Ингибиторы глюкозилцерамид-синтазы - RU2645675C2
Код документа: RU2645675C2
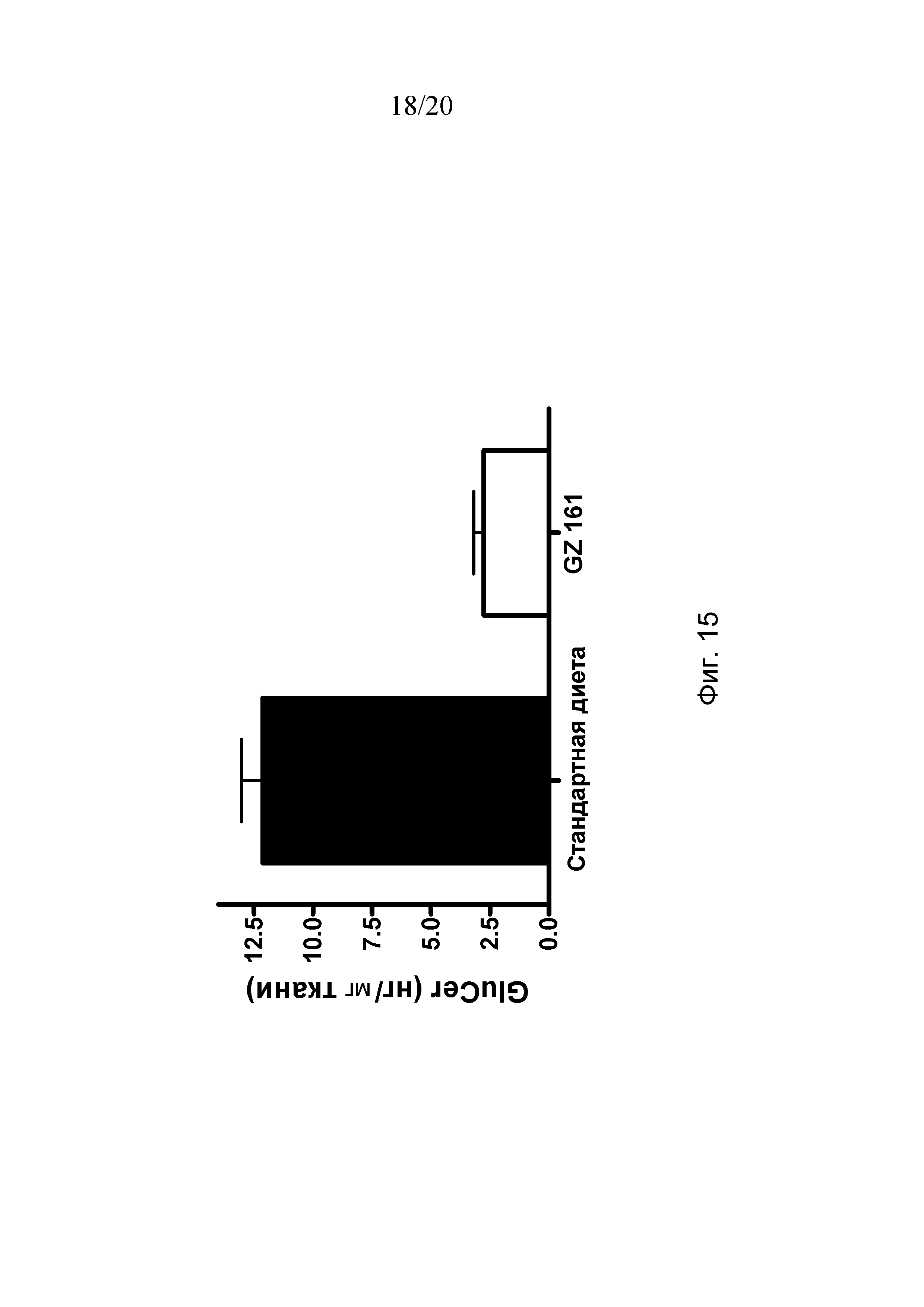
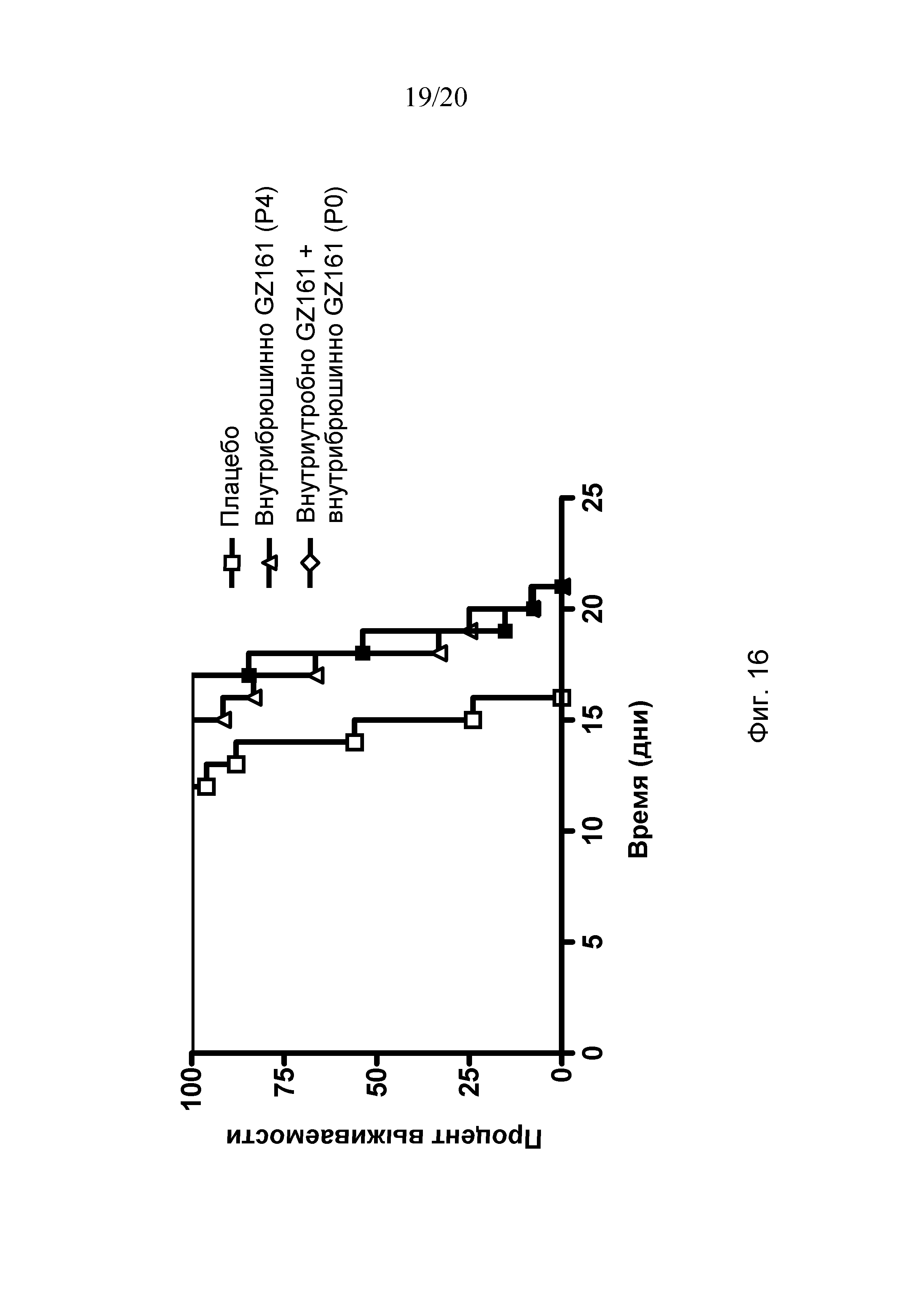
Чертежи
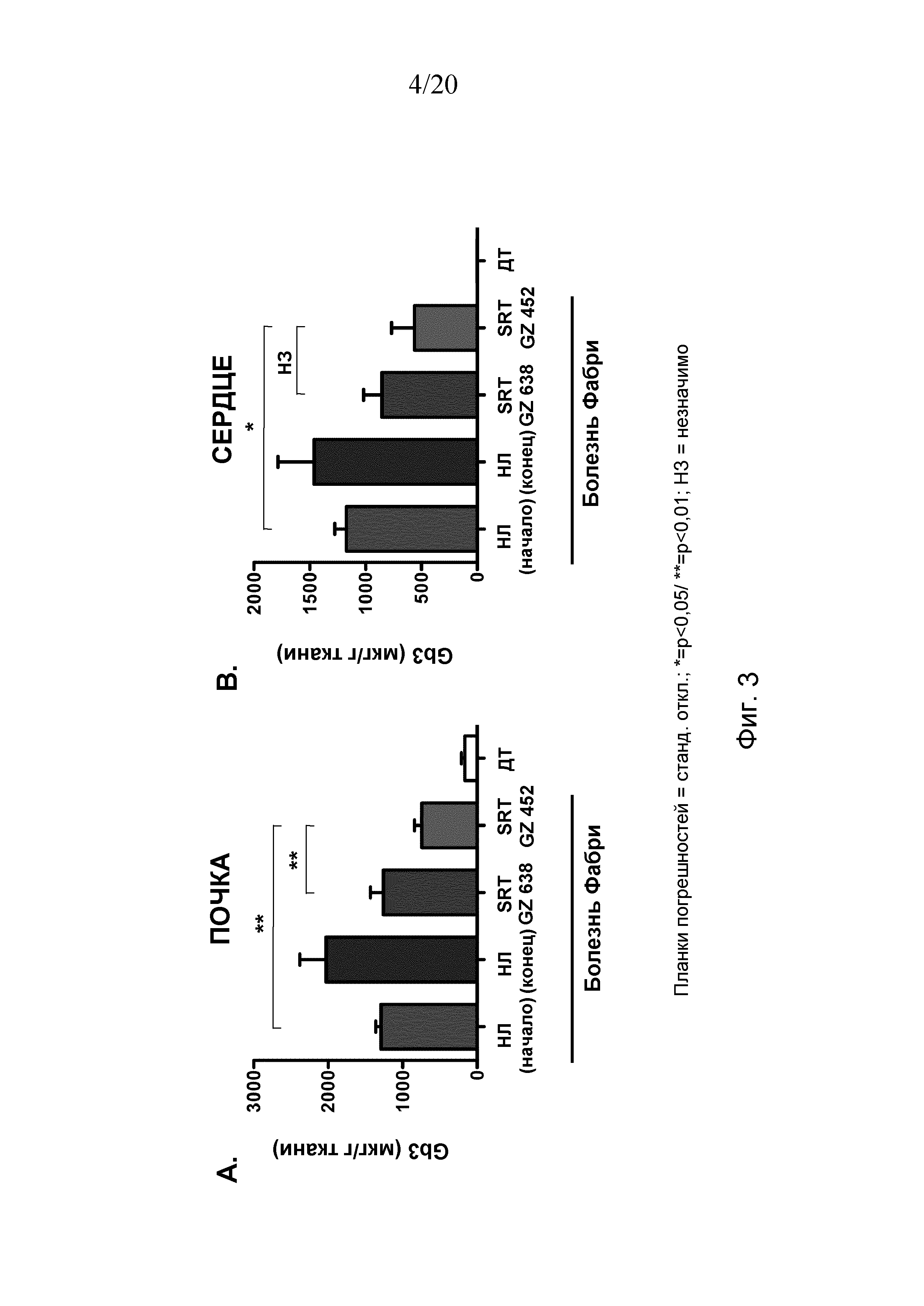










Описание
Предпосылки создания изобретения
Область техники, к которой относится изобретение
В общем смысле настоящее изобретение относится к области терапии рака и метаболических заболеваний. Более конкретно, настоящее изобретение относится к ингибиторам глюкозилцерамид-синтазы (GCS), которые можно применять для лечения метаболических заболеваний, таких как болезни лизосомного накопления (в виде монотерапии или в сочетании с ферментозаместительной терапией), а также для лечения рака.
Описание предшествующего уровня техники
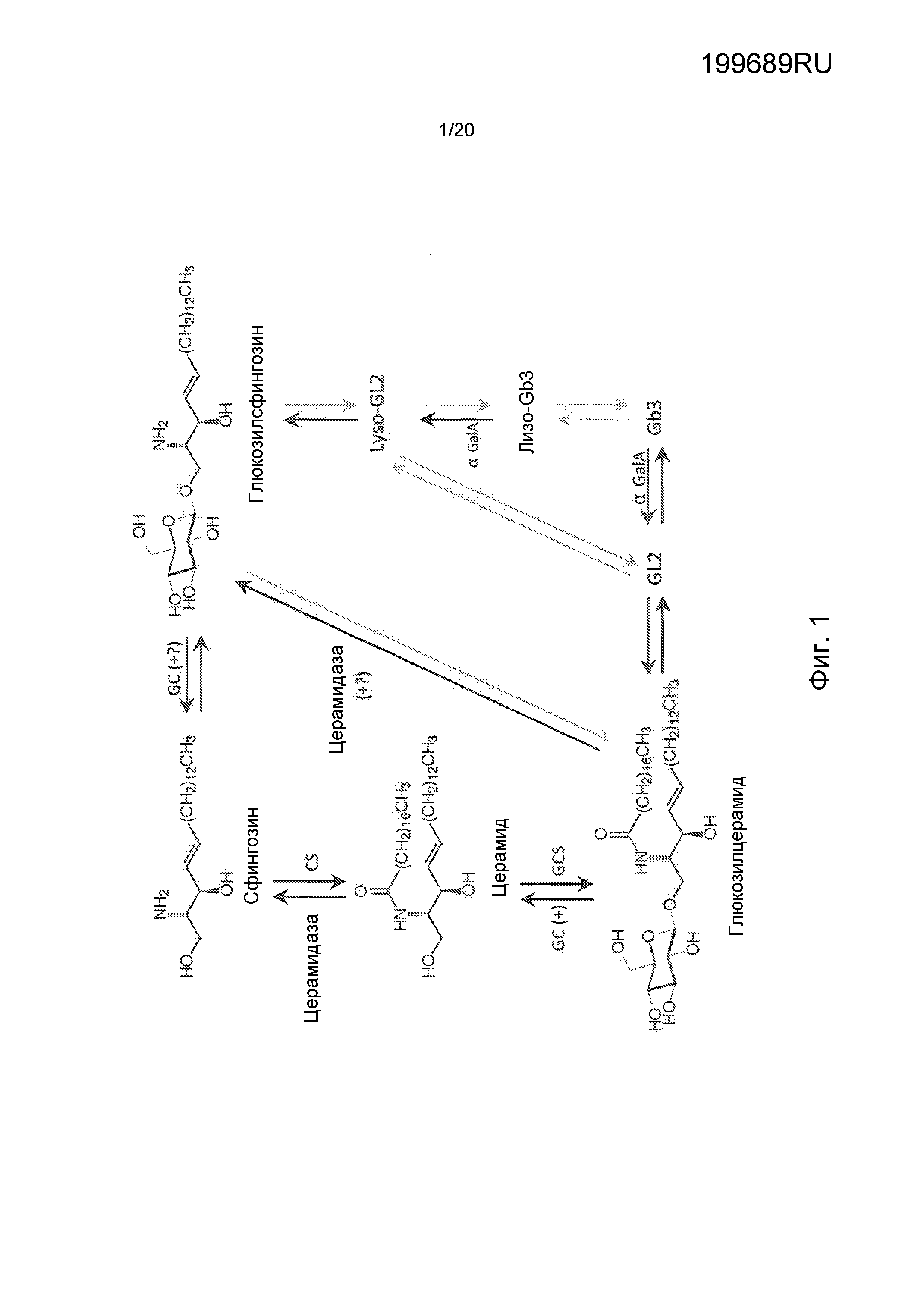
Глюкозилцерамид-синтаза (GCS) представляет собой ключевой фермент, катализирующий стадию начального гликозилирования в биосинтезе гликосфинголипидов (GSLs) на основе глюкозилцерамидов, а именно, путем ключевого переноса глюкозы от UDP-глюкозы (UDP-Glc) к церамиду для образования глюкозилцерамида (см. Фиг. 1). GCS является трансмембранным интегральным белком типа III, локализованным в цис/медиальных цистернах аппарата Гольджи. Гликосфинголипиды (GSLs), как полагают, представляют собой интегральную часть динамики многих клеточных событий, включая клеточные взаимодействия, передачу сигналов и транспорт. Было показано (см. Yamashita et al., Proc. Natl. Acad. Sci. USA 1999, 96(16), 9142-9147), что синтез структур GSL является важным для эмбрионального развития и дифференцировки некоторых тканей. Церамид играет центральную роль в метаболизме сфинголипидов, а понижающая регуляция активности GCS, как было показано, оказывает заметные эффекты на состав сфинголипидов, понижая экспрессию гликосфинголипидов. Сфинголипиды (SLs) играют биомодуляторную роль в физиологических и патофизиологических сердечнососудистых состояниях. В частности, сфинголипиды и их регуляторные ферменты, по-видимому, играют некоторую роль в адаптивных реакциях на хроническую гипоксию в сердце новорожденных крыс (см. El Alwanit et al., Prostaglandins and other lipid mediators 2005, 78(1-4), 249-263).
Ингибиторы GCS были предложены для лечения многих заболеваний (см., например, WO 2005068426). Такие способы лечения включают в себя лечение болезней накопления гликолипидов (например, болезни Тея-Сакса, Сандхоффа, дефицита активатора GM2, GM1-ганглиозидоза и болезни Фабри), заболеваний, связанных с накоплением гликолипидов (например, болезни Гоше; миглустат (Zavesca), ингибитор GCS, был одобрен для применения при лечении пациентов с болезнью Гоше типа 1, см. Treiber et al., Xenobiotica 2007, 37(3), 298-314), заболеваний, вызывающих ренальную гипертрофию или гиперплазию, такую как диабетическая нефропатия; заболеваний, вызывающих гипергликемию или гиперинсулинемию; форм рака с аномальным синтезом гликолипидов, инфекционных заболеваний, вызванных микроорганизмами, использующими поверхностные клеточные гликолипиды в качестве рецепторов, инфекционных заболеваний, при которых существенным или важным фактором является синтез глюкозилцерамида, других заболеваний, при которых существенным или важным фактором является синтез глюкозилцерамида, заболеваний, при которых имеет место избыточный синтез гликолипидов (таких как атеросклероз, поликистозная болезнь почек и гипертрофия почек), неврологических расстройств, неврологических травм, воспалительных заболеваний или расстройств, связанных с рекрутингом и активацией макрофагов (например, ревматоидного артрита, болезни Крона, астмы и сепсиса) и сахарного диабета и ожирения (см. WO 2006053043).
В частности, было показано, что чрезмерная экспрессия GCS является частью механизма множественной лекарственной резистентности и, кроме того, она прерывает апоптоз, индуцированный церамидами. Например, Turzanski et al., (Experimental Hematology 2005, 33 (1), 62-72 показали, что церамид индуцирует апоптоз клеток при остром миелоидном лейкозе (AML) и что P-гликопротеин (p-gp) придает резистентность к апоптозу, индуцированному церамидами, причем эту резистентность клеток TF-1 в значительной степени определяет модулирование церамид-глюкозилцерамидного пути. Таким образом, ингибиторы GCS могут быть полезными для лечения пролиферативных расстройств посредством индуцирования апоптоза больных клеток.
Сущность изобретения
Настоящее изобретение относится к соединению, представленному следующей структурной формулой:

или его фармацевтически приемлемой соли или пролекарству, где:
n равно 1, 2 или 3;
m равно 0 или 1;
p равно 0 или 1;
t равно 0, 1 или 2;
y равно 1 или 2;
z равно 0, 1 или 2;
E представляет собой S, O, NH, NOH, NNO2, NCN, NR, NOR или NSO2R;
X1 представляет собой CR1, когда m равно 1, или N, когда m равно 0;
X2 представляет собой О, -NH, -CH2-, SO2, NH-SO2; CH(C1-C6)-алкил или -NR2;
X3 представляет собой О, -NH, -CH2-, CO, -CH(C1-C6)-алкил, SO2NH, -CO-NH- или -NR3;
X4 представляет собой CR4R5, CH2CR4R5или CH2-(C1-C6)-алкил-CR4R5;
X5 представляет собой прямую связь, O, S, SO2, CR4R5; (C1-C6)-алкил, (C1-C6)-алкилоксигруппу, (C1-C6)-алкенил, (C1-C6)-алкенилоксигруппу;
R представляет собой (С6-C12)-арил, (C2-C9)-гетероарил, (C1-C6)-алкил, (C2-C9)-гетероарил-(C1-C6)-алкил;
R1 представляет собой Н, CN, (C1-C6)-алкилкарбонил или (C1-C6)-алкил;
каждый из радикалов R2 и R3 независимо представляет собой -H, (C1-C6)-алкил, необязательно замещенный одним или более из заместителей, выбранных из группы, включающей в себя галоген, (C1-C6)-алкил, (C6-C12)-арил, (C2-C9)-гетероарил, (C1-C6)-алкил-(C6-C12)-арил, галоген-(C6-C12)-арил и галоген-(C2-C9)-гетероарил или, необязательно, когда X2 представляет собой -NR2, а X3 представляет собой -NR3, тогда R2 и R3 вместе с атомом азота, к которому они присоединены, образуют неароматическое гетероциклическое кольцо, необязательно, замещенное одним или более заместителями, выбранными из группы, включающей в себя галоген, (C1-C6)-алкил, (C6-C12)-арил, (C2-C9)-гетероарил, (C1-C6)-алкил-(C6-C12)-арил, галоген-(C6-C12)-арил и галоген-(C2-C9)-гетероарил;
R4 и R5 являются независимо выбранными из H, (C1-C6)-алкила или совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо;
R6 представляет собой -H, галоген, -CN, (C6-C12)-арил, (C6-C12)-арилоксигруппу, (C1-C6)-алкилоксигруппу; (C1-C6)-алкил, необязательно, замещенный 1-4 галогенами, или (C1-C6)-алкил;
A1 представляет собой (С2-C6)-алкинил; (C6-C12)-арил, (C2-C9)-гетероарил, (C2-C9)-гетероциклоалкил или бензо-(C2-C9)-гетероциклоалкил, необязательно, замещенный одним или более заместителей, выбранных из группы, включающей в себя галоген, (C1-C6)-алкил, необязательно, замещенный 1-3 галогенами; (C1-C6)-алкенил, аминогруппу, (C1-C6)-алкиламиногруппу, (C1-C6)-диалкиламиногруппу, (C1-C6)-алкоксигруппу, нитрогруппу, CN, -OH, (C1-C6)-алкилоксигруппу, необязательно, замещенную 1-3 галогенами; (C1-C6)-алкоксикарбонил и (C1-C6)-алкилкарбонил;
A2 представляет собой Н, (C6-C12)-арил, (C2-C9)-гетероарил, (C2-C9)-гетероциклоалкил или бензо-(C2-C9)-гетероциклоалкил, необязательно, замещенный одним или более заместителей, выбранных из группы, включающей в себя галоген, (C1-C6)-алкил, необязательно, замещенный 1-3 галогенами; (C1-C6)-алкиленил, аминогруппу, (C1-C6)-алкиламиногруппу, (C1-C6)-диалкиламиногруппу, (C1-C6)-алкоксигруппу, O(C3-C6-циклоалкил), (C3-C6)-циклоалкоксигруппу, нитрогруппу, CN, OH, (C1-C6)-алкилоксигруппу, необязательно, замещенную 1-3 галогенами; (C3-C6)-циклоалкил, (C1-C6)-алкоксикарбонил, (C1-C6)-алкилкарбонил, (C1-C6)-галогеналкил;
при условии, что сумма n + t + y + z не превышает 6;
при условии, что когда p равно 0; X2 представляет собой NH-SO2 и X3 представляет собой NH;
при условии, что когда n равно 1; t равно 0; y равно 1; z равно 1; X2 представляет собой NH; E представляет собой О; X3 представляет собой NH; A2 представляет собой Н и X5 представляет собой прямую связь; A1 не является незамещенным фенилом, галогенофенилом или изопропенилфенилом;
при условии, что когда n равно 1; t равно 0; y равно 1; z равно 1; X2 представляет собой О; E представляет собой О; X3 представляет собой NH; A1 представляет собой (С6-C12)-арил и X5 представляет собой прямую связь; A2 представляет собой Н и R4представляет собой Н, тогда R5 не является циклогексилом; и
при условии, что когда n равно 1; t равно 0; y равно 1; z равно 1; X2 представляет собой NH; E представляет собой О; X3 представляет собой CH2; оба радикала R4 и R5 представляют собой водород; A2 представляет собой Н и X5 представляет собой прямую связь; тогда A1 не является незамещенным фенилом. Определенные аспекты настоящего изобретения включают в себя введение вышеуказанного соединения пациенту в качестве части комбинированной терапии, включающей в себя ферментозаместительную терапию (ERT) и терапию малыми молекулами (SMT) для уменьшения количества и/или ингибирования накопления субстрата у пациента с поставленным диагнозом болезни лизосомного накопления.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1; t равно 0; y равно 1 и z равно 1.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1; t равно 1; y равно 1 и z равно 1.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 2; t равно 0; y равно 1 и z равно 1.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 2; t равно 1; y равно 1 и z равно 1.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 3; t равно 0; y равно 1 и z равно 1.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1; t равно 2; y равно 1 и z равно 1.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1; t равно 0; y равно 1 и z равно 0.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1; t равно 1; y равно 1 и z равно 0.
Настоящее изобретение, кроме того, относится к соединению Формулы I,где n равно 2; t равно 0; y равно 1 и z равно 0.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 2; t равно 1; y равно 1 и z равно 0.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 3; t равно 0; y равно 1 и z равно 0.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1; t равно 2; y равно 1 и z равно 0.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1; t равно 1; y равно 2 и z равно 0.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 2; t равно 0; y равно 2 и z равно 0.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где m равно 1 и X1 представляет собой CR1.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где m равно 0 и X1 представляет собой N.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где m равно 1; E представляет собой О; X2 представляет собой О и X3представляет собой NH.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где m равно 1; E представляет собой О; X2 представляет собой NH и X3представляет собой NH.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где m равно 1; E представляет собой О; X2 представляет собой CH2 и X3представляет собой NH.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где m равно 1; E представляет собой О; X2 представляет собой NH и X3представляет собой CH2.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где m равно 1; E представляет собой S; X2 представляет собой NH и X3представляет собой NH.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где m равно 0; E представляет собой О; X1 представляет собой NH и X3представляет собой NH.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где m равно 1; E представляет собой О; X2 представляет собой NH и X3представляет собой CO-NH.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где m равно 1; p равно 0; X2 представляет собой NH-SO2 и X3представляет собой NH.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где каждый радикал R4 и R5 представляет собой (C1-C6)-алкил или совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где каждый радикал R4 и R5 представляет собой метил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-циклопропильное кольцо.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкоксильное кольцо.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где A1 представляет собой (С2-C6)-алкинил или (C6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где A1 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где A1 представляет собой тиофен, тиазол, изотиазол, фуран, оксазол, изоксазол, пиррол, имидазол, пиразол, триазол, пиридин, пиримидин, пиридазин, индол, бензотиазол, бензоизоксазол, бензопиразол, бензоимидазол, бензофуран, бензооксазол или бензоизоксазол.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где A1 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где A1 представляет собой пирролидинил, тетрагидрофуранил, дигидрофуранил, тетрагидропиранил, пиранил, тиопиранил, азиридинил, азетидинил, оксиранил, метилендиоксил, хроменил, барбитурил, изоксазолидинил, 1,3-оксазолидин-3-ил, изотиазолидинил, 1,3-тиазолидин-3-ил, 1,2-пиразолидин-2-ил, 1,3-пиразолидин-1-ил, пиперидинил, тиоморфолинил, 1,2-тетрагидротиазин-2-ил, 1,3-тетрагидротиазин-3-ил, тетрагидротиадиазинил, морфолинил, 1,2-тетрагидродиазин-2-ил, 1,3-тетрагидродиазин-1-ил, тетрагидроазепинил, пиперазинил, пиперизин-2-онил, пиперизин-3-онил, хроманил, 2-пирролинил, 3-пирролинил, имидазолидинил, 2-имидазолидинил, 1,4-диоксанил, 8-азабицикло[3.2.1]октанил, 3-азабицикло[3.2.1]октанил, 3,8-диазабицикло[3.2.1]октанил, 2,5-диазабицикло[2.2.1]гептанил, 2,5-диазабицикло[2.2.2]октанил, октагидро-2H-пиридо[1,2-a]пиразинил, 3-азабицикло[4.1.0]гептанил, 3-азабицикло[3.1.0]гексанил, 2-азаспиро[4,4]нонанил, 7-окса-1-аза-спиро[4,4]нонанил, 7-азабицикло[2.2.2]гептанил или октагидро-1H-индолил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где A1 представляет собой бензо-(C2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где A1 представляет собой 2,3-дигидробензо[b][1,4]диоксин или 2,2-дифторбензо[d][1,3]диоксол.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где R6 представляет собой Н.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где X5 представляет собой прямую связь.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где X5 представляет собой CR4R5.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где каждый радикал R4 и R5 представляет собой метил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-циклопропильное кольцо.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкоксильное кольцо.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где A2 представляет собой пиридин.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где A2 представляет собой пирролидинил, тетрагидрофуранил, дигидрофуранил, тетрагидропиранил, пиранил, тиопиранил, азиридинил, азетидинил, оксиранил, метилендиоксил, хроменил, барбитурил, изоксазолидинил, 1,3-оксазолидин-3-ил, изотиазолидинил, 1,3-тиазолидин-3-ил, 1,2-пиразолидин-2-ил, 1,3-пиразолидин-1-ил, пиперидинил, тиоморфолинил, 1,2-тетрагидротиазин-2-ил, 1,3-тетрагидротиазин-3-ил, тетрагидротиадиазинил, морфолинил, 1,2-тетрагидродиазин-2-ил, 1,3-тетрагидродиазин-1-ил, тетрагидроазепинил, пиперазинил, пиперизин-2-онил, пиперизин-3-онил, хроманил, 2-пирролинил, 3-пирролинил, имидазолидинил, 2-имидазолидинил, 1,4-диоксанил, 8-азабицикло[3.2.1]октанил, 3-азабицикло[3.2.1]октанил, 3,8-диазабицикло[3.2.1]октанил, 2,5-диазабицикло[2.2.1]гептанил, 2,5-диазабицикло[2.2.2]октанил, октагидро-2H-пиридо[1,2-a]пиразинил, 3-азабицикло[4.1.0]гептанил, 3-азабицикло[3.1.0]гексанил, 2-азаспиро[4,4]нонанил, 7-окса-1-аза-спиро[4,4]нонанил, 7-азабицикло[2.2.2]гептанил или октагидро-1H-индолил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где A2 представляет собой бензо-(C2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где A2 представляет собой.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где R1 представляет собой водород или метил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; p равно 1; E представляет собой О; X2 представляет собой О; X3 представляет собой NH; R1 представляет собой Н; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; p равно 1; E представляет собой О; X2 представляет собой О; X3 представляет собой NH; R1 представляет собой Н; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; p равно 1; E представляет собой О; X2 представляет собой О; X3 представляет собой NH; R1 представляет собой Н; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; p равно 1; E представляет собой О; X2 представляет собой О; X3 представляет собой NH; R1 представляет собой Н; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; p равно 1; E представляет собой О; X2 представляет собой О; X3 представляет собой NH; R1 представляет собой Н; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; p равно 1; E представляет собой О; X2 представляет собой О; X3 представляет собой NH; R1 представляет собой Н; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CH2; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CH2; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CH2; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CH2; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CH2; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CH2; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой CH2; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой CH2; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой CH2; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой CH2; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой CH2; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой CH2; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой S; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой S; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой S; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой S; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой S; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой S; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой SO2; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой SO2; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой SO2; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой SO2; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)- гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой SO2; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой SO2; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой N; m равно 0; E представляет собой О; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой N; m равно 0; E представляет собой О; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой N; m равно 0; E представляет собой О; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой N; m равно 0; E представляет собой О; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой N; m равно 0; E представляет собой О; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой N; m равно 0; E представляет собой О; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CO-NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CO-NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CO-NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CO-NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CO-NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CO-NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; p равно 1; E представляет собой О; X2 представляет собой О; X3 представляет собой NH; R1 представляет собой Н; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; p равно 1; E представляет собой О; X2 представляет собой О; X3 представляет собой NH; R1 представляет собой Н; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; p равно 1; E представляет собой О; X2 представляет собой О; X3 представляет собой NH; R1 представляет собой Н; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; p равно 1; E представляет собой О; X2 представляет собой О; X3 представляет собой NH; R1 представляет собой Н; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CH2; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CH2; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CH2; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CH2; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой CH2; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой CH2; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой CH2; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой CH2; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой S; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой S; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой S; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой S; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой SO2; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой SO2; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой SO2; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой SO2; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой N; m равно 0; E представляет собой О; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой N; m равно 0; E представляет собой О; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой N; m равно 0; E представляет собой О; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой N; m равно 0; E представляет собой О; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CO-NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CO-NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CO-NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CO-NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; p равно 1; E представляет собой О; X2 представляет собой О; X3 представляет собой NH; R1 представляет собой Н; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; p равно 1; E представляет собой О; X2 представляет собой О; X3 представляет собой NH; R1 представляет собой Н; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; p равно 1; E представляет собой О; X2 представляет собой О; X3 представляет собой NH; R1 представляет собой Н; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; p равно 1; E представляет собой О; X2 представляет собой О; X3 представляет собой NH; R1 представляет собой Н; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CH2; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CH2; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CH2; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CH2; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой CH2; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой CH2; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой CH2; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой CH2; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой S; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой S; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой S; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой S; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой SO2; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой SO2; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой SO2; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой SO2; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой N; m равно 0; E представляет собой О; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой N; m равно 0; E представляет собой О; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой N; m равно 0; E представляет собой О; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой N; m равно 0; E представляет собой О; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CO-NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CO-NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероциклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CO-NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CO-NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероциклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; p равно 1; E представляет собой О; X2 представляет собой О; X3 представляет собой NH; R1 представляет собой Н; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; p равно 1; E представляет собой О; X2 представляет собой О; X3 представляет собой NH; R1 представляет собой Н; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CH2; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CH2; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой CH2; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой CH2; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой S; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой S; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой SO2; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой SO2; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой N; m равно 0; E представляет собой О; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой N; m равно 0; E представляет собой О; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CO-NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CO-NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; p равно 1; E представляет собой О; X2 представляет собой О; X3 представляет собой NH; R1 представляет собой Н; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; p равно 1; E представляет собой О; X2 представляет собой О; X3 представляет собой NH; R1 представляет собой Н; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; p равно 1; E представляет собой О; X2 представляет собой О; X3 представляет собой NH; R1 представляет собой Н; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; p равно 1; E представляет собой О; X2 представляет собой О; X3 представляет собой NH; R1 представляет собой Н; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CH2; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CH2; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CH2; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CH2; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой CH2; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой CH2; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой CH2; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой CH2; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой S; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой S; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой S; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой S; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой SO2; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой SO2; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой SO2; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой SO2; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой N; m равно 0; E представляет собой О; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой N; m равно 0; E представляет собой О; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой N; m равно 0; E представляет собой О; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой N; m равно 0; E представляет собой О; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CO-NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CO-NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С6-C12)-арил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CO-NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CO-NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С6-C12)-арил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; p равно 1; E представляет собой О; X2 представляет собой О; X3 представляет собой NH; R1 представляет собой Н; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; p равно 1; E представляет собой О; X2 представляет собой О; X3 представляет собой NH; R1 представляет собой Н; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; p равно 1; E представляет собой О; X2 представляет собой О; X3 представляет собой NH; R1 представляет собой Н; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; p равно 1; E представляет собой О; X2 представляет собой О; X3 представляет собой NH; R1 представляет собой Н; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)- гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CH2; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CH2; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CH2; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CH2; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой CH2; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой CH2; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой CH2; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой CH2; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой S; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой S; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой S; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой S; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой SO2; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой SO2; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой SO2; X2 представляет собой NH; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой SO2; X2 представляет собой NH; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой N; m равно 0; E представляет собой О; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой N; m равно 0; E представляет собой О; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой N; m равно 0; E представляет собой О; X3 представляет собой NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой N; m равно 0; E представляет собой О; X3 представляет собой NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CO-NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CO-NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С2-C9)-гетероарил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CO-NH; R4 и R5 совместно с углеродом, к которому они присоединены, образуют спиро-(C3-C10)-циклоалкильное кольцо или спиро-(C3-C10)-циклоалкоксильное кольцо; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где n равно 1, 2 или 3; t равно 0, 1 или 2; y равно 0 или 1; z равно 0, 1 или 2; X1 представляет собой CR1; m равно 1; E представляет собой О; X2 представляет собой NH; X3 представляет собой CO-NH; каждый из радикалов R4 и R5 независимо представляет собой метил; R6 представляет собой водород или метил; A1 представляет собой (С3-C10)-циклоалкил; X5 представляет собой прямую связь, O или CR4R5и A2 представляет собой (С2-C9)-гетероарил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где A1 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I, где A2 представляет собой (С3-C10)-циклоалкил.
Настоящее изобретение, кроме того, относится к соединению Формулы I или его фармацевтически приемлемой соли или пролекарству, выбранным из группы, состоящей из:
1-азабицикло[2.2.2]окт-3-ил-[2-(2,4'-дифторбифенил-4-ил)-пропан-2-ил]-карбамата;
1-азабицикло[2.2.2]окт-3-ил-{2-[4-(1,3-бензотиазол-6-ил)-фенил]-пропан-2-ил}-карбамата;
1-азабицикло[3.2.2]нон-4-ил-{1-[5-(4-фторфенил)-пиридин-2-ил]-циклопропил}-карбамата;
1-азабицикло[2.2.2]окт-3-ил-{1-[3-(4-фторфенокси)-фенил]-циклопропил}-карбамата;
1-азабицикло[2.2.2]окт-3-ил-{1-[4-(1,3-бензотиазол-5-ил)-фенил]-циклопропил}-карбамата;
1-азабицикло[2.2.2]окт-3-ил-[1-(4'-фтор-3'-метоксибифенил-4-ил)-циклопропил]-карбамата;
1-азабицикло[2.2.2]окт-3-ил-[3-(4'-фторбифенил-4-ил)-оксетан-3-ил]-карбамата;
1-азабицикло[2.2.2]окт-3-ил-{1-[6-(4-фторфенокси)-пиридин-2-ил]-циклопропил}-карбамата;
1-азабицикло[2.2.2]окт-3-ил-[3-(4'-фторбифенил-4-ил)пентан-3-ил]-карбамата;
1-азабицикло[2.2.2]окт-3-ил-{2-[2-(4-фторфенил)-2H-индазол-6-ил]-пропан-2-ил}-карбамата;
1-азабицикло[2.2.2]окт-3-ил-{2-[2-(1H-пиррол-1-ил)-пиридин-4-ил]-пропан-2-ил}-карбамата;
1-(3-этил-1-азабицикло[2.2.2]окт-3-ил)-3-[1-(4'-фторбифенил-4-ил)-циклопропил]-мочевины;
N-(1-азабицикло[2.2.2]окт-3-ил)-N'-[1-(4'-фторбифенил-4-ил)-циклопропил]-этандиамида;
1-азабицикло[2.2.2]окт-3-ил-(1-{4[(4,4-дифторциклогексил)-окси]-фенил}-циклопропил)-карбамата;
1-(4-метил-1-азабицикло[3.2.2]нон-4-ил)-3-[1-(5-фенилпиридин-2-ил)-циклопропил]-мочевины;
1-[1-(4'-фторбифенил-4-ил)-циклопропил]-1-метил-3-(3-метил-1-азабицикло[2.2.2]окт-3-ил)-мочевины;
1-[1-(4'-фторбифенил-4-ил)-циклопропил]-1-метил-3-(3-метил-1-азабицикло[2.2.2]окт-3-ил)-мочевины;
1-{2-[4'-(2-метоксиэтокси)бифенил-4-ил]-пропан-2-ил}-3-(3-метил-1-азабицикло[2.2.2]окт-3-ил)-мочевины;
2-(1-азабицикло[3.2.2]нон-4-ил)-N-[1-(5-фенилпиридин-2-ил)-циклопропил]-ацетамида;
3-(4'-фторбифенил-4-ил)-3-метил-N-(4-метил-1-азабицикло[3.2.2]нон-4-ил)-бутанамида;
диамида N-[2-(бифенил-4-ил)-пропан-2-ил]-N'-(3-метил-1-азабицикло[2.2.2]окт-3-ил)-серной кислоты;
диамида N-[2-(4'-фторбифенил-4-ил)-пропан-2-ил]-N'-(3-метил-1-азабицикло[2.2.2]окт-3-ил)-серной кислоты;
1-(3-бутил-1-азабицикло[2.2.2]окт-3-ил)-3-{2-[1-(4-фторфенил)-1H-пиразол-4-ил]-пропан-2-ил}-мочевины;
1-азабицикло[2.2.2]окт-3-ил-[4-(4-фторфенил)-2-метилбут-3-ин-2-ил]-карбамата;
1-(3-бутил-1-азабицикло[2.2.2]окт-3-ил)-3-[4-(4-фторфенил)-2-метилбут-3-ин-2-ил]-мочевины;
N-[1-(4'-фторбифенил-4-ил)-циклопропил]-1,4-диазабицикло[3.2.2]нонан-4-карбоксамида;
1-(2-(4'-фтор-[1,1'-бифенил]-4-ил)-пропан-2-ил)-3-(3-метил-1-азабицикло[3.2.2]нонан-3-ил)-мочевины;
1-(2-(4'-фтор-[1,1'-бифенил]-4-ил)-пропан-2-ил)-3-(4-метил-1-азабицикло[4.2.2]декан-4-ил)-мочевины;
1-(2-(4'-фтор-[1,1'-бифенил]-4-ил)-пропан-2-ил)-3-(3-метил-1-азабицикло[4.2.2]декан-3-ил)-мочевины; и
1-(2-(4'-фтор-[1,1'-бифенил]-4-ил)-пропан-2-ил)-3-(5-метил-1-азабицикло[4.2.2]декан-5-ил)-мочевины.
Настоящее изобретение, кроме того, относится к фармацевтической композиции для лечения заболевания или расстройства, опосредуемого глюкозилцерамид-синтазой (GCS), или заболевания или расстройства, к которому GCS имеет отношение, у субъекта, нуждающегося в таком лечении, включающем в себя введение указанному субъекту эффективного количества соединения Формулы I.
Настоящее изобретение, кроме того, относится к способу лечения заболевания или расстройства, опосредуемого глюкозилцерамид-синтазой (GCS), или заболевания или расстройства, к которому GCS имеет отношение, у субъекта, нуждающегося в таком лечении, включающем в себя введение указанному субъекту эффективного количества соединения Формулы I.
Настоящее изобретение, кроме того, относится к способу лечения заболевания или расстройства, такого как рак.
Настоящее изобретение, кроме того, относится к способу лечения заболевания или расстройства, такого как метаболическое расстройство.
Настоящее изобретение, кроме того, относится к способу лечения заболевания или расстройства, такого как неврологическое заболевание.
Настоящее изобретение, кроме того, относится к способу, где указанное неврологическое заболевание представляет собой болезнь Альцгеймера.
Настоящее изобретение, кроме того, относится к способу, где указанное неврологическое заболевание представляет собой болезнь Паркинсона.
Настоящее изобретение, кроме того, относится к способу индуцирования пониженной каталитической активности глюкозилцерамид-синтазы в клетке in vitro, включающему в себя контактирование клетки с эффективным количеством соединения Формулы I.
Настоящее изобретение, кроме того, относится к соединению Формулы I, представленному следующей структурной формулой:
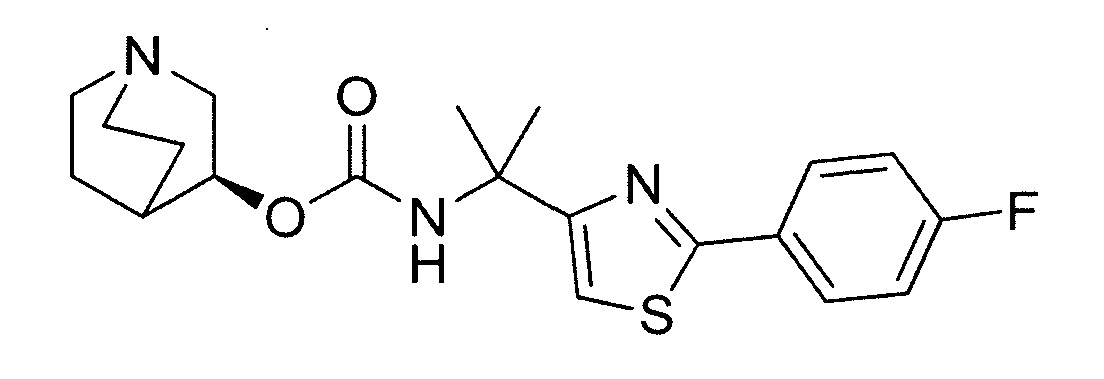
или его фармацевтически приемлемой соли или пролекарству.
Настоящее изобретение, кроме того, относится к соединению формулы I, представленному следующей структурной формулой:
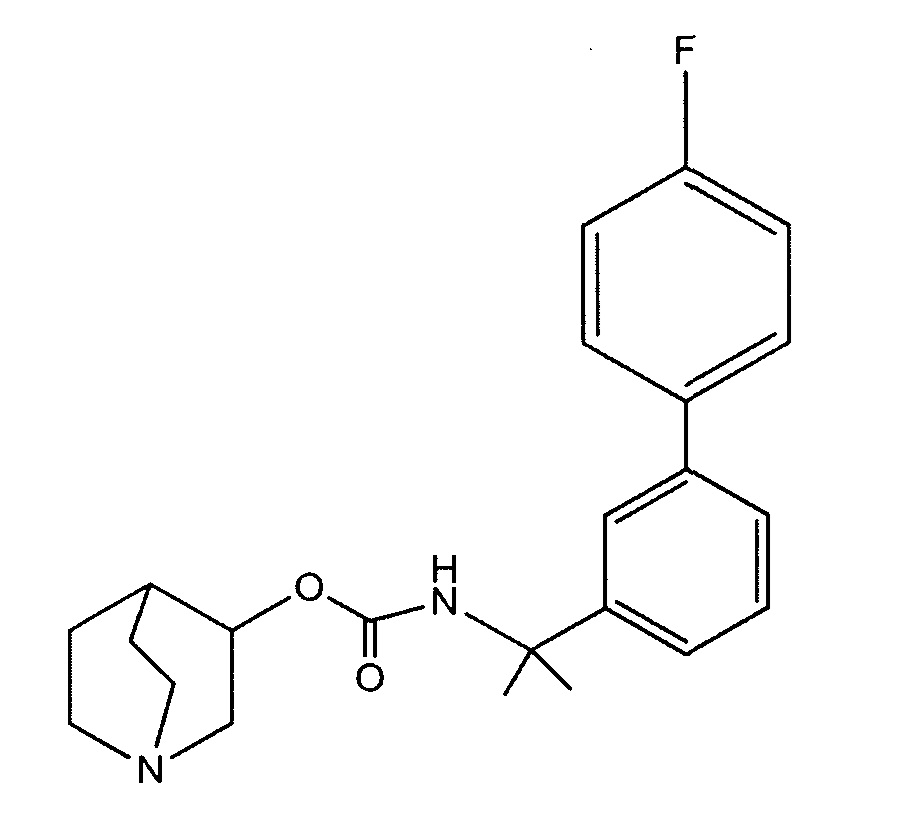
или его фармацевтически приемлемой соли или пролекарству.
Настоящее изобретение, кроме того, относится к способу лечения субъекта, с поставленным диагнозом болезни лизосомного накопления, причем указанный способ включает в себя введение указанному субъекту эффективного количества соединения Формулы I, и в определенных вариантах осуществления настоящего изобретения указанное соединение представлено следующей структурной формулой:
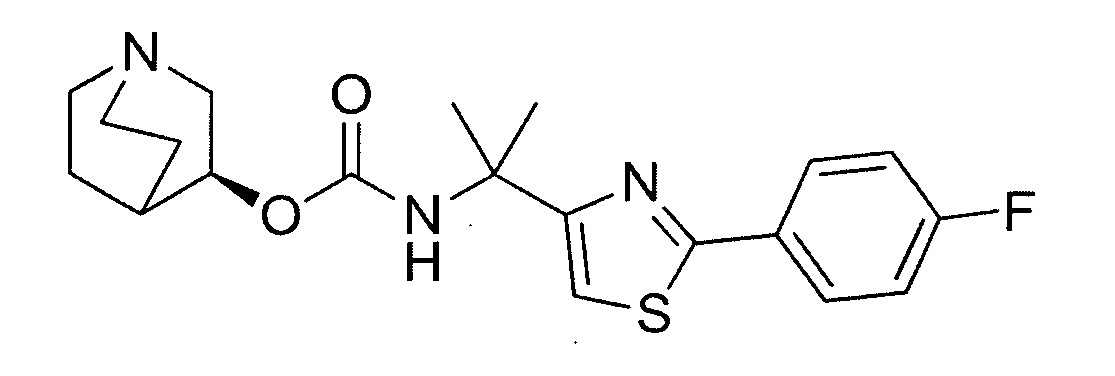
или его фармацевтически приемлемой солью или пролекарством или
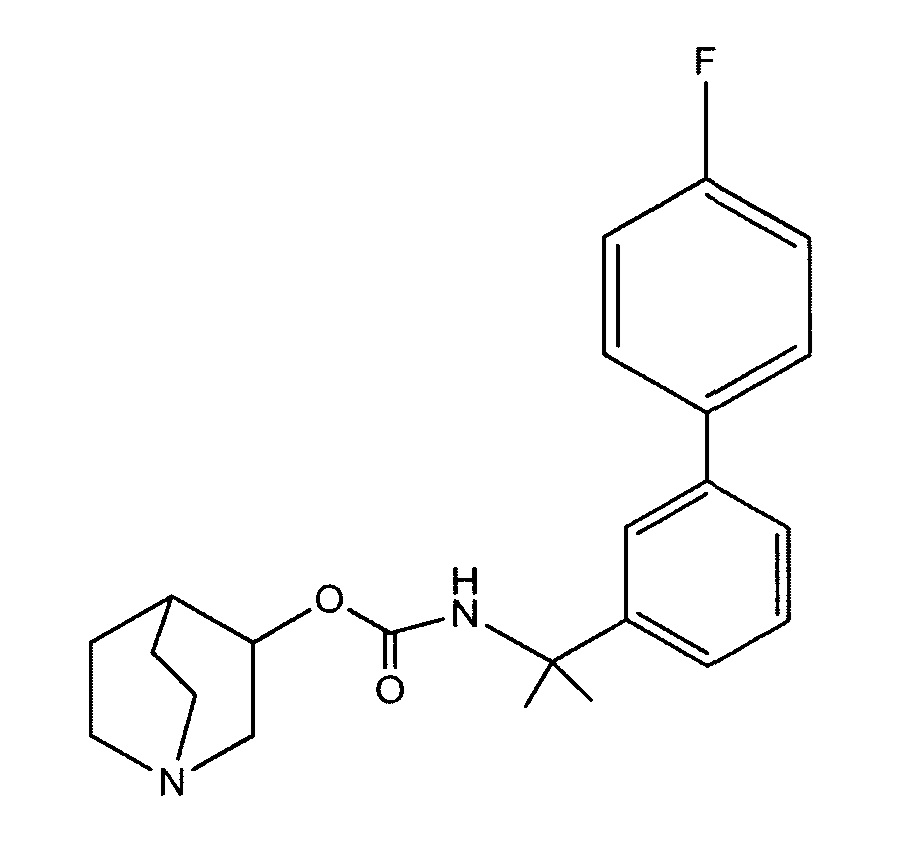
или его фармацевтически приемлемой солью или пролекарством.
В определенных вариантах осуществления настоящего изобретения указанная болезнь лизосомного накопления является следствием дефекта в гликосфинголипидном пути.
В определенных вариантах осуществления настоящего изобретения указанная болезнь лизосомного накопления представляет собой болезни Гоше, Фабри, GM1-ганглиозидоз, дефицит активатора GM2, болезни Тея-Сакса или Сандхоффа.
Настоящее изобретение, кроме того, относится к способу лечения субъекта, с поставленным диагнозом болезни лизосомного накопления, причем указанный способ включает в себя введение указанному субъекту эффективного количества соединения Формулы I и введение указанному субъекту терапевтически эффективного количества лизосомального фермента.
В определенных вариантах осуществления настоящего изобретения указанный лизосомальный фермент представляет собой глюкоцереброзидазу, альфа-галактозидазу A, гексозаминидазу A, гексозаминидазу B или GM1-ганглиозид-β-галактозидазу.
В определенных вариантах осуществления настоящего изобретения субъект до проведения лечения имеет повышенные уровни лизосомального субстрата, а после проведения лечения этот субъект имеет более низкие объединенные количества лизосомального субстрата в моче и плазме, чем субъект, лечение которого проводили либо одним лизосомальным ферментом, либо одним указанным соединением.
В определенных вариантах осуществления настоящего изобретения указанный субстрат представляет собой глоботриаозилцерамид или лизо-глоботриаозилцерамид и их комбинации.
Настоящее изобретение, кроме того, относится к способу уменьшения активности глюкозилцерамид-синтазы (GCS) у субъекта с поставленным диагнозом болезни лизосомного накопления, включающий в себя введение указанному субъекту эффективного количества соединения Формулы I - либо в виде монотерапии, либо в сочетании с ферментозаместительной терапией.
Настоящее изобретение, кроме того, относится к способу уменьшения накопления материала, производимого GCS, у субъекта с поставленным диагнозом болезни лизосомного накопления, включающему в себя введение указанному субъекту эффективного количества соединения Формулы I, либо в виде монотерапии, либо в сочетании с ферментозаместительной терапией.
Настоящее изобретение предоставляет способ комбинированной терапии для лечения субъекта с поставленным диагнозом болезни лизосомного накопления, включающий в себя поочередное применение ферментозаместительной терапии и терапии малыми молекулами.
Настоящее изобретение предоставляет способ комбинированной терапии для лечения субъекта с поставленным диагнозом болезни лизосомного накопления, включающий в себя одновременное применение ферментозаместительной терапии и терапии малыми молекулами.
При разнообразных вариантах комбинирования способов терапии согласно настоящему изобретению следует понимать, что терапию малыми молекулами можно проводить до, одновременно или после проведения ферментозаместительной терапии. Аналогичным образом, ферментозаместительную терапию можно проводить до, одновременно или после проведения терапии малыми молекулами.
Определения
Термин «фармацевтически приемлемая соль», используемый в настоящем документе, означает фармацевтически приемлемую кислотно-аддитивную соль или фармацевтически приемлемую основно-аддитивную соль соединения, раскрытого в настоящем документе, которую можно применять без существенного нежелательного биологического эффекта (одного или более), являющегося следствием ее применения, или вредного взаимодействия (одного или более), также являющегося следствием ее применения, с другим компонентом фармацевтической композиции, в которой она может содержаться.
