Производное пиридона, имеющее тетрагидропиранилметильную группу - RU2707953C2
Код документа: RU2707953C2
Чертежи










Описание
[Область техники]
Настоящее изобретение относится к соединению или его соли, обладающему ингибирующей активностью в отношении Axl.
[Уровень техники]
Axl представляет собой тирозинкиназный рецептор, относящийся к семейству тирозинкиназных рецепторов Tyro3-Axl-Mer (TAM), лиганд которого представляет собой белок, кодируемый геном блокировки роста 6 (Gas6). Ген этой киназы первоначально был идентифицирован как трансформирующий ген при хроническом миелоидном лейкозе (непатентный документ 1).
Сообщалось, что сигнальный путь системы Gas6/Axl регулирует различные клеточные реакции, такие как выживание клеток, клеточное деление, аутофагию, миграцию клеток, ангиогенез, агрегацию тромбоцитов и дифференциацию природных клеток-киллеров (непатентный документ 2). Кроме того, многие отчеты показывают сверхэкспрессию Axl в тканях злокачественных новообразований, таких как первичный рак толстой кишки (непатентный документ 3), рак желудка (непатентный документ 4), рак пищевода (непатентный документ 5), меланома (непатентный документ 6), рак яичника (непатентный документ 7), рак почки (непатентный документ 8), рак эндометрия (непатентный документ 9) и рак щитовидной железы (непатентный документ 10). Было установлено, что присутствие Axl тесно связано с поражением лимфатических узлов и стадией рака легкого и экспрессией ER при раке молочной железы (непатентный документ 11).
Было дополнительно установлено, что Axl играет роль в иммунитете (непатентный документ 12), функциях тромбоцитов (непатентный документ 13), сперматогенезе (непатентный документ 14), сосудистой кальцификации (непатентный документ 15), тромбин-индуцированной пролиферации гладкомышечной клетки сосудов (VSMC) (непатентный документ 16), и различных заболеваниях почек, например, остром и хроническом гломерулонефрите, диабетической нефропатии и хроническом отторжении аллотрансплантата (непатентный документ 17). Предполагают, что ингибиторы Axl обеспечат терапевтические эффекты для многих заболеваний, включая раковые заболевания (включая солидные опухоли, такие как карцинома и саркома, лейкоз, и лимфоидные злокачественные заболевания), а также сосудистых заболеваний (включая, но ими не ограничиваясь, тромбоз, атеросклероз и рестеноз), заболевания почек (включая, но ими не ограничиваясь, острый и хронический гломерулонефрит, диабетическую нефропатию и отторжение трансплантата), и заболевания, при которых дезорганизованное образование кровеносных сосудов имеет серьезные последствия (включая, но ими не ограничиваясь, диабетическую ретинопатию, ретинопатию, псориаз, ревматоидный артрит, атерому, саркому Капоши и ангиому).
В то же время, как сообщалось, Mer, еще один член семейства тирозинкиназных рецепторов TAM, к которому принадлежит Axl, вызывает аутосомно-рецессивный пигментный ретинит посредством его гомозиготной мутации (непатентный документ 24). Также сообщалось, что определенная мутация в Mer была связана с детской палочко-колбочковой дистрофией (непатентный документ 25).
Соединения, имеющие сульфонамидную структуру (патентный документ 3), соединения, имеющие пирролопиримидиновую структуру (патентные документы 4 и 5), соединения, имеющие пиридиновую и пиразиновую структуры (патентный документ 6), соединения, имеющие пиразиновую структуру (патентный документ 7), соединения, имеющие пиразинилбензимидазольную структуру (патентный документ 8), соединения, имеющие индолиноновую (патентный документ 9), соединения, имеющие триазолопиридиновую и триазолопиримидиновую структуры (патентный документ 10), соединения, имеющие имидазольную структуру (патентный документ 11), соединения, имеющие триазольную структуру (патентные документы 12, 13, 14, 15, 16, 17, 20, 24, 25, 26, 27 и 28), соединения, имеющие пиримидиндиаминовую структуру (патентный документ 18), соединения, имеющие пиримидиновую структуру (патентный документ 19 и непатентные документы 18 и 22), соединения, имеющие хинолинилоксифенилсульфонамидную структуру (патентный документ 21), соединения, имеющие хинолиновую структуру (патентные документы 22 и 30 и непатентный документ 21), соединения, имеющие пиридиновую структуру (патентные документы 23 и 33 и непатентный документ 19), соединения, имеющие структуру мочевины (патентный документ 29), соединения, имеющие 2,4-дизамещенную ариламидную структуру (непатентный документ 20), соединения, имеющие секостероидную структуру (непатентный документ 23), соединения, имеющие бициклическую пиримидиновую структуру (патентные документы 31, 32 и 34), и тому подобное были описаны в качестве соединений, ингибирующих Axl.
[Список ссылок]
[Патентные документы]
[Патентный документ 1] Международная публикация No. WO2008/025820
[Патентный документ 2] Международная публикация No. WO2008/074997
[Патентный документ 3] Международная публикация No. WO2008/128072
[Патентный документ 4] Опубликованная патентная заявка США No. 20100204221
[Патентный документ 5] Международная публикация No. WO2010/090764
[Патентный документ 6] Международная публикация No. WO2009/053737
[Патентный документ 7] Международная публикация No. WO2009/007390
[Патентный документ 8] Международная публикация No. WO2009/024825
[Патентный документ 9] Международная публикация No. WO2007/057399
[Патентный документ 10] Международная публикация No. WO2009/047514
[Патентный документ 11] Международная публикация No. WO2009/058801
[Патентный документ 12] Международная публикация No. WO2008/083367
[Патентный документ 13] Международная публикация No. WO2008/083353
[Патентный документ 14] Международная публикация No. WO2010/005879
[Патентный документ 15] Международная публикация No. WO2008/083357
[Патентный документ 16] Международная публикация No. WO2008/083356
[Патентный документ 17] Международная публикация No. WO2008/083354
[Патентный документ 18] Международная публикация No. WO2008/045978
[Патентный документ 19] Международная публикация No. WO2007/070872
[Патентный документ 20] Международная публикация No. WO2007/030680
[Патентный документ 21] Международная публикация No. WO2011/045084
[Патентный документ 22] Международная публикация No. WO2009/127417
[Патентный документ 23] Международная публикация No. WO2007/066187
[Патентный документ 24] Международная публикация No. WO2009/054864
[Патентный документ 25] Международная публикация No. WO2010/005876
[Патентный документ 26] Международная публикация No. WO2009/054864
[Патентный документ 27] Опубликованная патентная заявка США No. 20090111816
[Патентный документ 28] Опубликованная патентная заявка США No. 20100168416
[Патентный документ 29] Международная публикация No. WO2009/138799
[Патентный документ 30] Опубликованная патентная заявка США No. 20090274693
[Патентный документ 31] Опубликованная патентная заявка США No. 20100069369
[Патентный документ 32] Опубликованная патентная заявка США No. 20070142402
[Патентный документ 33] Международная публикация No. WO2013/115280
[Патентный документ 34] Международная публикация No. WO2013/162061
[Непатентные документы]
[Непатентный документ 1] O'Bryan et al., Mol. Cell. Biol., 11, 5031 (1991)
[Непатентный документ 2] Rachel MA Linger et al., Expert Opin. Ther. Targets, 14, 1073 (2010)
[Непатентный документ 3] Craven et al., Int. J. Cancer., 60, 791 (1995)
[Непатентный документ 4] Sawabu et al., Mol. Carcinog., 46, 155 (2007)
[Непатентный документ 5] Nemoto et al., Pathobiology, 65, 195 (1997)
[Непатентный документ 6] Quong et al., Melanoma Res., 4, 313 (1994)
[Непатентный документ 7] Sun et al., Oncology, 66, 450 (2004)
[Непатентный документ 8] Chung et al., DNA Cell Biol., 22, 533 (2003)
[Непатентный документ 9] Sun et al., Ann. Oncol., 14, 898 (2003)
[Непатентный документ 10] Ito et al., Thyroid, 9, 563 (1999)
[Непатентный документ 11] Berclaz et al., Ann. Oncol., 12, 819 (2001)
[Непатентный документ 12] Lu et al., Science, 293, 306 (2001)
[Непатентный документ 13] Angelillo-Scherrer et al., Nat. Med., 7, 215 (2001)
[Непатентный документ 14] Lu et al., Nature, 398, 723 (1999)
[Непатентный документ 15] Son et al., Eur. J. Pharmacol., 556, 1 (2007)
[Непатентный документ 16] Nakano et al., J. Biol. Chem., 270, 5702 (1995)
[Непатентный документ 17] Yanagita et al., J. Clin. Invest., 110, 239 (2002)
[Непатентный документ 18] AlexisMollard et al., Med. Chem. Lett., 2, 907 (2011)
[Непатентный документ 19] Gretchen M. Schroeder et al., J. Med. Chem., 52, 1251 (2009)
[Непатентный документ 20] Carl R. Illig et al., Bioorg. Med. Chem. Lett., 18, 1642 (2008)
[Непатентный документ 21] Yi-Xiang Zhang et al., Cancer Res., 68, 1905 (2008)
[Непатентный документ 22] D Mahadevan et al., Oncogene, 26, 3909 (2007)
[Непатентный документ 23] Daowan Lai et al., Bioorg. Med. Chem., 19, 6873 (2011)
[Непатентный документ 24] Ksantini M.,Eur J Ophthalmol., 22, 647 (2012)
[Непатентный документ 25] Mackay et al., Molecular Vision., 16, 369 (2010)
[Сущность изобретения]
[Техническая задача]
Задачей настоящего изобретения является обеспечение Axl-ингибирующим соединением, обладающим высокой ингибирующей специфичностью по отношению к Axl, и более высокой безопасностью. Другой задачей настоящего изобретения является создание терапевтического средства для заболевания, вызванного гиперфункцией Axl, терапевтического средства для заболевания, связанного с гиперфункцией Axl, и/или терапевтического средства для заболевания, сопровождающегося гиперфункцией Axl, например, противоракового средства, содержащего Axl-ингибирующее соединение.
[Решение задачи]
Соединение примера 9 патентного документа 33 (далее в настоящем документе называемое как соединение А) обладает высокой ингибирующей активностью в отношении Axl, но было подтверждено, что вызывает необратимую дегенерацию фоторецепторов сетчатки в испытании при длительном введении с использованием мышей. Сообщалось, что инактивация Mer, члена семейства TAM, как с Axl, связана с дегенерацией клеток сетчатки, и соединение примера 9 из патентного документа 33 также обладает ингибирующей активностью в отношении Mer. Принимая во внимание эти факты, авторы настоящего изобретения считают, что ингибирование Mer этим соединением приводит к ретинальной токсичности.
В результате проведения кропотливых исследований, авторы настоящего изобретения обнаружили, что соединение, имеющее структуру, представленную общей формулой (I), приведенной ниже, или его соль обладает высокой Axl ингибирующей специфичностью и низкой ингибирующей активностью в отношении Mer, и также обнаружили, что это соединение или его соль не вызывает ретинальную токсичность в испытании при длительном введении с использованием мышей.
В частности, настоящее изобретение относится к следующим пунктам [1]-[53]:
[1]
Соединение, представленное общей формулой (I), или его фармацевтически приемлемая соль:
[Формула 1]

где
W, X и Y каждый независимо друг от друга представляет собой атом азота, C-H, C-F или C-Cl,
Z представляет собой атом азота, C-H, C-F, C-Cl, C-C1-C6 алкил или C-C1-C6 алкоксигруппу,
R1 представляет собой группу, представленную следующей формулой (II-1) или (II-2):
[Формула 2]

где в формуле (II-1),
Q представляет собой атом азота, C-H или C-F,
T представляет собой атом азота или C-H,
U представляет собой атом азота или C-H, и
R6 представляет собой атом галогена, C1-C6 алкильную группу, C3-C6 циклоалкильную группу, цианогруппу или трифторметоксильную группу, и
в формуле (II-2),
V представляет собой атом серы или атом кислорода, и
R7 представляет собой C1-C6 алкильную группу,
R2 представляет собой атом водорода, атом галогена, C1-C6 алкильную группу, C1-C6 алкоксигруппу или цианогруппу,
R3 представляет собой атом водорода или C1-C6 алкильную группу,
R4 представляет собой атом водорода, атом галогена или C1-C6 алкильную группу, и
R5представляет собой атом водорода или группу, представленную следующей формулой (III-1), (III-2), (III-3) или (III-4):
[Формула 3]

где в формуле (III-1),
R8 и R12 каждый независимо друг от друга представляет собой атом водорода или атом дейтерия,
R9 представляет собой атом водорода, атом галогена или C1-C6 алкоксигруппу,
R10 представляет собой атом водорода, C1-C6 алкильную группу, необязательно замещенную гетероциклоалкильной группой, или C1-C6 алкоксигруппу, необязательно замещенную гетероциклоалкильной группой, необязательно замещенной C1-C6 алкильной группой, и
R11 представляет собой атом водорода, C1-C6 алкоксигруппу или дейтерий-замещенную C1-C6 алкоксигруппу,
в формуле (III-2),
R13 представляет собой C1-C6 алкильную группу, необязательно замещенную гетероциклоалкильной группой, или гетероциклоалкильную группу,
в формуле (III-3),
R14 представляет собой атом водорода или C1-C6 алкильную группу,
R15 представляет собой атом водорода, C1-C6 алкильную группу или C1-C6 алкоксигруппу, и
R16 представляет собой атом водорода или атом галогена, и
в формуле (III-4),
R17 представляет собой C1-C6алкоксигруппу, и
R18 представляет собой C1-C6алкоксигруппу.
[2]
N-[4-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид, представленный следующей формулой:
[Формула 4]

или его фармацевтически приемлемая соль.
[3]
N-[4-(2-Амино-5-{4-[(2S)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид, представленный следующей формулой:
[Формула 5]

или его фармацевтически приемлемая соль.
[4]
N-{4-[2-Амино-5-(3,4-диметоксифенил)пиридин-3-ил]фенил}-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид, представленный следующей формулой:
[Формула 6]

или его фармацевтически приемлемая соль.
[5]
N-[4-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамид, представленный следующей формулой:
[Формула 7]

или его фармацевтически приемлемая соль.
[6]
N-[4-(2-Амино-5-{4-[(2S)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамид, представленный следующей формулой:
[Формула 8]

или его фармацевтически приемлемая соль.
[7]
N-{4-[2-Амино-5-(1-этил-1H-пиразол-4-ил)пиридин-3-ил]-3-фторфенил}-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид, представленный следующей формулой:
[Формула 9]

или его фармацевтически приемлемая соль.
[8]
N-(4-{2-Амино-5-[1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-ил]пиридин-3-ил}-3-фторфенил)-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид, представленный следующей формулой:
[Формула 10]

или его фармацевтически приемлемая соль.
[9]
N-[6-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)пиридазин-3-ил]-5-(5-метилтиофен-2-ил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид, представленный следующей формулой:
[Формула 11]

или его фармацевтически приемлемая соль.
[10]
N-[6-(2-Амино-5-{3-метокси-4-[2-(морфолин-4-ил)этокси]фенил}пиридин-3-ил)пиридазин-3-ил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид, представленный следующей формулой:
[Формула 12]

или его фармацевтически приемлемая соль.
[11]
Гидробромид, нитрат, сульфат, фосфат, метансульфонат, этансульфонат, бензолсульфонат или п-толуолсульфонат соединения по пункту [2].
[12]
Метансульфонат соединения по пункту [4].
[13]
Метансульфонат, фосфат, нафталин-1,5-дисульфонат или сульфат соединения по пункту [5].
[14]
Кристалл метансульфоната по пункту [11], где кристалл проявляет характеристические пики при углах дифракции 2θ=3,74, 7,56, 8,96, 11,38, 12,36, 14,78, 15,60, 16,16, 18,70 и 24,10 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
[15]
Кристалл гидробромида по пункту [11], где кристалл проявляет характеристические пики при углах дифракции 2θ=3,84, 7,72, 9,40, 11,62, 14,92, 15,48, 16,70, 18,88, 19,32 и 24,40 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
[16]
Кристалл нитрата по пункту [11], где кристалл проявляет характеристические пики при углах дифракции 2θ=3,82, 7,66, 9,28, 9,52, 11,54, 15,26, 15,54, 16,62, 19,24 и 24,56 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
[17]
Кристалл сульфата по пункту [11], где кристалл проявляет характеристические пики при углах дифракции 2θ=3,74, 7,56, 8,92, 9,58, 11,36, 12,38, 14,68, 15,64, 16,06 и 24,38 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
[18]
Кристалл фосфата по пункту [11], где кристалл проявляет характеристические пики при углах дифракции 2θ=3,74, 7,56, 8,80, 9,56, 11,34, 14,56, 15,74, 23,68, 24,34 и 24,68 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
[19]
Кристалл этансульфоната по пункту [11], где кристалл проявляет характеристические пики при углах дифракции 2θ=6,72, 7,90, 12,02, 13,40, 16,90, 17,88, 19,00, 19,80, 21,26 и 24,18 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
[20]
Кристалл бензолсульфоната по пункту [11], где кристалл проявляет характеристические пики при углах дифракции 2θ=9,22, 10,60, 10,82, 11,10, 13,40, 15,78, 17,50, 18,66, 21,02 и 26,10 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
[21]
Кристалл п-толуолсульфоната по пункту [11], где кристалл проявляет характеристические пики при углах дифракции 2θ=4,18, 5,12, 13,44, 14,98, 16,96, 17,44, 18,92, 19,72, 20,16 и 23,04 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
[22]
Кристалл фосфата по пункту [13], где кристалл проявляет характеристические пики при углах дифракции 2θ=4,28, 8,42, 8,64, 10,54, 12,72, 13,48, 15,90, 17,00, 17,46 и 21,26 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
[23]
Кристалл сульфата по пункту [13], где кристалл проявляет характеристические пики при углах дифракции 2θ=3,66, 6,42, 7,32, 9,76, 11,00, 12,88, 18,42, 19,62, 20,54 и 24,22 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
[24]
Кристалл сульфата по пункту [13], где кристалл проявляет характеристические пики при углах дифракции 2θ=3,64, 6,40, 7,32, 9,76, 17,38, 18,42, 19,64, 20,56, 22,90 и 24,20 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
[25]
Кристалл сульфата по пункту [13], где кристалл проявляет характеристические пики при углах дифракции 2θ=3,64, 6,40, 7,30, 9,76, 17,34, 18,38, 19,34, 20,56, 21,52 и 22,94 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
[26]
Кристалл сульфата по пункту [13], где кристалл проявляет характеристические пики при углах дифракции 2θ=3,62, 6,38, 7,28, 9,74, 17,30, 18,36, 19,54, 20,52, 22,86 и 24,14 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
[27]
Кристалл сульфата по пункту [13], где кристалл проявляет характеристические пики при углах дифракции 2θ=3,64, 6,40, 7,30, 9,76, 12,86, 18,40, 19,62, 20,54, 22,92 и 24,20 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
[28]
Кристалл сульфата по пункту [13], где кристалл проявляет характеристические пики при углах дифракции 2θ=3,64, 6,36, 7,30, 18,36, 19,04, 19,42, 19,70, 20,12, 20,42 и 21,32 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
[29]
Кристалл сульфата по пункту [13], где кристалл проявляет характеристические пики при углах дифракции 2θ=5,62, 7,18, 9,22, 10,36, 15,56, 16,40 и 20,86 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
[30]
Кристалл нафталин-1,5-дисульфоната по пункту [13], где кристалл проявляет характеристические пики при углах дифракции 2θ=6,14, 6,98, 11,24, 14,84, 17,48, 19,54, 20,94, 22,38, 23,20 и 24,70 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
[31]
Кристалл нафталин-1,5-дисульфоната по пункту [13], где кристалл проявляет характеристические пики при углах дифракции 2θ=9,24, 9,58, 14,00, 14,46, 16,70, 17,02, 18,22, 20,24, 21,64 и 25,52 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
[32]
Ингибитор Axl, содержащий соединение по любому из пунктов [1]-[10] или его фармацевтически приемлемую соль.
[33]
Лекарственное средство, содержащее соединение по любому из пунктов [1]-[10] или его фармацевтически приемлемую соль в качестве активного ингредиента.
[34]
Лекарственное средство для лечения заболевания, вызванного гиперфункцией Axl киназы, заболевания, связанного с гиперфункцией Axl киназы, и/или заболевания, сопровождающегося гиперфункцией Axl киназы, содержащее соединение по любому из пунктов [1]-[10] или его фармацевтически приемлемую соль в качестве активного ингредиента.
[35]
Лекарственное средство для лечения гиперпролиферативных заболеваний, содержащее соединение по любому из пунктов [1]-[10] или его фармацевтически приемлемую соль в качестве активного ингредиента.
[36]
Лекарственное средство для лечения рака, содержащее соединение по любому из пунктов [1]-[10] или его фармацевтически приемлемую соль в качестве активного ингредиента.
[37]
Лекарственное средство для профилактики метастазирования рака, содержащее соединение по любому из пунктов [1]-[10] или его фармацевтически приемлемую соль в качестве активного ингредиента.
[38]
Лекарственное средство для преодоления лекарственной устойчивости рака, содержащее соединение по любому из пунктов [1]-[10] или его фармацевтически приемлемую соль в качестве активного ингредиента.
[39]
Лекарственное средство для ингибирования приобретения лекарственной устойчивости рака, содержащее соединение по любому из пунктов [1]-[10] или его фармацевтически приемлемую соль в качестве активного ингредиента.
[40]
Лекарственное средство по любому из пунктов [36]-[39], где рак выбран из рака молочной железы, рака толстой кишки, рака предстательной железы, рака легкого, рака желудка, рака яичника, рака эндометрия, рака почки, гепатоцеллюлярного рака, рака щитовидной железы, рака матки, рака пищевода, плоскоклеточного рака, лейкоза, остеосаркомы, меланомы, глиобластомы, нейробластомы и рака поджелудочной железы.
[41]
Фармацевтическая композиция, содержащая соединение по любому из пунктов [1]-[10] или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
[42]
Способ лечения заболевания, вызванного гиперфункцией Axl киназы, заболевания, связанного с гиперфункцией Axl киназы, и/или заболевания, сопровождающегося гиперфункцией Axl киназы, включающий использование соединения по любому из пунктов [1]-[10] или его фармацевтически приемлемой соли.
[43]
Способ лечения гиперпролиферативных заболеваний, включающий использование соединения по любому из пунктов [1]-[10] или его фармацевтически приемлемой соли.
[44]
Способ лечения рака, включающий использование соединения по любому из пунктов [1]-[10] или его фармацевтически приемлемой соли.
[45]
Способ профилактики метастазирования рака, включающий использование соединения по любому из пунктов [1]-[10] или его фармацевтически приемлемой соли.
[46]
Способ преодоления лекарственной устойчивости рака, включающий использование соединения по любому из пунктов [1]-[10] или его фармацевтически приемлемой соли.
[47]
Способ ингибирования приобретения лекарственной устойчивости рака, включающий использование соединения по любому из пунктов [1]-[10] или его фармацевтически приемлемой соли.
[48]
Способ по любому из пунктов [44]-[49], где рак выбран из рака молочной железы, рака толстой кишки, рака предстательной железы, рака легкого, рака желудка, рака яичника, рака эндометрия, рака почки, гепатоцеллюлярного рака, рак щитовидной железы, рака матки, рака пищевода, плоскоклеточного рака, лейкоза, остеосаркомы, меланомы, глиобластомы и нейробластомы.
[49]
Применение соединения по любому из пунктов [1]-[10] или его фармацевтически приемлемой соли для получения лекарственного средства.
[50]
Применение соединения по любому из пунктов [1]-[10] или его фармацевтически приемлемой соли для лечения рака.
[51]
Применение соединения по любому из пунктов [1]-[10] или его фармацевтически приемлемой соли для профилактики метастазирования рака.
[52]
Применение соединения по любому из пунктов [1]-[10] или его фармацевтически приемлемой соли для преодоления лекарственной устойчивости рака.
[53]
Применение соединения по любому из пунктов [1]-[10] или его фармацевтически приемлемой соли для ингибирования приобретения лекарственной устойчивости рака.
[Преимущественные эффекты изобретения]
Настоящее изобретение обеспечивает соединение, представленное формулой (I), обладающее ингибирующей активностью в отношении Axl. Соединение является полезным в качестве терапевтического средства для лечения заболевания, вызванного гиперфункцией Axl, заболевания, связанного с гиперфункцией Axl, и/или заболевания, сопровождающегося гиперфункцией Axl, например, противоракового средства.
[Краткое описание фигур]
[Фигура 1] На фигуре 1 представлена порошковая рентгеновская дифрактограмма кристалла, полученного в примере 130. На этой фигуре, на оси ординат показана интенсивность при дифракции (интенсивность) в единице измерения имп/сек (импульсов в секунду), и на оси абсцисс показаны значения угла дифракции 2θ.
[Фигура 2] На фигуре 2 представлена порошковая рентгеновская дифрактограмма кристалла, полученного в примере 131. На этой фигуре, на оси ординат показана интенсивность при дифракции (интенсивность) в единице измерения имп/сек (импульсов в секунду), и на оси абсцисс показаны значения угла дифракции 2θ.
[Фигура 3] На фигуре 3 представлена порошковая рентгеновская дифрактограмма кристалла, полученного в примере 132. На этой фигуре, на оси ординат показана интенсивность при дифракции (интенсивность) в единице измерения имп/сек (импульсов в секунду), и на оси абсцисс показаны значения угла дифракции 2θ.
[Фигура 4] На фигуре 4 представлена порошковая рентгеновская дифрактограмма кристалла, полученного в примере 133. На этой фигуре, на оси ординат показана интенсивность при дифракции (интенсивность) в единице измерения имп/сек (импульсов в секунду), и на оси абсцисс показаны значения угла дифракции 2θ.
[Фигура 5] На фигуре 5 представлена порошковая рентгеновская дифрактограмма кристалла, полученного в примере 134. На этой фигуре, на оси ординат показана интенсивность при дифракции (интенсивность) в единице измерения имп/сек (импульсов в секунду), и на оси абсцисс показаны значения угла дифракции 2θ.
[Фигура 6] На фигуре 6 представлена порошковая рентгеновская дифрактограмма кристалла, полученного в примере 135. На этой фигуре, на оси ординат показана интенсивность при дифракции (интенсивность) в единице измерения имп/сек (импульсов в секунду), и на оси абсцисс показаны значения угла дифракции 2θ.
[Фигура 7] На фигуре 7 представлена порошковая рентгеновская дифрактограмма кристалла, полученного в примере 136. На этой фигуре, на оси ординат показана интенсивность при дифракции (интенсивность) в единице измерения имп/сек (импульсов в секунду), и на оси абсцисс показаны значения угла дифракции 2θ.
[Фигура 8] На фигуре 8 представлена порошковая рентгеновская дифрактограмма кристалла, полученного в примере 137. На этой фигуре, на оси ординат показана интенсивность при дифракции (интенсивность) в единице измерения имп/сек (импульсов в секунду), и на оси абсцисс показаны значения угла дифракции 2θ.
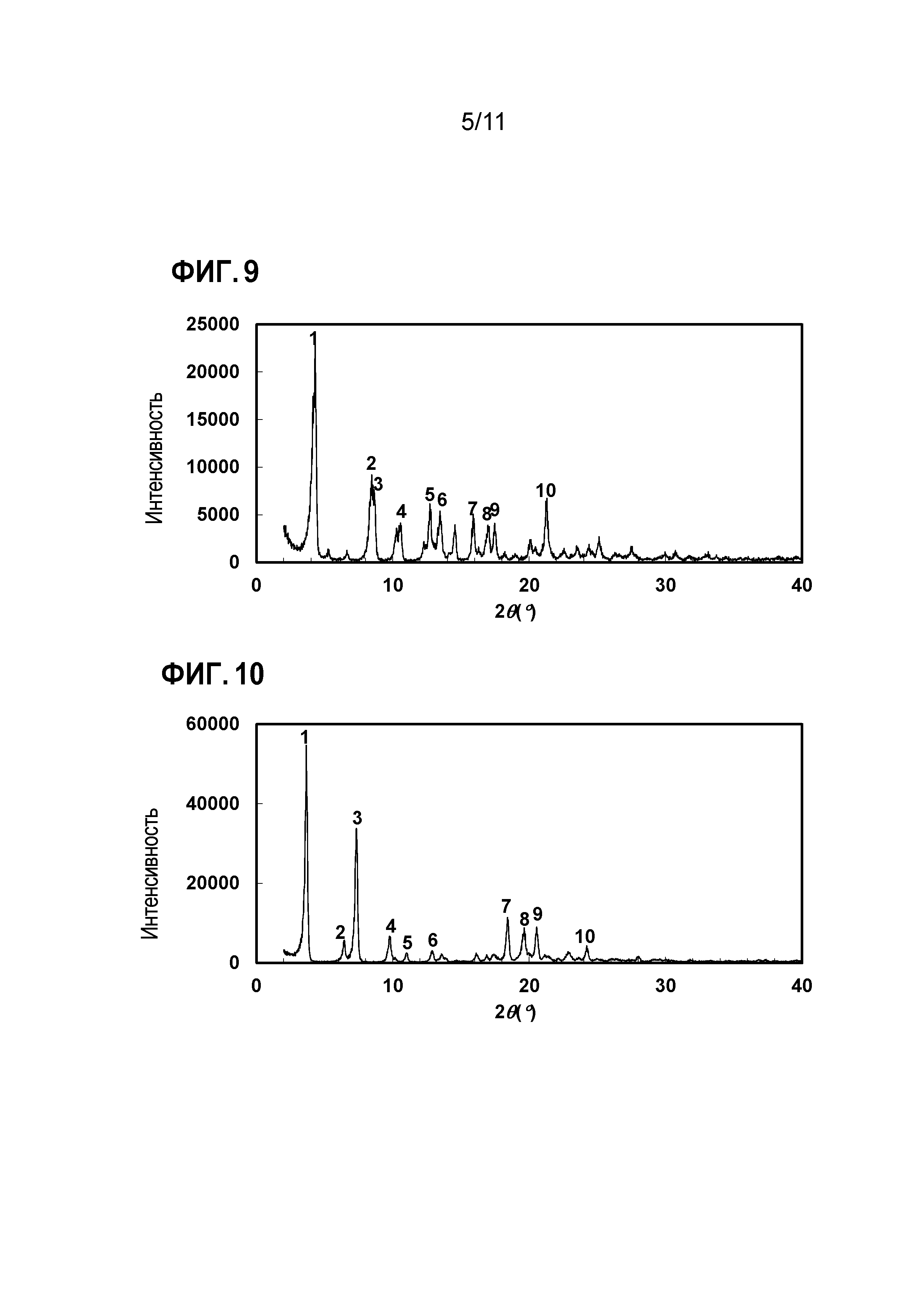
[Фигура 9] На фигуре 9 представлена порошковая рентгеновская дифрактограмма кристалла, полученного в примере 138. На этой фигуре, на оси ординат показана интенсивность при дифракции (интенсивность) в единице измерения имп/сек (импульсов в секунду), и на оси абсцисс показаны значения угла дифракции 2θ.
[Фигура 10] На фигуре 10 представлена порошковая рентгеновская дифрактограмма кристалла, полученного в примере 139. На этой фигуре, на оси ординат показана интенсивность при дифракции (интенсивность) в единице измерения имп/сек (импульсов в секунду), и на оси абсцисс показаны значения угла дифракции 2θ.
[Фигура 11] На фигуре 11 представлена порошковая рентгеновская дифрактограмма кристалла, полученного в примере 140. На этой фигуре, на оси ординат показана интенсивность при дифракции (интенсивность) в единице измерения имп/сек (импульсов в секунду), и на оси абсцисс показаны значения угла дифракции 2θ.
[Фигура 12] На фигуре 12 представлена порошковая рентгеновская дифрактограмма кристалла, полученного в примере 141. На этой фигуре, на оси ординат показана интенсивность при дифракции (интенсивность) в единице измерения имп/сек (импульсов в секунду), и на оси абсцисс показаны значения угла дифракции 2θ.
[Фигура 13] На фигуре 13 представлена порошковая рентгеновская дифрактограмма кристалла, полученного в примере 142. На этой фигуре, на оси ординат показана интенсивность при дифракции (интенсивность) в единице измерения имп/сек (импульсов в секунду), и на оси абсцисс показаны значения угла дифракции 2θ.
[Фигура 14] На фигуре 14 представлена порошковая рентгеновская дифрактограмма кристалла, полученного в примере 143. На этой фигуре, на оси ординат показана интенсивность при дифракции (интенсивность) в единице измерения имп/сек (импульсов в секунду), и на оси абсцисс показаны значения угла дифракции 2θ.
[Фигура 15] На фигуре 15 представлена порошковая рентгеновская дифрактограмма кристалла, полученного в примере 144. На этой фигуре, на оси ординат показана интенсивность при дифракции (интенсивность) в единице измерения имп/сек (импульсов в секунду), и на оси абсцисс показаны значения угла дифракции 2θ.
[Фигура 16] На фигуре 16 представлена порошковая рентгеновская дифрактограмма кристалла, полученного в примере 145. На этой фигуре, на оси ординат показана интенсивность при дифракции (интенсивность) в единице измерения имп/сек (импульсов в секунду), и на оси абсцисс показаны значения угла дифракции 2θ.
[Фигура 17] На фигуре 17 представлена порошковая рентгеновская дифрактограмма кристалла, полученного в примере 146. На этой фигуре, на оси ординат показана интенсивность при дифракции (интенсивность) в единице измерения имп/сек (импульсов в секунду), и на оси абсцисс показаны значения угла дифракции 2θ.
[Фигура 18] На фигуре 18 представлена порошковая рентгеновская дифрактограмма кристалла, полученного в примере 147. На этой фигуре, на оси ординат показана интенсивность при дифракции (интенсивность) в единице измерения имп/сек (импульсов в секунду), и на оси абсцисс показаны значения угла дифракции 2θ.
[Фигура 19-1] На фигуре 19-1 представлена диаграмма, показывающая эффект, вызванный комбинированным использованием Axl-ингибирующего соединения по настоящему изобретению с эрлотинибом, на опухоли, полученные у мышей с подкожно трансплантированным HCC827 (который имеет делеционную мутацию в 19 экзоне гена EGFR и проявляет высокую чувствительность к ингибиторам EGFR). Символ х представляет собой группу, получавшую носитель три раза в день (Tid). Символ незакрашенный кружок представляет собой группу, получавшую 50 мг/кг гидрата сульфата соединения B два раза в день (bid). Символ закрашенный кружок представляет собой группу, получавшую 25 мг/кг эрлотиниба один раз в сутки (qd). Символ незакрашенный треугольник представляет собой группу, получавшую 50 мг/кг гидрата сульфата соединения B (два раза в день) и 25 мг/кг эрлотиниба (один раз в сутки). На оси абсцисс представлено количество дней после начала введения. На оси ординат представлен предполагаемый объем опухоли, вычисленный из осей опухоли.
[Фигура 19-2] На фигуре 19-2 представлена диаграмма, показывающая среднюю скорость изменения массы тела индивидуумов в испытании при комбинированном использовании Axl-ингибирующего соединения по настоящей заявке с эрлотинибом (использовали те же пояснения условных обозначений, как на фигуре 19-1).
[Фигура 20-1] На фигуре 20-1 представлена диаграмма, показывающая противоопухолевый эффект эрлотиниба на опухоли, полученные у мышей с подкожно трансплантированным HCC827. Символ х представляет собой группу введения носителя. Символ незакрашенный кружок представляет собой группу, получавшую 25 мг/кг эрлотиниба. Символ закрашенный кружок представляет собой группу, получавшую 12,5 мг/кг эрлотиниба. Символ незакрашенный треугольник представляет собой группу, получавшую 6,25 мг/кг эрлотиниба. На оси абсцисс представлено количество дней после начала введения. На оси ординат представлен предполагаемый объем опухоли, вычисленный из осей опухоли.
[Фигура 20-2] На фигуре 20-2 представлена диаграмма, показывающая изменение уровня экспрессии белка Axl, когда эрлотиниб вводили в опухоли, полученные у мышей с подкожно трансплантированным HCC827.
[Описание вариантов осуществления]
В настоящем изобретении, Axl относится к белку, кодируемому геном Axl. ʺAxlʺ включает, например, белки Axl, кодируемые полноразмерным геном Axl, или белки Axl, кодируемые вариантом гена Axl (включая вариант делеции, вариант замены или вариант вставки). В настоящем изобретении ʺAxlʺ также включает гомологи, полученные от различных видов животных.
В настоящем изобретении термин ʺингибитор Axlʺ относится к ингибитору функции Axl как тирозинкиназы.
В настоящем изобретении термины ʺопухольʺ и ʺракʺ используются взаимозаменяемо. В настоящем изобретении опухоль, злокачественная опухоль, рак, злокачественное новообразование, карцинома, саркома и тому подобное, также совместно именуются как ʺопухольʺ или ʺракʺ.
В настоящем изобретении,
ʺC1-C6 алкильная группаʺ означает линейную или разветвленную алкильную группу, имеющую от 1 до 6 атомов углерода. Примеры ʺC1-C6 алкильной группыʺ включают метильную группу, этильную группу, пропильную группу, изопропильную группу, бутильную группу и трет-бутильную группу.
ʺC1-C6 алкоксигруппаʺ означает алкоксигруппу, имеющую линейную или разветвленную алкильную группу, имеющую от 1 до 6 атомов углерода. Примеры ʺC1-C6 алкоксигруппыʺ включают метоксигруппу, этоксигруппу, пропоксигруппу, изопропоксигруппу и бутоксигруппу.
Примеры ʺатома галогенаʺ включают атом фтора, атом хлора, атом брома и атом йода.
ʺЦиклоалкильная группаʺ означает циклическую алкильную группу, имеющую от 3 до 8 атомов углерода, если не указано иное. Соответствующие примеры включают циклопропильную группу, циклобутильную группу, циклопентильную группу и циклогексильную группу.
ʺГетероциклоалкильная группаʺ означает одновалентную насыщенную гетероциклическую группу. Соответствующие примеры включают насыщенную гетероциклическую группу, имеющую атом азота в кольце, и насыщенную гетероциклическую группу, имеющую атом кислорода в кольце и, в частности, включают одновалентные группы, полученные из пирролидина, имидазолина, пиперидина, пиперазина, азетидина, морфолина, диоксана, оксетана, тетрагидропирана и хинуклидина.
ʺЦиклоалкенильная группаʺ относится к вышеупомянутой ʺциклоалкильной группеʺ, имеющей одну или несколько ненасыщенных связей, таких как двойные связи. Соответствующие примеры включают циклопентенильную группу и циклогексенильную группу.
ʺГетероциклоалкенильная группаʺ относится к вышеупомянутой ʺгетероциклоалкильной группеʺ, имеющей одну или несколько ненасыщенных связей, таких как двойные связи. Соответствующие примеры включают тетрагидропиридинильную группу и дигидропиранильную группу. Далее в настоящем документе, каждый заместитель в формуле (I) будет описан.
В следующей общей формуле (I),
[Формула 13]

