Производные 2-пиперидона в качестве агонистов простагландина - RU2311409C2
Код документа: RU2311409C2
Описание
Настоящее изобретение относится к некоторым производным 2-пирролидона и ассоциированным фармацевтическим композициям, способам применения в качестве селективных агонистов простагландина и к способам их получения.
В литературе приводится множество публикаций по простагландинам или простаноидам (PG), которыми принято называть природные и синтетические простагландины и простагландинподобные соединения, причем хорошо известно, что даже небольшие изменения в химической структуре или в стереохимической конфигурации оказывают существенное влияние на биологическую активность.
Простагландины или простаноиды (PG) представляют собой группу биологически активных соединений, образующихся из мембранных фосфолипидов, и построены из незаменимых жирных кислот, содержащих 20 атомов углерода, и включают циклопентановое кольцо. Эти соединения образуют несколько основных классов, которые обозначаются разными буквами, и различаются заместителями в циклопентановом цикле. Кроме того, основные классы подразделяются на группы, которые обозначаются индексами 1, 2 или 3, соответствующими их жирнокислотным предшественникам.
Например, варианты простагландина Е обозначаются PGE2 и имеют следующую структуру:
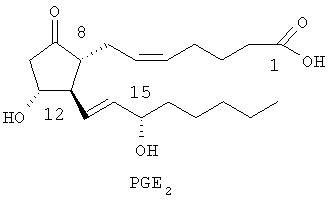
В настоящее время известны четыре различных подвида рецептора PGE2, которые обозначаются ЕР1, ЕР2, ЕР3 и ЕР4.
Области применения соединений, обладающих высоким сродством к рецепторам PGE2, включают профилактику и/или лечение иммунологических заболеваний (аутоиммунные заболевания, пересадка органов и т.п.), астмы, аномального формирования костной ткани, гибели (нейронных) клеток нервной ткани, тромбоза и инсульта, гепатопатии, аборта, мужской и женской половой дисфункции, преждевременные роды, воспаления, такого, как ревматоидный артрит, или невропатическое нарушение сетчатки, такое, как глаукома.
Простагландины и ассоциированные с ними рецепторы более подробно описаны, например, в следующей статье: М. Abramovitz и др.. The Utilization of Recombinant Prostanoid Receptors to Determine the Affinities and Selectivities of Prostaglandins and Related Analogs, Biochimica et Biophysica Acta, 1483, 285-293 (2000).
Участие агонистов рецептора простагландина Е в резорбции костной ткани описано, например, в статьях Т. Suzawa и др., The Role of Prostaglandin E Receptor Subtypes in Bone Resorption: An Analysis Using Specific Agonists for the Respective EPs, Endocrinology, 141, 1554-1559 (2000); К.Ono и др., Important Role of EP4, a Subtype of Prostaglandin (PG) E Receptor, in Osteoclast-like Cell Formation from Mouse Bone Marrow Cells Induced by PGE2, J. of Endocrinology, 158, R1-R5 (1998); M. Suda и др., Prostaglandin E Receptor Subtypes in Mouse Osteoblastic Cell Line, Endocrinology, 137, 1698-1705 (1996).
Селективные агонисты рецептора простагландина Е используются также для лечения патологических изменений ткани желудка, см., например, статьи Н.Araki и др., The Roles of Prostaglandin E Receptor Subtypes in the Cytoprotective Action of Prostaglandin E2 in Rat Stomach, Aliment. Pharmacol. Ther., 14 (Suppl. 1), 116-124 (2000); Т.Kunikata и др., Е Type Prostaglandin Inhibits Indomethacin-Induced Small Intestinal Lesions Through ЕР3 and EP4 Receptors: A Study Using Rats and Knockout Mice, Gastroenterology 118, abstract №3787.
Другой областью применения агонистов рецептора простагландина Е является улучшение функции почек, как описано, например, в статье М.D.Breyer и др., Prostaglandin Е Receptors and the Kidney, Am. J. Physiol., 279, F12-F23 (2000), и К.Е.Purdy и др., ЕР1 and EP4 Receptors Mediate Prostaglandin E2 Actions in the Microcirculation of Rat Kidney, Am. J. Physiol., 279, F755-F764 (2000); лечение тромбоза и инсульта, а также других состояний, при которых лечебное действие достигается за счет агрегации тромбоцитов, как описано, например, в статье В.Z.S.Paul и др., Distribution of Prostaglandin IP and EP Receptor Subtypes and Isoforms in Platelets and Human Umbilical Artery Smooth Muscle Cells, Br.J.Haematol., 102, 1204-1211 (1998); использование в качестве противовоспалительных агентов благодаря ингибированию синтеза TNF-α, как описано, например, в статьях К.К.Meja и др., Characterization of prostanoid receptor(s) on human blood monocytes at which prostaglandin E2 inhibits lipopolysaccharide-induced tumor necrosis factor-alpha generation, Br.J.Pharmacol., 122, 149-157 (1997), и A.Eigler и др., Anti-inflammatory activities of cAMP-elevating agents: enhancement of IL-10 synthesis and concurrent suppression of TNF production, J. Leukoc. Biol, 63, 101-107 (1998); или лечение глаукомы, как описано, например, в статьях М. Takamatsu и др., Localization of Prostaglandin E Receptor Subtypes in The Ciliary Body of Mouse Eye, Exp. Eye Res., 70, 623-628 (2000), и D.F.Woodward и др., Molecular Characterization and Ocular Hypotensive Properties of the Prostanoid EP2 Receptor, J. Ocul. Pharmacol. Ther., 11, 447 (1995).
Лечение импотенции и/или эректильной дисфункции при использовании простагландинов, которые являются селективными лигандами рецептора EP2 и/или EP4 описано в Международной опубликованной заявке WO 99/02164 (заявитель фирма Pharmacia и Upjohn АВ).
Дополнительная информация, относящаяся к простагландинам и их рецепторам, приводится в монографии Goodman и Gillman's, The Pharmacologica Basis of Therapeutics, 9 изд., McGraw-Hill, New York, гл.26, стр.601-616 (1996).
8-Аза-11-дезоксипростагландиновые аналоги, соответствующие PGE2, характеризуются следующей структурой:

8-Аза-11-дезоксипростагландин
Замещение по атому азота и атому углерода С8 приводит к изменению трехмерной структуры полученного простагландина, а поскольку структура соединения тесно связана с его биологической активностью, такое изменение конформации оказывает существенное влияние на биологическую активность. 8-Аза-11-дезоксипростагландин Е с природными боковыми цепями описан в литературе (см., например, BE 841165 (заявитель фирма Syntex США, Inc.).
Настоящее изобретение относится к соединениям формулы I

