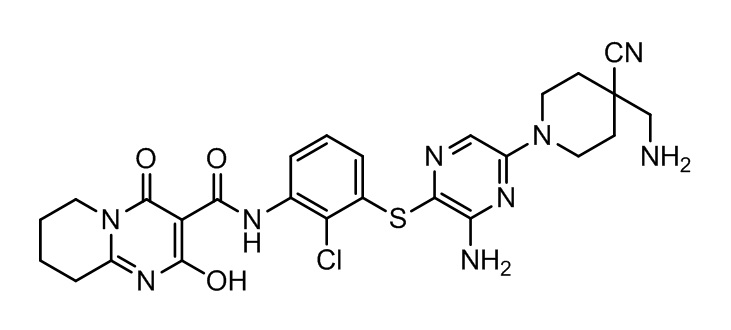
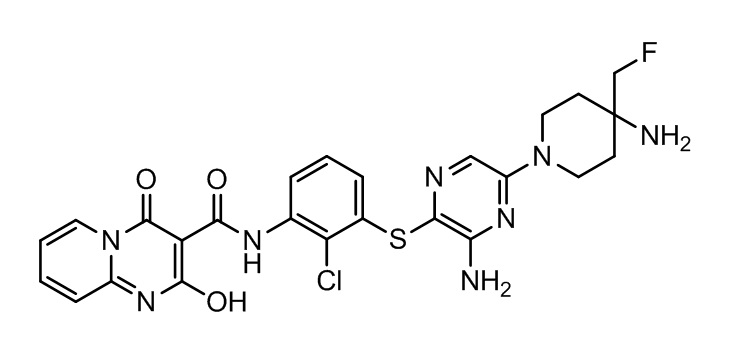
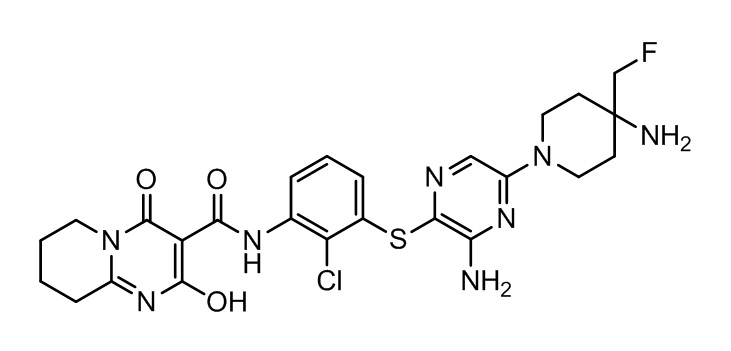
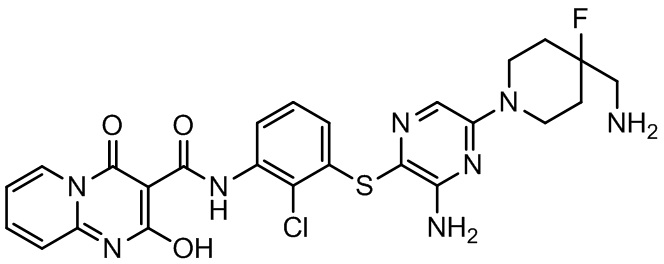

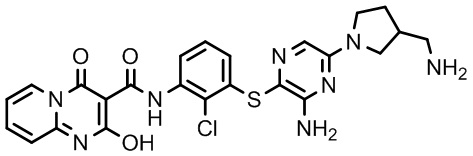
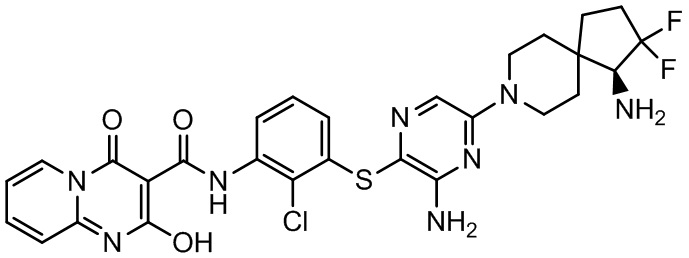





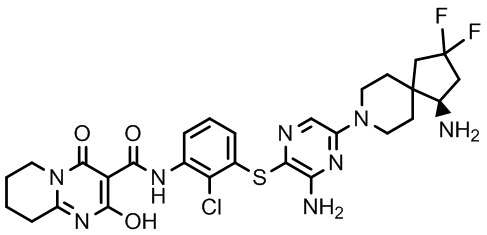

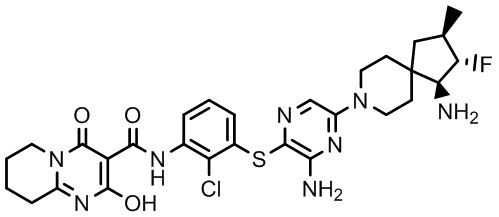





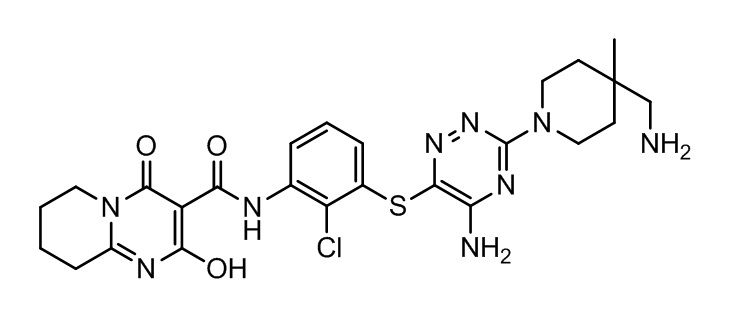
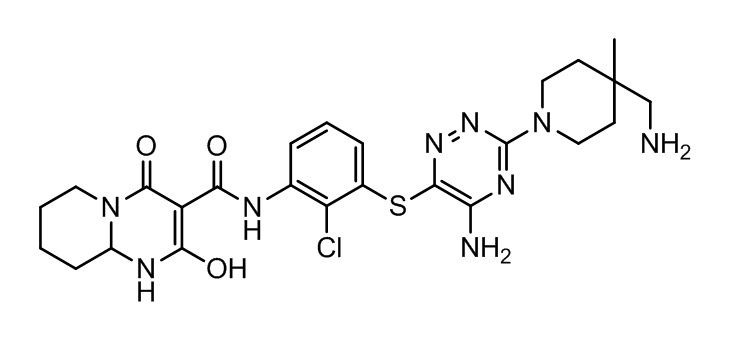
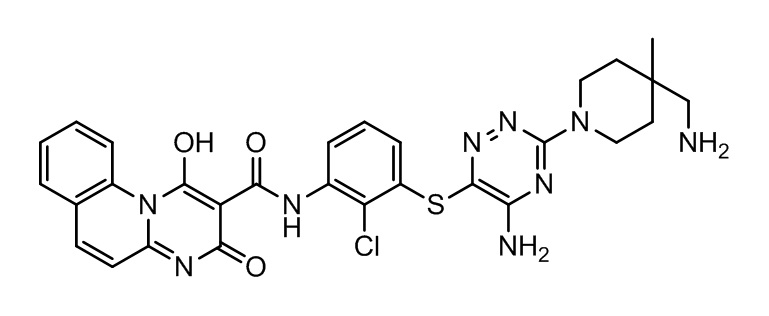
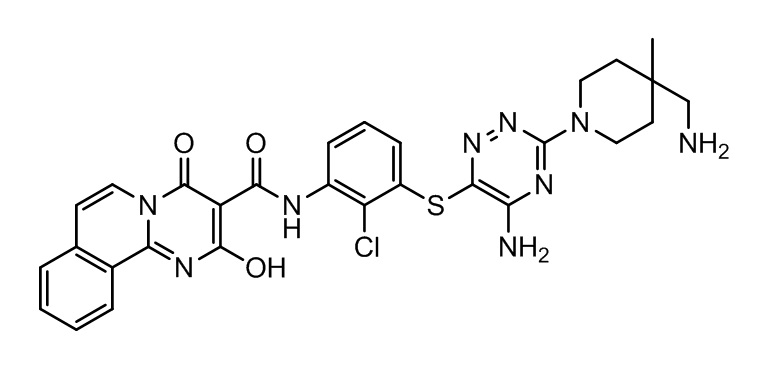

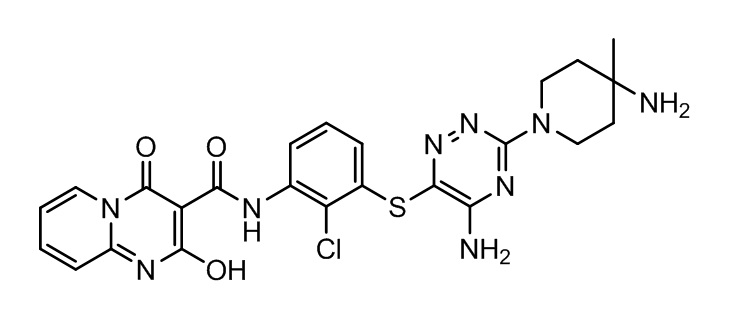



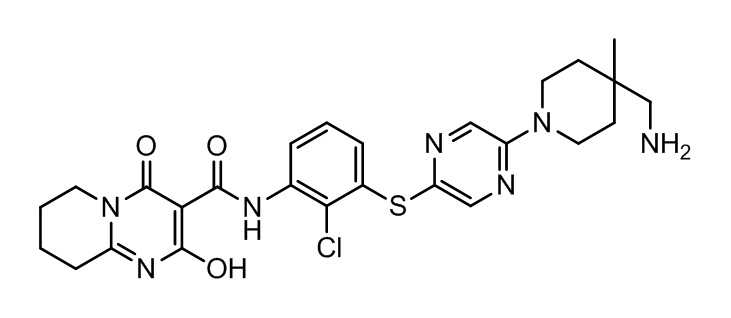
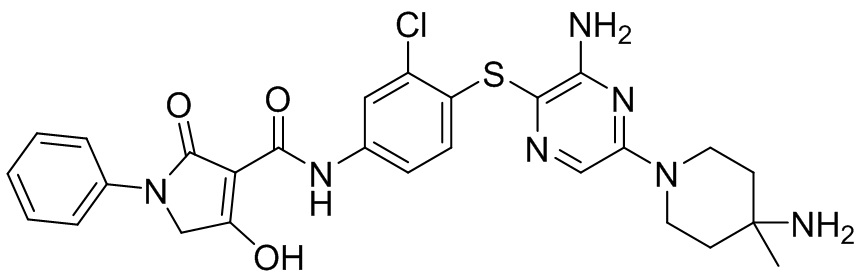

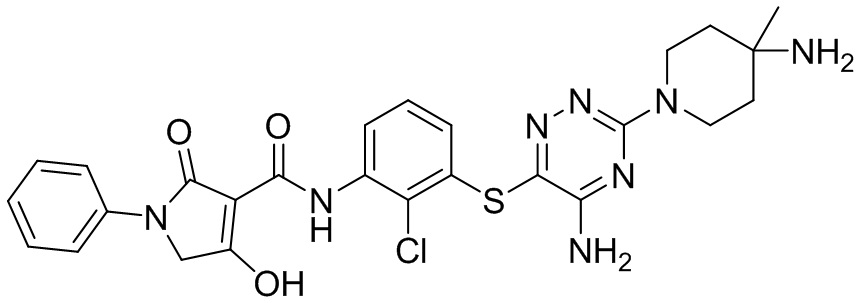
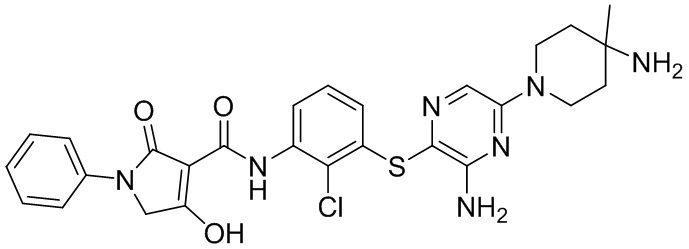
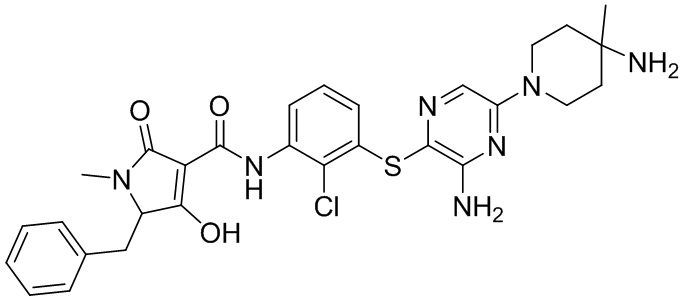
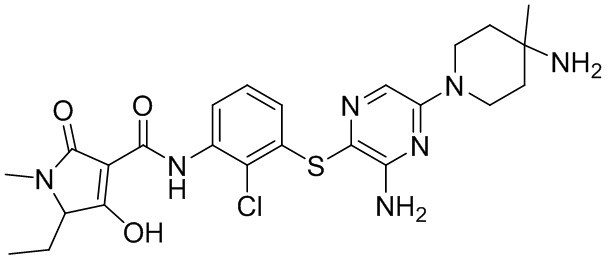


