Производное циклического амина и его медицинское применение - RU2696862C1
Код документа: RU2696862C1
Чертежи



Описание
Предпосылки создания изобретения
Область техники, к которой относится изобретение
[0001]
Настоящее изобретение относится к производному циклического амина и его медицинскому применению.
Уровень техники
[0002]
Аутоиммунное заболевание является общим термином для заболеваний, при которых у индивидуума обостряются чрезмерные иммунные реакции собственных нормальных клеток и тканей, что приводит к симптомам, примеры которых включают рассеянный склероз, ревматоидный артрит, псориаз, красную волчанку, болезнь Бехтерева, увеит или ревматическую полимиалгию.
[0003]
Аллергическое заболевание представляет собой заболевание, которое возникает из-за чрезмерной иммунной реакции на конкретные антигены, и его примеры включают аллергический дерматит, атопический дерматит, аллергический ринит (поллиноз), аллергический конъюнктивит, аллергический гастроэнтерит, бронхиальную астму, детскую астму или пищевую аллергию.
[0004]
Были предложены различные механизмы начала и хода аутоиммунных заболеваний и аллергических заболеваний. Известен один из этих механизмов, при котором Th17 клетки, являющиеся одним из подмножества хелперных Т-клеток и IL-17, которые являются воспалительными цитокинами, продуцированными Th17 клетками, играют важную роль в наступлении и ходе аутоиммунных заболеваний и аллергических заболеваний (непатентные документы 1 и 2).
[0005]
IL-17 воздействует на различные клетки, такие как фибробласты, эпителиальные клетки, сосудистые эндотелиальные клетки и макрофаги, и участвует в индуцировании воспалительных цитокинов, хемокинов, металлопротеаз и других воспалительных медиаторов, и миграции нейтрофилов. Таким образом, считается, что мощные противовоспалительные эффекты обнаруживаются, если продуцирование или функция IL-17 может быть подавлена, и были проведены клинические исследования анти-IL-17 антител с показаниями для различных аутоиммунных заболеваний.
[0006]
Недавно стало ясно, что ретиноидный орфанный рецептор γ (здесь и далее называемый RORγ), который является ядерным рецептором, функционирует как транскрипционный фактор, необходимый для дифференциации и пролиферации Th17 клеток и экспрессии IL-17 (непатентный документ 3), и было показано, что подавление экспрессии или функции RORγ является результатом подавления дифференциации и активации Th17 клеток и продуцирования IL-17 (непатентный документ 4).
[0007]
Сообщалось, что уровень экспрессии RORγ в периферической крови мононуклеаров или кожных тканях у пациентов с аутоиммунными заболеваниями (рассеянным склерозом, псориазом, системной красной волчанкой и т.д.) или у пациентов с аллергическими заболеваниями (аллергическим дерматитом и т.д.) выше, чем у здоровых лиц (непатентные документы 5-7). Сообщалось, что у нокаутированных RORγ мышей подавляется патологическое состояние на мышиной модели экспериментального аутоиммунного энцефаломиелита, которая представляет собой животную модель рассеянного склероза, и что подавляются симптомы аутоиммунных заболеваний, таких как колит, и симптомы аллергических заболеваний, таких как астма (непатентные документы 3, 8 и 9).
[0008]
Кроме того предполагается, что связь между RORγ и коактиватором необходима для функционирования RORγ в качестве транскрипционного фактора (непатентный документ 10). Таким образом, антагонист RORγ, которым является соединение, которое подавляет связь между RORγ и коактиватором, как ожидается, будет полезным в качестве терапевтического средства или профилактического средства против аутоиммунных заболеваний или аллергических заболеваний.
[0009]
С другой стороны, ранее сообщалось об антагонистах RORγ, таких как N-(5-(N-(4-(1,1,1,3,3,3-гексафтор-2-гидроксипропан-2-ил)фенил)сульфамоил)-4-метилтиазол-2-ил)ацетамид (непатентный документ 11), производных замещенного азола (патентный документ 1), таких как 6-(2-хлор-4-метилфенил)-3-(4-циклопропил-5-(3-неофенилциклобутил)изоксазол-3-ил)-5-оксогексановая кислота, и производных сульфонилбензола (патентный документ 2), таких как N-(5-(2-хлорбензоил)-4-(3-хлорфенил)тиазол-2-ил)-2-(4-(этилсульфонил)фенил)ацетамид, но соединения, имеющие структуру циклического амина, такие как 1-замещенный пиперидин-2-карбоксамид, не были раскрыты.
[0010]
В качестве соединения, имеющего структуру циклического амина, такого как 1-замещенный пиперидин-2-карбоксамид, сообщалось о (S)-1-(2-(3,3-дифторпирролидин-1-ил)ацетил)-N-(1-этил-5-фенил-1H-1,2,4-триазол-3-ил)пиперидин-2-карбоксамиде и т.д., в качестве агониста каннабиноидных 2 рецепторов (патентный документ 3), и сообщалось о (R)-N-(5-бензил-4-фенилтиазол-2-ил)-1-(2-циклопентилацетил)пиперидин-2-карбоксамиде и т.д. в качестве ацил-коэнзима A: ингибитора диацилглицерол ацилтрансферазы 1 (патентный документ 4), но воздействие этих соединений на RORγ не было ни раскрыто, ни предложено.
[0011]
В качестве терапевтического средства или профилактического средства против рассеянного склероза сообщалось о (S)-(3-(5-(4-фторбензил)-1,2,4-оксадиазол-3-ил)пиперидин-1-ил)(4-фторфенил)метаноне и т.д. как позитивном аллостерическом модуляторе рецептора метаботропного глутамата (патентный документ 5), но отсутствуют какие-либо конкретные данные об эффективности и полезности препарата на основе этих соединений в отношении рассеянного склероза. В патентном документе 5 эффективность этих соединений в отношении RORγ не была ни раскрыта, ни предложена, и соединения, имеющие структуру 1-замещенного пиперидин-2-карбоксамида, не были раскрыты.
Документы предшествующего уровня техники
[Патентные документы]
[0012]
[Патентный документ 1] JP 2012-236822 A
[Патентный документ 2] WO 2012/027965
[Патентный документ 3] WO 2010/096371
[Патентный документ 4] WO 2010/007046
[Патентный документ 5] WO 2006/129199
[Непатентные документы]
[0013]
[Непатентный документ 1] Chen et al., International Immunopharmacology, 2011, Vol. 11, p. 536-542
[Непатентный документ 2] Hofmann et al., Current Opinion in Allergy and Clinical Immunology, 2016, Vol. 16, p. 451-457
[Непатентный документ 3] Ivanov et al., Cell, 2006, Vol. 126, p. 1121-1133
[Непатентный документ 4] Jetten, Nuclear Receptor Signaling, 2009, Vol. 7, e003
[Непатентный документ 5] Hamzaoui et al., Medical Science Monitor, 2011, Vol. 17, p.CR227-234
[Непатентный документ 6] Ma et al., Journal of the European Academy of Dermatology and Venereology, 2014, Vol. 28, p. 1079-1086
[Непатентный документ 7] Zhao et al., British Journal of Dermatology, 2009, Vol. 161, p. 1301-1306
[Непатентный документ 8] Leppkes et al., Gastroenterology, 2009, Vol. 136, p. 257-267
[Непатентный документ 9] Jetten et al., Journal of Immunology, 2007, Vol. 178, p. 3208-3218
[Непатентный документ 10] Li et al., Molecular Endocrinology, 2010, Vol. 24. p. 923-929
[Непатентный документ 11] Burris et al., Nature, 2011, Vol. 472, p. 491-494
Сущность изобретения
[0014]
Для эффективного лечения аутоиммунных заболеваний и аллергических заболеваний используются стероиды или иммунодепрессанты, действующие на всю иммунную систему как внутренние лекарственные средства. Однако из-за опасения серьезных побочных эффектов, таких как инфекции, в настоящее время существует множество клинических случаев, при которых их введение должно быть прекращено, прежде чем получить достаточную эффективность препарата. Поэтому желательно разработать новое лекарственное средство, нацеленное на молекулы, играющие важную роль в механизме начала и прогрессирования аутоиммунных заболеваний и аллергических заболеваний.
[0015]
Поэтому, задачей настоящего изобретения является предоставление нового соединения, обладающего RORγ антагонистической активностью. Следующей задачей настоящего изобретения является предоставление терапевтического средства или профилактического средства против аутоиммунного заболевания или аллергического заболевания, основанного на эффекте подавления функции RORγ посредством антагонистической активности в отношении RORγ.
[0016]
С тем чтобы решить указанные выше задачи и найти новое производное циклического амина, обладающее антагонистической активностью в отношении RORγ, настоящие авторы провели тщательное исследование и осуществили настоящее изобретение.
[0017]
Таким образом, настоящее изобретение предоставляет производное циклического амина, представленное следующей общей формулой (I):
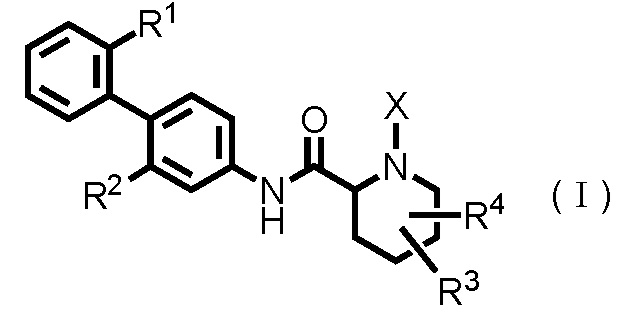
где
R1 представляет собой алкоксигруппу, имеющую 1-3 атома углерода, любые 1-3 атома водорода которой необязательно заменены атомом(ами) галогена;
R2 представляет собой атом галогена;
R3 представляет собой атом водорода, атом галогена или гидроксигруппу;
R4 представляет собой атом водорода или атом галогена;
X представляет собой -C(=O)-(CH2)n-R5 или -S(=O)2-R6;
n равен целому числу от 0 до 5;
R5 представляет собой атом водорода, -OR7, -SR7, -S(=O)2-R7, -C(=O)-OR7, -N(R7)R8, алкильную группу, имеющую 1-3 атома углерода, любые 1-3 атома водорода которой необязательно заменены атомом(ами) галогена, или гетероарильную группу, любой(ые) атом(ы) водорода которой необязательно замещены алкильной группой(ами), имеющими 1-3 атома углерода;
R6 представляет собой алкильную группу, имеющую 1-5 атомов углерода;
R7 представляет собой атом водорода или алкильную группу, имеющую 1-3 атома углерода, любые 1-3 атома водорода которой необязательно заменены атомом(ами) галогена; и
R8 представляет собой атом водорода, алкильную группу, имеющую 1-3 атома углерода, ацильную группу, имеющую 2-4 атома углерода, или алкилсульфонильную группу, имеющую 1-3 атома углерода,
или его фармацевтически приемлемую соль.
[0018]
В производном циклического амина, представленном общей формулой (I), приведенной выше, предпочтительным является, когда R1 представляет собой алкоксигруппу, имеющую 1-3 атома углерода, любые 1-3 атома водорода которой необязательно заменены атомом(ами) фтора или атомом(ами) хлора, R2 представляет собой атом фтора или атом хлора, R3 представляет собой атом водорода, атом фтора, атом хлора или гидроксигруппу, R4 представляет собой атом водорода, атом фтора или атом хлора, R5 представляет собой атом водорода, -OR7, -SR7, -S(=O)2-R7, -C(=O)-OR7, -N(R7)R8, алкильную группу, имеющую 1-3 атома углерода, любые 1-3 атома водорода которой необязательно заменены атомом(ами) фтора или атомом(ами) хлора, или гетероарильную группу, любой атом(ы) водорода которой необязательно заменен метильной группой(ами), R6 представляет собой алкильную группу, имеющую 1-3 атома углерода, и R7 представляет собой атом водорода или алкильную группу, имеющую 1-3 атома углерода, любые 1-3 атома водорода которой необязательно заменены атомом(ами) фтора или атомом(ами) хлора.
[0019]
В этом случае, можно ожидать более высокую антагонистическую активность в отношении RORγ.
[0020]
В производном циклического амина, представленном общей формулой (I), приведенной выше, более предпочтительным является, когда R1 представляет собой метоксигруппу, любые 1-3 атома водорода которой необязательно заменены атомом(ами) фтора, R2 представляет собой атом фтора или атом хлора, R3 представляет собой атом водорода, атом фтора или гидроксигруппу, R4 представляет собой атом водорода или атом фтора, n равен целому числу от 0 до 4, R5 представляет собой атом водорода, -OR7, -N(R7)R8, алкильную группу, имеющую 1-3 атома углерода, любые 1-3 атома водорода которой необязательно заменены атомом(ами) фтора, или 5-членную кольцевую гетероарильную группу, любой атом(ы) водорода которой необязательно заменен метильной группой(ами), R6 представляет собой метильную группу или этильную группу, R7 представляет собой атом водорода или алкильную группу, имеющую 1-3 атома углерода, любые 1-3 атома водорода которой необязательно заменены атомом(ами) фтора, и R8 представляет собой атом водорода, метильную группу, ацильную группу, имеющую 2-4 атома углерода, или алкилсульфонильную группу, имеющую 1-3 атома углерода.
[0021]
В этом случае можно ожидать более высокую RORγ антагонистическую активность и, кроме того, можно ожидать отличный терапевтический эффект или профилактический эффект в отношении аутоиммунных заболеваний, таких как рассеянный склероз или псориаз, или аллергических заболеваний, таких как аллергический дерматит.
[0022]
В производном циклического амина, представленном общей формулой (I), приведенной выше, еще более предпочтительным является, когда R1 представляет собой трифторметоксигруппу, R2 представляет собой атом хлора, R3 представляет собой атом водорода, R4 представляет собой атом водорода, X представляет собой -C(=O)-(CH2)n-R5, n равен целому числу от 0 до 3, R5 представляет собой метильную группу, трифторметильную группу, -N(R7)R8 или имидазолильную, триазолильную или тетразолильную группу, любой атом(ы) водорода которой необязательно заменен метильной группой(ами), R7 представляет собой атом водорода, метильную группу или этильную группу, и R8 представляет собой атом водорода, метильную группу, ацетильную группу, пропионильную группу, метилсульфонильную группу или этилсульфонильную группу.
[0023]
В этом случае можно ожидать более высокую RORγ антагонистическую активность и, кроме того, можно ожидать отличный терапевтический эффект или профилактический эффект в отношении аутоиммунных заболеваний, таких как рассеянный склероз или псориаз, или аллергических заболеваний, таких как аллергический дерматит.
[0024]
Настоящее изобретение предоставляет лекарственное средство и антагонист RORγ, каждый из которых содержит производное циклического амина, представленное общей формулой (I), приведенной выше, или его фармацевтически приемлемую соль в качестве активного ингредиента.
[0025]
Указанное выше лекарственное средство предпочтительно представляет собой терапевтическое средство или профилактическое средство против аутоиммунного заболевания или аллергического заболевания, более предпочтительно терапевтическое средство или профилактическое средство против рассеянного склероза или псориаза в качестве указанного выше терапевтического средства или профилактического средства против аутоиммунного заболевания, более предпочтительно терапевтическое средство или профилактическое средство против аллергического дерматита в качестве указанного выше терапевтического средства или профилактического средства против аллергического заболевания, и более предпочтительно терапевтическое средство или профилактическое средство против контактного дерматита или атопического дерматита в качестве указанного выше терапевтического средства или профилактического средства против аллергического дерматита.
[0026]
Поскольку производное циклического амина настоящего изобретения или его фармацевтически приемлемая соль обладает RORγ антагонистической активностью, оно может эффективно подавлять функцию RORγ и может быть использовано в качестве терапевтического средства или профилактического средства против аутоиммунных заболеваний или аллергических заболеваний.
Краткое описание фигур
[0027]
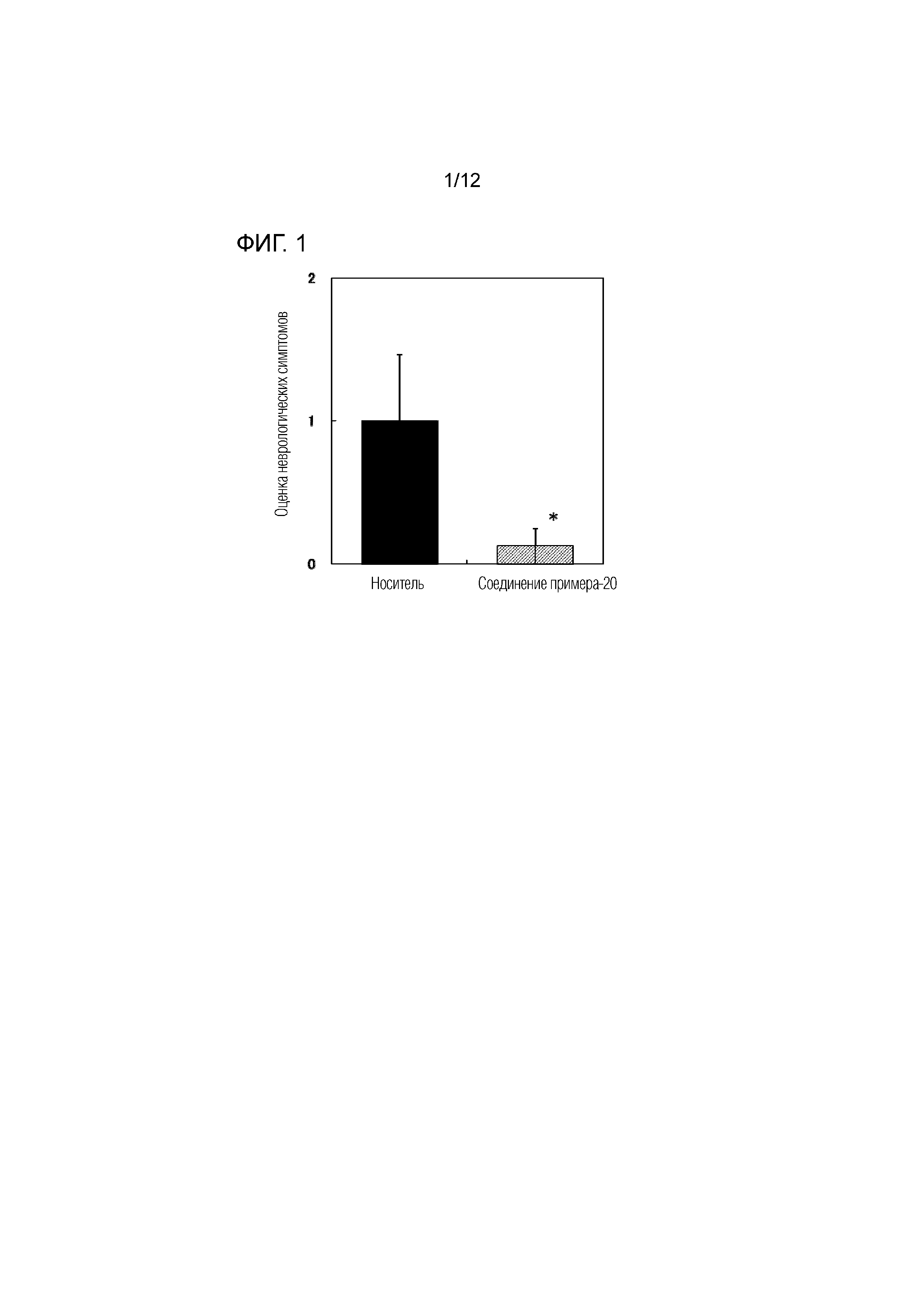
На фиг.1 представлена диаграмма, показывающая возрастание подавляющего воздействия соединения примера 20 при оценке неврологической симптоматики на экспериментальной мышиной модели аутоиммунного энцефаломиелита.
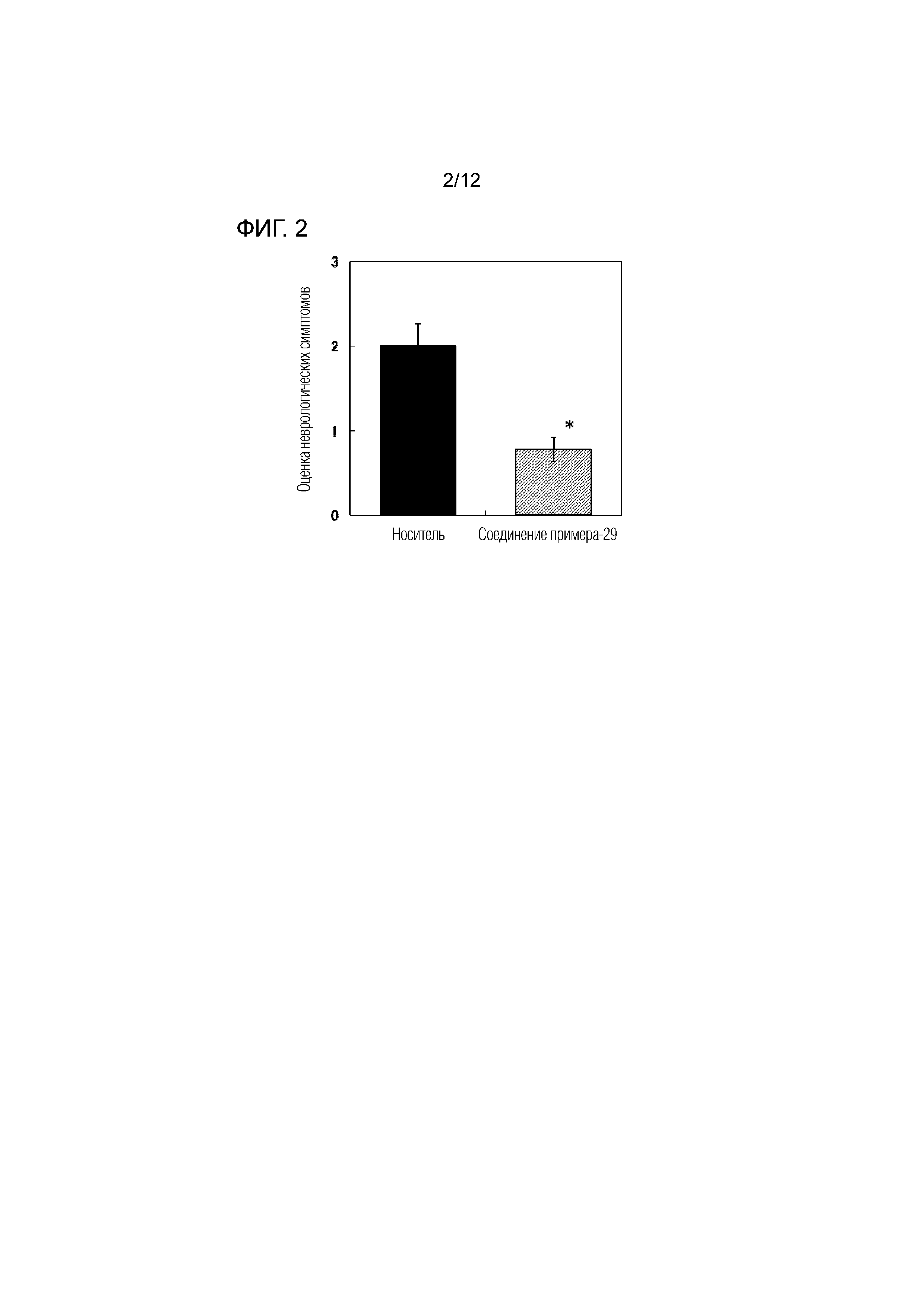
На фиг.2 представлена диаграмма, показывающая возрастание подавляющего воздействия соединения примера 29 при оценке неврологической симптоматики на экспериментальной мышиной модели аутоиммунного энцефаломиелита.
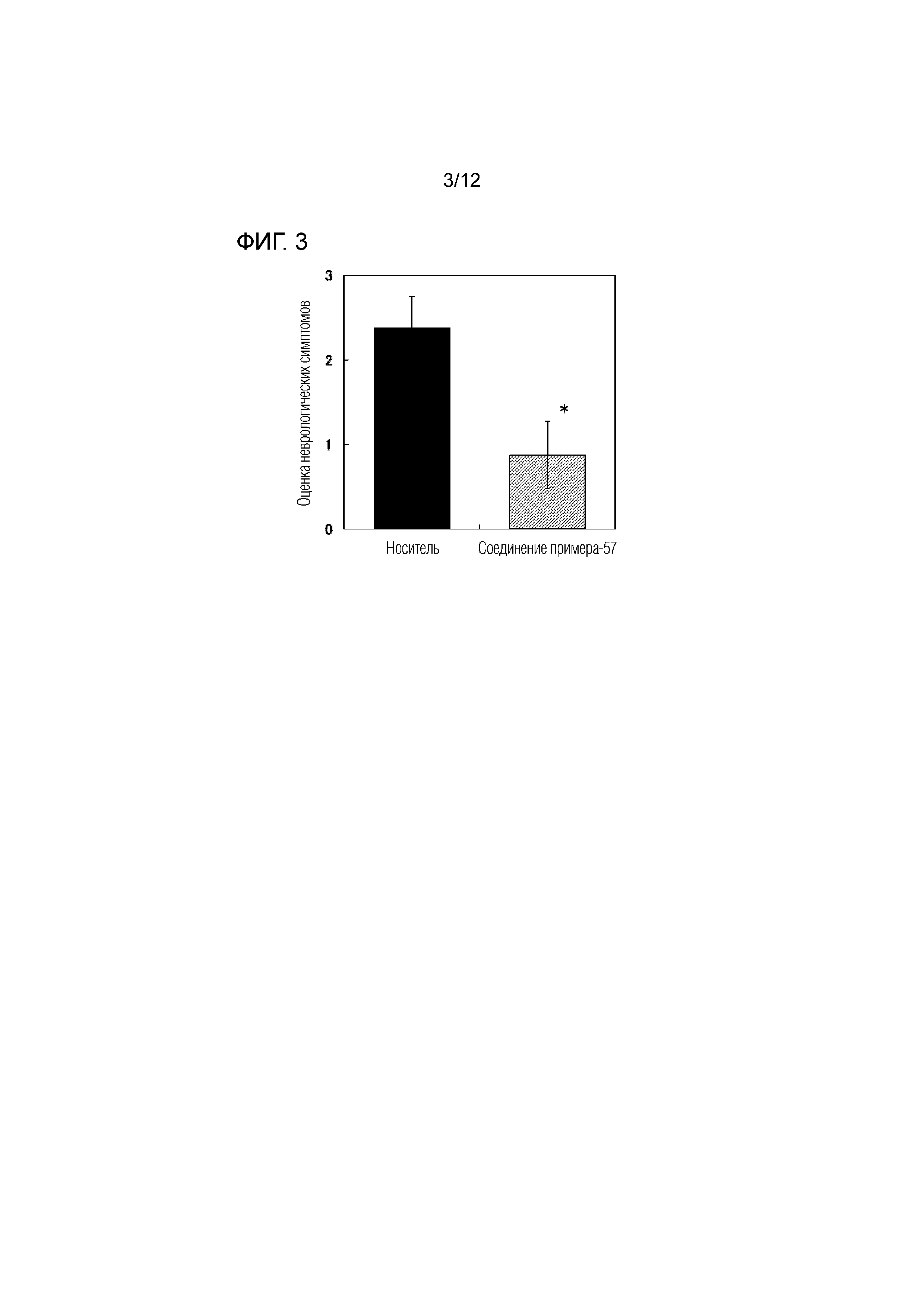
На фиг.3 представлена диаграмма, показывающая возрастание подавляющего воздействия соединения примера 57 при оценке неврологической симптоматики на экспериментальной мышиной модели аутоиммунного энцефаломиелита.
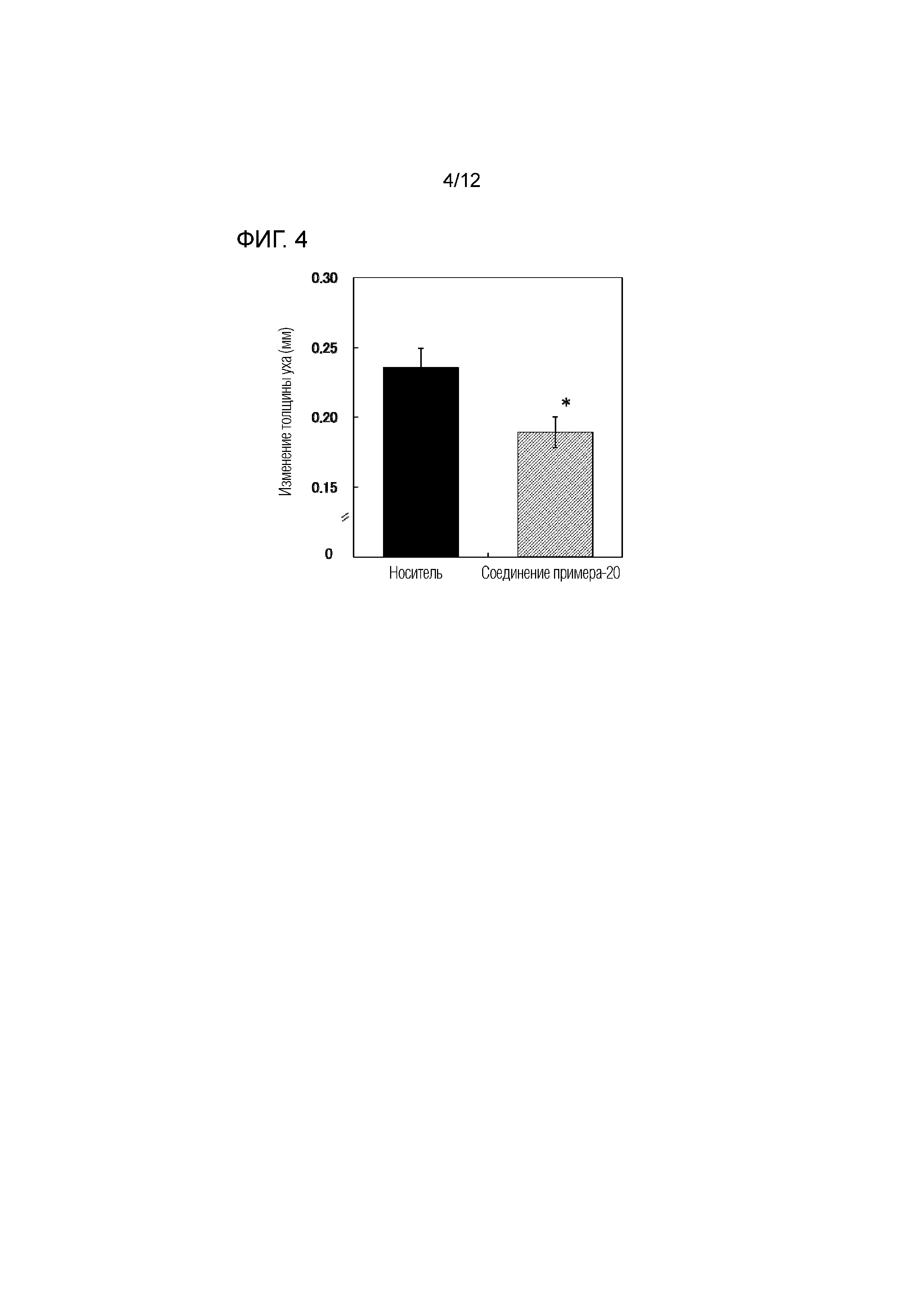
На фиг.4 представлена диаграмма, показывающая возрастание подавляющего воздействия соединения примера 20 на толщину ушей на мышиной модели индуцированного имиквимодом псориаза.
На фиг.5 представлена диаграмма, показывающая возрастание подавляющего воздействия соединения примера 29 на толщину ушей на мышиной модели индуцированного имиквимодом псориаза.
На фиг.6 представлена диаграмма, показывающая возрастание подавляющего воздействия соединения примера 57 на толщину ушей на мышиной модели индуцированного имиквимодом псориаза.
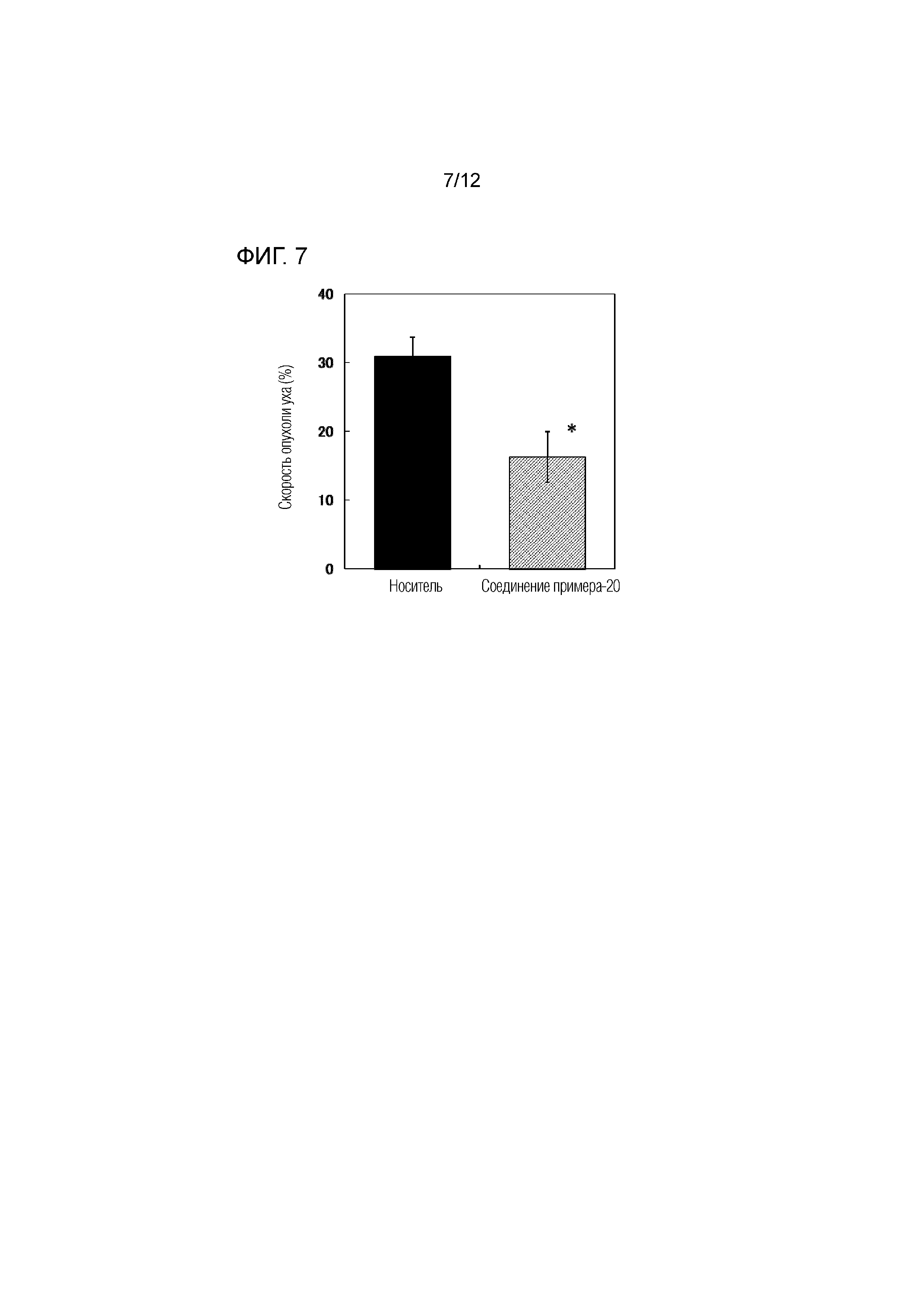
На фиг.7 представлена диаграмма, показывающая возрастание подавляющего воздействия соединения примера 20 на скорость отека уха на мышиной модели индуцированного динитрофторбензолом аллергического дерматита.
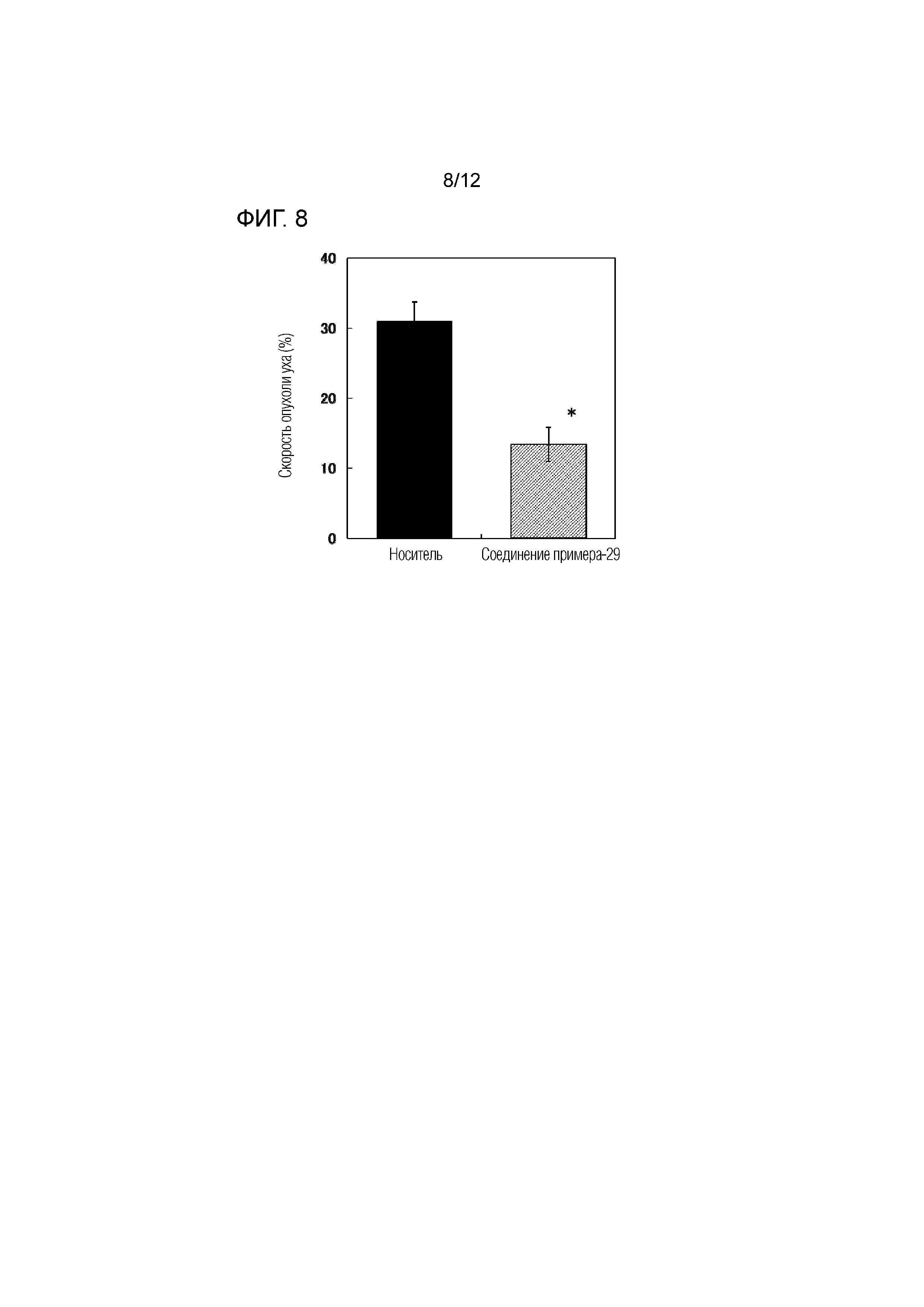
На фиг.8 представлена диаграмма, показывающая возрастание подавляющего воздействия соединения примера 29 на скорость отека уха на мышиной модели индуцированного динитрофторбензолом аллергического дерматита.