где m равно 1-4,
n равно 0-4,
А означает алкил, арил, гетероарил, арилалкил, арилциклоалкил,
циклоалкилалкил или арилоксиалкил,
Е означает -СНОН- или -С(O)-,
Х означает -(СН2)2- или -СН=СН-,
Y означает -СН2 -, арилен, гетероарилен, -СН=СН-, -О-, -S(O)p -, где р равно 0-2, или -NRa-, где Ra означает водород или алкил,
Z означает -СН2ОН, -СНО, тетразол-5-ил или -COORb, где Rb означает водород или алкил, а
R1, R2, R3, R4, R5, R6, R7, R8, R9 и R10 каждый независимо означает водород или алкил,
или к их фармацевтически приемлемой соли или сольвату, пролекарству, индивидуальному изомеру или рацемической или нерацемической смеси изомеров.
Соединения, которые являются объектом изобретения, обладают высокой селективностью как агонисты рецептора ЕР4. Увеличение селективности позволяет снизить острые побочные действия, часто наблюдаемые после введения неселектиных простагландиновых агонистов. Следовательно, в настоящее время существует необходимость в соединениях по настоящему изобретению.
Другим объектом настоящего изобретения являются фармацевтические композиции, содержащие терапевтически эффективное количество по меньшей мере одного соединения формулы I или его фармацевтически приемлемой соли или сольвата, пролекарства, индивидуального изомера или рацемической или нерацемической смеси изомеров в смеси по меньшей мере с одним пригодным носителем, разбавителем или эксципиентом.
Еще одним объектом изобретения является способ лечения заболевания, прежде всего заболевания костной ткани, у млекопитающего, которое излечивается при введении агониста рецептора простагландина ЕР4, причем способ включает введение терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли.
Еще одним объектом изобретения является способ получения соединений формулы I.
Если не указано иное, термины, используемые в описании и формуле изобретения, имеют следующие значения.
"Алкокси" означает радикал OR, где R означает алкил, указанный ниже, например метокси, этокси, пропокси, бутокси и т.п.
«Алкил» означает насыщенный одновалентный углеводородный радикал с прямой цепью, содержащий от одного до шести атомов углерода, или насыщенный одновалентный углеводородный радикал с разветвленной цепью, содержащий от трех до шести атомов углерода, например, метил, этил, пропил, 2-пропил, н-бутил, изобутил, трет-бутил, пентил и т.п.
"Алкилен" означает насыщенный двухвалентный углеводородный радикал с прямой цепью, содержащий от одного до шести атомов углерода, или насыщенный двухвалентный углеводородный радикал с разветвленной цепью, содержащий от трех до шести атомов углерода, например, метилен, этилен, 2,2-диметилэтилен, пропилен, 2-метилпропилен, бутилен, пентилен и т.п.
«Алкилтио» означает радикал SR, где R означает алкил, указанный выше, например, метилтио, этилтио, пропилтио, бутилтио и т.п.
"Арил" означает одновалентный моноциклический или бициклический ароматический углеводородный радикал, необязательно и независимо замещенный одним или более заместителями, предпочтительно одним, двумя или тремя заместителями, выбранными из группы, включающей алкил, галогеналкил, галоген, нитро, циано, амино, метилендиокси, этилендиокси, необязательно замещенный фенил, арил, арилалкил, гетероарил, гетероарилалкил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, -V-OR', -V-NR'R", -V-C(O)-R', -V-S(O)0-2-R'; -V-N-SO2-R', -V-SO2-NR'R", -V-N-C(O)-NR'R", где V означает химическую связь или C1-С3алкиленовую группу, а R' и R" каждый независимо друг от друга означают водород, алкил, галогеналкил, гидрокси, алкокси, необязательно замещенный фенил, гетероарил, циклоалкил или гетероциклил. Более конкретно термин арил включает, без ограничения перечисленными, фенил, хлорфенил, метоксифенил, метоксиметилфенил, фенилоксифенил, 1-нафтил, 2-нафтил и их производные.
"Арилен" означает двухвалентный моноциклический или бициклический ароматический углеводородный радикал и включает двухвалентные варианты арильных радикалов, указанных выше.
"Арилалкил" и "аралкил", которые используются взаимозаменяемым образом, означают радикал -RaRb где Ra означает алкиленовую группу, a Rbозначает арильную группу, указанную выше, например бензил, фенилэтил, 3-(3-хлорфенил)-2-метилпентил и т.п.
"Арилциклоалкил" означает радикал -RaRb, где Ra означает циклоалкилен, указанный выше, а Rb означает арил, указанный выше.
"Арилокси" означает радикал -ORa, где Ra означает арил, указанный выше.
"Арилоксиалкил" означает радикал -RaRb, где Ra означает алкилен, указанный выше, a Rb означает арилокси, указанный выше.
"Циклоалкил" означает одновалентный насыщенный карбоциклический остаток, включающий один или два цикла. Циклоалкил необязательно замещен одним или более заместителями, каждый из которых независимо означает, если не указано иное, гидрокси, алкил, алкокси, галоген, галогеналкил, амино, моноалкиламино или диалкиламино. Примеры циклоалкильной группы включают, без ограничения перечисленным, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и т.п., включая их частично ненасыщенные производные.
"Циклоалкилен" означает двухвалентный насыщенный карбоциклический остаток, содержащий моно- или бициклические кольца и включающий двухвалентные варианты циклоалкильных групп, указанных выше.
"Циклоалкилалкил" означает остаток формулы -R'-R", где R' означает алкилен, а R" означает циклоалкил, указанный выше.
"Галоген" означает фтор, хлор, бром или иод, предпочтительно фтор и хлор.
"Галогеналкил" означает алкил, замещенный одним или более идентичными или различными атомами галогена, например -CH2Cl, -CF3, -СН2CF3, -СН2CCl3 и т.п.
"Гетероарил" означает одновалентный моноциклический или бициклический радикал, содержащий от 5 до 12 атомов в цикле, и по меньшей мере один ароматический цикл, содержащий в цикле один, два или три гетероатома, выбранных из N, О или S, а остальные атомы в цикле являются атомами углерода, причем подразумевается, что гетероарильный радикал присоединен к ароматическому циклу. Гетероарильный радикал необязательно и независимо друг от друга замещен одним или более заместителями, предпочтительно одним или двумя заместителями, выбранными из группы, включающей алкил, галогеналкил, галоген, нитро, циано, амино, метилендиокси, необязательно замещенный фенил, арил, арилалкил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, -V-OR', -V-NR'R", -V-C(O)-R', VO-C(O)-R', -V-S(O)0-2-R'; -V-N-SO2-R', -V-SO2-NR'R", -V-N-C(O)-NR'R", где V отсутствует или означает C1-С3алкиленовую группу, а R' и R" каждый независимо друг от друга означают водород, алкил, галогеналкил, гидрокси, алкокси, необязательно замещенный фенил, циклоалкил, гетероциклил. Более конкретно термин гетероарил включает, без ограничения перечисленным, пиридил, фуранил, тиенил, тиазолил, изотиазолил, триазолил, имидазолил, изоксазолил, пирролил, пиразолил, пиримидинил, бензофуранил, тетрагидробензофуранил, изобензофуранил, бензотиазолил, бензоизотиазолил, бензотриазолил, индолил, изоиндолил, бензоксазолил, хинолил, тетрагидрохинолинил, изохинолил, бензимидазолил, бензизоксазолил или бензотиенил, имидазо[1,2-а]пиридинил, имидазо[2,1-b]тиазолил и их производные. "Гетероарилен" означает двухвалентный моноциклический или бициклический радикал, содержащий от 5 до 12 атомов в цикле, и по меньшей мере один ароматический цикл, содержащий в цикле один, два или три гетероатома, выбранных из N, О или S, а остальные атомы в цикле являются атомами углерода, причем подразумевается, что гетероариленовый радикал присоединен к ароматическому циклу. Гетероарилен включает двухвалентные варианты гетероарильных радикалов, указанных выше.
"Гетероарилалкил" означает остаток формулы -R'-R", где R' означает алкилен, а R" означает гетероарил, указанный выше.
"Гетероциклил" означает насыщенный или ненасыщенный неароматический циклический радикал, содержащий от 3 до 8 атомов в цикле, из которых один или два атома являются гетероатомами, выбранными из N, О или S(O)0-2, а остальные атомы являются атомами углерода, причем один или два атома углерода необязательно заменены карбонильной группой. Гетероциклильный цикл необязательно и независимо друг от друга замещен одним, двумя или тремя заместителями, выбранными из группы, включающей алкил, галогеналкил, галоген, нитро, циано, необязательно -V-замещенный фенил, арил, арилалкил, гетероарил, гетероарилалкил, циклоалкил, циклоалкилалкил, -V-OR', -V-NR'R", -V-C(O)-R', -V-S(O)0-2 -R'; -V-NR'-SO2-R", -V-SO2-NR'R", -V-N-C(O)-NR'R", где V отсутствует или означает C1-С3алкиленовую группу, а R' и R" каждый независимо друг от друга означают водород, алкил, галогеналкил, гидрокси, алкокси, необязательно замещенный фенил, гетероарил или циклоалкил. Более конкретно термин гетероциклил включает, без ограничения перечисленным, тетрагидропиранил, пиперидинил, N-метилпиперидин-3-ил, пиперазинил, N-метилпирролидин-3-ил, 3-пирролидинил, морфолинил, тиоморфолинил, тиоморфолино-1-оксид, тиоморфолино-1,1-диоксид, пирролинил, имидазолинил, N-метансульфонилпиперидин-4-ил и их производные.
"Гетероциклоалкил" означает остаток формулы -R'-R", где R' означает алкилен, а R" означает гетероциклил, указанный выше.
«Уходящая группа» означает группу, название которой обычно ассоциируется с ее использованием в синтетической органической химии, т.е. означает атом или группу, которая замещается нуклеофилом, и включает галоген (такой, как хлор, бром и иод), алкилсульфонилокси, арилсульфонилокси, алкилкарбонилокси (например, ацетокси), арилкарбонилокси, мезилокси, тозилокси, трифторметансульфонилокси, арилокси (например, 2,4-динитрофенокси), метокси, N,O-диметилгидроксиламино и т.п.
"Необязательно замещенный фенил" означает фенильный цикл, который необязательно и независимо друг от друга замещен одним или более заместителями, предпочтительно одним или двумя заместителями, выбранными из группы, включающей алкил, гидрокси, алкокси, галогеналкил, галогеналкокси, гетероалкил, галоген, нитро, циано, амино, метилендиокси, этилендиокси и ацил.
«Изомеры» означает соединения с одинаковой молекулярной формулой, но отличающиеся природой или последовательностью химических связей или пространственным расположением атомов. Изомеры, которые различаются пространственным расположением атомов, называются «стереоизомерами». Стереоизомеры, которые не являются зеркальными изображениями друг друга, называются диастереоизомерами, а стереоизомеры, которые являются несовместимыми зеркальными изображениями друг друга, называются «энантиомерами», или иногда оптическими изомерами. Атом углерода, связанный с четырьмя различными заместителями, называется «хиральным центром».
«Хиральный изомер» означает соединение с одним хиральным центром. Это соединение имеет две энантиомерные формы с противоположной хиральностью и может существовать как отдельный энантиомер или как смесь энантиомеров. Смесь, содержащая равные количества индивидуальных энантиомерных форм противоположной хиральности, называется «рацемической смесью». Соединения, содержащие более одного хирального центра, образуют 2n-1 энантиомерных пар, где n означает количество хиральных центров. Соединения, содержащие более одного хирального центра, могут существовать в виде индивидуального диастереомера или в виде смеси диастереомеров, которая называется «диастереомерной смесью». При наличии одного хирального центра стереоизомер можно охарактеризовать абсолютной конфигурацией (R или S) этого хирального центра. Абсолютная конфигурация означает пространственное расположение заместителей у хирального центра. Указанные заместители, связанные с хиральным центром, классифицируются по правилу Кана, Ингольда и Прелога ((Cahn и др., Angew. Chem., Inter. Edit., 5, 385, errata 511 (1966), Cahn и др., Angew. Chem., 78, 413 (1966), Cahn и Ingold., J. Chem. Soc. (London), 612 (1951), Cahn и др., Experientia, 12, 81, (1956), Cahn, J. Chem. Educ., 41, 116 (1964)).
«Геометрические изомеры» означают диастереомеры, образующиеся благодаря заторможенному вращению вокруг двойных связей. Эти конфигурации обозначаются приставками в названиях цис- и транс- или буквами Z и Е, которые означают, что группы располагаются по одну или по разные стороны плоскости двойных связей согласно правилам Кана-Ингольда-Прелога.
«Атропические изомеры» означают изомеры, образующиеся благодаря заторможенному вращению объемных групп вокруг центральной связи.
Соединения по настоящему изобретению могут существовать в стереоизомерных формах, следовательно, их можно получать в виде индивидуальных стереоизомеров или в виде их смесей.
«Фармацевтически приемлемый эксципиент» означает носитель, который используют при получении фармацевтической композиции и который обычно безопасен, не токсичен и является совместимым в биологическом или ином отношении, а также включает носитель, приемлемый как в ветеринарии, так и в фармацевтике. «Фармацевтически приемлемый эксципиент», используемый в описании и пунктах формулы изобретения, включает один или более одного носителя.
«Фармацевтически приемлемая соль» соединения означает соль, которая является фармацевтически приемлемой и которая обладает необходимой фармакологической активностью исходного соединения. Например, такие соли включают:
(1) кислотно-аддитивные соли неорганических кислот, таких, как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, и т.п., или органических кислот, таких, как уксусная кислота, пропионовая кислота, гексановая кислота, циклопентанпропионовая кислота, гликолевая кислота, пировиноградная кислота, молочная кислота, малоновая кислота, янтарная кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, 3-(4-гидроксибензоил)бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, 1,2-этандисульфоновая кислота, 2-гидроксиэтансульфоновая кислота, бензолсульфоновая кислота, 4-хлорбензолсульфоновая кислота, 2-нафталинсульфоновая кислота, 4-толуолсульфоновая кислота, камфорсульфоновая кислота, 4-метилбицикло[2.2.2]окт-2-ен-1-карбоновая кислота, глюкогептоновая кислота, 3-фенилпропионовая кислота, триметилуксусная кислота, трет-бутилуксусная кислота, лаурилсерная кислота, глюконовая кислота, глутаминовая кислота, гидроксинафтоевая кислота, салициловая кислота, стеариновая кислота, муконовая кислота и т.п., или
(2) соли, образующиеся при замене кислотного протона, присутствующего в исходном соединении, на ион металла, например ион щелочного металла, ион щелочно-земельного металла или ион алюминия, или при образовании протоном координационного соединения с органическим основанием, таким, как этаноламин, диэтаноламин, триэтаноламин, трометамин, N-метилглюкамин и т.п.
Подразумевается, что все ссылки на фармацевтически приемлемые соли включает сольватированные формы (сольваты) или кристаллические формы (полиморфы), указанные в описании, соответствующей кислотно-аддитивной соли. «Кристаллические формы» (или полиморфные модификации) означают кристаллические структуры с различной формой упаковки кристаллов, которые образуются при кристаллизации соединения, причем все формы имеют одинаковый элементный состав. Различные кристаллические формы обычно характеризуются разными рентгеновскими дифракционными картинами, разными инфракрасными спектрами, имеют разные температуры плавления, плотность, твердость и форму кристаллов, оптические и электрические свойства, устойчивость и растворимость. На образование преобладающей кристаллической формы влияют различные факторы, такие, как растворитель, используемый для перекристаллизации, скорость кристаллизации и температура хранения.
"Сольваты" означают сольватированные формы, содержащие стехиометрическое или нестехиометрическое количество растворителя. Некоторые соединения способны удерживать в кристаллической решетке фиксированное количество молекул растворителя, образуя сольват. Гидраты образуются в том случае, если в качестве растворителя используется вода, а алкоголяты образуются в том случае, если растворителем является спирт. Гидраты образуются благодаря комбинации одной или более молекул воды с одной молекулой соединения, причем в указанной комбинации вода сохраняет молекулярную форму, такую как Н2О, и такие комбинации могут существовать в виде одного или более гидратов.
"Пролекарство" означает любое соединение, из которого in vivo высвобождается активное исходное соединение формулы I при введении такого пролекарства млекопитающему субъекту. Пролекарства соединения формулы I получают модификацией одной или более функциональных групп, присутствующих в соединении формулы I, таким образом, что модифицированные группы могут расщепляться in vivo с высвобождением исходного соединения. Пролекарства включают соединения формулы I, в которых гидрокси, амино, сульфгидрил, карбокси или карбонилгруппа соединения (I) связана с любой группой, которая может отщепляться in vivo с образованием свободной гидроксил, амино или сульфгидрилгруппы соответственно. Примеры пролекарств включают, без ограничения перечисленным, сложные эфиры (например, ацетат, диалкиламиноацетаты, формиаты, фосфаты, сульфаты и бензоаты) и карбаматы (например, N,N-диметиламинокарбонил) гидроксильных функциональных групп, сложноэфирные группы (например, этиловые эфиры, морфолиноэтанольные эфиры) карбоксильных функциональных групп, N-ацилпроизводные (например, N-ацетил), N-основания Манниха, Шиффовы основания и енаминоны функциональных аминогрупп, оксимы, ацетали, кетали и енольные сложные эфиры кетонных и альдегидных функциональных групп в соединениях формулы I и т.п. (см. Bundegaard H., Design of Prodrugs, стр.1-92, Elesevier, New York-Oxford (1985).
"Защитная группа" (PG) означает группу атомов, которая при связывании с реакционноспособной группой в молекуле маскирует, снижает или блокирует реакционную способность группы. Примеры защитных групп можно найти в монографиях T.W.Greene и P.G.Futs, Protective Groups in Organic Chemistry, Wiley, 3 ed. (1999) и Harrison и Harrison и др., Compendium of Synthetic Organic Methods, т.1-8, J.Wiley и Sons (1971-1996). Типичные аминозащитные группы включают формил, ацетил, трифторацетил, бензил, бензилоксикарбонил (CBZ), трет-бутоксикарбонил (ВОС), триметилсилил (TMS), 2-триметилсилилэтансульфонил (SES), тритил и замещенные тритильные группы, аллилоксикарбонил, 9-флуоренилметилоксикарбонил (FMOC), нитровератрилоксикарбонил (NVOC) и т.п. Типичные гидроксизащитные группы включают такие группы, в которых гидроксильная группа ацилирована или алкилирована, такие как бензиловые и тритильные эфиры, а также алкильные эфиры, тетрагидропиранильные эфиры, триалкилсилильные эфиры и аллильные эфиры.
"Терапия" или "лечение" заболевания включает (1) профилактику заболевания, т.е. предупреждение развития клинических симптомов заболевания у субъекта, который подвержен или предрасположен к заболеванию, но у которого не обнаруживаются или не проявляются симптомы заболевания; (2) подавление заболевания, т.е. остановку или подавление развития заболевания или его клинических симптомов, или (3) ослабление заболевания, например, стимуляцию временной или долгосрочной регрессии заболевания или его клинических симптомов.
«Терапевтически эффективное количество» означает количество соединения, которое при введении млекопитающему для лечения заболевания, является достаточным для излечения такого заболевания. Терапевтически эффективное количество варьирует в зависимости от типа соединения, болезни и ее тяжести, возраста, массы тела и т.п. млекопитающего, проходящего курс лечения.
"Аналог простагландина" означает неприродное соединение, которое в структурном отношении подобно простагландину.
"Рецептор простагландина" или «простаноидный рецептор» означает природный белок, который связывает простагландины и после связывания с лигандом модулирует функционирование клетки. Простагландиновые рецепторы могут оказывать как стимулирующее, так и расслабляющее действие. Такие рецепторы включают, без ограничения перечисленным, ЕР1, ЕР2, ЕР3, ЕР4, DP, FP, IP, TP1 и ТР2. Кроме того указанные рецепторы подробно обсуждаются в обзоре Coleman и др., Pharmacological Reviews, т.6, №2, с.205-229 (1994).
Номенклатура и структуры
В общем случае номенклатура, использованная в описании заявки, основана на программном обеспечении AUTONOMTM версия 4.0 компьютерной системы института Бельштейна для формирования систематической номенклатуры ИЮПАК. Приведенные в описании химические структуры получены с использованием программы ISIS® версия 4.0. Любые свободные валентности у атомов углерода, кислорода или азота в приведенных структурах означают присутствие атомов водорода.
Соединения
В изобретении предлагаются соединения формулы I

где m равно 1-4, предпочтительно m равно 2,
n означает химическую связь или n равно 1-4, предпочтительно n равно 3,
А означает алкил, арил, гетероарил, арилалкил, арилциклоалкил, циклоалкилалкил или арилоксиалкил,
Е означает -СНОН- или -С(O)- (т.е. Е означает гидроксиметилен или оксо), предпочтительно Е означает -СНОН-(гидроксиметилен),
Х означает -(СН2)2-или -СН=СН-,
Y означает -СН2-, арилен, гетероарилен, -СН=СН-, -О-, -S(O)p-, где р равно 0-2, или -NRa-, где Ra означает водород или алкил, предпочтительно Y означает -СН2- или -S(O)p -, где р предпочтительно равно 0,
Z означает -CH2OH, -CHO, тетразол-5-ил или -COORb, где Rb означает водород или алкил, предпочтительно Z означает -COORb, где Rb означает водород,
R1, R2, R3, R4, R5, R6, R7, R8, R9 и R10 каждый независимо означает водород или алкил, предпочтительно R1, R2, R3, R4, R5, R6, R7, R8, R9 и R10 означают водород,
или их фармацевтически приемлемая соль или сольват, пролекарство, индивидуальный изомер или рацемическая или нерацемическая смесь изомеров.
Если любой из R1, R2, R3, R4, R5, R6, R7, R8, R9, R10, Ra и Rb означает алкил, то такой алкил предпочтительно означает (низш.)алкил, т.е. C1-С6алкил, более предпочтительно С1-С4 алкил.
В некоторых вариантах изобретения m равно 2, n равно 3, Е означает -СНОН-, Y означает -S- или -CH2-, Z означает -COORb, а R1, R2, R3, R4, R5, R6, R7, R8, R9 и R10 означают водород. В таких вариантах изобретения соединения формулы I характеризуются формулой II

где А, Х и Rb имеют значения, указанные выше. В предпочтительных вариантах осуществления изобретения стереохимия может быть иной, и соединения формулы II характеризуются формулой III

В других вариантах изобретения m равно 2, n равно 0, Е означает -СНОН-, Y означает арилен или фенилен, Z означает -COORb, a R1, R2, R3, R4, R5, R6, R7, R8, R9 и R10 означают водород. В таких вариантах соединения формулы I характеризуются формулой IV

В зависимости от стереохимической структуры соединения формулы IV могут характеризоваться формулой V

Типичные соединения по изобретению приводятся в таблице 1.




