Бициклосульфониловая кислота (bcsa) и ее применение в качестве терапевтических агентов - RU2472784C2
Код документа: RU2472784C2
Описание
РОДСТВЕННАЯ ЗАЯВКА
Настоящая заявка является родственной Заявке на Патент США номер 60/924518, зарегистрированной 18 мая 2007 г., содержание которой полностью включено в настоящую заявку посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение в целом имеет отношение к области терапевтических соединений и более конкретно к некоторым соединениям, которые являются производными бициклосульфонильной кислоты (BCSA), действующим как ингибиторы фермента, катализирующего образование фактора некроза опухоли-α, (Tumour Necrosis Factor-α Converting Enzyme, TACE). Соединения согласно настоящему изобретению являются полезными для лечения состояний, опосредуемых фактором некроза опухоли-α (TNF-α), таких как ревматоидный артрит; воспаление; псориаз; септический шок; отторжение имплантата; кахексия; анорексия; застойная сердечная недостаточность; постишемическая травма, связанная с реперфузией; воспалительное заболевание центральной нервной системы; воспалительное заболевание кишечника; резистентность к инсулину; инфекция ВИЧ; рак; хроническое обструктивное заболевание легких (COPD) или астма. Настоящее изобретение также относится к фармацевтическим композициям, которые содержат указанные соединения и применению указанных соединений и композиций как in vitro, так и in vivo, для ингибирования ТАСЕ и лечения состояний, течение которых улучшается при ингибировании ТАСЕ.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
В целях более полного описания и раскрытия изобретения и предшествующего уровня техники, к которому относится настоящая заявка, в настоящем описании процитирован ряд патентов и публикаций. Содержание каждой из этих публикаций и патентов полностью включено в настоящее описание посредством ссылки, как если бы независимо в данное описание было включено содержание каждого индивидуального процитированного документа.
На протяжении настоящего описания, включая нижеследующую формулу изобретения, если из контекста не следует обратное, слово «включать/содержать» и его варианты, такие как «включает/содержит» и «включающий/содержащий» следует понимать как включение указанной сущности или этапа, или группы сущностей или этапов, но не в качестве исключения любой другой сущности или этапа, или группы сущностей или этапов.
Следует отметить, что в описании и прилагаемой формуле изобретения применяемые формы единственного числа охватывают также соответствующие множественные формы, если контекст явно не указывает на обратное. Так, например, ссылка на "фармацевтический носитель" включает смеси из двух или более таких носителей, и тому подобное.
В настоящем описании диапазоны часто выражены в виде от "приблизительно" одного конкретного значения и/или до "приблизительно" другого конкретного значения. Когда диапазон представлен в таком виде, другие варианты реализации включают диапазон от указанного конкретного значения и/или до другого указанного значения. Подобным образом, если значения выражены приближенно, применение оборота "приблизительно" следует трактовать таким образом, что конкретные значения включены в другие варианты реализации настоящего изобретения.
ТАСЕ
ТАСЕ (фермент, конвертирующий в TNF-α) катализирует образование TNF-α (фактора некроза опухоли-α) из мембранно-связанного белка предшественника TNF-α. TNF-α представляет собой провоспалительный цитокин, который, как считают, играет роль в многочисленных заболеваниях, включая следующие:
ревматоидный артрит (см., например, Shire et al., 1998; Isomaki et al., 1997; Camussi et al., 1998);
воспаление (см., например, Ksontini et al., 1988);
псориаз (см., например, Le et al., 2005; Palladino et al., 2003);
септический шок (см., например, Mathison et al.,1988, Miethke et al., 1992);
отторжение имплантата (см., например, Piguet et al., 1987;
кахексию (см., например, Beutler et al., 1988);
анорексию (см., например, Schattneret al., 1990);
застойную сердечную недостаточность (см., например, Packer et al., 1995; Ferrari et al., 1995);
постишемическую травму, связанную с реперфузией (см., например, Gu et al., 2006);
воспалительное заболевание центральной нервной системы (см., например, Grau et al., 1987);
воспалительное заболевание кишечника (см., например, McDonald et al., 1990);
резистентность к инсулину (см., например, Hotamisligil et al., 1993);
инфекцию ВИЧ (см., например, Peterson et al., 1992; Pallares-Trujillo et al., 1995);
рак (см., например, Old, 1985);
хроническое обструктивное заболевание легких (COPD) или астму (см., например, Trifilieff et al., 2002).
Дополнительные примеры таких заболеваний включают: остеоартрит, язвенный колит, болезнь Крона, рассеянный склероз и дегенеративную потерю хрящевой ткани.
Несколько исследовательских групп синтезировали производные гидроксамовой кислоты, содержащие сульфонамидную группу, в качестве потенциальных антипролиферативных и противовоспалительных агентов (см., например, Levin et al., 1999; Ohtani et al., 1993; Owen et al., 2000, Yu et al., 2006).
Хотя известен ряд ингибиторов ТАСЕ, многие из этих соединений являются пептидами или аналогами пептидов, к недостаткам которых относятся проблемы с биодоступностью и фармакокинетическим профилем. Кроме того, многие из таких соединений демонстрируют отсутствие селективности, являясь мощными ингибиторами металлопротеаз матрикса, в частности ММР-1 (коллагеназы 1). На основании клинических испытаний ингибиторов металлопротеаз предполагают, что ММР-1 вызывает боль в суставах (см., например, Scrip, 1988).
Таким образом, обладающие длительным действием, селективные, биодоступные при пероральном введении, непептидные ингибиторы ТАСЕ являются крайне желательными для лечения вышеуказанных состояний.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Один из аспектов настоящего изобретения относится к некоторым производным "бициклосульфонильной кислоты" (BCSA), описанным в настоящей заявке.
Еще один аспект изобретения, раскрытого в настоящей заявке, относится к фармацевтической композиции, которая содержит производное BCSA, а также фармацевтически приемлемый носитель, разбавитель или наполнитель.
Еще один аспект настоящего изобретения, раскрытого в настоящей заявке, относится к способу приготовления фармацевтической композиции, включающему смешивание производного BCSA, описанного в настоящей заявке, и фармацевтически приемлемого носителя, разбавителя или наполнителя.
Еще один аспект настоящего изобретения, описанный в настоящей заявке, относится к производному BCSA для применения в способе лечения (например, заболевания или расстройства) организма человека или животного посредством терапии.
Еще один аспект настоящего изобретения, описанный в настоящей заявке, относится к применению производного BCSA в изготовлении лекарственного средства для лечения (например, заболевания или расстройства) организма человека или животного.
Еще один аспект настоящего изобретения, описанный в настоящей заявке, относится к способу лечения (например, заболевания или расстройства), включающему введение пациенту, нуждающемуся в указанном лечении, терапевтически эффективного количества BCSA, предпочтительно в виде фармацевтической композиции.
В одном варианте реализации настоящего изобретения указанное лечение представляет собой лечение заболевания или расстройства, опосредуемого ТАСЕ, например заболевания или расстройства, про которое известно, что оно опосредуется ТАСЕ.
В одном варианте реализации указанное лечение представляет собой лечение заболевания или расстройства, симптоматика которого улучшается при ингибировании ТАСЕ, например заболевания или расстройства, про которое известно, что симптоматика указанного состояния улучшается при ингибировании ТАСЕ.
В одном варианте реализации указанное лечение представляет собой лечение заболевания или расстройства, которое лечат ингибитором ТАСЕ, например заболевания или расстройства, про которое известно, что его лечат ингибитором ТАСЕ.
В одном варианте реализации указанное лечение представляет собой лечение ревматоидного артрита; воспаления; псориаза; септического шока; отторжения имплантата; кахексии; анорексии; застойной сердечной недостаточности; постишемической травмы, связанной с реперфузией; воспалительного заболевания центральной нервной системы; воспалительного заболевания кишечника; резистентности к инсулину; инфекции ВИЧ; рака; хронического обструктивного заболевания легких (COPD) или астмы.
В одном варианте реализации указанное лечение представляет собой лечение остеоартрита, язвенного колита, болезни Крона, рассеянного склероза или дегенеративной потери хряща.
В одном варианте реализации указанное лечение представляет собой лечение воспаления.
В одном варианте реализации указанное лечение представляет собой лечение ревматоидного артрита.
В одном варианте реализации указанное лечение представляет собой лечение псориаза.
Другой аспект настоящего изобретения относится к способу ингибирования ТАСЕ в клетке in vitro или in vivo, который включает осуществление контакта указанной клетки с эффективным количеством производного BCSA, описанного в настоящей заявке.
Еще один аспект настоящего изобретения относится к способу регулирования (например, ингибирования) высвобождения в клетке цитокинов (например, высвобождения TNF-α), in vitro или in vivo, который включает приведение указанной клетки в контакт с эффективным количеством производного BCSA, описанного в настоящей заявке.
Еще один аспект настоящего изобретения относится к набору, который содержит: (а) производное BCSA, описанное в настоящей заявке, предпочтительно в составе фармацевтической композиции и в подходящем контейнере и/или в подходящей упаковке и (b) инструкции по применению, например письменные инструкции о том, как принимать указанное производное/композицию.
Еще один аспект настоящего изобретения относится к соединениям, которые можно получать описанным в настоящей заявке способом синтеза или способом, включающим описанный в настоящей заявке способ синтеза.
Еще один аспект настоящего изобретения относится к соединениям, полученным посредством описанного в настоящей заявке способа синтеза или способом, который включает описанный в настоящей заявке способ синтеза.
Еще один аспект настоящего изобретения относится к новым промежуточным соединениям, описанным в настоящей заявке, пригодным для применения в способах синтеза, описанных в настоящей заявке.
Еще один аспект настоящего изобретения относится к применению таких новых промежуточных соединений, описанных в настоящей заявке, пригодных для применения в способах синтеза, описанных в настоящей заявке.
Специалист в данной области примет во внимание, что особенности и предпочтительные варианты реализации одного аспекта изобретения будут также относиться к другим аспектам изобретения.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Соединения
Один из аспектов настоящего изобретения относится к соединениям нижеследующей формулы и их фармацевтически приемлемым солям, гидратам и сольватам (которые собирательно называют в настоящей заявке «соединениями (BCSA) бициклосульфонильной кислоты»:
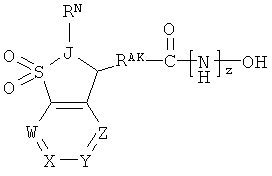
где:
W независимо представляет собой -N= или -CRPW=;
X независимо представляет собой -N= или -CRPX=;
Y независимо представляет собой -N= или -CRPY=;
Z независимо представляет собой -N= или -CRPZ=;
каждый из -RPW, -RPX, -RPY и -RPZ, если присутствует, независимо представляет собой -Н или -RRS1;
где каждый -RRS1, если присутствует, независимо представляет собой заместитель при кольце;
и где z равно 0 или 1;
и где -J< независимо представляет собой -N< или -СН<;
и где:
-RAK- независимо представляет собой:
ковалентную связь,
-RAK1-, -RAK2-, -RAK3-,
-RAK4-, -RAK1-RAK4-, -RAK4-RAK1-, -RAK1-RAK4-RAK1-,
-RAK5-, -RAK1-RAK5-, -RAK5-RAK1-, или -RAK1-RAK5-RAK1-.
где:
каждый -RAK1- независимо представляет собой насыщенный алифатический C1-6 алкилен, который может иметь заместители;
-RAK2- независимо представляет собой алифатический С2-6 алкенилен, который может иметь заместители;
-RAK3- независимо представляет собой алифатический С2-6 алкинилен, который может иметь заместители;
каждый -RAK4- независимо представляет собой насыщенный С3-6 циклоалкилен, который может иметь заместители и
каждый -RAK5- независимо представляет собой С3-6 циклоалкенилен, который может иметь заместители;
и где:
-RN независимо представляет собой -Н, -RNN, -RNNN или -LN-RNNN; где:
-LN- независимо представляет собой насыщенный алифатический C1-6 алкилен, который может иметь заместители;
-RNNN независимо представляет собой С1-6алкил, который может иметь заместители
и
-RNNN независимо представляет собой С3-6 циклоалкил, С3-7 гетероциклил, С6-10 карбоарил или С5-10 гетероарил, которые могут иметь заместители.
Стереохимия
Многие из приведенных в настоящем описании химических структур показаны в одной или более различных стереоизомерных конфигурациях. Аналогично, во многих показанных в настоящем описании химических структурах их стереизомерная конфигурация не показана. Аналогично, во многих, описанных в настоящей заявке структурах показаны конкретные стереоизомерные конфигурации в одном или более положениях, но они не показаны в одном или более других положениях. Если у приведенной в настоящей заявке химической структуры стереоизомерная конфигурация в определенном положении указанной структуры не показана, подразумевается что такая изображенная структура охватывает все возможные стереоизомерные конфигурации в данном положении, как конкретно для данной конфигурации, и так как если бы каждая стереоизомерная конфигурация, равно как и смесь стереоизомеров (то есть рацемическая смесь) для данного положения были упомянуты индивидуально.
Отметим, что углеродный атом в кольце, присоединенный к группе J (то есть атом, помеченный звездочкой (*) в следующей формуле) обязательно является хиральным центром.

В одном варианте реализации углеродный атом в кольце, присоединенный к группе J (то есть атом, помеченный звездочкой (*)) имеет конфигурацию, описываемую следующей формулой:

В одном варианте реализации углеродный атом в кольце, присоединенный к группе J (то есть атом, помеченный звездочкой (*)) имеет конфигурацию, описываемую следующей формулой:

В одном варианте реализации углеродный атом в кольце, присоединенный к группе J (то есть атом, помеченный звездочкой (*)) находится в (R) конфигурации.
В одном варианте реализации углеродный атом в кольце, присоединенный к группе J (то есть атом, помеченный звездочкой (*)) находится в (S) конфигурации.
Группы W, X, Y и Z
В одном варианте реализации:
W независимо представляет собой -N= или -CRPW=,
X независимо представляет собой -N= или -CRPX=,
Y независимо представляет собой -N= или -CRPY= и
Z независимо представляет собой -N= или -CRPZ=;
причем только одна или только две группы из W, X, Y и Z представляют собой -N=.
В одном варианте реализации:
W независимо представляет собой -N= или -CRPW=,
X независимо представляет собой -N= или -CRPX=,
Y независимо представляет собой -N= или -CRPY=, и
Z независимо представляет собой -N= или -CRPZ=;
причем только одна группа из W, X, Y и Z представляет собой -N=.
В одном варианте реализации:
W независимо представляет собой -CRPW=,
X независимо представляет собой -CRPX=,
Y независимо представляет собой -CRPY= и
Z независимо представляет собой -CRPZ=
В одном варианте реализации:
W независимо представляет собой -N=,
X независимо представляет собой -CRPX=,
Y независимо представляет собой -CRPY= и
Z независимо представляет собой -CRPZ=.
В одном варианте реализации:
W независимо представляет собой -CRPW=,
X независимо представляет собой -N=,
Y независимо представляет собой -CRPY= и
Z независимо представляет собой -CRPZ=.
В одном варианте реализации:
W независимо представляет собой -CRPW=
X независимо представляет собой -CRPX=,
Y независимо представляет собой -N= и
Z независимо представляет собой -CRPZ=.
В одном варианте реализации:
W независимо представляет собой -CRPW=,
X независимо представляет собой -CRPX=,
Y независимо представляет собой -CRPY= и
Z независимо представляет собой -N=.
В одном варианте реализации, каждый из -RPW, -RPX, -RPY и -RPZ, если присутствует, независимо представляет собой -Н.
Группа -[NH]z-
В одном варианте реализации z независимо равно 1.
В одном варианте реализации z независимо равно 0.
Группа J
В одном варианте реализации -J< независимо представляет собой -N<.
В одном варианте реализации -J< независимо представляет собой -СН<.
Группа -RAK-
В одном варианте реализации -RAK- независимо представляет собой: ковалентную связь,
-RAK1-, -RAK2-, -RAK3-
-RAK4-, -RAK1-RAK4-, -RAK4-RAK1-, -RAK1-RAK4-RAK1-,
-RAK5-, -RAK1-RAK5-, -RAK5-RAK1-, -RAK1-RAK5-RAK1-.
В одном варианте реализации -RAK- независимо представляет собой:
-RAK1, -RAK2-, -RAK3-
-RAK4-, -RAK1-RAK4-, -RAK4-RAK1-, -RAK1-RAK4-RAK1-,
-RAK5-, -RAK1-RAK5-, -RAK5-RAK1-, -RAK1-RAK5-RAK1-.
В одном варианте реализации -RAK- представляет собой независимо:
-RAK1-, -RAK2-, -RAK3-,
-RAK4-, -RAK1-RAK4-, -RAK4-RAK1-, -RAK1-RAK4-RAK1-.
В одном варианте реализации -RAK- независимо представляет собой -RAK1-, -RAK2- или -RAK3-.
В одном варианте реализации -RAK- независимо представляет собой -RAK1- или RAK2-.
В одном варианте реализации -RAK- независимо представляет собой -RAK1-.
В одном варианте реализации -RAK- независимо представляет собой -RAK2-.
В одном варианте реализации -RAK- независимо представляет собой -RAK3-.
В одном варианте реализации -RAK- независимо представляет собой -RAK1- или ковалентную связь.
В одном варианте реализации -RAK- независимо представляет собой ковалентную связь.
В одном варианте реализации -RAK- независимо представляет собой:
-RAK4-, -RAK1-RAK4-, -RAK4-RAK1- или -RAK1-RAK4-RAK1-.
В одном варианте реализации -RAK- независимо представляет собой -RAK4-.
В одном варианте реализации -RAK- независимо представляет собой -RAK1-RAK4-.
В одном варианте реализации -RAK- независимо представляет собой -RAK4-RAK1-.
В одном варианте реализации -RAK- независимо представляет собой
-RAK1-RAK4-RAK1-.
Группа -RAK1-
В одном варианте реализации, каждый -RAK1-, если присутствует, независимо представляет собой насыщенный алифатический С1-6 алкилен, который может иметь заместители.
В одном варианте реализации, каждый -RAK1-, если присутствует, независимо представляет собой насыщенный алифатический С1-4 алкилен, который может иметь заместители.
В одном варианте реализации, каждый -RAK1-, если присутствует, может независимо не иметь или иметь заместители, например один или более заместитель (например, 1, 2, 3) -RG1.
В одном варианте реализации, у каждого -RAK1-, если он присутствует, заместители независимым образом отсутствуют.
В одном варианте реализации, каждый -RAK1-, если присутствует, независимо представляет собой -(CH2)q-, где q независимо равен 1, 2, 3, 4, 5 или 6.
В одном варианте реализации, каждый -RAK1-, если присутствует, независимо представляет собой -(CH2)-, -(CH2)2-, -(CH2)3- или -(CH2)4-.
В одном варианте реализации, каждый -RAK1-, если присутствует, независимо представляет собой -(CH2)-, -(CH2)2- или -(CH2)3-.
В одном варианте реализации, каждый -RAK1-, если присутствует, независимо представляет собой -(CH2)- или -(CH2)2-.
В одном варианте реализации, каждый -RAK1-, если присутствует, независимо представляет собой -(CH2)-.
Группа -RAK2-
В одном варианте реализации, -RAK2-, если присутствует, независимо представляет собой алифатический С2-6алкенилен, который может иметь заместители.
Термин "С2-6 алкенилен", применяемый в настоящей заявке, относится к двухвалентной бидентатной алифатической гидрокарбильной группе, которая содержит от 2 до 6 атомов углерода и имеет по меньшей мере одну углерод-углеродную связь, но не имеет тройных углерод-углеродных связей.
В одном варианте реализации, -RAK2-, если присутствует, независимо представляет собой алифатический С2-4 алкенилен, который может иметь заместители.
В одном варианте реализации, -RAK2-, если присутствует, независимо может не иметь или иметь заместители, например один или более заместитель, например один или более (например, 1, 2, 3) заместителя -RG1.
В одном варианте реализации, -RAK2-, если присутствует, независимо не имеет заместители.
В одном варианте реализации, -RAK2-, если присутствует, независимо представляет собой:
-СН=СН-,
-С(СН3)=СН-, -СН=С(СН3)-,
-СН=СН-CH2-,
-С(СН3)=СН-CH2-, -СН=С(СН3)-CH2-, -СН=СН-СН(СН3)-,
-CH2-СН=СН-,
-СН(СН3)-СН=СН-, -CH2-С(СН3)=СН-, -CH2-СН=С(СН3)-,
-СН=СН-CH2-CH2-, -CH2-СН=СН-CH2- или -CH2-CH2-СН=СН-.
Группа -RAK3-
В одном варианте реализации, -RAK3-, если присутствует, независимо представляет собой алифатический С2-6алкинилен, который может иметь заместители.
Термин "С2-6алкинилен", применяемый в настоящем описании, относится к двухвалентной бидентатной алифатической гидрокарбильной группе, которая содержит по меньшей мере одну углерод-углеродную тройную связь и, возможно, также одну или более двойные углерод-углеродные связи.
В одном варианте реализации, -RAK3-, если присутствует, независимо представляет собой алифатический С2-4 алкинилен, который может иметь заместители.
В одном варианте реализации, группа -RAK3-, если присутствует, независимо не имеет или имеет заместители, например один или более заместитель, например один или более (например, 1, 2, 3) заместитель -RG1.
В одном варианте реализации, -RAK3-, если присутствует, независимо не имеет заместители.
В одном варианте реализации, -RAK3-, если присутствует, независимо представляет собой:
-C≡C-,
-С≡С-CH2-, -С≡С-СН(СН3)-,
-CH2-С≡С-, -СН(СН3)-С≡С-,
-С≡С-CH2-CH2-, -С≡С-СН(СН3)-CH2-, -С≡С-CH2-СН(СН3)-,
-CH2-С≡С-CH2-, -СН(СН3)-С≡С-CH2-, -CH2-С≡С-СН(СН3)-,
-CH2-CH2-С≡С-, -СН(СН3)-CH2-С≡С-, -CH2-СН(СН3)-С≡С-,
-С≡С-СН=СН-, -С≡С-С(СН3)=СН-, -С≡С-СН=С(СН3)-,
-СН=СН-С≡С-, -С(СН3)=СН-С≡С- или -СН=С(СН3)-С≡С-.
Группы -RAK4-, -RAK1-RAK4-, -RAK4-RAK1- и -RAK1-RAK4-RAK1-
В одном варианте реализации, каждый -RAK4-, если присутствует, независимо представляет собой насыщенный С3-6 циклоалкилен, который может иметь заместители.
Термин "насыщенный С3-6 циклоалкилен", применяемый в настоящей заявке, относится к двухвалентной бидентатной насыщенной карбоциклической группе, которая содержит в кольце от 3 до 6 атомов, причем указанные атомы кольца представляют собой атомы углерода, и где один или два указанных атомов кольца являются точками присоединения.
В одном варианте реализации, каждый -RAK4-, если присутствует, независимо представляет собой насыщенный С3-5 циклоалкилен, который может иметь заместители.
В одном варианте реализации, каждый -RAK4-, если присутствует, независимо представляет собой насыщенный и возможно замещенный С3-4 циклоалкилен, который может иметь заместители.
В одном варианте реализации, каждый -RAK4-, если присутствует, независимо представляет собой насыщенный и возможно замещенный С4-6 циклоалкилен, который может иметь заместители.
В одном варианте реализации, каждый -RAK4-, если присутствует, независимо представляет собой насыщенный и возможно замещенный С5-6 циклоалкилен, который может иметь заместители.
В одном варианте реализации, каждый -RAK4-, если присутствует, независимо может не иметь или иметь заместители, например один или более заместитель, например один или более (например, 1, 2, 3) заместитель -RG1.
В одном варианте реализации, каждый -RAK4-, если присутствует, независимо не имеет заместители.
В одном варианте реализации, каждый -RAK4-, если присутствует, независимо представляет собой:
циклопропил-диил, циклобутил-диил, циклопентил-диил или циклогексил-диил.
В одном варианте реализации, каждый -RAK4-, если присутствует, независимо представляет собой циклопропил-диил.
В одном варианте реализации, каждый -RAK4-, если присутствует, независимо представляет собой циклопропил-1,1-диил.
В одном варианте реализации, каждый -RAK1-RAK4-, если присутствует, независимо представляет собой:
метиленциклопропил-диил, метиленциклобутил-диил, метиленциклопентил-диил или метиленциклогексил-диил.
В одном варианте реализации, каждый -RAK4-RAK1-, если присутствует, независимо представляет собой:
циклопропил-диил-метилен, циклобутил-диил-метилен,
циклопентил-диил-метилен или циклогексил-диил-метилен.
В одном варианте реализации, -RAK1-RAK4-RAK1-, если присутствует, независимо представляет собой:
метилен-циклопропил-диил-метилен, метилен-циклобутил-диил-метилен,
метилен-циклопентил-диил-метилен или метилен-циклогексил-диил-метилен.
Группа -RAK5-
В одном варианте реализации, каждый -RAK5-, если присутствует, независимо представляет собой С3-6 циклоалкенилен, который может иметь заместители.
Термин "С3-6 циклоалкенилен", применяемый в настоящей заявке, относится к двухвалентной бидентатной карбоциклической группе, которая содержит в кольце от 3 до 6 атомов и имеет в кольце по меньшей мере одну углерод-углеродную двойную связь, но не имеет в кольце тройные углерод-углеродные связи, причем указанные атомы кольца представляют собой атомы углерода, и один или два указанных атома кольца являются точками присоединения.
В одном варианте реализации, каждый -RAK5-, если присутствует, независимо представляет собой С3-5 циклоалкенилен, который может иметь заместители.
В одном варианте реализации, каждый -RAK5-, если присутствует, независимо представляет собой С3-4 циклоалкенилен, который может иметь заместители.
В одном варианте реализации, каждый -RAK5-, если присутствует, независимо представляет собой С4-6 циклоалкенилен, который может иметь заместители.
В одном варианте реализации, каждый -RAK5-, если присутствует, независимо представляет собой С5-6 циклоалкенилен, который может иметь заместители.
В одном варианте реализации, каждый -RAK5-, если присутствует, независимо может не иметь или иметь заместители, например один или более заместитель, например один или более (например, 1, 2, 3) заместитель -RG1.
В одном варианте реализации, каждый -RAK5-, если присутствует, независимо не имеет заместители.
В одном варианте реализации, каждый -RAK5-, если присутствует, независимо представляет собой:
циклопропенил-диил, циклобутенил-диил, циклопентенил-диил или циклогексенил-диил.
В одном варианте реализации, каждый -RAK1-RAK5-, если присутствует, независимо представляет собой:
Метилен-циклопропенил-диил, метилен-циклобутенил-диил,
Метилен-циклопентенил-диил или метилен-циклогексенил-диил.
В одном варианте реализации, каждый -RAK5-RAK1-, если присутствует, независимо представляет собой:
циклопропенил-диил-метилен, циклобутенил-диил-метилен,
циклопентенил-диил-метилен или циклогексенил-диил-метилен.
В одном варианте реализации, -RAK1-RAK5-RAK1-, если присутствует, независимо представляет собой:
метилен-циклопропенил-диил-метилен, метилен-циклобутенил-диил-метилен,
метилен-циклопентенил-диил-метилен или метилен-циклогексенил-диил-метилен.
Заместители -RG1
В одном варианте реализации, каждый -RG1, если присутствует, независимо представляет собой -F, -Cl, -Br, -I, -ОН, -ORA1, -OCF3, -С(=O)ОН, -C(=O)ORA1, -NH2, -NHRA1, -NRA12, -NRA2RA3, -C(=O)-NH2, -C(=O)-NHRA1, -C(=O)-NRA12, -C(=O)-NRA2RA3, фенил или бензил; где каждый радикал RA1 независимо представляет собой алкил, фенил или бензил; и каждый -NRA2RA3 независимо представляет собой пирролидино-, пиперидино-, пиперизино- или морфолино-группу, которая может не иметь или иметь заместители, выбранные из С1-3алкила и -CF3.
В одном варианте реализации, каждый -RG1, если присутствует, независимо представляет собой -F, -Cl, -Br, -I, -ОН, -ОМе, -OEt, или -OCF3.
Группа -RN
В одном варианте реализации -RN независимо представляет собой -Н, -RNN, -RNN, или -LN-RNNN.
В одном варианте реализации, -RN независимо представляет собой -Н, -RNNN или
-LN-RNNN.
В одном варианте реализации, -RN независимо представляет собой -Н или -RNN.
В одном варианте реализации, -RN независимо представляет собой -RNNN или -LN-RNNN.
В одном варианте реализации, -RN независимо представляет собой -Н.
В одном варианте реализации, -RN независимо представляет собой -RNN.
В одном варианте реализации, -RN независимо представляет собой -RNNN.
В одном варианте реализации, -RN независимо представляет собой -LN-RNNN.
Группа -LN-
В одном варианте реализации -LN-, если присутствует, независимо представляет собой насыщенный алифатический С1-6алкилен, который может иметь заместители.
В одном варианте реализации, -LN-, если присутствует, независимо представляет собой насыщенный алифатический С1-3алкилен, который может иметь заместители.
В одном варианте реализации -LN-, если присутствует, независимо может не иметь или иметь заместители, например один или более заместитель, например один или более (например, 1, 2, 3) заместитель -RG2.
В одном варианте реализации, -LN-, если присутствует, независимо не имеет заместителей.
В одном варианте реализации, -LN-, если присутствует, независимо представляет собой -CH2-, -CH2CH2- или -CH2CH2CH2-.
В одном варианте реализации, -LN-, если присутствует, независимо представляет собой -CH2- или -CH2CH2-.
В одном варианте реализации, -LN-, если присутствует, независимо представляет собой -CH2-.
Заместители -RG2
В одном варианте реализации, каждый -RG2, если присутствует, независимо представляет собой -F, -Cl, -Br, -I, -ОН, -ORA1, -OCF3, -С(=O)ОН, -C(=O)ORA1,
-NH2, -NHRA1,


В одном варианте реализации, каждый -RG2, если присутствует, независимо представляет собой -F, -Cl, -Br, -I, -ОН, -ОМе, -OEt или -OCF3.
Группа -RNN
В одном варианте реализации, -RNN, если присутствует, независимо представляет собой С1-6 алкил, который может иметь заместители.
В одном варианте реализации, -RNN, если присутствует, независимо представляет собой С1-4алкил, который может иметь заместители.
В одном варианте реализации, -RNN, если присутствует, независимо не имеет или имеет заместители, например один или более заместитель, например один или более (например, 1, 2, 3) заместитель -RG3.
В одном варианте реализации, -RNN, если присутствует, независимо не имеет заместителей.
В одном варианте реализации, -RNN, если присутствует, независимо представляет собой -Me, -Et, -н-Pr, или -изо-Pr.
Заместители -RG3
В одном варианте реализации, каждый -RG3, если присутствует, независимо представляет собой -F, -Cl, -Br, -I, -ОН, -ORA1, -OCF3, -С(=O)ОН, -C(=O)ORA1,
-NH2, -NHRA1,

