Способ получения промежуточного соединения для синтеза лекарственного средства - RU2741389C1
Код документа: RU2741389C1
Описание
ОБЛАСТЬ ТЕХНИКИ
Перекрестная ссылка на родственные заявки
В данной заявке заявлен приоритет к заявке на патент Кореи № 10-2017-0153334, поданной 16 ноября 2017, и заявке на патент Кореи № 10-2018-0126663, поданной 23 октября 2018, и полное содержание, описанное в заявках па патент Кореи, включено сюда в качестве ссылки.
Данное изобретение относится к способу получения химической формулы 1, которая является необходимым промежуточным соединением для синтеза противодиабетических средств, ингибирующих фермент дипептидилпептидазу IV (ниже также обозначенную ‘ДПП-IV’).
УРОВЕНЬ ТЕХНИКИ
Известно, что соединение, которое применяют в качестве противодиабетического средства, ингибирующего фермент дипептидилпептидазу IV (ДПП-IV), описано в публикации международной заявки на патент WO 12/030106 (см. соединение химической формулы 1 в публикации международной заявки на патент WO 12/030106), обладает превосходным ингибирующим действием на фермент ДПП-IV, и поэтому может эффективно применяться для лечения и профилактики заболеваний, вызванных ферментом, включая диабет, ожирение и т.д. Для получения такого ДПП-IV ингибирующего соединения в публикации международной заявки на патент WO 12/030106 описан способ получения его из соединения следующей химической формулы 1 в качестве необходимого промежуточного соединения.
[Химическая формула 1]
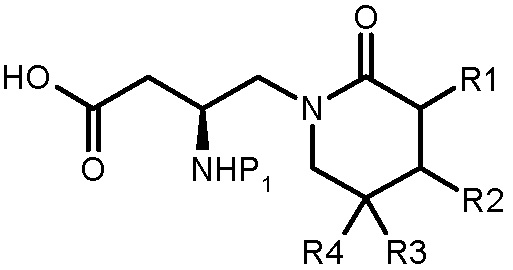
С другой стороны, ранее, для получения соединения химической формулы 1, защитную группу карбоновой кислоты (P2) соединения химической формулы 2 снимают для его сбора. Более конкретно, в случае соединения химической формулы 2, где защитной группой является бутилоксикарбонил (P1, Boc), и уходящей группой является трет-бутильная группа (P2), его получают (1) гидролизом защитной группы P2 с применением кислотных условий, более конкретно, сильных кислот, таких как серная кислота и т.д., и дихлорметана водного раствора гидроксида натрия и ди-трет-бутилдикарбоната (Boc2O) для снятия защиты, или (2) гидролизом защитной группы P2 с применением основных условий, более конкретно, водного раствора гидроксида натрия, этанола, условий кипячения воды с обратным холодильником для снятия защиты. В частности, если P2 является бензильной группой, метильной группой, этильной группой и изопропильной группой, применяют условия гидролиза с применением основания, указанного в (2), из двух вариантов условий.
[Химическая формула 2]
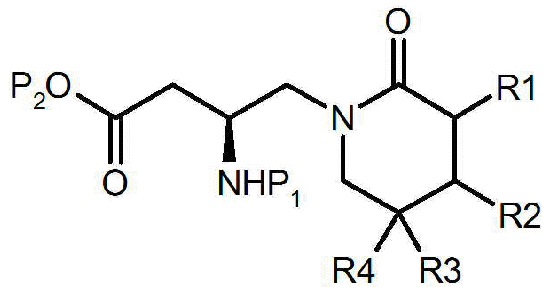
Однако такой способ получения соединения химической формулы 1 имеет недостатки, заключающиеся в том, что реакция проходит в немного тяжелых условиях и необходимо применять большое количество реакционного растворителя, и требуется дополнительный процесс концентрации.
ОПИСАНИЕ
ТЕХНИЧЕСКАЯ ПРОБЛЕМА
Следовательно, авторы данного изобретения провели интенсивные исследования для преодоления указанных выше недостатков известного уровня техники, и в результате подтвердили, что если в качестве основания применять конкретно гидроксид натрия в твердой форме, выход может быть значительно улучшен в мягких условиях, а экономическая осуществимость и производительность становятся высокими, поскольку способ становится экономичным за счет использования небольшого количества реакционного растворителя и отсутствия необходимости в дополнительном процессе концентрации.