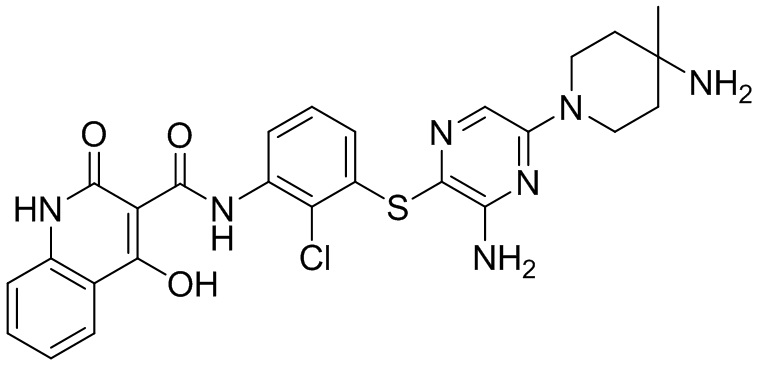

Фармакология и полезность
[0039] Фосфатаза с Src-гомологичным доменом 2 (SHP2) представляет собой тирозинфосфатазу белка, кодируемую геном PTPN11, который участвует во многих клеточных функциях, включая пролиферацию, дифференциацию, поддержание клеточного цикла и миграцию. SHP2 участвует в сигнальном процессе через каскадные пути Ras-митоген-активированной протеинкиназы, JAK-STAT или фосфоинозитол-3-киназы-AKT. SHP2 участвует как посредник в активации Erkl и Erk2 (Erkl/2, Erk) MAP киназ с помощью рецепторных тирозинкиназ, таких как ErbBl, ErbB2 и c-Met.
[0040] SHP2 имеет два N-концевых Src-гомологичных домена 2 (N-SH2 и C-SH2), каталитичский домен (PTP) и C-концевой хвост. Два SH2 домена контролируют субклеточную локализацию и функциональную регуляцию SHP2. Молекула существует в неактивной конформации, ингибирующей ее собственную активность посредством связывающей сети, в которой участвуют остатки из обоих доменов N-SH2 и PTP. В ответ на стимулирование фактором роста SHP2 связывается со специфическими тирозин-фосфорилированными сайтами на стыковочных белках, таких как Gab1 и Gab2, посредством своих SH2 доменов. Это вызывает изменение конформации, которое приводит к активации SHР2.
[0041] Мутации в PTPN11 были обнаружены в нескольких болезнях человека, таких как синдром Нунан, синдром Leopard, ювенильные миеломоноцитарные лейкозы, нейробластома, меланома, острый миелоидный лейкоз и раковые заболевания молочной железы, легкого и толстой кишки. SHP2 представляет собой важную сигнальную молекулу нисходящего пути для различных рецепторных тирозинкиназ, включая рецепторы тромбоцитарного фактора роста (PDGF-R), фибробластного фактора роста (FGF-R) и эпидермального фактора роста (EGF-R). SHP2 также представляет собой важную сигнальную молекулу нисходящего пути активации митоген-активированной протеинкиназы (MAP), который может приводить к перерождению клетки - необходимому условию развития онкологического заболевания. Нокдаун SHP2 в значительной степени подавлял клеточный рост в клеточных линиях рака легкого с мутацией SHP2 или транслокациями EML4/ALK, а также при усиленных EGFR типах рака молочной железы и пищевода. SHP2 также активируется по нисходящему от онкогенов пути при карциноме желудка, анапластической крупноклеточной лимфоме и глиобластоме.
[0042] Синдром Нунан (NS) и синдром Leopard (LS) -мутации PTPN11 вызывают LS (множественные лентигины, аномалии электрокардиографической проводимости, глазной гипертелоризм, стеноз легочного ствола, аномальные гениталии, задержку роста, перцептивную глухоту) и NS (врожденные аномалии, включая пороки сердца, черепно-лицевые аномалии и низкорослость). Оба нарушения являются частью семейства аутосомно-доминантных синдромов, вызванных мутациями зародышевой линии в компонентах сигнального пути RAS/RAF/MEK/ERK митоген-активированной протеинкиназы, необходимых для нормального роста и дифференциации клеток. Аберрантное регулирование этого сигнального пути имеет серьезные последствия, особенно для развития сердца, приводя к различным нарушениям, включая дефекты клапанной перегородки и/или гепертрофическую кардиомиопатию (НСМ). Было установлено, что искажение сигнального пути MAPK является основополагающей причиной этих нарушений, и несколько генов-кандидатов на протяжении этого сигнального пути были идентифицированы у человека, включая мутации в KRAS, NRAS, SOS1, RAF1, BRAF, MEK1, MEK2, SHOC2 и CBL. Наиболее часто мутирующим геном в случае NS и LS является PTPN11. Мутации зародышевой линии в PTPN11 (SHP2) найдены в ~50% случаев NS и почти у всех пациентов с LS, некоторые признаки которого сходны с NS. В случае NS, замены Y62D и Y63C в белке в основном инвариантны и являются одними из наиболее распространенных мутаций. Обе эти мутации негативно влияют на каталитически неактивную конформацию SHP2 без нарушения связывания фосфатазы с ее фосфорилированными сигнальными партнерами.
[0043] При ювенильных миеломоноцитарных лейкозах (JMML) соматические мутации в PTPN11 (SHP2) случаются у примерно 35% пациентов, страдающих JMML, вызывая миелопролиферативное нарушение детского возраста (MPD). Эти мутации приобретения функции обычно являются точечными мутациями в домене N-SH2 или в фосфатазном домене, что предотвращает самоингибирование между каталитическим доменом и доменом N-SH2, вызывая активность SHP2.
[0044] При остром миелоидном лейкозе мутации PTPN11 были обнаружены в: ~10% случаев детских острых лейкемий, таких как миелодиспластический синдром (MDS); ~7% случаев B-клеточного острого лимфобластного лейкоза (B-ALL); и ~4% случаев острого миелоидного лейкоза (AML).
[0045] Мутации при NS и лейкемии вызывают изменения в аминокислотах, расположенных на границе раздела, образованного доменами N-SH2 и PTP, в самоингибированной SHP2-конформации, нарушая ингибиторное внутримолекулярное взаимодействие, приводя к гиперактивности каталитического домена.
[0046] SHP2 действует как позитивный регулятор в сигнальном пути рецепторной тирозинкиназы (RTK). Онкологические заболевания, при которых встречаются изменения RTK (EGFRamp, Her2amp, FGFRamp, Metamp,транслокация/активация RTK, т.е. ALK, BCR/ABL) включают рак пищевода, молочной железы, легкого, толстой кишки, желудка, глиому, рак головы и шеи.
[0047] Рак пищевода - это злокачественное заболевание пищевода. Оно разделяется на различные подтипы, преимущественно на плоскоклеточный рак (<50%) и аденокарциному. При плоскоклеточном раке и аденокарциноме наблюдаются высокие уровни экспрессии RTK. Ингибитор SHP2 данного изобретения может, таким образом, применяться для передовых стратегий лечения.
[0048] Рак молочной железы является одним из основных онкологических заболеваний с летальным исходом у женщин, при котором развивается устойчивость к современным лекарственным средствам. Существует четыре основных типа рака молочной железы, которые включают люминальный A, люминальный B, Her2 и трижды негативный/базальный. Трижды негативный рак молочной железы (ТНРМЖ) представляет собой агрессивный рак молочной железы, для которого не существует достаточно целенаправленной терапии. Рецептор эпидермального фактора роста I (EGFR) был выделен как перспективная мишень при ТНРМЖ. Ингибирование Her2, также как и EGFR посредством SHP2 может оказаться перспективной терапией при раке молочной железы.
[0049] При раке легкого, NSCLC в настоящее время является основной причиной вызванных раком смертей, на долю которых приходится примерно 85% раков легкого (в основном аденокарциномы и плоскоклеточные карциномы). Хотя цитотоксическая химиотерапия остается важной частью лечения, целевые терапии, основанные на генетических изменениях, таких как EGFR и ALK в опухоли, скорее всего, выиграют от целевой терапии.
[0050] При раке толстой кишки известно, что примерно от 30% до 50% случаев колоректальных опухолей содержат мутировавший (ненормальный) KRAS, а мутации BRAF происходят в от 10 до 15% случаев колоректального рака. У пациентов с выявленной сверхэкпрессией EGFR в колоректальных опухолях наблюдается благоприятный клинический ответ на терапию анти-EGFR.
[0051] Рак желудка является одним из наиболее распространенных видов рака. В области техники известна аберрантная экспрессия тирозинкиназ, что выявлено наблюдением аберрантной фосфориляции тирозина в раковых клетках желудка. В желудочных карциномах часто амплифицированы три рецептора тирозинкиназ - c-met (HGF-рецептор), FGF-рецептор 2 и erbB2/neu. Таким образом, нарушение различных сигнальных путей может способствовать прогрессированию различных видов рака желудка.
[0052] Нейробластома является опухолью детского возраста развивающейся симпатической нервной системы, на которую приходится около 8% детских онкологических заболеваний. Было сделано предположение о том, что геномные изменения гена киназы анапластической лимфомы (ALK) способствуют патогенезу нейробластомы.
[0053] Плоскоклеточная карцинома головы и шеи (SCCHN). Высокие уровни экспрессии EGFR коррелируют с неблагоприятным прогнозом и устойчивостью к лучевой терапии при различных типах рака, в основном при плоскоклеточной карциноме головы и шеи (SCCHN). Блокировка сигнального пути EGFR приводит к подавлению стимуляции рецептора, клеточной пролиферации и снижает инвазивность и метастазирование. Поэтому EGFR является первой мишенью для новой противоопухолевой терапии при SCCHN.
[0054] Настоящее изобретение относится к соединениям, способным подавлять активность SHP2. В изобретении также предложен способ получения соединений данного изобретения и фармацевтические препараты, содержащие такие соединения. Другой аспект настоящего изобретения относится к способу лечения SHP2-опосредованных нарушений, включающему стадию введения пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы I, как определено в кратком изложении сущности изобретения.
[0055] В некоторых вариантах осуществления настоящее изобретение относится к вышеупомянутому способу, в котором упомянутые SHP2-опосредованные нарушения представляют собой онкологические заболевания, выбранные из, но не ограничиваясь ими: JMML; AML; MDS; B-ALL; нейробластомы; рака пищевода; рака молочной железы; рака легкого; рака толстой кишки; рака желудка, рака головы и шеи.
[0056] Соединения настоящего изобретения также могут быть полезны при лечении других заболеваний или состояний, связанных с отличающейся от нормальной активностью SHP2. Так в следующем аспекте изобретение касается способа лечения расстройства, выбранного из: NS; LS; JMML; AML; MDS; B-ALL; нейробластомы; рака пищевода; рака молочной железы; рака легкого; рак толстой кишки; рака желудка; рака головы и шеи.
[0057] Ингибитор SHP2 настоящего изобретения может быть с успехом объединен с другим фармакологически активным соединением или с двумя или более другими фармакологически активными соединениями, особенно при лечении рака. Например, соединение формулы (I) или его фармацевтически приемлемую соль, как определено выше, можно вводить одновременно, последовательно или отдельно в комбинации с одним или несколькими средствами, выбранными из химиотерапевтических средств, например, ингибиторов митоза, таких как таксан, алкалоид барвинка, паклитаксел, доцетаксел, винкристин, винбластин, винорелбин или винфлунин, и другими противоопухолевыми средствами, как например, цисплатин, 5-фторурацил или 5-фтор-2-4(1H,3H)-пиримидиндион (5FU), флутамид или гемцитабин.
[0058] При лечении такие комбинации могут иметь значительные преимущества, включая синергическое действие.
[0059] В некоторых вариантах осуществления настоящее изобретение относится к вышеупомянутому способу, в котором указанное соединение вводят парентерально.
[0060] В некоторых вариантах осуществления настоящее изобретение относится к вышеупомянутому способу, где указанное соединение вводят внутримышечно, внутривенно, подкожно, перорально, легочно, интратекально, местно или интраназально.
[0061] В некоторых вариантах осуществления настоящее изобретение относится к вышеупомянутому способу, в котором указанное соединение вводят системно.
[0062] В некоторых вариантах осуществления настоящее изобретение относится к вышеупомянутому способу, в котором указанный пациент является млекопитающим.
[0063] В некоторых вариантах осуществления настоящее изобретение относится к вышеупомянутому способу, в котором указанный пациент является приматом.
[0064] В некоторых вариантах осуществления настоящее изобретение относится к вышеупомянутому способу, в котором указанный пациент является человеком.
[0065] В другом аспекте настоящее изобретение относится к способу лечения SHP2-опосредованного нарушения, включающему: стадию введения пациенту, нуждающемуся в этом, терапевтически эффективного количества химиотерапевтического средства в комбинации с терапевтически эффективным количеством соединения формулы I, как определено в кратком изложении сущности изобретения.
Фармацевтические композиции
[0066] В другом аспекте настоящее изобретение относится к фармацевтически приемлемым композициям, которые содержат терапевтически эффективное количество одного или нескольких соединений, описанных выше, составленных вместе с одним или несколькими фармацевтически приемлемыми носителями (добавками) и/или разбавителями. Как подробно описано ниже, фармацевтические композиции настоящего изобретения могут быть специально приготовлены для введения в твердой или жидкой форме, включая те, которые приспособлены для следующего: (1) перорального введения, например, капель (водных или неводных растворов или суспензий), таблеток, например, предназначенных для трансбуккальной, сублингвальной и системной абсорбции, болюсов, порошков, гранул, паст для нанесения на язык; (2) парентерального введения, например, путем подкожной, внутримышечной, внутривенной или эпидуральной инъекции, например, в виде стерильного раствора или суспензии или композиции с замедленным высвобождением; (3) местного введения, например, в виде крема, мази, или пластыря с контролируемым высвобождением, или спрея, наносимого на кожу; (4) интравагинального или внутриректального введения, например, в виде пессария, крема или пены; (5) подъязычного введения; (6) введения через глаза; (7) чрескожного введения; (8) введения через нос; (9) легочного введения; или (10) интратекального введения.
[0067] Используемая здесь фраза «терапевтически эффективное количество» означает количество соединения, материала или композиции, включающих соединение настоящего изобретения, которое эффективно для получения какого-либо желаемого терапевтического эффекта, по меньшей мере, в субпопуляции клеток у животного при разумном соотношении пользы и риска, применимом к любому медицинскому лечению.
[0068] Фраза «фармацевтически приемлемый» используется здесь для обозначения тех соединений, материалов, композиций и/или лекарственных форм, которые по результатам тщательной медицинской оценки подходят для использования при контакте с тканями людей и животных без чрезмерной токсичности, раздражения, аллергической реакции или другой проблемы или осложнения, соизмеримых с разумным соотношением пользы и риска.
[0069] Используемая здесь фраза «фармацевтически приемлемый носитель» означает фармацевтически приемлемый материал, композицию или носитель, такие как жидкий или твердый наполнитель, разбавитель, формообразующее, вспомогательное вещество для изготовления (например, смазка, тальк, стеарат магния, кальция или цинка или стеариновая кислота) или материал для инкапсулирования растворителя, которые участвуют в переносе или транспортировке данного соединения из одного органа или части тела в другой орган или часть тела. Каждый носитель должен быть «приемлемым» в смысле совместимости с другими ингредиентами состава и не причинять вреда пациенту. Некоторые примеры материалов, которые могут служить в качестве фармацевтически приемлемых носителей, включают: (1) сахара, такие как лактоза, глюкоза и сахароза; (2) крахмалы, такие как кукурузный крахмал и картофельный крахмал; (3) целлюлозу и ее производные, такие как натрийкарбоксиметилцеллюлоза, этилцеллюлоза и ацетат целлюлозы; (4) порошкообразный трагакант; (5) солод; (6) желатин; (7) тальк; (8) формообразующие, такие как масло какао и суппозиторные воски; (9) масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; (10) гликоли, такие как пропиленгликоль; (11) многоатомные спирты, такие как глицерин, сорбит, маннит и полиэтиленгликоль; (12) сложные эфиры, такие как этилолеат и этиллаурат; (13) агар; (14) буферные агенты, такие как гидроксид магния и гидроксид алюминия; (15) альгиновую кислоту; (16) апирогенную воду; (17) изотонический солевой раствор; (18) раствор Рингера; (19) этиловый спирт; (20) буферные растворы для pH; (21) полиэфиры, поликарбонаты и/или полиангидриды; и (22) другие нетоксичные совместимые вещества, используемые в фармацевтических композициях.
[0070] Как указано выше, некоторые варианты осуществления настоящих соединений могут содержать основную функциональную группу, такую как амино или алкиламино, и, таким образом, способны образовывать фармацевтически приемлемые соли с фармацевтически приемлемыми кислотами. Термин «фармацевтически приемлемые соли» в этом смысле относится к относительно нетоксичным, неорганическим и органическим солям присоединения кислоты соединений настоящего изобретения. Эти соли могут быть получены in situ в носителе для доставки или в процессе изготовления лекарственной формы или путем индивидуальной реакции очищенного соединения данного изобретения в форме его свободного основания с подходящей органической или неорганической кислотой и выделения соли, полученной таким образом, во время последующей очистки. Типичные соли включают гидробромид, гидрохлорид, сульфат, бисульфат, фосфат, нитрат, ацетат, валерат, олеат, пальмитат, стеарат, лаурат, бензоат, лактат, фосфат, тозилат, цитрат, малеат, фумарат, сукцинат, тартрат, нафтилат, мезилат, глюкогептонат, лактобионат и лаурилсульфонатные соли и тому подобное. (См., например, Berge et al. (1977) «Pharmaceutical Salts», J. Pharm. Sci. 66:1-19).
[0071] Фармацевтически приемлемые соли указанных соединений включают обычные нетоксичные соли или соли четвертичного аммония соединений, например, из нетоксичных органических или неорганических кислот. Например, такие обычные нетоксичные соли включают соли, полученные из неорганических кислот, таких как гидрохлоридная, бромистоводородная, серная, сульфаминовая, фосфорная, азотная и тому подобное; и соли, полученные из органических кислот, таких как уксусная, пропионовая, янтарная, гликолевая, стеариновая, молочная, яблочная, винная, лимонная, аскорбиновая, пальмитиновая, малеиновая, гидроксималеиновая, фенилуксусная, глутаминовая, бензойная, салициловая, сульфанильная, 2-ацетоксибензойная, фумаровая, толуолсульфоновая, метансульфоновая, этандисульфоновая, щавелевая, изотионовая и тому подобное.
[0072] В других случаях соединения настоящего изобретения могут содержать одну или несколько кислотных функциональных групп и, таким образом, способны образовывать фармацевтически приемлемые соли с фармацевтически приемлемыми основаниями. Термин «фармацевтически приемлемые соли» в этих случаях относится к относительно нетоксичным, неорганическим и органическим солям присоединения основания соединений настоящего изобретения. Эти соли сходным образом можно получать in situ в носителе для доставки или в ходе производства лекарственной формы, или путем осуществления отдельной реакции очищенного соединения в форме свободной кислоты с подходящим основанием, таким как гидроксид, карбонат или бикарбонат катиона фармацевтически приемлемого металла, с аммиаком или с фармацевтически приемлемым органическим первичным, вторичным или третичным амином. Типичные соли щелочных или щелочноземельных металлов включают соли лития, натрия, калия, кальция, магния и алюминия и т.п. Типичные органические амины, пригодные для образования солей присоединения основания, включают в себя этиламин, диэтиламин, этилендиамин, этаноламин, диэтаноламин, пиперазин и т.п. (См., например, Berge et al., выше).
[0073] В композициях также могут присутствовать смачивающие средства, эмульгаторы и смазывающие вещества, такие как лаурилсульфат натрия и стеарат магния, а также красящие средства, противоадгезивные средства, средства для покрытия, подсластители, ароматизаторы и отдушки, консерванты и антиоксиданты.
[0074] Примеры фармацевтически приемлемых антиоксидантов включают: (1) водорастворимые антиоксиданты, такие как аскорбиновая кислота, гидрохлорид цистеина, бисульфат натрия, метабисульфит натрия, сульфит натрия и т.п.; (2) жирорастворимые антиоксиданты, такие как аскорбилпальмитат, бутилированный гидроксианизол (BHA), бутилированный гидрокситолуол (BHT), лецитин, пропилгаллат, альфа-токоферол и т.п.; и (3) металл-комплексообразующие средства, такие как лимонная кислота, этилендиаминтетрауксусная кислота (EDTA), сорбит, винная кислота, ортофосфорная кислота и т.п.
[0075] Составы настоящего изобретения включают составы, пригодные для перорального, назального, местного (включая трансбуккальное и подъязычное), ректального, вагинального и/или парентерального введения. С целью удобства составы могут быть представлены в виде стандартной лекарственной формы и могут быть получены с помощью любых способов, хорошо известных в области фармации. Количество активного ингредиента, который можно объединять с материалом носителя для получения стандартной лекарственной формы, будет варьироваться в зависимости от реципиента, подлежащего лечению, конкретного способа введения. Количество активного ингредиента, которое можно объединять с материалом носителя для получения единичной лекарственной формы, обычно будет представлять собой такое количество соединения, которое обеспечивает терапевтический эффект. Как правило, из расчета сто процентов данное количество будет варьироваться от приблизительно 0,1 процента до приблизительно девяноста девяти процентов активного ингредиента, предпочтительно от приблизительно 5 процентов до приблизительно 70 процентов, наиболее предпочтительно от приблизительно 10 процентов до приблизительно 30 процентов.
[0076] Согласно определенным вариантам осуществления состав настоящего изобретения содержит формообразующее, выбранное из группы, состоящей из циклодекстринов, целлюлоз, липосом, мицеллообразующих средств, например, желчных кислот, и полимерных носителей, например, сложных полиэфиров и полиангидридов; и соединение настоящего изобретения. Согласно определенным вариантам осуществления вышеуказанный состав придает биологическую доступность при пероральном введении соединению по настоящему изобретению.
[0077] Способы получения этих составов или композиций включают стадию объединения соединения по настоящему изобретению с носителем и, необязательно, с одним или несколькими вспомогательными ингредиентами. Как правило, составы получают путем равномерного и тщательного объединения соединения настоящего изобретения с жидкими носителями, или тонкоизмельченными твердыми носителями, или обоими, а затем, при необходимости, придания продукту формы.
[0078] Составы данного изобретения, пригодные для перорального введения, могут быть в форме капсул, облаток, пилюль, таблеток, леденцов (с применением ароматизированной основы, обычно - сахарозы и аравийской камеди или трагаканта), порошков, гранул или в виде раствора или суспензии в жидкости на основе воды или в жидкости не на основе воды, или в виде жидкой эмульсии типа масло-в-воде или типа вода-в-масле, или в виде настоя или сиропа, или в виде пастилок (с применением инертного основания, такого как желатин и глицерин или сахароза и аравийская камедь), и/или в виде жидкостей для полоскания рта и подобных, при этом каждое содержит предварительно установленное количество соединения настоящего изобретения в качестве активного ингредиента. Соединение настоящего изобретения также можно вводить в виде болюса, лекарственной кашки или пасты.
[0079] В твердых лекарственных формах данного изобретения для перорального введения (капсулах, таблетках, пилюлях, драже, порошках, гранулах, таблетках для рассасывания и т.п.) активный ингредиент смешан с одним или несколькими фармацевтически приемлемыми носителями, такими как цитрат натрия или фосфат дикальция, и/или чем-либо из следующего: (1) наполнителями или добавками, такими как крахмалы, лактоза, сахароза, глюкоза, маннит и/или кремниевая кислота; (2) связующими, такими как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и/или аравийская камедь; (3) увлажнителями, такими как глицерин; (4) разрыхляющими средствами, такими как агар-агар, карбонат кальция, картофельный или маниоковый крахмал, альгиновая кислота, некоторые силикаты и карбонат натрия; (5) замедлителями растворения, такими как парафин; (6) ускорителями впитывания, такими как соединения четвертичного аммония и ПАВ, такими как полоксамер и лаурилсульфат натрия; (7) смачивающими средствами, такими как, например, цетиловый спирт, глицерин моностеарат и неионные ПАВ; (8) абсорбентами, такими как каолин и бентонитовая глина; (9) смазывающими средствами, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия, стеарат цинка, стеарат натрия, стеариновая кислота и их смеси; (10) красителями; и (11) веществами для контролируемого высвобождения, такими как кросповидон или этилцеллюлоза. В случае капсул, таблеток и пилюль фармацевтические композиции также могут содержать буферные средства. Твердые композиции подобного типа также можно использовать в качестве наполнителей в мягких и твердых желатиновых капсулах, используя такие наполнители, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и т.п.
[0080] Таблетку можно получить путем прессования или формования, необязательно с одним или несколькими вспомогательными ингредиентами. Прессованные таблетки можно получать с применением связующего (например, желатина или гидроксипропилметилцеллюлозы), смазывающего средства, инертного разбавителя, консерванта, разрыхлителя (например, крахмалгликолята натрия или сшитой карбоксиметилцеллюлозы натрия), ПАВ и/или диспергирующего средства. Формованные таблетки можно получить формованием в подходящей машине смеси порошкообразного соединения, смоченного инертным жидким разбавителем.
[0081] Таблетки и другие твердые лекарственные формы фармацевтических композиций настоящего изобретения, такие как драже, капсулы, пилюли и гранулы, могут необязательно быть делимыми или получаемыми с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие покрытия, хорошо известные из области составления фармацевтических препаратов. Их также можно составлять таким образом, чтобы обеспечивать медленное или контролируемое высвобождение активного ингредиента, содержащегося в них, с применением, например, гидроксипропилметилцеллюлозы в различных соотношениях для обеспечения желаемого профиля высвобождения, других полимерных матриц, липосом и/или микросфер. Их можно составлять для быстрого высвобождения, например, они могут быть высушенные сублимацией. Их можно стерилизовать, например, посредством фильтрования через фильтр для удерживания бактерий или посредством включения в их состав стерилизующих средств в форме стерильных твердых композиций, которые можно растворять в стерильной воде или некоторых других стерильных инъекционных средах непосредственно перед применением. Эти композиции также могут необязательно содержать контрастные средства и могут представлять собой композицию, из которой высвобождается(ются) активный(ые) ингредиент(ы) исключительно, или предпочтительно, в определенном отделе желудочно-кишечного тракта необязательно с задержкой. Примеры капсулирующих композиций, которые можно применять, включают полимерные вещества и воски. Активный ингредиент также может быть в форме микрокапсул, если это необходимо, с одним или несколькими из описанных выше формообразующих.
[0082] Жидкие лекарственные формы для перорального введения соединений данного изобретения включают в себя фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к активному ингредиенту жидкие лекарственные формы могут содержать инертные разбавители, традиционно применяемые в данной области, такие как, например, вода или другие растворители, солюбилизирующие средства и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, масла (в частности, хлопковое, арахисовое масло, кукурузное масло, масло зародышей, оливковое масло, касторовое масло и кунжутное масло), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот и сорбитана, а также их смеси.
[0083] Помимо инертных разбавителей, композиции для перорального применения могут также содержать вспомогательные вещества, такие как смачивающие средства, эмульгаторы и суспендирующие средства, подсластители, ароматизаторы, красящие вещества, отдушки и консерванты.
[0084] Суспензии, помимо активных соединений, могут содержать суспендирующие средства, такие как, например, этоксилированные изостеариловые спирты, полиоксиэтиленсорбит и сложные эфиры сорбитана, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант и их смеси.
[0085] Составы и фармацевтические композиции настоящего изобретения для ректального или вагинального введения могут быть представлены в виде суппозитория, который можно получать смешиванием одного или нескольких соединений данного изобретения с одним или несколькими подходящими нераздражающими формообразующими или носителями, содержащими, например, масло какао, полиэтиленгликоль, воск для суппозиториев или салицилат, и который является твердым при комнатной температуре, но жидким при температуре тела, и, следовательно, плавится в прямой кишке или вагинальной полости и высвобождает активное соединение.
[0086] Составы настоящего изобретения, пригодные для вагинального введения, также включают пессарии, тампоны, кремы, гели, пасты, пены или аэрозольные составы, содержащие такие допустимые носители, которые известны в данной области техники.
[0087] Лекарственные формы для местного или трансдермального введения соединения по настоящему изобретению включают порошки, распыляемые растворы, мази, пасты, крема, лосьоны, гели, растворы, пластыри и средства для ингаляции. Активное соединение можно смешивать в стерильных условиях с фармацевтически приемлемым носителем и любыми консервантами, буферами или газами-вытеснителями, которые могут быть необходимы.
[0088] Помимо активного соединения данного изобретения мази, пасты, кремы и гели могут содержать формообразующие, такие как животные и растительные жиры, масла, воски, парафины, крахмал, трагакант, производные целлюлозы, полиэтиленгликоли, силиконы, бентониты, кремниевая кислота, тальк и оксид цинка или их смеси.
[0089] Порошки и спреи могут содержать, помимо соединения данного изобретения, формообразующие, такие как лактоза, тальк, кремниевая кислота, гидроксид алюминия, силикаты кальция и полиамидный порошок или смеси данных веществ. Спреи могут дополнительно содержать традиционные пропелленты, такие как хлорфторуглеводороды, и летучие незамещенные углеводороды, такие как бутан и пропан.
[0090] Трансдермальные пластыри обладают дополнительным преимуществом в обеспечении контролируемой доставки соединения данного изобретения в тело. Такие лекарственные формы можно получать посредством растворения или диспергирования соединения в соответствующей среде. Средства, улучшающие всасывание, можно также применять для увеличения потока соединения через кожу. Скорость такого потока можно контролировать либо обеспечением мембраны, регулирующей скорость, либо посредством диспергирования соединения в полимерной матрице или геле.
[0091] Офтальмологические составы, глазные мази, порошки и растворы и т.п. также рассматриваются как находящиеся в пределах объема настоящего изобретения.
[0092] Фармацевтические композиции данного изобретения, пригодные для парентерального введения, содержат одно или несколько соединений данного изобретения в сочетании с одним или несколькими фармацевтически приемлемыми стерильными изотоническими водными или неводными растворами, дисперсиями, суспензиями или эмульсиями или стерильными порошками, которые можно восстанавливать с получением стерильных инъекционных растворов или дисперсий непосредственно перед применением, которые могут содержать сахара, спирты, антиоксиданты, буферы, бактериостатические средства и растворенные вещества, которые делают состав изотоничным с кровью предполагаемого реципиента, или суспендирующие средства и загустители.
[0093] Примеры подходящих водных и неводных носителей, которые можно использовать в фармацевтических композициях согласно настоящему изобретению, включают воду, этанол, полиолы (такие как глицерин, пропиленгликоль, полиэтиленгликоль и т.п.) и их подходящие смеси, растительные масла, такие как оливковое масло, и инъецируемые органические сложные эфиры, такие как этилолеат. Надлежащую текучесть можно поддерживать, например, путем применения материалов для покрытия, таких как лецитин, путем поддержания необходимого размера частиц в случае дисперсий и путем применения ПАВ.
[0094] Эти композиции могут также содержать вспомогательные вещества, такие как консерванты, смачивающие средства, эмульгаторы и диспергирующие средства. Предупреждение воздействия микроорганизмов на интересующие соединения можно обеспечивать путем включения различных антибактериальных и противогрибковых средств, например, парабена, хлорбутанола, фенолсорбиновой кислоты и т.п. Также может быть необходимым включать в композиции средства, регулирующие изотоничность, такие как сахара, хлорид натрия и т.п. Кроме того, пролонгированное всасывание инъекционной фармацевтической формы может быть обусловлено включением средств, которые задерживают всасывание, таких как моностеарат алюминия и желатин.
[0095] В ряде случаев для пролонгирования эффекта лекарственного средства требуется замедлить всасывание лекарственного средства при подкожной или внутримышечной инъекции. Этого можно достичь путем использования жидкой суспензии на основе кристаллического или аморфного материала, имеющего слабую растворимость в воде. К тому же скорость всасывания лекарственного средства зависит от его скорости растворения, которая, в свою очередь, может зависеть от размера кристалла и от кристаллической формы. В качестве альтернативы замедленного всасывания парентерально вводимой формы лекарственного средства достигают путем растворения или суспендирования лекарственного средства в масляном носителе.
[0096] Инъекционные формы депо создают путем получения микроинкапсулированных матриц интересующих соединений в биоразлагаемых полимерах, таких как полилактид-полигликолид. В зависимости от соотношения лекарственного средства и полимера, а также природы конкретного используемого полимера, можно регулировать скорость высвобождения лекарственного средства. Примеры других биоразлагаемых полимеров включают сложные поли(ортоэфиры) и поли(ангидриды). Составы инъекционных депо также получают путем захвата лекарственного средства в липосомы или микроэмульсии, которые совместимы с тканью тела.
[0097] Когда соединения по настоящему изобретению вводят в качестве лекарственных средств людям или животным, их можно давать в чистом виде или в качестве фармацевтической композиции, содержащей, например, от 0,1% до 99% (более предпочтительно от 10 до 30%) активного ингредиента в комбинации с фармацевтически приемлемым носителем.
[0098] Составы настоящего изобретения можно принимать перорально, парентерально, местно или ректально. Их, разумеется, принимают в формах, подходящих для каждого пути введения. Например, их вводят в виде таблеток или капсул, путем инъекции, ингаляции, глазного лосьона, мази, суппозиториев и т.д., путем инъекции, инфузии или ингаляции; местного применения лосьона или мази; и ректальных суппозиториев. Предпочтительным является пероральное введение.
[0099] Фразы «парентеральное введение» и «вводимый парентерально», используемые в данном документе, относятся к способам введения, отличным от энтерального и местного введения, обычно с помощью инъекции, и включают без ограничения внутривенную, внутримышечную, внутриартериальную, интратекальную, внутрикапсульную, интраорбитальную, внутрисердечную, внутрикожную, внутрибрюшинную, транстрахеальную, подкожную, субкутикулярную, внутрисуставную, подкапсулярную, субарахноидальную, интраспинальную и интрастернальную инъекцию и инфузию.
[00100] Используемые здесь фразы «системное введение», «введенное системно», «периферическое введение» и «введенное периферически» означают введение соединения, лекарственного средства или другого материала, кроме непосредственного введения в центральную нервную систему, таким образом, чтобы оно поступало в организм пациента и, таким образом, подвергалось метаболизму и другим подобным процессам, например, подкожное введение.
[00101] Эти соединения можно вводить людям и другим животным для лечения любым подходящим способом введения, включая перорально, назально, например, с помощью распыления, ректально, интравагинально, парентерально, интрацистернально и местно, в виде порошков, мазей или капель, в том числе буккально и подъязычно.
[00102] Независимо от выбранного способа введения соединения настоящего изобретения, которые могут быть использованы в подходящей гидратированной форме, и/или фармацевтические композиции настоящего изобретения составляют в фармацевтически приемлемые лекарственные формы обычными способами, известными специалистам в данной области.
[00103] Фактические уровни дозировки активных ингредиентов в фармацевтических композициях данного изобретения можно изменять таким образом, чтобы получить количество активного ингредиента, эффективное для достижения необходимого терапевтического эффекта для конкретного пациента, композиции и способа введения, которое не является токсичным для пациента.
[00104] Выбранный уровень дозировки будет зависеть от разнообразных факторов, включая активность конкретного используемого соединения по настоящему изобретению или его сложного эфира, соли или амида, способ введения, время введения, скорость экскреции или метаболизма конкретного используемого соединения, скорость и степень абсорбции, длительность лечения, другие лекарственные средства, соединения и/или материалы, применяемые в комбинации с конкретными используемыми соединениями, возраст, пол, вес, состояние, общее состояние здоровья и предшествующую историю болезни пациента, подлежащего лечению, и подобных факторов, хорошо известных в области медицины.
[00105] Врач или ветеринар со стандартной квалификацией в данной области техники легко может определить и назначить эффективное количество необходимой фармацевтической композиции. Например, врач или ветеринар мог бы начинать вводить дозы соединений по настоящему изобретению, применяемых в фармацевтической композиции, при уровнях ниже, чем это необходимо для достижения требуемого терапевтического эффекта, и постепенно увеличивать дозировку до достижения требуемого эффекта.
[00106] В целом, подходящей суточной дозой соединения данного изобретения будет такое количество соединения, которое является наиболее низкой дозой, эффективной для получения терапевтического эффекта. Такая эффективная доза обычно будет зависеть от описанных выше факторов. Как правило, пероральные, внутривенные, интрацеребровентрикулярные и подкожные дозы соединений настоящего изобретения для пациента при использовании для указанных обезболивающих эффектов будут варьироваться от примерно 0,0001 до примерно 100 мг на килограмм массы тела в день.
[00107] При желании эффективную суточную дозу активного соединения можно вводить в виде двух, трех, четырех, пяти, шести или более частей дозы, вводимых по отдельности с соответствующими интервалами в течение дня, необязательно в виде стандартных лекарственных форм. Предпочтительным дозированием является одно введение в день.
[00108] Хотя соединение по настоящему изобретению можно вводить отдельно, предпочтительно вводить соединение в виде фармацевтического состава (композиции).
[00109] Соединения настоящего изобретения можно составлять для введения любым общепринятым способом для применения в медицине или в ветеринарной медицине аналогично другим фармацевтическим препаратам.
[00110] В другом аспекте настоящее изобретение относится к фармацевтически приемлемым композициям, которые содержат терапевтически эффективное количество одного или нескольких интересующих соединений, как описано выше, составленных вместе с одним или несколькими фармацевтически приемлемыми носителями (добавками) и/или разбавителями. Как подробно описано ниже, фармацевтические композиции настоящего изобретения могут быть специально приготовлены для введения в твердой или жидкой форме, включая те, которые приспособлены для следующего: (1) перорального введения, например, жидкие лекарственные формы (водные или неводные растворы или суспензии), таблетки, болюсы, порошки, гранулы, пасты для нанесения на язык; (2) парентерального введения, например, посредством подкожной, внутримышечной или внутривенной инъекции, как, например, стерильный раствор или суспензия; (3) местного нанесения, например, в виде крема, мази или распыляемого раствора, наносимых на кожу, легкие или слизистые оболочки; (4) интравагинального или внутрипрямокишечного введения, например, в виде пессария, крема или пены; (5) подъязычного или буккального введения; (6) глазного введения; (7) трансдермального или (8) интраназального введения.
[00111] Подразумевается, что термин «лечение» охватывает также профилактику, терапию и излечение.
[00112] Пациент, получающий данный вид лечения, представляет собой любое животное, требующее этого, включая приматов, в частности, людей, а также других млекопитающих, таких как лошади, крупный рогатый скот, свинья и овца; а также домашняя птица и домашние питомцы в общем смысле.
[00113] Соединение данного изобретения можно вводить само по себе или в смесях с фармацевтически приемлемыми носителями, а также можно вводить в сочетании с противомикробными агентами, такими как пенициллины, цефалоспорины, аминогликозиды и гликопептиды. Совместная терапия, таким образом, включает последовательное, одновременное и раздельное введение активного соединения так, чтобы терапевтические эффекты от первого вводимого соединения не полностью исчезали к моменту введения следующего вещества.
[00114] Технология микроэмульсификации может улучшить биодоступность некоторых липофильных (нерастворимых в воде) фармацевтических агентов. Примеры включают Trimetrine (Dordunoo, S. K., et al., Drug Development and Industrial Pharmacy, 17(12), 1685-1713, 1991) и REV 5901 (Sheen, P. C., et al., J Pharm Sci 80(7), 712-714, 1991). Среди прочего, микроэмульсификация обеспечивает повышенную биодоступность путем предпочтительного направления абсорбции в лимфатическую систему вместо кровеносной системы, что, таким образом, позволяет обходить печень и предотвращать разрушение соединений при гепатобилиарной циркуляции.
[00115] И хотя предполагаются все подходящие амфифильные носители, предпочтительными в настоящее время носителями являются, как правило, те, которые имеют статус «общепризнанно безопасный» (GRAS), и которые могут одновременно солюбилизировать соединение настоящего изобретения и микроэмульсифицировать его на более поздней стадии, когда раствор вступает в контакт со сложной водной фазой (например, в желудочно-кишечном тракте человека). Обычно амфифильные ингредиенты, которые удовлетворяют этим требованиям, имеют значения HLB (гидрофильно-липофильный баланс) в размере 2-20, а их структуры содержат алифатические радикалы с прямой цепью длиной от C-6 до C-20. Примерами являются полиэтиленгликолизированные жирные глицериды и полиэтиленгликоли.
[00116] Особенно предпочтительны поступающие в продажу амфифильные носители, включая Gelucire-series, Лабрафил, Лабрасол или Лаурогликоль (все производятся и распространяются компанией Gattefosse Corporation, Сен-Прист, Франция), ПЭГ-моно-олеат, ПЭГ-ди-олеат, ПЭГ-моно-лаурат и ди-лаурат, Лецитин, Полисорбат 80 и т.п. (производятся и распространяются рядом компаний в США и по всему миру).
[00117] Гидрофильные полимеры, подходящие для использования в настоящем изобретении, представляют собой те, которые легко растворимы в воде, могут быть ковалентно присоединены к образующему везикулы липиду и которые переносятся in vivo без токсических эффектов (то есть являются биосовместимыми). Подходящие полимеры включают полиэтиленгликоль (ПЭГ), полимер молочной кислоты (также называемй полилактидом), полимер гликолевой кислоты (также называемый полигликолидом), сополимер молочной и гликолевой кислот и поливиниловый спирт. Предпочтительные полимеры характеризуются молекулярной массой от примерно 100 или 120 Дальтон до примерно 5000 или 10000 Дальтон, и более предпочтительно - от примерно 300 Дальтон до примерно 5000 Дальтон. В особенно предпочтительном варианте осуществления полимер представляет собой полиэтиленгликоль, имеющий молекулярную массу от примерно 100 до примерно 5000 Дальтон, и более предпочтительно имеющий молекулярную массу от примерно 300 до примерно 5000 Дальтон. В особенно предпочтительном варианте осуществления полимер представляет собой полиэтиленгликоль с массой 750 Дальтон (ПЭГ(750)). Полимеры могут также определяться числом мономеров, содержащихся в них; в предпочтительном варианте осуществления данного изобретения используются полимеры, состоящие из не менее чем примерно трех мономеров, такие как полимеры ПЭГ, состоящие из трех мономеров (примерно 150 Дальтон).
[00118] Другие гидрофильные полимеры, которые могут подходить для применения в настоящем изобретении, включают поливинилпирролидон, полиметоксазолин, полиэтилоксазолин, полигидроксипропилметакриламид, полиметакриламид, полидиметилакриламид и производные целлюлоз, такие как гидроксиметилцеллюлоза или гидроксиэтилцеллюлоза.
[00119] В некоторых вариантах осуществления состав согласно настоящему изобретению содержит биосовместимый полимер, выбранный из группы, состоящей из полиамидов, поликарбонатов, полиалкиленов, полимеров акриловых и метакриловых сложных эфиров, поливиниловых полимеров, полигликолидов, полисилоксанов, полиуретанов и их сополимеров, целлюлоз, полипропилена, полиэтиленов, полистирола, полимеров молочной кислоты и гликолевой кислоты, полиангидридов, сложных поли(орто)эфиров, поли(масляной кислоты), поли(валериановой кислоты), поли(лактида с капролактоном), полисахаридов, белков, полигиалуроновых кислот, полицианоакрилатов и их смесей или сополимеров.
[00120] Циклодекстрины представляют собой циклические олигосахариды, состоящие из 6, 7 или 8 глюкозных звеньев, обозначаемых буквами греческого алфавита альфа, бета или гамма, соответственно. О существовании циклодекстринов с менее чем шестью глюкозными звеньями не известно. Глюкозные звенья соединены альфа-1,4-глюкозидными связями. Как следствие конформации стула у звеньев сахаров, все вторичные гидроксильные группы (при С-2, С-3) расположены с одной стороны кольца, тогда как все первичные гидроксильные группы на С-6 расположены на другой стороне. В результате внешние поверхности являются гидрофильными, что делает циклодекстрины водорастворимыми. Напротив, внутренние полости циклодекстринов являются гидрофобными, так как на них находятся водород атомов С-3 и С-5 и кислород простых эфирных групп. Использование таких матриц способствует комплексообразованию с различными относительно гидрофобными соединениями, включая, например, стероидные соединения, такие как 17.бета.-эстрадиол (см., например, van Uden et al. Plant Cell Tiss. Org. Cult. 38:1-3-113 (1994)). Комплексообразование происходит за счет сил Ван дер Ваальса и образования водородных связей. Обзор химии циклодекстринов см. в Wenz, Agnew. Chem. Int. Ed. Engl., 33:803-822 (1994).
[00121] Физико-химические свойства производных циклодекстрина сильно зависят от вида и степени замещения. Например, их растворимость в воде изменяется от нерастворимых (например, триацетил-бета-циклодекстрин) до 147% растворимости (вес./об.) (G-2-бета-циклодекстрин). Кроме того, они растворимы во многих органических растворителях. Свойства циклодекстринов позволяют регулировать растворимость различных компонентов состава путем увеличения или уменьшения их растворимости.
[00122] Описаны многочисленные циклодекстрины и способы их получения. Например, у Parmeter (I), et al. (патент США № 3453259) и Gramera, et al. (патент США № 3459731) описаны электронейтральные циклодекстрины. Другие производные включают циклодекстрины с катионными свойствами [Parmeter (II), патент США № 3453257], нерастворимые сшитые циклодекстрины (Solms, патент США № 3420788) и циклодекстрины с анионными свойствами [Parmeter (III), патент США № 3426011]. Среди производных циклодекстрина с анионными свойствами к исходному циклодекстрину присоединены карбоновые кислоты, фосфористые кислоты, фосфиновые кислоты, фосфоновые кислоты, фосфорные кислоты, тиофосфоновые кислоты, тиосульфиновые кислоты и сульфокислоты [см., Parmeter (III), выше]. Кроме того, производные сульфоалкилового эфира циклодекстрина описаны у Stella, et al. (патент США № 5134127).
[00123] Липосомы состоят, по меньшей мере, из одной липидной двуслойной мембраны, содержащей водную внутреннюю везикулу. Липосомы можно охарактеризовать типом мембраны и размером. Малые однослойные везикулы (SUV) имеют одну мембрану и их диаметр обычно составляет от 0,02 до 0,05 μм; большие однослойные везикулы (LUV) обычно больше чем 0,05 μм. Олиголамеллярные большие везикулы и многослойные везикулы имеют несколько, как правило концентрических, мембранных слоев и их размер обычно больше чем 0,1 μм. Липосомы с несколькими неконцентрическими мембранами, то есть несколько малых везикул, содержащихся в большей везикуле, называются мультивезикулярными везикулами.
[00124] Один аспект настоящего изобретения относится к составам, содержащим липосомы, содержащие соединение настоящего изобретения, где липосомная мембрана выполнена так, чтобы создать липосому с увеличенным переносимым объемом. Альтернативно или дополнительно соединение настоящего изобретения может содержаться внутри липосомного двойного слоя липосомы или адсорбироваться на нем. Соединение по настоящему изобретению можно агрегировать с липидным поверхностно-активным веществом (ПАВ) и переносить внутри внутреннего пространства липосомы; в этих случаях липосомная мембрана выполнена таким образом, чтобы противостоять разрушающему действию со стороны агрегата активного начала-ПАВ.
[00125] Согласно одному варианту осуществления настоящего изобретения липидный двойной слой липосомы содержит липиды, дериватизированные полиэтиленгликолем (ПЭГ), так что цепи ПЭГ отходят от внутренней поверхности липидного двойного слоя во внутреннее пространство, инкапсулированное липосомой, и вытянуты от внешней стороны липидного двойного слоя в окружающую среду.
[00126] Активное начало, содержащееся внутри липосом настоящего изобретения, находится в солюбилизированной форме. Агрегаты ПАВ и активного начала (такие как эмульсии или мицеллы, содержащие активное начало, представляющее интерес) могут быть заключены во внутреннем пространстве липосом согласно настоящему изобретению. Действие ПАВ используется для диспергирования и солюбилизации активного начала, и его можно выбирать из любого подходящего алифатического, циклоалифатического или ароматического ПАВ, включая, но не ограничиваясь ими, биосовместимые лизофосфатидилхолины (LPC) с различной длиной цепи (например, примерно от менее C 14 до примерно менее C 20). Полимерные дериватизированные липиды, такие как ПЭГ-липиды, также можно использовать для формирования мицелл, поскольку они будут предотвращать слияние мицелл/мембран, и поскольку добавление полимера к молекулам ПАВ снижает ККМ ПАВ и способствует образованию мицелл. Предпочтительными являются ПАВ с ККМ в микромолярном диапазоне; ПАВ с более высоким ККМ можно использовать для получения мицелл, заключенных внутри липосом настоящего изобретения, однако мономеры ПАВ мицелл могут влиять на стабильность липосомного двойного слоя, и это влияние необходимо учитывать при создании липосом с требуемой стабильностью.
[00127] Липосомы в соответствии с настоящим изобретением можно получать любым из множества методов, известных в данной области. См., например, патент США № 4235871; публикацию заявки РСТ WO 96/14057; New RRC, Liposomes: A practical approach, IRL Press, Oxford (1990), стр. 33-104; Lasic DD, Liposomes from physics to applications, Elsevier Science Publishers BV, Amsterdam, 1993.
[00128] Например, липосомы настоящего изобретения можно получать путем диффузии липида, дериватизированного гидрофильным полимером, в предварительно сформированные липосомы, например, путем воздействия на предварительно сформированные липосомы мицеллами, состоящими из липид-привитых полимеров, при концентрациях липидов, соответствующих конечным мольным процентам дериватизированного липида, который должен содержаться в липосоме. Липосомы, содержащие гидрофильный полимер, также могут быть образованы путем способов гомогенизации, гидратации липидного поля или экструзии, как известно в данной области техники.
[00129] В одном аспекте настоящего изобретения липосомы получают, чтобы они имели по существу одинаковые размеры в выбранном диапазоне размеров. Один эффективный способ получения одинаковых размеров включает экструзию водной суспензии липосом через ряд поликарбонатных мембран, имеющих выбранный одинаковый размер пор; размер пор мембраны будет примерно соответствовать максимальным размерам липосом, полученных экструзией через эту мембрану. См., например, патент США № 4737323 (12 апреля 1988г.).
[00130] Характеристики высвобождения состава настоящего изобретения зависят от инкапсулирующего материала, концентрации инкапсулированного лекарственного средства и наличия модификаторов высвобождения. Например, можно регулировать высвобождение в зависимости от рН, например, с использованием чувствительного к рН покрытия, которое высвобождается только при низком рН, таком как в желудке, или при более высоком рН, как в кишечнике. Энтеросолюбильное покрытие может использоваться, чтобы предотвратить высвобождение в момент прохождения через желудок. Можно использовать несколько покрытий или смеси цианамида, инкапсулированного в разные материалы, для обеспечения начального высвобождения в желудке с последующим высвобождением в кишечнике. Высвобождение можно регулировать включением солей или порообразующих агентов, которые могут увеличить поглощение воды, или высвобождать лекарственное средство путем диффузии из капсулы. Формообразующие, которые модифицируют растворимость лекарственного средства, также могут использоваться для контроля над скоростью высвобождения. Также могут быть включены агенты, которые усиливают разложение матрицы или высвобождение из матрицы. Они могут быть добавлены к лекарственному средству, добавлены в виде отдельной фазы (т.е. в виде частиц) или могут быть совместно растворены в полимерной фазе в зависимости от соединения. Во всех случаях количество должно составлять от 0,1 до тридцати процентов (вес/вес полимера). Типы усилителей разложения включают неорганические соли, такие как сульфат аммония и хлорид аммония, органические кислоты, такие как лимонная кислота, бензойная кислота и аскорбиновая кислота, неорганические основания, такие как карбонат натрия, карбонат калия, карбонат кальция, карбонат цинка и гидроксид цинка, и органические основания, такие как протаминсульфат, спермин, холин, этаноламин, диэтаноламин и триэтаноламин, и ПАВ, такие как Tween® и Pluronic®. В качестве частиц добавляют порообразующие агенты, которые привносят микроструктуру в матрицы (то есть водорастворимые соединения, такие как неорганические соли и сахара). Массовая доля добавки должна составлять от одного до тридцати процентов (вес/вес полимера).
[00131] Поглощением также можно манипулировать путем изменения времени пребывания частиц в кишечнике. Это может быть достигнуто, например, путем нанесения на частицы или выбора в качестве инкапсулирующего материала прилипающего к слизистым оболочкам полимера. Примеры включают большинство полимеров со свободными карбоксильными группами, такими как хитозан, целлюлозы и особенно полиакрилаты (в контексте данного документа полиакрилаты относятся к полимерам, включающим акрилатные группы и модифицированные акрилатные группы, таким как цианоакрилаты и метакрилаты).
Фармацевтические комбинации
[00132] Настоящее изобретение особенно относится к применению соединения формулы I (или фармацевтической композиции, содержащей соединение формулы I) для лечения одного или нескольких упомянутых в данном документе заболеваний; причем ответ на лечение благоприятен, как продемонстрировано, например, частичным или полным удалением одного или нескольких симптомов заболевания вплоть до полного излечения или ремиссии.
[00133] Соединение формулы (I) также может использоваться в комбинации со следующими соединениями и конъюгатами антитело/лекарственное средство:
[00134] Ингибиторы BCR-ABL: иматиниб (Gleevec®); инилотиниб гидрохлорид; нилотиниб (Tasigna®); дазатиниб (BMS-345825); босутиниб (SKI-606); понатиниб (AP24534); бафетиниб (INNO406); данусертиб (PHA-739358), AT9283 (CAS 1133385-83-7); саракатиниб (AZD0530); и N-[2-[(1S,4R)-6-[[4-(циклобутиламино)-5-(трифторметил)-2-пиримидинил]амино]-1,2,3,4-тетрагидронафталин-1,4-имин-9-ил]-2-оксоэтил]-ацетамид (PF-03814735, CAS 942487-16-3).
[00135] Ингибиторы ALK: PF-2341066 (XALKORI®; кризотиниб); 5-хлор-N4-(2-(изопропилсульфонил)фенил)-N2-(2-метокси-4-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)фенил)пиримидин-2,4-диамин; GSK1838705A; и CH5424802.
[00136] Ингибиторы BRAF: вемурафаниб (PLX4032); и дабрафениб.
[00137] Ингибиторы FLT3 - сунитиниба малат (продаваемый под торговым названием Sutent® от компании Pfizer); PKC412 (мидостаурин); танутиниб, сорафениб, сунитиниб, мидостаурин, лестауртиниб, KW-2449, квизартиниб (AC220) и креноланиб.
[00138] Ингибиторы MEK - траметиниб.
[00139] Ингибиторы рецептора фактора роста эндотелия сосудов (VEGF): бевацизумаб (продаваемый под торговой маркой Avastin® от компании Genentech/Roche), акситиниб, (N-метил-2-[[3-[(E)-2-пиридин-2-илэтенил]-1H-индазол-6-ил]сульфанил]бензамид, также известный как AG013736, и описанный в патентной заявке РСТ № WO 01/002369), бриваниб аланинат ((S)-((R)-1-(4-(4-фтор-2-метил-1H-индол-5-илокси)-5-метилпирроло[2,1-f][1,2,4]триазин-6-илокси)пропан-2-ил)2-аминопропаноат, также известный как BMS-582664), мотесаниб (N-(2,3-дигидро-3,3-диметил-1H-индол-6-ил)-2-[(4-пиридинилметил)амино]-3-пиридинкарбоксамид, и описанный в патентной заявке РСТ № WO 02/066470), пасиреотид (также известный как SOM230, и описанный в патентной заявке РСТ № WO 02/010192), сорафениб (продаваемый под торговым названием Nexavar®);
[00140] Ингибиторы HER2-рецептора: Трастузумаб (продаваемый под торговой маркой Herceptin® компанией Genentech/Roche), нератиниб (также известный как HKI-272, (2E)-N-[4-[[3-хлор-4-[(пиридин-2-ил)метокси]фенил]амино]-3-циано-7-этоксихинолин-6-ил]-4-(диметиламино)бут-2-енамид и описанный в патентной заявке РСТ № WO 05/028443), лапатиниб или лапатиниба дитозилат (продаваемый под торговой маркой Tykerb® компанией GlaxoSmithKline); трастузумаб эмтанзин (в США - адо-трастузумаб эмтанзин, торговое название Kadcyla) - конъюгат антитело-лекарственное средство, состоящий из моноклонального антитела трастузумаба (герцептин), связанного с цитотоксическим средством мертанзином (DM1);
[00141] Антитела к CD20: Ритуксимаб (продаваемый под торговыми марками Riuxan® и MabThera® компанией Genentech/Roche), тозитумомаб (продаваемый под торговыми марками Bexxar® компанией GlaxoSmithKline), офатумумаб (продаваемый под торговой маркой Arzerra® компанией GlaxoSmithKline);
[00142] Ингибиторы тирозинкиназ: Эрлотиниба гидрохлорид (продаваемый под торговой маркой Tarceva® компанией Genentech/Roche), линифаниб (N-[4-(3-амино-1H-индазол-4-ил)фенил]-N'-(2-фтор-5-метилфенил)мочевина, также известный как ABT 869, можно приобрести у Genentech), сунитиниба малат (продаваемый под торговым названием Sutent® компанией Pfizer), босутиниб (4-[(2,4-дихлор-5-метоксифенил)амино]-6-метокси-7-[3-(4-метилпиперазин-1-ил)пропокси]хинолин-3-карбонитрил, также известный как SKI-606 и описанный в патенте США № 6780996), дасатиниб (продаваемый под торговым названием Sprycel® компанией Bristol-Myers Squibb), армала (также известный как пазопаниб, продаваемый под торговым названием Votrient® компанией GlaxoSmithKline), иматиниб и иматиниб мезилат (продаваемый под торговыми названиями Gilvec® и Gleevec® компанией Novartis);
[00143] Ингибиторы синтеза ДНК: Капецитабин (продаваемый под торговой маркой Xeloda® компанией Roche), гемцитабина гидрохлорид (продаваемый под торговой маркой Gemzar® компанией Eli Lilly and Company), неларабин ((2R,3S,4R,5R)-2-(2-амино-6-метокси-пурин-9-ил)-5-(гидроксиметил)оксолан-3,4-диол, продаваемый под торговыми названиями Arranon® и Atriance® компанией GlaxoSmithKline);
[00144] Противоопухолевые средства: оксалиплатин (продаваемый под торговым названием Eloxatin® компанией Sanofi-Aventis и описанный в патенте США № 4169846);
[00145] Ингибиторы рецептора эпидермального фактора роста (EGFR): Гефитиниб (продаваемый под торговым названием Iressa®), N-[4-[(3-хлор-4-фторфенил)амино]-7-[[(3''S'')-тетрагидро-3-фуранил]окси]-6-хиназолинил]-4-(диметиламино)-2-бутенамид, продаваемый под торговым названием Tovok® компанией Boehringer Ingelheim), цетуксимаб (продаваемый под торговым названием Erbitux® компанией Bristol-Myers Squibb), панитумумаб (продаваемый под торговым названием Vectibix® компанией Amgen);
[00146] Ингибиторы димеризации HER: Пертузумаб (продаваемый под торговой маркой Omnitarg® компанией Genentech);
[00147] Модуляторы человеческого гранулоцитарного колониестимулирующего фактора (G-CSF): Филграстим (продаваемый под торговым названием Neupogen® компанией Amgen);
[00148] Иммуномодуляторы: Афутузумаб (можно приобрести у Roche®), пегфилграстим (продаваемый под торговым названием Neulasta® компанией Amgen), леналидомид (также известный как CC-5013, продаваемый под торговым названием Revlimid®), талидомид (продаваемый под торговым названием Thalomid®);
[00149] Ингибиторы CD40: Дацетузумаб (также известный как SGN-40 или huS2C6, можно приобрести у Seattle Genetics, Inc);
[00150] Агонисты проапоптического рецептора (PARA): Дуланермин (также известный как AMG-951, можно приобрести у Amgen/Genentech);
[00151] Антагонисты Hedgehog: 2-хлор-N-[4-хлор-3-(2-пиридинил)фенил]-4-(метилсульфонил)-бензамид (также известный как GDC-0449 и описанный в публикации заявки РСТ № WO 06/028958);
[00152] Ингибиторы PI3K: 4-[2-(1H-индазол-4-ил)-6-[[4-(метилсульфонил)пиперазин-1-ил]метил]тиено[3,2-d]пиримидин-4-ил]морфолин (также известный как GDC 0941 и описанный в публикациях заявок РСТ №№ WO 09/036082 и WO 09/055730), 2-метил-2-[4-[3-метил-2-оксо-8-(хинолин-3-ил)-2,3-дигидроимидазо[4,5-c]хинолин-1-ил]фенил]пропионитрил (также известный как BEZ 235 или NVP-BEZ 235 и описанный в публикации заявки РСТ № WO 06/122806);
[00153] Ингибиторы фосфолипазы А2: Анагрелид (продаваемый под торговым названием Agrylin®);
[00154] Ингибиторы BCL-2: 4-[4-[[2-(4-хлорфенил)-5,5-диметил-1-циклогексен-1-ил]метил]-1-пиперазинил]-N-[[4-[[(1R)-3-(4-морфолинил)-1-[(фенилтио)метил]пропил]амино]-3-[(трифторметил)сульфонил]фенил]сульфонил]бензамид (также известный как ABT-263 и описанный в публикации заявки РСТ № WO 09/155386);
[00155] Ингибиторы киназы митоген-активированной протеинкиназы (MEK): XL-518 (Cas № 1029872-29-4, можно приобрести у ACC Corp.);
[00156] Ингибиторы ароматазы: Экземестан (продаваемый под торговой маркой Aromasin® компанией Pfizer), летрозол (продаваемый под торговым названием Femara® компанией Novartis), анастрозол (продаваемый под торговым названием Arimidex®);
[00157] Ингибиторы топоизомеразы I: Иринотекан (продаваемый под торговой маркой Camptosar® компанией Pfizer), топотекана гидрохлорид (продаваемый под торговым названием Hycamtin® компанией GlaxoSmithKline);
[00158] Ингибиторы топоизомеразы II: этопозид (также известный как VP-16 и этопозида фосфат, продаваемый под торговыми названиями Toposar®, VePesid® и Etopophos®), тенипозид (также известный как VM-26, продаваемый под торговым названием Vumon®);
[00159] Ингибиторы mTOR: Темсиролимус (продаваемый под торговым названием Torisel® компанией Pfizer), ридаферолимус (ранее известный как деферолимус, (1R,2R,4S)-4-[(2R)-2 [(1R,9S,12S,15R,16E,18R,19R,21R, 23S,24E,26E,28Z,30S,32S,35R)-1,18-дигидрокси-19,30-диметокси-15,17,21,23, 29,35-гексаметил-2,3,10,14,20-пентаоксо-11,36-диокса-4-азатрицикло[30.3.1.04,9]гексатриаконта-16,24,26,28-тетраен-12-ил]пропил]-2-метоксициклогексил-диметилфосфинат, также известный как AP23573 и MK8669, и описанный в публикации заявки РСТ № WO 03/064383), эверолимус (продаваемый под торговым названием Afinitor® компанией Novartis);
[00160] Ингибиторы остеокластической ресорбции костной ткани: 1-Гидрокси-2-имидазол-1-ил-фосфоноэтил)-фосфоновой кислоты моногидрат (продаваемый под торговым названием Zometa® компанией Novartis);
[00161] Конъюгаты антитело к CD33-лекарственное средство: Гемтузумаб озогамицин (продаваемый под торговым названием Mylotarg® компанией Pfizer/Wyeth);
[00162] Конъюгаты антитело к CD22-лекарственное средство: Инотузумаб озогамицин (также называемый CMC-544 и WAY-207294, можно приобрести у Hangzhou Sage Chemical Co., Ltd.);
[00163] Конъюгаты антитело к CD20-лекарственное средство: Ибритумомаб тиуксетан (продаваемый под торговым названием Zevalin®);
[00164] Аналоги соматостатина: октреотид (также известный как октреотида ацетат, продаваемый под торговыми названиями Sandostatin® и Sandostatin LAR®);
[00165] Синтетический интерлейкин-11 (IL-11): опрелвекин (продаваемый под торговым названием Neumega® компанией Pfizer/Wyeth);
[00166] Синтетический эритропоэтин: Дарбэпоэтин альфа (продаваемый под торговым названием Aranesp® компанией Amgen);
[00167] Ингибиторы рецептора активатора ядерного фактора κ B (RANK): Деносумаб (продаваемый под торговым названием Prolia® компанией Amgen);
[00168] Пептидные антитела-подражатели тромбопоэтина: Ромиплостим (продаваемый под торговым названием Nplate® компанией Amgen);
[00169] Стимуляторы клеточного роста: Палифермин (продаваемый под торговым названием Kepivance® компанией Amgen);
[00170] Антитела к рецептору инсулиноподобного фактора роста-1 (IGF-1R): Фигитумумаб (также известный как CP-751,871, можно приобрести у ACC Corp), робатумумаб (CAS № 934235-44-6);
[00171] Антитела к CS1: Элотузумаб (HuLuc63, CAS № 915296-00-3);
[00172] Антитела к CD52: Алемтузумаб (продаваемый под торговым названием Campath®);
[00173] Ингибиторы CTLA-4: Тремелимумаб (IgG2 моноклональное антитело можно приобрести у Pfizer, ранее известный как тицилимумаб, CP-675,206), ипилимумаб (антитело CTLA-4, также известное как MDX-010, CAS № 477202-00-9);
[00174] Ингибиторы гистондезацетилазы (HDI): Вониностат (продаваемый под торговым названием Zolinza® компанией Merck);
[00175] Алкилирующие средства: Темозоломид (продаваемый под торговыми названиями Temodar® и Temodal® компанией Schering-Plough/Merck), дактиномицин (также известный как актиномицин-D и продаваемый под торговым названием Cosmegen®), мелфалан (также известный как L-PAM, L-сарколизин и фенилаланин-иприт, продаваемый под торговым названием Alkeran®), алтретамин (также известный как гексаметилмеламин (HMM), продаваемый под торговым названием Hexalen®), кармустин (продаваемый под торговым названием BiCNU®), бендамустин (продаваемый под торговым названием Treanda®), бусульфан (продаваемый под торговыми названиями Busulfex® и Myleran®), карбоплатин (продаваемый под торговым названием Paraplatin®), ломустин (также известный как CCNU, продаваемый под торговым названием CeeNU®), цисплатин (также известный как CDDP, продаваемый под торговыми названиями Platinol® и Platinol®-AQ), хлорамбуцил (продаваемый под торговым названием Leukeran®), циклофосфамид (продаваемый под торговыми названиями Cytoxan® и Neosar®), дакарбазин (также известный как DTIC, DIC и имидазолкарбоксамид, продаваемый под торговым названием DTIC-Dome®), алтретамин (также известный как гексаметилмеламин (HMM), продаваемый под торговым названием Hexalen®), ифосфамид (продаваемый под торговым названием Ifex®), прокарбазин (продаваемый под торговым названием Matulane®), мехлоретамин (также известный как азотистый иприт, мустин и мехлорэтамина гидрохлорид, продаваемый под торговым названием Mustargen®), стрептозоцин (продаваемый под торговым названием Zanosar®), тиотепа (также известный как тиофосфоамид, TESPA и TSPA, продаваемый под торговым названием Thioplex®);
[00176] Модификаторы биологического ответа: бациллы Кальметта-Герена (продаваемые под торговыми названиями theraCys® и TICE® BCG), денилейкин-дифтитокс (продаваемый под торговым названием Ontak®);
[00177] Противоопухолевые антибиотики: доксорубицин (продаваемый под торговыми названиями Adriamycin® и Rubex®), блеомицин (продаваемый под торговым названием lenoxane®), даунорубицин (также известный как даунорубицина гидрохлорид, дауномицин и рубидомицина гидрохлорид, продаваемый под торговым названием Cerubidine®), липосомный даунорубицин (липосома с даунорубицина цитратом, продаваемая под торговым названием DaunoXome®), митоксантрон (также известный как DHAD, продаваемый под торговым названием Novantrone®), эпирубицин (продаваемый под торговым названием Ellence™), идарубицин (продаваемый под торговыми названиями Idamycin®, Idamycin PFS®), митомицин C (продаваемый под торговым названием Mutamycin®);
[00178] Средства против микротрубочек: Эстрамустин (продаваемый под торговым названием Emcyl®);
[00179] Ингибиторы катепсина K: Оданакатиб (также известный как MK-0822, N-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)бифенил-4-ил]этил}-L-лейцинамид, поставляемый компаниями Lanzhou Chon Chemicals, ACC Corp. и ChemieTek, и описанный в PCT публикации № WO 03/075836);
[00180] Аналоги эпотилона B: Иксабепилон (продаваемый под торговым названием Lxempra® компанией Bristol-Myers Squibb);
[00181] Ингибиторы белка теплового шока (HSP): Танеспимицин (17-аллиламино-17-деметоксигелданамицин, также известный как KOS-953 и 17-AAG, поставляемый компанией SIGMA и описанный в Патенте США № 4261989);
[00182] Агонисты TpoR: Элтромбопаг (продаваемый под торговыми названиями Promacta® и Revolade® компанией GlaxoSmithKline);
[00183] Антимитотические средства: Доцетаксел (продаваемый под торговым названием Taxotere® компанией Sanofi-Aventis);
[00184] Ингибиторы синтеза гормонов надпочечников: аминоглютетимид (продаваемый под торговым названием Cytadren®);
[00185] Анти-андрогенные препараты: Нилутамид (продаваемый под торговыми названиями Nilandron® и Anandron®), бикалутамид (продаваемый под торговым названием Casodex®), флутамид (продаваемый под торговым названием Fulexin™);
[00186] Андрогены: Флуоксиместерон (продаваемый под торговым названием Halotestin®);
[00187] Ингибиторы протеасомы: Бортезомиб (продаваемый под торговым названием Velcade®);
[00188] Ингибиторы CDK1 Алвоцидиб (также известный как фловопирдол или HMR-1275, 2-(2-хлорфенил)-5,7-дигидрокси-8-[(3S,4R)-3-гидрокси-1-метил-4-пиперидил]-4-хроменон, и описанный в Патенте США № 5621002);
[00189] Агонисты рецепторов гонадотропин-высвобождающего гормона (GnRH): Лейпролид или лейпролида ацетат (продаваемый под торговыми названиями Viadure® компанией Bayer AG, Eligard® компанией Sanofi-Aventis и Lupron® компанией Abbott Lab);
[00190] Таксановые противоопухолевые средства: Кабазитаксел (1-гидрокси-7β,10β-диметокси-9-оксо-5β,20-эпокситакс-11-ен-2α,4,13α-триил-4-ацетат-2-бензоат-13-[(2R,3S)-3-{[(трет-бутокси)карбонил]амино}-2-гидрокси-3-фенилпропаноат), ларотаксел ((2α,3ξ,4α,5β,7α,10β,13α)-4,10-бис(ацетилокси)-13-({(2R,3S)-3- [(трет-бутоксикарбонил)амино]-2-гидрокси-3-фенилпропаноил}окси)-1- гидрокси-9-оксо-5,20-эпокси-7,19-циклотакс-11-ен-2-ил-бензоат);
[00191] Агонисты 5HT1a-рецепторов: Ксалипроден (также известный как SR57746, 1-[2-(2-нафтил)этил]-4-[3-(трифторметил)фенил]-1,2,3,6-тетрагидропиридин, как описано в патенте США № 5266573);
[00192] Вакцины против HPC: Cervarix®, поступает в продажу от компании GlaxoSmithKline, Gardasil®, поступает в продажу от компании Merck;
[00193] Хелаторы железа: Деферазинокс (продаваемый под торговым названием Exjade® от компании Novartis);
[00194] Антиметаболиты: Кларибин (2-хлордеоксиаденозин, продаваемый под торговым названием leustatin®), 5-фторурацил (продаваемый под торговым названием Adrucil®), 6-тиогуанин (продаваемый под торговым названием Purinethol®), пеметрексед (продаваемый под торговым названием Alimta®), цитарабин (также известный как арабинозилцитозин (Ara-C), продаваемый под торговым названием Cytosar-U®), цитарабин липосомальный (также известный как липосомальный Ara-C, продаваемый под торговым названием DepoCyt™), децитабин (продаваемый под торговым названием Dacogen®), гидроксимочевина (продаваемая под торговыми названиями Hydrea®, Droxia™ и Mylocel™), флударабин (продаваемый под торговым названием Fludara®), флоксуридин (продаваемый под торговым названием FUDR®), кладрибин (также известный как 2-хлордезоксиаденозин (2-CdA), продаваемый под торговым названием Leustatin™), метотрексат (также известный как аметоптерин, метотрексат натрия (MTX), продаваемый под торговыми названиями Rheumatrex® и Trexall™), пентостатин (продаваемый под торговым названием Nipent®);
[00195] Бисфосфонаты: Памидронат (продаваемый под торговым названием Aredia®), золедроновая кислота (продаваемая под торговым названием Zometa®);
[00196] Деметилирующие средства: 5-азацитидин (продаваемый под торговым названием Vidaza®), децитабин (продаваемый под торговым названием Dacogen®);
[00197] Растительные алкалоиды: Паклитаксел, связанный с белком (продаваемый под торговым названием Abraxane®), винбластин (также известный как винбластина сульфат, винкалейкобластин и VLB, продаваемый под торговыми названиями Alkaban-AQ® и Velban®), винкристин (также известный как винкристина сульфат, LCR и VCR, продаваемый под торговыми названиями Oncovin® и Vincasar Pfs®), винорелбин (продаваемый под торговым названием Navelbine®), паклитаксел (продаваемый под торговыми названиями Taxol и Onxal™);
[00198] Ретиноиды: Алитретиноин (продаваемый под торговым названием Panretin®), третиноин (полностью транс-ретиноевая кислота, также известная как ATRA, продаваемая под торговым названием Vesanoid®), Изотретиноин (13-цис-ретиноевая кислота, продаваемая под торговыми названиями Accutane®, Amnesteem®, Claravis®, Clarus®, Decutan®, Isotane®, Izotech®, Oratane®, Isotret® и Sotret®), бексаротен (продаваемый под торговым названием Targretin®);
[00199] Глюкокортикостероиды: Гидрокортизон (также известный как кортизон, гидрокортизон натрия сукцинат, гидрокортизон натрия фосфат, и продаваемый под торговыми названиями Ala-Cort®, Hydrocortisone Phosphate, Solu-Cortef®, Hydrocort Acetate® и Lanacort®), дексаметазон ((8S,9R,10S,11S,13S,14S,16R,17R)-9-фтор-11,17-дигидрокси-17-(2-гидроксиацетил)-10,13,16-триметил-6,7,8,9,10,11,12,13,14,15,16,17-додекагидро-3H-циклопента[a]фенантрен-3-он), преднизолон (продаваемый под торговыми названиями Delta-Cortel®, Orapred®, Pediapred® и Prelone®), преднизон (продаваемый под торговыми названиями Deltasone®, Liquid Red®, Meticorten® и Orasone®), метилпреднизолон (также известный как 6-метилпреднизолон, метилпреднизолона ацетат, метилпреднизолона натрия сукцинат, продаваемый под торговыми названиями Duralone®, Medralone®, Medrol®, M-Prednisol® и Solu-Medrol®);
[00200] Цитокины: интерлейкин-2 (также известный как альдеслейкин и IL-2, продаваемый под торговым названием Proleukin®), интерлейкин-11 (также известный как опревелкин, продаваемый под торговым названием Neumega®), альфа интерферон альфа (также известный как IFN-альфа, продаваемый под торговыми названиями Intron® A и Roferon-A®);
[00201] Супрессоры экстрогеновых рецепторов: Фулвестрант (продаваемый под торговым названием Faslodex®);
[00202] Антиэстрогены: тамоксифен (продаваемый под торговым названием Novaldex®);
[00203] Торемифен (продаваемый под торговым названием Fareston®);
[00204] Селективные модуляторы эстрогеновых рецепторов (SERM): Ралоксифен (продаваемый под торговым названием Evista®);
[00205] Агонисты высвобождающего фактора лютеинизирующего гормона (LHRH): Гозерелин (продаваемый под торговым названием Zoladex®);
[00206] Прогестероны: мегестрол (также известный как мегестрола ацетат, продаваемый под торговым названием Megace®);
[00207] Различные цитотоксические средства: Триоксид мышьяка (продаваемый под торговым названием Trisenox®), аспарагиназа (также известная как L-аспарагиназа, эрвиния L-аспарагиназа, продаваемая под торговыми названиями Elspar® и Kidrolase®);
[00208] Соединение формулы (I) также может использоваться в комбинации со следующими видами вспомогательной терапии:
[00209] Противорвотные средства: Антагонисты рецептора NK-1: Касопитант (продаваемый под торговыми названиями Rezonic® и Zunrisa® компанией GlaxoSmithKline); и
[00210] Цитопротекторные средства: Амифостин (продаваемый под торговым названием Ethyol®), лейковорин (также известный как лейковорин кальция, цитроворум-фактор и фолиновая кислота).
[00211] Ни одну из цитат ссылок, сделанных в настоящем раскрытии, не следует понимать как признание того, что упомянутые ссылки известны из уровня техники и отрицательно влияют на патентоспособность настоящего изобретения.
Процессы для получения соединений данного изобретения
[00212] Настоящее изобретение также включает способы получения соединений данного изобретения. В описанных реакциях может потребоваться защита реакционноспособных функциональных групп, например, гидрокси-, амино-, имино-, тио- или карбоксигрупп, при этом они желательны в конечном продукте, во избежание нежелательного участия их в реакциях. Можно применять традиционные защитные группы в соответствии со стандартной практикой, например, см. T.W. Greene and P. G. M. Wuts в «Protective Groups in Organic Chemistry», John Wiley and Sons, 1991.
[00213] Соединения формулы I можно получать, следуя изложенной ниже схеме реакций I:
Схема реакций I:

