Пролекарства альвоцидиба, имеющие повышенную биодоступность - RU2752729C2
Код документа: RU2752729C2
Чертежи










Описание
Уровень техники
Область техники
Данное изобретение в общем относится к фосфатным пролекарствам альвоцидиба и их применению для лечения рака.
Описание известного уровня техники
Циклинзависимые киназы (ЦЗК) являются важными регуляторами, которые контролируют временной паттерн и координацию клеточного цикла. ЦЗК образуют обратимые комплексы с их обязательными циклиновыми партнерами для контроля перехода через ключевые точки клеточного цикла. Например, активированный ЦЗК4-циклин D1 комплекс контролирует развитие через G1 фазу клеточного цикла, а ЦЗК1-циклин B1 комплекс контролирует вход в митотическую фазу клеточного цикла. Известно, что эндогенные, ингибирующие циклинзависимую киназу белки (ИЦЗК) связывают или ЦЗК или циклиновый компонент и ингибируют киназную активность комплекса. При многих опухолях, таких как меланомы, рак поджелудочной железы и пищевода, эти природные ИЦЗК либо отсутствуют, либо мутированы. Таким образом, селективные ингибиторы ЦЗК могут показать себя как эффективные химиотерапевтические агенты.
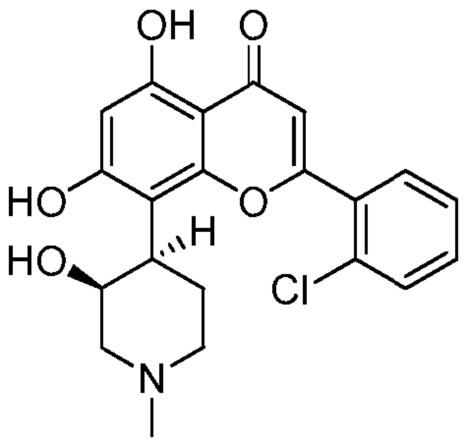
Альвоцидиб (также известный как Флавопиридол) является синтетическим флавоном, имеющим следующую структуру:
Альвоцидиб является мощным и селективным ингибитором ЦЗК и обладает противоопухолевым действием против различных колоний опухолевых клеток, таких как человеческая карцинома легких и карцинома молочной железы, а также ингибирует рост опухоли в моделях ксенотрансплантатов. Было показано, что Альвоцидиб вызывает остановку в обеих G1 и G2 фазах клеточного цикла, а также ингибирует транскрипцию, управляемую полимеразой II через ингибирование ЦЗК9. Через ингибирование ЦЗК9, которая образует часть комплекса, известного как позитивный транскрипционный фактор элонгации или P-TEFb, лечение альвоцидибом восстанавливает экспрессию ключевых онкогенов, таких как MYC, и ключевых анти-апоптозных белков, таких как MCL1. Следовательно, альвоцидиб является привлекательным терапевтическим агентом для рака и в настоящее время проходит клинические исследования у пациентов с рецидивирующей/трудно поддающейся лечению ОМЛ.
Пероральное введение альвоцидиба ограничено желудочно-кишечной токсичностью и ограниченной пероральной биодоступностью. Далее, предклинические исследования показали, что пролонгированное воздействие может быть важным для получения максимальной активности альвоцидиба. Следовательно, непрерывные внутривенные вливания широко применяют в исследованиях на людях. Альтернативное гибридное введение, включая внутривенные болюсы с последующим медленным вливанием, также применяется, но до сих пор не ни одного доклада о пероральном введении терапевтически эффективного количества альвоцидиба.
Хотя достигнуто определенное развитие, все еще остается необходимость в данной области техники в повышении пероральной биодоступности альвоцидиба. Данное изобретение удовлетворяет эту потребность и предоставляет соответствующие преимущества.
Сущность изобретения
Коротко, варианты данного изобретения представляют фосфатные пролекарства альвоцидиба, имеющие повышенную биодоступность по отношению к исходному соединению альвоцидиба. Следовательно, в одном варианте представлено соединение, имеющее следующую структуру (I):

(I)
или его стереоизомер, таутомер или фармацевтически приемлемая соль, где:
один из R1, R2 или R3 является -P(=O)(OH)2, и другие два из R1, R2 и R3 каждый являются H.
Другие варианты относятся к фармацевтической композиции, включающей фармацевтически приемлемый носитель или наполнитель и соединение структуры (I). Также представлен способ применения соединения структуры (I) и содержащих его фармацевтических композиций для лечения заболевания, связанного с сверхэкспрессией циклинзависимой киназы (ЦЗК) у млекопитающего, нуждающегося в таковом.
Эти и другие аспекты изобретения будут понятны при прочтении следующего подробного описания. Для этого здесь представлены различные ссылки, которые более подробно описывают определенную информацию известного уровня техники, методики, соединения и/или композиции, каждая из которых включена сюда в качестве ссылки полностью.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фиг. 1 показан фармакокинетический профиль альвоцидиба и соединения IB после введения соединения IB крысам Sprague Dawley.
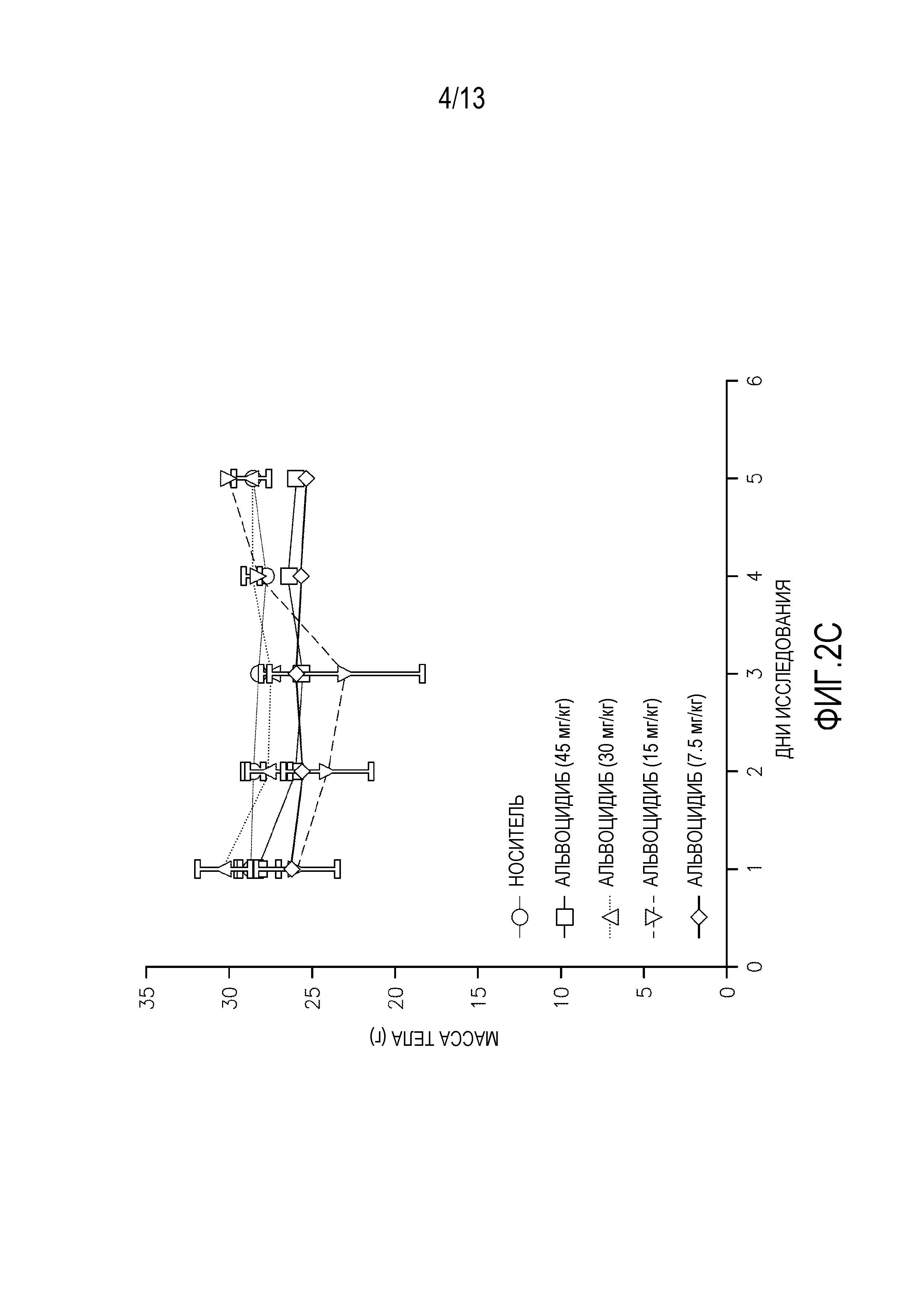
На фиг. 2A-D изображены массы тела мышей, леченных одной дозой (фиг. 2A-B перорально, фиг. 2C-D внутривенно) альвоцидиба или соединения IB.
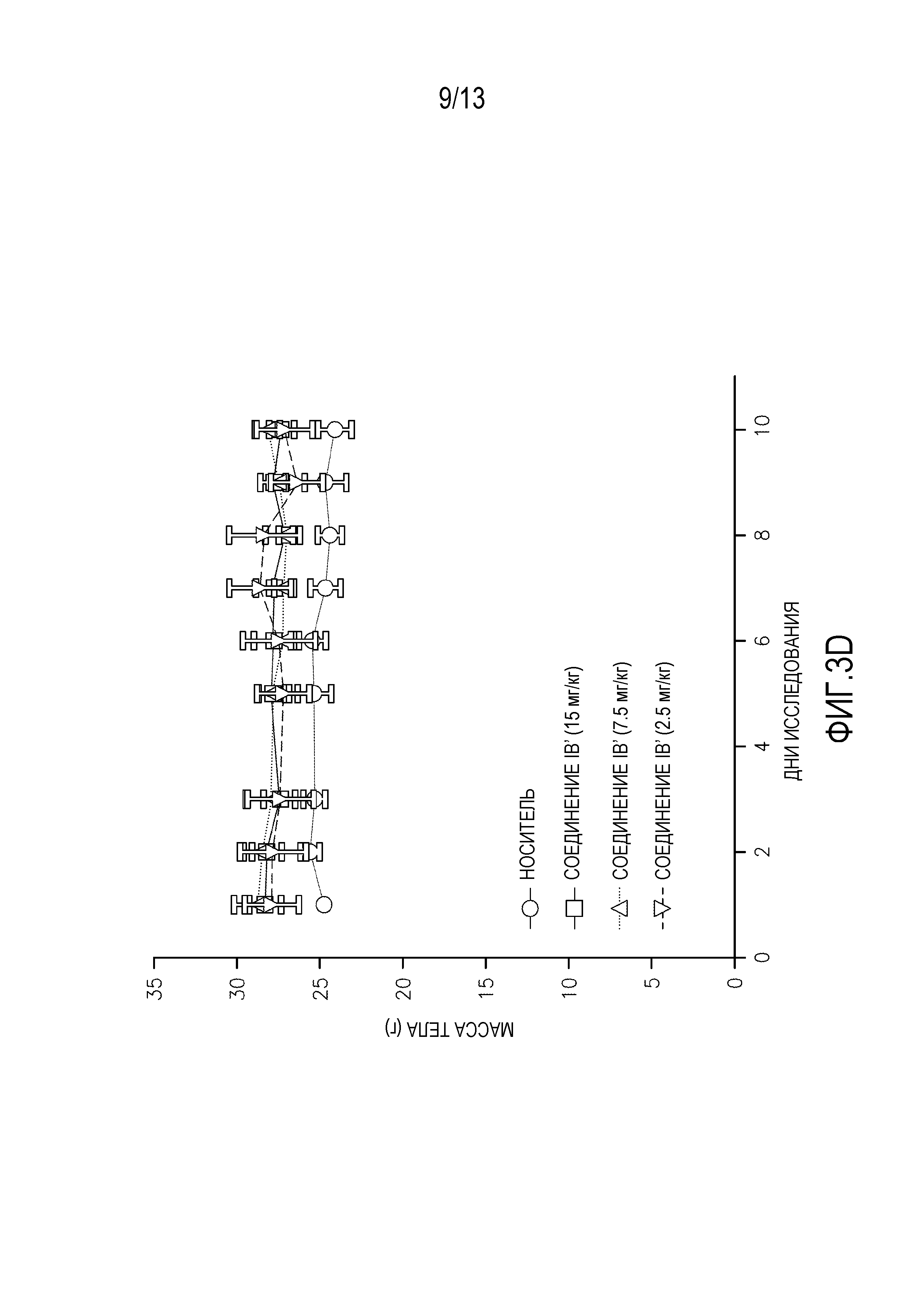
На фиг. 3A-D показаны массы тела мышей, леченных суточными дозами (фиг. 3A-B перорально, фиг. 3C-D внутривенно) альвоцидиба или соединения IB.