Термин «пролекарство», используемый в настоящем документе, означает фармакологическое производное молекулы исходного лекарственного вещества, которое, для высвобождения из него активного лекарственного вещества, требует осуществления некоторой биотрансформации (спонтанной или ферментативной), происходящей с ним в организме. Например, пролекарства представляют собой варианты или производные соединения Формулы I, которые имеют группы, способные к отщеплению и/или расщеплению при определенных метаболических условиях, которые после отщепления и/или расщепления превращаются в соединения Формулы 1. Такие пролекарства становятся активными in vivo, после того как они подвергнутся сольволизу или ферментативной деградации. В настоящем документе возможно употребление названий «единичное», «двойное», «тройное пролекарство» и т.д., в зависимости от числа стадий биотрансформации, требующихся для высвобождения активного лекарственного вещества в организме и числа функциональных элементов, присутствующих в форме, являющейся предшественником. Пролекарственные формы часто обладают лучшей растворимостью и совместимостью с тканями или отличаются замедленным высвобождением в организме млекопитающего (см. Bundgard, Design of prodrugs, pp. 7-9, 21-24, Elsevier, Amsterdam 1985 и Silverman, The organic chemistry of drug design and drug action, pp. 352-401, Academic Press, San Diego, Calif., 1992). Пролекарства, широко известные в данной области, включают в себя хорошо известные производные кислот (такие как, например, сложные эфиры, получаемые по реакции исходных кислот с подходящим спиртом), амиды (получаемые по реакции исходного кислотного соединения с амином), основные группы (прореагировавшие с образованием ацилированных производных оснований) и т.д. Очевидно, что для повышения биодоступности можно комбинировать и другие пролекарственные производные, обладающие другими особенностями, раскрытыми в настоящем документе. Поэтому квалифицированные специалисты в данной области признают, что определенные соединения, раскрытые в настоящем документе и обладающие свободной аминогруппой, амидной группой, гидроксильной или карбоксильной группой, могут быть преобразованы в пролекарства. Пролекарства включают в себя соединения, имеющие остаток аминокислоты или полипептидную цепь, состоящую из двух или более (например, двух, трех или четырех) аминокислотных остатков, которые пептидными связями ковалентно присоединены к свободной аминогруппе, гидроксильной группе или карбоксильной кислотной группе соединений, раскрытых в настоящем документе. К указанным аминокислотным остаткам относятся 20 природных аминокислот, обычно обозначаемые трехбуквенными символами, а также 4-гидроксипролин, гидроксилизин, десмозин, изодесмозин, 3-метилгистидин, норвалин, бета-аланин, гамма-аминомасляная кислота, цитруллин, гомоцистеин, гомосерин, орнитин и метионинсульфон. Пролекарства также включают в себя соединения, имеющие карбонатный, карбаматный, амидный или алкильный сложноэфирный фрагмент, ковалентно связанный с любым из вышеуказанных заместителей, раскрытых в настоящем документе.
Термин «(C1-C6)-алкил», используемый в настоящем документе, означает насыщенный линейный или разветвленный свободный радикал, состоящий существенно из 1-6 атомов углерода и соответствующего числа атомов водорода. Примеры (C1-C6)-алкильных групп включают в себя метил, этил, н-пропил, изопропил, н-бутил, изобутил и т.д. Конечно, квалифицированный специалист в данной области может легко представить и другие (C1-C6)-алкильные группы, полезные для настоящего изобретения.
Термин «(C3-C10)-циклоалкил», используемый в настоящем документе, означает неароматический насыщенный свободный радикал, образующий, по меньшей мере, одно кольцо, состоящее существенно из 3-10 атомов углерода и соответствующего числа атомов водорода. Сами по себе (C3-C10)-циклоалкильные группы могут быть моноциклическими или полициклическими. Индивидуальные кольца таких полициклических групп могут иметь разные виды соединений, например, конденсированные, мостовые, спиро и т.д., в дополнение к замещению ковалентных связей. Примеры (C3-C10)-циклоалкильных групп включают в себя циклопропил, циклобутил, циклопентил, циклогексил, норборнанил, бицикло[3.2.1]октанил, октагидропенталенил, спиро[4.5]деканил, циклопропил, замещенный циклобутилом, циклобутил, замещенный циклопентилом, циклогексил, замещенный циклопропилом, и т.д. Конечно, квалифицированный специалист в данной области может легко представить и другие (C3-C10)-циклоалкильные группы, полезные для настоящего изобретения.
Термин «(C2-C9)-гетероциклоалкил», используемый в настоящем документе, означает неароматический свободный радикал, имеющий от 3 до 10 атомов (т.е. кольцевых атомов), которые образуют, по меньшей мере, одно кольцо, где от 2 до 9 кольцевых атомов представляют собой атомы углерода, а остальные кольцевые атомы (один или более) (т.е. гетероатомы в кольце) являются выбранными из группы, состоящей из азота, серы и кислорода. Сами по себе, (C2-C9)-гетероциклоалкильные группы могут быть моноциклическими или полициклическими. Индивидуальные кольца таких полициклических групп могут иметь разные виды соединений, например, конденсированные, мостовые, спиро и т.д., в дополнение к замещению ковалентных связей. Примеры (C2-C9)-гетероциклоалкильных групп включают в себя пирролидинил, тетрагидрофуранил, дигидрофуранил, тетрагидропиранил, пиранил, тиопиранил, азиридинил, азетидинил, оксиранил, метилендиоксил, хроменил, барбитурил, изоксазолидинил, 1,3-оксазолидин-3-ил, изотиазолидинил, 1,3-тиазолидин-3-ил, 1,2-пиразолидин-2-ил, 1,3-пиразолидин-1-ил, пиперидинил, тиоморфолинил, 1,2-тетрагидротиазин-2-ил, 1,3-тетрагидротиазин-3-ил, тетрагидротиадиазинил, морфолинил, 1,2-тетрагидродиазин-2-ил, 1,3-тетрагидродиазин-1-ил, тетрагидроазепинил, пиперазинил, пиперизин-2-онил, пиперизин-3-онил, хроманил, 2-пирролинил, 3-пирролинил, имидазолидинил, 2-имидазолидинил, 1,4-диоксанил, 8-азабицикло[3.2.1]октанил, 3-азабицикло[3.2.1]октанил, 3,8-диазабицикло[3.2.1]октанил, 2,5-диазабицикло[2.2.1]гептанил, 2,5-диазабицикло[2.2.2]октанил, октагидро-2H-пиридо[1,2-a]пиразинил, 3-азабицикло[4.1.0]гептанил, 3-азабицикло[3.1.0]гексанил, 2-азаспиро[4,4]нонанил, 7-окса-1-аза-спиро[4,4]нонанил, 7-азабицикло[2.2.2]гептанил, октагидро-1H-индолил и т.д. Как правило, (C2-C9)-гетероциклоалкильная группа обычно является присоединенной к главной структуре через атом углерода или атом азота. Конечно, квалифицированный специалист в данной области может легко представить и другие (C2-C9)- гетероциклоалкильные группы, полезные для настоящего изобретения.
Термин «(C2-C9)-гетероарил», используемый в настоящем документе, означает ароматический свободный радикал, имеющий от 5 до 10 атомов (т.е. кольцевых атомов), которые образуют, по меньшей мере, одно кольцо, где от 2 до 9 кольцевых атомов представляют собой атомы углерода, а остальные кольцевые атомы (один или более) (т.е. кольцевые гетероатомы) являются выбранными из группы, состоящей из азота, серы и кислорода. Сами по себе, (C2-C9)-гетероарильные группы могут быть моноциклическими и полициклическими. Индивидуальные кольца таких полициклических гетероарильных групп могут иметь разные соединения, например, конденсированные и т.п., в дополнение к замещению ковалентной связи. Примеры (C2-C9)-гетероарильных групп включают в себя фурил, тиенил, тиазолил, пиразолил, изотиазолил, оксазолил, изоксазолил, пирролил, триазолил, тетразолил, имидазолил, 1,3,5-оксадиазолил, 1,2,4-оксадиазолил, 1,2,3-оксадиазолил, 1,3,5-тиадиазолил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, пиридил, пиримидил, пиразинил, пиридазинил, 1,2,4-триазинил, 1,2,3-триазинил, 1,3,5-триазинил, пиразоло[3,4-b]пиридинил, циннолинил, птеридинил, пуринил, 6,7-дигидро-5H-[1]пиридинил, бензо[b]тиофенил, 5,6,7,8-тетрагидрохинолин-3-ил, бензоксазолил, бензотиазолил, бензизотиазолил, бензизоксазолил, бензимидазолил, тианафтенил, изотианафтенил, бензофуранил, изобензофуранил, изоиндолил, индолил, индолизинил, индазолил, изохинолил, хинолил, фталазинил, хиноксалинил, хиназолинил и бензоксазинил и т.д. Как правило, (C2-C9)-гетероарильная группа обычно присоединяется к главной структуре посредством атома углерода; однако квалифицированный специалист в данной области признает, что к главной структуре могут присоединяться и определенные другие атомы, например, кольцевые гетероатомы. Конечно, квалифицированный специалист в данной области может легко представить и другие (C2-C9)-гетероарильные группы, полезные для настоящего изобретения.
Термин «(C6-C10)-арил», используемый в настоящем документе, означает фенил или нафтил.
Термин «галоген», используемый в настоящем документе, означает фтор, хлор, бром или йод.
Термин «амино», используемый в настоящем документе, означает свободный радикал, имеющий атом азота и от 1 до 2 атомов водорода. Сам по себе, термин «амино», как правило, относится к первичным и вторичным аминам. В связи с этим, в настоящем документе и прилагаемых пунктах формулы изобретения принято представлять третичный амин общей формулой RR'N-, где R и R' представляют собой углеродные радикалы, которые могут быть одинаковыми или разными. Тем не менее, термин «амино», как правило, используется в настоящем документе для описания первичного, вторичного или третичного амина, и квалифицированный специалист в данной области легко сможет их идентифицировать в соответствии с тем контекстом, в котором этот термин используется в настоящем раскрытии.
Термин «комбинированная терапия», используемый в настоящем документе, означает лечение пациента двумя или более терапевтическими способами (например, применяя ферментозаместительную терапию и терапию малыми молекулами), осуществляемыми по циклическим, чередующимся и/или одновременным схемам. Примеры схем лечения включают в себя, но не ограничиваются ими: (1) сначала ферментозаместительную терапию, затем терапию малыми молекулами; (2) сначала терапию малыми молекулами, затем ферментозаместительную терапию; (3) ферментозаместительную терапию, проводимую одновременно с терапией малыми молекулами и (4) любую комбинацию вышеуказанных вариантов. При необходимости, комбинированная терапия может представлять временное перекрывание применения терапевтических способов в зависимости от клинического течения данной болезни накопления у данного субъекта.
Термин «ферментозаместительная терапия» или «ERT (enzyme replacement therapy)», используемый в настоящем документе, означает введение пациенту, нуждающемуся в этом, экзогенно произведенного природного или рекомбинантного фермента. В случае болезни лизосомного накопления, например, пациент накапливает вредные уровни субстрата (т.е. сохраняемого материала) в лизосомах вследствие дефицита или дефекта фермента, ответственного за метаболизм данного субстрата, или вследствие дефицита активатора фермента, необходимого для адекватного функционирования фермента. Ферментозаместительную терапию пациенту проводят для снижения уровней (т.е. для уменьшения массы) накопленного субстрата в затронутых тканях. Таблица 1 предоставляет список болезней лизосомного накопления и для каждого такого заболевания указывает соответствующий дефицитный фермент и накапливаемый субстрат. В данной области известны некоторые способы ферментозаместительной терапии, применяемые для лечения болезней лизосомного накопления. В соответствии с комбинированной терапией согласно настоящему изобретению, лизосомальные ферменты, указанные в Таблице 1, можно применять для ферментозаместительной терапии в целях снижения уровней соответствующего субстрата у пациента с поставленным диагнозом соответствующей болезни лизосомного накопления.
Термин «эффективное количество», используемый в настоящем документе в отношении некоторого фермента или малой молекулы, вводимых субъекту при комбинированной терапии согласно настоящему изобретению, представляет собой количество, достаточное для улучшения клинического течения болезни лизосомного накопления, причем указанное клиническое улучшение измеряют по любому из множества определенных параметров, хорошо известных квалифицированному специалисту в данной области.
Принятые сокращения
ACN означает ацетонитрил.
DMF означает N,N-диметилформамид.
DMSO означает диметилсульфоксид.
EtOAc означает этилацетат.
EtOH означает этанол.
Основание Хунига означает диизопропилэтиламин («DIPEA»).
MeOH означает метанол.
NaOH означает гидроксид натрия.
THF означает тетрагидрофуран.
TFA означает трифторуксусную кислоту.
Дополнительные особенности и преимущества соединений, раскрытых в настоящем документе, будут видны из нижеследующего подробного описания определенных вариантов осуществления настоящего изобретения.
Краткое описание чертежей
Фиг. 1 представляет метаболический путь возможного синтеза Gb3 и лизо-Gb3. Документированные пути синтеза показаны черными стрелками, недокументированные (возможные) пути показаны серыми стрелками.
Фиг. 2A. Химическая структура (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)-тиазол-4-ил)-пропан-2-ил)-карбамата
Фиг. 2B. Химическая структура хинуклидин-3-ил-(2-(4'-фтор-[1,1'-бифенил]-3-ил)-пропан-2-ил)-карбамата
Фиг. 3. Концентрация Gb3 в почке (A) и сердце (B) у 12-месячных мышей с болезнью Фабри, которых лечили, вводя по 300 мг/кг/день соли L-винной кислоты с [2-(2',3'-дигидробензо[1,4]диоксин-6'-ил)-2-гидрокси-1-пирролидин-1-илметилэтил]-амидом (1R,2R)-октановой кислоты («GZ 638») или по 60 мг/кг/день (S)-2-гидроксисукцинатной соли (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)-тиазол-4-ил)-пропан-2-ил)-карбамата («GZ 452»).
Фиг. 4A. Календарный план исследования, показывающий начальное лечение мышей с болезнью Фабри, начатое в возрасте 3, 8 и 12 месяцев, которых лечили, вводя по 60 мг/кг/день GZ 452. Периодические заборы крови и мочи и испытания в тестах с горячей пластиной и активной камерой проводили в дни, указанные на плане.
Фиг. 4B. Концентрация Gb3 в моче (A) и в плазме (B) у 3- или 8-месячных мышей с болезнью Фабри, которым проводили начальное лечение, вводя по 60 мг/кг/день GZ 452. Лечение лекарственным средством (Rx) продолжали в течение 2 или 4 месяцев.
Фиг. 5. Концентрация Gb3 (A) и лизо-Gb3 (B) в ткани почки 12-месячных мышей с болезнью Фабри, которых либо не лечили (НЛ), либо лечили, вводя по 60 мг/кг/день GZ 452 в течение 4 месяцев (SRT - substrate reduction therapy, субстрат-редуцирующая терапия).
Фиг. 6A. Календарный план исследования, показывающий мышей с болезнью Фабри, которых лечили, вводя альфа-галактозидазу А (1 мг/кг каждые 2 месяца) или вводя по 60 мг/кг/день GZ 452, или сочетая 2 способа лечения, начиная с 3-месячного возраста. Периодические заборы крови и мочи и испытания в тестах с горячей пластиной проводили в дни, указанные на плане.
Фиг. 6B. Концентрации Gb3 (A и C) и лизо-Gb3 (C и D) в плазме (A и C) и в моче (B и D) у 5-месячных мышей с болезнью Фабри, которых лечили, вводя одну альфа-галактозидазу А (ERT - ферментозаместительная терапия), вводя одно соединение GZ 452 (SRT) или сочетая 2 способа (E+S) в течение 2 месяцев.
Фиг. 7. Анализ изоформ N-связанной ацильной цепи Gb3, выделенного из плазмы, мочи и почки мыши с болезнью Фабри.
Фиг. 8. Латентность (время до реакции) действия теплового раздражителя (горячая пластина при 55°С) у 10-месячных мышей с болезнью Фабри после 7 месяцев лечения с введением альфа-галактозидазы А (ERT), GZ 452 (SRT) или при сочетании двух способов (E+S) в сравнении с мышами, лечение которых не проводили (НЛ), и мышами дикого типа (ДТ).
Фиг. 9 Глюкозилцерамид (GluCer) и глюкозилсфингозин (GluSph) значительно повышены в мозге новорожденных мышей К14. Масс-спектрометрический анализ глюкозил- и галактозилцерамидов показывает, что (A) GluCer у мышей К14 (с моделью болезни Гоше у новорожденных животных, также известной как болезнь Гоше типа 2) был повышен в 10 раз по сравнению с мышами дикого типа в течение первых двух недель жизни, (B) уровни GalCer были в течение этого времени одинаковыми у мышей К14 и мышей дикого типа, (С) уровни GluSph в течение первых двух недель жизни у мышей К14 были не менее чем в 10 раз более высокими, чем у животных дикого типа того же возраста; уровни GluSph у животных дикого типа были ниже порога детектирования (<0,3 нг/мг). Точки на графике представляют средние значения и планки погрешности (стандартной ошибки среднего) для N=4.
Фиг. 10. Системное введение хинуклидин-3-ил-(2-(4'-фтор-[1,1'-бифенил]-3-ил)-пропан-2-ил)-карбамата (GZ 161) снижает уровни GluCer и GluSph в мозге мышей К14. Мышам К14 и мышам дикого типа, начиная с дня P4, ежедневно внутрибрюшинно вводили либо плацебо, либо 5 мг/кг соединения GZ 161 и в день P10 в мозге анализировали GluCer и GluSph. Животные, которых лечили соединением GZ 161, в это время были бессимптомными. Лечение мышей K14 соединением GZ 161 снижало уровни GluCer (A) и GluSph (В) приблизительно на 70 и 60%, соответственно. После лечения уровни обоих гликосфинголипидов оставались достоверно повышенными по сравнению с мышами дикого типа такого же возраста; генотип был подтвержден посмертным анализом ДНК. * p <0,05. N = 4 на группу.
Фиг. 11. Системное введение соединения GZ 161 уменьшает окрашивание CD68 во всем мозге мышей K14. Верхние панели: типичные картины иммуногистохимического окрашивания CD68, полученные в день P10 в гиппокампе, таламусе, стволе мозга и мозжечке мышей К14, которых, начиная с 4-го дня после рождения, лечили, ежедневно внутрибрюшинно вводя либо плацебо, либо соединение GZ 161, а также у мышей дикого типа. Нижние панели: количественные оценки интенсивности окрашивания в группах, показанных на верхних снимках, демонстрирующие, что результатом системного лечения соединением GZ 161 является значительное уменьшение числа CD68-положительных клеток во всех участках мозга. Похожее уменьшение наблюдали и в других структурах, таких как обонятельные луковицы и лобная кора (не показано). **p <0,01. N = 4 на группу.
Фиг. 12. Системное введение соединения GZ 161 уменьшает окрашивание F4/80 в некоторых участках мозга мышей K14. Верхние панели: типичные картины иммуногистохимического окрашивания CD68, полученные в день P10 в гиппокампе, таламусе, стволе мозга и мозжечке мышей К14, которых, начиная с 4-го дня, лечили, ежедневно внутрибрюшинно вводя либо плацебо, либо соединение GZ 161, а также у мышей дикого типа. Нижние панели: количественные оценки интенсивности окрашивания в группах, показанных на верхних снимках, демонстрирующие, что результатом системного лечения соединением GZ 161 является значительное уменьшение числа F4/80-положительных клеток в таламусе и лобной коре. Похожее уменьшение наблюдали и в других структурах, таких как обонятельные луковицы и лобная кора; в обеих структурах наблюдались статистически достоверные различия (не показано). * p <0,05. N = 4 на группу.
Фиг. 13. Системное введение соединения GZ 161 уменьшает глиоз у мышей K14. Верхние панели: типичные картины иммуногистохимического окрашивания GFAP, полученные в день P10 в гиппокампе, таламусе, стволе мозга и мозжечке мышей К14, которых, начиная с 4-го дня, лечили, ежедневно внутрибрюшинно вводя либо плацебо, либо соединение GZ 161, а также у мышей дикого типа. Нижние панели: количественные оценки интенсивности окрашивания в группах, показанных на верхних снимках, демонстрирующие, что результатом системного лечения соединением GZ 161 является значительное уменьшение числа GFAP-положительных клеток в гиппокампе и мозжечке; в обеих структурах наблюдались статистически достоверные различия (на показано).
Фиг. 14. Системное введение соединения GZ 161 увеличивает медиану продолжительности жизни мышей K14. Мышам K14 ежедневно, начиная с 4-го дня, внутрибрюшинно инъецировали либо плацебо, либо соединение GZ 161, или проводили комбинированное лечение, состоявшее из трех интрацеребровентрикулярных (ICV) инъекций rhGC, выполненных в дни P1, 2, 3, совместно с ежедневными внутрибрюшинными инъекциями соединения GZ 161, начиная с дня P4. У мышей, получавших плацебо, медиана продолжительности жизни составляла 15 дней (N = 25); у мышей, которых лечили соединением GZ 161, медиана продолжительности жизни составляла 18 дней (N = 12; p<0,0001 по сравнению с введением плацебо); у мышей, которым совместно вводили GZ 161 и rhGC медиана продолжительности жизни составляла 26 дней (N = 13).
Фиг. 15. По-видимому, соединение GZ 161 проникает через гематоэнцефалический барьер. Системное введение (по 20 мг/кг/день в корме) соединения GZ 161 беременным мышам дикого типа уменьшает количество GluCer в гомогенатах целого мозга новорожденных мышей (в день Р0). N = 7; p <0,0001)
Фиг. 16. Лечение мышей K14 соединением GZ 161, вводимым внутриматочно, оказывало минимальное влияние на выживаемость. У мышей К14, которым ежедневно, начиная с дня Р4, внутрибрюшинно вводили плацебо, медиана продолжительности жизни составляла 14 дней (N = 13). Системное введение (по 20 мг/кг/день в корме) соединения GZ 161 беременным мышам K14, а затем ежедневное, начиная с дня Р0, внутрибрюшинное введение соединения GZ 161 (5 мг/кг) новорожденным продлевало продолжительность жизни до 19 дней (N = 13) - результат, аналогичный полученному при лечении новорожденных крыс с ежедневным системным введением 5 мг/кг соединения GZ 161, начиная с дня P4 (N = 12).
Фиг. 17. Уровни Gb3 в ткани почки 12-месячных самцов и самок крыс с болезнью Фабри, которых лечили соединениями GZ 452, GZ 161 и GZ 638. Лечение мышей начинали приблизительно в 8-месячном возрасте и продолжали в течение 4 месяцев, вводя 60 мг/кг/день GZ 452, 120 мг/кг/день GZ 452, 20 мг/кг/день GZ 161, 300 мг/кг/день GZ 638, параллельно с контрольными группами мышей дикого типа и мышей, лечение которых не проводили.
Подробное описание
Хотя в данном разделе будут описаны конкретные варианты осуществления настоящего раскрытия со ссылками на препаративные процедуры и схемы, следует понимать, что такие варианты осуществления настоящего изобретения приведены только для примера и являются лишь иллюстрациями не более чем малого числа из многих возможных конкретных вариантов осуществления настоящего изобретения, которые могут представлять области применения принципов настоящего раскрытия. Квалифицированным специалистам в данной области техники будут очевидны возможности разнообразных изменений и модификаций, полезных для настоящего изобретения, которые считаются входящими в объем настоящего изобретения и соответствующими его сущности, как дополнительно определено в прилагаемых пунктах формулы изобретения.
Если не определено иначе, все технические и научные термины, используемые в настоящем документе, имеют смысл, понимаемый специалистом с обычной квалификацией в той области, к которой принадлежит настоящее раскрытие. Хотя при практическом применении и испытаниях можно использовать и другие соединения и методики, в контексте нижеследующих препаративных процедур и схем в настоящем разделе описаны определенные предпочтительные методики.
ПРЕПАРАТИВНАЯ ПРОЦЕДУРА A
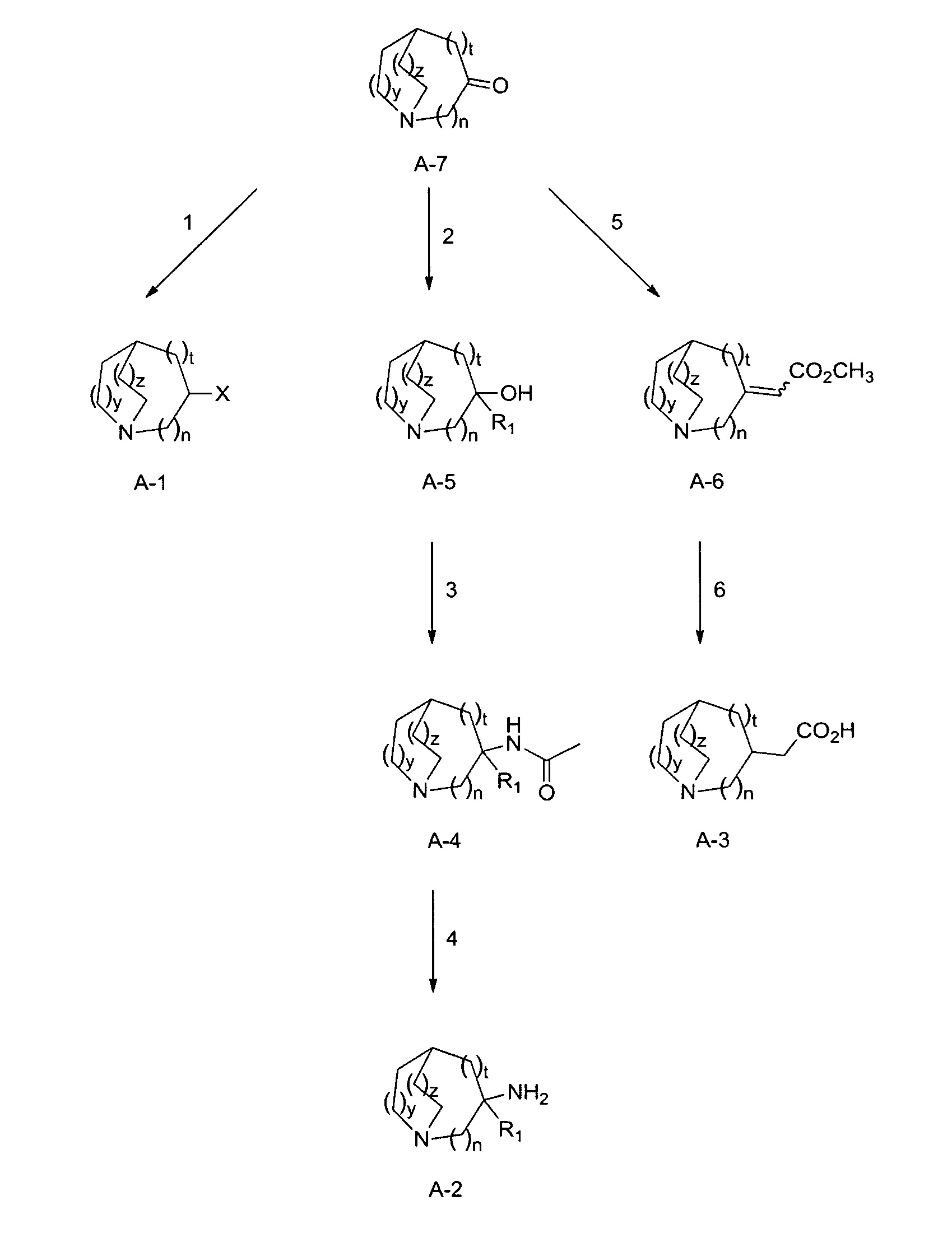
ПРЕПАРАТИВНАЯ ПРОЦЕДУРА B
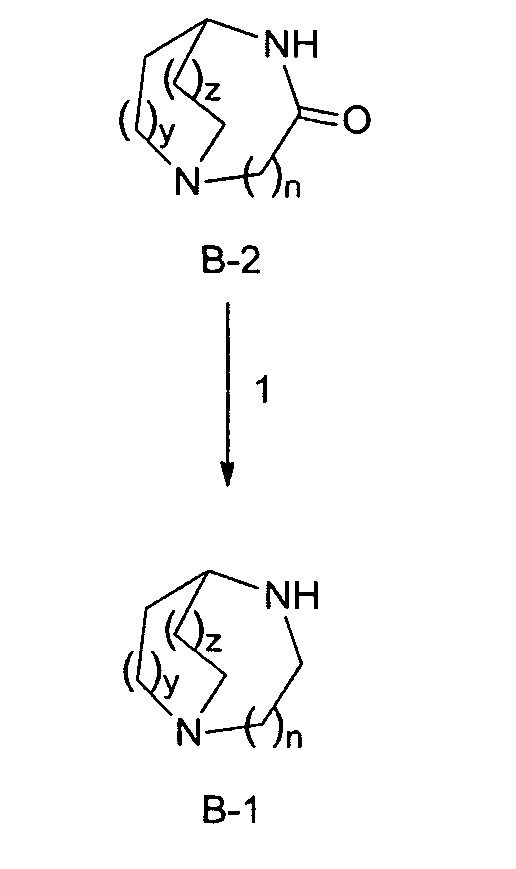
ПРЕПАРАТИВНАЯ ПРОЦЕДУРА C

ПРЕПАРАТИВНАЯ ПРОЦЕДУРА D
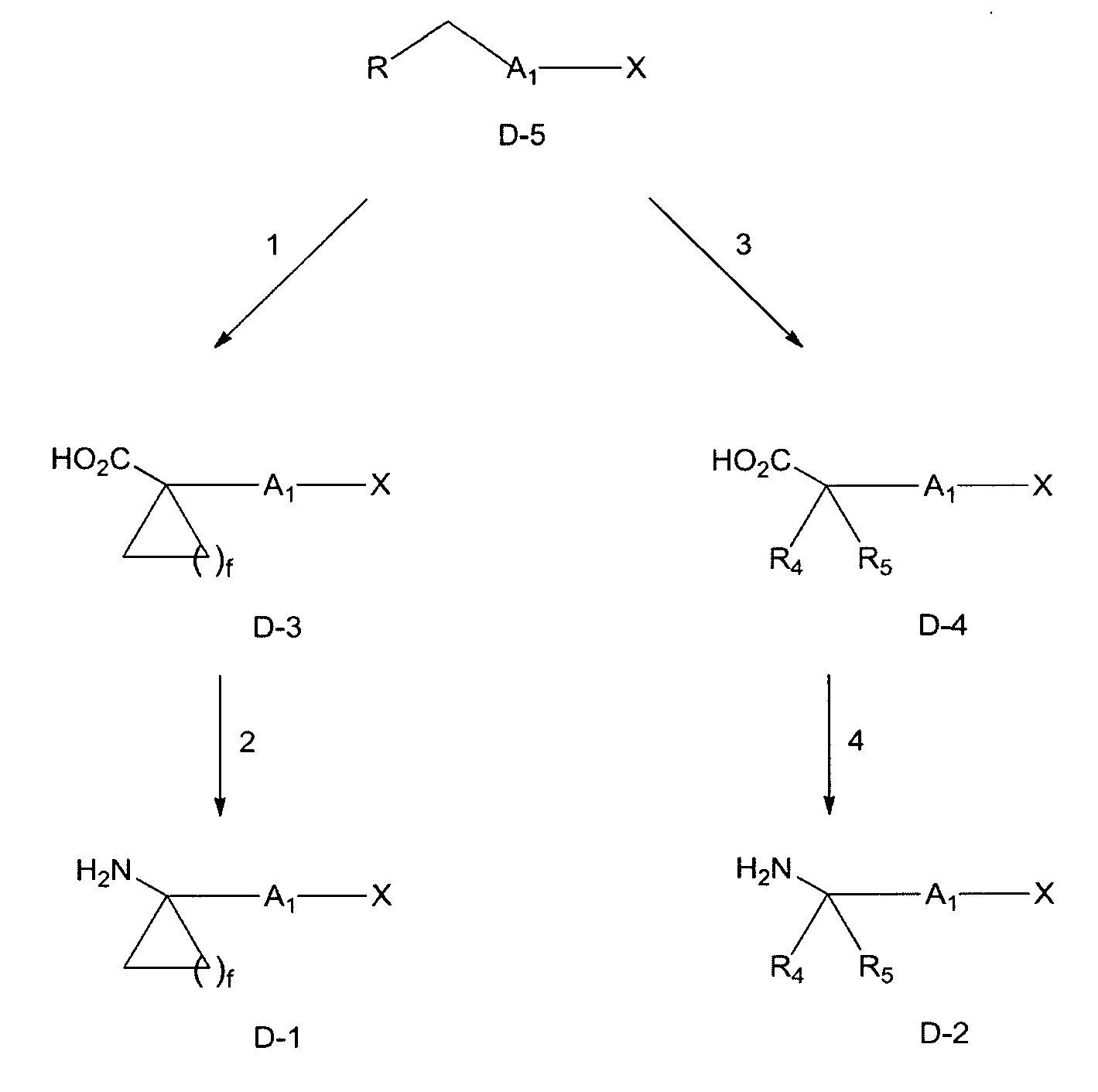
ПРЕПАРАТИВНАЯ ПРОЦЕДУРА E
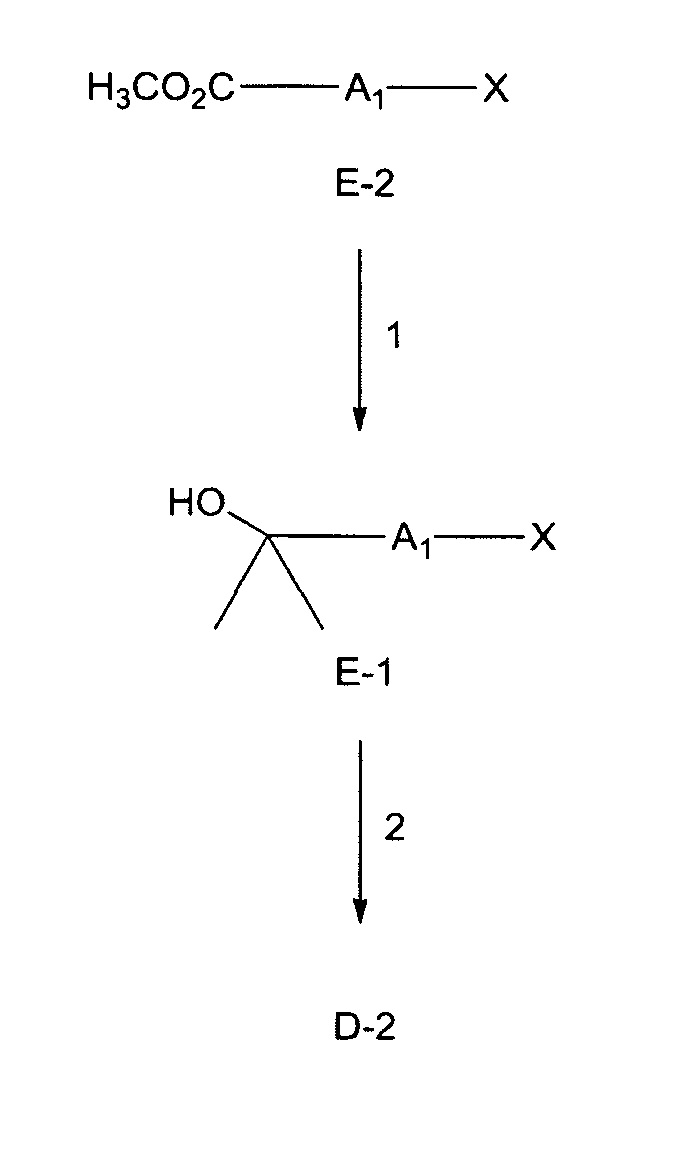
СХЕМА 1
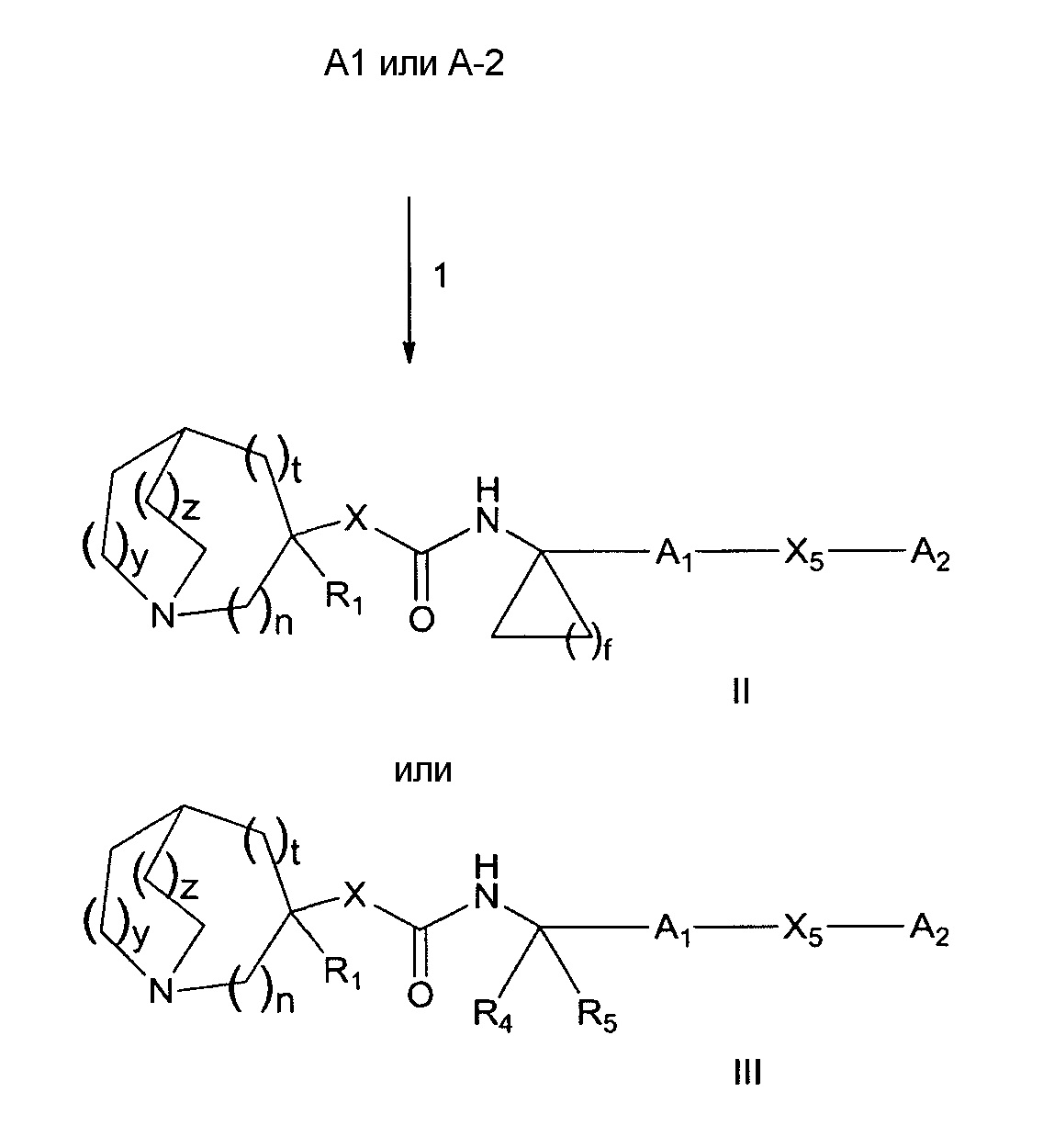
СХЕМА 2
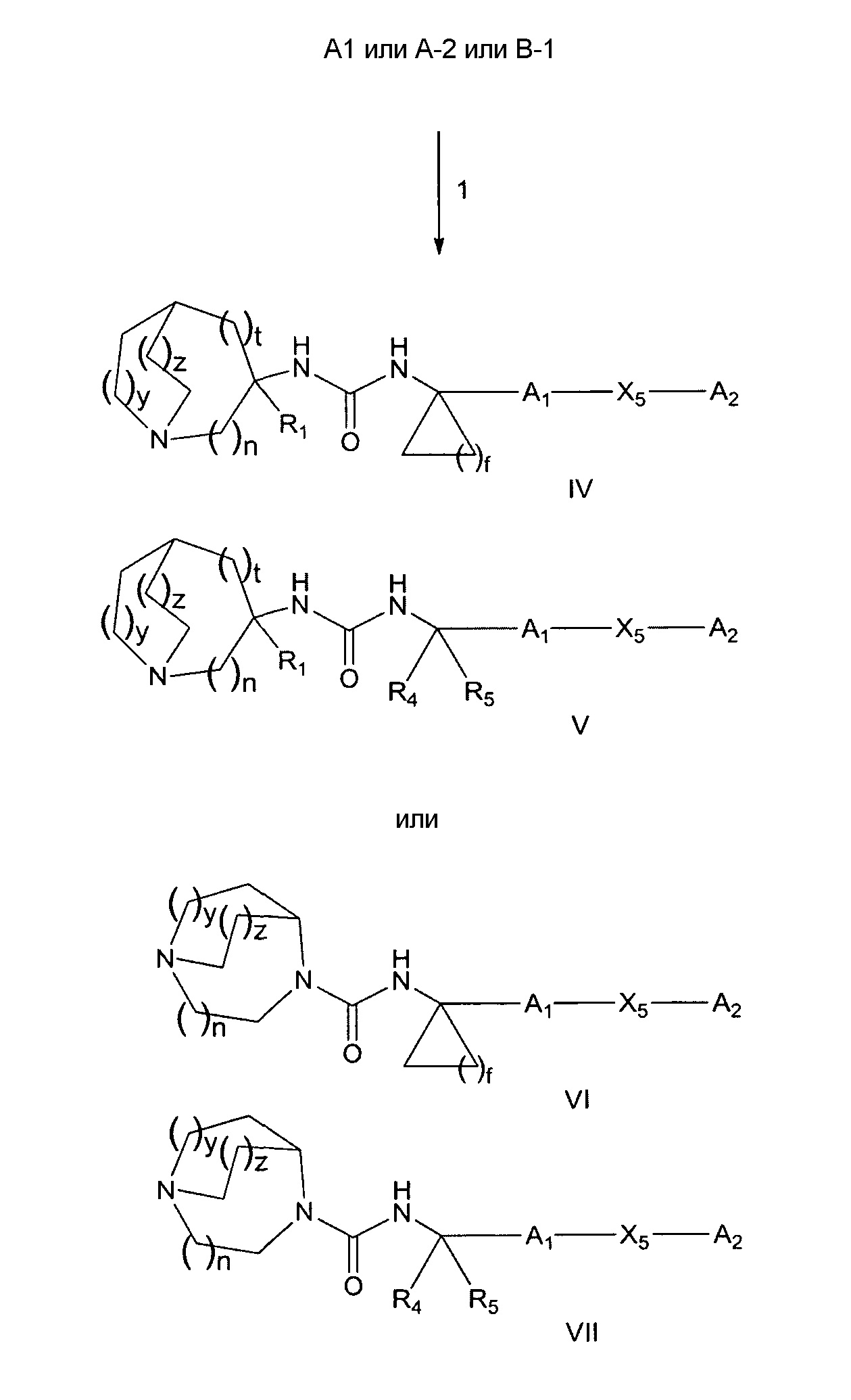
СХЕМА 3
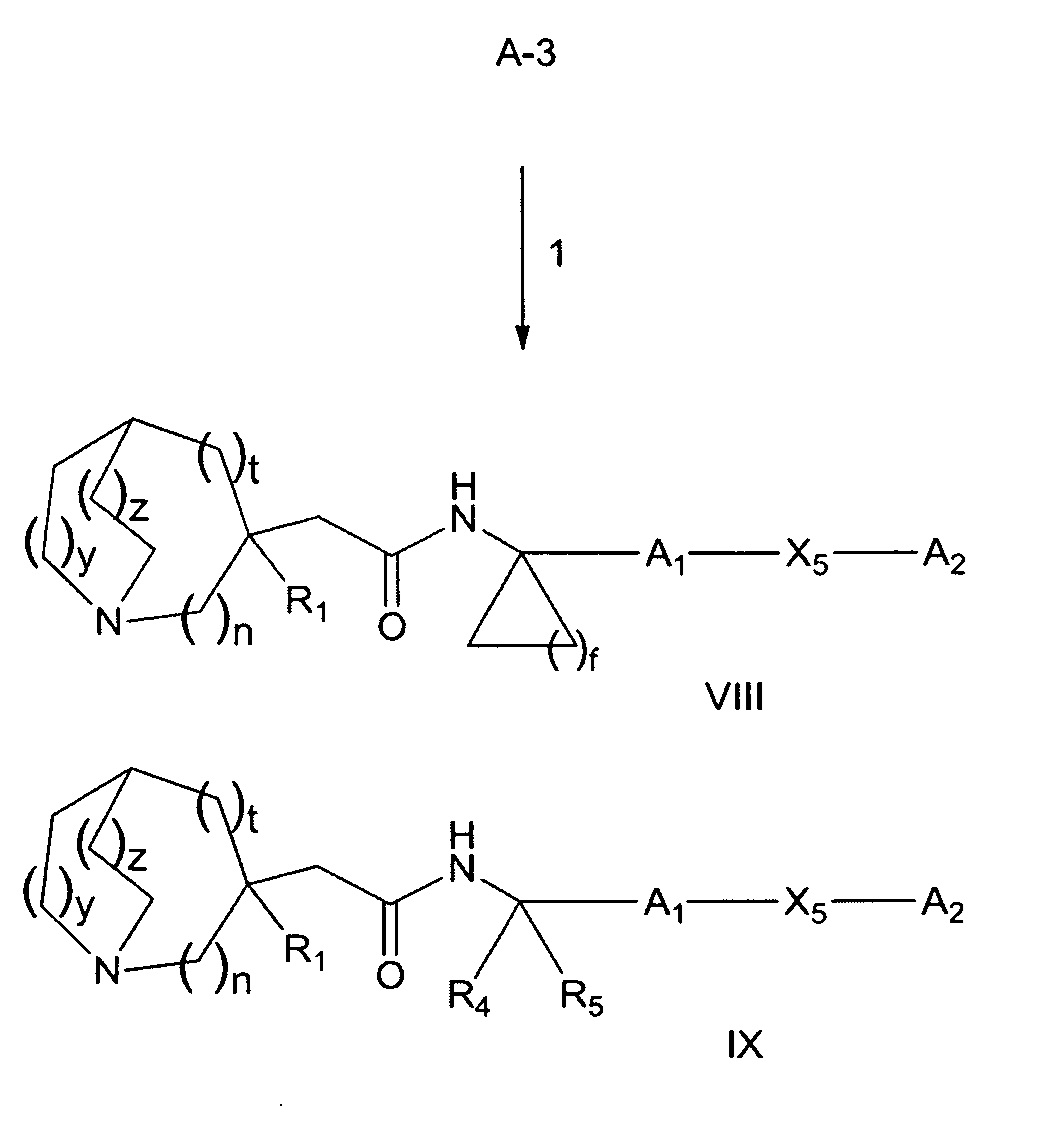
В реакции 1 Препаративной процедуры A соединение Формулы A-7 преобразуют в соответствующее соединение Формулы A-1, где X представляет собой OH, восстанавливая A-7 некоторым восстановителем (предпочтительно, литийалюминийгидридом) в апротонном растворителе, таком как тетрагидрофуран. Реакционную смесь перемешивают при температуре от 0°C до комнатной температуры в течение времени от приблизительно 15 минут до приблизительно 2 часов (предпочтительно, приблизительно 30 минут). В качестве альтернативы, соединение Формулы A-7 преобразуют в соответствующее соединение Формулы A-1, где X представляет собой OH, восстанавливая A-7 водородом под давлением приблизительно 1 атм в присутствии катализатора (предпочтительно, оксида платины) и полярного растворителя, такого как метанол или этанол, в течение 2-6 часов (предпочтительно, 4 часов). В качестве альтернативы, соединение Формулы A-7 преобразуют в соответствующее соединение Формулы A-1, где X представляет собой NH, проводя реакцию A-7 с гидрохлоридом гидроксиламина и ацетатом натрия в полярном растворителе, таком как этанол, метанол, изопропанол (предпочтительно, в изопропаноле). Реакционную смесь перемешивают при температуре 50-80°C в течение 2-7 часов (предпочтительно, 3 часов). После этого соединение, образованное, как указано выше, действуя восстановителем, преобразуют в соединение Формулы A-1 - предпочтительно, применяя металлический натрий в полярном протонном растворителе, таком как этанол, метанол, пропанол (предпочтительно, в н-пропаноле). Реакционную смесь перемешивают в течение ночи при 50-80°C (предпочтительно, при температуре кипячения растворителя с обратным холодильником).
В реакции 2 Препаративной процедуры A соединение Формулы A-7 преобразуют в соответствующее соединение Формулы A-5, где R1, n и z являются такими, как определено выше, добавляя раствор R1-магнийбромида в простом эфире к раствору A-7 в апротонном растворителе, таком как простой эфир, при температуре от приблизительно -60°C до приблизительно -90°C (предпочтительно, приблизительно при -78°C) в течение времени от приблизительно 1 часа до приблизительно 4 часов (предпочтительно, приблизительно 2 часа). В качестве альтернативы, соединение Формулы A-7 может реагировать с R1-литием, образуя соединение Формулы A-5.
В реакции 3 Препаративной процедуры A, соединение Формулы A-5 преобразуют в соответствующее соединение Формулы A-4, где R1, n и z являются такими, как определено выше, обрабатывая A-5 сильной кислотой (предпочтительно, серной кислотой) в присутствии ацетонитрила. Реакционную смесь перемешивают в течение ночи при комнатной температуре.
В реакции 4 Препаративной процедуры A, соединение Формулы A-4 преобразуют в соответствующее соединение Формулы A-3, где R1, n и z являются такими, как определено выше, обрабатывая A-4 кислотой (предпочтительно, хлористоводородной кислотой). Реакционную смесь перемешивают при кипячении с обратным холодильником в течение времени от 18 часов до 72 часов (предпочтительно, 24 часа) и подщелачивают до pH 8, обрабатывая неорганическим основанием, таким как гидроксид натрия, в водном растворе.
В реакции 5 Препаративной процедуры A, соединение Формулы A-7 преобразуют в соответствующее соединение Формулы A-6, где R1, n и z являются такими, как определено выше, проводя реакцию A-7 с илидом трифенилфосфония и получая соответствующее алкеновое соединение Формулы A-6. Реакционную смесь перемешивают при комнатной температуре в течение ночи.
В реакции 6 Препаративной процедуры A, соединение Формулы A-6 преобразуют в соответствующее соединение Формулы A-3, где R1, n и z являются такими, как определено выше, восстанавливая A-6 водородом под давлением приблизительно 1 атм в присутствии катализатора (предпочтительно, палладия на угле) и полярного растворителя, такого как метанол, этанол или этилацетат. Реакционную смесь перемешивают при комнатной температуре в течение времени от приблизительно 2 часов до приблизительно 24 часов (предпочтительно, приблизительно 18 часов). После этого образованное таким путем соединение обрабатывают основанием (предпочтительно, гидроксидом лития) в смеси растворителя, такого как тетрагидрофуран, метанол и вода, получая соединение A-3. Реакционную смесь перемешивают в течение ночи при комнатной температуре.
В реакции 1 Препаративной процедуры B соединение Формулы B-2 преобразуют в соответствующее соединение Формулы B-1, восстанавливая B-2 некоторым восстановителем (предпочтительно, литийалюминийгидридом) в апротонном растворителе, таком как тетрагидрофуран. Реакционную смесь перемешивают при температуре от 0°C до комнатной в течение времени от приблизительно 15 минут до приблизительно 2 часов (предпочтительно, приблизительно 30 минут).
В реакции 1 Препаративной процедуры C соединение C-4 преобразуют в соответствующее соединение Формулы C-3, где X представляет собой бром или хлорид, проводя реакцию C-4 с бороновой кислотой в присутствии катализатора, предпочтительно, 1,1'-бис-(дифенилфосфино)ферроцена/дихлорида палладия(II) и карбоната калия. Реакцию проводят в микроволновой печи в смеси диметоксиэтана и воды при температуре от приблизительно 130°C до приблизительно 170°C (предпочтительно, приблизительно при 150°C) в течение времени от приблизительно 15 мин до приблизительно 1 часа (предпочтительно, приблизительно 30 мин). В качестве альтернативы, реакцию можно проводить, применяя растворитель, такой как диоксан, и перемешивая в течение ночи при 100°C при обычном нагревании.
В реакции 2 Препаративной процедуры C соединение C-3 преобразуют в соответствующее соединение Формулы C-1, где f равно 1-8 и A1, X5 и A2 являются такими, как определено выше, добавляя этилмагнийбромид по каплям к смеси C-3 и изопропоксида титана в простом эфире. Реакционную смесь перемешивают при температуре от приблизительно -50°C до приблизительно -90°C (предпочтительно, приблизительно при -70°C). Смеси, полученной в результате этого, дают возможность нагреваться приблизительно до 20-30°C (предпочтительно, приблизительно до 25°C) и дополнительно перемешиваться в течение времени от приблизительно 30 минут до приблизительно 2 часов (предпочтительно, приблизительно 1 час). Затем к смеси по каплям добавляют диэтилэфират трифторида бора при температуре от приблизительно 20°C до приблизительно 30°C (предпочтительно, приблизительно при 25°C).
В реакции 3 Препаративной процедуры C соединение C-3 преобразуют в соответствующее соединение Формулы C-2, где A1, X5 и A2 являются такими, как определено выше, сначала перемешивая суспензию хлорида церия (III) в апротонном растворителе, таком как тетрагидрофуран, при комнатной температуре в течение времени от приблизительно 30 минут до приблизительно 2 часов (предпочтительно, приблизительно 1 час). Суспензию, полученную в результате этого, охлаждают до температуры от приблизительно -60°C до приблизительно -90°C (предпочтительно, приблизительно -78°C) и добавляют литийорганическое соединение (предпочтительно, метиллитий) в эфирном растворе. Получающемуся в результате этого церийорганическому комплексу дают возможность формироваться в течение времени от приблизительно 30 минут до приблизительно 2 часов (предпочтительно, приблизительно 1 час), после чего добавляют C-3 в апротонном растворителе, таком как тетрагидрофуран. Затем смесь, полученную в результате этого, нагревают до комнатной температуры и перемешивают в течение времени от приблизительно 16 часов до приблизительно 20 часов (предпочтительно, приблизительно 18 часов).
В реакции 1 Препаративной процедуры D соединение D-5, где R представляет собой CO2Et или CN, а X представляет собой бром или хлорид, преобразуют в соответствующее соединение Формулы D-3, проводя реакцию D-5 с дигалоидным алкилом, таким как 1,2-дибромэтан. После этого образованное таким путем соединение обрабатывают неорганическим основанием, таким как гидроксид лития или гидроксид калия, в смеси растворителя, такого как тетрагидрофуран, метанол, гликоль и вода, получая соединение D-3, где f равно 1-8. Реакционную смесь перемешивают в течение ночи при температуре от 25°C до 130°C. В качестве альтернативы, для образования соответствующего соединения Формулы D-3, где X представляет собой X5-A2, соединение D-5 должно сначала прореагировать по процедуре, обсуждавшейся выше для реакции 1 Препаративной процедуры C.
В реакции 2 Препаративной процедуры D соединение D-3 преобразуют в соответствующее соединение Формулы D-1, проводя реакцию D-3 с основанием, таким как триэтиламин, и дифенилфосфорилазидом в апротонном растворителе, таком как толуол. Реакционную смесь нагревают до температуры в диапазоне 80-110°C (предпочтительно, при 110°C) в течение времени от 15 мин до 1 часа (предпочтительно, 30 минут). Образованное таким путем промежуточное соединение затем обрабатывают трет-бутиловым спиртом в течение ночи при 60-110°C (предпочтительно, при 90°C). После этого образованный таким путем карбамат преобразуют в соответствующее соединение Формулы D-1, где f равно 1-8, обрабатывая его в кислых средах, используя, предпочтительно, трифторуксусную кислоту в дихлорметане, при комнатной температуре в течение времени от 30 мин до 5 часов (предпочтительно, 2 часа).
В реакции 3 Препаративной процедуры D соединение D-5, где R представляет собой CO2Et или CN, а X представляет собой бром или хлорид, преобразуют в соответствующее соединение Формулы D-4, проводя реакцию D-5 с галоидным алкилом, таким как метилйодид (MeI). После этого образованное таким путем соединение обрабатывают неорганическим основанием, таким как гидроксид лития или гидроксид калия, в смеси растворителя, такого как тетрагидрофуран, метанол, гликоль и вода, получая соединение D-4. Реакционную смесь перемешивают в течение ночи при температуре 25-130°C. В качестве альтернативы, для образования соответствующего соединения Формулы D-4, где X представляет собой X5-A2, соединение D-5 должно сначала прореагировать согласно процедуре, обсуждавшейся выше для реакции 1 Препаративной процедуры C.