[00214] в которой Q является уходящей группой, а Y1, Y2, R1, R2 и R3 такие, как определено в Кратком изложении сущности изобретения. Соединения формулы I можно получать реакцией соединения формулы 2 с соединением формулы 3 в присутствии подходящего основания (например, DIPEA или подобного) и подходящего растворителя (например, DMSO, NMP или подобного). Реакция протекает при температурном режиме от примерно 80°C до примерно 140°C и ее продолжительность может составлять от примерно 1 часа до примерно 24 часов до ее завершения. Последующая реакция с соединением формулы 4 с подходящей уходящей группой Х может проходить в подходящем растворителе при повышенной температуре в присутствии основания или без него. Эти процессы могут происходить с использованием подходящих защитных групп, которые впоследствии могут быть удалены, и стадии могут происходить в обратном порядке.
[00215] Соединения формулы II можно получать, следуя изложенной ниже схеме реакций II:
Схема реакций II:
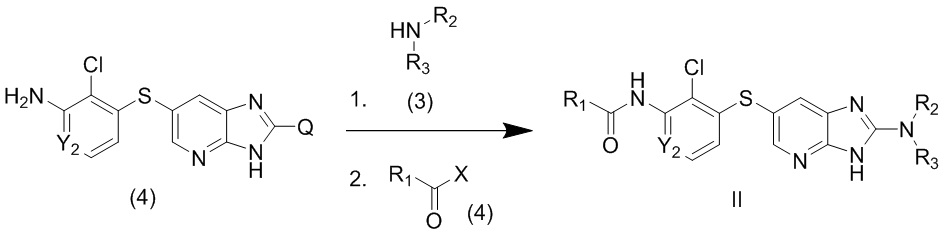
[00216] в которой Q является уходящей группой, а Y1, Y2, R1, R2 и R3 такие, как определено в Кратком изложении сущности изобретения. Соединения формулы II можно получать реакцией соединения 4 с соединением формулы 3 в присутствии подходящего основания (например, DIPEA или подобного) и подходящего растворителя (например, DMSO, NMP или подобного). Реакция протекает при температурном режиме от примерно 80°C до примерно 140°C и ее продолжительность может составлять от примерно 1 часа до примерно 24 часов до ее завершения. Последующая реакция с соединением формулы 4 с подходящей уходящей группой Х может проходить в подходящем растворителе при повышенной температуре в присутствии основания или без него. Эти процессы могут происходить с использованием подходящих защитных групп, которые впоследствии могут быть удалены, и стадии могут происходить в обратном порядке.
[00217] Подробные примеры синтеза соединений формулы I можно найти в примерах ниже.
Дополнительные процессы для получения соединений данного изобретения
[00218] Соединение данного изобретения можно получать в виде фармацевтически приемлемой соли присоединения кислоты взаимодействием соединения в форме свободного основания с фармацевтически приемлемой неорганической или органической кислотой. В альтернативном случае, фармацевтически приемлемую соль присоединения основания соединения данного изобретения можно получать взаимодействием соединения в форме свободной кислоты с фармацевтически приемлемым неорганическим или органическим основанием.
[00219] Соединения формулы I также могут быть модифицированы путем добавления соответствующих функциональных групп для усиления избирательных биологических свойств. Модификации такого типа известны в данной области и включают те, которые увеличивают проникновение в данную биологическую систему (например, кровь, лимфатическую систему, центральную нервную систему, яички), увеличивают биодоступность, повышают растворимость, позволяя проводить парентеральное введение (например, инъекцию, инфузию), изменять метаболизм и/или изменять скорость выведения. Примеры такого типа модификаций включают, но не ограничиваются ими, этерификацию, например, с полиэтиленгликолями, дериватизацию с помощью пивалоилокси- или жирнокислотных заместителей, превращение в карбаматы, гидроксилирование ароматических колец и замещение гетероатомов в ароматических кольцах. Любое упоминание соединений формулы I и/или их N-оксидов, таутомеров и/или (предпочтительно фармацевтически приемлемых) солей включает такие модифицированные формулы, при этом предпочтительно подразумеваются молекулы, соответствующие формуле I, их N-оксиды, таутомеры и/или соли.
[00220] В альтернативном случае соединения данного изобретения в форме солей можно получать, используя соли исходных материалов или промежуточных соединений. Вследствие близкой связи между новыми соединениями формулы I в свободной форме и соединениями в форме их солей, включая те соли, которые можно использовать в качестве промежуточных соединений, например, при очистке или идентификации новых соединений, любое упоминание соединений или соединения формулы I ранее и далее в данном документе следует понимать как относящееся к соединению в свободной форме и/или также к одной или нескольким его солям, по мере необходимости и целесообразности, а также к одному или нескольким сольватам, например, гидратам.
[00221] Соли, особенно фармацевтически приемлемые соли, образуются, например, в виде солей присоединения кислоты, предпочтительно с органическими или неорганическими кислотами, из соединений формулы I с основным атомом азота. Подходящими неорганическими кислотами являются, например, галогеновые кислоты, такие как хлористоводородная кислота, серная кислота или фосфорная кислота Подходящими органическими кислотами являются, например, карбоновые, фосфоновые, сульфоновые или сульфаминовые кислоты, например, уксусная кислота, пропионовая кислота, октановая кислота, декановая кислота, додекановая кислота, гликолеваякислота, молочная кислота, фумаровая кислота, янтарная кислота, малоновая кислота, адипиновая кислота, пимелиновая кислота, субериновая кислота, азелаиновая кислота, яблочная кислота, винная кислота, лимонная кислота, аминокислоты, такие как глутаминовая кислота или аспарагиновая кислота, малеиновая кислота, гидроксималеиновая кислота, метилмалеиновая кислота, циклогексанкарбоновая кислота, адамантанкарбоновая кислота, бензойная кислота, салициловая кислота, 4-аминосалициловая кислота, фталевая кислота, фенилуксусная кислота, миндальная кислота, коричная кислота, метан- или этансульфокислота, 2-гидроксиэтансульфокислота, этан-1,2-дисульфокислота, бензолсульфокислота, 4-толуолсульфокислота, 2-нафталинсульфокислота, 1,5-нафталин-дисульфокислота, 2- или 3-метилбензолсульфокислота, метилсерная кислота, этилсерная кислота, додецилсерная кислота, N-циклогексилсульфамовая кислота, N-метил-, N-этил- или N-пропил-сульфамовая кислота, или другие органические протонные кислоты, такие как аскорбиновая кислота.
[00222] Для целей выделения или очистки также можно использовать фармацевтически неприемлемые соли, например пикраты или перхлораты. Для терапевтического применения используются только фармацевтически приемлемые соли или несвязанные соединения (где это применимо для формы фармацевтических препаратов), и поэтому они предпочтительны.
[00223] Несвязанные кислотные или основные формы соединений данного изобретения могут быть получены из соответствующей соли присоединения основания или кислоты, соответственно. Например, соединение данного изобретения в форме соли присоединения кислоты может быть превращено в соответствующее свободное основание обработкой подходящим основанием (например, раствором гидроксида аммония, гидроксидом натрия и т.п.). Соединение данного изобретения в форме соли присоединения основания может быть превращено в соответствующую свободную кислоту обработкой подходящей кислотой (например, хлористоводородной кислотой и т.п.).
[00224] Соединения данного изобретения в неокисленной форме могут быть получены из N-оксидов соединений данного изобретения путем обработки восстановителем (например, серой, диоксидом серы, трифенилфосфином, боргидридом лития, боргидридом натрия, трихлоридом фосфора, трибромидом или т.п.) в подходящем инертном органическом растворителе (например, ацетонитриле, этаноле, водном диоксане и т.п.) при температуре от 0 до 80°C.
[00225] Пролекарственные производные соединений данного изобретения могут быть получены способами, известными специалистам в данной области техники (например, дополнительно см. Saulnier et al., (1994), Bioorganic and Medicinal Chemistry Letters, Vol. 4, p. 1985). Например, подходящие пролекарства могут быть получены путем взаимодействия немодифицированного соединения данного изобретения с подходящим карбамилирующим агентом (например, 1,1-ацилоксиалкилкарбанохлоридатом, пара-нитрофенилкарбонатом и т.п.).
[00226] Защищенные производные соединений данного изобретения можно получать способами, известными специалистам в данной области техники. Подробное описание методик, применимых для создания защитных групп и их удаления, можно найти в T. W. Greene, «Protecting Groups in Organic Chemistry», 3rd edition, John Wiley and Sons, Inc., 1999.
[00227] Соединения настоящего изобретения для удобства можно готовить, или получать способом настоящего изобретения, в виде сольватов (например, гидратов). Гидраты соединений настоящего изобретения можно легко получать перекристаллизацией из смеси водного/органического растворителя с использованием таких органических растворителей, как диоксин, тетрагидрофуран или метанол.
[00228] Соединения настоящего изобретения можно получать в виде индивидуальных стереоизомеров при взаимодействии рацемической смеси соединения с оптически активным разделяющим реагентом с образованием пары диастереомеров, разделения диастереомеров и выделения оптически чистых энантиомеров. Разделение энантиомеров можно проводить с использованием ковалентных диастереомерных производных соединений данного изобретения, причем предпочтительными являются диссоциирующие комплексы (например, кристаллических солей диастереомеров). Диастереомеры характеризуются присущими им физическими свойствами (например, температурой плавления, точкой кипения, растворимостью, реакционной способностью и т.п.), и их легко можно разделить, используя различия в этих свойствах. Диастереомеры можно разделять хроматографически или, предпочтительно, с использованием методик разделения/выделения, основанных на разной их растворимости. Оптически чистые энантиомеры затем выделяют вместе с разделяющим реагентом любым практическим методом, не приводящим к рацемизации. Более подробное описание методик выделения стереоизомеров соединений из их рацемической смеси приведено в Jean Jacques, Andre Collet, Samuel H. Wilen, «Enantiomers, Racemates and Resolutions», John Wiley and Sons, Inc., 1981.
[00229] Таким образом, соединения Формулы I можно получать способом, который включает:
(a) реакции, указанные на схемах реакций I и II; и
(b) необязательно превращение соединения данного изобретения в фармацевтически приемлемую соль;
(c) необязательно превращение соли соединения данного изобретения в соединение в несолевой форме;
(d) необязательно превращение неокисленной формы соединения данного изобретения в фармацевтически приемлемый N-оксид;
(e) необязательно превращение соединения данного изобретения в форме N-оксида в его неокисленную форму;
(f) необязательно выделение индивидуального изомера соединения данного изобретения из смеси изомеров;
(g) необязательно превращение немодифицированного соединения данного изобретения в фармацевтически приемлемое пролекарственное производное; и
(h) необязательно превращение пролекарственного производного соединения данного изобретения в его немодифицированную форму.
[00230] Получение исходных материалов не описано подробно, поскольку эти соединения либо известны, либо их получают известными в данной области способами или как описано в примерах ниже.
[00231] Специалисту в данной области техники представляется очевидным, что описанные выше превращения приведены только для иллюстрации способов получения соединений настоящего изобретения, и что можно использовать также другие хорошо известные способы.
Примеры
[00232] Приведенные ниже промежуточные соединения и примеры иллюстрируют данное изобретение, но не ограничивают его объем. Некоторые сокращения, используемые в приведенных ниже примерах, перечислены ниже: уксусная кислота (AcOH); ацетонитрил (MeCN); триэтиламин (TEA); тетрагидрофуран (THF); водный (водн.); атмосферное/атмосфера (атм.); 2,2'-бис-дифенилфосфанил-[1,1']бинафталенил (BINAP); 4-диметиламинопиридин (DMAP); трет-бутоксикарбонил (Boc); 1,1-карбонилдиимидазол (CDI); ди-трет-бутилдикарбонат (Boc2O); бензотриазол-1-ил-окси-трис-(диметиламино)-фосфония гексафторфосфат (BOP); дихлорметан (DCM); диэтиловый эфир (Et2O); п-толуолсульфокислота (PTSA); этилацетат (EtOAc); этанол (EtOH); бис(триметилсилил)амид лития (LHMDS); диизопропилазодикарбоксилат (DIAD); N,N-диизопропил-этиламин (DIEA или DIPEA); N,N-диметилформамид (DMF); диметилсульфоксид (DMSO); дифенилфосфорилазид (DPPA); час(ы) (ч); 2-(1H-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат (HATU); высокоэффективная жидкостная хроматография (ВЭЖХ); изопропиловый спирт (IPA); алюмогидрид лития (LAH); жидкостная хроматография в сочетании с масс-спектрометрией (ЖХМС); диизопропиламид лития (LDA); метанол (MeOH); миллилитр(ы) (мл); минута(ы) (мин); микроволновой (MW); бис(триметилсилил)амид натрия (NHMDS); н-бутиллитий (н-BuLi); 1,1-бис(дифенилфосфино)-ферроцендихлорпалладий(II) (PdCl2(dppf)); трис(дибензилиденацетон)дипалладий (0) (Pd2(dba)3); дихлорбис(трифенилфосфин)палладий (II) (PdCl2(PPh3)2); комнатная температура (к.т.); тетра-н-бутиламмония фторид (TBAF); трет-бутилдиметилсилилхлорид (TBSCl); трифторуксусная кислота (TFA); тетрагидрофуран (THF); тонкослойная хроматография (ТСХ); время удерживания (TR); (S)-(-)-2,2'-бис(ди-п-толилфосфино)-1,1'-бинафтил ((S)-TolBINAP); и 4,5-бис(дифенилфосфино)-9,9-диметилксантен (XantPhos).
Промежуточное соединение 1
трет-Бутил-((1-(6-амино-5-((3-амино-2-хлорфенил)тио)пиразин-2-ил)-4-метилпиперидин-4-ил)метил)карбамат
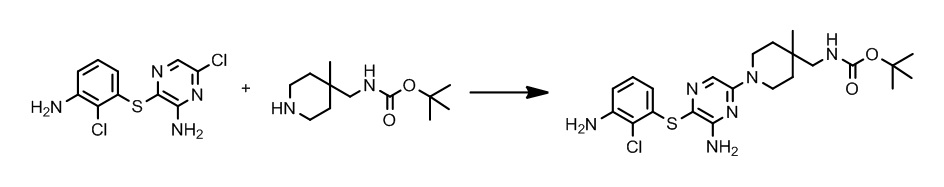
[00233] Как описано в Примере 1, аналогичную процедуру применяли, используя 3-((3-амино-2-хлорфенил)тио)-6-хлорпиразин-2-амин (1,5 г, 5,2 ммоль) и трет-бутил-((4-метилпиперидин-4-ил)метил)карбамат (1,9 г, 8,4 ммоль), получая трет-бутил-((1-(6-амино-5-((3-амино-2-хлорфенил)тио)пиразин-2-ил)-4-метилпиперидин-4-ил)метил)карбамат (2,4 г). MS масса/заряд 479,3 (M+H)+.
Промежуточное соединение 2
Метил-2-гидрокси-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-a]пиримидин-3-карбоксилат
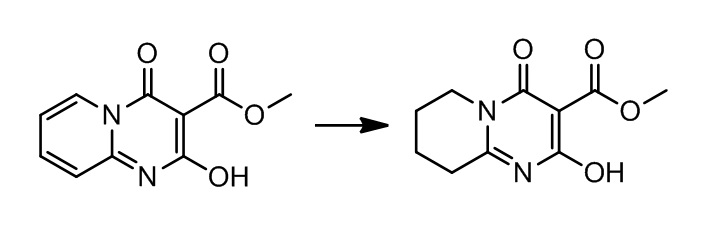
[00234] В раствор метил-2-гидрокси-4-оксо-4H-пиридо[1,2-a]пиримидин-3-карбоксилата (2 г, 9,08 ммоль) в МеОН (50 мл) добавляли 10% Pd/C (1,9 г, 1,8 ммоль). Реакционную смесь перемешивали при к.т. в атмосфере H2 (баллон) в течение 2,5 ч. Затем реакционную смесь фильтровали через Celite® и концентрировали при пониженном давлении до сухого состояния, получая указанное в заголовке соединение (2,15 г). MS масса/заряд 225,1 (M+H)+.
Промежуточное соединение 3
Этил-2-гидрокси-4-оксо-4,6,7,8,9,9а-гексагидро-1H-пиридо[1,2-a]пиримидин-3-карбоксилат
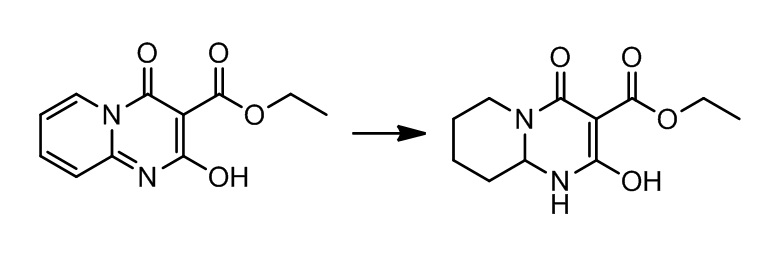
[00235] В раствор этил-2-гидрокси-4-оксо-4H-пиридо[1,2-a]пиримидин-3-карбоксилата (0,05 г, 0,21 ммоль) в МеОН (1 мл) добавляли 10% Pd/C (0,045 г, 0,04 ммоль). Реакционную смесь перемешивали в течение ночи при к.т. в атмосфере H2 (баллон). Затем реакционную смесь фильтровали через Celite® и концентрировали при пониженном давлении до сухого состояния, получая этил-2-гидрокси-4-оксо-1,6,7,8,9,9а-гексагидро-4H-пиридо[1,2-a]пиримидин-3-карбоксилат (0,043 г). MS масса/заряд 241,1 (M+H)+.
Промежуточное соединение 4
8-трет-Бутил-3-этил-2-гидрокси-4-оксо-6,7-дигидро-4H-пиразино[1,2-a]пиримидин-3,8(9H)-дикарбоксилат

