Производные полизамещенных 2-амидо-4-фенилтиазолов, способ их получения, промежуточные соединения синтеза и фармацевтическая композиция на их основе - RU2125569C1
Код документа: RU2125569C1
Чертежи



Описание
Настоящее изобретение относится к новым производным тиазола, обладающим биологической активностью, и к применению известных производных тиазола, оно относится к способу их получения и к содержащим их фармацевтическим композициям.
Более конкретно, предметом настоящего изобретения являются новые агонисты рецепторов холецистокинина /ХЦК/ в тесте с панкреатической амилазой.
ХЦК представляет собой пептид, находящийся в мозгу, особенно в коре головного мозга, полосатом теле, гиппокампе, брюшной оболочке, диафрагме и гипоталамусе.
ХЦК также выделяется на периферическом уровне тонкой кишкой; его действие проявляется в стимуляции везикулярного сокращения, повышении желчной секреции, контроле панкреатической ферментативной секреции, воздействии на сокращения желудка, воздействия на кишечную подвижность. В некоторых случаях он также может воздействовать на артериальное давление и влиять на иммунные системы.
ХЦК в некоторых центральных нейронах находится вместе с допамином. Он также принимает участие в механизмах, связанные с ацетилхолином, 4-аминомасляной кислотой, серотонином, опиоидом, соматостатином, веществом P и ионным каналом.
Его введение приводит к физиологическим изменениям, таким как пальцебральный птоз, гипотермия, гипергликемия, каталепсия; и к поведенческим изменениям, таким как гипоподвижность, снижение исследовательской функции, анальгезия изменяет способности к обучению, сексуальное поведение и чувство сытости.
Следовательно, агонисты рецепторов ХЦК могут быть использованы при лечении некоторых расстройств, вызванных питанием, тучности, диабета; при расстройствах эмоционального, сексуального и мнезического поведения; при шизофрении, психозах, болезни Паркинсона и в случае различных расстройств желудочно-кишечной сферы (Drugs of the future, 1992, 17 (3), 197 - 206).
Агонисты рецептора ХЦК описаны в литературе. Например, некоторые продукты, обладающие такими свойствами, описаны в европейском патенте А-0383690, ВОИС 90/06937 и европейском патенте А-0376849.
В европейской патентной заявке А-0432040 описываются ациламинотиазолы, обладающие сродством к рецептору ХЦК А и рецептору ХЦК Б. Некоторые из соединений, заявленных в этой заявке А-0432040, описаны в качестве антагонистов рецепторов ХЦК А и ХЦК Б.
В настоящее время неожиданно найдено, что ряд ациламинотиазолов, некоторые из которых входят в европейский патент А-432040, обладают сильной агонистической активностью по отношению к рецепторам ХЦК и, следовательно, пригодны для приготовления лекарств на основе агонистов ХЦК.
Таким образом, согласно одному из аспектов изобретения предметом изобретения является
использование
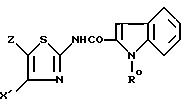
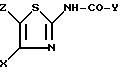
N-тиазол-2-ил-индокарбоксамидов или N-тиазол-2-ил-хинолинкарбоксамида формулы I
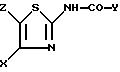
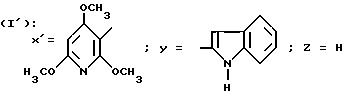
в которой Y обозначает 3-хинолинильную группу или 2-индолильную группу формулы
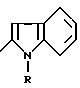
в которой R обозначает водород, ацетильную группу или группу CH2COOR', причем R' - водород или C1-C4-алкил;
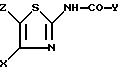
X обозначает (гетеро)арильный радикал, выбираемый из группы, включающей 4-хлор-2,6-диметоксифенильную, 2,6-диметокси-4-метилфенильную, 2,4,5-триметоксифенильную, 4-метил-2,3,6-триметоксифенильную, 2,6-диметокси-4-этилфенильную, 2,4,6-триметокси-5-хлорфенильную, 2,4, 6-триметокси-3-пиридинильную, 2,4-диметокси-6-метил-3-пиридинильную. 6-хлор-2,4-диметокси-5-пиримидинильную, 2,4,6-триметокси-5-пиримидинильную, 5-хлор-2,4-диметоксифенильную: 5-хлор-2-метокси-4-метилфенильную, 2,5-диметокси-4-метилфенильную, 4-трифторметил-2,6-диметоксифенильную, 2,4-диметокси-5-метилфенильную, 5-этил-2,4-диметоксифенильную, 2,4-диметоксифенильную группы;
Z обозначает водород, C1-C4-алкил или бензил, при условии, что Z обозначает обязательно водород, когда X - фенильный радикал, замещенный одновременно в положениях 2 и 6, или когда X обозначает 3-пиридинильный радикал, замещенный одновременно в положениях 2 и 4, или когда X обозначает 5-пиримидинальный радикал, замещенный одновременно в положениях 4 и 6,
а также их фармацевтически приемлемых солей и сольватов, в качестве полного или частичного агониста рецепторов холецистокинина.
Из соединений формулы (I) некоторые не описаны в литературе и, следовательно, составляют другой предмет настоящего изобретения.
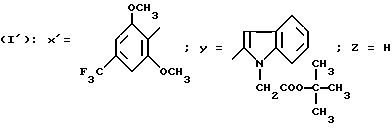
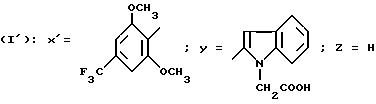
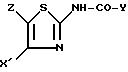
Так, согласно другому из своих аспектов изобретение относится к новым производным ациламинотиазолов
формулы I'
в которой Y обозначает 3-хинолинильную группу (а) или 2-индолильную группу (b) формулы
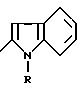
в которой R обозначает водород, ацетильную группу или группу CH2COOR', причем R обозначает водород или C1-C4-алкил;
X' обозначает (гетеро)арильный радикал, выбираемый из группы, включающей 4-хлор-2,6-диметоксифенильную, 2, 6-диметокси-4-метилфенильную, 2,4,5-триметоксифенильную, 4-метил-2,3,6-триметоксифенильную, 2,4,6-триметокси-5-хлорфенильную, 2,4.6-триметокси-3-пиридинильную, 2,4,6-триметокси-5-пиримидинильную, 2, 4-диметокси-6-метил-3-пиридинильную, 6-хлор-2,4-диметокси-5-пиримидинильную, 5-хлор-2,4-диметоксифенильную, 5-хлор-2-метокси-4-метилфенильную, 2,5-диметокси-4-метилфенильную, 4-трифторметил-2, 6-диметоксифенильную, 2,4-диметокси-5-метилфенильную, 5-этил-2,4-диметоксифенильную группы;
Z - водород, C1-C4-алкил или бензил,
при условии, что Z - водород, когда X обозначает фенильный радикал, замещенный одновременно в положениях 2 и 6, или когда X - 3-пиридинильный радикал, замещенный одновременно в положениях 2 и 4, или когда X - 5-пиримидинильный радикал, замещенный одновременно в положениях 4 и 6,
и к их солям и сольватам.
Соли присоединения этих соединений представляют собой соли, получаемые с неорганическими или органическими кислотами или основаниями; предпочтительны нетоксичные, фармацевтически приемлемые соли. Другие соли, используемые для выделения или очистки соединений формулы I' также составляют предмет изобретения.
Особенно предпочтительны соединения формулы I', в которой Y обозначает группу (b), где R представляет собой водород или группу CH2COOH.
Соединения формулы I', в которой Z - водород или метил, особенно предпочтительны.
Также предпочтительны соединения формулы I', в которой Y обозначает радикал (b), где R представляет собой водород или группу CH2COOH, Z - водород или метил, и X' - арильный радикал, выбираемый из группы, включающей 4-хлор-2,6-диметоксифенильную; 5-хлор-2,4-диметоксифенильную, 5-хлор-2-метокси-4-метилфенильную, 2,6-диметокси-4-метильфенильную, 2,4-диметокси-5-метилфенильную и 2,4,5-триметоксифенильную группы (причем Z обязательно обозначает водород, когда X' обозначает 4-хлор-2,6-диметоксифенильную или 2,6-диметокси-4-метилфенильную группу).
Из соединений формулы I' особенно предпочтительны
- N-/4-/4-хлор-2,
6-диметоксифенил/тиазол-2-ил/-1-/карбоксиметил/- индол-2-карбоксамид, его фармацевтически приемлемые соли и сольваты, особенно хлоргидрат;
- N-/4-/5-хлор-2,
4-диметоксифенил/тиазол-2-ил/-1H-индол-2-карбоксамид, его фармацевтически приемлемые соли и сольваты;
- N-/4-/5-хлор-2,4-диметоксифенил/-5-метилтиазол-2-ил/-1H-индол- 2-карбоксамид, его
фармацевтически приемлемые соли и сольваты;
- N-/4-/5-хлор-2,4-диметоксифенил/-5-метилтиазол-2-ил/-1-/карбоксиметил/ -индол-2-карбоксамид, его фармацевтически приемлемые соли и сольваты,
особенно трифторацетат;
- N-/4-/5-хлор-2-метокси-4-метилфенил/тиазол-2-ил/-1H-индол-2- карбоксамид, его фармацевтически приемлемые соли и сольваты;
- N-/4-/2,
6-диметокси-4-метилфенил/тиазол-2-ил/-1H-индол-2- карбоксамид, его фармацевтически приемлемые соли и сольваты, особенно моногидратированный хлоргидрат;
- N-/4-/2,
4-диметокси-5-метилфенил/-5-метилтиазол-2-ил/-1H-индол- 2-карбоксамид, его фармацевтически приемлемые соли и сольваты;
- N-/4-/2,4,5-триметоксифенил/-5-метилтиазол-2-ил/-1H-индол-2
карбоксамид, его фармацевтически приемлемые соли и сольваты.
Предметом изобретения также является способ получения соединений формулы I', отличающийся тем, что кислоту формулы II
Y' = COOH,
в которой Y' - 3-хинолинильный радикал (а)

2-индольльный радикал (в0)
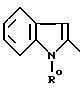
или 2-индолинильный радикал (с0)
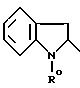
причем R0 обозначает защитную или N-группы или группу CH2COOR'', где R''-C1-C4 -алкил;
или функциональное производное вышеуказанной кислоты
II конденсируют с 2-аминотиазолом формулы III
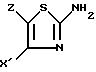
в которой
X' и Z имеют указанные значения,
в присутствии основания, с получением соединения формулы I''
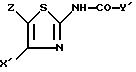
в которой
X' и Z имеют указанные значения;
Y' представляет собой один из радикалов (а), (b0) или (c0),
затем, когда в соединении I'' Y' обозначает радикал (b0), то, при желании, полученный продукт формулы I'' b0
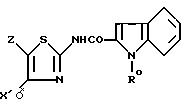
подвергают реакции удаления N-защитной группы, или омылению или кислотному гидролизу; когда в соединении I'' Y' обозначает радикал (c0), то полученный продукт формулы
подвергают дегидрированию, в известных случаях при предварительном удалении N-защитной группы, омыления или кислотном гидролизе, с получением соединения формулы I', в которой Y обозначает радикал (b), где R представляет собой водород или группу CH2COOR', причем Z, X' и R' имеют указанные значения, и выделяют продукт формулы I' таким, какой есть, или в виде одного из его сольватов, или одной из его фармацевтически приемлемых солей.
В качестве функционального производного кислоты II можно использовать саму кислоту, в известных случаях активированную, ее ангидрид, один из ее смешанных ангидридов или ее сложных активированных эфиров.
Конденсация аминотиазола III с кислотой II в виде сложного активированного эфира, полученного, например, путем воздействия I-гидроксибензотриазола на кислоту в присутствии дициклогексилкарбодиимида согласно способу работы; описанному в J. Am. Chem. Soc. 1971, 93, 6318 - 6319, или путем воздействия гексафторфосфата 1-бензотриазолилокси-трисдиметиламинофосфония (БОФ) согласно методике, описанной в Synthesis, 1976, 751-752, может быть осуществлена в растворителе, природа которого выбирается в зависимости от растворимости соединений и типа активации кислотной функции, предпочтительно в присутствии основания, например, как третичный амин, такой как триэтиламин; реакция обычно осуществляется при температуре 0 - 30oC.
На первой стадии способа согласно изобретению получают соединение I'', где Z, X' и Y' имеют указанные значения. Когда в соединении формулы I'' Y' обозначает либо радикал (а), либо радикал (b0), в котором R0 представляет собой группу CH2COOR'', то указанное соединение также может представлять собой целевой продукт формулы I', где Y обозначает либо радикал (а), либо радикал (b), в котором R представляет собой группу CH2COOR', где R' - C1-C4-алкил, причем Z и X' имеют указанные значения.