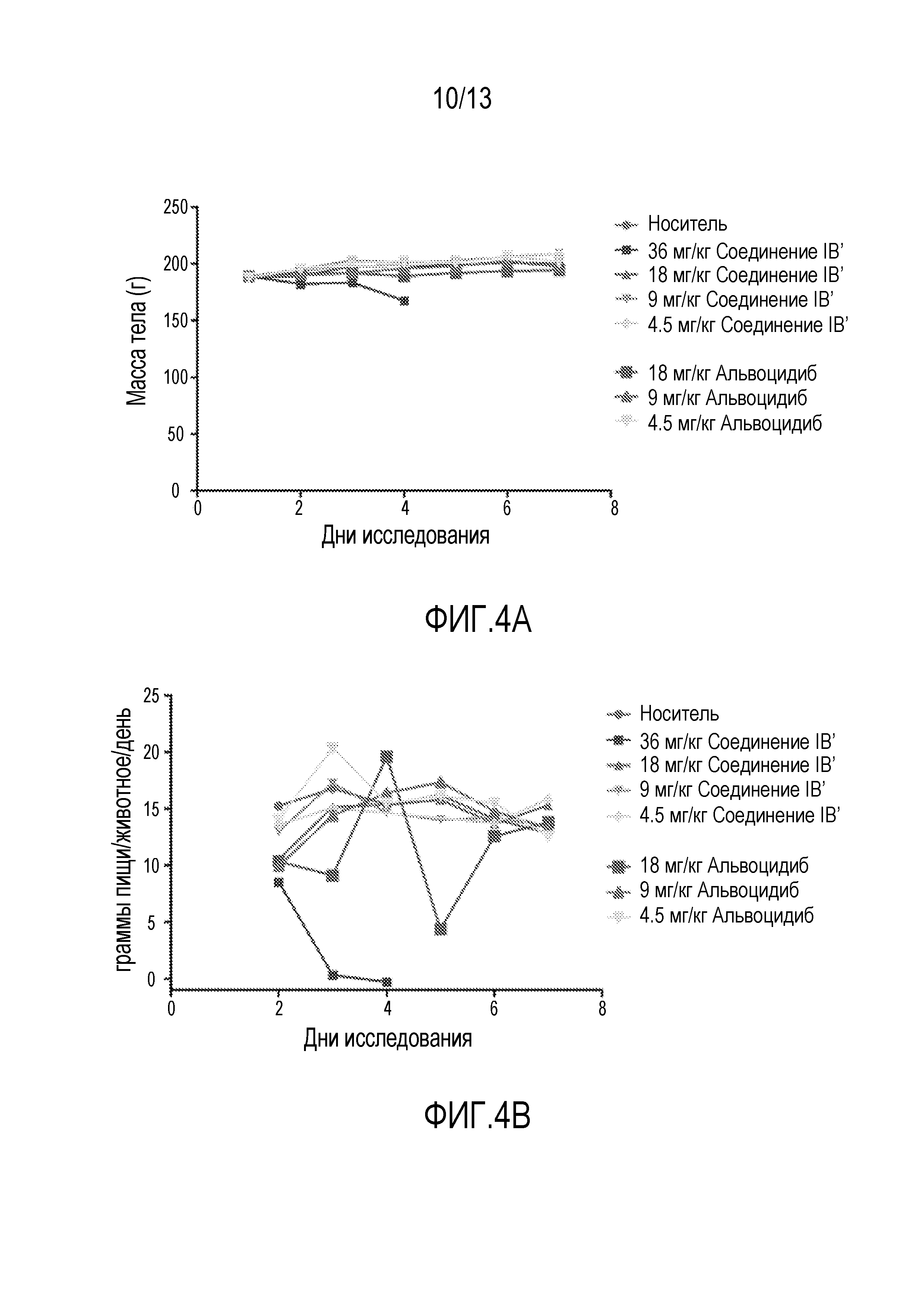
На фиг. 4A-B показаны массы тела и потребление пищи крыс, леченных одной дозой (перорально) альвоцидиба или соединения IB.
На фиг. 5A-B показан in vivo объем опухоли и масса тела после введения соединения IB во время исследования эффективности ксенотрансплантата.
На фиг. 6A-B изображено снижение экспрессии белка MCL-1 после лечения соединением IB во время фармакодинамического исследования ксенотрансплантата.
ПОДРОБНОЕ ОПИСАНИЕ
В следующем описании представлены определенные конкретные подробности для получения четкого понимания различных вариантов изобретения. Однако специалист в данной области техники поймет, что изобретение может быть осуществлено на практике без этих подробностей.
Если контекст не требует иного, в данном описании и формуле изобретения слово ʺвключатьʺ и его варианте, такие как ʺвключаетʺ и ʺвключающийʺ должно рассматриваться в открытом включительном смысле, то есть, как ʺвключающий, но не ограниченный имиʺ.
В данном описании ссылка на ʺодин вариантʺ или ʺвариантʺ означает, что конкретная особенность, структура или характеристика, описанная связи с вариантом, включена в, по крайней мере, один вариант данного изобретения. Таким образом, присутствие фраз ʺв одном вариантеʺ или ʺв вариантеʺ в различных местах данного описания не обязательно относится к одному и тому же варианту. Более того, конкретные особенности или характеристики могут быть объединены любым подходящим образом водном или более вариантах.
Варианты данного изобретения включают фосфатные пролекарства альвоцидиба. ʺФосфатʺ относится к -OP(=O)(OH)2 части. Для простоты иллюстрации, фосфатные части здесь часто изображены в дипротонированной форме, но также существуют в монопротонированной (-OP(=O)(OH)(O-)) и непротонированной формах (-OP(=O)(O-)2), в зависимости от pH. Моно- и непротонированные формы обычно связаны с противоионом так, что соединения имеют форму фармацевтически приемлемой соли. Такие моно- или непротонированные формы и их фармацевтически приемлемые соли включены в объем изобретения, даже если это отдельно не показано на химических структурах.
ʺПролекарствоʺ означает соединение, которое может быть превращено в физиологических условиях или сольволизом в биологически активное соединение, описанное здесь (например, соединение структуры (I)). Таким образом, термин "пролекарство" относится к предшественнику биологически активного соединения, которое фармацевтически приемлемо. В некоторых аспектах, пролекарство является неактивным при введении пациенту, но превращается in vivo в активное соединение, например, гидролизом. Пролекарство часто предоставляет преимущества растворимости, совместимости с тканями или отложенного выделения в организме млекопитающего (см., например, Bundgard, H., Design of Prodrugs (1985), pp. 7-9, 21-24 (Elsevier, Amsterdam). Обсуждение пролекарств представлено в Higuchi, T., et al., "Prodrugs as Novel Delivery Systems," A.C.S. Symposium Series, Vol. 14, и в Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987, обе которых включены сюда полностью в качестве ссылок.
ʺСоединение в соответствии с данным изобретениемʺ относится к соединению структуры (I) и его подструктурам, как определено здесь.
Варианты изобретения, описанные здесь, также охватывают все фармацевтически приемлемые соединения структуры (I) меченные изотопами через замену одного или более атомов атомом, имеющим другую атомную массу или массовое число. Примеры изотопов, которые могут быть введены в описываемые соединения, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора, хлора и йода, такие как2H,3H,11C,13C,14C,13N,15N,15O,17O,18O,31P,32P,35S,18F,36Cl,123I и125I, соответственно. Эти радиомеченные соединения могут применяться для определения или измерения эффективности соединений через анализ, например, места или способа действия, или сродства связывания с фармакологически важным местом действия. Определенные изотопно-меченные соединения структуры (I), например, включающие радиоактивный изотоп, применяют в анализе распределения лекарственного средства и/или субстрата в ткани. Радиоактивные изотопы тритий, т.е.3H, и углерод-14, т.е.14C, особенно полезны для этой цели благодаря простоте их введения и готовым средствам определения.
Замещение более тяжелыми изотопами, такими как дейтерий, т.е.2H, может дать определенные терапевтические преимущества благодаря большей метаболической стабильности, например, повышенному периоду полураспада in vivo или пониженной дозировке, и, следовательно, может быть предпочтительным в определенных обстоятельствах.
Замещение позитронно-активными изотопами, такими как11C,18F,15O и13N, может применяться в исследованиях позитронно-эмиссионной томографии (ПЭТ) для исследования степени занятости рецептора субстратом. Изотопно-меченные соединения структуры (I) в общем могут быть получены обычными методами, известными специалистам в данной области техники, или способами, аналогичными тем, которые описаны в разделе Препараты и примеры ниже, с применением подходящего изотопно-меченного реагента вместо не меченого реагента, применяемого ранее.
Варианты данного изобретения, описанные здесь, также охватывают in vivo метаболические продукты описанных соединений. Такие продукты могут получаться, например, при окислении, восстановлении, гидролизе, амидировании, эстерификации и подобных превращениях вводимого соединения, преимущественно, в результате ферментных процессов. Следовательно, варианты данного изобретения включают соединения, полученные способом, включающим введение соединения в соответствии с данным изобретением млекопитающему в течение времени, достаточного для получения метаболического продукта. Такие продукты обычно идентифицируют введением радиомеченного соединения в соответствии с данным изобретением в определяемой дозе животному, такому как крыса, мышь, морская свинка, обезьяна или человек, в течение времени, достаточного для прохождения метаболизма, и выделением продуктов его превращения из мочи, крови или других биологических образцов.