[00236] В раствор этил-2-гидрокси-4-оксо-4H-пиразино[1,2-a]пиримидин-3-карбоксилата (0,10 г, 0,44 ммоль) в МеОН (5 мл) добавляли Pd/C (10%, 0,09 г, 0,09 ммоль), Boc2O (0,15 мл, 0,66 ммоль) and DIPEA (0,23 мл, 1,31 ммоль). Реакционную смесь перемешивали при к.т. в атмосфере H2 (баллон) в течение 1 ч. Реакционную смесь фильтровали через Celite® и концентрировали при пониженном давлении до сухого состояния. Остаток очищали при помощи флеш-хроматографии на силикагеле (0-3% MeOH/DCM), получая указанное в заголовке соединение (0,072 г). MS масса/заряд 340,2 (M+H)+.
Промежуточное соединение 5
трет-Бутил-(1-(6-амино-5-((3-амино-2-хлорфенил)тио)пиразин-2-ил)-4-(фторметил)пиперидин-4-ил)карбамат
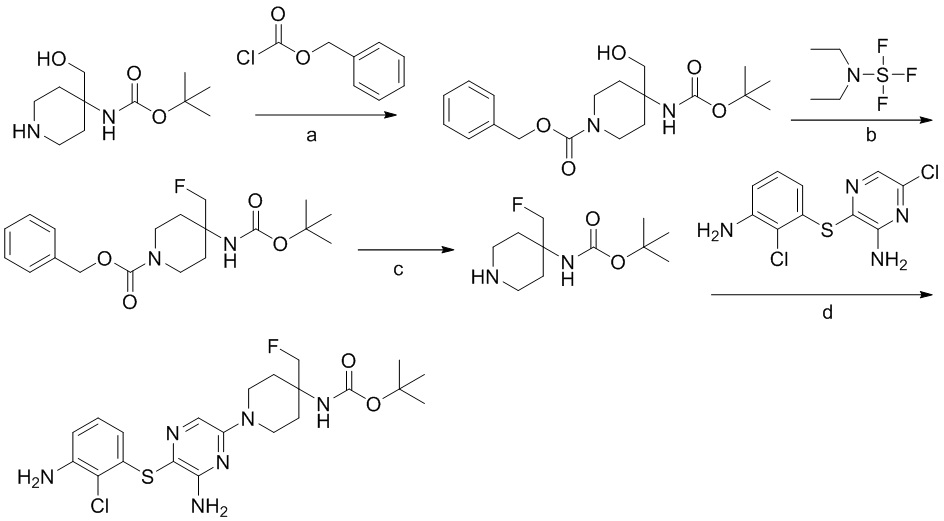
[00237] Стадия a: К суспензии трет-бутил-(4-гидроксиметил)пиперидин-4-ил)карбамата (4,8 г, 20,8 ммоль) в DCM (100 мл) при 0°C добавляли триэтиламин (8,7 мл, 6,3 г, 62,3 ммоль), а затем добавляли бензилхлорформиат (5,3 г, 31,1 ммоль). Реакционную смесь нагревали до к.т. и перемешивали в течение 3 ч. Реакционную смесь разбавляли DCM (150 мл), органические слои промывали водой и соляным раствором, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Сырой продукт очищали хроматографией на силикагеле (0-20% MeOH/DCM), получая 4,6 г бензил-4-((трет-бутоксикарбонил)амино)-4-(гидроксиметил)пиперидин-1-карбоксилата. MS масса/заряд 387,3 (M+Na)+.
[00238] Стадия b: К раствору бензил-4-((трет-бутоксикарбонил)амино)-4-(гидроксиметил)пиперидин-1-карбоксилата (4,6 г, 12,6 ммоль) в DCM (80 мл) при -78°C по каплям добавляли трифторид диэтиламиносеры (3,3 мл, 4,1 г, 25,2 ммоль). Реакционную смесь перемешивали при -78°C в течение 1 ч, а затем нагревали до к.т. за 30 мин. Реакционную смесь разбавляли, используя DCM (200 мл), дважды промывали насыщенным водн. Na2CO3, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении до получения мутного желтого масла. Сырой продукт очищали хроматографией на силикагеле (20-40% EtOAc/гептана), получая 1,9 г бензил-4-((трет-бутоксикарбонил)амино)-4-(фторметил)пиперидин-1-карбоксилата в виде прозрачного масла. MS масса/заряд 367,1 (M+H)+.
[00239] Стадия c: Суспензию бензил-4-((трет-бутоксикарбонил)амино)-4-(фторметил)пиперидин-1-карбоксилата (0,26 г, 0,71 ммоль) и Pd/C (10%, 0,08 г) в MeOH (15 мл) продували N2 в течение 10 мин, а затем перемешивали в атмосфере H2 (баллон) в течение 12 ч при к.т. Реакционную смесь фильтровали через подушку из Celite®, используя MeOH (100 мл). Объединенные органические слои концентрировали при пониженном давлении до получения прозрачного масла - трет-бутил-(4-(фторметил)пиперидин-4-ил)карбамата (0,16 г). MS масса/заряд 233,1 (M+H)+.
[00240] Стадия d: Как описано в Примере 1, аналогичную процедуру применяли, используя 3-((3-амино-2-хлорфенил)тио)-6-хлорпиразин-2-амин (0,37 г, 1,29 ммоль) и трет-бутил-(4-(фторметил)пиперидин-4-ил)карбамат (0,30 г, 1,29 ммоль), получая указанное в заголовке соединение (0,26 г). MS масса/заряд 483,1 (M+H)+.
Промежуточное соединение 6
трет-Бутил-((1-(6-амино-5-((3-амино-2-хлорфенил)тио)пиразин-2-ил)-4-фторпиперидин-4-ил)метил)карбоксилат

[00241] Как описано для Промежуточного соединения 1, аналогичную процедуру применяли, используя 3-((3-амино-2-хлорфенил)тио)-6-хлорпиразин-2-амин (0,2 г, 0,70 ммоль) и трет-бутил-((4-фторпиперидин-4-ил)метил)карбамат (0,24 г, 1,05 ммоль), получая указанное в заголовке соединение (0,29 г). MS масса/заряд 483,1 (M+H)+.
Промежуточное соединение 7
трет-Бутил-((1-(6-амино-5-((3-амино-2-хлорфенил)тио)пиразин-2-ил)пирролидин-3-ил)метил)карбамат
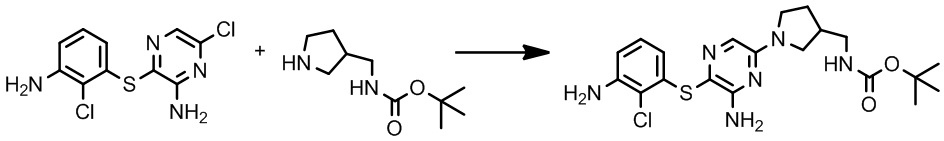
[00242] Как описано для Промежуточного соединения 1, аналогичную процедуру применяли, используя 3-((3-амино-2-хлорфенил)тио)-6-хлорпиразин-2-амин (0,20 г, 0,70 ммоль) и трет-бутил-(пирролидин-3-илметил)карбамат (0,21 г, 1,05 ммоль), получая указанное в заголовке соединение (0,19 г). MS масса/заряд 451,1 (M+H)+.
Промежуточное соединение 8
трет-Бутил-((1-(6-амино-5-((3-амино-2-хлорфенил)тио)пиразин-2-ил)пирролидин-3-ил)-2,2,2-трифторэтил)карбоксилат

[00243] Как описано для Промежуточного соединения 1, аналогичную процедуру применяли, используя 3-((3-амино-2-хлорфенил)тио)-6-хлорпиразин-2-амин (0,20 г, 0,70 ммоль) и трет-бутил-(2,2,2-трифтор-1-(пирролидин-3-ил)этил)карбамат (0,28 г, 1,05 ммоль), получая указанное в заголовке соединение (0,29 г). MS масса/заряд 519,2 (M+H)+.
Промежуточное соединение 9
3-((3-Амино-2-хлорфенил)тио)-6-(4-амино-4-(трифторметил)пиперидин-1-ил)пиразин-2-амин

[00244] Стадия a: К суспензии трет-бутил-4-амино-4-(трифторметил)пиперидин-1-карбоксилата (0,50 г, 1,86 ммоль) в диоксане (6 мл) и МеОН (1 мл) при к.т. добавляли 4 М HCl в диоксане (9,32 ммоль, 2,3 мл). После перемешивания при к.т. в течение 16 ч реакционную смесь концентрировали при пониженном давлении до белого твердого вещества (0,43 г) - бис-HCl соли.1H ЯМР (400 МГц, DMSO-d6) δ ppm 9,40-9,01 (m, 2 H), 4,16 (br.s, 3 H), 3,48 (d, J=11,5 Гц, 2 H), 3,31 (d, J=12,1 Гц, 2 H), 2,16 (t, J=10,9 Гц, 2 H), 1,99 (d, J=14,1 Гц, 2 H).
[00245] Стадия b: Как описано для Промежуточного соединения 2, аналогичную процедуру применяли, используя 3-((3-амино-2-хлорфенил)тио)-6-хлорпиразин-2-амина дигидрохлорид (0,23 г, 0,80 ммоль) и 4-(трифторметил)пиперидин-4-амин (0,25 г), получая указанное в заголовке соединение (0,32 г). MS масса/заряд 419,3 (M+H)+.
Промежуточное соединение 10
5-((3-Амино-2-хлорфенил)тио)-N2-метил-N2-(2,2,6,6-тетраметилпиперидин-4-ил)пиразин-2,6-диамин

[00246] Как описано для Промежуточного соединения 1, аналогичную процедуру применяли, используя 3-((3-амино-2-хлорфенил)тио)-6-хлорпиразин-2-амин (0,29 г, 1,00 ммоль) и N,2,2,6,6-тетраметилпиперидин-4-амин (0,51 г), получая указанное в заголовке соединение в виде соли HCl (0,48 г). MS масса/заряд 421,3 (M+H)+.
Промежуточное соединение 11
N-((S)-8-(6-Амино-5-((3-амино-2-хлорфенил)тио)пиразин-2-ил)-2,2-дифтор-8-азаспиро[4.5]декан-1-ил)-2-метилпропан-2-сульфинамид
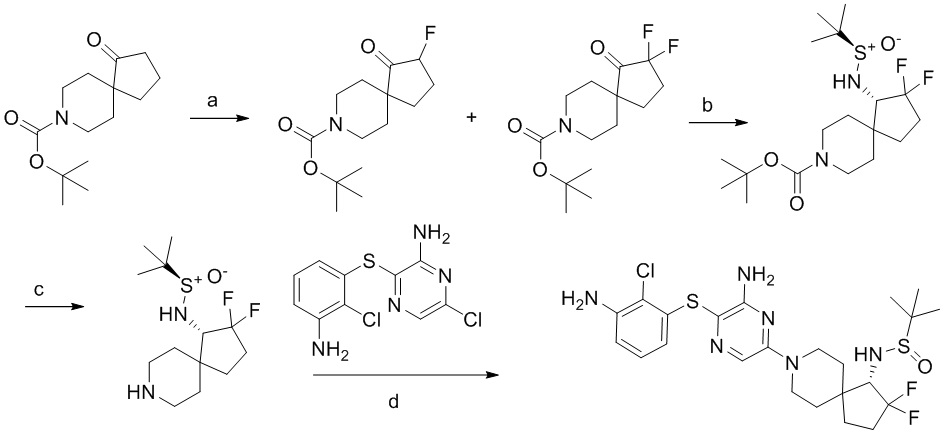
[00247] Стадия a: При -78°C к раствору NaHMDS (1 M в THF, 8,68 мл, 8,68 ммоль) добавляли раствор трет-бутил-1-оксо-8-азаспиро[4.5]декан-8-карбоксилата (2,0 г, 7,89 ммоль) в THF (5 мл). После перемешивания в течение 30 мин при этой температуре добавляли раствор N-фторбензолсульфонамида (2,49 г, 7,89 ммоль) в THF (10 мл). После 3 часов перемешивания при -78°C смесь разбавляли насыщ. водн. NaHCO3 (100 мл) и экстрагировали, используя DCM (3×100 мл). Объединенные органические фазы промывали соляным раствором, высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (0-25% EtOAc/гептана), получая рацемический трет-бутил-2-фтор-1-оксо-8-азаспиро[4.5]декан-8-карбоксилат (351 мг, 1,29 ммоль). MS масса/заряд 272,1 (M+H)+. Дифторкетон вымывался вместе с исходным материалом. Элюированные совместно фракции дифторкетона/исходного материала повторно очищали хроматографией на силикагеле (0-5% MeOH/DCM), получая трет-бутил-2,2-дифтор-1-оксо-8-азаспиро[4.5]декан-8-карбоксилат (573 мг). MS масса/заряд 290,1 (M+H)+.
[00248] Стадия b: В микроволновую ампулу объемом 10 мл, содержащую трет-бутил-2,2-дифтор-1-оксо-8-азаспиро[4.5]декан-8-карбоксилат (224 мг, 0,774 ммоль) в THF (4 мл), добавляли твердый (R)-2-метилпропан-2-сульфинамид (188 мг, 1,55 ммоль), а затем чистый тетраэтоксид титана (0,649 мл, 3,1 ммоль). Реакционный сосуд укупоривали колпачком и нагревали при 90°C в течение 1 ч. Реакционную смесь охлаждали (0°C) и добавляли LiBH4 (34 мг, 1,6 ммоль) непосредственно в THF раствор. Реакционную смесь перемешивали в течение 30 мин и затем гасили, медленно добавляя МеОН до прекращения вспенивания. Реакционную смесь разбавляли избытком МеОН, затем концентрировали при пониженном давлении, еще раз добавляя МеОН. Смесь разбавляли соляным раствором (при обработке ультразвуком) и экстрагировали, используя EtOAc (4×10 мл), органическую фазу концентрировали при пониженном давлении. С помощью флеш-хроматографии на силикагеле (0-50% EtOAc/гептана) получали трет-бутил-(S)-1-(((R)-трет-бутилсульфинил)амино)-2,2-дифтор-8-азаспиро[4.5]декан-8-карбоксилат в виде бесцветного масла, которое кристаллизовалось при отстаивании (180 мг), MS масса/заряд 395,2 (M+H)+.
[00249] Стадия c: В охлажденный на льду раствор трет-бутил-(S)-1-(((R)-трет-бутилсульфинил)амино)-2,2-дифтор-8-азаспиро[4.5]декан-8-карбоксилата (180 мг, 0,46 ммоль) в DCM (2 мл) вносили TFA (1 мл) и смесь охлаждали до 0°C в течение 20 мин. Смесь концентрировали при пониженном давлении, три раза добавляя DCM, и использовали без последующей очистки. MS масса/заряд 295,2 (M+H)+.
[00250] Стадия d: К смеси 3-((3-амино-2-хлорфенил)тио)-6-хлорпиразин-2-амина (42 мг, 0,15 ммоль) и N-((S)-2,2-дифтор-8-азаспиро[4.5]декан-1-ил)-2-метилпропан-2-сульфинамида (60 мг, 0,15 ммоль) в пробирке для использования под давлением добавляли DIPEA (0,3 мл, 1,8 ммоль). Пробирку укупоривали и нагревали на масляной бане при 100°C в течение 16 ч. После охлаждения до к.т. реакционную смесь разбавляли соляным раствором и EtOAc и слои разделяли. Органический слой промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (20-100% EtOAc/гептана), получая указанное в заголовке соединение (18 мг) в виде коричневого масла. MS масса/заряд 545,1 (M+H)+.
Промежуточное соединение 12
(R)-8-(6-Амино-5-((3-амино-2-хлорфенил)тио)пиразин-2-ил)-N-((R)-1-(4-метилоксифенил)этил)-8-азаспиро[4.5]декан-1-амин
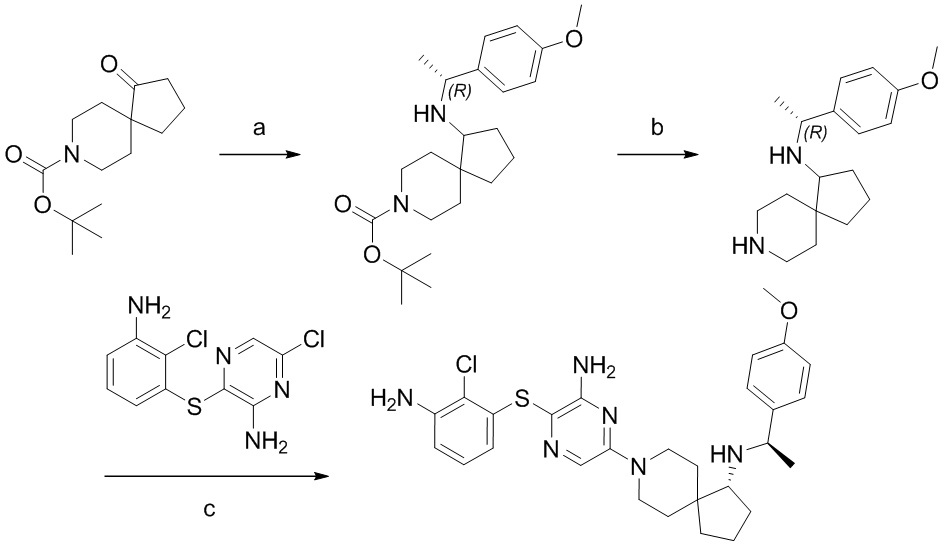
[00251] Стадия a: К раствору трет-бутил-1-оксо-8-азаспиро[4.5]декан-8-карбоксилата (1,15 г, 4,54 ммоль) и (R)-1-(4-метоксифенил)этанамина (961 мг, 6,36 ммоль) в DCE (3 мл) порциями добавляли цианоборогидрид натрия и перемешивали 16 ч при к.т. Смесь разбавляли насыщ. водным раствором NaHCO3 (5 мл) и экстрагировали с помощью EtOAc (3×10 мл). Объединенные органические фазы промывали солевым раствором и концентрировали при пониженном давлении. Полученный остаток, содержащий 9:1 смесь диастереомеров, очищали хроматографией на силикагеле (0-20% EtOAc/гептана), получая трет-бутил-1-(((R)-1-(4-метоксифенил)этил)амино)-8-азаспиро[4.5]декан-8-карбоксилат (основной диастереомер; 431 мг).1H ЯМР (400 МГц, DMSO-d6) δ ppm 7,18-7,24 (m, 2 H), 6,81-6,86 (m, 2 H), 3,76 (d, J=13,64 Гц, 1 H), 3,72 (s, 3 H), 3,64-3,70 (m, 2 H), 2,65-2,92 (m, 2 H), 2,05-2,14 (m, 1 H), 1,80-1,91 (m, 1 H), 1,65-1,75 (m, 1 H), 1,42-1,60 (m, 4 H), 1,40 (s, 9 H), 1,28-1,35 (m, 1 H), 1,20 (d, J=6,6 Гц, 3 H), 1,09-1,17 (m, 2 H), 0,80 (d, J=11,4 Гц, 1 H). MS масса/заряд 389,6 (M+H)+.
[00252] Стадия b: К раствору трет-бутил-1-(((R)-1-(4-метоксифенил)этил)амино)-8-азаспиро[4.5]декан-8-карбоксилат (основной диастереомер; 431 мг, 1,11 ммоль) в DCM (2 мл) добавляли TFA (2 мл) и перемешивали в течение 10 мин при к.т. Реакционную смесь концентрировали при пониженном давлении, еще раз добавляя DCM, затем разбавляли насыщ. водным раствором NaHCO3. Смесь экстрагировали, используя DCM (3×20 мл). Органическую фазу промывали соляным раствором, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая N-((R)-1-(4-метоксифенил)этил)-8-азаспиро[4.5]декан-1-амин, который использовали без последующей очистки.1H ЯМР (400 МГц, Метанол-d4) δ ppm 7,25 (d, J=8,6 Гц, 2 H), 6,87 (d, J=8,7 Гц, 2 H), 3,85-3,78 (m, 1 H), 3,78 (s, 3 H), 3,35 (m, 1 H), 3,28 (m, 1 H), 3,03 (m, 2 H), 2,63 (dd, J=9,6, 7,3 Гц, 1 H), 2,06-1,85 (m, 2 H), 1,83-1,69 (m, 2 H), 1,62 (m, 1 H), 1,54-1,38 (m, 4 H), 1,33 (d, J=6,6 Гц, 3 H), 1,31-1,23 (m, 1 H). MS масса/заряд 289,5 (M+H)+.
[00253] Стадия c: Как описано для Промежуточного соединения 1, аналогичную процедуру применяли, используя 3-((3-амино-2-хлорфенил)тио)-6-хлорпиразин-2-амин (0,77 г, 2,63 ммоль) и (R)-N-((R)-1-(4-метоксифенил)этил)-8-азаспиро[4.5]декан-1-амин (0,75 г, 2,60 ммоль), получая указанное в заголовке соединение (0,95 г). MS масса/заряд 539,3 (M+H)+.
Промежуточное соединение 13
N-(3-((3-Амино-5-хлорпиразин-2-ил)тио)-2-хлорфенил)-2-гидрокси-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-a]пиримидин-3-карбоксамид
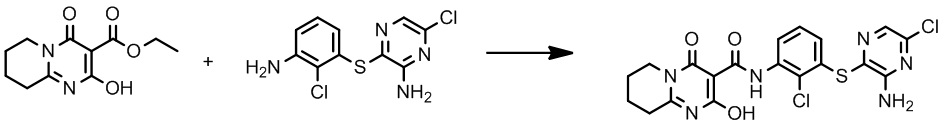
[00254] Суспензию этил-2-гидрокси-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-a]пиримидин-3-карбоксилата (1,5 г, 5,6 ммоль) и 3-((3-амино-2-хлорфенил)тио)-6-хлорпиразин-2-амина (1,0 г, 3,5 ммоль) в DMF (10 мл) нагревали при 160°C в течение 2,5 ч. После охлаждения реакционную смесь разделяли между EtOAc и соляным раствором и слои разделяли. Органический слой промывали водой и водн. слой экстрагировали еще раз, используя EtOAc. Объединенные органические слои высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Концентрат растирали с MeOH, получая указанное в заголовке соединение (1,36 г, чистота 90%) в виде желтого твердого вещества. MS масса/заряд 479,2 (M+H)+.
Промежуточное соединение 14
(3S,4S)-3-Метил-2-окса-8-азаспиро[4.5]декан-4-амин