Когда в соединении формулы I'' Y' обозначает группу (b0), в которой R0 представляет собой N-защитную группу или группу CH2COOR'', то указанное соединение может быть подвергнуто операции удаления N-защитной группы с получением соединений формулы I', в которой Y обозначает группу (b), где R - водород, или оно может быть подвергнуто омылению или кислотному гидролизу для получения соединения формулы I', в которой Y обозначает группу (b), где R представляет собой группу CH2COOH.
Когда в соединении формулы I'' Y' обозначает группу (c0), то указанное соединение подвергают операциям удаления защитных групп, к которым добавляется дегидрирование.
Кислоты Y'COOH, у которых R0 в радикалах (b0) и (c0) обозначает защитную ацильную группу, такую как ацетил, могут быть получены путем воздействия ацетилхлорида или уксусного ангидрида, например, на Y'COOH, в котором R0 = H, и в присутствии эквивалента триэтиламина или 4-диметиламинопиридина, например, в дихлорметане.
Когда функциональное производное кислоты II представляет собой смешанный ангидрид, то его можно получать путем воздействия алкилхлорформиата на кислоту, в присутствии основания, обычно третичного амина, такого как триэтиламин; эта реакция наиболее часто осуществляется в растворителе, таком как дихлорметан, дихлорэтан или хлороформ.
Когда хотят получить индол-2-карбоксамид формулы I', где R - водород, то R0 обозначает N-защитную группу в радикалах (b0) и (c0).
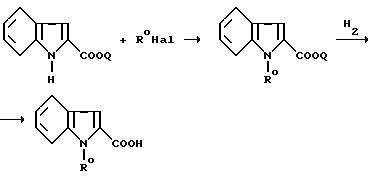
Так, производные I', в которых Y обозначает
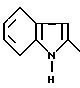
могут быть получены из соединений, получаемых конденсацией аминотиазола III с функциональным производным индол-2-карбоновой кислоты формулы
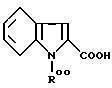
в которой R00 обозначает N-защитную группу, обычно используемую для защиты NH2-групп в реакциях конденсации аминокислот, такую как - COOC(CH3)3; - COOCH2C6H5; -CO-CH3; N-защитная группа затем может быть удалена при использовании классических методов удаления защитных групп.
Те же самые соединения также могут быть получены из соединений, получаемых путем конденсации
аминотиазола III с производными
индолин-2-карбоновой кислоты формулы
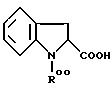
в которой R00 обозначает N-защитную группу, такую как -COOC(CH3)3,
для получения соединения формулы IV
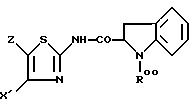
причем группа R00 может быть удалена из соединения IV путем воздействия сильной кислоты в безводной среде, такой как трифторуксусная кислота в дихлорметане или соляная кислота в диэтиловом эфире.
Таким образом полученное соединение затем дегидрируют.
Реакцию осуществляют путем воздействия на индолиновый остаток классических дегидрирующих реагентов, таких как 2,3,5,6-тетрахлор-1,4-бензохинон, 2,3-дихлор-5,6-дициано-1,4-бензохинон (ДДХ) или циклогексил, в присутствии палладия, в инертных высококипящих растворителях, таких как дифениловый эфир, ксилол 1, 2-диметоксиэтил или 2-метоксиэтиловый эфир, при повышенной температуре, и предпочтительно при температуре кипения с обратным холодильником растворителя.
Когда защитная группа, обозначается R00, представляет собой ацетил, то также можно получать группу R заместителя Y целевого продукта формулы I.
Гидролиз сложного C1-C4 -алкилового эфира группы R0 для получения продуктов формулы I', где Y обозначает CH2COOH, осуществляют либо в кислой среде, либо предпочтительно в основной среде, например, путем воздействия неорганического основания, такого как гидроксид щелочного металла, в водно-спиртовой среде.
Аминотиазолы формулы 2-амино-4-/2,4,5-триметоксифенил/-5-метилтиазола и 2-амино-4-/2,4,5-триметоксифенил/-тиазола описаны в Rev. Latinoam. Quim, 1990, 21 (3-4), 102-105.
Аминотиазолы формулы III'

в которой X'' обозначает (гетеро)арильный радикал, выбираемый из группы, включающей 4-хлор-2, 6-диметоксифенил, 2,6-диметокси-4-метилфенил, 4-метил-2,3,6-триметоксифенил, 2,4,6-триметокси-5-хлорфенил, 2,4,6-триметокси-3-пиридинил, 2,4,6-триметокси-5-пиримидинил, 2, 4-диметокси-6-метил-3-пиридинил, 6-хлор-2,4-диметокси-5-пиримидинил, 5-хлор-2,4-диметоксифенил, 5-хлор-2-метокси-4-метилфенил, 2,5-диметокси-4-метил-фенил, 4-трифторметил-2,6-диметокси-фенил, 2, 4-диметокси-5-метилфенил, 5-этил-2,4-диметоксифенил;
Z обозначает водород, C1-C4-алкил или бензил, при условии, что Z - водород, когда X'' обозначает фенильный радикал, замещенный одновременно в положениях 2 и 6, или когда X'' обозначает 3-пиридинильный радикал, замещенный одновременно в положениях 2 и 4, или когда X'' - 5-пиримидинильный радикал, замещенный одновременно в положениях 4 и 6,
являются новыми и составляют часть изобретения.
Из соединений формулы III' особенно предпочтительны -2-амино-4-[4-хлор-2,
6-диметоксифенил]-тиазол;
2-амино-4-[5-хлор-2,4-диметоксифенил] -тиазол;
2-амино-4-[5-хлор-2,4-диметоксифенил]-5-метилтиазол; 2-амино-4-[5-хлор-2-метокси-4-метилфенил]-тиазол; 2-амино-4-[2,
6-диметокси-4-метилфенил] -тиазол; 2-амино-4-[2,
4-диметокси-5-метилфенил]-5-метилтиазол.
Их можно получать по одному из описанных способов, особенно в Bull. Soc. Chim. (C), 1963, 2498 - 2503.
Обычно тиомочевину
вводят во взаимодействие с альфа-галогенированным кетоном, и предпочтительно с альфа-хлорированным кетоном, согласно следующей реакционной схеме I
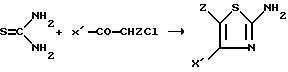
причем X' и Z имеют указанное значение.
Кетоны (Y) могут быть получены, например:
(1) путем реакции
Фриделя-Крафтса:

(2) путем реакции с использованием литийорганического соединения:
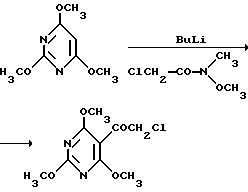
согласно европейскому патенту А-0432040.
Аминотиазолы III также могут быть получены в одну стадию, используя реакцию Hoesch (согласно Dubois. Organic Reactions, 1949, 5, 387, или согласно Satchell и др., The Chemistry of the Carbonyl Group изд. S.Patai, Interscience 1966, I, 5, 233 - 302) в отношении замещенного бензольного производного, с последующей циклизацией, с тиомочевиной.
Аминотиазолы III также
могут быть получены в одну стадию из ароматических кетонов согласно следующей реакционной схеме 2:
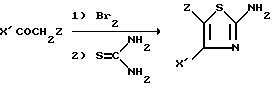
Исходные ароматические кетоны формулы VII получают путем реакции Фриделя-Крафтса из производных X'H. Производные X'H известны или получаются известными способами.
Некоторые кислоты Y'COOH известны и даже имеются в продаже; другие кислоты получаются при использовании способов, известных для молекул аналогов. Все они проиллюстрированы в европейском патенте А-0432040.
Так, индол-2-карбоновые кислоты формулы
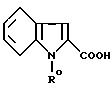
в которой R0 обозначает алкоксикарбонилметильную группу с 1-4 C-атомами, могут быть получены из имеющихся в продаже или получаемых классическими способами согласно нижеприведенной схеме 3 индол-2-карбоновых кислот
в которой Hal обозначает атом галогена;
Q обозначает бензильную группу.
Исходные сложные бензоловые эфиры схему 3 получают путем воздействия соответствующей кислоты на бензиловый спирт в присутствии одного из активаторов кислотной функции, обычно используемых в синтезе пептидов и таких как описанные в европейском патенте А-0432040.
Соли соединения формулы I' с органическими или неорганическими кислотами или основаниями получают обычным образом путем введения кислоты или основания в раствор соединения формулы I'. Соль выделяют согласно ее характеристикам растворимости после выпаривания растворителя или добавления нерастворителя (осадителя).
Согласно другому из аспектов изобретения предметом изобретения также являются фармацевтические композиции, содержащие вышеуказанные соединения формулы I'.
Более конкретно,
соединения формулы I являются объектом исследований связывания ин витро, относящихся к рецепторам ХЦК,
Изучение агонистического воздействия
соединений на выделение амилазы реализуется
следующим образом. Панкреатические ацинусы получают путем ферментативного переваривания (коллагенеза) поджелудочной железы крысы, подвергнутой голоданию в
течение 18 часов. Аликвоты (485 мкм)
инкубируют при 30oC в течение 30 минут в присутствии возрастающих концентраций агониста согласно Jensen и др., J. Biol. Chem. 1982, 257 (10), 5554.
Инкубацию прекращают путем
центрифугирования в течение 15 секунд. Надосадочную жидкость хранят в ледяной бане для измерения содержания амилазы согласно способу Ceska и др., Clin. Chim. Acta. 1969, 26,
437 (phadebas
-реактив):амилазный тест согласно Pharmacia Diagnostic. Испытуемые соединения растворяют в диметилсульфоксиде, затем в буфере для инкубации.
Соединения формулы I ведут себя как агонисты рецепторов ХЦК с ЭД50 (эффективная доза, вызывающая 50% выделения амилазы по сравнению с максимальным эффектом в присутствии ХЦК) около 10-9 М.
Исследование агонистического воздействия в отношении ХЦК соединений на потребление пищи реализуется следующим образом. Самцов крыс (200-240 г) Sprague Dawley (Charles River, Франция) изолируют за 10 дней до эксперимента и каждый день последовательно подвергают 18 часов голоданию и 6 часов голоданию и 6 часов кормлению: пища доступна от 10 до 16 часов, вода доступна по желанию. В день эксперимента, интраперитонально вводят продукты ( в виде суспензии в 0,6%-ном растворе метилцеллюлозы) или эксципиент. Спустя 30 минут после обработки (в 10 часов) в клетку вводят известное количество корма: измеряют потребление пищи спустя 1 час и 3 часа.
Соединения формулы I уменьшают прием пищи и, следовательно, ведут себя как агонисты рецепторов ХЦК (Gibbs J. и др.,
J.Comp. Physiol.
Psychol., 1973, 84, 488-495), в особенности:
N-/4-/5-хлор-2,4-диметоксифенил/тиазол/2-ил-/1H-индол-2- карбоксамид;
N-/4-/5-хлор-2,
4-диметоксифенил/-5-метилтиазол-2-ил/-1H-индол- 2-карбоксамид,
N-/-4/5-хлор-2,4-диметоксифенил/-5-метилтиазол-2-ил/-1-/ карбоксиметил/-индол-2-карбоксамид-трифторацетат,
N-/4-/5-хлор-2-метокси-4-метилфенил/-тиазол-2-ил/-1H-индол-2- карбоксамид,
являются активными в дозе 3 мг/кг, дозе, при которой они уменьшают потребление пищи на 30 - 40% по сравнению с
контрольным животным.
Следовательно, соединения формулы I могут использоваться, в качестве агонистов рецепторов ХЦК, предназначенных для борьбы с заболеваниями, лечение которых требует стимуляции путем полного или частичного агонизма рецепторов холециктокина.
Соединения формулы I мало токсичны; их токсичность совместима с их использованием в качестве фармацевтических композиций.
Новые соединения формулы I' могут быть сформулированы в виде фармацевтических композиций для введения млекопитающим, включая человека, для лечения заболеваний, в которых задействованы рецепторы ХЦК.
Дозировка изменяется в зависимости от лечения и причины болезненного состояния, и может составлять, например, 0,05 - 100 мг в день для взрослого перорально.
Фармацевтические композиции содержат в качестве действующего начала одно из вышеуказанных соединений. Эти композиции выпускаются в форме удобной для введения их пищеварительным путем или парентерально.