Следовательно, объектом данного изобретения является обеспечение способа получения соединения химической формулы 1, которое является промежуточным соединением, применяемым для синтеза противодиабетических агентов, ингибирующих ДПП-IV, который является способом синтеза, имеющим высокую экономическую осуществимость и производительность, который может значительно улучшить выход даже при умеренных условиях, заключающихся в применении гидроксида натрия и основных условиях, и является экономичным через применение небольшого количества реакционного растворителя и отсутствие необходимости в дополнительном процессе концентрации, в отличие от обычных способов.
ТЕХНИЧЕСКОЕ РЕШЕНИЕ
В одном аспекте решения проблемы, данное изобретение относится к способу получения соединения химической формулы 1, и отличается тем, что способ получения включает стадию селективного снятия защиты защитной группы карбоновой кислоты (P2) из двух видов защитных групп соединения химической формулы 2, P1 и P2 защитных групп, и затем, снятие защиты проводят с применением основания в твердой форме и низшего спирта.
ПРЕИМУЩЕСТВЕННЫЕ ЭФФЕКТЫ
Способ получения в соответствии с данным изобретением очень полезен, так как он имеет преимущества, заключающиеся в 1) возможности получать соединение химической формулы 1, которое является промежуточным соединением для пероральных лекарственных средств для инсулин-независимого диабета, ингибирующих ДПП-IV, с высокой высоким выходом даже у умеренных условиях, 2) экономичности вследствие снижения количества применяемого реакционного растворителя для снижения производственных затрат, и 3) возможности достигать улучшения, такого как снижение, повышение производительности, благодаря отсутствию процесса концентрации.
СПОСОБ ПРОВЕДЕНИЯ ИЗОБРЕТЕНИЯ
Далее настоящее изобретение подробно описано на основе формулы реакции. Однако следующая формула реакции предназначена для содействия пониманию данного изобретения и не предназначена для ограничения данного изобретения в любом смысле.
Для объяснения способа получения в соответствии с данным изобретением представлена следующая формула реакции 1:
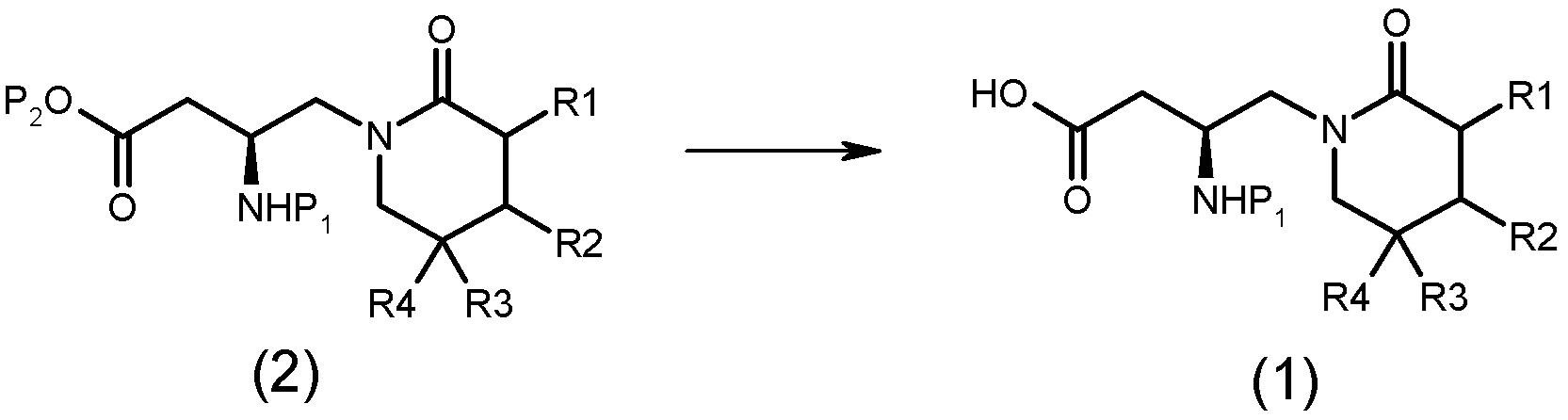
В формуле R1, R2, R3 и R4 независимо являются водородом, галогеном или замещенным или незамещенным C1-C4 алкилом, соответственно. P1 является защитной аминовой группой, и она является карбонильной группой, ацильной группой, сульфонильной группой, ацетильной группой или бензильной группой и, предпочтительно, P1 является Boc (бутилоксикарбонилом), Cbz (бензилоксикарбонилом) или Fmoc (9-флуоренилметилоксикарбонилом), более предпочтительно, Boc. P2 является защитной группой карбоновой кислоты и, предпочтительно, является бензильной группой, этильной группой, изопропильной группой или трет-бутильной группой, более предпочтительно, трет-бутильной группой.