[00255] Стадия a: В раствор диизопропиламина (23,4 мл, 166 ммоль) в THF (220 мл) при -10°C по каплям добавляли nBuLi (2,5 M раствор в гексане, 64,1 мл, 160 ммоль). После перемешивания в течение 30 мин по каплям добавляли 1-трет-бутил-4-этил-пиперидин-1,4-дикарбоксилат (27,5 г, 107 ммоль) в THF (50 мл) и полученную смесь перемешивали в течение 30 мин при 0°C. Добавляли (S)-2-((трет-бутилдиметилсилил)окси)пропаналь (20,47 мл, 102 ммоль) и смесь перемешивали в течение 1 ч при t 0°C и 1 ч при к.т. Реакционную смесь разбавляли насыщ. водн. раствором NaHCO3/H2O (1:4, 125 мл), добавляли EtOAc (50 мл) и фазы разделяли. Водную фазу дополнительно экстрагировали, используя EtOAc (3×100 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и растворитель удаляли при пониженном давлении. Полученный остаток использовали на следующей стадии без дополнительной очистки. MS масса/заряд 346,4 (M+H-Boc)+.
[00256] Стадия b: К раствору неочищенного 1-трет-бутил-4-этил-4-((2S)-2-((трет-бутилдиметилсилил)окси)-1-гидроксипропил)пиперидин-1,4-дикарбоксилата (95 г, 214 ммоль) в THF (600 мл) порциями добавляли LiBH4 (7,0 г, 321 ммоль) и полученную смесь перемешивали в течение 16 ч при к.т. После охлаждения до 0°C, добавляли насыщ. водн. NaHCO3/H2O (1:2, 150 мл) и полученную смесь интенсивно перемешивали до прекращения вспенивания. Добавляли EtOAc (100 мл), смесь отфильтровывали, фазы разделяли и водную фазу экстрагировали, используя EtOAc (3×50 мл). Объединенные органические фазы промывали соляным раствором, высушивали над Na2SO4, фильтровали и летучие соединения удаляли при пониженном давлении, получая трет-бутил-4-((2S)-2-((трет-бутилдиметилсилил)окси)-1-гидроксипропил)-4-(гидроксиметил)пиперидин-1-карбоксилат (64,8 г), который использовали на следующей стадии без дополнительной очистки.
[00257] Стадия c: Раствор трет-бутил-4-((2S)-2-((трет-бутилдиметилсилил)окси)-1-гидроксипропил)-4-(гидроксиметил)пиперидин-1-карбоксилата (64,8 г, 161 ммоль) и TBAF (1 М в THF, 242 мл, 242 ммоль) в THF (500 мл) перемешивали в течение 2 ч при к.т. Добавляли насыщ. водн. NaHCO3/H2O (1:2, 150 мл), фазы разделяли и водную фазу дополнительно экстрагировали, используя EtOAc (3×100 мл). Объединенные органические фазы промывали соляным раствором, высушивали над Na2SO4, фильтровали и летучие соединения удаляли при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (20-100% EtOAc/гептана), получая трет-бутил-4-((2S)-1,2-дигидроксипропил)-4-(гидроксиметил)пиперидин-1-карбоксилат (39,25 г) в виде полутвердого бесцветного масла.
[00258] Стадия d: К суспензии NaH (10,60 г, 424 ммоль) в THF (600 мл) при 0°C по каплям добавляли раствор трет-бутил-4-((2S)-1,2-дигидроксипропил)-4-(гидроксиметил)пиперидин-1-карбоксилата (35,06 г, 121 ммоль) и TsCl (23,10 г, 121 ммоль) в THF (200 мл). Полученную смесь перемешивали в течение 1 ч при 0°C. Медленно добавляли насыщ. водн. NH4Cl (~5 мл) при -20°C и реакционную смесь интенсивно перемешивали до прекращения вспенивания. Добавляли насыщ. водн. NH4Cl (100 мл), а затем соляной раствор (100 мл) и смесь экстрагировали, используя EtOAc (3×100 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая (3S)-трет-бутил-4-гидрокси-3-метил-2-окса-8-азаспиро[4.5]декан-8-карбоксилат (32,19 г), который использовали на следующей стадии без дополнительной очистки. MS масса/заряд 171,1 (M-Boc)-.
[00259] Стадия e: Раствор (3S)-трет-бутил-4-гидрокси-3-метил-2-окса-8-азаспиро[4.5]декан-8-карбоксилата (32,19 г, 119 ммоль) и периодинана Десса-Мартина (67,4 г, 154 ммоль) в DCM (300 мл) перемешивали в течение 2 ч при 0°C. После разогрева реакционной смеси до к. т. летучие удаляли при пониженном давлении и полученный остаток очищали хроматографией на силикагеле (0-40% EtOAc/гептана), получая (S)-трет-бутил-3-метил-4-оксо-2-окса-8-азаспиро[4.5]декан-8-карбоксилат (27,68 г) в виде бледно-желтого масла.1H ЯМР (400 МГц, Хлороформ-d) δ ppm 4,09 (d, J=9,60 Гц, 1 H), 3,66-3,86 (m, 4 H), 3,03 (ddd, J=13,8, 9,7, 3,8 Гц, 1 H), 2,90 (ddd, J=13,6, 10,2, 3,4 Гц, 1 H), 1,68 (ddd, J=13,8, 9,9, 4,3 Гц, 1 H), 1,41-1,59 (m, 2 H), 1,30-1,40 (m, 10 H), 1,20-1,25 (m, 3 H).
[00260] Стадия f: Раствор (3S)-трет-бутил-3-метил-4-оксо-2-окса-8-азаспиро[4.5]декан-8-карбоксилата (22,52 г мг, 84 ммоль), этоксида титана(IV) (70,1 мл, 334 ммоль), и (R)-2-метилпропан-2-сульфинамида (21 г, 173 ммоль) в THF (300 мл) перемешивали в течение 21 ч при 90°C. После охлаждения до -4°C добавляли MeOH (30 мл), а затем по каплям добавляли (поддерживая температуру реакции ниже 2°C) борогидрид лития (1,82 г, 84 ммоль) и полученную смесь перемешивали в течение 1 ч при -4°C. Медленно добавляли насыщ. водн. NH4Cl, а затем вносили EtOAc (500 мл). Полученную смесь интенсивно перемешивали в течение 15 мин при к.т., а затем фильтровали через подушку из Celite® (EtOAc). Летучие вещества удаляли при пониженном давлении и полученный остаток очищали хроматографией на силикагеле (0-100% EtOAc/гептана), получая (3S,4S)-трет-бутил-4-((R)-1,1-диметилэтилсульфинамидо)-3-метил-2-окса-8-азаспиро[4.5]декан-8-карбоксилат в виде диастереомерной смеси 95:5 (неосновным диастереомером является (3R,4S)-трет-бутил-4-((R)-1,1-диметилэтилсульфинамидо)-3-метил-2-окса-8-азаспиро[4.5]декан-8-карбоксилат).
[00261] Стадия g: Разделение диастереомеров выполняли с использованием хиральной SFC, как описано ниже: колонка: LC-4 30×250 мм, расход: 100 г в минуту, подвижная фаза: 30% MeOH в CO2, детектирование: 225 нм, Rt: 0,95 мин (неосновной диастереомер Rt: 0,55 мин), получая (3S,4S)-трет-бутил-4-((R)-1,1-диметилэтилсульфинамидо)-3-метил-2-окса-8-азаспиро[4.5]декан-8-карбоксилат (19 г). MS масса/заряд 375,2.
[00262] Стадия h: Смесь (3S,4S)-трет-бутил 4-((R)-1,1-диметилэтилсульфинамидо)-3-метил-2-окса-8-азаспиро[4.5]декан-8-карбоксилата (51 мг, 0,136 ммоль) и HCl (4 M в диоксане, 340 мкл, 1,362 ммоль) в MeOH (5 мл) перемешивали в течение 1 ч при 40°C. После охлаждения до к.т. летучие соединения удаляли при пониженном давлении, получая (3S,4S)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин, который использовали на следующей стадии без дополнительной очистки. MS масса/заряд 171,1 (M+H)+.
Промежуточное соединение 15
(R)-3,3-Дифтор-8-азаспиро[4.5]декан-1-амин

трет-бутил-1-оксо-8-азаспиро[4.5]дец-2-ен-8-карбоксилат

[00263] Стадия a: Смесь трет-бутил-4-формилпиперидин-1-карбоксилата (35,0 г, 164 ммоль), трет-бутоксида лития (15,77 г, 197 ммоль) и аллилбромида (11,54 мл, 189 ммоль) в DMF (328 мл) перемешивали в течение 1 ч при 0°C. Смесь выливали в разделительную воронку, содержащую насыщ. водн. NH4Cl/H2O (1:1, 500 мл), и экстрагировали, используя Et2O (5×50 мл). Объединенные органические фазы сушили над MgSO4, фильтровали и удаляли летучие вещества при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (0-25% EtOAc/гептана), получая трет-бутил-4-аллил-4-формилпиперидин-1-карбоксилат (24 г) в виде бесцветного масла.1H ЯМР (400 МГц, Хлороформ-d) δ ppm 9,52 (s, 1 H), 5,53-5,76 (m, 1 H), 4,96-5,19 (m, 2 H), 3,80 (br. s., 2 H), 2,97 (t, J=11,5 Гц, 2 H), 2,26 (d, J=7,3 Гц, 2 H), 1,95 (dt, J=13,7, 3,1 Гц, 2 H), 1,38-1,58 (m, 11 H).
[00264] Стадия b: К раствору трет-бутил-4-аллил-4-формилпиперидин-1-карбоксилата (24 г, 95 ммоль) в THF (300 мл) при -78°C в атмосфере N2 добавляли винилмагния бромид (1 M в THF, 118 мл, 118 ммоль). Полученный раствор медленно разогревали до к.т. в течение 1 ч. Смесь выливали в разделительную воронку, содержащую насыщ. водн. NH4Cl (250 мл), и экстрагировали, используя EtOAc (4×50 мл). Объединенные органические фазы сушили над MgSO4, фильтровали и удаляли летучие вещества при пониженном давлении, получая трет-бутил-4-аллил-4-(1-гидроксиаллил)пиперидин-1-карбоксилат (26,7 г, 95 ммоль) в виде бесцветного масла.1H ЯМР (400 МГц, Хлороформ-d) δ ppm 9,52 (s, 1 H), 5,56-5,75 (m, 1 H), 5,05-5,18 (m, 2 H), 3,80 (br. s., 2 H), 2,97 (t, J=11,5 Гц, 2 H), 2,26 (d, J=7,3 Гц, 2 H), 1,96 (dt, J=13,8, 3,1 Гц, 2 H), 1,49-1,60 (m, 2 H), 1,41-1,49 (m, 9 H). Это соединение использовали на следующей стадии без дополнительной очистки.
[00265] Стадия c: Смесь трет-бутил-4-аллил-4-(1-гидроксиаллил)пиперидин-1-карбоксилата (26,7 г, 95 ммоль) и периодинана Десса-Мартина (44,3 г, 105 ммоль) в DCM (380 мл) перемешивали в течение 1 ч при к.т. Смесь выливали в разделительную воронку, содержащую насыщ. водн. NaHCO3/Na2SO3 (1:1, 300 мл) и экстрагировали, используя DCM (4×50 мл). Объединенные органические фазы сушили над MgSO4, фильтровали и удаляли летучие вещества при пониженном давлении. Полученный твердый продукт суспендировали в гептане (250 мл) и обрабатывали ультразвуком в течение 5 минут. Белую суспензию отфильтровывали через подушку из Celite® и удаляли летучие вещества при пониженном давлении, получая трет-бутил-4-акрилоил-4-аллилпиперидин-1-карбоксилат (26,5 г) в виде желтого масла.1H ЯМР (400 МГц, Хлороформ-d) δ ppm 6,81 (dd, J=16,9, 10,4 Гц, 1 H), 6,40 (dd, J=16,8, 1,9 Гц, 1 H), 5,71 (dd, J=10,4, 2,0 Гц, 1 H), 5,46-5,66 (m, 1 H), 4,91-5,14 (m, 2 H), 3,78 (br. s., 2 H), 2,96 (br. s., 2 H), 2,25-2,39 (m, 2 H), 1,97-2,15 (m, 2 H), 1,37-1,57 (m, 11 H). Это соединение использовали на следующей стадии без дополнительной очистки.
[00266] Стадия d: К раствору трет-бутил-4-акрилоил-4-аллилпиперидин-1-карбоксилата (26,5 г, 95 ммоль) в толуоле (дегазированный, 850 мл) добавляли катализатор Граббса II (2,02 г, 2,38 ммоль) в толуоле (дегазированный, 100 мл). Полученную смесь перемешивали 45 мин при 85°C. Растворитель удаляли при пониженном давлении и полученный остаток очищали хроматографией на силикагеле (0-40% EtOAc/гептана), получая трет-бутил-1-оксо-8-азаспиро[4.5]дец-2-ен-8-карбоксилат (20,76 г, 83 ммоль) в виде коричневого твердого вещества. Раствор этого соединения и DDQ (2,3-дихлор-5,6-дициано-1,4-бензохинона) (565 мг, 2,49 ммоль) в толуоле (540 мл) перемешивали в течение 15 мин при к.т. Полученный красный раствор фильтровали через слой Сelite®. Добавляли уголь (200 г) и полученную суспензию перемешивали в течение 2 ч при к.т. Смесь фильтровали через подушку из Celite® и летучие соединения удаляли при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (0-40% EtOAc/гептана), получая трет-бутил-1-оксо-8-азаспиро[4.5]дец-2-ен-8-карбоксилат (15,6 г) в виде белого твердого вещества.1H ЯМР (400 МГц, Хлороформ-d) δ ppm 7,63-7,74 (m, 1 H), 6,20 (dt, J=5,8, 2,2 Гц, 1 H), 3,99-4,25 (m, 2 H), 2,92 (t, J=11,6 Гц, 2 H), 2,63 (s, 2 H), 1,72-1,86 (m, 2 H), 1,49 (s, 9 H), 1,29 (d, J=12,9 Гц, 2 H).
[00267] (1R,3R)-трет-Бутил-3-((трет-бутилдиметилсилил)окси)-1-((R)-1,1-диметилэтилсульфинамидо)-8-азаспиро[4.5]декан-8-карбоксилат)

[00268] Стадия a: Смесь Cu(I)Cl (142 мг, 1,432 ммоль), (S)-TolBINAP (972 мг, 1,432 ммоль) и трет-бутоксида натрия (138 мг, 1,432 ммоль) в THF (60 мл) перемешивали в течение 30 мин при к.т. Добавляли B2pin2 (13,34 г, 52,5 ммоль) в THF (20 мл) и полученную смесь перемешивали в течение 10 мин при к.т. Добавляли трет-бутил-1-оксо-8-азаспиро[4.5]дец-2-ен-8-карбоксилат (12,0 г, 47,7 ммоль) в THF (50 мл), а затем - MeOH (3,9 мл, 95 ммоль). Полученную смесь перемешивали в течение 16 ч при к.т. Добавляли H2O (150 мл), а затем - перборат натрия (36,7 г, 239 ммоль) и полученную смесь интенсивно перемешивали в течение 1 ч при к.т. Полученную суспензию зеленого цвета фильтровали через подушку из Celite®, выливали в разделительную воронку, содержащую насыщ. водн. NaHCO3/насыщ. водн. Na2SO3 (1:1, 300 мл), и экстрагировали, используя EtOAc (4×40 мл). Объединенные органические фазы сушили над MgSO4, фильтровали и удаляли летучие вещества при пониженном давлении, получая сырой (R)-трет-бутил-3-гидрокси-1-оксо-8-азаспиро[4.5]декан-8-карбоксилат. Исследование энантиомерного состава этой смеси показало 90% ee (Rt(S): 1,59 мин, Rt(R): 1,80 мин; хиральная SFC; колонка: IA 4,6×100 мм, расход: 70 г в минуту, подвижная фаза: 5-55% MeOH в CO2, детектирование: 220 мн УФ).
[00269] Смесь неочищенного (R)-трет-бутил-3-гидрокси-1-оксо-8-азаспиро[4.5]декан-8-карбоксилата (теор. 47,7 ммоль), имидазола (4,87 г, 71,6 ммоль) и TBSCl (8,99 г, 59,6 ммоль) в DMF (120 мл) перемешивали в течение 16 ч при к.т. Реакционную смесь выливали в разделительную воронку, содержащую насыщ. водн. NH4Cl/H2O (1:1, 250 мл), и экстрагировали, используя Et2O (5×50 мл). Объединенные органические фазы сушили над MgSO4, фильтровали и удаляли летучие вещества при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (0-30% EtOAc/гептана), получая (R)-трет-бутил-3-((трет-бутилдиметилсилил)окси)-1-оксо-8-азаспиро[4.5]декан-8-карбоксилат (13,11 г) в виде бесцветного масла, которое отвердевало при отстаивании.
[00270] Стадия b: Раствор (R)-трет-бутил-3-((трет-бутилдиметилсилил)окси)-1-оксо-8-азаспиро[4.5]декан-8-карбоксилата (8 г, 20,86 ммоль), этоксида титана(IV) (17,49 мл, 83,0 ммоль) и (R)-2-метилпропан-2-сульфинамида (5,06 г, 41,7 ммоль) в THF (100 мл) перемешивали в течение 16 ч при 65°C. После охлаждения до -78°C добавляли MeOH (15 мл), а затем борогидрид лития (1,363 г, 62,6 ммоль). Полученную смесь перемешивали в течение 16 ч при -78°C. Медленно добавляли насыщ. водн. NH4Cl, чтобы нейтрализовать избыток борогидрида, а затем вносили EtOAc (100 мл). Полученную смесь интенсивно перемешивали в течение 15 мин, а затем фильтровали через подушку из Celite®. Летучие удаляли при пониженном давлении и полученный остаток очищали хроматографией на силикагеле (0-100% EtOAc/гептана), получая (1R,3R)-трет-бутил-3-((трет-бутилдиметилсилил)окси)-1-((R)-1,1-диметилэтилсульфинамидо)-8-азаспиро[4.5]декан-8-карбоксилат (5,3 г) в виде белого твердого вещества. MS масса/заряд 489,3 (M+H)+.
[00271] 3,3-Дифтор-8-азаспиро[4.5]декан-1-амин

[00272] Стадия a: Смесь трет-бутил-3-((трет-бутилдиметилсилил)окси)-1-(1,1-диметилэтилсульфинамидо)-8-азаспиро[4.5]декан-8-карбоксилата (365 мг, 0,746 ммоль) и HCl (4 M в диоксане, 1,86 мл, 7,46 ммоль) в MeOH (4 мл) перемешивали 1 ч при 40°C. После охлаждения до к.т. летучие вещества удаляли при пониженном давлении, получая белое твердое вещество. MS масса/заряд 171,1 (M+H)+. Смесь этого остатка, DIPEA (2,6 мл, 14,92 ммоль) и Boc2O (407 мг, 1,865 ммоль) в THF (15 мл) перемешивали в течение 16 ч при к.т. Смесь выливали в разделительную воронку, содержащую насыщ. водн. NH4Cl и экстрагировали, используя Et2O (5×10 мл). Объединенные органические фазы сушили над MgSO4, фильтровали и удаляли летучие вещества при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (10-80% EtOAc/гептана), получая трет-бутил-1-((трет-бутоксикарбонил)амино)-3-гидрокси-8-азаспиро[4.5]декан-8-карбоксилат (275 мг, 0,742 ммоль). МS масса/заряд 271,3 (M+H-Boc)+.
[00273] Стадия b: Смесь трет-бутил-1-((трет-бутоксикарбонил)амино)-3-гидрокси-8-азаспиро[4.5]декан-8-карбоксилата (275 мг, 0,742 ммоль) и периодинана Десса-Мартина (472 мг, 1,113 ммоль) в DСM (7,5 мл) перемешивали в течение 2 ч при 0°C. Смесь выливали в разделительную воронку, содержащую насыщ. водн. NаHCO3, и экстрагировали, используя DСM (3×10 мл). Объединенные органические фазы сушили над MgSO4, фильтровали и удаляли летучие вещества при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (5-75% EtOAc/гептана), получая трет-бутил-1-((трет-бутоксикарбонил)амино)-3-оксо-8-азаспиро[4.5]декан-8-карбоксилат (135 мг).1H ЯМР (400 МГц, Хлороформ-d) δ ppm 4,57 (d, J=9,1 Гц, 1 H), 4,16 (d, J=8,1 Гц, 1 H), 3,89-4,08 (m, 2 H), 2,77-2,93 (m, 2 H), 2,71 (dd, J=19,0, 8,1 Гц, 1 H), 2,50 (d, J=18,2 Гц, 1 H), 2,07-2,24 (m, 2 H), 1,76 (td, J=12,8, 4,7 Гц, 1 H), 1,58-1,70 (m, 1 H), 1,42-1,53 (m, 18 H), 1,25-1,38 (m, 1 H).
[00274] Стадия c: Смесь трет-бутил-1-((трет-бутоксикарбонил)амино)-3-оксо-8-азаспиро[4.5]декан-8-карбоксилата (95 мг, 0,258 ммоль) и DeoxoFluor (190 мкл, 1,031 ммоль) в DСM (1 мл) перемешивали в течение 48 ч при 50°C. Смесь выливали в разделительную воронку, содержащую насыщ. водн. NаHCO3/лед, и экстрагировали, используя EtOAc (3×5 мл). Объединенные органические фазы сушили над MgSO4, фильтровали и удаляли летучие вещества при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (0-30% EtOAc/гептана), получая трет-бутил-1-((трет-бутоксикарбонил)амино)-3,3-дифтор-8-азаспиро[4.5]декан-8-карбоксилат (52 мг, 0,133 ммоль).1H ЯМР (400 МГц, Хлороформ-d) δ ppm 4,55 (d, J=9,4 Гц, 1 H), 3,78-4,02 (m, 3 H), 2,64-2,86 (m, 2 H), 2,38-2,59 (m, 1 H), 2,10-2,32 (m, 1 H), 1,79-2,10 (m, 2 H), 1,58 (qd, J=12,7, 3,8 Гц, 1 H), 1,27-1,52 (m, 21 H).
[00275] трет-Бутил-(R)-1-((трет-бутоксикарбонил)амино)-3,3-дифтор-8-азаспиро[4.5]декан-8-карбоксилат синтезировали, используя вышеуказанную процедуру или модификации вышеуказанной процедуры с использованием оптически чистого хирального (1R,3R)-трет-бутил-3-((трет-бутилдиметилсилил)окси)-1-((R)-1,1-диметилэтилсульфинамидо)-8-азаспиро[4.5]декан-8-карбоксилата в качестве исходного материала.
[00276] Стадия d: К суспензии трет-бутил-(R)-1-((трет-бутоксикарбонил)амино)-3,3-дифтор-8-азаспиро[4.5]декан-8-карбоксилата (252 мг, 0,644 ммоль) в 1,4-диоксане (4 мл) добавляли HCl (1,6 мл, 4 M в 1,4-диоксане). Смесь перемешивали при к.т. в течение 16 часов, а затем концентрировали при пониженном давлении до получения белого твердого вещества. В небольшой колбе Эрленмейера растворяли Na2CO3 (400 мг) в 2 мл воды и вносили неочищенное твердое вещество, перемешивая до полного растворения. Раствор переносили в разделительную воронку, разбавляли, используя 6 мл CF3CH2OH (20% в DCM), и смесь энергично встряхивали. Верхний водный слой имел щелочной pH (~10). Слои разделяли и водн. слой экстрагировали, используя DCM х 3 (20% трифторэтанола, 8 мл). Объединенные органические экстракты сушили и концентрировали при пониженном давлении до получения полутвердого вещества коричневого цвета, которое использовали на следующей стадии без дополнительной очистки. МS 191,2 масса/заряд (M+H)+.
Промежуточное соединение 16
(1S,2S,3R)-2-Фтор-3-метил-8-азаспиро[4.5]декан-1-амин

[00277] Стадия a: К суспензии трет-бутил-1-оксо-8-азаспиро[4.5]дец-2-ен-8-карбоксилата (4,2 г, 16,71 ммоль) при 0°C в атмосфере N2 и Cu(I)I (6,37 г, 33,4 ммоль) в Et2O (100 мл) добавляли MeLi (1,6 M в THF, 31,3 мл, 50,1 ммоль). После перемешивания в течение 90 мин при 0°C смесь выливали в разделительную воронку, содержащую насыщ. водн. NH4Cl и экстрагировали, используя EtOАс (3×15 мл). Объединенные органические фазы сушили над MgSO4, фильтровали и удаляли летучие вещества при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (0-50% EtOAc/гептана), получая трет-бутил-3-метил-1-оксо-8-азаспиро[4.5]дец-2-ен-8-карбоксилат (4,23 г) в виде бесцветного масла.1H ЯМР (400 МГц, Хлороформ-d) δ ppm 3,89-4,00 (m, 1 H), 3,83 (d, J=13,4 Гц, 1 H), 3,11 (ddd, J=13,6, 10,4, 3,3 Гц, 1 H), 2,99 (ddd, J=13,6, 10,4, 3,5 Гц, 1 H), 2,47-2,59 (m, 1 H), 2,19-2,36 (m, 2 H), 1,74-1,97 (m, 2 H), 1,50-1,65 (m, 2 H), 1,48 (s, 9 H), 1,33-1,44 (m, 2 H), 1,17 (d, J=6,3 Гц, 3 H).
[00278] Стадия b: Смесь трет-бутил-3-метил-1-оксо-8-азаспиро[4.5]дец-2-ен-8-карбоксилата (4,23 г, 15,82 ммоль) и TFA (17 мл) в DCM (80 мл) перемешивали в течение 30 мин при к.т. Летучие вещества удаляли при пониженном давлении. Смесь полученного остатка, DIPEA (13,82 мл, 79 ммоль) и бензилхлорформиата (3,39 мл, 23,73 ммоль) в DCM (80 мл) перемешивали в течение 16 ч при к.т. Смесь выливали в разделительную воронку, содержащую насыщ. водн. NH4Cl и экстрагировали, используя DCM (3×25 мл). Объединенные органические фазы сушили над MgSO4, фильтровали и удаляли летучие вещества при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (0-40% EtOAc/гептана), получая бензил-3-метил-1-оксо-8-азаспиро[4.5]декан-8-карбоксилат (4,58 г) в виде светло-желтого масла. MS масса/заряд 302,2 (M+H)+.
[00279] Стадия c: Бензил-3-метил-1-оксо-8-азаспиро[4.5]декан-8-карбоксилат (4,58 г, 15,20 ммоль) дополнительно очищали с помощью хиральной SFC, как указано далее: колонка: IA 21×250 мм, расход: 70 г в минуту, подвижная фаза: 45% (9:1 EtOH/MeCN) в CO2, детектирование: 220 нм УФ, получая (R)-бензил-3-метил-1-оксо-8-азаспиро[4.5]декан-8-карбоксилат (2,02 г, 6,70 ммоль), Rt: 2,0 мин; и (S)-бензил-3-метил-1-оксо-8-азаспиро[4.5]декан-8-карбоксилат (2,11 г, 7,0 ммоль), Rt: 3,6 мин.
[00280] Стадия d: Раствор LiHMDS (1 M в THF, 8,87 мл, 8,87 ммоль) вносили в THF (36 мл). Раствор охлаждали до -78°C, затем добавляли раствор (R)-бензил-3-метил-1-оксо-8-азаспиро[4.5]декан-8-карбоксилата (2,43 г, 8,06 ммоль) в THF (12 мл) к вышеуказанному раствору при -78°C. Полученную смесь перемешивали при -78°C в течение 30 мин. Затем по каплям добавляли раствор N-фтор-N-(фенилсульфонил)бензолсульфонамида (2,80 г, 8,87 ммоль) при -78°C и полученный раствор перемешивали при -78°C в течение 30 мин. К этому раствору добавляли смесь 1:1 воды и насыщ. раствора NaHCO3 (20 мл) и смеси давали нагреться до к.т. Раствор экстрагировали, используя EtOAc х 3, и объединенную органическую фазу промывали соляным раствором. Органическую фазу сушили над MgSO4, фильтровали и удаляли летучие вещества при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (0-50% EtOAc/гептана), получая (2S,3R)-бензил-2-фтор-3-метил-1-оксо-8-азаспиро[4.5]декан-8-карбоксилат (1,89 г, содержит 40% диастереомера). MS масса/заряд 320,2 (M+H)+.
[00281] Стадия e: К раствору (2S,3R)-бензил-2-фтор-3-метил-1-оксо-8-азаспиро[4.5]декан-8-карбоксилата (1,89 г, 5,92 ммоль, содержит 40% диастереомера) в THF/MeOH (9:1, 20 мл) добавляли раствор тетрагидробората лития (2 M в THF, 11,84 мл, 23,67 ммоль) при -78°C. Полученную смесь перемешивали при -78°C в течение 30 мин. Медленно прибавляли раствор насыщ. водн. NH4Cl и давали нагреться до к.т. Раствор экстрагировали, используя EtOAc х 3, и объединенную органическую фазу промывали соляным раствором. Органическую фазу сушили над MgSO4, фильтровали и удаляли летучие вещества при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (0-50% EtOAc/гептана), получая (1R,2S,3R)-бензил-2-фтор-1-гидрокси-3-метил-8-азаспиро[4.5]декан-8-карбоксилат (970 мг). MS масса/заряд 322,2 (M+H)+.1H ЯМР (400 МГц, Метанол-d4) δ ppm 7,37-7,28 (m, 5 H), 5,10 (s, 2 H), 4,47 (dt, J=54,4, 4,7 Гц, 1 H), 3,86 (d, J=12,9 Гц, 2 H), 3,65 (dd, J=18,0, 4,7 Гц, 1 H), 3,20-3,03 (m, 2 H), 2,39-2,21 (m, 1 H), 2,20-2,10 (m, 1 H), 1,75-1,60 (m, 2 H), 1,45 (d, J=13,4 Гц, 1 H), 1,29 (d, J=13,1 Гц, 1 H), 1,08 (d, J=7,1 Гц, 3 H), 0,96 (dd, J=13,3, 8,5 Гц, 1 H).
[00282] Стадия f: К раствору бензил(1R,2S,3R)-2-фтор-1-гидрокси-3-метил-8-азаспиро[4.5]декан-8-карбоксилата (760 мг, 2,365 ммоль) в THF (16,5 мл) добавляли трифенилфосфин (744 мг, 2,85 ммоль) и DIAD (0,557 мл, 2,84 ммоль). Полученную смесь перемешивали при 0°C в течение 20 мин. В реакционную смесь добавляли дифенилфосфоразидат (0,787 мл, 3,55 ммоль). Реакционной смеси давали нагреться до к.т. и перемешивали в течение 18 ч при к.т. Реакционную смесь выливали в разделительную воронку, содержащую EtOAc (30 мл). Органическую фазу промывали, используя насыщ. водн. NH4Cl и соляной раствор. Органические фазы сушили над MgSO4, фильтровали и удаляли летучие вещества при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (0-50% EtOAc/гептана), получая бензил-(1S,2S,3R)-1-азидо-2-фтор-3-метил-8-азаспиро[4.5]декан-8-карбоксилат (432 мг). MS масса/заряд 347,2 (M+H)+.1H ЯМР (400 МГц, Хлороформ-d) δ ppm 7,43-7,31 (m, 5 H), 5,15 (s, 2 H), 4,48 (dt, J=54,4, 7,5 Гц, 1 H), 3,93 (s, 2 H), 3,61 (dd, J=16,1, 6,9 Гц, 1 H), 3,13-2,95 (m, 2 H), 2,31-2,13 (m, 1 H), 1,96 (dd, J=13,1, 9,3 Гц, 1 H), 1,81-1,64 (m, 2 H), 1,47 (s, 1 H), 1,32-1,19 (m, 2 H), 1,16 (d, J=6,7 Гц, 3 H).
[00283] Стадия g: Суспензию Pd/C (10 (вес.)%, 65 мг) и бензил-(1S,2S,3R)-1-азидо-2-фтор-3-метил-8-азаспиро[4.5]декан-8-карбоксилата (423 мг, 1,221 ммоль) в EtOH (12,2 мл) интенсивно перемешивали в атмосфере H2 (баллон) в течении 16 ч. Реакционную смесь фильтровали через подушку из Celite® и удаляли летучие вещества при пониженном давлении, получая (1S,2S,3R)-2-фтор-3-метил-8-азаспиро[4.5]декан-1-амин (235 мг). MS масса/заряд 187,2 (M+H)+.1H ЯМР (400 МГц, Хлороформ-d) δ ppm 4,15 (dt, J=55,5, 8,1 Гц, 1 H), 2,95 (dt, J=12,5, 3,7 Гц, 2 H), 2,87 (dd, J=16,6, 8,0 Гц, 1 H), 2,74 (tdd, J=12,4, 7,3, 2,8 Гц, 2 H), 2,19-2,02 (m, 1 H), 1,95 (dd, J=13,4, 8,4 Гц, 1 H), 1,71-1,48 (m, 4 H), 1,34-1,23 (m, 3 H), 1,18-1,09 (m, 4 H).
Промежуточное соединение 17
трет-Бутил-((1-(5-амино-6-((3-амино-2-хлорфенил)тио)-1,2,4-триазин-3-ил)-4-метилпиперидин-4-ил)метил)карбамат
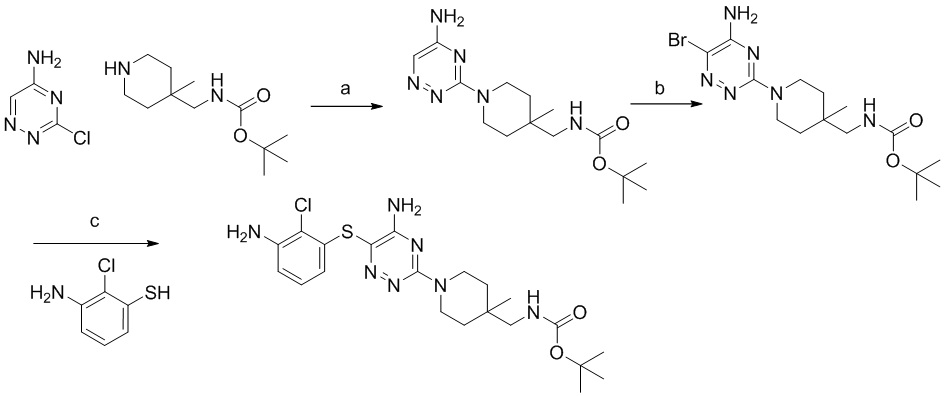
[00284] Стадия a: Раствор 3-хлор-1,2,4-триазин-5-амина (1,3 г, 9,96 ммоль), трет-бутил-((4-метилпиперидин-4-ил)метил)карбамата (2,7 г, 11,82 ммоль) и N-метилморфолина (3,3 мл, 30,0 ммоль) в NMP (20 мл) нагревали в укупоренном стеклянном реакторе при 130°C в течение 2 ч. После охлаждения реакционную смесь разбавляли, используя EtOAc (250 мл), промывали водой и соляным раствором, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Концентрат очищали хроматографией на силикагеле (0-50% EtOAc/DCM), получая трет-бутил-((1-(5-амино-1,2,4-триазин-3-ил)-4-метилпиперидин-4-ил)метил)карбамат (2,43 г). MS масса/заряд 323,5 (M+H)+.
[00285] Стадия b: К суспензии трет-бутил-((1-(5-амино-1,2,4-триазин-3-ил)-4-метилпиперидин-4-ил)метил)карбамата (2,4 г, 7,44 ммоль) в хлороформе (100 мл) при 0°C добавляли N-бромсукцинимид (1,5 г, 8,53 г). Через 1 ч при 0°C реакционную смесь концентрировали при пониженном давлении, растворяли в теплом DCM (50 мл), разбавляли, используя EtOAc (300 мл), промывали 5%-ным водн. раствором K2CO3, водой и соляным раствором. Органические фракции высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Используя хроматографию на силикагеле (0-10% MeOH/DCM), получали трет-бутил-((1-(5-амино-6-бром-1,2,4-триазин-3-ил)-4-метилпиперидин-4-ил)метил)карбамат (2,88 г). MS масса/заряд 401,5 (M+H)+.
[00286] Стадия c: Смесь 3-амино-2-хлорбензолтиола гидрохлорида (1,71 г, 8,72 ммоль), трет-бутил-(1-(5-амино-6-бром-1,2,4-триазин-3-ил)-4-метилпиперидин-4-ил)карбамата (2,80 г, 6,98 ммоль), Cu(I)I (0,27 г, 1,40 ммоль), TMEDA (4,21 мл, 27,9 ммоль) в диоксане (30 мл) облучали в микроволновом реакторе в течение 2 ч при 125°C. После охлаждения реакционную смесь разбавляли, используя DCM/MeOH (9:1, 100 мл) и фильтровали через пробку из Celite®. Фильтрат концентрировали при пониженном давлении, повторно суспендировали в EtOAc (200 мл), промывали водой, соляным раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Концентрат очищали хроматографией на силикагеле (0-50% EtOAc/гептана), получая указанное в заголовке соединение (3,29 г). MS масса/заряд 480,4 (M+H)+.
Промежуточное соединение 18
Этил-4-гидрокси-2-оксо-2H-пиримидо[2,1-a]изохинолин-3-карбоксилат