В реакции 4 Препаративной процедуры D соединение D-4 преобразуют в соответствующее соединение Формулы D-2, проводя реакцию D-4 с основанием, таким как триэтиламин, и дифенилфосфорилазидом в апротонном растворителе, таком как толуол. Реакционную смесь нагревают до температуры 80-110°C (предпочтительно, при 110°C) в течение времени от 15 мин до 1 часа (предпочтительно, 30 минут). Образованное таким путем промежуточное соединение затем обрабатывают трет-бутиловым спиртом в течение ночи при 60-110°C (предпочтительно, при 90°C). После этого образованный таким путем карбамат преобразуют в соответствующее соединение Формулы D-1, обрабатывая его в кислых средах, используя, предпочтительно, трифторуксусную кислоту в дихлорметане при комнатной температуре в течение времени от 30 мин до 5 часов (предпочтительно, 2 часа).
В реакции 1 Препаративной процедуры E соединение Формулы E-2, где X представляет собой бромид или хлорид, преобразуют в соответствующее соединение Формулы E-1, проводя реакцию E-2 с метилмагнийбромидом в простом эфире при температуре от приблизительно -60°C до приблизительно -90°C (предпочтительно, приблизительно при -78°C) в течение времени от приблизительно 30 мин до приблизительно 3 часов (предпочтительно, приблизительно 2 часа). В качестве альтернативы, для образования соответствующего соединения Формулы E-1, где X представляет собой X5-A2, соединение E-2 должно сначала прореагировать согласно процедуре, обсуждавшейся выше для реакции 1 Препаративной процедуры C.
В реакции 2 Препаративной процедуры E соединение Формулы E-1 преобразуют до соответствующего соединения D-2, обрабатывая E-1 сильной кислотой (предпочтительно, серной кислотой) в присутствии хлорацетонитрила. Реакционную смесь перемешивают в течение ночи при комнатной температуре. После этого образованное таким путем соединение обрабатывают тиомочевиной в полярном протонном растворителе, таком как этанол, в течение ночи при 80°C для образования соответствующего соединения Формулы D-2. В качестве альтернативы, E-1 обрабатывают азидом натрия и трифторуксусной кислотой в апротонном растворителе, таком как дихлорметан, при температуре от -10°C до комнатной (предпочтительно, при 0°C). Образованное таким путем соединение восстанавливают в присутствии трифенилфосфина в растворе тетрагидрофурана и воды, образуя соответствующее соединение Формулы D-2. Реакционную смесь перемешивают при температуре 25-80°C (предпочтительно, при комнатной температуре) в течение времени от 2 часов до 24 часов (предпочтительно, 18 часов).
В реакции 1 Схемы 1 соединения Формулы A-1 или A-2 преобразуют в соответствующие соединения Формулы II, где f равно 1-8, или Формулы III, соответственно, добавляя трифосген к суспензии C-1 или C-2 и триэтиламина в апротонном растворителе, таком как тетрагидрофуран. Реакционную смесь перемешивают при комнатной температуре в течение приблизительно 5-20 минут (предпочтительно, приблизительно 15 минут) и добавляют небольшое количество простого эфира. Отфильтровывают образовавшуюся триэтиламмонийную соль. Отдельно, при 0°C или комнатной температуре, к суспензии A-1 или A-2, где X представляет собой OH или NH, в апротонном растворителе, таком как тетрагидрофуран, добавляют гидрид натрия. Реакционную смесь перемешивают при комнатной температуре в течение приблизительно 5-20 минут (предпочтительно, приблизительно 15 минут) и по каплям добавляют раствор изоцианата в смеси тетрагидрофурана с простым эфиром, полученный как описано выше. В качестве альтернативы, соединения Формулы II и III можно получать, проводя реакцию соединения D3 или D4 с A-1 и A-2 в присутствии основания, такого как триэтиламин, и дифенилфосфорилазида в апротонном растворителе, таком как толуол, как описано в процедуре, обсуждавшейся выше для реакции 4 Препаративной процедуры D.
В реакции 1 Схемы 2 соединения Формулы A-1, A-2 или B-1 преобразуют в соответствующие соединения Формулы IV, V, VI и VII, где f равно 1-8, соответственно, добавляя трифосген к суспензии C-1, C-2, D-1 или D-2 и триэтиламина в апротонном растворителе, таком как тетрагидрофуран или толуол. Реакционную смесь перемешивают при комнатной температуре в течение приблизительно 5-20 минут (предпочтительно, приблизительно 15 минут) и добавляют небольшое количество простого эфира. После этого A-1 или A-2, где X представляет собой NH, добавляют к раствору изоцианата, полученному как описано выше, и реакционную смесь перемешивают при температуре 25-100°C (предпочтительно, при комнатной температуре) приблизительно в течение 2-24 часов (предпочтительно, 18 часов).
В реакции 1 Схемы 3 соединение Формулы A-3 преобразуют в соответствующие соединения Формулы VIII, где f равно 1-8, и Формулы IX, соответственно, проводя реакцию A3 с C1, C-2, D-1 или D-2 путем пептидной конденсации с использованием карбодиимидного связующего средства, такого как 1-этил-3-(3-диметиламинопропил)-карбодиимид, и 1-гидроксибензотриазола или гексафторфосфата 2-(1H-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония в растворителе, таком как тетрагидрофуран или диметилформамид. Реакционную смесь перемешивают при комнатной температуре в течение ночи.
Хотя в настоящем разделе будут описаны конкретные варианты осуществления настоящего раскрытия со ссылками на препаративные процедуры и схемы, следует понимать, что такие варианты осуществления настоящего изобретения приведены только для примера и являются лишь иллюстрациями не более чем малого числа из многих возможных конкретных вариантов осуществления настоящего изобретения, которые могут представлять области применения принципов настоящего раскрытия. Квалифицированным специалистам в данной области техники будут очевидны возможности разнообразных изменений и модификаций, полезных для настоящего изобретения, которые следует считать входящими в объем настоящего изобретения и соответствующими его сущности, как дополнительно определено в прилагаемых пунктах формулы изобретения.
Если не определено иначе, все технические и научные термины, используемые в настоящем документе, имеют смысл, понимаемый специалистом с обычной квалификацией в той области, к которой принадлежит настоящее раскрытие. Хотя при практическом применении и испытаниях можно использовать и другие соединения и методики, в контексте нижеследующих препаративных процедур и схем в настоящем разделе описаны определенные предпочтительные методики.
Все фармацевтически приемлемые соли, пролекарства, таутомеры, гидраты и сольваты соединений, раскрытых в настоящем документе, также входят в объем настоящего раскрытия.
Раскрытые в настоящем документе соединения, которые по свой природе являются основными, обычно способны образовывать множество разных солей с разнообразными неорганическими и/или органическими кислотами. Хотя такие соли, как правило, являются фармацевтически приемлемыми для введения животным и людям, на практике часто желательно сначала выделять соединение из реакционной смеси в виде фармацевтически неприемлемой соли, а затем просто преобразовывать ее назад в свободное основание этого соединения, обрабатывая щелочным реагентом, после чего преобразовывать это свободное основание в фармацевтически приемлемую кислотно-аддитивную соль. Кислотно-аддитивные соли основных соединений можно легко получать, применяя традиционные технические приемы - например, обрабатывая основное соединение существенно эквивалентным количеством выбранной минеральной или органической кислоты в среде водного растворителя или в подходящем органическом растворителе, таком как, например, метанол или этанол. После осторожного выпаривания растворителя получают желаемую твердую соль.
Кислоты, которые можно применять для получения фармацевтически приемлемых кислотно-аддитивных солей основных соединений, являются такими, которые образуют нетоксичные кислотно-аддитивные соли, т.е. соли, содержащие фармакологически приемлемые анионы, такие как хлоридные, бромидные, йодидные, нитратные, сульфатные или бисульфатные, фосфатные или кислые фосфатные, ацетатные, лактатные, цитратные или кислые цитратные, тартратные или битартратные, сукцинатные, малеатные, фумаратные, глюконатные, сахаратные, бензоатные, метансульфонатные и памоатные [т.е. 1,1'-метилен-бис-(2-гидрокси-3-нафтоатные)] соли.
Раскрытые в настоящем документе соединения, которые по свой природе являются кислотными (например, содержат СООН или тетразольный фрагмент), как правило, способны образовывать множество разных солей с разнообразными неорганическими и/или органическими основаниями. Хотя такие соли, как правило, являются фармацевтически приемлемыми для введения животным и людям, на практике часто желательно сначала выделять соединение из реакционной смеси в виде фармацевтически неприемлемой соли, а затем просто преобразовывать ее назад в свободную кислоту этого соединения, обрабатывая кислотным реагентом, после чего преобразовывать это свободную кислоту в фармацевтически приемлемую основно-аддитивную соль. Эти основно-аддитивные соли можно легко получать, применяя традиционные технические приемы - например, обрабатывая соответствующие кислотные соединения водным раствором, содержащим желаемые фармакологически приемлемые катионы, и затем досуха выпаривая раствор, полученный в результате этого (предпочтительно, при пониженном давлении). В качестве альтернативы, их также можно получать, смешивая растворы кислотных соединений в низших спиртах с алкоксидом желательного щелочного металла, а затем так же, как описано выше, досуха выпаривая раствор, полученный в результате. В обоих случаях, для обеспечения полноты реакции и максимального выхода продукта в виде желаемой твердой соли рекомендуется применять стехиометрические количества реагентов.
Основания, которые можно применять для получения фармацевтически приемлемых основно-аддитивных солей основных соединений, являются такими, которые могут образовывать нетоксичные основно-аддитивные соли, т.е. соли, содержащие фармацевтически приемлемые катионы, такие как катионы щелочных металлов (например, калия и натрия), катионы щелочноземельных металлов (например, кальция и магния), аммоний или другие водорастворимые амино-аддитивные соли, такие как N-метилглюкамин-(меглумин), низшие алканоламмонийные катионы и другие такие основания органических аминов.
В объем настоящего раскрытия входят и изотопно меченые соединения. Термин «изотопно меченые соединения», используемый в настоящем документе, относится к раскрытым в настоящем документе соединениям, включая их фармацевтические соли и пролекарства (каждое из которых являются таким, как описано в настоящем документе), в которых один или более из атомов заменен атомом, имеющим атомную массу или массовое число, отличное от той атомной массы или того массового числа, которое обычно находят в природе. Примеры изотопов, которые можно вводить в соединения, раскрытые в настоящем документе, включают в себя изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как2H,3H,13C,14C,15N,18O,17O,31P,32P,35S,18F и36Cl, соответственно.
Изотопное мечение соединений, раскрытых в настоящем документе, дает возможность применять их в исследованиях тканевого распределения лекарственных средств и/или субстратов. Благодаря легкости их получения и детектирования, особенно предпочтительны в этом отношении соединения, меченые тритием (3H) и углеродом-14 (14C). Кроме того, замена более тяжелыми изотопами, такими как дейтерий (2H), может предоставить и определенные терапевтические преимущества (например, более долгий период полуэлиминации in vivo или пониженные требования к дозированию) и поэтому может быть предпочтительной в некоторых обстоятельствах. Изотопно меченые соединения, раскрытые в настоящем документе, включая их фармацевтические соли и пролекарства, можно получать любыми средствами, известными в данной области техники.
В пределах объема настоящего раскрытия находятся стереоизомеры (например, цис- и транс-изомеры) и все оптические изомеры соединения, раскрытого в настоящем документе (например, R- и S-энантиомеры), а также рацемические, диастереомерные и другие смеси таких изомеров.
Соединения, соли, пролекарства, гидраты и сольваты, раскрытые в настоящем документе, могут существовать в нескольких таутомерных формах, включая енольную и иминную форму, а также кето- и енаминную форму и их геометрические изомеры и смеси. В растворе таутомеры существуют в виде смесей набора таутомерных форм. В твердой форме обычно преобладает один таутомер. Но даже если может быть описан лишь один таутомер, в пределах объема настоящего раскрытия находятся все таутомеры.
В пределах объема настоящего раскрытия находятся и атропоизомеры. Атропоизомерами называют соединения, которые можно разделить на ротационно ограниченные изомеры.
Настоящее раскрытие также предоставляет фармацевтические композиции, содержащие, по меньшей мере, одно раскрытое в настоящем документе соединение и, по меньшей мере, один фармацевтически приемлемый носитель. Указанный фармацевтически приемлемый носитель может представлять собой любой такой носитель, известный в данной области техники, включая те, которые описаны, например, в Remington's Pharmaceutical Sciences, Mack Publishing Co., (A. R. Gennaro edit. 1985). Фармацевтические композиции соединений, раскрытых в настоящем документе, можно изготавливать традиционными средствами, известными в данной области техники, включая, например, смешивание, по меньшей мере, одного раскрытого в настоящем документе соединения с фармацевтически приемлемым носителем.
Раскрытые в настоящем документе фармацевтические композиции можно применять для животного или человека. В частности, раскрытое в настоящем документе соединение можно вводить в рецептуру фармацевтической композиции для перорального, буккального, парентерального (например, внутривенного, внутримышечного или подкожного), местного, ректального или интраназального введения или для введения посредством ингаляции или инсуффляции.
Соединения, раскрытые в настоящем документе, можно также предоставлять в виде препаратов для пролонгированной доставки, изготавливаемых посредством способов, хорошо известных специалистам с обычной квалификацией в данной области техники. Примеры таких препаратов можно найти в патентах США №№ 3119742, 3492397, 3538214, 4060598 и 4173626.
Фармацевтическая композиция для перорального введения может принимать форму, например, таблетки или капсулы, изготавливаемых традиционными средствами с фармацевтически приемлемыми эксципиентами (одним или более), такими как связующие средства (например, пептизированный кукурузный крахмал, поливинилпирролидон или гидроксипропилметилцеллюлоза); наполнитель (например, лактоза, микрокристаллическая целлюлоза или фосфат кальция); смазывающее средство (например, стеарат магния, тальк или силикагель); дезинтегрант (например, картофельный крахмал или натриевая соль гликолата крахмала); и/или смачивающее средство (например, лаурилсульфат натрия). Применяя способы, хорошо известные в данной области техники, на таблетки можно наносить покрытия. Жидкие препараты для перорального введения могут принимать форму, например раствора, сиропа или суспензии или они могут быть представлены в виде сухого продукта для разведения водой или другой подходящей основой перед использованием. Такие жидкие препараты можно изготавливать традиционными средствами с фармацевтически приемлемыми добавками (одной или более), такими как суспендирующее средство (например, сироп сорбита, метилцеллюлоза или гидрогенизированные пищевые жиры); эмульгатор (например, лецитин или камедь акации); неводные основы (например, миндальное масло, масляные сложные эфиры или этиловый спирт); и/или консервант (например, метил- или пропил-п-гидроксибензоаты или сорбиновая кислота).
Для буккального применения композиция может принимать форму таблеток или леденцов для рассасывания, изготавливаемых традиционным образом.
Раскрытые в настоящем документе соединения могут быть изготовлены в виде препаратов для парентерального введения посредством инъекции, включая применение традиционной техники катетеризации или вливания. Препараты для инъекции могут быть представлены в единичной дозированной форме (например, в ампулах) или в многодозовых контейнерах с добавленным консервантом. Композиции могут принимать такие формы, как суспензии, растворы или эмульсии в масляных или водных основах, и могут содержать формообразующее средство, такое как суспендирующие, стабилизирующие и/или диспергирующие средства, известные квалифицированным специалистам в данной области техники. В качестве альтернативы, активный ингредиент может быть в форме порошка для восстановления с подходящей основой (например, с апирогенной водой) перед использованием.
Для местного применения раскрытое в настоящем документе соединение может быть приготовлено в виде мази или крема.
Раскрытые в настоящем документе соединения можно также вводить в рецептуры ректальных композиций, таких как суппозитории или удерживаемые клизмы (например, содержащие традиционные основы суппозиториев, такие как масло какао или другие глицериды).
Для интраназального введения или для введения посредством ингаляции раскрытые в настоящем документе соединения можно удобным образом доставлять в форме раствора или суспензии из контейнера с пульверизатором, который пациент сжимает или накачивает, или в форме аэрозольного спрея из контейнера под давлением или из небулайзера, использующего подходящий пропеллент (например, дихлордифторметан, трихлорфторметан, дихлортетрафторэтан, диоксид углерода или другой подходящий газ). В случае аэрозоля под давлением дозируемая единица может быть определена предоставлением клапана, поставляющего некоторое отмеренное количество. Контейнер под давлением или небулайзер может содержать раствор или суспензию соединения, раскрытого в настоящем документе. Капсулы или картриджи (изготовленные, например, из желатина), предназначенные для применения в ингаляторе или инсуффляторе, могут быть изготовлены с содержимым, представляющим собой смесь соединения, раскрытого в настоящем документе, с порошковой основой, такой как лактоза или крахмал.
Доза соединения, раскрытого в настоящем документе, предлагаемая для перорального, парентерального или буккального введения среднему взрослому человеку для лечения или профилактики состояния заболевания, связанного с TPO, составляет приблизительно 0,1-2000 мг. В определенных вариантах осуществления настоящего изобретения предполагаемая доза составляет приблизительно 0,1-200 мг активного ингредиента на единичную дозу. Независимо от количественного состава предлагаемой дозы, введение соединения может происходить, например, от 1 до 4 раз в день.
Аэрозольные препараты для лечения или предупреждения указанных выше состояний у среднего взрослого человека, предпочтительно, составляют так, чтобы каждая отмеренная доза или «впрыск» аэрозоля содержали приблизительно 20-10000 мг (предпочтительно, приблизительно 20-1000 мг) соединения, раскрытого в настоящем документе. Общая суточная доза, вводимая с аэрозолем, будет находиться в диапазоне от приблизительно 100 мг до приблизительно 100 мг. В определенных вариантах осуществления настоящего изобретения общая суточная доза, вводимая с аэрозолем, будет, как правило, находиться в диапазоне от приблизительно 100 мг до приблизительно 10 мг. Введение можно производить несколько раз в день (например 2, 3, 4 или 8 раз), давая, например, 1, 2 или 3 дозы каждый раз.
Комбинированные аэрозольные препараты для лечения или предупреждения указанных выше состояний у среднего взрослого человека, предпочтительно, составляют так, чтобы каждая отмеренная доза или «впрыск» аэрозоля содержали от приблизительно 0,01 мг до приблизительно 1000 мг комбинации, содержащей соединение, раскрытое в настоящем документе. В определенных вариантах осуществления настоящего изобретения каждая отмеренная доза или «впрыск» аэрозоля содержит от приблизительно 0,01 мг до приблизительно 100 мг комбинации, содержащей соединение, раскрытое в настоящем документе. В определенных вариантах осуществления настоящего изобретения каждая отмеренная доза или «впрыск» аэрозоля содержит от приблизительно 1 мг до приблизительно 10 мг комбинации, содержащей соединение, раскрытое в настоящем документе. Введение можно производить несколько раз в день (например 2, 3, 4 или 8 раз), давая, например, 1, 2 или 3 дозы каждый раз.
Фармацевтические композиции и способы лечения и профилактики, включающие в себя введение пролекарств, по меньшей мере, одного раскрытого в настоящем документе соединения, также находятся в пределах объема настоящего раскрытия.
Анализ глюкозилцерамид-синтазы
В качестве источника активности глюкозилцерамид-синтазы в ферментативном анализе используют микросомы. К связанному с мембранами ферменту добавляют флуоресцентный церамидный субстрат в виде комплекса с альбумином. После реакции церамид и глюкозилцерамид разделяют и количественно определяют посредством обращенно-фазовой ВЭЖХ с флуоресцентным детектированием.
Процедура
Получение микросом из клеток меланомы человека A375
Суспензию клеток на льду обрабатывали ультразвуком до полного лизиса клеток, после чего центрифугировали при 10000 g в течение 10 мин при 4°C.
Супернатант осветляли посредством повторного центрифугирования при 100000 g в течение 1 часа при 4°C.
Осадок ресуспендировали в буфере лизиса, разливали на аликвоты и хранили при -80°С.
Анализ глюкозилцерамид-синтазы
Субстрат и микросомы соединяли в соотношении 1:1, тщательно перемешивали на планшетном шейкере, планшет закрывали и инкубировали 1 час при комнатной температуре в темноте.
Реакцию останавливали, добавляя останавливающий раствор в лунки реакционного планшета и смесь переносили на аналитический планшет.
Анализ с использованием обращенно-фазовой ВЭЖХ
- колонка: сменный картридж MercuryMSTM (Phenomenex) (Luna C8, 3 мкм, 20×4 мм)
- система: Agilent 1100 с флуоресцентным детектором серии Agilent 1200
- подвижная фаза: 1% муравьиной кислоты в смеси 81% метанола и 19% воды, скорость потока 0,5 мл/мин, изократический режим, 4 мин
- разбавитель образца: 0,1 мМ C8-церамид (блокатор адсорбции) в смеси 50% изопропанола и 50% воды (по объему)
- детектирование флуоресценции: λex = 470 нм, λem = 530 нм
- при этих условиях NBD C6 GluCer имел время удерживания приблизительно 1,7 мин, а NBD C6 Cer выходил приблизительно при 2,1 мин; пики четко разделялись вплоть до базовой линии, их интегрирование производилось автоматически программным обеспечением системы ВЭЖХ.
- чтобы избежать вариабельности, обусловленной ошибками при разбавлении и испарением образцов, в качестве регистрируемого параметра при испытаниях ингибиторов использовали % преобразования субстрата в продукт.
При анализе «Reporter Assay» все представленные соединения имели значения IC50 менее 5 мкМ
Экспериментальная часть
Общая процедура A: Образование карбамата/мочевины с трифосгеном
К суспензии гидрохлорида амина А (1 экв) и триэтиламина (3-4 экв) в THF (концентрация ~0,2 M) при комнатной температуре добавляли трифосген (0,35 экв). Реакционную смесь перемешивали в течение 10 мин и добавляли небольшое количество простого эфира (1-2 мл). Триэтиламмонийную соль отфильтровывали и получали прозрачный раствор изоцианата в смеси THF с простым эфиром.
К раствору спирта (1,5 экв) в THF (концентрация ~0,2 M) при комнатной температуре добавляли NaH [60%, масло] (1,5 экв). Реакционную смесь перемешивали в течение 15 мин и по каплям добавляли вышеуказанный раствор (изоцианат в смеси THF/эфир).
При стандартной обработке реакцию гасили насыщенным солевым раствором. Раствор экстрагировали EtOAc и органический слой сушили над Na2SO4, фильтровали и концентрировали. Неочищенный материал очищали посредством комбифлэш-хроматографии (SiO2-картридж, CHCl3 и 2 н NH3 в MeOH), получая соответствующий карбамат.
В качестве альтернативы: К суспензии гидрохлорида амина A (1 экв) и триэтиламина (3-4 экв) в THF (концентрация ~0,2 M) при комнатной температуре добавляли трифосген (0,35 экв). Реакционную смесь перемешивали в течение 10 мин и добавляли небольшое количество простого эфира (1-2 мл). Триэтиламмонийную соль отфильтровывали и получали прозрачный раствор изоцианата в смеси THF/эфир.
К раствору амина B (1 экв) в THF (концентрация ~1,0 M) при комнатной температуре по каплям добавляли вышеуказанный раствор (изоцианат в смеси THF/эфир). Реакционную смесь перемешивали в течение 18 часов и концентрировали. Неочищенный материал очищали посредством комбифлэш-хроматографии (SiO2-картридж, CHCl3 и 2 н NH3 в MeOH), получая соответствующую мочевину.
Общая процедура B: Алкилирование органическим соединением церия
Суспензию CeCl3 (4 экв) в THF (концентрация ~0,2 M) перемешивали при комнатной температуре в течение 1 часа. Суспензию охлаждали до -78°C и по каплям добавляли MeLi в простом эфире (1,6 M, 4 экв). Церийорганическому комплексу давали возможность образовываться в течение 1 часа и по каплям добавляли раствор нитрила (1 экв) в THF (концентрация 2,0 M). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 18 часов. Раствор охлаждали до 0°C и гасили водой (~1 мл), после чего добавляли 50%-ный водный раствор гидроксида аммония (~3 мл) до тех пор, пока не образовывался осадок, оседавший на дно колбы. Смесь фильтровали через слой целита и концентрировали. Неочищенный материал обрабатывали раствором HCl в диоксане (4,0 M). Промежуточный гидрохлорид арилпропан-2-амина растирали в простом эфире и в таком виде использовали на следующей стадии. В качестве альтернативы неочищенное свободное основание амина очищали посредством комбифлэш-хроматографии (SiO2-картридж, CHCl3 и 2 н NH3 в MeOH), получая соответствующий арилпропиламин.
Общая процедура C: Образование мочевины с карбонилдиимидазолом (CDI)
Раствор амина A (1 экв) и CDI (1,3 экв) в THF (концентрация ~0,15 M) перемешивали при кипячении с обратным холодильником в течение 1 часа. Добавляли раствор амина B (1,3 экв) в THF и реакционную смесь перемешивали в течение дополнительных 1,5 часов. Реакционную смесь охлаждали до комнатной температуры и разбавляли простым эфиром. Желаемое соединение осаждали и отфильтровывали. Неочищенный материал очищали посредством комбифлэш-хроматографии (картридж основного Al2O3, CHCl3 и MeOH) или (SiO2-картридж, CHCl3 и 2 н NH3 в MeOH), получая соответствующую мочевину.
Общая процедура D: Образование мочевины с трифосгеном
К суспензии амина A (1 экв) и триэтиламина (4 экв) в THF (концентрация ~0,15 M) при комнатной температуре добавляли трифосген (0,35 экв). Реакционную смесь перемешивали в течение 15 мин и добавляли амин B (1,1 экв). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов и затем разбавляли EtOAc. Органический слой промывали водным NaOH (1,0 M), сушили над Na2SO4, фильтровали и концентрировали. Неочищенный материал очищали посредством комбифлэш-хроматографии (SiO2-картридж, CHCl3 и 2 н NH3 в MeOH), получая соответствующую мочевину.
Общая процедура E: Реакция сочетания по Сузуки
К раствору арилгалогенида (1 экв) в смеси DME с водой (4:1) (концентрация ~0,2 M) добавляли бороновую кислоту (2 экв), палладиевый катализатор (0,1-0,25 экв) и карбонат натрия (2 экв). Реакционную смесь нагревали в микроволновой печи в течение 25 мин при 150°C. После фильтрования через слой целита и концентрирования неочищенный продукт очищали посредством комбифлэш-хроматографии (SiO2-картридж, CHCl3 и 2 н NH3 в MeOH), получая соответствующий аддукт сочетания.
В качестве альтернативы: К раствору арилгалогенида (1 экв) в смеси толуола с водой [20:1] (концентрация ~0,2 M) добавляли бороновую кислоту (1,3-2,5 экв), палладиевый катализатор (0,05-0,15 экв), трициклогексилфосфин (0,15-0,45 экв) и фосфат калия (5 экв). Реакционную смесь нагревали в микроволновой печи в течение 25 мин при 150°C. После фильтрования через слой целита и концентрирования неочищенный продукт очищали посредством комбифлэш-хроматографии (SiO2-картридж, CHCl3 и 2 н NH3 в MeOH), получая соответствующий аддукт сочетания.
Общая процедура F: Гидрогенизация
К раствору субстрата в метаноле, этаноле или EtOAc (концентрация ~0,2 M) добавляли палладиевый катализатор (20% от массы субстрата). Реакционную смесь перемешивали при комнатной температуре при 1 атм H2 до завершения реакции. Реакционную смесь фильтровали через слой целита, который затем два раза промывали хлороформом. Неочищенный продукт концентрировали и очищали посредством комбифлэш-хроматографии (SiO2-картридж, CHCl3 и 2 н NH3 в MeOH), получая гидрированный продукт. В качестве альтернативы, конечный материал очищали посредством осаждения или перекристаллизации.
Общая процедура G: Циклопропанирование
К смеси арилнитрила (1 экв) и Ti(Oi-Pr)4 (1,7 экв), перемешиваемой при -70°C, добавляли по каплям EtMgBr [3,0 M в простом эфире] (1,1 экв). Реакционной смеси давали возможность нагреваться до 25°C и перемешивание продолжали в течение 1 часа. К вышеуказанной смеси по каплям добавляли BF3·Et2O (3 экв) при 25°C. После этого добавления смесь перемешивали еще в течение 2 часов и затем гасили водным раствором HCl [2 M]. Раствор, полученный в результате этого, подщелачивали, добавляя водный NaOH [2 M]. Органический материал экстрагировали этиловым эфиром. Органические слои объединяли, сушили над Na2SO4, фильтровали и концентрировали. Неочищенный материал очищали посредством хроматографии на колонке с силикагелем, элюируя смесью петролейного эфира с EtOAc (от 10/1 до 1/1) и получая соответствующий 1-арил-циклопропанамин.
Общая процедура H: Реакция сочетания посредством перегруппировки Курциуса in situ
Смесь кислоты (1 экв), триэтиламина (2,5 экв), DPPA (1,0 экв) в толуоле (концентрация ~0,3 M) кипятили с обратным холодильником в течение 30 мин. Смесь охлаждали до комнатной температуры и добавляли спирт (1 экв). После этого добавления смесь нагревали при 90°C в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли EtOAc и насыщенным водным раствором бикарбоната натрия. Органическую фазу сушили над Na2SO4, концентрировали и очищали посредством препаративной TLC (смесь EtOAc/MeOH 5:1, содержавшая 1% TEA), получая соответствующий карбамат.
Общая процедура I: Образование амида с использованием EDCI
К раствору амина (1 экв) в DMF или THF (концентрация ~0,3 M) добавляли EDCI (1,2-2,5 экв), HOBT (1,2-2,5 экв), DIPEA (1,2-2,5 экв) и триэтиламин (несколько капель). Реакционную смесь перемешивали и добавляли кислоту (1,2 экв). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов и затем концентрировали. Остаток растворяли в EtOAc и промывали насыщенным солевым раствором. Органический слой сушили над Na2SO4 и концентрировали. Неочищенный материал очищали посредством препаративной ВЭЖХ-МС или комбифлэш-хроматографии (SiO2-картридж, CHCl3 и 2 н NH3 в MeOH).
Препаративная процедура A
Промежуточное соединение 1
Гидрохлорид 2-(3-бромфенил)-пропан-2-амина
К раствору метил-3-бромбензоата (15,0 г, 69,8 ммоль) в THF (140 мл) при -78°C добавляли по каплям раствор MeMgBr в диэтиловом простом эфире [3,0 M] (58 мл). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 2 часов. Раствор выливали в насыщенный водный раствор хлорида аммония и органический материал экстрагировали EtOAc. Органический слой сушили над Na2SO4, фильтровали и концентрировали, получая соответствующий спирт (14,9 г), который использовали без дальнейшей очистки.
К раствору 2-(3-бромфенил)-пропан-2-ола (17,2 г, 79,8 ммоль) в хлорацетонитриле (160 мл) добавляли уксусную кислоту (14 мл). Реакционную смесь охлаждали до 0°C и по каплям добавляли H2SO4 (14 мл). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 18 часов. Затем реакционную смесь выливали на лед и экстрагировали EtOAc. Органический слой промывали водным раствором NaOH [1,0 M] и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали, получая соответствующий хлорацетамид (21,4 г), который использовали без дальнейшей очистки.
К раствору N-(2-(3-бромфенил)-пропан-2-ил)-2-хлорацетамида (20,3 г) в этаноле (120 мл) добавляли уксусную кислоту (20 мл). Реакционную смесь перемешивали при кипячении с обратным холодильником в течение 18 часов. Раствор охлаждали до комнатной температуры и осадок отфильтровывали на слое целита. Фильтрат концентрировали и остаток растворяли в EtOAc. Органический слой обрабатывали водным раствором NaOH [1,0 M], сушили над Na2SO4 и концентрировали. Неочищенный материал обрабатывали раствором HCl в диоксане [4 M]. Промежуточное соединение 2-(3-бромфенил)-пропан-2-амин гидрохлорид растирали в простом эфире и в таком виде использовали на следующей стадии (7,50 г, 43%).1H ЯМР (400 МГц, CD3OD) δ 7,69 (кв., J=1,8 Гц, 1H), 7,55 (ддд, J=1,0, 1,8, 7,9 Гц, 1H), 7,49 (ддд, J=1,0, 2,0, 8,0 Гц, 1H), 7,38 (т, J=8,0 Гц, 1H), 1,71 (с, 6H) м.д.
Препаративная процедура B
Промежуточное соединение 2
Гидрохлорид 2-(5-бром-2-фторфенил)-пропан-2-амина
К раствору 5-бром-2-фторбензойной кислоты (4,85 г, 22,8 ммоль) в метаноле (45 мл) добавляли H2SO4 (4,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов и раствор концентрировали. Остаток обрабатывали водным раствором NaOH [10%, масса/объем] и органический материал экстрагировали CHCl3. Органический слой сушили над Na2SO4, фильтровали и концентрировали, получая соответствующий сложный эфир (4,69 г, 91%), который использовали без дальнейшей очистки.
Указанное сложноэфирное промежуточное соединение (4,69 г, 20,1 ммоль) преобразовывали в промежуточное соединение 2, применяя ту же процедуру, что и в примере с промежуточным соединением 1, получая соответствующую аммонийную соль (3,94 г, общий выход 67%) в виде белого твердого вещества.1H ЯМР (400 МГц, CD3OD) δ 7,67-7,57 (м, 2H), 7,21 (дд, J=8,7, 12,3 Гц, 1H), 1,77 (с, 6H) м.д.
Промежуточное соединение 3
Гидрохлорид 2-(3-бром-4-фторфенил)-пропан-2-амина
5-Бром-2-фторбензойную кислоту преобразовывали в промежуточное соединение 3, применяя ту же процедуру, что и в примере с промежуточным соединением 2, получая соответствующую аммонийную соль (2,79 г, общий выход 49%) в виде белого твердого вещества.
Промежуточное соединение 4
2-(3-бром-2-фторфенил)-пропан-2-амин
3-Бром-2-фторбензойную кислоту преобразовывали в промежуточное соединение 4, применяя ту же процедуру, что и в примере с промежуточным соединением 2, получая соответствующий амин в виде светло-желтого масла.
Промежуточное соединение 5
Гидрохлорид 2-(4-бромфенил)-пропан-2-амина
Применяя общую процедуру B, преобразовывали бромбензонитрил (2,00 г, 11,0 ммоль) в соответствующий 2-(4-бромфенил)-пропан-2-амин, который получали в виде коричневого масла (1,20 г, 51%).
Препаративная процедура C
Промежуточное соединение 6
1,4-Диазабицикло[3.2.2]нонан
К перемешиваемому раствору 1,4-диазабицикло[3.2.2]нонан-3-она (1,0 г, 7,2 ммоль) в 1,4-диоксане (7,2 мл) при комнатной температуре добавляли литийалюминийгидрид [2,0 M в THF] (4,1 мл, 8,2 ммоль). Реакционную смесь затем нагревали при кипячении с обратным холодильником в течение 6 часов, после чего охлаждали до комнатной температуры. Реакцию гасили, последовательно добавляя 200 мкл H2O, 200 мкл 15%-ного водного раствора NaOH и 600 мкл H2O. Смесь фильтровали через целит, который затем промывали EtOAc. Объединенный фильтрат концентрировали в вакууме, получая продукт (0,82 г, 90%), который использовали без дальнейшей очистки.1H ЯМР (400 МГц, CDCl3) δ 3,28-3,25 (м, 1H), 2,99-2,95 (м, 8H), 1,86-1,80 (м, 3H), 1,69-1,64 (м, 2H) м.д.
Препаративная процедура D
Промежуточное соединение 7
2-Метилхинуклидин-3-ол
Раствор карбоната калия (11,4 г, 82,8 ммоль) и хинуклидингидрата (5,00 г, 20,4 ммоль) растворяли в H2O (15,6 мл). После полного растворения добавляли дихлорметан (20,4 мл) и реакционную смесь премешивали при комнатной температуре в течение ночи. Органическую фазу отделяли и водную фазу экстрагировали хлороформом (3×50 мл). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Продукт использовали без дальнейшей очистки.1H ЯМР (400 МГц, CDCl3) 2,79 (с, 1H), 5,19 (с, 1H), 3,14-3,06 (м, 2H), 2,99-2,91 (м, 2H), 2,57-2,55 (м, 1H), 1,98-1,93 (м, 4H) м.д.
2-Метиленхинуклидин-3-он (3,50 г) в этаноле (30 мл) восстанавливали над 10% Pd/C (50% масс.) в атмосфере водорода. Когда на основании TLC делали вывод о завершении реакции (~3 дня), отфильтровывали катализатор и слой на фильтре промывали этилацетатом. Растворитель удаляли в вакууме, получая желаемый продукт (2,80 г, 80%), который использовали без дальнейшей очистки.1H ЯМР (400 МГц, CDCl3) 3,37-3,31 (м, 1H), 3,21-3,13 (м, 2H), 3,09-3,00 (м, 1H), 2,97-2,89 (м, 1H), 2,46-2,43 (м, 1H), 2,05-1,91 (м, 4H), 1,34 (д, J=7,6 Гц, 3H) м.д.
К 2-метилхинуклидин-3-ону (0,50 г, 3,60 ммоль) в 1,4-диоксане (18 мл) при комнатной температуре добавляли литийалюминийгидрид [1,0 M в THF] (4,1 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 минут. Реакцию гасили, последовательно добавляя 116 мкл H2O, 116 мкл 15%-ного водного раствора NaOH и 348 мкл H2O. Смесь фильтровали через целит, который затем промывали EtOAc. Растворитель удаляли в вакууме, получая продукт (0,48 г, 95%) в виде смеси диастереомеров (2:1), которую использовали без дальнейшей очистки.
Препаративная процедура E
Промежуточное соединение 8
1-Азабицикло[3.2.2]нонан-4-ол
К 1-азабицикло[3.2.2]нонан-4-ону (0,20 г, 1,4 ммоль) в 1,4-диоксане (2,8 мл) при 0°C добавляли литийалюминийгидрид [1,0 M в THF] (1,7 мл, 1,7 ммоль). Реакционную смесь поддерживали при 0°C в течение 15 минут. Реакцию гасили, последовательно добавляя 46 мкл H2O, 46 мкл 15%-ного водного раствора NaOH и 138 мкл H2O. Смесь фильтровали через целит, который затем промывали EtOAc. Растворитель удаляли в вакууме, получая продукт (0,19 г, 96%), который использовали без дальнейшей очистки.1H ЯМР (400 МГц, CDCl3) δ 3,90-3,86 (м, 1H), 3,09-3,03 (м, 1H), 2,96-2,91 (дд, J=9,2, 6,8 Гц, 1H), 2,86-2,75 (м, 3H), 2,71-2,64 (м, 1H), 2,34-2,27 (bs s, 1H), 1,98-1,86 (м, 3H), 1,71-1,59 (м, 3H), 1,51-1,35 (м, 1H) м.д.
Препаративная процедура F
Промежуточное соединение 9
1-Азабицикло[2.2.1]гептан-3-ол
К смеси метоксида натрия (2,00 г, 37,9 ммоль) в метаноле (9 мл) при 0°C добавляли гидрохлорид метилового сложного эфира глицина (4,76 г, 37,9 ммоль) и диметилитаконат (5,00 г, 31,6 ммоль.) Реакционную смесь нагревали при кипячении с обратным холодильником в течение 16 часов, после чего охлаждали до комнатной температуры. Твердое вещество отфильтровывали и промывали дихлорметаном. Фильтрат концентрировали и остаток разбавляли 5 н HCl (50 мл). Водный слой экстрагировали дихлорметаном (4×50 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме. Продукт использовали без дальнейшей очистки.1H ЯМР (400 МГц, CDCl3) δ 4,04 (дд, J=82,0, 17,6 Гц), 3,74-3,64 (м, 8H), 3,32-3,24 (м, 1H), 2,77-2,63 (м, 2H) м.д.
К метил-1-(2-метокси-2-оксоэтил)-5-оксопирролидин-3-карбоксилату (3,40 г, 16,0 ммоль) в THF (20 мл) при 0°C добавляли боран-THF [1,0 M В THF] (32,0 мл, 32,0 ммоль). Реакционную смесь перемешивали при кипячении с обратным холодильником в течение 1 часа и затем охлаждали до комнатной температуры, после чего ей давали возможность перемешиваться в течение дополнительных 12 часов. Реакцию гасили, добавляя насыщенный раствор карбоната калия (5,52 г в 20 мл H2O), и нагревали при кипячении с обратным холодильником в течение одного дополнительного часа, после чего охлаждали до комнатной температуры. Растворитель удаляли в вакууме и остаток подкисляли, добавляя 5 н HCl (25 мл). Водный слой экстрагировали дихлорметаном (2×30 мл). Затем рН водного слоя делали щелочным, добавляя твердый карбонат калия. Водный слой дополнительно экстрагировали дихлорметаном (5×30 мл). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме. Продукт использовали без дальнейшей очистки.1H ЯМР (400 МГц, CDCl3) δ 3,66 (с, 3H), 3,63 (с, 3H), 3,29 (ABq, 2H, J=24,0, 16,8 Гц), 3,06-3,02 (м, 2H), 2,87-2,81 (м, 1H), 2,71-2,65 (м, 1H), 2,56-2,50 (м, 1H), 2,09-2,04 (м, 2H) м.д.
К кипящему с обратным холодильником раствору трет-бутоксида калия (2,46 г, 22,0 ммоль) в толуоле (32 мл) по каплям в течение 1 часа добавляли раствор метил-1-(2-метокси-2-оксоэтил)-пирролидин-3-карбоксилата (2,00 г, 10,0 ммоль) в толуоле (10 мл). Реакционной смеси давали возможность перемешиваться в течение дополнительных 3 часов при кипячении с обратным холодильником, после чего ее сначала охлаждали до комнатной температуры, а затем до -10°C. После этого при перемешивании добавляли уксусную кислоту (1,3 мл). Толуольный слой экстрагировали 5 н HCl (4×50 мл). Объединенные водные слои нагревали при 110°C в течение 8 часов. Реакционную смесь затем охлаждали до комнатной температуры и в вакууме вдвое уменьшали ее объем. pH реакционной смеси делали щелочным, добавляя твердый карбонат калия. Водный слой экстрагировали дихлорметаном (5×50 мл) и объединенные органические слои концентрировали в вакууме. К неочищенному продукту добавляли этиловый простой эфир. Твердое вещество отфильтровывали, получая желаемый продукт (0,30 г, 27%), который использовали без дальнейшей очистки.1H ЯМР (400 МГц, CDCl3) δ 3,05-2,96 (м, 3H), 2,76 (с, 2H), 2,72-2,66 (м, 2H), 2,09-2,01 (м, 1H), 1,78-1,71 (м, 1H) м.д.
1-Азабицикло[2.2.1]гептан-3-он (0,30 г, 2,7 ммоль) в этаноле (2-3 мл) восстанавливали над PtO2 (50% масс.) в атмосфере водорода. После перемешивания в течение 4 часов катализатор отфильтровывали и слой на фильтре промывали этанолом. Этанол удаляли в вакууме, получая желаемый продукт (0,29 г, 95%).1H ЯМР (400 МГц, CDCl3) δ 4,36-4,35 (м, 1H), 3,10-3,05 (м, 1H), 2,95-2,88 (м, 1H), 2,72-2,66 (м, 1H), 2,63-2,57 (м, 2H), 2,48-2,44 (дд, J=10,0, 3,2 Гц, 1H), 2,11-2,05 (м, 2H), 1,51-1,44 (м, 1H) м.д.
Препаративная процедура G
Промежуточное соединение 10
(R)-3-метилхинуклидин-3-амин и (S)-3-метилхинуклидин-3-амин
К хорошо перемешиваемому раствору MeLi [3,0 M в 150 мл безводного диэтилового простого эфира] (67,0 мл, 201 ммоль) при -78°C по каплям добавляли раствор хинуклидин-3-она (12,5 г, 100 ммоль) в диэтиловом простом эфире (100 мл). Раствор, полученный в результате этого, поддерживали при -78°C в течение 1 часа, затем при комнатной температуре в течение 18 часов. По каплям при 0°C добавляли воду (60 мл) и смесь концентрировали в вакууме, получая остаток, который очищали посредством хроматографии на колонке с нейтральным оксидом алюминия (0-20% MeOH в CHCl3), получая 3-метилхинуклидин-3-ол (10,0 г, 71%) в виде светло-желтого твердого вещества. К перемешиваемому ацетонитрилу (250 мл) при 0°C медленно добавляли концентрированную серную кислоту (100 мл). Раствор, полученный в результате этого, добавляли по каплям к смеси 3-метилхинуклидин-3-ола (9,10 г, 64,5 ммоль) в ацетонитриле (250 мл) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 60 часов, затем охлаждали на ледяной бане и подщелачивали водным раствором гидроксида натрия до pH 10. Смесь экстрагировали смесью CHCl3/i-PrOH (5:1 об./об.). Органический слой концентрировали, получая остаток, который разбавляли 2 н водной HCl и промывали смесью CHCl3/i-PrOH (5:1 об./об.). Оставшийся водный слой затем подщелачивали 2 н NaOH и экстрагировали смесью CHCl3/i-PrOH (5:1 об./об.). Объединенные органические слои промывали водой, сушили (Na2SO4) и концентрировали, получая 9,5 г (82%) желаемого соединения в виде светло-желтого масла. Два энантиомера вышеуказанного промежуточного соединения разделяли, используя хиральную колонку в системе сверхкритической жидкостной хроматографии (SFC).
Раствор вышеуказанного хирального ацетамидного промежуточного соединения (9,50 г, 52,0 ммоль) в концентрированной HCl (100 мл) кипятили с обратным холодильником в течение 3 дней, охлаждали на ледяной бане и нейтрализовали водным раствором гидроксида натрия до pH 1. Смесь промывали смесью CHCl3/i-PrOH (5:1 об./об.). Водный слой затем подщелачивали 2 н NaOH и экстрагировали смесью CHCl3/i-PrOH (5:1 об./об.). Объединенные экстракты промывали водой, сушили (Na2SO4) и концентрировали, получая 5,00 г (69%) желаемого хирального соединения в виде светло-желтого полужидкого вещества.1H ЯМР (500 МГц, DMSO-d6) δ 2,72-2,39 (м, 6H), 2,01-1,96 (м, 1H), 1,67-1,61 (м, 1H), 1,43-1,36 (м, 2H), 1,23-1,17 (м, 1H), 1,09 (с, 3H) м.д.13C ЯМР (125 МГц, DMSO-d6) δ 65,3, 48,3, 46,6, 46,4, 34,2, 30,0, 24,8, 22,8 м.д. Чистота более 99% (GC-MS); время удерживания 6,63 мин; (M) 140,1.
Препаративная процедура H
Промежуточное соединение 11
2-(3-(4-Фторфенил)-изотиазол-5-ил)-пропан-2-амин
К перемешиваемой суспензии 4-фторбензамида (70,00 г, 503,1 ммоль) в толуоле (900 мл) добавляли хлоркарбонилсульфенилхлорид (83,0 мл, 1,00 моль). Смесь нагревали в течение ночи при 60°C и концентрировали. Полученное в результате этого желтовато-коричневое твердое вещество растирали с метиленхлоридом (200 мл), собирали посредством фильтрования с отсасыванием и дополнительно промывали метиленхлоридом (4×70 мл). Неочищенным продуктом пропитывали силикагель (100 г), который в сухом виде загружали в большую фильтровальную воронку, после чего проводили хроматографическую процедуру, используя гексан/этилацетатный градиент. Продукт, представлявший собой 5-(4-фторфенил)-1,3,4-оксатиазол-2-он, получали в виде светло-серого твердого вещества (55,98 г, 56%).
К перемешиваемому раствору 5-(4-фторфенил)-1,3,4-оксатиазол-2-она (42,80 г, 217,1 ммоль) в o-дихлорбензоле (600 мл) добавляли этилпропиолат (66,0 мл, 651 ммоль). Смесь нагревали в течение ночи при 135°C и концентрировали. Маслянистый остаток очищали посредством флэш-хроматографии, используя гексан/этилацетатный градиент и получая этил-3-(4-фторфенил)-изотиазол-5-карбоксилат в виде светло-золотистого твердого вещества (17,35 г, 32%). Более полярный изомер - этил-3-(4-фторфенил)-изотиазол-4-карбоксилат (образуемый в приблизительном соотношении 57/43 с желаемым продуктом) - отбрасывали.
К перемешиваемому и охлаждаемому (0°C) раствору этил-3-(4-фторфенил)-изотиазол-5-карбоксилата (38,50 г, 153,2 ммоль) в THF (400 мл) в течение 20 минут по каплям добавляли раствор метилмагнийбромида в диэтиловом простом эфире (3,0 M, 128 мл, 384 ммоль). После следующих 1,5 часов при 0°С реакцию гасили медленным добавлением этилацетата (20 мл) и концентрировали. Остаток растворяли в водном NH4Cl (400 мл) и экстрагировали этилацетатом (2×150 мл). Объединенные экстракты сушили (Na2SO4) и концентрировали. Полученный в результате этого сироп янтарного цвета очищали посредством флэш-хроматографии, используя гексан/этилацетатный градиент и получая 2-(3-(4-фторфенил)-изотиазол-5-ил)-пропан-2-ол в виде полужидкого вещества золотистого цвета (29,02 г, 80%).
2-(3-(4-Фторфенил)-изотиазол-5-ил)-пропан-2-ол (29,00 г, 122,2 ммоль) смешивали с тионилхлоридом (75 мл). Смесь быстро охлаждали (на ледяной бане) и перемешивали. Спустя 4 часа реакционную смесь концентрировали и остаток распределяли между этилацетатом (200 мл) и водным NaHCO3 (300 мл). Органический слой объединяли с обратным экстрактом водного слоя (этилацетат, 1×100 мл), сушили (Na2SO4) и концентрировали, получая смесь желаемого продукта, представлявшего собой 5-(2-хлорпропан-2-ил)-3-(4-фторфенил)-изотиазол, и отбрасываемого побочного продукта, представлявшего собой 3-(4-фторфенил)-5-(проп-1-ен-2-ил)-изотиазол (в приблизительном соотношении 63/39)), в виде темного масла янтарного цвета (29,37 г). Этот материал использовали без очистки в следующей реакции.
К перемешиваемому раствору продукта предыдущей стадии в DMSO (80 мл) добавляли азид натрия (14,89 г, 229,0 ммоль). Смесь нагревали при 50°C в течение ночи, разбавляли этилацетатом (250 мл) и промывали водой (6×400 мл). Органический слой сушили (Na2SO4) и концентрировали, получая смесь 5-(2-азидопропан-2-ил)- 3-(4-фторфенил)-изотиазола и 3-(4-фторфенил)-5-(проп-1-ен-2-ил)-изотиазола (в приблизительном соотношении 56/44) в виде темного масла янтарного цвета (29,10 г). Этот материал использовали без очистки в следующей реакции.
Продукт предыдущей стадии смешивали с 10%-ным палладием на угле (50% воды; 7,50 г) и суспендировали в метаноле (350 мл). Перемешиваемую суспензию подвергали трем циклам чередования вакуума и продувки азотом. После дополнительного вауумирования реактор заполняли газообразным водородом (с надувным резервуаром) и перемешивали в течение ночи. Реакционную смесь фильтровали через целит. Фильтрат объединяли с метанолом, использованным для промывания целита, и концентрировали. Темное масло янтарного цвета, полученное в результате этого, очищали посредством флэш-хроматографии, используя метиленхлорид/метанольный градиент и получая 2-(3-(4-фторфенил)-изотиазол-5-ил)-пропан-2-амин в виде вязкого масла янтарного цвета (14,23 г, 49% за 3 стадии).
Для лечения болезней лизосомного накопления (LSD) применяют несколько подходов, большинство из которых фокусируется на ферментозаместительной терапии, применяемой в виде монотерапевтического ведения больного. Для лечения LSD имеется много коммерчески доступных средств ферментозаместительной терапии (например, Myozyme® для болезни Помпе, Aldurazyme® для мукополисахаридоза I, Cerezyme® для болезни Гоше и Fabrazyme® для болезни Фабри). Кроме того, авторы настоящего изобретения идентифицировали ряд малых молекул, применимых для монотерапевтического лечения LSD. Терапевтические способы согласно настоящему изобретению, описанные в настоящем документе, предоставляют практическому врачу, сталкивающемуся с проблемой ведения больных, страдающих различными формами болезней лизосомного накопления, варианты лечения, подробно описанные ниже.
В определенных аспектах настоящего изобретения соединения согласно настоящему изобретению можно применять для лечения метаболического заболевания, такого как болезнь лизосомного накопления (LSD), либо в виде монотерапии, либо в сочетании с ферментозаместительной терапией. В других аспектах настоящего изобретения, соединения согласно настоящему изобретению можно применять для ингибирования или уменьшения активности GCS у субъекта с поставленным диагнозом метаболического заболевания, такого как LSD, либо в виде монотерапии, либо в сочетании с ферментозаместительной терапией. В других аспектах настоящего изобретения, соединения согласно настоящему изобретению можно применять для уменьшения или ингибирования накопления сохраняемого материала (например, лизосомного субстрата) у субъекта с поставленным диагнозом метаболического заболевания, такого как LSD. В определенных вариантах осуществления вышеуказанных аспектов указанное LSD представляет собой болезнь Гоше (типа 1, типа 2 или типа 3), болезнь Фабри, GM1-ганглиозидоз или GM2-ганглиозидозы (например, дефицит активатора GM2, болезнь Тея-Сакса и болезнь Сандхоффа). В Таблице 1 перечислены многие формы LSD и указаны соответствующие дефицитные ферменты, которые можно использовать в качестве ферментозаместительной терапии ERT в вышеуказанных аспектах настоящего изобретения.
При других ситуациях и планах действий, пациенту, состояние которого требует снижения уровней субстратов в мозге, что невозможно осуществить посредством системного введения средств ERT, может оказаться необходимым предоставление терапии малыми молекулами (SMT). Хотя прямое интрацеребровентрикулярное или интратекальное введение может понизить уровни субстратов в мозге, однако при LSD с вовлеченностью центральной нервной системы (ЦНС) системное введение средств ERT не является эффективным вследствие их неспособности проникать через гематоэнцефалический барьер (ГЭБ), и поэтому для пациентов с остаточной ферментативной активностью в ЦНС может оказаться полезной терапия малыми молекулами (SMT).
Согласно настоящему изобретению, SMT предоставляют пациенту для лечения рака и/или метаболического заболевания, такого как болезнь лизосомного накопления. SMT может включать в себя одну или более малых молекул. SMT включает в себя введение пациенту соединения согласно настоящему изобретению. В конкретных вариантах осуществления настоящего изобретения указанное соединение представляет собой (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)-тиазол-4-ил)-пропан-2-ил)-карбамат или хинуклидин-3-ил-(2-(4'-фтор-[1,1'-бифенил]-3-ил)-пропан-2-ил)-карбамат или их комбинации.
В определенных вариантах осуществления настоящего изобретения соединения согласно настоящему изобретению, такие как, например, (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)-тиазол-4-ил)-пропан-2-ил)-карбамат и хинуклидин-3-ил-(2-(4'-фтор-[1,1'-бифенил]-3-ил)-пропан-2-ил)-карбамат, можно применять для лечения практически любой болезни накопления, причиной которой является дефект на гликосфинголипидном пути (например, болезнь Гоше типа 1, типа 2 и типа 3, болезнь Фабри, GM1-ганглиозидоз, GM2-ганглиозидозы (например, дефицит активатора GM2, болезнь Тея-Сакса и болезнь Сандхоффа)). В особо предпочтительном варианте осуществления настоящего изобретения (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)-тиазол-4-ил)-пропан-2-ил)-карбамат или его фармацевтически приемлемую соль или пролекарство применяют для ингибирования и/или уменьшения накопления Gb3 и/или лизо-Gb3 у пациента с болезнью Фабри, либо в виде монотерапии, либо в виде комбинированного лечения с ферментозаместительной терапией (см. Примеры). В предпочтительном варианте осуществления настоящего изобретения ферментозаместительная терапия включает в себя введение пациенту с болезнью Фабри альфа-галактозидазы А. Фактически, примеры, приведенные ниже, демонстрируют, что ингибитор GCS согласно настоящему изобретению эффективно уменьшает накопление Gb3 и лизо-Gb3 в модели болезни Фабри у мышей, что подтверждает обоснованность его применения в качестве целесообразного способа лечения болезни Фабри. Кроме того, приведенные в Примерах данные, полученные при комбинированной терапии in vivo, убедительно свидетельствуют о том, что комбинированный терапевтический подход может быть как аддитивным, так и комплементарным.