В одном варианте реализации, каждый -RG3, если присутствует, независимо представляет собой -F, -Cl, -Br, -I, -ОН, -ОМе, -OEt или -OCF3.
Группа -RNNN
В одном варианте реализации, -RNNN, если присутствует, независимо представляет собой С3-6 циклоалкил, С3-7 гетероциклил, С6-10 карбоарил или С5-10 гетероарил, которые могут иметь заместители.
В одном варианте реализации, -RNNN, если присутствует, независимо представляет собой циклопропил, циклобутил, циклопентил, циклогексил, пирролидинил, имидазолидинил, пиразолидинил, пиперидинил, пиперизинил, морфолинил, тиоморфолинил, азепинил, диазепинил, фенил, нафтил, фуранил, тиенил, пирролил, имидазолил, пиразолил, триазолил, оксазолил, изоксазолил, тиазолил, изотиазолил, пиридил, пиразинил, пиримидинил, пиридазинил, бензофуранил, изобензофуранил, индазолил, пуринил, хинолинил, изохинолинил, нафтиридинил, хиноксалинил, хиназолинил, циннолинил, индолил, изоиндолил, карбазолил, карболинил, акридинил, феноксазинил или фенотиазинил, которые могут иметь заместители.
В одном варианте реализации, -RNNN, если присутствует, независимо представляет собой С6-10 карбоарил или С5-10 гетероарил, которые могут иметь заместители.
В одном варианте реализации, -RNNN, если присутствует, независимо представляет собой фенил, нафтил, фуранил, тиенил, пирролил, имидазолил, пиразолил, триазолил, оксазолил, изоксазолил, тиазолил, изотиазолил, пиридил, пиразинил, пиримидинил, пиридазинил, бензофуранил, изобензофуранил, индазолил, пуринил, хинолинил, изохинолинил, нафтиридинил, хиноксалинил, хиназолинил, циннолинил, индолил, изоиндолил, карбазолил, карболинил, акридинил, феноксазинил или фенотиазинил, которые могут иметь заместители.
В одном варианте реализации, -RNNN, если присутствует, независимо представляет собой фенил, нафтил, фуранил, тиенил, пирролил, имидазолил, пиразолил, триазолил, оксазолил, изоксазолил, тиазолил, изотиазолил, пиридил, пиразинил, пиримидинил или пиридазинил, которые могут иметь заместители.
В одном варианте реализации, -RNNN, если присутствует, независимо представляет собой фенил, нафтил, пиридил, пиразинил, пиримидинил, пиридазинил или пиразолил, которые могут иметь заместители.
В одном варианте реализации, -RNNN, если присутствует, независимо представляет собой возможно замещенные фенил, нафтил, пиридил или пиразолил, которые могут иметь заместители.
В одном варианте реализации, -RNNN, если присутствует, независимо представляет собой возможно замещенные фенил или нафтил, которые могут иметь заместители.
В одном варианте реализации, -RNNN, если присутствует, независимо представляет собой возможно замещенный фенил, который может иметь заместитель.
В одном варианте реализации, -RNNN, если присутствует, независимо может не иметь или иметь заместители, например, не иметь или иметь один или более (например, 1, 2, 3) заместитель.
В одном варианте реализации, -RNNN, если присутствует, независимо представляет собой фенил, который может иметь заместитель в параположении и быть незамещенным по всем другим положениям.
В одном варианте реализации, каждый заместитель у -RNNN, если присутствует, независимо представляет собой -RS.
В одном варианте реализации, -RNNN, если присутствует, независимо не имеет заместители.
Заместители -RRS1
В одном варианте реализации, каждый -RRS1, если присутствует, независимо представляет собой такую же группу, как определены для -RS.
В одном варианте реализации, каждый -RRS1, если присутствует, независимо представляет собой -F, -Cl, -Br, -I, -RA1, -CF3, -ОН, -ORA1, -OCF3, -С(=O)ОН, -C(=O)ORA1, -NH2, -NHRA1,
В одном варианте реализации, каждый -RRS1, если присутствует, независимо представляет собой -F, -Cl, -Br, -I, -Me, -Et, -CF3, -ОН, -ОМе, -OEt, -OCF3, или фенил; и дополнительно две смежные группы -RRS1, если присутствуют, могут образовывать -OCH2CH2O-.
В одном варианте реализации, каждый -RRS1, если присутствует, представляет собой независимо -F, -Cl, -Br, -Me, -CF3, -ОМе, -OEt или фенил и дополнительно две смежные группы -RRS1, если присутствуют, могут образовать -OCH2CH2O-.
Заместители -RS
В одном варианте реализации, каждый -RS, если присутствует, независимо представляет собой:
-F, -Cl, -Br, -I,
-RD1,
-CF3, -CH2CF3, -CF2CF2H,
-ОН,
-L1-OH,
-O-L1-OH,
-ORD1,
-L1-ORD1,
-O-L1-ORD1,
-OCF3, -OCH2CF3, -OCF2CF2H,
-SH,
-SRD1, -SCF3,
-CN,
-NO2,
-NH2, -NHRD1, -NRD12, -NRN1RN2,
-L1-NH2, -L1-NHRD1, -L1-NRD12, -L1-NRN1RN2,
-O-L1-NH2, -O-L1-NHRD1, -O-L1-NRD12, -O-L1-NRN1RN2,
-NH-L1-NH2, -NH-L1-NHRD1, -NH-L1-NRD12, -NH-L1-NRN1RN2,
-NRD1-L1-NH2, -NRD1-L1-NHRD1, -NRD1-L1-NRD12, -NRD1-L1-NRN1RN2,
-C(=O)OH,
-C(=O)ORD1,
-C(=O)NH2, -C(=O)NHRD1, -C(=O)NRD12, -C(=O)NRN1RN2,
-NHC(=O)RD1, -NRD1C(=O)RD1,
-NHC(=O)ORD1, -NRD1C(=O)ORD1,
-OC(=O)NH2, -OC(=O)NHRD1, -OC(=O)NRD12, -OC(=O)NRN1RN2,
-OC(=O)RD1,
-C(=O)RD1,
-NHC(=O)NH2, -NHC(=O)NHRD1, -NHC(=O)NRD12, -NHC(=O)NRN1RN2,
-NRD1C(=O)NH2, -NRD1C(=O)NHRD1, -NRD1C(=O)NRD12, -NRD1C(=O)NRN1RN2,
-NHS(=O)2RD1, -NRD1S(=O)2RD1,
-S(=O)2NH2, -S(=O)2NHRD1, -S(=O)2NRD12, -S(=O)2NRN1RN2,
-S(=O)RD1,
-S(=O)2RD1,
-OS(=O)2RD1,
-S(=O)2ORD1,
=O,
=NRD1,
=NOH или
=NORD1
и дополнительно две близко расположенные к кольцу группы -RS, если присутствуют, совместно могут образовать группу -O-L2-O-;
где:
каждый -L1- независимо представляет собой насыщенный алифатический С1-5 алкилен, алифатический С2-5 алкенилен или алифатический С2-5 алкинилен;
каждый -L2- независимо представляет собой насыщенный алифатический С1-3 алкилен;
каждая из групп -NRN1RN2, -RN1 и -RN2 совместно с атомом азота, к которому она присоединена, образует 5-, 6- или 7-членное неароматическое кольцо, которое имеет только 1 гетероатом в кольце или только 2 гетероатома в кольце, причем один из указанных только двух гетероатомов в кольце представляет собой N, а другой из указанных только двух гетероатомов в кольце независимо представляет собой N, О или S;
каждый -RD1 независимо представляет собой:
-RE1, -RE2, -RE3, -RE4, -RE5, -RE6, -RE7, -RE8,
-L3-RE4, -L3-RE5, -L3-RE6, -L3-RE7 или -L3-RE8;
где:
каждый -RE1 независимо представляет собой насыщенный алифатический С1-6 алкил;
каждый -RE2 независимо представляет собой алифатический С2-6 алкенил;
каждый -RE3 независимо представляет собой алифатический С2-6алкинил;
каждый -RE4 независимо представляет собой насыщенный С3-6 циклоалкил;
каждый -RE5 независимо представляет собой С3-7 циклоалкенил;
каждый -RE6 независимо представляет собой неароматический С3-7 гетероциклил;
каждый -RE7 независимо представляет собой C6-14 карбоарил;
каждый -RE8 независимо представляет собой C5-14 гетероарил;
каждый -L3- независимо представляет собой насыщенный алифатический C1-3 алкилен;
и где:
каждый C1-6 алкил, С2-6 алкенил, С2-6 алкинил, С3-6 циклоалкил, С3-6 циклоалкенил, неароматический С3-7 гетероциклил, C6-14 карбоарил, С5-14 гетероарил и C1-3 алкилен может иметь заместитель, например один или более (например, 1, 2, 3) заместитель -RG4, где каждый -RG4 независимо представляет собой:
-F, -Cl, -Br, -I,
-RF1,
-CF3, -CH2CF3, -CF2CF2H, -ОН,
-L4-OH,
-O-L4-OH,
-ORF1,
-L4-ORF1,
-O-L4-ORF1,
-OCF3, -OCH2CF3, -OCF2CF2H,
-SH,
-SRF1, -SCF3,
-CN,
-NO2,
-NH2, -NHRF1, -NRF12, -NRN3RN4,
-L4-NH2, -L4-NHRF1, -L4-NRF12 или -L4-NRN3RN4,
-O-L4-NH2, -O-L4-NHRF1, -O-L4-NRF12, -O-L4-NRN3RN4,
-NH-L4-NH2, -NH-L4-NHRF1, -NH-L4-NRF12, -NH-L4-NRN3RN4,
-NRF1-L4-NH2, -NRF1-L4-NHRF1, -NRF1-L4-NRF12, -NRF1-L4-NRN3RN4,
-C(=O)OH,
-C(=O)ORF1,
-C(=O)NH2, -C(=O)NHRF1, -C(=O)NRF12 или -C(=O)NRN3RN4;
где:
каждый -RF1 независимо представляет собой насыщенный алифатический С1-4 алкил, фенил или бензил;
каждый -L4- независимо представляет собой насыщенный алифатический С1-5 алкилен и
каждая группа -NRN3RN4, -RN3 и -RN4, совместно с атомом азота, к которому она присоединена, образует 5-, 6- или 7-членное неароматическое кольцо, имеющее только 1 гетероатом в кольце или только 2 гетероатома а кольце, причем один из указанных только двух гетероатомов в кольце представляет собой N, а другой из указанных только двух гетероатомов в кольце независимо представляет собой N, О или S.
В одном варианте реализации, каждый -RS, если присутствует, независимо представляет собой:
-F, -Cl, -Br, -I,
-RD1,
-CF3, -CH2CF3, -CF2CF2H,
-ОН,
-L1-ОН,
-O-L1-OH,
-ORD1,
-L1-ORD1,
-O-L1-ORD1,
-OCF3, -OCH2CF3, -OCF2CF2H,
-SH,
-SRD1, -SCF3,
-CN,
-NO2,
-NH2, -NHRD1, -NRD12, -NRN1RN2,
-L1-NH2, -L1-NHRD1, -L1-NRD12, -L1-NRN1RN2,
-O-L1-NH2, -O-L1-NHRD1, -O-L1-NRD12, -O-L1-NRN1RN2,
-NH-L1-NH2, -NH-L1-NHRD1, -NH-L1-NRD12, -NH-L1-NRN1RN2,
-NRD1-L1-NH2, -NRD1-L1-NHRD1, -NRD1-L1-NRD12, -NRD1-L1-NRN1RN2,
-C(=O)OH,
-C(=O)ORD1,
-C(=O)NH2, -C(=O)NHRD1, -C(=O)NRD12, -C(=O)NRN1RN2,
-NHC(=O)RD1, -NRD1C(=O)RD1,
-OC(=O)RD1,
-C(=O)RD1,
-NHS(=O)2RD1, -NRD1S(=O)2RD1,
-S(=O)2NH2, -S(=O)2NHRD1, -S(=O)2NRD12 или -S(=O)2NRN1RN2;
и дополнительно две примыкающие к кольцу группы -RS, если присутствуют, могут совместно образовывать группу -O-L2-O-.
В одном варианте реализации, каждый -RS, если присутствует, независимо представляет собой -ORD1.
В одном варианте реализации, каждая группа -NRN1RN2, если присутствует, независимо представляет собой пирролидино-, имидазолидино-, пиразолидино-, пиперидино-, пиперизино-, морфолино-, тиоморфолино-, азепино- или диазепиногруппу и независимо может не иметь или иметь заместители, например одну или более (например, 1, 2, 3) группы, которая выбрана из С1-3 алкила и -CF3.
В одном варианте реализации, каждая группа -NRN1RN2, если присутствует, независимо представляет собой пирролидино-, пиперидино-, пиперизино- или морфолиногруппу и независимо может не иметь или иметь заместители, например одну или более (например, 1, 2, 3) группу, которая выбрана из С1-3 алкила и -CF3.
В одном варианте реализации, каждый -RD1, если присутствует, независимо представляет собой:
-RE1, -RE3, -RE4, -RE7, -RE8,
-L3-RE4, -L3-RE7 или -L3-RE8.
В одном варианте реализации, каждый радикал -RD1, если присутствует, независимо представляет собой:
-RE1, -RE3, -RE7, -RE8, -L3-RE7 или -L3-RE8.
В одном варианте реализации, каждый -RD1, если присутствует, независимо представляет собой -L3-RE7 или -L3-RE8.
В одном варианте реализации, каждый -RD1, если присутствует, независимо представляет собой -RE3.
В одном варианте реализации, каждый -RE1, если присутствует, независимо представляет собой метил, этил, н-пропил, изо-пропил, н-бутил, изо-бутил, втор-бутил или трет-бутил, которые могут иметь заместители.
В одном варианте реализации, каждый -RE2, если присутствует, независимо представляет собой алифатический С2-4 алкенил, который может иметь заместители.
В одном варианте реализации, каждый -RE2, если присутствует, независимо представляет собой -CH2-СН=CH2, который может иметь заместители.
В одном варианте реализации, каждый -RE3, если присутствует, независимо представляет собой алифатический С3-5 алкинил, который может иметь заместители.
В одном варианте реализации, каждый -RE3, если присутствует, независимо представляет собой -CH2-С≡СН, -СН(СН3)-С≡СН, -CH2-С≡С-СН3, -СН(СН3)-С≡С-СН3, -CH2-С≡С-CH2-СН3 или -CH2-CH2-С≡СН, которые могут иметь заместители.
В одном варианте реализации, каждый -RE4, если присутствует, независимо представляет собой возможно замещенный циклопропил, циклобутил, циклопентил или циклогексил, которые могут иметь заместители.
В одном варианте реализации, каждый -RE6, если присутствует, независимо представляет собой азетидинил, пирролидинил, имидазолидинил, пиразолидинил, пиеридинил, пиперазинил, морфолинил, азепинил, диазепинил, тетрагидрофуранил, тетрагидропиранил, диоксанил, которые могут иметь заместители.
В одном варианте реализации, каждый -RE6, если присутствует, независимо представляет собой пирролидинил, пиперидинил, пиперазинил, морфолинил, тетрагидрофуранил или тетрагидропиранил, которые могут иметь заместители.
В одном варианте реализации, каждый -RE7, если присутствует, независимо представляет собой фенил или нафтил, которые могут иметь заместители.
В одном варианте реализации, каждый -RE7, если присутствует, независимо представляет собой возможно замещенный фенил, который может иметь заместители.
В одном варианте реализации, каждый -RE8, если присутствует, независимо представляет собой фуранил, тиенил, пирролил, имидазолил, пиразолил, триазолил, оксазолил, изоксазолил, тиазолил, изотиазолил, пиридил, пиразинил, пиримидинил, пиридазинил, бензофуранил, изобензофуранил, индазолил, пуринил, хинолинил, изохинолинил, нафтиридинил, хиноксалинил, хиназолинил, циннолинил, индолил, изоиндолил, карбазолил, карболинил, акридинил, феноксазинил или фенотиазинил, которые могут иметь заместители.
В одном варианте реализации, каждый -RE8, если присутствует, независимо представляет собой фуранил, тиенил, пирролил, имидазолил, пиразолил, триазолил, оксазолил, изоксазолил, тиазолил, изотиазолил, пиридил, пиразинил, пиримидинил, пиридазинил, хинолинил или изохинолинил, которые могут иметь заместители.
В одном варианте реализации, каждый -RE8, если присутствует, независимо представляет собой фуранил, пирролил, пиразолил, триазолил, оксазолил, изоксазолил, тиазолил, изотиазолил, пиридил, хинолинил или изохинолинил, которые могут иметь заместители.
В одном варианте реализации, каждый -L1-, если присутствует, независимо представляет собой насыщенный алифатический C1-5 алкилен или алифатический С2-5 алкинилен.
В одном варианте реализации, каждый -L1-, если присутствует, независимо представляет собой насыщенный алифатический C1-5 алкилен.
В одном варианте реализации, каждый -L1-, если присутствует, независимо представляет собой насыщенный алифатический С2-5 алкилен.
В одном варианте реализации, каждый -L2-, если присутствует, независимо представляет собой -CH2- или -CH2CH2-
В одном варианте реализации, каждый -L2-, если присутствует, представляет собой независимо -CH2CH2-
В одном варианте реализации, каждый -L3-, если присутствует, независимо представляет собой -CH2-.
В одном варианте реализации, каждый -RG4, если присутствует, независимо выбран из:
-F, -Cl, -Br, -I,
-RF1,
-CF3, -CH2CF3, -CF2CF2H,
-ОН,
-L4-OH,
-O-L4-OH,
-ORF1,
-L4-ORF1,
-O-L4-ORF1,
-OCF3, -OCH2CF3, -OCF2CF2H,
-SRF1,
-NH2, -NHRF1, -NRF12, -NRN3RN4,
-L4-NH2, -L4-NHRF1, -L4-NRF12 или -L4-NRN3RN4,
-O-L4-NH2, -O-L4-NHRF1, -O-L4-NRF12, -O-L4-NRN3RN4,
-NH-L4-NH2, -NH-L4-NHRF1, -NH-L4-NRF12, -NH-L4-NRN3RN4,
-NRF1-L4-NH2, -NRF1-L4-NHRF1, -NRF1-L4-NRF12 или -NRF1-L4-NRN3RN4.
В одном варианте реализации, каждый -RG4, если присутствует, независимо выбран
из:
-F, -Cl, -Br, -I,
-RF1,
-ОН,
-ORF1,
-NH2, -NHRF1, -NRF12 и -NRN3RN4.
В одном варианте реализации, каждая группа -NRN3RN4, если присутствует, независимо представляет собой пирролидино-, имидазолидино-, пиразолидино-, пиперидино-, пиперизино-, морфолино-, тиоморфолино-, азепино- или диазепиногруппу, которые независимо могут не иметь или иметь в качестве заместителей, например, одну или более (например, 1, 2, 3) группу, которая выбрана из C1-3 алкила и -CF3.
В одном варианте реализации, каждая группа -NRN3RN4, если присутствует, независимо представляет собой пирролидино-, пиперидино-, пиперизино- или морфолиногруппу, которые независимо могут не иметь или иметь заместители, например одну или более (например, 1, 2, 3) группу, которая выбрана из С1-3 алкила и -CF3.
В одном варианте реализации, каждый -RF1, если присутствует, независимо представляет собой насыщенный алифатический С1-4 алкил.
В одном варианте реализации, каждый -L4-, если присутствует, независимо представляет собой насыщенный алифатический С2-5 алкилен.
Некоторые предпочтительные комбинации
В одном предпочтительном варианте реализации:
W независимо представляет собой -CRPW=;
X независимо представляет собой -CRPX=;
Y независимо представляет собой -CRPY=;
Z независимо представляет собой -CRPZ=;
каждая из групп -RPW, -RPX, -RPY и -RPZ, если присутствует, независимо представляет собой -Н или -RRS1;
z равно 1;
-J< независимо представляет собой -N<;
-RAK- независимо представляет собой -RAK1-;
-RAK1- независимо представляет собой -CH2- и
-RN независимо представляет собой -RNNN.
В одном предпочтительном варианте реализации, дополнительно, каждая группа
-RRS1, если присутствует, независимо представляет собой -F, -Cl, -Br, -I, -Me, -Et, -CF3,
-ОН, -ОМе, -OEt, -OCF3 или фенил; и дополнительно, две соседние группы -RRS1, если присутствуют, могут образовать -OCH2CH2O-.
В одном предпочтительном варианте реализации, дополнительно, каждая группа
-RNNN независимо представляет собой фенил, который может иметь заместители, например один или более (например, 1, 2, 3) заместитель -RS.
В одном предпочтительном варианте реализации, дополнительно, -RNNN независимо представляет собой фенил, который может иметь заместитель в параположении, например заместитель -RS; и быть незамещенным во всех других положениях.
В одном предпочтительном варианте реализации, дополнительно, RNNN независимо представляет собой фенил, который может иметь в качестве заместителя -RS, где -RS независимо представляет собой -ORD1.
В одном предпочтительном варианте реализации, дополнительно, RNNN независимо представляет собой фенил, возможно замещенный в параположении заместителем -RS и незамещенный во всех других положениях, где -RS независимо представляет собой -ORD1.
В одном предпочтительном варианте реализации, дополнительно, RNNN независимо представляет собой фенил, который может иметь в качестве заместителя -RS, где -RS независимо представляет собой -ORD1, где -RD1 независимо представляет собой -L3-RE7 или -L3-RE8, где -L3- независимо представляет собой -CH2-.
В одном предпочтительном варианте реализации, дополнительно, RNNN независимо представляет собой фенил, который может иметь в качестве заместителя в параположении группу -RS, и не иметь заместителей во всех других положениях, где -RS независимо представляет собой -ORD1, где -RD1 независимо представляет собой -L3-RE7 или -L3-RE8, где -L3- независимо представляет собой -CH2-.
В одном предпочтительном варианте реализации, дополнительно, RNNN независимо представляет собой фенил, который может иметь в качестве заместителя -RS, где -RS независимо представляет собой -ORD1, где -RD1 независимо представляет собой -RE3.
В одном предпочтительном варианте реализации, дополнительно, RNNN независимо представляет собой фенил, который может иметь в качестве заместителя в параположении заместитель -RS, и быть незамещенным во всех других положениях, где -RS независимо представляет собой -ORD1, где -RD1 независимо представляет собой -RE3.
Молекулярная масса
В одном варианте реализации, соединение - производное BCSA имеет молекулярную массу от 227 до 1200.
В одном варианте реализации, нижняя граница дипазона составляет от 240, 250, 275, 300 или 350.
В одном варианте реализации, верхняя граница диапазона составляет 1100, 1000, 900, 800, 700 или 600.
В одном варианте реализации диапазон составляет от 240 до 600.
Комбинации
Все совместимые комбинации вышеописанных вариантов реализации однозначно раскрыты в настоящем описании, как если бы каждая и любая комбинация была индивидуально и однозначно описана.
Примеры конкретных вариантов реализации
В одном варианте реализации настоящего изобретения соединения выбраны из соединений нижеследующих формул и фармацевтически приемлемых солей, гидратов и сольватов указанных соединений:
































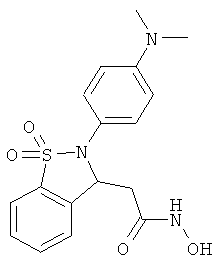










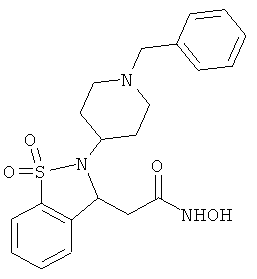


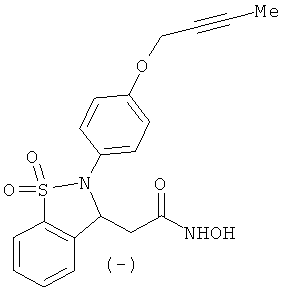









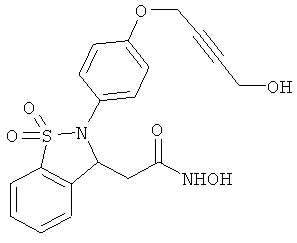

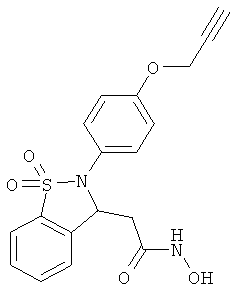













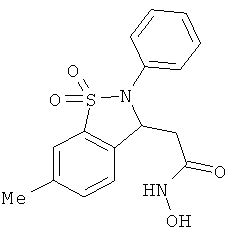




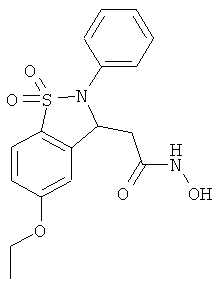

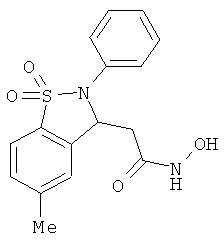






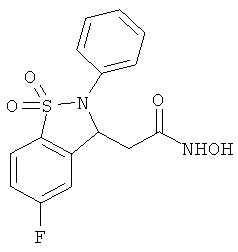








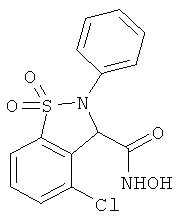
В одном варианте реализации настоящего изобретения соединения выбраны из соединений нижеследующих формул и фармацевтически приемлемых солей, гидратов и сольватов указанных соединений:

В одном варианте реализации соединения выбраны из соединений нижеследующих формул и их фармацевтически приемлемых солей, гидратов и сольватов:

В одном варианте реализации соединения выбраны из соединений нижеследующих формул и фармацевтически приемлемых солей, гидратов и сольватов указанных соединений:
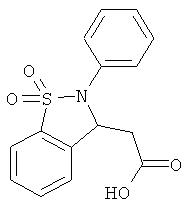


Дополнительные примеры
В одном варианте реализации настоящего изобретения соединения выбраны из соединений нижеследующих формул и фармацевтически приемлемых солей, гидратов и сольватов указанных соединений:


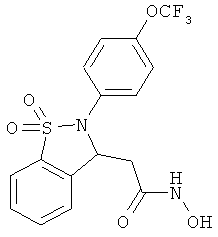









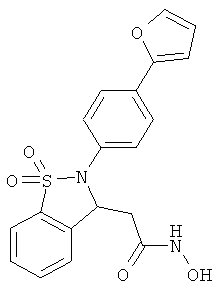

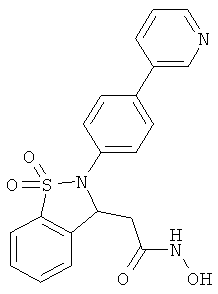








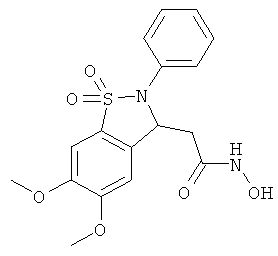






В одном варианте реализации настоящего изобретения соединения выбраны из соединений нижеследующих формул и фармацевтически приемлемых солей, гидратов и сольватов указанных соединений:





В одном варианте реализации соединения выбраны из соединений нижеследующих формул и фармацевтически приемлемых солей, гидратов и сольватов указанных соединений:



В одном варианте реализации настоящего изобретения соединения выбраны из соединений нижеследующих формул и фармацевтически приемлемых солей, гидратов и сольватов указанных соединений:

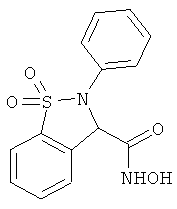
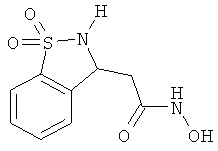
В одном варианте реализации соединения согласно настоящему изобретению выбраны из соединений нижеследующих формул и фармацевтически приемлемых солей, гидратов и сольватов указанных соединений:







По существу очищенные формы
Один из аспектов настоящего изобретения относится к производным соединениям BCSA, описанным в настоящей заявке в существенно очищенной форме и/или в форме, практически свободной от загрязнений.
В одном варианте реализации, в существенной степени очищенная форма является чистой по меньшей мере 50% по массе, например, по меньшей мере 60% по массе, например, по меньшей мере 70% по массе, например, по меньшей мере 80% по массе, например, по меньшей мере 90% по массе, например, по меньшей мере 95% по массе, например, по меньшей мере 97% по массе, например, по меньшей мере 98% по массе, например, по меньшей мере 99% по массе.
Если специально не оговорено, по существу очищенная форма относится к соединению в любой стереоизомерной или энантиомерной форме. Например, в одном варианте реализации, по существу очищенной формой называется смесь стереоизомеров, то есть очищенной от других соединений. В одном варианте реализации, под существенно очищенной формой подразумевают форму, относящуюся к одному стереоизомеру, например оптически чистому стереоизомеру. В одном варианте реализации под по существу чистой формой понимают смесь энантиомеров. В одном варианте реализации к существенно чистой форме относят эквимолярную смесь энантиомеров (то есть рацемическую смесь или рацемат). В одном варианте реализации под существенно очищенной формой понимают один энантиомер, например оптически чистый энантиомер.
В одном варианте реализации, примеси составляют не более чем 50% по массе, например, не более чем 40% по массе, например, не более чем 30% по массе, например, не более чем 20% по массе, например, не более чем 10% по массе, например, не более чем 5% по массе, например, не более чем 3% по массе, например, не более чем 2% по массе, например, не более чем 1% по массе.
Если специально не оговорено, под примесями понимают другие соединения, то есть не стереоизомеры или энантиомеры. В одном варианте реализации, под примесями понимают другие соединения и другие стереоизомеры. В одном варианте реализации, под примесями понимают другие соединения и другой энантиомер.
В одном варианте реализации по существу очищенная форма является оптически чистой по меньшей мере на 60% (то есть 60% соединения, по расчету в молях, представляет собой желаемый стереоизомер или энантиомер и 40% представляет собой нежелательный стереоизомер или энантиомер), например, оптически чистой по меньшей мере на 70%, например, оптически чистой по меньшей мере на 80%, например, оптически чистой по меньшей мере на 90%, например, оптически чистой по меньшей мере на 95%, например, оптически чистой по меньшей мере на 97%, например, оптически чистой по меньшей мере на 98%, например, оптически чистой по меньшей мере на 99%.
Изомеры
Некоторые соединения согласно настоящему изобретению могут существовать в одной или более различных геометрических, оптических, энантиомерных, диастереомерных, эпимерных, атропических, стереоизомерных, таутомерных, конформационных или аномерных формах, включая, но не ограничиваясь перечисляемыми, цис- и транс-формы; Е- и Z-формы; с-, t- и r-формы; эндо- и экзо-формы; R-, S-, и мезо-формы; D- и L-формы; d- и I-формы; (+) и (-) формы; кето-, енольные- и енолятные формы; син- и анти-формы; синклинные и антиклинные формы; α- и β-формы; аксиальные и экваториальные формы; формы «лодки», «кресла», «скрутки», «конверта» и «полукресла» и их комбинации, далее объединяемые под названием "изомеры" (или "изомерные формы").
Авторы настоящего изобретения отмечают, что, за исключением обсуждаемых ниже таутомерных форм, в настоящем описании намеренно из понятия «изомеры» исключены структурные (или системные) изомеры, (то есть изомеры, различающиеся соединениями между атомами, отличными от различий в пространственных позициях). Например, ссылка на метоксигруппу, -ОСН3, не должна толковаться как ссылка на ее структурный изомер, гидроксиметильную группу, -СН2ОН. Аналогично, ссылка на ортохлорфенил не должна толковаться как ссылка на его структурный изомер метахлорфенил. Однако ссылка на класс структур может включать структурно-изомерные формы, подпадающие под этот класс (например, С1-7 алкил включает н-пропил и изо-пропил; бутил включает н-, изо-, втор- и трет-бутил; метоксифенил включает орто-, мета-, и параметоксифенил).
Вышеописанное исключение не относится к таутомерным формам, например, кето-, енольным и енолятным формам, как, например, в следующих таутомерных парах: кетон/енол (показана ниже), имин/енамин, амид/иминоспирт, амидин/амидин, нитрозо/оксим, тиокетон/ентиол, N-нитрозо/гидроксиазо, и нитро/ацинитро.