ʺФармацевтически приемлемый носитель, разбавитель или наполнительʺ включает, без ограничений, любой адъювант, носитель, наполнитель, глидант, подсластитель, разбавитель, консервант, краситель, вкусовую добавку, поверхностно-активное вещество, смачивающий агент, диспергирующий агент, суспендирующий агент, стабилизатор, изотонический агент, растворитель или эмульгатор, который одобрен Управление по контролю за продуктами питания и лекарствами США как приемлемый для применения у человека или домашних животных.
ʺФармацевтически приемлемая сольʺ включает кислотно- и основно-аддитивные соли.
ʺФармацевтически приемлемая кислотно-аддитивная сольʺ относится к солям, которые сохраняют биологическую эффективность и свойства свободных оснований, которые не являются биологически или другим образом нежелательными, и которые получают с неорганическими кислотами, такими как, но не ограниченными ими, хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и подобные, и органическими кислотами, такими как, но не ограниченными ими, уксусная кислота, 2,2-дихлоруксусная кислота, адипиновая кислота, альгиновая кислота, аскорбиновая кислота, аспарагиновая кислота, бензолсульфоновая кислота, бензойная кислота, 4-ацетамидобензойная кислота, камфорная кислота, камфор-10-сульфоновая кислота, каприновая кислота, капроновая кислота, каприловая кислота, карбоновая кислота, коричная кислота, лимонная кислота, цикламиновая кислота, додецилсерная кислота, этан-1,2-дисульфоновая кислота, этансульфоновая кислота, 2-гидроксиэтансульфоновая кислота, муравьиная кислота, фумаровая кислота, галактаровая кислота, гентизиновая кислота, глюкогептоновая кислота, глюконовая кислота, глюкуроновая кислота, глутаминовая кислота, глутаровая кислота, 2-оксоглутаровая кислота, глицерофосфорная кислота, гликолевая кислота, гиппуровая кислота, изомасляная кислота, молочная кислота, лактобионовая кислота, лауриновая кислота, малеиновая кислота, яблочная кислота, малоновая кислота, миндальная кислота, метансульфоновая кислота, муциновая кислота, нафталин-1,5-дисульфоновая кислота, нафталин-2-сульфоновая кислота, 1-гидрокси-2-нафтойная кислота, никотиновая кислота, олеиновая кислота, оротовая кислота, щавелевая кислота, пальмитиновая кислота, памовая кислота, пропионовая кислота, пироглутаминовая кислота, пировиноградная кислота, салициловая кислота, 4-аминосалициловая кислота, себациновая кислота, стеариновая кислота, янтарная кислота, винная кислота, тиоциановая кислота, п-толуолсульфоновая кислота, трифторуксусная кислота, ундециленовая кислота и подобные.
ʺФармацевтически приемлемые основно-аддитивные солиʺ относятся к солям, которые сохраняют биологическую эффективность и свойства свободных кислот, которые не являются биологически или другим образом нежелательными. Эти соли получают добавлением неорганического основания или органического основания к свободной кислоте. Соли, получаемые из неорганических оснований, включают, но не ограничены ими, соли натрия, калия, лития, аммония, кальция, магния, железа, цинка, меди марганца, алюминия и подобные. Предпочтительные неорганические соли включают соли аммония, натрия, калия, кальция и магния. Соли, полученные из органических оснований, включают, но не ограничены ими, соли первичных, вторичных и третичных аминов, замещенных амино, включая природные замещенные амины, циклических аминов и основных ионообменных смол, таких как аммиак, изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин, диэтаноламин, этаноламин, деанол, 2-диметиламиноэтанол, 2-диэтиламиноэтанол, дициклогексиламин, лизин, аргинин, гистидин, кофеин, прокаин, гидрабамин, холин, бетаин, бенетамин, бензатин, этилендиамин, глюкозамин, метилглюкамин, теобромин, триэтаноламин, трометамин, пурины, пиперазин, пиперидин, N-этилпиперидин, полиаминовые смолы и подобные. Особенно предпочтительными органическими основаниями являются изопропиламин, диэтиламин, этаноламин, триметиламин, дициклогексиламин, холин и кофеин.
Часто кристаллизация дает сольват соединения в соответствии с данным изобретением. В данном описании, ʺсольватʺ относится к агрегату, который содержит одну или более молекул соединения в соответствии с данным изобретением с одной или более молекулами растворителя. Растворителем может быть вода, в этом случае сольватом может быть гидрат. Альтернативно, растворителем может быть органический растворитель. Таким образом, варианты соединений в соответствии с данным изобретением могут существовать в виде гидрата, включая моногидрат, дигидрат, полугидрат, полуторагидрат, тригидрат, тетрагидрат и подобные, а также соответствующие сольватированные формы. Варианты соединения в соответствии с данным изобретением могут быть истинными сольватами, а в других случаях, соединение в соответствии с данным изобретением может только удерживать случайную воду или быть смесью воды и некоторого случайного растворителя.
ʺФармацевтическая композицияʺ относится к композиции соединения в соответствии с данным изобретением и среды, принятой в данной области техники для доставки биологически активного соединения млекопитающему, например, человеку. Такая среды включает все фармацевтически приемлемые носители, разбавители или наполнители.
ʺМлекопитающееʺ включает человека и домашних животных, таких как лабораторные животные и домашние животные (например, кошки, собаки, свиньи, крупный рогатый скот, овцы, козы, лошади, кролики) и не домашних животных, таких как дикие животные и подобные.
ʺЭффективное количествоʺ или ʺтерапевтически эффективное количествоʺ относится к количеству соединения в соответствии с данным изобретением, которое, при введении млекопитающему, предпочтительно, человеку, достаточно для проведения лечения, как определено ниже, заболевания, связанного со сверхэкспрессией циклинзависимой киназы (ЦЗК) у млекопитающего, предпочтительно, человека. Количество соединения в соответствии с данным изобретением, которое составляет ʺтерапевтически эффективное количествоʺ варьируется в зависимости от соединения, состояния и его тяжести, способа введения и возраста лечимого млекопитающего, но может быть определено обычным образом специалистом в данной области техники на основе его знаний и этого описания.
ʺЛечитьʺ или ʺлечениеʺ в данном описании включает лечение рассматриваемого заболевания или состояния у млекопитающего, предпочтительно, человека, страдающего рассматриваемым заболеванием или состоянием, и включает:
(i) профилактику заболевания или состояния у млекопитающего, в частности, если это млекопитающее предрасположено к состоянию, но оно еще не диагностировано;
(ii) ингибирование заболевания или состояния, т.е. остановку его развития;
(iii) облегчение заболевания или состояния, т.е. регрессию заболевания или состояния; или
(iv) облегчение симптомов заболевания или состояния, т.е. облегчение боль без устранения основного заболевания или состояния. В данном описании термины ʺзаболеваниеʺ и ʺсостояниеʺ могут применяться взаимозаменяемо или могут отличаться в том, что конкретный недуг или состояние может не иметь известного возбудителя (так, что этиология еще не разработана) и поэтому еще не распознано как заболевание, а только как нежелательное состояние или синдром, где более или менее определенный набор симптомов идентифицирован клиницистами.
Соединения в соответствии с данным изобретением или их фармацевтически приемлемые соли могут содержать один или более асимметрических центров и поэтому могут образовывать энантиомеры, диастереомеры и другие стереоизомерные формы, которые могут быть определены, с точки зрения абсолютной стереохимии, как (R)- или (S)- или как (D)- или (L)- для аминокислот. Данное изобретение включает все возможные изомеры, а также их рацемические и оптически чистые формы. Оптически активные (+) и (-), (R)- и (S)- или (D)- и (L)- изомеры могут быть получены с применением хиральных синтонов или хиральных реагентов, или разделены с помощью обычных методов, хроматографии и фракционной кристаллизации. Обычные методы получения/выделения отдельных энантиомеров включают хиральный синтез из подходящего оптически чистого предшественника или разделение рацемата (или рацемата соли или производного) с помощью, например, хиральной жидкостной хроматографии высокого давления (ЖХВД). Если описанные здесь соединения содержат олефиновые двойные связи или другие центры геометрической асимметрии, и если не указано иначе, полагают, что соединения включают оба E и Z геометрических изомера. Также включены все таутомерные формы.
ʺСтереоизомерʺ относится к соединению, полученному из одинаковых атомов, связанных одинаковыми связями, но имеющих различные трехмерные структуры, которые не являются взаимозаменяемыми. Варианты данного изобретения охватывают различные стереоизомеры и их смеси и включают ʺэнантиомерыʺ, которые относятся к двум стереоизомерам, молекулы которых является неналагаемыми зеркальными изображениями друг друга.
ʺТаутомерʺ относится к протонному сдвигу от одного атома молекулы к другому атому той же молекулы. Варианты данного изобретения включают таутомеры любых указанных соединений.
I. Соединения
Как отмечено выше, варианты данного описания относятся к пролекарствам альвоцидиба, имеющим повышенную биодоступность по отношению к исходному соединению. Неожиданно, эксперименты, проведенные в поддержку данного изобретения, демонстрируют, что монофосфатный аналог альвоцидиба имеет биодоступность приблизительно в 1,3 раза больше, чем у исходного соединения альвоцидиба при пероральном введении мышам CD-1, и больше, чем в 8 раз по сравнению с соответствующими дифосфатными пролекарствами. Описанные монофосфатные соединения метаболизируют до альвоцидиба in vivo и, не будучи связанными теорией, полагают, что повышение биодоступности альвоцидиба, выделенного из монофосфатных пролекарств, по сравнению с исходным соединением альвоцидиба, связано с меньшей скоростью метаболизма пролекарства по сравнению с альвоцидибом. Другие ожидаемые преимущества данных соединений включают повышенную растворимость в типовых фармацевтических композициях, в воде и жидкостях тела, и пониженную токсичность по отношению к исходному соединению альвоцидиба при пероральном введении.
Следовательно, в одном варианте представлено соединение, имеющее следующую структуру (I):

(I)
или его стереоизомер, таутомер или фармацевтически приемлемая соль, где:
один из R1, R2 или R3 является -P(=O)(OH)2, и два другие из R1, R2 и R3 каждый является H.