[00287] Стадия a: К суспензии гидроксиламина гидрохлорида (5,2 г, 75 ммоль) в EtOH/воде (3:2, 20 мл) добавляли мелко перемолотый KOH (4,2 г, 75 ммоль). После перемешивания в течение 1 ч при к.т. суспензию охлаждали до 0°C и отфильтровывали, чтобы удалить KCl. К фильтрату при 0°C по каплям добавляли диэтил-(2-этоксиметилен)малонат (5,4 г, 25 ммоль), растворенный в EtOH (4 мл). Реакционную смесь перемешивали в течение ночи при к.т., а затем нагревали при 75°C в течение 2 ч. После охлаждения до 50°C реакционную смесь подкисляли конц. HCl (25 мл) и перемешивали при 50°C в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении до половины ее исходного объема и полученный осадок отфильтровывали и сушили с получением этил-5-оксо-2,5-дигидроизоксазол-4-карбоксилата в виде белого порошка (3,5 г). MS масса/заряд 158,0 (M+H)+.
[00288] Стадия b: Суспензию 1-хлоризохинолина (0,82 г, 5,0 ммоль) и этил-5-оксо-2,5-дигидроизоксазол-4-карбоксилата (0,79 г, 5,0 ммоль) в EtOH (20 мл) облучали в микроволновом реакторе в течение 1 ч при 100oC. После охлаждения полученный осадок отфильтровывали и высушивали. Сырой продукт растворяли в 5% MeOH в DCM (15 мл) и очищали хроматографией на силикагеле (0-20% EtOAc/DCM) с получением этил-2-(изохинолин-1-ил)-5-оксо-2,5-дигидроизоксазол-4-карбоксилата (0,43 г). MS масса/заряд 285,1 (M+H)+.
[00289] Стадия c: К раствору этил-2-(изохинолин-1-ил)-5-оксо-2,5-дигидроизоксазол-4-карбоксилата (0,79 г, 2,78 ммоль) в THF (20 мл) при к.т. добавляли раствор Na2CO3 (1,4 г, 2,8 ммоль) в воде (20 мл). Полученную реакционную смесь, состоящую из двух фаз, перемешивали при к.т. в течение 3 ч. THF удаляли и оставшуюся реакционную смесь подкисляли до pH 1, используя 6 М водн. HCl. Подкисленную реакционную смесь экстрагировали, используя EtOAc (2х), и объединенные органические слои промывали водой, соляным раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Сырой продукт растворяли в EtOAc/DCM (10%) и очищали хроматографией на силикагеле (0-20% EtOAc/DCM), получая указанное в заголовке соединение (0,43 г). MS масса/заряд 285,1 (M+H)+.
Промежуточное соединение 19
Этил-1-гидрокси-3-оксо-6а,10а-дигидро-3H-пиримидо[1,2-а]хинолин-2-карбоксилат
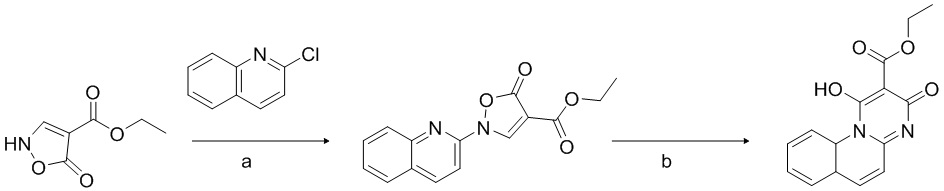
[00290] Стадия a: Суспензию 2-хлоризохинолина (0,82 г, 5,0 ммоль) и этил-5-оксо-2,5-дигидроизоксазол-4-карбоксилата (0,79 г, 5,0 ммоль) в EtOH (20 мл) облучали в микроволновом реакторе в течение 1 ч при 100oC. После охлаждения до к.т. реакционную смесь концентрировали при пониженном давлении, повторно растворяли в 5% MeOH/DCM (15 мл) и очищали хроматографией на силикагеле (0-20% EtOAc/DCM), получая 0,78 г этил-5-оксо-2-(хинолин-2-ил)-2,5-дигидроизоксазол-4-карбоксилата. MS масса/заряд 285,1 (M+H)+.
[00291] Стадия b: К раствору этил-5-оксо-2-(хинолин-2-ил)-2,5-дигидроизоксазол-4-карбоксилата (0,20 г, 0,70 ммоль) в THF (50 мл) при к.т. добавляли раствор Na2CO3 (0,85 г, 0,80 ммоль) в воде (5 мл). Полученную смесь перемешивали при к.т. в течение 3 ч. THF удаляли при пониженном давлении и реакционную смесь подкисляли до pH 1, используя 2М водн. HCl. Реакционную смесь экстрагировали, используя EtOAc (2х), и объединенные органические слои промывали водой, соляным раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Сырой продукт растворяли в 5% МеOН/DCM и очищали хроматографией на силикагеле (0-10% EtOAc/DCM), получая указанное в заголовке соединение (0,054 г). MS масса/заряд 285,1 (M-H)+.
Промежуточное соединение 20
трет-Бутил-(1-(5-амино-6-((3-амино-2-хлорфенил)тио)-1,2,4-триазин-3-ил)-4-метилпиперидин-4-ил)карбамат
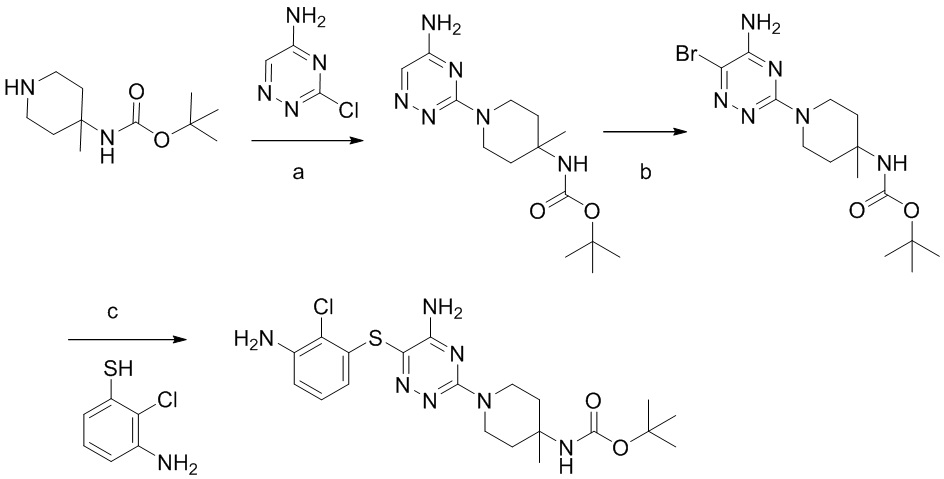
[00292] Стадия a: Как описано для Промежуточного соединения 17, аналогичную процедуру применяли, используя 3-хлор-1,2,4-триазин-5-амин (3,0 г, 23,0 ммоль) и трет-бутил-(4-метилпиперидин-4-ил)карбамат (9,9 г), получая 7,4 г. MS масса/заряд 309,4 (M+H)+.
[00293] Стадия b: Как описано для Промежуточного соединения 17, аналогичную процедуру применяли, используя трет-бутил-(1-(5-амино-1,2,4-триазин-3-ил)-4-метилпиперидин-4-ил)карбамат (3,8 г, 12,3 ммоль), получая трет-бутил-(1-(5-амино-6-бром-1,2,4-триазин-3-ил)-4-метилпиперидин-4-ил)карбамат (3,5 г). MS масса/заряд 389,2 (M+H)+.
[00294] Стадия c: Как описано для Промежуточного соединения 17, аналогичную процедуру применяли, используя 3-амино-2-хлорбензолтиол (0,32 г, 1,62 ммоль) и трет-бутил-(1-(5-амино-6-бром-1,2,4-триазин-3-ил)-4-метилпиперидин-4-ил)карбамат (0,50 г, 1,30 ммоль), получая указанное в заголовке соединение (0,41 г). MS масса/заряд 466,4 (M+H)+.
Промежуточное соединение 21
(R)-8-(5-Амино-6-((3-амино-2-хлорфенил)тио)-1,2,4-триазин-3-ил)-N-((R)-1-(4-метоксифенил)этил)-8-азаспиро[4.5]декан-1-амин
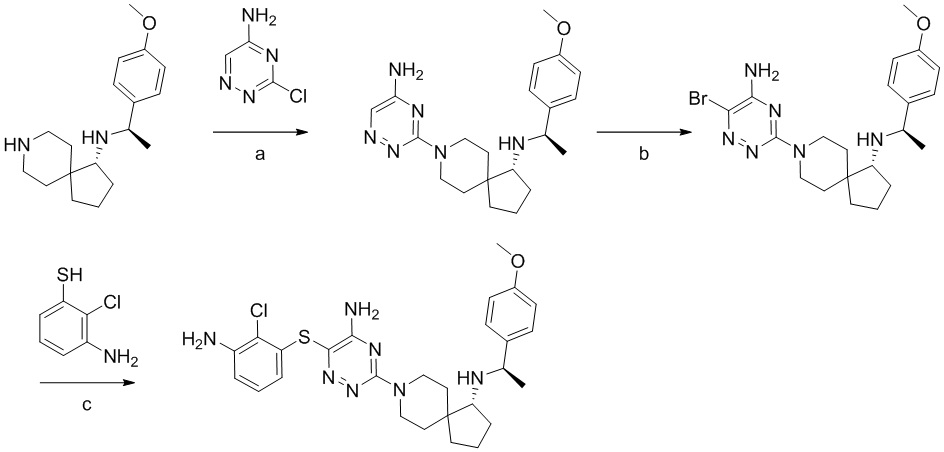
[00295] Стадия a: Как описано для Промежуточного соединения 17, аналогичную процедуру применяли, используя 3-хлор-1,2,4-триазин-5-амин (0,51 г, 3,93 ммоль) и (R)-N-((R)-1-(4-метоксифенил)этил)-8-азаспиро[4.5]декан-1-амин (1,08 г, 3,74 ммоль), получая 0,78 г (R)-8-(5-амино-1,2,4-триазин-3-ил)-N-((R)-1-(4-метоксифенил)этил)-8-азаспиро[4.5]декан-1-амина. MS масса/заряд 383,3 (M+H)+.
[00296] Стадия b: Как описано для Промежуточного соединения 17, аналогичную процедуру применяли, используя (R)-8-(5-амино-1,2,4-триазин-3-ил)-N-((R)-1-(4-метоксифенил)этил)-8-азаспиро[4.5]декан-1-амин (0,48 г, 1,26 ммоль), получая 0,23 г (0,49 ммоль). MS масса/заряд 461,1 (M+H)+.
[00297] Стадия c: Как описано для Промежуточного соединения 17, аналогичную процедуру применяли, используя 3-амино-2-хлорбензолтиол (0,07 г, 0,35 ммоль) и (R)-8-(5-амино-6-бром-1,2,4-триазин-3-ил)-N-((R)-1-(4-метоксифенил)этил)-8-азаспиро[4.5]декан-1-амин (0,13 г, 0,27 ммоль), получая 0,12 г (R)-8-(5-амино-6-((3-амино-2-хлорфенил)тио)-1,2,4-триазин-3-ил)-N-((R)-1-(4-метоксифенил)этил)-8-азаспиро[4.5]декан-1-амина. MS масса/заряд 540,3 (M+H)+.
Промежуточное соединение 22
трет-Бутил-(1-(5-((3-амино-2-хлорфенил)тио)пиразин-2-ил)-4-метилпиперидин-4-ил)карбамат
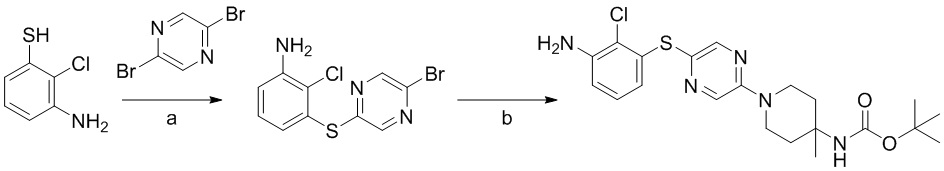
[00298] Стадия a: Смесь 3-амино-2-хлорбензолтиола гидрохлорида (0,21 г, 1,05 ммоль), 2,5-дибромпиразина (0,62 г, 2,61 ммоль), Cu(I) иодида (0,04 г, 0,21 ммоль), 1,10-фенантролина (0,08 г, 0,42 ммоль) и фосфата калия (0,44 г, 2,10 ммоль) в диоксане (7 мл) облучали в микроволновом реакторе в течение 1 ч при 100oC. После охлаждения до к.т. реакционную смесь фильтровали через подушку из Celite® (EtOAc) и фильтрат концентрировали при пониженном давлении. Концентрат очищали хроматографией на силикагеле (0-100% EtOAc/гептана), получая 0,17 г 3-((5-бромпиразин-2-ил)тио)-2-хлоранилина. MS масса/заряд 317,9 (M+H)+.
[00299] Стадия b: К смеси 3-((5-бромпиразин-2-ил)тио)-2-хлоранилин (0,70 г, 2,21 ммоль), трет-бутил-((4-метилпиперидин-4-ил)метил)карбамат (0,76 г, 3,32 ммоль), дициклогексил(2',6'-диизопропоксибифенил-2-ил)фосфин (52 мг, 0,11 ммоль), хлор(2-дициклогексилфосфино-2',6'-диизопропокси-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладий(II), (81 мг, 0,11 ммоль) в диоксане (25 мл) добавляли при к.т. трет- бутоксид натрия (0,30 г, 3,10 ммоль). После перемешивания в течение 14 ч при 90°C, реакционную смесь охлаждали до к.т., объединяли с силикагелем и концентрировали при пониженном давлении до сухого состояния. Полученный остаток очищали хроматографией на силикагеле (0-100% EtOAc/гептана), получая указанное в заголовке соединение (0,50 г). MS масса/заряд 450,2 (M+H)+.
Промежуточное соединение 23
трет-Бутил-((1-(5-((3-амино-2-хлорфенил)тио)пиразин-2-ил)-4-метилпиперидин-4-ил)метил)карбамат

[00300] Как описано для Промежуточного соединения 1, аналогичную процедуру применяли, используя 3-((5-бромпиразин-2-ил)тио)-2-хлоранилин (0,70 г, 2,21 ммоль) и трет-бутил-((4-метилпиперидин-4-ил)метил)карбамат (0,76 г, 3,32 ммоль), получая указанное в заголовке соединение (0,46 г). MS масса/заряд 464,2 (M+H)+.
Промежуточное соединение 24
трет-Бутил-(1-(6-амино-5-((4-амино-2-хлорфенил)тио)пиразин-2-ил)-4-метилпиперидин-4-ил)карбамат
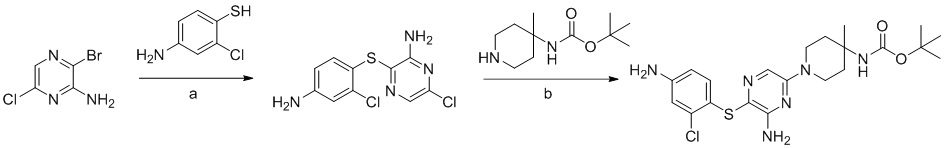
[00301] Стадия a: Как описано для Промежуточного соединения 1, аналогичную процедуру применяли, используя 4-амино-2-хлорбензолтиол (1,0 г, 6,3 ммоль), 3-бром-6-хлорпиразин-2-амин (1,3 г, 6,26 ммоль), Cu(I) иодид (0,239 г, 1,3 ммоль), 1,10-фенантролин (0,45 г, 2,5 ммоль) и фосфат калия (3,32 г, 15,7 ммоль), получая 3-((4-амино-2-хлорфенил)тио)-6-хлорпиразин-2-амин (0,95 г). MS масса/заряд 287,0 (M+H)+.
[00302] Стадия b: Как описано для Промежуточного соединения 2, аналогичную процедуру применяли, используя 3-((4-амино-2-хлорфенил)тио)-6-хлорпиразин-2-амин (0,74 г, 3,5 ммоль) и трет-бутил-((4-метилпиперидин-4-ил)метил)карбамат (0,83 г, 2,9 ммоль), получая указанное в заголовке соединение (0,30 г). MS масса/заряд 465,0 (M+H)+.
Промежуточное соединение 25
Этил-4-гидрокси-1,5,5-триметил-2-оксо-2,5-дигидро-1H-пиррол-3-карбоксилат

[00303] Стадия a: К раствору THF (20 мл) метил-2-((трет-бутоксикарбонил)амино)-2-метилпропаноата (1,49 г, 6,86 ммоль) при 0°C добавляли гидрид натрия (60% в минеральном масле), 0,49 г, 20,57 ммоль). Реакционную смесь перемешивали в течение 5 мин и по каплям добавляли иодометан (0,52 мл, 8,23 ммоль). Реакционную смесь нагревали до к.т. и перемешивали в течение ночи. Реакционную смесь гасили водой пока не прекратилось пенообразование. THF удаляли при пониженном давлении и сырой продукт экстрагировали, используя EtOAc, и очищали флеш-хроматографией на силикагеле (0-25% EtOAc/гептана), получая метил-2-((трет-бутоксикарбонил)(метил)амино)-2-метилпропаноат. (585 мг).1H ЯМР (400 МГц, Хлороформ-d) δ ppm 3,69 (s, 3 H), 2,90 (s, 3 H), 1,42 (s, 15 H).
[00304] Стадия b: В раствор метил-2-((трет-бутоксикарбонил)(метил)амино)-2-метилпропаноата (563 мг, 2,43 ммоль) в DCM (5 мл) добавляли TFA (5,0 мл, 64,9 ммоль). Реакционную смесь перемешивали при к.т. в течение 3 часов, а затем концентрировали при пониженном давлении до получения 2-метил-2-(метиламино)пропаноата - белого кристаллического вещества. (597 мг, 2,43 ммоль).1H ЯМР (400 МГц, Метанол-d4) δ ppm 3,88 (s, 3 H), 2,70 (s, 3 H), 1,59 (s, 6 H).
[00305] Стадия c: К суспензии 2-метил-2-(метиламино)пропаноата (319 мг, 2,43 ммоль) в THF (10 мл) добавляли триэтиламин (1,02 мл, 7,29 ммоль). Полученную смесь охлаждали до 0°C и добавляли этил-3-хлор-3-оксопропаноат (530 мг, 3,52 ммоль). Реакционную смесь нагревали до к.т. и перемешивали в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении и очищали флеш-хроматографией на силикагеле (0-50% EtOAc/гептана), получая этил-3-((1-метокси-2-метил-1-оксопропан-2-ил)(метил)амино)-3-оксопропаноат в виде бледно-желтого масла (455 мг). MS масса/заряд 246,3 (M+H)+.1H ЯМР (400 МГц, Хлороформ-d) δ ppm 4,27-4,11 (m, 2 H), 3,68 (d, J=0,9 Гц, 3 H), 3,44 (s, 2 H), 2,97 (s, 3 H), 1,43 (s, 6 H), 1,26 (t, J=7,1 Гц, 3 H).
[00306] Стадия d: К раствору этил-3-((1-метокси-2-метил-1-оксопропан-2-ил)(метил)амино)-3-оксопропаноата (450 мг, 1,83 ммоль) в EtOH (6 мл) добавляли этанолят натрия (1,03 мл, 2,75 ммоль) при к.т. Реакционную смесь нагревали при 85°C в течение 5 мин, охлаждали и разбавляли, используя Et2O. Осадок отфильтровывали и промывали, используя Et2O. Высушенное твердое вещество растворяли в воде и подкисляли, используя 2 M HCl. Смесь концентрировали при пониженном давлении и очищали флеш-хроматографией на силикагеле (0-10% MeOH/DCM), получая этил-4-гидрокси-1,5,5-триметил-2-оксо-2,5-дигидро-1H-пиррол-3-карбоксилат (306 мг). MS масса/заряд 214,1 (M+H)+.1H ЯМР (400 МГц, Хлороформ-d) δ ppm 11,27 (s, 1 H), 4,37 (q, J=7,0 Гц, 2 H), 2,97-2,72 (m, 3 H), 1,49-1,22 (m, 9 H).
Промежуточное соединение 26
(S)-Этил-4-гидрокси-2-оксо-5-фенил-2,5-дигидро-1H-пиррол-3-карбоксилат
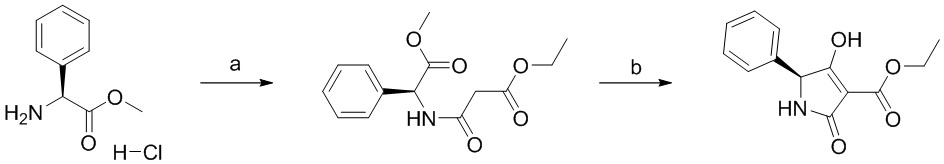
[00307] Стадия a: К суспензии (S)-метил-2-амино-2-фенилацетата гидрохлорида (1,5 г, 7,44 ммоль) в THF (14,9 мл) добавляли триэтиламин (2,6 мл, 18,6 ммоль). Полученную смесь охлаждали до 0°C и добавляли этил-3-хлор-3-оксопропаноат (1,17 г, 7,81 ммоль). Реакционную смесь нагревали до к.т. и перемешивали в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении и очищали флеш-хроматографией на силикагеле (0-50% EtOAc/гептана), получая (S)-этил-3-((2-метокси-2-оксо-1-фенилэтил)амино)-3-оксопропаноат в виде бледно-желтого твердого вещества (1,72 г). MS масса/заряд 280,1 (M+H)+.
[00308] Стадия b: К раствору (S)-этил-3-((2-метокси-2-оксо-1-фенилэтил)амино)-3-оксопропаноата (1,0 г, 3,58 ммоль) в EtOH (12 мл) добавляли этанолят натрия (1,27 мл, 3,4 ммоль) при к.т. Реакционную смесь нагревали при 85°C в течение 5 мин, охлаждали и разбавляли, используя Et2O. Осадок отфильтровывали и промывали, используя Et2O. Высушенное твердое вещество растворяли в воде и подкисляли, используя 2 M HCl. Смесь концентрировали при пониженном давлении полученное твердое вещество промывали водой, получая (S)-этил-4-гидрокси-2-оксо-5-фенил-2,5-дигидро-1H-пиррол-3-карбоксилат (600 мг). MS масса/заряд 248,1 (M+H)+.
Пример 1
N-(3-((3-Амино-5-(4-амино-4-метилпиперидин-1-ил)пиразин-2-ил)тио)-2-хлорфенил)-2-гидрокси-4-оксо-4H-пиридо[1,2-a]пиримидин-3-карбоксамид

[00309] Стадия a: Суспензию 3-амино-2-хлорбензолтиол гидрохлорида (8,0 г, 40,8 ммоль), 3-бром-6-хлорпиразин-2-амина (6,0 г, 28,8 ммоль), Cu(I) иодида (1,1 г, 5,8 ммоль), 1,10-фенантролина (2,1 г, 11,7 ммоль) и фосфата калия (12,5 г, 58,9 ммоль) в диоксане (80 мл) нагревали при 85°C в течение 4 ч. После охлаждения до к.т. реакционную смесь разбавляли, используя EtOAc (100 мл), фильтровали через подушку из Celite®, концентрировали при пониженном давлении до получения масла и суспендировали в DCM (100 мл). Полученный осадок грязно-белого цвета отфильтровывали и сушили, получая 3-((3-амино-2-хлорфенил)тио)-6-хлорпиразин-2-амин (6,5 г). MS масса/заряд 287,1 (M+H)+.
[00310] Стадия b: Суспензию 3-((3-амино-2-хлорфенил)тио)-6-хлорпиразин-2-амина (0,29 г, 1,0 ммоль), трет-бутил-(4-метилпиперидин-4-ил)карбамата (0,43 г, 2,0 ммоль) и DIPEA (0,87 мл, 5,0 ммоль) в NMP (5 мл) облучали в микроволновом реакторе при 150°C в течение 2 ч. После охлаждения до к.т. реакционную смесь разбавляли, используя EtOAc (100 мл), промывали водой и соляным раствором, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Сырой продукт очищали хроматографией на силикагеле (10-50% EtOAc/гептана), получая 0,44 г трет-бутил-(1-(6-амино-5-((3-амино-2-хлорфенил)тио)пиразин-2-ил)-4-метилпиперидин-4-ил)карбамата. MS масса/заряд 465,2 (M+H)+.
[00311] Стадия c: Суспензию трет-бутил-(1-(6-амино-5-((3-амино-2-хлорфенил)тио)пиразин-2-ил)-4-метилпиперидин-4-ил)карбамата (0,13 г, 0,26 ммоль) и метил-2-гидрокси-4-оксо-4H-пиридо[1,2-a]пиримидин-3-карбоксилата (69,1 мг, 0,314 ммоль) в бромбензоле (2 мл) нагревали при 170°C в микроволновом реакторе в течение 1 ч. После охлаждения до к.т. реакционную смесь концентрировали при пониженном давлении и полученный остаток очищали с помощью ВЭЖХ (15-40% MeCN в воде, 0,1% NH4OH модификатора), получая трет-бутил-(1-(6-амино-5-((2-хлор-3-(2-гидрокси-4-оксо-4H-пиридо[1,2-a]пиримидин-3-карбоксамидо)фенил)тио)пиразин-2-ил)-4-метилпиперидин-4-ил)карбамат (0,09 г). MS масса/заряд 653,3 (M+H)+.
[00312] Стадия d: К раствору трет-бутил-(1-(6-амино-5-((2-хлор-3-(2-гидрокси-4-оксо-4H-пиридо[1,2-a]пиримидин-3-карбоксамидо)фенил)тио)пиразин-2-ил)-4-метилпиперидин-4-ил)карбамата (0,09 г, 0,14 ммоль) в DCM (0,5 мл) добавляли TFA (0,5 мл). После перемешивания при к.т. в течение 2 ч летучие соединения удаляли при пониженном давлении и полученный остаток очищали с помощью ВЭЖХ (15-40% MeCN в воде, 0,1% TFA модификатора). Лиофилизированный продукт растворяли в MeOH, содержащем HCl (1,2 M) и сушили, получая N-(3-((3-амино-5-(4-амино-4-метилпиперидин-1-ил)пиразин-2-ил)тио)-2-хлорфенил)-2-гидрокси-4-оксо-4H-пиридо[1,2-a]пиримидин-3-карбоксамид в виде соли HCl (0,085 г).1H ЯМР (400 МГц, Метанол-d4) δ ppm 9,19 (d, J=6,8 Гц, 1 H), 8,34 (d, J=8,0 Гц, 1 H), 8,24 (t, J=7,9 Гц, 1 H), 7,81 (s, 1 H), 7,61 (d, J=8,7 Гц, 1 H), 7,52 (d, J=7,1 Гц, 1 H), 7,29 (t, J=8,3 Гц, 1 H), 6,74 (d, J=7,9 Гц, 1 H), 4,26 (d, J=14,2 Гц, 2 H), 3,52 (m, 2 H), 1,93 (m, 4 H), 1,53 (s, 3 H). HRMS рассч. для C25H26ClN8O3S (M+H)+ 553,1537, найденное значение 553,1524. IC50=0,006 мкM.
[00313] Следующие соединения, приведенные в Таблице 1, были получены с использованием процедуры или модифицированной процедуры согласно Примеру 1.
Таблица 1
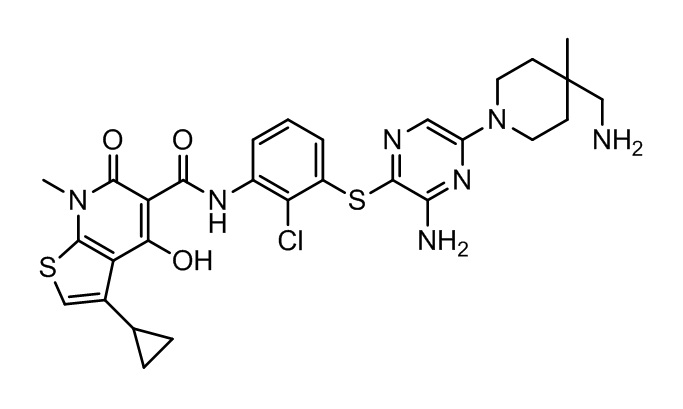
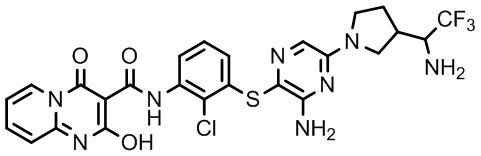

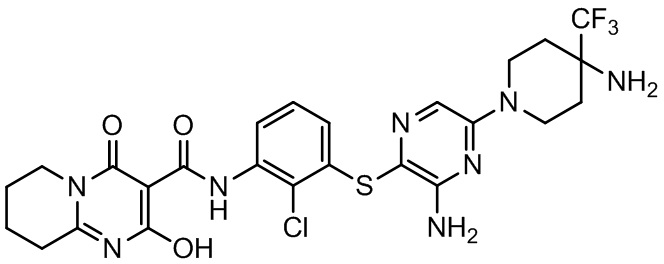
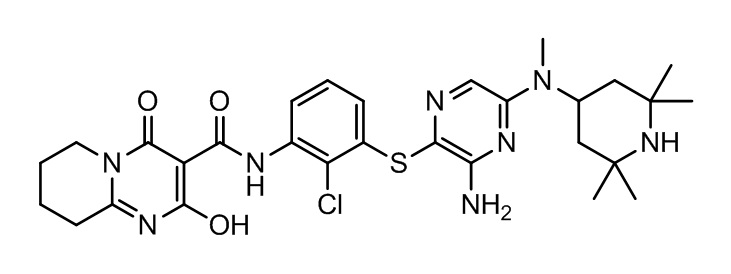
Пример 25
(5-Метил-2-оксо-1,3-диоксол-4-ил)метил-((1-(6-амино-5-((2-хлор-3-(2-гидрокси-4-оксо-4H-пиридо[1,2-a]пиримидин-3-карбоксамидо)фенил)тио)пиразин-2-ил)-4-метилпиперидин-4-ил)метил)карбамат