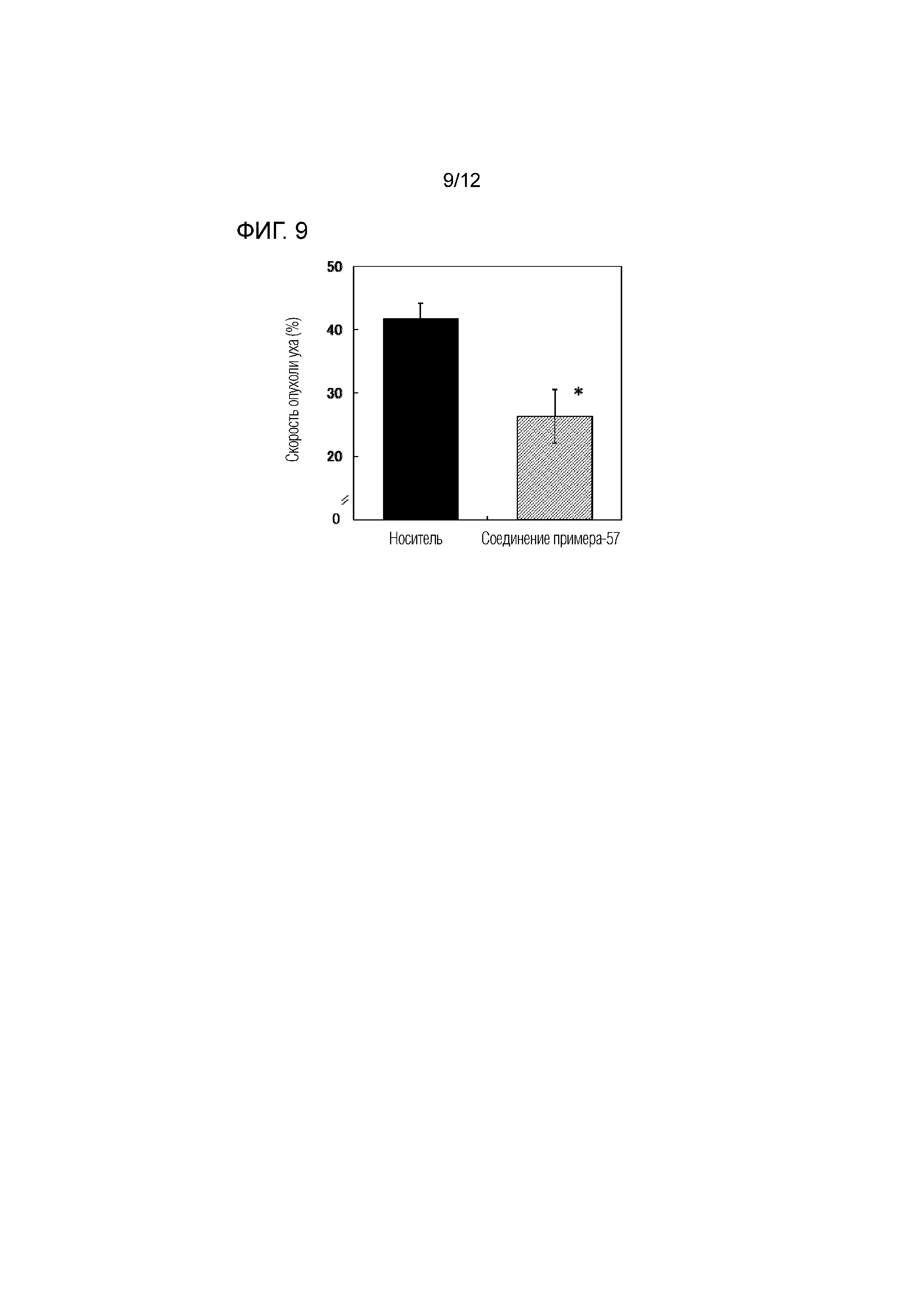
На фиг.9 представлена диаграмма, показывающая возрастание подавляющего воздействия соединения примера 57 на скорость отека уха на мышиной модели индуцированного динитрофторбензолом аллергического дерматита.
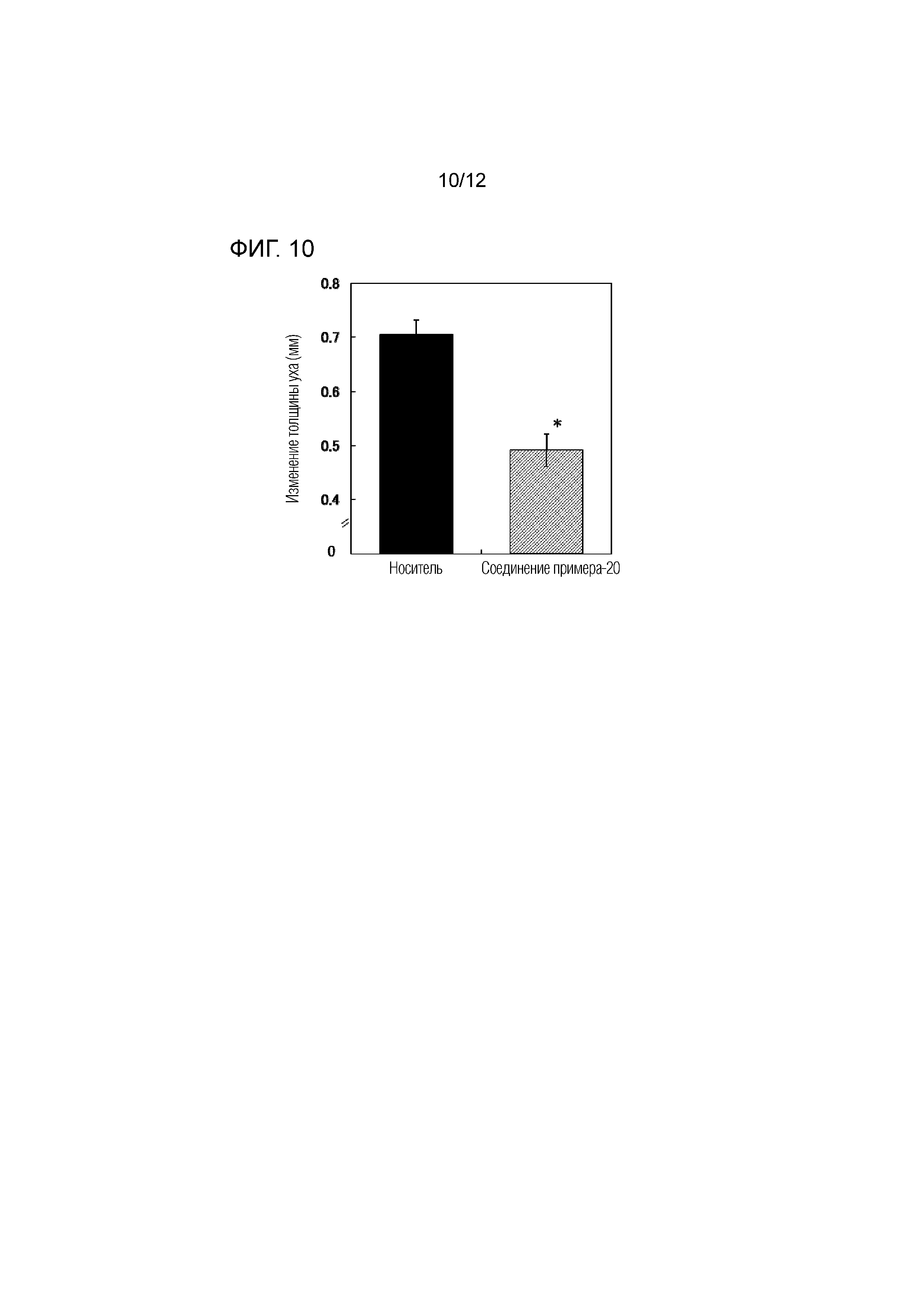
На фиг.10 представлена диаграмма, показывающая возрастание подавляющего воздействия соединения примера 20 на толщину ушей на мышиной модели индуцированного оксазолоном атопического дерматита.
На фиг.11 представлена диаграмма, показывающая возрастание подавляющего воздействия соединения примера 29 на толщину ушей на мышиной модели индуцированного оксазолоном атопического дерматита.
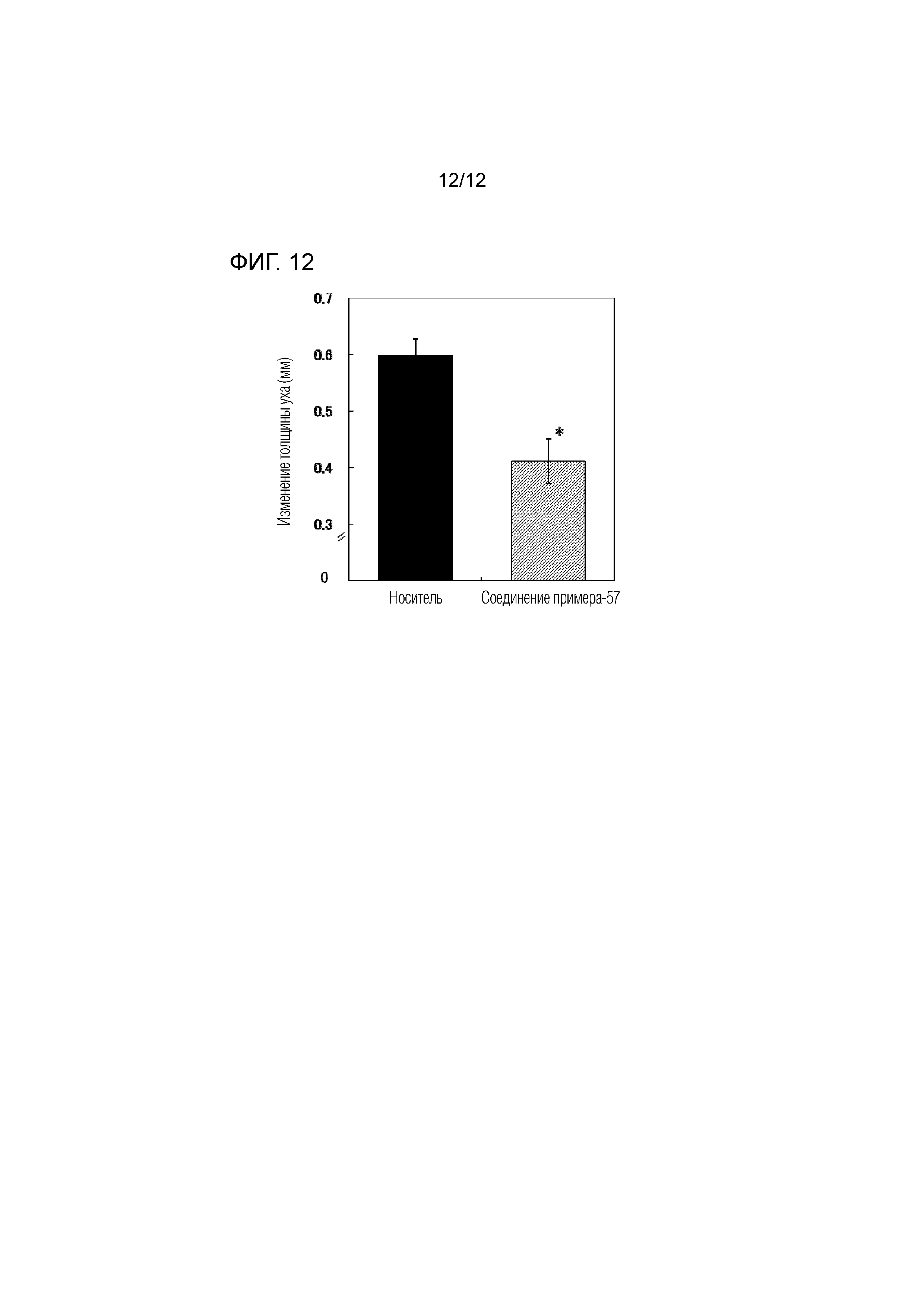
На фиг.12 представлена диаграмма, показывающая возрастание подавляющего воздействия соединения примера 57 на толщину ушей на мышиной модели индуцированного оксазолоном атопического дерматита.
Подробно описание изобретения
[0028]
Производное циклического амина настоящего изобретения характеризуется представленной ниже следующей общей формулой (I):
где
R1 представляет собой алкоксигруппу, имеющую 1-3 атома углерода, любые 1-3 атома водорода которой необязательно заменены атомом(ами) галогена;
R2 представляет собой атом галогена;
R3 представляет собой атом водорода, атомом галогена или гидроксигруппу;
R4 представляет собой атом водорода или атом галогена;
X представляет собой -C(=O)-(CH2)n-R5 или -S(=O)2-R6;
n равен целому числу от 0 до 5;
R5 представляет собой атом водорода, -OR7, -SR7, -S(=O)2-R7, -C(=O)-OR7, -N(R7)R8, алкильную группу, имеющую 1-3 атома углерода, любые 1-3 атома водорода которой необязательно заменены атомом(ами) галогена, или гетероарильную группу, любой атом(ы) водорода которой необязательно заменен алкильной группой, имеющей 1-3 атома углерода;
R6 представляет собой алкильную группу, имеющую 1-5 атомов углерода;
R7 представляет собой атом водорода или алкильную группу, имеющую 1-3 атома углерода, любые 1-3 атома водорода которой необязательно заменены атомом(ами) галогена; и
R8 представляет собой атом водорода, алкильную группу, имеющую 1-3 атома углерода, ацильную группу, имеющую 2-4 атома углерода, или алкилсульфонильную группу, имеющую 1-3 атома углерода.
[0029]
Следующие термины, использованные в данном описании, определяются следующим образом, если специально не указано иное.
[0030]
Термин ʺалкильная группа, имеющая 1-3 атома углеродаʺ означает метильную группу, этильную группу, пропильную группу или изопропильную группу.
[0031]
Термин ʺалкильная группа, имеющая 1-5 атомов углеродаʺ означает линейную насыщенную углеводородную группу, имеющую 1-5 атомов углерода или разветвленную насыщенную углеводородную группу, имеющую 3-5 атомов углерода, и их примеры включают метильную группу, этильную группу, пропильную группу, изопропильную группу, бутильную группу, изобутильную группу, втор-бутильную группу, трет-бутильную группу, пентильную группу, изопентильную группу, неофенильную группу или трет-пентильную группу.
[0032]
Термин ʺалкоксигруппа, имеющая 1-3 атома углеродаʺ означает метоксигруппу, этоксигруппу, пропилоксигруппу или изопропилоксигруппу.
[0033]
Термин ʺацильная группа, имеющая 2-4 атома углеродаʺ означает ацетильную группу, пропионильную группу, бутаноильную группу или 2-метилпропаноильную группу.
[0034]
Термин ʺалкилсульфонильная группа, имеющая 1-3 атома углеродаʺ означает метилсульфонильную группу, этилсульфонильную группу, пропилсульфонильную группу или изопропилсульфонильную группу.
[0035]
Термин ʺгетероарильная группаʺ означает гетероциклическую арильную группу, содержащую 1-4 гетероатома, независимо выбранных из группы, состоящей из атома азота, атома кислорода и атома серы, и ее примеры включают тиенильную группу, пирролильную группу, фурилильную группу, тиазолильную группу,
имидазолильную группу, оксазолильную группу, пиразолильную группу, изотиазолильную группу, изоксазолильную группу, триазолильную группу, оксадиазолильную группу, тетразолильную группу, пиридильную группу, пиридазинильную группу, пиримидинильную группу, пиразинильную группу или триазинильную группу.
[0036]
Термин ʺ5-членная кольцевая гетероарильная группаʺ означает гетероциклическую ароматическую группу, имеющую 5 составляющих кольцо атомов, содержащую 1-4 гетероатома, независимо выбранных из группы, состоящей из атома азота, атома кислорода и атома серы, и ее примеры включают тиенильную группу, пирролильную группу, фурилильную группу, тиазолильную группу, имидазолильную группу, оксазолильную группу, пиразолильную группу, изотиазолильную группу, изоксазолильную группу, триазолильную группу, оксадиазолильную или тетразолильную группу.
[0037]
Термин ʺатом галогенаʺ означает атом фтора, атом хлора, атом брома или атом йода.
[0038]
Термин ʺалкильная группа, имеющая 1-3 атома углерода, любые 1-3 атома водорода которой необязательно заменены атомом(ами) галогенаʺ означает алкильную группу, имеющую 1-3 атома углерода, как описано выше, любые 1-3 атома водорода которой, каждый независимо, может быть заменен атомом галогена, как описано выше, и ее примеры включают метильную группу, этильную группу, пропильную группу, изопропильную группу, фторметильную группу, дифторметильную группу, трифторметильную, 2-фторэтильную группу, трифторэтильную группу, трихлорметильную группу или трихлорэтильную группу.
[0039]
Термин ʺалкильная группа, имеющая 1-3 атома углерода, любые 1-3 атома водорода которой необязательно заменены атомом(ами) фтора или атом(ами) хлораʺ означает алкильную группу, имеющую 1-3 атома углерода, как описано выше, любые 1-3 атома водорода которой, каждый независимо, может быть заменен атомом фтора или атомом хлора, и ее примеры включают метильную группу, этильную группу, пропильную группу, изопропильную группу, фторметильную группу, дифторметильную группу, трифторметильную группу, 2-фторэтильную группу, трифторэтильную группу, трихлорметильную группу или трихлорэтильную группу.
[0040]
Термин ʺалкильная группа, имеющая 1-3 атома углерода, любые 1-3 атома водорода которой необязательно заменены атомом(ами) фтораʺ означает алкильную группу, имеющую 1-3 атома углерода, как описано выше, любые 1-3 атома водорода которой могут быть заменены атомом(ами) фтора, и ее примеры включают метильную группу, этильную группу, пропильную группу, изопропильную группу, фторметильную группу, дифторметильную группу, трифторметильную группу, 2-фторэтильную группу или трифторэтильную группу.
[0041]
Термин ʺалкоксигруппа, имеющая 1-3 атома углерода, любые 1-3 атома водорода которой необязательно заменены атомом(ами) галогенаʺ означает алкоксигруппу, имеющую 1-3 атома углерода, как описано выше, любые 1-3 атома водорода которой, каждый независимо, может быть заменен атомом галогена, как описано выше, и ее примеры включают метоксигруппу, этоксигруппу, пропилоксигруппу, изопропилоксигруппу, фторметоксигруппу, дифторметоксигруппу, трифторметоксигруппу, 2-фторэтоксигруппу, трифторэтоксигруппу, трихлорметоксигруппу или трихлорэтоксигруппу.
[0042]
Термин ʺалкоксигруппа, имеющая 1-3 атома углерода, любые 1-3 атома водорода которой необязательно заменены атомом(ами) фтора или хлораʺ означает алкоксигруппу, имеющую 1-3 атома углерода, как описано выше, любые 1-3 атома водорода которой, каждый независимо, может быть заменен атомом фтора или атомом хлора, и ее примеры включают метоксигруппу, этоксигруппу, пропилоксигруппу, изопропилоксигруппу, фторметоксигруппу, дифторметоксигруппу, трифторметоксигруппу, 2-фторэтоксигруппу, трифторэтоксигруппу, трихлорметоксигруппу или трихлорэтоксигруппу.
[0043]
Термин ʺметоксигруппа, любые 1-3 атома водорода которой необязательно заменены атомом(ами) фтораʺ означает метоксигруппу, фторметоксигруппу, дифторметоксигруппу или трифторметоксигруппу.
[0044]
Термин ʺгетероарильная группа, любой атом(ы) водорода которой необязательно замещен(ы) алкильной группой(ами), имеющими 1-3 атома углеродаʺ означает гетероарильную группу, как описано выше, любой атом(ы) водорода которой, каждый независимо, может быть заменен алкильной группой, имеющей 1-3 атома углерода, как описано выше, и ее примеры включают тиенильную группу, пирролильную группу, фурилильную группу, тиазолильную группу, имидазолильную группу, оксазолильную группу, пиразолильную группу, изотиазолильную группу, изоксазолильную группу, триазолильную группу, оксадиазолильную группу, тетразолильную группу, пиридильную группу, пиридазинильную группу, пиримидинильную группу, пиразинильную группу, триазинильную группу, метилтиенильную группу, диметилтиенильную группу, этилтиенильную группу, метилпирролильную группу, диметилпирролильную группу, этилпирролильную группу, фурильную группу, дифурильную группу, этилфурильную группу, метилтиазолильную группу, диметилтиазолильную группу, этилтиазолильную группу, метилимидазолильную группу, диметилимидазолильную группу, этилимидазолильную группу, метилоксазолильную группу, диметилоксазолильную группу, этилоксазолильную группу, метилпиразолильную группу, диметилпиразолильную группу, этилпиразолильную группу, метилизотиазолильную группу, диметилизотиазолильную группу, этилизотиазолильную группу, метилизоксазолильную группу, диметилизоксазолильную группу, этилизоксазолильную группу, метилтриазолильную группу, диметилтриазолильную группу, этилтриазолильную группу, метилоксадиазолильную группу, диметилоксадиазолильную группу, этилоксадиазолильную группу, метилтетразолильную группу, этилтетразолильную группу, метилпиридильную группу, диметилпиридильную группу, этилпиридильную группу, метилпиридазинильную группу, диметилпиридазинильную группу, этилпиридазинильную группу, метилпиримидинильную группу, диметилпиримидинильную группу, этилпиримидинильную группу, метилпиразинильную группу, диметилпиразинильную группу, этилпиразинильную группу, метилтриазинильную группу, диметилтриазинильную группу или этилтриазинильную группу.
[0045]
Термин ʺгетероарильная группа, любой атом(ы) водорода которой необязательно заменен метильной группой(ами)ʺ означает гетероарильную группу, как описано выше, любой атом(ы) водорода которой, каждый независимо, может быть заменен метильной группой, и ее примеры включают тиенильную группу, пирролильную группу, фурилильную группу, тиазолильную группу, имидазолильную группу, оксазолильную группу, пиразолильную группу, изотиазолильную группу, изоксазолильную группу, триазолильную группу, оксадиазолильную группу, тетразолильную группу, пиридильную группу, пиридазинильную группу, пиримидинильную группу, пиразинильную группу, триазинильную группу, метилтиенильную группу, диметилтиенильную группу, метилпирролильную группу, диметилпирролильную группу, фурильную группу, дифурильную группу, метилтиазолильную группу, диметилтиазолильную группу, метилимидазолильную группу, диметилимидазолильную группу, метилоксазолильную группу, диметилоксазолильную группу, метилпиразолильную группу, диметилпиразолильную группу, метилизотиазолильную группу, диметилизотиазолильную группу, метилизоксазолильную группу, диметилизоксазолильную группу, метилтриазолильную группу, диметилтриазолильную группу, метилоксадиазолильную группу, диметилоксадиазолильную группу, метилтетразолильную группу, метилпиридильную группу, диметилпиридильную группу, метилпиридазинильную группу, диметилпиридазинильную группу, метилпиримидинильную группу, диметилпиримидинильную группу, метилпиразинильную группу, диметилпиразинильную группу, метилтриазинильную группу или диметилтриазинильную группу.
[0046]
Термин ʺ5-членная кольцевая гетероарильная группа, любой атом(ы) водорода которой необязательно заменен метильной группой(ами)ʺ означает 5-членную кольцевую гетероарильную группу, как описано выше, любой атом(ы) водорода которой, каждый независимо, может быть заменен метильной группой, и ее примеры включают тиенильную группу, пирролильную группу, фурилильную группу, тиазолильную группу, имидазолильную группу, оксазолильную группу, пиразолильную группу, изотиазолильную группу, изоксазолильную группу, триазолильную группу, оксадиазолильную группу, тетразолильную группу, метилтиенильную группу, диметилтиенильную группу, метилпирролильную группу, диметилпирролильную группу, фурильную группу, дифурильную группу, метилтиазолильную группу, диметилтиазолильную группу, метилимидазолильную группу, диметилимидазолильную группу, метилоксазолильную группу, диметилоксазолильную группу, метилпиразолильную группу, диметилпиразолильную группу, метилизотиазолильную группу, диметилизотиазолильную группу, метилизоксазолильную группу, диметилизоксазолильную группу, метилтриазолильную группу, диметилтриазолильную группу, метилоксадиазолильную группу, диметилоксадиазолильную группу или метилтетразолильную группу.
[0047]
Что касается указанного выше производного циклического амина, в общей формуле (I) R1 предпочтительно представляет собой алкоксигруппу, имеющую 1-3 атома углерода, любые 1-3 атома водорода которой необязательно заменены атомом(ами) фтора или атомом(ами) хлора, более предпочтительно метоксигруппу, любые 1-3 атома водорода которой необязательно заменены атомом(ами) фтора, и еще более предпочтительно трифторметоксигруппу.
[0048]
R2 предпочтительно представляет собой атом фтора или атом хлора, и более предпочтительно атом хлора.
[0049]
R3 предпочтительно представляет собой атом водорода, атом фтора, атом хлора или гидроксигруппу, более предпочтительно атом водорода, атом фтора или гидроксигруппу, и еще более предпочтительно атом водорода.
[0050]
R4 предпочтительно представляет собой атом водорода, атом фтора или атом хлора, более предпочтительно атом водорода или атом фтора, и еще более предпочтительно атом водорода.
[0051]
X предпочтительно представляет собой -C(=O)-(CH2)n-R5.
[0052]
n предпочтительно равен целому числу от 0 до 4, и более предпочтительно целому числу от 0 до 3.
[0053]
R5 предпочтительно представляет собой атом водорода, -OR7, -SR7, -S(=O)2-R7, -C(=O)-OR7, -N(R7)R8, алкильную группу, имеющую 1-3 атома углерода, любые 1-3 атома водорода которой необязательно заменены атомом(ами) фтора или атомом(ами) хлора, или гетероарильную группу, любой атом(ы) водорода которой необязательно заменен метильной группой(ами), более предпочтительно атом водорода, -OR7, -N(R7)R8, алкильную группу, имеющую 1-3 атома углерода, любые 1-3 атома водорода которой необязательно заменены атомом(ами) фтора, или 5-членную кольцевую гетероарильную группу, любой атом(ы) водорода которой необязательно заменен метильной группой(ами), и еще более предпочтительно метильную группу, трифторметильную группу, -N(R7)R8 или имидазолильную, триазолильную или тетразолильную группу, любой атом(ы) водорода которой необязательно заменен метильной группой(ами).
[0054]
R6 предпочтительно представляет собой алкильную группу, имеющую 1-3 атома углерода, и более предпочтительно метильную группу или этильную группу.
[0055]
R7 предпочтительно представляет собой атом водорода или алкильную группу, имеющую 1-3 атома углерода, любые 1-3 атома водорода которой необязательно заменены атомом(ами) фтора или атомом(ами) хлора, более предпочтительно атом водорода или алкильную группу, имеющую 1-3 атома углерода, любые 1-3 атома водорода которой необязательно заменены атомом(ами) фтора, и еще более предпочтительно атом водорода, метильную группу или этильную группу.
[0056]
R8 более предпочтительно представляет собой атом водорода, метильную группу, ацильную группу, имеющую 2-4 атома углерода, или алкилсульфонильную группу, имеющую 1-3 атома углерода, и еще более предпочтительно атом водорода, метильную группу, ацетильную группу, пропионильную группу, метилсульфонильную группу или этилсульфонильную группу.
[0057]
Конкретные примеры предпочтительных соединений производного циклического амина, представленного общей формулой (I), показаны в таблицах (1-1)-(1-3), но настоящее изобретение ими не ограничено.
[0058]
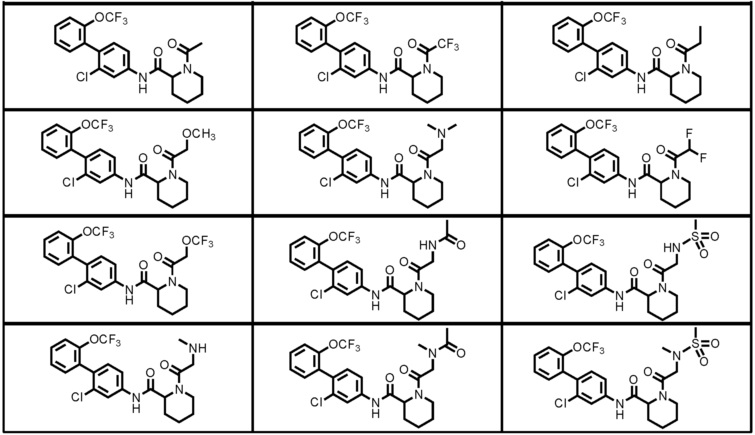
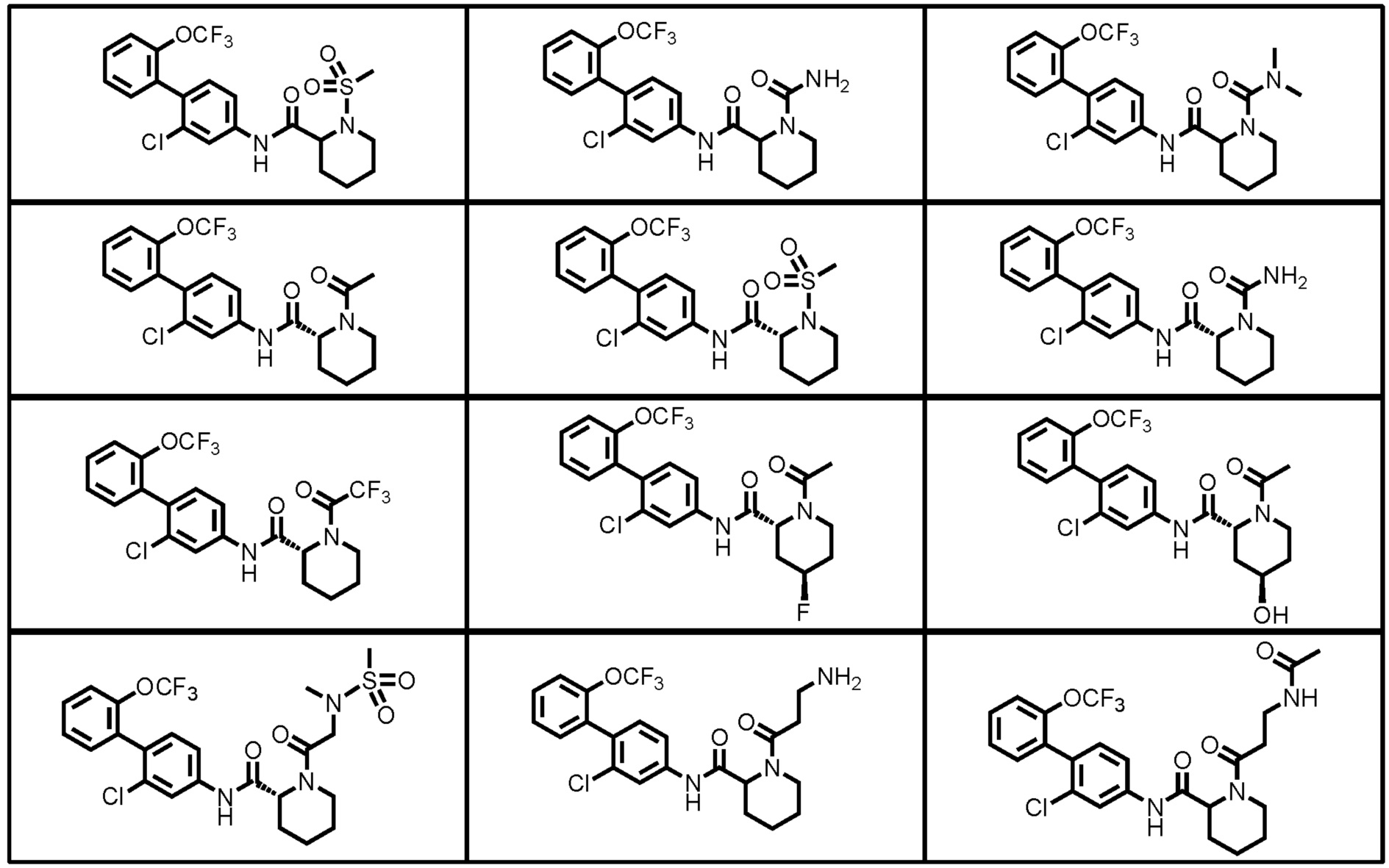
[0059]
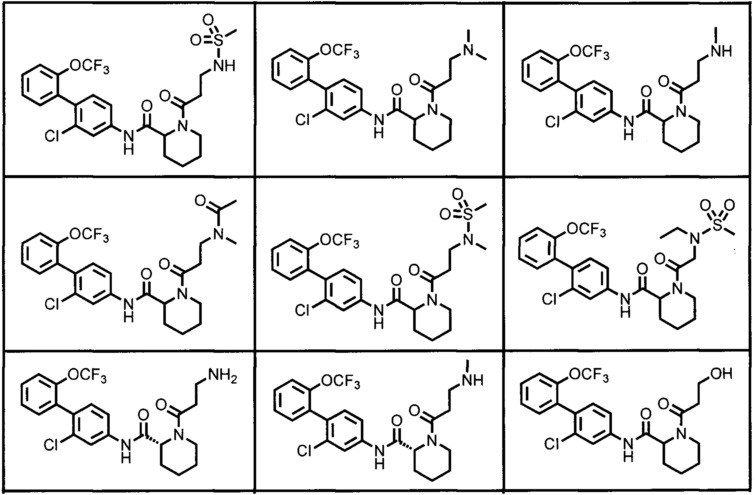
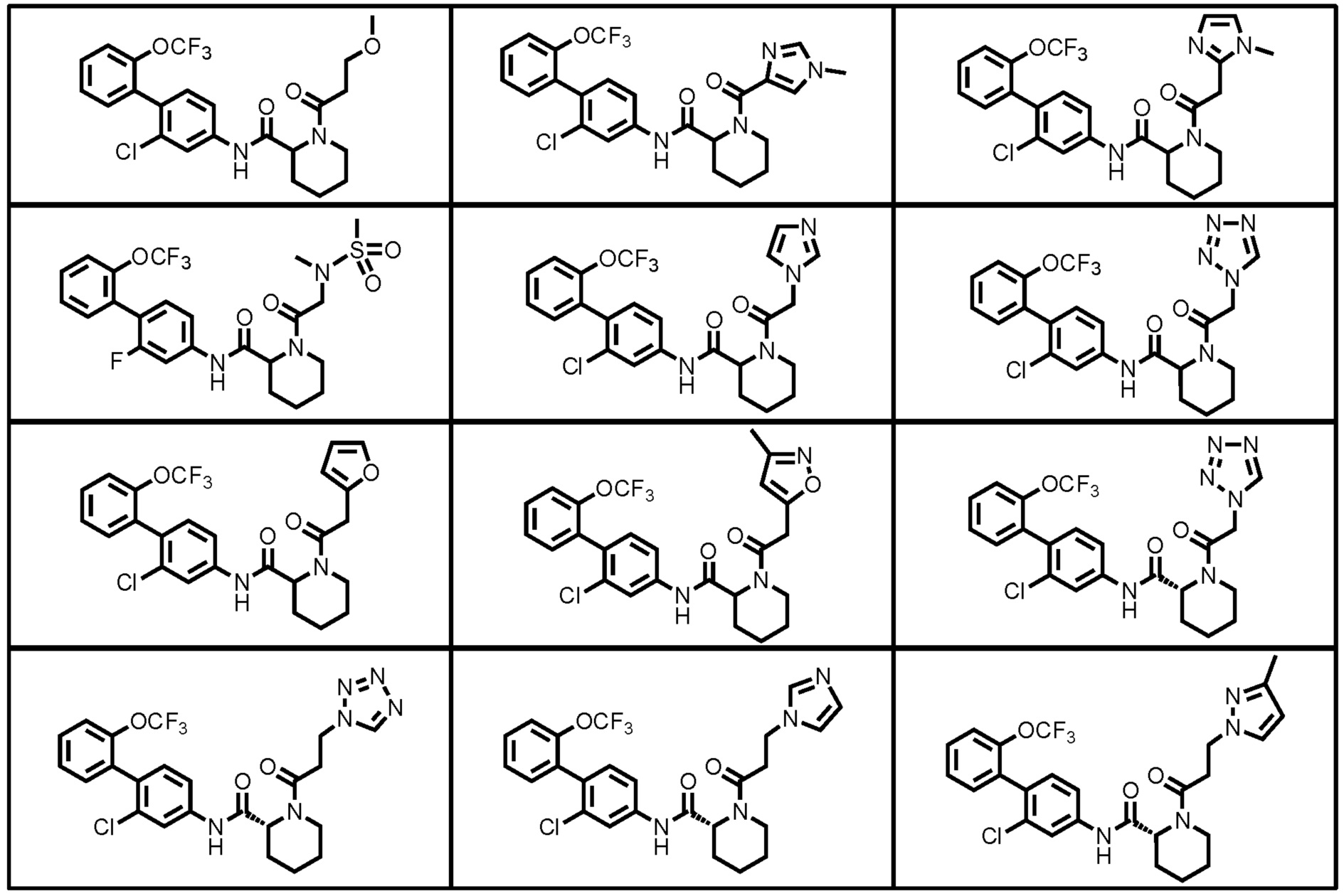
[0060]
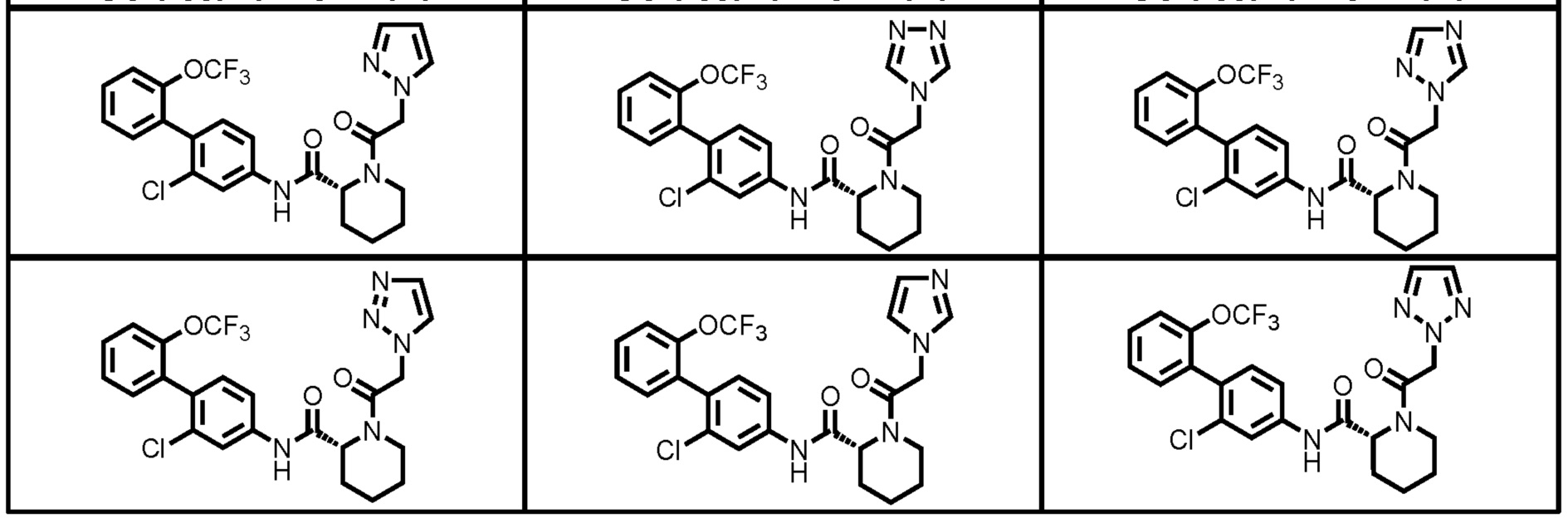
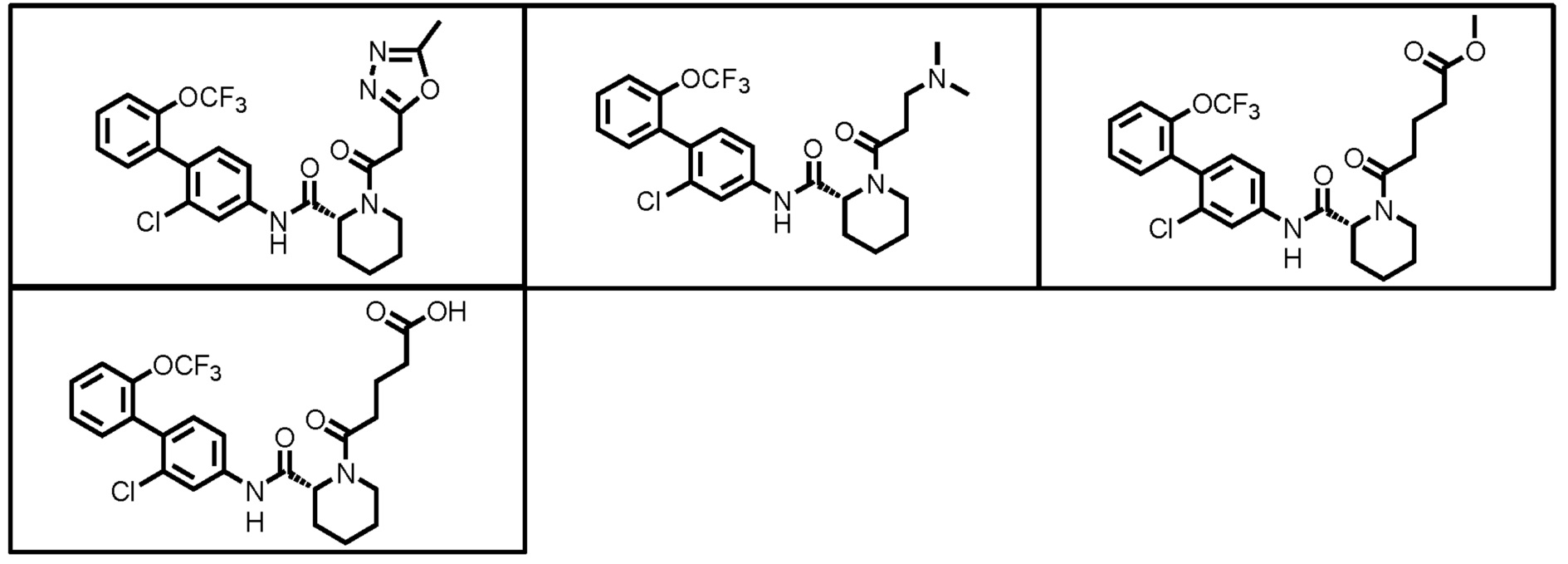
[0061]
Соединения, представленные в таблицах (1-1)-(1-3) также включают их фармацевтически приемлемые соли.