В определенных вариантах, соединение имеет следующую структуру (I′):

(I′)
В других вариантах, соединение имеет следующую структуру (IA):
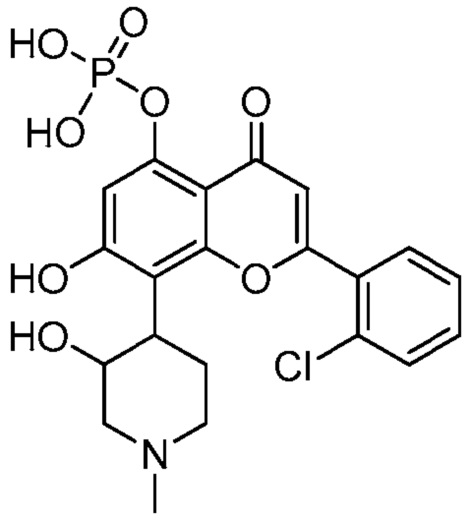
(IA)
В некоторых других вариантах, соединение имеет следующую структуру (IA′):

(IA′)
В еще других вариантах, соединение имеет следующую структуру (IB):

(IB)
В других различных вариантах, соединение имеет следующую структуру (IB′):

(IB′)
В других вариантах, соединение имеет следующую структуру (IC):
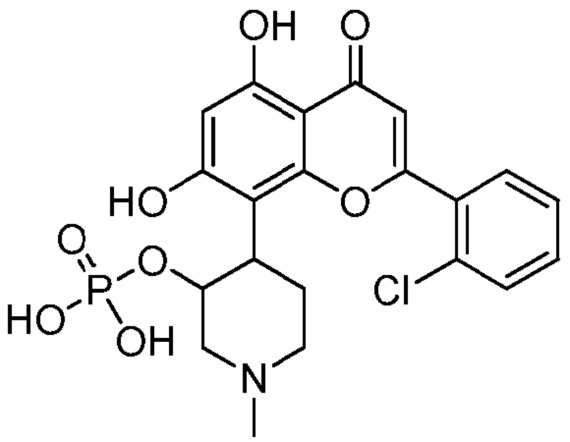
(IC)
В некоторых других различных вариантах, соединение имеет следующую структуру (IC′):
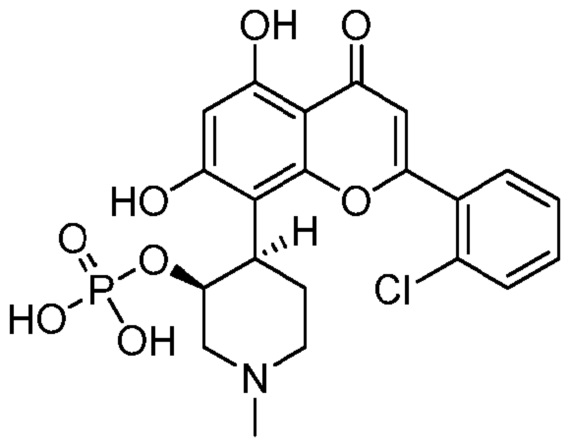
(IC′)
В некоторых вариантах любое из указанных выше соединений имеет форму фармацевтически приемлемой соли. Солью может быть кислотно-аддитивная соль или основно-аддитивная соль. Например, солью может быть соль амина, полученная протонированием N-метилпиперазиновой части (например, соль HCl и подобные). В других вариантах, соль получают на фосфате, и соединения имеют форму моно- или дисолей фосфатной группы (например, моно- иди динатрийфосфат и подобные). Все фармацевтически приемлемые соли указанных выше соединений включены в объем изобретения.
Также представлены фармацевтические композиции, содержащие фармацевтически приемлемый носитель или наполнитель и любое из представленных выше соединений (т.е., соединение структуры (I), (I′), (IA), (IA′), (IB), (IB′), (IC) или (IC′)). Предпочтительно, описанные здесь соединения имеют повышенную биодоступность относительно исходного соединения альвоцидиба, и эти определенные варианты относятся к фармацевтическим композициям, составленным для перорального введения. Любой носитель и/или наполнитель, известный в данной области техники для пероральных композиций, может применяться в этих вариантах, в дополнение к другим носителям и/или наполнителям, полученным специалистом в данной области техники.
Для целей введения, соединения в соответствии с данным изобретением могут вводиться в виде химического сырья или могут быть составлены в фармацевтические композиции. Варианты фармацевтических композиций в соответствии с данным изобретением включают соединение структуры (I) и фармацевтически приемлемый носитель, разбавитель или наполнитель. Соединение структуры (I) присутствует в композиции в количестве, которое эффективно для лечения конкретного рассматриваемого заболевания или состояния - то есть, обычно в количестве, достаточном для лечения заболевания, связанного со сверхэкспрессией циклинзависимой киназы (ЦЗК), и, предпочтительно, имеет приемлемую для пациента токсичность. Биодоступность соединений структуры (I) может быть определена специалистом в данной области техники, например, как описано в примерах ниже. Подходящие концентрации и дозы могут быть легко определены специалистом в данной области техники.
Введение соединений в соответствии с данным изобретением или их фармацевтически приемлемых солей, в чистой форме или в подходящей фармацевтической композиции, может проводиться любым приемлемым способом введения агентов для подобных целей. Фармацевтические композиции из вариантов в соответствии с данным изобретением могут быть получены объединением соединения в соответствии с данным изобретением с подходящим фармацевтически приемлемым носителем, разбавителем или наполнителем, и могут быть составлены в препараты в твердой, полутвердой, жидкой или газообразной формах, такие как таблетки, капсулы, порошки, гранулы, мази, растворы, суппозитории, инъекции, препараты для ингаляции, гели, микросферы и аэрозоли. Типовые способы введения таких фармацевтических композиций включают, без ограничения, пероральный, местный, чрезкожный, ингаляционный, парентеральный, подъязычный, буккальный, ректальный, вагинальный и интраназальный. Термин парентеральный в данном описании включает подкожные инъекции, внутривенные, внутримышечные, внутригрудинные инъекции или вливания. Фармацевтические композиции в соответствии с данным изобретением составлены так, чтобы обеспечить биодоступность содержащихся в них активных ингредиентов при введении композиции пациенту. Композиции, вводимые субъекту или пациенту, имеют форму одной или более стандартной лекарственной формы где, например, таблетка может быть единичной лекарственной формой, и контейнер с соединением в соответствии с данным изобретением в аэрозоли может содержать множество доз. Актуальные способы получения таких лекарственных форм известны или очевидны специалистам в данной области техники; например, см. Remington: The Science and Practice of Pharmacy, 20th Edition (Philadelphia College of Pharmacy and Science, 2000). Вводимая композиция, в любом случае, содержит терапевтически эффективное количество соединения в соответствии с данным изобретением или его фармацевтически приемлемой соли для лечения рассматриваемого заболевания или состояния в соответствии с положениями данного изобретения.
Фармацевтическая композиция из некоторых вариантов изобретения может быть в форме твердого вещества или жидкости. В одном аспекте, носители имеют форму частиц так, что композиции принимают, например, форму таблетки или порошка. Носители могут быть жидкими, при этом композиция является, например, пероральным сиропом, жидкостью для инъекций или аэрозолью, которую применяют, например, для ингаляций.
Если она предназначена для перорального введения, фармацевтическая композиция предпочтительно имеет твердую или жидкую форму, где в качестве рассматриваемых здесь твердых или жидких форм рассматриваются полутвердые, полужидкие, суспендированные или гелевые формы.
В качестве твердой композиции для перорального введения, фармацевтическая композиция может быть составлена в виде порошка, гранул, прессованных таблеток, пилюль, капсул, жевательной резинки, вафель или подобных форм. Такие твердые композиции обычно содержат один или более инертных разбавителей или съедобных носителей. Такая твердая композиция обычно содержит один или более инертных разбавителей или съедобных носителей. Кроме того, может присутствовать один или более из: связующих агентов, таких как карбоксиметилцеллюлоза, этилцеллюлоза, микрокристаллическая целлюлоза, трагакант или желатин; наполнителей, таких как крахмал, лактоза или декстрины, разрыхляющих агентов, таких как альгиновая кислота, альгинат натрия, Примогель, кукурузный крахмал и подобные; смазывающих агентов, таких как стеарат магния или Стеротекс; глидантов, таких как коллоидная двуокись кремния; подсластителей, таких как сахароза или сахарин; вкусовых добавок, таких как перечная мята, метилсалицилат или апельсин; и красителя.
Если фармацевтическая композиция имеет форму капсулы, например, желатиновой капсулы, она может содержать, кроме материалов указанного выше типа, жидкий носитель, такой как полиэтиленгликоль или масло.
Фармацевтическая композиция может быть в форме жидкости, например, эликсира, сиропа, раствора, эмульсии или суспензии. Жидкость может быть для перорального введения или для введения инъекцией, в качестве двух примеров. Если она предназначена для перорального введения, предпочтительная композиция содержит, кроме соединений в соответствии с данным изобретением, один или более из подсластителя, консервантов, красителя и вкусовой добавки. В композиции для введения инъекцией может быть добавлен один или более из поверхностно-активного вещества, консерванта, смачивающего агента, диспергирующего агента, суспендирующего агента, буфера, стабилизатора и изотонического агента.
Жидкие фармацевтические композиции из некоторых вариантов изобретения, являются ли они растворами, суспензиями или другими подобными формами, могут включать один или более из следующих адъювантов: стерильные разбавители, такие как вода для инъекций, солевой раствор, предпочтительно, физиологический раствор, раствор Рингера, изотонический хлорид натрия, нелетучие масла, такие как синтетические моно- или диглицериды, которые могут служить в качестве растворителя или суспендирующей среде, полиэтиленгликоли, глицерин, пропиленгиколь или другие растворители; антибактериальные агенты, такие как бензиловый спирт или метилпарабен; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатирующие агенты, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетаты, цитраты или фосфаты, и агенты для корректировки тоничности, такие как хлорид натрия или декстроза. Парентеральные препараты могут быть заключены в ампулы, одноразовые шприцы или многоразовые флаконы из стекла или пластика. Физиологический раствор является предпочтительным адъювантом. Фармацевтическая композиция для инъекций предпочтительно является стерильной.
Жидкая фармацевтическая композиция из определенных вариантов изобретения, предназначенная для парентерального или перорального введения, должна содержать такое количество соединения в соответствии с данным изобретением, чтобы получить подходящую дозу.
В некоторых вариантах, фармацевтическая композиция в соответствии с данным изобретением может быть предназначена для местного введения, в этом случае носитель может подходящим образом содержать основу для раствора, эмульсии, мази или геля. Основа, например, может содержать один или более из следующих: вазелин, ланолин, полиэтиленгликоли, пчелиный воск, минеральное масло, разбавители, такие как вода и спирт, и эмульгаторы и стабилизаторы. Загущающие агенты могут присутствовать в фармацевтической композиции для местного введения. Если она предназначена для чрезкожного введения, композиция может включать чрезкожный пластырь или устройство для ионофореза.
Фармацевтическая композиция из различных вариантов изобретения может быть предназначена для ректального введения в форме, например, суппозиториев, которые могут плавиться в прямой кишке и выделять лекарственное средство. Композиция для ректального введения может содержать масличное основание в качестве подходящего не раздражающего наполнителя. Такие основания включают, без ограничений, ланолин, масло какао и полиэтиленгликоль.
Варианты фармацевтической композиции в соответствии с данным изобретением могут включать различные материалы, которые модифицируют физическую форму твердой или жидкой лекарственной формы. Например, композиция может включать материалы, которые образуют покрытие вокруг активных ингредиентов. Материалы, которые образуют покрытие, обычно являются инертными и могут быть выбраны из, например, сахара, шеллака и других энтеросолюбильных покрытий. Альтернативно, активные ингредиенты могут находиться в желатиновой капсуле.