В определенных вариантах осуществления настоящего изобретения соединения согласно настоящему изобретению, такие как, например, (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)-тиазол-4-ил)-пропан-2-ил)-карбамат и хинуклидин-3-ил-(2-(4'-фтор-[1,1'-бифенил]-3-ил)-пропан-2-ил)-карбамат, можно применять для снижения уровня GluCer и GluSph в мозге субъекта, которому поставлен диагноз нейропатической болезни Гоше, - либо в виде монотерапии, либо в комбинации с ERT (например, в сочетании с введением глюкоцереброзидазы).
Режимы дозирования для того компонента комбинированной терапии согласно настоящему изобретению, которым является терапия малыми молекулами, как правило, определяет квалифицированный клиницист; они могут значительно варьировать в зависимости от конкретной болезни накопления, лечение которой проводят, и от клинического состояния конкретного больного индивида. Общие принципы определения режима дозирования для данной формы терапии малыми молекулами согласно настоящему изобретению при лечении любой болезни накопления хорошо известны квалифицированному специалисту. Руководящие указания по режимам дозирования можно получить в любом из многих хорошо известных справочных изданий по этой теме. Дополнительные рекомендации доступны, в частности, в обзорах по конкретным предметам, указанных в настоящем документе. В определенных вариантах осуществления настоящего изобретения такие дозы могут находиться в диапазонах от приблизительно 0,5 мг/кг до приблизительно 300 мг/кг, предпочтительно, от приблизительно 5 мг/кг до приблизительно 60 мг/кг (например, 5 мг/кг, 10 мг/кг, 15 мг/кг, 20 мг/кг, 25 мг/кг, 30 мг/кг, 35 мг/кг, 40 мг/кг, 45 мг/кг, 50 мг/кг, 55 мг/кг и 60 мг/кг) при внутрибрюшинном, пероральном или эквивалентном введении от одного до пяти раз в день. Такие дозы могут находиться в диапазонах от приблизительно 5 мг/кг до приблизительно 5 г/кг (предпочтительно, от приблизительно 10 мг/кг до приблизительно 1 г/кг) при пероральном, внутрибрюшинном или эквивалентном введении от одного до пяти раз в день. В одном варианте осуществления настоящего изобретения диапазоны доз составляют от приблизительно 10 мг/день до приблизительно 500 мг/день (например, 10 мг/день, 20 мг/день, 30 мг/день, 40 мг/день, 50 мг/день, 60 мг/день, 70 мг/день, 80 мг/день, 90 мг/день, 100 мг/день, 110 мг/день, 120 мг/день, 130 мг/день, 140 мг/день, 150 мг/день, 160 мг/день, 170 мг/день, 180 мг/день, 190 мг/день, 200 мг/день, 210 мг/день, 220 мг/день, 230 мг/день, 240 мг/день, 250 мг/день, 260 мг/день, 270 мг/день, 280 мг/день, 290 мг/день, 300 мг/день). Особо предпочтительный диапазон пероральных доз составляет от приблизительно 50 мг до приблизительно 100 мг при введении дозы дважды в день. Конкретный диапазон пероральных доз для соединения согласно настоящему изобретению составляет от приблизительно 5 мг/кг/день до приблизительно 600 мг/кг/день. В частности, диапазон пероральных доз для соединения согласно настоящему изобретению составляет от приблизительно 1 мг/кг/день до приблизительно 120 мг/кг/день (например, 1 мг/кг/день, 5 мг/кг/день, 10 мг/кг/день, 15 мг/кг/день, 20 мг/кг/день, 25 мг/кг/день, 30 мг/кг/день, 35 мг/кг/день, 40 мг/кг/день, 45 мг/кг/день, 50 мг/кг/день, 55 мг/кг/день или 60 мг/кг/день, 65 мг/кг/день, 70 мг/кг/день, 75 мг/кг/день, 80 мг/кг/день, 85 мг/кг/день, 90 мг/кг/день, 95 мг/кг/день, 100 мг/кг/день, 105 мг/кг/день, 110 мг/кг/день, 115 мг/кг/день или 120 мг/кг/день).
В определенных вариантах осуществления настоящее изобретение относится к формам комбинированной терапии с проведением SMT с применением соединений согласно настоящему изобретению и ERT для лечения болезней лизосомного накопления. Частичный список известных болезней лизосомного накопления, которые можно лечить согласно настоящему изобретению, представлен в Таблице 1, включающей тривиальные названия заболеваний, накапливаемый материал и соответствующий дефицитный фермент (адаптировано из Таблицы 38-4 в публикации Kolodny et al., 1998, Id.).
Для мониторирования состояния заболевания и эффективности комбинированной терапии согласно настоящему изобретению можно применять любой способ, известный квалифицированному специалисту в данной области. Клинический мониторинг состояния заболевания может включать в себя, не ограничиваясь этим, регистрацию объема органа (например, печени, селезенки), гемоглобин, численность эритроцитов, гематокрит, тромбоцитопению, кахексию (истощение) и уровни хитиназы (например, хитотриозидазы) в плазме. Хитотриозидаза, фермент из семейства хитиназы, как известно, продуцируется в больших количествах макрофагами у субъектов с болезнями лизосомного накопления (см. Guo et al., 1995, J. Inherit. Metab. Dis. 18, 717-722; den Tandt et al., 1996, J. Inherit. Metab. Dis. 19, 344-350; Dodelson de Kremer et al., 1997, Medicina (Buenos Aires) 57, 677-684; Czartoryska et al., 2000, Clin. Biochem. 33, 147-149; Czartoryska et al., 1998, Clin. Biochem. 31, 417-420; Mistry et al., 1997, Baillieres Clin. Haematol. 10, 817-838; Young et al., 1997, J. Inherit. Metab. Dis. 20, 595-602; Hollak et al., 1994, J. Clin. Invest. 93, 1288-1292). Предпочтительно, хитотриозидазу измеряют вместе с ангиотензин-превращающим ферментом и кислой фосфатазой, ингибируемой тартратом, для мониторирования реакции на лечение у пациентов с болезнью Гоше.
Способы и препараты для проведения комбинированной терапии согласно настоящему изобретению включают в себя все способы и препараты, хорошо известные в данной области (см., например, Remington's Pharmaceutical Sciences, 1980 и последующие годы, 16th ed. и последующие издания, A. Oslo editor, Easton Pa.; Controlled Drug Delivery, 1987, 2nd rev., Joseph R. Robinson and Vincent H. L. Lee, eds., Marcel Dekker, ISBN: 0824775880; Encyclopedia of Controlled Drug Delivery, 1999, Edith Mathiowitz, John Wiley and Sons, ISBN: 0471148288; U.S. Pat. No. 6066626 и ссылки, указанные в этой публикации; см. также ссылки, цитируемые в разделах, представленных ниже).
Согласно настоящему изобретению, предоставлены следующие общие способы комбинированной терапии при лечении болезней лизосомного накопления. Каждый общий способ включает в себя комбинирование ферментозаместительной терапии с терапией малыми молекулами, соответствующее оптимизации клинической эффективности при минимизации неблагоприятных эффектов, связанных с применением каждого способа лечения в виде монотерапии.
В одном варианте осуществления настоящего изобретения ферментозаместительную терапию (в виде монотерапии или в комбинации с терапией малыми молекулами) проводят для начала лечения (т.е. для «разгрузки» субъекта), а терапию малыми молекулами проводят после фазы «разгрузки» для достижения и поддержания стабильного длительного терапевтического эффекта без необходимого проведения частых внутривенных инъекций средств ERT. Например, препараты ферментозаместительной терапии можно вводить внутривенно (например, в течение периода длительностью от одного до двух часов) один раз в неделю, один раз каждые две недели или один раз каждые два месяца в течение нескольких недель или месяцев или более долго (например, до тех пор, пока не уменьшится размер индикаторного органа, такого как селезенка или печень). Кроме того, ERT-фазу начального разгрузочного лечения можно проводить в виде монотерапии или в комбинации с терапией малыми молекулами. Компонент комбинированной терапии, которым является терапия малыми молекулами, особенно желателен тогда, когда малую молекулу можно вводить перорально, благодаря чему можно дополнительно уменьшить частоту внутривенного вмешательства.
Поочередное применение ферментозаместительной терапии и терапии малыми молекулами или дополнение терапии, использующей малые молекулы, при необходимости, ферментозаместительной терапией предоставляет стратегию, при которой одновременно используются достоинства и преодолеваются слабости, характерные для каждого такого способа лечения как монотерапии. Преимуществом ферментозаместительной терапии, как применяемой для «разгрузки», так и для более длительного лечения, является более широкий клинический опыт, доступный для информирования практических врачей, принимающих решения в реальных ситуациях. Кроме того, субъекту можно эффективно подбирать дозу ERT при проведении разгрузочной фазы - например, мониторируя биохимические метаболиты в моче или других биологических образцах или измеряя объем пораженного органа. Недостатком ферментозаместительной терапии, однако, является частота требующегося воздействия, которое обычно включает в себя внутривенные инъекции, выполняемые еженедельно или раз в две недели, что обусловлено постоянным повторным накоплением субстрата. Применение терапии малыми молекулами для уменьшения количества субстрата или для ингибирования его накопления у пациента может, в свою очередь, уменьшить частоту процедур ферментозаместительной терапии. Например, в двухнедельный режим дозирования ферментозаместительной терапии может быть включен период «выходных дней» (например, когда применяют терапию малыми молекулами), так что устраняется необходимость в частых инъекциях ферментов. Кроме того, лечение болезни лизосомного накопления посредством комбинированной терапии может предоставить некоторые комплементарные терапевтические подходы. Фактически, как продемонстрировано ниже в разделе «Примеры», комбинированная терапия SMT и ERT может значительно усовершенствовать каждый из этих терапевтических способов сам по себе. Эти данные свидетельствуют, что комбинированная терапия с применением SMT и ERT может быть и аддитивной, и комплементарной. В одном варианте осуществления настоящего изобретения ERT можно применять как способ «разгрузки» (т.е. для начального лечения) с последующим или одновременным дополнительным применением SMT, при которой используются соединения согласно настоящему изобретению. В другом варианте осуществления настоящего изобретения пациента сначала лечат, применяя SMT с использованием соединения согласно настоящему изобретению, а после нее или одновременно с ней дополнительно проводят ERT. В других вариантах осуществления настоящего изобретения SMT применяют для ингибирования или уменьшения дальнейшего накопления субстрата (или повторного накопления субстрата, если SMT применяют после «разгрузки», выполненной с применением ERT) у пациента с болезнью лизосомного накопления и, необязательно, при необходимости проводят ERT для дальнейшего снижения накопления субстрата. В одном варианте осуществления настоящее изобретение предоставляет способ комбинированной терапии для лечения субъекта с поставленным диагнозом болезни лизосомного накопления, включающий в себя поочередное применение ферментозаместительной терапии и терапии малыми молекулами. В другом варианте осуществления настоящее изобретение предоставляет способ комбинированной терапии для лечения субъекта с поставленным диагнозом болезни лизосомного накопления, включающий в себя одновременное применение ферментозаместительной терапии и терапии малыми молекулами. Следует понимать, что в различных вариантах комбинированной терапии согласно настоящему изобретению терапию малыми молекулами можно проводить до проведения ферментозаместительной терапии, одновременно с ней или после нее. Аналогичным образом, ферментозаместительную терапию можно проводить до проведения терапии малыми молекулами, одновременно с ней или после нее.
В любом из вариантов осуществления настоящего изобретения указанная болезнь лизосомного накопления является выбранной из группы, включающей в себя болезнь Гоше (типы 1, 2 и 3), болезнь Ниманна-Пика, болезнь Фарбера, GM1-ганглиозидоз, GM2-ганглиозидозы (например, дефицит активатора GM2, болезнь Тея-Сакса и болезнь Сандхоффа), болезнь Краббе, болезнь Гурлера-Шейе (MPS I), болезнь Хантера (MPS II), болезнь Санфилиппо (MPS III) Типа A, болезнь Санфилиппо (MPS III) Типа B, болезнь Санфилиппо (MPS III) Типа C, болезнь Санфилиппо (MPS III) Типа D, болезнь Моркио (MPS IV) Типа A, болезнь Моркио (MPS IV) Типа B, болезнь Марото-Лами (MPS VI), болезнь Слая (MPS VII), мукосульфатидоз, сиалидозы, муколипидоз II, муколипидоз III, муколипидоз IV, болезнь Фабри, болезнь Шиндлера, болезнь Помпе, болезнь накопления сиаловой кислоты, фукозидоз, маннозидоз, аспартилглюкозаминурию, болезнь Вольмана и неврональные цероидные липофусцинозы.
Кроме того, ферментозаместительная терапия предоставляет эффективное количество, по меньшей мере, одного из следующих ферментов: глюкоцереброзидазы, сфингомиелиназы, церамидазы, GM1-ганглиозид-бета-галактозидазы, гексозаминидазы A, гексозаминидазы B, бета-галактоцероброзидазы, альфа-L-идуронидазы, идуронатсульфатазы, гепаран-N-сульфатазы, N-ацетил-альфа-глюкозаминидазы, ацетил-CoA:альфа-глюкозаминид-ацетилтрансферазы, N-ацетил-альфа-глюкозамин-6-сульфатазы, галактозамин-6-сульфатазы, бета-галактозидазы, галактозамин-4-сульфатазы (арилсульфатазы B), бета-глюкуронидазы, арилсульфатазы A, арилсульфатазы C, альфа-нейраминидазы, N-ацетилглюкозамин-1-фосфат-трансферазы, альфа-галактозидазы A, альфа-N-ацетилгалактозаминидазы, альфа-глюкозидазы, альфа-фукозидазы, альфа-маннозидазы, аспартилглюкозаминамидазы, кислой липазы, пальмитоилпротеин-тиоэстеразы (CLN-1), PPT1, TPP1, CLN3, CLN5, CLN6, CLN8, NPC1 или NPC2.
Согласно настоящему изобретению, указанные SMT и/или ERT понижают уровень, по меньшей мере, одного из следующих накапливаемых материалов: глюкоцереброзида, сфингомиелина, церамида, GM1-ганглиозида, GM2-ганглиозида, глобозида, галактозилцерамида, дерматансульфата, гепарансульфата, кератансульфата, сульфатидов, мукополисахаридов, сиалилолигосахаридов, гликопротеинов, сиалилолигосахаридов, гликолипидов, глоботриаозилцерамида, O-связанных гликопептидов, гликогена, свободной сиаловой кислоты, фукогликолипидов, фукозилолигосахаридов, маннозилолигосахаридов, аспартилглюкозамина, холестерильных сложных эфиров, триглицеридов, гранулярных осмофильных отложений - сапозинов A и D, с-субъединицы ATP-синтазы, NPC1 или NPC2.
В определенных вариантах осуществления настоящего изобретения терапия малыми молекулами включает в себя введение указанному субъекту эффективного количества (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)-тиазол-4-ил)-пропан-2-ил)-карбамата (см. Фиг. 2A). В других вариантах осуществления настоящего изобретения терапия малыми молекулами включает в себя введение указанному субъекту эффективного количества хинуклидин-3-ил-(2-(4'-фтор-[1,1'-бифенил]-3-ил)-пропан-2-ил)-карбамата (см. Фиг. 2B). Терапия малыми молекулами может включать в себя введение субъекту одного или более соединений. В определенных вариантах осуществления настоящего изобретения по меньшей мере, одно из указанных соединений представляет собой соединение согласно настоящему изобретению, такое как те, которые показаны на Фигурах 2A и/или 2B.
Ферментозаместительная терапия может спровоцировать нежелательные иммунные реакции. Поэтому совместно с ферментозаместительным компонентом комбинированной терапии согласно настоящему изобретению можно применять иммуносупрессивные средства. Такие средства могут также применяться и с терапией малыми молекулами, но в этом случае потребность такого вмешательства, как правило, является менее вероятной. Совместно с комбинированной терапией согласно настоящему изобретению можно применять любой иммуносупрессант, известный квалифицированному специалисту в данной области. К таким имуносупрессантам относятся, но не ограничиваются ими, циклоспорин, FK506, рапамицин, CTLA4-Ig и препараты против TNF, такие как этанерцепт (см. например Moder, 2000, Ann. Allergy Asthma Immunol. 84, 280-284; Nevins, 2000, Curr. Opin. Pediatr. 12, 146-150; Kurlberg et al., 2000, Scand. J. Immunol. 51, 224-230; Ideguchi et al., 2000, Neuroscience 95, 217-226; Potteret al., 1999, Ann. N.Y. Acad. Sci. 875, 159-174; Slavik et al., 1999, Immunol. Res. 19, 1-24; Gaziev et al., 1999, Bone Marrow Transplant. 25, 689-696; Henry, 1999, Clin. Transplant. 13, 209-220; Gummert et al., 1999, J. Am. Soc. Nephrol. 10, 1366-1380; Qi et al., 2000, Transplantation 69, 1275-1283). В качестве иммуносупрессанта можно также применять антитела против альфа-субъединицы рецептора IL2 (препараты даклизумаб, зенапаксТМ), которые показали свою эффективность у пациентов, перенесших трансплантацию органов (см., например, Wiseman et al., 1999, Drugs 58, 1029-1042; Beniaminovitz et al., 2000, N. Engl J. Med. 342, 613-619; Ponticelli et al., 1999, Drugs R. D. 1, 55-60; Berard et al., 1999, Pharmacotherapy 19, 1127-1137; Eckhoff et al., 2000, Transplantation 69, 1867-1872; Ekberg et al., 2000, Transpl. Int. 13, 151-159). Дополнительные иммуносупрессанты включают в себя, но не ограничиваются ими, анти-CD2-препараты (Branco et al., 1999, Transplantation 68, 1588-1596; Przepiorka et al., 1998, Blood 92, 4066-4071), анти-CD4-препараты (Marinova-Mutafchieva et al., 2000, Arthritis Rheum. 43, 638-644; Fishwild et al., 1999, Clin. Immunol. 92, 138-152) и анти-CD40-лиганд (Hong et al., 2000, Semin. Nephrol. 20, 108-125; Chirmule et al., 2000, J. Virol. 74, 3345-3352; Ito et al., 2000, J. Immunol. 164, 1230-1235).
Совместно с комбинированной терапией согласно настоящему изобретению можно применять любую комбинацию иммуносупрессантов, известных квалифицированному специалисту в данной области. Одной из особенно полезных комбинаций иммуносупрессантов является комбинация такролимуса (FK506) с сиролимусом (рапамицин) и даклизумабом (антитела к альфа-субъединице рецептора IL2). Эта комбинация показала себя эффективной в качестве альтернативы стероидам и циклоспорину, особенно в отношении печени. Кроме того, эта комбинация, как было недавно показано, дает возможность выполнять успешную трансплантацию клеток островков поджелудочной железы. См. Denise Grady, The New York Times, Saturday, May 27, 2000, pages A1 and A11. См. также A. M. Shapiro et al., Jul. 27, 2000, «Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen», N. Engl. J. Med. 343, 230-238; Ryan et al., 2001, Diabetes 50, 710-719. Для удаления или снижения уровня антител, которые могут возникнуть против различных компонентов комбинированной терапии, можно также применять плазмаферез, проводимый любым способом, известным в данной области.
Показатели иммунного статуса, которые можно применять согласно настоящему изобретению, включают в себя, но не ограничиваются ими, антитела и любые цитокины, известные квалифицированному специалисту в данной области, - например, интерлейкины, CSF и интерфероны (см., главным образом, Leonard et al., 2000, J. Allergy Clin. Immunol. 105, 877-888; Oberholzer et al., 2000, Crit. Care Med. 28 (4 Suppl.), N3-N12; Rubinstein et al., 1998, Cytokine Growth Factor Rev. 9, 175-181). Например, для определения иммунного статуса субъекта можно следить за уровнем антител, специфично иммунореактивных к заместительному ферменту. Среди десятков известных интерлейкинов особенно предпочтительными индикаторами иммунного статуса являются IL-1-альфа, IL-2, IL-4, IL-8 и IL-10. Среди колониестимулирующих факторов (CSF) особенно предпочтительными индикаторами иммунного статуса являются G-CSF, GM-CSF и M-CSF. Среди интерферонов предпочтительными индикаторами иммунного статуса являются один или более из членов группы, включающей в себя альфа-, бета- или гамма-интерфероны.
В нижеследующих разделах указаны разнообразные компоненты, которые можно применять при восьми конкретных болезнях лизосомного накопления (болезнь Гоше (включая типы 1, 2 и 3), болезнь Фабри, болезнь Ниманна-Пика B, болезнь Хантера, болезнь Моркио, болезнь Марото-Лами, болезнь Помпе и болезнь Гурлера-Шейе). В последующих разделах предоставлено дальнейшее раскрытие компонентов, делающих возможными ферментозаместительную терапию и терапию малыми молекулами согласно настоящему изобретению.
Болезнь Гоше
Как отмечено выше, болезнь Гоше вызывается дефицитом фермента глюкоцереброзидазы (бета-D-глюкозил-N-ацилсфингозин-глюкогидролаза, КФ 3.2.1.45) и накоплением глюкоцереброзида (глюкозилцерамида). Для ферментозаместительной терапии как компонента комбинированной терапии согласно настоящему изобретению для лечения болезни Гоше доступен ряд литературных источников, которые информируют о достаточных дозах и режимах дозирования и содержат другие полезные сведения, относящиеся к лечению (см. Morales, 1996, Gaucher's Disease: A Review, The Annals of Pharmacotherapy 30, 381-388; Rosenthal et al., 1995, Enzyme Replacement Therapy for Gaucher Disease: Skeletal Responses to Macrophage-targeted Glucocerebrosidase, Pediatrics 96, 629-637; Barton et al., 1991, Replacement Therapy for Inherited Enzyme Deficiency--Macrophage-targeted Glucocerebrosidase for Gaucher's Disease, New England Journal of Medicine 324, 1464-1470; Grabowski et al., 1995, Enzyme Therapy in Type 1 Gaucher Disease: Comparative Efficacy of Mannose-terminated Glucocerebrosidase from Natural and Recombinant Sources, Annals of Internal Medicine 122, 33-39; Pastores et al., 1993, Enzyme Therapy in Gaucher Disease Type 1: Dosage Efficacy and Adverse Effects in 33 Patients treated for 6 to 24 Months, Blood 82, 408-416); и Weinreb et al., Am. J. Med.;113 (2): 112-9 (2002).
В одном варианте осуществления настоящего изобретения предоставлен режим дозирования ERT, при котором вводят от 2,5 единиц фермента на килограмм (Ед/кг) три раза в неделю до 60 Ед/кг один раз каждые две недели посредством внутривенного вливания продолжительностью 1-2 часа. Единица глюкоцереброзидазы определена как количество фермента, которое катализирует гидролиз одного микромоля синтетического субстрата (пара-нитрофенил-п-D-глюкопиранозида) в минуту при 37°C. В другом варианте осуществления настоящего изобретения предоставлен режим дозирования, при котором вводят от 1 Ед/кг три раза в неделю до 120 Ед/кг один раз каждые две недели. В еще одном другом варианте осуществления настоящего изобретения предоставлен режим дозирования, при котором вводят от 0,25 Ед/кг ежедневно или три раза в неделю до 600 Ед/кг один раз каждые 2-6 недель.
С 1991 года стал доступным препарат алглуцераза (цередаза®) от Genzyme Corporation. Алглуцераза представляет собой модифицированную форму глюкоцереброзидазы, выделенной из плаценты. В 1994 году стал доступным и препарат имиглуцераза (церезим®), также от Genzyme Corporation. Имиглуцераза является модифицированной формой глюкоцереброзидазы, производимой посредством экспрессии рекомбинантной ДНК в системе культуры клеток млекопитающего (клетки яичника китайского хомячка). Имиглуцераза представляет собой мономерный гликопротеин, состоящий из 497 аминокислот и содержащий четыре сайта N-гликозилирования. Имиглуцераза выгодна тем, что, теоретически, ее поставки не имеют ограничений, и, в отличие от плацентарной аглуцеразы, для нее характерна меньшая вероятность наличия биологических загрязнений. Эти ферменты модифицируют по сайтам гликозилирования, экспонируя остатки маннозы - прием, который повышает степень ее попадания в лизосомы, опосредуемого рецептором маннозо-6-фосфата. Имиглуцераза отличается от плацентарной глюкоцереброзидазы одной аминокислотой в положении 495, где аргинин заменен гистидином. Схемы введения эффективных доз известны (см. Morales, 1996, Id.; Rosenthal et al., 1995, Id.; Barton et al., 1991, Id.; Grabowski et al., 1995, Id.; Pastores et al., 1993, Id.). Например, клинически эффективен режим дозирования, при котором субъектам с умеренной и тяжелой формой заболевания вводят по 60 Ед/кг один раз каждые две недели. Квалифицированному практическому врачу следует ознакомиться с вышеуказанными источниками и с инструкцией по применению, прилагаемой к упаковке препарата, для получения информации о дополнительных режимах дозирования и способах введения. См. также патенты США №№ 5236838 и 5549892, выданные Genzyme Corporation.
Как отмечено выше, болезнь Гоше представляет собой результат дефицита лизосомного фермента глюкоцереброзидазы (GC). В наиболее распространенном фенотипе болезни Гоше (тип 1) патология ограничена ретикулоэндотелиальной и скелетной системами без неврологических симптомов. См. Barranger, Glucosylceramide lipidosis: Gaucher disease. In: Scriver CR BA, Sly WS, Valle D, editor. The Metabolic Basis of Inherited Disease. New York: McGraw-Hill. pp. 3635-3668 (2001). При невропатической болезни Гоше (nGD), подразделяемой на болезнь Гоше типа 2 и типа 3, дефицит глюкоцереброзидазы (GC) вызывает накопление глюкозилцерамида (GluCer; GL-1) и глюкозилсфингозина (GluSph) в мозге, приводя к неврологическим нарушениям. Болезнь Гоше типа 2 характеризуется ранним началом, быстрым прогредиентным течением, обширной патологией внутренних органов и центральной нервной системы; смерть наступает в возрасте 2 лет. Болезнь Гоше типа 3, также известная как подострая форма nGD, представляет собой промежуточный фенотип с разным возрастом начала заболевания при разной степени тяжести и скорости прогредиентного течения. Goker-Alpan et al., The Journal of Pediatrics 143: 273-276 (2003). Недавно была создана модель болезни Гоше типа 2 у мышей K14 lnl/lnl (далее «мыши K14»); эта модель у мышей, характеризующемуся атаксией, судорогами, спастичностью и медианой продолжительности жизни, уменьшенной всего лишь до 14 дней, весьма сходна с соответствующим заболеванием людей. Enquist et al., PNAS 104: 17483-17488 (2007).
Как и у пациентов с nGD, некоторые модели этого заболевания у мышей имеют повышенные уровни GluCer и GluSph в мозге вследствие недостаточной активности GC. Liu et al., PNAS 95: 2503-2508 (1998) и Nilsson, J. Neurochem 39: 709-718 (1982). Мыши «K14» демонстрируют невропатический фенотип, который сходен с болезнью Гоше типа 2 по многим патологическим признакам (таким как нейродегенерация, астроглиоз, микроглиальная пролиферация и повышенные уровни GluCer и GluSph в специфических участках мозга. Enquist et al. (2007).
Клиническое ведение пациентов, страдающих nGD, является для лечащих врачей сложной проблемой, как по причине тяжести заболевания типа 2, так и вследствие неспособности средств, применяемых в современной терапии, проникать через гематоэнцефалический барьер (ГЭБ). Современное лечение других форм заболевания (отличных от nGD) основывается на внутривенном введении рекомбинантной глюкоцереброзидазы человека (имиглуцераза, церезим™) для восполнения недостатка фермента или на введении ингибиторов глюкозилцерамид-синтазы для уменьшения продуцирования субстрата (GL-1). Однако эти лекарственные средства не проникают через гематоэнцефалический барьер и поэтому не могут быть эффективными у пациентов с nGD. Современные низкомолекулярные ингибиторы глюкозилцерамид-синтазы, применяемые в клинике, по-видимому, неэффективны в отношении невропатических фенотипов nGD. Испытание одного из соединений согласно настоящему изобретению - хинуклидин-3-ил-(2-(4'-фтор-[1,1'-бифенил]-3-ил)-пропан-2-ил)-карбамата (далее «Gz161») - на модели болезни Гоше типа 2 у мышей K14 продемонстрировало, что оно действительно снижает уровни GluCer и GluSph в мозге (см. Примеры 122-125). Кроме того, в этой модели оно ослабляет невропатологию мозга и продлевает продолжительность жизни. Более того, комбинированный подход, при котором ферментозаместительная терапия и терапия малыми молекулами, снижают уровень субстрата, может оказаться наилучшим способом лечения болезни Гоше типа 2.
Болезнь Фабри
Как отмечено выше, болезнь Фабри вызывается дефицитом лизосомального фермента альфа-галактозидазы A. Этот энзимологический дефект приводит к системному отложению гликосфинголипидов, имеющих альфа-галактозильные концевые фрагменты (преимущественно, глоботриаозилцерамидные (GL3 или Gb3) и, в меньшей степени, церамидные), и гликосфинголипиды группы крови B.
Для мониторирования течения заболевания и определения момента, когда необходимо сменить один способ лечения другим, имеется несколько доступных аналитических методик. В одном варианте осуществления настоящего изобретения можно применять методику определения удельной активности альфа-галактозидазы A в образце ткани. В другом варианте осуществления настоящего изобретения можно применять методику определения накопления Gb3. В другом варианте осуществления настоящего изобретения практический врач может исследовать накопление гликосфинголипидных субстратов в биологических жидкостях организма и в лизосомах сосудистых эндотелиальных, перителиальных и гладкомышечных клеток кровеносных сосудов. Другие клинические проявления, которые могут быть полезными показателями при ведении заболевания, включают в себя протеинурию и другие признаки поражения почек, такие как эритроциты и липидные глобулы в моче и повышенная скорость оседания эритроцитов. Можно также мониторировать анемию, пониженную концентрацию железа в сыворотке, высокую концентрацию бета-тромбоглобулина и увеличенную численность ретикулоцитов или агрегацию тромбоцитов. Фактически, для мониторирования течения заболевания можно использовать любой способ, известный квалифицированному специалисту в данной области (см. например, Desnick RJ et al., 1995, Alpha-galactosidase A Deficiency: Fabry Disease, In: The Metabolic and Molecular Bases of Inherited Disease, Scriver et al., eds., McGraw-Hill, N.Y., 7.sup.th ed., pages 2741-2784). Предпочтительным суррогатным маркером для мониторирования ведения болезни Фабри является боль. К другим предпочтительным способам относятся измерение общего клиренса фермента и/или субстрата и жидкостей организма или анализ образцов, получаемых при биопсии. Предпочтительный режим дозирования для ферментозаместительной терапии при болезни Фабри - 1-10 мг/кг внутривенно через день. Можно применять и режим дозирования, при котором внутривенно вводят от 0,1 до 100 мг/кг с частотой от одного раза в два дня до одного раза еженедельно или каждые две недели.
Болезнь Ниманна-Пика B
Как отмечено выше, болезнь Ниманна-Пика B вызывается пониженной активностью лизосомального фермента кислой сфингомиелиназы и накоплением мембранного липида, в первую очередь, сфингомиелина. Эффективная доза заместительной кислой сфингомиелиназы может быть в диапазоне от приблизительно 0,01 мг/кг до приблизительно 10 мг/кг массы тела, при введении от одного раза в два дня до одного раза еженедельно, одного раза каждые две недели или одного раза каждые два месяца. В других вариантах осуществления настоящего изобретения эффективная доза может быть в диапазоне от приблизительно 0,03 мг/кг до приблизительно 1 мг/кг; от приблизительно 0,03 мг/кг до приблизительно 0,1 мг/кг; и/или от приблизительно 0,3 мг/кг до приблизительно 0,6 мг/кг. В конкретном варианте осуществления настоящего изобретения пациенту вводят кислую сфингомиелиназу в режиме возрастающих доз в следующей последовательности: 0,1 мг/кг; 0,3 мг/кг; 0,6 мг/кг; и 1,0 мг/кг, когда каждую дозу кислой сфингомиелиназы вводят, по меньшей мере, дважды и каждую дозу вводят с двухнедельными интервалами, проводя мониторинг пациента для выявления токсических побочных эффектов перед повышением дозы до следующего уровня (см. опубликованную заявку на патент США № 2011/0052559).
Болезнь Гурлера-Шейе (MPS I)
Болезнь Гурлера, болезнь Шейе и болезнь Гурлера-Шейе, также известные как MPS I, вызываются инактивацией альфа-идуронидазы и накоплением дерматансульфата и гепарансульфата. Для мониторирования течения MPS I имеется несколько доступных аналитических методик. Например, можно мониторировать ферментативную активность альфа-идуронидазы в образцах ткани, извлеченных при биопсии, или в культивируемых клетках, полученных из периферийной крови. Кроме того, традиционным средством наблюдения за течением заболевания при MPS I и других мукополисахаридозах является определение экскреции гликозаминогликанов (дерматансульфата и гепарансульфата) с мочой (см. Neufeld et al., 1995, Id.). В конкретном варианте осуществления настоящего изобретения фермент альфа-идуронидазу вводят один раз в неделю в виде внутривенного вливания в дозе 0,58 мг/кг массы тела.
Болезнь Хантера (MPS II)
Болезнь Хантера (или MPS II) вызывается инактивацией идуронатсульфатазы и накоплением дерматансульфата и гепарансульфата. Болезнь Хантера имеет клинически тяжелые и легкие формы. Предпочтительным является режим дозирования терапевтического фермента, при котором вводят от 1,5 мг/кг каждые две недели до 50 мг/кг каждую неделю.
Болезнь Моркио (MPS IV)
Синдром Моркио (или MPS IV) является результатом накопления кератансульфата вследствие инактивации любого из двух ферментов. При MPS IVA инактивированным ферментом является галактозамин-6-сульфатаза, а при MPS IVB инактивированным ферментом является бета-галактозидаза. Предпочтительным является режим дозирования терапевтического фермента, при котором вводят от 1,5 мг/кг каждые две недели до 50 мг/кг каждую неделю.
Болезнь Марото-Лами (MPS VI)
Синдром Марото-Лами (или MPS VI) вызывается инактивацией галактозамин-4-сульфатазы (арилсульфатазы B) и накоплением дерматансульфата. Режимом дозирования, предоставляемым ферментозаместительной терапией, является введение эффективного терапевтического фермента в предпочтительном диапазоне от 1,5 мг/кг каждые две недели до 50 мг/кг каждую неделю. В необязательном варианте, применяемая доза составляет не более 10 мг/кг в неделю. Предпочтительным суррогатным маркером для наблюдения за течением заболевания при MPS VI являются уровни протеогликанов.
Болезнь Помпе
Болезнь Помпе вызывается инактивацией фермента кислой альфа-глюкозидазы и накоплением гликогена. Ген кислой альфа-глюкозидазы человека (обозначаемой GAA) находится на хромосоме 17. H.G.Hers первым предложил концепцию врожденного лизосомного заболевания, основываясь на своих исследованиях этого заболевания, которое он называл болезнью накопления гликогена типа II (GSD II) и которое в настоящее время называют дефицитом кислой мальтазы (AMD) (см. Hers, 1965, Gastroenterology 48, 625). В конкретном варианте осуществления настоящего изобретения GAA вводят каждые 2 недели в виде внутривенного вливания в дозе 20 мг/кг массы тела.
Для мониторирования течения болезни Помпе имеется несколько доступных аналитических методик. Можно применять любой аналитический способ, известный квалифицированному специалисту в данной области. Например, можно исследовать накопление гранул гликогена внутри лизосом, особенно в сердечной мышце, печени и в волокнах скелетных мышц, полученных при биопсии. Ферментативную активность альфа-глюкозидазы можно также прослеживать в образцах биопсии или в культивируемых клетках периферийной крови. В качестве показателя течения заболевания можно мониторировать повышение креатинкиназы (СК) в сыворотке. У пациентов с началом заболевания в детском возрасте сывороточная СК может быть повышенной до десятикратного уровня; у пациентов, заболевание которых началось во взрослом возрасте, она обычно повышается в меньшей степени. См. Hirschhorn R, 1995, Glycogen Storage Disease Type II: Acid alpha-Glucosidase (Acid Maltase) Deficiency, In: The Metabolic and Molecular Bases of Inherited Disease, Scriver et al., eds., McGraw-Hill, N.Y., 7.sup.th ed., pages 2443-2464.
Ферментозаместительная терапия
Нижеследующие разделы описывают конкретное раскрытие и альтернативные варианты осуществления ферментозаместительной терапии как компонента комбинированной терапии согласно настоящему изобретению. Как правило, режимы дозирования для ферментозаместительной терапии как компонента комбинированной терапии согласно настоящему изобретению обычно определяет квалифицированный специалист в данной области. Некоторые примеры режимов дозирования при лечении болезни Гоше глюкоцереброзидазой предоставлены выше. Общие принципы определения режима дозирования для любого данного способа ERT как компонента комбинированной терапии согласно настоящему изобретению для лечения любой формы LSD будут понятны квалифицированному специалисту в данной области из общедоступной информации - например из обзоров конкретных источников, указанных в разделах, посвященных каждой конкретной форме LSD. Средства ERT можно вводить пациенту путем внутривенного вливания. Для проведения ERT у пациента с поставленным диагнозом болезни лизосомного накопления с проявлениями в ЦНС можно также применять интрацеребровентрикулярное и/или интратекальное вливание (например, в дополнение к внутривенному вливанию).
Для изготовления препаратов ферментов, применяемых при ферментозаместительной терапии, проводимой в качестве компонента комбинированной терапии согласно настоящему изобретению, можно применять любой способ, известный в данной области техники. Известно много таких способов, они включают в себя, но не ограничиваются этим, технологию активации генов, разработанную Shire plc (см. Патенты США №№ 5968502 и 5272071).
Терапия малыми молекулами
Следующий раздел также описывает конкретные раскрытия и альтернативные варианты осуществления, доступные для терапии малыми молекулами, проводимой в качестве компонента комбинированной терапии согласно настоящему изобретению. Режимы дозирования при терапии малыми молекулами, проводимой в качестве компонента комбинированной терапии согласно настоящему изобретению, обычно определяет квалифицированный клиницист, причем они будут значительно варьировать в зависимости от конкретной болезни накопления, лечение которой необходимо проводить, и клинического состояния конкретного больного индивида. Общие принципы определения режима дозирования для данного способа терапии малыми молекулами, проводимой в качестве компонента любой комбинированной терапии любой болезни накопления согласно настоящему изобретению, хорошо известны квалифицированному специалисту в данной области. Руководящие указания по режимам дозирования можно получить в любом из многих хорошо известных источников по этой теме в данной области. Дополнительные рекомендации доступны, между прочим, в обзоре конкретных источников, ссылки на которые даны в настоящем документе.
Как правило, соединения согласно настоящему изобретению, такие как, например, (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)-тиазол-4-ил)-пропан-2-ил)-карбамат и хинуклидин-3-ил-(2-(4'-фтор-[1,1'-бифенил]-3-ил)-пропан-2-ил)-карбамат можно применять в комбинированной терапии согласно настоящему изобретению для лечения практически любой болезни накопления, обусловленной нарушениями в гликосфинголипидном пути (например, болезни Гоше, болезни Фабри, GM1-ганглиозидоза и GM2-ганглиозидозов (например, дефицита активатора GM2, болезни Тея-Сакса и болезни Сандхоффа)). Аналогичным образом, аминогликозиды (например, гентамицин, G418) можно применять в комбинированной терапии согласно настоящему изобретению для лечения любого индивида с болезнью накопления, имеющего мутацию с преждевременным стоп-кодоном (т.е. нонсенс-мутацию). Такие мутации особенно преобладают при синдроме Гурлера. Терапия малыми молекулами как компонент комбинированной терапии согласно настоящему изобретению является особенно предпочтительной для лечения болезни накопления с признаками нарушений в центральной нервной системе (например, при болезни Сандхоффа, болезни Тея-Сакса, болезни Ниманна-Пика типа A и болезни Гоше типов 2 и 3), поскольку малые молекулы обычно могут легче проникать через гематоэнцефалический барьер по сравнению со средствами, применяемыми при других способах лечения.
Предпочтительные дозы ингибиторов накопления субстратов, применяемые в комбинированной терапии согласно настоящему изобретению, может легко определить квалифицированный специалист в данной области. В определенных вариантах осуществления настоящего изобретения такие дозы могут быть в диапазоне от приблизительно 0,5 мг/кг до приблизительно 300 мг/кг, предпочтительно, от приблизительно 5 мг/кг до приблизительно 60 мг/кг (например, 5 мг/кг, 10 мг/кг, 15, мг/кг, 20 мг/кг, 25 мг/кг, 30 мг/кг, 35 мг/кг, 40 мг/кг, 45 мг/кг, 50 мг/кг, 55 мг/кг и 60 мг/кг) при внутрибрюшинном, пероральном или эквивалентном введении от одного до пяти раз в день. Такие дозы могут быть в диапазоне от приблизительно 5 мг/кг до приблизительно 5 г/кг, предпочтительно, от приблизительно 10 мг/кг до приблизительно 1 г/кг при пероральном, внутрибрюшинном или эквивалентном введении от одного до пяти раз в день. В одном варианте осуществления настоящего изобретения дозы находятся в диапазоне от приблизительно 10 мг/день до приблизительно 500 мг/день (например, 10 мг/день, 20 мг/день, 30 мг/день, 40 мг/день, 50 мг/день, 60 мг/день, 70 мг/день, 80 мг/день, 90 мг/день, 100 мг/день, 110 мг/день, 120 мг/день, 130 мг/день, 140 мг/день, 150 мг/день, 160 мг/день, 170 мг/день, 180 мг/день, 190 мг/день, 200 мг/день, 210 мг/день, 220 мг/день, 230 мг/день, 240 мг/день, 250 мг/день, 260 мг/день, 270 мг/день, 280 мг/день, 290 мг/день, 300 мг/день). Особо предпочтительная пероральная доза находится в диапазоне от приблизительно 50 мг до приблизительно 100 мг, причем эту дозу вводят два раза в день. Конкретный диапазон пероральных доз соединения согласно настоящему изобретению составляет от приблизительно 5 мг/кг/день до приблизительно 600 мг/кг/день. В частности, диапазон пероральных доз для соединения согласно настоящему изобретению составляет от приблизительно 1 мг/кг/день до приблизительно 100 мг/кг/день, например, 1 мг/кг/день, 5 мг/кг/день, 10 мг/кг/день, 15 мг/кг/день, 20 мг/кг/день, 25 мг/кг/день, 30 мг/кг/день, 35 мг/кг/день, 40 мг/кг/день, 45 мг/кг/день, 50 мг/кг/день, 55 мг/кг/день или 60 мг/кг/день, 65 мг/кг/день, 70 мг/кг/день, 75 мг/кг/день, 80 мг/кг/день, 85 мг/кг/день, 90 мг/кг/день, 95 мг/кг/день или 100 мг/кг/день.
Предпочтительным является циклическое чередование способов лечения (т.е. ферментозаместительной терапии и терапии малыми молекулами). Однако при необходимости, определяемой квалифицированным клиницистом, субъектов можно также лечить с перекрыванием периодов применения обоих способов. Примеры схем лечения могут включать в себя, не ограничиваясь ими, следующие: (1) SMT с последующей ERT; (2) ERT с последующей SMT; и (3) ERT и SMT, проводимые приблизительно в одно и то же время. Как отмечено выше, при необходимости можно также осуществлять перекрывание периодов применения разных способов лечения в зависимости от клинического течения данной болезни накопления у данного субъекта.
Лечебные интервалы для различных форм комбинированной терапии могут широко варьировать и быть различными при разных болезнях накопления и у разных индивидов, в зависимости от того, насколько интенсивно аккумулируются продукты накопления. Например, аккумулирование продуктов накопления при болезни Фабри может быть медленным по сравнению с быстрым их накоплением при болезни Помпе. Квалифицированный специалист в данной области, прослеживая клинические признаки течения заболевания и успех лечения, осуществляет постепенный подбор режима лечения конкретной болезни накопления у конкретного индивида.
Разнообразные макромолекулы, которые накапливаются при болезнях лизосомного накопления, распределяются не однородно, а образуют отложения в определенных предпочтительных анатомических участках, специфичных для каждого заболевания. Однако эндогенно вводимый фермент, как правило, поглощается клетками ретикулоэндотелиальной системы и направляется в лизосомальный компартмент, где он действует, гидролизуя накопленный субстрат. Кроме того, клеточное поглощение терапевтического фермента можно усиливать определенными специфическими приемами, повышающими степень его попадания в лизосомы (см., например, патент США № 5549892 (Friedman et al.), выданный Genzyme Corporation, который описывает рекомбинантную глюкоцереброзидазу, фармакокинетика которой улучшена благодаря перемоделированию олигосахаридных боковых цепей, узнаваемых маннозными рецепторами на поверхностях клеток, осуществляющих эндоцитоз и транспорт в лизосомы).
Некоторые способы лечения действуют на некоторые пораженные органы лучше, чем на другие. Например, при болезни Фабри, если ERT недостаточно клинически эффективно действует на почки, для понижения уровней субстрата в почках можно применять SMT. Как показано в Примере 112 и на Фиг. 6В, SMT более эффективно, чем ERT, понижала уровни Gb3 (субстрата, накапливаемого у пациентов с болезнью Фабри) в моче мышей с моделью болезни Фабри. Считается, что почки являются главным источником Gb3, присутствующего в моче. В отличие от этого, Фиг. 6В показывает, что ERT более эффективно, чем SMT, понижала уровни Gb3 в плазме. Эти результаты демонстрируют, что комбинированная терапия ERT и SMT предоставляет комплементарную терапевтическую стратегию, которая использует преимущества и эффективность каждого из этих способов лечения, применяемых в виде монотерапии, и преодолевает их слабости. Средства, применяемые при SMT, способны проникать через ГЭБ, благодаря чему, в сочетании с ERT, предоставляется эффективный способ лечения болезней лизосомного накопления с проявлениями патологии ЦНС, таких как тип А болезни Ниманна-Пика и невропатическая болезнь Гоше (nGD). Более того, уменьшение количества субстрата, осуществляемое SMT, комбинируемой с ферментозаместительной терапией, решает проблему накопления, осуществляя вмешательство в разных специфических точках, благодаря чему возможно повышение клинической эффективности.
Следует понимать, что ссылки на одновременное или совпадающее проведение двух или более способов лечения не требует того, чтобы их проводили в одно и то же время, т.е. чтобы они действовали на субъекта в одно и то же время.
Пример 1
Хинуклидин-3-ил-1-фенилциклобутилкарбамат
Применяя общую процедуру A, из гидрохлорида 1-фенилциклобутанамина (100 мг, 0,540 ммоль) и хинуклидин-3-ола (103 мг, 0,810 ммоль) получали хинуклидин-3-ил-1-фенилциклобутилкарбамат (76 мг, 47%) в виде белого твердого вещества.1H ЯМР (400 МГц, CDCl3) δ 7,43 (д, J=7,9 Гц, 2H), 7,34 (т, J=7,7 Гц, 2H), 7,23 (т, J=7,3 Гц, 1H), 5,75-5,25 (м, 1H), 4,60 (шир.с, 1H), 3,25-2,22 (м, 9H), 2,16-2,03 (м, 1H), 2,02- 0,94 (м, 6H), 0,88 (т, J=6,8 Гц, 1H) м.д.13C ЯМР (100 МГц, CDCl3) δ 158,1, 128,5, 126,9, 125,6, 71,4, 59,4, 55,7, 47,5, 46,6, 34,0, 31,8, 29,9, 25,5, 24,7, 22,9, 19,7, 15,3, 14,4 м.д. Чистота: >99,9% UPLCMS (210 нм); время удерживания 0,62 мин; (M+1) 331.
Пример 2
Хинуклидин-3-ил-2-(бензо[d][1,3]диоксол-5-ил)-пропан-2-илкарбамат
Применяя общую процедуру B, бензо[d][1,3]диоксол-5-карбонитрил (1,00 г, 6,81 ммоль) преобразовывали до гидрохлорида 2-(бензо[d][1,3]диоксол-5-ил)-пропан-2-амина (692 мг, 47%).
Применяя общую процедуру A, из вышеуказанного аммонийхлоридного промежуточного соединения (150 мг, 0,695 ммоль) и хинуклидин-3-ола получали хинуклидин-3-ил-2-(бензо[d][1,3]диоксол-5-ил)-пропан-2-илкарбамат (125 мг, 54%) в виде белого твердого вещества.1H ЯМР (400 МГц, CDCl3) δ 6,91 (дд, J=1,9 Гц, 1H), 6,87 (дд, J=1,9, 8,2 Гц, 1H), 6,75 (д, J=8,2 Гц, 1H), 5,93 (с, 2H), 5,12 (с, 1H), 4,69-4,66 (м, 1H), 3,26-2,11 (м, 7H), 2,03-1,07 (м, 4H), 1,63 (с, 6H) м.д.13C ЯМР (100 МГц, CDCl3) δ 156,7, 147,9, 118,0, 108,1, 106,1, 101,2, 71,2, 55,9, 55,3, 47,6, 46,7, 29,9, 29,7, 25,6, 24,8, 19,8 м.д. Чистота: 97,5% UPLCMS (210 нм); время удерживания 0,65 мин; (M+1) 333.
Пример 3
Хинуклидин-3-ил-2-(нафталин-1-ил)-пропан-2-илкарбамат
Применяя общую процедуру A, из гидрохлорида 2-(нафталин-1-ил)-пропан-2-амина (100 мг, 0,450 ммоль) и хинуклидин-3-ола получали хинуклидин-3-ил-2-(нафталин-1-ил)-пропан-2-илкарбамат (115 мг, 59%) в виде белого твердого вещества.1H ЯМР (400 МГц, CDCl3) δ 8,79-8,46 (м, 1H), 7,99-7,72 (м, 2H), 7,69-7,36 (м, 4H), 5,86-5,37 (м, 1H), 4,72-4,34 (м, 1H), 3,25-2,20 (м, 6H), 2,16-0,41 (м, 5H), 1,93 (с, 6H) м.д.13C ЯМР (100 МГц, CDCl3) δ 154,6, 135,2, 130,7, 129,7, 128,8, 125,9, 125,3, 123,9, 72,2, 71,1, 56,5, 55,7, 47,6, 46,6, 31,8, 31,2, 25,5, 24,8, 22,9, 19,7, 14,4 м.д. Чистота: 100% UPLCMS (210 нм); время удерживания 0,80 мин; (M+1) 339.
Препаративная процедура I
Пример 4
(R)-хинуклидин-3-ил-2-(3-(проп-1-ен-2-ил)-фенил)-пропан-2-илкарбамат
К раствору (R)-хинуклидин-3-ола (194 мг, 1,52 ммоль) в THF (5 мл) при комнатной температуре добавляли NaH [60%, масло] (64 мг, 1,6 ммоль). Реакционную смесь перемешивали в течение 15 мин и по каплям добавляли 1-(2-изоцианатопропан-2-ил)-3-(проп-1-ен-2-ил)-бензол (302 мкл, 1,53 ммоль). Реакционную смесь перемешивали в течение 30 мин и гасили насыщенным солевым раствором. Раствор экстрагировали EtOAc и органический слой сушили над Na2SO4 и концентрировали. Неочищенный материал очищали посредством комбифлэш-хроматографии (SiO2-картридж, CHCl3 и 2 н NH3 в MeOH), получая соответствующий карбамат (475 мг, 95%) в виде прозрачного масла.1H ЯМР (400 МГц, CDCl3) δ 7,49 (с, 1H), 7,31 (шир.с, 3H), 5,33 (с, 1H), 5,17 (с, 1H), 5,08 (с, 1H), 4,77-4,61 (м, 1H), 3,33-2,27 (м, 5H), 2,14 (с, 3H), 2,25-0,75 (м, 6H), 1,68 (шир.с, 6H) м.д.13C ЯМР (100 МГц, CDCl3) δ 154,7, 147,2, 143,7, 141,6, 128,5, 124,2, 122,1, 112,8, 70,9, 55,7, 55,5, 47,5, 46,6, 32,2, 31,5, 29,9, 29,6, 25,5, 24,6, 22,9, 22,2, 19,6 м.д. Чистота: 100% UPLCMS (210 нм); время удерживания 0,84 мин; (M+1) 329,2. Анал. расчет для C20H28N2O2·0,06 (CHCl3): C, 71,59; H, 8,40; N, 8,58. Найдено: C, 71,51; H, 9,05; N, 8,60.
Препаративная процедура J
Пример 5
Хинуклидин-3-ил-2-(3-изопропоксифенил)-пропан-2-илкарбамат
Раствор 3-цианофенола (1,00 г, 8,39 ммоль), 2-йодпропана (839 мкл, 8,39 ммоль) и карбоната цезия (2,73 г, 8,39 ммоль) в смеси CH2Cl2/CH3CN (1:1, 16 мл) перемешивали при кипячении с обратным холодильником в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали через целит. Фильтрат концентрировали и неочищенный материал очищали посредством комбифлэш-хроматографии (SiO2-картридж, CH2Cl2), получая соответствующий простой эфир (763 мг, 57%) в виде белого твердого вещества.
Применяя общую процедуру B, 3-изопропоксибензонитрил (763 мг, 4,24 ммоль) преобразовывали в соответствующий 2-(3-изопропоксифенил)-пропан-2-амин (362 мг, 45%) в виде прозрачного масла.
Применяя общую процедуру A, из вышеуказанного амина (100 мг, 0,520 ммоль) и хинуклидин-3-ола получали хинуклидин-3-ил-2-(3-изопропоксифенил)-пропан-2-илкарбамат (110 мг, 61%) в виде белого твердого вещества.1H ЯМР (400 МГц, CDCl3) δ 7,17 (т, J=7,9 Гц, 1H), 6,92 (д, J=7,8 Гц, 1H), 6,89 (т, J=2,1 Гц, 1H), 6,70 (д, J=8,1 Гц, 1H), 5,38-5,13 (м, 1H), 4,58 (шир.с, 1H), 4,49 (гепт, J=6,1 Гц, 1H), 3,31-2,04 (м, 6H), 2,00-0,79 (м, 5H) 1,60 (шир.с, 6H), 1,28 (д, J=6,1 Гц, 6H) м.д.13C ЯМР (100 МГц, CDCl3) δ 158,1, 129,5, 117,2, 113,6, 71,1, 69,9, 55,8, 55,4, 47,6, 46,6, 29,4, 25,6, 24,8, 22,3, 19,7 м.д. Чистота: >99,9% UPLCMS (210 нм); время удерживания 0,83 мин; (M+1) 347.
Пример 6
Хинуклидин-3-ил-2-(3-бром-2-фторфенил)-пропан-2-илкарбамат
Применяя общую процедуру A, из 2-(3-бром-2-фторфенил)-пропан-2-амина (1,0 г, 4,3 ммоль) и хинуклидин-3-ола получали хинуклидин-3-ил-2-(3-бром-2-фторфенил)-пропан-2-илкарбамат (957 мг, 58%) в виде белого твердого вещества.1H ЯМР (400 МГц, CDCl3) δ 7,45 (ддд, J=1,6, 6,3, 7,9 Гц, 1H), 7,31 (тд, J=1,6, 7,7 Гц, 1H), 6,99 (тд, J=1,0, 8,0 Гц, 1H), 5,31-5,15 (шир.с, 1H), 4,59 (шир.с, 1H), 3,25-2,19 (м, 6H), 2,06-0,81 (м, 5H), 1,73 (с, 3H), 1,71 (с, 3H) м.д.13C ЯМР (100 МГц, CDCl3) δ 158,1, 155,6, 132,5, 127,1, 127,0, 124,8, 124,8, 110,6, 110,4, 71,5, 55,7, 54,2, 47,5, 46,7, 29,9, 28,4, 25,5, 24,8, 19,7 м.д. Чистота: >99,9% UPLCMS (210 нм); время удерживания 0,79 мин; (M+1) 385.
