Соединения бензолсульфонамидов и их использование в качестве терапевтических средств - RU2760303C2
Код документа: RU2760303C2
Описание
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение направлено на соединения бензолсульфонамидов и фармацевтические композиции, содержащие соединения, а также на способы использования соединений и фармацевтических композиций при лечении опосредованных натриевыми каналами заболеваний или состояний, таких как эпилепсия и/или эпилептическое пароксизмальное нарушение, а также других заболеваний и состояний, ассоциированных с посредничеством натриевых каналов.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Потенциалзависимые натриевые каналы (NaV) являются ключевыми определяющими факторами клеточной возбудимости в мышцах и нервах (Hille, B, Ion Channels of Excitable Membranes (2001), Sunderland, MA, Sinauer Associates, Inc.). Четыре изоформы, в частности, NaV1.1, NaV1.2, NaV1.3 и NaV1.6, отвечают за основной натриевый ток в нейронах центральной нервной системы. Экспрессия NaV1.3 впервые происходит у эмбриона. После неонатального этапа NaV1.1, NaV1.2 и NaV1.6 представляют собой ключевые изоформы, которые регулируют передачу нейрональных сигналов в головном мозге (Catterall, W.A., Annual Review of Pharmacology and Toxicology (2014), том 54, стр. 317-338).
NaV1.5 экспрессирован преимущественно в кардиомиоцитах (Raymond, C.K. et al., J. Biol. Chem. (2004), том 279, № 44, стр. 46234-41), включая предсердия, желудочки, синусно-предсердный узел, предсердно-желудочковый узел и сердечные волокна Пуркинье. Мутации в NaV1.5 человека ведут к множеству аритмических синдромов, включая, например, длинный QT3 (LQT3), синдром Бругада (BS), наследственный дефект сердечной проводимости, синдром внезапной неожиданной ночной смерти (SUNDS) и синдром внезапной детской смерти (SIDS) (Liu, H., et al., Am. J. Pharmacogenomics (2003), том 3, № 3, стр. 173-9). Терапию блокаторами натриевые каналов широко использовали при лечении сердечных аритмий.
Эпилепсия представляет собой состояние, отличающееся чрезмерной синхронной возбудимостью в головном мозге, которая возникает при нарушении шаткого равновесия возбуждающих и ингибирующих сигналов в головном мозге. Это может происходить из-за избытка возбуждения или дефицита ингибирования. Мутации в генах, кодирующих каналы NaV, связаны с нарушением равновесия обоих типов.
Установлено, что NaV1.1 является основной изоформой NaV в ингибирующих вставочных нейронах (Yu, F.H. et al., Nat. Neurosci. (2006), том 9, стр. 1142-1149). Эти вставочные нейроны образуют синапсы со многими другими нейронами, включая возбуждающие глутаматергические нейроны. Потенциалы действия во вставочных нейронах индуцируют высвобождение нейротрансмиттера GABA к другим нейронам, гиперполяризуя их и, таким образом, смягчая возбуждение. Это ведет к отрицательной обратной связи, которая делает возможной управляемую передачу сигналов и предотвращает распространение локальных сигналов в виде вол возбуждения, которые охватывают большие области головного мозга. В силу этой решающей роли ингибирующих вставочных нейронов, мутации, которые ослабляют функцию канала NaV1.1, могут вести к неспособности этих нейронов к активации и высвобождению GABA (Ogiwara, I. et al., J. Neurosci. (2007), том 27, стр. 5903-5914; Martin, M.S. et al., J. Biol. Chem. (2010), том 285, стр. 9823-9834; Cheah, C.S. et al., Channels (Austin) (2013), том 7, стр. 468-472; и Dutton, S.B., et al., (2013), том 49, стр. 211-220). Результатом является утрата ингибирующего тонуса головного мозга и неспособность вмещать возбудимость глутаматергических нейронов. Эта неспособность ингибирующих вставочных нейронов может вести к аберрантным обширным синхронным импульсам нейронов, охватывающим области головного мозга (эпилепсия).
Мутации в гене, кодирующем NaV1.1 (SCN1A), делятся на два обширных класса, те, которые вызывают генерализованную эпилепсию с фебрильными пароксизмами плюс (GEFS+), и те, которые вызывают тяжелую детскую миоклоническую эпилепсию (SMEI), также известную как синдром Драве или ранняя детская эпилептическая энцефалопатия 6 (EIEE6) (McKusik, V.K. et al., A Epileptic Encephalopathy, Early Infantile 6, EIEE6 (2012), Online Mendelian Inheritance in Man: John Hopkins University). Мутации SMEI представляют собой гетерозиготные аутосомно-доминантные мутации и часто обусловлены делецией или усечением гена, которые ведут к каналу с небольшой или нулевой функцией. Мутации возникают de novo, или в некоторых случаях показано, что они возникают у бессимптомных мозаичных родителей (Tuncer, F.N. et al., Epilepsy Research (2015), том 113, стр. 5-10). Пациенты рождаются фенотипически нормальными и соответствуют основным вехам развития до начала пароксизмов, обычно в возрасте между 6 месяцами и 1 годом. Полагают, что это время начала является последствием нормального снижения экспрессии эмбриональной изоформы NaV1.3 и совпадающего повышения NaV1.1. Когда каналы NaV1.1 не способны достичь нормальных уровней, происходит проявление фенотипа (Cheah, C.S. et al., Channels (Austin) (2013), том 7, стр. 468-472). Начальный пароксизм часто запускается фебрильным эпизодом и может быть манифестирован в виде status epilepticus. Пароксизмы продолжаются и нарастают по частоте и тяжести в течение первых нескольких лет жизни и могут достигать частот более 100 эпизодов в сутки. Пароксизмы могут запускаться лихорадкой или могут возникать самопроизвольно без видимой причины. После начала пароксизмов, пациенты начинают отставать в развитии, и происходит нарастание значимой когнитивной и поведенческой недостаточности (Dravet, C. and Oguni, H., Handbook of Clinical Neurology (2013), том 111, стр. 627-633). Полагают, что от 80 до 85% фенотипически диагностированных пациентов с синдромом Драве имеют ответственную мутацию в SCN1A, тогда как другие 15-20% пациентов имеют другие мутации или имеют неизвестную этиологию. Имеет место высокая доля внезапной неожиданной смерти при эпилепсии (SUDEP) у пациентов с SMEI, причем оценочно 37% пациентов умирают при SUDEP, но механизм этого катастрофического исхода остается неясным (Massey, C.A., et al., Nature Reviews Neurology (2014), том 10, стр. 271-282). Клинически эффективные противоэпилептические лекарственные средства, которые неизбирательно направлены на потенциалзависимые натриевые каналы, подобно карбамазепину и фенитоину, противопоказаны для пациентов с SMEI, поскольку они могут усугублять пароксизмы у этих пациентов (Wilmshurst, J.M. et al., Epilepsia (2015), том 56, стр. 1185-1197). Предполагают, что это обусловлено неспособностью пациентов переносить дальнейшее снижение функции NaV1.1.
GEFS+ часто обусловлена миссенс-мутациями SCN1A, которые индуцируют относительно мягкое нарушение функции каналов, в соответствии с относительно более мягким пароксизмальным фенотипом. Идентифицировано большое и растущее число мутаций, при которых значительно варьирует как тяжесть, так и пенетрантность фенотипа. Многие пациенты GEFS+ перерастают пароксизмальный фенотип, но не все, и пациенты с GEFS+ с детской эпилепсией значительно более склонны к эпилепсии во взрослом возрасте, чем общая популяция. Мутации, которые вызывают недостаточность других генов, участвующих в GABA-ергической передаче сигналов, например, SCN1B, который кодирует вспомогательную субъединицу натриевого канала, и GABRG2, который кодирует субъединицу рецепторов GABAA, также могут давать начало GEFS+ (Helbig, I., Seminars in Neurology (2015), том 35, стр. 288-292).
Созданы трансгенные мыши, которые несут те же мутации, которые идентифицированы у пациентов с SMEI и GEFS+. В обоих случаях мыши хорошо воспризводят человеческий фенотип, хотя на пенетрантность фенотипа может значительно влиять генетический фон. Некоторые штаммы мышей переносят мутации относительно хорошо, тогда как в других штаммах те же мутации могут вызывать выраженные пароксизмальные фенотипы. Предположительно эти различия обусловлены различающимися уровнями экспрессии других генов, которые модулируют фенотип возбуждения (Miller, A.R. et al., Genes, Brain, and Behavior (2014), том 13, стр. 163-172; Mistry, A.M. et al., Neurobiology of Disease (2014), том 65, стр. 1-11; и Hawkins, N.A. et al., Epilepsy Research (2016), том 119, стр. 20-23).
В головном мозге NaV1.2 и NaV1.6 в первую очередь экспрессированы в возбуждающих глутаматергических нейронах. Оба канала имеют особенно высокую плотность в начальном сегменте действия (AIS), области нейрона, смежной с телом нейрона, который функционирует для того, чтобы интегрировать входные сигналы и инициирует распространение потенциала действия на тело и дистальные дендриты (Royeck, M. et al., J. Neurophysiol. (2008), том 100, стр. 2361-2380; Vega, A.V. et al., Neurosci. Lett. (2008), том 442, стр. 69-73; и Hu, W. et al., Nat. Neurosci. (2009), том 12, стр. 996-1002). NaV1.6 обычно бывает особенно плотно локализован в раннем AIS (дистально от тела), где он предположительно функционирует для того, чтобы запускать инициацию потенциала действия. NaV1.2 больше локализован в сегменте AIS, наиболее проксимальном относительно тела. Мутации в SCN2A (NaV1.2) и SCN8A (NaV1.6) связаны с эпилепсией и когнитивной задержкой. Эффекты мутации разнообразны как по уровню влияния на функцию канала, так и по фенотипу пациента. Оба NaV1.2 и NaV1.6 также экспрессированы в периферических нейронах. NaV1.6 имеет особенно высокую плотность в перехватах Ранвье на миелинизированных нейронах, где он необходим для поддержания волны проводимости и высокой скорости передачи нейрональных сигналов.
Описана лишь часть мутаций NaV1.2, но они в первую очередь связаны с патологиями центральной нервной системы, в частности, с эпилепсией (Kearney, J.A. et al., Neuroscience (2001), том 102, стр. 307-317; Zerem, A. et al., European Journal of Paediatric Neurology: EJPN: Official Journal of the European Paediatric Neurology Society (2014), том 18, стр. 567-571; Fukasawa, T. et al., Brain & Development (2015), том 37, стр. 631-634; Howell, K.B. et al., Neurology (2015), том 85, стр. 958-966; Saitoh, M. et al., Epilepsy Research (2015), том 117, стр. 1-6; Samanta, D. et al., Acta Neurologica Belgica (2015), том 115, стр. 773-776; Carroll, L.S. et al., Psychiatric Genetics (2016), том 26, стр. 60-65; и Schwarz, N. et al., Journal of Neurology (2016), том 263, стр. 334-343). Предполагают, что эпилептические мутации в первую очередь представляют собой мутации с приобретением функции, что обозначает, что они ведут к увеличению количества натриевого тока и, тем самым, к увеличению возбудимости. Установление влияния на функцию канала in vivo при отсутствии обоснованного сомнения является сложным, и некоторые из этих мутаций также могут вести к фенотипам с утратой функции.
Аналогично, сообщалось, что мутации в SCN8A демонстрируют ряд эффектов с приобретением и утратой функции, оказываемых на канал NaV1.6, хотя для NaV1.6 большинство исследованных мутаций ассоциировано с фенотипами с приобретением функции. Мутации в NaV1.6 связаны с эпилепсием и нарушениями аутистического спектра (Trudeau, M.M. et al., Journal of Medical Genetics (2006), том 43, стр. 527-530; Veeramah, K.R. et al., Am. J. Hum. Genet. (2012), том 90, стр. 502-510; Vaher, U. et al., Journal of Child Neurology (2013); de Kovel, C.G. et al., Epilepsy Research (2014); Estacion, M. et al., Neurobiology of Disease (2014), том 69, стр. 117-123; Ohba, C. et al., Epilepsia (2014), том 55, стр. 994-1000; Wagnon, J.L. et al., Human Molecular Genetics (2014); Kong, W. et al., Epilepsia (2015), том 56, стр. 431-438; и Larsen, J. et al., Neurology (2015), том 84, стр. 480-489). Наилучшим образом описанные пациенты с мутантным SCN8A имеют синдром, известный как ранняя детская эпилептическая энцефалопатия, 13 (EIEE13). Идентифицировано более 100 пациентов с EIEE13. Пациенты обычно имеют некупируемые пароксизмы в возрасте от рождения до 18 месяцев. Пациенты имеют задержку развития и когнитивных функций, а также двигательную недостаточность, часто ассоциированную с хронической мышечной гипотонией. Пациенты с наиболее тяжелыми поражениями никогда не приобретают достаточного контроля моторики, чтобы ходить. Многие не вербальны. Менее тяжелые фенотипы позволяют учиться ходить и говорить, но с двигательной и когнитивной недостаточностью и потерей социальных навыков. Большинство идентифицированных мутаций представляют собой миссенс-мутации, и допускают, что конкретное функциональное влияние мутации вносит вклад в вариабельность фенотипа, хотя генетический фон также вероятно участвует (Larsen, J. et al., Neurology (2015), том 84, стр. 480-489). В отличие от SMEI пациентов, случаи из практики подсказывают, что противоэпилептические лекарственные средства, которые неизбирательно направлены на потенциалзависимые натриевые каналы, могут улучшать симптомы у пациентов с EIEE13, хотя контролируемые клинические исследования не выполнены (Boerma, R.S. et al., Neurotherapeutics: The Journal of the American Society for Experimental NeuroTherapeutics (2016), том 13, стр. 192-197). Хотя фенитоин, похоже, вызывает эффект у пациентов с EIEE13, это имеет определенную цену. Эффекта достигают только при очень высоких дозах, при которых значимые нежелательные эффекты могут терпеть только пациенты, находящиеся в такой крайней нужде. Нежелательные эффекты, обычно ассоциированные с терапией фенитоином, включают некроз печени, гипертрихоз, нервозность, тремор рук, онемение, головокружение, сонливость, тремор, депрессию, спутанность сознания, утомление, констипацию, вертиго, атаксию, изменения психического статуса, миастению, изменения настроения, возбужденное состояние, раздражительность и возбуждение. Похоже, что лекарственное средство, которое избирательно направлено на NaV1.6, будет сохранять эффект, при этом снижая бремя его нежелательных явлений.
Мутации с утратой функции в SCN8A у мышей ведут к фенотипу, известному как болезнь двигательной концевой пластинки (med), и несколько мутаций и фенотипов связаны с областью гена med прежде идентификации гена SCN8A (Burgess, D.L. et al., Nat. Genet. (1995), том 10, стр. 461-465). Мыши с мутациями SCN8Amed имеют различные степени мышечной гипотониии, в соответствии со степенью нарушения функции NaV1.6. Мыши с SCN8Amed/jo имеют каналы NaV1.6, которые имеют фенотип с утратой функции, но не нулевой. Мыши SCN8Amed и SCN8Amed/jo устойчивы к пароксизмам, индуцируемым посредством химического инсульта (флуротил, каиновая кислота и пикротоксин) (Martin, M.S. et al., Human Molecular Genetics (2007), том 16, стр. 2892-2899; Hawkins, N.A. et al., Neurobiology of Disease (2011), том 41, стр. 655-660; и Makinson, C.D. et al., Neurobiology of Disease (2014), том 68, стр. 16-25). Что интересно, когда мышей SCN8Amed/jo скрещивают с мутантными мышами SCN1Anull для получения мыши, которая гетерозиготна как по аллелю SCN1Anull, так и по аллелю SCN8Amed/jo, мыши с двойной мутацией имеют значительно улучшенный пароксизмальный и когнитивный фенотип, чем те, которые имеют только мутацию SCN1Anull (Martin, M.S. et al., Human Molecular Genetics (2007), том 16, стр. 2892-2899). Такие мыши имеют уровень самопроизвольных пароксизмов и смертности, схожий с мышами дикого типа, а их пароксизмальный порог после химического инсульта также увеличен. Схожий результат возникает при скрещивании мышей с миссенс-мутациями SCN1A (модель для GEFS+) и мышей с мутациями с утратой функции SCN8A. Наличие одного аллеля SCN8Amed/jo защищало мышей с моделью GEFS+ от пароксизмов и преждевременной смерти (Hawkins, N.A. et al., Neurobiology of Disease (2011), том 41, стр. 655-660). Способность нокдауна SCN8A улучшать устойчивость к пароксизмам не ограничена нокаутами, где ген глобально отсутствует на всем протяжении развития животного. Нокдаун SCN8A у взрослых мышей или глобально или конкретно в гиппокампе через подход CRE-LOX индуцибельного нокаута также улучшал устойчивость к электрически и химически индуцированным пароксизмам Makinson, C.D. et al., Neurobiology of Disease (2014), том 68, стр. 16-25). Эти данные показывают, что супрессию передачи ингибирующих сигналов, обусловленную сниженным током в NaV1.1, можно уравновешивать, по меньшей мере отчасти, подавлением передачи возбуждающих сигналов через снижение тока в NaV1.6.
Антагонизм потенциалзависимых натриевых каналов является наиболее распространенным механизмом широко назначаемых противоэпилептических лекарственных средств (AED) (Ochoa, J.R. et al., Sodium Channel Blockers. In: Antiepileptic Drugs (2016), том (Benbadis, S., ed) Medscape News & Perspectives). Полагают, что карбамазепин, эсликарбазепин, окскарбазепин, лакозамид, ламотригин, фенитоин, руфинамид и зонизамид действуют, в первую очередь, посредством блокирования этой функции каналов NaV. Несмотря на предполагаемый механизм действия, эти лекарственные средства относительно неразборчивы. Они блокируют все изоформы каналов NaV без разбора, таким образом, следует ожидать, что блокирование NaV1.1 будет проконвульсантным. Блокирование NaV1.6 и, возможно. NaV1.2 будет антиконвульсантным. В дополнение к натриевым каналам, эти соединения также блокируют другие мишени, в том числе потенциалзависимые кальциевые каналы. Ожидают, что избирательные антагонисты NaV, которые щадят NaV1.1 и другие нецелевые рецепторы, обладают улучшенными эффектом и терапевтическим индексом по сравнению с доступными в настоящее время блокирующими NaV лекарственными средствами.
Следовательно, существует неудовлетворенная медицинская потребность в лечении эпилепсии и других ассоциировнных с NaV1.6 патологических состояний эффективно и без нежелательных побочных эффектов из-за блокирования других натриевых каналов, таких как NaV1.1 и/или NaV1.5. Настоящее изобретение предусматривает способы удовлетворения этих критических потребностей.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение направлено на соединения бензолсульфонамидов и фармацевтические композиции, содержащие эти соединения, а также на способы использования соединений и фармацевтических композиций по изобретению для лечения заболеваний или состояний, ассоциированных с активностью потенциалзависимых натриевых каналов, в частности, с активностью NaV1.6, таких как эпилепсия и/или эпилептическое пароксизмальное нарушение.
Соответственно, в одном из аспектов, это изобретение направлено на соединения бензолсульфонамидов формулы (I):

в которой:
A представляет собой непосредственную связь или (CH2)m C(R4)(R5) (CH2)n, где m и n представляют собой независимо 0, 1, 2, 3 или 4;
q представляет собой 1, 2 или 3;
R1 представляет собой необязательно замещенный циклоалкил, необязательно замещенный арил, необязательно замещенный моноциклический гетероарил или необязательно замещенный бициклический гетероарил;
R2 представляет собой необязательно замещенный 5-членный N-гетероарил или необязательно замещенный 6 членный N-гетероарил;
R3 представляет собой O, N(R8) или S(O)t (где t представляет собой 0, 1 или 2);
R4 и R5 представляют собой каждый независимо водород, алкил, галогеналкил, необязательно замещенный циклоалкил, необязательно замещенный циклоалкилалкил, необязательно замещенный гетероциклил, необязательно замещенный гетероциклилалкил, необязательно замещенный гетероарил или необязательно замещенный гетероарилалкил, R9 OR10 или R9 N(R10)R11;
или R4 и R5, вместе с углеродом, к которому они прикреплены, образуют необязательно замещенный циклоалкил или необязательно замещенный гетероциклил;
каждый R6 представляет собой независимо водород, алкил, галоген, галогеналкил, циано или OR12;
R7 представляет собой алкил, алкенил, галоген, галогеналкил, циано или OR12;
каждый R8, R10, R11 и R12 представляет собой независимо водород, алкил, галогеналкил, необязательно замещенный циклоалкил, необязательно замещенный циклоалкилалкил, необязательно замещенный гетероциклил, необязательно замещенный гетероциклилалкил; и
каждый R9 представляет собой независимо непосредственную связь или необязательно замещенную неразветвленную или разветвленную алкиленовую цепь;
в виде их индивидуального стереоизомера, энантиомера или таутомера или их смеси;
или их фармацевтически приемлемой соли, сольвата или пролекарственного средства;
при условии, что:
(a) когда A представляет собой непосредственную связь, R1 не представляет собой необязательно замещенный циклоалкил;
(b) когда A представляет собой непосредственную связь и R3 представляет собой -O- или -S(O)t- (где t представляет собой 0, 1 или 2), R1 не представляет собой необязательно замещенный фенил;
(c) когда A представляет собой непосредственную связь и R3 представляет собой -N(R8)-, R1 не представляет собой необязательно замещенный фенил или необязательно замещенный 2,4,5,6-тетрагидроциклопента[c]пиразолил;
(d) когда A представляет собой (CH2)m C(R4)(R5) (CH2)n, где m и n оба представляют собой 0 и R4 и R5 оба представляют собой водород и R3 представляет собой -O-, R2 не представляет собой необязательно замещенный тиадиазолил; и
(e) когда A представляет собой непосредственную связь и R3 представляет собой -N(R8)-, R1 не представляет собой необязательно замещенный моноциклический гетероарил.
Соединения по изобретению, которые представляют собой соединения формулы (I) как описано выше, в виде их индивидуальных стереоизомеров, энантиомеров или таутомеров или их смесей; или в виде их фармацевтически приемлемых солей, сольватов или пролекарственных средств, можно использовать при лечении заболеваний или состояний, ассоциированных с потенциалзависимыми натриевыми каналами, предпочтительно NaV1.6. Предпочтительно, соединения по изобретению представляют собой ингибиторы NaV1.6. Более предпочтительно, соединения по изобретению проявляют избирательность ингибирования NaV1.6 по сравнению с ингибированием NaV1.5 и/или NaV1.1. Не желая ограничиваться теорией, такую избирательность считают полезно снижающей любые побочные эффекты, которые могут быть ассоциированы с ингибированием NaV1.5 и/или NaV1.1.
В другом аспекте изобретение относится к фармацевтическим композициям, содержащим фармацевтически приемлемый эксципиент и соединение формулы (I), как описано выше, в виде его стереоизомера, энантиомера или таутомера или их смесей; или его фармацевтически приемлемой соли, сольвата или пролекарственного средства.
В другом аспекте изобретение относится к способам лечения эпилепсии и/или эпилептического пароксизмального нарушения у млекопитающего, предпочтительно человека, где способы включают введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения по изобретению, как изложено выше, в виде его стереоизомера, энантиомера или таутомера или их смесей; или его фармацевтически приемлемой соли, сольвата или пролекарственного средства, или фармацевтической композиции, содержащей терапевтически эффективное количество соединения по изобретению, как изложено выше, в виде его стереоизомера, энантиомера или таутомера или их смесей или его фармацевтически приемлемой соли, сольвата или пролекарственного средства и фармацевтически приемлемый эксципиент.
В другом аспекте настоящее изобретение предусматривает способ лечения или уменьшения тяжести заболевания, состояния или нарушения у млекопитающего, где активация или гиперактивность NaV1.6 вовлечена в заболевание, состояние или нарушение, где способ включает введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения по изобретению, как изложено выше, в виде его стереоизомера, энантиомера или таутомера или их смесей; или его фармацевтически приемлемой соли, сольвата или пролекарственного средства, или фармацевтической композиции, содержащей терапевтически эффективное количество соединения по изобретению, как изложено выше, в виде его стереоизомера, энантиомера или таутомера или их смесей или его фармацевтически приемлемой соли, сольвата или пролекарственного средства и фармацевтически приемлемый эксципиент.
В другом аспекте изобретение относится к способам лечения или улучшения, но не предотвращения, эпилепсии и/или эпилептического пароксизмального нарушения у млекопитающего, где способы включают введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения по изобретению, как изложено выше, в виде его стереоизомера, энантиомера или таутомера или их смесей или его фармацевтически приемлемой соли, сольвата или пролекарственного средства или фармацевтической композиции, содержащей терапевтически эффективное количество соединения по изобретению, как изложено выше, в виде его стереоизомера, энантиомера или таутомера или их смесей или его фармацевтически приемлемой соли, сольвата или пролекарственного средства и фармацевтически приемлемый эксципиент.
В другом аспекте изобретение относится к фармацевтической терапии в комбинации с одним или несколькими другими соединениями по изобретению или одной или несколькими другими общепринятыми терапиями или в виде любого их сочетания для увеличения удельной активности существующей или будущей лекарственной терапии или для уменьшения нежелательных явлений, ассоциированных с общепринятой терапией. В одном из вариантов осуществления настоящее изобретение относится к фармацевтической композиции, объединяющей соединения по настоящему изобретению с существующими или будущими терапиями для показаний, перечисленных в настоящем описании.
В другом аспекте это изобретение направлено на способы избирательного ингибирования первого потенциалзависимого натриевого канала у млекопитающего относительно второго потенциалзависимого натриевого канала, где способ включает введение млекопитающему ингибирующего количества соединения по изобретению, как изложено выше, в виде его стереоизомера, энантиомера или таутомера или их смесей; или его фармацевтически приемлемой соли, сольвата или пролекарственного средства, или фармацевтической композиции, содержащей ингибирующее количество соединения по изобретению, как изложено выше, в виде его стереоизомера, энантиомера или таутомера или их смесей или его фармацевтически приемлемой соли, сольвата или пролекарственного средства и фармацевтически приемлемый эксципиент.
В другом аспекте это изобретение направлено на использование соединений по изобретению, как изложено выше, в виде их стереоизомера, энантиомера или таутомера или их смесей или их фармацевтически приемлемой соли, сольвата или пролекарственного средства или на использование фармацевтической композиции, содержащей фармацевтически приемлемый эксципиент и соединение по изобретению, как изложено выше, в виде его стереоизомера, энантиомера или таутомера или их смесей или его фармацевтически приемлемой соли, сольвата или пролекарственного средства, в препарате лекарственного средства для лечения заболевания или состояния, ассоциированного с активностью потенциалзависимого натриевого канала, предпочтительно NaV1.6, у млекопитающего, и где предпочтительно заболевание или состояние представляет собой эпилепсию и/или эпилептическое пароксизмальное нарушение.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
Определенным химическим группам, названным в настоящем описании, может предшествовать сокращенное обозначение, указывающее общее число атомов углерода, которые должны находиться в указанной химической группе. Например; C7 C12алкил описывает алкильную группу, как определено ниже, имеющую всего от 7 до 12 атомов углерода, и C4 C12циклоалкилалкил описывает циклоалкилалкильную группу, как определено ниже, имеющую всего от 4 до 12 атомов углерода. Общее число углеродов в сокращенном обозначении не включает углероды, которые могут существовать в заместителях описанной группы.
В дополнение к вышеуказанному, как используют в описании и приложенной формуле изобретения, пока конкретно не определено иное, следующие термины имеют указанные значения:
«Алкил» относится к радикалу неразветвленной или разветвленной углеводородной цепи, состоящему только из атомов углерода и водорода, не содержащему ненасыщенность, имеющему от одного до двенадцать атомов углерода, предпочтительно от одного до восьми атомов углерода, более предпочтительно от одного до шести атомов углерода, который прикрепляют к остальной части молекулы посредством одинарной связи, например, метил, этил, н пропил, 1 метилэтил (изо пропил), н бутил, н пентил, 1,1 диметилэтил (т бутил), 3 метилгексил, 2 метилгексил и т. п. Когда конкретно установлено в описании, алкильную группу можно необязательно замещать одной из следующих групп: алкил, алкенил, галоген, галогеналкенил, циано, нитро, арил, циклоалкил, гетероциклил, гетероарил, оксо, триметилсиланил, OR20, OC(O)R20, N(R20)2, C(O)R20, C(O)OR20, C(O)N(R20)2, N(R20)C(O)OR22, N(R20)C(O)R22, N(R20)S(O)pR22 (где p представляет собой от 1 до 2), S(O)pOR22 (где p представляет собой от 1 до 2), S(O)tR22 (где t представляет собой от 0 до 2) и S(O)pN(R20)2 (где p представляет собой от 1 до 2), где каждый R20 представляет собой независимо водород, алкил, галогеналкил, циклоалкил, циклоалкилалкил, арил, аралкил, гетероциклил, гетероциклилалкил, гетероарил или гетероарилалкил; и каждый R22 представляет собой алкил, галогеналкил, циклоалкил, циклоалкилалкил, арил, аралкил, гетероциклил, гетероциклилалкил, гетероарил или гетероарилалкил.
«Алкенил» относится к группе радикала неразветвленной или разветвленной углеводородной цепи, состоящей только из атомов углерода и водорода, содержащей по меньшей мере одну двойную связь, имеющей от двух до двенадцати атомов углерода, предпочтительно от двух до восьми атомов углерода, которую прикрепляют к остальной части молекулы посредством одинарной связи, например, этенил, проп 1 енил, бут 1 енил, пент 1 енил, пента 1,4 диенил и т. п. Когда конкретно установлено в описании, алкенильную группу можно необязательно замещать одной из следующих групп: галоген, циано, нитро, арил, циклоалкил, гетероциклил, гетероарил, оксо, триметилсиланил, -OR20, -OC(O)-R20, -N(R20)2, -C(O)R20, -C(O)OR20, C(O)N(R20)2, N(R20)C(O)OR22, -N(R20)C(O)R22, -N(R20)S(O)pR22 (где p представляет собой от 1 до 2), S(O)pOR22 (где p представляет собой от 1 до 2), -S(O)tR22 (где t представляет собой от 0 до 2) и -S(O)pN(R20)2 (где p представляет собой от 1 до 2), где каждый R20 представляет собой независимо водород, алкил, галогеналкил, циклоалкил, циклоалкилалкил, арил, аралкил, гетероциклил, гетероциклилалкил, гетероарил или гетероарилалкил; и каждый R22 представляет собой алкил, галогеналкил, циклоалкил, циклоалкилалкил, арил, аралкил, гетероциклил, гетероциклилалкил, гетероарил или гетероарилалкил.
«Алкилен» или «алкиленовая цепь» относится к неразветвленной или разветвленной двухвалентной углеводородной цепи, связывающей остальную часть молекулы с радикальной группой или связывающей две части молекулы, состоящей только из углерода и водорода, не содержащей ненасыщенность и имеющей от одного до двенадцати атомов углерода, например, метилен, этилен, пропилен, н бутилен и т. п. Алкиленовая цепь необязательно может содержать один или несколько гетероатомов, где углерод в алкиленовой цепи заменяют на гетероатом, выбранный из кислорода, азота или серы. Алкиленовую цепь прикрепляют к остальной части молекулы через одинарную связь и к радикальной группе через одинарную связь или прикрепляют к двумя частям молекулы через одинарную связь в каждой точке прикрепления. Когда конкретно установлено в описании, алкиленовую цепь можно необязательно замещать одной из следующих групп: алкил, алкенил, галоген, галогеналкенил, циано, нитро, арил, циклоалкил, гетероциклил, гетероарил, оксо, триметилсиланил, OR20, OC(O)R20, N(R20)2, C(O)R20, C(O)OR20, C(O)N(R20)2, N(R20)C(O)OR22, N(R20)C(O)R22, N(R20)S(O)pR22 (где p представляет собой от 1 до 2), S(O)pOR22 (где p представляет собой от 1 до 2), S(O)tR22 (где t представляет собой от 0 до 2) и S(O)pN(R20)2 (где p представляет собой от 1 до 2), где каждый R20 представляет собой независимо водород, алкил, галогеналкил, циклоалкил, циклоалкилалкил, арил, аралкил, гетероциклил, гетероциклилалкил, гетероарил или гетероарилалкил; и каждый R22 представляет собой алкил, галогеналкил, циклоалкил, циклоалкилалкил, арил, аралкил, гетероциклил, гетероциклилалкил, гетероарил или гетероарилалкил.
«Арил» относится к радикалу углеводородной кольцевой системы, содержащему водород, от 6 до 18 атомов углерода и по меньшей мере одно ароматическое кольцо. Для целей данного изобретения, арильный радикал может представлять собой моноциклическую, бициклическую, трициклическую или тетрациклическую кольцевую систему, которая может содержать конденсированные или мостиковые кольцевые системы. Арильные радикалы включают, но не ограничиваясь этим, арильные радикалы, полученные из ацеантрилена, аценафтилена, ацефенантрилена, антрацена, азулена, бензола, хризена, флуорантена, флуорена, as-индацена, s индацена, индана, индена, нафталина, феналена, фенантрена, плеиадена, пирена и трифенилена. Когда конкретно установлено в описании, арильную группу можно необязательно замещать одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, галогена, галогеналкила, галогеналкенила, циано, нитро, арила, аралкила, циклоалкила, циклоалкилалкила, гетероциклила, гетероциклилалкила, гетероарила, гетероарилалкила, R21OR20, R21OC(O)R20, R21N(R20)2, R21N(R20)R23OR20, R21C(O)R20, R21C(O)OR20, R21C(O)N(R20)2, R21N(R20)C(O)OR22, R21N(R20)C(O)R22, R21N(R20)S(O)pR22 (где p представляет собой от 1 до 2), R21N=C(OR20)R20, R21S(O)pOR22 (где p представляет собой от 1 до 2), R21S(O)tR22 (где t представляет собой от 0 до 2) и R21S(O)pN(R20)2 (где p представляет собой от 1 до 2), где каждый R20 представляет собой независимо водород, алкил, галогеналкил, циклоалкил, циклоалкилалкил, арил, аралкил, гетероциклил, гетероциклилалкил, гетероарил или гетероарилалкил; каждый R21 представляет собой независимо непосредственную связь или неразветвленную или разветвленную алкиленовую цепь; каждый R22 представляет собой алкил, галогеналкил, циклоалкил, циклоалкилалкил, арил, аралкил, гетероциклил, гетероциклилалкил, гетероарил или гетероарилалкил, и каждый R23 представляет собой непосредственную связь или неразветвленную или разветвленную алкиленовую цепь. Предпочтительно, необязательные заместители на необязательно замещенной арильной группе для R1 в настоящем описании представляют собой алкил, необязательно замещенный циклоалкил, галоген, галогеналкил, циано, необязательно замещенный гетероциклил, необязательно замещенный гетероциклилалкил, необязательно замещенный гетероарил -R21-OR20 и R21N(R20)2, (где R20 и R21 представляют собой то, что определено выше).
«Циклоалкил» относится к стабильному неароматическому моноциклическому или полициклическому углеводородному радикалу, состоящему только из атомов углерода и водорода, который может содержать конденсированные или мостиковые кольцевые системы, имеющие от трех до пятнадцати атомов углерода, предпочтительно имеющие от трех до десяти атомов углерода, который насыщен или ненасыщен и прикреплен к остальной части молекулы посредством одинарной связи. Моноциклические радикалы включают, например, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил. Полициклические радикалы включают, например, адамантил, норборнил, декалинил и т. п. Когда конкретно установлено в описании, циклоалкильную группу можно необязательно замещать одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, галогена, галогеналкила, галогеналкенила, циано, нитро, оксо, арила, аралкила, циклоалкила, циклоалкилалкила, гетероциклила, гетероциклилалкила, гетероарила, гетероарилалкила, R21OR20, R21OC(O)R20, R21N(R20)R23OR20, R21N(R20)2, R21C(O)R20, R21C(O)OR20, R21C(O)N(R20)2, R21N(R20)C(O)OR22, R21N(R20)C(O)R22, R21N(R20)S(O)pR22 (где p представляет собой от 1 до 2), R21N=C(OR20)R20, R21S(O)pOR22 (где p представляет собой от 1 до 2), R21S(O)tR22 (где t представляет собой от 0 до 2) и R21S(O)pN(R20)2 (где p представляет собой от 1 до 2), где каждый R20 представляет собой независимо водород, алкил, галогеналкил, циклоалкил, циклоалкилалкил, арил, аралкил, гетероциклил, гетероциклилалкил, гетероарил или гетероарилалкил; каждый R21 представляет собой независимо непосредственную связь или неразветвленную или разветвленную алкиленовую цепь; каждый R22 представляет собой алкил, галогеналкил, циклоалкил, циклоалкилалкил, арил, аралкил, гетероциклил, гетероциклилалкил, гетероарил или гетероарилалкил, и каждый R23 представляет собой непосредственную связь или неразветвленную или разветвленную алкиленовую цепь.
«Циклоалкилалкил» относится к радикалу формулы RbRg, где Rb представляет собой алкиленовую цепь, как определено выше, и Rg представляет собой циклоалкильный радикал, как определено выше. Когда конкретно установлено в описании, алкиленовую цепь и/или циклоалкильный радикал можно необязательно замещать, как определено выше, по необязательно замещенной алкиленовой цепи и необязательно замещенному циклоалкилу.
«Галоген» относится к брому, хлору, фтору или йоду.
«Галогеналкил» относится к алкильному радикалу, как определено выше, который замещен одним или несколькими галогеновыми радикалами, как определено выше, например, трифторметил, дифторметил, трихлорметил, 2,2,2 трифторэтил, 1 фторметил 2 фторэтил, 3 бром 2 фторпропил, 1 бромметил 2 бромэтил и т. п. Алкильную часть галогеналкильного радикала можно необязательно замещать, как определено выше, по алкильной группе.
«Гетероциклил» относится к стабильному 3 18 членному неароматическому кольцевому радикалу, который состоит из 2-12 атомов углерода и из 1-6 гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы. Если не установлено иное конкретно в описании, гетероциклильный радикал может представлять собой моноциклическую, бициклическую, трициклическую или тетрациклическую кольцевую систему, которая может включать конденсированные, мостиковые и спирокольцевые системы; и атомы азота, углерода или серы в гетероциклильном радикале необязательно могут быть окислены; атом азота необязательно может быть кватернизирован; и гетероциклильный радикал может быть частично или полностью насыщенным. Примеры таких гетероциклильных радикалов включают, но не ограничиваясь этим, азетидинил, 3-азабицикло[3.1.0]гексан-3-ил, 1азаспиро[3.3]гептан-1-ил, 5-азаспиро[2.3]гексан-5-ил, 2-окса-6-азаспиро[3.3]гептан-6-ил, 1-окса-6-азаспиро[3.4]октан-6-ил, 1-окса-6-азаспиро[3.3]гептан-6-ил, 6-окса-1-азаспиро[3.3]гептан-1-ил, 6-азаспиро[3.4]октан-6-ил, 7-окса-2-азаспиро[3.5]нонан-2-ил, 2,6-диазаспиро[3.3]гептан-2-ил, диоксоланил, диоксинил, тиенил[1.3]дитианил, декагидроизохинолил, имидазолинил, имидазолидинил, изотиазолидинил, изоксазолидинил, морфолинил, октагидроиндолил, октагидроизоиндолил, 2 оксопиперазинил, 2 оксопиперидинил, 2 оксопирролидинил, оксазолидинил, пиперидинил, пиперазинил, 4 пиперидонил, пирролидинил, пиразолидинил, хинуклидинил, тиазолидинил, 1,2,4-тиадиазол-5(4H)-илиден, тетрагидрофурил, триоксанил, тритианил, триазинанил, тетрагидропиранил, тиоморфолинил, тиаморфолинил, 1 оксо тиоморфолинил и 1,1 диоксо тиоморфолинил. Когда конкретно установлено в описании, гетероциклильную группу можно необязательно замещать одним или несколькими заместителями, выбранными из группы, состоящей из алкила, алкенила, галогена, галогеналкила, галогеналкенила, циано, оксо, тиоксо, нитро, арила, аралкила, циклоалкила, циклоалкилалкила, гетероциклила, гетероциклилалкила, гетероарила, гетероарилалкила, R21OR20, R21OC(O)R20, R21N(R20)R23OR20, R21N(R20)2, R21C(O)R20, R21C(O)OR20, R21C(O)N(R20)2, R21N(R20)C(O)OR22, R21N(R20)C(O)R22, R21N(R20)S(O)pR22 (где p представляет собой от 1 до 2), R21N=C(OR20)R20, R21S(O)pOR22 (где p представляет собой от 1 до 2), R21S(O)tR22 (где t представляет собой от 0 до 2) и R21S(O)pN(R20)2 (где p представляет собой от 1 до 2), где каждый R20 представляет собой независимо водород, алкил, алкенил, галогеналкил, циклоалкил, циклоалкилалкил, арил, аралкил, гетероциклил, гетероциклилалкил, гетероарил или гетероарилалкил; каждый R21 представляет собой независимо непосредственную связь или неразветвленную или разветвленную алкиленовую цепь; каждый R22 представляет собой алкил, алкенил, галогеналкил, циклоалкил, циклоалкилалкил, арил, аралкил, гетероциклил, гетероциклилалкил, гетероарил или гетероарилалкил, и каждый R23 представляет собой непосредственную связь или неразветвленную или разветвленную алкиленовую цепь.
«Гетероциклилалкил» относится к радикалу формулы RbRh, где Rb представляет собой алкиленовую цепь, как определено выше, и Rh представляет собой гетероциклильный радикал, как определено выше, и если гетероциклил представляет собой азот-содержащий гетероциклил, гетероциклил можно прикреплять к алкильному радикалу через атом азота. Когда конкретно установлено в описании, алкиленовую цепь гетероциклилалкильного радикала можно необязательно замещать, как определено выше, по необязательно замещенной алкиленовой цепи. Когда конкретно установлено в описании, гетероциклильную часть гетероциклилалкильного радикала можно необязательно замещать, как определено выше, по необязательно замещенной гетероциклильной группе. Предпочтительно необязательные заместители на необязательно замещенной гетероциклилалкильной группе для R5 в настоящем описании представляют собой галоген.
«Гетероарил» относится к радикалу 5 14 членной кольцевой системы, содержащему атомы водорода, 1-13 атомов углерода, 1-6 гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, и по меньшей мере одно ароматическое кольцо. Для целей данного изобретения, гетероарильный радикал может представлять собой моноциклическую, бициклическую, трициклическую или тетрациклическую кольцевую систему, которая может включать конденсированные или мостиковые кольцевые системы; и атомы азота, углерода или серы в гетероарильном радикале необязательно могут быть окисленными; атом азота необязательно может быть кватернизирован. Примеры включают, но не ограничиваясь этим, азепинил, акридинил, бензимидазолил, бензтиазолил, бензиндолил, бензодиоксолил, бензофуранил, бензооксазолил, бензотиазолил, бензотиадиазолил, бензо[b][1.4]диоксепинил, 1,4 бензодиоксанил, бензонафтофуранил, бензоксазолил, бензодиоксолил, бензодиоксинил, бензопиранил, бензопиранонил, бензофуранил, бензофуранонил, бензотиенил (бензотиофенил), бензотриазолил, бензо[4.6]имидазо[1.2 a]пиридинил, бензоксазолинонил, бензимидазолтионил, карбазолил, циннолинил, дибензофуранил, дибензотиофенил, фуранил, фуранонил, изотиазолил, имидазолил, индазолил, индолил, индазолил, изоиндолил, индолинил, изоиндолинил, изохинолил, индолизинил, изоксазолил, нафтиридинил, оксадиазолил, 2 оксоазепинил, оксазолил, оксиранил, 1 оксидопиридинил, 1 оксидопиримидинил, 1-оксидопиразинил, 1-оксидопиридазинил, 1 фенил 1H пирролил, феназинил, фенотиазинил, феноксазинил, фталазинил, птеридинил, птеридинонил, пуринил, пирролил, пиразолил, пиридинил, пиридинонил, пиразинил, пиримидинил, пиримидинонил, пиридазинил, пирролил, пиридо[2.3-d]пиримидинонил, хиназолинил, хиназолинонил, хиноксалинил, хиноксалинонил, хинолинил, изохинолинил, тетрагидрохинолинил, тиазолил, тиадиазолил, тиено[3,2-d]пиримидин-4-онил, тиено[2.3-d]пиримидин-4-онил, триазолил, тетразолил, триазинил и тиофенил (т. е. тиенил). Когда конкретно установлено в описании, гетероарильную группу можно необязательно замещать одним или несколькими заместителями, выбранным из группы, состоящей из алкила, алкенила, галогена, галогеналкила, галогеналкенила, циано, оксо, тиоксо, нитро, тиоксо, арила, аралкила, циклоалкила, циклоалкилалкила, гетероциклила, гетероциклилалкила, гетероарила, гетероарилалкила, R21OR20, R21OC(O)R20, R21N(R20)R23OR20, R21N(R20)2, R21C(O)R20, R21C(O)OR20, R21C(O)N(R20)2, R21N(R20)C(O)OR22, R21N(R20)C(O)R22, R21N(R20)S(O)pR22 (где p представляет собой от 1 до 2), R21N=C(OR20)R20, R21S(O)pOR22 (где p представляет собой от 1 до 2), R21S(O)tR22 (где t представляет собой от 0 до 2) и R21S(O)pN(R20)2 (где p представляет собой от 1 до 2), где каждый R20 представляет собой независимо водород, алкил, алкенил, галогеналкил, циклоалкил, циклоалкилалкил, арил, аралкил, гетероциклил, гетероциклилалкил, гетероарил или гетероарилалкил; каждый R21 представляет собой независимо непосредственную связь или неразветвленную или разветвленную алкиленовую цепь; каждый R22 представляет собой алкил, алкенил, галогеналкил, циклоалкил, циклоалкилалкил, арил, аралкил, гетероциклил, гетероциклилалкил, гетероарил или гетероарилалкил, и каждый R23 представляет собой непосредственную связь или неразветвленную или разветвленную алкиленовую цепь. Предпочтительно, необязательные заместители на необязательно замещенной бициклической гетероарильной группе для R1 в настоящем описании представляют собой галоген. Предпочтительно, необязательные заместители на необязательно замещенной моноциклической гетероарильной группе для R1 в настоящем описании представляют собой алкил.
«N гетероарил» относится к гетероарильному радикалу, как определено выше, содержащему по меньшей мере один азот. Точка прикрепления N-гетероарила к остальной части молекулы может представлять собой атом азота или атом углерода в N-гетероариле. Когда конкретно установлено в описании, N-гетероарильный радикал можно необязательно замещать, как описано выше, по необязательно замещенному гетероарильному радикалу. Предпочтительно необязательные заместители на необязательно замещенной 5-членной N-гетероарильной группе для R2 в настоящем описании представляют собой алкил и галоген. Предпочтительно необязательные заместители на необязательно замещенной 6-членной N-гетероарильной группе для R2 в настоящем описании представляют собой алкил, галоген и галогеналкил.
«Гетероарилалкил» относится к радикалу формулы RbRi, где Rb представляет собой алкиленовую цепь, как определено выше, и Ri представляет собой гетероарильный радикал, как определено выше. Когда конкретно установлено в описании, гетероарильную часть гетероарилалкильного радикала можно необязательно замещать, как определено выше, по необязательно замещенной гетероарильной группе. Когда конкретно установлено в описании, часть алкиленовой цепи гетероарилалкильного радикала можно необязательно замещать, как определено выше, по необязательно замещенной алкиленовой цепи.
«Пролекарственное средство» используют для обозначения соединения, которое может превращаться в физиологических условиях или посредством сольволиза в биологически активное соединение по изобретению. Таким образом, термин «пролекарственное средство» относится к метаболическому предшественнику соединения по изобретению, который является фармацевтически приемлемым. Пролекарственное средство может быть неактивным при введении нуждающемуся в этом субъекту, но превращается in vivo в активное соединение по изобретению. Пролекарственные средства обычно быстро превращаются in vivo и дают исходное соединение по изобретению, например, посредством гидролиза в крови. Соединение пролекарственного средства часто дает преимущества растворимости, тканевой совместимости или отсроченного высвобождения в организме млекопитающего (см., Bundgard, H., Design of Prodrugs (1985), стр. 7 9, 21 24 (Elsevier, Amsterdam)). Обсуждение пролекарственных средств приведено в Higuchi, T., et al., «Pro drugs as Novel Delivery Systems», A.C.S. Symposium Series, том 14, и в Bioreversible Carriers in Drug Design, ред. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987, оба полностью включены по ссылке в настоящее описание.
Значение термина «пролекарственное средство» также включает любые ковалентно связанные носители, которые высвобождают активное соединение по изобретению in vivo, когда такое пролекарственное средство вводят млекопитающему субъекту. Пролекарственные средства соединения по изобретению можно получать посредством модификации функциональных групп, присутствующих в соединении по изобретению таким образом, что происходит отщепление модификаций или при стандартных манипуляциях или in vivo до исходного соединения по изобретению. Пролекарственные средства включают соединения по изобретению, в которых гидрокси-, амино- или меркаптогруппу связывают с любой группой, которая при введении пролекарственного средства соединения по изобретению млекопитающему субъекту отщепляется для того, чтобы формировать свободную гидрокси-, свободную амино- или свободную меркаптогруппу, соответственно. Примеры пролекарственных средств включают, но не ограничиваясь этим, ацетатные, формиатные и бензоатные производные спиртовых или амидные производные аминных функциональных групп в соединении по изобретению и т. п.
Также подразумевают, что раскрытое в настоящем описании изобретение охватывает все фармацевтически приемлемые соединения формулы (I), которые метят изотопами, заменяя один или несколько атомов на атом, имеющий другую атомную массу или массовое число. Примеры изотопов, которые можно встраивать в раскрытые соединения, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора, хлора и йода, такие как2H,3H,11C,13C,14C,13N,15N,15O,17O,18O,31P,32P,35S,18F,36Cl,123I и125I, соответственно. Эти радиоактивно меченные соединения могут быть полезны для того, чтобы помогать определять или измерять эффективность соединений, посредством определения характеристик, например, места или механизма действия в натриевых каналах, или аффинности связывания с фармакологически важным местом приложения действия в натриевых каналах. Определенные изотопно меченные соединения формулы (I), например, те, которые содержат радиоактивный изотоп, можно использовать в исследованиях распределения лекарственных средств и/или субстратов в тканях. Радиоактивные изотопы тритий, т. е.3H, и углерод-14, т. е.14C, в частности, можно использовать с этой целью ввиду простоты их встраивания и готовых средств обнаружения.
Замена на более тяжелые изотопы, такие как дейтерий, т. е.2H, могут давать определенные терапевтические преимущества в результате более высокой метаболической стабильности, например, увеличенное время полужизни in vivo или сниженные требования к дозам, и, таким образом, могут быть предпочтительны в некоторых обстоятельствах. В одном из вариантов осуществления изобретения соединения формулы (I) обогащают дейтерием. Такие дейтерированные соединения можно получать способами, известными специалисту в данной области, такими как замена протонов на дейтерий или синтез молекулы с использованием обогащенных исходных материалов.
Замена с использованием изотопов, испускающих позитроны, таких как11C,18F,15O и13N, может быть полезна в исследованиях позитронно-эмиссионной томографии (PET) для изучения заполнения рецепторов субстратом. Изотопно меченные соединения формулы (I) в целом можно получать общепринятыми способами, известными специалистам в данной области, или с помощью процессов, аналогичных тем, что описаны в примерах и Получении, как изложено далее, используя подходящий изотопно меченный реактив вместо немеченного реактива, который использовали ранее.
Также подразумевают, что раскрытое в настоящем описании изобретение охватывает для раскрытых соединений метаболические продукты in vivo. Такие продукты могут быть результатом, например, окисления, восстановления, гидролиза, амидирования, этерификации и т. п. вводимого соединения, в первую очередь вследствие ферментативных процессов. Соответственно, изобретение включает соединения, получаемые с помощью процесса, включающего приведение в контакт соединения по данному изобретению с млекопитающим в течение определенного периода времени, достаточного для получения его метаболического продукта. Такие продукты обычно идентифицируют посредством введения радиоактивно меченного соединения по изобретению в поддающейся обнаружению дозе животному, такому как крыса, мышь, морская свинка, обезьяна, или человеку, давая достаточное время, чтобы прошел метаболизм, и выделения его продуктов превращения из мочи, крови или других биологических образцов.
«Стабильное соединение» и «стабильная структура» обозначают соединение, которое достаточно устойчиво, чтобы перенести выделение до полезной степени чистоты из реакционной смеси и формулирование в эффективном терапевтическом средстве.
«Млекопитающее» включает человека, а также домашних животных, таких как лабораторные животные, домашних питомцев (например, кошек, собак, свиней, крупный рогатый скот, овец, коз, лошадей, кроликов) и не одомашненных животных, например, из дикой природы, и т. п.
«Необязательный» или «необязательно» обозначает, что далее описанное событие или обстоятельства могут произойти или не произойти, и что описание включает случаи, когда указанное событие или обстоятельства происходят, и случаи, когда они не происходят. Например, «необязательно замещенный арил» обозначает, что арильный радикал может быть или может не быть замещенным и что описание включает как замещенные арильные радикалы, так и арильные радикалы, не имеющие замены («незамещенные»). Когда функциональную группу описывают как «необязательно замещенную», и, в свою очередь, заместители на функциональной группе также «необязательно замещены» и так далее, для целей данного изобретения, такие итерации ограничены пятью, предпочтительно такие итерации ограничены двумя.
«Фармацевтически приемлемый носитель, разбавитель или эксципиент» включает без ограничения любой адъювант, носитель, эксципиент, скользящее средство, подсластитель, разбавитель, консервант, краситель/красящее вещество, усилитель аромата, поверхностно-активное средство, смачивающее средство, диспергирующее средство, суспендирующее средство, стабилизатор, изотоническое средство, растворитель или эмульсификатор, которые одобрены в United States Food and Drug Administration в качестве приемлемых для использования у человека или домашних животных.
«Фармацевтически приемлемая соль» включает кислотно- и основно-аддитивные соли.
«Фармацевтически приемлемая кислотно-аддитивная соль» относится к тем солям, которые сохраняют биологическую эффективность и свойства свободных оснований, которые являются биологически или иным образом нежелательными и которые образуются с неорганическими кислотами, такими как, но не ограничиваясь этим, соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т. п., и органическими кислотами, такими как, но не ограничиваясь этим, уксусная кислота, 2,2-дихлоруксусная кислота, адипиновая кислота, альгиновая кислота, аскорбиновая кислота, аспарагиновая кислота, бензолсульфоновая кислота, бензойная кислота, 4-ацетамидобензойная кислота, камфорная кислота, камфор-10-сульфоновая кислота, каприновая кислота, капроновая кислота, каприловая кислота, угольная кислота, коричная кислота, лимонная кислота, цикламовая кислота, додецилсерная кислота, этан-1,2-дисульфоновая кислота, этансульфоновая кислота, 2-гидроксиэтансульфоновая кислота, муравьиная кислота, фумаровая кислота, галактаровая кислота, гентизиновая кислота, глюкогептоновая кислота, глюконовая кислота, глюкуроновая кислота, глутаминовая кислота, глутаровая кислота, 2-оксоглутаровая кислота, глицерофосфорная кислота, гликолевая кислота, гиппуровая кислота, изомасляная кислота, молочная кислота, лактобионовая кислота, лауриновая кислота, малеиновая кислота, яблочная кислота, малоновая кислота, миндальная кислота, метансульфоновая кислота, муциновая кислота, нафталин-1,5-дисульфоновая кислота, нафталин-2-сульфоновая кислота, 1-гидрокси-2-нафтойная кислота, никотиновая кислота, олеиновая кислота, оротовая кислота, щавелевая кислота, пальмитиновая кислота, памовая кислота, пропановая кислота, пироглутаминовая кислота, пировиноградная кислота, салициловая кислота, 4-аминосалициловая кислота, себациновая кислота, стеариновая кислота, янтарная кислота, винная кислота, тиоциановая кислота, п-толуолсульфоновая кислота, трифторуксусная кислота, ундециленовая кислота и т. п.
«Фармацевтически приемлемая основно-аддитивная соль» относится к тем солям, которые сохраняют биологическую эффективность и свойства свободных кислот, которые не являются биологически или иным образом нежелательными. Эти соли получают добавлением неорганического основания или органического основания к свободной кислоте. Соли, получаемые из неорганический оснований, включают, но не ограничиваясь этим, соли натрия, калия, лития, аммония, кальция, магния, железа, цинка, меди, марганца, алюминия и т. п. Предпочтительные неорганические соли представляют собой соли аммония, натрия, калия, кальция и магния. Соли, получаемые из органических оснований, включают, но не ограничиваясь этим, соли первичных, вторичных и третичных аминов, замещенных аминов, включая встречаемые в природе замещенные амины, циклические амины и основные ионообменные смолы, таких как аммиак, изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин, диэтаноламин, этаноламин, деанол, 2 диметиламиноэтанол, 2 диэтиламиноэтанол, дициклогексиламин, лизин, аргинин, гистидин, кофеин, прокаин, гидрабамин, холин, бетаин, бенетамин, бензатин, этилендиамин, глюкозамин, метилглюкамин, теобромин, триэтаноламин, трометамин, пурины, пиперазин, пиперидин, N этилпиперидин, полиаминные смолы и т. п. Особенно предпочтительными органическими основаниями являются изопропиламин, диэтиламин, этаноламин, триметиламин, дициклогексиламин, холин и кофеин.
Часто кристаллизацией получают сольватные соединения по изобретению. Как используют в настоящем описании, термин «сольват» относится к агрегату, который содержит одну или несколько молекул соединения по изобретению с одной или несколькими молекулами растворителя. Растворителем может быть вода, и в этом случае сольват может представлять собой гидрат. Альтернативно, растворителем может быть органический растворитель. Таким образом, соединения по настоящему изобретению могут существовать в виде гидрата, в том числе моногидрата, дигидрата, полугидрата, сесквигидрат, тригидрата, тетрагидрата и т. п., а также соответствующих сольватированных форм. Соединение по изобретению может быть истинным сольватом, тогда как в других случаях соединение по изобретению может лишь сохранять случайную воду или представлять собой смесь воды и некоторого случайного растворителя.
«Фармацевтическая композиция» относится к составу соединения по изобретению и среде, общепринятой в данной области для доставки биологически активного соединения млекопитающим, например, человеку. Для этого такая среда включает все фармацевтически приемлемые носители, разбавители или эксципиенты.
«Терапевтически эффективное количество» относится к такому количеству соединения по изобретению, которого при введении млекопитающему, предпочтительно человеку, достаточно для того, чтобы осуществлять лечение, как определено ниже, опосредованного натриевыми каналами заболевания или состояния у млекопитающего, предпочтительно у человека. Количество соединения по изобретению, которое составляет «терапевтически эффективное количество», варьирует в зависимости от соединения, состояния и его тяжести, способа введения и возраста млекопитающего, подлежащего лечению, но его может определять обычным образом специалист в данной области, исходя из своих знаний и этого раскрытия.
«Лечение», как используют в настоящем описании, охватывает лечение заболевания или состояния, представляющего интерес, у млекопитающего, предпочтительно человека, имеющего заболевание или состояние, представляющее интерес, и включает:
(a) предотвращение возникновения заболевания или состояния у млекопитающего, в частности, когда такое млекопитающе предрасположено к состоянию, но у него оно еще не диагностировано;
(b) ингибирование заболевания или состояния, т. е. торможение его развития;
(c) облегчение (или улучшение) заболевания или состояния, т. е. инициацию регрессии заболевания или состояния; или
(d) облечгение (или улучшение) симптомов в результате заболевания или состояния, например, облегчение эпилепсии, без воздействия на подлежащее заболевание или состояние.
Как используют в настоящем описании, термины «заболевание» и «состояние» можно использовать взаимозаменяемо, или они могут различаться в том отношении, что конкретное расстройство или состояние может не иметь известного причинного фактора (так что этиология до сих пор не проработана) и, следовательно, оно до сих пор не известно в качестве заболеваниа, но известно только в качестве нежелательного состояния или синдрома, при котором клиницистами идентифицирован более или менее специфический набор симптомов.
Соединения по изобретению или их фармацевтически приемлемые соли могут содержать один или несколько асимметричных центров и, таким образом, могут давтаь начало энантиомерам, диастереомерам и другим стереоизомерным формам, которые можно определять в терминах абсолютной стереохимии как (R) или (S) или как (D) или (L) для аминокислот. Подразумевают, что настоящее изобретение включает все такие возможные изомеры, а также их рацемически и оптически чистые формы. Оптически активные (+) и ( ), (R) и (S) или (D) и (L) изомеры можно получать с использованием хиральных синтонов или хиральных реактивов или разрешать с использованием общепринятых способов, например, хроматографии и фракционной кристаллизации. Общепринятые способы для получения/выделения индивидуальных энантиомеров включают хиральный синтез из подходящего оптическм чистого предшественника или разрешение рацемата (или рацемата соли или производного) с использованием, например, хиральной жидкостной хроматографии высого давления (HPLC). Когда соединения, описанные в настоящем описании, содержат олефиновые двойные связи или другие центры геометрической aсимметрии, и пока не определено иное, подразумевают, что соединения включают E и Z геометрические изомеры. Аналогичным образом, также предусмотрено, что включены все таутомерные формы.
«Стереоизомер» относится к соединению, состоящему из тех же атомов, связанных теми же связями, но имеющему отличающиеся трехмерные структуры, которые не являются взаимнозаменяемыми. Настоящее изобретение предусматривает различные стереоизомеры и ию смеси и включает энантиомеры, что относится к двум стереоизомерам, молекулых которых представляют собой зеркальные изображения друг друга, которые не совпадают при наложении. Подробное описание структуры и свойств энантиомеров и стереоизомеров см., например, в Smith, M.B. and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 6-е изд. (Wiley, 2007).
«Таутомер» относится к сдвигу протона от одного атома молекулы к другому атому той же молекулы. Настоящее изобретение включает таутомеры любых указанных соединений.
Использование круглых скобок и квадратных скобок в группах заместителей используют в настоящем описании для экономии места. Соответственно, использование круглой скобки в группе заместителя указывает на то, что группа, заключенная в круглые скобки, прикреплена непосредственно к атому, предшествующему круглой скобке. Использование квадратных скобок в группе заместителя указывает на то, что группу, заключенную в квадратные скобки, также прикрепляют непосредственно к атому, предшествующему круглой скобке.
Протокол наименования химических соединений и структурные диаграммы, используемые в настоящем описании, представляют собой модифицированную форму систематической номенклатуры IUPAC, с использованием программного обеспечения ChemBioDraw Ultra версии 14.0, где в настоящем описании соединения по изобретению называли в виде производных структуры центрального ядра, например, бензолсульфонамидной структуры. Для сложных химических названий, используемых в настоящем описании, группу заместителя называют перед группой, к которой его прикрепляют. Например, циклопропилэтил содержит этиловый остов с циклопропильным заместителем. На диаграммах химических структур идентифицированы все связи, за исключением некоторых атомов углерода, которые предполагают связанными с достаточным числом атомов водорода до полной валентности.
«Энантиомеры» относятся к асимметричным молекулам, которые могут существовать в двух различных изомерных формах, которые имеют различные конфигурации в пространстве. Другие термины, используемые для обозначения или отсылки к энантиомерам, включают «стереоизомеры» (по причине различного расположения или стереохимии вокруг хирального центра; несмотря на то, что все энантиомеры являются стереоизомерами, не все стереоизомеры являются энантиомерами) или «оптические изомеры» (по пичине оптической активности чистых энантиомеров, которая представляет собой способность различных чистых энантиомеров вращать плоско-поляризованный свет в различных направлениях).
Обозначения «R» и «S» для абсолютной конфигурации энантиомера по изобретению могут выглядеть как префикс или как суффикс в названии соединения; они могут быть или не быть отделены от названия энантиомера дефисом; они могут быть или не быть написанными через дефис; и они могут быть или не быть окружены круглыми скобками.
Следуя практике описания в стандартной химической литературе и как используют в этом описании, сплошная целая связь, как проиллюстрировано вверху структуры (A), и штриховая целая связь, как проиллюстрировано в образцовой структуре (A) внизу, обозначают, что заместители находятся в транс-конфигурации относительно плоскости кольца:

Аналогичным образом, связи в следующих образцовых структурах (Aa) и (Ab) находятся в цис-конфигурации относительно плоскости кольца:

Следуя практике описания в стандартной химической литературе и как используют в этом описании, сплошная клиновидная связь, как проиллюстрировано далее в структуре (B), обозначает, что заместитель, связанный с кольцом с помощью этой связи, в этом случае заместитель R30, находится выше плоскости кольца, как проиллюстрировано на странице в двухмерном представлении, а штриховая клиновидная связь, как проиллюстрировано ниже в структуре (B), обозначает, что заместитель, связанный с кольцом с помощью этой связи, в этом случае заместитель R31, находится ниже плоскости кольца, как показано на странице в двухмерном представлении;

Следуя практике описания в стандартной химической литературе и как используют в этом описании, волнистая связь, как проиллюстрировано ниже в структуре (C), указывает на то, что заместитель, в этом случае заместитель R30, находится или ниже плоскости кольца или выше плоскости кольца:

В формулах, изображенных в настоящем описании, связь с заместителем и/или связь, которая соединяет молекулярный фрагмент с остальной частью соединения, можно показывать пересекающей одну или несколько связей в кольцевой структуре. Это указывает на то, что связь можно прикреплять к любому одному из атомов, которые образуют кольцевую структуру, при условии, что атом водорода иначе мог бы присутствовать при этом атоме. Где для конкретного положения в структуре не идентифицируют конкретный заместитель(и), в этом положении присутствует водород(ы). Например, в следующей структуре (D), связь, прикрепляющая заместитель R30, может быть на любом из углеродов, включая углерод, к которому прикреплен R31, при условии, что валентность допускает такое прикрепление:

«Разрешение», когда используют в отношении рацемического соединения или рацемической смеси соединений по изобретению, относится к разделению рацемического соединения или рацемической смеси на две его энантиомерные формы (т. е. (+) и (-); формы (R) и (S)).
«Энантиомерный избыток» или «ee», как используют в настоящем описании, относится к продукту, в котором один энантиомер присутсвует в избытке относительно другого, и его определяют как абсолютную разность в молярной фракции каждого энантиомера. Энантиомерный избыток обычно выражают как процентную долю энантиомера, присутствующего в смеси, относительно другого энантиомера. Для целей данного изобретения, (S)-энантиомер соединения, полученный способами, раскрытыми в настоящем описании, считают «по существу свободным» от соответствующего (R) энантиомера, когда (S)-энантиомер присутствует в энантиомерном избытке больше 80%, предпочтительно больше 90%, более предпочтительно больше 95% и наиболее предпочтительно больше 99%.
Протокол наименования химических соединений и структурные диаграммы, используемые в настоящем описании, представляют собой модифицированную форму систематической номенклатуры IUPAC, с использованием программного обеспечения ChemBioDraw Ultra версии 14.0, где в настоящем описании соединения по изобретению называли в виде производных структуры центрального ядра, например, бензолсульфонамидной структуры. Для сложных химических названий, используемых в настоящем описании, группу заместителя называют перед группой, к которой его прикрепляют. Например, циклопропилэтил содержит этиловый остов с циклопропильным заместителем. На диаграммах химических структур идентифицированы все связи, за исключением некоторых атомов углерода, которые предполагают связанными с достаточным числом атомов водорода до полной валентности.
Соответственно, (R)-энантиомер соединения формулы (I), как описано выше в Сущности изобретения, где A представляет собой (CH2)m C(R4)(R5) (CH2)n (где m и n оба представляют собой 0, R4 представляет собой этил и R5 представляет собой водород); q представляет собой 1, R1 представляет собой незамещенный фенил, R2 представляет собой 1,2,4-тиадиазол-5-ил, R3 представляет собой -N(R8)-, где R8 представляет собой водород, R6 представляет собой водород и R7 представляет собой хлор, т. е., соединение следующей структуры:

называют в настоящем описании как (R)-3-хлор-4-(1-фенилпропиламино)-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамид.
Варианты осуществления изобретения
Один аспект изобретения представляет собой соединения формулы (I), как изложено выше в Сущности изобретения, в виде их индивидуального стереоизомера, энантиомера или таутомера или их смеси; или их фармацевтически приемлемой соли, сольвата или пролекарственного средства.
В одном из вариантов осуществления соединение формулы (I) представляет собой соединение формулы (I), в котором R3 представляет собой O, где соединение имеет следующую формулу (Ia):

где каждое из q, A, R1, R2, R4, R5, R6, R7, R9, R10, R11 и R12 представляет собой то, что определено выше в Сущности изобретения;
в виде его индивидуального стереоизомера, энантиомера или таутомера или их смесей;
или его фармацевтически приемлемой соли, сольвата или пролекарственного средства.
В другом варианте осуществления соединение формулы (I) представляет собой соединение формулы (Ia), как определено выше, где A представляет собой непосредственную связь, т. е. соединение формулы (Ia1):

где каждое из q, R1, R2, R6 и R7 представляет собой то, что определено выше в Сущности изобретения;
в виде его индивидуального стереоизомера, энантиомера или таутомера или их смесей; или его фармацевтически приемлемой соли, сольвата или пролекарственного средства.
По этому варианту осуществления, предпочтительное соединение формулы (Ia1) представляет собой 5-хлор-2-фтор-N-(тиазол-4-ил)-4-(4-(трифторметил)-2,3-дигидро-1H-инден-1-илокси)бензолсульфонамид.
В другом варианте осуществления соединение формулы (I) представляет собой соединение формулы (Ia), как определено выше, где A представляет собой (CH2)m C(R4)(R5) (CH2)n, т. е. соединение формулы (Ia2):

где каждое из m, n, R1, R2, R4, R5, R6 и R7 представляет собой то, что определено выше в Сущности изобретения;
в виде его индивидуального стереоизомера, энантиомера или таутомера или их смесей;
или его фармацевтически приемлемой соли, сольвата или пролекарственного средства.
По этому варианту осуществления, предпочтительные соединения формулы (Ia2) выбирают из:
4-((2-(азетидин-1-илметил)-3,6-дифторбензил)окси)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
(R)-3-хлор-4-(1-фенилэтокси)-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида;
(S)-3-хлор-4-(1-фенилэтокси)-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида;
(S)-3-хлор-2-фтор-4-(1-(2-фторфенил)этокси)-N-(тиазол-2-ил)бензолсульфонамида;
5-хлор-2-фтор-4-(изохинолин-8-илметокси)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-2,5-дифтор-4-(1-(2-фторфенил)этокси)-N-(тиазол-2-ил)бензолсульфонамида;
(R)-5-хлор-2-фтор-N-(тиазол-2-ил)-4-(2,2,2-трифтор-1-фенилэтокси)бензолсульфонамида;
(S)-5-хлор-4-(1-(5-хлор-2-фторфенил)этокси)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-(1-(2-фторфенил)этокси)-N-(тиазол-4-ил)бензолсульфонамида;
(S)-5-хлор-4-(1-(3,4-дихлорфенил)этокси)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-(1-(3-фторфенил)этокси)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-(1-фенилэтокси)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-(1-(2-фторфенил)этокси)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-4-(1-(2-хлорфенил)этокси)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-(1-(3-фторфенил)этокси)-N-(тиазол-4-ил)бензолсульфонамида;
(S)-5-хлор-4-(1-(2,6-дифторфенил)этокси)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-4-(1-(2,6-дифторфенил)этокси)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
(R)-5-хлор-2-фтор-4-(1-(2-фторфенил)этокси)-N-(тиазол-2-ил)бензолсульфонамида;
5-хлор-2-фтор-4-((2-фторбензил)окси)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-4-(1-(2-хлорфенил)этокси)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
(S)-5-хлор-4-(1-(5-хлор-2-фторфенил)этокси)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)бензил)окси)-5-хлор-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
(S)-2,6-дифтор-4-(1-(2-фторфенил)этокси)-N-(тиазол-4-ил)бензолсульфонамида;
5-хлор-2-фтор-4-(1-фенилциклопропокси)-N-(тиазол-4-ил)бензолсульфонамида; и
(S)-5-хлор-2-фтор-4-(1-(2-фторфенил)пропокси)-N-(тиазол-4-ил)бензолсульфонамида.
В другом варианте осуществления соединение формулы (I) представляет собой соединение формулы (I), в котором R3 представляет собой N(R8), где соединение имеет следующую формулу (Ib):

где каждое из q, A, R1, R2, R6, R7 и R8 представляет собой то, что определено выше в Сущности изобретения;
в виде его индивидуального стереоизомера, энантиомера или таутомера или их смесей;
или его фармацевтически приемлемой соли, сольвата или пролекарственного средства.
В другом варианте осуществления соединение формулы (I) представляет собой соединение формулы (Ib), как определено выше, где A представляет собой непосредственную связь, т. е. соединение формулы (Ib1):

где каждое из q, R1, R2, R6, R7 и R8 представляет собой то, что определено выше в Сущности изобретения;
в виде его индивидуального стереоизомера, энантиомера или таутомера или их смесей;
или его фармацевтически приемлемой соли, сольвата или пролекарственного средства.
По этому варианту осуществления, предпочтительные соединения формулы (Ib1) выбирают из:
5-хлор-2-фтор-4-((1-(пиридин-3-ил)пропан-2-ил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
(R)-3-хлор-4-(2,3-дигидро-1H-инден-1-иламино)-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида;
(S)-3-хлор-4-(2,3-дигидро-1H-инден-1-иламино)-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-(1,2,3,4-тетрагидронафтален-1-иламино)-N-(тиазол-2-ил)бензолсульфонамида;
(R)-5-хлор-2-фтор-4-(1,2,3,4-тетрагидронафтален-1-иламино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-3-хлор-4-((5,6,7,8-тетрагидрохинолин-8-ил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
2,2,2-трифторацетата 5-хлор-2-фтор-4-((5,6,7,8-тетрагидроизохинолин-8-ил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
2,2,2-трифторацетата 5-хлор-2-фтор-4-((5,6,7,8-тетрагидроизохинолин-5-ил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
соли 5-хлор-2-фтор-4-((5,6,7,8-тетрагидрохинолин-5-ил)амино)-N-(тиазол-2-ил)бензолсульфонамида и муравьиной кислоты
4-(((1R,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-ил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-(((1R,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-ил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида; и
4-(((1S,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-ил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамидформиата.
В другом варианте осуществления соединение формулы (I) представляет собой соединение формулы (Ib), как определено выше, где A представляет собой (CH2)m C(R4)(R5) (CH2)n, т. е. соединение формулы (Ib2):

где каждое из m, n, R1, R2, R4, R5, R6, R7 и R8 представляет собой то, что определено выше в п. 1;
в виде его индивидуального стереоизомера, энантиомера или таутомера или их смесей;
или его фармацевтически приемлемой соли, сольвата или пролекарственного средства.
По этому варианту осуществления, предпочтительным вариантом осуществления являются те соединения формулы (Ib2), в которых R2 представляет собой необязательно замещенный 5членный N-гетероарил.
По этому варианту осуществления, предпочтительным вариантом осуществления являются те соединения формулы (Ib2), в которых R2 выбирают из необязательно замещенного тиазолила, необязательно замещенного тиадиазолила, необязательно замещеннго изоксазолила, необязательно замещеннго изотиазолила или необязательно замещеннго оксазолила.
По этому варианту осуществления, предпочтительным вариантом осуществления являются те соединения формулы (Ib2), в которых:
R1 представляет собой необязательно замещенный циклоалкил;
или R1 представляет собой арил, необязательно замещенный одним или несколькими заместителями, выбранными из галогена, алкила, галогеналкила, необязательно замещенного циклоалкила, циано, R9OR12, R9N(R10)R11, R9N(R10)-R13-OR12, необязательно замещенного гетероциклила и необязательно замещенного гетероарила;
R2 представляет собой необязательно замещенный тиазолил; и
R13 представляет собой разветвленную или неразветвленную алкиленовую цепь.
По этому варианту осуществления, предпочтительные соединения формулы (Ib2) выбирают из:
(S)-5-хлор-4-((1-циклогексилэтил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
3-хлор-4-(1-фенилпропиламино)-N-(тиазол-2-ил)бензолсульфонамида;
5-хлор-2-фтор-4-(1-фенилпропиламино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-(1-фенилпропиламино)-N-(тиазол-4-ил)бензолсульфонамида;
(R)-5-хлор-2-фтор-4-(1-фенилпропиламино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-(1-фенилпропиламино)-N-(тиазол-2-ил)бензолсульфонамида;
5-хлор-2-фтор-4-(1-(2-фторфенил)этиламино)-N-(тиазол-2-ил)бензолсульфонамида;
5-хлор-4-(2-(диметиламино)-1-фенилэтиламино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
(R)-5-хлор-2-фтор-4-(1-фенилэтиламино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-(1-фенилэтиламино)-N-(тиазол-2-ил)бензолсульфонамида;
5-хлор-2-фтор-4-(1-фенилциклопропиламино)-N-(тиазол-2-ил)бензолсульфонамида;
5-хлор-2-фтор-N-(тиазол-2-ил)-4-(3,3,3-трифтор-1-фенилпропиламино)бензолсульфонамида;
(S)-5-бром-2-фтор-4-(1-фенилпропиламино)-N-(тиазол-2-ил)бензолсульфонамида;
5-хлор-4-(1-(2-хлорфенил)этиламино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-(1-(2-фторфенил)этиламино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-(1-(2-фторфенил)пропиламино)-N-(тиазол-2-ил)бензолсульфонамида;
3-хлор-4-(циклопропил(фенил)метиламино)-N-(тиазол-2-ил)бензолсульфонамида;
3-хлор-4-(метил(1-фенилпропил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-(1-(4-фторфенил)этиламино)-N-(тиазол-2-ил)бензолсульфонамида;
5-хлор-2-фтор-4-(2-морфолино-1-фенилэтиламино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-2-фтор-5-метил-4-(1-фенилпропиламино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-3-хлор-4-(1-фенилэтиламино)-N-(тиазол-2-ил)бензолсульфонамида;
5-хлор-2-фтор-4-(1-(5,6,7,8-тетрагидронафтален-2-ил)пропиламино)-N-(тиазол-2-ил)бензолсульфонамида;
(R)-3-хлор-4-(2-гидрокси-1-фенилэтиламино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-N-(тиазол-2-ил)-4-(1-о-толилпропиламино)бензолсульфонамида;
(R)-4-(2-(азетидин-1-ил)-1-фенилэтиламино)-5-хлор-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-4-(1-(2-хлорфенил)этиламино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
5-хлор-2-фтор-4-(1-фенилциклобутиламино)-N-(тиазол-2-ил)бензолсульфонамида;
5-хлор-2-фтор-4-(3-метил-1-фенилбутиламино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-(2-метокси-1-фенилэтиламино)-N-(тиазол-2-ил)бензолсульфонамида;
(R)-5-хлор-2-фтор-4-(2-метокси-1-фенилэтиламино)-N-(тиазол-2-ил)бензолсульфонамида;
3-хлор-4-(1-фенилциклобутиламино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-(1-фенилэтиламино)-N-(тиазол-4-ил)бензолсульфонамида;
3-хлор-4-(3-фенилоксетан-3-иламино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-((1-(нафтален-1-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-N-(тиазол-2-ил)-4-((1-(3-(трифторметил)фенил)этил)амино)бензолсульфонамида;
соли (R)-3-хлор-N-(тиазол-2-ил)-4-((2,2,2-трифтор-1-фенилэтил)амино)бензолсульфонамида и муравьиной кислоты;
(S)-5-хлор-4-((1-(5-хлор-2-фторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
(S)-5-хлор-4-((1-(3-хлор-2-фторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
(S)-4-((1-(3-бромфенил)этил)амино)-5-хлор-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-4-((1-(5-хлор-2-фторфенил)этил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
(S)-3-хлор-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-4-((1-(4-хлорфенил)этил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-((1-(нафтален-2-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
5-хлор-4-((2-цианобензил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
5-хлор-2-фтор-4-((1-фенилциклобутил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
5-хлор-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
(S)-3,5-дихлор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-3-хлор-4-((1-фенилэтил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
(S)-2,5-дифтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-2,6-дифтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
5-хлор-2-фтор-4-((1-(4-фторфенил)циклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-((1-(3-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-4-((1-(3-хлорфенил)этил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-4-((1-(3,5-дихлорфенил)этил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-4-((1-(2,4-дифторфенил)этил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-4-((1-(3,4-дифторфенил)этил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
2,2,2-трифторацетата (S)-5-хлор-4-((2-(диметиламино)-1-фенилэтил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
2,2,2-трифторацетата (R)-5-хлор-4-((2-(диметиламино)-1-фенилэтил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
2,2,2-трифторацетата (S)-4-((2-(азетидин-1-ил)-1-фенилэтил)амино)-5-хлор-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
2,2,2-трифторацетата (S)-5-хлор-4-((2-(3-фторазетидин-1-ил)-1-фенилэтил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-4-((2-(3,3-дифторазетидин-1-ил)-1-фенилэтил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-((1-фенил-2-(пирролидин-1-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
(R)-5-хлор-2-фтор-4-((1-фенил-2-(пирролидин-1-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-((1-фенил-2-(пирролидин-1-ил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
2,2,2-трифторацетата (S)-5-хлор-2-фтор-4-((2-морфолино-1-фенилэтил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-3-хлор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-4-((1-(2-хлорфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
5-хлор-4-((циклопропил(фенил)метил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
5-хлор-2-фтор-4-((1-(3-фторфенил)циклобутил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
5-хлор-2-фтор-4-((1-(2-фторфенил)циклобутил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-((3-метил-1-фенилбутил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
(R)-5-хлор-2-фтор-4-((3-метил-1-фенилбутил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-((1-фенилбутил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-4-((1-(2-((диметиламино)метил)фенил)этил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
(R)-5-хлор-4-((1-(2-((диметиламино)метил)фенил)этил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
4-((2-((трет-бутил(метил)амино)метил)бензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
(R)-2,6-дифтор-4-((2-фтор-6-(1-гидроксиэтил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((трет-бутил(метил)амино)метил)-6-фторбензил)амино)-2,3-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
2,6-дифтор-4-((2-фтор-6-((метил(трет-пентил)амино)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(((циклопропилметил)(метил)амино)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((трет-бутиламино)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((циклобутиламино)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
2,6-дифтор-4-((2-фтор-6-((изобутил(метил)амино)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
2,6-дифтор-4-((2-фтор-6-(2-метилпиридин-4-ил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((циклобутил(метил)амино)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((трет-бутил(метил)амино)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
2,6-дифтор-4-((2-фтор-6-((метил(оксетан-3-ил)амино)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
2,6-дифтор-4-((2-фтор-6-((изопропил(метил)амино)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
2,6-дифтор-4-((2-фтор-6-((1-метилазетидин-3-ил)окси)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((диэтиламино)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
2,6-дифтор-4-((2-фтор-6-((метил((3-метилоксетан-3-ил)метил)амино)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
2,6-дифтор-4-((2-фтор-6-(((2-метоксиэтил)(метил)амино)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((диметиламино)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
5-хлор-2-фтор-4-((2-фтор-6-(метоксиметил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-((1-(2-фтор-5-(оксетан-3-илметокси)фенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
5-хлор-4-((1-(2-((диметиламино)метил)фенил)циклопропил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
(S)-5-хлор-4-((1-(5-(2,2-дифторэтил)-2-фторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-((1-(2-фтор-5-((3-метилоксетан-3-ил)метокси)фенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
(S)-3-циано-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
(S)-2,6-дифтор-4-((1-(2-фтор-5-метоксифенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
(S)-4-((1-(5-циано-2-фторфенил)этил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((диметиламино)метил)бензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
(S)-4-((1-(2,5-дифторфенил)этил)амино)-2,6-дифтор-N-(изоксазол-3-ил)бензолсульфонамида;
(S)-4-((1-(5-(дифторметил)-2-фторфенил)этил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
2,6-дифтор-4-((1-фенилциклопропил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
(R)-5-хлор-4-((1-(3-((диметиламино)метил)фенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-((1-(2-фтор-5-гидроксифенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-((1-(2-фтор-5-(оксетан-3-илокси)фенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
(S)-2,6-дифтор-4-((1-(2-фтор-5-(метоксиметил)фенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
5-хлор-4-((2,5-дифторбензил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-((1-(2-фтор-5-(метоксиметил)фенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
(R)-5-хлор-4-((1-(2,5-дифторфенил)-2,2-дифторэтил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
5-хлор-4-((3,6-дифтор-2-(гидроксиметил)бензил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
5-хлор-4-((2-хлор-6-метилбензил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
5-хлор-4-((2-((диметиламино)метил)-6-фторбензил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
(S)-5-хлор-4-((1-(5-циано-2-фторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
(S)-5-хлор-4-((1-(3-((диметиламино)метил)фенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
(S)-5-хлор-4-((1-(5-(дифторметокси)-2-фторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
(S)-3-хлор-4-((1-(2,6-дифторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
5-хлор-2-фтор-4-((5-фтор-2-метилбензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
(S)-4-((1-(5-хлор-2-фторфенил)этил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-((1-(2-фтор-5-метоксифенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
(S)-4-((1-(2,5-дифторфенил)этил)амино)-5-этил-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
5-хлор-4-((1-(2,5-дифторфенил)циклопропил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
(S)-4-((1-(2,5-дифторфенил)этил)амино)-N-(тиазол-4-ил)-3-(трифторметил)бензолсульфонамида;
(S)-3-хлор-4-((1-(5-хлор-2-фторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
(S)-5-хлор-4-((1-(2-фторфенил)этил)амино)-2-метил-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-N-(тиазол-4-ил)-4-((1-(2,4,5-трифторфенил)этил)амино)бензолсульфонамида;
(S)-2,6-дифтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
5-хлор-4-((1-(2,4-дифторфенил)циклопропил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
(S)-5-хлор-4-((1-(5-циклопропил-2-фторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
(R)-5-хлор-2-фтор-N-(тиазол-2-ил)-4-((2,2,2-трифтор-1-(2-фторфенил)этил)амино)бензолсульфонамида;
(S)-5-хлор-4-((1-(3,5-дифторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
(S)-5-хлор-4-((1-(2-хлорфенил)пропил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
5-хлор-2-фтор-4-((1-(2-фторфенил)циклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-4-((1-(2,5-дифторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
(S)-5-хлор-4-((1-(2,5-дифторфенил)этил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-N-(5-хлортиазол-2-ил)-2-фтор-4-((1-(2-фторфенил)этил)амино)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(5-фтортиазол-2-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-N-(5-фтортиазол-2-ил)-4-((1-фенилпропил)амино)бензолсульфонамида;
(S)-3-хлор-4-((1-(2-хлор-6-фторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида; и
(S)-5-хлор-4-((1-(2,5-дифторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида.
По варианту осуществления соединений формулы (Ib2), в которой R2 выбирают из необязательно замещенного тиазолила, необязательно замещенного тиадиазолила, необязательно замещенного изоксазолила, необязательно замещенного изотиазолила или необязательно замещенного оксазолила, как описано выше, другим предпочтительным вариантом осуществления являются те соединения формулы (Ib2), в которых:
R1 представляет собой арил, замещенный необязательно замещенным гетероциклилалкилом и необязательно замещенный посредством одним или несколькими заместителями, выбранными из галогена, алкила, галогеналкила, необязательно замещенного циклоалкила, циано, R9OR12, R9N(R10)R11, R9N(R10)-R13-OR12, необязательно замещенного гетероциклила и необязательно замещенного гетероарила;
R2 представляет собой необязательно замещенный тиазолил; и
R13 представляет собой разветвленную или неразветвленную алкиленовую цепь.
По этому варианту осуществления, предпочтительные варианты осуществления для необязательно замещенного гетероциклилалкила выбирают из пирролидинилалкила, пиперазинилалкила, пиперидинилалкила, морфолинилалкила, азетидинилалкила, 3-азабицикло[3.1.0]гексан-3-илалкила, 1азаспиро[3.3]гептан-1-илалкила, 5-азаспиро[2.3]гексан-5-илалкила, 2-окса-6-азаспиро[3.3]гептан-6-илалкила, 1-окса-6-азаспиро[3.4]октан-6-илалкила, 1-окса-6-азаспиро[3.3]гептан-6-илалкила, 6-окса-1-азаспиро[3.3]гептан-1-илалкила, 6-азаспиро[3.4]октан-6-илалкила, 7-окса-2-азаспиро[3.5]нонан-2-илалкила, 2,6-диазаспиро[3.3]гептан-2-илалкила, всео ни могут быть необязательно замещены одним или несколькими заместителями, выбранными из алкила, галогена, галогеналкила, -R9OR12, где R9и R12, как определено в Сущности изобретения.
По этим вариантам осуществления, предпочтительные соединения формулы (Ib2) выбирают из:
2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)бензил)амино)-5-хлор-2-фтор-N-(тиазол-2-ил)бензолсульфонамида;
2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)бензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
5-хлор-4-((2-((2,2-диметилазетидин-1-ил)метил)-6-фторбензил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((2-азаспиро[3.3]гептан-2-ил)метил)-6-фторбензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-хлор-6-((3-фторазетидин-1-ил)метил)бензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
2-фтор-4-((2-фтор-6-((3-фторазетидин-1-ил)метил)бензил)амино)-3-метил-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-4-фторбензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
3-хлор-2-фтор-4-((2-фтор-6-((3-фторазетидин-1-ил)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-3,6-дифторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
2-фтор-4-((2-фтор-6-((3-фторазетидин-1-ил)метил)бензил)амино)-5-метил-N-(тиазол-4-ил)бензолсульфонамида; и
4-((2-((3-этокси-3-метилазетидин-1-ил)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
(S)-4-((2-(1-(азетидин-1-ил)этил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
(R)-4-((2-(1-(азетидин-1-ил)этил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)бензил)амино)-2,3-дифтор-6-метил-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-3,6-дифторбензил)амино)-2,3,6-трифтор-N-(тиазол-4-ил)бензолсульфонамида;
2,6-дифтор-4-((2-фтор-6-((3-гидрокси-3-(трифторметил)азетидин-1-ил)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-хлор-6-((2,2-диметилазетидин-1-ил)метил)бензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((2,2-диметилазетидин-1-ил)метил)-3-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((3-азабицикло[3.1.0]гексан-3-ил)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
2,6-дифтор-4-((2-((3-(2-метоксипропан-2-ил)азетидин-1-ил)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((1-азаспиро[3.3]гептан-1-ил)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-3,6-дифторбензил)амино)-2,3-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(2-(3,3-дифторазетидин-1-ил)этил)бензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
2,6-дифтор-4-((2-фтор-6-((3-метокси-3-метилазетидин-1-ил)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((5-азаспиро[2.3]гексан-5-ил)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
2,6-дифтор-4-((2-(2-(3-фторазетидин-1-ил)этил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((2,2-диметилазетидин-1-ил)метил)-6-фторбензил)амино)-2-фтор-3-метил-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((3-(дифторметил)азетидин-1-ил)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
2,6-дифтор-4-((2-фтор-6-((3-фтор-3-метилазетидин-1-ил)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((2,2-диметилазетидин-1-ил)метил)бензил)амино)-2,6-дифтор-3-метил-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-6-хлор-3-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-6-циклопропилбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
2,6-дифтор-4-((2-фтор-6-((3-гидрокси-3-метилазетидин-1-ил)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-6-этилбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-6-метилбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)бензил)амино)-2-фтор-N-(тиазол-4-ил)-5-винилбензолсульфонамида;
2,6-дифтор-4-((2-фтор-6-(пирролидин-1-илметил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((2-окса-6-азаспиро[3.3]гептан-6-ил)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
(R)-2,6-дифтор-4-((2-фтор-6-((2-(метоксиметил)пирролидин-1-ил)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((1-окса-6-азаспиро[3.4]октан-6-ил)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((1-окса-6-азаспиро[3.3]гептан-6-ил)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((6-окса-1-азаспиро[3.3]гептан-1-ил)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
(S)-2,6-дифтор-4-((2-фтор-6-((2-метилпирролидин-1-ил)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((6-азаспиро[3.4]октан-6-ил)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((6,6-дифтор-2-азаспиро[3.3]гептан-2-ил)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((1,1-дифтор-5-азаспиро[2.3]гексан-5-ил)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
(R)-2,6-дифтор-4-((2-фтор-6-((3-фторпирролидин-1-ил)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
(S)-2,6-дифтор-4-((2-фтор-6-((3-фторпирролидин-1-ил)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
2,6-дифтор-4-((2-фтор-6-((3-метоксиазетидин-1-ил)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
2,6-дифтор-4-((2-фтор-6-((3-метилазетидин-1-ил)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((3,3-диметилазетидин-1-ил)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
2,6-дифтор-4-((2-фтор-6-((4-метилпиперазин-1-ил)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
5-хлор-2-фтор-4-((2-фтор-5-(2-(3-фторазетидин-1-ил)этил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-3-фторбензил)амино)-3-хлор-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-3-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((5-(2-(азетидин-1-ил)этил)-2-фторбензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-6-метоксибензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((5-(азетидин-1-илметил)-2-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(2-(азетидин-1-ил)этил)бензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
(S)-4-((1-(5-(азетидин-1-илметил)-2-фторфенил)этил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-((1-(2-фтор-5-(пиперидин-1-илметил)фенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
(S)-4-((1-(5-(2-(азетидин-1-ил)этил)-2-фторфенил)этил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((2-азаспиро[3.3]гептан-2-ил)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,5-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-3-хлор-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-3-метил-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((2,2-диметилазетидин-1-ил)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)бензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
5-хлор-4-((2-((4,4-дифторпиперидин-1-ил)метил)-6-фторбензил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((7-окса-2-азаспиро[3.5]нонан-2-ил)метил)-3,6-дифторбензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-3,6-дифторбензил)(метил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
5-хлор-4-((2-хлор-6-((6-метил-2,6-диазаспиро[3.3]гептан-2-ил)метил)бензил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((2,6-диазаспиро[3.3]гептан-2-ил)метил)-6-хлорбензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
5-хлор-2-фтор-4-((2-фтор-6-(морфолинометил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-6-хлорбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
2,6-дифтор-4-((2-фтор-6-((3-фторазетидин-1-ил)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
5-хлор-2-фтор-4-((2-фтор-6-((3-метоксиазетидин-1-ил)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-((2-окса-6-азаспиро[3.3]гептан-6-ил)метил)-6-хлорбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-4,5-дифторбензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-3-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
5-хлор-2-фтор-4-((2-фтор-6-((3-фторазетидин-1-ил)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-3,6-дифторбензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида;
2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-хлор-3-фторбензил)амино)-2,6-дифтор-3-метил-N-(тиазол-4-ил)бензолсульфонамида; и
2,2,2-трифторацетата 4-((2-((3,3-диметилазетидин-1-ил)метил)-6-фторбензил)амино)-2,3-дифтор-6-метил-N-(тиазол-4-ил)бензолсульфонамида.
По варианту осуществления соединений формулы (Ib2), в которой R2 выбирают из необязательно замещенного тиазолила, необязательно замещенного тиадиазолила, необязательно замещенного изоксазолила, необязательно замещенного изотиазолила или необязательно замещенного оксазолила, предпочтительным вариантом осуществления является соединение формулы (Ib2), в которой:
R1 представляет собой арил, необязательно замещенный посредством одним или несколькими заместителями, выбранными из галогена, алкила, галогеналкила, необязательно замещенного циклоалкила, циано, R9OR12, R9N(R10)R11, R9N(R10)-R13-OR12, необязательно замещенного гетероциклила, необязательно замещенного гетероциклилалкила и необязательно замещенного гетероарила;
R2 представляет собой необязательно замещенный тиадиазолил, необязательно замещенный изотиазолил, необязательно замещенный оксазолил, необязательно замещенный изоксазолил; и
R13 представляет собой разветвленную или неразветвленную алкиленовую цепь.
По этому варианту осуществления, предпочтительные варианты осуществления для необязательно замещенного гетероциклилалкила выбирают из пирролидинилалкила, пиперазинилалкила, пиперидинилалкила, морфолинилалкила, азетидинилалкила, 3-азабицикло[3.1.0]гексан-3-илалкила, 1азаспиро[3.3]гептан-1-илалкила, 5-азаспиро[2.3]гексан-5-илалкила, 2-окса-6-азаспиро[3.3]гептан-6-илалкила, 1-окса-6-азаспиро[3.4]октан-6-илалкила, 1-окса-6-азаспиро[3.3]гептан-6-илалкила, 6-окса-1-азаспиро[3.3]гептан-1-илалкила, 6-азаспиро[3.4]октан-6-илалкила, 7-окса-2-азаспиро[3.5]нонан-2-илалкила, 2,6-диазаспиро[3.3]гептан-2-илалкила, все они могут быть необязательно замещены одним или несколькими заместителями, выбранными из алкила, галогена, галогеналкила, -R9OR12, где R9и R12 представляют собой то, что определено в Сущности изобретения.
По этим вариантам осуществления, предпочтительные соединения формулы (Ib2) выбирают из:
3-хлор-4-(1-фенилпропиламино)-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида;
(R)-3-хлор-4-(1-фенилпропиламино)-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида;
(S)-3-хлор-4-(1-фенилпропиламино)-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида;
3-хлор-4-(1-фенилэтиламино)-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида;
2,5-дифтор-4-(1-фенилпропиламино)-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида;
4-(бензиламино)-3-хлор-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида;
3-хлор-4-(2-фенилпропиламино)-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида;
2,6-дифтор-4-((2-фтор-6-((3-фтор-3-метилазетидин-1-ил)метил)бензил)амино)-N-(изоксазол-3-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2-фтор-N-(изоксазол-3-ил)-5-метилбензолсульфонамида;
2,6-дифтор-4-((2-фтор-6-((изопропил(метил)амино)метил)бензил)амино)-N-(изоксазол-3-ил)бензолсульфонамида;
2,3-дифтор-4-((2-фтор-6-(пирролидин-1-илметил)бензил)амино)-N-(изоксазол-3-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(изотиазол-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-3,6-дифторбензил)амино)-2,6-дифтор-N-(5-метилизоксазол-3-ил)бензолсульфонамида;
4-((2-((трет-бутил(метил)амино)метил)-6-фторбензил)амино)-2,6-дифтор-N-(изотиазол-3-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(изотиазол-3-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)бензил)амино)-2,6-дифтор-3-метил-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)бензил)амино)-3-хлор-2,6-дифтор-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(изоксазол-3-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)бензил)амино)-2,6-дифтор-N-(изоксазол-3-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)бензил)амино)-5-хлор-2-фтор-N-(изоксазол-3-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-5-хлор-2-фтор-N-(изоксазол-3-ил)бензолсульфонамида;
(S)-5-хлор-4-((1-(2,5-дифторфенил)этил)амино)-2-фтор-N-(оксазол-2-ил)бензолсульфонамида; и
2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2-фтор-N-(изоксазол-3-ил)-3-метилбензолсульфонамида.
В другом варианте осуществления соединение формулы (Ib2), как определено выше, представляет собой соединение формулы (Ib2), в котором:
R1 представляет собой необязательно замещенный арил; и
R2 представляет собой необязательно замещенный 6членный N-гетероарил.
По этому варианту осуществления, предпочтительным вариантом осуществления являются соединения формулы (Ib2), в которой R2 выбирают из необязательно замещенного пиридинила, необязательно замещенного пиримидинила, необязательно замещенного пиридазинила и необязательно замещенного пиразинила.
По этому варианту осуществления, предпочтительные соединения формулы (Ib2) выбирают из:
5-хлор-2-фтор-N-(6-фторпиридин-2-ил)-4-(1-фенилпропиламино)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(6-фторпиридин-2-ил)бензолсульфонамида;
(S)-5-хлор-4-((1-(2-хлорфенил)этил)амино)-2-фтор-N-(6-фторпиридин-2-ил)бензолсульфонамида;
2,2,2-трифторацетата (S)-5-хлор-2-фтор-N-(6-фторпиридин-2-ил)-4-((1-фенил-2-(пирролидин-1-ил)этил)амино)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-3,6-дифторбензил)амино)-5-хлор-2-фтор-N-(6-фторпиридин-2-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-5-хлор-2-фтор-N-(6-фторпиридин-2-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(пиримидин-2-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(5-фторпиридин-2-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(пиридазин-3-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)бензил)амино)-2,6-дифтор-N-(пиримидин-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)бензил)амино)-2,6-дифтор-3-метил-N-(пиримидин-4-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)бензил)амино)-2-фтор-N-(6-фторпиридин-2-ил)-5-метилбензолсульфонамида;
4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(пиридин-2-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(6-фторпиридин-2-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)бензил)амино)-2,6-дифтор-N-(6-фторпиридин-2-ил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-5-хлор-2-фтор-N-(пиридазин-3-ил)бензолсульфонамида;
(S)-5-хлор-4-((1-(2-хлорфенил)этил)амино)-2-фтор-N-(2-(трифторметил)пиримидин-4-ил)бензолсульфонамида;
(S)-5-хлор-4-((1-(2-хлорфенил)этил)амино)-2-фтор-N-(6-метилпиримидин-4-ил)бензолсульфонамида;
(S)-5-хлор-4-((1-(5-хлор-2-фторфенил)этил)амино)-2-фтор-N-(пиразин-2-ил)бензолсульфонамида;
(R)-5-хлор-4-((1-(5-хлор-2-фторфенил)-2,2,2-трифторэтил)амино)-2-фтор-N-(пиримидин-4-ил)бензолсульфонамида;
(S)-4-((1-(2,5-дифторфенил)этил)амино)-2,6-дифтор-N-(пиримидин-4-ил)бензолсульфонамида;
(S)-4-((1-(2-хлор-5-фторфенил)пропил)амино)-2,6-дифтор-N-(пиримидин-4-ил)бензолсульфонамида;
(S)-4-((1-(5-хлор-2-фторфенил)этил)амино)-2,6-дифтор-N-(пиримидин-4-ил)бензолсульфонамида;
5-хлор-2-фтор-4-((1-(2-фторфенил)циклопропил)амино)-N-(пиримидин-4-ил)бензолсульфонамида;
5-хлор-4-((1-(2,5-дифторфенил)циклопропил)амино)-2-фтор-N-(пиримидин-4-ил)бензолсульфонамида;
(S)-3-хлор-4-((1-(2-хлорфенил)этил)амино)-2-фтор-N-(пиримидин-4-ил)бензолсульфонамида;
(S)-4-((1-(5-хлор-2-фторфенил)этил)амино)-2-фтор-5-метил-N-(пиримидин-4-ил)бензолсульфонамида;
(S)-4-((1-(2,5-дифторфенил)этил)амино)-N-(пиримидин-4-ил)-3-(трифторметил)бензолсульфонамида;
4-((2-(азетидин-1-илметил)бензил)амино)-5-хлор-2-фтор-N-(6-фторпиридин-2-ил)бензолсульфонамида;
(S)-5-хлор-4-((1-(2-хлорфенил)пропил)амино)-2-фтор-N-(пиримидин-4-ил)бензолсульфонамида; и
(S)-5-хлор-2-фтор-4-((1-(2-фторфенил)пропил)амино)-N-(6-фторпиридин-2-ил)бензолсульфонамида.
В другом варианте осуществления соединение формулы (Ib2), как определено выше, представляет собой соединение формулы (Ib2), в котором R1 представляет собой необязательно замещенный моноциклический гетероарил или необязательно замещенный бициклический гетероарил.
По этому варианту осуществления, предпочтительные соединения формулы (Ib2) выбирают из:
5-хлор-2-фтор-N-(6-фторпиридин-2-ил)-4-((изохинолин-8-илметил)амино)бензолсульфонамида;
(S)-5-хлор-2-фтор-4-((1-(изохинолин-8-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
(R)-5-хлор-2-фтор-4-((1-(изохинолин-8-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
5-хлор-2-фтор-4-(изохинолин-8-илметиламино)-N-(тиазол-2-ил)бензолсульфонамида;
5-хлор-2-фтор-4-((1-(пиридин-3-ил)пропан-2-ил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
5-хлор-2-фтор-4-((1-(пиридин-3-ил)пропил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
5-хлор-2-фтор-4-((1-(пиридин-4-ил)пропил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
3-хлор-4-((1-(пиридин-2-ил)пропил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
(S)-3-хлор-4-((1-(пиридин-2-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
3-хлор-4-((1-(1-метил-1H-пиразол-4-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида;
2,2,2-трифторацетата 5-хлор-2-фтор-4-((изохинолин-8-илметил)амино)-N-(тиазол-4-ил)бензолсульфонамида;
соли (R)-5-хлор-2-фтор-4-((1-(пиридин-3-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида и муравьиной кислоты;
5-хлор-2-фтор-4-(((6-фтор-1H-индол-7-ил)метил)амино)-N-(тиазол-4-ил)бензолсульфонамида; и
5-хлор-2-фтор-4-(((6-фтор-1H-индазол-7-ил)метил)амино)-N-(тиазол-4-ил)бензолсульфонамида.
В другом варианте осуществления соединение формулы (I) представляет собой соединение формулы (I), в котором R3 представляет собой S(O)t (где t представляет собой 0, 1 или 2), где соединение имеет следующую формулу (Ic):
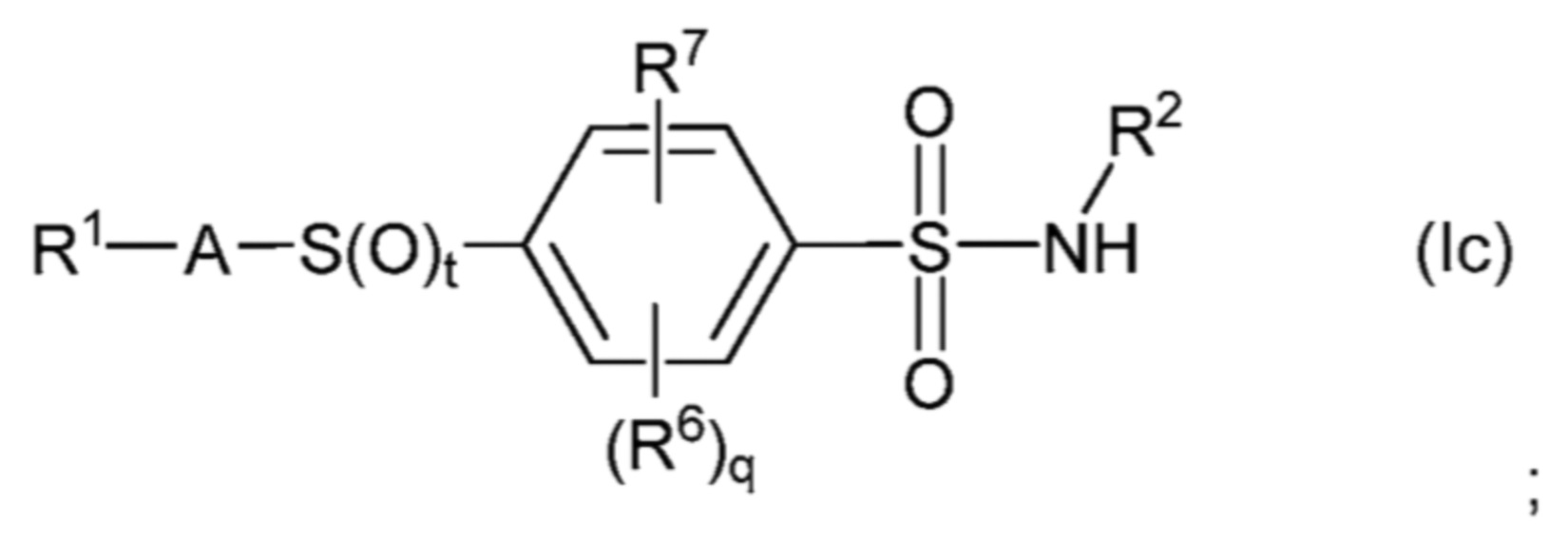
где каждое из q, t, A, R1, R2, R6 и R7 представляет собой то, что определено выше в п. 1;
в виде его индивидуального стереоизомера, энантиомера или таутомера или их смесей;
или его фармацевтически приемлемой соли, сольвата или пролекарственного средства.
В другом варианте осуществления соединение формулы (I) представляет собой соединение формулы (Ic), как определено выше, где A представляет собой непосредственную связь, т. е. соединение формулы (Ic1):

где каждое из q, t, R1, R2, R6 и R7 представляет собой то, что определено выше в п. 1;
в виде его индивидуального стереоизомера, энантиомера или таутомера или их смесей;
или его фармацевтически приемлемой соли, сольвата или пролекарственного средства.
В другом варианте осуществления соединение формулы (I) представляет собой соединение формулы (Ic), как определено выше, где A представляет собой (CH2)m C(R4)(R5) (CH2)n, т. е. соединение формулы (Ic2):