Авторы отмечают, что в понятие «изомер» специально включены соединения с одним или более изотопными замещениями. Например, Н может быть в любой изотопной форме, включая1Н,2Н (D) и3Н (Т); С может быть в любой изотопной группе, включая12С,13С и14С; О может быть в любой изотопной форме, включая16O и18O; и тому подобное.
Если специально не оговорено, ссылка на конкретное соединение охватывает все такие изомерные формы, включая их смеси (например, рацемические смеси). Способы приготовления (например, асимметрический синтез) и разделения (например, дробная кристаллизация и хроматография) таких изомерных форм либо известны специалистам в данной области, либо они легко могут быть получены посредством адаптации указанных в настоящем описании способов, посредством известных методик.
Соли
Может оказаться удобным или желательным приготовить, очистить и/или работать с соответствующей солью соединения согласно настоящему изобретению, например, с фармацевтически приемлемой солью. Примеры фармацевтически приемлемых солей обсуждаются в Berge et al., 1977, "Pharmaceutically Acceptable Salts," J.Pharm. Sci., Vol.66, pp.1-19.
Например, если соединение имеет анионную природу или имеет функциональную группу, которая может проявлять анионный характер (например, -СООН может стать -СОО-), тогда можно получить его соль с подходящим катионом. Примеры подходящих неорганических катионов включают, не ограничиваясь перечисляемыми, ионы щелочных металлов, такие как Na+ и K+, щелочноземельных металлов, такие как Са2+ и Mg2+, и другие катионы, такие как Al+3. Примеры подходящих органических катионов включают, не ограничиваясь перечисленными, ион аммония (то есть





Если соединение имеет катионную природу или содержит функциональную группу, которая может превратиться в катионную группу (например, -NH2 может стать

Примеры подходящих органических анионов включают, не ограничиваясь перечисляемыми, анионы, производные следующих органических кислот: 2-ацетоксибензойной, уксусной, аскорбиновой, аспарагиновой, бензойной, камфоросульфоновой, коричной, лимонной, этилендиаминотетрауксусной, этандисульфоновой, этансульфоновой, фумаровой, глюкогептоновой, глюконовой, глутаминовой, гликолевой, гидроксималеиновой, гидроксинафталинкарбоновой, изетионовой, молочной, лактобионовой, лауриновой, малеиновой, малоновой, метансульфоновой, слизевой, олеиновой, щавелевой, пальмитиновой, памоевой, пантотеновой, фенилуксусной, фенилсульфоновой, пропионовой, пировиноградной, салициловой, стеариновой, янтарной, сульфаниловой, винной, толуолсульфоновой и валериановой. Примеры подходящих полимерных органических анионов включают, не ограничиваясь перечисленными, анионы, производные следующих полимерных кислот: дубильной кислоты, карбоксиметилцеллюлозы.
Если специально не оговорено, ссылка на конкретное соединение также охватывает его солевые формы.
Сольваты и гидраты
Может оказаться удобным или желательным приготовить, очистить и/или работать с соответствующим сольватом соединения. Термин "сольват" в настоящем описании применяют в его традиционном смысле, он указывает на комплекс растворенного вещества (например, соединения, соли соединения) и растворитель. Если растворителем является вода, сольват может традиционно называться гидратом, например моногидратом, дигидратом, тригидратом и так далее.
Если специально не оговорено, ссылка на конкретное соединение также охватывает его сольватные и гидратные формы.
Химически защищенные формы
Может оказаться удобным или желательным приготовить, очистить и/или работать с соединением в химически защищенной форме. Понятие "химически защищенная форма" в настоящем описании употребляется в традиционном химическом смысле и относится к соединению, в котором одна или более из реакционноспособных функциональных групп защищены от нежелательных химических реакций при конкретных параметрах (например, pH, температуре, облучении, растворителе и тому подобное). На практике для обратимого придания функциональной группе, которая иначе при конкретных условиях была бы реакционноспособной, реакционной резистентности применяют хорошо известные химические способы. В химически защищенной форме одна или более реакционноспособных функциональных групп находится в форме защищенной или защитной группы (также известной под названием маскированной или маскирующей группы или блокированной или блокирующей группы). Защищая реакционноспособную функциональную группу можно проводить реакции, вовлекающие незащищенные реакционноспособные функциональные группы, не затрагивая защищенную группу, защитную группу можно удалять, обычно на более поздней стадии, существенно не влияя на остаток молекулы. См., например, Protective Groups in Organic Synthesis (Т.Green и P.Wuts; 3rd Edition; John Wiley and Sons, 1999).
Если специально не оговорено, ссылка на конкретное соединение также охватывает его химически защищенные формы.
В химическом синтезе хорошо известно и широко применяется большое многообразие способов такой "защиты," "блокирования" или "маскирования". Например, соединение, имеющее две неэквивалентные реакционноспособные функциональные группы, обе из которых в определенных условиях стали бы реакционноспособными, может быть дериватизировано таким образом, чтобы сделать «защищенной», и потому нереакционноспособной при указанных определенных условиях, одну из таких функциональных групп; защищенное таким образом соединение можно применять в качестве реагента, который фактически имеет только одну реакционноспособную функциональную группу. После завершения желаемой реакции (включающей другую функциональную группу), «защита» может быть удалена, при этом ранее защищенная функциональная группа приобретает свою присущую ему функциональность.
Например, гидроксильную группу можно защитить, либо (-OR, простой эфир) или (-OC(=O)R, сложный эфир), например, в виде: трет-бутилового эфира; бензилового, бензгидрилового (дифенилметилового) или тритилового (трифенилметилового) эфира; триметилсилилового или трет-бутилдиметилсилилового эфира; или ацетилового эфира (-ОС(=O)СН3, -ОАс).
Например, альдегидную или кетонную группу можно защищать в виде ацеталя (R-CH(OR)2) или кеталя (R2C(OR)2), соответственно, в которых карбонильная группа (>С=O) превращается в диэфир (>C(OR)2), посредством реакции, например, с первичным спиртом. Альдегидную или кетонную группу легко регенерировать гидролизом в большом избытке воды в присутствии кислоты.
Например, аминогруппу можно защищать преобразованием в амид (-NRCO-R) или уретан (-NRCO-OR), например, в виде: метиламида (-NHCO-CH3); бензилоксиамида
(-NHCO-OCH2C6H5, -NH-Cbz); такого как трет-бутоксиамид (-NHCO-OC(CH3)3, -NH-Boc); 2-дифенил-2-пропоксиамид (-NHCO-ОС(СН3)2С6Н4С6Н5, -NH-Bpoc), в виде 9-флуоренилметоксиамида (-NH-Fmoc), в виде 6-нитровератрилоксиамида (-NH-Nvoc), в виде 2-триметилсилилэтилоксиамида (-NH-Teoc), в виде 2,2,2-трихлорэтилоксиамида
(-NH-Troc), в виде аллилоксиамида (-NH-Alloc), в виде
2(-фенилсульфонил)этилоксиамида (-NH-Psec); или в подходящих случаях (например, для циклических аминов), в виде нитроксидного радикала (>N-O•).
Например, кислотную карбоксильную группу можно защитить в виде сложного эфира, например, С1-7 алкилового эфира (например, метилового эфира; трет-бутилового эфира); С1-7 галогеналкилового эфира (например, C1-7 тригалогеналкилового эфира); три С1-7алкилсилил-С1-7алкилового эфира или С5-20арил-С1-7алкилового эфира (например, бензилового эфира; нитробензилового эфира) или в виде амида, например метиламида.
Например, тиольную группу можно защищать как тиоэфир (-SR), например, в виде: бензилтиоэфира; ацетамидометилового эфира (-S-CH2NHC(=O)CH3).
Пролекарства
Может быть удобным или желательным приготовить, очистить и/или работать с соединением в форме пролекарства. Термин "пролекарство", применяемый в настоящем описании, относится к соединению, из которого при метаболизме (например, in vivo), образуется желаемое активное соединение. Типично, пролекарство является неактивным, или менее активным, чем желаемое активное соединение, но может обеспечить преимущества в обращении с ним, введении или метаболических свойствах.
Если специально не оговорено, ссылка на конкретное соединение также охватывает его пролекарство.
Например, некоторые пролекарства являются сложными эфирами активного соединения (например, физиологически приемлемым метаболически лабильным сложным эфиром). При метаболизме сложноэфирная группа (-C(=O)OR) расщепляется с образованием активного лекарственного средства. Такие сложные эфиры можно образовать путем этерификации, например, любой карбоксильной группы (-С(=O)ОН) в родительском соединении, при, если это необходимо, предварительной защите любой другой реакционноспособной группой, имеющейся в материнском соединении с последующей снятием защиты, если это требуется.
Также некоторые пролекарства активируются ферментативно, с образованием активного соединения, или соединения, которое после дополнительной химической реакции образует активное соединение (например, ADEPT, GDEPT, LIDEPT, и так далее). Например, пролекарство может представлять собой углеводное производное или другой конъюгат с гликозидом или может представлять собой производное сложного эфира аминокислоты.
Химический синтез
В настоящем описании описаны некоторые способы химического синтеза соединений - производных BCSA согласно настоящему изобретению. В рамках объема притязаний настоящего изобретения, с целью облегчения синтеза дополнительных соединений приведенные здесь и/или другие хорошо известные способы могут быть модифицированы и/или адаптированы.
Применение
Соединения - производные BCSA, раскрытые в настоящем описании, могут быть полезны, например, при лечении заболеваний и состояний, которые облегчаются при ингибировании ТАСЕ.
Применение в способах ингибирования ТАСЕ и способах регулирования высвобождения цитокинов
Один из аспектов настоящего изобретения относится к описанному в этой заявке способу ингибирования ТАСЕ в клетке in vitro или in vivo, включающему приведение в контакт указанной клетки и эффективного количества соединения -производного BCSA.
Подходящие системы анализа для определения ингибирования ТАСЕ известны специалистам в данной области и/или описаны в настоящей заявке.
Еще одной аспект настоящего изобретения относится к описанному в настоящей заявке способу регулирования (например, инигибирования) высвобождения цитокинов (например, высвобождения TNF-α) в клетке in vitro или in vivo, включающему приведение в контакт указанной клетки и эффективного количества соединения - производного BCSA.
Подходящие системы анализа для определения регулирования (например, ингибирования) высвобождения цитокинов известны специалистам в данной области и/или описаны в настоящей заявке.
В одном варианте реализации указанный способ осуществляют in vitro.
В одном варианте реализации указанный способ осуществляют in vivo.
В одном варианте реализации соединение - производное BCSA обеспечивают в форме фармацевтически приемлемой композиции.
Лечения можно применять в отношении любого типа клеток, включая, но не ограничиваясь перечисляемыми, клетки легких, желудочно-кишечные (включая, например, клетки кишечника, толстой кишки), молочной железы (относящиеся к молочной железе), яичника, предстательной железы, печени (гепатические), почек (ренальные), мочевого пузыря, поджелудочной железы, головного мозга и кожи.
Применение в способах терапии
Еще один аспект настоящего изобретения относится к применению соединения - производного BCSA, описанного в настоящей заявке, для лечения (например, заболевания или расстройства) в организме человека или животного посредством терапевтического воздействия.
Применение в производстве лекарственных средств
Еще один аспект настоящего изобретения относится к применению соединения - производного BCSA, описанного в настоящей заявке, в производстве лекарственного средства для лечения (например, заболевания или расстройства).
В одном варианте реализации лекарственное средство содержит соединение - производное BCSA.
Способы лечения
Еще один аспект настоящего изобретения относится к способу лечения (например, заболевания или расстройства), включающему введению пациенту, нуждающемуся в лечении, терапевтически эффективного количества соединения - производного BCSA, описанного в настоящей заявке, предпочтительно в форме фармацевтической композиции.
Излечиваемые состояния - состояния, опосредуемые ТАСЕ
В одном варианте реализации (например, при применении способов терапии, при производстве лекарственных средств или способов лечения) указанное лечение представляет собой лечение заболевания или расстройства, опосредуемого ТАСЕ, например заболевания или расстройства, про которое известно, что оно опосредовано ТАСЕ.
Заболевание или расстройство, опосредованное ТАСЕ, представляет собой, например, заболевание или расстройство, при котором ТАСЕ и/или активность ТАСЕ являются важными или необходимыми, например, для возникновения, развития, проявления, и так далее этого заболевания или расстройства.
Излечиваемые состояния - состояния, облегчаемые при ингибировании ТАСЕ
В одном варианте реализации (например, при применении способов терапии, при производстве лекарственных средств, способов лечения), указанное лечение представляет собой лечение заболевания или расстройства, которое облегчается при ингибирования ТАСЕ, например, заболевания или расстройства, про которое известно, что его облегчает ингибирование ТАСЕ.
Излечиваемые состояния - состояния, которые излечивают ингибиторы ТАСЕ
В одном варианте реализации (например, при применении способов терапии, при производстве лекарственных средств, способов лечения), лечение представляет собой лечение заболевания или расстройства, про которое известно, что его можно лечить ингибитором ТАСЕ.
Излечиваемые состояния - конкретные состояния
В одном варианте реализации (например, при применении способов терапии, при производстве лекарственных средств, способов лечения), указанное лечение представляет собой лечение ревматоидного артрита; воспаления; псориаза; септического шока; отторжения имплантата; кахексии; анорексии; застойной сердечной недостаточности; постишемической травмы, связанной с реперфузией; воспалительного заболевания центральной нервной системы; воспалительного заболевания кишечника; резистентности к инсулину; инфекции ВИЧ; рака; хронического обструктивного заболевания легких (COPD) или астмы.
В одном варианте реализации (например, при применении способов терапии, при производстве лекарственных средств, способов лечения), указанное лечение представляет собой лечение: остеоартрита, язвенного колита, болезни Крона, рассеянного склероза или дегенеративной потери хряща.
В одном варианте реализации настоящего изобретения, указанное лечение представляет собой лечение воспаления.
В одном варианте реализации настоящего изобретения, лечение представляет собой лечение ревматоидного артрита.
В одном варианте реализации, лечение представляет собой лечение псориаза.
Излечиваемые состояния - рак и т.п.
В одном варианте реализации настоящего изобретения, лечение представляет собой лечение: рака.
В одном варианте реализации, указанное лечение представляет собой лечение: рака легких, мелкоклеточного рака легких, немелкоклеточного рака легких, рака желудочно-кишечного тракта, рака желудка, рака кишечника, рака толстой кишки, рака прямой кишки, рака толстой и прямой кишки, рака щитовидной железы, рака молочной железы, рака яичников, рака эндометрия, рака предстательной железы, рака яичка, рака печени, рака почек, ренальноклеточной карциномы, рака яичников, рака поджелудочной железы, рака головного мозга, глиомы, саркомы, остеосаркомы, рака костей, рака кожи, чешуйчато-клеточного рака, саркомы Капоши, меланомы, злокачественной меланомы, лимфомы или лейкоза.
В одном варианте реализации, указанное лечение представляет собой лечение:
карциномы, например карциномы мочевого пузыря, молочной железы, толстой кишки (например, карцином толстой и прямой кишки, таких как аденокарцинома толстой кишки и аденома толстой кишки), карциномы почек, эпидермальной карциномы, карциномы печени, легких (например, аденокарциномы, мелкоклеточного рака легких и немелкоклеточного рака легких), пищевода, желчного пузыря, яичников, поджелудочной железы (например, экзокринной панкреатической карциномы), желудка, шейки матки, щитовидной железы, предстательной железы, кожи (например, чешуйчато-клеточной карциномы);
гематопоэтической опухоли лимфоидной системы, например лейкоза, острого лимфоцитарного лейкоза, В-клеточной лимфомы, Т-клеточной лимфомы, лимфогранулематоза, неходжкинской лимфомы, волосково-клеточной лимфомы или лимфомы Бэркитта;
гематопоэтической опухоли мелоидной системы, например острого и хронического миелогенного лейкоза, синдрома миелодисплазии или промиелоцитарного лейкоза;
опухоли мезенхимного происхождения, например фибросаркомы или рабдомиосаркомы;
опухоли центральной или периферической нервной системы, например астроцитомы, нейробластомы, глиомы или шванномы;
меланомы; семиномы; тератокарциномы; остеосаркомы; xenoderoma pigmentosum; кератоакантомы; фолликулярного рака щитовидной железы или саркомы Капоши.
В одном варианте реализации, указанное лечение представляет собой лечение солидного рака.
В одном варианте реализации (например, при применении способов терапии, при производстве лекарственных средств, способов лечения), указанное лечение представляет собой лечение гиперпролиферативного расстройства кожи.
В одном варианте реализации, лечение представляет собой лечение псориаза, старческого кератоза и/или рака кожи, не относящегося к меланоме.
Излечиваемые состояния - воспаление и т.п.
В одном варианте реализации (например, при применении способов терапии, при производстве лекарственных средств, способов лечения), указанное лечение представляет собой лечение воспалительного заболевания.
В одном варианте реализации, лечение представляет собой лечение: воспалительного заболевания, включающего патологическую активацию Т- и В-клеточных лимфоцитов, нейтрофилов и/или базофилов.
В одном варианте реализации, указанное лечение представляет собой лечение: воспалительного заболевания, такого как ревматоидный артрит, остеоартрит, ревматоидный спондилит, подагрический артрит, травматический артрит, краснуха, псориатический артрит и другие артритные состояния; болезни Альцгеймера; синдрома токсического шока, воспалительной реакции, индуцируемой эндотоксином или воспалительного заболевания кишечника; туберкулеза; атеросклероза; мышечной дегенерации; синдрома Рейтера; подагры; острого синовита; сепсиса; септического шока; эндотоксического шока; грам-негативного сепсиса; респираторного патологического синдрома взрослых; церебральной малярии; хронического воспаления легких; силикоза; саркоидоза легких; заболеваний, связанных с ресорбцией костной ткани; травмы, связанной с ишемической реперфузией; реакции отторжения имплантата; отторжения аллогенного трансплантата; лихорадки и миалгий, вызванных инфекцией, такой как грипп, кахексии, в частности кахексии вторичной по отношению к инфекции или злокачественности, кахексии, вторичной к приобретенному синдрому иммунодефицита (AIDS); AIDS; ARC (комплекс, относящийся к AIDS); образования келоида; образования рубцовой ткани; болезни Крона; язвенного колита; гипертермии; хронического обструктивного заболевания легких (COPD); синдрома острой дыхательной недостаточности (ARDS); астмы; фиброза легких; бактериальной пневмонии.
В одном из предпочтительных вариантов реализации указанное лечение представляет собой лечение: артритного состояния, включая ревматоидный артрит и ревматоидный спондилит; воспалительного заболевания кишечника, включая болезнь Крона и язвенного колита и хронического обструктивного заболевания легких (COPD).
В одном из предпочтительных вариантов реализации, указанное лечение представляет собой лечение: воспалительного расстройства, характеризующегося пролиферацией Т-клеток (активирование и рост Т-клеток), например, отторжения тканевого имплантата, эндотоксического шока и гломерулярного нефрита.
Лечение
Термин "лечение" применяемый в настоящем описании в контексте лечения состояния, в целом относится к лечению и терапии, как человека, так и животного (например, в ветеринарных приложениях), при котором достигают некоторый желаемый терапевтический эффект, например ингибирование прогрессирования состояния, и включает уменьшение скорости прогрессирования, остановку прогрессирования, облегчение симптомов состояния, улучшение состояния и излечивание состояния. Также включено лечение как профилактическая мера (то есть профилактика). Например, применение у пациентов, у которых состояние еще не развилось, но у которых имеется риск возникновения состояния, также охватывается термином "лечение".
Например, лечение рака включает профилактику рака, уменьшение заболеваемости раком, облегчение симптомов рака и т.п.
Выражение "терапевтически эффективное количество", применяемый в настоящем описании, относится к такому количеству соединения, или материала, или композиции, или лекарственной формы, содержащей соединение, которое после введения в соответствии с желаемым режимом лечения эффективно для создания некоторого желаемого терапевтического эффекта, соразмерного с разумным отношением польза/риск.
Сочетанная терапия
Термин "лечение" включает комбинированное лечение и сочетанную терапию, при которой, например, последовательно или одновременно, объединяют два или более курса лечения или терапии. Например, соединения - производные BCSA, описанные в настоящей заявке, также можно применять в курсах сочетанной терапии, например, в сочетании с другими агентами, например другими ингибиторами ТАСЕ, другими цитотоксическими агентами, другими противораковыми агентами и так далее. Примеры курсов лечения и терапии включают, не ограничиваясь перечисляемыми, химиотерапию (введение активных агентов, включая, например, лекарственные средства, антитела (например, при иммунотерапии), пролекарства (например, при фотодинамической терапии, GDEPT, ADEPT, и так далее); хирургию; радиационную терапию; фотодинамическую терапию и контролируемую диету.
Например, может быть полезным комбинировать лечение соединением - производным BCSA, описанным в настоящей заявке с одним или более (например, 1, 2, 3, 4) агентом или терапией, регулирующими рост клеток или выживание клеток, или дифференциацию клеток различным механизмом, влияя таким образом на несколько характерных особенностей развития рака.
Один аспект настоящего изобретения относится к описанному в настоящей заявке соединению - производному BCSA в комбинации с одним или более дополнительным терапевтическим агентом, как описано ниже.
Конкретное сочетание будет относиться к компетенции врача, который выберет дозировки, применяя общие познания, и режимы дозировки, известные практику в данной области.
Агенты (то есть соединения - производные BCSA, описанные в настоящей заявке, плюс один или более других агентов) можно вводить одновременно или последовательно, и можно вводить в индивидуально варьирующихся дозовых режимах и различными путями. Например, при последовательном введении допускается введение агентов в близко расположенные моменты времени (например, через период 5-10 минут) или через более длительные интервалы (например, 1, 2, 3, 4 или более часов друг от друга или даже, если это необходимо, через более длительные промежутки времени), соразмеряя точный режим введения со свойствами терапевтического агента(ов).
Агенты (то есть производные соединения BCSA, описанные в настоящей заявке, плюс один или более других агентов) можно совместно вводить в рецептуру одной и той же лекарственной формы или иначе, индивидуальные агенты можно вводить в рецептуры по отдельности и можно обеспечивать совместно в форме набора, возможно с инструкциями по их применению.
Другие применения
Описанные в настоящей заявке соединения - производные BCSA также можно применять в качестве добавок к клеточным культурам для ингибирования ТАСЕ, для ингибирования высвобождения цитокинов (например, высвобождения TNF-α) и так далее.
Описанные в настоящей заявке соединения - производные BCSA также можно применять в качестве составной части при исследованиях in vitro, например, для того чтобы определить вероятность того, что испытуемый объект получит пользу от лечения рассматриваемым соединением.
Описанные в настоящей заявке соединения - производные BCSA можно применять также в качестве стандарта при исследованиях по поиску других соединений, других ингибиторов ТАСЕ и так далее.
Наборы
Один из аспектов настоящего изобретения относится к набору, содержащему (а) описанное в настоящей заявке соединение - производное BCSA, или композицию, содержащую описанное в настоящей заявке соединение, например, предпочтительно в подходящем контейнере и/или в подходящей упаковке и (b) инструкции для применения, например письменные инструкции о том, как вводить указанное соединение или композицию.
Письменные инструкции могут также включать список показаний, при которых активный ингредиент обеспечит адекватное лечение.
Пути введения
Соединение - производное BCSA или фармацевтическую композицию, содержащую соединение - производное BCSA, можно вводить субъекту любыми удобными путями введения, или системно/периферическим образом, или местно (например, в месте желаемого действия).
Способы введения включают, не ограничиваясь перечисляемыми: пероральный способ (например, глотанием); трансбуккальный; трансдермальный (включая например, накладку, пластырь и так далее); трансмукозальный (включая например, накладку, пластырь и так далее); интраназальный (например, спреем для носа); окулярный (например, глазными каплями); пульмонарный (например, при ингаляционной или инсуффляционной терапии, применяя аэрозоль, например, через рот или нос); ректальный (например, суппозиторием или клистиром); вагинальный (например, пессарием); парентеральный, например, инъекцией, включая подкожную, чрескожную, внутримышечную, внутивенную, внутриартериальную, интракардиальную, интратекальную, интраспинальную, интракапсулярную, субкапсуларную, интраорбитальную, внутрибрюшинную, интратрахеальную, субкутикулярную, внутрисуставную, субарахноидальную и интрастернальную инъекции; путем имплантата депо или резервуара, например, подкожно или внутримышечно.
Субъект/пациент
Субъект/пациент может быть хордовым, позвоночным, млекопитающим, плацентарным млекопитающим, сумчатым (например, кенгуру, вомбатом), грызуном (например, морской свинкой, хомяком, крысой, мышью), мышиным (например, мышью), резцовым (например, кроликом), птичьим (например, птицей), псовым (например, собакой), кошачьим (например, котом), лошадиным (например, лошадью), свиноподобным (например, свиньей), овцам (например, овцой), бычьим (например, коровой), приматом или приматообразным (например, обезьяной или человекообразной обезьяной), обезьяной (например, мартышкой, бабуином), человекообразной обезьяной (например, гориллой, шимпанзе, орангутангом, гиббоном), или человеком.
Более того субъект/пациент может находиться в любой форме развития, например в виде плода.
В одном предпочтительном варианте реализации субъектом/пациентом является человек.
Композиции
Хотя можно вводить соединение - производное BCSA само по себе, предпочтительно обеспечивать его в виде фармацевтической рецептуры (например, композиции, препарата, лекарственного средства), содержащей по меньшей мере одно описанное в настоящей заявке соединение, совместно с одним или более фармацевтически приемлемыми ингредиентами, хорошо известными специалистам в данной области, включая, но не ограничиваясь: фармацевтически приемлемыми носителями, разбавителями, эксципиентами, адъювантами, наполнителями, буферами, консервантами, антиоксидантами, лубрикантами, стабилизаторами, солюбилизаторами, сурфактантами (например, смачивающими агентами), маскирующими агентами, красителями, ароматизаторами и подсластителями. Композиция может дополнительно содержать другие активные агенты, например другие терапевтические и профилактические агенты.
Таким образом, настоящее изобретение дополнительно обеспечивает вышеупомянутые фармацевтические композиции и способы приготовления фармацевтических композиций посредством смешивания по меньшей мере одного соединения - производного BCSA, описанного в настоящей заявке, совместно с одним или более фармацевтически приемлемым ингредиентом, хорошо известными специалистам в данной области, например носителем, разбавителем, эксципиентом и т.д. При введении в рецептуру в виде дискретных единиц (например, таблеток и так далее) каждая такая единица содержит заранее определенное количество (дозу) соединения.
Выражение "фармацевтически приемлемый", употребляемое в настоящей заявке, относится к соединениям, ингредиентам, материалам, композициям, лекарственным формам и т.д., которые согласно компетентному медицинскому суждению пригодны для осуществления их контакта с тканями рассматриваемого субъекта (например, человека) без опасности чрезмерной токсичности, раздражения, аллергической реакции или другой проблемы или осложнения, соразмерно с разумным отношением польза/риск. Каждый носитель, разбавитель, эксципиент и т.д. также должен быть "приемлемым" в смысле совместимости с другими ингредиентами указанной композиции.
Подходящие носители, разбавители, эксципиенты и так далее можно найти в стандартных фармацевтических текстах, например, Remington's Pharmaceutical Sciences, 18th edition, Mack Publishing Company, Easton, Pa., 1990; и Handbook of Pharmaceutical Excipients. 2nd edition, 1994.
Композиции можно приготовить любыми способами, хорошо известными специалистам в области фармацевтики. Такие способы включают стадию осуществления ассоциации соединения согласно настоящему изобретению с носителем, состоящим из одного или более вспомогательного ингредиента. В целом композиции готовят посредством равномерного и непосредственного объединения соединения с носителями (например, жидкими носителями, тонкодисперсными твердыми носителями и т.д.), и затем, если необходимо, приданием продукту формы.
Композицию можно приготовить таким образом, чтобы обеспечить быстрое или медленное высвобождение; мгновенное, замедленное, рассчитанное по времени или поддерживающее высвобождение или комбинацию различных указанны видов высвобождения.
Композиции в зависимости от потребности можно получать в форме жидкости, растворов (например, водных, неводных), суспензий (например, водных, неводных), эмульсий (например, масло в воде, вода в масле), эликсиров, сиропов, лекарственных кашек, полосканий, капель, таблеток (включая, например, покрытые таблетки), гранул, порошков, лепешек, пастилок, капсул (включая, например, твердые и мягкие желатиновые капсулы), крахмальных капсул, гранул, ампул, болюсов, суппозиториев, пессариев, настоек, гелей, паст, мазей, кремов, лосьонов, масел, пен, спреев, туманов или аэрозолей.
Композиции в зависимости от потребности можно получать в форме пластыря, адгезивного пластыря, бандажей, шин или тому подобного, импрегнированных одним или более соединением и возможно одним или более фармацевтически приемлемым ингредиентом, включая, например, усилители проникновения, просачивания и абсорбции. Композиции в зависимости от потребности можно получать в форме депо или резервуара.
Соединение согласно настоящему изобретению можно растворять, суспендировать или смешивать с одним или более фармацевтически приемлемым ингредиентом. Соединение можно обеспечить в виде липосом и других микрочастиц, предназначенных для целевой доставки соединения, например, в компоненты крови или один или более орган.
Композиции могут быть пригодными для перорального введения (например, глотанием) включают жидкости, растворы (например, водные, неводные), суспензии (например, водные, неводные), эмульсии (например, масло в воде, вода в масле), эликсиры, сиропы, лекарственные кашки, таблетки, гранулы, порошки, капсулы, крахмальные облатки, пилюли, ампулы, болюсы.
Композиции, пригодные для буккального введения, включают полоскания, лепешки, пастилки, а также накладки, адгезионные пластыри, депо и резервуары. Лепешки типично включают соединение на базе вкусовых ароматизирующих веществ, обычно сахарозы и камедей акации или трагаканта. Пастилки типично содержат соединение в инертной матрице, такой как желатин и глицеринили сахарозу и камедь акации. Полоскания типично содержат соединение в пригодном жидком носителе.
Композиции, пригодные для подъязычного введения, включают таблетки, лепешки, пастилки, капсулы и пилюли.
Композиции, пригодные для введения через слизистую оболочку рта, включают жидкости, растворы (например, водные, неводные), суспензии (например, водные, неводные), эмульсии (например, масло в воде, вода в масле), полоскания, лепешки, пастилки, а также накладки, адгезивные пластыри, депо и резервуары.
Композиции, пригодные для введения через слизистые оболочки, отличные от слизистой оболочки рта, включают жидкости, растворы (например, водные, неводные), суспензии (например, водные, неводные), эмульсии (например, масло в воде, вода в масле), суппозитории, пессарии, гели, пасты, мази, кремы, лосьоны, масла, а также накладки, адгезивные пластыри, депо и резервуары.
Композиции, пригодные для чрескожного введения, включают гели, пасты, мази, кремы, лосьоны и масла, а также накладки, адгезивные пластыри, бандажи, повязки, депо и резервуары.
Таблетки могут быть изготовлены традиционным способом, например компрессией или формованием, возможно с одним или более вспомогательным ингредиентом. Прессованные таблетки можно приготовить компрессией в подходящей машине соединения в свободно текучей форме, такой как порошок или гранулы, возможно смешанные с одним или более связующими агентами (например, повидоном, желатином, камедью акации, сорбитолом, трагакантом, гидроксипропилметилцеллюлозой); наполнителями или разбавителями (например, лактозой, микрокристаллической целлюлозой, гидрофосфатом кальция); лубрикантами (например, стеаратом магния, тальком, силикагелем); дезинтегрантами (например, натриевой солью гликолята крахмала, поперечно-связанным повидоном, поперечно-связанной натриевой солью карбоксиметилцеллюлозы); поверхностно-активными или диспергирующими, или смачивающими агентами (например, натриевой солью лаурилсульфата); консервантами (например, метиловым эфиром парагидроксибензойной кислоты, пропиловым эфиром парагидроксибензойной кислоты, сорбиновой кислотой); ароматизаторами, усилителями вкуса и аромата и подсластителями. Формованные таблетки можно изготавливать в подходящей машине отливкой смеси порошкообразного соединения, смоченной инертным жидким разбавителем. Таблетки можно дополнительно покрывать или делать рифлеными, таким образом, чтобы обеспечить из них медленное или контролируемое высвобождение, применяя в различных пропорциях, например, гидроксипропилметилцеллюлозу для того, чтобы обеспечить желаемый профиль высвобождения. Дополнительно таблетки можно обеспечивать покрытием, например, чтобы повлиять на высвобождение, например покрытием, растворяющимся в кишечнике, чтобы обеспечить высвобождение в участках желудочно-кишечного тракта иных, чем желудок.
Мази типично готовят из соединения на парафиновой или смешивающейся с водой мазевой основе.
Кремы типично изготавливают из соединения и масляно-водной кремовой основы. Если желательно, водная фаза кремовой основы может включать, например, по меньшей мере около 30% масс./масс., многоатомного спирта, то есть спирта, имеющего две или более гидроксильных группы, такого как пропиленгликоль, бутан-1,3-диол, маннитол, сорбит, глицерин и полиэтиленгликоль и их смеси. Композиции местного действия могут желательно включать соединение, увеличивающее абсорцию, или проникновение соединения через кожу или другие пораженные области. Примеры таких соединений, увеличивающих проникание через кожу, включают диметилсульфоксид и родственные аналоги.
Эмульсии типично готовят из соединения и масляной фазы, возможно содержащей только эмульгирующий агент (иначе известный как эмульгатор), или эта фаза может содержать смесь по меньшей мере одного эмульгатора с жиром или маслом, или как жиром, так и маслом. Предпочтительно гидрофильный эмульгатор включают совместно с липофильным эмульгатором, который действует как стабилизатор. Также предпочтительно включать как масло, так и жир. Вместе эмульгатор(ы), в присутствии или в отсутствии стабилизатора(ов) составляют так называемый эмульгирующий воск, а воск вместе с маслом и/или жиром составляет так называемую эмульгирующую основу мази, образующую масляную дисперсионную фазу кремовых композиций.
Подходящие эмульгаторы и стабилизаторы эмульсии включают Tween 60, Span 80, цетостеариловый спирт, миристиловый спирт, моностеарат глицерина и натриевую соль лаурилсульфата. Выбор пригодных масел или жиров для композиции основан на достижении желаемых косметических свойств, поскольку растворимость соединения в большинстве масел, которые принято применять в композициях фармацевтической эмульсии, может быть очень низкой. Так, крем должен представлять собой предпочтительно нежирный, не окрашивающий и смывающийся продукт с консистенцией, позволяющей избежать протекание из туб или других контейнеров. Можно применять неразветвленные или разветвленные моно- или диосновные алкиловые сложные эфиры, такие как диизоадипат, изоцетилстеарат, диосновный сложный эфир пропиленгликоля, диосновные сложные эфиры жирных кислот кокосового ореха, изопропилмиристат, децилолеат, изопропилпальмитат, бутилстеарат, 2-этилгексилпальмитат или можно применять смесь разветвленных сложных эфиров, известных под названием Crodamol САР, причем три из последних сложных эфиров являются предпочтительными. Их можно применять сами по себе или в комбинации, зависящей от требуемых свойств. Альтернативно, можно применять липиды с высокими точками плавления, такие как белый мягкий парафин или другие минеральные масла.
Композиции для интраназального введения с жидким носителем включают, например, спрей для носа, капли для носа или композиции для аэрозольного введения, содержащие распылитель, включающие водные или масляные растворы соединения.
Композиции для интраназального введения с твердым носителем включают, например, крупный порошок с диаметром частиц в диапазоне приблизительно от 20 до 500 мкм, который вводят так, как принято вводить лекарственный порошок для введения через нос, то есть быстрой ингаляцией через носовой проход из контейнера порошка, который держат расположенным близко от носа.
Композиции для легочного введения (например, при ингаляционной или инсуффляционной терапии) включают составы, полученные в виде аэрозольного спрея в упаковке под давлением с применением подходящего газа-вытеснителя (пропелланта), такого как дихлордифторметан, трихлорфторметан, дихлортетрафторэтан, диоксид углерода или других подходящих газов.
Композиции, пригодные для окулярного введения, включают глазные капли, в которых соединение растворяют или суспендируют в подходящем носителе, особенно в водном растворителе соединения.
Композиции, пригодные для ректального введения, могут быть представлены в виде суппозиториев с подходящей основой, содержащей, например, природные или гидрогенизированные масла, воска, жиры, полужидкие или жидкие полиолы, например масло какао или салицилат; или раствор или суспензию для лечения с помощью клистира.
Композиции, пригодные для вагинального введения, могут быть представлены в виде пессариев, тампонов, кремов, гелей, паст, пен или спреев, содержащих наряду с соединением такие носители, которые специалисты в данной области считают приемлемыми.
Композиции, пригодные для парентерального введения (например, инъекцией), включают водные или неводные, изотонические, апирогенные, стерильные жидкости (например, растворы, суспензии), в которых соединение растворено, суспендировано или заключено иначе (например, в липосоме или другой микрочастице). Такие жидкости дополнительно могут содержать другие фармацевтически приемлемые ингредиенты, такие как антиоксиданты, буферные смеси, консерванты, стабилизаторы, бактериостатики, суспендирующие агенты, загустители и растворенные вещества, сообщающие композиции изотоничность с кровью (или с другой, относящейся к настоящей заявке жидкостью организма) намеченного реципиента. Примеры эксципиентов включают, например, воду, спирты, полиолы, глицерин, растительные масла и тому подобное. Примеры подходящих изотонических носителей для применения в таких композициях включают раствор хлористого натрия для инъекций Рингера или лактатный раствор Рингера. Типично концентрация соединения в жидкости составляют от около 1 нг/мл до около 10 мкг/мл, например от около 10 нг/мл до около 1 мкг/мл. Композиции можно поставлять в стандартных контейнерах для однократного дозирования или мультидозных герметизированных контейнерах, например в ампулах или склянках, и можно хранить в состоянии сухой заморозки (лиофилизованном состоянии), требующем только добавление стерильного жидкого носителя, например воды для инъекций, непосредственно перед применением. Импровизированные инъекционные растворы и суспензии можно приготовить из стерильных порошков, гранул и таблеток.
Дозировка
Специалист в данной области техники примет во внимание, что подходящие дозировки соединений - производных BCSA, и композиций, содержащих соединения -производные BCSA, могут варьироваться от пациента к пациенту. Определение оптимальной дозировки в целом будет зависеть от баланса уровня терапевтической пользы относительно любого риска или вредных побочных эффектов. Выбранный уровень дозировки будет зависеть от многих факторов, включая, но не ограничиваясь перечисленными: активность конкретного соединения, способ введения, время введения, способ экскреции соединения, продолжительность лечения, другие лекарственные средства, соединения и/или материалы, применяемые в комбинации, тяжесть состояния, а также видовой принадлежности, пола, возраста, массы, состояния, общего состояния здоровья и предыдущей медицинской истории пациента. Количество соединения и способ введения, в конце концов, будет на усмотрении врача, ветеринара или клинициста, хотя в целом выбор дозы будет выбран таким образом, чтобы достичь в месте действия концентраций, обуславливающих желаемый эффект без проявления значительных токсичных или вредных побочных эффектов.
Во время курса лечения введение можно осуществлять в виде единичной дозы, непрерывно или периодически (например, разделенные дозы через определенные интервалы времени). Способы определения наиболее эффективных способов введения и дозировок хорошо известны специалистам в данной области и будут изменяться в зависимости от композиции, применяемой для терапии, назначения терапии, вида клеток, на которые нацелена терапия, и излечиваемого субъекта. По выбору лечащего врача, ветеринара или клинициста можно применять уровни дозировки и режим введения.
В целом пригодная суточная доза соединения - производного BCSA находится в диапазоне от около 100 мкг до около 250 мг (более типично от около 100 мкг до около 25 мг) на килограмм массы тела субъекта. Независимо от того, является ли соединение солью, сложным эфиром, амидом, пролекарством или тому подобным, вводимое количество рассчитывают на базе материнского соединения, пропорционально увеличивая расчетную массу телу.
ПРИМЕРЫ
Нижеследующие примеры, описанные в настоящей заявке, приведены только, чтобы проиллюстрировать настоящее изобретение, и не предназначены ограничить объем его притязаний.
Общий синтез
Циклические сульфонамидные производные (5.1)-(5.68) были приготовлены следующим образом (Схема 1). Сульфонилирование аминов (2.1)-(2.61) сульфонилхлоридами (1.1)-(1.8) осуществлялось нагревом, обеспечивающим циклизацию. Некоторые сложные эфиры (3) были выделены и гидролизованы в кислых условиях, образуя соответствующие карбоновые кислоты (4). Некоторые промежуточные сложные эфиры (3) трансформировали в карбоновые кислоты (4) без выделения путем продолжительного нагревания в том же реакционном сосуде, что привело к гидролизу сложноэфирной функции. Карбоновые кислоты (4.1)-(4.68) превращали в соответствующие гидроксамовые кислоты (5.1)-(5.68), применяя один из трех способов (Условия А-С, Схема 1).
Схема 1