W, X и Y каждый независимо друг от друга представляет собой атом азота, C-H, C-F или C-Cl.
Z представляет собой атом азота, C-H, C-F, C-C1-C6 алкоксигруппу, C-C1-C6 алкильную группу или C-Cl.
R1 представляет собой группу, представленную следующей формулой (II-1) или (II-2):
[Формула 14]

где в формуле (II-1), Q представляет собой атом азота, C-H или C-F, T представляет собой атом азота или C-H, U представляет собой атом азота или C-H, и R6 представляет собой атом галогена, C1-C6 алкильную группу, C3-C6 циклоалкильную группу, цианогруппу или трифторметоксильную группу, и в формуле (II-2), V представляет собой атом серы или атом кислорода, и R7 представляет собой C1-C6 алкильную группу.
R2 представляет собой атом водорода, атом галогена, C1-C6 алкильную группу, C1-C6 алкоксигруппу или цианогруппу. В данном контексте, C1-C6 алкильная группа предпочтительно представляет собой метильную группу, этильную группу или пропильную группу. C1-C6 алкоксигруппа предпочтительно представляет собой метоксигруппу или этоксигруппу.
R3 представляет собой атом водорода или C1-C6 алкильную группу. C1-C6 алкильная группа предпочтительно представляет собой метильную группу, этильную группу или пропильную группу.
R4 представляет собой атом водорода, атом галогена или C1-C6 алкильную группу. R4 предпочтительно представляет собой атом водорода, атом фтора, атом хлора, метильную группу или этильную группу.
R5 представляет собой атом водорода или группу, представленную следующей формулой (III-1), (III-2), (III-3) или (III-4):
[Формула 15]

где
в формуле (III-1),
R8 и R12 каждый независимо друг от друга представляет собой атом водорода или атом дейтерия,
R9 представляет собой атом водорода, атом галогена или C1-C6 алкоксигруппу,
R10 представляет собой атом водорода, C1-C6 алкильную группу, необязательно замещенную гетероциклоалкильной группой, или C1-C6 алкоксигруппу, необязательно замещенную гетероциклоалкильной группой, необязательно замещенной C1-C6 алкильной группой, и
R11 представляет собой атом водорода, C1-C6 алкоксигруппу или дейтерий-замещенную C1-C6 алкоксигруппу,
в формуле (III-2),
R13 представляет собой C1-C6 алкильную группу, необязательно замещенную гетероциклоалкильной группой, или гетероциклоалкильную группу,
в формуле (III-3),
R14 представляет собой атом водорода или C1-C6 алкильную группу,
R15 представляет собой атом водорода, C1-C6 алкильную группу или C1-C6 алкоксигруппу, и
R16 представляет собой атом водорода или атом галогена, и
в формуле (III-4),
R17 представляет собой C1-C6алкоксигруппу, и
R18 представляет собой C1-C6алкоксигруппу.
Когда R5 представляет собой группу, представленную формулой (III-1), гетероциклоалкильная группа R10 предпочтительно представляет собой диоксанильную группу, морфолиногруппу, пиперазинильную группу или тетрагидропиранильную группу, и C1-C6 алкильная группа предпочтительно представляет собой метоксигруппу или этоксигруппу. C1-C6 алкоксигруппа R11 предпочтительно представляет собой метоксигруппу или этоксигруппу.
Когда R5 представляет собой группу, представленную формулой (III-2), гетероциклоалкильная группа R13 предпочтительно представляет собой диоксанильную группу, морфолиногруппу, пиперазинильную группу или тетрагидропиранильную группу, и C1-C6 алкильная группа R13 предпочтительно представляет собой метильную группу или этильную группу.
Соединение, представленное общей формулой (I), или его фармацевтически приемлемая соль, более предпочтительно, представляет собой любое из следующих соединений или их фармацевтически приемлемых солей:
N-[4-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид, представленный следующей формулой:
[Формула 16]

N-[4-(2-Амино-5-{4-[(2S)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид, представленный следующей формулой:
[Формула 17]

N-{4-[2-Амино-5-(3,4-диметоксифенил)пиридин-3-ил]фенил}-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид, представленный следующей формулой:
[Формула 18]

N-[4-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамид, представленный следующей формулой:
[Формула 19]

N-[4-(2-Амино-5-{4-[(2S)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамид, представленный следующей формулой:
[Формула 20]

N-{4-[2-Амино-5-(1-этил-1H-пиразол-4-ил)пиридин-3-ил]-3-фторфенил}-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид, представленный следующей формулой:
[Формула 21]

N-(4-{2-Амино-5-[1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-ил]пиридин-3-ил}-3-фторфенил)-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид, представленный следующей формулой:
[Формула 22]

N-[6-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)пиридазин-3-ил]-5-(5-метилтиофен-2-ил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид, представленный следующей формулой:
[Формула 23]

и
N-[6-(2-Амино-5-{3-метокси-4-[2-(морфолин-4-ил)этокси]фенил}пиридин-3-ил)пиридазин-3-ил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид, представленный следующей формулой:
[Формула 24]

Соединения, представленные формулой (I), по настоящему изобретению могут иметь стереоизомеры или оптические изомеры, полученные из асимметрических атомов углерода. Все эти стереоизомеры и оптические изомеры, и их смеси включены в настоящее изобретение.
Соединения, представленные общей формулой (I) по настоящему изобретению, могут образовывать фармацевтически приемлемую соль, при необходимости, когда имеют основную группу, такую как аминогруппа. Примеры таких солей могут включать: гидрогалогениды, такие как гидрохлориды, гидробромиды и гидройодиды; соли неорганических кислот, такие как нитраты, перхлораты, сульфаты и фосфаты; низшие алкансульфонаты, такие как метансульфонаты, трифторметансульфонаты и этансульфонаты; арилсульфонаты, такие как бензолсульфонаты и п-толуолсульфонаты; соли органических кислот, такие как формиаты, ацетаты, малаты, фумараты, сукцинаты, цитраты, тартраты, оксалаты и малеаты; и соли аминокислот, такие как соли орнитина, глутаматы и аспартаты. Предпочтительными являются гидрогалогениды и соли органических кислот.
Соединения, представленные общей формулой (I) по настоящему изобретению, могут, как правило, образовывать основно-аддитивную соль, когда имеют кислотную группу, такую как карбоксигруппа. Примеры таких фармацевтически приемлемых солей могут включать: соли щелочных металлов, такие как натриевые соли, калиевые соли и литиевые соли; соли щелочноземельных металлов, такие как кальциевые соли и магниевые соли; неорганические соли, такие как аммониевые соли; и органические аминовые соли, такие как дибензиламиновые соли, морфолиновые соли, соли сложных фенилглициналкиловых эфиров, этилендиаминовые соли, N-метилглюкаминовые соли, диэтиламиновые соли, триэтиламиновые соли, циклогексиламиновые соли, дициклогексиламиновые соли, N,N'-дибензилэтилендиаминовые соли, диэтаноламиновые соли, N-бензил-N-(2-фенилэтокси)аминовые соли, пиперазиновые соли, тетраметиламмониевые соли и трис(гидроксиметил)аминометановые соли.
Соединения, представленные общей формулой (I) по настоящему изобретению, или их соли могут существовать в свободной форме или в форме сольвата. Соединения, представленные общей формулой (I) по настоящему изобретению, или их соли могут существовать в форме гидратов, например, посредством поглощения влаги из воздуха. Сольваты не имеют особых ограничений до тех пор, пока сольват является фармацевтически приемлемым. В частности, сольват, предпочтительно, представляет собой гидрат, сольват этанола или тому подобное. Соединения, представленные общей формулой (I) по настоящему изобретению могут быть в форме N-оксида, когда содержат атом азота. Эти сольватные и N-оксидные формы также включены в объем настоящего изобретения. Кристаллы соединений по настоящему изобретению или их соли также включены в объем настоящего изобретения.
Более конкретно, примеры соли включают гидробромид, нитрат, сульфат, фосфат, этансульфонат, бензолсульфонат и п-толуолсульфонат N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамида.
Их дополнительные примеры включают метансульфонат N-{4-[2-амино-5-(3,4-диметоксифенил)пиридин-3-ил]фенил}-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамида.
Их дополнительные примеры включают метансульфонат, фосфат и сульфат N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамида.
В другом аспекте настоящее изобретение относится к кристаллам соединений по настоящему изобретению или их солей. В контексте настоящего изобретения, кристаллы относятся к твердым кристаллам, чья внутренняя структура образована регулярным повторением в пространстве атомов, образующих молекулу (или их группы), и отличаются от твердого аморфного вещества, не имеющего такого правильного внутреннего строения регулярной внутренней структуры.
Кристаллы одного и того же соединения, имеющие множество различных внутренних структур и физико-химических свойств (кристаллические полиморфы), могут быть получены в зависимости от условий кристаллизации. Кристаллы по настоящему изобретению могут представлять собой любой из этих кристаллических полиморфов или могут представлять собой смесь двух или нескольких кристаллических полиморфов.
Кристаллы по настоящему изобретению могут поглощать влагу или адсорбировать воду при стоянии на воздухе или могут быть нагреты до 25-150°C при нормальных атмосферных условиях, например, для образования гидрата. Кристаллы по настоящему изобретению также могут содержать растворитель для кристаллизации в присоединенном остаточном растворителе или сольвате.
В настоящем описании кристаллы по настоящему изобретению могут быть представлены на основе данных порошковой рентгеновской дифракции. Дифракция рентгеновских лучей на порошке может быть измерена и проанализирована с помощью подхода, обычно используемого в данной области, и может быть осуществлена, например, способом, описанным в Примерах. Обычно, в гидратах и дегидратах, присоединение и отсоединение кристаллизационной воды может изменить их параметры решетки и, таким образом, изменять углы дифракции (2θ) в порошковой рентгеновской дифракции. Кроме того, интенсивность пика может изменяться в зависимости, например, от разницы в поверхности роста кристаллов или тому подобное (габитуса кристалла). Соответственно, когда кристаллы по настоящему изобретению, представлены на основе данных порошковой рентгеновской дифракции, кристаллы, чьи пики углов дифракции и порошковые рентгеновские дифрактограммы в порошковой рентгеновской дифракции, идентичны таковым кристаллам по настоящему изобретению, а также гидраты и дегидраты, полученные из этих кристаллов, также включены в объем настоящего изобретения.
В более конкретном аспекте настоящее изобретение относится к кристаллу метансульфоната N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамида. Более предпочтительно, настоящее изобретение относится к кристаллу метансульфоната N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамида, где кристалл проявляет характеристические пики при углах дифракции 2θ=3,74, 7,56, 8,96, 11,38, 12,36, 14,78, 15,60, 16,16, 18,70 и 24,10 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем). Кристалл имеет порошковую рентгеновскую дифрактограмму, как показано на фигуре 1, полученную при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
В более конкретном аспекте настоящее изобретение относится к кристаллу гидробромида N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамида. Более предпочтительно, настоящее изобретение относится к кристаллу гидробромида N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамида, где кристалл проявляет характеристические пики при углах дифракции 2θ=3,84, 7,72, 9,40, 11,62, 14,92, 15,48, 16,70, 18,88, 19,32 и 24,40 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем). Кристалл имеет порошковую рентгеновскую дифрактограмму, как показано на фигуре 2, полученную при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
В более конкретном аспекте настоящее изобретение относится к кристаллу нитрата N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамида. Более предпочтительно, настоящее изобретение относится к кристаллу нитрата N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамида, где кристалл проявляет характеристические пики при углах дифракции 2θ=3,82, 7,66, 9,28, 9,52, 11,54, 15,26, 15,54, 16,62, 19,24 и 24,56 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем). Кристалл имеет порошковую рентгеновскую дифрактограмму, как показано на фигуре 3, полученную при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
В более конкретном аспекте настоящее изобретение относится к кристаллу сульфата N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамида. Более предпочтительно, настоящее изобретение относится к кристаллу сульфата N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамида, где кристалл проявляет характеристические пики при углах дифракции 2θ=3,74, 7,56, 8,92, 9,58, 11,36, 12,38, 14,68, 15,64, 16,06 и 24,38 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем). Кристалл имеет порошковую рентгеновскую дифрактограмму, как показано на фигуре 4, полученную при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
В более конкретном аспекте настоящее изобретение относится к кристаллу фосфата N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамида. Более предпочтительно, настоящее изобретение относится к кристаллу фосфата N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамида, где кристалл проявляет характеристические пики при углах дифракции 2θ=3,74, 7,56, 8,80, 9,56, 11,34, 14,56, 15,74, 23,68, 24,34 и 24,68 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем). Кристалл имеет порошковую рентгеновскую дифрактограмму, как показано на фигуре 5, полученную при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
В более конкретном аспекте настоящее изобретение относится к кристаллу этансульфоната N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамида. Более предпочтительно, настоящее изобретение относится к кристаллу этансульфоната N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамида, где кристалл проявляет характеристические пики при углах дифракции 2θ=6,72, 7,90, 12,02, 13,40, 16,90, 17,88, 19,00, 19,80, 21,26 и 24,18 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем). Кристалл имеет порошковую рентгеновскую дифрактограмму, как показано на фигуре 6, полученную при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
В более конкретном аспекте настоящее изобретение относится к кристаллу бензолсульфоната N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамида. Более предпочтительно, настоящее изобретение относится к кристаллу бензолсульфоната N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамида, где кристалл проявляет характеристические пики при углах дифракции 2θ=9,22, 10,60, 10,82, 11,10, 13,40, 15,78, 17,50, 18,66, 21,02 и 26,10 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем). Кристалл имеет порошковую рентгеновскую дифрактограмму, как показано на фигуре 7, полученную при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
В более конкретном аспекте настоящее изобретение относится к кристаллу п-толуолсульфоната N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамида. Более предпочтительно, настоящее изобретение относится к кристаллу п-толуолсульфоната N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамида, где кристалл проявляет характеристические пики при углах дифракции 2θ=4,18, 5,12, 13,44, 14,98, 16,96, 17,44, 18,92, 19,72, 20,16 и 23,04 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем). Кристалл имеет порошковую рентгеновскую дифрактограмму, как показано на фигуре 8, полученную при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
В более конкретном аспекте настоящее изобретение относится к кристаллу фосфата N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамида. Более предпочтительно, настоящее изобретение относится к кристаллу фосфата N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамида, где кристалл проявляет характеристические пики при углах дифракции 2θ=4,28, 8,42, 8,64, 10,54, 12,72, 13,48, 15,90, 17,00, 17,46 и 21,26 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем). Кристалл имеет порошковую рентгеновскую дифрактограмму, как показано на фигуре 9, полученную при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
В более конкретном аспекте настоящее изобретение относится к кристаллу сульфата N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамида. Более предпочтительно, настоящее изобретение относится к кристаллу сульфата N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамида, где кристалл проявляет характеристические пики при углах дифракции 2θ=3,66, 6,42, 7,32, 9,76, 11,00, 12,88, 18,42, 19,62, 20,54 и 24,22 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем). Кристалл имеет порошковую рентгеновскую дифрактограмму, как показано на фигуре 10, полученную при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем). Также, предпочтительно, настоящее изобретение относится к кристаллу сульфата N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамида, где кристалл проявляет характеристические пики при углах дифракции 2θ=3,64, 6,40, 7,32, 9,76, 17,38, 18,42, 19,64, 20,56, 22,90 и 24,20 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем). Кристалл имеет порошковую рентгеновскую дифрактограмму, как показано на фигуре 11, полученную при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем). Также, предпочтительно, настоящее изобретение относится к кристаллу сульфата N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамида, где кристалл проявляет характеристические пики при углах дифракции 2θ=3,64, 6,40, 7,30, 9,76, 17,34, 18,38, 19,34, 20,56, 21,52, и 22,94 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем). Кристалл имеет порошковую рентгеновскую дифрактограмму, как показано на фигуре 12, полученную при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем). Также, предпочтительно, настоящее изобретение относится к кристаллу сульфата N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамида, где кристалл проявляет характеристические пики при углах дифракции 2θ=3,62, 6,38, 7,28, 9,74, 17,30, 18,36, 19,54, 20,52, 22,86 и 24,14 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем). Кристалл имеет порошковую рентгеновскую дифрактограмму, как показано на фигуре 13, полученную при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем). Также, предпочтительно, настоящее изобретение относится к кристаллу сульфата N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамида, где кристалл проявляет характеристические пики при углах дифракции 2θ=3,64, 6,40, 7,30, 9,76, 12,86, 18,40, 19,62, 20,54, 22,92 и 24,20 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем). Кристалл имеет порошковую рентгеновскую дифрактограмму, как показано на фигуре 14, полученную при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем). Также, предпочтительно, настоящее изобретение относится к кристаллу сульфата N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамида, где кристалл проявляет характеристические пики при углах дифракции 2θ=3,64, 6,36, 7,30, 18,36, 19,04, 19,42, 19,70, 20,12, 20,42 и 21,32 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем). Кристалл имеет порошковую рентгеновскую дифрактограмму, как показано на фигуре 15, полученную при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем). Также, предпочтительно, настоящее изобретение относится к кристаллу сульфата N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамида, где кристалл проявляет характеристические пики при углах дифракции 2θ=5,62, 7,18, 9,22, 10,36, 15,56, 16,40 и 20,86 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем). Кристалл имеет порошковую рентгеновскую дифрактограмму, как показано на фигуре 16, полученную при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
В более конкретном аспекте настоящее изобретение относится к кристаллу нафталин-1,5-дисульфоната N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамида. Более предпочтительно, настоящее изобретение относится к кристаллу нафталин-1,5-дисульфоната N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамида, где кристалл проявляет характеристические пики при углах дифракции 2θ=6,14, 6,98, 11,24, 14,84, 17,48, 19,54, 20,94, 22,38, 23,20 и 24,70 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем). Кристалл имеет порошковую рентгеновскую дифрактограмму, как показано на фигуре 17, полученную при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем). Также, предпочтительно, настоящее изобретение относится к кристаллу нафталин-1,5-дисульфоната N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамида, где кристалл проявляет характеристические пики при углах дифракции 2θ=9,24, 9,58, 14,00, 14,46, 16,70, 17,02, 18,22, 20,24, 21,64 и 25,52 в порошковой рентгеновской дифрактограмме, полученной при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем). Кристалл имеет порошковую рентгеновскую дифрактограмму, как показано на фигуре 18, полученную при облучении с использованием Kα-излучения меди (длина волны λ=1,54 ангстрем).
Соединения, представленные общей формулой (I), по настоящему изобретению могут существовать в виде различных изомеров, включая геометрические изомеры, такие как цис- и транс-изомеры, таутомеры или оптические изомеры, такие как d- и l-изомеры в зависимости от типов и комбинаций заместителей. Соединения по настоящему изобретению также включают все указанные изомеры и стереоизомеры, и смеси указанных изомеров и стереоизомеров в любом соотношении, если не указано иное.
Соединения, представленные общей формулой (I), по настоящему изобретению могут также содержать неестественные соотношения атомных изотопов у одного или нескольких атомов, которые составляют соединения. Примеры атомных изотопов включают дейтерий (2H), также называемый D в настоящем описании, тритий (3H), йод-125 (125I) и углерод-14 (14C). Эти соединения могут быть использованы в качестве терапевтических или профилактических средств, реактивов для исследования (например, реактивов для анализа) и диагностических средств (например, средств для диагностической визуализации in vivo). Все изотопные варианты соединений, представленных общей формулой (I), включены в объем настоящего изобретения, независимо от того являются они радиоактивными или нет.
Кроме того, настоящее изобретение охватывает соединения, которые преобразуются в соединения, представленные общей формулой (I), которые служат в качестве активных ингредиентов в фармацевтических композициях по настоящему изобретению в результате реакций с ферментом, желудочной кислотой и тому подобное в физиологических условиях in vivo, т.е. "фармацевтически приемлемые пролекарственные соединения", которые преобразуются в соединения, представленные общей формулой (I), вследствие ферментативного окисления, восстановления, гидролиза и тому подобного, или преобразуются в соединения, представленные общей формулой (I), вследствие гидролиза и тому подобного, желудочной кислотой или тому подобным.
Примеры пролекарств соединений, представленных общей формулой (I), в которых присутствует аминогруппа, могут включать соединения, в которых аминогруппа ацилирована, алкилирована или фосфорилирована (например, соединения, в которых аминогруппа эйкозаноилирована, аланилирована или пентилaминoкарбонилирована, (5-метил-2-оксо-1,3-диоксолен-4-ил)метоксикарбонилирована, тетрагидрофуранилирована, пирролидилметилирована, пивалоилоксиметилирована или трет-бутилирована). Примеры пролекарств соединений, представленных общей формулой (I), в которых присутствует гидроксигруппа может включать соединения, в которых гидроксигруппа ацилирована, алкилирована, фосфорилирована или боратирована (например, соединения, в которых гидроксигруппа ацетилирована, пальмитоилирована, пропаноилирована, пивалоилирована, сукцинилирована, фумарилирована, аланилирована или диметиламинометилкарбонилирована). Примеры пролекарств соединений, представленных общей формулой (I), в которых присутствует карбоксильная группа, включают соединения, в которых карбоксильная группа этерифицирована или амидирована (например, соединения, в которых карбоксильная группа этилэтерифицирована, фенилэтерифицирована, карбоксиметилэтерифицирована, диметиламинометилэтерифицирована, пивалоилоксиметилэтерифицирована, этоксикарбонилоксиэтилэтерифицирована, амидирована или метиламидирована).
Пролекарства соединений по настоящему изобретению могут быть получены из соединений, представленных общей формулой (I), способами, известными в данной области. Пролекарства соединений по настоящему изобретению также включают соединения, которые преобразуются в соединения, представленные общей формулой (I), в физиологических условиях, как описано в "Iyakuhin No Kaihatsu" [Development of Pharmaceuticals], Vol. 7, Bunshi Sekkei [Molecular Design], Hirokawa Shoten, 1990, pp. 163-198.
Далее будут описаны типичные способы получения соединений, представленных общей формулой (I). Соединения по настоящему изобретению могут быть получены с помощью различных способов получения. Способы получения, показанные ниже, приведены только в иллюстративных целях, и настоящее изобретение не следует рассматривать как ограничивающее объем таким образом. Каждую реакцию можно проводить с заместителями, защищенными соответствующими защитными группами, как это требуется, и тип защитной группы не имеет конкретных ограничений.
[Формула 25]

Можно предположить, что соединения общей формулы (I) образованы отдельными участками, т.е., участком A, участком B и участком C, как описано выше. Примеры основных способов синтеза включают, но конкретно ими не ограничиваются, способ, который включает сначала связывание соединения (В) с соединением (C-I) и связывание комплекса с соединением (A-I), способ, который включает сначала связывание соединения (A-I) с соединением (B) и связывание комплекса с соединением (C-I), способ, который включает сначала связывание соединения (B) с соединением (C-I), связывание комплекса с соединением (A-II), и затем превращение группы брома в R1, способ, который включает превращение группы брома в соединении (BC) в R5 и затем связывание полученного соединения с соединением (A-I) или соединением (A-II), способ, который включает сначала связывание соединения (A-I) с соединением (BC) и затем превращение группы брома в R5, и способ, который включает связывание комплекса соединение (A-I)-соединение (B) с соединением (C-II) и затем превращение группы брома в R5.
[Формула 26]

где R1-R5, W, X, Y и Z являются такими, как определено выше; R31 представляет собой заместитель, необходимый для связывания соединения (В) с соединением (С-I) или соединением (C-II) и представляет собой галогеновую группу, трифлатную группу, бороновую кислоту или эфир бороновой кислоты; R32 представляет собой заместитель, необходимый для связывания соединения (B) с соединением (C-I) и представляет собой галогеновую группу, трифлатную группу, бороновую кислоту или эфир бороновой кислоты; и R33 представляет собой заместитель, необходимый для связывания соединения (B) с соединением (C-II) и представляет собой бороновую кислоту или сложный эфир бороновой кислоты.
Сначала будет описан способ синтеза соединения (A-I).
[Способ получения 1]
Соединение (A-I) может быть синтезировано, но конкретно этим не ограничивается, способом, как показано на следующей схеме реакции 1:
Cхема реакции 1
[Формула 27]

где R1-R3,Q, T и U являются такими, как определено выше; R41 представляет собой галогеновую группу; R42 представляет собой защитную группу для карбоновой кислоты и представляет собой C1-C6 алкильную группу; R43 представляет собой бороновую кислоту или сложный эфир бороновой кислоты; и R44 представляет собой C1-C6 алкильную группу или цикло-С3-C6 алкильную группу. Соединение (1d) входит в соединение (1e).
Синтез соединения (1c)
Коммерчески доступное или соответствующим образом синтезированное соединение (1a) может быть подвергнуто реакции сочетания с коммерчески доступным соединением (1b) или соединением (1b), синтезированным способом, показанным в способе получения 4, способе получения 5 или способе получения 6 в присутствии органического или неорганического основания, такого как карбонат калия, карбонат натрия, карбонат цезия, ацетат калия, фосфат калия, трет-бутоксид или триэтиламин, и катализатора на основе переходного металла, такого как трис(дибензилиденацетон)дипалладий(0), тетракис(трифенилфосфин)палладий(0), палладий(II) бис(трифенилфосфин)хлорид, комплекс палладий(II) [1,1'-бис(дифенилфосфино)ферроцен]хлорид-дихлорметан, ацетат палладия(II), иодид меди(I) или хлорид меди(I) с получением соединения (1c). Реакцию также можно проводить в присутствии лиганда, такого как трифенилфосфин, трициклогексилфосфин, дибензилиденацетон, 1,3-бис(дифенилфосфино)пропан, 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил или 4,5-бис(дифенилфосфино)-9,9-диметилксантен. Реакцию можно также проводить с использованием двух или нескольких типов катализаторов на основе переходного металла в комбинации. Примеры растворителя включают, но конкретно ими не ограничиваются, 1,4-диоксан, 1,2-диметоксиэтан, метанол, толуол, воду и смеси этих растворителей. Температура реакции обычно находится в диапазоне от 0°C до 200°C или точки кипения растворителя, предпочтительно, в диапазоне от 0°C до точки кипения растворителя. Указанная выше обработка также может быть осуществлена при помощи микроволновой печи в герметично закрытой пробирке. Органическое или неорганическое основание может быть использовано в количестве от 1 до избыточных молярных эквивалентов, предпочтительно, от 1 до 5 молярных эквивалентов по отношению к соединению (1a). Катализатор на основе переходного металла или лиганд может быть использован в количестве от 0,001 до 1 молярных эквивалентов, предпочтительно, от 0,05 до 1 молярных эквивалентов по отношению к соединению (1c). Эфир бороновой кислоты может быть использован в количестве от 1 до избыточных молярных эквивалентов, предпочтительно, от 1 до 5 молярных эквивалентов по отношению к соединению (1c).
Синтез соединения (1d)
Соединение (1c) может быть гидролизовано кислотой, такой как серная кислота или хлористоводородная кислота, или щелочью, такой как гидроксид натрия или карбонат калия, с получением соединения (1d). Основание или кислота может быть использована в количестве от 1 до избыточных молярных эквивалентов по отношению к соединению (1c). Примеры растворителя для использования в реакции, включают, но конкретно ими не ограничиваются, метанол, этанол, пропанол, воду и смеси этих растворителей. Температура реакции обычно находится в диапазоне от 0°C до 200°C или точки кипения растворителя, предпочтительно, в диапазоне от 0°C до точки кипения растворителя.
Синтез соединения (1f)
Коммерчески доступное или соответствующим образом синтезированное соединение (1e) можно быть активировано и подвергнуто взаимодействию с моноэфиром малоновой кислоты или ее солью в присутствии хлорида магния с получением соединения (1f). Примеры способа для активации карбоновой кислоты могут включать способ с использованием 1,1'-карбонилдиимидазола и способ, опосредованный хлорангидридом. В случае необходимости, в реакционную систему может быть добавлено основание. Примеры основания могут включать триэтиламин. Основание может быть использовано в количестве от 1 до избыточных молярных эквивалентов, предпочтительно, от 2 до 10 молярных эквивалентов по отношению к соединению (1e). Примеры растворителя для использования в реакции, включают, но конкретно ими не ограничиваются, тетрагидрофуран, диоксан, ацетонитрил, N,N-диметилформамид, толуол и смеси этих растворителей. Температура реакции обычно находится в диапазоне от -78°C до 100°C или точки кипения растворителя, предпочтительно, в диапазоне от приблизительно комнатной температуры до 100°C.
Синтез соединения (1g)
Соединение (1f) может быть обработано диметилацеталем N,N-диметилформамида с получением соединения (1g). Диметилацеталь N,N-диметилформамида может быть использовано в количестве от 2 до избыточных молярных эквивалентов по отношению к соединению (1f). Примеры растворителя для использования в реакции, включают, но конкретно ими не ограничиваются, толуол, ксилол, дихлорметан, этанол, N,N-диметилформамид, этилацетат и смеси этих растворителей. Альтернативно, реакцию осуществляют без растворителя. Температура реакции обычно находится в диапазоне от 0°C до 300°C, предпочтительно, в диапазоне от приблизительно комнатной температуры до 130°C.
Синтез соединения (1i)
Соединение (1g) может быть обработано коммерчески доступным или соответствующим образом синтезированным соединением (1h) с получением соединения (1i). Примеры растворителя для использования в реакции, включают, но конкретно ими не ограничиваются, толуол, этилацетат, этанол, 1,4-диоксан, и смеси этих растворителей. Температура реакции обычно находится в диапазоне от -78°C до 130°C или точки кипения растворителя, предпочтительно, в диапазоне от приблизительно комнатной температуры до точки кипения растворителя. Реакцию можно проводить, при необходимости, путем добавления органического или неорганического основания, такого как карбонат калия, карбонат цезия, трет-бутоксид калия или триэтиламин, или кислоты, такой как уксусная кислота, хлористый водород, бромистый водород или серная кислота. Соединение (1h), основание или кислота может быть использована в количестве от 1 до избыточных молярных эквивалентов, предпочтительно, от 1 до 5 молярных эквивалентов по отношению к соединению (1g).
Синтез соединения (A-I)
Соединение (A-I) может быть получено таким же образом, как в синтезе соединения (1d).
[Способ получения 2]
Соединение (1i) также может быть синтезировано, но конкретно этим не ограничивается, способом, как показано на следующей схеме реакции 2:
Cхема реакции 2
[Формула 28]

где R1 и R42 являются такими, как определено выше; и R45 представляет собой галогеновую группу, метансульфонилоксигруппу, трифторметансульфонилоксигруппу или п-толуолсульфонилоксигруппу. Соединение (2c) входит в соединение (1i).
Синтез соединения (2a)
Соединение (2a) является коммерчески доступным или может быть синтезировано из соединения (1f) на основании предыдущих сообщений, таких как J. Heterocyclic Chem. 17, 359 (1980) and Chem. Pharm. Bull. 43, 450 (1995).
Синтез соединения (2c)
Соединение (2a) может быть обработано соединением (2b) в присутствии органического или неорганического основания, такого как карбонат калия, карбонат цезия, трет-бутоксид калия или триэтиламин с получением соединения (2c). Примеры растворителя для использования в реакции включают, но конкретно ими не ограничиваются, N,N-диметилформамид, диметилсульфоксид, ацетонитрил и смеси этих растворителей. Температура реакции обычно находится в диапазоне от 0°C до 200°C или точки кипения растворителя, предпочтительно, в диапазоне от 0°C до точки кипения растворителя. Соединение (2b) или основание может быть использовано в количестве от 1 до избыточных молярных эквивалентов, предпочтительно, от 1 до 5 молярных эквивалентов по отношению к соединению (2a).
Далее будет описан способ синтеза соединения (A-II).
[Способ получения 3]
Соединение (A-II) может быть синтезировано, но конкретно этим не ограничивается, способом, как показано на следующей схеме реакции 3:
Cхема реакции 3
[Формула 29]

где R42 и R45 являются такими, как определено выше.
Синтез соединения (3b)
Коммерчески доступное соединение (3a) или соединение (3а), которое могут быть легко синтезировано на основании предыдущих публикаций, таких как J. Med. Chem. 51, 5330 (2008), может быть обработано с соединением (2b) в присутствии органического или неорганического основания, такого как карбонат калия, карбонат цезия, трет-бутоксид калия или триэтиламин, с получением соединения (3b). Примеры растворителя для использования в реакции, включают, но конкретно ими не ограничиваются, N,N-диметилформамид, диметилсульфоксид, ацетонитрил и смеси этих растворителей. Температура реакции обычно находится в диапазоне от 0°C до 200°C или точки кипения растворителя, предпочтительно, в диапазоне от 0°C до точки кипения растворителя. Соединение (2b) или основание может быть использовано в количестве от 1 до избыточных молярных эквивалентов, предпочтительно, от 1 до 5 молярных эквивалентов по отношению к соединению (3a).
Синтез соединения (A-II)
Соединение (A-II) может быть получено таким же образом, как в синтезе соединения (A-I) в [способе получения 1].
Далее будет описан способ синтеза соединения (C-I).
Когда R5 представляет собой атом водорода, соединение (C-I) является коммерчески доступным или может быть легко синтезировано из коммерчески доступного сырья способом, известным в литературе.
Когда R5 представляет собой группу, отличную от атома водорода, соединение (С-I) может быть синтезировано с помощью способа, как показано в способе получения 4 - в способе получения 7, приводимые ниже. В частности, соединение (4c), соединение (5h) или соединение (6c), составляющие R5, синтезируют путем выбора любого из способов, показанных в способе получения 4 - способе получения 6. Затем, соединение (C-I) может быть синтезировано с помощью способа получения 7 с использованием соединения, соответственно, синтезированного, или коммерчески доступного соединения, хотя способ синтеза конкретно не ограничивается этим.
[Способ получения 4]
Cхема реакции 4
[Формула 30]

где R8, R9, R11, R12, R43 и R45 являются такими, как определено выше; и R51 представляет собой C1-C6 алкильную группу, необязательно замещенную гетероциклоалкильной группой.
Синтез соединения (4c)
Коммерчески доступное или соответствующим образом синтезированное соединение (4a) может быть обработано соединением (4b) в присутствии органического или неорганического основания, такого как карбонат калия, карбонат цезия, трет-бутоксид калия или триэтиламин, с получением соединения (4c). Примеры растворителя для использования в реакции, включают, но конкретно ими не ограничиваются, N,N-диметилформамид, диметилсульфоксид, ацетонитрил и смеси этих растворителей. Температура реакции обычно находится в диапазоне от 0°C до 200°C или точки кипения растворителя, предпочтительно, в диапазоне от 0°C до точки кипения растворителя. Соединение (4b) или основание может быть использовано в количестве от 1 до избыточных молярных эквивалентов, предпочтительно, от 1 до 5 молярных эквивалентов по отношению к соединению (4a).
[Способ получения 5]
Cхема реакции 5
[Формула 31]