где каждое из m, n, t, R1, R2, R4, R5, R6 и R7 каждое представляет собой то, что определено выше в п. 1; в виде его индивидуального стереоизомера, энантиомера или таутомера или их смесей; или его фармацевтически приемлемой соли, сольвата или пролекарственного средства.
По этому варианту осуществления, предпочтительным соединением формулы (Ic2) является (S)-5-хлор-2-фтор-4-((1-фенилэтил)тио)-N-(тиазол-2-ил)бензолсульфонамид.
Другой вариант осуществления изобретения представляет собой соединения формулы (I), в которой R7 находится в орто-положении относительно R3.
Другой вариант осуществления изобретения представляет собой соединения формулы (I), в которой R7 находится в орто-положении относительно R3 и представляет собой галоген.
Другой вариант осуществления изобретения представляет собой соединения формулы (I), в которой R7 представляет собой хлор или фтор.
Другой вариант осуществления изобретения представляет собой способ использования соединений формулы (I) в качестве стандартов или контролей в анализах in vitro или in vivo при определении эффекта тестируемых соединений, оказываемого на модуляцию потенциалзависимых натриевых каналов.
Понятно, что любой вариант осуществления соединений по изобретению, как изложено выше, и любой конкретный заместитель, приведенный в настоящем описании для конкретной группы A, q, t, R1, R2, R3, R4, R5, R6, R7, R8, R9, R10, R11 и R12 в соединениях по изобретению, как изложено выше, можно независимо комбинировать с другими вариантами осуществления и/или заместителями соединений по изобретению для того, чтобы формировать варианты осуществления изобретения, конкретно не изложенные выше. Кроме того, в том случае, если список заместителей раскрыт для любой конкретной группы A, q, t, R1, R2, R3, R4, R5, R6, R7, R8, R9, R10, R11 и R12 в конкретном варианте осуществления и/или пункте, понятно, что один или несколько заместителей можно удалить из списка и что остающийся список заместителей следует рассматривать в качестве варианта осуществления изобретения.
Также понятно, что условие, изложенное выше в сущности изобретения для соединений формулы (I), применимо ко всем релевантным вариантам осуществления соединений формулы (I), как описано выше.
Другой аспект изобретения представляет собой фармацевтическую композицию, содержащую фармацевтически приемлемый эксципиент и соединение по изобретению, как описано выше, в виде его стереоизомера, энантиомера или таутомера или их смеси; или его фармацевтически приемлемой соли, сольвата или пролекарственного средства.
Другой аспект изобретения представляет собой способ лечения заболевания или состояния, ассоциированного с активностью NaV1.6, у млекопитающего, где заболевание или состояние представляет собой эпилепсию и/или эпилептическое пароксизмальное нарушение и где способ включает введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения по изобретению, как описано выше, в виде его стереоизомера, энантиомера или таутомера или их смеси; или его фармацевтически приемлемой соли, сольвата или пролекарственного средства.
В одном из вариантов осуществления этого аспекта, эпилепсию или эпилептическое пароксизмальное нарушение выбирают из фоточувствительной эпилепсии, самопроизвольного синкопе, инкурабельной эпилепсии, синдрома Энгельмана, доброкачественной роландической эпилепсии, нарушения CDKL5, детской и ювенильной абсенс-эпилепсии, синдрома Драве, эпилепсии лобной доли, синдрома недостаточности Glut1, гипоталамической гамартомы, младенческих судорог/синдрома Веста, ювенильной миоклонической эпилепсии, синдрома Ландау-Клефнера, синдрома Леннокса-Гасто (LGS), эпилепсии с миоклоническими абсансами, синдрома Отахара, синдрома Панайотопулоса, эпилепсии PCDH19, прогрессирующей миоклонической эпилепсии, синдром Расмуссена, синдрома кольцевой 20-й хромосомы, рефлекторной эпилепсии, эпилепсии височной доли, прогрессирующей миоклонической эпилепсии Лафора, нейрокутанных синдромов, комплекса туберозного склероза, ранней детской эпилептической энцефалопатии, эпилептической энцефалопатии с ранним началом, генерализованной эпилепсии с фебрильными пароксизмами +, синдрома Ретта, рассеянного склероза, болезни Альцгеймера, аутизма, атаксии, гипотонии и пароксизмальной дискинезии.
В одном из вариантов осуществления по этому варианту осуществления, эпилепсию или эпилептическое пароксизмальное нарушение выбирают из синдрома Драве, младенческих судорог/синдрома Веста, эпилепсии височной доли, синдрома Леннокса-Гасто (LGS), генерализованной эпилепсии с фебрильными пароксизмами+и ранней детской эпилептической энцефалопатии.
Другой аспект изобретения представляет собой способ снижения ионного потока через NaV1.6 в клетке млекопитающего, где способ включает приведение в контакт клетки с соединением по изобретению, как описано выше, в виде его стереоизомера, энантиомера или таутомера или их смеси; или его фармацевтически приемлемой соли, сольвата или пролекарственного средства.
Другой аспект изобретения представляет собой способ избирательного ингибирования первого потенциалзависимого натриевого канала относительно второго потенциалзависимого натриевого канала у млекопитающего, где способ включает введение млекопитающему модулирующего количества соединения по изобретению, как описано выше, в виде его стереоизомера, энантиомера или таутомера или их смеси; или его фармацевтически приемлемой соли, сольвата или пролекарственного средства.
В одном из вариантов осуществления этого аспекта, первый потенциалзависимый натриевый канал представляет собой NaV1.6.
В другом варианте осуществления этого аспекта, первый потенциалзависимый натриевый канал представляет собой NaV1.6 и второй потенциалзависимый натриевый канал представляет собой NaV1.5.
В другом варианте осуществления этого аспекта, первый потенциалзависимый натриевый канал представляет собой NaV1.6 и второй потенциалзависимый натриевый канал представляет собой NaV1.1.
Конкретные варианты осуществления соединений по изобретению описаны более подробно далее в Получении соединений по изобретению и в Примерах.
ПОЛЕЗНОСТЬ И ТЕСТИРОВАНИЕ СОЕДИНЕНИЙ ПО ИЗОБРЕТЕНИЮ
Соединения по изобретению модулируют, предпочтительно ингибируют, ионный поток через потенциалзависимый натриевый канал, предпочтительно NaV1.6, у млекопитающего, в частности, у человека. Любую такую модуляцию, будь то частичное или полное ингибирование или предотвращение ионного потока, в настоящем описании иногда обозначают как «блокирование», а соответствующие соединения как «блокаторы» или «ингибиторы». В целом, соединения по изобретению модулируют активность потенциалзависимого натриевого канала вниз посредством ингибирования потенциалзависимой активности натриевого канала и/или снижают или предотвращают поток ионов натрия через клеточную мембрану, препятствуя активности натриевого канала, такой как ионный поток.
Соединения по изобретению ингибируют ионный поток через потенциалзависимый натриевый канал, предпочтительно NaV1.6. Соединения по изобретению зависящие от состояния или частоты модификаторы натриевого канала, обладающие низкой аффинностью к покоящемуся/закрытому состоянию, и высокой аффинностью к инактивированному состоянию. Эти соединения вероятно взаимодействуют с перекрывающимися участками, расположенными во внутренней полости натрий-проводящей поры канала, подобно тому, что описано для других зависящих от состояния блокаторов натриевых каналов (Cestèle, S., et al., op. cit.). Эти соединения также вероятно могут взаимодействовать с участками вне внутренней полости и имеют аллостерические эффекты, оказываемые на проводимость ионов натрия через пору канала.
Любые из этих последствий в конечном итоге могут отвечать за общий терапевтический эффект, обеспечиваемый этими соединениями.
Соответственно, соединения по изобретению представляют собой ингибиторы потенциалзависимых натриевых каналов, предпочтительно ингибиторы NaV1.6, и, следовательно, их можно использовать для лечения заболеваний и состояний, предпочтительно эпилепсии и/или эпилептического пароксизмального нарушения, у млекопитающих, предпочтительно у человека, и других организмов, включая все те заболевания и состояния человека, которые являются результатом аберрантной биологической активности потенциалзависимых натриевых каналов, предпочтительно аберрантной активности NaV1.6, или которые можно улучшать посредством модуляции биологической активности потенциалзависимых натриевых каналов. В частности, соединения по изобретению, т. е. соединения формулы (I), как изложено выше в Сущности изобретения, в виде их индивидуальных стереоизомеров, энантиомеров или таутомеров или их смесей; или в виде их фармацевтически приемлемых солей, сольватов или пролекарственных средств, можно использовать для лечения заболеваний и состояний у млекопитающих, предпочтительно у человека, которые являются результатом аберрантной биологической активности потенциалзависимых NaV1.6 или которые можно улучшать посредством модуляции, предпочтительно ингибирования, биологической активности NaV1.6. Предпочтительно соединения по изобретению избирательно ингибируют NaV1.6 по сравнению с NaV1.5 и/или NaV1.1.
Как определено в настоящем описании, заболевание, нарушениее или состояние, ассоциированное с активностью NaV1.6, включает, но не ограничиваясь этим, эпилепсию и/или эпилептическое пароксизмальное нарушение. Такие эпилепсия и/или эпилептические пароксизмальные нарушения включают, но не ограничиваясь этим, фоточувствительную эпилепсию, самопроизвольное синкопе, инкурабельную эпилепсию, синдром Энгельмана, доброкачественную роландическую эпилепсию, нарушение CDKL5, детскую и ювенильную абсенс-эпилепсию, синдром Драве, эпилепсию лобной доли, синдром недостаточности Glut1, гипоталамическую гамартому, младенческие судороги/синдром Веста, ювенильную миоклоническую эпилепсию, синдром Ландау-Клефнера, синдром Леннокса-Гасто (LGS), эпилепсию с миоклоническими абсансами, синдром Отахара, синдром Панайотопулоса, эпилепсию PCDH19, прогрессирующую миоклоническую эпилепсию, синдром Расмуссена, синдром кольцевой 20-й хромосомы, рефлекторную эпилепсию, эпилепсию височной доли, прогрессирующую миоклоническую эпилепсию Лафора, нейрокутанные синдромы, комплекс туберозного склероза, раннюю детскую эпилептическую энцефалопатию, эпилептическую энцефалопатию с ранним началом, генерализованную эпилепсию с фебрильными пароксизмами +, синдром Ретта, рассеянный склероз, болезнь Альцгеймера, аутизм, атаксию, гипотонию и пароксизмальную дискинезию.
Следовательно, настоящее изобретение относится к соединениям, фармацевтическим композициям и способам использования соединений и фармацевтических композиций для лечения заболеваний или состояний, ассоциированных с активностью NaV1.6, у млекопитающего, предпочтительно у человека, посредством введения млекопитающему, предпочтительно человеку, нуждающемуся в таком лечении, эффективного количества соединения по изобретению или фармацевтической композиции, содержащей соединение по изобретению.
Общее значение соединений по изобретению в ингибировании ионного потока NaV1.6 можно определять с использованием анализов, описанных далее в разделе о Биологических анализах. Альтернативно, общее значение соединений в лечении состояний и заболеваний у человека можно устанавливать на животных моделях по отраслевым стандартам, чтобы демонстрировать эффект соединений в лечении эпилепсии и/или эпилептического пароксизмального нарушения. Разработаны животные модели эпилептических состояний человека, которые ведут к воспроизводимому сенсорному дефициту в течение длительного периода времени, который можно оценивать с помощью сенсорного тестирования.
Например, многие модели на грызунах разработаны для того, чтобы оценивать предрасположенность к пароксизмам или эпилептиформной активности (Klein, B.R. et al.,(2016), «Models Currently in Active Use. In: Epilepsy Therapy Screening Program», том 2016, National Institute of Neurological Disorders and Stroke). Они включают острые химические или электрические инсульты, которые вызывают пароксизмы, а также хронические химические или генетические инсульты, которые создают животных, склонных к пароксизмам. Эти модели можно использовать для того, чтобы определять относительную способность соединения стимулировать или предотвращать пароксизмальную активность. Анализ максимальных электрошоковых пароксизмов (MES) и 6 Гц психомоторный пароксизмальный тест (6 Гц) представляют собой два примера анализов пароксизмов при остром инсульте, используемых для того, чтобы оценивать противосудорожные вмешательства (Suzuki, F. et al., Neuroscience (1995), том 64, стр. 665-674; Barton, M.E. et al., Epilepsy Research (2001), том 47, стр. 217-227). Оба анализа включают электрический инсульт с наложением электродов, помещаемых на роговицы или уши для того, чтобы провоцировать острый пароксизм. Острые пароксизмы также можно вызывать химически, например, посредством введения проконвульсантного соединения простого эфира флуротила (Makinson, C.D. et al., Exp. Neurol. (2016), том 275, Pt 1, стр. 46-58).
Генетическая эпилепсия связана со многими отдельными генами, включая несколько генов потенциалзависимых натриевых каналов. Можно создавать генетически модифицированных мышей, которые несут мутации, идентифицированные у пациентов-людей. В некоторых случаях эти генетические модификации ведут к животным, которые ведут себя очень похоже на пациентов-людей, у которых изначально идентифицированы генетические вариации. Мутантных мышей можно использовать для того, чтобы тестировать антиконвульсантные вмешательства. Такие эксперименты могут включать предотвращение самопроизвольных пароксизмов или использование провоцирующих пароксизмы стимулов, схожих с теми, которые используют у мышей дикого типа. Животные модели ранней детской эпилептической энцефалопатии 6 (EIEE6), также известной как тяжелая детская миоклоническая эпилепсия или синдром Драве, созданы посредством мутации в гене SCN1A, который кодирует потенциалзависимый натриевый канал NaV1.1 (Yu, F.H. et al., Nat. Neurosci. (2006), том 9, стр. 1142-1149). Модели EIEE13 аналогичным образом созданы посредством мутации в гене SCN6A, который кодирует потенциалзависимый натриевый канал NaV1.6 (Wagnon, J.L. et al., Human Molecular Genetics(2014)). Оба этих штамма мышей дают возможность оценивать потенциальные терапевтические вмешательства, что может доказать эффективность в популяциях клинических пациентов (Martin, M.S. et al., J. Biol. Chem. (2010), том 285, стр. 9823-9834; и Martin, M.S. et al., Human Molecular Genetics (2007), том 16, стр. 2892-2899).
Настоящее изобретение без труда предоставляет многие различные средства для идентификации ингибирующих NaV1.6 средств, которые можно использовать в качестве терапевтических средств. При идентификации, ингибиторы NaV1.6 можно оценивать с использованием различных анализов in vitro и in vivo, таких как измерение тока, измерение мембранного потенциала, измерение ионного потока, (например, натрия или гуанидиния), измерение концентрации натрия, измерение вторичных мессенджеров и уровней транскрипции и использование, например, потенциалчувствительных красителей, радиоактивных средств отслеживания и электрофизиологии «пэтч-клемп».
Один такой протокол включает скрининг химических средств по способности модулировать активность натриевого канала, тем самым, идентифицируя его в качестве модулирующего средства.
В типичном анализе, описанном в Bean et al., J. General Physiology (1983), 83:613-642, и Leuwer, M., et al., Br. J. Pharmacol (2004), 141(1):47-54, используют приемы пэтч-клемп для исследования поведения каналов. Такие приемы известны специалистам в данной области, и, исползуя существующие технологии, их можно превращать в анализы низкой или средней пропускной способности для оценки соединений по их способности модулировать поведение натриевых каналов.
Пропускная способность тестируемых соединений является важным фактором при выборе анализа скрининга, подлежащего использованию. В некоторых стратегиях, в которых сотни тысяч соединений подлежат тестированию, не желательно использовать средства с низкой пропускной способностью. Однако в других случаях низкая пропускная способность является удовлетворительной для того, чтобы идентифицировать важные различия между ограниченным числом соединений. Часто необходимо комбинировать типы анализов для того, чтобы идентифицировать конкретные соединения, модулирующие натриевые каналы.
Электрофизиологические анализы с использованием приемов пэтч-клемп являются общепринятыми в качестве золотого стандарта для подробного определения характеристик взаимодействия натриевых каналов и соединений, и как описано в Bean et al., op. cit. и Leuwer, M., et al., op. cit. Существует ручной способ скрининга с низкой пропускной способностью (LTS), который позволяет сравнивать 2-10 соединений в сутки; в последнее время разработана система для автоматизированного скрининга со средней пропускной способностью (MTS) на 20-50 пэтчей (т. е. соединений) в сутки; и технология Molecular Devices Corporation (Sunnyvale, CA), которая делает возможным автоматизированный высокопроизводительный скрининг (HTS) на 1000-3000 пэтчей (т. е. соединений) в сутки.
В одной автоматизированной системе пэтч-клемпа используют технологию плоских электродов для увеличения скорости разработки лекарственных средств. Плоские электроды позволяют достигать прикрепленной к клеткам изоляции высокого сопротивления, после чего следует стабильная малошумная регистрация всей клетки, которая сравнима со стандартной регистрацией. Подходящим прибором является PatchXpress 7000A (Axon Instruments Inc, Union City, CA). Различные клеточные линии и приемы культивирования, которые включают прилипающие клетки, а также клетки, растущие самопроизвольно в суспензии, ранжированы по степени успешности изоляции и стабильности. Иммортализованные клетки (например, HEK и CHO), стабильно экспрессирующие высокие уровни релевантных натриевых ионных каналов, можно адаптировать к суспензионным культурам высокой плотности.
Можно выбирать другие анализы, которые позволяют исследователю идентифицировать соединения, которые блокируют конкретные состояния канала, такие как открытое состояние, закрытое состояние или покоящееся состояние, или которые блокируют переход из открытого в закрытое, из закрытого в покоящееся или из покоящегося в открытое. Специалисты в данной области в целом знакомы с такими анализами.
Анализы связывания также доступны. Схемы включают традиционные анализы связывания на основе радиоактивных фильтров или конфокальную флуоресцентную систему, доступную в группе компаний Evotec OAI (Hamburg, Germany), и то и другое представляет собой HTS.
Также можно использовать анализ радиоактивного потока. В этом анализе каналы стимулируют к открытию вератридином или аконитином и удерживают в стабилизированном открытом состоянии с использованием токсина, и идентифицируют блокаторы каналов по их способности для того, чтобы предотвращать входящий ионный поток. В анализе можно использовать радиоактивные ионы22[Na] и14[C] гуанидиния в качестве средств отслеживания. Планшеты FlashPlate & Cytostar-T в живых клетках позволяют избегать стадий разделения и подходят для HTS. Технология сцинтилляционных планшетов также позволяет улучшить этот способ до пригодности для HTS. В силу функциональных аспектов анализа, информационное содержание является обоснованно хорошим.
В еще одном другом формате измеряют перераспределение мембранного потенциала с использованием набора для мембранного потенциала для системы FLIPR (HTS), доступного в Molecular Dynamics (подразделение Amersham Biosciences, Piscataway, NJ). Этот способ ограничен медленными изменениями мембранного потенциала. Некоторые проблемы могут быть результатом флуоресцентного фона соединений. Тестируемые соединения также могут непосредственно влиять на текучесть клеточной мембраны и вести к увеличению внутриклеточных концентраций красителей. Также, в силу функциональных аспектов анализа, информационное содержание является обоснованно хорошим.
Натриевые красители можно использовать для того, чтобы измерять скорость или количество входящего потока ионов натрия через канал. Анализ этого типа обеспечивает очень высокое информационное содержание в отношении потенциальных блокаторов каналов. Анализ является функциональным и позволяет измерять входящий поток Na+ непосредственно. CoroNa Red, SBFI и/или натриевый зеленый (Molecular Probes, Inc. Eugene OR) можно использовать для того, чтобы измерять входящий поток Na; все являются красителями, чувствительными к Na. Их можно использовать в комбинации с прибором FLIPR. Использование этих красителей в скрининге ранее не описано в литературе. Кальциевые красители также могут иметь потенциал в этом формате.
В другом анализе датчики напряжения на основе FRET используют для того, чтобы измерять способность тестируемого соединения непосредственно блокировать входящий поток Na. Коммерчески доступные HTS системы включают систему VIPR™ II FRET (Aurora Biosciences Corporation, San Diego, CA, подразделение Vertex Pharmaceuticals, Inc.), которую можно использовать в сочетании с красителями FRET, также доступными в Aurora Biosciences. В этом анализе измеряют субсекундные ответы на изменения напряжения. Требования к модификатору функции канала отсутствуют. В анализе измеряют деполяризацию и гиперполяризацию и для количественного определения предоставляют выходные данные о соотношениях. В несколько менее дорогостоящей версии MTS этого анализа используют FLEXstation™ (Molecular Devices Corporation) в сочетании с красителями FRET из Aurora Biosciences. Другие способы тестирования соединений, раскрытые в настоящем описании, также хорошо известны и легкодоступны специалистам в данной области.
Эти результаты предоставляют основу для анализа зависимости структура-активность (SAR) между тестируемыми соединениями и натриевым каналом. Определенные заместители на структуре ядра тестируемого соединения склонны давать более активные ингибирующие соединения. SAR анализ является одним из инструментов, которые специалисты в данной области могут сейчас использовать для того, чтобы идентифицировать предпочтительные варианты осуществления соединений по изобретению для применения в качестве терапевтических средств.
Модулирующие средства, идентифицированные таким образом, затем тестируют в различных моделях in vivo с тем, чтобы определять, можно ли их использовать при лечении заболевания или состояния, ассоциированного с активность натриевого канала, представляющего интерес, предпочтительно NaV1.6, при минимальных нежелательных явлениях. Анализы, описанные далее в разделе о Биологических анализах, можно использовать при оценке биологической активности данных соединений.
Обычно эффект соединения по изобретению выражают с помощью его значения IC50 («ингибирующая концентрация - 50%»), которое является мерой количества соединения, необходимого для того, чтобы добиваться 50% ингибирования активности целевого натриевого канала за конкретный период времени. Например, репрезентативные соединения по настоящему изобретению демонстрировали IC50 в диапазоне от меньше чем 100 наномоль до меньше чем 10 микромоль в электрофизиологическом анализе NaV1.6 с фиксацией напряжения, описанном в настоящем описании.
При альтернативном использовании по изобретению, соединения по изобретению можно использовать в исследованиях in vitro или in vivo в качестве образцовых средств для целей сравнения, чтобы находить другие соединения, которые также можно использовать при лечении или защите от различных заболеваний, раскрытых в настоящем описании.
Другой аспект изобретения относится к ингибирующей NaV1.6 активности в биологическом образце или у млекопитающего, предпочтительно у человека, этот способ включает введение млекопитающему, предпочтительно человеку, или приведение в контакт указанного биологического образца с соединением формулы (I) или фармацевтической композицией, содержащей соединение формулы (I). Термин «биологический образец», как используют в настоящем описании, включает, без ограничения, клеточные культуры или их экстракты; материал биопсии, полученный у млекопитающего, или его экстракты; и кровь, слюну, мочу, фекалии, семя, слезы или другие текучие вещества организма или их экстракты.
Ингибирование активности NaV1.6 в биологическом образце можно использовать для различных целей, которые известны специалисту в данной области. Примеры таких целей включают, но не ограничиваясь этим, исследование натриевых ионных каналов при биологических и патологических феноменах; и сравнительную оценку новых ингибиторов натриевых ионных каналов.
Соединения по изобретению, как изложено выше в Сущности изобретения, в виде их стереоизомеров, энантиомеров, таутомеров или их смесей или их фармацевтически приемлемых солей, сольватов или пролекарственных средств и/или фармацевтических композиций, описанных в настоящем описании, которые содержат фармацевтически приемлемый эксципиент и одно или несколько соединений по изобретению, как изложено выше в Сущности изобретения, в виде его стереоизомера, энантиомера или таутомера или их смесей или его фармацевтически приемлемой соли, сольвата или пролекарственного средства, можно использовать при получении лекарственного средства для лечения заболеваний или состояний, ассоциированных с активностью потенциалзависимого натриевого канала, предпочтительно с активностью NaV1.6, у млекопитающего.
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ПО ИЗОБРЕТЕНИЮ AND ADMINISTRATION
Настоящее изобретение также относится к фармацевтической композиции, содержащей соединения по изобретению, раскрытые в настоящем описании. В одном из вариантов осуществления настоящее изобретение относится к композиции, содержащей соединения по изобретению в фармацевтически приемлемом носителе, эксципиенте или разбавителе и в количестве, эффективном для того, чтобы модулировать, предпочтительно ингибировать, ионный поток через потенциалзависимый натриевый канал, чтобы лечить опосредованные натриевыми каналами заболевания, такие как эпилепсия и/или эпилептическое пароксизмальное нарушение, при введении животному, предпочтительно млекопитающему, наиболее предпочтительно пациенту-человеку.
Введение соединений по изобретению или их фармацевтически приемлемых солей, в чистой форме или в подходящей фармацевтической композиции, можно осуществлять с помощью любых общепринятых способов введения средств, служащих схожим практическим задачам. Фармацевтические композиции по изобретению можно получать посредством объединения соединения по изобретению с подходящим фармацевтически приемлемым носителем, разбавителем или эксципиентом, и можно формулировать в виде препаратов в твердой, полутвердой, жидкой или газообразной формах, таких как таблетки, капсулы, порошки, гранулы, мази, растворы, суппозитории, инъекции, средства для ингаляции, гели, микросферы и аэрозоли. Типичные пути введения таких фармацевтических композиций включают, без ограничения, оральный, местный, трансдермальный, ингаляционный, парентеральный, сублингвальный, ректальный, вагинальный и интраназальный. Термин «парентеральный», как используют в настоящем описании, включает подкожные инъекции, внутривенные, внутримышечные, интрастернальные инъекции или инфузионные приемы. Фармацевтические композиции по изобретению формулируют с тем, чтобы делать активные ингредиенты, содержащиеся в них, биодоступными при введении композиции пациенту. Композиции, которые следует вводить субъекту или пациенту, принимают форму одной или нескольких единиц дозирования, где, например, таблетка может представлять собой одну единицу дозирования, а контейнер с соединением по изобретению в аэрозольной форме может содержать множество единиц дозирования. Современные способы получения таких дозированных форм известны или будут видны специалистам в данной области; например, см. The Science and Practice of Pharmacy, 20-е изд. (Philadelphia College of Pharmacy and Science, 2000). Композиция, подлежащая введению, в любом случае, должна содержать терапевтически эффективное количество соединения по изобретению или его фармацевтически приемлемой соли для лечения заболевания или состояния, представляющего интерес, в соответствии с положениями данного изобретения.
Фармацевтические композиции, которые можно использовать в настоящем описании, также содержат фармацевтически приемлемый носитель, содержащий любой подходящий разбавитель или эксципиент, который включает любое фармацевтическое средство, которое само не вызывает образование антител, вредных для индивидуума, получающего композицию, и которое можно вводить без излишней токсичности. Фармацевтически приемлемые носители включают, но не ограничиваясь этим, жидкости, такие как вода, солевой раствор, глицерин и этанол, и т. п. Подробное обсуждение фармацевтически приемлемых носителей, разбавителей и других эксципиентов приведено в REMINGTON'S PHARMACEUTICAL SCIENCES (Mack Pub. Co., N.J., текущее издание).
Фармацевтическая композиция по изобретению может быть в форме твердого вещества или жидкости. В одном из аспектов, носитель(и) представляет собой частицы с тем, чтобы композиции были, например, в таблетированной или порошкообразной форме. Носитель(и) может представлять собой жидкость, причем композиции представляют собой, например, оральный сироп, инъецируемую жидкость или аэрозоль, который можно использовать, например, при ингаляционном введении.
Когда предназначена для орального введения, фармацевтическая композиция предпочтительно имеет твердую или жидкую форму, где полутвердые, полужидкие, суспензионные и гелевые формы включены в формы, рассматриваемые в настоящем описании в качестве твердых или жидких.
В качестве твердой композиции для орального введения, фармацевтическую композиция можно формулировать в виде порошка, гранулы, прессованной таблетки, пилюли, капсулы, жевательной резинки, пластинки или тому подобной форы. Такая твердая композициа обычно содержит один или несколько инертных разбавителей или съедобных носителей. Кроме того, могут присутствовать одно или несколько из следующих: связывающие средства, такие как карбоксиметилцеллюлоза, этилцеллюлоза, микрокристаллическая целлюлоза, трагантовая камедь или желатин; эксципиенты, такие как крахмал, лактоза или декстрины, средства для улучшения распадаемости, такие как альгиновая кислота, альгинат натрия, Primogel, кукурузный крахмал и т. п.; смазывающие средства, такие как стеарат магния или Sterotex; скользящие средства, такие как коллоидный диоксид кремния; подсластители, такие как сахароза или сахарин; ароматизатор, такой как перечная мята, метилсалицилат или оранжевого цветаовый ароматизатор; и краситель.
Когда фармацевтическая композиция имеет форму капсулы, например, желатиновой капсулы, она может содержать, в дополнение к материалам вышеуказанного типа, жидкий носитель, такой как полиэтиленгликоль или масло.
Фармацевтическая композиция может быть в форме жидкости, например, крепкого настоя, сиропа, раствора, эмульсии или суспензии. Жидкость может быть предназначена для орального введения или для доставки посредством инъекции, в качестве двух примеров. Когда предназначена для орального введения, предпочтительная композиция содержит, в дополнение к данным соединениям, одно или несколько из подсластителя, консервантов, красителя/красящего вещества и усилителя аромата. В композицию, предназначенную для введения посредством инъекции, можно включать одно или несколько из поверхностно-активного средства, консерванта, смачивающего средства, диспергирующего средства, суспендирующего средства, буфера, стабилизатора и изотонического средства.
Жидкие фармацевтические композиции по изобретению, будь то растворы, суспензии или другие подобные формы, могут содержать один или несколько из следующих адъювантов: стерильные разбавители, такие как вода для инъекций, солевой раствор, предпочтительно физиологический солевой раствор, раствор Рингера, изотонический хлорид натрия, жирные масла, такие как синтетические моно- или диглицериды, которые могут служить в качестве растворителя или суспендирующей среды, полиэтиленгликоли, глицерин, пропиленгликоль или другие растворители; антибактериальные средства, такие как бензиловый спирт или метилпарабен; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатирующие средства, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетаты, цитраты или фосфаты, и средства для корректировки тоничности, такие как хлорид натрия или декстроза. Парентеральный препарат можно заключать в ампулы, одноразовые шприцы или флаконы с несколькими дозами, выполненные из стекла или пластмассы. Физиологический солевой раствор является предпочтительным адъювантом. Инъецируемая фармацевтическая композиция предпочтительно является стерильной.
Жидкая фармацевтическая композиция по изобретению, предназначенная для парентерального или орального введения, должна содержать такое количество соединения по изобретению, чтобы получать подходящую дозу. Обычно это количество составляет по меньшей мере 0,01% соединения по изобретению в композиции. Когда предназначено для орального введения, это количество может варьировать между 0,1 и приблизительно 70% по массе композиции. Предпочтительные оральные фармацевтические композиции содержат между приблизительно 4% и приблизительно 50% соединения по изобретению. Предпочтительные фармацевтические композиции и препараты в соответствии с настоящим изобретением получают с тем, чтобы парентеральная единица дозирования содержала между 0,01 и 10% по массе соединения перед разведениеим по изобретению.
Фармацевтическая композиция по изобретению может быть предназначена для топического введения, и в этом случае носитель может подходящим образом содержать раствор, эмульсионную, мазевую или гелевую основу. Основа, например, может содержать одно или несколько из следующего: вазелин, ланолин, полиэтиленгликоли, пчелиный воск, минеральное масло, разбавители, такие как вода и спирт, и эмульсификаторы и стабилизаторы. Загустители могут присутствовать в фармацевтической композиции для топического введения. Если предназначена для трансдермального введения, композиция может включать трансдермальный пластырь или устройство для ионтофореза. Топические составы могут иметь концентрацию соединения по изобретению приблизительно от 0,1 приблизительно до 10% масс./об. (масса на единицу объема).
Фармацевтическая композиция по изобретению может быть предназначена для ректального введения, в форме, например, суппозитория, который будет плавиться в прямой кишке и высвобождать лекарственное средство. Композиция для ректального введения может содержать маслянистую основу в качестве подходящего нераздражающего эксципиента. Такие основания включают, без ограничения, ланолин, масло какао и полиэтиленгликоль.
Фармацевтическая композиция по изобретению может содержать различные материалы, которые модифицируют физическую форму твердой или жидкой единицы дозирования. Например, композиция может содержать материалы, которые формируют покрывающую оболочку вокруг активных ингредиентов. Материалы, которые формируют покрывающую оболочку, обычно являются инертными, и их можно выбирать например, из сахара, шеллака и других энтеросолюбильных покрывающих средств. Альтернативно, активные ингредиенты можно заключать в желатиновую капсулу.
Фармацевтическая композиция по изобретению в твердой или жидкой форме может содержать средство, которое связывается с соединением по изобретению и тем самым способствует доставке соединения. Подходящие средства, которые могут действовать в этом направлении, включают моноклональное или поликлональное антитело, белок или липосому.
Фармацевтическая композиция по изобретению может состоять из единиц дозирования, которые можно вводить в качестве аэрозоля. Термин аэрозоль используют для обозначения различных систем в диапазоне от таковых с коллоидными свойствами до систем, состоящих из упаковок под давлением. Доставка может быть опосредована сжиженным или сжатым газом или подходящей насосной системой, которая дозирует активные ингредиенты. Аэрозоли соединений по изобретению можно доставлять в виде одной фазы, двухфазных или трехфазных систем для того, чтобы доставлять активный ингредиент(ы). Доставка аэрозоля включает необходимый контейнер, активаторы, клапаны, подконтейнеры и т. п., которые вместе могут формировать набор. Специалист в данной области, без излишних экспериментов, может определять предпочтительные аэрозоли.
Фармацевтические композиции по изобретению можно получать способами, хорошо известными в фармацевтической области. Например, фармацевтическую композицию, предназначенную для введения посредством инъекции, можно получать посредством объединения соединения по изобретению со стерильной дистиллированной водой с тем, чтобы формировать раствор. Поверхностно-активное средство можно добавлять для того, чтобы содействовать формированию гомогенного раствора или суспензии. Поверхностно-активные средства представляют собой соединения, которые нековалентно взаимодействуют с соединением по изобретению с тем, чтобы содействовать растворению или гомогенной суспензии соединения в водной системе доставки.
Соединения по изобретению или их фармацевтически приемлемые соли вводят в терапевтически эффективном количестве, которое будет варьировать в зависимости от различных факторов, включая активность конкретного используемого соединения; метаболическую стабильность и длительность действия соединения; возраст, массу тела, общее состояние здоровья, пол и диету пациента; способ и время введения; скорость экскреции; комбинацию лекарственных средств; тяжесть конкретного нарушения или состояния; и субъекта, проходящего терапию. В целом, терапевтически эффективная суточная доза составляет (для 70 кг млекопитающего) приблизительно от 0,001 мг/кг (т. е. 0,07 мг) приблизительно до 100 мг/кг (т. е. 7,0 г); предпочтительно терапевтически эффективная доза составляет (для 70 кг млекопитающего) приблизительно от 0,01 мг/кг (т. е. 0,7 мг) приблизительно до 50 мг/кг (т. е. 3,5 г); более предпочтительно терапевтически эффективная доза составляет (для 70 кг млекопитающего) приблизительно от 1 мг/кг (т. е. 70 мг) приблизительно до 25 мг/кг (т. е. 1,75 г).
Диапазоны эффективных доз, предусмотренные в настоящем описании, не предназначены для того, чтобы ограничивать и представлять предпочтительные диапазоны доз. Однако наиболее предпочтительную дозу следует подбирать для индивидуального субъекта, как это понимает и может определять специалист в связанной области (см., например, Berkow et al., eds., The Merck Manual, 19-е изд., Merck and Co., Rahway, N.J., 2011; Brunton et al. ред., Goodman and Cilman's The Pharmacological Basis of Therapeuticals, 12-е изд., McGraw-Hill 2011; Avery's Drug Treatment: Principles and Practice of Clinical Pharmacology and Therapeutics, 3-е изд., ADIS Press, LTD., Williams and Wilkins, Baltimore, MD. (1987), Ebadi, Pharmacology, Little, Коричневого цвета and Co., Boston, (1985); Osolci al., ред., Remington's Pharmaceutical Sciences, текущее издание, Mack Publishing Co., Easton, PA; Katzung, Basic and Clinical Pharmacology, Appleton and Lange, Norwalk, CT (1992)).
Суммарную дозу, необходимую для каждого лечения, можно вводить с помощью нескольких доз или в однократной дозе в течение дня, при желании. В целом, лечение начинают с меньших доз, которые меньше оптимальной дозы соединения. После этого дозу увеличивают малыми приращениями до тех пор, пока не достигают оптимального эффекта при определенных условиях. Диагностическое фармацевтическое соединение или композицию можно вводить отдельно или в сочетании с другими диагностическими и/или фармацевтическими средствами, направленными на патологию или направленными на другие симптомы патологии. Реципиенты введения соединений и/или композиций по изобретению могут являться любым позвоночным животным, таким как млекопитающее. Среди млекопитающих предпочтительными реципиентами являются млекопитающие отрядов Primates (включая человека, человекообразных обезьян и мартышек), Arteriodactyla (включая лошадей, коз, коров, овец, свиней), Rodenta (включая мышей, крыс и хомяков), Lagamorpha (включая кроликов) и Carnivora (включая кошек и собак). Среди птиц предпочтительными реципиентами являются индюшки, куры и другие члены того же отряда. Наиболее предпочтительным реципиентом является человек.
Для топического нанесения предпочтительно вводить эффективное количество фармацевтической композиции в соответствии с изобретением на целевую область, например, кожные поверхности, слизистые оболочки и т. п., которые прилежат к периферическим нейронам, которые подлежат лечению. Это количество в целом находится в диапазоне приблизительно от 0,0001 мг приблизительно до 1 г соединения по изобретению на нанесение, в зависимости от области, подлежащей лечению, в зависимости от диагностического, профилактического или терапевтического использования, тяжести симптомов и свойств используемого топического носителя. Предпочтительным топическим препаратом является мазь, в которой приблизительно от 0,001 приблизительно до 50 мг активного ингредиента используют на кубический сантиметр мазевой основы. Фармацевтическую композицию можно формулировать в виде трансдермальных композиций или устройств трансдермальной доставки («пластырей»). Такие композиции включают, например, подложку, резервуар активного соединения, контролирующую мембрану, защитное покрытие и контактный адгезив. Такие трансдермальные пластыри можно использовать для обеспечения непрерывной импульсной доставки или доставки по требованию для соединений по настоящему изобретению, по желанию.
Композиции по изобретению можно формулировать с тем, чтобы обеспечивать быстое, замедленное или отсроченное высвобождение активного ингредиента после введения пациенту, используя процедуры, известные в данной области. Системы доставки лекарственных средств с контролируемым высвобождением включают системы осмотического насоса и растворимые системы, включающие резервуары с полимерным покрытием или составы полимерных матриц с лекарственным средством. Примеры систем с контролируемым высвобождением приведены в патентах США №№ 3,845,770 и 4,326,525 и в P. J. Kuzma et al., Regional Anesthesia 22 (6): 543-551 (1997), все они включены в настоящее описание посредством ссылки.
Композиции по изобретению также можно доставлять через интраназальные системы доставки лекарственных средств для местной, системной терапии и медицинской терапии из носа в головной мозг. Специалистам в данной области известно, что технология частиц с контролируемой дисперсией (CPD)TM, традиционные бутылки с назальными спреями, ингаляторы или небулайзеры обеспечивают эффективную местную и системную доставку лекарственных средств посредством направленного воздействия на обонятельную область и околоносовые синусы.
Изобретение также относится к интравагинальному устройству доставки лекарственного средства с ядром в оболочке, подходящему для введения самке человека или животного. Устройство может состоять из активного фармацевтического ингредиента в полимерной матрице, окруженного покрытием и способного высвобождать соединение ежедневно по паттерну по существу нулевого порядка, подобно устройствам, используемым для применения тестостерона, как описано в опубликованной патентной заявке PCT № WO 98/50016.
Современные способы глазной доставки включают топическое введение (глазные капли), субконъюктивальные инъекции, периокулярные инъекции, интравитреальные инъекции, хирургические импланты и ионтофорез (с использованием слабого электрического тока для транспортировки ионизированных лекарственных средств в и через ткани организма). Специалисты в данной области объединят наиболее подходящие эксципиенты с соединением для безопасного и эффективного внутриглазного введения.
Наиболее подходящий путь зависит от свойств и тяжести состояния, которое лечат. Специалисты в данной области также знакомы с определением способов введения (например, оральный, внутривенный, ингаляционный, подкожный, ректальный и т. д.), дозированных форм, подходящих фармацевтических эксципиентов и других вопросов, связанных с доставкой соединений нуждающемуся в этом субъекту.
КОМБИНИРОВАННАЯ ТЕРАПИЯ
Соединения по изобретению можно эффективно комбинировать с одним или несколькими другими соединениями по изобретению или одним или несколькими другими терапевтическими средствами или в виде любого их сочетания, при лечении заболеваний и состояний, ассоциированных с активностью потенциалзависимых натриевых каналов. Например, соединение по изобретению можно вводить одновременно, последовательно или отдельно в комбинации с другими терапевтическими средствами, включая в качестве неограничивающих примеров:
- опиоидные аналгетики, например, морфин, героин, кокаин, оксиморфин, леворфанол, леваллорфан, оксикодон, кодеин, дигидрокодеин, пропоксифен, налмефен, фентанил, гидрокодон, гидроморфон, мерипидин, метадон, налорфин, налоксон, налтрексон, бупренорфин, буторфанол, налбуфин и пентазоцин;
- неопиоидные аналгетики, например, ацетаминофен, салицилаты (например, аспирин);
- нестероидные противовоспалительные лекарственные средства (НПВС), например, ибупрофен, напроксен, фенопрофен, кетопрофен, целекоксиб, диклофенак, дифлузинал, этодолак, фенбуфен, фенопрофен, флуфенизал, флурбипрофен, ибупрофен, индометацин, кетопрофен, кеторолак, меклофенамовую кислоту, мефенамовую кислоту, мелоксикам, набуметон, напроксен, нимесулид, нитрофлурбипрофен, олсалазин, оксапрозин, фенилбутазон, пироксикам, сульфасалазин, сулиндак, толметин и зомепирак;
- антиконвульсанты, например, карбамазепин, окскарбазепин, ламотригин, вальпроат, топирамат, габапентин и прегабалин;
- антидепрессанты, такие как трициклические антидепрессанты, например, амитриптилин, кломипрамин, деспрамин, имипрамин и нортриптилин;
- избирательные ингибиторы ЦОГ-2, например, целекоксиб, рофекоксиб, парекоксиб, валдекоксиб, деракоксиб, эторикоксиб и люмиракоксиб;
- α-адренергические средства, например, доксазозин, тамсулозин, клонидин, гуанфацин, дексметатомидин, модафинил и 4-амино-6,7-диметокси-2-(5-метансульфонамидо-1,2,3,4-тетрагидроизохинол-2-ил)-5-(2-пиридил) хиназолин;
- барбитуратные седативные средства, например, амобарбитал, апробарбитал, бутабарбитал, бутабитал, мефобарбитал, метарбитал, метогекситал, пентобарбитал, фенобартитал, секобарбитал, талбутал, теамилал и тиопентал;
- антагонист тахикинина (NK), в частности, антагонист NK-3, NK-2 или NK-1, например, (αR, 9R)-7-[3,5-бис(трифторметил)бензил)]-8,9,10,11-тетрагидро-9-метил-5-(4-метилфенил)-7H-[1.4]диазоцино[2.1-g][1,7]-нафтиридин-6-13-дион (TAK-637), 5-[[2R,3S)-2-[(1R)-1-[3,5-бис(трифторметилфенил]этокси-3-(4-фторфенил)-4-морфолинил]-метил]-1,2-дигидро-3H-1,2,4-триазол-3-он (MK-869), апрепитант, ланепитант, дапитант или 3-[[2-метокси5-(трифторметокси)фенил]-метиламино]-2-фенилпиперидин (2S,3S);
- аналгетики из каменноугольной смолы, в частности парацетамол;
- ингибиторы обратного захвата серотонина, например, пароксетин, сертралин, норфлуоксетин (метаболит флуоксетиндесметила), метаболит деметилсертралин, '3 флувоксамин, пароксетин, циталопрам, десметилциталопрам метаболит циталопрама, эсциталопрам, d,l-фенфлурамин, фемоксетин, ифоксетин, цианодотиепин, литоксетин, дапоксетин, нефазодон, церикламин, тразодон и флуоксетин;
- ингибиторы обратного захвата норадреналина (норэпинефрина), например, мапротилин, лофепрамин, миртазепин, оксапротилин, фезоламин, томоксетин, миансерин, бупроприон, гидроксибупроприон метаболит бупроприона, номифензин и вилоксазин (вивалан®)), в частности, избирательный ингибитор обратного захвата норадреналина, такой как ребоксетин, в частности (S,S)-ребоксетин, и венлафаксин дулоксетин нейролептики седативные/анксиолитики;
- двойные ингибиторы обратного захвата серотонина и норадреналина, такие как венлафаксин, O-десметилвенлафаксин метаболит венлафаксина, кломипрамин, десметилкломипрамин метаболит кломипрамина, дулоксетин, милнаципран и имипрамин;
- ингибиторы ацетилхолинэстеразы, такие как донепезил;
- антагонисты 5-HT3, такие как ондансетрон;
- антагонисты метаботропных глутаматных рецепторов (mGluR);
- локальный анестетик, такой как мексилетин и лидокаин;
- кортикостероид, такой как дексаметазон;
- антиаритмические средства, например, мексилетин и фенитоин;
- мускариновые антагонисты, например, толтеродин, пропиверин, троспия хлорид, дарифенацин, солифенацин, темиверин и ипратропий;
- каннабиоиды;
- агонисты (например, ресинферотоксин) или антагонисты (например, капсазепин) ваниллоидных рецепторов;
- седативные средства, например, глютетимид, мепробамат, метаквалон и дихлоралфеназон;
- анксиолитики, такие как бензодиазепины,
- антидепрессанты, такие как миртазапин,
- топические средства (например, лидокаин, капсайцин и ресинеферотоксин);
- мышечные релаксанты, такие как бензодиазепины, баклофен, карисопродол хлорзоксазон, циклобензаприн, метокарбамол и орфренадин;
- антигистаминные средства или антагонисты H1;
- антагонисты NMDA рецепторов;
- агонисты/антагонисты 5-HT рецепторов;
- ингибиторы PDEV;
- трамадол®;
- холинергические (никотиновые) аналгетики;
- α-2-δ лиганды;
- антагонисты простагландинов подтипа E2;
- антагонисты лейкотриена B4;
- ингибиторы; и
- антагонисты 5-HT3.
Как используют в настоящем описании, «комбинация» относится к любой смеси или перестановке одного или нескольких соединений по изобретению и одного или нескольких других соединений по изобретению или одного или нескольких дополнительных терапевтических средств. Пока контекст явно не указывает на иное, «комбинация» может включать одновременную или последовательную доставку соединения по изобретению с одним или несколькими терапевтическими средствами. Пока контекст явно не указывает на иное, «комбинация» может включать дозированные формы соединения по изобретению с другим терапевтическим средством. Пока контекст явно не указывает на иное, «комбинация» может включать пути введения соединения по изобретению с другим терапевтическим средством. Пока контекст явно не указывает на иное, «комбинация» может содержать составы соединения по изобретению с другим терапевтическим средством. Дозированные формы, пути введения и фармацевтические композиции включают, но не ограничиваясь этим, то, что описано в настоящем описании.
НАБОРЫ ЧАСТЕЙ
Настоящее изобретение также относится к наборам, которые содержат фармацевтическую композицию, которая содержит одно или несколько соединений по изобретению. Набор также содержит инструкции по использованию фармацевтической композиции для ингибирования активности потенциалзависимых натриевых каналов, предпочтительно NaV1.6, для лечения эпилепсии, а также другой пользы, как раскрыто в настоящем описании. Предпочтительно, коммерческая упаковка содержит одну или несколько стандартных доз фармацевтической композиции. Например, такая стандартная доза может представлять собой количество, достаточное для получения внутривенной инъекции. Специалистам в данной области будет очевидно, что соединения, которые чувствительны к свету и/или воздуху, могут требовать специальной упаковки и/или состава. Например, можно использовать упаковку, которая непрозрачна для света и/или герметизирована от контакта с окружающим воздухом и/или сформулирована с подходящими покрытиями или эксципиентами.
ПОЛУЧЕНИЕ СОЕДИНЕНИЙ ПО ИЗОБРЕТЕНИЮ
Следующие схемы реакций иллюстрируют способы получения соединений по данному изобретению, т. е. соединений формулы (I), в виде их индивидуальных стереоизомеров, энантиомеров или таутомеров или их смесей; или в виде их фармацевтически приемлемых солей, сольватов или пролекарственных средств
Также понятно, что специалист в данной области сможет получать соединения по изобретению с помощью схожих способов или способов, известных специалисту в данной области. Также понятно, что специалист в данной области сможет получать схожим образом, как описано ниже, другие соединения по изобретению, конкретно не проиллюстрированные далее, используя подходящие исходные компоненты и модифицируя параметры синтеза, при необходимости. Также понятно, что простые превращения функциональных групп (см., например, Larock, R.C. Comprehensive Organic Transformations, 2-е изд. (Wiley, 1999) можно осуществлять способами, известными специалисту в данной области. В целом, исходные компоненты можно получать из таких источников, как Sigma Aldrich, Combi-Blocks, Oakwood Chemicals, Inc., Maybridge, Matrix Scientific, TCI и Fluorochem USA и т. д., или синтезировать в соответствии с источниками, известными специалистам в данной области (см., например, Smith, M.B. и J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 6-е изд. (Wiley, 2007)), или получать как раскрыто в настоящем описании.
Также понятно, что в дальнейшем описании, комбинации заместителей и/или переменных в изображенных формулах допустимы, только если такие вклады ведут к стабильным соединениям.
Также специалисты в данной области примут во внимание, что в процессе, описанном далее, функциональные группы промежуточных соединений могут требовать защиты с помощью подходящих защитных групп. Такие функциональные группы включают гидрокси, амино, меркапто и карбоновую кислоту. Подходящие защитные группы для гидрокси включают триалкилсилил или диарилалкилсилил (например, т-бутилдиметилсилил, т-бутилдифенилсилил или триметилсилил), тетрагидропиранил, бензил и т. п. Подходящие защитные группы для амино, амидино и гуанидино включают т-бутоксикарбонил, бензилоксикарбонил и т. п. Подходящие защитные группы для меркапто включают C(O) R'' (где R'' представляет собой алкил, арил или аралкил), п метоксибензил, тритил и т. п. Подходящие защитные группы для карбоновой кислоты включают сложные алкиловые, ариловые или арилалкиловые эфиры.
Защитные группы можно добавлять или удалять в соответствии со стандартными приемами, которые известны специалисту в данной области и как раскрыто в настоящем описании.
Использование защитных групп подробно описано в Greene, T.W. and P.G.M. Wuts, Greene's Protective Groups in Organic Synthesis (2006), 4-е изд., Wiley. Защитная группа также может представлять собой полимерную смолу, такую как смола Ванга или 2-хлортритил-хлоридная смола.
Также специалисты в данной области примут во внимание, что, несмотря на то, что такие защищенные производные соединений по данному изобретению могут не обладать фармакологической активностью сами по себе, их можно вводить млекопитающем и после этого подвергать метаболизму в организме для того, чтобы формировать соединения по изобретению, которые фармакологически активны. Следовательно, такие производные можно описывать как «пролекарственные средства». Все пролекарственные средства соединений по данному изобретению включены в объем изобретения.
Соединения формулы (I) могут содержать по меньшей мере один асимметричный атом углерода и, таким образом, могут существовать в виде рацематов, энантиомеров и/или диастереоизомеров. Конкретные энантиомеры или диастереоизомеры можно получать с использованием подходящего хирального исходного материала. Альтернативно, диастереоизомерные смеси или рацемические смеси соединений формулы (I) можно разрешать на соответствующие им энантиомеры или диастереоизомеры. Способы разрешения диастереоизомерных смесей или рацемических смесей соединений формулы (I), как раскрыто в настоящем описании, или промежуточных соединений, полученных в настоящем описании, хорошо известны в данной области (например, E.L. Eliel and S.H. Wilen, в Stereochemistry of Organic Compounds; John Wiley & Sons: New York, 1994; глава 7, и цитируемые в них ссылки). Подходящие процессы, такие как кристаллизация (например, предпочтительная кристаллизация, предпочтительная кристаллизация в присутствии добавок), асимметричное превращение рацематов, химическое разделение (например, формирование и разделение диастереомеров, таких как смеси диастереомерных солей, или использование других разрешающих средств; разделение через комплексы и соединения включения), кинетическое разрешение (например, с катализатором тартратом титана), ферментативное разрешение (например, опосредованное липазой) и хроматографическое разделение (например, HPLC с хиральной стационарной фазой и/или с использованием технологии имитации подвижного слоя или сверхкритическая жидкостная хроматография и родственные приемы) являются некоторыми из примеров, которые можно применять (см., например, T.J. Ward, Analytical Chemistry, 2002, 2863-2872).
Получение соединений формулы (I)
В целом, соединения формулы (I), как описано выше в Сущности изобретения, можно синтезировать, придерживаясь общей процедуры, описанной далее на схеме реакции 1, где q, A, R1, R2, R3, R6 и R7 представляют собой то, что описано выше в Сущности изобретения для соединений формулы (I), X представляет собой бром, хлор или фтор, и Z1 представляет собой защитную группу водорода или азота, например, но не ограничиваясь этим, трет-бутилоксикарбонил, 2,4-диметоксибензил или 4-метоксибензил:
СХЕМА РЕАКЦИИ 1

Соединения формул (101), (102) и (103) являются коммерчески доступными, или их можно получать в соответствии со способамии, известными специалисту в данной области, или с помощью способов, раскрытых в настоящем описании. В целом, соединения формулы (I) получают, как описано выше на схеме реакции 1, следующим образом:
Соединение формулы (101) вступает в реакцию с сульфонамидом (102) при стандартных условиях реакции, таких как, но не ограничиваясь этим, использование полярного апротонного растворителя, такого как, но не ограничиваясь этим, диметилсульфоксид или N,N-диметилформамид, в присутствии основания, такого как, но не ограничиваясь этим, карбонат калия, гидрид натрия или N,N-диизопропилэтиламин, при температуре между приблизительно 0°C и 80°C, в течение приблизительно от 1 до 48 часов для того, чтобы получать соединение формулы (103). Соединение формулы (103) после этого обрабатывают кислотой, такой как, но не ограничиваясь этим, трифторуксусная кислота, в полярном апротонном растворителе, таком как, но не ограничиваясь этим, дихлорметан, при температуре между приблизительно 0°C и температурой окружающей среды для того, чтобы создавать соединение формулы (I), которое можно выделять из реакционной смеси стандартными приемами.
Альтернативно, соединения формулы (I), как описано выше в Сущности изобретения, можно синтезировать, придерживаясь общей процедуры, описанной далее на схеме реакции 2, где q, A, R1, R2, R3, R6 и R7 представляют собой то, что описано выше в Сущности изобретения для соединений формулы (I), X представляет собой бром, хлор или йод и Z1 представляет собой защитную группу водорода или азота, например, но не ограничиваясь этим, трет-бутилоксикарбонил, 2,4-диметоксибензил или 4-метоксибензил:
СХЕМА РЕАКЦИИ 2

Соединения формул (201), (202) и (203) являются коммерчески доступными, или их можно получать в соответствии со способамии, известными специалисту в данной области, или способами, раскрытыми в настоящем описании. В целом, соединения формулы (I) получают как описано выше на схеме реакции 2 следующим образом:
Соединение формулы (201) вступает в реакцию с сульфонамидом (202) при стандартных условиях перекрестного сочетания Бухвальда-Хартвига, таких как, но не ограничиваясь этим, использование растворителя, такого как, но не ограничиваясь этим, толуол, в присутствии основания, такого как, но не ограничиваясь этим, трет-бутоксид натрия, и в присутствии палладиевого катализатора, состоящего, например, но не ограничиваясь этим, из 2-ди-трет-бутилфосфино-2′,4′,6′-триизопропилбифенил и бис(дибензилиденацетон)палладия(0), при температуре между приблизительно температурой окружающей среды и 150°C, в течение приблизительно от 30 минут до 72 часов для того, чтобы создавать соединение формулы (203). Соединение формулы (203) затем можно обрабатывать с использованием, например, но не ограничиваясь этим, кислоты, такой как, но не ограничиваясь этим, трифторуксусная кислота, в полярном апротонном растворителе, таком как, но не ограничиваясь этим, дихлорметан, при температуре между приблизительно 0°C и температурой окружающей среды для того, чтобы создавать соединение формулы (I), которое можно выделять из реакционной смеси стандартными приемами.
При определенных условиях, вышеуказанное превращение соединения формулы (201) будет давать соединение формулы (I) вместо соединения формулы (303). В этом случае, соединение формулы (I) можно выделять из реакционной смеси стандартными приемами.
Альтернативно, соединения формулы (I), как описано выше в Сущности изобретения, можно синтезировать, придерживаясь общей процедуры, описанной далее на схеме реакции 3, где q, A, R1, R2, R3, R6 представляют собой то, что описано выше в Сущности изобретения для соединений формулы (I), X представляет собой бром, хлор или йод, R7b представляет собой алкил, алкенил, галогеналкил или циано и Z1 представляет собой защитную группу водорода или азота, например, но не ограничиваясь этим, трет-бутилоксикарбонил, 2,4-диметоксибензил или 4-метоксибензил:
СХЕМА РЕАКЦИИ 3

Соединение формулы (301) можно получать в соответствии со способамии, известными специалисту в данной области, или способами, раскрытыми в настоящем описании. В целом, соединения формулы (I) получают как описано выше на схеме реакции 3 следующим образом:
Соединение формулы (301) вступает в реакцию с сульфонамидом (302) при стандартных условиях реакции, таких как, но не ограничиваясь этим, использование полярного апротонного растворителя, такого как, но не ограничиваясь этим, диметилсульфоксид или N,N-диметилформамид, в присутствии основания, такого как, но не ограничиваясь этим, карбонат калия, гидрид натрия или N,N-диизопропилэтиламин, при температуре между приблизительно 0°C и 80°C, в течение приблизительно от 1 до 48 часов для того, чтобы получать соединение формулы (303). Соединение формулы (303) вступает в реакцию с производным бороновой кислоты, таким как, но не ограничиваясь этим, R7b-B(OH)2 или R7b-BF3K, при стандартных условиях перекрестного сочетания Сузуки-Мияуры, таких как, но не ограничиваясь этим, использование растворителя, такого как, но не ограничиваясь этим, 1,4-диоксан, в присутствии основания, такого как, но не ограничиваясь этим, водный карбонат натрия, и в присутствии палладиевого катализатора, состоящего из, например, но не ограничиваясь этим, тетракис(трифенилфосфин)палладия(0) или ацетата палладия(II) и трициклогексилфосфинтетрафторбората, при температуре между приблизительно температурой окружающей среды и 150°C, в течение приблизительно от 30 минут до 16 часов для того, чтобы создавать соединение формулы (304).
Альтернативно, соединение формулы (303) можно обрабатывать восстанавливающим средством, таким как, но не ограничиваясь этим, формиат натрия, в присутствии палладиевого катализатора, состоящего из, например, но не ограничиваясь этим, трис(дибензилиденацетон)дипалладия(0) и три-трет-бутилфосфина, в полярном растворителе, таком как, но не ограничиваясь этим, диметилсульфоксид, при температуре между приблизительно температурой окружающей среды и 120°C, в течение приблизительно от 1 до 20 часов для того, чтобы получать соединение формулы (304).
Альтернативно, соединение формулы (303) можно обрабатывать восстанавливающим средством, таким как, но не ограничиваясь этим, водород, в присутствии палладиевого катализатора, например, но не ограничиваясь этим, палладия на углероде, в полярном растворителе, таком как, но не ограничиваясь этим, метанол, в присутствии основания, такого как, но не ограничиваясь этим, триметиламин, при температуре между приблизительно температурой окружающей среды и 120°C, в течение приблизительно от 1 до 20 часов для того, чтобы получать соединение формулы (304).
Соединение формулы (304) затем можно обрабатывать, например, но не ограничиваясь этим, кислотой, такой как, но не ограничиваясь этим, трифторуксусная кислота, в полярном апротонном растворителе, таком как, но не ограничиваясь этим, дихлорметан, при температуре между приблизительно 0°C и температурой окружающей среды для того, чтобы создавать соединение формулы (I), которое можно выделять из реакционной смеси стандартными приемами.
При определенных условиях, вышеуказанные превращения соединения формулы (303) будут давать соединение формулы (I) вместо соединения формулы (304). В этих случаях соединение формулы (I) можно выделять из реакционной смеси стандартными приемами.
Альтернативно, соединения формулы (I), как описано выше в Сущности изобретения, можно синтезировать, придерживаясь общей процедуры, описанной далее на схеме реакции 4, где q, A, A1, R2, R3, R6 и R7 представляют собой то, что описано выше в Сущности изобретения для соединений формулы (I), и X представляет собой фтор, Y представляет собой кислород или серу и Z1 представляет собой защитную группу водорода или азота, например, но не ограничиваясь этим, трет-бутилоксикарбонил или 2,4-диметоксибензил:
СХЕМА РЕАКЦИИ 4

Соединения формул (401), (402) и (203) являются коммерчески доступными, или их можно получать в соответствии со способамии, известными специалисту в данной области, или способами, раскрытыми в настоящем описании. В целом, соединения формулы (I) получают как описано выше на схеме реакции 4 следующим образом:
Сульфонамид формулы (401) вступает в реакцию при стандартных условиях реакции, таких как, но не ограничиваясь этим, использование полярного апротонного растворителя, такого как, но не ограничиваясь этим, диметилсульфоксид или N,N-диметилформамид, в присутствии нуклеофила, такого как, но не ограничиваясь этим, сульфид натрия, гидроксид натрия или бензоат натрия, при температуре между приблизительно 0°C и 80°C, в течение приблизительно от 1 до 48 часов для того, чтобы получать соединение формулы (402). Соединение формулы (402) после этого вступает в реакцию со спиртом формулы (403) при условиях реакции Мицунобу, таких как, но не ограничиваясь этим, использование растворителя, такого как, но не ограничиваясь этим, простой диэтиловый эфир, в присутствии фосфинового реактива, такого как, но не ограничиваясь этим, трифенилфосфин, и в присутствии азодикарбоксилатного реактива, такого как, но не ограничиваясь этим, диизопропилазодикарбоксилат, при температуре между приблизительно 0°C и 80°C, в течение приблизительно от 1 до 48 часов для того, чтобы получать соединение формулы (403). Соединение формулы (404) после этого обрабатывают кислотой, такой как, но не ограничиваясь этим, трифторуксусная кислота, в полярном апротонном растворителе, таком как, но не ограничиваясь этим, дихлорметан, при температуре между приблизительно 0°C и температурой окружающей среды для того, чтобы создавать соединение формулы (I), которое можно выделять из реакционной смеси стандартными приемами.
Альтернативно, соединения формулы (Ib), как описано выше в Сущности изобретения, можно синтезировать, придерживаясь общей процедуры, описанной далее на схеме реакции 5, где q, A, R1, R2, R6, R7 и R8 представляют собой то, что описано выше в Сущности изобретения для соединений формулы (Ib), Z1 представляет собой защитную группу водорода или азота, например, но не ограничиваясь этим, трет-бутилоксикарбонил, 2,4-диметоксибензил или 4-метоксибензил, и Z2 представляет собой защитную группу водорода или азота, например, но не ограничиваясь этим, трет-бутилоксикарбонил:
СХЕМА РЕАКЦИИ 5

Соединения формул (501), (502) и (503) являются коммерчески доступными, или их можно получать в соответствии со способамии, известными специалисту в данной области, или способами, раскрытыми в настоящем описании. В целом, соединения формулы (Ib) получают как описано выше на схеме реакции 5 следующим образом:
Соединение формулы (501) вступает в реакцию с сульфонамидом (502) при стандартных условиях реакции, таких как, но не ограничиваясь этим, использование полярного апротонного растворителя, такого как, но не ограничиваясь этим, диметилсульфоксид или N,N-диметилформамид, в присутствии основания, такого как, но не ограничиваясь этим, карбонат калия, гидрид натрия или N,N-диизопропилэтиламин, при температуре между приблизительно 0°C и 80°C, в течение приблизительно от 1 до 48 часов для того, чтобы получать соединение формулы (503). Соединение формулы (103) после этого обрабатывают кислотой, такой как, но не ограничиваясь этим, трифторуксусная кислота, в полярном апротонном растворителе, таком как, но не ограничиваясь этим, дихлорметан, при температуре между приблизительно 0°C и температурой окружающей среды для того, чтобы создавать соединение формулы (Ib), которое можно выделять из реакционной смеси стандартными приемами.
Альтернативно, соединения формулы (Ib), как описано выше в Сущности изобретения, можно синтезировать, придерживаясь общей процедуры, описанной далее на схеме реакции 6, где q, A, R1, R2 и R6 представляют собой то, что описано выше в Сущности изобретения для соединений формулы (I), X представляет собой бром, хлор или йод, R7b представляет собой алкил, алкенил, галогеналкил или циано, Z1 представляет собой защитную группу водорода или азота, например, но не ограничиваясь этим, трет-бутилоксикарбонил, 2,4-диметоксибензил или 4-метоксибензил, и Z2представляет собой защитную группу водорода или азота, например, но не ограничиваясь этим, трет-бутилоксикарбонил:
СХЕМА РЕАКЦИИ 6

Соединения формул (601), (602), (603) и (604) являются коммерчески доступными, или их можно получать в соответствии со способамии, известными специалисту в данной области, или способами, раскрытыми в настоящем описании. В целом, соединения формулы (Ib) получают как описано выше на схеме реакции 6 следующим образом:
Соединение формулы (601) вступает в реакцию с сульфонамидом (602) при стандартных условиях реакции, таких как, но не ограничиваясь этим, использование полярного апротонного растворителя, такого как, но не ограничиваясь этим, диметилсульфоксид или N,N-диметилформамид, в присутствии основания, такого как, но не ограничиваясь этим, карбонат калия, гидрид натрия или N,N-диизопропилэтиламин, при температуре между приблизительно 0°C и 80°C, в течение приблизительно от 1 до 48 часов для того, чтобы получать соединение формулы (603).
Соединение формулы (603) вступает в реакцию с производным бороновой кислоты, таким как, но не ограничиваясь этим, R7b-B(OH)2 или R7b-BF3K, при стандартных условиях перекрестного сочетания Сузуки-Мияуры, таких как, но не ограничиваясь этим, использование растворителя, такого как, но не ограничиваясь этим, 1,4-диоксан, в присутствии основания, такого как, но не ограничиваясь этим, водный карбонат натрия, и в присутствии палладиевого катализатора, состоящего из, например, но не ограничиваясь этим, тетракис(трифенилфосфин)палладия(0) или ацетата палладия(II) и трициклогексилфосфинтетрафторбората, при температуре между приблизительно температурой окружающей среды и 150°C, в течение приблизительно от 30 минут до 16 часов для того, чтобы создавать соединение формулы (604).
Альтернативно, соединение формулы (603) можно обрабатывать восстанавливающим средством, таким как, но не ограничиваясь этим, формиат натрия, в присутствии палладиевого катализатора, состоящего из, например, но не ограничиваясь этим, трис(дибензилиденацетон)дипалладия(0) и три-трет-бутилфосфина, в полярном растворителе, таком как, но не ограничиваясь этим, диметилсульфоксид, при температуре между приблизительно температурой окружающей среды и 120°C, в течение приблизительно от 1 до 20 часов для того, чтобы получать соединение формулы (604).
Альтернативно, соединение формулы (603) можно обрабатывать восстанавливающим средством, таким как, но не ограничиваясь этим, водород, в присутствии палладиевого катализатора например, но не ограничиваясь этим, палладий на углероде, в полярном растворителе, таком как, но не ограничиваясь этим, метанол, в присутствии основания, такого как, но не ограничиваясь этим, триметиламин, при температуре между приблизительно температурой окружающей среды и 120°C, в течение приблизительно от 1 до 20 часов для того, чтобы получать соединение формулы (604).
Соединение формулы (604) затем можно обрабатывать, например, но не ограничиваясь этим, кислотой, такой как, но не ограничиваясь этим, трифторуксусная кислота, в полярном апротонном растворителе, таком как, но не ограничиваясь этим, дихлорметан, при температуре между приблизительно 0°C и температурой окружающей среды для того, чтобы создавать соединение формулы (Ib), которое можно выделять из реакционной смеси стандартными приемами.
При определенных условиях, вышеуказанные превращения соединения формулы (603) будут давать соединение формулы (I) вместо соединения формулы (604). В этих случаях, соединение формулы (I) можно выделять из реакционной смеси стандартными приемами.
Альтернативно, соединения формулы (Ib), как описано выше в Сущности изобретения, может синтезировать специалист в данной области посредством простых превращений функциональных групп. По существу, но не ограничиваясь этим, соединение формулы (I), в котором R7b представляет собой алкенил, можно превращать в соединение формулы (Ib), в котором R7b представляет собой алкил, посредством обработки водородом в присутствии, но не ограничиваясь этим, палладия на углероде, в растворителях, таких как, но не ограничиваясь этим, метанол и этилацетат.
Альтернативно, соединения формулы (I), как описано выше в Сущности изобретения, можно синтезировать, придерживаясь общей процедуры, описанной далее на схеме реакции 7, где q, A, R2 и R6 представляют собой то, что описано выше в Сущности изобретения для соединений формулы (I), R1b представляет собой необязательно замещенный арил или необязательно замещенный гетероарил, X представляет собой бром, хлор или йод, Z1 представляет собой защитную группу водорода или азота, например, но не ограничиваясь этим, трет-бутилоксикарбонил, 2,4-диметоксибензил или 4-метоксибензил, R13 и R14 представляют собой независимо водород, необязательно замещенный алкил или необязательно замещенный циклоалкил, или R13 и R14, вместе с азотом, к которому их прикрепляют, образуют необязательно замещенный гетероциклил, и Z2 представляет собой производное бороновой кислоты, такое как, но не ограничиваясь этим, B(OH)2 или BF3K:
СХЕМА РЕАКЦИИ 7

Соединения формул (701), (702) и (703) являются коммерчески доступными, или их можно получать в соответствии со способамии, известными специалисту в данной области, или способами, раскрытыми в настоящем описании. В целом, соединения формулы (I) получают как описано выше на схеме реакции 7 следующим образом:
Соединение формулы (701) вступает в реакцию с производным бороновой кислоты (702), при стандартных условиях перекрестного сочетания Сузуки-Мияуры, таких как, но не ограничиваясь этим, использование смеси растворителей, такой как, но не ограничиваясь этим, 1,4-диоксан и вода, в присутствии основания, такого как, но не ограничиваясь этим, карбонат цезия, и в присутствии палладиевого катализатора, состоящего из, например, но не ограничиваясь этим, ацетата палладия(II) и ди(1-адамантил)-н-бутилфосфина, при температуре между приблизительно температурой окружающей среды и 150°C, в течение приблизительно от 30 минут до 20 часов для того, чтобы создавать соединение формулы (703).
Соединение формулы (703) затем можно обрабатывать, например, но не ограничиваясь этим, кислотой, такой как, но не ограничиваясь этим, трифторуксусная кислота, в полярном апротонном растворителе, таком как, но не ограничиваясь этим, дихлорметан, при температуре между приблизительно 0°C и температурой окружающей среды для того, чтобы создавать соединение формулы (I), которое можно выделять из реакционной смеси стандартными приемами.
При определенных условиях, вышеуказанное превращение соединения формулы (703) будет давать соединение формулы (I) вместо соединения формулы (703). В этих случаях, соединение формулы (I) можно выделять из реакционной смеси стандартными приемами.
Альтернативно, соединения формулы (I), как описано выше в Сущности изобретения, можно синтезировать, придерживаясь общей процедуры, описанной далее на схеме реакции 8, где q, A, R2 и R6 представляют собой то, что описано выше в Сущности изобретения для соединений формулы (I), R1b представляет собой необязательно замещенный арил или необязательно замещенный гетероарил, X представляет собой бром, хлор или йод, Z1 представляет собой защитную группу водорода или азота, например, но не ограничиваясь этим, трет-бутилоксикарбонил, 2,4-диметоксибензил или 4-метоксибензил, R13 и R14 представляют собой независимо водород, необязательно замещенный алкил или необязательно замещенный циклоалкил, или R13 и R14, вместе с азотом, к которому их прикрепляют, образуют необязательно замещенный гетероциклил, и Z2 представляет собой производное бороновой кислоты, такое как, но не ограничиваясь этим, B(OH)2 или BF3K:
СХЕМА РЕАКЦИИ 8

Соединения формул (801), (802) и (803) являются коммерчески доступными, или их можно получать в соответствии со способамии, известными специалисту в данной области, или способами, раскрытыми в настоящем описании. В целом, соединения формулы (I) получают как описано выше на схеме реакции 8 следующим образом:
Соединение формулы (801) вступает в реакцию с производным бороновой кислоты (802), при стандартных условиях перекрестного сочетания Сузуки-Мияуры, таких как, но не ограничиваясь этим, использование смеси растворителей, такой как, но не ограничиваясь этим, 1,4-диоксан и вода, в присутствии основания, такого как, но не ограничиваясь этим, карбонат цезия, и в присутствии палладиевого катализатора, состоящего из, например, но не ограничиваясь этим, метансульфоната (2-дициклогексилфосфино-2′,6′-диметоксибифенил) [2-(2′-амино-1,1′-бифенил)]палладия(II) и 2-дициклогексилфосфино-2′,6′-диметоксибифенила, при температуре между приблизительно температурой окружающей среды и 150°C, в течение приблизительно от 30 минут до 20 часов для того, чтобы создавать соединение формулы (I), которое можно выделять из реакционной смеси стандартными приемами.
При определенных условиях, вышеуказанное превращение соединения формулы (801) будет давать соединение формулы (803) вместо соединения формулы (I). В этих случаях, соединение формулы (803) затем можно обрабатывать, например, но не ограничиваясь этим, кислотой, такой как, но не ограничиваясь этим, трифторуксусная кислота, в полярном апротонном растворителе, таком как, но не ограничиваясь этим, дихлорметан, при температуре между приблизительно 0°C и температурой окружающей среды для того, чтобы создавать соединение формулы (I), которое можно выделять из реакционной смеси стандартными приемами.
Альтернативно, соединение формулы (801) может вступать в реакцию с бис(пинаколато)дибороном при условиях перекрестного сочетания Сузуки-Мияуры, таких как, но не ограничиваясь этим, использование смеси растворителей, такой как, но не ограничиваясь этим, 1,4-диоксан и вода, в присутствии основания, такого как, но не ограничиваясь этим, ацетат калия, и в присутствии палладиевого катализатора, состоящего из, например, но не ограничиваясь этим, [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладия(II), при температуре между приблизительно температурой окружающей среды и 150°C, в течение приблизительно от 30 минут до 20 часов для того, чтобы создавать соединение. Соединение, которое можно выделять из реакционной смеси стандартными приемами, затем можно обрабатывать, например, но не ограничиваясь этим, окислителем, таким как, но не ограничиваясь этим, пероксид водорода, в присутствии основания, такого как, но не ограничиваясь этим, гидроксид натрия, в полярном апротонном растворителе, таком как, но не ограничиваясь этим, тетрагидрофуран, при температуре между приблизительно 0°C и температурой окружающей среды в течение приблизительно от 1 до 20 часов для того, чтобы создавать соединение (801), в котором X представляет собой гидроксил и Z1 представляет собой водород.
Альтернативно, соединения формулы (Ib), как описано выше в Сущности изобретения, можно синтезировать, придерживаясь общей процедуры, описанной далее на схеме реакции 9, где m, n, q, A, R2 и R6 представляют собой то, что описано выше в Сущности изобретения для соединений формулы (Ib), R8 представляет собой водород, A представляет собой (CH2)m C(R4)(R5) (CH2)n, R1b представляет собой необязательно замещенный арил или необязательно замещенный гетероарил, X представляет собой бром, хлор или йод, Z1 представляет собой защитную группу водорода или азота, например, но не ограничиваясь этим, трет-бутилоксикарбонил, 2,4-диметоксибензил или 4-метоксибензил, R13 и R14 представляют собой независимо водород, необязательно замещенный алкил или необязательно замещенный циклоалкил, или R13 и R14, вместе с азотом, к которому их прикрепляют, образуют необязательно замещенный гетероциклил, LG представляет собой уходящую группу, такую как, но не ограничиваясь этим, бром, хлор или йод, и Z2 представляет собой представляет собой защитную группу водорода или азота, такую как, но не ограничиваясь этим, трет-бутилоксикарбонил:
СХЕМА РЕАКЦИИ 9

Соединения формул (901), (902), (903), (904), (905), (906), (907) и (908) являются коммерчески доступными, или их можно получать в соответствии со способамии, известными специалисту в данной области, или способами, раскрытыми в настоящем описании. В целом, соединения формулы (Ib) получают как описано выше на схеме реакции 9 следующим образом:
Соединение формулы (901) вступает в реакцию с азотосодержащим нуклеофилом, таким как, но не ограничиваясь этим, азид натрия, при стандартных условиях реакции, таких как, но не ограничиваясь этим, использование полярного апротонного растворителя, такого как, но не ограничиваясь этим, диметилсульфоксид или N,N-диметилформамид, при температуре между приблизительно 0°C и 80°C, в течение приблизительно от 1 до 48 часов. Соединение, которое можно выделять из реакционной смеси стандартными приемами, затем обрабатывают восстанавливающим средством, таким как, но не ограничиваясь этим, цинковая пыль, в полярном апротонном растворителе, таком как, но не ограничиваясь этим, тетрагидрофуран, в присутствии слабой кислоты, такой как, но не ограничиваясь этим, водный хлорид аммония, чтобы получать соединение формулы (902). Соединение формулы (902) затем может вступать в реакцию с, например, но не ограничиваясь этим, ди-трет-бутилдикарбонатом, в полярном апротонном растворителе, таком как, но не ограничиваясь этим, дихлорметан, в присутствии основания, такого как, но не ограничиваясь этим, 4-(диметиламино)пиридин, приблизительно при температуре окружающей среды, в течение приблизительно от 1 до 20 часов для того, чтобы создавать соединение формулы (903). Соединение формулы (903) после этого вступает в реакцию с соединением формулы (904) при стандартных условиях нуклеофильного замещения в полярном апротонном растворителе, таком как, но не ограничиваясь этим, N,N-диметилформамид, в присутствии основания, такого как, но не ограничиваясь этим, карбонат калия, при температуре окружающей среды в течение 1-20 ч для того, чтобы получать соединение формулы (905). Соединение формулы (905) после этого вступает в реакцию с трет-бутилизоцианидом в присутствии палладиевого катализатора, состоящего из, например, но не ограничиваясь этим, ацетата палладия и 2-(ди-трет-бутилфосфино)бифенила, и с использованием растворителя, такого как, но не ограничиваясь этим, N,N-диметилформамид, в присутствии основания, такого как, но не ограничиваясь этим, карбонат натрия, и в присутствии восстанавливающего средства, такого как, но не ограничиваясь этим, триэтилсилан, при температуре между приблизительно температурой окружающей среды и 120°C, в течение приблизительно от 1 до 20 часов для того, чтобы создавать соединение формулы (906).
Альтернативно, соединение формулы (905) может вступать в реакцию с производным бороновой кислоты, таким как, но не ограничиваясь этим, сложный эфир винилбороновой кислоты и пинакола, при стандартных условиях сочетания Сузуки в присутствии палладиевого катализатора, состоящего из, например, но не ограничиваясь этим, тетракис(трифенилфосфин)палладия(0), в смеси растворителей, например, но не ограничиваясь этим, N,N-диметилформамиде и воде, и в присутствии основания, например, но не ограничиваясь этим, карбоната натрия, при температуре между приблизительно температурой окружающей среды и 120°C, в течение приблизительно от 1 до 20 часов. Соединение, которое можно выделять из реакционной смеси стандартными приемами, затем можно обрабатывать окисляющим реактивом, таким как, но не ограничиваясь этим, озон, в полярном апротонном растворителе, таком как, но не ограничиваясь этим, дихлорметан, при температуре приблизительно -78°C в течение 1-4 часов, чтобы создавать соединение формулы (906).
Соединение формулы (906) после этого вступает в реакцию с, например, но не ограничиваясь этим, амином формулы (907) в присутствии восстанавливающего средства, такого как, но не ограничиваясь этим, цианоборогидрид натрия, в протонном растворителе, таком как, но не ограничиваясь этим, метанол, в присутствии кислоты, такой как, но не ограничиваясь этим, уксусная кислота, при температуре между приблизительно 0°C и температурой окружающей среды, в течение приблизительно от 1 до 20 часов для того, чтобы создавать соединение формулы (908).
Альтернативно, соединение формулы (906) может вступать в реакцию с амином формулы (907) в присутствии изопропоксида титана(IV) в апротонном полярном растворителе, таком как, но не ограничиваясь этим, дихлорметан, и в присутствии восстановителя, такого как, но не ограничиваясь этим, триацетоксиборгидрид натрия, при температуре между приблизительно 0°C и температурой окружающей среды, в течение приблизительно от 1 до 20 часов для того, чтобы создавать соединение формулы (908).
Соединение формулы (908) затем можно обрабатывать, например, но не ограничиваясь этим, кислотой, такой как, но не ограничиваясь этим, трифторуксусная кислота, в полярном апротонном растворителе, таком как, но не ограничиваясь этим, дихлорметан, при температуре между приблизительно 0°C и температурой окружающей среды для того, чтобы создавать соединение формулы (Ib), которое можно выделять из реакционной смеси стандартными приемами.
Альтернативно, соединение формулы ((b), в котором R8 представляет собой водород, и R13 и R14 не представляют собой водород, можно обрабатывать альдегидом, таким как, но без ограничения, параформальдегид, в кислоте в качестве растворителя, такой как, но не ограничиваясь этим, трифторуксусная кислота, в присутствии восстанавливающего средства, такого как, но не ограничиваясь этим, триацетоксиборгидрид натрия, при температуре приблизительно 0°C в течение от 10 минут до 2 часов, чтобы создавать соединение формулы (Ib), в котором R8 представляет собой алкил, которое можно выделять из реакционной смеси стандартными приемами.
Альтернативно, соединения формулы (Ib), как описано выше в Сущности изобретения, можно синтезировать, придерживаясь общей процедуры, описанной далее на схеме реакции 10, где m, n, q, A, R2 и R6 представляют собой то, что описано выше в Сущности изобретения для соединений формулы (Ib), R8 представляет собой водород, R1b представляет собой необязательно замещенный арил или необязательно замещенный гетероарил, LG представляет собой уходящую группу, такую как, но не ограничиваясь этим, бром, хлор или йод, Z1 представляет собой защитную группу водорода или азота, например, но не ограничиваясь этим, трет-бутилоксикарбонил, 2,4-диметоксибензил или 4-метоксибензил, R13 и R14 представляют собой независимо водород, необязательно замещенный алкил или необязательно замещенный циклоалкил, или R13 и R14, вместе с азотом, к которому их прикрепляют, образуют необязательно замещенный гетероциклил, LG представляет собой уходящую группу, такую как, но не ограничиваясь этим, бром, хлор или йод, и Z2 представляет собой представляет собой защитную группу водорода или азота, такую как, но не ограничиваясь этим, трет-бутилоксикарбонил:
СХЕМА РЕАКЦИИ 10

Соединения формул (1001), (1002), (1003), (1004) и (1005) являются коммерчески доступными, или их можно получать в соответствии со способамии, известными специалисту в данной области, или способами, раскрытыми в настоящем описании. В целом, соединения формулы (Ib) получают как описано выше на схеме реакции 10 следующим образом:
Соединение формулы (1001) вступает в реакцию с восстанавливающим средством, например, но не ограничиваясь этим, цианоборогидрид натрия, в полярном растворителе, таком как, но не ограничиваясь этим, метанол, при температуре между приблизительно 0°C и окружающей температурой, в течение приблизительно от 1 до 20 часов, чтобы создавать соединение формулы (1002). Соединение формулы (1002) затем можно обрабатывать реактивом, образованным из, например, но не ограничиваясь этим, тетрабромида углерода и трифенилфосфина, в полярном апротонном растворителе, таком как дихлорметан, при температуре между приблизительно 0°C и окружающей температурой, в течение приблизительно от 1 до 20 часов, чтобы создавать соединение формулы (1003).
Соединение формулы (1006) после этого вступает в реакцию с, например, но не ограничиваясь этим, амином формулы (1004) в присутствии основания, такого как, но не ограничиваясь этим, карбонат калия, в полярном апротонном растворителе, таком как, но не ограничиваясь этим, N,N-диметилформамид, при температуре между приблизительно 0°C и температурой окружающей среды, в течение приблизительно от 1 до 20 часов для того, чтобы создавать соединение формулы (1005).
Соединение формулы (1005) затем можно обрабатывать, например, но не ограничиваясь этим, кислотой, такой как, но не ограничиваясь этим, трифторуксусная кислота, в полярном апротонном растворителе, таком как, но не ограничиваясь этим, дихлорметан, при температуре между приблизительно 0°C и температурой окружающей среды для того, чтобы создавать соединение формулы (Ib), которое можно выделять из реакционной смеси стандартными приемами.
Альтернативно, соединения формулы (Ib), как описано выше в Сущности изобретения, можно синтезировать, придерживаясь общей процедуры, описанной далее на схеме реакции 11, где m, n, q, A, R2 и R6 представляют собой то, что описано выше в Сущности изобретения для соединений формулы (Ib), R8 представляет собой водород, R1b и R13 представляют собой необязательно замещенные арилы или необязательно замещенные гетероарилы, X1 представляет собой бром, хлор или йод, X2 представляет собой производное бороновой кислоты, такое как, но не ограничиваясь этим, B(OH)2 или BF3K, Z1 представляет собой защитную группу водорода или азота, например, но не ограничиваясь этим, трет-бутилоксикарбонил, 2,4-диметоксибензил или 4-метоксибензил, и Z2 представляет собой представляет собой защитную группу водорода или азота, такую как, но не ограничиваясь этим, трет-бутилоксикарбонил:
СХЕМА РЕАКЦИИ 11

Соединения формул (1101), (112) и (1103) являются коммерчески доступными, или их можно получать в соответствии со способамии, известными специалисту в данной области, или способами, раскрытыми в настоящем описании. В целом, соединения формулы (Ib) получают как описано выше на схеме реакции 11 следующим образом:
Соединение формулы (1101) вступает в реакцию с производными бороновой кислоты (1102) при стандартных условиях сочетания Сузуки в присутствии палладиевого катализатора, состоящего из, например, но не ограничиваясь этим, [1,1'-бис(дифенилфосфино)ферроцен]-дихлорпалладия(II), в смеси растворителей, например, но не ограничиваясь этим, 1,4-диоксане и воде, и в присутствии основания, например, но не ограничиваясь этим, карбоната натрия, при температуре между приблизительно температурой окружающей среды и 120°C, в течение приблизительно от 1 до 20 часов для того, чтобы создавать соединение формулы (1103). Соединение формулы (1103) затем можно обрабатывать, например, но не ограничиваясь этим, кислотой, такой как, но не ограничиваясь этим, трифторуксусная кислота, в полярном апротонном растворителе, таком как, но не ограничиваясь этим, дихлорметан, при температуре между приблизительно 0°C и температурой окружающей среды для того, чтобы создавать соединение формулы (Ib), которое можно выделять из реакционной смеси стандартными приемами.
Альтернативно, соединение формулы (Ib), как описано выше в Сущности изобретения, можно синтезировать, придерживаясь общей процедуры, описанной далее на схеме реакции 12, где q, R2 и R6 представляют собой то, что описано выше в Сущности изобретения для соединений формулы (Ib), R8 представляет собой водород, A представляет собой метилен, R1b представляет собой необязательно замещенный арил или необязательно замещенный гетероарил, Z1 представляет собой защитную группу водорода или азота, например, но не ограничиваясь этим, трет-бутилоксикарбонил, 2,4-диметоксибензил или 4-метоксибензил, R13 и R14 представляют собой независимо водород, необязательно замещенный алкил или необязательно замещенный циклоалкил, или R13 и R14, вместе с азотом, к которому их прикрепляют, образуют необязательно замещенный гетероциклил, PG представляет собой защитную группу, такую как, но не ограничиваясь этим, трет-бутилдиметилсилил, и Z2 представляет собой представляет собой защитную группу водорода или азота, такую как, но не ограничиваясь этим, трет-бутилоксикарбонил:
СХЕМА РЕАКЦИИ 12

Соединения формул (1201), (1202), (1203), (1204), (1205), (1206), (1207) и (1208) являются коммерчески доступными, или их можно получать в соответствии со способамии, известными специалисту в данной области, или способами, раскрытыми в настоящем описании. В целом, соединения формулы (Ib) получают как описано выше на схеме реакции 12 следующим образом:
Соединение формулы (1201) вступает в реакцию с альдегидом формулы (1202) в присутствии восстанавливающего средства, такого как, но не ограничиваясь этим, триацетоксиборгидрид натрия, в смеси растворителей, такой как, но не ограничиваясь этим, трифторуксусная кислота и дихлорметан, при температуре между приблизительно 0°C и температурой окружающей среды, в течение приблизительно от 1 до 20 часов для того, чтобы создавать соединение формулы (1203). Соединение формулы (1203) затем может вступать в реакцию с, например, но не ограничиваясь этим, ди-трет-бутилдикарбонатом, в полярном апротонном растворителе, таком как, но не ограничиваясь этим, дихлорметан, в присутствии основания, такого как, но не ограничиваясь этим, 4-(диметиламино)пиридин, приблизительно при температуре окружающей среды, в течение приблизительно от 1 до 20 часов для того, чтобы создавать соединение формулы (1204). Соединение формулы (1204) затем можно обрабатывать реактивом, таким как, но не ограничиваясь этим, фторид тетра-н-бутиламмония, в полярном апротонном растворителе, таком как, но не ограничиваясь этим, тетрагидрофуран, при температуре окружающей среды в течение от 30 минут до 20 ч для того, чтобы получать соединение формулы (1205). Соединение формулы (1205) затем окисляют реактивом, таким как, но не ограничиваясь этим, реактив периодинан Десса-Мартина, в полярном апротонном растворителе, таком как, но не ограничиваясь этим, дихлорметан, при температуре окружающей среды в течение от 30 минут до 20 ч для того, чтобы получать соединение формулы (1206). Соединение формулы (1206) после этого вступает в реакцию с, например, но не ограничиваясь этим, амином формулы (1207) в присутствии восстанавливающего средства, такого как, но не ограничиваясь этим, триацетоксиборгидрид натрия, в полярном апротонном растворителе, таком как, но не ограничиваясь этим, дихлорметан, при температуре между приблизительно 0°C и температурой окружающей среды, в течение приблизительно от 1 до 20 часов для того, чтобы создавать соединение формулы (1208). Соединение формулы (1208) затем можно обрабатывать, например, но не ограничиваясь этим, кислотой, такой как, но не ограничиваясь этим, трифторуксусная кислота, в полярном апротонном растворителе, таком как, но не ограничиваясь этим, дихлорметан, при температуре между приблизительно 0°C и температурой окружающей среды для того, чтобы создавать соединение формулы (Ib), которое можно выделять из реакционной смеси стандартными приемами.
Все соединения, описанные далее в качестве получаемых, которые могут существовать в форме свободного основания или кислоты, можно превращать в их фармацевтически приемлемые соли посредством обработки подходящим неорганическим или органическим основанием или кислотой. Соли соединений, получаемых далее, можно превращать в их форму свободного основания или кислоты стандартными приемами. Кроме того, все соединения по изобретению, которые содержат кислотную или сложноэфирную группу, можно превращать в соответствующий сложный эфир или кислоту, соответственно, с помощью способов, известных специалисту в данной области, или с помощью способов, описанных в настоящем описании.
Следующие Примеры, которые направлены на синтез соединений по изобретению; и следующие Биологические примеры предоставлены в качестве ориентира для того, чтобы содействовать проактической реализации изобретения, и не предназначены в качестве ограничения объема изобретения.
В нижеследующих примерах, если не указано иное, все температуры приведены в градусах Цельсия. Коммерчески доступные реактивы приобретали у поставщиков, таких как Aldrich Chemical Company, Combi-Blocks, TCI или Oakwood Chemicals, и использовали без дополнительной очистки, если не указано иное. Реакции, приведенные далее, выполняли в целом при положительном давлении азота или аргона или с осушительной трубкой (если не указано иное) в безводных растворителях, а реакционные колбы обычно снабжали резиновыми пробками для введения субстратов и реактивов через шприц. Стеклянную посуду сушили в печи и/или сушили теплом. Выходы не оптимизировали. Температуры плавления определяли на аппарате с горячим столом Büchi и не корректировали. Данные1H ЯМР,19F и13C ЯМР получали в растворах дейтерированных растворителей CDCI3, DMSO-d6, CD3OD, CD3CN или ацетона-d6 с химическими сдвигами (δ), приведенными в миллионных долях (м. д.) относительно пиков триметилсилана (TMS) или остаточного недейтерированного растворителя в качестве эталонного стандарта. Данные приведены следующим образом, если применимо: химический сдвиг, мультиплетность, константа взаимодействия в Гц и число протонов, атомов фтора или углерода. Когда приведены мультиплетности пиков, используют следующие сокращения: с (синглет), д (дублет), т (триплет), к (квартет), м (мультиплет, ушир. (уширенный), дд (дублет дублетов), дт (дублет триплетов). Константы взаимодействияя, когда даны, приведены в Гц (герцы).
ПРИМЕР 1
Синтез (S)-5-бром-2-фтор-4-((1-фенилпропил)амино)-N-(тиазол-2-ил)бензолсульфонамида
Стадия 1. Получение 5-бром-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамида
В смесь N-(2,4-диметоксибензил)тиазол-2-амина (8,60 г, 34,4 ммоль, получен в соответствии с WO 2013063459) в безводном тетрагидрофуране (80 мл) добавляли 1 M раствор бис(триметилсилил)амида лития в тетрагидрофуране (39,6 мл, 39,6 ммоль) при -78°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 1 ч. Реакционную смесь охлаждали до -78°C и добавляли в нее раствор 5-бром-2,4-дифторбензолсульфонилхлорида (9,12 г, 31,3 ммоль) в безводном тетрагидрофуране (20 мл). Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 16 ч. Реакционную смесь гасили добавлением насыщенного раствора хлорида аммония (100 мл) и туда добавляли этилацетат (400 мл). Водную фазу экстрагировали этилацетатом (2×50 мл) и объединенную органическую фазу промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 40% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (6,3 г, выход 40%):1H ЯМР (300 МГц, CDCl3) δ 8,06 (т, J=7,3 Гц, 1H), 7,42 (д, J=3,6 Гц, 1H), 7,19 (д, J=8,2 Гц, 1H), 7,03 (д, J 3,9 Гц, 1H), 6,97 (дд, J=9,1, 8,2 Гц, 1H), 6,38-6,34 (м, 2H), 5,19 (с, 2H), 3,37 (с, 3H), 3,73 (с, 3H); MS (ES+) m/z 504,9 (M+1), 506,9 (M+1).
Стадия 2. Получение (S)-5-бром-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилпропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

В смесь 5-бром-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамида (1,50 г, 2,97 ммоль) и карбоната цезия (1,94 г, 5,94 ммоль) в безводном диметилсульфоксиде (20 мл) добавляли (S)-2-этилбензиламин (0,40 г, 2,97 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч. Смесь разводили этилацетатом (200 мл) и промывали водой (2×20 мл) и солевым раствором (20 мл). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата при пониженном давлении получали остаток, который очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 40% этилацетата в гексанах, чтобы получать указанное в заголовке соединение в виде бесцветного аморфного твердого вещества (1,51 г, выход 82%): MS (ES+) m/z 620,0 (M+1), 622,0 (M+1).
Стадия 3. Получение (S)-5-бром-2-фтор-4-((1-фенилпропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

В смесь (S)-5-бром-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилпропил)амино)-N-(тиазол-2-ил)бензолсульфонамида (1,51 г, 2,43 ммоль) в дихлорметане (20 мл) добавляли трифторуксусную кислоту (5 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Концентрированием при пониженном давлении получали остаток, который растирали в метаноле (20 мл) и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток растирали в простом диэтиловом эфире (10 мл) для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (1,0 г, выход 88%):1H ЯМР (300 МГц, DMSO-d6) δ 12,73 (с, 1H), 7,73 (д, J=7,3 Гц, 1H), 7,43-7,17 (м, 5H), 6,83-6,78 (м, 1H), 6,41 (д, J=13,1 Гц, 1H), 6,09 (д, J=6,8 Гц, 1H), 4,49-4,39 (м, 1H), 3,33 (с, 1H), 2,07-1,91 (м, 1H), 1,86-1,72 (м, 1H), 0,89 (т, J=6,4 Гц, 3H); MS (ES+) m/z 469,9 (M+1), 471,9 (M+1).
ПРИМЕР 2
Синтез (S)-5-хлор-2-фтор-4-((1-(нафтален-1-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамида

Раствор N-(2,4-диметоксибензил)тиазол-2-амина (20,86 г, 83,3 ммоль, получен в соответствии с WO2013063459) в безводном тетрагидрофуране (350 мл) обрабатывали 1 M раствором бис(триметилсилил)амида в тетрагидрофуране (100,0 мл, 100,0 ммоль) при -78°C. Получаемую смесь нагревали до температуры окружающей среды и перемешивали в течение 1 ч. Реакционную смесь охлаждали до -78°C и туда добавляли раствор 5-хлор-2,4-дифторбензолсульфонилхлорида (20,58 г, 83,3 ммоль) в безводном тетрагидрофуране (75 мл). Реакционную смесь оставляли нагреваться до температуры окружающей среды, перемешивали в течение 2 ч и разводили этилацетатом (700 мл). Органическую фазу промывали насыщенным бикарбонатом натрия (200 мл), насыщенным хлоридом аммония (2×150 мл), солевым раствором (2×150 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата при пониженном давлении получали остаток, который растирали с метанолом (80 мл) для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (12,7 г, выход 33%):1H ЯМР (300 МГц, CDCl3) δ 7,94 (т, J=7,4 Гц, 1H), 7,44 (д, J=3,6 Гц, 1H) 7,21 (д, J=8,1 Гц, 1H), 7,06-6,99 (м, 2H), 6,41-6,36 (м, 2H), 5,20 (с, 2H), 3,78 (с, 3H), 3,74 (с, 3H); MS (ES+) m/z 461,0 (M+1), 463,0 (M+1).
Стадия 2. Получение (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(нафтален-1-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида
В смесь 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамида (0,46 г, 1,0 ммоль) и карбоната цезия (0,65 г, 2,0 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли (S)-1-(1-нафтил)этиламин (0,17 г, 1,0 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч. Затем в смесь добавляли воду (20 мл) и отфильтровывали осадок. Осадок растворяли в дихлорметане (100 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата при пониженном давлении и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 40% этилацетата в гексанах, получали указанное в заголовке соединение в виде масла желтоватого цвета (0,55 г, выход 90%): MS (ES+) m/z 612,1 (M+1), 614,0 (M+1).
Стадия 3. Получение (S)-5-хлор-2-фтор-4-((1-(нафтален-1-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

В смесь (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(нафтален-1-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида (0,55 г, 0,89 ммоль) в дихлорметане (20 мл) добавляли трифторуксусную кислоту (0,5 мл) при 0°C и реакционную смесь перемешивали в течение 30 минут. Концентрированием при пониженном давлении получали остаток, который очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 50% этилацетата в гексанах для того, чтобы получать указанное в заголовке соединение в виде твердого вещества грязно-белого цвета (0,31 г, выход 74%):1H ЯМР (300 МГц, DMSO-d6) δ 12,73 (ушир. с, 1H), 8,29 (д, J=8,3 Гц, 1H), 7,97 (дд, J=8,1, 1,3 Гц, 1H), 7,82 (д, J=7,5 Гц, 1H), 7,65-7,41 (м, 5H), 7,23 (д, J=4,6 Гц, 1H), 6,80 (д, J=4,6 Гц, 1H), 6,70 (дд, J=6,7, 1,2 Гц, 1H), 6,13 (д, J=13,0 Гц, 1H), 5,54-5,44 (м, 1H), 1,65 (д, J=6,6 Гц, 3H); MS (ES+) m/z 462,1 (M+1), 464,0 (M+1).
ПРИМЕР 3
Синтез (S)-5-хлор-2-фтор-N-(тиазол-2-ил)-4-((1-(3-(трифторметил)фенил)этил)амино)бензолсульфонамида

Стадия 1. Получение (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)-4-((1-(3-(трифторметил)фенил)этил)амино)бензолсульфонамида

Следуя процедуре как описано для примера 2, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять (S)-1-(1-нафтил)этиламин на (S)-1-(3-(трифторметил)фенил)этан-1-амин, указанное в заголовке соединение получали в виде масла желтоватого цвета (0,67 г, выход 80%): MS (ES+) m/z 630,1 (M+1), 632,1 (M+1).
Стадия 2. Получение (S)-5-хлор-2-фтор-N-(тиазол-2-ил)-4-((1-(3-(трифторметил)фенил)этил)амино)бензолсульфонамида

Следуя процедуре как описано для примера 2, стадии 3 и выполняя некритические вариации при необходимости, чтобы заменять (S)-5-бром-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилпропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)-4-((1-(3-(трифторметил)фенил)этил)-амино)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,37 г, выход 72%):1H ЯМР (300 МГц, DMSO-d6) δ 12,76 (ушир. с, 1H), 7,84 (с, 1H), 7,77-7,71 (м, 1H), 7,62-7,52 (м, 3H), 7,25 (д, J=4,6 Гц, 1H), 6,81 (д, J=4,6 Гц, 1H), 6,68 (дд, J=7,8, 1,3 Гц, 1H), 6,50 (д, J=13,1 Гц, 1H), 4,91-4,80 (м, 1H), 1,54 (д, J=6,8 Гц, 3H);19F ЯМР (282 МГц, DMSO-d6) δ -60,9 (с, 3F), -109,3 (с, 1F); MS (ES+) m/z 480,0 (M+1), 482,0 (M+1).
ПРИМЕР 4
Синтез формиата (R)-3-хлор-N-(тиазол-2-ил)-4-((2,2,2-трифтор-1-фенилэтил)амино)бензолсульфонамида

Стадия 1. Получение 4-бром-3-хлор-N-(тиазол-2-ил)бензолсульфонамида

В смесь 2-аминотиазола (7,1 г, 70,7 ммоль) в дихлорметане (190 мл) и пиридине (62 мл) добавляли 4-бром-3-хлорбензолсульфонилхлорид (20,5 г, 70,7 ммоль) при 0°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 16 ч. Смесь концентрировали при пониженном давлении и остаток выпаривали совместно с толуолом (2×100 мл). Растиранием в метаноле (50-100 мл) получали твердое вещество, которое отфильтровывали. Твердое вещество промывали в метаноле (50 мл) для того, чтобы получать указанное в заголовке соединение в виде порошка бежевого цвета (14,6 г, выход 58%):1H ЯМР (300 МГц, DMSO-d6) δ 12,07 (ушир. с, 1H), 7,96 (д, J=8,4 Гц, 1H), 7,90 (д, J=2,1 Гц, 1H), 7,65 (дд, J=8,4, 2,1 Гц, 1H), 7,32 (д, J=4,6 Гц, 1H), 6,90 (д, J 4,6 Гц, 1H); MS (ES+) m/z 352,9 (M+1), 354,9 (M+1).
Стадия 2. Получение формиата (R)-3-хлор-N-(тиазол-2-ил)-4-((2,2,2-трифтор-1-фенилэтил)амино)бензолсульфонамида
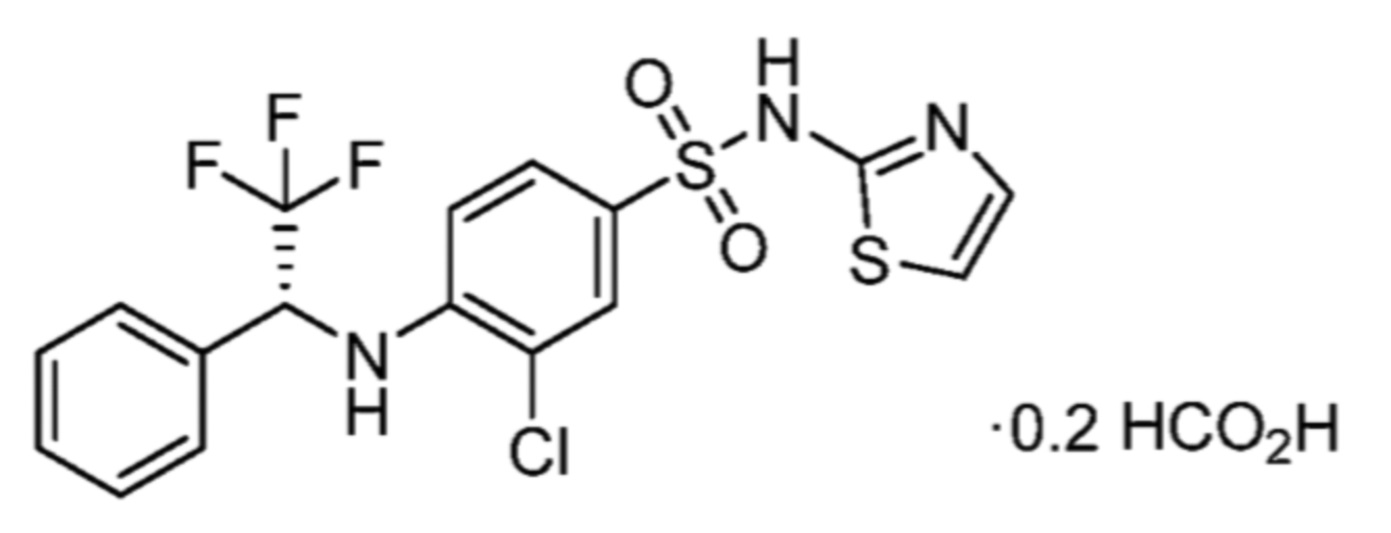
Смесь 4-бром-3-хлор-N-(тиазол-2-ил)бензолсульфонамида (0,35 г, 1,0 ммоль), (R)-2,2,2-трифтор-1-фенилэтиламина (0,35 г, 2,0 ммоль), 2-дициклогексилфосфино-2′,4′,6′-триизопропилбифенила (0,058 г, 0,12 ммоль), трис(дибензилиденацетон)дипалладия(0) (0,036 г, 0,04 ммоль) и трет-бутоксида натрия (0,24 г, 2,5 ммоль) в диоксане (5 мл) дегазировали пропусканием потока аргона через нее и затем нагревали в герметизированном флаконе при 150°C в течение 30 минут с использованием микроволн. Реакционную смесь оставляли остывать температура окружающей среды и разводили дихлорметаном (200 мл) и насыщенным раствором хлорида аммония (100 мл). Органическую фазу промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo и очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% муравьиную кислоту, в качестве элюента. Указанное в заголовке соединение получали в виде твердого вещества бежевого цвета (0,032 г, выход 7%):1H ЯМР (300 МГц, DMSO-d6) δ 8,14 (с, 0,2H), 7,70-7,63 (м, 3H), 7,52 (дд, J=8,7, 2,1 Гц, 1H), 7,47-7,36 (м, 3H), 7,18 (д, J=4,5 Гц, 1H), 7,07 (д, J=8,8 Гц, 1H), 6,74 (д, J=4,5 Гц, 1H), 6,11 (д, J=9,8 Гц, 1H), 5,90-5,77 (м, 1H), способные к обмену протоны не наблюдали;19F ЯМР (282 МГц, DMSO-d6) δ -72,3 (s); MS (ES-) m/z 446,0 (M - 1), 448,0 (M - 1).
ПРИМЕР 5
Синтез (S)-5-хлор-4-((1-(5-хлор-2-фторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил (S)-((5-хлор-4-((1-(5-хлор-2-фторфенил)этил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата

В смесь гидрохлорида (S)-1-(5-хлор-2-фторфенил)этан-1-амина (0,092 г, 0,44 ммоль) и трет-бутил ((5-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,18 г, 0,44 ммоль, получен в соответствии с WO2010079443) в безводном диметилсульфоксиде (1,0 мл) добавляли карбонат калия (0,18 г, 1,3 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 12 ч. Смесь выливали в смесь лед/вода 1:1 (50 мл) и экстрагировали этилацетатом (3×60 мл). Объединенную органическую фазу промывали солевым раствором (50 мл), сушили над безводным Na2SO4, фильтровали и концентрировали in vacuo. Остаток очищали с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 25% этилацетата в петролейном эфире, чтобы получать указанное в заголовке соединение в виде бесцветного масла (0,18 г, выход 73%): MS (ES+) m/z 563,9 (M+1), 565,9 (M+1).
Стадия 2. Получение (S)-5-хлор-4-((1-(5-хлор-2-фторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

К трет-бутил (S)-((5-хлор-4-((1-(5-хлор-2-фторфенил)этил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамату (0,18 г, 0,32 ммоль) добавляли 4 M раствор хлороводорода в диоксане (10,0 мл, 40,0 ммоль) и реакционная смесь находилась при температуре окружающей среды в течение 12 ч. Смесь концентрировали in vacuo и очищали остаток. Остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,2% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,082 г, выход 54%):1H ЯМР (400 МГц, CDCl3) δ 10,32 (с, 1H), 8,68 (д, J=2,0 Гц, 1H), 7,78 (д, J=6,8 Гц, 1H), 7,20-7,25 (м, 1H), 7,18-7,16 (м, 1H), 7,03 (т, J=9,2 Гц, 1H), 6,90 (д, J=2,0 Гц, 1H), 6,06 (д, J=12,4 Гц, 1H), 5,15 (д, J=5,2 Гц, 1H), 4,73 (квин, J=6,4 Гц, 1H), 1,61 (д, J=6,8 Гц, 3H); MS (ES+) m/z 464,0 (M+1), 466,0 (M+1).
ПРИМЕР 6
Синтез (S)-5-хлор-4-((1-(3-хлор-2-фторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил (S)-((5-хлор-4-((1-(3-хлор-2-фторфенил)этил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата
Следуя процедуре как описано для примера 5, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять гидрохлорид (S)-1-(5-хлор-2-фторфенил)этан-1-амина на (S)-1-(3-хлор-2-фторфенил)этан-1-амин, указанное в заголовке соединение выделяли в виде бесцветного твердого вещества (0,15 г, выход 44%): MS (ES+) m/z 563,9 (M+1), 565,9 (M+1).
Стадия 2. Получение (S)-5-хлор-4-((1-(3-хлор-2-фторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано для примера 5, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять трет-бутил (S)-((5-хлор-4-((1-(5-хлор-2-фторфенил)этил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамат на трет-бутил (S)-((5-хлор-4-((1-(3-хлор-2-фторфенил)этил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамат, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,058 г, выход 54%):1H ЯМР (400 МГц, CDCl3) δ 9,19 (с, 1H), 8,62 (д, J=2,4 Гц, 1H), 7,75 (д, J=7,2 Гц, 1H), 7,34 (тд, J=7,2, 2,0 Гц, 1H), 7,02-7,16 (м, 2H), 6,94 (д, J=2,4 Гц, 1H), 6,07 (д, J=12,0 Гц, 1H), 5,19 (д, J=6,0 Гц, 1H), 4,78 (квин, J=6,4 Гц, 1H), 1,63 (д, J=6,8 Гц, 3H); MS (ES+) m/z 464,0 (M+1), 466,0 (M+1).
ПРИМЕР 7
Синтез (S)-4-((1-(3-бромфенил)этил)амино)-5-хлор-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение (S)-4-((1-(3-бромфенил)этил)амино)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

В смесь 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамида (0,50 г, 1,1 ммоль) и (S)-1-(3-бромфенил)этан-1-амина (0,26 г, 1,3 ммоль) в безводном диметилсульфоксиде (4 мл) добавляли карбонат калия (0,30 г, 2,2 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 12 ч. После добавления воды (20 мл), смесь экстрагировали этилацетатом (3×20 мл). Объединенную органическую фазу промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата при пониженном давлении и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с 30% этилацетатом в петролейном эфире, получали указанное в заголовке соединение в виде твердого вещества желтоватого цвета (0,40 г, выход 58%):1H ЯМР (400 МГц, CDCl3) δ 7,74-7,68 (д, J=8,0 Гц, 1H), 7,46-7,41 (м, 2H), 7,40-7,36 (д, J=4,0 Гц, 1H), 7,26-7,16 (м, 3H), 6,98-6,94 (д, J=4,0 Гц, 1H), 6,39-6,34 (дд, J=8,0 Гц, 4,0 Гц, 1H), 6,33-6,30 (м, 1H), 6,08-6,02 (д, J=12,0 Гц, 1H), 5,19-5,12 (м, 3H), 4,49-4,39 (м, 1H), 3,77 (с, 3H), 3,67 (с, 3H), 1,63-1,59 (д, J=6,8 Гц, 3H).
Стадия 2. Получение (S)-4-((1-(3-бромфенил)этил)амино)-5-хлор-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

В смесь (S)-4-((1-(3-бромфенил)этил)амино)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида (0,14 г, 0,22 ммоль) в дихлорметане (5,0 мл) добавляли трифторуксусную кислоту (0,5 мл) и смесь перемешивали при температуре окружающей среды в течение 30 минут. Смесь концентрировали in vacuo и остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,2% муравьиную кислоту, в качестве элюента для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,067 г, выход 62%):1H ЯМР (400 МГц, CDCl3) δ 12,80 (ушир. с, 1H), 7,89 (д, J=8,0 Гц, 1H), 7,48-7,39 (м, 2H), 7,26-7,20 (м, 2H), 7,13 (д, J=4,0 Гц, 1H), 6,49 (д, J=4,0 Гц, 1H), 6,06 (д, J=12,0 Гц, 1H), 5,16-5,08 (м, 1H), 4,50-4,38 (м, 1H), 1,61 (д, J=8,0 Гц, 3H); MS (ES+) m/z 490,1 (M+1), 492,1 (M+1).
ПРИМЕР 8
Синтез (S)-5-хлор-4-((1-(5-хлор-2-фторфенил)этил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение (R,E)-N-(5-хлор-2-фторбензилиден)-2-метилпропан-2-сульфинамида

В раствор 5-хлор-2-фторбензальдегида (5,00 г, 31,5 ммоль) в дихлорметане (10,0 мл) добавляли п-толуолсульфонат пиридиния (0,40 г, 1,59 ммоль), (R)-(+)-2-метил-2-пропансульфинамид (3,82 г, 31,5 ммоль) и безводный сульфат магния (19,0 г, 158 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 12 ч. Фильтрованием реакционной смеси и концентрированием фильтрата при пониженном давлении получали остаток, который очищали с помощью колоночной хроматографии, осуществляя элюирование с 10% этилацетатом в петролейном эфире, чтобы получать указанное в заголовке соединение в виде масла желтого цвета (2,45 г, выход 30%):1H ЯМР (400 МГц, CDCl3) δ 8,85-8,84 (м, 1H), 7,96-7,94 (дд, J=6,0, 2,8 Гц, 1H), 7,47-7,43 (м, 1H), 7,14-7,10 (т, J=8,0 Гц, 1H), 1,28 (с, 9H).
Стадия 2. Получение (R)-N-((S)-1-(5-хлор-2-фторфенил)этил)-2-метилпропан-2-сульфинамида
В смесь (R,E)-N-(5-хлор-2-фторбензилиден)-2-метилпропан-2-сульфинамида (2,10 г, 8,02 ммоль) в дихлорметане (5 мл) добавляли 3 M раствор бромида метилмагния в простом диэтиловом эфире (5,35 мл, 16,05 ммоль) по частям при -48°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 12 ч. Реакционную смесь выливали в воду (30 мл) и экстрагировали этилацетатом (2×30 мл). Объединенную органическую фазу промывали солевым раствором (2×30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата при пониженном давлении получали остаток, который очищали с помощью колоночной хроматографии, осуществляя элюирование с 25% этилацетата в петролейном эфире для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (1,20 г, выход 54%):1H ЯМР (400 МГц, CDCl3) δ 7,35-7,33 (дд, J=8,0, 2,0 Гц, 1H), 7,22 (м, 1H), 7,02-6,98 (т, J=8,0 Гц, 1H), 4,79-4,72 (м, 1H), 1,54-1,52 (д, J=8,0 Гц, 3H), 1,70 (с, 1H), 1,24 (с, 9H).
Стадия 3. Получение (S)-1-(5-хлор-2-фторфенил)этан-1-амина

В смесь (R)-N-((S)-1-(5-хлор-2-фторфенил)этил)-2-метилпропан-2-сульфинамида (0,5 г, 1,80 ммоль) в метаноле (5 мл) добавляли 4 M раствор хлороводорода в диоксане (0,9 мл, 3,6 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч и затем концентрировали при пониженном давлении. Остаток растирали в петролейном эфире для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,25 г, выход 80%):1H ЯМР (400 МГц, CD3OD) δ 7,60-7,59 (м, 1H), 7,50-7,48 (м, 1H), 7,30-7,25 (дд, J=12,0, 8,0 Гц, 1H), 4,77-4,72 (м, 1H), 1,68-1,64 (м, 3H), способные к обмену протоны не наблюдали.
Стадия 4. Получение (S)-5-хлор-4-((1-(5-хлор-2-фторфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 7, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять (S)-1-(3-бромфенил)этан-1-амин на (S)-1-(5-хлор-2-фторфенил)этан-1-амина, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,12 г, выход 53%):1H ЯМР (400 МГц, CDCl3) δ 7,72 (д, J=8,0 Гц, 1H), 7,39 (д, J=4,0 Гц, 1H), 7,22-7,18 (м, 3H), 7,08-7,05 (м, 1H), 6,96 (д, J=4,0 Гц, 1H), 6,36 (дд, J=8,0, 4,0 Гц, 1H), 6,32 (д, J=4,0 Гц, 1H), 6,08 (д, J=12,0 Гц, 1H), 5,16-5,20 (м, 3H), 4,78-4,71 (м, 1H), 3,77-3,68 (м, 3H), 3,68 (с, 3H), 1,62 (д, J=8,0 Гц, 3H).
Стадия 5. Получение (S)-5-хлор-4-((1-(5-хлор-2-фторфенил)этил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 7, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять (S)-4-((1-(3-бромфенил)этил)амино)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид на (S)-5-хлор-4-((1-(5-хлор-2-фторфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,057 г, выход 63%):1H ЯМР (400 МГц, CD3OD) δ 7,76 (д, J=4,0 Гц, 1H), 7,36 (дд, J=4,0, 2,0 Гц, 1H), 7,28-7,32 (м, 1H), 7,18-7,10 (м, 2 H), 6,73 (д, J=4,0 Гц, 1H), 6,25 (д, J=12,0 Гц, 1H), 4,80-4,95 (м, 1H), 1,64-1,62 (д, J=8,0 Гц, 3H), способные к обмену протоны не наблюдали; MS (ES+) m/z 464,1 (M+1), 466,1 (M+1).
ПРИМЕР 9
Синтез (S)-3-хлор-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение 3-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамида

В раствор N-(2,4-диметоксибензил)тиазол-2-амина (1,24 г, 4,94 ммоль) в безводном тетрагидрофуране (11 мл) добавляли 1 M раствор бис(триметилсилил)амида лития в тетрагидрофуране (4,9 мл, 4,9 ммоль) при -78°C. Реакционную смесь перемешивали при -78°C в течение 30 мин, затем оставляли нагреваться до температуры окружающей среды и перемешивали в течение 1 ч. Реакционную смесь охлаждали до -78°C и туда добавляли раствор 3-хлор-2,4-дифторбензолсульфонилхлорида (1,11 г, 4,49 ммоль) в безводном тетрагидрофуране (5 мл). Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 16 ч. Реакционную смесь разводили этилацетатом (50 мл) и насыщенным водным раствором хлорида аммония (20 мл), и водную фазу экстрагировали этилацетатом (3×50 мл). Объединенные органические фазы промывали солевым раствором (3×20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 20% этилацетата в гексанах, получали указанное в заголовке соединение в виде масла желтого цвета (0,992 г, выход 48%):1H ЯМР (300 МГц, CDCl3) δ 7,81 (ддд, J=9,0, 7,4, 5,8, 1H), 7,40 (д, J=3,6 Гц, 1H), 7,18 (дд, J=7,8, 0,7 Гц, 1H), 7,04 (ддд, J=9,0, 7,7, 1,8 Гц, 1H), 7,00 (д, J=3,6 Гц, 1H), 6,39-6,34 (м, 2H), 5,20 (с, 2H), 3,78 (с, 3H), 3,75 (с, 3H); MS (ES+) m/z 461,0 (M+1), 463,0 (M+1).
Стадия 2. Получение (S)-3-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

В суспензию 3-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамида (0,375 г, 0,814 ммоль) и (S)-1-(2-фторфенил)этан-1-амина (0,136 г, 0,976 ммоль) в безводном диметилсульфоксиде (4 мл) добавляли карбонат цезия (0,665 г, 2,04 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 18 ч. Реакционную смесь разводили этилацетатом (50 мл) и водой (20 мл) и водную фазу экстрагировали этилацетатом (2×50 мл). Объединенные органические фазы промывали солевым раствором (3×20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с 0-40% этилацетата в гексанах, получали указанное в заголовке соединение в виде пены желтого цвета (0,352 г, выход 75%): MS (ES+) m/z 580,1 (M+1), 582,1 (M+1).
Стадия 3. Получение (S)-3-хлор-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

В раствор (S)-3-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида (0,352 г, 0,607 ммоль) в дихлорметане (4 мл) добавляли трифторуксусную кислоту (0,140 мл, 1,82 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 30 минут. После концентрирования in vacuo, реакционную смесь разводили метанолом (20 мл) и перемешивали при температуре окружающей среды в течение 1 ч. Осадок удаляли посредством фильтрования и промывали метанолом (2×15 мл). Объединенный фильтрат концентрировали in vacuo и остаток растирали в простом диэтиловом эфире (10 мл) для того, чтобы получать указанное в заголовке соединение в виде твердого вещества бледно-желтого цвета (0,123 г, выход 47%):1H ЯМР (300 МГц, DMSO-d6) δ 12,76 (ушир. с, 1H), 7,46 (т, J=8,5 Гц, 1H), 7,38 (дт, J=7,8, 1,7 Гц, 1H), 7,32-7,11 (м, 4H), 6,81 (д, J=4,6 Гц, 1H), 6,52 (д, J=7,6 Гц, 1H), 6,32 (д, J=9,2 Гц, 1H), 4,99-4,89 (м, 1H), 1,56 (д, J=6,7 Гц, 3H); MS (ES+) m/z 430,0 (M+1), 432,0 (M+1).
ПРИМЕР 10
Синтез (S)-3-хлор-2-фтор-4-(1-(2-фторфенил)этокси)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение (S)-3-хлор-N-(2,4-диметоксибензил)-2-фтор-4-(1-(2-фторфенил)этокси)-N-(тиазол-2-ил)бензолсульфонамида

В раствор 3-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамида (0,250 г, 0,56 ммоль) и (S)-1-(2-фторфенил)этан-1-ола (0,075 г, 0,540 ммоль) в безводном N,N-диметилформамиде (5 мл) добавляли гидрид натрия (60% дисперсия в минеральном масле, 0,043 г, 1,08 ммоль) при 0°C. Реакцию оставляли нагреваться до температуры окружающей среды и перемешивали в течение 2 ч. Реакцию разводили этилацетатом (50 мл) и насыщенным водным раствором хлорида аммония (30 мл) и водную фазу экстрагировали этилацетатом (2×50 мл). Объединенные органические фазы промывали солевым раствором (3×20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 40% этилацетата в гексанах, получали указанное в заголовке соединение в виде масла желтого цвета (0,250 г, выход 80%):1H ЯМР (300 МГц, CDCl3) δ 7,65-7,59 (м, 1H), 7,41-7,36 (м, 2H), 7,33-7,25 (м, 2H), 7,18-7,04 (м, 2H), 6,95 (дд, J=3,6, 1,4, 1H), 6,60-6,57 (м, 1H), 6,37-6,31 (м, 2H), 5,79-5,72 (м, 1H), 5,18 (с, 2H), 3,75 (д, J=1,3 Гц, 3H), 3,69 (д, J=1,2 Гц, 3H), 1,72 (д, J=6,4 Гц, 3H); MS (ES+) m/z 581,1 (M+1), 583,1 (M+1).
Стадия 2. Получение (S)-3-хлор-2-фтор-4-(1-(2-фторфенил)этокси)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 9, стадии 3 и выполняя некритические вариации при необходимости, чтобы заменять (S)-3-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамид на (S)-3-хлор-N-(2,4-диметоксибензил)-2-фтор-4-(1-(2-фторфенил)этокси)-N-(тиазол-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде «a ???» (0,029 г, выход 16%);1H ЯМР (300 МГц,DMSO-d6) δ 12,93 (ушир. с, 1H), 7,71-7,65 (м, 1H), 7,46-7,18 (м, 5H), 7,02 (д, J=9,0 Гц, 1H), 6,87 (д, J=4,6 Гц, 1H), 5,98-5,92 (м, 1H), 1,64 (д, J=6,3 Гц, 3H);19F ЯМР (282 МГц, DMSO-d6) δ -108,9 (с, 1F), -118,5 (с, 1F); MS (ES+) m/z 431,0 (M+1), 433,0 (M+1).
ПРИМЕР 11
Синтез 5-хлор-2-фтор-4-(изохинолин-8-илметокси)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-(изохинолин-8-илметокси)-N-(тиазол-2-ил)бензолсульфонамида
В суспензию 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамида (0,475 г, 1,03 ммоль) и изохинолин-8-илметанола (0,164 г, 1,03 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли карбонат цезия (0,671 г, 2,06 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч и затем нагревали при 75°C в течение 4 ч. Реакционную смесь оставляли остывать до температуры окружающей среды, разводили этилацетатом (50 мл) и водой (20 мл) и водную фазу экстрагировали этилацетатом (2×50 мл). Объединенные органические фазы промывали солевым раствором (3×20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 40% этилацетата в гексанах, получали указанное в заголовке соединение в виде твердого вещества бежевого цвета (0,106 г, выход 17%): MS (ES+) m/z 600,1 (M+1), 602,1 (M+1).
Стадия 2. Получение 5-хлор-2-фтор-4-(изохинолин-8-илметокси)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 9, стадии 3 и выполняя некритические вариации при необходимости, чтобы заменять (S)-3-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамид на 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-(изохинолин-8-илметокси)-N-(тиазол-2-ил)бензолсульфонамида, указанное в заголовке соединение получали в виде «???» (0,074 г, выход 90%):1H ЯМР (300 МГц, DMSO-d6) δ 13,01 (ушир. с, 1H), 9,78 (с, 1H), 8,69 (д, J=6,0 Гц, 1H), 8,25 (д, J=6,0 Гц, 1H), 8,19 (дд, J=7,4, 1,6 Гц, 1H), 8,07-7,98 (м, 2H), 7,82 (д, J=7,4 Гц, 1H), 7,67 (д, J=11,8 Гц, 1H), 7,33 (д, J=4,6 Гц, 1H), 6,90 (д, J=4,6 Гц, 1H), 5,90 (с, 2H);19F ЯМР (282 МГц, DMSO-d6) δ -74,46 (s); MS (ES+) m/z 450,0 (M+1), 452,0 (M+1).
ПРИМЕР 12
Синтез (S)-5-хлор-2-фтор-4-((1-(изохинолин-8-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение (R)-N-(изохинолин-8-илметилен)-2-метилпропан-2-сульфинамида

В раствор изохинолин-8-карбальдегида (1,00 г, 6,36 ммоль) и (R)-2-метилпропан-2-сульфинамида (0,700 г, 5,78 ммоль) в безводном тетрагидрофуране (11 мл) добавляли этоксид титана(IV) (2,42 мл, 11,56 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 4 ч. Реакционную смесь разводили солевым раствором (20 мл) и получаемую суспензию перемешивали в течение 15 минут. Суспензию фильтровали через слой целита и фильтровальный слой промывали этилацетатом (2×20 мл). Фильтрат промывали солевым раствором (2×20 мл) и объединенные водные слои экстрагировали этилацетатом (2×50 мл). Объединенные органические слои сушили над сульфатом магния и фильтровали. Фильтрат концентрировали in vacuo и остаток растирали с простым диэтиловым эфиром (20 мл) для того, чтобы получать указанное в заголовке соединение в виде твердого вещества желтоватого цвета (1,52 г, количественный выход): MS (ES+) m/z 261,3 (M+1).
Стадия 2. Получение (R)-N-((S)-1-(изохинолин-8-ил)этил)-2-метилпропан-2-сульфинамида

В раствор (R)-N-(изохинолин-8-илметилен)-2-метилпропан-2-сульфинамида (0,414 г, 1,59 ммоль) в безводном дихлорметане (10 мл) добавляли 3 M раствор бромида метилмагния в простом диэтиловом эфире (1,23 мл, 3,69 ммоль) при -45°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 16 ч. Реакцию гасили добавлением насыщенного водного раствора хлорида аммония (15 мл) и разводили водой (50 мл). Водный слой экстрагировали дихлорметаном (3×50 мл) и объединенные органические слои сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 5% метанола в дихлорметане, получали указанное в заголовке соединение в 67% диастереомерном избытке (как определяют посредством1H ЯМР) в виде твердого вещества желтого цвета (0,361 г, выход 82%). Данные для основного диастереомера:1H ЯМР (300 МГц, CDCl3) δ 9,69 (с, 1H), 8,55 (д, J=5,7 Гц, 1H), 7,79-7,74 (м, 1H), 7,70-7,66 (м, 3H), 5,41 (дд, J=6,8, 4,7 Гц, 1H), 3,47 (д, J=4,5 Гц, 1H), 1,80 (д, J=6,8 Гц, 3H), 1,20 (с, 9H); MS (ES+) m/z 277,2 (M+1).
Стадия 3. Получение гидрохлорида (S)-1-(изохинолин-8-ил)этан-1-амина

В раствор (R)-N-((S)-1-(изохинолин-8-ил)этил)-2-метилпропан-2-сульфинамида (0,361 г, 1,31 ммоль) в метаноле (15 мл) добавляли 4 M раствор хлороводорода в диоксане (5 мл, 20,0 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч. Реакцию концентрировали in vacuo и остаток растирали с простым диэтиловым эфиром (количество) для того, чтобы получать указанное в заголовке соединение в виде твердого вещества бледно-желтого цвета (0,247 г, выход 90%):1H ЯМР (300 МГц, DMSO-d6) δ 10,13 (с, 1H), 8,73 (д, J=6,3 Гц, 1H), 8,49 (д, J=6,3 Гц, 1H), 8,33-8,20 (м, 3H), 5,61-5,53 (м, 1H), 1,71 (д, J=6,7 Гц, 3H), способные к обмену протоны не наблюдали; MS (ES+) m/z 173,2.
Стадия 4. Получение (S)-5-хлор-2-фтор-4-((1-(изохинолин-8-ил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида

В суспензию трет-бутил ((5-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,225 г, 0,548 ммоль) и гидрохлорида (S)-1-(изохинолин-8-ил)этан-1-амина (0,106 г, 0,508 ммоль) в безводном диметилсульфоксиде добавляли карбонат калия (0,210 г, 1,52 ммоль) и реакционную смесь нагревали при 80°C в течение 48 ч. Реакционную смесь оставляли остывать до температуры окружающей среды и разводили этилацетатом (50 мл) и насыщенным водным раствором хлорида аммония (20 мл). Водную фазу экстрагировали этилацетатом (3×50 мл). Объединенную органическую фазу промывали солевым раствором (3×20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью HPLC с обращенной фазой, осуществляя элюирование ацетонитрилом в воде, содержащей 0,1% трифторуксусную кислоту, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,050 г, выход 21%):1H ЯМР (300 МГц, DMSO-d6) δ 11,11 (ушир. с, 1H), 9,99 (с, 1H), 8,85 (д, J=2,2 Гц, 1H), 8,70 (д, J=6,2 Гц, 1H), 8,36 (д, J=6,2 Гц, 1H), 8,12 (д, J=8,1 Гц, 1H), 8,04-7,97 (м, 1H), 7,82 (д, J=7,0 Гц, 1H), 7,64 (д, J=7,4 Гц, 1H), 7,01-6,95 (м, 2H), 6,42 (д, J=13,1 Гц, 1H), 5,75-5,66 (м, 1H), 1,70 (д, J=6,7 Гц, 3H);19F ЯМР (282 МГц, DMSO-d6) δ -74,4 (s); MS (ES+) m/z 463,0 (M+1), 465,0 (M+1).
ПРИМЕР 13
Синтез (R)-5-хлор-2-фтор-4-((1-(изохинолин-8-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение (S)-N-(изохинолин-8-илметилен)-2-метилпропан-2-сульфинамида