Соединения по изобретению могут так же образовывать фармацевтически приемлемые основно-аддитивные соли. Подразумевается, что все указанные формы включены в объем настоящего изобретения.
Более конкретно изобретение относится к соединениям формулы I
где m равно 1-4,
n равно 0-4,
А означает алкил, арил, гетероарил, арилалкил, арилциклоалкил,
циклоалкилалкил или арилоксиалкил,
Е означает -СНОН- или -С(O)-,
Х означает -(CH2)2- или -СН=СН-,
Y означает -СН2-, -СН=СН-, арилен, гетероарилен, -О-, -S(O)p -, где р равно 0-2, или -NRa-, где Ra означает водород или алкил,
Z означает -СН2ОН, -СНО, тетразол-5-ил или -COORb, где Rb означает водород или алкил, а
R1, R2, R3, R4, R5, R6, R7, R8, R9 и R10 каждый независимо означает водород или алкил,
или к их фармацевтически приемлемой соли, сольвату, пролекарству, индивидуальному изомеру или рацемической или нерацемической смеси изомеров.
Более предпочтительно в вышеуказанных соединениях Е означает -СНОН-. Другой предпочтительный вариант настоящего изобретения включает соединения, где Z означает -COORb. В вышеуказанных соединениях Y означает -СН2- или в других предпочтительных соединениях Y означает -S(O)p-, где р равно 0, или Y означает арилен. В еще одних предпочтительных соединениях Х означает -СН=СН- или Х означает -(CH2)2-. В другом предпочтительном варианте настоящего изобретения в вышеуказанных соединениях А независимо выбирают из группы, включающей алкил, арил, гетероарил, арилалкил, арилциклоалкил, циклоалкилалкил и арилоксиалкил. Более предпочтительно А означает алкил, или А означает арил, или А означает гетероарил, или А означает арилалкил, или А означает арилциклоалкил, или А означает циклоалкилалкил, или А означает арилоксиалкил.
Предпочтительными соединениями по настоящему изобретению являются соединения формулы II
где Y означает -СН2- или -S-, а А, Х и Rb имеют значения, указанные выше.
Кроме того, предпочтительны соединения формулы III
где А, X, Y и Rb имеют значения, указанные выше. В этих соединениях Х предпочтительно означает -СН=СН-, а Y предпочтительно означает арилен. Предпочтительные соединения выбирают из группы, включающей следующие:
а) 4-{2-[(R)-2-((S)-(Е)-5-циклопропил-3-гидроксипент-1-енил)-6-оксопиперидин-1-ил]этилсульфанил}масляная кислота,
б) 4-(2-{2R-[3R-(4'-хлор-2'-метилбифенил-3-ил)-3-гидроксипропил]-6-оксопиперидин-1-ил}этилсульфанил)масляная кислота,
в) 7-{2R-[3S-гидрокси-4-(4-гидрокси-3-метилфенил)бут-1Е-енил]-6-оксопиперидин-1-ил}гептановая кислота,
г) 7-{2R-[3S-гидрокси-4-(3-метоксиметилфенил)бут-1Е-енил]-6-оксопиперидин-1-ил}гептановая кислота,
д) 7-{2R-[3S-гидрокси-4-(4-гидрокси-3-изопропилфенил)бут-1Е-енил]-6-оксопиперидин-1-ил}гептановая кислота,
е) 4-(2-{2R-[3-гидрокси-3-(1-фенилциклопропил)проп-1Е-енил]-6-оксопиперидин-1-ил}этилсульфанил)масляная кислота,
ж) 4-(2-{2R-[3R-3-гидрокси-3-(трифторметилфуран-2-ил)пропил]-6-оксопиперидин-1-ил}этилсульфанил)масляная кислота,
з) 4-(2-{2R-[3R-гидрокси-3-(1-фенилциклопропил)пропил]-6-оксопиперидин-1-ил}этилсульфанил)масляная кислота,
и) 4-(2-{2R-[3S-гидрокси-4-(3-метоксиметилфенил)бут-1Е-енил]-6-оксопиперидин-1-ил}этилсульфанил)масляная кислота,
к) 7-{2R-[3R-(4'-гидрокси-2'-метилбифенил-3-ил)-3-гидроксипропил]-6-оксопиперидин-1-ил}гептановая кислота,
л) 7-{2R-[3-гидрокси-3-(4'-гидрокси-2'-метилбифенил-3-ил)проп-1Е-енил]-6-оксопиперидин-1-ил}гептановая кислота,
м) 7-(2-{2R-[3R-(4'-гидрокси-2'-метилбифенил-3-ил)-3-оксопропил]-6-оксопиперидин-1-ил}гептановая кислота,
н) 4-{2-[2R-(5-циклобутил-3S-гидроксипент-1Е-енил)-6-оксопиперидин-1-ил]этилсульфанил}масляная кислота,
о) 4-(2-{2R-[3R-(3'-фторфеноксифенил-3-ил)-3-гидроксипропил]-6-оксопиперидин-1-ил}этилсульфанил)-3-метилмасляная кислота,
п) 4-{2-[2R-(3-гидрокси-4, 4-диметилокт-1Е-енил)-6-оксопиперидин-1-ил]этилсульфанил}масляная кислота,
р) 7-{2R-[3-гидрокси-3-(2,5-диметилфенил-3-ил)проп-1Е-енил]-6-оксопиперидин-1-ил}гептановая кислота,
с) 7-[2R-(3-гидрокси-4-феноксибут-1Е-енил)-6-оксопиперидин-1-ил]гептановая кислота,
т) 7-{2R-[3-гидрокси-3-(3'-хлорбифенил-3-ил)проп-1Е-енил]-6-оксопиперидин-1-ил}гептановая кислота,
у) 7-{2R-[3R-(3'-хлорбифенил-3-ил)-3-гидроксипропил]-6-оксопиперидин-1-ил}гептановая кислота,
ф) 4-(2-{2-[3-гидрокси-4-(3-трифторметилфенил)бут-1Е-енил]-6-оксопиперидин-1-ил}этил)бензойная кислота,
х) 4-(2-{2-[3-гидрокси-4-(3-трифторметилфенил)бут-1Е-енил]-6-оксопиперидин-1-ил}этил)бензойная кислота и
ц) 3, 4-(2-{2-[3-(4'-хлор-2'-метилбифен-3-ил)-3-гидроксипропил]-6-оксопиперидин-1-ил}этил)бензойная кислота.
Изобретение также включает фармацевтическую композицию, включающую терапевтически эффективное количество соединения, указанного выше, в смеси по меньшей мере с одним пригодным носителем, разбавителем или эксципиентом. Кроме того, изобретение включает способ лечения заболевания у млекопитающего, излечиваемого введением селективного агониста простагландина ЕР4, причем указанный способ включает введение млекопитающему терапевтически эффективного количества соединения, указанного выше. Заболевание можно ассоциировать с нарушением костной ткани, например с остеопорозом. Соединения по настоящему изобретению можно использовать в терапии, например, для получения лекарственных средств, предназначенных, например, для лечения заболевания костной ткани, например, остеопороза. Изобретение также относится к способу получения описанных выше соединений по реакциям, показанным на схемах 1-3.
Синтез соединений по изобретению
Соединения по изобретению можно получить различными методами, показанными на схемах и описанными ниже.
Исходные материалы и реагенты, которые используются при получении указанных соединений, являются коммерческими препаратами, поставляемыми коммерческими фирмами, такими, как Aldrich Chemical Co, или их можно получить методами, известными специалисту в данной области, по методикам, описанным в литературе, такой, как Fieser and Fieser's Reagents for Organic Synthesis, Wiley & Sons, New York, т.1-15 (1991), Rodd's Chemistry of Carbon Compounds, Elsevier Science Publishers, т.1-5, и Supplementals (1989) и Organic Reactions, т.1-40, Wiley and Sons, New York (1991). Следующие схемы реакций лишь иллюстрируют некоторые методы синтеза соединений по настоящему изобретению, а различные модификации этих схем реакций могут быть разработаны и предложены специалистом в данной области со ссылкой на материалы настоящей заявки.
Исходные материалы и промежуточные соединения в указанных схемах реакций можно получить и при необходимости очистить соответствующими способами, включая, без ограничения перечисленным, фильтрование, перегонку, кристаллизацию, хроматографию и т.п. Такие материалы можно охарактеризовать соответствующими методами, включая физические константы и спектральные данные.
Если не указано иное, реакции, описанные в заявке, предпочтительно проводят в атмосфере инертного газа при нормальном (атмосферном) давлении и при температуре от приблизительно -78°С до приблизительно 150°С, более предпочтительно от приблизительно 0°С до приблизительно 125°С, наиболее предпочтительно и обычно при приблизительно комнатной температуре, например приблизительно при 20°С.
Ниже на схемах А, Б и В иллюстрируются методики получения соединений формулы I, где LG означает уходящую группу, PG означает защитную группу, R означает любой (низш.)алкил и в каждом случае может быть идентичным или различным, а m, n, А, X, Y, Z, R1, R2, R4, R6, R7, R8, R9 и R10 имеют значения, указанные в описании заявки. Конкретные примеры методики, показанной на схеме А, приводятся в разделе Примеры.
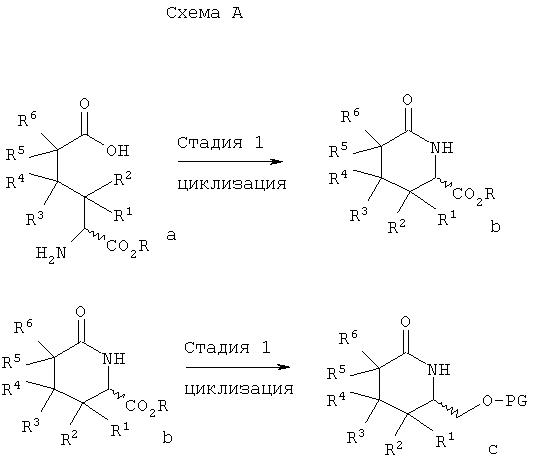
В схеме А на стадии 1 аминоадипиновую кислоту а циклизуют с образованием δ-лактама b. Используемые на стадии 1 различные аминоадипиновые кислоты, имеющие R- и S-конфигурацию, являются коммерческими препаратами или их можно получить известными способами. Циклизацию на этой стадии проводят, например, обычным нагреванием аминоадипиновой кислоты в уксусной кислоте или в другом полярном протонном растворителе, предпочтительно в кислотной среде.
Восстановление карбоксильной группы в δ-лактаме b проводят на стадии 2, а затем введением защитной группы получают защищенный спирт с. Восстановление на стадии 2 проводят, например, при обработке δ -лактама b борогидридом или цианоборогидридом щелочного металла в мягких условиях в полярном протонном растворителе. Затем в полученный спирт (на схеме не показан) вводят защитную группу, например, при обработке алкилвиниловым простым эфиром в присутствии трифторуксусной кислоты из спирта получают ацеталь. Защита гидроксильных групп превращением в ацеталь описана в статье S.Saijo и др., Chem. Pharm. Bull., 28, 1449-1458 (1980). Гидроксильную групп в соединении с можно также защитить силилированием, как описано в монографии Т.W.Green и Р.G.М.Futs, Protective Groups in Organic Chemistry, Wiley, 3 изд. (1999).
Получение соединений формулы I иллюстрируется ниже на схеме Б. Методику, показанную на схеме Б, можно использовать в вариантах изобретения, где Y означает О, S или NRa, или где Y может выступать в качестве нуклеофила.
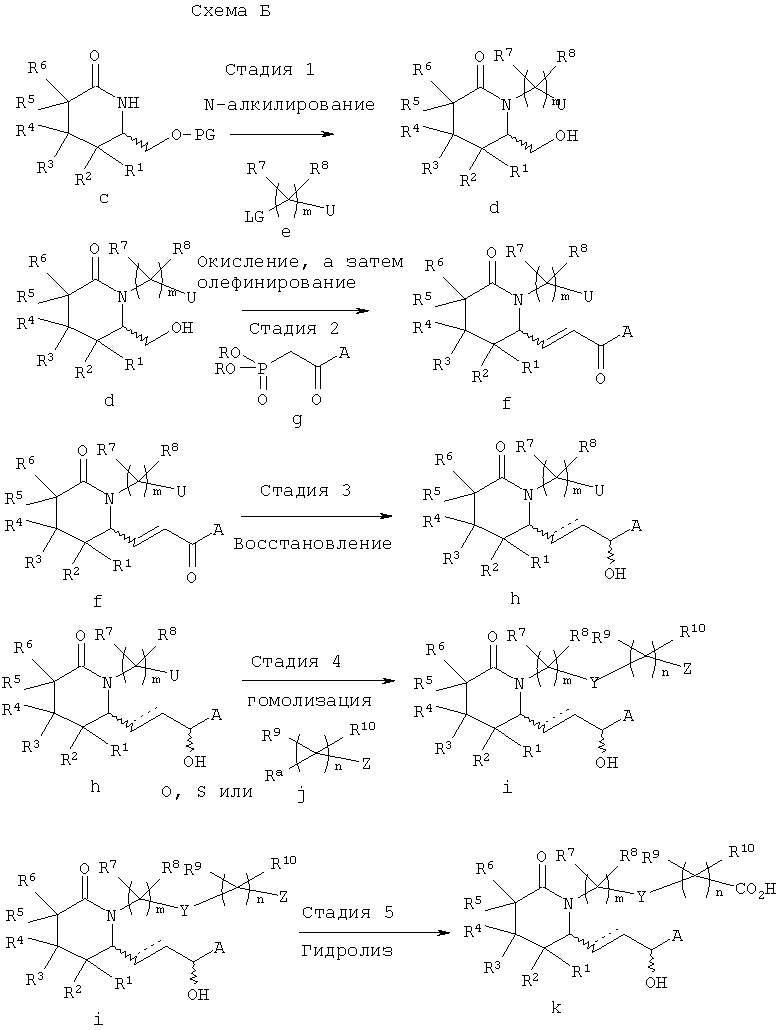
В схеме Б на стадии 1 проводят N-алкилирование азота лактамного цикла в защищенном спирте с, полученном, как показано на схеме А. Алкилирование проводят обработкой защищенного спирта с, основанием, таким, как гидрид натрия или гексаметилдисилазид калия при пониженной температуре в атмосфере инертного газа, при последующем взаимодействии с алкилирующим агентом е. Затем в полученном алкилированном продукте (на схеме не показан) при обработке кислотой удаляют защитную группу с образованием спирта d. Уходящая группа LG включает галоген, тозил или другую пригодную уходящую группу. На схеме Б группа U может означать защищенную гидроксигруппу (-O-PG) или группу R9R10CnZ.
На стадии 2 спирт е, полученный на стадии 1, окисляют с образованием альдегида (на схеме не показан) в мягких условиях при обработке комбинацией диметилсульфоксид/оксалилхлорид, периодинаном Десс-Мартина (Dess-Martin), комбинацией ТЕМРО/гипохлорит натрия, РСС, PDC или т.п. Альдегид немедленно вводят в реакцию с диалкиловым эфиром фосфоновой кислоты g в присутствии основания в полярном апротонном растворителе, при этом получают продукт конденсации - енон f.
На стадии 3 в продукте конденсации f, полученном на стадии 5, необязательно восстанавливают карбонильную группу и/или непредельную связь с образованием соединения h. Стереоселективное восстановление карбонильной группы в соединении f c использованием CBS-реагентов описано в статье Е.J.Corey и др., J. Am. Chem. Soc., 109, 7925-7926 (1987), или на этой стадии можно использовать другие стереоселективные восстанавливающие агенты. Если предпочтительно образование одного из диастереомеров, такого, как S-гидроксильный изомер формулы I, где А означает алкил, арилалкил, циклоалкилалкил или арилоксиалкил, то можно использовать стехиометрическую комбинацию литийалюминийгидрид/этанол-(S)-(-)-бинафтол, как описано в работе R.Noyori и др., J. Am. Chem. Soc., 106, 6717-6725 (1984), или, если необходим R-гидроксильный изомер, то используется комбинация каталитических количеств (R)-2-метил-CBS-оксазаборолидина со стехиометрическим количеством борандиметилсульфида, как описано в статье Е.J.Corey и др., J. Am. Chem. Soc., 109, 7925-7926 (1987), или стехиометрические количества (R)-3-пинанил-9-борабицикло[3.3.1]нонана, как описано в статье М.М.Midland и др., J. Am. Chem. Soc., 102, 867-869 (1980). 1,2-Восстановление можно проводить гидридом, таким, как борогидрид натрия, например, в растворителе, таком, как дихлорметан, толуол, этанол или тетрагидрофуран. Если А означает арил или гетероарил, то можно также использовать комбинацию соли лантанида, такой, как хлорид церия (III), с борогидридом натрия.
Насыщенную боковую цепь получают при каталитическом гидрировании двойной связи в присутствии никеля Ренея или палладия на угле.
На стадии 4 проводят гомологизацию соединения h, при которой замещают группу U по реакции с нуклеофильным соединением j с образованием соединения i. Если Z означает карбоксилат, то гидролиз соответствующей кислоты k необязательно проводят по известным методикам, таким, как добавление основания, такого, как гидроксид лития, натрия или калия, или добавление кислоты, такой, как серная кислота или соляная кислота, в протонном растворителе или в эфире, содержащем воду, или используют липазу типа VII в 0, 05 М фосфатном буферном растворе (рН 6,8), как описано в статье С.Luthy и др., J. Am. Chem. Soc., 100, 6211-6217 (1978).
Различные варианты методики показаны на схеме Б. Например, в некоторых вариантах изобретения δ-лактам с N-алкилируют на стадии 1, а затем из N-алкилированного лактама последовательно получают альдегид, алкилируют и селективно восстанавливают с образованием соединения формулы I. В другом варианте енон f вводят в реакцию с металлом или галогенидом магния общей формулы R11M, где R11 означает алкил, для введения дополнительной группы по атому углерода, связанному с группой А. Возможны и другие варианты синтеза, показанного на схеме Б, которые известны специалисту в данной области. При необходимости с использованием известных защитных групп можно вводить защитные группы A, Y и Z.
Другие схемы синтеза соединений по изобретению показаны ниже на схеме В, которые предпочтительны для получения соединений, где n равно 0, Y означает арил или гетероарил, а А, X, Z, R1, R2, R4, R6, R7, R8, R9 и R10 имеют значения, указанные в описании заявки.