где R8, R9, R12, R43, R45 и R51 являются такими, как определено выше; R61 представляет собой дейтерий или атом водорода; R62 представляет собой гидроксильную группу или дейтерий-замещенную защитную группу для гидроксильной группы, где примеры защитной группы включают простые эфирные группы, представленные тетрагидропиранильной группой, силильные группы, представленные трет-бутилдифенилсилильной группой, сложноэфирные группы, представленные ацетильной группой, и карбонаты, представленные винилкарбонатом; и R63 представляет собой дейтерий-замещенную C1-C6 алкильную группу.
Синтез соединения (5b)
Только одна из двух гидроксильных групп в коммерчески доступном или соответствующим образом синтезированном соединении (5a) может быть защищена для получения соединения (5b). Например, соединение (5a) может быть обработано 3,4-дигидро-2Н-пираном в присутствии кислоты, такой как пиридиниевая соль п-толуолсульфоновой кислоты, с получением соединения (5b) только с одной из гидроксильных групп, защищенной тетрагидропиранильной группой, хотя способ защиты гидроксильной группы конкретно не ограничивается этим. Примеры растворителя для использования в реакции, включают, но конкретно ими не ограничиваются, дихлорметан, хлороформ, толуол и смеси этих растворителей. Температура реакции обычно находится в диапазоне от 0°C до 200°C или точки кипения растворителя, предпочтительно, в диапазоне от 0°C до точки кипения растворителя. Соединение (4b) или кислота может быть использовано в количестве от 0,01 до избыточных молярных эквивалентов, предпочтительно, от 0,01 до 1 молярных эквивалентов по отношению к соединению (5a).
Синтез соединения (5d)
Соединение (5b) может быть обработано соединением (5c) в присутствии органического или неорганического основания, такого как карбонат калия, карбонат цезия, трет-бутоксид калия или триэтиламин, с получением соединения (5d). Примеры растворителя для использования в реакции, включают, но конкретно ими не ограничиваются, N,N-диметилформамид, диметилсульфоксид, ацетонитрил, ацетон и смеси этих растворителей. Температура реакции обычно находится в диапазоне от 0°C до 200°C или точки кипения растворителя, предпочтительно, в диапазоне от 0°C до точки кипения растворителя. Соединение (5c) или основание может быть использовано в количестве от 1 до избыточных молярных эквивалентов, предпочтительно, от 1 до 5 молярных эквивалентов по отношению к соединению (5b).
Синтез соединения (5e)
Соединение (5d) может быть обработано кислотой, такой как хлористоводородная кислота, уксусная кислота или пиридиниевая соль п-толуолсульфоновой кислоты, с получением соединения (5e). Примеры растворителя для использования в реакции, включают, но конкретно ими не ограничиваются, этанол, метанол, изопропанол, воду и смеси этих растворителей. Температура реакции обычно находится в диапазоне от 0°C до 200°C или точки кипения растворителя, предпочтительно, в диапазоне от 0°C до точки кипения растворителя. Соединение (4b) или кислота может быть использована в количестве от 0,01 до избыточных молярных эквивалентов, предпочтительно, от 0,01 до 1 молярных эквивалентов по отношению к соединению (5d).
Синтез соединения (5f)
Соединение (5e) может быть обработано бромирующим реагентом, таким как N-бромсукцинимид или бромат калия, с получением соединения (5f). Примеры растворителя для использования в реакции, включают, но конкретно ими не ограничиваются, N,N-диметилформамид, 30% раствор бромистоводородной кислоты в уксусной кислоте, уксусную кислоту и смеси этих растворителей. Температура реакции обычно находится в диапазоне от 0°C до 200°C или точки кипения растворителя, предпочтительно, в диапазоне от 0°C до точки кипения растворителя. Бромирующий реагент может быть использован в количестве от 0,1 до избыточных молярных эквивалентов, предпочтительно, от 0,1 до 5 молярных эквивалентов по отношению к соединению (5e).
Синтез соединения (5g)
Соединение (5g) может быть получено таким же образом, как в [способе получения 4].
Синтез соединения (5h)
Соединение (5g) может быть обработано сложным эфиром бороновой кислоты, таким как бис(пинаколато)диборон или пинакол борон, в присутствии органического или неорганического основания, такого как карбонат калия, карбонат натрия, карбонат цезия, ацетат калия, фосфат калия, трет-бутоксид или триэтиламин, катализатора на основе переходного металла, такого как трис(дибензилиденацетон)-дипалладий(0), бис(трифенилфосфин)хлорид палладий(II), комплекс палладий(II) [1,1'-бис(дифенилфосфино)ферроцен]хлорид-дихлорметан, комплекс палладий(II) [1,1'-бис(дифенилфосфино)ферроцен]хлорид-дихлорметан или никель(II) 1,3-бис(дифенилфосфино)пропан]хлорид, и лиганда, такого как трифенилфосфин, трициклогексилфосфин, дибензилиденацетон, 1,3-бис(дифенилфосфино)пропан, 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил или 4,5-бис(дифенилфосфино)-9,9-диметилксантен, с получением соединения (5h). Примеры растворителя включают, но конкретно ими не ограничиваются, 1,4-диоксан, 1,2-диметоксиэтан, метанол, толуол и смеси этих растворителей. Температура реакции обычно находится в диапазоне от 0°C до 200°C или точки кипения растворителя, предпочтительно, в диапазоне от 0°C до точки кипения растворителя. Указанная выше обработка также может быть осуществлена при помощи микроволновой печи в герметично закрытой пробирке. Органическое или неорганическое основание может быть использовано в количестве от 1 до избыточных молярных эквивалентов, предпочтительно от 1 до 5 молярных эквивалентов по отношению к соединению (5g). Катализатор на основе переходного металла или лиганд может быть использован в количестве от 0,001 до 1 молярных эквивалентов, предпочтительно, от 0,05 до 1 молярных эквивалентов по отношению к соединению (5g). Эфир бороновой кислоты может быть использован в количестве от 1 до избыточных молярных эквивалентов, предпочтительно, от 1 до 5 молярных эквивалентов по отношению к соединению (5g).
[Способ получения 6]
Cхема реакции 6
[Формула 32]

где R13, R43 и R45 являются такими, как определено выше.
Синтез соединения (6c)
Соединение (6c) может быть получено таким же образом, как в [способе получения 4] с использованием коммерчески доступных или соответствующим образом синтезированных соединений (6a) и (6b).
[Способ получения 7]
Cхема реакции 7
[Формула 33]

где R5 и R43 являются такими, как определено выше; и R81 представляет собой галогеновую группу. Соединение (7a) представляет собой соединение, содержащее бороновую кислоту или эфир бороновой кислоты, соединение (4c), соединение (5h) или соединение (6c). Соединение (7c) и соединение (7d) входят в соединение (C-I).
Синтез соединения (7c)
Соединение (7b) может быть подвергнуто реакции сочетания с коммерчески доступным соединением (7а) или соединением (7а), синтезированным способом, показанным в способе получения 4, способе получения 5 или способе получения 6, таким же образом, как в синтезе соединения (1c) в [способ получения 1] с получением соединения (7c).
Синтез соединения (7d)
Соединение (7d) может быть получено таким же образом, как в синтезе соединения (5h) в [способе получения 5] с использованием коммерчески доступного или соответствующим образом синтезированного соединения (7c).
Далее будет описан способ синтеза комплекса соединение (В)-соединение (С-I).
Комплекс соединение (В)-соединение (С-I) может быть синтезирован посредством способа превращения группы брома в соединении (BC) в R5, как показано в [способе получения 8], хотя способ синтеза конкретно не ограничивается.
[Способ получения 8]
Cхема реакции 8
[Формула 34]

где R4, R5, W, X, Y, Z и R43 являются такими, как определено выше. Соединение (8) входит в комплекс соединение (B)-соединение (C-I).
Соединение (8) может быть получено таким же образом, как в синтезе соединения (1c) в [способе получения 1] с использованием коммерчески доступного или соответствующим образом синтезированного соединения (BC) или соединения (8).
Соединение (8) также может быть синтезировано с помощью способа, показанного в [способе получения 9] или [способе получения 10], хотя способ синтеза конкретно не ограничивается.
[Способ получения 9]
Cхема реакции 9
[Формула 35]

где R4, R5, W, X, Y, Z и R81 являются такими, как определено выше. Соединение (8) входит в комплекс соединение (B)-соединение (C-I).
Соединение (8) может быть получено таким же образом, как в синтезе соединения (1c) в [способе получения 1] с использованием коммерчески доступных или соответствующим образом синтезированных соединений (9a) и (9b).
[Способ получения 10]
Cхема реакции 10
[Формула 36]

где R4, R5, W, X, Y, Z и R81 являются такими, как определено выше. Соединение (8) входит в комплекс соединение (B)-соединение (C-I).
Соединение (8) может быть получено таким же образом, как в синтезе соединения (1c) в [способе получения 1] с использованием коммерчески доступного или соответствующим образом синтезированного соединения (7d) и соединения (10).
Далее будет описан способ синтеза для связывания комплекса соединение (В)-соединение (С-I) с соединением (A-I).
Комплекс соединение (В)-соединение (С-I) может быть связан с соединением (А-I) в соответствии со следующим способом получения 11 или способом получения 12:
[Способ получения 11]
Соединение (11) может быть синтезировано, но конкретно этим не ограничивается, способом, как показано на следующей схеме реакции 11:
Cхема реакции 11
[Формула 37]

где R1-R5, W, X, Y и Z являются такими, как определено выше. Соединение (8) входит в комплекс соединение (B)-соединение (C-I).
Соединение (А-I) может быть подвергнуто взаимодействию с соединением (8) в присутствии конденсирующего средства с получением соединения (11). Примеры конденсирующего средства включают, но не ограничиваются ими, N-[1-(циано-2-этокси-2-оксоэтилиденаминоокси)диметиламино(морфолино)]урония гексафторфосфат (COMU). Примеры растворителя для использования в реакции, включают, но конкретно ими не ограничиваются, N,N-диметилформамид, дихлорметан, и смеси этих растворителей. Температура реакции обычно находится в диапазоне от -78°C до 200°C или точки кипения растворителя, предпочтительно, в диапазоне от 0°C до 100°C. Конденсирующее средство может быть использовано в количестве от 1 до избыточных молярных эквивалентов, предпочтительно, от 1 до 5 молярных эквивалентов по отношению к соединению (A-I).
[Способ получения 12]
Соединение (11) может быть синтезировано, но конкретно этим не ограничивается, способом, как показано на следующей схеме реакции 12:
Cхема реакции 12
[Формула 38]

где R1-R5, W, X, Y и Z являются такими, как определено выше. Соединение (8) входит в комплекс соединение (B)-соединение (C-I).
Соединение (А-I) может быть обработано кислотным галогенирующим реагентом, таким как тионилхлорид или оксалилхлорид, с получением соединения (12), которое затем обрабатывают соединением (8) с получением соединения (11). Примеры растворителя для использования в реакции, включают, но конкретно ими не ограничиваются, дихлорметан, дихлорэтан и смеси этих растворителей. Кислотный галогенирующий реагент может быть использован в количестве от 0,9 до избыточных молярных эквивалентов, предпочтительно, от 0,9 до 2 молярных эквивалентов по отношению к соединению (A-I).
Далее будет описан способ синтеза комплекса соединение (A-I)-соединение (В).
[Способ получения 13]
Соединение (13) может быть синтезировано, но конкретно этим не ограничивается, способом, как показано на следующей схеме реакции 13.
Cхема реакции 13
[Формула 39]

где R1-R4, W, X, Y, и R31 являются такими, как определено выше. Соединение (13) входит в комплекс соединение (A-I)-соединение (B).
Синтез соединения (13)
Соединение (B) является коммерчески доступным или может быть легко синтезировано из коммерчески доступного сырья способом, известным в литературе. Соединение (13) может быть получено таким же образом, как в [способе получения 11] с использованием соединения (A-I) и коммерчески доступного или соответствующим образом синтезированного соединения (B).
Далее будет описан способ синтеза для связывания комплекса соединение (А-I)-соединение (В) с соединением (C-I).
[Способ получения 14]
Соединение (11) может быть синтезировано, но конкретно этим не ограничивается, способом, как показано на следующей схеме реакции 14:
Cхема реакции 14
[Формула 40]

где R1-R5, W, X, Y и Z являются такими, как определено выше; когда R31 представляет собой галогеновую группу, трифлатную группу, или тому подобное, R32 представляет собой бороновую кислоту или сложный эфир бороновой кислоты; и когда R31 представляет собой бороновую кислоту или сложный эфир бороновой кислоты, R32 представляет собой галогеновую группу, трифлатную группу, или тому подобное. Соединение (13) входит в комплекс соединение (A-I)-соединение (B).
Синтез соединения (11)
Соединение (11) может быть получено таким же образом, как в синтезе соединения (1c) в [способе получения 1] с использованием соединения (13) и коммерчески доступного или соответствующим образом синтезированного соединения (C-I).
Далее будет описан способ синтеза для связывания комплекса соединение (В)-соединение (С-I) с соединением (A-II).
[Способ получения 15]
Соединение (15) может быть синтезировано, но конкретно этим не ограничивается, способом, как показано на следующей схеме реакции 15:
Cхема реакции 15
[Формула 41]

где R4-R5, W, X, Y и Z являются такими, как определено выше. Соединение (8) входит в комплекс соединение (B)-соединение (C-I).
Синтез соединения (15)
Соединение (15) может быть получено таким же образом, как в [способе получения 11] с использованием соединения (A-II) и соединения (8).
Далее будет описан способ синтеза для превращения брома в комплексе соединение (A-II)-соединение (В)-соединение (С-I) в R1.
[Способ получения 16]
Соединение (16b) может быть синтезировано, но конкретно этим не ограничивается, способом, как показано на следующей схеме реакции 16:
Cхема реакции 16
[Формула 42]
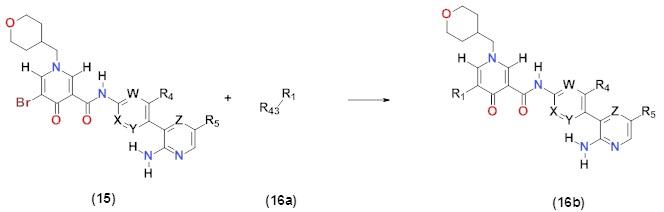
где R1, R4-R5, W, X, Y, Z и R43 являются такими, как определено выше. Соединение (15) входит в комплекс соединение (A-I)-соединение (В)-соединение (C-I).
Синтез соединения (16b)
Соединение (16b) может быть получено таким же образом, как в синтезе соединения (1c) в [способе получения 1] с использованием соединения (15) и коммерчески доступного или соответствующим образом синтезированного соединения (16a).
Далее будет описан способ синтеза для превращения галогеновой группы в комплексе соединение (A-I)-соединение (В)-соединение (C-I) в алкильную группу.
[Способ получения 17]
Соединение (17b) может быть синтезировано, но конкретно этим не ограничивается, способом, как показано на следующей схеме реакции 17:
Cхема реакции 17
[Формула 43]

где R2-R5, Q, т, U, W, X, Y, Z, R43 и R44 являются такими, как определено выше. Соединение (17a) и соединение (17b) входят в комплекс соединение (A-I)-соединение (B)-соединение (C-I).
Синтез соединения (17b)
Соединение (17b) может быть получено таким же образом, как в синтезе соединения (1c) в [способе получения 1] с использованием соединения (17a) и коммерчески доступного или соответствующим образом синтезированного соединения (1b).
Далее будет описан способ синтеза для связывания соединения (А-I) с соединением (ВС) и затем превращение группы брома в R5.
[Способ получения 18]
Соединение (11) может быть синтезировано, но конкретно этим не ограничивается, способом, как показано на следующей схеме реакции 18:
Cхема реакции 18
[Формула 44]

где R1-R5, W, X, Y, Z и R43 являются такими, как определено выше.
Синтез соединения (18)
Соединение (18) может быть получено таким же образом, как в [способе получения 11] с использованием соединения (A-I) и соединения (BC).
Синтез соединения (11)
Соединение (11) может быть получено таким же образом, как в синтезе соединения (1c) в [способе получения 1] с использованием соединения (18) и коммерчески доступного или соответствующим образом синтезированного соединения (7a).
Соединение (18) может также быть синтезировано способом, как показано в [способе получения 19].
[Способ получения 19]
Cхема реакции 19
[Формула 45]

где R1-R4, R33, R81, W, X, Y и Z являются такими, как определено выше. Соединение (19) входит в соединение (13).
Соединение (С-II) является коммерчески доступным или может быть легко синтезировано из коммерчески доступного вещества способом, известным в литературе. Соединение (18) может быть получено таким же образом, как в синтезе соединения (1c) в [способе получения 1] с использованием соединения (19) и коммерчески доступного или соответствующим образом синтезированного соединения (C-II).
Соединение (11) также может быть получено в соответствии со способом получения 20.
[Способ получения 20]
Cхема реакции 20
[Формула 46]

где R1-R4, R43, W, X, Y и Z являются такими, как определено выше; R201 представляет собой атом водорода или галоген; и R202 представляет собой C1-C6 алкиламино-группу, необязательно образующую кольцо или окси-C1-C6 алкиламино-группу, необязательно образующую кольцо. Соединение (20d) входит в соединение (11).
Синтез соединения (20b)
Соединение (20b) может быть получено таким же образом, как в синтезе соединения (1c) в [способе получения 1] с использованием соединения (18) и коммерчески доступного или соответствующим образом синтезированного соединения (20a).
Синтез соединения (20d)
Соединение (20b) может быть подвергнуто взаимодействию с коммерчески доступным или соответствующим образом синтезированным соединением (20в) в присутствии восстанавливающего агента с получением соединения (20d). Примеры используемого восстанавливающего агента могут включать триацетоксиборгидрид натрия, цианоборгидрид натрия и боргидрид натрия. Примеры используемого растворителя включают, но конкретно ими не ограничиваются, метанол, этанол, воду, тетрагидрофуран, диоксан, N,N-диметилформамид, метиленхлорид, уксусную кислоту, и смеси этих растворителей. Температура реакции обычно находится в диапазоне от -78°C до 100°C или точки кипения растворителя, предпочтительно, в диапазоне от 0°C до 100°C.
Как упоминалось выше, сообщалось, сигнальный путь системы Gas6/Axl регулирует различные клеточные реакции, такие как выживание клеток, клеточное деление, аутофагию, миграцию клеток, ангиогенез, агрегацию тромбоцитов и дифференциацию природных клеток-киллеров (непатентный документ 2). Поэтому, ингибиторы Axl являются полезными для лечения заболевания, вызванного гиперфункцией Axl киназы, заболевания, связанного с гиперфункцией Axl киназы, и/или заболевания, сопровождающегося гиперфункцией Axl киназы.
В частности, соединения по настоящему изобретению, могут быть использованы более безопасно, поскольку соединения по настоящему изобретению обладают высокой Axl ингибирующей специфичностью и не вызывают ретинальную токсичность.
Примеры заболеваний, вызванных гиперфункцией Axl-киназы, заболеваний, связанных с гиперфункцией Axl-киназы, и заболеваний, сопровождающихся гиперфункцией Axl-киназы, включают заболевания, имеющие ткани, содержащие сверхэкспрессированные гены и/или белки Axl, и заболевания, имеющие ткани с повышенной фосфорилирующей активностью Axl.
Примеры вышеуказанных заболеваний включают гиперпролиферативные заболевания. Примеры гиперпролиферативных заболеваний включают, но ими не ограничиваются, гиперплазию эндометрия, тромбин-индуцированную пролиферацию гладкомышечной клетки сосудов (VSMC), доброкачественную опухоль, злокачественную опухоль (рак), острый и хронический гломерулонефрит и диабетическую нефропатию.
Было дополнительно установлено, что Axl играет роль в иммунитете (непатентный документ 12), функциях тромбоцитов (непатентный документ 13), сперматогенезе (непатентный документ 14), сосудистой кальцификации (непатентный документ 15), (непатентный документ 16) и различных заболеваниях почек и хроническом отторжении аллотрансплантата (непатентный документ 17). Таким образом, ингибиторы Axl полезны при лечении многих заболеваний, включая сосудистые заболевания (включая, но ими не ограничиваясь, тромбоз, атеросклероз и рестеноз) и заболевания, при которых дезорганизованное образование кровеносных сосудов имеет серьезные последствия (включая, но ими не ограничиваясь, диабетическую ретинопатию, ретинопатию, псориаз, ревматоидный артрит, атерому, саркому Капоши и ангиому).
Соединения по настоящему изобретению ингибируют Axl и, следовательно, полезны при лечении заболеваний, описанных выше.
Более предпочтительно, соединения по настоящему изобретению могут быть использованы при лечении различных видов рака. Примеры видов рака включают, но ими не ограничиваются, рак молочной железы, рак толстой кишки, рак предстательной железы, рака легкого, рак желудка, рак яичника, рак шейки матки, рак эндометрия, рак тела матки, рак почки, гепатоцеллюлярный рак, рак щитовидной железы, рак пищевода, плоскоклеточный рак, лейкоз, остеосаркома, меланома, глиобластома, нейробластома, рак яичника, рак головы и шеи, тестикулярный рак, колоректальный рак, рак крови, ретинобластома и рак поджелудочной железы.
Различные доклады об отношении Axl к видам рака были сделаны с точки зрения ингибирования роста, ингибирования метастазирования, миграции или инвазии, и преодоления лекарственной устойчивости.
Как сообщалось, доминантно-негативные мутации Axl ингибируют рост опухоли головного мозга (Vajkoczy et al., PNAS 2006, 103, 5799). Сообщалось, что опухоли в тканях у пациента с глиобластомой, имеющие экспрессию Axl или коэкспрессию Axl/Gas6, растут значительно быстрее и в результате укорачивает время жизни пациентов (Hutterer et al., Clin Cancer Res 14, 130 (2008)). Сообщалось, что кшРНК Axl ингибируют рост клеток рака молочной железы (Yi-Xiang et al., Cancer Res 68, 1905 (2008)). Как видно из этих сообщений, ингибиторы Axl полезны для ингибирования роста клеток при раковых заболеваниях.
С другой стороны, как сообщалось, доминантно-негативные мутации Axl ингибируют миграцию клеток и инвазию (Zhang et al., Cancer Res 68, 1905 (2008); Vajkoczy et al., PNAS 103, 5799 (2006); and Holland et al., Cancer Res 65, 9294 (2005)). Сообщалось, что кшРНК Axl ингибируют метастазирование in vivo (Li et al., oncogene 28, 3442 (2009)). Сообщалось, что антитела против Axl и миРНК ингибируют рост опухоли и метастазирование в мышиной модели (Li et al., Oncogene 28, 3442 (2009); Ye et al., Oncogene 29, 5254 (2010)). Сообщалось, что Axl активирует инвазию клеток (Tai et al., Oncogene 27, 4044 (2008)). Сообщалось, что R-428, ингибитор Axl, ингибирует диффузную форму метастатического рака молочной железы (Holland et al., Cancer Res 70, 1544 (2010)). Сообщалось, что антитела против Axl, кшРНК Axl и ингибитор NA80×1 Axl ингибируют миграцию и инвазию клеток рака молочной железы (Yi-Xiang et al., Cancer Res 68, 1905 (2008)). Кроме того, есть множество отчетов об участии Axl в метастазировании и злокачественном прогрессировании рака простаты, рака селезенки, метастатического рака яичника, рака вилочковой железы и тому подобное. Как видно из этих сообщений, ингибиторы Axl являются полезными, например, для ингибирования, лечения и профилактики метастазирования рака, миграции клеток и инвазии клеток.
Кроме того, сообщалось, что ингибиторы Axl преодолели устойчивость к иматинибу при раке желудочно-кишечного тракта (Mahadevan et al., Oncogene 26, 3909 (2007)). Было обнаружено, что Axl индуцирует устойчивость острого миелоидного лейкоза к химиотерапевтическим средствам, таким как доксорубицин, VP16 и цисплатин (Hong et al., Cancer Letters 268, 314 (2008)). Axl, как сообщается, активируется в устойчивости к лапатинибу в клетках HER-2 положительного рака молочной железы (Liu et al., Cancer Res 69, 6871 (2009)). Сообщалось, что Axl участвует в механизме устойчивости к PLX4032 (вемурафениб) (Johannessen et al., Nature 468, 968 (2010)). Кроме того, сообщалось, что Axl участвует в устойчивости к темозоломиду, карбоплатину и винкристину (AK Keeating et al., Mol Cancer Ther 9 (5), 1298 (2010)). Как видно из этих сообщений, ингибиторы Axl полезны в преодолении лекарственной устойчивости, например, преодолении устойчивости к различным противораковым средствам.
Ингибиторы Axl по настоящей заявке, как показано в тестовых примерах по настоящей заявке, ингибируют устойчивость опухолей к эрлотинибу путем введения в комбинации с эрлотинибом. Наблюдалась значительная индукция белка Axl при введении эрлотиниба, что указывает на то, что Axl участвует в приобретении лекарственной устойчивости злокачественных заболеваний.
Комбинированное использование с ингибиторами Axl по настоящей заявке в случае опухолей, резистентных к эрлотинибу, восстановило чувствительность к эрлотинибу, что указывает на то, что ингибиторы Axl являются эффективными для преодоления лекарственной устойчивости злокачественных заболеваний.
Также сообщалось, что Axl участвует в заболеваниях почек, таких как образование фибрилл в почках и диабетическая нефропатия (Национальная публикация международной патентной заявки No. 2005-517412). Таким образом, ингибиторы Axl, очевидно, полезны при лечении таких заболеваний почек, а также фиброзирующих заболеваний, таких как идиопатический фиброз легких.
Axl ингибирующая активность соединений может быть измерена, но ими не ограничивается, способами, описанными, например, в Тестовых примерах настоящей заявки.
Ингибирующая активность в отношении роста клеток может быть исследована с помощью способа анализа ингибирования роста, обычно используемого специалистами в данной области. Ингибирующая активность в отношении роста клеток может быть определена сравнением степени роста клеток (например, опухолевых клеток) в присутствии и в отсутствие тестируемого соединения. Степень роста может быть исследована с использованием, например, тест-системы для измерения живых клеток. Примеры способа измерения живых клеток включают анализ захвата [3H]-тимидина, BrdU-метод и МТТ-анализ.
In vivo противоопухолевую активность можно определять, используя метод тестирования противоопухолевой активности, обычно используемый специалистом в данной области техники. In vivo противоопухолевую активность в соответствии с настоящим изобретением, подтверждали, например: трансплантировали различные типы опухолевых клеток мышам, крысам или им подобным; после подтверждения жизнеспособности трансплантированных клеток, вводили соединения по настоящему изобретению животному посредством перорального введения, внутривенного введения, или тому подобное; спустя время от нескольких дней до нескольких недель, сравнивали рост опухоли в группе, которой вводили носитель, с ростом опухоли в группе, которой вводили соединение.
Кроме того, ингибирующая активность в отношении метастазирования, ингибирующая активность в отношении инвазии, ингибирующая активность в отношении миграции и активность преодоления устойчивости к лекарственному средству может быть измерена с помощью методов испытаний, описанных выше, в литературе, представляющей информацию об отношении Axl к каждой активности.
Фармацевтические композиции по настоящему изобретению содержат соединение по настоящему изобретению и фармацевтически приемлемый носитель и могут быть введены в виде различных инъекций, таких как внутривенные инъекции, внутримышечные инъекции или подкожные инъекции, либо различными способами, такими как пероральное введение или трансдермальное введение. Фармацевтически приемлемый носитель означает фармацевтически приемлемое вещество (например, эксципиент, разбавитель, вспомогательное вещество или растворитель), связанное с транспортом соединения по настоящему изобретению или композиции, содержащей соединение по настоящему изобретению, от одного органа к другому органу.
Лекарственные формы (например, пероральные лекарственные формы или инъекции) могут быть соответствующим образом выбраны в соответствии со способом введения и получены способами, обычно используемыми для получения различных лекарственных форм. Примеры пероральных лекарственных форм могут включать таблетки, порошки, гранулы, капсулы, пилюли, пастилки, растворы, сиропы, эликсиры, эмульсии и масляные или водные суспензии. Эти лекарственные формы могут быть введены перорально или в свободной форме или в форме соли. Водные лекарственные формы могут быть получены путем образования аддуктов кислоты с фармацевтически приемлемыми кислотами, или путем образования солей щелочных металлов, таких как натрий. В случае инъекций, стабилизаторы, консерванты, солюбилизирующие средства и тому подобное, могут быть использованы в лекарственных формах. Растворы, которые могут содержать эти адъюванты и тому подобное, могут храниться в контейнерах и затем подвергали лиофилизации, например, с образованием твердых композиций, которые готовят перед использованием. Одна доза может храниться в одном контейнере, или многократное число доз может храниться в одном контейнере.
Примеры твердых лекарственных форм включают таблетки, порошки, гранулы, капсулы, пилюли и лепешки. Указанные твердые лекарственные формы могут содержать фармацевтически приемлемые добавки вместе с соединениями по настоящему изобретению. Примеры добавок включают наполнители, объемообразующие агенты, связующие вещества, разрыхлители, вещества, способствующие растворению, смачивающие агенты и лубриканты, которые могут быть выбраны и смешаны, при необходимости, для получения лекарственных форм.
Примеры жидких лекарственных форм включают растворы, сиропы, эликсиры, эмульсии и суспензии. Указанные жидкие лекарственные формы могут содержать фармацевтически приемлемые добавки вместе с соединениями по настоящему изобретению. Примеры добавок включают суспендирующие вещества и эмульгаторы, которые могут быть выбраны и смешаны, при необходимости, для получения лекарственных форм.
Соединения по настоящему изобретению могут быть использованы для лечения рака у млекопитающих, в частности, у человека. Доза и интервал между введением дозы лекарственного препарата могут быть выбраны должным образом на основании заключения врача, в соответствии с локализацией заболевания и ростом, массой тела, полом или историей болезни пациента. Когда соединение по настоящему изобретению вводят человеку, доза находится в диапазоне приблизительно от 0,01 мг/кг массы тела до приблизительно 500 мг/кг массы тела в день, предпочтительно, приблизительно от 0,1 мг/кг массы тела до приблизительно 100 мг/кг массы тела в день. При введении человеку, дозу, предпочтительно, вводят, или разовой дозой или раздельными дозами количеством от двух до четырех в день, и введение предпочтительно повторяют через определенные промежутки времени. Суточная доза может превышать вышеуказанную дозу, если это необходимо на основании заключения врача.
Соединения по настоящему изобретению могут быть использованы в комбинации с дополнительными противоопухолевыми средствами. Примеры дополнительных противоопухолевых средств включают ингибиторы тирозинкиназы (ИТК), такие как эрлотиниб, перечисленные выше, а также противоопухолевые антибиотики, противоопухолевые растительные компоненты, BRM (модификаторы биологической реакции), гормоны, витамины, противоопухолевые антитела, лекарственные средства, действующие прицельно на молекулярном уровне, и другие противоопухолевые средства.
Более конкретно, примеры алкилирующих средств включают: алкилирующие средства, такие как азотистый иприт, N-оксид азотистого иприта и хлорамбуцил; азиридиновые алкилирующие средства, такие как карбоквон и тиотепа; эпоксидные алкилирующие средства, такие как дибромманнит и дибромдулцитол; алкилирующие средства из производных нитрозомочевины, такие как кармустин, ломустин, семустин, нимустингидрохлорид, стрептозоцин, хлорзотоцин и ранимустин; и другие, такие как бусульфан, импросульфан тозилат и дакарбазин.
Примеры различных антиметаболитов включают: пуриновые антиметаболиты, такие как 6-меркаптопурин, 6-тиогуанин и тиоинозин; пиримидиновые антиметаболиты, такие как фторурацил, тегафур, тегафур-урацил, кармофур, доксифлуридин, броксуридин, цитарабин и эноцитабин; и антифолаты, такие как метотрексат и триметрексат.
Примеры противоопухолевых антибиотиков включают: антрациклиновые противоопухолевые средства, такие как митомицин С, блеомицин, пепломицин, даунорубицин, акларубицин, доксорубицин, пирарубицин, THP-адриамицин, 4'-эпидоксорубицин и эпирубицин; и другие, такие как хромомицин A3 и актиномицин D.
Примеры противоопухолевых растительных компонентов включают: алкалоиды барвинка, такие как виндезин, винкристин и винбластин; таксаны, такие как паклитаксел и доцетаксел; и эпиподофиллотоксины, такие как этопозид и тенипозид.
Примеры BRM включают факторы некроза опухоли и индометацин.
Примеры гормонов включают гидрокортизон, дексаметазон, метилпреднизолон, преднизолон, прастерон, бетаметазон, триамцинолон, оксиметолон, нандролон, метенолон, фосфестрол, этинилэстрадиол, хлормадинон и медроксипрогестерон.
Примеры витаминов включают витамин С и витамин А.
Примеры противоопухолевых антител и лекарственных средств, прицельно действующих на молекулярном уровне, включают трастузумаб, ритуксимаб, цетуксимаб, нимотузумаб, денозумаб, бевацизумаб, инфликсимаб, иматиниб мезилат, гефитиниб, эрлотиниб, сунитиниб, лапатиниб, сорафениб, дазатиниб, нилотиниб, вемурафениб и тивантиниб.
Примеры других противоопухолевых средств включают цисплатин, карбоплатин, оксалиплатин, тамоксифен, камптотецин, ифосфамид, циклофосфамид, мелфалан, L-аспарагиназу, ацеглатон, сизофиран, пицибанил, прокарбазин, пипоброман, неокарциностатин, гидроксимочевину, убенимекс и крестин.
Настоящее изобретение также относится к способу профилактики и/или лечения рака, включающему введение соединения по настоящему изобретению или его соли.
Настоящее изобретение дополнительно включает применение соединения по настоящему изобретению или его соли, или сольвата соединения или соли для получения вышеуказанного лекарственного средства.
Настоящее изобретение будет подробно описано со ссылкой на Примеры, представленные ниже. Тем не менее, настоящее изобретение не ограничивается этими примерами, и эти примеры не следует рассматривать как ограничивающие в каком-либо смысле. Реагенты, растворители и исходные вещества, описанные в настоящем описании, легко доступны из коммерческих источников, если не указано иное.
[Примеры]
Аббревиатуры
DMF: N,N-диметилформамид
THF: тетрагидрофуран
HATU: N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония гексафторфосфат
HOAt: 1-гидрокси-7-азабензотриазол
DIPEA: N,N-диизопропилэтиламин
COMU: (1-циано-2-этокси-2-оксоэтилиденаминоокси)диметиламино-морфолино-карбений гексафторфосфат
TFA: трифторуксусная кислота
EDC⋅HCl: 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид
HOBt: 1-гидроксибензотриазол
DMAP: 4-диметиламинопиридин
ПТСХ: препаративная тонкослойная хроматография
ВЭЖХ: высокоэффективная жидкостная хроматография
(Промежуточное соединение 1a) 5-(4-Метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоновая кислота
[Формула 47]

(Стадия 1) Этил 4-(4-метилфенил)-3-оксобутаноат
(4-Метилфенил)уксусную кислоту (75,0 г, 499 ммоль) растворяли в тетрагидрофуране (1,00 л). К раствору добавляли карбонилдиимидазол (105 г, 649 ммоль) и смесь перемешивали при комнатной температуре в течение 40 минут. К реакционной смеси добавляли калиевую соль сложного моноэтилового эфира малоновой кислоты (111 г, 649 ммоль) и безводный хлорид магния (57,1 г, 599 ммоль), и смесь нагревали при перемешивании при температуре 60°C в течение 6 часов. Реакционную смесь охлаждали до комнатной температуры, и добавляли к этому этилацетат (2,00 л), затем промывали 1 н хлористоводородной кислотой (1,00 л). Органический слой промывали насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором в данном порядке. Органический слой сушили над безводным сульфатом натрия, и затем, растворитель отгоняли при пониженном давлении. Полученное масло подвергали азеотропной перегонке с толуолом при пониженном давлении с получением указанного в заголовке соединения (86,8 г, выход: 78,8%) в виде масла.
1Н-ЯМР (CDCl3) g(47,18-7,07 (4H, м), 4,17 (2H, кв, J=7,5 Гц), 3,78 (2H, с), 3,43 (2H, с), 2,33 (3H, с), 1,26 (3H, т, J=7,5 Гц).
МС (ESI) m/z:221 [(M+H)+].
(Стадия 2) Этил 5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксилат
Этил 4-(4-метилфенил)-3-оксобутаноат (43,0 г, 195 ммоль) растворяли в толуоле (390 мл). К раствору добавляли диметилацеталь N,N-диметилформамида (130 мл, 976 ммоль) и смесь нагревали при перемешивании при температуре 100°C в течение 5 часов, в то время как образовавшийся метанол отделяли в аппарате Дина-Старка. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток растворяли в этаноле (390 мл). К раствору добавляли тетрагидро-2H-пиран-4-илметанамин (26,0 мл, 215 ммоль) и смесь нагревали при перемешивании при температуре 60°C в течение 9 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (Shoko Scientific Co., Ltd., растворитель для элюирования: дихлорметан/этилацетат=75/25 -> 20/80) с получением указанного в заголовке соединения (36,9 г, выход: 53,2%) в виде карамель-подобного вещества.
1Н-ЯМР (CDCl3) δ: 8,10 (1H, д, J=2,4 Гц), 7,48 (2H, д, J=8,0 Гц), 7,32 (1H, д, J=2,4 Гц), 7,20 (2H, д, J=8,0 Гц), 4,38 (2H, кв, J=7,0 Гц), 4,02 (2H, дд, J=11,5, 4,0 Гц), 3,74 (2H, д, J=7,3 Гц), 3,38 (2H, тд, J=11,5, 2,0 Гц), 2,08-2,00 (1H, м), 1,63-1,56 (2H, м), 1,35-1,42 (5H, м).
МС (APCI) m/z:356 [(M+H)+].
(Стадия 3) 5-(4-Метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоновая кислота
Этил 5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксилат (36,9 г, 104 ммоль) растворяли в метаноле (519 мл). К раствору добавляли 1 н водного раствора гидроксида натрия (311 мл, 311 ммоль) и смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении. К остатку добавляли 1 н хлористоводородную кислоту (400 мл), затем экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия, и затем, растворитель отгоняли при пониженном давлении. Остаток промывали метанолом с помощью суспензионного способа с получением указанного в заголовке соединения (31,6 г, выход: 92,9%) в виде твердого вещества.
1Н-ЯМР (CDCl3) δ: 8,46 (1H, д, J=2,4 Гц), 7,56 (1H, д, J=2,4 Гц), 7,48 (2H, д, J=8,5 Гц), 7,28 (2H, д, J=8,5 Гц), 4,03 (2H, дд, J=11,3, 4,0 Гц), 3,89 (2H, д, J=7,5 Гц), 3,38 (2H, т, J=11,3 Гц), 2,40 (3H, с), 2,16-2,04 (1H, м), 1,62-1,53 (3H, м), 1,49-1,37 (2H, м).
МС (APCI) m/z:328 [(M+H)+].
Подобным образом, промежуточные соединения в таблице 1, были синтезированы из соответствующих исходных веществ А и В.
[Таблица 1-1]