Следуя процедуре как описано для примера 12, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять (R)-2-метилпропан-2-сульфинамид на (S)-2-метилпропан-2-сульфинамид (0,700 г, 5,78 ммоль), указанное в заголовке соединение получали в виде твердого вещества желтого цвета (1,36 г, выход 90%): MS (ES+) m/z 261,3.
Стадия 1. Получение (S)-N-((R)-1-(изохинолин-8-ил)этил)-2-метилпропан-2-сульфинамида
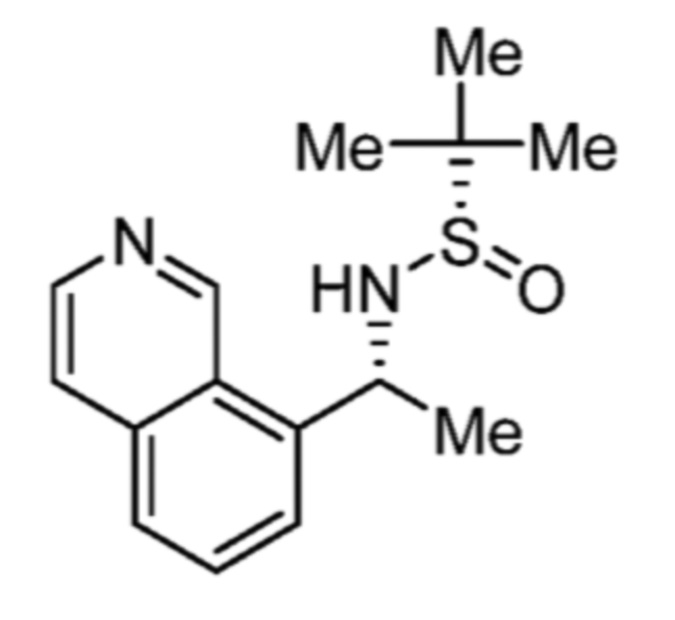
Следуя процедуре как описано для примера 12, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять (R)-N-(изохинолин-8-илметилен)-2-метилпропан-2-сульфинамид на (S)-N-(изохинолин-8-илметилен)-2-метилпропан-2-сульфинамид, указанное в заголовке соединение получали в 67% диастереомерном избытке (как определяют по1H ЯМР) в виде твердого вещества желтого цвета (0,498 г, выход 89%):1H ЯМР (300 МГц, CDCl3) δ 9,69 (с, 1H), 8,55 (д, J=5,6 Гц, 1H), 7,77 (дд, J=4,0, 5,4 Гц, 1H), 7,69-7,66 (м, 3H), 5,41 (дд, J=4,6, 6,8 Гц, 1H), 3,47 (д, J=4,5 Гц, 1H), 1,80 (д, J=6,8 Гц, 3H), 1,20 (с, 9H); MS (ES+) m/z 277,2 (M+1).
Стадия 3. Получение гидрохлорида (R)-1-(изохинолин-8-ил)этан-1-амина
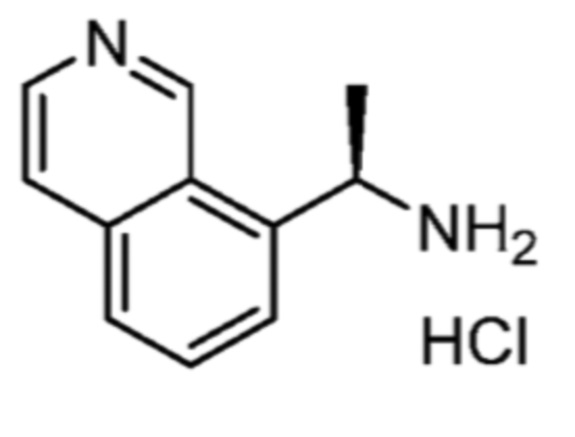
Следуя процедуре как описано для примера 12, стадии 3 и выполняя некритические вариации при необходимости, чтобы заменять (R)-N-((S)-1-(изохинолин-8-ил)этил)-2-метилпропан-2-сульфинамид на (S)-N-((R)-1-(изохинолин-8-ил)этил)-2-метилпропан-2-сульфинамид, указанное в заголовке соединение получали в виде ??? (0,360 г, выход 96%):1H ЯМР (300 МГц, DMSO-d6) δ 10,10 (с, 1H), 8,73 (д, J=6,3 Гц, 1H), 8,46 (д, J=6,3 Гц, 1H), 8,31-8,18 (м, 3H), 5,60-5,52 (м, 1H), 1,70 (д, J=6,7 Гц, 3H), способные к обмену протоны не наблюдали; MS (ES+) m/z 173,2.
Стадия 4. Получение (R)-5-хлор-2-фтор-4-((1-(изохинолин-8-ил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано для примера 12, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять гидрохлорид (S)-1-(изохинолин-8-ил)этан-1-амина на гидрохлорид (R)-1-(изохинолин-8-ил)этан-1-амина, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,096 г, выход 20%):1H ЯМР (300 МГц, DMSO-d6) δ 11,10 (ушир. с, 1H), 9,95 (с, 1H), 8,85 (д, J=2,2 Гц, 1H), 8,68 (д, J=6,1 Гц, 1H), 8,30 (д, J=6,2 Гц, 1H), 8,09 (д, J=8,1 Гц, 1H), 8,01-7,93 (м, 1H), 7,79 (д, J=7,2 Гц, 1H), 7,64 (д, J=7,4 Гц, 1H), 6,98-6,96 (м, 2H), 6,41 (д, J=13,2 Гц, 1H), 5,75-5,66 (м, 1H), 1,70 (д, J=6,7 Гц, 3H);19F ЯМР (282 МГц, DMSO-d6) δ -74,3 (с); MS (ES+) m/z 463,0 (M+1), 465,0 (M+1).
ПРИМЕР 14
Синтез (S)-5-хлор-4-((1-(4-хлорфенил)этил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение (S)-5-хлор-4-((1-(4-хлорфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида
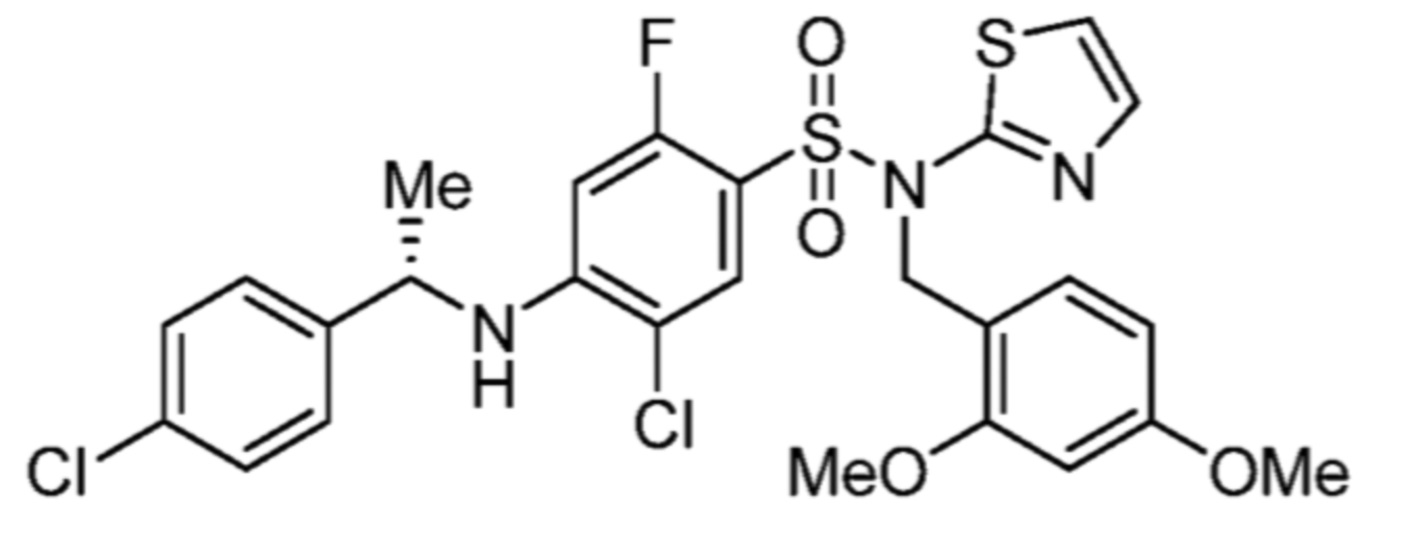
В смесь 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамида (0,35 г, 0,76 ммоль) и (S)-1-(4-хлорфенил)этиламина (0,107 мл, 0,76 ммоль) в безводном диметилсульфоксиде (4 мл) добавляли карбонат калия (0,210 г, 1,52 ммоль) и реакционную смесь нагревали при 75°C в течение 16 ч. Реакционную смесь оставляли остывать до температуры окружающей среды, разводили насыщенным водным раствором хлорида аммония (20 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали водой (40 мл), солевым раствором (40 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием от 0% до 50% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,16 г, выход 35%): MS (ES+) m/z 596,1 (M+1), 598,1 (M+1).
Стадия 2. Получение (S)-5-хлор-4-((1-(4-хлорфенил)этил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

В раствор (S)-5-хлор-4-((1-(4-хлорфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида (0,16 г, 0,27 ммоль) в безводном дихлорметане (5 мл) добавляли трифторуксусную кислоту (62 мкл, 0,81 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 30 минут и концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с использованием от 0% до 35% ацетона в гексанах, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,089 г, выход 74%):1H ЯМР (300 МГц, DMSO-d6) δ 12,75 (с, 1H), 7,58 (д, J=7,3 Гц, 1H), 7,44-7,35 (м, 4H), 7,25 (д, J=4,6 Гц, 1H), 6,81 (д, J=4,6 Гц, 1H), 6,58-6,53 (м, 1H), 6,40 (д, J=13,2 Гц, 1H), 4,77-4,67 (м, 1H), 1,51 (д, J=6,7 Гц, 3H); MS (ES+) m/z 446,0 (M+1), 448,0 (M+1).
ПРИМЕР 15
Синтез (S)-5-хлор-2-фтор-4-((1-(нафтален-2-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(нафтален-2-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида
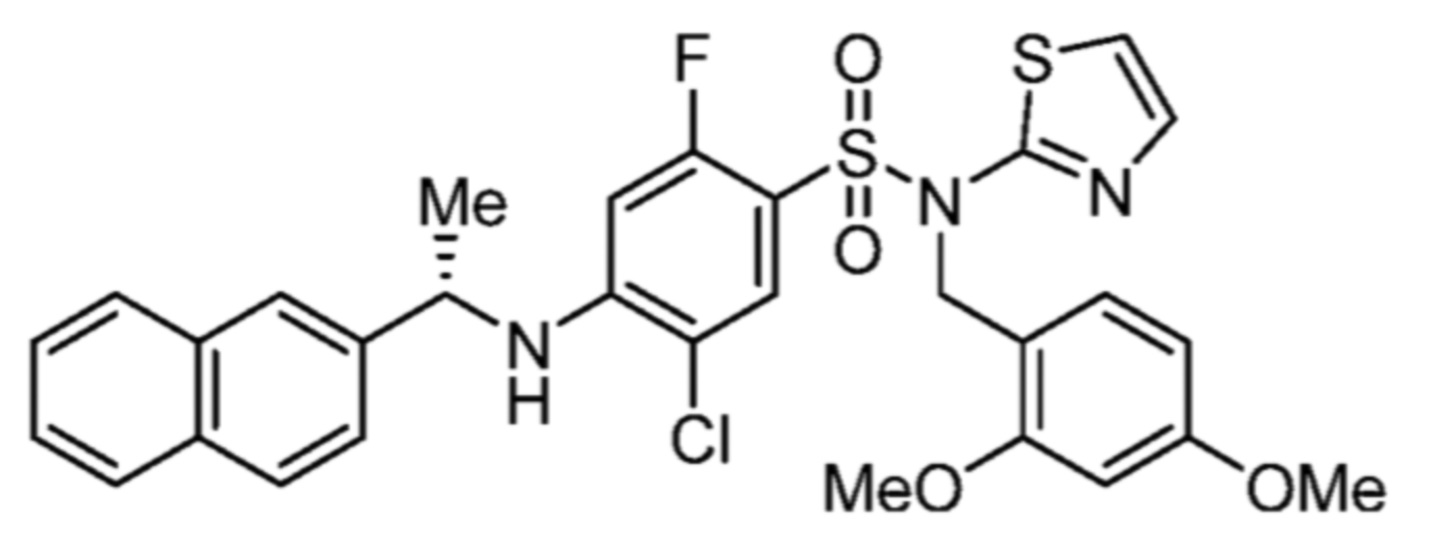
Следуя процедуре как описано в примере 14, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять (S)-1-(4-хлорфенил)этиламин на (S)-1-(нафтален-2-ил)этан-1-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,16 г, выход 34%): MS (ES+) m/z 612,2 (M+1), 614,1 (M+1).
Стадия 2. Получение (S)-5-хлор-2-фтор-4-((1-(нафтален-2-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 14, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять (S)-5-хлор-4-((1-(4-хлорфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид на (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(нафтален-2-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,087 г, выход 73%):1H ЯМР (300 МГц; DMSO-d6) δ 12,70 (с, 1H), 7,91-7,83 (м, 4H), 7,61-7,56 (м, 2H), 7,52-7,44 (м, 2H), 7,22-7,20 (м, 1H), 6,80-6,78 (м, 1H), 6,64-6,60 (м, 1H), 6,46 (д, J=13,2 Гц, 1H), 4,91-4,83 (м, 1H), 1,61 (д, J=6,8 Гц, 3H); MS (ES+) m/z 462,1 (M+1), 464,1 (M+1).
ПРИМЕР 16
Синтез 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)бензил)амино)-5-хлор-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение 2-(азетидин-1-илметил)бензонитрила

В раствор 2-(бромметил)бензонитрила (1,8 г, 8,16 ммоль) и азетидина (0,466 г, 8,16 ммоль) в безводном дихлорметане (60 мл) добавляли N,N-диизопропилэтиламин (1,78 мл, 10,2 ммоль) при 0°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 16 ч. Реакционную смесь разводили насыщенным водным раствором хлорида аммония (20 мл) и экстрагировали дихлорметаном (3×40 мл). Объединенные органические слои промывали солевым раствором (80 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием от 0% до 20% метанола в дихлорметане, получали указанное в заголовке соединение в виде масла бледно-желтого цвета (0,61 г, выход 43%): MS (ES+) m/z 173,2 (M+1).
Стадия 2. Получение (2-(азетидин-1-илметил)фенил)метанамина

В раствор 2-(азетидин-1-илметил)бензонитрила (0,6 г, 3,46 ммоль) в безводном тетрагидрофуране (35 мл) добавляли 1 M раствор гидрида лития алюминия в тетрагидрофуране (5,2 мл, 5,2 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 3 ч и декагидрат сульфата натрия (5 г) добавляли малыми частями. Смесь перемешивали при 0°C в течение 30 минут и затем при температуре окружающей среды в течение 1 ч. Смесь фильтровали и фильтрат сушили над безводным сульфатом магния. Фильтрованием и концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде масла бледно-коричневого цвета (0,55 г, выход 90%): MS (ES+) m/z 177,2 (M+1).
Стадия 3. Получение 4-((2-(азетидин-1-илметил)бензил)амино)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 14, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять (S)-1-(4-хлорфенил)этиламин на (2-(азетидин-1-илметил)фенил)метанамин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,07 г, выход 15%): MS (ES+) m/z 617,1 (M+1), 619,1 (M+1).
Стадия 4. Получение 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)бензил)амино)-5-хлор-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 1, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять (S)-5-хлор-4-((1-(4-хлорфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид на 4-((2-(азетидин-1-илметил)бензил)амино)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,014 г, выход 27%):1H ЯМР (300 МГц, DMSO-d6) δ 12,78 (с, 1H), 9,97 (с, 1H), 7,63 (д, J=7,3 Гц, 1H), 7,44-7,31 (м, 3H), 7,27-7,25 (м, 2H), 7,11-7,03 (м, 1H), 6,83 (д, J=4,6 Гц, 1H), 6,48 (д, J=12,9 Гц, 1H), 4,58-4,49 (м, 4H), 4,22-4,01 (м, 4H), 2,45-2,25 (м, 2H); MS (ES+) m/z 467,0 (M+1), 469,0 (M+1).
ПРИМЕР 17
Синтез 5-хлор-4-((2-цианобензил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение 5-хлор-4-((2-цианобензил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 14, стадии 3 и выполняя некритические вариации при необходимости, чтобы заменять (S)-1-(4-хлорфенил)этиламин на гидрохлорид 2-(аминометил)бензонитрила, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,33 г, выход 66%): MS (ES+) m/z 573,0 (M+1), 575,0 (M+1).
Стадия 2. Получение 5-хлор-4-((2-цианобензил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 1, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять (S)-5-хлор-4-((1-(4-хлорфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид на 5-хлор-4-((2-цианобензил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,065 г, выход 29%):1H ЯМР (300 МГц, DMSO-d6) δ 12,78 (с, 1H), 7,86 (дд, J=7,7, 1,2 Гц, 1H), 7,69-7,62 (м, 2H), 7,50-7,42 (м, 1H), 7,38-7,33 (м, 1H), 7,26 (д, J=4,6 Гц, 1H), 7,19-7,13 (м, 1H), 6,83 (д, J=4,6 Гц, 1H), 6,52 (д, J=12,8 Гц, 1H), 4,69-4,63 (м, 2H); MS (ES+) m/z 423,0 (M+1), 425,0 (M+1).
ПРИМЕР 18
Синтез 5-хлор-2-фтор-4-((1-фенилциклобутил)амино)-N-(тиазол-4-ил)бензолсульфонамида

В смесь гидрохлорида 1-фенилциклобутан-1-амина (0,16 г 0,87 ммоль) и трет-бутил ((5-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,358 г, 0,87 ммоль) в безводном диметилсульфоксиде (4 мл) добавляли карбонат калия (0,240 г, 1,74 ммоль) и реакционную смесь нагревали при 75°C в течение 24 ч. Реакционную смесь оставляли остывать до температуры окружающей среды, разводили насыщенным водным раствором хлорида аммония (20 мл), и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали водой (40 мл), солевым раствором (40 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием от 0% до 35% ацетона в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,067 г, выход 19%):1H ЯМР (300 МГц, CDCl3) δ 9,56 (с, 1H), 8,62 (с, 1H), 7,75-7,71 (м, 1H), 7,42-7,30 (м, 4H), 7,28-7,22 (м, 1H), 6,92-6,87 (м, 1H), 5,77-5,72 (м, 1H), 5,59 (с, 1H), 2,74-2,60 (м, 2H), 2,44-2,31 (м, 2H), 2,23-2,03 (м, 2H); MS (ES+) m/z 438,0 (M+1), 440,0 (M+1).
ПРИМЕР 19
Синтез 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)бензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида
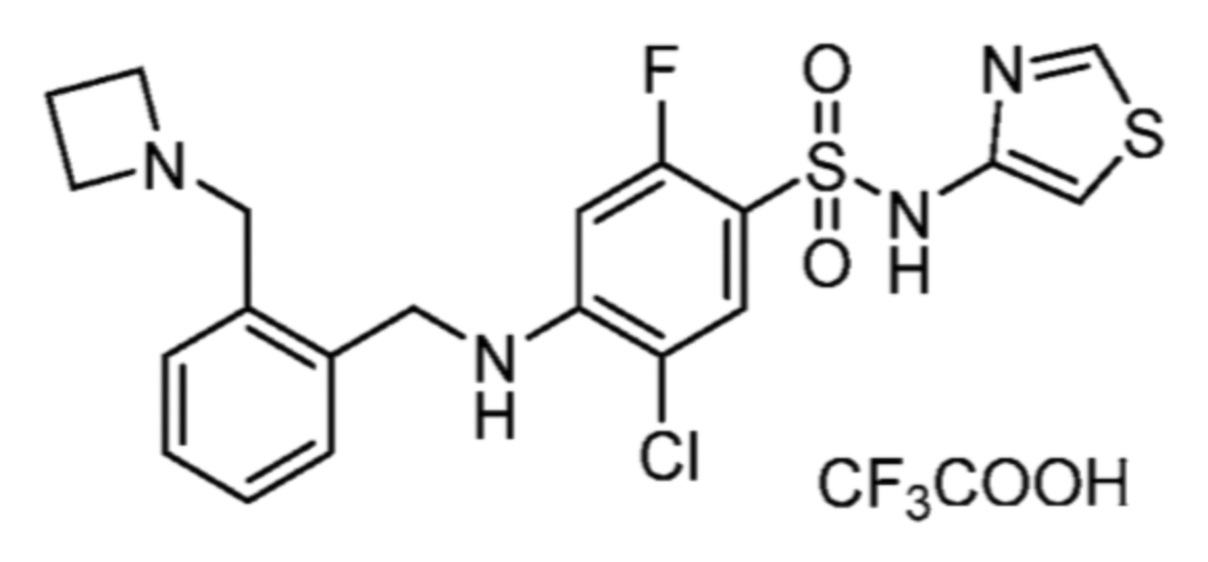
Следуя процедуре как описано для примера 18 и выполняя некритические вариации при необходимости, чтобы заменять гидрохлорид 1-фенилциклобутан-1-амина на (2-(азетидин-1-илметил)фенил)метанамин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,02 г, выход 3%):1H ЯМР (300 МГц; DMSO-d6) δ 11,12 (с, 1H), 10,05 (с, 1H), 8,88 (д, J=2,2 Гц, 1H), 7,64 (д, J=7,4 Гц, 1H), 7,48-7,17 (м, 5H), 6,99 (д, J=2,1 Гц, 1H), 6,51 (д, J=13,0 Гц, 1H), 4,59-4,46 (м, 4H), 4,22-4,01 (м, 4H), 2,46-2,26 (м, 2H); MS (ES+) m/z 467,0 (M+1), 469,0 (M+1).
ПРИМЕР 20
Синтез of 5-хлор-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-4-ил)бензолсульфонамид

Следуя процедуре как описано для примера AZ5 и выполняя некритические вариации при необходимости, чтобы заменять гидрохлорид 1-фенилциклобутан-1-амина на 1-фенилциклопропан-1-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,021 г, выход 2%):1H ЯМР (300 МГц, CDCl3) δ 9,78 (с, 1H), 8,70-8,63 (м, 1H), 7,76 (д, J=7,1 Гц, 1H), 7,33-7,24 (м, 2H), 7,23-7,16 (м, 1H), 7,05-7,00 (м, 2H), 6,92 (дд, J=4,2, 1,7 Гц, 1H), 6,43 (д, J=12,3 Гц, 1H), 5,69-5,64 (м, 1H), 1,45-1,27 (м, 4H); MS (ES+) m/z 424,1 (M+1), 426,0 (M+1).
ПРИМЕР 21
Синтез (S)-3,5-дихлор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение 3,5-дихлор-N-(2,4-диметоксибензил)-4-фтор-N-(тиазол-2-ил)бензолсульфонамида
В смесь N-(2,4-диметоксибензил)тиазол-2-амина (8,60 г, 34,4 ммоль, полученного в соответствии с WO 2013063459) в безводном тетрагидрофуране (100 мл) добавляли 1 M раствор бис(триметилсилил)амида лития в тетрагидрофуране (18,2 мл, 18,2 ммоль) при -78°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 1 ч. Реакционную смесь охлаждали до -78°C и туда добавляли 3,5-дихлор-4-фторбензолсульфонилхлорид (4,0 г, 15,2 ммоль). Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 16 ч. Реакционную смесь гасили добавлением насыщенного раствора хлорида аммония (50 мл) и разводили этилацетатом (300 мл). Органическую фазу промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 50% этилацетата в гексанах, и растиранием с метанолом (10 мл) получали указанное в заголовке соединение в виде твердого вещества грязно-белого цвета (5,5 г, выход 76%):1H ЯМР (300 МГц, CDCl3) δ 7,75 (дд, J=6,0 Гц, 2H), 7,50 (дд, J=3,6 Гц, 1H), 7,15 (д, J=8,0 Гц, 1H), 7,12 (дд, J=3,6 Гц, 1H), 6,40-6,34 (м, 2H), 5,07 (с, 2H), 3,79 (с, 3H), 3,68 (с, 3H).
Стадия 2. Получение (S)-3,5-дихлор-N-(2,4-диметоксибензил)-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 14, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять (S)-1-(4-хлорфенил)этиламин на (S)-1-(2-фторфенил)этан-1-амин и 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамид на 3,5-дихлор-N-(2,4-диметоксибензил)-4-фтор-N-(тиазол-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,2 г, выход 41%): MS (ES+) m/z 596,1 (M+1), 598,1 (M+1).
Стадия 3. Получение (S)-3,5-дихлор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида
Следуя процедуре как описано в примере 14, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять (S)-5-хлор-4-((1-(4-хлорфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид на (S)-3,5-дихлор-N-(2,4-диметоксибензил)-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,085 г, выход 42%):1H ЯМР (300 МГц; DMSO-d6) δ 12,84 (с, 1H), 7,58 (с, 2H), 7,53-7,47 (м, 1H), 7,30-7,21 (м, 2H), 7,18-7,06 (м, 2H), 6,86 (д, J=4,5 Гц, 1H), 5,46-5,31 (м, 2H), 1,54 (д, J=6,4 Гц, 3H); MS (ES+) m/z 446,0 (M+1), 448,0 (M+1).
ПРИМЕР 22
Синтез (S)-5-хлор-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(6-фторпиридин-2-ил)бензолсульфонамида

Стадия 1. Получение 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(6-фторпиридин-2-ил)бензолсульфонамида

В раствор N-(2,4-диметоксибензил)-6-фторпиридин-2-амина (получен в соответствии с WO2014066490, 20,00 г, 76,25 ммоль) в безводном тетрагидрофуране (200 мл) добавляли 1,6 M раствор метиллития в простом диэтиловом эфире (66,7 мл, 106,7 ммоль) по каплям при -78°C. Реакционную смесь оставляли нагреваться до 0°C и перемешивали в течение 30 минут. Реакционную смесь охлаждали до -78°C и туда по каплям добавляли раствор 5-хлор-2,4-дифторбензолсульфонилхлорида (33,9 г, 137,3 ммоль) в безводном тетрагидрофуране (100 мл). Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 12 ч. Смесь разводили водой (200 мл) и экстрагировали этилацетатом (3×200 мл). Объединенную органическую фазу промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и растиранием остатка в смеси метанола и дихлорметана (20:1, 2×150 мл) получали указанное в заголовке соединение в виде бесцветного твердого вещества (12,1 г, выход 32%):1H ЯМР (300 МГц, CDCl3) δ 8,02 (т, J=8,0 Гц, 1H), 7,77-7,66 (м, 1H), 7,27-7,15 (м, 2H), 7,01 (т, J=8,0 Гц, 1H), 6,72 (дд, J=8,0, 2,8 Гц, 1H), 6,43-6,35 (м, 2H), 5,07 (с, 2H), 3,78 (с, 3H), 3,73 (с, 3H).
Стадия 2. Получение (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(6-фторпиридин-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 14, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять (S)-1-(4-хлорфенил)этиламин на (S)-1-(2-фторфенил)этан-1-амин и 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамид на 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(6-фторпиридин-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,34 г, выход 70%): MS (ES+) m/z 592,2 (M+1), 594,2 (M+1).
Стадия 3. Получение (S)-5-хлор-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(6-фторпиридин-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 14, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять (S)-5-хлор-4-((1-(4-хлорфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид на (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(6-фторпиридин-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,095 г, выход 38%):1H ЯМР (300 МГц, DMSO-d6) δ 11,50 (с, 1H), 7,86-7,74 (м, 2H), 7,43-7,37 (м, 1H), 7,33-7,25 (м, 1H), 7,22-7,12 (м, 2H), 6,83 (дд, J=7,9, 2,1 Гц, 1H), 6,74-6,68 (м, 2H), 6,34 (д, J=13,4 Гц, 1H), 4,99-4,87 (м, 1H), 1,55 (д, J=6,7 Гц, 3H); MS (ES+) m/z 442,1 (M+1), 444,1 (M+1).
ПРИМЕР 23
Синтез (S)-5-хлор-4-((1-(2-хлорфенил)этил)амино)-2-фтор-N-(6-фторпиридин-2-ил)бензолсульфонамида

Стадия 1. Получение (S)-5-хлор-4-((1-(2-хлорфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(6-фторпиридин-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 14, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять (S)-1-(4-хлорфенил)этиламин на гидрохлорид (S)-1-(2-хлорфенил)этан-1-амина и 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамид на 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(6-фторпиридин-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,26 г, выход 51%): MS (ES+) m/z 608,1 (M+1), 610,0 (M+1).
Стадия 2. Получение (S)-5-хлор-4-((1-(2-хлорфенил)этил)амино)-2-фтор-N-(6-фторпиридин-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 14, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять (S)-5-хлор-4-((1-(4-хлорфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид на (S)-5-хлор-4-((1-(2-хлорфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(6-фторпиридин-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,041 г, выход 21%):1H ЯМР (300 МГц, DMSO-d6) δ 11,52 (с, 1H), 7,87-7,74 (м, 2H), 7,50-7,44 (м, 2H), 7,34-7,24 (м, 2H), 6,99-6,93 (м, 1H), 6,82 (дд, J=7,9, 2,1 Гц, 1H), 6,70 (дд, J=8,0, 2,4 Гц, 1H), 6,05 (д, J=13,3 Гц, 1H), 4,96-4,83 (м, 1H), 1,53 (д, J=6,7 Гц, 3H); MS (ES+) m/z 458,1 (M+1), 460,1 (M+1).
ПРИМЕР 24
Синтез (S)-3-хлор-4-((1-фенилэтил)амино)-N-(тиазол-4-ил)бензолсульфонамида

В смесь 4-бром-3-хлор-N-(тиазол-4-ил)бензолсульфонамида (0,25 г, 0,71 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантена (0,025 г, 0,042 ммоль) и трис(дибензилиденацетон)дипалладия (0,013 мг, 0,014 ммоль) в безводном 1,4-диоксане (6 мл) добавляли трет-бутоксид натрия (0,163 г, 1,70 ммоль) и смесь продували аргоном в течение 15 минут. Затем в смесь добавляли (S)-1-фенилэтан-1-амин (0,11 мл, 0,85) и реакционную смесь нагревали до 100°C в течение 16 ч. Реакционную смесь оставляли остывать до температуры окружающей среды и фильтровали через подушку из целита. Фильтрат разводили этилацетатом (30 мл) и насыщенным водным раствором бикарбоната натрия (15 мл) и водный слой экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали солевым раствором (40 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием от 0% до 50% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,116 г, выход 42%):1H ЯМР (300 МГц, CDCl3) δ 9,47 (с, 1H), 8,65 (д, J=2,1 Гц, 1H), 7,68 (д, J=2,2 Гц, 1H), 7,39-7,25 (м, 6H), 6,95 (д, J=2,2 Гц, 1H), 6,33 (д, J=8,9 Гц, 1H), 5,12 (с, 1H), 4,56-4,49 (м, 1H), 1,58 (д, J=6,8 Гц, 3H); MS (ES+) m/z 394,1 (M+1), 396,1 (M+1).
ПРИМЕР 25
Синтез (S)-2,5-дифтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение (S)-N-(2,4-диметоксибензил)-2,5-дифтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 14, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять (S)-1-(4-хлорфенил)этиламин на (S)-1-(2-фторфенил)этан-1-амин и 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамид на N-(2,4-диметоксибензил)-2,4,5-трифтор-N-(тиазол-2-ил)бензолсульфонамид (получен в соответствии с WO 2013118854), указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,26 г, выход 41%): MS (ES+) m/z 564,2 (M+1).
Стадия 2. Получение (S)-2,5-дифтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 14, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять (S)-5-хлор-4-((1-(4-хлорфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид на (S)-N-(2,4-диметоксибензил)-2,5-дифтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,15 г, выход 79%):1H ЯМР (300 МГц, CDCl3) δ 13,04 (с, 1H), 7,56 (дд, J=10,8, 6,3 Гц, 1H), 7,31-7,20 (м, 2H), 7,15-7,02 (м, 3H), 6,46 (д, J=4,5 Гц, 1H), 6,09 (дд, J=11,7, 6,6 Гц, 1H), 4,83-4,69 (м, 2H), 1,59 (д, J=6,0 Гц, 3H); MS (ES+) m/z 414,1 (M+1).
ПРИМЕР 26
Синтез (S)-2,5-дифтор-4-(1-(2-фторфенил)этокси)-N-(тиазол-2-ил)бензолсульфонамида
Стадия 1. Получение (S)-N-(2,4-диметоксибензил)-2,5-дифтор-4-(1-(2-фторфенил)этокси)-N-(тиазол-2-ил)бензолсульфонамида

В смесь (S)-1-(2-фторфенил)этан-1-ола (0,14 г, 1,0 ммоль) и N-(2,4-диметоксибензил)-2,4,5-трифтор-N-(тиазол-2-ил)бензолсульфонамида (0,49 г, 1,1 ммоль) в безводном N,N-диметилформамиде (10 мл) добавляли гидрид натрия (60% дисперсия в минеральном масле, 0,08 г, 2,0 ммоль) при 0°C. Смесь перемешивали при температуре окружающей среды в течение 2 ч, охлаждали до 0°C и гасили добавлением насыщенного раствора хлорида аммония (20 мл). Смесь представляла собой этилацетат (3×30 мл). Объединенные органические слои промывали водой (40 мл), солевым раствором (40 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием от 0% до 50% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,56 г, выход 99%): MS (ES+) m/z 565,1 (M+1).
Стадия 2. Получение (S)-2,5-дифтор-4-(1-(2-фторфенил)этокси)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 14, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять (S)-5-хлор-4-((1-(4-хлорфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид на (S)-N-(2,4-диметоксибензил)-2,5-дифтор-4-(1-(2-фторфенил)этокси)-N-(тиазол-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,165 г, выход 40%):1H ЯМР (300 МГц, DMSO-d6) δ 12,91 (с, 1H), 7,59 (дд, J=10,4, 6,7 Гц, 1H), 7,47 (тд, J=7,7, 1,8 Гц, 1H), 7,42-7,34 (м, 1H), 7,32-7,16 (м, 4H), 6,87 (д, J=4,6 Гц, 1H), 5,95-5,87 (м, 1H), 1,63 (д, J=6,3 Гц, 3H); MS (ES+) m/z 415,0 (M+1).
ПРИМЕР 27
Синтез (S)-2,6-дифтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида


В раствор N-(2,4-диметоксибензил)тиазол-2-амина (3,0 г, 11,98 ммоль) в безводном тетрагидрофуране (23 мл) добавляли 1 M раствор бис(триметилсилил)амида лития в тетрагидрофуране (14,4 мл, 14,4 ммоль) при -78°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 1 ч. Реакционную смесь охлаждали до -78°C и туда по каплям добавляли раствор 2,4,6-трифторбензолсульфонилхлорида (2,76 г, 11,98 ммоль) в безводном тетрагидрофуране (10 мл). Реакционную смесь оставляли нагреваться до температуры окружающей среды, перемешивали в течение 16 ч и разводили насыщенным раствором хлорида аммония (50 мл). Смесь экстрагировали этилацетатом (3×60 мл). Объединенные органические слои промывали солевым раствором (100 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием от 0% до 25% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (1,28 г, выход 24%): MS (ES+) m/z 445,1 (M+1).
Стадия 2. Получение (S)-N-(2,4-диметоксибензил)-2,6-дифтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 14, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять (S)-1-(4-хлорфенил)этиламин на (S)-1-(2-фторфенил)этан-1-амин и 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамид на N-(2,4-диметоксибензил)-2,4,6-трифтор-N-(тиазол-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,21 г, выход 33%): MS (ES+) m/z 564,1 (M+1).
Стадия 3. Получение (S)-2,6-дифтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида
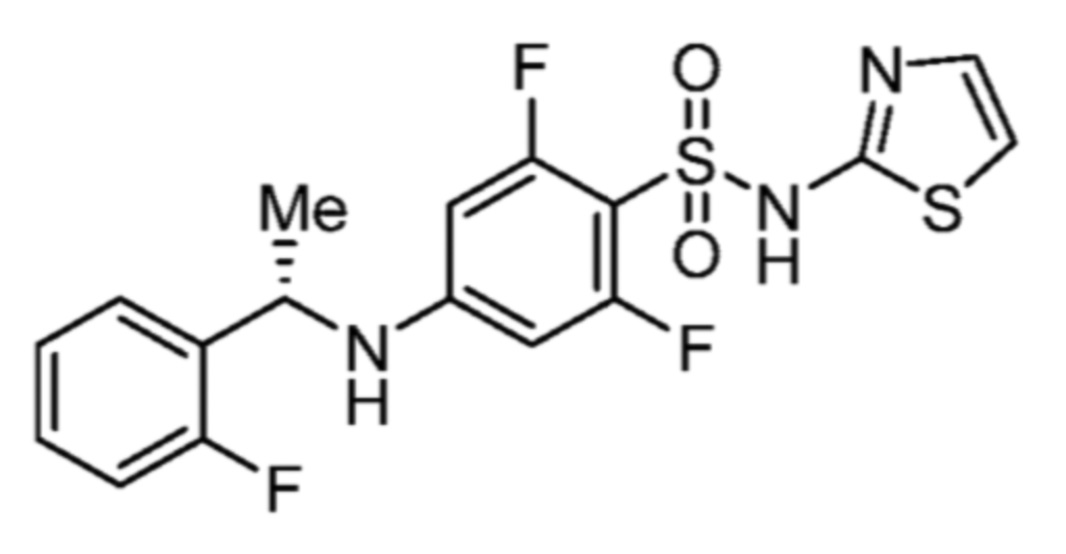
Следуя процедуре как описано в примере 14, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять (S)-5-хлор-4-((1-(4-хлорфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид на (S)-N-(2,4-диметоксибензил)-2,6-дифтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,105 г, выход 68%):1H ЯМР (300 МГц, DMSO-d6) δ 12,73 (с, 1H), 7,48 (д, J=7,0 Гц, 1H), 7,36-7,25 (м, 3H), 7,22-7,13 (м, 2H), 6,81 (д, J=4,6 Гц, 1H), 6,12 (д, J=12,4 Гц, 2H), 4,83-4,72 (м, 1H), 1,44 (д, J=6,7 Гц, 3H); MS (ES+) m/z 414,0 (M+1).
ПРИМЕР 28
Синтез (S)-5-хлор-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил (S)-((5-хлор-2-фтор-4-((1-(2-фторфенил)этил)амино)фенил)сульфонил)(тиазол-4-ил)карбамата
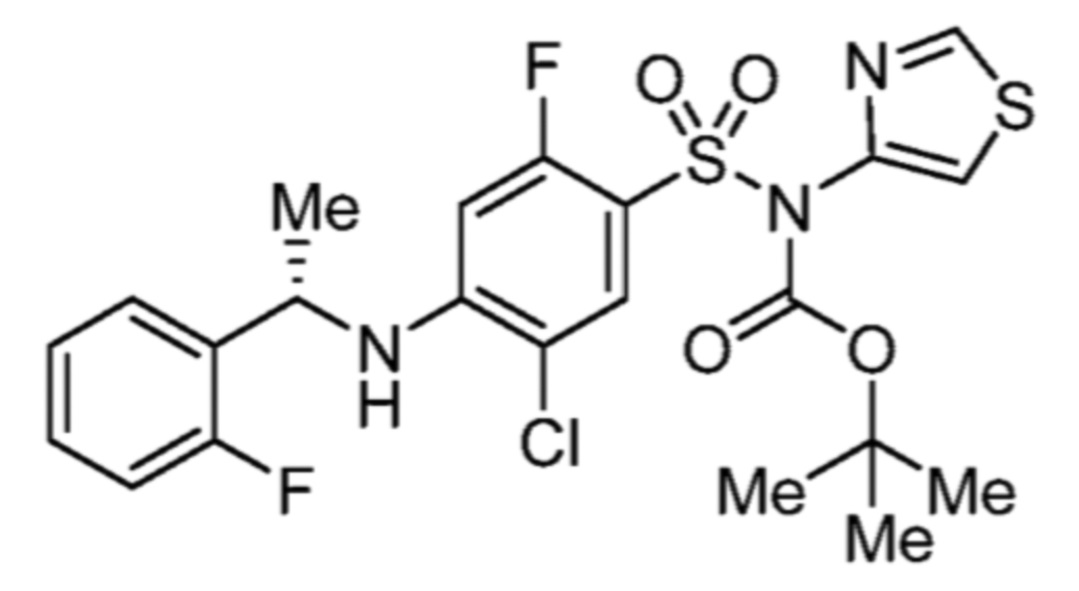
В смесь трет-бутил ((5-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,54 г, 1,31 ммоль) и (S)-1-(2-фторфенил)этан-1-амина (0,15 г, 1,09 ммоль) в безводном диметилсульфоксиде (10 мл) добавляли карбонат цезия (0,82 г, 2,51 ммоль) и реакционную смесь перемешивали при 50°C в течение 16 ч. Реакционную смесь оставляли остывать до температуры окружающей среды, разводили насыщенным раствором хлорида аммония (20 мл), и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали водой (40 мл), солевым раствором (40 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием от 0% до 25% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,18 г, выход 31%): MS (ES+) m/z 530,1 (M+1), 532,1 (M+1).
Стадия 2. Получение (S)-5-хлор-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано в примере 14, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять (S)-5-хлор-4-((1-(4-хлорфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид на трет-бутил (S)-((5-хлор-2-фтор-4-((1-(2-фторфенил)этил)амино)фенил)сульфонил)(тиазол-4-ил)карбамата, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,093 г, выход 64%):1H ЯМР (300 МГц, CDCl3) δ 9,67 (с, 1H), 8,63 (д, J=2,3 Гц, 1H), 7,73 (д, J=7,2 Гц, 1H), 7,29-7,16 (м, 2H), 7,12-7,03 (м, 2H), 6,91 (д, J=2,3 Гц, 1H), 6,09 (д, J=12,3 Гц, 1H), 5,21-5,17 (м, 1H), 4,82-4,72 (м, 1H), 1,61 (д, J=6,7 Гц, 3H); MS (ES+) m/z 430,0 (M+1), 432,0 (M+1).
ПРИМЕР 29
Синтез 5-хлор-2-фтор-N-(тиазол-4-ил)-4-((4-(трифторметил)-2,3-дигидро-1H-инден-1-ил)окси)бензолсульфонамида

В раствор 4-(трифторметил)-2,3-дигидро-1H-инден-1-ола (0,24 г, 1,19 ммоль, получен в соответствии с WO 2009157418) в безводном N,N-диметилформамиде (8 мл) добавляли гидрид натрия (60% дисперсия в минеральном масле, 0,057 г, 1,43 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 30 мин и при температуре окружающей среды в течение 15 мин. Затем реакционную смесь охлаждали до 0°C и туда добавляли трет-бутил ((5-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамат (0,49 г, 1,19 ммоль). Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 2 ч. Затем реакционную смесь разводили водой (20 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали водой (40 мл), солевым раствором (40 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием от 0% до 70% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,02 г, выход 3%):1H ЯМР (300 МГц, CDCl3) δ 9,90 (с, 1H), 8,72 (д, J=2,0 Гц, 1H), 7,90 (д, J=7,5 Гц, 1H), 7,61 (дд, J=10,5, 7,9 Гц, 2H), 7,42-7,37 (м, 1H), 7,03 (д, J=2,0 Гц, 1H), 6,88 (д, J=11,3 Гц, 1H), 5,77-5,73 (м, 1H), 3,40-3,27 (м, 1H), 3,18-3,07 (м, 1H), 2,73-2,61 (м, 1H), 2,29-2,18 (м, 1H); MS (ES-) m/z 491,0 (M - 1), 493,0 (M - 1).
ПРИМЕР 30
Синтез (S)-5-хлор-2-фтор-4-((1-фенилэтил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил (S)-((5-хлор-2-фтор-4-((1-фенилэтил)амино)фенил)сульфонил)(тиазол-4-ил)карбамата
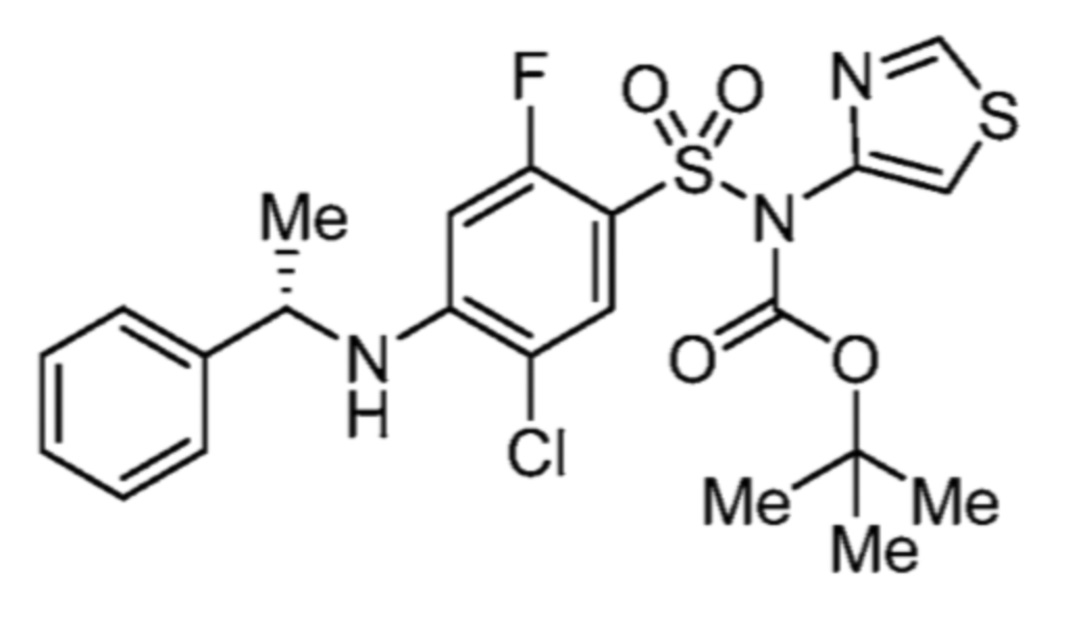
В смесь трет-бутил ((5-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,4 г, 0,97 ммоль) и (S)-1-фенилэтан-1-амина (0,12 мл, 0,97 ммоль) в безводном диметилсульфоксиде (10 мл) добавляли карбонат калия (0,32 г, 2,33 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч и затем нагревали при 50°C в течение 16 ч. Реакционную смесь оставляли остывать до температуры окружающей среды, разводили водой (30 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали водой (40 мл), солевым раствором (40 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием от 0% до 100% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,11 г, выход 22%): MS (ES+) m/z 512,0 (M+1), 514,1 (M+1).
Стадия 2. Получение (S)-5-хлор-2-фтор-4-((1-фенилэтил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано в примере 14, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять (S)-5-хлор-4-((1-(4-хлорфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид на трет-бутил (S)-((5-хлор-2-фтор-4-((1-фенилэтил)амино)фенил)сульфонил)(тиазол-4-ил)карбамат, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,047 г, выход 53%):1H ЯМР (300 МГц, CDCl3) δ 9,59 (с, 1H), 8,61 (с, 1H), 7,73 (д, J=7,2 Гц, 1H), 7,36-7,24 (м, 5H), 6,89 (с, 1H), 6,08 (д, J=12,4 Гц, 1H), 5,21-5,18 (м, 1H), 4,50-4,41 (м, 1H), 1,59 (д, J=6,8 Гц, 3H); MS (ES+) m/z 412,0 (M+1), 414,0 (M+1).
ПРИМЕР 31
Синтез (R)-3-хлор-4-((1-фенилпропил)амино)-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида

В смесь 3-хлор-N-(2,4-диметоксибензил)-4-фтор-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида (0,250 г, 0,563 ммоль) и (R)-1-фенилпропан-1-амина (0,115 г, 0,563 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли карбонат цезия (0,440 г, 1,35 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 17 ч. Реакционную смесь разводили этилацетатом (5 мл) и водой (5 мл) и водную фазу экстрагировали этилацетатом (3×3 мл). Объединенные органические фазы промывали солевым раствором (1×5 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo, растворяли в дихлорметане (5 мл) и туда добавляли трифторуксусную кислоту (1 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч и затем туда добавляли метанол (10 мл). Суспензию фильтровали и фильтрат концентрировали in vacuo. Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 12 до 80% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,026 г, выход 9%):1H ЯМР (300 МГц, DMSO-d6) δ 8,38 (с, 1H), 7,54 (д, J=2,1 Гц, 1H), 7,38-7,34 (м, 3H), 7,31-7,24 (м, 2H), 7,20-7,13 (м, 1H), 6,60 (д, J=9,0 Гц, 1H), 6,26 (д, J=7,5 Гц, 1H), 4,39 (к, J=7,5 Гц, 1H), 2,01-1,90 (м, 1H), 1,83-1,66 (м, 1H), 0,86 (т, J=7,2 Гц, 3H), NH сульфонамида не наблюдали; MS (ES-) m/z 407,0 (M - 1), 409,0 (M - 1).
ПРИМЕР 32
Синтез (S)-3-хлор-4-((1-фенилпропил)амино)-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида

Следуя процедуре как описано для примера 31 и выполняя некритические вариации при необходимости, чтобы заменять (R)-1-фенилпропан-1-амин на (S)-1-фенилпропан-1-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,118 г, выход 43%):1H ЯМР (300 МГц, DMSO-d6) δ 8,38 (с, 1H), 7,54 (д, J=2,1 Гц, 1H), 7,38-7,34 (м, 3H), 7,30-7,25 (м, 2H), 7,20-7,13 (м, 1H), 6,60 (д, J=9,0 Гц, 1H), 6,26 (д, J=7,5 Гц, 1H), 4,39 (к, J=7,2 Гц, 1H), 2,01-1,89 (м, 1H), 1,82-1,68 (м, 1H), 0,86 (т, J=7,2 Гц, 3H); NH сульфонамида не наблюдали; MS (ES-) m/z 407,0 (M - 1), 409,0 (M - 1).
ПРИМЕР 33
Синтез 4-(бензиламино)-3-хлор-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида

Следуя процедуре как описано для примера 31 и выполняя некритические вариации при необходимости, чтобы заменять (R)-1-фенилпропан-1-амин на бензиламин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,086 г, выход 40%):1H ЯМР (300 МГц, DMSO-d6) δ 8,40 (с, 1H), 7,55 (д, J=2,1 Гц, 1H), 7,39 (дд, J=8,7, 2,1 Гц, 1H), 7,31-7,25 (м, 4H), 7,22-7,15 (м, 1H), 7,02 (т, J=6,0 Гц, 1H), 6,58 (д, J=9,9 Гц, 1H), 4,43 (д, J=6,0 Гц, 2H), NH сульфонамида не наблюдали; MS (ES+) m/z 381,0 (M+1), 383,0 (M+1).
ПРИМЕР 34
Синтез 3-хлор-4-((2-фенилпропил)амино)-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида

Следуя процедуре как описано для примера 31 и выполняя некритические вариации при необходимости, чтобы заменять (R)-1-фенилпропан-1-амин на 2-фенилпропан-1-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,088 г, выход 38%):1H ЯМР (300 МГц, DMSO-d6) δ 8,42 (с, 1H), 7,52-7,47 (м, 2H), 7,31-7,27 (м, 4H), 7,19-7,13 (м, 1H), 6,82 (д, J=8,7 Гц, 1H), 6,04 (т, J=5,7 Гц, 1H), 3,32 (м, 2H), 3,12-2,98 (м, 1H), 1,19 (д, J=6,9 Гц, 3H), NH сульфонамида не наблюдали; MS (ES+) m/z 409,0 (M+1), 411,0 (M+1).
ПРИМЕР 35
Синтез (R)-3-хлор-4-((2,3-дигидро-1H-инден-1-ил)амино)-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида

Следуя процедуре как описано для примера 31 и выполняя некритические вариации при необходимости, чтобы заменять (R)-1-фенилпропан-1-амин на (R)-2,3-дигидро-1H-инден-1-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,046 г, выход 20%):1H ЯМР (300 МГц, DMSO-d6) δ 8,46 (с, 1H), 7,62 (д, J=2,1 Гц, 1H), 7,57 (дд, J=8,7, 2,1 Гц, 1H), 7,30-7,15 (м, 4H), 7,07 (д, J=9,0 Гц, 1H), 6,18 (д, J=8,1 Гц, 1H), 5,20 (к, J=7,8 Гц, 1H), 3,03-2,94 (м, 1H), 2,90-2,79 (м, 1H), 2,56-2,45 (м, 1H), 2,08-1,96 (м, 1H), NH сульфонамида не наблюдали; MS (ES-) m/z 405,0 (M - 1), 407,0 (M - 1).
ПРИМЕР 36
Синтез (S)-3-хлор-4-((2,3-дигидро-1H-инден-1-ил)амино)-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида

Следуя процедуре как описано для примера 31 и выполняя некритические вариации при необходимости, чтобы заменять (R)-1-фенилпропан-1-амин на (S)-2,3-дигидро-1H-инден-1-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,061 г, выход 27%):1H ЯМР (300 МГц, DMSO-d6) δ 8,46 (с, 1H), 7,62 (д, J=2,1 Гц, 1H), 7,57 (дд, J=8,7, 2,1 Гц, 1H), 7,31-7,17 (м, 4H), 7,06 (д, J=9,0 Гц, 1H), 6,17 (д, J=8,1 Гц, 1H), 5,20 (к, J=7,8 Гц, 1H), 3,03-2,93 (м, 1H), 2,90-2,78 (м, 1H), 2,56-2,45 (м, 1H), 2,08-1,95 (м, 1H), NH сульфонамида не наблюдали; MS (ES-) m/z 405,0 (M - 1), 407,0 (M - 1).
ПРИМЕР 37
Синтез (R)-5-хлор-2-фтор-4-((1-фенилэтил)амино)-N-(тиазол-2-ил)бензолсульфонамида
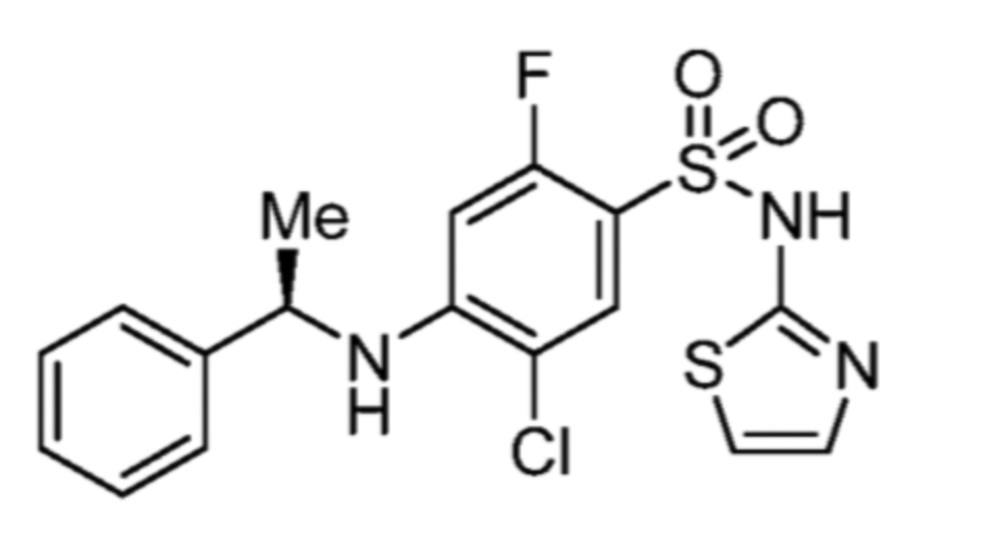
В смесь 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамида (0,250 г, 0,543 ммоль) и (R)-1-фенилэтан-1-амина (0,065 мг, 0,54 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли карбонат цезия (0,424 г, 1,30 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 17 ч. Реакционную смесь разводили этилацетатом (5 мл) и водой (5 мл) и водную фазу экстрагировали этилацетатом (3×3 мл). Объединенные органические фазы промывали солевым раствором (5 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo, остаток растворяли в дихлорметане (5 мл) и туда добавляли трифторуксусную кислоту (1 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч и затем туда добавляли метанол (10 мл). Суспензию фильтровали и фильтрат концентрировали in vacuo. Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 12 до 80% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,111 г, выход 50%):1H ЯМР (300 МГц, DMSO-d6) δ 12,73 (с, 1H), 7,58 (д, J=7,2 Гц, 1H), 7,42-7,35 (м, 2H), 7,34-7,29 (м, 2H), 7,25-7,17 (м, 2H), 6,81 (д, J=4,5 Гц, 1H), 6,49 (дд, J=7,2, 1,5 Гц, 1H), 6,39 (д, J=13,2 Гц, 1H), 4,69 (дк, J=7,2, 6,9 Гц, 1H), 1,52 (д, J=6,9 Гц, 3H); MS (ES-) m/z 410,0 (M - 1), 412,0 (M - 1).
ПРИМЕР 38
Синтез (S)-5-хлор-2-фтор-4-((1-фенилэтил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 37 и выполняя некритические вариации при необходимости, чтобы заменять (R)-1-фенилэтан-1-амин на (S)-1-фенилэтан-1-амин, и очисткой с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,062 г, выход 28%):1H ЯМР (300 МГц, DMSO-d6) δ 12,74 (с, 1H), 7,58 (д, J=7,5 Гц, 1H), 7,43-7,36 (м, 2H), 7,36-7,29 (м, 2H), 7,27-7,16 (м, 2H), 6,81 (д, J=4,5 Гц, 1H), 6,49 (дд, J=7,2, 1,2 Гц, 1H), 6,39 (д, J=13,2 Гц, 1H), 4,69 (дк, J=7,2, 6,9 Гц, 1H), 1,52 (д, J=6,9 Гц, 3H); MS (ES-) m/z 410,0 (M - 1), 412,0 (M - 1).
ПРИМЕР 39
Синтез (R)-5-хлор-2-фтор-4-((1-фенилпропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 37 и выполняя некритические вариации при необходимости, чтобы заменять (R)-1-фенилэтан-1-амин на (R)-1-фенилпропан-1-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,116 г, выход 50%):1H ЯМР (300 МГц, DMSO-d6) δ 12,74 (с, 1H), 7,56 (д, J=7,5 Гц, 1H), 7,44-7,38 (м, 2H), 7,35-7,26 (м, 2H), 7,26-7,17 (м, 2H), 6,81 (д, J=4,5 Гц, 1H), 6,49-6,45 (м, 1H), 6,44 (д, J=13,2 Гц, 1H), 4,42 (к, J=7,2 Гц, 1H), 2,07-1,90 (м, 1H), 1,84-1,60 (м, 1H), 0,88 (т, J=7,2 Гц, 3H); MS (ES-) m/z 424,1 (M - 1), 426,1 (M - 1).
ПРИМЕР 40
Синтез (S)-5-хлор-2-фтор-4-((1-фенилпропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 37 и выполняя некритические вариации при необходимости, чтобы заменять (R)-1-фенилэтан-1-амин на (S)-1-фенилпропан-1-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,134 г, выход 58%):1H ЯМР (300 МГц, DMSO-d6) δ 12,73 (с, 1H), 7,56 (д, J=7,5 Гц, 1H), 7,44-7,38 (м, 2H), 7,35-7,26 (м, 2H), 7,25-7,17 (м, 2H), 6,81 (д, J=4,5 Гц, 1H), 6,46-6,43 (м, 1H), 6,44 (д, J=13,2 Гц, 1H), 4,42 (к, J=7,2 Гц, 1H), 2,07-1,90 (м, 1H), 1,84-1,60 (м, 1H), 0,88 (т, J=7,2 Гц, 3H); MS (ES-) m/z 424,1 (M - 1), 426,1 (M - 1).
ПРИМЕР 41
Синтез 5-хлор-2-фтор-N-(тиазол-2-ил)-4-((3,3,3-трифтор-1-фенилпропил)амино)бензолсульфонамида

Следуя процедуре как описано для примера 37 и выполняя некритические вариации при необходимости, чтобы заменять (R)-1-фенилэтан-1-амин на 3,3,3-трифтор-1-фенилпропан-1-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,062 г, выход 24%)1H ЯМР (300 МГц, DMSO-d6) δ 12,76 (с, 1H), 7,58 (д, J=7,5 Гц, 1H), 7,52 (д, J=7,2 Гц, 2H), 7,38-7,30 (м, 2H), 7,29-7,21 (м, 2H), 6,85 (д, J=7,5 Гц, 1H), 6,81 (д, J=4,5 Гц, 1H), 6,68 (д, J=12,9 Гц, 1H), 5,06-4,94 (м, 1H), 3,39-3,18 (м, 1H), 2,85-2,62 (м, 1H);19F ЯМР (282 МГц, DMSO-d6) δ -62,1 (с, 3F), -109,2 (с, 1F); MS (ES-) m/z 478,1 (M - 1), 480,1 (M - 1).
ПРИМЕР 42
Синтез (S)-5-хлор-4-((1-циклогексилэтил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 37 и выполняя некритические вариации при необходимости, чтобы заменять (R)-1-фенилэтан-1-амин на (S)-1-циклогексилэтан-1-амин, и очисткой с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,007 г, выход 3%);1H ЯМР (300 МГц, DMSO-d6) δ 12,76 (с, 1H), 7,55 (д, J=7,5 Гц, 1H), 7,26 (д, J=4,8 Гц, 1H), 6,82 (д, J=4,5 Гц, 1H), 6,68 (д, J=13,5 Гц, 1H), 5,67 (д, J=9,0 Гц, 1H), 3,51-3,34 (м, 1H), 1,80-1,41 (м, 5H), 1,26-1,03 (м, 3H), 1,10 (д, J=6,3 Гц, 3H), 1,01-0,82 (м, 2H); NH сульфонамида не наблюдали;19F ЯМР (282 МГц, DMSO-d6) δ -109,1 (с); MS (ES-) m/z 416,1 (M - 1), 418,1 (M - 1).
ПРИМЕР 43
Синтез 5-хлор-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

В раствор 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамида (0,250 г, 0,543 ммоль) и 1-фенилциклопропан-1-амина (0,072 мг, 0,543 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли карбонат цезия (0,424 г, 1,30 ммоль) и реакционная смесь находилась при температуре окружающей среды в течение 17 ч. Реакционную смесь разводили этилацетатом (5 мл) и водой (5 мл) и водную фазу экстрагировали этилацетатом (3×3 мл). Объединенные органические фазы промывали солевым раствором (5 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 12 до 80% этилацетата, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,253 г, выход 81%):1H ЯМР (300 МГц, CDCl3) δ 7,71 (д, J=6,6 Гц, 1H), 7,40-7,23 (м, 3H), 7,22-7,13 (м, 2H), 7,05 (д, J=7,5 Гц, 2H), 6,97-6,89 (м, 1H), 6,41 (д, J=12,3 Гц, 1H), 6,34 (д, J=8,4 Гц, 1H), 6,31 (с, 1H), 5,67 (с, 1H), 5,16 (с, 2H), 3,74 (с, 3H), 3,67 (с, 3H), 1,42 (с, 2H), 1,37 (с, 2H).
Стадия 2. Получение 5-хлор-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

В смесь 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамида в дихлорметане (5 мл) добавляли трифторуксусную кислоту (1 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Метанол (10 мл) добавляли в смесь и получаемую суспензию фильтровали. Фильтрат концентрировали in vacuo и остаток очищали посредством растирания с метанолом (3×5 мл) для того, чтобы получать бесцветное твердое вещество (0,075 г, выход 40%):1H ЯМР (300 МГц, DMSO-d6) δ 12,76 (с, 1H), 7,61 (д, J=7,2 Гц, 1H), 7,56-7,50 (м, 1H), 7,33-7,23 (м, 3H), 7,20-7,09 (м, 3H), 6,82 (д, J=4,5 Гц, 1H), 6,37 (д, J=12,0 Гц, 1H), 1,44-1,33 (м, 2H), 1,33-1,22 (м, 2H);19F ЯМР (282 МГц, DMSO-d6) δ -109,7 (с); MS (ES-) m/z 422,0 (M - 1), 424,0 (M - 1).
ПРИМЕР 44
Синтез 5-хлор-2-фтор-4-((1-(4-фторфенил)циклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(4-фторфенил)циклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на 1-(4-фторфенил)циклопропан-1-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,246 г, выход 77%): MS (ES+) m/z 592,4 (M+1), 594,4 (M+1).
Стадия 2. Получение 5-хлор-2-фтор-4-((1-(4-фторфенил)циклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамида
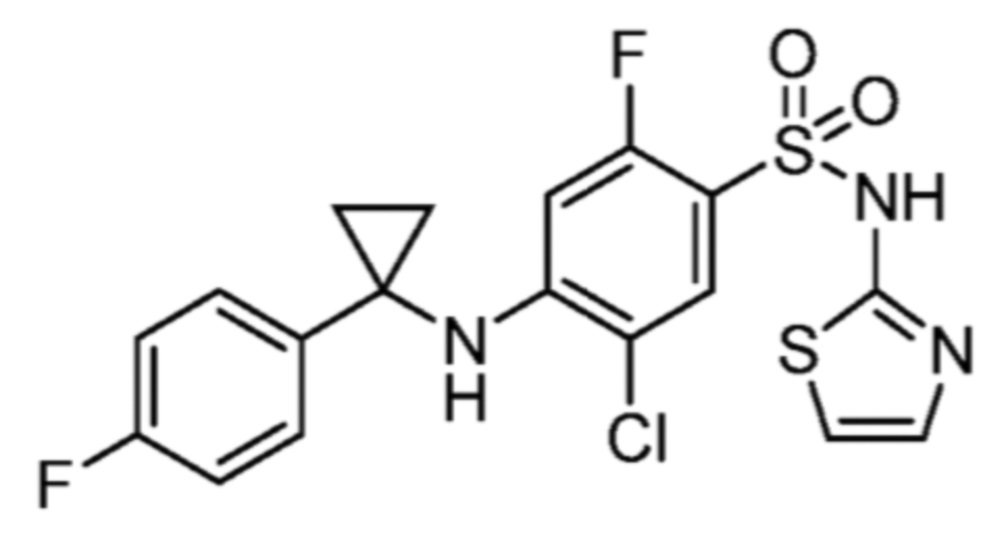
Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(4-фторфенил)циклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,095 г, выход 40%):1H ЯМР (300 МГц, DMSO-d6) δ 12,77 (с, 1H), 7,60 (д, J=7,2 Гц, 1H), 7,55 (д, J=1,2 Гц, 1H), 7,26 (д, J=4,2 Гц, 1H), 7,23-7,15 (м, 2H), 7,14-7,05 (м, 2H), 6,82 (д, J=4,5 Гц, 1H), 6,40 (д, J=12,9 Гц, 1H), 1,41-1,33 (м, 2H), 1,30-1,22 (м, 2H);19F ЯМР (282 МГц, DMSO-d6) δ -109,5 (с, 1F), -117,3 (с, 1F); MS (ES+) m/z 441,9 (M+1), 443,9 (M+1).
ПРИМЕР 45
Синтез (S)-5-хлор-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1: (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на (S)-1-(2-фторфенил)этан-1-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,257 г, выход 82%): MS (ES+) m/z 580,1 (M+1), 582,1 (M+1).
Стадия 2: (S)-5-хлор-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамид

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамид, растиранием с ацетонитрилом (2×1 мл) получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,037 г, выход 20%):1H ЯМР (300 МГц, DMSO-d6) δ 12,76 (с, 1H), 7,60 (д, J=7,2 Гц, 1H), 7,45-7,36 (м, 1H), 7,34-7,16 (м, 4H), 6,81 (д, J=4,8 Гц, 1H), 6,49 (д, J=6,6 Гц, 1H), 6,32 (д, J=12,9 Гц, 1H), 4,98-4,84 (м, 1H), 1,56 (д, J=6,9 Гц, 3H);19F ЯМР (282 МГц, DMSO-d6) δ -109,2 (с, 1F), -119,7 (с, 1F); MS (ES-) m/z 428,1 (M - 1), 430,1 (M - 1).
ПРИМЕР 46
Синтез (S)-5-хлор-2-фтор-4-((1,2,3,4-тетрагидронафтален-1-ил)амино)-N-(тиазол-2-ил)бензолсульфонамида

В смесь (S)-1,2,3,4-тетрагидронафтален-1-амина (0,096 г, 0,65 ммоль) и 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамида (0,300 г, 0,652 ммоль) в безводном диметилсульфоксиде (2,6 мл) добавляли карбонат цезия (0,509 г, 1,56 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 17 ч. Реакционную смесь разводили этилацетатом (5 мл) и водой (5 мл) и водную фазу экстрагировали этилацетатом (3×3 мл). Объединенные органические фазы промывали солевым раствором (5 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 30% этилацетата в гексанах. Затем получаемый материал растворяли в дихлорметане (5 мл) и туда добавляли трифторуксусную кислоту (0,5 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 10 минут, концентрировали in vacuo и туда добавляли метанол. Суспензию фильтровали и фильтрат концентрировали in vacuo. Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 10% метанола в дихлорметане, за которой следовала очистка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,011 г, выход 4%):1H ЯМР (300 МГц, DMSO-d6) δ 12,79 (с, 1H), 7,60 (д, J=7,0 Гц, 1H), 7,28 (с, 1H), 7,15 (с, 4H), 6,91-6,79 (м, 2H), 6,27 (д, J=8,2 Гц, 1H), 4,91-4,79 (м, 1H), 2,89-2,65 (м, 2H), 2,04-1,67 (м, 4H); MS (ES+) m/z 438,0, 440,0 (M+1).
ПРИМЕР 47
Синтез (S)-5-хлор-2-фтор-4-((1-(2-фторфенил)пропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(2-фторфенил)пропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на гидрохлорид (S)-1-(2-фторфенил)пропан-1-амина, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,265 г, выход 82%): MS (ES+) m/z 594,2 (M+1), 596,2 (M+1).
Стадия 2. Получение (S)-5-хлор-2-фтор-4-((1-(2-фторфенил)пропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(2-фторфенил)пропил)амино)-N-(тиазол-2-ил)бензолсульфонамид, очисткой посредством перекристаллизации из ацетонитрила (5 мл) получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,045 г, выход 23%):1H ЯМР (300 МГц, DMSO-d6) δ 12,76 (с, 1H), 7,59 (д, J=6,9 Гц, 1H), 7,50-7,41 (м, 1H), 7,35-7,12 (м, 4H), 6,82 (д, J=3,6 Гц, 1H), 6,78 (д, J=7,8 Гц, 1H), 6,36 (д, J=12,9 Гц, 1H), 4,66 (к, J=7,2 Гц, 1H), 2,13-1,95 (м, 1H), 1,90-1,75 (м, 1H), 1,56 (д, J=7,2 Гц, 3H);19F ЯМР (282 МГц, DMSO-d6) δ -109,16 (с, 1F), -119,67 (с, 1F); MS (ES-) m/z 442,1 (M - 1), 444,1 (M - 1).
ПРИМЕР 48
Синтез (S)-5-хлор-2-фтор-4-((1-(4-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(4-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на (S)-1-(4-фторфенил)этан-1-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,235 г, выход 75%): MS (ES+) m/z 580,1 (M+1), 582,1 (M+1).
Стадия 2. Получение (S)-5-хлор-2-фтор-4-((1-(4-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(4-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамид, очисткой с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 6 до 80% этилацетата в гексанах, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,037 г, выход 20%):1H ЯМР (300 МГц, DMSO-d6) δ 12,75 (с, 1H), 7,57 (д, J=7,2 Гц, 1H), 7,45 (дд, J=8,7, 5,7 Гц, 2H), 7,24 (д, J=4,5 Гц, 1H), 7,14 (дд, J=9,0, 8,7 Гц, 2H), 6,81(д, J=4,5 Гц, 1H), 6,52 (д, J=6,9 Гц, 1H), 6,42 (д, J=13,2 Гц, 1H), 4,72 (дк, J=7,2, 6,9 Гц, 1H), 1,51 (д, J=6,9 Гц, 3H); MS (ES-) m/z 428,0 (M - 1), 430,0 (M - 1).
ПРИМЕР 49
Синтез (S)-5-хлор-2-фтор-4-((1-(3-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(3-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на (S)-1-(3-фторфенил)этан-1-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,220 г, выход 70%): MS (ES+) m/z 580,1 (M+1), 582,0 (M+1).
Стадия 2. Получение (S)-5-хлор-2-фтор-4-((1-(3-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(3-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамид, перекристаллизацией из ацетонитрила (10 мл) получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,119 г, выход 51%):1H ЯМР (300 МГц, DMSO-d6) δ 12,76 (с, 1H), 7,58 (д, J=6,0 Гц, 1H), 7,43-7,31 (м, 1H), 7,31-7,20 (м, 3H), 7,10-6,97 (м, 1H), 6,85-6,77 (м, 1H), 6,56 (д, J=6,6 Гц, 1H), 6,44 (д, J=12,6 Гц, 1H), 4,81-4,67 (м, 1H), 1,52 (д, J=5,4 Гц, 3H); MS (ES-) m/z 428,0 (M - 1), 430,0 (M - 1).
ПРИМЕР 50
Синтез (S)-5-хлор-4-((1-(3-хлорфенил)этил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение (S)-5-хлор-4-((1-(3-хлорфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на (S)-1-(3-хлорфенил)этан-1-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,280 г, выход 87%): MS (ES+) m/z 595,1 (M+1), 597,9 (M+1).
Стадия 2. Получение (S)-5-хлор-4-((1-(3-хлорфенил)этил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на (S)-5-хлор-4-((1-(3-хлорфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,152 г, выход 63%):1H ЯМР (300 МГц, DMSO-d6) δ 12,76 (с, 1H), 7,59 (д, J=7,2 Гц, 1H), 7,53-7,50 (м, 1H), 7,41-7,31 (м, 2H), 7,31-7,26 (м, 1H), 7,25 (д, J=4,8 Гц, 1H), 6,81 (д, J=4,8 Гц, 1H), 6,59 (дд, J=7,5, 1,2 Гц, 1H), 6,46 (д, J=13,2 Гц, 1H) 4,78-4,69 (м, 1H), 1,51 (д, J=6,9 Гц, 3H); MS (ES-) m/z 443,9 (M - 1), 446,0 (M - 1).
ПРИМЕР 51
Синтез (S)-5-хлор-4-((1-(3,5-дихлорфенил)этил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение (S)-5-хлор-4-((1-(3,5-дихлорфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на (S)-1-(3,5--дихлорфенил)этан-1-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,234 г, выход 71%): MS (ES+) m/z 630,0 (M+1), 632,0 (M+1).
Стадия 2: (S)-5-хлор-4-((1-(3,5-дихлорфенил)этил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на (S)-5-хлор-4-((1-(3,5-дихлорфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,152 г, выход 63%):1H ЯМР (300 МГц, DMSO-d6) δ 12,77 (с, 1H), 7,59 (д, J=7,5 Гц, 1H), 7,54 (д, J=1,8 Гц, 2H), 7,46 (т, J=1,8 Гц, 1H), 7,25 (д, J=4,5 Гц, 1H), 6,82 (д, J=4,5 Гц, 1H), 6,66 (д, J=13,3 Гц, 1H), 6,53 (д, J=12,9 Гц, 1H), 4,83-4,68 (м, 1H), 1,51 (д, J=6,9 Гц, 3H); MS (ES+) m/z 479,7 (M+1); 481,7 (M+1).
ПРИМЕР 52
Синтез (S)-5-хлор-4-((1-(2,4-дифторфенил)этил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение (S)-5-хлор-4-((1-(2,4-дифторфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на (S)-1-(2,4-дифторфенил)этан-1-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,220 г, выход 70%): MS (ES+) m/z 598,0 (M+1), 600,0 (M+1).
Стадия 2. Получение (S)-5-хлор-4-((1-(2,4-дифторфенил)этил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на (S)-5-хлор-4-((1-(2,4-дифторфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,088 г, выход 36%):1H ЯМР (300 МГц, DMSO-d6) δ 12,78 (с, 1H), 7,60 (д, J=7,2 Гц, 1H), 7,44 (дт, J=6,9, 8,7 Гц, 1H), 7,29-7,18 (м, 2H), 7,05 (дт, J=2,1, 8,4 Гц, 1H), 6,82 (д, J=3,2 Гц, 1H), 6,52 (д, J=8,7 Гц, 1H), 6,36 (д, J=12,9 Гц, 1H), 4,94-4,85 (м, 1H), 1,54 (д, J=6,9 Гц, 3H);19F ЯМР (282 МГц, DMSO-d6) δ -109,1 (с, 1F), -111,7 (д, J=7,3 Гц, 1F), -115,3 (д, J=7,3 Гц, 1F); MS (ES-) m/z 446,0 (M - 1), 447,9 (M - 1).
ПРИМЕР 53
Синтез (S)-5-хлор-4-((1-(3,4-дифторфенил)этил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение (S)-5-хлор-4-((1-(3,4-дифторфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на гидрохлорид (S)-1-(3,4-дифторфенил)этан-1-амина, указанное в заголовке соединение получали в виде бесцветного твердого вещества (257 г, выход 79%): MS (ES+) m/z 598,4 (M+1), 600,4 (M+1).
Стадия 2. Получение (S)-5-хлор-4-((1-(3,4-дифторфенил)этил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на (S)-5-хлор-4-((1-(3,4-дифторфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,088 г, выход 36%):1H ЯМР (300 МГц, DMSO-d6) δ 12,77 (с, 1H), 7,58 (д, J=7,2 Гц, 1H), 7,52 (ддд, J=12,0, 8,1, 2,1 Гц, 1H), 7,42-7,31 (м, 1H), 7,31-7,27 (м, 1H), 7,25 (д, J=4,5 Гц, 1H), 6,81 (д, J=4,8 Гц, 1H), 6,57 (дд, J=7,8, 1,2 Гц, 1H), 6,48 (д, J=13,2 Гц, 1H) 4,80-4,65 (м, 1H), 1,51 (д, J=6,9 Гц, 3H);19F ЯМР (282 МГц, DMSO-d6) δ -109,22 (с, 1F), -138,21 (д, J=22 Гц, 1F), -140,82 (д, J=23,7 Гц, 1F); MS (ES+) m/z 447,9 (M+1), 449,9 (M+1).
ПРИМЕР 54
Синтез (S)-5-хлор-2-фтор-N-(тиазол-2-ил)-4-((1-(о-толил)пропил)амино)бензолсульфонамида

Стадия 1. Получение (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)-4-((1-(о-толил)пропил)амино)бензолсульфонамида
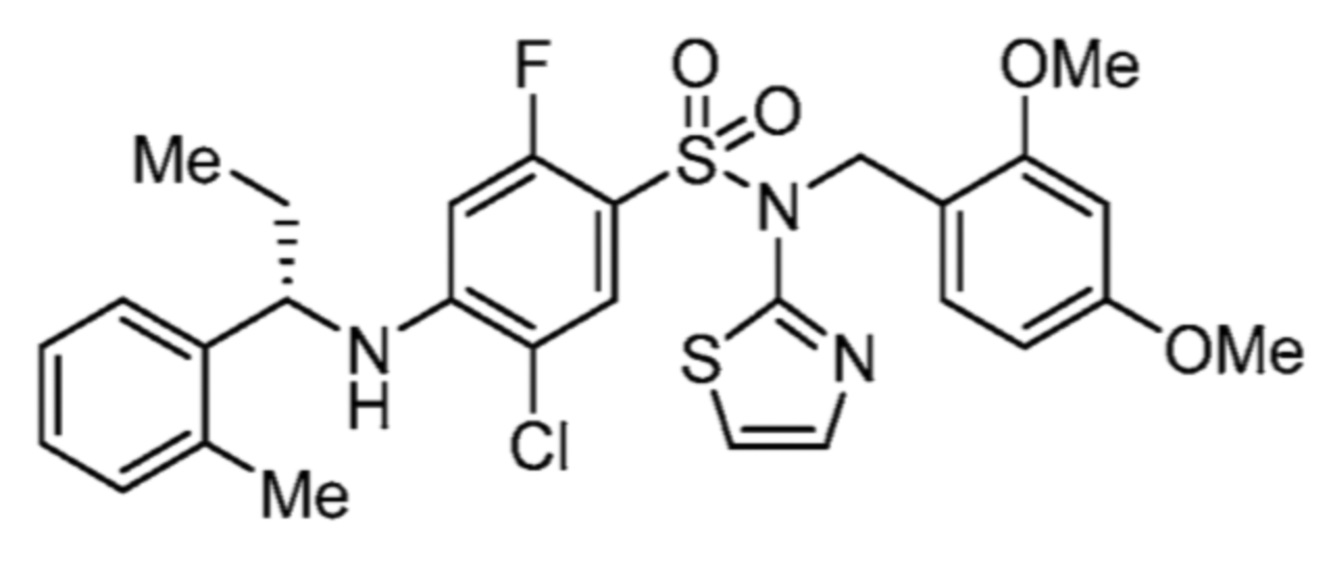
Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на гидрохлорид (S)-1-(о-толил)пропан-1-амина, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,096 г, выход 30%): MS (ES+) m/z 590,0 (M+1), 592,0 (M+1).
Стадия 2. Получение (S)-5-хлор-2-фтор-N-(тиазол-2-ил)-4-((1-(о-толил)пропил)амино)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)-4-((1-(о-толил)пропил)амино)бензол-сульфонамид, очисткой с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,010 г, выход 4%):1H ЯМР (300 МГц, DMSO-d6) δ 12,74 (с, 1H), 7,58 (д, J=7,2 Гц, 1H), 7,31-7,22 (м, 2H), 7,20-7,08 (м, 3H), 6,81 (д, J=4,5 Гц, 1H), 6,40 (д, J=7,5 Гц, 1H), 6,11 (д, J=13,2 Гц, 1H), 4,58-4,47 (м, 1H), 2,40 (с, 3H), 2,02-1,86 (м, 1H), 1,80-1,64 (м, 1H), 0,97 (д, J=7,2 Гц, 3H); MS (ES-) m/z 438,0 (M - 1), 440,0 (M - 1).
ПРИМЕР 55
Синтез 5-хлор-2-фтор-4-((1-(5,6,7,8-тетрагидронафтален-2-ил)пропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(5,6,7,8-тетрагидронафтален-2-ил)пропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на 1-(5,6,7,8-тетрагидронафтален-2-ил)пропан-1-амин, указанное в заголовке соединение получали в виде бесцветного масла (0,272 г, выход 80%): MS (ES+) m/z 630,2 (M+1), 632,2 (M+1).
Стадия 2. Получение 5-хлор-2-фтор-4-((1-(5,6,7,8-тетрагидронафтален-2-ил)пропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(5,6,7,8-тетрагидронафтален-2-ил)пропил)амино)-N-(тиазол-2-ил)бензолсульфонамид, очисткой с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 12 до 80% этилацетата в гексанах, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,036 г, выход 14%):1H ЯМР (300 МГц, DMSO-d6) δ 12,74 (с, 1H), 7,56 (д, J=7,5 Гц, 1H), 7,24 (д, J=4,8 Гц, 1H), 7,13-7,05 (м, 2H), 6,97 (д, J=8,4 Гц, 1H), 6,80 (д, J=4,8 Гц, 1H), 6,44 (д, J=13,2 Гц, 1H), 6,37 (дд, J=7,8, 1,2 Гц, 1H), 4,30 (к, J=6,9 Гц, 1H), 2,73-2,56 (м, 4H), 2,03-1,86 (м, 1H), 1,78-1,60 (м, 5H), 0,87 (т, J=7,2 Гц, 3H);19F ЯМР (282 МГц, DMSO-d6) δ -109,3 (с); MS (ES-) m/z 478,1 (M - 1), 480,1 (M - 1).
ПРИМЕР 56
Синтез 5-хлор-2-фтор-4-((1-(пиридин-3-ил)пропан-2-ил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(пиридин-3-ил)пропан-2-ил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на 1-(пиридин-3-ил)пропан-2-амин, указанное в заголовке соединение получали в виде бесцветного масла (0,248 г, выход 79%): MS (ES+) m/z 577,1 (M+1), 579,0 (M+1).
Стадия 2. Получение 5-хлор-2-фтор-4-((1-(пиридин-3-ил)пропан-2-ил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(пиридин-3-ил)пропан-2-ил)амино)-N-(тиазол-2-ил)бензолсульфонамид, очисткой с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 12 до 80% этилацетата в гексанах, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,127 г, выход 53%):1H ЯМР (300 МГц, DMSO-d6) δ 12,78 (с, 1H), 8,65 (д, J=1,8 Гц, 1H), 8,60 (дд, J=5,4, 1,4 Гц, 1H), 8,15-8,12 (м, 1H), 7,69 (дд, J=7,8, 5,4 Гц, 1H), 7,53 (д, J=7,4 Гц, 1H), 7,27 (д, J=4,6 Гц, 1H), 6,83 (д, J=4,6 Гц, 1H), 6,69 (д, J=13,5 Гц, 1H), 6,04 (ддд, J=9,3, 1,4, 0,5 Гц, 1H), 4,03-3,93 (м, 1H), 3,07 (дд, J=13,8, 1,8 Гц, 1H), 2,94 (дд, J=13,5, 1,8 Гц, 1H), 1,18 (д, J=6,3 Гц, 3H); MS (ES+) m/z 426,9 (M+1), 428,9 (M+1).
ПРИМЕР 57
Синтез 5-хлор-2-фтор-4-((1-(пиридин-3-ил)пропан-2-ил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((5,6,7,8-тетрагидрохинолин-6-ил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на 5,6,7,8-тетрагидрохинолин-6-амин, указанное в заголовке соединение получали в виде бесцветного масла (0,180 г, выход 56%): MS (ES+) m/z 589,0 (M+1), 591,0 (M+1).
Стадия 2. Получение 5-хлор-2-фтор-4-((1-(пиридин-3-ил)пропан-2-ил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((5,6,7,8-тетрагидрохинолин-6-ил)амино)-N-(тиазол-2-ил)бензолсульфонамида, очисткой с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 12 до 80% этилацетата в гексанах, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,043 г, выход 19%):1H ЯМР (300 МГц, DMSO-d6) δ 12,82 (с, 1H), 8,62 (д, J=5,1 Гц, 1H), 8,13 (д, J=7,8 Гц, 1H), 7,69 (дд, J=7,8, 5,7 Гц, 1H), 7,62 (д, J=7,5 Гц, 1H), 7,27 (д, J=4,5 Гц, 1H), 6,90 (д, J=13,2 Гц, 1H), 6,84 (д, J=4,5 Гц, 1H), 6,22 (д, J=7,8 Гц, 1H), 3,98-3,90 (м, 1H), 3,26-3,14 (м, 3H), 3,08-2,98 (м, 1H), 2,18-2,11 (м, 1H), 1,99-1,85 (м, 1H); MS (ES+) m/z 438,9 (M+1), 440,9 (M+1).
ПРИМЕР 58
Синтез 5-хлор-2-фтор-4-((1-(пиридин-3-ил)пропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(пиридин-3-ил)пропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на 1-(пиридин-3-ил)пропан-1-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,215 г, выход 69%):1H ЯМР (300 МГц, CDCl3) δ 8,64-8,53 (м, 2H), 7,72 (д, J=7,2 Гц, 1H), 7,70-7,61 (м, 1H), 7,46-7,38 (м, 1H), 7,36 (д, J=4,5 Гц, 1H), 7,17 (д, J=8,4 Гц, 1H), 6,95 (дд, J=3,6, 0,6 Гц, 1H), 6,35 (дд, J=8,1, 2,1 Гц, 1H), 6,32-6,29 (м, 1H), 6,02 (д, J=12,0 Гц, 1H), 5,26 (д, J=5,1 Гц, 1H), 5,13 (с, 2H), 4,30 (к, J=7,2 Гц, 1H), 3,76 (с, 3H), 3,67 (с, 3H), 1,99-1,85 (м, 2H), 1,04 (т, J=7,5 Гц, 3H); MS (ES+) m/z 577,0 (M+1), 579,0 (M+1).
Стадия 2. Получение 5-хлор-2-фтор-4-((1-(пиридин-3-ил)пропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(пиридин-3-ил)пропил)амино)-N-(тиазол-2-ил)бензолсульфонамид, растиранием с ацетонитрилом (3×объем), указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,159 г, выход 69%):1H ЯМР (300 МГц, DMSO-d6) δ 12,76 (с, 1H), 8,85 (с, 1H), 8,68-8,59 (м, 1H), 8,26 (д, J=7,8 Гц, 1H), 7,76-7,66 (м, 1H), 7,61 (д, J=6,9 Гц, 1H), 7,29-7,21 (м, 1H), 6,86-6,77 (м, 1H), 6,67 (д, J=8,4 Гц, 1H), 6,62 (д, J=13,5 Гц, 1H), 4,66 (к, J=6,9 Гц, 1H), 2,13-1,96 (м, 1H), 1,93-1,75 (м, 1H), 0,91 (т, J=6,9 Гц, 3H); MS (ES+) m/z 427,0 (M+1), 427,9 (M+1).
ПРИМЕР 59
Синтез 5-хлор-2-фтор-4-((1-(пиридин-3-ил)пропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(пиридин-3-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на (S)-1-(пиридин-3-ил)этан-1-амин, очисткой с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 12 до 80% этилацетата в гексанах, указанное в заголовке соединение получали в виде бесцветного масла (0,145 г, выход 47%): MS (ES+) m/z 563,0 (M+1), 565,0 (M+1).
Стадия 2. Получение 5-хлор-2-фтор-4-((1-(пиридин-3-ил)пропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(пиридин-3-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамид, растиранием с ацетонитрилом (3×5 мл), указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,080 г, выход 28%):1H ЯМР (300 МГц, DMSO-d6) δ 12,78 (с, 1H), 8,82 (д, J=1,5 Гц, 1H), 8,64 (д, J=4,5 Гц, 1H), 8,23 (д, J=8,1 Гц, 1H), 7,71 (дд, J=7,8, 2,4 Гц, 1H), 7,60 (д, J=7,2 Гц, 1H), 7,26 (д, J=4,5 Гц, 1H), 6,82 (д, J=4,8 Гц, 1H), 6,70 (д, J=7,5 Гц, 1H), 6,57 (д, J=12,9 Гц, 1H), 4,92 (дк, J=6,9, 7,2 Гц, 1H), 1,58 (д, J=6,6 Гц, 3H); MS (ES+) m/z 412,9 (M+1), 414,9 (M+1).
ПРИМЕР 60
Синтез 5-хлор-2-фтор-4-((1-(пиридин-4-ил)пропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(пиридин-4-ил)пропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на 1-(пиридин-4-ил)пропан-1-амин, очисткой с помощью колоночной хроматографии, осуществляя элюирование с градиентом 5-25% метанола в дихлорметане, указанное в заголовке соединение получали в виде бесцветного масла (0,235 г, выход 75%):1H ЯМР (300 МГц, CDCl3) δ 8,59 (д, J=6,0 Гц, 2H), 7,70 (д, J=6,9 Гц, 1H), 7,35 (д, J=3,6 Гц, 1H), 7,19 (д, J=6,0 Гц, 2H), 7,15 (д, J=8,1 Гц, 1H), 6,93 (д, J=3,6 Гц, 1H), 6,33 (дд, J=8,4, 2,1 Гц, 1H), 6,30-6,26 (м, 1H), 5,96 (д, J=12,0 Гц, 1H), 5,26 (д, J=5,4 Гц, 1H), 5,11 (с, 2H), 4,21 (к, J=6,3 Гц, 1H), 3,73 (с, 3H), 3,65 (с, 3H), 1,97-1,82 (м, 2H), 1,01 (т, J=7,5 Гц, 3H); MS (ES+) m/z 577,0 (M+1), 579,0 (M+1).
Стадия 2. Получение 5-хлор-2-фтор-4-((1-(пиридин-4-ил)пропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(пиридин-4-ил)пропил)амино)-N-(тиазол-2-ил)бензолсульфонамид, растиранием с ацетонитрилом (3×объем), указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,077 г, выход 33%):1H ЯМР (300 МГц, DMSO-d6) δ 12,79 (с, 1H), 8,76 (д, J=5,7 Гц, 2H), 7,90 (д, J=6,3 Гц, 2H), 7,61 (д, J=7,2 Гц, 1H), 7,25 (д, J=4,5 Гц, 1H), 6,82 (д, J=4,5 Гц, 1H), 6,76 (д, J=7,8 Гц, 1H), 6,52 (д, J=13,2 Гц, 1H), 4,80-4,69 (м, 1H), 2,12-1,96 (м, 1H), 1,93-1,75 (м, 1H), 0,94 (т, J=7,2 Гц, 3H); MS (ES+) m/z 427,0 (M+1), 429,0 (M+1).
ПРИМЕР 61
Синтез 5-хлор-2-фтор-4-((изохинолин-8-илметил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((изохинолин-8-илметил)амино)-N-(тиазол-2-ил)бензолсульфонамида

В смесь 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамида (0,250 г, 0,543 ммоль) и изохинолин-8-илметанамина (0,086 мг, 0,54 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли карбонат цезия (0,424 г, 1,30 ммоль) и реакционную смесь нагревали при 90°C в течение 17 ч. Реакционную смесь оставляли остывать до температуры окружающей среды, разводили этилацетатом (5 мл) и водой (5 мл) и водную фазу экстрагировали этилацетатом (3×3 мл). Объединенные органические фазы промывали солевым раствором (1×5 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 6 до 80% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного масла (0,062 г, выход 19%): MS (ES+) m/z 599,0 (M+1), 601,0 (M+1).
Стадия 2. Получение 5-хлор-2-фтор-4-((изохинолин-8-илметил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((изохинолин-8-илметил)амино)-N-(тиазол-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,033 г, выход 13%):1H ЯМР (300 МГц, DMSO-d6) δ 12,77 (с, 1H), 9,87 (с, 1H), 8,66 (д, J=6,3 Гц, 1H), 8,24 (д, J=6,0 Гц, 1H), 8,06 (д, J=8,1 Гц, 1H), 7,94 (дд, J=7,2, 7,2 Гц, 1H), 7,63 (д, J=7,2 Гц, 2H), 7,33 (т, J=4,8 Гц, 1H), 7,26 (д, J=4,8 Гц, 1H), 6,82 (д, J=4,5 Гц, 1H), 6,59 (д, J=12,9 Гц, 1H), 5,13 (д, J=5,7 Гц, 2H); MS (ES+) m/z 449,0 (M+1), 451,0 (M+1).
ПРИМЕР 62
Синтез 5-хлор-2-фтор-N-(6-фторпиридин-2-ил)-4-((изохинолин-8-илметил)амино)бензолсульфонамида

Стадия 1. Получение 5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(6-фторпиридин-2-ил)-4-((изохинолин-8-илметил)амино)бензолсульфонамида

В смесь 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(6-фторпиридин-2-ил)бензолсульфонамида (0,256 г, 0,543 ммоль) и изохинолин-8-илметанамина (0,086 мг, 0,54 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли карбонат калия (0,233 г, 1,69 ммоль) и реакционную смесь нагревали до 110°C в течение 18 ч. Реакционную смесь оставляли остывать до температуры окружающей среды, разводили этилацетатом (5 мл) и водой (5 мл) и водную фазу экстрагировали этилацетатом (3×3 мл). Объединенные органические фазы промывали солевым раствором (1×5 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 6 до 80% этилацетата в гексанах, получали указанное в заголовке соединение в виде масла коричневого цвета (0,062 г, выход 19%): MS (ES+) m/z 611,1 (M+1), 613,0 (M+1).
Стадия 2. Получение 5-хлор-2-фтор-N-(6-фторпиридин-2-ил)-4-((изохинолин-8-илметил)амино)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на 5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(6-фторпиридин-2-ил)-4-((изохинолин-8-илметил)амино)бензолсульфонамид, очисткой с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 25% метанола в дихлорметане, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,044 г, выход 17%):1H ЯМР (300 МГц, DMSO-d6) δ 11,48 (с, 1H), 9,65 (с, 1H), 8,56 (д, J=5,7 Гц, 1H), 7,90-7,80 (м, 3H), 7,77 (д, J=7,5 Гц, 1H), 7,69 (дд, J=7,2, 7,2 Гц, 1H), 7,51-7,40 (м, 1H), 7,47 (д, J=6,9 Гц, 1H), 6,85 (дд, J=7,8, 2,1 Гц, 1H), 6,71 (дд, J=8,1, 2,4 Гц, 1H), 6,61 (д, J=13,2 Гц, 1H), 5,09 (д, J=5,7 Гц, 2H); MS (ES+) m/z 460,9 (M+1), 462,9 (M+1).
ПРИМЕР 63
Синтез 2,2,2-трифторацетата 5-хлор-4-((2-(диметиламино)-1-фенилэтил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение 5-хлор-N-(2,4-диметоксибензил)-4-((2-(диметиламино)-1-фенилэтил)амино-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на N1,N1-диметил-2-фенилэтан-1,2-диамин, указанное в заголовке соединение получали в виде бесцветного масла (0,158 г, выход 48%): MS (ES+) m/z 605,2 (M+1), 607,2 (M+1).
Стадия 2. Получение 2,2,2-трифторацетата 5-хлор-4-((2-(диметиламино)-1-фенилэтил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на 5-хлор-N-(2,4-диметоксибензил)-4-((2-(диметиламино)-1-фенилэтил)амино-2-фтор-N-(тиазол-2-ил)бензолсульфонамид, растиранием с ацетонитрилом (3×5 мл) указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,072 г, выход 29%):1H ЯМР (300 МГц, DMSO-d6) δ 7,63 (д, J=7,2 Гц, 1H), 7,48-7,34 (м, 4H), 7,33-7,28 (м, 1H), 7,26 (д, J=4,8 Гц, 1H), 6,99 (д, J=9,0 Гц, 1H), 6,89-6,77 (м, 2H), 5,32-5,19 (м, 1H), 3,84 (т, J=11,7 Гц, 1H), 3,33 (дд, J=13,5, 3,0 Гц, 1H), 2,87 (с, 6H); NH и COOH сульфонамида не наблюдали; MS (ES+) m/z 455,0 (M+1), 457,0 (M+1).
ПРИМЕР 64
Синтез 2,2,2-трифторацетата (S)-5-хлор-4-((2-(диметиламино)-1-фенилэтил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение гидрохлорида (S)-N1,N1-диметил-2-фенилэтан-1,2-диамина

К 2,2-диоксиду трет-бутил (S)-4-фенил-1,2,3-оксатиазолидин-3-карбоксилата (0,200 г, 0,67 ммоль, получен как описано в James et al., Org. Lett. 2013; 15 (23):6094-6097) добавляли 2 M раствор в метаноле (1,7 мл, 3,33 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 3 ч. Реакционную смесь концентрировали in vacuo, остаток растворяли в диоксане (3 мл) и туда добавляли 4 M раствор хлороводорода в диоксане (0,50 мл, 2,0 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 17 ч. Концентрированием in vacuo получали указанное в заголовке соединение в виде гигроскопического твердого вещества коричневатого цвета (0,080 г, выход 77%): MS (ES+) m/z 165,1 (M+1).
Стадия 2. Получение (S)-5-хлор-N-(2,4-диметоксибензил)-4-((2-(диметиламино)-1-фенилэтил)амино-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на гидрохлорид (S)-N1,N1-диметил-2-фенилэтан-1,2-диамина, указанное в заголовке соединение получали в виде бесцветного масла (0,085 г, выход 21%): MS (ES+) m/z 605,0 (M+1), 607,0 (M+1).
Стадия 3: 2,2,2-трифторацетат (S)-5-хлор-4-((2-(диметиламино)-1-фенилэтил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида
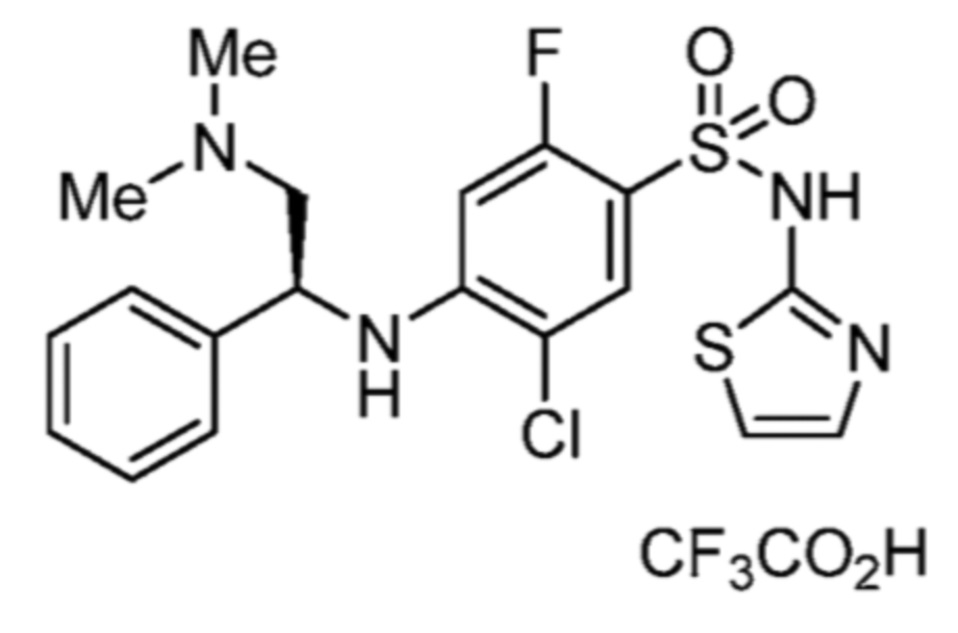
Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на (S)-5-хлор-N-(2,4-диметоксибензил)-4-((2-(диметиламино)-1-фенилэтил)амино-2-фтор-N-(тиазол-2-ил)бензолсульфонамид, очисткой с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,017 г, выход 6%):1H ЯМР (300 МГц, DMSO-d6) δ 12,80 (с, 1H), 9,25 (с, 1H), 7,62 (д, J=7,2 Гц, 1H), 7,48-7,34 (м, 4H), 7,33-7,29 (м, 1H), 7,25 (д, J=4,5 Гц, 1H), 7,00 (д, J=8,7 Гц, 1H), 6,83 (д, J=4,5 Гц, 1H), 6,83 (д, J=12,9 Гц, 1H), 5,32-5,19 (м, 1H), 3,85 (т, J=11,7 Гц, 1H), 3,40-3,28 (м, 1H), 2,86 (с, 6H);19F ЯМР (282 МГц, DMSO-d6) δ -73,7 (с, 3F), -109,4 (с, 1F); MS (ES+) m/z 454,9 (M+1), 456,9 (M+1).
ПРИМЕР 65
Синтез 2,2,2-трифторацетата (R)-5-хлор-4-((2-(диметиламино)-1-фенилэтил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение гидрохлорида (R)-N1,N1-диметил-2-фенилэтан-1,2-диамина:

Следуя процедуре как описано для примера 64, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 2,2-диоксид трет-бутил (S)-4-фенил-1,2,3-оксатиазолидин-3-карбоксилата на 2,2-диоксид трет-бутил (R)-4-фенил-1,2,3-оксатиазолидин-3-карбоксилата, указанное в заголовке соединение получали в виде масла коричневого цвета (0,133 г, количественный выход): MS (ES+) m/z 165,1 (M+1).
Стадия 2. Получение (S)-5-хлор-N-(2,4-диметоксибензил)-4-((2-(диметиламино)-1-фенилэтил)амино-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на гидрохлорид (R)-N1,N1-диметил-2-фенилэтан-1,2-диамина, указанное в заголовке соединение получали в виде бесцветного масла (0,075 г, выход 18%): MS (ES+) m/z 605,0 (M+1), 607,0 (M+1).
Стадия 3: 2,2,2-трифторацетат (S)-5-хлор-4-((2-(диметиламино)-1-фенилэтил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на (S)-5-хлор-N-(2,4-диметоксибензил)-4-((2-(диметиламино)-1-фенилэтил)амино-2-фтор-N-(тиазол-2-ил)бензолсульфонамид, очисткой с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,027 г, выход 8%):1H ЯМР (300 МГц, DMSO-d6) δ 12,79 (с, 1H), 9,20 (с, 1H), 7,62 (д, J=7,2 Гц, 1H), 7,47-7,35 (м, 4H), 7,29-7,27 (м, 1H), 7,26 (д, J=4,5 Гц, 1H), 6,99 (д, J=8,7 Гц, 1H), 6,83 (д, J=4,5 Гц, 1H), 6,83 (д, J=12,9 Гц, 1H), 5,33-5,19 (м, 1H), 3,85 (т, J=11,7 Гц, 1H), 3,40-3,29 (м, 1H), 2,88 (с, 3H), 2,86 (с, 3H);19F ЯМР (282 МГц, DMSO-d6) δ -73,7 (с, 3F), -109,4 (с, 1F); MS (ES+) m/z 455,0 (M+1), 457,0 (M+1).
ПРИМЕР 66
Синтез 2,2,2-трифторацетата (R)-4-((2-(азетидин-1-ил)-1-фенилэтил)амино)-5-хлор-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил (R)-(2-(азетидин-1-ил)-1-фенилэтил)карбамата

В смесь (R)-2-((трет-бутоксикарбонил)амино)-2-фенилэтилметансульфоната (0,50 г, 1,58 ммоль, получен в соответствии с WO2009013171) и азетидина (0,45 мл, 7,9 ммоль) в безводном тетрагидрофуране (4,5 мл) добавляли N,N-диизопропилэтиламин (0,83 мл, 4,8 ммоль) и реакционную смесь перемешивали при 60°C в течение 17 ч. Суспензию разводили водой (10 мл) и этилацетатом (10 мл) и водную фазу экстрагировали этилацетатом (3×10 мл). Объединенные органические фазы промывали солевым раствором (5 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и растиранием остатка с гексанами (20 мл) получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,136 г, выход 31%): MS (ES+) m/z 277,3 (M+1).
Стадия 2. Получение гидрохлорида (R)-2-(азетидин-1-ил)-1-фенилэтан-1-амина:

В смесь трет-бутил (R)-(2-(азетидин-1-ил)-1-фенилэтил)карбамата (0,136 г, 0,492 ммоль) в безводном диоксане (3 мл) добавляли 4,0 M раствор хлороводорода в диоксане (0,37 мл, 1,5 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч. Реакционную смесь разводили простым диэтиловым эфиром (10 мл) и образованный преципитат собирали фильтрованием для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,279 г, выход 77%): MS (ES+) m/z 177,2 (M+1).
Стадия 3. Получение (R)-4-((2-(азетидин-1-ил)-1-фенилэтил)амино)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на гидрохлорид (R)-2-(азетидин-1-ил)-1-фенилэтан-1-амина, указанное в заголовке соединение получали в виде бесцветного масла (0,163 г, выход 75%): MS (ES+) m/z 617,1 (M+1), 619,1 (M+1).
Стадия 4. 2,2,2-трифторацетат (R)-4-((2-(азетидин-1-ил)-1-фенилэтил)амино)-5-хлор-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на (R)-4-((2-(азетидин-1-ил)-1-фенилэтил)амино)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид, растиранием с ацетонитрилом (5 мл) указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,084 г, выход 39%):1H ЯМР (300 МГц, DMSO-d6) δ 12,76 (с, 1H), 9,70 (с, 1H), 7,58 (д, J=7,2 Гц, 1H), 7,45-7,39 (м, 2H), 7,38-7,31 (м, 2H), 7,30-7,26 (м, 1H), 7,22 (д, J=4,5 Гц, 1H), 6,86 (д, J=8,1 Гц, 1H), 6,79 (д, J=4,8 Гц, 1H), 6,63 (д, J=12,9 Гц, 1H), 5,00-5,89 (м, 1H), 4,27-3,99 (м, 2H), 3,85-3,67 (м, 2H), 3,64-3,46 (м, 2H), 2,41-2,18 (м, 2H); MS (ES+) m/z 467,0 (M+1), 469,0 (M+1).
ПРИМЕР 67
Синтез 2,2,2-трифторацетата (S)-4-((2-(азетидин-1-ил)-1-фенилэтил)амино)-5-хлор-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение гидрохлорида (S)-2-(азетидин-1-ил)-1-фенилэтан-1-амина

Следуя процедуре как описано для примера 43, стадии 1 и стадии 2, получали указанное в заголовке соединение в виде твердого вещества коричневатого цвета (0,279 г, выход 83%): MS (ES+) m/z 177,2 (M+1).
Стадия 2. Получение (S)-4-((2-(азетидин-1-ил)-1-фенилэтил)амино)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на гидрохлорид (S)-2-(азетидин-1-ил)-1-фенилэтан-1-амина, указанное в заголовке соединение получали в виде бесцветного масла (0,106 г, выход 32%): MS (ES+) m/z 617,1 (M+1), 619,1 (M+1).
Стадия 3: 2,2,2-трифторацетат (S)-4-((2-(азетидин-1-ил)-1-фенилэтил)амино)-5-хлор-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на (S)-4-((2-(азетидин-1-ил)-1-фенилэтил)амино)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,095 г, выход 29%):1H ЯМР (300 МГц, DMSO-d6) δ 12,80 (с, 1H), 9,72 (с, 1H), 7,62 (д, J=7,2 Гц, 1H), 7,48-7,43 (м, 2H), 7,42-7,35 (м, 2H), 7,33-7,29 (м, 1H), 7,26 (д, J=4,5 Гц, 1H), 6,89 (д, J=8,4 Гц, 1H), 6,83 (д, J=4,5 Гц, 1H), 6,67 (д, J=12,9 Гц, 1H), 5,04-4,91 (м, 1H), 4,27-4,04 (м, 4H), 3,89-3,75 (м, 1H), 3,64-3,49 (м, 1H), 2,46-2,20 (м, 2H); MS (ES+) m/z 467,0 (M+1), 469,0 (M+1).
ПРИМЕР 68
Синтез 2,2,2-трифторацетата (S)-5-хлор-4-((2-(3-фторазетидин-1-ил)-1-фенилэтил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение гидрохлорида (S)-2-(3-фторазетидин-1-ил)-1-фенилэтан-1-амина

В раствор 2,2-диоксида трет-бутил (S)-4-фенил-1,2,3-оксатиазолидин-3-карбоксилата (0,200 г, 0,67 ммоль) в безводном ацетонитриле (3 мл) добавляли 3-фторазетидин (0,750 г, 6,70 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 3 ч. Реакционную смесь концентрировали in vacuo, остаток растворяли в диоксане (3 мл) и туда добавляли 4 M раствор хлороводорода диоксан (0,84 мл, 3,4 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч. Концентрированием in vacuo получали указанное в заголовке соединение в виде твердого вещества коричневатого цвета (0,137 г, выход 89%): MS (ES+) m/z 195,1 (M+1).
Стадия 2. Получение (S)-4-((2-(3-фторазетидин-1-ил)-1-фенилэтил)амино)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на гидрохлорид (S)-2-(3-фторазетидин-1-ил)-1-фенилэтан-1-амина, указанное в заголовке соединение получали в виде бесцветного масла (0,045 г, выход 11%): MS (ES+) m/z 635,4 (M+1), 637,4 (M+1).
Стадия 3: 2,2,2-трифторацетат (S)-5-хлор-4-((2-(3-фторазетидин-1-ил)-1-фенилэтил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости для процедуры как описано для примера 43, стадии 2 для того, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на (S)-4-((2-(3-фторазетидин-1-ил)-1-фенилэтил)амино)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид, очисткой с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,013 г, выход 4%):1H ЯМР (300 МГц, DMSO-d6) δ 12,79 (с, 1H), 10,54 (ушир. с, 0,5H), 9,71 (ушир. с, 0,5H), 7,62 (д, J=7,5 Гц, 1H), 7,49-7,42 (м, 2H), 7,40-7,35 (м, 2H), 7,34-7,30 (м, 1H), 7,26 (д, J=4,8 Гц, 1H), 6,90 (д, J=7,5 Гц, 1H), 6,83 (д, J=4,5 Гц, 1H), 6,61 (д, J=12,9 Гц, 1H), 5,59-5,19 (м, 1H), 5,08-4,92 (м, 1H), 4,69-4,24 (м, 4H), 4,02-3,80 (м, 1H), 3,70-3,55 (м, 1H); MS (ES+) m/z 485,0 (M+1), 487,0 (M+1).
ПРИМЕР 69
Синтез (S)-5-хлор-4-((2-(3,3-дифторазетидин-1-ил)-1-фенилэтил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение гидрохлорида (S)-2-(3-фторазетидин-1-ил)-1-фенилэтан-1-амина:

Следуя процедуре как описано для примера 68, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 3-фторазетидин на 3,3-дифторазетидин, указанное в заголовке соединение получали в виде бесцветного масла (0,164 г, выход 99%): MS (ES+) m/z 213,1 (M+1).
Стадия 2. Получение (S)-4-((2-(3,3-дифторазетидин-1-ил)-1-фенилэтил)амино)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на гидрохлорид (S)-2-(3,3-дифторазетидин-1-ил)-1-фенилэтан-1-амина, указанное в заголовке соединение получали в виде бесцветного масла (0,032 г, выход 9%): MS (ES+) m/z 653,4 (M+1), 655,4 (M+1).
Стадия 3: (S)-5-хлор-4-((2-(3-фторазетидин-1-ил)-1-фенилэтил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на (S)-4-((2-(3,3-дифторазетидин-1-ил)-1-фенилэтил)амино)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид, очисткой с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,010 г, выход 4%):1H ЯМР (300 МГц, DMSO-d6) δ 12,78 (с, 1H), 7,60 (д, J=6,9 Гц, 1H), 7,47-7,40 (м, 2H), 7,39-7,30 (м, 2H), 7,30-7,22 (м, 2H), 6,86-6,79 (м, 1H), 6,66-6,56 (м, 1H), 6,44 (д, J=12,9 Гц, 1H), 4,79-4,63 (м, 1H), 4,20-3,79 (м, 4H), 3,53-3,30 (м, 1H), 3,25-3,03 (м, 1H); MS (ES+) m/z 503,0 (M+1), 505,0 (M+1).
ПРИМЕР 70
Синтез (S)-5-хлор-2-фтор-4-((1-фенил-2-(пирролидин-1-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение гидрохлорида (S)-1-фенил-2-(пирролидин-1-ил)этан-1-амина

Следуя процедуре как описано для примера 66, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять (R)-2-((трет-бутоксикарбонил)амино)-2-фенилэтилметансульфонат на (S)-2-((трет-бутоксикарбонил)амино)-2-фенилэтилметансульфонат и азетидин на пирролидин, указанное в заголовке соединение получали в виде твердого вещества коричневатого цвета (0,198 г, выход 55%): MS (ES+) m/z 191,2 (M+1).
Стадия 2. Получение (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенил-2-(пирролидин-1-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на гидрохлорид (S)-1-фенил-2-(пирролидин-1-ил)этан-1-амина, указанное в заголовке соединение получали в виде бесцветного масла (147 г, выход 43%): MS (ES+) m/z 631,1 (M+1), 633,1 (M+1).
Стадия 3. Получение (S)-5-хлор-2-фтор-4-((1-фенил-2-(пирролидин-1-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида
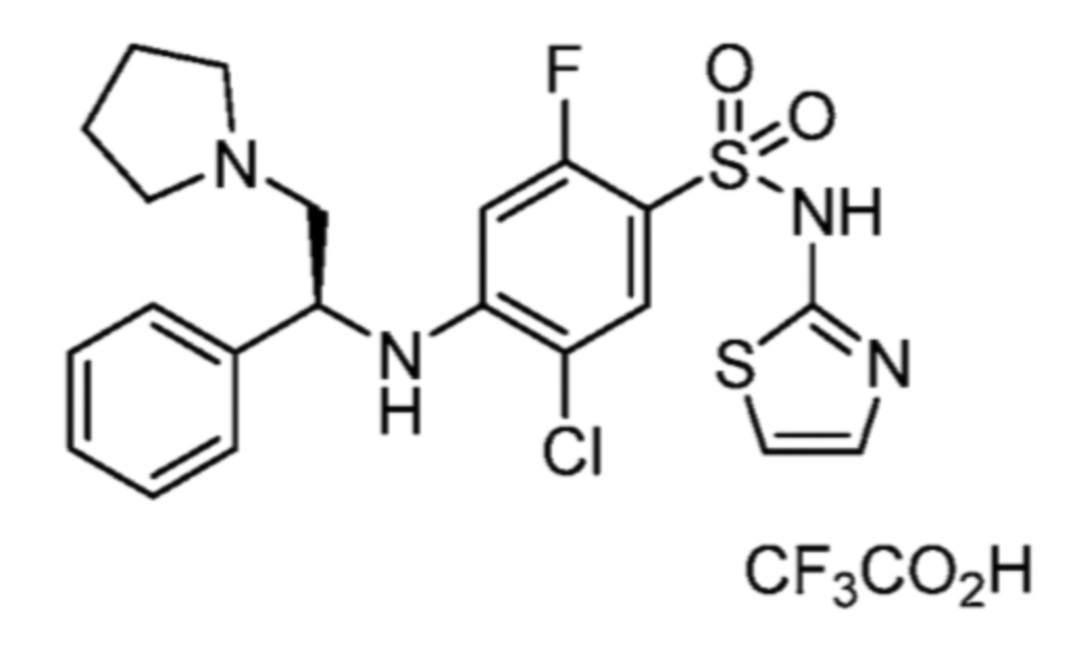
Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенил-2-(пирролидин-1-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамид, очисткой с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,023 г, выход 9%):1H ЯМР (300 МГц, DMSO-d6) δ 12,80 (с, 1H), 9,45 (с, 1H), 7,62 (д, J=7,2 Гц, 1H), 7,52-7,43 (м, 2H), 7,43-7,34 (м, 2H), 7,33-7,29 (м, 1H), 7,26 (д, J=4,8 Гц, 1H), 6,99 (д, J=8,1 Гц, 1H), 6,87-6,75 (м, 2H), 5,26-5,13 (м, 1H), 3,97 (т, J=12,3 Гц, 1H), 3,64-3,46 (м, 2H), 3,45-3,34 (м, 1H), 3,33-3,22 (м, 1H), 3,21-3,06 (м, 1H), 2,12-1,82 (м, 4H);19F ЯМР (282 МГц, DMSO-d6) δ -73,7 (с, 3F), -109,3 (с, 1F); MS (ES+) m/z 480,9 (M+1), 482,9 (M+1).
ПРИМЕР 71
Синтез (R)-5-хлор-2-фтор-4-((1-фенил-2-(пирролидин-1-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение гидрохлорида (R)-1-фенил-2-(пирролидин-1-ил)этан-1-амина

Следуя процедуре как описано для примера 66, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять азетидин на пирролидин, указанное в заголовке соединение получали в виде твердого вещества коричневатого цвета (0,313 г, количественный выход): MS (ES+) m/z 191,2 (M+1).
Стадия 2. Получение (R)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенил-2-(пирролидин-1-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на гидрохлорид (R)-1-фенил-2-(пирролидин-1-ил)этан-1-амина, указанное в заголовке соединение получали в виде бесцветного масла (0,169 г, выход 49%): MS (ES+) m/z 631,1 (M+1), 633,1 (M+1).
Стадия 3. Получение (R)-5-хлор-2-фтор-4-((1-фенил-2-(пирролидин-1-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на (R)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенил-2-(пирролидин-1-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамид, очисткой с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,045 г, выход 17%):1H ЯМР (300 МГц, DMSO-d6) δ 12,80 (с, 1H), 9,48 (с, 1H), 7,62 (д, J=6,3 Гц, 1H), 7,51-7,43 (м, 2H), 7,42-7,34 (м, 2H), 7,31 (д, J=6,3 Гц, 1H), 7,28-7,22 (м, 1H), 6,99 (д, J=8,1 Гц, 1H), 6,86-6,73 (м, 2H), 5,25-5,12 (м, 1H), 3,97 (т, J=12,3 Гц, 1H), 3,63-3,47 (м, 2H), 3,56-3,34 (м, 1H), 3,33-3,24 (м, 1H), 3,21-3,06 (м, 1H), 2,12-1,81 (м, 4H); MS (ES+) m/z 480,9 (M+1), 482,9 (M+1).
ПРИМЕР 72
Синтез 2,2,2-трифторацетата (S)-5-хлор-2-фтор-N-(6-фторпиридин-2-ил)-4-((1-фенил-2-(пирролидин-1-ил)этил)амино)бензолсульфонамида

Стадия 1. Получение (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(6-фторпиридин-2-ил)-4-((1-фенил-2-(пирролидин-1-ил)этил)амино)бензолсульфонамида

В раствор 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(6-фторпиридин-2-ил)бензолсульфонамида (0,457 г, 0,969 ммоль) в безводном диметилсульфоксиде (10 мл) добавляли гидрохлорид (S)-1-фенил-2-(пирролидин-1-ил)этан-1-амина (0,220 г, 0,969 ммоль) и карбонат калия (0,669 г, 4,85 ммоль) и реакционную смесь перемешивали при 75°C в течение 18 ч. Реакционную смесь оставляли остывать до температуры окружающей среды, разводили этилацетатом (5 мл) и водой (5 мл). Водную фазу экстрагировали этилацетатом (3×3 мл). Объединенные органические фазы промывали солевым раствором (1×5 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 12-80% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного масла (0,312 г, выход 49%): MS (ES+) m/z 643,1 (M+1), 645,1 (M+1).
Стадия 2. Получение 2,2,2-трифторацетата (S)-5-хлор-2-фтор-N-(6-фторпиридин-2-ил)-4-((1-фенил-2-(пирролидин-1-ил)этил)амино)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(6-фторпиридин-2-ил)-4-((1-фенил-2-(пирролидин-1-ил)этил)амино)бензолсульфонамид, очисткой с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,074 г, выход 15%):1H ЯМР (300 МГц, DMSO-d6) δ 11,55 (с, 1H), 9,45 (ушир. с, 1H), 7,83 (дд, J=16,5, 8,1 Гц, 1H), 7,79 (д, J=7,5 Гц, 1H), 7,48-7,45 (м, 2H), 7,41-7,34 (м, 2H), 7,32-7,27 (м, 1H), 7,19 (д, J=9,3 Гц, 1H), 6,84 (дд, J=7,8, 1,8 Гц, 1H), 6,82 (д, J=13,5 Гц, 1H), 6,71 (дд, J=8,0, 2,5 Гц, 1H), 5,23-5,15 (м, 1H), 4,03-3,94 (м, 1H), 3,95-3,51 (м, 2H), 3,44-3,35 (м, 1H), 3,29-3,22 (м, 1H), 3,17-3,10 (м, 1H), 2,05-1,97 (м, 2H), 1,92-1,86 (м, 2H);19F ЯМР (282 МГц, DMSO-d6) δ -69,0 (с, 1F), -73,8 (с, 3F), -110,1 (с, 1F); MS (ES+) m/z 493,0 (M+1), 495,0 (M+1).
ПРИМЕР 73
Синтез (S)-5-хлор-2-фтор-4-((1-фенил-2-(пирролидин-1-ил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил (S)-((5-хлор-2-фтор-4-((1-фенил-2-(пирролидин-1-ил)этил)амино)фенил)сульфонил)(тиазол-4-ил)карбамата

Следуя процедуре как описано для примера 72, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(6-фторпиридин-2-ил)бензолсульфонамид на трет-бутил ((5-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамат, указанное в заголовке соединение получали в виде масла оранжевого цвета (0,062 г, выход 11%); MS (ES+) m/z 581,1 (M+1), 583,1 (M+1).
Стадия 2. Получение (S)-5-хлор-2-фтор-4-((1-фенил-2-(пирролидин-1-ил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на трет-бутил (S)-((5-хлор-2-фтор-4-((1-фенил-2-(пирролидин-1-ил)этил)амино)фенил)сульфонил)(тиазол-4-ил)карбамат, очисткой с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,025 г, выход 5%):1H ЯМР (300 МГц, DMSO-d6) δ 11,18 (с, 1H), 9,47 (ушир. с, 1H), 8,85 (д, J=2,2 Гц, 1H), 7,64 (д, J=7,4 Гц, 1H), 7,48-7,46 (м, 2H), 7,41-7,36 (м, 2H), 7,33-7,28 (м, 1H), 7,14-7,11 (м, 1H), 6,98 (д, J=2,2 Гц, 1H), 6,82 (д, J=13,3 Гц, 1H), 5,23-5,15 (м, 1H), 4,02-3,93 (м, 1H), 3,60-3,50 (м, 2H), 3,44-3,36 (м, 1H), 3,32-3,21 (м, 1H), 3,17-3,10 (м, 1H), 2,06-1,97 (м, 2H), 1,95-1,87 (м, 2H); MS (ES+) m/z 481,0 (M+1), 483,0 (M+1).
ПРИМЕР 74
Синтез 2,2,2-трифторацетата 5-хлор-2-фтор-4-((2-морфолино-1-фенилэтил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((2-морфолино-1-фенилэтил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на 2-морфолино-1-фенилэтан-1-амин, указанное в заголовке соединение получали в виде бесцветного масла (0,310 г, выход 88%): MS (ES+) m/z 647,1 (M+1), 649,1 (M+1).
Стадия 2. Получение 2,2,2-трифторацетата 5-хлор-2-фтор-4-((2-морфолино-1-фенилэтил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((2-морфолино-1-фенилэтил)амино)-N-(тиазол-2-ил)бензолсульфонамид, растиранием с ацетонитрилом (2×5 мл) указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,075 г, выход 28%):1H ЯМР (300 МГц, DMSO-d6) δ 12,78 (с, 1H), 9,81 (с, 1H), 7,62 (д, J=7,5 Гц, 1H), 7,48-7,34 (м, 4H), 7,31 (д, J=7,2 Гц, 1H), 7,25 (д, J=4,5 Гц, 1H), 6,97-6,86 (м, 1H), 6,82 (д, J=4,5 Гц, 1H), 6,78-6,65 (м, 1H), 5,37-5,13 (м, 1H), 4,10-3,86 (м, 3H), 3,86-3,62 (м, 3H), 3,52-3,31 (м, 2H), 3,29-3,05 (м, 2H); MS (ES+) m/z 497,1 (M+1), 499,1 (M+1).
ПРИМЕР 75
Синтез 2,2,2-трифторацетата (S)-5-хлор-2-фтор-4-((2-морфолино-1-фенилэтил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение гидрохлорида (S)-2-морфолино-1-фенилэтан-1-амина