Сульфонилхлорид (1.1), (где R1=R2=R3=Н), используемый при синтезе сульфонамидов (5.1)-(5.61), приготовили по известной методике (см., например, Finn et al., 2005). Сульфонилхлорид (1.2)-(1.6), требующийся для синтеза сульфонамидов (5.62)-(5.66), приготовили региоселективным хлорсульфонилированием известных ненасыщенных сложных эфиров (7.1)-(7.5) (см., например, Imashiro, 2004; Westman et al., 2001; El-Batta et al., 2007; Mahajan et al., 2005; Skretas et al., 2007).
Схема 2

Сульфонилхлорид (1.7)-(1.8), требуемый для синтеза сульфонамидов (5.67)-(5.68), приготовили исходя из аминобензолсульфоновых кислот (8.1)-(8.2) (Схема 3). Их переводили в соли диазония (9.1)-(9.2), которые затем применяли в реакции Хека, получая ненасыщенные сложные эфиры (10.1)-(10.2). Промежуточные соединения (10.1)-(10.2) реакцией с хлористым тионилом переводили в сульфонилхлорид (1.7)-(1.8).
Схема 3

Амины (2.1)-(2.42), применяемые в синтезе соединений (5.1)-(5.42), имелись в продаже. Амины (2.43)-(2.44), требующиеся в синтезе сульфонамидов (5.43)-(5.44), получали О-алкилированием парагидроксианилина (11) бут-2-ин-1-ил метансульфонатом (12) (см., например, Brummond et al., 2004) и 4-хлорметил-2-метилхинолином (13) (см., например, Duan et al., 2002) с образованием анилинов (2.43) и (2.44), соответственно (Схема 4).
Схема 4

Амины (2.45)-(2.61), требующиеся в синтезе сульфонамидов (5.45)-(5.61), получали О-алкилированием парагидроксинитробензола (14) алкилирующими агентами (15.1)-(15.17) и последующим восстановлением нитрогруппы в полученных промежуточных соединениях (16.1)-(16.17), применяя для восстановления одно из трех условий реакции (Схема 5, условия А-С).
Схема 5

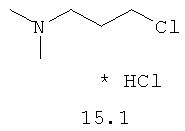



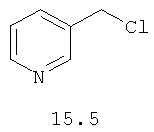







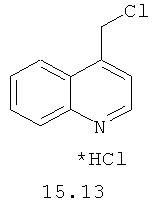




Алкилирующиеся агенты (15.1)-(15.7), требующиеся в синтезе анилинов (2.45)-(2.51), имелись в продаже. Алкилирующие агенты (15.8)-(15.11), требующиеся в синтезе анилинов (2.52)-(2.55), приготовили описанными в литературе способами (см., например, White et al., 1982; Jackson et al., 1988; Thibault et al., 2006; Marshall et. al., 2000).
Алкилирующий агент (15.12), требующийся в синтезе анилина (2.56), приготовили способом, показанным на Схеме 6. 2-Метил-4-гидроксиметилхинолин (17) окисляли периодинаном Dess-Martin с образованием альдегида. Добавление бромистого метилмагния к промежуточному альдегиду привело к вторичному спирту, который обрабатывали метансульфонилхлоридом с образованием алкилирующего агента (15.12).
Схема 6

4-Хлорметилхинолин (15.13), требующийся в синтезе анилина (2.57), приготовили из известного 4-гидроксиметилхинолина (18) (см., например, Boutros et.al., 2000) (Схема 7).
Схема 7

Синтез алкилирующих агентов (15.14) и (15.15), требующихся для приготовления анилинов (2.58) и (2.59), начинали из карбоновых кислот (19.1) и (19.2), которые приготовили способами, описанными в литературе (см., например, Yen et al., 1958; Buchman et al., 1946) (Схема 8). Карбоновые кислоты (19.1) и (19.2) переводили в их эфиры, которые затем восстанавливали до спиртов. Эти промежуточные продукты переводили в искомые хлорметилхинолины (15.14) и (15.15) реакцией с хлористым тионилом.
Схема 8

Синтез 4-хлорметилпиридинов (15.16) и (15.17), требующихся для приготовления анилинов (2.60) и (2.61), провели исходя из производных 4-метилпиридина (20.1) и (20.2) (Схема 9). Спирты (21.1) и (21.2) приготовили по известной схеме (см., например, Ragan et al., 2002) и переводили в 4-хлорметилпиридины (15.16) и (15.17).
Схема 9

Для приготовления гидроксамовой кислоты (24) сульфонилхлорид (1.1) сначала переводили в ненасыщенный сложный эфир (23) реакцией с замещенным анилином (22) (Схема 10). Реакция сложного эфира (23) с гидроксиламином в основных условиях привела к внутримолекулярной циклизации и образованию гидроксамовой кислоты (24).
Схема 10

Синтез гидроксамовой кислоты (29) показан на Схеме 11. Свободную гидроксильную группу промежуточного соединения (16.7) (Схема 5) мезитилировали. Метансульфонатную группу замещали азидогруппой, азид восстанавливали и в полученном амине защищали аминогруппу трет-бутоксикарбонильной группой, получая промежуточное соединение (25). Восстановление нитрогруппы привело к анилину (26), реакция которого с сульфонилхлоридом (1.1) привела к циклической карбоновой кислоте (27). Карбоновую кислоту трансформировали в гидроксамовую кислоту (28), N-трет-бутоксикарбонильную защитную группу, в которой отщепляли с образованием конечного продукта (29) в виде хлористоводородной соли.
Схема 11

Гидроксамовую кислоту (36) приготовили по Схеме 12. Известный ненасыщенный сложный эфир (30) (см., например, Eberbach et al., 1986) региоселективно хлорсульфонилировали и продукт (31) применяли для реакции с анилином (2.1) с образованием циклического сложного эфира (32). Фенольную гидроксильную группу сульфонилировали ангидридом трифторметансульфоновой кислоты и полученный продукт (33) применяли в реакции сочетания по Сузуки-Мияура с фенилбороновой кислотой. Сложноэфирную группу в промежуточном соединении (34) гидролизовали, и карбоновую кислоту (35) переводили в гидроксамовую кислоту (36).
Схема 12

Гидроксамовую кислоту (39) приготовили из циклического сложного эфира (32). Его О-алкилировали и продукт (37) гидролизовали с образованием карбоновой кислоты (38), которую, в свою очередь, переводили в гидроксамовую кислоту (39).
Схема 13

Синтез гидроксамовой кислоты (43) описывается Схемой 14. Реакция сульфонилхлорида (1.8) с анилином (2.1) привела к ненасыщенному сложному эфиру (40). Его применили в сочетании с фенилбороновой кислотой по Сузуки-Мияура с образованием промежуточного соединения (41), которое подвергали циклизации с последующим гидролизом с образованием карбоновой кислоты (42), в свою очередь, трансформируемой в гидроксамовую кислоту (43).
Схема 14

Гидроксамовые кислоты (48.1) и (48.2) приготовили, исходя из имеющихся в продаже сульфонамидов (44.1) и (44.2) (Схема 15). Они были обработаны литием в ортопозиции и функционально переведены в сульфонамид (см., например, MacNeil et al., 2001) с последующим йодированием, которое привело к промежуточным продуктам (45.1) и (45.2). Реакция Хека арилиодидов (45.1) и (45.2) с метилакрилатом привела к циклическим сложным эфирам (46.1) и (46.2). Их гидролизовали до карбоновых кислот (47.1) и (47.2) и дополнительно трансформировали в гидроксамовые кислоты (48.1) и (48.2).
Схема 15

Гидроксамовые кислоты (54.1)-(54.9) приготовили, применяя другой подход (Схема 16). Сульфонамиды (50.1)-(50.9) получали из имеющегося в продаже сульфонилхлорида (49.1)-(49.9) и применяли для реакции прямого ортолитирования - формилирования с образованием промежуточных соединений (51.1)-(51.9). Реакция олефинирования этих промежуточных соединений привела к циклическим эфирам (52.1)-(52.9), которые были гидролизованы до кислот (53.1)-(53.9), далее трансфомированных в гидроксамовые кислоты (54.1)-(54.9).
Схема 16

Сульфонамид (50.10) приготовили из сульфонилхлорида (49.10) и применяли для реакции ортолитирования - формилирования. Это привело к дегалогенированному продукту (51.10), который в дальнейшем трансформировали в гидроксамовую кислоту (54.10), применяя уже охарактеризованный синтетический путь (Схема 17).
Схема 17

Гидроксамовую кислоту (54.11) получали по Схеме 18. Сульфонамид (50.11) приготовили из сульфонилхлорида. (49.11) подвергли реакции ортолитирования-формилирования с образованием прмежуточного соединения (51.11). Последний применяли в реакции олефинирования с образованием продукта 52.11, в котором группа фтора замещена метосигруппой. Последняя была дополнительно трансформирована в гидроксамовую кислоту (54.11) по известным процедурам.
Схема 18

Циклический промежуточный продукт (51.5) привел к продукту (52.12), содержащему группу фтора, по реакции олефинирования замещенную кроме продукта (52.5) метоксигруппой (Схема 19). Циклический сложный эфир (52.12) трансформировали в гидроксамовую кислоту (54.12), применяя общепризнанные процедуры.
Схема 19
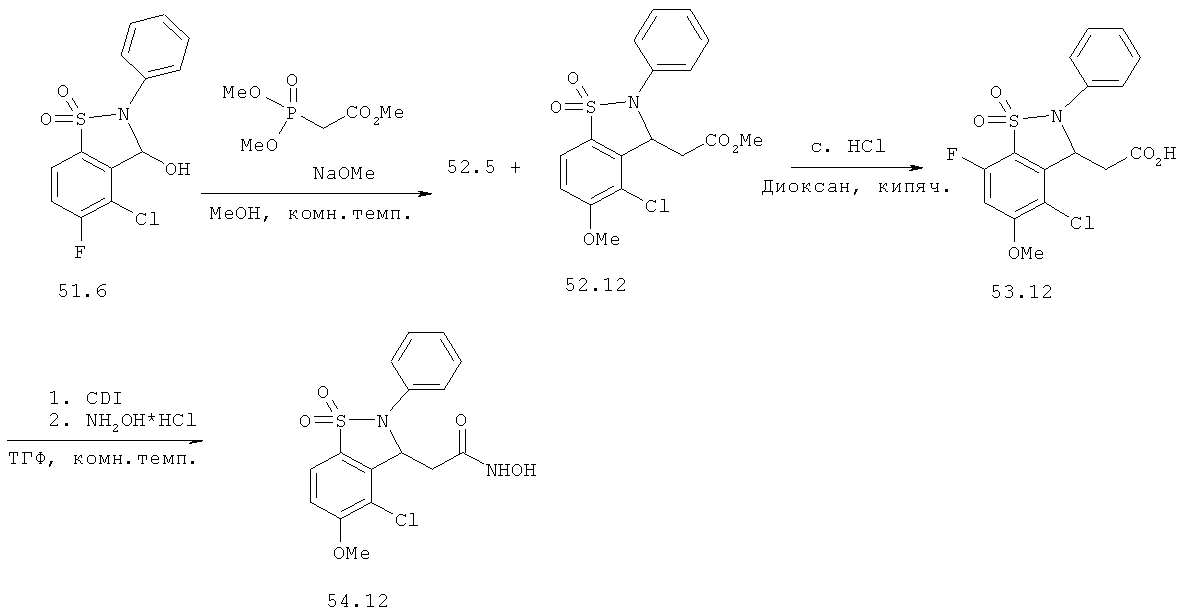
Гидроксамовую кислоту (57) приготовили из сложного эфира (3.1) (Схема 20). Его восстанавливали и полученный первичный спирт трансформировали в хлорид. Хлорид замещали на цианид с образованием промежуточного продукта (55), который был гидролизован и полученную карбоновую кислоту (56) дополнительно трансформировали в гидроксамовую кислоту (57).
Схема 20

Синтез гидроксамовых кислот (62.1)-(62.2) проводили по Схеме 21. Сульфонамиды (59.1)-(59.2), приготовленные из сульфонилхлорида (58.1)-(58.2), трансформировали в эфиры карбоновых кислот (60.1)-(60.2), согласно ранее опубликованной последовательности (см., например, Takahashi et al., 2003). Сложные эфиры (60.1)-(60.2) гидролизовали, и полученные карбоновые кислоты (61.1)-(61.2) превращали в гидроксамовые кислоты (62.1)-(62.2).
Схема 21

Стереоизомеры циклических сульфонамидов (5.1), (5.43) и (5.44) были приготовлены в энантиомерно чистой форме (Схема 22). Для этой цели (R)-фенилглицинол ацетилировали рацемическими кислотами (4.1), (4.43) и (4.44) с образованием соответствующих амидов в виде смеси диастереомеров (S,R)-(63.1), (63.2), (63.3) и (RR)-(63.1), (62.3), (63.3), которую разделяли хроматографически. Разделенные амиды (S,R)-(63.1), (63.2), (63.3) и (R,R)-(63.1), (63.2), (63.3) были гидролизованы до энантиомерно чистых изомеров кислот (S)-(4.1), (4.43), (4.44) и (S)-(4.1), (4.43), (4.44), которые далее превращали в энантиомерно чистые гидроксамовые кислоты (+)-(5.1), (+)-(5.43), (+)-(5.44) и (-)-(5.1), (-)-(5.43) и (-)-(5.44).
Схема 22

Гидроксамовые кислоты (72) приготовили следующим образом (Схема 23). Салициловый альдегид (64) обрабатывали N,N-диметилтиокарбамоилхлоридом с образованием тиокарбамата (65). Его вводили в перегруппировку Ньюмана-Кварта, получая S-карбамоилтиосалициловый альдегид (66). Карбамоильную группу в (66) отщепляли MeONa, и образовавшийся тиолят in situ алкилировали бромистым бензилом, получая S-бензилтиосалициловый альдегид (67). Последующая реакция Виттига с альдегидом (67) привела к ненасыщенному сложному эфиру (68). Сульфидную группу в сложном эфире (68) окисляли с образованием сульфона (69), который трансформировали в результате поддерживаемой NaHCO3 внутримолекулярной реакции Михаэля в циклический продукт (70). Гидролиз сложного эфира (70) в кислых условиях привело к карбоновой кислоте (71), которую переводили в гидроксамовую кислоту (72).
Схема 23
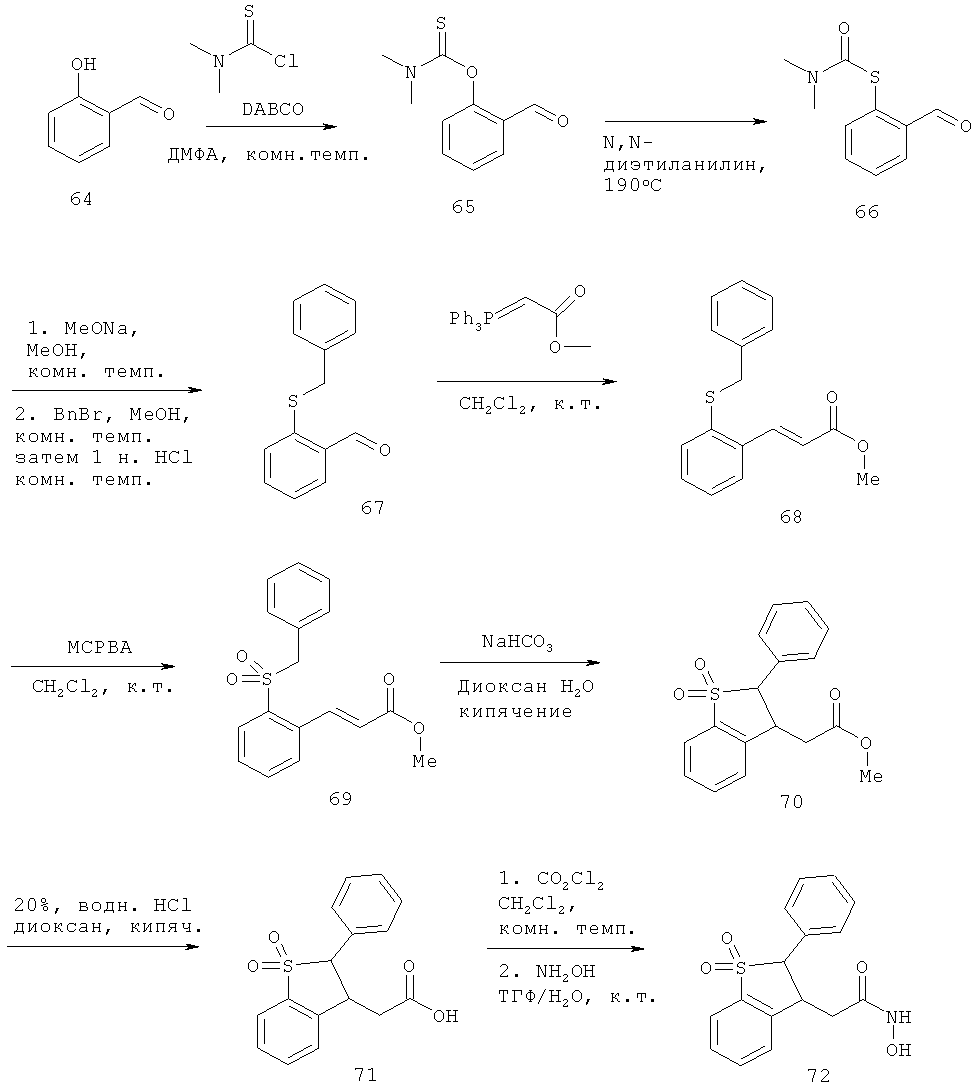
Гидроксамовую кислоту (77) приготовили исходя из известного сульфонамида (73) (см., например, Goulaouic-Dubois et al., 1995). Последовательность реакций ортолитирования и йодирования привела к йодиду (74), который применяли в реакции Хека с метилакрилатом с образованием циклического сложного эфира (75). Его гидролизовали до карбоновой кислоты (76), в дальнейшем трансформируемой в гидроксамовую кислоту (77).
Схема 24

Общая процедура приготовления метиловых эфиров(Е)-3-(2-хлорсульфонилфенил)акриловой кислоты (1.2)-(1.6)
Способ А: Хлорсульфоновую кислоту (3,5 мл, 52 мМоль) охлаждали на ледяной бане и добавляли к ней ненасыщенный сложный эфир (7) (1,0 г, 5,2 мМоль). Охлаждаемую смесь перемешивали до исчезновения исходного материала (контроль по ТСХ, от 30 минут до 6 часов) и осторожно выливали в ледяную воду. В случае образования осадка, его собирали на фильтре, промывали водой и сушили под вакуумом с образованием продуктов (1). Если осадок не выпадал, водную фазу экстрагировали CHCl3, объединенную органическую фазу сушили над Na2SO4, а растворитель удаляли под вакуумом с образованием неочищенного продукта (1), который применяли на следующей стадии без дополнительной очистки.
Следуя методике, аналогичной способу А, в виде неочищенных продуктов были получены следующие соединения.




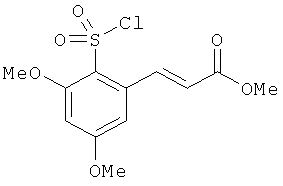
Соединение (1.2): Сероватый порошок (0,88 г, 59%).1Н-ЯМР (CDCl3, ТМС (тетраметилсилан)) δ: 3.85 (3Н, s); 3.94 (3H, s); 6.41 (1Н, d, 15 Гц); 7.01 (1Н, dd, 2 Гц и 9 Гц); 7.16 (1Н, d, 2 Гц); 8.07 (1Н, d, 9 Гц) и 8.46 ppm (миллионных долей) (1Н, d, 15 Гц).
Общая процедура синтеза метиловых эфиров (Е)-3-(2-хлорсульфонилфенил)акриловой кислоты (1.7) и (1.8)
Способ В: 2-Аминобензолсульфоновую кислоту (8) (10 мМоль) суспендировали в серной кислоте (5 мл) и реакционную смесь охлаждали на ледяной бане. К смеси добавляли 40% водный раствор NaNO2 (2 мл), и смесь перемешивали в течение 1 часа. Добавляли Et2O, и осадок собирали на фильтре. Получали неочищенный продукт (9) (2,35 г), который суспендировали в ДМФА (диметилформамиде) (7 мл) в инертной атмосфере, и добавляли Pd2(dba)3 (30 мг), а затем метиловый эфир акриловой кислоты (2,7 мМоль, 30 мМоль). Реакционную смесь перемешивали при комнатной температуре в течение 10 ч. Растворитель упаривали под вакуумом, получая сырую сульфоновую кислоту (10). К ней добавляли толуол (7 мл) и хлористый тионил (5,5 мл, 80 мМоль). Полученную смесь кипятили с обратным холодильником в течение 4 ч, охлаждали до комнатной температуры и фильтровали. Раствор концентрировали под вакуумом с образованием неочищенных продуктов (1.7) или (1.8).
Следуя методике, аналогичной способу В, в виде неочищенных продуктов были получены следующие соединения.


Синтез 8
4-(Бут-2-ин-1-илокси)фениламин (2.43)

Смесь сернокислой соли парагидроксианилина (11) (395 мг, 2,5 мМоль), бут-2-ин-1-илметансульфоната (12) (370 мг, 2,5 мМоль) и Cs2CO3 (2,44 г, 7,5 мМоль) в ДМФА (10 мл) нагревали при 60°С в течение 6 ч. Смесь выливали в воду (50 мл) и экстрагировали EtOAc (20 мл). Органическую фазу отделяли и сушили над Na2SO4. Раствор фильтровали и упаривали с образованием неочищенного продукта (2.43) (140 мг, 35%) в виде темного масла, применяемого на следующей стадии без дополнительной очистки.1Н-ЯМР (CDCl3, ТМС) δ: 1.84 (3Н, t, 2 Гц); 4.53 (2Н, m); 6.62 (2Н, d, 8 Гц) и 6.78 ppm (2Н, d, 8 Гц).
Синтез 9
4-(2-Метилхинолин-4-метилокси)анилин (2.44)

Смесь сернокислой соли парагидроксианилина (11) (207 мг, 1 мМоль), хлористоводородной соли 4-хлорметил-2-метил-хинолина (13) (228 мг, 1 мМоль) и Cs2CO3 (1.63 г, 5 мМоль) в ДМФА (5 мл) перемешивали при комнатной температуре в течение 3 ч. Смесь выливали в воду (50 мл) и экстрагировали EtOAc (20 мл). Органическую фазу отделяли и сушили над Na2SO4. Раствор фильтровали и упаривали. Остаток очищали флэш-хроматографией на силикагеле, элюируя EtOAc, с образованием продукта (2.44) (165 мг, 63%).1Н-ЯМР (CDCl3, ТМС) δ: 2.73 (3Н, s); 3.9 (2Н, br s); 5.40 (2Н, s); 6.65 (2Н, d, 9 Гц); 6.85 (2Н, d, 9 Гц); 7.44 (1Н, s); 7.50 (1Н, t, 8 Гц); 7.68 (1Н, t, 8 Гц); 7.90 (1Н, d) и 8.07 ppm (1Н, d, 8 Гц).
Общая методика приготовления анилинов (2.45)-(2.61)
Способ С: 4-Нитрофенол (14) (3,1 г, 2,2 мМоль), алкилирующий агент (15) и K2CO3 (920 мг, 6,7 мМоль) суспендировали в ДМФА (7 мл). Полученную суспензию перемешивали при комнатной температуре в течение 48 ч и выливали в воду (70 мл). Продукт обрабатывали EtOAc (70 мл). Органическую фазу отделяли и промывали насыщенным солевым раствором (70 мл). Экстракт сушили над Na2SO4, фильтровали и растворитель удаляли под вакуумом с образованием практически чистого промежуточного продукта (16).
Для синтеза анилинов (2.45) и (2.46), промежуточные продукты (16.1) и (16.2) (6,5 мМоль) растворяли в EtOH (15 мл), и к раствору добавляли 10% Pd/C (95 мг). Смесь перемешивали в атмосфере Н2 до полного превращения исходного материала (около 4 ч). Смесь пропускали через колонку с целитом, и растворитель удаляли под вакуумом с образованием неочищенных анилинов (2.45) и (2.46). В синтезе анилина (2.48), в качестве катализатора гидрирования применяли никель Ренея.
В синтезе анилинов (2.47) (2.49)-(2.61) соответствущие промежуточные продукты (16) (0,3 мМоль) растворяли в метаноле (2 мл), к раствору добавляли Na2S×9H2O (1 мМоль), и смесь нагревали до образования флегмы до полного превращения исходного материала (около 2 ч). Растворитель удаляли под вакуумом и остаток распределяли между водой и EtOAc. Органическую фазу сушили над Na2SO4, фильтровали и упаривали. Остаток очищали при помощи колоночной хроматографии на силикагеле или применяли на следующей стадии без очистки.
Следуя методике, аналогичной способу С, следующие соединения были получены в виде неочищенных продуктов.