Пример 7
(+/-)-Хинуклидин-3-ил-(1R,2S)-2-фенилциклопропилкарбамат
Применяя общую процедуру A, из (+/-)((1S,2R)-2-изоцианатоциклопропил)-бензола (117 мкл, 0,780 ммоль) и хинуклидин-3-ола получали (+/-)-хинуклидин-3-ил-(1R,2S)-2-фенилциклопропилкарбамат (63 мг, 28%) в виде белого твердого вещества.1H ЯМР (400 МГц, CDCl3) δ 7,30-7,05 (м, 5H), 5,43 (шир.с, 1H), 4,77 (шир.с, 1H), 3,23 (дд, J=9,0, 14,0 Гц, 1H), 2,97-2,65 (м, 6H), 2,15-1,12 (м, 8H) м.д.13C ЯМР (100 МГц, CDCl3) δ 157,1, 140,7, 140,2, 128,5, 126,8, 126,3, 71,5, 55,7, 47,5, 46,6, 32,7, 25,6, 25,2, 24,5, 19,5, 16,2 м.д. Чистота: >99,9% UPLCMS (210 нм); время удерживания 0,67 мин; (M+1) 287.
Пример 8
Хинуклидин-3-ил-1-фенилциклогексилкарбамат
Применяя общую процедуру A, из 1-фенилциклогексанамина (36 мг, 0,21 ммоль) и хинуклидин-3-ола получали хинуклидин-3-ил-1-фенилциклогексилкарбамат (40 мг, 58%) в виде белого твердого вещества.1H ЯМР (400 МГц, CDCl3) δ 7,41 (д, J=7,4 Гц, 2H), 7,32 (т, J=7,7 Гц, 2H), 7,21 (т, J=7,3 Гц, 1H), 5,19-4,98 (шир.с, 1H), 4,70-4,56 (с, 1H), 3,34-0,83 (м, 21H) м.д.13C ЯМР (100 МГц, CDCl3) δ 158,4, 128,3, 126,5, 124,9, 57,2, 46,3, 36,1, 25,4, 24,2, 22,0, 19,2, 15,2 м.д. Чистота: >99,9% UPLCMS (210 нм); время удерживания 0,84 мин; (M+1) 329.
Препаративная процедура K
Пример 9
(R)-1-(2-(3-(проп-1-ен-2-ил)-фенил)-пропан-2-ил)-3-(хинуклидин-3-ил)-мочевина
К раствору дигидрохлорида (R)-хинуклидин-3-амина (120 мг, 0,603 ммоль) и 1-(2-изоцианатопропан-2-ил)-3-(проп-1-ен-2-ил)-бензола (119 мг, 0,597 ммоль) в THF (3 мл) добавляли триэтиламин (168 мкл, 1,21 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов и затем гасили насыщенным солевым раствором. Смесь экстрагировали CHCl3 и органический слой сушили (Na2SO4) и концентрировали. Неочищенный материал очищали посредством комбифлэш-хроматографии (SiO2-картридж, CHCl3 и 2 н NH3 в MeOH), получая соответствующую мочевину (163 мг, 50%) в виде белого твердого вещества.1H ЯМР (400 МГц, CDCl3) δ 7,62 (т, J=1,6 Гц, 1H), 7,44 (дт, J=1,9, 7,0 Гц, 1H), 7,41-7,33 (м, 2H), 5,35 (шир.с, 1H), 5,11 (п, J=1,4 Гц, 1H), 4,84 (с, 1H), 4,21 (д, J=7,5 Гц, 1H), 3,70-3,61 (м, 1H), 3,13 (ддд, J=2,3, 9,3, 14,2 Гц, 1H), 2,71-2,54 (м, 3H), 2,30-2,22 (м, 1H), 2,15 (дд, J=0,8, 1,4, 3H), 2,05-1,96 (м, 1H), 1,65 (с, 3H), 1,64 (с, 3H), 1,65-1,60 (м, 1H) 1,54-1,45 (м, 2H), 1,22-1,12 (м, 1H), 0,95-0,80 (м, 1H) м.д.13C ЯМР (400 МГц, CDCl3) δ 157,4, 148,5, 143,8, 141,3, 128,4, 124,4, 123,8, 122,2, 112,6, 55,2, 53,4, 46,2, 46,1, 44,5, 30,5, 30,4, 25,0, 22,2, 17,7, 8,9 м.д. Чистота: 97,5% UPLCMS (210 нм); время удерживания 0,83 мин; (M+1) 328.
Пример 10
1-(2-(Нафталин-2-ил)-пропан-2-ил)-3-(хинуклидин-3-ил)-мочевина
Применяя общую процедуру B, нафталин-2-карбонитрил (1,00 г, 6,53 ммоль) преобразовывали в соответствующий 2-(нафталин-2-ил)-пропан-2-амин (294 мг, 25%), получаемый в виде прозрачного масла.
Применяя общую процедуру C, из хинуклидин-3-амина (102 мг, 0,808 ммоль), CDI (131 мг, 0,808 ммоль) и 2-(нафталин-2-ил)-пропан-2-амина (150 мг, 0,819 ммоль) получали 1-(2-(нафталин-2-ил)-пропан-2-ил)-3-(хинуклидин-3-ил)-мочевину (132 мг, 49%) в виде белого твердого вещества.1H ЯМР (400 МГц, CDCl3) δ 7,94-7,78 (м, 4H), 7,69 (дд, J=2,0, 8,7 Гц, 1H), 7,53-7,46 (м, 2H), 4,84 (с, 1H), 4,23 (д, J=8,0 Гц, 1H), 3,68-3,54 (м, 1H), 3,07 (ддд, J=2,3, 9,3, 14,1 Гц, 1H), 2,61-2,51 (м, 2H), 2,42-2,32 (м, 1H), 1,95-1,83 (м, 2H), 1,75 (с, 3H), 1,74 (с, 3H), 1,58-1,54 (м, 1H), 1,46-1,40 (м, 2H), 1,03-0,91 (м, 1H), 0,72-0,60 (м, 1H) м.д.13C ЯМР (100 МГц, CDCl3) δ 157,5, 143,7, 133,4, 132,8, 129,3, 128,1, 127,8, 126,9, 126,7, 124,4, 124,0, 57,0, 55,0, 47,1, 47,1, 46,6, 30,5, 30,3, 26,0, 25,9, 20,0 м.д. Чистота: >99,9% UPLCMS (210 нм); время удерживания 0,71 мин; (M+1) 338.
Пример 11
1-(2-метил-2-(м-толил)-пропил)-3-(3-метилхинуклидин-3-ил)-мочевина
Применяя общую процедуру D, из гидрохлорида 2-метил-2-(м-толил)-пропан-1-амина (100 мг, 0,501 ммоль), триэтиламина (279 мкл, 2,00 ммоль), трифосгена (47 мг, 0,18 ммоль) и 2,2,2-трифторацетата 3-метилхинуклидин-3-амина (140 мг, 0,550 ммоль) получали 1-(2-метил-2-(м-толил)-пропил)-3-(3-метилхинуклидин-3-ил)-мочевину (41 мг, 25%) в виде белого твердого вещества.1H ЯМР (400 МГц, CDCl3) δ 7,23 (т, J=7,7 Гц, 1H), 7,18-7,12 (м, 2H), 7,04 (д, J=7,5 Гц, 1H), 4,12 (с, 1H), 4,08 (т, J=6,0 Гц, 1H), 3,39-3,22 (м, 2H), 2,81-2,62 (м, 6H), 2,34 (с, 3H), 1,98-1,89 (м, 1H), 1,80-1,63 (м, 2H), 1,51-1,23 (м, J=26,9 Гц, 2H), 1,37 (с, 3H), 1,30 (с, 6H) м.д.13C ЯМР (100 МГц, CDCl3) δ 157,9, 147,1, 138,2, 128,6, 127,1, 127,1, 123,3, 64,0, 52,2, 52,1, 46,9, 46,7, 39,2, 31,2, 27,1, 26,8, 25,4, 23,5, 22,7, 21,9. Чистота: >99,9% UPLCMS (210 нм); время удерживания 0,79 мин; (M+1) 330.
Пример 12
1-(2-(3-метоксифенил)-пропан-2-ил)-3-(хинуклидин-3-ил)- мочевина
Применяя общую процедуру C, из хинуклидин-3-амина (380 мг, 3,01 ммоль), CDI (489 мг, 3,01 ммоль) и 2-(3-метоксифенил)-пропан-2-амина (506 мг, 3,07 ммоль) получали 1-(2-(3-метоксифенил)-пропан-2-ил)-3-(хинуклидин-3-ил)-мочевину (560 мг, 59%) в виде белого твердого вещества.1H ЯМР (400 МГц, CDCl3) δ 7,35-7,29 (м, 1H), 7,14-7,07 (м, 2H), 6,87-6,81 (ддд, 1H), 4,76 (с, 1H), 4,19 (д, 1H), 3,81 (с, 3H), 3,70-3,62 (м, 1H), 3,19-3,10 (м, 1H), 2,74-2,59 (м, 3H), 2,37-2,26 (м, 1H), 2,07-1,98 (дд, 1H), 1,80 (шир.с, 1H), 1,69-1,63 (м, 1H), 1,63 (с, 3H), 1,62 (с, 3H), 1,58-1,44 (м, 2H), 1,28-1,14 (м, 1H), 1,02-0,90 (м, 1H) м.д.13C ЯМР (100 МГц, CDCl3) δ 160,0, 157,5, 148,4, 129,9, 117,7, 112,0, 111,9, 56,7, 55,3, 54,6, 47,2, 46,8, 46,4, 30,1, 25,8, 20,0 м.д. Чистота: >99,4% UPLCMS (210 нм); время удерживания 1,73 мин; (M+1) 318.
Пример 13
Хинуклидин-3-ил-2-(3-метоксифенил)-пропан-2-илкарбамат
Применяя общую процедуру A, из 1-(3-метоксифенил)-пропан-2-амина (327 мг, 1,98 ммоль) и хинуклидин-3-ола получали хинуклидин-3-ил-2-(3-метоксифенил)-пропан-2-илкарбамат (370 мг, 59%) в виде белого твердого вещества.1H ЯМР (400 МГц, CDCl3) δ 7,30-7,20 (м, 1H), 7,03-6,97 (м, 1H), 6,97-6,93 (м, 1H), 6,80-6,74 (дд, 1H), 5,18-5,00 (шир.с, 1H), 4,67-4,57 (м, 1H), 3,80 (с, 3H), 3,30-2,12 (шир.м, 7H), 2,02-1,00 (м, 10H) м.д.13C ЯМР (100 МГц, CDCl3) δ 159,7, 154,5, 149,0, 129,3, 117,2, 111,4, 111,0, 70,9, 55,7, 55,1, 47,4, 46,5, 29,4, 25,4, 24,6, 19,6 м.д. Чистота: >99,9% UPLCMS (210 нм); время удерживания 1,85 мин; (M+1) 319.
Пример 14
Хинуклидин-3-ил-2-(3-метоксифенил)-пропан-2-илкарбамат
Применяя общую процедуру A, из гидрохлорида 1-(4-метоксифенил)-пропан-2-амина (316 мг, 1,57 ммоль) и хинуклидин-3-ола получали хинуклидин-3-ил-2-(3-метоксифенил)-пропан-2-илкарбамат (370 мг, 59%) в виде белого твердого вещества.1H ЯМР (400 МГц, CDCl3) δ 7,33 (д, 2H), 6,86 (д, 2H), 5,15-5,01 (шир.с, 1H), 4,66-4,57 (м, 1H), 3,79 (с, 3H), 3,33-2,12 (м, 7H), 2,10-0,96 (м, 10H) м.д.13C ЯМР (100 МГц, CDCl3) δ 158,1, 154,5, 139,2, 125,8, 113,5, 70,7, 55,7, 55,2, 54,6, 47,2, 46,3, 31,2, 29,4, 25,3, 24,5, 19,4 м.д. Чистота: >94,1% UPLCMS (210 нм); время удерживания 1,81 мин; (M+1) 319.
Пример 15
Хинуклидин-3-ил-2-(4-трет-бутилфенил)-пропан-2-илкарбамат
Применяя общую процедуру A, из 1-(4-трет-бутилфенил)-пропан-2-амина (348 мг, 1,82 ммоль) и хинуклидин-3-ола получали хинуклидин-3-ил-2-(4-трет-бутилфенил)-пропан-2-илкарбамат (427 мг, 68%) в виде белого твердого вещества.1H ЯМР (400 МГц, CDCl3) δ 7,34 (с, 4H), 5,09 (шир.с, 1H), 4,69-4,52 (м, 1H), 3,47-2,05 (м, 7H), 3,33-2,12 (м, 7H), 2,00-0,80 (м, 20H) м.д.13C ЯМР (100 МГц, CDCl3) δ 156,2, 149,4, 144,5, 125,3, 124,5, 70,9, 55,8, 55,1, 47,5, 46,6, 34,4, 31,4, 29,8, 29,3, 25,5, 24,6, 19,6 м.д. Чистота: >98,2% UPLCMS (210 нм); время удерживания 2,29 мин; (M+1) 345.
Пример 16
Хинуклидин-3-ил-2-(4-изопропилфенил)-пропан-2-илкарбамат
Применяя общую процедуру A, из 1-(4-изопропилфенил)-пропан-2-амина (158 мг, 0,891 ммоль) и хинуклидин-3-ола получали хинуклидин-3-ил-2-(4-изопропилфенил)-пропан-2-илкарбамат (205 мг, 70%) в виде белого твердого вещества.1H ЯМР (400 МГц, CDCl3) δ 7,33 (д, J=7,3 Гц, 2H), 7,19 (с, 1H), 7,17 (с, 1H), 5,09 (с, 1H) 4,69-4,51 (шир.с, 1H) 3,30-1,30 (м, 17H), 1,24 (с, 3H), 1,22 (с, 3H), 1,06-0,77 (м, 1H) м.д.13C ЯМР (100 МГц, CDCl3) δ 156,2, 147,1, 144,4, 126,4, 124,7, 70,9, 55,7, 55,0, 47,4, 46,5, 33,6, 29,8, 29,4, 25,4, 24,6, 24,0, 19,5 м.д. Чистота: >98,3% UPLCMS (210 нм); время удерживания 2,19 мин; (M+1) 331.
Пример 17
Хинуклидин-3-ил-2-(4-этилфенил)-пропан-2-илкарбамат
Применяя общую процедуру A, из 1-(4-этилфенил)-пропан-2-амина (230 мг, 1,41 ммоль) и хинуклидин-3-ола получали хинуклидин-3-ил-2-(4-этилфенил)-пропан-2-илкарбамат (248 мг, 56%) в виде белого твердого вещества.1H ЯМР (400 МГц, CDCl3) δ 7,33 (д, J=7,3 Гц, 2H), 7,19 (с, 1H), 7,17 (с, 1H), 5,09 (с, 1H) 4,69-4,51 (шир.с, 1H) 3,34-0,73 (м, 22H) м.д.13C ЯМР (100 МГц, CDCl3) δ 154,5, 144,3, 142,4, 127,8, 124,7, 71,0, 55,6, 55,1, 47,4, 46,5, 29,6, 28,3, 25,4, 24,6, 19,5, 15,8, 15,4 м.д. Чистота: >99,5% UPLCMS (210 нм); время удерживания 2,07 мин; (M+1) 317.
Пример 18
Хинуклидин-3-ил-2-o-толилпропан-2-илкарбамат
Применяя общую процедуру A, из 2-o-толилпропан-2-амина (230 мг, 1,52 ммоль) и хинуклидин-3-ола получали хинуклидин-3-ил-2-o-толилпропан-2-илкарбамат (200 мг, 44%) в виде белого твердого вещества.1H ЯМР (400 МГц, CDCl3) (ротамеры) δ 7,33 (шир.с, 1H), 7,15-7,10 (м, 3H), 5,35-5,20 (м, 1H), 4,60 (шир.с, 1H), 3,20-2,60 (м, 5H), 2,5 (с, 3H), 2,15 (шир.с, 1H), 1,80-1,30 (м, 10H).13C ЯМР (100 МГц, CDCl3) (ротамеры) δ 154,2, 144,5, 140,2, 133,0, 127,1, 126,2, 126,1, 72,2, 71, 56,0, 46,6, 46,7, 31,0, 29,0, 26,0, 24,7, 22,3, 19,7. Чистота: >95 % UPLCMS (210 нм); (M+1) 303.
Пример 19
хинуклидин-3-ил-2-(2-метоксифенил)-пропан-2-илкарбамат
Применяя общую процедуру A, из 2-(2-метоксифенил)-пропан-2-амина (150 мг, 0,908 ммоль) и хинуклидин-3-ола получали хинуклидин-3-ил-2-(2-метоксифенил)-пропан-2-илкарбамат (60 мг, 21%) в виде белого твердого вещества.1H ЯМР (400 МГц, CDCl3) (ротамеры) δ 7,3 (м, 1H), 7,2 (м, 1H), 6,9 (м, 2H), 5,4 (шир.с, 1H), 4,6 (м, 1H), 3,8 (с, 1H), 3,1 (м, 1H), 2,4-2,8 (м, 5H), 1,9 (с, 1H), 1,3-1,7 (м, 10H).13C ЯМР (100 МГц, CDCl3) (ротамеры) δ 157, 155, 140, 134, 129, 127, 121, 111, 70, 56, 55, 48, 47, 29, 26, 25, 20. Чистота: >99 % UPLCMS (210 нм); (M+1) 319.
Препаративная процедура L
Пример 20
1-(3-Цианохинуклидин-3-ил)-3-(2-(3-(проп-1-ен-2-ил)-фенил)-пропан-2-ил)-мочевина
3-Амино-3-цианохинуклидин получали, как описано в литературе (Fernandez, M. A.; Gonzalez, G.; Martinez, M.; Galvez, E. Anales de la Real Academia de Farmacia 1988, 54, 502).
К раствору 3-амино-3-цианохинуклидина(100 мг, 0,661 ммоль) в CH2Cl2 (5 мл) добавляли по каплям 3-изопренил-2,2-диметилбензилизоцианат (0,13 мл, 0,66 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов, концентрировали и подвергали флэш-хроматографии на силикагеле (19:1 CH2Cl2/7 M NH3(CH3OH)). Продукт, указанный в заголовке, был получен в виде белого твердого вещества (155 мг, 67%).1H ЯМР (400 МГц, CD3OD) δ 7,52 (с, 1H), 7,35-7,25 (м, 3H), 5,34 (с, 1H), 5,05 (с, 1H), 2,60-3,41 (м, 6H), 2,25- 2,32 (м, 1H), 2,13 (с, 3H), 1,42-2,10 (м, 4H), 1,64 (с, 6H) м.д.13C ЯМР (100 МГц, CD3OD) δ 157,0, 147,9, 144,0, 141,4, 128,1, 124,0, 123,4, 121,9, 121,7, 111,5, 61,0, 55,1, 50,4, 30,9, 23,3, 22,5, 21,0 19,0 м.д. Чистота: >99,9% UPLCMS (210 нм); время удерживания 0,82 мин; (M+1) 353.
Препаративная процедура M
Пример 21
1-[(3S)-1-азабицикло[2.2.2]окт-3-ил]-3-{2-[3-(проп-1-ен-2-ил)-фенил]-пропан-2-ил}-мочевина
К суспензии дигидрохлорида (S)-(-)-3-аминохинуклидина (120 мг, 0,603 ммоль) и триэтиламина (168 мкл, 1,21 ммоль) в THF (2 мл) при комнатной температуре добавляли 3-изопропенил-2,2-диметилбензилизоцианат (121 мг, 0,601 ммоль). Реакционную смесь перемешивали в течение 18 часов и затем промывали насыщенным водным раствором NaHCO3.Органическую фазу сушили над Na2SO4 и концентрировали. Неочищенный материал очищали посредством комбифлэш-хроматографии (SiO2-картридж, CHCl3 и 2 н NH3 в MeOH), получая соединение, указанное в заголовке, (29 мг, 47%) в виде светло-серого твердого вещества.1H ЯМР (400 МГц, CD3OD) δ 7,51 (дт, J=2,5, 1,2 Гц, 1H), 7,41-7,14 (м, 3H), 5,32 (дд, J=1,6, 0,8 Гц, 1H), 5,06 (с, 1H), 3,74-3,60 (м, 1H), 3,31-3,29 (м, 2H), 3,19 (ддд, J=13,7, 9,5, 1,6 Гц, 1H), 2,88-2,50 (м, 4H), 2,37 (ддд, J=14,0, 4,9, 2,2 Гц, 1H), 2,14 (ддд, J=2,3, 1,8, 1,0 Гц, 3H), 1,81-1,63 (м, 4H), 1,62 (д, J=6,2 Гц, 6H), 1,55-1,38 (м, 1H) м.д.13C ЯМР (100 МГц, CD3OD) δ 158,4, 148,4, 144,0, 141,3, 128,0, 124,1, 123,3, 121,9, 111,4, 55,7, 54,7, 46,7, 46,4, 46,0, 29,6, 29,4, 28,4, 26,1, 25,1, 21,0, 19,4 м.д. Чистота: >96% UPLCMS (210 нм); время удерживания 0,81 мин; (M+1) 329,5.
Препаративная процедура N
Пример 22
1-(1-Азабицикло[2.2.2]окт-3-ил)-3-{2-[3-(пропан-2-ил)-фенил]-пропан-2-ил}-мочевина
К раствору 3-аминохинуклидина (150 мг, 1,19 ммоль) в THF (5 мл) добавляли 3-изопропенил-2,2-диметилбензилизоцианат. Раствор перемешивали при комнатной температуре в течение 30 мин, затем концентрировали силикагеле и очищали посредством комбифлэш-хроматографии (SiO2-картридж, CHCl3 и 2 н NH3 в MeOH), получая светло-серое твердое вещество (299 мг, 77%).
Применяя общую процедуру F, из вышеуказанной изопренилмочевины (150 мг, 1,19 ммоль) и гидроксида палладия (30 мг, 20% масс. на угле) получали 1-(1-азабицикло[2.2.2]окт-3-ил)-3-{2-[3-(пропан-2-ил)-фенил]-пропан-2-ил}-мочевину (116 мг, 77%) в виде светло-серого твердого вещества.1H ЯМР (400 МГц, CDCl3) δ 7,38-7,28 (м, 3H), 7,16 (дт, J=6,9, 1,6 Гц, 1H), 4,93 (с, 1H), 4,26 (д, J=7,5 Гц, 1H), 3,70-3,58 (м, 1H), 3,11 (ддд, J=14,1, 9,4, 2,3 Гц, 1H), 2,90 (гепт, J=6,9 Гц, 1H), 2,71-2,52 (м, 4H), 2,31-2,19 (м, 1H), 1,98 (дд, J=14,2, 2,9 Гц, 1H), 1,61 (д, J=2,0 Гц, 6H), 1,52-1,43 (м, 2H), 1,23 (д, J=6,9 Гц, 6H), 1,19-1,09 (м, 1H), 0,92-0,79 (м, 1H) м.д.13C ЯМР (100 МГц, CDCl3) δ 157,6, 150,1, 146,1, 129,2, 125,8, 124,1, 123,3, 57,1, 54,9, 47,4, 47,0, 46,6, 34,5, 30,7, 30,5, 26,1, 26,0, 24,3, 24,2, 20,3 м.д. Чистота: 94% UPLCMS (210 нм); время удерживания 0,87 мин; (M+1) 329,3.
Пример 23
1-(1-Азабицикло[2.2.2]окт-3-ил)-3-[1-(нафталин-1-ил)этил]-мочевина
Дигидрохлорид 3-аминохинуклидина (150 мг, 0,753 ммоль) перемешивали с THF (3 мл) и триэтиламином (152 мг, 1,50 ммоль), после чего добавляли 1-(1-нафтил)-этилизоцианат (149 мг, 0,752 ммоль). Смесь перемешивали 48 часов при комнатной температуре. Реакционный раствор концентрировали и очищали посредством комбифлэш-хроматографии (SiO2-картридж, CHCl3 и 2 н NH3 в MeOH), получая соединение, указанное в заголовке, в виде светло-серого твердого вещества (46 мг, 19%).1H ЯМР (400 МГц, CDCl3) δ 8,20-8,05 (м, 1H), 7,85 (дд, J=7,9, 1,5 Гц, 1H), 7,75 (д, J=8,1 Гц, 1H), 7,59-7,34 (м, 4H), 5,55 (гепт, 1H), 5,35-5,19 (м, 1H), 4,84 (дд, 1H), 3,70-3,53 (м, 1H), 3,09 (ддд, 1H), 2,74-2,28 (м, 4H), 2,17 (ддд, J=1,8, 4,5, 14,1 Гц, 1H), 1,75-1,62 (м, 1H), 1,55 (дд, J=1,8, 6,8 Гц, 3H), 1,52-1,06 (м, 4H) м.д.13C ЯМР (100 МГц, CDCl3) δ 157,7, 140,0, 134,1, 130,9, 129,2, 128,3, 126,8, 126,7, 126,7, 126,0, 125,6, 123,2, 123,1, 122,8, 122,7, 56,9, 56,7, 47,4, 47,3, 46,7, 46,4, 26,1, 25,9, 22,7, 22,6, 20,1, 20,0 м.д. Чистота: 97% UPLCMS (210 нм); время удерживания 0,68 мин; (M+1) 324,2
Пример 24
1-(1-Азабицикло[2.2.2]окт-3-ил)-3-[2-(3-бромфенил)-пропан-2-ил]-мочевина
Применяя общую процедуру C, из хинуклидин-3-амина (100 мг, 0,792 ммоль), CDI (128 мг, 0,789 ммоль) и 2-(3-бромфенил)-пропан-2-амина (170 мг, 0,791 ммоль) получали соединение, указанное в заголовке, в виде белого твердого вещества (166 мг, 75%).1H ЯМР (400 МГц, CDCl3) δ 7,50 (с, 1H), 7,30 (т, J=7,2 Гц, 2H), 7,15 (т, J=7,9 Гц, 1H), 5,54 (д, J=22,7 Гц, 1H), 5,16 (д, J=29,7 Гц, 1H), 3,60 (с, 1H), 3,14 (ддд, J=13,3, 9,4, 1,6 Гц, 1H), 2,61 (д, J=52,6 Гц, 4H), 2,18 (дд, J=14,1, 2,8 Гц, 1H), 1,66 (д, J=3,0 Гц, 2H), 1,51 (д, J=7,6 Гц, 6H), 1,28 (с, 3H) м.д.13C ЯМР (100 МГц, CDCl3) δ 157,1, 150,4, 130,3, 130,0, 128,6, 124,0, 123,0, 57,0, 54,6, 47,6, 47,2, 46,8, 30,5, 30,3, 26,3, 26,2, 20,2 м.д. Чистота: 100% UPLCMS (210 нм); время удерживания 0,66 мин; (M+1) 367,8.
Пример 25
1-(1-Азабицикло[2.2.2]окт-3-ил)-3-[2-(бифенил-3-ил)-пропан-2-ил]-мочевина
Применяя общую процедуру E, из 1-(1-азабицикло[2.2.2]окт-3-ил)-3-[2-(3-бромфенил)-пропан-2-ил]-мочевины (111 мг, 0,301 ммоль), фенилбороновой кислоты (78,8 мг, 0,606 ммоль) и тетракис-(трифенилфосфин)-палладия(0) получали соединение, указанное в заголовке, в виде светло-серого твердого вещества (21 мг, 11%).1H ЯМР (400 МГц, CDCl3) δ 7,74 (с, 1H), 7,52-7,40 (м, 8H), 4,89 (с, 1H), 4,28 (д, J=7,3 Гц, 1H), 3,75-3,59 (м, 1H), 3,15 (ддд, J=1,9, 9,3, 13,9 Гц, 1H), 2,46 (м, 4H), 2,05 (дд, J=3,5, 14,0 Гц, 1H), 1,68 (д, J=4,7 Гц, 6H), 1,66-0,76 (м, 5H) м.д.13C ЯМР (100 МГц, CDCl3) δ 157,4, 146,9, 142,5, 141,1, 129,7, 129,1, 127,8, 127,4, 126,7, 124,8, 124,7, 57,1, 55,1, 30,7, 30,1, 26,1, 26,0, 20,2 м.д. Чистота: 100% UPLCMS (210 нм); время удерживания 0,78 мин; (M+1) 364,0.
Пример 26
1-Азабицикло[2.2.2]окт-3-ил-{2-[3-(пропан-2-ил)-фенил]-пропан-2-ил}-карбамат
Применяя общую процедуру F, из 1-азабицикло[2.2.2]окт-3-ил-{2-[3-(проп-1-ен-2-ил)-фенил]-пропан-2-ил}-карбамата (48,8 мг, 0,146 ммоль) и гидроксида палладия (30 мг, 20% масс. на угле) получали соединение, указанное в заголовке, в виде светло-серого твердого вещества (16 мг, 33%).1H ЯМР (400 МГц, CDCl3) δ 7,24 (д, J=5,1Hz, 3H), 7,10 (д, 1H), 5,12 (с, 1H), 4,63 (с, 1H), 3,54-2,96 (м, 1H), 2,89 (с, 1H), 2,68 (с, 5H), 2,17-1,75 (м, 2H), 1,67 (с, 6H), 1,62-1,30 (м, 2H), 1,24 (д, J=6,9 Гц, 6H), 1,15-0,85 (м, 1H) м.д.13C ЯМР (100 МГц, CDCl3) δ 149,1, 128,5, 124,9, 123,1, 122,5, 55,8, 55,6, 46,6, 34,5, 25,6, 24,6, 24,3, 19,7 м.д. Чистота: 94% UPLCMS (210 нм); время удерживания 0,89 мин; (M+1) 331,1.
Пример 27
1-Азабицикло[2.2.2]окт-3-ил-[2-(3-бромфенил)-пропан-2-ил]-карбамат
Применяя общую процедуру A, из гидрохлорида 2-(3-бромфенил)-пропан-2-амина (2,00 г, 7,89 ммоль) и хинуклидин-3-ола получали соединение, указанное в заголовке, в виде белого твердого вещества (2,23 г, 76%).1H ЯМР (400 МГц, CDCl3) δ 7,54 (с, 1H), 7,41-7,30 (м, 2H), 7,19 (т, J=7,9 Гц, 1H), 5,11 (с, 1H), 4,68-4,54 (м, 1H), 3,51-2,11 (м, 6H), 2,04-1,68 (м, 2H), 1,63 (д, J=10,2 Гц, 6H), 1,51-0,67 (м, 3H) м.д.13C ЯМР (100 МГц, CDCl3) δ 156,0, 154,7, 150,6, 149,7, 130,2, 130,0, 128,4, 123,7, 72,5, 71,6, 71,5, 55,8, 55,1, 47,6, 46,7, 31,2, 29,9, 29,8, 29,5, 25,6, 24,8, 19,7 м.д. Чистота: 100% UPLCMS (210 нм); время удерживания 0,69 мин; (M+1) 368,8.
Пример 28
1-Азабицикло[2.2.2]окт-3-ил-[2-(3-циклопропилфенил)-пропан-2-ил]-карбамат
Применяя общую процедуру E, из 11-азабицикло[2.2.2]окт-3-ил-[2-(3-бромфенил)-пропан-2-ил]-карбамата (44,3 мг, 0,121 ммоль), циклопропилбороновой кислоты (14 мг, 0,16 ммоль) и ацетата палладия (II) получали соединение, указанное в заголовке, в виде светло-серого твердого вещества (21 мг, 11%).1H ЯМР (400 МГц, CDCl3) δ 7,54 (с, 1H), 7,41-7,30 (м, 2H), 7,19 (т, J=7,9 Гц, 1H), 5,11 (с, 1H), 4,68-4,54 (м, 1H), 3,51-2,11 (м, 6H), 2,04-1,68 (м, 2H), 1,63 (д, J=10,2 Гц, 6H), 1,36 (д, J=9,5 Гц, 3H) м.д.13C ЯМР (100 МГц, CDCl3) δ 147,2, 144,2, 128,6, 128,4, 125,0, 123,7, 122,8, 122,1, 110,0, 72,2, 71,4, 55,9, 55,4, 47,7, 47,3, 46,7, 33,1, 31,6, 30,0, 29,6, 25,6, 24,8, 19,8, 19,3, 15,8, 9,5 м.д. Чистота: 91% UPLCMS (210 нм); время удерживания 0,75 мин; (M+1) 329,0.
Пример 29
1-Азабицикло[2.2.2]окт-3-ил-[2-(бифенил-3-ил)-пропан-2-ил]-карбамат
Применяя общую процедуру E, из 1-азабицикло[2.2.2]окт-3-ил-[2-(3-бромфенил)-пропан-2-ил]-карбамата (600 мг, 1,63 ммоль), фенилбороновой кислоты (398 мг, 3,27 ммоль) и ацетата палладия (II) получали соединение, указанное в заголовке, в виде белого твердого вещества (379 мг, 64%).1H ЯМР (400 МГц, CDCl3) δ 7,61 (с, 1H), 7,56 (д, J=7,4 Гц, 2H), 7,50-7,38 (м, 4H), 7,34 (м, 2H), 5,16 (с, 1H), 4,63 (с, 1H), 3,39-2,09 (м, 6H), 1,72 (с, 6H), 2,02-0,73 (м, 5H) м.д.13C ЯМР (100 МГц, CDCl3) δ 154,8, 147,8, 141,6, 129,0, 129,0, 128,6, 127,5, 125,8, 125,0, 124,0, 71,6, 71,3, 55,9, 55,5, 47,6, 46,8, 31,5, 30,2, 30,0, 29,5, 25,6, 24,8, 19,8 м.д. Чистота: 99% UPLCMS (210 нм); время удерживания 0,84 мин; (M+1) 365,0. Анал. расчет для C23H28N2O2·0,29(CHCl3): C, 70,02; H, 7,14; N, 7,01. Найдено: C, 70,02; H, 7,37; N, 6,84.
Пример 30
1-Азабицикло[2.2.2]окт-3-ил-{2-[3-(2-метилпропил)-фенил]-пропан-2-ил}-карбамат
Применяя общую процедуру E, из 1-азабицикло[2.2.2]окт-3-ил-[2-(3-бромфенил)-пропан-2-ил]-карбамата (75 мг, 0,20 ммоль), 2-метилпропилбороновой кислоты (28,1 мг, 0,276 ммоль) и ацетата палладия (II) получали соединение, указанное в заголовке, в виде белого твердого вещества (50 мг, 71%).1H ЯМР (400 МГц, CDCl3) δ 7,21 (д, J=4,9 Гц, 2H), 7,16 (с, 1H), 7,00 (с, 1H), 5,17 (с, 1H), 4,60 (с, 1H), 3,35-2,10 (м, 6H), 2,45 (д, J=7,1 Гц, 2H), 1,82 (дт, J=6,8, 13,5 Гц, 1H), 2,03-0,94 (м, 5H), 1,65 (с, 6H), 0,89 (д, J=6,6 Гц, 6H) м.д.13C ЯМР (100 МГц, CDCl3) δ 172,6, 172,1, 170,8, 170,2, 160,1, 160,0, 157,8, 157,7, 140,4, 139,8, 130,5, 130,4, 130,0, 129,8, 129,5, 129,3, 127,9, 127,7, 120,8, 120,7, 120,3, 113,9, 113,6, 113,2, 113,0, 110,5, 110,4, 66,6, 66,5, 56,8, 56,3, 55,4, 55,4, 54,0, 53,7, 51,1, 46,6, 43,8, 43,7, 42,0, 38,4, 37,8, 37,7, 33,8, 33,2, 27,4, 27,0, 25,7, 25,5, 20,9, 20,9 м.д. Чистота: 90% UPLCMS (210 нм); время удерживания 0,89 мин; (M+1) 345.
Пример 31
1-Азабицикло[2.2.2]окт-3-ил-[2-(5-бром-2-фторфенил)-пропан-2-ил]-карбамат
Применяя общую процедуру A, из гидрохлорида 2-(5-бром-2-фторфенил)-пропан-2-амина (100 мг, 0,372 ммоль) и хинуклидин-3-ола получали соединение, указанное в заголовке, в виде белого твердого вещества (90,3 мг, 98%).1H ЯМР (400 МГц, CDCl3) δ 7,45 (дд, J=2,3, 7,3 Гц, 1H), 7,31 (ддд, J=2,5, 4,2, 8,6 Гц, 1H), 6,88 (дд, J=8,6, 11,9 Гц, 1H), 5,38 (с, 1H), 4,82-4,33 (м, 1H), 3,28-2,28 (м, 6H), 1,68 (д, J=9,0 Гц, 6H), 1,98-1,27 (м, 5H) м.д.13C ЯМР (100 МГц, CDCl3) δ 161,1, 158,6, 131,7, 131,6, 131,0, 131,0, 118,6, 118,3, 116,8, 55,8, 54,0, 47,6, 46,7, 28,5, 25,6, 24,8, 19,7 м.д.Чистота: 100% UPLCMS (210 нм); время удерживания 0,81 мин; (M+1) 386,7.Анал. расчет для C17H22BrFN2O2·0,37(CHCl3): C, 52,20; H, 5,66; N, 7,14. Найдено: C, 52,21; H, 5,57; N, 7,13.
Пример 32
1-Азабицикло[2.2.2]окт-3-ил-[2-(4'-фторбифенил-3-ил)-пропан-2-ил]-карбамат
Применяя общую процедуру E, из 1-азабицикло[2.2.2]окт-3-ил-[2-(3-бромфенил)-пропан-2-ил]-карбамата (600 мг, 1,63 ммоль), 4-фторфенилбороновой кислоты (457 мг, 3,27 ммоль) и ацетата палладия (II) получали соединение, указанное в заголовке, в виде белого твердого вещества (373 мг, 60%).1H ЯМР (400 МГц, CDCl3) δ 7,56 (с, 1H), 7,52 (дд, J=5,4, 8,4 Гц, 2H), 7,42-7,38 (м, 3H), 7,12 (м, 2H), 5,18 (с, 1H), 4,62 (с, 1H), 2,66 (м, 6H), 1,72 (с, 6H), 2,01-0,83 (м, 5H) м.д.13C ЯМР (100 МГц, CDCl3) δ 125,0, 124,0, 123,8, 116,0, 116,0, 71,3, 55,9, 55,5, 47,6, 46,7, 29,6, 25,6, 24,8, 19,8 м.д. Чистота: 98,0% UPLCMS (210 нм); время удерживания 0,95 мин; (M+1) 382,9. Анал. расчет для C23H27FN2O2·0,37(CHCl3): C, 65,86; H, 6,47; N, 6,57. Найдено: C, 65,85; H, 6,69; N, 6,49.
Пример 33
1-Азабицикло[2.2.2]окт-3-ил-[2-(4-фторбифенил-3-ил)-пропан-2-ил]-карбамат
Применяя общую процедуру E, из 1-азабицикло[2.2.2]окт-3-ил-[2-(5-бром-2-фторфенил)-пропан-2-ил]-карбамата (990 мг, 2,57 ммоль), фенилбороновой кислоты (209 мг, 1,71 ммоль) и ацетата палладия (II) получали соединение, указанное в заголовке, в виде белого твердого вещества (257 мг, 26%).1H ЯМР (400 МГц, CDCl3) δ 7,58-7,49 (м, 3H), 7,44-7,38 (м, 3H), 7,35-7,29 (м, 1H), 7,08 (дд, J=8,4, 12,1 Гц, 1H), 5,30 (с, 1H), 4,75-4,42 (м, 1H), 2,89 (д, J=10,2 Гц, 6H), 1,81-1,66 (м, 6H), 2,04-1,18 (м, 5H) м.д.13C ЯМР (100 МГц, CDCl3) δ 161,7, 159,3, 140,7, 137,3, 137,3, 131,7, 131,7, 131,0, 129,0, 127,5, 127,3, 126,7, 117,1, 116,9, 71,4, 55,8, 54,3, 47,6, 46,7, 28,6, 25,6, 24,8, 19,8 м.д.Чистота: 92,0% UPLCMS (210 нм); время удерживания 0,95 мин; (M+1) 382,9.Анал. расчет для C23H27FN2O2·0,4(CHCl3): C, 65,39; H, 6,43; N, 6,52. Найдено: C, 65,39; H, 6,51; N, 6,42.
Пример 34
1-Азабицикло[2.2.2]окт-3-ил-{2-[2-фтор-5-(2-метилпропил)-фенил]-пропан-2-ил}-карбамат
Применяя общую процедуру E, из 1-азабицикло[2.2.2]окт-3-ил-[2-(5-бром-2-фторфенил)-пропан-2-ил]-карбамата (120 мг, 0,312 ммоль), 2-метилпропилбороновой кислоты (79,4 мг, 0,779 ммоль) и ацетата палладия (II) получали соединение, указанное в заголовке, в виде белого твердого вещества (37 мг, 33%).1H ЯМР (400 МГц, CDCl3) δ 7,08 (дд, J=2,0, 8,2 Гц, 1H), 6,95 (д, J=4,9 Гц, 1H), 6,93-6,85 (м, 1H), 5,23 (с, 1H), 4,72-4,52 (м, 1H), 3,20-2,47 (м, 6H), 2,41 (д, J=7,1 Гц, 2H), 1,89-1,76 (м, 1H), 2,02-1,26 (м, 5H), 1,70 (д, J=7,6 Гц, 6H), 0,88 (д, J=6,6 Гц, 6H) м.д.13C ЯМР (100 МГц, CDCl3) δ 160,4, 158,0, 137,1, 137,1, 129,2, 129,1, 128,1, 116,2, 116,0, 71,2, 55,8, 54,2, 47,6, 46,7, 45,1, 30,5, 29,9, 28,6, 27,0, 25,6, 24,8, 22,5, 19,8, 19,5 м.д. Чистота: 95,0% UPLCMS (210 нм); время удерживания 1,02 мин; (M+1) 363.
Пример 35
1-Азабицикло[2.2.2]окт-3-ил-[2-(5-циклопропил-2-фторфенил)-пропан-2-ил]-карбамат
Применяя общую процедуру E, из 1-азабицикло[2.2.2]окт-3-ил-[2-(5-бром-2-фторфенил)-пропан-2-ил]-карбамата (750 мг, 0,649 ммоль), циклопропилбороновой кислоты (139 мг, 1,62 ммоль) и ацетата палладия (II) получали соединение, указанное в заголовке, в виде белого твердого вещества (727 мг, 86%).1H ЯМР (400 МГц, CDCl3) δ 7,08 (д, J=6,4 Гц, 1H), 6,97-6,78 (м, 2H), 5,19 (с, 1H), 4,65-4,57 (м, 1H), 2,66 (с, 6H), 1,85 (тт, J=5,1, 8,4 Гц, 1H), 2,00-1,17 (м, 5H), 1,71 (д, J=8,7 Гц, 6H), 0,92 (ддд, J=4,6, 6,3, 8,4 Гц, 2H), 0,62 (дт, J=4,7, 6,4 Гц, 2H) м.д.13C ЯМР (100 МГц, CDCl3) δ 160,2, 157,8, 139,2, 139,2, 125,6, 125,5, 125,4, 116,5, 116,3, 71,3, 55,8, 54,2, 47,6, 46,7, 29,9, 29,6, 28,6, 25,6, 24,8, 19,6, 15,2, 9,1 м.д. Чистота: 100% UPLCMS (210 нм); время удерживания 0,87 мин; (M+1) 347,2. Анал. расчет для C20H27FN2O2·0,07(CHCl3): C, 68,00; H, 7,70; N, 7,90. Найдено: C, 67,99; H, 7,86; N, 7,81.
Пример 36
1-Азабицикло[2.2.2]окт-3-ил-[2-(3-бром-4-фторфенил)-пропан-2-ил]-карбамат
Применяя общую процедуру A, из гидрохлорида 2-(3-бром-4-фторфенил)-пропан-2-амина (1,00 г, 3,72 ммоль) и хинуклидин-3-ола получали соединение, указанное в заголовке, в виде белого твердого вещества (434 мг, 30%).1H ЯМР (400 МГц, CDCl3) δ 7,57 (с, 1H), 7,38-7,25 (м, 1H), 7,06 (т, J=8,5 Гц, 1H), 5,62 (с, 1H), 4,86-4,32 (м, 1H), 3,33-2,12 (м, 6H), 1,73 (т, J=7,2 Гц, 5H), 1,61 (д, J=9,6 Гц, 6H) м.д.13C ЯМР (100 МГц, CDCl3) δ 159,1, 156,7, 154,6, 130,4, 125,8, 125,7, 116,4, 116,2, 109,1, 108,9, 71,3, 55,7, 54,7, 47,4, 46,5, 29,9, 29,6, 25,5, 24,6, 22,9, 19,6 м.д. Чистота: 100% UPLCMS (210 нм); время удерживания 0,79 мин; (M+1) 387,8. Анал. расчет для C17H22BrFN2O2·0,27(CHCl3): C, 49,68; H, 5,38; N, 6,71. Найдено: C, 49,67; H, 5,39; N, 6,74.
Пример 37
1-Азабицикло[2.2.2]окт-3-ил-[2-(6-фторбифенил-3-ил)-пропан-2-ил]-карбамат
Применяя общую процедуру E, из 1-азабицикло[2.2.2]окт-3-ил-[2-(3-бром-4-фторфенил)-пропан-2-ил]-карбамат (750 мг, 1,95 ммоль), фенилбороновой кислоты (418 мг, 4,87 ммоль) и ацетата палладия (II) получали соединение, указанное в заголовке, в виде белого твердого вещества (195 мг, 29%).1H ЯМР (400 МГц, CDCl3) δ 7,49 (с, 2H), 7,46-7,38 (м, 3H), 7,35 (дд, J=4,3, 11,7 Гц, 2H), 7,08 (дд, J=8,6, 10,1 Гц, 1H), 5,10 (с, 1H), 4,60 (с, 1H), 3,33-2,10 (м, 6H), 1,67 (д, J=7,9 Гц, 6H), 1,67 (м, 5H) м.д.13C ЯМР (100 МГц, CDCl3) δ 159,9, 157,4, 136,2, 129,3, 129,0, 128,7, 127,9, 127,6, 125,7, 125,6, 71,0, 66,1, 55,7, 55,1, 47,5, 46,6, 29,9, 29,6, 25,5, 24,5, 19,5, 15,5 м.д. Чистота: 98% UPLCMS (210 нм); время удерживания 0,95 мин; (M+1) 382,9. Анал. расчет для C23H27FN2O2·0,29(CHCl3): C, 67,08; H, 6,60; N, 6,72. Найдено: C, 67,09; H, 6,95; N, 6,37.
Пример 38
1-Азабицикло[2.2.2]окт-3-ил-{2-[4-фтор-3-(2-метилпропил)-фенил]-пропан-2-ил}-карбамат
Применяя общую процедуру E, из 1-азабицикло[2.2.2]окт-3-ил-[2-(3-бром-4-фторфенил)-пропан-2-ил]-карбамата (125 мг, 0,324 ммоль), 2-метилпропилбороновой кислоты (66 мг, 0,65 ммоль) и ацетата палладия (II) получали соединение, указанное в заголовке, в виде белого твердого вещества (27 мг, 23%).1H ЯМР (400 МГц, CDCl3) δ 7,23-7,11 (м, 2H), 7,04-6,82 (м, 1H), 5,11 (с, 1H), 4,59 (с, 1H), 3,32-2,12 (м, 6H), 2,48 (д, J=7,2 Гц, 2H), 1,86 (д, J=6,7, Гц, 1H), 2,05-0,96 (м, 5H), 1,62 (д, J=5,8 Гц, 6H), 0,90 (д, J=6,6 Гц, 6H) м.д.13C ЯМР (100 МГц, CDCl3) δ 161,4, 159,0, 154,7, 142,5, 128,3, 124,0, 115,1, 114,9, 71,2, 66,1, 55,8, 55,0, 47,6, 46,7, 38,7, 29,9, 29,6, 25,6, 24,8, 22,6, 19,7 м.д. Чистота: 85% UPLCMS (210 нм); время удерживания 1,0 мин; (M+1) 362,9.
Пример 39
1-Азабицикло[2.2.2]окт-3-ил-[2-(4',6-дифторбифенил-3-ил)-пропан-2-ил]-карбамат
Применяя общую процедуру E, из 1-азабицикло[2.2.2]окт-3-ил-[2-(3-бром-4-фторфенил)-пропан-2-ил]-карбамата (125 мг, 0,324 ммоль), 4-фторбороновой кислоты (64 мг, 0,46 ммоль) и ацетата палладия (II) получали соединение, указанное в заголовке, в виде белого твердого вещества (76 мг, 56%).1H ЯМР (400 МГц, CDCl3) δ 7,48 (т, 2H), 7,44-7,29 (м, 2H), 7,21-7,00 (м, 3H), 5,27 (с, 1H), 4,68-4,55 (м, 1H), 3,29-2,10 (м, 6H), 1,67 (д, J=9,4 Гц, 6H), 2,01-0,69 (м, 5H) м.д.13C ЯМР (100 МГц, CDCl3) δ 163,9, 161,4, 159,8, 157,3, 154,8, 143,5, 132,2, 130,9, 127,9, 127,8, 127,4, 125,9, 125,8, 116,2, 116,0, 115,7, 115,5, 71,4, 66,1, 55,9, 47,6, 46,7, 30,0, 39,7, 25,6, 24,8, 19,8 м.д. Чистота: 98% UPLCMS (210 нм); время удерживания 0,96 мин; (M+1) 400,9.
Пример 40
1-Азабицикло[2.2.2]окт-3-ил-{2-[4-фтор-3-(пиримидин-5-ил)-фенил]-пропан-2-ил}-карбамат
Применяя общую процедуру E, из 1-азабицикло[2.2.2]окт-3-ил-[2-(3-бром-4-фторфенил)-пропан-2-ил]-карбамата (150 мг, 0,389 ммоль), пиримидин-5-бороновой кислоты (75,9 мг, 0,613 ммоль) и трис-(дибензилиденацетон)-дипалладия(0) получали соединение, указанное в заголовке, в виде белого твердого вещества (49 мг, 31%).1H ЯМР (400 МГц, CDCl3) δ 9,22 (с, 1H), 8,92 (с, 2H), 7,55-7,41 (м, 2H), 7,19 (дд, J=8,7, 9,9 Гц, 1H), 5,37 (с, 1H), 4,72-4,49 (м, 1H), 3,34-2,04 (м, 6H), 2,04-0,98 (м, 5H), 1,66 (т, J=10,9 Гц, 6H) м.д.13C ЯМР (100 МГц, CDCl3) δ 159,9, 157,9, 157,4, 156,6, 154,7, 130,2, 127,85, 127,77, 126,9, 122,0, 121,8, 116,7, 116,5, 116,2, 71,6, 55,9, 54,9, 47,6, 46,7, 30,3, 29,6, 25,6, 24,8, 19,8 м.д. Чистота: 93% UPLCMS (210 нм); время удерживания 0,63 мин; (M+1) 384,9
Пример 41
1-Азабицикло[2.2.2]окт-3-ил-{2-[4-фтор-3-(пиридин-3-ил)-фенил]-пропан-2-ил}-карбамат
Применяя общую процедуру E, из 1-азабицикло[2.2.2]окт-3-ил-[2-(3-бром-4-фторфенил)-пропан-2-ил]-карбамата (110 мг, 0,286 ммоль), пиридин-3-бороновой кислоты (53 мг, 0,43 ммоль) и трис-(дибензилиденацетон)-дипалладия(0) получали соединение, указанное в заголовке, в виде белого твердого вещества (42 мг, 39%).1H ЯМР (400 МГц, CDCl3) δ 8,78 (с, 1H), 8,62 (д, J=3,5 Гц, 1H), 7,86 (с, 1H), 7,46 (д, J=7,2 Гц, 2H), 7,38 (дд, J=4,9, 7,9 Гц, 1H), 7,15 (дд, J=8,7, 9,9 Гц, 1H), 5,32 (с, 1H), 4,69-4,57 (м, 1H), 2,68 (с, 6H), 1,70 (д, J=11,3 Гц, 6H), 2,08-0,94 (м, 5H) м.д.13C ЯМР (100 МГц, CDCl3) δ 160,0, 157,5, 149,9, 149,0, 136,6, 132,1, 127,3, 126,8, 126,7, 125,5, 125,3, 123,5, 116,4, 116,2, 71,5, 55,9, 55,0, 47,6, 46,7, 30,1, 29,66, 25,6, 24,8, 19,7 м.д. Чистота: 100% UPLCMS (210 нм); время удерживания 0,54 мин; (M+1) 367,8.
Пример 42
1-Азабицикло[2.2.2]окт-3-ил-{2-[4-фтор-3-(фуран-3-ил)-фенил]-пропан-2-ил}-карбамат
Применяя общую процедуру E, из 1-азабицикло[2.2.2]окт-3-ил-[2-(3-бром-4-фторфенил)-пропан-2-ил]-карбамата (110 мг, 0,296 ммоль), фуран-3-бороновой кислоты (47,9 мг, 0,428 ммоль) и трис-(дибензилиденацетон)-дипалладия(0) получали соединение, указанное в заголовке, в виде белого твердого вещества (47 мг, 44%).1H ЯМР (400 МГц, CDCl3) δ 7,88 (ддд, J=0,9, 1,5, 2,5 Гц, 1H), 7,53 (дд, J=2,5, 7,1 Гц, 1H), 7,50 (т, J=1,7 Гц, 1H), 7,31-7,23 (м, 1H), 7,08 (дд, J=8,6, 10,6 Гц, 1H), 6,76 (дт, J=0,8, 1,7 Гц, 1H), 5,22 (с, 1H), 4,62 (с, 1H), 3,40-2,11 (м, 6H), 2,02-0,87 (м, 5H), 1,59 (дд, J=11,6, 70,3 Гц, 6H) м.д.13C ЯМР (100 МГц, CDCl3) δ 160,0, 157,5, 150,2, 143,9, 127,44, 127,36, 127,0, 126,1, 123,8, 116,6, 116,4, 71,5, 55,9, 55,0, 47,6, 46,7, 30,1, 29,9, 25,6, 24,8, 19,8 м.д. Чистота: 96% UPLCMS (210 нм); время удерживания 0,85 мин; (M+1) 372,9.
Пример 43
1-Азабицикло[2.2.2]окт-3-ил-{2-[4-фтор-3-(пиридин-4-ил)-фенил]-пропан-2-ил}-карбамат
Применяя общую процедуру E, из 1-азабицикло[2.2.2]окт-3-ил-[2-(3-бром-4-фторфенил)-пропан-2-ил]-карбамата (130 мг, 0,291 ммоль), пиридин-4-бороновой кислоты (54 мг, 0,43 ммоль) и трис-(дибензилиденацетон)-дипалладия(0) получали соединение, указанное в заголовке, в виде белого твердого вещества (46 мг, 41%).1H ЯМР (400 МГц, CDCl3) δ 8,68 (дд, J=1,6, 4,5 Гц, 2H), 7,61-7,39 (м, 4H), 7,15 (дд, J=8,6, 10,2 Гц, 1H), 5,31 (с, 1H), 4,69-4,57 (м, 1H), 3,40-2,07 (м, 6H), 1,69 (д, J=10,8 Гц, 6H), 2,06-0,74 (м, 5H) м.д.13C ЯМР (100 МГц, CDCl3) δ 160,0, 157,6, 143,3, 141,6, 141,5, 126,7, 126,6, 124,9, 124,8, 120,1, 119,9, 116,1, 115,9, 115,4, 115,1, 109,2, 71,4, 55,9, 55,0, 47,6, 46,7, 29,9, 25,6, 24,8, 19,8 м.д. Чистота: 97% UPLCMS (210 нм); время удерживания 0,82 мин; (M+1) 384,6.
Пример 44
1-(1-Азабицикло[2.2.2]окт-3-ил)-3-(2-фенилпропан-2-ил)-мочевина
Применяя общую процедуру C, из хинуклидин-3-амина (102 мг, 0,6 ммоль), CDI (131 мг, 0,789 ммоль) и кумиламина (95 мг, 0,70 ммоль) получали соединение, указанное в заголовке, в виде белого твердого вещества (21 мг, 10%).1H ЯМР (400 МГц, CDCl3) δ 7,61-7,47 (м, 2H), 7,44-7,37 (м, 2H), 7,34-7,28 (м, 1H), 4,86 (с, 1H), 4,20 (д, J=7,3 Гц, 1H), 3,71-3,60 (м, 1H), 3,14 (ддд, J=2,3, 9,4, 14,2 Гц, 1H), 2,79-1,89 (м, 6H), 1,64 (д, J=3,3 Гц, 6H), 1,58-1,10 (м, 5H) м.д.13C ЯМР (100 МГц, CDCl3) δ 157,4, 146,2, 129,3, 127,8, 125,8, 57,1, 54,9, 47,7, 47,1, 46,7, 30,6, 30,5, 26,0, 20,3 м.д. Чистота: 79% UPLCMS (210 нм); время удерживания 0,61 мин; (M+1) 288,2.