Следуя процедуре как описано для примера 66, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять (R)-2-((трет-бутоксикарбонил)амино)-2-фенилэтилметансульфонат на (S)-2-((трет-бутоксикарбонил)амино)-2-фенилэтилметансульфонат и азетидин на морфолин, указанное в заголовке соединение получали в виде твердого вещества коричневатого цвета (0,186 г, выход 48%): MS (ES+) m/z 207,2 (M+1).
Стадия 2. Получение (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((2-морфолино-1-фенилэтил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклопропан-1-амин на гидрохлорид (S)-2-морфолино-1-фенилэтан-1-амина, указанное в заголовке соединение получали в виде бесцветного масла (0,138 г, выход 39%): MS (ES+) m/z 647,1 (M+1), 649,1 (M+1).
Стадия 3. Получение 2,2,2-трифторацетата (S)-5-хлор-2-фтор-4-((2-морфолино-1-фенилэтил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((2-морфолино-1-фенилэтил)амино)-N-(тиазол-2-ил)бензолсульфонамид, очисткой с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,042 г, выход 16%):1H ЯМР (300 МГц, DMSO-d6) δ 12,79 (с, 1H), 9,86 (с, 1H), 7,61 (д, J=7,2 Гц, 1H), 7,48-7,34 (м, 4H), 7,31 (д, J=6,9 Гц, 1H), 7,25 (д, J=4,5 Гц, 1H), 6,97-6,86 (м, 1H), 6,82 (д, J=4,5 Гц, 1H), 6,80-6,66 (м, 1H), 5,37-5,13 (м, 1H), 4,10-3,86 (м, 3H), 3,86-3,62 (м, 3H), 3,52-3,31 (м, 2H), 3,29-3,05 (м, 2H);19F ЯМР (282 МГц, DMSO-d6) δ -73,8 (с, 3F), -109,5 (с, 1F); MS (ES+) m/z 496,9 (M+1), 498,9 (M+1).
ПРИМЕР 76
Синтез (S)-3-хлор-4-((1-фенилэтил)амино)-N-(тиазол-2-ил)бензолсульфонамида

В раствор 5-хлор-N-(2,4-диметоксибензил)-4-дифтор-N-(тиазол-2-ил)бензолсульфонамида (0,250 г, 0,543 ммоль) и (S)-1-фенилэтан-1-амина (0,065 мг, 0,54 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли карбонат цезия (0,424 г, 1,30 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 17 ч. Реакционную смесь разводили этилацетатом (5 мл) и водой (5 мл) и водную фазу экстрагировали этилацетатом (3×3 мл). Объединенные органические фазы промывали солевым раствором (5 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo, остаток растворяли в дихлорметане (5 мл) и туда добавляли трифторуксусную кислоту (1 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч и затем разводили метанолом (10 мл). Суспензию фильтровали и фильтрат концентрировали in vacuo. Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 12-80% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,106 г, выход 49%):1H ЯМР (300 МГц, DMSO-d6) δ 12,60 (с, 1H), 7,56 (д, J=2,1 Гц, 1H), 7,42-7,34 (м, 3H), 7,30 (т, J=7,8 Гц, 2H), 7,21 (д, J=4,8 Гц, 1H), 7,20-7,16 (м, 1H), 6,77 (д, J=4,5 Гц, 1H), 6,56 (д, J=8,7 Гц, 1H), 6,19 (д, J=7,2 Гц, 1H), 4,68 (дк, J=7,2, 6,6 Гц, 1H), 1,53 (д, J=6,6 Гц, 3H); MS (ES-) m/z 392,1 (M - 1), 394,1 (M - 1).
ПРИМЕР 77
Синтез (S)-3-хлор-4-((5,6,7,8-тетрагидрохинолин-8-ил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение (S)-3-хлор-N-(2,4-диметоксибензил)-4-((5,6,7,8-тетрагидрохинолин-8-ил)амино)-N-(тиазол-2-ил)бензолсульфонамида

В смесь 5-хлор-N-(2,4-диметоксибензил)-4-дифтор-N-(тиазол-2-ил)бензолсульфонамида (0,250 г, 0,566 ммоль) и дигидрохлорида (S)-5,6,7,8-тетрагидрохинолин-8-амина (0,124 мг, 0,566 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли карбонат калия (0,390 г, 2,83 ммоль) и реакционную смесь перемешивали при 60°C в течение 17 ч. Реакционную смесь оставляли остывать до температуры окружающей среды и разводили этилацетатом (5 мл) и водой (5 мл). Водную фазу экстрагировали этилацетатом (3×3 мл). Объединенные органические фазы промывали солевым раствором (1×5 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 6-80% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,226 г, выход 95%): MS (ES+) m/z 571,1 (M+1), 573,1 (M+1).
Стадия 2. Получение (S)-3-хлор-4-((5,6,7,8-тетрагидрохинолин-8-ил)амино)-N-(тиазол-2-ил)бензолсульфонамида

В раствор (S)-3-хлор-N-(2,4-диметоксибензил)-4-((5,6,7,8-тетрагидрохинолин-8-ил)амино)-N-(тиазол-2-ил)бензолсульфонамида (226 мг, 0,396 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (0,092 мл, 1,2 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 10 минут и затем разводили метанолом (10 мл). Полученную суспензию фильтровали и фильтрат концентрировали in vacuo. Очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,016 мг, выход 7%):1H ЯМР (300 МГц, DMSO-d6) δ 12,68 (с, 1H), 8,53 (д, J=4,5 Гц, 1H), 8,00 (д, J=7,5 Гц, 1H), 7,64 (д, J=2,1 Гц, 1H), 7,57 (дд, J=8,4, 1,8 Гц, 2H), 7,26 (д, J=4,5 Гц, 1H), 7,06 (д, J=9,0 Гц, 1H), 6,81 (д, J=4,5 Гц, 1H), 6,54 (д, J=7,5 Гц, 1H), 5,05-4,93 (м, 1H), 2,96-2,85 (м, 2H), 2,25-2,09 (м, 1H), 1,98-1,75 (м, 3H); MS (ES+) m/z 421,0 (M+1), 423,0 (M+1).
ПРИМЕР 78
Синтез (S)-5-хлор-4-((1-(2-хлорфенил)этил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида
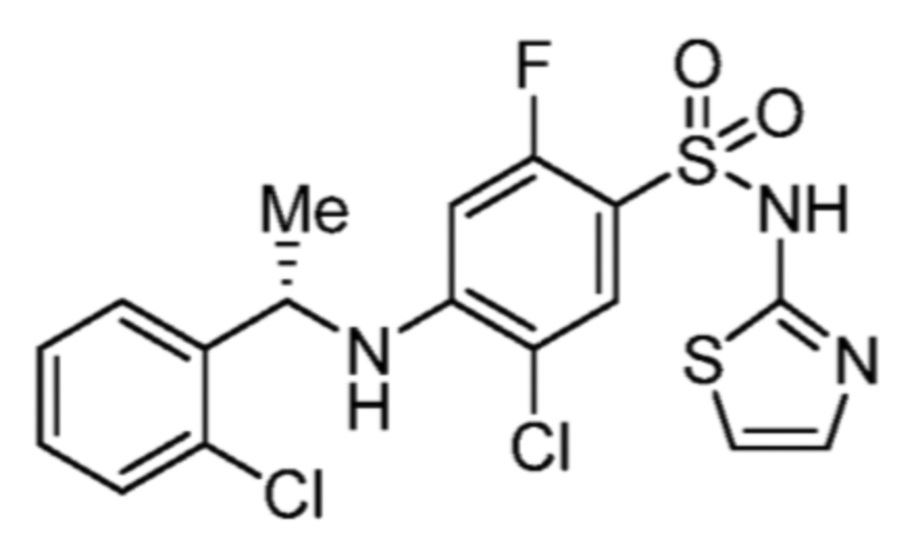
Стадия 1. Получение (S)-5-хлор-4-((1-(2-хлорфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 77, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять дигидрохлорид (S)-5,6,7,8-тетрагидрохинолин-8-амина на гидрохлорид (S)-1-(2-хлорфенил)этан-1-амина, указанное в заголовке соединение получали в виде бесцветного масла (0,137 г, выход 83%): MS (ES+) m/z 596,0 (M+1), 598,0 (M+1).
Стадия 2. Получение (S)-5-хлор-4-((1-(2-хлорфенил)этил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на (S)-5-хлор-4-((1-(2-хлорфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,042 г, выход 17%):1H ЯМР (300 МГц, DMSO-d6) δ 12,76 (с, 1H), 7,60 (д, J=7,5 Гц, 1H), 7,45 (дд, J=6,0, 1,5 Гц, 2H), 7,36-7,26 (м, 2H), 7,25 (д, J=4,5 Гц, 1H), 6,81 (д, J=4,8 Гц, 1H), 6,73 (д, J=6,0 Гц, 1H), 6,05 (д, J=12,9 Гц, 1H), 4,97-4,81 (м, 1H), 1,54 (д, J=6,6 Гц, 3H);19F ЯМР (282 МГц, DMSO-d6) δ -109,2 (s); MS (ES-) m/z 444,0 (M - 1), 446,0 (M - 1).
ПРИМЕР 79
Синтез (S)-3-хлор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение (S)-3-хлор-N-(2,4-диметоксибензил)-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида
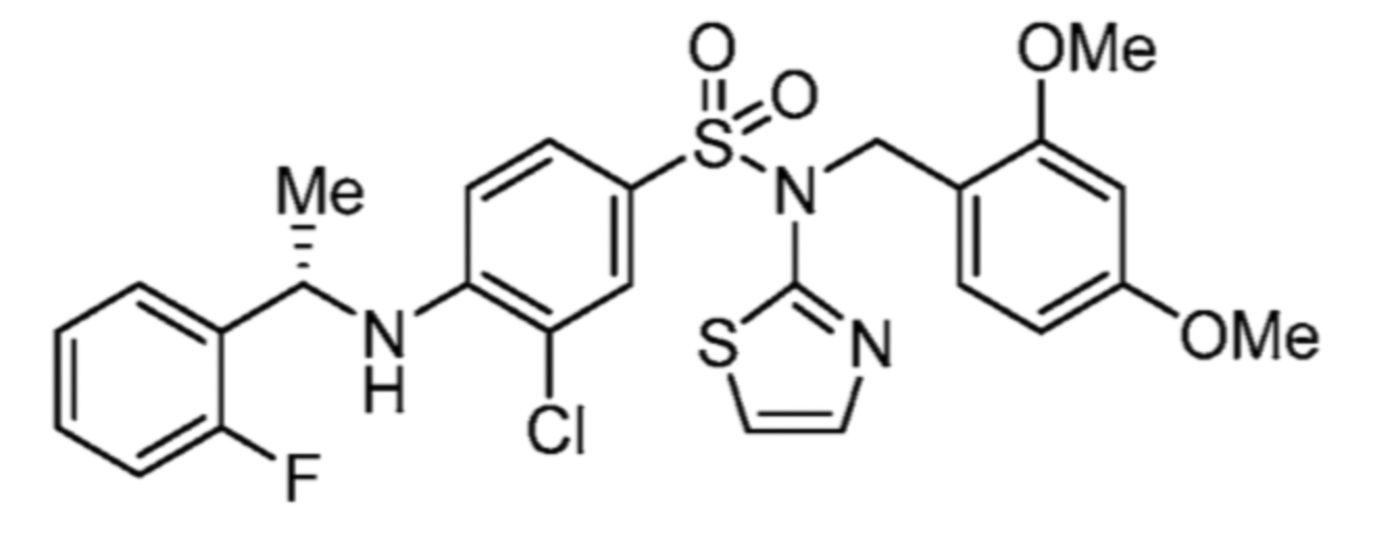
В смесь 5-хлор-N-(2,4-диметоксибензил)-4-дифтор-N-(тиазол-2-ил)бензолсульфонамида (0,300 г, 0,679 ммоль) и (S)-(2-фторфенил)этиламина (0,94 мг, 0,679 ммоль) в безводном диметилсульфоксиде (6 мл) добавляли карбонат калия (0,224 г, 1,63 ммоль) и реакционную смесь перемешивали при 75°C в течение 17 ч. Реакционную смесь оставляли остывать до температуры окружающей среды и разводили этилацетатом (5 мл) и водой (5 мл). Водную фазу экстрагировали этилацетатом (3×3 мл). Объединенные органические фазы промывали солевым раствором (1×5 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 5-60% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного масла (0,305 г, выход 80%): MS (ES+) m/z 561,9 (M+1), 563,9 (M+1).
Стадия 2. Получение (S)-3-хлор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида
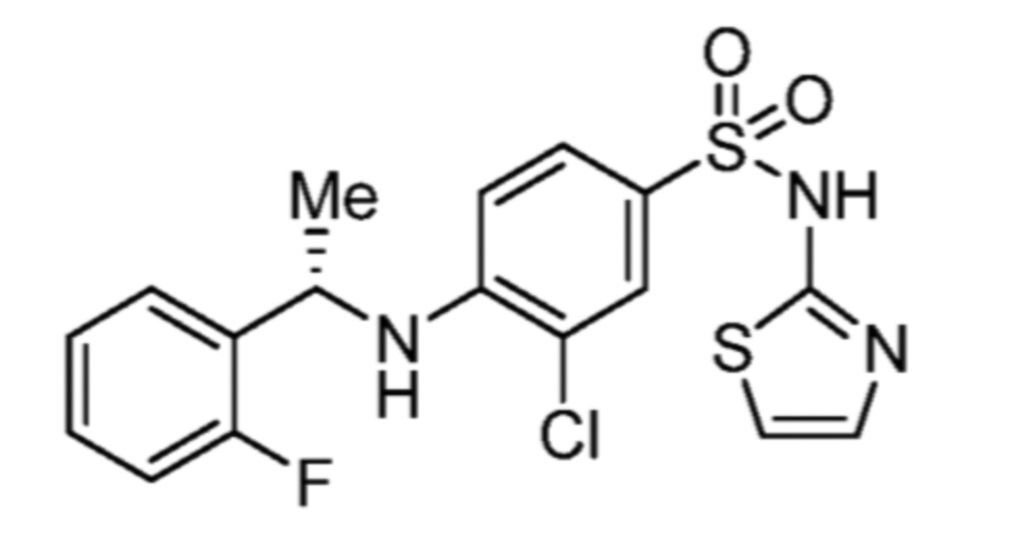
Следуя процедуре как описано для примера 43, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилциклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на (S)-3-хлор-N-(2,4-диметоксибензил)-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамид, очисткой с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,084 мг, выход 30%):1H ЯМР (300 МГц, DMSO-d6) δ 12,61 (с, 1H), 7,59 (д, J=2,1 Гц, 1H), 7,42 (дд, J=8,7, 1,8 Гц, 1H), 7,36 (дд, J=7,5, 1,5 Гц, 1H), 7,31-7,10 (м, 4H), 6,78 (д, J=4,5 Гц, 1H), 6,49 (д, J=8,8 Гц, 1H), 6,23 (д, J=7,5 Гц, 1H), 4,96-4,87 (м, 1H), 1,56 (д, J=6,7 Гц, 3H);19F ЯМР (282 МГц, DMSO-d6) δ -120,1 (с); MS (ES+) m/z 411,9 (M+1), 413,9 (M+1).
ПРИМЕР 80
Синтез (R)-5-хлор-2-фтор-N-(тиазол-2-ил)-4-(2,2,2-трифтор-1-фенилэтокси)бензолсульфонамида

В раствор (R)-2,2,2-трифтор-1-фенилэтан-1-ола (0,114 г, 0,648 ммоль) и 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамида (0,300 г, 0,652 ммоль) в безводном диметилсульфоксиде (2,5 мл) добавляли карбонат цезия (0,509 г, 1,56 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 65 ч. Реакционную смесь разводили этилацетатом (5 мл) и водой (5 мл) и водную фазу экстрагировали этилацетатом (5 мл). Объединенные органические экстракты промывали солевым раствором (2×5 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с использованием 0-60% этилацетата в гексанах. Получаемый остаток растворяли в дихлорметане (4 мл) и туда добавляли трифторуксусную кислоту (0,7 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 10 минут и затем концентрировали in vacuo. Остаток растирали в метаноле (5 мл), используя древесный уголь (0,3 г), и получаемую суспензию фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,074 г, выход 24%):1H ЯМР (300 МГц, DMSO-d6) δ 12,96 (ушир. с, 1H), 7,83 (д, J=7,3 Гц, 1H), 7,60-7,52 (м, 2H), 7,52-7,44 (м, 3H), 7,38-7,27 (м, 2H), 6,88 (д, J=4,6 Гц, 1H), 6,59 (к, J=6,5 Гц, 1H); MS (ES+) m/z 467,0, 469,0 (M+1).
ПРИМЕР 81
Синтез 3-хлор-4-((1-(пиридин-2-ил)пропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

В смесь дигидрохлорида1-(пиридин-2-ил)пропан-1-амина (0,129 г, 0,625 ммоль), 4-бром-3-хлор-N-(тиазол-2-ил)бензолсульфонамида (0,200 г, 0,568 ммоль), 2-ди-трет-бутилфосфино-2′,4′,6′-триизопропилбифенила (0,012 г, 0,028 ммоль), трис(дибензилиденацетон)дипалладия(0) (0,013 г, 0,014 ммоль) и трет-бутоксида натрия (0,273 г, 2,84 ммоль) добавляли безводный толуол (3 мл) и реакционную смесь дегазировали в течение 10 минут посредством пропускания потока азота через нее. Реакционную смесь перемешивали при 100°C в течение 72 ч. Реакционную смесь оставляли остывать до температуры окружающей среды, разводили этилацетатом (5 мл) и фильтровали через подушку из целита. Фильтровальную подушку промывали этилацетатом (20 мл) и объединенный фильтрат концентрировали in vacuo. Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-25% метанола в дихлорметане, и растиранием в метаноле (2×5 мл) получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,074 г, выход 3%):1H ЯМР (300 МГц, DMSO-d6) δ 12,60 (с, 1H), 8,55 (д, J=4,2 Гц, 1H), 7,77 (дт, J=1,5, 7,5 Гц, 1H), 7,59 (д, J=2,1 Гц, 1H), 7,49-7,39 (м, 2H), 7,31-7,25 (м, 1H), 7,22 (д, J=4,8 Гц, 1H), 6,78 (д, J=4,5 Гц, 1H), 6,73 (д, J=9,0 Гц, 1H), 6,29 (д, J=7,8 Гц, 1H), 4,61 (к, J=7,2 Гц, 1H), 2,00-1,80 (м, 2H), 0,91 (т, J=7,2 Гц, 3H); MS (ES+) m/z 409,1 (M+1), 411,1 (M+1).
ПРИМЕР 82
Синтез (S)-3-хлор-4-((1-(пиридин-2-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 81 и выполняя некритические вариации при необходимости, чтобы заменять дигидрохлорид 1-(пиридин-2-ил)пропан-1-амина на дигидрохлорид (S)-1-(пиридин-2-ил)этан-1-амина, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,018 г, 8,выход 1%):1H ЯМР (300 МГц, DMSO-d6) δ 12,97 (с, 1H), 7,78 (дт, J=7,5, 1,5 Гц, 1H), 7,60 (д, J=2,1 Гц, 1H), 7,49-7,40 (м, 2H), 7,28 (ддд, J=7,2, 3,6, 1,2 Гц, 1H), 7,22 (д, J=4,5 Гц, 1H), 6,79 (д, J=4,5 Гц, 1H), 6,68 (д, J=9,0 Гц, 1H), 6,70 (д, J=7,5 Гц, 1H), 6,39 (д, J=7,2 Гц, 1H), 4,80 (дк, J=6,9, 7,2 Гц, 1H), 1,51 (д, J=6,9 Гц, 3H); MS (ES+) m/z 395,0 (M+1), 397,0 (M+1).
ПРИМЕР 83
Синтез 3-хлор-4-((1-(1-метил-1H-пиразол-4-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 81 и выполняя некритические вариации при необходимости, чтобы заменять дигидрохлорид 1-(пиридин-2-ил)пропан-1-амина на 1-(1-метил-1H-пиразол-4-ил)этан-1-амин, очисткой с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% гидроксид аммония, в качестве элюента, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,012 г, выход 5%):1H ЯМР (300 МГц, DMSO-d6) δ 7,57 (с, 1H), 7,56 (д, J=2,1 Гц, 1H), 7,46 (дд, J=8,7, 2,1 Гц, 1H), 7,37 (с, 1H), 7,13 (д, J=4,2 Гц, 1H), 6,77 (д, J=8,7 Гц, 1H), 6,68 (д, J=4,2 Гц, 1H), 5,69 (д, J=7,8 Гц, 1H), 4,67 (дк, J=7,2, 6,6 Гц, 1H), 3,75 (с, 3H), 1,52 (д, J=6,6 Гц, 3H), NH сульфонамида не наблюдали; MS (ES+) m/z 398,0 (M+1), 400,0 (M+1).
ПРИМЕР 84
Синтез (S)-4-((1-фенилпропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение N-(2,4-диметоксибензил)-4-фтор-N-(тиазол-2-ил)бензолсульфонамида

В смесь (N-(2,4-диметоксибензил)тиазол-2-амина (1,251 г, 5,03 ммоль) в безводном тетрагидрофуране (20 мл) добавляли 1 M раствор бис(триметилсилил)амида лития в тетрагидрофуране (5,0 мл, 5,03 ммоль) при -78°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 1 ч. После охлаждения реакционной смеси до -78°C, туда добавляли раствор 4-фторбензолсульфонилхлорида (0,750 г, 3,86 ммоль) в безводном тетрагидрофуране (20 мл). Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 16 ч. Реакционную смесь гасили добавлением насыщенного раствора хлорида аммония (50 мл) и разводили этилацетатом (100 мл). Водную фазу экстрагировали этилацетатом (2×50 мл) и объединенную органическую фазу промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 12 до 80% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,841 г, выход 53%):1H-ЯМР (300 МГц, CDCl3): δ 7,88-7,83 (м, 2H), 7,44-7,42 (м, 1H), 7,29-7,27 (м, 1H), 7,20-7,14 (м, 2H), 7,05-7,04 (м, 1H), 6,39-6,36 (м, 2H), 5,06 (с, 2H), 3,78 (с, 3H), 3,74 (с, 3H).
Стадия 2. Получение (S)-N-(2,4-диметоксибензил)-4-((1-фенилпропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

В смесь N-(2,4-диметоксибензил)-4-фтор-N-(тиазол-2-ил)бензолсульфонамида (0,200 г, 0,490 ммоль) и (S)-1-фенилпропан-1-амина (0,066 г, 0,490 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли карбонат калия (0,162 г, 1,18 ммоль) и реакционную смесь перемешивали при 90°C в течение 17 ч. Реакционную смесь оставляли остывать до температуры окружающей среды и разводили этилацетатом (5 мл) и водой (5 мл). Водную фазу экстрагировали этилацетатом (3×5 мл). Объединенные органические фазы промывали солевым раствором (5 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 5-60% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного масла (0,084 г, выход 33%): MS (ES+) m/z 524,1 (M+1).
Стадия 3. Получение (S)-4-((1-фенилпропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

В раствор (S)-N-(2,4-диметоксибензил)-4-((1-фенилпропил)амино)-N-(тиазол-2-ил)бензолсульфонамида (0,084 г, 0,16 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (0,5 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч. Концентрированием реакционной смеси in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 10-100% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,029 г, выход 16%):1H ЯМР (300 МГц, DMSO-d6) δ 12,40 (с, 1H), 7,39-7,26 (м, 6H), 7,20-7,15 (м, 2H), 6,93 (д, J=7,4 Гц, 1H), 6,46-6,43 (м, 1H), 6,71 (д, J=4,7 Гц, 1H), 6,53 (д, J=8,8 Гц, 1H), 4,26 (к, J=6,5 Гц, 1H), 1,843-1,62 (м, 2H), 0,88 (т, J=7,3 Гц, 3H); MS (ES+) m/z 374,1 (M+1).
ПРИМЕР 85
Синтез 2,5-дифтор-4-((1-фенилпропил)амино)-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида

В смесь 2,4,5-трифтор-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида (0,40 г, 0,90 ммоль) и 1-фенилпропан-1-амина (0,13 мл, 0,90 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли карбонат цезия (0,703 г, 2,16 ммоль), и реакционная смесь находилась при температуре окружающей среды в течение 17 ч. Реакционную смесь разводили этилацетатом (5 мл) и водой (5 мл) и водную фазу экстрагировали этилацетатом (3×5 мл). Объединенные органические фазы промывали солевым раствором (1×5 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo, растворяли в дихлорметане (5 мл) и туда добавляли трифторуксусную кислоту (1 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч и затем туда добавляли метанол (10 мл). Суспензию фильтровали и фильтрат концентрировали in vacuo. Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 12 до 100% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (195 г, выход 53%):1H ЯМР (300 МГц, DMSO-d6) δ 8,42 (с, 1H), 7,46-7,22 (м, 5H), 7,21-7,06 (м, 2H), 6,51-6,36 (м, 1H), 4,40-4,23 (м, 1H), 2,00-1,81 (м, 1H), 1,78-1,58 (м, 1H), 0,84 (т, J=6,9 Гц, 3H), NH сульфонамида не наблюдали;19F ЯМР (282 МГц, DMSO-d6) δ -112,6 (д, J=15 Гц, 1F), -134,7 (д, J=15 Гц, 1F); MS (ES+) m/z 411,0 (M+1), 412,0 (M+1).
ПРИМЕР 86
Синтез 5-хлор-2-фтор-N-(6-фторпиридин-2-ил)-4-((1-фенилпропил)амино)бензолсульфонамида

Следуя процедуре как описано для примера 85 и выполняя некритические вариации при необходимости, чтобы заменять 2,4,5-трифтор-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамид на 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(6-фторпиридин-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,251 г, выход 68%):1H ЯМР (300 МГц, DMSO-d6) δ 11,43 (с, 1H), 7,83-7,74 (м, 1H), 7,69 (д, J=7,5 Гц, 1H), 7,39 (д, J =7,5 Гц, 2H), 7,30-7,25 (м, 2H), 7,20-7,15 (м, 1H), 6,81 (д, J=7,8 Гц, 1H), 6,68-6,61 (м, 2H), 6,44 (д, J=13,5 Гц, 1H), 4,43-4,32 (м, 1H), 2,00-1,87 (м, 1H), 1,81-1,65 (м, 1H), 0,84 (т, J=6,9 Гц, 3H);19F ЯМР (282 МГц, DMSO-d6) δ -69,0 (с, 1F), -110,1 (с, 1F); MS (ES+) m/z 436,0 (M+1), 438,0 (M+1).
ПРИМЕР 87
Синтез (R)- и (S)-3-хлор-4-(1-фенилэтокси)-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида

В смесь 3-хлор-N-(2,4-диметоксибензил)-4-фтор-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида (0,20 г, 0,45 ммоль) и (R)-1-фенилэтан-1-ола (0,055 мл, 0,45 ммоль) в безводном диметилсульфоксиде (2 мл) добавляли карбонат цезия (0,352 г, 1,08 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 24 ч. Смесь разводили этилацетатом (5 мл) и водой (5 мл), и водная фаза была с этилацетатом (3×5 мл). Объединенные органические фазы промывали солевым раствором (5 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo, растворяли в дихлорметане (5 мл) и туда добавляли трифторуксусную кислоту (0,025 мл мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 15 минут ч и затем туда добавляли метанол (10 мл). Суспензию фильтровали и фильтрат концентрировали in vacuo. Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 20% метанола в дихлорметане, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,036 г, выход 20%):1H ЯМР (300 МГц, DMSO-d6) δ 8,46 (с, 1H), 7,75 (д, J=2,1 Гц, 1H), 7,61 (дд, J=8,7, 2,4 Гц, 1H), 7,43-7,24 (м, 5H), 7,20 (д, J=8,7 Гц, 1H), 5,75 (к, J=6,3 Гц, 1H), 1,60 (д, J=6,3 Гц, 3H), NH сульфонамида не наблюдали; MS (ES-) m/z 394,0 (M - 1), 396,0 (M - 1).
ПРИМЕР 88
Синтез (S)-3-хлор-4-(1-фенилэтокси)-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида

Следуя процедуре как описано для примера 87 и выполняя некритические вариации при необходимости, чтобы заменять (R)-1-фенилэтан-1-ол на (S)-1-фенилэтан-1-ол, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,014 г, выход 8%):1H ЯМР (300 МГц, DMSO-d6) δ 8,42 (с, 1H), 7,75 (д, J=2,1 Гц, 1H), 7,60 (дд, J=8,7, 2,4 Гц, 1H), 7,41-7,23 (м, 5H), 7,19 (д, J=8,7, 1H), 5,73 (к, J=6,3 Гц, 1H), 1,59 (д, J=6,3 Гц, 3H), NH сульфонамида не наблюдали; MS (ES-) m/z 394,0 (M - 1), 396,0 (M - 1).
ПРИМЕР 89
Синтез (S)-5-хлор-2-фтор-4-((1-фенилпропил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил (S)-5-хлор-2-фтор-4-((1-фенилпропил)амино)фенил)сульфонил)(тиазол-4-ил)карбамата

В смесь трет-бутил ((5-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,317 г, 0,732 ммоль) и (S)-1-фенилпропан-1-амина (0,098 г, 0,73 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли карбонат цезия (0,573 г, 1,76 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 17 ч. Реакционную смесь разводили этилацетатом (5 мл) и водой (5 мл) и водную фазу экстрагировали этилацетатом (3×5 мл). Объединенные органические фазы промывали солевым раствором (5 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде бесцветного масла (0,143 г, выход 37%): MS (ES-) m/z 524,1 (M - 1), 526,1 (M - 1).
Стадия 2. Получение (S)-5-хлор-2-фтор-4-((1-фенилпропил)амино)-N-(тиазол-4-ил)бензолсульфонамида

В смесь трет-бутил (S)-5-хлор-2-фтор-4-((1-фенилпропил)амино)фенил)-сульфонил)(тиазол-4-ил)карбамата (0,143 г, 0,273 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (1 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Реакционную смесь разводили метанолом (10 мл) и получаемую суспензию фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 12 до 70% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,060 г, выход 19%):1H ЯМР (300 МГц, DMSO-d6) δ 11,04 (с, 1H), 8,82 (д, J=2,1 Гц, 1H), 7,54 (д, J=7,2 Гц, 1H), 7,38 (д, J=7,2 Гц, 1H), 7,28 (т, J=6,9 Гц, 1H), 7,20-7,16 (м, 1H), 6,92 (д, J=2,1 Гц, 1H), 6,58 (д, J=7,8 Гц, 1H), 6,44 (д, J=13,5 Гц, 1H), 4,40 (к, J=6,9 Гц, 1H), 2,04-1,89 (м, 1H), 1,80-1,66 (м, 1H), 0,84 (т, J=7,2 Гц, 3H); MS (ES-) m/z 424,1 (M - 1), 426,1 (M - 1).
ПРИМЕР 90
Синтез 3-хлор-4-(метил(1-фенилпропил)амино)-N-(тиазол-2-ил)бензолсульфонамида
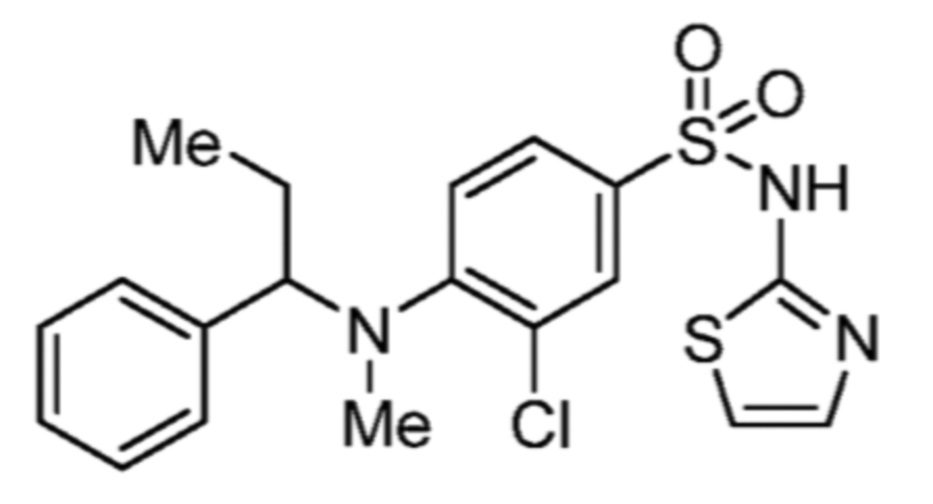
В смесь 4-бром-3-хлор-N-(тиазол-2-ил)бензолсульфонамида (0,200 г, 0,568 ммоль), 2-ди-трет-бутилфосфино-2′,4′,6′-триизопропилбифенила (0,012 г, 0,028 ммоль), трис(дибензилиденацетон)дипалладия (0,013 г, 0,014 ммоль) и трет-бутоксида натрия (0,164 г, 1,70 ммоль) в безводном толуоле (3 мл) добавляли N-метил-1-фенилпропан-1-амин (0,093 г, 0,625 ммоль). Реакционную смесь дегазировали пропусканием потока азота через нее и затем нагревали до 100°C в течение 18 ч. Реакционную смесь оставляли остывать до температуры окружающей среды, разводили этилацетатом (10 мл) и фильтровали через подушку из целита. Фильтрат концентрировали in vacuo и остаток очищали два раза с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 20 до 100% этилацетата в гексанах, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,011 г, выход 4%):1H ЯМР (300 МГц, DMSO-d6) δ 12,80 (с, 1H), 7,75-7,69 (м, 1H), 7,55 (д, J=8,4 Гц, 1H), 7,35-7,19 (м, 4H), 7,17-7,09 (м, 2H), 6,93 (д, J=8,7 Гц, 1H), 6,86-6,80 (м, 1H), 4,57-4,47 (м, 1H), 2,52 (с, 3H), 2,14-1,89 (м, 2H), 0,81 (т, J=6,6 Гц, 3H); MS (ES-) m/z 420,1 (M - 1), 422,1 (M - 1).
ПРИМЕР 91
Синтез (R)-3-хлор-4-((2-гидрокси-1-фенилэтил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение (R)-3-хлор-N-(2,4-диметоксибензил)-4-((2-гидрокси-1-фенилэтил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 2, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять (S)-1-(1-нафтил)этиламин на (R)-2-амино-2-фенилэтан-1-ол, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,23 г, выход 7%):1H ЯМР (300 МГц, CDCl3) δ 7,67 (дд, J=2,2, 0,8 Гц, 1H), 7,43-7,32 (м, 8H), 7,14 (д, J=9,0 Гц, 1H), 6,96 (дд, J=3,6, 0,9 Гц, 1H), 6,39-6,34 (м, 2H), 5,78 (д, J=5,5 Гц, 1H), 5,03 (с, 2H), 4,60-4,54 (м, 1H), 4,07-4,02 (м, 1H), 3,91-3,84 (м, 2H), 3,77 (с, 3H), 3,69 (с, 3H); MS (ES+) m/z 560,1 (M+1), 562,1 (M+1).
Стадия 2. Получение (R)-3-хлор-4-((2-гидрокси-1-фенилэтил)амино)-N-(тиазол-2-ил)бензолсульфонамида
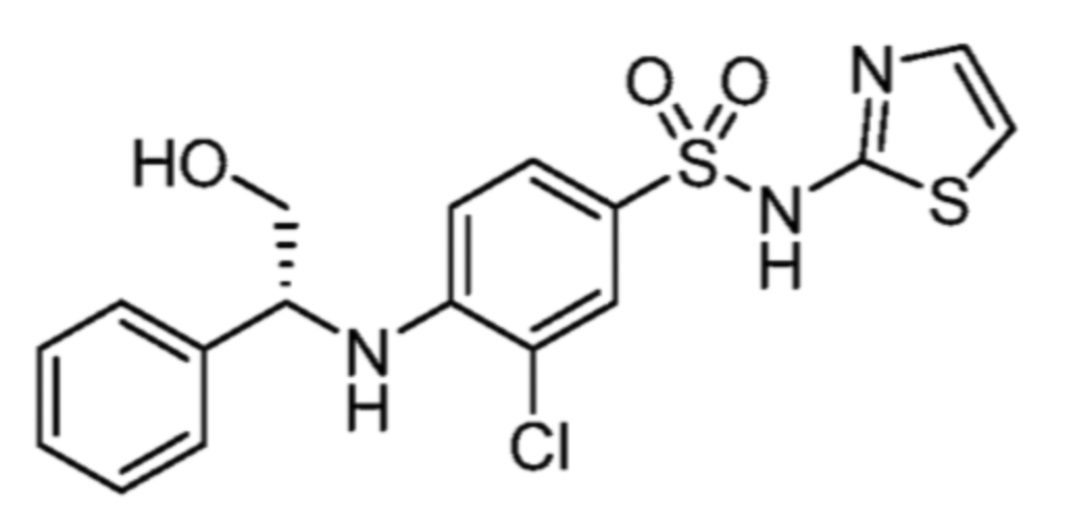
Следуя процедуре как описано для примера 2, стадии 3 и выполняя некритические вариации при необходимости, чтобы заменять (S)-5-бром-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилпропил)амино)-N-(тиазол-2-ил)бензолсульфонамид на (R)-3-хлор-N-(2,4-диметоксибензил)-4-((2-гидрокси-1-фенилэтил)амино)-N-(тиазол-2-ил)бензолсульфонамида, очисткой с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,10 г, выход 59%):1H ЯМР (300 МГц, DMSO-d6) δ 12,57 (ушир. с, 1H), 7,57 (д, J=2,2 Гц, 1H), 7,38-7,24 (м, 5H), 7,22-7,15 (м, 2H), 7,73 (д, J=4,5 Гц, 1H), 6,46 (д, J=8,7 Гц, 1H), 6,07 (д, J=5,8 Гц, 1H), 4,57-4,49 (м, 1H), 3,75-3,58 (м, 2H), OH не наблюдали; MS (ES+) m/z 410,1 (M+1), 412,1 (M+1).
ПРИМЕР 92
Синтез (S)-5-хлор-4-((1-(2-хлорфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано для примера 5, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять гидрохлорид (S)-1-(5-хлор-2-фторфенил)этан-1-амина на (S)-1-(2-хлорфенил)этан-1-амин, очисткой с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,085 г, выход 28%):1H ЯМР (300 МГц, DMSO-d6) δ 11,10 (с, 1H), 8,82 (д, J=2,2 Гц, 1H), 7,58 (д, J=7,6 Гц, 1H), 7,45-7,39 (м, 2H), 7,31-7,20 (м, 2H), 6,93 (д, J=2,2 Гц, 1H), 6,87-6,81 (м, 1H), 6,02 (д, J=13,1 Гц, 1H), 4,92-4,80 (м, 1H), 1,50 (д, J=6,7 Гц, 3H); MS (ES+) m/z 446,0 (M+1), 448,0 (M+1).
ПРИМЕР 93
Синтез 2,2,2-трифторацетата 5-хлор-2-фтор-4-((5,6,7,8-тетрагидроизохинолин-8-ил)амино)-N-(тиазол-2-ил)бензолсульфонамида

В смесь 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамида (0,46 г, 1,0 ммоль) и 5,6,7,8-тетрагидроизохинолин-8-амина (0,15, 1,0 ммоль) в безводном диметилсульфоксиде (8 мл) добавляли карбонат калия (0,41 г, 3,0 ммоль) и смесь нагревали до 60°C в течение 3 ч. Реакционную смесь оставляли остывать до температуры окружающей среды, разводили этилацетатом (60 мл) и промывали насыщенным хлоридом аммония (50 мл) и солевым раствором (30 мл). Органический слой сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo и остаток растворяли в дихлорметане (20 мл). Затем в этот раствор добавляли трифторуксусную кислоту (5 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 40 минут. Реакционную смесь концентрировали in vacuo и к остатку добавляли метанол (20 мл). Суспензию фильтровали и фильтрат концентрировали in vacuo. Остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,075 г, выход 14%):1H ЯМР (300 МГц, DMSO-d6) δ 12,84 (ушир. с, 1H), 8,62 (с, 1H), 8,57 (д, J=5,8 Гц, 1H), 7,68 (д, J=5,8 Гц, 1H), 7,64 (д, J=7,4 Гц, 1H), 7,29 (д, J=4,6 Гц, 1H), 7,01 (д, J=13,4 Гц, 1H), 6,85 (д, J=4,6 Гц, 1H), 6,72-6,69 м, 1H), 5,07-4,99 (м, 2H), 2,03-1,79 (м, 4H), NH и COOH не наблюдали;19F ЯМР (282 МГц, DMSO-d6) δ 73,9 (с, 3F), 109,2 (с, 1F); MS (ES+) m/z 439,0 (M+1), 441,0 (M+1).
ПРИМЕР 94
Синтез 2,2,2-трифторацетата 5-хлор-2-фтор-4-((5,6,7,8-тетрагидроизохинолин-5-ил)амино)-N-(тиазол-2-ил)бензолсульфонамида
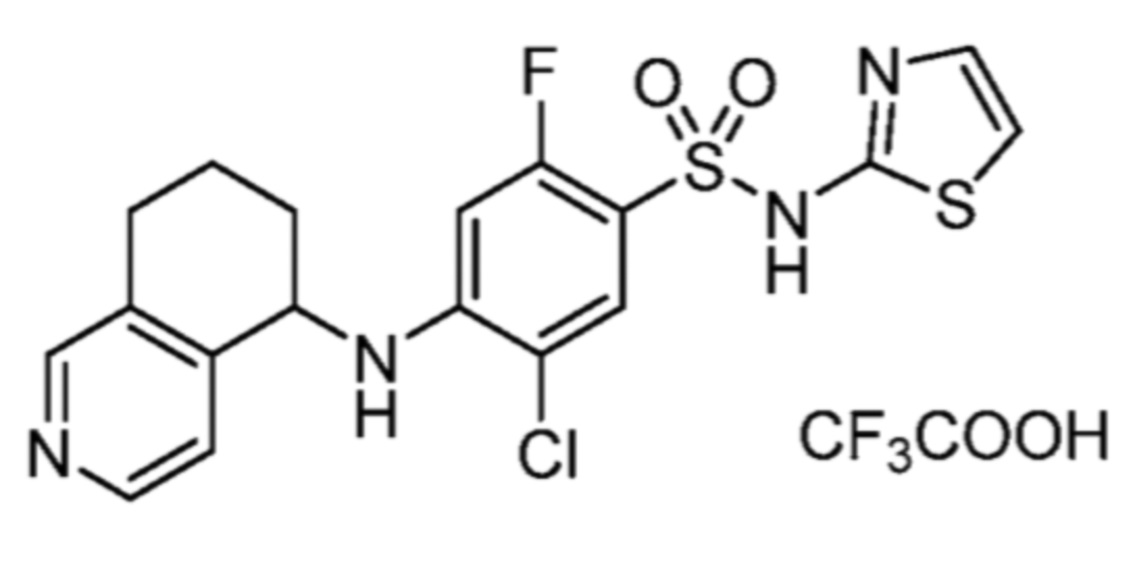
Следуя процедуре как описано для примера 93 и выполняя некритические вариации при необходимости, чтобы заменять 5,6,7,8-тетрагидроизохинолин-8-амин на 5,6,7,8-тетрагидроизохинолин-5-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,070 г, выход 13%):1H ЯМР (300 МГц, DMSO-d6) δ 12,86 (ушир. с, 1H), 8,68 (с, 1H), 8,55 (д, J=5,8 Гц, 1H), 7,67-7,61 (м, 2H), 7,29 (д, J=4,6 Гц, 1H), 6,97 (д, J=13,3 Гц, 1H), 6,85 (д, J=4,6 Гц, 1H), 6,97 (дд, J=9,0, 1,4 Гц, 1H), 5,08-5,00 (м, 2H), 2,96-2,83 (м, 2H), 2,13-1,83 (м, 4H), NH и COOH не наблюдали;19F ЯМР (282 МГц, DMSO-d6) δ -73,9 (с, 3F), -109,1 (с, 1F); MS (ES+) m/z 439,0 (M+1), 441,0 (M+1).
ПРИМЕР 95
Синтез (S)-5-хлор-4-(1-(5-хлор-2-фторфенил)этокси)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 93 и выполняя некритические вариации при необходимости, чтобы заменять 5,6,7,8-тетрагидроизохинолин-8-амин на (S)-1-(5-хлор-2-фторфенил)этан-1-ол, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,10 г, выход 43%):1H ЯМР (300 МГц, DMSO-d6) δ 12,95 (ушир. с, 1H), 7,79 (д, J=7,4 Гц, 1H), 7,53 (дд, J=6,2, 2,7 Гц, 1H), 7,45 (ддд, J=8,8, 4,5, 2,7 Гц, 1H), 7,35-7,29 (м, 3H), 6,88 (д, J=4,6 Гц, 1H), 5,94 (к, J=6,3 Гц, 1H), 1,63 (д, J=6,3 Гц, 3H); MS (ES+) m/z 465,0 (M+1), 467,0 (M+1).
ПРИМЕР 96
Синтез 2,2,2-трифторацетата 5-хлор-2-фтор-4-((изохинолин-8-илметил)амино)-N-(тиазол-4-ил)бензолсульфонамида

В смесь трет-бутил ((5-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,82 г, 2,00 ммоль) и изохинолин-8-илметанамина (0,32 г, 2,00 ммоль) в безводном DMSO (12 мл) добавляли карбонат калия (0,28 г, 2,00 ммоль) и реакционную смесь нагревали до 80°C в течение 2 ч. Реакционную смесь оставляли остывать до температуры окружающей среды и разводили этилацетатом (100 мл). Смесь промывали насыщенным хлоридом аммония (2×100 мл), солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo, остаток растворяли в дихлорметане (30 мл) и туда добавляли трифторуксусную кислоту (10 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 40 минут и затем концентрировали in vacuo. Получаемый остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, после чего следовало растирание с метанолом (35 мл) для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,30 г, выход 33%):1H ЯМР (300 МГц, DMSO-d6) δ 11,08 (ушир. с, 1H), 9,85 (с, 1H), 8,84 (д, J=2,2 Гц, 1H), 8,64 (д, J=6,1 Гц, 1H), 8,23 (д, J=6,1 Гц, 1H), 8,04 (д, J=8,3 Гц, 1H), 7,94-7,87 (м, 1H), 7,64-7,57 (м, 2H), 7,46-7,38 (м, 1H), 6,95 (д, J=2,0 Гц, 1H), 6,59 (д, J=13,3 Гц, 1H), 5,10 (д, J=6,0 Гц, 2H), COOH не наблюдали;19F ЯМР (282 МГц, DMSO-d6) δ -74,1 (с, 3F), -109,6 (с, 1F); MS (ES+) m/z 449,0 (M+1), 451,0 (M+1).
ПРИМЕР 97
Синтез формиата (R)-5-хлор-2-фтор-4-((1-(пиридин-3-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 93 и выполняя некритические вариации при необходимости, чтобы заменять 5,6,7,8-тетрагидроизохинолин-8-амин на (R)-1-(пиридин-3-ил)этан-1-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,125 г, выход 30%):1H ЯМР (300 МГц, DMSO-d6) δ 12,78 (ушир. с, 1H), 8,65 (д, J=1,9 Гц, 1H), 8,43 (дд, J=4,7, 1,5 Гц, 1H), 8,14 (с, 0,3H), 7,81 (дт, J=8,0, 1,9 Гц, 1H), 7,59 (д, J=7,3 Гц, 1H), 7,34 (дд, J=7,8, 4,8 Гц, 1H), 7,25 (д, J=4,6 Гц, 1H), 6,81 (д, J=4,5 Гц, 1H), 6,60 (дд, J=7,7, 1,4 Гц, 1H), 6,51 (д, J=13,1 Гц, 1H), 4,86-4,76 (м, 1H), 1,56 (д, J=6,8 Гц, 3H), COOH не наблюдали; MS (ES+) m/z 413,0 (M+1), 415,0 (M+1).
ПРИМЕР 98
Синтез формиата 5-хлор-2-фтор-4-((5,6,7,8-тетрагидрохинолин-5-ил)амино)-N-(тиазол-2-ил)бензолсульфонамида
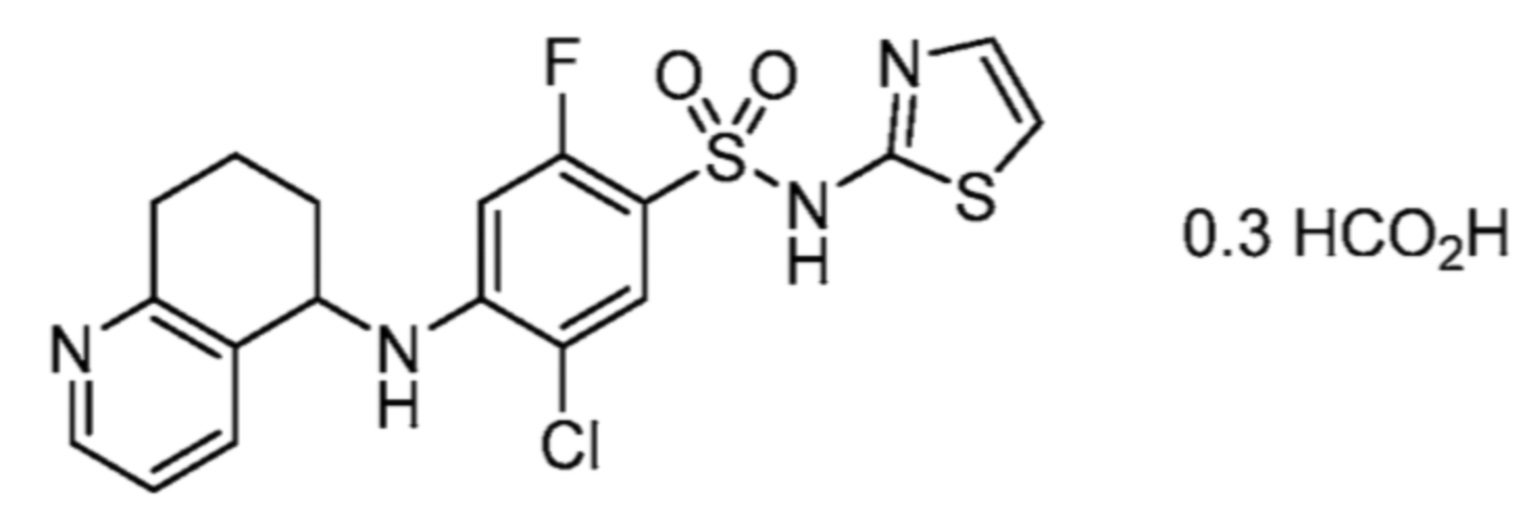
Следуя процедуре как описано для примера 93 и выполняя некритические вариации при необходимости, чтобы заменять 5,6,7,8-тетрагидроизохинолин-8-амин на 5,6,7,8-тетрагидрохинолин-5-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,195 г, выход 44%):1H ЯМР (300 МГц, DMSO-d6) δ 12,84 (ушир. с, 1H), 8,44 (дд, 4,9, 1,5 Гц, 1H), 8,14 (с, 0,3 H), 7,67 (дд, J=7,8, 0,9 Гц, 1H), 7,32-7,27 (м, 2H), 6,93 (д, 13,4 Гц, 1H), 6,84 (д, J=4,6 Гц, 1H), 6,57-6,53 (м, 1H), 4,98-4,92 (м, 1H), 2,98-2,82 (м, 2H), 2,04-1,78 (м, 4H), NH и COOH не наблюдали; MS (ES+) m/z 439,0 (M+1), 441,0 (M+1).
ПРИМЕР 99
Синтез 3-хлор-4-((циклопропил(фенил)метил)амино)-N-(тиазол-2-ил)бензолсульфонамида

В раствор 4-бром-3-хлор-N-(тиазол-2-ил)бензолсульфонамида (0,200 г, 0,568 ммоль), 2-ди-трет-бутилфосфино-2′,4′,6′-триизопропилбифенила (0,012 г, 0,028 ммоль), трис(дибензилиденацетон)дипалладия(0) (0,013 г, 0,014) и трет-бутоксида натрия (0,273 г, 2,85 ммоль) в безводном толуоле (3 мл) добавляли циклопропил(фенил)метанамин (0,096 г, 0,62 ммоль) ммоль). Реакционную смесь продували аргоном и затем нагревали до 100°C в течение 17 ч. Реакционную смесь оставляли остывать до температуры окружающей среды, разводили этилацетатом (5 мл) и фильтровали через подушку из целита. Фильтрат концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 80% этилацетата в гексанах. Дополнительная очистка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, давала указанное в заголовке соединение в виде бесцветного твердого вещества (0,010 г, выход 4%):1H ЯМР (300 МГц, DMSO-d6) δ 12,58 (ушир. с, 1H), 7,57 (с, 1H), 7,42 (д, J=7,5 Гц, 2H), 7,32 (дд, J=6,5, 8,2 Гц, 3H), 7,26-7,16 (м, 2H), 6,80-6,73 (м, 1H), 6,47 (д, J=8,9 Гц, 1H), 6,31 (д, J=6,4 Гц, 1H), 3,80 (т, J=7,7 Гц, 1H), 1,51-1,35 (м, 1H), 0,66-0,53 (м, 1H), 0,53-0,31 (м, 3H); MS (ES+) m/z 420,0 (M+1), 422,0 (M+1).
ПРИМЕР 100
Синтез 5-хлор-4-((циклопропил(фенил)метил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

В раствор циклопропил(фенил)метанамина (0,096 г, 0,65 ммоль) и 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамида (0,300 г, 0,652 ммоль) в безводном диметилсульфоксиде (2,6 мл) добавляли карбонат цезия (0,509 г, 1,56 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 17 ч. Реакционную смесь разводили этилацетатом (5 мл) и водой (5 мл) и водную фазу экстрагировали этилацетатом (2×5 мл). Объединенные органические слои промывали солевым раствором (2×5 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали in vacuo и остаток растворяли в дихлорметане (9 мл). В этот раствор добавляли трифторуксусную кислоту (0,14 мл, 1,8 ммоль) при 0°C и реакционную смесь перемешивали при 0°C в течение 30 мин. Реакционную смесь гасили добавлением насыщенного раствора бикарбоната натрия (6 мл) и водную фазу экстрагировали дихлорметаном (2×5 мл). Объединенные органические слои промывали водой (5 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали in vacuo и остаток растирали в метаноле (7 мл). Получаемую суспензию фильтровали и фильтрат концентрировали in vacuo. Очистка остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% гидроксида аммония, в качестве элюента, давала указанное в заголовке соединение в виде бесцветного твердого вещества (0,025 г, выход 9%):1H ЯМР (300 МГц, DMSO-d6) δ 12,71 (ушир. с, 1H), 7,57 (д, J=7,1 Гц, 1H), 7,48-7,40 (м, 2H), 7,32 (т, J=7,3 Гц, 2H), 7,26-7,18 (м, 2H), 6,78 (д, J=4,5 Гц, 1H), 6,62 (д, J=7,1 Гц, 1H), 6,30 (д, J=13,3 Гц, 1H), 3,87-3,75 (м, 1H), 1,53-1,36 (м, 1H), 0,65-0,53 (м, 1H), 0,53-0,31 (м, 3H); MS (ES+) m/z 438,0 (M+1), 440,0 (M+1).
ПРИМЕР 101
Синтез (S)-5-хлор-2-фтор-4-(1-(2-фторфенил)этокси)-N-(тиазол-4-ил)бензолсульфонамида

В раствор (S)-1-(2-фторфенил)этан-1-ола (0,102 г, 0,730 ммоль) и трет-бутил ((5-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,30 г, 0,730 ммоль) в безводном диметилсульфоксиде (3 мл) добавляли карбонат цезия (0,571 г, 1,75 ммоль) и реакционную смесь нагревали при 75°C в течение 17 ч. Реакционную смесь разводили этилацетатом (5 мл) и водой (5 мл) и водную фазу экстрагировали этилацетатом (2×5 мл). Объединенные органические слои промывали солевым раствором (5 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,062 г, выход 20%):1H ЯМР (300 МГц, DMSO-d6) δ 11,34 (с, 1H), 8,88 (д, J=2,2 Гц, 1H), 7,80 (д, J=7,5 Гц, 1H), 7,51-7,33 (м, 2H), 7,30-7,18 (м, 3H), 7,06 (д, J=2,2 Гц, 1H), 5,97 (к, J=6,4 Гц, 1H), 1,63 (д, J=6,4 Гц, 3H); MS (ES+) m/z 431,0 (M+1), 432,9 (M+1).
ПРИМЕР 102
Синтез (R)-5-хлор-2-фтор-4-((1,2,3,4-тетрагидронафтален-1-ил)амино)-N-(тиазол-2-ил)бензолсульфонамида

В раствор (R)-1,2,3,4-тетрагидронафтален-1-амина (0,096 г, 0,65 ммоль) и 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамида (0,300 г, 0,652 ммоль) в безводном диметилсульфоксиде (2,6 мл) добавляли карбонат цезия (0,509 г, 1,56 ммоль) и получаемую суспензию перемешивали при температуре окружающей среды в течение 17 ч. Реакционную смесь разводили этилацетатом (5 мл) и водой (5 мл) и водную фазу экстрагировали этилацетатом (2×5 мл). Объединенные органические слои промывали солевым раствором (2×5 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 30% этилацетата в гексанах. Затем получаемый остаток растворяли в дихлорметане (7 мл) и туда добавляли трифторуксусную кислоту (0,12 мл, 1,6 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 10 минут и затем концентрировали in vacuo. Остаток растирали в метаноле (5 мл) и получаемую суспензию фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 15% метанола в дихлорметане, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,041 г, выход 14%):1H ЯМР (300 МГц, DMSO-d6) δ 12,78 (с, 1H), 7,61 (д, J=7,7 Гц, 1H), 7,27 (д, J=5,1 Гц, 1H), 7,22-7,08 (м, 4H), 6,91-6,78 (м, 2H), 6,33-6,22 (м, 1H), 4,92-4,78 (м, 1H), 2,88-2,65 (м, 2H), 2,03-1,68 (м, 4H); MS (ES+) m/z 438,0 (M+1), 440,0 (M+1).
ПРИМЕР 103
Синтез 3-хлор-4-((3-фенилоксетан-3-ил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 99 и выполняя некритические вариации при необходимости, чтобы заменять циклопропил(фенил)метанамин на 3-фенилоксетан-3-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,015 г, выход 6%):1H ЯМР (300 МГц, DMSO-d6) δ 12,59 (ушир. с, 1H), 7,63 (д, J=2,2 Гц, 1H), 7,58-7,50 (м, 2H), 7,44-7,35 (м, 2H), 7,33-7,25 (м, 3H), 7,19 (д, J=4,5 Гц, 1H), 6,74 (д, J=4,5 Гц, 1H), 5,74 (д, J=8,6 Гц, 1H), 4,94 (д, J=6,4 Гц, 2H), 4,82 (д, J=6,4 Гц, 2H); MS (ES+) m/z 422,1 (M+1), 424,1 (M+1).
ПРИМЕР 104
Синтез 3-хлор-4-((1-фенилциклобутил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 99 и выполняя некритические вариации при необходимости, чтобы заменять циклопропил(фенил)метанамин на 1-фенилциклобутан-1-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,015 г, выход 5%):1H ЯМР (300 МГц, DMSO-d6) δ 7,57 (д, J=2,1 Гц, 1H), 7,53-7,46 (м, 2H), 7,37-7,28 (м, 2H), 7,28-7,12 (м, 3H), 6,70 (д, J=4,5 Гц, 1H), 6,59 (с, 1H), 6,03 (д, J=8,7 Гц, 1H), 2,65-2,40 (м, 4H), 2,12-1,85 (м, 2H), NH сульфонамида не наблюдали; MS (ES+) m/z 420,1 (M+1), 422,1 (M+1).
ПРИМЕР 105
Синтез 5-хлор-2-фтор-4-((1-фенилциклобутил)амино)-N-(тиазол-2-ил)бензолсульфонамида

В раствор 1-фенилциклобутан-1-амина (0,120 г, 0,65 ммоль) и 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамида (0,300 г, 0,652 ммоль) в безводном диметилсульфоксиде (2,5 мл) добавляли карбонат цезия (0,509 г, 1,56 ммоль) и получаемую суспензию перемешивали при температуре окружающей среды в течение 17 ч. Реакционную смесь разводили этилацетатом (5 мл) и водой (5 мл) и водную фазу экстрагировали этилацетатом (2×5 мл). Объединенные органические слои промывали солевым раствором (2×5 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали in vacuo и получаемый остаток растворяли в дихлорметане (6 мл). Туда добавляли трифторуксусную кислоту (0,16 мл, 2,1 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 20 минут и затем концентрировали in vacuo. Остаток растирали в метаноле (5 мл) и получаемую суспензию фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% муравьиную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,016 г, выход 6%):1H ЯМР (300 МГц, DMSO-d6) δ 7,57 (д, J=7,3 Гц, 1H), 7,51 (д, J=7,5 Гц, 2H), 7,35 (т, J=7,8 Гц, 2H), 7,26-7,17 (м, 2H), 6,96 (с, 1H), 6,75 (д, J=4,6 Гц, 1H), 5,71 (д, J=12,7 Гц, 1H), 2,63-2,43 (м, 4H), 2,09-1,84 (м, 2H), NH сульфонамида не наблюдали; MS (ES+) m/z 438,1 (M+1), 440,1 (M+1).
ПРИМЕР 106
Синтез 5-хлор-2-фтор-4-((1-(3-фторфенил)циклобутил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 105 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклобутан-1-амин на 1-(3-фторфенил)циклобутан-1-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,011 г, выход 4%):1H ЯМР (300 МГц, DMSO-d6) δ 12,75 (с, 1H), 7,59 (д, J=7,4 Гц, 1H), 7,45-7,30 (м, 3H), 7,25 (д, J=4,6 Гц, 1H), 7,08-7,02 (м, 1H), 6,82 (д, J=4,6 Гц, 1H), 6,56 (с, 1H), 5,74 (д, J=12,7 Гц, 1H), 2,66-2,44 (м, 4H), 2,07-1,86 (м, 2H); MS (ES+) m/z 456,0 (M+1), 458,0 (M+1).
ПРИМЕР 107
Синтез 5-хлор-2-фтор-4-((1-(2-фторфенил)циклобутил)амино)-N-(тиазол-2-ил)бензолсульфонамида
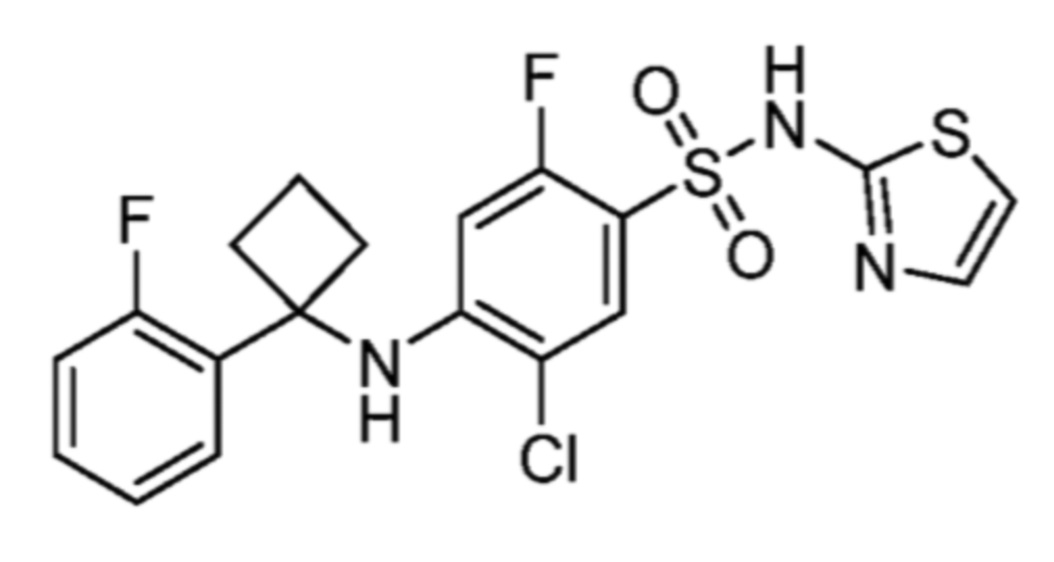
Следуя процедуре как описано в примере 105 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклобутан-1-амин на 1-(2-фторфенил)циклобутан-1-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,008 г, выход 3%):1H ЯМР (300 МГц, DMSO-d6) δ 12,75 (с, 1H), 7,64 (т, J=8,0 Гц, 1H), 7,57 (д, J=7,3 Гц, 1H), 7,36-7,10 (м, 4H), 6,82 (д, J=4,8 Гц, 1H), 6,59-6,52 (м, 1H), 6,08 (д, J=12,9 Гц, 1H), 2,81-2,66 (м, 2H), 2,66-2,54 (м, 2H), 2,13-1,96 (м, 1H), 1,96-1,78 (м, 1H); MS (ES+) m/z 456,0 (M+1), 458,0 (M+1).
ПРИМЕР 108
Синтез (S)-5-хлор-4-(1-(3,4-дихлорфенил)этокси)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида
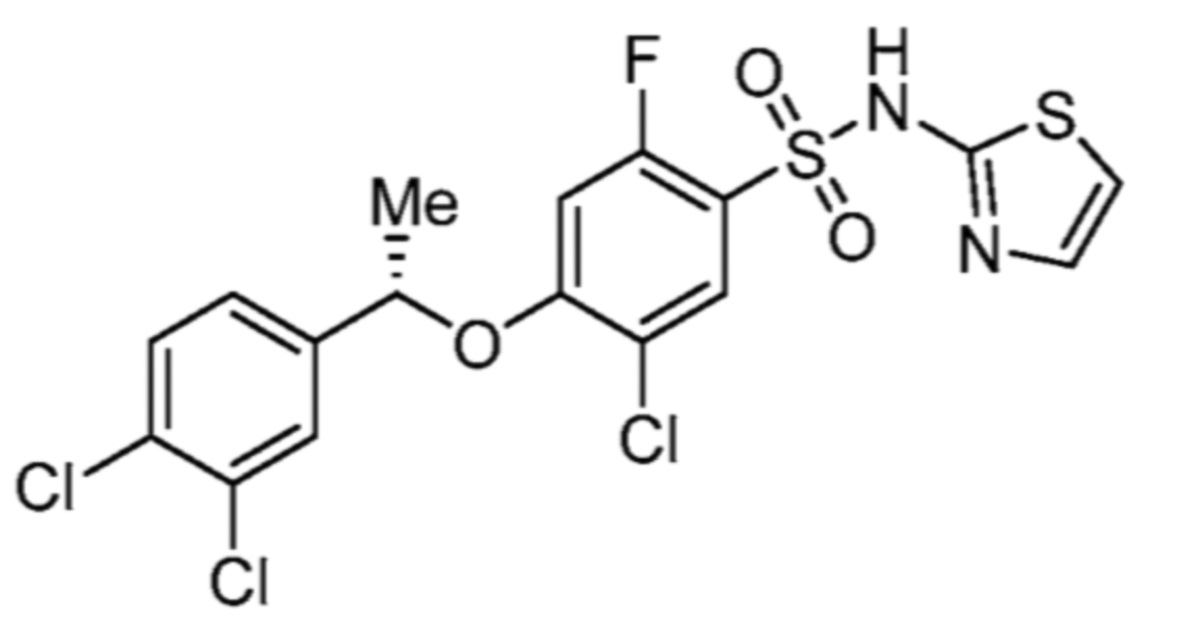
Следуя процедуре как описано в примере 105 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклобутан-1-амин на (S)-1-(3,4-дихлорфенил)этан-1-ол, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,106 г, выход 34%):1H ЯМР (300 МГц, DMSO-d6) δ 12,93 (с, 1H), 7,77 (д, J=7,4 Гц, 1H), 7,70 (д, J=2,1 Гц, 1H), 7,65 (д, J=8,4 Гц, 1H), 7,38 (дд, J=2,1, 8,4 Гц, 1H), 7,30 (д, J=4,6 Гц, 1H), 7,26 (д, J=11,9 Гц, 1H), 6,88 (д, J=4,6 Гц, 1H), 5,79 (к, J=6,4 Гц, 1H), 1,59 (д, J=6,4 Гц, 3H); MS (ES+) m/z 480,9 (M+1), 482,9 (M+1), 484,9 (M+1).
ПРИМЕР 109
Синтез (S)-5-хлор-2-фтор-4-((3-метил-1-фенилбутил)амино)-N-(тиазол-2-ил)бензолсульфонамида
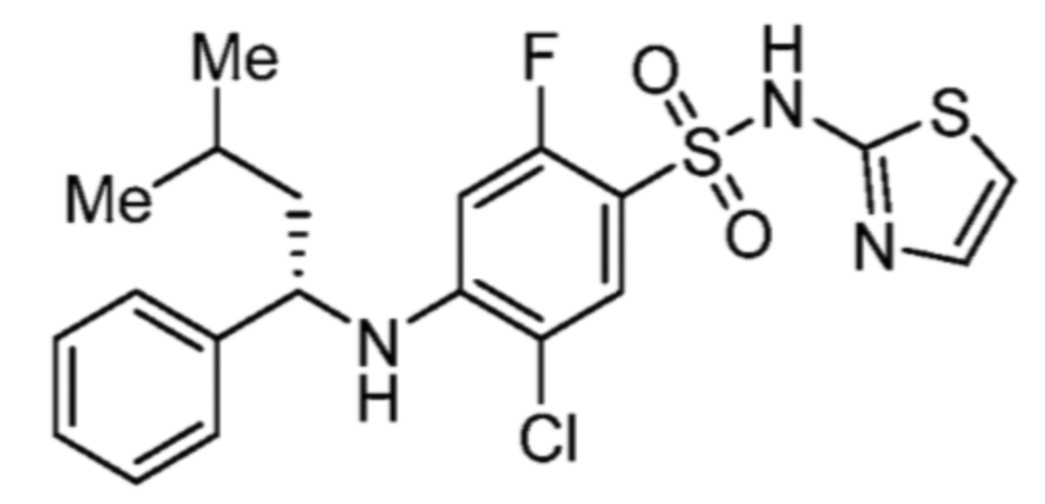
Следуя процедуре как описано в примере 105 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклобутан-1-амин на (S)-3-метил-1-фенилбутан-1-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,038 г, выход 10%):1H ЯМР (300 МГц, DMSO-d6) δ 12,74 (с, 1H), 7,55 (д, J=7,4 Гц, 1H), 7,48-7,39 (м, 2H), 7,35-7,26 (м, 2H), 7,26-7,16 (м, 2H), 6,81 (д, J=4,5 Гц, 1H), 6,54 (д, J=13,3 Гц, 1H), 6,51-6,44 (м, 1H), 4,63-4,50 (м, 1H), 2,04-1,89 (м, 1H), 1,68-1,43 (м, 2H), 0,93 (д, J=6,3 Гц, 3H), 0,87 (д, J=6,3 Гц, 3H); MS (ES+) m/z 454,1 (M+1), 456,1 (M+1).
ПРИМЕР 110
Синтез (R)-5-хлор-2-фтор-4-((3-метил-1-фенилбутил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 105 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклобутан-1-амин на (R)-3-метил-1-фенилбутан-1-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,056 г, выход 14%):1H ЯМР (300 МГц, DMSO-d6) δ 12,73 (с, 1H), 7,55 (д, J=7,4 Гц, 1H), 7,47-7,40 (м, 2H), 7,34-7,27 (м, 2H), 7,26-7,16 (м, 2H), 6,81 (д, J=4,5 Гц, 1H), 6,54 (д, J=13,3 Гц, 1H), 6,51-6,44 (м, 1H), 4,63-4,49 (м, 1H), 2,05-1,89 (м, 1H), 1,68-1,43 (м, 2H), 0,93 (д, J=6,3 Гц, 3H), 0,87 (д, J=6,3 Гц, 3H); MS (ES+) m/z 454,1 (M+1), 456,1 (M+1).
ПРИМЕР 111
Синтез (S)-5-хлор-2-фтор-4-((1-фенилбутил)амино)-N-(тиазол-2-ил)бензолсульфонамида

В раствор (S)-1-фенилбутан-1-амина (0,130 г, 0,872 ммоль) и 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамида (0,400 г, 0,870 ммоль) в безводном диметилсульфоксиде (3,5 мл) добавляли карбонат цезия (0,685 г, 2,10 ммоль) и получаемую суспензию перемешивали при температуре окружающей среды в течение 17 ч. Реакционную смесь разводили этилацетатом (5 мл) и водой (5 мл) и водную фазу экстрагировали этилацетатом (2×5 мл). Объединенные органические слои промывали солевым раствором (5 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали in vacuo и очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 50% этилацетата в гексанах. Затем получаемый остаток растворяли в дихлорметане (7,5 мл) и туда добавляли трифторуксусную кислоту (0,12 мл, 1,6 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 30 минут и затем концентрировали in vacuo. Остаток растирали в метаноле (5 мл) и получаемую суспензию фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% муравьиную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,004 г, выход 1%):1H ЯМР (300 МГц, DMSO-d6) δ 12,66 (ушир. с, 1H), 7,55 (д, J=7,3 Гц, 1H), 7,45-7,38 (м, 2H), 7,35-7,26 (м, 2H), 7,25-7,15 (м, 2H), 6,75 (д, J=4,4 Гц, 1H), 6,58-6,40 (м, 2H), 4,50 (дт, J=7,7, 6,6 Гц, 1H), 2,07-1,90 (м, 1H), 1,80-1,60 (м, 1H), 1,48-1,31 (м, 1H), 1,31-1,16 (м, 1H), 0,88 (т, J=7,3 Гц, 3H); MS (ES+) m/z 440,0 (M+1), 442,0 (M+1).
ПРИМЕР 112
Синтез (S)-5-хлор-2-фтор-4-((2-метокси-1-фенилэтил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 105 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклобутан-1-амин на (S)-2-метокси-1-фенилэтан-1-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,014 г, выход 5%):1H ЯМР (300 МГц, DMSO-d6) δ 12,71 (ушир. с, 1H), 7,59 (д, J=7,4 Гц, 1H), 7,45-7,39 (м, 2H), 7,37-7,29 (м, 2H), 7,28-7,21 (м, 1H), 7,19 (д, J=4,5 Гц, 1H), 6,75 (д, J=4,5 Гц, 1H), 6,40 (д, J=13,0 Гц, 1H), 6,37-6,32 (м, 1H), 4,87-4,76 (м, 1H), 3,77 (дд, J=10,0, 8,0 Гц, 1H), 3,56 (дд, J=10,0, 4,6 Гц, 1H), 3,29 (с, 3H); MS (ES+) m/z 442,1 (M+1), 444,1 (M+1).
ПРИМЕР 113
Синтез (R)-5-хлор-2-фтор-4-((2-метокси-1-фенилэтил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 111 и выполняя некритические вариации при необходимости, чтобы заменять (S)-1-фенилбутан-1-амин на (R)-2-метокси-1-фенилэтан-1-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,037 г, выход 13%):1H ЯМР (300 МГц, DMSO-d6) δ 12,74 (ушир. с, 1H), 7,59 (д, J=7,3 Гц, 1H), 7,45-7,38 (м, 2H), 7,37-7,29 (м, 2H), 7,29-7,20 (м, 2H), 6,79 (д, J=4,6 Гц, 1H), 6,47-6,35 (м, 2H), 4,88-4,77 (м, 1H), 3,78 (дд, J=10,1, 8,0 Гц, 1H), 3,56 (дд, J=10,1, 4,6 Гц, 1H), 3,29 (с, 3H); MS (ES+) m/z 442,0 (M+1), 444,0 (M+1).
ПРИМЕР 114
Синтез (S)-5-хлор-2-фтор-4-(1-(3-фторфенил)этокси)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 105 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклобутан-1-амин на (S)-1-(3-фторфенил)этан-1-ол, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,105 г, выход 38%):1H ЯМР (300 МГц, DMSO-d6) δ 12,92 (с, 1H), 7,77 (д, J=7,4 Гц, 1H), 7,30 (д, J=4,6 Гц, 1H), 7,28-7,20 (м, 3H), 7,18-7,08 (м, 1H), 6,87 (д, J=4,6 Гц, 1H), 6,88 (д, J=4,6 Гц, 1H), 5,79 (к, J=6,4 Гц, 1H), 1,59 (д, J=6,4 Гц, 3H); MS (ES+) m/z 431,0 (M+1), 433,0 (M+1).
ПРИМЕР 115
Синтез (S)-5-хлор-2-фтор-4-(1-фенилэтокси)-N-(тиазол-2-ил)бензолсульфонамида
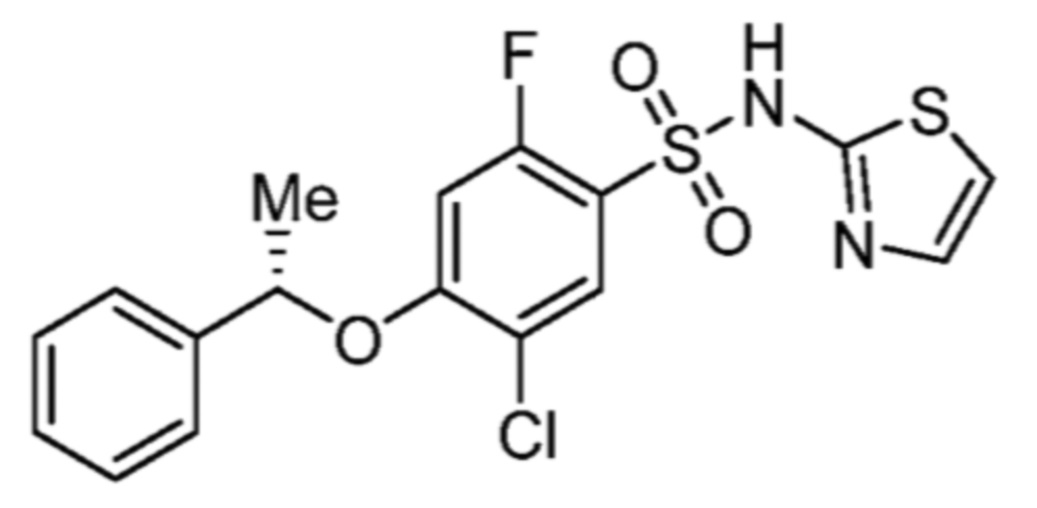
Следуя процедуре как описано в примере 102 и выполняя некритические вариации при необходимости, чтобы заменять (R)-1,2,3,4-тетрагидронафтален-1-амин на (S)-1-фенилэтан-1-ол, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,041 г, выход 15%):1H ЯМР (300 МГц, DMSO-d6) δ 12,90 (ушир. с, 1H), 7,75 (д, J=7,5 Гц, 1H), 7,45-7,22 (м, 6H), 7,17 (д, J=11,9 Гц, 1H), 6,83 (ушир. с, 1H), 5,77 (к, J=6,4 Гц, 1H), 1,59 (д, J=6,3 Гц, 3H); MS (ES+) m/z 413,0 (M+1), 415,0 (M+1).
ПРИМЕР 116
Синтез (S)-5-хлор-2-фтор-4-(1-(2-фторфенил)этокси)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 111 и выполняя некритические вариации при необходимости, чтобы заменять (S)-1-фенилбутан-1-амин на (S)-1-(2-фторфенил)этан-1-ол, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,027 г, выход 10%):1H ЯМР (300 МГц, DMSO-d6) δ 12,92 (ушир. с, 1H), 7,77 (д, J=7,4 Гц, 1H), 7,51-7,42 (м, 1H), 7,42-7,32 (м, 1H), 7,30-7,15 (м, 4H), 6,86 (д, J=4,5 Гц, 1H), 5,95 (к, J=6,3 Гц, 1H), 1,63 (д, J=6,3 Гц, 3H); MS (ES+) m/z 431,0 (M+1), 433,0 (M+1).
ПРИМЕР 117
Синтез (S)-5-хлор-4-(1-(2-хлорфенил)этокси)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 105 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклобутан-1-амин на (S)-1-(2-хлорфенил)этан-1-ол, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,015 г, выход 5%):1H ЯМР (300 МГц, DMSO-d6) δ 12,94 (с, 1H), 7,79 (д, J=7,4 Гц, 1H), 7,53-7,45 (м, 2H), 7,42-7,31 (м, 2H), 7,30 (д, J=4,6 Гц, 1H), 6,96 (д, J=11,8 Гц, 1H), 6,88 (д, J=4,6 Гц, 1H), 5,94 (к, J=6,3 Гц, 1H), 1,62 (д, J=6,3 Гц, 3H); MS (ES+) m/z 447,0 (M+1), 449,0 (M+1).
ПРИМЕР 118
Синтез (S)-5-хлор-2-фтор-4-(1-(3-фторфенил)этокси)-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано в примере 101 и выполняя некритические вариации при необходимости, чтобы заменять (S)-1-(2-фторфенил)этан-1-ол на (S)-1-(3-фторфенил)этан-1-ол, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,039 г, выход 12%):1H ЯМР (300 МГц, DMSO-d6) δ 11,33 (с, 1H), 8,88 (с, 1H), 7,80 (д, J=7,5 Гц, 1H), 7,48-7,37 (м, 1H), 7,32-7,20 (3H), 7,18-7,09 (м, 1H), 7,06 (д, J=2,2 Гц, 1H), 5,81 (к, J=6,4 Гц, 1H), 1,59 (к, J=6,4 Гц, 3H); MS (ES+) m/z 431,0 (M+1), 432,9 (M+1).
ПРИМЕР 119
Синтез (S)-5-хлор-4-(1-(2,6-дифторфенил)этокси)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 105 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклобутан-1-амин на (S)-1-(2,6-дифторфенил)этан-1-ол, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,046 г, выход 16%):1H ЯМР (300 МГц, DMSO-d6) δ 12,94 (с, 1H), 7,76 (д, J=7,4 Гц, 1H), 7,51-7,38 (м, 1H), 7,30 (д, J=4,6 Гц, 1H), 7,19-7,07 (м, 3H), 6,88 (д, J=4,6 Гц, 1H), 6,02 (к, J=6,5 Гц, 1H), 1,74 (д, J=6,5 Гц, 3H); MS (ES+) m/z 449,0 (M+1), 451,0 (M+1).
ПРИМЕР 120
Синтез (S)-5-хлор-4-(1-(2,6-дифторфенил)этокси)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано в примере 101 и выполняя некритические вариации при необходимости, чтобы заменять (S)-1-(2-фторфенил)этан-1-ол на (S)-1-(2,6-дифторфенил)этан-1-ол, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,028 г, выход 9%):1H ЯМР (300 МГц, DMSO-d6) δ 11,35 (с, 1H), 8,88 (с, 1H), 7,78 (д, J=7,5 Гц, 1H), 7,52-7,39 (м, 1H), 7,24-7,08 (м, 3H), 7,06 (д, J=2,2 Гц, 1H), 6,04 (к, J=6,5 Гц, 1H), 1,74 (д, J=6,5 Гц, 3H); MS (ES+) m/z 448,9 (M+1), 450,8 (M+1).
ПРИМЕР 121
Синтез (R)-5-хлор-2-фтор-4-(1-(2-фторфенил)этокси)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 105 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклобутан-1-амин на (R)-1-(2-фторфенил)этан-1-ол, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,056 г, выход 20%):1H ЯМР (300 МГц, DMSO-d6) δ 12,93 (с, 1H), 7,77 (д, J=7,4 Гц, 1H), 7,51-7,43 (м, 1H), 7,42-7,33 (м, 1H), 7,30 (д, J=4,6 Гц, 1H), 7,28-7,16 (м, 3H), 6,87 (д, J=4,6 Гц, 1H), 5,96 (к, J=6,4 Гц, 1H), 1,63 (д, J=6,4 Гц, 3H); MS (ES+) m/z 431,0 (M+1), 433,0 (M+1).
ПРИМЕР 122
Синтез 5-хлор-2-фтор-4-((2-фторбензил)окси)-N-(тиазол-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 105 и выполняя некритические вариации при необходимости, чтобы заменять 1-фенилциклобутан-1-амин на (2-фторфенил)метанол, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,003 г, выход 1%):1H ЯМР (300 МГц, DMSO-d6) δ 12,98 (с, 1H), 7,80 (д, J=7,3 Гц, 1H), 7,59 (дт, J=1,8, 7,6 Гц, 1H), 7,53-7,41 (м, 2H), 7,34-7,23 (м, 3H), 6,89 (д, J=4,6 Гц, 1H), 5,32 (с, 2H); MS (ES+) m/z 417,1 (M+1), 419,0 (M+1).
ПРИМЕР 123
Синтез (S)-5-хлор-4-(1-(2-хлорфенил)этокси)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида
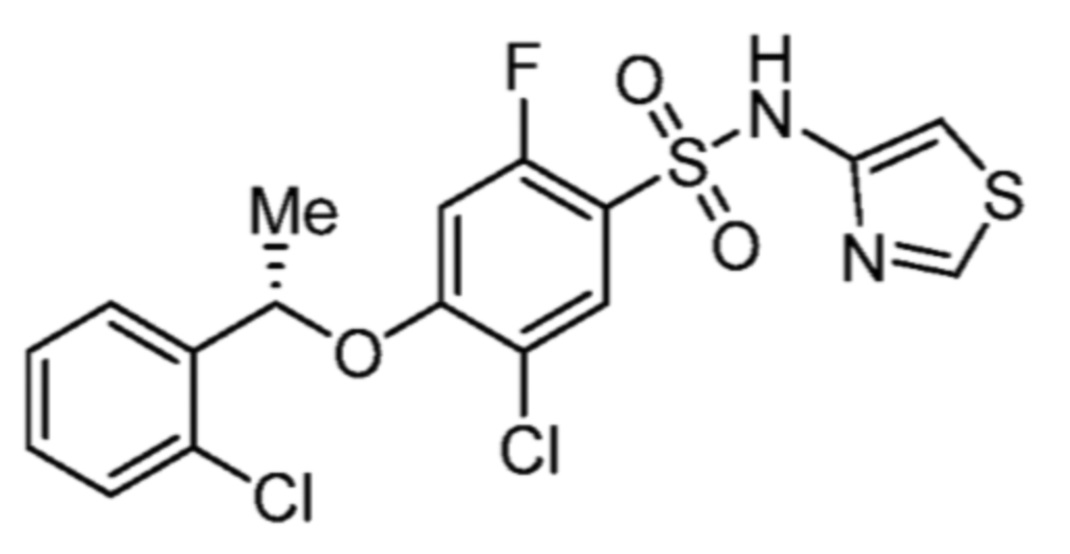
Следуя процедуре как описано в примере 101 и выполняя некритические вариации при необходимости, чтобы заменять (S)-1-(2-фторфенил)этан-1-ол на (S)-1-(2-хлорфенил)этан-1-ол, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,041 г, выход 13%):1H ЯМР (300 МГц, DMSO-d6) δ 11,36 (с, 1H), 8,88 (д, J=2,2 Гц, 1H), 7,82 (д, J=7,5 Гц, 1H), 7,53-7,45 (м, 2H), 7,43-7,32 (м, 2H), 7,06 (д, J=2,2 Гц, 1H), 7,02 (д, J=12,0 Гц, 1H), 5,96 (к, J=6,4 Гц, 1H), 1,62 (д, J=6,4 Гц, 3H); MS (ES+) m/z 447,0 (M+1), 449,0 (M+1).
ПРИМЕР 124
Синтез (S)-5-хлор-4-(1-(5-хлор-2-фторфенил)этокси)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано для примера 96 и выполняя некритические вариации при необходимости, чтобы заменять изохинолин-8-илметанамин на (S)-1-(5-хлор-2-фторфенил)этан-1-ол, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,135 г, выход 58%):1H ЯМР (300 МГц, DMSO-d6) δ 11,36 (ушир. с, 1H), 8,88 (д, J=2,2 Гц, 1H), 7,82 (д, J=7,5 Гц, 1H), 7,52 (дд, J=6,2, 2,7 Гц, 1H), 7,46 (ддд, J=8,8, 4,5, 2,7 Гц, 1H), 7,39-7,29 (м, 2H), 7,06 (д, J=2,2 Гц, 1H), 5,96 (к, J=6,3 Гц, 1H), 1,63 (д, J=6,3 Гц, 3H); MS (ES+) m/z 465,0 (M+1), 467,0 (M+1).
ПРИМЕР 125
Синтез (S)-5-хлор-2-фтор-4-((1-фенилэтил)тио)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилэтил)тио)-N-(тиазол-2-ил)бензолсульфонамида.
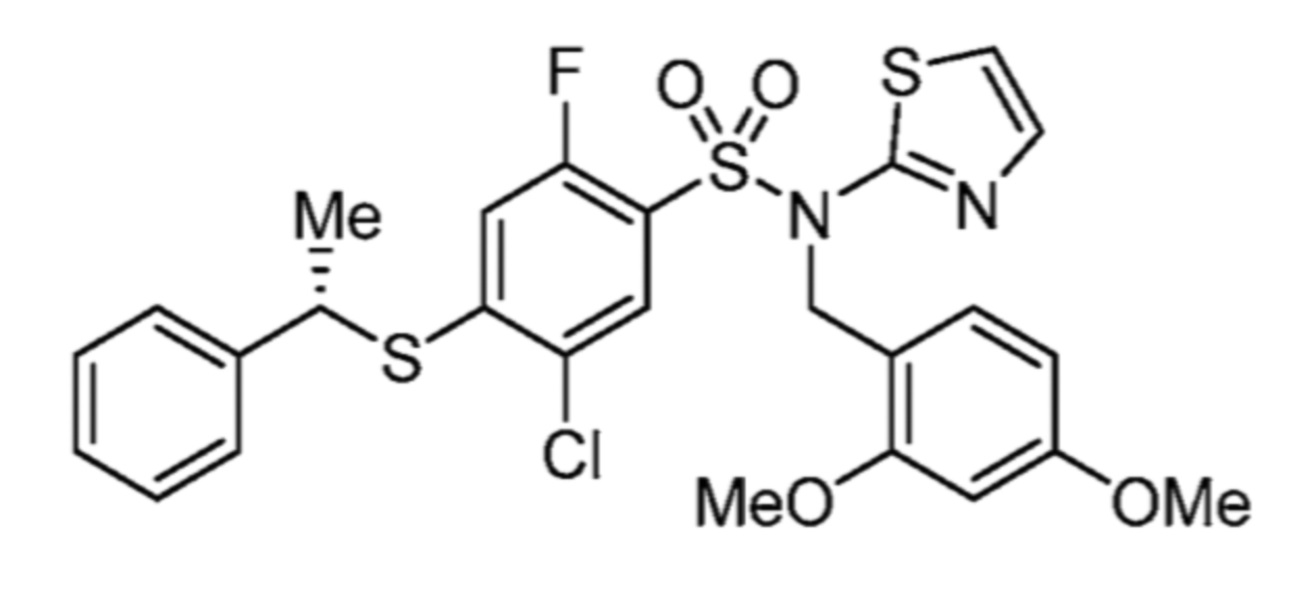
В раствор 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамида (0,23 г, 0,50 ммоль) в безводном DMF (4 мл) добавляли сульфид натрия (0,04 г, 0,55 ммоль) и реакционную смесь перемешивали в течение 3 ч. Реакционную смесь гасили добавлением насыщенного водного раствора хлорида аммония (10 мл) и экстрагировали простым диэтиловым эфиром (2×10 мл). Органические слои сушили над безводным сульфатом натрия и фильтровали. После концентрирования фильтрата in vacuo, остаток растворяли в безводном простом диэтиловом эфире (2 мл) и туда добавляли трифенилфосфин (0,24 г, 0,90 ммоль) и (R)-1-фенилэтан-1-ол (0,07 г, 0,60 ммоль). Реакционную смесь охлаждали до 0°C и по каплям в течение 10 минут добавляли диизопропилазодикарбоксилат (0,18 мл, 0,9 ммоль). Реакционную смесь перемешивали при 0°C в течение 2 ч и затем концентрировали in vacuo. Получаемый остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-30% этилацетата в гексанах для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (0,14 г, выход 48%): MS (ES+) m/z 579,0 (M+1), 581,0 (M+1).
Стадия 2. Получение (S)-5-хлор-2-фтор-4-((1-фенилэтил)тио)-N-(тиазол-2-ил)бензолсульфонамида.

Следуя процедуре как описано для примера 9, стадии 3 и выполняя некритические вариации при необходимости, чтобы заменять (S)-3-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамид на (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-фенилэтил)тио)-N-(тиазол-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,07 г, выход 33%):1H ЯМР (300 МГц, DMSO d6) δ 12,87 (ушир. с, 1H), 7,92 (д, J=6,6 Гц, 1H), 7,45-7,42 (м, 2H), 7,38-7,30 (м, 3H), 7,18 (д, J=4,6 Гц, 1H), 6,90 (д, J=10,6 Гц, 1H), 6,57 (д, J=4,5 Гц, 1H), 4,51-4,43 (м, 1H), 1,72 (д, J=7,0 Гц, 3H); MS (ES+) m/z 431,0 (M+1), 429,0 (M+1).
ПРИМЕР 126
Синтез (S)-2-фтор-5-метил-4-((1-фенилпропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

В смесь (S)-5-бром-2-фтор-4-((1-фенилпропил)амино)-N-(тиазол-2-ил)бензолсульфонамида (0,410 г, 0,87 ммоль), тетракис(трифенилфосфин)палладия(0) (0,10 г, 0,087 ммоль) и метилбороновой кислоты (0,208 г, 3,48 ммоль) в 1,4-диоксане (10,4 мл) добавляли 2 M раствор карбоната натрия (2,6 мл, 5,2 ммоль). Реакционную смесь дегазировали в течение 10 минут посредством пропускания потока азота через нее и затем нагревали при 100°C в течение 16 ч. Реакционную смесь оставляли остывать до температуры окружающей среды, корректировали до pH 5-6 посредством добавления 1 Н раствора соляной кислоты и экстрагировали этилацетатом (3×40 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 100% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,065 г, выход 18%):1H ЯМР (300 МГц, DMSO-d6) δ 12,54 (с, 1H), 7,42-7,36 (м, 2H), 7,36-7,26 (м, 3H), 7,22-7,15 (м, 2H), 6,75 (д, J=4,6 Гц, 1H), 6,13 (д, J=13,8 Гц, 1H), 6,01 (д, J=6,6 Гц, 1H), 4,36-4,27 (м, 1H), 2,19 (с, 3H), 2,01-1,88 (м, 1H), 1,81-1,66 (м, 1H), 0,91 (т, J=7,3 Гц, 3H).; MS (ES+) m/z 406,2 (M+1).
ПРИМЕРЫ 127-144
Схожим образом, как описано в примерах выше, используя надлежащим образом замещенные исходные материалы и промежуточные соединения, получали следующие соединения:
ПРИМЕР 145
Синтез 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(пиримидин-2-ил)бензолсульфонамида

Стадия 1. Получение N-(2,4-диметоксибензил)пиримидин-2-амина

Смесь 2-хлорпиримидина (2,00 г, 17,50 ммоль), (2,4-диметоксифенил)метанамина (2,92 г, 17,50 ммоль) и карбоната калия (2,90 г, 21,00 ммоль) в безводном ацетонитриле (20 мл) дегазировали барботированием азота и затем перемешивали до 80°C в течение 10 ч. После охлаждения до температуры окружающей среды, реакционную смесь концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с использованием 50% петролейного эфира в этилацетате, чтобы получать указанное в заголовке соединение в виде твердого вещества желтого цвета (2,10 г, выход 49%):1H ЯМР (400 МГц, CDCl3) δ 8,27 (д, J=4,5 Гц, 2H), 7,24 (д, J=8,3 Гц, 1H), 6,50 (т, J=4,8 Гц, 1H), 6,47 (д, J=2,5 Гц, 1H), 6,43 (дд, J=8,3, 2,5 Гц, 1H), 5,58 (ушир. с, 1H), 4,55 (д, J=6,0 Гц, 2H), 3,83 (с, 3H), 3,80 (с, 3H).
Стадия 2. Получение N-(2,4-диметоксибензил)-2,4,6-трифтор-N-(пиримидин-2-ил)бензолсульфонамида

В раствор N-(2,4-диметоксибензил)пиримидин-2-амина (1,00 г, 4,10 ммоль) в безводном тетрагидрофуране (10 мл) добавляли 1,6 M раствор метиллития в тетрагидрофуране (3,57 мл, 5,80 ммоль) при -78°C. Реакционную смесь перемешивали при 0°C в течение 30 минут, после чего туда добавляли 2,4,6-трифторбензолсульфонилхлорид (1,00 г, 4,48 ммоль) в безводном тетрагидрофуране (2 мл). Реакционную смесь перемешивали при 0°C в течение 4 ч, нагревали до температуры окружающей среды и гасили водой (10 мл). Смесь экстрагировали этилацетатом (3×30 мл). Объединенные органические экстракты промывали солевым раствором (2×40 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием 50% петролейного эфира в этилацетате, получали указанное в заголовке соединение в виде твердого вещества желтого цвета (0,30 г, выход 17%):1H ЯМР (400 МГц, CDCl3) δ 8,42 (д, J=4,8 Гц, 2H), 7,24 (д, J=8,3 Гц, 1H), 6,89 (т, J=4,8 Гц, 1H), 6,81-6,72 (м, 2H), 6,48 (д, J=2,3 Гц, 1H), 6,43 (дд, J=8,4, 2,4 Гц, 1H), 5,42 (с, 2H), 3,87 (с, 3H), 3,79 (с, 3H).
Стадия 3. Получение трет-бутил (2-(азетидин-1-илметил)-6-фторбензил)карбамата

В раствор (2-(азетидин-1-илметил)-6-фторфенил)метанамина (0,30 г, 1,50 ммоль) в дихлорметане (4 мл) добавляли ди-трет-бутилдикарбонат (0,37 г, 1,70 ммоль) и триэтиламин (0,43 мл, 3,10 ммоль). Смесь перемешивали при температуре окружающей среды в течение 2 ч, и затем концентрировали in vacuo. Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием 50% петролейного эфира в этилацетате, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,38 г, выход 84%):1H ЯМР (400 МГц, CDCl3) δ 7,22-7,14 (м, 1H), 7,06-6,96 (м, 2H), 4,39 (ушир. с, 2H), 3,62 (ушир. с, 2H), 3,21 (т, J=6,8 Гц, 4H), 2,14-2,00 (м, 2H), 1,46 (с, 9H), NH не наблюдали.
Стадия 4. Получение трет-бутил (2-(азетидин-1-илметил)-6-фторбензил)(4-(N-(2,4-диметоксибензил)-N-(пиримидин-2-ил)сульфамоил)-3,5-дифторфенил)карбамата

В раствор трет-бутил 2-(азетидин-1-илметил)-6-фторбензилкарбамата (0,10 г, 0,34 ммоль) в безводном N,N-диметилформамиде (3 мл) добавляли 60% дисперсию гидрида натрия в минеральном масле (0,016 г, 0,41 ммоль) при 0°C. Смесь перемешивали в течение 5 мин при 0°C и затем туда по каплям добавляли раствор N-(2,4-диметоксибензил)-2,4,6-трифтор-N-(пиримидин-2-ил)бензолсульфонамида (0,15 г, 0,34 ммоль) в безводном N,N-диметилформамиде (1 мл). Получаемую смесь перемешивали при температуре окружающей среды в течение 1 ч и затем гасили добавлением воды (2 мл). Смесь экстрагировали этилацетатом (2×30 мл). Объединенные органические экстракты промывали солевым раствором (2×30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом ацетонитрила в воде (содержащей 0,05% гидроксида аммония), получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,10 г, выход 41%):1H ЯМР (400 МГц, CDCl3) δ 8,58 (д, J=4,9 Гц, 1H), 8,50-8,37 (м, 1H), 8,04-7,93 (м, 2H), 7,35 (д, J=4,9 Гц, 1H), 7,23-7,07 (м, 1H), 7,06-6,99 (м, 1H), 6,92-6,71 (м, 2H), 6,68-6,27 (м, 2H), 4,90 (с, 2H), 4,74-4,51 (м, 2H), 4,38 (ушир. с, 2H), 4,11-3,96 (м, 3H), 3,88-3,77 (м, 5H), 3,74-3,60 (м, 2H), 2,71 (д, J=7,8 Гц, 1H), 2,52 (с, 1H), 1,47-1,33 (м, 9H).
Стадия 5. Получение 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(пиримидин-2-ил)бензолсульфонамида

В трет-бутил (2-(азетидин-1-илметил)-6-фторбензил)(4-(N-(2,4-диметоксибензил)-N-(пиримидин-2-ил)сульфамоил)-3,5-дифторфенил)карбамат (0,090 г, 0,13 ммоль) добавляли 4 M раствор хлороводорода в 1,4-диоксане (1,50 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Реакционную смесь концентрировали in vacuo и остаток очищали с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом ацетонитрила в воде (содержащей 0,1% трифторуксусную кислоту), чтобы получать 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(пиримидин-2-ил)бензолсульфонамид в виде бесцветного твердого вещества (0,061 г, выход 97%):1H ЯМР (400 МГц, DMSO-d6) δ 11,92 (ушир. с, 1H), 10,90 (ушир. с, 1H), 8,51 (д, J=4,9 Гц, 2H), 7,54-7,47 (м, 1H), 7,46-7,41 (м, 1H), 7,41-7,32 (м, 2H), 7,06 (т, J=4,8 Гц, 1H), 6,39 (д, J=12,5 Гц, 2H), 4,46 (д, J=6,1 Гц, 2H), 4,38 (д, J=3,9 Гц, 2H), 4,15-4,06 (м, 2H), 4,04-3,96 (м, 2H), 2,43-2,35 (м, 1H), 2,27 (тд, J=9,5, 4,4 Гц, 1H); MS (ES+) m/z 464,2 (M+1).
ПРИМЕР 146
Синтез (S)-5-хлор-4-((1-(5-циклопропил-2-фторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 5-циклопропил-2-фторбензальдегида

Смесь 5-бром-2-фтор-бензальдегида (10,00 г, 49,20 ммоль), циклопропилбороновой кислоты (21,10 г, 246,30 ммоль), фосфата калия (41,80 г, 197,00 ммоль), ацетата палладия (2,2 г, 9,80 ммоль) и тетрафторбората трициклогексилфосфония (3,60 г, 9,80 ммоль) в безводном толуоле (120 мл) дегазировали посредством барботирования азотом и затем нагревали до 90°C в течение 12 ч. После охлаждения до температуры окружающей среды, реакционную смесь разводили водой (100 мл) и экстрагировали этилацетатом (3×200 мл). Объединенные органические экстракты концентрировали in vacuo и получаемый остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 2 до 10% этилацетата в петролейном эфире, чтобы получать указанное в заголовке соединение в виде масла желтого цвета (6,50 г, выход 80%):1H ЯМР (400 МГц, CDCl3) δ 10,26 (с, 1H), 7,46 (дд, J=6,4, 2,4 Гц, 1H), 7,30-7,22 (м, 1H), 6,99 (дд, J=10,0, 8,4 Гц, 1H), 1,91-1,80 (м, 1H), 0,98-0,88 (м, 2H), 0,67-0,58 (м, 2H).
Стадия 2. Получение (R)-N-(5-циклопропил-2-фторбензилиден)-2-метилпропан-2-сульфинамида

В раствор 5-циклопропил-2-фторбензальдегида (4,00 г, 24,30 ммоль) и (R)-2-метилпропан-2-сульфинамида (5,90 г, 48,70 ммоль) в безводном дихлорметане (40 мл) добавляли пара-толуолсульфонат пиридиния (0,31 г, 1,20 ммоль) и безводный сульфат магния (14,60 г, 121,80 ммоль). Смесь перемешивали при температуре окружающей среды в течение 12 ч и затем фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 1 до 2% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного масла (1,80 г, выход 28%):1H ЯМР (400 МГц, CDCl3) δ 8,88 (с, 1H), 7,71 (дд, J=6,4, 2,0 Гц, 1H), 7,24-7,15 (м, 1H), 7,11-7,01 (м, 1H), 2,02-1,88 (м, 1H), 1,30 (с, 9H), 1,08-0,94 (м, 2H), 0,77-0,64 (м, 2H).
Стадия 3. Получение (R)-N-((S)-1-(5-циклопропил-2-фторфенил)этил)-2-метилпропан-2-сульфинамида

В раствор (R)-N-(5-циклопропил-2-фторбензилиден)-2-метилпропан-2-сульфинамида (1,50 г, 5,60 ммоль) в безводном дихлорметане (20 мл) добавляли 3,0 M раствор бромида метилмагния в простом диэтиловом эфире (3,70 мл) по каплям при -48°C. Реакционную смесь нагревали до температуры окружающей среды и перемешивали в течение 12 ч и затем гасили добавлением насыщенного хлорида аммония (10 мл). Смесь экстрагировали дихлорметаном (3×20 мл) и объединенные органические экстракты концентрировали in vacuo. Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 5 до 20% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного масла (0,80 г, выход 50%):1H ЯМР (400 МГц, CDCl3) δ 7,06 (д, J=7,6 Гц, 1H), 6,98-6,89 (м, 2H), 4,87-4,75 (м, 1H), 1,96-1,82 (м, 1H), 1,59 (д, J=6,8 Гц, 3H), 1,22 (с, 9H), 0,96 (д, J=8,0 Гц, 2H), 0,73-0,59 (м, 2H), NH не наблюдали.
Стадия 4. Получение гидрохлорида (S)-1-(5-циклопропил-2-фторфенил)этан-1-амина

К (R)-N-((S)-1-(5-циклопропил-2-фторфенил)этил)-2-метилпропан-2-сульфинамиду (0,70 г, 2,40 ммоль) добавляли 4 M раствор хлороводорода в 1,4-диоксане (2,6 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении. Получаемый остаток разводили метанолом (1 мл) и кристаллизовали из простого метил-трет-бутилового эфира (30 мл) для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества: (0,35 г, выход 68%).
Стадия 5. Получение трет-бутил (S)-((5-хлор-4-((1-(5-циклопропил-2-фторфенил)этил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата

В раствор трет-бутил (5-хлор-2,4-дифторфенил)сульфонил(тиазол-4-ил)карбамата (0,25 г, 0,61 ммоль) и гидрохлорида (S)-1-(5-циклопропил-2-фторфенил)этан-1-амина (0,11 г, 0,51 ммоль) в N,N-диметилформамиде (5 мл) добавляли карбонат калия (0,25 г, 1,80 ммоль). Смесь перемешивали при температуре окружающей среды в течение 12 ч. Реакционную смесь гасили добавлением воды (50 мл) и затем экстрагировали этилацетатом (3×80 мл). Объединенные органические слои концентрировали при пониженном давлении. Остаток очищали с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 33% петролейного эфира в этилацетате, чтобы получать указанное в заголовке соединение в виде бесцветного масла (0,12 г, выход 41%): MS (ES+) m/z 470,1 (M - 100+1)
Стадия 6. Получение (S)-5-хлор-4-((1-(5-циклопропил-2-фторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

К (S)-трет-бутил(5-хлор-4-((1-(5-циклопропил-2-фторфенил)этил)амино)-2-фторфенил)сульфонил(тиазол-4-ил)карбамату (0,11 г, 0,19 ммоль) добавляли 4 M раствор хлороводорода в 1,4-диоксане (6,6 мл) и смесь перемешивали при температуре окружающей среды в течение 12 ч. Реакционную смесь концентрировали при пониженном давлении. Получаемый остаток очищали с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом ацетонитрила в воде (содержащей 0,05% гидроксида аммония), чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,043 г, выход 47%):1H ЯМР (400 МГц, CDCl3) δ 8,59 (с, 1H), 7,73 (д, J=7,2 Гц, 1H), 7,03-6,81 (м, 4H), 6,15 (д, J=12,4 Гц, 1H), 5,20 (д, J=6,0 Гц, 1H), 4,73 (квин, J=6,4 Гц, 1H), 1,89-1,76 (м, 1H), 1,60 (д, J=6,8 Гц, 3H), 0,93 (дд, J=8,4, 1,6 Гц, 2H), 0,65-0,48 (м, 2H), NH не наблюдали; MS (ES+) m/z 470,1 (M+1).
ПРИМЕР 147
Синтез 5-хлор-2-фтор-4-((5-фтор-2-метилбензил)амино)-N-(тиазол-4-ил)бензолульфонамида

Стадия 1. Получение трет-бутил ((5-хлор-2-фтор-4-((5-фтор-2-метилбензил)амино)фенил)сульфонил)(тиазол-4-ил)карбамата

Следуя процедуре как описано в примере 146, стадии 5 и выполняя некритические вариации, чтобы заменять гидрохлорид (S)-1-(5-циклопропил-2-фторфенил)этан-1-амина на (5-фтор-2-метилфенил)метанамин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,25 г, выход 65%):1H ЯМР (400 МГц, CDCl3) δ 8,79 (д, J=2,2 Гц, 1H), 8,04-8,00 (м, 1H), 7,50 (д, J=2,2 Гц, 1H), 7,23-7,18 (м, 1H), 6,97-6,91 (м, 2H), 6,31 (д, J=12,0 Гц, 1H), 5,30 (ушир. с, 1H), 4,39 (д, J=5,6 Гц, 2H), 2,33 (с, 3H), 1,39 (с, 9H); MS (ES+) m/z 530,1 (M+1).
Стадия 2. Получение 5-хлор-2-фтор-4-((5-фтор-2-метилбензил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано в примере 5, стадии 2 и выполняя некритические вариации, чтобы заменять трет-бутил (S)-((5-хлор-4-((1-(5-хлор-2-фторфенил)этил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамат на трет-бутил ((5-хлор-2-фтор-4-((5-фтор-2-метилбензил)амино)фенил)сульфонил)(тиазол-4-ил)карбамат, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,15 г, выход 73%):1H ЯМР (400 МГц, CDCl3) δ 9,03 (ушир. с, 1H), 8,64 (д, J=2,2 Гц, 1H), 7,77 (д, J=7,2 Гц, 1H), 7,18 (дд, J=8,0, 5,8 Гц, 1H), 6,99 (д, J=2,2 Гц, 1H), 6,97-6,88 (м, 2H), 6,25 (д, J=12,0 Гц, 1H), 5,14 (ушир. с, 1H), 4,31 (д, J=5,6 Гц, 2H), 2,30 (с, 3H); MS (ES+) m/z 430,0 (M+1).
ПРИМЕР 148
Синтез (S)-3-хлор-4-((1-(2-хлор-6-фторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение (R,E)-N-(2-хлор-6-фторбензилиден)-2-метилпропан-2-сульфинамида

В раствор 2-хлор-6-фторбензальдегида (10,43 г, 65,8 ммоль) и (R)-2-метилпропан-2-сульфинамида (7,97 г, 65,8 ммоль) в безводном дихлорметане (100 мл) добавляли карбонат цезия (22,1 г, 67,8 ммоль). Смесь перемешивали при температуре окружающей среды в течение 17 ч, затем фильтровали через подушку диатомовой земли. Фильтровальную подушку промывали дихлорметаном (150 мл). Объединенный фильтрат концентрировали in vacuo для того, чтобы получать указанное в заголовке соединение в виде масла светло-коричневого цвета (17,4 г, количественный выход):1H ЯМР (300 МГц, CDCl3) δ 8,89 (с, 1H), 7,42-7,35 (м, 1H), 7,29-7,26 (м, 1H), 7,12-7,06 (м, 1H), 1,28 (с, 9H); MS (ES+) m/z 262,1 (M+1), 264,1 (M+1).
Стадия 2. Получение (R)-N-((S)-1-(2-хлор-6-фторфенил)этил)-2-метилпропан-2-сульфинамида

В холодных (-78°C) раствор (R,E)-N-(2-хлор-6-фторбензилиден)-2-метилпропан-2-сульфинамида (5,34 г, 20,4 ммоль) в безводном дихлорметане (75 мл) добавляли бромид метилмагния (3 M в простом диэтиловом эфире, 10,0 мл, 30,0 ммоль) по каплям в течение 30 минут. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 5 суток. Затем реакционную смесь гасили насыщенным хлоридом аммония (5 мл). Реакционную смесь разводили насыщенным хлоридом аммония (75 мл), солевым раствором (75 мл) и экстрагировали дихлорметаном (2×150 мл). Объединенные органические слои сушили над безводным сульфатом магния, фильтровали и фильтрат концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с использованием 0-70% градиента этилацетата в гексанах, чтобы получать указанное в заголовке соединение в виде бесцветного сиропа (0,38 г, выход 7%):1H ЯМР (300 МГц, CDCl3) δ 7,19-7,15 (м, 2H), 7,01-6,94 (м, 1H), 5,21-5,11 (м, 1H), 3,91 (д, J=8,3 Гц, 1H), 1,67 (дд, J=1,0, 7,0 Гц, 3H), 1,14 (с, 9H); MS (ES+) m/z 278,1 (M+1), 280,1 (M+1).
Стадия 3. Получение гидрохлорида (S)-1-(2-хлор-6-фторфенил)этан-1-амина

В раствор (R)-N-((S)-1-(2-хлор-6-фторфенил)этил)-2-метилпропан-2-сульфинамида (0,90 г, 3,24 ммоль) в безводном метаноле (10 мл) добавляли 4 M раствор хлороводорода (2,0 мл, 8,0 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч и затем концентрировали in vacuo для того, чтобы получать указанное в заголовке соединение в виде бесцветного сиропа (1,03 г, количественный выход):1H ЯМР (300 МГц, CDCl3) δ 8,88 (ушир. с, 3H), 7,27-7,19 (м, 2H), 7,08-7,02 (м, 1H), 5,10-5,00 (м, 1H), 1,76 (д, J=6,9 Гц, 3H); MS (ES+) m/z 174,1 (M+1), 176,1 (M+1).
Стадия 4. Получение трет-бутил ((3-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

В раствор трет-бутил N-тиазол-4-илкарбамата (110 г, 549 ммоль) в безводном тетрагидрофуране (1000 мл) добавляли бис(триметилсилил)амид лития (1 M в тетрагидрофуране, 659 мл, 659 ммоль) при -78°C. Смесь нагревали до 5°C прежде, чем туда по каплям добавляли охлажденный (-78°C) раствор 3-хлор-2,4-дифтор-бензолсульфонилхлорида (163 г, 659 ммоль) в тетрагидрофуране (300 мл). Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 12 ч. После разведения насыщенным водным хлоридом аммония (200 мл) смесь экстрагировали этилацетатом (3×1000 мл). Объединенные органические слои промывали солевым раствором (3×1000 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и растиранием остатка с метанолом (300 мл) получали указанное в заголовке соединение в виде бесцветного твердого вещества (75 г, выход 33%)1H ЯМР (400 МГц, CDCl3) δ 9,14 (с, 1H), 8,26-8,09 (м, 1H), 8,03 (с, 1H), 7,66 (т, J=8,6 Гц, 1H), 1,27 (с, 9H); MS (ES+) m/z 432,8 (M+23), 434,8 (M+23).
Стадия 5. Получение трет-бутил (S)-((3-хлор-4-((1-(2-хлор-6-фторфенил)этил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата

В раствор гидрохлорида (S)-1-(2-хлор-6-фторфенил)этан-1-амина (0,19 г, 0,91 ммоль) и трет-бутил ((3-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,26 г, 0,64 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли N,N-диизопропилэтиламин (0,56 мл, 3,2 ммоль). Раствор перемешивали при температуре окружающей среды в течение 18 ч и затем гасили насыщенным хлоридом аммония (15 мл). Реакционную смесь разводили солевым раствором (100 мл) и экстрагировали этилацетатом (2×100 мл). Объединенные органические слои сушили над безводным сульфатом магния, фильтровали и фильтрат концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с использованием 0-30% градиента этилацетата в гексанах, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,11 г, выход 32%):1H ЯМР (300 МГц, CDCl3) δ 8,80 (д, J=2,3 Гц, 1H), 7,77 (дд, J=9,0, 7,7 Гц, 1H), 7,52 (дд, J=2,2, 0,3 Гц, 1H), 7,22-7,18 (м, 2H), 7,02-6,96 (м, 1H), 6,49 (дд, J=9,2, 1,1 Гц, 1H), 5,67 (д, J=8,9 Гц, 1H), 5,36-5,26 (м, 1H), 1,76 (д, J=6,9 Гц, 3H), 1,29 (с, 9H);MS (ES+) m/z 564,1 (M+1), 566,1 (M+1).
Стадия 6. Получение (S)-3-хлор-4-((1-(2-хлор-6-фторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил (S)-((3-хлор-4-((1-(2-хлор-6-фторфенил)этил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата (0,11 г, 0,20 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (2 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч и затем концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с использованием 0-30% градиента этилацетата (содержащего 0,1% муравьиную кислоту) в гексанах, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,047 г, выход 50%):1H ЯМР (300 МГц, DMSO-d6) δ 11,16 (с, 1H), 8,85 (д, J=2,2 Гц, 1H), 7,51 (т, J=8,5 Гц, 1H), 7,36-7,31 (м, 2H), 7,23-7,16 (м, 1H), 6,95 (д, J=2,2 Гц, 1H), 6,37-6,31 (м, 2H), 5,23-5,13 (м, 1H), 1,65 (д, J=6,9 Гц, 3H); MS (ES+) m/z 464,1 (M+1), 466,1 (M+1).
ПРИМЕР 149
Синтез (S)-3-хлор-4-((1-(2,6-дифторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида
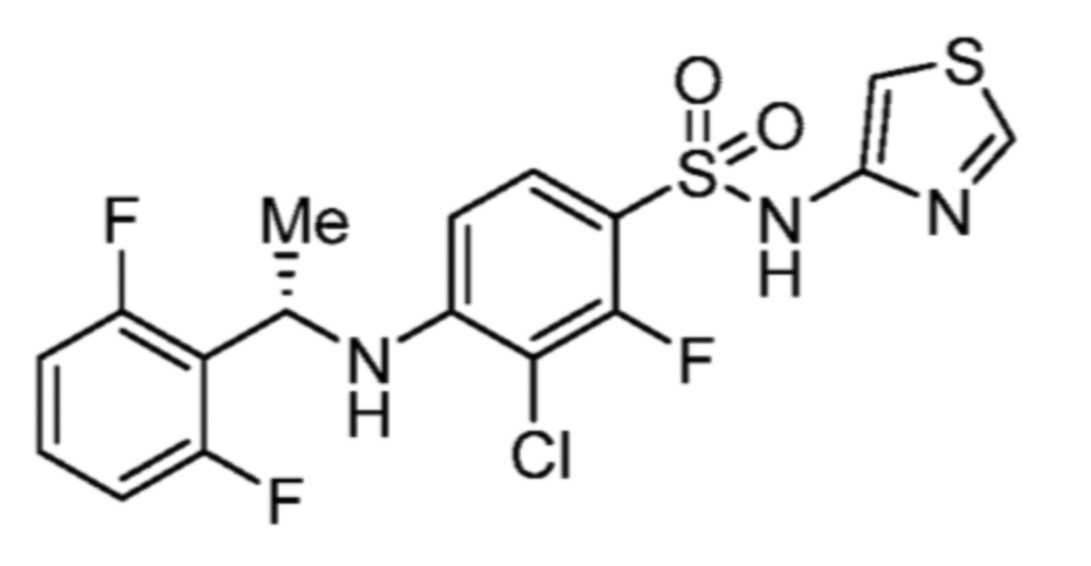
Стадия 1. Получение (R,E)-N-(2,6-дифторбензилиден)-2-метилпропан-2-сульфинамида

Следуя процедуре как описано в примере 148, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 2-хлор-6-фторбензальдегид на 2,6-дифторбензальдегид, указанное в заголовке соединение получали в виде масла светло-желтого цвета (19,2 г, количественный выход):1H ЯМР (300 МГц, CDCl3) δ 8,81 (с, 1H), 7,50-7,41 (м, 1H), 7,03-6,95 (м, 2H), 1,27 (с, 9H); MS (ES+) m/z 246,1 (M+1).
Стадия 2. Получение (R)-N-((S)-1-(2,6-дифторфенил)этил)-2-метилпропан-2-сульфинамида

Следуя процедуре как описано в примере 148, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять (R,E)-N-(2-хлор-6-фторбензилиден)-2-метилпропан-2-сульфинамид на (R,E)-N-(2,6-дифторбензилиден)-2-метилпропан-2-сульфинамид, указанное в заголовке соединение получали в виде бесцветного сиропа (0,68 г, выход 13%):1H ЯМР (300 МГц, CDCl3) δ 7,24-7,16 (м, 1H), 6,90-6,84 (м, 2H), 5,03-4,93 (м, 1H), 3,75 (д, J=8,0 Гц, 1H), 1,67 (д, J=6,9 Гц, 3H), 1,15 (с, 9H); MS (ES+) m/z 262,2 (M+1).
Стадия 3. Получение гидрохлорида (S)-1-(2,6-дифторфенил)этан-1-амина

Следуя процедуре как описано в примере 148, стадии 3 и выполняя некритические вариации при необходимости, чтобы заменять (R)-N-((S)-1-(2-хлор-6-фторфенил)этил)-2-метилпропан-2-сульфинамид на (R)-N-((S)-1-(2,6-дифторфенил)этил)-2-метилпропан-2-сульфинамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,73 г, количественный выход):1H ЯМР (300 МГц, CDCl3) δ 8,83 (ушир. с, 3H), 7,34-7,24 (м, 1H), 6,97-6,86 (м, 2H), 4,88-4,84 (м, 1H), 1,74 (д, J=6,9 Гц, 3H); MS (ES+) m/z 158,1 (M+1).
Стадия 4. Получение трет-бутил (S)-((3-хлор-4-((1-(2,6-дифторфенил)этил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата

Следуя процедуре как описано в примере 148, стадии 5 и выполняя некритические вариации при необходимости, чтобы заменять гидрохлорид (S)-1-(2-хлор-6-фторфенил)этан-1-амина на гидрохлорид (S)-1-(2,6-дифторфенил)этан-1-амина, указанное в заголовке соединение получали в виде сиропа светло-желтого цвета (0,10 г, выход 37%):1H ЯМР (300 МГц, CDCl3) δ 8,80 (д, J=2,3 Гц, 1H), 7,79 (дд, J=9,0, 7,7 Гц, 1H), 7,51 (дд, J=2,3, 0,7 Гц, 1H), 7,26-7,20 (м, 1H), 6,93-6,87 (м, 2H), 6,53 (дд, J=9,2, 1,2 Гц, 1H), 5,54 (д, J=8,8 Гц, 1H), 5,20-5,10 (м, 1H), 1,76 (д, J=6,9 Гц, 3H), 1,30 (с, 9H);MS (ES+) m/z 548,2 (M+1), 550,2 (M+1).
Стадия 5. Получение (S)-3-хлор-4-((1-(2,6-дифторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано в примере 148, стадии 6 и выполняя некритические вариации при необходимости, чтобы заменять трет-бутил (S)-((3-хлор-4-((1-(2-хлор-6-фторфенил)этил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамат на трет-бутил (S)-((3-хлор-4-((1-(2,6-дифторфенил)этил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамат, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,045 г, выход 56%):1H ЯМР (300 МГц, DMSO-d6) δ 11,15 (с, 1H), 8,85 (д, J=2,2 Гц, 1H), 7,52 (т, J=8,5 Гц, 1H), 7,41-7,31 (м, 1H), 7,11-7,05 (м, 2H), 6,95 (д, J=2,2 Гц, 1H), 6,46 (д, J=8,3 Гц, 1H), 6,28 (д, J=7,8 Гц, 1H), 5,14-5,04 (м, 1H), 1,65 (д, J=6,9 Гц, 3H); MS (ES+) m/z 448,0 (M+1), 450,0 (M+1).
ПРИМЕР 150
Синтез 4-((2-(азетидин-1-илметил)-3,6-дифторбензил)окси)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил ((4-((2-(азетидин-1-илметил)-3,6-дифторбензил)окси)-5-хлор-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата

В раствор (2-(азетидин-1-илметил)-3,6-дифторфенил)метанола (0,39 г, 0,94 ммоль) в безводном N,N-диметилформамиде (3 мл) добавляли трет-бутил ((5-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамат (0,30 г, 1,41 ммоль) и гидрид натрия (60% дисперсия в минеральном масле, 0,075 г, 1,88 ммоль). Раствор перемешивали при температуре окружающей среды в течение 16 ч и затем очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 10 до 60% этилацетата в гексанах, чтобы получать указанное в заголовке соединение в виде сиропа желтого цвета (выход не определяли): MS (ES+) m/z 604,3 (M+1), 606,3 (M+1).
Стадия 2. Получение 4-((2-(азетидин-1-илметил)-3,6-дифторбензил)окси)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил ((4-((2-(азетидин-1-илметил)-3,6-дифторбензил)окси)-5-хлор-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата в дихлорметане (2 мл) добавляли трифторуксусную кислоту (0,70 мл) и получаемый раствор перемешивали в течение 16 ч. Реакционную смесь концентрировали in vacuo, растирали с метано (3 мл) и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью HPLC с обращенной фазой, используя градиент ацетонитрила в воде, содержащей 0,5% муравьиную кислоту, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,004 г, выход 1% на две стадии):1H-ЯМР (300 МГц, DMSO-d6) δ 11,46-11,44 (м, 1H), 8,92 (д, J=2,2 Гц, 1H), 7,85 (д, J=7,4 Гц, 1H), 7,61-7,54 (м, 3H), 7,11 (д, J=2,2 Гц, 1H), 5,40 (с, 2H), 4,57-4,56 (м, 2H), 4,14-4,01 (м, 4H), 2,38-2,21 (м, 2H); MS (ES+) m/z 504,2, 506,2 (M+1).
ПРИМЕР 151
Синтез формиата 2,6-дифтор-4-((2-фтор-6-((4-метилпиперазин-1-ил)метил)- бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил тиазол-4-ил((2,4,6-трифторфенил)-сульфонил)карбамата

В раствор трет-бутил тиазол-4-илкарбамата (140,0 г, 699,1 ммоль) в безводном тетрагидрофуране (700 мл) добавляли 1 M раствор бис(триметилсилил)амида лития в тетрагидрофуране (758,9 мл, 758,0 ммоль) при -78°C. Реакционную смесь оставляли нагреваться до 0°C и перемешивали в течение 20 минут. После охлаждения реакционной смеси до -78°C, туда медленно добавляли раствор 2,4,6-трифторбензолсульфонилхлорида (175,0 г, 758,9 ммоль) в безводном тетрагидрофуране (200 мл). Реакционную смесь оставляли нагреваться до температуры окружающей среды, перемешивали в течение 12 ч и затем гасили добавлением насыщенного хлорида аммония (200 мл). Смесь экстрагировали этилацетатом (3×1000 мл). Органическую фазу промывали солевым раствором (3×1000 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием in vacuo и растираниемостатка в метаноле (100 мл) получали указанное в заголовке соединение в виде бесцветного твердого вещества (140,0 г, выход 58%):1H ЯМР (400 МГц, CDCl3) δ 8,81 (д, J=2,1 Гц, 1H), 7,53 (д, J=2,1 Гц, 1H), 6,85 (ушир. т, J=8,4 Гц, 2H), 1,39 (с, 9H); MS (ES+) m/z 417,0 (M+23).
Стадия 2. Получение трет-бутил ((4-азидо-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

В раствор трет-бутил тиазол-4-ил((2,4,6-трифторфенил)сульфонил)карбамата (10,0 г, 25,3 ммоль) в безводном N,N-диметилформамиде (200 мл) добавляли азид натрия (1,81 г, 27,9 ммоль) небольшими частями при 0°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды, перемешивали в течение 3 ч, и затем выливали в воду (300 мл). Осадок собирали посредством фильтрования для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (15,0 г, количественный выход):1H ЯМР (400 МГц, CDCl3) δ 8,72 (д, J=2,2 Гц, 1H), 7,44 (д, J=2,2 Гц, 1H), 6,68-6,62 (м, 2H), 1,31 (с, 9H).
Стадия 3. Получение трет-бутил ((4-амино-2,6-дифторфенил)сульфонил)-(тиазол-4-ил)карбамата

В смесь трет-бутил (4-азидо-2,6-дифторфенил)сульфонил(тиазол-4-ил)карбамата (10,0 г, 23,9 ммоль) в тетрагидрофуране (180 мл) и насыщенного хлорида аммония (50 мл) добавляли порошок цинка (4,7 г, 71,8 ммоль) при 0°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 12 ч. Смесь фильтровали через подушку из целита и фильтрат разводили этилацетатом (200 мл). Органический слой промывали солевым раствором (3×100 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде бесцветного твердого вещества (9,0 г, выход 96%):1H ЯМР (400 МГц, DMSO-d6) δ 9,12 (д, J=2,2 Гц, 1H), 7,81 (д, J=2,2 Гц, 1H), 6,93 (с, 2H), 6,36 (д, J=12,4 Гц, 2H), 1,35 (с, 9H); MS (ES+) m/z 291,5 (M - 100).
Стадия 4. Получение трет-бутил ((4-((трет-бутоксикарбонил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

В смесь трет-бутил N-(4-амино-2,6-дифтор-фенил)сульфонил-N-тиазол-4-ил-карбамата (11,5 г, 29,3 ммоль) и ди-трет-бутилдикарбоната (7,7 г, 35,3 ммоль) в дихлорметане (100 мл) добавляли 4-(диметиламино)пиридин (0,717 мг, 5,88 ммоль) и триэтиламин (5,95 г, 58,7 ммоль) и смесь перемешивали при температуре окружающей среды в течение 12 ч. Концентрированием in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием 30% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (7,30 г, выход 50%):1H ЯМР (400 МГц, CDCl3) δ 8,79 (д, J=2,2 Гц, 1H), 7,50 (д, J=2,2 Гц, 1H), 7,28 (с, 1H), 7,19-7,14 (м, 2H), 1,51 (с, 9H), 1,38 (с, 9H); MS (ES+) m/z 392,0 (M - 99).
Стадия 5. Получение трет-бутил ((4-((2-бром-6-фторбензил)(трет-бутоксикарбонил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

В смесь трет-бутил(4-((трет-бутоксикарбонил)амино)-2,6-дифторфенил)-сульфонил(тиазол-4-ил)карбамата (7,30 г, 14,8 ммоль) и 1-бром-2-(хлорметил)-3-фторбензола (6,64 г, 29,7 ммоль) в безводном N,N-диметилформамиде (100 мл) добавляли карбонат калия (8,21 г, 59,4 ммоль) и смесь перемешивали при температуре окружающей среды в течение 12 ч. Добавляли воду (100 мл) и смесь экстрагировали этилацетатом (3×100 мл). Объединенную органическую фазу промывали солевым раствором (3×50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием 6% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (7,0 г, выход 69%):1H ЯМР (400 МГц, CDCl3) δ 8,80 (д, J=2,2 Гц, 1H), 7,50 (д, J=2,2 Гц, 1H), 7,32 (д, J=8,0 Гц, 1H), 7,11 (дт, J=6,0, 8,2 Гц, 1H), 7,03-6,94 (м, 3H), 5,18 (с, 2H), 1,50 (с, 9H), 1,32 (с, 9H); MS (ES+) m/z 521,9 (M - 155), 523,9 (M - 155).
Стадия 6. Получение трет-бутил ((4-((трет-бутоксикарбонил)(2-фтор-6-формилбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата
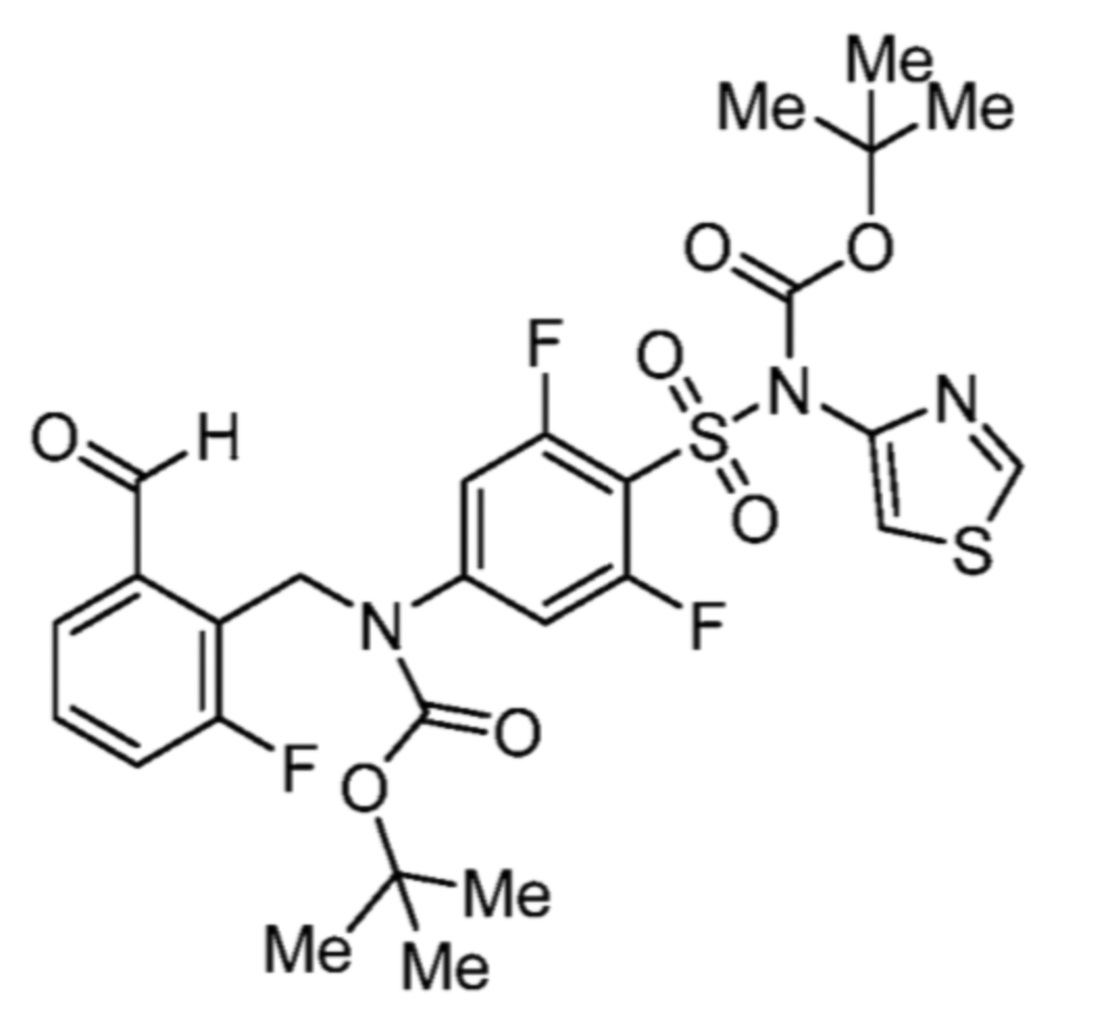
В смесь трет-бутил ((4-((2-бром-6-фторбензил)(трет-бутоксикарбонил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (3,50 г, 5,16 ммоль ), трет-бутилизоцианида (0,643 г, 7,74 ммоль), ацетата палладия(II) (0,115 г, 0,516 ммоль), карбоната натрия (0,546 г, 5,16 ммоль) и 2-(ди-трет-бутилфосфино)бифенила (0,307 г, 1,03 ммоль) в безводном N,N-диметилформамиде (30 мл) добавляли триэтилсилан (1,80 г, 15,48 ммоль). Реакционную смесь дегазировали азотом и затем нагревали до 65°C в течение 12 ч. Добавляли воду (30 мл) и смесь экстрагировали этилацетатом (3×30 мл). Объединенную органическую фазу промывали солевым раствором (3×30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием 30% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде твердого вещества желтого цвета (1,00 г, выход 30%):1H ЯМР (400 МГц, CDCl3) δ 10,13 (с, 1H), 8,72 (д, J=1,0 Гц, 1H), 7,55 (д, J=7,4 Гц, 1H), 7,43 (с, 1H), 7,41-7,35 (м, 1H), 7,22-7,15 (м, 1H), 6,88 (д, J=10,8 Гц, 2H), 5,43 (с, 2H), 1,40 (с, 9H), 1,25 (с, 9H);19F ЯМР (376,5 МГц, CDCl3) δ -105,0 (с, 2F), -115,6 (с, 1F); MS (ES+) m/z 471,9 (M - 155).
Стадия 7. Получение трет-бутил (4-(N-(трет-бутоксикарбонил)-N-(тиазол-4-ил)сульфамоил)-3,5-дифторфенил)(2-фтор-6-((4-метилпиперазин-1-ил)метил)бензил)карбамата

В смесь трет-бутил ((4-((трет-бутоксикарбонил)(2-фтор-6-формилбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,10 г, 0,159 ммоль), 1-метилпиперазина (0,015 г, 0,159 ммоль) и уксусной кислоты (0,009 г, 0,159 ммоль) в метаноле (1 мл) добавляли цианоборогидрид натрия (0,020 г, 0,318 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч и концентрировали in vacuo. К остатку добавляли воду (5 мл) и смесь экстрагировали этилацетатом (3×10 мл). Объединенную органическую фазу промывали солевым раствором (20 мл), сушили над сульфатом натрия и фильтровали. Концентрированием in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 50% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде масла желтого цвета (0,050 г, выход 44%): MS (ES+) m/z 712,2 (M+1).
Стадия 8. Получение формиата 2,6-дифтор-4-((2-фтор-6-((4-метилпиперазин-1-ил)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида

В смесь трет-бутил (4-(N-(трет-бутоксикарбонил)-N-(тиазол-4-ил)сульфамоил)-3,5-дифторфенил)(2-фтор-6-((4-метилпиперазин-1-ил)метил)бензил)карбамата (0,040 г, 0,056 ммоль) в дихлорметане (2 мл) добавляли трифторуксусную кислоту (0,83 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 12 ч. Концентрированием in vacuo и очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,011 г, выход 36%):1H ЯМР (400 МГц, CD3OD) δ 8,76 (д, J=2,2 Гц, 1H), 8,49 (с, 1H), 7,34 (дт, J=5,8, 7,8 Гц, 1H), 7,21-7,09 (м, 2H), 6,98 (д, J=2,2 Гц, 1H), 6,34-6,28 (м, 2H), 4,45 (с, 2H), 3,64 (с, 2H), 2,86 (с, 4H), 2,59 (с, 7H), NH и COOH не наблюдали;19F ЯМР (376,5 МГц, CD3OD) δ -109,6 (ушир. с, 2F), 119,3 (с, 1F); MS (ES+) m/z 512,0 (M+1).
ПРИМЕР 152
Синтез формиата 4-((2-((3-этокси-3-метилазетидин-1-ил)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида
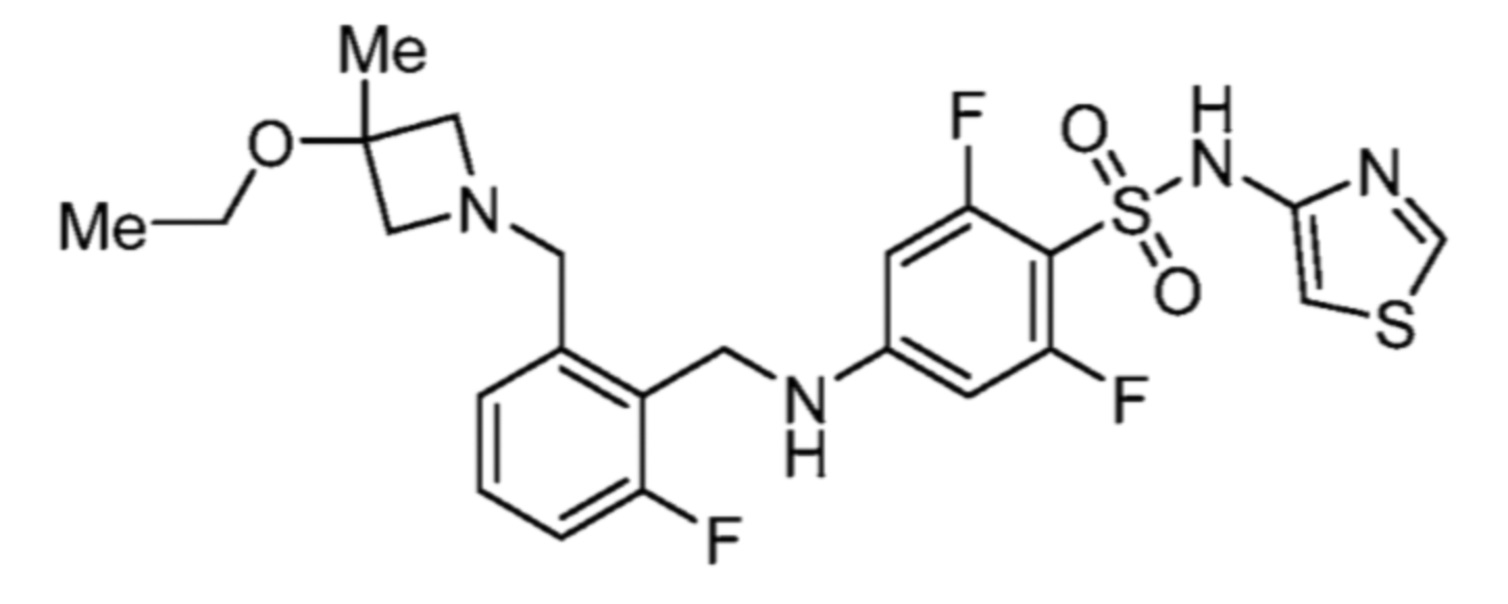
Стадия 1. Получение 2-фтор-6-((3-гидрокси-3-метилазетидин-1-ил)метил)бензонитрила

В смесь гидрохлорида 3-метилазетидин-3-ола (1,40 г, 11,33 ммоль) и 2-(бромметил)-6-фтор-бензонитрила (1,21 г, 5,67 ммоль) в дихлорметане (20 мл) добавляли триэтиламин (2,29 г, 22,6 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 12 ч. Концентрированием in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием 50% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,90 г, выход 72%):1H ЯМР (400 МГц, CDCl3) δ 7,55 (дт, J =5,6, 8,2 Гц, 1H), 7,32 (д, J=7,6 Гц, 1H), 7,10 (т, J=8,6 Гц, 1H), 3,85 (с, 2H), 3,43-3,31 (м, 2H), 3,15 (д, J=8,2 Гц, 2H), 2,28 (ушир. с, 1H), 1,52 (с, 3H).
Стадия 2. Получение 2-((3-этокси-3-метилазетидин-1-ил)метил)-6-фторбензонитрила

В раствор 2-фтор-6-((3-гидрокси-3-метилазетидин-1-ил)метил)бензонитрила (0,200 г, 0,908 ммоль) в безводном N,N-диметилформамиде (5 мл) добавляли 60% дисперсию гидрида натрия в минеральном масле (0,072 г, 1,82 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 1 ч и затем туда добавляли йодэтан (0,283 г, 1,82 ммоль). Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали 11 ч. Добавляли воду (10 мл) и смесь экстрагировали этилацетатом (3×10 мл). Объединенную органическую фазу промывали солевым раствором (3×10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 50% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде масла желтого цвета (0,100 г, выход 44%):1H ЯМР (400 МГц, CDCl3) δ 7,55 (дт, J=5,6, 8,2 Гц, 1H), 7,34 (д, J=7,8 Гц, 1H), 7,10 (т, J=8,4 Гц, 1H), 3,88 (с, 2H), 3,40 (к, J=7,0 Гц, 2H), 3,35-3,28 (м, 2H), 3,17 (д, J=7,6 Гц, 2H), 1,53 (с, 3H), 1,22 (т, J=7,0 Гц, 3H).
Стадия 3. Получение (2-((3-этокси-3-метилазетидин-1-ил)метил)-6-фторфенил)метанамина

В раствор 2-((3-этокси-3-метилазетидин-1-ил)метил)-6-фторбензонитрила (0,100 г, 0,402 ммоль) в метаноле (20 мл) и гидроксиде аммония (5 мл) добавляли ренеевский никель (0,100 г). Суспензию дегазировали в вакууме и продували водородом несколько раз. Реакционную смесь перемешивали в атмосфере водорода (50 фунтов/дюйм2) при температуре окружающей среды в течение 12 ч. Фильтрованием и концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде масла желтого цвета (0,100 г, выход 98%):1H ЯМР (400 МГц, CDCl3) δ 7,38-7,30 (м, 1H), 7,21-7,10 (м, 2H), 6,2 (ушир. с, 2H), 4,39 (с, 2H), 4,15 (с, 2H), 3,61 (д, J=8,4 Гц, 2H), 3,45 (д, J=7,8 Гц, 2H), 3,38 (к, J=6,8 Гц, 2H), 1,50 (с, 3H), 1,21 (ушир. т, J=6,8 Гц, 3H); MS (ES+) m/z 253,3 (M+1).
Стадия 4. Получение трет-бутил ((4-((2-((3-этокси-3-метилазетидин-1-ил)метил)-6-фторбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

В смесь трет-бутил тиазол-4-ил((2,4,6-трифторфенил)-сульфонил)карбамата (0,156 г, 0,396 ммоль) и (2-((3-этокси-3-метилазетидин-1-ил)метил)-6-фторфенил)метанамина (0,100 г, 0,396 ммоль) в безводном N,N-диметилформамиде (5 мл) добавляли карбонат калия (0,109 г, 0,792 ммоль). Реакционную смесь нагревали до 30°C в течение 12 ч. Добавляли воду (10 мл) и смесь экстрагировали этилацетатом (3×10 мл). Объединенную органическую фазу промывали солевым раствором (3×10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,05% гидроксида аммония, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,050 г, выход 20%):1H ЯМР (400 МГц, CDCl3) δ 8,81 (д, J=2,2 Гц, 1H), 7,89 (ушир. с, 1H), 7,52 (д, J=2,2 Гц, 1H), 7,25 (дд, J=5,8, 8,0 Гц, 1H), 7,11-7,05 (м, 2H), 6,33 (д, J=12,0 Гц, 2H), 4,40 (ушир. д, J=4,2 Гц, 2H), 3,72 (с, 2H), 3,49-3,37 (м, 2H), 3,25-3,21 (м, 2H), 3,16-3,11 (м, 2H), 1,51 (с, 3H), 1,41 (с, 9H), 1,26 (т, J=7,0 Гц, 3H); MS (ES+) m/z 627,3 (M+1).
Стадия 5. Получение 4-((2-((3-этокси-3-метилазетидин-1-ил)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил ((4-((2-((3-этокси-3-метилазетидин-1-ил)метил)-6-фторбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,050 г, 0,079 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (0,770 г, 6,75 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Концентрированием in vacuo и очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,255% муравьиной кислоты, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,034 г, выход 71%):1H ЯМР (400 МГц, CD3OD) δ 8,75 (д, J=2,2 Гц, 1H), 8,38 (ушир. с, 0,6H), 7,41-7,32 (м, 1H), 7,21 (д, J=7,6 Гц, 1H), 7,14 (т, J=9,0 Гц, 1H), 6,98 (д, J=2,2 Гц, 1H), 6,37-6,29 (м, 2H), 4,40 (д, J=1,0 Гц, 2H), 3,94 (ушир. с, 2H), 3,47-3,37 (м, 6H), 1,46 (с, 3H), 1,18 (т, J=7,0 Гц, 3H), NH и COOH не наблюдали;19F ЯМР (376 МГц, CD3OD) δ -109,6 (с, 2F), -118,6 (с, 1F); MS (ES+) m/z 527,2 (M+1).
ПРИМЕРЫ 153-182
Схожим образом, как описано в примере 151, используя надлежащим образом замещенные исходные материалы и промежуточные соединения, получали следующие соединения:
ПРИМЕРЫ 183-184
Схожим образом, как описано в примере 152, используя надлежащим образом замещенные исходные материалы и промежуточные соединения, получали следующие соединения:
ПРИМЕР 185
Синтез формиата 2,6-дифтор-4-((2-фтор-6-((изопропил(метил)амино)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил (4-(N-(трет-бутоксикарбонил)-N-(тиазол-4-ил)сульфамоил)-3,5-дифторфенил)(2-фтор-6-(гидроксиметил)бензил)карбамата

В смесь трет-бутил ((4-((трет-бутоксикарбонил)(2-фтор-6-формилбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,100 г, 0,159 ммоль) и N,2-диметилпропан-2-амина (0,020 г, 0,238 ммоль) в метаноле (1 мл) добавляли цианоборогидрид натрия (0,010 г, 0,159 ммоль) и реакционную смесь перемешивали при температуре окружающей среды 1 ч. Концентрированием in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 50% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,160 г, количественный выход):1H ЯМР (400 МГц, CDCl3) δ 8,83 (д, J=2,2 Гц, 1H), 7,55 (д, J=2,2 Гц, 1H), 7,28-7,14 (м, 3H), 6,97 (д, J=10,2 Гц, 2H), 5,14 (с, 2H), 4,77 (с, 2H), 1,46 (с, 9H), 1,37 (с, 9H), OH не наблюдали; MS (ES+) m/z 474,1 (M - 155).
Стадия 2. Получение трет-бутил (2-(бромметил)-6-фторбензил)(4-(N-(трет-бутоксикарбонил)-N-(тиазол-4-ил)сульфамоил)-3,5-дифторфенил)карбамата

В раствор трет-бутил (4-(N-(трет-бутоксикарбонил)-N-(тиазол-4-ил)сульфамоил)-3,5-дифторфенил)(2-фтор-6-(гидроксиметил)бензил)карбамата (0,160 г, 0,254 ммоль) и тетрабромида углерода (0,168 г, 0,508 ммоль) в дихлорметане (2,00 мл) добавляли трифенилфосфин (0,133 г, 0,508 ммоль) при 0°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 2 ч. Концентрированием in vacuo и очисткой с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 30% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,100 г, 0,090 ммоль, выход 35%):1H ЯМР (400 МГц, CDCl3) δ 8,83 (д, J=2,2 Гц, 1H), 7,54 (д, J=2,2 Гц, 1H), 7,26-7,20 (м, 2H), 7,04 (д, J=10,4 Гц, 2H), 6,93-6,89 (м, 1H), 5,18 (с, 2H), 4,70 (с, 2H), 1,51 (с, 9H), 1,34 (с, 9H); MS (ES+) m/z 536,0 (M - 155), 538,0 (M - 155).
Стадия 3. Получение трет-бутил (4-(N-(трет-бутоксикарбонил)-N-(тиазол-4-ил)сульфамоил)-3,5-дифторфенил)(2-фтор-6-((изопропил(метил)амино)метил)-бензил)карбамата

В смесь N-метилпропан-2-амина (0,010 г, 0,144 ммоль) и трет-бутил (2-(бромметил)-6-фторбензил)(4-(N-(трет-бутоксикарбонил)-N-(тиазол-4-ил)сульфамоил)-3,5-дифторфенил)карбамата (0,050 г, 0,072 ммоль) в безводном N,N-диметилформамиде (2 мл) добавляли карбонат калия (0,019 г, 0,144 ммоль) при 0°C. Реакционную смесь перемешивали при температуре окружающей среды в течение 12 ч и затем туда добавляли воду (10 мл). Смесь экстрагировали этилацетатом (3×10 мл). Объединенную органическую фазу промывали солевым раствором (3×10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой получали указанное в заголовке соединение в виде твердого вещества желтого цвета (0,050 мг, количественный выход): MS (ES+) m/z 685,3 (M+1).
Стадия 4. Получение формиата 2,6-дифтор-4-((2-фтор-6-((изопропил(метил)амино)-метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил (4-(N-(трет-бутоксикарбонил)-N-(тиазол-4-ил)сульфамоил)-3,5-дифторфенил)(2-фтор-6-((изопропил(метил)амино)метил)бензил)карбамата (0,050 г, 0,073 ммоль) в дихлорметане (2 мл) добавляли трифторуксусную кислоту (6,16 г, 54,03 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 3 ч. Концентрированием in vacuo и очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,255% муравьиной кислоты, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,012 г, выход 29%):1H ЯМР (400 МГц, CD3OD) δ 8,75 (д, J=2,2 Гц, 1H), 8,50-8,41 (м, 1H), 7,46-7,37 (м, 1H), 7,33-7,25 (м, 1H), 7,25-7,16 (м, 1H), 6,98 (д, J=2,2 Гц, 1H), 6,32 (д, J=12,3 Гц, 2H), 4,42 (д, J=1,3 Гц, 2H), 4,00-3,89 (м, 2H), 3,30-3,16 (м, 1H), 2,42-2,30 (м, 3H), 1,27-1,13 (м, 6H), NH и COOH не наблюдали; MS (ES+) 485,2 (M+1).
ПРИМЕР 186
Синтез формиата 4-((2-((5-азаспиро[2.3]гексан-5-ил)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано для примера 201, стадий 3-4 и выполняя некритические вариации при необходимости, чтобы заменять N-метилпропан-2-амин на 5-азаспиро[2.3]гексан, указанное в заголовке соединение получали в виде бесцветного твердого вещества (3,5 мг, выход 22%):1H ЯМР (400 МГц, CD3OD) δ 8,75 (д, J=2,2 Гц, 1H), 8,51-8,47 (м, 1H), 7,44-7,39 (м, 1H), 7,27-7,26 (м, 1H), 7,21-7,16 (м, 1H), 6,98 (д, J=2,2 Гц, 1H), 6,32 (д, J=12,3 Гц, 2H), 4,40 (д, J=0,8 Гц, 2H), 4,17 (с, 2H), 3,77 (с, 4H), 0,67 (с, 4H), NH и COOH не наблюдали; MS (ES+) m/z 495,3 (M+1).
ПРИМЕР 187
Синтез формиата 4-((2-(((циклопропилметил)(метил)амино)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил (4-(N-(трет-бутоксикарбонил)-N-(тиазол-4-ил)сульфамоил)-3,5-дифторфенил)(2-(((циклопропилметил)(метил)амино)метил)-6-фторбензил)карбамата

В смесь гидрохлорида 1-циклопропил-N-метилметанамина (0,038 г, 0,318 ммоль) и трет-бутил ((4-((трет-бутоксикарбонил)(2-фтор-6-формилбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,100 г, 0,159 ммоль) в дихлорметане (2 мл) добавляли изопропоксид титана(IV) (0,090 г, 0,318 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Затем туда добавляли триацетоксиборгидрид натрия (0,135 г, 0,637 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 11 ч. Добавляли насыщенный бикарбонат натрия (1 мл) и смесь перемешивали в течение 30 минут. После разведения водой (10 мл) смесь экстрагировали дихлорметаном (3×10 мл). Объединенную органическую фазу промывали солевым раствором (3×10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием in vacuo получали указанное в заголовке соединение в виде маслянистого остатка (0,072 г, выход 65%), который использовали без дополнительной очистки: MS (ES+) m/z 697,2 (M+1).
Стадия 2. Получение формиата 4-((2-(((циклопропилметил)(метил)амино)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил (4-(N-(трет-бутоксикарбонил)-N-(тиазол-4-ил)сульфамоил)-3,5-дифторфенил)(2-(((циклопропилметил)(метил)амино)метил)-6-фторбензил)карбамата (0,070 г, 0,100 ммоль) в дихлорметане (2 мл) добавляли трифторуксусную кислоту (3,08 г, 27,0 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 12 ч. Концентрированием in vacuo и очисткой остатка in vacuo и очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,05% гидроксида аммония, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,032 г, выход 62%):1H ЯМР (400 МГц, DMSO-d6) δ 8,78 (ушир. с, 1H), 7,37-7,24 (м, 1H), 7,20-7,07 (м, 3H), 6,64 (ушир. с, 1H), 6,30 (д, J=12,4 Гц, 2H), 4,34 (д, J=3,8 Гц, 2H), 3,51 (с, 2H), 2,18 (д, J=6,6 Гц, 2H), 2,09 (с, 3H), 0,75-0,80 (м, 1H), 0,35 (к, J=5,0, 2H), -0,01 (к, J=5,0 Гц, 2H), NH и COOH не наблюдали; MS (ES+) m/z 497,3 (M+1).
ПРИМЕР 188
Синтез 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(изотиазол-3-ил)бензолсульфонамида
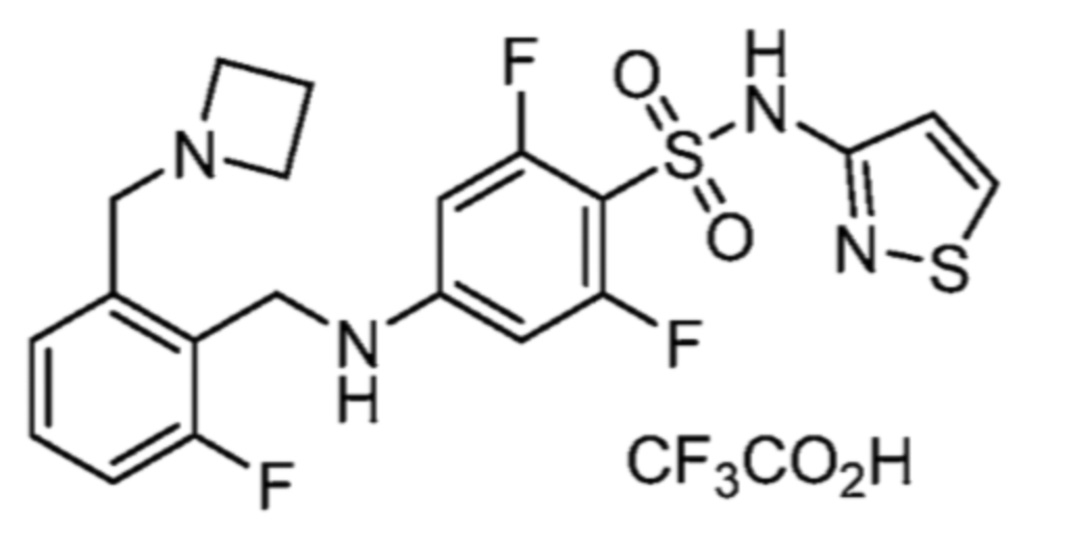
Стадия 1. Получение трет-бутил изотиазол-3-илкарбамата

Во взвесь изотиазол-3-карбоновой кислоты (5,0 г, 38,7 ммоль) в трет-бутаноле (194 мл) добавляли триэтиламин (4,3 г, 42,6 ммоль), после чего следовал дифенилфосфорилазид (11,9 г, 43,3 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 9 ч. После охлаждения до температуры окружающей среды, реакционную смесь концентрировали in vacuo и остаток растворяли в этилацетате (300 мл). Органический слой промывали водой (100 мл), 1 Н раствором гидроксида натрия (50 мл), водой (100 мл), солевым раствором (50 мл) и сушили над безводным сульфатом магния. Фильтрованием и концентрированием фильтрата in vacuo получали остаток. Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 10% этилацетата в гептане, получали указанное в заголовке соединение в виде бесцветного твердого вещества (6,16 г, выход 79%):1H ЯМР (300 МГц, CDCl3) δ 9,03-8,98 (м, 1H), 8,58 (д, J=4,9 Гц, 1H), 7,70 (д, J=4,9 Гц, 1H), 1,53 (д, J=0,7 Гц, 9H).
Стадия 2. Получение 3-бром-2,4,6-трифторбензолсульфонилхлорида

К 4-бром-1,3,5-трифторбензолу (25 г, 0,118 ммоль) добавляли хлорсульфоновую кислоту (24 мл) и реакционную смесь нагревали до 80°C в течение 72 ч. Реакционную смесь оставляли остывать до температуры окружающей среды и медленно добавляли на лед. Получаемое твердое вещество отфильтровывали и растворяли в дихлорметане (200 мл). Органическую фазу промывали водой (2×50 мл), солевым раствором (50 мл), сушили над безводным сульфатом магния. Фильтрованием через подушку из целита и концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде бесцветного твердого вещества (28,6 г, выход 78%):1H ЯМР (300 МГц, CDCl3) δ 7,06 (ддд, J=9,9, 7,8, 2,2 Гц, 1H).
Стадия 3. Получение трет-бутил ((3-бром-2,4,6-трифторфенил)сульфонил)-(изотиазол-3-ил)карбамата

В раствор трет-бутил изотиазол-3-илкарбамата (0,9 г, 4,49 ммоль) в безводном тетрагидрофуране (12 мл) добавляли 1 M раствор бис(триметилсилил)амида лития в тетрагидрофуране (4,94 мл, 4,94 ммоль) при -78°C. Реакционную смесь перемешивали в течение 10 минут при -78°C и затем оставляли нагреваться до температуры окружающей среды и перемешивали в течение 1 ч. После охлаждения реакционной смеси до -78°C, туда добавляли раствор 5-бром-2,4,6-трифторбензолсульфонилхлорида (1,39 г, 4,49 ммоль) в безводном тетрагидрофуране (2,6 мл). Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 12 ч. Реакционную смесь гасили добавлением насыщенного раствора хлорида аммония (50 мл) и водный слой экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали водой (50 мл), солевым раствором (50 мл), сушили над сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 5% этилацетата в гептане, получали указанное в заголовке соединение в виде твердого вещества бежевого цвета (1,08 г, выход 97%):1H ЯМР (300 МГц, CDCl3) δ 8,76 (д, J=4,7 Гц, 1H), 7,33 (д, J=4,7 Гц, 1H), 6,99 (ддд, J=9,9, 7,9, 2,1 Гц, 1H), 1,44-1,35 (м, 9H).
Стадия 4: получение трет-бутил ((4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-3-бром-2,6-дифторфенил)сульфонил)(изотиазол-3-ил)карбамата

В раствор трет-бутил ((3-бром-2,4,6-трифторфенил)сульфонил)(изотиазол-3-ил)карбамата (0,50 г, 1,1 ммоль) в безводном N,N-диметилформамиде (5,3 мл) добавляли (2-(азетидин-1-илметил)-6-фторфенил)метанамин (0,25 г, 1,3 ммоль), после чего следовал N,N-диизопропилэтиламин (0,27 г, 2,12 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч. Реакционную смесь разводили этилацетатом (150 мл). Органический слой промывали водой (2×50 мл), солевым раствором (50 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 10 до 50% этилацетата в гептане, получали указанный в заголовке продукт в виде бесцветного твердого вещества (0,37 г, выход 54%): LCMS (ES+) m/z 647,4 (M+1), 649,4 (M+1).
Стадия 5: получение 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(изотиазол-3-ил)бензолсульфонамида

В раствор трет-бутил ((4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-3-бром-2,6-дифторфенил)сульфонил)(изотиазол-3-ил)карбамата (0,33 г, 0,50 ммоль) в безводном диметилсульфоксиде (2 мл) добавляли формиат натрия (0,068 г, 1,0 ммоль) и смесь дегазировали аргоном в течение 10 минут. Затем туда добавляли трис(дибензилиденацетон)дипалладий(0) (0,014 г, 0,015 ммоль) и три-трет-бутилфосфин (0,006 г, 0,03 ммоль) и реакционную смесь нагревали до 80°C в течение 16 ч. После охлаждения до температуры окружающей среды, реакционную смесь разводили этилацетатом (100 мл). Органическую фазу промывали водой (4×30 мл), солевым раствором (30 мл), сушили над сульфатом магния и фильтровали через подушку из целита. Концентрированием in vacuo и очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,037 г, выход 13%):1H ЯМР (300 МГц, DMSO-d6) δ 11,76 (с, 1H), 10,06 (с, 1H), 8,93 (д, J=4,8 Гц, 1H), 7,56-7,49 (м, 1H), 7,41-7,32 (м, 2H), 7,29-7,25 (м, 1H), 6,93 (д, J=4,8 Гц, 1H), 6,42-6,35 (м, 2H), 4,50-4,41 (м, 2H), 4,37-4,30 (м, 2H), 4,17-3,94 (м, 4H), 2,46-2,22 (м, 2H);19F ЯМР (282 МГц, DMSO-d6) δ -73,6 (с, 3F), -108,3 (с, 2F), -115,3 (с, 1F); MS (ES+) m/z 469,1 (M+1).
ПРИМЕР 189
Синтез формиата 4-((2-((трет-бутил(метил)амино)метил)-6-фторбензил)амино)-2,6-дифтор-N-(изотиазол-3-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил ((3-бром-4-((2-((трет-бутил(метил)амино)метил)-6-фторбензил)амино)-2,6-дифторфенил)сульфонил)(изотиазол-3-ил)карбамата

В раствор трет-бутил ((3-бром-2,4,6-трифторфенил)сульфонил)(изотиазол-3-ил)карбамата (0,88 г, 1,85 ммоль) в безводном N,N-диметилформамиде (10 мл) добавляли N-(2-(аминометил)-3-фторбензил)-N,2-диметилпропан-2-амин (0,83 г, 3,7 ммоль), после чего следовал N,N-диизопропилэтиламин (0,72 г, 5,5 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 12 ч, и затем разводили этилацетатом (150 мл). Органический слой промывали водой (3×50 мл), солевым раствором (50 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 10 до 50% этилацетата в гептане, получали указанное в заголовке соединение в виде масла оранжевого цвета (0,42 г, выход 34%): LCMS (ES+) m/z 577,4 (M - 99), 579,4 (M - 99).
Стадия 2: получение формиата 4-((2-((трет-бутил(метил)амино)метил)-6-фторбензил)амино)-2,6-дифтор-N-(изотиазол-3-ил)бензолсульфонамида

В раствор трет-бутил ((3-бром-4-((2-((трет-бутил(метил)амино)метил)-6-фторбензил)амино)-2,6-дифторфенил)сульфонил)(изотиазол-3-ил)карбамата (0,42 г, 0,62 ммоль) в безводном диметилсульфоксиде (1 мл) добавляли формиат натрия (0,13 г, 1,86 ммоль) и смесь дегазировали аргоном в течение 10 минут. Затем туда добавляли трис(дибензилиденацетон)дипалладий(0) (0,057 г, 0,062 ммоль) и добавляли три-трет-бутилфосфин (0,025 г, 0,124 ммоль) и реакционную смесь нагревали до 80°C в течение 16 ч. После охлаждения до температуры окружающей среды, реакционную смесь разводили этилацетатом (100 мл). Органическую фазу промывали водой (4×30 мл), солевым раствором (30 мл) и сушили над безводным сульфатом магния. Фильтрованием и концентрированием фильтрата in vacuo получали остаток. Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 10 до 80% этилацетата в гептане, после чего следовала очистка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,5% муравьиной кислоты, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,013 г, выход 4%):1H-ЯМР (300 МГц, DMSO-d6) δ 8,79 (т, J=3,3 Гц, 1H), 8,19 (с, 1H), 7,38-7,25 (м, 2H), 7,16-7,10 (м, 2H), 6,87 (д, J=4,8 Гц, 1H), 6,35 (д, J=12,6 Гц, 2H), 4,34 (с, 2H), 3,57 (с, 2H), 1,96 (с, 3H), 1,03 (с, 9H), NH не наблюдали; MS (ES+) m/z 499,1 (M+1).
ПРИМЕР 190
Синтез (S)-3-хлор-4-((1-(5-хлор-2-фторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

В смесь гидрохлорида (S)-1-(5-хлор-2-фторфенил)этан-1-амина (0,24 г, 1,15 ммоль) и трет-бутил ((3-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,47 г, 1,15 ммоль) в безводном диметилсульфоксиде (6 мл) добавляли карбонат калия (0,48 г, 3,45 ммоль) и реакционную смесь перемешивали при 75°C в течение 1 ч. Реакционную смесь оставляли остывать до температуры окружающей среды и затем разводили водой (20 мл) и этилацетат (30 мл). Водный слой экстрагировали этилацетатом (2×30 мл). Объединенную органическую фазу промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали остаток, который растворяли в безводном дихлорметане (6 мл). Туда добавляли трифторуксусную кислоту (0,26 мл, 3,46 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 5 ч. Реакционную смесь концентрировали in vacuo и остаток очищали с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом ацетонитрила в воде, содержащей 0,1% муравьиной кислоты, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,059 г, выход 11% на 2 стадии):1H-ЯМР (300 МГц, CDCl3) δ 10,09 (с, 1H), 8,71 (д, J=2,2 Гц, 1H), 7,59-7,53 (м, 1H), 7,26-7,18 (м, 2H), 7,05 (т, J=9,2 Гц, 1H), 6,87 (д, J=2,2 Гц, 1H), 6,18-6,15 (м, 1H), 5,13 (д, J=5,9 Гц, 1H), 4,88-4,79 (м, 1H), 1,63 (д, J=6,7 Гц, 3H); MS (ES+) m/z 463,9 (M+1), 465,9 (M+1), 467,9 (M+1)
ПРИМЕР 191
Синтез (S)-3-хлор-4-((1-(2-хлорфенил)этил)амино)-2-фтор-N-(пиримидин-4-ил)бензолсульфонамида

Стадия 1. Получение N-(2,4-диметоксибензил)пиримидин-4-амина

В раствор 4-аминопиримидина (1,50 г, 15,8 ммоль) и 2,4-диметоксибензальдегида (2,62 г, 15,8 ммоль) в толуоле (80 мл) добавляли уксусную кислоту (0,090 мл, 1,6 ммоль) и смесь нагревали с обратным холодильником в течение 23 ч с использованием ловушки Дина-Старка для азеотропного удаления воды. После охлаждения до температуры окружающей среды, смесь концентрировали in vacuo и к остатку добавляли безводный метанол (50 мл). Затем в смесь добавляли борогидрид натрия (1,2 г, 32 ммоль) при температуре окружающей среды в течение периода 40 минут. Смесь перемешивали при температуре окружающей среды в течение 16 ч и затем концентрировали in vacuo. Остаток распределялся между этилацетатом (50 мл) и 1 M гидроксидом натрия (30 мл). Водную фазу экстрагировали этилацетатом (40 мл) и объединенные органические слои сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 25 до 50% этилацетата (содержащего 10% изопропанола и 10% триэтиламина) в гептане, получали указанное в заголовке соединение в виде масла желтого цвета (2,69 г, выход 70%):1H ЯМР (300 МГц, CDCl3) δ 8,52 (с, 1H), 8,12 (д, J=6,0 Гц, 1H), 7,17 (д, J=8,2 Гц, 1H), 6,46 (д, J=2,3 Гц, 1H), 6,42 (дд, J=8,2, 2,4 Гц, 1H), 6,32 (д, J=6,0 Гц, 1H), 5,54 (ушир. с, 1H), 4,43-4,41 (м, 2H), 3,82 (с, 3H), 3,79 (с, 3H); MS (ES+) m/z 246,3 (M+1).
Стадия 2. Получение 3-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(пиримидин-4-ил)бензолсульфонамида

В смесь N-(2,4-диметоксибензил)пиримидин-4-амина (получен в соответствии с WO2012004743, 3,28 г, 13,4 ммоль) в безводном тетрагидрофуране (60 мл) добавляли 1 M раствор бис(триметилсилил)амида лития в тетрагидрофуране (13,4 мл, 13,4 ммоль) при -50°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 1 ч. Затем реакционную смесь добавляли в холодный (-78°C) раствор 3-хлор-2,4-дифторбензолсульфонилхлорида (3,00 г, 12,1 ммоль) в безводном тетрагидрофуране (60 мл). Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 16 ч. Затем реакционную смесь гасили добавлением насыщенного раствора хлорида аммония (80 мл) и туда добавляли этилацетат (100 мл). Водную фазу экстрагировали этилацетатом (2×100 мл). Объединенную органическую фазу промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 50% этилацетата в гексанах, получали указанное в заголовке соединение в виде масла желтого цвета (0,86 г, выход 15%):1H ЯМР (300 МГц, CDCl3) δ 8,75 (т, J=0,6 Гц, 1H), 8,46 (дд, J=5,9, 0,4 Гц, 1H), 8,03 (ддд, J=9,0, 7,6, 5,7 Гц, 1H), 7,23-7,20 (м, 1H), 7,17-7,13 (м, 1H), 7,11-7,09 (м, 1H), 6,42 (дд, J=6,9, 2,3 Гц, 2H), 5,25 (с, 2H), 3,81 (с, 3H), 3,78 (с, 3H); MS (ES+) m/z 456,1 (M+1), 458,1 (M+1).
Стадия 3. Получение (S)-3-хлор-4-((1-(2-хлорфенил)этил)амино)-2-фтор-N-(пиримидин-4-ил)бензолсульфонамида

В смесь (S)-1-(2-хлорфенил)этан-1-амина (0,18 г, 0,93 ммоль) и 3-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(пиримидин-4-ил)бензолсульфонамида (0,42 г, 0,93 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли карбонат калия (0,39 г, 2,80 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч. Реакционную смесь разводили водой (20 мл) и этилацетатом (20 мл). Водный слой экстрагировали этилацетатом (2×30 мл). Объединенную органическую фазу промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo для того, чтобы получать остаток, который растворяли в безводном дихлорметане (5 мл). Туда добавляли трифторуксусную кислоту (0,80 мл, 11,6 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 5 ч. Реакционную смесь разводили метанолом (20 мл) и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом ацетонитрила в воде, содержащей 0,1% муравьиной кислоты, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,10 г, выход 24% на 2 стадии):1H-ЯМР (300 МГц, DMSO-d6) δ 8,54 (с, 1H), 8,26 (д, J=7,2 Гц, 1H), 7,56 (т, J=8,5 Гц, 1H), 7,48-7,43 (м, 2H), 7,33-7,25 (м, 2H), 6,94 (д, J=6,6 Гц, 1H), 6,79 (д, J=7,2 Гц, 1H), 6,15 (д, J=9,3 Гц, 1H), 5,00-4,92 (м, 1H), 1,55 (д, J=6,7 Гц, 3H), NH не наблюдали; MS (ES+) m/z 441,0 (M+1), 443,0 (M+1), 445,0 (M+1).
ПРИМЕР 192
Синтез формиата 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-3-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

В смесь (2-(азетидин-1-илметил)-6-фторфенил)метанамина (0,10 г, 0,51 ммоль) и трет-бутил ((3-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,21 г, 0,51 ммоль) в безводном диметилсульфоксиде (3 мл) добавляли карбонат калия (0,14 г, 1,03 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч. Реакционную смесь разводили водой (20 мл) и этилацетатом (20 мл). Водный слой экстрагировали этилацетатом (2×30 мл). Объединенную органическую фазу промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали остаток, который растворяли в безводном дихлорметане (3 мл). Туда добавляли трифторуксусную кислоту (0,47 мл, 6,18 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 2,5 ч. Реакционную смесь концентрировали in vacuo и остаток очищали с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом ацетонитрила в воде, содержащей 0,5% муравьиной кислоты, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,074 г, выход 30% на 2 стадии):1H-ЯМР (300 МГц, DMSO-d6) δ 11,17-11,01 (м, 1H), 8,88 (д, J=2,2 Гц, 1H), 8,14 (с, 0,5H), 7,59 (т, J=8,6 Гц, 1H), 7,37-7,34 (м, 1H), 7,24 (д, J=7,8 Гц, 2H), 6,97 (д, J=2,2 Гц, 1H), 6,82 (д, J=9,6 Гц, 1H), 4,50-4,49 (м, 2H), 4,06-3,99 (м, 2H), 3,58-3,44 (м, 4H), 2,19-2,10 (м, 2H), два способных к обмену протона не наблюдали; MS (ES+) m/z 485,0 (M+1), 487,0 (M+1).
ПРИМЕР 193
Синтез формиата 4-((2-(азетидин-1-илметил)-4,5-дифторбензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 2-(азетидин-1-илметил)-4,5-дифторбензонитрила

Следуя процедуре как описано в примере 16, стадии 1 и выполняя вариации при необходимости, чтобы заменять 2-(бромметил)бензонитрил на (2-(бромметил)-4,5-дифторбензонитрил, указанное в заголовке соединение получали в виде масла желтого цвета (6,99 г, выход 79%):1H ЯМР (300 МГц, CDCl3) δ 7,61 (дд, J=8,6, 5,4 Гц, 1H), 7,02 (ддд, J=8,5, 7,9, 2,6 Гц, 1H), 3,33-3,28 (м, 4H), 2,18-2,09 (м, 2H); MS (ES+) m/z 191,2 (M+1).
Стадия 2. Получение формиата 4-((2-(азетидин-1-илметил)-4,5-дифторбензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано в примере 16, стадии 2 и выполняя вариации при необходимости, чтобы заменять 2-(азетидин-1-илметил)бензонитрил на 2-(азетидин-1-илметил)-4,5-дифторбензонитрил, получали масло красного цвета в качестве остатка. В смесь этого остатка в безводном диметилсульфоксиде (5 мл) добавляли трет-бутил ((5-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамат (0,39 г, 0,94 ммоль) и карбонат калия (0,26 г, 1,88 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч. Реакционную смесь разводили водой (20 мл) и этилацетатом (20 мл). Водный слой экстрагировали этилацетатом (2×30 мл). Объединенную органическую фазу промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали остаток, который растворяли в безводном дихлорметане (5 мл). Туда добавляли трифторуксусную кислоту (1,1 мл, 14,1 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Реакционную смесь концентрировали in vacuo и остаток очищали с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом ацетонитрила в воде, содержащей 0,5% муравьиной кислоты, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,057 г, выход 11% на 3 стадии):1H-ЯМР (300 МГц, DMSO-d6) δ 11,09 (с, 1H), 8,88 (д, J=2,2 Гц, 1H), 8,14 (с, 1H), 7,63 (д, J=7,4 Гц, 1H), 7,52-7,45 (м, 2H), 7,30 (дд, J=11,7, 8,3 Гц, 1H), 6,99 (д, J=2,2 Гц, 1H), 6,79 (д, J=13,7 Гц, 1H), 4,49-4,47 (м, 2H), 3,97 (с, 2H), 3,56 (с, 4H), 2,20-2,13 (м, 2H), один способный к обмену протон не наблюдали; MS (ES+) m/z 501,0 (M+1), 503,1 (M+1).
ПРИМЕР 194
Синтез формиата 2,6-дифтор-4-((2-фтор-6-((3-фторазетидин-1-ил)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида

В смесь (2-фтор-6-((3-фторазетидин-1-ил)метил)фенил)метанамина (0,50 г, 2,36 ммоль) и трет-бутил тиазол-4-ил((2,4,6-трифторфенил)сульфонил)карбамата (0,93 г, 2,36 ммоль) в безводном диметилсульфоксиде (12 мл) добавляли N,N-диизопропилэтиламин (2,1 мл, 11,8 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч. Реакционную смесь разводили водой (20 мл) и этилацетатом (20 мл). Водный слой экстрагировали этилацетатом (2×30 мл). Объединенную органическую фазу промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали остаток, который растворяли в безводном дихлорметане (12 мл). Туда добавляли трифторуксусную кислоту (5,4 мл, 70,7 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч. Реакционную смесь концентрировали in vacuo и остаток очищали с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом ацетонитрила в воде, содержащей 0,5% муравьиной кислоты, чтобы получать указанное в заголовке соединение в виде твердого вещества бледно-желтого цвета (0,062 г, выход 5% на 2 стадии):1H-ЯМР (300 МГц, DMSO-d6) δ 11,24-11,17 (м, 1H), 8,90 (д, J=2,1 Гц, 1H), 8,14 (с, 1H), 7,41-7,15 (м, 4H), 6,91-6,90 (м, 1H), 6,40-6,36 (м, 2H), 5,29-5,07 (м, 1H), 4,32-4,30 (м, 2H), 3,11-3,03 (м, 2H), 1,29-1,21 (м, 4H), один способный к обмену протон не наблюдали; MS (ES+) m/z 487,0 (M+1).
ПРИМЕР 195
Синтез формиата 4-((5-(азетидин-1-илметил)-2-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 5-(азетидин-1-илметил)-2-фторбензонитрила

Следуя процедуре как описано в примере 16, стадии 1 и выполняя вариации при необходимости, чтобы заменять 2-(бромметил)бензонитрил на 5-(бромметил)-2-фторбензонитрил, указанное в заголовке соединение получали в виде бесцветного масла (1,73 г, выход 89%):1H ЯМР (300 МГц, CDCl3) δ 7,57-7,50 (м, 2H), 7,16 (т, J=8,6 Гц, 1H), 5,27-5,01 (м, 2H), 3,71-3,61 (м, 4H), 3,26-3,13 (м, 2H); MS (ES+) m/z 209,2 (M+1).
Стадия 2. Получение (5-(азетидин-1-илметил)-2-фторфенил)метанамина

Следуя процедуре как описано в примере 16, стадии 2 и выполняя вариации при необходимости, чтобы заменять (2-фтор-6-((3-фторазетидин-1-ил)метил)фенил)-метанамин на (5-(азетидин-1-илметил)-2-фторфенил)метанамин, указанное в заголовке соединение получали в виде масла желтого цвета (1,37 г, количественный выход):1H ЯМР (300 МГц, CDCl3) δ 7,36-7,25 (м, 1H), 7,24-7,12 (м, 1H), 7,02-6,94 (м, 1H), 3,89-3,86 (м, 2H), 3,60-3,56 (м, 2H), 3,30-3,24 (м, 4H), 2,17-2,10 (м, 2H), NH не наблюдали; MS (ES+) m/z 195,2 (M+1).
Стадия 3. Получение формиата 4-((5-(азетидин-1-илметил)-2-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано в примере 194, и выполняя вариации при необходимости, чтобы заменять (2-фтор-6-((3-фторазетидин-1-ил)метил)фенил)метанамин на (5-(азетидин-1-илметил)-2-фторфенил)метанамин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,13 г, выход 27%):1H-ЯМР (300 МГц, DMSO-d6): δ 10,80 (с, 1H), 8,89 (д, J=2,2 Гц, 1H), 8,15 (с, 1H), 7,62-7,58 (м, 1H), 7,40-7,25 (м, 3H), 6,89 (д, J=2,2 Гц, 1H), 6,34-6,30 (м, 2H), 4,36 (д, J=5,6 Гц, 2H), 4,12 (с, 2H), 3,80-3,74 (м, 4H), 2,28-2,17 (м, 2H), один способный к обмену протон не наблюдали; MS (ES+) m/z 469,2 (M+1).
ПРИМЕР 196
Синтез 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-3-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 2-(азетидин-1-илметил)-3-фторбензонитрила

Следуя процедуре как описано в примере 16, стадии 1 и выполняя вариации при необходимости, чтобы заменять 2-(бромметил)бензонитрил на 2-(бромметил)-3-фторбензонитрил, указанное в заголовке соединение получали в виде масла желтого цвета (0,72 г, выход 81%):1H ЯМР (300 МГц, CDCl3) δ 7,47-7,44 (м, 1H), 7,39-7,28 (м, 2H), 3,78 (д, J=1,8 Гц, 2H), 3,36-3,31 (м, 4H), 2,11-2,01 (м, 2H); MS (ES+) m/z 191,2 (M+1).
Стадия 2. Получение (2-(азетидин-1-илметил)-3-фторфенил)метанамина

Следуя процедуре как описано в примере 16, стадии 2 и выполняя вариации при необходимости, чтобы заменять (2-фтор-6-((3-фторазетидин-1-ил)метил)фенил)-метанамин на 2-(азетидин-1-илметил)-3-фторбензонитрил, указанное в заголовке соединение получали в виде масла оранжевого цвета (0,84 г, количественный выход):1H ЯМР (300 МГц, CDCl3) δ 7,23-7,12 (м, 2H), 6,97 (дддд, J=9,6, 8,1, 4,5, 1,6 Гц, 1H), 4,40-4,39 (м, 1H), 3,91-3,89 (м, 1H), 3,67 (дд, J=4,3, 1,6 Гц, 1H), 3,65-3,62 (м, 1H), 3,29-3,21 (м, 4H), 2,10-1,97 (м, 2H), NH не наблюдали; MS (ES+) m/z 195,2 (M+1).
Стадия 3. Получение 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-3-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

В смесь (2-(азетидин-1-илметил)-3-фторфенил)метанамина (0,25 г, 1,29 ммоль) и трет-бутил тиазол-4-ил((2,4,6-трифторфенил)сульфонил)карбамата (0,51 г, 1,29 ммоль) в безводном диметилсульфоксиде (6 мл) добавляли N,N-диизопропилэтиламин (1,1 мл, 6,44 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч. Реакционную смесь разводили водой (20 мл) и этилацетатом (20 мл). Водный слой экстрагировали этилацетатом (2×30 мл). Объединенную органическую фазу промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 50% этилацетата (содержащего 10% триэтиламин и 10% 2-пропанол) в гексанах, получали трет-бутил ((4-((2-(азетидин-1-илметил)-3-фторбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамат (выход не определяли): MS (ES+) m/z 569,0 (M+1). Затем туда добавляли безводный дихлорметан (5 мл) и трифторуксусную кислоту (2,2 мл, 28,3 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч и затем концентрировали in vacuo для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,15 г, выход 25% на 2 стадии):1H-ЯМР (300 МГц, DMSO-d6) δ 11,21 (с, 1H), 10,03-9,89 (м,1H) 8,90 (д, J=2,2 Гц, 1H), 7,55-7,46 (м, 2H), 7,32-7,26 (м, 1H), 7,20-7,17 (м, 1H), 6,91 (д, J=2,2 Гц, 1H), 6,33 (д, J=12,4 Гц, 2H), 4,49-4,46 (м, 4H), 4,23-4,02 (м, 4H), 2,41-2,23 (м, 2H); MS (ES+) m/z 469,1 (M+1).
ПРИМЕР 197
Синтез 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-3-фторбензил)амино)-3-хлор-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 3-хлор-2,4,6-трифторбензолсульфонилхлорида

К хлорсульфоновой кислоте (18,0 мл, 270,3 ммоль) добавляли 2-хлор-1,3,5-трифторбензол (7,20 г, 43,3 ммоль) при 0°C. Получаемую смесь перемешивали в течение 18 ч при температуре окружающей среды и затем нагревали до 65°C. Реакционную смесь оставляли остывать до температуры окружающей среды и затем добавляли по каплям в смесь льда (400 г) и концентрированной соляной кислоты (125 мл), поддерживая температуру ниже 5°C. После завершения добавления, смесь энергично перемешивали при 0°C в течение 1 ч. Осадок отфильтровывали и споласкивали водой (250 мл) для того, чтобы получать указанное в заголовке соединение в виде бесцветного аморфного твердого вещества (8,02 г, выход 70%):1H ЯМР (300 МГц, CDCl3) δ 7,07 (ддд, J=9,8, 8,3, 2,3 Гц, 1H).
Стадия 2. Получение трет-бутил ((3-хлор-2,4,6-трифторфенил)сульфонил)(тиазол-4-ил)карбамата

В раствор трет-бутил тиазол-4-илкарбамата (3,32 г, 16,6 ммоль) в безводном тетрагидрофуране (210 мл) добавляли 1 M раствор бис(триметилсилил)амида лития в тетрагидрофуране (16,6 мл, 16,6 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 1 ч, охлаждали до -78°C и затем туда по каплям добавляли раствор 3-хлор-2,4,6-трифторбензолсульфонилхлорида (4,00 г, 15,09 ммоль) в безводном тетрагидрофуране (15 мл). Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 16 ч. Реакционную смесь концентрировали in vacuo до объема приблизительно 50 мл. После разведения этилацетатом (160 мл), органический слой промывали насыщенным хлоридом аммония (150 мл), насыщенным бикарбонатом натрия (150 мл), солевым раствором (50 мл) и сушили над безводным сульфатом натрия. Фильтрованием и концентрированием фильтрата in vacuo получали остаток, который очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 10 до 50% этилацетата в гексанах, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (3,35 г, выход 52%):1H ЯМР (300 МГц, CDCl3) δ 8,83 (д, J=2,2 Гц, 1H), 7,55 (д, J=2,2 Гц, 1H), 6,99 (ддд, J=10,0, 8,2, 2,0 Гц, 1H), 1,40 (с, 9H); MS (ES+) m/z 329,0 (M - 99), 331,0 (M - 99).
Стадия 3. Получение 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-3-фторбензил)амино)-3-хлор-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида
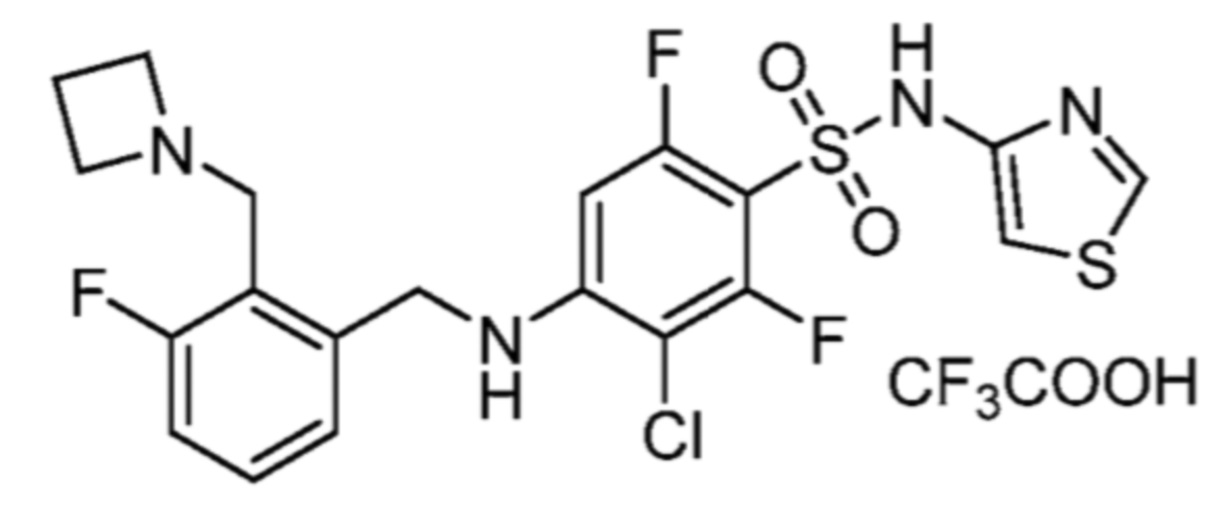
Следуя процедуре как описано в примере 196, стадии 3 и выполняя вариации при необходимости, чтобы заменять трет-бутил тиазол-4-ил((2,4,6-трифторфенил)сульфонил)карбамат на трет-бутил ((3-хлор-2,4,6-трифторфенил)сульфонил)(тиазол-4-ил)карбамат, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,24 г,выход 47%):1H-ЯМР (300 МГц, DMSO-d6) δ 11,39-11,37 (м, 1H), 10,08-9,90 (м, 1H), 8,92-8,89 (м, 1H), 7,55-7,43 (м, 2H), 7,29-7,23 (м, 1H), 7,12-7,09 (м, 1H), 7,02-6,98 (м, 1H), 6,47-6,42 (м, 1H), 4,62-4,53 (м, 4H), 4,22-4,07 (м, 4H), 2,41-2,26 (м, 2H); MS (ES+) m/z 503,1 (M+1), 505,1 (M+1).
ПРИМЕР 198
Синтез формиата 4-((2-((2,2-диметилазетидин-1-ил)метил)-3-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 2-((2,2-диметилазетидин-1-ил)метил)-3-фторбензонитрила

В раствор 2-(бромметил)-3-фторбензонитрила (0,35 г, 1,6 ммоль) и 2,2-диметилазетидина (0,15 г, 1,7 ммоль) в безводном дихлорметане (12 мл) добавляли N,N-диизопропилэтиламин (0,37 мл, 2,1 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч. Реакционную смесь разводили насыщенным раствором хлорида аммония (20 мл) и экстрагировали дихлорметаном (3×40 мл). Объединенные органические слои промывали солевым раствором (80 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде масла желтого цвета (0,34 г, выход 96%):1H ЯМР (300 МГц, CDCl3) δ 7,44 (дт, J=7,5, 2,0 Гц, 1H), 7,36-7,27 (м, 1H), 7,26-7,23 (м, 1H), 3,77-3,67 (м, 2H), 3,33-3,22 (м, 2H), 1,98-1,87 (м, 2H), 1,35-1,25 (м, 6H); MS (ES+) m/z 219,2 (M+1).
Стадия 2. Получение (2-((2,2-диметилазетидин-1-ил)метил)-3-фторфенил)-метанамина

В смесь ренеевского никеля (0,7 мл) и 2-((2,2-диметилазетидин-1-ил)метил)-3-фторбензонитрила (0,34 г, 1,6 ммоль) в этаноле (22 мл) добавляли концентрированный гидроксид аммония (5,4 мл). Реакционную смесь перемешивали в атмосфере водорода (1 атм.) при температуре окружающей среды в течение 16 ч. Реакционную смесь фильтровали через целит и фильтрат сушили над безводным сульфатом магния. Фильтрованием и концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде масла оранжевого цвета (0,38 г, количественный выход):1H ЯМР (300 МГц, CDCl3) δ 7,20 (д, J=1,5 Гц, 2H), 7,03-6,99 (м, 1H), 4,02 (с, 2H), 3,69 (д, J=1,2 Гц, 2H), 3,10 (с, 2H), 1,85 (д, J=7,4 Гц, 2H), 1,30 (с, 6H), NH не наблюдали; MS (ES+) m/z 223,2 (M+1).
Стадия 3. Получение трет-бутил ((3-бром-2,4,6-трифторфенил)сульфонил)(тиазол-4-ил)карбамата

В раствор трет-бутил тиазол-4-илкарбамата (26,90 г, 134,00 ммоль) в безводном тетрагидрофуране (500 мл) добавляли бис(триметилсилил)амид лития (1 M в тетрагидрофуране, 168 мл, 168,0 ммоль) при -78°C. Реакционную смесь перемешивали при -78°C в течение 20 минут, после чего при -78°C по каплям добавляли раствор 3-бром-2,4,6-трифторбензол-1-сульфонилхлорида (50,00 г, 161,00 ммоль) в безводном тетрагидрофуране (100 мл). Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 12 ч. Реакционную смесь концентрировали in vacuo и остаток разводили этилацетатом (3×400 мл). Органическую фазу промывали водой (3×400 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и растиранием остатка в метаноле (100 мл) получали указанное в заголовке соединение в виде бесцветного твердого вещества (40,00 г, выход 62%):1H ЯМР (400 МГц, CDCl3) δ 8,81 (д, J=2,3 Гц, 1H), 7,54 (д, J=2,2 Гц, 1H), 6,97 (ддд, J=9,8, 8,0, 2,2 Гц, 1H), 1,47-1,34 (м, 9H); MS (ES+) m/z 496,9 (M+23).
Стадия 4. Получение трет-бутил ((3-бром-4-((2-((2,2-диметилазетидин-1-ил)метил)-3-фторбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

В смесь трет-бутил ((3-бром-2,4,6-трифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,34 г, 0,71 ммоль) и карбоната калия (0,20 г, 1,42 ммоль) в безводном диметилсульфоксиде (4 мл) медленно добавляли раствор (2-((2,2-диметилазетидин-1-ил)метил)-3-фторфенил)метанамина (0,19 г, 0,85 ммоль) в безводном диметилсульфоксиде (3 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч. Реакционную смесь разводили водой (20 мл) и этилацетатом (20 мл). Водный слой экстрагировали этилацетатом (2×30 мл). Объединенную органическую фазу промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 40% этилацетата (содержащего 0,2% гидроксида аммония) в гептане, получали указанное в заголовке соединение в виде твердого вещества желтого цвета (0,19 г, выход 40%):1H ЯМР (300 МГц, CDCl3) δ 8,80 (д, J=2,3 Гц, 1H), 7,51 (д, J=2,2 Гц, 1H), 7,22-7,17 (м, 1H), 7,08-6,88 (м, 3H), 6,62-6,57 (м, 1H), 4,52-4,50 (м, 2H), 3,67-3,66 (м, 2H), 3,14-3,10 (м, 2H), 1,95-1,90 (м, 2H), 1,38 (с, 9H), 1,27 (с, 6H); MS (ES+) m/z 675,2 (M+1), 677,2 (M+1).
Стадия 5. Получение формиата 4-((2-((2,2-диметилазетидин-1-ил)метил)-3-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

В смесь трет-бутил ((3-бром-4-((2-((2,2-диметилазетидин-1-ил)метил)-3-фторбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,19 г, 0,29 ммоль) и палладия на углероде (4,5% палладия, влажн., 0,034 г) в метаноле (6 мл) добавляли триэтиламин (0,16 мл, 1,15 ммоль). Реакционную смесь перемешивали в атмосфере водорода (1 атм.) при 60°C в течение 7 ч. Реакционную смесь оставляли остывать до температуры окружающей среды, фильтровали через подушку целлиту и фильтрат концентрировали in vacuo. К остатку добавляли метанол (6 мл), палладий на углероде (4,5% палладия, влажн., 0,034 г) и триэтиламин (0,16 мл, 1,15 ммоль). Реакционную смесь перемешивали в атмосфере водорода (1 атм.) при 60°C в течение 2 ч. После охлаждения до температуры окружающей среды, смесь фильтровали через подушку из целита. Концентрированием фильтрата in vacuo получали остаток, который растворяли в безводном дихлорметане (7 мл). Туда добавляли трифторуксусную кислоту (0,52 мл, 6,73 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 3,5 ч. Реакционную смесь концентрировали in vacuo и остаток очищали с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом ацетонитрила в воде, содержащей 0,5% муравьиной кислоты, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,097 г, выход 58% на 2 стадии):1H-ЯМР (300 МГц, DMSO-d6) δ 8,88 (д, J=2,1 Гц, 1H), 8,14 (с, 0,8H), 7,74 (с, 1H), 7,29-7,22 (м, 1H), 7,10-7,04 (м, 2H), 6,89 (д, J=2,2 Гц, 1H), 6,42 (д, J=12,6 Гц, 2H), 4,50 (д, J=3,3 Гц, 2H), 3,66 (с, 2H), 3,14-3,08 (м, 2H), 1,85 (т, J=6,9 Гц, 2H), 1,23 (с, 6H), NH и COOH не наблюдали; MS (ES+) m/z 497,2 (M+1).
ПРИМЕР 199
Синтез формиата 4-((2-хлор-6-((2,2-диметилазетидин-1-ил)метил)бензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 2-хлор-6-((2,2-диметилазетидин-1-ил)метил)бензонитрила

Следуя процедуре как описано в примере 198, стадии 1 и выполняя вариации при необходимости, чтобы заменять 2-(бромметил)-3-фторбензонитрил на 2-(бромметил)-6-хлорбензонитрил, указанное в заголовке соединение получали в виде масла желтого цвета (1,06 г, количественный выход):1H ЯМР (300 МГц, CDCl3) δ 7,48-7,34 (м, 3H), 3,73 (с, 2H), 3,19-3,14 (м, 2H), 1,95-1,90 (м, 2H), 1,61-1,43 (м, 2H), 1,33-1,23 (м, 6H);MS (ES+) m/z 235,1 (M+1), 237,1 (M+1).
Стадия 2. Получение (2-хлор-6-((2,2-диметилазетидин-1-ил)метил)фенил)-метанамина
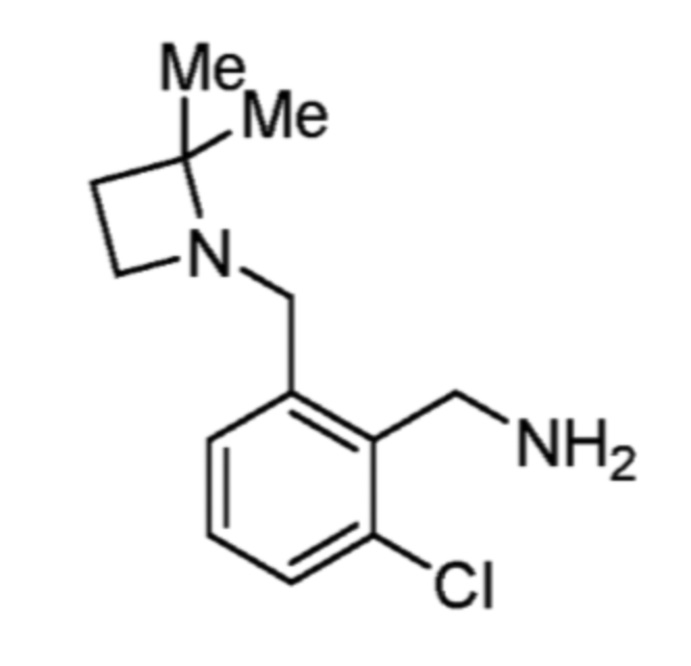
Следуя процедуре как описано в примере 198, стадии 2 и выполняя вариации при необходимости, чтобы заменять 2-((2,2-диметилазетидин-1-ил)метил)-3-фторбензонитрил на 2-хлор-6-((2,2-диметилазетидин-1-ил)метил)бензонитрил, указанное в заголовке соединение получали в виде масла оранжевого цвета (0,99 г, количественный выход): MS (ES+) m/z 239,2 (M+1), 241,2 (M+1).
Стадия 3. Получение трет-бутил ((3-бром-4-((2-хлор-6-((2,2-диметилазетидин-1-ил)метил)бензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

Следуя процедуре как описано в примере 198, стадии 3 и выполняя вариации при необходимости, чтобы заменять (2-((2,2-диметилазетидин-1-ил)метил)-3-фторфенил)метанамин на (2-хлор-6-((2,2-диметилазетидин-1-ил)метил)фенил)метанамин, указанное в заголовке соединение получали в виде твердого вещества бежевого цвета (0,41 г, выход 71%):1H ЯМР (300 МГц, CDCl3) δ 8,81 (д, J=2,2 Гц, 1H), 7,52 (д, J=2,3 Гц, 1H), 7,37 (дд, J=7,6, 1,7 Гц, 1H), 7,23-7,14 (м, 2H), 6,93-6,87 (м, 1H), 6,66-6,61 (м, 1H), 4,58 (д, J=5,6 Гц, 2H), 3,63 (с, 2H), 3,07 (т, J=7,0 Гц, 2H), 1,95-1,91 (м, 2H), 1,40 (с, 9H), 1,24 (с, 6H); MS (ES+) m/z 691,1 (M+1), 693,1 (M+1), 695,1 (M+1).
Стадия 4. Получение формиата 4-((2-хлор-6-((2,2-диметилазетидин-1-ил)метил)бензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

В смесь трет-бутил ((3-бром-4-((2-хлор-6-((2,2-диметилазетидин-1-ил)метил)бензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,40 г, 0,59 ммоль) и палладия на углероде (4,5% палладия, влажн., 0,070 г) в метаноле (6 мл) добавляли триэтиламин (0,33 мл, 2,37 ммоль). Реакционную смесь перемешивали в атмосфере водорода (1 атм.) при 60°C в течение 7 ч. После охлаждения до температуры окружающей среды, смесь фильтровали через подушку из целита. Концентрированием фильтрата in vacuo получали остаток, который растворяли в безводном дихлорметане (14 мл). Туда добавляли трифторуксусную кислоту (1,08 мл, 14,1 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 3,5 ч. Реакционную смесь концентрировали in vacuo и остаток очищали с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом ацетонитрила в воде, содержащей 0,5% муравьиной кислоты, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,14 г, выход 39%):1H-ЯМР (300 МГц, DMSO-d6) δ 8,89 (д, J=2,2 Гц, 1H), 8,19 (с, 1H), 7,42-7,27 (м, 3H), 7,18-7,15 (м, 1H), 6,87 (д, J=2,2 Гц, 1H), 6,40 (д, J=12,6 Гц, 2H), 4,44 (с, 2H), 3,51 (с, 2H), 2,96 (т, J=6,8 Гц, 2H), 1,76 (т, J=6,9 Гц, 2H), 1,09 (с, 6H), NH и COOH не наблюдали; MS (ES+) m/z 513,2 (M+1), 515,2 (M+1).
ПРИМЕР 200
Синтез 4-((2-(азетидин-1-илметил)бензил)амино)-5-хлор-2-фтор-N-(6-фторпиридин-2-ил)бензолсульфонамида

Стадия 1. Получение 4-((2-(азетидин-1-илметил)бензил)амино)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(6-фторпиридин-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 72, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять гидрохлорид (S)-1-фенил-2-(пирролидин-1-ил)этан-1-амина на (2-(азетидин-1-илметил)фенил)метанамин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,21 г, выход 30%): MS (ES+) m/z 629,2, 631,2 (M+1).
Стадия 2. Получение 4-((2-(азетидин-1-илметил)бензил)амино)-5-хлор-2-фтор-N-(6-фторпиридин-2-ил)бензолсульфонамида

Следуя процедуре как описано в примере 14, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять (S)-5-хлор-4-((1-(4-хлорфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид на 4-((2-(азетидин-1-илметил)бензил)амино)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(6-фторпиридин-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,012 г, выход 13%):1H ЯМР (300 МГц, DMSO-d6) δ 11,33 (с, 1H), 7,83-7,70 (м, 3H), 7,32-7,19 (м, 4H), 6,90-6,80 (м, 2H), 6,68-6,61 (м, 1H), 4,49-4,47 (м, 2H), 3,69 (с, 2H), 3,29-3,19 (м, 4H), 2,09-1,97 (м, 2H); MS (ES +) m/z 479,1 (M+1), 481,1 (M+1).
ПРИМЕР 201
Синтез 2,2,2-трифторацетата 2,6-дифтор-4-((2-фтор-6-((изопропил(метил)амино)метил)бензил)амино)-N-(изоксазол-3-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил изоксазол-3-ил((2,4,6-трифторфенил)сульфонил)карбамата

Следуя процедуре как описано в примере 27, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять N-(2,4-диметоксибензил)тиазол-2-амин на трет-бутил изоксазол-3-илкарбамат (получен в соответствии с WO2001040222), указанное в заголовке соединение получали в виде бесцветного твердого вещества (2,56 г, выход 38%): MS (ES+) m/z 379,1 (M+1).
Стадия 2. Получение трет-бутил ((2,6-дифтор-4-((2-фтор-6-((изопропил(метил)амино)метил)бензил)амино)фенил)сульфонил)(изоксазол-3-ил)карбамата

В смесь трет-бутил изоксазол-3-ил((2,4,6-трифторфенил)сульфонил)карбамата (1,0 г, 2,64 ммоль), N-(2-(аминометил)-3-фторбензил)-N-метилпропан-2-амина (0,56 г, 2,64 ммоль) и в безводном диметилсульфоксиде (25 мл) добавляли карбонат калия (0,73 г, 5,28 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч. Затем реакционную смесь разводили насыщенным хлоридом аммония (20 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали водой (40 мл), солевым раствором (40 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 40% этилацетата (содержащего 10% изопропанола и 10% триэтиламина) в гептане, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,21 г, выход 11%): MS (ES+) m/z 469,2 (M - 99).
Стадия 3. Получение 2,2,2-трифторацетата 2,6-дифтор-4-((2-фтор-6-((изопропил(метил)амино)метил)-бензил)амино)-N-(изоксазол-3-ил)бензолсульфонамида

В раствор трет-бутил ((2,6-дифтор-4-((2-фтор-6-((изопропил(метил)амино)метил)-бензил)амино)фенил)сульфонил)(изоксазол-3-ил)карбамата (0,2 г, 0,35 ммоль) в безводном дихлорметане (3 мл) добавляли трифторуксусную кислоту (0,4 мл, 5,28 ммоль) при 0°C. Реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч и затем концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 20% метанола в дихлорметане, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,089 г, выход 54%):1H ЯМР (300 МГц, DMSO-d6) δ 11,77 (с, 1H), 9,33 (с, 1H), 8,75-8,72 (м, 1H), 7,65-7,28 (м, 4H), 6,46-6,36 (м, 2H), 6,33-6,30 (м, 1H), 4,45-4,10 (м, 4H), 3,62-3,49 (м, 1H), 2,54 (с, 3H), 1,27-1,17 (м, 6H); MS (ES+) m/z 469,0 (M+1).
ПРИМЕР 202
Синтез 2,2,2-трифторацетата 4-((2-((трет-бутил(метил)амино)метил)бензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 2-((трет-бутил(метил)амино)метил)бензонитрила

В смесь 2-(бромметил)бензонитрила (3,37 г, 17,21 ммоль) и N-трет-бутилметиламина (1,5 г, 17,21 ммоль) в безводном диметилсульфоксиде (28 мл) добавляли карбонат калия (4,76 г, 34,42 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч. Затем смесь разводили водой (40 мл) и экстрагировали простым диэтиловым эфиром (3×40 мл). Объединенные органические слои промывали водой (30 мл), солевым раствором (40 мл), сушили над безводным сульфатом магния, фильтровали и фильтрат концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 10% метанола в дихлорметане, чтобы получать указанное в заголовке соединение в виде бесцветного масла (2,38 г, выход 51%): MS (ES+) m/z 203,3 (M+1).
Стадия 2. Получение N-(2-(аминометил)бензил)-N,2-диметилпропан-2-амина

Следуя процедуре как описано в примере 16, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 2-(азетидин-1-илметил)бензонитрил на 2-((трет-бутил(метил)амино)метил)бензонитрил, указанное в заголовке соединение получали в виде твердого вещества бледно-желтого цвета (2,1 г, выход 86%): MS (ES+) m/z 207,3 (M+1).
Стадия 3. Получение 2,4,6-трифтор-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил тиазол-4-ил((2,4,6-трифторфенил)сульфонил)карбамата (21,0 г, 53,1 ммоль) в дихлорметане (25 мл) добавляли трифторуксусную кислоту (12 мл). Смесь перемешивали при температуре окружающей среды в течение 16 ч и затем концентрировали in vacuo для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (15,5 г, выход 99%): MS (ES+) m/z 295,2 (M+1).
Стадия 4. Получение 2,4,6-трифтор-N-(4-метоксибензил)-N-(тиазол-4-ил)бензолсульфонамида

В раствор 2,4,6-трифтор-N-(тиазол-4-ил)бензолсульфонамида (15,5 г, 52,5 ммоль) в безводном диметилсульфоксиде (75 мл) добавляли 4-метоксибензилхлорид (12,3 г, 78,8 ммоль) и бикарбонат натрия (22,1 г, 262,5 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч. Реакционную смесь разводили этилацетатом (200 мл), промывали насыщенным хлоридом аммония (2×150 мл), солевым раствором (100 мл) и сушили над безводным сульфатом натрия. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 40% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (18,6 г, выход 85%):1H ЯМР (300 МГц, CDCl3) δ 8,57 (д, J=2,3 Гц, 1H), 7,25-7,21 (м, 3H), 6,81-6,72 (м, 4H), 5,07 (с, 2H), 3,77 (с, 3H); MS (ES+) m/z 415,0 (M+1).
Стадия 3. Получение 4-((2-((трет-бутил(метил)амино)метил)бензил)амино)-2,6-дифтор-N-(4-метоксибензил)-N-(тиазол-4-ил)бензолсульфонамида

В смесь 2,4,6-трифтор-N-(4-метоксибензил)-N-(тиазол-4-ил)бензолсульфонамида (1,05 г, 2,54 ммоль) и N-(2-(аминометил)бензил)-N,2-диметилпропан-2-амина (0,52 г, 2,54 ммоль) в безводном диметилсульфоксиде (20 мл) добавляли карбонат калия (0,70 г, 5,08 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч. Реакционную смесь разводили насыщенным хлоридом аммония (20 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали водой (40 мл), солевым раствором (40 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 40% этилацетата (содержащего 10% изопропанола и 10% триэтиламина) в гептане, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,52 г, выход 34%): MS (ES+) m/z 601,6 (M+1).
Стадия 4. Получение 2,2,2-трифторацетата 4-((2-((трет-бутил(метил)амино)метил)бензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида
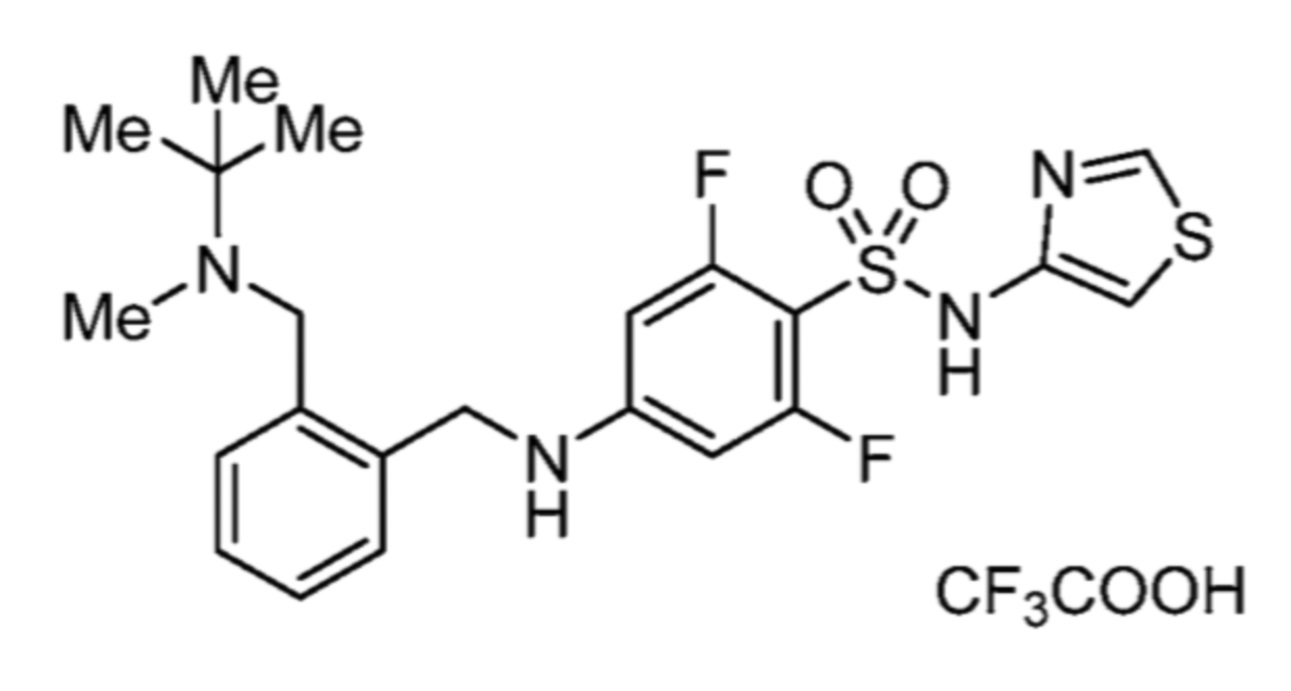
В раствор 4-((2-((трет-бутил(метил)амино)метил)бензил)амино)-2,6-дифтор-N-(4-метоксибензил)-N-(тиазол-4-ил)бензолсульфонамида (0,52 г, 0,87 ммоль) в безводном дихлорметане (7 мл) добавляли трифторуксусную кислоту (7 мл) и смесь нагревали с обратным холодильником в течение 16 ч. После охлаждения до температуры окружающей среды, смесь концентрировали in vacuo. Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 20% метанола в дихлорметане, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,495 г, выход 96%):1H ЯМР (300 МГц, DMSO-d6) δ 11,20 (с, 1H), 9,01-8,89 (м, 2H), 7,64-7,34 (м, 5H), 6,90 (д, J=2,2 Гц, 1H), 6,33 (д, J=12,6 Гц, 2H), 4,75-4,65 (м, 1H), 4,52-4,45 (м, 2H), 4,04-3,92 (м, 1H), 2,65-2,55 (м, 3H), 1,43 (с, 9H); MS (ES +) m/z 481,1 (M+1).
ПРИМЕР 203
Синтез 2,2,2-трифторацетата 4-((2-((трет-бутил(метил)амино)метил)-6-фторбензил)амино)-2,3-дифтор-N-(тиазол-4-ил)бензолсульфонамида
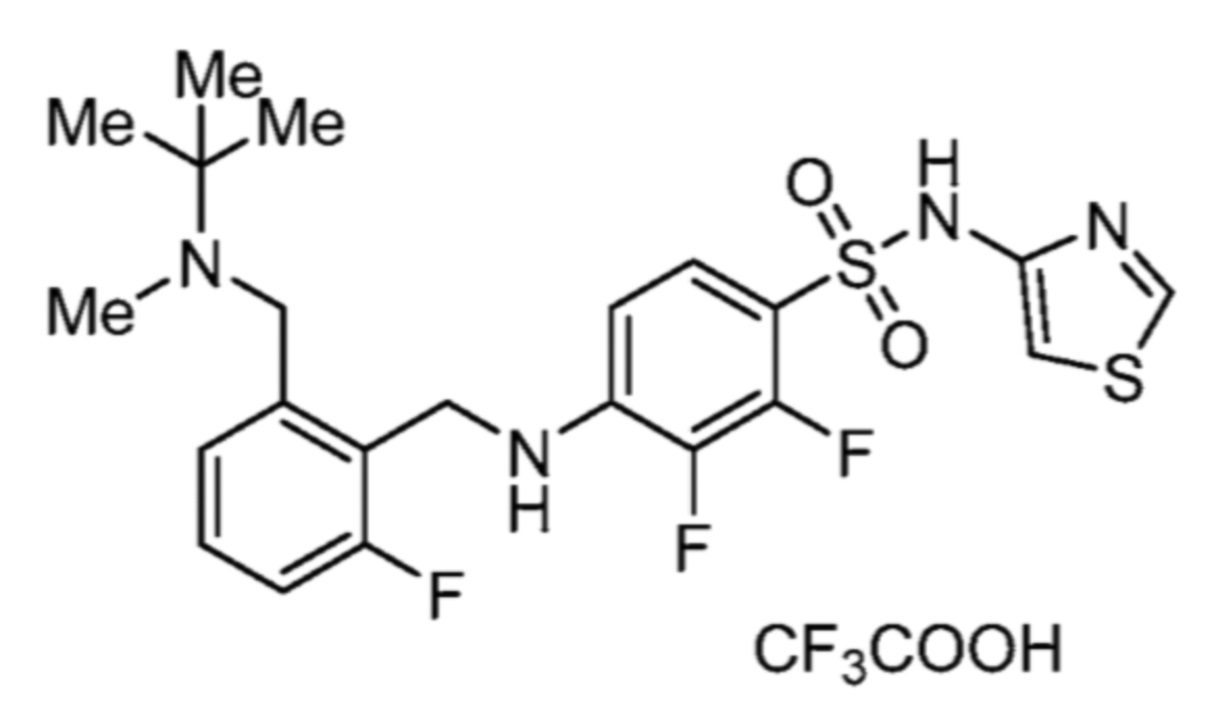
Стадия 1. Получение трет-бутил ((4-((трет-бутоксикарбонил)(2-((трет-бутил(метил)амино)метил)-6-фторбензил)амино)-2,3-дифторфенил)сульфонил)(тиазол-4-ил)карбамата
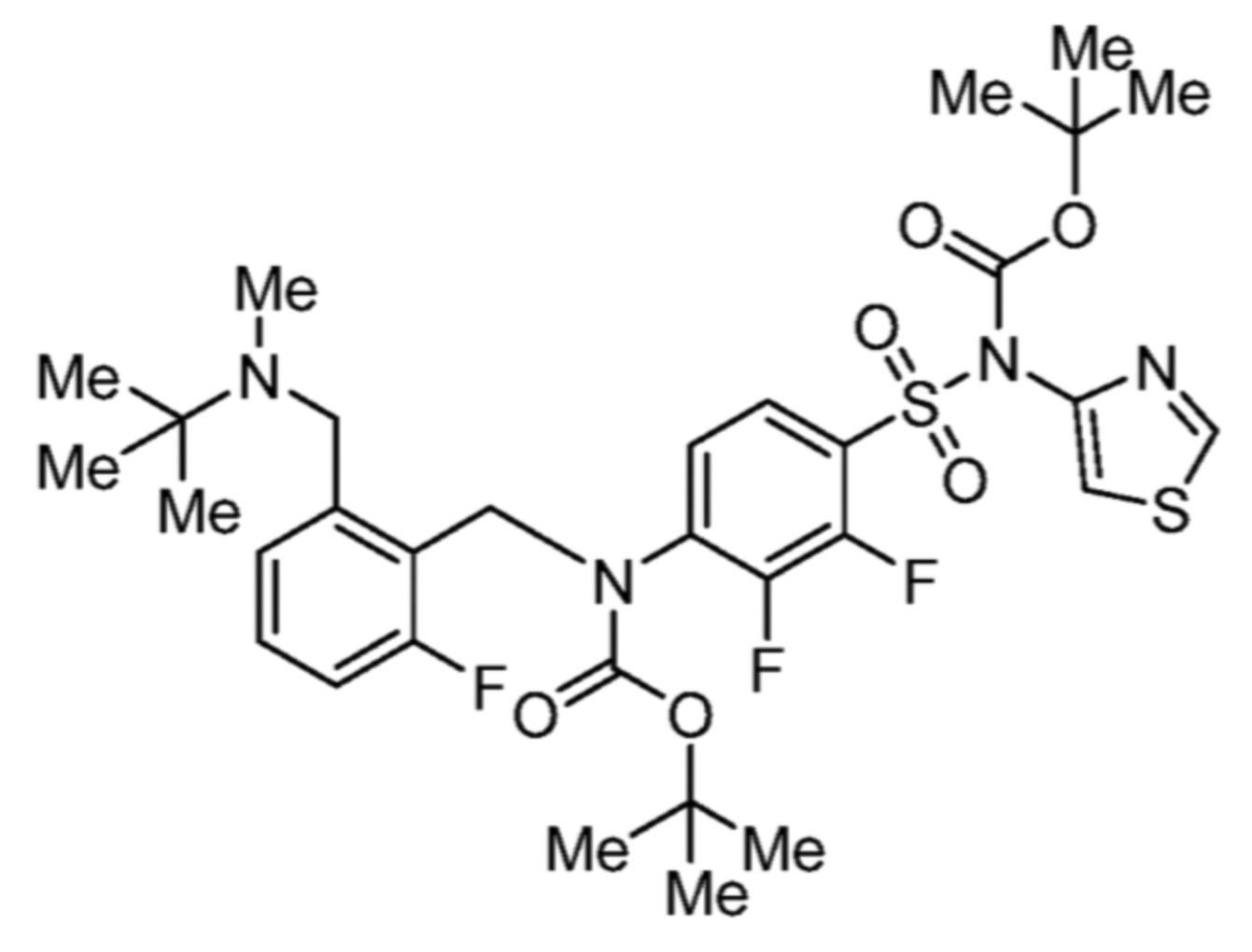
В раствор трет-бутил (2-((трет-бутил(метил)амино)метил)-6-фторбензил)карбамата (0,82 г, 2,54 ммоль) в безводном N,N-диметилформамиде (25 мл) добавляли гидрид натрия (60% дисперсия в минеральном масле, 0,122 г, 3,05 ммоль) при 0°C. Смесь перемешивали при 0°C в течение 30 минут и затем добавляли по каплям в перемешиваемый раствор трет-бутил тиазол-4-ил((2,3,4-трифторфенил)сульфонил)карбамата (1,0 г, 2,54 ммоль) в безводном N,N-диметилформамиде (10 мл) при 0°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 6 ч. Затем смесь охлаждали до 0°C, гасили добавлением насыщенного хлорида аммония (20 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали водой (40 мл), солевым раствором (40 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 40% этилацетата (содержащего 10% изопропанола и 10% триэтиламина) в гептане, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,13 г, выход 7%): MS (ES+) m/z 699,3 (M+1).
Стадия 2. Получение 2,2,2-трифторацетата 4-((2-((трет-бутил(метил)амино)метил)-6-фторбензил)амино)-2,3-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано в примере 14, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять (S)-5-хлор-4-((1-(4-хлорфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид на трет-бутил ((4-((трет-бутоксикарбонил)(2-((трет-бутил(метил)амино)метил)-6-фторбензил)амино)-2,3-дифторфенил)сульфонил)(тиазол-4-ил)карбамат, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,099 г, выход 87%):1H ЯМР (300 МГц, DMSO-d6) δ 11,19 (с, 1H), 8,98 (с, 1H), 8,89 (д, J=2,2 Гц, 1H), 7,59-7,35 (м, 4H), 7,06-6,99 (м, 2H), 6,77-6,69 (м, 1H), 4,73-4,62 (м, 1H), 4,48-4,38 (м, 2H), 4,13-4,01 (м, 1H), 2,58 (д, J=4,7 Гц, 3H), 1,39 (с, 9H); MS (ES+) m/z 499,4 (M+1).
ПРИМЕР 204
Синтез 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(изоксазол-3-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил ((4-((2-(азетидин-1-илметил)-6-фторбензил)(трет-бутоксикарбонил)амино)-2,6-дифторфенил)сульфонил)(изоксазол-3-ил)карбамата

В раствор трет-бутил (2-(азетидин-1-илметил)-6-фторбензил)карбамата (0,78 г, 2,65 ммоль) в безводном N,N-диметилформамиде (27 мл) добавляли гидрид натрия (60% дисперсия в минеральном масле, 0,127 г, 3,18 ммоль) при 0°C. Смесь перемешивали при 0°C в течение 30 минут и затем добавляли по каплям в перемешиваемый раствор трет-бутил изоксазол-3-ил((2,4,6-трифторфенил)сульфонил)карбамата (1,0 г, 2,65 ммоль) в безводном N,N-диметилформамиде (15 мл) при 0°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 18 ч. Затем смесь охлаждали до 0°C, гасили добавлением насыщенного хлорида аммония (20 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали водой (40 мл), солевым раствором (40 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 60% этилацетата (содержащего 10% изопропанола и 10% триэтиламина) в гептане, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,37 г, выход 21%): MS (ES+) m/z 653,3 (M+1).
Стадия 2. Синтез 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(изоксазол-3-ил)бензолсульфонамида

Следуя процедуре как описано в примере 14, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять (S)-5-хлор-4-((1-(4-хлорфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид на трет-бутил ((4-((2-(азетидин-1-илметил)-6-фторбензил)(трет-бутоксикарбонил)амино)-2,6-дифторфенил)сульфонил)(изоксазол-3-ил)карбамат, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,22 г, выход 52%):1H ЯМР (300 МГц, DMSO-d6) δ 11,76 (с, 1H), 10,19 (с, 1H), 8,73 (д, J=1,8 Гц, 1H), 7,57-7,47 (м, 1H), 7,39-7,33 (м, 3H), 6,44-6,36 (м, 2H), 6,32 (д, J=1,8 Гц, 1H), 4,49-4,42 (м, 2H), 4,38-4,31 (м, 2H), 4,15-3,98 (м, 4H), 2,45-2,21 (м, 2H); MS (ES +) m/z 453,4 (M+1).
ПРИМЕР 205
Синтез 2,2,2-трифторацетата (S)-2,6-дифтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил (S)-((2,6-дифтор-4-((1-(2-фторфенил)этил)амино)-фенил)сульфонил)(тиазол-4-ил)карбамата

В раствор трет-бутил тиазол-4-ил((2,4,6-трифторфенил)сульфонил)карбамата (1,20 г, 3,03 ммоль) и гидрохлорида (S)-1-(2-фторфенил)этан-1-амина (0,464 г, 3,34 ммоль) в безводном диметилсульфоксиде (20 мл) добавляли карбонат калия (1,03 мл, 7,50 ммоль) и смесь перемешивали при температуре окружающей среды в течение 12 ч. Затем туда добавляли насыщенный хлорид аммония (20 мл) и смесь экстрагировали этилацетатом (3×30 мл). Объединенные органические фазы промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-50% этилацетата в гексанах, получали указанное в заголовке соединение в виде масла желтого цвета (0,750 г, выход 48%): MS (ES+) m/z 514,1 (M+1).
Стадия 2: получение 2,2,2-трифторацетата (S)-2,6-дифтор-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида

К трет-бутил (S)-((2,6-дифтор-4-((1-(2-фторфенил)этил)амино)фенил)-сульфонил)(тиазол-4-ил)карбамату (0,75 г, 1,46 ммоль) в дихлорметане (6 мл) добавляли трифторуксусную кислоту (3 мл). Реакционную смесь перемешивали в течение 1 ч и затем концентрировали in vacuo Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 60% этилацетата (содержащего 0,1% трифторуксусной кислоты) в гексане, получали указанное в заголовке соединение в виде бесцветной пены (0,42 г, выход 69%):1H-ЯМР (300 МГц, DMSO-d6) δ 11,17 (с, 1H), 8,87 (д, J=2,1 Гц, 1H), 7,64 (д, J=6,7 Гц, 1H), 7,36-7,26 (м, 2H), 7,22-7,13 (м, 2H), 6,87 (д, J=2,1 Гц, 1H), 6,16 (д, J=13,5 Гц, 2H), 4,79 (квинтет, J=7,0 Гц, 1H), 1,44 (д, J=6,8 Гц, 3H), один способный к обмену протон не наблюдали; MS (ES+) m/z 414,1 (M+1).
ПРИМЕР 206
Синтез 2,2,2-трифторацетата (S)-4-((1-(5-хлор-2-фторфенил)этил)амино)-2,6-дифтор-N-(пиримидин-4-ил)бензолсульфонамида

Стадия 1. Получение N-(2,4-диметоксибензил)-2,4,6-трифтор-N-(пиримидин-4-ил)бензолсульфонамида

В раствор N-(2,4-диметоксибензил)пиримидин-4-амина (2,35 г, 9,59 ммоль) в безводном тетрагидрофуране (50 мл) добавляли 1 M раствор бис(триметилсилил)амида лития в тетрагидрофуране (11,5 мл, 11,5 ммоль) при -78°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 1 ч. Реакционную смесь охлаждали до -78°C и туда добавляли раствор 2,4,6-трифторбензолсульфонилхлорида (2,20 г, 9,59 ммоль) в безводном тетрагидрофуране (10 мл). Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 3 ч. Смесь разводили этилацетатом (100 мл), промывали насыщенным хлоридом аммония (2×100 мл), солевым раствором (2×50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 50% этилацетата в гептане, получали указанное в заголовке соединение в виде бесцветного твердого вещества (3,70 г, выход 87%): MS (ES+) m/z 440,1 (M+1).
Стадия 2. Получение (S)-4-((1-(5-хлор-2-фторфенил)этил)амино)-N-(2,4-диметоксибензил)-2,6-дифтор-N-(пиримидин-4-ил)бензолсульфонамида

В раствор N-(2,4-диметоксибензил)-2,4,6-трифтор-N-(пиримидин-4-ил)бензолсульфонамида (0,20 г, 0,45 ммоль) и (S)-1-(5-хлор-2-фторфенил)этан-1-амина (0,085 г, 0,49 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли карбонат калия (0,155 г, 1,125 ммоль). Смесь перемешивали при температуре окружающей среды в течение 16 ч и затем разводили водой (20 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 60% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,120 г, выход 45%): MS (ES+) m/z 593,1 (M+1), 595,1 (M+1).
Стадия 3. Получение 2,2,2-трифторацетата (S)-4-((1-(5-хлор-2-фторфенил)этил)амино)-2,6-дифтор-N-(пиримидин-4-ил)бензолсульфонамида

В раствор (S)-4-((1-(5-хлор-2-фторфенил)этил)амино)-N-(2,4-диметоксибензил)-2,6-дифтор-N-(пиримидин-4-ил)бензолсульфонамида (0,12 г, 0,20 ммоль) в дихлорметане (6 мл) добавляли трифторуксусную кислоту (3 мл). Смесь перемешивали при температуре окружающей среды в течение 1 ч и затем концентрировали in vacuo. Остаток очищали с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом от 10 до 60% ацетонитрила в воде, содержащей 0,1% трифторуксусной кислоты, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,024 г, выход 27%):1H ЯМР (300 МГц, DMSO-d6) δ 8,63 (с, 1H), 8,54-8,33 (м, 1H), 7,62 (с, 1H), 7,47-7,24 (м, 3H), 6,96 (с, 1H), 6,23 (т, J=9,9 Гц, 2H), 4,79 (с, 1H), 1,44-1,42 (м, 3H), COOH и NH не наблюдали; MS (ES+) m/z 443,1 (M+1), 445,1 (M+1).
ПРИМЕР 207
Синтез 2,2,2-трифторацетата (S)-4-((1-(5-хлор-2-фторфенил)этил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил (S)-((4-((1-(5-хлор-2-фторфенил)этил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

В раствор трет-бутил тиазол-4-ил((2,4,6-трифторфенил)сульфонил)карбамата (0,20 г, 0,50 ммоль) и (S)-1-(5-хлор-2-фторфенил)этан-1-амина (0,065 г, 0,55 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли карбонат калия (0,172 г, 1,25 ммоль). Смесь перемешивали при температуре окружающей среды в течение 16 ч и затем разводили водой (20 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 60% этилацетата в гексанах, и получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,142 г, выход 50%): MS (ES+) m/z 548,2 (M+1), 550,2 (M+1).
Стадия 2. Получение 2,2,2-трифторацетата (S)-4-((1-(5-хлор-2-фторфенил)этил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил (S)-((4-((1-(5-хлор-2-фторфенил)этил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,142 г, 0,26 ммоль) в дихлорметане (6,0 мл) добавляли трифторуксусную кислоту (3,0 мл). Смесь перемешивали при температуре окружающей среды в течение 1 ч и затем концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 60% этилацетата в гексанах, и получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,081 г, выход 69%):1H ЯМР (300 МГц, DMSO-d6): δ 11,18 (с, 1H), 8,87 (д, J=2,2 Гц, 1H), 7,60 (д, J=7,4 Гц, 1H), 7,39-7,34 (м, 2H), 7,30-7,24 (м, 1H), 6,88 (д, J=2,2 Гц, 1H), 6,19 (д, J=13,0 Гц, 2H), 4,79 (квинтет, J=6,9 Гц, 1H), 1,43 (д, J=6,7 Гц, 3H). Примечание: NH не наблюдали; MS (ES+) m/z 448,1 (M+1), 450,1 (M+1).
ПРИМЕР 208
Синтез 2,2,2-трифторацетата (S)-4-((1-(2,5-дифторфенил)этил)амино)-2,6-дифтор-N-(пиримидин-4-ил)бензолсульфонамида

Стадия 1. Получение (S)-4-((1-(2,5-дифторфенил)этил)амино)-N-(2,4-диметоксибензил)-2,6-дифтор-N-(пиримидин-4-ил)бензолсульфонамида

В раствор N-(2,4-диметоксибензил)-2,4,6-трифтор-N-(пиримидин-4-ил)бензолсульфонамида (0,16 г, 0,36 ммоль) и (S)-1-(2,5-дифторфенил)этан-1-амина (0,057 г, 0,36 ммоль) в безводном диметилсульфоксиде (3,0 мл) добавляли карбонат калия (0,248 г, 1,80 ммоль). Смесь перемешивали при температуре окружающей среды в течение 16 ч и затем разводили водой (10 мл) и экстрагировали этилацетатом (3×20 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 60% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,080 г, выход 38%): MS (ES+) m/z 577,2 (M+1).
Стадия 2. Получение 2,2,2-трифторацетата (S)-4-((1-(2,5-дифторфенил)этил)амино)-2,6-дифтор-N-(пиримидин-4-ил)бензолсульфонамида

В раствор (S)-4-((1-(2,5-дифторфенил)этил)амино)-N-(2,4-диметоксибензил)-2,6-дифтор-N-(пиримидин-4-ил)бензолсульфонамида (0,080 г, 0,14 ммоль) в дихлорметане (5,0 мл) добавляли трифторуксусную кислоту (2,0 мл). Смесь перемешивали при температуре окружающей среды в течение 1 ч и затем концентрировали in vacuo. Остаток очищали с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом от 10 до 50% ацетонитрила в воде, содержащей 0,1% трифторуксусной кислоты, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,022 г, выход 36%):1H ЯМР (300 МГц, DMSO-d6) δ 8,60 (с, 1H), 8,37 (с, 1H), 7,62 (д, J=5,9 Гц, 1H), 7,28-7,14 (м, 3H), 6,95 (с, 1H), 6,20 (д, J=12,7 Гц, 2H), 4,81-4,76 (м, 1H), 1,45-1,43 (м, 3H), COOH и NH не наблюдали; MS (ES+) m/z 427,2 (M+1).
ПРИМЕР 209
Синтез 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-метоксибензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида
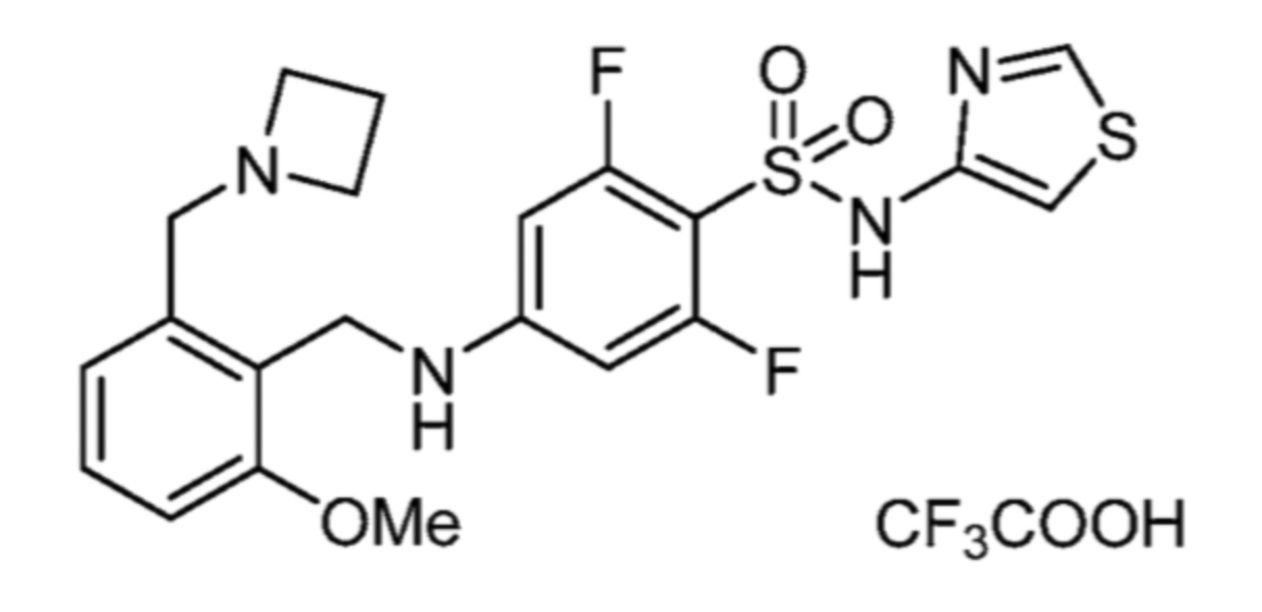
Стадия 1. Получение 2-бром-3-метоксибензальдегида

В смесь 2-бром-3-гидроксибензальдегида (1,30 г, 6,50 ммоль) и карбоната калия (2,70 г, 19,5 ммоль) в N,N-диметилформамиде (60 мл) добавляли иодометан (0,80 г, 12,9 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч и затем разводили этилацетатом (60 мл). Смесь промывали насыщенным хлоридом аммония (2×50 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 30% этилацетата в гексанах, чтобы получать указанное в заголовке соединение в виде масла желтого цвета (1,50 г, количественный выход): MS (ES+) m/z 215,1 (M+1).
Стадия 2. Получение 1-(2-бром-3-метоксибензил)азетидина

В смесь 2-бром-3-метоксибензальдегида (1,50 г, 6,50 ммоль) и азетидина (0,44 г, 7,80 ммоль) в безводном 1,2-дихлорэтане (10 мл) добавляли триацетоксиборгидрид натрия (2,70 г, 13,0 ммоль) и получаемую смесь перемешивали в течение 18 ч. Смесь разводили этилацетатом (30 мл), промывали насыщенным хлоридом аммония (2×30 мл) и концентрировали органическую фазу in vacuo. Остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 10% метанола в дихлорметане, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (1,35 г, выход 81%): MS (ES+) m/z 256,2 (M+1).
Стадия 3. Получение 2-(азетидин-1-илметил)-6-метоксибензальдегида

В раствор 1-(2-бром-3-метоксибензил)азетидина (0,27 г, 1,64 ммоль) в безводном тетрагидрофуране (10 мл) добавляли 1,6 M раствор N-бутиллития в тетрагидрофуране (1,30 мл, 1,97 ммоль) по каплям при -78°C. Реакционную смесь перемешивали -78°C в течение 20 минут и туда добавляли N,N-диметилформамид (0,663 мл, 8,20 ммоль). Реакционную смесь оставляли нагреваться до температуры окружающей среды. После перемешивания в течение 30 минут при температуре окружающей среды, реакционную смесь разводили дихлорметаном (50 мл). Смесь промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали бесцветное масло (0,22 г, выход 65%): MS (ES+) m/z 206,2 (M+1).
Стадия 4. Получение 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-метоксибензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

В смесь трет-бутил ((4-амино-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,38 г, 0,975 ммоль) и 2-(азетидин-1-илметил)-6-метоксибензальдегида (0,20 г, 0,975 ммоль) в трифторуксусной кислоте (2,0 мл) добавляли триацетоксиборгидрид натрия (0,41 г, 1,95 ммоль) при 0°C. Получаемую смесь перемешивали при температуре окружающей среды в течение 15 минут. Реакционную смесь разводили этилацетатом (30 мл), промывали насыщенным хлоридом аммония (2×30 мл) и концентрировали органическую фазу in vacuo. Остаток очищали с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом от 10 до 50% ацетонитрила в воде, содержащей 0,1% трифторуксусной кислоты, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,014 г, выход 3%):1H ЯМР (300 МГц, DMSO-d6) δ 8,74 (д, J=2,2 Гц, 1H), 8,43 (с, 1H), 7,42 (т, J=8,0 Гц, 1H), 7,14 (д, J=8,4 Гц, 1H), 7,03 (д, J=7,5 Гц, 1H), 6,95 (д, J=2,2 Гц, 1H), 6,29 (д, J=12,3 Гц, 2H), 4,39 (с, 2H), 4,35 (с, 2H), 4,09 (т, J=8,1 Гц, 4H), 3,86 (с, 3H), 2,45 (квинтет, J=8,1 Гц, 2H), COOH и NH не наблюдали; MS (ES+) m/z 481,2 (M+1).
ПРИМЕР 210
Синтез 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-этилбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение (S)-4-((1-(2,5-дифторфенил)этил)амино)-N-(2,4-диметоксибензил)-2,6-дифтор-N-(пиримидин-4-ил)бензолсульфонамида

В раствор трет-бутил ((3-бром-2,4,6-трифторфенил)сульфонил)(тиазол-4-ил)карбамата (11,6 г, 24,7 ммоль) и (2-(азетидин-1-илметил)-6-хлорфенил)метанамина (5,20 г, 24,7 ммоль) в безводном диметилсульфоксиде (250 мл) добавляли карбонат калия (6,80 г, 49,4 ммоль). Смесь перемешивали при температуре окружающей среды в течение 16 ч и затем разводили водой (200 мл) и экстрагировали этилацетатом (3×300 мл). Объединенные органические слои промывали солевым раствором (200 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и растиранием остатка в простом диэтиловом эфире (50 мл) получали указанное в заголовке соединение в виде бесцветного твердого вещества (5,0 г, выход 36%): MS (ES+) m/z 563,0 (M+1), 565,0 (M+1).
Стадия 2. Получение трет-бутил ((4-((2-(азетидин-1-илметил)-6-хлорбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