Фармацевтическая композиция из некоторых вариантов изобретения в твердой и жидкой форме могут включать агент, который связывается с соединением изобретения и, тем самым, способствует доставке соединения. Подходящие агенты, которые могут действовать таким образом, включают моноклональное или поликлональное антитело, белок или липосому.
Фармацевтическая композиция из других вариантов изобретения может включать лекарственные формы, которые могут вводиться в виде аэрозоля. Термин аэрозоль применяют для обозначения множества систем, от систем коллоидной природы до систем, состоящих из упаковок под давлением. Доставка может осуществляться сжиженным или сжатым газом или подходящей системой пульверизации, которые распределяют активные ингредиенты. Аэрозоли соединений в соответствии с данным изобретением могут доставляться в однофазных, двухфазных или трехфазных системах для доставки активного ингредиента. Доставка аэрозоля включают необходимый контейнер, активаторы, клапаны, субконтейнеры и подобные, которые вместе могут формировать набор. Специалист в данной области техники без лишних экспериментов может определить предпочтительные аэрозоли.
В некоторых вариантах, фармацевтические композиции в соответствии с данным изобретением могут быть получены методами, хорошо известными в области фармацевтики. Например, фармацевтическая композиция, предназначенная для введения инъекцией, может быть получена объединением соединения в соответствии с данным изобретением со стерильной дистиллированной водой с получением раствора. Может быть добавлено поверхностно-активное вещество для облегчения получения гомогенного раствора или суспензии. Поверхностно-активные соединения являются соединения, которые не ковалентно взаимодействуют с соединением в соответствии с данным изобретением для облегчения растворения или гомогенной суспензии соединения в водной системе доставки.
Соединения в соответствии с данным изобретением или их фармацевтически приемлемые соли вводят в терапевтически эффективном количестве, которое варьируется в зависимости от множества факторов, включая активность определенного применяемого соединения; метаболическую стабильность и длительность действия соединения; возраст, массу тела, общее состояние здоровья, пол и питание пациента; способ и время введения; скорость выведения; сочетание лекарственных средств; тяжесть конкретного расстройства или состояния; и пациент, проходящий терапию.
Соединения в соответствии с данным изобретением или их фармацевтически приемлемые производные также могут вводиться одновременно с, до или после введения одного или более других терапевтических агентов. Такая комбинированная терапия включает введение единой фармацевтической дозированной композиции, которая содержит соединение в соответствии с данным изобретением и один или более дополнительных активных агентов, а также введение соединения в соответствии с данным изобретением и каждого активного агента в отдельных фармацевтических дозированных композициях. Например, соединение в соответствии с данным изобретением и другой активный агент может вводиться пациенту вместе в единой пероральной лекарственной форме, такой как таблетка или капсула, или каждый агент вводят в отдельных пероральных лекарственных формах. Если применяют раздельные лекарственные формы, соединение в соответствии с данным изобретением и один или более дополнительных активных агентов могут вводиться практически в одно и то же время, т.е., одновременно, или в разное время по очереди, т.е., последовательно; понимается, что комбинированная терапия включает все такие режимы.
В некоторых вариантах концентрация соединения структуры (I), представленного в фармацевтических композициях в соответствии с данным изобретением, меньше чем 100%, 90%, 80%, 70%, 60%, 50%, 40%, 30%, 20%, 19%, 18%, 17%, 16%, 15%,14%, 13%, 12%, 11%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0,5%, 0,4%, 0,3%, 0,2%, 0,1%, 0,09%, 0,08%, 0,07%, 0,06%, 0,05%, 0,04%, 0,03%, 0,02%, 0,01%, 0,009%, 0,008%, 0,007%, 0,006%, 0,005%, 0,004%, 0,003%, 0,002%, 0,001%, 0,0009%, 0,0008%, 0,0007%, 0,0006%, 0,0005%, 0,0004%, 0,0003%, 0,0002% или 0,0001% масс./масс., масс./об. или об./об.
В некоторых вариантах, концентрация соединения структуры (I), представленная в фармацевтических композициях в соответствии с данным изобретением, больше 90%, 80%, 70%, 60%, 50%, 40%, 30%, 20%, 19,75%, 19,50%, 19,25% 19%, 18,75%, 18,50%, 18,25% 18%, 17,75%, 17,50%, 17,25% 17%, 16,75%, 16,50%, 16,25% 16%, 15,75%, 15,50%, 15,25% 15%, 14,75%, 14,50%, 14,25% 14%, 13,75%, 13,50%, 13,25% 13%, 12,75%, 12,50%, 12,25% 12%, 11,75%, 11,50%, 11,25% 11%, 10,75%, 10,50%, 10,25% 10%, 9,75%, 9,50%, 9,25% 9%, 8,75%, 8,50%, 8,25% 8%, 7,75%, 7,50%, 7,25% 7%, 6,75%, 6,50%, 6,25% 6%, 5,75%, 5,50%, 5,25% 5%, 4,75%, 4,50%, 4,25%, 4%, 3,75%, 3,50%, 3,25%, 3%, 2,75%, 2,50%, 2,25%, 2%, 1,75%, 1,50%, 125%, 1%, 0,5%, 0,4%, 0,3%, 0,2%, 0,1%, 0,09%, 0,08%, 0,07%, 0,06%, 0,05%, 0,04%, 0,03%, 0,02%, 0,01%, 0,009%, 0,008%, 0,007%, 0,006%, 0,005%, 0,004%, 0,003%, 0,002%, 0,001%, 0,0009%, 0,0008%, 0,0007%, 0,0006%, 0,0005%, 0,0004%, 0,0003%, 0,0002% или 0,0001% масс./масс., масс./об. или об./об.
В некоторых вариантах, концентрация соединения структуры (I), представленная в фармацевтических композициях в соответствии с данным изобретением, составляет от приблизительно 0,0001% до приблизительно 50%, от приблизительно 0,001% до приблизительно 40%, от приблизительно 0,01% до приблизительно 30%, от приблизительно 0,02% до приблизительно 29%, от приблизительно 0,03% до приблизительно 28%, от приблизительно 0,04% до приблизительно 27%, от приблизительно 0,05% до приблизительно 26%, от приблизительно 0,06% до приблизительно 25%, от приблизительно 0,07% до приблизительно 24%, от приблизительно 0,08% до приблизительно 23%, от приблизительно 0,09% до приблизительно 22%, от приблизительно 0,1% до приблизительно 21%, от приблизительно 0,2% до приблизительно 20%, от приблизительно 0,3% до приблизительно 19%, от приблизительно 0,4% до приблизительно 18%, от приблизительно 0,5% до приблизительно 17%, от приблизительно 0,6% до приблизительно 16%, от приблизительно 0,7% до приблизительно 15%, от приблизительно 0,8% до приблизительно 14%, от приблизительно 0,9% до приблизительно 12%, от приблизительно 1% до приблизительно 10% масс./масс., масс./об. или об./об.
В некоторых вариантах, концентрация соединения структуры (I), представленная в фармацевтических композициях в соответствии с данным изобретением, составляет от приблизительно 0,001% до приблизительно 10%, от приблизительно 0,01% до приблизительно 5%, от приблизительно 0,02% до приблизительно 4,5%, от приблизительно 0,03% до приблизительно 4%, от приблизительно 0,04% до приблизительно 3,5%, от приблизительно 0,05% до приблизительно 3%, от приблизительно 0,06% до приблизительно 2,5%, от приблизительно 0,07% до приблизительно 2%, от приблизительно 0,08% до приблизительно 1,5%, от приблизительно 0,09% до приблизительно 1%, от приблизительно 0,1% до приблизительно 0,9% масс./масс., масс./об. или об./об.
В некоторых вариантах, количество соединения структуры (I), представленное в фармацевтических композициях в соответствии с данным изобретением, равно или менее 10 г, 9,5 г, 9,0 г, 8,5 г, 8,0 г, 7,5 г, 7,0 г, 6,5 г, 6,0 г, 5,5 г, 5,0 г, 4,5 г, 4,0 г, 3,5 г, 3,0 г, 2,5 г, 2,0 г, 1,5 г, 1,0 г, 0,95 г, 0,9 г, 0,85 г, 0,8 г, 0,75 г, 0,7 г, 0,65 г, 0,6 г, 0,55 г, 0,5 г, 0,45 г, 0,4 г, 0,35 г, 0,3 г, 0,25 г, 0,2 г, 0,15 г, 0,1 г, 0,09 г, 0,08 г, 0,07 г, 0,06 г, 0,05 г, 0,04 г, 0,03 г, 0,02 г, 0,01 г, 0,009 г, 0,008 г, 0,007 г, 0,006 г, 0,005 г, 0,004 г, 0,003 г, 0,002 г, 0,001 г, 0,0009 г, 0,0008 г, 0,0007 г, 0,0006 г, 0,0005 г, 0,0004 г, 0,0003 г, 0,0002 г или 0,0001 г.
В некоторых вариантах, количество соединения структуры (I), представленное в фармацевтических композициях в соответствии с данным изобретением, более 0,0001 г, 0,0002 г, 0,0003 г, 0,0004 г, 0,0005 г, 0,0006 г, 0,0007 г, 0,0008 г, 0,0009 г, 0,001 г, 0,0015 г, 0,002 г, 0,0025 г, 0,003 г, 0,0035 г, 0,004 г, 0,0045 г, 0,005 г, 0,0055 г, 0,006 г, 0,0065 г, 0,007 г, 0,0075 г, 0,008 г, 0,0085 г, 0,009 г, 0,0095 г, 0,01 г, 0,015 г, 0,02 г, 0,025 г, 0,03 г, 0,035 г, 0,04 г, 0,045 г, 0,05 г, 0,055 г, 0,06 г, 0,065 г, 0,07 г, 0,075 г, 0,08 г, 0,085 г, 0,09 г, 0,095 г, 0,1 г, 0,15 г, 0,2 г, 0,25 г, 0,3 г,, 0,35 г, 0,4 г,, 0,45 г, 0,5 г, 0,55 г, 0,6 г, 0,65 г, 0,7 г, 0,75 г, 0,8 г, 0,85 г, 0,9 г, 0,95 г, 1 г, 1,5 г, 2 г, 2,5, 3 г, 3,5, 4 г, 4,5 г, 5 г, 5,5 г, 6 г, 6,5 г, 7 г, 7,5 г, 8 г, 8,5 г, 9 г, 9,5 г или 10 г.
В некоторых вариантах, количество соединения структуры (I), представленное в фармацевтических композициях в соответствии с данным изобретением, составляет 0,0001-10 г, 0,0005-9 г, 0,001-8 г, 0,005-7 г, 0,01-6 г, 0,05-5 г, 0,1-4 г, 0,5-4 г или 1-3 г.