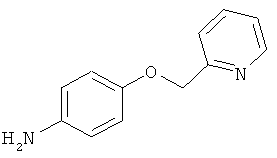

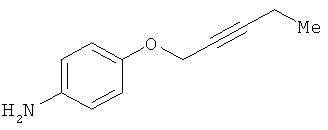



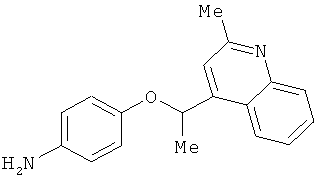
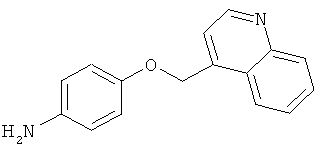




Общая процедура приготовления метиловых эфиров (1,1-диоксо-2-фенил-2,3-дигидро-1Н-бензо[d]изотиазол-3-ил)уксусной кислоты (3.1)-(3.3), (3.26)-(3.29), (3.44)-(3.46), (3.48)-(3.50), (3.56)-(3.66)
Способ D: К раствору хлористого сульфонила (1) (1 мМоль) и амина (2) (1 мМоль) в диоксане (5 мл) добавляли 1М водный раствор NaHCO3 (3 мл). Полученную смесь перемешивали при комнатной температуре в течение 2 часов и затем кипятили с обратным холодильником в течение 2 часов. После охлаждения до комнатной температуры добавляли воду (20 мл) и EtOAc (20 мл). Органическую фазу отделяли и промывали насыщенным солевым раствором (20 мл) и сушили над Na2SO4. Раствор фильтровали и упаривали, и остаток очищали флэш-хроматографией на силикагеле, элюируя смесью легкого петролейного эфира и EtOAc.
Следуя методике, аналогичной способу D, в виде неочищенных продуктов были получены следующие соединения.




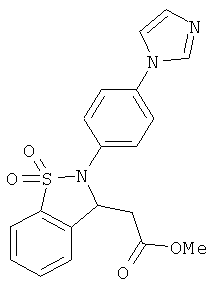

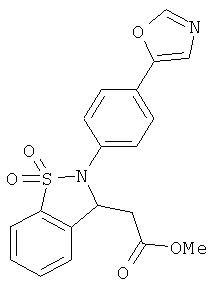














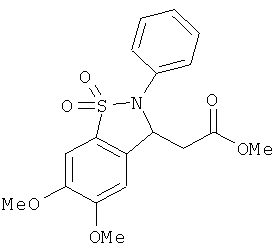

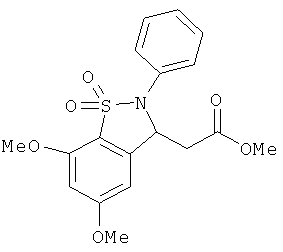
Соединение (3.1): Выход 61%,1Н-ЯМР (CDCl3, ТМС) δ: 2.74 (1Н, dd, 8 Гц и 16 Гц); 2.97 (1Н, dd, 4 Гц, полоса 16 Гц); 3.59 (3H, s); 5.58 (1Н, dd, 4 Гц и 8 Гц); 7.3-7.7 (8Н, m) и 7.89 ppm (1Н, d, 8 Гц).
Соединение (3.2): Выход 71%,1Н-ЯМР (ДМСО-d6, ТМС) δ: 3.01 (2Н, d, 5 Гц); 3.29 (3H, s); 5.92 (1Н, t, 5 Гц) и 7.5-8.0 ppm (11Н, m).
Соединение (3.3): Выход 23%,1Н-ЯМР (CDCl3, ТМС) δ: 2.40 (3H, s); 2.77 (1Н, dd, 8 Гц и 16 Гц); 2.97 (1Н, dd, 4 Гц и 16 Гц); 3.61 (3H, s); 5.56 (1Н, dd, 4 Гц и 8 Гц); 7.1-7.7 (7Н, m) и 7.89 ppm (1Н, d, 8 Гц).
Соединение (3.62): Выход 66%,1Н-ЯМР (CDCl3, ТМС) δ: 2.76 (1Н, dd, 8 Гц и 16 Гц); 2.96 (1Н, dd, 4 Гц и 16 Гц); 3.60 (3H, s); 3.70 (3H, s); 3.89 (3H, s); 5.53 (1Н, dd, 4 Гц и 8 Гц); 6.9-7.5 (7Н, m) и 7.77 ppm (1Н, d, 8 Гц).
Общие методики приготовления 2-(1,1-диоксо-2-фенил-2,3-дигидро-1Н-бензо[d]изотиазол-3-ил)уксусных кислот (4)
Способ Е: Из сложных эфиров (3). Раствор сложного эфира (3) (1 мМоль) в смеси диоксана (20 мл) и концентрированной водной HCl (5 мл) перемешивали при комнатной температуре в течение 2 суток. Растворители упаривали и заменяли свежим диоксаном (20 мл) и концентрированной водной HCl (5 мл). Перемешивание продолжали дополнительно еще двое суток, до полного исчезновения исходного материала (контроль по TCXI; если необходимо, систему растворителей меняли еще один раз). Растворители упаривали с образованием продукта (4).
Способ F: Из сульфонилхлорида (1) и аминов (2). К раствору хлористого сульфонила (1) (1 мМоль) и амина (2) (1 мМоль) в диоксане (5 мл) добавляли 1 М водный раствор NaHCO3 (3 мл). Полученную смесь перемешивали при комнатной температуре в течение 2 часов и затем кипятили с обратным холодильником в течение 8 часов. После охлаждения до комнатной температуры добавляли воду (20 мл) и EtOAc (20 мл). Водную фазу отделяли и подкисляли до значения pH приблизительно 2 концентрированной водной HCl и экстрагировали EtOAc (20 мл). Органическую фазу промывали насыщенным солевым раствором (20 мл) и сушили над Na2SO4. Раствор фильтровали и упаривали с образованием в остатке продукта (4) (содержание приблизительно 30-80%). В большинстве случаев его применяли в дальнейших реакциях без очистки.
Следуя способам, аналогичным способу Е или способу F, были получены сырые продукты следующих соединений.


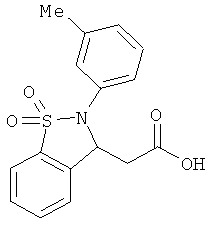








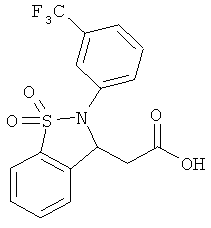




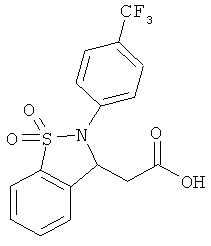

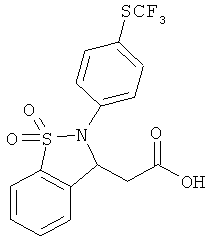








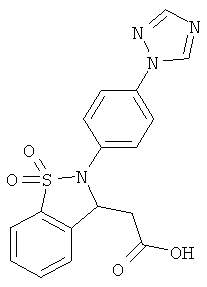




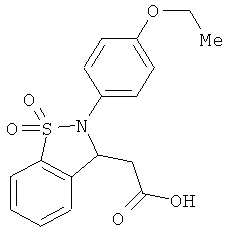


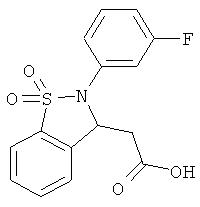



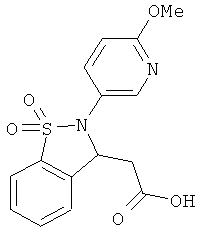


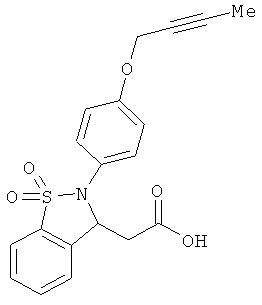



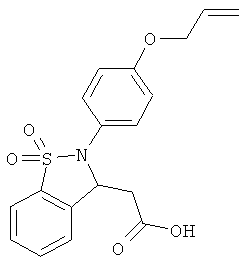



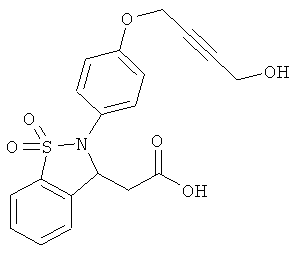




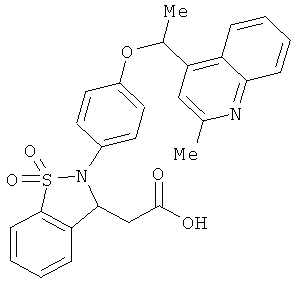








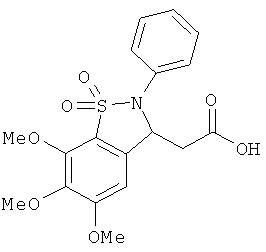



Соединение (4.1): Выход 65%, точка плавления 178-179°С (с разложением),1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.73 (1Н, dd, 7 Гц и 16 Гц); 2.91 (1Н, dd, 4 Гц и 16 Гц); 5.70 (1Н, t, 5 Гц); 7.3-7.6 (5Н, m) и 7.6-8.1 ppm (4Н, m).
Соединение (4.2): Выход 64%,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.83 (1Н, dd, 5 Гц и 16 Гц); 2.94 (1Н, dd, 5 Гц и 16 Гц); 5.86 (1Н, t, 5 Гц) и 7.5-8.1 ppm (11Н, m).
Соединение (4.3): Выход 23%,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.35 (3H, s); 2.72 (1Н, dd, 4 Гц и 16 Гц); 2.90 (1Н, dd, 7 Гц и 16 Гц); 5.67 (1Н, t, 5 Гц); 7.16 (1Н, d, 7 Гц); 7.2-7.4 (3H, m); 7.6-7.9 (3H, m) и 7.95 ppm (1Н, d, 7 Гц).
Соединение (4.43): Выход 61%, точка плавления 160-161°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 1.86 (3H, t, 2 Гц); 2.73 (1Н, dd, 16 Гц и 6 Гц); 2.85 (1Н, dd, 16 Гц и 4 Гц); 4.79 (2Н, d, 2 Гц); 5.50 (1Н, t, 5 Гц); 7.06 (2Н, d, 9 Гц); 7.42 (2Н, d, 9 Гц); 7.6-7.9 (3H, m); 7.94 (1Н, d, 8 Гц) и 12.42 ppm (менее 1Н, br s).
Соединение (4.44): Выход 32%,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.67 (3H, s); 2.79 (2Н, m); 5.51 (1Н, br t, ~5 Гц); 5.63 (2Н, s); 7.26 (2Н, d, 9 Гц); 7.45 (2Н, d, 9 Гц); 7.5-7.8 (6Н, m); 7.95 (2Н, t, 7 Гц) и 8.11 ppm (1Н, d, 8 Гц).
Соединение (4.62): Выход 37%,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.78 (1Н, dd, 6 Гц и 16 Гц); 2.91 (1Н, dd, 4 Гц и 16 Гц); 3.88 (3H, s); 5.63 (1Н, t, 5 Гц); 7.2-7.6 (7Н, m) и 7.95 ppm (1Н, d, 7 Гц).
Общие методики приготовления (1,1-диоксо-2-фенил-2,3-дигидро-1Н-бензо[d]изотиазол-3-ил)-N-гидроксиацетамидов (5)
Способ G: К раствору карбоновой кислоты (4) (1 мМоль) в CH2Cl2 (10 мл) добавляли хлористый ангидрид щавелевой кислоты (0,43 мл, 5 мМоль) и каплю ДМФА. Полученную смесь перемешивали при комнатной температуре и упаривали. К остатку добавляли смесь, полученную растворением хлористоводородной соли гидроксиламина (347 мг, 5 мМоль) в смеси ТГФ, (THF, тетрагидрофурана), (5 мл) и 1М водного NaHCO3 (5 мл). Полученную суспензию перемешивали в течение 15 минут и распределяли между EtOAc (50 мл) и водой (30 мл). Органическую фазу отделяли и промывали насыщенным NaHCO3 (20 мл) и насыщенным солевым раствором (20 мл). Раствор сушили над Na2SO4, фильтровали и упаривали. Продукт очищали при помощи препаративной обращеннофазной хроматографии и/или кристаллизацией.
Способ Н: CDI (4,5 мМоль, 1,5 эквивалента) добавляли к раствору карбоновой кислоты (4) (3,0 мМоль) в сухом ТГФ (5 мл). Реакционную смесь перемешивали в течение 1 ч. К смеси добавляли тонко растертую хлористоводородную соль гидроксиламина (417 мг, 6 мМоль). Полученную гетерогенную смесь перемешивали в течение ночи (около 16 ч). Смесь разбавляли 5% водн. KHSO4 (30 мл) и экстрагировали EtOAC (2×30 мл). Объединенную органическую фазу промывали насыщенным солевым раствором (30 мл) и сушили над Na2SO4. Экстракт фильтровали и концентрировали под вакуумом с образованием неочищенного продукта. Продукт (5) очищали с помощью препаративной обращеннофазной хроматографии и/или кристаллизацией.
Способ I: Смесь карбоновой кислоты (4) (0,24 мМоль), О-тритилгидроксиламина (66 мг, 0,24 эквивалента), EDCl, 1-этил-3-(3-диметилпропиламинопропил)карбодиимида, (33 мг, 0.24 мг) и HOBt, гидроксибензотриазола (46 мг, 0,24 мМоль) в ДМФА (2,4 мл) перемешивали в течение ночи и затем разбавляли насыщенным водным NaHCO3 (25 мл). Полученную смесь экстрагировали EtOAc (3×20 мл) и объединенную органическую фазу промывали насыщенным солевым раствором (20 мл). Экстракт сушили над Na2SO4, фильтровали и упаривали. Остаток очищали посредством флэш-хроматографии на силикагеле, элюируя легким петролейным эфиром и EtOAc. Полученную гидроксамовую кислоту, защищенную О-тритилом (0,17 мМоль), и 10% (объем/объем) трифторметансульфоновую кислоту растворяли в DCM (дихлорметане). Смесь перемешивали при комнатной температуре в течение 1 ч и добавляли МеОН (1 мл). Растворители удаляли под вакуумом, и остаток очищали с помощью препаративной обращеннофазной хроматографии с образованием продукта (5).
Следуя методикам, аналогичным Способу G, Способу Н или Способу I, получали следующие соединения в виде неочищенных продуктов.

