В смесь (S)-4-((1-(2,5-дифторфенил)этил)амино)-N-(2,4-диметоксибензил)-2,6-дифтор-N-(пиримидин-4-ил)бензолсульфонамида (3,0 г, 5,33 ммоль) в этаноле (30 мл) добавляли триэтиламин (2,90 мл, 21,3 ммоль) и 15% палладий на углероде (500 мг). Суспензию дегазировали в вакууме и продували водородом несколько раз. Смесь перемешивали в атмосфере водорода под давлением (50 фунтов/дюйм2) при 80°C в течение 16 ч. После охлаждения до температуры окружающей среды, реакционную смесь фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде бесцветной пены (3,1 г, количественный выход): MS (ES+) m/z 585,2 (M+1).
Стадия 3. Получение 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-хлорбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида
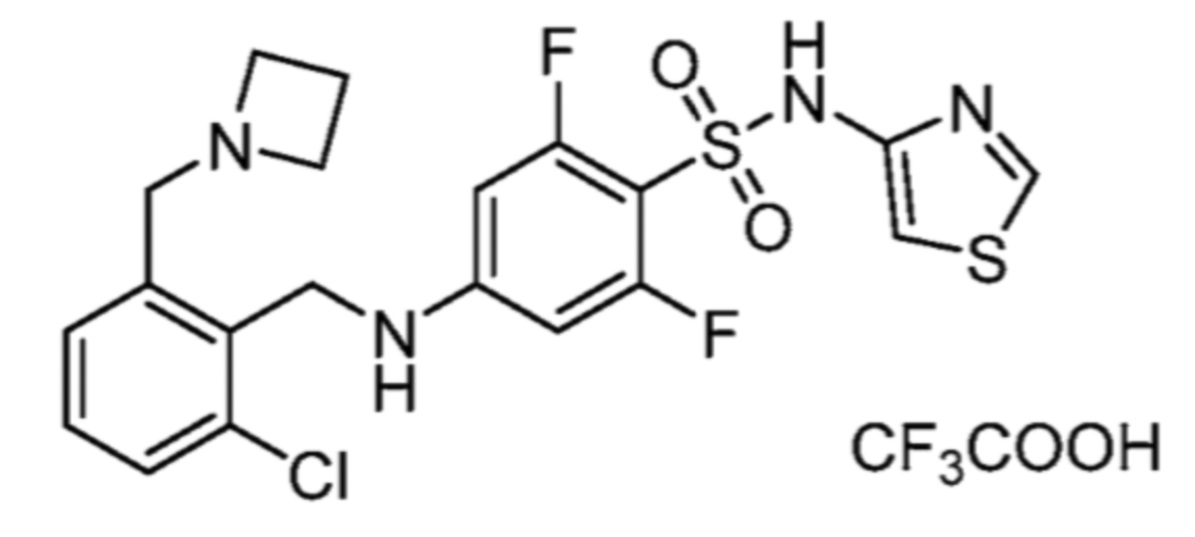
В раствор трет-бутил ((4-((2-(азетидин-1-илметил)-6-хлорбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (3,10 г, 5,29 ммоль) в дихлорметане (10 мл) добавляли трифторуксусную кислоту (5 мл). Смесь перемешивали при температуре окружающей среды в течение 1 ч и затем концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 15% метанола в дихлорметане, чтобы получать указанное в заголовке соединение в виде бесцветной пены (1,0 г, выход 39%): MS (ES+) m/z 485,2 (M+1), 487,2 (M+1).
Стадия 4. Получение 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-этилбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида
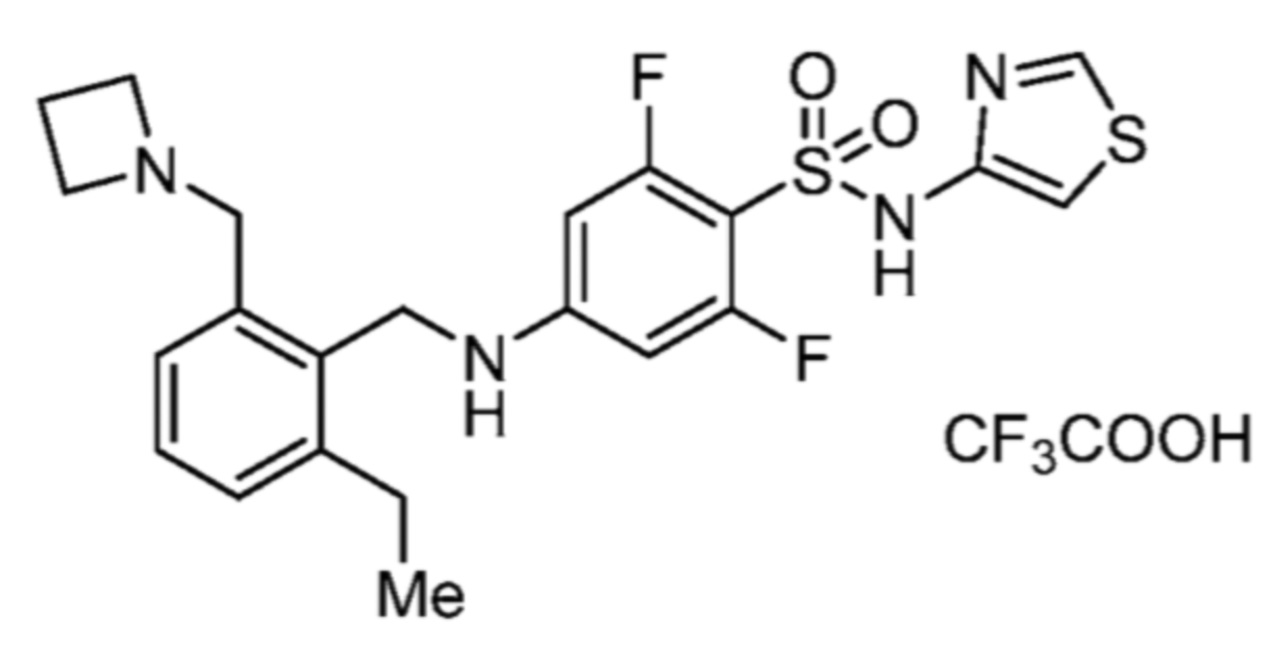
В смесь 4-((2-(азетидин-1-илметил)-6-хлорбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида (0,053 г, 0,11 ммоль) в 1,4-диоксане (1,5 мл) добавляли этилбороновую кислоту (0,049 г, 0,66 ммоль), метансульфонат (2-дициклогексилфосфино-2′,6′-диметоксибифенил) [2-(2′-амино-1,1′-бифенил)]палладия(II) (3,7 мг, 0,003 ммоль), 2-дициклогексилфосфино-2′,6′-диметоксибифенил (2,2 мг, 0,003 ммоль) и фосфат калия (0,088 г, 0,24 ммоль). Получаемую смесь дегазировали пропусканием потока аргона через нее в течение 15 минут и затем нагревали до 160°C в течение 45 минут микроволнами. Реакционную смесь оставляли остывать до температуры окружающей среды и фильтровали через подушку из целита. Фильтровальную подушку промывали этилацетатом (30 мл) и объединенный фильтрат концентрировали in vacuo. Очисткой остатка с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом от 10 до 50% ацетонитрила в воде, содержащей 0,1% трифторуксусной кислоты, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,021 г, выход 40%):1H ЯМР (300 МГц, DMSO-d6) δ 11,24 (с, 1H), 10,19 (с, 1H), 8,92 (д, J=2,2 Гц, 1H), 7,42-7,31 (м, 3H), 7,04 (с, 1H), 6,92 (д, J=2,2 Гц, 1H), 6,39 (д, J=12,6 Гц, 2H), 4,38 (д, J=5,5 Гц, 2H), 4,24 (д, J=3,4 Гц, 2H), 4,14-3,97 (м, 4H), 2,68-2,60 (м, 2H), 2,46-2,21 (м, 2H), 1,13 (т, J=7,5 Гц, 3H); MS (ES+) m/z 479,1 (M+1).
ПРИМЕР 211
Синтез 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-циклопропилбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано в примере 210, стадии 4 и выполняя вариации при необходимости, чтобы заменять этилбороновую кислоту на циклопропилбороновую кислоту, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,015 г, выход 5%):1H ЯМР (300 МГц, DMSO-d6) δ 11,23 (с, 1H), 10,34 (с, 1H), 8,94-8,92 (м, 1H), 7,38-7,31 (м, 2H), 7,11 (тд, J=5,8, 2,3 Гц, 2H), 6,92 (д, J=7,7 Гц, 1H), 6,39 (д, J=12,6 Гц, 2H), 4,43-4,40 (м, 4H), 4,14-3,98 (м, 4H), 2,42-2,18 (м, 2H), 2,00-1,91 (м, 1H), 0,91-0,85 (м, 2H), 0,67-0,62 (м, 2H); MS (ES+) m/z 491,2 (M+1).
ПРИМЕР 212
Синтез 2,2,2-трифторацетата 4-((2-((трет-бутил(метил)амино)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 2-((трет-бутил(метил)амино)метил)-6-фторбензонитрила

В раствор 2-(бромметил)-6-фторбензонитрила (0,737 г, 3,40 ммоль) в N,N-диметилформамиде (5 мл) добавляли N,2-диметилпропан-2-амин (0,30 г, 3,40 ммоль) и N,N-диизопропилэтиламин (1,10 мл, 6,80 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч и затем разводили этилацетатом (20 мл). Смесь промывали насыщенным хлоридом аммония (2×20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 10% метанола в дихлорметане, чтобы получать указанное в заголовке соединение в виде масла желтого цвета (0,45 г, выход 60%): MS (ES+) m/z 221,3 (M+1).
Стадия 2. Получение N-(2-(аминометил)-3-фторбензил)-N,2-диметилпропан-2-амина
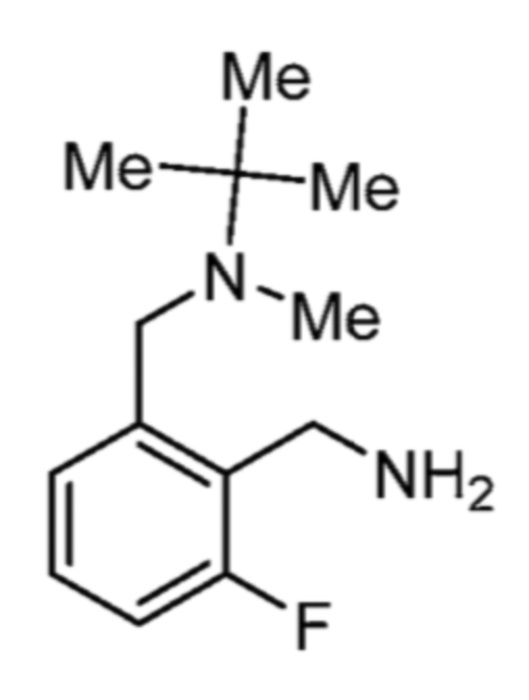
В раствор 2-((трет-бутил(метил)амино)метил)-6-фторбензонитрила (0,450 г, 2,04 ммоль) в метаноле (20,0 мл) и гидроксиде аммония (5,00 мл) добавляли ренеевский никель (0,175 г, 2,04 ммоль). Суспензию дегазировали и продували водородом три раза. Смесь перемешивали в атмосфере водорода (50 фунтов/дюйм2) при температуре окружающей среды в течение 12 ч. Реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении для того, чтобы получать указанное в заголовке соединение в виде масла желтого цвета (0,300 г, выход 65%): MS (ES+) m/z 225,3 (M+1).
Стадия 3. Получение трет-бутил ((3-бром-4-((2-((трет-бутил(метил)амино)метил)-6-фторбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

В раствор трет-бутил ((3-бром-2,4,6-трифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,30 г, 1,33 ммоль) и N-(2-(аминометил)-3-фторбензил)-N,2-диметилпропан-2-амина (0,632 г, 1,33 ммоль) в безводном диметилсульфоксиде (6 мл) добавляли карбонат калия (0,369 г, 2,68 ммоль). Смесь перемешивали при температуре окружающей среды в течение 16 ч и затем разводили водой (20 мл) и экстрагировали этилацетатом (2×30 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 10% метанола в дихлорметане, получали указанное в заголовке соединение в виде бесцветной пены (0,35 г, выход 38%): MS (ES+) m/z 677,4 (M+1), 679,4 (M+1).
Стадия 4. Получение трет-бутил ((4-((2-((трет-бутил(метил)амино)метил)-6-фторбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата
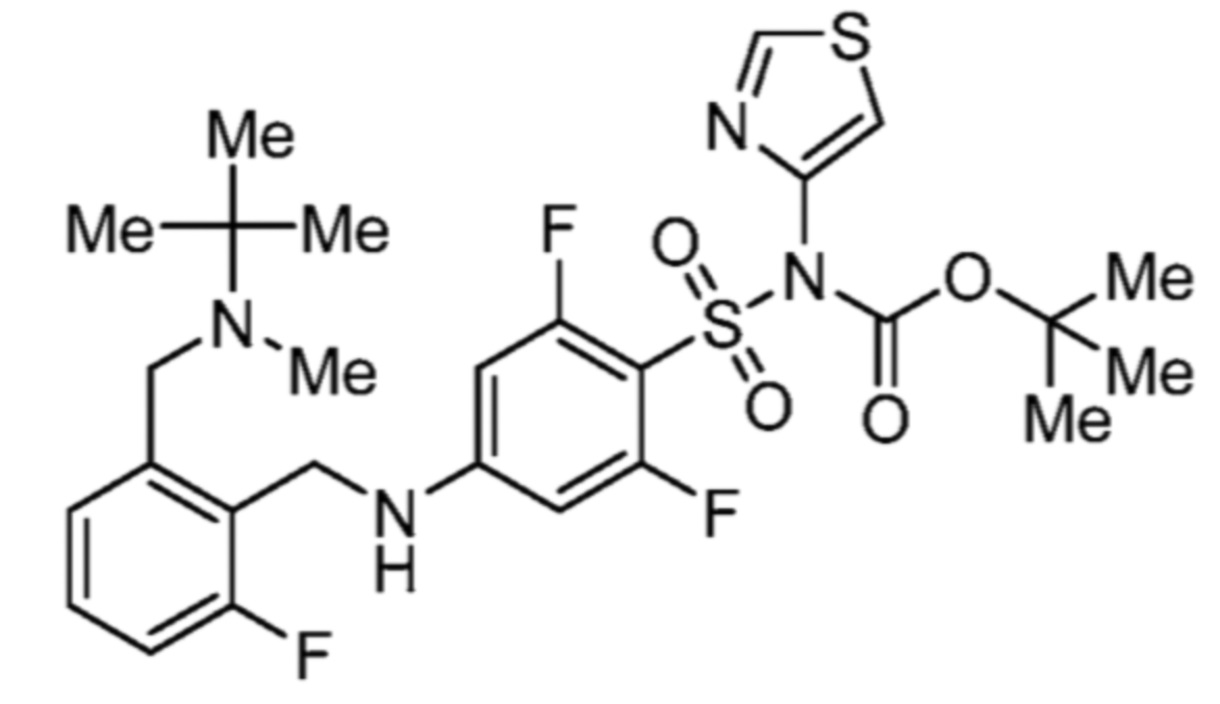
В смесь трет-бутил ((3-бром-4-((2-((трет-бутил(метил)амино)метил)-6-фторбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,35 г, 0,51 ммоль) в этаноле (5 мл) добавляли триэтиламин (0,288 мл, 2,07 ммоль) и 15% палладий на углероде (51 мг). Суспензию дегазировали в вакууме и продували водородом несколько раз. Реакционную смесь перемешивали в атмосфере водорода (50 фунтов/дюйм2) при 70°C в течение 16 ч. Реакционную смесь фильтровали и фильтрат концентрировали in vacuo для того, чтобы получать указанное в заголовке соединение в виде бесцветной пены (0,21 г, выход 68%): MS (ES+) m/z 599,2 (M+1).
Стадия 5. Получение 2,2,2-трифторацетата 4-((2-((трет-бутил(метил)амино)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил ((4-((2-((трет-бутил(метил)амино)метил)-6-фторбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,21 г, 0,35 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (2 мл). Смесь перемешивали при температуре окружающей среды в течение 1 ч и затем концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 12% метанола в дихлорметане, чтобы получать указанное в заголовке соединение в виде бесцветной пены (0,090 г, выход 51%):1H ЯМР (300 МГц, DMSO-d6) δ 11,24 (с, 1H), 9,52 (с, 1H), 8,91 (с, 1H), 7,57-7,34 (м, 4H), 6,92 (с, 1H), 6,39 (д, J=12,7 Гц, 2H), 4,65-4,57 (м, 1H), 4,35 (с, 2H), 4,08-3,97 (м, 1H), 2,54 (с, 3H), 1,39 (с, 9H); MS (ES+) m/z 499,2 (M+1).
ПРИМЕР 213
Синтез 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-3,6-дифторбензил)амино)-2,3-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил тиазол-4-ил((2,3,4-трифторфенил)сульфонил)карбамата

В раствор трет-бутил тиазол-4-илкарбамата (4,90 г, 34,7 ммоль) в безводном тетрагидрофуране (150 мл) добавляли 1 M раствор бис(триметилсилил)амида лития в тетрагидрофуране (41,6 мл, 41,6 ммоль) при -78°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 1 ч. Реакционную смесь охлаждали до -78°C и туда добавляли раствор 2,3,4-трифторбензолсульфонилхлорида (8,0 г, 34,7 ммоль) в безводном тетрагидрофуране (20 мл). Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 3 ч. Смесь разводили этилацетатом (200 мл), промывали насыщенным хлоридом аммония (2×200 мл), солевым раствором (2×100 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 10 до 50% этилацетата в гептане, получали указанное в заголовке соединение в виде бесцветного твердого вещества (6,70 г, выход 50%): MS (ES+) m/z 395,1 (M+1).
Стадия 2. Получение трет-бутил ((4-((2-(азетидин-1-илметил)-3,6-дифторбензил)амино)-2,3-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

В раствор трет-бутил тиазол-4-ил((2,3,4-трифторфенил)сульфонил)карбамата (0,256 г, 0,64 ммоль) и (2-(азетидин-1-илметил)-3,6-дифторфенил)метанамина (0,165 г, 0,77 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли карбонат калия (0,76 г, 1,28 ммоль). Смесь перемешивали при температуре окружающей среды в течение 16 ч и затем разводили водой (20 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 8% метанола в дихлорметане, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,095 г, выход 25%): MS (ES+) m/z 587,2 (M+1).
Стадия 3. Получение 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-3,6-дифторбензил)амино)-2,3-дифтор-N-(тиазол-4-ил)бензолсульфонамида
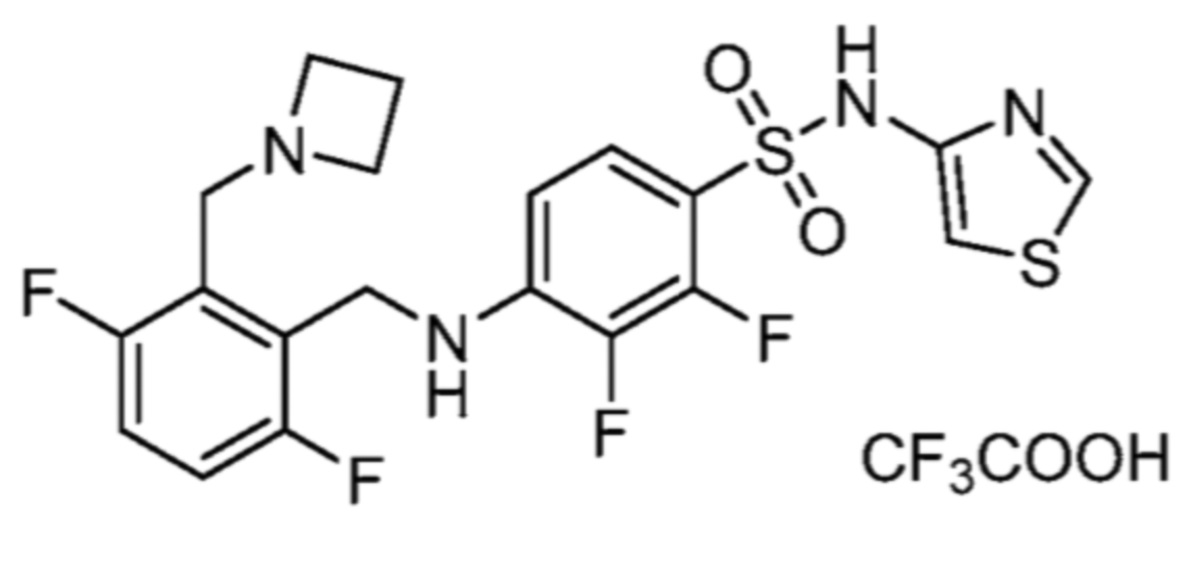
В раствор трет-бутил ((4-((2-(азетидин-1-илметил)-3,6-дифторбензил)амино)-2,3-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,095 г, 0,16 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (2 мл). Смесь перемешивали при температуре окружающей среды в течение 1 ч и затем концентрировали in vacuo. Остаток очищали с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом от 10 до 50% ацетонитрила в воде, содержащей 0,1% трифторуксусной кислоты, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,060 г, выход 77%):1H ЯМР (300 МГц, DMSO-d6) δ 11,20 (с, 1H), 10,33 (с, 1H), 8,89 (д, J=2,2 Гц, 1H), 7,48-7,40 (м, 3H), 7,05 (с, 1H), 6,99 (д, J=2,1 Гц, 1H), 6,73-6,68 (м, 1H), 4,53 (с, 2H), 4,47-4,46 (м, 2H), 4,21-4,12 (м, 2H), 4,09-3,99 (м, 2H), 2,37 (дд, J=18,6, 10,3 Гц, 1H), 2,30-2,17 (м, 1H); MS (ES+) m/z 487,2 (M+1).
ПРИМЕР 214
Синтез 2,2,2-трифторацетата 2,6-дифтор-4-((2-((3-(2-метоксипропан-2-ил)азетидин-1-ил)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил 3-(2-гидроксипропан-2-ил)азетидин-1-карбоксилата
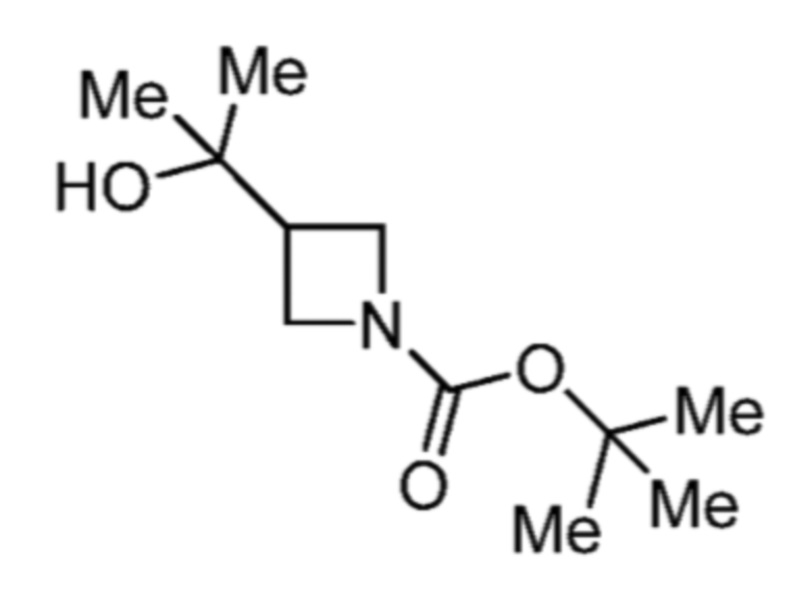
В раствор 1-(трет-бутил) 3-метил азетидин-1,3-дикарбоксилата (3,00 г, 13,9 ммоль) в безводном тетрагидрофуране (30 мл) при 0°C добавляли 3 M раствор бромида метилмагния в простом диэтиловом эфире (11,1 мл, 33,4 ммоль). Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 16 ч. Затем реакционную смесь разводили насыщенным хлоридом аммония (20 мл) и экстрагировали этилацетатом (2×30 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде пены желтого цвета (2,85 г, выход 95%):1H ЯМР (300 МГц, CDCl3) δ 3,89 (дт, J=14,4, 7,3 Гц, 4H), 2,57 (дд, J=10,4, 4,3 Гц, 1H), 1,44-1,43 (м, 9H), 1,19 (с, 6H), OH не наблюдали.
Стадия 2. Получение трет-бутил 3-(2-метоксипропан-2-ил)азетидин-1-карбоксилата

В раствор трет-бутил 3-(2-гидроксипропан-2-ил)азетидин-1-карбоксилата (0,67 г, 3,11 ммоль) в безводном тетрагидрофуране (6 мл) при -78°C добавляли 1,6 M раствор н-бутиллития в гексане (2,5 мл, 4,0 ммоль). Смесь перемешивали при -78°C в течение 30 минут и затем туда добавляли метилйодид (0,50 мл, 8,10 ммоль). Смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 16 ч. Затем реакционную смесь разводили насыщенным хлоридом аммония (10 мл) и экстрагировали простым диэтиловым эфиром (2×20 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде пены желтого цвета (1,06 г, выход 99%):1H-ЯМР (300 МГц, CDCl3) δ 3,91-3,83 (м, 4H), 3,21 (с, 3H), 2,63-2,56 (м, 1H), 1,45 (с, 9H), 1,19 (с, 3H), 1,12 (с, 3H).
Стадия 3. Получение 2-((3-(2-метоксипропан-2-ил)азетидин-1-ил)метил)бензонитрила

В раствор трет-бутил 3-(2-метоксипропан-2-ил)азетидин-1-карбоксилата (1,06 г, 4,65 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (5 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Смесь концентрировали in vacuo и остаток разводили в дихлорметане (5 мл). В этот раствор добавляли 2-формилбензонитрил (0,61 г, 4,65 ммоль) и триацетоксиборгидрид натрия (2,76 г, 13,6 ммоль) одной частью. Смесь перемешивали при температуре окружающей среды в течение 1 ч. Смесь разводили насыщенным бикарбонатом натрия (30 мл) и экстрагировали дихлорметаном (3×20 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде пены желтого цвета (1,00 г, выход 88%): MS (ES+) m/z 245,2 (M+1).
Стадия 4. Получение (2-((3-(2-метоксипропан-2-ил)азетидин-1-ил)метил)фенил)метанамина

В раствор 2-((3-(2-метоксипропан-2-ил)азетидин-1-ил)метил)бензонитрила (0,72 г, 3,00 ммоль) в безводном тетрагидрофуране (10 мл) добавляли 1,0 M раствор гидрида лития алюминия в тетрагидрофуране (3,00 мл, 3,00 ммоль) при 0°C. Смесь перемешивали при 0°C в течение 15 минут и затем при температуре окружающей среды в течение 1 ч. Затем смесь гасили добавлением декагидрата сульфата натрия (3,00 г) и затем смесь перемешивали в течение 16 ч. Смесь фильтровали через подушку диатомовой земли и фильтровальный осадок споласкивали дихлорметаном (30 мл). Концентрированием объединенного фильтрата при пониженном давлении получали указанное в заголовке соединение в виде бесцветного масла (0,59 г, 79%): MS (ES+) m/z 249,1 (M+1).
Стадия 5. Получение трет-бутил ((3-бром-2,6-дифтор-4-((2-((3-(2-метоксипропан-2-ил)азетидин-1-ил)метил)бензил)амино)фенил)сульфонил)(тиазол-4-ил)карбамата

В раствор трет-бутил ((3-бром-2,4,6-трифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,247 г, 0,52 ммоль) и (2-((3-(2-метоксипропан-2-ил)азетидин-1-ил)метил)фенил)метанамина (0,13 г, 0,52 ммоль) в безводном диметилсульфоксиде (6 мл) добавляли карбонат калия (0,072 г, 1,08 ммоль). Смесь перемешивали при температуре окружающей среды в течение 16 ч и затем разводили водой (20 мл) и экстрагировали этилацетатом (2×30 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над сульфатом натрия, фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 10% метанола в дихлорметане, получали указанное в заголовке соединение в виде бесцветной пены (0,040 г, выход 10%): MS (ES+) m/z 701,4 (M+1), 703,4 (M+1).
Стадия 6. Получение трет-бутил ((2,6-дифтор-4-((2-((3-(2-метоксипропан-2-ил)азетидин-1-ил)метил)бензил)амино)фенил)сульфонил)(тиазол-4-ил)карбамата

В смесь трет-бутил ((3-бром-2,6-дифтор-4-((2-((3-(2-метоксипропан-2-ил)азетидин-1-ил)метил)бензил)амино)фенил)сульфонил)(тиазол-4-ил)карбамата (0,040 г, 0,05 ммоль) в этаноле (5 мл) добавляли триэтиламин (0,028 мл, 0,20 ммоль) и 15% палладий на углероде (5 мг). Суспензию дегазировали в вакууме и продували водородом несколько раз. Смесь перемешивали в атмосфере водорода (50 фунтов/дюйм2) при 70°C в течение 16 ч. Реакционную смесь фильтровали и фильтрат концентрировали in vacuo для того, чтобы получать указанное в заголовке соединение в виде бесцветной пены (0,036 г, количественный выход): MS (ES+) m/z 623,3 (M+1).
Стадия 7. Получение 2,2,2-трифторацетата 2,6-дифтор-4-((2-((3-(2-метоксипропан-2-ил)азетидин-1-ил)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил ((2,6-дифтор-4-((2-((3-(2-метоксипропан-2-ил)азетидин-1-ил)метил)бензил)амино)фенил)сульфонил)(тиазол-4-ил)карбамата (0,036 г, 0,057 ммоль) в дихлорметане (4 мл) добавляли трифторуксусную кислоту (2 мл). Смесь перемешивали при температуре окружающей среды в течение 1 ч и затем концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 10% метанола в дихлорметане, чтобы получать указанное в заголовке соединение в виде бесцветной пены (0,023 г, выход 77%):1H ЯМР (300 МГц, DMSO-d6) δ 11,22 (с, 1H), 10,37 (с, 1H), 8,90 (д, J=2,2 Гц, 1H), 7,52-7,36 (м, 5H), 6,91 (д, J=2,2 Гц, 1H), 6,35 (д, J=12,6 Гц, 2H), 4,50-4,32 (м, 4H), 4,20-4,04 (м, 2H), 3,96-3,84 (м, 2H), 3,18-3,10 (м, 3H), 2,89-2,77 (м, 1H), 1,04-0,98 (м, 6H); MS (ES+) m/z 523,3 (M+1).
ПРИМЕР 215
Синтез 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-3,6-дифторбензил)амино)-2,3,6-трифтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил ((2,3,4,6-тетрафторфенил)сульфонил)(тиазол-4-ил)карбамата

В раствор трет-бутил тиазол-4-илкарбамата (0,80 г, 4,03 ммоль) в безводном тетрагидрофуране (20 мл) добавляли 1 M раствор бис(триметилсилил)амида лития в тетрагидрофуране (4,83 мл, 4,83 ммоль) при -78°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 1 ч. Реакционную смесь охлаждали до -78°C и туда добавляли раствор 2,3,4,6-тетрафторбензолсульфонилхлорида (1,0 г, 4,03 ммоль) в безводном тетрагидрофуране (10 мл). Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 3 ч. Смесь разводили этилацетатом (50 мл), промывали насыщенным хлоридом аммония (2×50 мл), солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и растиранием остатка в метаноле (20 мл) получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,80 г, выход 63%): MS (ES+) m/z 313,2 (M - 99).
Стадия 2. Получение трет-бутил ((4-((2-(азетидин-1-илметил)-3,6-дифторбензил)амино)-2,3,6-трифторфенил)сульфонил)(тиазол-4-ил)карбамата

В раствор трет-бутил ((2,3,4,6-тетрафторфенил)сульфонил)(тиазол-4-ил)карбамата (0,20 г, 0,48 ммоль) и (2-(азетидин-1-илметил)-3,6-дифторфенил)метанамина (0,103 г, 0,48 ммоль) в безводном диметилсульфоксиде (3 мл) добавляли карбонат калия (0,132 г, 0,96 ммоль). Смесь перемешивали при температуре окружающей среды в течение 16 ч и затем разводили водой (20 мл) и экстрагировали этилацетатом (2×15 мл). Объединенные органические слои промывали солевым раствором (15 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 10% метанола в дихлорметане, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,075 г, выход 26%): MS (ES+) m/z 605,0 (M+1).
Стадия 3. Получение 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-3,6-дифторбензил)амино)-2,3,6-трифтор-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил ((4-((2-(азетидин-1-илметил)-3,6-дифторбензил)амино)-2,3,6-трифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,075 г, 0,12 ммоль) в дихлорметане (4 мл) добавляли трифторуксусную кислоту (2 мл). Смесь перемешивали при температуре окружающей среды в течение 1 ч и затем концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 10% метанола в дихлорметане, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,052 г, выход 85%):1H ЯМР (300 МГц, DMSO-d6) δ 11,40 (с, 1H), 8,92 (д, J=2,1 Гц, 1H), 7,45-7,39 (м, 3H), 7,00 (д, J=2,1 Гц, 1H), 6,67 (дд, J=13,5, 6,5 Гц, 1H), 4,54-4,44 (м, 4H), 4,13-4,00 (м, 4H), 2,36-2,21 (м, 2H), один способный к обмену протон не наблюдали; MS (ES+) m/z 505,0.
ПРИМЕР 216
Синтез 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-3,6-дифторбензил)амино)-2,6-дифтор-N-(5-метилизоксазол-3-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил (5-метилизоксазол-3-ил)((2,4,6-трифторфенил)сульфонил)-карбамата

В раствор трет-бутил (5-метилизоксазол-3-ил)карбамата (1,98 г, 10,0 ммоль) в безводном тетрагидрофуране (20 мл) при -78°C добавляли 1M раствор бис(триметилсилил)амид лития в тетрагидрофуране (11,0 мл, 11,0 ммоль). Реакционную смесь перемешивали при -78°C в течение 15 минут, затем нагревали до температуры окружающей среды и перемешивали в течение 10 минут. Реакционную смесь охлаждали до -78°C и туда добавляли раствор 2,4,6-трифторбензолсульфонилхлорида (2,30 г, 10,0 ммоль) в безводном тетрагидрофуране (20 мл). Реакционную смесь перемешивали при -78°C в течение 2 ч и затем оставляли нагреваться до температуры окружающей среды. После перемешивания при температуре окружающей среды в течение 16 ч, реакционную смесь разводили насыщенным хлоридом аммония (50 мл) и экстрагировали этилацетатом (2×40 мл). Объединенные органические фазы промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и растиранием остатка в метаноле (10 мл) получали указанное в заголовке соединение в виде бесцветного твердого вещества (1,51 г, выход 38%): MS (ES+) m/z 393,1 (M+1).
Стадия 2. Получение трет-бутил (2-(азетидин-1-илметил)-3,6-дифторбензил)(4-(N-(трет-бутоксикарбонил)-N-(5-метилизоксазол-3-ил)сульфамоил)-3,5-дифторфенил)карбамата

В раствор трет-бутил (2-(азетидин-1-илметил)-3,6-дифторбензил)карбамата (0,30 г, 0,95 ммоль) в безводном N,N-диметилформамиде (20 мл) добавляли дисперсию 60% гидрида натрия в минеральном масле (0,115 г, 2,87 ммоль) при 0°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 25 минут. Затем туда добавляли трет-бутил (5-метилизоксазол-3-ил)((2,4,6-трифторфенил)сульфонил)карбамат (0,372 г, 0,95 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Реакционную смесь гасили добавлением воды (50 мл) и экстрагировали этилацетатом (70 мл). Органический слой промывали насыщенным хлоридом аммония (2×50 мл), солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали остаток, который очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 5% метанола в дихлорметане, чтобы получать указанное в заголовке соединение в виде бесцветной пены (0,12 г, выход 18%): MS (ES+) m/z 685,2 (M+1).
Стадия 3. Получение 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-3,6-дифторбензил)амино)-2,6-дифтор-N-(5-метилизоксазол-3-ил)бензолсульфонамида

В раствор трет-бутил (2-(азетидин-1-илметил)-3,6-дифторбензил)(4-(N-(трет-бутоксикарбонил)-N-(5-метилизоксазол-3-ил)сульфамоил)-3,5-дифторфенил)карбамата (0,12 г, 0,17 ммоль) в дихлорметане (4 мл) добавляли трифторуксусную кислоту (2 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч и затем концентрировали in vacuo. Остаток очищали с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом от 10 до 50% ацетонитрила в воде, содержащей 0,1% муравьиной кислоты, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,072 г, выход 87%):1H ЯМР (300 МГц, DMSO-d6) δ 11,62 (с, 1H), 10,35 (с, 1H), 7,53-7,43 (м, 2H), 7,38-7,36 (м, 1H), 6,39 (д, J=12,6 Гц, 2H), 6,03 (д, J=0,9 Гц, 1H), 4,49 (д, J=0,4 Гц, 2H), 4,38-4,37 (м, 2H), 4,19-4,01 (м, 4H), 2,46-2,13 (м, 5H); MS (ES+) m/z 485,1 (M+1).
ПРИМЕР 217
Синтез 2,2,2-трифторацетата 5-хлор-4-((1-(2-((диметиламино)метил)фенил)циклопропил)-амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил ((4-((1-(2-бромфенил)циклопропил)амино)-5-хлор-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата

В раствор трет-бутил ((5-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (1,23 г, 3,00 ммоль) и диизопропилэтиламина (1,00 мл, 6,00 ммоль) в безводном N,N-диметилформамиде (10 мл) добавляли 1-(2-бромфенил)циклопропан-1-амин (0,64 г, 3,00 ммоль). Получаемую смесь перемешивали при температуре окружающей среды в течение 18 ч и затем разводили этилацетатом (200 мл). Смесь промывали 1 M соляной кислотой (2×20 мл), солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 40% этилацетата в гексанах, чтобы получать указанное в заголовке соединение в виде твердого вещества грязно-белого цвета (0,60 г, выход 33%): MS (ES+) m/z 602,1 (M+1), 604,1 (M+1), 606,1 (M+1).
Стадия 2. Получение 2,2,2-трифторацетата 5-хлор-4-((1-(2-((диметиламино)метил)фенил)циклопропил)-амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

В смесь трет-бутил ((4-((1-(2-бромфенил)циклопропил)амино)-5-хлор-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата (0,60 г, 1,00 ммоль), диметиламинометилтрифторбороната калия (0,20 г, 1,20 ммоль) и 2 M карбонат натрия (1,50 мл, 3,00 ммоль) в диоксане (6 мл) добавляли ацетат палладия (0,023 г, 0,10 ммоль) и ди(1-адамантил)-н-бутилфосфин (0,070 г, 0,02 ммоль). Смесь дегазировали и нагревали до 100°C в течение 16 ч. После охлаждения до температуры окружающей среды, смесь разводили этилацетатом (50 мл) и фильтровали через подушку из целита. Фильтрат концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 100% этилацетата (содержащего 10% изопропанол и 10% триэтиламин) в гексанах. Дополнительной очисткой с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом от 20 до 80% ацетонитрила в воде, содержащей 0,1% трифторуксусной кислоты, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,015 г, выход 3%):1H ЯМР (300 МГц, DMSO-d6) δ 11,20-11,17 (м, 1H), 9,61-9,57 (м, 1H), 8,86 (д, J=2,2 Гц, 1H), 7,83-7,79 (м, 1H), 7,56 (д, J=7,3 Гц, 2H), 7,42-7,39 (м, 3H), 6,97-6,94 (м, 2H), 4,74-4,69 (м, 2H), 2,79-2,76 (м, 6H), 1,34-1,30 (м, 4H); MS (ES+) m/z 481,3 (M+1), 483,3 (M+1).
ПРИМЕР 218
Синтез 5-хлор-2-фтор-4-((2-фтор-6-(метоксиметил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 2-фтор-6-(метоксиметил)бензонитрила
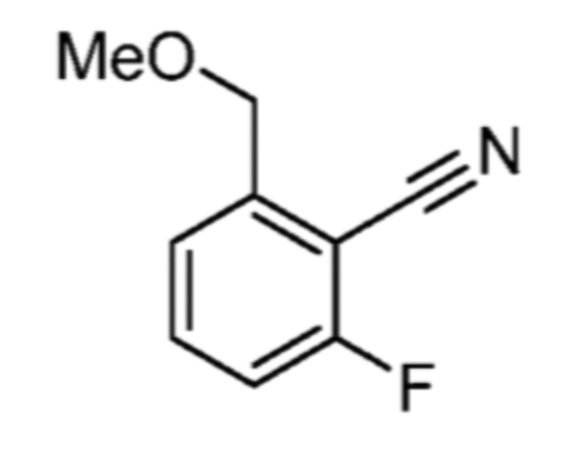
К безводному метанолу (20 мл) добавляли дисперсию 60% гидрида натрия в минеральном масле (0,21 г, 5,13 ммоль) при 0°C. Смесь перемешивали в течение 1 ч и затем туда добавляли 2-(бромметил)-6-фторбензонитрил (1,10 г, 5,13 ммоль). Смесь перемешивали при 0°C в течение 5 ч и при температуре окружающей среды в течение 16 ч. Затем смесь гасили добавлением насыщенного хлорида аммония (5 мл) и концентрировали in vacuo. Остаток разводили водой (5 мл) и экстрагировали этилацетатом (3×5 мл). Объединенный органический слой промывали солевым раствором (5 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 20% этилацетата в гексанах, чтобы получать указанное в заголовке соединение в виде бесцветного масла (0,40 г, выход 47%):1H ЯМР (300 МГц, CDCl3) δ 7,64-7,57 (м, 1H), 7,39 (дд, J=7,8, 0,6 Гц, 1H), 7,19-7,13 (м, 1H), 4,65 (с, 2H), 3,50 (с, 3H).
Стадия 2. Получение (2-фтор-6-(метоксиметил)фенил)метанамина

В раствор 2-фтор-6-(метоксиметил)бензонитрила (0,40 г, 2,40 ммоль) в безводном тетрагидрофуране (16 мл) добавляли 1,0 M раствор гидрида лития алюминия в тетрагидрофуране (3,60 мл, 3,60 ммоль) при 0°C. Смесь перемешивали при 0°C в течение 15 минут и затем при температуре окружающей среды в течение 18 ч. Затем смесь гасили добавлением декагидрата сульфата натрия (8,30 г) и затем смесь перемешивали в течение 18 ч. Смесь фильтровали через подушку из целита и фильтровальный осадок споласкивали этилацетатом (100 мл). Концентрированием объединенного фильтрата получали указанное в заголовке соединение в виде бесцветного масла (0,31 г, 75%):1H ЯМР (300 МГц, CDCl3) δ 7,20 (тд, J=7,8, 5,6 Гц, 1H), 7,11-7,00 (м, 2H), 4,51 (с, 2H), 3,88 (д, J=1,4 Гц, 2H), 3,41 (с, 3H), 1,76 (с, 2H); MS (ES+) m/z 170,2 (M+1).
Стадия 3. Получение 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((2-фтор-6-(метоксиметил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида

В смесь 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-4-ил)бензолсульфонамида (0,41 г, 0,89 ммоль) и (2-фтор-6-(метоксиметил)фенил)метанамина (0,15 г, 0,89 ммоль) в безводном диметилсульфоксиде (3,5 мл) добавляли диизопропилэтиламин (0,40 мл, 2,13 ммоль). Смесь перемешивали в течение 18 ч, затем разводили водой (2 мл) и экстрагировали этилацетатом (3×5 мл). Объединенные органические фракции промывали солевым раствором (5 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 50% этилацетата в гексанах, чтобы получать указанное в заголовке соединение в виде бесцветного масла (0,36 г, 66%): MS (ES+) m/z 610,3 (M+1), 612,3 (M+1).
Стадия 3. Получение 5-хлор-2-фтор-4-((2-фтор-6-(метоксиметил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида

В раствор 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((2-фтор-6-(метоксиметил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида (0,36 г, 0,59 ммоль) в дихлорметане (6 мл) добавляли трифторуксусную кислоту (1,5 мл). Смесь перемешивали в течение 1 ч и затем концентрировали in vacuo. К остатку добавляли метанол (20 мл) и смесь фильтровали. Фильтрат концентрировали in vacuo и остаток очищали с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом от 20 до 80% ацетонитрила в воде, содержащей 0,1% трифторуксусной кислоты, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,055 г, выход 20%):1H ЯМР (300 МГц, DMSO-d6) δ 11,14-11,12 (м, 1H), 8,88 (д, J=2,2 Гц, 1H), 7,59 (д, J=7,3 Гц, 1H), 7,36 (тд, J=7,9, 5,8 Гц, 1H), 7,25-7,16 (м, 2H), 6,99 (д, J=2,1 Гц, 1H), 6,86 (д, J=13,3 Гц, 1H), 6,49-6,46 (м, 1H), 4,54 (с, 2H), 4,48-4,47 (м, 2H), 3,30 (с, 3H); MS (ES+) m/z 460,1 (M+1), 462,1 (M+1).
ПРИМЕР 219
Синтез 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2-фтор-N-(изоксазол-3-ил)-5-метилбензолсульфонамида

Стадия 1. Получение 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-5-хлор-2-фтор-N-(изоксазол-3-ил)-N-(4-метоксибензил)бензолсульфонамида

В смесь (2-(азетидин-1-илметил)-6-фторфенил)метанамина (0,95 г, 4,89 ммоль) и N,N-диизопропилэтиламина (6,32 г, 48,9 ммоль) в N,N-диметилформамиде (20 мл) добавляли 5-хлор-2,4-дифтор-N-(изоксазол-3-ил)-N-(4-метоксибензил)бензолсульфонамид (2,03 г, 4,89 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 72 ч. Реакционную смесь корректировали до pH 6 с использованием 1 M раствора гидрохлорида и смесь экстрагировали этилацетатом (2×80 мл). Объединенные органические фракции промывали насыщенным хлоридом аммония (30 мл), солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo. Кристаллизацией остатка из этилацетата (60 мл) получали указанное в заголовке соединение в виде бесцветного твердого вещества (2,30 г, 80%): MS (ES+) m/z 589,2 (M+1), 591,2 (M+1).
Стадия 2. Получение 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2-фтор-N-(изоксазол-3-ил)-N-(4-метоксибензил)-5-метилбензолсульфонамида

Во флакон для микроволновой обработки добавляли 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-5-хлор-2-фтор-N-(изоксазол-3-ил)-N-(4-метоксибензил)-бензолсульфонамид (0,20 г, 0,34 ммоль), метилбороновую кислоту (0,08 г, 1,36 ммоль), трехосновный фосфат калия (0,22 г, 1,02 ммоль), трициклогексилфосфинтетрафторборат (0,03 г, 0,07 ммоль), ацетат палладия (0,01 г, 0,03 ммоль) и безводный 1,4-диоксан (2 мл). Смесь дегазировали и затем нагревали до 130°C в течение 30 минут в микроволновом реакторе. Реакционную смесь оставляли остывать до температуры окружающей среды, разводили этилацетатом (100 мл) и насыщенным хлоридом аммония (30 мл) и фильтровали. Собирали фильтрат и разделяли слои. Органический слой промывали насыщенным хлоридом аммония (20 мл), солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 20 до 100% этилацетата (содержащего 20% этанол и 0,2% гидроксида аммония) в гептане для того, чтобы получать указанное в заголовке соединение в виде вязкого масла (0,14 г, выход 70%):1H ЯМР (300 МГц, CDCl3) δ 8,14 (д, J=1,8 Гц, 1H), 7,45-7,39 (м, 3H), 7,29-7,22 (м, 1H), 7,12-7,02 (м, 3H), 6,83 (д, J=8,7 Гц, 2H), 6,67 (д, J=1,8 Гц, 2H), 5,04 (с, 2H), 4,40 (с, 2H), 3,77 (с, 3H), 3,64 (с, 2H), 3,25 (т, J=7,1 Гц, 4H), 2,16 (с, 3H), 2,14 (с, 2H); MS(ES+) m/z 568,9 (M+1).
Стадия 2. Получение 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2-фтор-N-(изоксазол-3-ил)-5-метилбензолсульфонамида

В смесь 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2-фтор-N-(изоксазол-3-ил)-N-(4-метоксибензил)-5-метилбензолсульфонамида (0,12 г, 0,21 ммоль) в безводном 1,2-дихлорэтане (2 мл) добавляли трифторуксусную кислоту (1 мл). Реакционную смесь перемешивали при 65°C в течение 1 ч и затем концентрировали in vacuo. Очисткой остатка с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом от 15 до 60% ацетонитрила в воде, содержащей 0,5% муравьиной кислоты, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,05 г, выход 30%):1H ЯМР (300 МГц, DMSO-d6) δ 11,52-11,31 (м, 1H), 8,64 (д, J=1,7 Гц, 1H), 7,46-7,39 (м, 1H), 7,37-7,27 (м, 1H), 7,24-7,11 (м, 2H), 7,04-6,88 (м, 1H), 6,73 (д, J=14,0 Гц, 1H), 6,31 (д, J=1,7 Гц, 1H), 4,41 (с, 2H), 3,73 (с, 2H), 3,32-3,17 (м, 4H), 2,16-2,09 (м, 3H), 2,06-1,94 (м, 2H); MS(ES+) m/z 449,2 (M+1).
ПРИМЕР 220
Синтез 5-хлор-2-фтор-4-((1-(2-фторфенил)циклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(2-фторфенил)циклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

В раствор 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(тиазол-2-ил)бензолсульфонамида (0,250 г, 0,543 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли 1-(2-фторфенил)циклопропан-1-амин (0,082 г, 0,54 ммоль) и карбонат калия (0,180 г, 1,30 ммоль). Получаемую суспензию перемешивали при 60°C в течение 16 ч. Смесь разводили этилацетатом (5 мл) и водой (5 мл). Разделяли слои и экстрагировали водную фазу этилацетатом (3×3 мл). Объединенную органическую фазу промывали солевым раствором (1×5 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрирование фильтрата in vacuo и очистка остатка. Остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 5 до 60% этилацетата в гексанах, чтобы получать указанное в заголовке соединение в виде бесцветного масла (0,153 г, выход 48%): MS (ES+) m/z 592,0 (M+1), 594,0 (M+1).
Стадия 2. Получение 5-хлор-2-фтор-4-((1-(2-фторфенил)циклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамида

В раствор 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(2-фторфенил)циклопропил)амино)-N-(тиазол-2-ил)бензолсульфонамида (0,153 г, 0,258 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (1 мл) и смесь перемешивали при температуре окружающей среды в течение 1 ч. Затем туда добавляли метанол (10 мл) и получаемый белый осадок удаляли посредством фильтрования. Фильтрат концентрировали in vacuo и остаток растирали с метанолом (2×5 мл) для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,040 г, выход 32%):1H-ЯМР (300 МГц, DMSO-d6) δ 12,77 (д, J=0,4 Гц, 1H), 7,60-7,54 (м, 2H), 7,31-7,23 (м, 2H), 7,20-7,10 (м, 3H), 6,82 (д, J=4,6 Гц, 1H), 6,77 (д, J=12,7 Гц, 1H), 1,39-1,37 (м, 2H), 1,28-1,23 (м, 2H); MS (ES+) m/z 441,9 (M+1), 443,9 (M+1).
ПРИМЕР 221
Синтез 5-хлор-2-фтор-4-(1-фенилциклопропокси)-N-(тиазол-4-ил)бензолсульфонамида

В смесь трет-бутил ((5-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,223 г, 0,543 ммоль) и 1-фенилциклопропан-1-ола (0,087 г, 0,65 ммоль) в безводном N,N-диметилформамиде (5 мл) добавляли гидрид натрия (60% дисперсия в минеральном масле, 0,073 г, 1,19 ммоль) при температуре окружающей среды и реакционную смесь перемешивали в течение 17 ч. Затем реакционную смесь разводили этилацетатом (5 мл) и туда добавляли насыщенный хлорид аммония (5 мл). Слои разделяли и водную фазу экстрагировали этилацетатом (3×5 мл). Объединенную органическую фазу промывали солевым раствором (5 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 5-100% этилацетата в гексанах, после чего следовала препаративная HPLC с обращенной фазой, используя ацетонитрил в воде (содержащей 0,1% трифторуксусную кислоту) в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,014 г, выход 6%):1H-ЯМР (300 МГц, DMSO-d6) δ 11,38 (с, 1H), 8,89-8,88 (м, 1H), 7,84 (д, J=7,4 Гц, 1H), 7,34 (т, J=7,4 Гц, 2H), 7,25 (д, J=7,3 Гц, 1H), 7,22-7,16 (м, 2H), 7,04 (д, J=2,1 Гц, 1H), 6,98 (д, J=11,5 Гц, 1H), 1,48 (с, 4H); MS (ES+) m/z 424,9 (M+1), 426,9 (M+1).
ПРИМЕР 222
Синтез (S)-5-хлор-4-((1-(2-хлорфенил)пропил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил (S)-((5-хлор-4-((1-(2-хлорфенил)пропил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата
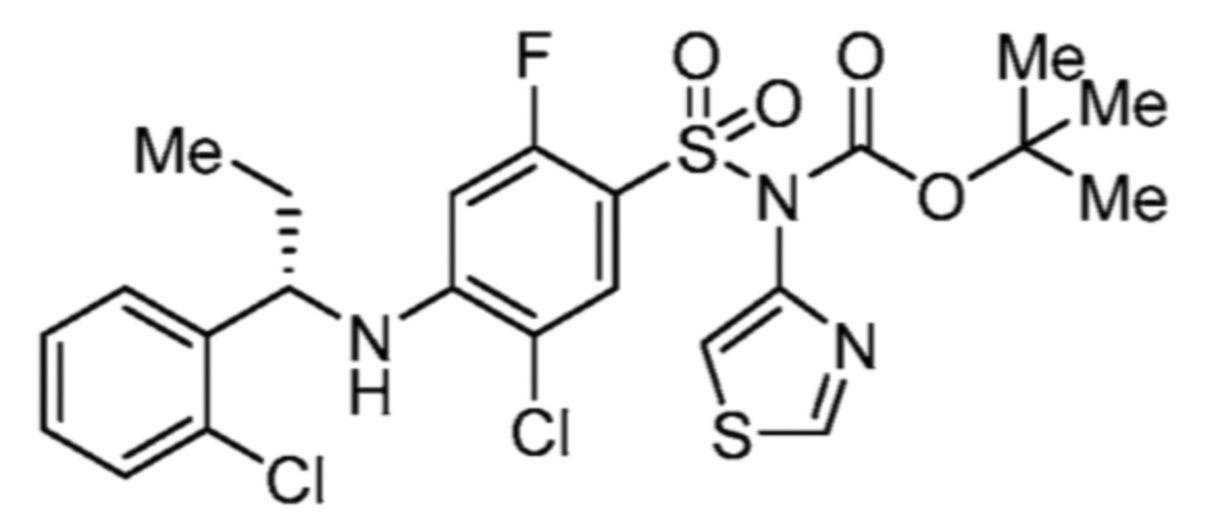
В раствор трет-бутил ((5-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,250 г, 0,543 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли гидрохлорид (S)-1-(2-хлорфенил)пропан-1-амина (0,223 г, 0,543 ммоль) и карбонат калия (0,254 г, 1,85 ммоль). Получаемую суспензию перемешивали при 75°C в течение 18 ч. Смесь разводили этилацетатом (5 мл) и водой (5 мл). Слои разделяли и водную фазу экстрагировали этилацетатом (3×3 мл). Объединенную органическую фазу промывали солевым раствором (1×5 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 5 до 70% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного масла (0,112 г, выход 37%): MS (ES+) m/z 459,9 (M - 99), 461,9 (M - 99).
Стадия 2. Получение (S)-5-хлор-4-((1-(2-хлорфенил)пропил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил (S)-((5-хлор-4-((1-(2-хлорфенил)пропил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата (0,112 г, 0,200 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (1 мл) и смесь перемешивали при температуре окружающей среды в течение 1 ч. Туда добавляли метанол (10 мл) добавляли и получаемый осадок удаляли фильтрованием. Фильтрат концентрировали in vacuo. Получаемый остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде (содержащей 0,1% трифторуксусную кислоту) в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,051 г, выход 55%):1H-ЯМР (300 МГц, DMSO-d6) δ 11,14 (с, 1H), 8,86 (д, J=2,2 Гц, 1H), 7,61 (д, J=7,4 Гц, 1H), 7,52-7,44 (м, 2H), 7,35-7,24 (м, 2H), 6,96 (д, J=2,2 Гц, 1H), 6,86-6,83 (м, 1H), 6,12 (д, J=13,2 Гц, 1H), 4,71-4,63 (м, 1H), 2,09-1,94 (м, 1H), 1,87-1,73 (м, 1H), 0,94 (т, J=7,3 Гц, 3H); MS (ES+) m/z 459,9 (M+1), 462,0 (M+1).
ПРИМЕР 223
Синтез (S)-5-хлор-4-((1-(3,5-дифторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Синтез трет-бутил (S)-((5-хлор-4-((1-(3,5-дифторфенил)пропил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата

Следуя процедуре как описано для примера 222, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять гидрохлорид (S)-1-(2-хлорфенил)пропан-1-амина на гидрохлорид (S)-1-(3,5-дифторфенил)этан-1-амина, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,087 г, выход 29%): MS (ES) m/z 448,2 (M - 99), 450,2 (M - 99).
Стадия 2. Получение (S)-5-хлор-4-((1-(3,5-дифторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано для примера 222, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять трет-бутил (S)-((5-хлор-4-((1-(2-хлорфенил)пропил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамат на трет-бутил (S)-((5-хлор-4-((1-(3,5-дифторфенил)пропил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамат, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,010 г, выход 4%):1H-ЯМР (300 МГц, DMSO-d6) δ 11,12 (с, 1H), 8,87 (д, J=2,1 Гц, 1H), 7,61 (д, J=7,4 Гц, 1H), 7,24-7,19 (м, 1H), 7,13-6,98 (м, 1H), 6,97-6,96 (м, 1H), 6,77-6,74 (м, 1H), 6,54 (д, J=13,3 Гц, 1H), 6,49-6,46 (д, J=7,8 Гц, 1H), 4,79-4,67 (м, 1H), 1,52 (д, J=6,7 Гц, 3H); MS (ES+) m/z 447,9 (M+1), 449,9 (M+1).
ПРИМЕР 224
Синтез 5-хлор-4-((1-(2,4-дифторфенил)циклопропил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил ((5-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,250 г, 0,543 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли гидрохлорид 1-(2,4-дифторфенил)циклопропан-1-амина (0,223 г, 0,543 ммоль) и карбонат цезия (0,792 г, 2,49 ммоль). Получаемую суспензию перемешивали при 75°C в течение 18 ч. После охлаждения до температуры окружающей среды, смесь разводили этилацетатом (5 мл) и водой (5 мл). Слои разделяли и водную фазу экстрагировали этилацетатом (3×3 мл). Объединенную органическую фазу промывали солевым раствором (1×5 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали остаток, который растворяли в дихлорметане (10 мл). Туда добавляли трифторуксусную кислоту (1 мл) и смесь перемешивали при температуре окружающей среды в течение 18 ч. Раствор концентрировали in vacuo и получаемый остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 5 до 80% этилацетата в гексанах. Дополнительной очисткой с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,014 г, выход 4%):1H-ЯМР (300 МГц, DMSO-d6) δ 11,16 (с, 1H), 8,87 (д, J=2,2 Гц, 1H), 7,68 (тд, J=8,9, 6,7 Гц, 1H), 7,59 (д, J=7,3 Гц, 1H), 7,34 (д, J=1,4 Гц, 1H), 7,19 (ддд, J=11,6, 9,2, 2,5 Гц, 1H), 7,07-7,01 (м, 1H), 6,98 (д, J=2,1 Гц, 1H), 6,80 (д, J=12,9 Гц, 1H), 1,39-1,34 (м, 2H), 1,27-1,24 (м, 2H); MS (ES+) m/z 460,0 (M+1), 462,0 (M+1).
ПРИМЕР 225
Синтез 5-хлор-4-((1-(2,5-дифторфенил)циклопропил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано для примера 224 и выполняя некритические вариации при необходимости, чтобы заменять гидрохлорид 1-(2,4-дифторфенил)циклопропан-1-амина на гидрохлорид 1-(2,5-дифторфенил)циклопропан-1-амина, очисткой с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 5 до 80% этилацетата в гексанах, после чего следовало растирание с метанолом (2×5 мл), получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,037 г, выход 15%):1H-ЯМР (300 МГц, DMSO-d6) δ 11,16 (с, 1H), 8,86 (д, J=2,2 Гц, 1H), 7,72-7,64 (м, 1H), 7,58 (д, J=7,3 Гц, 1H), 7,34-7,34 (м, 1H), 7,18 (ддд, J=11,7, 9,1, 2,6 Гц, 1H), 7,07-7,00 (м, 1H), 6,97 (д, J=2,2 Гц, 1H), 6,80 (д, J=13,0 Гц, 1H), 1,36-1,34 (м, 2H), 1,25-1,22 (м, 2H); MS (ES+) m/z 460,0 (M+1), 462,0 (M+1).
ПРИМЕР 226
Синтез 5-хлор-4-((2-хлор-6-метилбензил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида
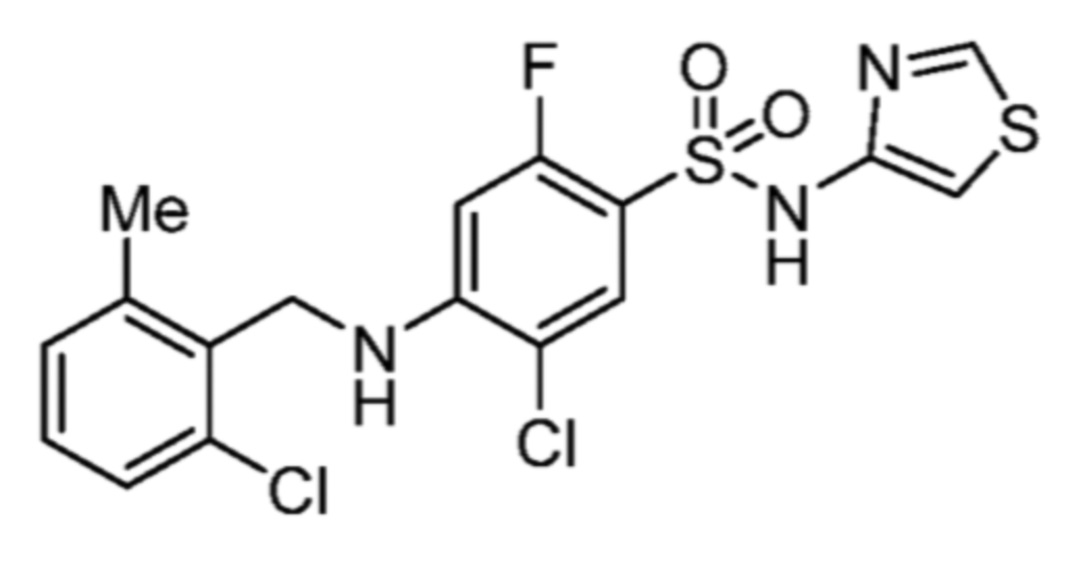
Стадия 1. Получение трет-бутил ((5-хлор-4-((2-хлор-6-метилбензил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата

В раствор трет-бутил ((5-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,250 г, 0,610 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли (2-хлор-6-метилфенил)метанамин (0,095 г, 0,610 ммоль) и триэтиламин (0,34 мл, 2,43 ммоль). Получаемый раствор перемешивали при температуре окружающей среды в течение 18 ч. Смесь разводили этилацетатом (5 мл) и водой (5 мл). Слои разделяли и водную фазу экстрагировали этилацетатом (3×5 мл). Объединенную органическую фазу промывали солевым раствором (1×5 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 5 до 60% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного масла (0,261 г, выход 78%): MS (ES+) m/z 546,1 (M+1), 548,1 (M+1).
Стадия 2. Получение 5-хлор-4-((2-хлор-6-метилбензил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил ((5-хлор-4-((2-хлор-6-метилбензил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата (0,261 г, 0,478 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (1 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч. Смесь концентрировали in vacuo для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,145 г, выход 68%):1H-ЯМР (300 МГц, DMSO-d6) δ 11,14 (с, 1H), 8,89 (д, J=2,2 Гц, 1H), 7,60 (д, J=7,4 Гц, 1H), 7,33 (дд, J=7,8, 1,3 Гц, 1H), 7,28-7,15 (м, 2H), 7,00-6,99 (м, 1H), 6,80 (д, J=13,2 Гц, 1H), 6,39-6,36 (м, 1H), 4,47 (д, J=4,6 Гц, 2H), 2,36 (с, 3H); MS (ES+) m/z 446,0 (M+1), 448,0 (M+1).
ПРИМЕР 227
Синтез (S)-5-хлор-4-((1-(5-(2,2-дифторэтил)-2-фторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида
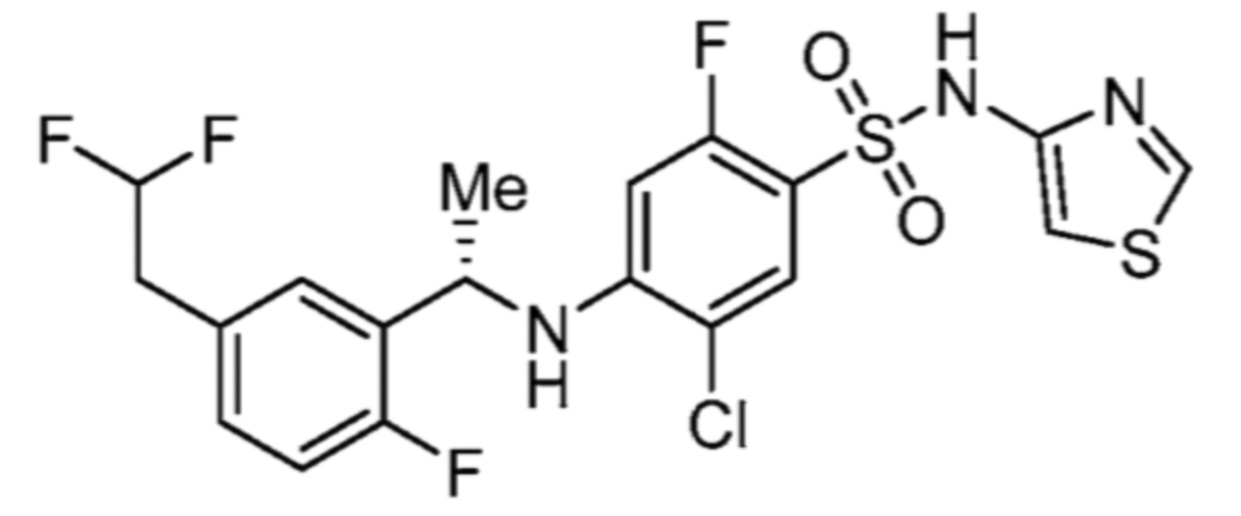
Стадия 1. Получение трет-бутил (S,E)-((5-хлор-4-((1-(5-(2-этоксивинил)-2-фторфенил)этил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата
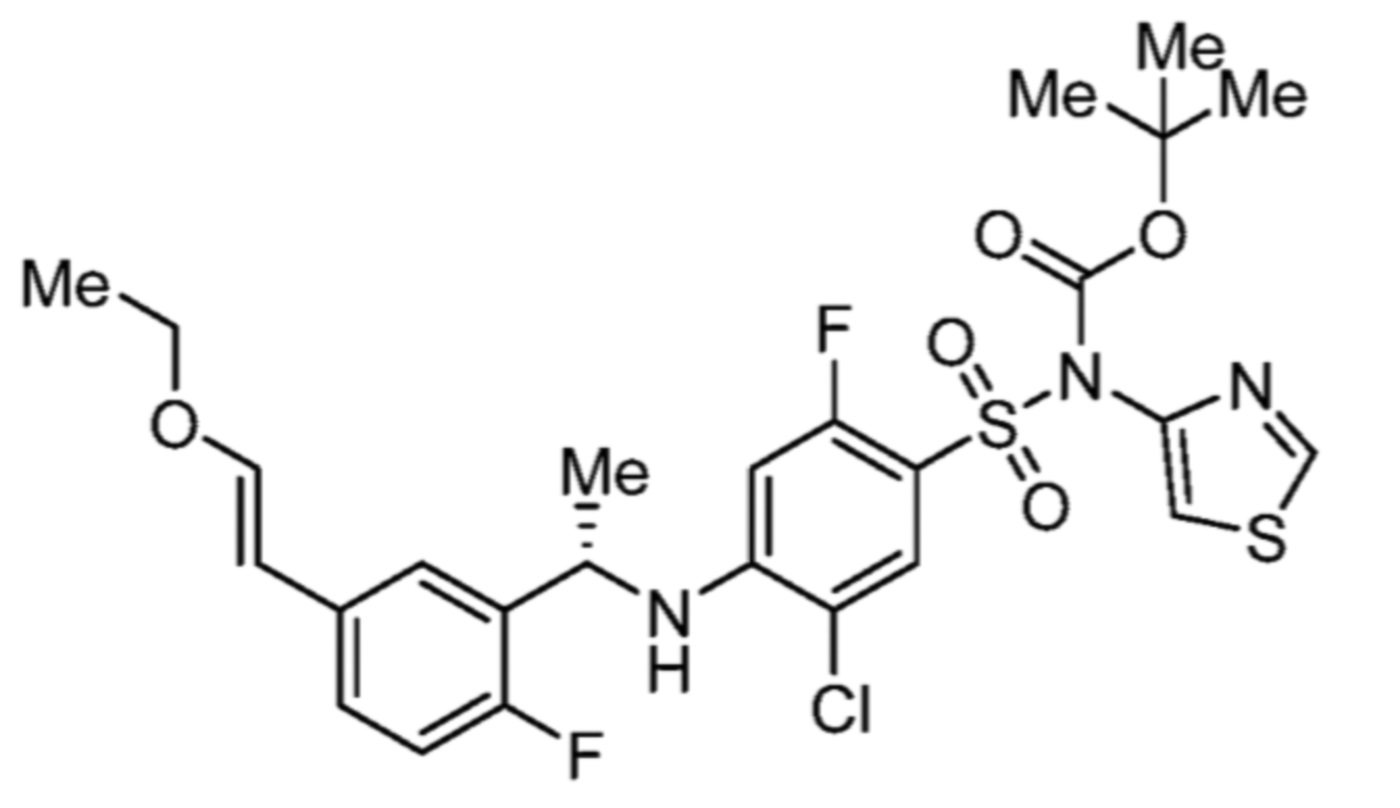
В раствор (S)-трет-бутил (4-((1-(5-бром-2-фторфенил)этил)амино)-5-хлор-2-фторфенил)сульфонил(тиазол-4-ил)карбамата (1,00 г, 1,64 ммоль), (E)-2-(2-этоксивинил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (0,65 г, 3,3 ммоль) и карбоната натрия (0,35 г, 3,3 ммоль) в толуоле (5 мл), этаноле (5 мл) и воде (5 мл) добавляли тетракис(трифенилфосфин)-палладий(0) (0,38 г, 0,33 ммоль). Реакционную смесь нагревали 90°C в течение 12 ч. После охлаждения до температуры окружающей среды, реакционную смесь разводили водой (50 мл) и экстрагировали этилацетатом (3×50 мл). Объединенный органический слой промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью хроматографии на силикагеле, осуществляя элюирование с градиентом от 10 до 33% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного масла (0,90 г, выход 91%):1H ЯМР (400 МГц, CDCl3) δ 8,77 (д, J=2,0 Гц, 1H), 7,96 (д, J=7,2 Гц, 1H), 7,47 (д, J=2,0 Гц, 1H), 7,14-7,09 (м, 1H), 7,04-6,97 (м, 2H), 6,86 (д, J=12,8 Гц, 1H), 6,21 (д, J=12,0 Гц, 1H), 5,74 (д, J=12,8 Гц, 1H), 4,81 (т, J=6,4 Гц, 1H), 3,87 (к, J=7,2 Гц, 3H), 1,65 (д, J=6,4 Гц, 3H), 1,34 (с, 9H), 1,33-1,30 (м, 3H); MS (ES+) m/z 499,9 (M - 99).
Стадия 2. Получение (S)-5-хлор-2-фтор-4-((1-(2-фтор-5-(2-оксоэтил)фенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида
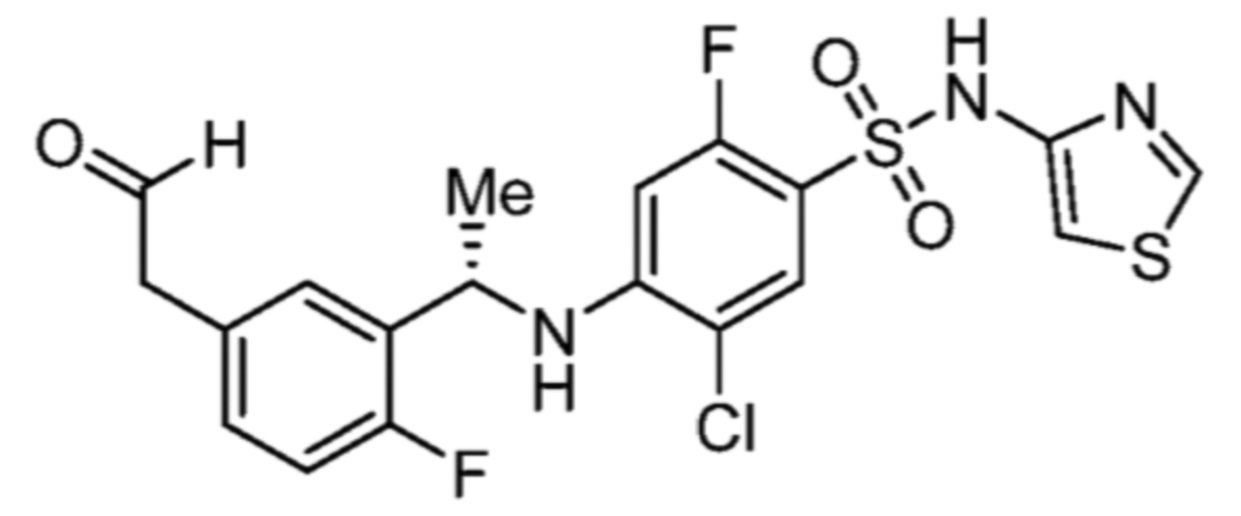
Раствор (S)-трет-бутил(5-хлор-4-((1-(5-(2-этоксивинил)-2-фторфенил)этил)амино)-2-фторфенил)сульфонил(тиазол-4-ил)карбамата (0,90 г, 1,5 ммоль) в муравьиной кислоте (10 мл) перемешивали при температуре окружающей среды в течение 30 минут. Затем смесь концентрировали in vacuo. К остатку добавляли водный бикарбонат натрия (30 мл) и смесь экстрагировали этилацетатом (3×30 мл). Объединенный органический слой промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 5-50% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде твердого вещества желтого цвета (0,23 г, выход 32%):1H ЯМР (400 МГц, CDCl3) δ 9,71 (т, J=2,0 Гц, 1H), 9,06 (с, 1H), 8,63 (д, J=2,0 Гц, 1H), 7,75 (д, J=7,2 Гц, 1H), 7,14-7,10 (м, 2H), 7,04 (д, J=5,6 Гц, 1H), 6,96 (д, J=2,0 Гц, 1H), 6,11 (д, J=12,4 Гц, 1H), 5,25-5,18 (м, 1H), 4,78 (т, J=6,4 Гц, 1H), 3,66 (с, 2H), 1,63 (д, J= 6,8 Гц, 3H); MS (ES+) m/z 472,0 (M+1), 474,0 (M+1).
Стадия 3. Получение (S)-5-хлор-4-((1-(5-(2,2-дифторэтил)-2-фторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

В раствор (S)-5-хлор-2-фтор-4-((1-(2-фтор-5-(2-оксоэтил)фенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида (0,100 г, 0,212 ммоль) в безводном дихлорметане (4 мл) добавляли трифторид (диэтиламино)серы (0,068 г, 0,424 ммоль) по каплям при -78°C. Смесь перемешивали при 0°C в течение 30 минут. Смесь гасили водным бикарбонатом натрия (30 мл) и экстрагировали дихлорметаном (3×30 мл). Объединенную органическую фазу промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,016 г, выход 15%):1H ЯМР (400 МГц, CDCl3) δ 8,60 (д, J=2,4 Гц, 1H), 7,75 (д, J=7,2 Гц, 1H), 7,21-7,14 (м, 1H), 7,11-7,04 (м, 2H), 6,97 (д, J=2,0 Гц, 1H), 6,12 (д, J=12,0 Гц, 1H), 6,03-5,68 (м, 1H), 5,21 (д, J=5,6 Гц, 1H), 4,77 (к, J=6,4 Гц, 1H), 3,08 (тд, J=17,2, 4,4 Гц, 2H), 1,63 (д, J=6,4 Гц, 3H), NH не наблюдали; MS (ES+) m/z 494,0 (M+1), 496,0 (M+1).
ПРИМЕР 228
Синтез (S)-2,6-дифтор-4-((1-(2-фтор-5-(метоксиметил)фенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 2-бром-1-фтор-4-(метоксиметил)бензола

В раствор (3-бром-4-фторфенил)метанола (5,00 г, 24,4 ммоль) в N,N-безводном N,N-диметилформамиде (50 мл) добавляли 60% суспензию гидрида натрия в минеральном масле (1,37 г, 34,2 ммоль) по частям при 0°C и смесь перемешивали при 0°C в течение 30 минут. Затем туда добавляли иодометан (4,15 г, 29,3 ммоль) при 0°C. Смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 1 ч. Реакцию гасили добавлением воды (200 мл) и экстрагировали этилацетатом (3×200 мл). Объединенные органические слои промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием от 2 до 5% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного масла (4,50 г, выход 84%):1H ЯМР (400 МГц, CDCl3) δ 7,57 (дд, J=6,4, 2,0 Гц, 1H), 7,28-7,23 (м, 1H), 7,11 (т, J=8,0 Гц, 1H), 4,42 (с, 2H), 3,41 (с, 3H).
Стадия 2. Получение 2-фтор-5-(метоксиметил)бензальдегида

В раствор 2-бром-1-фтор-4-(метоксиметил)бензола (4,50 г, 20,54 ммоль) в безводном тетрагидрофуране (50 мл) добавляли 2,5 M раствор н-бутиллития в простом диэтиловом эфире (9,86 мл, 3,94 ммоль) -78°C. Реакционную смесь перемешивали в течение 30 минут -78°C и затем туда добавляли безводный N,N-диметилформамид (3,00 г, 41,09 ммоль). Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 1 ч, и затем гасили водой (50 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием от 2 до 10% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного масла (2,30 г, выход 66%):1H ЯМР (400 МГц, CDCl3) δ 10,30 (с, 1H), 7,76 (дд, J=6,4, 2,0 Гц, 1H), 7,59-7,50 (м, 1H), 7,10 (дд, J=10,0, 8,4 Гц, 1H), 4,39 (с, 2H), 3,33 (с, 3H); MS (ES+) m/z 169,1 (M+1)
Стадия 3. Получение (R)-N-(2-фтор-5-(метоксиметил)бензилиден)-2-метилпропан-2-сульфинамида

В раствор 2-фтор-5-(метоксиметил)бензальдегида (2,30 г, 13,7 ммоль) и (R)-2-метилпропан-2-сульфинамида (1,82 г, 15,1 ммоль) в безводном дихлорметане (40 мл) добавляли карбонат цезия (8,91 г, 27,4 ммоль). Смесь перемешивали при температуре окружающей среды в течение 12 ч. Смесь фильтровали и фильтрат концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с использованием от 3 до 17% этилацетата в петролейном эфире, чтобы получать указанное в заголовке соединение в виде бесцветного масла (3,50 г, выход 94%):1H ЯМР (400 МГц, CDCl3) δ 8,92 (с, 1H), 7,96 (дд, J=6,4, 2,0 Гц, 1H), 7,54-7,47 (м, 1H), 7,17 (дд, J=9,6, 8,4 Гц, 1H), 4,48 (с, 2H), 3,44 (с, 3H), 1,30 (с, 9H).
Стадия 4. (R)-N-((S)-1-(2-фтор-5-(метоксиметил)фенил)этил)-2-метилпропан-2-сульфинамид

В раствор (R)-N-(2-фтор-5-(метоксиметил)бензилиден)-2-метилпропан-2-сульфинамида (3,50 г, 12,9 ммоль) в дихлорметане (40 мл) добавляли 3 M раствор бромида метилмагния в простом диэтиловом эфире (8,60 мл, 25,80 ммоль) при -50°C. Реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч и затем гасили насыщенным хлоридом аммония (100 мл) и экстрагировали дихлорметаном (3×100 мл). Объединенные органические слои промывали солевым раствором (50 мл) и сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием от 10 до 50% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного масла (2,20 г, выход 59%):1H ЯМР (400 МГц, CDCl3) δ 7,34 (дд, J=7,2, 2,0 Гц, 1H), 7,26-7,20 (м, 1H), 7,03 (дд, J=10,0, 8,4 Гц, 1H), 4,90-4,81 (м, 1H), 4,42 (с, 2H), 3,40 (с, 3H), 1,61 (д, J=6,4 Гц, 3H), 1,21 (с, 9H), NH не наблюдали.
Стадия 5. Получение гидрохлорида (S)-1-(2-фтор-5-(метоксиметил)фенил)этанамина

К (R)-N-((S)-1-(2-фтор-5-(метоксиметил)фенил)этил)-2-метилпропан-2-сульфинамиду (2,20 г, 7,66 ммоль) добавляли 4 M раствор хлороводорода в метаноле (20 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч. Смесь концентрировали in vacuo, разводили водой (50 мл) и экстрагировали этилацетатом (3×30 мл). К водной фазе добавляли насыщенный бикарбонат натрия (5 мл) и затем смесь экстрагировали этилацетатом (3×30 мл). Объединенные органические экстракты промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде бесцветного масла (1,30 г, выход 93%):1H ЯМР (400 МГц, CDCl3) δ 7,40 (дд, J=7,2, 1,6 Гц, 1H), 7,22-7,16 (м, 1H), 7,00 (дд, J=10,4, 8,4 Гц, 1H), 4,43 (с, 2H), 4,41-4,35 (м, 1H), 3,41 (с, 3H), 1,43 (д, J=6,8 Гц, 3H), способные к обмену протоны не наблюдали; MS (ES+) m/z 184,0 (M+1).
Стадия 6. Получение (S)-трет-бутил(2,6-дифтор-4-((1-(2-фтор-5-(метоксиметил)фенил)этил)амино)фенил)сульфонил(тиазол-4-ил)карбамата

В раствор трет-бутил тиазол-4-ил((2,4,6-трифторфенил)сульфонил)карбамата (0,30 г, 0,76 ммоль) и гидрохлорида (S)-1-(2-фтор-5-(метоксиметил)фенил)этанамина (0,15 г, 0,68 ммоль) в безводном диметилсульфоксиде (8 мл) добавляли карбонат цезия (0,49 г, 1,5 ммоль) одной частью. Смесь перемешивали при температуре окружающей среды в течение 12 ч и затем разводили водой (30 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 33% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного масла (0,10 г, выход 26%):1H ЯМР (400 МГц, CDCl3) δ 8,77 (д, J=2,0 Гц, 1H), 7,47 (д, J=2,0 Гц, 1H), 7,25-7,19 (м, 2H), 7,06 (т, J=9,6 Гц, 1H), 6,09 (д, J=12,0 Гц, 2H), 4,94 (д, J=6,4 Гц, 1H), 4,87-4,75 (м, 1H), 4,38 (с, 2H), 3,37 (с, 3H), 1,59 (д, J=6,4 Гц, 3H), 1,34 (с, 9H); MS (ES+) m/z 558,1 (M+1).
Стадия 7. Получение (S)-2,6-дифтор-4-((1-(2-фтор-5-(метоксиметил)фенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида

К (S)-трет-бутил(2,6-дифтор-4-((1-(2-фтор-5-(метоксиметил)фенил)этил)-амино)фенил)сульфонил(тиазол-4-ил)карбамата (0,10 г, 0,18 ммоль) добавляли 4 M раствор хлороводорода в этилацетате (5 мл) и смесь перемешивали при температуре окружающей среды в течение 1 ч. Смесь концентрировали in vacuo и остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,021 г, выход 26%):1H ЯМР (400 МГц, CDCl3) δ 9,47-8,92 (м, 1H), 8,64 (с, 1H), 7,26-7,20 (м, 2H), 7,12-7,04 (м, 1H), 7,03-7,00 (м, 1H), 6,01 (д, J=11,6 Гц, 2H), 4,82-4,70 (м, 2H), 4,39 (с, 2H), 3,36 (с, 3H), 1,56 (д, J=6,4 Гц, 3H); MS (ES+) m/z 458,1 (M+1), 459,1 (M+1).
ПРИМЕР 229
Синтез (S)-5-хлор-4-((1-(5-циано-2-фторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение (R)-N-(5-бром-2-фторбензилиден)-2-метилпропан-2-сульфинамида

Смесь 5-бром-2-фтор-бензальдегида (3,00 г, 14,8 ммоль), (R)-2-метилпропан-2-сульфинамида (2,15 г, 17,7 ммоль) и карбоната цезия (7,22 г, 22,1 ммоль) в безводном дихлорметане (30 мл) перемешивали при температуре окружающей среды в течение 10 ч. Реакционную смесь фильтровали и фильтрат концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с использованием 25% этилацетата в гексанах, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (3,70 г, выход 82%):1H ЯМР (400 МГц, CDCl3) δ 8,84 (с, 1H), 8,11 (дд, J=6,0, 2,8 Гц, 1H), 7,63-7,58 (м, 1H), 7,12-7,04 (м, 1H), 1,29 (с, 9H); MS (ES+) m/z 305,9 (M+1), 307,9 (M+1).
Стадия 2. Получение (R)-N-((S)-1-(5-бром-2-фторфенил)этил)-2-метилпропан-2-сульфинамида

В раствор (R)-N-(5-бром-2-фторбензилиден)-2-метилпропан-2-сульфинамида (2,00 г, 6,53 ммоль) в безводном дихлорметане (20 мл) добавляли 3,0 M раствор бромида метилмагния в тетрагидрофуране (3,3 мл, 9,9 ммоль) по каплям при -50°C. Смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 1 ч, и затем гасили добавлением водного хлорида аммония (50 мл). Смесь экстрагировали этилацетатом (2×50 мл). Объединенные органические слои промывали солевым раствором (2×30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного масла (1,40 г, выход 67%):1H ЯМР (400 МГц, CDCl3) δ 7,50 (дд, J=6,8, 2,8 Гц, 1H), 7,41-7,34 (м, 1H), 7,00-6,91 (м, 1H), 4,85 (м, 1H), 3,35 (ушир. д, J=4,4 Гц, 1H), 1,58 (д, J=6,8 Гц, 3H), 1,23 (с, 9H); MS (ES+) m/z 322,0 (M+1), 324,0 (M+1).
Стадия 3. Получение (R)-N-((S)-1-(5-циано-2-фторфенил)этил)-2-метилпропан-2-сульфинамида

Смесь (R)-N-((S)-1-(5-бром-2-фторфенил)этил)-2-метилпропан-2-сульфинамида (1,50 г, 4,65 ммоль), порошка цинка (0,030 г, 0,465 ммоль), цианида цинка (0,546 г, 4,65 ммоль), 1,1'-бис(дифенилфосфино)ферроцена (0,516 г, 0,93 ммоль) и трис(дибензилиденацетон)дипалладия(0) (0,426 г, 0,465 ммоль) в безводном N,N-диметилацетамиде (10 мл) перемешивали при 120°C в течение 12 ч. После охлаждения до температуры окружающей среды, смесь разводили этилацетатом (50 мл) и промывали солевым раствором (2×20 мл). Органический слой сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 9 до 50% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде масла коричневого цвета (1,50 г, выход 96%):1H ЯМР (400 МГц, CDCl3) δ 7,74 (дд, J=6,7, 2,2 Гц, 1H), 7,65-7,56 (м, 1H), 7,24-7,14 (м, 1H), 4,99-4,87 (м, 1H), 3,39 (ушир. д, J=3,8 Гц, 1H), 1,59 (д, J=6,8 Гц, 3H), 1,24 (с, 9H); MS (ES+) m/z 269,1 (M+1).
Стадия 4. Получение (S)-3-(1-аминоэтил)-4-фторбензонитрила
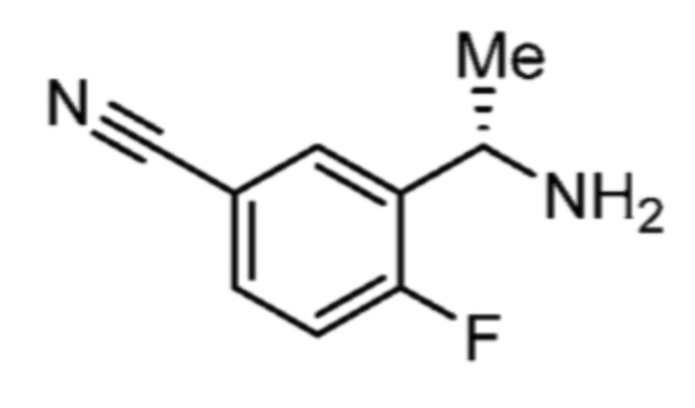
В смесь (R)-N-((S)-1-(5-циано-2-фторфенил)этил)-2-метилпропан-2-сульфинамида (1,50 г, 4,47 ммоль) в простом диэтиловом эфире (10 мл) добавляли 1 M раствор хлороводорода в диоксане (10 мл) и смесь перемешивали при температуре окружающей среды в течение 12 ч. Получаемое твердое вещество отфильтровывали. Туда добавляли насыщенный бикарбонат натрия (10 мл) и смесь экстрагировали дихлорметаном (3×20 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали in vacuo, чтобы получать указанное в заголовке соединение в виде масла коричневого цвета (1,00 г, выход 90%):1H ЯМР (400 МГц, CDCl3) δ 7,88 (дд, J=6,8, 1,8 Гц 1H), 7,56 (ддд, J=8,6, 4,8, 2,2 Гц, 1H), 7,14 (дд, J=10,0, 8,6 Гц, 1H), 4,46 (к, J=6,8 Гц, 1H), 1,43 (д, J=6,6 Гц, 3H), NH не наблюдали; MS (ES+) m/z 165,0 (M+1).
Стадия 5. Получение (S)-трет-бутил (5-хлор-4-((1-(5-циано-2-фторфенил)этил)амино)-2-фторфенил)сульфонил(тиазол-4-ил)карбамата
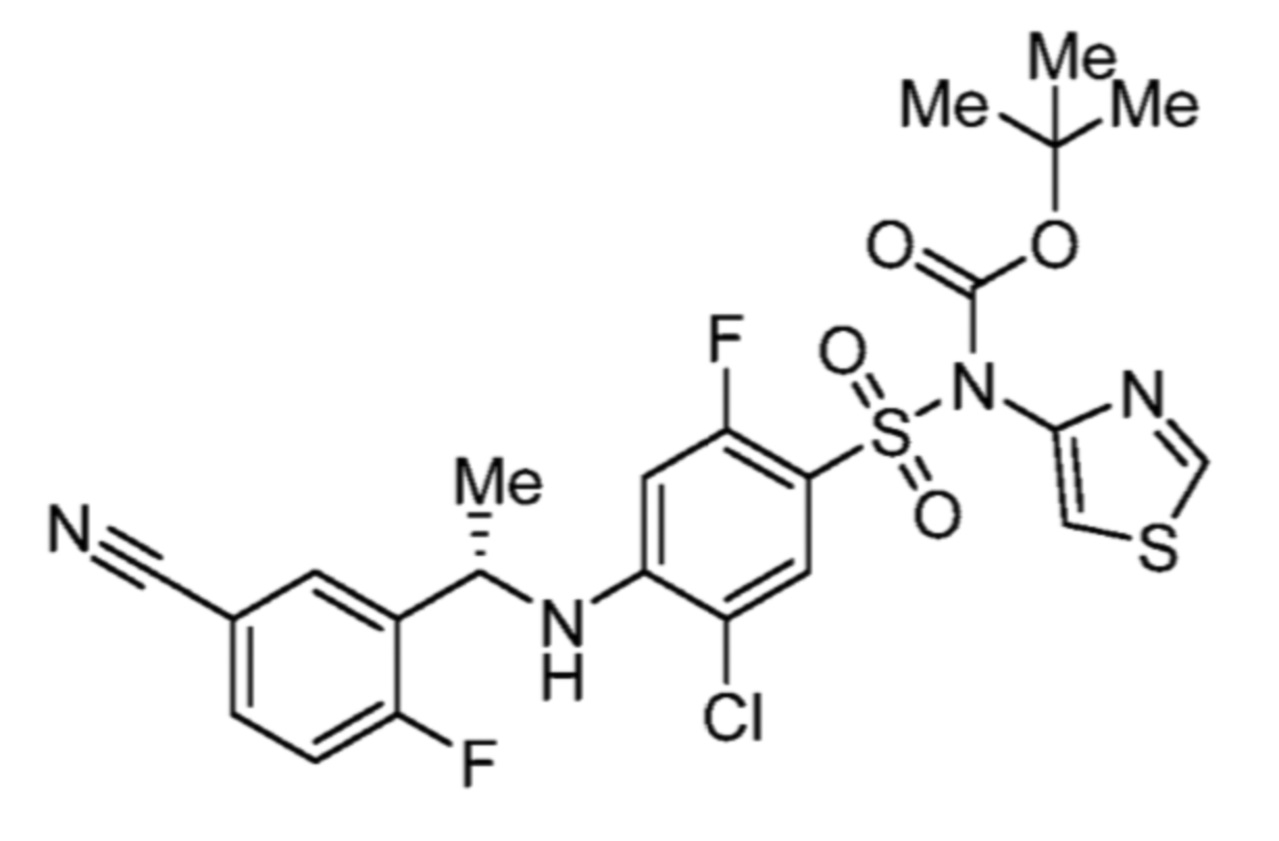
В смесь (S)-3-(1-аминоэтил)-4-фторбензонитрила (0,100 г, 0,609 ммоль) и трет-бутил N-(5-хлор-2,4-дифтор-фенил)сульфонил-N-тиазол-4-ил-карбамата (0,300 г, 0,730 ммоль) в безводном N,N-диметилформамиде (2 мл) добавляли карбонат цезия (0,396 г, 1,22 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 10 ч. Смесь разводили этилацетатом (80 мл) и фильтровали. Фильтрат промывали солевым раствором (2×30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 25% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,130 г, выход 38%):1H ЯМР (400 МГц, CDCl3) δ 8,77 (д, J=2,0 Гц, 1H), 8,01 (д, J=6,8 Гц, 1H), 7,68-7,60 (м, 1H), 7,58 (ушир. д, J=6,4 Гц, 1H), 7,48 (с, 1H), 7,29-7,23 (м, 1H), 6,08 (д, J=12,0 Гц, 1H), 5,34-5,26 (м, 1H), 4,91-4,85 (м, 1H), 1,68 (д, J=6,8 Гц, 3H), 1,36 (с, 9H).
Стадия 6. Получение (S)-5-хлор-4-((1-(5-циано-2-фторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида
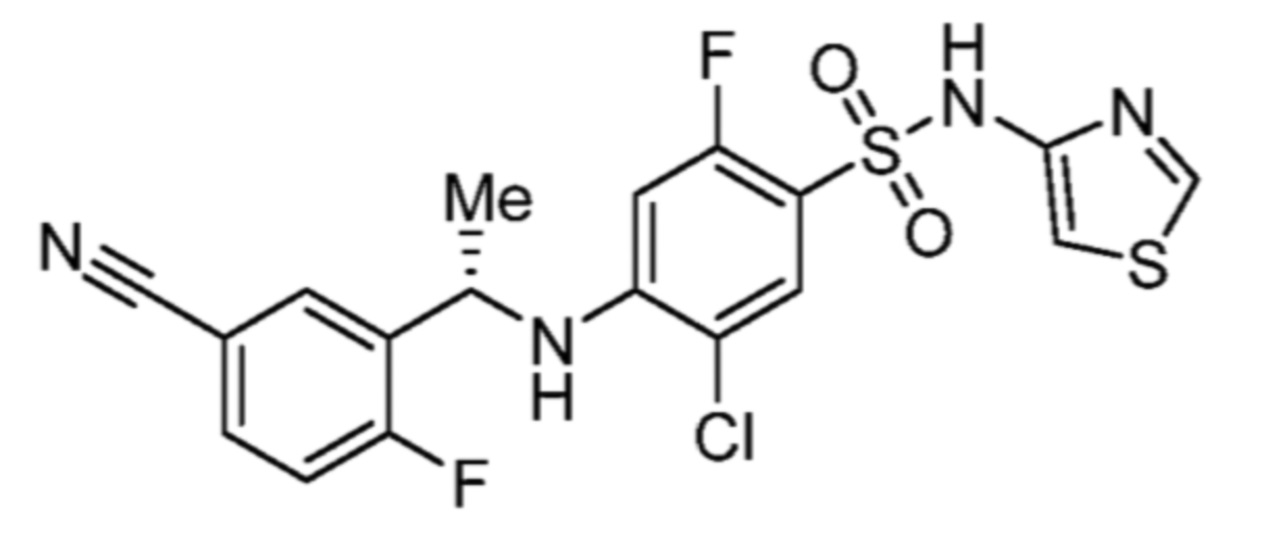
В смесь (S)-трет-бутил (5-хлор-4-((1-(5-циано-2-фторфенил)этил)амино)-2-фторфенил)сульфонил(тиазол-4-ил)карбамата (0,100 мг, 0,180 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (1 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 10 ч. Смесь концентрировали in vacuo и остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,0616 мг, выход 75%):1H ЯМР (400 МГц, CD3OD) δ 8,68 (д, J=2,0 Гц, 1H), 7,76-7,65 (м, 3H), 7,33 (дд, J=10,0, 8,0 Гц, 1H), 6,95 (д, J=2,0 Гц, 1H), 6,23 (д, J=12,4 Гц, 1H), 4,98-4,92 (м, 1H), 1,61 (д, J=6,8 Гц, 3H), способные к обмену протоны не наблюдали; MS (ES+) m/z 454,8 (M+1), 456,8 (M+1).
ПРИМЕР 230
Синтез (S)-4-((1-(5-циано-2-фторфенил)этил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение (S)-трет-бутил (4-((1-(5-циано-2-фторфенил)этил)амино)-2,6-дифторфенил)сульфонил(тиазол-4-ил)карбамата

В смесь (S)-3-(1-аминоэтил)-4-фторбензонитрила (0,150 г, 0,914 ммоль) и трет-бутил тиазол-4-ил((2,4,6-трифторфенил)сульфонил)карбамата (0,342 г, 0,868 ммоль) в безводном диметилсульфоксиде (6 мл) добавляли N,N-диизопропилэтиламин (0,191 мл, 1,10 ммоль). Реакционную смесь перемешивали при 36°C в течение 12 ч. Остаток выливали в воду со льдом (30 мл) и водную фазу экстрагировали этилацетатом (3×30 мл). Объединенную органическую фазу промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 33% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного масла (0,180 г, выход 37%):1H ЯМР (400 МГц, CDCl3) δ 8,79 (д, J=2,4 Гц, 1H), 7,70-7,61 (м, 2H), 7,50 (д, J=2,4 Гц, 1H), 7,25 (ушир. д, J=8,6 Гц, 1H), 6,09 (д, J=11,2 Гц, 2H), 4,93 (ушир. д, J=5,6 Гц, 1H), 4,88-4,80 (м, 1H), 1,62 (м, 3H), 1,37 (с, 9H); MS (ES+) m/z 438,9 (M - 99).
Стадия 2. Получение (S)-4-((1-(5-циано-2-фторфенил)этил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

В смесь (S)-трет-бутил (4-((1-(5-циано-2-фторфенил)этил)амино)-2,6-дифторфенил)сульфонил(тиазол-4-ил)карбамата (0,180 г, 0,334 ммоль) в дихлорметане (10 мл) добавляли трифторуксусную кислоту (2 мл). Концентрированием in vacuo и очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,127 г, выход 87%):1H ЯМР (400 МГц, CD3OD) δ 8,72 (д, J=2,4 Гц, 1H), 7,77-7,65 (м, 2H), 7,35 (дд, J=10,0, 8,4 Гц, 1H), 6,95 (д, J=2,4 Гц, 1H), 6,10 (ушир. д, J=12,4 Гц, 2H), 4,86-4,81 (м, 1H), 1,54 (д, J=6,8 Гц, 3H), способные к обмену протоны не наблюдали; MS (ES+) m/z 439,0 (M+1), 441,0 (M+1).
ПРИМЕР 231
Синтез 2,6-дифтор-4-[(1-фенилциклопропил)амино]-N-тиазол-4-ил-бензолсульфонамида

Стадия 1. Получение трет-бутил (2,6-дифтор-4-((1-фенилциклопропил)амино)фенил)сульфонил(тиазол-4-ил)карбамата

В раствор трет-бутил N-тиазол-4-ил-N-(2,4,6-трифторфенил)сульфонил-карбамата (0,236 г, 0,590 ммоль) в безводном N,N-диметилформамиде (5 мл) добавляли карбонат калия (0,332 г, 2,36 ммоль) и 1-фенилциклопропанамин (0,080 г, 0,60 ммоль). Смесь перемешивали при 60°C в течение 12 ч. После охлаждения до температуры окружающей среды смесь разводили насыщенным хлоридом аммония (5 мл) и экстрагировали этилацетатом (3×20 мл). Объединенную органическую фазу промывали солевым раствором (3×20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 40% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде масла желтого цвета (0,100 г, выход 43%):1H ЯМР (400 МГц, CDCl3) δ 8,78 (д, J=2,4 Гц, 1H), 7,48 (д, J=2,4 Гц, 1H), 7,33-7,28 (м, 2H), 7,25-7,19 (м, 1H), 7,12-7,06 (м, 2H), 6,26 (д, J=11,4 Гц, 2H), 5,32 (ушир. с, 1H), 1,46-1,42 (м, 2H), 1,37 (с, 9H), 1,35-1,31 (м, 2H).
Стадия 2. Получение 2,6-дифтор-4-[(1-фенилциклопропил)амино]-N-тиазол-4-ил-бензолсульфонамида

В раствор трет-бутил (2,6-дифтор-4-((1-фенилциклопропил)амино)фенил)-сульфонил(тиазол-4-ил)карбамата (0,080 г, 0,16 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (1 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 12 ч. Реакционную смесь концентрировали in vacuo и получаемый остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,428 г, выход 67%):1H ЯМР (400 МГц, CDCl3) δ 8,83 (д, J=1,8 Гц, 1H), 8,02 (ушир. с, 1H), 7,33-7,22 (м, 2H), 7,17 (д, J=7,3 Гц, 1H), 7,15-7,10 (м, 2H), 6,76 (ушир. с, 1H), 6,16 (ушир. с, 2H), 1,37-1,30 (м, 2H), 1,16 (ушир. д, J=1,9 Гц, 2H), NH не наблюдали; MS (ES+) m/z 408,0 (M+1).
ПРИМЕР 232
Синтез (S)-2,6-дифтор-4-(1-(2-фторфенил)этокси)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение (S)-1-(2-фторфенил)этанола

К безводному тетрагидрофурану (20 мл) добавляли (R)-2-метил-CBS-оксазаборолидин (1,0 M, 2,9 мл) и борандиметилсульфидный комплекс (10,0 M, 1,88 мл) и смесь перемешивали при температуре окружающей среды в течение 1 ч. Затем в эту смесь добавляли по каплям раствор 1-(2-фторфенил)этанон (2,00 г, 14,5 ммоль, 1,75 мл) в безводном тетрагидрофуране (5 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч. Смесь гасили добавлением метанола (20 мл) и концентрировали in vacuo для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (2,00 г, выход 98%), которое использовали без дополнительной очистки:1H ЯМР (400 МГц, CDCl3) δ 7,53-7,49 (м, 1H), 7,28-7,24 (м, 1H), 7,20-7,16 (м, 1H), 7,04 (ддд, J=10,8, 8,2, 1,2 Гц, 1H), 5,23 (к, J=6,4 Гц, 1H), 1,54 (д, J=6,4 Гц, 3H), OH не наблюдали.
Стадия 2. Получение (S)-трет-бутил (2,6-дифтор-4-(1-(2-фторфенил)этокси)фенил)сульфонил(тиазол-4-ил)карбамата

В раствор трет-бутил тиазол-4-ил((2,4,6-трифторфенил)сульфонил)карбамата (0,150 г, 0,380 ммоль) в безводном диметилсульфоксиде (2 мл) добавляли (S)-1-(2-фторфенил)этанол (0,106 г, 0,760 ммоль) и карбонат цезия (0,248 г, 0,760 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 12 ч. Реакционную смесь разводили водой (10 мл) и экстрагировали этилацетатом (2×10 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 20% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде масла желтого цвета (0,100 г, выход 51%): MS (ES+) m/z 414,9 (M - 99).
Стадия 3. Получение (S)-2,6-дифтор-4-(1-(2-фторфенил)этокси)-N-(тиазол-4-ил)бензолсульфонамида

К (S)-трет-бутил(2,6-дифтор-4-(1-(2-фторфенил)этокси)фенил)сульфонил(тиазол-4-ил)карбамату (0,100 г, 0,194 ммоль) добавляли 3 M хлороводород в метаноле (5 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 30 минут. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,020 г, выход 25%):1H ЯМР (400 МГц, CD3OD) δ 8,72 (д, J=2,4 Гц, 1H), 7,46-7,29 (м, 2H), 7,24-7,10 (м, 2H), 6,94 (ушир. с, 1H), 6,65-6,54 (м, 2H), 5,77 (к, J=6,4 Гц, 1H), 1,66 (д, J=6,4 Гц, 3H), NH не наблюдали; MS (ES+) m/z 415,0 (M+1).
ПРИМЕР 233
Синтез (S)-4-((1-(5-(дифторметил)-2-фторфенил)этил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида
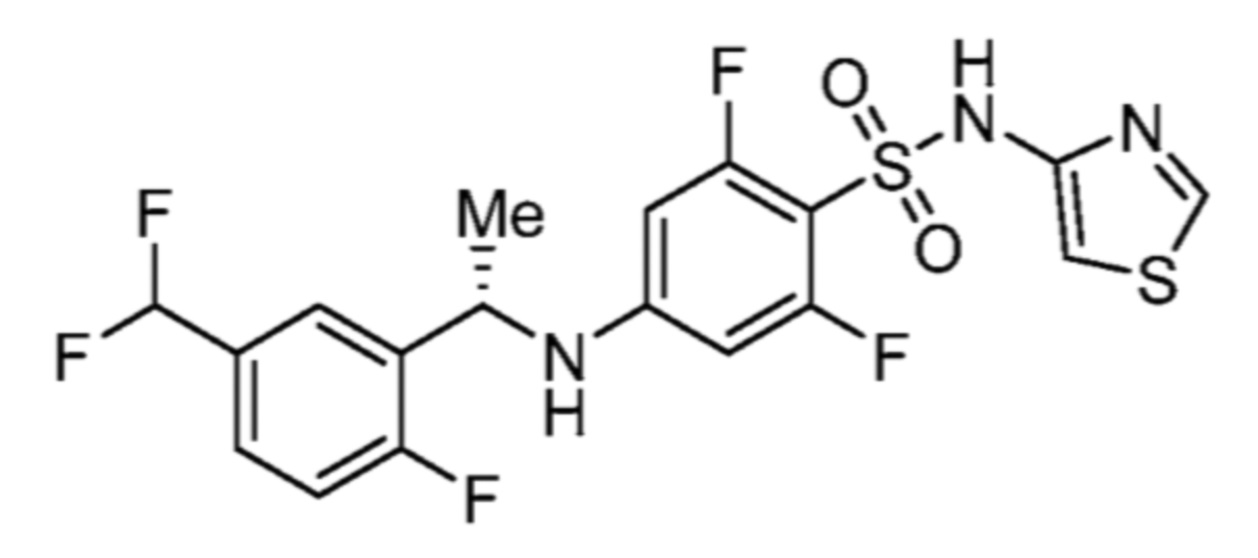
Стадия 1. Получение (R)-N-(5-(дифторметил)-2-фторбензилиден)-2-метилпропан-2-сульфинамида

В раствор 5-(дифторметил)-2-фторбензальдегида (получен в соответствии с WO2008051494, 2,50 г, 14,4 ммоль) и (R)-2-метилпропан-2-сульфинамида (1,91 г, 15,8 ммоль) в безводном дихлорметане (40 мл) добавляли карбонат цезия (7,02 г, 21,5 ммоль). Получаемую смесь перемешивали при температуре окружающей среды в течение 12 ч. Реакционную смесь разводили водой (20 мл) и экстрагировали дихлорметаном (3×20 мл). Объединенную органическую фазу промывали солевым раствором (3×20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 1 до 15% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде масла желтого цвета (3,45 г, выход 87%):1H ЯМР (300 МГц, CDCl3) δ 8,92 (с, 1H), 8,15 (ушир. д, J=5,2 Гц, 1H), 7,68 (дт, J=5,4, 2,4 Гц, 1H), 7,31-7,24 (м, 1H), 6,69 (т, J=56,0 Гц, 1H), 1,29 (с, 9H).
Стадия 2. Получение (R)-N-((S)-1-(5-(дифторметил)-2-фторфенил)этил)-2-метилпропан-2-сульфинамида

В раствор (R)-N-(5-(дифторметил)-2-фторбензилиден)-2-метилпропан-2-сульфинамида (3,45 г, 12,4 ммоль) в безводном дихлорметане (30 мл) добавляли по каплям 3,0 M раствор бромида метилмагния в простом диэтиловом эфире (8,29 мл) при -50°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды, перемешивали в течение 1 ч и затем тщательно гасили добавлением насыщенного хлорида аммония (30 мл). Слои разделяли и водный слой экстрагировали дихлорметаном (2×30 мл). Объединенные органические слои сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 10 до 25% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде масла желтого цвета (1,71 г, выход 47%):1H ЯМР (400 МГц, CDCl3) δ 7,57 (ушир. д, J=7,0 Гц, 1H), 7,43 (ушир. с, 1H), 7,15 (т, J=9,2 Гц, 1H), 6,64 (т, J=56,4 Гц, 1H), 5,01-4,87 (м, 1H), 3,39 (ушир. д, J=3,4 Гц, 1H), 1,62 (с, 3H), 1,23 (с, 9H).
Стадия 3. Получение гидрохлорида (S)-1-(5-(дифторметил)-2-фторфенил)этанамина

К (R)-N-((S)-1-(5-(дифторметил)-2-фторфенил)этил)-2-метилпропан-2-сульфинамиду (1,71 г, 5,83 ммоль) добавляли 4 M раствор хлороводорода в метаноле (20 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Реакционную смесь концентрировали in vacuo для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества, которое использовали без очистки (1,00 г, выход 76%).
Стадия 4. Получение (S)-трет-бутил (4-((1-(5-(дифторметил)-2-фторфенил)этил)амино)-2,6-дифторфенил)сульфонил(тиазол-4-ил)карбамата
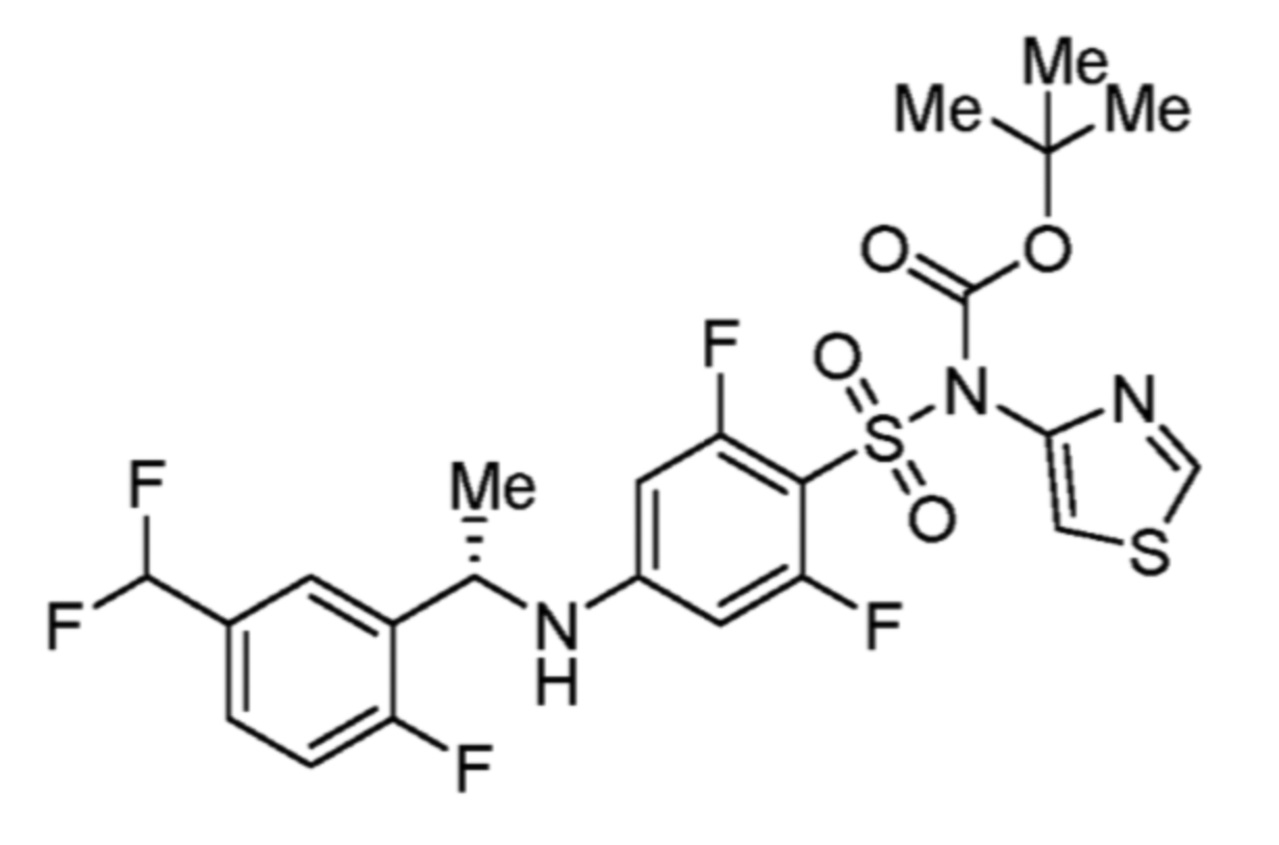
В раствор трет-бутил тиазол-4-ил ((2,4,6-трифторфенил)сульфонил)карбамата (0,200 г, 0,507 ммоль) в безводном N,N-диметилформамиде (8 мл) добавляли карбонат цезия (0,330 г, 1,01 ммоль) и гидрохлорид (S)-1-(5-(дифторметил)-2-фторфенил)этанамина (0,200 г, 0,89 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 12 ч и затем разводили солевым раствором (30 мл) и экстрагировали этилацетатом (2×30 мл). Объединенные органические слои промывали солевым раствором (2×20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде бесцветного масла, которое использовали без дополнительной очистки (0,150 г, выход 30%): MS (ES+) m/z 564,1 (M+1).
Стадия 5. Получение (S)-4-((1-(5-(дифторметил)-2-фторфенил)этил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

К (S)-трет-бутил (4-((1-(5-(дифторметил)-2-фторфенил)этил)амино)-2,6-дифторфенил)сульфонил(тиазол-4-ил)карбамату (0,130 г, 0,231 ммоль) добавляли 5 M раствор хлороводорода в этилацетате (5 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 30 минут. Растворитель удаляли in vacuo и остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,410 г, выход 38%):1H ЯМР (400 МГц, CDCl3) δ 10,38-9,77 (ушир. с, 1H), 8,68 (д, J=2,2 Гц, 1H), 7,51-7,35 (м, 2H), 7,19 (т, J=9,2 Гц, 1H), 6,99 (д, J=2,2 Гц, 1H), 6,60 (т, J=56,4 Гц, 1H), 6,00 (д, J=11,6 Гц, 2H), 4,89-4,69 (м, 2H), 1,58 (д, J=6,2 Гц, 3H); MS (ES+) m/z 463,9 (M+1).
ПРИМЕР 234
Синтез (S)-5-хлор-2-фтор-4-((1-(2-фтор-5-гидроксифенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение (R)-N-(5-бром-2-фторбензилиден)-2-метилпропан-2-сульфинамида

В раствор 5-бром-2-фторбензальдегида (10,5 г, 51,7 ммоль) и (R)-2-метилпропан-2-сульфинамида (7,52 г, 62,1 ммоль) в дихлорметане (50 мл) добавляли карбонат цезия (33,7 г, 103 ммоль) одной частью и реакционную смесь перемешивали при температуре окружающей среды в течение 3 ч. Смесь фильтровали и фильтрат концентрировали in vacuo. Получаемый остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 2 до 10% этилацетата в петролейном эфире, чтобы получать указанное в заголовке соединение в виде бесцветного масла (15,0 г, выход 95%):1H ЯМР (400 МГц, CDCl3) δ 8,85 (с, 1H), 8,12 (дд, J=6,0, 2,4 Гц, 1H), 7,61 (ддд, J=8,8, 4,4, 2,4 Гц, 1H), 7,08 (т, J=8,0 Гц, 1H), 1,30 (с, 9H).
Стадия 2. Получение (R)-N-((S)-1-(5-бром-2-фторфенил)этил)-2-метилпропан-2-сульфинамида

В раствор (R)-N-(5-бром-2-фторбензилиден)-2-метилпропан-2-сульфинамида (15,0 г, 49,0 ммоль) в безводном дихлорметане (200 мл) добавляли по каплям 3,0 M раствор бромида метилмагния в простом диэтиловом эфире (32,7 мл, 98,1 ммоль) при -45°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 1 ч и затем гасили добавлением водного хлорида аммония (200 мл). Смесь экстрагировали дихлорметаном (2×200 мл). Объединенные органические слои промывали солевым раствором (50 мл) и сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью хроматографии на колонке с гелем, осуществляя элюирование с градиентом от 10 до 33% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного масла (11,0 г, выход 70%):1H ЯМР (400 МГц, CDCl3) δ 7,41 (дд, J=6,4, 2,4 Гц, 1H), 7,28 (ддд, J=8,4, 4,4, 2,4 Гц, 1H), 6,86 (дд, J=9,6, 8,8 Гц, 1H), 4,82-4,69 (м, 1H), 3,27 (д, J=4,0 Гц, 1H), 1,49 (д, J=6,8 Гц, 3H), 1,14 (с, 9H).
Стадия 3. Получение гидрохлорида (S)-1-(5-бром-2-фторфенил)этанамина

К (R)-N-((S)-1-(5-бром-2-фторфенил)этил)-2-метилпропан-2-сульфинамиду (10,0 г, 31,0 ммоль) добавляли 4 M раствор хлороводорода в метаноле (100 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Смесь концентрировали in vacuo. Остаток разводили метанолом (5 мл) и очищали посредством кристаллизации из простого трет-бутилметилового эфира (300 мл) для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (5,00 г, выход 63%):1H ЯМР (400 МГц, CDCl3) δ 7,59 (дд, J=6,4, 2,4 Гц, 1H), 7,33 (ддд, J=8,4, 4,4, 2,4 Гц, 1H), 6,92 (дд, J=10,0, 8,8 Гц, 1H), 4,38 (к, J=6,4 Гц, 1H), 1,42 (д, J=6,4 Гц, 3H), способные к обмену протоны не наблюдали; MS (ES+) m/z 217,9 (M+1), 219,9 (M+1).
Стадия 4. Получение (S)-трет-бутил (4-((1-(5-бром-2-фторфенил)этил)амино)-5-хлор-2-фторфенил)сульфонил(тиазол-4-ил)карбамата

В раствор трет-бутил (5-хлор-2,4-дифторфенил)сульфонил(тиазол-4-ил)карбамата (5,00 г, 12,2 ммоль) и (S)-1-(5-бром-2-фторфенил)этанамина (3,18 г, 14,6 ммоль) в безводном диметилсульфоксиде (50 мл) добавляли карбонат цезия (7,93 г, 24,3 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 12 ч и затем разводили водой (200 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде твердого вещества желтого цвета (1,80 г, выход 24%):1H ЯМР (400 МГц, CDCl3) δ 8,79 (д, J=2,0 Гц, 1H), 8,00 (д, J=7,2 Гц, 1H), 7,50 (д, J=2,0 Гц, 1H), 7,41 (ддд, J=8,4, 4,4, 2,4 Гц, 1H), 7,35 (дд, J=6,4, 2,4 Гц, 1H), 7,03 (дд, J=10,0, 8,8 Гц, 1H), 6,16 (д, J=12,0 Гц, 1H), 5,29 (д, J=5,6 Гц, 1H), 4,84 (к, J=6,4 Гц, 1H), 1,67 (д, J=6,8 Гц, 3H), 1,37 (с, 9H); MS (ES+) m/z 507,9 (M - 99), 509,9 (M - 99).
Стадия 5. Получение трет-бутил (S)-((5-хлор-2-фтор-4-((1-(2-фтор-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)амино)фенил)сульфонил)(тиазол-4-ил)карбамата

В раствор (S)-трет-бутил (4-((1-(5-бром-2-фторфенил)этил)амино)-5-хлор-2-фторфенил)сульфонил(тиазол-4-ил)карбамата (0,30 г, 0,49 ммоль) в безводном диоксане (10 мл) добавляли бис(пинаколато)диборон (0,25 г, 0,99 ммоль), ацетат калия (0,097 г, 0,99 ммоль) и [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (0,036 г, 0,049 ммоль). Реакционную смесь перемешивали при 100°C в течение 12 ч. После охлаждения до температуры окружающей среды, реакционную смесь разводили водой (30 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 10 до 33% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного масла (0,32 г, количественный выход): MS (ES+) m/z 556,0 (M - 99).
Стадия 6. Получение трет-бутил (S)-((5-хлор-2-фтор-4-((1-(2-фтор-5-гидроксифенил)этил)амино)фенил)сульфонил)(тиазол-4-ил)карбамата

В смесь трет-бутил (S)-((5-хлор-2-фтор-4-((1-(2-фтор-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)амино)фенил)сульфонил)(тиазол-4-ил)карбамата (0,32 г, 0,49 ммоль) и гидроксида натрия (1,0 M, 0,73 мл) в тетрагидрофуране (5 мл) добавляли по каплям пероксид водорода (0,050 г, 1,46 ммоль) при 0°C. Смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 2 ч. Затем туда добавляли 2 M соляную кислоту для того, чтобы корректировать смесь до pH 7. Смесь разводили водой (30 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 50% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,10 г, выход 38%):1H ЯМР (400 МГц, CDCl3) δ 8,75 (д, J=2,0 Гц, 1H), 7,92 (д, J=7,2 Гц, 1H), 7,48 (д, J=2,0 Гц, 1H), 6,92 (т, J=9,2 Гц, 1H), 6,73-6,63 (м, 2H), 6,55-6,30 (м, 1H), 6,20 (д, J=12,4 Гц, 1H), 5,35 (д, J=6,0 Гц, 1H), 4,78 (к, J=6,4 Гц, 1H), 1,62 (д, J=6,4 Гц, 3H), 1,33 (с, 9H); MS (ES+) m/z 446,0 (M - 99), 447,9 (M - 99).
Стадия 7. Получение (S)-5-хлор-2-фтор-4-((1-(2-фтор-5-гидроксифенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида

К (S)-трет-бутил (5-хлор-2-фтор-4-((1-(2-фтор-5-гидроксифенил)этил)амино)фенил)-сульфонил(тиазол-4-ил)карбамату (0,10 г, 0,19 ммоль) добавляли 4 M раствор хлороводорода в 1,4-диоксане (5 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Смесь концентрировали in vacuo и остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,05 г, выход 59%):1H ЯМР (400 МГц, CDCl3) δ 9,85-9,45 (м, 1H), 8,65 (д, J=2,0 Гц, 1H), 7,70 (д, J=7,2 Гц, 1H), 7,00-6,92 (м, 2H), 6,73-6,67 (м, 1H), 6,65 (дд, J=5,6, 3,2 Гц, 1H), 6,07 (д, J=12,4 Гц, 1H), 5,16 (д, J=6,0 Гц, 1H), 4,72 (к, J=6,4 Гц, 1H), 1,61 (д, J=6,4 Гц, 3H), один способный к обмену протон не наблюдали; MS (ES+) m/z 446,0 (M+1), 448,0 (M+1).
ПРИМЕР 235
Синтез (S)-5-хлор-2-фтор-4-((1-(2-фтор-5-(оксетан-3-илокси)фенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида

В раствор (S)-5-хлор-2-фтор-4-((1-(2-фтор-5-гидроксифенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида (0,09 г, 0,2 ммоль) в безводном N,N-диметилформамиде (3 мл) добавляли 3-йодоксетан (0,074 г, 0,40 ммоль) и карбонат калия (0,07 г, 0,5 ммоль). Смесь перемешивали при 60°C в течение 3 ч. После охлаждения до температуры окружающей среды, реакционную смесь фильтровали и фильтрат концентрировали in vacuo. Остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,065 г, выход 64%):1H ЯМР (400 МГц, CDCl3) δ 8,64 (д, J=2,0 Гц, 1H), 7,76 (д, J=7,2 Гц, 1H), 7,01 (т, J=9,2 Гц, 1H), 6,96 (д, J=2,4 Гц, 1H), 6,57 (дд, J=6,0, 3,2 Гц, 1H), 6,48 (тд, J=8,8, 3,6 Гц, 1H), 6,08 (д, J=12,2 Гц, 1H), 5,16 (ушир. д, J=5,2 Гц, 1H), 5,10 (квин, J=5,6 Гц, 1H), 4,92 (т, J=6,8 Гц, 1H), 4,86 (т, J=6,8 Гц, 1H), 4,74 (к, J=6,2 Гц, 2H), 4,67-4,61 (м, 1H), 1,62 (д, J=6,8 Гц, 3H), NH не наблюдали; MS (ES+) m/z 502,0 (M+1), 504,0 (M+1).
ПРИМЕР 236
Синтез (S)-5-хлор-2-фтор-4-((1-(2-фтор-5-(оксетан-3-илметокси)фенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение оксетан-3-илметил метансульфоната

В раствор оксетан-3-илметанола (0,300 г, 3,41 ммоль) в безводном дихлорметане (10 мл) добавляли триэтиламин (0,516 г, 5,11 ммоль), после чего следовал метансульфонилхлорид (0,468 г, 4,09 ммоль) при 0°C. Смесь перемешивали при температуре окружающей среды в течение 1 ч. Затем реакционную смесь гасили добавлением воды (10 мл) и экстрагировали дихлорметаном (10 мл). Органический слой промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата при пониженном давлении получали указанное в заголовке соединение в виде бесцветного масла (0,500 г, выход 88%):1H ЯМР (400 МГц, CDCl3) δ 4,86 (дд, J=6,6, 7,6 Гц, 2H), 4,55-4,44 (м, 4H), 3,48-3,35 (м, 1H), 3,08 (с, 3H).
Стадия 2. Получение (S)-5-хлор-2-фтор-4-((1-(2-фтор-5-(оксетан-3-илметокси)фенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида
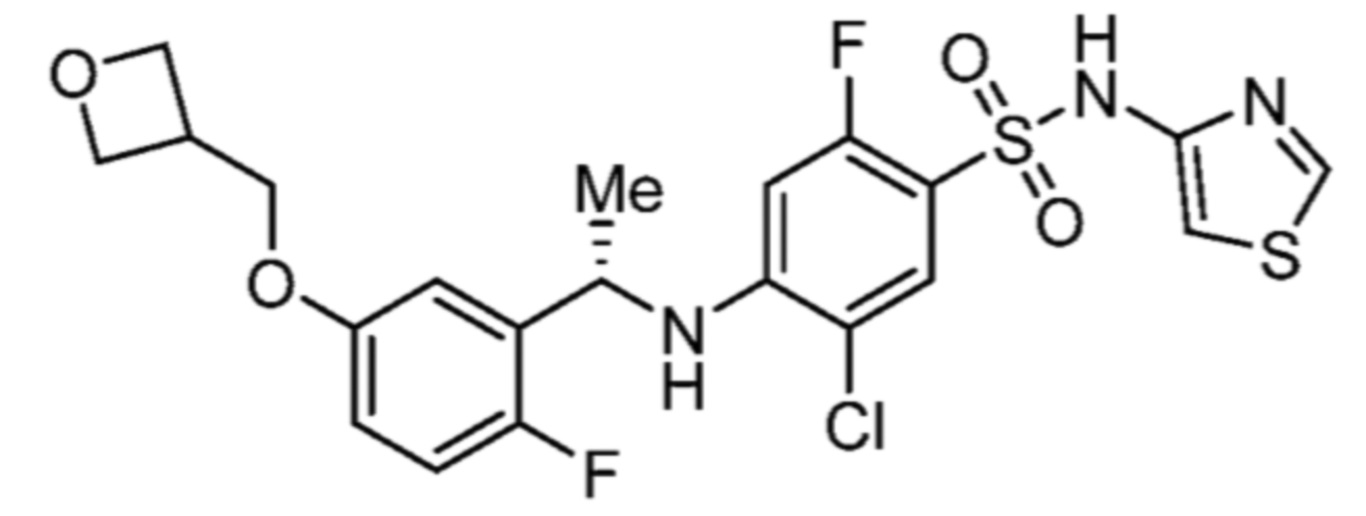
В раствор (S)-5-хлор-2-фтор-4-((1-(2-фтор-5-гидроксифенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида (0,080 г, 0,18 ммоль) в безводном N,N-диметилформамиде (5 мл) добавляли оксетан-3-илметил метансульфонат (0,030 г, 0,18 ммоль) и карбонат калия (0,049 г, 0,36 ммоль). Смесь перемешивали при 60°C в течение 12 ч. После охлаждения до температуры окружающей среды, реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении. Получаемый остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,012 г, выход 13%):1H ЯМР (400 МГц, CDCl3) δ 8,53 (д, J=2,2 Гц, 1H), 7,66 (д, J=7,2 Гц, 1H), 6,94 (т, J=9,2 Гц, 1H), 6,87 (д, J=2,2 Гц, 1H), 6,69 (тд, J=3,6, 8,8 Гц, 1H), 6,65 (дд, J=3,0, 6,0 Гц, 1H), 6,04 (д, J=12,2 Гц, 1H), 5,10 (ушир. д, J=5,2 Гц, 1H), 4,79 (т, J=7,0 Гц, 2H), 4,65 (квин, J=6,6 Гц, 1H), 4,47 (т, J=6,0 Гц, 2H), 4,08-3,98 (м, 2H), 3,38-3,24 (м, 1H), 1,52 (ушир. с, 3H), NH не наблюдали; MS (ES+) m/z 516,0 (M+1), 518,0 (M+1).
ПРИМЕР 237
Синтез (S)-5-хлор-2-фтор-4-((1-(2-фтор-5-((3-метилоксетан-3-ил)метокси)фенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение (3-метилоксетан-3-ил)метил метансульфоната

В раствор (3-метилоксетан-3-ил)метанола (0,400 г, 3,92 ммоль) в безводном дихлорметане (10 мл) добавляли триэтиламин (0,594 г, 5,87 ммоль, 0,814 мл), после чего следовал метансульфонилхлорид (0,538 г, 4,70 ммоль, 0,363 мл) при 0°C. Смесь перемешивали при температуре окружающей среды в течение 1 ч и затем гасили добавлением воды (10 мл). Смесь экстрагировали дихлорметаном (10 мл). Органический слой промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали in vacuo для того, чтобы получать указанное в заголовке соединение в виде масла желтого цвета (0,500 г, выход 71%):1H ЯМР (400 МГц, CDCl3) δ 4,54 (д, J=6,4 Гц, 2H), 4,45 (д, J=6,4 Гц, 2H), 4,34 (с, 2H), 3,09 (с, 3H), 1,41 (с, 3H).
Стадия 2. Получение (S)-5-хлор-2-фтор-4-((1-(2-фтор-5-((3-метилоксетан-3-ил)метокси)фенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида TF057E

В раствор (S)-5-хлор-2-фтор-4-((1-(2-фтор-5-гидроксифенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида (0,080 г, 0,179 ммоль) в безводном N,N-диметилформамиде (5 мл) добавляли (3-метилоксетан-3-ил)метил метансульфонат (0,032 г, 0,179 ммоль) и карбонат калия (0,049 г, 0,36 ммоль). Смесь перемешивали при 60°C в течение 12 ч. После охлаждения до температуры окружающей среды, реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,015 г, выход 16%):1H ЯМР (400 МГц, CDCl3) δ 9,43 (ушир. с, 1H), 8,64 (д, J=2,4 Гц, 1H), 7,76 (д, J=7,2 Гц, 1H), 7,03 (т, J=9,2 Гц, 1H), 6,95 (д, J=2,0 Гц, 1H), 6,85-6,70 (м, 2H), 6,14 (д, J=12,4 Гц, 1H), 5,19 (ушир. д, J=5,4 Гц, 1H), 4,75 (квин, J=6,6 Гц, 1H), 4,61 (д, J=6,0 Гц, 2H), 4,46 (д, J=6,0 Гц, 2H), 4,00-3,88 (м, 2H), 1,62 (д, J=6,8 Гц, 3H), 1,42 (с, 3H), NH не наблюдали; MS (ES+) m/z 530,0 (M+1), 532,0 (M+1).
ПРИМЕР 238
Синтез (S)-2,6-дифтор-4-((1-(2-фтор-5-метоксифенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение (R)-N-(2-фтор-5-метоксибензилиден)-2-метилпропан-2-сульфинамида

В раствор 2-фтор-5-метоксибензальдегида (2,00 г, 13,0 ммоль) в безводном дихлорметане (20 мл) добавляли карбонат цезия (8,46 г, 26,0 ммоль) и (R)-2-метилпропан-2-сульфинамид (2,36 г, 19,5 ммоль). Смесь перемешивали при температуре окружающей среды в течение 12 ч и затем фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 5 до 50% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (2,50 г, выход 75%):1H ЯМР (400 МГц, CDCl3) δ 8,88 (с, 1H), 7,46 (дд, J=3,0, 5,2 Гц, 1H), 7,12-7,06 (м, 1H), 7,06-7,01 (м, 1H), 3,85 (с, 3H), 1,28 (с, 9H).
Стадия 2. Получение (R)-N-((S)-1-(2-фтор-5-метоксифенил)этил)-2-метилпропан-2-сульфинамида

В раствор (R)-N-(2-фтор-5-метоксибензилиден)-2-метилпропан-2-сульфинамида (1,50 г, 5,83 ммоль) в безводном дихлорметане (20 мл) добавляли по каплям бромид метилмагния (3,0 M, 3,9 мл, 11,7 ммоль) при -50°C. Реакционную смесь нагревали до температуры окружающей среды и перемешивали в течение 12 ч. Смесь разводили насыщенным хлоридом аммония (20 мл) и экстрагировали дихлорметаном (3×20 мл). Объединенную органическую фазу промывали солевым раствором (3×20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 5 до 25% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде масла желтого цвета (1,00 г, выход 63%):1H ЯМР (400 МГц, CDCl3) δ 6,95 (т, J=9,3 Гц, 1H), 6,87 (дд, J=5,8, 3,2 Гц, 1H), 6,74 (тд, J=8,9, 3,5 Гц, 1H), 4,85-4,74 (м, 1H), 3,77 (с, 3H), 3,38 (ушир. д, J=4,3 Гц, 1H), 1,56 (д, J=6,7 Гц, 3H), 1,20 (с, 9H).
Стадия 3. Получение гидрохлорид (S)-1-(2-фтор-5-метоксифенил)этанамина
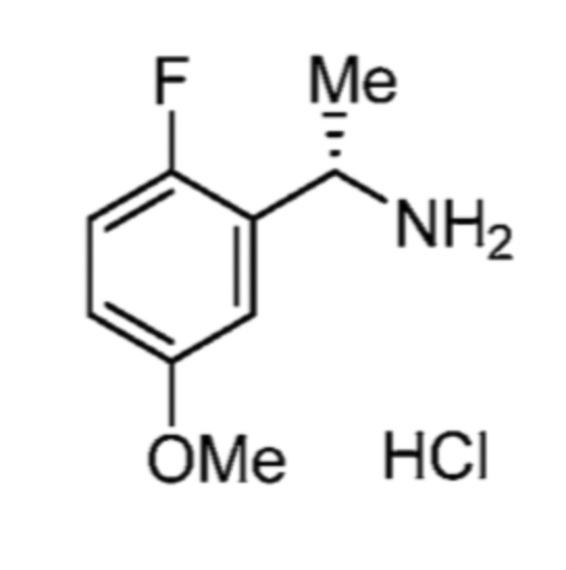
К (R)-N-((S)-1-(2-фтор-5-метоксифенил)этил)-2-метилпропан-2-сульфинамиду (0,90 г, 3,3 ммоль) добавляли 4 M раствор хлороводорода в 1,4-диоксане (10 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 30 минут. Реакционную смесь концентрировали in vacuo. К остатку добавляли две капли метанола и простого трет-бутилметилового эфира (10 мл) и смесь перемешивали в течение 6 ч. Затем твердое вещество отфильтровывали и сушили in vacuo для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,40 г, выход 59%):1H ЯМР (400 МГц, CDCl3) δ 8,87 (ушир. с, 3H), 7,23 (дд, J=5,7, 2,8 Гц, 1H), 6,99 (т, J=9,4 Гц, 1H), 6,82 (тд, J=8,8, 3,6 Гц, 1H), 4,76 (ушир. с, 1H), 3,68 (с, 3H), 1,69 (д, J=6,8 Гц, 3H).
Стадия 4. Получение (S)-трет-бутил (2,6-дифтор-4-((1-(2-фтор-5-метоксифенил)этил)амино)фенил)сульфонил(тиазол-4-ил)карбамата

В раствор гидрохлорида (S)-1-(2-фтор-5-метоксифенил)этанамина (0,15 г, 0,73 ммоль) в безводном N,N-диметилформамиде (5 мл) добавляли трет-бутил тиазол-4-ил((2,4,6-трифторфенил)сульфонил)карбамат (0,29 г, 0,73 ммоль) и карбонат калия (0,40 г, 2,9 ммоль). Смесь перемешивали при 60°C в течение 12 ч. После охлаждения до температуры окружающей среды, реакционную смесь затем разводили насыщенным хлоридом аммония (5 мл) и экстрагировали этилацетатом (3×20 мл). Объединенную органическую фазу промывали солевым раствором (3×20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 40% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,08 г, выход 20%):1H ЯМР (400 МГц, CDCl3) δ 8,77 (д, J=2,4 Гц, 1H), 7,47 (д, J=221 Гц, 1H), 7,05-6,97 (м, 1H), 6,78-6,69 (м, 2H), 6,09 (д, J=11,6 Гц, 2H), 5,10 (ушир. д, J=6,2 Гц, 1H), 4,79-4,69 (м, 1H), 3,73 (с, 3H), 1,56 (д, J=6,8 Гц, 3H), 1,33 (с, 9H).
Стадия 5. Получение (S)-2,6-дифтор-4-((1-(2-фтор-5-метоксифенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида
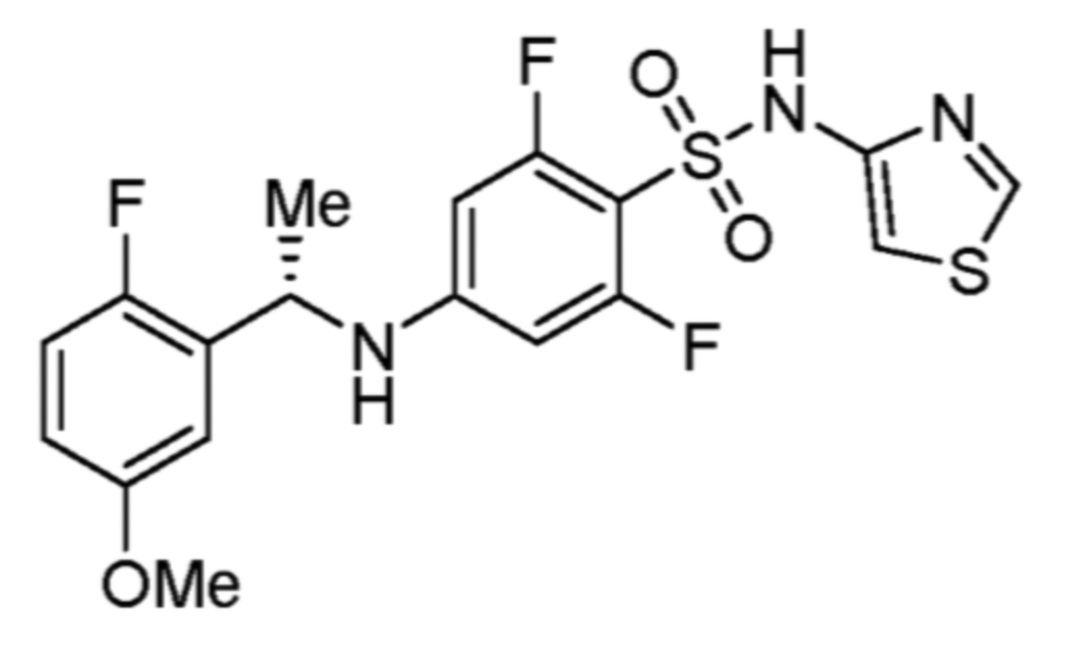
В раствор (S)-2,6-дифтор-4-((1-(2-фтор-5-метоксифенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида (0,10 г, 0,18 ммоль) в дихлорметане (10 мл) добавляли трифторуксусную кислоту (3,08 г, 27,0 ммоль, 2 мл). Смесь перемешивали при температуре окружающей среды в течение 12 ч и затем концентрировали in vacuo. Остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,063 г, выход 69%):1H ЯМР (400 МГц, CDCl3) δ 10,92 (ушир. с, 1H), 8,73 (д, J=2,1 Гц, 1H), 7,03-6,97 (м, 1H), 6,96 (д, J=2,3 Гц, 1H), 6,78-6,72 (м, 2H), 6,00 (д, J=11,7 Гц, 2H), 4,86 (ушир. д, J=6,1 Гц, 1H), 4,69 (квин, J=6,6 Гц, 1H), 3,74 (с, 3H), 1,54 (д, J=6,7 Гц, 3H); MS (ES+) m/z 444,0 (M+1).
ПРИМЕР 239
Синтез (S)-3-циано-4-((1-(2-фторфенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил ((3-циано-4-фторфенил)сульфонил)(тиазол-4-ил)карбамата

В раствор трет-бутил тиазол-4-илкарбамата (0,500 г, 2,50 ммоль) в безводном N,N-диметилформамиде (20 мл) добавляли гидрид натрия (60% дисперсия в минеральном масле, 0,120 г, 3,00 ммоль) при 0°C. Смесь нагревали до 10°C и перемешивали в течение 1 ч и охлаждали до 0°C. Туда при 0°C добавляли 3-циано-4-фторбензолсульфонилхлорид (0,713 г, 3,25 ммоль). Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 4 ч. Смесь разводили насыщенным хлоридом аммония (20 мл) и экстрагировали этилацетатом (3×20 мл). Объединенную органическую фазу промывали солевым раствором (3×20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 5 до 50% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,500 г, выход 52%):1H ЯМР (400 МГц, CDCl3) δ 8,80 (д, J=2,2 Гц, 1H), 8,49 (дд, J=2,2, 5,8 Гц, 1H), 8,46-8,42 (м, 1H), 7,56 (д, J=2,2 Гц, 1H), 7,43 (т, J=8,6 Гц, 1H), 1,36 (с, 9H).
Стадия 2. Получение трет-бутил (S)-((3-циано-4-((1-(2-фторфенил)этил)амино)фенил)сульфонил)(тиазол-4-ил)карбамата

В раствор трет-бутил ((3-циано-4-фторфенил)сульфонил)(тиазол-4-ил)карбамата (0,200 г, 0,521 ммоль) в безводном N,N-диметилформамиде (3 мл) добавляли карбонат калия (0,288 г, 2,09 ммоль) и гидрохлорид (S)-1-(2-фторфенил)этанамина (0,916 г, 0,521 ммоль). Смесь перемешивали при температуре окружающей среды в течение 12 ч. Затем ее разводили насыщенным хлоридом аммония (20 мл) и экстрагировали этилацетатом (3×20 мл). Объединенные органические слои промывали солевым раствором (3×20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 40% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,120 г, выход 46%):1H ЯМР (400 МГц, CDCl3) δ 8,78 (д, J=2,4 Гц, 1H), 8,19 (д, J=2,4 Гц, 1H), 7,98 (дд, J=9,0, 2,0 Гц, 1H), 7,50 (д, J=2,4 Гц, 1H), 7,31-7,27 (м, 2H), 7,17-7,08 (м, 2H), 6,58 (д, J=9,2 Гц, 1H), 5,47 (ушир. д, J=6,2 Гц, 1H), 5,01 (т, J=6,6 Гц, 1H), 1,69 (д, J=6,8 Гц, 3H), 1,34 (с, 9H).
Стадия 3. Получение (S)-3-циано-4-((1-(2-фторфенил)этил)амино)- N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил (S)-((3-циано-4-((1-(2-фторфенил)этил)амино)фенил)сульфонил)(тиазол-4-ил)карбамата (0,120 г, 0,239 ммоль) в дихлорметане (10 мл) добавляли трифторуксусную кислоту (3,08 г, 27,0 ммоль, 2 мл). Смесь перемешивали при температуре окружающей среды в течение 12 ч и затем концентрировали in vacuo. Остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,074 г, выход 69%):1H ЯМР (400 МГц, CDCl3) δ 10,28 (с, 1H), 8,72 (д, J=2,4 Гц, 1H), 7,82 (д, J=2,0 Гц, 1H), 7,59 (дд, J=9,0, 1,8 Гц, 1H), 7,28-7,21 (м, 2H), 7,16-7,04 (м, 2H), 6,97 (д, J=2,2 Гц, 1H), 6,44 (д, J=9,0 Гц, 1H), 5,34 (ушир. д, J=6,2 Гц, 1H), 4,91 (квин, J=6,6 Гц, 1H), 1,63 (д, J=6,8 Гц, 3H); MS (ES+) m/z 403,0 (M+1), 425,0 (M+23).
ПРИМЕР 240
Синтез (S)-4-((1-(2,5-дифторфенил)этил)амино)-2,6-дифтор-N-(изоксазол-3-ил)бензолсульфонамида

Стадия 1. Получение 2,4,6-трифтор-N-(изоксазол-3-ил)бензолсульфонамида
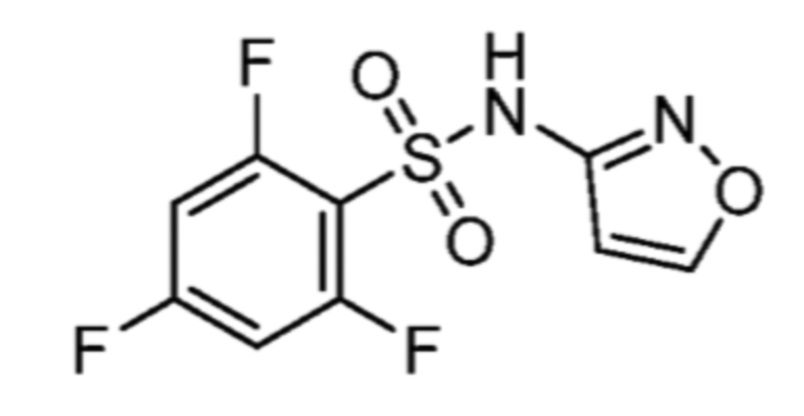
В смесь изоксазол-3-амина (1,00 г, 11,9 ммоль, 0,877 мл), 4-(диметиламино)пиридина (0,291 г, 2,38 ммоль) и пиридина (1,88 г, 23,8 ммоль, 1,92 мл) в безводном дихлорметане (20 мл) добавляли раствор 2,4,6-трифторбензолсульфонилхлорида (3,02 г, 13,1 ммоль) в дихлорметане (4 мл) при 0°C. Смесь перемешивали при 0°C в течение 30 минут и температуре окружающей среды в течение 12 ч. Смесь концентрировали in vacuo и остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде твердого вещества желтого цвета (1,18 г, выход 36%):1H ЯМР (400 МГц, CDCl3) δ 8,28 (д, J=1,8 Гц, 1H), 6,81 (т, J=8,4 Гц, 2H), 6,61 (д, J=1,8 Гц, 1H), NH не наблюдали.
Стадия 2. Получение 2,4,6-трифтор-N-(изоксазол-3-ил)-N-((2-(триметилсилил)этокси)метил)-бензолсульфонамида

В смесь 2,4,6-трифтор-N-(изоксазол-3-ил)бензолсульфонамида (1,18 г, 4,24 ммоль) и карбоната калия (1,17 г, 8,48 ммоль) в безводном N,N-диметилформамиде (10 мл) добавляли (2-(хлорметокси)этил)триметилсилан (0,849 г, 5,09 ммоль, 0,902 мл) при 0°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 1 ч. Остаток выливали в воду со льдом (50 мл) и водную фазу экстрагировали этилацетатом (3×30 мл). Объединенную органическую фазу промывали солевым раствором (3×30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием 10% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного масла (1,60 г, выход 92%):1H ЯМР (400 МГц, CDCl3) δ 8,30 (д, J=1,8 Гц, 1H), 6,78 (т, J=8,6 Гц, 2H), 6,65 (д, J=1,8 Гц, 1H), 5,44 (с, 2H), 3,77-3,69 (м, 2H), 0,98-0,86 (м, 2H), 0,05 (с, 9H); MS (ES+) m/z 430,9 (M+23).
Стадия 3. Получение (S)-4-((1-(2,5-дифторфенил)этил)амино)-2,6-дифтор-N-(изоксазол-3-ил)-N-((2-(триметилсилил)этокси)метил)бензолсульфонамида

Смесь 2,4,6-трифтор-N-(изоксазол-3-ил)-N-((2-(триметилсилил)этокси)метил)бензолсульфонамида (0,250 г, 0,612 ммоль), гидрохлорида (S)-1-(2,5-дифторфенил)этанамина (0,142 г, 0,734 ммоль) и карбоната калия (0,338 г, 2,45 ммоль) в безводном N,N-диметилформамиде (6 мл) перемешивали при 60°C в течение 12 ч. Смесь выливали в воду со льдом (30 мл) и водную фазу экстрагировали этилацетатом (3×30 мл). Объединенную органическую фазу промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 20% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного масла (0,150 г, выход 45%):1H ЯМР (400 МГц, CDCl3) δ 8,24 (д, J=2,0 Гц, 1H), 7,07 (тд, J=9,2, 4,6 Гц, 1H), 7,02-6,93 (м, 2H), 6,65 (д, J=1,8 Гц, 1H), 6,02 (д, J=11,6 Гц, 2H), 5,44 (с, 2H), 4,75 (ушир. с, 1H), 3,75-3,72 (м, 2H), 3,04-2,90 (м, 1H), 1,57 (ушир. с, 3H), 0,97-0,91 (м, 2H), 0,00 (с, 9H); MS (ES+) m/z 546,1 (M+1).
Стадия 4. Получение (S)-4-((1-(2,5-дифторфенил)этил)амино)-2,6-дифтор-N-(изоксазол-3-ил)бензолсульфонамида

В раствор (S)-4-((1-(2,5-дифторфенил)этил)амино)-2,6-дифтор-N-(изоксазол-3-ил)-N-((2-(триметилсилил)этокси)метил)бензолсульфонамида (0,150 г, 0,275 ммоль) в диоксане (5 мл) добавляли 4 M раствор HCl в диоксане (15 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 12 ч. Смесь концентрировали in vacuo и остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,049 г, выход 43%):1H ЯМР (400 МГц, CDCl3) δ 8,21 (д, J=1,6 Гц, 1H), 7,07 (дт, J=4,4, 9,2 Гц, 1H), 6,98-6,87 (м, 2H), 6,58 (д, J=1,6 Гц, 1H), 6,02 (д, J=11,6 Гц, 2H), 4,82 (ушир. д, J=5,6 Гц, 1H), 4,72 (квин, J=6,6 Гц, 1H), 1,56 (д, J=6,8 Гц, 3H), NH не наблюдали; MS (ES+) m/z 416,0 (M+1).
ПРИМЕР 241
Синтез 5-хлор-4-((1-(2,5-дифторфенил)циклопропил)амино)-2-фтор-N-(пиримидин-4-ил)бензолсульфонамида

Стадия 1. Получение 5-хлор-4-((1-(2,5-дифторфенил)циклопропил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(пиримидин-4-ил)бензолсульфонамида

В смесь 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(пиримидин-4-ил)бензолсульфонамида (0,200 г, 0,441 ммоль) и гидрохлорида 1-(2,5-дифторфенил)циклопропан-1-амина (0,091 г, 0,44 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли карбонат калия (0,207 г, 1,50 ммоль) и реакционную смесь перемешивали при 70°C в течение 18 ч. Смесь охлаждали до температуры окружающей среды и разводили этилацетатом (5 мл) и водой (5 мл). Слои разделяли и водную фазу экстрагировали этилацетатом (3×5 мл). Объединенную органическую фазу промывали солевым раствором (1×5 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 60% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного масла (0,107 г, выход 40%): MS (ES+) m/z 605,4 (M+1), 607,4 (M+1).
Стадия 2. Получение 5-хлор-4-((1-(2,5-дифторфенил)циклопропил)амино)-2-фтор-N-(пиримидин-4-ил)бензолсульфонамида

Следуя процедуре как описано для примера 222, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять трет-бутил (S)-((5-хлор-4-((1-(2-хлорфенил)пропил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамат на 5-хлор-4-((1-(2,5-дифторфенил)циклопропил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(пиримидин-4-ил)бензолсульфонамид, очисткой с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,029 г, выход 35%):1H-ЯМР (300 МГц, DMSO-d6) δ 8,58 (с,1H), 8,30 (ушир. с, 1H), 7,72-7,69 (м, 1H), 7,53-7,47 (м, 1H), 7,33-7,29 (м, 1H), 7,25-7,10 (м, 2H), 7,00-6,94 (м, 1H), 6,83-6,79 (м, 1H), 1,43-1,40 (м, 2H), 1,28-1,25 (м, 2H), NH не наблюдали; MS (ES+) m/z 455,2 (M+1), 457,2 (M+1).
ПРИМЕР 242
Синтез 5-хлор-2-фтор-4-((1-(2-фторфенил)циклопропил)амино)-N-(пиримидин-4-ил)бензолсульфонамида

Стадия 1. Получение 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(2-фторфенил)циклопропил)амино)-N-(пиримидин-4-ил)бензолсульфонамида

Следуя процедуре как описано для примера 241, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять гидрохлорид 1-(2,5-дифторфенил)циклопропан-1-амина на гидрохлорид 1-(2-фторфенил)циклопропан-1-амина, указанное в заголовке соединение получали в виде бесцветного масла (0,101 г, выход 39%): MS (ES+) m/z 587,4 (M+1), 589,4 (M+1).
Стадия 2. Получение 5-хлор-2-фтор-4-((1-(2-фторфенил)циклопропил)амино)-N-(пиримидин-4-ил)бензолсульфонамида

Следуя процедуре как описано для примера 241, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 5-хлор-4-((1-(2,5-дифторфенил)циклопропил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(пиримидин-4-ил)бензолсульфонамид на 5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(2-фторфенил)циклопропил)амино)-N-(пиримидин-4-ил)бензолсульфонамид, очисткой посредством растирания с метанолом (3×5 мл) указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,047 г, выход 63%):1H-ЯМР (300 МГц, DMSO-d6) δ 8,57 (с, 1H), 8,30 (ушир. с, 1H), 7,69 (д, J=7,3 Гц, 1H), 7,61-7,55 (м, 1H), 7,31-7,23 (м, 2H), 7,16-7,10 (м, 2H), 6,98-6,95 (м, 1H), 6,77 (д, J=12,9 Гц, 1H), 1,38-1,37 (м, 2H), 1,27-1,23 (м, 2H), NH не наблюдали; MS (ES+) m/z 437,2 (M+1), 439,2 (M+1).
ПРИМЕР 243
Синтез (S)-5-хлор-4-((1-(2-хлор-5-фторфенил)этил)амино)-2-фтор-N-(пиразин-2-ил)бензолсульфонамида

Стадия 1. Получение N-(2,4-диметоксибензил)пиразин-2-амина

Смесь пиразин-2-амин (2,000 г, 21,03 ммоль), 2,4-диметоксибензальдегида (3,851 г, 23,19 ммоль) и триацетоксиборгидрида натрия (6,233 г, 29,54 ммоль) в дихлорметане (90 мл) перемешивали при температуре окружающей среды в течение 18 ч. Затем туда добавляли воду (50 мл) и разделяли слои. Водную фазу экстрагировали дихлорметаном (3×40 мл). Объединенную органическую фазу промывали солевым раствором (50 мл), сушили с использованием сульфата натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 10 до 80% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного масла (2,914 г, выход 56%):1H-ЯМР (300 МГц, CDCl3) δ 7,97 (дд, J=2,8, 1,5 Гц, 1H), 7,88 (д, J=1,5 Гц, 1H), 7,76 (д, J=2,8 Гц, 1H), 7,21 (д, J=8,2 Гц, 1H), 6,47 (д, J=2,4 Гц, 1H), 6,42 (дд, J=8,2, 2,4 Гц, 1H), 5,07-5,06 (м, 1H), 4,46 (д, J=5,8 Гц, 2H), 3,83 (с, 3H), 3,79 (с, 3H).
Стадия 2. Получение 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(пиразин-2-ил)бензолсульфонамида

В раствор N-(2,4-диметоксибензил)пиразин-2-амина (2,411 г, 9,840 ммоль) в безводном тетрагидрофуране (30 мл) добавляли 1,0 M раствор бис(триметилсилил)амида лития в тетрагидрофуране (10,3 мл, 10,3 ммоль) при -78°C. Реакционную смесь нагревали до температуры окружающей среды в течение 30 минут, охлаждали до -78°C и затем туда добавляли раствор 5-хлор-2,4-дифторбензолсульфонилхлорида (2,430 г, 9,841 ммоль) в безводном тетрагидрофуране (10 мл). Смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 18 ч. Затем туда добавляли насыщенный хлорид аммония (20 мл) и смесь экстрагировали этилацетатом (3×20 мл). Объединенную органическую фазу промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 5 до 80% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного масла (1,408 г, выход 31%):1H-ЯМР (300 МГц, CDCl3) δ 8,63 (д, J=1,3 Гц, 1H), 8,37 (д, J=2,5 Гц, 1H), 8,29 (дд, J=2,5, 1,5 Гц, 1H), 7,94 (дд, J=7,8, 7,0 Гц, 1H), 7,18 (д, J=8,3 Гц, 1H), 6,99 (дд, J=9,2, 8,4 Гц, 1H), 6,38-6,33 (м, 2H), 5,03 (с, 2H), 3,75 (с, 3H), 3,65 (с, 3H).
Стадия 3. Получение (S)-5-хлор-4-((1-(5-хлор-2-фторфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(пиразин-2-ил)бензолсульфонамида

В смесь 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(пиразин-2-ил)бензолсульфонамида (0,250 г, 0,548 ммоль) и (S)-1-(5-хлор-2-фторфенил)этан-1-амина (0,115 г, 0,548 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли карбонат цезия (0,178 г, 1,86 ммоль). Реакционную смесь перемешивали при 70°C в течение 18 ч. Смесь охлаждали до температуры окружающей среды и разводили этилацетатом (5 мл) и водой (5 мл). Слои разделяли и водную фазу экстрагировали этилацетатом (3×5 мл). Объединенную органическую фазу промывали солевым раствором (1×5 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 5 до 60% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного масла (0,084 г, выход 25%): MS (ES+) m/z 609,4 (M+1), 611,4 (M+1).
Стадия 4. Получение (S)-5-хлор-4-((1-(2-хлор-5-фторфенил)этил)амино)-2-фтор-N-(пиразин-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 222, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять трет-бутил (S)-((5-хлор-4-((1-(2-хлорфенил)пропил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамат на (S)-5-хлор-4-((1-(5-хлор-2-фторфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(пиразин-2-ил)бензолсульфонамид, очисткой с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 5 до 60% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,050 г, выход 20%):1H-ЯМР (300 МГц, DMSO-d6) δ 11,70 (с, 1H), 8,29 (д, J=1,4 Гц, 1H), 8,20 (д, J=2,7 Гц, 1H), 8,19-8,17 (м, 1H), 7,76 (д, J=7,5 Гц, 1H), 7,50 (дд, J=6,5, 2,7 Гц, 1H), 7,36 (ддд, J=8,7, 4,6, 2,7 Гц, 1H), 7,26 (дд, J=9,8, 8,8 Гц, 1H), 6,79-6,76 (м, 1H), 6,41 (д, J=13,4 Гц, 1H), 4,98-4,88 (м, 1H), 1,54 (д, J=6,7 Гц, 3H); MS (ES+) m/z: 459,0 (M+1), 461,0 (M+1).
ПРИМЕР 244
Синтез (S)-5-хлор-4-((1-(2-хлорфенил)этил)амино)-2-фтор-N-(6-метилпиримидин-4-ил)бензолсульфонамида

Стадия 1. Получение 2-хлор-N-(2,4-диметоксибензил)-6-метилпиримидин-4-амина

Раствор 2,4-дихлор-6-метилпиримидина (3,931 г, 24,12 ммоль), (2,4-диметоксифенил)метанамина (4,430 г, 26,53 ммоль) и триэтиламина (17 мл, 121 ммоль) в ацетонитриле (92 мл) перемешивали при температуре окружающей среды в течение 18 ч. Получаемую суспензию фильтровали и фильтрат концентрировали in vacuo. Остаток растворяли в дихлорметане (50 мл), промывали водой (3×30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 10 до 80% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (4,020 г, выход 57%):1H-ЯМР (300 МГц, CDCl3) δ 7,17-7,14 (м, 1H), 6,47-6,44 (м, 1H), 6,42 (д, J=2,3 Гц, 1H), 6,10 (ушир. с, 1H), 4,39 (ушир. с, 2H), 3,82 (с, 3H), 3,80 (с, 3H), 2,30 (с, 3H), NH не наблюдали.
Стадия 2. Получение N-(2,4-диметоксибензил)-6-метилпиримидин-4-амина

В смесь 2-хлор-N-(2,4-диметоксибензил)-6-метилпиримидин-4-амина (4,020 г, 13,41 ммоль) в этаноле (30 мл) добавляли Pd/C (10% влажн., 0,402 г) и формиат аммония (1,246 г. 19,78 ммоль). Смесь затем нагревали до 80°C и перемешивали в течение 18 ч. После охлаждения до температуры окружающей среды, смесь фильтровали через подушку из целита. Фильтрат концентрировали in vacuo для получения остатка, который разделялся между водой (30 мл) и этилацетатом (30 мл). Органическую фазу промывали водой (2×20 мл), солевым раствором (1×20 мл), сушили с использованием сульфата натрия и фильтровали. Концентрированием in vacuo получали указанное в заголовке соединение в виде бесцветного порошка (2,787 г, выход 78%):1H-ЯМР (300 МГц, CDCl3) δ 8,30 (с, 1H), 7,71-7,67 (м, 1H), 7,08 (д, J=8,3 Гц, 1H), 6,55 (д, J=2,4 Гц, 1H), 6,46 (дд, J=8,3, 2,4 Гц, 1H), 6,38-6,37 (м, 1H), 4,38-4,35 (м, 2H), 3,79 (с, 3H), 3,79-3,72 (с, 3H), 2,19 (с, 3H).
Стадия 3. Получение 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(6-метилпиримидин-4-ил)бензолсульфонамида

В раствор N-(2,4-диметоксибензил)-6-метилпиримидин-4-амина (1,002 г, 3,85 ммоль) в тетрагидрофуране (15 мл) добавляли 1,0 M раствор бис(триметилсилил)амида лития в тетрагидрофуране (4 мл, 4,0 ммоль) при -78°C. Смесь перемешивали в течение 10 минут при -78°C и затем нагревали до температуры окружающей среды в течение 30 минут. Суспензию охлаждали до -78°C и туда добавляли раствор 5-хлор-2,4-дифторбензолсульфонилхлорида (0,951 г, 3,85 ммоль) в тетрагидрофуране (5 мл). Смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 18 ч. Туда добавляли насыщенный хлорид аммония (20 мл) и смесь экстрагировали этилацетатом (3×20 мл). Объединенную органическую фазу промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 10 до 70% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного масла (1,275 г, выход 70%):1H-ЯМР (300 МГц, CDCl3) δ 8,66 (с, 1H), 8,12 (т, J=7,5 Гц, 1H), 7,22 (д, J=9,0 Гц, 1H), 7,00-6,96 (м, 2H), 6,44-6,40 (м, 2H), 5,19 (с, 2H), 3,79 (д, J=5,3 Гц, 6H), 2,42 (с, 3H).
Стадия 4. Получение (S)-5-хлор-4-((1-(2-хлорфенил)этил)амино)-2-фтор-N-(6-метилпиримидин-4-ил)бензолсульфонамида

В раствор 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(6-метилпиримидин-4-ил)бензолсульфонамида (0,250 г, 0,531 ммоль) и гидрохлорида (S)-1-(2-хлорфенил)этан-1-амина (0,101 г, 0,526 ммоль) в диметилсульфоксиде (5 мл) добавляли триэтиламин (0,30 мл, 2,1 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 20 ч. Смесь разводили этилацетатом (5 мл) и водой (5 мл). Слои разделяли и водную фазу экстрагировали этилацетатом (3×3 мл). Объединенную органическую фазу промывали солевым раствором (1×5 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 5 до 60% этилацетата в гексанах, получали бесцветное масло. Затем в него добавляли дихлорметан (5 мл) и трифторуксусную кислоту (1 мл) и смесь перемешивали при температуре окружающей среды в течение 1 ч. Смесь концентрировали in vacuo и очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 20 до 80% этилацетата в гексанах, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,177 г, выход 73%):1H-ЯМР (300 МГц, DMSO-d6) δ 8,45 (с, 1H), 7,73 (д, J=7,4 Гц, 1H), 7,48-7,44 (м, 2H), 7,34-7,24 (м, 2H), 6,87-6,80 (м, 2H), 6,03 (д, J=13,0 Гц, 1H), 4,93-4,85 (м, 1H), 2,29 (с, 3H), 1,54 (д, J=6,7 Гц, 3H), NH не наблюдали; MS (ES+) m/z 455,1 (M+1), 457,1 (M+1).
ПРИМЕР 245
Синтез (S)-5-хлор-4-((1-(2-хлорфенил)этил)амино)-2-фтор-N-(2-(трифторметил)пиримидин-4-ил)бензолсульфонамида

Стадия 1. Получение 5-хлор-N-(2,4-диметоксибензил)-2,4-дифторбензолсульфонамида

Смесь 5-хлор-2,4-дифторбензолсульфонилхлорида (5,000 г, 20,2 ммоль), (2,4-диметоксифенил)метанамина (3,38 г, 20,2 ммоль) и N,N-диизопропилэтиламина (4,2 мл, 24 ммоль) в безводном дихлорметане (100 мл) перемешивали при температуре окружающей среды в течение 18 ч. Затем смесь промывали 1 Н соляной кислотой (50 мл), насыщенным хлоридом аммония (50 мл) и солевым раствором (50 мл). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и кристаллизацией остатка из дихлорметана и гексанов получали указанное в заголовке соединение в виде кристаллов бледно-желтого цвета (5,26 г, выход 69%): MS (ES-) m/z 376,2 (M - 1), 378,2 (M - 1).
Стадия 2. Получение 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(2-(трифторметил)пиримидин-4-ил)бензолсульфонамида

Смесь 5-хлор-N-(2,4-диметоксибензил)-2,4-дифторбензолсульфонамида (1,000 г, 2,65 ммоль), 4-хлор-2-(трифторметил)пиримидина (0,482 г, 2,65 ммоль) и карбоната калия (0,549 г, 3,98 ммоль) в безводном диметилсульфоксиде (20 мл) перемешивали при 50°C в течение 18 ч. Туда добавляли насыщенный хлорид аммония (10 мл) и смесь экстрагировали этилацетатом (3×10 мл). Объединенную органическую фазу промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 5 до 60% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,919 г, выход 66%):1H-ЯМР (300 МГц, DMSO-d6) δ 8,94 (д, J=5,1 Гц, 1H), 8,13 (т, J=7,6 Гц, 1H), 7,86 (т, J=9,8 Гц, 1H), 7,65 (д, J=4,9 Гц, 1H), 7,04 (д, J=8,2 Гц, 1H), 6,59 (д, J=2,2 Гц, 1H), 6,49 (дд, J=8,4, 2,2 Гц, 1H), 5,30 (с, 2H), 3,77 (с, 3H), 3,74 (с, 3H).
Стадия 3. Получение (S)-5-хлор-4-((1-(2-хлорфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(2-(трифторметил)пиримидин-4-ил)бензолсульфонамида

В смесь 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(2-(трифторметил)пиримидин-4-ил)бензолсульфонамида (0,250 г, 0,478 ммоль) и гидрохлорида (S)-1-(2-хлорфенил)этан-1-амина (0,092 г, 0,48 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли триэтиламин (0,27 мл, 1,9 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 22 ч. Смесь разводили этилацетатом (5 мл) и водой (5 мл). Слои разделяли и водную фазу экстрагировали этилацетатом (3×3 мл). Объединенную органическую фазу промывали солевым раствором (1×5 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 60% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного масла (0,198 г, выход 62%): MS (ES) m/z 659,3 (M+1), 661,2 (M+1).
Стадия 4. Получение (S)-5-хлор-4-((1-(2-хлорфенил)этил)амино)-2-фтор-N-(2-(трифторметил)пиримидин-4-ил)бензолсульфонамида

В смесь (S)-5-хлор-4-((1-(2-хлорфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(2-(трифторметил)пиримидин-4-ил)бензолсульфонамида (0,198 г, 0,301 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (1 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Туда добавляли метанол (10 мл) и получаемый осадок удаляли фильтрованием. Фильтрат концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 10 до 80% этилацетата в гексанах, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,104 г, выход 68%):1H-ЯМР (300 МГц, DMSO-d6) δ 12,57 (с, 1H), 8,84 (д, J=4,8 Гц, 1H), 7,79 (д, J=7,4 Гц, 1H), 7,49 (д, J=4,8 Гц, 1H), 7,47-7,43 (м, 2H), 7,32-7,23 (м, 2H), 6,96-6,93 (м, 1H), 6,05 (д, J=13,4 Гц, 1H), 4,95-4,85 (м, 1H), 1,53 (д, J=6,7 Гц, 3H); MS (ES+) m/z 509,0 (M+1), 511,0 (M+1).
ПРИМЕР 246
Синтез 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Синтез 2-(азетидин-1-илметил)-6-фторбензонитрила

В смесь азетидина (0,266 г, 7,01 ммоль) и 2-(бромметил)-6-фторбензонитрила (1,00 г, 4,67 ммоль) в дихлорметане (30 мл) добавляли N,N-диизопропилэтиламин (1,2 мл, 7,0 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 18 ч. Затем туда добавляли насыщенный хлорид аммония (20 мл) и смесь экстрагировали дихлорметаном (3×20 мл). Объединенную органическую фазу промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 20% метанола в дихлорметане, получали указанное в заголовке соединение в виде бесцветного масла (0,819 г, выход 92%);1H-ЯМР (300 МГц, CDCl3) δ 7,56-7,50 (м, 1H), 7,33-7,31 (м, 1H), 7,11-7,05 (м, 1H), 3,78 (с, 2H), 3,33-3,29 (м, 4H), 2,17-2,10 (м, 2H).
Стадия 2. Синтез (2-(азетидин-1-илметил)-6-фторфенил)метанамина

В раствор 2-(азетидин-1-илметил)-6-фторбензонитрила (0,819 г, 4,51 ммоль) в безводном тетрагидрофуране (40 мл) добавляли гидрид лития алюминия (1,0 M в тетрагидрофуране, 6,8 мл, 6,8 ммоль) при 0°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 4 ч. Затем туда добавляли декагидрат сульфата натрия (5,0 г) частями при 0°C. Смесь перемешивали в течение 30 минут и затем фильтровали. Фильтровальный осадок промывали этилацетатом (20 мл). Объединенный фильтрат концентрировали in vacuo для того, чтобы получать указанное в заголовке соединение в виде масла красного цвета (0,811 мг, выход 99%):1H-ЯМР (300 МГц, CDCl3) δ 7,13 (тд, J=7,8, 5,6 Гц, 1H), 7,01-6,93 (м, 2H), 3,85 (д, J=1,9 Гц, 2H), 3,60 (с, 2H), 3,16 (т, J=7,0 Гц, 4H), 2,03 (квинтет, J=7,0 Гц, 2H).
Стадия 3. Синтез трет-бутил ((4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-5-хлор-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата

Смесь (2-(азетидин-1-илметил)-6-фторфенил)метанамина (0,152 г, 0,780 ммоль), трет-бутил ((5-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,321 г, 0,0.780 ммоль) и карбоната калия (0,258 г, 1,46 ммоль) в безводном диметилсульфоксиде (5 мл) перемешивали при температуре окружающей среды в течение 3 ч. Затем туда добавляли воду (5 мл) и этилацетат (5 мл). Водную фазу экстрагировали этилацетатом (3×5 мл). Объединенную органическую фазу промывали солевым раствором (5 мл), сушили с использованием безводного сульфата натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 20 до 80% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного масла (0,223 г, выход 49%): MS (ES+) m/z 585,4 (M+1), 587,4 (M+1).
Стадия 4: синтез 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил ((4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-5-хлор-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата (0,223 г, 0,382 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (1 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Смесь концентрировали in vacuo для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,197 г, выход 86%):1H-ЯМР (300 МГц, DMSO-d6) δ 11,18 (ушир. с, 1H), 10,10 (ушир. с, 1H), 8,89 (д, J=2,1 Гц, 1H), 7,62 (д, J=7,4 Гц, 1H), 7,52-7,45 (м, 1H), 7,37-7,30 (м, 2H), 7,00 (д, J=2,4 Гц, 1H), 6,84-6,82 (м, 1H), 6,77-6,72 (д, J=12,9 Гц, 1H), 4,53-4,48 (м, 4H), 4,12-4,01 (м, 4H), 2,39-2,27 (м, 2H); MS (ES+) m/z 485,1 (M+1), 487,1 (M+1).
ПРИМЕР 247
Синтез 5-хлор-2-фтор-4-((2-фтор-6-((3-фторазетидин-1-ил)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Синтез 2-фтор-6-((3-фторазетидин-1-ил)метил)бензонитрила

Следуя процедуре как описано для примера 246, стадии 1 и выполняя некритические вариации, чтобы заменять азетидин на гидрохлорид 3-фторазетидина, указанное в заголовке соединение выделяли в виде бесцветного масла (0,815 г, выход 93%);1H-ЯМР (300 МГц, CDCl3) δ 7,56 (тд, J=8,1, 5,7 Гц, 1H), 7,31 (д, J=7,8 Гц, 1H), 7,11 (т, J=8,5 Гц, 1H), 5,16 (дквинтет, J=57,2, 5,3 Гц, 1H), 3,87 (с, 2H), 3,77-3,68 (м, 2H), 3,38-3,25 (м, 2H).
Стадия 2. Синтез (2-фтор-6-((3-фторазетидин-1-ил)метил)фенил)метанамина

Следуя процедуре как описано для примера 246, стадии 2 и выполняя некритические вариации, чтобы заменять 2-(азетидин-1-илметил)-6-фторбензонитрил на 2-фтор-6-((3-фторазетидин-1-ил)метил)бензонитрил, указанное в заголовке соединение выделяли в виде масла оранжевого цвета (0,776 г, выход 89%):1H-ЯМР (300 МГц, CDCl3) δ 7,31-7,24 (м, 1H), 7,19-7,14 (м, 1H), 7,05-6,99 (м, 1H), 5,22-4,97 (м, 1H), 3,88-3,85 (м, 2H), 3,74-3,71 (м, 2H), 3,66-3,57 (м, 2H), 3,26-3,13 (м, 2H), NH не наблюдали.
Стадия 3. Синтез трет-бутил ((5-хлор-2-фтор-4-((2-фтор-6-((3-фторазетидин-1-ил)метил)бензил)амино)фенил)сульфонил)(тиазол-4-ил)карбамата

Следуя процедуре как описано для примера 246, стадии 3 и выполняя некритические вариации при необходимости, чтобы заменять (2-(азетидин-1-илметил)-6-фторфенил)метанамин на (2-фтор-6-((3-фторазетидин-1-ил)метил)фенил)метанамин, указанное в заголовке соединение выделяли в виде бесцветного масла (0,187 г, выход 44%): MS (ES-) m/z 603,4 (M-1), 605,4 (M-1).
Стадия 4. Синтез 5-хлор-2-фтор-4-((2-фтор-6-((3-фторазетидин-1-ил)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида
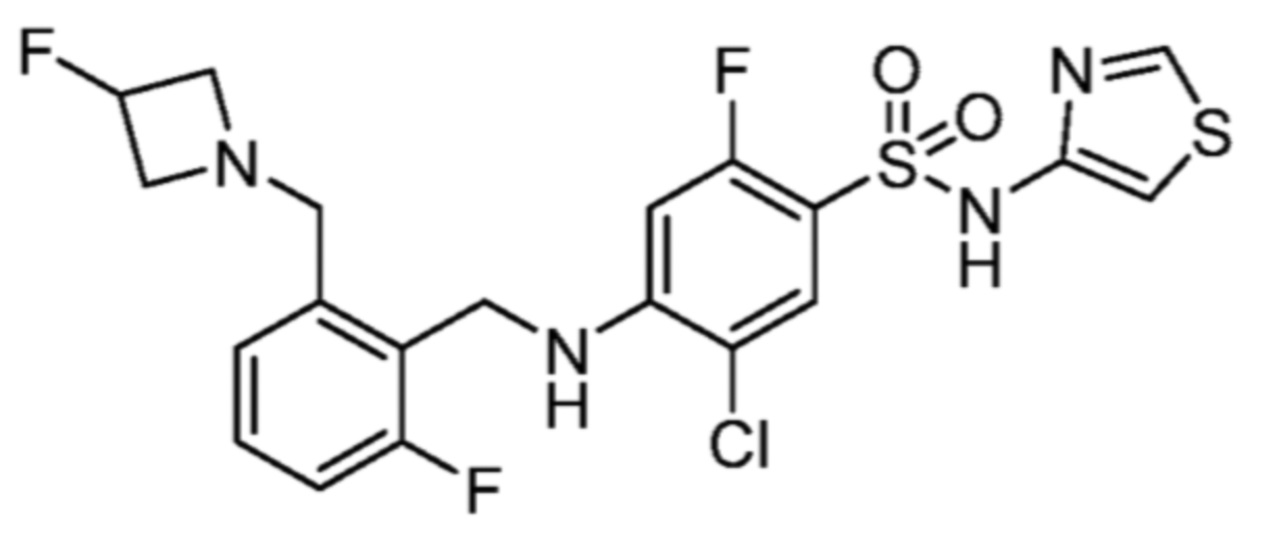
Следуя процедуре как описано для примера 246, стадии 4 и выполняя некритические вариации при необходимости, чтобы заменять трет-бутил ((4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-5-хлор-2-фторфенил)сульфонил)(тиазол-4-ил)карбамат на трет-бутил ((5-хлор-2-фтор-4-((2-фтор-6-((3-фторазетидин-1-ил)метил)бензил)амино)фенил)сульфонил)(тиазол-4-ил)карбамат, очисткой с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,506 г, выход 14%):1H-ЯМР (300 МГц, DMSO-d6) δ 11,16 (с, 1H), 8,89 (д, J=2,2 Гц, 1H), 7,61 (д, J=7,4 Гц, 1H), 7,51-7,35 (м, 1H), 7,33-7,30 (м, 2H), 7,00 (д, J=2,2 Гц, 1H), 6,87-6,71 (м, 2H), 5,50-5,22 (м, 2H) 4,76-4,19 (м, 7H); MS (ES+) m/z 503,2 (M+1)., 505,2 (M+1).
ПРИМЕР 248
Синтез 2,2,2-трифторацетата 5-хлор-2-фтор-4-((2-фтор-6-((3-метоксиазетидин-1-ил)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 2-фтор-6-((3-метоксиазетидин-1-ил)метил)бензонитрила

Следуя процедуре как описано для примера 246, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять азетидин на гидрохлорид 3-метоксиазетидина, указанное в заголовке соединение выделяли в виде бесцветного масла (0,287 г, выход 55%):1H-ЯМР (300 МГц, CDCl3) δ 7,56 (тд, J=8,1, 5,7 Гц, 1H), 7,33 (д, J=7,4, Гц, 1H), 7,14-7,08 (м, 1H), 4,15-4,06 (м, 1H), 3,86 (с, 2H), 3,71-3,66 (м, 2H), 3,27 (с, 3H), 3,12-3,08 (м, 2H).
Стадия 2. Получение (2-фтор-6-((3-метоксиазетидин-1-ил)метил)фенил)метанамина

Следуя процедуре как описано для примера 246, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 2-(азетидин-1-илметил)-6-фторбензонитрил на азетидин на 2-фтор-6-((3-метоксиазетидин-1-ил)метил)бензонитрил, указанное в заголовке соединение выделяли в виде бесцветного масла, которое использовали без дополнительной очистки.
Стадия 3. Получение трет-бутил ((5-хлор-2-фтор-4-((2-фтор-6-((3-метоксиазетидин-1-ил)метил)бензил)амино)фенил)сульфонил)(тиазол-4-ил)карбамата

Следуя процедуре как описано для примера 246, стадии 3 и выполняя некритические вариации при необходимости, чтобы заменять (2-(азетидин-1-илметил)-6-фторфенил)метанамин метанамин на (2-фтор-6-((3-метоксиазетидин-1-ил)метил)фенил)метанамин, указанное в заголовке соединение выделяли в виде бесцветного масла (0,159 г, выход 42%): MS (ES+) m/z 615,2 (M+1), 617,2 (M+1).
Стадия 4. Получение 2,2,2-трифторацетата 5-хлор-2-фтор-4-((2-фтор-6-((3-метоксиазетидин-1-ил)метил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано для примера 246, стадии 4 и выполняя некритические вариации при необходимости, чтобы заменять трет-бутил ((4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-5-хлор-2-фторфенил)сульфонил)(тиазол-4-ил)карбамат на трет-бутил ((5-хлор-2-фтор-4-((2-фтор-6-((3-метоксиазетидин-1-ил)метил)бензил)амино)фенил)сульфонил)(тиазол-4-ил)карбамат, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,068 г, выход 42%):1H-ЯМР (300 МГц, DMSO-d6) δ 11,19 (с, 1H), 10,61-10,45 (м, 0,5H), 10,14-9,98 (м, 0,5H), 8,89 (д, J=2,2 Гц, 1H), 7,61 (д, J=7,2 Гц, 1H), 7,51-7,44 (м, 1H), 7,37-7,30 (м, 2H), 7,00 (м, J=2,1 Гц, 1H), 6,84-6,77 (м, 1H), 6,74 (д, J=12,6 Гц, 1H), 4,59-4,53 (м, 2H), 4,49-4,45 (м, 2H), 4,33-4,21 (м, 3H), 4,04-3,97 (м, 2H), 3,24-3,21 (м, 3H); MS (ES+) m/z 515,2, 517,2 (M+1).
ПРИМЕР 249
Синтез 2,2,2-трифторацетата 5-хлор-4-((2-((диметиламино)метил)-6-фторбензил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил ((4-((2-бром-6-фторбензил)амино)-5-хлор-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата

В смесь (2-бром-6-фторфенил)метанамина (0,995 г, 4,88 ммоль) и трет-бутил ((5-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (2,000 г, 4,88 ммоль) в безводном диметилсульфоксиде (40 мл) добавляли карбонат калия (1,643 г, 11,91 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 4 ч. Затем туда добавляли насыщенный хлорид аммония (20 мл) и смесь экстрагировали этилацетатом (3×30 мл). Объединенную органическую фазу промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 10 до 80% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (1,078 г, выход 37%):1H-ЯМР (300 МГц, CDCl3) δ 8,79 (д, J=2,3 Гц, 1H), 7,93 (д, J=7,1 Гц, 1H), 7,50 (д, J=2,3 Гц, 1H), 7,43 (дд, J=7,9, 0,8 Гц, 1H), 7,24-7,19 (м, 1H), 7,14-7,08 (м, 1H), 6,69-6,65 (м, 1H), 5,60-5,55 (м, 1H), 4,61-4,59 (м, 2H), 1,36 (с, 9H); MS (ES+) m/z 494,2 (M+1), 496,2 (M+1).
Стадия 2. Получение 2,2,2-трифторацетата 5-хлор-4-((2-((диметиламино)метил)-6-фторбензил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

В смесь трет-бутил ((4-((2-бром-6-фторбензил)амино)-5-хлор-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата (0,200 г, 0,338 ммоль), диметиламинометилтрифторбороната калия (0,067 г, 0,41 ммоль), карбоната цезия (0,330 г, 1,01 ммоль), ацетата палладия(II) (0,008 г, 0,03 ммоль) и ди(1-адамантил)-н-бутилфосфина (0,024 г, 0,068 ммоль) добавляли дегазированную смесь воды (0,53 мл) и диоксана (2,6 мл). Реакционную смесь до 85°C и перемешивали в течение 18 ч. После охлаждения до температуры окружающей среды, смесь фильтровали через подушку из целита. Фильтровальный осадок промывали этилацетатом (10 мл) и объединенный фильтрат концентрировали in vacuo. К остатку добавляли дихлорметан (5 мл) и трифторуксусную кислоту (1 мл) и реакционную смесь перемешивали в течение 5 ч. Концентрированием in vacuo получали остаток, который очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 20 до 100% этилацетата (содержащего 10% изопропанола и 10% триэтиламина) в гексанах. Дополнительной очисткой с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,008 г, выход 4%):1H-ЯМР (300 МГц, DMSO-d6) δ 11,17 (с, 1H), 9,67 (ушир. с, 1H), 8,89 (д, J=2,2 Гц, 1H), 7,61 (д, J=7,5 Гц, 1H), 7,56-7,48 (м, 1H), 7,45-7,29 (м, 2H), 7,00 (д, J=2,2 Гц, 1H), 6,80-6,75 (м, 2H), 4,50-4,43 (м, 4H), 2,78-2,76 (м, 6H); MS (ES+) m/z 473,1 (M+1), 475,1 (M+1).
ПРИМЕР 250
Синтез 5-хлор-2-фтор-4-((2-фтор-6-(морфолинометил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано для примера 249, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять диметиламинометилтрифторборонат калия на (морфолин-4-ил)метилтрифторборат калия, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,046 г, выход 26%):1H-ЯМР (300 МГц, DMSO-d6) δ 11,21 (с, 1H), 8,86 (д, J=2,2 Гц, 1H), 7,60 (д, J=7,4 Гц, 1H), 7,34-7,28 (м, 1H), 7,19-7,12 (м, 2H), 6,97-6,90 (м, 2H), 6,41-6,35 (м, 1H), 4,54-4,52 (м, 2H), 3,56-3,53 (м, 6H), 2,37-2,34 (м, 4H); MS (ES+) m/z 515,1 (M+1), 517,1 (M+1).
ПРИМЕР 251
Синтез 5-хлор-4-((2-((4,4-дифторпиперидин-1-ил)метил)-6-фторбензил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано для примера 249, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять диметиламинометилтрифторборонат калия на (4,4-дифторпипериденил)метилтрифторборат калия, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,043 г, выход 23%):1H-ЯМР (300 МГц, DMSO-d6) δ 11,1 (с, 1H), 8,88 (д, J=2,2 Гц, 1H), 7,61 (д, J=7,4 Гц, 1H), 7,36-7,31 (м, 1H), 7,20-7,15 (м, 2H), 6,99-6,94 (м, 2H), 6,51-6,47 (м, 1H), 4,55-4,52 (м, 2H), 3,65-3,63 (м, 2H), 3,40-3,38 (м, 2H), 2,58-2,43 (м, 2H), 1,99-1,87 (м, 4H); MS (ES+) m/z 549,0 (M+1), 551,0 (M+1).
ПРИМЕР 252
Формиат 4-((2-((2-азаспиро[3.3]гептан-2-ил)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Синтез 2-((2-азаспиро[3.3]гептан-2-ил)метил)-6-фторбензонитрила

Раствор 2-азаспиро[3.3]гептана (0,250 г, 2,34 ммоль), 2-(бромметил)-6-фторбензонитрила (0,500 г, 2,34 ммоль) и N,N-диизопропилэтиламина (610 мкл, 3,50 ммоль) в дихлорметане (10 мл) перемешивали при температуре окружающей среды в течение 18 ч. Затем туда добавляли насыщенный хлорид аммония (10 мл) и смесь экстрагировали дихлорметаном (3×10 мл). Объединенную органическую фазу промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 10 до 80% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного масла (0,413 г, выход 75%);1H-ЯМР (300 МГц, CDCl3) δ 7,53 (тд, J=8,1, 5,8 Гц, 1H), 7,33 (д, J=7,8 Гц, 1H), 7,07 (т, J=8,5 Гц, 1H), 3,77 (с, 2H), 3,28 (с, 4H), 2,12 (т, J=7,5 Гц, 4H), 1,85-1,75 (м, 2H).
Стадия 2. Синтез (2-((2-азаспиро[3.3]гептан-2-ил)метил)-6-фторфенил)метанамина:

В раствор 2-((2-азаспиро[3.3]гептан-2-ил)метил)-6-фторбензонитрила (0,413 г, 1,75 ммоль) в безводном тетрагидрофуране (20 мл) при 0°C добавляли 1,0 M раствор гидрида лития алюминия в тетрагидрофуране (5,3 мл, 5,3 ммоль). Реакционную смесь перемешивали в течение 4 ч. Затем туда частями добавляли декагидрат сульфата натрия (2,5 г) при 0°C. Смесь оставляли нагреваться до температуры окружающей среды, перемешивали в течение 30 минут и затем фильтровали. Фильтровальный осадок промывали этилацетатом (50 мл). Объединенный фильтрат концентрировали in vacuo для того, чтобы получать указанное в заголовке соединение в виде масла желтого цвета;1H-ЯМР (300 МГц, CDCl3): δ 7,16-7,09 (м, 1H), 6,99-6,93 (м, 2H), 3,84-3,81 (м, 2H), 3,69-3,66 (м, 1H), 3,58 (д, J=1,2 Гц, 2H), 3,11 (д, J=1,8 Гц, 3H), 2,29-2,17 (м, 2H), 2,05 (т, J=7,5 Гц, 4H), 1,82-1,74 (м, 2H).
Стадия 3. Синтез формиата 4-((2-((2-азаспиро[3.3]гептан-2-ил)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

В раствор (2-((2-азаспиро[3.3]гептан-2-ил)метил)-6-фторфенил)метанамина (0,269 г, 1,14 ммоль) и трет-бутил тиазол-4-ил((2,4,6-трифторфенил)сульфонил)карбамата (0,449 г, 1,14 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли N,N-диизопропилэтиламин (0,5 мл, 3 ммоль) и смесь перемешивали при температуре окружающей среды в течение 12 ч. Затем туда добавляли воду (10 мл) и смесь экстрагировали этилацетатом (3×10 мл). Объединенную органическую фазу промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали остаток, который растворяли в дихлорметане (10 мл). В него добавляли трифторуксусную кислоту (2 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 4 ч. Смесь концентрировали in vacuo и остаток очищали с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с использованием ацетонитрила в воде, содержащей 0,5% муравьиной кислоты, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,099 г, выход 17%):1H-ЯМР (300 МГц, DMSO-d6) δ 8,89 (д, J=2,2 Гц, 1H), 8,15 (с, 0,8H), 7,36-7,28 (м, 2H), 7,18-7,10 (м, 2H), 6,88 (д, J=2,2 Гц, 1H), 6,34 (д, J=12,8 Гц, 2H), 4,30 (с, 2H), 3,56 (с, 2H), 3,10 (с, 4H), 2,00 (т, J=7,4 Гц, 4H), 1,77-1,67 (м, 2H), NH не наблюдали; MS (ES+) m/z 509,2 (M+1).
ПРИМЕР 253
Синтез формиата (S)-5-хлор-4-((1-(3-((диметиламино)метил)фенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил (S)-((4-((1-(3-бромфенил)этил)амино)-5-хлор-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата
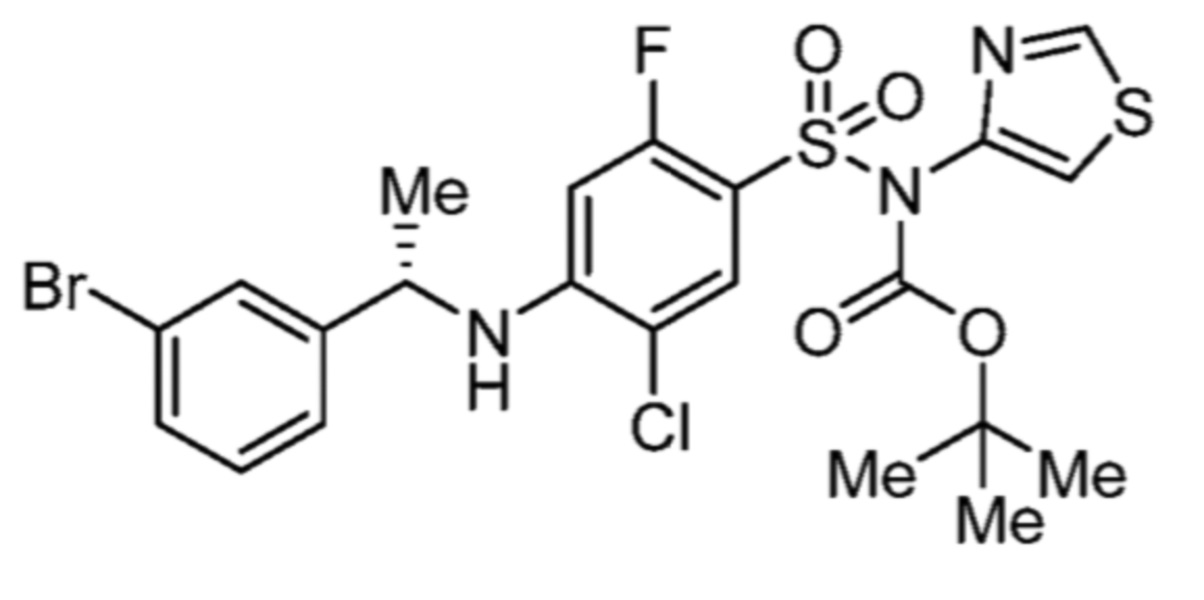
Раствор трет-бутил ((5-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,500 г, 2,44 ммоль), (S)-1-(3-бромфенил)этан-1-амин (0,244 г, 2,44 ммоль) и триэтиламина (0,65 мл, 9,8 ммоль) в безводном диметилсульфоксиде (10 мл) перемешивали при температуре окружающей среды в течение 3 ч. Добавляли насыщенный хлорид аммония (10 мл) и смесь экстрагировали этилацетатом (3×10 мл). Объединенные органические фазы промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 5 до 60% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,504 г, выход 70%): MS (ES+) m/z 592,0 (M+1), 594,0 (M+1).
Стадия 2. Получение формиата (S)-5-хлор-4-((1-(3-((диметиламино)метил)фенил)-этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

В смесь трет-бутил (S)-((4-((1-(3-бромфенил)этил)амино)-5-хлор-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата (0,200 г, 0,338 ммоль), диметиламинометилтрифторбороната калия (0,067 г, 0,41 ммоль), карбоната цезия (0,330 г, 1,01 ммоль), ацетата палладия (0,008 г, 0,03 ммоль) и ди(1-адамантил)-н-бутилфосфина (0,024 г, 0,068 ммоль) добавляли раствор воды (0,53 мл) и 1,4-диоксана (2,6 мл), который дегазировали продуванием азотом. Реакционную смесь нагревали до 85°C в течение 18 ч. После охлаждения до температуры окружающей среды, смесь фильтровали через подушку из целита. Фильтровальный осадок промывали этилацетатом (10 мл) и объединенный фильтрат концентрировали in vacuo. К остатку добавляли дихлорметан (5 мл) и трифторуксусную кислоту (1 мл) и смесь перемешивали при температуре окружающей среды в течение 5 ч. Затем смесь концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 20% метанола в дихлорметане. Дополнительной очисткой с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% муравьиную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,035 г, выход 22%):1H-ЯМР (300 МГц, DMSO-d6) δ 8,82 (д, J=2,1 Гц, 1H), 8,20 (с, 0,6H), 7,57 (д, J=7,4 Гц, 1H), 7,33-7,25 (м, 3H), 7,13-7,10 (м, 1H), 6,82 (д, J=2,1 Гц, 1H), 6,59-6,56 (м, 1H), 6,37 (д, J=12,9 Гц, 1H), 4,75-4,59 (м, 1H), 3,37 (с, 2H), 2,08 (с, 6H), 1,51 (д, J=6,9 Гц, 3H), NH и COOH не наблюдали; MS (ES+) m/z 469,1 (M+1), 471,1 (M+1).
ПРИМЕР 254
Синтез формиата (R)-5-хлор-4-((1-(3-((диметиламино)метил)фенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил (R)-((4-((1-(3-бромфенил)этил)амино)-5-хлор-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата

Следуя процедуре как описано для примера 253, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять (S)-1-(3-бромфенил)этан-1-амин на (R)-1-(3-бромфенил)этан-1-амин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,978 г, выход 68%): MS (ES+) m/z 592,0 (M+1), 594,0 (M+1).
Стадия 2. Получение формиата (R)-5-хлор-4-((1-(3-((диметиламино)метил)фенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано для примера 253, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять трет-бутил (S)-((4-((1-(3-бромфенил)этил)амино)-5-хлор-2-фторфенил)сульфонил)(тиазол-4-ил)карбамат на трет-бутил (R)-((4-((1-(3-бромфенил)этил)амино)-5-хлор-2-фторфенил)сульфонил)(тиазол-4-ил)карбамат, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,023 г, выход 15%):1H-ЯМР (300 МГц, DMSO-d6) δ 8,83 (д, J=2,2 Гц, 1H), 8,17 (с, 0,4H), 7,57 (д, J=7,4 Гц, 1H), 7,33-7,26 (м, 3H), 7,13-7,10 (м, 1H), 6,87 (д, J=2,2 Гц, 1H), 6,61-6,59 (м, 1H), 6,38 (д, J=13,5 Гц, 1H), 4,73-4,66 (м, 1H), 3,38 (с, 2H), 2,09 (с, 6H), 1,52-1,50 (д, J=6,3 Гц, 3H), NH и COOH не наблюдали; MS (ES+) m/z 469,1 (M+1), 471,1 (M+1).
ПРИМЕР 255
Синтез (S)-4-((1-(5-(азетидин-1-илметил)-2-фторфенил)этил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение (S)-трет-бутил (5-хлор-2-фтор-4-((1-(2-фтор-5-винилфенил)этил)амино)фенил)сульфонил(тиазол-4-ил)карбамата
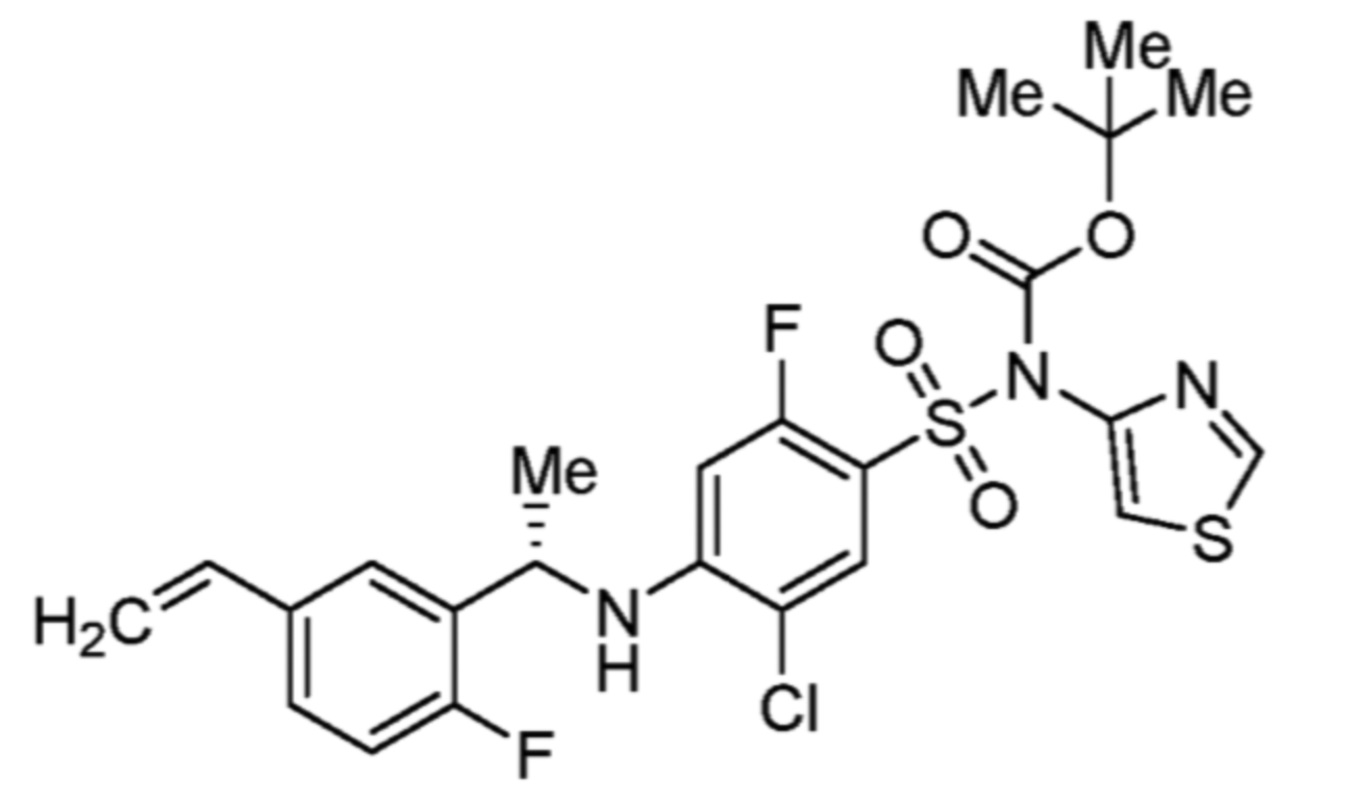
В раствор (S)-трет-бутил (4-((1-(5-бром-2-фторфенил)этил)амино)-5-хлор-2-фторфенил)сульфонил(тиазол-4-ил)карбамата (0,80 г, 1,3 ммоль), сложного эфира винилбороновой кислоты и пинакола (0,40 г, 2,6 ммоль) и карбоната натрия (0,56 г, 5,2 ммоль) в N,N-диметилформамиде (10 мл) и воде (2 мл) добавляли тетракис(трифенилфосфин)палладий(0) (0,30 г, 0,26 ммоль). Реакционную смесь перемешивали при 80°C в течение 12 ч. После охлаждения до температуры окружающей среды, смесь разводили водой (30 мл) и экстрагировали этилацетатом (3×30 мл). Объединенный органический слой промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 10 до 33% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного масла (0,65 г, выход 89%):1H ЯМР (400 МГц, CDCl3) δ 8,79-8,76 (м, 1H), 7,98 (д, J=7,2 Гц, 1H), 7,48 (д, J=2,4 Гц, 1H), 7,38-7,32 (м, 1H), 7,23 (дд, J=7,2, 2,4 Гц, 1H), 7,11-7,05 (м, 1H), 6,63 (дд, J=17,6, 10,8 Гц, 1H), 6,22 (д, J=12,4 Гц, 1H), 5,64 (д, J=17,6 Гц, 1H), 5,35 (д, J=5,6 Гц, 1H), 5,24 (д, J=10,8 Гц, 1H), 4,90-4,82 (м, 1H), 1,68 (д, J=6,8 Гц, 3H), 1,35 (с, 9H); MS (ES+) m/z 456,0 (M - 99), 458,0 (M - 99).
Стадия 2. Получение (S)-трет-бутил (5-хлор-2-фтор-4-((1-(2-фтор-5-формилфенил)этил)амино)фенил)сульфонил(тиазол-4-ил)карбамата

Раствор (S)-трет-бутил(5-хлор-2-фтор-4-((1-(2-фтор-5-винилфенил)этил)амино)фенил)сульфонил(тиазол-4-ил)карбамата (0,65 г, 1,2 ммоль) в дихлорметане (10 мл) барботировали озоном при -78°C до тех пор, пока цвет реакционной смеси не становился голубым. Поток озона останавливали и затем в реакционную смесь частями добавляли трифенилфосфин (0,61 г, 2,3 ммоль) при -78°C. Затем смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 2 ч. Концентрированием in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с градиентом 33% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,15 г, выход 23%):1H ЯМР (400 МГц, CDCl3) δ 9,93 (с, 1H), 8,76 (д, J=2,0 Гц, 1H), 7,98 (д, J=6,8 Гц, 1H), 7,89-7,80 (м, 2H), 7,47 (д, J=2,0 Гц, 1H), 7,31 (д, J=9,6 Гц, 1H), 6,13 (д, J=12,0 Гц, 1H), 5,35 (д, J=5,6 Гц, 1H), 4,99-4,86 (м, 1H), 1,70 (д, J=6,8 Гц, 3H), 1,34 (с, 9H); MS (ES+) m/z 458,0 (M - 99), 460,0 (M - 99).
Стадия 3. Получение (S)-трет-бутил (4-((1-(5-(азетидин-1-илметил)-2-фторфенил)этил)амино)-5-хлор-2-фторфенил)сульфонил(тиазол-4-ил)карбамата
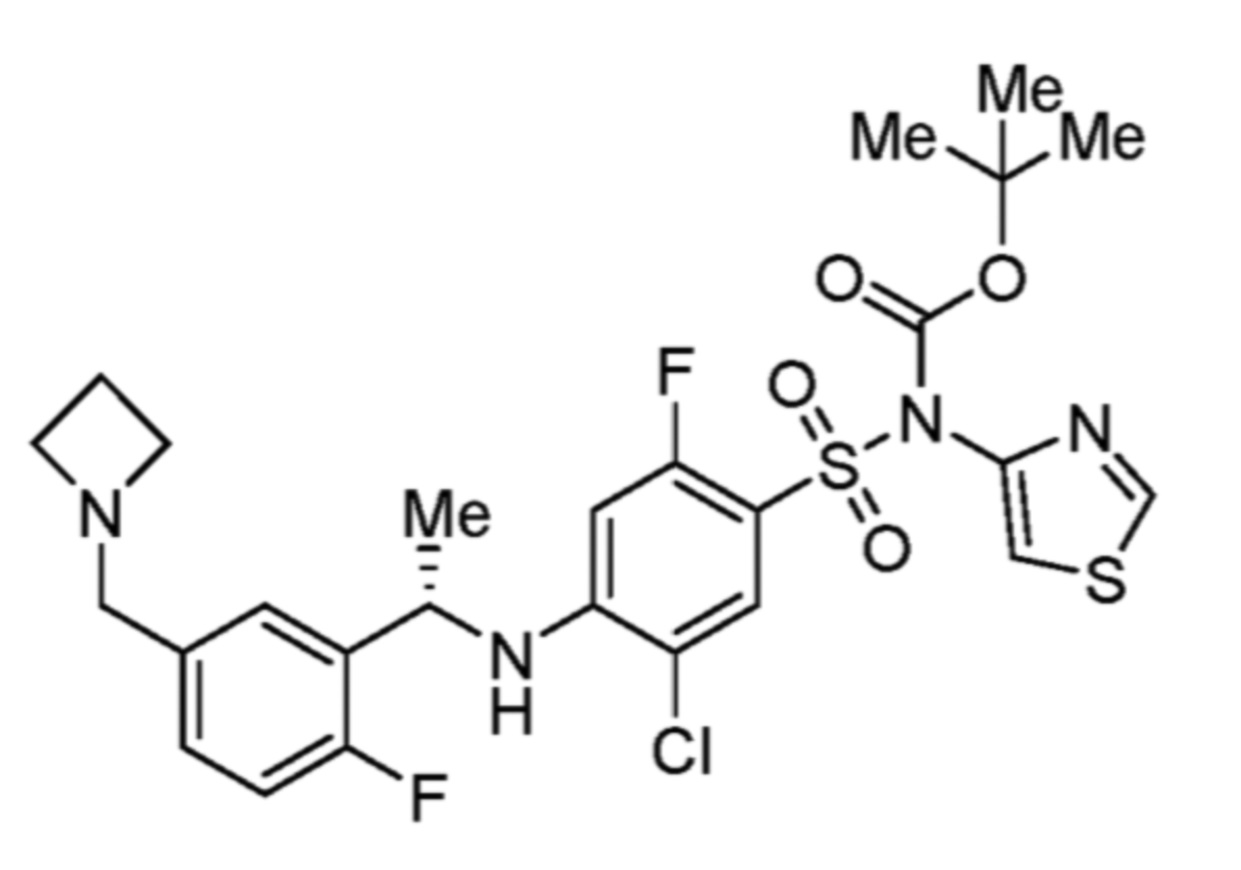
В раствор (S)-трет-бутил(5-хлор-2-фтор-4-((1-(2-фтор-5-формилфенил)этил)амино)фенил)сульфонил(тиазол-4-ил)карбамата (0,070 г, 0,13 ммоль), гидрохлорида азетидина (0,023 г, 0,25 ммоль) и уксусной кислоты (0,0015 г, 0,025 ммоль) в дихлорметане (3 мл) добавляли триацетоксиборгидрид натрия (0,053 г, 0,25 ммоль) одной частью. Смесь перемешивали при температуре окружающей среды в течение 2 ч. Смесь разводили водой (30 мл) и экстрагировали дихлорметаном (3×20 мл). Объединенный органический слой промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде бесцветного масла (0,07 г, выход 90%): MS (ES+) m/z 599,0 (M+1), 601,1 (M+1).
Стадия 4. Синтез формиата (S)-4-((1-(5-(азетидин-1-илметил)-2-фторфенил)этил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

К (S)-трет-бутил(4-((1-(5-(азетидин-1-илметил)-2-фторфенил)этил)амино)-5-хлор-2-фторфенил)сульфонил(тиазол-4-ил)карбамату (0,070 г, 0,12 ммоль) добавляли 4,0 M раствор хлороводорода в 1,4-диоксане (5 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 30 минут. Смесь концентрировали in vacuo и остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,0317 г, выход 54%):1H ЯМР (400 МГц, CDCl3) δ 8,58 (д, J=2,0 Гц, 1H), 8,51 (с, 0,4 H), 7,75 (д, J=7,2 Гц, 1H), 7,31-7,29 (м, 1H), 7,26-7,19 (м, 1H), 7,09-7,03 (м, 1H), 6,92 (д, J=2,0 Гц, 1H), 6,10 (д, J=12,4 Гц, 1H), 5,30 (д, J=6,0 Гц, 1H), 4,76 (квин, J=6,4 Гц, 1H), 3,78 (к, J=12,8 Гц, 2H), 3,49 (т, J=7,6 Гц, 4H), 2,25 (квин, J=7,6 Гц, 2H), 1,63 (д, J=6,8 Гц, 3H), NH и COOH не наблюдали; MS (ES+) m/z 499,1 (M+1).
ПРИМЕР 256
Синтез формиата (S)-5-хлор-2-фтор-4-((1-(2-фтор-5-(пиперидин-1-илметил)фенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение (S)-трет-бутил(5-хлор-2-фтор-4-((1-(2-фтор-5-(пиперидин-1-илметил)фенил)этил)амино)фенил)сульфонил(тиазол-4-ил)карбамата

В раствор (S)-трет-бутил(5-хлор-2-фтор-4-((1-(2-фтор-5-формилфенил)этил)амино)фенил)сульфонил(тиазол-4-ил)карбамата (0,070 г, 0,13 ммоль), пиперидина (0,0214 г, 0,251 ммоль) и трифторуксусной кислоты (0,0043 г, 0,038 ммоль) в тетрагидрофуране (2 мл) добавляли триацетоксиборгидрид натрия (0,0532 г, 0,251 ммоль) одной частью. Смесь перемешивали при температуре окружающей среды в течение 12 ч. Смесь разводили водой (30 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием in vacuo получали указанное в заголовке соединение в виде бесцветного масла (0,08 г, выход 98%): MS (ES+) m/z 527,1 (M - 99), 529,0 (M - 99).
Стадия 2. Получение формиата (S)-5-хлор-2-фтор-4-((1-(2-фтор-5-(пиперидин-1-илметил)фенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида

В раствор (S)-трет-бутил(5-хлор-2-фтор-4-((1-(2-фтор-5-(пиперидин-1-илметил)фенил)этил)амино)фенил)сульфонил(тиазол-4-ил)карбамата (0,070 г, 0,112 ммоль) в дихлорметане (3 мл) добавляли трифторуксусную кислоту (0,77 г, 6,8 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 30 минут и затем концентрировали in vacuo. Остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,018 г, выход 30%):1H ЯМР (400 МГц, CDCl3) δ 8,64 (д, J=2,0 Гц, 1H), 8,49 (с, 0,7H), 7,76 (д, J=7,2 Гц, 1H), 7,41 (д, J=5,6 Гц, 1H), 7,34-7,29 (м, 1H), 7,08 (дд, J=9,6, 8,4 Гц, 1H), 6,91 (д, J=2,0 Гц, 1H), 6,08 (д, J=12,4 Гц, 1H), 5,34 (д, J=6,0 Гц, 1H), 4,83-4,74 (м, 1H), 3,96-3,77 (м, 2H), 2,71 (с, 4H), 1,80-1,71 (м, 4H), 1,65 (д, J=6,4 Гц, 3H), 1,51 (с, 2H), NH и COOH не наблюдали; MS (ES+) m/z 527,1 (M+1).
ПРИМЕР 257
Синтез формиата (S)-4-((1-(5-(2-(азетидин-1-ил)этил)-2-фторфенил)этил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

В раствор (S)-5-хлор-2-фтор-4-((1-(2-фтор-5-(2-оксоэтил)фенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида (0,080 г, 0,17 ммоль), гидрохлорида азетидина (0,032 г, 0,34 ммоль) и уксусной кислоты (0,015 г, 0,25 ммоль) в метаноле (3 мл) добавляли цианоборогидрид натрия (0,021 г, 0,34 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 12 ч и затем концентрировали in vacuo. Остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,022 г, выход 25%):1H ЯМР (400 МГц, CDCl3) δ 8,61 (д, J=2,0 Гц, 1H), 8,58 (с, 0,6H), 7,77 (д, J=7,2 Гц, 1H), 7,16-7,10 (м, 2H), 7,07-6,99 (м, 1H), 6,90 (д, J=2,0 Гц, 1H), 6,10 (д, J=12,4 Гц, 1H), 5,31 (д, J=5,6 Гц, 1H), 4,75 (к, J=6,40 Гц, 1H), 3,82-3,72 (м, 4H), 3,10 (т, J=7,6 Гц, 2H), 2,85 (т, J=7,6 Гц, 2H), 2,38 (к, J=8,0 Гц, 2H), 1,63 (д, J=6,8 Гц, 3H), NH и COOH не наблюдали.
ПРИМЕР 258
Синтез 4-((2-(азетидин-1-илметил)бензил)амино)-5-хлор-2-фтор-N-(изоксазол-3-ил)бензолсульфонамида

Стадия 1. Получение 5-хлор-2,4-дифтор-N-изоксазол-3-ил-N-(2-триметилсилилэтоксиметил)-бензолсульфонамида

В раствор 5-хлор-2,4-дифтор-N-(изоксазол-3-ил)бензолсульфонамида (1,00 г, 3,39 ммоль) и карбоната калия (0,937 г, 6,78 ммоль) в безводном N,N-диметилформамиде (10 мл) добавляли 2-(триметилсилил)этоксиметилхлорид (0,678 г, 4,07 ммоль) при 0°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 1 ч. Смесь выливали в воду со льдом (50 мл) и водную фазу экстрагировали этилацетатом (3×30 мл). Объединенную органическую фазу промывали солевым раствором (3×30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием 10% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного масла (1,40 г, выход 97%):1H ЯМР (400 МГц, CDCl3) δ 8,30 (д, J=1,8 Гц, 1H), 8,06 (т, J=7,4 Гц, 1H), 7,03 (дд, J=9,2, 8,2 Гц, 1H), 6,61 (д, J=1,8 Гц, 1H), 5,40 (с, 2H), 3,75-3,66 (м, 2H), 0,94-0,85 (м, 2H), 0,00 (с, 9H).
Стадия 2. Получение 4-((2-(азетидин-1-илметил)бензил)амино)-5-хлор-2-фтор-N-(изоксазол-3-ил)-N-((2-(триметилсилил)этокси)метил)бензолсульфонамида

Смесь (2-(азетидин-1-илметил)фенил)метанамина (0,103 г, 0,588 ммоль), 5-хлор-2,4-дифтор-N-изоксазол-3-ил-N-(2-триметилсилилэтоксиметил)бензолсульфонамида (0,250 г, 0,588 ммоль) и карбоната калия (0,243 г, 1,77 ммоль) в безводном N,N-диметилформамиде (5 мл) перемешивали при температуре окружающей среды в течение 12 ч. Затем туда добавляли воду (10 мл) и смесь экстрагировали этилацетатом (3×10 мл). Объединенные органические слои промывали солевым раствором (3×10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата при пониженном давлении получали указанное в заголовке соединение в виде масла желтого цвета (0,300 г, выход 71%): MS (ES+) m/z 581,4 (M+1), 583,4 (M+1).
Стадия 2. Получение 4-((2-(азетидин-1-илметил)бензил)амино)-5-хлор-2-фтор-N-(изоксазол-3-ил)бензолсульфонамида

В раствор 4-((2-(азетидин-1-илметил)бензил)амино)-5-хлор-2-фтор-N-(изоксазол-3-ил)-N-((2-(триметилсилил)этокси)метил)бензолсульфонамида (0,300 г, 0,516 ммоль) в 1,4-диоксане (2 мл) добавляли 4,0 M раствор хлороводорода в 1,4-диоксане (6 мл) и смесь перемешивали при температуре окружающей среды в течение 12 ч. Реакционную смесь концентрировали in vacuo. Остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,066 г, выход 28%):1H ЯМР (400 МГц, CDCl3) δ 8,21 (д, J=1,8 Гц, 1H), 7,72 (д, J=7,2 Гц, 1H), 7,39-7,32 (м, 4H), 6,54-6,48 (м, 2H), 4,52 (с, 2H), 3,93 (с, 2H), 3,63 (т, J=7,4 Гц, 4H), 2,35-2,28 (м, 2H), способные к обмену протоны не наблюдали; MS (ES+) m/z 451,0 (M+1), 453,0 (M+1).
ПРИМЕР 259
Синтез 4-((2-(азетидин-1-илметил)бензил)амино)-2,6-дифтор-N-(изоксазол-3-ил)бензолсульфонамида

Стадия 1. Получение 4-((2-(азетидин-1-илметил)бензил)амино)-2,6-дифтор-N-(изоксазол-3-ил)-N-((2-(триметилсилил)этокси)метил)бензолсульфонамида

Смесь (2-(азетидин-1-илметил)фенил)метанамина (0,200 г, 1,13 ммоль), 2,4,6-трифтор-N-(изоксазол-3-ил)-N-((2-(триметилсилил)этокси)метил)бензолсульфонамида (0,461 г, 1,13 ммоль) и карбоната калия (0,468 г, 3,39 ммоль) в N,N-диметилформамиде (5 мл) перемешивали при температуре окружающей среды в течение 12 ч. Смесь разводили водой (30 мл) и экстрагировали этилацетатом (3×10 мл). Объединенные органические фракции промывали солевым раствором (3×10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 25% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде масла желтого цвета (0,130 г, выход 20%):1H ЯМР (400 МГц, CDCl3) δ 8,24 (д, J=1,6 Гц, 1H), 7,36-7,27 (м, 4H), 6,69-6,65 (м, 1H), 6,32-6,20 (м, 2H), 5,46 (с, 2H), 4,33 (с, 2H), 3,88 (с, 2H), 3,77-3,70 (м, 2H), 3,61 (с, 4H), 2,37-2,24 (м, 2H), 0,98-0,91 (м, 2H), 0,00 (с, 9H), NH не наблюдали; MS (ES+) m/z 565,1 (M+1).
Стадия 2. Синтез 4-((2-(азетидин-1-илметил)бензил)амино)-2,6-дифтор-N-(изоксазол-3-ил)бензолсульфонамида
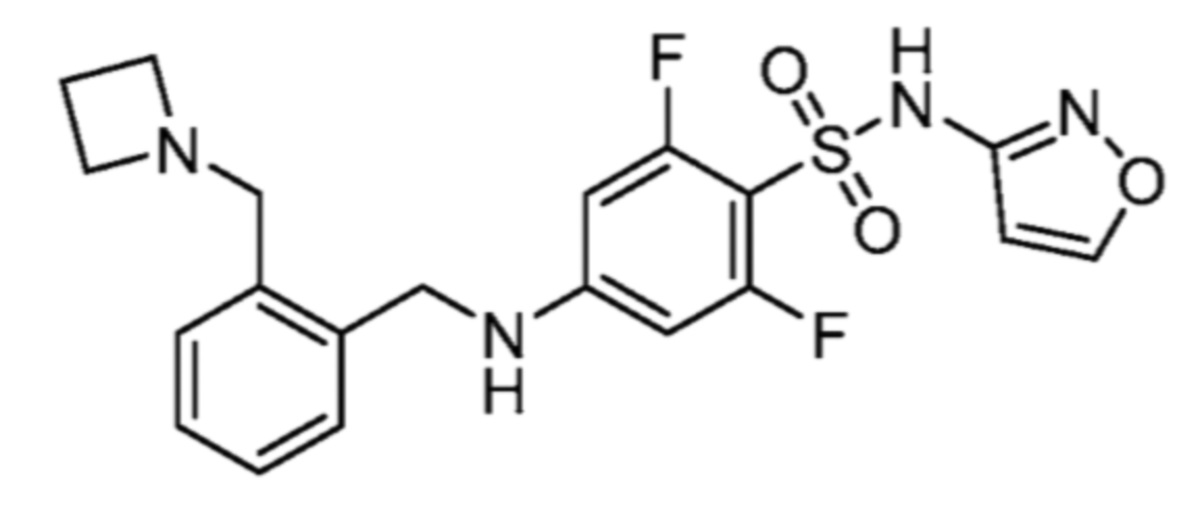
В раствор 4-((2-(азетидин-1-илметил)бензил)амино)-2,6-дифтор-N-(изоксазол-3-ил)-N-((2-(триметилсилил)этокси)метил)бензолсульфонамида (0,130 г, 0,230 ммоль) в 1,4-диоксане (2 мл) добавляли 4,0 M раствор хлороводорода в 1,4-диоксане (2 мл) и смесь перемешивали при температуре окружающей среды в течение 4 ч. Реакционную смесь концентрировали при пониженном давлении. Получаемый остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,021 г, выход 21%):1H ЯМР (400 МГц, DMSO-d6) δ 8,39 (с, 1H), 7,33-7,19 (м, 4H), 6,27 (д, J=12,4 Гц, 2H), 6,21 (с, 1H), 4,37 (с, 2H), 3,69 (с, 2H), 3,28-3,25 (м, 4H), 2,10-1,99 (м, 2H), способные к обмену протоны не наблюдали; MS (ES+) m/z 435,0 (M+1).
ПРИМЕР 260
Синтез 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-5-хлор-2-фтор-N-(пиридазин-3-ил)бензолсульфонамида

Стадия 1. Получение 5-хлор-2,4-дифтор-N-(пиридазин-3-ил)бензолсульфонамида

В раствор пиридазин-3-амина (1,00 г, 10,5 ммоль) в ацетонитриле (15 мл) добавляли 1,4-диазабицикло[2.2.2]октан (2,36 г, 21,0 ммоль) и 5-хлор-2,4-дифторбензолсульфонилхлорид (3,12 г, 12,6 ммоль). Смесь перемешивали при температуре окружающей среды в течение 12 ч и затем концентрировали in vacuo. Остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде твердого вещества желтого цвета (0,400 г, выход 12%):1H ЯМР (400 МГц, CDCl3) δ 8,19 (дд, J=4,0, 1,6 Гц, 1H), 8,14 (т, J=7,6 Гц, 1H), 7,45 (дд, J=9,4, 4,0 Гц, 1H), 7,33 (дд, J=9,4, 1,4 Гц, 1H), 7,06-6,99 (м, 1H), NH не наблюдали.
Стадия 2. Получение 5-хлор-2,4-дифтор-N-(пиридазин-3-ил)-N-((2-(триметилсилил)этокси)метил)бензолсульфонамида
В раствор 5-хлор-2,4-дифтор-N-пиридазин-3-ил-бензолсульфонамида (0,200 г, 0,654 ммоль) в безводном N,N-диметилформамиде (1 мл) добавляли 2-(триметилсилил)этокси-метил хлорид (0,131 г, 0,785 ммоль, 0,139 мл) и карбонат калия (0,181 г, 1,31 ммоль) при 0°C. Смесь перемешивали при температуре окружающей среды в течение 30 минут. Затем в смесь добавляли воду (10 мл) и этилацетат (10 мл). Водную фазу экстрагировали этилацетатом (3×10 мл). Объединенную органическую фазу промывали солевым раствором (2×10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,080 г, выход 82%):1H ЯМР (400 МГц, CDCl3) δ 8,39 (дд, J=9,6, 1,4 Гц, 1H), 8,16-8,11 (м, 1H), 8,10 (дд, J=4,0, 1,6 Гц, 1H), 7,40 (дд, J=9,6, 4,0 Гц, 1H), 7,00 (т, J=8,8 Гц, 1H), 5,62 (с, 2H), 3,72-3,62 (м, 2H), 0,98-0,80 (м, 2H), 0,11-0,01 (м, 9H).
Стадия 3. Получение 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-5-хлор-2-фтор-N-(пиридазин-3-ил)-N-((2-(триметилсилил)этокси)метил)бензолсульфонамида

В раствор (2-(азетидин-1-илметил)-6-фторфенил)метанамина (0,047 г, 0,24 ммоль) и 5-хлор-2,4-дифтор-N-(пиридазин-3-ил)-N-((2-(триметилсилил)этокси)метил)бензолсульфонамида (0,070 г, 0,16 ммоль) в безводном N,N-диметилформамиде (1 мл) добавляли карбонат калия (0,044 г, 0,32 ммоль). Смесь перемешивали при 60°C в течение 12 ч. Затем в смесь добавляли воду (10 мл) и этилацетат (10 мл) и разделяли слои. Водную фазу экстрагировали этилацетатом (3×10 мл). Объединенную органическую фазу промывали солевым раствором (2×10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 33% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,080 г, выход 82%):1H ЯМР (400 МГц, CD3OD) δ 8,39 (дд, J=9,6, 1,5 Гц, 1H), 8,03 (дд, J=4,0, 1,6 Гц, 1H), 7,87 (д, J=7,4 Гц, 1H), 7,59-7,41 (м, 1H), 7,31 (дд, J=9,6, 4,0 Гц, 1H), 7,24 (тд, J=8,0, 5,8 Гц, 1H), 7,11-6,94 (м, 2H), 6,70 (д, J=12,6 Гц, 1H), 5,65 (с, 2H), 4,42 (с, 2H), 3,77-3,68 (м, 2H), 3,65 (с, 2H), 3,25 (т, J=7,0 Гц, 4H), 2,14-2,09 (м, 2H), 0,86-1,00 (м, 2H), 0,08-0,00 (м, 9H); MS (ES+) m/z 610,1 (M+1), 612,1 (M+1).
Стадия 4. Синтез 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-5-хлор-2-фтор-N-(пиридазин-3-ил)бензолсульфонамида

К 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-5-хлор-2-фтор-N-(пиридазин-3-ил)-N-((2-(триметилсилил)этокси)метил)бензолсульфонамиду (0,0800 г, 0,131 ммоль) добавляли 4,0 M раствор хлороводорода в 1,4-диоксане (2 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Смесь концентрировали in vacuo и получаемый остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,019 г, выход 30%):1H ЯМР (400 МГц, CDCl3) δ 8,15 (дд, J=4,0, 1,6 Гц, 2H), 7,86 (д, J=7,2 Гц, 1H), 7,38-7,32 (м, 1H), 7,27-7,23 (м, 1H), 7,10-7,04 (м, 2H), 6,69 (д, J=13,0 Гц, 1H), 4,44 (с, 2H), 3,73 (с, 2H), 3,37-3,32 (м, 4H), 2,17 (дд, J=14,4, 7,8 Гц, 2H), способные к обмену протоны не наблюдали; MS (ES+) m/z 480,0 (M+1), 482,1 (M+1).
ПРИМЕР 261
Синтез формиата 4-((2-(2-(азетидин-1-ил)этил)бензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение (E)-2-(2-этоксивинил)бензонитрила

В смесь 2-бромбензонитрила (1,00 г, 5,49 ммоль), эфира транс-2-этоксивинилбороновой кислоты и пинакола (1,20 г, 6,04 ммоль) и карбоната натрия (1,16 г, 11,0 ммоль) в толуоле (5 мл), этаноле (5 мл) и воде (5 мл) добавляли тетракис(трифенилфосфин)палладий(0) (0,63 г, 0,55 ммоль) и смесь нагревали до 80°C в течение 3 ч. После охлаждения до температуры окружающей среды, реакционную смесь разводили водой (30 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 2% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде масла желтого цвета (0,80 г, выход 84%);1H ЯМР (400 МГц, CDCl3) δ 7,58 (д, J=7,6 Гц, 1H), 7,47-7,43 (м, 2H), 7,25-7,17 (м, 2H), 6,14 (д, J=12,8 Гц, 1H), 4,00 (к, J=7,2 Гц, 2H), 1,40 (т, J=7,2 Гц, 3H).
Стадия 2. Получение 2-(2-оксоэтил)бензонитрила

Раствор (E)-2-(2-этоксивинил)бензонитрила (0,40 г, 2,3 ммоль) в муравьиной кислоте (0,11 г, 2,3 ммоль, 5 мл) перемешивали при температуре окружающей среды в течение 30 минут. Смесь концентрировали in vacuo и остаток разводили водным бикарбонатом натрия (30 мл). Смесь экстрагировали этилацетатом (3×30 мл). Объединенные органические экстракты промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде масла желтого цвета (0,3 г, выход 90%): MS (ES+) m/z 146,1 (M+1).
Стадия 3. Получение 2-(2-(азетидин-1-ил)этил)бензонитрила

В раствор 2-(2-оксоэтил)бензонитрила (0,30 г, 2,1 ммоль), уксусной кислоты (0,025 мг, 0,41 ммоль) и гидрохлорида азетидина (0,387 г, 4,14 ммоль) в метаноле (5 мл) частями добавляли цианоборогидрид натрия (0,26 г, 4,1 ммоль). Смесь перемешивали при температуре окружающей среды в течение 3 ч и затем концентрировали in vacuo. К остатку добавляли воду (30 мл) и смесь экстрагировали дихлорметаном (3×30 мл). Объединенную органическую фазу промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного масла (0,15 г, выход 39%):1H ЯМР (400 МГц, CDCl3) δ 7,63 (д, J=7,6 Гц, 1H), 7,57-7,49 (м, 1H), 7,39-7,29 (м, 2H), 3,27 (т, J=7,2 Гц, 4H), 2,93- 2,85 (м, 2H), 2,79-2,68 (м, 2H), 2,18-2,02 (м, 2H); MS (ES+) m/z 187,1 (M+1).
Стадия 4. Получение (2-(2-(азетидин-1-ил)этил)фенил)метанамина
В смесь 2-(2-(азетидин-1-ил)этил)бензонитрила (0,15 г, 0,81 ммоль) в метаноле (20 мл) и концентрированном гидроксиде аммония (5 мл) добавляли ренеевский никель (0,014 г, 0,161 ммоль) одной частью. Смесь перемешивали при температуре окружающей среды в атмосфере водорода (50 фунтов/дюйм2) в течение 12 ч. Смесь затем фильтровали и фильтрат концентрировали in vacuo для того, чтобы получать указанное в заголовке соединение в виде масла желтого цвета (0,120 г, выход 78%):1H ЯМР (400 МГц, CDCl3) δ 7,34-7,29 (м, 1H), 7,18-7,08 (м, 3H), 3,92 (с, 2H), 3,18 (т, J=7,2 Гц, 4H), 2,66 (с, 4H), 2,05 (к, J=7,2 Гц, 2H), NH не наблюдали; MS (ES+) m/z 191,1 (M+1).
Стадия 5. Получение трет-бутил (4-((2-(2-(азетидин-1-ил)этил)бензил)амино)-5-хлор-2-фторфенил)сульфонил(тиазол-4-ил)карбамата

В раствор (2-(2-(азетидин-1-ил)этил)фенил)метанамина (0,11 г, 0,58 ммоль) и трет-бутил(5-хлор-2,4-дифторфенил)сульфонил(тиазол-4-ил)карбамата (0,238 г, 0,578 ммоль) в безводном N,N-диметилформамиде (4 мл) добавляли карбонат калия (0,16 г, 1,2 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 12 ч. Смесь разводили водой (30 мл) и экстрагировали этилацетатом (3×30 мл). Объединенную органическую фазу промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде масла желтого цвета (0,20 г, выход 59%); MS (ES+) m/z 581,1 (M+1).
Стадия 6. Получение формиата 4-((2-(2-(азетидин-1-ил)этил)бензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

К трет-бутил(4-((2-(2-(азетидин-1-ил)этил)бензил)амино)-5-хлор-2-фторфенил)сульфонил(тиазол-4-ил)карбамату (0,19 г, 0,33 ммоль) добавляли 4,0 M раствор хлороводорода в 1,4-диоксане (20 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 30 минут. Смесь концентрировали in vacuo и остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,068 г, выход 42%):1H ЯМР (400 МГц, CDCl3) δ 8,63 (д, J=2,0 Гц, 1H), 8,54 (с, 1H), 7,74 (д, J=7,2 Гц, 1H), 7,34-7,29 (м, 2H), 7,28-7,24 (м, 1H), 7,24-7,20 (м, 1H), 6,95-6,91 (м, 1H), 6,40 (д, J=12,4 Гц, 1H), 5,88 (с, 1H), 4,43 (д, J=2,8 Гц, 2H), 3,87 (т, J=8,0 Гц, 4H), 3,18-3,10 (м, 2H), 3,06-2,96 (м, 2H), 2,46 (квин, J=8,0 Гц, 2H), NH и COOH не наблюдали; MS (ES+) m/z 481,1 (M+1), 483,1 (M+1).
ПРИМЕР 262
Синтез формиата 4-((5-(2-(азетидин-1-ил)этил)-2-фторбензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение (E)-5-(2-этоксивинил)-2-фторбензонитрила

Следуя процедуре как описано для примера 261, стадии 1 и выполняя некритическое при необходимости, чтобы заменять 2-бромбензонитрил на 5-бром-2-фторбензонитрил, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,74 г, выход 88%):1H ЯМР (400 МГц, CDCl3) δ 7,43-7,39 (м, 2H), 7,15-7,07 (м, 1H), 6,96 (д, J=12,8 Гц, 1H), 5,78 (д, J=12,8 Гц, 1H), 3,93 (к, J=7,2 Гц, 2H), 1,37 (т, J=7,2 Гц, 3H).
Стадия 2. Получение 2-фтор-5-(2-оксоэтил)бензонитрила

Следуя процедуре как описано для примера 261, стадии 2 и выполняя некритическое при необходимости, чтобы заменять (E)-2-(2-этоксивинил)бензонитрил на (E)-5-(2-этоксивинил)-2-фторбензонитрил, указанное в заголовке соединение получали в виде масла желтого цвета (0,54 г, выход 97%): MS (ES+) m/z 164,1 (M+1).
Стадия 3. Получение 5-(2-(азетидин-1-ил)этил)-2-фторбензонитрила

Следуя процедуре как описано для примера 261, стадии 3 и выполняя некритическое при необходимости, чтобы заменять 2-(2-оксоэтил)бензонитрил на 2-фтор-5-(2-оксоэтил)бензонитрил, указанное в заголовке соединение получали в виде бесцветного масла (0,15 г, выход 22%):1H ЯМР (400 МГц, CDCl3) δ 7,50-7,41 (м, 2H), 7,14 (т, J=8,4 Гц, 1H), 3,22 (т, J=7,2 Гц, 4H), 2,66 (с, 4H), 2,16-2,06 (м, 2H).
Стадия 4. Получение (5-(2-(азетидин-1-ил)этил)-2-фторфенил)метанамина

Следуя процедуре как описано для примера 261, стадии 4 и выполняя некритическое при необходимости, чтобы заменять 2-(2-(азетидин-1-ил)этил)бензонитрил на 5-(2-(азетидин-1-ил)этил)-2-фторбензонитрил, указанное в заголовке соединение получали в виде масла желтого цвета (0,1 г, выход 65%): MS (ES+) m/z 209,1 (M+1).
Стадия 5. Получение трет-бутил (4-((5-(2-(азетидин-1-ил)этил)-2-фторбензил)амино)-5-хлор-2-фторфенил)сульфонил(тиазол-4-ил)карбамата

Следуя процедуре как описано для примера 261, стадии 5 и выполняя некритическое при необходимости, чтобы заменять (2-(2-(азетидин-1-ил)этил)фенил)метанамин на (5-(2-(азетидин-1-ил)этил)-2-фторфенил)метанамин, указанное в заголовке соединение получали в виде масла желтого цвета (0,080 г, выход 70%): MS (ES+) m/z 599,1 (M+1), 601,1 (M+1).
Стадия 6. Получение формиата 4-((5-(2-(азетидин-1-ил)этил)-2-фторбензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано для примера 261, стадии 6 и выполняя некритическое при необходимости, чтобы заменять трет-бутил(4-((2-(2-(азетидин-1-ил)этил)бензил)амино)-5-хлор-2-фторфенил)сульфонил(тиазол-4-ил)карбамат на трет-бутил(4-((5-(2-(азетидин-1-ил)этил)-2-фторбензил)амино)-5-хлор-2-фторфенил)сульфонил(тиазол-4-ил)карбамат, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,024 г, выход 35%):1H ЯМР (400 МГц, CDCl3) δ 8,55 (д, J=2,4 Гц, 1H), 8,46 (с, 1H), 7,69 (д, J=7,2 Гц, 1H), 7,12-7,05 (м, 2H), 6,95 (т, J=9,2 Гц, 1H), 6,84 (д, J=2,4 Гц, 1H), 6,23 (д, J=12,0 Гц, 1H), 5,43 (т, J=5,2 Гц, 1H), 4,34 (д, J=5,6 Гц, 2H), 3,70 (т, J=8,0 Гц, 4H), 3,03 (т, J=7,2 Гц, 2H), 2,77 (т, J=7,6 Гц, 2H), 2,30 (к, J=8,0 Гц, 2H), NH и COOH не наблюдали; MS (ES+) m/z 499,0 (M+1), 501,0 (M+1).
ПРИМЕР 263
Синтез формиата 5-хлор-2-фтор-4-((2-фтор-5-(2-(3-фторазетидин-1-ил)этил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 2-фтор-5-(2-(3-фторазетидин-1-ил)этил)бензонитрила

В раствор 2-фтор-5-(2-оксоэтил)бензонитрила (0,55 г, 3,4 ммоль), 3-фторазетидина (0,564 г, 5,06 ммоль) и уксусной кислоты (0,04 г, 0,7 ммоль) в метаноле (4 мл) добавляли цианоборогидрид натрия (0,424 г, 6,74 ммоль) одной частью. Смесь перемешивали при температуре окружающей среды в течение 12 ч. Смесь разводили водой (50 мл) и экстрагировали этилацетатом (3×50 мл). Объединенную органическую фазу промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии с обращенной фазой, осуществляя элюирование с использованием ацетонитрила в воде, получали указанное в заголовке соединение в виде масла желтого цвета (0,30 г, выход 40%):1H ЯМР (400 МГц, CDCl3) δ 7,41-7,31 (м, 2H), 7,06 (т, J=8,4 Гц, 1H), 5,15-4,93 (м, 1H), 3,65-3,51 (м, 2H), 3,14-2,98 (м, 2H), 2,73-2,53 (м, 4H); MS (ES+) m/z 222,9 (M+1).
Стадия 2. Получение (2-фтор-5-(2-(3-фторазетидин-1-ил)этил)фенил)метанамина

В раствор 2-фтор-5-(2-(3-фторазетидин-1-ил)этил)бензонитрила (0,30 г, 1,4 ммоль) и концентрированного гидроксида аммония (2 мл) в метаноле (8 мл) добавляли ренеевский никель (0,023 г, 0,27 ммоль). Смесь перемешивали при температуре окружающей среды в атмосфере водорода (50 Фунтов/дюйм2) в течение 12 ч. Смесь фильтровали и фильтрат концентрировали in vacuo для того, чтобы получать указанное в заголовке соединение в виде масла желтого цвета (0,3 г, выход 95%): MS (ES+) m/z 227,1 (M+1).
Стадия 3. Получение трет-бутил (5-хлор-2-фтор-4-((2-фтор-5-(2-(3-фторазетидин-1-ил)этил)бензил)амино)фенил)сульфонил(тиазол-4-ил)карбамата

Следуя процедуре как описано для примера 261, стадии 5 и выполняя некритическое при необходимости, чтобы заменять (2-(2-(азетидин-1-ил)этил)фенил)метанамин на (2-фтор-5-(2-(3-фторазетидин-1-ил)этил)фенил)метанамин, указанное в заголовке соединение получали в виде масла желтого цвета (0,06 г, выход 22%): MS (ES+) m/z 617,1 (M+1), 619,0 (M+1).
Стадия 4. Синтез формиата 5-хлор-2-фтор-4-((2-фтор-5-(2-(3- фторазетидин-1-ил)этил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано для примера 261, стадии 6 и выполняя некритическое при необходимости, чтобы заменять трет-бутил(4-((2-(2-(азетидин-1-ил)этил)бензил)амино)-5-хлор-2-фторфенил)сульфонил(тиазол-4-ил)карбамат на трет-бутил(5-хлор-2-фтор-4-((2-фтор-5-(2-(3-фторазетидин-1-ил)этил)бензил) амино)фенил)сульфонил(тиазол-4-ил)карбамат, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,034 г, выход 50%):1H ЯМР (400 МГц, CDCl3) δ 8,66 (д, J=2,2 Гц, 1H), 8,26 (с, 1H), 7,77 (д, J=7,2 Гц, 1H), 7,19-7,10 (м, 2H), 7,08-7,01 (м, 1H), 6,97 (д, J=2,2 Гц, 1H), 6,34 (д, J=12,0 Гц, 1H), 5,38 (т, J=5,2 Гц, 1H), 5,33-5,10 (м, 1H), 4,42 (д, J=5,6 Гц, 2H), 4,07-3,97 (м, 2H), 3,42-3,39 (м, 1H), 3,37-3,34 (м, 1H), 3,04-2,96 (м, 2H), 2,79-2,73 (м, 2H), NH и COOH не наблюдали; MS (ES+) m/z 517,0 (M+1), 519,0 (M+1).
ПРИМЕР 264
Синтез формиата 4-(((1S,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-ил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида
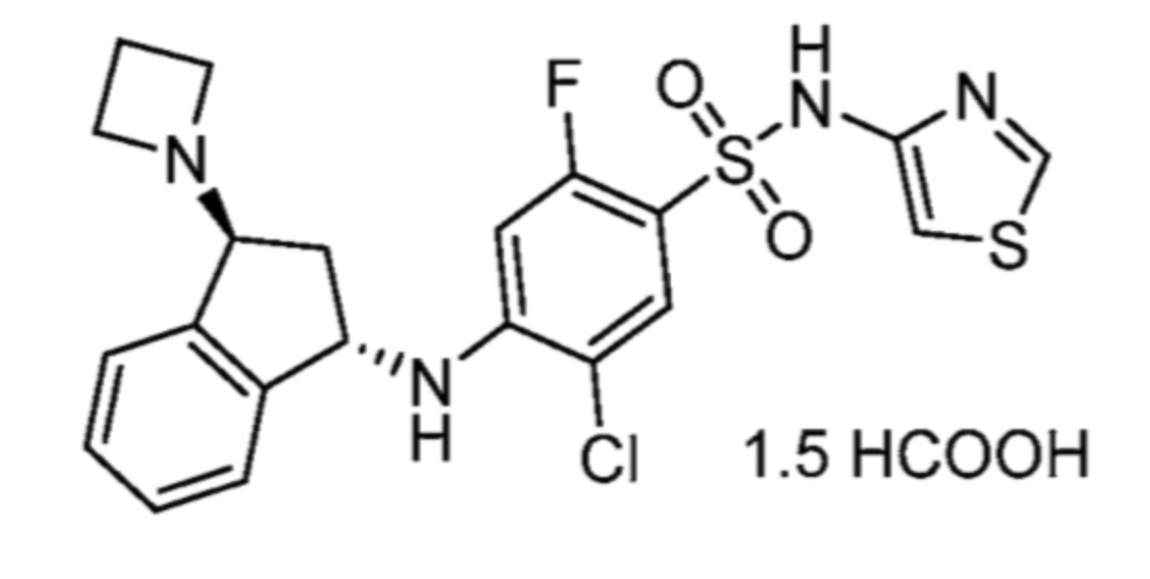
Стадия 1. Получение (S)-3-фенил-3-(2,2,2-трифторацетамидо)пропановой кислоты

Смесь (S)-3-амино-3-фенилпропановой кислоты (1,00 г, 6,05 ммоль) в трифторуксусном ангидриде (3,75 мл) перемешивали при температуре окружающей среды в течение 12 ч. Смесь концентрировали in vacuo и остаток растирали в простом эфире (15 мл) для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (1,40 г, выход 59%):1H ЯМР (400 МГц, DMSO-d6) δ 12,44 (ушир. с, 1H), 9,96 (д, J=8,4 Гц, 1H), 7,40-7,35 (м, 5H), 5,25 (дт, J=8,8, 5,8 Гц, 1H), 2,93-2,74 (м, 2H); MS (ES+) m/z 284,1 (M+23).
Стадия 2. Получение (S)-3-фенил-3-(2,2,2-трифторацетамидо)пропанoилхлорида
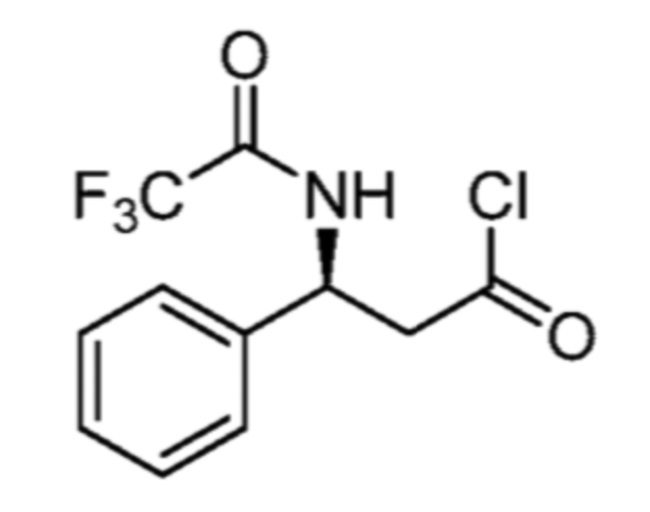
Раствор (S)-3-фенил-3-(2,2,2-трифторацетамидо)пропановой кислоты (3,00 г, 11,5 ммоль) в тионилхлориде (30 мл) нагревали до 80°C в течение 12 ч. Концентрированием in vacuo и растиранием остатка в петролейном эфире получали указанное в заголовке соединение в виде твердого вещества желтого цвета (3,00 г, выход 93%):1H ЯМР (300 МГц, DMSO-d6) δ 9,97 (ушир. д, J=8,1 Гц, 1H), 7,40-7,36 (м, 3H), 7,34-7,23 (м, 2H), 5,27-5,20 (м, 1H), 2,97-2,84 (м, 1H), 2,84-2,71 (м, 1H).
Стадия 3. Получение (S)-2,2,2-трифтор-N-(3-оксо-2,3-дигидро-1H-инден-1-ил)ацетамида

В раствор (S)-3-фенил-3-(2,2,2-трифторацетамидо)пропанoилхлорида (3,00 г, 10,7 ммоль) в безводном дихлорметане (20 мл) добавляли раствор трихлорида алюминия (2,86 г, 21,5 ммоль) в безводном дихлорметане (20 мл) при 0°C. Затем смесь нагревали до 40°C в течение 12 ч. Концентрированием in vacuo получали твердое вещество коричневого цвета, которое растирали в воде (30 мл). Твердое вещество отфильтровывали, промывали водой (3×30 мл) и сушили при пониженном давлении для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (2,50 г, выход 96%):1H ЯМР (400 МГц, DMSO-d6) δ 10,01 (ушир. д, J=8,0 Гц, 1H), 7,81-7,74 (м, 1H), 7,71 (д, J=7,8 Гц, 1H), 7,63-7,54 (м, 2H), 5,63 (дт, J=8,0, 3,4 Гц, 1H), 3,17-3,10 (м, 1H), 2,65-2,60 (м, 1H).
Стадия 4. Получение N-((1S,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-ил)-2,2,2-трифторацетамида

В смесь (S)-2,2,2-трифтор-N-(3-оксо-2,3-дигидро-1H-инден-1-ил)ацетамида (1,00 г, 4,11 ммоль), гидрохлорида азетидина (0,480 г, 5,14 ммоль) и триэтиламина (2,28 мл, 16,4 ммоль) в безводном дихлорметане (30 мл) добавляли изопропоксид титана(IV) (2,34 г, 8,22 ммоль, 2,43 мл) и смесь перемешивали при температуре окружающей среды в течение 1 ч. Затем частями добавляли триацетоксиборгидрид натрия (2,18 г, 10,3 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 47 ч. Смесь разводили дихлорметаном (20 мл) и туда добавляли насыщенный хлорид аммония (30 мл). Органический слой промывали солевым раствором (20 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде твердого вещества желтого цвета (0,200 г, выход 17%):1H ЯМР (400 МГц, CDCl3) δ 7,38-7,29 (м, 4H), 6,41 (ушир. с, 1H), 5,71 (к, J=7,6 Гц, 1H), 3,94 (дд, J=6,8, 2,0 Гц, 1H), 3,36 (к, J=6,8 Гц, 2H), 3,24 (к, J=6,8 Гц, 2H), 2,54 (ддд, J=13,4, 7,4, 2,0 Гц, 1H), 2,09 (квин, J=7,0 Гц, 2H), 1,92 (тд, J=13,6, 7,0 Гц, 1H); MS (ES+) m/z 285,0 (M+1).
Стадия 5. Получение (1S,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-амина

В раствор N-((1S,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-ил)-2,2,2-трифторацетамида (0,150 г, 0,527 ммоль) в метаноле (10 мл) добавляли 1,0 M гидроксид натрия (2,6 мл) и реакционную смесь нагревали до 80°C в течение 24 ч. После охлаждения до температуры окружающей среды, реакционную смесь разводили водой (30 мл) и экстрагировали этилацетатом (3×30 мл). Объединенную органическую фазу промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата при пониженном давлении получали указанное в заголовке соединение в виде твердого вещества желтого цвета (0,100 г, количественный выход): MS (ES+) m/z 189,2 (M+1).
Стадия 6. Получение трет-бутил (4-(((1S,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-ил)амино)-5-хлор-2-фторфенил)сульфонил(тиазол-4-ил)карбамата

В раствор трет-бутил (5-хлор-2,4-дифторфенил)сульфонил(тиазол-4-ил)карбамата (0,100 г, 0,243 ммоль) в безводном N,N-диметилформамиде (5 мл) добавляли (1S,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-амин (0,080 г, 0,43 ммоль) и карбонат калия (0,067 г, 0,49 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 12 ч. Реакционную смесь разводили водой (10 мл) и экстрагировали этилацетатом (2×10 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата под давлением и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с градиентом 33% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде масла желтого цвета (0,100 г, выход 71%): MS (ES+) m/z 579,0 (M+1), 581,0 (M+1).
Стадия 7. Синтез формиата 4-(((1S,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-ил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

К трет-бутил (4-(((1S,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-ил)амино)-5-хлор-2-фторфенил)сульфонил(тиазол-4-ил)карбамату (0,100 г, 0,208 ммоль) добавляли 4 M раствор хлороводорода в 1,4-диоксане (5 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении и остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,035 г, выход 35%):1H ЯМР (400 МГц, DMSO-d6) δ 8,86 (д, J=2,0 Гц, 1H), 8,24 (с, 1,5H), 7,61 (д, J=7,2 Гц, 1H), 7,36-7,32 (м, 1H), 7,29-7,25 (м, 2H), 7,19-7,11 (м, 1H), 6,91-6,81 (м, 2H), 6,39 (д, J=9,2 Гц, 1H), 5,27 (к, J=8,0 Гц, 1H), 3,86 (д, J=6,0 Гц, 1H), 3,35-3,21 (м, 2H), 3,14-3,07 (м, 2H), 2,32-2,27 (м, 1H), 2,12-2,06 (м, 1H), 1,93 (квин, J=6,8 Гц, 2H), NH и COOH не наблюдали; MS (ES+) m/z 478,9 (M+1), 479,9 (M+1).
ПРИМЕР 265
Синтез (S)-5-хлор-2-фтор-N-(5-фтортиазол-2-ил)-4-((1-фенилпропил)амино)бензолсульфонамида

Стадия 1. Получение 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(5-фтортиазол-2-ил)бензолсульфонамида

В раствор N-(2,4-диметоксибензил)-5-фтортиазол-2-амина (0,30 г, 1,12 ммоль) в безводном тетрагидрофуране (15 мл) добавляли 1 M раствор бис(триметилсилил)амида лития в тетрагидрофуране (1,68 мл, 1,68 ммоль) при -78°C. Реакционную смесь перемешивали в течение 50 минут при -78°C. Затем туда добавляли раствор 5-хлор-2,4-дифторбензол-1-сульфонилхлорида (0,33 г, 1,34 ммоль) в безводном тетрагидрофуране (3 мл) при -78°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 2 часов. Реакционную смесь разводили водой (30 мл) и экстрагировали этилацетатом (2×20 мл). Объединенные органические слои промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 20% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,30 г, выход 81%):1H ЯМР (400 МГц, CDCl3) δ 7,89 (т, J=7,2 Гц, 1H), 7,18 (д, J=8,4 Гц, 1H), 7,01-7,08 (м, 2H), 6,38-6,42 (м, 1H), 6,36 (д, J=2,4 Гц, 1H), 5,06 (с, 2H), 3,80 (с, 3H), 3,75 (с, 3H).
Стадия 2. Получение (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(5-фтортиазол-2-ил)-4-((1-фенилпропил)амино)бензолсульфонамида

В раствор 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(5-фтортиазол-2-ил) бензолсульфонамида (0,12 г, 0,25 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли карбонат калия (0,069 г, 0,50 ммоль) и (S)-1-фенилпропан-1-амин (0,067 г, 0,50 ммоль). Смесь перемешивали при температуре окружающей среды в течение 12 часов. Реакционную смесь разводили водой (10 мл) и экстрагировали этилацетатом (2×10 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата при пониженном давлении получали указанное в заголовке соединение в виде неочищенного масла желтого цвета (0,15 г, количественный выход), которое использовали без дополнительной очистки: MS (ES+) m/z 594,1 (M+1), 596,1 (M+1).
Стадия 3. Получение (S)-5-хлор-2-фтор-N-(5-фтортиазол-2-ил)-4-((1-фенилпропил)амино)бензолсульфонамида.