Также специалисту в данной области техники понятно, что в способах получения соединений структуры (I), описанных здесь, может понадобиться защищать функциональные группы промежуточных соединений подходящими защитными группами. Такие функциональные группы включают гидрокси, амино, меркапто и карбоновую кислоту. Подходящие защитные группы для гидрокси включают триалкилсилил или диарилалкилсилил (например, трет-бутилдиметилсилил, трет-бутилдифенилсилил или триметилсилил), тетрагидропиранил, бензил и подобные. Подходящие защитные группы для амино, амидино и гуанидино включают трет-бутоксикарбонил, бензилоксикарбонил и подобные. Подходящие защитные группы для меркапто включают -C(O)-Rʺ (где Rʺ является алкилом, арилом или арилалкилом), п-метоксибензил, тритил и подобные. Подходящие защитные группы для карбоновой кислоты включают сложные эфиры алкила, арила или арилалкила. Защитные группы могут быть добавлены или удалении стандартными методами, которые известны специалисту в данной области техники и которые описаны здесь. Применение защитных групп подробно описано в Green, T.W. and P.G.M. Wutz, Protective Groups in Organic Synthesis (1999), 3rd Ed., Wiley. Специалист в данной области техники поймет, что защитной группой также может быть полимерная смола, такая как смола Ванга, смола Ринка или 2-хлортритилхлоридная смола.
Также специалисту в данной области техники понятно, что хотя такие защищенные производные соединений в соответствии с данным изобретением могут не обладать фармакологическим действием как таковым, они могут вводиться млекопитающим и затем метаболизировать в теле с получением соединений в соответствии с данным изобретением, которые фармакологически активны. Такие производные, поэтому могут быть описаны как ʺпролекарстваʺ. Все пролекарства соединений в соответствии с данным изобретением включены в объем изобретения.
Более того, все соединения в соответствии с данным изобретением, которые существуют в форме свободного основания или кислоты, могут быть превращены в их фармацевтически приемлемые соли обработкой подходящим неорганическим или органическим основанием или кислотой способами, известными специалисту в данной области техники. Соли соединений в соответствии с данным изобретением могут быть превращены в их свободные основания или кислоты стандартными методами.
Соединения структуры (I) могут быть получены добавлением фосфатной группы к одному из трех гидроксилов альвоцидиба. Исходное соединение альвоцидиба (и его соли и сольваты) может быть куплено у коммерческих источников или получено способами, известными в данной области техники, например, как описано в патентах США №№: 6,136,981; 6,225,473; 6,406,912; 6,576,647; и 6,821,990; полное описание которых включено в качестве ссылки полностью.
Представленные ниже общие схемы реакции иллюстрируют способ получения соединений в соответствии с данным изобретением, т.е., соединения структуры (I):

(I)
или его стереоизомера, таутомера или фармацевтически приемлемой соли, где R1, R2 и R3 такие, как определены выше. Понятно, что специалист в данной области техники сможет получить эти соединения подобными способами или объединением других способов, известных специалисту в данной области техники. Также понятно, что специалист в данной области техники сможет получить, так же, как описано ниже, другие соединения структуры (I), которые отдельно не показаны ниже, с применением подходящих исходных компонентов и модификацией параметров синтеза при необходимости. В общем, исходные компоненты могут быть получены из источников, таких как Sigma Aldrich, Lancaster Synthesis, Inc., Maybridge, Matrix Scientific, TCI и Fluorochem USA, ит.д. или синтезированы согласно источникам, известным специалисту в данной области техники (см., например, Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 5th edition (Wiley, December 2000)) или получены как описано в данном изобретении.
Общая схема реакции 1

Как показано на общей схеме реакции 1, HCl соль альвоцидиба A сначала подвергают взаимодействию с подходяще защищенным хлорфосфатом (т.е., B, где R является защитной группой, такой как этил). Затем снятие защиты дает желаемое соединение структуры (I). Специалисту в данной области техники очевидно, что соединения структуры (I), имеющие один фосфат на любой из трех гидроксильных групп альвоцидиба, могут быть получены по указанно выше схеме, и желаемый региоизомер отделяют обычными методами, такими как хроматография. Выбор защитной группы для оптимизации выхода желаемого региоизомера также очевиден специалисту в данной области техники.
Способы
В различных вариантах, в изобретении представлен способ лечения заболевания у млекопитающего, нуждающегося в таковом, введением соединения структуры (I) или содержащей его фармацевтической композиции млекопитающему. В некоторых определенных вариантах, способ предназначен для лечения заболевания, связанного со сверхэкспрессией циклинзависимой киназы (ЦЗК) у млекопитающего, нуждающегося в таковом, где способ включает введение терапевтически эффективного количества любого из представленных выше соединений структуры (I) или содержащей его фармацевтической композиции млекопитающему.
В некоторых других вариантах, заболеванием является рак, например, гемобластоз. В некоторых из этих вариантов, гемобластоз выбирают из острого миелоидного лейкоза (ОМЛ), множественной миеломы, фолликулярной лимфомы, острого лимфобластного лейкоза (ОЛЛ), хронического лимфоцитарного лейкоза (ХЛЛ) и неходжкинской лимфомы. В других вариантах, гемобластозом является острый миелогенный лейкоз (ОМЛ). В других вариантах, гемобластозом является хронический лимфоцитарный лейкоз (ХЛЛ). В других вариантах, гемобластозом является миелодиспластический синдром (МДС).
В некоторых других конкретных вариантах представленных выше способов, способ включает пероральное введение соединения структуры (I) или содержащей его фармацевтической композиции, млекопитающему.
Кроме представленных выше типовых болезней, множество видов рака, включая солидные опухоли и лейкозы (например, острый миелоидный лейкоз) поддаются описанным здесь способам. Типы рака, которые могут быть лечены в различных вариантах, включают, но не ограничены ими: аденокарциному молочной железы, простаты и прямой кишки; все формы бронхогенной карциномы легких; миелоид; меланому; гепатому; нейробластому; папиллому; апудому; хористому; бранхиому; злокачественный карциноидный синдром; карциноидную болезнь сердца; и карциному (например, Уолкера, базальных клеток, базальную плоскоклеточную, Брауна-Пирса, проточную, опухоль Эрлиха, Кребса 2, осязательных клеток, слизеобразующую, немелкоклеточный рак легких, овсяноклеточную, папиллярную, цирроз, бронхиолярную, бронхогенную, плоскоклеточную и переходноклеточную). Дополнительные виды рака, которые могут быть лечены, включают: гистиоцитозы; лейкоз; злокачественный гистиоцитоз; болезнь Ходжкина; средиземноморскую лимфому; неходжкинскую лимфому; плазмацитому; ретикулоэндотелиоз; меланому; хондробластому; хондрому; хондросаркому; фиброму; фибросаркому; гигантоклеточные опухоли; гистиоцитому; липому; липосаркому; мезотелиому; миксому; миксосаркому; остеому; остеосаркому; хондрому; краниофарингиому; дисгерминому; гамартому; мезенхимому; мезонефрому; миосаркому; амелобластому; цементому; одонтому; тератому; тимому; трофобластную опухоль. Далее, следующие типы рака также рассматриваются как поддающиеся лечению: аденома; холангиома; холестеатома; циклиндрома; цистаденокарцинома; цистаденома; фолликулома; гинандробластома; гепатома; гидраденома; инсулома; лейдигома; папиллома; андробластома; текома; леймиома; лейомиосаркома; миобластома; миома; миосаркома; рабдомиома; рабдомиосаркома; эпендимома; ганглионеврома; глиома; медуллобластома; менингиома; невринома; нейробластома; нейроэпителиома; нейрофиброма; неврома; параганглиома; нехромаффинная параганглиома. типы рака, которые могут быть лечены, также включают, но не ограничены ими, ангиокератому; ангиофимфоидную гиперплазию с эозинофиллией; склерозирующую ангиому; ангиоматоз; гломангиому; гемангиоэндотелиому; гемангиому; гемангиоперицитому; гемангиосаркому; лимфангиому; лимфангиомиому; лимфангиосаркому; пинеалому; карциносаркому; хондросаркому; филлоидную цистосаркому; фибросаркому; гемангиосаркому; лейомиосаркому; лейкосаркому; липосаркому; лимфангиосаркому; миосаркому; миксосаркому; карциному яичников; рабдомиосаркому; саркому; новообразования; нейрофиброматоз; и дисплазию шейки матки.
Соединения в соответствии с данным изобретением являются эффективными в широком интервале доз. Например, при лечении взрослых людей, дозы от 0,01 до 1000 мг, от 0,5 до 100 мг, от 1 до 50 мг в сутки и от 5 до 40 мг в сутки являются примерами доз, которые применяют в некоторых вариантах. Типовые дозы составляют от 10 до 30 мг в сутки. Точная доза зависит от способа введения, формы, в которой вводят соединение, лечимого пациента, массы тела лечимого пациента, и предпочтений и опыта лечащего врача.
В некоторых вариантах, соединение в соответствии с данным изобретением вводят в однократной дозе. Однократная доза соединения в соответствии с данным изобретением также может применяться для лечения острого состояния.
В некоторых вариантах, соединение в соответствии с данным изобретением вводят несколькими дозами. В некоторых вариантах, дозу дают один, два, три раза, четыре раза, пять раз, шесть раз или более шести раз в сутки. В других вариантах, дозу дают один раз в месяц, один раз каждые две недели, один раз в неделю или один раз через день. В другом варианте, соединение в соответствии с данным изобретением и другой агент вводят вместе от около одного раза в сутки до около 6 раз в сутки. В другом варианте, введение соединения в соответствии с данным изобретением и агента продолжается менее около 7 дней. В еще одном варианте, введение продолжают в течение более 6, 10, 14, 28 дней, двух месяцев, шести месяцев или одного года. В которых случаях, непрерывное введение достигается и сохраняется так долго, как это необходимо.
Введение соединений в соответствии с данным изобретением может продолжаться так долго, как это необходимо. В некоторых вариантах, соединение в соответствии с данным изобретением вводят в течение более 1, 2, 3, 4, 5, 6, 7, 14 или 28 дней. В некоторых вариантах, соединение в соответствии с данным изобретением вводят в течение менее 28, 14, 7, 6, 5, 4, 3, 2 или 1 дня. В некоторых вариантах, соединение в соответствии с данным изобретением вводят постоянно, например, для лечения хронический проявлений.
В некоторых вариантах, соединения в соответствии с данным изобретением вводят дозами. Из-за межиндивидуальной вариабельности фармакокинетики соединения, индивидуализация режима введения обеспечивается в определенных вариантах. Введение соединения в соответствии с данным изобретением может быть определено обычными экспериментами с учетом данного описания и/или может быть определено специалистом в данной области техники.
ПРИМЕРЫ
Пример 1
Получение типовых фосфатных пролекарств (IB′)

Диэтилфосфат 2-(2-хлорфенил)-5-гидрокси-8-(3-гидрокси-1-метилпиперидин-4-ил)-4-оксо-4H-хромен-7-ила
Суспензию альвоцидиба HCl (2 г, 4,56 ммоль, 1 экв.) в 1,2-дихлорэтане (40 мл) охлаждают до 0°C. К этому раствору добавляют триэтиламин (1,9 мл, 13,7 ммоль, 3 экв.), затем диэтилхлорфосфат (0,78 г, 4,56 ммоль, 1 экв.). Реакционную смесь перемешивают при 0°C в течение 30-45 мин. Затем реакционную смесь выливают на лед и экстрагируют дихлорметаном (3×25 мл). Объединенные органические слои сушат над безводным Na2SO4 и концентрируют с получением неочищенного остатка. Неочищенный остаток очищают флэш-хроматографией на колонке с применением 10-15% метанола в дихлореметане с получением диэтилфосфата 2-(2-хлорфенил)-5-гидрокси-8-(3-гидрокси-1-метилпиперидин-4-ил)-4-оксо-4H-хромен-7-ил (550 мг, 1,02 ммоль; 22%).