На стадии 1 5-гексеновую кислоту m вводят в реакцию с амином I, затем полученный продукт дигидроксилируют по двойной связи с образованием амида n. 5-Гексеновые кислоты, используемые на стадии 1, являются коммерческими препаратами или их можно получить известными методами.
На стадии 2 амид n циклизуют с образованием спирта δ-лактама q. Циклизацию можно проводить после введения в соединение n защитной группы по первичной гидроксильной группе. Силильные или бензильные защитные группы вводят с использованием соответствующих реагентов, таких, как трет-бутилдиметилсилилтрифторметансульфонат или бензил-2,2,2-трихлорацетимидат (бензиловый эфир 2,2,2-трихлориминоуксусной кислоты) соответственно. Затем в соединении n активируют вторичную гидроксильную группу и вводят заместитель при обработке алкил- или арилсульфонилхлоридом, таким, как метансульфонилхлорид. Затем соответствующий втор-амидосульфонат (на схеме не показан) циклизуют при обработке основанием, таким, как гидрид щелочного металла или алкоксид в полярном растворителе, таком, как метанол, тетрагидрофуран или N,N-диметилформамид. Затем защитную эфирную группу (по первичной гидроксигруппе) удаляют, в случае силильного эфира обработкой фторидом (фторидом тетрабутиламмония), а в случае бензильного эфира каталитическим гидрированием (водородом в присутствии Pd/C). Затем из соединений q через промежуточные стадии 2-5 получают соединения формулы I. Если необходим R-энантиомер q, то используют асимметрическое дигидроксилирование по методике, описанной в статье М. Shipman и др., Synthesis, 1141-1144 (1998).
Способы введения и фармацевтические композиции
Настоящее изобретение включает фармацевтические композиции, содержащие по меньшей мере одно соединение по настоящему изобретению или его индивидуальный изомер, рацемическую или нерацемическую смесь изомеров или фармацевтически приемлемую соль или ее сольват, в смеси по меньшей мере с одним фармацевтически приемлемым носителем и необязательно с другими терапевтическими и/или профилактическими ингредиентами.
В общем случае соединения по настоящему изобретению вводят в терапевтически эффективном количестве любыми приемлемыми способами введения, принятыми для агентов аналогичного назначения. Пригодные дозы обычно составляют 0,001-50 мг в сутки, предпочтительно 0,005-10 мг в сутки и наиболее предпочтительно 0,010-1,0 мг в сутки, в зависимости от множества факторов, таких, как тяжесть излечиваемого заболевания, возраст и относительное состояние здоровья субъекта, активность используемого соединения, способ и форма введения, показания, на основании которых назначается введение лекарственного средства, опыт и квалификация лечащего врача. Специалист по указанным заболеваниям без особых экспериментов, на основании своего опыта и описания настоящего изобретения, сможет определить терапевтически эффективное количество соединений по настоящему изобретению, необходимое для данного заболевания.
В общем случае соединения по настоящему изобретению вводят в виде фармацевтических композиций, включающих композиции, пригодные для перорального (включая трансбуккальный и сублингвальный (подъязычный)), ректального, назального, местного, легочного, вагинального или парентерального (включая внутримышечный, внутриартериальный, подоболочечный, подкожный и внутривенный) способов введения или в форме, пригодной для введения ингаляционным способом или вдуванием. Предпочтительным способом введения обычно является пероральный способ при соответствующей суточной схеме приема лекарственного средства, которая назначается в соответствии с тяжестью заболевания.
Соединение или соединения по настоящему изобретению в смеси с одним или более соответствующих адъювантов, носителей или разбавителей можно изготовлять в форме фармацевтических композиций и стандартных доз. Фармацевтические композиции и стандартные дозы могут включать соответствующие ингредиенты в соответствующих пропорциях, могут содержать или не содержать дополнительные активные соединения или составные части, а стандартные дозы могут содержать любое приемлемое эффективное количество активного ингредиента в соответствии с назначенной суточной схемой применения лекарственного средства. Фармацевтические композиции могут использоваться в виде твердых форм, таких, как таблетки или заполненные капсулы, в виде полутвердых, порошкообразных составов пролонгированного действия, или жидкостей, таких, как растворы, суспензии, эмульсии, эликсиры, или заполненных капсул для перорального введения, или в форме суппозиториев для ректального или вагинального введения, или в форме стерильных инъекционных растворов для парентерального введения. Для типичных стандартных доз пригодны составы, содержащие в таблетке приблизительно 1 мг активного ингредиента или в более общем виде от приблизительно 0,01 до приблизительно 100 мг.
Соединения по настоящему изобретению можно изготовить в виде множества форм для перорального введения. Фармацевтические композиции и дозировки могут включать в качестве активного компонента соединение или соединения по настоящему изобретению или их фармацевтически приемлемые соли. Фармацевтически приемлемые носители могут иметь твердую или жидкую консистенцию. Твердые формы препаратов включают порошки, таблетки, пилюли, капсулы, крахмальные облатки, суппозитории и диспергируемые гранулы. Твердый носитель может представлять собой одно или более веществ, которые могут действовать как разбавители, ароматизаторы, солюбилизаторы, замасливатели, суспендирующие агенты, связующие агенты, консерванты, дезинтегрирующие агенты или инкапсулирующий материал. При изготовлении в виде порошков носитель обычно представляет собой тонко измельченное твердое вещество, которое находится в смеси с тонко измельченным активным компонентом. При изготовлении таблеток активный компонент обычно смешивают с носителем, содержащим связующий компонент в соответствующей пропорции, и прессуют в таблетки необходимой формы и размера. Порошки и таблетки предпочтительно содержат от приблизительно 1 до приблизительно 70% активного соединения. Пригодные носители включают, без ограничения перечисленным, карбонат магния, стеарат магния, тальк, сахарозу, лактозу, пектин, декстрин, крахмал, желатин, трагакант, метилцеллюлозу, натриевую соль карбоксиметилцеллюлозы, низкоплавкий воск, масло какао и т.п. Подразумевается, что термин «препарат» включает композицию активного соединения с инкапсулирующим материалом-носителем, при условии, что капсула содержит активный компонент в смеси с носителями или без них, причем активный компонент окружен носителем, который контактирует с активным компонентом. Аналогичное определение относится к облаткам и лепешкам. Таблетки, порошки, капсулы, пилюли, облатки и лепешки представляют собой твердые формы, пригодные для перорального введения.
Другие формы, пригодные для перорального введения, включают жидкие формы препаратов, включающие эмульсии, сиропы, эликсиры, водные растворы, водные суспензии или твердые формы препаратов, которые непосредственно перед применением переводят в жидкую форму. Эмульсии можно получать в растворах, например в водных растворах пропиленгликоля, или они могут содержать эмульгирующие агенты, например, такие, как лецитин, моноолеат сорбита или аравийская камедь. Водные растворы получают, растворяя активный компонент в воде и добавляя соответствующие красители, ароматизаторы, стабилизаторы и загустители. Водные суспензии готовят диспергированием тонко измельченного активного компонента в воде, содержащей вязкий материал, такой, как природные или синтетические камеди, смолы, метилцеллюлоза, натриевая соль карбоксиметилцеллюлозы и прочие известные суспендирующие агенты. Жидкие формы препаратов включают растворы, суспензии и эмульсии и могут содержать, наряду с активным компонентом, красители, ароматизаторы, стабилизирующие агенты, буферные вещества, искусственные и природные подсластители, диспергирующие агенты, загустители, солюбилизирующие агенты и т.п.
Соединения по настоящему изобретению можно получать в форме для парентерального введения (например, инъекцией, например инъекцией струйным вливанием или непрерывным вливанием) и можно поставлять в форме стандартной дозы в ампулах, одноразовых шприцах, небольших контейнерах для вливания или контейнерах с несколькими дозами, содержащих консервант.
Композиции могут иметь форму суспензий, растворов или эмульсий в масляном или водном связующем, например растворов в водном полиэтиленгликоле. Примеры масляных или неводных носителей, разбавителей, растворов или связующих включают пропиленгликоль, полиэтиленгликоль, растительные масла (например, оливковое масло) и сложные эфиры органических кислот, пригодные для инъекций, (например, этилолеат), и могут содержать дополнительные компоненты, такие, как консервирующие, смачивающие, эмульгирующие или суспендирующие, стабилизирующие и/или диспергирующие агенты. В другом варианте активный ингредиент может находиться в порошкообразной форме, полученной выделением стерильного твердого вещества в асептических условиях или лиофилизацией раствора, для смешивания перед употреблением с соответствующим носителем, например стерильной, апирогенной водой.
Соединения по настоящему изобретению можно получать для местного нанесения на эпидермис в виде мазей, кремов или лосьонов или в виде чрескожного пластыря. Например, мази и кремы получают на водной или масляной основе при добавлении пригодных загустителей и/или гелеобразующих агентов. Лосьоны получают на водной или на масляной основе, кроме того, обычно содержащей один или более эмульгирующих агентов, стабилизирующих агентов, диспергирующих агентов, суспендирующих агентов, загустителей или красителей. Составы, пригодные для местного введения в ротовую полость, представляют собой лепешки, включающие активные ингредиенты в смеси с ароматизированной основой, обычно с сахарозой и аравийской камедью или трагакантом; пастилки, включающие активный ингредиент на инертной основе, такой, как желатин и глицерин или сахароза и аравийская камедь, и раствор для полоскания, включающий активный ингредиент в соответствующем жидком носителе.
Соединения по настоящему изобретению можно получать в форме суппозиториев. Низкоплавкий воск, такой, как смесь глицеридов жирных кислот или масло какао, сначала расплавляют, а затем в нем равномерно диспергируют активный компонент, например, перемешиванием. Расплавленную гомогенную смесь выливают в формы соответствующего размера и охлаждают для приобретения твердой консистенции.
Соединения по настоящему изобретению можно получать для вагинального введения. Пригодными являются вагинальные суппозитории, тампоны, кремы, гели, пасты, пены и спреи, содержащие наряду с активным ингредиентом носители, известные в данной области техники.
Соединения по настоящему изобретению можно получать для назального введения. Растворы или суспензии вводят непосредственно в носовую полость обычными способами, например, капельницей, пипеткой или спреем. Составы могут поставляться в форме, содержащий одну или несколько доз. В случае капельницы или пипетки пациенту вводят определенный объем раствора или суспензии. Аэрозоли вводят дозирующим пульверизатором.
Соединения по настоящему изобретению можно получать для аэрозольного введения, прежде всего в дыхательные пути, включая интраназальное введение. Обычно частицы соединения имеют небольшие размеры, например приблизительно 5 мкм или менее. Такой размер частиц получают известными методами, например измельчением на микронной коллоидной мельнице (микронизацией). Активный ингредиент получают в герметичной упаковке с соответствующим газом-вытеснителем, таким, как хлорфторуглерод (ХФУ), например, дихлордифторметан, трихлорфторметан или дихлортетрафторэтан или диоксид углерода или другой приемлемый газ. Аэрозоль может также содержать ПАВ, такое, как лецитин. Доза лекарственного средства регулируется дозирующим клапаном. В другом варианте активные ингредиенты готовят в форме сухого порошка, например порошкообразной смеси соединения с соответствующей порошкообразной основой, такой, как лактоза, крахмал, производные крахмала, такие, как гидроксипропилметилцеллюлоза, и поливинилпирролидон (ПВП). В полости носа порошкообразный носитель образует гель. Порошкообразную композицию можно изготовить в виде стандартной дозы, например, в капсулах или картриджах, например, в желатиновой или блистерной упаковке, из которой порошок можно вводить ингалятором.
При необходимости составы можно получать с энтеросолюбильным покрытием для введения с замедленным или регулируемым высвобождением активного ингредиента. Например, соединения по настоящему изобретению можно получать в виде систем для чрескожной или подкожной доставки. Эти системы обладают преимуществом в тех случаях, когда необходимо замедленное высвобождение соединения, или если решающим фактором является согласие пациента с назначенным курсом лечения. В чрескожных системах доставки соединение часто фиксируют на твердой подложке, которую наклеивают на кожу. Указанное соединение может также находиться в комбинации с усилителем всасывания, например азоном (1-додецилазациклогептан-2-он). Системы замедленного высвобождения вводятся подкожно в субдермальный слой хирургом или с использованием инъекции. В подкожных имплантатах соединение включено в липидную растворимую мембрану, например в силиконовый каучук, или биодеградируемый полимер, например полимолочную кислоту.
Фармацевтические препараты предпочтительно представляют собой стандартные лекарственные формы. В такой форме препарат разделен на стандартные дозы, содержащие соответствующие количества активного компонента. Стандартная форма может представлять собой упакованный препарат, причем упаковка содержит дискретные количества препарата, такие, как упакованные таблетки, капсулы, и порошки во флаконах или ампулах. Кроме того, стандартная форма может представлять собой капсулу, таблетку, облатку или лепешку или может представлять собой упаковку, содержащую определенное количество любой из указанных форм.
Прочие пригодные фармацевтические носители и составы по изобретению описаны в примере 4.
Примеры
Следующие препараты и примеры приведены с целью предоставить возможность специалисту в данной области более подробно ознакомится с предлагаемым изобретением и использовать его на практике. Примеры приведены с целью иллюстрации и не ограничивают объема изобретения.
Методика 1
Диметиловый эфир (4-циклопропил-2-оксобутил)фосфоновой кислоты
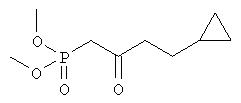
К суспензии гидрида натрия (95%, 760 мг, 31,6 ммоля, фирма Aldrich) в безводном тетрагидрофуране (90 мл) при комнатной температуре в течение 40 мин добавляли диметиловый эфир (2-оксопропил)фосфоновой кислоты (5,0 г, 30,6 ммоля, фирма Sigma) в тетрагидрофуране (10 мл) и смесь охлаждали до 0°С. К суспензии добавляли н-бутиллитий (2,5 М раствор в гексане, 13,2 мл, 33 ммоля) и полученный раствор перемешивали при 0°С в течение 2 ч. Затем к смеси добавляли бромциклопропан (3,1 мл, 32 ммоля) в тетрагидрофуране (10 мл), перемешивали при 0°С в течение 1,5 ч и при комнатной температуре в течение 1 ч, а затем добавляли этанол (2 мл). Смесь распределяли между 0,3 М HCl (150 мл) и дихлорметаном (2×100 мл). Объединенные органические экстракты промывали солевым раствором и сушили над безводным сульфатом натрия. Продукт очищали хроматографией на силикагеле (элюент: этилацетат/гексан, 2:1), при этом получали диметиловый эфир (4-циклопропил-2-оксобутил)фосфоновой кислоты в виде масла (2,11 г).
1H-ЯМР (300 МГц, CDCl3): δ 3,74 (d, J 11,4 Гц, 6Н), 3,06 (d, J 22,8 Гц, 2Н), 2,67 (t, J 7,2 Гц, 2Н), 1,43 (q, J 7,2 Гц, 2Н), 0,61-0,72 (m, 1H), 0,35-0,42 (m, 2H), 0,03-0,08 (m, 2H).
13С-ЯМР (75 МГц, CDCl3): 202,0 (d, J 6,0 Гц), 53,1 (d, J 6,3 Гц), 44,2, 41,4 (d, J 128 Гц), 28,6, 10,3, 4,5.
Методика 2
Диметиловый эфир {2-[3-('3-фторфенокси)фенил]-2-оксоэтил}фосфоновой кислоты