В данном изобретении одной из особенностей изобретения является то, что основание в твердой форме применяют в качестве реакционного основания, в отличие от используемого ранее основания в водном растворе или жидкой форме, такой как водный раствор гидроксида натрия, и т.д., когда соединение химической формулы 1 собирают через снятие защиты защитной группы карбоновой кислоты (P2) в соединении химической формулы 2. Основанием в твердой форме, применяемым в соответствии с данным изобретением, может быть гидроксид натрия, гидроксид лития, гидроксид калия, гидроксид кальция или их сочетания, и, предпочтительно, соединение химической формулы 1 собирают с применением твердого гидроксида натрия.
Применяемое количество реакционного основания предпочтительно составляет от 1 эквивалента до 4 эквивалентов, предпочтительно, от 1 эквивалента до 2 эквивалентов, по отношению к соединению химической формулы 2.
Кроме того, в качестве реакционного растворителя, применяемого в реакции, применяют низший спирт, имеющий 1-6 атомов углерода и его смешанный растворитель. В частности, низшим спиртом, имеющим 1-6 атомов углерода, может быть один или более видов, выбранных из группы, состоящей из метилового спирта, этилового спирта, изопропилового спирта и их смешанного спирта (сорастворителя), и, предпочтительно, может применяться этиловый спирт. Количество используемого реакционного растворителя составляет от 1 раза (мл/г) до 7 раз (мл/г), предпочтительно, от 2 раз (мл/г) до 3 раз (мл/г) для соединения химической формулы 2. Реакционный растворитель в соответствии с данным изобретением отличается использованием небольшого количества реакционного растворителя, в отличие от способа получения химической формулы 1 в соответствии с обычными основными условиями.
В частности, температура реакции во время снятия защиты может различаться в соответствии с условиями реакции, но в случае данного изобретения, реакция может проводиться при температуре ниже температуры кипения с обратным холодильником, например, 30-80°С, благодаря техническим особенностям. Время реакции предпочтительно составляет от 1 часа до 6 часов, более предпочтительно, 3 часа и менее, но не ограничено им.
В качестве дополнительного аспекта, способ получения в соответствии с данным изобретением может дополнительно включать стадию кристаллизации соединения химической формулы 1, собранного, как описано выше. Растворителем для кристаллизации может быть один или более видов растворителей, выбранных из группы, состоящей из воды, метилового спирта, этилового спирта, изопропилового спирта и их смешанного растворителя (сорастворителя), но не ограничен ими, и предпочтительно им является вода или смешанный растворитель этилового спирта и воды. На стадии кристаллизации кристаллы могут быть получены путем регулирования рН с использованием кислоты, и рН предпочтительно составляет от 2,5 до 3,0.
Далее данное изобретение будет описано более подробно со ссылкой на примеры получения и примеры, но они предназначены для содействия пониманию настоящего изобретения, и объем данного изобретения никаким образом не ограничивается ими.
[Пример]
Пример 1: Синтез (3S)-3-трет-бутоксикарбониламино-4-(5,5-дифтор-2-оксопиперидино)бутановой кислоты

После добавления исходного материала, 462,3 кг трет-бутил (3S)-3-трет-бутоксикарбониламино-4-(5,5-дифтор-2-оксопиперидинобутаноата, 729,6 кг этилового спирта и 82,2 кг гидроксида натрия в реактор при комнатной температуре, температуру повышают в интервале 40~50°С для взаимодействия в течение 3 часов. После завершения реакции добавляют 3699 кг воды и 3N водный раствор хлористоводородной кислоты добавляют по каплям для контроля pH 2,5~3,0 и проводят кристаллизацию. Указанное в заголовке соединение получают в виде твердого вещества, фильтруют и промывают смешанным раствором воды и этилового спирта, трет-бутилметиловым эфиром и затем сушат с получением 347,6 кг указанного в заголовке соединения (содержание: 97,5%, выход: 85,5%).
1H ЯМР (500 МГц, ДМСО-d6) δ 1,32 (с, 9H), 2,20-2,43 (м, 6H), 3,26-3,31 (м, 2H), 3,61 (м, 1H), 3,81 (м, 1H), 4,02 (м, 1H), 6,73 (д, J=8,6 Гц, 1H), 12,16 (с, 1H).