В фармацевтических композициях для орального, подъязычного, подкожного, внутримышечного, внутривенного, чрескожного, локального или ректального введения активный ингредиент может содержаться в терапевтически эффективной дозе вместе с классическими фармацевтическими носителями. Соответствующие единичные формы введения могут выпускаться для перорального введения, такие как таблетки, желатиновые капсулы с лекарством, порошки, гранулы или в виде оральных растворов или суспензий; формы для подъязычного и орального введения формы для подкожного, внутримышечного, внутривенного, внутриглазного или внутриносового введения и формы для ректального введения.
Когда готовят твердую композицию в форме таблеток, то действующее начало смешивают с фармацевтическим эксципиентом, таким как желатина, крахмал, лактоза, стеарат магния, тальк, гуммиарабик или аналогичные вещества. Таблетки можно покрывать сахарозой или другими соответствующими материалами или можно их обрабатывать таким образом, чтобы они обладали пролонгированной или замедленной активностью и непрерывно высвобождали заранее определенное количество действующего начала.
Формулировку в виде желатиновых капсул с лекарством получают путем смешения активного ингредиента с разбавителем и путем внесения полученной смеси в мягкие или жесткие желатиновые капсулы.
Формулировка в форме сиропа или эликсира может содержать действующее начало вместе с подслащивающим веществом, предпочтительно некалорийным, метилпарабеном и пропилпарабеном в качестве антисептических средств, или с придающим вкус агентом и соответствующим красителем.
Диспергируемые в воде порошки или гранулы могут содержать действующее начало в смеси с диспергаторами или смачивателями, или с суспендирующими агентами, как поливинилпирролидон, точно так же как с подслащивающими веществами или улучшающими вкус веществами.
Для ректального (кишечного) введения прибегают к свечам, которые готовят со связующими, плавящимися при ректальной температуре, например, как масло какао или полиэтиленгликоли.
Для парентерального, внутриносового или внутриглазного введения используют водные суспензии, солевые изотонические растворы или стерильные растворы, которые содержат фармакологически приемлемые диспергаторы и/или смачиватели, например, как пропиленгликоль или бутиленгликоль.
Действующее начало может быть сформулировано также в виде микрокапсул, в известных случаях с одним или несколькими носителями или добавками.
Действующее начало также может находиться в форме комплекса с циклодекстрином, например, как α-, β - или γ - циклодекстрин, 2-гидроксипропил -β- циклодекстрин или метил -β- циклодекстрин.
Композиция может содержать единичную дозу, в количестве 0,05 - 100 мг действующего начала.
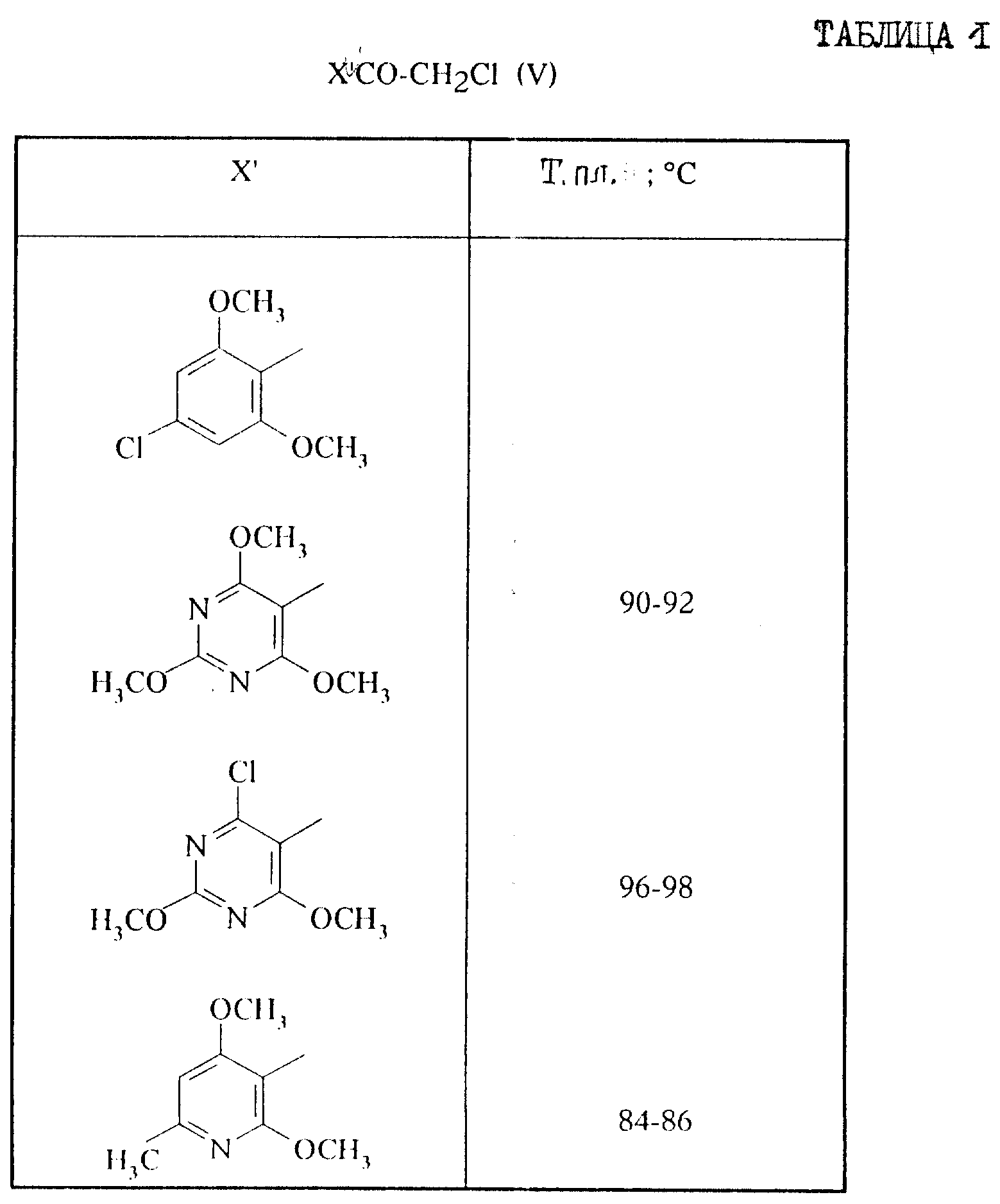
Ниже описываются примеры осуществления изобретения и способы получения некоторых промежуточных соединений синтеза формулы X' H, V, VII, III, и II. Указанные температуры плавления определяются в капилляре. ЯМР-спектр регистрируются по отношению к тетраметилсилану.
В приготовлениях в примерах, используются следующие сокращения: ДМХ - дихлорметан; эфир - диэтиловый эфир; изоэфир - диизопропиловый эфир; CCl4 - четыреххлористый углерод; MeOH - метанол; EtOH -этанол; AcOEt - этилацетат; ДМФ - диметилформамид; ТГФ - тетрагидрофуран; CHCl3 -хлороформ; AlCl3 -хлорид алюминия; ZnCl2 - хлорид цинка; TiCl4 - тетрахлорид титана; HCl -соляная кислота; H2SO4 - серная кислота; ТКФ - трифторуксусная кислота; KHSO4 - гидросульфат калия; NaOH -гидросульфат натрия; диоксид кремния H - силикагель 6OH, выпускаемый в продаже фирмой MER CK (Дармштадт); tBu -третбутил; F - температура плавления; Tкип -температура кипения; ТК - комнатная температура; ЯМР - ядерный магнитный резонанс; с. - синглет; с.ш. - уширенный синглет; м. - массив.
Приготовление 1. Соединения X'H.
А. 2,4,6-Триметоксипиримидин.
Это соединение получают согласно методике, описанной в J. Am. Chem. Soc. 1932, 54, 727-733.
Б. 2,4-Диметокси-6-метилпиридин.
Сначала получают 1,2-дигидро-4-гидрокси-6-метил-2-оксопиридин согласно методике, описанной в J.Heterocycl. Chem 1975, 12 (5), 963-967.
Смесь 7,51 г вышеполученного соединения и 75 мл оксихлорида фосфора нагревают при 120oC в течение 2,5 часов. Оставляют на ночь при комнатной температуре, реакционную смесь выпаривают в вакууме, остаток поглощают льдом, добавляют насыщенный раствор гидрокарбоната натрия до pH 10, экстрагируют эфиром, сушат над сульфатом натрия и выпаривают растворитель в вакууме. Получают 9,7 г 2,4-дихлор-6-метилпиридина в виде масла.
В течение 36 часов при температуре 130 - 140oC, в реакторе, под давлением 5 бар, нагревают смесь 9,7 г вышеполученного соединения, 7,13 г метилата натрия в 15 мл метанола. После охлаждения, добавляют 200 мл эфира, отфильтровывают и фильтрат выпаривают в вакууме. Хроматографируют на диоксиде кремния, элюируя ДХМ. Получают 4 г монометоксилированного продукта, который снова водят в реакцию. В течение 20 часов при температуре 132-140oC, в реакторе под давлением 5 бар, нагревают смесь 4 г вышеполученного продукта с раствором метилата натрия, полученным из 0,7 г натрия и 15 мл метанола. После охлаждения, добавляют 200 мл эфира, отфильтровывают и фильтрат выпаривают при атмосферном давлении. Остаток перегоняют в вакууме и получают 2,5 г целевого продукта. Ткип = 101-103oC при 0,02 бара.
В. 1-Хлор-2,4-диметоксибензол
К раствору 20 г
4-хлоррезорцина в 200 мл этанола добавляют 82,6 г 50 вес. %-ного раствора гидроксида цезия в воде. Выпаривают в вакууме, остаток обрабатывают изопропанолом, снова выпаривают в вакууме и эту операцию
повторяют 3 раза. Таким образом полученную соль цезия растворяют в 100 мл ДМФ, добавляют 40 мл метилиодида и нагревают при 80oC в течение 3 часов. Реакционную смесь выпаривают в вакууме,
остаток поглощают ДХМ, промывают насыщенным раствором гидрокарбоната натрия, сушат над сульфатом магния и растворитель выпаривают в вакууме. Хроматографируют на диоксане кремния, элюируя толуолом.
Перегоняют в вакууме и получают 13 г целевого продукта. Ткип=138oC при 0,02 бара.
г. 2-хлор-5-метокситолуол
К раствору 20 г 4-хлор-3-метилфенола в 200
мл
этанола добавляют 42,05 г 50 вес.%-ного водного раствора гидроксида цезия. Выпаривают в вакууме, остаток поглощают изопропанолом, снова выпаривают в вакууме и эту операцию повторяют 3 раза. Таким
образом полученную соль цезия растворяют в 100 мл ДМФ, добавляют 3- мл метилиодида и нагревают при 80oC в течение 3 часов. Реакционную смесь выпаривают в вакууме, остаток поглощают ДХМ,
промывают водой, насыщенным раствором карбоната натрия, сушат над сульфатом магния и растворитель выпаривают в вакууме. Хроматографируют на диоксиде кремния, элюируя толуолом. Перегоняют в вакууме и
получают 14 г целевого продукта. Ткип=105oC при давлении 0,02 бара.
Д. 2,5-Диметокситолуол.
Смесь 12 г метилгидрохинона, 45 г карбоната калия, 45 г диметилсульфата в 300 мл безводного ацетона кипятят с обратным холодильником в течение 4 дней. После охлаждения реакционную смесь отфильтровывают и выпаривают фильтра в вакууме. Остаток поглощают 150 мл концентрированного раствора аммиака, перемешивают 2 часа, разбавляют водой, экстрагируют ДХМ, сушат над сульфатом магния и выпаривают растворитель в вакууме. Остаток хроматографируют на диоксане кремния H, элюируя смесью гептана с ДХМ (50:50 по объему). Получают 12 г целевого продукта. ЯМР-спектр (200 МГц, DMCO), δ : 2,05 (с., 3H), 3,60 (с.,3H), 3,65 (с., 3H), 6,5 - 6,9 (м., 3H).
Приготовление II. Альфа-хлоркетоны формулы V.
А. 1-(2,6-Диметокси-4-метилфенил)-2-хлор-1-этанон.