[0062]
Производное циклического амина, представленное общей формулой (I), может включать конформационные изомеры, ротамеры, таутомеры, оптические изомеры, диастереомеры и т.п., и включает не только индивидуальные изомеры, но также рацематы и диастереомерные смеси.
[0063]
Производное циклического амина, представленное общей формулой (I), может быть помечено одним или более изотопами, и примеры меченого изотопа включают2H,3H,13C,14C,15N,15O,18O и/или125I.
[0064]
Примеры ʺфармацевтически приемлемой солиʺ производного циклического амина, представленного общей формулой (I), включают соли с неорганическим основанием, соли с органическим основанием, соли с неорганической кислотой и соли с органической кислотой. Примеры соли с неорганическим основанием включают соли щелочного металла, такие как натриевая соль или калиевая соль, соли щелочноземельного металла, такие как соль кальция или соль магния, аммониевая соль, соль алюминия или соль цинка, и примеры соли с органическим основанием включают соли с органическим амином, таким как триэтиламин, этаноламин, морфолин, пиперидин или дициклогексиламин, или соли с основной аминокислотой, такой как аргинин или лизин. Примеры солей с неорганической кислотой включают гидрохлорид, сульфат, нитрат, гидробромид, гидройодид, фосфат или т.п., и примеры солей с органической кислотой включают оксалат, малонат, цитрат, фумарат, лактат, малат, сукцинат, тартрат, ацетат, трифторацетат, малеат, глюконат, бензоат, аскорбат, глутарат, манделат, фталат, метансульфонат, этансульфонат, бензосульфонат, п-толуолсульфонат, камфорсульфонат, аспартат, глютамат, циннамат или т.п.
[0065]
Производное циклического амина, представленное приведенной выше формулой (I), или его фармацевтически приемлемая соль может быть в форме ангидрида или сольвата, таком как гидрат. В данном описании сольват предпочтительно является фармакологически приемлемым сольватом. Фармакологически приемлемый сольват может быть гидратом или негидратом, и предпочтительно представляет собой гидрат. Примеры растворителя, образующего сольват, включают растворители на основе спиртов, такие как метанол, этанол или н-пропанол, N,N-диметилформамид (здесь и далее сокращенно ДМФА), диметилсульфоксид (здесь и далее сокращенно ДМСО) или воду.
[0066]
Производное циклического амина, представленное приведенной выше формулой (I) (здесь и далее называемое производным циклического амина (I)), может быть получено соответствующим способом, основанном на свойствах основного скелета производного и типов заместителей. Исходные вещества и реагенты, которые используются в получении данных соединений, обычно могут быть коммерчески доступными или получены известными способами.
[0067]
Производное циклического амина (I) и промежуточные, и исходные соединения, используемые при их получении, могут быть выделены и очищены известными методами. Примеры известных методов выделения и очистки включают экстракцию растворителем, перекристаллизацию или хроматографию.
[0068]
Когда производное циклического амина (I) содержит оптический изомер или стереоизомер, каждый из изомеров может быть получен в виде отдельного соединения известным способом. Примеры известного способа включают кристаллизацию, ферментативное разделение или хиральную хроматографию.
[0069]
В каждом из реакционных способов получения, приведенных ниже, когда исходное соединение имеет аминогруппу или карбоксильную группу, при необходимости, в указанные группы могут быть введены защитные группы, и после проведения основной реакции, данные защитные группы могут быть удалены, с получением целевого соединения.
[0070]
Примеры аминозащитных групп включают алкилкарбонильную группу, имеющую 2-6 атомов углерода (например, ацетильную группу), бензоильную группу, алкилоксикарбонильную группу, имеющую 2-8 атомов углерода (например, трет-бутоксикарбонильную группу или бензилоксикарбонильную группу), аралкильную группу, имеющую 7-10 атомов углерода (например, бензильную группу), или фталоильную группу.
[0071]
Примеры карбоксилзащитных групп включают алкильную группу, имеющую 1-6 атомов углерода (например, метильную группу, этильную группу или трет-бутильную группу), или аралкильную группу, имеющую 7-10 атомов углерода (например, бензильную группу).
[0072]
Удаление защитных групп варьируется в зависимости от типа защитных групп, но удаление защиты может быть проведено в соответствии с известными способами (например, Greene, TW, ʺGreene's Protective Groups in Organic Synthesisʺ, Wiley-Interscience) или аналогичными способами.
[0073]
Как показано, например, на схеме 1, производное циклического амина (I) может быть получено реакцией сочетания (первая стадия) производного бороновой кислоты (II) с производным арилгалогенида (III) в присутствии металлического катализатора и основания, с последующей реакцией конденсации (вторая стадия) производного бифениламина (IV), полученного на первой стадии, с производным пипеколиновой кислоты (V) в присутствии конденсирующего агента и основания, с последующей реакцией удаления защиты (третья стадия) у производного амида N-трет-бутоксикарбонилпипеколиновой кислоты (VI), полученного на второй стадии, в присутствии кислоты, и с последующей реакцией конденсации (четвертая стадия) производного амида пипеколиновой кислоты (VII), полученного на третьей стадии, с производным ангидрида органической кислоты (VIII) в присутствии основания. Производное циклического амина (I) также может быть получено реакцией конденсации производного амида пипеколиновой кислоты (VII) с производным эфира органической кислоты (IX). Производное циклического амина (I) также может быть получено реакцией конденсации производного амида пипеколиновой кислоты (VII) с производным хлорангидрида органической кислоты (X) в присутствии основания. Производное циклического амина (I) также может быть получено реакцией конденсации производного амида пипеколиновой кислоты (VII) и производного органической кислоты (XI) в присутствии конденсирующего агента и основания. Производное циклического амина (I) также может быть получено реакцией конденсации производного амида пипеколиновой кислоты (VII) с триметилсилилизоцианатом в присутствии основания.
[0074]
Когда производное циклического амина (I) содержит, например, аминогруппу, данная аминогруппа может быть преобразована в амидную группу, сульфонамидную группу или т.п. или N-алкильное производное реакцией конденсации, реакцией восстановительного аминирования или т.п. Когда производное циклического амина (I) содержит сульфидную группу, данная сульфидная группа может быть преобразована в сульфонильную группу реакцией оксидирования. Когда производное циклического амина (I) содержит сложноэфирную группу, данная сложноэфирная группа может быть преобразована в карбоксильную группу реакцией гидролиза.
Схема 1
На схеме 1 Q представляет собой атом галогена, и R1-R4 и X являются такими, как описано выше.
[0075]
(Первая стадия)
Количество производного арилгалогенида (III), используемого в реакции сочетания, предпочтительно составляет от 0,5 до 10 эквивалентов, и более предпочтительно от 0,7 до 3 эквивалентов, из расчета на производное бороновой кислоты (II).
[0076]
Примеры металлического катализатора, используемого в реакции сочетания, включают дихлорметановый аддукт 1,1'-бис(дифенилфосфино)ферроцендихлорпалладия(II), палладий(II)хлорид, бис(дибензилиденацетон)палладий(0), тетракистрифенилфосфинпалладий(0) или дихлорбистрифенилфосфинпалладий(0), и дихлорметановый аддукт 1,1'-бис(дифенилфосфино)ферроцендихлорпалладия(II) является предпочтительным.
[0077]
Количество металлического катализатора, используемого для реакции сочетания, предпочтительно составляет от 0,01 до 5 эквивалентов, и более предпочтительно от 0,05 до 0,5 эквивалентов, из расчета на производное бороновой кислоты (II).
[0078]
Примеры основания, используемого в реакции сочетания, включают органическое основание, такое как триэтиламин или диизопропилэтиламин, неорганическое основание, такое как карбонат натрия или карбонат калия, амид лития, такой как литийгексаметилдисилазид или литийдиизопропиламид, или алкоксид металла, такой как трет-бутилоксид натрия, трет-бутилоксид калия или их смесь, и неорганическое основание, такое как карбонат натрия или карбонат калия, является предпочтительным.
[0079]
Количество основания, используемого в реакции сочетания, предпочтительно составляет от 0,5 до 10 эквивалентов, и более предпочтительно от 1 до 3 эквивалентов, из расчета на производное бороновой кислоты (II).
[0080]
Реакционный растворитель, используемый для реакции сочетания, надлежащим образом выбирают в соответствии с типом используемого реагента или т.п., но особенно не ограничивают до тех пор, пока он не препятствует проведению реакции, и его примеры включают растворители на основе простых эфиров, такие как тетрагидрофуран, 1,4-диоксан, простой диметиловый эфир этиленгликоля или диметоксиэтан, растворители на основе нитрила, такие как ацетонитрил или пропионитрил, ароматические углеводородные растворители, такие как бензол или толуол, апротонные полярные растворители, такие как ДМФА или ДМСО, воду или их смесь. Смешанный растворитель из растворителей на основе нитрила, таких как ацетонитрил или пропионитрил, и воды является предпочтительным.
[0081]
Температура реакции сочетания предпочтительно составляет от 0 до 200°C, и более предпочтительно от 50 до 150°C.
[0082]
Время реакции сочетания надлежащим образом выбирают в соответствии с реакционными условиями, такими как температура реакции, и время реакции предпочтительно составляет от 1 до 30 часов.
[0083]
Концентрация производного бороновой кислоты (II), используемого в реакции сочетания, в начале реакции предпочтительно составляет от 1 ммоль/л до 1 моль/л.
[0084]
Производное бороновой кислоты (II) и производное барилгалогенида (III), используемые в реакции сочетания, могут быть приобретены или получены известными способами.
[0085]
(Стадия 2)
Количество производного пипеколиновой кислоты (V), используемого в реакции конденсации, предпочтительно составляет от 0,1 до 10 эквивалентов, и более предпочтительно от 0,5 до 3 эквивалентов, из расчета на производное бифениламина (IV).
[0086]
Примеры конденсирующего агента, используемого в реакции конденсации, включают N,N'-дициклогексилкарбодиимид, гидрохлорид N-этил-N'-3-диметиламинопропилкарбодиимида, N,N'-карбодиимидазол, гексафторфосфат {{[(1-циано-2-этокси-2-оксоэтилиден)амино]окси}-4-морфолинометилен}диметиламмония (здесь и далее сокращенно COMU), гексафторфосфат O-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония (здесь и далее сокращенно HATU) или гексафторфосфат O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (здесь и далее сокращенно HBTU), и HATU или HBTU являются предпочтительными.
[0087]
Количество конденсирующего агента, используемого в реакции конденсации, предпочтительно составляет от 0,5 до 10 эквивалентов, и более предпочтительно от 1 до 3 эквивалентов, из расчета на производное бифениламина (IV).
[0088]
Примеры основания, используемого в реакции конденсации, включают органическое основание, такое как триэтиламин или диизопропилэтиламин, неорганическое основание, такое как гидрокарбонат натрия или карбонат калия, гидрогенизированное соединение металла, такое как гидрид натрия, гидрид калия или гидрид кальция, алкиллитий, такой как метиллитий или бутиллитий, амид лития, такой как литийгексаметилдисилазид или литийдиизопропиламид или их смесь, и органическое основание, такое как триэтиламин или диизопропилэтиламин, является предпочтительным.
[0089]
Количество основания, используемого в реакции конденсации, предпочтительно составляет от 0,5 до 10 эквивалентов, и более предпочтительно от 1 до 5 эквивалентов, из расчета на производное бифениламина (IV).
[0090]
Производное бифениламина (IV), используемое в реакции конденсации, может быть в свободной форме или форме соли, такой как гидрохлорид.
[0091]
Реакционный растворитель, используемый в реакции конденсации, надлежащим образом выбирают в соответствии с типом используемого реагента или т.п., но особенно не ограничивают до тех пор, пока он не препятствует проведению реакции, и его примеры включают растворители на основе простых эфиров, такие как тетрагидрофуран, 1,4-диоксан, простой диметиловый эфир этиленгликоля или диметоксиэтан, растворители на основе галогенов, такие как дихлорметан, хлороформ или 1,2-дихлорэтан, апротонные полярные растворители, такие как ДМФА или ДМСО, или растворители на основе нитрила, такие как ацетонитрил или пропионитрил, и растворители на основе галогенов, такие как дихлорметан, хлороформ или 1,2-дихлорэтан, или апротонные полярные растворители, такие как ДМФА или ДМСО, являются предпочтительными.
[0092]
Температура реакции конденсации предпочтительно составляет от 0 до 200°C, и более предпочтительно 20 до 100°C.
[0093]
Время реакции конденсации надлежащим образом выбирают в соответствии с реакционными условиями, такими как температура реакции, и предпочтительно оно составляет от 0,5 до 100 часов.
[0094]
Концентрация производного бифениламина (IV), используемого в реакции конденсации, в начале реакции предпочтительно составляет от 1 ммоль/л до 1 моль/л.
[0095]
Производное пипеколиновой кислоты (V), используемое в реакции конденсации, может быть приобретено или получено известными способами или аналогичными им способами.
[0096]
(Третья стадия)
Примеры кислоты, используемой в реакции удаления защиты, включают хлористоводородную кислоту, трифторуксусную кислоту или фтористоводородную кислоту, и хлористоводородная кислота или трифторуксусная кислота являются предпочтительными.
[0097]
Количество кислоты, используемой в реакции удаления защиты, предпочтительно составляет от 0,5 до 100 эквивалентов, и более предпочтительно от 1 до 30 эквивалентов, из расчета на производное амида N-трет-бутоксикарбонилпипеколиновой кислоты (VI).
[0098]
Реакционный растворитель, используемый в реакции удаления защиты, надлежащим образом выбирают в соответствии с типом используемого реагента, но особенно не ограничивают до тех пор, пока он не препятствует проведению реакции, и его примеры включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диметоксиэтан или 1,4-диоксан, растворители на основе сложных эфиров, такие как этилацетат или пропилацетат, растворители на основе хлора, такие как дихлорметан, хлороформ или 1,2-дихлорэтан, растворители на основе спиртов, такие как метанол или этанол, апротонные полярные растворители, такие как ДМФА или ДМСО, или их смешанные растворители, и растворители на основе галогенов, такие как дихлорметан, хлороформ или 1,2-дихлорэтан, или апротонные полярные растворители, такие как ДМФА или ДМСО, являются предпочтительными.
[0099]
Температура реакции удаления защиты предпочтительно составляет от -78°C до 200°C, и более предпочтительно от -20°C до 100°C.
[0100]
Время реакции удаления защиты надлежащим образом выбирают в соответствии с условиями, такими как температура реакции, и время реакции предпочтительно составляет от 1 до 50 часов.
[0101]
Концентрация производного амида N-трет-бутоксикарбонилпипеколиновой кислоты (VI), используемого в реакции удаления защиты, в начале реакции предпочтительно составляет от 1 ммоль/л до 1 моль/л.
[0102]
(Стадия 4)
Количество производного ангидрида органической кислоты (VIII), производного эфира органической кислоты (IX), производного хлорангидрида органической кислоты (X), производного органической кислоты (XI) или триметилсилилизоцианата, используемых в реакции конденсации, предпочтительно составляет от 1 до 200 эквивалентов, и более предпочтительно от 1 до 80 эквивалентов, из расчета на производное амида пипеколиновой кислоты (VII).
[0103]
Примеры конденсирующего агента, используемого в реакции конденсации, включают N,N'-дициклогексилкарбодиимид, гидрохлорид N-этил-N'-3-диметиламинопропилкарбодиимида, N,N'-карбодиимидазол, COMU, HATU или HBTU, и HATU или HBTU являются предпочтительными.
[0104]
Количество конденсирующего агента, используемого в реакции конденсации, предпочтительно составляет от 0 до 10 эквивалентов, и более предпочтительно от 0 до 3 эквивалентов, из расчета на производное амида пипеколиновой кислоты (VII).
[0105]
Примеры основания, используемого в реакции конденсации, включают органическое основание, такое как триэтиламин или диизопропилэтиламин, неорганическое основание, такое как гидрокарбонат натрия или карбонат калия, гидрогенизированное соединение металла, такое как гидрид натрия, гидрид калия или гидрид кальция, алкиллитий, такой как метиллитий или бутиллитий, литийамид, такой как литийгексаметилдисилазид или литийдиизопропиламид, или их смесь, и органическое основание, такое как триэтиламин или диизопропилэтиламин, являются предпочтительными.
[0106]
Количество основания, используемого в реакции конденсации, предпочтительно составляет от 0 до 10 эквивалентов, и более предпочтительно от 0 до 5 эквивалентов, из расчета на производное амида пипеколиновой кислоты (VII).
[0107]
Производное амида пипеколиновой кислоты (VII), используемое в реакции конденсации, может быть в свободной форме или форме соли, такой как гидрохлорид.
[0108]
Реакционный растворитель, используемый в реакции конденсации, надлежащим образом выбирают в соответствии с типом используемого реагента или т.п., но особенно не ограничивают до тех пор, пока он не препятствует проведению реакции, и его примеры включают растворители на основе простых эфиров, такие как тетрагидрофуран, 1,4-диоксан, простой диметиловый эфир этиленгликоля или диметоксиэтан, растворители на основе хлора, такие как дихлорметан, хлороформ или 1,2-дихлорэтан, апротонные полярные растворители, такие как ДМФА или ДМСО, или растворители на основе нитрила, такие как ацетонитрил или пропионитрил, и растворители на основе галогенов, такие как дихлорметан, хлороформ или 1,2-дихлорэтан, или апротонные полярные растворители, такие как ДМФА или ДМСО, являются предпочтительными.
[0109]
Температура реакции конденсации предпочтительно составляет от -78°C до 200°C, и более предпочтительно от -20°C до 100°C.
[0110]
Время реакции конденсации надлежащим образом выбирают в соответствии с условиями, такими как температура реакции, и время реакции предпочтительно составляет от 0,5 до 100 часов.
[0111]
Концентрация производного амида пипеколиновой кислоты (VII), используемого в реакции сочетания, в начале реакции предпочтительно составляет от 1 ммоль/л до 1 моль/л.
[0112]
Производное ангидрида органической кислоты (VIII), производное эфира органической кислоты (IX), производное хлорангидрида органической кислоты (X), производное органической кислоты (XI) и триметилсилилизоцианат, используемые в реакции конденсации, могут быть приобретены или получены известными способами или аналогичными им способами.
[0113]
Лекарственное средство, RORγ антагонист и терапевтическое средство или профилактическое средство против аутоиммунного заболевания или аллергического заболевания по настоящему изобретению отличаются содержанием производного циклического амина (I) или его фармацевтически приемлемой соли в качестве активного ингредиента. Аутоиммунным заболеванием, приведенным выше, предпочтительно является рассеянный склероз или псориаз, и аллергическим заболеванием, приведенным выше, предпочтительно является аллергический дерматит, и более предпочтительно контактный дерматит или атопический дерматит.
[0114]
ʺRORγ антагонистʺ означает соединение, обладающее эффектом подавления функции RORγ, таким образом, устраняя или ослабляя его активность.
[0115]
ʺАутоиммунное заболеваниеʺ является общим термином для заболеваний, при которых у индивидуума обостряются чрезмерные иммунные реакции собственных нормальных клеток и тканей, что приводит к симптомам, примеры которых включают рассеянный склероз, псориаз, ревматоидный артрит, системную красную волчанку, анкилозирующий спондилоартрит, увеит, ревматическую полимиалгию, склеродермию, васкулит, пузырчатку, пемфигоид или дерматомиозит. В дополнение, аутоиммунное заболевание по настоящему изобретению включает акне или витилиго.
[0116]
ʺАллергическое заболеваниеʺ представляет собой заболевание, являющееся результатом чрезмерной иммунной реакцией на конкретные антигены, и его примеры включают аллергический дерматит, контактный дерматит, атопический дерматит, аллергический ринит (поллиноз), аллергический конъюнктивит, аллергический гастроэнтерит, бронхиальную астму, детскую астму или пищевую аллергию.
[0117]
ʺРассеянный склерозʺ представляет собой заболевание, характеризуемое демиелинизацией, при которой миелиновые оболочки, покрывающие нервные волокна, например, головного мозга, спинного мозга и зрительного нерва, разрушаются, и характеризуется прогрессированием расстройства с повторяемыми рецидивами и ремиссией. Симптомы варьируются в зависимости от места поражения и представляют собой различные неврологические симптомы, такие как зрительные нарушения, квадриплегия, сенсорные нарушения и нарушения походки. Примеры рассеянного склероза включают возвратно-ремиттирующий рассеянный склероз, первично-прогрессирующий рассеянный склероз и вторично-прогрессирующий рассеянный склероз.
[0118]
ʺПсориазʺ представляет собой воспалительное заболевание кожи, связанное с инвазией и активацией иммунных клеток, и возникающему в результате акантозу. Как правило, симптом, называемый шелушением, при котором возникает плотное отрубевидное высыпание, сливающееся в красную сыпь в различных частях тела и затем отшелушивающуюся. Примеры псориаза включают бляшковидный псориаз, пустулезный псориаз, артропатический псориаз, каплевидный псориаз и эритродермический псориаз.
[0119]
ʺАллергический дерматитʺ является общим термином для кожных заболеваний, вызванных аллергическими реакциями, и характеризуется хроническим зудом и сыпью на лице, шее, локте и/или колене. Примеры аллергического дерматита включают контактный дерматит, атопический дерматит и т.п.
[0120]
ʺКонтактный дерматитʺ представляет собой экзематозное воспалительное заболевание, развивающееся, когда экзогенный антиген приходит в контакт с кожей, и его примеры включают аллергический контактный дерматит, фотоконтактный дерматит, системный контактный дерматит и контактную крапивницу. Примеры антигена включают металлические аллергены (кобальт, никель и т.д.), растительные аллергены (ядовитый дуб, примула и т.д.) и пищевые аллергены (манго, орехи гинкго и т.д.).
[0121]
ʺАтопический дерматитʺ представляет собой кожное заболевание, возникающее у многих пациентов с атопической предрасположенностью. Оно характеризуется симметричной системной экземой, при которой повторяются обострения и ремиссия. Его примеры включают диффузный нейродермит, атопическую экзему, атопический нейродермит, пруриго Бенье, острую детскую экзему, экзему сгибательных поверхностей, детскую экзему в конечностях, педиатрическую атопическую экзему, детскую сухую экзему, детскую экзему, атопический дерматит у взрослых, эндогенную экзему, детский дерматит и хроническую детскую экзему.
[0122]
Производное циклического амина (I) или его фармацевтически приемлемая соль характеризуются подавлением функции RORγ путем ингибирования связи между RORγ и коактиватором. Поскольку известно, что RORγ вовлечен в различные заболевания, и что можно ожидать проявления улучшения на стадии патологического состояния или ремиссии путем подавления функции RORγ, производное циклического амина (I) или его фармацевтически приемлемая соль могут быть использованы в качестве лекарственного средства против заболеваний, при которых можно ожидать проявления улучшения на стадии патологического состояния или ремиссии путем подавления функции RORγ, в частности, в качестве терапевтического средства или профилактического средства против аутоиммунных заболеваний или аллергических заболеваний. Терапевтическое средство или профилактическое средство против аутоиммунных заболеваний, приведенных выше, могут быть предпочтительно использованы в качестве терапевтического средства или профилактического средства против рассеянного склероза, псориаза, ревматоидного артрита, системной красной волчанки, анкилозирующего спондилоартрита, увеита, ревматической полимиалгии, склеродермии, васкулита, пузырчатки, пемфигоида, дерматомиозита, акне или витилиго, и более предпочтительно использовать в качестве терапевтического средства или профилактического средства против рассеянного склероза или псориаза. Терапевтическое средство или профилактическое средство против аллергических заболеваний, приведенных выше, могут быть предпочтительно использованы в качестве терапевтического средства или профилактического средства против аллергического дерматита, атопического дерматита, аллергического ринита (поллиноз), аллергического конъюнктивита, аллергического гастроэнтерита, бронхиальной астмы, детской астмы или пищевой аллергии, и более предпочтительно использовать в качестве терапевтического средства или профилактического средства против контактного дерматита или атопического дерматита.
[0123]
Это дает возможность квалифицировать производное циклического амина (I) или его фармацевтически приемлемую соль, как обладающие RORγ антагонистической активностью, которая подавляет связывание между RORγ и коактиватором, используя in vitro исследование. Примеры in vitro исследований включают метод оценки связывания между RORγ и агонистом (например, холестерином) (WO 2012/158784, WO 2013/018695) и метод оценки связывания между лиганд-связывающим доменом RORγ и коактиватором (WO 2012/064744, WO 2013/018695). Ингибирующий эффект на активность транскрипции RORγ можно оценить с помощью различных анализов репортерного гена (WO 2012/158784, WO 2012/064744, WO 2013/018695).
[0124]
Тот факт, что производное циклического амина (I) или его фармацевтически приемлемая соль подавляют функцию RORγ, может быть оценен при использовании лимфоцитарных клеток, полученных из различных органов, таких как селезенка, или периферической крови продуцированием IL-17 или дифференциацией Th17 клеток в качестве показателя. Примеры способов, использующих продуцирование IL-17 в качестве показателя, включают способ измерения продуцирования IL-17 путем стимуляции IL-23 с помощью мышиных спленоцитов (The Journal of Biological Chemistry, 2003, Vol. 278, No. 3, p. 1910-1914). Примеры способов, использующих дифференциацию Th17 клеток в качестве показателя, включают способ измерения количества продуцированного IL-17 или пропорции IL-17-позитивных клеток и т.д. путем стимуляции CD4-позитивных интактных T клеток, полученных из мышиных спленоцитов или PBMC человека различными цитокинами (например, IL-1β, IL-6, IL-23, и/или TGF-β) и различными антителами (например, анти-CD3 антителом, анти-CD28 антителом, анти-IL-4 антителом, анти-IFN-γ антителом и/или анти-IL-2 антителом) для дифференциации в Th17 (WO 2012/158784, WO 2013/018695).
[0125]
Тот факт, что производное циклического амина (I) или его фармацевтически приемлемая соль эффективны при лечении или профилактике аутоиммунных заболеваний, может быть оценен при использовании модели заболевания. Примеры модели заболевания включают экспериментальную модель аутоиммунного энцефаломиелита (Journal of Neuroscience Research, 2006, Vol. 84, p. 1225-1234), модель имиквимод-индуцированного псориаза (Journal of Immunology, 2009, Vol. 182, p. 5836-5845), модель коллаген-индуцированного артрита (Annual Review of Immunology, 1984, Vol. 2, p. 199-218), спонтанную модель системной красной волчанки (Nature, 2000, Vol. 404, p. 995-999), модель анкилозирующего спондилита (Arthritis Research & Therapy, 2012, Vol. 14, p. 253-265), экспериментальную модель аутоиммунного увеита (Journal of Immunology, 2006, Vol. 36, p. 3071-3081), модель склеродермии (Journal of Investigative Dermatology, 1999, Vol. 112, p. 456-462), модель васкулита (The Journal of Clinical Investigation, 2002, Vol. 110, p. 955-963), модель пузырчатки (The Journal of Clinical Investigation, 2000, Vol. 105, p. 625-631), модель пемфигоида (Experimental Dermatology, 2012, Vol. 21, p. 901-905), модель дерматомиозита (American Journal of Pathology, 1985, Vol. 120, p. 323-325), спонтанную модель акне (European Journal of Dermatology, 2005, Vol. 15, p. 459-464) или модель витилиго (Pigment Cell & Melanoma Research, 2014, Vol. 27, p. 1075-1085). Экспериментальную модель аутоиммунного энцефаломиелита обычно используют в качестве модели рассеянного склероза. Модель имиквимод-индуцированного псориаза, как правило, используют в качестве модели псориаза.
[0126]
Тот факт, что производное циклического амина (I) или его фармацевтически приемлемая соль эффективны при лечении или профилактике аллергических заболеваний, может быть оценен при использовании модели данных заболеваний. Примеры модели данных заболеваний включают модель динитрофторбензол-индуцированного (здесь и далее сокращенно DNFB) аллергического дерматита (Pharmacological Reports, 2013, Vol. 65, p. 1237-1246), модель оксазолон-индуцированного атопического дерматита (Journal of Investigative Dermatology, 2014, Vol. 134, p. 2122-2130), модель овальбумин-индуцированного аллергического ринита (Journal of Animal Science, 2010, Vol. 81, p. 699-705), модель IgE-индуцированного аллергического конъюнктивита (British Journal of Ophthalmology, 2012, Vol. 96, p. 1332-1336), модель аллергического гастроэнтерита (Gastroenterology, 1997, Vol. 113, p. 1560-1569), модель овальбумин-индуцированной астмы (American Journal of Respiratory and Critical Care Medicine, 1997, Vol. 156, p. 766-775) или модель овальбумин-индуцированной пищевой аллергии (Clinical & Experimental Allergy, 2005, Vol. 35, p. 461-466). Модель DNFB-индуцированного аллергического дерматита обычно используют в качестве модели аллергического дерматита, особенно в качестве модели контактного дерматита. Модель оксазолон-индуцированного атопического дерматита обычно используют в качестве модели атипического дерматита.
[0127]
Эффективность производного циклического амина (I) или его фармацевтически приемлемой соли при лечении или профилактике аутоиммунных заболеваний или аллергических заболеваний может быть оценена при использовании показателей в in vitro исследованиях, приведенных выше, например, по уменьшению количества связываний между лиганд-связывающим доменом RORγ и коактиватором, или по уменьшению количества продуцирования IL-17, которое является показателем функции RORγ. Эффективность лечения или профилактики рассеянного склероза может быть оценена при использовании показателей на экспериментальной модели аутоиммунного энцефаломиелита, приведенной выше, например, по уменьшению при оценке неврологической симптоматики, которая является характеристическим показателем рассеянного склероза. Эффективность лечения или профилактики псориаза может быть оценена при использовании показателей на модели имиквимод-индуцированного псориаза, приведенной выше, например, по уменьшению толщины кожи, например, на модели ушной раковины, которая увеличивается с прогрессированием симптомов псориаза. Эффективность лечения или профилактики аллергического дерматита, в частности контактного дерматита, может быть оценена при использовании показателей на модели DNFB-индуцированного аллергического дерматита, приведенной выше, например, по уменьшению толщины кожи, на модели ушной раковины, которая увеличивается с прогрессированием симптомов дерматита. Эффективность лечения или профилактике атопического дерматита может быть оценена при использовании показателей на модели оксазолон-индуцированного атопического дерматита, приведенной выше, например, по уменьшению толщины кожи, на модели ушной раковины, которая увеличивается с прогрессированием симптомов дерматита, в качестве показателя.
[0128]
Производное циклического амина (I) или его фармацевтически приемлемая соль могут быть использованы в качестве полезного лекарственного средства (в частности, терапевтического средства или профилактического средства против аутоиммунного заболевания или аллергического заболевания) при введении млекопитающему (например, мыши, крысе, хомяку, кролику, собаке, обезьяне, корове, овце или человеку), в частности, человеку. Когда производное циклического амина (I) или его фармацевтически приемлемую соль используют клинически в качестве лекарственного средства, производное циклического амина (I) или его фармацевтически приемлемая соль могут быть использованы как таковые или могут быть надлежащим образом смешаны с добавками, такими как эксципиенты, стабилизаторы, консерванты, буферирующие агенты, солюбилизаторы, эмульгаторы, разбавители или придающие изотоничность агенты. Лекарственное средство, приведенное выше, может быть изготовлено обычными способами при использовании соответствующих носителей для лекарственных средств. Примеры дозированных форм лекарственных средств, приведенных выше, включают пероральные препараты, такие как таблетки, капсулы, гранулы, порошки или сиропы; парентеральные препараты, такие как ингаляции, инъекции, суппозитории или жидкости; или мази, кремы или пластыри для местного введения. Кроме того, могут быть использованы известные препараты длительного действия.
[0129]
Лекарственное средство, приведенное выше, предпочтительно содержит от 0,00001 до 90% по массе, и более предпочтительно от 0,01 до 70% по массе производного циклического амина (I) или его фармацевтически приемлемой соли. Дозу надлежащим образом выбирают в соответствии с симптомами, возрастом и массой тела пациента, и способом введения. В качестве количества активного ингредиента для взрослого, предпочтительным является 0,1 мкг до 1 г в день для инъекций, 1 мкг до 10 г в день для пероральных препаратов, и 1 мкг до 10 г в день для пластырей, каждое из которых может вводиться один раз в единой или несколько раз в разделенных дозах.
[0130]
Примеры фармакологически приемлемого носителя или разбавителя приведенного выше лекарственного средства включают связующее (сироп, желатин, аравийскую камедь, сорбитол, поливинилхлорид, трагакант или т.п.), эксципиент (сахар, лактоза, кукурузный крахмал, фосфат кальция, сорбитол, глицин или т.п.) или лубрикант (стеарат магния, полиэтиленгликоль, тальк, диоксид кремния или т.п.).
[0131]
Для дополнения или усиления терапевтического или профилактического эффекта или уменьшения дозы, лекарственное средство может использоваться в смеси или комбинации с другими лекарственными средствами в подходящем количестве.
[0132]
Настоящее изобретение будет более подробно раскрыто при помощи следующих ссылочных примеров и примеров, но настоящее изобретение ими не ограничивается.
Примеры
[0133]
В качестве соединений, использованных в синтезе соединений ссылочных примеров и примеров, использовали коммерчески доступные соединения, способ синтеза которых не приводится. ʺКомнатная температураʺ в следующих ссылочных примерах и примерах обычно означает температуру окружающей среды, составляющую примерно от 10°C до примерно 35°C. Процент (%) указан в % моль/моль для выхода, % по объему для растворителей, использованных в колоночной хроматографии и высокоэффективной жидкостной хроматографии, и % по массе для других применений, если специально не указано иное. Название растворителя, показанного в данных ЯМР, обозначает растворитель, используемый при измерении. 400 МГц ЯМР спектр измеряли при использовании JNM-AL 400 спектрометра ядерно-магнитного резонанса (JEOL Ltd.) и JNM-ECS 400 спектрометра ядерно-магнитного резонанса (JEOL Ltd.). Химический сдвиг указан как δ (единица: м.д.) с тетраметилсиланом в качестве стандарта, сигналы обозначены как с (синглет), д (дублет), т (триплет), кв (квартет), квин. (квинтет), септ. (септет), м (мультиплет), шир. (широкий), дд (дублет дублета), дт (дублет триплета), ддд (дублет дублета дублета), дкв (дублет квартета), тд (триплет дублета) и тт (триплет триплета). Когда протоны гидроксильной группы, аминогруппы или т.п. имеют очень слабый пик, они не указываются. ESI-МС спектры измеряли при использовании Agilent Technologies 1200 Series, G6130A (Agilent Technologies). В качестве силикагеля использовали силикагель 60 (Merck), в качестве амино-силикагеля использовали амино-силикагель DM 1020 (Fuji Silysia Chemical Ltd.), и в качестве хроматографии использовали YFLC W-prep2XY (Yamazen Corporation).