Пример 2: Синтез (3S)-3-трет-Бутоксикарбониламино-4-(5,5-дифтор-2-оксопиперидино)бутановой кислоты

После добавления исходного материала, 412,2 кг трет-бутил (3S)-3-трет-бутоксикарбониламино-4-(5,5-дифтор-2-оксопиперидинобутаноата, 2049,0 кг этилового спирта и 299,7 кг 6N водного раствора гидроксида натрия в реактор, температуру повышают до кипения реакционной смеси с обратным холодильником. После завершения реакции смесь концентрируют и добавляют 1649 кг воды для ее растворения. Водный слой промывают 1221,8 кг трет-бутилметилового эфира, и 3N водный раствор хлористоводородной кислоты добавляют по каплям для контроля pH 3,0~3,5, и проводят кристаллизацию. Указанное в заголовке соединение, полученное в виде твердого вещества, фильтруют и промывают водой и трет-бутилметиловым эфиром и затем сушат с получением 309,8 кг указанного в заголовке соединения (содержание: 97,4%, выход: 85,4%).
Пример 3: Синтез (3S)-3-трет-Бутоксикарбониламино-4-(5,5-дифтор-2-оксопиперидино)бутановой кислоты
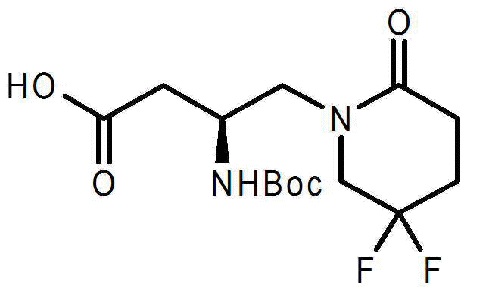
После добавления исходного материала, 449,2 кг трет-бутил (3S)-3-трет-бутоксикарбониламино-4-(5,5-дифтор-2-оксопиперидинобутаноата, 2033,0 кг этилового спирта и 361,9 кг 6N водного раствора гидроксида натрия, температуру повышают до кипения реакционной смеси с обратным холодильником. После завершения реакции смесь концентрируют и добавляют 1796,9 кг воды для ее растворения. Добавляют 354,4 кг этилового спирта и 3N водный раствор хлористоводородной кислоты добавляют по каплям для контроля pH, первоначально, 4,1~5,0, и потом 2,5~3,0, и проводят кристаллизацию. Указанное в заголовке соединение, полученное в виде твердого вещества, фильтруют и промывают смешанным раствором воды и этилового спирта и затем сушат с получением 325,0 кг указанного в заголовке соединения (содержание: 95,5%, выход: 80,6%).
Пример 4: Синтез (3S)-3-трет-Бутоксикарбониламино-4-(5,5-дифтор-2-оксопиперидино)бутановой кислоты
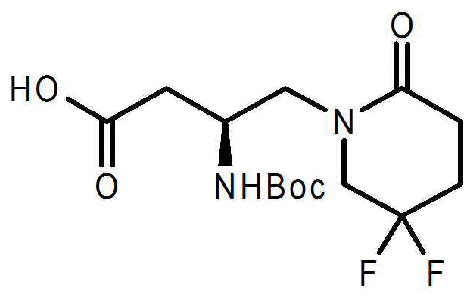
После добавления исходного материала, 43,0 г трет-бутил (3S)-3-трет-бутоксикарбониламино-4-(5,5-дифтор-2-оксопиперидинобутаноата, 213,7 г этилового спирта и 8,8 г 6N водного раствора гидроксида натрия, температуру повышают до кипения реакционной смеси с обратным холодильником. После завершения реакции смесь концентрируют и добавляют 172,0 г воды для ее растворения. Добавляют 33,9 г этилового спирта и 3N водный раствор хлористоводородной кислоты добавляют по каплям для контроля pH, первоначально, 4,1~5,0, и потом 2,5~3,0, и проводят кристаллизацию. Указанное в заголовке соединение, полученное в виде твердого вещества, фильтруют и промывают смешанным раствором воды и этилового спирта и затем сушат с получением 35,5 г указанного в заголовке соединения (содержание: 93,0%, выход: 89,6%).