7,61 г 3,5-Диметокситолуола и 6,10 г тетраметилэтилендиамина растворяют, в атмосфере азота, в 150 мл гексана. Раствор охлаждают до 0oC, добавляют 32,8 мл 1,6 М раствора бутиллития в гексане, смесь перемешивают при 10oC в течение 20 минут, затем при 20oC в течение 1 часа. К охлажденному до -10oC производному лития в течение 20 минут добавляют охлажденный до 0oC раствор 6,13 г N-метокси-N-метилхлорацетамида в 50 мл ТГФ. Реакционную смесь оставляют на 1 час при температуре 0 - 5oC, на 1 час при 20oC, затем выливают в 100 мл воды. Экстрагируют 2 раза с помощью 300 мл диэтилового эфира, эфирные фазы промывают насыщенным раствором хлорида натрия, органические фазы сушат над сульфатом магния, отфильтровывают и концентрируют в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью ДХМ/гексан (50: 50 по объему). Концентрирование чистых фракций дает 1,6 г целевого продукта. Т.пл. = 82 - 84oC.
Б. 1-(2, 4,6-Триметокси-3-пиридинил)-2-хлор-1-этанон.
(согласно Chem. Pharm. Bull 1986, 34, 3658 и J. Am. Chem. Soc. 1932, 54, 727)
24 г 2,6-Дихлорпиридина, 200 мл трифторуксусной
кислоты и 28 мл 33%-ного пероксида водорода нагревают при 100oC в течение 4 часов. Охлаждают, затем добавляют 600 мл воды и концентрируют в вакууме до объема 50 - 100 мл. Подщелачивают с
помощью гидрокарбоната натрия, затем экстрагируют с помощью ДХМ, органическую фазу декантируют и сушат над сульфатом натрия. Отфильтровывают, концентрируют в вакууме и остаток перекристаллизуют из
этилацетата с получением 18,8 г 2,6-дихлорпиридин-N-оксида; т.пл. = 138 - 140oC.
18,8 г вышеполученного соединения кипятят с обратным холодильником в течение 6 часов в 40 мл оксихлорида фосфора и оставляют на ночь при комнатной температуре, затем смесь концентрируют в вакууме. Остаток выливают в холодную воды, затем последовательно нейтрализуют карбонатом натрия, экстрагируют эфиром, эфирную фазу декантируют, сушат ее над сульфатом натрия, отфильтровывают и концентрируют в вакууме. Остаток хроматографируют на силикагеле, элюируя смесью ДХМ с гептаном (60:40 по объему). Концентрирование чистых фракций дает 14,6 г 2,4,6-трихлорпиридина.
Смесь 14,6 г вышеполученного продукта и 129,7 г метилата натрия в 400 мл метанола кипятят с обратным холодильником в течение ночи. Добавляют 0,7 л воды, затем, последовательно, экстрагируют с помощью ДХМ, органический экстракт промывают водой и сушат над сульфатом натрия. Концентрируют в вакууме и остаток перекристаллизуют из пентана, получая 9,5 г 2,4,6-триметоксипиридина. Т.пл. = 47 - 49oC.
В атмосфере азота, при -40oC, добавляют к 15 мл безводного ТГФ 7, 5 мл 1,6 М раствора метиллития в эфире, 0,02 мл диизопропиламина, после чего перемешивают в течение 5 минут и при -40oC, в течение 10 минут, добавляют 1,13 г вышеполученного производного пиридина в виде раствора в 10 мг ТГФ. Смесь перемешивают в течение 3 часов при 0oC. Затем охлаждают до -70oC и в течение 5 минут добавляют 0,824 г N-метил-N-метоксихлорацетамида в виде раствора в 20 мл ТГФ и оставляют на час до повышения температуры вплоть до 10oC. Реакционную смесь выливают в 300 мл холодной воды, насыщенной хлоридом натрия, и экстрагируют эфиром. Органический экстракт, последовательно промывают насыщенным раствором хлорида натрия, декантируют, сушат над сульфатом натрия, отфильтровывают и концентрируют в вакууме. Остаток хроматографируют на силикагеле, элюируя смесью циклогексана с этилацетатом (80:20 по объему). Концентрирование фракций чистого продукта дает 0,61 г целевого этанона; т. пл. = 85 - 87oC.
В. 1-(2, 4-Диметокси-5-метилфенил)-2-хлор-1-этанон.
Это соединение получают согласно Chem. Pharm. Bull.1991, 39, (9) 2400-2407.
Суспензию 5,24 г AlCl3, 0,52 г ZnCl2 в 40 мл 1,2-дихлорэтана охлаждают до 0oC и прикапывают раствор 5,0 г 2,4-диметокситолуола в 20 мл 1,2-дихлорэтана. Затем охлаждают до - 10oC и прикапывают раствор 2,9 мл хлорацетилхлорида в 1,5 мл 1,2-дихлорэтана, поддерживая температуру реакционной среды при от -10oC до 7oC. Оставляют стоять при перемешивании до повышения температуры до комнатной, реакционную смесь выливают в смесь льда с концентрированной HCl, экстрагируют с помощью ДХМ, объединенные органические фазы промывают водой, сушат над сульфатом и выпаривают растворители в вакууме. Остаток обрабатывают гептаном и отфильтровывают образовавшийся осадок. Получают 3,0 г целевого продукта. Т.пл. = 166 - 167oC.
Г 1-(4-Трифторметил-2, 6-диметоксифенил)-2-хлор-1-этанон.
Раствор 9,73 г 3-амино-5-метокси-1-трифторметилбензола в 400 мл 2 н. HCl охлаждают до 10oC и в течение 10 минут добавляют раствор натрия 3, 80 г нитрата в 20 мл воды. Оставляют на 30 минут при перемешивании при 10oC и добавляют раствор 800 мл концентрированной серной кислоты в 800 мл воды, поддерживая температуру ниже 20oC. Затем в течение 2 часов нагревают при 95oC и оставляют на ночь при комнатной температуре. Добавляют 1000 г льда к реакционной смеси, экстрагируют эфиром, промывают насыщенным раствором хлорида натрия, сушат над сульфатом натрия и выпаривают растворитель в вакууме. Получают 9,8 г 3-гидрокси-5-метокси-1- трифторметилбензола. Т. пл.= 75oC (согласно J.Chem. Soc., 1951, 2013).
К раствору 9,8 г вышеполученного соединения в 100 мл ацетона добавляют 7,90 г карбоната калия и нагревают до 50oC. Затем прикапывают, при этой температуре и в течение 20 минут, 6,74 г диметилсульфата и кипятят с обратным холодильником в течение 2 часов. Реакционную смесь выпаривают в вакууме, остаток обрабатывают с помощью 30 мл 20%-ного раствора аммиака и 50 мл воды, экстрагируют эфиром, промывают насыщенным раствором хлорида натрия, сушат над сульфатом, натрия и растворитель выпаривают в вакууме. Получают 8, 7 г 3,5-диметокси-1-трифторметилбензола, после вакуумной перегонки. Tкип.= 92 - 94oC при давлении 0,02 бара.
К раствору 8,6 г вышеполученного соединения в 100 мл гексана добавляют 5,09 г тетраметилэтилендиамина. Охлаждают до -5oC и, в атмосфере азота и в течение 15 минут, добавляют 27,4 г 1,6 М раствора бутиллития в гексане, затем оставляют при перемешивании в течение 1,5 часов при температуре от -5oC до +5oC. После этого добавляют раствор литиевого производного к охлажденному до -25oC раствору 5,41 г N-метоки-N-метилхлорацетамида в 45 мл ТГФ и оставляют на 2 часа при перемешивании вплоть до повышения температуры до +5oC. Добавляют 100 мл воды, экстрагируют эфиром, промывают насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Получают 3,6 г целевого продукта, после кристаллизации из гексана. Т. пл.= 120 - 122oC.
Хлорсодержащие кетоны формулы V,
описанные в таблице I, получают согласно одному из вышеприведенных способов осуществления и используя соответствующие исходные продукты:
Приготовление III. Ароматические кетоны формулы
VII
А. 1-(5-Хлор-2,4-диметоксифенил)-1-этанон.
Смесь 2 г 1-хлор-2,4-диметоксибензола, 0,9 г ацетилхлорида в 20 мл CCl4 охлаждают до 0oC и прикапывают раствор 1,3 мл TiCl4 в 7 мл CCl4. Оставляют при перемешивании в течение 2 часов до повышения температуры до комнатной. Реакционную смесь выливают в смесь концентрированной HCl со льдом, экстрагируют с помощью ДХМ, сушат над сульфатом магния и растворители выпаривают в вакууме. Хроматографируют на диоксиде кремния H, элюируя смесью ДХМ с гептаном (70:30 по объему). Получают 1, 19 г целевого продукта. Т.пл. - 138oC.
Б. 1-(5-Хлор-2,4-диметоксифенил)-1-пропанон.
Смесь 2,01 г 1-хлор-2,4-диметоксибензола, 1,08 г пропионилхлорида в 20 мл CCl4 охлаждают до 0oC и прикапывают раствор 1,3 мл TiCl4 в 7 мл CCl4. Оставляют при перемешивании в течение 2 часов до повышения температуры до комнатной. Реакционную смесь выливают в смесь концентрированной HCl со льдом, экстрагируют с помощью ДХМ, сушат над сульфатом магния и выпаривают растворители в вакууме. Хроматографируют на диоксиде кремния H, элюируя смесью ДХМ (80: 20 по объему). Получают 1,14 г целевого продукта. Т. пл. 115oC.
В. 1-(5-Хлор-2-метокси-4-метилфенил)-1-этанон.
Суспензию 2,12 г AlCl3 в 20 мл CCl4 охлаждают до 0oC, в атмосфере азота, и прикапывают раствор 1,25 г ацетилхлорида в 10 мл CCl4. Затем прикапывают раствор 2,5 г 2-хлор-5-метокситолуола в 10 мл CCl4 и оставляют при перемешивании в течение 2 часов до повышения температуры до комнатной. Выливают в смесь концентрированной HCl со льдом, экстрагируют с помощью ДХМ, сушат над сульфатом магния и выпаривают растворитель в вакууме. Хроматографируют на диоксиде кремния H, элюируя смесь ДХМ с гептаном (70:30 по объему). Получают 0,68 г целевого продукта. Т.пл = 83oC.
Г. 1-(5-Хлор-2-метокси-4-метилфенил)-1-пропанон.
Суспензию 2,55 г AlCl3 в 30 мл ДХМ охлаждают до 0oC, в атмосфере азота, и прикапывают раствор 1,77 г пропионилхлорида в 15 мл ДХМ. Затем прикапывают раствор 3 г 2-хлор-5-метокситолуола в 15 мл ДХМ и оставляют при перемешивании на 2 часа. Реакционную смесь выливают в смесь концентрированной HCl со льдом, экстрагируют с помощью ДХМ, сушат над сульфатом магния и растворитель выпаривают в вакууме. Хроматографируют на диоксиде кремния, элюируя смесью ДХМ с гептаном (70:30 по объему). Получают 2,2 г целевого продукта. Tпл.= 79oC.
Д. 1-(5-Этил-2, 4-диметоксифенил)-1-пропанон.
Суспензию 10 г 4-этил-резорцинола в 20 мл эфирата трифторида бора охлаждают до +4oC и прикапывают 11,7 г пропионового ангидрида. Нагревают при 75oC в течение 6 часов и, после охлаждения, реакционную смесь выливают в смесь воды со льдом. Оставляют на 2 часа при перемешивании, отфильтровывают образовавшийся осадок, промывают его водой, обрабатывают его этилацетатом, органическую фазу промывают водой, сушат над сульфатом натрия и выпаривают растворитель в вакууме. Хроматографируют на диоксиде кремния, элюируя с помощь. ДХМ, затем с помощью смеси ДХМ/этилацетат (90:10 по объему). Получают 9,32 г 1-(5-этил-2, 4-дигидроксифенил)-1-пропанона. Т.пл. 74 - 75oC.
В течение 48 часов кипятят с обратным холодильником суспензию 5 г вышеполученного соединения, 30 г карбоната калия, 30 мл карбоната калия, 30 мл диметилсульфата в 500 мл ацетона. После охлаждения, отфильтровывают нерастворимую часть, фильтрат выпаривают в вакууме и остаток поглощают с помощью 100 мл концентрированного раствора аммиака. После перемешивания в течение 1 часа, добавляют 400 мл воды, отфильтровывают образовавшийся осадок, промывают его водой, поглощают его с помощью ДХМ, органическую фазу промывают водой, сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 5,68 г целевого продукта. Т.пл. = 64 - 65oC.
Е. 1-(2, 4-Диметоксифенил)-3-фенил-1-пропанон.
К суспензии 43,2 г AlCl3, 37,5 г 1,3-диметоксибензола в 210 мл CCl4 прикапывают раствор 45,5 г хлорангидрида 3-фенилпропановой кислоты в 50 мл CCl4. Оставляют при перемешивании и при комнатной температуре в течение 1 часа и реакционную среду выливают в смесь 400 г льда со 150 мл концентрированной HCl. После перемешивания в течение 30 минут, экстрагируют с помощью ДХМ, объединенные органические фазы промывают насыщенным раствором гидрокарбоната натрия, сушат над сульфатом натрия и растворители выпаривают в вакууме. Получают 66,5 г целевого продукта в виде масла, который используют таким, какой есть.