[Таблица 1-2]












[Таблица 1-3]




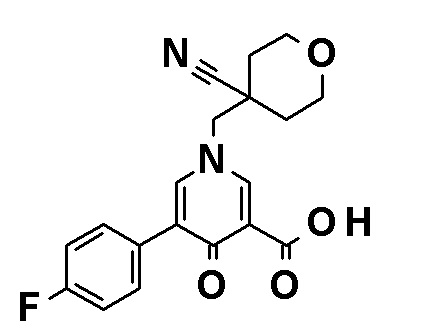


[Таблица 1-4]










[Таблица 1-5]




(Промежуточное соединение 2) 5-(4-Циклопропил-2-фторфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоновая кислота
[Формула 48]

(Стадия 1) Этил (4-бром-2-фторфенил)ацетат
К раствору (4-бром-2-фторфенил)уксусной кислоты (2,00 г, 8,58 ммоль) в метиленхлориде (40,0 мл) добавляли этанол (0,969 мл, 0,791 г, 17,2 ммоль), 4-диметиламинопиридин (0,105 г, 0,858 ммоль) и 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (EDAC⋅HCl) (1,97 г, 10,3 ммоль), и смесь перемешивали в течение ночи при комнатной температуре. К реакционной смеси добавляли 1 н хлористоводородную кислоту, затем экстрагировали метиленхлоридом три раза. Органический слой промывали насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором в указанном порядке и затем сушили над безводным сульфатом натрия. После фильтрования и концентрирования при пониженном давлении, полученный остаток очищали при помощи колоночной хроматографии на силикагеле (Yamazen Corp., н-гексан/этилацетат=(100/0 -> 87/13) с получением указанного в заголовке соединения (1,96 г, выход: 87,5%) в виде твердого вещества.
1Н-ЯМР (CDCl3) δ: 7,27-7,24 (2H, м), 7,18-7,14 (1H, м), 4,17 (2H, кв, J=7,0 Гц), 3,62 (2H, с), 1,26 (3H, т, J=7,0 Гц).
(Стадия 2) Этил (4-циклопропил-2-фторфенил)ацетат
К суспензии этил (4-бром-2-фторфенил)ацетата (1,95 г, 7,47 ммоль), синтезированного на стадии 1, в толуоле (30,0 мл), циклопропилбороновую кислоту (0,962 г, 11,2 ммоль), трициклогексилфосфин (0,419 г, 1,49 ммоль), трикалийфосфат (5,55 г, 26,1 ммоль), и воду (6,00 мл) добавляли, и реакционную смесь продували азотом. Затем, добавляли ацетат палладия(II) (0,168 г, 0,747 ммоль) и смесь перемешивали при 100°C в течение 2 часов. Реакционную смесь оставляли охлаждаться до комнатной температуры, и затем, к смеси добавляли воду, с последующей экстракцией три раза этилацетатом. Органический слой промывали насыщенным солевым раствором и затем сушили над безводным сульфатом натрия. После фильтрования и концентрирования при пониженном давлении полученный остаток очищали при помощи колоночной хроматографии на силикагеле (Yamazen Corp., н-гексан/этилацетат=100/0 -> 87/13) и затем очищали при помощи колоночной хроматографии на аминосиликагеле (Yamazen Corp., н-гексан/этилацетат=100/0 -> 87/13) с получением указанного в заголовке соединения (1,59 г, выход: 95,8%) в виде масла.
1Н-ЯМР (CDCl3) δ: 7,12 (1H, т, J=7,9 Гц), 6,84-6,82 (1H, м), 6,76-6,72 (1H, м), 4,16 (2H, кв, J=7,0 Гц), 3,61 (2H, с), 1,90-1,83 (1H, м), 1,25 (3H, т, J=7,0 Гц), 1,00-0,95 (2H, м), 0,70-0,66 (2H, м).
МС (APCI) m/z:223 [(M+H)+].
(Стадия 3) (4-Циклопропил-2-фторфенил)уксусная кислота
К раствору этил (4-циклопропил-2-фторфенил)ацетата (1,59 г, 7,15 ммоль), синтезированного на стадии 2, в тетрагидрофуране (30,0 мл), добавляли метанол (15,0 мл) и 1 моль/л водный раствор гидроксида натрия (14,3 мл, 14,3 ммоль), и смесь перемешивали в течение ночи при комнатной температуре. Органический растворитель отгоняли при пониженном давлении, и затем добавляли воду к остатку, с последующей промывкой диэтиловым эфиром. Водный слой подкисляли путем добавления 1 н хлористоводородной кислоты, с последующей экстракцией три раза этилацетатом. Органический слой промывали насыщенным солевым раствором и затем сушили над безводным сульфатом натрия. Проводили фильтрование и концентрацию при пониженном давлении с получением указанного в заголовке соединения (1,36 г, 7,00 ммоль) в виде твердого вещества.
1Н-ЯМР (CDCl3) δ: 7,11 (1H, т, J=7,9 Гц), 6,83 (1H, дд, J=7,9, 1,8 Гц), 6,75 (1H, дд, J=11,2, 1,8 Гц), 3,65 (2H, с), 1,90-1,83 (1H, м), 1,00-0,95 (2H, м), 0,70-0,66 (2H, м).
(Стадия 4) 5-(4-Циклопропил-2-фторфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоновая кислота
Указанное в заголовке соединение получали в виде твердого вещества с использованием (4-циклопропил-2-фторфенил)уксусной кислоты вместо (4-метилфенил)уксусной кислоты на стадии 1 промежуточного соединения 1а и проведения тех же последующих реакций как на стадии 1 - стадии 3 промежуточного соединения 1а.
1Н-ЯМР (ДМСО-D6) δ: 8,80 (1H, д, J=2,4 Гц), 8,31 (1H, д, J=2,4 Гц), 7,34 (1H, т, J=8,2 Гц), 7,03-6,99 (2H, м), 4,12 (2H, д, J=7,3 Гц), 3,85 (2H, дд, J=11,2, 3,3 Гц), 3,24 (2H, т, J=11,2 Гц), 2,12-1,97 (2H, м), 1,43 (2H, ушир. д, J=10,9 Гц), 1,33-1,22 (2H, м), 1,04-0,99 (2H, м), 0,77-0,73 (2H, м).
МС (APCI) m/z:372 [(M+H)+].
(Промежуточное соединение 3) 5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоновая кислота
[Формула 49]

(Стадия 1) Этил 4-(5-метилпиридин-2-ил)-3-оксобутаноат
К раствору (5-метилпиридин-2-ил)уксусной кислоты (0,500 г, 3,31 ммоль) в тетрагидрофуране (15,0 мл) добавляли 1,1'-карбонилбис-1H-имидазол (0,805 г, 4,96 ммоль) и смесь перемешивали при комнатной температуре в течение 2,5 часов. В другом реакционном сосуде моноэтил калий малонат (1,69 г, 9,92 ммоль) и хлорид магния (0,945 г, 9,92 ммоль) суспендировали в тетрагидрофуране (25,0 мл). К суспензии добавляли триэтиламин (2,38 мл, 1,74 г, 17,2 ммоль) при охлаждении льдом и смесь перемешивали при комнатной температуре в течение 2,5 часов и затем снова охлаждали льдом. Раствор активного сложного эфира, полученного выше, добавляли к этому, и смесь перемешивали в течение ночи при комнатной температуре. К реакционной смеси добавляли 10%-ный водный раствор лимонной кислоты, затем экстрагировали этилацетатом пять раз. Органический слой промывали насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором в указанном порядке и затем сушили над безводным сульфатом натрия. После фильтрования и концентрирования при пониженном давлении полученный остаток очищали с помощью колоночной хроматографии на силикагеле (Yamazen Corp., растворитель для элюирования: н-гексан/этилацетат=57/43 -> 36/64) с получением указанного в заголовке соединения (0,634 г, выход: 86,6%) в виде масла.
МС (APCI) m/z:222 [(M+H)+].
(Стадии 2 и 3) 5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоновая кислота
Указанное в заголовке соединение получали в виде твердого вещества с использованием этил 4-(5-метилпиридин-2-ил)-3-оксобутаноата вместо этил 4-(4-метилфенил)-3-оксобутаноата на стадии 2 промежуточного соединения 1a и проведения таких же последующих реакций как на стадии 2 и стадии 3 промежуточного соединения 1а.
1Н-ЯМР (ДМСО-D6) δ: 8,86 (1H, д, J=2,4 Гц), 8,80 (1H, д, J=2,4 Гц), 8,53 (1H, с), 8,38 (1H, д, J=8,5 Гц), 7,72 (1H, дд, J=8,5, 2,1 Гц), 4,24 (2H, д, J=7,3 Гц), 3,84 (2H, дд, J=11,2, 3,3 Гц), 3,24 (2H, т, J=11,2 Гц), 2,35 (3H, с), 2,11-2,01 (1H, м), 1,44 (2H, ушир. д, J=10,9 Гц), 1,35-1,24 (2H, м).
МС (APCI) m/z:329 [(M+H)+].
Подобным образом, промежуточное соединение в таблице 2 было синтезировано из соответствующего исходного вещества.
[Таблица 2]
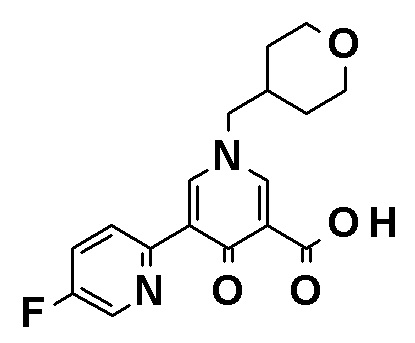
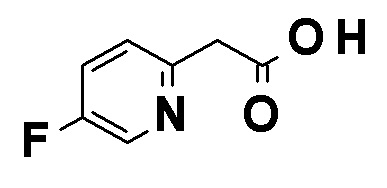
(Промежуточное соединение 4a) 5-(4-Бромфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоновая кислота
[Формула 50]

(Стадия 1) Этил 4-(4-бромфенил)-3-оксобутаноат
Указанное в заголовке соединение получали в виде масла с использованием 2-(4-бромфенил)уксусной кислоты вместо (4-метилфенил)уксусной кислоты на стадии 1 промежуточного соединения 1a и проведения последующих реакций с помощью той же операции.
1Н-ЯМР (CDCl3) δ: 7,47 (2H, д, J=8,5 Гц), 7,09 (2H, д, J=8,5 Гц), 4,18 (2H, кв, J=7,1 Гц), 3,81 (2H, с), 3,46 (2H, с), 1,27 (3H, т, J=7,1 Гц).
(Стадия 2) Этил 5-(4-бромфенил)-4-оксо-1,4-дигидропиридин-3-карбоксилат
К раствору этил 4-(4-бромфенил)-3-оксобутаноата (5,00 г, 17,5 ммоль), синтезированного на стадии 1, в этаноле (50,0 мл), добавляли 1,3,5-триазин (1,56 г, 19,3 ммоль) и этоксид натрия (1,43 г, 21,0 ммоль), и смесь перемешивали при 85°C в течение 4 часов. Реакционную смесь оставляли охлаждаться до комнатной температуры и затем концентрировали при пониженном давлении. Полученный остаток подкисляли добавлением 1 н хлористоводородной кислоты, и затем, осажденное твердое вещество собирали путем фильтрации, затем промывали ацетоном и сушили с получением сырого продукта указанного в заголовке соединения (3,73 г) в виде твердого вещества.
МС (APCI) m/z:322 [(M+H)+].
(Стадия 3) Этил 5-(4-бромфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксилат
К суспензии сырого продукта этил 5-(4-бромфенил)-4-оксо-1,4-дигидропиридин-3-карбоксилата (0,322 г), синтезированного на стадии 2, в N,N-диметилформамиде (45,0 мл) добавляли карбонат цезия (0,977 г, 3,00 ммоль) и смесь перемешивали при 80°C в течение 15 минут. Затем, добавляли к этому метансульфоновую кислоту тетрагидро-2H-пиран-4-илметил эфира (0,583 г, 3,00 ммоль), и смесь перемешивали при 80°C в течение 4 часов. Реакционную смесь оставляли охлаждаться до комнатной температуры, и затем, к смеси добавляли воду, с последующей экстракцией три раза этилацетатом. Органический слой промывали насыщенным солевым раствором и затем сушили над безводным сульфатом натрия. После фильтрования и концентрирования при пониженном давлении, полученный остаток очищали при помощи колоночной хроматографии на силикагеле (Yamazen Corp., растворитель для элюирования: н-гексан/этилацетат=57/43 -> 36/64) с получением указанного в заголовке соединения (0,234 г, выход в 2 стадии: 36,7%) в виде аморфного твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 8,30 (1H, д, J=2,4 Гц), 8,02 (1H, д, J=2,4 Гц), 7,59 (4H, с), 4,20 (2H, кв, J=7,0 Гц), 3,92 (2H, д, J=7,3 Гц), 3,85 (2H, дд, J=10,9, 3,0 Гц), 3,25 (2H, т, J=10,9 Гц), 2,09-1,99 (1H, м), 1,43 (2H, ушир. д, J=10,9 Гц), 1,32-1,24 (5H, м).
МС (APCI) m/z:420 [(M+H)+].
(Стадия 4) 5-(4-Бромфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоновая кислота
Указанное в заголовке соединение получали в виде твердого вещества с использованием этил 5-(4-бромфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксилата вместо этил 5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксилата на стадии 3 промежуточного соединения 1a и проведения последующих реакций с помощью той же операции.
1Н-ЯМР (ДМСО-D6) δ: 8,79 (1H, д, J=1,8 Гц), 8,45 (1H, д, J=1,8 Гц), 7,70-7,65 (4H, м), 4,14 (2H, д, J=7,3 Гц), 3,85 (2H, дд, J=11,2, 3,3 Гц), 3,24 (2H, т, J=11,2 Гц), 2,16-2,06 (1H, м), 1,43 (2H, ушир. д, J=10,9 Гц), 1,34-1,24 (2H, м).
МС (APCI) m/z:392 [(M+H)+].
Подобным образом, промежуточные соединения в таблице 3, были синтезированы из соответствующих исходных веществ.
[Таблица 3]




(Промежуточное соединение 5a) (2S)-2-{[2-Метокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]метил}-1,4-диоксан
[Формула 51]

2-Метокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенол (0,500 г, 1,99 ммоль) растворяли в N,N-диметилформамиде (10,0 мл). К раствору добавляли (2S)-1,4-диоксан-2-илметил метансульфоновую кислоту(0,401 г, 2,04 ммоль) и смесь перемешивали при температуре 90°C в течение 10 часов. Реакционную смесь охлаждали до комнатной температуры и разделяли на органический и водный слои посредством добавления этилацетата и воды. Органический слой промывали насыщенным солевым раствором и затем сушили над безводным сульфатом магния. Растворитель отгоняли при пониженном давлении, и остаток очищали при помощи колоночной хроматографии на силикагеле (Biotage Japan Ltd., растворитель для элюирования: гексан/этилацетат=100/0 -> 50/50) с получением указанного в заголовке соединения (0,451 г, выход: 64,4%) в виде масла.
1Н-ЯМР (CDCl3) δ: 7,39 (1H, д, J=7,9 Гц), 7,28 (1H, с), 6,89 (1H, д, J=7,9 Гц), 4,09-4,03 (2H, м), 4,00-3,93 (2H, м), 3,89 (3H, с), 3,86-3,63 (4H, м), 3,57-3,50 (1H, м), 1,34 (12H, с).
Подобным образом, промежуточное соединение в таблице 4 было синтезировано из соответствующего исходного вещества.
[Таблица 4]
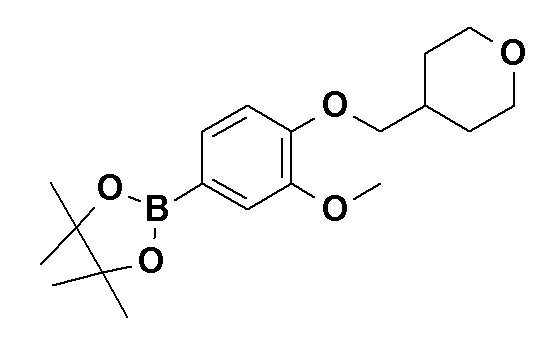

(Промежуточное соединение 6) 1-(Тетрагидро-2H-пиран-4-илметил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол
[Формула 52]

Указанное в заголовке соединение получали таким же образом, как и в промежуточном соединении 5а, используя 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол и 4-(бромметил)тетрагидропиран в качестве исходных веществ.
1Н-ЯМР (CDCl3) δ: 7,79 (1H, с), 7,65 (1H, с), 4,00 (2H, д, J=7,5 Гц), 3,96 (2H, дд, J=11,5, 4,5 Гц), 3,35 (2H, тд, J=11,5, 2,0 Гц), 2,23-2,11 (1H, м), 1,48 (2H, д, J=11,5 Гц), 1,40-1,28 (14H, м).
МС (APCI) m/z:293 [(M+H)+].
(Промежуточное соединение 7) (2R)-2-[({2-[(2H3)Метокси]-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)(2H3)фенил}окси)метил}-1,4-диоксан
[Формула 53]

(Стадия 1) 4-Бром-2,3,5-тридейтерио-6-(тридейтериометокси)фенол
4-бром-2-[(2H3)метокси](2H3)фенол
(2H4)Бензол-1,2-диол (12,0 г, 105 ммоль) суспендировали в дихлорметане (105 мл). К суспензии добавляли 3,4-дигидро-2H-пиран (9,53 мл, 105 ммоль) и пиридиниевую соль п-толуолсульфоновой кислоты (396 мг, 1,58 ммоль) и смесь перемешивали при комнатной температуре в течение 3 часов. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия, и смесь перемешивали в течение 10 минут. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Полученное масло растворяли в ацетоне (207 мл). К раствору добавляли карбонат калия (45,8 г, 330 ммоль) и тридейтерометилиодид (14,4 мл, 228 ммоль) и смесь нагревали при перемешивании при температуре 40°C в течение 24 часов в потоке азота. Реакционную смесь охлаждали до комнатной температуры и затем фильтровали через воронку Kiriyama, и фильтрат концентрировали при пониженном давлении. К остатку добавляли гексан и смесь снова фильтровали через воронку Kiriyama. Затем фильтрат концентрировали при пониженном давлении. Полученное масло растворяли в этаноле (181 мл). К раствору добавляли пиридиниевую соль п-толуолсульфоновой кислоты (341 мг, 1,36 ммоль) и смесь нагревали при перемешивании при температуре 65°C в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Остаток растворяли в N,N-диметилформамиде (90,0 мл). К раствору добавляли N-бромсукцинимид (16,1 г, 90,6 ммоль) небольшими порциями при охлаждении льдом, и смесь перемешивали в течение 1 часа при охлаждении льдом. К реакционной смеси добавляли диэтиловый эфир (500 мл), и затем реакцию останавливали добавлением 8,85 моль/л водного раствора тиосульфата натрия (50 мл). Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на аминосиликагеле (Shoko Scientific Co., Ltd., растворитель для элюирования: гексан/этилацетат=90/10 -> 55/45) с получением указанного в заголовке соединения (6,97 г, выход: 28,3%) в виде масла.
1Н-ЯМР (CDCl3) δ: 5,57 (1H, с).
МС (CI) m/z: 207 [(M-H)+].
(Стадия 2) (2R)-2-[({4-Бром-2-[(2H3)метокси](2H3)фенил}окси)метил]-1,4-диоксан
4-Бром-2-[(2H3)метокси](2H3)фенол (2,00 г, 9,57 ммоль) растворяли в N,N-диметилформамиде (48,0 мл). К раствору добавляли карбонат калия (2,64 г, 19,1 ммоль) и (2R)-1,4-диоксан-2-илметил метансульфоновую кислоту (1,88 г, 9,57 ммоль) и смесь нагревали при перемешивании при температуре 90°C в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры, и добавляли к смеси этилацетат. Нерастворимое вещество отфильтровывали. Фильтрат концентрировали при пониженном давлении, и остаток очищали при помощи колоночной хроматографии на силикагеле (Shoko Scientific Co., Ltd., растворитель для элюирования: гексан/этилацетат=100/0 -> 70/30) с получением указанного в заголовке соединения (1,75 г, выход: 59,2%) в виде твердого вещества.
1Н-ЯМР (CDCl3) δ: 4,06-3,48 (9H, м).
МС (APCI) m/z:309 [(M+H)+].
(Стадия 3) (2R)-2-[({2-[(2H3)Метокси]-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)(2H3)фенил}окси)метил}-1,4-диоксан
(2R)-2-[({4-Бром-2-[(2H3)метокси](2H3)фенил}окси)метил]-1,4-диоксан (800 мг, 2,59 ммоль) суспендировали в толуоле (5,20 мл). К суспензии добавляли никель(II) 1,3-бис(дифенилфосфино)пропан]хлорид (140 мг, 259 мкмоль), 1,3-бис(дифенилфосфино)пропан (107 мг, 0,259 ммоль), пинакол борон (0,744 мл, 5,17 ммоль) и триэтиламин (1,10 мл, 7,76 ммоль), и смесь нагревали при перемешивании при температуре 100°C в течение 16 часов в потоке азота. Реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом, и затем, нерастворимое вещество отфильтровывали через целит. Фильтрат концентрировали при пониженном давлении, и остаток очищали при помощи колоночной хроматографии на силикагеле (Shoko Scientific Co., Ltd., растворитель для элюирования: гексан/этилацетат=100/0 -> 85/15) с получением указанного в заголовке соединения (840 мг, 91,1%) в виде твердого вещества.
1Н-ЯМР (CDCl3) δ: 4,15-4,02 (2H, м), 4,00-3,93 (2H, м), 3,87-3,62 (4H, м), 3,57-3,49 (1H, м), 1,34 (12H, с).
МС (ESI) m/z:357 [(M+H)+].
(Промежуточное соединение 8)
3-(4-Аминофенил)-5-{4-[(2S)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-2-амин
[Формула 54]

3-(4-Аминофенил)-5-бромпиридин-2-амин (0,510 г, 1,93 ммоль), синтезированный способом, описанным в патенте (WO2013/115280 A1), растворяли в 1,4-диоксане (5,00 мл). К раствору добавляли воду (1,00 мл), (2S)-2-{[2-метокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]метил}-1,4-диоксан (0,451 г, 1,29 ммоль), тетракис(трифенилфосфин)палладий(0) (0,0744 г, 0,0644 ммоль), и карбонат калия (0,356 г, 2,58 ммоль) и смесь перемешивали при 100°C в течение 5 часов в атмосфере азота. Реакционную смесь разделяли на органический и водный слои путем добавления дихлорметана и воды. Органический слой сушили над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (Biotage Japan Ltd., растворитель для элюирования: этилацетат/метанол=99/1 -> 90/10) с получением указанного в заголовке соединения (0,139 г, выход: 26,5%) в виде масла.
1Н-ЯМР (CDCl3) δ: 8,24-8,22 (1H, м), 7,53-7,51 (1H, м), 7,32-7,27 (2H, м), 7,07-6,99 (2H, м), 6,98-6,93 (1H, м), 6,81-6,76 (2H, м), 4,62-4,58 (2H, м), 4,14-4,01 (3H, м), 4,01-3,93 (3H, м), 3,92-3,63 (7H, м), 3,59-3,50 (1H, м).
МС (ESI) m/z:408 [(M+H)+]
(Промежуточное соединение 9a) 3-Бром-5-{4-[(2S)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-2-амин
[Формула 55]

К 3-бром-5-иодпиридин-2-амину (0,533 г, 1,79 ммоль), (2S)-2-{[2-метокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]метил}-1,4-диоксану (0,625 г, 1,79 ммоль), карбонат натрия (0,378 г, 3,57 ммоль), и тетракис(трифенилфосфин)палладий(0) (0,103 г, 0,0892 ммоль), 1,4-диоксан (9,00 мл) и воду (1,80 мл) добавляли, и смесь перемешивали при 90°C в течение 7 часов в атмосфере азота. Реакционную смесь оставляли охлаждаться до комнатной температуры, и затем, к смеси добавляли воду, с последующей экстракцией метиленхлоридом три раза. Органический слой сушили над безводным сульфатом натрия. После фильтрования и концентрирования при пониженном давлении, полученный остаток очищали при помощи колоночной хроматографии на силикагеле (Yamazen Corp., растворитель для элюирования: хлороформ/этилацетат=59/41 -> 38/62) с получением указанного в заголовке соединения (0,388 г, выход: 55,0%) в виде твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 8,28 (1H, д, J=1,8 Гц), 8,05 (1H, д, J=2,4 Гц), 7,17 (1H, д, J=2,4 Гц), 7,10 (1H, дд, J=8,5, 2,4 Гц), 6,99 (1H, д, J=8,5 Гц), 6,28 (2H, с), 3,98-3,74 (8H, м), 3,69-3,60 (2H, м), 3,52-3,46 (1H, м), 3,42-3,36 (1H, м).
МС (APCI) m/z:395 [(M+H)+].
Подобным образом, промежуточные соединения в таблице 5, были синтезированы из соответствующих исходных веществ.
[Таблица 5-1]






[Таблица 5-2]




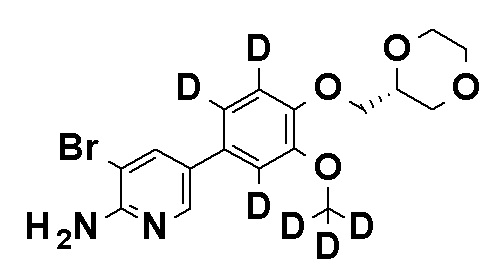

(Промежуточное соединение 10a)
3-(4-Амино-2-фторфенил)-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-2-амин
[Формула 56]
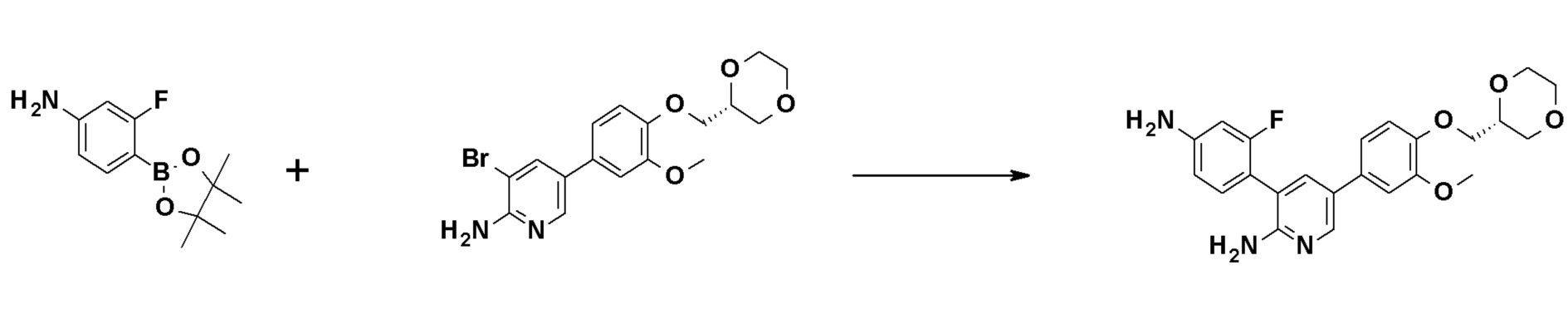
Указанное в заголовке соединение получали таким же образом, как и в промежуточном соединении 8, используя 3-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин и 3-бром-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-2-амин, синтезированный способом, описанным в патенте (WO2013/115280 A1), в качестве исходных веществ, 1,4-диоксан и воду в качестве растворителя, тетракис(трифенилфосфин)палладий(0) в качестве катализатора, и карбонат калия в качестве основания, и осуществляя нагревание с перемешиванием при температуре реакции 100°C в течение 5 часов.
1Н-ЯМР (CDCl3) δ: 8,27 (1H, д, J=2,4 Гц), 7,54 (1H, д, J=2,4 Гц), 7,17 (1H, т, J=8,0 Гц), 7,06-7,01 (2H, м), 6,95 (1H, д, J=8,0 Гц), 6,55 (1H, дд, J=8,0, 2,4 Гц), 6,51 (1H, дд, J=11,5, 2,4 Гц), 4,52 (2H, ушир. с), 4,13-3,52 (12H, м).
МС (APCI) m/z:426 [(M+H)+].
Подобным образом, промежуточные соединения в таблице 6, были синтезированы из соответствующих исходных веществ А и В.
[Таблица 6]


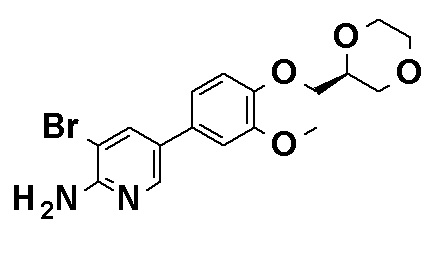


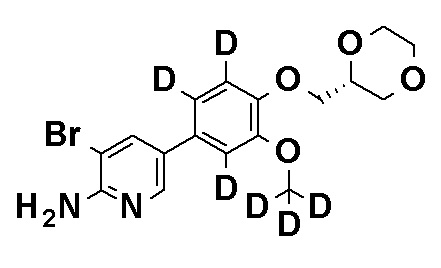



(Промежуточное соединение 11a) 3-(4-Амино-3-фторфенил)-5-(3,4-диметоксифенил)пиридин-2-амин
[Формула 57]
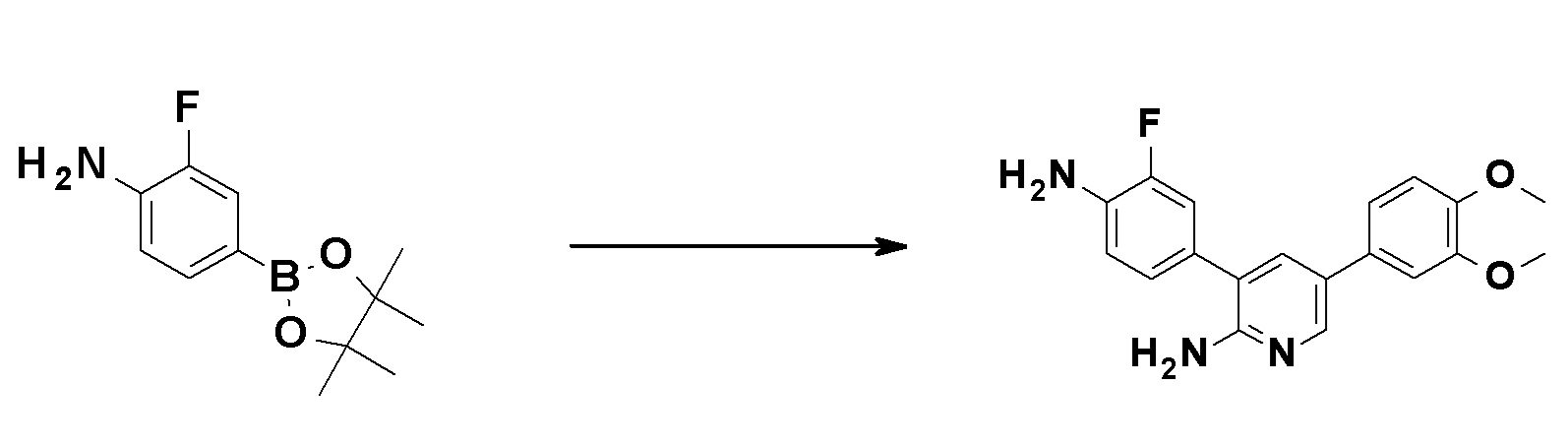
К 3-бром-5-(3,4-диметоксифенил)пиридин-2-амину (1,30 г, 4,20 ммоль), синтезированному способом, описанным в патенте (WO2013/115280 A1), 2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин (1,20 г, 5,06 ммоль), комплекс [1,1'-бис(дифенилфосфино)ферроцен]палладий(II) дихлорид-дихлорметан (1:1) (0,340 г, 0,416 ммоль), и карбонат цезия (4,10 г, 12,6 ммоль), 1,4-диоксан (25,0 мл) и воду (5,00 мл) добавляли, и смесь перемешивали при 80°C в течение 5 часов. Реакционную смесь оставляли охлаждаться до комнатной температуры, и затем, к смеси добавляли воду и метиленхлорид. Нерастворимое вещество отфильтровывали через целит, и фильтрат подвергали экстракции метиленхлоридом три раза. Затем, органический слой сушили над безводным сульфатом натрия. После фильтрования и концентрирования при пониженном давлении полученный остаток очищали с помощью колоночной хроматографии на силикагеле (Yamazen Corp., хлороформ/этилацетат=50/50 -> 0/100) и затем очищали при помощи колоночной хроматографии на аминосиликагеле (Yamazen Corp., н-гексан/этилацетат=25/75 -> 0/100 -> этилацетат/метанол=100/0 -> 90/10). После концентрирования при пониженном давлении, полученное твердое вещество суспендировали в диэтиловом эфире, собирали фильтрованием и сушили с получением указанного в заголовке соединения (0,928 г, выход: 65,1%) в виде твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 8,20 (1H, д, J=2,4 Гц), 7,52 (1H, д, J=2,4 Гц), 7,18-7,11 (3H, м), 7,06 (1H, дд, J=7,9, 2,4 Гц), 6,98 (1H, д, J=8,5 Гц), 6,87-6,83 (1H, м), 5,61 (2H, с), 5,30 (2H, с), 3,82 (3H, с), 3,77 (3H, с).
МС (APCI) m/z:340 [(M+H)+].
Подобным образом, промежуточное соединение в таблице 7 было синтезировано из соответствующего исходного вещества.
[Таблица 7]


(Промежуточное соединение 12a) 3-(4-Амино-3-фторфенил)-4-метоксипиридин-2-амин
[Формула 58]

К 3-иод-4-метоксипиридин-2-амину (0,500 г, 2,00 ммоль), 2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилину (0,498 г, 2,10 ммоль), тетракис(трифенилфосфин)палладий(0) (0,231 г, 0,200 ммоль), и карбонат натрия (0,636 г, 6,00 ммоль), 1,4-диоксан (10,0 мл) и воду (2,00 мл) добавляли, и смесь перемешивали при 80°C в течение 12 часов в атмосфере азота. Реакционную смесь оставляли охлаждаться до комнатной температуры, и затем, к смеси добавляли воду, с последующей экстракцией три раза этилацетатом. Органический слой промывали насыщенным солевым раствором и затем сушили над безводным сульфатом натрия. После фильтрования и концентрирования при пониженном давлении, полученный остаток очищали при помощи колоночной хроматографии на силикагеле (Yamazen Corp., этилацетат/метанол=100/0 -> 90/10) и затем очищали при помощи колоночной хроматографии на аминосиликагеле (Yamazen Corp., этилацетат/метанол=100/0 -> 90/10) с получением указанного в заголовке соединения (0,177 г, выход: 38,0%) в виде твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 7,83 (1H, д, J=5,5 Гц), 7,65-7,53 (1H, м), 6,83-6,72 (3H, м), 6,39 (1H, д, J=6,1 Гц), 5,18 (2H, с), 5,10 (2H, с), 3,65 (3H, с).
МС (APCI) m/z:234 [(M+H)+].
Подобным образом, промежуточные соединения в таблице 8, были синтезированы из соответствующих исходных веществ А и В.
[Таблица 8]










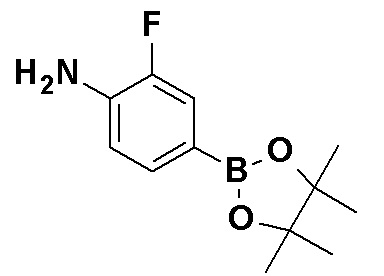

(Промежуточное соединение 13) 3-(4-Аминофенил)-5-(3,4-диметоксифенил)пиразин-2-амин
[Формула 59]