Препаративная процедура O
Пример 45
3-Циано-1-азабицикло[2.2.2]окт-3-ил-{2-[3-(проп-1-ен-2-ил)-фенил]-пропан-2-ил}-карбамат
К раствору 3-гидроксихинуклидин-3-карбонитрила (38 мг, 0,25 ммоль) в смеси ацетонитрила с диоксаном (3 мл) при комнатной температуре добавляли триэтиламин (7,0 мкл, 0,05 ммоль). Реакционную смесь перемешивали в течение 15 мин и по каплям добавляли 1-(2-изоцианатопропан-2-ил)-3-(проп-1-ен-2-ил)-бензол (49,0 мкл, 0,248 ммоль). Реакционную смесь перемешивали в течение 18 часов при 65°C и концентрировали. Неочищенный материал очищали посредством комбифлэш-хроматографии (SiO2-картридж, CHCl3 и 2 н NH3 в MeOH), получая соответствующий карбамат в виде прозрачного масла (57 мг, 65%).1H ЯМР (400 МГц, CDCl3) δ 7,42-7,20 (с, 5H), 6,61 (с, 1H), 5,11 (с, 1H), 3,29 (д, J=12,0 Гц, 1H), 3,09 (д, J=12,0 Гц, 1H), 2,93 (д, J=4,0 Гц, 2H), 2,79-2,68 (м, 2H), 2,13 (с, 6H) 2,05-2,00 (м, 2H), 1,91 (с, 3H), 1,87 (с, 2H), 1,50-1,37 (м, 2H) м.д.13C ЯМР (100 МГц, CDCl3) δ 135,4, 128,7, 125,2, 124,5, 124,0, 122,0, 112,9, 61,2, 47,0, 46,2, 32,4, 31,8, 29,9, 29,4, 29,2, 26,6, 25,7, 23,7, 22,8, 22,2, 19,2 м.д. Чистота: >99% UPLCMS (210 нм); время удерживания 0,81 мин; (M+1) 354.
Препаративная процедура P
Пример 46
N-(2-(3-(проп-1-ен-2-ил)-фенил)-пропан-2-ил)-1,4-диазабицикло[3.2.2]нонан-4-карбоксамид
К раствору 1,4-диазабицикло[3.2.2]нонана (350 мг, 2,77 ммоль) и 1-(2-изоцианатопропан-2-ил)-3-(проп-1-ен-2-ил)-бензола (1,09 мл, 5,55 ммоль) в хлороформе (2 мл) добавляли 3-4 гранулы молекулярных сит. Реакционную смесь перемешивали при комнатной температуре в течение 18 часов и затем концентрировали. Неочищенный материал очищали посредством комбифлэш-хроматографии (SiO2-картридж, CHCl3 и 2 н NH3 в MeOH), получая соответствующую мочевину в виде светло-серого твердого вещества (650 мг, 36%).1H ЯМР (400 МГц, CDCl3) δ 7,48 (с, 1H), 7,31-7,26 (м, 3H), 5,34 (с, 1H), 5,07 (с, 1H), 4,73 (шир.с, 1H), 4,03 (шир.с, 1H), 3,64 (м, 2H), 3,14-3,03 (м, 6H), 2,15 (с, 3H) 2,06 (м, 2H), 1,72 (м, 8H) м.д.13C ЯМР (100 МГц, CDCl3) δ 155,7, 148,3, 143,8, 141,3, 128,5, 124,1, 123,9, 122,0, 112,5, 57,8, 55,8, 48,1, 46,4 (2×), 41,2, 30,2 (2×), 27,3 (2×), 22,1 м.д. Чистота: >98% UPLCMS (210 нм); время удерживания 0,71 мин; (M+1) 328
Пример 47
Бифенил-2-ил-1,4-диазабицикло[3.2.2]нонан-4-карбоксилат
Бифенил-2-илкарбонохлоридат (83,0 мг, 0,358 ммоль) обрабатывали 1,4-диазабицикло[3.2.2]нонаном (113 мг, 0,896 ммоль), применяя ту же процедуру, что и в Примере 46, получая соединение, указанное в заголовке, в виде светло-серого твердого вещества (17 мг, 15%). Чистота: >99% UPLCMS (210 нм); время удерживания 0,75 мин; (M+1) 323.
Пример 48
N-{2-[3-(пропан-2-ил)-фенил]-пропан-2-ил}-1,4-диазабицикло[3.2.2]нонан-4-карбоксамид
Применяя общую процедуру F, из N-(2-(3-(проп-1-ен-2-ил)-фенил)-пропан-2-ил)-1,4-диазабицикло[3.2.2]нонан-4-карбоксамида (100 мг, 0,305 ммоль) и палладия (20 мг, 20% масс. на угле) получали соединение, указанное в заголовке, в виде светло-серого твердого вещества (60 мг, 57%).1H ЯМР (400 МГц, CDCl3) δ 7,28-7,21 (м, 3H), 7,11 (д, J=8,0 Гц, 1H), 4,71 (с, 1H), 4,02 (с, 1H), 3,66 (т, J=8,0 Гц, 2H) 3,15 (м, 7H), 2,06 (шир.с, 2H), 1,77 (с, 7H) 1,26 (д, J=4,0 Гц, 6H) м.д.13C ЯМР (100 МГц, CDCl3) δ 155,8, 148,9, 148,5, 128,5,1, 124,7, 123,1, 122,4, 57,8, 56,0, 48,1, 46,4, 41,2, 34,5, 32,2, 30,4, 30,0, 29,9, 27,3, 24,3, 22,9 м.д. Чистота: >91% UPLCMS (210 нм); время удерживания 0,74 мин; (M+1) 330.
Препаративная процедура Q
Пример 49
(+/-)-(3S,4S)-1-азабицикло[2.2.1]гептан-3-ил-2-(3-(проп-1-ен-2-ил)-фенил)-пропан-2-илкарбамат
К раствору (+/-)-(3S,4S)-1-азабицикло[2.2.1]гептан-3-ола (294 мг, 2,6 ммоль) в THF (5 мл) при комнатной температуре добавляли NaH [60%, масло] (107 мг, 2,67 ммоль). Реакционную смесь перемешивали в течение 15 мин и по каплям добавляли 1-(2-изоцианатопропан-2-ил)-3-(проп-1-ен-2-ил)-бензол (344 мкл, 1,73 ммоль). Реакционную смесь перемешивали в течение 30 мин и гасили насыщенным солевым раствором. Раствор экстрагировали EtOAc и органический слой сушили над Na2SO4 и концентрировали. Неочищенный материал очищали посредством комбифлэш-хроматографии (SiO2-картридж, CHCl3 и 2 н NH3 в MeOH), получая соответствующий карбамат в виде прозрачного масла (140 мг, 26%).1H ЯМР (400 МГц, CDCl3) δ 7,26-7,20 (м, 3H), 7,11 (м, 1H), 5,18 (с, 1H), 5,19 (шир.с, 1H), 5,01 (с, 1H), 4,81 (шир.с, 1H), 2,99 (шир.с, 1H), 2,82 (шир.с, 1H), 2,70 (шир.с, 1H), 2,53 (шир.с, 2H), 2,33 (шир.с, 1H), 2,02 (с, 3H), 1,76 (шир.с, 1H) 1,61 (шир.с, 6H), 1,52 (шир.с, 1H), 1,37 (шир.с, 1H) м.д.13C ЯМР (100 МГц, CDCl3) δ 147,1, 143,7, 141,5, 128,5, 124,1, 122,1, 112,7, 75,6, 60,6, 59,4, 55,4, 54,3, 53,9, 41,5, 29,9, 29,8, 29,4, 22,2, 21,6 м.д. Чистота: >98% UPLCMS (210 нм); время удерживания 0,83 мин; (M+1) 315
Пример 50
(+/-)-(3S,4S)-1-азабицикло[2.2.1]гепт-3-ил-{2-[3-(пропан-2-ил)-фенил]-пропан-2-ил}-карбамат
Применяя общую процедуру F, из (+/-)-(3S,4S)-1-азабицикло[2.2.1]гептан-3-ил-2-(3-(проп-1-ен-2-ил)-фенил)- пропан-2-илкарбамата (110 мг, 0,350 ммоль) и палладия (20 мг, 20% масс. на угле) получали соединение, указанное в заголовке, в виде светло-серого твердого вещества (36 мг, 46%).1H ЯМР (400 МГц, CDCl3) δ 7,25-7,19 (м, 3H), 7,11 (д, J=8,0 Гц, 1H), 5,19 (с, 1H), 4,91 (с, 1H), 3,44 (т, J=4,0 Гц, 2H) 3,19 (шир.с, 1H), 3,02 (шир.с, 1H), 2,89 (м, 2H), 2,69 (шир.с, 1H), 2,39 (шир.с, 1H), 1,91 (шир.с, 1H), 1,66 (шир.с, 7H) 1,26 (д, J=4,0 Гц, 6H) м.д.13C ЯМР (100 МГц, CDCl3) δ 128,5 (2×), 124,8, 123,1 (2×), 122,4 (2×), 77,4, 60,6, 59,4, 55,5, 41,5, 34,5, 29,9, 29,9, 29,5, 24,3 (2×), 21,6 м.д. Чистота: >95% UPLCMS (210 нм); время удерживания 0,88 мин; (M+1) 317.
Пример 51
N-[2-(3-бром-4-фторфенил)-пропан-2-ил]-1,4-диазабицикло[3.2.2]нонан-4-карбоксамид
Применяя общую процедуру A, из гидрохлорида 2-(3-бром-4-фторфенил)-пропан-2-амина (1,00 г, 3,72 ммоль) и 1,4-диазабицикло[3.2.2]нонана получали соединение, указанное в заголовке, в виде белого твердого вещества (265 мг, 18%).1H ЯМР (400 МГц, CDCl3) δ 7,54-7,52 (м, 1H), 7,31-7,25 (м, 1H), 7,04 (т, J=8,0 Гц, 1H), 4,71 (с, 1H), 3,99 (шир.с, 1H), 3,59 (т, J=8,0 Гц, 2H), 3,13-2,95 (м, 6H), 2,04-1,97 (м, 2H) 1,77-1,67 (м, 2H), 1,65 (с, 6H) м.д.13C ЯМР (100 МГц, CDCl3) δ 158,9, 156,4, 155,4, 145,9, 130,2, 125,6, 116,2, 57,8, 54,9, 48,3, 46,6, 46,6, 41,6, 30,5, 30,5, 27,6, 27,6 м.д. Чистота: >99% UPLCMS (210 нм); время удерживания 0,73 мин; (M+1) 384.
Пример 52
N-[2-(6-фторбифенил-3-ил)-пропан-2-ил]-1,4-диазабицикло[3.2.2]нонан-4-карбоксамид
Применяя общую процедуру E, из N-[2-(3-бром-4-фторфенил)-пропан-2-ил]-1,4-диазабицикло[3.2.2]нонан-4-карбоксамида (100 мг, 0,261 ммоль), фенилбороновой кислоты (79 мг, 0,65 ммоль) и ацетата палладия (II) получали соединение, указанное в заголовке, в виде светло-серого твердого вещества (66 мг, 66%).1H ЯМР (400 МГц, CDCl3) δ 7,54 (м, 2H), 7,44-7,40 (м, 5H), 7,08 (м, 1H), 4,78 (шир.с, 1H), 4,00 (шир.с, 1H), 3,60 (м, 2H), 3,11-2,92 (м, 6H), 2,00 (м, 2H) 1,67 (м, 7H), 1,26 (с, 1H) м.д.13C ЯМР (100 МГц, CDCl3) δ 159,6, 155,6, 144,6, 136,5, 129,3, 128,6, 128,5, 127,8, 127,5, 125,7, 125,6, 116,1, 115,9, 57,9, 55,3, 48,2, 46,4, 46,4, 41,6, 30,6, 30,5, 29,9, 27,6 м.д. Чистота: >99% UPLCMS (210 нм); время удерживания 0,88 мин; (M+1) 382.
Пример 53
N-[2-(4',6-дифторбифенил-3-ил)-пропан-2-ил]-1,4-диазабицикло[3.2.2]нонан-4-карбоксамид
Применяя общую процедуру E, из N-[2-(3-бром-4-фторфенил)-пропан-2-ил]-1,4-диазабицикло[3.2.2]нонан-4-карбоксамида (100 мг, 0,261 ммоль), 4-фторфенилбороновой кислоты (91 мг, 0,65 ммоль) и ацетата палладия (II) получали соединение, указанное в заголовке, в виде светло-серого твердого вещества (64 мг, 62%).1H ЯМР (400 МГц, CDCl3) δ 7,48 (м, 2H), 7,37-7,30 (м, 2H), 7,10-7,03 (м, 3H), 4,77 (с, 1H), 3,99 (шир.с, 1H), 3,58 (м, 2H), 3,10-2,90 (м, 6H), 1,98 (м, 2H) 1,71 (м, 8H) м.д.13C ЯМР (100 МГц, CDCl3) δ 163,7, 161,3, 159,5, 157,1, 155,6, 144,6, 131,0, 130,9, 127,4, 127,2, 125,7, 116,1, 115,6, 57,9, 55,2, 48,2, 46,4, 46,4, 41,5, 30,6, 30,6, 27,6, 27,6 м.д. Чистота: >99% UPLCMS (210 нм); время удерживания 0,90 мин; (M+1) 400.
Пример 54
N-[2-(нафталин-1-ил)-пропан-2-ил]-1,4-диазабицикло[3.2.2]нонан-4-карбоксамид
Применяя общую процедуру A, из гидрохлорида 2-(нафталин-1-ил)-пропан-2-амина (227 мг, 1,23 ммоль) и 1,4-диазабицикло[3.2.2]нонана получали соединение, указанное в заголовке, в виде белого твердого вещества (206 мг, 50%).1H ЯМР (400 МГц, CDCl3) δ 8,63-8,60 (м, 1H), 7,90-7,87 (м, 1H), 7,78 (д, J=8,0 Гц, 1H), 7,63 (д, J=8,0 Гц, 1H), ), 7,47-7,42 (м, 3H), 4,86 (с, 1H), 3,94 (шир.с, 1H), 3,61 (м, 2H), 3,11-2,88 (м, 7H), 2,01-1,91 (м, 7H), 1,68-1,62 (м, 2H) м.д.13C ЯМР (100 МГц, CDCl3) δ 176,2, 155,5, 142,6, 135,3, 130,6, 129,9, 128,8, 126,3, 125,4, 125,2, 123,9, 57,3, 57,1, 47,7, 45,8, 45,8, 40,7, 29,4, 29,4, 26,9, 26,9 м.д. Чистота: >99% UPLCMS (210 нм); время удерживания 0,72 мин; (M+1) 338.
Пример 55
N-(2-(5-бром-2-фторфенил)-пропан-2-ил)-1,4-диазабицикло[3.2.2]нонан-4-карбоксамид
Применяя общую процедуру A, из гидрохлорида 2-(5-бром-2-фторфенил)-пропан-2-амина (100 мг, 0,372 ммоль) и 1,4-диазабицикло[3.2.2]нонана получали соединение, указанное в заголовке, в виде белого твердого вещества (70 мг, 49%).1H ЯМР (400 МГц, CDCl3) δ 7,48 (м, 1H), 7,28 (м, 1H), 6,86 (м, 1H), 4,85 (с, 1H), 3,98 (шир.с, 1H), 3,56 (м, 2H), 3,14-2,91 (м, 7H), 1,99 (м, 2H) 1,71 (м, 7H) м.д.13C ЯМР (100 МГц, CDCl3) δ 161,1, 158,6, 155,7, 131,3, 113,1, 118,3, 118,0 57,8, 54,0, 48,1, 46,4, 46,4, 41,5, 29,1, 29,1, 27,5, 27,5 м.д. Чистота: >99% UPLCMS (210 нм); время удерживания 0,73 мин; (M+1) 384.
Пример 56
N-[2-(4-фторбифенил-3-ил)-пропан-2-ил]-1,4-диазабицикло[3.2.2]нонан-4-карбоксамид
Применяя общую процедуру E, из N-(2-(5-бром-2-фторфенил)-пропан-2-ил)-1,4-диазабицикло[3.2.2]нонан-4-карбоксамида (100 мг, 0,261 ммоль), фенилбороновой кислоты (30 мг, 0,25 ммоль) и ацетата палладия (II) получали соединение, указанное в заголовке, в виде светло-серого твердого вещества (27 мг, 39%).1H ЯМР (400 МГц, CDCl3) δ 7,56-7,51 (м, 3H), 7,41-7,37 (м, 3H), 7,32-7,30 (м, 1H), 7,03 (м, 1H), 4,90 (с, 1H), 4,00 (шир.с, 1H), 3,59 (м, 2H), 3,11-2,92 (м, 6H), 2,04-1,98 (м, 2H) 1,78 (с, 6H), 1,73-1,67 (м, 2H) м.д.13C ЯМР (100 МГц, CDCl3) δ 161,8, 159,4, 155,9, 140,9, 137,2, 134,6, 128,9, 127,4, 127,3, 127,2, 127,1, 127,0, 116,9, 57,9, 54,4, 48,1, 46,5, 46,5, 41,4, 29,9, 29,3, 27,5, 27,5 м.д. Чистота: >99% UPLCMS (210 нм); время удерживания 0,90 мин; (M+1) 400.
Пример 57
N-(2-(3-изопропоксифенил)-пропан-2-ил)-1,4-диазабицикло[3.2.2]нонан-4-карбоксамид
Применяя общую процедуру A, из гидрохлорида 2-(3-изопропоксифенил)-пропан-2-амина (60 мг, 0,31 ммоль) и 1,4-диазабицикло[3.2.2]нонана получали соединение, указанное в заголовке, в виде белого твердого вещества (70 мг, 57%).1H ЯМР (400 МГц, CDCl3) δ 7,17 (т, J=8,0 Гц, 1H), 6,92-6,87 (м, 2H), 6,69 (д, J=8,0 Гц, 1H) 4,66 (шир.с, 1H), 4,48 (м, 1H), 3,94 (шир.с, 1H) 3,56 (м, 2H), 3,08-2,90 (м, 5H), 1,96 (м, 2H) 1,69-1,60 (м, 7H), 1,27 (д, J=8,0 Гц, 6H), 1,17 (шир.с, 2H) м.д.13C ЯМР (100 МГц, CDCl3) δ 158,0, 155,7, 150,3, 129,4, 117,1, 113,4, 113,1, 77,5, 69,8, 55,7, 48,2, 46,4, 46,4, 46,3, 41,5, 30,3, 30,0, 29,9, 27,6, 22,3 м.д. Чистота: >99% UPLCMS (210 нм); время удерживания 0,75 мин; (M+1) 346.
Пример 58
N-(бифенил-3-ил)-1,4-диазабицикло[3.2.2]нонан-4-карбоксамид
Применяя общую процедуру A, из бифенил-3-амина (100 мг, 0,592 ммоль) и 1,4-диазабицикло[3.2.2]нонана получали соединение, указанное в заголовке, в виде светло-серого твердого вещества (93 мг, 49%).1H ЯМР (400 МГц, CDCl3) δ 7,60 (м, 1H), 7,56-53 (м, 2H), 7,39-7,21 (м, 6H), 6,67 (шир.с, 1H), 4,85 (с, 1H), 4,16 (шир.с, 1H), 3,66-3,61 (м, 2H), 3,07-2,86 (м, 6H), 1,97 (м, 2H) 1,68 (м, 2H) м.д.13C ЯМР (100 МГц, CDCl3) δ 154,7, 142,0, 141,1, 139,9, 129,3, 128,8. 128,8, 127,5, 127,3, 127,3, 122,0, 119,4, 119,2, 57,5, 48,4, 46,3, 46,3, 42,1, 27,5, 27,5 м.д. Чистота: >96% UPLCMS (210 нм); время удерживания 0,75 мин; (M+1) 333.
Пример 59
N-{2-[2-фтор-5-(2-метилпропил)-фенил]-пропан-2-ил}-1,4-диазабицикло[3.2.2]нонан-4-карбоксамид
Применяя общую процедуру E, из N-(2-(5-бром-2-фторфенил)-пропан-2-ил)-1,4-диазабицикло[3.2.2]нонан-4-карбоксамида (100 мг, 0,261 ммоль), изопропилбороновой кислоты (66 мг, 0,65 ммоль) и ацетата палладия (II) получали соединение, указанное в заголовке, в виде светло-серого твердого вещества (27 мг, 39%).1H ЯМР (400 МГц, CDCl3) δ 7,08 (д, J=8,0 Гц, 1H), 6,93-6,81 (м, 2H), 4,85 (с, 1H), 3,96 (шир.с, 1H), 3,65 (кв., J=8,0 Гц, 1H), 3,55 (т, J=8,0 Гц, 2H), 3,09-2,90 (м, 5H), 2,40 (д, J=4,0 Гц, 1H), 2,01-1,92 (м, 2H) 1,81-1,61 (м, 8H), 1,22-1,17 (м, 2H), 0,87 (д, J=8,0 Гц, 6H) м.д.13C ЯМР (100 МГц, CDCl3) δ 160,5, 158,0, 155,9, 137,0, 133,6, 128,8, 116,0, 57,9, 54,3, 48,1, 46,4 (2×), 45,2, 41,4, 30,5, 29,9, 29,2, 29,2, 27,5, 22,6, 22,6 м.д. Чистота: >99% UPLCMS (210 нм); время удерживания 0,94 мин; (M+1) 362.
Пример 60
N-(бифенил-2-ил)-1,4-диазабицикло[3.2.2]нонан-4-карбоксамид
Применяя общую процедуру A, из 1-изоцианатобифенила (50 мг, 0,26 ммоль) и 1,4-диазабицикло[3.2.2]нонана получали соединение, указанное в заголовке, в виде белого твердого вещества (55 мг, 65%).1H ЯМР (400 МГц, CDCl3) δ 8,15 (д, J=8,0 Гц, 1H), 7,45-7,31 (м, 6H), 7,16 (д, J=8,0 Гц, 1H), 7,04 (т, J=8,0 Гц, 1H), 6,47 (шир.с, 1H), 3,63 (шир.с, 1H), 3,57 (т, J=8,0 Гц, 2H), 3,00-2,80 (м, 6H), 1,68 (м, 2H) 1,43 (м, 2H) м.д.13C ЯМР (100 МГц, CDCl3) δ 154,2, 138,9, 136,6, 131,7, 129,7, 129,5, 129,5, 129,3, 129,3, 128,7, 128,1, 122,6, 120,5, 57,6, 48,5, 46,2, 46,2, 41,6, 29,9, 27,3 м.д. Чистота: >99% UPLCMS (210 нм); время удерживания 0,64 мин; (M+1) 322.
Пример 61
N-(нафталин-1-ил)-1,4-диазабицикло[3.2.2]нонан-4-карбоксамид
Применяя общую процедуру A, из 1-изоцианатонафталина (208 мг, 1,23 ммоль) и 1,4-диазабицикло[3.2.2]нонана получали соединение, указанное в заголовке, в виде белого твердого вещества (150 мг, 48%).1H ЯМР (400 МГц, CDCl3) δ 7,80-7,72 (м, 2H), 7,64 (д, J=8,0 Гц, 1H), 7,56 (д, J=8,0 Гц, 1H), 7,44-7,35 (м, 3H), 6,65 (шир.с, 1H), 4,18 (шир.с, 1H), 3,64 (т, J=8,0 Гц, 2H), 3,09-2,91 (м, 6H), 2,08-1,93 (м, 2H) 1,74-1,66 (м, 2H) м.д.13C ЯМР (100 МГц, CDCl3) δ 155,3, 134,4, 134,3, 128,9, 128,2, 126,2, 126,1, 126,0, 125,1, 121,2, 121,0, 57,6, 48,7, 46,4, 46,4, 42,2, 27,6, 27,6 м.д. Чистота: >99% UPLCMS (210 нм); время удерживания 0,53 мин; (M+1) 296.
Пример 62
(S)-хинуклидин-3-ил-2-(бифенил-4-ил)-пропан-2-илкарбамат
Применяя общую процедуру B, бромбензонитрил (2,00 г, 11,0 ммоль) преобразовывали в соответствующий 2-(4-бромфенил)-пропан-2-амин (1,20 г, 51%), получаемый в виде коричневого масла.
Применяя общую процедуру A, из 2-(4-бромфенил)-пропан-2-амина (1,0 г, 4,7 ммоль) и (S)-хинуклидин-3-ола получали (S)-хинуклидин-3-ил-2-(4-бромфенил)-пропан-2-илкарбамат (1,0 г, 58%) в виде коричневого масла.
Применяя общую процедуру E, из вышеуказанного бромида (200 мг, 0,540 ммоль), фенилбороновой кислоты (133 мг, 1,10 ммоль) и [PdCl2(pddf)]CH2Cl2 получали соединение, указанное в заголовке, в виде белого твердого вещества (70 мг, 35%).1H ЯМР (500 МГц, CDCl3) δ 7,60-7,53 (м, 4H), 7,47 (д, J=8,5 Гц, 2H), 7,42 (т, J=7,5 Гц, 2H), 7,33 (т, J=7,5 Гц, 1H), 5,26 (шир.с, 1H), 4,64 (м, 1H), 3,33-3,15 (м, 1H), 3,10-2,45 (м, 5H), 2,40-1,80 (м, 2H), 1,78-1,58 (м, 7H), 1,55-1,33 (м, 2H) м.д.13C ЯМР (125 МГц, CDCl3) δ 154,5, 146,1, 140,8, 139,5, 128,7, 127,2, 127,1, 127,1, 125,2, 70,9, 55,5, 55,1, 47,4, 46,4, 31,1, 29,5, 25,3, 24,5, 19,5 м.д. Чистота: 100 % LCMS (214 нм и 254 нм); время удерживания 1,56 мин; (M+1) 365.
Пример 63
Хинуклидин-3-ил-2-(4-(пиримидин-5-ил)-фенил)-пропан-2-илкарбамат
Применяя общую процедуру E, из хинуклидин-3-ил-2-(4-бромфенил)-пропан-2-илкарбамата (200 мг, 0,540 ммоль), пиримидин-5-илбороновой кислоты (136 мг, 1,12 ммоль) и [PdCl2(pddf)]CH2Cl2 получали соединение, указанное в заголовке, в виде белого твердого вещества (80 мг, 40%).1H ЯМР (500 МГц, CDCl3) δ 9,17 (с, 1H), 8,92 (с, 2H), 7,58-7,51 (м, 4H), 5,34 (с, 1H), 4,61 (м, 1H), 3,20-3,10 (м, 1H), 2,92-2,41 (м, 5H), 2,00-1,76 (м, 2H), 1,72-1,53 (м, 7H), 1,52-1,32 (м, 2H) м.д.13C ЯМР (125 МГц, CDCl3) δ 157,4, 154,8, 154,5, 148,2, 134,0, 132,5, 127,0, 126,0, 71,2, 55,6, 55,0, 47,4, 46,3, 29,7, 29,4, 25,4, 24,5, 19,5 м.д. Чистота: >96 % LCMS (214 нм и 254 нм); время удерживания 1,34 мин; (M+1) 366.
Пример 64
Хинуклидин-3-ил-1-(бифенил-4-ил)-циклопропилкарбамат
Применяя общую процедуру G, бромбензонитрил (3,00 г, 16,5 ммоль) преобразовывали в соответствующий 1-(4-бромфенил)-циклопропанамин (1,80 г, 51 %), получаемый в виде желтого твердого вещества.
Применяя общую процедуру A, из 1-(4-бромфенил)-циклопропанамина (1,0 г, 4,7 ммоль) и хинуклидин-3-ола получали хинуклидин-3-ил-1-(4-бромфенил)-циклопропилкарбамат (1,3 г, 75 %) в виде белого полужидкого вещества.
Применяя общую процедуру E, из вышеуказанного карбамата (400 мг, 1,12 ммоль), фенилбороновой кислоты (267 мг, 2,22 ммоль) и [PdCl2(pddf)]CH2Cl2 получали соединение, указанное в заголовке, в виде вязкого масла (100 мг, 25%).1H ЯМР (500 МГц, CDCl3) δ 7,47 (д, J=7,5 Гц, 2H), 7,43 (д, J=8,0 Гц, 2H), 7,33 (т, J=7,5 Гц, 2H), 7,26-7,15 (м, 3H), 5,93 (шир.с, 0,6H), 5,89 (шир.с, 0,4H), 4,67 (м, 1H), 3,20-3,06 (м, 1H), 2,88-2,42 (м, 5H), 1,98-1,08 (м, 9H) м.д.13C ЯМР (125 МГц, CDCl3) δ 155,0, 141,0, 139,7, 138,2, 127,7, 126,1, 126,0, 124,8, 124,1, 70,0, 54,5, 46,3, 45,4, 34,1, 24,3, 23,2, 18,3, 17,0 м.д. Чистота: 100 % LCMC (214 нм и 254 нм); время удерживания 1,52 мин; (M+1) 363.
Препаративная процедура R
Пример 65
Хинуклидин-3-ил-1-(4-(пиридин-2-ил)-фенил)-циклопропилкарбамат
К раствору хинуклидин-3-ил-1-(4-бромфенил)-циклопропилкарбамата (870 мг, 2,43 ммоль) в 30 мл 1,4-диоксана, добавляли бис-(пинаколато)-диборан (1,81 г, 7,22 ммоль), CH3COOK (2,10 г, 21,4 ммоль) и [PdCl2(pddf)]CH2Cl2 (97 мг, 0,12 ммоль). Смесь перемешивали при 80°C в течение 18 часов. Растворитель выпаривали и остаток экстрагировали EtOAc. Экстракты концентрировали и очищали посредством хроматографии на колонке с силикагелем (элюируя смесью EtOAc/метанол от 20/1 до 10/1 с 1% TEA), получая хинуклидин-3-ил-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-фенил)-циклопропилкарбамат (260 мг, 33%) в виде коричневого полужидкого вещества.
Применяя общую процедуру E, из вышеуказанного бороната (260 мг, 0,632 ммоль), 2-бромпиридина (149 мг, 0,941 ммоль) и Pd2(dba)3 (32,0 мг, 0,036 ммоль) получали соединение, указанное в заголовке, в виде белого полужидкого вещества (70 мг, 31%).1H ЯМР (500 МГц, CDCl3) δ 8,58 (д, J=4,5 Гц, 1H), 7,82 (д, J=7,0 Гц, 2H), 7,66-7,57 (м, 2H), 7,23-7,15 (м, 2H), 7,11 (т, J=5,0 Гц, 1H), 6,16 (шир.с, 0,6H), 5,97 (шир.с, 0,4H), 4,63 (м, 1H), 3,17-3,02 (м, 1H), 2,90-2,38 (м, 5H), 1,90-1,10 (м, 9H) м.д.13C ЯМР (125 МГц, CDCl3) δ 156,1, 155,2, 148,6, 143,0, 136,3, 135,7, 125,9, 124,5, 120,9, 119,4, 70,3, 54,6, 46,3, 45,4, 34,1, 24,4, 23,5, 18,5, 17,3 м.д. Чистота: 100 % LCMS (214 нм и 254 нм); время удерживания 1,18 (M+H) 364.
Пример 66
Хинуклидин-3-ил-1-(4-(пиримидин-5-ил)-фенил)-циклопропилкарбамат
Применяя общую процедуру E, из вышеуказанного хинуклидин-3-ил-1-(4-бромфенил)-циклопропилкарбамата (400 мг, 1,10 ммоль), пиримидин-5-илбороновой кислоты (204 мг, 1,64 ммоль) и [PdCl2(pddf)]CH2Cl2 получали соединение, указанное в заголовке, в виде вязкого масла (110 мг, 28 %).1H ЯМР (500 МГц, CDCl3) δ (с, 1H), (с, 2H), 7,44 (д, J=8,5 Гц, 2H), 7,33-7,25 (м, 2H), 6,02 (шир.с, 0,7H), 6,02 (шир.с, 0,3H), 4,65 (м, 1H), 3,20-3,05 (м, 1H), 2,86-2,40 (м, 5H), 1,98-1,12 (м, 9H) м.д.13C ЯМР (125 МГц, CDCl3) δ 156,3, 155,1, 153,7, 143,3, 132,9, 131,1, 126,0, 125,3, 70,5, 54,7, 46,4, 45,4, 34,1, 24,4, 23,5, 18,5, 17,5 м.д. Чистота: 100 % LCMS (214 нм и 254 нм); время удерживания 1,29 мин; (M+1) 365.
Пример 67
(S)-хинуклидин-3-ил-1-(4'-фторбифенил-4-ил)-циклопропилкарбамат
Применяя общую процедуру E, из (S)-хинуклидин-3-ил-1-(4-бромфенил)-циклопропилкарбамата, 4-F-фенилбороновой кислоты и [PdCl2(pddf)]CH2Cl2 получали соединение, указанное в заголовке, в виде белого твердого вещества (45%).1H ЯМР (500 МГц, DMSO-d6) δ 8,06-7,83 (д, 1H), 7,69-7,66 (м, 2H), 7,59-7,55 (м, 2H), 7,29-7,22 (м, 4H), 4,56-4,54 (м, 1H), 3,13-2,32 (м, 6H), 1,91-1,19 (м, 9H) м.д.13C ЯМР (125 МГц, DMSO-d6) δ 163,2, 161,2, 156,4, 143,7, 136,9, 128,9, 128,8, 126,8, 125,6, 116,2, 116,0, 70,7, 55,8, 47,4, 46,4, 34,8, 25,7, 24,6, 19,6, 18,7, 18,6 м.д. Чистота: >97 % LCMS (214 нм и 254 нм); время удерживания 1,96 мин; (M+1) 381,2.
Пример 68
1-Азабицикло[3.2.2]нонан-4-ил-1-(4'-фторбифенил-4-ил)-циклопропилкарбамат
Применяя общую процедуру E, из 1-азабицикло[3.2.2]нонан-4-ил-1-(4-бромфенил)-циклопропилкарбамата, 4-F-фенилбороновой кислоты и [PdCl2(pddf)]CH2Cl2 получали соединение, указанное в заголовке, в виде белого твердого вещества (27%).1H ЯМР (500 МГц, CDCl3) δ 7,52-7,48 (м, 4H), 7,33-7,28 (м, 2H), 7,14-7,11 (т, J=8,5 Гц, 2H), 5,47-5,33 (д, 1H), 4,93-4,89 (м, 1H), 3,15-2,75 (м, 6H), 2,10-0,88 (м, 11H) м.д.13C ЯМР (125 МГц, CDCl3) δ 163,4, 161,4, 155,7, 142,1, 138,3, 136,9, 128,5, 128,5, 127,0, 125,9, 125,4, 115,7, 115,5, 78,8, 51,7, 48,3, 44,9, 35,2, 33,7, 30,6, 29,7, 24,8, 22,2, 18,1 м.д. Чистота: >99 % LCMS (214 нм и 254 нм); время удерживания 1,56 мин; (M+1) 395,2.
Пример 69
(S)-хинуклидин-3-ил-1-(4-(5-фторпиридин-2-ил)-фенил)-циклопропилкарбамат
Применяя общую процедуру E, из (S)-хинуклидин-3-ил-(1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-фенил)-циклопропил)-карбамата, 2-бром-5-фторпиридина и [PdCl2(pddf)]CH2Cl2 получали соединение, указанное в заголовке, в виде белого твердого вещества (34%).1H ЯМР (500 МГц, CDCl3) δ 8,51-8,52 (д, J=3,5 Гц, 1H), 7,87-7,85 (д, J=10,5 Гц, 2H), 7,69-7,67 (м, 1H), 7,47-7,42 (м, 1H), 7,32-7,27 (м, 2H), 5,79-5,66 (д, 1H), 4,73-4,71 (т, J=5,0 Гц,1H), 3,22-3,19 (м, 1H), 2,87-2,61 (м, 5H), 2,01-1,22 (м, 9H) м.д.13C ЯМР (125 МГц, CDCl3) δ 160,8, 157,4, 156,1, 153,5, 144,4, 137,8, 136,3, 126,7, 125,7, 124,9, 123,6, 121,1, 71,6, 55,7, 47,4, 46,5, 35,3, 29,7, 25,4, 24,8, 19,4, 18,2 м.д. Чистота: >99 % LCMS (214 нм и 254 нм); время удерживания 1,41 мин; (M+1) 382,2.
Препаративная процедура S
Пример 70
(S)-1-(1-(4'-фторбифенил-4-ил)-циклопропил)-3-(3-метилхинуклидин-3-ил)-мочевина
В трехгорлой круглодонной колбе, снабженной двумя выравнивающими давление капельными воронками и резиновой трубкой, соединенной с газовым расходомером, в течение 10 мин перемешивали суспензию 1-(4'-фтор-[1,1'-бифенил]-4-ил)-циклопропанамина (1,50 г, 7,07 ммоль) в смеси 20 мл воды и 1 мл конц. HCl. Добавляли толуол (10 мл) и при интенсивном перемешивании раствор охлаждали до 0°C. В течение 1 часа по каплям добавляли раствор трифосгена в 20 мл толуола и 40 мл насыщенного водного NaHCO3. Реакционную смесь перемешивали в течение дополнительных 30 мин. Перемешивание прекращали, отделяли верхний толуольный слой, сушили (Na2SO4) и концентрировали, получая соответствующий изоцианат, который использовали на следующей стадии без дальнейшей очистки.
К раствору вышеуказанного изоцианата (134 мг, 0,571 ммоль) в 15 мл толуола добавляли (S)-3-метилхинуклидин-3-амин (80 мг, 0,57 ммоль). Смесь, полученную в результате этого, нагревали при кипячении с обратным холодильником в течение ночи, охлаждали до температуры окружающей среды и концентрировали в вакууме, получая остаток, который очищали посредством обращенно-фазовой хроматографий на комбифлэш-картридже (0-20% MeCN в воде), получая соединение, указанное в заголовке, в виде белого твердого вещества (73 мг, 33%).1H ЯМР (500 МГц CDCl3) δ 7,52-7,48 (м, 4H), 7,27-7,25 (д, J=10,0 Гц, 2H), 7,13-7,09 (м, 2H), 5,39 (с, 1H), 4,78 (с, 1H), 2,95-2,71 (м, 5H), 2,65-2,64 (м, 1H), 1,94-1,93 (м, 1H), 1,69-1,68 (м, 1H), 1,46-1,38 (м, 5H), 1,36-1,33 (м, 4H), 1,26-1,23 (м, 1H) м.д.13C ЯМР (125 МГц CDCl3) δ 163,5, 161,5, 157,5, 141,5, 138,5, 136,6, 136,6, 128,5, 128,4, 127,2, 124,7, 115,8, 115,6, 63,8, 52,3, 46,6, 46,3, 34,9, 31,0, 25,0, 23,2, 22,5, 20,2, 20,0 м.д. Чистота: >99 % LCMS (214 нм и 254 нм); время удерживания 1,51 мин; (M+H+) 394,2.
Пример 71
(S)-1-азабицикло[2.2.2]окт-3-ил-[1-(2',4'-дифторбифенил-4-ил)-циклопропил]-карбамат
Применяя общую процедуру E, из (S)-хинуклидин-3-ил-1-(4-бромфенил)-циклопропилкарбамата (0,446 г, 1,22 ммоль), 2,4-дифторфенилбороновой кислоты (0,386 г, 2,44 ммоль) и Pd(OAc)2 (0,015 г, 0,067 ммоль) получали соединение, указанное в заголовке, в виде коричнево-желтого твердого вещества (0,111 г, 23%).1H ЯМР (CDCl3) δ 7,43 (дд, J=8,4, 1,6 Гц, 2H), 7,40-7,33 (м, 1H), 7,31 (д, J=7,7 Гц, 2H), 6,99-6,81 (м, 2H), 5,54 (д, J=48,0 Гц, 1H), 4,82-4,65 (м, 1H), 3,30-3,07 (м, 1H), 2,98-2,44 (м, 5H), 1,97 (д, J=32,7 Гц, 1H), 1,83 (д, J=10,3 Гц, 1H), 1,64 (с, 1H), 1,52 (с, 1H), 1,39 (с, 1H), 1,31 (д, J=6,8 Гц, 4H) м.д.13C ЯМР главного ротамера (CDCl3) δ 162,2 (дд, J=12,8, 249,1 Гц), 159,8 (дд, J=11,8, 251,0 Гц), 156,9, 156,0, 142,6, 133,1, 131,3 (м), 128,9, 125,6, 124,9, 111,5 (дд, J=3,9, 21,2 Гц) 104,4 (дд, J=25,2, 29,4 Гц), 72,1, 71,6, 55,7, 47,4, 46,5, 35,7, 35,3, 25,5, 24,6, 24,4, 19,5, 18,1 м.д. Чистота: LCMS >99,3 % (214 нм и 254 нм); время удерживания 0,90 мин; (M+1) 399,0
Пример 72
1-Азабицикло[2.2.2]окт-3-ил-[1-(4'-метоксибифенил-4-ил)-циклопропил]-карбамат
Применяя общую процедуру E, из хинуклидин-3-ил-1-(4-бромфенил)-циклопропилкарбамата (0,485 г, 1,33 ммоль), 4-метоксифенилбороновой кислоты (0,404 г, 2,66 ммоль) и Pd(OAc)2 (0,016 г, 0,071 ммоль) получали соединение, указанное в заголовке, в виде серого твердого вещества (0,337 мг, 65%).1H ЯМР (CDCl3) δ 7,48 (дд, J=8,6, 5,5 Гц, 4H), 7,29 (д, J=7,6 Гц, 2H), 6,96 (д, J=8,8 Гц, 2H), 5,58 (д, J=48,7 Гц, 1H), 4,83-4,63 (м, 1H), 3,84 (с, 3H), 3,20 (дд, J=24,0, 15,5 Гц, 1H), 2,97-2,42 (м, 5H), 1,97 (д, J=30,9 Гц, 1H), 1,81 (с, 1H), 1,75-1,33 (м, 3H), 1,28 (д, J=6,8 Гц, 4H) м.д.13C ЯМР основной ротомер (CDCl3) δ 159,1, 156,0, 141,4, 139,0, 133,4, 128,0, 126,7, 125,9, 114,2, 71,5, 55,7, 55,3, 47,4, 46,5, 35,3, 25,5, 24,6, 19,6, 17,8 м.д. Чистота: LCMS >97,1 % (214 нм и 254 нм); время удерживания 0,88 мин; (M+1) 393,4.
Препаративная процедура T
Пример 73
Хинуклидин-3-ил-2-(5-(4-фторфенил)-тиофен-3-ил)-пропан-2-илкарбамат
К перемешиваемому и охлаждаемому (0°С) раствору этил-5-бромтиофен-3-карбоксилата (13,30 г, 56,57 ммоль) в THF (100 мл) в течение 20 минут по каплям добавляли раствор метилмагнийбромида в диэтиловом простом эфире [3,0 M] (55,0 мл, 165 ммоль). Спустя 2 часа реакционный раствор концентрировали. Остаток смешивали с водным NH4Cl (200 мл) и экстрагировали этилацетатом (2×100 мл). Объединенные экстракты сушили (Na2SO4) и концентрировали. Масло янтарного цвета, полученное в результате этого, очищали посредством флэш-хроматографии, используя гексан-этилацетатный градиент, получая 2-(5-бромтиофен-3-ил)-пропан-2-ол в виде масла светло-янтарного цвета (8,05 г, 64%).
К перемешиваемому раствору 2-(5-бромтиофен-3-ил)-пропан-2-ола (8,03 г, 36,3 ммоль) в метиленхлориде (80 мл) добавляли азид натрия (7,08 г, 109 ммоль), а затем трифторуксусную кислоту (8,0 мл; по каплям в течение 5-6 минут). Густеющую суспензию перемешивали в течение 1,5 часов, после чего разбавляли водой (350 мл) и экстрагировали этилацетатом (1×200 мл). Органический слой промывали водным NaHCO3 (1×250 мл), сушили (Na2SO4) и концентрировали, получая продукт в виде неочищенного азида. К перемешиваемому раствору этого материла в THF (160 мл) добавляли воду (11 мл), затем трифенилфосфин (23,8 г, 90,7 ммоль). Реакционную смесь премешивали в течение 2 дней и затем концентрировали. Остаток, полученный в результате этого, растворяли в этилацетате (250 мл) и экстрагировали 1 н водной HCl (4×75 мл). Объединенные экстракты подщелачивали концентрированным NH4OH и экстрагировали этилацетатом (2×100 мл). Эти экстракты, в свою очередь, сушили (Na2SO4) и концентрировали. Масло янтарного цвета, полученное в результате этого, очищали посредством флэш-хроматографии, используя метиленхлорид-метанол-аммиачный градиент, получая смесь 2-(5-бромтиофен-3-ил)-пропан-2-амина и трифенилфосфиноксида (в приблизительном соотношении 70/30) в виде вязкого масла янтарного цвета (1,32 г, 17%).
К перемешиваемому раствору 3-хинуклидинола (3,00 г, 23,6 ммоль) в THF (100 мл) добавляли 4-нитрофенилхлорформиат (5,94 г, 29,5). После перемешивания в течение 4 часов, осадок отфильтровывали, ополаскивали THF и сушили на воздухе на фильтре в неглубоком вакууме. Слой на фильтре растворяли в этилацетате (150 мл) и промывали водным NaHCO3 (1×150 мл) и водой (2×150 мл). Органический слой сушили (Na2SO4) и концентрировали, получая продукт в виде неочищенного 4-нитрофенилхинуклидин-3-илкарбоната, который без очистки использовали на следующей стадии.
К перемешиваемому раствору 2-(5-бромтиофен-3-ил)-пропан-2-амина (0,366 г, 1,66 ммоль) в THF (10 мл) добавляли 4-нитрофенилхинуклидин-3-илкарбонат (0,571 г, 1,95 ммоль) и несколько гранул 4-(диметиламино)-пиридина. Смесь кипятили с обратным холодильником в течение ночи, концентрировали и распределяли между этилацетатом (50 мл) и водным NaHCO3 (50 мл). Органический слой опять промывали водным NaHCO3 (1×50 мл), сушили (Na2SO4) и концентрировали. Полученную в результате этого грязновато-желтую смолу очищали посредством флэш-хроматографии, используя хлороформ-метанол-аммиачный градиент и получая хинуклидин-3-ил-(1-(5-бромтиофен-3-ил)-циклопропил)-карбамат в виде светло-серого твердого вещества (0,305 г, 49%).
Применяя общую процедуру E, из хинуклидин-3-ил-(1-(5-бромтиофен-3-ил)-циклопропил)-карбамата (0,227 г, 0,742 ммоль), 4-фторфенилбороновой кислоты (0,208 г, 1,49 ммоль), трициклогексилфосфина (0,021 г, 0,075 ммоль), фосфата калия (0,866, 4,08 ммоль) и ацетат палладия (8,0 мг, 36 мкмоль) получали соединение, указанное в заголовке, в виде твердого вещества серого цвета (0,142 г, 49%).1H ЯМР (400 МГц, CDCl3) δ 7,60-7,45 (м, 2H), 7,24-7,19 (м, 1H), 7,10-6,97 (м, 3H), 5,23 (шир.с, 1H), 4,72-4,61 (м, 1H), 3,30-3,04 (м, 1H), 3,03-2,25 (м, 5H), 2,09-1,02 (м, 11H) м.д.13C ЯМР (400 МГц, CDCl3) δ 162,3 (д, J=247,1 Гц), 154,5, 149,8, 143,6, 130,7, 127,4 (д, J=8,1 Гц), 121,8, 118,9, 115,8 (д, J=21,6 Гц), 70,8, 55,5, 53,4, 47,3, 46,4, 29,0, 25,4, 24,4, 19,4 м.д. Чистота: 95,8 % UPLCMS (210 нм и 254 нм); время удерживания 0,90 мин; (M+1) 389.
Препаративная процедура U
Пример 74
(S)-хинуклидин-3-ил-2-(3-(4-фторфенил)-изотиазол-5-ил)-пропан-2-илкарбамат
К перемешиваемому раствору 2-(3-(4-фторфенил)-изотиазол-5-ил)-пропан-2-амина (1,21 г, 5,12 ммоль) в толуоле добавляли раствор фосгена в толуоле [~1,9 M] (10,8 мл, 20,5 ммоль). Реакционную смесь нагревали при кипячении с обратным холодильником в течение двух часов и затем концентрировали. Остаток выпаривали с толуолом (2×15 мл), получая неочищенное изоцианатное промежуточное соединение в виде масла золотистого цвета. Этот материал смешивали с толуолом (10 мл) и обрабатывали (S)-3-хинуклидинолом (0,749 г, 5,89 ммоль). Реакционную смесь нагревали при кипячении с обратным холодильником в течение ночи и концентрировали. Остаток очищали посредством флэш-хроматографии, используя хлороформ-метанол-аммиачный градиент, получая соединение, указанное в заголовке, в виде белого твердого вещества (0,971 г, 49%).1H ЯМР (400 МГц, DMSO-d6) δ 8,09-8,00 (м, 2H), 7,87 (шир.с, 1H), 7,75 (с, 1H), 7,35-7,25 (м, 2H), 4,54-4,45 (м, 1H), 3,14-2,92 (м, 1H), 2,87-2,17 (м, 5H), 1,98-0,98 (м, 11H) м.д.13C ЯМР (400 МГц, DMSO-d6) δ 180,1, 165,6, 162,6 (д, J=246,4 Гц), 154,7, 131,2 (д, J=3,0 Гц), 128,7 (д, J=8,4 Гц), 118,2, 115,7 (д, J=21,8 Гц), 70,6, 55,3, 52,8, 46,9, 45,9, 29,9, 25,2, 24,2, 19,2 м.д. Чистота: 100 % UPLCMS (210 нм и 254 нм); время удерживания 0,82 мин; (M+1) 390.
Препаративная процедура V
Пример 75
(S)-хинуклидин-3-ил-2-(2-(4-фторфенил)-тиазол-4-ил)-пропан-2-илкарбамат
К перемешиваемому раствору 4-фтортиобензамида (8,94 г, 57,6 ммоль) в этаноле (70 мл) добавляли этил-4-хлорацетоацетат (7,8 мл, 58 ммоль). Реакционную смесь нагревали при кипячении с обратным холодильником в течение 4 часов, обрабатывали добавочной аликвотой этил-4-хлорацетоацетата (1,0 мл, 7,4 ммоль) и кипятили с обратным холодильником в течение дополнительных 3,5 часов. Реакционную смесь затем концентрировали и остаток распределяли между этилацетатом (200 мл) и водным NaHCO3 (200 мл). Органический слой объединяли с обратным экстрактом водного слоя (этилацетат, 1×75 мл), сушили (Na2SO4) и концентрировали. Масло янтарного цвета, полученное в результате этого, очищали посредством флэш-хроматографии, используя гексан-этилацетатный градиент, получая этил-2-(2-(4-фторфенил)-тиазол-4-ил)-ацетат в виде низкоплавкого почти бесцветного твердого вещества (13,58 г, 89%).
К перемешиваемому раствору этил-2-(2-(4-фторфенил)-тиазол-4-ил)-ацетата (6,28 г, 23,7 ммоль) в DMF (50 мл) добавляли гидрид натрия [60%-ная дисперсия в минеральном масле] (2,84 г, 71,0 ммоль). Пенистую смесь перемешивали в течение 15 минут, после чего охлаждали на ледяной бане и добавляли йодметан (4,4 мл, 71 ммоль). Реакционную смесь перемешивали в течение ночи, давая охлаждающей бане возможность медленно нагреваться до комнатной температуры. Затем смесь концентрировали и остаток распределяли между этилацетатом (80 мл) и водой (200 мл). Органический слой промывали второй порцией воды (1×200 мл), сушили (Na2SO4) и концентрировали. Масло янтарного цвета, полученное в результате этого, очищали посредством флэш-хроматографии, используя гексан-этилацетатный градиент, получая этил-2-(2-(4-фторфенил)-тиазол-4-ил)-2-метилпропаноат в виде бесцветного масла (4,57 г, 66%).
К перемешиваемому раствору этил-2-(2-(4-фторфенил)-тиазол-4-ил)-2-метилпропаноата (4,56 г, 15,5 ммоль) в смеси THF с этанолом и водой (1:1:1, 45 мл) добавляли моногидрат гидроксида лития (2,93 г, 69,8 ммоль). Реакционную смесь перемешивали в течение ночи, концентрировали и повторно растворяли в воде (175 мл). Раствор промывали простым эфиром (1×100 мл), подкисляли, добавляя 1,0 н HCl (80 мл) и экстрагировали этилацетатом (2×70 мл). Объединенные экстракты сушили (Na2SO4) и концентрировали, получая 2-(2-(4-фторфенил)-тиазол-4-ил)-2-метилпропановую кислоту в виде белого твердого вещества (4,04 г, 98%). Этот материал без очистки использовали на следующей стадии.
К перемешиваемому и охлаждаемому (0°С) раствору 2-(2-(4-фторфенил)-тиазол-4-ил)-2-метилпропановой кислоты (4,02 г, 15,2 ммоль) в THF (100 мл) добавляли триэтиламин (4,2 мл, 30 ммоль), за которым следовал изобутилхлорформиат (3,0 мл, 23 ммоль). Реакционную смесь перемешивали на холоду в течение еще одного часа, после чего добавляли раствор азида натрия (1,98 г, 30,5 ммоль) в воде (20 мл). Реакционную смесь перемешивали в течение ночи, давая охлаждающей бане возможность медленно нагреваться до комнатной температуры. Затем смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (2×60 мл). Объединенные экстракты промывали водным NaHCO3 (1×150 мл) и насыщенным солевым раствором (1×100 мл), сушили (Na2SO4) и концентрировали. После выпаривания с толуолом (2×50 мл) твердое вещество, полученное в результате этого, смешивали с толуолом (100 мл) и кипятили с обратным холодильником в течение 4 часов. Затем добавляли (S)-3-хинуклидинол (3,87 г, 30,4 ммоль) и кипячение с обратным холодильником продолжали в течение ночи. Реакционную смесь концентрировали и остаток распределяли между этилацетатом (100 мл) и водным NaHCO3 (150 мл). Органический слой промывали водой (1×150 мл), сушили (Na2SO4) и концентрировали. Светло-серое твердое вещество, полученное в результате этого, очищали посредством флэш-хроматографии, используя хлороформ-метанол-аммиачный градиент, получая соединение, указанное в заголовке, в виде белого твердого вещества (4,34 г, 73%).1H ЯМР (400 МГц, CDCl3) δ 7,96-7,88 (м, 2H), 7,16-7,04 (м, 3H), 5,55 (шир.с, 1H), 4,69-4,62 (м, 1H), 3,24-3,11 (м, 1H), 3,00-2,50 (м, 5H), 2,01-1,26 (м, 11H) м.д.13C ЯМР (400 МГц, CDCl3) δ 166,4, 165,1, 163,8 (д, J=250,3 Гц), 162,9, 155,0, 130,1 (д, J=3,3 Гц), 128,4 (д, J=8,5 Гц), 115,9 (д, J=22,3 Гц), 112,5, 71,2, 55,7, 54,2, 47,5, 46,5, 28,0, 25,5, 24,7, 19,6 м.д. Чистота: 100 % UPLCMS (210 нм и 254 нм); время удерживания 0,83 мин; (M+1) 390.
Препаративная процедура W
Пример 76
(S)-хинуклидин-3-ил-2-(4-(4-фторфенил)-тиазол-2-ил)-пропан-2-илкарбамат
К перемешиваемому раствору этил-3-амино-3-тиоксопропаноата (20,00 г, 135,9 ммоль) в этаноле (120 мл) добавляли 2-бром-4'-фторацетофенон (29,49 г, 135,9 ммоль). Смесь кипятили с обратным холодильником в течение 1 часа, концентрировали и распределяли между этилацетатом (300 мл) и водным NaHCO3 (400 мл). Органический слой объединяли с обратным экстрактом водного слоя (этилацетат, 1×100 мл), сушили (Na2SO4) и концентрировали. Светло-коричневое твердое вещество, полученное в результате этого, очищали посредством флэш-хроматографии, используя гексан-этилацетатный градиент, получая этил-2-(4-(4-фторфенил)-тиазол-2-ил)-ацетат в виде светло-серого твердого вещества (29,92 г, 83%).