ЖХМС: колонка: XBridge C8 (50 × 4,6 мм × 3,5 мкм); подвижная фаза: A: 10 мМ NH4CO3 в H2O; B: АЦН; ВУ: 5,97; чистота: (макс.: 67,63); M+H: 538,0.
дигидрофосфат 2-(2-хлорфенил)-5-гидрокси-8-(3-гидрокси-1-метилпиперидин-4-ил)-4-оксо-4H-хромен-7-ила (IB′)
К раствору диэтилфосфата 2-(2-хлорфенил)-5-гидрокси-8-(3-гидрокси-1-метилпиперидин-4-ил)-4-оксо-4H-хромен-7-ила (0,55 г, 1,02 ммоль, 1 экв.) в дихлорметане (4 мл) при 0°C добавляют триметилсилилбромид (2,0 мл, 15,1 ммоль, 15 экв.). Затем реакционную смесь нагревают при 36°C в герметичных условиях в течение 20 ч. Реакционную смесь выпаривают. Полученный неочищенный остаток очищают препаративной ВЭЖХ с получением дигидрофосфата 2-(2-хлорфенил)-5-гидрокси-8-(3-гидрокси-1-метилпиперидин-4-ил)-4-оксо-4H-хромен-7-ила (35 мг; 0,073 ммоль; 7%).
ЖХМС: колонка: XBridge C8 (50 × 4,6 мм × 3,5 мкм); подвижная фаза: A: 10 мМ NH4CO3 в H2O; B: АЦН; ВУ: 3,11; чистота: (макс.: 93,56); M+H: 482,0.
ВЭЖХ: колонка: XBridge C8 (50 × 4,6 мм × 3,5 мкм); подвижная фаза: A: 0,1% ТФК в H2O; B: АЦН; ВУ: 2,55; чистота: (макс.: 96,39; 254 нм: 96,57).
1H ЯМР (ДМСО-d6-D2O обмен): δ 7,84 (д, J=7,20 Гц, 1H), 7,71-7,70 (м, 1H), 7,65-7,62 (м, 1H), 7,59-7,55 (м, 1H), 7,07 (с, 1H), 6,62 (с, 1H), 4,12 (с, 1H), 3,60-3,54 (м, 1H), 3,30-3,26 (м, 3H), 3,13-3,11 (м, 2H), 2,71 (с, 3H), 1,83-1,80 (м, 1H).
Пример 2
Фармакокинетический профиль Пролекарств Альвоцидиба
Получают следующие соединения и из фармакокинетический профиль определяют и сравнивают с фармакокинетическим профилем соединения (IB′) как описано ниже.
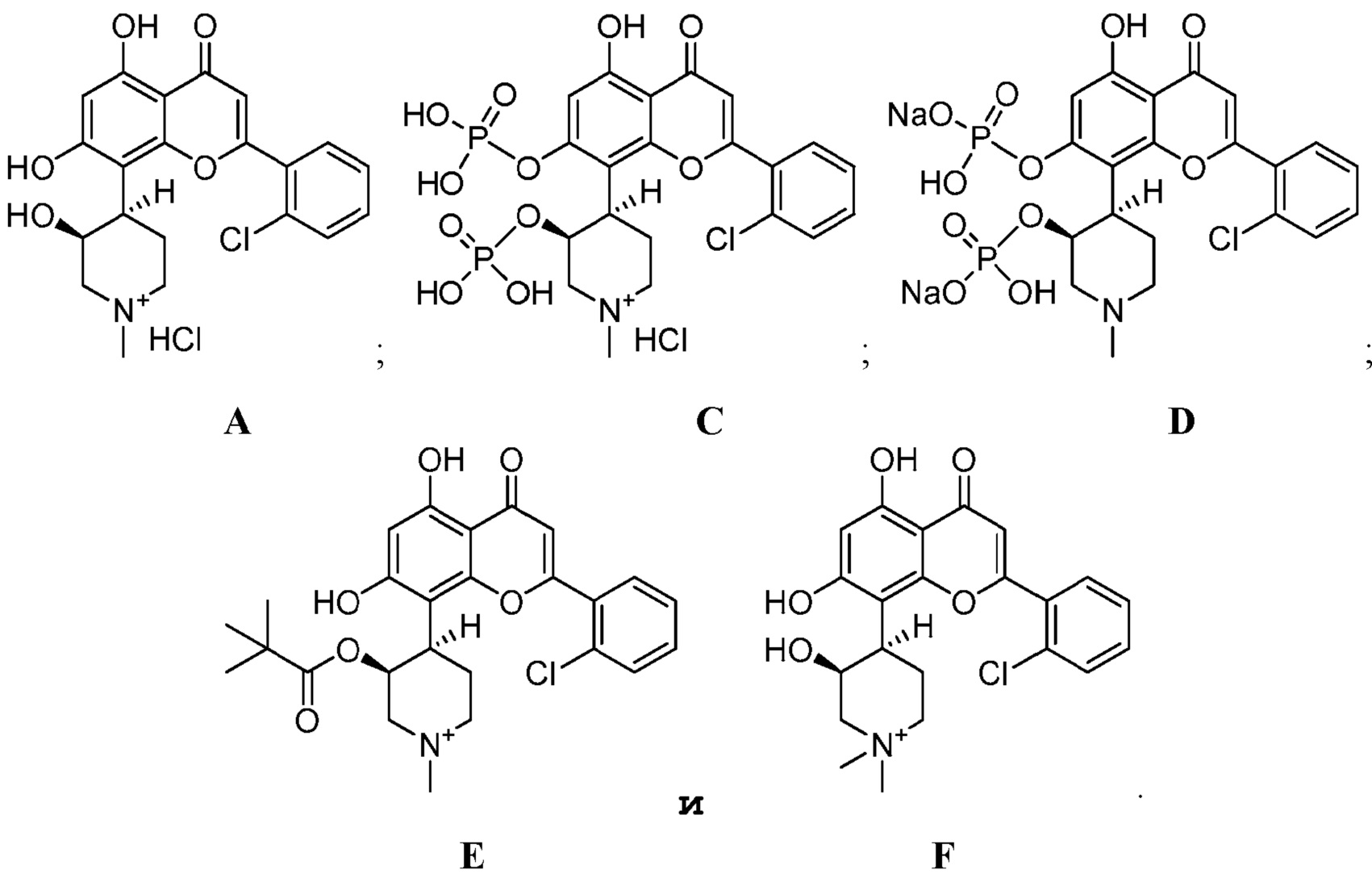
Соединения получают и вводят мышам CD-1 внутривенно (ВВ) или перорально (ПО) как суммировано в таблице 1. Концентрацию исходного соединения альвоцидиба в плазме определяют в разные интервалы времени (таблица 2) и рассчитывают фармакокинетические параметры (таблица 3). Соединения E и F не превращаются в альвоцидиб in vivo (т.е., альвоцидиб не определяется в образцах плазмы мышей, получивших эти соединения), и их фармакокинетические параметры далее не исследуют. Как можно видеть из таблицы 3, биодоступность соединения (IB′) превосходит исходное соединение альвоцидиба (A) и два дифосфатных соединения (C и D).
Таблица 1. Разработка экспериментов по получению фармакокинетического профиля
Таблица 2. Концентрация Альвоцидиба в плазме
Примечание: результаты даны как среднее ± СО, n=3 животных/группу
н.о.=не оценивали
Таблица 3. Фармакокинетические профили
Примечание: результаты даны как среднее ± СО, n=3 животных/группу
Пример 3
Профили кинетической растворимости
Водную кинетическую растворимость соединения IB′ определяют в широком спектре pH (т.е. pH 2,2-pH 8,7) и сравнивают с водной кинетической растворимостью альвоцидиба при том же интервале pH. Было найдено, что растворимость соединения IB′ превышает 1 мг/мл при наименьшем тестируемом pH (pH 2,2), повышаясь до выше 5 мг/мл при pH 6,8 и pH 8,7. По сравнению, растворимость альвоцидиба превышает 1 мг/мл при pH 2,2 и pH 4,5, но падает до 0,02 мг/мл при pH 6,8 и pH 8,7.
Таблица 4. Профили кинетической растворимости
Пример 4
Профили стабильности в плазме
Стабильность в плазме соединения IB′ определяют с применением плазмы от четырех видов. Результаты для мышей, крыс, собак и человека показаны в таблицах 5, 6, 7 и 8, соответственно. В качестве контроля применяют Альвоцидиб и флумазенил. У мышей, крыс и человека соединение IB′ сохраняет 100% стабильность через 5 часов инкубирования. В плазме собак через 5 часов остается приблизительно 90% соединения IB′. По сравнению, альвоцидиб сохраняет 100% стабильность для всех четырех видов через 5 часов, и флумазенил не стабилен в плазме мышей и крыс.
Таблица 5. Профили стабильности в плазме мышей
Таблица 6. Профили стабильности в плазме крыс
Таблица 7. Профили стабильности в плазме собак
Таблица 8. Профили стабильности в плазме человека
Пример 5
Фармакокинетика у крыс Sprague Dawley
Концентрации альвоцидиба в плазме при пероральном и внутривенном (ВВ) введении соединения IB′ и абсорбированного самого соединения IB′ определяют у самцов крыс Sprague Dawley (SD) (см. фигуру 1). Образцы плазмы берут в 8 временных интервалов (ВВ) или 7 временных интервалов (перорально) в течение 24 часов после единственной дозы соединения IB′ (3 животных в группе). Рассчитанные фармакокинетические параметры показаны в таблице 9 и таблице 10. ВВ и пероральное введение соединения IB′ дает значительное количество альвоцидиба. При внутривенном введении соединение IB′ (1 мг/кг) метаболизирует до альвоцидиба с C0 270,3 нг/мл, который выводится с периодом полувыведения 1,6 часов. При пероральном введении соединение IB′ (10 мг/кг) метаболизирует до альвоцидиба с Cmax 178,6 нг/мл и Tmax 2,92 часа, который выводится с периодом полувыведения 4,4 часа. Биодоступность альвоцидиба (99,03%) рассчитывают из отношения площади под кривой (ППК) для альвоцидиба, полученной при пероральном и ВВ введении соединения IB′. Образцы плазмы также анализируют на присутствие соединения IB′. Концентрации в плазме соединения IB′ у крыс SD также показано на фигуре 1 и в таблице 11. При ВВ и пероральном введении у крыс SD уровни соединения IB′ в плазме падали ниже количественных уровней через 2 часа после введения.