[0134]
(Ссылочный пример 1) Синтез 2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-амина:
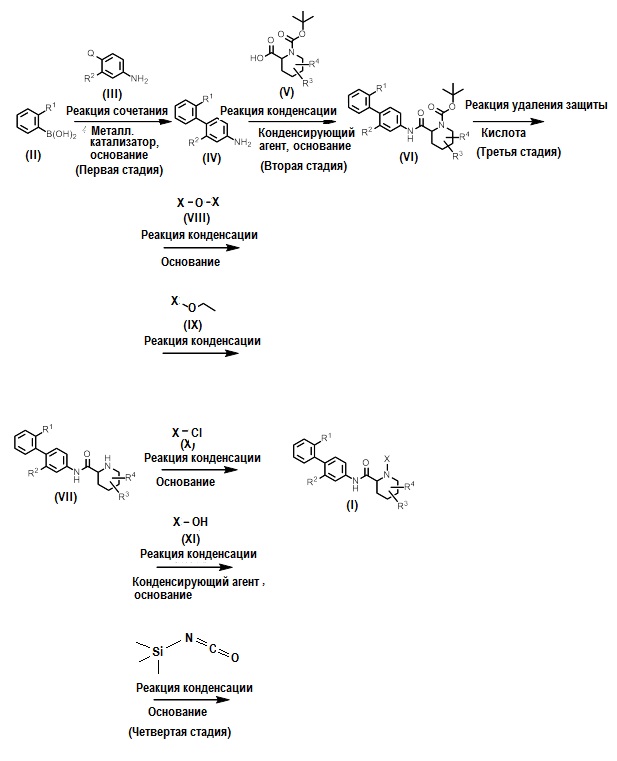
К ацетонитрильному раствору (9,0 мл) 2-трифторметоксифенилбороновой кислоты (1,10 г, 5,33 ммоль) при комнатной температуре добавляли 4-бром-3-хлоранилин (1,00 г, 4,84 ммоль), карбонат калия (1,00 г, 7,27 ммоль), дихлорметановый аддукт 1,1'-бис(дифенилфосфино)ферроцендихлорпалладия(II) (0,396 г, 0,484 ммоль) и дистиллированную воду (3,0 мл), и температуру поднимали до 90°C, с последующим перемешиванием в течение 18 часов. Полученный реакционный раствор фильтровали через силикагель, и затем фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, н-гексан/этилацетат=85/15 до 67/33), с получением 2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-амина (здесь и далее называемого соединением ссылочного примера 1) (1,03 г, 3,57 ммоль, 73,6%) в виде желтого маслянистого продукта.
1H-ЯМР (400 МГц, CDCl3) δ: 3,79 (с, 2H), 6,62 (дд, J=8,3, 2,3 Гц, 1H), 6,80 (д, J=2,3 Гц, 1H), 7,05 (д, J=8,3 Гц, 1H), 7,30-7,41 (м, 4H).
ESI-МС:m/z=288(M+H)+.
[0135]
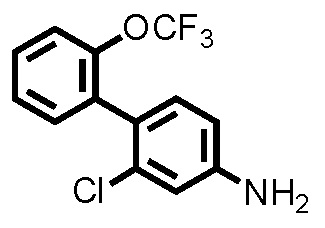
(Ссылочный пример 2) Синтез трет-бутил 2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)пиперидин-1-карбоксилата:
К ДМФА раствору (2,0 мл) 1-(трет-бутоксикарбонил)пиперидин-2-карбоновой кислоты (0,263 г, 1,15 ммоль) при комнатной температуре добавляли ДМФА раствор (2,0 мл) соединения ссылочного примера 1 (0,300 г, 1,04 ммоль), HATU (0,436 г, 1,15 ммоль) и диизопропилэтиламин (0,273 мл, 1,56 ммоль), с последующим перемешиванием при этой же температуре в течение 16 часов. К полученному реакционному раствору добавляли дистиллированную воду, и полученный раствор экстрагировали смешанным растворителем н-гексан/этилацетат=20/80(об./об.). Органический слой промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, н-гексан/этилацетат=90/10 до 67/33), с получением трет-бутил 2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)пиперидин-1-карбоксилата (здесь и далее называемого соединением ссылочного примера 2) (0,483 г, 0,968 ммоль, 92,8%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,43-1,51 (м, 2H), 1,53 (с, 9H), 1,60-1,75 (м, 3H), 2,35 (д, J=12,7 Гц, 1H), 2,80-2,89 (м, 1H), 4,03-4,13 (м, 1H), 4,86-4,89 (м, 1H), 7,22 (д, J=8,3 Гц, 1H), 7,29-7,45 (м, 6H), 7,80 (шир., 1H).
ESI-МС:m/z=499(M+H)+.
[0136]
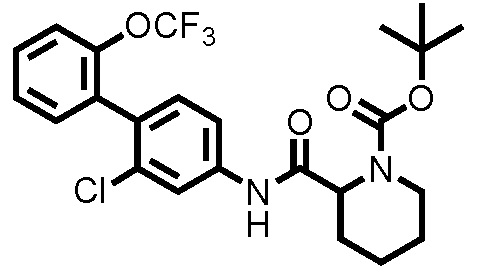
(Ссылочный пример 3) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида:
К дихлорметановому раствору (5,0 мл) соединения ссылочного примера 2 (0,483 г, 0,968 ммоль) при комнатной температуре добавляли трифторуксусную кислоту (0,522 мл, 6,78 ммоль), с последующим перемешиванием при этой же температуре в течение 20 часов. Полученный реакционный раствор концентрировали при пониженном давлении, нейтрализовали водным раствором карбоната калия и затем экстрагировали хлороформом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (амино-силикагель, н-гексан/этилацетат=60/40 до 20/80), с получением N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида (здесь и далее называемого соединением ссылочного примера 3) (0,309 г, 0,775 ммоль, 80,0%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,53(ддд, J=36,8, 17,9, 8,8 Гц, 4H), 1,78-1,86 (м, 1H), 2,00-2,07 (м, 1H), 2,74-2,82 (м, 1H), 3,03-3,10 (м, 1H), 3,38 (дд, J=9,6, 3,5 Гц, 1H), 7,23 (д, J=8,3 Гц, 1H), 7,31-7,37 (м, 3H), 7,40-7,45 (м, 1H), 7,53 (дд, J=8,3, 2,0 Гц, 1H), 7,82 (д, J=2,2 Гц, 1H), 9,02 (шир., 1H).
ESI-МС:m/z=399(M+H)+.
[0137]
(Пример 1) Синтез 1-ацетил-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида:
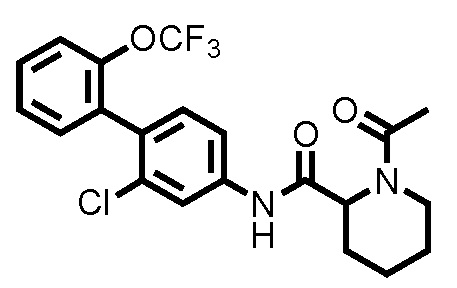
К дихлорметановому раствору (3,0 мл) соединения ссылочного примера 3 (0,0700 г, 0,176 ммоль) при 0°C добавляли триэтиламин (0,0367 мл, 0,263 ммоль) и уксусный ангидрид (0,0182 мл, 0,193 ммоль), и температуру поднимали до комнатной температуры, с последующим перемешиванием в течение 1 часа. К полученному реакционному раствору добавляли дистиллированную воду, и полученный раствор экстрагировали хлороформом. Органический слой промывали дистиллированной водой и насыщенным раствором соли, сушили над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, хлороформ/метанол=95/5), с получением 1-ацетил-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида (здесь и далее называемого соединением примера 1) (0,0730 г, 0,166 ммоль, 94,3%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,46-1,63 (м, 1H), 1,67 (д, J=7,8 Гц, 1H), 1,89-2,02 (м, 2H), 2,22 (с, 3H), 2,29 (д, J=12,9 Гц, 1H), 3,22 (т, J=13,2 Гц, 1H), 3,78 (д, J=12,7 Гц, 1H), 5,29 (д, J=5,1 Гц, 1H), 7,20 (д, J=8,3 Гц, 1H), 7,29-7,37 (м, 3H), 7,40-7,44 (м, 2H), 7,80 (шир., 1H), 8,65 (шир., 1H).
ESI-МС:m/z=441(M+H)+.
[0138]
(Пример 2) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2,2,2-трифторацетил)пиперидин-2-карбоксамида:
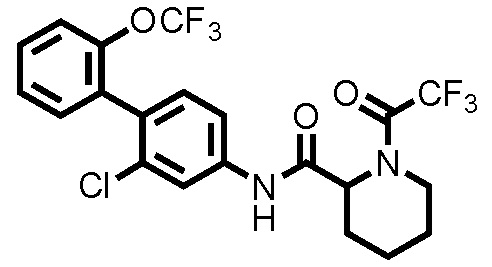
Следуя методике, аналогичной примеру 1, за исключением того, что трифторуксусный ангидрид использовали вместо уксусного ангидрида, N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2,2,2-трифторацетил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 2) (0,0500 г, 0,101 ммоль, 99,0%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,56-1,86 (м, 4H), 1,98 (дт, J=11,2, 4,6 Гц, 1H), 2,36 (д, J=14,1 Гц, 1H), 3,37 (тд, J=13,4, 2,6 Гц, 1H), 4,01 (д, J=13,9 Гц, 1H), 5,18 (д, J=5,4 Гц, 1H), 7,21 (д, J=8,3 Гц, 1H), 7,30-7,46 (м, 5H), 7,79 (шир., 1H), 7,89 (шир., 1H).
ESI-МС:m/z=495(M+H)+.
[0139]
(Пример 3) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-пропионилпиперидин-2-карбоксамида:
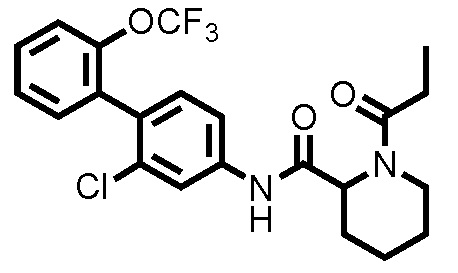
К дихлорметановому раствору (2,0 мл) соединения ссылочного примера 3 (0,0300 г, 0,0752 ммоль) при 0°C добавляли триэтиламин (0,0157 мл, 0,113 ммоль) и пропионилхлорид (0,00719 мл, 0,0828 ммоль), и температуру поднимали до комнатной температуры, с последующим перемешиванием в течение 30 минут. К полученному реакционному раствору добавляли метанол, и полученный раствор концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, хлороформ/метанол=100/0 до 90/10), с получением N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-пропионилпиперидин-2-карбоксамида (здесь и далее называемого соединением примера 3) (0,0340 г, 0,0747 ммоль, 99,4%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,22 (т, J=7,3 Гц, 3H), 1,55 (шир., 2H), 1,76 (шир., 2H), 1,97 (т, J=13,2 Гц, 1H), 2,30 (д, J=12,7 Гц, 1H), 2,48 (дкв, J=6,6, 2,0 Гц, 2H), 3,12 (тд, J=13,2, 2,8 Гц, 1H), 3,83 (д, J=13,2 Гц, 1H), 5,29 (д, J=5,4 Гц, 1H), 7,20 (д, J=8,3 Гц, 1H), 7,29-7,36 (м, 4H), 7,39-7,45 (м, 1H), 7,84 (шир., 1H), 8,56 (шир., 1H).
ESI-МС:m/z=455(M+H)+.
[0140]
(Пример 4) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-метоксиацетил)пиперидин-2-карбоксамида:
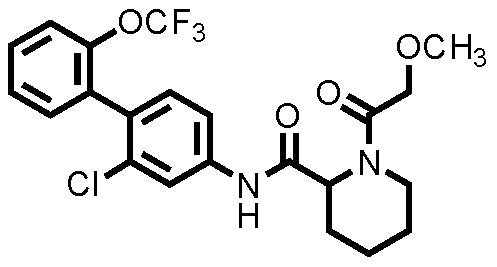
К ДМФА раствору (0,5 мл) 2-метоксиуксусной кислоты (0,00693 мл, 0,0903 ммоль) при комнатной температуре добавляли ДМФА раствор (0,5 мл) соединения ссылочного примера 3 (0,0300 г, 0,0752 ммоль), HATU (0,0343 г, 0,0902 ммоль) и диизопропилэтиламин (0,0197 мл, 0,113 ммоль), с последующим перемешиванием при этой же температуре в течение 3 часов. К полученному реакционному раствору добавляли дистиллированную воду, и полученный раствор экстрагировали смешанным растворителем н-гексан/этилацетат=20/80. Органический слой промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, н-гексан/этилацетат=50/50 до 0/100), с получением N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-метоксиацетил)пиперидин-2-карбоксамида (здесь и далее называемого соединением примера 4) (0,0266 г, 0,0565 ммоль, 74,6%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,58-2,00 (м, 5H), 2,33 (д, J=14,4 Гц, 0,8H), 2,48 (д, J=12,7 Гц, 0,2H), 2,63 (т, J=12,7 Гц, 0,2H), 3,14 (т, J=13,0 Гц, 0,8H), 3,48 (с, 2,4H), 3,51 (с, 0,6H), 3,82 (д, J=12,7 Гц, 0,8H), 4,12 (д, J=11,7 Гц, 0,2H), 4,18 (д, J=13,9 Гц, 0,8H), 4,26 (д, J=13,9 Гц, 0,8H), 4,34 (д, J=11,7 Гц, 0,2H), 4,52-4,60 (м, 0,2H), 4,64-4,68 (м, 0,2H), 5,23 (д, J=6,1 Гц, 0,8H), 7,20 (д, J=8,3 Гц, 1H), 7,28-7,45 (м, 5H), 7,65-7,90 (м, 1H), 8,46 (шир., 0,8H), 8,57 (шир., 0,2H).
ESI-МС:m/z=471(M+H)+.
[0141]
(Пример 5) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-гидроксиацетил)пиперидин-2-карбоксамида:
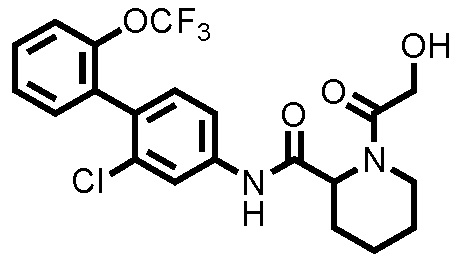
Следуя методике, аналогичной примеру 4, за исключением того, что гликолевую кислоту использовали вместо 2-метоксиуксусной кислоты, N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-гидроксиацетил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 5) (0,0114 г, 0,0250 ммоль, 33,2%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,48-1,55 (м, 1H), 1,63-1,71 (м, 1H), 1,75-1,85 (м, 2H), 1,91-2,02 (м, 1H), 2,32 (д, J=13,4 Гц, 1H), 3,17-3,25 (м, 1H), 3,43-3,53 (м, 2H), 4,28-4,32 (м, 2H), 5,26 (д, J=5,6 Гц, 1H), 7,22 (д, J=8,3 Гц, 1H), 7,30-7,45 (м, 5H), 7,77 (шир., 1H), 8,14 (шир., 1H).
ESI-МС:m/z=457(M+H)+.
[0142]
(Пример 6) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-(диметиламино)ацетил)пиперидин-2-карбоксамида:
Следуя методике, аналогичной примеру 4, за исключением того, что гидрохлорид N,N-диметилглицина использовали вместо 2-метоксиуксусной кислоты, N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-(диметиламино)ацетил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 6) (0,0273 г, 0,0564 ммоль, 90,0%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,40-2,20 (м, 6H), 2,34 (с, 3H), 2,47-2,50 (м, 3,4H), 2,56-2,64 (м, 0,4H), 2,92 (д, J=12,6 Гц, 0,6H), 3,06-3,14 (м, 0,4H), 3,24 (с, 0,6H), 3,67 (д, J=12,6 Гц, 0,6H), 4,03-4,07 (м, 0,4H), 4,54-4,62 (м, 1,2H), 5,24-5,27 (м, 0,4H), 7,19-7,23 (м, 1H), 7,30-7,46 (м, 5H), 7,73-7,75 (м, 1H), 8,53 (шир., 0,4H), 10,69 (шир., 0,6H).
ESI-МС:m/z=484(M+H)+.
[0143]
(Пример 7) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2,2-дифторацетил)пиперидин-2-карбоксамида:
Следуя методике, аналогичной примеру 4, за исключением того, что дифторуксусную кислоту использовали вместо 2-метоксиуксусной кислоты, N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2,2-дифторацетил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 7) (0,0212 г, 0,0444 ммоль, 59,1%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, ДМСО-D6) δ: 1,40-1,80 (м, 5,0H), 2,21-2,24 (м, 0,7H), 2,29-2,34 (м, 0,3H), 2,65-2,68 (м, 0,3H), 3,46-3,55 (м, 0,7H), 3,82-3,88 (м, 0,7H), 4,28-4,34 (м, 0,3H), 4,79-4,81 (м, 0,3H), 5,07-5,10 (м, 0,7H), 6,73 (т, J=52,6 Гц, 0,3H), 6,83 (т, J=52,7 Гц, 0,7H), 7,32 (д, J=8,3 Гц, 1H), 7,39-7,43 (м, 1H), 7,45-7,51 (м, 2H), 7,53-7,60 (м, 2H), 7,90-7,93 (м, 1H), 10,19 (шир., 0,3H), 10,25 (шир., 0,7H).
ESI-МС:m/z=477(M+H)+.
[0144]
(Пример 8) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-(трифторметокси)ацетил)пиперидин-2-карбоксамида:
Следуя методике, аналогичной примеру 4, за исключением того, что 2-трифторметоксиуксусную кислоту использовали вместо 2-метоксиуксусной кислоты, N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-(трифторметокси)ацетил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 8) (0,00890 г, 0,0170 ммоль, 16,9%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,55-1,71 (м, 2H), 1,76-1,84 (м, 2H), 1,94-2,03 (м, 1H), 2,32 (д, J=14,5 Гц, 1H), 3,29 (тд, J=13,1,2,7 Гц, 1H), 3,67 (д, J=12,7 Гц, 1H), 4,68-4,76 (м, 2H), 5,22 (д, J=5,4 Гц, 1H), 7,21-7,45 (м, 6H), 7,81 (шир., 1H), 8,26 (с, 1H).
ESI-МС:m/z=523(M-H)-.
[0145]
(Ссылочный пример 4) Синтез трет-бутил(2-(2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)пиперидин-1-ил)-2-оксоэтил)карбамата:
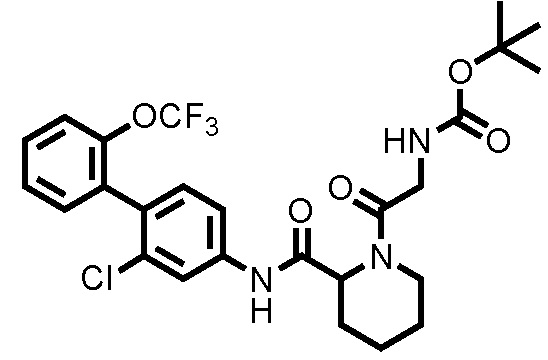
Следуя методике, аналогичной примеру 4, за исключением того, что 2-((трет-бутоксикарбонил)амино)уксусную кислоту использовали вместо 2-метоксиуксусной кислоты, трет-бутил(2-(2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)пиперидин-1-ил)-2-оксоэтил)карбамат (здесь и далее называемый соединением ссылочного примера 4) (0,116 г, 0,208 ммоль, количественный) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,46 (с, 9H), 1,45-1,90 (м, 5H), 2,38 (д, J=13,2 Гц, 1H), 3,21 (т, J=12,1 Гц, 1H), 3,75 (д, J=13,9 Гц, 1H), 3,95 (дд, J=16,7, 5,0 Гц, 1H), 4,09-4,15 (м, 1H), 5,32 (д, J=4,9 Гц, 1H), 5,42 (шир., 1H), 7,21 (д, J=8,3 Гц, 1H), 7,29-7,36 (м, 3H), 7,40-7,45 (м, 1H), 7,52 (шир., 1H), 7,80 (шир., 1H), 8,31 (шир., 1H).
ESI-МС:m/z=556(M+H)+.
[0146]
(Ссылочный пример 5) Синтез 1-(2-аминоацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида:
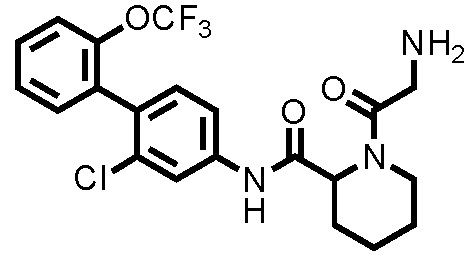
К дихлорметановому раствору (1,0 мл) соединения ссылочного примера 4 (0,115 г, 0,207 ммоль) при комнатной температуре добавляли трифторуксусную кислоту (0,112 мл, 1,45 ммоль), с последующим перемешиванием при этой же температуре в течение 15 часов. Полученный реакционный раствор концентрировали при пониженном давлении, нейтрализовали водным раствором карбоната калия и затем экстрагировали хлороформом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (амино-силикагель, хлороформ/метанол=100/0 до 96/4), с получением 1-(2-аминоацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида (здесь и далее называемого соединением ссылочного примера 5) (0,0613 г, 0,134 ммоль, 65,0%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,45-1,63 (м, 2H), 1,70-1,81 (м, 2H), 1,89-1,99 (м, 1H), 2,31 (д, J=14,0 Гц, 1H), 3,16 (тд, J=14,0,2,3 Гц, 1H), 3,60 (д, J=1,0 Гц, 2H), 3,68 (д, J=14,0 Гц, 1H), 5,27 (д, J=5,4 Гц, 1H), 7,21 (д, J=8,3 Гц, 1H), 7,30-7,45 (м, 5H), 7,77 (шир., 1H), 8,39 (шир., 1H).
ESI-МС:m/z=456(M+H)+.
[0147]
(Ссылочный пример 6) Синтез трет-бутил(2-(2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)пиперидин-1-ил)-2-оксоэтил)(метил)карбамата:
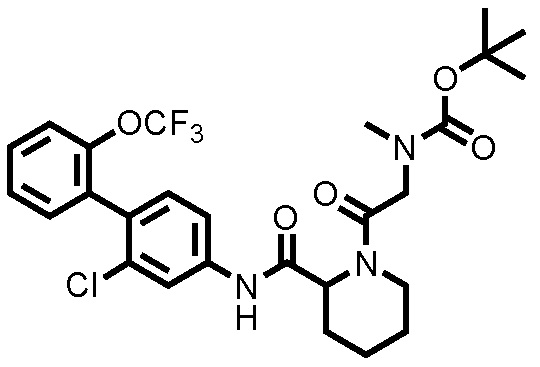
Следуя методике, аналогичной примеру 4, за исключением того, что N-(трет-бутоксикарбонил)-N-метилглицин использовали вместо 2-метоксиуксусной кислоты, трет-бутил(2-(2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)пиперидин-1-ил)-2-оксоэтил)(метил)карбамат (здесь и далее называемый соединением ссылочного примера 6) (0,132 г, 0,232 ммоль, 92,6%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,57 (с, 9H), 1,38-1,80 (м, 5H), 2,46-2,53 (м, 1H), 3,05 (с, 3H), 3,15-3,22 (м, 1H), 3,66 (д, J=15,7 Гц, 1H), 3,77-3,84 (м, 1H), 4,41 (д, J=15,7 Гц, 1H), 5,41-5,44 (м, 1H), 7,19 (д, J=8,3 Гц, 1H), 7,28-7,44 (м, 4H), 7,70 (шир., 1H), 7,88 (шир., 1H), 8,61 (шир., 1H).
ESI-МС:m/z=571(M+H)+.
[0148]
(Пример 9) Синтез 1-(2-ацетамидацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида:
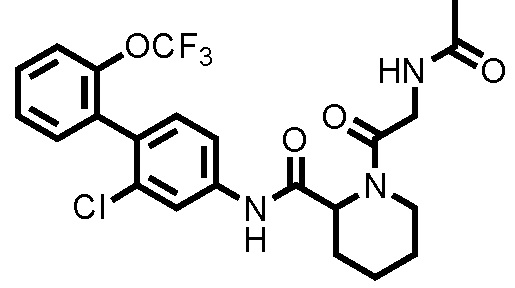
Следуя методике, аналогичной примеру 3, за исключением того, что соединение ссылочного примера 5 использовали вместо соединения ссылочного примера 3, и ацетилхлорид использовали вместо пропионилхлорида, 1-(2-ацетамидацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 9) (0,0274 г, 0,0550 ммоль, 80,9%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,50-1,90 (м, 5H), 2,09 (с, 3H), 2,37 (д, J=14,4 Гц, 1H), 3,25 (тд, J=13,0,2,4 Гц, 1H), 3,75 (д, J=12,4 Гц, 1H), 4,11 (дд, J=17,2, 4,0 Гц, 1H), 4,21 (дд, J=17,2, 4,0 Гц, 1H), 5,29 (д, J=5,1 Гц, 1H), 6,53 (шир., 1H), 7,21 (д, J=8,5 Гц, 1H), 7,29-7,37 (м, 3H), 7,40-7,48 (м, 2H), 7,80 (шир., 1H), 8,26 (шир., 1H).
ESI-МС:m/z=498(M+H)+.
[0149]
(Пример 10) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-(метилсульфонамид)ацетил)пиперидин-2-карбоксамида:
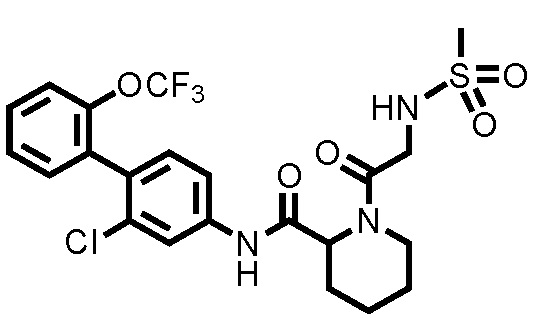
Следуя методике, аналогичной примеру 3, за исключением того, что соединение ссылочного примера 5 использовали вместо соединения ссылочного примера 3, и метансульфонилхлорид использовали вместо пропионилхлорида, N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-(метилсульфонамид)ацетил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 10) (0,0202 г, 0,0378 ммоль, 79,2%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,52-1,95 (м, 5H), 2,32 (д, J=14,1 Гц, 1H), 3,02 (с, 3H), 3,33 (т, J=12,8 Гц, 1H), 3,64 (д, J=13,0 Гц, 1H), 4,08 (д, J=4,6 Гц, 2H), 5,25 (д, J=4,6 Гц, 1H), 5,48 (шир., 1H), 7,22 (д, J=8,3 Гц, 1H), 7,29-7,45 (м, 5H), 7,81 (шир., 1H), 8,09 (шир., 1H).
ESI-МС:m/z=534(M+H)+.
[0150]
(Пример 11) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-(метиламино)ацетил)пиперидин-2-карбоксамида:
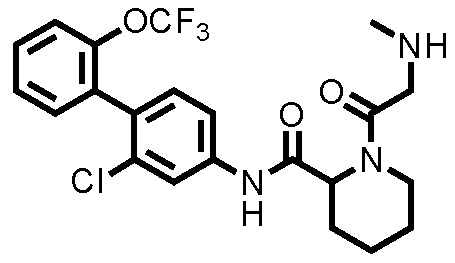
Следуя методике, аналогичной ссылочному примеру 5, за исключением того, что соединение ссылочного примера 6 использовали вместо соединения ссылочного примера 4, N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-(метиламино)ацетил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 11) (0,0799 г, 0,170 ммоль, 73,4%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,26-1,76 (м, 5H), 1,90-2,06 (м, 1H), 2,28-2,42 (м, 1H), 2,50 (с, 2,4H), 2,60-2,63 (м, 0,8H), 3,15 (т, J=12,2 Гц, 0,8H), 3,43 (д, J=12,7 Гц, 0,2H), 3,52 (с, 1,6H), 3,70-3,76 (м, 1H), 4,58-4,63 (м, 0,4H), 5,28 (д, J=4,9 Гц, 0,8H), 7,19-7,23 (м, 1H), 7,30-7,45 (м, 5H), 7,75-7,77 (м, 1H), 8,44 (с, 0,8H), 10,49 (с, 0,2H).
ESI-МС:m/z=470(M+H)+.
[0151]
(Пример 12) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-(N-метилацетамид)ацетил)пиперидин-2-карбоксамида:
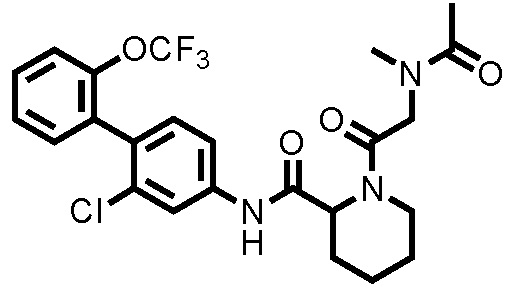
Следуя методике, аналогичной примеру 3, за исключением того, что соединение примера 11 использовали вместо соединения ссылочного примера 3, и ацетилхлорид использовали вместо пропионилхлорида, N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-(N-метилацетамид)ацетил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 12) (0,0261 г, 0,0510 ммоль, 95,8%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,48-1,76 (м, 5H), 2,20 (с, 3H), 2,50-2,65 (м, 1H), 3,24 (с, 2,1H), 3,30 (с, 0,9H), 3,20-3,31 (м, 1H), 3,34 (д, J=15,0 Гц, 0,3H), 3,63 (д, J=15,0 Гц, 0,7H), 3,83-3,89 (м, 0,7H), 4,60 (д, J=15,0 Гц, 0,7H), 4,64-4,70 (м, 0,6H), 4,78 (д, J=15,0 Гц, 0,3H), 5,41 (д, J=4,5 Гц, 0,7H), 7,20-7,22 (м, 1H), 7,30-7,35 (м, 3H), 7,39-7,44 (м, 1H), 7,60-7,75 (м, 0,7H), 7,77 (дд, J=8,4, 2,0 Гц, 0,3H), 7,98-8,07 (м, 0,7H), 8,14 (д, J=1,8 Гц, 0,3H), 8,64 (шир., 0,7H), 9,63 (шир., 0,3H).
ESI-МС:m/z=512(M+H)+.
[0152]
(Пример 13) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-(N-метилметилсульфонамид)ацетил)пиперидин-2-карбоксамида:
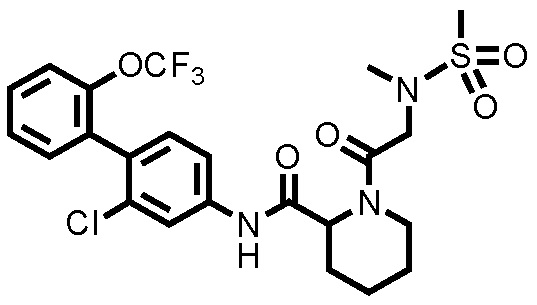
Следуя методике, аналогичной примеру 3, за исключением того, что соединение примера 11 использовали вместо соединения ссылочного примера 3, и метансульфонилхлорид использовали вместо пропионилхлорида, N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-(N-метилметилсульфонамид)ацетил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 13) (0,0264 г, 0,0482 ммоль, 90,6%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,52-1,89 (м, 5H), 2,35-2,38 (м, 1H), 3,03-3,07 (м, 6H), 3,20-3,31 (м, 1H), 3,67-3,76 (м, 1H), 4,16-4,27 (м, 2H), 5,25-5,26 (м, 1H), 7,21-7,23 (м, 1H), 7,30-7,45 (м, 5H), 7,83 (с, 1H), 8,22 (шир., 1H).
ESI-МС:m/z=548(M+H)+.
[0153]
(Пример 14) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(этилсульфонил)пиперидин-2-карбоксамида:
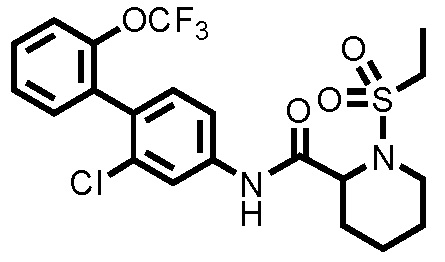
Следуя методике, аналогичной примеру 3, за исключением того, что этансульфонилхлорид использовали вместо пропионилхлорида, N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(этилсульфонил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 14) (0,0660 г, 0,134 ммоль, 99,3%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,47 (т, J=7,4 Гц, 3H), 1,58-1,68 (м, 2H), 1,69-1,84 (м, 3H), 2,59 (д, J=12,4 Гц, 1H), 3,06-3,21 (м, 4H), 3,88 (д, J=12,0 Гц, 1H), 4,56 (д, J=8,3 Гц, 1H), 7,24 (д, J=8,3 Гц, 1H), 7,31-7,38 (м, 3H), 7,40-7,50 (м, 2H), 7,85 (с, 1H), 8,53 (шир., 1H).
ESI-МС:m/z=491(M+H)+.
[0154]
(Пример 15) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(метилсульфонил)пиперидин-2-карбоксамида:
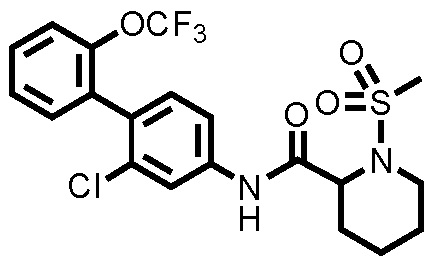
Следуя методике, аналогичной примеру 3, за исключением того, что метансульфонилхлорид использовали вместо пропионилхлорида, N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(метилсульфонил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 15) (0,0800 г, 0,168 ммоль, 66,9%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,47-1,61 (м, 1H), 1,62-1,81 (м, 4H), 2,45 (д, J=10,4 Гц, 1H), 3,04 (с, 3H), 3,23 (тд, J=13,3, 2,4 Гц, 1H), 3,93 (т, J=7,0 Гц, 1H), 4,64 (шир., 1H), 7,25 (д, J=8,5 Гц, 1H), 7,30-7,38 (м, 3H), 7,43 (дт, J=10,8, 3,7 Гц, 2H), 7,84 (д, J=2,2 Гц, 1H), 8,29 (шир., 1H).
ESI-МС:m/z=477(M+H)+.
[0155]
(Пример 16) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-формилпиперидин-2-карбоксамида:
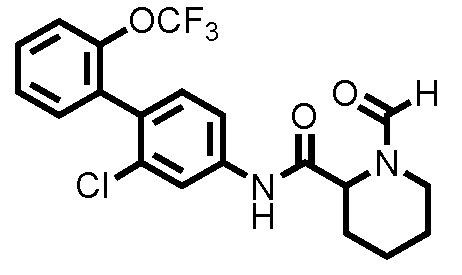
К дихлорметановому раствору (1,0 мл) соединения ссылочного примера 3 (0,0400 г, 0,100 ммоль) при 0°C добавляли этилформиат (0,567 мл, 7,02 ммоль), и температуру поднимали до 90°C, с последующим перемешиванием в течение 18 часов. Полученный реакционный раствор концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией (силикагель, хлороформ/метанол=100/0 до 90/10), с получением N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-формилпиперидин-2-карбоксамида (здесь и далее называемого соединением примера 16) (0,0300 г, 0,0703 ммоль, 70,1%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,42-1,64 (м, 2H), 1,82 (д, J=10,0 Гц, 2H), 1,95 (дт, J=8,8, 4,3 Гц, 1H), 2,35 (д, J=13,9 Гц, 1H), 3,29 (тд, J=13,2, 2,8 Гц, 1H), 3,63 (д, J=9,5 Гц, 1H), 5,12 (д, J=5,6 Гц, 1H), 7,21 (д, J=8,3 Гц, 1H), 7,28-7,45 (м, 5H), 7,80 (шир., 1H), 8,21 (д, J=9,2 Гц, 1H), 8,32 (шир., 1H).
ESI-МС:m/z=427(M+H)+.
[0156]
(Пример 17) Синтез N2-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-1,2-дикарбоксамида:
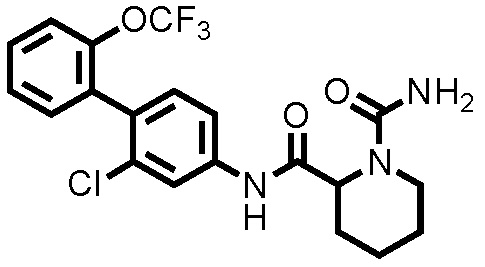
К дихлорметановому раствору (3,0 мл) соединения ссылочного примера 3 (0,100 г, 0,251 ммоль) при 0°C добавляли триметилсилилизоцианат (0,0333 мл, 0,251 ммоль) и триэтиламин (0,0349 мл, 0,251 ммоль), и температуру поднимали до комнатной температуры, с последующим перемешиванием в течение 72 часов. К полученному реакционному раствору добавляли метанол, и полученный раствор концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, хлороформ), с получением N2-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-1,2-дикарбоксамида (здесь и далее называемого соединением примера 17) (0,0300 г, 0,0679 ммоль, 27,1%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,50-1,68 (м, 2H), 1,73 (шир., 2H), 1,81-1,92 (м, 1H), 2,30 (д, J=12,9 Гц, 1H), 3,21 (дт, J=12,8, 2,6 Гц, 1H), 3,52 (д, J=13,2 Гц, 1H), 4,81 (шир., 2H), 5,03 (д, J=4,6 Гц, 1H), 7,20 (д, J=8,3 Гц, 1H), 7,29-7,37 (м, 3H), 7,42 (дт, J=10,8, 3,8 Гц, 2H), 7,81 (шир., 1H), 8,95 (шир., 1H).
ESI-МС:m/z=442(M+H)+.
[0157]
(Пример 18) Синтез N2-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-N1,N1-диметилпиперидин-1,2-дикарбоксамида:
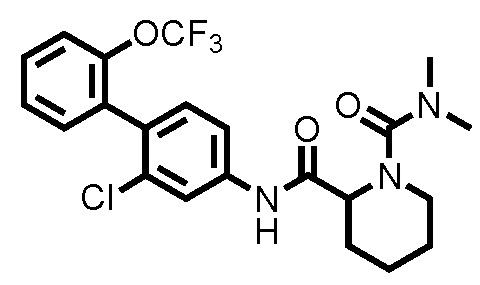
Следуя методике, аналогичной примеру 3, за исключением того, что диметилкарбамоилхлорид использовали вместо пропионилхлорида, N2-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-N1,N1-диметилпиперидин-1,2-дикарбоксамид (здесь и далее называемый соединением примера 18) (0,0231 г, 0,0492 ммоль, 65,4%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,48-1,78 (м, 4H), 1,92-2,05 (м, 1H), 2,27-2,35 (м, 1H), 2,94 (с, 6H), 2,87-2,99 (м, 1H), 3,40-3,46 (м, 1H), 4,47-4,51 (м, 1H), 7,20 (д, J=8,6 Гц, 1H), 7,30-8,00 (м, 6H), 10,60 (шир., 1H).
ESI-МС:m/z=470(M+H)+.
[0158]
(Пример 19) Синтез метил 2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)пиперидин-1-карбоксилата:
Следуя методике, аналогичной примеру 3, за исключением того, что метилхлорформиат использовали вместо пропионилхлорида, метил 2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)пиперидин-1-карбоксилат (здесь и далее называемый соединением примера 19) (0,0316 г, 0,0692 ммоль, 92,0%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,46-1,78 (м, 5H), 2,30-2,41 (м, 1H), 2,92 (т, J=12,1 Гц, 1H), 3,81 (с, 3H), 4,05-4,20 (шир., 1H), 4,93 (д, J=4,6 Гц, 1H), 7,23 (д, J=8,5 Гц, 1H), 7,31-7,45 (м, 5H), 7,74-7,86 (м, 1H), 8,21 (шир., 1H).
ESI-МС:m/z=457(M+H)+.