(Стадия 1) 3-Хлор-5-(3,4-диметоксифенил)пиразин-2-амин
К 3-хлор-5-иодпиразин-2-амину (0,510 г, 2,00 ммоль), синтезированному в соответствии со способом, описанным в патенте (WO2011/110545 A1), (3,4-диметоксифенил)бороновой кислоте (0,363 г, 2,00 ммоль), комплексу [1,1'-бис(дифенилфосфино)ферроцен]палладий(II) дихлорид-дихлорметан (1:1) (0,163 г, 0,200 ммоль) и карбонату цезия (1,30 г, 4,00 ммоль), добавляли 1,4-диоксан (7,50 мл) и воду (2,50 мл), и смесь перемешивали при 80°C в течение 4 часов. Реакционную смесь оставляли охлаждаться до комнатной температуры, и затем, к смеси добавляли воду, с последующей экстракцией метиленхлоридом три раза. Органический слой сушили над безводным сульфатом натрия. После фильтрования и концентрирования при пониженном давлении, полученный остаток очищали при помощи колоночной хроматографии на силикагеле (Yamazen Corp., хлороформ/этилацетат=100/0 -> 90/10) и затем очищали при помощи колоночной хроматографии на аминосиликагеле (Yamazen Corp., н-гексан/этилацетат=25/75 -> 0/100) с получением указанного в заголовке соединения (0,412 г, выход: 77,7%) в виде твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 8,57 (1H, с), 7,47-7,45 (2H, м), 7,01 (1H, д, J=7,9 Гц), 6,85 (2H, с), 3,83 (3H, с), 3,79 (3H, с).
МС (APCI) m/z:266 [(M+H)+].
(Стадия 2) 3-(4-Аминофенил)-5-(3,4-диметоксифенил)пиразин-2-амин
К 3-хлор-5-(3,4-диметоксифенил)пиразин-2-амину (0,0800 г, 0,301 ммоль), синтезированному на стадии 1, 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилину (0,0990 г, 0,452 ммоль), комплексу [1,1'-бис(дифенилфосфино)ферроцен]палладий(II) дихлорид-дихлорметан (1:1) (0,0246 г, 0,030 ммоль) и карбонату цезия (0,294 г, 0,903 ммоль), добавляли 1,4-диоксан (2,00 мл) и воду (0,200 мл) и смесь перемешивали при 100°C в течение 4 часов. Реакционную смесь оставляли охлаждаться до комнатной температуры, и затем, к этому добавляли воду, с последующей экстракцией метиленхлоридом три раза. Органический слой сушили над безводным сульфатом натрия. После фильтрования и концентрирования при пониженном давлении, полученный остаток очищали при помощи колоночной хроматографии на силикагеле (Yamazen Corp., только этилацетат) с получением указанного в заголовке соединения (0,0940 г, выход: 96,8%) в виде аморфного твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 8,39 (1H, с), 7,54-7,50 (4H, м), 7,01 (1H, д, J=8,5 Гц), 6,67 (2H, д, J=8,5 Гц), 6,00 (2H, с), 5,44 (2H, с), 3,83 (3H, с), 3,79 (3H, с).
МС (APCI) m/z:323 [(M+H)+].
(Промежуточное соединение 14a) N-(6-хлорпиридазин-3-ил)-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид
[Формула 60]

5-(4-Метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоновую кислоту (3,00 г, 9,16 ммоль) растворяли в N,N-диметилформамиде (30,0 мл). К раствору добавляли N-[1-(циано-2-этокси-2-оксоэтилиденаминоокси)диметиламино(морфолино)]урония гексафторфосфат (COMU) (4,32 г, 10,1 ммоль) и смесь перемешивали при комнатной температуре в течение 1 часа. Затем, добавляли к этому 3-амино-6-хлорпиридазин (1,31 г, 10,1 ммоль) и смесь перемешивали при комнатной температуре в течение 3 дней. Реакционную смесь разбавляли этилацетатом, и затем, к этому добавляли 1 н хлористоводородную кислоту. Органический слой промывали насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором в данном порядке. Органический слой сушили над безводным сульфатом натрия. Растворитель концентрировали при пониженном давлении, и остаток очищали при помощи колоночной хроматографии на силикагеле (Shoko Scientific Co., Ltd., этилацетат/метанол=100/0 -> 96/4) с получением аморфного твердого вещества. Это твердое вещество промывали метанолом с помощью суспензионного способа с получением указанного в заголовке соединения (2,10 г, выход: 52,2%) в виде твердого вещества.
1Н-ЯМР (CDCl3) δ: 13,68 (1H, с), 8,59 (1H, д, J=9,0 Гц), 8,48 (1H, д, J=2,0 Гц), 7,51-7,46 (4H, м), 7,26 (2H, д, J=8,0 Гц), 4,03 (2H, дд, J=11,5, 4,0 Гц), 3,87 (2H, д, J=7,5 Гц), 3,39 (2H, т, J=11,5 Гц), 2,40 (3H, с), 2,16-2,06 (1H, м), 1,64-1,55 (2H, м), 1,50-1,38 (2H, м).
МС (APCI) m/z:439 [(M+H)+].
Подобным образом, промежуточные соединения в таблице 9, были синтезированы из соответствующих исходных веществ А и В.
[Таблица 9-1]









[Таблица 9-2]

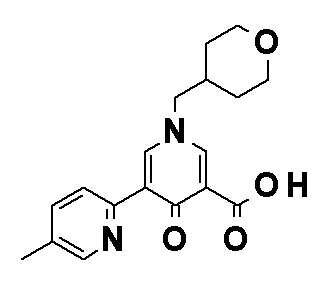




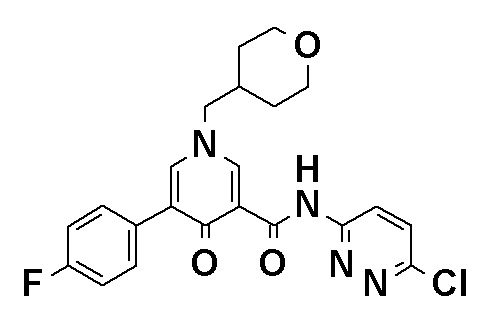


(Промежуточное соединение 16a) N-(5-иодпиридин-2-ил)-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид
[Формула 61]

К раствору 5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоновой кислоты (300 мг, 0,916 ммоль) в диметилацетамиде (1 мл), добавляли тионилхлорид (0,087 мл, 1,19 ммоль) на бане с ледяной водой. После перемешивания в течение 0,5 часов, 5-иодпиридин-2-амин (201 мг, 0,916 ммоль) и диизопропилэтиламин (0,23 мл, 1,37 ммоль) добавляли к этому на бане с ледяной водой, и температуру реакционной смеси постепенно повышали до комнатной температуры. Реакционную смесь перемешивали при комнатной температуре в течение 5 часов, и затем, к этому добавляли воду. Нерастворимое вещество собирали фильтрованием. Полученный продукт промывали гексаном и затем сушили при 50°C в течение 1 часа при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (хлороформ/метанол=99/1 -> 95/5) с получением указанного в заголовке соединения (302 мг, выход: 62,3%) в виде твердого вещества.
1Н-ЯМР (CDCl3) δ: 13,19 (1H, с), 8,53 (1H, д, J=2,4 Гц), 8,49 (1H, д, J=2,4 Гц), 8,15 (1H, д, J=8,5 Гц), 7,95 (1H, дд, J=8,5, 2,4 Гц), 7,51 (2H, д, J=7,9 Гц), 7,46 (1H, д, J=2,4 Гц), 7,28-7,23 (2H, м), 4,03 (2H, дд, J=11,0, 3,7 Гц), 3,85 (2H, д, J=7,3 Гц), 3,42-3,34 (2H, м), 2,40 (3H, с), 2,15-2,03 (1H, м), 1,63-1,51 (2H, м), 1,49-1,37 (2H, м).
МС (ESI/APCI) m/z:530 [(M+H)+].
Подобным образом, промежуточное соединение в таблице 10 было синтезировано из соответствующего исходного вещества.
[Таблица 10]


(Промежуточное соединение 17a) 5-(3,4-Диметоксифенил)-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амин
[Формула 62]
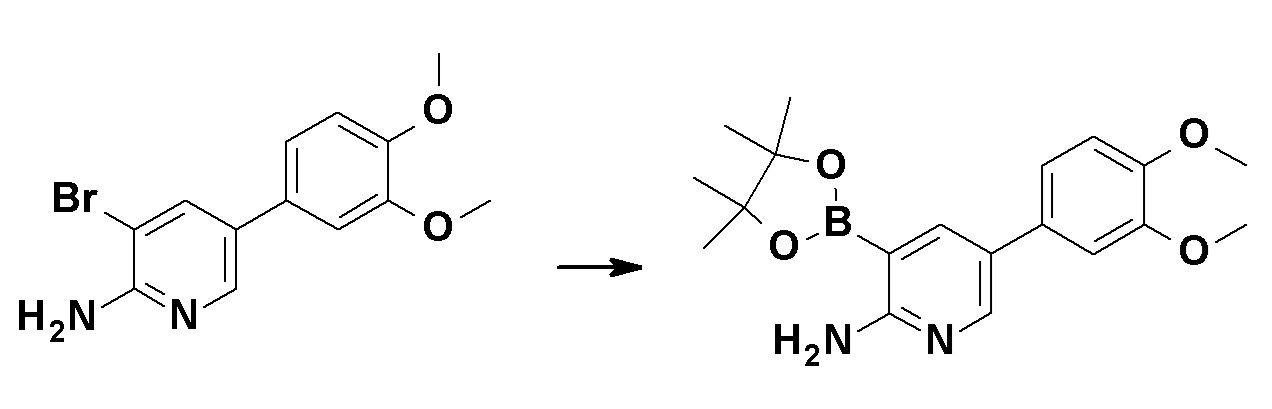
К 3-бром-5-(3,4-диметоксифенил)пиридин-2-амину (2,48 г, 8,02 ммоль), синтезированному способом, описанным в патенте (WO2013/115280 A1), бис(пинаколато)диборон (3,05 г, 12,0 ммоль), трис(дибензилиденацетон)палладий(0) (0,441 г, 0,481 ммоль), трициклогексилфосфин (0,315 г, 1,12 ммоль), и ацетат калия (1,18 г, 12,0 ммоль), 1,4-диоксан (40,0 мл) добавляли, и смесь перемешивали при 80°C в течение 6,5 часов в атмосфере азота. Реакционную смесь оставляли охлаждаться до комнатной температуры, и затем, к смеси добавляли воду, с последующей экстракцией этилацетатом три раза. Органический слой промывали насыщенным солевым раствором. После фильтрования и концентрирования при пониженном давлении, полученное твердое вещество суспендировали в диэтиловом эфире, собирали фильтрованием и сушили с получением указанного в заголовке соединения (2,20 г, выход: 77,0%) в виде желтого твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 8,36 (1H, д, J=3,0 Гц), 7,86 (1H, д, J=3,0 Гц), 7,09-6,97 (3H, м), 6,18 (2H, с), 3,83 (3H, с), 3,77 (3H, с), 1,32 (12H, с).
Подобным образом, промежуточные соединения в таблице 11, были синтезированы из соответствующих исходных веществ.
[Таблица 11-1]






[Таблица 11-2]




(Промежуточное соединение 18) 5'-(3,4-Диметоксифенил)-3-фтор-2,3'-бипиридин-2',5-диамин
[Формула 63]

К 6-хлор-5-фторпиридин-3-амину (0,0300 г, 0,205 ммоль), 5-(3,4-диметоксифенил)-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амину (0,109 г, 0,307 ммоль), комплексу [1,1'-бис(дифенилфосфино)ферроцен]палладий(II) дихлорид-дихлорметан (1:1) (0,0167 г, 0,0205 ммоль) и карбонату цезия (0,200 г, 0,614 ммоль), добавляли 1,4-диоксан (2,00 мл) и воду (0,200 мл), и смесь перемешивали при 100°C в течение 3 часов. Реакционную смесь оставляли охлаждаться до комнатной температуры, и затем, к этому добавляли воду, с последующей экстракцией метиленхлоридом три раза. Органический слой сушили над безводным сульфатом натрия. После фильтрования и концентрирования при пониженном давлении, полученный остаток очищали при помощи колоночной хроматографии на силикагеле (Shoko Scientific Co., Ltd., этилацетат/метанол=100/0 -> 85/15) с получением указанного в заголовке соединения (0,0690 г, выход: 99,0%) в виде масла.
1Н-ЯМР (CDCl3) δ: 8,27 (1H, д, J=2,4 Гц), 7,98-7,95 (2H, м), 7,10-7,04 (2H, м), 6,94 (1H, д, J=7,9 Гц), 6,85 (1H, дд, J=12,1, 2,4 Гц), 5,99 (2H, с), 3,99-3,92 (8H, м).
МС (APCI) m/z:341 [(M+H)+].
(Промежуточное соединение 19) 5-Бром-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоновая кислота
[Формула 64]

(Стадия 1) Метил 5-бром-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксилат
К метил 5-бром-4-гидроксипиридин-3-карбоксилату (2,36 г, 10,2 ммоль), синтезированному в соответствии со способом, описанным в литературе (J. Med. Chem. 2008, 51, 5330-5341), добавляли карбонат цезия (9,94 г, 30,5 ммоль) и N,N-диметилформамид (30,0 мл), и смесь перемешивали при 80°C в течение 15 минут. Затем, к этому добавляли 4-(бромметил)тетрагидро-2H-пиран (5,46 г, 30,5 ммоль) и смесь перемешивали при 80°C в течение 11 часов и затем оставляли на ночь при комнатной температуре. К реакционной смеси добавляли воду и насыщенный солевой раствор, затем экстрагировали три раза этилацетатом и затем экстрагировали метиленхлоридом пять раз. Органические слои объединяли и сушили над безводным сульфатом натрия. После фильтрования и концентрирования при пониженном давлении, полученный остаток очищали при помощи колоночной хроматографии на аминосиликагеле (Yamazen Corp., растворитель для элюирования: этилацетат/метанол=100/0 -> 85/15) с получением сырого продукта указанного в заголовке соединения (2,83 г) в виде масла.
МС (APCI) m/z:330 [(M+H)+].
(Стадия 2) 5-Бром-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоновая кислота
К раствору сырого продукта метил 5-бром-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксилата (2,83 г), синтезированного на стадии 1 в тетрагидрофуране (50,0 мл), добавляли метанол (25,0 мл) и 1 н водный раствор гидроксида натрия (25,7 мл, 25,7 ммоль), и смесь перемешивали в течение ночи при комнатной температуре. Органический растворитель отгоняли при пониженном давлении, и затем добавляли воду к остатку, с последующей промывкой этилацетатом дважды. Водный слой подкисляли путем добавления 1 н хлористоводородной кислоты, и затем, к смеси добавляли этилацетат. Осажденное твердое вещество собирали путем фильтрации, и затем, фильтрат подвергали экстракции этилацетатом три раза. Органический слой промывали насыщенным солевым раствором и затем сушили над безводным сульфатом натрия. После фильтрования и концентрирования при пониженном давлении, полученное твердое вещество суспендировали в диэтиловом эфире и собирали фильтрацией. Это твердое вещество объединяли с предварительно полученным твердым веществом и сушили с получением указанного в заголовке соединения (1,93 г, выход в 2 стадии: 60,0%) в виде твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 8,79 (1H, д, J=1,8 Гц), 8,75 (1H, д, J=1,8 Гц), 4,08 (2H, д, J=7,9 Гц), 3,84 (2H, дд, J=11,6, 3,4 Гц), 3,23 (2H, т, J=11,6 Гц), 2,10-2,00 (1H, м), 1,40 (2H, ушир. д, J=10,4 Гц), 1,30-1,20 (2H, м).
МС (APCI) m/z:316 [(M+H)+].
(Промежуточное соединение 20) N-{4-[2-Амино-5-(3,4-диметоксифенил)пиридин-3-ил]фенил}-5-бром-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид
[Формула 65]

К 5-бром-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоновой кислоте (0,754 г, 2,39 ммоль), 3-(4-аминофенил)-5-(3,4-диметоксифенил)пиридин-2-амину (0,730 г, 2,27 ммоль), синтезированному способом, описанным в патенте (WO2013/115280 A1), и N-[1-(циано-2-этокси-2-оксоэтилиденаминоокси)диметиламино(морфолино)]урония гексафторфосфату (COMU) (1,27 г, 2,95 ммоль), N,N-диметилформамид (12,0 мл) добавляли, затем добавляли N,N-диизопропилэтиламин (0,593 мл, 0,440 г, 3,41 ммоль), и смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении, и затем насыщенный водный раствор бикарбоната натрия добавляли к остатку, с последующим экстрагированием этилацетатом три раза. Органический слой промывали насыщенным солевым раствором и затем сушили над безводным сульфатом натрия. После фильтрования и концентрирования при пониженном давлении, полученный остаток очищали при помощи колоночной хроматографии на силикагеле (Yamazen Corp., растворитель для элюирования: этилацетат/метанол=100/0 -> 85/15). После концентрирования при пониженном давлении, остаток отверждали добавлением диэтилового эфира, и твердое вещество собирали путем фильтрации и сушили с получением указанного в заголовке соединения (0,724 г, выход: 51,5%) в виде твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 12,61 (1H, с), 8,74 (1H, д, J=2,4 Гц), 8,63 (1H, д, J=2,4 Гц), 8,26 (1H, д, J=2,4 Гц), 7,82 (2H, д, J=8,5 Гц), 7,64 (1H, д, J=2,4 Гц), 7,55 (2H, д, J=8,5 Гц), 7,20-7,14 (2H, м), 6,99 (1H, д, J=8,5 Гц), 5,77 (2H, с), 4,07 (2H, д, J=7,3 Гц), 3,87-3,83 (5H, м), 3,77 (3H, с), 3,26 (2H, т, J=10,7 Гц), 2,11-2,02 (1H, м), 1,42 (2H, ушир. д, J=10,9 Гц), 1,33-1,23 (2H, м).
МС (APCI) m/z:619 [(M+H)+].
Подобным образом, промежуточное соединение в таблице 12 было синтезировано из соответствующего исходного вещества.
[Таблица 12]


(Промежуточное соединение 21) N-[4-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-бром-1'-[(4-цианотетрагидро-2Н-пиран-4-ил)метил]-4'-оксо-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамид
[Формула 66]

К суспензии 5-бром-1'-[(4-цианотетрагидро-2Н-пиран-4-ил)метил]-4'-оксо-1',4'-дигидро-2,3'-бипиридин-5'-карбоновой кислоты (0,168 г, 0,401 ммоль) в N,N-диметилформамиде (5,00 мл) добавляли N-[1-(циано-2-этокси-2-оксоэтилиденаминоокси)диметиламино(морфолино)]урония гексафторфосфат (COMU) (0,172 г, 0,401 ммоль) и N,N-диизопропилэтиламин (0,0825 мл, 0,0612 г, 0,474 ммоль), и смесь перемешивали при комнатной температуре в течение 1 часа. В этой операции, реакционная смесь становилась гомогенной. К смеси добавляли 3-(4-амино-2-фторфенил)-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-2-амин (0,155 г, 0,364 ммоль) и смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении, и затем насыщенный водный раствор бикарбоната натрия добавляли к остатку, с последующим экстрагированием этилацетатом три раза. Органический слой промывали насыщенным солевым раствором и затем сушили над безводным сульфатом натрия. После фильтрования и концентрирования при пониженном давлении, полученный остаток очищали при помощи колоночной хроматографии на силикагеле (Yamazen Corp., этилацетат/метанол=99/1 -> 85/15) и затем очищали при помощи колоночной хроматографии на аминосиликагеле (Yamazen Corp., этилацетат/метанол=99/1 -> 93/7) с получением указанного в заголовке соединения (0,238 г, выход: 79,1%) в виде аморфного твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 12,95 (1H, с), 8,89-8,80 (3H, м), 8,59 (1H, д, J=8,5 Гц), 8,31 (1H, д, J=2,4 Гц), 8,18 (1H, дд, J=8,5, 2,4 Гц), 7,96-7,92 (1H, м), 7,63 (1H, д, J=2,4 Гц), 7,51-7,42 (2H, м), 7,19 (1H, д, J=2,4 Гц), 7,11 (1H, дд, J=8,5, 2,4 Гц), 6,99 (1H, д, J=8,5 Гц), 5,71 (2H, с), 4,71 (2H, с), 3,97-3,75 (10H, м), 3,69-3,59 (2H, м), 3,53-3,30 (4H, м), 1,88-1,78 (4H, м).
МС (APCI) m/z:825 [(M+H)+].
(Пример 1)
N-[4-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид
[Формула 67]

5-(4-Метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоновую кислоту (0,121 г, 0,367 ммоль) растворяли в N,N-диметилформамиде (3,00 мл). К раствору добавляли N-[1-(циано-2-этокси-2-оксоэтилиденаминоокси)диметиламино(морфолино)]урония гексафторфосфат (COMU) (0,236 г, 0,367 ммоль) и смесь перемешивали при комнатной температуре в течение 30 минут. Затем, к смеси добавляли 3-(4-аминофенил)-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-2-амин (0,149 г, 0,367 ммоль), синтезированный способом, описанным в патенте (WO2013/115280 A1), и смесь перемешивали при комнатной температуре в течение 4 часов. К реакционной смеси добавляли воду и осажденное твердое вещество собирали путем фильтрации. Это твердое вещество очищали при помощи колоночной хроматографии на силикагеле (Biotage Japan Ltd., растворитель для элюирования: дихлорметан/метанол=99/1 -> 95/5) с получением указанного в заголовке соединения (0,18 г, выход: 69,8%) в виде пены.
1Н-ЯМР (CDCl3) δ: 12,85 (1H, с), 8,57 (1H, д, J=2,4 Гц), 8,25 (1H, д, J=1,8 Гц), 7,87 (2H, д, J=8,5 Гц), 7,57 (1H, д, J=1,8 Гц), 7,47 (5H, дд, J=8,2, 5,8 Гц), 7,29 (2H, д, J=8,5 Гц), 7,08-7,02 (2H, м), 6,98-6,94 (1H, м), 4,71 (2H, с), 4,11-3,94 (6H, м), 3,93-3,79 (8H, м), 3,79-3,63 (1H, м), 3,59-3,51 (1H, м), 3,44-3,35 (2H, м), 2,41 (3H, с), 2,18-2,04 (1H, м), 1,64-1,58 (2H, м), 1,51-1,35 (2H, м).
МС (ESI) m/z:717 [(M+H)+]
Подобным образом, промежуточные соединения в таблице 13, были синтезированы из соответствующих исходных веществ А и В.
[Таблица 13-1]
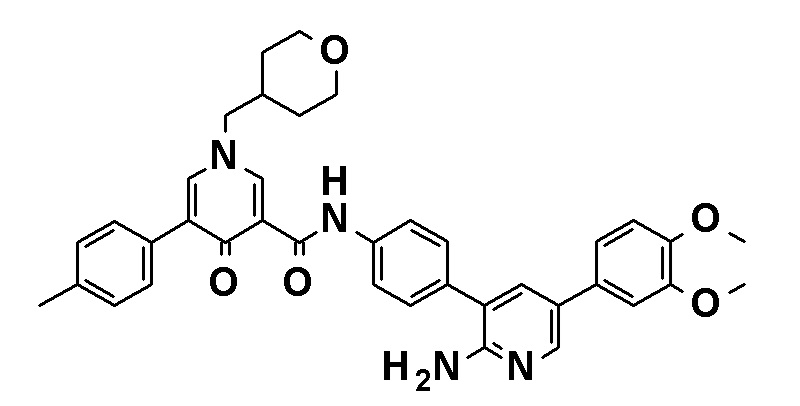





[Таблица 13-2]






[Таблица 13-3]






[Таблица 13-4]





[Таблица 13-5]






[Таблица 13-6]






[Таблица 13-7]









[Таблица 13-8]



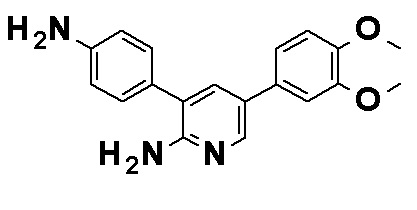



[Таблица 13-9]




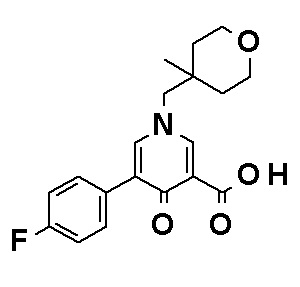




[Таблица 13-10]






[Таблица 13-11]






[Таблица 13-12]
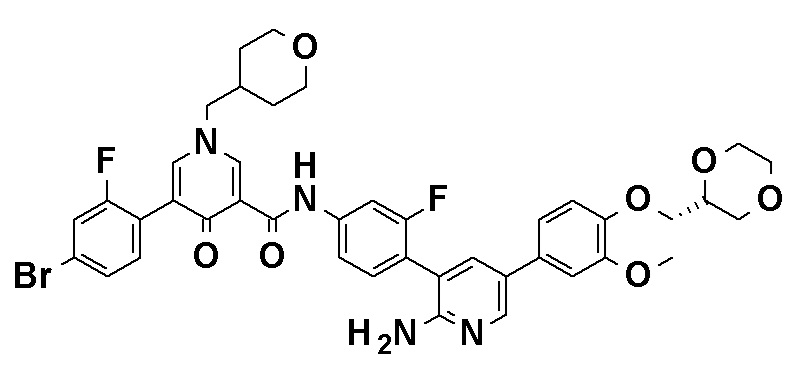




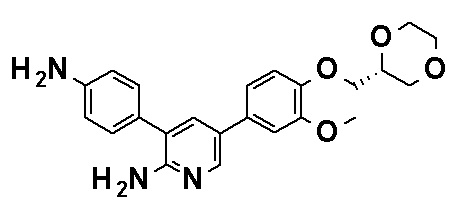
[Таблица 13-13]
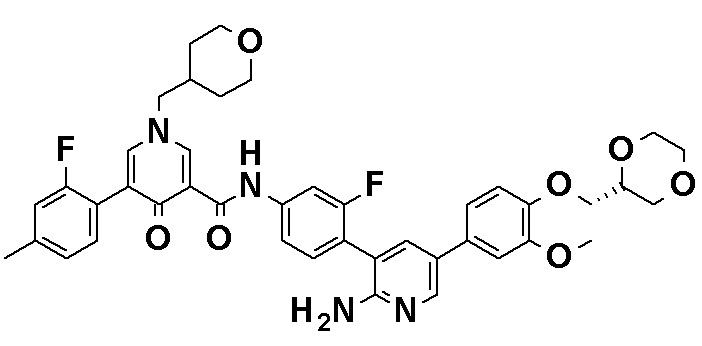








[Таблица 13-14]








[Таблица 13-15]



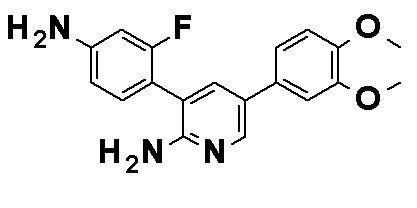
[Таблица 13-16]






[Таблица 13-17]





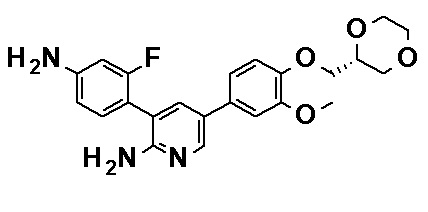



[Таблица 13-18]


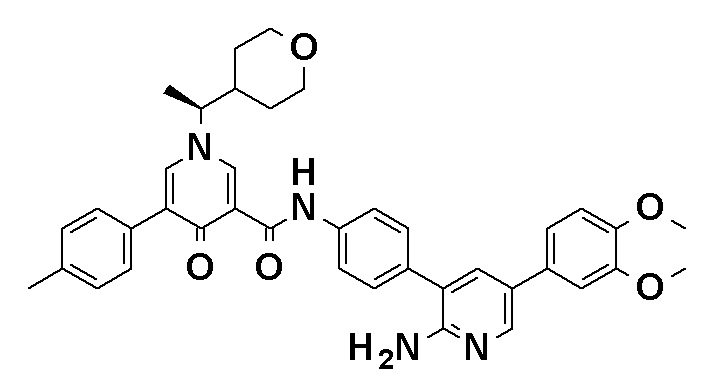





[Таблица 13-19]








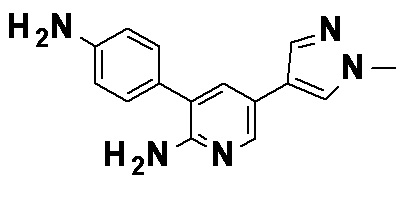
[Таблица 13-20]








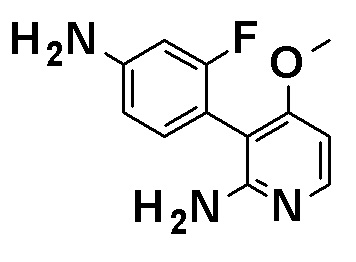
[Таблица 13-21]









[Таблица 13-22]





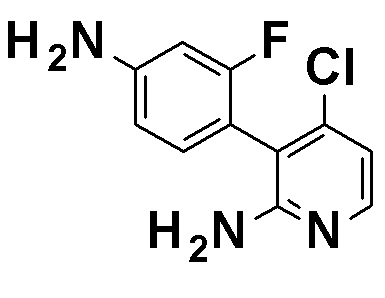



[Таблица 13-23]

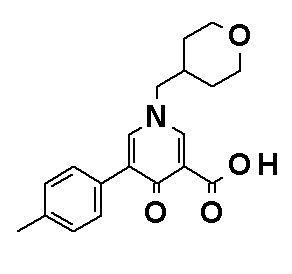

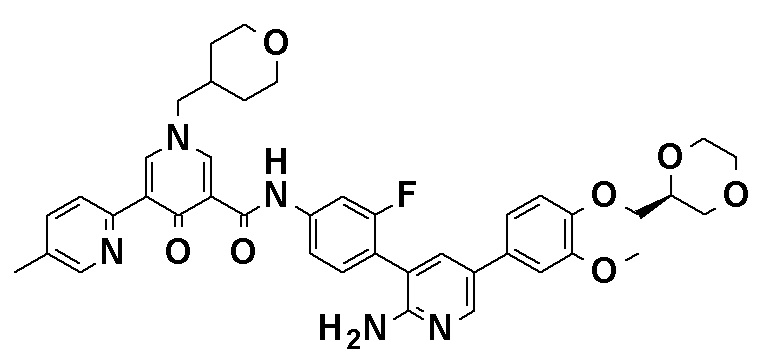


[Таблица 13-24]









[Таблица 13-25]




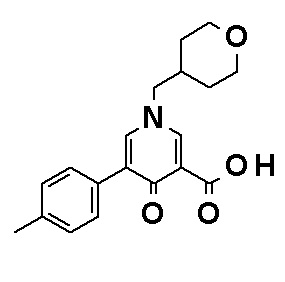




[Таблица 13-26]

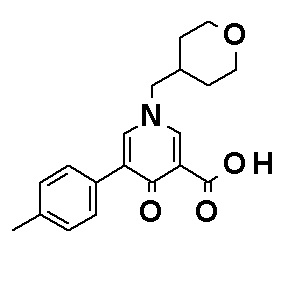







(Пример 68)
N-{4-[2-Амино-5-(3,4-диметоксифенил)пиридин-3-ил]-3-фторфенил}-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид метансульфонат
[Формула 68]

5-(4-Метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоновую кислоту (0,127 г, 0,389 ммоль) растворяли в N-метил-2-пирролидоне (3,00 мл). К раствору добавляли тионилхлорид (0,0277 мл, 0,382 ммоль) и смесь перемешивали при комнатной температуре в течение 30 минут. Затем, к смеси добавляли 3-(4-амино-2-фторфенил)-5-(3,4-диметоксифенил)пиридин-2-амин (0,121 г, 0,354 ммоль) при охлаждении льдом, и смесь перемешивали при комнатной температуре в течение 5 часов. Реакционную смесь снова охлаждали льдом и нейтрализовали насыщенным раствором бикарбоната натрия, затем экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором и затем сушили над безводным сульфатом магния, и растворитель отгоняли при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле (Biotage Japan Ltd., растворитель для элюирования: дихлорметан/метанол=99/1 -> 96/4) с получением указанного в заголовке соединения (0,106 г, выход: 46,2%) в виде пены.
1Н-ЯМР (CDCl3) δ: 12,98 (1H, с), 8,56 (1H, д, J=2,4 Гц), 8,31 (1H, д, J=2,4 Гц), 7,92 (1H, дд, J=12,1, 1,8 Гц), 7,59 (1H, д, J=2,4 Гц), 7,49-7,43 (4H, м), 7,36 (1H, т, J=8,2 Гц), 7,29 (2H, д, J=7,9 Гц), 7,08 (1H, дд, J=8,2, 2,1 Гц), 7,03 (1H, д, J=1,8 Гц), 6,93 (1H, д, J=7,9 Гц), 4,57-4,52 (2H, м), 4,07-4,00 (2H, м), 3,94 (3H, с), 3,92 (3H, с), 3,88 (2H, д, J=7,3 Гц), 3,44-3,35 (2H, м), 2,41 (3H, с), 2,19-2,04 (1H, м), 1,65-1,57 (2H, м), 1,51-1,38 (2H, м).
МС (ESI) m/z:649 [(M+H)+]
Подобным образом, промежуточные соединения в таблице 14, были синтезированы из соответствующих исходных веществ.
[Таблица 14]



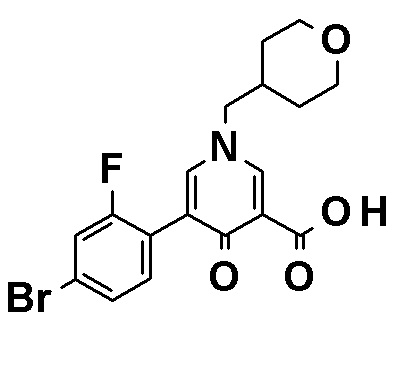
(Пример 71) N-[4-(2-Амино-5-{4-[(4-метилпиперазин-1-ил)метил]фенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид метансульфонат
[Формула 69]

(Стадия 1) N-[4-(2-Амино-5-бромпиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид
Указанное в заголовке соединение получали таким же образом, как и в Примере 1, используя 3-(4-аминофенил)-5-бромпиридин-2-амин в качестве исходного вещества.
1Н-ЯМР (CDCl3) δ: 12,88-12,85 (1H, м), 8,56 (1H, д, J=2,4 Гц), 8,08 (1H, д, J=2,4 Гц), 7,87-7,84 (2H, м), 7,49-7,45 (4H, м), 7,43-7,39 (2H, м), 7,31-7,27 (2H, м), 4,63-4,58 (2H, м), 4,06-4,00 (2H, м), 3,89-3,86 (2H, м), 3,43-3,35 (2H, м), 2,41 (3H, с), 2,17-2,06 (1H, м), 1,65-1,38 (4H, м).
МС (ES+APCI) m/z:573 [(M+H)+].
(Стадия 2) N-[4-(2-Амино-5-{4-[(4-метилпиперазин-1-ил)метил]фенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид
Указанное в заголовке соединение получали таким же образом, как и в промежуточном соединении 8, используя N-[4-(2-амино-5-бромпиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид и 1-метил-4-[[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]метил]пиперазин в качестве исходных веществ, 1,4-диоксан и воду в качестве растворителя, тетракис(трифенилфосфин)палладий(0) в качестве катализатора, и карбонат калия в качестве основания, и осуществляя нагревание с перемешиванием при температуре реакции 100°C в течение 2 часов.
1Н-ЯМР (CDCl3) δ: 12,87-12,85 (1H, м), 8,57 (1H, д, J=2,4 Гц), 8,31 (1H, д, J=2,4 Гц), 7,87 (2H, д, J=7,9 Гц), 7,61 (1H, д, J=2,4 Гц), 7,52-7,45 (7H, м), 7,37 (2H, д, J=7,9 Гц), 7,29 (2H, д, J=7,9 Гц), 4,69-4,64 (2H, м), 4,06-4,00 (2H, м), 3,87 (2H, д, J=7,3 Гц), 3,54 (2H, с), 3,43-3,35 (2H, м), 2,63-2,34 (8H, м), 2,41 (3H, с), 2,29 (3H, с), 2,15-2,07 (1H, м), 1,64-1,57 (2H, м), 1,50-1,38 (2H, м).
МС (ES+APCI) m/z:683 [(M+H)+].
(Стадия 3) N-[4-(2-Амино-5-{4-[(4-метилпиперазин-1-ил)метил]фенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамида метансульфонат
N-[4-(2-Амино-5-{4-[(4-метилпиперазин-1-ил)метил]фенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид (34,0 мг, 49,8 мкмоль) растворяли в хлороформе (3,00 мл). К раствору добавляли 1 моль/л раствор метансульфоновой кислоты в этаноле (49,8 мкл, 49,8 мкмоль) при комнатной температуре, и затем, растворитель отгоняли при пониженном давлении. К полученному остатку добавляли диизопропиловый эфир, и осажденное твердое вещество собирали путем фильтрации с получением указанного в заголовке соединения (25,7 мг, выход: 66,3%).
1Н-ЯМР (CDCl3) δ: 12,92-12,90 (1H, м), 8,57 (1H, д, J=2,4 Гц), 8,20 (1H, д, J=2,4 Гц), 7,91-7,87 (2H, м), 7,69 (1H, д, J=2,4 Гц), 7,52-7,44 (7H, м), 7,39-7,35 (2H, м), 7,31-7,27 (2H, м), 4,07-4,00 (2H, м), 3,88 (2H, д, J=7,3 Гц), 3,69-3,63 (2H, м), 3,44-3,35 (2H, м), 3,08-2,61 (8H, м), 2,86 (3H, с), 2,41 (3H, с), 2,18 (3H, с), 2,16-2,06 (1H, м), 1,65-1,58 (2H, м), 1,51-1,39 (2H, м).
МС (ES+APCI) m/z:683 [(M+H)+].
Подобным образом, промежуточные соединения в таблице 15, были синтезированы из соответствующих исходных веществ.
[Таблица 15-1]