Ароматические кетоны формулы VII, описанные в таблице II, получают согласно одному из вышеприведенных способов осуществления и используя соответствующие исходные продукты.
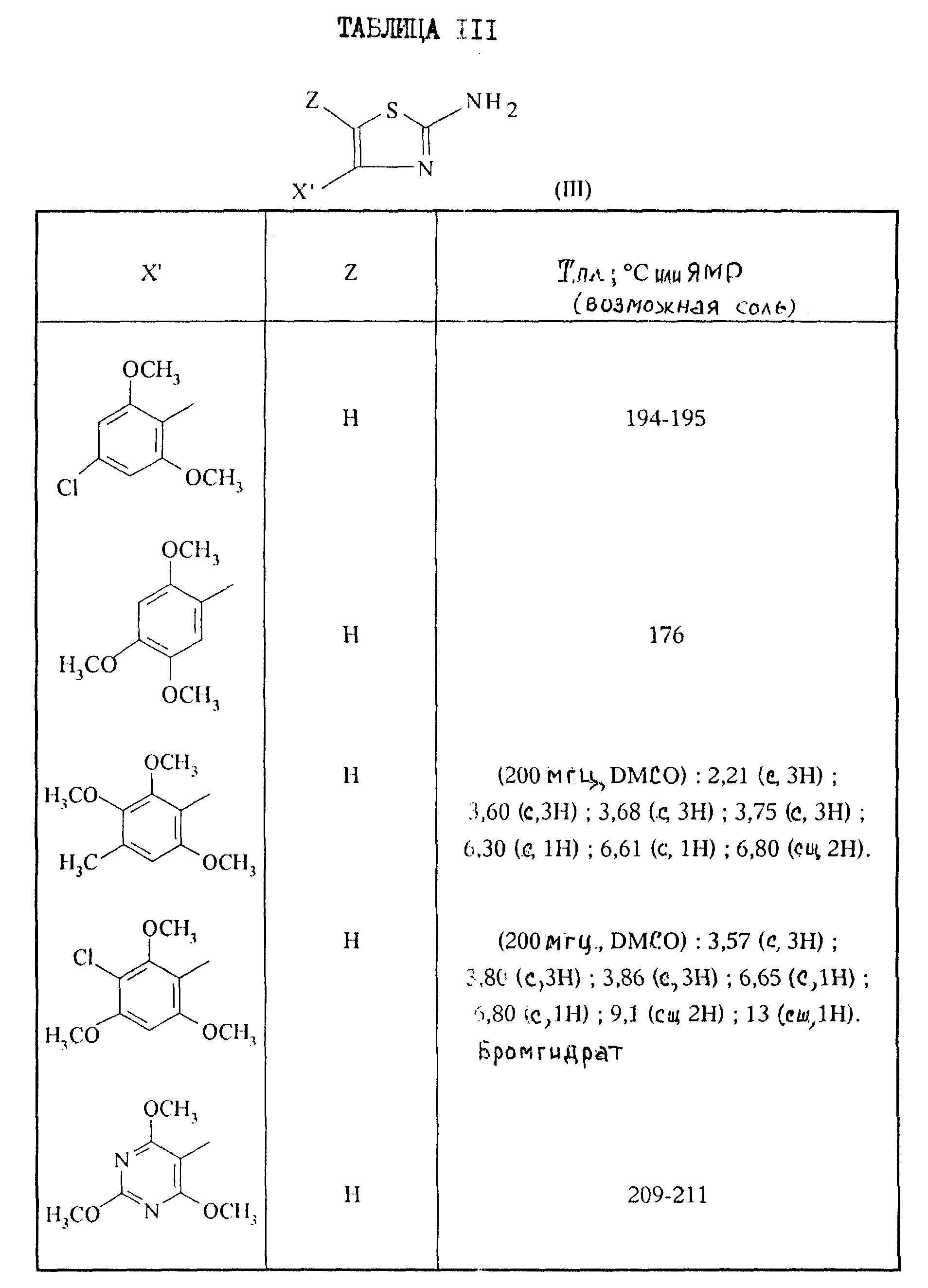
Приготовление IV 2-Аминотиазолы формулы III.
А. 2-Амино-4-(2, 6-диметокси-4-метилфенил)-тиазол.
0,41 г вышеполученного, согласно приготовлению II, А/, продукта и 0, 164 г тиомочевины растворяют в 50 мл абсолютного этанола. Реакционную смесь кипятят с обратным холодильником в течение 18 часов, затем концентрируют в вакууме. Остаток поглощают с помощью 100 мл 2 н. раствора NaOH, затем экстрагируют 2 раза с помощью 200 мл ДХМ, органические фазы декантируют, сушат над сульфатом натрия, отфильтровывают и концентрируют в вакууме. Остаток кристаллизуют из эфира, получая 0,34 г целевого аминотизола. Т.пл. 204 - 206oC.
Б.2-Амино-4-(2,4,6-триметокси-3-пиридил)-тиазол.
Смесь 0,55 г полученного согласно приготовлению II, Б/, кетона, и 0,21 г тиомочевины в 25 мл абсолютного этанола кипятят с обратным холодильником в течение 24 часов. Реакционную смесь концентрируют в вакууме, остаток обрабатывают водой и добавляют 10%-ный раствор карбоната натрия. Экстрагируют этилацетатом, органическую фазу сушат над сульфатом натрия, отфильтровывают и концентрируют в вакууме. Остаток кристаллизуют из минимального количества изо-эфира. Получают 0,51 г целевого тиазола. Т.пл. = 191oC.
В. 2-Амино-4-(5-хлор-2,4-диметоксифенил)-тиазол.
К раствору 1,08 г полученного в приготовлении III, А/, соединения в 20 мл CCl4 прикапывают, при комнатной температуре, раствор 0,26 мл брома в 10 мл CCl4. Органическую фазу промывают водой, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток поглощают с помощью 20 мл этанола, добавляют 2 г тиомочевины и кипятят с обратным холодильником в течение 3 часов. Выпаривают в вакууме, экстрагируют с помощью ДХМ, промывают насыщенным раствором карбоната натрия, сушат над сульфатом магния и выпаривают в вакууме. Хроматографируют на диоксиде кремния, элюируя смесью ДХМ/метанол (100:1 по объему). Получают 0,92 г целевого продукта. Т. пл. =162oC.
Г. 2-Амино-4-(5-хлор-2,4-диметоксифенил)-5-метилтиазол.
К раствору 1,12 г полученного в приготовлении III, Б/, соединения в 20 мл ДХМ, прикапывают, при комнатной температуре, раствор 0, 25 мл брома в 5 мл ДХМ. Органическую фазу промывают водой, сушат сульфатом магния и растворитель выпаривают в вакууме. Остаток поглощают с помощью 20 мл этанола, добавляют 1,0 г тиомочевины и кипятят с обратным холодильником в течение 2 часов. Выпаривают в вакууме, остаток обрабатывают с помощью насыщенного раствора карбоната натрия, экстрагируют с помощью ДХМ, сушат над сульфатом магния и выпаривают в вакууме. Остаток обрабатывают эфиром и отфильтровывают образовавшийся осадок. Получают 1,26 г целевого продукта. Т.пл. = 188oC.
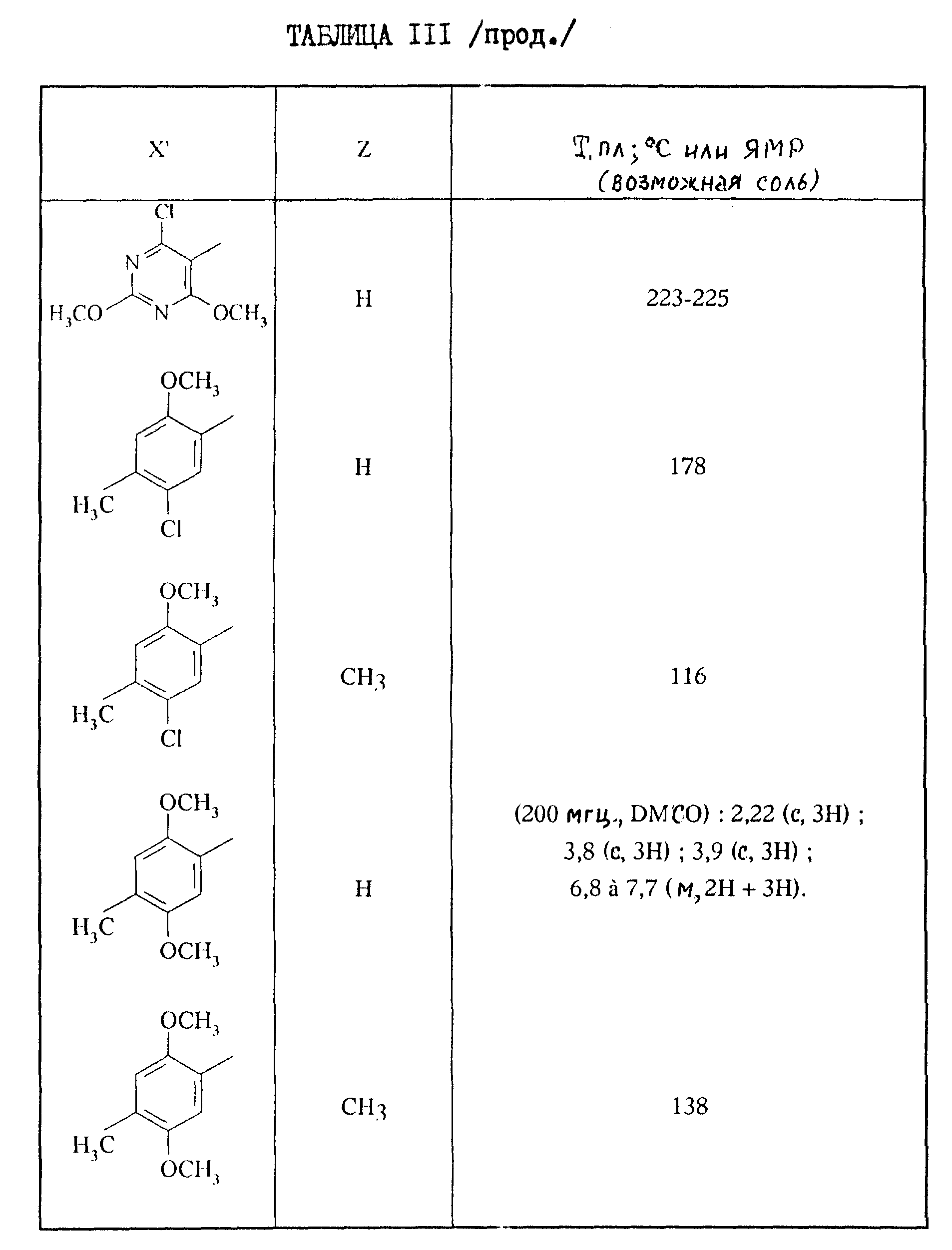
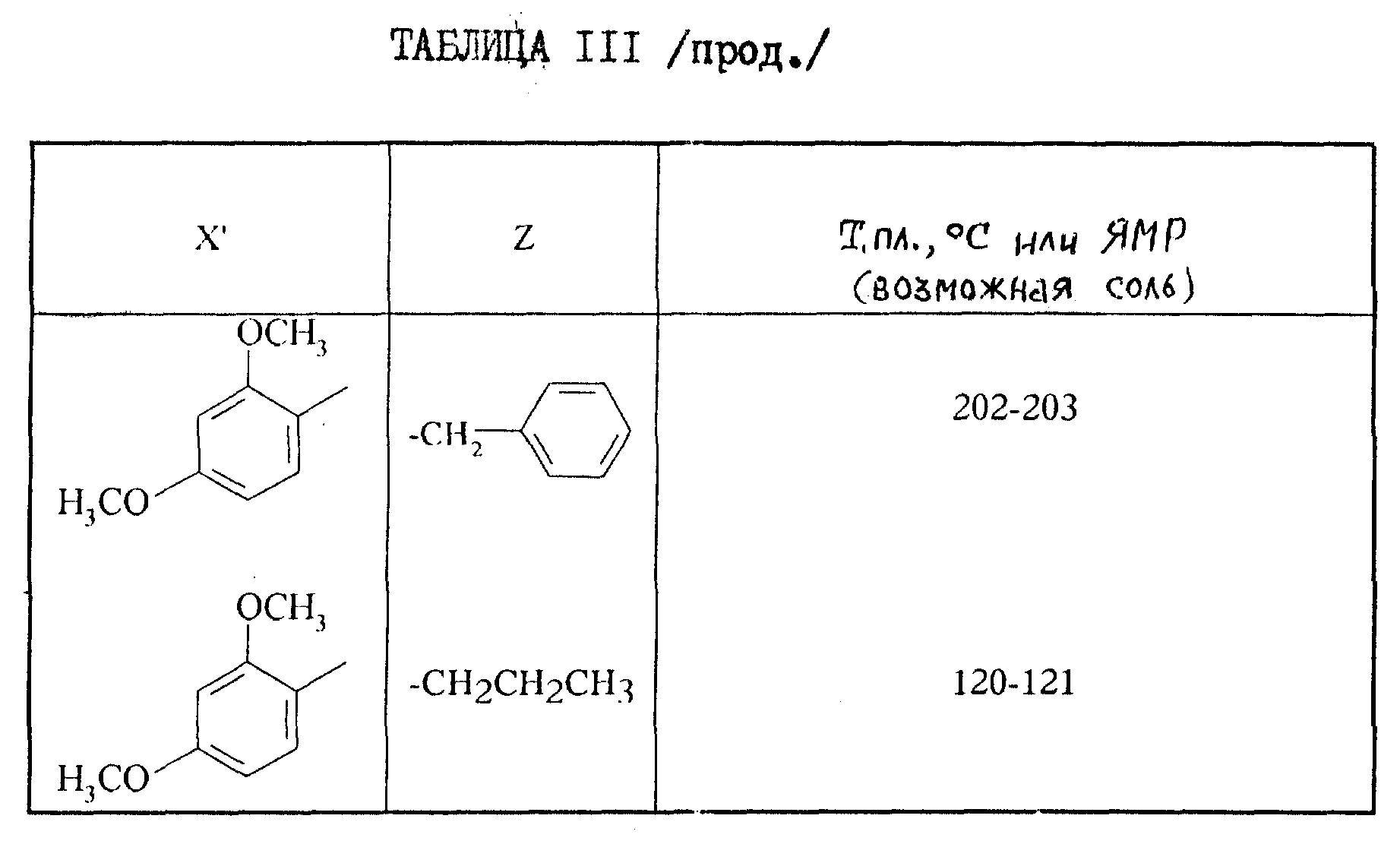
2-Аминотиазолы формулы III, описанные в таблице III, приведенной ниже, синтезируются при применении предыдущих способов.