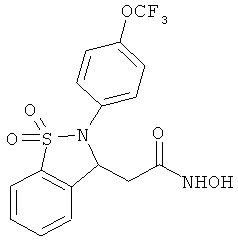




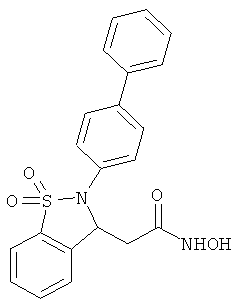




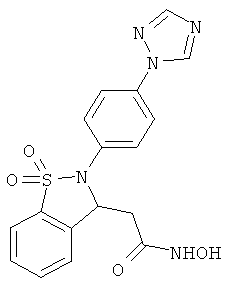










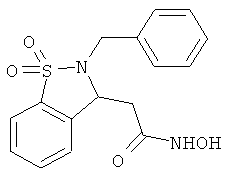
















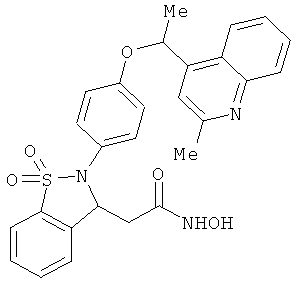


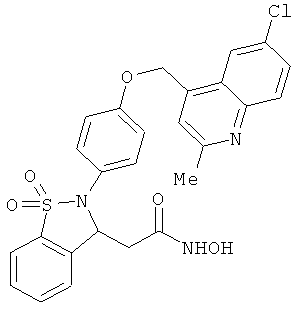







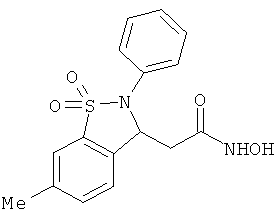

Соединение (5.1): Выход 85%, точка плавления 141-143°С,1Н-ЯМР (ДМСО-d6) ТМС) δ: 2.32 (1Н, dd, 9 Гц и 15 Гц); 2.64 (1Н, dd, 4 Гц и 15 Гц); 5.73 (1Н, dd, 4 Гц и 9 Гц); 7.2-7.4 (1Н, m); 7.4-7.6 (4Н, m); 7.6-7.9 (3H, m); 7.98 (1Н, d, 8 Гц); 8.9 (1Н, br s) и 10.5 ppm (1Н, brs).
Соединение (5.2): Выход 12%, аморфное,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.39 (1Н, dd, 15 Гц и 9 Гц); 2.69 (1Н, dd, 15 Гц и 4 Гц); 5.84 (1Н, dd, 8 Гц и 4 Гц); 7.5-8.1 (11Н, m); 8.89 (1H, s) и 10.49 ppm (1Н, s).
Соединение (5.3): Выход 45%, аморфное,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.29 (1Н, dd, 9 Гц и 15 Гц); 2.35 (3H, s); 2.63 (1Н, dd, 4 Гц и 15 Гц); 5.67 (1Н, dd, 4 Гц и 9 Гц); 7.15 (1Н, d, 7 Гц); 7.2-7.9 (6Н, m); 7.97 (1Н, d, 8 Гц); 8.94 (1Н, br s) и 10.52 ppm (1Н, br s).
Соединение (5.4): Выход 12%, аморфное,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.30 (1Н, dd, 8 Гц и 15 Гц); 2.58 (1Н, dd, 4 Гц и 15 Гц); 5.60 (1Н, dd, 4 Гц и 8 Гц); 7.29 (2Н, d, 8 Гц); 7.37 (2H, d, 8 Гц); 7.6-7.9 (3H, m); 7.96 (1Н, d, 8 Гц); 8.92 (1Н, br s) и 10.51 ppm (1Н, br s).
Соединение (5.5): Выход 86%, аморфное,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.1-2.6 (5Н, m); 5.34 (1Н, t, 7 Гц); 7.2-7.9 (7Н, m); 7.97 (1Н, d, 8 Гц); 8.90 (1Н, br s) и 10.55 ppm (1Н, br s).
Соединение (5.6): Выход 62%, аморфное,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.34 (1Н, dd, 15 Гц и 8 Гц); 2.4-2.6 (1Н, перекрывание с ДМСО); 3.76 (3H, s); 5.49 (1Н, dd, 8 Гц и 5 Гц); 7.05 (1Н, t, 7 Гц); 7.19 (1Н, d, 8 Гц); 7.4-7.9 (5Н, m); 7.95 (1Н, d, 7 Гц); 8.86 (1Н, s) и 10.51 ppm (1Н, s).
Соединение (5.7): Выход 62%, точка плавления 147-148°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.31 (1Н, dd, 15 Гц и 9 Гц); 2.66 (1Н, dd, 15 Гц и 4 Гц); 3.79 (3H, s); 5.71 (1Н, dd, 9 Гц и 4 Гц); 6.91 (1Н, d, 7 Гц); 7.05 (1Н, s); 7.07 (1Н, d, 7 Гц); 7.45 (1Н, t, 8 Гц); 7.6-7.8 (3H, m); 7.98 (1Н, d, 7 Гц); 8.93 (1Н, s) и 10.52 ppm (1Н, s).
Соединение (5.8): Выход 51%, точка плавления 185-186°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.33 (1Н, dd, 15 Гц и 8 Гц); 2.5-2.6 (1Н, перекрывание с ДМСО); 3.80 (1Н, s); 5.47 (1Н, dd, 8 Гц и 4 Гц); 7.05 (2Н, d, 9 Гц); 7.43 (2Н, d, 9 Гц); 7.6-7.9 (3H, m); 7.94 (1Н, d, 8 Гц); 8.89 (1Н, s) и 10.50 ppm (1Н, s).
Соединение (5.9): Выход 54%, точка плавления 181-182°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.34 (1Н, dd, 15 Гц и 8 Гц); 2.68 (1Н, dd, 15 Гц и 4 Гц); 5.73 (1Н, dd, 9 Гц и 4 Гц); 6.88 (1Н, d, 8 Гц); 7.0-7.9 (11Н, m); 7.97 (1Н, d, 8 Гц); 8.93 (1Н, s) и 10.51 ppm (1Н, s).
Соединение (5.10): Выход 10%, точка плавления 184-185°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.36 (1Н, dd, 14 Гц и 8 Гц); 2.62 (1Н, dd, 14 Гц и 8 Гц); 5.72 (1Н, dd, 8 Гц и 4 Гц); 7.50 (2Н, d, 8 Гц); 7.57 (2Н, d, 8 Гц); 7.6-7.9 (3H, m); 7.99 (1Н, d, 7 Гц); 8.89 (1Н, s) и 10.49 ppm (1Н, s).
Соединение (5.11): Выход 53%, аморфное,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.33 (1Н, dd, 15 Гц и 8 Гц); приблизительно 2.5 (3H, перекрывание с ДМСО); 2.65 (1Н, dd, 15 Гц и 4 Гц); 5.75 (1Н, dd, 8 Гц и 4 Гц); 7.2-7.9 (7Н, m); 7.98 (1Н, d, 8 Гц); 8.93 (1Н, s) и 10.51 ppm (1H,s).
Соединение (5.12): Выход 12%, аморфное,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.40 (1Н, dd, 15 Гц и 8 Гц); 2.66 (1Н, dd, 15 Гц и 4 Гц); 5.90 (1Н, dd, 8 Гц и 4 Гц); 7.6-7.9 (7Н, m); 8.01 (1Н, d, 7 Гц); 8.89 (1Н, s) и 10.51 ppm (1Н, s).
Соединение (5.13): Выход 19%, аморфное,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.36 (1Н, dd, 15 Гц и 8 Гц); 2.67 (1Н, dd, 15 Гц и 4 Гц); 5.84 (1Н, dd, 8 Гц и 4 Гц); 7.31 (1Н, d, 8 Гц); 7.4-7.9 (1Н, m); 8.00 (1Н, d, 8 Гц); 8.92 (1Н, s) и 10.51 ppm (1Н, s).
Соединение (5.14): Выход 27%, аморфное,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.36 (1Н, dd, 8 Гц и 15 Гц); 2.62 (1Н, dd, 4 Гц и 15 Гц); 5.70 (1Н, dd, 4 Гц и 8 Гц); 7.27 (2Н, d, 8 Гц); 7.6-7.9 (5Н, m); 7.97 (1Н, d, 8 Гц); 8.89 (1Н, br s) и 10.48 ppm (1Н, br s).
Соединение (5.15): Выход 37%, аморфное,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.37 (1Н, dd, 15 Гц и 8 Гц); 2.70 (1Н, dd, 15 Гц и 4 Гц); 5.83 (1Н, dd, 8 Гц и 4 Гц); 7.5-7.8 (12Н, m); 7.98 (1Н, d, 7 Гц); 8.91 (1Н, s) и 10.52 ppm (1Н, s).
Соединение (5.16): Выход 52%, аморфное,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.36 (1Н, dd, 15 Гц и 8 Гц); 2.56 (1Н, dd, 15 Гц и 4 Гц); 5.60 (1Н, dd, 8 Гц и 4 Гц); 7.33 (2Н, t, 9 Гц); 7.52 (2Н, dd, 9 Гц и 5 Гц); 7.61- 7.83 (3H, m); 7.96 (1Н, d, 7 Гц); 8.86 (1Н, s) и 10.48 ppm (1Н, s).
Соединение (5.17): Выход 22%, аморфное,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.36 (1Н, dd, 15 Гц и 9 Гц); 2.73 (1Н, dd, 15 Гц и 4 Гц); 5.93 (1Н, dd, 9 Гц и 4 Гц); 7.54-7.90 (7Н, m); 8.03 (1Н, d, 8 Гц); 8.94 (1Н, s) и 10.52 ppm (1Н, s).
Соединение (5.18): точка плавления 178-179°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.37 (1Н, dd, 15 Гц и 8 Гц); 2.64 (1Н, dd, 15 Гц и 4 Гц); 5.75 (1Н, dd, 8 Гц, и 4 Гц); 7.50-7.83 (7Н, m); 8.00 (1Н, d, 8 Гц); 8.91 (1Н, s) и 10.51 ppm (1Н, s).
Соединение (5.19): Выход 9%, аморфное,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.32 (1Н, dd, 14.7 Гц и 8.8 Гц); 2.70 (1Н, dd, 14.7 Гц и 3.9 Гц); 5.86 (1Н, dd, 8.8 Гц и 3.9 Гц); 7.5-7.9 (7Н, m); 8.00 (1Н, d, 7.8 Гц), 8.96 (1Н, s) и 10.54 ppm (1Н, s).
Соединение (5.20): Выход 85%, точка плавления, 197-198°С (dec.)1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.32 (1Н, dd, 8.8 и 14.7 Гц); 2.49 (3H, s); 2.59 (1Н, dd, 4.4 и 14.7); 5.61 (1Н, dd, 4.4 и 8.1 Гц); 7.39 (2Н, d, 8.8 Гц); 7.42 (2Н, d, 8.8 Гц); 7.5-7.9 (3H, m); 7.95 (1Н, d, 8.1 Гц); 8.89 (1Н, s) и 10.49 ppm (1Н, s).
Соединение (5.21): Выход 48% аморфное,1Н-ЯМР (ДМСО-d6, ТМС) δ: 1.19 (3H, t, 7.3 Гц); 2.28 (1Н, dd, 14.7 и 8.8 Гц); 2.5-2.7 (1Н, m перекрывание с ДМСО); 2.62 (2Н, q, 8.1 Гц); 5.59 (1Н, dd, 8.8 и 3.7 Гц); 7.31 (2Н, d, 8.8 Гц); 7.39 (2Н, d, 8.8 Гц); 7.5-7.9 (3H, m); 7.95 (1Н, d, 7.3 Гц); 8.91 (1Н, s) и 10.50 ppm (1Н, s).
Соединение (5.22): Выход 42%, точка плавления 160-161°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 1.23 (6Н, d, 7 Гц); 2.29 (1Н, dd, 15 Гц и 9 Гц); 2.62 (1Н, dd, 15 Гц и 4 Гц); 2.93 (1Н, m); 5.61 (1Н, dd, 9 Гц, и 4 Гц); 7.34-7.44 (4Н, m); 7.59-7.84 (3H, m); 7.97 (1Н, d, 7 Гц); 8.95 (1Н, s) и 10.53 ppm (1Н, s).
Соединение (5.23): Выход 27%, точка плавления 207-208°С (dec.),1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.2-2.9 (2Н, m, перекрывание с ДМСО); 5.7-5.8 (1Н, m); 7.3-7.9 (12Н, m); 7.99 (1Н, d, 6.8 Гц); 8.93 (1Н, s) и 10.53 ppm (1Н, s).
Соединение (5.24): Выход 47%, точка плавления, 174-175°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.35 (1Н, dd, 15 Гц и 8 Гц); 2.60 (1Н, dd, 15 Гц и 4 Гц); 5.57 (1Н, dd, 8 Гц и 4 Гц); 7.06-7.21 (5Н, m); 7.38-7.52 (4Н, m); 7.73-7.84 (3H, m); 7.97 (1Н, d, 8 Гц); 8.91 (1Н, s) и 10.51 ppm (1Н, s).
Соединение (5.25): Выход 86%, точка плавления, 186-187°С1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.32 (1Н, dd, 15 Гц и 8 Гц); приблизительно 2.5 (1Н, перекрывание с ДМСО); 5.14 (2Н, s); 5.48 (1Н, dd, 8 Гц и 4 Гц); 7.13 (2Н, d, 9 Гц); 7.37-7.82 (10H, m); 7.95 (1Н, d, 8 Гц); 8.92 (1Н, s) и 10.52 ppm (1Н, s).
Соединение (5.26): Выход 47%, точка плавления, 192-194°С (dec.),1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.32-2.67 (2Н, m, 1Н, перекрывание с ДМСО); 5.70 (1Н, m); 6.29 (2Н, s); 7.41 (2Н, s); 7.40-7.95 (7Н, m); 7.98 (1Н, d, 7 Гц); 8.91 (1Н, s) и 10.52 ppm (1Н, s).
Соединение (5.27): Выход 22%, точка плавления, 199-201°С (dec.),1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.34-2.63 (1Н, m, перекрывание с ДМСО); 5.76 (1Н, m); 7.13 (1Н, s); 7.60-7.78 (8Н, m); 7.99 (1Н, d, 7.3 Гц); 8.29 (1Н, s); 8.91 (1Н, s) и 10.55 ppm (1Н, s).
Соединение (5.28): Выход 60%, точка плавления, 205-207°С (dec.),1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.41 (1Н, dd, 14 Гц и 8 Гц); 2.67 (1Н, dd, 14 Гц и 5 Гц); 5.80 (1Н, dd, 8 Гц, и 5 Гц); 7.65-7.87 (5Н, m); 7.99 (2Н, d, 9 Гц); 8.00 (1Н, d, 7 Гц); 8.27 (1Н, s); 8.90 (1Н, s); 9.33 (1Н, s)n 10.51 ppm (1Н, s).
Соединение (5.29): Выход 27%, точка плавления, 138-140°С (dec.),1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.37 (1Н, dd, 15 Гц и 8 Гц); 2.68 (1Н, dd, 15 Гц и 4 Гц); 5.81 (1Н, dd, 8 Гц, и 4 Гц); 7.56-7.87 (8Н, m); 7.99 (1Н, d, 7 Гц); 8.48 (1Н, s); 8.92 (1Н, s) и 10.52 ppm (1Н, s).
Соединение (5.30): Выход 38%, точка плавления,179-181°С (dec.),1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.37 (1Н, dd, перекрывание с ДМСО); 2.71 (3H, s); 5.74 (1Н, dd, 8.1 Гц и 3.7 Гц); 7.51 (2Н, d, 8.8 Гц); 7.61- 7.84 (3H, m); 7.95-8.05 (4Н, m); 8.91 (1Н, s) и 10.53 ppm (1Н, s).
Соединение (5.31): Выход 39%. Точка плавления: 185-186°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.27 (1Н, dd, 15 Гц и 9 Гц); ~2.5 (1Н, перекрывание с ДМСО); 2.94 (6Н, s); 5.33 (1Н, dd, 8 Гц и 4 Гц); 6.77 (2Н, d, 9 Гц); 7.27 (2Н, d, 9 Гц); 7.59-7.82 (3H, m); 7.92 (1Н, d, 7 Гц); 8.92 (1Н, s) и 10.52 ppm (1Н, s).
Соединение (5.32): Выход 61%, точка плавления 204-206°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.29 (1Н, dd, 15 Гц и 9 Гц); приблизительно 2.5 (1Н, перекрывание с ДМСО); 3.16 (4Н, m); 3.75 (4Н, m); 5.43 (1Н, dd, 9 Гц и 4 Гц); 7.04 (2Н, d, 9 Гц); 7.33 (2Н, d, 9 Гц); 7.58-7.82 (3H, m); 7.94 (1Н, d, 7 Гц); 8.92 (1Н, s) и 10.52 ppm (1Н, s).
Соединение (5.33): Выход 60%, точка плавления 177-178°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 1.35 (3H, t, 7 Гц); 2.31 (1Н, dd, 15 Гц и 8 Гц); приблизительно 2.5 (1Н, перекрывание с ДМСО); 4.06 (2Н, q, 7 Гц); 5.46 (1Н, dd, 8 Гц, и 4 Гц); 7.03 (2Н, d, 9 Гц); 7.40 (2Н, d, 9 Гц); 7.59-7.82 (3H, m); 7.97 (1Н, d, 7 Гц); 8.89 (1Н, s) и 10.51 ppm (1Н, s).
Соединение (5.34): Выход 63%, точка плавления 165-166°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 0.94 (3H, t, 7 Гц); 1.39-1.50 (2Н, m); 1.65-1.75 (2Н, m); 2.28 (1Н, dd, 15 Гц и 8 Гц); 2.55 (1Н, dd, 15 Гц и 4 Гц); 4.00 (2Н, t, 7 Гц); 5.47 (1Н, dd, 8 Гц и 4 Гц); 7.04 (2Н, d, 9 Гц); 7.39 (2Н, d, 9 Гц); 7.60- 7.82 (3H, m); 7.95 (1Н, d, 7 Гц); 8.91 (1Н, s) и 10.51 ppm (1Н, s).
Соединение (5.35): Выход 63%, аморфное,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.33 (1Н, dd, 15 Гц и 8 Гц); 2.56 (1Н, dd, 15 Гц и 4 Гц); 4.82 (2Н, q, 9 Гц); 5.54 (1Н, dd, 8 Гц, и 4 Гц); 7.18 (2Н, d, 9 Гц); 7.46 (2Н, d, 9 Гц); 7.61-7.84 (3H, m); 7.96 (1Н, d, 7 Гц); 8.90 (1Н, s) и 10.51 ppm (1Н, s).
Соединение (5.36): Выход 49% точка плавления 145-147°С (dec.),1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.34 (1Н, dd, 8 Гц и 15 Гц); 2.66 (1Н, dd, 4 Гц и 15 Гц); 5.77 (1Н, dd, 4 Гц и 8 Гц); 7.1-7.3 (1Н, m); 7.3-7.5 (2Н, m); 7.5-7.9 (4Н, m); 7.99 (1Н, d, 7 Гц); 8.91 (1Н, s) и 10.55 ppm (1Н, s).
Соединение (5.37): Выход 7%, точка плавления, 152-154°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.37 (1Н, dd, 15 Гц и 9 Гц); 2.60 (1Н, dd, 15 Гц и 6 Гц); 4.98 (1Н, t, 7 Гц); 7.54-7.82 (3H, m); 7.80 (1Н, d, 9 Гц); 8.80 (1Н, br s); 8.92 (1Н, br s) и 10.54 ppm (1Н, s).
Соединение (5.38): аморфное,1Н-ЯМР (ДМСО-d6, ТМС) δ: приблизительно 2.5 (1Н, перекрывание с ДМСО); 2.67 (1Н, dd, 15 Гц и 6 Гц); 2.79 (3H, s); 4.76 (1Н, t, 6 Гц); 7.60 -7.77 (3H, m); 7.87 (1Н, d, 7 Гц); 8.90 (1Н, s) и 10.65 ppm (1Н, s).
Соединение (5.39): Выход 55%, аморфное,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.32 (1Н, dd, 15 Гц и 8 Гц); 2.71 (1Н, dd, 15 Гц и 6 Гц); 4.37 (1Н, d, 15 Гц); 4.63 (1Н, d, 15 Гц); 4.81 (1Н, dd, 8 Гц и 6 Гц); 7.23-7.51 (6Н, m); 7.51-7.75 (2Н, m); 7.91 (1Н, d, 8 Гц); 8.98 (1Н, s) и 10.61 ppm (1Н, s).
Соединение (5.40): Выход 39% точка плавления 189-190°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.40-2.62 (2Н, m, перекрывание с ДМСО); 3.89 (3H, s); 5.51 (1Н, t, 6 Гц); 6.96 (1Н, d, 8 Гц); 7.67-7.88 (4Н, m); 7.98 (1Н, d, 8 Гц); 8.24 (1Н, d, 2 Гц); 8.85 (1Н, s) и 10.50 ppm (1Н, s).
Соединение (5.41): Выход 58%, аморфное,1Н-ЯМР (ДМСО-d6, ТМС) δ: 0.94-2.36 (10H, m); 2.31 (1Н, dd, 14.7 Гц и 8.1 Гц); 2.72 (1Н, dd, 14.7 Гц, 5.9 Гц); 3.42-3.54 (1Н, m); 5.06 (1Н, dd, 8.1 Гц, 5.1 Гц); 7.47-7.71 (3H, m); 7.79 (1Н, d, 8.1 Гц); 8.95 (1Н, s) и 10.57 ppm (1H, s).
Соединение (5.42): Выход 58%, точка плавления 141-143°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 1.7-2.1 (6Н, m); 2.30 (1Н, dd, 8.1 Гц и 14.7 Гц); 2.69 (1Н, dd, 5.1 Гц и 14.7 Гц); 2.8-3.0 (2Н, m); 3.44 (2Н, s); 3.3.2-3.6 (1Н, m); 5.05 (1Н, dd, 5.9 и 7.3 Гц); 7.2- 7.4 (5Н, m); 7.4-7.7 (3H, m); 7.78 (1Н, d, 8.1 Гц); 8.96 (1Н, s) и 10.57 ppm (1Н, s).
Соединение (5.43): Выход 25%, аморфное,1Н-ЯМР (ДМСО-d6, ТМС) δ: 1.84 (3H, s); 2.30 (1Н, dd, 14 Гц и 7 Гц); 2.5-2.6 (1Н, перекрывание с ДМСО); 4.77 (2Н, d, 2 Гц); 5.47 (1Н, dd, 8 Гц и 4 Гц); 7.06 (2Н, d, 9 Гц); 7.40 (2Н, d, 9 Гц); 7.5-7.8 (3H, m); 7.94 (1Н, d, 7 Гц); 8.89 (1Н, s) и 10.50 ppm (1Н, s).
Соединение (5.44): Выход 31%, точка плавления 195-197°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.1-2.6 (2Н, m); 2.68 (3H, s); 5.4-5.6 (1Н, m); 5.65 (2Н, s); 7.28 (2Н, d, 9 Гц); 7.47 (2Н, d, 9 Гц); 7.5-7.9 (6Н, m); 7.9-8.1 (2Н, m); 8.13 (1Н, d, 7 Гц); 8.90 (1Н, s) и 10.52 ppm (1Н, s).
Соединение (5.45): Выход 14%, аморфное,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.1-2.6 (2Н, m); 2.21 (6Н, s); 2.62 (2Н, t, 6 Гц); 4.07 (2Н, t, 6 Гц); 5.45 (2Н, dd, 4 и 8 Гц); 7.04 (2Н, d, 9 Гц); 7.39 (2Н, d, 9 Гц); 7.5-7.9 (3H, m); 7.94 (1Н, d, 7 Гц); 8.91 (1Н, br. s) и 10.50 ppm (1Н, br. s).
Соединение (5.46): Выход 30%, аморфное,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.1-2.6 (2Н, m); 2.21 (6Н, s); 2.62 (2Н, t, 6 Гц); 4.07 (2Н, t, 6 Гц); 5.45 (2Н, dd, 4 и 8 Гц); 7.04 (2Н, d, 9 Гц); 7.39 (2Н, d, 9 Гц); 7.5-7.9 (3H, m); 7.94 (1Н, d, 7 Гц); 8.91 (1Н, br. s) и 10.50 ppm (1Н, br. s).
Соединение (5.47): Выход 55%, точка плавления 166-168°С (dec.),1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.34-2.63 (2Н, m, перекрывание с ДМСО); 4.59 (2Н, d, 5.1 Гц); 2.29 (2Н, dt, 13.9 Гц, 1.5 Гц); 5.45 (1Н, dd, 7.8 Гц, 2.9 Гц); 5.95-6.14 (1Н, m); 7.04 (2Н, dd, 8.8 Гц, 2.0 Гц); 7.38 (2Н, dd, 8.8 Гц, 2.0 Гц); 7.61-7.96 (4Н, m); 8.89 (1Н, s); 10.50 ppm (1Н, s).
Соединение (5.48): Точка плавления 212-214°С (dec.),1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.2-2.7 (2Н, m, перекрывание с ДМСО); 5.23 (2Н, s); 5.4-5.6 (1Н, m); 7.14 (2Н, d, 8.8 Гц); 7.35-7.55 (4Н, m); 7.55-7.90 (3H, m); 7.95 (1Н, d, 7.3 Гц); 8.59 (2Н, d, 5.1 Гц); 8.91 (1Н, br s) и 10.52 ppm (1Н, s).
Соединение (5.49): Аморфный порошок,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.2-2.6 (2Н, m, перекрывание с ДМСО); 5.33 (2Н, s); 5.4-5.6 (1Н, m); 7.17 (2Н, d, 8.8 Гц); 7.45 (2Н, d, 8.8 Гц); 7.5-7.9 (3H, m); 7.9-8.1 (2Н, m); 8.58 (1Н, d, 8.1 Гц); 8.8-8.9 (1Н, m); 8.9-9.1 (1Н, m) и 10.55 ppm (1Н, s).
Соединение (5.50): Точка плавления: более 170°С (dec.),1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.2-2.6 (2Н, m, перекрывание с ДМСО); 5.21 (2Н, s); 5.4-5.6 (1Н, m); 7.14 (2Н, d, 8.8 Гц); 7.41 (2Н, d, 8.8 Гц); 7.3-8.0 (7Н, m); 8.58 (1Н, d, 4.4 Гц); 8.89 (1Н, s) и 10.49 ppm (1Н, s).
Соединение (5.51): Выход 23%, аморфное,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.50-2.56 (2Н, m, перекрывание с ДМСО); 4.11 (2Н, d, 5.8 Гц); 4.86 (2Н, s); 5.25 (1Н, t, 5.8 Гц); 5.48 (1Н, dd, 8.4 Гц и 4.4 Гц); 7.09 (2Н, dd, 8.7 Гц и 1.8 Гц); 7.43 (2Н, dd, 9.1 Гц и 2.2 Гц); 7.62 (1Н, d, 8.0 Гц); 7.69 (1Н, t, 7.3 Гц); 7.79 (1Н, t, 7.3 Гц); 7.95 (1Н, d, 8.0 Гц); 8.89 (1Н, s) и 10.50 ppm (1Н, s).
Соединение (5.52): Выход 7%, точка плавления, 135-137°С (dec.),1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.22-2.26 (2Н, m); 2.36-2.58 (2Н, m, перекрывание с ДМСО); 4.78 (2Н, d, 2.0 Гц); 5.48 (1Н, dd, 7.8 Гц, 2.9 Гц); 7.08 (2Н, dd, 8.8 Гц, 2.0 Гц); 7.42 (2Н, dd, 8.8 Гц, 2.0 Гц); 7.59-7.97 (4Н, m); 8.90 (1Н, s); 10.51 ppm (1Н, s).
Соединение (5.53): Выход 2%, точка плавления, 155-157°С (dec.),1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.37-2.59 (3H, m, перекрывание с ДМСО); 4.85 (2Н, d, 2.2 Гц); 5.50 (1Н, dd, 7.8 Гц, 2.9 Гц); 7.14 (2Н, dd, 8.8 Гц, 2.0 Гц); 7.44 (2Н, dd, 8.8 Гц, 2.0 Гц); 7.61-7.99 (4Н, m); 8.91 (1Н, s); 10.52 ppm (1Н, s).
Соединение (5.54): Выход 64%, точка плавления, 148-150°С (dec.),1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.30-2.50 (1Н, m, перекрывание с ДМСО); 2.50-2.68 (3H, m, перекрывание с ДМСО); 4.10 (2Н, t, 5.9 Гц); 5.50 (1Н, dd, 7.8 Гц и 2.9 Гц); 7.06 (2Н, dd, 8.8 Гц и 1.95 Гц); 7.40 (2Н, dd, 8.8 Гц и 1.95 Гц); 7.59-7.97 (4Н, m); 8.89 (1Н, s) и 10.50 ppm (1Н, s).
Соединение (5.55): Выход 75%, точка плавления 152-154°С (dec.),1Н-ЯМР (ДМСО-d6, ТМС) δ: 1.55 (3H, d, 5.9 Гц); 1.82 (3H, s); 2.30-2.50 (2Н, m, перекрывание с ДМСО); 5.00-5.20 (1Н, m); 5.50 (1Н, dd, 7.8 Гц и 2.9 Гц); 7.08 (2Н, dd, 8.8 Гц и 1.95 Гц); 7.41 (2Н, dd, 8.8 Гц и 1.95 Гц); 7.60-7.98 (4Н, m); 8.91 (1Н, s) и 10.52 ppm (1Н, s).
Соединение (5.56): Выход 10%, точка плавления 200-204°С (dec.),1Н-ЯМР (ДМСО-d6, ТМС) δ: 1.70 (3H, d, 5.8 Гц); 2.1-2.6 (2Н, m, перекрывание с ДМСО); 2.63 (3H, s); 5.3-5.5 (1Н, m); 6.2-6.4 (1Н, s); 7.05 (2Н, d, 8.8 Гц); 7.34 (2Н, d, 8.8 Гц); 7.4-7.9 (6Н, m); 7.9-8.1 (2Н, m); 8.33 (1Н, d, 8.0 Гц); 8.90 (1Н, s) и 10.49 ppm (1Н, s).
Соединение (5.57): Выход 36%, точка плавления 218-220°С (dec.),1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.2-2.7 (2Н, m); 5.4-5.6 (1Н, m); 5.71 (2Н, s); 7.27 (2Н, d, 8.8 Гц); 7.46 (2Н, d, 8.8 Гц); 7.5-7.9 (6Н, m); 7.96 (1Н, d, 7.3 Гц); 8.09 (1Н, d, 8.8 Гц); 8.20 (1Н, d, 8.0 Гц); 8.89 (1Н, s); 8.93 (1Н, d, 4.4 Гц) и 10.51 ppm (1Н, s).
Соединение (5.58): Выход 18%, точка плавления: более 140°С (dec.),1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.32 (1Н, dd, 8.7 и 14.6 Гц, частичное перекрывание с ДМСО); 2.66 (3H, s); 5.51 (1Н, dd, 3.7 и 8.1 Гц); 5.60 (2Н, s); 7.28 (2Н, d, 8.8 Гц); 7.46 (2Н, d, 8.8 Гц); 7.5-7.8 (5Н, m); 7.8-8.1 (3H, m); 8.89 (1Н, s) и 10.49 ppm (1Н, s).
Соединение (5.59): Точка плавления более 215°С (dec.),1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.2-2.7 (2Н, m, перекрывание с ДМСО); 2.67 (3H, s); 4.9-5.6 (1Н, m); 5.63 (2Н, s); 7.20 (2Н, d, 8.8 Гц); 7.46 (2Н, d, 8.8 Гц); 7.5-7.9 (5Н, m); 7.9-8.1 (2Н, m); 8.22 (1Н, d, 1.9 Гц); 8.88 (1Н,8) и 10.50 ppm (1H, s).
Соединение (5.60): Выход 76%, точка плавления более 204°С (dec.),1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.30 (1Н, dd, 8.1 и 14.7 Гц), 2.4-2.6 (1Н, частичное перекрывание с ДМСО); 3.29 (3H, s, перекрывание с ДМСО, Н20); 5.16 (2Н, s); 5.47 (1Н, dd, 4.4 и 8.8 Гц); 7.12 (2Н, d, 8.8 Гц); 7.23 (1Н, d, 4.4 Гц); 7.31 (1Н, s); 7.41 (2Н, d, 8.8 Гц); 7.5-7.8 (3H, m); 7.93 (1Н, d, 7.3 Гц); 8.43 (1Н, d, 5.1 Гц); 8.88 (1Н, d, 1.5 Гц) и 10.48 ppm (1Н, s).
Соединение (5.61): Выход 78%, точка плавления более 210°С (dec.)1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.32 (1Н, dd, 8.8 и 14.7 Гц), 2.43 (6Н, s, перекрывание с ДМСО); 2.4-2.7 (1Н, частичное перекрывание с ДМСО); 5.12 (2Н, s); 5.49 (1Н, dd, 3.7 и 8.1 Гц); 7.0-7.2 (4Н, m); 7.43 (2Н, d, 8.8 Гц); 7.5-7.9 (3H, m); 7.95 (1Н, d, 7.3 Гц); 8.89 (1Н, s) и 10.50 ppm (1Н, s).
Соединение (5.62): Выход 68%, точка плавления 180-182°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.34 (1Н, dd, 9 Гц и 15 Гц); 2.62 (1Н, dd, 4 Гц и 15 Гц); 5.62 (1Н, dd, 4 Гц и 9 Гц); 7.11 (1Н, s); 7.2-7.6 (6Н, m); 7.89 (1Н, d, 9 Гц); 8.96 (1Н, s) и 10.53 ppm (1Н, s).
Соединение (5.63): Точка плавления 202-204°С (dec.),1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.23 (1Н, dd, 9 Гц и 15 Гц); 2.4-2.7 (1Н, m, перекрывание с ДМСО); 4.33 (4Н, m); 5.52 (1Н, dd, 4 Гц и 9 Гц); 7.04 (1Н, s); 7.2-7.6 (6Н, m); 8.93 (1Н, s) и 10.50 ppm (1Н, s).
Соединение (5.64): Точка плавления 197-199°С (dec.),1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.2-2.7 (2Н, m перекрывание с ДМСО); 3.84 (3H, s); 3.86 (3H, s); 5.55 (1Н, dd, 4 Гц и 9 Гц); 7.07 (1Н, s); 7.2-7.4 (1Н, m); 7.4-7.6 (5Н, m); 8.94 (1Н, s) и 10.52 ppm (1Н, s).
Соединение (5.65): Выход 68%, аморфное,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.2-2.7 (2Н, m перекрывание с ДМСО); 3.82 (3H, s); 3.89 (3H, s); 4.00 (3H, s); 5.51 (1Н, dd, 3.7 Гц и 8.1 Гц); 6.90 (1Н, s); 7.2-7.6 (6Н, m); 8.93 (1Н, s) и 10.51 ppm (1Н, s).
Соединение (5.66): Выход 75%, аморфное,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.2-2.7 (2Н, m перекрывание с ДМСО); 3.86 (3H, s); 3.93 (3H, s); 5.52 (1Н, dd, 3.7 Гц и 8.1 Гц); 6.63 (1Н, s); 6.77 (1Н, s); 7.3-7.6 (5Н, m); 8.92 (1Н, d, 1.5 Гц) и 10.50 ppm (1Н, d, 1.5 Гц).
Соединение (5.67): Выход 46%, аморфное,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.27 (1Н, dd, 14.7 Гц, 9.5 Гц); 2.45 (3H, s); 2.61 (1Н, dd, 14.7 Гц, 3.7 Гц); 5.64 (1Н, dd, 9.5 Гц, 3.7 Гц); 7.27-7.38 (1Н, m); 7.44-7.54 (5Н, m); 7.61 (1Н, d, 8.1 Гц); 7.79 (1Н, s); 8.93 (1Н, s) и 10.53 ppm (1Н, s).
Соединение (5.68): Выход 38%, точка плавления 182-183°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.33 (1Н, dd, 8.8 Гц и 14.6 Гц); 2.65 (1Н, dd, 3.7 Гц и 14.6 Гц); 5.65 (1Н, dd, 3.7 и 8.1 Гц); 7.2-7.6 (5Н, m); 7.58 (1Н, d, 8.1 Гц); 7.78 (1Н, d, 8.1 Гц); 8.31 (1Н, s); 8.89 (1Н, s) и 10.48 ppm (1Н, s).
Синтез 187
Этиловый эфир 1-(2-метил-хинолин-4-ил)метансульфоновой кислоты (15.12)

Раствор гидроксиметилхинолина (17) (1,7 г, 10 мМоль) в DCM (5 мл) охлаждали на ледяной бане и затем добавляли 0,39 М раствор периодинана по Дессу-Мартину в DCM (31 мл, 12 мМоль). Полученный раствор перемешивали при охлаждении в течение 1,5 ч и затем добавляли водный раствор NaHCO3 (15 мл). Смесь перемешивали до тех пор, пока и органическая, и водная фаза не стали гомогенными. Органическую фазу промывали водным Na2S2O3 и насыщенным солевым раствором и сушили над Na2SO4. Экстракт фильтровали и растворитель удаляли под вакуумом. Остаток очищали посредством флэш-хроматографии на силикагеле, элюируя смесью легкого петролейного эфира и EtOAc (2:1, 1:1) с образованием кристаллического материала (0,51 г). Его растворяли в ТГФ (10 мл) и охлаждали на ледяной бане. По каплям при охлаждении добавляли 1,4 М раствор MeMrBr в ТГФ (4,3 мл, 6 мМоль). Смесь перемешивали в течение 30 мин, при охлаждении и затем к ней добавляли насыщенный водный NH4Cl (50 мл) и воду (50 мл). Смесь экстрагировали EtOAc (100+50 мл). Объединенную органическую фазу промывали насыщенным солевым раствором (50 мл) сушили над Na2SO4, фильтровали и упаривали. Остаток очищали посредством флэш-хроматографии на силикагеле, элюируя смесью легкого петролейного эфира и EtOAc (1:1, 1:0), с образованием кристаллического материала (0,325 г). Этот продукт (300 мг) растворяли в DCM (5 мл) и раствор охлаждали на ледяной бане. К нему одной порцией добавляли триэтиламин (0,45 мл, 3,2 мМоль), а затем по каплям добавляли мезилхлорид (0,25 мл, 3,2 мМоль). Охлаждающую баню отставляли и полученную смесь перемешивали при комнатной температуре в течение 30 минут. Смесь разбавляли DCM (30 мл) и промывали насыщенным солевым раствором (2×30 мл). Органическую фазу сушили над Na2SO4, фильтровали и упаривали с образованием (5.12) (0,47 г) в виде неочищенного продукта.
Синтез 188
Хлористоводородная соль 4-хлорметилхинолина (15.13)

Хлористый тионил (1,45 мл, 20 мМоль) добавляли по каплям к раствору карбинола (18) (1,5 г, 9,4 мМоль) в DCM при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч и упаривали с образованием (15.13) (2,0 г) в виде неочищенного продукта.
Общая процедура синтеза хлорметилхинолинов (15.14) и (15.15)
Способ J: Хинолинкарбоновую кислоту (19.1) или (19.2) (4 мМоль) кипятили с обратным холодильником в смеси МеОН (13 мл) и H2O4 (2,5 мл) в течение 3 ч. Реакционную смесь охлаждали до комнатной температуры и к ней добавляли воду (25 мл). Добавляли насыщенный водный NaHCO3, чтобы подогнать значение pH до приблизительно 8. Смесь экстрагировали EtOAc (2×35 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и упаривали с образованием неочищенного сложного эфира. Его растворяли в (25 мл) и порциями добавляли NaBH4 (0,76 г, 20 мМоль), таким образом, чтобы поддерживать слабое кипение. После окончания добавления реакционную смесь перемешивали в течение 1 ч при комнатной температуре и добавляли воду (100 мл). Реакционную смесь экстрагировали EtOAc (100 мл), промывали насыщенным солевым раствором (50 мл) и сушили над Na2SO4. Экстракт фильтровали и упаривали с образованием гидроксиметилхинолинового производного. Его растворяли в DCM (30 мл) и к раствору по каплям добавляли хлористый тионил (0,47 мл, 6,4 мМоль). Реакционную смесь кипятили с обратным холодильником в течение 3 ч и упаривали с образованием (15.14) или (15.15) в виде неочищенного продукта.
Следуя методике, аналогичной Способу J, были получены соединения в виде неочищенного продукта.
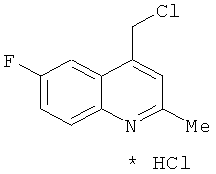

Общая процедура синтеза хлорметилпиридинов (15.16) и (15.17)
Способ K: Раствор 4-метилпиридинового производного (20.1) или (20.2) (40 мМоль) в сухом ТГФ охлаждали до -70°С в инертной атмосфере, и к нему по каплям добавляли 1,6 М н-BuLi в гексанах (28 мл, 44 мМоль). После завершения добавления раствор дополнительно перемешивали еще 30 минут при -70°С и добавляли ДМФА (6,2 мл, 80 мМоль). Смесь дополнительно перемешивали в течение 1 ч 30 мин при -70°С и «гасили» насыщенным водным NH4Cl (10 мл) и нагревали до комнатной температуры. Смесь частично упаривали, к остатку добавляли воду (100 мл) и экстрагировали CHCl3 (3×100 мл). Объединенную органическую фазу промывали насыщенным солевым раствором (100 мл), сушили над Na2SO4 фильтровали и упаривали. Остаток растворяли в МеОН (30 мл) и к суспензии по каплям добавляли NaIO4 (25,7 г, 120 мМоль) в МеОН (30 мл) с такой скоростью, чтобы поддерживалось слабое кипение. Смесь пропускали через короткую целитовую колонку и к раствору добавляли NaBH4 (4,54 г, 120 мМоль). Смесь перемешивали в течение 30 мин и добавляли незначительное количество SiO2. Растворитель удаляли под вакуумом, и остаток наносили на силикагелевую колонку. Элюирование DCM, содержащим 5% МеОН, привело к гидроксиметилпиридинам (20.1) или (20.2). Промежуточное соединение (20.1) или (20.2) (7 мМоль) растворяли в DCM (70 мл), и к раствору по каплям добавляли хлористый тионил (1,05 мл, 14,4 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 3 ч и упаривали с образованием (15.16) или (15.17) в виде неочищенного продукта.
Следуя методикам, аналогичным Способу J, были получены следующие соединения в виде неочищенного продукта.


Синтез 193
2-(1,1-Диоксо-2-(3-гидроксиметилфенил)-2,3-дигидро-1Н-бензо[d]изотиазол-3-ил)-N-гидроксиацетамид (24)

К раствору хлористого сулльфонила (1.1) (2 мМоль) и 3-гидроксиметиланилина (22) (2 мМоль) в диоксане (10 мл) добавляли 1 М водный раствор NaHCO3 (4 мл). Полученную смесь перемешивали при комнатной температуре в течение 2 часов и разбавляли водой (50 мл). Осадок собирали на фильтре, промывали водой и под вакуумом сушили над P2O5, получая сложный эфир (23) (427 мг, 62%). К раствору сложного эфира (23) (174 мг, 0,5 мМоль) в метаноле (2 мл) добавляли раствор хлористоводородной соли гидроксиламина (174 мг, 2,5 мМоль) и KOH (278 мг, 4 мМоль) в метаноле (3 мл). Смесь оставляли на ночь перемешиваться при комнатной температуре и упаривали. К остатку добавляли воду и затем 20% KHSO4 до достижения нейтрального значения pH. Смесь экстрагировали этилацетатом (20 мл). Органическую фазу отделяли, промывали насыщенным солевым раствором (20 мл) и сушили над Na2SO4. Раствор фильтровали и упаривали. Остаток обрабатывали ацетонитрилом, осадок собирали на фильтре и сушили, получая (24) (15 мг, 9%), точка плавления 165-166°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.29 (1Н, dd, 9 Гц и 15 Гц); 2.63 (1Н, dd, 4 Гц и 15 Гц); 4.54 (1Н, d, 5 Гц); 5.34 (1Н, t, 5 Гц); 5.67 (1Н, dd, 4 Гц и 9 Гц); 7.2-7.8 (7Н, m); 7.98 (1Н, d, 8 Гц); 8.9 (1Н, br s) и 10.5 ppm (1Н, br s).
Синтез 194
N-трет-Бутоксикарбонил-4-(4-нитрофенокси)-бут-2-иниламин (25)

Раствор спирта (16.7) (278 мг, 1,34 мМоль) и триэтиламина (0,37 мл, 2,68 мМоль) в бензоле (11 мл) охлаждали на ледяной бане в атмосфере аргона. К этому раствору добавляли мезилхлорид (0,21 мл, 2,68 мМоль). Смесь оставляли до достижения комнатной температуры и перемешивали в течение 2 ч. Ее фильтровали через короткую силикагелевую колонку, элюируя бензолом. Раствор промывали насыщенным водным NaHCO3 (50 мл) и насыщенным водным солевым раствором (50 мл), сушили над Na2SO4 и упаривали. Остаток (382 мг) растворяли в ДМФА (11 мл) и NaN3 (231 мг, 3,55 мМоль). Реакционную смесь перемешивали в течение 4 суток при комнатной температуре и разбавляли водой (30 мл). Смесь экстрагировали Et2O (3×25 мл). Объединенную органическую фазу промывали водой (30 мл), сушили над Na2SO4, и растворитель удаляли под вакуумом. Остаток (311 мг) растворяли в Et2O (10 мл), и смесь охлаждали на ледяной бане. К ней добавляли трифенилфосфин (352 мг, 1,34 мМоль) и смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 1 ч 30 мин. Добавляли воду (1,25 мл) и смесь оставляли на ночь перемешиваться. Органическую фазу отделяли декантацией, сушили над Na2SO4 и растворитель удаляли под вакуумом. Остаток (276 мг) растворяли в DCM, и к раствору добавляли Boc2O. Смесь оставляли на ночь перемешиваться, растворитель удаляли под вакуумом. Остаток очищали посредством флэш-хроматографии на силикагеле, элюируя смесью легкого петролейного эфира и EtOAc (4:1, 2:1) с образованием соединения 25 (182 мг), указанного в заголовке.
Синтез 195
N-трет-Бутоксикарбонил-4-(4-аминофенокси)-бут-2-иниламин (26)

Нитробензольное соединение (25) (182 мг, 0,6 мМоль) растворяли в метаноле (5 мл) и к раствору добавляли Na2S×9H2O (576 мг, 2,4 мМоль) и смесь поставили кипятиться с обратным холодильником на 3 ч. Растворитель удаляли под вакуумом, и остаток распределяли между водой и Et2O (30 мл). Органическую фазу экстрагировали 1 М водным HCl. Кислый водный экстракт отделяли и подщелачивали 5 М водным NaOH до значения pH приблизительно 10. Смесь экстрагировали Et2O (3×30 мл) и объединенную органическую фазу промывали насыщенным солевым раствором (30 мл). Экстракт сушили над Na2SO4, фильтровали и упаривали, получая соединение (26) (40 мг), указанное в заголовке, в виде неочищенного продукта.
Синтез 196
{2-[4-(4-трет-Бутоксикарбониламино-бут-2-инилокси)-фенил]-1,1-диоксо-2,3-дигидро-1H-бензо[а]изотиазол-3-ил}-уксусная кислота (27)

Следуя методике, аналогичной Способу F (для приготовления соединения 4), соединение, указанное в заголовке, было получено из хлористого сульфонила (1.1) и анилина (26) в виде неочищенного продукта.
Синтез 197
2-{2-[4-(4-трет-Бутоксикарбониламино-бут-2-инилокси)-фенил]-1,1-диоксо-2,3-дигидро-1H-бензо[d]изотиазол-3-ил}-N-гидроксиацетамид (28)

Следуя методике, аналогичной Способу Н (для приготовления соединений 5) из карбоновой кислоты (27), соединение, укзанное в заголовке, было получено в виде неочищенного продукта.
Синтез 198
Хлористоводородная соль 2-{2-[4-(4-Амино-бут-2-инилокси)-фенил]-1,1-диоксо-2,3-дигидро-1H-бензо[d]изотиазол-3-ил}-N-гидроксиацетамида (29)

К раствору соединения (28) (40 мг, 0,09 мМоль) в DCM (2,8 мл), защищенном N-Вос, по каплям при охлаждении на ледяной бане добавляли трифторуксусную кислоту (1,4 мл). После завершения добавления ледяную баню удаляли и смесь перемешивали при комнатной температуре в течение 45 мин. Растворитель и избыток трифторуксусной кислоты удаляли под вакуумом и остаток обрабатывали 2 М HCl в Et2O. Смесь упаривали, и остаток повторно обрабатывали 2 М HCl in Et2O и вновь упаривали. Остаток обрабатывали Et2O, и осадок собирали на фильтре, получая соединение (29). Выход 68%, аморфное,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.29-2.50 (2Н, m, перекрывание с ДМСО); 3.79 -3.83 (2Н, m); 4.94 (2Н, s); 5.46-5.53 (1Н, m); 7.11 (2Н, d, 8.1 Гц); 7.44 (2Н, d, 8.1 Гц); 7.60-7.80 (3H, m); 7.95 (1Н, d, 6.6 Гц); 8.40 (4Н, s) и 10.59 ppm (1Н, s).
Синтез 199
Метиловый эфир (Е)-3-(2-хлорсульфонил-5-гидроксифенил)акриловой кислоты (31)