[Таблица 15-2]


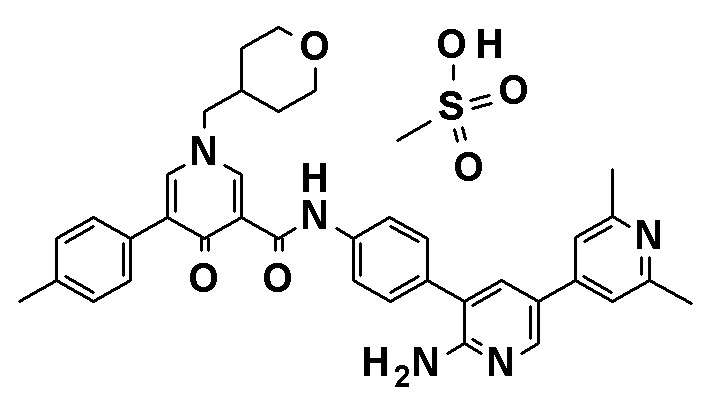


[Таблица 15-3]






[Таблица 15-4]


(Пример 83) N-(4-{2-Амино-5-[3-фтор-4-(морфолин-4-илметил)фенил]пиридин-3-ил}фенил)-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид метансульфонат
[Формула 70]
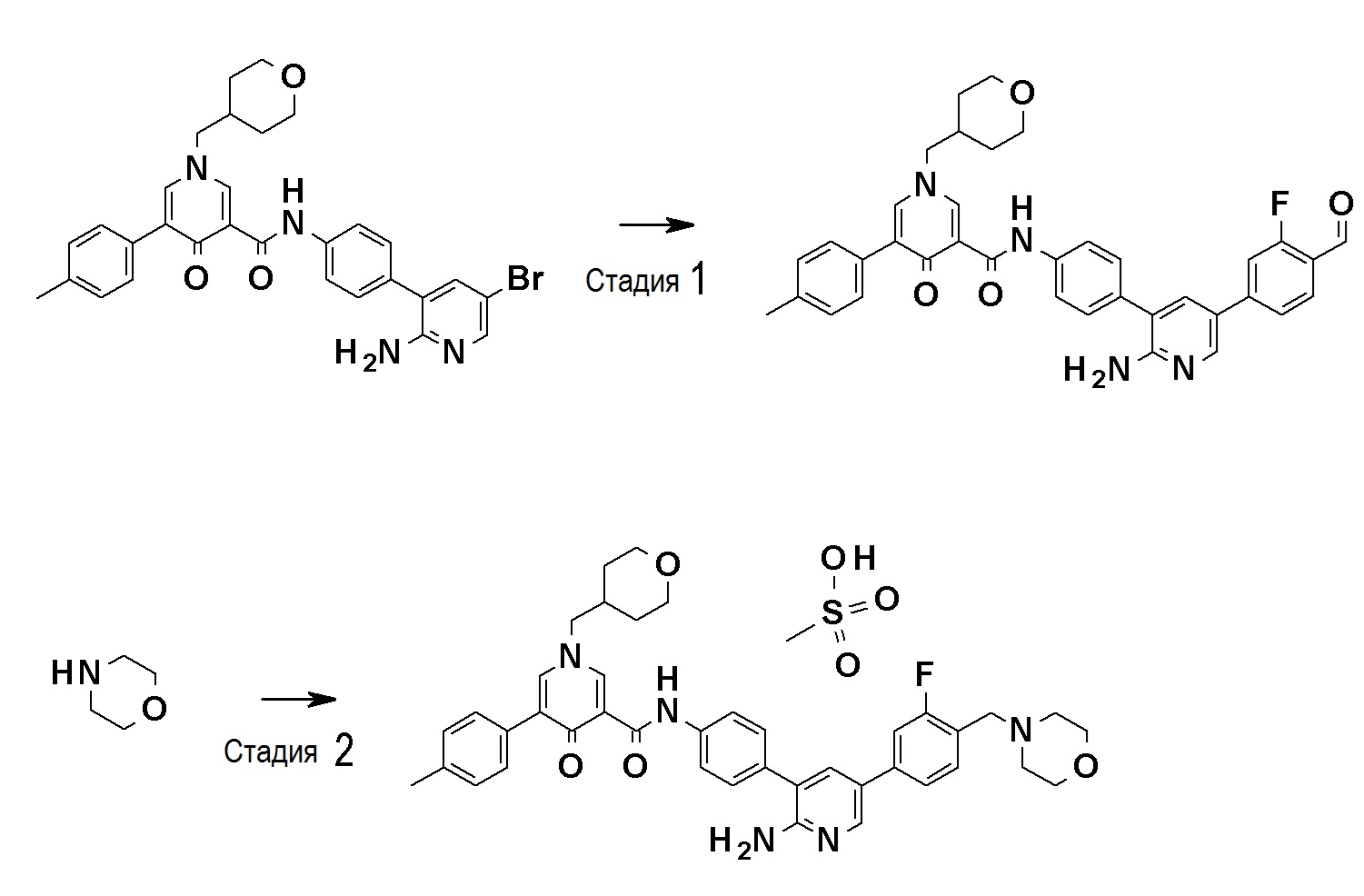
(Стадия 1) N-{4-[2-Амино-5-(3-фтор-4-формилфенил)пиридин-3-ил]фенил}-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид
Указанное в заголовке соединение получали таким же образом, как и в промежуточном соединении 8, используя N-[4-(2-амино-5-бромпиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид и 3-фтор-4-формилфенилборную кислоту, 1,4-диоксан и воду (отношение: 10:1) в качестве растворителя, тетракис(трифенилфосфин)палладий(0) в качестве катализатора, и карбонат калия в качестве основания и осуществляя нагревание с перемешиванием при температуре реакции 100°C в течение 7 часов.
1Н-ЯМР (CDCl3) δ: 12,89 (1H, с), 10,37-10,35 (1H, м), 8,58-8,57 (1H, м), 8,37 (1H, д, J=2,4 Гц), 7,94-7,87 (3H, м), 7,64 (1H, д, J=2,4 Гц), 7,49-7,45 (6H, м), 7,36 (1H, дд, J=12,1, 1,8 Гц), 7,31-7,28 (2H, м), 4,86-4,81 (2H, м), 4,07-4,01 (2H, м), 3,88 (2H, д, J=7,3 Гц), 3,43-3,35 (2H, м), 2,41 (3H, с), 2,17-2,07 (1H, м), 1,64-1,53 (2H, м), 1,51-1,39 (2H, м).
МС (ES+APCI) m/z:617 [(M+H)+].
(Стадия 2) N-(4-{2-Амино-5-[3-фтор-4-(морфолин-4-илметил)фенил]пиридин-3-ил}фенил)-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид метансульфонат
N-{4-[2-Амино-5-(3-фтор-4-формилфенил)пиридин-3-ил]фенил}-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид (70,0 мг, 0,114 ммоль) суспендировали в дихлорэтане (2,27 мл). К суспензии добавляли морфолин (19,8 мкл, 0,227 ммоль) и смесь перемешивали при комнатной температуре в течение 5 минут. К реакционной смеси добавляли триацетоксиборгидрид натрия (48,1 мг, 0,227 ммоль) и смесь перемешивали при комнатной температуре в течение 3 часов. К реакционной смеси добавляли воду и бикарбонат натрия, затем экстрагировали хлороформом. Экстракты сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении, и полученный остаток очищали при помощи колоночной хроматографии на силикагеле (Biotage Japan Ltd., дихлорметан/этанол=100/1 -> 50/1) с получением твердого вещества. Это твердое вещество растворяли в хлороформе (8,00 мл). К раствору добавляли 1 M раствора метансульфоновой кислоты в этаноле (91,2 мкл, 91,2 мкмоль) и смесь концентрировали при пониженном давлении. К полученному остатку добавляли диэтиловый эфир, и осажденное твердое вещество собирали путем фильтрации с получением указанного в заголовке соединения (30,5 мг, 34,7%) в виде твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 13,21-13,18 (1H, м), 10,01-9,83 (1H, м), 8,72 (1H, д, J=2,4 Гц), 8,42 (1H, д, J=2,4 Гц), 8,19 (1H, д, J=2,4 Гц), 8,10-8,03 (1H, м), 7,91-7,81 (3H, м), 7,78-7,73 (1H, м), 7,69-7,63 (1H, м), 7,61-7,55 (4H, м), 7,27 (2H, д, J=7,9 Гц), 4,49-4,36 (2H, м), 4,12 (2H, д, J=7,3 Гц), 4,03-3,92 (2H, м), 3,90-3,84 (2H, м), 3,72-3,59 (2H, м), 3,47-3,10 (8H, м), 2,36 (3H, с), 2,32 (3H, с), 2,17-2,05 (1H, м), 1,50-1,42 (2H, м), 1,38-1,25 (2H, м).
МС (ES+APCI) m/z:688 [(M+H)+].
Подобным образом, окончательное соединение в таблице 16 было синтезировано из соответствующего исходного вещества.
[Таблица 16]


(Пример 85) N-(4-{2-Амино-5-[1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-ил]пиридин-3-ил}-3-фторфенил)-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид метансульфонат
[Формула 71]

(Стадия 1) N-[4-(2-Амино-5-бромпиридин-3-ил)-3-фторфенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид
Указанное в заголовке соединение получали таким же образом, как и в Примере 1 с использованием 3-(4-амино-2-фторфенил)-5-бром-пиридин-2-амина, синтезированного способом, описанным в патенте (WO2013/115280 A1), в качестве исходного вещества.
1Н-ЯМР (CDCl3) δ: 13,01-12,98 (1H, м), 8,55 (1H, д, J=2,4 Гц), 8,13 (1H, д, J=2,4 Гц), 7,91 (1H, дд, J=12,1, 2,4 Гц), 7,51-7,41 (5H, м), 7,32-7,25 (3H, м), 4,54-4,50 (2H, м), 4,07-4,01 (2H, м), 3,88 (2H, д, J=7,3 Гц), 3,43-3,35 (2H, м), 2,41 (3H, с), 2,16-2,05 (1H, м), 1,64-1,54 (2H, м), 1,51-1,38 (2H, м).
МС (ES+APCI) m/z:591 [(M+H)+].
(Стадия 2) N-(4-{2-Амино-5-[1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-ил]пиридин-3-ил}-3-фторфенил)-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид метансульфонат
N-(4-{2-Амино-5-[1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-ил]пиридин-3-ил}-3-фторфенил)-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид получали таким же образом, как и в промежуточном соединении 8 с использованием N-[4-(2-амино-5-бромпиридин-3-ил)-3-фторфенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамида, полученного на стадии 1, и 1-(тетрагидро-2H-пиран-4-ил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола в качестве исходных веществ, 1,4-диоксана и воды (отношение: 10:1) в качестве растворителя, тетракис(трифенилфосфин)палладия(0) в качестве катализатора, и карбоната калия в качестве основания и проведением нагревания при перемешивании при температуре реакции 100°C в течение 7 часов. Указанное в заголовке соединение получали с помощью той же операции, что и на стадии 3 примера 71 с использованием этого соединения.
1Н-ЯМР (ДМСО-D6) δ: 13,38-13,36 (1H, м), 8,73 (1H, д, J=2,4 Гц), 8,42-8,40 (1H, м), 8,26-8,19 (3H, м), 8,03-7,98 (2H, м), 7,61-7,44 (6H, м), 7,27 (2H, д, J=7,9 Гц), 4,44-4,35 (1H, м), 4,12 (2H, д, J=6,7 Гц), 3,99-3,93 (2H, м), 3,90-3,84 (2H, м), 3,52-3,43 (2H, м), 3,31-3,23 (2H, м), 2,36 (3H, с), 2,31 (3H, с), 2,17-2,06 (1H, м), 2,05-1,98 (2H, м), 1,97-1,84 (2H, м), 1,49-1,42 (2H, м), 1,37-1,25 (2H, м).
МС (ES+APCI) m/z:663 [(M+H)+].
Подобным образом, промежуточные соединения в таблице 17, были синтезированы из соответствующих исходных веществ.
[Таблица 17]
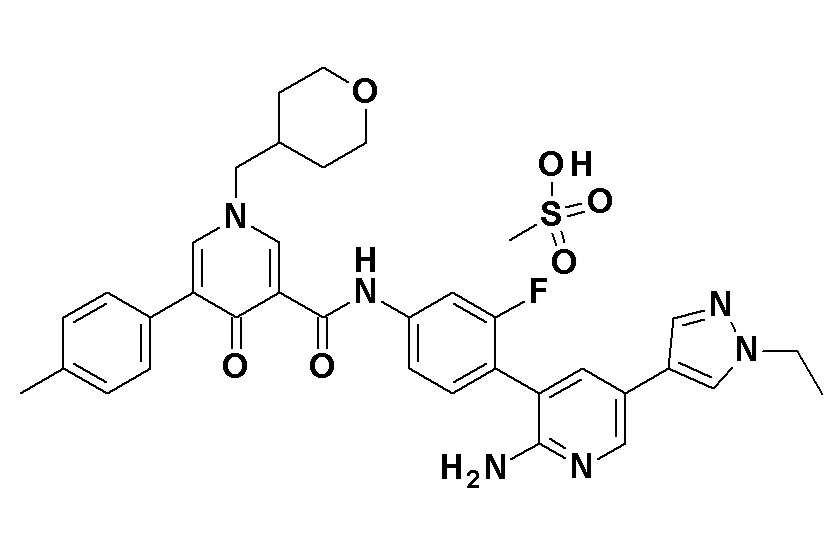



(Пример 88) N-[2'-Амино-5'-(3,4-диметоксифенил)-2,3'-бипиридин-5-ил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид метансульфонат
[Формула 72]

(Стадия 1) N-(2'-Амино-5'-бром-2,3'-бипиридин-5-ил)-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид
К раствору 5-бром-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амина (200 мг, 0,669 ммоль) в диоксане (10,0 мл) и воде (1,00 мл), N-(6-иодпиридин-3-ил)-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид (354 мг, 0,669 ммоль), карбонат калия (277 мг, 2,01 ммоль), и тетракис(трифенилфосфин)палладий(0) (38,0 мг, 0,0344 ммоль) добавляли, и смесь перемешивали при 90°C в течение 3 часов в атмосфере азота. К полученному дополнительно добавляли 5-бром-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амин (200 мг, 0,669 ммоль) и смесь перемешивали при 90°C в течение 5 часов. Реакционную смесь оставляли охлаждаться и затем разделяли на органический и водный слои путем добавления воды, метиленхлорида и метанола. Органический слой сушили над безводным сульфатом натрия и затем фильтровали, и фильтрат концентрировали. Остаток превращали в суспензию этилацетатом с получением указанного в заголовке соединения (352 мг, 91,6%) в виде твердого вещества.
1Н-ЯМР (CDCl3) δ: 13,05 (1H, с), 8,94 (1H, д, J=2,4 Гц), 8,56 (1H, д, J=2,4 Гц), 8,32 (1H, дд, J=8,5, 2,4 Гц), 8,10 (1H, д, J=1,8 Гц), 7,92 (1H, д, J=2,4 Гц), 7,68 (1H, д, J=9,2 Гц), 7,50-7,44 (3H, м), 7,29 (2H, д, J=7,9 Гц), 6,76 (2H, ушир. с), 4,04 (2H, дд, J=11,9, 3,4 Гц), 3,89 (2H, д, J=7,3 Гц), 3,39 (2H, т, J=11,0 Гц), 2,42 (3H, с), 2,16-2,06 (1H, м), 1,65-1,57 (2H, м), 1,51-1,38 (2H, м).
МС (APCI) m/z:574 [(M+H)+].
(Стадия 2) N-[2'-Амино-5'-(3,4-диметоксифенил)-2,3'-бипиридин-5-ил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамида метансульфонат
К суспензии N-(2'-амино-5'-бром-2,3'-бипиридин-5-ил)-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамида (130 мг, 0,226 ммоль) и 3,4-диметоксифенилбороновой кислоты (50 мг, 0,272 ммоль) в диоксане (10 мл) и воде (1 мл), добавляли карбонат калия (94 мг, 0,679 ммоль) и тетракис(трифенилфосфин)палладий(0) (13 мг, 0,0113 ммоль), и смесь перемешивали при 100°C в течение 2 часов в атмосфере азота. Реакционную смесь разбавляли метиленхлоридом и затем промывали водой. Органический слой сушили над безводным сульфатом натрия и фильтровали, и затем растворитель отгоняли. Остаток очищали при помощи колоночной хроматографии на силикагеле (метиленхлорид/метанол=19/1; колонку с аминосиликагелем использовали в качестве предварительной колонки) с получением N-[2'-амино-5'-(3,4-диметоксифенил)-2,3'-бипиридин-5-ил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамида (69,0 мг, 48,3%) в виде твердого вещества. К раствору этого соединения в метиленхлориде (1,00 мл) и этаноле (1,00 мл), добавляли метансульфоновую кислоту (10,5 мг, 0,192 ммоль), и растворитель отгоняли. Остаток суспендировали в метиленхлориде/этилацетате/изопропиловом эфире, и нерастворимое вещество собирали фильтрованием с получением указанного в заголовке соединения (65,0 мг, 81,8%) в виде твердого вещества.
1Н-ЯМР (CDCl3) δ: 14,93 (1H, с), 13,27 (1H, с), 9,03 (1H, д, J=2,4 Гц), 8,57 (1H, с), 8,41-8,36 (2H, м), 7,91-7,85 (2H, м), 7,52 (1H, д, J=2,4 Гц), 7,46 (2H, д, J=8,5 Гц), 7,30 (2H, д, J=7,9 Гц), 7,05 (1H, дд, J=8,2, 2,1 Гц), 7,00-6,94 (2H, м), 4,04 (2H, дд, J=11,6, 3,7 Гц), 3,96 (3H, с), 3,95 (3H, с), 3,91 (2H, д, J=7,3 Гц), 3,40 (2H, т, J=11,0 Гц), 2,95 (3H, с), 2,42 (3H, с), 2,18-2,07 (1H, м), 1,65-1,55 (2H, м), 1,52-1,39 (2H, м).
МС (ESI) m/z:632 [(M+H)+].
Подобным образом, промежуточные соединения в таблице 18, были синтезированы из соответствующих исходных веществ А и В.
[Таблица 18-1]






[Таблица 18-2]
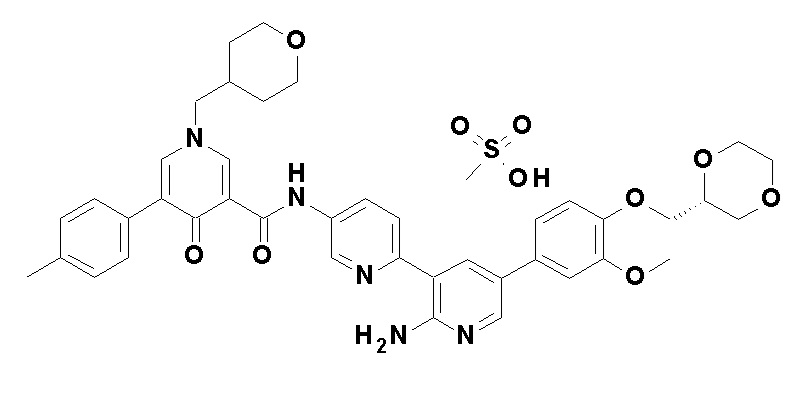





[Таблица 18-3]









[Таблица 18-4]






[Таблица 18-5]





(Пример 100) N-{6-[2-Амино-5-(3,4-диметоксифенил)пиридин-3-ил]пиридазин-3-ил}-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид
[Формула 73]

К 5-(3,4-диметоксифенил)-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амину (0,0800 г, 0,182 ммоль), N-(6-хлорпиридазин-3-ил)-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамиду (0,0974 г, 0,273 ммоль), трис(дибензилиденацетон)палладию(0) (0,0167 г, 0,0182 ммоль), 2-дициклогексилфосфино-2,4',6'-триизопропилбифенилу (XPhos) (0,0348 г, 0,0729 ммоль), и карбонату натрия (0,0229 г, 0,547 ммоль), добавляли 1,4-диоксан (2,00 мл) и воду (0,200 мл) и смесь перемешивали при 100°C в течение 4 часов. Реакционную смесь оставляли охлаждаться до комнатной температуры, и затем, к этому добавляли воду, затем экстрагировали метиленхлоридом три раза. Органический слой сушили над безводным сульфатом натрия. После фильтрования и концентрирования при пониженном давлении, полученный остаток очищали при помощи колоночной хроматографии на силикагеле (Yamazen Corp., этилацетат/метанол=100/0 -> 80/20). После концентрирования при пониженном давлении, полученное твердое вещество суспендировали в этаноле, собирали фильтрованием и сушили с получением указанного в заголовке соединения (0,067 г, выход: 58,1%) в виде твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 13,94 (1H, с), 8,78 (1H, д, J=2,4 Гц), 8,64 (1H, д, J=9,7 Гц), 8,52 (1H, д, J=9,7 Гц), 8,42 (1H, д, J=2,4 Гц), 8,25-8,21 (2H, м), 7,61 (2H, д, J=7,9 Гц), 7,48 (2H, с), 7,28-7,23 (4H, м), 7,02 (1H, д, J=8,5 Гц), 4,12 (2H, д, J=7,3 Гц), 3,91-3,83 (5H, м), 3,79 (3H, с), 3,27 (2H, т, J=10,9 Гц), 2,37 (3H, с), 2,16-2,07 (1H, м), 1,47 (2H, ушир. д, J=10,9 Гц), 1,37-1,26 (2H, м).
МС (APCI) m/z:633 [(M+H)+].
Подобным образом, промежуточные соединения в таблице 19, были синтезированы из соответствующих исходных веществ А и В.
[Таблица 19-1]






[Таблица 19-2]









[Таблица 19-3]

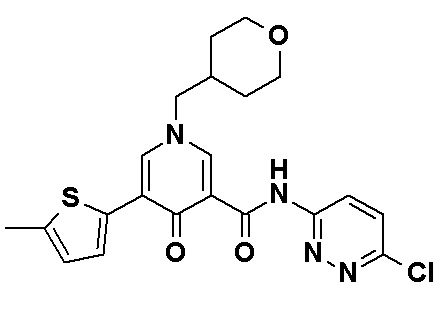



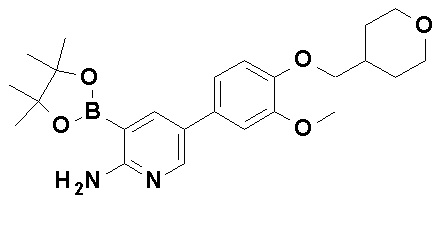
[Таблица 19-4]





(Пример 110) N-[6-(2-Амино-5-{3-метокси-4-[2-(морфолин-4-ил)этокси]фенил}пиридин-3-ил)пиридазин-3-ил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид
[Формула 74]
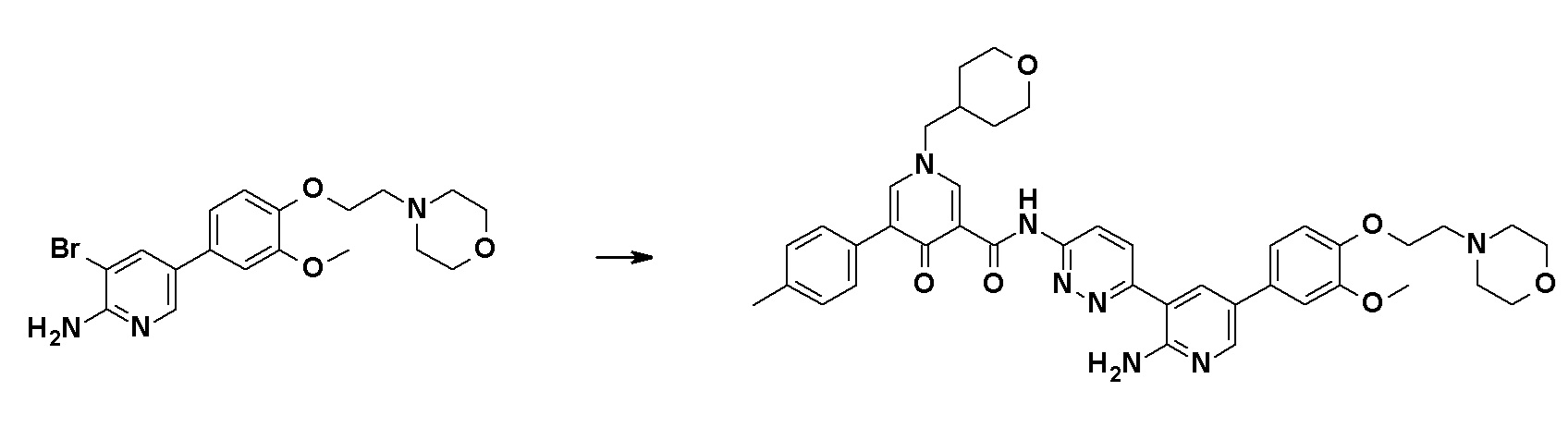
К 3-бром-5-{3-метокси-4-[2-(морфолин-4-ил)этокси]фенил}пиридин-2-амину(0,245 г, 0,600 ммоль), бис(пинаколато)диборон (0,229 г, 0,900 ммоль), трис(дибензилиденацетон)дипалладий (0) (0,0330 г, 0,0360 ммоль), трициклогексилфосфин (0,0236 г, 0,0840 ммоль), и ацетат калия (0,0883 г, 0,900 ммоль), 1,4-диоксан (3,00 мл) добавляли, и смесь перемешивали при 80°C в течение 6 часов в атмосфере азота. Реакционную смесь оставляли охлаждаться до комнатной температуры и затем фильтровали, и фильтрат концентрировали при пониженном давлении. К полученному остатку N-(6-хлорпиридазин-3-ил)-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид (0,175 г, 0,399 ммоль), комплекс [1,1'-бис(дифенилфосфино)ферроцен]палладий(II) дихлорид-метиленхлорид (1:1) (0,0326 г, 0,0399 ммоль), карбонат цезия (0,390 г, 1,20 ммоль), 1,4-диоксан (4,00 мл) и воду (0,400 мл) добавляли, и смесь перемешивали при 100°C в течение 4 часов в атмосфере азота. Реакционную смесь оставляли охлаждаться до комнатной температуры, и затем к смеси добавляли насыщенный водный раствор бикарбоната натрия, затем экстрагировали метиленхлоридом три раза. Органический слой сушили над безводным сульфатом натрия. После фильтрования и концентрирования при пониженном давлении, полученный остаток очищали при помощи колоночной хроматографии на аминосиликагеле (Yamazen Corp., этилацетат/метанол=100/0 -> 80/20). После концентрирования при пониженном давлении, полученное твердое вещество суспендировали в этилацетате, собирали путем фильтрации и сушили с получением указанного в заголовке соединения (0,153 г, выход: 52,4%) в виде твердого вещества.
1Н-ЯМР (CDCl3) δ: 13,77 (1H, с), 8,67 (1H, д, J=9,7 Гц), 8,51 (1H, д, J=2,4 Гц), 8,35 (1H, д, J=2,4 Гц), 7,96-7,94 (2H, м), 7,53-7,48 (3H, м), 7,27 (2H, д, J=7,9 Гц), 7,08-7,05 (2H, м), 6,98 (1H, д, J=7,9 Гц), 6,89 (2H, с), 4,21 (2H, т, J=6,1 Гц), 4,03 (2H, дд, J=11,2, 3,3 Гц), 3,94 (3H, с), 3,87 (2H, д, J=7,3 Гц), 3,76 (4H, т, J=4,9 Гц), 3,39 (2H, т, J=11,2 Гц), 2,88 (2H, т, J=6,1 Гц), 2,63-2,61 (4H, м), 2,41 (3H, с), 2,15-2,07 (1H, м), 1,62-1,60 (2H, ушир. д, J=11,2 Гц), 1,49-1,39 (2H, м).
МС (APCI) m/z:732 [(M+H)+].
Подобным образом, окончательное соединение в таблице 20 было синтезировано из соответствующего исходного вещества.
[Таблица 20]
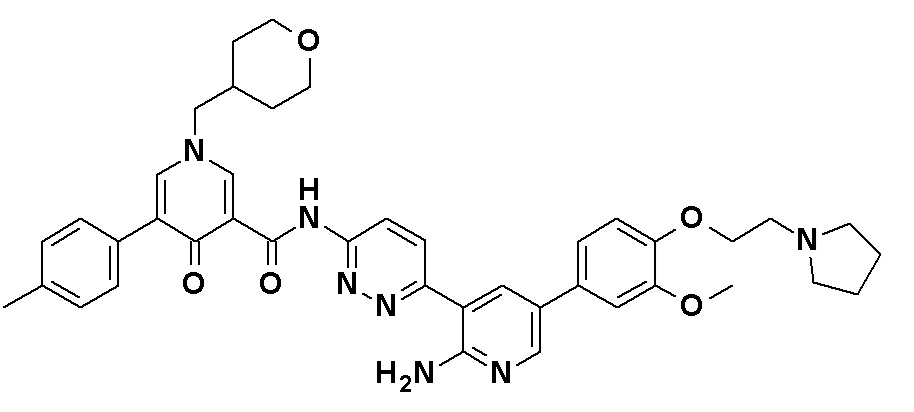

(Пример 112) N-{5-[2-Амино-5-(3,4-диметоксифенил)пиридин-3-ил]пиразин-2-ил}-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид
[Формула 75]

К 5-(3,4-диметоксифенил)-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амину (0,111 г, 0,310 ммоль), N-(5-бромпиразин-2-ил)-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамиду (0,100 г, 0,207 ммоль), комплексу [1,1'-бис(дифенилфосфино)ферроцен]палладий(II) дихлорид-метиленхлорид (1:1) (0,0169 г, 0,0207 ммоль), и карбонату цезия (0,202 г, 0,621 ммоль), добавляли 1,4-диоксан (2,00 мл) и воду (0,400 мл), и смесь перемешивали при 80°C в течение 1 часа. Реакционную смесь оставляли охлаждаться до комнатной температуры, и затем, к смеси добавляли воду, с последующей экстракцией три раза этилацетатом. Органический слой промывали насыщенным солевым раствором и затем сушили над безводным сульфатом натрия. После фильтрования и концентрирования при пониженном давлении, полученный остаток очищали при помощи колоночной хроматографии на аминосиликагеле (Yamazen Corp., этилацетат/метанол=100/0 -> 90/10). После концентрирования при пониженном давлении, остаток отверждали с использованием этилацетата и диэтилового эфира, и твердое вещество собирали путем фильтрации и сушили с получением указанного в заголовке соединения (0,111 г, выход: 84,8%) в виде твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 13,61 (1H, с), 9,58 (1H, д, J=1,2 Гц), 9,17 (1H, д, J=2,4 Гц), 8,79 (1H, д, J=2,4 Гц), 8,39 (1H, д, J=2,4 Гц), 8,32 (1H, д, J=2,4 Гц), 8,20 (1H, д, J=2,4 Гц), 7,59 (2H, д, J=8,5 Гц), 7,29-7,23 (6H, м), 7,02 (1H, д, J=8,5 Гц), 4,13 (2H, д, J=6,7 Гц), 3,89-3,85 (5H, м), 3,79 (3H, с), 3,27 (2H, т, J=10,9 Гц), 2,36 (3H, с), 2,16-2,07 (1H, м), 1,49-1,44 (2H, м), 1,37-1,27 (2H, м).
МС (APCI) m/z:633 [(M+H)+].
(Пример 113) N-{4-[2-Амино-5-(3,4-диметоксифенил)пиридин-3-ил]фенил}-5-(4-циклопропилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид
[Формула 76]

К суспензии N-{4-[2-амино-5-(3,4-диметоксифенил)пиридин-3-ил]фенил}-5-(4-бромфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамида (0,350 г, 0,503 ммоль) в толуоле (5,00 мл), циклопропилбороновую кислоту (0,0648 г, 0,755 ммоль), трициклогексилфосфин (0,0282 г, 0,101 ммоль), трикалийфосфат (0,374 г, 1,76 ммоль), и воду (1,00 мл) добавляли, и реакционную смесь продували азотом. Затем, к смеси добавляли ацетат палладия(II) (0,0113 г, 0,0503 ммоль), и смесь нагревали до 100°C. Затем, к смеси добавляли 1,4-диоксан (2,00 мл), и смесь перемешивали при той же температуре, что и выше в течение 4 часов. Реакционную смесь оставляли охлаждаться до комнатной температуры, и затем, к этому добавляли воду, затем экстрагировали метиленхлоридом три раза. Органический слой промывали насыщенным солевым раствором и затем сушили над безводным сульфатом натрия. После фильтрования и концентрирования при пониженном давлении, полученный остаток очищали при помощи колоночной хроматографии на силикагеле (Yamazen Corp., этилацетат/метанол=100/0 -> 85/15) и далее очищали с помощью высокоэффективной жидкостной хроматографии (NOMURA Develosil Combi, ацетонитрил/вода/0,1% муравьиная кислота). Органический растворитель отгоняли при пониженном давлении и затем насыщенный водный раствор бикарбоната натрия добавляли к остатку. Осажденное твердое вещество собирали фильтрованием и сушили с получением указанного в заголовке соединения (0,200 г, выход: 60,5%) в виде твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 13,11 (1H, с), 8,71 (1H, д, J=2,4 Гц), 8,25 (1H, д, J=2,4 Гц), 8,16 (1H, д, J=2,4 Гц), 7,82 (2H, д, J=8,5 Гц), 7,63 (1H, д, J=2,4 Гц), 7,58-7,52 (4H, м), 7,20-7,14 (4H, м), 6,99 (1H, д, J=8,5 Гц), 5,76 (2H, с), 4,10 (2H, д, J=7,3 Гц), 3,88-3,83 (5H, м), 3,77 (3H, с), 3,27 (2H, т, J=10,9 Гц), 2,16-2,04 (1H, м), 2,00-1,93 (1H, м), 1,46 (2H, ушир. д, J=10,9 Гц), 1,35-1,26 (2H, м), 1,02-0,97 (2H, м), 0,73-0,69 (2H, м).
МС (APCI) m/z:657[(M+H)+].
(Пример 114) N-{4-[2-Амино-5-(3,4-диметоксифенил)пиридин-3-ил]фенил}-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамид
[Формула 77]

К N-{4-[2-амино-5-(3,4-диметоксифенил)пиридин-3-ил]фенил}-5-бром-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамиду (0,620 г, 1,00 ммоль), 5-метилпиридин-2-бороновой кислоте N-фенилдиэтаноламинового эфира (0,480 г, 1,70 ммоль), и иодиду меди(I) (0,0762 г, 0,400 ммоль), толуол (6,00 мл), метанол (1,85 мл), и раствор карбоната калия (1,84 г, 13,3 ммоль) в воде (1,20 мл) добавляли, и реакционную смесь продували азотом. Затем, к смеси добавляли тетракис(трифенилфосфин)палладий(0) (0,116 г, 0,100 ммоль), и смесь перемешивали при 100°C в течение 4 часов. Реакционную смесь оставляли охлаждаться до комнатной температуры, и затем к смеси добавляли этилацетат и воду. Нерастворимое вещество отфильтровывали. Фильтрат подвергали экстракции этилацетатом три раза. Органический слой промывали насыщенным солевым раствором и затем сушили над безводным сульфатом натрия. После фильтрования и концентрирования при пониженном давлении, полученный остаток очищали при помощи колоночной хроматографии на силикагеле (Yamazen Corp., этилацетат/метанол=99/1 -> 85/15). После концентрирования при пониженном давлении, остаток отверждали путем добавления этилацетата, и твердое вещество собирали путем фильтрации и сушили с получением указанного в заголовке соединения (0,229 г, выход: 36,2%) в виде твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 12,98 (1H, с), 8,75 (1H, д, J=2,4 Гц), 8,71 (1H, д, J=2,4 Гц), 8,30 (1H, д, J=2,4 Гц), 8,25 (1H, д, J=2,4 Гц), 8,02 (1H, дд, J=8,5, 2,4 Гц), 7,82 (2H, д, J=8,5 Гц), 7,61 (1H, д, J=2,4 Гц), 7,53 (2H, д, J=8,5 Гц), 7,34 (1H, д, J=8,5 Гц), 7,19-7,13 (2H, м), 6,98 (1H, д, J=8,5 Гц), 5,69 (2H, с), 4,12 (2H, д, J=7,3 Гц), 3,88-3,83 (5H, м), 3,77 (3H, с), 3,27 (2H, т, J=11,0 Гц), 2,52 (3H, с), 2,17-2,07 (1H, м), 1,47 (2H, ушир. д, J=11,0 Гц), 1,37-1,26 (2H, м).
МС (APCI) m/z: 632 [(M+H)+].
Подобным образом, окончательное соединение в таблице 21 было синтезировано из соответствующего исходного вещества.
[Таблица 21]


(Пример 116) N-{4-[2-Амино-5-(3,4-диметоксифенил)пиридин-3-ил]фенил}-6'-метил-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидро-3,3'-бипиридин-5-карбоксамид
[Формула 78]