Приготовление V. Индолкарбоновые кислоты III.
Индолкарбоновые кислоты получают согласно европейскому патенту А-0432040.
Пример 1.
Моногидрат N-/4-(2, 6-диметокси-4-метилфенил)тиазол-2-ил/-1H-индол-2- карбоксамид-хлоргидрата (Способ А).

0,33 г вышеполученного, согласно приготовлению IV, А/, амина, 0,29 г N-ацетилиндол-2-карбоновой кислоты, 0,7 г БОФ и 0,16 г триэтиламина растворяют в 40 мл ДХМ. Реакционную смесь перемешивают в течение 48 часов при комнатной температуре, последовательно добавляют 50 мл буферного раствора с pH 2, органическую фазу декантируют, сушат над сульфатом натрия, отфильтровывают и концентрируют в вакууме. Остаток обрабатывают с помощью 80 мл 96o этанола, добавляют 10 мл 2 н. раствора NaOH и реакционную смесь перемешивают при комнатной температуре в течение 2,5 часов. Раствор нейтрализуют с помощью 1,8 мл концентрированной HCl. Образовавшийся осадок отделяют путем отфильтровывания, промывают водой и сушат в вакууме при 60oC, получая 0,45 г целевого соединения. Т.пл. = 250-252oC.
Пример 2.
N-/-4-(2,4,
6-Триметокси-3-пиридинил)-тиозол-2-ил/ -1H-индол-2-карбоксамид (Способ А)
Перемешивают в течение 24 часов при комнатной температуре раствор 0,5 г аминотиазола, полученного согласно приготовлению IV, Б/, 25 мл ДХМ, 0,40 г N-ацетилиндол-2-карбоновой кислоты, 0,99 г БОФ и 0,23 г триэтиламина. Добавляют 20 мл воды, органическую фазу декантируют, сушат над сульфатом натрия, отфильтровывают и концентрируют в вакууме. Остаток хроматографируют на силикагеле H, элюируя смесью ДХМ с метанолом (100:1 по объему). Сначала удаляют примеси в виде головной части, затем продукт сочетания, соответствующий производному, ацетилированному на индольном азоте. Эти фракции концентрируют в вакууме и остаток растворяют в 50 мл абсолютного этанола. К этому раствору добавляют 5 мл 2 н. раствора NaOH и реакционную смесь перемешивают при комнатной температуре в течение 1,5 часов. Нейтрализуют добавлением 0,85 мл концентрированной HCl и концентрируют в вакууме. Остаток обрабатывают с помощью воды, к которой добавлен карбонат натрия, отфильтровывают и осадок промывают последовательно водой, затем абсолютным эталоном, получая 0,44 г целевого соединения. Т. пл.=285 - 287oC.
Пример 3
N-/-4-(2,
6-Диметокси-4-метилфенил)-тиазол-2-ил/-хинолин-3- карбоксамид (Способ Б).
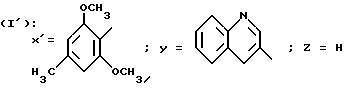
1 г. 2-Амино-4-(2,6-диметокси-4-метилфенил)-тиазола, 0,76 г хинолин-3-карбоновой кислоты, 0,65 мл триэтиламина и 2, 15 г БОФ растворяют в 10 мл ДМФ и оставляют стоять реакционную смесь в течение 48 часов при комнатной температуре. Затем ее выливают в буферный раствор с pH 2, осадок отделяют путем отфильтровывания и желтого цвета твердое вещество последовательно промывают водой, перемешивают в 5%-ном растворе карбоната натрия, отфильтровывают, затем растворяют в ДХМ. Промывают 5%-ным раствором карбоната натрия, затем последовательно органическую фазу декантируют, сушат над сульфатом магния, отфильтровывают и концентрируют в вакууме. Остаток перемешивают в эфире, отфильтровывают и высушивают, получая 1,58 г целевого соединения. Т.пл. = 245 - 246oC.
Пример 4.
N-/4-(4-Хлор-2,6-диметоксифенил)тиазол-2-ил/-1-(карбоксиметил) индол-2-карбоксамид (Способ В).
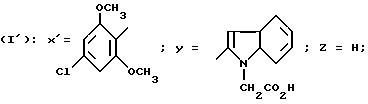
0,7 г 2-Амино-4-(2,6-диметокси-4-хлорфенил)-тиазола, 0,61 г N-(метоксикарбонилметил)-индол-2-карбоновой кислоты, 0,42 мл триэтиламина и 1,4 г БОФ растворяют в 5 мл ДМФ, затем реакционную смесь оставляют стоять при комнатной температуре в течение 48 часов. Смесь выливают в сульфатный буфер с pH 2, затем отфильтровывают осадок, который после этого промывают водой и растворяют в ДХМ. Раствор промывают 5%-ным раствором гидрокарбоната натрия, затем сульфатным буфером с pH 2, органическую фазу декантируют, сушат над сульфатом натрия, отфильтровывают и концентрируют в вакууме. Остаток хроматографируют на силикагеле Н. Концентрирование фракций чистого продукта дает 1,08 г целевого сложного метилового эфира. Т. пл.= 236 - 237oC.
1,08 г вышеполученного сложного метилового эфира растворяют в 100 мл 95o этанола в присутствии 1,5 мл н. раствора NaOH. Реакционную смесь перемешивают при комнатной температуре в течение 48 часов и концентрируют в вакууме. Остаток обрабатывают водой, затем прикапывают концентрированную HCl вплоть до pH 1. Осадок отфильтровывают и сушат его, получая 0,84 г целевого хлоргидрата. Т.пл. выше 300oC.
Пример 5.
N-/-4-(2, 6-Диметокси-4-метилфенил)тиазол-2-ил/-1-(карбоксиметил)- индол-2-карбоксамид-трифторацетат (способ Г).
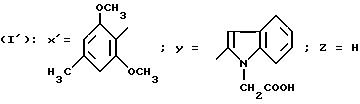
1,07 г N-/-4-(2,6-Диметокси-4-метилфенил)-2-тиазолил/- 1-(трет. - бутоксикарбонилметил)индол-2-карбоксамида (получен согласно европейскому патенту А-0432040) растворяют в смеси 2 мл анизола и 20 мл ТФК. Реакционную смесь выдерживают 3/4 часа при комнатной температуре, затем концентрируют в вакууме. Остаток обрабатывают эфиром, затем осадок отфильтровывают и высушивают в сушильном шкафу, получая 1,13 г целевого соединения. Т. пл.=223 - 224o C.
Пример 6
N-/4-(2,3,
6-Триметокси-4-метилфенил)-тиазол-2-ил/-1H-индол-2- карбоксамид (Способ Д).
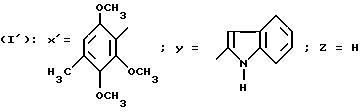
0,16 г 2-Амино-4-(2,3,6-триметокси-4-метилфенил)-тиазола растворяют в 10 мл ДМФ. Добавляют 0,18 г N-(трет.-бутилоксикарбонилметил)-индолин-2-карбоновой кислоты, 0,2 мл триэтиламина, 0,38 г БОФ и оставляют реакционную смесь при перемешивании в течение 48 часов. Добавляют 100 мл воды, экстрагируют с помощью этилацетата, органическую фазу декантируют, сушат над сульфатом натрия и концентрируют в вакууме. Остаток растворяют в 10 мл хлороформа, затем добавляют 10 мл ТФК и реакционную смесь перемешивают при комнатной температуре в течение 2,5 часов. Концентрируют в вакууме, добавляя 3 раза по 20 мл бензола. Остаток растворяют в 20 мл диметоксиэтана, затем добавляют 0,1 мл триэтиламина и 0,112 г ДДХ и реакционную смесь оставляют стоять в течение ночи при комнатной температуре. Концентрируют в вакууме, остаток обрабатывают с помощью этилацетата и промывают последовательно с помощью 1 н. раствора NaOH, раствора KHSO4 и раствора хлорида натрия, органическую фазу декантируют, сушат над сульфатом натрия, отфильтровывают и концентрируют в вакууме. Остаток хроматографируют на силикагеле, элюируя смесью хлороформа с этилацетатом (50:50 по объему). Фракции чистых продуктов концентрируют в вакууме и остаток конкретизируют в пентане, получая 0,08 г целевого продукта. Т.пл = 200oC.
Пример 7.
N-/4-(5-Хлор-2, 4-диметоксифенил)-тиазол-2-ил/-1H-индол-2- карбоксамид (Способ Е).
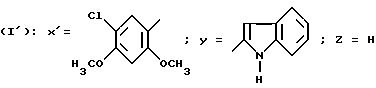
Смесь 0,9 г соединения, полученного в приготовлении IV, В, 0,67 г П-ацетилиндол-2-карбоновой кислоты, 1,5 г БОФ, 0,46 мл триэтиламина в 4 мл ДМФ перемешивают при комнатной температуре в течение ночи. Реакционную смесь выливают в буферный раствор с pH 2, образовавшийся осадок отфильтровывают и промывают его водой. Осадок обрабатывают с помощью ДХМ, промывают насыщенным раствором гидрокарбоната натрия, сушат над сульфатом магния и выпаривают в вакууме. Хроматографируют на диоксиде кремния, элюируя смесью ДХМ/этилацетат (100: 1 по объему). Ацетилированное на индольном азоте производное, которое получается, обрабатывают с помощью 30 мл этанола, добавляют 1 г карбоната натрия и перемешивают в течение ночи при комнатной температуре. Реакционную смесь выпаривают в вакууме, остаток обрабатывают водой, образовавшийся осадок отфильтровывают, промывают водой и высушивают в вакууме в сушильном шкафу. Получают 0,56 г целевого продукта. Т. пл = 293oC.
Пример 8.
N-/4-(5-Хлор-2,4-диметоксифенил)-5-метилтиазол-2-ил/-1H-индол-2- карбоксамид (Способ Е).