Следуя методике, аналогичной Способу А (для синтеза (1)) из ненасыщенного сложного эфира (30), было получено указанное в заголовке соединение в виде неочищенного продукта.
Синтез 200
Метиловый эфир (5-гидрокси-1,1-диоксо-2-фенил-2,3-дигидро-1Н-бензо[d]изотиазол-3-ил)уксусной кислоты (32)

Раствор хлористого сульфонила (31) (7,05 г, 25,5 мМоль) и анилина (2.1) (4,75 г, 51 мМоль) в DCM (200 мл) перемешивали в течение 17 ч при комнатной температуре. Раствор промывали 1 М водным HCl (200 мл) и насыщенным солевым раствором (3×100 мл) и сушили над Na2SO4. Раствор фильтровали и упаривали с образованием промежуточного продукта (8,13 г). Его растворяли в ДМФА (40 мл) и добавляли K2CO3 (6,74 г, 48,9 мМоль). Полученную смесь нагревали при 80°С в течение 5 ч, охлаждали до комнатной температуры и выливали в воду (300 мл). Водную фазу экстрагировали EtOAc (2×250 мл). Объединенную органическую фазу промывали насыщенным солевым раствором (3×100 мл), сушили над Na2SO4, фильтровали и упаривали. Остаток очищали посредством флэш-хроматографии на силикагеле, элюируя смесью легкого петролейного эфира EtOAc (2:1, 1:1) с образованием продукта (32) (1,52 г).
Синтез 201
Метиловый эфир (1,1-диоксо-2-фенил-5-трифторметансульфонилокси-2,3-дигидро-1H-бензо[d]изотиазол-3-ил)уксусной кислоты (33)

К раствору (32) (333 мг, 1 мМоль) в DCM (5 мл) добавляли пиридин (0,16 мл, 2 мМоль), и раствор охлаждали на ледяной бане. К раствору порциями добавляли ангидрид трифторметансульфоновой кислоты (0,2 мл, 1,2 мМоль) и полученный раствор оставляли нагреваться до комнатной температуры. Добавляли 1М водный HCl (100 мл), и смесь экстрагировали EtOAc (2×150 мл). Объединенную органическую фазу промывали насыщенным водным NaHCO3 (100 мл) и насыщенным солевым раствором (100 мл) и сушили над Na2SO4. Экстракт фильтровали, и растворитель удаляли под вакуумом с образованием (33) (460 мг).
Синтез 202
Метиловый эфир (1,1-диоксо-2,5-дифенил-2,3-дигидро-1Н-бензо[d]изотиазол-3-ил)уксусной кислоты (34)

К раствору соединения (33) (460 мг, 1 мМоль) в DME (10 мл) добавляли Pd(PhP)4 (28 мг, 0,03 мМоль), фенилбороновую кислоту (133 мг, 1,1 мМоль) и 2 М водный Na2CO3 (1,3 мл, 2,6 мМоль). Полученную смесь нагревали при 85°С в течение 2 ч и охлаждали до комнатной температуры. Ее растворяли в воде (20 мл) и экстрагировали EtOAc (2×20 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и упаривали. Остаток очищали посредством флэш-хроматографии на силикагеле, элюируя смесью легкого петролейного эфира и EtOAc (4:1) с образованием (34) (340 мг).
Синтез 203
(1,1-Диоксо-2,5-дифенил-2,3-дигидро-1Н-бензо[b]изотиазол-3-ил)уксусной кислота
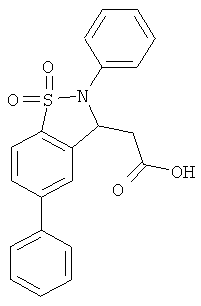
Следуя методике, аналогичной Способу Е (для синтеза (4)) из сложного эфира (34), соединение, указанное в заголовке, было получено в виде неочищенного продукта.
Синтез 204
2-(1,1-Диоксо-2,5-дифенил-2,3-дигидро-1Н-бензо[d]изотиазол-3-ил)-N-гидроксиацетамид (36)

Следуя методике, аналогичной Способу G (для синтеза (5)) из карбоновой кислоты было получено соединение, указанное в заголовке. Выход 80%, точка плавления: более 115°С (dec.),1Н-ЯМР спектр (ДМСО-d6, ТМС) δ: 2.3-2.5 (1Н, m, перекрывание с ДМСО); 2.69 (1Н, dd, 3.7 Гц и 14.6 Гц); 5.75 (1Н, dd, 4.4 Гц и 8.8 Гц); 7.34 (1Н, m); 7.4-7.8 (9Н, m); 7.86 (1Н, s); 7.9-8.2 (2Н, m); 8.97 (1Н, s) и 10.55 ppm (1Н, s).
Синтез 205
Метиловый эфир (5-этокси-1,1-диоксо-2-фенил-2,3-дигидро-1H-бензо[d]изотиазол-3-ил)уксусной кислоты (37)

Смесь соединения (32) (167 мг, 0,5 мМоль), йодистый этил (0,08 мл, 1,0 мМоль) и K2CO3 (415 мг, 1,5 мМоль) в ДМФА (3 мл) перемешивали при комнатной температуре в течение 2 ч 30 мин. Смесь разбавляли водой (20 мл) и экстрагировали EtOAc (2×20 мл). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл) и сушили над Na2SO4. Экстракт фильтровали и растворитель удаляли под вакуумом с образованием соединения (37) (180 мг), указанного в заголовке, в виде неочищенного продукта.
Синтез 206
(5-Этокси-1,1-диоксо-2-фенил-2,3-дигидро-1H-бензо[d]изотиазол-3-ил)уксусная кислота (38)

Следуя методике Способа Е (для синтеза (4)) из сложного эфира (37), соединение, указанное в заголовке, было получено в виде неочищенного продукта.
Синтез 207
2-(5-Этокси-1,1-диоксо-2-фенил-2,3-дигидро-1Н-бензо[d]изотиазол-3-ил)-N-гидроксиацетамид (39)

Следуя методике, аналогичной Способу G (для синтеза (5)) из карбоновой кислоты (38), было получено соединение, указанное в заголовке. Выход 35%, аморфное,1Н-ЯМР (ДМСО-d6, ТМС): δ 1.36 (3H, t, 7 Гц); 2.31 (1Н, dd, 8 Гц и 15 Гц); 2.60 (1Н, dd, 4 Гц и 15 Гц); 4.13 (2Н, q, 7 Гц); 5.60 (1Н, dd, 7 Гц и 8 Гц); 7.0-7.4 (3H, m); 7.4-7.6 (4Н, m); 7.86 (1Н, d, 8 Гц) 8.94 (1Н, s) и 10.51 ppm (1Н, s).
Синтез 208
Метиловый эфир (Е)-3-(4-бром-2-фенилсульфамоилфенил)акриловой кислоты (40)

К раствору анилина (2.1) (1,68 г, 18 мМоль) в диоксане (20 мл) добавляли 1 М водный NaHCO3 (15 мл). К смеси добавляли раствор хлористого сульфонила (1.8) (3,3 г, 9,7 мМоль) в диоксане (20 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, разбавляли 5% водным KHSO4 (100 мл) и экстрагировали EtOAc (100 мл). Органическую фазу промывали насыщенным водным NaHCO3 (100 мл) и насыщенным солевым раствором (100 мл). Экстракт сушили над Na2SO4, фильтровали и растворитель удаляли под вакуумом. Остаток обрабатывали смесью гексана и EtOAc (2:1). Образовавшийся осадок собирали на фильтре, получая (40) (1,8 г).
Синтез 209
Метиловый эфир (Е)-3-(3-Фенилсульфамоил-дифенил-4-ил) акриловой кислоты (41)

Смесь бромистого арила (40) (0,4 г, 1,0 мМоль), фенилбороновой кислоты (0,146 г, 1,2 мМоль), Pd(Ph3P)4 (35 мг, 0,03 мМоль) и Cs2CO3 (0,456 г, 1,4 мМоль) нагревали в диоксане (12 мл) при 90°С в течение 7 ч. Реакционную смесь охлаждали до комнатной температуры и разбавляли EtOAc (50 мл). Смесь промывали 5% водным KHSO4 (50 мл), насыщенным водным NaHCO3 (50 мл) и насыщенным солевым раствором (50 мл). Органическую фазу сушили над Na2SO4, фильтровали и упаривали. Остаток обрабатывали смесью гексана и EtOAc (3:1). Образовавшийся осадок собирали на фильтре, получая (41) (0,21 г).
Синтез 210
(1,1-Диоксо-2,6-дифенил-2,3-дигидро-1H-бензо[d]изотиазол-3-ил)уксусная кислота (42)

Смесь соединения (41) (0,21 г, 0,54 мМоль) в диоксане (4 мл) и 1 М водного NaHCO3 (3 мл) кипятили с обратным холодильником в течение 8 ч. Охлаждали до комнатной температуры и разбавляли водой (40 мл). Продукт экстрагировали EtOAc (50 мл) и органическую фазу промывали насыщенным солевым раствором (50 мл). Экстракт сушили над Na2SO4, фильтровали и упаривали, получая соединение (42) (0,18 г), указанное в заголовке, в виде неочищенного продукта.
Синтез 211
2-(1,1-Диоксо-2,6-дифенил-2,3-дигидро-1H-бензо[d]изотиазол-3-ил)-N-гидроксиацетамид (43)

Следуя методике, аналогичной Способу G (для приготовления соединений (5)) из карбоновой кислоты (42), было получено соединение, указанное в заголовке. Выход 45%, точка плавления: 191-193°С,1Н-ЯМР (ДМСО-d6, ТМС): δ: 2.36 (1Н, dd, 9.5 Гц и 14.7 Гц); 2.67 (1Н, dd, 3.7 Гц и 14.7 Гц); 5.74 (1Н, dd, 3.7 и 8.1 Гц); 7.3-7.6 (8Н, m); 7.72 (1Н, d, 8.1 Гц); 7.82 (2Н, d, 6.6 Гц); 8.12 (1Н, d, 8.1 Гц); 8.24 (1Н, s); 8.96 (1Н, s) и 10.56 ppm (1Н, s).
Общий способ синтеза 2-йод-N-фенилбензилсульфонамидов (45.1) и (45.2)
Способ L: Раствор сульфонамида (44) (3,7 мМоль) в ТГФ (20 мл) охлаждали до 0°С в атмосфере аргона. По каплям добавляли 1,4 М н-BuLi в гексанах (5,7 мл, 7,9 мМоль), и смесь оставляли нагреваться до комнатной температуры. После перемешивания при комнатной температуре в течение 1 ч устанавливали температуру реакционной смеси -78°С и к раствору добавляли l2 (1,04 г, 4,11 мМоль) в ТГФ (12 мл). Смесь перемешивали при -78°С в течение 1 ч, и затем оставляли нагреваться до комнатной температуры. Добавляли концентрированный водный Na2S2O3 до исчезновения окраски, и смесь экстрагировали EtOAc (3×50 мл). Объединенную органическую фазу промывали насыщенным солевым раствором (100 мл) и сушили над Na2SO4. После упаривания растворителя образовался неочищенный продукт (45).
Следуя методике, аналогичной Способу L, были в виде продуктов получены следующие соединения.


Общая методика синтеза метиловых эфиров 2-(1,1-диоксо-2-фенил-2,3-дигидро-1Н-бензо[d]изотиазол-3-ил)уксусной кислоты (46.1) и (46.2)
Способ М: Смесь 2-йод-N-фенилбензолсульфонамида (45) (1,3 мМоль), Pd(OAc)2 (28 мг, 0,13 мМоль), три-ортотолилфосфина (77,3 мг, 0,25 мМоль), триэтиламина (1 мл, 7,2 мМоль) и метилакрилата (2,37 мл, 25,4 мМоль) в ДМФА (3 мл) нагревали при 110°С в течение 3 ч. После охлаждения до комнатной температуры добавляли воду (50 мл), и смесь экстрагировали EtOAc (3×30 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и упаривали. Остаток очищали посредством флэш-хроматографии на силикагеле, элюируя смесью легкого петролейного эфира и EtOAc (5:1) с образованием (46).
Следуя методике, аналогичной способу М, были получены следующие соединения в виде неочищенного продукта.


Синтез 216
2-(5-Метил-1,1-диоксо-2-фенил-2,3-дигидро-1H-бензо[d]изотиазол-3-ил)уксусная кислота (47.1)

Следуя методике, аналогичной Способу Е (для синтеза (4)) из сложного эфира (46.1), соединение, указанное в заголовке, было получено в виде неочищенного продукта.
Синтез 217
2-(5-Хлор-1,1-диоксо-2-фенил-2,3-дигидро-1H-бензо[d]изотиазол-3-ил) уксусная кислота (47.2)
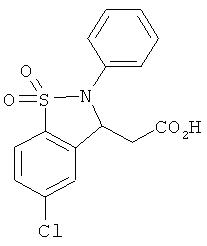
Следуя методике, аналогичной Способу Е (для синтеза (4)) из сложного эфира (46.2), соединение, указанное в заголовке, было получено в виде неочищенного продукта.
Синтез 218
2-(5-Метил-1,1-диоксо-2-фенил-2,3-дигидро-1Н-бензо[d]изотиазол-3-ил)-N-гидроксиацетамид (48.1)

Следуя методике, аналогичной Способу G (для синтеза (5)) из карбоновой кислоты (47.1), было получено соединение, указанное в заголовке. Выход 45%, точка плавления: 150-155°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.30 (dd, J=14.7 и 9.0 Гц, 1Н); 2.46 (s, перекрывание с ДМСО, 3H); 2.62 (dd, J=14.7 3.8 Гц, 1Н, перекрывание с ДМСО); 5.57-5.70 (m, 1Н); 7.26-7.38 (m, 1Н); 7.40-7.57 (m, 6Н); 7.86 (d, J=7.9 Гц, 1Н); 8.95 (s, 1Н); 10.52 ppm (s, 1Н).
Синтез 219
(5-Хлор-1,1-диоксо-2-фенил-2,3-дигидро-1Н-бензо[d]изотиазол-3-ил)-N-гидроксиацетамид (48.2)

Следуя методике, аналогичной Способу Н (для синтеза (5)) из карбоновой кислоты (47.1), было получено соединение, указанное в заголовке. Выход 80%, точка плавления: 192-194°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.41 (dd, J=15.0 и 8.3 Гц, 1Н); 2.70 (dd, J=15.0 и 4.0 Гц, 1Н); 5.70 (dd, J=8.0 и 4.0 Гц, 1Н); 7.30-7.41 (m, 1Н); 7.44-7.52 (m, 4Н); 7.73-7.82 (m, 2Н); 8.05 (d, J=8.9 Гц, 1Н); 8.94 (s, 1Н) и 10.49 ppm (s, 1Н).
Общий способ приготовления N-фенилбензолсульфонамидов (50.1)-(50.11)
Способ N: Анилин (2.1) (0,70 г, 7,5 мМоль) суспендировали в 1 М водного NaHCO3 (15 мл). К суспензии добавляли раствор хлористого сульфонила (49) (5 мМоль) в диоксане (15 мл), и смесь перемешивали при комнатной температуре в течение 22 ч. Его разбавили 5% водным KHSO4 (40 мл). Образовавшийся осадок собирали на фильтре и промывали большим количеством воды. Материал тщательно сушили под вакуумом над P2O5 с образованием (50).
Следуя методике, аналогичной Способу N, были получены следующие соединения в виде неочищенного продукта.



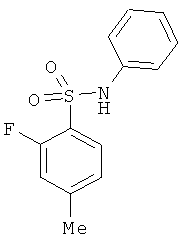







Общий способ приготовления 1,1-диоксо-2-фенил-2,3-дигидро-1Н-бензо[d]изотиазол-3-ола (51.1)-(51.11)
Способ О: Раствор сульфонамида (50) (2,5 мМоль) в ТГФ (25 мл) охлаждали до 0°С для синтеза соединений 51.1, 51.3, или до -78°С для синтеза соединений 51.2, 51.4-51.11). По каплям добавляли 1,6 М н-BuLi в гексанах (3,5 мл, 5,5 мМоль) и смесь выдерживали при охлаждении в течение 2 ч. Устанавливали температуру смеси -78°С и одной порцией добавляли ДМФА (0,39 мл, 5,0 мМоль). Отставляли охлаждающую баню, и смесь оставляли нагреваться до достижения комнатной температуры, и перемешивали в течение 2 ч. Добавляли 5% водный KHSO4 (100 мл), и смесь экстрагировали EtOAc (200 мл). Органическую фазу отделяли и промывали насыщенным солевым раствором (100 мл). Экстракт сушили над Na2SO4, фильтровали и упаривали. Остаток очищали посредством флэш-хроматографии на силикагеле, элюируя смесью легкого петролейного эфира и EtOAc.
Следуя методике, аналогичной Способу О, следующие соединения были получены в виде неочищенного продукта.





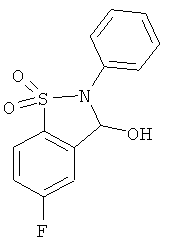
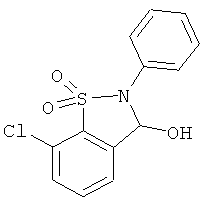


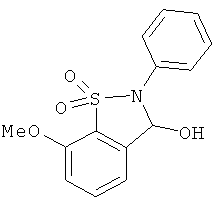

Общий способ приготовления метиловых эфиров 2-(1,1-диоксо-2-фенил-2,3-дигидро-1H-бензо[d]изотиазол-3-ил)уксусной кислоты (52.1)-(52.12)
Способ Р: К раствору соединение (51) (215 мг, 0,75 мМоль) и триметилфосфоноацетата (0,16 мл, 1,1 мМоль) добавляли 1 М NaOMe в МеОН (1,5 мл, 1,5 мМоль). Смесь оставляли на ночь перемешиваться при комнатной температуре и разбавляли водой (50 мл). Обычно образовывался белый осадок, который собирали на фильтре, промывали на фильтре водой и сушили под вакуумом над P2O5 с образованием 52. Если фильтруемый осадок не образовывался, продукт экстрагировали EtOAc, и органическую фазу промывали насыщенным солевым раствором. Сушка над Na2SO4, фильтрование и удаление растворителя приводили к остатку, который очищали флэш-хроматографией на силикагеле, элюируя смесью легкого петролейного эфира и EtOAc с образованием 52.
Следуя методике, аналогичной Способу Р, следующие соединения были получены в виде неочищенного продукта.

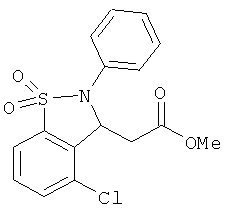

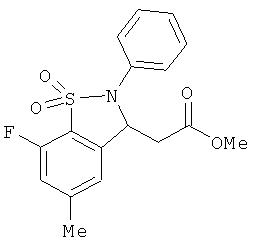








Следуя методике, аналогичной Способу Е (для синтеза (4)), и применяя указанные выше сложные эфиры, следующие соединения были получены в виде неочищенных продуктов.












Следуя методике, аналогичной Способу Н (для синтеза (5)), и применяя вышеуказанные карбоновые кислоты, были получены следующие соединения в виде неочищенных продуктов.
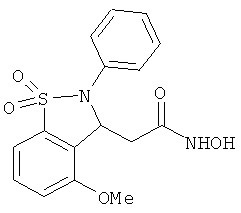





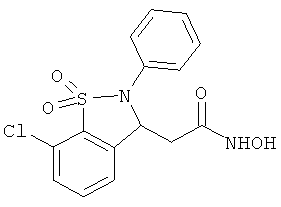





Соединение (54.1): Выход 64%, точка плавления 188-190°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.4-2.6 (2Н, перекрывание с ДМСО); 3.90 (3H, s); 5.66 (1Н, t, 5.0 Гц); 7.23-7.46 (7Н, гл); 7.66 (1Н, 18.0 Гц); 8.66 (1Н, s) и 10.29 ppm (1Н, s).
Соединение (54.2): Выход 54%, точка плавления 204-206°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.63 (1Н, dd, 15.4 Гц, 4.4 Гц); 2.75 (1Н, dd, 15.4 Гц, 5.9 Гц); 5.81 (1Н, t, 4.4 Гц); 7.27-7.51 (5Н, m); 7.71 (1Н, t, 7.7 Гц); 7.88 (1Н, d, 7.3 Гц); 7.95 (1Н, d, 7.3 Гц); 8.70 (1Н, s) и 10.37 ppm (1Н, s).
Соединение (54.3): Выход 48%, точка плавления 187-189°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.3-2.5 (1Н, m, перекрывание с ДМСО); 2.70 (1Н, dd, 4.4 и 14.7 Гц); 5.74 (1Н, dd, 4.4 Гц и 8.1 Гц); 7.33-7.56 (5Н, m); 7.95-8.14 (3H, m); 8.90 (1Н, s) и 10.47 ppm (1Н, s).
Соединение (54.4): Выход 37%, аморфный порошок,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.34 (1Н, dd, 8.1 и 14.7 Гц, частичное перекрывание с ДМСО); 2.45 (3H, s, перекрывание с ДМСО); 2.61 (1Н, dd, 4.4 и 8.1 Гц, частичное перекрывание с ДМСО); 5.64 (1Н, dd, 3.7 Гц и 8.8 Гц); 7.2-7.6 (7Н, m); 8.90 (1Н, s) и 10.46 ppm (1Н, s). MS (масс-спектроскопия): 350.9 (М+).
Соединение (54.5): Выход 50%, аморфный порошок,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.6-2.9 (2Н, m, частичное перекрывание с ДМСО); 5.82 (1Н, t, 4.4 Гц); 7.2-7.5 (5Н, m); 7.7-7.9 (1Н, m); 8.0-8.2 (1Н, m); 8.71 (1Н, d, 1.5 Гц) и 10.37 ppm (1Н, d, 1.5 Гц). MS: 370.9 (М+).
Соединение (54.6): Выход 80%, аморфный порошок,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.3-2.8 (2Н, m, частичное перекрывание с ДМСО); 5.64 (1Н, dd, 3.7 Гц и 8.1 Гц); 7.2-7.4 (1Н, m); 7.4-7.7 (7Н, m); 8.08 (1Н, dd, 5.1 Гц и 8.8 Гц); 8.90 (1Н, s) и 10.47 ppm (1Н, s). MS: 337.0 (М+).
Соединение (54.7): Выход 99%, точка плавления: более 188°С (dec.),1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.33 (1Н, dd, 8.1 Гц и 14.7 Гц); 2.64 (1Н, dd, 3.7 Гц, 14,7 Гц); 5.67 (1Н, dd, 4.4 Гц и 8.1 Гц); 7.3-7.9 (8Н, гл); 8.88 (1Н, s) и 10.46 ppm (1Н, s). MS: 352.9 (М+).
Соединение (54.8): Выход 60%, точка плавления: более 150°С (dec.),1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.4-2.7 (1Н, m, частичное перекрывание с ДМСО); 2.79 (1Н, dd, 16.1 Гц и 5.1 Гц); 5.77 (1Н, t, 4.4 Гц); 7.2-7.4 (1Н, m); 7.46 (4Н, m); 8.08 (1Н, d, 2.0 Гц); 8.23 (1Н, d, 2.0 Гц); 8.69 (1Н, s) и 10.36 ppm (1Н, s). MS: 386.8 (М+).
Соединение (54.9): Выход 60%, аморфный порошок1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.3-2.8 (2Н, m, перекрывание с ДМСО); 5.79 (1Н, dd, 3.9 Гц и 7.8 Гц); 7.3-7.6 (5Н, m); 7.88 (1Н, d, 7.8 Гц); 8.19 (1Н, d, 7.8 Гц); 8.52 (1Н, s); 8.89 (1Н, s) и 10.45 ppm (1Н, s). MS: 387.0 (М+).
Соединение (54.10): Выход 40%, точка плавления: более 161°С (dec.),1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.2-2.7 (2Н, m, перекрывание с ДМСО); 3.95 (3H, s); 5.59 (1Н, dd, 2.9 Гц и 8.8 Гц); 7.10 (1Н, d, 6.7 Гц); 7.2-7.6 (8Н, m); 7.72 (1Н, t, 8.8 Гц); 8.90 (1Н, s) и 10.49 ppm (1Н, s). MS: 348.9 (М+).
Соединение (54.11): Выход 65%, точка плавления более 170°С (dec.)1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.2-2.7 (2Н, m, частичное перекрывание с ДМСО); 3.98 (3H, s); 5.59 (1Н, dd, 3.7 Гц и 8.1 Гц); 7.2-7.6 (7Н, m); 8.90 (1Н, d, 1.5 Гц) и 10.46 ppm (1Н, d, 1.5 Гц). MS: 382.8 (М+).
Соединение (54.12): Выход 42%, точка плавления 184-188°С (dec.),1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.5-2.7 (2Н, m, частичное перекрывание с ДМСО); 3.99 (3H, s); 5.74 (1Н, t, 4.4 Гц), 7.2-7.6 (6Н, m); 7.7-7.9 (1Н, m); 7.94 (1Н, d, 8.1 Гц); 8.68 (1Н, s) и 10.37 ppm (1Н, s). MS (масс-спектр): 382.9 (М+).
Синтез 278
3-(1,1-диоксо-2-фенил-2,3-дигидро-1H-бензо[d]изотиазол-3-ил)пропионитрил (55)

Раствор сложного эфира (3.1) (2,9 г, 9,14 мМоль) в ТГФ (70 мл) охлаждали на ледяной бане и к нему несколькими порциями добавляли LiAlH4 (1,04 г, 27,4 мМоль). Смесь оставляли нагреваться до комнатной температуры и дополнительно перемешивали 1 ч. Остаточный LiAlH4 уничтожали добавлением воды по каплям до образования геля. Добавляли насыщенный раствор калий-натриевой соли винной кислоты (100 мл), и полученную суспензию экстрагировали EtOAc (300 мл). Органическую фазу промывали насыщенным солевым раствором (100 мл) и сушили над Na2SO4. Раствор фильтровали и упаривали с образованием промежуточного спирта. Полученный промежуточный продукт (1,16 г) растворяли в DCM (70 мл) и к раствору добавляли PCl5 (0,92 г, 4,4 мМоль). Смесь перемешивали при комнатной температуре в течение 1 ч, промывали насыщенным водным NaHCO3 (100 мл). Органическую фазу упаривали, и остаток очищали флэш-хроматографией на силикагеле, элюируя смесью легкого петролейного эфира и EtOAc (1:1), получая промежуточный хлорид (610 мг). Его растворяли в ДМФА (15 мл) и добавляли KCN (258 мг, 3,96 мМоль). Смесь перемешивали при 50°С в течение 20 ч и разбавляли водой (100 мл). Продукт обрабатывали EtOAc (100 мл), и органическую фазу промывали насыщенным солевым раствором (100 мл). Раствор сушили над Na2SO4, фильтровали и упаривали. Остаток очищали посредством флэш-хроматографии на силикагеле, элюируя смесью легкого петролейного эфира и EtOAc (1:1), получая соединение, указанное в заголовке (55) (450 мг).
Синтез 279
3-(1,1-Диоксо-2-фенил-2,3-дигидро-1H-бензо[d]изотиазол-3-ил)пропионовая кислота (56)

К раствору нитрила (55) (136 мг, 0,46 мМоль) в диоксане (7 мл) добавляли водную концентрированную HCl (1,2 мл, 14,4 мМоль). Смесь нагревали при 115°С в течение 72 ч и упаривали. Остаток очищали посредством флэш-хроматографии на силикагеле, элюируя смесью легкого петролейного эфира и EtOAc (1:10), получая соединение (56) (138 мг).
Синтез 280
3-(1,1-Диоксо-2-фенил-2,3-дигидро-1H-бензо[d]изотиазол-3-ил)-N-гидроксипропионамид (57)

Следуя методике, аналогичной Способу G (для синтеза (5)) из карбоновой кислоты (56) было получено соединение, указанное в заголовке. Выход 63%, вязкое масло,1Н-ЯМР (ДМСО-d6, ТМС) δ: 1.38-1.58 (m, 1Н); 1.76-1.96 (m, 1Н); 2.03-2.35 (m, 2Н); 5.68 (t, J=3.1 Гц, 1Н); 7.28-7.41 (m, 1Н); 7.45-7.57 (m, 4Н); 7.65-7.90 (m, 3H); 7.98 (d, J=7.7 Гц, 1Н); 8.62 (s, 1Н); 10.29 ppm (s, 1Н).
Синтез 281
2-Метил-N-фенилбензолсульфонамид (59.1)
Следуя методике, аналогичной Способу N (для синтеза соединений (50)), из хлористого сульфонила (58.1) и анилина (2.1) было получено соединение, указанное в заголовке, в виде неочищенного продукта.
Синтез 282
2-Метил-3-хлор-N-фенилбензолсульфонамид (59.2)

Следуя методике, аналогичной Способу N (для синтеза соединений (50)), из хлористого сульфонила (58.2) и анилина (2.1) было получено соединение, указанное в заголовке, в виде неочищенного продукта.
трет-бутиловые эфиры 1,1-диоксо-2-фенил-2,3-дигидро-1Н-бензо[d]изотиазол-3-карбоновой кислоты (60.1) и (60.2)
Способ Q: Раствор сульфонамида (58) (2,8 мМоль), Вос2O (1,2 г, 5,54 мМоль) и DMAP (338 мг, 2,8 мМоль) в ТГФ (35 мл) оставляли на ночь перемешиваться при комнатной температуре. Растворитель удаляли под вакуумом, и остаток обрабатывали EtOAc (100 мл). Органическую фазу промывали 10% водным HCl, насыщенным водным NaHCO3 и насыщенным солевым раствором. Экстракт сушили над Na2SO4, фильтровали и упаривали. Остаток очищали посредством флэш-хроматографии на силикагеле, элюируя смесью легкого петролейного эфира и EtOAc (4:1). Промежуточное соединение (2,0 мМоль) растворяли в ТГФ и к нему добавляли TMEDA (0,66 мл, 4,4 мМоль), и смесь охлаждали до -78°С. При этой температуре по каплям добавляли 1,5 М трет-BuLi в гексанах (2,9 мл, 4,4 мМоль), и смесь перемешивали при -78°С дополнительно 30 мин. Добавляли хлористый ангидрид диметиламиносульфоновой кислоты (0,24 мл, 2,2 мМоль), смесь перемешивали при
-78°С в течение 1 ч, оставляли нагреваться до достижения комнатной температуры и затем, на ночь, перемешиваться. Смесь разбавляли EtOAc и промывали насыщенным солевым раствором. Органическую фазу сушили над Na2SO4, фильтровали и упаривали. Остаток очищали посредством флэш-хроматографии на силикагеле, элюируя смесью легкого петролейного эфира и EtOAc (4:1), получая соединение (60), указанное в заголовке.
Следуя методике, аналогичной Способу Q, были получены следующие соединения в виде неочищенного продукта.


Следуя методике, аналогичной Способу Е (для синтеза (4)), и применяя вышеприведенный сложный эфир, следующее соединение было получено в виде неочищенных продуктов.


Следуя методике, аналогичной Способу G (для синтеза (5)), и применяя вышеуказанную карбоновую кислоту, были получены соединения в виде неочищенных продуктов.