К перемешиваемому и охлаждаемому (-78°C) раствору этил-2-(4-(4-фторфенил)-тиазол-2-ил)-ацетата (10,00 г, 37,69 ммоль) в THF (250 мл) в течение 15 минут добавляли по каплям раствор трет-бутоксида калия в THF [1,0 M] (136 мл, 136 ммоль), за которым следовал 18-краун-6 (1,6 мл, 7,5 ммоль). Спустя еще 30 минут при -78°C в течение 5 минут добавляли по каплям йодметан (8,5 мл). Реакционную смесь перемешивали на холоду еще в течение 2 часов, после чего выливали в воду (450 мл) и экстрагировали этилацетатом (2×150 мл). Объединенные экстракты промывали насыщенным солевым раствором (1×200 мл), сушили (Na2SO4) и концентрировали. Коричневое масло, полученное в результате этого, очищали посредством флэш-хроматографии, используя гексан-этилацетатный градиент, получая этил-2-(4-(4-фторфенил)-тиазол-2-ил)-2-метилпропаноат в виде масла светло-янтарного цвета (8,64 г, 78%).
К перемешиваемому раствору этил-2-(4-(4-фторфенил)-тиазол-2-ил)-2-метилпропаноата (0,900 г, 3,07 ммоль) в смеси THF с этанолом и водой (1:1:1, 15 мл) добавляли моногидрат гидроксида лития (0,451 г, 10,7 ммоль). После перемешивания в течение ночи реакционную смесь концентрировали и повторно растворяли в воде (80 мл). Раствор промывали простым эфиром (1×50 мл), подкисляли, добавляя 1 н HCl (15 мл) и экстрагировали этилацетатом (2×50 мл). Объединенные экстракты сушили (Na2SO4) и концентрировали, получая 2-(4-(4-фторфенил)-тиазол-2-ил)-2-метилпропановую кислоту в виде твердого вещества светло-золотистого цвета (0,808 г, 99%).
К перемешиваемому и охлаждаемому (0°С) раствору 2-(4-(4-фторфенил)-тиазол-2-ил)-2-метилпропановой кислоты (0,784 г, 2,96 ммоль) в THF (25 мл) добавляли триэтиламин (0,82 мл, 5,9 ммоль), за которыми следовал изобутилхлорформиат (0,58 мл, 4,4 ммоль). Реакционную смесь перемешивали на холоду в течение еще одного часа, после чего добавляли раствор азида натрия (0,385 г, 5,92 ммоль) в воде (7 мл). Реакционную смесь перемешивали в течение ночи, давая охлаждающей бане медленно нагреваться до комнатной температуры. Смесь затем разбавляли водой (100 мл) и экстрагировали этилацетатом (2×60 мл). Объединенные экстракты промывали водным NaHCO3 (1×150 мл) и насыщенным солевым раствором (1×100 мл), сушили (Na2SO4) и концентрировали. После выпаривания с толуолом (2×30 мл) твердое вещество светло-серого цвета, полученное в результате этого, смешивали с толуолом (25 мл) и кипятили с обратным холодильником в течение 4 часов. Затем добавляли (S)-3-хинуклидинол (0,753 г, 5,92 ммоль) и кипячение с обратным холодильником продолжали в течение 3 часов. Реакционную смесь концентрировали и остаток очищали посредством флэш-хроматографии, используя хлороформ-метанол-аммиачный градиент, получая соединение, указанное в заголовке, в виде белого твердого вещества (0,793 г, 69%).1H ЯМР (400 МГц, CDCl3) δ 7,90-7,81 (м, 2H), 7,32 (с, 1H), 7,14-7,05 (м, 2H), 5,76 (шир.с, 1H), 4,72-4,65 (м, 1H), 3,26-3,10 (м, 1H), 3,03-2,37 (м, 5H), 2,05-1,23 (м, 11H) м.д.13C ЯМР (400 МГц, CDCl3) δ 177,6, 162,6 (д, J=248,4 Гц), 154,8, 153,6, 130,8 (д, J=3,2 Гц), 128,1 (д, J=8,1 Гц), 115,9 (д, J=21,7 Гц), 112,2, 71,6, 55,7, 47,4, 46,5, 29,1, 25,4, 24,7, 19,6 м.д. Чистота: 100 % UPLCMS (210 нм и 254 нм); время удерживания 0,82 мин; (M+1) 390.
Пример 77
Хинуклидин-3-ил-1-(4-(бензилокси)-фенил)-циклопропилкарбамат
Смесь 4-цианофенола (5,0 г, 42 ммоль), бензилбромида (8,6 г, 50 ммоль), карбоната калия (11,6 г, 84,0 ммоль) в DMF (40 мл) перемешивали при 100°C в течение 3 часов. Осадок отфильтровывали и фильтрат разбавляли EtOAc и промывали водой. Органический слой сушили (Na2SO4) и концентрировали. Неочищенный продукт, полученный в результате этого, очищали посредством хроматографии на колонке с силикагелем (элюируя смесью петролейного эфира с EtOAc от 20/1 до 5/1), получая 4-(бензилокси)-бензонитрил в виде белого твердого вещества (8,1 г, 92%).
Применяя общую процедуру G, 4-(бензилокси)-бензонитрил (6,00 г, 28,7 ммоль) преобразовывали в соответствующий 1-(4-(бензилокси)-фенил)-циклопропанамин, получаемый в виде желтого твердого вещества (1,8 г, 26%).
Применяя общую процедуру A, из 1-(4-(бензилокси)-фенил)-циклопропанамина (600 мг, 2,51 ммоль) и хинуклидин-3-ола получали соединение, указанное в заголовке, в виде вязкого масла (170 мг, 17%).1H ЯМР (500 МГц, CDCl3) δ 7,34 (д, J=7,0 Гц, 2H), 7,30 (т, J=7,0 Гц, 2H), 7,25 (т, J=7,0 Гц, 1H), 7,16 (д, J=8,5 Гц, 1H), 7,07 (д, J=7,0 Гц, 1H), 6,83 (д, J=8,0 Гц, 2H), 5,50 (шир.с, 0,6H), 5,40 (шир.с, 0,4H), 4,96 (с, 2H), 4,64 (м, 1H), 3,20-3,15 (м, 1H), 2,88-2,50 (м, 5H), 1,95-1,05 (м, 9H) м.д.13C ЯМР (125 МГц, CDCl3) δ 156,6, 154,8, 136,0, 134,2, 127,6, 126,9, 126,4, 125,4, 113,7, 70,0, 69,0, 54,5, 46,3, 45,4, 34,2, 24,3, 23,3, 18,3, 15,8 м.д. Чистота: >90 % LCMS (214 нм и 254 нм); время удерживания 1,57 мин; (M+1) 393.
Пример 78
Хинуклидин-3-илбифенил-3-илметилкарбамат
К перемешиваемому и охлаждаемому (0°С) раствору трифосгена (0,80 г, 2,7 ммоль) в THF (20 мл) в течение 2 часов добавляли по каплям смесь (3-бромфенил)-метанамина (1,0 г, 5,4 ммоль) и триэтиламина (1,08 г, 10,7 ммоль) в THF (30 мл). После завершения этого добавления смесь кипятили с обратным холодильником в течение 1 часа и затем охлаждали до комнатной температуры. Добавляли хинуклидин-3-ол (1,40 г, 10,7 ммоль) и смесь кипятили с обратным холодильником в течение 18 часов. Растворитель удаляли в вакууме, и остаток растворяли в EtOAc, промывали водой, сушили над Na2SO4 и концентрировали. Неочищенный остаток очищали посредством хроматографии на колонке с силикагелем (элюируя смесью EtOAc/метанол, 10/1), получая хинуклидин-3-ил-3-бромбензилкарбамат в виде бесцветной жидкости (0,68 г, 37%).
Применяя общую процедуру E, из хинуклидин-3-ил-3-бромбензилкарбамата (237 мг, 0,700 ммоль), фенилбороновой кислоты (171 мг, 1,4 ммоль) и [PdCl2(pddf)]CH2Cl2 получали соединение, указанное в заголовке, в виде вязкого полужидкого вещества (110 мг, 47 %).1H ЯМР (500 МГц, CDCl3) δ 7,57 (д, J=7,5 Гц, 2H), 7,50 (м, 2H), 7,62-7,38 (м, 3H), 7,35 (т, J=7,5 Гц, 1H), 7,28 (д, J=8,0 Гц, 1H), 5,32-5,17 (м, 1H), 7,78 (м, 1H), 4,42 (д, J=6,0 Гц, 2H), 3,26 (м, 1H), 2,95-2,65 (м, 5H), 2,05 (м, 1H), 1,84 (м, 1H), 1,70 (м, 1H), 1,58 (м, 1H), 1,42 (м, 1H) м.д.13C ЯМР (125 МГц, CDCl3) δ 155,2, 140,7, 139,8, 138,0, 128,1, 127,8, 126,4, 126,1, 125,5, 125,4, 125,3, 70,1, 54,4, 46,2, 45,3, 44,1, 24,3, 23,1, 18,2 м.д. Чистота: >98 % LCMS (214 нм и 254 нм); время удерживания 1,44 мин; (M+1) 337.
Пример 79
Хинуклидин-3-ил-3-(пиримидин-5-ил)-бензилкарбамат
Применяя общую процедуру E, из хинуклидин-3-ил-3-бромбензилкарбамата (203 мг, 0,600 ммоль), пиримидин-5-илбороновой кислоты (149 мг, 1,2 ммоль) и [PdCl2(pddf)]CH2Cl2 получали соединение, указанное в заголовке, в виде вязкого полужидкого вещества (110 мг, 54%).1H ЯМР (500 МГц, CDCl3) δ 9,20 (с, 1H), 8,94 (с, 2H), 7,51 (м, 3H), 7,40 (м, 1H), 5,62 (м, 1H), 4,81 (м, 1H), 4,50-4,40 (м, 2H), 3,30 (м, 1H), 2,97-2,65 (м, 5H), 2,12 (м, 1H), 1,92-1,82 (м, 1H), 1,79-1,69 (м, 1H), 1,65-1,56 (м, 1H), 1,50-1,42 (м, 1H) м.д.13C ЯМР (125 МГц, CDCl3) δ 157,6, 156,2, 154,9, 140,1, 134,7, 134,1, 129,8, 128,2, 126,2, 70,9, 55,2, 47,2, 46,2, 44,8, 25,2, 23,8, 19,0 м.д. Чистота: >95 % LCMS (214 нм и 254 нм); время удерживания 1,22 мин; (M+1) 339.
Пример 80
Хинуклидин-3-ил-3-(бензилокси)-бензилкарбамат
Смесь 2-(3-гидроксифенил)-уксусной кислоты (0,6 г, 3,95 ммоль), бензил бромида (0,710 г, 4,14 ммоль), гидроксида калия (0,550 г, 9,87 ммоль) и KI (13 мг, 0,079 ммоль) в THF (20 мл) кипятили с обратным холодильником в течение 18 часов. Растворитель удаляли и остаток растворяли в 50 мл воды и экстрагировали простым эфиром. Водный слой подкисляли 1 н водной HCl и отфильтровывали образовавшийся белый осадок, получая 2-(3-(бензилокси)-фенил)-уксусную кислоту в виде твердого вещества серого цвета (0,87 г, 91%).
Применяя общую процедуру H, из 2-(3-(бензилокси)-фенил)-уксусной кислоты (242 мг, 1,00 ммоль) и хинуклидин-3-ола получали соединение, указанное в заголовке, в виде вязкого полужидкого вещества (200 мг, 55%).1H ЯМР (500 МГц, CDCl3) δ 7,42 (д, J=7,5 Гц, 2H), 7,38 (т, J=7,5 Гц, 2H), 7,32 (т, J=7,0 Гц, 1H), 7,24 (д, J=7,5 Гц, 1H), 6,92 (с, 1H), 6,88 (д, J=7,0 Гц, 2H), 5,30 (м, 1H), 5,05 (с, 2H), 4,75 (м, 1H), 4,32 (д, J=6,0 Гц, 2H), 3,23 (м, 1H), 2,93-2,60 (м, 5H), 2,08-1,96 (м, 1H), 1,88-1,75 (м, 1H), 1,72-1,62 (м, 1H), 1,60-1,50 (м, 1H), 1,42-1,34 (м, 1H) м.д.13C ЯМР (125 МГц, CDCl3) δ 159,1, 156,3, 140,2, 136,8, 129,7, 128,6, 128,0, 127,5, 120,0, 114,1, 113,6, 71,3, 70,0, 55,5, 47,3, 46,4, 45,0, 25,4, 24,3, 19,3 м.д. Чистота: >95 % LCMS (214 нм и 254 нм); время удерживания 1,51 мин; (M+1) 367
Пример 81
Хинуклидин-3-ил-4-феноксибензилкарбамат
Применяя общую процедуру H, из 2-(3-феноксифенил)-уксусной кислоты (228 мг, 1,00 ммоль) и хинуклидин-3-ола получали соединение, указанное в заголовке, в виде вязкого полужидкого вещества (70 мг, 20%).1H ЯМР (500 МГц, CDCl3) δ 7,29-7,18 (м, 3H), 7,03 (т, J=7,5 Гц, 1H), 6,96-6,90 (м, 3H), 6,86 (с, 1H), 6,82 (д, J=8,5 Гц, 1H), 5,40-5,15 (м, 1H), 4,70 (м, 1H), 4,25 (д, J=6,0 Гц, 2H), 3,18 (м, 1H), 2,90-2,60 (м, 5H), 2,03-1,92 (м, 1H), 1,82-1,74 (м, 1H), 1,68-1,60 (м, 1H), 1,57-1,45 (м, 1H), 1,40-1,32 (м, 1H) м.д.13C ЯМР (125 МГц, CDCl3) δ 156,6, 155,9, 155,1, 139,6, 129,0, 128,8, 122,4, 121,1, 118,0, 116,7, 69,7, 54,1, 46,1, 45,2, 43,7, 24,2, 22,7, 18,0 м.д. Чистота: 100 % LCMS (214 нм и 254 нм); время удерживания 1,50 мин; (M+1) 353.
Пример 82
Хинуклидин-3-ил-3-изопропоксибензилкарбамат
Смесь, содержавшую 2-(3-гидроксифенил)-уксусную кислоту (0,800 г, 5,26 ммоль), 2-бромпропан (0,971 г, 7,89 ммоль), гидроксид калия (0,740 г, 13,2 ммоль) и KI (18 мг, 0,11 ммоль) в 20 мл EtOH, кипятили с обратным холодильником в течение 18 часов. Растворитель удаляли и остаток растворяли в 50 мл воды и экстрагировали простым эфиром. Водный слой подкисляли 1 н водной HCl и экстрагировали EtOAc. Органические слои сушили (Na2SO4) и концентрировали, получая остаток, который очищали посредством хроматографии на силикагеле (петролейный эфир/EtOAc, 4:1), получая 2-(3-(бензилокси)-фенил)-уксусную кислоту в виде белого твердого вещества (0,45 г, 44%).
Применяя общую процедуру H, из 2-(3-изопропоксифенил)-уксусной кислоты (291 мг, 1,50 ммоль) и хинуклидин-3-ола получали соединение, указанное в заголовке, в виде вязкого полужидкого вещества (120 мг, 25%).1H-ЯМР (500 МГц, CDCl3) δ 7,23 (т, J=7,5 Гц, 1H), 6,86-6,77 (м, 3H), 5,16-5,00 (м, 1H), 4,78 (м, 1H), 4,55 (м, 1H), 4,32 (д, J=5,0 Гц, 2H), 3,26 (м, 1H), 2,95-2,70 (м, 5H), 2,10-2,05 (м, 1H), 1,90-1,80 (м, 1H), 1,75-1,65 (м, 1H), 1,63-1,53 (м, 1H), 1,47-1,37 (м, 1H), 1,33 (д, J=5,5 Гц, 6H) м.д.13C ЯМР (125 МГц, CDCl3) δ 158,1, 156,2, 140,1, 129,7, 119,6, 115,2, 114,6, 71,0, 69,8, 55,3, 47,2, 46,3, 45,0, 25,3, 24,1, 22,0, 19,2 м.д. Чистота: >90 % LCMS (214 нм и 254 нм); время удерживания 1,42 мин; (M+1) 319.
Пример 83
Хинуклидин-3-ил-3-изобутоксибензилкарбамат
Смесь, содержавшую 2-(3-гидроксифенил)-уксусную кислоту (1,0 г, 6,6 ммоль), 1-бром-2-метилпропан (1,08 г, 7,91 ммоль), гидроксид калия (0,920 г, 16,4 ммоль) и KI (22 мг, 0,13 ммоль) в EtOH (20 мл), кипятили с обратным холодильником в течение 18 часов. Растворитель удаляли и остаток растворяли в 50 мл воды и экстрагировали простым эфиром. Водный слой подкисляли 1 н водной HCl и экстрагировали EtOAc. Органические слои сушили (Na2SO4) и концентрировали, получая остаток, который очищали посредством хроматографии на силикагеле (петролейный эфир/EtOAc, 4:1), получая 2-(3-(бензилокси)-фенил)-уксусную кислоту в виде белого твердого вещества (0,42 г, 31%).
Применяя общую процедуру H, из 2-(3-изобутоксифенил)-уксусной кислоты (208 мг, 1,00 ммоль) и хинуклидин-3-ола получали соединение, указанное в заголовке, в виде вязкого полужидкого вещества (130 мг, 39%).1H ЯМР (500 МГц, CDCl3) δ 7,23 (т, J=7,5 Гц, 1H), 6,86-6,76 (м, 3H), 5,35-5,10 (м, 1H), 4,77 (м, 1H), 4,31 (д, J=5,5 Гц, 2H), 3,69 (д, J=6,5 Гц, 2H), 3,26 (м, 1H), 2,95-2,70 (м, 5H), 2,10-2,00 (м, 2H), 1,88-1,80 (м, 1H), 1,75-1,63 (м, 1H), 1,62-1,52 (м, 1H), 1,45-1,36 (м, 1H), 1,01 (д, J=6,5 Гц, 6H) м.д.13C ЯМР (125 МГц, CDCl3) δ 159,6, 156,1, 139,9, 129,7, 119,6, 113,9, 113,4, 74,4, 70,9, 55,3, 47,2, 46,3, 45,1, 28,3, 25,3, 23,9, 19,3, 19,1 м.д. Чистота: >95 % LCMS (214 нм и 254 нм); время удерживания 1,50 мин; (M+1) 333.
Пример 84
Хинуклидин-3-ил-3-(циклопропилметокси)-бензилкарбамат
Смесь, содержавшую 2-(3-гидроксифенил)-уксусную кислоту (1,0 г, 6,6 ммоль), (бромметил)-циклопропан (0,97 г, 7,2 ммоль), гидроксид калия (0,920 г, 16,4 ммоль) и KI (22 мг, 0,13 ммоль) в EtOH (20 мл), кипятили с обратным холодильником в течение 18 часов. Растворитель удаляли в вакууме и остаток растворяли в 50 мл воды и экстрагировали простым эфиром. Водный слой подкисляли 1 н водной HCl и экстрагировали EtOAc. Органические слои сушили (Na2SO4) и концентрировали, получая остаток, который очищали посредством хроматографии на силикагеле (петролейный эфир/EtOAc 4:1), получая 2-(3-(циклопропилметокси)-фенил)-уксусную кислоту в виде белого твердого вещества (0,80 г, 59%).
Применяя общую процедуру H, из 2-(3-(циклопропилметокси)-фенил)-уксусной кислоты (300 мг, 1,50 ммоль) и хинуклидин-3-ола получали соединение, указанное в заголовке, в виде вязкого масла (90 мг, 19%).1H ЯМР (500 МГц, CDCl3) δ 7,24 (т, J=7,5 Гц, 1H), 6,88-6,78 (м, 3H), 5,13-4,95 (м, 1H), 4,74 (м, 1H), 4,33 (д, J=6,0 Гц, 2H), 3,79 (д, J=7,0 Гц, 2H), 3,23 (м, 1H), 2,93-2,63 (м, 5H), 2,04-1,98 (м, 1H), 1,85-1,76 (м, 1H), 1,72-1,60 (м, 1H), 1,58-1,50 (м, 1H), 1,41-1,22 (м, 2H), 0,68-0,62 (м, 2H), 0,37-0,32 (м, 2H) м.д.13C ЯМР (125 МГц, CDCl3) δ 158,2, 155,5, 139,3, 128,6, 118,6, 112,8, 112,3, 71,7, 70,4, 54,5, 46,2, 45,3, 43,9, 24,4, 23,5, 18,5, 9,3, 2,2 м.д. Чистота: >95 % LCMS (214 нм и 254 нм); время удерживания 1,44 мин; (M+1) 331.
Препаративная процедура X
Пример 85
N-(2-(бифенил-4-ил)-пропан-2-ил)-2-(хинуклидин-3-ил)-ацетамид
К раствору гидрохлорида 2-(хинуклидин-3-ил)-уксусной кислоты (0,97 г, 4,7 ммоль) в DMF (30 мл) добавляли HATU (1,79 г, 4,72 ммоль), 2-(4-бромфенил)-пропан-2-амин (1,0 г, 4,7 ммоль) и триэтиламин (3,9 мл, 28 ммоль). Смесь, полученную в результате этого, перемешивали при 60°C в течение 16 часов. Смесь концентрировали в вакууме, разбавляли EtOAc и промывали насыщенным солевым раствором. Органический слой сушили над Na2SO4 и выпаривали, получая неочищенный продукт, который очищали посредством хроматографии на колонке с силикагелем (EtOAc/метанол, от 50/1 до 3/1), получая N-(2-(4-бромфенил)-пропан-2-ил)-2-(хинуклидин-3-ил)-ацетамид в виде желтого твердого вещества (1,3 г, 76%).
Применяя общую процедуру E, из N-(2-(4-бромфенил)-пропан-2-ил)-2-(хинуклидин-3-ил)-ацетамида (200 мг, 0,550 ммоль), фенилбороновой кислоты (134 мг, 1,00 ммоль) и [PdCl2(pddf)]CH2Cl2 получали соединение, указанное в заголовке, в виде вязкого коричневого масла (58 мг, 32%).1H ЯМР (500 МГц, CDCl3) δ 7,58-7,50 (м, 4H), 7,44-7,37 (м, 4H), 7,31 (т, J=7,0 Гц, 1H), 6,50 (с, 1H), 3,16 (м, 1H), 3,02 (м, 1H), 2,92-2,78 (м, 3H), 2,60 (м, 1H), 2,40-2,20 (м, 3H), 1,47-1,90 (м, 11H) м.д.13C ЯМР (125 МГц, CDCl3) δ 170,5, 146,1, 140,7, 139,2, 128,8, 127,2, 127,0 125,2, 55,6, 53,1, 46,8, 46,2, 40,3, 31,7, 29,3, 29,2, 26,0, 24,4, 19,7 м.д. Чистота: 100 % LCMS (214 нм и 254 нм); время удерживания 1,55 мин; (M+1) 363.
Пример 86
Хинуклидин-3-илбифенил-3-илкарбамат
К раствору хинуклидин-3-ола (635 мг, 5,00 ммоль) в THF (15 мл) добавляли NaH [60%-ная дисперсия в минеральном масле] (260 мг, 6,50 ммоль) при комнатной температуре. Смесь перемешивали в течение 15 мин и при перемешивании добавляли 3-бромфенилизоцианат (990 мг, 5,00 ммоль). Смесь, полученную в результате этого, перемешивали при комнатной температуре в течение 18 часов, гасили насыщенным солевым раствором и экстрагировали EtOAc. Органические слои объединяли, сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали посредством хроматографии на колонке с силикагелем (элюируя смесью EtOAc/метанол, 3:1), получая хинуклидин-3-ил-3-бромфенилкарбамат в виде белого твердого вещества (0,70 г, 43%).
Применяя общую процедуру E, из вышеуказанного карбаматного промежуточного соединения (130 мг, 0,402 ммоль), фенилбороновой кислоты (72 мг, 0,6 ммоль) и [PdCl2(pddf)]CH2Cl2 получали соединение, указанное в заголовке, в виде белого твердого вещества (75 мг, 58%).1H ЯМР (500 МГц, CDCl3) δ 7,67 (шир.с, 1H), 7,59 (д, J=7,5 Гц, 2H), 7,43 (т, J=7,5 Гц, 2H), 7,41-7,28 (м, 4H), 6,77 (шир.с, 1H), 4,85 (м, 1H), 3,30 (м, 1H), 2,98-2,75 (м, 5H), 2,12 (м, 1H), 1,93-1,68 (м, 2H), 1,64-1,55 (м, 1H), 1,47-1,40 (м, 1H) м.д.13C ЯМР (125 МГц, CDCl3) δ 153,3, 142,3, 140,7, 138,3, 129,5, 128,8, 127,5, 127,2, 122,3, 117,4, 72,1, 55,4, 47,4, 46,5, 30,9, 25,4, 24,5, 19,5 м.д. Чистота: 100 % LCMS (214 нм и 254 нм); время удерживания 1,53 мин; (M+1) 323.
Пример 87
Хинуклидин-3-ил-2'-метоксибифенил-3-илкарбамат
Применяя общую процедуру E, из хинуклидин-3-ил-3-бромфенилкарбамата, 2-метокси-фенилбороновой кислоты и [PdCl2(pddf)]CH2Cl2 получали соединение, указанное в заголовке, в виде белого твердого вещества (75 мг, 58%).1H ЯМР (500 МГц, CDCl3) δ 7,49 (шир.с, 1H), 7,41 (шир.с, 1H), 7,37-7,28 (м, 3H), 7,23 (д, J=7,5 Гц, 1H), 7,04-6,94 (м, 3H), 4,83 (м, 1H), 3,80 (с, 3H), 3,29 (м, 1H), 2,97-2,70 (м, 5H), 2,10 (м, 1H), 1,91-1,82 (м, 1H), 1,74-1,65 (м, 1H), 1,62-1,53 (м, 1H), 1,46-1,37 (м, 1H) м.д.13C ЯМР (125 МГц, CDCl3) δ 156,4, 153,4, 139,4, 137,6, 130,9, 130,2, 128,8, 128,6, 124,8, 120,8, 119,9, 117,3, 111,2, 72,0, 55,6, 55,4, 47,4, 46,5, 25,4, 24,5, 19,5 м.д. Чистота: >95 % LCMS (214 нм и 254 нм); время удерживания 1,52 мин; (M+1) 353.
Пример 88
Хинуклидин-3-ил-2'-этилбифенил-3-илкарбамат
Применяя общую процедуру E, из хинуклидин-3-ил-3-бромфенилкарбамата, 2-этилфенилбороновой кислоты и [PdCl2(pddf)]CH2Cl2 получали соединение, указанное в заголовке, в виде белого твердого вещества (110 мг, 78%).1H ЯМР (400 МГц, CDCl3) δ 7,42-7,28 (м, 5H), 7,25-7,16 (м, 2H), 7,03-7,00 (м, 1H), 6,88 (шир.с, 1H), 4,83 (м, 1H), 3,27 (м, 1H), 2,98-2,70 (м, 5H), 2,61 (кв., J=7,6 Гц, 2H), 2,08 (м, 1H), 1,92-1,80 (м, 1H), 1,75-1,65 (м, 1H), 1,63-1,55 (м, 1H), 1,46-1,37 (м, 1H), 1,10 (т, J=7,6 Гц, 3H) м.д.13C ЯМР (100 МГц, CDCl3) δ 152,1, 141,7, 140,4, 139,9, 136,4, 128,6, 127,5, 127,4, 126,4, 124,3, 123,2, 118,4, 115,9, 71,0, 54,3, 46,2, 45,3, 25,0, 24,2, 23,4, 18,3, 14,5 м.д. Чистота: 100 % LCMS (214 нм и 254 нм); время удерживания 1,61 мин; (M+1) 351.
Пример 89
Хинуклидин-3-ил-3'-метоксибифенил-3-илкарбамат
Применяя общую процедуру E, из хинуклидин-3-ил-3-бромфенилкарбамата, 3-метоксифенилбороновой кислоты и [PdCl2(pddf)]CH2Cl2 получали соединение, указанное в заголовке, в виде белого твердого вещества (100 мг, 71%).1H ЯМР (500 МГц, CDCl3) δ 7,63 (шир.с, 1H), 7,40-7,27 (м, 4H), 7,17 (д, J=8,0 Гц, 1H), 7,11 (м, 1H), 7,07 (шир.с, 1H), 6,89 (дд, J=8,0, 2,0 Гц, 1H), 4,85 (м, 1H), 3,85 (с, 3H), 3,30 (м, 1H), 2,99-2,70 (м, 5H), 2,12 (м, 1H), 1,92-1,84 (м, 1H), 1,75-1,68 (м, 1H), 1,62-1,55 (м, 1H), 1,48-1,40 (м, 1H) м.д.13C ЯМР (125 МГц, CDCl3) δ 159,9, 153,4, 142,3, 142,1, 138,4, 129,8, 129,4, 122,3, 119,7, 117,7, 112,9, 112,8, 72,0, 55,4, 55,3, 47,4, 46,5, 25,4, 24,5, 19,5 м.д. Чистота: >97 % LCMS (214 нм и 254 нм); время удерживания 1,52 мин; (M+1) 353
Пример 90
Хинуклидин-3-ил-3'-этилбифенил-3-илкарбамат
К раствору 1-бром-3-этилбензола (370 мг, 2,00 ммоль) в 5 мл 1,4-диоксана добавляли бис-(пинаколато)-диборан (609 мг, 2,40 ммоль), CH3COOK (589 мг, 6,02 ммоль) и [PdCl2(pddf)]CH2Cl2 (75 мг, 0,09 ммоль). Смесь перемешивали при 80°С в течение 5 часов. Смесь охлаждали, разбавляли водой и экстрагировали EtOAc. Объединенные экстракты сушили (Na2SO4) и концентрировали, получая неочищенный боронат (410 мг, >100%), который без очистки использовали на следующей стадии.
Применяя общую процедуру E, из хинуклидин-3-ил-3-бромфенилкарбамата, 3-этилфенилбороновой кислоты и [PdCl2(pddf)]CH2Cl2 получали соединение, указанное в заголовке, в виде белого твердого вещества (78 мг, 56%).1H ЯМР (500 МГц, CDCl3) δ 7,64 (шир.с, 1H), 7,43-7,27 (м, 6H), 7,24 (шир.с, 1H), 7,18 (д, J=8,0 Гц, 1H), 4,85 (м, 1H), 3,30 (м, 1H), 2,99-2,73 (м, 5H), 2,70 (кв., J=7,5 Гц, 2H), 2,12 (м, 1H), 1,92-1,84 (м, 1H), 1,75-1,67 (м, 1H), 1,62-1,55 (м, 1H), 1,48-1,38 (м, 1H), 1,27 (т, J=7,5 Гц, 3H) м.д.13C ЯМР (125 МГц, CDCl3) δ 153,5, 144,8, 142,4, 140,8, 138,4, 129,4, 128,8, 127,1, 126,8, 124,6, 122,3, 117,4, 72,1, 55,4, 47,4, 46,5, 29,0, 25,4, 24,5, 19,5, 15,7 м.д. Чистота: >98 % LCMS (214 нм и 254 нм); время удерживания 1,66 мин; (M+1) 351.
Пример 91
Хинуклидин-3-илбифенил-2-илкарбамат
К раствору хинуклидин-3-ола (382 мг, 3,00 ммоль) в THF (15 мл) при комнатной температуре добавляли NaH [60%-ная дисперсия в минеральном масле] (156 мг, 3,90 ммоль). Смесь перемешивали в течение 15 мин и при перемешивании добавляли 2-бромфенилизоцианат (594 мг, 3,00 ммоль). Смесь, полученную в результате этого, перемешивали при комнатной температуре в течение 18 часов, гасили насыщенным солевым раствором и экстрагировали EtOAc. Органические слои объединяли, сушили над Na2SO4 и концентрировали. Неочищенный продукт, полученный в результате этого, очищали посредством хроматографии на колонке с силикагелем (EtOAc/метанол, 3:1), получая в качестве продукта хинуклидин-3-ил-2-бромфенилкарбамат в виде вязкого масла (0,80 г, 82%).
Применяя общую процедуру E, из хинуклидин-3-ил-2-бромфенилкарбамата (130 мг, 0,400 ммоль), фенилбороновой кислоты (96 мг, 0,8 ммоль) и [PdCl2(pddf)]CH2Cl2 получали соединение, указанное в заголовке, в виде белого твердого вещества (112 мг, 87%).1H ЯМР (400 МГц, CDCl3) δ 8,07 (шир.с, 1H), 7,55-7,33 (м, 6H), 7,25-7,21 (дд, J=7,6 и 1,6 Гц, 1H), 7,43 (тд, J=8,0, 1,2 Гц, 1H), 6,65 (шир.с, 1H), 4,78 (м, 1H), 3,24 (м, 1H), 2,90-2,68 (м, 5H), 2,04 (м, 1H),1,80-1,62 (м, 2H), 1,61-1,50 (м, 1H), 1,41-1,30 (м, 1H) м.д.13C ЯМР (100 МГц, CDCl3) δ 151,2, 135,9, 132,5, 129,5, 128,0, 127,0, 126,9, 126,2, 125,7, 121,4, 117,9, 69,9, 53,1, 45,1, 44,3, 23,1, 22,3, 17,2 м.д. Чистота: 100 % LCMS (214 нм и 254 нм); время удерживания 1,47 мин; (M+1) 323.
Пример 92
Хинуклидин-3-ил-2'-метоксибифенил-2-илкарбамат
Применяя общую процедуру E, из хинуклидин-3-ил-2-бромфенилкарбамата, 2-метоксифенилбороновой кислоты и [PdCl2(pddf)]CH2Cl2 получали соединение, указанное в заголовке, в виде белого твердого вещества (102 мг, 72%).1H ЯМР (500 МГц, CDCl3) δ 7,95 (шир.с, 1H), 7,42 (т, J=7,5 Гц, 1H), 7,37 (т, J=7,5 Гц, 1H), 7,25 (д, J=7,5 Гц, 1H), 7,15 (т, J=7,5 Гц, 1H), 7,09 (т, J=7,5 Гц, 1H), 7,04 (д, J=8,5 Гц, 1H), 6,72 (шир.с, 1H), 4,76 (м, 1H), 3,82 (с, 3H), 3,23 (м, 1H), 2,90-2,64 (м, 5H), 1,98-2,08 (м, 1H), 1,81-1,63 (м, 2H), 1,60-1,50 (м, 1H), 1,42-1,30 (м, 1H) м.д.,13C ЯМР (125 МГц, CDCl3) δ 156,2, 153,8, 135,6, 132,1, 130,9, 129,7, 128,3, 127,1, 123,8, 121,5, 111,3, 71,8, 55,7, 55,5, 47,3, 46,5, 25,3, 24,5, 19,4 м.д. Чистота: 100 % LCMS (214 нм и 254 нм); время удерживания 1,48 мин; (M+1) 353.
Пример 93
Хинуклидин-3-ил-2'-этилбифенил-2-илкарбамат
Применяя общую процедуру E, из хинуклидин-3-ил-2-бромфенилкарбамата, 2-этилфенилбороновой кислоты и [PdCl2(pddf)]CH2Cl2 получали соединение, указанное в заголовке, в виде белого твердого вещества (71 мг, 51%).1H ЯМР (500 МГц, CDCl3) δ 8,11 (шир.с, 1H), 7,43-7,34 (м, 3H), 7,33-7,28 (м, 1H), 7,18-7,08 (м, 3H), 6,24 (шир.с, 1H), 4,75 (м, 1H), 3,23 (м, 1H), 2,85-2,65 (м, 5H), 2,40 (м, 2H), 2,02 (м, 1H), 1,73-1,62 (м, 2H), 1,61-1,50 (м, 1H), 1,40-1,30 (м, 1H), 1,05 (м, 3H) м.д.13C ЯМР (125 МГц, CDCl3) δ 153,3, 142,9, 136,4, 135,3, 130,6, 130,3, 130,1, 129,0, 128,7, 128,4, 126,4, 123,0, 119,1, 72,1, 55,2, 47,3, 46,4, 26,0, 25,3, 24,5, 19,3, 15,2 м.д. Чистота: >98 % LCMS (214 нм и 254 нм); время удерживания 1,55 мин; (M+1) 351.
Пример 94
Хинуклидин-3-ил-3'-метоксибифенил-2-илкарбамат
Применяя общую процедуру E, из хинуклидин-3-ил-2-бромфенилкарбамата, 3-метоксифенилбороновой кислоты и [PdCl2(pddf)]CH2Cl2 получали соединение, указанное в заголовке, в виде белого твердого вещества (120 мг, 85%).1H ЯМР (500 МГц, CDCl3) δ 8,08 (шир.с, 1H), 7,40 (т, J=7,5 Гц, 1H), 7,36 (т, J=7,5 Гц, 1H), 7,23 (дд, J=7,5, 1,5 Гц, 1H), 7,13 (тд, J=7,5, 1,5 Гц, 1H), 6,96 (дд, J=8,0, 2,0 Гц, 2H), 6,91 (т, J=1,5 Гц, 1H), 6,73 (шир.с, 1H), 4,79 (м, 1H), 3,84 (с, 3H), 3,24 (м, 1H), 2,90-2,70 (м, 5H), 2,05 (м, 1H), 1,80-1,70 (м, 2H), 1,62-1,52 (м, 1H), 1,41-1,32 (м, 1H) м.д.13C ЯМР (125 МГц, CDCl3) δ 160,1, 153,4, 139,5, 134,7, 131,5, 130,1, 130,1, 128,5, 123,5, 121,4, 119,9, 114,7, 113,6, 72,1, 55,3, 55,3, 47,3, 46,5, 25,3, 24,5, 19,4 м.д. Чистота: >98 % LCMS (214 нм и 254 нм); время удерживания 1,48 мин; (M+1) 353
Пример 95
Хинуклидин-3-ил-3'-этилбифенил-2-илкарбамат
Применяя общую процедуру E, из хинуклидин-3-ил-2-бромфенилкарбамата, 3-этилфенилбороновой кислоты и [PdCl2(pddf)]CH2Cl2 получали соединение, указанное в заголовке, в виде белого твердого вещества (120 мг, 86%).1H ЯМР (500 МГц, CDCl3) δ 8,09 (шир.с, 1H), 7,41 (т, J=7,5 Гц, 1H), 7,36 (т, J=7,5 Гц, 1H), 7,26-7,18 (м, 4H), 7,14 (т, J=7,5 Гц, 1H), 6,71 (шир.с, 1H), 4,79 (м, 1H), 3,25 (м, 1H), 2,90-2,65 (м, 7H), 2,05 (м, 1H), 1,80-1,64 (м, 2H), 1,62-1,52 (м, 1H), 1,40-1,32 (м, 1H), 1,28 (т, J=7,5 Гц, 3H) м.д.13C ЯМР (125 МГц, CDCl3) δ 153,4, 145,2, 138,1, 134,7, 131,8, 130,2, 129,1, 128,8, 128,4, 127,5, 126,6, 123,5, 120,1, 72,0, 55,3, 47,3, 46,4, 28,9, 25,3, 24,5, 19,4, 15,7 м.д. Чистота: >95 % LCMS (214 нм и 254 нм); время удерживания 1,55 мин; (M+1) 351.
Препаративная процедура Y
Пример 96
Хинуклидин-3-ил-2-изопропоксифенилкарбамат
К смеси 3-аминофенола (1,50 г, 13,8 ммоль), изопропанола (3,3 г, 55 ммоль) и трифенилфосфина (14,4 г, 54,9 ммоль) в THF (15 мл) в течение 30 мин добавляли по каплям диэтилазодикарбоксилат (9,60 г, 55,0 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов и концентрировали. Остаток разбавляли водой, подкисляли 2 н водной HCl и экстрагировали простым эфиром. Водную фазу подщелачивали 2 н водным NaOH и экстрагировали EtOAc. Объединенные органические слои сушили (Na2SO4) и концентрировали. Неочищенный продукт, полученный в результате этого, очищали посредством хроматографии на колонке с силикагелем (петролейный эфир/EtOAc, от 10:1 до 5:1), получая 3-изопропоксибензоламин в виде желтого масла (1,3 г, 64%).
Применяя общую процедуру A, из 3-изопропоксибензоламина (300 мг, 2,00 ммоль) и хинуклидин-3-ола получали соединение, указанное в заголовке, в виде вязкого масла (130 мг, 22%).1H ЯМР (500 МГц, CDCl3) δ 7,20 (шир.с, 1H), 7,09 (т, J=8,5 Гц, 1H), 7,05 (шир.с, 1H), 6,77 (д, J=8,0 Гц, 1H), 6,51 (д, J=8,5 Гц, 1H), 4,75 (м, 1H), 4,46 (м, 1H), 3,26-3,18 (м, 1H), 2,92-2,65 (м, 5H), 2,04 (м, 1H), 1,80 (м, 1H), 1,63 (м, 1H), 1,52 (м, 1H), 1,35 (м, 1H), 1,28 (д, J=5,5 Гц, 6H) м.д.13C ЯМР (125 МГц, CDCl3) δ 157,5, 152,2, 138,2, 128,7, 110,1, 109,6, 105,1, 70,7, 68,9, 54,3, 46,3, 45,4, 28,7, 24,3, 23,3, 21,0, 18,3 м.д. Чистота: >90 % LCMS (214 нм и 254 нм); время удерживания 1,43 мин; (M+1) 305.
Пример 97
Хинуклидин-3-ил-2-изобутоксифенилкарбамат
К смеси 3-аминофенола (500 мг, 4,60 ммоль), 2-метилпропан-1-ола (1,40 г, 18,9 ммоль) и трифенилфосфина (4,80 г, 16,2 ммоль) в THF (10 мл) в течение 30 мин добавляли по каплям диэтилазодикарбоксилат (3,20 г, 18,3 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов. Растворитель выпаривали и остаток разбавляли водой, подкисляли 2 н водной HCl и экстрагировали простым эфиром. Водную фазу подщелачивали 2 н водным NaOH и экстрагировали EtOAc. Объединенные органические слои сушили (Na2SO4) и концентрировали. Неочищенный продукт, полученный в результате этого, очищали посредством хроматографии на колонке с силикагелем (петролейный эфир/EtOAc, 15:1), получая 3-изобутоксибензоламин в виде желтого масла (330 мг, 45%).
Применяя общую процедуру A, из 3-изобутоксибензоламина (330 мг, 2,00 ммоль) и хинуклидин-3-ола получали соединение, указанное в заголовке, в виде вязкого масла (140 мг, 22%).1H ЯМР (500 МГц, CDCl3) δ 7,17 (т, J=8,0 Гц, 1H), 7,15 (шир.с, 1H), 6,81 (д, J=8,5 Гц, 1H), 6,80 (шир.с, 1H), 6,61 (д, J=8,5 Гц, 1H), 4,83 (м, 1H), 3,71 (д, J=6,5 Гц, 2H), 3,34-3,21 (м, 1H), 2,97-2,72 (м, 5H), 2,12-2,04 (м, 2H), 1,90-1,84 (м, 1H), 1,75-1,67 (м, 1H), 1,55-1,63 (м, 1H), 1,46-1,38 (м, 1H), 1,01 (д, J=6,5 Гц, 6H) м.д.13C ЯМР (100 МГц, CDCl3) δ 159,9, 153,4, 139,3, 129,6, 110,6, 109,7, 105,0, 74,4, 72,0, 55,4, 47,3, 46,5, 28,3, 25,4, 24,5, 19,5, 19,3 м.д. Чистота: 100 % LCMS (214 нм и 254 нм); время удерживания 1,56 мин; (M+1) 319.
Пример 98
Хинуклидин-3-ил-2-(циклопропилметокси)-фенилкарбамат
К смеси 3-аминофенола (300 мг, 2,70 ммоль), циклопропилметанола (793 мг, 11,0 ммоль) и трифенилфосфина (2,90 г, 11,0 ммоль) в THF (6 мл) в течение 30 мин добавляли по каплям диэтилазодикарбоксилат (1,90 г, 11,0 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов. Растворитель выпаривали и остаток разбавляли водой, подкисляли 2 н водной HCl и экстрагировали простым эфиром. Водную фазу подщелачивали 2 н водным NaOH и экстрагировали EtOAc. Объединенные органические слои сушили (Na2SO4) и концентрировали. Неочищенный продукт, полученный в результате этого, очищали посредством хроматографии на колонке с силикагелем (петролейный эфир/EtOAc, 15:1), получая 3-(циклопропилметокси)-бензоламин в виде коричневого масла (260 мг, 58%).
Применяя общую процедуру A, из 3-(циклопропилметокси)-бензоламина (260 мг, 1,60 ммоль) и хинуклидин-3-ола получали соединение, указанное в заголовке, в виде вязкого масла (80 мг, 16%).1H ЯМР (400 МГц, CDCl3) δ 7,72 (шир.с, 1H), 7,14 (шир.с, 1H), 7,13 (т, J=8,0 Гц, 1H), 6,85 (д, J=8,0 Гц, 1H), 6,57 (дд, J=8,0, 2,0 Гц, 1H), 4,85 (м, 1H), 3,75 (д, J=6,8 Гц, 2H), 3,35-3,26 (м, 1H), 3,05-2,78 (м, 5H), 2,18-2,12 (м, 1H), 1,97-1,86 (м, 1H), 1,80-1,67 (м, 1H), 1,66-1,55 (м, 1H), 1,52-1,42 (м, 1H), 1,26-1,15 (м, 1H), 0,61-0,55 (м, 2H), 0,31-0,26 (м, 2H) м.д.13C ЯМР (100 МГц, CDCl3) δ 158,6, 152,1, 138,3, 128,6, 109,9, 108,9, 104,0, 71,7, 69,7, 53,8, 46,0, 45,2, 28,7, 24,1, 22,4, 17,8, 9,2, 2,2 м.д. Чистота: 100 % LCMS (214 нм и 254 нм); время удерживания 1,46 мин; (M+1) 317.
Пример 99
1-Бензил-3-(хинуклидин-3-ил)-имидазолидин-2-он
К перемешиваемому раствору гидрохлорида хинуклидин-3-амина (324 мг, 0,199 ммоль) в DMF (30 мл) добавляли триэтиламин (3 капли), после которого осторожно добавляли (изоцианатометил)- бензол (275 мг, 2,10 ммоль). Смесь, полученную в результате этого, перемешивали в течение 18 часов при 25°C. После разделения с использованием HPLC получали 1-бензил-3-(хинуклидин-3-ил)-мочевину (283 мг, 55%).
К раствору 1-бензил-3-(хинуклидин-3-ил)-мочевины (260 мг, 1,00 ммоль) в DMF (30 мл) добавляли NaH [60%-ная дисперсия в минеральном масле] (96 мг, 2,4 ммоль) при охлаждении на ледяной бане. Смесь, полученную в результате этого, перемешивали в течение 2 часов, после чего осторожно добавляли BrCH2CH2Br (0,75 г, 4,0 ммоль). Реакционную смесь премешивали в течение дополнительных 18 часов приблизительно при 25°C. После разделения с использованием HPLC водный слой лиофилизировали и очищали посредством препаративной TLC (от CHCl3 до 5% MeOH в CHCl3 до 5% 2 н NH3(MeOH) в CHCl3), получая соединение, указанное в заголовке, (81 мг, 28%).1H ЯМР (400 МГц, CDCl3) δ 7,19-7,22 (м, 4H), 7,11-7,14 (м, 1H), 6,09 (дд, J=15,2, 8,4 Гц, 1H), 5,45 (дд, J=15,6, 4,0 Гц, 1H), 5,30 (дд, J=8,0, 3,6 Гц, 1H), 4,17-4,29 (м, 4H), 3,66-3,75 (м, 2H), 3,47 (д, J=12,4 Гц, 1H), 3,19-3,27 (м, 3H), 2,34 (шир.с, 1H), 2,22 (д, J=2,4 Гц, 1H), 1,92 (шир.с, 2H), 1,75 (шир.с, 1H) м.д.13C ЯМР (400 МГц, CDCl3) δ 158,9, 141,1, 140,4, 128,7, 127,4, 127,3, 113,6, 63,6, 57,1, 56,0, 45,6, 43,9, 25,2, 23,0, 18,8 м.д. Чистота: 93,8% HPLCMS (210 нм); время удерживания 1,84 мин; (M+1) 286.
Пример 100
N-(1-азабицикло[2.2.2]окт-3-ил)-4-п-толилбутирамид
Применяя общую процедуру I, из 1-азабицикло[2.2.2]окт-3-иламина (200 мг, 1,00 ммоль) и п-толилмасляной кислоты (220 мг, 1,2 ммоль) получали N-(1-азабицикло[2.2.2]окт-3-ил)-4-п-толилбутирамид (114 мг, 40%) в виде белого твердого вещества.1H ЯМР (400 МГц, CDCl3) δ 7,06 (с, 4H), 4,19 (м, 1H), 3,66-3,73 (т, J=8,4 Гц, 1H), 3,29-3,33 (м, 4H), 2,91 (дд, J=8,0, J=3,6 Гц, 1H), 2,59 (т, J=7,6 Гц, 2H), 2,28 (с, 3H), 2,24 (т, J=7,6 Гц, 2H), 2,15-2,16 (м, 1H), 2,02-2,14 (м, 1H), 1,92-2,01 (м, 2H), 1,81-1,91 (м, 3H) м.д.13C ЯМР (400 МГц, CDCl3) δ 175,1, 138,6, 135,1, 128,8, 128,2, 52,7, 47,4, 47,3, 44,5, 34,8, 27,3, 24,3, 21,6, 19,8, 17,1 м.д. Чистота: 99,7% HPLCMS (210 нм); время удерживания 1,76 мин; (M+1) 287.
Пример 101
N-(1-азабицикло[2.2.2]окт-3-ил)-4-(4-метоксифенил)-бутирамид
Применяя общую процедуру I, из 1-азабицикло[2.2.2]окт-3-иламина (200 мг, 1,00 ммоль) и 4-(4-метоксифенил)-масляной кислоты получали соединение, указанное в заголовке, в виде белого твердого вещества (85 мг, 28%).1H ЯМР (400 МГц, CD3OD) δ 7,08 (д, J=8,4 Гц, 2H), 6,81 (д, J=8,4 Гц, 2H), 4,18 (шир.с, 1H), 3,74 (с, 3H), 3,68 (д, J=11,6 Гц, 1H), 3,24-3,33 (м, 4H), 2,98-3,03 (м, 1H), 2,57 (т, J=7,6 Гц, 2H), 2,24 (т, J=7,6 Гц, 2H), 2,03-2,16 (м, 2H), 2,02 (шир.с, 2H), 1,85-1,91 (м, 3H) м.д.13C ЯМР (400 МГц, CD3OD) δ 175,2, 158,3, 133,6, 129,2, 113,6, 54,5, 52,6, 47,2, 46,4, 44,5, 34,9, 34,2, 27,5, 24,4, 21,5, 17,1 м.д. Чистота: 96,4% HPLCMS (210 нм); время удерживания 1,76 мин; (M+1) 303.
Пример 102
(1-Азабицикло[2.2.2]окт-3-ил)-амид бифенил-3-карбоновй кислоты
Применяя общую процедуру I, из 1-азабицикло[2.2.2]окт-3-иламина (200 мг, 1,00 ммоль) и бифенил-3-карбоновй кислоты получали соединение, указанное в заголовке, в виде белого твердого вещества (211 мг, 68%).1H ЯМР (400 МГц, CD3OD) δ 7,86 (с, 1H), 7,59 (д, J=7,6 Гц, 1H), 7,53 (д, J=8,0 Гц, 1H), 7,40 (д, J=7,6 Гц, 2H), 7,28 (т, J=7,6 Гц, 1H), 7,19 (т, J=7,6 Гц, 2H), 7,11 (т, J=7,6 Гц, 1H), 4,21 (шир.с, 1H), 3,56 (т, J=11,6 Гц, 1H), 3,11-3,22 (м, 1H), 3,05-3,10 (м, 4H), 2,10 (кв., J=3,2 Гц, 1H), 1,95 (шир.с, 1H), 1,79-1,83 (м, 2H), 1,59-1,20 (м, 1H) м.д.13C ЯМР (400 МГц, CD3OD) δ 169,6, 141,6, 140,2, 134,5, 130,3, 129,0, 127,7, 126,9, 126,3, 125,9, 51,9, 46,4, 46,0, 45,6, 24,6, 21,6, 17,3 м.д. Чистота: 99,8% HPLCMS (210 нм); время удерживания 1,60 мин; (M+1) 307.
Пример 103
N-(1-азабицикло[2.2.2]окт-3-ил)-2-бифенил-4-илацетамид
Применяя общую процедуру I, из 1-азабицикло[2.2.2]окт-3-иламина (200 мг, 1,00 ммоль) и бифенил-4-илуксусной кислоты получали соединение, указанное в заголовке, в виде белого твердого вещества (140 мг, 44%).1H ЯМР (400 МГц, CD3OD) δ 7,56 (т, J=8,0 Гц, 4H), 7,29-7,41 (м, 5H), 4,19 (шир.с, 1H), 3,70 (т, J=7,2 Гц, 1H), 3,58 (с, 2H), 3,24-3,31 (м, 5H), 3,12-3,19 (м, 1H), 2,16-2,17 (м, 2H), 1,95-1,98 (м, 2H), 1,82 (шир.с, 1H) м.д.13C ЯМР (400 МГц, CD3OD) δ 173,1, 140,8, 140,0, 134,7, 129,4, 128,7, 126,9, 52,3, 46,4, 45,9, 44,3, 41,9, 24,4, 21,5, 17,1 м.д. Чистота: 93,9% HPLCMS (210 нм); время удерживания 2,87 мин; (M+1) 321.
Препаративная процедура Z
Пример 104
2-(Хинуклидин-3-ил)-N-(1-п-толилциклопропил)-ацетамид
К раствору метил-2-(диметоксифосфорил)-ацетата (2,70 г, 14,8 ммоль) в THF (200 мл) при 0°C добавляли NaH [60%-ная дисперсия в минеральном масле] (600 мг, 15,0 ммоль). После перемешивания в течение 1 часа добавляли хинуклидин-3-он (2,00 г, 12,4 ммоль) и смесь, полученную в результате этого, перемешивали при комнатной температуре в течение 18 часов. Реакцию гасили, добавляя 50 мл воды при 0°C, и смесь экстрагировали EtOAc. Органические слои объединяли и концентрировали при пониженном давлении, получая неочищенный метил-2-(хинуклидин-3-илиден)-ацетат, который без очистки использовали на следующей стадии (1,2 г, 70%).
Смесь метил-2-(хинуклидин-3-илиден)-ацетата (70 мг, 0,38 ммоль) и Pd/C (100 мг, 20% масс.) в EtOH (10 мл) перемешивали в атмосфере H2 (20 фунт/кв.дюйм, 1,4 атм) при комнатной температуре в течение 18 часов. Реакционный раствор фильтровали через целит и фильтрат концентрировали при пониженном давлении, получая неочищенный метил-2-(хинуклидин-3-ил)-ацетат (60 мг, 85%), который использовали с очисткой на следующей стадии.
Смесь метил-2-(хинуклидин-3-ил)-ацетата (1,1 г, 6,0 ммоль) и 50 мл концентрированной HCl [12 M] перемешивали при 70°C в течение 18 часов. Реакционную смесь концентрировали при пониженном давлении, получая неочищенную 2-(хинуклидин-3-ил)-уксусную кислоту, которую без очистки использовали на следующей стадии (900 мг, 86%).
Применяя общую процедуру I, из 2-(хинуклидин-3-ил)-уксусной кислоты (169 мг, 1,00 ммоль) и 1-п-толилциклопропанамина (149 мг, 1,10 ммоль) получали соединение, указанное в заголовке, в виде белого твердого вещества (60 мг, 18%).1H ЯМР (400 МГц, CDCl3) δ 7,86 (с, 1H), 6,96-7,07 (м, 4H), 3,22-3,37 (м, 2H), 2,85-3,05 (м, 4H), 2,39-2,45 (м, 2H), 2,21 (с, 3H), 1,45-1,92 (м, 5H), 1,07-1,23 (м, 5H) м.д.13C ЯМР (400 МГц, CDCl3) δ 171,6, 139,8, 136,1, 129,2, 125,6, 52,1, 50,9, 46,5, 46,0, 39,2, 34,9, 30,8, 24,5, 21,1, 18,7, 17,6 м.д. Чистота: 96,2% HPLCMS (210 нм); время удерживания 1,21 мин; (M+1) 299.