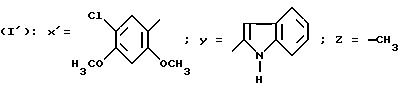
Смесь 1,24 г соединения, полученного в приготовлении IV, Г/, 0,88 г N-ацетилиндол-2-карбоновой кислоты, 1,95 г БОФ, 0,60 мл триэтиламина в 4 мл ДМФ перемешивают в течение ночи при комнатной температуре. Реакционную смесь выливают в буферный раствор с pH 2, отфильтровывают образовавшийся осадок и промывают осадок водой. Осадок обрабатывают с помощью ДХМ, промывают насыщенным раствором гидрокарбоната натрия, сушат над сульфатом магния и выпаривают в вакууме. Хроматографируют на диоксиде кремния, элюируя смесью ДХМ с этилацетатом (100:1 по объему). Полученное, ацетилированное на индольном азоте, производное обрабатывают с помощью 30 мл этанола, добавляют 2 г карбоната натрия и перемешивают в течение ночи при комнатной температуре. Реакционную смесь выпаривают в вакууме, остаток обрабатывают водой, отфильтровывают образовавшийся осадок, промывают водой, затем эфиром и высушивают в вакуумном сушильном шкафу. Получают 0,81 г целевого продукта. Т. пл. = 249o C.
Пример 9.
N-/4-(5-Хлор-2,4-диметоксифенил)-5-метилтиазол-2-ил/-1- (карбоксиметил)-индол-2-карбоксамид-трифторацетат (Способ Ж.).
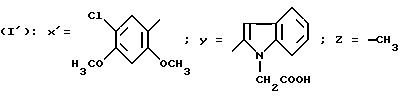
Смесь 1 г соединения, полученного в приготовлении IV,Г/, 0,96 г N-(трет. -бутоксикарбонилметил)-индол-2-карбоновой кислоты, 1,6 г БОФ, 0,49 мл триэтиламина в 6 мл ДМФ перемешивают в течение ночи при комнатной температуре. Реакционную смесь выливают в буферный раствор с pH 2, образовавшийся осадок отфильтровывают и промывают его водой. Остаток обрабатывают с помощью ДХМ, промывают буферным раствором с pH 2, насыщенным раствором гидрокарбоната натрия, сушат над сульфатом магния и выпаривают в вакууме. Хроматографируют на диоксиде кремния, элюируя смесью ДХМ с метанолом (100:1,5 по объему). Полученный сложный трет.-бутиловый эфир обрабатывают с помощью 10 мл ТФК и оставляют в течение 1,5 часов при перемешивании при комнатной температуре. Выпаривают в вакууме, остаток обрабатывают водой, образовавшийся осадок отфильтровывают, промывают водой и высушивают в вакуумном сушильном шкафу. Получают 1,37 г целевого продукта. Т.пл. = 167oC.
Пример 10.
N-/-4-(2, 5-Диметокси-4-метилфенил)тиазол-2-ил/-1-карбоксиметил) -индол-2-карбоксамид (Способ 3).
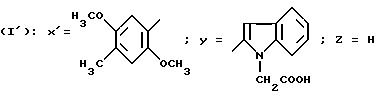
Смесь 1,0 г 2-амино-4-(2, 5-диметокси-4-метилфенил)тиазола, 1,09 г N-(трет. -бутоксикарбонилметил)индол-2-карбоновой кислоты, 2,0 г БОФ, 0,55 мл триэтиламина в 5 мл ДМФ перемешивают в течение 48 часов при комнатной температуре. Реакционную смесь выливают в буферный раствор с pH 2, образовавшийся осадок отфильтровывают и промывают его водой. Осадок обрабатывают с помощью ДХМ, промывают насыщенным раствором гидрокарбоната натрия, буферным раствором с pH 2, сушат над сульфатом магния и выпаривают в вакууме. Хроматографируют на диоксиде кремния, элюируя смесью ДХМ/этилацетат (100:1,5 по объему). Полученный сложный трет.-бутиловый эфир обрабатывают с помощью 10 мл ТФК и оставляют при перемешивании в течение 3 часов. Выпаривают в вакууме, остаток обрабатывают 2 н. раствором NaOH, промывают с помощью ДХМ, водную фазу подкисляют путем добавления концентрированной HCl, образовавшийся осадок отфильтровывают и высушивают в вакуумном сушильном шкафу. Получают 1,4 г целевого продукта. Т. пл. = 206oC.
Пример 11
N-/4-/4-Трифторметилл-2,6-диметоксифенил/-тиазол-2-ил/-1H- индол-2-карбоксамид (Способ Е).
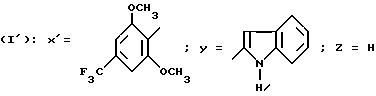
Смесь 0,609 г 2-амино-4-(4-трифторметил-2,6-диметоксифенил)-тиазола, 0,447 г N-ацетилиндол-2-карбоновой кислоты, 1,062 г БОФ, 0,243 г триэтиламина в 30 мл ДХМ перемешивают в течение 48 часов при комнатной температуре. Добавляют 100 мл воды, после декантации органическую фазу сушат над сульфатом натрия и выпаривают в вакууме. Полученное, ацетилированное на индольном азоте, производное обрабатывают с помощью 50 мл метанола, добавляют 2 г карбоната натрия и перемешивают в течение ночи при комнатной температуре. Выпаривают в вакууме, остаток обрабатывают с помощью 100 мл воды, экстрагируя с помощью ДХМ, сушат над сульфатом натрия и выпаривают в вакууме. Получают 0,54 г целевого продукта, после кристаллизации из ДХМ. Т. пл. выше 260oC.
Пример 12.
N-/4-(4-Трифторметил-2,
6-диметоксифенил)-тиазол-2-ил/-1- (трет-бутокси-карбонилметил)-индол-2-карбоксамид (Способ И)
Смесь 0,609 г 2-амино-4-(4-трифторметил-2,6-диметоксифенил)-тиазола, 0,606 г N-(трет.-бутоксикарбонилметил)-индол-2-карбоновой кислоты, 1,062 г БОФ и 0,243 г триэтиламина в 30 мл ДХМ перемешивают в течение 24 часов при комнатной температуре. Затем добавляют 50 мл воды, после декантации органическую фазу сушат над сульфатом натрия и выпаривают в вакууме. Хроматографируют на диоксиде кремния H, элюируя смесью ДХМ с этилацетатом (100:5 по объему). Получают 0,79 г целевого продукта, после кристаллизации из эфира. Т.пл. = 214 - 216oC.
Пример 13.
N-/4-(4-Трифторметил-2,
6-диметоксифенил)-тиазол-2-ил/-1- (карбоксиметил)-индол-2-карбоксамид-трифторацетат (Способ
К)
Охлаждают до 10oC 20 мл ТФК, добавляют 0,5 г соединения, полученного в примере 12, и оставляют при перемешивании при 10oC в течение 3 часов. Выпаривают в вакууме, остаток обрабатывают водой, экстрагируют этилацетатом, сушат над сульфатом натрия и выпаривают в вакууме. Получают 0,47 г целевого продукта, после кристаллизации из эфира. Т. пл. = 230 - 232oC.
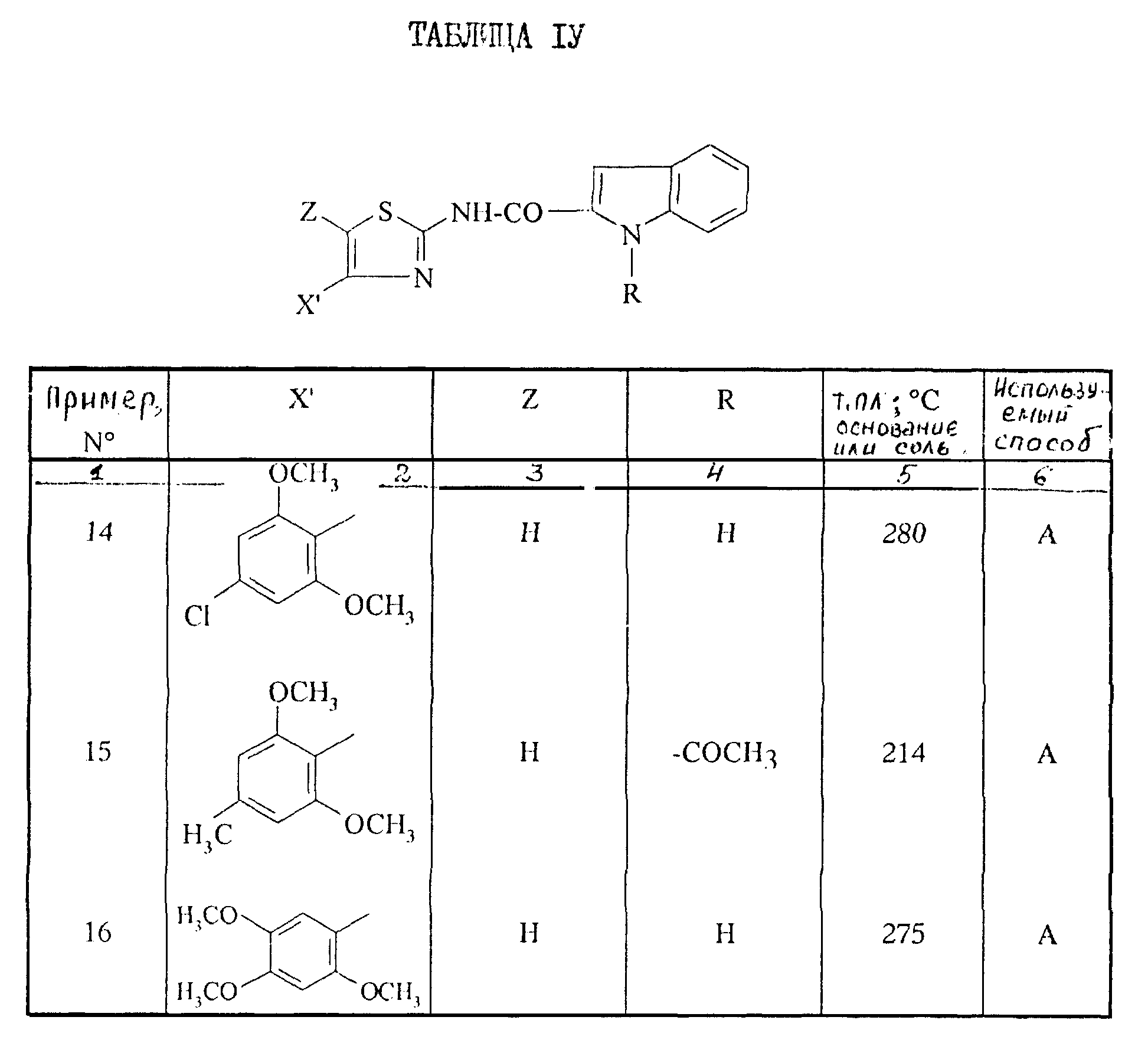
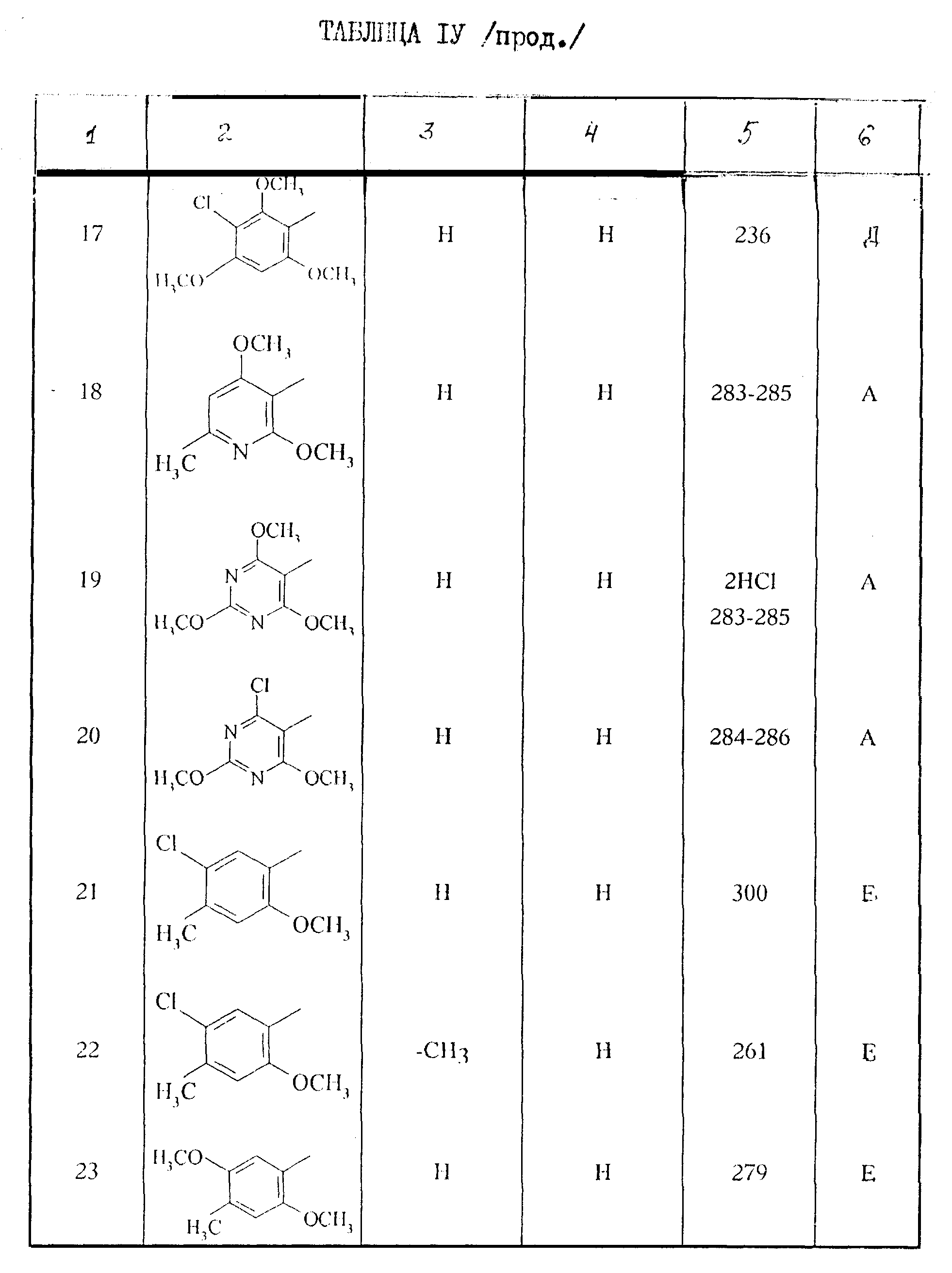
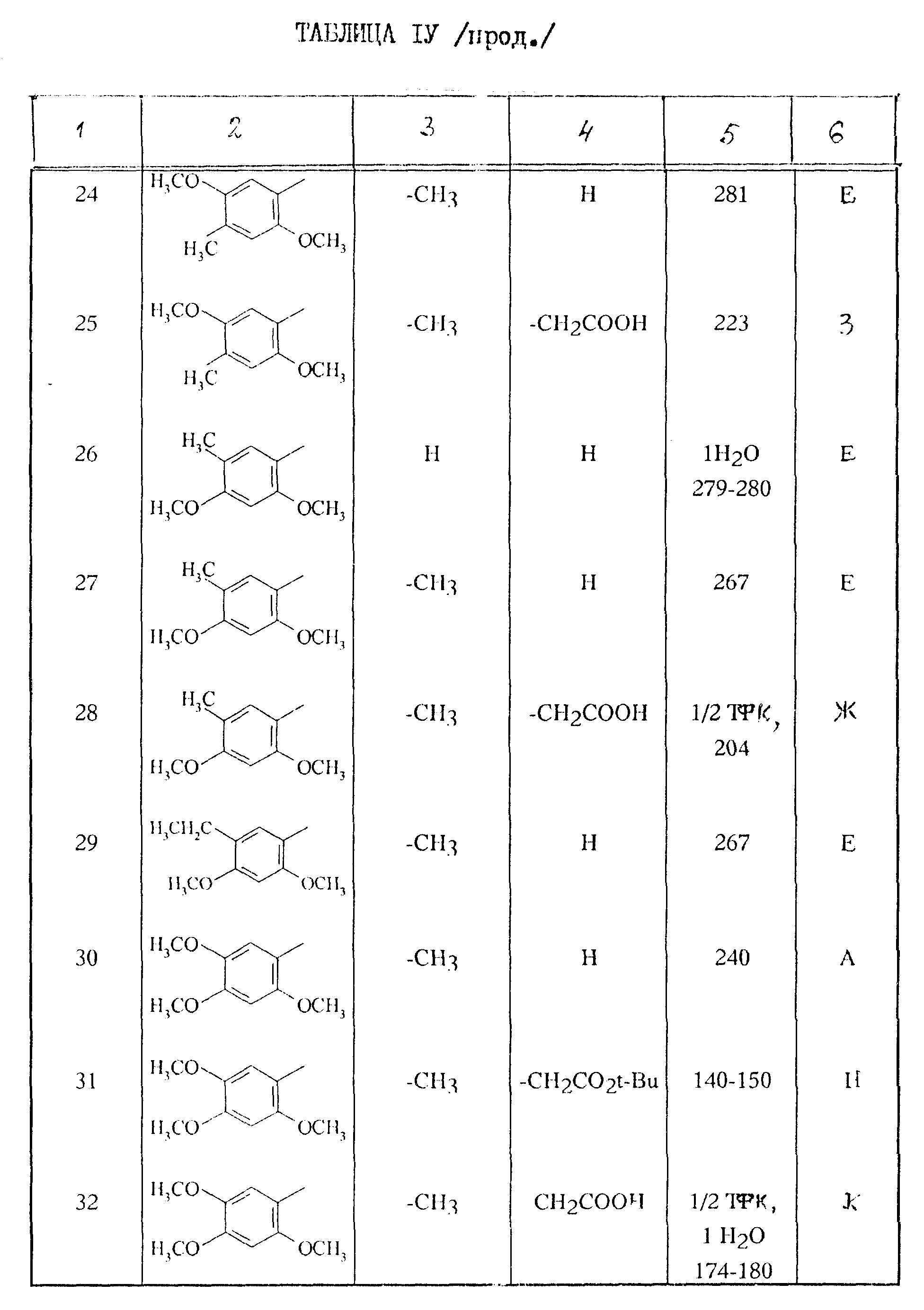
Поступая согласно способам работы, описанным выше, получают соединения формулы I, описанные в таблице IV.
Реферат