Стадия 1
Метиловый эфир 3-(3-фторфенокси)бензойной кислоты
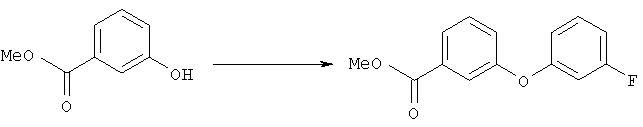
Суспензию метилового эфира 3-гидроксибензойной кислоты (5,4 г, 35,5 ммоля), 3-фторфенилбороновой кислоты (5,5 г, 35,5 ммоля), ацетата меди (7,1 г, 35,5 ммоля), молекулярных сит 3Å (9 г), пиридина (12 мл, 145 ммолей) в дихлорметане (220 мл) перемешивали при комнатной температуре при нормальном давлении в течение 11 сут. Затем смесь фильтровали через слой целита® и фильтрат упаривали. Продукт очищали хроматографией на колонке с силикагелем (элюент: гексан/этилацетат, 5:1), при этом получали метиловый эфир 3-(3-фторфенокси)бензойной кислоты (3,68 г), который использовали на следующей стадии.
Стадия 2
Диметиловый эфир {2-[3-(3-фторфенокси)фенил]-2-оксоэтил}фосфоновой кислоты
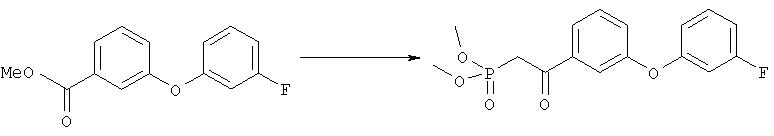
Раствор диметилового эфира метилфосфоновой кислоты (4,0 мл, 37,5 ммоля) в тетрагидрофуране (100 мл) охлаждали до -78°С в атмосфере аргона, добавляли н-бутиллитий (15,0 мл, 2,5 М раствор в гексане, 37,5 ммоля) и перемешивали в течение 45 мин. Эфир, полученный на стадии 1 (4,62 г, 18,7 ммоля), растворяли в тетрагидрофуране (15 мл), добавляли к вышеуказанному раствору при -78°С и полученную смесь перемешивали при 0°С в течение 1 ч. Затем раствор желтого цвета распределяли между водным раствором хлорида аммония (100 мл) и этиловым эфиром (200 мл). Органический слой промывали водой (3×30 мл), солевым раствором, сушили над безводным сульфатом натрия, фильтровали и упаривали в вакууме, при этом получали требуемый кетофосфонат (5,8 г) в виде вязкого масла.
1H-ЯМР (300 МГц, CDCl3): δ 7,78 (dt, J 0,6, 0,9, 7,8 Гц, 1Н), 7,63 (t, J 2, 1 Гц, 1 Н), 7,48 (t, J 8,1 Гц, 1Н), 7,32-7,26 (m, 2Н), 6,90-6,78 (m, 2H), 6,70 (dt, J 2,4, 9,9 Гц, 1H), 3,80 (d, J 11,2 Гц, 6Н), 3,61 (d, J 22,6 Гц, 2H).
Методика 3
Метиловый эфир 4-[(2-хлорэтил)тио]масляной кислоты

К раствору 4-меркаптомасляной кислоты (3,85 г, 20 ммолей) в изопропаноле (70 мл) при 0°С в течение 20 мин четырьмя порциями добавляли гидрид натрия (95%, всего 1,56 г, 65 ммолей) и смесь нагревали до комнатной температуры. Затем к смеси быстро добавляли 1-бром-2-хлорэтан (11 мл, 128 ммолей) и полученную суспензию интенсивно перемешивали в течение 2 суток. Затем смесь упаривали и остаток распределяли между 5% уксусной кислотой и этилацетатом. Объединенные органические экстракты промывали солевым раствором, сушили над сульфатом натрия, фильтровали и упаривали в вакууме. Остаток растворяли в метаноле (60 мл) и охлаждали до 0°С в атмосфере аргона. К раствору добавляли по каплям тионилхлорид (5 мл, 69 ммолей) и перемешивали при комнатной температуре в течение 2-3 ч. Летучие компоненты упаривали, к остатку добавляли толуол и упаривали вторично. После очистки продукта хроматографией получали метиловый эфир 4-[(2-хлорэтил)тио]масляной кислоты (2,93 г, 14 ммолей) в виде бесцветного масла. МС (NH3): m/z 199 (M+1++37Cl), 197 (М+1+35Cl).
Пример 1
6R-(1-Этоксиэтоксиметил)пиперидин-2-он

Стадия 1
Метиловый эфир 6-оксопиперидин-2R-карбоновой кислоты

Раствор R-2-аминоадипиновой кислоты (5 г, 31 ммоль, фирма Sigma Chemical Co.) в уксусной кислоте (30 мл) кипятили с обратным холодильником в течение 6 ч. Смесь охлаждали, летучие компоненты удаляли на роторном испарителе, а затем перегонкой азеотропной смеси с толуолом (2×25 мл). Остаток растворяли в метаноле (15 мл) и дихлорметане (30 мл) при комнатной температуре. К раствору добавляли (триметилсилил)диазометан (30 мл, 2 М раствор в гексане, фирма Aldrich) и раствор золотистого цвета перемешивали в течение 4 ч. К раствору добавляли по каплям уксусную кислоту до исчезновения золотистого цвета и летучие компоненты удаляли на роторном испарителе. Остаток наносили на слой силикагеля и промывали дихлометаном/этилацетатом, 1:1, продукт элюировали этилацетатом. Метиловый эфир 6-оксопиперидин-2R-карбоновой кислоты получали в виде бесцветного масла (4,3 г). [α]D +12,0° (с 1,0, CHCl3) и спектр1Н-ЯМР соответствуют литературным данным (см. С.E.Davies и др., Synthetic Commun., 26, 687-696 (1996).
Стадия 2
6R-(1-Этоксиэтоксиметил)пиперидин-2-он