[0159]
(Ссылочный пример 7) Синтез трет-бутил(R)-2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)пиперидин-1-карбоксилата:
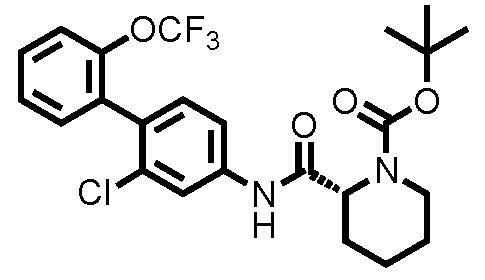
К ДМФА раствору (18 мл) (R)-(+)-1-(трет-бутоксикарбонил)пиперидин-2-карбоновой кислоты (0,840 г, 3,66 ммоль) при комнатной температуре добавляли соединение ссылочного примера 1 (1,05 г, 3,66 ммоль), HATU (1,53 г, 4,03 ммоль) и диизопропилэтиламин (0,768 мл, 4,40 ммоль), с последующим перемешиванием при этой же температуре в течение 18 часов. К полученному реакционному раствору добавляли дистиллированную воду, и полученный раствор экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, н-гексан/этилацетат=80/20), с получением трет-бутил(R)-2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)пиперидин-1-карбоксилата (здесь и далее называемого соединением ссылочного примера 7) (1,60 г, 3,20 ммоль, 87,3%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,43-1,51 (м, 2H), 1,53 (с, 9H), 1,60-1,75 (м, 3H), 2,35 (д, J=12,7 Гц, 1H), 2,80-2,89 (м, 1H), 4,03-4,13 (м, 1H), 4,86-4,89 (м, 1H), 7,22 (д, J=8,3 Гц, 1H), 7,29-7,45 (м, 6H), 7,80 (шир., 1H).
ESI-МС:m/z=499(M+H)+.
[0160]
(Ссылочный пример 8) Синтез (R)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида:
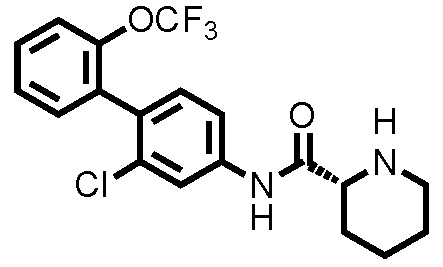
К дихлорметановому раствору (30 мл) соединения ссылочного примера 7 (1,60 г, 3,21 ммоль) при комнатной температуре добавляли трифторуксусную кислоту (8,02 мл, 104 ммоль), с последующим перемешиванием при этой же температуре в течение 2 часов. К полученному реакционному раствору добавляли дистиллированную воду, и полученный раствор экстрагировали хлороформом. Водный слой нейтрализовали добавлением 1М водного раствора гидроксида натрия и затем экстрагировали хлороформом. Органический слой промывали дистиллированной водой и насыщенным раствором соли, сушили над безводным сульфатом натрия, и фильтровали, и затем фильтрат концентрировали при пониженном давлении, с получением (R)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида (здесь и далее называемого соединением ссылочного примера 8) (1,13 г, 2,84 ммоль, 88,6%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,53(ддд, J=36,8,17,9,8,8 Гц, 4H), 1,78-1,86 (м, 1H), 2,00-2,07 (м, 1H), 2,74-2,82 (м, 1H), 3,03-3,10 (м, 1H), 3,38 (дд, J=9,6, 3,5 Гц, 1H), 7,23 (д, J=8,3 Гц, 1H), 7,31-7,37 (м, 3H), 7,40-7,45 (м, 1H), 7,53 (дд, J=8,3, 2,0 Гц, 1H), 7,82 (д, J=2,2 Гц, 1H), 9,02 (шир., 1H).
ESI-МС:m/z=399(M+H)+.
[0161]
(Пример 20) Синтез (R)-1-ацетил-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида:
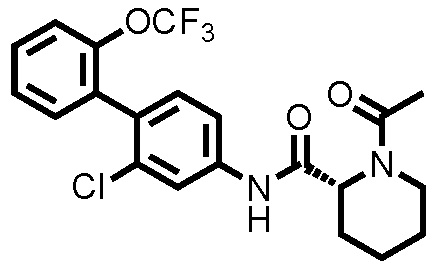
К дихлорметановому раствору (36 мл) соединения ссылочного примера 8 (1,43 г, 3,59 ммоль) при 0°C добавляли триэтиламин (0,750 мл, 5,38 ммоль) и уксусный ангидрид (0,338 мл, 3,59 ммоль), и температуру поднимали до комнатной температуры, с последующим перемешиванием в течение 30 минут. К полученному реакционному раствору добавляли дистиллированную воду, и полученный раствор экстрагировали хлороформом. Органический слой промывали дистиллированной водой и насыщенным раствором соли, сушили над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, хлороформ/метанол=95/5), с получением (R)-1-ацетил-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида (здесь и далее называемого соединением примера 20) (1,02 г, 2,32 ммоль, 64,6%) в виде белого твердого вещества. По результатам анализа с использованием хиральной колонки, время удерживания полученного таким образом соединения примера 20 составляло 32,8 минут, и оптическая чистота при таком времени составляла 99,0% ee. Условия анализа с использованием хиральной колонки были следующими.
Измерительное оборудование: Высокоэффективный жидкостной хроматограф LC-2010CHT, производства фирмы Shimadzu Corporation
Колонка: CHIRALCEL OD-RH 0,46 см × 15 см, размер частиц 5 мкм, производства фирмы Daicel Chemical Industries Ltd.
Температура колонки: 40°C
Подвижная фаза: (Раствор A) 20мМ водный раствор дигидрофосфата калия, (Раствор B) ацетонитрил
Состав подвижной фазы: Раствор A:Раствор B=60:40 до 50:50 (0 до 40 минут, линейный градиент)
Раствор A:Раствор B=50:50 до 60:40 (40 до 41 минут, линейный градиент)
Раствор A:Раствор B=60:40 (41 до 50 минут)
Скорость потока: 0,5 мл/минута
Детекция: УФ (210 нм)
1H-ЯМР (400 МГц, CDCl3) δ: 1,46-1,63 (м, 1H), 1,67 (д, J=7,8 Гц, 1H), 1,89-2,02 (м, 2H), 2,22 (с, 3H), 2,29 (д, J=12,9 Гц, 1H), 3,22 (т, J=13,2 Гц, 1H), 3,78 (д, J=12,7 Гц, 1H), 5,29 (д, J=5,1 Гц, 1H), 7,20 (д, J=8,3 Гц, 1H), 7,29-7,37 (м, 3H), 7,40-7,44 (м, 2H), 7,80 (шир., 1H), 8,65 (шир., 1H).
ESI-МС:m/z=441(M+H)+.
[0162]
(Пример 21) Синтез (R)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(метилсульфонил)пиперидин-2-карбоксамида:
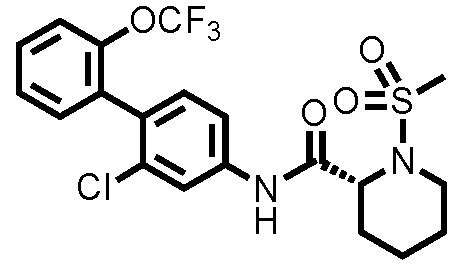
Следуя методике, аналогичной примеру 3, за исключением того, что соединение ссылочного примера 8 использовали вместо соединения ссылочного примера 3, и метансульфонилхлорид использовали вместо пропионилхлорида, (R)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(метилсульфонил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 21) (0,0600 г, 0,126 ммоль, 99,0%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,47-1,61 (м, 1H), 1,62-1,81 (м, 4H), 2,45 (д, J=10,4 Гц, 1H), 3,04 (с, 3H), 3,23 (тд, J=13,3, 2,4 Гц, 1H), 3,93 (т, J=7,0 Гц, 1H), 4,64 (шир., 1H), 7,25 (д, J=8,5 Гц, 1H), 7,30-7,38 (м, 3H), 7,43 (дт, J=10,8, 3,7 Гц, 2H), 7,84 (д, J=2,2 Гц, 1H), 8,29 (шир., 1H).
ESI-МС:m/z=477(M+H)+.
[0163]
(Пример 22) Синтез (R)-N2-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-1,2-дикарбоксамида:
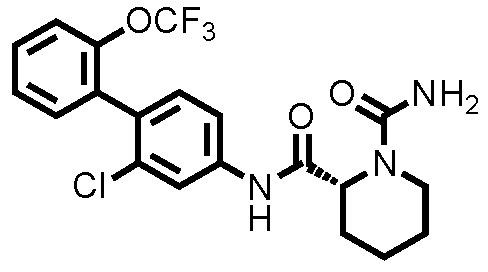
К дихлорметановому раствору (30 мл) соединения ссылочного примера 8 (3,00 г, 7,52 ммоль) при 0°C добавляли триметилсилилизоцианат (2,00 мл, 15,04 ммоль) и триэтиламин (1,05 мл, 7,57 ммоль), и температуру поднимали до комнатной температуры, с последующим перемешиванием в течение 18 часов. К полученному реакционному раствору добавляли метанол, и полученный раствор концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, хлороформ), с получением (R)-N2-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-1,2-дикарбоксамида (здесь и далее называемого соединением примера 22) (2,50 г, 5,66 ммоль, 75,2%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,50-1,68 (м, 2H), 1,73 (шир., 2H), 1,81-1,92 (м, 1H), 2,30 (д, J=12,9 Гц, 1H), 3,21 (дт, J=12,8, 2,6 Гц, 1H), 3,52 (д, J=13,2 Гц, 1H), 4,81 (шир., 2H), 5,03 (д, J=4,6 Гц, 1H), 7,20 (д, J=8,3 Гц, 1H), 7,29-7,37 (м, 3H), 7,42 (дт, J=10,8, 3,8 Гц, 2H), 7,81 (шир., 1H), 8,95 (шир., 1H).
ESI-МС:m/z=442(M+H)+.
[0164]
(Пример 23) Синтез (R)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2,2,2-трифторацетил)пиперидин-2-карбоксамида:
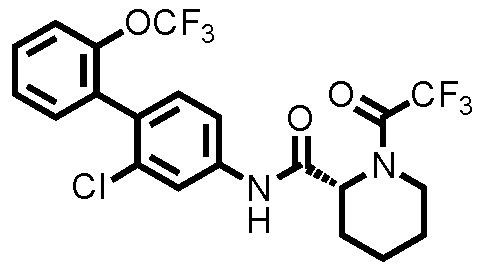
К дихлорметановому раствору (75 мл) соединения ссылочного примера 8 (3,00 г, 7,52 ммоль) при 0°C добавляли триэтиламин (1,57 мл, 11,28 ммоль) и трифторуксусный ангидрид (1,17 мл, 8,27 ммоль), и температуру поднимали до комнатной температуры, с последующим перемешиванием в течение 30 минут. К полученному реакционному раствору добавляли дистиллированную воду, и полученный раствор экстрагировали хлороформом. Органический слой промывали дистиллированной водой и насыщенным раствором соли, сушили над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, н-гексан/этилацетат=20/80), с получением (R)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2,2,2-трифторацетил)пиперидин-2-карбоксамида (здесь и далее называемого соединением примера 23) (2,50 г, 5,05 ммоль, 67,2%) в виде белого твердого вещества. По результатам анализа с использованием хиральной колонки, время удерживания полученного таким образом соединения примера 23 составляло 33,6 минут, и оптическая чистота при таком времени составляла 95,0% ee. Условия анализа с использованием хиральной колонки были следующими.
Измерительное оборудование: Высокоэффективный жидкостной хроматограф LC-2010CHT, производства фирмы Shimadzu Corporation
Колонка: CHIRALCEL OD-RH 0,46 см × 15 см, размер частиц 5 мкм, производства фирмы Daicel Chemical Industries Ltd.
Температура колонки: 40°C
Подвижная фаза: (Раствор A) 20мМ водный раствор дигидрофосфата калия, (Раствор B) ацетонитрил
Состав подвижной фазы: Раствор A:Раствор B=60:40 до 50:50 (0 до 40 минут, линейный градиент)
Раствор A:Раствор B=50:50 до 60:40 (40 до 41 минут, линейный градиент)
Раствор A:Раствор B=60:40 (41 до 50 минут)
Скорость потока: 0,5 мл/минута
Детекция: УФ (210 нм)
1H-ЯМР (400 МГц, CDCl3) δ: 1,56-1,86 (м, 4H), 1,98 (дт, J=11,2, 4,6 Гц, 1H), 2,36 (д, J=14,1 Гц, 1H), 3,37 (тд, J=13,4, 2,6 Гц, 1H), 4,01 (д, J=13,9 Гц, 1H), 5,18 (д, J=5,4 Гц, 1H), 7,21 (д, J=8,3 Гц, 1H), 7,30-7,46 (м, 5H), 7,79 (шир., 1H), 7,89 (шир., 1H).
ESI-МС:m/z=495(M+H)+.
[0165]
(Ссылочный пример 9) Синтез (1R,5S)-2-((R)-1-фенилэтил)-6-окса-2-азабицикло[3,2,1]октан-7-она:
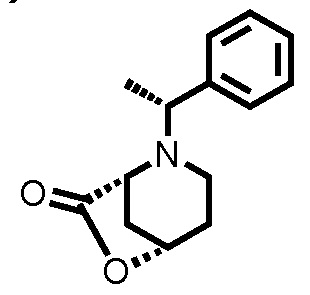
К ДМФА раствору (30 мл) (R)-α-метилбензиламина (3,77 мл, 29,6 ммоль) при комнатной температуре добавляли карбонат калия (4,09 г, 29,6 ммоль) и 4-бром-1-бутен (3,01 мл, 29,6 ммоль), с последующим перемешиванием при этой же температуре в течение 24 часов. К полученному реакционному раствору добавляли дистиллированную воду, и полученный раствор экстрагировали диэтиловым эфиром. Органический слой промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. К тетрагидрофурановому раствору (12 мл) остатка при 0°C добавляли глиоксалевую кислоту (4,09 мл, 36,8 ммоль), и температуру поднимали до 60°C, с последующим перемешиванием в течение 9 часов. К полученному реакционному раствору добавляли дистиллированную воду и 1М водный раствор гидроксида натрия, и полученный раствор экстрагировали этилацетатом. Органический слой промывали водным насыщенным раствором гидрокарбоната натрия, дистиллированной водой и насыщенным раствором соли, сушили над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, н-гексан/этилацетат=91/9 до 85/15), с получением (1R,5S)-2-((R)-1-фенилэтил)-6-окса-2-азабицикло[3,2,1]октан-7-она (здесь и далее называемого соединением ссылочного примера 9) (1,73 г, 7,48 ммоль, 25,3%) в виде бледно-желтого маслянистого продукта.
1H-ЯМР (400 МГц, CDCl3) δ: 1,33 (д, J=6,6 Гц, 3H), 1,82 (д, J=11,7 Гц, 1H), 1,87-1,95 (м, 1H), 2,03-2,12 (м, 2H), 2,47 (тд, J=11,8, 5,1 Гц, 1H), 3,19 (д, J=5,1 Гц, 1H), 3,35 (дд, J=12,0, 6,6 Гц, 1H), 3,70 (кв, J=6,6 Гц, 1H), 4,78 (т, J=5,1 Гц, 1H), 7,23-7,27 (м, 1H), 7,31-7,35 (м, 2H), 7,39-7,41 (м, 2H).
ESI-МС:m/z=232(M+H)+.
[0166]
(Ссылочный пример 10) Синтез (1R,5S)-2-(трет-бутоксикарбонил)-6-окса-2-азабицикло[3,2,1]-октан-7-она:
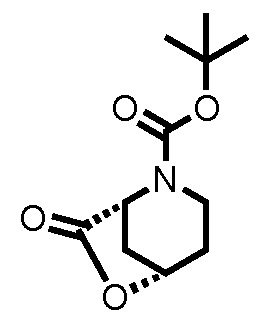
К этилацетатному раствору (25 мл) соединения ссылочного примера 9 (1,73 г, 7,48 ммоль) при комнатной температуре добавляли 20% по массе гидроксид палладия на углероде (содержащий 50% по массе воды, 0,210 г) и ди-трет-бутилдикарбонат (1,80 г, 8,23 ммоль), с последующим перемешиванием в атмосфере водорода при этой же температуре в течение 36 часов. Полученный реакционный раствор фильтровали через целит, и затем фильтрат концентрировали при пониженном давлении. Остаток суспендировали в смеси диэтиловый эфир/н-гексан=1/9(об./об.), и полученное твердое вещество отделяли фильтрованием и сушили, с получением (1R,5S)-2-(трет-бутоксикарбонил)-6-окса-2-азабицикло[3,2,1]-октан-7-она (здесь и далее называемого соединением ссылочного примера 10) (1,63 г, 7,17 ммоль, 95,9%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,48 (с, 9H), 1,84-1,93 (м, 1H), 1,95 (д, J=12,0 Гц, 1H), 2,03-2,06 (м, 1H), 2,29-2,32 (м, 1H), 3,18-3,21 (м, 1H), 4,06 (м, 1H), 4,70-4,85 (м, 1H), 4,97 (т, J=5,1 Гц, 1H).
ESI-МС:m/z=228(M+H)+.
[0167]
(Ссылочный пример 11) Синтез трет-бутил(2R,4S)-2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)-4-гидроксипиперидин-1-карбоксилата:
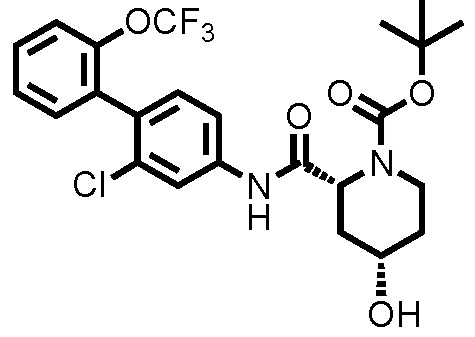
К толуольному раствору (2,3 мл) соединения ссылочного примера 10 (0,320 г, 1,41 ммоль) при 0°C добавляли раствор триметилалюминия в толуоле (1,4M, 1,31 мл, 1,83 ммоль), и температуру поднимали до комнатной температуры, с последующим перемешиванием в течение 30 минут. Добавляли толуольный раствор (2,3 мл) соединения ссылочного примера 1 (0,486 г, 1,690 ммоль), и температуру поднимали до 50°C, с последующим перемешиванием в течение 4 часов. К полученному реакционному раствору добавляли 1M хлористоводородную кислоту, и полученный раствор экстрагировали этилацетатом. Органический слой промывали дистиллированной водой и насыщенным раствором соли, сушили над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, н-гексан/этилацетат=80/20 до 50/50), с получением трет-бутил(2R,4S)-2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)-4-гидроксипиперидин-1-карбоксилата (здесь и далее называемого соединением ссылочного примера 11) (0,654 г, 1,27 ммоль, 90,2%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,54 (с, 9H), 1,66-1,69 (м, 1H), 1,78-1,82 (м, 1H), 1,93-2,00 (м, 1H), 2,40 (д, J=13,4 Гц, 1H), 3,25 (тд, J=13,2, 2,4 Гц, 1H), 3,86-3,88 (м, 1H), 4,12-4,14 (м, 1H), 4,98-5,00 (м, 1H), 5,20 (шир., 1H), 7,24 (д, J=8,3 Гц, 1H), 7,30-7,46 (м, 5H), 7,75-7,78 (м, 1H), 9,08 (шир., 1H).
ESI-МС:m/z=515(M+H)+.
[0168]
(Ссылочный пример 12) Синтез гидрохлорида (2R,4S)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-4-гидроксипиперидин-2-карбоксамида:
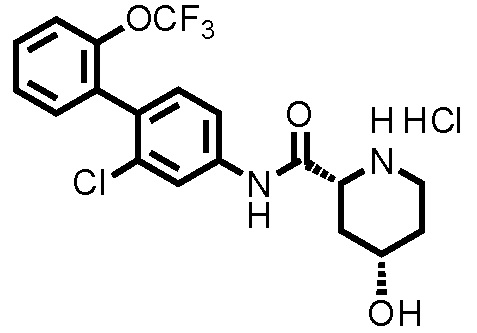
К этилацетатному раствору (0,5 мл) соединения ссылочного примера 11 (0,0500 г, 0,0973 ммоль) при 0°C добавляли раствор хлористого водорода в этилацетате (4,0M, 0,486 мл, 1,94 ммоль), и температуру поднимали до комнатной температуры, с последующим перемешиванием в течение 3 часов. Полученный реакционный раствор фильтровали, и полученное отфильтрованное твердое вещество промывали этилацетатом и затем сушили, с получением гидрохлорида (2R,4S)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-4-гидроксипиперидин-2-карбоксамида (здесь и далее называемого соединением ссылочного примера 12) (0,0409 г, 0,0908 ммоль, 93,3%) в виде белого твердого вещества.
1H-ЯМР(ДМСО-D6) δ: 1,46-1,62 (м, 2H), 1,91-1,94 (м, 1H), 2,42-2,45 (м, 1H), 3,01 (т, J=12,2 Гц, 1H), 3,28-3,32 (м, 1H), 3,69-3,78 (м, 1H), 4,00 (д, J=12,0 Гц, 1H), 5,28 (д, J=4,9 Гц, 1H), 7,38 (д, J=8,3 Гц, 1H), 7,42 (дд, J=7,8, 1,7 Гц, 1H), 7,48-7,52 (м, 2H), 7,56-7,64 (м, 2H), 7,94 (с, 1H), 8,93 (шир., 1H), 11,00 (с, 1H).
ESI-МС:m/z=415(M+H)+.
[0169]
(Ссылочный пример 13) Синтез трет-бутил(2R,4R)-2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)-4-фторпиперидин-1-карбоксилата:
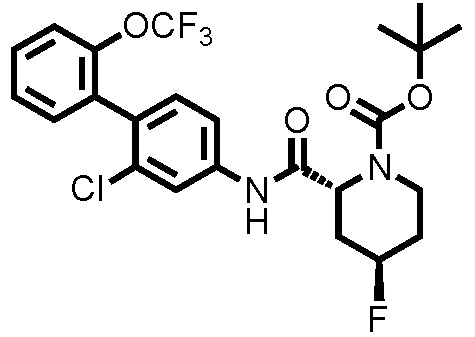
К дихлорметановому раствору (1,9 мл) соединения ссылочного примера 11 (0,100 г, 0,194 ммоль) при -78°C добавляли трифторид (диэтиламино)серы (0,0380 мл, 0,291 ммоль), и температуру поднимали до комнатной температуры, с последующим перемешиванием в течение 24 часов. К полученному реакционному раствору добавляли водный насыщенный раствор гидрокарбоната натрия, и полученный раствор экстрагировали хлороформом. Органический слой промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, н-гексан/этилацетат=90/10 до 80/20), с получением трет-бутил(2R,4R)-2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)-4-фторпиперидин-1-карбоксилата (здесь и далее называемого соединением ссылочного примера 13) (0,0272 г, 0,0527 ммоль, 27,2%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,48-1,56 (м, 1H), 1,54 (с, 9H), 1,59-1,86 (м, 2H), 2,07-2,19 (м, 1H), 2,65-2,71 (м, 1H), 2,93 (т, J=12,8 Гц, 1H), 4,10-4,13 (м, 1H), 5,04-5,06 (м, 1H), 7,22 (д, J=8,3 Гц, 1H), 7,29-7,36 (м, 4H), 7,41-7,45 (м, 1H), 7,76 (шир., 1H).
ESI-МС:m/z=517(M+H)+.
[0170]
(Ссылочный пример 14) Синтез гидрохлорида (2R,4R)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-4-фторпиперидин-2-карбоксамида:
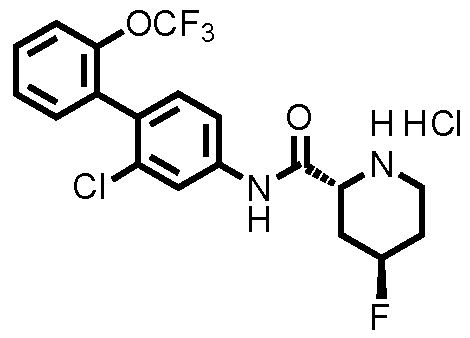
Следуя методике, аналогичной примеру 12, за исключением того, что соединение ссылочного примера 13 использовали вместо соединения ссылочного примера 11, гидрохлорид (2R,4R)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-4-фторпиперидин-2-карбоксамида (здесь и далее называемый соединением ссылочного примера 14) (0,0186 г, 0,0410 ммоль, 84,9%) получали в виде белого твердого вещества.
1H-ЯМР(ДМСО-D6) δ: 1,89-2,06 (м, 2H), 2,24-2,33 (м, 1H), 3,12-3,27 (м, 2H), 4,18 (д, J=12,4 Гц, 1H), 5,16 (д, J=47,1 Гц, 1H), 7,38 (д, J=8,5 Гц, 1H), 7,42 (дд, J=7,9, 1,8 Гц, 1H), 7,48-7,52 (м, 2H), 7,56-7,61 (м, 2H), 7,95 (шир., 1H), 9,12 (шир., 1H), 10,97 (с, 1H).
ESI-МС:m/z=417(M+H)+.
[0171]
(Ссылочный пример 15) Синтез трет-бутил(2R,4R)-2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)-4-(формилокси)пиперидин-1-карбоксилата:
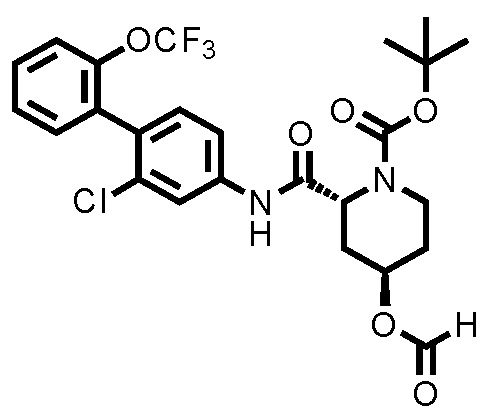
К тетрагидрофурановому раствору (1,0 мл) трифенилфосфина (0,153 г, 0,583 ммоль) при 0°C добавляли диизопропилазодикарбоксилат (0,113 мл, 0,583 ммоль), и после перемешивания при этой же температуре в течение 1 часа, добавляли муравьиную кислоту (0,0220 мл, 0,583 ммоль), и полученную смесь перемешивали при этой же температуре в течение 30 минут. По каплям добавляли тетрагидрофурановый раствор (1,00 мл) соединения ссылочного примера 11 (0,200 г, 0,388 ммоль), и температуру поднимали до комнатной температуры, с последующим перемешиванием в течение 12 часов. Полученный реакционный раствор концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией (силикагель, н-гексан/этилацетат=85/15 до 70/30), с получением трет-бутил(2R,4R)-2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)-4-(формилокси)пиперидин-1-карбоксилата (здесь и далее называемого соединением ссылочного примера 15) (0,0939 г, 0,173 ммоль, 44,5%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,28-1,37 (м, 2H), 1,54 (с, 9H), 1,67-1,74 (м, 1H), 2,07-2,12 (м, 1H), 2,61-2,63 (м, 1H), 2,96-3,02 (м, 1H), 4,12-4,14 (м, 1H), 5,06 (шир., 1H), 5,43 (шир., 1H), 7,22 (д, J=8,3 Гц, 1H), 7,32-7,37 (м, 3H), 7,41-7,45 (м, 2H), 7,80 (шир., 1H), 8,06 (с, 1H).
ESI-МС:m/z=543(M+H)+.
[0172]
(Ссылочный пример 16) Синтез трет-бутил(2R,4R)-2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)-4-гидроксипиперидин-1-карбоксилата:
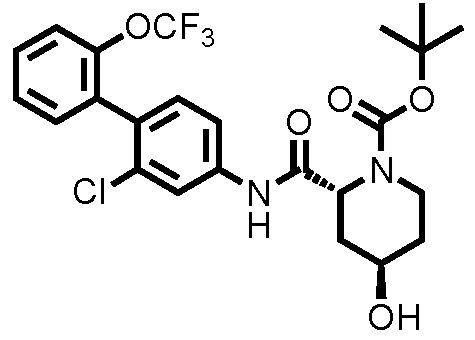
К метанольному раствору (1,1 мл) соединения ссылочного примера 15 (0,0900 г, 0,166 ммоль) при 0°C добавляли метанольный раствор метоксида натрия (4,0M, 0,0207 мл, 0,0828 ммоль), с последующим перемешиванием при этой же температуре в течение 15 минут. К полученному реакционному раствору добавляли 1M хлористоводородную кислоту, и полученный раствор экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, н-гексан/этилацетат=80/20 до 50/50), с получением трет-бутил(2R,4R)-2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)-4-гидроксипиперидин-1-карбоксилата (здесь и далее называемого соединением ссылочного примера 16) (0,0844 г, 0,164 ммоль, 99,3%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,46-1,54 (м, 2H), 1,54 (с, 9H), 1,68-1,71 (м, 1H), 1,94-1,97 (м, 1H), 2,54-2,56 (м, 1H), 2,86-2,93 (м, 1H), 4,13-4,22 (м, 2H), 5,04 (шир., 1H), 7,22 (д, J=8,3 Гц, 1H), 7,30-7,45 (м, 5H), 7,78 (шир., 1H), 8,52 (с, 1H).
ESI-МС:m/z=515(M+H)+.
[0173]
(Ссылочный пример 17) Синтез трет-бутил(R)-2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)-4-оксопиперидин-1-карбоксилата:
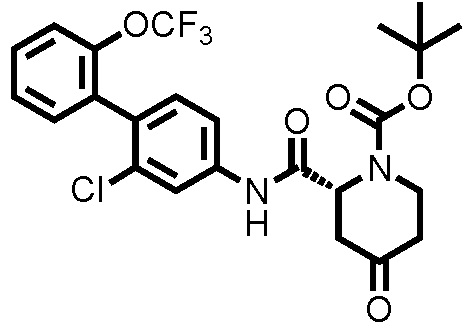
К дихлорметановому раствору (2,0 мл) соединения ссылочного примера 11 (0,210 г, 0,408 ммоль) при 0°C добавляли периодинан Десса-Мартина (0,190 г, 0,449 ммоль), и температуру поднимали до комнатной температуры, с последующим перемешиванием в течение 3 часов. К полученному реакционному раствору добавляли водный раствор тиосульфата натрия, и полученный раствор экстрагировали этилацетатом. Органический слой промывали дистиллированной водой и насыщенным раствором соли, сушили над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, н-гексан/этилацетат=90/10 до 60/40), с получением трет-бутил(R)-2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)-4-оксопиперидин-1-карбоксилата (здесь и далее называемого соединением ссылочного примера 17) (0,190 г, 0,370 ммоль, 90,9%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,56 (с, 9H), 2,46-2,53 (м, 1H), 2,61-2,73 (м, 2H), 3,00 (дд, J=16,5,3,3 Гц, 1H), 3,66-3,73 (м, 1H), 3,82 (шир., 1H), 5,05 (с, 1H), 7,22 (д, J=8,3 Гц, 1H), 7,29-7,36 (м, 3H), 7,41-7,45 (м, 2H), 7,78 (шир., 1H), 9,14 (шир., 1H).
ESI-МС:m/z=513(M+H)+.
[0174]
(Ссылочный пример 18) Синтез трет-бутил(R)-2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)-4,4-дифторпиперидин-1-карбоксилата:
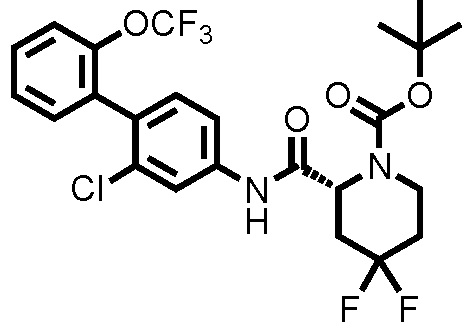
К дихлорметановому раствору (1,9 мл) соединения ссылочного примера 17 (0,190 г, 0,370 ммоль) при 0°C добавляли трифторид (диэтиламино)серы (0,108 мл, 0,815 ммоль), и температуру поднимали до комнатной температуры, с последующим перемешиванием в течение 24 часов. К полученному реакционному раствору добавляли водный насыщенный раствор гидрокарбоната натрия, и полученный раствор экстрагировали хлороформом. Органический слой промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, н-гексан/этилацетат=85/15 до 70/30), с получением трет-бутил(R)-2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)-4,4-дифторпиперидин-1-карбоксилата (здесь и далее называемого соединением ссылочного примера 18) (0,0393 г, 0,0735 ммоль, 19,8%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,54 (с, 9H), 1,86-2,15 (м, 3H), 2,99-3,07 (м, 1H), 3,21 (тд, J=13,3,2,7 Гц, 1H), 4,22-4,25 (м, 1H), 5,07 (шир., 1H), 7,24 (д, J=8,5 Гц, 1H), 7,30-7,37 (м, 3H), 7,41-7,46 (м, 2H), 7,76 (с, 1H), 7,97 (шир., 1H).
ESI-МС:m/z=535(M+H)+.
[0175]
(Пример 24) Синтез (2R,4S)-1-ацетил-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-4-гидроксипиперидин-2-карбоксамида:
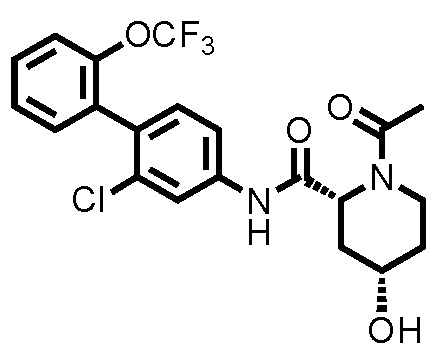
Следуя методике, аналогичной примеру 3, за исключением того, что соединение ссылочного примера 12 использовали вместо соединения ссылочного примера 3, и ацетилхлорид использовали вместо пропионилхлорида, (2R,4S)-1-ацетил-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-4-гидроксипиперидин-2-карбоксамид (здесь и далее называемый соединением примера 24) (0,0166 г, 0,0363 ммоль, 91,1%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,68-1,76 (м, 1H), 1,87-1,98 (м, 2H), 2,24 (с, 3H), 2,39 (д, J=14,6 Гц, 1H), 3,50-3,57 (м, 1H), 3,61-3,66 (м, 1H), 4,14-4,17 (м, 1H), 5,42 (д, J=6,8 Гц, 1H), 5,52 (д, J=6,8 Гц, 1H), 7,23 (д, J=8,5 Гц, 1H), 7,31-7,37 (м, 3H), 7,41-7,46 (м, 2H), 7,77 (шир., 1H), 9,15 (с, 1H).
ESI-МС:m/z=457(M+H)+.
[0176]
(Пример 25) Синтез (2R,4R)-1-ацетил-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-4-фторпиперидин-2-карбоксамида:
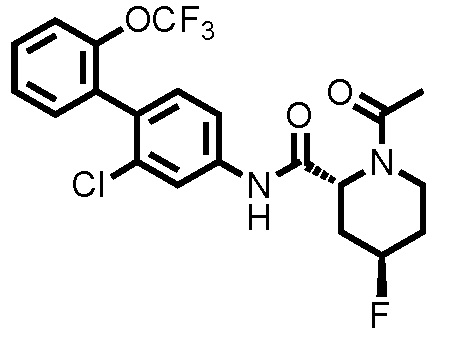
Следуя методике, аналогичной примеру 3, за исключением того, что соединение ссылочного примера 14 использовали вместо соединения ссылочного примера 3, и ацетилхлорид использовали вместо пропионилхлорида, (2R,4R)-1-ацетил-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-4-фторпиперидин-2-карбоксамид (здесь и далее называемый соединением примера 25) (0,0108 г, 0,0235 ммоль, 62,8%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,69-1,85 (м, 2H), 2,22-2,25 (м, 1H), 2,25 (с, 3H), 2,62-2,69 (м, 1H), 3,21-3,28 (м, 1H), 3,85 (дд, J=13,4,2,9 Гц, 1H), 5,24-5,43 (м, 1H), 5,43 (д, J=5,4 Гц, 1H), 7,21 (д, J=8,3 Гц, 1H), 7,30-7,36 (м, 3H), 7,41-7,45 (м, 2H), 7,70-7,82 (м, 1H), 8,72 (с, 1H).
ESI-МС:m/z=459(M+H)+.
[0177]
(Пример 26) Синтез (2R,4S)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-4-гидрокси-1-(метилсульфонил)пиперидин-2-карбоксамида:
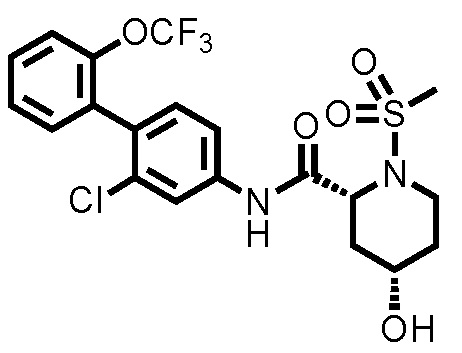
Следуя методике, аналогичной примеру 3, за исключением того, что соединение ссылочного примера 12 использовали вместо соединения ссылочного примера 3, и метансульфонилхлорид использовали вместо пропионилхлорида, (2R,4S)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-4-гидрокси-1-(метилсульфонил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 26) (0,00841 г, 0,0171 ммоль, 42,7%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,68-1,76 (м, 1H), 1,85-1,88 (м, 1H), 1,99(ддд, J=14,9,6,9,3,1 Гц, 1H), 2,61 (д, J=14,9 Гц, 1H), 3,03 (с, 3H), 3,55-3,62 (м, 1H), 3,72 (д, J=4,6 Гц, 1H), 3,75-3,79 (м, 1H), 4,22-4,24 (м, 1H), 4,66 (д, J=6,6 Гц, 1H), 7,25-7,27 (м, 1H), 7,31-7,37 (м, 3H), 7,42-7,46 (м, 2H), 7,81 (д, J=2,0 Гц, 1H), 8,58 (с, 1H).
ESI-МС:m/z=493(M+H)+.
[0178]
(Пример 27) Синтез (2R,4R)-1-ацетил-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-4-гидроксипиперидин-2-карбоксамида:
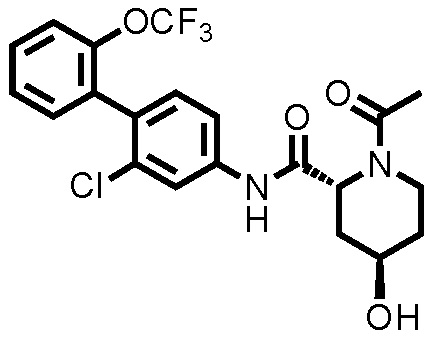
К этилацетатному раствору (0,4 мл) соединения ссылочного примера 16 (0,0200 г, 0,0388 ммоль) при 0°C добавляли раствор хлористого водорода в этилацетате (4,0M, 0,194 мл, 1,94 ммоль), и температуру поднимали до комнатной температуры, с последующим перемешиванием в течение 3 часов. Полученный реакционный раствор концентрировали при пониженном давлении, и остаток растворяли в дихлорметане (0,8 мл), и затем при 0°C добавляли триэтиламин (0,00135 мл, 0,0970 ммоль) и ацетилхлорид (0,00359 мл, 0,0504 ммоль), с последующим перемешиванием при этой же температуре в течение 1 часа. К полученному реакционному раствору добавляли метанол, и полученный раствор концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, этилацетат/метанол=100/0 до 97/3), с получением (2R,4R)-1-ацетил-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-4-гидроксипиперидин-2-карбоксамида (здесь и далее называемого соединением примера 27) (0,0106 г, 0,0232 ммоль, 59,7%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,47-1,57 (м, 2H), 1,81 (д, J=3,6 Гц, 1H), 2,04-2,09 (м, 1H), 2,25 (с, 3H), 2,50 (ддт, J=13,1, 5,0, 1,8 Гц, 1H), 3,22 (тд, J=13,4, 2,6 Гц, 1H), 3,85 (д, J=13,4 Гц, 1H), 4,40-4,48 (м, 1H), 5,43 (д, J=5,9 Гц, 1H), 7,21 (д, J=8,2 Гц, 1H), 7,30-7,37 (м, 3H), 7,41-7,45 (м, 2H), 7,69-7,83 (м, 1H), 8,65 (с, 1H).