[00314] К раствору N-(3-((3-амино-5-(4-(аминометил)-4-метилпиперидин-1-ил)пиразин-2-ил)тио)-2-хлорфенил)-2-гидрокси-4-оксо-4H-пиридо[1,2-a]пиримидин-3-карбоксамида (50 мг, 0,078 ммоль) в DMSO (1 мл) добавляли DIPEA (0,055 мл, 0,313 ммоль) и (5-метил-2-оксо-1,3-диоксол-4-ил)метил(4-нитрофенил)карбонат (24,2 мг, 0,082 ммоль). После перемешивания при к.т. в течение 1 ч, сырой продукт отфильтровали и очищали с помощью ВЭЖХ (45-70% MeCN/воды, 0,1% муравьиной кислоты), получая указанное в заголовке соединение (28 мг).1H ЯМР (400 МГц, Ацетонитрил-d3) δ ppm 15,15 (s, 1 H), 12,14 (s, 1 H), 9,00 (d, J=7,1 Гц, 1 H), 8,19 (d, J=8,2 Гц, 1 H), 8,02 (ddd, J=8,9, 6,9, 1,8 Гц, 1 H), 7,62 (s, 1 H), 7,54 (d, J=8,9 Гц, 1 H), 7,32 (t, J=7,0 Гц, 1 H), 7,19 (t, J=8,1 Гц, 1 H), 6,54 (dd, J=8,0, 1,7 Гц, 1 H), 5,78 (t, J=6,8 Гц, 1 H), 5,20 (s, 2 H), 4,81 (s, 2 H), 3,83 (dt, J=13,4, 5,1 Гц, 2 H), 3,41 (ddd, J=13,5, 9,4, 3,6 Гц, 2 H), 3,06 (d, J=6,7 Гц, 2 H), 2,12 (s, 3 H), 1,48 (ddd, J=13,3, 8,7, 3,8 Гц, 2 H), 1,36 (dt, J=13,6, 4,7 Гц, 2 H), 0,97 (s, 3 H). HRMS рассч. для C32H32ClN8O8S (M+H)+ 723,1752, найденное значение 723,1728. IC50=0,08 мкM.
Пример 26
(S)-N-(3-((3-Амино-5-(4-амино-2-окса-8-азаспиро[4.5]декан-8-ил)пиразин-2-ил)тио)-2-хлорфенил)-2-гидрокси-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-a]пиримидин-3-карбоксамид

[00315] Стадия a: В микроволновую ампулу объемом 20 мл, содержащую раствор трет-бутил-4-оксо-2-окса-8-азаспиро[4.5]декан-8-карбоксилата (220 мг, 0,86 ммоль) в THF (4 мл), добавляли (R)-2-метилпропан-2-сульфинамид (209 мг, 1,72 ммоль), а затем тетраэтоксид титана (0,723 мл, 3,45 ммоль). Ампулу укупоривали колпачком и нагревали при 90°C в течение 1 ч. Смесь охлаждали до 0°C, затем вносили LiBH4 (23 мг, 1,06 ммоль). Реакционную смесь перемешивали в течение 30 мин и затем гасили, медленно добавляя МеОН до прекращения вспенивания. Смесь разбавляли, используя МеОН, и затем концентрировали при пониженном давлении. Остаток разбавляли соляным раствором (при обработке ультразвуком). Смесь экстрагировали, используя EtOAc (4×10 мл), затем органическую фазу промывали соляным раствором, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. После флеш-хроматографии на силикагеле (0-100% EtOAc/гептана) получали (S)-трет-бутил-4-((R)-1,1-диметилэтилсульфинамидо)-2-окса-8-азаспиро[4.5]декан-8-карбоксилат (170 мг), MS масса/заряд 361,1 (M+H)+.
[00316] Стадия b: К раствору (S)-трет-бутил-4-((R)-1,1-диметилэтилсульфинамидо)-2-окса-8-азаспиро[4.5]декан-8-карбоксилата (800 мг, 2,22 ммоль) в MeOH (20 мл) добавляли 4 M HCl в диоксане (3,33 мл, 13,3 ммоль) и нагревали при 40°C в течение 20 мин. Реакционную смесь концентрировали при пониженном давлении, еще раз добавляя MeOH, получая HCl соль (S)-2-окса-8-азаспиро[4.5]декан-4-амина, MS масса/заряд 157,2 (M+H)+. Сырой остаток переносили на следующую стадию без дополнительной очистки.
[00317] Альтернативная процедура: К раствору (S)-трет-бутил-4-((R)-1,1-диметилэтилсульфинамидо)-2-окса-8-азаспиро[4.5]декан-8-карбоксилата (891 мг, 2,47 ммоль) в MeOH (9 мл) добавляли 4 M HCl в диоксане (3,71 мл, 14,8 ммоль) и нагревали при 40°C в течение 20 мин. Реакционную смесь концентрировали при пониженном давлении и перетирали с MeCN при обработке ультразвуком. Смесь фильтровали через 0,45 мкм PTFE мембрану, получая (S)-2-окса-8-азаспиро[4.5]декан-4-амин в виде двойной HCl соли (510 мг).1H ЯМР (400 МГц, Метанол-d4) δ ppm 4,19 (dd, J=10,8, 5,5 Гц, 1 H), 3,92 (d, J=9,4 Гц, 1 H), 3,88-3,81 (m, 2 H), 3,69 (dd, J=5,6, 2,6 Гц, 1 H), 3,48-3,34 (m, 2 H), 3,20-3,03 (m, 2 H), 2,10-1,97 (m, 2 H), 1,94-1,84 (m, 2 H). MS масса/заряд 157,2 (M+H)+.
[00318] Стадия c: Суспензию 3-((3-амино-2-хлорфенил)тио)-6-хлорпиразин-2-амина (0,10 г, 0,24 ммоль), (S)-2-окса-8-азаспиро[4.5]декан-4-амина дигидрохлорида (0,084 г, 0,29 ммоль) и DIPEA (0,5 мл, 2,7 ммоль) в DMSO (0,8 мл) нагревали при 120°C в течение 1 ч. После охлаждения до к.т. и внесения MeCN отфильтровывали коричневый осадок и очищали его перетиранием с MeCN/MeOH (примерно 4 мл, 9:1), получая сырой (S)-8-(6-амино-5-((3-амино-2-хлорфенил)тио)пиразин-2-ил)-2-окса-8-азаспиро[4.5]декан-4-амин в виде коричневого твердого вещества. Этот материал суспендировали в смеси DCM/DMF (1,1 мл, 10:1) и вносили в раствор DIPEA (0,04 мл, 0,23 ммоль) и Boc2O (0,10 г, 0,48 ммоль). После перемешивания в течение 60 ч при к. т., а затем при 40°C в течение 5 ч, реакционную смесь концентрировали при пониженном давлении. Полученное твердое вещество растворяли в EtOAc, промывали водн. насыщ. NH4Cl, а затем водой. Органическую фазу сушили над MgSO4, фильтровали и концентрировали при пониженном давлении, получая трет-бутил-(S)-(8-(6-амино-5-((3-амино-2-хлорфенил)тио)пиразин-2-ил)-2-окса-8-азаспиро[4.5]декан-4-ил)карбамат в виде коричневого твердого вещества (0,11 г), который использовали без дополнительной очистки. MS масса/заряд 507,3 (M+H)+.
[00319] Стадии d и e выполняли согласно Примеру 1, используя 4 M HCl в диоксане, получая (S)-N-(3-((3-амино-5-(4-амино-2-окса-8-азаспиро[4.5]декан-8-ил)пиразин-2-ил)тио)-2-хлорфенил)-2-гидрокси-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-a]пиримидин-3-карбоксамид в виде HCl соли.1H ЯМР (400 МГц, Метанол-d4) δ ppm 8,25 (d, J=7,1 Гц, 1 H), 7,60 (s, 1 H), 7,16 (t, J=8,1 Гц, 1 H), 6,51 (d, J=8,0 Гц, 1 H), 4,36-4,13 (m, 3 H), 4,02-3,96 (m, 2 H), 3,90 (d, J=9,2 Гц, 1 H), 3,82 (dd, J=10,6, 2,6 Гц, 1 H), 3,61-3,54 (m, 1 H), 3,37-3,34 (m, 1 H), 3,27-3,25 (m, 1 H), 3,23-3,14 (m, 1 H), 2,95 (t, J=6,5 Гц, 2 H), 2,06-1,99 (m, 2 H), 1,97-1,89 (m, 2 H), 1,81-1,67 (m, 3 H). HRMS рассч. для: C27H32ClN8O4S (M+H)+ 599,1956, найденное значение 599,1947. IC50=0,018 мкM.
Пример 27
N-(3-((3-Амино-5-((3S,4S)-4-амино-3-метил-2-окса-8-азаспиро[4.5]декан-8-ил)пиразин-2-ил)тио)-2-хлорфенил)-2-гидрокси-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-a]пиримидин-3-карбоксамид

[00320] Раствор N-(3-((3-амино-5-хлорпиразин-2-ил)тио)-2-хлорфенил)-2-гидрокси-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-a]пиримидин-3-карбоксамида (0,13 г, 0,27 ммоль), (3S,4S)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амина (0,20 г, 0,81 ммоль, чистота примерно 70%) и DIPEA (0,1 мл, 0,55 ммоль) в DMF (2 мл) облучали в микроволновом реакторе в течение 90 мин при 100°C. После охлаждения до к.т. реакционную смесь смешивали с диэтиловым эфиром и полученный коричневый осадок отфильтровывали. Осадок очищали с помощью ВЭЖХ с обращенной фазой (15-40% MeCN в воде, 0,1% TFA модификатора) с получением указанного в заголовке соединения в виде TFA соли. Чистые фракции концентрировали при пониженном давлении, а затем разбавляли, используя HCl (1,25 M в MeOH), и упаривали (3x), получая указанное в заголовке соединение в виде HCl соли (56 мг).
[00321] Следующие соединения, приведенные в Таблице 2, были получены с использованием процедуры или модифицированной процедуры согласно Примеру 27 с применением N-(3-((3-амино-5-хлорпиразин-2-ил)тио)-2-хлорфенил)-2-гидрокси-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-a]пиримидин-3-карбоксамида и соответствующего амина.
Таблица 2
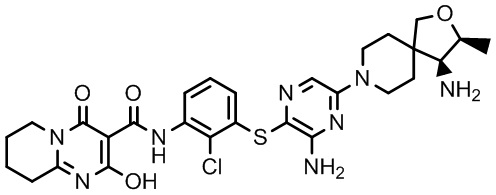
[00322] Следующие соединения, приведенные в Таблице 3, были получены с использованием процедуры или модифицированной процедуры согласно Примеру 1.
Таблица 3
[00323] Следующие соединения, приведенные в Таблице 4, были получены с использованием процедуры или модифицированной процедуры согласно Примеру 1 с соответствующим пиримидин-3-карбоновым эфиром и трет-бутил-(1-(5-((3-амино-2-хлорфенил)тио)пиразин-2-ил)-4-метилпиперидин-4-ил)карбаматом.
Таблица 4
Пример 46
N-(3-((2-(4-Амино-4-метилпиперидин-1-ил)-3H-имидазо[4,5-b]пиридин-5-ил)тио)фенил)-4-гидрокси-2-оксо-1-фенил-2,5-дигидро-1H-пиррол-3-карбоксамид
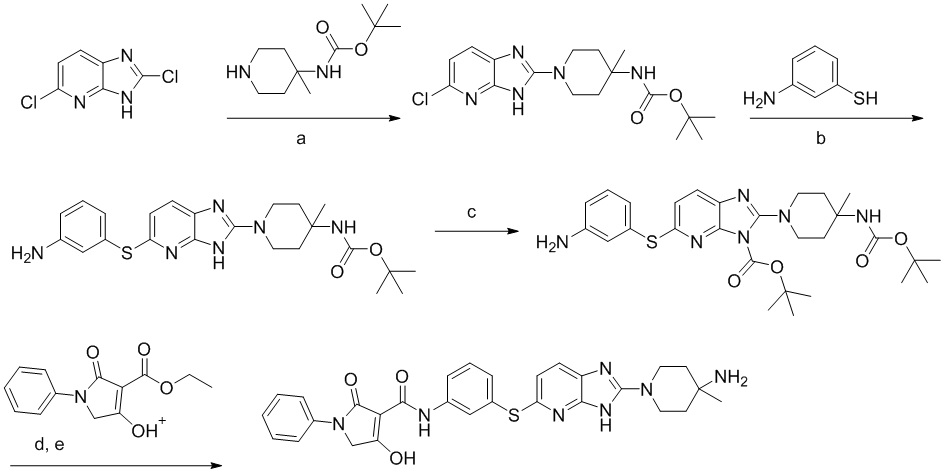
[00324] Стадия a: Смесь 2,5-дихлор-3 H-имидазо[4,5-b]пиридина (2,0 г, 10,64 ммоль), трет-бутил-(4-метилпиперидин-4-ил)карбамата (3,19 г, 14,89 ммоль) и N-метилморфолина (1,29 мл, 11,70 ммоль) в NMP (13 мл) облучали в микроволновом реакторе при 110°C в течение 1 ч. После охлаждения до к.т. реакционную смесь выливали в ледяную воду (800 мл) и отфильтровывали. Твердый остаток промывали водой и сушили при пониженном давлении при 45oC, получая трет-бутил-(1-(5-хлор-3H-имидазо[4,5-b]пиридин-2-ил)-4-метилпиперидин-4-ил)карбамат (3,11 г). MS масса/заряд 366,3 (M+H)+.
[00325] Стадия b: Суспензию трет-бутил-(1-(5-хлор-3H-имидазо[4,5-b]пиридин-2-ил)-4-метилпиперидин-4-ил)карбамата (0,350 г, 0,957 ммоль), 3-аминобензолтиола (0,599 г, 4,78 ммоль) и N-метилморфолина (1,578 мл, 14,35 ммоль) в NMP (3 мл) облучали в микроволновом реакторе при 230°C в течение 45 мин. После охлаждения реакционную смесь выливали в DCM и отфильтровывали. Сырой твердый продукт очищали, используя ВЭЖХ (15-40% MeCN в воде, 5 мM NH4OH модификатор), получая 61 мг трет-бутил-(1-(5-((3-аминофенил)тио)-3H-имидазо[4,5-b]пиридин-2-ил)-4-метилпиперидин-4-ил)карбамата. MS масса/заряд 355,2 (M+H)+.
[00326] Стадия c: К раствору трет-бутил-(1-(5-((3-аминофенил)тио)-3H-имидазо[4,5-b]пиридин-2-ил)-4-метилпиперидин-4-ил)карбамата (60 мг, 0,169 ммоль), Boc2O (85 мг, 0,389 ммоль) в THF (4 мл) добавляли TEA (0,083 мл, 0,592 ммоль) и нагревали при 70°C в течение 4 ч. Реакционную смесь концентрировали при пониженном давлении с получением 94 мг трет-бутил-5-((3-аминофенил)тио)-2-(4-((трет-бутоксикарбонил)амино)-4-метилпиперидин-1-ил)-3H-имидазо[4,5-b]пиридин-3-карбоксилата. MS масса/заряд 554,8 (M+H)+.
[00327] Стадия d: Суспензию трет-бутил-5-((3-аминофенил)тио)-2-(4-((трет-бутоксикарбонил)амино)-4-метилпиперидин-1-ил)-3H-имидазо[4,5-b]пиридин-3-карбоксилата (90 мг, 0,162 ммоль) и этил-4-гидрокси-2-оксо-1-фенил-2,5-дигидро-1H-пиррол-3-карбоксилата (48,1 мг, 0,195 ммоль) в THF (2 мл) подвергали облучению в микроволновом реакторе при 125°C в течение 20 мин. После охлаждения реакционной смеси до к.т. осадок отфильтровывали и промывали, используя MeOH, получая трет-бутил-2-(4-((трет-бутоксикарбонил)амино)-4-метилпиперидин-1-ил)-5-((3-(4-гидрокси-2-оксо-1-фенил-2,5-дигидро-1H-пиррол-3-карбоксамидо)фенил)тио)-3H-имидазо[4,5-b]пиридин-3-карбоксилат (0,095 г). MS масса/заряд 555,3 (M+H-2Boc)+.
[00328] Стадия e: К раствору трет-бутил-2-(4-((трет-бутоксикарбонил)амино)-4-метилпиперидин-1-ил)-5-((3-(4-гидрокси-2-оксо-1-фенил-2,5-дигидро-1H-пиррол-3-карбоксамидо)фенил)тио)-3H-имидазо[4,5-b]пиридин-3-карбоксилата (0,091 г, 0,120 ммоль) в диоксане (0,2 мл) добавляли 4 M HCl в диоксане (0,121 мл). После перемешивания при температуре окружающей среды в течение 14 ч летучие продукты удаляли при пониженном давлении. Сырой остаток перетирали с MeOH несколько раз и твердый продукт очищали с помощью ВЭЖХ (15-40% MeCN в воде, 5 мМ NH4OH модификатор) с получением N-(3-((2-(4-амино-4-метилпиперидин-1-ил)-3H-имидазо[4,5-b]пиридин-5-ил)тио)фенил)-4-гидрокси-2-оксо-1-фенил-2,5-дигидро-1H-пиррол-3-карбоксамида (3,5 мг).
[00329] Следующие соединения, приведенные в Таблице 5, были получены с использованием процедуры или модифицированной процедуры согласно Примеру 46.
Таблица 5
Пример 50
N-(4-((3-Амино-5-(4-амино-4-метилпиперидин-1-ил)пиразин-2-ил)тио)-3-хлорфенил)-4-гидрокси-2-оксо-1-фенил-2,5-дигидро-1H-пиррол-3-карбоксамид

[00330] Суспензию трет-бутил-(1-(6-амино-5-((4-амино-2-хлорфенил)тио)пиразин-2-ил)-4-метилпиперидин-4-ил)карбамата (50 мг, 0,108 ммоль) и метил-4-гидрокси-2-оксо-1-фенил-2,5-дигидро-1H-пиррол-3-карбоксилата (30 мг, 0,140 ммоль) в THF (1 мл) облучали при 115°C в микроволновом реакторе в течение 1,5 ч. После охлаждения реакционной смеси летучие продукты удаляли при пониженном давлении и сырую смесь растворяли в DCM (2 мл). Добавляли раствор 4 M HCl (в диоксане, 2 мл) и полученную смесь нагревали при 40°C в течение 1 ч. После охлаждения до комнатной температуры раствор концентрировали при пониженном давлении и полученный остаток очищали с помощью ВЭЖХ (15-40% MeCN/воды, 0,1% NH4OH модификатора), получая указанное в заголовке соединение (12 мг). MS масса/заряд 566,0 (M+H)+.
[00331] Следующие соединения, приведенные в Таблице 6, были получены с использованием процедуры или модифицированной процедуры согласно Примеру 46.
Таблица 6

Пример 58
N-(3-((3-Амино-5-(4-амино-4-метилпиперидин-1-ил)пиразин-2-ил)тио)-2-хлорфенил)-4-гидрокси-2-оксо-1,2-дигидрохинолин-3-карбоксамид
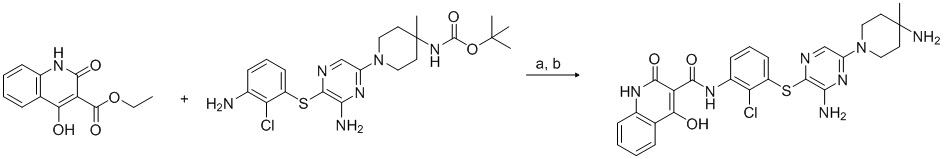
[00332] Суспензию трет-бутил-(1-(6-амино-5-((3-амино-2-хлорфенил)тио)пиразин-2-ил)-4-метилпиперидин-4-ил)карбамата (0,140 г, 0,30 ммоль) и этил-4-гидрокси-2-оксо-1,2-дигидрохинолин-3-карбоксилата (91 мг, 0,39 ммоль) в THF (1,5 мл) облучали при 115°C в микроволновом реакторе в течение 1,5 ч. После охлаждения реакционной смеси до к. т. летучие вещества удаляли при пониженном давлении и сырую смесь растворяли в DCM (2 мл). Добавляли раствор HCl (4 M в диоксане, 2 мл) и полученную смесь нагревали при 40°C в течение 1 ч. После охлаждения до к.т. раствор концентрировали при пониженном давлении и полученный остаток очищали с помощью ВЭЖХ (15-40% MeCN/воды, 0,1% NH4OH модификатора), получая указанное в заголовке соединение (12 мг). MS масса/заряд 552,0 (M+H)+.1H ЯМР (400 МГц, DMSO-d6) δ ppm 8,23-8,37 (m, 1 H), 7,91-8,03 (m, 1 H), 7,59-7,71 (m, 2 H), 7,40-7,52 (m, 1 H), 7,16-7,25 (m, 1 H), 7,09 (br. s, 2 H), 6,77-6,89 (m, 1 H), 6,51-6,61 (m, 1 H), 6,26-6,35 (m, 1 H), 6,17 (br. s, 2 H), 5,76-5,87 (m, 1 H), 5,38-5,49 (m, 1 H), 3,81-4,03 (m, 2 H), 3,38-3,49 (m, 2 H), 1,57-1,80 (m, 4 H), 1,33 (s, 3 H). HRMS рассч. для C26H27ClN7O3S (M+H)+ 552,1585, найденное значение 552,1602. IC50=0,072 мкM.
[00333] Следующие соединения, приведенные в Таблице 7, были получены с использованием процедуры или модифицированной процедуры согласно Примеру 50.
Таблица 7
Пример 66
N-(3-((3-Амино-5-(4-амино-4-метилпиперидин-1-ил)пиразин-2-ил)тио)фенил)-4-гидрокси-2-оксо-1-фенил-2,5-дигидро-1H-пиррол-3-карбоксамид

[00334] Как описано для Промежуточного соединения 1, аналогичную процедуру применяли, используя 3-((3-аминофенил)тио)-6-хлорпиразин-2-амин (0,150 г, 0,594 ммоль) и трет-бутил-(4-метилпиперидин-4-ил)карбамат (0,191 г, 0,890 ммоль), получая трет-бутил-(1-(6-амино-5-((3-аминофенил)тио)пиразин-2-ил)-4-метилпиперидин-4-ил)карбамат (0,162 г, 0,376 ммоль). МS масса/заряд 431,4 (M+H)+. N-(3-((3-амино-5-(4-амино-4-метилпиперидин-1-ил)пиразин-2-ил)тио)фенил)-4-гидрокси-2-оксо-1-фенил-2,5-дигидро-1H-пиррол-3-карбоксамид получали, используя процедуру или модифицированную процедуру согласно Примеру 50. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 11,05 (s, 1 H), 7,69 (d, J=7,8 Гц, 2 H), 7,63 (s, 1 H), 7,55 (s, 2 H), 7,33 (s, 1 H), 7,26 (dd, J=8,6, 7,4 Гц, 2 H), 7,15 (d, J=8,3 Гц, 1 H), 7,10 (t, J=7,8 Гц, 1 H), 6,91 (t, J=7,3 Гц, 1 H), 6,70 (d, J=7,6 Гц, 1 H), 6,08 (s, 2 H), 4,05 (d, J=13,9 Гц, 2 H), 3,85 (s, 2 H), 3,26 (d, J=10,4 Гц, 2 H), 1,79-1,62 (m, 4 H), 1,37 (s, 3 H). HRMS рассч. для C27H30N7O3S (M+H)+ 532,2131, найденное значение 532,2122. IC50=0,017 мкM.
Пример 67
N-(3-((3-Амино-5-(4-амино-4-метилпиперидин-1-ил)пиразин-2-ил)тио)-2-(трифторметил)фенил)-4-гидрокси-2-оксо-1-фенил-2,5-дигидро-1H-пиррол-3-карбоксамид

[00335] Стадия a: Суспензию 3-фтор-2-(трифторметил)анилина (9,7 г, 54,1 ммоль), 2-метилпропан-2-тиола (18,31 мл, 162 ммоль) и Cs2CO3 (52,9 г, 162 ммоль) в DMF (100 мл) нагревали при 130°C в течение 14 ч. После охлаждения реакционную смесь разбавляли водой (500 мл) и экстрагировали, используя EtOAc (500 мл). Органический слой затем промывали водой (3×400 мл), соляным раствором (3×300 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 12,52 г 3-(трет-бутилтио)-2-(трифторметил)анилина. MS масса/заряд 250,2 (M+H)+.
[00336] Стадия b: Суспензию 3-(трет-бутилтио)-2-(трифторметил)анилина (2,2 г, 8,82 ммоль) в конц. HCl (80 мл) нагревали при 85°C с течение 3 ч. После охлаждения реакционную смесь концентрировали при пониженном давлении почти до сухого состояния и твердое вещество отфильтровывали. Твердое вещество промывали, используя EtOAc и гептан, и сушили еще раз при пониженном давлении при 40oC, получая 3-амино-2-(трифторметил)бензолтиол в виде соли HCl, 1,02 г (4,44 ммоль). MS масса/заряд 194,1 (M+H)+.
[00337] Стадия c: Как описано для Промежуточного соединения 1, аналогичную процедуру применяли, используя 3-амино-2-(трифторметил)бензолтиол (0,45 г, 1,94 ммоль) и 3-бром-6-хлорпиразин-2-амид (0,27 г, 1,30 ммоль), получая 3-((3-амино-2-(трифторметил)фенил)тио)-6-хлорпиразин-2-амин (0,211 г, 0,66 ммоль). MS масса/заряд 321,1 (M+H)+.
[00338] Стадия d: Как описано для Промежуточного соединения 1, аналогичную процедуру применяли, используя 3-((3-амино-2-(трифторметил)фенил)тио)-6-хлорпиразин-2-амин (100 мг, 0,31 ммоль), трет-бутил-(4-метилпиперидин-4-ил)карбамат (66,7 мг, 0,31 ммоль), получая трет-бутил-(1-(6-амино-5-((3-амино-2-(трифторметил)фенил)тио)пиразин-2-ил)-4-метилпиперидин-4-ил)карбамат (100 мг, 0,201 ммоль). MS масса/заряд 499,3 (M+H)+.
[00339] Стадия e: N-(3-((3-Амино-5-(4-амино-4-метилпиперидин-1-ил)пиразин-2-ил)тио)-2-(трифторметил)фенил)-4-гидрокси-2-оксо-1-фенил-2,5-дигидро-1H-пиррол-3-карбоксамид получали, используя процедуру или модификации процедуры согласно Примеру 50. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 11,11 (s, 1 H), 7,91-7,80 (m, 3 H), 7,72-7,68 (m, 2 H), 7,66 (s, 1 H), 7,29-7,19 (m, 3 H), 6,92 (t, J=7,3 Гц, 1 H), 6,44 (d, J=8,0 Гц, 1 H), 6,14 (s, 2 H), 4,10-3,95 (m, 2 H),3,86 (s, 2 H), 3,35-3,25 (m, 2 H), 1,82-1,66 (m, 4 H), 1,37 (s, 3 H). HRMS рассч. для C28H29F3N7O3S (M+H)+ 600,2032, найденное значение 600,2004. IC50=0,004 мкM.
Анализы
[00340] Соединения данного изобретения оценивали в отношении их способности избирательно ингибировать активность SHP2. Ингибирующие свойства соединений данного изобретения, описанных в данном документе, можно подтвердить путем испытания любым из следующих анализов.
Анализ аллостерического ингибирования SHP2
[00341] SHP2 аллостерически активировали путем связывания бис-тирозил-фосфорилированных пептидов с их Src-гомологичными доменами 2 (SH2). Последняя стадия активации приводит к высвобождению самоингибирующей поверхности взаимодействия SHP2, что в свою очередь делает SHP2 белковую тирозинфосфатазу (PTP) активной и доступной для распознавания субстрата и реакционного катализа. За каталитической активностью SHP2 наблюдали, используя суррогатный субстрат DiFMUP в режиме немедленного флуоресцентного анализа.
[00342] Более конкретно, фосфатазные реакции проводили при комнатной температуре в 384-луночных черных полистирольных планшетах, плоскодонных, с низким бордюром, несвязывающей поверхностью (Corning, Кат. № 3575), используя конечный реакционный объем 25 μл и следующие буферные условия при анализе: 60 мМ HEPES, pH 7,2, 75 мМ NaCl, 75 мМ KCl, 1 мМ ЕDTА, 0,05% P-20, 5 мM DTT.
[00343] За ингибированием SHP2 соединениями данного изобретения (концентрация изменяется в пределах 0,003-100 μM) наблюдали, используя анализ, в котором 0,5 нM SHP2 инкубировали с 0,5 мкM пептида IRS1_pY1172(dPEG8)pY1222 (последовательность: H2N-LN(pY)IDLDLV(dPEG8)LST(pY)ASINFQK-амид) (SEQ ID NO:1). После 30-60 минут инкубирования при 25oC, в реакцию добавляли суррогатный субстрат DiFMUP (Invitrogen, Кат.№ D6567) и инкубировали при 25°C в течение 30 минут. Реакцию погасили, добавляя 5 мкл 160 мкM раствора bpV(Phen) (Enzo Life Sciences Кат.№ ALX-270-204). За флуоресцентным сигналом следили, используя считывающее устройство для микропланшетов (Envision, Perki-Elmer) с возбуждающими и испускаемыми длинами волн 340 нм и 450 нм, соответственно. Кривые зависимости доза-реакция ингибитора анализировали, используя для построения нормированную кривую регрессии IC50 с нормализацией по контролю. Результаты IC50 для соединений данного изобретения показаны в примерах и таблицах 1-7 выше.
p-ERK клеточный анализ
[00344] p-ERK клеточный анализ с использованием набора AlphaScreen® SureFire™ Phospho-ERK 1/2 Kit (PerkinElmer): клетки KYSE-520 (30000 клеток/лунку) выращивали в культуральной среде в 96-луночном планшете в течение ночи и обрабатывали ингибиторами Shp2 при концентрациях 20, 6,6, 2,2, 0,74, 0,24, 0,08, 0,027 мкM в течение 2 часов при 37°C. Инкубирование останавливали добавлением 30 мкл буферного раствора для лизиса (PerkinElmer), поставляемого вместе с набором SureFire фосфорилированной киназы, регулируемой внеклеточными сигналами (pERK), для анализа (PerkinElmer). Образцы обрабатывали согласно инструкциям производителя. Флуоресцентный сигнал от pERK измеряли в двух повторностях, используя многоканальное считывающее устройство 2101 multilabel reader (Perkin Elmer Envision). Процент ингибирования нормализовали по общему сигналу ERK и сравнивали с контрольным раствором DMSO-носителя.
Анализ образования колоний и анализ пролиферации клеток
[00345] Клетки KYSE-520 (1500 клеток/лунку) вносили в 24-луночные планшеты в 300 мкл среды (RPMI-1640, содержащей 10% FBS (фетальной бычьей сыворотки), Lonza). Для обработки лекарственным средством добавляли соединения данного изобретения при различных концентрациях (20, 10, 5, 2,5, 1,25 мкМ) в течение 24 часов и 5 дней после внесения клеток. На 11 день колонии окрашивали 0,2% кристаллическим фиолетовым (MP Biomedicals), а затем растворяли в 20% уксусной кислоте для подсчета с использованием считывающего устройства Spectramax (Thermo Scientific). Для анализа клеточной пролиферации клетки (1500 клеток/лунку) вносили в 96-луночные планшеты в 100 мкл среды (RPMI-1640, содедержащей 10% FBS, Lonza). На 6 день добавляли 50 мкл реагента Celltiter-Glo (Promega) и люминисцентный сигнал регистрировали согласно инструкции производителя (Promega).
Понятно, что описанные в данном документе примеры и варианты осуществления служат только в качестве иллюстрации, и что различные модификации или изменения с их учетом будут понятны специалистам в данной области техники, и они должны быть включены в сущность и содержание данной заявки, а также объем прилагаемой формулы изобретения.
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> Novartis AG
Chen, Zhuoliang
Fortanet, Jorge Garcia
Karki, Rajesh
LaMarche, Matthew J
Majumdar, Dyuti
Perez, Lawrence Blas
Sendzik, Martin
Smith, Troy Douglas
Yang, Fan
Yu, Bing
<120> СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ ИНГИБИРОВАНИЯ АКТИВНОСТИ SHP2
<130> PAT057289-WO-PCT
<160> 1
<170> PatentIn версия 3.5
<210> 1
<211> 21
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> бифосфорилированный пептид, полученный из субстрата инсулинового рецептора-1 (IRS-1)
<220>
<221> МОДИФИЦИРОВАННЫЙ_ОСТАТОК
<222> (3)..(3)
<223> ФОСФОРИЛИРОВАННЫЙ ТИРОЗИН
<220>
<221> ДРУГОЙ_ПРИЗНАК
<222> (10)..(10)
<223> dPEG8
<220>
<221> МОДИФИЦИРОВАННЫЙ_ОСТАТОК
<222> (14)..(14)
<223> ФОСФОРИЛИРОВАННЫЙ ТИРОЗИН
<220>
<221> МОДИФИЦИРОВАННЫЙ_ОСТАТОК
<222> (21)..(21)
<223> АМИДИРОВАННЫЙ ЛИЗИН
<400> 1
Leu Asn Tyr Ile Asp Leu Asp Leu Val Xaa Leu Ser Thr Tyr Ala Ser
1 5 10 15
Ile Asn Phe Gln Lys
20
<---
Реферат
Изобретение относится к области органической химии, а именно гетероциклическому соединению формулы I и II, где R1выбирают из:R2и R3вместе с атомом азота, к которому присоединены оба R2и R3, образуют кольцо, выбранное из пиперидинила, пиперазинила, 2-окса-8-азаспиро[4.5]декан-8-ила, 8-азаспиро[4.5]декан-8-ила и пирролидинила; где указанные пирролидинил, пиперазинил, 2-окса-8-азаспиро[4.5]декан-8-ил, 8-азаспиро[4.5]декан-8-ил или пиперидинил не замещены или замещены 1-3 группами, независимо выбранными из аминогруппы, метила, этила, амино-метила, метил-аминогруппы, гидроксила, цианогруппы, фтор-метила, фтора и ((((5-метил-2-оксо-1,3-диоксол-4-ил)метокси)карбонил)амино)метила; R4представляет собой гидроксил; R5выбран из Н и метила; R6выбран из водорода, метила и фенила; R7выбран из водорода, метила, этила, фенила и бензила; R8выбран из водорода и метила; Y1выбран из N и CH; Y2представляет собой CH; Y3выбран из NН и CH2; Y4представляет собой N; Y5представляет собой N. Также изобретение относится к фармацевтической композиции на основе соединения формулы I или II, способу лечения заболевания или расстройства, опосредованного активностью SHP2, основанному на использовании указанного соединения, и к конкретным гетероциклическим соединениям. Технический результат: получены новые гетероциклические соединения, способные ингибировать SHP2. 4 н. и 6 з.п. ф-лы, 7 табл., 67 пр.
Формула
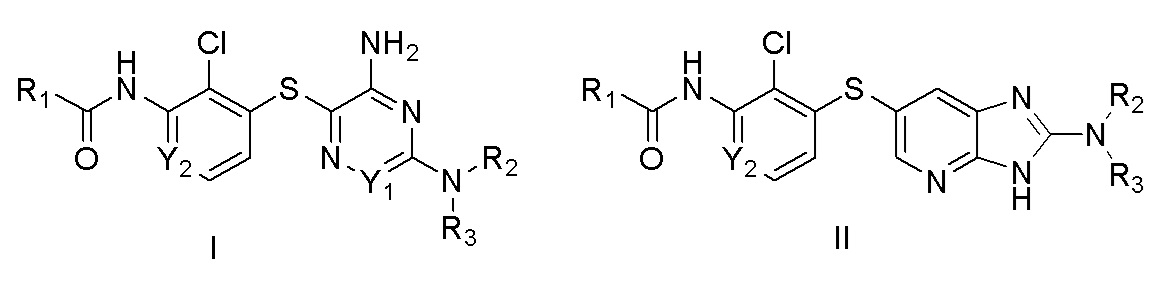
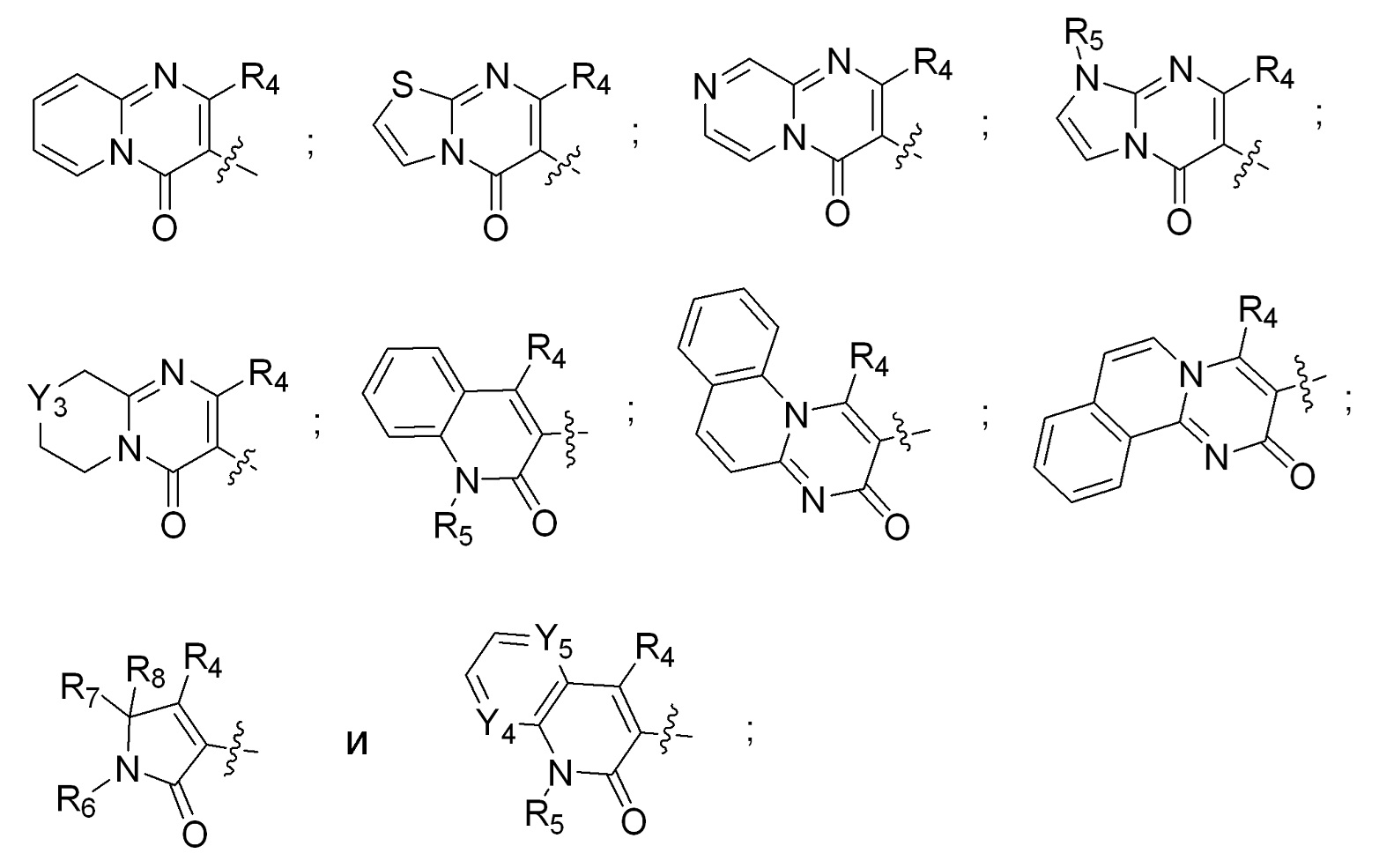