Раствор метилового эфира 6-оксопиперидин-2R-карбоновой кислоты (13,7 г, 87 ммолей) в этаноле (400 мл) охлаждали на бане с проточной водой в атмосфере аргона и в течение 15 мин тремя равными порциями добавляли борогидрид натрия (всего 4,2 г, 109 ммолей). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч, а затем добавляли уксусную кислоту до рН 4. Летучие компоненты удаляли на роторном испарителе, остаток наносили на слой силикагеля. Продукт элюировали 5% метанолом в дихлорметане, при этом получали 6R-гидроксиметилпиперидин-2-он в виде масла (6, 3 г, 49 ммолей), к которому немедленно добавляли этилвиниловый эфир (7,0 мл, 73 ммоля) и трифторуксусную кислоту (1 мл) в дихлорметане (200 мл) и смесь выдерживали при комнатной температуре в течение 2 ч. Затем к раствору добавляли раствор бикарбоната натрия, экстрагировали дихлорметаном (2×50 мл), сушили над безводным сульфатом натрия, фильтровали и упаривали в вакууме, при этом получали 6R-(1-этоксиэтоксиметил)пиперидин-2-он в виде масла (6,9 г, 29 ммолей).
1Н-ЯМР (300 МГц, CDCl3): δ 6,14-6,24 (ушир. s, 1H), 4,67-4,76 (m, 1H), 3,18-3,67 (m, 5H), 2,23-2,47 (m, 3Н), 1,64-1,98 (m, 3Н), 1,31 (dd, 3Н), 1,20 (dt, 3H).
Пример 2
4-{2-[(R)-2-((S)-(Е)-5-Циклопропил-3-гидроксипент-1-енил)-6-оксопиперидин-1-ил]этилсульфанил}масляная кислота

Стадия 1
6R-Гидроксиметил-1-(2-триизопропилсиланилоксиэтил)пиперидин-2-он
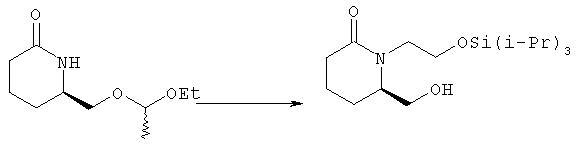
Раствор 6R-(1-этоксиэтоксиметил)пиперидин-2-она (пример 1) (6,9 г, 34 ммоля) и иодида калия (5,7 г, 34 ммоля) в диметилформамиде (70 мл) охлаждали до 0°С в атмосфере аргона, одной порцией добавляли гидрид натрия (95%, 910 мг, 36 ммолей) и выделяющую пузырьки смесь нагревали до комнатной температуры. Через 1,5 ч к смеси добавляли триизопропилсилиловый эфир 2-бромэтанола (10,5 г, 37,2 ммоля) в диметилформамиде (15 мл) и смесь нагревали при 50°С в течение 40 ч. Летучие компоненты удаляли перегонкой с коротким холодильником (5 мм рт. ст., температура смеси 75°С) и кубовый остаток распределяли между водой (100 мл) и гексаном/этилацетатом (1:1, 4×100 мл). Объединенные органические экстракты промывали водой (2×25 мл), солевым раствором, сушили над безводным сульфатом натрия, фильтровали и упаривали на роторном испарителе, при этом получали масло желтовато-коричневого цвета (15,3 г), к которому немедленно добавляли этанол (150 мл), пара-толуолсульфонат пиридиния (800 мг, 3,2 ммоля) и смесь кипятили с обратным холодильником в течение 60 мин. Затем раствор охлаждали, добавляли 5% раствор бикарбоната натрия (20 мл) и летучие компоненты удаляли на роторном испарителе. Остаток наносили на слой силикагеля и промывали этилацетатом/гексаном, 2:1. Продукт элюировали этилацетатом, при этом получали 6R-гидроксиметил-1 -(2-триизопропилсиланилоксиэтил)пиперидин-2-он (5,22 г, 15,8 ммоля). [α]D - 37,5° (с 1,0, CH3CN). МС (ES): m/z 330 (M+1 )+.
ИК (см-1): 3373, 2943, 2865, 1617, 1465.
1Н-ЯМР (300 МГц, CDCl3): δ 4,11-4,20 (m, 1H), 3,61-3,90 (m, 5H), 3,42-3, 53 (m, 2Н), 2,36-2,41 (m, 2H), 1,63-1,99 (m, 4H), 1,07 (s, 21H).
13С-ЯМР (75 МГц, CDCl3): 172,1, 64,8, 62,7, 61,0, 50,1, 32,8, 26,4, 18,7, 18,3, 12,2.
Стадия 2
6R-(5-Циклопропил-3-оксопент-1Е-енил)-1-(2-триизопропилсиланилоксиэтил)пиперидин-2-он
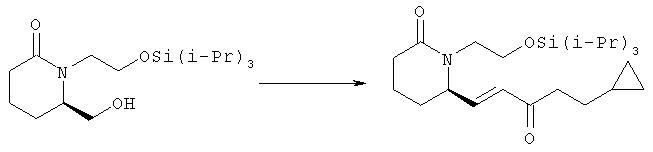
К раствору безводного диметилсульфоксида (0,87 мл, 11,2 ммоля) в дихлорметане при -78°С в атмосфере аргона в течение 3 мин добавляли оксалилхлорид (2 М раствор в СН2Cl2, 3,9 мл, 7,8 ммоля). Через 20 мин к смеси добавляли по каплям раствор 6R-гидроксиметил-1-(2-триизопропилсиланилоксиэтил)пиперидин-2-она (1,85 г, 5,6 ммоля, полученного на стадии 1) в дихлорметане (10 мл), полученный раствор желтого цвета перемешивали при -78°С в течение 15 мин, а затем быстро добавляли триэтиламин (2,3 мл, 16,8 ммоля) и удаляли охлаждающую баню. Через 30 мин суспензию выливали в раствор бикарбоната и экстрагировали дихлорметаном (3×50 мл). Объединенные органические экстракты сушили над безводным сульфатом натрия, фильтровали и упаривали, при этом получали альдегид в виде масла желтовато-коричневого цвета (1,75 г), который использовали на следующей стадии.
Неочищенный альдегид (875 мг, 2,65 ммоля) растворяли в ацетонитриле (25 мл), добавляли диметиловый эфир (4-циклопропил-2-оксобутил)фосфоновой кислоты (670 мг, 3,05 ммоля), хлорид лития (135 мг, 3,2 ммоля) и диизопропилэтиламин (0,51 мл, 2,9 ммоля) и полученную суспензию перемешивали при комнатной температуре в течение 2,5 ч. Затем смесь выливали в эфир и раствор хлорида аммония, водный слой экстрагировали этилацетатом (4×30 мл), сушили над безводным сульфатом натрия, фильтровали и упаривали в вакууме. Остаток очищали хроматографией на силикагеле, при этом получали 6R-(5-циклопропил-3-оксопент-1Е-енил)-1-(2-триизопропилсиланилоксиэтил)пиперидин-2-он (793 мг, 1,85 ммоля) в виде масла.
1H-ЯМР (300 МГц, CDCl3): δ 6,70 (dd, J 5,5, 15,8 Гц, 1Н), 6,11 (dd, J 0,6, 15,8 Гц, 1H), 4,52-4,59 (m, 1H), 4,06-4,14 (m, 1H), 3,95 (dt, J 3,6, 9,3 Гц, 1Н), 3,71-3,80 (m, 1H), 2,70-2,79 (m, 1H), 2,66 (t, J 6,9 Гц, 1H), 2,37-2,44 (m, 1H), 1,96-2,05 (m, 1H), 1,49-1,88 (m, 6H), 1,05 (s, 22H), 0,64-0,73 (m, 1H), 0,39-0,44 (m, 2Н), 0,02-0,07 (m, 2H).
Стадия 3
6R-(5-Циклопропил-3S-гидроксипент-1Е-енил)-1-(2-гидроксиэтил)пиперидин-2-он

(R)-2-Метил-CBS-оксазаборолидин (0,20 мл, 1,3 ммоля, 5 М раствор в этиловом эфире) при 0°С в атмосфере аргона разбавляли толуолом (10 мл). К полученному раствору добавляли по каплям раствор 6R-(5-циклопропил-3-оксопент-1Е-енил)-1-(2-триизопропилсиланилоксиэтил)пиперидин-2-она (793 мг, 1,85 ммоля) в безводном толуоле (5 мл) и смесь перемешивали при 0°С в течение 20 мин. Затем реакцию останавливали добавлением HCl (1,5 мл, 2 М раствор в метаноле) и летучие компоненты удаляли в вакууме, при этом получали остаток в виде твердого вещества. Остаток повторно растворяли в метаноле и раствор упаривали в вакууме. Остаток очищали хроматографией на колонке с силикагелем (элюент: градиент изопропанола от 2 до 6% в гексане/этилацетате, 3:1), при этом получали 6R-(5-циклопропил-3S-гидроксипент-1Е-енил)-1-(2-триизопропилсиланилоксиэтил)пиперидин-2-он (541 мг), который использовали на следующей стадии.
6R-(5-Циклопропил-3S-гидроксипент-1Е-енил)-1-(2-триизопропилсиланилоксиэтил)пиперидин-2-он (541 мг, 1,26 ммоля) растворяли в ТГФ (10 мл), добавляли гидрат фторида тетрабутиламмония (480 мг, 1,5 ммоля) и полученный раствор перемешивали при комнатной температуре в течение 2,5 ч. Затем к смеси добавляли 10 мл гексана и наносили на слой силикагеля. Продукт элюировали 10% этанолом в этилацетате, при этом получали 6R-(5-циклопропил-3S-гидроксипент-1Е-енил)-1-(2-гидроксиэтил)пиперидин-2-он (328 мг, 1,23 ммоля) в виде масла. МС: m/z 268 (M+1)+, 250 (М+1-Н2 O)+.
1H-ЯМР (300 МГц, CDCl3/D2O, частичный спектр): δ 5,64-5,59 (m, 2H), 4,22-4,18 (m, 1H), 4,03-3,98 (m, 1H), 3,80-3,69 (m, 3Н), 3, 31-3,20 (m, 1H), 0,72-0,61 (m, 1H), 0,48-0,40 (m, 2H), 0,08-0,00 (m, 2H).
Стадия 4
Метиловый эфир 4-{2-[(R)-2-((S)-(Е)-5-циклопропил-3-гидроксипент-1-енил)-6-оксопиперидин-1-ил]этилсульфанил}масляной кислоты

6R-(5-Циклопропил-3S-гидроксипент-1Е-енил)-1-(2-гидроксиэтил)пиперидин-2-он (328 мг, 1,23 ммоля, полученный на стадии 3) растворяли в тетрагидрофуране (10 мл) и охлаждали до -20°С в атмосфере аргона. К полученному раствору последовательно добавляли триэтиламин (0,21 мл, 1,48 ммоля) и метансульфонилхлорид (0,095 мл, 1,23 ммоля), при этом получали суспензию. В отдельном сосуде к смеси безводного метанола (1 мл) и безводного тетрагидрофурана (5 мл) в атмосфере аргона добавляли трет-бутоксид калия (3,7 мл, 1 М раствор в тетрагидрофуране, фирма Aldrich) и теплый раствор перемешивали в течение 10 мин. Затем к раствору одной порцией добавляли тиобутиролактон (0,26 мл, 3,1 ммоля, фирма Aldrich Chemical Со.) и смесь перемешивали при комнатной температуре в течение 10 мин. К полученному раствору тиолята калия через трубочку добавляли суспензию мезилата и смесь перемешивали при комнатной температуре в течение 18 ч. Затем смесь распределяли между раствором хлорида аммония и этилацетатом (4×25 мл). Объединенные органические экстракты сушили над безводным сульфатом натрия и упаривали на роторном испарителе. Остаток очищали хроматографией на силикагеле (элюент: этилацетат/гексан, 4:1), при этом получали метиловый эфир 4-{2-[(R)-2-((S)-(Е)-5-циклопропил-3-гидроксипент-1-енил)-6-оксопиперидин-1-ил]этилсульфанил} масляной кислоты (98 мг, 0,25 ммоля) в виде масла.
1Н-ЯМР (300 МГц, CDCl3): δ 5,58-5,64 (m, 2H), 4,16-4,22 (m, 1H), 4,02-4,07 (m, 1H), 3,87-3,98 (m, 1H), 3, 68 (s, 3H), 2,95-3,04 (m, 1H), 2,63-2,76 (m, 2H), 2,58 (t, J 7,2 Гц, 2H), 2,44 (t, J 7,2 Гц, 2H), 2,34-2,41 (m, 2H), 1,58-1,99 (m, 9H), 1,22-1,30 (m, 2H), 0,61-0,72 (m, 1H), 0,40-0,47 (m, 2H), 0,02-0, 07 (m, 2H).
Стадия 5
4-{2-[(R)-2-((S)-(Е)-5-Циклопропил-3-гидроксипент-1-енил)-6-оксопиперидин-1-ил]этилсульфанил}масляная кислота