ESI-МС:m/z=457(M+H)+.
[0179]
(Пример 28) Синтез (R)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-4,4-дифтор-1-(метилсульфонил)пиперидин-2-карбоксамида:
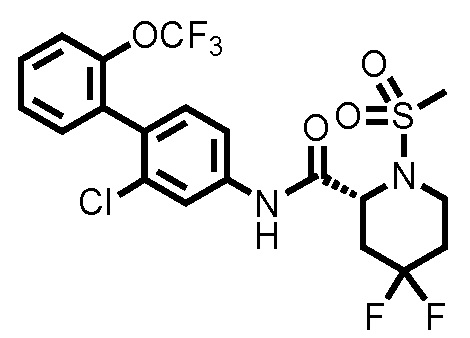
Следуя методике, аналогичной примеру 27, за исключением того, что соединение ссылочного примера 18 использовали вместо соединения ссылочного примера 16, и метансульфонилхлорид использовали вместо ацетилхлорида, (R)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-4,4-дифтор-1-(метилсульфонил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 28) (0,0127 г, 0,0248 ммоль, 77,0%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,95-2,12 (м, 1H), 2,15-2,32 (м, 2H), 2,95-3,02 (м, 1H), 3,10 (с, 3H), 3,60 (тд, J=13,5, 3,0 Гц, 1H), 4,02-4,08 (м, 1H), 4,89 (д, J=7,2 Гц, 1H), 7,26 (д, J=8,2 Гц, 1H), 7,30-7,37 (м, 3H), 7,42-7,46 (м, 2H), 7,76 (с, 1H), 7,93 (с, 1H).
ESI-МС:m/z=513(M+H)+.
[0180]
(Ссылочный пример 19) Синтез трет-бутил 2-(N-метилметилсульфонамид)ацетата:
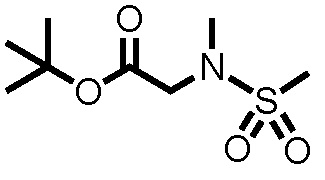
К дихлорметановому раствору (2,0 мл) гидрохлорида трет-бутил 2-(метиламино)ацетата (0,100 г, 0,550 ммоль) при 0°C добавляли триэтиламин (0,192 мл, 1,385 ммоль) и метансульфонилхлорид (0,0515 мл, 0,661 ммоль), и температуру поднимали до комнатной температуры, с последующим перемешиванием в течение 3 часов. К полученному реакционному раствору добавляли насыщенный водный раствор хлорида аммония, и полученный раствор экстрагировали хлороформом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, н-гексан/этилацетат=90/10 до 70/30), с получением трет-бутил 2-(N-метилметилсульфонамид)ацетата (здесь и далее называемого соединением ссылочного примера 19) (0,117 г, 0,524 ммоль, 95,2%) в виде бесцветного маслянистого продукта.
1H-ЯМР (400 МГц, CDCl3) δ: 1,48 (с, 9H), 2,98 (с, 3H), 3,00 (с, 3H), 3,98 (с, 2H).
[0181]
(Ссылочный пример 20) Синтез 2-(N-метилметилсульфонамид)уксусной кислоты:
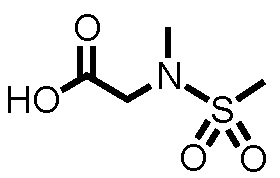
К ацетонитрильному раствору (1,5 мл) соединения ссылочного примера 19 (0,117 г, 0,524 ммоль) при 0°C добавляли раствор хлористого водорода в этилацетате (4,0M, 1,31 мл, 5,24 ммоль), и температуру поднимали до комнатной температуры, с последующим перемешиванием в течение 16 часов. Полученный реакционный раствор концентрировали при пониженном давлении, с получением неочищенной 2-(N-метилметилсульфонамид)уксусной кислоты (здесь и далее называемой соединением ссылочного примера 20) (0,0855 г) в виде бесцветного маслянистого продукта. Соединение ссылочного примера 20 использовали непосредственно в последующей реакции.
1H-ЯМР (400 МГц, CDCl3) δ: 2,99 (с, 3H), 3,01 (с, 3H), 4,14 (с, 2H).
ESI-МС:m/z=168(M+H)+.
[0182]
(Пример 29) Синтез (R)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-(N-метилметилсульфонамид)ацетил)пиперидин-2-карбоксамида:
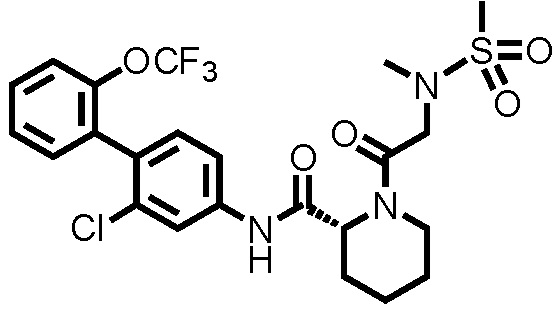
К ДМФА раствору (2,0 мл) соединения ссылочного примера 20 (0,0850 г, 0,508 ммоль) при комнатной температуре добавляли ДМФА раствор (1,0 мл) соединения ссылочного примера 8 (0,184 г, 0,462 ммоль), HATU (0,193 г, 0,508 ммоль) и диизопропилэтиламин (0,121 мл, 0,693 ммоль), с последующим перемешиванием при этой же температуре в течение 18 часов. К полученному реакционному раствору добавляли дистиллированную воду, и полученный раствор экстрагировали смешанным растворителем н-гексан/этилацетат=20/80(об./об.). Органический слой промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, н-гексан/этилацетат=50/50 до 30/70), с получением (R)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-(N-метилметилсульфонамид)ацетил)пиперидин-2-карбоксамида (здесь и далее называемого соединением примера 29) (0,209 г, 0,380 ммоль, 82,0%) в виде белого твердого вещества. По результатам анализа с использованием хиральной колонки, время удерживания полученного таким образом соединения примера 29 составляло 34,5 минут, и оптическая чистота при таком времени составляла 98,2% ee. Условия анализа с использованием хиральной колонки были следующими.
Измерительное оборудование: Высокоэффективный жидкостной хроматограф LC-2010CHT, производства фирмы Shimadzu Corporation
Колонка: CHIRALCEL OD-RH 0,46 см × 15 см, размер частиц 5 мкм, производства фирмы Daicel Chemical Industries Ltd.
Температура колонки: 40°C
Подвижная фаза: (Раствор A) 20мМ водный раствор дигидрофосфата калия, (Раствор B) ацетонитрил
Состав подвижной фазы: Раствор A:Раствор B=60:40 до 50:50 (0 до 40 минут, линейный градиент)
Раствор A:Раствор B=50:50 до 60:40 (40 до 41 минут, линейный градиент)
Раствор A:Раствор B=60:40 (41 до 50 минут)
Скорость потока: 0,5 мл/минута
Детекция: УФ (210 нм)
1H-ЯМР (400 МГц, CDCl3) δ: 1,52-1,89 (м, 5H), 2,35-2,38 (м, 1H), 3,03-3,07 (м, 6H), 3,20-3,31 (м, 1H), 3,67-3,76 (м, 1H), 4,16-4,27 (м, 2H), 5,25-5,26 (м, 1H), 7,21-7,23 (м, 1H), 7,30-7,45 (м, 5H), 7,83 (с, 1H), 8,22 (шир., 1H).
ESI-МС:m/z=548(M+H)+.
[0183]
(Ссылочный пример 21) Синтез трет-бутил(3-(2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)пиперидин-1-ил)-3-оксопропил)карбамата:
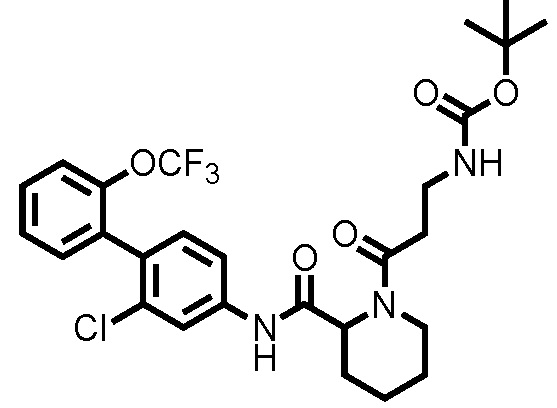
Следуя методике, аналогичной примеру 4, за исключением того, что 3-((трет-бутоксикарбонил)амино)пропановую кислоту использовали вместо 2-метоксиуксусной кислоты, трет-бутил(3-(2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)пиперидин-1-ил)-3-оксопропил)карбамат (здесь и далее называемый соединением ссылочного примера 21) (0,288 г, 0,505 ммоль, количественный) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,39 (с, 9H), 1,50-1,90(5H,m), 2,35 (д, J=13,7 Гц, 1H), 2,55-2,75 (м, 2H), 3,20 (т, J=12,8 Гц, 1H), 3,41-3,49 (м, 1H), 3,50-3,60 (м, 1H), 3,80 (д, J=13,7 Гц, 1H)5,17 (шир., 1H), 5,33 (д, J=4,9 Гц, 1H), 7,20 (д, J=8,3 Гц, 1H), 7,29-7,60 (м, 5H), 7,70-7,89 (м, 1H), 8,65 (шир., 1H).
ESI-МС:m/z=570(M+H)+.
[0184]
(Пример 30) Синтез 1-(3-аминопропаноил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида:
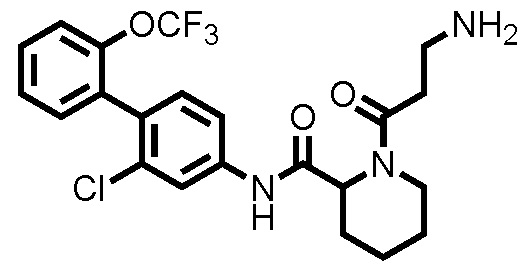
Следуя методике, аналогичной ссылочному примеру 5, за исключением того, что соединение ссылочного примера 21 использовали вместо соединения ссылочного примера 4, 1-(3-аминопропаноил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 30) (0,155 г, 0,329 ммоль, 65,6%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,46-1,82 (м, 5H), 2,43 (д, J=13,1 Гц, 1H)2,56 (дт, J=15,3, 6,2 Гц, 1H), 2,71-2,79 (м, 1H), 3,09-3,21 (м, 3H), 3,88 (д, J=13,1 Гц, 1H), 5,43 (д, J=5,0 Гц, 1H), 7,19 (д, J=8,2 Гц, 1H), 7,29-7,36 (м, 3H), 7,40-7,85 (м, 3H), 8,96 (шир., 1H).
ESI-МС:m/z=470(M+H)+.
[0185]
(Пример 31) Синтез 1-(3-ацетамидпропаноил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида:
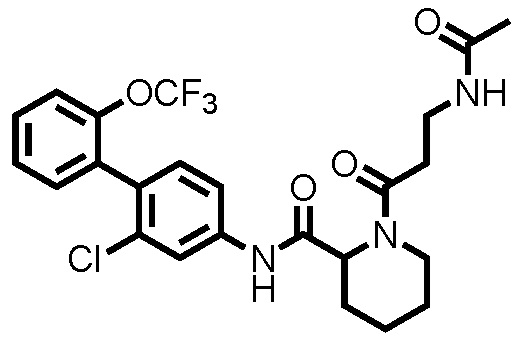
Следуя методике, аналогичной примеру 3, за исключением того, что соединение примера 30 использовали вместо соединения ссылочного примера 3, и ацетилхлорид использовали вместо пропионилхлорида, 1-(3-ацетамидпропаноил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 31) (0,0218 г, 0,0420 ммоль, 99,1%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,46-1,94 (м, 5H), 1,98 (с, 3H), 2,34 (д, J=13,3 Гц, 1H), 2,60-2,73 (м, 2H), 3,20 (тд, J=13,3, 2,4 Гц, 1H), 3,52-3,70 (м, 2H), 3,79 (д, J=13,3 Гц, 1H), 5,30 (д, J=4,5 Гц, 1H), 6,23 (шир., 1H), 7,21 (д, J=8,2 Гц, 1H), 7,30-7,90 (м, 6H), 8,51 (шир., 1H).
ESI-МС:m/z=512(M+H)+.
[0186]
(Пример 32) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(3-(метилсульфонамид)пропаноил)пиперидин-2-карбоксамида:
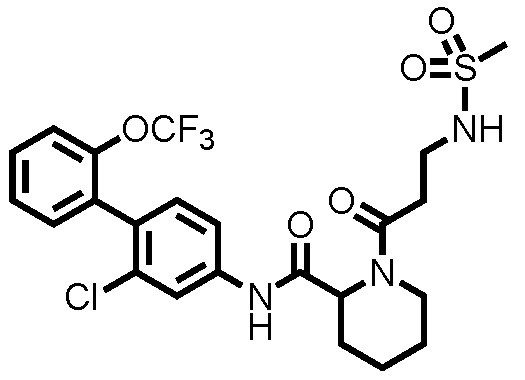
Следуя методике, аналогичной примеру 3, за исключением того, что соединение примера 30 использовали вместо соединения ссылочного примера 3, и метансульфонилхлорид использовали вместо пропионилхлорида, N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(3-(метилсульфонамид)пропаноил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 32) (0,0224 г, 0,0409 ммоль, 96,1%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,50-1,90 (м, 5H), 2,33 (д, J=13,5 Гц, 1H), 2,75-2,80 (м, 2H)3,00 (с, 3H), 3,22 (т, J=13,5 Гц, 1H), 3,45-3,51 (м, 2H), 3,77 (д, J=13,5 Гц, 1H), 5,26-5,30 (м, 2H), 7,22 (д, J=8,3 Гц, 1H), 7,30-7,44 (м, 5H), 7,80 (шир., 1H), 8,22 (шир., 1H).
ESI-МС:m/z=548(M+H)+.
[0187]
(Пример 33) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(3-(диметиламино)пропаноил)пиперидин-2-карбоксамида:
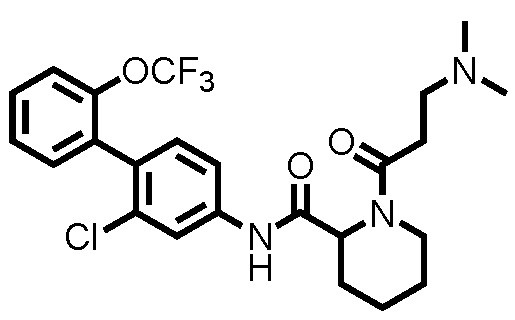
Следуя методике, аналогичной примеру 4, за исключением того, что гидрохлорид 3-(диметиламино)проионовой кислоты использовали вместо 2-метоксиуксусной кислоты, N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(3-(диметиламино)пропаноил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 33) (0,0274 г, 0,0550 ммоль, 73,2%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,49-1,83 (м, 5H), 2,27 (с, 6H), 2,38-2,45 (м, 1H), 2,56-2,61 (м, 1H), 2,66-2,80 (м, 3H), 3,12-3,20 (м, 1H), 3,85-3,93 (м, 1H), 5,42 (д, J=5,4 Гц, 1H), 7,21 (д, J=8,3 Гц, 1H), 7,27-7,45 (м, 6H), 8,73 (шир., 1H).
ESI-МС:m/z=498(M+H)+.
[0188]
(Ссылочный пример 22) Синтез трет-бутил(3-(2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)пиперидин-1-ил)-3-оксопропил)(метил)карбамата:
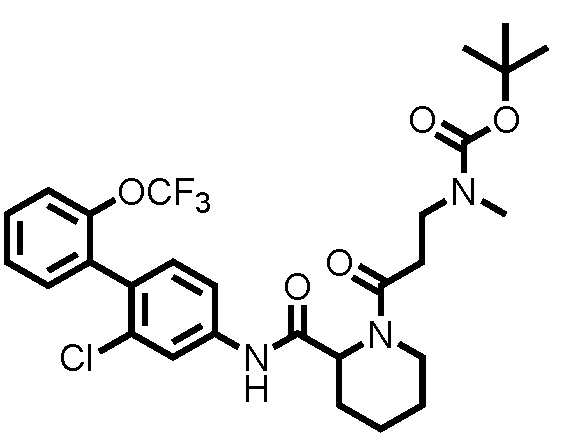
Следуя методике, аналогичной примеру 4, за исключением того, что 3-((трет-бутоксикарбонил)(метил)амино)пропановую кислоту использовали вместо 2-метоксиуксусной кислоты, трет-бутил(3-(2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)пиперидин-1-ил)-3-оксопропил)(метил)карбамат (здесь и далее называемый соединением ссылочного примера 22) (0,130 г, 0,223 ммоль, 89,0%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,35 (с, 9H), 1,30-1,77 (м, 5H), 2,37-2,80 (м, 3H), 2,93 (с, 3H), 3,18-3,30 (м, 2H)3,84 (д, J=13,7 Гц, 1H), 3,95-4,03 (м, 1H), 5,38-5,42 (м, 1H), 7,20 (д, J=8,3 Гц, 1H), 7,31-7,46 (м, 4H), 7,66-7,72 (м, 1H), 7,90-7,92 (м, 1H), 9,10 (шир., 1H).
ESI-МС:m/z=584(M+H)+.
[0189]
(Пример 34) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(3-(метиламино)пропаноил)пиперидин-2-карбоксамида:
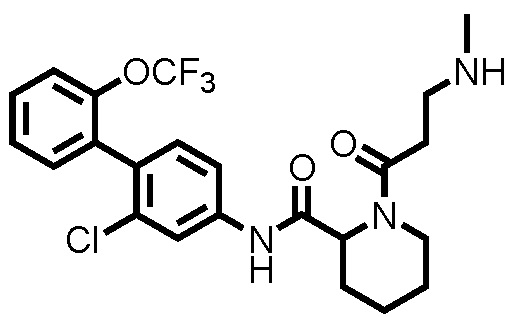
Следуя методике, аналогичной ссылочному примеру 5, за исключением того, что соединение ссылочного примера 22 использовали вместо соединения ссылочного примера 4, N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(3-(метиламино)пропаноил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 34) (0,804 г, 0,166 ммоль, 74,6%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,46-1,83 (м, 5H), 2,47 (с, 3H), 2,40-2,48 (м, 1H), 2,75-2,83 (м, 1H), 2,94-2,99 (м, 2H), 3,16 (тд, J=13,1, 2,6 Гц, 1H), 3,88 (д, J=13,1 Гц, 1H), 5,41 (д, J=5,4 Гц, 1H), 7,21 (д, J=8,3 Гц, 1H), 7,30-7,81 (м, 6H), 8,80 (шир., 1H).
ESI-МС:m/z=484(M+H)+.
[0190]
(Пример 35) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(3-(N-метилацетамид)пропаноил)пиперидин-2-карбоксамида:
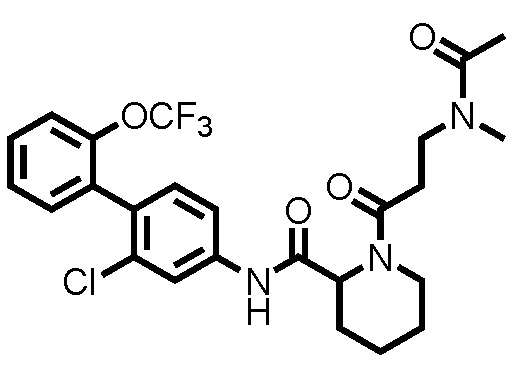
Следуя методике, аналогичной примеру 3, за исключением того, что соединение примера 34 использовали вместо соединения ссылочного примера 3, и ацетилхлорид использовали вместо пропионилхлорида, N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(3-(N-метилацетамид)пропаноил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 35) (0,0316 г, 0,0601 ммоль, 74,5%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,48-1,77 (м, 5H), 2,09 (с, 3H), 2,45 (д, J=13,7 Гц, 1H), 2,59-2,79 (м, 2H), 3,11 (с, 3H), 3,20-3,29 (м, 2H), 3,84 (д, J=13,7 Гц, 1H), 4,21-4,28 (м, 1H), 5,35 (д, J=5,1 Гц, 1H), 7,20 (д, J=8,3 Гц, 1H), 7,31-7,36 (м, 3H), 7,38-7,44 (м, 1H), 7,70-7,80 (м, 1H), 7,97-8,06 (м, 1H), 9,09 (шир., 1H).
ESI-МС:m/z=526(M+H)+.
[0191]
(Пример 36) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(3-(N-метилметилсульфонамид)пропаноил)пиперидин-2-карбоксамида:
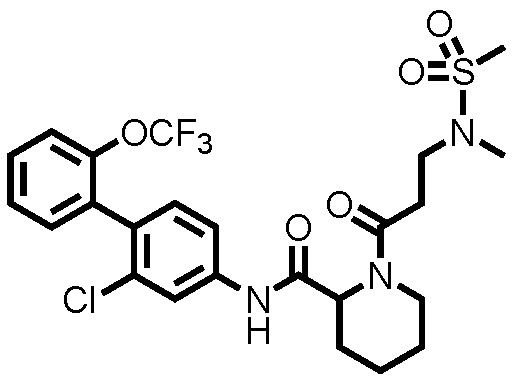
Следуя методике, аналогичной примеру 3, за исключением того, что соединение примера 34 использовали вместо соединения ссылочного примера 3, и метансульфонилхлорид использовали вместо пропионилхлорида, N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(3-(N-метилметилсульфонамид)пропаноил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 36) (0,0380 г, 0,0676 ммоль, 86,2%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,50-1,98 (м, 5H), 2,35 (д, J=13,5 Гц, 1H), 2,76 (дт, J=15,9, 7,0 Гц, 1H), 2,86 (с, 3H), 2,83-2,89 (м, 1H), 2,95 (с, 3H), 3,20 (тд, J=13,5, 2,7 Гц, 1H), 3,50-3,57 (м, 2H), 3,85 (д, J=13,5 Гц, 1H), 5,31 (д, J=5,0 Гц, 1H), 7,21 (д, J=8,2 Гц, 1H), 7,30-7,36 (м, 3H), 7,40-7,52 (м, 2H), 7,80-7,83 (м, 1H), 8,42 (шир., 1H).
ESI-МС:m/z=584(M+H)+.
[0192]
(Ссылочный пример 23) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-(этиламино)ацетил)пиперидин-2-карбоксамида:
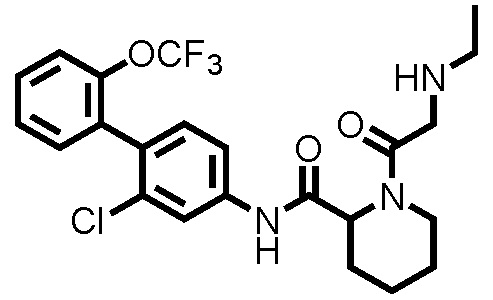
К дихлорметановому раствору (1,0 мл) соединения ссылочного примера 5 (0,0400 г, 0,0877 ммоль) при 0°C добавляли дихлорметановый раствор (0,0600 мл) ацетальдегида (0,00464 г, 0,105 ммоль), уксусную кислоту (0,000502 мл, 0,00877 ммоль) и триацетоксиборогидрид натрия (0,0279m г, 0,132 ммоль), и температуру поднимали до комнатной температуры, с последующим перемешиванием в течение 2,5 часов. К полученному реакционному раствору добавляли водный насыщенный раствор гидрокарбоната натрия, и полученный раствор экстрагировали хлороформом. Органический слой промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (амино-силикагель, н-гексан/этилацетат=40/60 до 0/100), с получением N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-(этиламино)ацетил)пиперидин-2-карбоксамида (здесь и далее называемого соединением ссылочного примера 23) (0,0193 г, 0,00399 ммоль, 45,5%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,17 (т, J=7,1 Гц, 3H), 1,38-1,80 (м, 5H), 1,90-2,00 (м, 1H), 2,30-2,45 (м, 1H), 2,55-2,90 (м, 2H), 3,16 (т, J=13,3 Гц, 1H), 3,56 (с, 2H), 3,70-3,76 (м, 1H), 5,29 (д, J=4,9 Гц, 1H), 7,20-7,45 (м, 6H), 7,70-7,90 (м, 1H), 8,46 (с, 1H).
ESI-МС:m/z=484(M+H)+.
[0193]
(Пример 37) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-(N-этилметилсульфонамид)ацетил)пиперидин-2-карбоксамида:
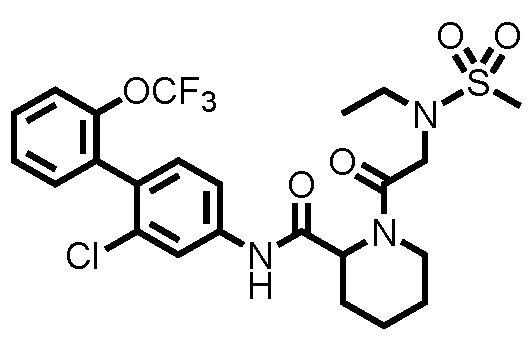
Следуя методике, аналогичной примеру 3, за исключением того, что соединение ссылочного примера 23 использовали вместо соединения ссылочного примера 3, и метансульфонилхлорид использовали вместо пропионилхлорида, N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-(N-этилметилсульфонамид)ацетил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 37) (0,0175 г, 0,0311 ммоль, 78,1%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,25 (т, J=7,1 Гц, 3H), 1,50-1,90 (м, 5H), 2,38 (д, J=13,0 Гц, 1H), 3,07 (с, 3H), 3,25 (т, J=13,0 Гц, 1H), 3,43 (кв, J=7,1 Гц, 2H), 3,76 (д, J=13,0 Гц, 1H), 4,14 (д, J=17,0 Гц, 1H), 4,30 (д, J=17,0 Гц, 1H), 5,27 (д, J=4,6 Гц, 1H), 7,21 (д, J=8,3 Гц, 1H), 7,29-7,36 (м, 3H), 7,40-7,45 (м, 2H), 7,83-7,85 (м, 1H), 8,23 (шир., 1H).
ESI-МС:m/z=584(M+Na)+.
[0194]
(Ссылочный пример 24) Синтез трет-бутил(R)-(3-(2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)пиперидин-1-ил)-3-оксопропил)карбамата:
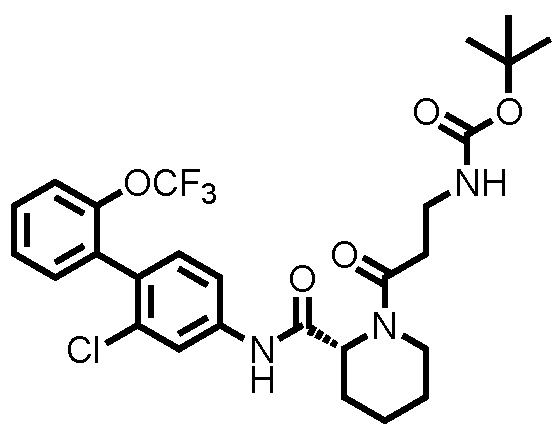
Следуя методике, аналогичной примеру 4, за исключением того, что 3-((трет-бутоксикарбонил)амино)пропановую кислоту использовали вместо 2-метоксиуксусной кислоты, и соединение ссылочного примера 8 использовали вместо соединения ссылочного примера 3, трет-бутил(R)-(3-(2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)пиперидин-1-ил)-3-оксопропил)карбамат (здесь и далее называемый соединением ссылочного примера 24) (0,104 г, 0,182 ммоль, 96,7%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,39 (с, 9H), 1,50-1,90(5H, m), 2,35 (д, J=13,7 Гц, 1H), 2,55-2,75 (м, 2H), 3,20 (т, J=12,8 Гц, 1H), 3,41-3,49 (м, 1H), 3,50-3,60 (м, 1H), 3,80 (д, J=13,7 Гц, 1H), 5,17 (шир., 1H), 5,33 (д, J=4,9 Гц, 1H), 7,20 (д, J=8,3 Гц, 1H), 7,29-7,60 (м, 5H), 7,70-7,89 (м, 1H), 8,65 (шир., 1H).
ESI-МС:m/z=570(M+H)+.
[0195]
(Пример 38) Синтез (R)-1-(3-аминопропаноил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида:
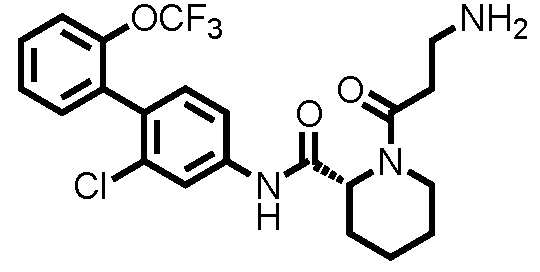
Следуя методике, аналогичной ссылочному примеру 5, за исключением того, что соединение ссылочного примера 24 использовали вместо соединения ссылочного примера 4, (R)-1-(3-аминопропаноил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 38) (0,497 г, 0,106 ммоль, 58,5%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,46-1,82 (м, 5H), 2,43 (д, J=13,1 Гц, 1H)2,56 (дт, J=15,3, 6,2 Гц, 1H), 2,71-2,79 (м, 1H), 3,09-3,21 (м, 3H), 3,88 (д, J=13,1 Гц, 1H), 5,43 (д, J=5,0 Гц, 1H), 7,19 (д, J=8,2 Гц, 1H), 7,29-7,36 (м, 3H), 7,40-7,85 (м, 3H), 8,96 (шир., 1H).
ESI-МС:m/z=470(M+H)+.
[0196]
(Пример 39) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(3-(метилтио)пропаноил)пиперидин-2-карбоксамида:
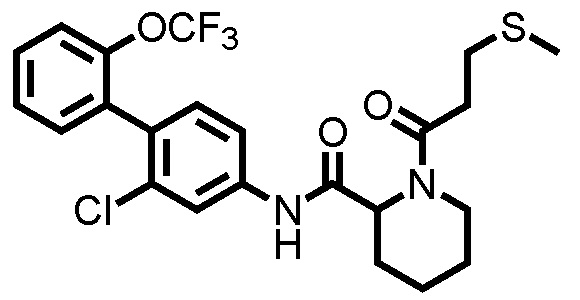
Следуя методике, аналогичной примеру 4, за исключением того, что 3-(метилтио)пропановую кислоту использовали вместо 2-метоксиуксусной кислоты, N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(3-(метилтио)пропаноил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 39) (0,0489 г, 0,0976 ммоль, 97,4%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,51-1,90 (м, 5H), 2,18 (с, 3H), 2,36 (д, J=13,9 Гц, 1H), 2,68-2,77 (м, 1H), 2,80-2,99 (м, 3H), 3,17 (тд, J=13,2, 2,6 Гц, 1H), 3,86 (д, J=12,4 Гц, 1H), 5,37 (д, J=4,9 Гц, 1H), 7,20 (д, J=8,3 Гц, 1H), 7,30-7,52 (м, 5H), 7,66-7,90 (м, 1H), 8,49 (шир., 1H).
ESI-МС:m/z=501(M+H)+.
[0197]
(Пример 40) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(3-(метилсульфонил)пропаноил)пиперидин-2-карбоксамида:
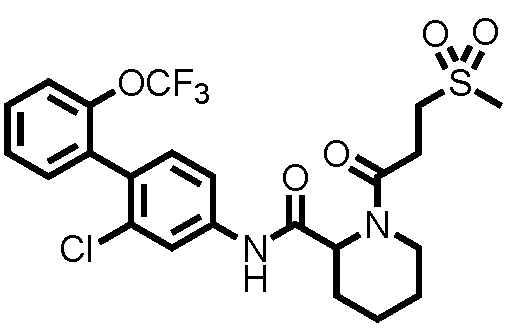
К дихлорметановому раствору (1,0 мл) соединения примера 39 (0,0480 г, 0,0958 ммоль) при 0°C добавляли 3-хлорнадбензойную кислоту (0,0496 г, 0,287 ммоль), и температуру поднимали до комнатной температуры, с последующим перемешиванием в течение 17 часов. К полученному реакционному раствору добавляли водный насыщенный раствор тиосульфата натрия и насыщенный гидрокатбонат натрия, и полученный раствор экстрагировали хлороформом. Органический слой промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, н-гексан/этилацетат=50/50 до 25/75), с получением N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(3-(метилсульфонил)пропаноил)пиперидин-2-карбоксамида (здесь и далее называемого соединением примера 40) (0,0412 г, 0,0773 ммоль, 80,6%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,50-1,82 (м, 5H), 2,45-2,48 (м, 1H), 2,79 (тд, J=11,0,5,9 Гц, 1H), 3,06 (с, 3H), 3,18-3,27 (м, 2H), 3,40 (дт, J=13,8, 5,5 Гц, 1H), 3,77-3,79 (м, 1H), 3,93-3,96 (м, 1H), 5,41 (д, J=5,4 Гц, 1H), 7,20 (д, J=8,5 Гц, 1H), 7,30-7,36 (м, 3H), 7,40-7,44 (м, 1H), 7,51-7,53 (м, 1H), 7,86-7,89 (м, 1H), 8,28 (шир., 1H).
ESI-МС:m/z=533(M+H)+.
[0198]
(Ссылочный пример 25) Синтез трет-бутил(R)-(3-(2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)пиперидин-1-ил)-3-оксопропил)(метил)карбамата:
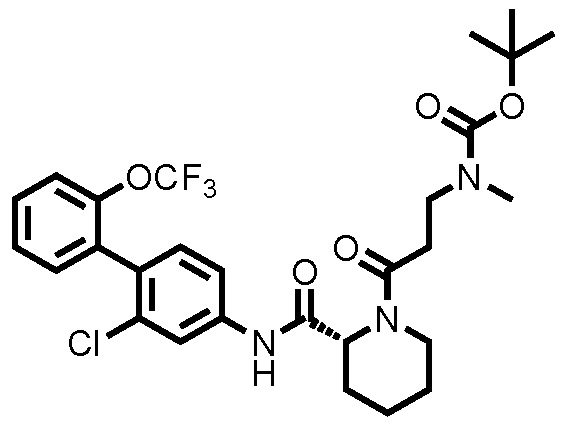
Следуя методике, аналогичной примеру 4, за исключением того, что 3-((трет-бутоксикарбонил)(метил)амино)пропановую кислоту использовали вместо 2-метоксиуксусной кислоты, и соединение ссылочного примера 8 использовали вместо соединения ссылочного примера 3, трет-бутил(R)-(3-(2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)пиперидин-1-ил)-3-оксопропил)(метил)карбамат (здесь и далее называемый соединением ссылочного примера 25) (0,117 г, 0,201 ммоль, 91,5%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,35 (с, 9H), 1,30-1,77(m5H,), 2,37-2,80 (м, 3H), 2,93 (с, 3H), 3,18-3,30 (м, 2H), 3,84 (д, J=13,7 Гц, 1H), 3,95-4,03 (м, 1H), 5,38-5,42 (м, 1H), 7,20 (д, J=8,3 Гц, 1H), 7,31-7,46 (м, 4H), 7,66-7,72 (м, 1H), 7,90-7,92 (м, 1H), 9,10 (шир., 1H).
ESI-МС:m/z=584(M+H)+.
[0199]
(Пример 41) Синтез (R)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(3-(метиламино)пропаноил)пиперидин-2-карбоксамида:
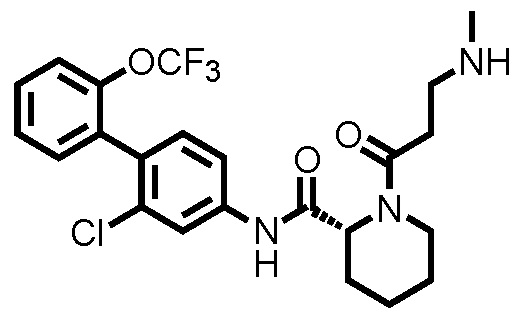
Следуя методике, аналогичной ссылочному примеру 5, за исключением того, что соединение ссылочного примера 25 использовали вместо соединения ссылочного примера 4, (R)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(3-(метиламино)пропаноил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 41) (0,748 г, 0,155 ммоль, 77,1%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,46-1,83 (м, 5H), 2,47 (с, 3H), 2,40-2,48 (м, 1H), 2,75-2,83 (м, 1H), 2,94-2,99 (м, 2H), 3,16 (тд, J=13,1, 2,6 Гц, 1H), 3,88 (д, J=13,1 Гц, 1H), 5,41 (д, J=5,4 Гц, 1H), 7,21 (д, J=8,3 Гц, 1H), 7,30-7,81 (м, 6H), 8,80 (шир., 1H).
ESI-МС:m/z=484(M+H)+.
[0200]
(Пример 42) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(3-гидроксипропаноил)пиперидин-2-карбоксамида:
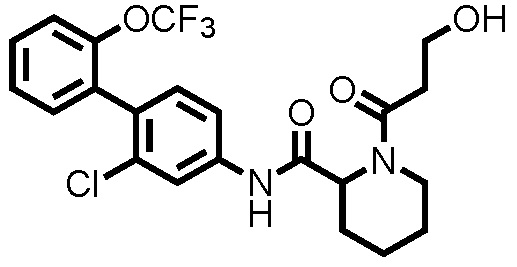
Следуя методике, аналогичной примеру 4, за исключением того, что 3-гидроксипропановую кислоту использовали вместо 2-метоксиуксусной кислоты, N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(3-гидроксипропаноил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 42) (0,212 г, 0,450 ммоль, 59,9%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,50-1,92 (м, 4H), 2,35 (д, J=14,1 Гц, 1H), 2,64-2,80 (м, 2H), 3,06 (т, J=6,3 Гц, 1H), 3,19 (тд, J=13,2, 2,4 Гц, 1H), 3,83 (д, J=14,1 Гц, 1H), 3,98 (кв, J=5,4 Гц, 2H), 5,34 (д, J=5,4 Гц, 1H), 7,21 (д, J=8,0 Гц, 1H), 7,30-7,44 (м, 6H), 7,70-7,90 (шир.м,1H), 8,39 (шир., 1H).
ESI-МС:m/z=471(M+H)+.
[0201]
(Пример 43) Синтез метил 3-(2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)пиперидин-1-ил)-3-оксопропаноата:
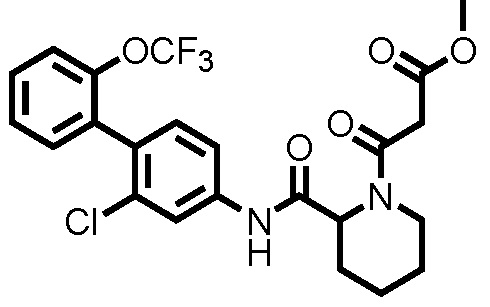
Следуя методике, аналогичной примеру 3, за исключением того, что метил 3-хлор-3-оксопропаноат использовали вместо пропионилхлорида, метил 3-(2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)пиперидин-1-ил)-3-оксопропаноат (здесь и далее называемый соединением примера 43) (0,0500 г, 0,100 ммоль, 80,0%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,50-1,80 (м, 5H), 2,57-2,62 (м, 1H), 3,16-3,25 (м, 1H), 3,57 (д, J=17,2 Гц, 1H), 3,59-3,65 (м, 1H), 3,84 (с, 3H), 3,85 (д, J=17,2 Гц, 2H), 5,49 (с, 1H), 7,22 (д, J=8,2 Гц, 1H), 7,30-7,37 (м, 3H), 7,40-7,44 (м, 1H), 7,92-7,95 (м, 1H), 8,87 (шир., 1H).