Соединение (62.1): Выход 95%, точка плавления 188-191°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 5.78 (s, 1Н); 7.22-7.32 (m, 1Н); 7.41-7.51 (m, 4Н); 7.62-7.91 (m, 3H); 8.00-8.08 (m, 1Н); 9.34 (s, 1Н); 11.35 ppm (s, 1Н).
Соединение (62.2): Выход 43%, точка плавления 167-170°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 5.72 (s, 1Н); 7.30-7.43 (m, 1Н); 7.46-7.55 (m, 4Н); 7.78 (dd, J=8.0 и 8.0 Гц, 1Н); 7.91 (dd, J=0.9 и 8.0 Гц, 1Н); 8.05 (dd, J=0.9 и 8.0 Гц, 1Н); 9.35 (d, J=1.0 Гц, 1Н); 11.31 ppm (s, 1Н).
Синтез 289
(+)- и (-)-1,1-Диоксо-2-фенил-2,3-дигидро-1H-бензо[d]изотиазол-3-ил)-N-гидроксил-ацетамиды (+)-(S)-(5.1) и (-)-(R) (5.1)


Раствор карбоновой кислоты (4.1) (606 мг, 2 мМоль), (R)-фенилглицинол (274 мг, 2 мМоль), HOBt (270 мг, 2 мМоль) и EDCl (383 мг, 2 мМоль) в ДМФА (2 мл) оставляли на ночь перемешиваться при комнатной температуре. Раствор распределяли между EtOAc (30 мл) и водой (30 мл). Органическую фазу отделяли и промывали насыщенным солевым раствором (20 мл), насыщенным водным NaHCO3 (20 мл) и насыщенным солевым раствором (20 мл). Раствор сушили над Na2SO4, фильтровали и упаривали. Диастереомерные амиды разделяли хроматографией с вращающимся электродом на силикагеле, элюируя смесью гексан-этилацетат (1:2), с образованием быстро элюирующегося диастереомера (S,R)-(63.1) (структура определена рентгеновской спектроскопией) и медленно элюирующегося диастереомера (R,R)-(63.1).
Каждый из диастереомерных амидов (S,R)-(63.1) и (R,R)-(63.1) был гидролизован в 20% водном HCl при 80°С в течение 7 часов. Продукт экстрагировали CHCl3, и раствор сушили над Na2SO4. Раствор фильтровали и упаривали с образованием карбоновых кислот (S)-(4.1) и (R)-(4.1). Карбоновые кислоты (S)-(4.1) и (R)-(4.1) переводили в гидроксамовые кислоты (+)-(S)-(5.1) ([α]D20=+80° (с=1, ацетон)) и (-)-(R)-(5.1) ([α]D20=-92° (с=1, ацетон)), следуя общей процедуре, описанной для синтеза рацемической гидроксамовой кислоты (5.1) и имели данные1Н-ЯМР, идентичные данным для рацемической гидроксамовой кислоты (5.1).
Синтез 290
(+)- и (-)-2-[2-(4-Бут-2-инилоксифенил)-1,1-диоксо-2,3-дигидро-1H-бензо[d]изотиазол-3-ил]-N-гидроксиацетамид (+)-(5.43) и (-)-(5.43)


Раствор карбоновой кислоты (4.43) (1,11 г, 3,0 мМоль), (R)-фенилглицинол (0,45 г, 3,3 мМоль), HOBt (0,45 г, 3,3 мМоль) и EDCl (0,63 г, 3,3 мМоль) в ДМФА (15 мл) перемешивали при комнатной температуре в течение 24 ч. Раствор распределяли между EtOAc (70 мл) и водой (100 мл). Органическую фазу отделяли и промывали водой (2×100 мл), и насыщенным солевым раствором (100 мл), раствор сушили над Na2SO4, фильтровали и упаривали. Диастереомерные амиды отделяли флэш-хроматографией на силикагеле, элюируя EtOAc с образованием быстро элюирующегося диастереомерного амида Е7-(63.2) (0,67 г) и E2-(63.2) (0,56 г) медленно элюирующегося диастереомерного амида. Каждый из диастереомерных амидов E1-(63.2) (343 мг) и E2-(63.2) (343 мг) гидролизовали смесью 1 М водного H2SO4 (12 мл) и диоксана (12 мл) при температуре кипячения с обратным холодильником в течение 30 ч. Диоксан удаляли под вакуумом и добавляли воду (30 мл). Смесь экстрагировали EtOAc (50 мл + 30 мл), и объединенную органическую фазу промывали насыщенным солевым раствором (50 мл), раствор сушили над Na2SO4, фильтровали и упаривали с образованием карбоновых кислот Е1-(4.43) (242 мг) и Е2-(4.43) (269 мг), соответственно. Карбоновые кислоты Е7-(4.43) и Е2-(4.43) переводили в гидроксамовые кислоты (+)-(5.43) ([α]D20=+71° (с=0,86, ацетон)) и (-)-(5.43) ([α]D20=-69° (с=0,84, ацетон)), следуя общей процедуре, описанной для синтеза рацемической гидроксамовой кислоты (5.43), и имели данные по1Н-ЯМР, идентичные данным для рацемической гидроксамовой кислоты (5.43).
Синтез 291
(+)- и (-)-2-{2-[4-(2-Метилхинолин-4-илметокси)фенил]-1,1-диоксо-2,3-дигидро-1H-бензо[d]изотиазол-3-ил}-N-гидроксиацетамид (+)-(5.44) и (-)-(5.44)

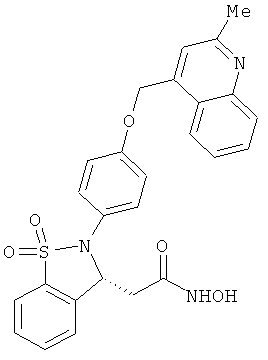
Раствор карбоновой кислоты (4.44) (1,02 г, 2,0 мМоль), (R)-фенилглицинол (0,27 г, 2,0 мМоль), HOBt (0,27 г, 2,0 мМоль) и EDCl (0,38 г, 2,0 мМоль) в ДМФА (4 мл) перемешивали при комнатной температуре в течение 15 ч. Смесь разбавляли насыщенным водным NaHCO3 (100 мл) и экстрагировали EtOAc (2×100 мл). Объединенную органическую фазу отделяли и промывали насыщенным солевым раствором (100 мл). Раствор сушили над Na2SO4, фильтровали и упаривали. Диастереомерные амиды разделяли флэш-хроматографией на силикагеле, элюируя EtOAc с образованием быстро элюирующегося диастереомерного амида Е7-(63.3) (0,30 г) и медленно элюирущегося диастереомерного амида Е2-(63.3) (0,27 г). Каждый из диастереомерных амидов Е7-(63.3) (140 мг) и Е2-(63.3) (150 мг) гидролизовали в смеси 10% водного HCl (0,92 мл) и диоксана (0,92 мл) при 110°С в течение 2 ч. Диоксан удаляли под вакуумом и добавляли воду (4 мл). Осадок отделяли центрифугированием и несколько раз промывали водой. Остаток сушили над Р2O5 под вакуумом с образованием карбоновых кислот Е1-(4.44) (0,11 г) и Е2-(4.44) (0,10 г), соответственно. Карбоновую кислоту Е1-(4.44) (102 мг, 0,2 мМоль) растворяли в диоксане (2 мл) и к ней добавляли хлористый ангидрид щавелевой кислоты (0,35 мл, 4 мМоль) и затем каплю ДМФА. Смесь нагревали при 50°С в течение 2 ч и упаривали. К остатку добавляли раствор О-ТНР гидроксиламина (117 мг, 1 мМоль) в ДМФА (1 мл), и полученную смесь перемешивали при комнатной температуре в течение 30 мин. Смесь разбавляли насыщенным водным NaHCO3 (10 мл) и экстрагировали EtOAc (15 мл). Экстракт сушили над Na2SO4, фильтровали и упаривали. Остаток очищали посредством флэш-хроматографии на силикагеле, элюируя EtOAc с образованием защищенной О-ТНР гидроксамовой кислоты (64 мг). Промежуточное соединение растворяли в диоксане (1,2 мл) и к раствору добавляли 1 М водный HCl (0,6 мл). Смесь оставляли на ночь перемешиваться при комнатной температуре и нейтрализовали водным 1 М NaHCO3 (10 мл). Образовавшийся осадок собирали на фильтре и несколько раз промывали водой. Остаток сушили над P2O5 под вакуумом и обрабатывали MeCN (2 мл). Осадок собирали на фильтре и сушили над P2O5 под вакуумом с образованием гидроксамовой кислоты (+)-5.44 ([α]D20=+62° (с=0.5, ДМСО-d6)), имеющей спектр1Н-ЯМР, идентичный спектру рацемической гидроксамовой кислоты (5.44).
Следуя вышеописанной процедуре, карбоновую кислоту Е2-(4.44) (180 мг, 0,35 мМоль) переводили в гидроксамовую кислоту (-)-5.44 ([α]D20=-48° (с=0.5, ДМСО-d6)), имеющей спектр1Н-ЯМР, идентичный спектру рацемической гидроксамовой кислоты (5.44).
Синтез 292
O-(N,N-Диметилтиокарбамоил)-2-гидроксибензальдегид (65)

N,N-Диметилтиокарбамоилхлорид (7,42 г, 60 мМоль) добавляли к раствору салицилового альдегида (64 (4,89 г, 40 мМоль) и DABCO, 1,4-диазабицикло[2.2.2]октан, (8,96 г, 80 мМоль) в ДМФА (80 мл). Полученную смесь оставляли на ночь перемешиваться при комнатной температуре и выливали в воду (250 мл). Осадок собирали на фильтре и промывали большим количеством воды. После сушки над NaOH под вакуумом соединение (65) (7,34 г, 87%) получали в виде светло-серых кристаллов.1Н-ЯМР (ДМСО-d6) δ: 3.38 и 3.40 (всего 6Н, оба s); 7.24 (1Н, d, 8 Гц); 7.46 (1Н, t, 7 Гц); 7.74 (1Н, dt, 7 Гц и 2 Гц); 7.86 (1Н, dd, 8 Гц и 2 Гц) и 10.00 ppm (1Н, s).
Синтез 293
8-(N,N-Диметилтиокарбамоил)-2-тиобензальдегид (66)

Соединение (65) (1.04 г, 5 мМоль) нагревали в N,N-диэтиланилине (1 мл) при 190°С в течение 5 ч. После охлаждения добавляли воду (10 мл), и смесь подкисляли 20% водным KHSO4. Продукт обрабатывали этилацетатом (20 мл). Органическую фазу отделяли и промывали насыщенным солевым раствором (20 мл). После сушки над Na2SO4 растворитель удаляли под вакуумом, и остаток очищали флэш-хроматографией на силикагеле, элюируя смесью легкого петролейного эфира и этилацетата (2:1) с образованием соединения (66) (495 мг, 48%).1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.91 (3H, br s) и 3.09 (3H, br s); 7.5-7.8 (3H, m); 7.8-8.0 (2Н, m); и 10.14 ppm (1Н, s).
Синтез 294
8-Бензил-2-тиобензальдегид (67)

Соединение (66) (495 мг, 2,4 мМоль) растворяли в растворе 1М метанольного NaOMe (10 мл). Смесь оставляли на ночь перемешиваться при комнатной температуре и к ней добавляли бромистый бензил (0,35 мл, 2,9 мМоль). перемешивание продолжали в течение 2 ч, и смесь выливали в ледяную воду (50 мл). Продукт обрабатывали CH2Cl2 (3×20 мл). Объединенную органическую фазу промывали насыщенным солевым раствором и сушили над Na2SO4. Раствор фильтровали и упаривали с образованим диметилацеталя альдегида. Его растворяли в смеси диоксана (2 мл) и 1 н. водного HCl (1 мл) и перемешивали в течение 2 часов 30 минут при комнатной температуре, добавляли воду (20 мл), и смесь экстрагировали EtOAc (30 мл). Органическую фазу отделяли, промывали насыщенным солевым раствором (20 мл) и сушили над Na2SO4. Раствор фильтровали и упаривали с образованием (67) (417 мг, 76%) в виде масла.1Н-ЯМР (ДМСО-d6, ТМС) δ: 4.26 (2Н, s); 7.2-7.5 (6Н, m); 7.6-7.7 (2Н, m); 7.87 (1Н, d, 8 Гц) и 10.10 ppm (1Н, s).
Синтез 295
Метиловый эфир (E)-3-(2-Бензилсульфанилфенил)акриловой кислоты (68)

Раствор альдегида (67) (1,24 г, 5,5 мМоль) и метил(трифенилфосфоранилиден)ацетата (1,93 г, 5,8 мМоль) в CH2Cl2 (30 мл) перемешивали при комнатной температуре в течение 2 часов. Добавляли силикагель (3 небольшие порции), и растворитель упаривали. Остаток выливали на короткую силикагелевую колонку, и продукт элюировали смесью гексана и EtOAc (10:1) с образованием сложного эфира (68) в виде бесцветных кристаллов (985 мг, 63%).1Н-ЯМР (CDCl3, ТМС) δ: 3.81 (3H, s); 4.03 (2Н, s); 6.32 (1Н, d, 15 Гц); 7.2-7.7 (9Н, m) и 8.20 ppm (1Н, d, 15 Гц).
Синтез 296
Метиловый эфир (E)-3-(2-фенилметансульфонилфенил)акриловой кислоты (69)

К раствору соединения (68) (853 мг, 3 мМоль) в CH2Cl2 (30 мл) добавляли 70% МСРВА, метахлорпероксибензойную кислоту (1,84 г, 7,5 мМоль). Смесь перемешивали при комнатной температуре в течение 1 часа и дополнительно добавляли CH2Cl2 (30 мл). Органическую фазу промывали насыщенным водным Na2S2O3 (20 мл) и насыщенным водным NaHCO3. Раствор сушили над Na2SO4 и упаривали. Остаток кристаллизовали из смеси гексана и EtOAc (4:1) с образованием соединения (69) (613 мг, 65%) в виде бесцветных кристаллов.1Н-ЯМР (ДМСО-d6, ТМС) δ: 3.73 (3H, s); 4.61 (2Н, s); 6.46 (1Н, d, 16 Гц); 7.0-8.0 (9Н, m) и 8.17 ppm (1Н, d, 16 Гц).
Синтез 297
Метиловый эфир 2-(1,1-диоксо-2-фенил-2,3-дигидро-1Н-бензо [b]тиофен-3-ил)уксусной кислоты (70)

1 М водный NaHCO3 (0,64 мл) добавляли к раствору соединения (69) (0,32 мМоль, 100 мг) в диоксане (1,2 мл). Смесь кипятили с обратным холодильником в течение 45 минут и упаривали. Остаток распределяли между EtOAc (20 мл) и водой (20 мл). Органическую фазу отделяли и промывали насыщенным солевым раствором (20 мл). Раствор сушили над Na2SO4 фильтровали и упаривали. Остаток обрабатывали гексаном и фильтровали с образованием соединения (70) (78 мг, 78%) в виде бесцветных кристаллов.1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.84 (1Н, dd, 16 Гц и 6 Гц); 3.02 (1Н, dd, 16 Гц и 6 Гц); 3.40 (3H, s); 4.15 (1Н, m); 4.88 (1Н, d, 9 Гц); 7.45 (5Н, s) и 7.5-7.8 ppm (4Н, m).
Синтез 298
2-(1,1-Диоксо-2-фенил-2,3-дигидро-1H-бензо[b]тиофен-3-ил)уксусная кислота (71)

Раствор сложного эфира (70) (175 мг, 0,55 мМоль) в смеси диоксана (3,3 мл) и концентрированной водной HCl (1,1 мл) перемешивали при комнатной температуре в течение 2 суток. Растворители упаривали и замещали свежим диоксаном (3,3 мл) и концентрированной водной HCl (3,3 мл). Перемешивание продолжали дополнительно в течение 2 суток, до полного исчезновения исходного материала. Растворители удаляли под вакуумом, остаток распределяли между EtOAc (30 мл) и насыщенным водным NaHCO3 (30 мл). Водную фазу отделяли и подкисляли концентрированной водной HCl. Продукт обрабатывали EtOAc (30 мл), органическую фазу отделяли и промывали насыщенным солевым раствором (20 мл). Раствор сушили над Na2SO4, фильтровали и упаривали с образованием соединения (71) (120 мг, 72%).1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.79 (2Н, m); 4.10 (1Н, m); 4.90 (1Н, d, 9 Гц); 7.45 (5Н, s) и 7.5-7.8 ppm (4Н, m).
Синтез 299
2-(1,1-Диоксо-2-фенил-2,3-дигидро-1Н-бензо[b]тиофен-3-ил)-N-гидроксиацетамид (72)

К раствору карбоновой кислоты (71) (120 мг, 0,4 мМоль) в CH2Cl2 (2 мл) добавляли хлористый ангидрид щавелевой кислоты (0,17 мл, 2 мМоль) и каплю ДМФА. Полученную смесь перемешивали при комнатной температуре и упаривали. К остатку добавляли смесь, приготовленную растворением хлористоводородной соли гидроксиламина (347 мг, 5 мМоль) в смеси ТГФ (5 мл) и 1М водного NaHCO3 (5 мл). Полученную суспензию перемешивали в течение 1 часа и распределяли между EtOAc (20 мл) и водой (20 мл). Органическую фазу отделяли и промывали насыщенным NaHCO3 (10 мл) и насыщенным солевым раствором (10 мл), раствор сушили над Na2SO4, фильтровали и упаривали. Остаток кристаллизовали из EtOAc с образованием гидроксамовой кислоты (72) (22 мг, 17%) в виде бесцветных кристаллов с точкой плавления 113-114°С.1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.2-2.7 (2Н, m, перекрывание с ДМСО); 4.13 (1Н, m); 5.05 (1Н, d, 8 Гц); 7.3-7.9 (9Н, m); 8.9 (1Н, br s) и 10.6 ppm (1Н, br s).
Синтез 300
Фениламид 2-йод-пиридин-3-сульфоновой кислоты (74)

Следуя методике, аналогичной Способу L (для синтеза соединений (45)) из сульфонамида (73), было получено соединение, указанное в заголовке в виде неочищенного продукта.
Синтез 301
Метиловый эфир (1,1-диоксо-2-фенил-2,3-дигидро-1Н-изотиазоло[4,5-b]пиридин-3-ил)уксусной кислоты (75)

Следуя методике, аналогичной Способу М (для синтеза соединений (46)) из йодида (74), было получено соединение, указанное в заголовке в виде неочищенного продукта.
Синтез 302
(1,1-Диоксо-2-фенил-2,3-дигидро-1Н-изотиазоло[4,5-b]пиридин-3-ил)уксусная кислота (76)

Следуя методике, аналогичной Способу Е (для синтеза (4)) из сложного эфира (75), было получено соединение, указанное в заголовке в виде неочищенного продукта.
Синтез 303
(1,1-Диоксо-2-фенил-2,3-дигидро-1H-изотиазоло [4,5-b]пиридин-3-ил)уксусная кислота (77)

Следуя методике, аналогичной Способу G (для синтеза (5)), из карбоновой кислоты (76), было получено соединение, указанное в заголовке. Выход 30%, точка плавления: 123-128°С,1Н-ЯМР (ДМСО-d6, ТМС) δ: 2.37 (1H, dd перекрывание с ДМСО, J=14.9 8.4 Гц); 2.71 (1H, dd перекрывание с ДМСО, J=14.9 4.0 Гц); 5.87-5.65 (1Н, m); 7.45-7.31 (1Н, m); 7.67-7.45 (4Н, m); 7.95-7.79 (1Н, m) 8.17 (1Н, d, J=7.7 Гц); 9.02-8.77 (2Н, m) и 10.48 ppm (1Н, s).
Биологические способы анализа
Система анализа ТАСЕ
Активность соединений в качестве ингибиторов ТАСЕ определяли, применяя имеющийся в продаже пептидный субстрат (М-2255, Bachem UK Ltd, St. Helens, UK) и рекомбинантный фермент ТАСЕ (930-ADB, R and D Systems, Abingdon, UK). Рекомбинантный фермент ТАСЕ человека (5 нг/30 мкл) инкубировали в течение 3,5 часов при 37°С в буфере для испытаний (25 мМ Трис. HCl, 2,5 мкМ ZnCl2, 0,005% Brij 35, pH 8,0) при концентрации субстрата 5 мкМ в присутствии тестового соединения (ингибитора ТАСЕ). Степень активности ТАСЕ определяли по измерению флуоресценции (возбуждение при 355 нм, эмиссия при 460 нм).
Активность, выраженную в процентах (% активности) в каждом тесте рассчитывали следующим образом:
% активности = {(SC-В)/(S°-В)}×100
где SC обозначает сигнал, измеренный в присутствии фермента и испытуемого соединения, S° обозначает сигнал, измеренный в присутствии фермента, но при отсутствии испытуемого соединения, а В обозначает фоновый сигнал при отсутствии и фермента, и испытуемого соединения. Значение IC50 соответствует концентрации, при которой достигается 50% активность.
Исследование селективности: активность по отношению к HDAC (деацетилазе гистонов): анализ по флуоресценции
Альтернативно, активность соединений, в качестве ингибиторов HDAC определяли, применяя имеющийся в продаже набор для анализа флуоресцентного (Fluor de Lys™, BioMol Research Labs, Inc., Plymouth Meeting, USA). Экстракт клеток HeLa инкубировали в течение 1 часа при 37°С в буфере для испытаний (25 мМ HEPES, 4-(2-Гидроксиэтил)пиперазин-1-этансульфоновая кислота, 137 мМ NaCl, 2,7 мМ KCl, 1 мМ MgCl2, pH 8,0) с 15 мкМоль ацетилированного субстрата в присутствии испытуемого соединения (ингибитора HDAC). Степень дезацетилирования определяли добавлением 50 мкл компонента-проявителя в разбавлении 1 к 500, и последующим измерением флуоресценции (возбуждение при 355 нм, эмиссия при 460 нм), в соответствии с инструкциями, приложенными к набору.
Экстракт клеток HeLa
Экстракт клеток HeLa приготовили из клеток HeLa (АТСС Ref. No.CCL-2) путем трехкратного замораживания-оттаивания в 60 мМ трис.HCl, pH 8,0, 450 мМ NaCl, 30% глицерина. Применяли буфер для экстракции в двойном по отношению к клеткам объеме, и материал из твердых частиц центрифугировали (20800 g, 4°С, 10 минут) и отбрасывали. Экстракт в супернатанте, обладающий дезацетилазной активностью, разделили на аликвоты и заморозили для хранения.
Активность, выраженную в процентах, (% активности) для каждого испытуемого соединения рассчитывали следующим образом:
% активности = {(SC-В)/(S°-В)}×100
где SC обозначает сигнал, измеренный в присутствии фермента и испытуемого соединения, S° обозначает сигнал, измеренный в присутствии фермента, но при отсутствии испытуемого соединения, и В обозначает фоновый сигнал при отсутствии и фермента, и испытуемого соединения. Значение IC50 соответствует концентрации, при которой достигается 50% активность.
Измерение жизнеспособности клеток в присутствии испытуемого соединения в увеличивающейся концентрации в различные моменты времени применяли для оценки как цитотоксичности, так и влияния соединения на клеточную пролиферацию.
Биологические данные
В нижеследующих таблицах представлены значения IC50 при анализе с ТАСЕ для некоторых соединений согласно настоящему изобретению.
Некоторые соединения протестировали в одной концентрации по отношению к набору металлопротеаз матрикса, включая фермент, конвертирующий ангиотензин (АСЕ), и определили ингибирование в процентах. Данные суммированы в нижеследующей таблице.
Эти данные показывают, что внутри класса соединений - производных BCSAs, раскрытых в описании настоящего изобретения, возможно достижение улучшенной селективности ингибиторов ТАСЕ по сравнению с родственными Zn-металлопротеазами, такими как деацетилазы гистонов или матричные металлопротеазы. Поэтому ожидается, что возможно применение этих ингибиторов ТАСЕ без побочных эффектов, которые являются следствием ингибирования HDAC или ММР.
В вышеизложенном описании описаны принципы, предпочтительные варианты реализации и способы осуществления настоящего изобретения. Однако обсуждавшиеся частные варианты реализации не следует толковать как ограничительные для объема притязаний согласно настоящему изобретению. Вместо этого вышеописанные варианты реализации следует рассмативать как иллюстративные, а не ограничительные, и следует принять во внимание изменения в реализации, которые могли бы сделать работники, являющиеся специалистами в данной области без выхода за рамки объема притязаний настоящего изобретения.
ССЫЛКИ
Для более полного описания и раскрытия идеи изобретения выше цитирован ряд патентов и публикаций в области знаний, к которой относится настоящее изобретение. Ниже приводятся полные ссылки на эти публикации. Содержание каждой из этих ссылок вводится в описание настоящей заявки во всей своей полноте, в такой же мере, как если бы указывалось, что каждая отдельная публикация конкретно и индивидуально вводится посредством ссылки.



Реферат
Изобретение относится к соединениям бициклосульфонильной кислоты (BCSA) формулы:где: каждый из -R, -R, -Rи -Rнезависимо представляет собой -Н или -R; каждый -Rнезависимо представляет собой -F, -Cl, -Br, -I, -R, -CF, -ОН, -OCFили -OR; при этом каждый Rнезависимо представляет собой Салкил, фенил или бензил; и дополнительно, две соседние группы -Rмогут вместе образовать -ОСНО-, -ОСНСНО- или -ОСНСНСНО-; -Rнезависимо представляет собой ковалентную связь, -(СН)- или -(СН)-; -Rнезависимо представляет собой -R, или -L-R; остальные значения радикалов представлены в п.1 формулы изобретения, действующим как ингибиторы фермента, конвертирующего фактор некроза опухоли -α (ТАСЕ). Соединения являются полезными для лечения состояний, опосредуемых TNF-α. 5 н. и 31 з.п. ф-лы, 303 пр.
Формула

в котором каждый из -RPW, -RPX, -RPY и -RPZ независимо представляет собой -H или -RRS1;
каждый -RRS1 независимо представляет собой -F, -Cl, -Br, -I, -RA1, -CF3, -ОН, -OCF3 или -ORA1; при этом каждый RA1 независимо представляет собой C1-4алкил, фенил или бензил; и дополнительно две соседние группы -RRS1 могут вместе образовать -OCH2O-, -OCH2CH2O- или -OCH2CH2CH2O-;
-RAK независимо представляет собой ковалентную связь, -(СН2)- или -(СН2)2-; -RN независимо представляет собой -RNNN, или -LN-RNNN;
LN - независимо представляет собой -СН2- или -СН2СН2-;
RNNN независимо представляет собой фенил, нафтил или пиридил и возможно замещен одним или более заместителем RS; и
каждый -RS представляет собой независимо -F, -Cl, -Br, -I, -RD1, -RE7, -RE8, -CF3, -CH2CF3, -CF2CF2H, -OCF3, -OCH2CF3, -OCF2CF2H, -ОН, -ORD1, -L1-OH, -
L1-ORD1, -SRD1,-SCF3, -NH2, -NHRD1,
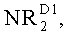

каждый -L1 представляет собой насыщенный алифатический С1-5алкилен;
каждая группа -NRN1RN2 независимо представляет собой пирролидино, пиперидино, или морфолино, и возможно замещена одной или более C1-3алкильной группой;
каждый -RD1 независимо представляет собой насыщенный алифитический С1-6алкил, фенил или бензил:
каждый -RE2 независимо представляет собой алифатический С2-6 алкенил и возможно замещен одним или более -ОН, ORF1, -NH2, -NH RF1,

каждый -RE3 независимо представляет собой алифатический Cz-s алкинил и возможно замещен одним или более -ОН, ORF1, -NH2, -NH RF1,

каждый -RЕ7 независимо представляет собой фенил и возможно замещен одним или несколькими -F, -Cl, -Br, -I, - RF1, -ОН, -ORF1, -NH2, -NH RF1,
каждый -RE8 независимо представляет собой фуранил, тиенил, пирролил, имидазолил, паразолил, триазолил, оксазолил, изоксазолил, изотиозолил, пиридил, пиразинил, пиримидил, пиридазинил, хинолинил или изохинолинил и возможно замещен одним или более -F, -Cl, -Br, -I, - RF1, -ОН, -ORF1, -NH2, -NH RF1,
каждый -L3 независимо представляет собой насыщенный алифатический С1-3алкилен;
где каждый -RF1 независимо представляет собой насыщенный алифатический С1-4алкил, фенил или бензил; и
каждая группа -NRN3RN4 независимо представляют собой пирролидино, пиперидино, пиперизино или морфолино и возможно замещена одним или более C1-3алкилом.
каждый из -RPW, -RPX, -RPY и -RPZ представляет собой -Н.


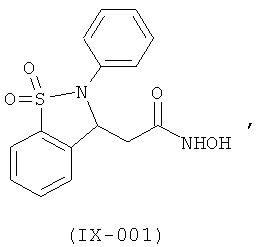
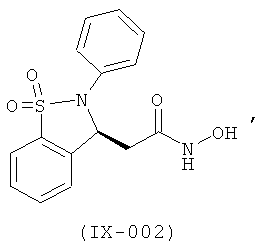



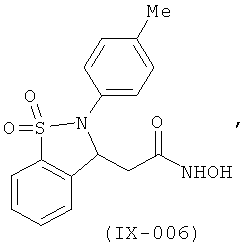




















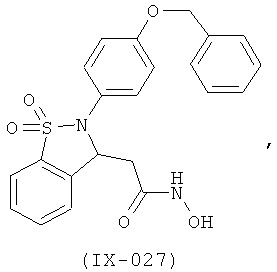




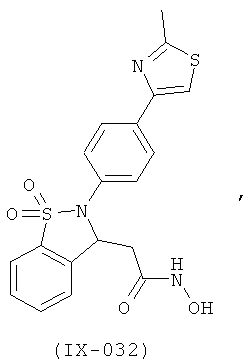








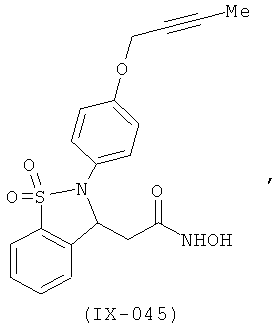



























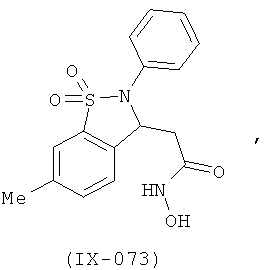





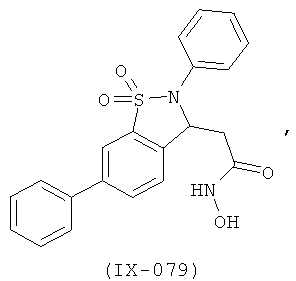














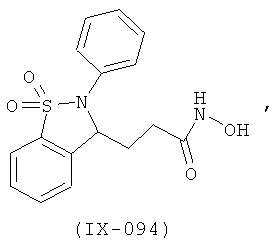

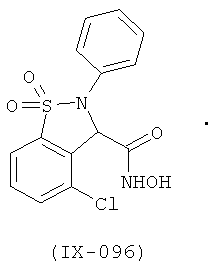
ревматоидного артрита; воспаления; псориаза; септического шока; отторжения трансплантата; кахексии; анорексии; застойной сердечной недостаточности; постишемической реперфузионной травмы; воспалительного заболевания центральной нервной системы; воспалительной болезни кишечника; резистентности к инсулину; инфекции ВИЧ (вируса иммунодефицита человека); рака; хронического обструктивного заболевания легких (COPD) или астмы; остеоартрита, язвенного колита, болезни Крона, рассеянного склероза или дегенеративной потери хряща, которая содержит соединение по любому из пп.1-31 и фармацевтически приемлемый носитель, разбавитель или эксципиент.
ревматоидного артрита; воспаления; псориаза; септического шока; отторжения трансплантата; кахексии; анорексии; застойной сердечной недостаточности; постишемической реперфузионной травмы; воспалительного заболевания центральной нервной системы; воспалительной болезни кишечника; резистентности к инсулину; инфекции ВИЧ (вируса иммунодефицита человека); рака; хронического обструктивного заболевания легких (COPD) или астмы; остеоартрита, язвенного колита, болезни Крона, рассеянного склероза или дегенеративной потери хряща.
ревматоидного артрита; воспаления; псориаза; септического шока; отторжения трансплантата; кахексии; анорексии; застойной сердечной недостаточности; постишемической реперфузионной травмы; воспалительного заболевания центральной нервной системы; воспалительной болезни кишечника; резистентности к инсулину; инфекции ВИЧ (вируса иммунодефицита человека); рака; хронического обструктивного заболевания легких (COPD) или астмы; или остеоартрита, язвенного колита, болезни Крона, рассеянного склероза или дегенеративной потери хряща.
ревматоидного артрита; воспаления; псориаза; септического шока; отторжения трансплантата; кахексии; анорексии; застойной сердечной недостаточности; постишемической реперфузионной травмы; воспалительного заболевания центральной нервной системы; воспалительной болезни кишечника; резистентности к инсулину; инфекции ВИЧ (вируса иммунодефицита человека); рака; хронического обструктивного заболевания легких (COPD) или астмы, или остеоартрита, язвенного колита, болезни Крона, рассеянного склероза или дегенеративной потери хряща, включающий введение пациенту, нуждающемуся в лечении, терапевтически эффективного количества соединения по любому из пп.1-31.
Комментарии