К раствору метилового эфира 4-{2-[(R)-2-((S)-(Е)-5-циклопропил-3-гидроксипент-1-енил)-6-оксопиперидин-1-ил]этилсульфанил} масляной кислоты (4,98 мг, 0,25 ммоля, полученного на стадии 4) в метаноле (10 мл) добавляли гидроксид натрия (0,3 мл, 5 М раствор в воде) и смесь перемешивали при комнатной температуре в течение 3 ч. Летучие компоненты удаляли в атмосфере азота и смесь распределяли между водой и этиловым эфиром. Водный слой подкисляли добавлением соляной кислоты (12 М раствор) и экстрагировали этилацетатом (3×15 мл). Объединенные органические экстракты сушили над безводным сульфатом натрия, фильтровали и упаривали, при этом получали 4-{2-[(R)-2-((S)-(Е)-5-циклопропил-3-гидроксипент-1-енил)-6-оксопиперидин-1-ил]этилсульфанил} масляную кислоту в виде масла. МС: m/z 370 (М+1).
1H-ЯМР (300 МГц, CDCl3): δ 6,7 (ушир. s, 1H), 5,58-5,63 (m, 2H), 4,18-4,22 (m, 1H), 4,02-4,07 (m, 1H), 3,87-3,98 (m, 1H), 2,98-3,08 (m, 1H), 2,62-2,72 (m, 2H), 2, 59 (t, J 6,9 Гц, 2 Н), 2,36-2,49 (m, 4Н), 1,61-1,99 (m, 8H), 1,22-1,30 (m, 2H), 0,59-0,71 (m, 1H), 0,40-0,46 (m, 2H), 0,00-0,06 (m, 2H).
Следующие соединения формулы I получали по методикам, описанным в примере 2, при внесении следующих изменений.
4-(2-{2R-[3R-(4'-Хлор-2'-метилбифенил-3-ил)-3-гидроксипропил]-6-оксопиперидин-1-ил}этилсульфанил)масляную кислоту получали при использовании на стадии 2 диметилового эфира [2-(4'-хлор-2'-метилбифен -3-ил)-2-оксоэтил]фосфоновой кислоты; на стадии 3 енон гидрировали при 1 атм водорода в присутствии каталитического количества 10% Pd/C в течение 1,5 ч с последующей обработкой (S)-2-метил-CBS и борандиметилсульфидом при 0°С. МС: m/z 504 и 506 (М+1).
7-{2R-[3S-Гидрокси-4-(4-гидрокси-3-метилфенил)-бут-1Е-енил]-6-оксопиперидин-1-ил}гептановую кислоту получали при использовании на стадии 2 диметилового эфира 3-[(4-гидрокси-3-метилфенил)-2-оксопропил]фосфоновой кислоты и при исключении стадии 4. МС: m/z 404 (М+1).
7-{2R-[3S-Гидрокси-4-(3-метоксиметилфенил)бут-1Е-енил]-6-оксопиперидин-1-ил}гептановую кислоту получали при использовании на стадии 2 диметилового эфира 3-[(3-метоксиметилфенил)-2-оксопропил]фосфоновой кислоты и при исключении стадии 4. МС: m/z 418 (М+1).
7-{2R-[3S-Гидрокси-4-(4-гидрокси-3-изопропилфенил)бут-1Е-енил]-6-оксопиперидин-1-ил}гептановую кислоту получали при использовании на стадии 2 диметилового эфира 3-[(3-гидрокси-3-изопропилфенил)-2-оксопропил]фосфоновой кислоты и при исключении стадии 4. МС: m/z 432 (M+1).
4-(2-{2R-[3-Гидрокси-3-(1-фенилциклопропил)проп-1Е-енил]-6-оксопиперидин-1-ил}этилсульфанил)масляную кислоту получали при использовании на стадии 2 диметилового эфира 2-(фенилциклопропил)-2-оксоэтилфосфоновой кислоты и при использовании на стадии 3 комбинации хлорида церия (III) и борогидрида натрия вместо стереоселективного реагента CBS. MC:m/z 418(M+1 ).
4-(2-{2R-[3R-3-Гидрокси-3-(трифторметилфуран-2-ил)пропил]-6-оксопиперидин-1-ил}этилсульфанил)масляную кислоту получали при использовании на стадии 2 диметилового эфира 2-[(5-трифторметилфуран-2-ил)-2-оксоэтил]фосфоновой кислоты; на стадии 3 енон гидрировали при 1 атм водорода в присутствии каталитического количества 10% Pd/C в течение 1,5 ч с последующей обработкой (S)-2-метил-CBS и борандиметилсульфидом при 0°С. МС:m/z 438 (М+1).
4-(2-{2R-[3R-Гидрокси-3-(1-фенилциклопропил)пропил]-6-оксопиперидин-1-ил}этилсульфанил)масляную кислоту получали при использовании на стадии 2 диметилового эфира 2-[(фенилциклопропил)-2-оксоэтил]фосфоновой кислоты; на стадии 3 енон гидрировали при 1 атм водорода в присутствии каталитического количества 10% Pd/C в течение 1,5 ч с последующей обработкой (S)-2-метил-CBS и борандиметилсульфидом при 0°С. МС: m/z 420 (М+1).
4-(2-{2R-[3S-Гидрокси-4-(3-метоксиметилфенил)бут-1Е-енил]-6-оксопиперидин-1-ил}этилсульфанил)масляную кислоту получали при использовании на стадии 2 диметилового эфира 3-[(3-метоксиметилфенил)-2-оксопропил]фосфоновой кислоты. МС: m/z 436 (М+1).
7-{2R-[3R-(4'-Гидрокси-2'-метилбифенил-3-ил)-3-гидроксипропил]-6-оксопиперидин-1-ил}гептановую кислоту получали при использовании на стадии 2 диметилового эфира [2-(4'-гидрокси-2'-метилбифен-3-ил)-2-оксоэтил]фосфоновой кислоты; при гидрировании енона на стадии 3 при 1 атм водорода в присутствии каталитического количества 10% Pd/C в течение 1,5 ч с последующей обработкой (S)-2-метил-CBS и борандиметилсульфидом при 0°С и при исключении стадии 4. МС: m/z 468 (М+1).
7-{2R-[3-Гидрокси-3-(4'-гидрокси-2'-метилбифенил-3-ил)проп-1Е-енил]-6-оксопиперидин-1-ил}гептановую кислоту получали при использовании на стадии 2 диметилового эфира [2-(4'-гидрокси-2'-метилбифен-3-ил)-2-оксоэтил]фосфоновой кислоты; при использовании на стадии 3 комбинации хлорида церия (III) и борогидрида натрия при 0°С и при исключении стадии 4. МС: m/z 466 (М+1).
7-(2-{2R-[3R-(4'-Гидрокси-2'-метилбифенил-3-ил)-3-оксопропил]-6-оксопиперидин-1-ил}гептановую кислоту получали при использовании на стадии 2 диметилового эфира [2-(4'-гидрокси-2'-метилбифен-3-ил)-2-оксоэтил]фосфоновой кислоты; при гидрировании енона на стадии 3 при 1 атм водорода в присутствии каталитического количества 10% Pd/C в течение 2 ч и при исключении стадии 4. МС: m/z 466 (М+1).
4-{2-[2R-(5-Циклобутил-3S-гидроксипент-1Е-енил)-6-оксопиперидин-1-ил]этилсульфанил} масляную кислоту получали при использовании на стадии 2 диметилового эфира (4-циклобутил-2-оксобутил)фосфоновой кислоты. МС: m/z 384 (М+1).
4-(2-{2R-[3R-(3'-Фторфеноксифенил-3-ил)-3-гидроксипропил]-6-оксопиперидин-1-ил}этилсульфанил)-3-метилмасляную кислоту получали при использовании на стадии 2 диметилового эфира [2-(3-фторфеноксифен-3-ил)-2-оксоэтил]фосфоновой кислоты; при гидрировании енона на стадии 3 при 1 атм водорода в присутствии каталитического количества 10% Pd/C в течение 1,5 ч с последующей обработкой (S)-2-метил-CBS и борандиметилсульфидом при 0°С и при использовании на стадии 4 метилового эфира 4-меркапто-3-метилмасляной кислоты вместо тиолактона. МС: m/z 504 (М+1 ).
4-{2-[2R-(3-Гидрокси-4,4-диметилокт-1Е-енил)-6-оксопиперидин-1-ил]этилсульфанил} масляную кислоту получали при использовании на стадии 2 диметилового эфира (3, 3-диметил-2-оксогептил)фосфоновой кислоты и комбинации хлорида церия (III) и борогидрида натрия на стадии 3. МС: m/z 400 (М+1).
7-{2R-[3-Гидрокси-3-(2, 5-диметилфенил-3-ил)проп-1Е-енил]-6-оксопиперидин-1-ил}гептановую кислоту получали при использовании на стадии 2 диметилового эфира [2-(2,5-диметилфен-3-ил)-2-оксоэтил]фосфоновой кислоты; при использовании на стадии 3 комбинации хлорида церия (III) и борогидрида натрия при 0°С и при исключении стадии 4. МС: m/z 388 (М+1).
7-[2R-(3-Гидрокси-4-феноксибут-1Е-енил)-6-оксопиперидин-1-ил]гептановую кислоту получали при использовании на стадии 2 диметилового эфира (3-фенокси-2-оксопропил)фосфоновой кислоты, при использовании на стадии 3 комбинации хлорида церия (III) и борогидрида натрия при 0°С и при исключении стадии 4. МС: m/z 390 (M+1).
7-{2R-[3-Гидрокси-3-(3'-хлорбифенил-3-ил)проп-1Е-енил]-6-оксопиперидин-1-ил}гептановую кислоту получали при использовании на стадии 2 диметилового эфира [2-(3'-хлорбифен-3-ил)-2-оксоэтил]фосфоновой кислоты, при использовании на стадии 3 комбинации хлорида церия (III) и борогидрида натрия при 0°С и при исключении стадии 4. МС: m/z 470 и 472 (М+1).
7-{2R-[3R-(3'-Хлорбифенил-3-ил)-3-гидроксипропил]-6-оксопиперидин-1-ил}гептановую кислоту получали при использовании на стадии 2 диметилового эфира [2-(3'-хлорбифен-3-ил)-2-оксоэтил]фосфоновой кислоты, при гидрировании енона на стадии 3 при 1 атм водорода в присутствии каталитического количества 10% Pd/C в течение 2 ч с последующей обработкой (S)-2-метил-СВS и бораном при 0°С и при исключении стадии 4. МС: m/z 472 и 474 (М+1).
Пример 3
4-(2-{2-[3-Гидрокси-4-(3-трифторметилфенил)бут-1Е-этил]-6-оксопиперидин-1-ил}этил)бензойная кислота

Стадия 1
Метиловый эфир 4-[2-(5, 6-дигидроксигексаноиламино)этил]бензойной кислоты

К раствору 5-гексеновой кислоты (8,45 г, 73,8 ммоля, фирма Aldrich) в N,N-диметилформамиде (110 мл) добавляли карбонилдиимидазол (12 г, 7,4 ммоля) и смесь нагревали при 50°С в течение 3 ч. Затем раствор охлаждали, добавляли диизопропилэтиламин (18,4 мл, 105 ммолей) и метиловый эфир 4-(2-аминометил)бензойной кислоты (12,6 г, 70,3 ммоля, полученный, как описано в заявке Т. Takemoto и др., ЕРА 0544205, фирма Ajinomoto Co.), и нагревание при 50°С продолжали в течение еще 3 ч. Смесь распределяли между водой (400 мл) и этилацетатом (3×150 мл). Объединенные органические экстракты промывали 1 М HCl, насыщенным раствором NaHCO3, солевым раствором, сушили над безводным MgSO4, фильтровали и упаривали в вакууме. Полученное твердое вещество (11,4 г) растворяли в ацетоне (440 мл) и воде (10 мл), добавляли N-метилморфолин-N-оксид (5,35 г, 45,7 ммоля), тетраоксид осмия (приблизительно 20 мг) и кипятили с обратным холодильником в течение 3 ч, при этом результаты ТСХ свидетельствовали о потреблении алкена. Смесь охлаждали, добавляли раствор бисульфита натрия и подкисляли добавлением 1 М HCl. Затем смесь экстрагировали хлороформом (3×200 мл), экстракт сушили над безводным сульфатом натрия и упаривали. Остаток перекристаллизовывали из этилацетата и минимального объема метанола, при этом получали метиловый эфир 4-[2-(5, 6-дигидроксигексаноиламино)этил]бензойной кислоты в виде твердого вещества (12,85 г).
Стадия 2
Метиловый эфир 4-[2-(2-гидроксиметил-6-оксопиперидин-1-ил)этил]бензойной кислоты