ESI-МС:m/z=499(M+H)+.
[0202]
(Пример 44) Синтез метил 4-(2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)пиперидин-1-ил)-4-оксобутаноата:
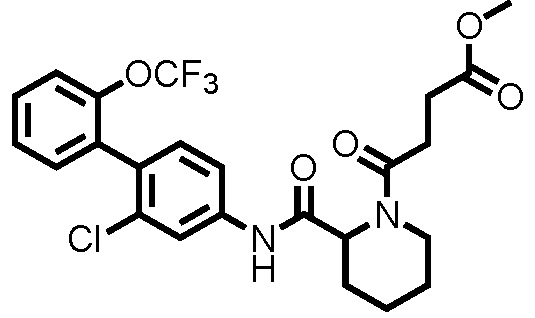
Следуя методике, аналогичной примеру 3, за исключением того, что метил 4-хлор-4-оксобутаноат использовали вместо пропионилхлорида, метил 4-(2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)пиперидин-1-ил)-4-оксобутаноат (здесь и далее называемый соединением примера 44) (0,0390 г, 0,0760 ммоль, количественный) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,52-1,60 (м, 5H), 2,42-2,60 (м, 2H), 2,62-2,70 (м, 1H), 2,88-2,96 (м, 1H), 2,99-3,08 (м, 1H), 3,22 (д, J=14,6 Гц, 1H), 3,74 (с, 3H), 3,97 (д, J=14,6 Гц, 1H), 5,46 (д, J=5,4 Гц, 1H), 7,21 (д, J=8,3 Гц, 1H), 7,28-7,44 (м, 6H), 8,49 (шир., 1H).
ESI-МС:m/z=513(M+H)+.
[0203]
(Пример 45) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(3-метоксипропаноил)пиперидин-2-карбоксамида:
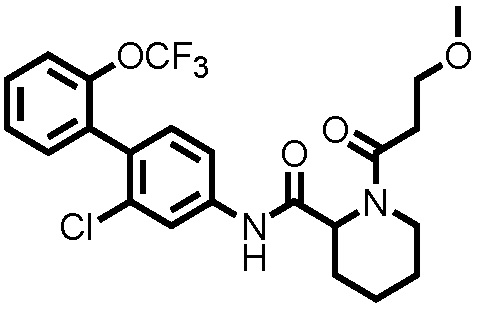
Следуя методике, аналогичной ссылочному примеру 2, за исключением того, что 1-(3-метоксипропаноил)пиперидин-2-карбоновую кислоту использовали вместо 1-(трет-бутоксикарбонил)пиперидин-2-карбоновой кислоты, N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(3-метоксипропаноил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 45) (0,0467 г, 0,0963 ммоль, 50,6%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,46-1,87 (м, 5H), 2,39 (д, J=13,9 Гц, 1H), 2,64 (дт, J=15,0, 5,7 Гц, 1H), 2,85-2,92 (м, 1H), 3,14 (тд, J=13,1, 2,3 Гц, 1H), 3,38 (с, 3H), 3,67-3,78 (м, 1H), 3,81-3,86 (м, 1H), 3,92 (д, J=13,9 Гц, 1H), 5,40 (д, J=4,9 Гц, 1H), 7,21 (д, J=8,3 Гц, 1H), 7,29-7,80 (м, 6H), 8,46 (с, 1H).
ESI-МС:m/z=483(M-H)-.
[0204]
(Пример 46) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(1-метил-1H-пиразол-4-карбонил)пиперидин-2-карбоксамида:
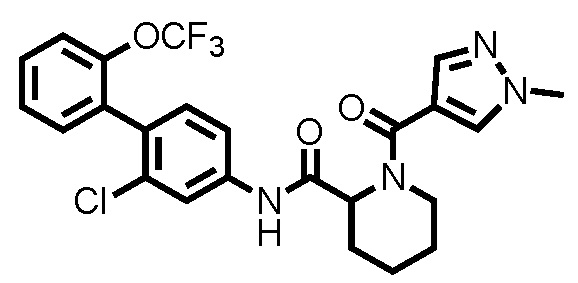
Следуя методике, аналогичной примеру 4, за исключением того, что 1-метил-1H-пиразол-4-карбоновую кислоту использовали вместо 2-метоксиуксусной кислоты, N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(1-метил-1H-пиразол-4-карбонил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 46) (0,0314 г, 0,0619 ммоль, 82,4%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,62-1,71 (м, 2H), 1,78-1,87 (м, 2H), 1,99-2,11 (м, 1H), 2,37 (д, J=12,9 Гц, 1H), 3,08-3,21 (м, 1H), 3,96 (с, 3H), 4,13-4,23 (м, 1H), 5,18-5,20 (м, 1H), 7,22 (д, J=8,3 Гц, 1H), 7,27-7,90 (м, 6H), 7,70 (с, 1H), 7,81 (с, 1H), 9,18 (шир., 1H).
ESI-МС:m/z=507(M+H)+.
[0205]
(Пример 47) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(1-метил-1H-имидазол-4-карбонил)пиперидин-2-карбоксамида:
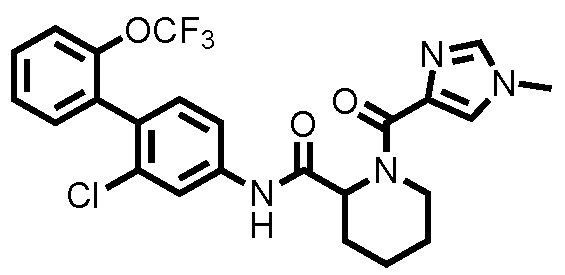
Следуя методике, аналогичной примеру 4, за исключением того, что 1-метил-1H-имидазол-4-карбоновую кислоту использовали вместо 2-метоксиуксусной кислоты, N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(1-метил-1H-имидазол-4-карбонил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 47) (0,0369 г, 0,0728 ммоль, 96,9%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,45-1,83 (м, 4H), 2,15-2,32 (м, 2H), 2,75-2,87 (м, 1H), 3,79 (с, 3H), 4,55-4,65 (м, 1H), 5,31-5,37 (м, 1H), 7,21 (д, J=8,3 Гц, 1H), 7,31-7,63 (м, 7H), 7,75-7,90 (м, 1H), 11,47 (шир., 1H).
ESI-МС:m/z=507(M+H)+.
[0206]
(Пример 48) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(1H-пиразол-4-карбонил)пиперидин-2-карбоксамида:
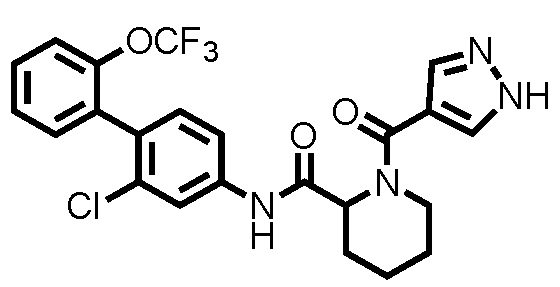
Следуя методике, аналогичной примеру 4, за исключением того, что 1H-пиразол-4-карбоновую кислоту использовали вместо 2-метоксиуксусной кислоты, N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(1H-пиразол-4-карбонил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 48) (0,0163 г, 0,0331 ммоль, 44,0%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,45-1,90 (м, 4H), 1,95-2,17 (м, 1H), 2,33-2,43 (м, 1H), 3,15-3,26 (м, 1H), 4,09-4,21 (м, 1H), 5,20-5,27 (м, 1H), 7,22 (д, J=8,5 Гц, 1H), 7,28-7,52 (м, 5H), 7,70-8,00 (м, 3H), 9,16 (шир., 1H), 10,79 (шир., 1H).
ESI-МС:m/z=493(M+H)+.
[0207]
(Ссылочный пример 26) Синтез метил 1-(2-(N-метилметилсульфонамид)ацетил)пиперидин-2-карбоксилата:
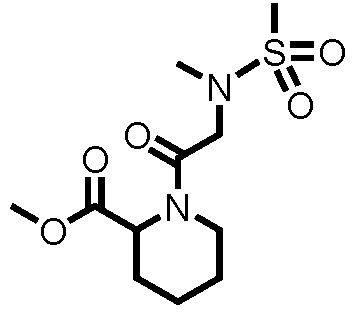
Следуя методике, аналогичной примеру 4, за исключением того, что соединение ссылочного примера 20 использовали вместо 2-метоксиуксусной кислоты, и метилпиперидин-2-карбоксилат гидрохлорид использовали вместо соединения ссылочного примера 3, метил 1-(2-(N-метилметилсульфонамид)ацетил)пиперидин-2-карбоксилат (здесь и далее называемый соединением ссылочного примера 26) (0,934 г, 3,19 ммоль, 82,0) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,24-1,76 (м, 5H), 2,25-2,32 (м, 1H), 2,99 (с, 3H), 3,00 (с, 3H), 3,25 (тд, J=13,0, 3,2 Гц, 1H), 3,58-3,64 (м, 1H), 3,75 (д, J=4,6 Гц, 3H), 4,11 (д, J=17,1 Гц, 1H), 4,30 (д, J=17,1 Гц, 1H), 5,25 (д, J=5,6 Гц, 1H).
ESI-МС:m/z=293(M+H)+.
[0208]
(Ссылочный пример 27) Синтез 1-(2-(N-метилметилсульфонамид)ацетил)пиперидин-2-карбоновой кислоты:
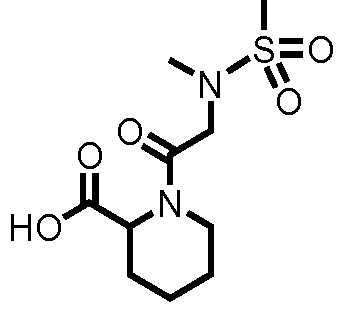
К метанольному раствору (10,0 мл) соединения ссылочного примера 26 (0,933 г, 3,19 ммоль) при 0°C добавляли 1М водный раствор гидроксида натрия (3,83 мл, 3,83 ммоль), и температуру поднимали до комнатной температуры, с последующим перемешиванием в течение 17 часов. К полученному реакционному раствору добавляли 1M хлористоводородную кислоту, и полученный раствор экстрагировали хлороформом. Органический слой промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении, с получением неочищенной 1-(2-(N-метилметилсульфонамид)ацетил)пиперидин-2-карбоновой кислоты (здесь и далее называемой соединением ссылочного примера 27) (0,812g) в виде белого твердого вещества. Соединение ссылочного примера 27 использовали непосредственно для последующей реакции.
1H-ЯМР (400 МГц, CDCl3) δ: 1,18-1,75 (м, 5H), 2,30 (д, J=13,1 Гц, 1H), 2,98 (с, 3H), 2,99 (с, 3H), 3,24 (т, J=12,0 Гц, 1H), 3,63 (д, J=13,1 Гц, 1H), 4,13 (д, J=17,2 Гц, 1H), 4,27 (д, J=17,2 Гц, 1H), 5,24 (д, J=4,1 Гц, 1H).
ESI-МС:m/z=279(M+H)+.
[0209]
(Ссылочный пример 28) Синтез N-(4-бром-3-хлорфенил)-1-(2-(N-метилметилсульфонамид)ацетил)пиперидин-2-карбоксамида:
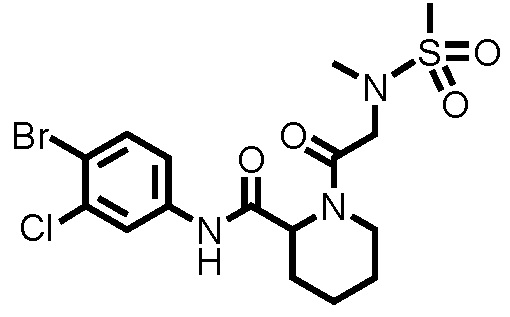
Следуя методике, аналогичной ссылочному примеру 2, за исключением того, что соединение ссылочного примера 27 использовали вместо 1-(трет-бутоксикарбонил)пиперидин-2-карбоновой кислоты, и 4-бром-3-хлоранилин использовали вместо соединения ссылочного примера 1, N-(4-бром-3-хлорфенил)-1-(2-(N-метилметилсульфонамид)ацетил)пиперидин-2-карбоксамид (здесь и далее называемый соединением ссылочного примера 28) (0,296 г, 0,634 ммоль, 58,8%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,49-1,85 (м, 5H), 2,34 (д, J=12,8 Гц, 1H), 3,03 (с, 3H), 3,03 (с, 3H), 3,20 (т, J=12,8 Гц, 1H), 3,71 (д, J=12,8 Гц, 1H), 4,12 (д, J=16,7 Гц, 1H), 4,23 (д, J=16,7 Гц, 1H), 5,22 (д, J=4,9 Гц, 1H), 7,20-7,24 (м, 1H), 7,50 (дд, J=8,5, 2,0 Гц, 1H), 7,84 (т, J=2,3 Гц, 1H), 8,20 (шир., 1H).
ESI-МС:m/z=467(M+H)+.
[0210]
(Пример 49) Синтез N-(2-хлор-2'-изопропокси-[1,1'-бифенил]-4-ил)-1-(2-(N-метилметилсульфонамид)ацетил)пиперидин-2-карбоксамида:
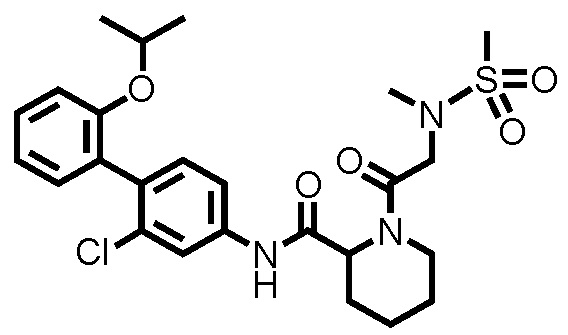
Следуя методике, аналогичной ссылочному примеру 1, за исключением того, что 2-изопропоксифенилбороновую кислоту использовали вместо 2-трифторметоксифенилбороновой кислоты, и соединение ссылочного примера 28 использовали вместо 4-бром-3-хлоранилина, N-(2-хлор-2'-изопропокси-[1,1'-бифенил]-4-ил)-1-(2-(N-метилметилсульфонамид)ацетил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 49) (0,0253 г, 0,0485 ммоль, 75,3%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,20 (д, J=6,0 Гц, 6H), 1,50-1,88 (м, 5H), 2,31-2,39 (м, 1H), 3,05 (с, 3H), 3,05 (с, 3H), 3,21-3,28 (м, 1H), 3,69-3,74 (м, 1H), 4,19 (д, J=16,8 Гц, 1H), 4,25 (д, J=16,8 Гц, 1H), 4,41 (т, J=6,0 Гц, 1H), 5,23-5,26 (м, 1H), 6,95-7,00 (м, 2H), 7,17 (дд, J=7,4,2,0 Гц, 1H), 7,22-7,37 (м, 3H), 7,76 (д, J=2,0 Гц, 1H), 8,11 (с, 1H).
ESI-МС:m/z=523(M+H)+.
[0211]
(Пример 50) Синтез N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-(1-метил-1H-имидазол-2-ил)ацетил)пиперидин-2-карбоксамида:
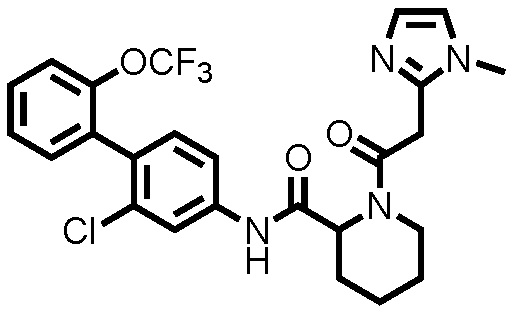
Следуя методике, аналогичной примеру 4, за исключением того, что 2-(1-метил-1H-имидазол-2-ил)уксусную кислоту использовали вместо 2-метоксиуксусной кислоты, N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-(1-метил-1H-имидазол-2-ил)ацетил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 50) (0,0341 г, 0,0654 ммоль, 87,0%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,49-1,80 (м, 5H), 2,67-2,74 (м, 1H), 3,20-3,27 (м, 1H), 3,54-3,62 (м, 1H), 3,63 (с, 3H), 3,73 (д, J=15,9 Гц, 1H), 4,05 (д, J=15,9 Гц, 1H), 5,59-5,63 (м, 1H), 6,89-6,98 (м, 2H), 7,20-7,29 (м, 1H), 7,32-7,37 (м, 3H), 7,39-7,45 (м, 1H), 7,71-7,99 (м, 2H), 10,64 (шир., 1H).
ESI-МС:m/z=521(M+H)+.
[0212]
(Ссылочный пример 29) Синтез N-(4-бром-3-фторфенил)-1-(2-(N-метилметилсульфонамид)ацетил)пиперидин-2-карбоксамида:
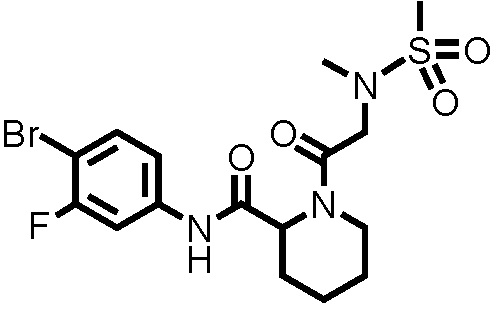
Следуя методике, аналогичной ссылочному примеру 2, за исключением того, что соединение ссылочного примера 27 использовали вместо 1-(трет-бутоксикарбонил)пиперидин-2-карбоновой кислоты, и 4-бром-3-фторанилин использовали вместо соединения ссылочного примера 1, N-(4-бром-3-фторфенил)-1-(2-(N-метилметилсульфонамид)ацетил)пиперидин-2-карбоксамид (здесь и далее называемый соединением ссылочного примера 29) (0,0253 г, 0,0562 ммоль, 52,1%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,48-1,83 (м, 5H), 2,31-2,39 (м, 1H), 3,03 (с, 3H), 3,03 (с, 3H), 3,16-3,23 (м, 1H), 3,68-3,75 (м, 1H), 4,10 (д, J=16,6 Гц, 1H), 4,24 (д, J=16,6 Гц, 1H), 5,21-5,24 (м, 1H), 7,06 (дд, J=9,0, 2,0 Гц, 1H), 7,43 (т, J=8,2 Гц, 1H), 7,63 (дд, J=10,5, 2,4 Гц, 1H), 8,24 (шир., 1H).
ESI-МС:m/z=451(M+H)+.
[0213]
(Пример 51) Синтез N-(2-фтор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-(N-метилметилсульфонамид)ацетил)пиперидин-2-карбоксамида:
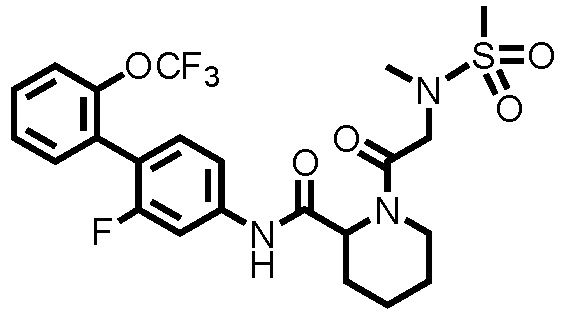
Следуя методике, аналогичной ссылочному примеру 1, за исключением того, что соединение ссылочного примера 29 использовали вместо 4-бром-3-хлоранилина, N-(2-фтор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-(N-метилметилсульфонамид)ацетил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 51) (0,0132 г, 0,0248 ммоль, 44,7%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,45-1,90 (м, 5H), 2,33-2,41 (м, 1H), 3,05 (с, 6H), 3,20-3,29 (м, 1H), 3,69-3,76 (м, 1H), 4,17 (д, J=16,8 Гц, 1H), 4,25 (д, J=16,8 Гц, 1H), 5,23-5,27 (м, 1H), 7,20-7,44 (м, 6H), 7,62 (дд, J=11,7, 2,0 Гц, 1H), 8,24 (шир., 1H).
[0214]
(Пример 52) Синтез 1-(2-(1H-имидазол-1-ил)ацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида:
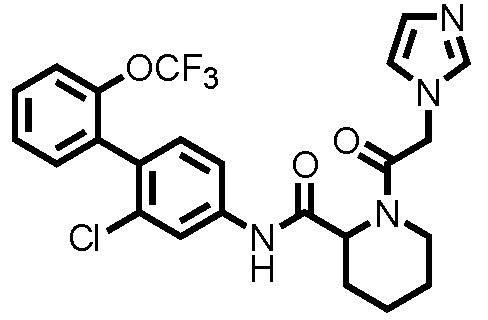
Следуя методике, аналогичной примеру 4, за исключением того, что 1-имидазолуксусную кислоту использовали вместо 2-метоксиуксусной кислоты, 1-(2-(1H-имидазол-1-ил)ацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 52) (0,0189 г, 0,0373 ммоль, 49,6%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,44-2,05 (м, 5H), 2,23-2,31 (м, 1H), 3,37-3,47 (м, 1H), 3,67-3,74 (м, 1H), 4,86 (д, J=16,6 Гц, 1H), 4,91 (д, J=16,6 Гц, 1H), 5,16-5,22 (м, 1H), 6,97 (с, 1H), 7,13 (с, 1H), 7,20 (д, J=8,3 Гц, 1H), 7,29-7,37 (м, 4H), 7,40-7,45 (м, 1H), 7,53 (с, 1H), 7,70-7,87 (м, 1H), 8,41 (шир., 1H).
ESI-МС:m/z=507(M+H)+.
[0215]
(Пример 53) Синтез 1-(2-(1H-тетразол-1-ил)ацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида:
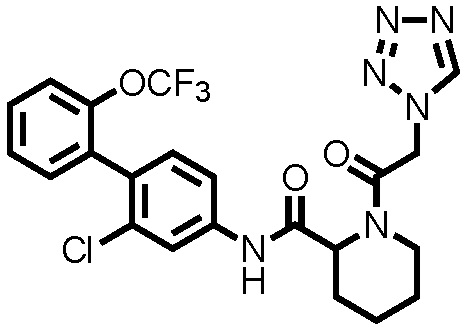
Следуя методике, аналогичной примеру 4, за исключением того, что 1H-тетразол-1-уксусную кислоту использовали вместо 2-метоксиуксусной кислоты, 1-(2-(1H-тетразол-1-ил)ацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 53) (0,0244 г, 0,0479 ммоль, 38,2%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,59-2,02 (м, 5H), 2,29-2,32 (м, 1H), 3,49-3,57 (м, 1H), 3,73-3,77 (м, 1H), 5,18-5,19 (м, 1H), 5,40 (д, J=16,8 Гц, 1H), 5,48 (д, J=16,8 Гц, 1H), 7,21-7,46 (м, 6H), 7,81 (шир., 1H), 8,02 (с, 1H), 8,86 (с, 1H).
ESI-МС:m/z=509(M+H)+.
[0216]
(Пример 54) Синтез 1-(2-(фуран-2-ил)ацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида:
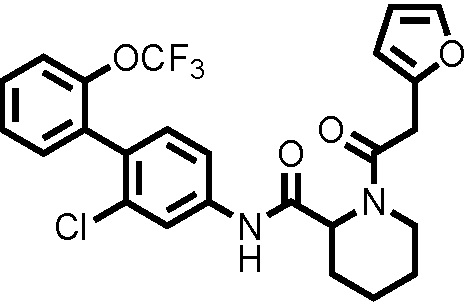
Следуя методике, аналогичной примеру 4, за исключением того, что 2-фурануксусную кислоту использовали вместо 2-метоксиуксусной кислоты, 1-(2-(фуран-2-ил)ацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 54) (0,0605 г, 0,119 ммоль, 95,2%) получали в виде белого амофного вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,41-1,95 (м, 5H), 2,33-2,38 (м, 1H), 3,08-3,16 (м, 1H), 3,85-3,97 (м, 3H), 5,34-5,36 (м, 1H), 6,22-6,23 (м, 1H), 6,35-6,37 (м, 1H), 7,19-7,44 (м, 8H), 8,33 (шир., 1H).
ESI-МС:m/z=505(M-H)-.
[0217]
(Пример 55) Синтез 1-(2-(3,5-диметил-1H-пиразол-1-ил)ацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида:
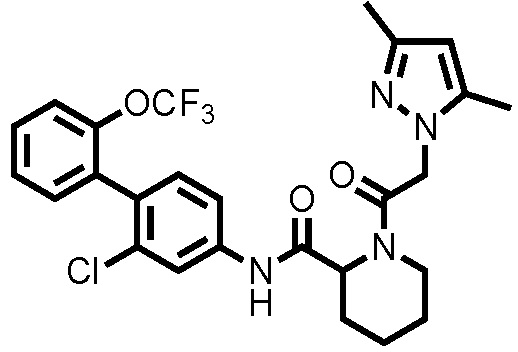
Следуя методике, аналогичной примеру 4, за исключением того, что 3,5-диметил-1H-пиразол-1-уксусную кислоту использовали вместо 2-метоксиуксусной кислоты, 1-(2-(3,5-диметил-1H-пиразол-1-ил)ацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 55) (0,0579 г, 0,108 ммоль, 86,3%) получали в виде белого амофного вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,46-1,66 (м, 2H), 1,74-1,79 (м, 3H), 2,08 (с, 3H), 2,27 (с, 3H), 2,47-2,50 (м, 1H), 3,11-3,19 (м, 1H), 3,69-3,74 (м, 1H), 4,83 (д, J=15,2 Гц, 1H), 4,96 (д, J=15,2 Гц, 1H), 5,35-5,36 (м, 1H), 5,89 (с, 1H), 7,21-7,45 (м, 7H), 8,86 (шир., 1H).
ESI-МС:m/z=535(M+H)+.
[0218]
(Пример 56) Синтез 1-(2-(3-метилизоксазол-5-ил)ацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида:
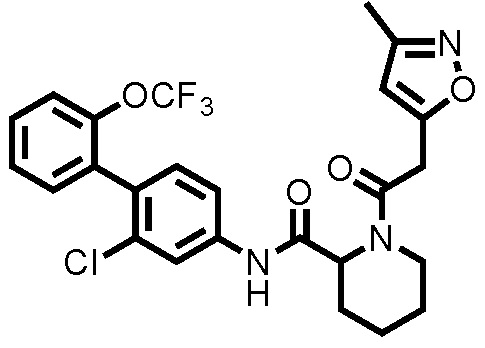
Следуя методике, аналогичной примеру 4, за исключением того, что 3-метил-5-изоксазолуксусную кислоту использовали вместо 2-метоксиуксусной кислоты, 1-(2-(3-метилизоксазол-5-ил)ацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 56) (0,0652 г, 0,125 ммоль, 99,6%) получали в виде белого амофного вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,47-1,96 (м, 5H), 2,29 (с, 3H), 2,33-2,39 (м, 1H), 3,21-3,28 (м, 1H), 3,82-3,87 (м, 1H), 3,95 (д, J=16,1 Гц, 1H), 4,01 (д, J=16,1 Гц, 1H), 5,32-5,33 (м, 1H), 6,08 (с, 1H), 7,21-7,45 (м, 7H), 8,27 (шир., 1H).
ESI-МС:m/z=520(M-H)-.
[0219]
(Пример 57) Синтез (R)-1-(2-(1H-тетразол-1-ил)ацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида:
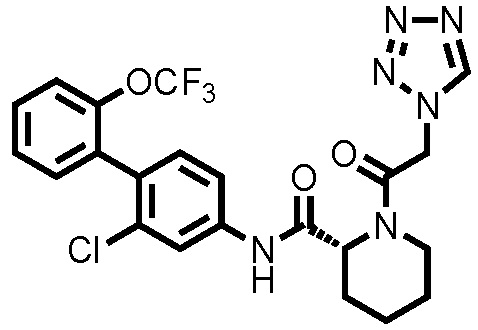
К ДМФА раствору (30 мл) 1H-тетразол-1-уксусную кислоту (1,67 г, 13,04 ммоль) при комнатной температуре добавляли ДМФА раствор (10 мл) соединения ссылочного примера 8 (4,00 г, 10,03 ммоль), HATU (4,96 г, 13,04 ммоль) и диизопропилэтиламин (2,63 мл, 15,04 ммоль), с последующим перемешиванием при этой же температуре в течение 17 часов. К полученному реакционному раствору добавляли дистиллированную воду, и полученный раствор экстрагировали толуолом. Органический слой концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией (амино-силикагель, н-гексан/этилацетат=40/60 до 0/100), с получением (R)-1-(2-(1H-тетразол-1-ил)ацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида (здесь и далее называемого соединением примера 57) (3,87 г, 7,60 ммоль, 75,9%) в виде белого аморфного вещества. По результатам анализа с использованием хиральной колонки, время удерживания полученного таким образом соединения примера 57 составляло 55,3 минут, и оптическая чистота при таком времени составляла 99,4% ee. Условия анализа с использованием хиральной колонки были следующими.
Измерительное оборудование: Высокоэффективный жидкостной хроматограф LC-2010CHT, производства фирмы Shimadzu Corporation
Колонка: CHIRALCEL OD-RH 0,46 см × 15 см, размер частиц 5 мкм, производства фирмы Daicel Chemical Industries Ltd.
Температура колонки: 40°C
Подвижная фаза: (Раствор A) дистиллированная вода, (Раствор B) ацетонитрил
Состав подвижной фазы: Раствор A:Раствор B=60:40 (0 до 75 минут)
Скорость потока: 0,5 мл/минута
Детекция: УФ (210 нм)
1H-ЯМР (400 МГц, CDCl3) δ: 1,62-2,00 (м, 5H), 2,27-2,31 (м, 1H), 3,52-3,58 (м, 1H), 3,73-3,76 (м, 1H), 5,18-5,19 (м, 1H), 5,40 (д, J=16,5 Гц, 1H), 5,48 (д, J=16,5 Гц, 1H), 7,21-7,45 (м, 6H), 7,81 (шир., 1H), 8,15 (с, 1H), 8,86 (с, 1H).
ESI-МС:m/z=509(M+H)+.
[0220]
(Пример 58) Синтез (R)-1-(3-(1H-тетразол-1-ил)пропаноил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида:
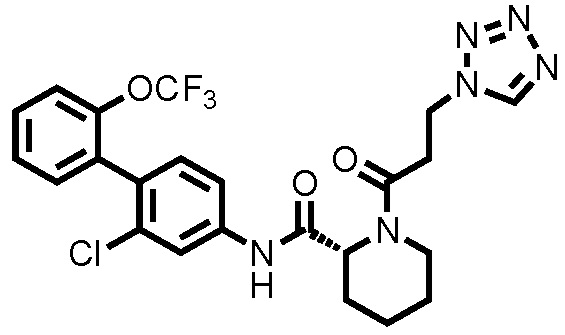
Следуя методике, аналогичной примеру 4, за исключением того, что 3-(тетразол-1-ил)пропионовую кислоту использовали вместо 2-метоксиуксусной кислоты, и соединение ссылочного примера 8 использовали вместо соединения ссылочного примера 3, (R)-1-(3-(1H-тетразол-1-ил)пропаноил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 58) (0,117 г, 0,224 ммоль, 89,1%) получали в виде белого амофного вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,43-1,97 (м, 5H), 2,25-2,29 (м, 1H), 3,04-3,19 (м, 2H), 3,25 (тд, J=13,0, 2,7 Гц, 1H), 3,70-3,74 (м, 1H), 4,79-4,92 (м, 2H), 5,18-5,19 (м, 1H), 7,22-7,44 (м, 6H), 7,79 (шир., 1H), 8,13 (шир., 1H), 8,84 (с, 1H).
ESI-МС:m/z=521(M-H)-.
[0221]
(Пример 59) Синтез (R)-1-(3-(1H-имидазол-1-ил)пропаноил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида:
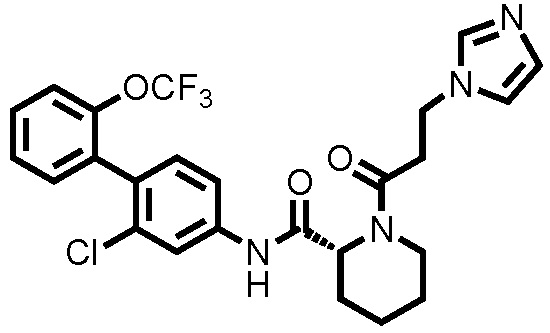
Следуя методике, аналогичной примеру 4, за исключением того, что 3-(имидазол-1-ил)пропионовую кислоту использовали вместо 2-метоксиуксусной кислоты, и соединение ссылочного примера 8 использовали вместо соединения ссылочного примера 3, (R)-1-(3-(1H-имидазол-1-ил)пропаноил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 59) (0,0796 г, 0,153 ммоль, 60,9%) получали в виде белого амофного вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,37-1,99 (м, 5H), 2,26-2,29 (м, 1H), 2,87-2,91 (м, 2H), 3,17 (тд, J=13,3, 2,7 Гц, 1H), 3,67-3,71 (м, 1H), 4,34-4,47 (м, 2H), 5,24-5,25 (м, 1H), 6,99 (с, 1H), 7,06 (с, 1H), 7,21-7,45 (м, 6H), 7,58 (шир., 1H), 7,71-7,82 (м, 1H), 8,33 (шир., 1H).
ESI-МС:m/z=521(M+H)+.
[0222]
(Пример 60) Синтез (R)-1-(3-(3-метил-1H-пиразол-1-ил)пропаноил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида:
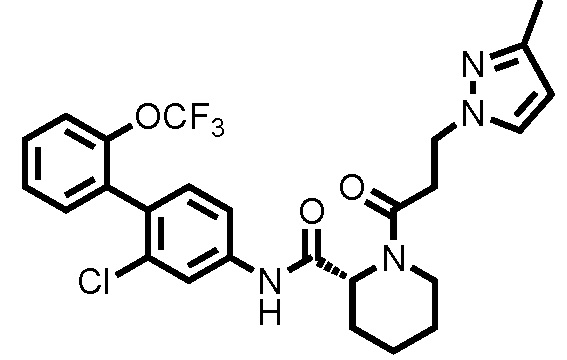
Следуя методике, аналогичной примеру 4, за исключением того, что 3-(3-метил-пиразол-1-ил)пропионовую кислоту использовали вместо 2-метоксиуксусной кислоты, и соединение ссылочного примера 8 использовали вместо соединения ссылочного примера 3, (R)-1-(3-(3-метил-1H-пиразол-1-ил)пропаноил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 60) (0,135 г, 0,252 ммоль, количественный) получали в виде белого аморфного вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,37-1,89 (м, 5H), 2,12 (с, 3H), 2,35-2,38 (м, 1H), 2,85-3,12 (м, 3H), 3,67-3,70 (м, 1H), 4,38-4,44 (м, 1H), 4,51-4,58 (м, 1H), 5,30-5,32 (м, 1H), 5,96-5,97 (м, 1H), 7,19-7,45 (м, 8H), 8,59 (шир., 1H).
ESI-МС:m/z=535(M+H)+.
[0223]
(Пример 61) Синтез (R)-1-(2-(1H-пиразол-1-ил)ацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида:
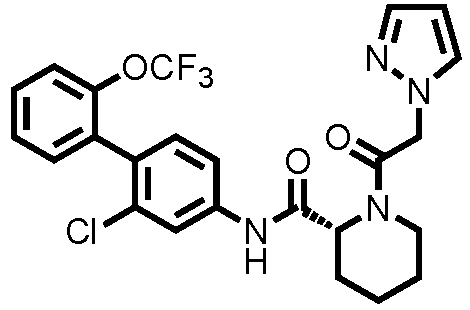
Следуя методике, аналогичной примеру 4, за исключением того, что 2-(1H-пиразол-1-ил)уксусную кислоту использовали вместо 2-метоксиуксусной кислоты, и соединение ссылочного примера 8 использовали вместо соединения ссылочного примера 3, (R)-1-(2-(1H-пиразол-1-ил)ацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 61) (0,0623 г, 0,123 ммоль, 98,0%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,46-1,74 (м, 5H), 2,52-2,55 (м, 1H), 3,04-3,12 (м, 1H), 3,65-3,69 (м, 1H), 5,01 (д, J=14,5 Гц, 1H), 5,22 (д, J=14,5 Гц, 1H), 5,43-5,44 (м, 1H), 6,39 (дд, J=2,3,2,0 Гц, 1H), 7,24 (д, J=8,2 Гц, 1H), 7,32-7,37 (м, 3H), 7,41-7,45 (м, 1H), 7,52-7,61 (м, 1H), 7,53 (д, J=2,3 Гц, 1H), 7,58 (д, J=2,0 Гц, 1H), 7,80-7,87 (шир.м, 1H), 9,02 (с, 1H).
ESI-МС:m/z=507(M+H)+.
[0224]
(Пример 62) Синтез (R)-1-(2-(4H-1,2,4-триазол-4-ил)ацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида:
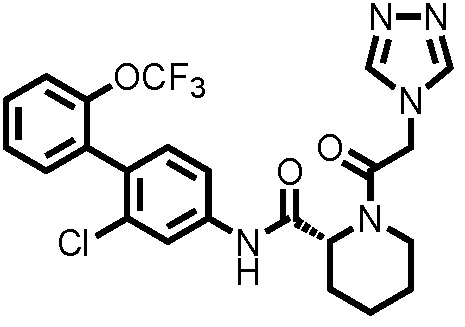
Следуя методике, аналогичной примеру 4, за исключением того, что 2-(4H-1,2,4-триазол-4-ил)уксусную кислоту использовали вместо 2-метоксиуксусной кислоты, и соединение ссылочного примера 8 использовали вместо соединения ссылочного примера 3, (R)-1-(2-(4H-1,2,4-триазол-4-ил)ацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 62) (0,0575 г, 0,113 ммоль, 90,3%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,55-1,90 (м, 5H), 2,29-2,32 (м, 1H), 3,57-3,71 (м, 2H), 4,93 (д, J=16,8 Гц, 1H), 5,02 (д, J=16,8 Гц, 1H), 5,22-5,23 (м, 1H), 7,19 (д, J=8,2 Гц, 1H), 7,28-7,37 (м, 4H), 7,40-7,45 (м, 1H), 7,81 (с, 1H), 8,21 (с, 2H), 8,84 (с, 1H).
ESI-МС:m/z=508(M+H)+.
[0225]
(Пример 63) Синтез (R)-1-(2-(1H-1,2,4-триазол-1-ил)ацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида:
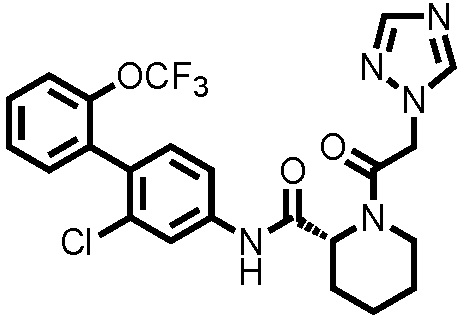
Следуя методике, аналогичной примеру 4, за исключением того, что 2-(1H-1,2,4-триазол-1-ил)ацетат натрия использовали вместо 2-метоксиуксусной кислоты, и соединение ссылочного примера 8 использовали вместо соединения ссылочного примера 3, (R)-1-(2-(1H-1,2,4-триазол-1-ил)ацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 63) (0,0587 г, 0,116 ммоль, 92,2%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,54-1,70 (м, 2H), 1,77-1,90 (м, 3H), 2,38-2,41 (м, 1H), 3,31-3,39 (м, 1H), 3,74-3,78 (м, 1H), 5,13 (д, J=15,4 Гц, 1H), 5,22 (д, J=15,4 Гц, 1H), 5,29 (д, J=5,0 Гц, 1H), 7,23 (д, J=8,2 Гц, 1H), 7,30-7,37 (м, 4H), 7,41-7,45 (м, 1H), 7,79 (шир.с, 1H), 8,02 (с, 1H), 8,26 (с, 1H), 8,39 (с, 1H).