Таблица 9. Фармакокинетические параметры Альвоцидиба после внутривенного введения соединения IB′ крысам Sprague Dawley
Таблица 10. Фармакокинетические параметры Альвоцидиба после перорального введения соединения IB′ крысам Sprague Dawley
Таблица 11. Концентрации в плазме соединения IB после внутривенного или перорального введения соединения IB′ крысам Sprague Dawley
# не измеряли
НПКО=ниже предела количественного определения
Пример 6
Максимально переносимая доза при кратковременном введении у мышей
Исследования кратковременной (т.е. однократная доза) токсикологии проводят на мышах. Кратковременные исследования проводят на самках мышей SHO SCID с применением трех животных на обрабатываемую группу. Животным дают одну дозу соединения IB′ в количестве 45, 30, 15 или 7,5 мг/кг. Для сравнения, дополнительным животным дают альвоцидиб в тех же дозах. Применяют измерения массы тела после перорального введения (фигура 2A-B) и внутривенного (ВВ) введения (фигура 2C-D), вместе со смертностью и клиническими наблюдениями для определения максимально переносимой дозы при кратковременном введении (МПДкратк.).
Результаты кратковременного исследования показали, что МПДкратк. соединения IB′, вводимого перорально, составляет 15 мг/кг. МПДкратк. соединения IB′, вводимого внутривенно, составляет 15 мг/кг. Потерю массы тела и повышенную летаргию наблюдают у мышей, которым вводят 30 мг/кг и 45 мг/кг. У животных, получавших дозу 45 мг/кг, одно животное умерло на второй день, и одно животное умерло на третий день. У животных, получавших дозу 30 мг/кг, одно животное умерло на четвертый день. У животных, получавших внутривенно 45 мг/кг, два животных умерли на второй день. У животных, получавших внутривенно 30 мг/кг, одно животное умерло на третий день.
Кратковременная МПДкратк. альвоцидиба при пероральном введении составляет 15 мг/кг. МПДкратк. альвоцидиба при внутривенном введении составляет 7,5 мг/кг. Некоторую потерю массы тела, повышенную летаргию и смерть животных наблюдают у животных, получавших альвоцидиб в дозе 30 и 45 мг/кг.
Потерю массы тела наблюдают у выживших животных при пероральном введении 45 мг/кг и 30 мг/кг соединения IB′, пик достигается при 17% в группе 30 мг/кг. Пик потери массы тела у выживших животных при внутривенном введении составляет 12%.
Очевидная токсичность не наблюдается у мышей, получавших перорально или внутривенно 15 мг/кг или 7,5 мг/кг. Незначительную потерю массы тела с пиком 3,3% в группе с внутривенным введением 15 мг/кг относят за счет нормальной флуктуации массы тела у тестируемых животных.
Соединение IB′ переносится лучше (МПДкратк.=15 мг/кг) у мышей при внутривенном введении по сравнению с альвоцидибом (МПДкратк.=7,5 мг/кг).
Пример 7
Максимально переносимая доза при многократном введении у мышей
Повторные исследования токсикологии дозы проводят на самках мышей SHO SCID, используя 3 животных на обрабатываемую группу. Животным дают пять суточных доз соединения IB′ в количестве 15, 7,5 или 2,5 мг/кг, и наблюдают в течение еще пяти дней после введения. Для сравнения, дополнительным животным дают альвоцидиб в тех же дозах и с тем же расписанием введения/наблюдения. Измерения массы тела животных, получающих дозу перорально (см. фигуру 3A-B) и внутривенно (см. фигуру 3C-D) в течение 5 дней повторяющегося введения и в течение последующего 5-дневного наблюдения, а также смертность и клинические наблюдения, применяют для определения расписания максимально переносимого введения (МПДповтор).
Результаты 5-дневого исследования повторяющегося введения показывают, что МПДповтор соединения IB при пероральном введении составляет 7,5 мг/кг. МПДповтор соединения IB′ при внутривенном введении составляет 15 мг/кг. Потеря массы тела наблюдается у животных, получающих перорально дозу 15 мг/кг. Среди животных, получающих перорально дозу 15 мг/кг, одно животное умирает на 5 день, и одно животное умирает на 7 день.
Для сравнения, МПДповтор, определенная для альвоцидиба при пероральном введении составляет 7,5 мг/кг. МПДповтор, определенная для альвоцидиба при внутривенном введении составляет 7,5 мг/кг. Летаргию, потерю массы тела и смерть наблюдают при введении дозы 15 мг/кг, и при пероральном, и при внутривенном введении альвоцидиба.
Потерю массы тела наблюдают у выживших животных при пероральном введении 15 мг/кг соединения IB′, пик составляет 12%. Никакой очевидной токсичности не наблюдают у животных, которые получают перорально 7,5 мг/кг или 2,5 мг/кг, или у животных, получающих любую из исследуемых доз при введении внутривенно.
Соединение IB′ переносится лучше (МПДповтор=15 мг/кг) у мышей при внутривенном введении по сравнению с альвоцидибом (МПДповтор=7,5 мг/кг).
Пример 8
Максимально переносимая доза при кратковременном введении у крыс
Исследования кратковременной (т.е. однократная доза) токсикологии проводят на крысах. Кратковременные исследования проводят на самках крыс Sprague Dawley с применением трех животных на обрабатываемую группу. Животным дают одну дозу соединения IB′ в количестве 36, 18, 9 или 4,5 мг/кг. Для сравнения, дополнительным животным дают альвоцидиб в дозе 18, 9 или 4,5 мг/кг. Применяют измерения массы тела после перорального введения (фигура 4A), вместе со смертностью, клиническими наблюдениями, потреблением пищи (см.фигуру 4В) и общим анализом крови (ОАК; см. таблицу 12) для определения максимально переносимой дозы при кратковременном введении (МПДкратк.).
Результаты кратковременного исследования показали, что МПДкратк. соединения IB′, вводимого перорально, составляет 15 мг/кг. МПДкратк. соединения IB′, у крыс составляет 18 мг/кг. Диарею, потерю массы тела и повышенную летаргию наблюдают у животных, получающих соединение IB в дозе 36 мг/кг. При такой дозе одно животное умирает на третий день, одно животное умирает на четвертый день и одно животное умирает на 5 день. Смерть не наблюдается в других обрабатываемых группах.
У обрабатываемых животных наблюдают потерю массы тела, перед смертью, достигающую 13,1% у животных, получивших дозу соединения IB′ 36 мг/кг (см. фигуру 4A). Такая потеря массы тела сопровождается значительной диареей и повышенной летаргией у этих животных. Никакой очевидной токсичности, включая потерю массы тела и диарею, не наблюдают у крыс, получивших 18, 9 или 4,5 мг/кг соединения IB′. По сравнению, животные, получившие 18 мг/кг альвоцидиба, показывают признаки диареи. Кроме того, аномальное потребление пищи наблюдается при введении 18 мг/кг альвоцидиба, но не наблюдается у животных, получивших соединение IB′ в тех же дозах.
У некоторых животных были аномальные ОАК (таблица 12). Конкретно, количество тромбоцитов было вне нормального интервала для носителя и 9 мг/кг дозы соединения IB′ и 4,5 мг/кг дозы альвоцидиба. Никакой дозозависимой закономерности не наблюдается у выживших обработанных животных. Незначительное снижение количества красных и белых кровяных телец наблюдается при дозе 18 мг/кг соединения IB′. Однако, незначительное повышение количества также наблюдается у некоторых не обработанных животных. Высокая вариабельность этих результатов относится на счет межиндивидуальной вариабельности, но не на счет лекарствозависимых механизмов. Так как животные, получившие 36 мг/кг соединения IB′, умерли в течение ночи, ОАК для них не доступны.
Основываясь на представленных выше данных было обнаружено, что пероральная МПДкратк. соединения IB′ у крыс отличает его профиль переносимости от профиля альвоцидиб, так как уровень отсутствия наблюдаемых побочных эффектов (NOAEL) составляет 18 мг/кг для соединения IB′ и 9 мг/кг для альвоцидиба.
Таблица 12. Анализ крови крыс, получавших одну дозу соединения IB
Пример 9
Исследование эффективности ксенотрансплантата у мышей
In vivo активность соединения IB′ определяют в модели ксенотрансплантата острого миелоидного лейкоза (ОМЛ) у мышей MV4-11. Инъекция 8×106 MV4-11 клеток/мышь вызывает рост опухолей до приблизительно 100 мм3. После того, как опухоли достигнут подходящего размера, мышей произвольно делят на следующие обрабатываемые группы: носитель, соединение IB′ (7,5 мг/кг, qdx5×3), соединение IB′ (2,5 мг/кг, qdx5×3), соединение IB′ (0,1 мг/кг, qdx5×3) и соединение IB (7,5 мг/кг, q7dx3). Носитель и соединение IB′ вводят перорально, за исключением последней дозы соединения IB′ (7,5 мг/кг, q7dx3), которую вводят внутривенно. Обработка дает значительное ингибирование роста опухоли (% ИРО; см. фигуры 5A-B и таблицу 13).
Таблица 13. Ингибирование роста опухоли в исследовании эффективности ксенотранспланата у мышей
Пример 10
Исследование фармакодинамики ксенотрансплантата у мышей
In vivo фармакодинамическую активность соединения IB′ определяют в модели ксенотранплантата ОМЛ у мышей MV4-11 (фигура 6A-B). Инъекция 8×106 клеток/мышь вызывает рост опухолей до приблизительно 100 мм3. После того, как опухоли достигнут подходящего размера, мышей произвольно делят на следующие обрабатываемые группы: носитель, соединение IB′ (2,5 мг/кг), соединение IB′ (0,5 мг/кг), соединение IB′ (0,1 мг/кг), соединение IB' 0,02 мг/кг). Мышам вводят одну дозу и опухоли собирают через 48 часов после лечения. Уровни белка MCL-1 оценивают на собранных опухолях с применением стандартного электрофореза в полиакриламидном геле и методики иммуноблоттинга (фигура 6A). Лечение дает снижение экспрессии белка MCL-1 (см. фигуру 6B и таблицу 14 ниже).
Таблица 14. Снижение экспрессии белка MCL-1
Все патенты США, публикации заявок на патенты США, заявки на патенты США, иностранные патенты, заявки на иностранные патенты и не патентные публикации, цитированные в данном описании, включая предварительную заявку на патент США № 62/163,188, поданную 18 мая 2015, включены сюда в качестве ссылок полностью до той степени, пока они не противоречат данному описанию.
Из представленного выше понятно, что, хотя конкретные примеры изобретения описаны здесь для целей иллюстрации, различные модификации могут быть сделаны, не выходя за суть и объем данного изобретения. Следовательно, изобретение не ограничено, за исключением представленной формулы изобретения.
Реферат
Изобретение относится к пригодному в медицине соединению формулы (IB′), его таутомеру или фармацевтически приемлемой соли, а также к фармацевтической композиции на его основе и способу лечения с его использованием(IB′). Предложено новое соединение, композиция и способ, эффективный для лечения заболевания, связанного со сверхэкспрессией циклинзависимой киназы (ЦЗК) у млекопитающих. 4 н. и 28 з.п. ф-лы, 10 пр., 14 табл., 15 ил.
Формула

Комментарии