К раствору метилового эфира 4-[2-(5, 6-дигидроксигексаноиламино)этил]бензойной кислоты (7,4 г) в дихлорметане (170 мл) и ДМФА (35 мл) при 0°С добавляли 2,6-лутидин (9,1 мл), трет-бутилдиметилсилиловый эфир трифторметансульфоновой кислоты (5,5 мл) и смесь перемешивали в течение ночи. Затем смесь распределяли между раствором бикарбоната натрия и этилацетататом 3×150 мл). Объединенные экстракты сушили и упаривали на роторном испарителе, остаток очищали хроматографией на силикагеле (элюент: гексан/этилацетат, 2:1). Полученный первичный силиловый эфир (5,85 г) растворяли в дихлорметане (50 мл) и триэтиламине (3,85 мл) и охлаждали до 0°С атмосфере аргона. К раствору добавляли метансульфонилхлорид (1,5 мл, 19,3 ммоля) и смесь перемешивали в течение 30 мин. Затем смесь выливали в раствор бикарбоната натрия, экстрагировали дихлорметаном (3×30 мл), экстракты сушили над безводным сульфатом натрия и упаривали в вакууме, при этом получали требуемый сульфонат по вторичной гидроксигруппе (6,6 г), который непосредственно использовали на следующей стадии. Неочищенный сульфонат (6,6 г, 13,2 ммоля) растворяли при комнатной температуре в безводном толуоле (106 мл) в атмосфере аргона и добавляли трет-бутоксид калия (1 М раствор в ТГФ, фирма Aldrich). Через 2 ч смесь коричневого цвета распределяли между раствором хлорида аммония и этилацетатом (4×50 мл), объединенные органические экстракты сушили над безводным сульфатом натрия и упаривали. Продукт очищали хроматографией на силикагеле (элюент: гексан/этилацетат, 4:1), при этом получали требуемый лактам (1,79 г, 4,6 ммоля), который непосредственно растворяли в тетрагидрофуране (23 мл). К полученному раствору добавляли гидрат фторида тетрабутиламмония (1,74 г, 5,5 ммоля) и смесь перемешивали при КТ в течение 1 ч. Затем смесь фильтровали через слой силикагеля и промывали этилацетатом/гексаном (1:1), при этом получали метиловый эфир 4-[2-(2-гидроксиметил-6-оксопиперидин-1-ил)этил]бензойной кислоты (1,055 г).
4-(2-{2-[3-Гидрокси-4-(3-трифторметилфенил)бут-1Е-енил]-6-оксопиперидин-1-ил}этил)бензойную кислоту получали из метилового эфира 4-[2-(2-гидроксиметил-6-оксопиперидин-1-ил)этил]бензойной кислоты, аналогично тому, как описано в примере 2, при использовании на стадии 2 диметилового эфира 4-[(3-трифторметилфенил)-2-оксобутил]фосфоновой кислоты (полученного по методике 2), борогидрида натрия на стадии 3, при исключении стадии 4. МС: m/z 462 (М+1).
4-(2-{2-[3-(4'-Хлор-2'-метилбифен-3-ил)-3-гидроксипропил]-6-оксопиперидин-1-ил}этил)бензойную кислоту получали аналогично тому, как описано в примере 3, при использовании на стадии 2 диметилового эфира [3-(4'-хлор-2'-метилбифен-3-ил)-2-оксопропил]фосфоновой кислоты (полученного по методике 2), при восстановлении двойной связи на стадии 3 водородом при 1 атм и КТ в присутствии каталитического количества 10% Pd/C в этилацетате с последующей обработкой борогидридом натрия и при исключении стадии 4. МС: m/z 507 и 509 (М+1).
Пример 4
Составы
Фармацевтические препараты для доставки в организм субъекта соединений различными способами получают, как показано ниже. "Активный ингредиент" означает одно или более соединений формулы I.
Композиция для перорального введения
Ингредиенты смешивают и смесь помещают в капсулы, содержащие каждая по 100 мг смеси; одна капсула приблизительно соответствует суточной дозе.
Композиция для перорального введения
Ингредиенты смешивают и гранулируют при использовании в качестве растворителя метанола. Затем состав высушивают и прессуют в таблетки (содержащие приблизительно 20 мг активного соединения) на соответствующем прессе.
Композиция для перорального введения
Ингредиенты смешивают с образованием суспензии для перорального введения
Состав для парентерального введения
Активный ингредиент растворяют в порции воды для инъекции. Затем при перемешивании добавляют хлорид натрия до получения изотонического раствора. Раствор доводят до необходимой массы добавлением остальной части воды для инъекции, фильтруют через мембранный фильтр с диаметром пор 0,2 микрона и пакуют в стерильных условиях.
Состав для суппозитория
Ингредиенты перемалывают, смешивают на паровой бане и выливают в формочки, содержащие 2,5 г состава.
Состав для местного применения
Все ингредиенты, за исключением воды, смешивают и при перемешивании нагревают до приблизительно 60°С. При интенсивном перемешивании добавляют небольшое количество воды, нагретой до 60°С, для эмульгирования ингредиентов, а затем добавляют воду до приблизительно 100 г.
Составы для назального спрея
Получено несколько водных суспензий, содержащих приблизительно 0,025-0,5% активного соединения, в качестве составов для назального спрея. Составы необязательно содержат неактивные ингредиенты, такие, например, как микрокристаллическая целлюлоза, натриевая соль карбоксиметилцеллюлозы, декстроза и т.п. Для регуляции величины рН добавляют соляную кислоту. Составы для назального спрея обычно вводят дозирующим пульверизатором, подающим при активации приблизительно 50-100 мкл состава. Типичным режимом дозирования является 2-4 спрея каждые 4-12 ч.
Пример 5
Функциональная активность рецептора ЕР4 (или ЕР2), определенная по активности люциферазы
а) Получение стабильно трансфектированных клонов ЕР4-люцифераза
кДНК, соответствующую полноразмерной последовательности простаноидного рецептора ЕР4, субклонировали в соответствующие сайты вектора экспрессии пкДНК 3.1(+)/Zeo млекопитающих (фирма Invitrogen). Кроме того, последовательность, содержащую САМР-респонсивный элемент (CRE) и ген люциферазы клонировали в вектор рХР1. Одновременную трансфекцию клеток СНО двумя векторами, пкДНК, содержащим EP4R, и рХР1, содержащим CRE-люциферазу, проводили при соотношении ДНК от 5 до 1 с использованием фугена (фирма Roche Molecular) в среде F-12 (фирма Gibco), дополненной 10% инактивированной нагреванием эмбриональной телячьей сывороткой (ЭТС, фирма Gibco). Через трое суток после трансфекции среду заменяли свежей средой, содержащей зеоцин. Клетки культивировали в течение одного месяца до получения стабильных клонов.
б) цАМФ-опосредованный анализ гена люциферазы
Функциональную активность лигандов-агонистов ЕР4 после связывания с рецептором определяли по продуцированию внутриклеточного цАМФ. В этом случае уровень цАМФ определяли косвенным способом по трансляции репортерного гена люциферазы в составе клонов ЕР4-люцифераза. Клетки клона ЕР4-люцифераза субкультивировали в 200 мкл среды F12 (фирма Gibco, BRL), содержащий 10% ЭТС (фирма Gibco, BRL) и 25 мМ Hepes, в 96-луночных планшетах (фирма Packard) с плотностью 40000 клеток в лунке. Клетки культивировали в СО2-инкубаторе (37°С, 5% СО2, 95% воздух) в течение ночи, на следующее утро культуральную среду удаляли, клетки дважды промывали 100 мкл буферного раствора Хенкса и ресуспендировали в 90 мкл среды F12, содержащей 0,1% БСА. После предварительной инкубации клеток в СО2-инкубаторе (37°С, 5% СО2, 95% воздух) в течение 1,5-3 ч в лунки добавляли по 10 мкл раствора анализируемых соединений (серийное 10-кратное разведение) и инкубировали при 37°С в течение еще 3 ч. Для определения максимальной стимуляции люциферазы, опосредованной рецептором ЕР4, в каждый анализ включали контрольный образец, содержащий в качество полного агониста 0,1 мкМ PGE2.
По завершении инкубации культуральную среду удаляли и лунки промокали салфетками. Готовые планшеты использовали для анализа люциферазы.
в) Количественное определение люциферазной активности
Для количественного анализа люциферазы использовали набор реактивов LucLite (фирма Packard). Субстрат LucLite и субстратный буферный раствор (фирма Packard) нагревали до комнатной температуры за 30 мин до завершения инкубации клеток. Субстрат растворяли в субстратном буферном растворе и перемешивали переворачиванием пробирки. Для применения на следующей стадии смешивали равные объемы приготовленного раствора субстрата и фосфатно-солевого буферного раствора Дальбекко (ДФСБ, фирма Gibco BRL), содержащего 1 мМ MgCl2 и 1 мМ CaCl2. В каждую лунку 96-луночного планшета добавляли по 100 мкл приготовленного смешанного раствора и планшет встряхивали при 300 об/мин на качалке для планшетов в течение 3 мин. Крышку планшета удаляли и заменяли липкой пленкой (фирма Packard) для анализа на сцинтилляционном счетчике. Затем определяли величину EC50соединения по графику с четырьмя переменными по программе KaleidaGraph. По результатам анализа соединения формулы I обладали активностью.
Пример 6
Анализ конкурентного связывания [3H]PGE2 с рецепторами rEP1, rEP2, rEP3и rEP4
а) Клеточная культура и трансфекции
Стабильно трансфектированные клетки, экспрессирующие ЕР3, культивировали в среде F-12 (фирма GIBCO), дополненной 10% инактивированной нагреванием ЭТС (фирма Gibco), и осаждали центрифугированием. кДНК, соответствующую полноразмерной последовательности простаноидного рецептора ЕР2 или ЕР4, субклонировали в соответствующие сайты вектора экспрессии pcDNA 3.1(+)/Zeo млекопитающих (фирма Invitrogen). Количество вектора, пригодное для трансфекции, получали с использованием набора Qiagen Endo-Free Plasmid Maxi Kit, а клетки COS-7 трансфектировали с использованием реагента FuGene 6 (фирма Roche Molecular) по рекомендациям фирмы-изготовителя (фирма Roche). Клетки COS-7 культивировали в среде DMEM (фирма GIBCO), дополненной 10% инактивированной нагреванием ЭТС (фирма Gibco) и гентамицином (фирма GIBCO), и собирали через 72 ч после трансфекции. Клетки осаждали центрифугированием, промывали ФСБ (фирма GIBCO), вторично осаждали центрифугированием и быстро замораживали смесью сухой лед/этанол или использовали немедленно для получения мембранной фракции.
б) Получение мембранной фракции
При получении мембранной фракции все операции проводили при 4°С. Клетки COS-7, трансфектированные простаноидным рецептором, или стабильно трансфектированные клетки СНО, гомогенизировали в буферном растворе для анализа (см. описание ниже) с использованием гомогенизатора Polytron (фирма Brinkman) и центрифугировали при 48000g в течение 30 мин. Осадки ресуспендировали в буферном растворе для анализа и обрабатывали ультразвуком с использованием ультразвукового дезинтегратора Branson. Концентрацию белка определяли с использованием набора для анализа белков BioRad DC по рекомендациям фирмы-изготовителя и полученные препараты хранили при -80°C.
в) Анализ связывания с простаноидным рецептором
Методы анализа конкурентного аффинного связывания с ЕР2, ЕР3 и ЕР4 описаны М. Abramovitz и др., The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs, Biochimica et Biophysica Acta, 1483, 285-293 (2000). Реакцию связывания проводили в конечном объеме 0,2 мл буферного раствора для анализа, следующего состава: 20 мМ HEPES (рН 7,4), 1 мМ EDTA и 10 мМ MgCl2 (ЕР3), или 10 мМ MES (+ NaOH до рН 6,0), 10 мМ MnCl2 и 1 мМ EDTA (EP2 и ЕР4) и радиоактивно меченый лиганд {2,25 нМ (ЕР3) или 2,5 нМ (EP2) [3H]-PGE2 (200 Ки/ммоль, NEN)}. Реакции инициировали добавлением мембранного белка (приблизительно 50 мкг/образец для ЕР3, 100 мкг для EP2 и ЕР4). Во всех образцах концентрация ДМСО (фирма Sigma) составляла 1% (об./об.), а соединения анализировали при конечных концентрациях 100 мкМ-0,3 нМ. Неспецифическое связывание определяли в присутствии 10 мкМ немеченого PGE2 (фирма Cayman Chemical). Образцы инкубировали при 30°С в течение 60 мин (ЕР3) или при 23°С в течение 45 мин (EP2 и ЕР4). Инкубацию останавливали быстрым фильтрованием на 96-луночном фильтре GF/B (фирма Packard) (который предварительно смачивали 10 мМ MES, 0,01% БСА, рН 6, 0 для EP2) при 4°С с использованием 96-луночного полуавтоматического коллектора клеток Filtermate 196 (фирма Packard). Фильтры промывали 3-4 мл буферного раствора для промывки (20 мМ HEPES, рН 7,4, для ЕР3, 10 мМ MES, 0,01% БСА, рН 6,0, для ЕР2 и ЕР4), высушивали при комнатной температуре в течение по меньшей мере 1 ч и после добавления 37,5 мкл сцинтиллятора Microscint 20 (фирма Packard) определяли остаточную радиоактивность на фильтрах после промывки с использованием сцитилляционного счетчика для микропланшетов Packard TopCount. Статистическую обработку результатов проводили с использованием программного обеспечения Prism версия 3.0 (GraphPad). По результатам анализа соединения формулы I обладали активностью.
Пример 7
Определение плотности костной ткани
Соединения по настоящему изобретению исследовали по их воздействию на массу костной ткани у крыс с удаленным яичником.
У взрослых самок крыс Sprague-Dawley или Wistar Hanover удаляли яичники по методу Charles River или симулировали операцию. После операции крыс помещали парами в помещении с контролируемой средой, где они проходили акклиматизацию в течение по меньшей мере одной недели. В ходе акклиматизации животных кормили парами.
Анализируемое соединение вводили подкожно один раз в сутки, начиная с 20 суток после проведения операции, в течение 5 недель. Соединение вводили в 10% EtOH/физрастворе или в 20 мМ фосфатном буферном растворе.
Перед лечением и после лечения животных исследовали на голографическом денситометре костной ткани высокого разрешения QDR-4500 с использованием пакета программного обеспечения для измерения минеральной составляющей костной ткани (BMD). Затем снимки анализировали в участках, подлежащих анализу, таких, как бедренная кость, середина бедра, диализ бедра, дистальная точка бедра, метафаз дистальной точки бедра, середина большеберцовой кости, метафиз середины большеберцовой кости, позвонки L2-L4, позвонок L5.
Для подтверждения последствий удаления яичника сравнивали массу костной ткани у оперированных (OVX) и ложно оперированных групп крыс, а также у группы животных, получавших носитель (плацебо), с использованием t-теста Стьюдента. Группу OVX и контрольную группу сравнивали однофакторным дисперсионным анализом (ANOA) с последующим анализом по LSD Фишера для сравнения каждой пролеченной группы с группой, получавшей носитель в том случае, если общий эффект был статистически достоверным. Данные можно было ранжировать до проведения вышеуказанного анализа и провести соответствующий непараметрический анализ (критерий суммы рангов по Вилкоксону или Крускал-Валлису).
Хотя настоящее изобретение описано со ссылкой на конкретные варианты его осуществления, для специалиста в данной области представляется очевидным, что в пределах сущности и объема изобретения возможны различные изменения и эквиваленты этих вариантов. Кроме того, в пределах сущности и объема настоящего изобретения возможны многочисленные модификации в соответствии с конкретной ситуацией, материалом, композицией материалов, способом, стадией или стадиями способа. Подразумевается, что все указанные модификации включены в объем пунктов прилагаемой формулы изобретения.
Реферат
Изобретение относится к новым соединениям формулы I
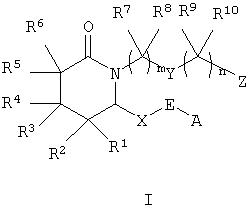
где m равно от 1 до 4; n равно от 0 до 4; А означает алкил, арил, трифторметилфурил, арилалкил, арилциклоалкил, циклоалкилалкил или арилоксиалкил; Е означает -СНОН- или -С(O)-; Х означает -(СН2)2- или -СН=СН-; Y означает -СН2-, арилен, -S-; Z означает -СООН; R1, R2, R3, R4, R5, R6, R7, R8, R9 и R10 каждый независимо означает водород или алкил, или его фармацевтически приемлемая соль или сольват, индивидуальный изомер или рацемическая или нерацемическая смесь изомеров. Изобретение также относится к фармацевтической композиции и к применению соединений на основе формулы I, обладающих активностью в качестве агонистов простагландина. Технический результат - получение новых биологически активных соединений и фармацевтических композиций на их основе, обладающих активностью в качестве агонистов простагландина. 3 н. и 21 з.п. ф-лы, 1 табл.
Формула

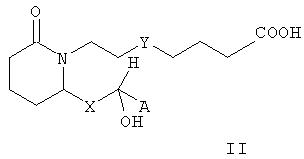

Комментарии