Пример 5: Синтез (3S)-3-трет-Бутоксикарбониламино-4-(5,5-дифтор-2-оксопиперидино)бутановой кислоты

После добавления исходного материала, 43,0 г трет-бутил (3S)-3-трет-бутоксикарбониламино-4-(5,5-дифтор-2-оксопиперидинобутаноата, 67,9 г этилового спирта и 8,8 г 6N водного раствора гидроксида натрия, температуру повышают до 70°С для реакции. После завершения реакции смесь охлаждают и добавляют 344,0 г воды. Добавляют по каплям 3N водный раствор хлористоводородной кислоты для контроля pH, первоначально, 4,1~5,0, и потом 2,5~3,0, и проводят кристаллизацию. Указанное в заголовке соединение, полученное в виде твердого вещества, фильтруют и промывают смешанным раствором воды и этилового спирта и затем сушат с получением 37,7 г указанного в заголовке соединения (содержание: 92,4%, выход: 94,4%).
Пример 6: Синтез (3S)-3-трет-Бутоксикарбониламино-4-(5,5-дифтор-2-оксопиперидино)бутановой кислоты
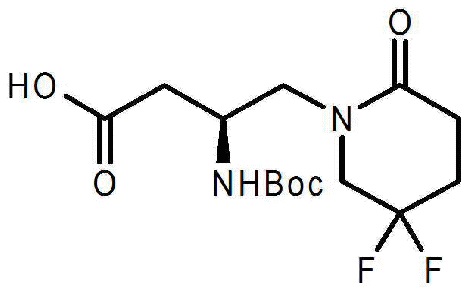
После добавления исходного материала, 43,0 г трет-бутил (3S)-3-трет-бутоксикарбониламино-4-(5,5-дифтор-2-оксопиперидинобутаноата, 67,9 г этилового спирта и 8,8 г 6N водного раствора гидроксида натрия, температуру повышают до 30°С для реакции. После завершения реакции смесь охлаждают и добавляют 344,0 г воды. Добавляют по каплям 3N водный раствор хлористоводородной кислоты для контроля pH, первоначально, 4,1~5,0, и потом 2,5~3,0, и проводят кристаллизацию. Указанное в заголовке соединение, полученное в виде твердого вещества, фильтруют и промывают смешанным раствором воды и этилового спирта и затем сушат с получением 37,7 г указанного в заголовке соединения (содержание: 94,1%, выход: 94,2%).
Экспериментальный пример: Сравнение выхода (3S)-3-трет-Бутоксикарбониламино-4-(5,5-дифтор-2-оксопиперидино)бутановой кислоты в зависимости от условий получения
Для сравнения выхода соединения химической формулы 1 в зависимости от используемого основания, температуры реакции и реакционного растворителя, соединение химической формулы 1 было получено из соединения химической формулы 2, и оно было получено в соответствии с условиями следующей таблицы 1, и результат также показан в таблице 1.
Таблица 1
Сравнение выхода соединения химической формулы 1 в зависимости от используемого основания, температуры реакции и реакционного растворителя
Как видно из результата сравнительного испытания, описанного в таблице 1, было подтверждено, что при использовании твердого основания, такого как твердый гидроксид натрия (заходы 4-6), количество растворителя реакции может быть уменьшено, и реакция можно проводить при температуре ниже, чем температура кипения с обратным холодильником, при этом получая соединение химической формулы 1 с более высоким выходом по сравнению с основным водным раствором, таким как водный раствор гидроксида натрия. Это является предпочтительным, поскольку производительность может быть улучшена, поскольку соединение химической формулы 1 может быть получено в виде твердого вещества путем подкисления с использованием кислоты в смешанном растворителе из этилового спирта и воды без концентрации растворителя после завершения реакции, когда количество растворителя реакции уменьшают.
Реферат
Данное изобретение относится к способу получения соединения химической формулы 1,включающему стадию селективного снятия защиты защитной группы карбоновой кислоты (P2) из P1и P2защитных групп на соединении химической формулы 2где при снятии защиты применяют твердое основание в качестве реакционного основания, и низший спирт в качестве реакционного растворителя. И где R1, R2, R3 и R4 независимо являются водородом или галогеном, P1является карбонильной группой, ацильной группой, сульфонильной группой, ацетильной группой или бензильной группой в качестве аминовой защитной группы, Р2является бензильной группой, этильной группой, изопропильной группой или трет-бутильной группой. Технический результат: описан имеющий высокую экономическую осуществимость и производительность, способ получения соединения, которое является промежуточным соединением для пероральных лекарственных средств для инсулиннезависимого диабета, который может значительно улучшить выход целевого продукта даже при умеренных условиях. 8 з.п. ф-лы, 6 пр., 1 табл.
Формула


Комментарии