К N-{4-[2-амино-5-(3,4-диметоксифенил)пиридин-3-ил]фенил}-5-бром-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамиду (0,400 г, 0,646 ммоль), 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридину (0,212 г, 0,969 ммоль), карбонату цезия (0,631 г, 1,94 ммоль) и комплексу [1,1'-бис(дифенилфосфино)ферроцен]палладий(II) дихлорид-метиленхлорид (1:1) (0,0527 г, 0,0646 ммоль), добавляли 1,4-диоксан (5,00 мл) и воду (1,00 мл) в атмосфере азота, и смесь перемешивали при 80°C в течение 4 часов. Реакционную смесь оставляли охлаждаться до комнатной температуры, и затем, к смеси добавляли воду и этилацетат. Нерастворимое вещество отфильтровывали. Фильтрат подвергали экстракции этилацетатом три раза. Органический слой промывали насыщенным солевым раствором и затем сушили над безводным сульфатом натрия. После фильтрования и концентрирования при пониженном давлении, полученный остаток очищали при помощи колоночной хроматографии на аминосиликагеле (Yamazen Corp., этилацетат/метанол=99/1 -> 90/10) и далее очищали при помощи колоночной хроматографии на силикагеле (Yamazen Corp., этилацетат/метанол=99/1 -> 5/95). После концентрирования при пониженном давлении, остаток очищали с помощью высокоэффективной жидкостной хроматографии (NOMURA Develosil Combi, ацетонитрил/вода/0,1% муравьиная кислота). Органический растворитель отгоняли при пониженном давлении и затем насыщенный водный раствор бикарбоната натрия добавляли к остатку, с последующим экстрагированием этилацетатом три раза. Органический слой промывали насыщенным солевым раствором и затем сушили над безводным сульфатом натрия. Проводили фильтрование и концентрацию при пониженном давлении с получением указанного в заголовке соединения (0,129 г, выход: 31,6%) в виде аморфного твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 13,08 (1H, с), 8,76 (1H, д, J=2,4 Гц), 8,69 (1H, д, J=2,4 Гц), 8,51-8,47 (2H, м), 8,26 (1H, д, J=2,4 Гц), 7,84 (2H, д, J=8,5 Гц), 7,71 (1H, дд, J=8,5, 2,4 Гц), 7,62 (1H, д, J=2,4 Гц), 7,55 (2H, д, J=8,5 Гц), 7,20-7,14 (2H, м), 6,99 (1H, д, J=8,5 Гц), 5,69 (2H, с), 4,20 (2H, д, J=7,3 Гц), 3,88-3,83 (5H, м), 3,77 (3H, с), 3,27 (2H, т, J=11,3 Гц), 2,36 (3H, с), 2,11-2,03 (1H, м), 1,47 (2H, ушир. д, J=11,0 Гц), 1,37-1,26 (2H, м).
МС (APCI) m/z: 632 [(M+H)+].
Подобным образом, промежуточные соединения в таблице 22, были синтезированы из соответствующих исходных веществ.
[Таблица 22]



(Пример 119) N-{4-[2-Амино-5-(3,4-диметоксифенил)пиридин-3-ил]фенил}-5-(5-метилтиофен-2-ил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид
[Формула 79]

К N-{4-[2-амино-5-(3,4-диметоксифенил)пиридин-3-ил]фенил}-5-бром-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамиду (0,200 г, 0,323 ммоль), (5-метил-2-тиенил)бороновой кислоте (0,055 г, 0,387 ммоль), карбонату калия (0,134 г, 0,969 ммоль), и тетракис(трифенилфосфин)палладий(0) (0,0373 г, 0,0323 ммоль), 1,2-диметоксиэтан (5,00 мл) и воду (0,500 мл) добавляли, и смесь перемешивали при температуре 90°C в течение 12 часов и затем оставляли охлаждаться. К реакционной смеси добавляли воду, затем экстрагировали метиленхлоридом три раза. Органический слой сушили над безводным сульфатом натрия. После фильтрования и концентрирования при пониженном давлении, полученный остаток очищали при помощи колоночной хроматографии на силикагеле (Yamazen Corp., этилацетат/метанол=99/1 -> 80/20) и очищали при помощи колоночной хроматографии на аминосиликагеле (Yamazen Corp., этилацетат/метанол=99/1 -> 93/7). Полученное соединение дополнительно очищали с помощью высокоэффективной жидкостной хроматографии (NOMURA Develosil Combi, ацетонитрил/вода/0,1% муравьиная кислота) с получением указанного в заголовке соединения (0,0860 г, выход: 41,8%) в виде твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 12,90 (1H, с), 8,69-8,66 (2H, м), 8,26 (1H, д, J=2,4 Гц), 7,84 (2H, д, J=8,5 Гц), 7,62 (1H, д, J=2,4 Гц), 7,57-7,54 (3H, м), 7,20-7,14 (2H, м), 6,99 (1H, д, J=8,5 Гц), 6,87-6,85 (1H, м), 5,70 (2H, с), 4,14 (2H, д, J=7,3 Гц), 3,88-3,83 (5H, м), 3,77 (3H, с), 3,27 (2H, т, J=10,9 Гц), 2,49 (3H, с), 2,16-2,10 (1H, м), 1,45 (2H, ушир. д, J=10,9 Гц), 1,38-1,27 (2H, м).
МС (APCI) m/z:637 [(M+H)+].
Подобным образом, окончательное соединение в таблице 23 было синтезировано из соответствующего исходного вещества.
[Таблица 23]


(Пример 121) N-[4-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-1'-[(4-цианотетрагидро-2Н-пиран-4-ил)метил]-5-метил-4'-оксо-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамид
[Формула 80]

К N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-бром-1'-[(4-цианотетрагидро-2Н-пиран-4-ил)метил]-4'-оксо-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамиду (0,230 г, 0,279 ммоль), комплексу [1,1'-бис(дифенилфосфино)ферроцен]палладий(II) дихлорид- метиленхлорид (1:1) (0,0228 г, 0,0279 ммоль), и карбонату цезия (0,272 г, 0,836 ммоль), добавляли 1,4-диоксан (5,00 мл), воду (0,500 мл), и 50% раствор триметилбороксина в тетрагидрофуране (3,5 моль/л, 0,0960 мл, 0,334 ммоль) в атмосфере азота, и смесь перемешивали при 100°C в течение 3,5 часов. Реакционную смесь оставляли охлаждаться до комнатной температуры, и затем к смеси добавляли насыщенный водный раствор бикарбоната натрия и этилацетат. Нерастворимое вещество отфильтровывали. Фильтрат подвергали экстракции этилацетатом три раза. Органический слой промывали насыщенным солевым раствором и затем сушили над безводным сульфатом натрия. После фильтрования и концентрирования при пониженном давлении, полученный остаток очищали при помощи колоночной хроматографии на силикагеле (Yamazen Corp., этилацетат/метанол=99/1 -> 80/20) и затем дополнительно очищали при помощи колоночной хроматографии на аминосиликагеле (Yamazen Corp., этилацетат/метанол=99/1 -> 93/7) с получением указанного в заголовке соединения (0,145 г, выход: 68,4%) в виде аморфного твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 13,07 (1H, с), 8,86 (1H, д, J=2,4 Гц), 8,80 (1H, д, J=2,4 Гц), 8,52-8,48 (2H, м), 8,31 (1H, д, J=2,4 Гц), 7,94 (1H, дд, J=12,1, 2,4 Гц), 7,72 (1H, дд, J=8,2, 2,4 Гц), 7,63 (1H, д, J=2,4 Гц), 7,50-7,42 (2H, м), 7,18 (1H, д, J=2,4 Гц), 7,11 (1H, дд, J=8,5, 2,4 Гц), 6,99 (1H, д, J=8,5 Гц), 5,71 (2H, с), 4,70 (2H, с), 3,97-3,74 (10H, м), 3,69-3,60 (2H, м), 3,52-3,32 (4H, м), 2,36 (3H, с), 1,88-1,77 (4H, м).
МС (APCI) m/z:761 [(M+H)+].
Подобным образом, промежуточные соединения в таблице 24, были синтезированы из соответствующих исходных веществ.
[Таблица 24]




(Пример 124) N-[4-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамида метансульфонат (аморфное твердое вещество)
[Формула 81]

К раствору N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамида (23,9 г, 32,5 ммоль) в метиленхлориде (110 мл) добавляли раствор метансульфоновой кислоты (2,11 мл, 3,12 г, 32,5 ммоль) в этаноле (36,0 мл) и смесь перемешивали при комнатной температуре в течение 15 минут. Реакционную смесь концентрировали при пониженном давлении. Затем, полученное аморфное твердое вещество суспендировали в этилацетате, собирали путем фильтрации и сушили с получением указанного в заголовке соединения (25,7 г, выход: 95,1%) в виде аморфного твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 13,27 (1H, с), 8,79 (1H, д, J=2,4 Гц), 8,72 (1H, д, J=2,4 Гц), 8,54 (1H, д, J=2,4 Гц), 8,46 (1H, д, J=8,5 Гц), 8,34 (1H, д, J=2,4 Гц), 8,29 (1H, д, J=2,4 Гц), 8,03-7,99 (1H, м), 7,78 (1H, д, J=8,5 Гц), 7,59-7,51 (4H, м), 7,31 (1H, д, J=2,4 Гц), 7,25 (1H, дд, J=8,5, 2,4 Гц), 7,06 (1H, д, J=8,5 Гц), 4,21 (2H, д, J=7,3 Гц), 4,01-3,93 (2H, м), 3,90-3,74 (8H, м), 3,69-3,24 (6H, м), 2,37 (3H, с), 2,34 (3H, с), 2,13-2,02 (1H, м), 1,47 (2H, ушир. д, J=10,9 Гц), 1,37-1,28 (2H, м).
МС (APCI) m/z:736[(M+H)+].
Данные элементного анализа для C41H42FN5O7⋅1,0CH3SO3H⋅1,0H2O
Вычислено: C, 59,35; H, 5,69; N, 8,24; F, 2,24; S, 3,77.
Найдено: C, 59,38; H, 5,80; N, 8,10; F, 2,23; S, 3,71.
Подобным образом, каждый мезилат из таблицы 25 получали в виде аморфного твердого вещества из соответствующих исходных веществ.
[Таблица 25-1]




[Таблица 25-2]


[Таблица 25-3]




Далее в настоящем документе, N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид представляет собой соединение примера 1, и N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамид представляет собой соединение примера 34.
(Пример 130) N-[4-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид метансульфонат гидрат
[Формула 82]

N-[4-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид (46,3 г, 62,6 ммоль) суспендировали в ацетоне (244 мл). К суспензии добавляли воду (27,0 мл). Метансульфоновую кислоту (4,27 мл, 65,8 ммоль) добавляли небольшими порциями к смеси. Реакционную смесь перемешивали при комнатной температуре в течение 30 часов и затем проводили вакуумное фильтрование через воронку Бюхнера. Твердое вещество, собранное путем фильтрации, промывали смесью вода/ацетон (вода/ацетон=1/9) с получением указанного в заголовке соединения (47,5 г, выход: 87,5%) в виде кристаллического твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 13,22 (1H, с), 8,72 (1H, д, J=2,0 Гц), 8,28 (1H, д, J=2,0 Гц), 8,21 (1H, д, J=2,0 Гц), 8,19 (1H, д, J=2,0 Гц), 7,90 (2H, д, J=8,5 Гц), 7,60-7,52 (6H, м), 7,32 (1H, д, J=2,0 Гц), 7,27 (3H, д, J=8,5 Гц), 7,06 (1H, д, J=8,5 Гц), 4,11 (2H, д, J=7,5 Гц), 4,01-3,93 (2H, м), 3,89-3,22 (14H, м), 2,36 (3H, с), 2,32 (3H, с), 2,17-2,04 (1H, м), 1,46 (2H, д, J=11,0 Гц), 1,37-1,25 (2H, м).
МС (APCI) m/z:717 [(M+H)+].
Данные элементного анализа для C42H44N4O7⋅1,0CH3SO3H⋅3,0H2O
Вычислено: C, 59,57; H, 6,28; N, 6,46; S, 3,70.
Найдено: C, 59,75; H, 6,29; N, 6,48; S, 3,76.
Порошковая рентгеновская дифрактограмма (CuKα, λ=1,54 ангстрем, скорость сканирования=20°/мин) показана на фиг. 1 и пики, имеющие относительную интенсивность 22 или более, с максимальной интенсивностью пика, определенной как 100, представлены в таблице 26.
[Таблица 26]
(Пример 131) N-[4-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид гидробромид гидрат
[Формула 83]

К N-[4-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамиду (250 мг, 349 мкмоль) добавляли метанол (3,99 мл) и воду (633 мкл). Затем, к смеси добавляли 1,00 моль/л водный раствор бромистоводородной кислоты (365 мкл, 365 мкмоль). Смесь перемешивали при 40°C в течение приблизительно 21 часа и затем при комнатной температуре в течение приблизительно 30 минут и проводили вакуумное фильтрование с воронкой Kiriyama, и осажденное твердое вещество собирали путем фильтрации. Затем твердое вещество сушили на воздухе с получением указанного в заголовке соединения (243 мг, выход: 81,4%) в виде кристаллического твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 13,22 (1H, с), 8,72 (1H, д, J=2,5 Гц), 8,28 (1H, д, J=2,5 Гц), 8,21 (1H, д, J=2,5 Гц), 8,19 (1H, д, J=2,5 Гц), 7,90 (2H, д, J=8,8 Гц), 7,60-7,55 (4H, м), 7,50 (2H, ушир. с), 7,31 (1H, д, J=2,5 Гц), 7,29-7,23 (3H, м), 7,06 (1H, д, J=8,8 Гц), 4,12 (2H, д, J=7,5 Гц), 4,01-3,93 (2H, м), 3,89-3,24 (14H, м), 2,36 (3H, с), 2,16-2,05 (1H, м), 1,46 (2H, д, J=12,5 Гц), 1,37-1,25 (2H, м).
Данные элементного анализа для C42H44N4O7⋅1HBr⋅3,3H2O
Вычислено: C, 58,85; H, 6,07; N, 6,53; Br, 9,32.
Найдено: C, 58,91; H, 5,98; N, 6,58; Br, 9,42.
Порошковая рентгеновская дифрактограмма (CuKα, λ=1,54 ангстрем, скорость сканирования=20°/мин) показана на фиг. 2, и пики, имеющие относительную интенсивность 19 или более, с максимальной интенсивностью пика, определенной как 100, представлены в таблице 27.
[Таблица 27]
(Пример 132) N-[4-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид нитрат гидрат
[Формула 84]

К N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамиду (249 мг, 347 мкмоль) добавляли метанол (1,49 мл) и воду (9,10 мкл). Затем, к смеси добавляли 1,00 моль/л водный раствор азотной кислоты (364 мкл, 364 мкмоль). Смесь перемешивали при 40°C в течение приблизительно 21 часа и затем при комнатной температуре в течение приблизительно 30 минут и проводили вакуумное фильтрование с воронкой Kiriyama, и осажденное твердое вещество собирали путем фильтрации. Затем твердое вещество сушили на воздухе с получением указанного в заголовке соединения (252 мг, выход: 87,0%) в виде кристаллического твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 13,22 (1H, с), 8,72 (1H, д, J=2,5 Гц), 8,27 (1H, д, J=2,5 Гц), 8,21 (1H, д, J=2,5 Гц), 8,19 (1H, д, J=2,5 Гц), 7,90 (2H, д, J=8,8 Гц), 7,60-7,55 (4H, м), 7,49 (2H, ушир. с), 7,31 (1H, д, J=2,4 Гц), 7,29-7,24 (3H, м), 7,06 (1H, д, J=8,8 Гц), 4,11 (2H, д, J=7,5 Гц), 4,01-3,93 (2H, м), 3,90-3,22 (14H, м), 2,36 (3H, с), 2,17-2,04 (1H, м), 1,46 (2H, д, J=12,5 Гц), 1,38-1,26 (2H, м).
Данные элементного анализа для C42H44N4O7⋅1HNO3⋅3H2O
Вычислено: C, 60,49; H, 6,16; N, 8,40.
Найдено: C, 60,58; H, 6,15; N, 8,43.
Порошковая рентгеновская дифрактограмма (CuKα, λ=1,54 ангстрем, скорость сканирования=20°/мин) показана на фиг. 3, и пики, имеющие относительную интенсивность 34 или более, с максимальной интенсивностью пика, определенной как 100, представлены в таблице 28.
[Таблица 28]
(Пример 133) N-[4-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид сульфат гидрат
[Формула 85]
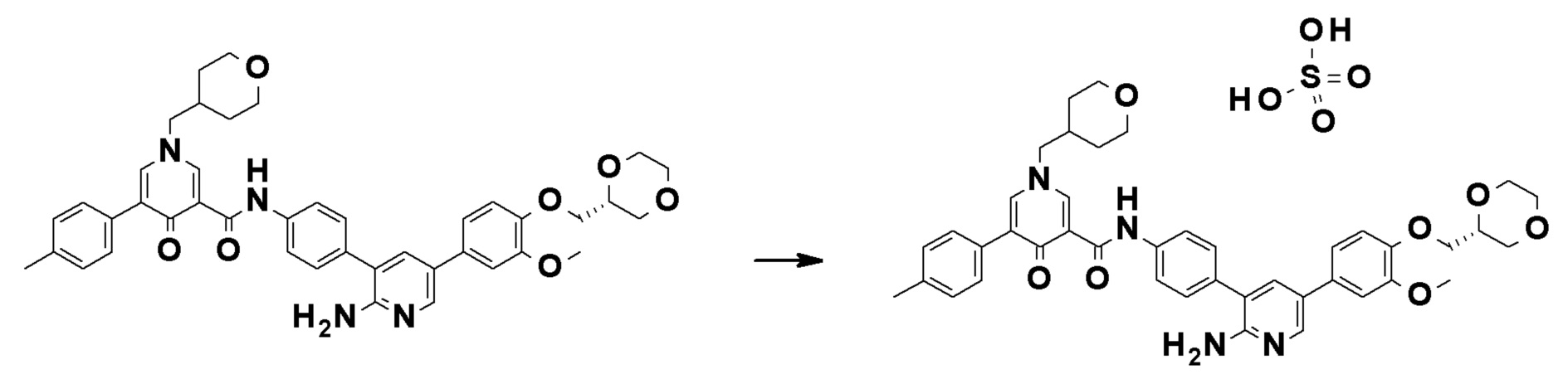
К N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамиду (249 мг, 347 мкмоль) добавляли метанол (3,98 мл) и воду (632 мкл). Затем, к смеси добавляли 1,00 моль/л водный раствор серной кислоты (363 мкл, 363 мкмоль). Смесь перемешивали при 40°C в течение приблизительно 21 часа и затем при комнатной температуре в течение приблизительно 30 минут и проводили вакуумное фильтрование с воронкой Kiriyama, и осажденное твердое вещество собирали путем фильтрации. Затем твердое вещество сушили на воздухе с получением указанного в заголовке соединения (271 мг, выход: 90,4%) в виде кристаллического твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 13,20 (1H, с), 8,72 (1H, д, J=2,4 Гц), 8,27 (1H, д, J=2,4 Гц), 8,18 (1H, д, J=2,4 Гц), 8,08 (1H, с), 7,88 (2H, д, J=8,5 Гц), 7,57 (4H, т, J=8,5 Гц), 7,30-7,03 (7H, м), 4,11 (2H, д, J=7,5 Гц), 4,01-3,92 (2H, м), 3,91-3,21 (14H, м), 2,36 (3H, с), 2,16-2,05 (1H, м), 1,46 (2H, д, J=12,5 Гц), 1,37-1,25 (2H, м).
Данные элементного анализа для C42H44N4O7⋅0,75H2SO4⋅4H2O
Вычислено: C, 58,49; H, 6,25; N, 6,50; S, 2,79.
Найдено: C, 58,53; H, 6,13; N, 6,52; S, 2,80.
Порошковая рентгеновская дифрактограмма (CuKα, λ=1,54 ангстрем, скорость сканирования=20°/мин) показана на фиг. 4, и пики, имеющие относительную интенсивность 13 или более, с максимальной интенсивностью пика, определенной как 100, представлены в таблице 29.
[Таблица 29]
(Пример 134) N-[4-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид фосфат гидрат
[Формула 86]

К N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамиду (251 мг, 350 мкмоль) добавляли метанол (501 мкл) и воду (1,28 мл). Затем, к смеси добавляли 0,504 моль/л водный раствор фосфорной кислоты (729 мкл, 367 мкмоль). Смесь перемешивали при 40°C в течение приблизительно 24 часов и затем при комнатной температуре в течение приблизительно 30 минут и проводили вакуумное фильтрование с воронкой Kiriyama, и осажденное твердое вещество собирали путем фильтрации. Затем твердое вещество сушили на воздухе с получением указанного в заголовке соединения (254 мг, выход: 82,8%) в виде кристаллического твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 13,11 (1H, с), 8,72 (1H, д, J=2,5 Гц), 8,25 (1H, д, J=2,5 Гц), 8,17 (1H, д, J=2,5 Гц), 7,82 (2H, д, J=8,5 Гц), 7,62 (1H, д, J=2,5 Гц), 7,58 (2H, д, J=8,5 Гц), 7,53 (2H, д, J=8,5 Гц), 7,26 (2H, д, J=8,5 Гц), 7,20 (1H, д, J=2,0 Гц), 7,13 (1H, дд, J=8,5, 2,0 Гц), 6,99 (1H, д, J=8,5 Гц), 5,73 (2H, ушир. с), 4,11 (2H, д, J=7,5 Гц), 3,98-3,90 (2H, м), 3,89-3,22 (14H, м), 2,36 (3H, с), 2,16-2,04 (1H, м), 1,46 (2H, д, J=11,5 Гц), 1,37-1,25 (2H, м).
Данные элементного анализа для C42H44N4O7⋅1,0H3PO4⋅3,5H2O
Вычислено: C, 57,46; H, 6,20; N, 6,38; P, 3,53.
Найдено: C, 57,41; H, 6,20; N, 6,41; P, 3,41.
Порошковая рентгеновская дифрактограмма (CuKα, λ=1,54 ангстрем, скорость сканирования=20°/мин) показана на фиг. 5, и пики, имеющие относительную интенсивность 20 или более, с максимальной интенсивностью пика, определенной как 100, представлены в таблице 30.
[Таблица 30]
(Пример 135) N-[4-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид этансульфонат гидрат
[Формула 87]

К N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамиду (251 мг, 350 мкмоль) добавляли метанол (1,00 мл) и воду (3,65 мл). Затем, к смеси добавляли 1,00 моль/л водный раствор этансульфоновой кислоты (368 мкл, 368 мкмоль). Небольшое количество затравочного кристалла, полученного способом, приведенным ниже, добавляли к смеси. Смесь перемешивали при 40°C в течение приблизительно 21 часа и затем при комнатной температуре в течение приблизительно 30 минут и проводили вакуумное фильтрование с воронкой Kiriyama, и осажденное твердое вещество собирали путем фильтрации. Затем твердое вещество сушили на воздухе с получением указанного в заголовке соединения (265 мг, выход: 81,8%) в виде кристаллического твердого вещества.
Способ получения затравочного кристалла
К N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамиду (10,4 мг, 14,5 мкмоль) добавляли метанол (41,6 мкл) и воду (151 мкл). Затем, к смеси добавляли 1,00 моль/л водный раствор этансульфоновой кислоты (15,2 мкл, 15,2 мкмоль). Смесь перемешивали при 40°C в течение приблизительно 24 часов и затем при комнатной температуре в течение приблизительно 30 минут, и проводили вакуумное фильтрование с воронкой Kiriyama, и осажденное твердое вещество собирали путем фильтрации. Затем твердое вещество сушили на воздухе с получением затравочного кристалла (10,9 мг).
1Н-ЯМР (ДМСО-D6) δ: 13,22 (1H, с), 8,72 (1H, д, J=2,0 Гц), 8,28 (1H, д, J=2,0 Гц), 8,20 (2H, дд, J=8,5, 2,0 Гц), 7,90 (2H, д, J=8,5 Гц), 7,61-7,49 (6H, м), 7,32 (1H, д, J=2,0 Гц), 7,29-7,24 (3H, м), 7,06 (1H, д, J=8,5 Гц), 4,11 (2H, д, J=7,5 Гц), 4,01-3,93 (2H, м), 3,87-3,24 (14H, м), 2,41-2,31 (5H, м), 2,16-2,04 (1H, м), 1,46 (2H, д, J=11,5 Гц), 1,37-1,25 (2H, м), 1,06 (3H, т, J=7,5 Гц).
Данные элементного анализа для C42H44N4O7⋅1,0C2H5SO3H⋅5,5H2O
Вычислено: C, 57,07; H, 6,64; N, 6,05; S, 3,46.
Найдено: C, 57,13; H, 6,78; N, 6,08; S, 3,60.
Порошковая рентгеновская дифрактограмма (CuKα, λ=1,54 ангстрем, скорость сканирования=20°/мин) показана на фиг. 6, и пики, имеющие относительную интенсивность 5 или более, с максимальной интенсивностью пика, определенной как 100, представлены в таблице 31.
[Таблица 31]
(Пример 136) N-[4-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид бензолсульфонат гидрат
[Формула 88]

К N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамиду (250 мг, 349 мкмоль) добавляли метанол (1,00 мл) и воду (3,64 мл). Затем, к смеси добавляли 1,00 моль/л водный раствор бензолсульфоновой кислоты (366 мкл, 366 мкмоль). Из этой смеси готовили суспензию с использованием ультразвуковой моющей машины, затем перемешивали при 40°C в течение приблизительно 21 часа, и затем при комнатной температуре в течение приблизительно 30 минут, и проводили вакуумное фильтрование с воронкой Kiriyama, и осажденное твердое вещество собирали путем фильтрации. Затем твердое вещество сушили на воздухе с получением указанного в заголовке соединения (287 мг, выход: 88,3%) в виде кристаллического твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 13,22 (1H, с), 8,72 (1H, д, J=2,0 Гц), 8,27 (1H, д, J=2,0 Гц), 8,19 (2H, д, J=2,0 Гц), 7,90 (2H, д, J=8,5 Гц), 7,61-7,55 (6H, м), 7,44 (2H, ушир. с), 7,35-7,24 (7H, м), 7,06 (1H, д, J=8,5 Гц), 4,11 (2H, д, J=7,5 Гц), 4,01-3,93 (2H, м), 3,90-3,22 (14H, м), 2,36 (3H, с), 2,17-2,04 (1H, м), 1,46 (2H, д, J=11,6 Гц), 1,31 (2H, тд, J=12,2, 8,5 Гц).
Данные элементного анализа для C42H44N4O7⋅1,0C6H5SO3H⋅3H2O
Вычислено: C, 62,05; H, 6,08; N, 6,03; S, 3,45.
Найдено: C, 62,18; H, 6,07; N, 6,08; S, 3,46.
Порошковая рентгеновская дифрактограмма (CuKα, λ=1,54 ангстрем, скорость сканирования=20°/мин) показана на фиг. 7, и пики, имеющие относительную интенсивность 29 или более, с максимальной интенсивностью пика, определенной как 100, представлены в таблице 32.
[Таблица 32]
(Пример 137) N-[4-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамид п-толуолсульфонат
[Формула 89]

К N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамиду (268 мг, 373 мкмоль) добавляли метанол (2,14 мл) и воду (145 мкл). Затем к смеси добавляли 1,00 моль/л водный раствор п-толуолсульфоновой кислоты (390 мкл, 390 мкмоль). Небольшое количество затравочного кристалла, полученного способом, приведенным ниже, добавляли к смеси. Смесь перемешивали при 40°C в течение приблизительно 21 часа и затем при комнатной температуре в течение приблизительно 30 минут и проводили вакуумное фильтрование с воронкой Kiriyama, и осажденное твердое вещество собирали путем фильтрации. Затем твердое вещество сушили на воздухе с получением указанного в заголовке соединения (255 мг, выход: 77,0%) в виде кристаллического твердого вещества.
Способ получения затравочного кристалла
К N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)фенил]-5-(4-метилфенил)-4-оксо-1-(тетрагидро-2H-пиран-4-илметил)-1,4-дигидропиридин-3-карбоксамиду (10,3 мг, 14,4 мкмоль) добавляли метанол (165 мкл) и воду (26,2 мкл). Затем к смеси добавляли 1,00 моль/л водный раствор п-толуолсульфоновой кислоты (15,0 мкл, 15,0 мкмоль). Смесь перемешивали при 40°C в течение приблизительно 24 часов и затем при комнатной температуре в течение приблизительно 30 минут и проводили вакуумное фильтрование с воронкой Kiriyama, и осажденное твердое вещество собирали путем фильтрации. Затем твердое вещество сушили на воздухе с получением затравочного кристалла (10,7 мг).
1Н-ЯМР (ДМСО-D6) δ: 13,22 (1H, с), 8,73 (1H, д, J=2,0 Гц), 8,28 (1H, д, J=2,0 Гц), 8,21 (1H, д, J=2,0 Гц), 8,19 (1H, д, J=2,0 Гц), 7,90 (2H, д, J=8,0 Гц), 7,60-7,45 (8H, м), 7,31 (1H, д, J=2,0 Гц), 7,29-7,24 (3H, м), 7,11 (2H, д, J=8,0 Гц), 7,06 (1H, д, J=8,0 Гц), 4,11 (2H, д, J=7,5 Гц), 4,02-3,93 (2H, м), 3,90-3,22 (14H, м), 2,36 (3H, с), 2,29 (3H, с), 2,16-2,04 (1H, м), 1,46 (2H, д, J=11,5 Гц), 1,37-1,25 (2H, м).
Данные элементного анализа для C42H44N4O7⋅1,0C7H7SO3H
Вычислено: C, 66,19; H, 5,89; N, 6,30; S, 3,61.
Найдено: C, 65,91; H, 5,97; N, 6,26; S, 3,59.
Порошковая рентгеновская дифрактограмма (CuKα, λ=1,54 ангстрем, скорость сканирования=20°/мин) показана на фиг. 8, и пики, имеющие относительную интенсивность 23 или более, с максимальной интенсивностью пика, определенной как 100, представлены в таблице 33.
[Таблица 33]
(Пример 138) N-[4-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамид фосфат гидрат
[Формула 90]

N-[4-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамид (50,0 мг, 67,9 мкмоль) суспендировали в ацетоне (400 мкл). К суспензии добавляли воду (64,4 мкл). Затем, к этому добавляли 4,00 моль/л водный раствор фосфорной кислоты (35,7 мкл, 143 мкмоль), и смесь перемешивали при 40°C в течение 3 часов. Затем, добавляли к смеси 20% водный раствор ацетона (500 мкл), и смесь дополнительно перемешивали в течение приблизительно 21 часа. Затем реакционную смесь перемешивали при комнатной температуре в течение приблизительно 30 минут и затем проводили вакуумное фильтрование с воронкой Kiriyama с получением указанного в заголовке соединения (42,5 мг, выход: 69,7%) в виде кристаллического твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 13,22 (1H, с), 8,77 (1H, д, J=2,0 Гц), 8,69 (1H, д, J=2,0 Гц), 8,51 (1H, д, J=2,0 Гц), 8,47 (1H, д, J=8,5 Гц), 8,30 (1H, д, J=2,0 Гц), 7,93 (1H, дд, J=12,5, 2,0 Гц), 7,71 (1H, дд, J=8,5, 2,0 Гц), 7,64 (1H, д, J=2,0 Гц), 7,49-7,41 (2H, м), 7,19 (1H, д, J=2,0 Гц), 7,12 (1H, дд, J=8,5, 2,0 Гц), 6,99 (1H, д, J=8,5 Гц), 5,77 (2H, ушир. с), 4,21 (2H, д, J=7,5 Гц), 3,98-3,24 (16H, м), 2,36 (3H, с), 2,13-2,01 (1H, м), 1,46 (2H, д, J=12,0 Гц), 1,38-1,26 (2H, м).
МС (APCI) m/z: 736[(M+H)+].
Данные элементного анализа для C41H42FN5O7⋅1,0H3PO4⋅3,5H2O
Вычислено: C, 54,91; H, 5,84; N, 7,81; F, 2,12; P, 3,45.
Найдено: C, 54,95; H, 5,54; N, 7,86; F, 2,20; P, 3,19.
Порошковая рентгеновская дифрактограмма (CuKα, λ=1,54 ангстрем, скорость сканирования=20°/мин) показана на фиг. 9, и пики, имеющие относительную интенсивность 18 или более, с максимальной интенсивностью пика, определенной как 100, представлены в таблице 34.
[Таблица 34]
(Пример 139) N-[4-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамид сульфат гидрат
[Формула 91]

N-[4-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамид (80,0 г, 109 ммоль) суспендировали в ацетоне (1,28 л). К суспензии добавляли воду (247 мл). К смеси добавляли по каплям 6,00 моль/л серную кислоту (71,7 мл, 430 ммоль) при 40°C. Реакционную смесь перемешивают при 40°C в течение 3 дней и затем проводили вакуумное фильтрование через воронку Бюхнера, и осажденное твердое вещество собирали путем фильтрации. Твердое вещество промывали смесью вода/ацетон (вода/ацетон=1/9) с получением указанного в заголовке соединения (92,3 г, выход: 88,3%) в виде кристаллического твердого вещества.
1Н-ЯМР (ДМСО-D6) δ: 13,04 (1H, ушир. с), 8,83 (2H, д, J=9,5 Гц), 8,63 (1H, с), 8,44 (1H, д, J=8,0 Гц), 8,34 (2H, с), 8,05-7,98 (2H, м), 7,72 (2H, ушир. с), 7,59-7,52 (2H, м), 7,32 (1H, д, J=2,0 Гц), 7,26 (1H, дд, J=8,0, 2,0 Гц), 7,07 (1H, д, J=8,0 Гц), 4,22 (2H, д, J=7,5 Гц), 4,01-3,93 (2H, м), 3,90-3,74 (8H, м), 3,70-3,59 (2H, м), 3,53-3,45 (1H, м), 3,40 (1H, дд, J=11,0, 10,0 Гц), 3,27 (2H, т, J=11,0 Гц), 2,43 (3H, с), 2,16-2,05 (1H, м), 1,51-1,43 (2H, м), 1,39-1,26 (2H, м).
МС (APCI) m/z:736[(M+H)+].
Данные элементного анализа для C41H42FN5O7⋅1,8H2SO4⋅3,0H2O
Вычислено: C, 50,96; H, 5,38; N, 7,25; F, 1,97; S, 5.97.
Найдено: C, 50,98; H, 5,36; N, 7,23; F, 1,97; S, 6,19.
Порошковая рентгеновская дифрактограмма (CuKα, λ=1,54 ангстрем, скорость сканирования=20°/мин) показана на фиг. 10, и пики, имеющие относительную интенсивность 6 или более, с максимальной интенсивностью пика, определенной как 100, представлены в таблице 35.
[Таблица 35]
(Пример 140) N-[4-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамид сульфат гидрат
К N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамиду (39,8 мг, 54,1 мкмоль) добавляли ацетон (637 мкл) и воду (78,3 мкл). Затем, к смеси добавляли 1,00 моль/л водный раствор серной кислоты (80,9 мкл, 80,9 мкмоль). Смесь перемешивали при 40°C в течение приблизительно 24 часов и затем при комнатной температуре в течение приблизительно 30 минут и проводили вакуумное фильтрование с воронкой Kiriyama, и осажденное твердое вещество собирали путем фильтрации. Затем твердое вещество сушили на воздухе с получением указанного в заголовке соединения (36,7 мг, выход: 71,6%) в виде кристаллического твердого вещества.
Данные элементного анализа для C41H42FN5O7⋅1,60H2SO4⋅3,0H2O
Вычислено: C, 52,01; H, 5,45; N, 7,40; F, 2,01; S, 5,42.
Найдено: C, 52,07; H, 5,24; N, 7,25; F, 2,09; S, 5,50.
Порошковая рентгеновская дифрактограмма (CuKα, λ=1,54 ангстрем, скорость сканирования=20°/мин) показана на фиг. 11, и пики, имеющие относительную интенсивность 12 или более, с максимальной интенсивностью пика, определенной как 100, представлены в таблице 36.
[Таблица 36]
(Пример 141) N-[4-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамид сульфат гидрат
К N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамиду (40,5 мг, 55,0 мкмоль) добавляли ацетон (648 мкл) и воду (24,8 мкл). Затем, к смеси добавляли 1,00 моль/л водный раствор серной кислоты (137 мкл, 137 мкмоль). Смесь перемешивали при 40°C в течение приблизительно 24 часов и затем при комнатной температуре в течение приблизительно 30 минут и проводили вакуумное фильтрование с воронкой Kiriyama, и осажденное твердое вещество собирали путем фильтрации. Затем твердое вещество сушили на воздухе с получением указанного в заголовке соединения (47,2 мг, выход: 88,8%) в виде кристаллического твердого вещества.
Данные элементного анализа для C41H42FN5O7⋅1,80H2SO4⋅3,0H2O
Вычислено: C, 50,96; H, 5,38; N, 7,25; F, 1,97; S, 5,97.
Найдено: C, 50,85; H, 5,20; N, 7,06; F, 2,09; S, 5,99.
Порошковая рентгеновская дифрактограмма (CuKα, λ=1,54 ангстрем, скорость сканирования=20°/мин) показана на фиг. 12, и пики, имеющие относительную интенсивность 7 или более, с максимальной интенсивностью пика, определенной как 100, представлены в таблице 37.
[Таблица 37]
(Пример 142) N-[4-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамид сульфат гидрат
К N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамиду (39,8 мг, 54,0 мкмоль) добавляли ацетон (636 мкл) и воду (112 мкл). Затем, к смеси добавляли 5,79 моль/л водный раствор серной кислоты (46,7 мкл, 270 мкмоль). Смесь перемешивали при 40°C в течение приблизительно 24 часов и затем при комнатной температуре в течение приблизительно 30 минут и проводили вакуумное фильтрование с воронкой Kiriyama, и осажденное твердое вещество собирали путем фильтрации. Затем твердое вещество сушили на воздухе с получением указанного в заголовке соединения (47,5 мг, выход: 89,2%) в виде кристаллического твердого вещества.
Данные элементного анализа для C41H42FN5O7⋅2,00H2SO4⋅3,0H2O
Вычислено: C, 49,94; H, 5,32; N, 7,10; F, 1,93; S, 6,50.
Найдено: C, 49,75; H, 5,08; N, 6,91; F, 2,20; S, 6,69.
Порошковая рентгеновская дифрактограмма (CuKα, λ=1,54 ангстрем, скорость сканирования=20°/мин) показана на фиг. 13, и пики, имеющие относительную интенсивность 11 или более, с максимальной интенсивностью пика, определенной как 100, представлены в таблице 38.
[Таблица 38]
(Пример 143) N-[4-(2-Амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамид сульфат гидрат
К N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамиду (40,2 мг, 54,6 мкмоль) добавляли ацетон (643 мкл) и воду (51,8 мкл). Затем, к смеси добавляли 1,00 моль/л водный раствор серной кислоты (109 мкл, 109 мкмоль). Смесь перемешивали при 40°C в течение приблизительно 24 часов и затем при комнатной температуре в течение приблизительно 30 минут и проводили вакуумное фильтрование с воронкой Kiriyama, и осажденное твердое вещество собирали путем фильтрации. Затем твердое вещество сушили на воздухе с получением указанного в заголовке соединения (44,4 мг, выход: 84,6%) в виде кристаллического твердого вещества.
Данные элементного анализа для C41H42FN5O7⋅1,75H2SO4⋅3,0H2O
Вычислено: C, 51,22; H, 5,40; N, 7,28; F, 1,98; S, 5,84.
Найдено: C, 50,92; H, 5,19; N, 7,11; F, 2,25; S, 5,81.
Порошковая рентгеновская дифрактограмма (CuKα, λ=1,54 ангстрем, скорость сканирования=20°/мин) показана на фиг. 14, и пики, имеющие относительную интенсивность 11 или более, с максимальной интенсивностью пика, определенной как 100, представлены в таблице 39.
[Таблица 39]
(Пример 144) N-{4-[2-Амино-5-(4-{[(2R)-1,4-диоксан-2-ил]метокси}-3-метоксифенил)-3-пиридил]-3-фторфенил}-5-(5-метил-2-пиридил)-4-оксо-1-(тетрагидропиран-4-илметил)пиридин-3-карбоксамид сульфат гидрат
[Формула 92]