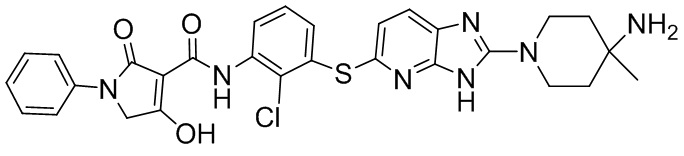
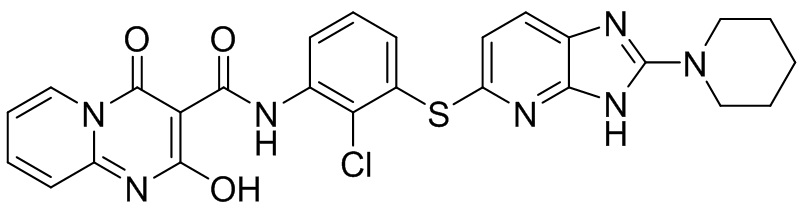
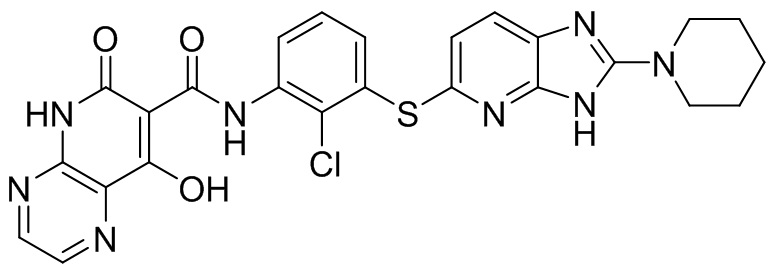

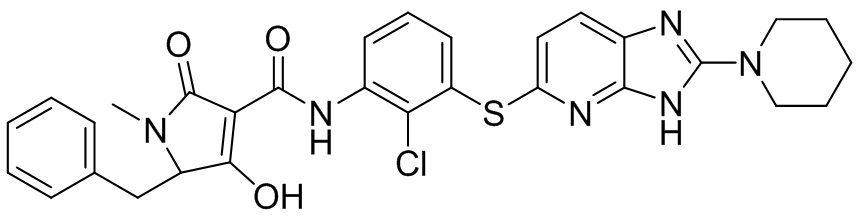

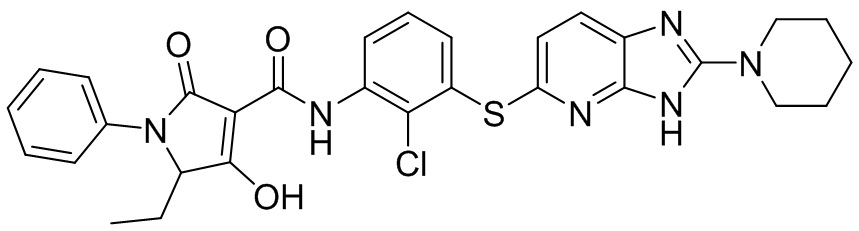


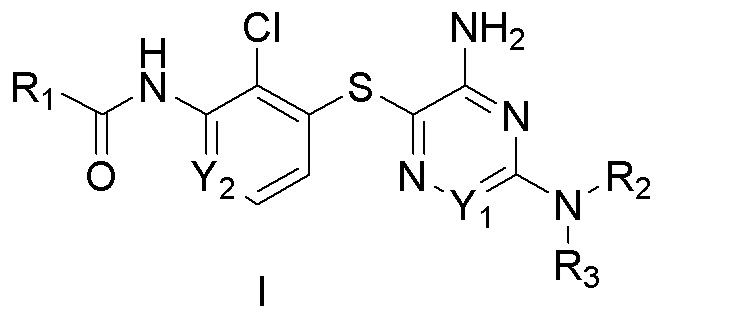
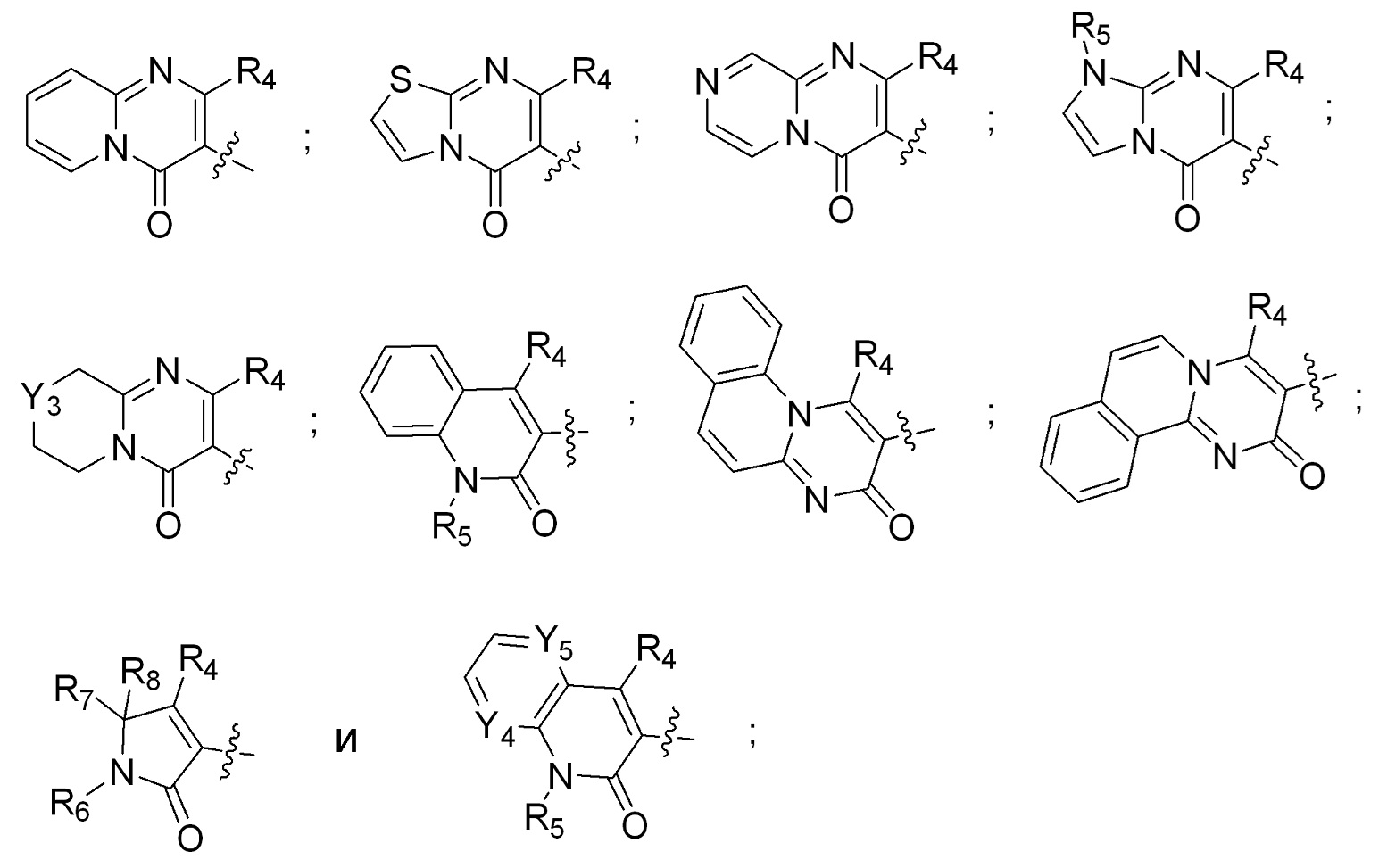
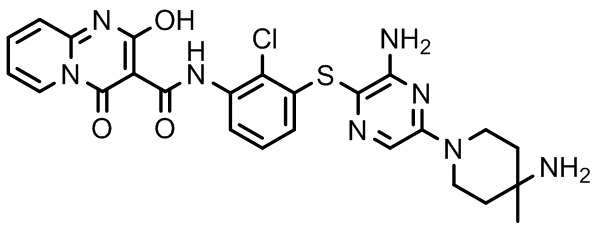



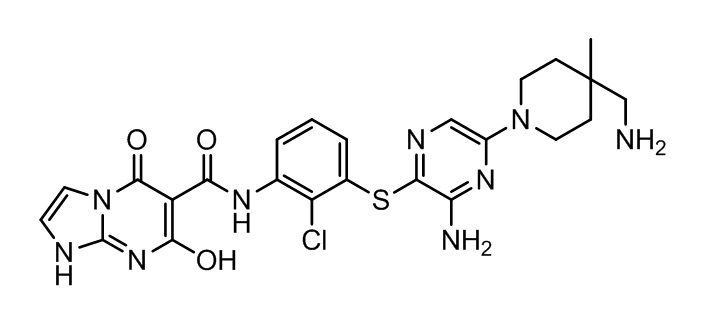



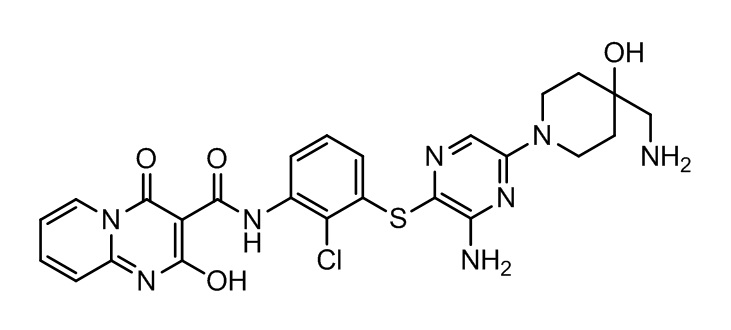
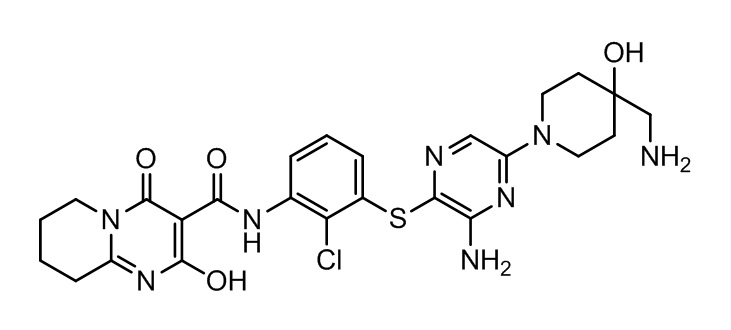
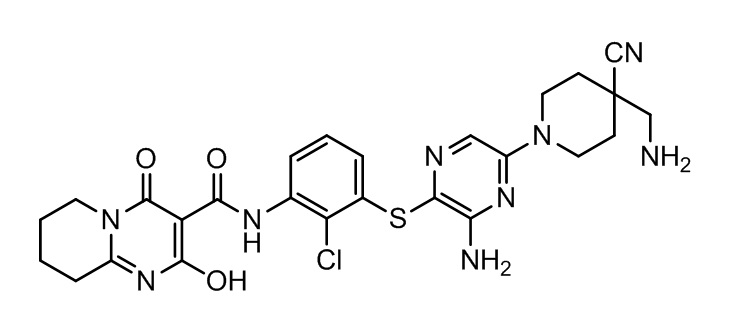

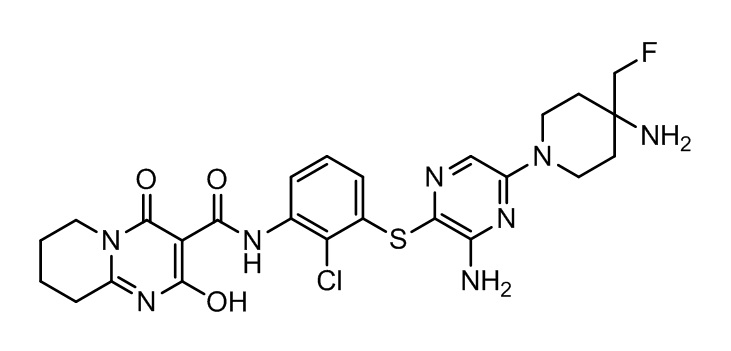



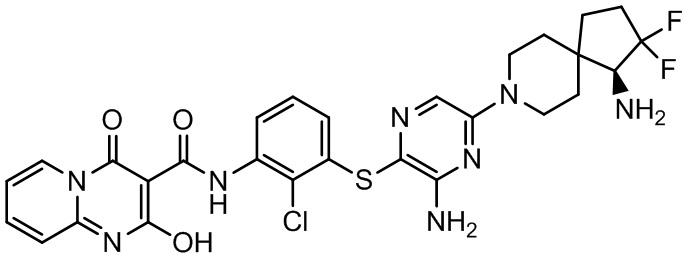




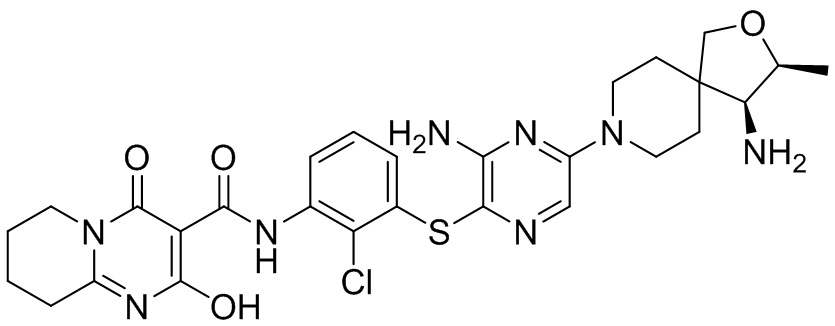


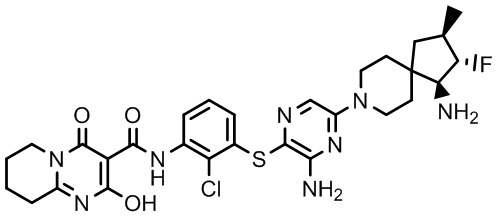
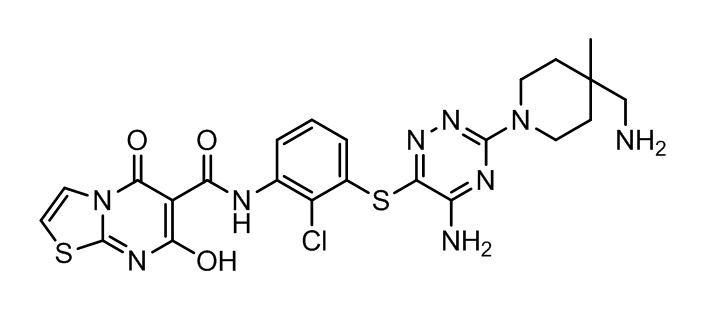
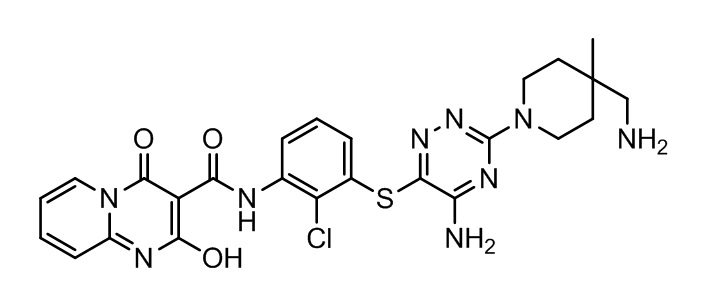
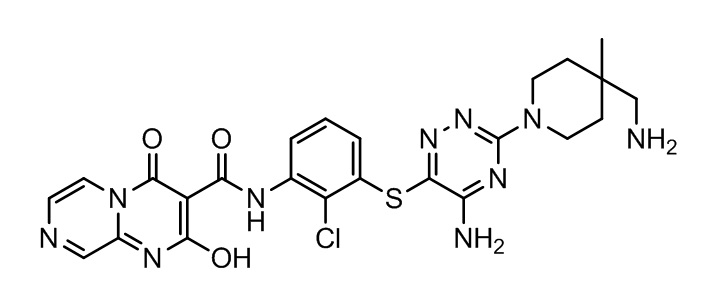
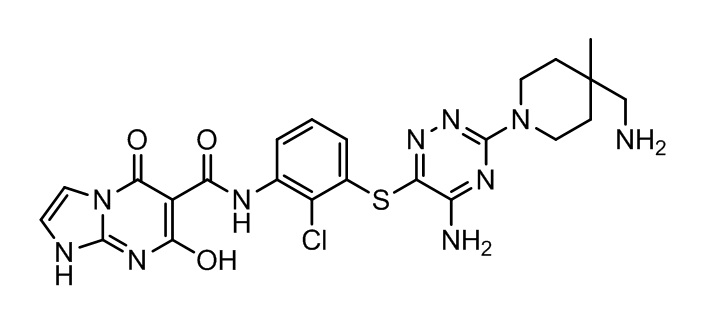



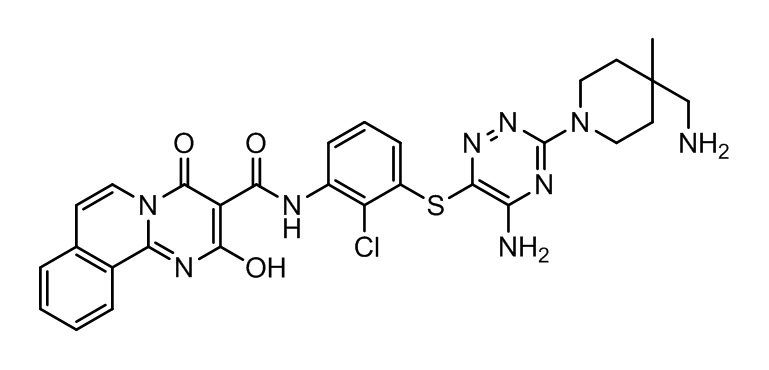




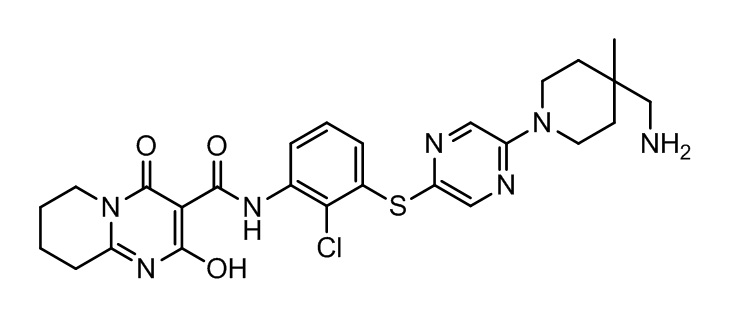

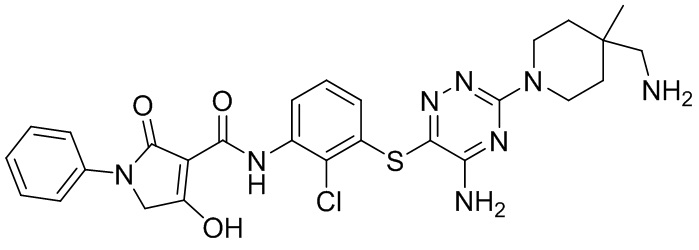

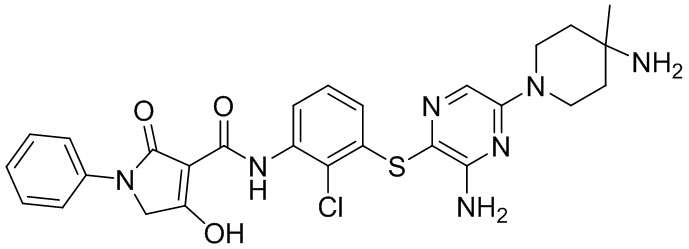
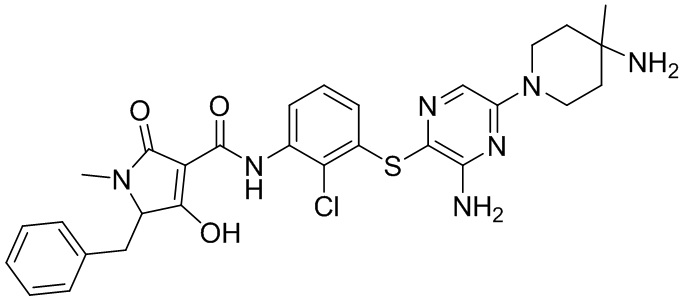

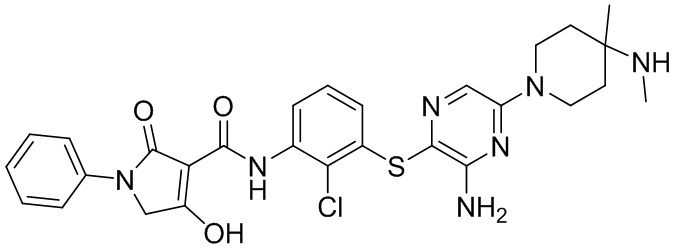
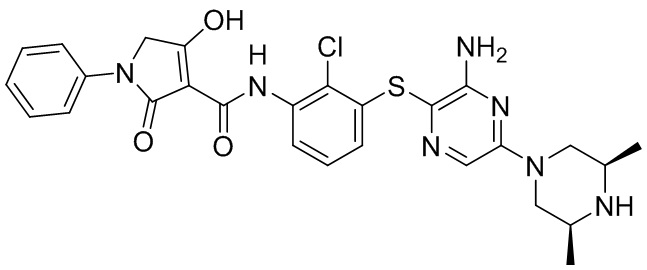
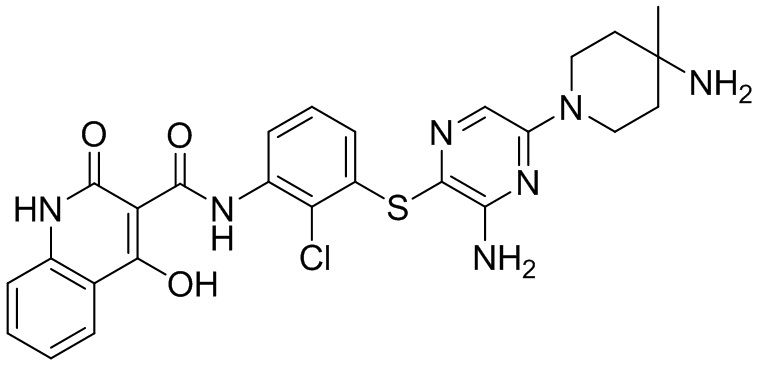
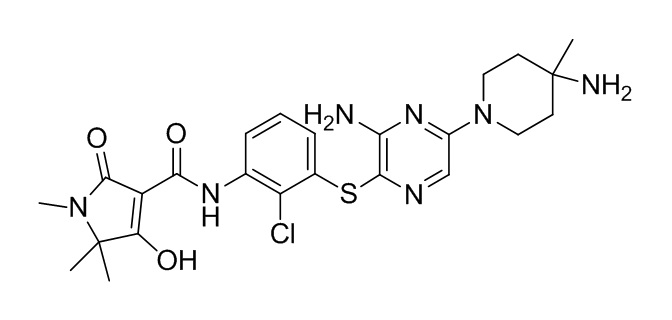
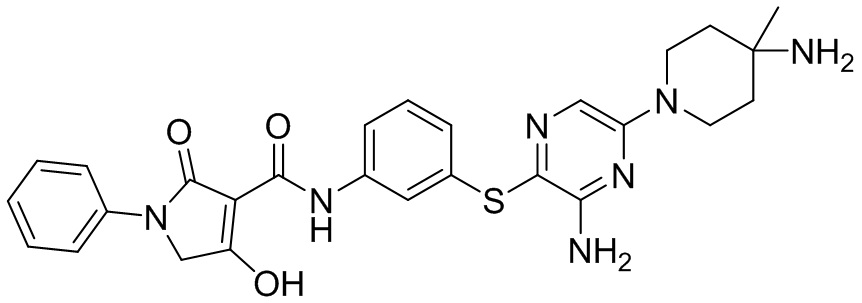
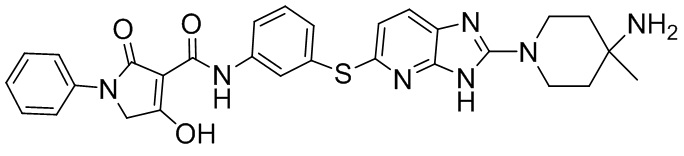
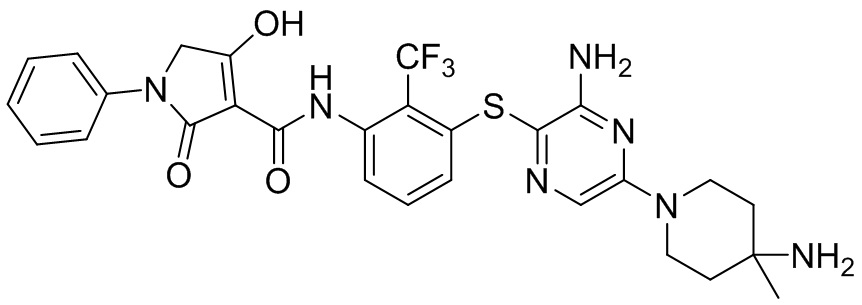
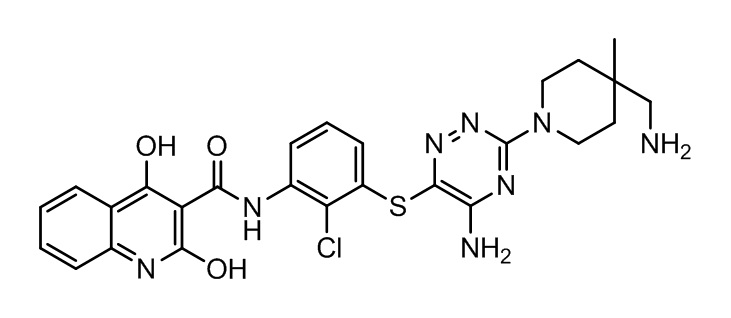

Комментарии