Пример 105
N-(2-(3-метоксифенил)-пропан-2-ил)-2-(хинуклидин-3-ил)-ацетамид
Применяя общую процедуру I, из 2-(хинуклидин-3-ил)-уксусной кислоты (169 мг, 1,00 ммоль) и 2-(3-метоксифенил)-пропан-2-амина (182 мг, 1,10 ммоль) получали соединение, указанное в заголовке, в виде белого твердого вещества (126 мг, 40%).1H ЯМР (400 МГц, CDCl3) δ 7,39 (с, 1H), 7,20 (т, J=8,0 Гц, 1H), 6,92 (д, J=8,0 Гц, 1H), 6,87 (д, J=2,0 Гц, 1H), 6,71 (дд, J=8,0, 2,0 Гц, 1H), 3,75 (с, 3H), 3,31-3,42 (м, 2H), 3,00-3,19 (м, 4H), 2,47-2,60 (м, 2H), 2,27 (дд, J=14,0, 6,0 Гц, 1H), 1,83-2,06 (м, 4H), 1,64-1,74 (м, 1H), 1,61 (д, J=12,4 Гц, 6H) м.д.13C ЯМР (400 МГц, CDCl3) δ 170,0, 159,7, 149,3, 129,5, 117,5, 111,8, 111,0, 55,9, 55,4, 52,0, 50,6, 46,6, 46,0, 39,7, 30,9, 29,7, 29,1, 24,3, 18,8 м.д. Чистота: 93,7% HPLCMS (210 нм); время удерживания 0,76 мин; (M+1) 317.
Пример 106
2-(1-Азабицикло[2.2.2]окт-3-ил)-N-[1-(3-изопропилфенил)-1-метилэтил]-ацетамид
К раствору 1-(1-изоцианато-1-метилэтил)-3-изопропенилбензола (10 г, 50 ммоль) в t-BuOH (1000 мл) добавляли KOH (40,0 г, 71,6 ммоль). Смесь перемешивали при кипячении с обратным холодильником в течение 3 часов. Смесь, полученную в результате этого, охлаждали до комнатной температуры, концентрировали и растворяли в CH2Cl2. Твердый остаток отфильтровывали и органический слой доводили до pH <7, используя концентрированную HCl. Аммонийную соль экстрагировали водой. Водный слой делали щелочным, используя водный раствор NaOH [5% масс., 200 мл] и затем метиленхлоридом экстрагировали амин, преобразованный в свободное основание. Органические слои объединяли, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 1-(3-изопропенилфенил)-1-метилэтиламин (3,3 г, 63%).
Раствор вышеуказанного соединения (8,5 г, 48 ммоль) и PtO2 (1,8 г, 8,0 ммоль) в EtOH (600 мл) перемешивали при комнатной температуре при 1 атм H2 в течение 18 часов. Реакционную смесь фильтровали через целит и концентрировали при пониженном давлении, получая 1-(3-изопропилфенил)-1-метилэтиламин (5,0 г, 58%).
Применяя общую процедуру I, из (1-азабицикло[2.2.2]окт-3-ил)-уксусной кислоты (200 мг, 1,20 ммоль) и 1-(3-изопропилфенил)-1-метилэтиламина получали соединение, указанное в заголовке, в виде белого твердого вещества (42 мг, 10%).1H ЯМР (400 МГц, DMSO-d6) δ 7,15-1,20 (м, 3H), 7,05 (д, J=8,4 Гц, 1H), 3,41-3,45 (м, 1H), 3,26 (с, 2H), 3,15-3,22 (м, 2H), 2,71-2,82 (м, 2H), 2,41-2,47 (м, 3H), 2,05-2,12 (м, 1H), 1,79-1,90 (м, 4H), 1,61 (д, J=8,0 Гц, 6H), 1,21 (д, J=6,4 Гц, 6H) м.д.13C ЯМР (400 МГц, DMSO-d6) δ 171,1, 148,7, 147,3, 128,1, 123,9, 122,8, 122,2, 55,6, 52,1, 46,5, 45,9, 38,8, 34,5, 30,7, 28,9, 28,5, 23,8, 23,4, 18,0 м.д. Чистота: 96,8% HPLCMS (210 нм); время удерживания 1,93 мин; (M+1) 329.
Пример 107
2-(1-Азабицикло[2.2.2]окт-3-ил)-N-[2-(2-метокси-фенил)-этил]-ацетамид
Применяя общую процедуру I, из (1-азабицикло[2.2.2]окт-3-ил)-уксусной кислоты (200 мг, 1,20 ммоль) и 2-(2-метоксифенил)-этиламина получали соединение, указанное в заголовке, в виде белого твердого вещества (60 мг, 15%).1H ЯМР (400 МГц, CD3OD) δ 7,17 (т, J=7,2 Гц, 1H), 7,08 (д, J=7,2 Гц, 1H), 6,89 (д, J=8,4 Гц, 1H), 6,84 (т, J=7,6 Гц, 1H), 3,89 (с, 3H), 3,35-3,45 (м, 3H), 3,21-3,31 (м, 3H), 2,76-2,83 (м, 3H), 2,29-2,45 (м, 3H), 1,82-2,01 (м, 3H), 1,72-1,81 (м, 2H) м.д.13C ЯМР (400 МГц, CD3OD) δ 172,0, 158,0, 130,4, 127,8, 127,2, 120,2, 110,4, 54,6, 52,0, 46,4, 45,9, 39,1, 38,3, 30,7, 30,2, 23,8, 17,9 м.д. Чистота: 92,4% HPLCMS (210 нм); время удерживания 1,59 мин; (M+1) 303.
Пример 108
1-(1-азабицикло[2.2.2]окт-3-ил)-3-[1-(3-изопропилфенил)-циклопропил]-мочевина
Смесь 3-изопропилбензойной кислоты (5,00 г, 30,4 ммоль) в SOCl2(50 мл) перемешивали при 100°C в течение 2 часов. Реакционную смесь концентрировали, получая 3-изопропилбензоилхлорид (5,00 г, 91%).
В раствор вышеуказанного хлорангидрида кислоты (5,00 г, 27,0 ммоль) в CH2Cl2(20 мл) при -70°C добавляли по каплям раствор NH3/CH2Cl2 (200 мл). Смесь перемешивали при комнатной температуре в течение 18 часов и затем концентрировали, получая 3-изопропилбензамид (4,2 г, 93%).
Раствор вышеуказанного амида (4,20 г, 25,7 ммоль) в POCl3(36,0 г, 236 ммоль) перемешивали при 80°C в течение 18 часов. Раствор концентрировали и остаток выливали в воду (100 мл). Смесь экстрагировали EtOAc. Органические слои объединяли, промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали, получая 3-изопропилбензонитрил (3,00 г, 80%).
Применяя общую процедуру G, 3-изопропилбензонитрил (3,00 г, 20,6 ммоль) преобразовывали в соответствующий 1-(3-изопропилфенил)-циклопропиламин (0,80 г, 22%).
Применяя общую процедуру C, из вышеуказанного амина (300 мг, 1,71 ммоль), хинуклидин-3-амина (215 мг, 1,71 ммоль) и CDI (290 мг, 2,05 ммоль) получали соединение, указанное в заголовке, в виде белого твердого вещества (88 мг, 46%).1H ЯМР (400 МГц, CD3OD) δ 7,18 (т, J=8,0 Гц, 1H), 7,12 (с, 1H), 7,01 (дд, J=19,2, 7,6 Гц, 2H), 3,74-3,77 (м, 1H), 3,18-3,24 (м, 1H), 2,71-2,87 (м, 5H), 2,42-2,47 (м, 1H), 1,63-1,82 (м, 4H), 1,45 (шир.с, 1H), 1,21-1,26 (м, 10H) м.д.13C ЯМР (400 МГц, CD3OD) δ 193,0, 168,7, 160,1, 128,2, 122,7, 121,9, 55,3, 46,8, 46,1, 34,1, 25,9, 25,0, 23,5, 19,6, 18,2 м.д. Чистота: 92,4% HPLCMS (210 нм); время удерживания 2,53 мин; (M+1) 328.
Пример 109
2-(1-Азабицикло[2.2.2]окт-3-ил)-N-[1-(3-изопропилфенил)-циклопропил]-ацетамид
Применяя общую процедуру I, из 1-(3-изопропилфенил)-циклопропиламина (278 мг, 1,58 ммоль) и (1-азабицикло[2.2.2]окт-3-ил)-уксусной кислоты (267 мг, 1,58 ммоль) получали соединение, указанное в заголовке, в виде белого твердого вещества (70 мг, 14%).1H ЯМР (400 МГц, CD3OD) δ 7,16 (т, J=8,0 Гц, 1H), 7,07 (с, 1H), 6,97 (дд, J=19,2, 7,6 Гц, 2H), 3,16-3,23 (м, 1H), 2,79-2,97 (м, 5H), 2,51-2,58 (м, 1H), 2,23-2,41 (м, 3H), 1,83-1,92 (м, 1H), 1,68-1,81 (м, 3H), 1,54-1,62 (м, 1H), 1,15-1,25 (м, 10H) м.д.13C ЯМР (400 МГц, CD3OD) δ 173,6, 148,5, 142,4, 127,8, 123,6, 123,1, 122,0, 73,0, 53,1, 46,6, 45,9, 39,3, 34,1, 32,3, 28,1, 26,4, 24,4, 19,7, 16,8 м.д. Чистота: 96,9% HPLCMS (210 нм); время удерживания 2,55 мин; (M+1) 327.
Пример 110
1-Азабицикло[2.2.2]окт-3-иловый сложный эфир [1-(3-изопропилфенил)-циклопропил]-карбаминовой кислоты
Применяя общую процедуру A, из 1-(3-изопропилфенил)-циклопропиламина (278 мг, 1,58 ммоль) и хинуклидин-3-ола получали соединение, указанное в заголовке, в виде белого твердого вещества (75 мг, 22%).1H ЯМР (400 МГц, CD3OD) δ 7,17 (м, 1H), 7,09 (с, 1H), 6,97-7,08 (м, 2H), 4,72-4,79 (м, 1H), 3,36-3,42 (м, 1H), 2,79-3,08 (м, 5H), 1,93-2,17 (м, 2H), 1,81-1,90 (м, 1H), 1,67-1,78 (м, 2H), 1,31-1,54 (м, 1H), 1,13-1,28 (м, 10H) м.д.13C ЯМР (400 МГц, CD3OD) δ 157,1, 148,4, 143,1, 128,1, 123,9, 123,0, 122,4, 69,2, 54,4, 46,7, 45,7, 34,7, 34,3, 24,9, 23,3, 22,0, 18,0, 17,3 м.д. Чистота: 99,2% HPLCMS (210 нм); время удерживания 1,83 мин; (M+1) 329.
Пример 111
Исследования эффективности терапии малыми молекулами с применением (S)-2-гидроксисукцинатной соли (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)-тиазол-4-ил)-пропан-2-ил)-карбамата, проведенные in vivo на модели болезни Фабри у мышей
В данном разделе описаны эксперименты, проведенные in vivo с использованием ингибитора GCS на модели болезни Фабри у мышей, которые демонстрируют, что субстрат-редуцирующая терапия (SRT) одинаково эффективна в отношении понижения уровней как Gb3, так и лизо-Gb3 в плазме, почке и моче мышей с болезнью Фабри. Данное исследование было разработано для того, чтобы оценить, может ли ингибирование образования субстрата (т.е. «субстрат-редуцирующая терапия»), осуществляемое с применением типов соединений согласно настоящему изобретению, уменьшать аккумулирование накапливаемого материала в виде глоботриаозилцерамиада (Gb3) и лизоглоботриаозилцерамида (лизо-Gb3). Недавно было высказано предположение, что лизо-Gb3 в моче может служить надежным биологическим маркером, клинически значимым для болезни Фабри (Aerts et al., PNAS USA 105: 2812-2817 (2008); и Auray-Blais et al., Clin Chim Acta 411: 1906-1914 (2010)). Метаболическое происхождение лизо-Gb3 неизвестно, возможно, он образуется либо посредством деацилирования Gb3, либо путем анаболического синтеза из глюкозилсфингозина.
На Фиг. 1 черные стрелки обозначают продемонстрированные пути, а серые стрелки - пути недокументированные. Как известно, ферментозаместительная терапия с использованием α-галактозидазы A разрушает как Gb3, так и лизо-Gb3. Соответственно, субстрат-редуцирующая терапия с использование ингибитора GCS была бы наиболее эффективной в отношении ограничения накопления лизо-Gb3, если лизо-Gb3 образуется преимущественно посредством деацилирования Gb3, т.е. по GCS-зависимому пути. Эти эксперименты демонстрируют, что субстрат-редуцирующая терапия, использующая ингибиторы GCS, понижает и Gb3, и лизо-Gb3 у мышей с моделью болезни Фабри, тем самым свидетельствуя в пользу применения соединений согласно настоящему изобретению в качестве эффективных терапевтических вариантов для пациентов с болезнью Фабри.
В следующих экспериментах мыши получали дозы ингибиторов GCS, составлявшие либо ~60 мг/кг/день для (S)-2-гидроксисукцинатной соли (S)-хинуклидин-3-ил-(2-(2-(4-фторфенил)-тиазол-4-ил)-пропан-2-ил)-карбамата (далее «GZ 452»), либо ~300 мг/кг/день для соли L-винной кислоты с [2-(2',3'-дигидробензо[1,4]дикосин-6'-ил)-2-гидрокси-1-пирролидин-1-илметилэтил]-амидом (1R,2R)-октановой кислоты (далее «GZ 638»), в виде компонента свободно доступного гранулированного корма. Анализ липидов проводили посредством ESI/MS, как описано у Marshall et al., PLoS ONE 5:e15033 (2010). Как более подробно обсуждается ниже, лечение начинали, когда возраст мышей составлял 3, 8 или 12 месяцев (для испытания эффективности при разной тяжести заболевания). Кровь и мочу собирали ежемесячно, периодический отбор образцов тканей предоставлял материал для оценки эффективности терапии (по уровням Gb3 и лизо-Gb3). Предшествующие исследования показали, что ингибиторы глюкозилцерамид-синтазы более раннего поколения (класса, подобного P4) могли сдерживать накопление Gb3; однако, как обсуждается ниже, лечение с применением Genz-452 было способно не только предотвращать или сдерживать дальнейшее накопление, но и эффективно понижать абсолютные уровни Gb3 и лизо-Gb3 в исследованных тканях (в печени, сердце, моче, плазме). Как дополнительно обсуждается ниже, эффективность терапии малыми молекулами зависит от возраста мышей в начале лечения. Как правило, чем старше мышь, тем выше уровни накопленного Gb3 и, таким образом, тем более длительное лечение требуется для того, чтобы вызвать одинаковый терапевтический эффект (см. Фиг. 4). Эксперименты и результаты дополнительно описаны ниже.
Ингибитор GCS понижает уровни Gb3 в тканях внутренних органов мышей с болезнью Фабри
В этом эксперименте мышей с болезнью Фабри лечили ингибиторами GCS, добавленными в их корм, в течение 4 месяцев, начиная с 8-месячного возраста. Авторы настоящего изобретения ранее уже сообщали, что тартрат элиглустата (GZ 638) в дозе 300 мг/кг/день (SRT GZ 638) эффективен в отношении ингибирования дальнейшего накопления Gb3 в ткани, о чем в данном эксперименте свидетельствует отсутствие значимых изменений Gb3 относительно начальных уровней (на диаграмме обозначено как «НЛ (начало)»). Как показано на Фиг. 3 (эксперимент SRT-Gz452), был испытан и более сильный ингибитор GCS (соединение GZ 452 в дозе 60 мг/кг/день), который, как было найдено, не только предотвращал дальнейшее накопление, но и значительно снижал уже накопленный Gb3 относительно начальных уровней («НЛ (начало)»). Показаны и его уровни у мышей дикого типа (ДТ) того же возраста. Эти результаты демонстрируют, что GZ 638 является сильным ингибитором GCS и эффективно понижает уровни Gb3 в тканях внутренних органов мышей с болезнью Фабри.
Субстрат-редуцирующая терапия понижает уровни Gb3 в моче и плазме молодых и старых мышей с болезнью Фабри
В этом эксперименте мышей с болезнью Фабри лечили соединением GZ 452 в корме (Rx:) в течение 2 или 4 месяцев, начиная с возраста 3 или 8 месяцев (Возраст:), как указано на Фиг. 4А и 4В. Уровни Gb3 в моче более молодых и более старых мышей были одинаково чувствительны к воздействию субстрат-редуцирующей терапии, достигая приблизительно 90%-ного снижения после 2 месяцев лечения. Уровни Gb3 в плазме отвечали на лечение более медленно, причем для ~50%-ного снижения этого уровня у более старых мышей требовалось более длительное лечение, чем у молодых (4 и 2 месяца, соответственно). Эти результаты демонстрируют, что GZ 638 эффективно понижает уровни Gb3 в моче и плазме молодых и старых мышей с болезнью Фабри.
Субстрат-редуцирующая терапия понижает уровни Gb3 и лизо-Gb3 в почке мыши с болезнью Фабри
В этом эксперименте мышей с болезнью Фабри лечили соединением Genz-452, добавленным в их корм, в течение 4 месяцев, начиная с 8-месячного возраста. Как показано на Фиг. 5, ткань почки анализировали на содержание (А) Gb3 и (B) лизо-Gb3 у мышей, не подвергавшихся лечению (НЛ), у мышей с болезнью Фабри, которых лечили соединением GZ 452 (SRT), и у контрольных мышей дикого типа (ДТ). Результатом проведения SRT было значимое в одинаковой степени снижение уровней как Gb3, так и лизо-Gb3 (60-70%). Эти результаты демонстрируют, что GZ 638 эффективно снижает уровни Gb3 и лизо-Gb3 в почках мышей с болезнью Фабри.
Пример 112
Исследования эффективности комбинированной терапии с применением GZ 452 и альфа-галактозидазы A, проведенные in vivo на модели болезни Фабри у мышей
Мышей с болезнью Фабри использовали для испытаний эффективности сочетания ферментозаместительной терапии с терапией малыми молекулами, проведенных in vivo в параллельном формате. Данное исследования было разработано для того, чтобы оценить, может ли ингибирование субстрата (т.е. «субстрат-редуцирующая терапия» с применением соединения GZ 452) уменьшить повторное накопление сохраняемого материала Gb3 и лизо-Gb3. Протокол исследования включал в себя три группы мышей 3-месячного возраста с болезнью Фабри, которых лечили разными способами (Фиг. 6А). Первая группа получала внутривенные инъекции фермента альфа-галактозидазы A (ERT) в дозе 1 мг/кг для понижения уровней Gb3, повторявшиеся каждые 2 месяца. Вторая группа получала такие же инъекции фермента, как и группа 1, но одновременно она с гранулированным кормом получала и соединение GZ 452 в дозе, составлявшей приблизительно 60 мг/кг/день. Третья группа получала только ежедневные дозы GZ 452 в своем корме. Четвертая группа не получала никакого лечения и служила плацебо-контролем, а пятая группа (животные дикого типа) предоставляла «нормальные» значения Gb3 и лизо-Gb3. Месячные коллекции мочи и крови и три месячных образца тканей предоставляли материалы для оценки относительной эффективности терапии (Фиг. 6А).
Спустя 2 месяца (возраст мышей составлял 5 месяцев) плазму (Фиг. 6B, диаграммы A и C) и мочу (Фиг. 6B, диаграммы B и D) анализировали на Gb3 (Фиг. 6B, диаграммы A и B) и лизо-Gb3 (Фиг. 6B, диаграммы C и D). В плазме ERT и SRT понижали уровни и Gb3 (диаграмма A), и лизо-Gb3 (диаграмма C), а результатом сочетания ERT и SRT были значительные улучшения по сравнению с каждым из этих способов лечения, проводившихся раздельно. ERT не влияла на уровни Gb3 в моче, которые, однако, значительно снижались под воздействием SRT (диаграмма B). Все виды терапии примерно одинаково понижали уровень лизо-Gb3 в моче (диаграмма D), на основании чего можно предположить, что Gb3 и лизо-Gb3, присутствующие в моче, могут происходить из разных источников. Результаты этих исследований показывают, что SMT была эффективной в отношении снижения уровня Gb3 в почках и моче. ERT была более эффективной, чем SMT, в отношении снижения уровня Gb3 в плазме, однако наиболее эффективным было лечение, сочетавшее оба способа терапии. Как монотерапия SMT, так и ее комбинация с ERT, были способны влиять на уровень лизо-Gb3 (уменьшая его накопление).
Пример 113
Профиль изоформ ацильной цепи Gb3
Относительное содержание амидно связанных ацильных групп с разной длиной углеродной цепи определяли для Gb3 из плазмы, мочи и почек мышей с болезнью Фабри. Как показано на Фиг. 7, в плазме главными изоформами были C16:0 и C24:1. Профили изоформ в моче и почках были почти одинаковыми, причем преобладали цепи с длиной C24:0 и C22:0. Эти данные согласуются с представлением о том, что Gb3, присутствующий в моче, происходит преимущественно из почек - вероятно, вследствие эпидермального экзосомального отделения. Наличие корреляции между этими результатами и результатами, представленными на Фиг. 6, где ERT понижала уровень лизо-Gb3 в плазме и моче, но не влияла на уровень Gb3 в моче, позволяет предположить, что лизо-Gb3, присутствующий в моче, происходит из фильтрата плазмы. Это различие источников Gb3 и лизо-Gb3, присутствующих в моче, если оно справедливо и для пациентов, может объяснить, почему лизо-Gb3 считают более точным предиктором тяжести заболевания и эффективности лечения, чем Gb3 в моче.
Пример 114
SRT, но не ERT, значительно сдвигает на более поздний срок потерю термической ноцицептивной реакции
Мышей 3-месячного возраста с болезнью Фабри лечили соединением GZ 452, вводимым с их кормом (SRT), α-галактозидазой, вводимой каждые 2 месяца (ERT), или комбинацией двух этих способов терапии (E+S), как описано выше. После 6 месяцев комбинированной терапии оценивали время (латентность) термической ноцицептивной реакции, помещая мышей на горячую плиту с температурой 55°С и записывая время до проявления реакции (четко видимое отдергивание задней лапы). Как показано на Фиг. 8, после 7 месяцев лечения (10-месячных мышей) группа, в которой проводили ферментозаместительную монотерапию, значительно отличалась от группы, не подвергавшейся никакому лечению (НЛ). Группы, получавшие лечение в виде субстрат-редуцирующей монотерапии или в виде комбинированной терапии, имели значительно более короткие времена проявления реакции на термический раздражитель. Эти результаты демонстрируют, что SRT (но не ERT) сдвигает на более поздний срок потерю термической ноцицептивной реакции, суррогатного маркера периферической невропатии, часто наблюдаемой у пациентов с болезнью Фабри.
Пример 115
Модель nGD у мышей для исследований SMT, проводимых in vivo с применением Gz161
Мышей K14 lnl/lnl (сокращенно K14) получали из Lund University (Enquist et al. (2007)) и разводили согласно протоколу, одобренному Institutional Animal Care and Use Committee. Детенышам, полученным при гетерозиготном спаривании, надрезали хвосты и проводили генотипирование в первый день после рождения (Р1). ДНК экстрагировали, используя буфер лизиса, содержавший 5 мМ EDTA, 0,2% SDS, 200 мМ NaCl, 100 мМ Tris pH 8,0 с добавлением 0,25 мг/мл протеиназы K (Invitrogen, Carlsbad, California), осаждали 100%-ным изопропанолом и повторно растворяли в буфере 1Х Tris-EDTA. Эту ДНК затем использовали в полимеразной цепной реакции (ПЦР) для выявления присутствия гена GC с промотором кератина К14 (CRE) (Enquist et al. (2007)). Чтобы обнаружить повреждение сайта резистентности к неомицину в гене мышиной глюкоцереброзидазы (NEO) авторы настоящего изобретения использовали методику с тремя праймерами: GC WT Fwd 5'-TGTTCCCCAACACAATGCTCTTT-3'; Rev 5'-TCTGTGACTCTGATGCCACCTTG-3' и Neo Rev 5'-AAGACAGAATAAAACGCACGG GTG-3', как описано ранее в публикации Cabrera-Salazar et al., Experimental Neurology 225: 436-444 (2010).
Новорожденные мыши получали ежедневно внутрибрюшинные инъекции хинуклидин-3-ил-(2-(4'-фтор-[1,1'-бифенил]-3-ил)-пропан-2-ил)-карбамата (далее «GZ 161») в дозе 5 мг/кг, растворенной в объеме, взятом из расчета 10 мкл на грамм массы тела, начиная с четвертого дня после рождения. Мышей К14 и мышей дикого типа того же возраста эвтаназировали на 10-й день после рождения (до появления симптомов) и в возрасте 14 дней (гуманная конечная точка) для оценки уровней гликосфинголипидов (GSL). Мышам вводили пентобарбитал (Euthasol, Virbac Inc, Forth Worth, TX) в дозе 150 мг/кг и проводили транскардиальную перфузию холодным 0,9%-ным раствором NaCl. Извлекали и разделяли мозг; одно полушарие использовали для анализа на GSL, а другое фиксировали в 4%-ном параформальдегиде в течение 96 часов и обрабатывали для гистологии.
Чтобы определить, возможно ли достижение дополнительных благоприятных эффектов посредством пренатального воздействия GZ 161, подгруппе беременных самок K14 его вводили с кормом, составленным таким образом, чтобы в течение последних 5-7 дней беременности суточная доза GZ 161 составляла 20 мг/кг. После родов самки, получавшие GZ 161, переводились на стандартную диету, а детеныши получали ежедневные внутрибрюшинные инъекции GZ 161 в дозе 5 мг/кг (10 мкл на грамм массы тела), начиная с дня Р1. Группу детенышей дикого типа, рожденных самками, получавшими лекарственное средство или стандартную рецептуру, умерщвляли сразу после рождения, чтобы определить, не могло ли внутриутробное воздействие GZ 161 понизить уровни GSL в мозге.
Пример 116
Количественное определение гликосфинголипидов
Количественный анализ на сфинголипиды проводили посредством жидкостной хроматографии и тандемной масс-спектрометрии (LC/MS/MS), как было описано ранее у Merrill et al., Methods 36: 207-224 (2005). В кратком изложении: 10 мкл гомогената ткани мозга (масса ткани/вода: 100 мг/мл) экстрагировали 1,00 мл смеси органических растворителей (97% ацетонитрила, 2% метанола и 1% уксусной кислоты по объему) и интенсивно перемешивали на вортекс-миксере в течение 10 мин. Экстрагированные сфинголипиды (GluCer и GluSph) разделяли прямым образом посредством гидрофильной жидкостной хроматографии (колонка Atlantis HILIC, Waters Corp.) и анализировали посредством тройной квадрупольной тандемной масс-спектрометрии (API 4000, Applied Biosystems/MDS SCIEX), сравнивая со стандартами сфинголипидов (Matreya, LLC; Pleasant Gap, PA)
Пример 117
Переработка рецептуры препарата рекомбинантной человеческой глюкоцереброзидазы
Рецептуру препарата человеческой глюкоцереброзидазы (rhGC) перерабатывали, как описано ранее у Cabrera-Salazar et al. (2010). В кратком изложении: rhGC связывали с использованием катионного обмена (CM Sepharose) и в элюат добавляли человеческий сывороточный альбумин (HSA) в качестве стабилизатора. Рецептура для ICV-введения: 2 мг/мл rhGC в 10 мМ натрий-фосфатном буфере при рН 7,2, содержащем 135 мМ хлорида натрия, 5 мг/мл HSA и 0,01% полисорбата 80.
Пример 118
Интрацеребровентрикулярные инъекции
Животному с моделью невропатической болезни Гоше (nGD), идентифицированному как K14, проводили криоанестезию и выполняли двусторонние интрацеребровентрикулярные (ICV) инъекции 2 мкл rhGC (2 мг/мл) или плацебо, как описано ранее (Cabrera-Salazar et al. (2010)). После этой процедуры инъецированных детенышей наблюдали до возвращения в обычное состояние и возвращали матерям.
Пример 119
Гистопатология
После подтверждения генотипа животных эвтаназировали в возрасте 10 дней. В этом возрасте мыши К14 были бессимптомными. Мышам делали внутрибрюшинную инъекцию 150 мг/кг пентобарбитала натрия (Euthasol, Virbac Inc, Forth Worth, TX) и интракардиальную перфузию охлажденным 0,9%-ным хлоридом натрия. Мозг извлекали и фиксировали в 4%-ном параформальдегиде в течение 72 часов. Ткань переносили в PBS и заливали парафином. Делали сагиттальные срезы толщиной 5 мкм, которые окрашивали, как описано ниже. Глиоз и присутствие клеток макрофагальной линии оценивали по окрашиванию глиального фибриллярного кислого протеина, а для выявления экспрессии панмакрофагальных маркеров CD68 и F4/80 использовали систему иммунохимических красителей Leica Bond Max (Leica Microsystems, Wetzlar, Germany).
Окрашивание GFAP: Парафиновые срезы помещали на предметные стекла и обрабатывали, используя систему Bond Polymer Refine IHC system (Leica Microsystems, Wetlzar, Germany) блокировали в течение 10 минут в бессывороточном белковом блоке (Dako systems, Glostrup, Denmark), инкубировали в течение 30 минут с первичными антителами, специфичными к GFAP, разведенными в отношении 1:1500 в Dako antibody diluent (Dako, Glostrup, Denmark), и окрашивали, используя Bond Polymer Refine detection kit (Leica Microsystems, Wetzlar, Germany).
Окрашивание F4/80: Парафиновые срезы помещали на предметные стекла и обрабатывали, используя систему Bond Polymer Refine IHC system (Leica Microsystems, Wetlzar, Germany), инкубировали в течение 30 минут с крысиными антителами, специфичными к мышиному F4/80 (eBioscience, San Diego, CA), в разбавлении 1:2500 или с крысиным IgG2a (eBioscience, San Diego, CA) в качестве контроля изотипа. После этого срезы инкубировали со вторичными кроличьими антителами против иммуноглобулинов крысы (Vector laboratories, Burlingame, CA) в разбавлении 1:250 и окрашивали, используя Bond Polymer Refine detection kit (Leica Microsystems, Wetzlar, Germany).
Окрашивание CD 68: Парафиновые срезы помещали на предметные стекла и обрабатывали, используя систему Bond Polymer Refine IHC system (Leica Microsystems, Wetlzar, Germany), инкубировали в течение 30 минут с крысиными антителами антителами клона FA-11, специфичными к мышиному CD68 (AbD Serotec, Oxford, UK), в разбавлении 1:2500 или с крысиным IgG2a для контроля изотипа (AbD Serotec, Oxford, UK). Затем срезы инкубировали со вторичными кроличьими антителами против иммуноглобулинов крысы (Vector laboratories, Burlingame, CA) в разбавлении 1:250 и окрашивали, используя Bond Polymer Refine detection kit (Leica Microsystems, Wetzlar, Germany).
Для каждой техники окрашивания, используя систему Aperio ScanScope XT (Aperio Technologies, Vista, CA), в каждой экспериментальной группе получали выровненные по экспозиции цифровые изображения сходных участков мозга. Окрашенные срезы оцифровывали с высоким разрешением и на каждом срезе выделяли по шесть участков, вызывающих интерес, которые независимо анализировали гистоморфометрически. Определяли положительно окрашенные участки и ядра, анализируя количественные данные по методике однофакторного дисперсионного анализа с последующим множественным сравнением по критерию Тьюки, используя Graph Pad Prism V 4.0 (GraphPad Software, San Diego, CA). Различия между групповыми средними считали достоверными при p <0,05.
Пример 119
Выживаемость
Мыши К14 получали ежедневные внутрибрюшинные инъекции GZ 161 в дозе 5 мг/кг массы тела, как описано выше. Отдельная группа животных получала также интрацеребровентрикулярные инъекции GC в первый, второй и третий дни после рождения, после которых следовали ежедневные внутрибрюшинные инъекции GZ 161. Животные, достигшие возраста отъема, получали GZ 161 в специальном корме, приготовленном так, чтобы предоставлять дозу 60 мг/кг/день.
Все животные находились под ежедневным наблюдением для выявления развития неврологических осложнений. Мышей эвтаназировали, когда они достигали гуманной конечной точки (когда они в течение 10 секунд не могут самостоятельно выправить положение тела после принудительного поворота на бок), инъецируя пентобарбитал натрия в дозе 150 мг/кг (Euthasol, Virbac Inc, Forth Worth, TX). Этот момент времени регистрировали как конец жизни, который использовали в анализе с графиками Каплана-Мейера.
Пример 120
Статистический анализ
Значения, показанные в настоящем документе, соответствуют средним величинам, а приведенные планки погрешностей представляют стандартную ошибку среднего. Для сравнения групп применяли однофакторный дисперсионный анализ с последующим множественным сравнением по критерию Тьюки. Для сравнения данных о внутриутробном уменьшении субстрата проводили анализ по двухвыборочному t-критерию для независимых выборок с поправкой Уэлча. Кривые выживаемости по Каплану-Мейеру анализировали, применяя логарифмический ранговый критерий, эквивалентный критерию Мантеля-Гензеля. Все статистические анализы проводили, используя Graph Pad Prism v4.0 (GraphPad Software, San Diego, CA). Различия между групповыми средними считали достоверными при p <0,05.
Пример 121
Накопление субстрата в мозге мыши К14
Прежде чем проводить оценку эффектов, оказываемых на липиды мозга, авторы настоящего изобретения сравнивали зависимые от времени изменения уровней GluCer, GalCer и GluSph в мозге мышей К14 с их уровнями в мозге контрольных мышей дикого типа. Диаграммы A и B Фигуры 9 показывают, что в мозге мышей дикого типа в течение нескольких первых дней жизни преобладающим изомером GL-1 являлся GluCer; к 14-му постнатальному дню (P14) преобладающим изомером становился GalCer. Эти результаты согласуются с результатами исследования мозга крысы, в которых было выявлено, что GluCer синтезируется в большей степени в течение первой недели жизни, а затем, начиная с дня Р8, следует повышенный синтез GalCer (Brenkert et al., Brain Research 36: 183-193 (1972)). Диаграмма A Фигуры 9 также показывает, что GluCer у мышей К14 повышен в 10 раз по сравнению с мышами дикого типа и что это повышение сохранялось в течение первых двух недель жизни вплоть до гибели мышей в день Р14.
В согласии с прежними моделями невропатической болезни Гоше у мышей (Liu et al., PNAS 95: 2503-2508 (1998)), диаграмма C Фигуры 9 показывает, что у мышей К14 в момент рождения лизогликосфинголипид GluSph был повышен более чем в 20 раз по сравнению с мышами дикого типа. Это повышение сохранялось в течение первых двух недель жизни и было даже более высоким у животных, эвтаназированных на конечной стадии (Фиг. 9, диаграмма C). У мышей дикого типа, соответствующих по возрасту мышам К14, уровни GluSph были ниже порога детектирования (0,3 нг/мг ткани). Диаграмма D Фигуры 9 показывает, что эти повышенные гликосфинголипиды и лизогликосфинголипиды у мышей K14, по-видимому, не влияют на массу мозга (по сравнению с мозгом мышей дикого типа). Учитывая известную токсичность GluSph, следует ожидать, что терапевтические стратегии, направленные на снижение накопления этих субстратов в мозге мышей К14, могут влиять и на особенности патологии данного заболевания, и на продолжительность жизни этих животных.
Пример 122
Внутрибрюшинное введение GZ 161 снижает уровни GluCer и GluSph в мозге мышей К14
Фигура 10 показывает, что по сравнению с мышами К14, которым вводили плацебо, ежедневное внутрибрюшинное введение соединения GZ 161 более чем на 60% снижало мозговые уровни GluCer и GluSph, измеряемые в гуманной конечной точке (т.е. в возрасте 14-15 дней). Мыши К14, которых лечили соединением GZ 161, в это время были бессимптомными. И хотя введение GZ 161 значительно снижало уровни этих гликосфинголипидов, Фиг. 10 показывает, что они, тем не менее, оставались в несколько раз более высокими, чем у мышей дикого типа того же возраста; в проанализированных образцах, полученных от мышей дикого типа или гетерозиготных особей того же возраста, GluSph не детектировался. Снижение мозговых гликосфинголипидов, обусловленное системным введением лекарственного средства, является веским свидетельством того, что соединение GZ 161 способно как проникать через ГЭБ, так и ингибировать свой целевой фермент (GCS).
Пример 123
Внутрибрюшинное введение Gz 161 уменьшает окрашивание микроглии/макрофагов по всему мозгу мышей К14
Клетки миелоидной линии можно детектировать в мышином мозге, используя антитела к таким антигенам, как F4/80 и CD68. F4/80 представляет собой трансмембранный гликопротеин, найденный на разветвленной (покоящейся) микроглии и макрофагах, а CD68 является лизосомным белком, экспрессированным на относительно высоких уровнях в макрофагах и активированной (реактивной) микроглии и на более низких уровнях в разветвленной микроглии. Увеличенное окрашивание F4/80 и CD68 в мозге может появляться при рекрутировании моноцитов или при микроглиальной пролиферации, оно является нормальной реакцией на травму и воспаление. Фиг. 11 качественно и количественно показывает, что, по сравнению с мышами дикого типа в 10-дневном возрасте (Р10), во многих участках мозга мышей К14 повышена численность CD68-положительных клеток (в гиппокампе, таламусе, стволе мозга, мозжечке). Самая высокая концентрация CD68-положительных клеток обнаружена в таламусе и стволе мозга - двух отделах мозга, патологичность которых наблюдается и у пациентов с болезнью Гоше типа 2. (Conradi et al., Acta Neuropathologica 65: 99-109 (1984); Conradi et al., Acta Neuropathologica 82: 152-157 (1991); и Wong et al., Molecular Genetics and Metabolism 82: 192-207 (2004)). Фиг. 11 также показывает, что системное введение GZ 161 уменьшает численность CD68-положительных клеток во всех этих отделах; лечение также уменьшало численность CD68-положительных клеток в обонятельной луковице и лобной коре (данные не показаны). В согласии с данными о гистопатологии CD68, Фиг. 12 показывает повышенное окрашивание F4/80 у мышей К14, получавших плацебо, наблюдаемое в день Р10, по сравнению с животными дикого типа. Ежедневные внутрибрюшинные инъекции GZ 161 уменьшали численность F4/80-положительных клеток в таламусе и стволе мозга, но почти не влияли на их численность в других участках мозга. В совокупности с данными по CD68, эти результаты свидетельствуют о том, что результатом системного лечения мышей К14 соединением GZ 161 является пониженная численность макрофагов/микроглии во многих отделах мозга.
Пример 124
Внутрибрюшинное введение GZ 161 уменьшает глиоз в нескольких отделах мозга мышей К14
В ответ на воспаление и повреждение или гибель нейронов может иметь место гипертрофия или пролиферация астроцитов - процесс, известный как астроглиоз. Глиальный фибриллярный кислый протеин (GFAP) является белком промежуточных филаментов, который очень сильно экспрессирован в активированных (реактивных) астроцитах, и поэтому его можно использовать для мониторинга астроглиоза. Фиг. 13 показывает, что в день Р10 окрашивание GFAP в нескольких отделах мозга крысы К14 (в гиппокампе, таламусе, стволе мозга, мозжечке) было более значительным, чем у животных дикого типа, что свидетельствует о присутствии реактивных астроцитов. Фиг. 13 также показывает, что системное лечение мышей К14 соединением GZ 161 приводило к уменьшенному окрашиванию GFAP в гиппокампе и мозжечке в день Р10; окрашивание было пониженным и в обонятельной луковице и лобной коре (данные не показаны). Таким образом, эти результаты, относящиеся к GFAP, согласуются с вышеописанными данными по макрофагам/микроглии, которые демонстрируют, что у мышей К14, вероятно, имеется текущий воспалительный процесс, который можно до некоторой степени ослабить системным введением GZ 161.
Пример 125
Внутрибрюшинное введение GZ 161 увеличивает выживаемость мышей К14
Учитывая положительные эффекты лечения соединением GZ 161 на гликосфинголипиды и гистопатологию мозга, авторы настоящего изобретения заинтересовались, не могут ли эти эффекты повысить и выживаемость мышей К14. Фиг. 14 демонстрирует, что мыши К14, которым давали плацебо, имели медиану продолжительности жизни, равную 15 дням, что согласуется с прежними результатами авторов настоящего изобретения, полученными с этой моделью у мышей (Cabrera-Salazar et al. (2010)). Результатом системного (внутрибрюшинного) введения мышам К14 соединения GZ 161 было увеличение медианы продолжительности жизни до 18 дней (p<0,0001), что согласуется с благоприятными молекулярными и клеточными эффектами этого лекарственного средства в мозге, показанными выше.
В более ранних экспериментах с мышами К14 было показано, что интрацеребровентрикулярные инъекции GC новорожденным (P1-P3) могли даже еще больше увеличить медиану выживаемости (до 23 дней) (Cabrera-Salazar et al. (2010)). Поскольку и GC, и GZ 161 обладают способностью понижать уровни одного и того же гликосфинголипида, GluCer (GC - разрушая GluCer; GZ 161 - ингибируя его синтез), авторы настоящего изобретения задались еще и вопросом, не может ли комбинация Gz161 и интрацеребровентрикулярного (ICV) введения GC оказаться более благоприятной в отношении выживаемости, чем индивидуальное применение каждого из этих средств. Фиг. 14 демонстрирует, что результатом комбинирования интрацеребровентрикулярного введения GC (в дни P1, 2, 3) с ежедневным внутрибрюшинным введением Gz161 была медиана выживаемости, равная 26 дням, что значительно больше, чем при применении только GZ 161 или только интрацеребровентрикулярной GC (p=0,0007). Таким образом, системное введение GZ 161, по-видимому, является аддитивным с интрацеребровентрикулярной GC и предоставляет дополнительный благоприятный эффект в отношении выживаемости.
Пример 126
Пренатальное введение GZ 161 не способно увеличить выживаемость мышей К14
Поскольку было обнаружено, что в день Р10 уровни GluSph в мозге мыши К14 повышены, по меньшей мере, в 10 раз и поскольку были получены документальные свидетельства того, что GluSph повышен в мозге мышей и людей с nGD даже еще до их рождения (Orvisky et al., Pediatric Research 48: 233-237 (2000)), было проведено исследование возможности благоприятного изменения выживаемости при внутриутробном воздействия GZ 161 на мышей К14. Фиг. 15 показывает, что введение GZ 161 беременным самкам мышей дикого типа приводило приблизительно к 5-кратному снижению уровней GluCer в мозге новорожденных мышей (Р0), свидетельствуя о способности GZ 161 проникать через гематоплацентарный барьер. Однако введение GZ 161 беременным самкам мышей К14 и последующее внутрибрюшинное введение GZ 161 рожденным детенышам оказалось неспособным увеличить выживаемость сверх того, что наблюдается у мышей, которые системно получали GZ 161 только постнатально (18 дней) (Фиг. 14 и 16). Таким образом, эти данные согласуются с результатами, описанными на Фиг. 14, и означают, что хотя GZ 161 снижает гликосфинголипиды и ослабляет патологию нервной системы, текущий режим лечения является недостаточным для защиты ЦНС. Эти результаты согласуются и с прежними результатами авторов настоящего изобретения, полученными в этой модели с применением интрацеребровентрикулярных инъекций рекомбинантной человеческой глюкоцереброзидазы (Cabrera-Salazar et al. (2010)), и вместе с ними свидетельствуют о том, что для дальнейшего улучшения выживаемости потребуется более сильное и непрерывное снижение гликосфинголипидов, таких как GluCer.
Эти данные показывают качественно и количественно, что системное (внутрибрюшинное) введение GZ 161 новорожденным мышам К14 значительно снижает субстратную нагрузку, ослабляет патологические симптомы заболевания и увеличивает медиану продолжительности жизни. При сочетании с интрацеребровентрикулярной доставкой rhGC системное введение GZ 161 приводит к аддитивному увеличению продолжительности жизни, свидетельствуя о том, что такое сочетание могло бы быть более эффективным у пациентов с nGD, чем каждый способ монотерапии, проводимой отдельно. С учетом того, что на основании этих исследований можно заключить, что GZ 161, по-видимому, может проникать через ГЭБ и ингибировать свой целевой фермент (глюкозилцерамид-синтазу), разумно предположить, что эту молекулу можно также применять и для лечения других болезней лизосомного накопления, обусловленных накоплением субстратов, метаболически следующих за GluCer.
Важно отметить, что в проводимых исследованиях соединение GZ 161 вводили мышам К14 в пределах временного интервала, в котором GluCer и GluSph продуцировались в развивающемся мозге мыши на относительно высоких уровнях по сравнению с мышами дикого типа (Фиг. 9; Brenkert et al., 1972). Ежедневное внутрибрюшинное введение GZ 161 успешно снижало, но не нормализировало уровни GluCer и GluSph в мозге мышей К14 (Фиг. 10). Имеется несколько свидетельств того, что GluSph и другие лизосфинголипиды, такие как галактозилсфингозин, могут вносить свой вклад в патологию ЦНС, инициируя продуцирование воспалительных медиаторов - см. Giri et al., Journal of lipid research 47: 1478-1492 (2006) и Gräler et al., Molecular and Cell Biology of Lipids 1582: 168-174 (2002). Способность GZ 161 понижать уровни GluSph и одновременно уменьшать окрашивание макрофагов/микроглии и астроцитов (Фиг. 11-13) согласуется с этой гипотезой. Поскольку GluSph обладает известными нейротоксичными свойствами (Schueler et al., Neurobiology of Disease 14: 595-601 (2003); Orvisky et al., Molecular Genetics and Metabolism 76: 262-270 (2002); Sun et al., Hum Mol Genet 19: 1088-1097 (2010); и Pelled et al., Journal of Inherited Metabolic Disease 23: 175-184 (2000)), неспособность терапии с применением GZ 161 нормализовать уровни GluSph согласуется с представлением о GluSph как о возможном действующем факторе, способствующем ранней смерти, характерной для этой модели.
В совокупности, преклинические результаты, полученные в настоящей работе на модели у мышей К14, свидетельствуют о том, что введение GZ 161 может сдерживать прогредиентное течение заболевания и развитие неврологических симптомов у пациентов с болезнью Гоше типа 2 и типа 3. Однако трудно прогнозировать возможную пользу от таких терапевтических подходов у пациентов с симптоматикой типа 2, поскольку известно, что их мозг содержит очень высокие уровни GluSph, возникшие еще в пренатальной жизни. Goker-Alpan et al., The Journal of Pediatrics 143: 273-276 (2003). Болезнь Гоше типа 3 может быть более чувствительной к лечению, поскольку в этом случае уровни GluSph в мозге являются более низкими (Nilsson, J Neurochem 39: 709-718 (1982)), а прогредиентное течение болезни является более медленным, несмотря на то, что она является частью фенотипического континуума (Goker-Alpan et al. (2003)) и в некоторых случаях можно выявить таких пациентов по результатам анализа мутаций еще до начала проявления невропатического фенотипа (Ida et al., Human Genetics 105: 120-126 (1999)). На основе получаемых результатов можно было бы заключить, что для лечения этих пациентов необходим более агрессивный способ терапии с ранним началом. Низкомолекулярные ингибиторы глюкозилцерамид-синтазы могут представлять собой одно из средств такого всестороннего подхода.
Пример 127
SMT самцов и самок мышей с болезнью Фабри, которых лечили соединениями GZ 452, GZ 161 и GZ 638
Лечение мышей с болезнью Фабри начинали в возрасте приблизительно 8 месяцев и продолжали в течение 4 месяцев, применяя: 60 мг/кг/день GZ 452 (Фаб 452 при 60 мг/кг/день), 120 мг/кг/день GZ 452 (Фаб 452 при 120 мг/кг/день), 20 мг/кг/день GZ 161 (Фаб 161 при 20 мг/кг/день), 300 мг/кг/день GZ 638 (Фаб 638 при 300 мг/кг/день). В ткани почек 12-месячных самцов и самок мышей с болезнью Фабри определяли уровни Gb3. Как показано на Фиг. 17, соединения GZ 161 и GZ 452 значительно уменьшали количество Gb3 в ткани почек по сравнению с контрольными животными, лечение которых не проводили (Фаб НЛ 12 мес).
Реферат
Настоящее изобретение относится к ингибиторам глюкозилцерамид-синтазы (GCS) формулы, а также к способу лечения с помощью указанных соединений. Технический результат: получены новые соединения, которые можно применять для лечения болезни лизосомного накопления, где указанная болезнь лизосомного накопления является результатом дефекта в гликосфинголипидном пути. 9 н. и 24 з.п. ф-лы, 20 фиг., 1 табл., 127 пр.
Формула

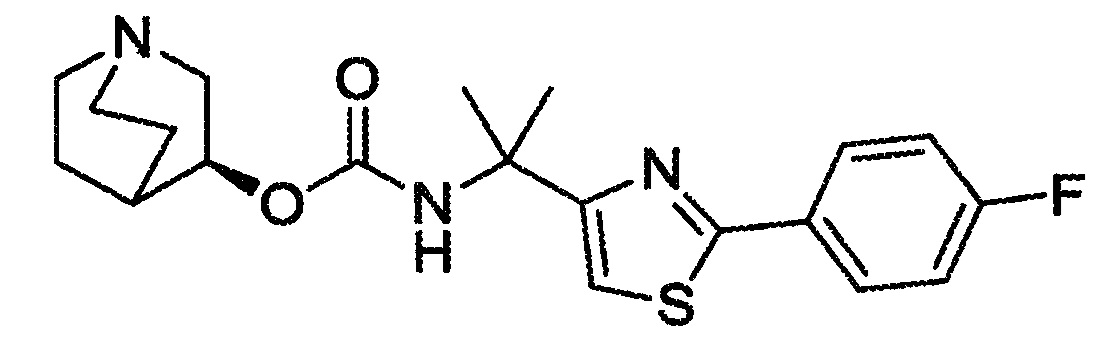
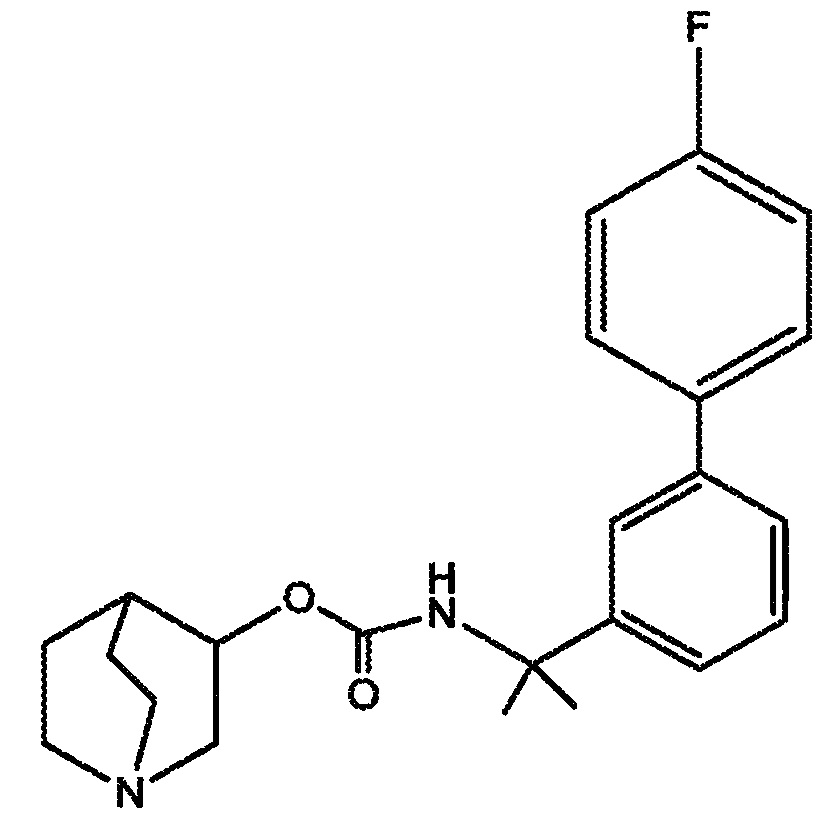
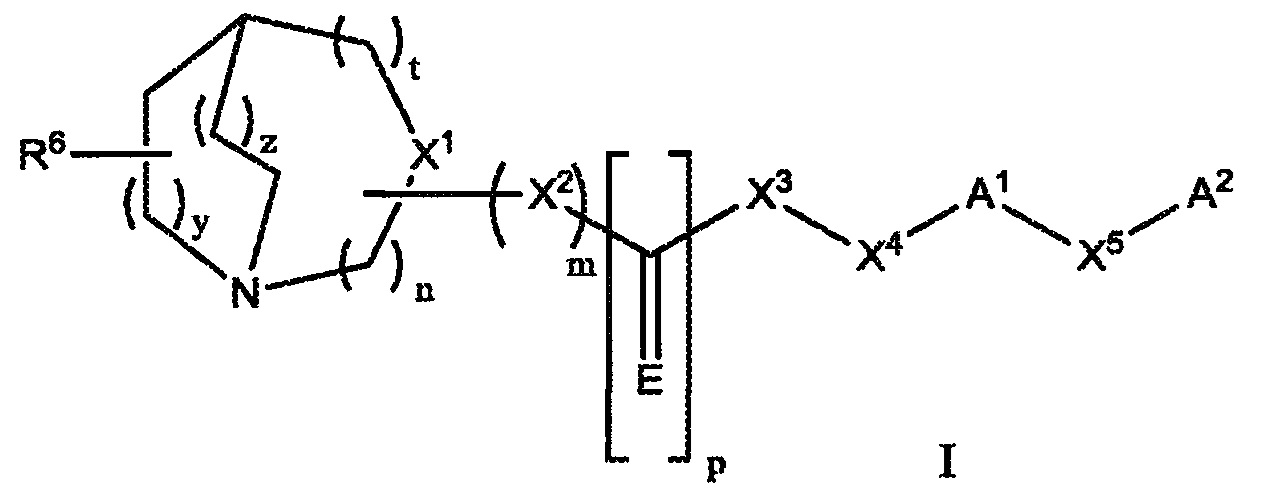
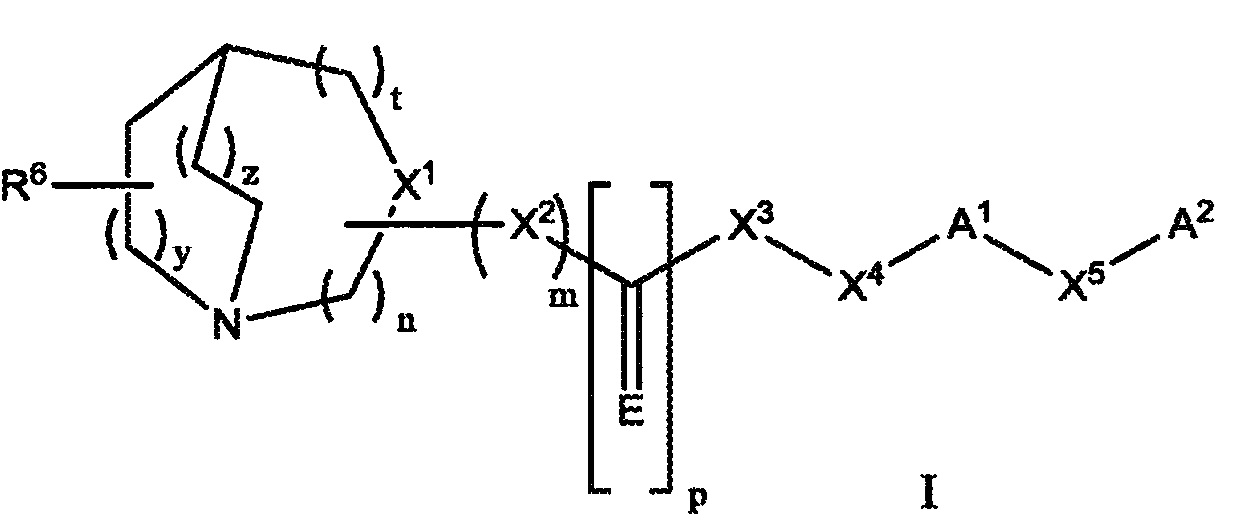


Комментарии