Использование: в химии гетероциклических соединений, обладающих агонизмом к рецепторам холестокинина. Раскрыты соединения формулы (I), где Y обозначает 3-хинолинильную группу или 2-индольную группу формулы (II), в которой R обозначает водород, ацетильную групп или группу CH2COOR1, причем R1 - водород или C1 - C4-алкил; Х - (гетеро)арильный радикал, выбираемый из группы, представленной в описании; Z - водород, C1 - C4-алкил или бензил, а также их соли или сольваты. Изобретение относится также к способу получения соединений формулы (I), промежуточным соединениям синтеза, фармацевтической композиции на их основе и может быть использовано при лечении расстройств, вызванных холецистокинином. 4 с. и 8 з.п. ф-лы, 4 табл.


Формула
в которой Y обозначает 3-хинолинильную или 2-индолильную группу формулы

в которой R обозначает водород, ацетильную группу или группу CH2COOR1, причем R1 - водород или C1 - C4 - алкил;
X обозначает (гетеро)арильный радикал, выбираемый из группы, включающей 4-хлор-2,6-диметоксифенильную, 2,6-диметокси-4-метилфенильную, 2,4,5-триметоксифенильную, 4-метил-2,3,6-триметоксифенильную, 2,6-диметокси-4-этилфенильную, 2, 4,6-триметокси-5-хлорфенильную, 2,4,6-триметокси-3-пиридинильную, 2,4-диметокси-6-метил-3-пиридинильную, 6-хлор-2,4-диметокси-5-пиримидинильную, 2,4,6-триметокси-5-пиримидинильную, 5-хлор-2, 4-диметоксифенильную, 5-хлор-2-метокси-4-метилфенильную, 2,5-диметокси-4-метилфенильную, 4-трифторметил-2,6-диметоксифенильную, 2,4-диметокси-5-метилфенильную, 5-этил-2,4-диметоксифенильную, 2, 4-диметоксифенильную группы;
Z обозначает водород, C1 - C4-алкил или бензил, при условии, что Z обязательно обозначает водород, когда X обозначает фенильный радикал, замещенный одновременно в положениях 2 и 6, или когда X - 3-пиридинильный радикал, замещенный одновременно в положениях 2 и 4, или когда X - 5-пиримидинильный радикал, замещенный одновременно в положениях 4 и 6;
или их фармацевтически приемлемые соли или сольваты в качестве полного или частичного агониста рецепторов холецистокинина.
в которой Y обозначает 3-хинолинильную группу (a) или 2-индолильную группу (b) формулы
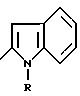
в которой R обозначает водород, ацетильную группу или группу CH2 COOR1, причем R1 - водород или C1 - C4 - алкил;
X' обозначает (гетеро)арильный радикал, выбираемый из группы, включающей 4-хлор-2, 6-диметоксифенильную, 2, 6-диметокси-4-метилфенильную, 2,4,5-триметоксифенильную, 4-метил-2,3,6-триметоксифенильную, 2,4,6-триметокси-5-хлорфенильную, 2,4,6-триметокси-3-пиридинильную, 2,4, 6-триметокси-5-пиримидинильную, 2, 4-диметокси-6-метил-3-пиридинильную, 6-хлор-2,4-диметокси-5-пиримидинильную, 5-хлор-2,4-диметоксифенильную, 5-хлор-2-метокси-4-метилфенильную, 2, 5-диметокси-4-метилфенильную, 4-трифторметил-2, 6-диметоксифенильную, 2,4-диметокси-5-метилфенильную, 5-этил-2,4-диметоксифенильную группы;
Z обозначает водород, C1 - C4 -алкил или бензил, при условии, что Z - обязательно водород, когда X' - фенильный радикал, замещенный одновременно в положениях 2 и 6, или когда X' - 3-пиридинильный радикал, замещенный одновременно в положениях 2 и 4, или когда X' - 5-пиримидинильный радикал, замещенный одновременно в положениях 4 и 6,
или их соли, или сольваты.
Y′-COOH,
в которой Y' обозначает 3-хинолинильный радикал (a)
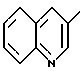
2-индолильный радикал (b0)
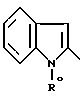
или 2-индолинильный радикал (c0)
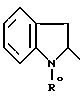
причем R0 обозначает N - защитную группу или группу CH2COOR'', где R'' - C1 - C4-алкил;
или функциональное производное кислоты формулы II конденсируют с 2-аминотиазолом формулы III
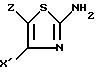
в которой X' и Z имеют указанные в п.2 значения,
в присутствии основания с получением соединения формулы I''
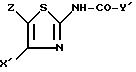
в которой X и Z имеют указанные в п.2 значения,
Y' - один из радикалов формул (a), (b0) или (c0),
затем, когда в соединения I'' Y' обозначает радикал (b0), при желании, полученный продукт формулы I'' (b0)
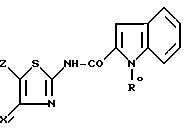
подвергают реакции удаления N-защитной группы, или омылению, или кислотному гидролизу; когда в соединении I'' Y' обозначает радикал (c0), полученный продукт формулы I'' c0
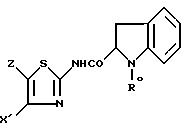
подвергают дегидрированию, в известных случаях при предварительном удалении N-защитной группы, омылении или кислотном гидролизе, с получением соединения формулы I', в которой Y - радикал (b), где R - водород или группа CH2COOR', причем X', Z и R' имеют указанные в п.2 значения, и продукт формулы I' выделяют таким, какой есть, или в форме одного из его сольватов, или одной из его солей.
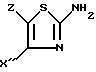
в которой X'' - (гетеро)арильный радикал, выбираемый из группы, включающей 4-хлор-2, 6-диметоксифенильную, 2,6-диметокси-4-метилфенильную, 4-метил-2,3,6-триметокси-фенильную, 2,4,6-триметокси-5-хлорфенильную, 2,4,6-триметокси-3-пиридинильную, 2,4,6-триметокси-5-пиримидинильную, 2, 4-диметокси-6-метил-3-пиридинильную, 6-хлор-2,4-диметокси-5-пиримидинильную, 5-хлор-2,4-диметоксифенильную, 5-хлор-2-метокси-4-метилфенильную, 2,5-диметокси-4-метил-фенильную, 4-трифторметил-2, 6-диметоксифенильную, 2,4-диметокси-5-метилфенильную, 5-этил-2,4-диметоксифенильную группы;
Z - водород, C1 - C4-алкил или бензил, с ограничением, что Z - водород, когда X'' - фенильный радикал, замещенный одновременно в положениях 2 и 6, или когда X'' - 3-пиридинильный радикал, замещенный одновременно в положениях 2 и 4, или когда X'' - 5-пиримидинильный радикал, замещенный одновременно в положениях 4 и 6,
в качестве промежуточных соединений синтеза.
Комментарии