ESI-МС:m/z=508(M+H)+.
[0226]
(Пример 64) Синтез (R)-1-(2-(1H-1,2,3-триазол-1-ил)ацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида:
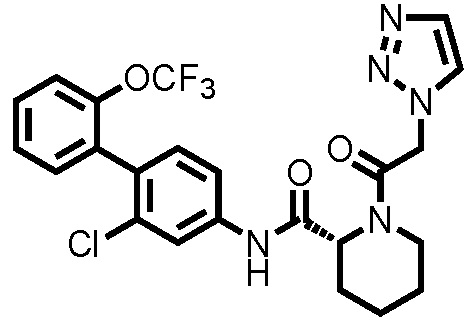
Следуя методике, аналогичной примеру 4, за исключением того, что 2-(1H-1,2,3-триазол-1-ил)уксусную кислоту использовали вместо 2-метоксиуксусной кислоты, и соединение ссылочного примера 8 использовали вместо соединения ссылочного примера 3, (R)-1-(2-(1H-1,2,3-триазол-1-ил)ацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 64) (0,0619 г, 0,122 ммоль, 97,2%) получали в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,51-1,70 (м, 2H), 1,76-1,88 (м, 3H), 2,39-2,42 (м, 1H), 3,32-3,39 (м, 1H), 3,74-3,78 (м, 1H), 5,30-5,31 (м, 1H), 5,34 (д, J=15,4 Гц, 1H), 5,41 (д, J=15,4 Гц, 1H), 7,23 (д, J=8,6 Гц, 1H), 7,30-7,37 (м, 3H), 7,41-7,45 (м, 1H), 7,52 (шир.с, 1H), 7,76 (д, J=0,9 Гц, 1H), 7,82 (д, J=0,9 Гц, 1H), 7,91 (шир.с, 1H), 8,43 (с, 1H).
ESI-МС:m/z=508(M+H)+.
[0227]
(Пример 65) Синтез (R)-1-(2-(1H-имидазол-1-ил)ацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида:
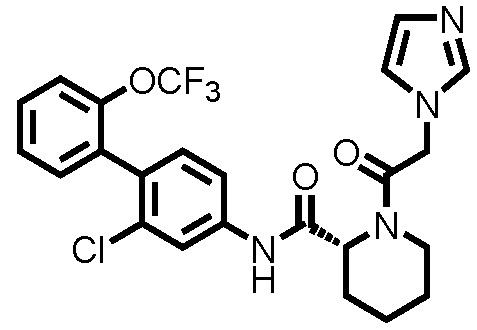
Следуя методике, аналогичной примеру 4, за исключением того, что 1-имидазолуксусную кислоту использовали вместо 2-метоксиуксусной кислоты, и соединение ссылочного примера 8 использовали вместо соединения ссылочного примера 3, (R)-1-(2-(1H-имидазол-1-ил)ацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 65) (0,635 г, 1,25 ммоль, 63,5%) получали в виде белого амофного вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,44-2,05 (м, 5H), 2,23-2,31 (м, 1H), 3,37-3,47 (м, 1H), 3,67-3,74 (м, 1H), 4,86 (д, J=16,6 Гц, 1H), 4,91 (д, J=16,6 Гц, 1H), 5,16-5,22 (м, 1H), 6,97 (с, 1H), 7,13 (с, 1H), 7,20 (д, J=8,3 Гц, 1H), 7,29-7,37 (м, 4H), 7,40-7,45 (м, 1H), 7,53 (с, 1H), 7,70-7,87 (м, 1H), 8,41 (шир.с, 1H).
ESI-МС:m/z=507(M+H)+.
[0228]
(Пример 66) Синтез (R)-1-(2-(2H-1,2,3-триазол-2-ил)ацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамида:
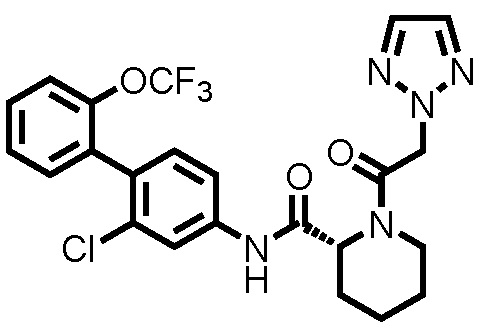
Следуя методике, аналогичной примеру 4, за исключением того, что 2-(2H-1,2,3-триазол-2-ил)уксусную кислоту использовали вместо 2-метоксиуксусной кислоты, и соединение ссылочного примера 8 использовали вместо соединения ссылочного примера 3, (R)-1-(2-(2H-1,2,3-триазол-2-ил)ацетил)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 66) (0,0321 г, 0,0632 ммоль, 50,4%) получали в виде белого амофного вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,47-1,65 (м, 2H), 1,68-1,77 (м, 3H), 2,49-2,52 (м, 1H), 2,98-3,06 (м, 1H), 3,55-3,58 (м, 1H), 5,33 (д, J=15,0 Гц, 1H), 5,42 (д, J=5,0 Гц, 1H), 5,57 (д, J=15,0 Гц, 1H), 7,24 (д, J=8,2 Гц, 1H), 7,32-7,37 (м, 3H), 7,41-7,46 (м, 1H), 7,42-7,88 (шир.м, 2H), 7,73 (с, 2H), 8,63 (с, 1H).
ESI-МС:m/z=530(M+Na)+.
[0229]
(Ссылочный пример 30) Синтез этил 2-(5-метил-1,3,4-оксадиазол-2-ил)ацетата:
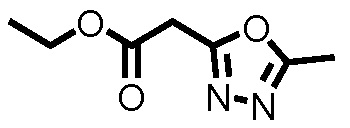
К дихлорметановому раствору (6,4 мл) этил 2-(1H-тетразол-5-ил)ацетата (0,500 г, 3,20 ммоль) при комнатной температуре добавляли уксусный ангидрид (0,393 мл, 4,16 ммоль), и температуру поднимали до 100°C, с последующим перемешиванием в течение 11 часов. К полученному реакционному раствору добавляли 1М водный раствор гидроксида натрия, и полученный раствор экстрагировали этилацетатом. Органический слой промывали 1М водным раствором гидроксида натрия и насыщенным раствором соли, сушили над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, н-гексан/этилацетат=70/30 до 40/60), с получением этил 2-(5-метил-1,3,4-оксадиазол-2-ил)ацетата (здесь и далее называемого соединением ссылочного примера 30) (0,0908 г, 0,534 ммоль, 16,7%) в виде бесцветного маслянистого продукта.
1H-ЯМР (400 МГц, CDCl3) δ: 1,29 (т, J=7,2 Гц, 3H), 2,55 (с, 3H), 3,92 (с, 2H), 4,23 (кв, J=7,2 Гц, 2H).
ESI-МС:m/z=171(M+H)+.
[0230]
(Ссылочный пример 31) Синтез 2-(5-метил-1,3,4-оксадиазол-2-ил)ацетата натрия:
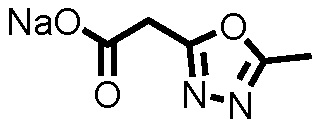
К тетрагидрофурановому раствору (1,0 мл) соединения ссылочного примера 30 (0,0900 г, 0,529 ммоль) при комнатной температуре добавляли 1М водный раствор гидроксида натрия (1,06 мл, 1,06 ммоль) и этанол (1,0 мл), с последующим перемешиванием при этой же температуре в течение 2 часов. Полученный реакционный раствор концентрировали при пониженном давлении, с получением неочищенного 2-(5-метил-1,3,4-оксадиазол-2-ил)ацетата натрия (здесь и далее называемого соединением ссылочного примера 31) (0,0835 г) в виде белого твердого вещества. Соединение ссылочного примера 31 использовали непосредственно для последующей реакции.
1H-ЯМР (400 МГц, ДМСО-D6) δ: 2,41 (с, 3H), 3,38 (с, 2H).
ESI-МС:m/z=143(M+H)+.
[0231]
(Пример 67) Синтез (R)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-(5-метил-1,3,4-оксадиазол-2-ил)ацетил)пиперидин-2-карбоксамида:
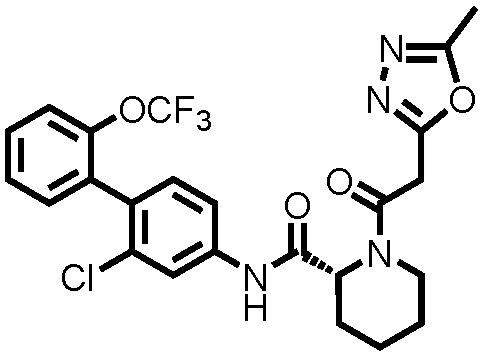
Следуя методике, аналогичной примеру 4, за исключением того, что соединение ссылочного примера 31 использовали вместо 2-метоксиуксусной кислоты, и соединение ссылочного примера 8 использовали вместо соединения ссылочного примера 3, (R)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(2-(5-метил-1,3,4-оксадиазол-2-ил)ацетил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 67) (0,0524 г, 0,0910 ммоль, 90,8%) получали в виде белого амофного вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,50-1,66 (м, 3H), 1,74-1,77 (м, 2H), 2,57 (с, 3H), 2,62-2,65 (м, 1H), 3,27-3,34 (м, 1H), 3,61-3,64 (м, 1H), 3,94 (д, J=17,4 Гц, 1H), 4,21 (д, J=17,4 Гц, 1H), 5,54-5,55 (м, 1H), 7,25-7,27 (м, 1H), 7,33-7,36 (м, 3H), 7,40-7,44 (м, 1H), 7,77 (шир.с, 1H), 8,18 (шир.с, 1H), 9,38 (с, 1H).
ESI-МС:m/z=545(M+Na)+.
[0232]
(Пример 68) Синтез (R)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(3-(диметиламино)пропаноил)пиперидин-2-карбоксамида:
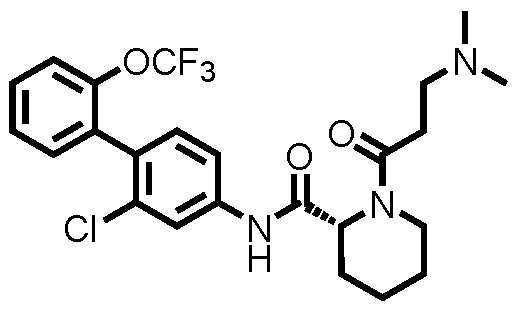
Следуя методике, аналогичной примеру 4, за исключением того, что гидрохлорид 3-(диметиламино)проионовой кислоты использовали вместо 2-метоксиуксусной кислоты, и соединение ссылочного примера 8 использовали вместо соединения ссылочного примера 3, (R)-N-(2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)-1-(3-(диметиламино)пропаноил)пиперидин-2-карбоксамид (здесь и далее называемый соединением примера 68) (0,0826 г, 0,166 ммоль, 66,2%) получали в виде белого амофного вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,46-1,83 (м, 5H), 2,26 (с, 0,6H), 2,28 (с, 5,4H), 2,40-2,44 (м, 1H), 2,55-2,62 (м, 1H), 2,65-2,81 (м, 3H), 2,99-3,05 (м, 0,1H), 3,13-3,20 (м, 0,9H), 3,88-3,91 (м, 0,9H), 4,69 (д, J=5,0 Гц, 0,1H), 4,73-4,76 (м, 0,1H), 5,43 (д, J=5,0 Гц, 0,9H), 7,21 (д, J=8,6 Гц, 1H), 7,31-7,86 (м, 6H), 8,76 (шир., 0,9H), 9,33 (шир., 0,1H).
ESI-МС:m/z=498(M+H)+.
[0233]
(Пример 69) Синтез метил (R)-5-(2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)пиперидин-1-ил)-5-оксопентаноата:
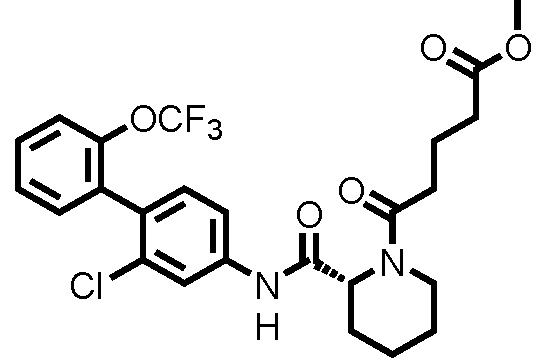
Следуя методике, аналогичной примеру 3, за исключением того, что метил 4-(хлороформил)бутират использовали вместо пропионилхлорида, и соединение ссылочного примера 8 использовали вместо соединения ссылочного примера 3, метил (R)-5-(2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)пиперидин-1-ил)-5-оксопентаноат (здесь и далее называемый соединением примера 69) (0,130 г, 0,247 ммоль, 98,4%) получали в виде белого амофного вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,44-1,79 (м, 4H), 1,84-1,95 (м, 1H), 1,96-2,13 (м, 2H), 2,35 (д, J=13,7 Гц, 1H), 2,40-2,62 (м, 4H), 2,63-2,70 (м, 0,1H), 3,14-3,21 (м, 0,9H), 3,68 (с, 2,7H), 3,69 (с, 0,3H), 3,84-3,88 (м, 0,9H), 4,66-4,69 (м, 0,2H), 5,34 (д, J=5,0 Гц, 0,9H), 7,20 (д, J=8,2 Гц, 1H), 7,29-7,94 (м, 6H), 8,68 (с, 0,9H), 8,90 (с, 0,1H).
[0234]
(Пример 70) Синтез (R)-5-(2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)пиперидин-1-ил)-5-оксопентановой кислоты:
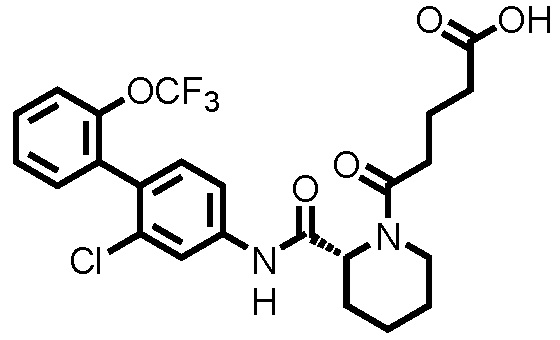
К метанольному раствору (2,5 мл) соединения примера 69 (0,130 г, 0,247 ммоль) при 0°C добавляли 1М водный раствор гидроксида натрия (2,47 мл, 2,47 ммоль) и тетрагидрофуран (2,5 мл), и температуру поднимали до комнатной температуры, с последующим перемешиванием в течение 5 часов. К полученному реакционному растворудобавляли 1M хлористоводородную кислоту, и полученный раствор экстрагировали хлороформом. Органический слой промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (силикагель, хлороформ/метанол=98/2 до 90/10), с получением (R)-5-(2-((2-хлор-2'-(трифторметокси)-[1,1'-бифенил]-4-ил)карбамоил)пиперидин-1-ил)-5-оксопентановой кислоты (здесь и далее называемой соединением примера 70) (0,0592 г, 0,115 ммоль, 46,7%) в виде белого аморфного вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,49-1,79 (м, 4H), 1,84-1,94 (м, 1H), 2,00-2,10 (м, 2H), 2,32 (д, J=13,6 Гц, 1H), 2,44-2,63 (м, 4H), 3,20 (тд, J=13,3, 2,6 Гц, 1H), 3,81-3,87 (м, 1H), 5,30 (д, J=4,5 Гц, 1H), 7,20 (д, J=8,6 Гц, 1H), 7,29-7,36 (м, 3H), 7,40-7,45 (м, 2H), 7,75-7,88 (м, 1H), 8,62 (с, 1H).
ESI-МС:m/z=535(M+Na)+.
[0235]
(Пример 71) Ингибирующий эффект на связывание RORγ-коактиватор
Ингибирующий эффект производного циклического амина (I) или его фармацевтически приемлемой соли на связывание между лиганд-связывающим доменом RORγ (здесь и далее называемым RORγ-LBD) и коактиватором оценивали с использованием набора для инвитрогенного анализа LanthaScreenTM TR-FRET гамма коактиватора ретиноидного орфанного рецептора (ROR), основанного на резонансном переносе энергии флюоресценции с временным разрешением (TR-FRET).
[0236]
Тестируемое соединение растворяли в ДМСО и разводили TR-FRET корегуляторным буфером D (Invitrogen), содержащим 5 ммоль/л DTT, таким образом, чтобы конечная концентрация ДМСО составляла 1% перед использованием. В каждую лунку 384-луночного черного планшета (Corning Inc.) добавляли 4 нмоль/л GST-слитого RORγ-LBD (Invitrogen) разведенного буфером, приведенным выше, и тестируемое соединение. Получали лунки без добавления тестируемого соединения и без добавления GST-слитого RORγ-LBD (фоновый), и лунки без добавления тестируемого соединения и с добавлением GST-слитого RORγ-LBD (контроль). Затем в каждую лунку добавляли 150 нмоль/л меченного флуоресцеином TRAP220/DRIP-2 (Invitrogen), разведенного буфером, приведенным выше, и в каждую лунку добавляли 32 нмоль/л меченного тербием анти-GST антитела (Invitrogen). После инкубации планшета при комнатной температуре в течение 16-24 часов, измеряли флуоресценцию при 495 нм и 520 нм при возбуждении в 320 нм для каждой лунки, и рассчитывали соотношение (значение флуоресценции при 520 нм/значение флуоресценции при 495 нм).
[0237]
Рассчитывали кратность изменения с добавлением тестируемого соединения (отношение с добавлением тестируемого соединения/отношение фоновое), кратность изменения контроля (отношение контроля/отношение фоновое) и кратность изменения фона (отношение фоновое/отношение фоновое), и затем скорость ингибирования связывания между RORγ-LBD и коактиватором (здесь и далее называемая скоростью ингибирования связывания RORγ-коактиватор) (%) рассчитывали по следующей формуле 1:
[Формула 1]
Скорость ингибирования связывания RORγ-коактиватор (%)=(1-((Кратность изменения с добавлением тестируемого соединения)-(Кратность фонового изменения))/((Кратность изменения контроля)-(Кратность фонового изменения)))×100
[0238]
Скорость ингибирования связывания RORγ-коактиватор (%) при 33 мкмоль/л тестируемого соединения показана в таблицах 2-1 и 2-2.
[0239]
[0240]
[0241]
Эти результаты показали, что производное циклического амина (I) или его фармацевтически приемлемая соль заметно тормозят связывание между RORγ-LBD и коактиватором.
[0242]
(Пример 72) Подавляющий эффект на продуцирование IL-17 в мышиных спленоцитах:
Используя мышиные спленоциты, подавляющий эффект производного циклического амина (I) или его фармацевтически приемлемой соли на продуцирование IL-17 при помощи стимуляции IL-23 оценивали частично модифицированным методом, приведенным в The Journal of Biological Chemistry, 2003, Vol. 278, No. 3, p. 1910-1914.
[0243]
Одноклеточную суспензию получали из селезенки мышей C57BL/6J (самцы, 7-23 недельного возраста) (Charles River Laboratories Japan, Inc.), и спленоциты получали, используя Histopaque-1083 (Sigma-Aldrich Japan). Использовали культуральную среду при добавлении 10% FBS (Gibco), 50 Ед/мл пенициллина.50 мкг/мл стрептомицина (Gibco), 50 мкмоль/л 2-меркаптоэтанола (Gibco) и 100 Ед/мл IL-2 человека (Cell Science & Technology Institute, Inc.) к среде RPMI1640 (Gibco). Тестируемое соединение растворяли в ДМСО и затем разводили полученной культуральной средой таким образом, чтобы конечная концентрация ДМСО перед использованием составляла 0,1%. Спленоциты (3×105 клеток/лунка), полученные в культуральной среде, высевали в лунки 96-луночного планшета с плоским дном (Corning Incorporated), туда добавляли тестируемое соединение и 10 нг/мл IL-23 человека (R & D systems, Inc.), и клетки культивировали при 37°C в атмосфере 5% CO2 в течение 3 дней. Получали лунки без добавления IL-23 человека и без добавления тестируемого соединения, и лунки с добавлением IL-23 человека и без добавления тестируемого соединения. После завершения культивирования, культуральный супернатант собирали, и определяли количество продуцированного IL-17 в супернатанте методом ELISA (R & D systems, Inc.).
[0244]
Ингибирование скорости продуцирования IL-17 (%) рассчитывали по следующей формуле 2.
[Формула 2]
Ингибирование скорости продуцирования IL-17 (%)=(1-((количество продуцированного IL-17 с добавлением IL-23 и с добавлением тестируемого соединения)-(количество продуцированного IL-17 без добавления IL-23 и без добавления тестируемого соединения))/((количество продуцированного IL-17 с добавлением IL-23 и без добавления тестируемого соединения)-(количество продуцированного IL-17 без добавления IL-23 и без добавления тестируемого соединения)))×100
[0245]
Ингибирование скорости продуцирования IL-17 (%) при 5 мкмоль/л тестируемого соединения показано в таблицах 3-1 и 3-2.
[0246]
[0247]
[0248]
Эти результаты показали, что производное циклического амина (I) или его фармацевтически приемлемая соль подавляют продуцирование IL-17.
[0249]
(Пример 73) Проявление подавляющего эффекта на экспериментальной мышиной модели аутоиммунного энцефаломиелита:
Используя увеличение неврологической симптоматики в качестве показателя обострения симптомов, эффект производного циклического амина (I) или его фармацевтически приемлемой соли оценивали на экспериментальной мышиной модели аутоиммунного энцефаломиелита. Экспериментальную мышиную модель аутоиммунного энцефаломиелита получали модифицированным способом, описанным Hindinger et al. (Journal of Neuroscience Research, 2006, Vol. 84, p. 1225-1234).
[0250]
В общей сложности 0,1 мл (по 0,05 мл на каждую из сторон) вводимого раствора MOG35-55, полученного смешиванием PBS раствора, содержащего полусинтетические пептидные миелиновые олигодендроцитарные гликопротеины (MOG35-55; CSBio, Inc.) в концентрации 4 мг/мл, с полным адъювантом Фрейнда в на равном количестве, внутрикожно инокулировали в боковую и подвздошную области живота мышей C57BL/6J (самцы, 8 недельного возраста, от Charles River Laboratories Japan, Inc.). Кроме того, 200 мкл коклюшного токсина (Sigma-Aldrich Japan), полученного в концентрации 1 мкг/мл, внутрибрюшинно вводили мышам в день инокуляции и через 2 дня после инокуляции вводимого раствора MOG35-55.
[0251]
После инокуляции вводимого раствора MOG35-55, мышам вводили тестируемое соединение. В качестве тестируемого соединения использовали соединение примера 20, соединение примера 29 и соединение примера 57. Соединение примера 20 суспендировали в 0,5% масс./об. метилцеллюлозном растворе и перорально вводили один раз в день при дозе 3 мг/кг каждый день из 2-х дней после инокуляции вводимого раствора MOG35-55. Соединение примера 29 и соединение примера 57 суспендировали в 0,5% масс./об. метилцеллюлозном растворе и перорально вводили один раз в день при дозе 1 мг/кг каждый день из 13 дней после инокуляции вводимого раствора MOG35-55. Группу мышей, которым вводили соединение примера 20, определяли как группу введения соединения примера-20, группу мышей, которым вводили соединение примера 29, определяли как группу введения соединения примера-29, и группу мышей, которым вводили соединение примера 57, определяли как группу введения соединения примера-57. В группе введения носителя, носитель каждого из тестируемых соединений (0,5% масс./об. метилцеллюлозный раствор) вводили аналогичным образом.
[0252]
После инокуляции вводимого раствора MOG35-55, оценивали неврологические симптомы в группах, которым вводили тестируемое соединение, и в группах, которым вводили носитель (0: нормально, 1: вялый хвост или слабая задняя конечность, 2: вялый хвост и слабая задняя конечность, 3: частичный паралич задних конечностей, 4: полный паралич задних конечностей, 5: агональное состояние). В качестве метода оценки использовали существующий в иммунологии протокол метода (John Wiley & Sons. Inc, 2000, p. 15.1.1-15.1.20).
[0253]
Результаты показаны на фиг.1, 2 и 3. На вертикальной оси указана оценка неврологического симптома (среднее±стандартная ошибка, n=8-9). ʺНосительʺ на горизонтальной оси обозначает группу введения носителя, ʺСоединение примера-20ʺ обозначает группу введения соединения примера-20, ʺСоединение примера-29ʺ обозначает группу введения соединения примера-29, и ʺСоединение примера-57ʺ обозначает группу введения соединения примера-57. На фиг.1 показана оценка неврологического симптома на 13-ый день после инокуляции вводимого раствора MOG35-55, на фиг.2 показана оценка неврологического симптома на 23-ий день после инокуляции вводимого раствора MOG35-55, и на фиг.3 показана оценка неврологического симптома на 30-ый день после инокуляции вводимого раствора MOG35-55. Знак звездочки (*) указывает статистическую значимость по сравнению с группой введения носителя (критерий Вилкоксона) (*: P<0,05).
[0254]
Инокуляция вводимого раствора MOG35-55 увеличивала показатель неврологических симптомов в группе введения носителя до 1,0-2,4. Такое увеличение показателя неврологической симптоматики статистически значимо подавлялось введением соединения примера 20, соединения примера 29 или соединения примера 57.
[0255]
Эти результаты показали, что производное циклического амина (I) или его фармацевтически приемлемая соль проявляют заметный подавляющий эффект на рассеянный склероз.
[0256]
(Пример 74) Проявление подавляющего эффекта на мышиной модели имиквимод-индуцированного псориаза:
Используя увеличение толщины уха в качестве показателя обострения симптомов, эффект производного циклического амина (I) или его фармацевтически приемлемой соли оценивали на мышиной модели индуцированного имиквимодом псориаза. Мышиную модель имиквимод-индуцированного псориаза получали частично модифицированным способом, описанным Schaper et al. (The Journal of Dermatological Science, 2013, Vol. 71, No. 1, p. 29-36).
[0257]
BALB/c мышей (самцы, 7 недельного возраста, от Charles River Laboratories Japan, Inc.) использовали в 8 недельном возрасте после предварительного скрещивания. Для индуцирования псориаз-подобных симптомов мышам наносили по 5 мг на каждую сторону правой и левой ушных раковин 5% крем BESELNA один раз в день в течение 8 дней от дня первого введения имиквимода (здесь и далее называемого днем индуцирования) до 7 дня после индуцирования (доза имиквимода, 0,5 мг/тела/день).
[0258]
Тестируемое соединение при дозе 10 мг/кг мышам вводили раз в день в течение 5 дней от 3 дня после индуцирования до 7 дня после индуцирования. В качестве тестируемого соединение использовали соединение примера 20, соединение примера 29 и соединение примера 57. Соединение примера 20, соединение примера 29 и соединение примера 57 суспендировали в 0,5% масс./об. метилцеллюлозном растворе и вводили перорально. Группу мышей, которым вводили соединение примера 20, определяли как группу введения соединения примера-20, группу мышей, которым вводили соединение примера 29, определяли как группу введения соединения примера-29, и группу мышей, которым вводили соединение примера 57, определяли как группу введения соединения примера-57. В группе введения носителя, носитель каждого из тестируемых соединений (0,5% масс./об. метилцеллюлозный раствор) вводили аналогичным образом.
[0259]
Толщину правого и левого уха измеряли перед введением имиквимода (перед индуцированием) в день индуцирования, и толщину правого и левого уха измеряли на 8-ой день после индуцирования при помощи цифрового микрометра (Mitutoyo Corporation). Среднюю толщину правого и левого уха рассматривали как толщину уха, и изменение толщины уха (толщина уха на 8-ой день после индуцирования - толщина уха перед индуцированием) использовали в качестве показателя оценки эффективности лекарственного средства.
[0260]
Результаты показаны на фиг.4, 5 и 6. На вертикальной оси представлено изменение толщины уха (мм) (среднее±стандартная ошибка, n=6). ʺНосительʺ на горизонтальной оси обозначает группу введения носителя, ʺСоединение примера-20ʺ обозначает группу введения соединения примера-20, ʺСоединение примера-29ʺ обозначает группу введения соединения примера-29, и ʺСоединение примера-57ʺ обозначает группу введения соединения примера-57. Знак звездочки (*) указывает статистическую значимость по сравнению с группой введения носителя (T критерий Стьюдента) (*: P<0,05).
[0261]
Индицирование имиквимодом увеличивает толщину уха на 8-ой день после индуцирования в группе введения носителя на 0,23 мм до 0,27 мм по сравнению с толщиной уха перед индуцированием. Такое увеличение толщины уха статистически значимо подавлялось введением соединения примера 20, соединения примера 29 или соединения примера 57.
[0262]
Эти результаты показали, что производное циклического амина (I) или его фармацевтически приемлемая соль проявляют заметный подавляющий эффект на псориаз.
[0263]
(Пример 75) Проявление подавляющего эффекта на мышиной модели DNFB-индуцированного аллергического дерматита:
Используя увеличение скорости отека уха в качестве показателя обострения симптомов, эффект производного циклического амина (I) или его фармацевтически приемлемой соли оценивали на мышиной модели DNFB-индуцированного аллергического дерматита. Мышиную модель DNFB-индуцированного аллергического дерматита получали частично модифицированным способом, описанным Curzytek et al. (Pharmacological Reports, 2013, Vol. 65, p. 1237-1246).
[0264]
BALB/c мышей (самки, 6 недельного возраста, от Charles River Laboratories Japan, Inc.) использовали в 7 недельном возрасте после предварительного скрещивания. В заднюю часть мышей вводили 25 мкл 0,5% об./об. раствора DNFB в смеси ацетон:оливковое масло (4:1). На следующий день, такую же самую операцию повторяли для сенсибилизации мышей. На четвертый день после сенсибилизации на обе стороны правой ушной раковины сенсибилизированных мышей наносили по 10 мкл на каждую сторону 0,2% об./об. раствора DNFB в смеси ацетон:оливковое масло (4:1), чтобы вызвать воспаление.
[0265]
За один день перед индуцированием мышам вводили соединение примера 20, соединение примера 29 или соединение примера 57 в дозе 10 мг/кг мышам. Соединение примера 20, соединение примера 29 и соединение примера 57 суспендировали в 0,5% масс./об. метилцеллюлозном растворе и вводили перорально. Группу мышей, которым вводили соединение примера 20, определяли как группу введения соединения примера-20, группу мышей, которым вводили соединение примера 29, определяли как группу введения соединения примера-29, и группу мышей, которым вводили соединение примера 57, определяли как группу введения соединения примера-57. В группе введения носителя, носитель каждого из тестируемых соединение (0,5% масс./об. метилцеллюлозном растворе) вводили аналогичным образом.
[0266]
Толщину правого уха перед нанесением раствора DNFB (перед индуцированием) в день индуцирования и толщину правого уха через 24 часа после индуцирования измеряли при помощи цифрового микрометра (Mitutoyo Corporation). Скорость отека ушной раковины рассчитывали по следующей формуле 3 и использовали в качестве показателя оценки эффективности лекарственного средства.
[Формула 3]
Скорость отека ушной раковины (%)=((Толщина правого уха через 24 часа после индуцирования)-(Толщина правого уха перед индуцированием))/Толщина правого уха перед индуцированием×100
[0267]
Результаты показаны на фиг.7, 8 и 9. На вертикальной оси указана скорость отека ушной раковины (%) (среднее±стандартная ошибка, n=6-8). ʺНосительʺ на горизонтальной оси обозначает группу введения носителя, ʺСоединение примера-20ʺ обозначает группу введения соединения примера-20, ʺСоединение примера-29ʺ обозначает группу введения соединения примера-29, и ʺСоединение примера-57ʺ обозначает группу введения соединения примера-57. Знак звездочки (*) указывает статистическую значимость по сравнению с группой введения носителя (T критерий Стьюдента) (*: P<0,05).
[0268]
Нанесение раствора DNFB на ушную раковину увеличивало скорость отека ушной раковины в группе введения носителя на 30,9% до 41,7%. Такое увеличение скорости отека уха статистически значимо подавлялось введением соединения примера 20, соединения примера 29 или соединения примера 57.
[0269]
Эти результаты показали, что производное циклического амина (I) или его фармацевтически приемлемая соль проявляют заметный подавляющий эффект в отношении аллергического дерматита, в частности, на контактный дерматит.
[0270]
(Пример 76) Проявление подавляющего эффекта на мышиной модели оксазолон-индуцированного атопического дерматита:
Используя увеличение толщины уха в качестве показателя обострения симптомов, эффект производного циклического амина (I) или его фармацевтически приемлемой соли оценивали на мышиной модели индуцированного оксазолоном атопического дерматита. Мышиную модель оксазолон-индуцированного атопического дерматита получали частично модифицированным способом, описанным Nakajima et al. (Journal of Investigative Dermatology, 2014, Vol. 134, p. 2122-2130).
[0271]
BALB/c мышей (самки, 7 недельного возраста, Charles River Laboratories Japan, Inc.) использовали в 8 или 9 недельного возраста после предварительного скрещивания. В заднюю часть мышей вводили 25 мкл 3% масс./об. раствор оксазолона в этаноле для сенсибилизации мышей. Каждый день от 5 дня до 13 дня после сенсибилизации, на обе стороны правой ушной раковины сенсибилизированных мышей наносили по 10 мкл на каждую сторону 0,6% масс./об. раствора оксазолона в этаноле, чтобы вызвать воспаление.
[0272]
Тестируемое соединение при дозе 10 мг/кг мышам вводили раз в день в течение 15 дней от дня сенсибилизации до 14 дня после сенсибилизации. В качестве тестируемого соединение использовали соединение примера 20, соединение примера 29 и соединение примера 57. Соединение примера 20, соединение примера 29 и соединение примера 57 суспендировали в 0,5% масс./об. метилцеллюлозном растворе и вводили перорально. Группу мышей, которым вводили соединение примера 20, определяли как группу введения соединения примера-20, группу мышей, которым вводили соединение примера 29, определяли как группу введения соединения примера-29, и группу мышей, которым вводили соединение примера 57, определяли как группу введения соединения примера-57. В группе введения носителя, носитель каждого из тестируемых соединение (0,5% масс./об. метилцеллюлозном растворе) вводили аналогичным образом.
[0273]
Толщину правого уха перед нанесением раствора оксазолона (перед сенсибилизацией) на день сенсибилизации и толщину правого уха на следующий день конечного индицирования измеряли при помощи цифрового микрометра (Mitutoyo Corporation). Изменение толщины уха (толщина правого уха следующий день конечного индицирования - толщина правого уха перед сенсибилизацией) использовали в качестве показателя оценки эффективности лекарственного средства.
[0274]
Результаты показаны на фиг.10, 11 и 12. На вертикальной оси указано изменение толщины уха (мм) (среднее±стандартная ошибка, n=7). ʺНосительʺ на горизонтальной оси обозначает группу введения носителя, ʺСоединение примера-20ʺ обозначает группу введения соединения примера-20, ʺСоединение примера-29ʺ обозначает группу введения соединения примера-29, и ʺСоединение примера-57ʺ обозначает группу введения соединения примера-57. Знак звездочки (*) указывает статистическую значимость по сравнению с группой введения носителя (T критерий Стьюдента) (*: P<0,05).
[0275]
Нанесение раствора оксазолона на ушную раковину увеличивает толщину уха на следующий день конечного индицирования в группе введения носителя на 0,60 мм до 0,70 мм по сравнению с толщиной уха перед сенсибилизацией. Такое увеличение толщины уха статистически значимо подавлялось введением соединения примера 20, соединения примера 29 или соединения примера 57.
[0276]
Эти результаты показали, что производное циклического амина (I) или его фармацевтически приемлемая соль проявляют заметный подавляющий эффект на аллергический дерматит, в частности, атопический дерматит.
Промышленная применимость
[0277]
Поскольку производное циклического амина настоящего изобретения или его фармацевтически приемлемая соль обладают великолепной RORγ антагонистической активностью, они могут применяться в качестве лекарственного средства против заболеваний, при которых можно ожидать проявления улучшения патологического состояния или ремиссии, путем подавления функции RORγ. В частности, производное циклического амина настоящего изобретения или его фармацевтически приемлемая соль могут быть использованы в качестве терапевтического средства или профилактического средства против аутоиммунных заболеваний, таких как рассеянный склероз или псориаз, или аллергических заболеваний, включая аллергический дерматит или т.п., такой как контактный дерматит или атопический дерматит.
Реферат
Изобретение относится к производному циклического амина формулы (I)где Rпредставляет собой алкоксигруппу, имеющую 1-3 атома углерода, любые 1-3 атома водорода которой необязательно заменены атомом(ами) галогена; Rпредставляет собой атом галогена; Rпредставляет собой атом водорода, атом галогена или гидроксигруппу; Rпредставляет собой атом водорода или атом галогена; X представляет собой -C(=O)-(CH)-Rили -S(=O)-R; n равен целому числу от 0 до 5; Rпредставляет собой атом водорода, -OR, -SR, -S(=O)-R, -C(=O)-OR, -N(R)R, алкильную группу, имеющую 1-3 атома углерода, любые 1-3 атома водорода которой необязательно заменены атомом(ами) галогена, или 5-членную кольцевую гетероарильную группу, содержащую от 1 до 4 гетероатомов, независимо выбранных из группы, состоящей из атома азота, атома кислорода и атома серы, любой атом(ы) водорода которой необязательно заменен(ы) алкильной группой(ами), имеющей(ими) 1-3 атома углерода; Rпредставляет собой алкильную группу, имеющую 1-5 атомов углерода; Rпредставляет собой атом водорода или алкильную группу, имеющую 1-3 атома углерода, любые 1-3 атома водорода которой необязательно заменены атомом(ами) галогена; и Rпредставляет собой атом водорода, алкильную группу, имеющую 1-3 атома углерода, ацильную группу, имеющую 2-4 атома углерода, или алкилсульфонильную группу, имеющую 1-3 атома углерода, или его фармацевтически приемлемой соли, которое обладает антагонистической активностью в отношении ретиноидного орфанного рецептора γ и оказывает терапевтический эффект или профилактический эффект на аутоиммунные заболевания. 8 н. и 3 з.п. ф-лы,12 ил., 3 табл., 74 пр.
Формула
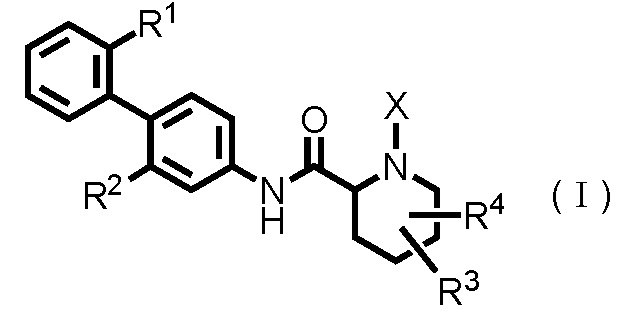
Документы, цитированные в отчёте о поиске
Гетероциклическое соединение
Комментарии