К N-{4-[2-амино-5-(4-{[(2R)-1,4-диоксан-2-ил]метокси}-3-метоксифенил)-3-пиридил]-3-фторфенил}-5-(5-метил-2-пиридил)-4-оксо-1-(тетрагидропиран-4-илметил)пиридин-3-карбоксамиду (2,11 г, 2,88 ммоль) добавляли метанол (12,7 мл) и воду (2,72 мл), и 1,00 моль/л серную кислоту (5,72 мл, 5,72 ммоль) добавляли по каплям. Реакционную смесь перемешивали при температуре 25°C в течение приблизительно 21 часа, и затем, осажденное твердое вещество собирали путем фильтрации. Твердое вещество сушили при пониженном давлении при комнатной температуре в течение приблизительно 18 часов с получением указанного в заголовке соединения (2,39 г, выход: 85,8%).
Данные элементного анализа для C41H42N5O7F⋅1,75H2SO4⋅3,5H2O
Вычислено: C, 50,74; H, 5,45; N, 7,22; F, 1,96; S, 5,78.
Найдено: C, 50,70; H, 5,33; N, 7,13; F, 2,01; S, 5,82.
Порошковая рентгеновская дифрактограмма (CuKα, λ=1,54 ангстрем, скорость сканирования=20°/мин) показана на фиг. 15, и пики, имеющие относительную интенсивность 18 или более, с максимальной интенсивностью пика, определенной как 100, представлены в таблице 40.
[Таблица 40]
(Пример 145) N-{4-[2-Амино-5-(4-{[(2R)-1,4-диоксан-2-ил]метокси}-3-метоксифенил)-3-пиридил]-3-фторфенил}-5-(5-метил-2-пиридил)-4-оксо-1-(тетрагидропиран-4-илметил)пиридин-3-карбоксамид сульфат гидрат
[Формула 93]

К сульфат гидрату (2,44 г), описанному в примере 139, добавляли 20% водный раствор метанола (24,4 мл), и смесь перемешивали при 5°C в течение приблизительно 27 часов. Твердое вещество собирали путем фильтрации и затем сушили при пониженном давлении при комнатной температуре в течение 4,5 часов с получением указанного в заголовке соединения (2,30 г, выход: 93,5%).
Данные элементного анализа для C41H42N5O7F⋅1,5H2SO4⋅5,0H2O
Вычислено: C, 50,61; H, 5,70; N, 7,20; F, 1,95; S, 4,94.
Найдено: C, 50,59; H, 5,55; N, 7,24; F, 2,04; S, 5,09.
Порошковая рентгеновская дифрактограмма (CuKα, λ=1,54 ангстрем, скорость сканирования=20°/мин) показана на фиг. 16, и пики, имеющие относительную интенсивность 13 или более, с максимальной интенсивностью пика, определенной как 100, представлены в таблице 41.
[Таблица 41]
(Пример 146) N-{4-[2-Амино-5-(4-{[(2R)-1,4-диоксан-2-ил]метокси}-3-метоксифенил)-3-пиридил]-3-фторфенил}-5-(5-метил-2-пиридил)-4-оксо-1-(тетрагидропиран-4-илметил)пиридин-3-карбоксамид нафталин-1,5-дисульфонат гидрат
[Формула 94]

К N-{4-[2-амино-5-(4-{[(2R)-1,4-диоксан-2-ил]метокси}-3-метоксифенил)-3-пиридил]-3-фторфенил}-5-(5-метил-2-пиридил)-4-оксо-1-(тетрагидропиран-4-илметил)пиридин-3-карбоксамиду (199,34 мг, 271 мкмоль) добавляли метанол (3,19 мл), 1 моль/л водный раствор нафталин-1,5-дисульфоновой кислоты (285 мкл, 284 мкмоль) и воду (513 мкл). Реакционную смесь перемешивали при 40°C в течение приблизительно 26 часов, и затем при комнатной температуре в течение приблизительно 0,5 часа. Твердое вещество собирали путем фильтрации и затем сушили на воздухе в течение ночи с получением указанного в заголовке соединения (285,7 мг, выход: 94,6%).
1Н-ЯМР (ДМСО-D6) δ: 12,97 (1H, ушир.с), 8,90-8,80 (4H, м), 8,64 (1H, с), 8,42 (1H, д, J=8,5 Гц), 8,33 (2H, с), 8,11-7,97 (2H, м), 7,92 (2H, д, J=7,0 Гц), 7,71 (2H, ушир.с), 7,59-7,52 (2H, м), 7,40 (2H, дд, J=8,5, 7,0 Гц), 7,31 (1H, д, J=2,5 Гц), 7,26 (1H, дд, J=8,5, 2,5 Гц), 7,06 (1H, д, J=8,5 Гц), 4,22 (2H, д, J=7,0 Гц), 3,99-3,24 (16H, м), 2,43 (3H, с), 2,18-2,05 (1H, м), 1,47 (2H, д, J=10,5 Гц), 1,39-1,26 (2H, м).
МС (APCI) m/z:736 [(M+H)+].
Данные элементного анализа для C41H42N5O7F⋅1,0C10H8O6S2⋅5,0H2O
Вычислено: C, 54,98; H, 5,43; N, 6,29; F, 1,71; S, 5,76.
Найдено: C, 54,74; H, 5,37; N, 6,24; F, 1,92; S, 5,82.
Порошковая рентгеновская дифрактограмма (CuKα, λ=1,54 ангстрем, скорость сканирования=20°/мин) показана на фиг. 17, и пики, имеющие относительную интенсивность 45 или более, с максимальной интенсивностью пика, определенной как 100, представлены в таблице 42.
[Таблица 42]
(Пример 147) N-{4-[2-Амино-5-(4-{[(2R)-1,4-диоксан-2-ил]метокси}-3-метоксифенил)-3-пиридил]-3-фторфенил}-5-(5-метил-2-пиридил)-4-оксо-1-(тетрагидропиран-4-илметил)пиридин-3-карбоксамид нафталин-1,5-дисульфонат гидрат
[Формула 95]

К N-{4-[2-амино-5-(4-{[(2R)-1,4-диоксан-2-ил]метокси}-3-метоксифенил)-3-пиридил]-3-фторфенил}-5-(5-метил-2-пиридил)-4-оксо-1-(тетрагидропиран-4-илметил)пиридин-3-карбоксамиду (199,50 мг, 271 мкмоль) добавляли ацетон (3,19 мл), 1 моль/л водный раствор нафталин-1,5-дисульфоновой кислоты (285 мкл, 284 мкмоль), и воду (513 мкл). Реакционную смесь перемешивали при 40°C в течение приблизительно 26 часов и и затем при комнатной температуре в течение приблизительно 0,5 часа. Твердое вещество собирали путем фильтрации и затем сушили на воздухе с получением указанного в заголовке соединения (290,9 мг, выход: 97,9%).
Данные элементного анализа для C41H42N5O7F⋅1,0C10H8O6S2⋅4,0H2O
Вычислено: C, 55,88; H, 5,33; N, 6,39; F, 1,73; S, 5,85.
Найдено: C, 55,71; H, 5,45; N, 6,18; F, 1,82; S, 5,62.
Порошковая рентгеновская дифрактограмма (CuKα, λ=1,54 ангстрем, скорость сканирования=20°/мин) показана на фиг.18, и пики, имеющие относительную интенсивность 42 или более, с максимальной интенсивностью пика, определенной как 100, представлены в таблице или более, с максимальной интенсивностью пика, определенной как 100, представлены в таблице 43.
[Таблица 43]
(Тестовый пример 1 Бесклеточный анализ ингибирующей активности в отношении киназы Axl)
Разбавленный раствор киназы, содержащий 170 нг/мл AXL (464-885 аминокислоты внутриклеточного домена человеческого AXL, экспрессированного в виде слитого белка с глутатион-трансферазой в бакуловирусной системе экспрессии и очищенного с помощью хроматографии на глутатион-сефарозе; CARNA Biosciences, Inc., номер по каталогу No. 08-107), получали с использованием киназного реакционного буферного раствора (100 мМ HEPES (pH 7,4), 0,003% Brij-35, 0,004% Tween-20, 1 мМ DTT, и 10 мМ MgCl2) и добавляли в 384-луночный планшет в количестве 19 мкл/лунку.
Далее, каждое тестируемое соединение разбавляли ДМСО, и этот разбавленный раствор добавляли в планшет в количестве 1 мкл/лунку.
После предварительной инкубации при комнатной температуре в течение 30 минут, раствор, содержащий пептидный субстрат (FL-пептид 30 (5FAM-KKKKEEIYFFF-CONH2), Caliper Life Sciences, номер по каталогу No. 760430) и АТФ при 1,5 мкM и 10 мкM, соответственно, получали и добавляли в планшет при 5 мкл/лунку для начала киназной реакции. Планшет инкубировали при 28°C в течение 1,5 часов, и реакцию останавливали добавлением терминирующего буферного раствора (100 мМ HEPES (рН 7,4), 0,015% Brij-35, 40 мМ ЭДТА, и 0,1% покрывающий реагент 3) при 40 мкл/лунку.
Пептидный субстрат и фосфорилированный пептид в реакционной смеси разделяли и количественно определяли с использованием EZ Reader II (Caliper Life Sciences).
Киназную реакцию оценивали на основе соотношения продукта (P/(P+S)), рассчитанного из высоты пика (S) пептидного субстрата и высоты пика (Р) фосфорилированного пептида.
Скорость ингибирования (ингибирование) определяли в соответствии со следующим выражением (вычисляли автоматически с помощью программного обеспечения EZ Reader II system).
Ингибирование (%)=100×(1-Ci/C0)
где Ci представляет собой соотношение продукта при добавлении тестируемого соединения, и C0 представляет собой соотношение продукта при добавлении ДМСО вместо тестируемого соединения.
IC50 определяли по данным о скоростях ингибирования при 12 концентрациях тестируемого соединения методом нелинейной регрессии (4-параметрическая логистическая регрессия) в соответствии со следующим выражением:
Ингибирование (%)=Ниж.+(Верх.-Ниж.)/(1+([Соединение]/IC50)наклон)
(Тестовый пример 2 Бесклеточный анализ ингибирующей активности в отношении киназы Mer)
Разбавленный раствор киназы, содержащий 20 нг/мл Mer (528-999 аминокислоты внутриклеточного домена человеческого MER, экспрессированного в виде слитого белка с глутатион-трансферазой в бакуловирусной системе экспрессии и очищенного с помощью хроматографии на глутатион-сефарозе и ионнообменной хроматографии; Carna Biosciences, Inc., номер по каталогу No. 08-108), получали с использованием киназного реакционного буферного раствора (100 мМ HEPES (pH 7,4), 0,003% Brij-35, 0,004% Tween-20, 1 мМ DTT, и 10 мМ MgCl2) и добавляли в 384-луночный планшет в количестве 19 мкл/лунку.
Далее, каждое тестируемое соединение разбавляли ДМСО, и этот разбавленный раствор добавляли в планшет в количестве 1 мкл/лунку.
После предварительной инкубации при комнатной температуре в течение 20 минут, раствор, содержащий пептидный субстрат (FL-пептид 27 (5FAM-EFPIYDFLPAKKK-CONH2), Caliper Life Sciences, номер по каталогу No. 760424) и АТФ при 5 мМ, получали и добавляли в планшет при 5 мкл/лунку для начала киназной реакции. Планшет инкубировали при 28°C в течение 45 минут и реакцию останавливали добавлением терминирующего буферного раствора (100 мМ HEPES (рН 7,4), 0,015% Brij-35, 40 мМ ЭДТА, и 0,1% покрывающий реагент 3) при 40 мкл/лунку.
Пептидный субстрат и фосфорилированный пептид в реакционной смеси разделяли и количественно определяли с использованием EZ Reader II (Caliper Life Sciences).
Киназную реакцию оценивали на основе соотношения продукта (P/(P+S)), рассчитанного из высоты пика (S) пептидного субстрата и высоты пика (Р) фосфорилированного пептида.
Скорость ингибирования (ингибирование) определяли в соответствии со следующим выражением (вычисляли автоматически с помощью программного обеспечения EZ Reader II system).
Ингибирование (%)=100×(1-Ci/C0)
где Ci представляет собой соотношение продукта при добавлении тестируемого соединения, и C0 представляет собой соотношение продукта при добавлении ДМСО вместо тестируемого соединения.
IC50 определяли по данным о скоростях ингибирования при 12 концентрациях тестируемого соединения методом нелинейной регрессии (4-параметрическая логистическая регрессия) в соответствии со следующим выражением:
Ингибирование (%)=Ниж.+(Верх.-Ниж.)/(1+([Соединение]/IC50)наклон)
В таблице 44 показаны ингибирующая активность в отношении киназы Axl в виде IC50 (нМ) и ингибирование киназы Mer IC50 (нМ) при условиях, включающих концентрацию АТФ в 1 мМ, и, также, селективность киназы Axl (кратная) относительно киназы Mer.
[Таблица 44-1]
[Таблица 44-2]
[Таблица 44-3]
[Таблица 44-4]
[Таблица 44-5]
[Таблица 44-6]
[Таблица 44-7]
(Тестовый пример 3 Ингибирующая активность в отношениии внутриклеточного фосфорилирования Axl)
Тест на ингибирование фосфорилированного Axl (далее в настоящем документе, также называемый pAxl) проводили с использованием линии клеток немелкоклеточного рака легкого человека NCI-H1299.
Клетки NCI-H1299 суспендировали в среде (среда RPMI1640, содержащая 10% эмбриональную бычью сыворотку), а затем инокулировали при 15000 клеток/100 мкл/лунку для каждого 96-луночного многослойного планшета, и культивировали при 37°C в течение 1 дня, в присутствии 5% CO2. На следующий день среду отбрасывали и среду добавляли при 100 мкл/лунку в планшет, и затем культивировали при 37°C в течение 1 дня в присутствии 5% CO2. Каждое тестируемое соединение растворяли в ДМСО и разбавляли со средой без FBS для получения раствора образца (концентрация ДМСО: 2%). Среду или среду с добавлением образца добавляли при 25 мкл/лунку (концентрация ДМСО: 0,4%) в планшет и инкубировали при 37°C в течение 1 часа в присутствии 5% CO2.
GAS6 (R&D Systems Inc., модель: 885-GS) разбавляли до 6 мкг/мл средой без FBS, затем добавляли в планшет при 25 мкл/лунку, и инкубировали при 37°C в течение 10 минут в присутствии 5% CO2 после перемешивания.
Супернатант отбрасывали и 37% раствор формалина, разбавленный до 4% фосфатно-солевым буферным раствором (PBS) (далее в настоящем документе, 4% раствор формалина) добавляли при 0,1 мл/лунку в планшет, который затем оставляли стоять при комнатной температуре в течение 10 минут. Затем 4% раствор формалина удаляли, и раствор Triton X-100, разбавленный до 0,1% с PBS (далее в настоящем документе, также называемый промывочный буфер) добавляли в планшет при 0,2 мл/лунку и удаляли путем декантации. Избыток воды удаляли на бумажное полотенце.
Затем, 10% NaN3 и 110 мкл H2O2 добавляли в 10,7 мл промывочного буфера (далее в настоящем документе, также называемый гасящий буфер), и этот гасящий буфер добавляли при 0,1 мл/лунку в планшет, который затем оставляли стоять при комнатной температуре в течение 15 минут.
Гасящий буфер удаляли и добавляли промывочный буфер при 0,2 мл/лунку в планшет и отбрасывали путем декантации. Избыток воды удаляли на бумажное полотенце. Добавляли обезжиренное молоко (WAKO #198-10605) (конечная концентрация 5%) в промывочный буфер (далее в настоящем документе, также называемый блокирующий буфер), и этот блокирующий буфер добавляли в планшет при 0,25 мл/лунку, который затем оставляли стоять при комнатной температуре в течение 1 часа.
Блокирующий буфер удаляли, и моноклональное кроличье анти-фосфо-Axl-антитело (Y702) (D12B2) (Cell Signaling Technology, Inc., No. по каталогу 5724) подвергали взаимодействию в концентрации 1/1000 с планшетом, который затем оставляли стоять в течение ночи при 4°C. Каждую лунку повторно промывали буфером для промывки пять раз, и конъюгированные с пероксидазой аффинно-очищенные антитела осла к кроличьим IgG (H+L) (Jackson ImmunoResearch Inc., номер по каталогу No. 711-035-152) подвергали реакции при концентрации 1/2000 в планшете при комнатной температуре в течение 1 часа. Проводили аналогичную операцию промывки, и хемилюминесцентный субстрат Super Signal ELISA pico (Thermo Fisher Scientific, Inc., номер по каталогу No. 37069) добавляли в планшет в количестве 0,05 мл/лунку и осторожно перемешивали, затем инкубировали в течение 20 минут. Затем, сформировавшийся свет измеряли с помощью ARVO sx (PerkinElmer Inc.) для измерения уровня pAxl (Y702).
Ингибирующую активность в отношении pAxl определяли в соответствии со следующим выражением:
Ингибирование % =100-(A-B)×100/(T-B)
A: Измеренное значение тестируемого соединения
B: Интенсивность излучения реакционной смеси, дополненной положительным контрольным соединением, имеющим концентрацию, которая ингибирует почти 100% фосфорилирование (например, интенсивность излучения реакционной смеси с добавлением 1 мкМ BMS-777607)
T: Интенсивность излучения реакционной смеси без добавления соединения
Концентрацию 50% ингибирования (IC50) определяли из данных о pAxl ингибирующей активности при совокупности концентраций с помощью программы GraphPad Prism 4.
В таблице 45 показана ингибирующая активность в отношениии внутриклеточного фосфорилирования Axl как IC50 (нМ).
[Таблица 45-1]
[Таблица 45-2]
(Тестовый пример 4 Офтальмологическое гистопатологическое исследование)
1. Животное: Самка мыши NOG
2. Лекарственное средство:
Соединение A моногидрохлорид дигидрат
Соединение примера 124
Соединение примера 128
Соединение примера 129
3. Получение раствора для введения:
Соединение A, соединение примера 124, соединение примера 128 и соединение примера 129 каждое суспендировали в 0,5% растворе метилцеллюлозы (0,5% MC, Wako Pure Chemical Industries, Ltd.).
4. Способ введения:
Четыре постоянные дозы, 1 ʺлекарственные каникулыʺ, 5 постоянных доз, 2 ʺлекарственных каникулʺ, 5 постоянных доз, 2 ʺлекарственных каникулʺ, 5 постоянных доз, 2 ʺлекарственных каникулʺ и 4 постоянные дозы.
5. Медико-патологическое исследование
На следующий день после даты окончания периода введения, мышей подвергали эвтаназии путем обескровливания под анестезией изофлураном. Затем их глазные яблоки собирали, фиксировали в растворах Дэвидсона, затем заливали в парафин и окрашивали гематоксилин-эозином для получения препаратов, которые затем подвергали гистопатологическому исследованию.
Легкая-умеренная дегенерация и истончение внешнего ядерного слоя и слоя палочковидных и колбочковидных зрительных клеток в сетчатке были обнаружены у всех мышей (n=8) в группе, получавшей 100 мг/кг соединения А моногидрохлорида дигидрата.
Никакое гистологическое изменение в сетчатке глаза не было найдено ни у одной из мышей (n=8) в группах, получавших 100 или 200 мг/кг соединения примера 124, примера 128 или примера 129.
(Тестовый пример 5: Тестирование противоопухолевой активности)
NIH-3T3-Axl #7 (клетки, полученные путем трансфекции клеток NIH3T3 с ретровирусом pLXSN, имеющим вставку полноразмерной кДНК Axl) трансплантировали блоком самке мыши NOG. Когда их расчетные объемы опухоли достигали приблизительно 500 мм3, соединение A моногидрохлорид дигидрат (25, 4,2 или 0,7 мг/кг), соединение примера 124 (50, 12,5, 3,1 или 0,8 мг/кг) и соединение примера 128 (50, 12,5, 3,1 или 0,8 мг/кг) каждое, вводили перорально два раза в день (bid) при 5 постоянных дозах. Соединение А, вводимое при 25 и 4,2 мг/кг, продемонстрировало явный эффект регрессии опухоли. Соединения примера 124 и примера 128, вводимые при 3,1 мг/кг, почти полностью ингибировали рост опухоли в течение периода введения, и эти соединения, вводимые при 50 и 12,5 мг/кг, продемонстрировали явный эффект регрессии опухоли.
(Тестовый пример 6: Исследование эффекта in vivo, вызванного комбинированным использованием с эрлотинибом)
Клетки рака легкого HCC827, имеющие делеционную мутацию в 19 экзоне гена EGFR и проявляющие высокую чувствительность к ингибиторам EGFR, суспендировали при 5×107 клеток/мл в фосфатно-солевом буферном растворе. 0,1 мл приготовленной суспензии клеток подкожно трансплантировали каждой бестимусной мыши (самка, 5 недельного возраста). Когда расчетные объемы опухоли большинства мышей достигли приблизительно 450 мм3 (52 дня после трансплантации опухоли), мышей разделяли на группы в соответствии с их объемами опухоли и мыши принудительно перорально получали 25 мг/кг эрлотиниб (LC Laboratories) один раз в день (qd) или 50 мг/кг N-[4-(2-амино-5-{4-[(2R)-1,4-диоксан-2-илметокси]-3-метоксифенил}пиридин-3-ил)-3-фторфенил]-5-метил-4'-оксо-1'-(тетрагидро-2H-пиран-4-илметил)-1',4'-дигидро-2,3'-бипиридин-5'-карбоксамид (соединение B) сульфат гидрат два раза в день (bid) (эти средства вводили отдельно или в комбинации). Введение начинали в день (День 1) после дня распределения по группам и осуществляли введение пять раз в неделю (суббота и воскресенье были приняты как ʺлекарственные каникулыʺ). Введение продолжали до дня 46 для групп, получавших носитель или гидрат сульфата соединения B отдельно, до дня 86 для группы, получавшей эрлотиниб отдельно, и до дня 106 для группы, получавшей эти средства в комбинации. Большую ось (мм) и малую ось (мм) каждой опухоли измеряли с течением времени с использованием электронного цифрового штангенциркуля. Расчетный объем опухоли рассчитывали согласно выражению (1), приведенному ниже, и наносили на график. Кроме того, массу тела измеряли с течением времени, используя автоматические весы для мелких животных. Скорость изменения массы тела (изменение массы тела %) рассчитывали согласно выражению (2), приведенному ниже. Таким образом, было изучено влияние введения лекарственного средства на массу тела, в то время как результаты последнего измерения массы тела использовали при расчете дозы.
Расчетный объем опухоли (мм3)=Средний расчетный объем опухоли индивидуумов... (1)
Расчетный объем опухоли каждого индивидуума=An×Bn2/2
An: Большая ось опухоли в День n
Bn: Малая ось опухоли в День n
Изменение массы тела (%)=Средняя скорость изменения масс тела индивидуумов... (2)
Скорость изменения массы тела каждого индивидуума=(1-BWn/BWs)×100
BWn: Масса тела в День n
BWs: Масса тела в первый день введения
Было подтверждено, что эрлотиниб, вводимый отдельно, имеет эффект регрессии опухоли сразу после введения. Однако, опухоль затем приобрела устойчивость и стала больше, чем ее размер в начале введения в день 77. В отличие от этого, размер опухоли в группе, получавшей эрлотиниб и гидрат сульфата соединения B в комбинации, не достиг такового в начале введения даже в день 107 (фиг. 19-1). Никакой существенной потери массы не было обнаружено в группе, получавшей эрлотиниб и гидрат сульфата соединения B в комбинации (фиг. 19-2).
(Тестовый пример 7: Исследование на повышение экспрессии AXL путем введения эрлотиниба)
Клетки рака легкого HCC827 суспендировали при 5×107 клеток/мл в фосфатно-солевом буферном растворе. 0,1 мл приготовленной суспензии клеток подкожно трансплантировали каждой бестимусной мыши (самка, 5 недельного возраста). Когда расчетные объемы опухоли большинства мышей достигли приблизительно 450 мм3 (52 дня после трансплантации опухоли), мышей разделяли на группы в соответствии с их объемами опухоли (день 0) и мыши принудительно перорально получали 25 мг/кг, 12,5 мг/кг или 6,25 мг/кг эрлотиниба (LC Laboratories) пять дней в неделю (суббота и воскресенье были приняты как ʺлекарственные каникулыʺ). Введение продолжали до дня 46 для группы с введением носителя и до дня 86 для групп с введением эрлотиниба. Большую ось (мм) и малую ось (мм) каждой опухоли измеряли с течением времени с использованием электронного цифрового штангенциркуля. Расчетный объем опухоли рассчитывали согласно выражению (1), приведенному ниже, и наносили на график (фиг. 20-1). Клеточный лизат получали из масс опухоли 2-6 мышей в каждой группе, и оценивали экспрессию AXL путем Вестерн-блоттинга, и каждый отдельный бэнд количественно оценивали с использованием ImageQuant LAS4000 (GE Healthcare Japan Corp.) (фиг. 20-2). Получение клеточного лизата и Вестерн-блот проводили следующим образом: приблизительно 100 мг опухолевых масс гомогенизировали в буфере для лизиса (Cell Signaling Technology, Inc., 9803), содержащем ингибитор фосфатазы (Roche Diagnostics., 04 906 837 001) и ингибитор протеазы (Roche Diagnostics, 11 836 153 001), и супернатант использовали в качестве образца лизата. Белки в этом образце фиксировали с помощью буферов (Life Technologies Corp, NP0008 и NP0009) и тепловой денатурации и подвергали Вестерн-блоттингу. 15 мкг образца подвергали электрофорезу и переносили на нитроцеллюлозную мембрану. Мембрану блокировали в течение 1 часа и затем подвергали взаимодействию в течение ночи с кроличьим анти-AXL-антителом (Cell Signaling Technology, Inc., 9803, 1/1000) или кроличьим анти-актин-антителом (Santa Cruz Biotechnology, Inc., SC-1616, 1/2000) при низких температурах. Мембрану промывали трис-буферным солевым раствором и затем подвергали взаимодействию с HRP-меченным антителом против кроличьего IgG (Cell Signaling Technology, Inc., 7074, 1/2000) при комнатной температуре в течение 1 часа. Мембрану промывали трис-буферным солевым раствором и затем обнаруживали излучение с субстратом HRP (Merck Millipore Corp., WBLUF0500).
Экспрессию AXL подтверждали с помощью Вестерн-блоттинга, и каждый отдельный бэнд количественно оценивали с использованием ImageQuant LAS4000. В результате, значительное повышение экспрессии AXL в группах с введением эрлотиниба было подтверждено с помощью теста Велша.
(Тестовый пример 8: Исследование эффекта in vivo, вызванного комбинированным использованием с эрлотинибом - 2)
Клетки рака легкого HCC827 суспендировали при 4×107 клеток/мл в фосфатно-солевом буферном растворе. 0,1 мл приготовленной суспензии клеток подкожно трансплантировали каждой бестимусной мыши (самка, 5 недельного возраста). Через 52 дня после трансплантации опухолевых клеток, мыши, имеющие расчетный объем опухоли 200 мм3 или более и менее чем 700 мм3 (86 мышей/129 мышей) принудительно перорально получали 25 мг/кг эрлотиниба (LC Laboratories) один раз в день (qd). Введение начинали в день (53 дня после трансплантации) после дня распределения по группам и проводили пять раз в неделю (суббота и воскресенье были приняты как ʺлекарственные каникулыʺ). Мыши, несущие рак, с временной регрессией и явным возобновлением роста опухоли, были разбиты на 3 группы (6 мышей/группу, 115 дней после трансплантации): группа 1: группа с введением эрлотиниб+носитель (средний расчетный объем опухоли: 275 мм3), группа 2: эрлотиниб+50 мг/кг гидрат сульфат соединения B (средний расчетный объем опухоли: 284 мм3) и группа 3: эрлотиниб+25 мг/кг гидрат сульфат соединения B (средний расчетный объем опухоли: 268 мм3). Исследование было сделано о том, восстановит ли комбинированное использование с гидратом сульфатом соединения B действие эрлотиниба на опухоли, устойчивые к эрлотинибу. Гидрат сульфат соединения В вводили дважды в день (bid) через 116 дней после трансплантации, и это введение продолжали пять раз в неделю (суббота и воскресенье были приняты как ʺлекарственные каникулыʺ). Опухоль выросла до расчетного объема опухоли на том же уровне как первоначальный объем на момент трансплантации в группе 1, тогда как явный рост ингибирующего эффекта эрлотиниба был подтвержден в группе 2 и группе 3. В группе 2 расчетный объем опухоли при 116 днях после трансплантации, когда было начато комбинированное введение, сохранялся даже при 140 днях после трансплантации.
Реферат
Изобретение относится к производному пиридона, имеющему тетрагидропиранилметильную группу, представленную следующей формулой (I), или к его фармацевтически приемлемой соли, где W, X и Y каждый независимо друг от друга представляет собой атом азота, C-H, C-F или C-Cl, Z представляет собой атом азота, C-H, C-F, C-Cl, C-C-Cалкил или C-C-Cалкоксигруппу, Rпредставляет собой группу, представленную следующей формулой (II-1) или (II-2):, где в формуле (II-1) Q представляет собой атом азота, C-H или C-F, T представляет собой атом азота или C-H, U представляет собой атом азота или C-H и Rпредставляет собой атом галогена, C-Cалкильную группу, C-Cциклоалкильную группу, цианогруппу или трифторметоксильную группу, и в формуле (II-2) V представляет собой атом серы или атом кислорода и Rпредставляет собой C-Cалкильную группу, Rпредставляет собой атом водорода, атом галогена, C-Cалкильную группу, C-Cалкоксигруппу или цианогруппу, Rпредставляет собой атом водорода или C-Cалкильную группу, Rпредставляет собой атом водорода, атом галогена или C-Cалкильную группу и Rпредставляет собой атом водорода или группу, представленную следующей формулой (III-1), (III-2), (III-3) или (III-4):, где в формуле (III-1) Rи Rкаждый независимо друг от друга представляет собой атом водорода или атом дейтерия, Rпредставляет собой атом водорода, атом галогена или C-Cалкоксигруппу, Rпредставляет собой атом водорода, C-Cалкильную группу, необязательно замещенную гетероциклоалкильной группой, которая является морфолинильной или метил-замещенной пиридинильной группой, или C-Cалкоксигруппу, необязательно замещенную гетероциклоалкильной группой, которая является диоксанильной, морфолинильной или пирролидинильной группой, и Rпредставляет собой атом водорода, C-Cалкоксигруппу или дейтерий-замещенную C-Cалкоксигруппу, в формуле (III-2) Rпредставляет собой C-Cалкильную группу, необязательно замещенную гетероциклоалкильной группой, которая является диоксанильной или тетрагидропиранильной группой, или гетероциклоалкильную группу, которая является тетрагидропиранильной группой, в формуле (III-3) Rпредставляет собой атом водорода или C-Cалкильную группу, Rпредставляет собой атом водорода, C-Cалкильную группу или C-Cалкоксигруппу и Rпредставляет собой атом водорода или атом галогена, и в формуле (III-4) Rпредставляет собой C-Cалкоксигруппу и Rпредставляет собой C-Cалкоксигруппу. Изобретение также относится к кристаллическим формам указанного соединения, фармацевтической композиции, обладающей Axl ингибирующей активностью, и лекарственным средствам на их основе. Технический результат – получены новые соединения и фармацевтические композиции на их основе, которые могут найти применение в медицине при лечении рака, вызванного гиперфункцией Axl, заболевания, связанного с гиперфункцией Axl, и/или заболевания, сопровождающегося гиперфункцией Axl. 51 н. и 2 з.п. ф-лы, 45 табл., 20 ил., 123 пр.
Формула


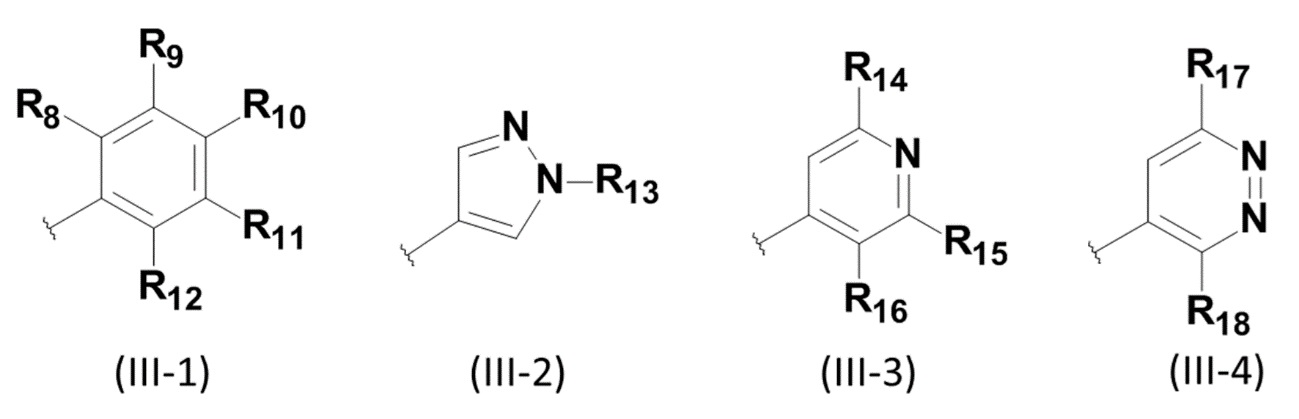


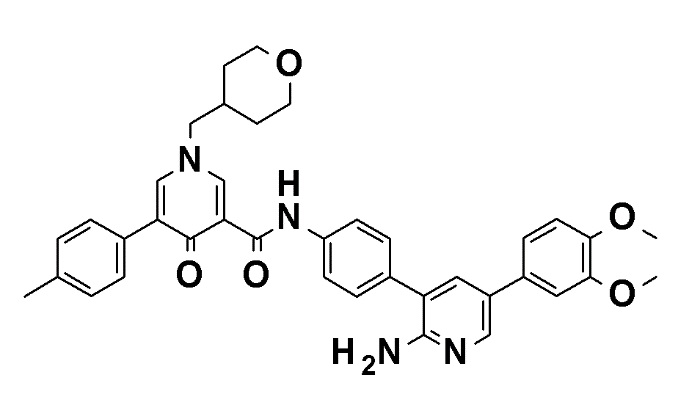






Комментарии