В смесь (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(5-фтортиазол-2-ил)-4-((1-фенилпропил)амино)бензолсульфонамида (0,13 г, 0,22 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (1 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 15 минут. Реакционную смесь концентрировали in vacuo и остаток растирали с метанолом (5 мл). Фильтрованием и концентрированием фильтрата получали остаток, который очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,080 г, выход 82%):1H ЯМР (400 МГц, CDCl3) δ 7,80 (д, J=7,2 Гц, 1H), 7,33-7,41 (м, 2H), 7,29-7,33 (м, 1H), 7,26 (с, 2H), 6,77 (с, 1H), 6,12 (д, J=12,4 Гц, 1H), 5,27 (д, J=4,8 Гц, 1H), 4,23 (к, J=6,4 Гц, 1H), 1,93 (дк, J=11,6, 7,2 Гц, 2H), 1,01 (т, J=7,2 Гц, 3H), NH не наблюдали; MS (ES+) m/z 444,0 (M+1), 446,0 (M+1).
ПРИМЕР 266
Синтез (S)-5-хлор-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(5-фтортиазол-2-ил)бензолсульфонамида

Стадия 1. Получение (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(5-фтортиазол-2-ил)бензолсульфонамида

В раствор 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(5-фтортиазол-2-ил) бензолсульфонамида (0,10 г, 0,21 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли карбонат калия (0,058 г, 0,42 ммоль) и (S)-1-(2-фторфенил)этанамин (0,058 г, 0,42 ммоль). Смесь перемешивали при температуре окружающей среды в течение 12 часов. Реакционную смесь разводили водой (10 мл) и экстрагировали этилацетатом (2×10 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 20% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,060, выход 48%): MS (ES+) m/z 598,1 (M+1), 600,1 (M+1).
Стадия 2. Получение (S)-5-хлор-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(5-фтортиазол-2-ил)бензолсульфонамида

В смесь (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(5-фтортиазол-2-ил)бензолсульфонамида (0,060 г, 0,10 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (1 мл) и смесь перемешивали при температуре окружающей среды в течение 15 минут. Концентрированием in vacuo получали остаток, который растирали с метанолом (5 мл). Смесь фильтровали и фильтрат концентрировали при пониженном давлении. Очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,025 г, выход 55%):1H ЯМР (400 МГц, CDCl3) δ 7,82 (д, J=7,2 Гц, 1H), 7,31 (с, 1H), 7,22-7,27 (м, 1H), 7,05-7,17 (м, 2H), 6,77 (с, 1H), 6,13 (д, J=12,4 Гц, 1H), 5,21 (д, J=5,6 Гц, 1H), 4,82 (т, J=6,4 Гц, 1H), 1,65 (д, J=6,8 Гц, 3H), NH не наблюдали; MS (ES+) m/z 448,0 (M+1), 450,0 (M+1).
ПРИМЕР 267
Синтез (S)-5-хлор-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(5-хлортиазол-2-ил)бензолсульфонамида

Стадия 1. Получение (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(5-хлортиазол-2-ил)бензолсульфонамида

В раствор 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(5-хлортиазол-2-ил)бензолсульфонамида (получен в соответствии с WO 2015077905, 0,10 г, 0,20 ммоль) в безводном диметилсульфоксиде (3 мл) добавляли карбонат калия (0,084 г, 0,61 ммоль) и (S)-1-(2-фторфенил)этанамин (0,039 г, 0,22 ммоль). Смесь перемешивали при температуре окружающей среды в течение 12 часов. Остаток выливали на воду со льдом (30 мл) и водную фазу экстрагировали этилацетатом (3×50 мл). Объединенные органические экстракты промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде масла желтого цвета (0,15, количественный выход): MS (ES+) m/z 614,0 (M+1), 616,0 (M+1).
Стадия 2. Получение (S)-5-хлор-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(5-хлортиазол-2-ил)бензолсульфонамида

В смесь (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(2-фторфенил)этил)амино)-N-(5-хлортиазол-2-ил)бензолсульфонамида (0,15 г, 0,17 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (0,50 мл). Смесь перемешивали при температуре окружающей среды в течение 12 часов. Концентрированием in vacuo и очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,043 г, выход 54%):1H ЯМР (400 МГц, CDCl3) δ 7,81 (д, J=6,8 Гц, 1H), 7,29 (м, 1H), 7,25-7,21 (м, 1H), 7,13-7,06 (м, 2H), 6,99 (с, 1H), 6,12 (д, J=8,4 Гц, 1H), 5,20 (д, J=5,6 Гц, 1H), 4,83-4,77 (м, 1H), 1,64 (д, J=6,8 Гц, 3H), NH не наблюдали; MS (ES+) m/z 464,0 (M+1), 486,0 (M+1+23).
ПРИМЕР 268
Синтез (S)-5-хлор-4-((1-(2,5-дифторфенил)этил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение (R)-N-(2,5-дифторбензилиден)-2-метилпропан-2-сульфинамида.
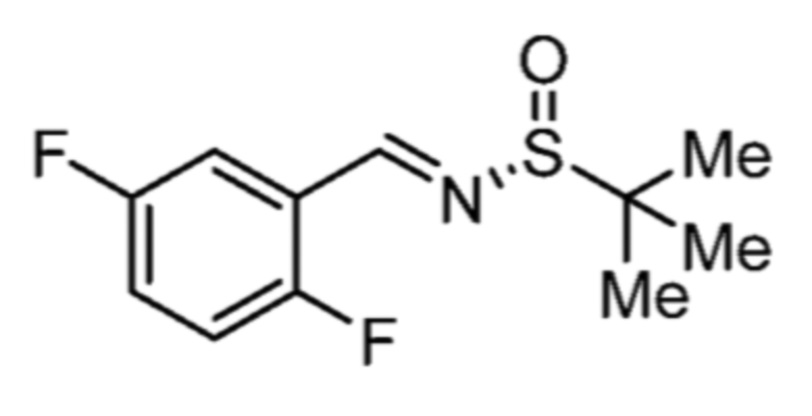
В раствор 2,5-дифторбензальдегида (3,00 г, 21,1 ммоль) в безводном дихлорметане (20 мл) добавляли п-толуолсульфонат пиридиния (0,26 г, 1,06 ммоль), (R)-2-метилпропан-2- улфинамид (2,81 г, 23,2 ммоль) и безводный сульфат магния (12,7 г, 105 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 12 часов. Реакционную смесь фильтровали и фильтрат концентрировали in vacuo. Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 1-2% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного масла (3,20 г, выход 61%):1H ЯМР (400 МГц, CDCl3) δ 8,85 (д, J=2,0 Гц, 1H), 7,66 (ддд, J=8,4, 5,2, 3,2 Гц, 1H), 7,23-7,10 (м, 2H), 1,27 (с, 9H).
Стадия 2. Получение (R)-N-((S)-1-(2,5-дифторфенил)этил)-2-метилпропан-2-сульфинамида

В раствор (R)-N-(2,5-дифторбензилиден)-2-метилпропан-2-сульфинамида (3,20 г, 13,0 ммоль) в безводном дихлорметане (20 мл) добавляли по каплям 3 M раствор бромида метилмагния в простом диэтиловом эфире (8,7 мл, 26 ммоль) при -50°C. Реакционную смесь нагревали до температуры окружающей среды и перемешивали в течение 12 часов. Реакционную смесь разводили хлоридом аммония (50 мл), водой (100 мл) и экстрагировали дихлорметаном (3×80 мл). Объединенные органические слои промывали солевым раствором (3×60 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием 20% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (1,50 г, выход 43%):1H ЯМР (400 МГц, CDCl3) δ 7,09-6,88 (м, 3H), 4,90-4,79 (м, 1H), 3,41 (д, J=4,0 Гц, 1H), 1,55 (д, J=6,8 Гц, 3H), 1,21 (с, 9H).
Стадия 3. Получение (S)-1-(2,5-дифторфенил)этан-1-амина

К (R)-N-((S)-1-(2,5-дифторфенил)этил)-2-метилпропан-2-сульфинамиду (1,00 г, 3,83 ммоль) добавляли 0,4 M раствор хлороводорода в метаноле (10 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 часа. Туда добавляли насыщенный бикарбонат натрия (100 мл) и смесь перемешивали при температуре окружающей среды в течение 30 минут. Затем смесь экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата при пониженном давлении получали указанное в заголовке соединение в виде бесцветного масла (0,40 г, выход 66%):1H ЯМР (400 МГц, CDCl3) δ 7,36 (ддд, J=8,8, 5,6, 3,2 Гц, 1H), 7,30-7,17 (м, 2H), 4,74 (к, J=6,8 Гц, 1H), 1,66 (д, J=6,8 Гц, 3H), NH не наблюдали.
Стадия 4. Получение (S)-5-хлор-4-((1-(2,5-дифторфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида

В смесь 5-хлор-N-[(2,4-диметоксифенил)метил]-2,4-дифтор-N-тиазол-2-ил-бензолсульфонамида (0,30 г, 0,65 ммоль) и (S)-1-(2,5-дифторфенил)этанамина (0,10 г, 0,65 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли карбонат калия (0,18 г, 1,3 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 12 часов. Затем туда добавляли воду (80 мл) и смесь экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали солевым раствором (3×50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата при пониженном давлении и очисткой с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 25% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,16 г, выход 41%): MS (ES+) m/z 598,2 (M+1), 600,2 (M+1).
Стадия 5. Получение (S)-5-хлор-4-((1-(2,5-дифторфенил)этил)амино)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида
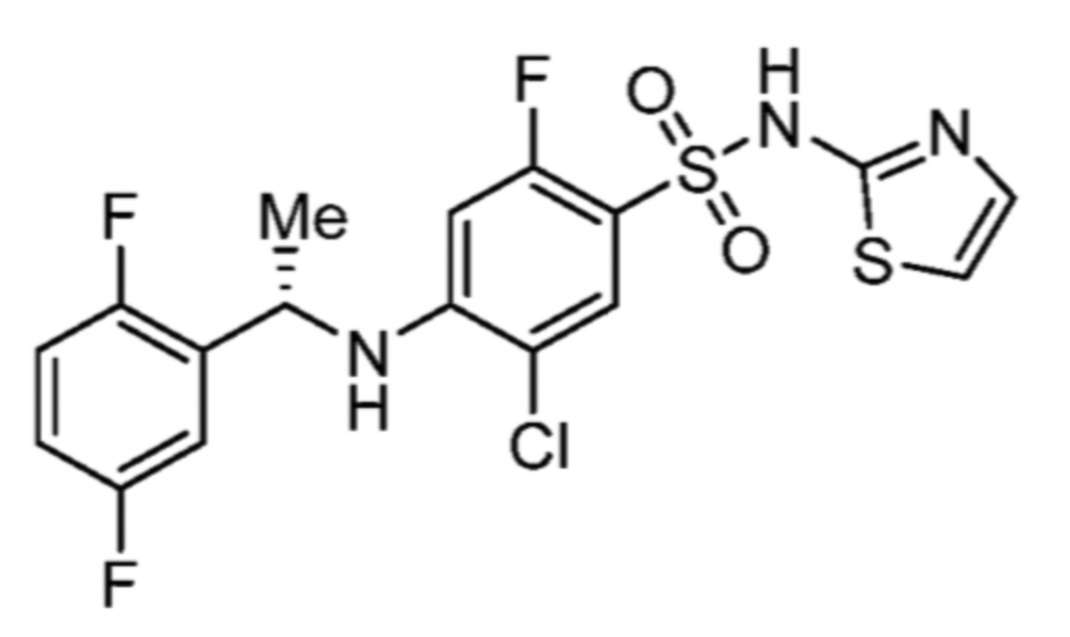
В раствор (S)-5-хлор-4-((1-(2,5-дифторфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида (0,16 г, 0,27 ммоль) в дихлорметане (2 мл) добавляли трифторуксусную кислоту (0,31 г, 2,70 ммоль) и смесь перемешивали при температуре окружающей среды в течение 1 часа. Добавляли воду (20 мл) и смесь экстрагировали дихлорметаном (3×10 мл). Объединенные органические экстракты промывали солевым раствором (3×10 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,068 г, выход 56%):1H ЯМР (400 МГц, CDCl3) δ 7,88 (д, J=7,2 Гц, 1H), 7,12 (д, J=4,4 Гц, 1H), 7,09-7,01 (м, 1H), 6,98-6,89 (м, 2H), 6,49 (д, J=4,4 Гц, 1H), 6,06 (д, J=12,0 Гц, 1H), 5,09 (д, J=5,6 Гц, 1H), 4,75 (квин, J=6,4 Гц, 1H), 1,62 (д, J=6,8 Гц, 3H), потерян сигнал сульфонамидного N-H ; MS (ES+) m/z 448,1 (M+1), 450,1 (M+1).
ПРИМЕР 269
Синтез (S)-5-хлор-4-((1-(2-фторфенил)этил)амино)-2-метил-N-(тиазол-2-ил)бензолсульфонамида

Стадия 1. Получение 4-бром-5-хлор-2-метилбензолсульфонилхлорида

Насыщенный раствор диоксида серы формировали посредством барботирования уксусной кислоты (20 мл) газообразным диоксидом серы при 0°C в течение 15 минут, и туда добавляли хлорид меди(II) (0,67 г, 6,8 ммоль). В суспензию 4-бром-5-хлор-2-метил-анилина (5,0 г, 23 ммоль) в концентрированной соляной кислоте (20,0 мл) добавляли раствор нитрита натрия (2,3 г, 34 ммоль) в воде (10 мл) при 0°C. После перемешивания в течение 30 минут, реакционную смесь добавляли в раствор диоксида серы при 0°C в течение периода 15 минут. Затем реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 30 минут. Остаток выливали на воду со льдом (30 мл) и водную фазу экстрагировали этилацетатом (3×100 мл). Объединенную органическую фазу промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием 10% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде твердого вещества желтого цвета (4,0 г, выход 58%):1H ЯМР (400 МГц, CDCl3) δ 8,13 (с, 1H), 7,73 (с, 1H), 2,74 (с, 3H).
Стадия 3. Получение 4-бром-5-хлор-N-(2,4-диметоксибензил)-2-метил-N-(тиазол-2-ил)бензолсульфонамида
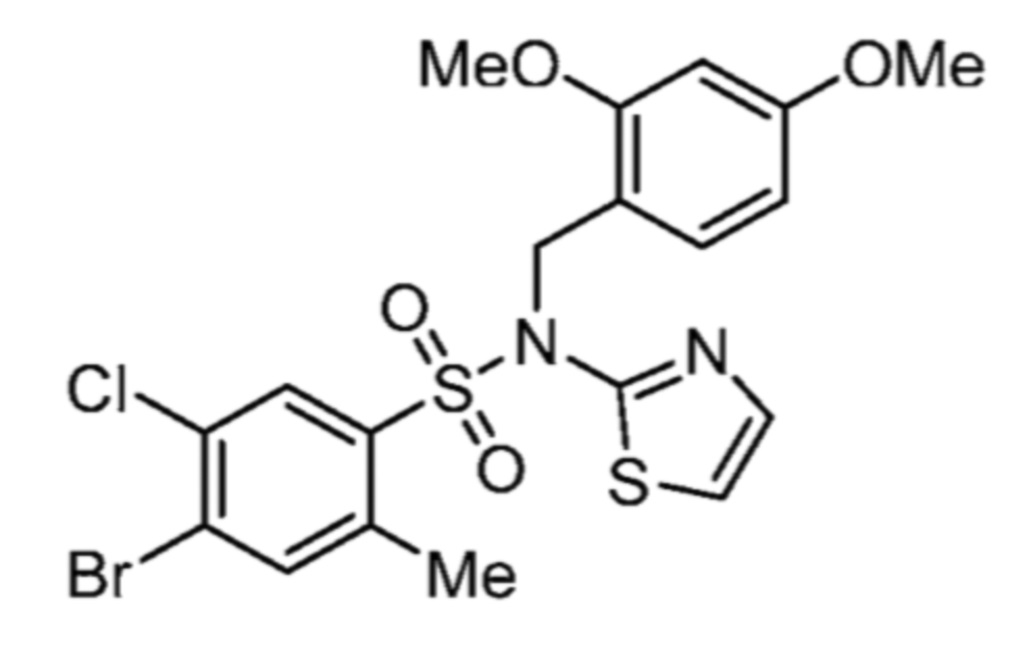
В смесь N-(2,4-диметоксибензил)тиазол-2-амина (0,30 г, 1,2 ммоль) в безводном тетрагидрофуране (10 мл) добавляли 1 M раствор бис(триметилсилил)амида лития в тетрагидрофуране (1,7 мл, 1,7 ммоль) при -78°C. Смесь нагревали до 0°C и перемешивали в течение 30 минут. Затем смесь охлаждали до -78°C и туда добавляли раствор 4-бром-5-хлор-2-метилбензолсульфонилхлорида (0,40 г, 1,3 ммоль) в безводном тетрагидрофуране (2 мл). Смесь перемешивали при -78°C в течение 30 минут, нагревали до температуры окружающей среды и перемешивали в течение 11 часов. Смесь выливали в насыщенный раствор хлорида аммония (10 мл) и водную фазу экстрагировали этилацетатом (3×30 мл). Объединенную органическую фазу промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, используя 20% этилацетат в петролейном эфире в качестве элюента, получали указанное в заголовке соединение в виде твердого вещества желтого цвета (0,26 г, выход 26%):1H ЯМР (400 МГц, CDCl3) δ 8,01 (с, 1H), 7,52 (с, 1H), 7,46 (д, J=3,6 Гц, 1H), 7,18 (д, J=8,4 Гц, 1H), 7,04 (д, J=3,6 Гц, 1H), 6,39-6,31 (м, 2H), 5,13 (с, 2H), 3,80-3,67 (м, 6H), 2,46 (с, 3H); MS (ES+) m/z 519,0 (M+1), 521,0 (M+1).
Стадия 4. Получение (S)-5-хлор-N-(2,4-диметоксибензил)-4-((1-(2-фторфенил)этил)амино)-2-метил-N-(тиазол-2-ил)бензолсульфонамида

Раствор 4-бром-5-хлор-N-(2,4-диметоксибензил)-2-метил-N-(тиазол-2-ил)бензолсульфонамида (0,14 г, 0,27 ммоль), (S)-1-(2-фторфенил)этанамина (0,071 г, 0,40 ммоль), бис(дибензилиденацетон)палладия(0) (0,031 г, 0,054 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантена (0,031 г, 0,054 ммоль) и карбоната цезия (0,35 г, 1,1 ммоль) в безводном толуоле (2,0 мл) дегазировали посредством барботирования азотом и затем нагревали до 100°C в течение 12 часов. Смесь выливали в воду (10 мл) и водную фазу экстрагировали этилацетатом (3×30 мл). Объединенную органическую фазу промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 25% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде масла желтого цвета (0,12 г, выход 77%): MS (ES+) m/z 576,1 (M+1), 578,1 (M+1).
Стадия 5. Получение (S)-5-хлор-4-((1-(2-фторфенил)этил)амино)-2-метил-N-(тиазол-2-ил)бензолсульфонамида

К (S)-5-хлор-N-(2,4-диметоксибензил)-4-((1-(2-фторфенил)этил)амино)-2-метил-N-(тиазол-2-ил)бензолсульфонамиду (0,11 г, 0,19 ммоль) добавляли 4 M раствор хлороводорода в 1,4-диоксане (110 мл, 440 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Концентрированием in vacuo и очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,038 г, выход 46%):1H ЯМР (400 МГц, CDCl3) δ 12,41 (с, 1H), 8,01 (с, 1H), 7,27-7,20 (м, 2H), 7,14-7,04 (м, 2H), 7,01 (д, J=4,4 Гц, 1H), 6,43 (д, J=4,4 Гц, 1H), 6,24 (с, 1H), 5,01 (д, J=6,4 Гц, 1H), 4,90 (квин, J=6,4 Гц, 1H), 2,32 (с, 3H), 1,62 (д, J=6,4 Гц, 3H); MS (ES+) m/z 426,1 (M+1), 428,1 (M+1).
ПРИМЕР 270
Синтез (R)-5-хлор-4-((1-(5-хлор-2-фторфенил)-2,2,2-трифторэтил)амино)-2-фтор-N-(пиримидин-4-ил)бензолсульфонамида

Стадия 1. Получение 4-бром-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(пиримидин-4-ил)бензолсульфонамида

В раствор N-(2,4-диметоксибензил)пиримидин-4-амина (1,65 г, 6,71 ммоль) и 1,4-диазабицикло[2.2.2]октана (1,51 г, 13,43 ммоль) в ацетонитриле (20 мл) добавляли раствор 4-бром-5-хлор-2-фторбензол-1-сульфонилхлорида (3,10 г, 10,07 ммоль) в ацетонитриле (10 мл) при 0°C. Смесь перемешивали при 15°C в течение 12 ч и затем разводили водой (30 мл). Смесь экстрагировали этилацетатом (3×30 мл) и органические слои объединяли. Органический слой промывали солевым раствором (3×10 мл), сушили над сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с 50% этилацетатом в петролейном эфире для того, чтобы получать указанное в заголовке соединение в виде твердого вещества желтого цвета (2,00 г, выход 58%):1H ЯМР (400 МГц, CDCl3) δ 8,80 (д, J=0,8 Гц, 1H), 8,49 (д, J=6,0 Гц, 1H), 8,10 (д, J=6,6 Гц, 1H), 7,44 (д, J=9,0 Гц, 1H), 7,21 (д, J=9,0 Гц, 1H), 7,16 (дд, J=5,8, 1,2 Гц, 1H), 6,44-6,39 (м, 2H), 5,23 (с, 2H), 3,78 (д, 6H); MS (ES+) m/z 538 (M+23), 540 (M+23).
Стадия 1. Получение 1-(5-хлор-2-фторфенил)-2,2,2-трифторэтан-1-ола

В смесь 5-хлор-2-фторбензальдегида (3,00 г, 18,9 ммоль) в безводном тетрагидрофуране (30 мл) добавляли триметил(трифторметил)силан (3,50 г, 24,6 ммоль) и смесь перемешивали при 0°C в течение 10 минут. Затем туда добавляли 1 M раствор фторида тетрабутиламмония в тетрагидрофуране (0,95 мл, 0,95 ммоль) при 0°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 12 часов. Смесь разводили водой (50 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 1-2% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (4,20 г, выход 97%):1H ЯМР (400 МГц, CDCl3) δ 7,54 (дд, J=6,0, 2,4 Гц, 1H), 7,28 (ддд, J=8,8, 4,4, 2,8 Гц, 1H), 6,98 (т, J=9,2 Гц, 1H), 5,32 (квин, J=6,0 Гц, 1H), 2,76 (д, J=5,2 Гц, 1H).
Стадия 2. Получение 1-(5-хлор-2-фторфенил)-2,2,2-трифторэтан-1-она
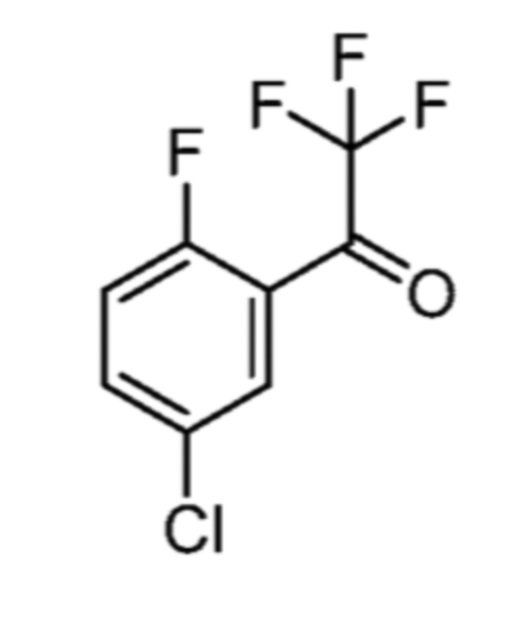
В раствор 1-(5-хлор-2-фторфенил)-2,2,2-трифторэтанола (4,20 г, 18,4 ммоль) в этилацетате (50 мл) частями добавляли 2-йодоксибензойную кислоту (15,4 г, 55,1 ммоль). Смесь нагревали с обратным холодильником в течение 36 ч. После охлаждения до температуры окружающей среды, смесь фильтровали и фильтрат концентрировали in vacuo для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (4,00 г, выход 96%):1H ЯМР (400 МГц, CDCl3) δ 7,86 (дд, J=5,6, 2,4 Гц, 1H), 7,66 (ддд, J=8,8, 4,0, 2,8 Гц, 1H), 7,23 (дд, J=10,0, 8,8 Гц, 1H).
Стадия 3. Получение (R)-N-(1-(5-хлор-2-фторфенил)-2,2,2-трифторэтилиден)-2-метилпропан-2-сульфинамида

В раствор 1-(5-хлор-2-фторфенил)-2,2,2-трифторэтанона (2,00 г, 8,83 ммоль) и (R)-2-метилпропан-2-сульфинамида (1,28 г, 10,6 ммоль) в безводном простом диэтиловом эфире (20 мл) добавляли изопропоксид титана(IV) (6,27 г, 22,1 ммоль) по каплям. Затем реакционную смесь нагревали до 50°C в течение 12 часов. После охлаждения до температуры окружающей среды, реакционную смесь разводили водой (30 мл) и фильтровали. Фильтрат экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 5 до 20% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (1,50 г, выход 52%):1H ЯМР (400 МГц, CDCl3) δ 7,46 (ддд, J=8,8, 4,4, 2,4 Гц, 1H), 7,34-7,30 (м, 1H), 7,11 (т, J=8,8 Гц, 1H), 1,35 (с, 9H).
Стадия 4. Получение (R)-N-((R)-1-(5-хлор-2-фторфенил)-2,2,2-трифторэтил)-2-метилпропан-2-сульфинамида

В раствор (R,Z)-N-(1-(5-хлор-2-фторфенил)-2,2,2-трифторэтилиден)-2-метилпропан-2-сульфинамида (1,50 г, 4,55 ммоль) и изопропоксида титана(IV) (1,29 г, 4,55 ммоль) в простом диэтиловом эфире (15 мл) частями добавляли борогидрид натрия (0,52 мг, 13 ммоль) при -78°C. Реакционную смесь перемешивали при -78°C в течение 5 часов. Затем смесь гасили водой (30 мл) и фильтровали. Фильтрат экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 10-25% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного масла (1,00 г, выход 66%):1H ЯМР (400 МГц, CDCl3) δ 7,45-7,36 (м, 2H), 7,12 (т, J=9,2 Гц, 1H), 5,29-5,20 (м, 1H), 3,94 (д, J=4,4 Гц, 1H), 1,26 (с, 9H).
Стадия 5. Получение гидрохлорида (R)-1-(5-хлор-2-фторфенил)-2,2,2-трифторэтан-1-амина

К (R)-N-((R)-1-(5-хлор-2-фторфенил)-2,2,2-трифторэтил)-2-метилпропан-2-сульфинамиду (1,00 г, 3,01 ммоль) добавляли 4 M раствор хлороводорода в метаноле (10 мл, 40 ммоль) и смесь перемешивали при температуре окружающей среды в течение 2 часов. Смесь концентрировали in vacuo. Остаток разводили метанолом (1 мл) и очищали посредством перекристаллизации из простого трет-бутилметилового эфира (30 мл) для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,63 г, выход 79%):1H ЯМР (400 МГц, CD3OD) δ 7,75-7,63 (м, 2H), 7,41 (т, J=9,4 Гц, 1H), 5,69 (к, J=7,2 Гц, 1H), способные к обмену протоны не наблюдали.
Стадия 6. Получение (R)-5-хлор-4-((1-(5-хлор-2-фторфенил)-2,2,2-трифторэтил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(пиримидин-4-ил)бензолсульфонамида

В раствор (R)-1-(5-хлор-2-фторфенил)-2,2,2-трифторэтанамина (0,10 г, 0,38 ммоль) и 4-бром-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(пиримидин-4-ил)бензолсульфонамида (0,16 г, 0,32 ммоль) в безводном толуоле (2,0 мл) добавляли трет-бутоксид калия (0,14 г, 1,26 ммоль) и хлор(2-дициклогексилфосфино-2′,6′-диметокси-1,1′-бифенил)[2-(2′-амино-1,1′-бифенил)]палладий(II) (0,048 г, 0,063 ммоль). Смесь затем нагревали до 60°C в течение 12 ч. После охлаждения до температуры окружающей среды, реакционную смесь концентрировали in vacuo и разводили водой (30 мл). Смесь экстрагировали этилацетатом (3×20 мл). Объединенные органические экстракты промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 50% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде масла желтого цвета (0,016 г, выход 8%):1H ЯМР (400 МГц, CDCl3) δ 8,81 (с, 1H), 8,47 (д, J=6,0 Гц, 1H), 7,97 (д, J=6,8 Гц, 1H), 7,46-7,39 (м, 2H), 7,25 (дд, J=6,0, 1,2 Гц, 1H), 7,23-7,14 (м, 2H), 6,44-6,38 (м, 2H), 6,31 (д, J=11,6 Гц, 1H), 5,62 (д, J=7,6 Гц, 1H), 5,30-5,25 (м, 1H), 5,24 (с, 2H), 3,78 (д, J=4,0 Гц, 6H); MS (ES+) m/z 663,1 (M+1), 665,1 (M+1).
Стадия 7. Получение (R)-5-хлор-4-((1-(5-хлор-2-фторфенил)-2,2,2-трифторэтил)амино)-2-фтор-N-(пиримидин-4-ил)бензолсульфонамида.

В раствор (R)-5-хлор-4-((1-(5-хлор-2-фторфенил)-2,2,2-трифторэтил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(пиримидин-4-ил)бензолсульфонамида (0,020 г, 0,030 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (1,54 г, 13,5 ммоль) при температуре окружающей среды и получаемую смесь перемешивали в течение 30 минут. Концентрированием in vacuo и очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,011 г, выход 71%):1H ЯМР (400 МГц, CDCl3) δ 8,72 (с, 1H), 8,32 (д, J=6,0 Гц, 1H), 7,89 (д, J=7,2 Гц, 1H), 7,37-7,28 (м, 2H), 7,16 (д, J=5,6 Гц, 1H), 7,07 (т, J=9,2 Гц, 1H), 6,26 (д, J=11,6 Гц, 1H), 5,51 (д, J=7,6 Гц, 1H), 5,23-5,12 (м, 1H), NH не наблюдали; MS (ES+) m/z 513,0 (M+1), 515,0 (M+1).
ПРИМЕР 271
Синтез 4-((2-((диметиламино)метил)бензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида
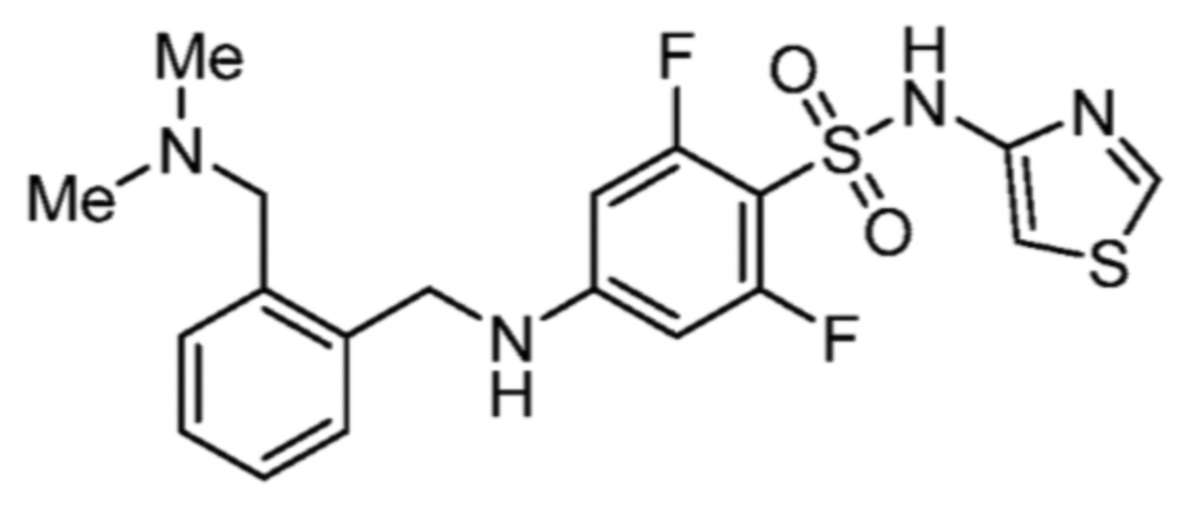
Стадия 1. Получение 2-((диметиламино)метил)бензонитрила

Следуя процедуре как описано для примера 16, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять азетидин на гидрохлорид диметиламина, указанное в заголовке соединение получали в виде масла желтого цвета (0,600 г, выход 73%):1H ЯМР (400 МГц, CDCl3) δ 7,64 (д, J=7,4 Гц, 1H), 7,60-7,54 (м, 2H), 7,39-7,33 (м, 1H), 3,63 (с, 2H), 2,30 (с, 6H); MS (ES+) m/z 161,0 (M+1).
Стадия 2. Получение 1-(2-(аминометил)фенил)-N,N-диметилметанамина

В смесь 2-((диметиламино)метил)бензонитрила (0,600 г, 3,74 ммоль) в концентрированном гидроксиде аммония (4 мл) и метаноле (20 мл) добавляли ренеевский никель (0,320 г, 3,74 ммоль). Суспензию дегазировали в вакууме и продували водородом несколько раз. Смесь перемешивали в атмосфере водорода (50 фунтов/дюйм2) при температуре окружающей среды в течение 12 ч. Реакционную смесь фильтровали и фильтрат концентрировали in vacuo для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (0,300 г, выход 49%):1H ЯМР (400 МГц, CDCl3) δ 7,19-7,12 (м, 2H), 7,11-7,05 (м, 2H), 3,71 (с, 2H), 3,31 (с, 2H), 2,09 (с, 6H), NH не наблюдали; MS (ES+) m/z 147,9 (M+1-14).
Стадия 3. Получение трет-бутил (4-((2-((диметиламино)метил)бензил)амино)-2,6-дифторфенил)сульфонил(тиазол-4-ил)карбамата

В раствор трет-бутил тиазол-4-ил((2,4,6-трифторфенил)сульфонил)карбамата (0,721 г, 1,83 ммоль) и 1-(2-(аминометил)фенил)-N,N-диметилметанамина (0,300 г, 1,83 ммоль) в безводном N,N-диметилформамиде (10 мл) добавляли карбонат калия (0,505 г, 3,66 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 3 ч. Затем туда добавляли воду (10 мл) и смесь экстрагировали этилацетатом (3×10 мл). Объединенные органические слои промывали солевым раствором (3×10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 25% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,080 г, выход 8%):1H ЯМР (400 МГц, CDCl3) δ 8,81 (д, J=2,2 Гц, 1H), 7,51 (д, J=2,2 Гц, 1H), 7,37-7,30 (м, 3H), 7,27-7,25 (м, 1H), 6,18 (д, J=11,8 Гц, 2H), 4,34 (с, 2H), 3,47 (с, 2H), 2,30 (с, 6H), 1,40 (с, 9H), NH не наблюдали; MS (ES+) m/z 438,9 (M - 99).
Стадия 4. Получение 4-((2-((диметиламино)метил)бензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

К (4-((2-((диметиламино)метил)бензил)амино)-2,6-дифторфенил)сульфонил(тиазол-4-ил) карбамату (0,080 г, 0,148 ммоль) добавляли 4 M раствор хлороводорода в диоксане (2 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 8 ч. Затем реакционную смесь концентрировали in vacuo и остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиной кислоты в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,012 г, выход 19%):1H ЯМР (400 МГц, CD3OD) δ 8,74 (д, J=2,2 Гц, 1H), 7,49-7,35 (м, 4H), 6,97 (д, J=2,2 Гц, 1H), 6,30-6,23 (м, 2H), 4,45 (с, 2H), 4,04 (с, 2H), 2,64 (с, 6H), NH не наблюдали; MS (ES+) m/z 439,0 (M+1).
ПРИМЕР 272
Синтез формиата 4-((2-(азетидин-1-илметил)бензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида
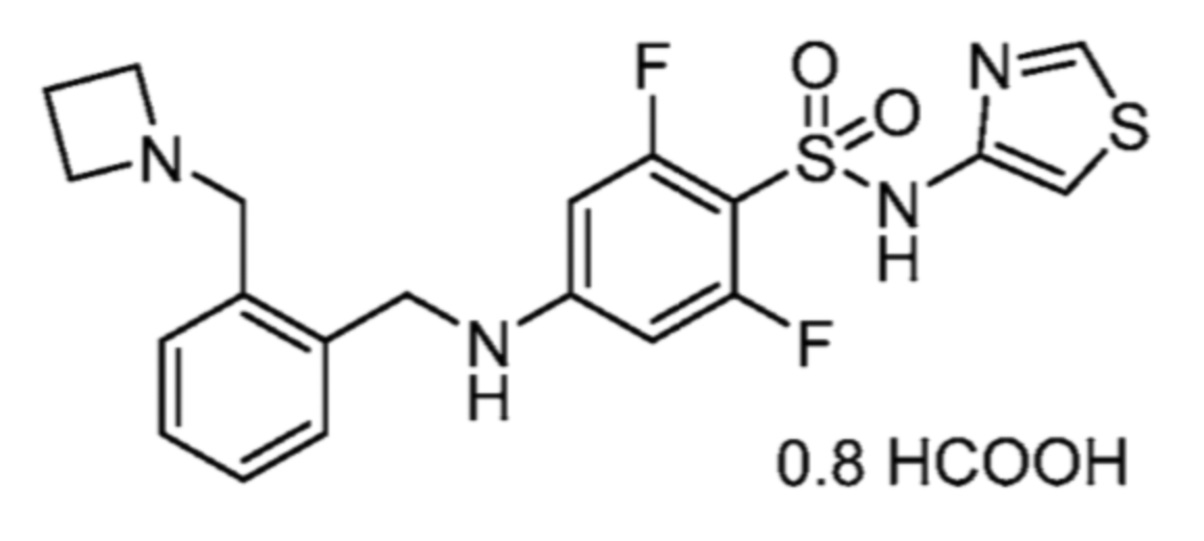
Раствор 2,4,6-трифтор-N-(тиазол-4-ил)бензолсульфонамида (1,00 г, 2,54 ммоль), (2-(азетидин-1-илметил)фенил)метанамина (0,446 г, 2,53 ммоль) и N,N-диизопропилэтиламина (1,0 мл, 6,1 ммоль) в безводном диметилсульфоксиде (10 мл) перемешивали при температуре окружающей среды в течение 2 ч. Затем в смесь добавляли насыщенный хлорид аммония (10 мл) и смесь экстрагировали этилацетатом (3×10 мл). Объединенную органическую фазу промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали остаток, который растворяли в дихлорметане (5 мл). Затем туда добавляли трифторуксусную кислоту (2 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 18 ч. Реакционную смесь концентрировали in vacuo и остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,05% муравьиной кислоты в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,128 г, выход 11%):1H-ЯМР (300 МГц, DMSO-d6) δ 8,89 (д, J=2,2 Гц, 1H), 8,16 (с, 0,8 H), 7,65-7,63 (м, 1H), 7,30-7,29 (м, 1H), 7,27-7,21 (м, 3H), 6,88 (д, J=2,1 Гц, 1H), 6,37 (д, J=12,6 Гц, 2H), 4,39 (с, 2H), 3,65 (с, 2H), 3,22 (т, J=7,1 Гц, 4H), 2,07-2,00 (м, 2H), NH и COOH не наблюдали; MS (ES+) m/z 451,1 (M+1).
ПРИМЕР 273
Синтез (S)-4-((1-(2,5-дифторфенил)этил)амино)-N-(пиримидин-4-ил)-3-(трифторметил)бензолсульфонамида

Стадия 1. Получение N-(2,4-диметоксибензил)-4-фтор-N-(пиримидин-4-ил)-3-(трифторметил)бензолсульфонамида
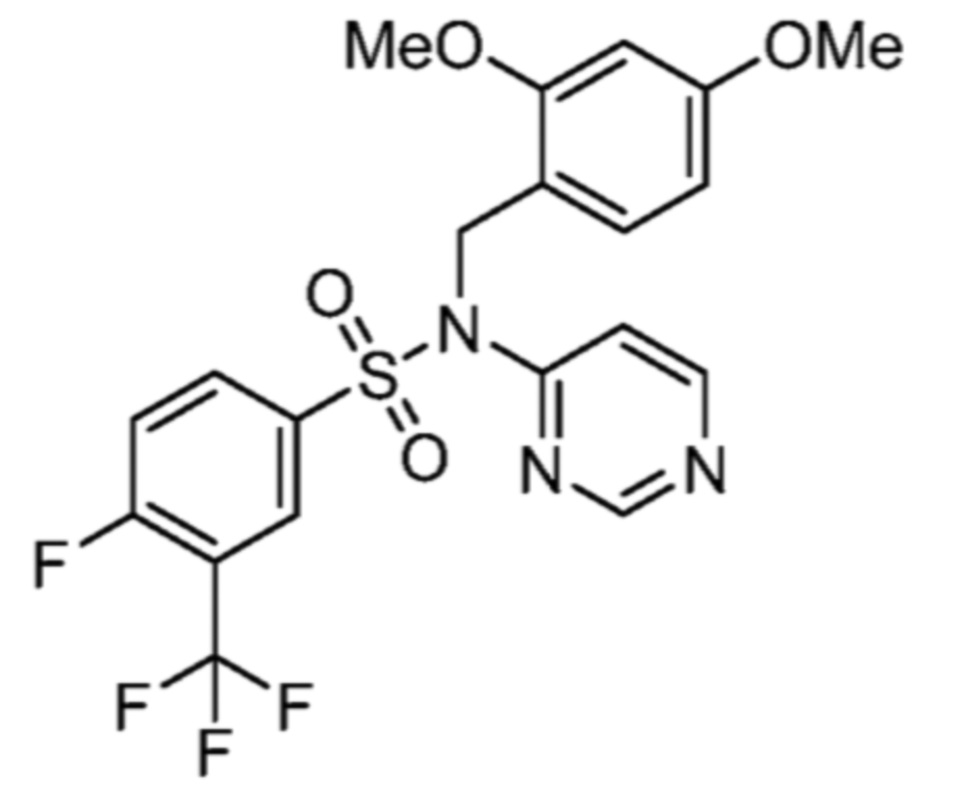
В раствор N-(2,4-диметоксибензил)пиримидин-4-амина (0,70 г, 2,80 ммоль) и 1,4-диазабицикло[2.2.2]октана (0,64 г, 5,70 ммоль) в безводном ацетонитриле (30 мл) добавляли 4-фтор-3-(трифторметил)бензолсульфонилхлорид (1,50 г, 5,70 ммоль) по каплям при 0°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 12 ч. Затем реакционную смесь фильтровали и фильтрат концентрировали in vacuo. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием 10% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,90 г, выход 67%):1H ЯМР (400 МГц, CDCl3) δ 8,86 (д, J=0,8 Гц, 1H), 8,53 (д, J=6,0 Гц, 1H), 8,08-8,22 (м, 2H), 7,27-7,34 (м, 1H), 7,10-7,17 (м, 2H), 6,41 (дд, J=8,4, 2,4 Гц, 1H), 6,37 (д, J=2,4 Гц, 1H), 5,17 (с, 2H), 3,78 (с, 3H), 3,65 (с, 3H).
Стадия 2. Получение (S)-4-((1-(2,5-дифторфенил)этил)амино)-N-(2,4-диметоксибензил)-N-(пиримидин-4-ил)-3-(трифторметил)бензолсульфонамида

В раствор N-(2,4-диметоксибензил)-4-фтор-N-(пиримидин-4-ил)-3-(трифторметил)бензолсульфонамида (0,40 г, 0,85 ммоль) и гидрохлорида (S)-1-(2,5-дифторфенил)этанамина (0,20 г, 1,00 ммоль) в безводном N,N-диметилформамиде (5 мл) добавляли карбонат калия (0,47 г, 3,3 ммоль). Смесь перемешивали при температуре окружающей среды в течение 12 ч. Реакцию гасили добавлением воды (50 мл) и экстрагировали этилацетатом (3×40 мл). Объединенные органические слои концентрировали при пониженном давлении, чтобы получать остаток, который очищали посредством препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 33% этилацетата в петролейном эфире, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,090 г, выход 17%): MS (ES+) m/z 609,1 (M+1).
Стадия 2. Получение (S)-4-((1-(2,5-дифторфенил)этил)амино)-N-(пиримидин-4-ил)-3-(трифторметил)бензолсульфонамида
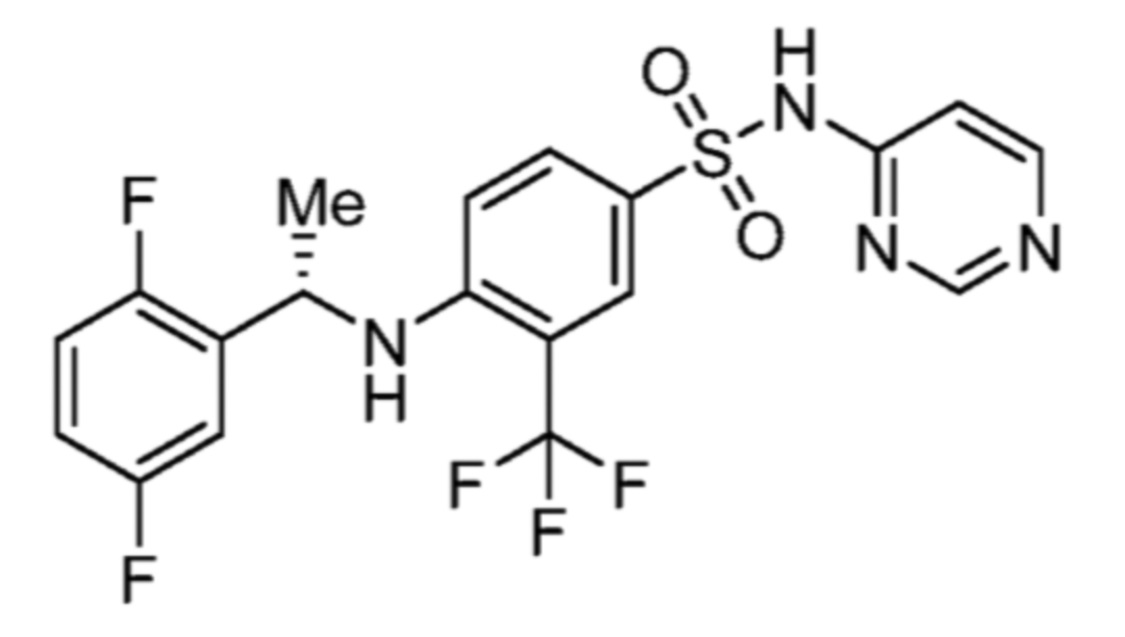
В смесь (S)-4-((1-(2,5-дифторфенил)этил)амино)-N-(2,4-диметоксибензил)-N-(пиримидин-4-ил)-3-(трифторметил)бензолсульфонамида (0,080 г, 0,13 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (1 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении, разводили метанолом (1 мл) и фильтровали. Фильтрат концентрировали in vacuo для того, чтобы получать остаток, который очищали с помощью HPLC с обращенной фазой, осуществляя элюирование с градиентом ацетонитрила в воде (содержащей 0,225% муравьиную кислоту), чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,018 г, выход 29%):1H ЯМР (400 МГц, CDCl3) δ 8,87 (с, 1H), 8,50 (д, J=6,0 Гц, 1H), 8,06 (д, J=2,0 Гц, 1H), 7,80 (дд, J=8,8, 2,0 Гц, 1H), 7,23 (д, J=5,6 Гц, 1H), 7,08 (тд, J=9,2, 4,4 Гц, 1H), 6,90-7,00 (м, 2H), 6,53 (д, J=8,8 Гц, 1H), 5,17 (ушир. с, 1H), 4,83-4,96 (м, 1H), 1,63 (д, J=6,8 Гц, 3H), NH не наблюдали; MS (ES+) m/z 459,0 (M+1).
ПРИМЕР 274
Синтез (S)-4-((1-(2,5-дифторфенил)этил)амино)-N-(тиазол-4-ил)-3-(трифторметил)бензолсульфонамида

Стадия 1. Получение трет-бутил ((4-бром-3-(трифторметил)фенил)сульфонил)(тиазол-4-ил)карбамата

В раствор трет-бутил тиазол-4-илкарбамата (1,00 г, 4,99 ммоль) в безводном тетрагидрофуране (15 мл) по каплям добавляли 1 M раствор бис(триметилсилил)амида лития в тетрагидрофуране (6,99 мл, 6,99 ммоль) при -78°C. Реакционную смесь нагревали до 0°C, перемешивали в течение 30 минут и охлаждали до -78°C. Затем туда добавляли раствор 4-бром-3-(трифторметил)бензолсульфонилхлорида (2,26 г, 6,99 ммоль) в тетрагидрофуране (8 мл) при -78°C. Реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч и затем разводили водой (50 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью HPLC с обращенной фазой, осуществляя элюирование с использованием ацетонитрила в воде (содержащей 0,225% муравьиную кислоту), получали указанное в заголовке соединение в виде твердого вещества желтого цвета (1,10 г, выход 45%):1H ЯМР (400 МГц, CDCl3) δ 8,81 (д, J=2,0 Гц, 1H), 8,48 (д, J=2,0 Гц, 1H), 8,20 (дд, J=8,4, 2,0 Гц, 1H), 7,96 (д, J=8,4 Гц, 1H), 7,58 (д, J=2,0 Гц, 1H), 1,37 (с, 9H); MS (ES+) m/z 387,0 (M - 99), 389,0 (M - 99).
Стадия 2. Получение трет-бутил (S)-((4-((1-(2,5-дифторфенил)этил)амино)-3-(трифторметил)фенил)сульфонил)(тиазол-4-ил)карбамата
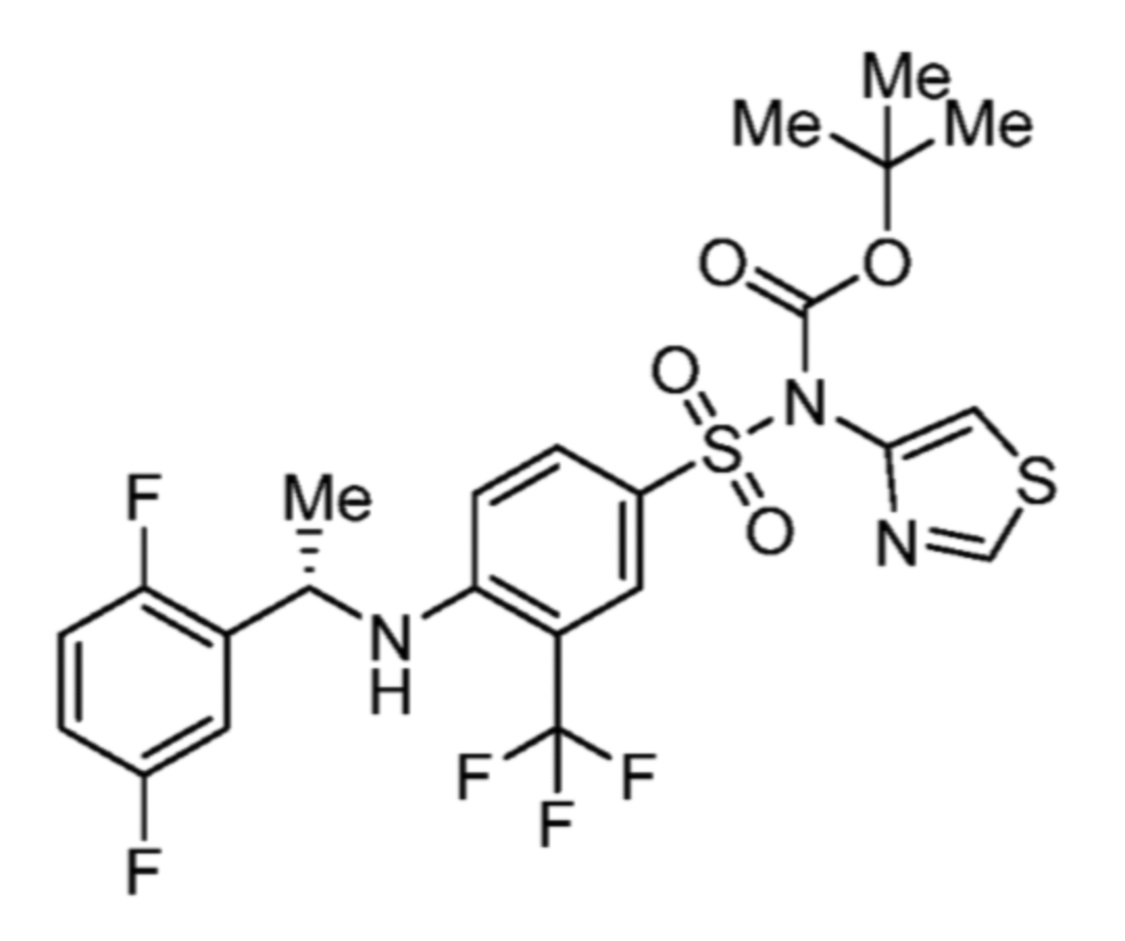
В раствор трет-бутил (4-бром-3-(трифторметил)фенил)сульфонил(тиазол-4-ил)карбамата (0,20 г, 0,41 ммоль), гидрохлорида (S)-1-(2,5-дифторфенил)этанамина (0,095 г, 0,49 ммоль) и трет-бутоксида калия (0,18 г, 1,64 ммоль) в безводном тетрагидрофуране (3 мл) добавляли аддукт метил-т-бутилового эфира хлор(2-дициклогексилфосфино-2',6'-ди-и-пропокси-1,1'-бифенил)[2-(2-аминоэтилфенил)]палладия(II) (0,062 г, 0,082 ммоль). Смесь нагревали до 80°C в течение 12 ч и затем концентрировали in vacuo. Остаток очищали с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 25% этилацетата в петролейном эфире, чтобы получать указанное в заголовке соединение в виде масла желтого цвета (0,10 г, выход 43%):1H ЯМР (400 МГц, CDCl3) δ 8,78 (д, J=2,0 Гц, 1H), 8,24 (д, J=2,0 Гц, 1H), 8,00 (дд, J=9,2, 2,0 Гц, 1H), 7,51 (д, J=2,0 Гц, 1H), 7,13-7,04 (м, 1H), 7,00-6,90 (м, 2H), 6,57 (д, J=8,6 Гц, 1H), 5,20 (д, J=4,4 Гц, 1H), 4,97 (к, J=6,4 Гц, 1H), 1,65 (д, J=6,4 Гц, 3H), 1,33 (с, 9H).
Стадия 2. Получение (S)-4-((1-(2,5-дифторфенил)этил)амино)-N-(тиазол-4-ил)-3-(трифторметил)бензолсульфонамида
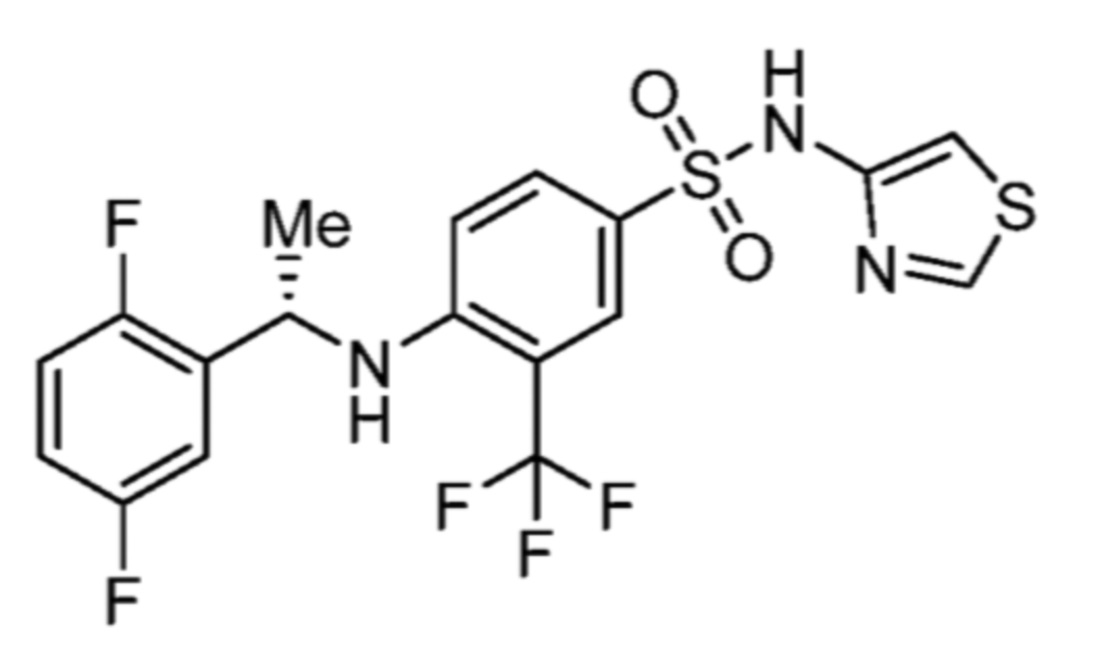
К (S)-трет-бутил(4-((-(2,5-дифторфенил)этил)амино)-3-(трифторметил)фенил)сульфонил-(тиазол-4-ил)карбамату (0,10 г, 0,18 ммоль) добавляли 4 M раствор хлороводорода в 1,4-диоксане (8 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 12 ч. Концентрированием in vacuo и очисткой остатка с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом ацетонитрила в воде (содержащей 0,225% муравьиную кислоту), получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,047 г, выход 54%):1H ЯМР (400 МГц, CDCl3) δ 9,06 (с, 1H), 8,66 (д, J=2,0 Гц, 1H), 7,90 (д, J=2,0 Гц, 1H), 7,64 (дд, J=8,4, 2,0 Гц, 1H), 7,10-7,04 (м, 1H), 7,03 (д, J=2,0 Гц, 1H), 6,99-6,88 (м, 2H), 6,43 (д, J=8,8 Гц, 1H), 5,07 (д, J=4,0 Гц, 1H), 4,93-4,81 (м, 1H), 1,61 (д, J=6,4 Гц, 3H).
ПРИМЕР 275
Синтез (S)-4-((1-(5-хлор-2-фторфенил)этил)амино)-2-фтор-5-метил-N-(пиримидин-4-ил)бензолсульфонамида

Стадия 1. Получение 4-бром-N-(2,4-диметоксибензил)-2-фтор-5-метил-N-(пиримидин-4-ил)бензолсульфонамида

В смесь N-(2,4-диметоксибензил)пиримидин-4-амина (3,00 г, 12,20 ммоль) и 1,4-диазабицикло[2.2.2]октана (2,74 г, 24,5 ммоль) в ацетонитриле (30 мл) добавляли раствор 4-бром-2-фтор-5-метилбензолсульфонилхлорида (5,28 г, 18,40 ммоль) в ацетонитриле (2 мл) при 0°C. Реакционную смесь перемешивали при 0°C в течение 30 минут и затем при температуре окружающей среды в течение 12 ч. Реакционную смесь концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 10 до 30% этилацетата в петролейном эфире, чтобы получать указанное в заголовке соединение в виде масла желтого цвета (4,82 г, выход 79%):1H ЯМР (400 МГц, CDCl3) δ 8,78 (с, 1H), 8,46 (д, J=5,8 Гц, 1H), 7,87 (д, J=7,2 Гц, 1H), 7,36 (д, J=9,4 Гц, 1H), 7,22 (дд, J=5,6, 4,8 Гц, 2H), 6,43-6,40 (м, 2H), 5,25 (с, 2H), 3,79 (с, 3H), 3,78 (с, 3H), 2,44 (с, 3H); MS (ES+) m/z 496,0 (M+1).
Стадия 2. Получение (S)-4-((1-(5-хлор-2-фторфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-5-метил-N-(пиримидин-4-ил)бензолсульфонамида

Смесь 4-бром-N-(2,4-диметоксибензил)-2-фтор-5-метил-N-(пиримидин-4-ил)бензолсульфонамида (2,00 г, 4,03 ммоль), гидрохлорида (S)-1-(5-хлор-2-фторфенил)этанамина (1,27 г, 6,05 ммоль), бис(дибензилиденацетон)палладия(0) (0,46 г, 0,81 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантена (0,47 г, 0,81 ммоль) и карбоната цезия (5,25 г, 16,10 ммоль) в безводном толуоле (20 мл) дегазировали и затем нагревали до 100°C в течение 12 часов. Смесь выливали в воду со льдом (20 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические экстракты промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 25% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде масла желтого цвета (2,37 г, количественный выход):1H ЯМР (400 МГц, CDCl3) δ 8,76 (с, 1H), 8,40 (д, J=5,8 Гц, 1H), 7,62 (д, J=7,8 Гц, 1H), 7,33 (д, J=6,2 Гц, 1H), 7,25-7,16 (м, 3H), 7,04 (т, J=9,2 Гц, 1H), 6,40-6,38 (м, 2H), 5,98 (д, J=13,0 Гц, 1H), 5,25 (д, J=2,8 Гц, 2H), 4,73 (к, J=6,4 Гц, 1H), 4,40 (д, J=5,2 Гц, 1H), 3,77 (с, 3H), 3,76 (с, 3H), 2,22 (с, 3H), 1,60 (д, J=6,6 Гц, 3H); MS (ES+) m/z 589,1 (M+1).
Стадия 3. Получение (S)-4-((1-(5-хлор-2-фторфенил)этил)амино)-2-фтор-5-метил-N-(пиримидин-4-ил)бензолсульфонамида

Следуя процедуре как описано в примере 7, стадии 2 и выполняя некритические вариации, чтобы заменять (S)-4-((1-(3-бромфенил)этил)амино)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамид на (S)-4-((1-(5-хлор-2-фторфенил)этил)амино)-N-(2,4-диметоксибензил)-2-фтор-5-метил-N-(пиримидин-4-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,83 г, выход 79%):1H ЯМР (400 МГц, CDCl3) δ 11,80 (ушир. с, 1H), 8,90 (с, 1H), 8,41 (д, J=6,0 Гц, 1H), 7,64 (д, J=8,0 Гц, 1H), 7,27-7,17 (м, 3H), 7,02 (т, J=9,2 Гц, 1H), 6,01 (д, J=12,4 Гц, 1H), 4,73 (к, J=6,4 Гц, 1H), 4,41 (д, J=5,4 Гц, 1H), 2,23 (с, 3H), 1,60 (д, J=6,8 Гц, 3H).
ПРИМЕР 276
Синтез (S)-4-((1-(2-хлор-5-фторфенил)пропил)амино)-2,6-дифтор-N-(пиримидин-4-ил)бензолсульфонамида

Стадия 1. Получение N-(2,4-диметоксибензил)-2,4,6-трифтор-N-(пиримидин-4-ил)бензолсульфонамида

В смесь N-(2,4-диметоксибензил)пиримидин-4-амина (1,12 г, 4,56 ммоль) и 1,4-диазабицикло[2.2.2]октана (1,02 г, 9,11 ммоль) в безводном ацетонитриле (2 мл) добавляли раствор 2,4,6-трифторбензолсульфонилхлорида (2,10 г, 9,11 ммоль) в безводногом ацетонитриле (1 мл) при 0°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 10 ч. Затем туда добавляли воду (15 мл) и этилацетат (100 мл). Органический слой промывали солевым раствором (2×30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием 33% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде твердого вещества желтого цвета (0,35 г, выход 17%):1H ЯМР (400 МГц, CDCl3) δ 8,79 (с, 1H), 8,48-8,46 (д, J=5,6 Гц, 1H), 7,26-7,24 (д, J=8,0 Гц, 1H), 7,07-7,06 (д, J=5,6 Гц, 1H), 6,81-6,76 (т, J=8,4 Гц, 2H), 6,47-6,43 (м, 2H), 5,26 (с, 2H), 3,84 (с, 3H), 3,79 (с, 3H).
Стадия 2. Получение (S)-4-((1-(2-хлор-5-фторфенил)пропил)амино)-N-(2,4-диметоксибензил)-2,6-дифтор-N-(пиримидин-4-ил)бензолсульфонамида

Раствор N-(2,4-диметоксибензил)-2,4,6-трифтор-N-(пиримидин-4-ил)бензолсульфонамида (0,17 г, 0,39 ммоль), (S)-1-(2-хлор-5-фторфенил)пропан-1-амина (0,073 г, 0,39 ммоль) и карбоната цезия (0,19 г, 0,58 ммоль) в безводном N,N-диметилформамиде (2 мл) нагревали до 80°C в течение 3 ч. После охлаждения до температуры окружающей среды, реакционную смесь гасили добавлением воды (10 мл) и экстрагировали этилацетатом (50 мл). Органический экстракт промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 33% петролейного эфира в этилацетате, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,070 г, выход 30%): MS (ES+) m/z 607,0 (M+1).
Стадия 2. Получение (S)-4-((1-(2-хлор-5-фторфенил)пропил)амино)-2,6-дифтор-N-(пиримидин-4-ил)бензолсульфонамида

Следуя процедуре как описано в примере 2, стадии 3 и выполняя некритические вариации, чтобы заменять (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(нафтален-1-ил)этил)амино)-N-(тиазол-2-ил)бензолсульфонамид на (S)-4-((1-(2-хлор-5-фторфенил)пропил)амино)-N-(2,4-диметоксибензил)-2,6-дифтор-N-(пиримидин-4-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,028 г, выход 52%):1H ЯМР (400 МГц, CD3OD) δ 8,57 (с, 1H), 8,31 (д, J=6,0 Гц, 1H), 7,44 (дд, J=4,8, 8,8 Гц, 1H), 7,07-7,00 (м, 3H), 6,07-6,04 (д, J=12,4 Гц, 2H), 4,65-4,62 (т, J=6,4 Гц, 1H), 1,87-1,75 (м, 2H), 1,04-1,00 (т, J=7,6 Гц, 3H), способные к обмену протоны не наблюдали; MS (ES+) m/z 457,1 (M+1).
ПРИМЕР 277
Синтез 2,2,2-трифторацетата (S)-5-хлор-4-((1-(2-хлорфенил)пропил)амино)-2-фтор-N-(пиримидин-4-ил)бензолсульфонамида

Стадия 1. Получение 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(пиримидин-4-ил)бензолсульфонамида

Следуя процедуре как описано в примере 191, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 3-хлор-2,4-дифторбензолсульфонилхлорид на 5-хлор-2,4-дифторбензолсульфонилхлорид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,9 г, выход 35%): MS (ES+) m/z 456,1 (M+1), 458,1 (M+1).
Стадия 2. Получение (S)-5-хлор-4-((1-(2-хлорфенил)пропил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(пиримидин-4-ил)бензолсульфонамида

Смесь 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(пиримидин-4-ил)бензолсульфонамида (0,225 г, 0,492 ммоль), (S)-1-(2-хлорфенил)пропан-1-амина (0,111 г, 0,54 ммоль) и карбоната калия (0,169 г, 1,23 ммоль) в безводном диметилсульфоксиде (4 мл) нагревали до 65°C в течение 3 ч. После охлаждения до температуры окружающей среды, туда добавляли насыщенный хлорид аммония (20 мл) и смесь экстрагировали этилацетатом (2×20 мл). Объединенную органическую фазу промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-60% этилацетата в гексанах, получали указанное в заголовке соединение в виде масла желтого цвета (0,140 г, выход 47%): MS (ES+) m/z 605,0 (M+1), 607,0 (M+1).
Стадия 3. Получение 2,2,2-трифторацетата (S)-5-хлор-4-((1-(2-хлорфенил)пропил)амино)-2-фтор-N-(пиримидин-4-ил)бензолсульфонамида

В смесь (S)-5-хлор-4-((1-(2-хлорфенил)пропил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(пиримидин-4-ил)бензолсульфонамида (0,140 г, 0,231 ммоль) в дихлорметанеа (4 мл) добавляли трифторуксусную кислоту (2 мл) и смесь перемешивали при температуре окружающей среды в течение 1 ч и затем концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 60% этилацетата (содержащего 0,1% трифторуксусной кислоты) в гексанах, чтобы получать указанное в заголовке соединение в виде бесцветной пены (0,089 г, выход 85%):1H-ЯМР (300 МГц, DMSO-d6) δ 8,59 (с, 1H), 8,29 (д, J=6,1 Гц, 1H), 7,73 (д, J=7,4 Гц, 1H), 7,51 (дд, J=7,5, 1,9 Гц, 1H), 7,45 (дд, J=7,6, 1,6 Гц, 1H), 7,34-7,23 (м, 2H), 6,95 (д, J=6,3 Гц, 1H), 6,81 (д, J=7,0 Гц, 1H), 6,12 (д, J=13,1 Гц, 1H), 4,67 (к, J=6,9 Гц, 1H), 2,10-1,95 (м, 1H), 1,87-1,73 (м, 1H), 0,94 (т, J=7,3 Гц, 3H), NH и COOH не наблюдали;19F ЯМР (282 МГц, DMSO-d6) δ -74,8 (с, 3F), -109,6 (с, 1F); MS (ES+) m/z 455,0 (M+1), 457,0 (M+1).
ПРИМЕР 278
Синтез формиата (S)-4-((2-(1-(азетидин-1-ил)этил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение (2-бром-6-фторфенил)метанол

В раствор 2-бром-6-фторбензальдегида (35,0 г, 172 ммоль) в метаноле (200 мл) добавляли борогидрид натрия (3,26 г, 86,2 ммоль) при 0°C. Смесь перемешивали при 0°C в течение 1 ч, разводили водой (100 мл) и затем концентрировали при пониженном давлении. Оставшуюся водную фазу экстрагировали этилацетатом (3×200 мл). Объединенные органические слои промывали солевым раствором (3×100 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 10-20% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (33,4 г, выход 94%):1H ЯМР (400 МГц, CDCl3) δ 7,45-7,33 (м, 1H), 7,23-6,98 (м, 2H), 4,86 (ушир. с, 2H), OH не наблюдали.
Стадия 2. Получение ((2-бром-6-фторбензил)окси)триизопропилсилана

В раствор (2-бром-6-фторфенил)метанола (57,0 г, 278 ммоль) и имидазола (37,9 г, 556 ммоль) в дихлорметане (300 мл) добавляли хлортриизопропилсилан (107 г, 556 ммоль) и смесь перемешивали при температуре окружающей среды в течение 12 ч. Реакционную смесь концентрировали при пониженном давлении и остаток разводили водой (400 мл) и экстрагировали этилацетатом (3×400 мл). Объединенные органические слои промывали солевым раствором (3×100 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-5% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде масла желтого цвета (95,0 г, выход 94%):1H ЯМР (400 МГц, CDCl3) δ 7,39 (д, J=7,8 Гц, 1H), 7,15 (дт, J=6,0, 8,0 Гц, 1H), 7,07-6,99 (м, 1H), 4,94 (д, J=1,8 Гц, 2H), 1,24-1,15 (м, 3H), 1,14-1,10 (м, 18H).
Стадия 3. Получение 3-фтор-2-(((триизопропилсилил)окси)метил)бензальдегида

В смесь ((2-бром-6-фторбензил)окси)триизопропилсилана (65,0 г, 180 ммоль), 2-изоциано-2-метилпропана (22,4 г, 270 ммоль), ацетата палладия(II) (4,04 г, 18,0 ммоль), карбоната натрия (19,1 г, 180 ммоль) и (2-бифенил)ди-трет-бутилфосфина (10,7 г, 36,0 ммоль) в N,N-диметилформамиде (500 мл) добавляли триэтилсилан (62,8 г, 540 ммоль). Реакционную смесь дегазировали и затем нагревали до 65°C в течение 12 ч. После охлаждения до температуры окружающей среды, реакционную смесь разводили водой (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали солевым раствором (3×50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-5% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде масла желтого цвета (28,0 г, выход 50%):1H ЯМР (400 МГц, CDCl3) δ 10,45 (с, 1H), 7,70-7,64 (м, 1H), 7,35-7,27 (м, 2H), 5,10 (д, J=1,4 Гц, 2H), 1,03 (с, 3H), 0,99-0,95 (м, 18H).
Стадия 4. Получение (R)-N-(3-фтор-2-(((триизопропилсилил)окси)метил)бензилиден)-2-метилпропан-2-сульфинамида

В раствор 3-фтор-2-(((триизопропилсилил)окси)метил)бензальдегида (14,0 г, 45,1 ммоль) и (R)-2-метил-2-пропансульфинамида (6,01 г, 49,6 ммоль) в дихлорметане (150 мл) добавляли карбонат цезия (29,4 г, 90,2 ммоль) и смесь перемешивали при температуре окружающей среды в течение 12 ч. Затем реакционную смесь фильтровали и фильтрат концентрировали in vacuo. Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием петролейного эфира, получали указанное в заголовке соединение в виде бесцветного масла (9,00 г, выход 48%):1H ЯМР (400 МГц, CDCl3) δ 9,11 (с, 1H), 7,90 (д, J=7,6 Гц, 1H), 7,42-7,33 (м, 1H), 7,25-7,15 (м, 1H), 5,11 (кд, J=12,0, 1,6 Гц, 2H), 1,30 (с, 9H), 1,22-1,13 (м, 3H), 1,09-1,05 (м, 18H); MS (ES+) m/z 414,4 (M+1).
Стадия 5. Получение (R)-N-((S)-1-(3-фтор-2-(((триизопропилсилил)окси)метил)фенил)этил)-2-метилпропан-2-сульфинамида

В раствор (R)-N-(3-фтор-2-(((триизопропилсилил)окси)метил)бензилиден)-2-метилпропан-2-сульфинамида (9,00 г, 21,8 ммоль) в дихлорметане (100 мл) добавляли по каплям 3,0 M раствор бромида метилмагния в простом диэтиловом эфире (14,5 мл, 43,5 ммоль) при -45°C. Смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 2 ч. Затем туда добавляли насыщенный хлорид аммония (500 мл) и смесь экстрагировали дихлорметаном (3×500 мл). Объединенные органические слои промывали солевым раствором (200 мл) и сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 5-30% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного масла (7,60 г, выход 63%):1H ЯМР (400 МГц, CDCl3) δ 7,32-7,28 (м, 1H), 7,27-7,22 (м, 1H), 6,97 (ддд, J=9,6, 8,0, 1,6 Гц, 1H), 5,13 (к, J=6,4 Гц, 1H), 5,06-4,80 (м, 2H), 3,38 (ушир. с, 1H), 1,59-1,56 (м, 3H), 1,19 (с, 9H), 1,18-1,13 (м, 3H), 1,10-1,05 (м, 18 H).
Стадия 6. Получение гидрохлорида (S)-(2-(1-аминоэтил)-6-фторфенил)метанол

К (R)-N-((S)-1-(3-фтор-2-(((триизопропилсилил)окси)метил)фенил)этил)-2-метилпропан-2-сульфинамиду (7,60 г, 17,7 ммоль) добавляли 4,0 M раствор хлороводорода в метаноле (100 мл, 400,0 ммоль) и смесь перемешивали при температуре окружающей среды в течение 12 ч. Смесь концентрировали in vacuo для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (3,60 г, выход 100%):1H ЯМР (400 МГц, DMSO-d6) δ 7,31-7,26 (м, 2H), 7,02 (ддд, J=9,6, 8,0, 1,6 Гц, 1H), 4,57 (д, J=2,0 Гц, 2H), 4,36 (к, J=6,4 Гц, 1H), 1,32 (д, J=6,4 Гц, 3H), способный к обмену протон не наблюдали.
Стадия 7. Получение трет-бутил (S)-(1-(3-фтор-2-(гидроксиметил)фенил)этил)карбамата

В раствор (S)-(2-(1-аминоэтил)-6-фторфенил)метанола (3,60 г, 17,5 ммоль) в дихлорметане (50,0 мл) добавляли триэтиламин (7,08 г, 70,0 ммоль) и ди-трет-бутилдикарбонат (4,20 г, 19,2 ммоль). Смесь перемешивали при температуре окружающей среды в течение 1 ч и затем разводили водой (200 мл) и экстрагировали дихлорметаном (3×100 мл). Объединенные органические слои промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде бесцветного твердого вещества (5,00 г, количественный выход): MS (ES+) m/z 292,3 (M+23).
Стадия 8. Получение (S)-2-(1-((трет-бутоксикарбонил)амино)этил)-6-фторбензилметансульфоната

В раствор (S)-трет-бутил (1-(3-фтор-2-(гидроксиметил)фенил)этил)карбамата (5,00 г, 18,6 ммоль) в безводном дихлорметане (50,0 мл) добавляли триэтиламин (3,76 г, 37,1 ммоль) при 0°C после чего следовало добавление метансульфонилхлорида (3,40 г, 29,7 ммоль) по каплям. Смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 1 ч. После добавления воды (100 мл), смесь экстрагировали дихлорметаном (3×100 мл). Объединенные органические слои промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде твердого вещества желтого цвета (6,50 г, количественный выход): MS (ES+) m/z 370,3 (M+23).
Стадия 9. Получение трет-бутил (S)-(1-(2-((1,3-диоксоизоиндолин-2-ил)метил)-3-фторфенил)этил)карбамата

В раствор (S)-2-(1-((трет-бутоксикарбонил)амино)этил)-6-фторбензилметансульфоната (6,50 г, 18,7 ммоль) в N,N-диметилформамиде (70 мл) добавляли карбонат калия (5,17 г, 37,4 ммоль) и фталимид (4,13 г, 28,1 ммоль). Смесь перемешивали при температуре окружающей среды в течение 12 ч и затем разводили водой (200 мл) и экстрагировали этилацетатом (3×200 мл). Объединенные органические слои промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии с обращенной фазой, осуществляя элюирование с градиентом ацетонитрила в воде, содержащей 0,1% гидроксида аммония, получали указанное в заголовке соединение в виде твердого вещества желтого цвета (4,00 г, выход 54%):1H ЯМР (400 МГц, CDCl3) δ 7,91-7,75 (м, 2H), 7,70-7,63 (м, 2H), 7,33-7,25 (м, 1H), 7,16 (ушир. д, J=7,6 Гц, 1H), 6,96 (ушир. т, J=9,2 Гц, 1H), 5,42-5,27 (м, 1H), 5,12-5,02 (м, 2H), 4,93-4,85 (м, 1H), 1,53-1,44 (м, 3H), 1,35 (с, 9H); MS (ES+) m/z 421,4 (M+23).
Стадия 10. Получение гидрохлорида (S)-2-(2-(1-аминоэтил)-6-фторбензил)изоиндолин-1,3-диона

К (S)-трет-бутил(1-(2-((1,3-диоксоизоиндолин-2-ил)метил)-3-фторфенил)этил)карбамату (4,00 г, 10,0 ммоль) добавляли 4,0 M раствор хлороводорода в метаноле (40 мл, 160 ммоль), и смесь перемешивали при температуре окружающей среды в течение 1 ч. Концентрированием in vacuo получали указанное в заголовке соединение в виде бесцветного твердого вещества (3,40 г, выход 100%): MS (ES+) m/z 299,3 (M+1).
Стадия 11. Получение (S)-2-(2-(1-(азетидин-1-ил)этил)-6-фторбензил)изоиндолин-1,3-диона

В раствор гидрохлорида (S)-2-(2-(1-аминоэтил)-6-фторбензил)изоиндолин-1,3-диона (3,40 г, 10,2 ммоль) и 1,3-дибромпропана (2,26 г, 11,2 ммоль) в N,N-диметилформамиде (40 мл) добавляли карбонат калия (5,62 г, 40,6 ммоль) и смесь перемешивали при 80°C в течение 12 ч. Затем реакционную смесь разводили водой (200 мл) и экстрагировали этилацетатом (3×200 мл). Объединенные органические слои промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии с обращенной фазой, осуществляя элюирование с градиентом ацетонитрила в воде, содержащей 0,1% гидроксида аммония, получали указанное в заголовке соединение в виде масла желтого цвета (0,46 г, выход 13%):1H ЯМР (400 МГц, CDCl3) δ 7,80-7,72 (м, 2H), 7,62-7,58 (м, 2H), 7,30 (ушир. д, J=8,0 Гц, 1H), 7,13-7,10 (м, 1H), 6,83 (т, J=9,2 Гц, 1H), 5,04 (д, J=14,8 Гц, 1H), 4,82 (ушир. д, J=14,8 Гц, 1H), 3,82 (ушир. с, 1H), 3,20-2,98 (м, 4H), 1,90-1,89 (м, 2H), 1,07 (ушир. д, J=6,4 Гц, 3H); MS (ES+) m/z 339,3 (M+1).
Стадия 12. Получение (S)-(2-(1-(азетидин-1-ил)этил)-6-фторфенил)метанамина

В раствор (S)-2-(2-(1-(азетидин-1-ил)этил)-6-фторбензил)изоиндолин-1,3-диона (0,20 г, 0,59 ммоль) в этаноле (5 мл) добавляли моногидрат гидразина (0,15 г, 2,96 ммоль) и смесь перемешивали при 80°C в течение 12 ч. Смесь оставляли остывать до температуры окружающей среды и затем фильтровали. Концентрированием фильтрата in vacuo получали остаток, который разводили 1 M гидроксидом натрия (20 мл) и экстрагировали дихлорметаном (3×200 мл). Объединенный органический слой промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде бесцветного масла (0,10 г, выход 81%): MS (ES+) m/z 209,3 (M+1).
Стадия 13. Получение трет-бутил (S)-((4-((2-(1-(азетидин-1-ил)этил)-6-фторбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

В раствор (S)-(2-(1-(азетидин-1-ил)этил)-6-фторфенил)метанамина (0,18 г, 0,86 ммоль) и трет-бутил тиазол-4-ил((2,4,6-трифторфенил)сульфонил)карбамата (0,34 г, 0,86 ммоль) в N,N-диметилформамиде (3 мл) добавляли карбонат калия (0,24 г, 1,73 ммоль) и смесь перемешивали при температуре окружающей среды в течение 12 ч. Смесь разводили водой (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 10% метанола в дихлорметане, получали указанное в заголовке соединение в виде бесцветного масла (0,10 г, выход 20%):1H ЯМР (400 МГц, CDCl3) δ 8,81 (д, J=2,2 Гц, 1H), 7,52 (д, J=2,2 Гц, 1H), 7,28-7,25 (м, 1H), 7,23-7,20 (м, 1H), 7,07-7,00 (м, 1H), 6,61-6,55 (ушир. с, 1H), 6,30 (д, J=11,6 Гц, 2H), 4,63-4,48 (м, 2H), 3,60-3,53 (м, 1H), 3,21-3,17 (м, 2H), 3,12-3,07 (м, 2H), 2,08-2,05 (м, 2H), 1,41 (с, 9H), 1,29 (д, J=6,8 Гц, 3H); MS (ES+) m/z 583,4 (M+1).
Стадия 14. Получение формиата (S)-4-((2-(1-(азетидин-1-ил)этил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

В раствор (S)-трет-бутил(4-((2-(1-(азетидин-1-ил)этил)-6-фторбензил)амино)-2,6-дифторфенил)сульфонил(тиазол-4-ил)карбамата (0,10 г, 0,17 ммоль) в дихлорметане (2 мл) добавляли трифторуксусную кислоту (1,0 мл) и смесь перемешивали при температуре окружающей среды в течение 1 ч. Затем смесь концентрировали при пониженном давлении, чтобы получать остаток, который очищали с помощью препаративной колоночной хроматографии с обращенной фазой, осуществляя элюирование с градиентом ацетонитрила в воде, содержащей 0,1% муравьиной кислоты, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,05 г, выход 52%):1H ЯМР (400 МГц, CDCl3) δ 8,68 (д, J=2,0 Гц, 1H), 8,43 (с, 0,3H), 7,38-7,32 (м, 1H), 7,32-7,30 (м, 1H), 7,11-7,06 (м, 1H), 7,05 (д, J=2,0 Гц, 1H), 6,23 (д, J=11,6 Гц, 2H), 4,48-4,37 (м, 2H), 4,06-3,95 (м, 1H), 3,48 (к, J=7,2 Гц, 2H), 3,39 (к, J=7,2 Гц, 2H), 2,17 (квин, J=7,2 Гц, 2H), 1,37 (д, J=6,8 Гц, 3H), NH и COOH не наблюдали; MS (ES+) m/z 483,1 (M+1).
ПРИМЕР 279
Синтез формиата (R)-4-((2-(1-(азетидин-1-ил)этил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение (S)-N-(3-фтор-2-(((триизопропилсилил)окси)метил)бензилиден)-2-метилпропан-2-сульфинамида

Следуя процедуре как описано в примере 278, стадии 4 и выполняя некритические вариации при необходимости, чтобы заменять (S)-2-метилпропан-2-сульфинамид на ((R)-2-метилпропан-2-сульфинамид, указанное в заголовке соединение получали в виде масла желтого цвета (7,80 г, выход 41%):1H ЯМР (400 МГц, CDCl3) δ 9,01 (с, 1H), 7,81 (д, J=7,8 Гц, 1H), 7,35-7,24 (м, 1H), 7,15-7,07 (м, 1H), 5,01 (дк, J=1,6, 11,8 Гц, 2H), 1,20 (с, 9H), 1,10-1,03 (м, 3H), 1,00-0,95 (м, 18H).
Стадия 2. Получение (S)-N-((R)-1-(3-фтор-2-(((триизопропилсилил)окси)метил)фенил)этил)-2-метилпропан-2-сульфинамида
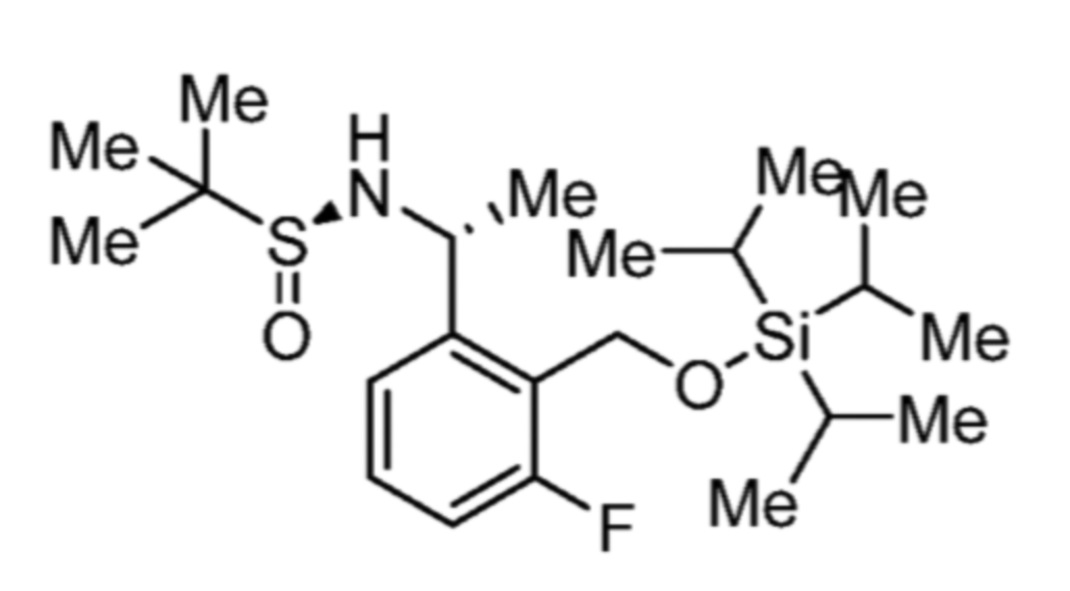
Следуя процедуре как описано в примере 278, стадии 5 и выполняя некритические вариации при необходимости, чтобы заменять (R)-N-(3-фтор-2-(((триизопропилсилил)окси)метил)бензилиден)-2-метилпропан-2-сульфинамид на (S)-N-(3-фтор-2-(((триизопропилсилил)окси)метил)бензилиден)-2-метилпропан-2-сульфинамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (6,00 г, выход 73%):1H ЯМР (400 МГц, CDCl3) δ 7,19-7,11 (м, 2H), 6,91-6,84 (м, 1H), 5,04 (к, J=6,6 Гц, 1H), 4,96-4,72 (м, 2H), 3,29 (ушир. с, 1H), 1,49 (д, J=6,8 Гц, 3H), 1,10 (с, 9H), 1,09-1,04 (м, 3H), 1,02-0,97 (м, 18H).
Стадия 3. Получение гидрохлорида (R)-(2-(1-аминоэтил)-6-фторфенил)метанола

Следуя процедуре как описано в примере 278, стадии 6 и выполняя некритические вариации при необходимости, чтобы заменять ((R)-N-((S)-1-(3-фтор-2-(((триизопропилсилил)окси)метил)фенил)этил)-2-метилпропан-2-сульфинамид на (S)-N-((R)-1-(3-фтор-2-(((триизопропилсилил)окси)метил)фенил)этил)-2-метилпропан-2-сульфинамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (3,00 г, количественный выход), которое использовали без очистки
Стадия 4. Получение трет-бутил (R)-(1-(3-фтор-2-(гидроксиметил)фенил)-этил)карбамата

Следуя процедуре как описано в примере 278, стадии 7 и выполняя некритические вариации при необходимости, чтобы заменять (S)-(2-(1-аминоэтил)-6-фторфенил)метанол на (R)-(2-(1-аминоэтил)-6-фторфенил)метанол, указанное в заголовке соединение получали в виде бесцветного твердого вещества (2,42 г, выход 66%):1H ЯМР (400 МГц, CDCl3) δ 7,34-7,29 (м, 1H), 7,12 (ушир. д, J=7,8 Гц, 1H), 7,00 (т, J=8,8 Гц, 1H), 5,16-5,03 (м, 2H), 4,86 (ушир. с, 2H), 1,48 (ушир. д, J=6,3 Гц, 3H), 1,39 (с, 9H), OH не наблюдали.
Стадия 5. Получение (R)-2-(1-((трет-бутоксикарбонил)амино)этил)-6-фторбензилметансульфоната

Следуя процедуре как описано в примере 278, стадии 8 и выполняя некритические вариации при необходимости, чтобы заменять (S)-трет-бутил (1-(3-фтор-2-(гидроксиметил)фенил)этил)карбамат на (R)-трет-бутил (1-(3-фтор-2-(гидроксиметил)фенил)этил)карбамат, указанное в заголовке соединение получали в виде масла (3,40 г, количественный выход), которое использовали без очистки.
Стадия 6. Получение трет-бутил (R)-(1-(2-((1,3-диоксоизоиндолин-2-ил)метил)-3-фторфенил)этил)карбамата

Следуя процедуре как описано в примере 278, стадии 9 и выполняя некритические вариации при необходимости, чтобы заменять (S)-2-(1-((трет-бутоксикарбонил)амино)этил)-6-фторбензилметансульфонат на (R)-2-(1-((трет-бутоксикарбонил)амино)этил)-6-фторбензилметансульфонат, указанное в заголовке соединение получали в виде твердого вещества желтого цвета (2,40 г, выход 65%):1H ЯМР (400 МГц, CDCl3) δ 7,89-7,79 (м, 2H), 7,70 (дд, J=3,0, 5,4 Гц, 2H), 7,38-7,29 (м, 1H), 7,16 (д, J=7,8 Гц, 1H), 7,02-6,95 (м, 1H), 5,43-5,27 (м, 1H), 5,15-5,02 (м, 2H), 4,92 (ушир. с, 1H), 1,51 (ушир. д, J=6,4 Гц, 3H), 1,36 (с, 9H); MS (ES+) m/z 299,3 (M - 100+1).
Стадия 7. Получение гидрохлорида
(R)-2-(2-(1-аминоэтил)-6-фторбензил)изоиндолин-1,3-диона

Следуя процедуре как описано в примере 278, стадии 10 и выполняя некритические вариации при необходимости, чтобы заменять (S)-трет-бутил(1-(2-((1,3-диоксоизоиндолин-2-ил)метил)-3-фторфенил)этил)карбамат на (R)-трет-бутил(1-(2-((1,3-диоксоизоиндолин-2-ил)метил)-3-фторфенил)этил)карбамат, указанное в заголовке соединение получали в виде твердого вещества желтого цвета (2,00 г, количественный выход): MS (ES+) m/z 299,3 (M+1).
Стадия 8. Получение (R)-2-(2-(1-(азетидин-1-ил)этил)-6-фторбензил)изоиндолин-1,3-диона

Следуя процедуре как описано в примере 278, стадии 11 и выполняя некритические вариации при необходимости, чтобы заменять гидрохлорид (S)-2-(2-(1-аминоэтил)-6-фторбензил)изоиндолин-1,3-диона на гидрохлорид (R)-2-(2-(1-аминоэтил)-6-фторбензил)изоиндолин-1,3-дион, указанное в заголовке соединение получали (0,40 г, выход 20%):1H ЯМР (400 МГц, CDCl3) δ 7,86 (дд, J=5,6, 2,8 Гц, 2H), 7,73 (дд, J=5,6, 3,2 Гц, 2H), 7,49-7,40 (м, 2H), 6,95-6,91 (м, 1H), 5,14 (дд, J=14,8, 1,4 Гц, 1H), 4,92-4,80 (м, 2H), 3,43-3,22 (м, 4H), 2,15-2,04 (м, 2H), 1,21 (д, J=6,4 Гц, 3H); MS (ES+) m/z 339,3 (M+1)
Стадия 9. Получение (R)-(2-(1-(азетидин-1-ил)этил)-6-фторфенил)метанамина

Следуя процедуре как описано в примере 278, стадии 12 и выполняя некритические вариации при необходимости, чтобы заменять (S)-2-(2-(1-(азетидин-1-ил)этил)-6-фторбензил)изоиндолин-1,3-дион на (R)-2-(2-(1-(азетидин-1-ил)этил)-6-фторбензил)изоиндолин-1,3-дион, указанное в заголовке соединение получали в виде твердого вещества желтого цвета (0,21 г, 85%): MS (ES+) m/z 192,0 (M - 16).
Стадия 10. Получение трет-бутил (R)-((4-((2-(1-(азетидин-1-ил)этил)-6-фторбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

Следуя процедуре как описано в примере 278, стадии 13 и выполняя некритические вариации при необходимости, чтобы заменять (S)-(2-(1-(азетидин-1-ил)этил)-6-фторфенил)метанамин на (R)-(2-(1-(азетидин-1-ил)этил)-6-фторфенил)метанамин, указанное в заголовке соединение получали в виде твердого вещества желтого цвета (0,05 г, выход 11%):1H ЯМР (400 МГц, CDCl3) δ 8,81 (д, J=2,4 Гц, 1H), 7,52 (д, J=2,4 Гц, 1H), 7,28-7,21 (м, 2H), 7,08-7,01 (м, 1H), 6,33-6,24 (м, 2H), 4,60-4,47 (м, 2H), 4,46-4,36 (м, 1H), 3,25-3,06 (м, 4H), 2,08 (квин, J=7,0 Гц, 2H), 1,42-1,40 (м, 9H), 1,29 (д, J=6,8 Гц, 3H), протон NH не наблюдали; MS (ES+) m/z 583,4 (M+1).
Стадия 11. Получение формиата (R)-4-((2-(1-(азетидин-1-ил)этил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано в примере 278, стадии 14 и выполняя некритические вариации при необходимости, чтобы заменять (S)-трет-бутил(4-((2-(1-(азетидин-1-ил)этил)-6-фторбензил)амино)-2,6-дифторфенил)сульфонил(тиазол-4-ил)карбамат на (R)-трет-бутил(4-((2-(1-(азетидин-1-ил)этил)-6-фторбензил)амино)-2,6-дифторфенил)сульфонил(тиазол-4-ил)карбамат, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,02 г, выход 45%):1H ЯМР (400 МГц, CDCl3) δ 8,68 (д, J=2,4 Гц, 1H), 8,39 (с, 1H), 7,41-7,30 (м, 2H), 7,14-7,03 (м, 2H), 6,24 (д, J=11,8 Гц, 2H), 4,49-4,32 (м, 2H), 4,21-4,12 (м, 1H), 3,58 (к, J=7,8 Гц, 2H), 3,47 (к, J=7,6 Гц, 2H), 2,20 (квин, J=7,6 Гц, 2H), 1,41 (д, J=6,8 Гц, 3H), NH и COOH не наблюдали; MS (ES+) m/z 483,1(M+1).
ПРИМЕР 280
Синтез (S)-5-хлор-2-фтор-4-((1-(2-фтор-5-(метоксиметил)фенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида
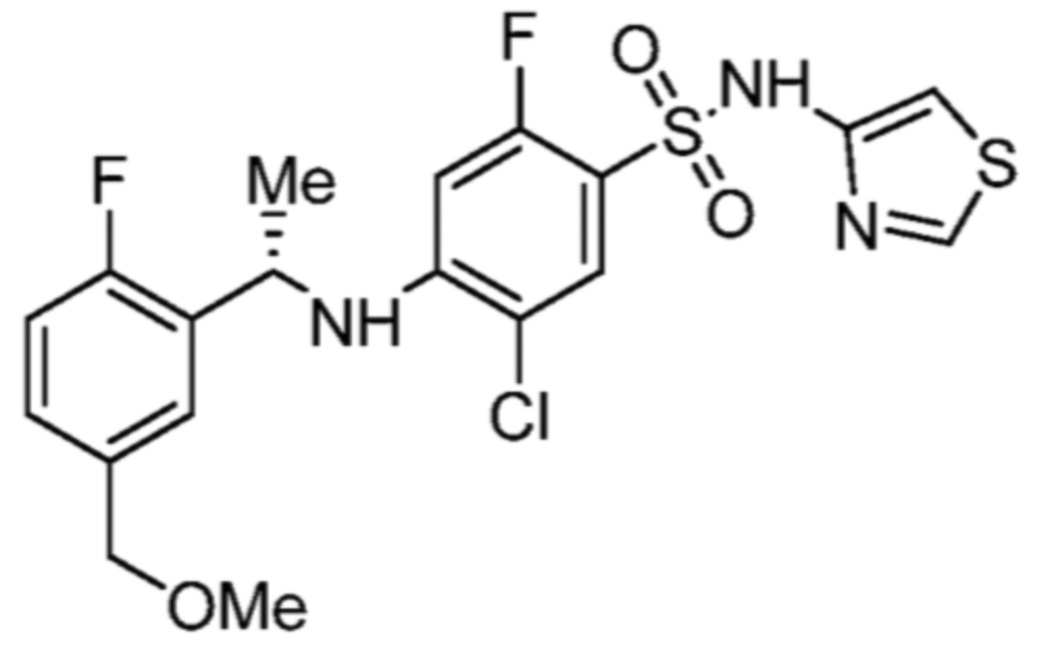
Стадия 1. Получение трет-бутил (S)-((5-хлор-2-фтор-4-((1-(2-фтор-5-(метоксиметил)фенил)этил)амино)фенил)сульфонил)(тиазол-4-ил)карбамата
В раствор трет-бутил (5-хлор-2,4-дифторфенил)сульфонил(тиазол-4-ил)карбамата (0,30 г, 0,73 ммоль) и гидрохлорида (S)-1-(2-фтор-5-(метоксиметил)фенил)этанамина (0,16 г, 0,73 ммоль) в безводном N,N-диметилформамиде (5 мл) добавляли карбонат цезия (0,48 г, 1,46 ммоль). Смесь перемешивали при температуре окружающей среды в течение 12 ч, разводили водой (30 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические экстракты промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 33% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного масла (0,15 г, выход 36%):1H ЯМР (400 МГц, CDCl3) δ 8,77 (д, J=2,0 Гц, 1H), 7,96 (д, J=7,2 Гц, 1H), 7,48 (д, J=2,0 Гц, 1H), 7,26-7,20 (м, 2H), 7,08 (дд, J=10,0, 8,4 Гц, 1H), 6,20 (д, J=12,4 Гц, 1H), 5,36 (д, J=5,6 Гц, 1H), 4,85 (к, J=6,4 Гц, 1H), 4,38 (с, 2H), 3,37 (с, 3H), 1,67 (д, J=6,4 Гц, 3H), 1,36 (с, 9H); MS (ES+) m/z 574,0 (M+1), 576,0 (M+1).
Стадия 2. Получение (S)-5-хлор-2-фтор-4-((1-(2-фтор-5-(метоксиметил)фенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано в примере 5, стадии 2 и выполняя некритические вариации, чтобы заменять трет-бутил (S)-((5-хлор-4-((1-(5-хлор-2-фторфенил)этил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамат на трет-бутил (S)-((5-хлор-2-фтор-4-((1-(2-фтор-5-(метоксиметил)фенил)этил)амино)фенил)сульфонил)(тиазол-4-ил)карбамат, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,032 г, выход 26%):1H ЯМР (400 МГц, CDCl3) δ 10,22 (с, 1H), 8,68 (д, J=2,0 Гц, 1H), 7,77 (д, J=7,2 Гц, 1H), 7,26-7,20 (м, 1H), 7,19 (д, J=6,0 Гц, 1H), 7,06 (т, J=9,2 Гц, 1H), 6,91 (д, J=2,0 Гц, 1H), 6,12 (д, J=12,4 Гц, 1H), 5,22 (д, J=5,6 Гц, 1H), 4,78 (к, J=6,4 Гц, 1H), 4,37 (с, 2H), 3,35 (с, 3H), 1,63 (д, J=6,4 Гц, 3H); MS (ES+) m/z 474,0 (M+1), 476,0 (M+1).
ПРИМЕР 281
Синтез (S)-5-хлор-4-((1-(5-(дифторметокси)-2-фторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 5-(дифторметокси)-2-фторбензальдегида

В раствор 2-фтор-5-гидроксибензальдегида (0,30 г, 2,14 ммоль) в 1,4-диоксане (10 мл) и воде (2 мл) добавляли гидроксид натрия (0,25 г, 6,42 ммоль) и через смесь барботировали хлор(дифтор)метан. Смесь перемешивали при температуре окружающей среды в течение 30 минут. Смесь разводили насыщенным хлоридом аммония (20 мл) и экстрагировали этилацетатом (3×20 мл). Объединенные органические экстракты промывали солевым раствором (3×20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием от 2 до 20% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде масла желтого цвета (0,18 г, выход 44%):1H ЯМР (400 МГц, CDCl3) δ 10,34 (с, 1H), 7,62 (дд, J=5,4, 3,2 Гц, 1H), 7,45-7,36 (м, 1H), 7,21 (т, J=9,2 Гц, 1H), 6,52 (т, J=72,4 Гц, 1H).
Стадия 2. Получение (R)-N-(5-(дифторметокси)-2-фторбензилиден)-2-метилпропан-2-сульфинамида

В раствор 5-(дифторметокси)-2-фторбензальдегида (0,90 г, 4,73 ммоль) и (R)-2-метилпропан-2-сульфинамида (0,68 г, 5,68 ммоль) в дихлорметане (20 мл) добавляли карбонат цезия (3,08 г, 9,46 ммоль) и смесь перемешивали при температуре окружающей среды в течение 12 ч. Реакционную смесь фильтровали и фильтрат концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 5 до 50% петролейного эфира в этилацетате, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,90 г, выход 64%):1H ЯМР (400 МГц, CDCl3) δ 8,86 (с, 1H), 7,75 (дд, J=5,6, 3,0 Гц, 1H), 7,31-7,27 (м, 1H), 7,20-7,15 (м, 1H), 6,52 (т, J=73,2 Гц, 1H), 1,28 (с, 9H); MS (ES+) m/z 294,1 (M+1).
Стадия 3. Получение (R)-N-((S)-1-(5-(дифторметокси)-2-фторфенил)этил)-2-метилпропан-2-сульфинамида

Следуя процедуре как описано в примере 146, стадии 3 и выполняя некритические вариации, чтобы заменять (R)-N-(5-циклопропил-2-фторбензилиден)-2-метилпропан-2-сульфинамид на (R)-N-(5-(дифторметокси)-2-фторбензилиден)-2-метилпропан-2-сульфинамид), указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,20 г, выход 63%):1H ЯМР (400 МГц, CDCl3) δ 7,20-7,12 (м, 1H), 7,05-6,97 (м, 2H), 6,45 (т, J=73,2 Гц, 1H), 4,87 (дд, J=6,8, 4,2 Гц, 1H), 1,55 (д, J=6,8 Гц, 3H), 1,24-1,19 (м, 9H), NH не наблюдали; MS (ES+) m/z 332,1 (M+23).
Стадия 4. Получение гидрохлорида (S)-1-(5-(дифторметокси)-2-фторфенил)этан-1-амина

Следуя процедуре как описано в примере 146, стадии 4 и выполняя некритические вариации, чтобы заменять ((R)-N-((S)-1-(5-циклопропил-2-фторфенил)этил)-2-метилпропан-2-сульфинамид на (R)-N-((S)-1-(5-(дифторметокси)-2-фторфенил)этил)-2-метилпропан-2-сульфинамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,12 г, выход 90%):1H ЯМР (400 МГц, CDCl3) δ 8,91 (с, 3H), 7,47 (д, J=5,0 Гц, 1H), 7,19-7,02 (м, 2H), 6,55 (т, J=73,2 Гц, 1H), 4,74 (д, J=6,8 Гц, 1H), 1,70 (д, J=6,8 Гц, 3H); MS (ES+) m/z 206,0 (M+1).
Стадия 5. Получение трет-бутил (S)-((5-хлор-4-((1-(5-(дифторметокси)-2-фторфенил)этил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата

Следуя процедуре как описано в примере 146, стадии 5 и выполняя некритические вариации, чтобы заменять гидрохлорид (S)-1-(5-циклопропил-2-фторфенил)этан-1-амина на гидрохлорид (S)-1-(5-(дифторметокси)-2-фторфенил)этан-1-амина, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,15 г, выход 100%):1H ЯМР (400 МГц, CDCl3) δ 8,76 (д, J=2,0 Гц, 1H), 7,97 (д, J=7,0 Гц, 1H), 7,47 (д, J=2,0 Гц, 1H), 7,10 (д, J=9,4 Гц, 1H), 7,07-7,03 (м, 1H), 7,00-6,97 (м, 1H), 6,42 (т, J=73,2 Гц, 1H), 6,15 (д, J=12,0 Гц, 1H), 5,28 (д, J=5,6 Гц, 1H), 4,83 (т, J=6,6 Гц, 1H), 1,66 (д, J=6,8 Гц, 3H), 1,34 (с, 9H); MS (ES+) m/z 595,9 (M+1).
Стадия 6. Получение (S)-5-хлор-4-((1-(5-(дифторметокси)-2-фторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано в примере 5, стадии 2 и выполняя некритические вариации, чтобы заменять трет-бутил (S)-((5-хлор-4-((1-(5-хлор-2-фторфенил)этил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамат на трет-бутил (S)-((5-хлор-4-((1-(5-(дифторметокси)-2-фторфенил)этил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамат, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,056 г, выход 45%):1H ЯМР (400 МГц, CDCl3) δ 9,90 (с, 1H), 8,64 (д, J=2,0 Гц, 1H), 7,75 (д, J=7,2 Гц, 1H), 7,11-7,05 (м, 1H), 7,04-6,99 (м, 1H), 6,95 (дд, J=2,8, 6,0 Гц, 1H), 6,91 (д, J=2,4 Гц, 1H), 6,40 (т, J=73,2 Гц, 1H), 6,05 (д, J=12,0 Гц, 1H), 5,15 (д, J=5,6 Гц, 1H), 4,74 (к, J=6,6 Гц, 1H), 1,61 (д, J=6,8 Гц, 3H); MS (ES+) m/z 517,9 (M+23).
ПРИМЕР 282
Синтез 2,2,2-трифторацетата 4-((2-((7-окса-2-азаспиро[3.5]нонан-2-ил)метил)-3,6-дифторбензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 4-амино-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил ((5-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (1,23 г, 3,00 ммоль) в N,N-диметилформамиде (10 мл) добавляли азид натрия (0,19 г, 3,00 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 20 минут. Затем в реакционную смесь добавляли цинковую пыль (1,00 г, 16,0 ммоль) и насыщенный хлорид аммония (10 мл) и реакционную смесь перемешивали в течение 16 ч. Реакцию разводили водой (20 мл) и этилацетатом (30 мл) и фильтровали через диатомовую землю. Фильтрат экстрагировали этилацетатом (2×30 мл). Объединенные органические слои промывали солевым раствором (2×40 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде бесцветного твердого вещества (1,22 г, выход 99%): MS (ES+) m/z 308,2 (M - Boc+1), 310,2 (M - Boc+1).
Стадия 2. Получение 4-((2-(((трет-бутилдиметилсилил)окси)метил)-3,6-дифторбензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

К трифторуксусной кислоте (5 мл) добавляли трет-бутил ((4-амино-5-хлор-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата (1,22 г, 3,00 ммоль) при 0°C, после чего следовал триацетоксиборгидрид натрия (1,02 г, 4,80 ммоль), и смесь перемешивали в течение 5 минут. Затем туда добавляли при 0°C раствор 2-(((трет-бутилдиметилсилил)окси)метил)-3,6-дифторбензальдегида (0,86 г, 3,00 ммоль) в дихлорметане (10 мл) и реакционную смесь перемешивали при 0°C в течение 10 минут. Реакционную смесь выливали в холодных насыщенный бикарбонат натрия (50 мл) и экстрагировали этилацетатом (3×30 мл). Объединенную органическую фазу сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-50% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного масла (0,98 г, выход 57%): MS (ES+) m/z 580,2 (M+1), 578,2 (M+1).
Стадия 3. Получение трет-бутил (2-(((трет-бутилдиметилсилил)окси)метил)-3,6-дифторбензил)(2-хлор-5-фтор-4-(N-(тиазол-4-ил)сульфамоил)фенил)карбамата

В раствор 4-((2-(((трет-бутилдиметилсилил)окси)метил)-3,6-дифторбензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида (0,98 г, 1,72 ммоль) в дихлорметане (10 мл) добавляли ди-трет-бутилдикарбонат (0,75 г, 3,45 ммоль) и 4-(диметиламино)пиридина (0,42 г, 3,45 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч. Концентрированием реакционной смеси in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-50% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного масла (0,28 г, выход 24%): MS (ES+) m/z 680,2 (M+1), 678,2 (M+1).
Стадия 4. Получение трет-бутил (2-хлор-5-фтор-4-(N-(тиазол-4-ил)сульфамоил)фенил)(3,6-дифтор-2-(гидроксиметил)бензил)карбамата

В раствор трет-бутил (2-(((трет-бутилдиметилсилил)окси)метил)-3,6-дифторбензил)(2-хлор-5-фтор-4-(N-(тиазол-4-ил)сульфамоил)фенил)карбамата (0,28 г, 0,41 ммоль) в безводном тетрагидрофуране (4 мл) добавляли 1,0 M раствор фторида тетра-н-бутиламмония в тетрагидрофуране (0,63 мл, 0,63 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 45 минут. Концентрированием реакционной смеси in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-80% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного масла (0,23 г, выход 100%): MS (ES+) m/z 566,2 (M+1), 564,2 (M+1).
Стадия 5. Получение трет-бутил (2-хлор-5-фтор-4-(N-(тиазол-4-ил)сульфамоил)фенил)(3,6-дифтор-2-формилбензил)карбамата

В раствор трет-бутил (2-хлор-5-фтор-4-(N-(тиазол-4-ил)сульфамоил)фенил)(3,6-дифтор-2-(гидроксиметил)бензил)карбамата (0,23 г, 0,41 ммоль) в дихлорметане (3 мл) добавляли реактив периодинан Десса-Мартина (0,27 г, 0,63 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Реакционную смесь фильтровали через подушку диатомовой земли и фильтрат концентрировали in vacuo для того, чтобы получать указанное в заголовке соединение в виде масла, которое использовали без дополнительной очистки: MS (ES+) m/z 564,2 (M+1), 562,2 (M+1).
Стадия 6. Получение трет-бутил (2-((7-окса-2-азаспиро[3.5]нонан-2-ил)метил)-3,6-дифторбензил)(2-хлор-5-фтор-4-(N-(тиазол-4-ил)сульфамоил)фенил)карбамата

В раствор трет-бутил (2-хлор-5-фтор-4-(N-(тиазол-4-ил)сульфамоил)фенил)(3,6-дифтор-2-формилбензил)карбамата (0,08 г, 0,14 ммоль) в дихлорметане (2 мл) добавляли соль 8-окса-2-азаспиро[4.5]декана и щавелевой кислоты (0,02 г, 0,14 ммоль) и триацетоксиборгидрид натрия (0,07 г, 0,35 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч. Затем туда добавляли насыщенный бикарбонат натрия (5 мл) и смесь экстрагировали этилацетатом (2×10 мл). Объединенную органическую фазу промывали солевым раствором (5 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-10% метанола в дихлорметане, получали указанное в заголовке соединение в виде масла (0,01 г, выход 10%): MS (ES+) m/z 675,2 (M+1), 673,2 (M+1).
Стадия 7. Получение 2,2,2-трифторацетата 4-((2-((7-окса-2-азаспиро[3.5]нонан-2-ил)метил)-3,6-дифторбензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил (2-((7-окса-2-азаспиро[3.5]нонан-2-ил)метил)-3,6-дифторбензил)(2-хлор-5-фтор-4-(N-(тиазол-4-ил)сульфамоил)фенил)карбамата (0,01 г, 0,01 ммоль) в дихлорметане (3 мл) добавляли трифторуксусную кислоту (3 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Затем смесь концентрировали in vacuo для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,01 г, выход 100%):1H ЯМР (300 МГц, DMSO d6) δ 11,18-11,17 (ушир. с, 1H), 10,35-10,31 (ушир. с, 1H), 8,90-8,88 (м, 1H), 7,65-7,60 (м, 1H), 7,48-7,41 (м, 2H), 7,02-6,99 (м, 1H), 6,86-6,73 (м, 2H), 4,64-4,60 (м, 2H), 4,55-4,51 (м, 2H), 4,01-3,97 (м, 4H), 3,52-3,46 (м, 4H), 1,83-1,72 (м, 4H); MS (ES+) m/z 575,2 (M+1), 573,2 (M+1).
ПРИМЕР 283
Синтез 4-((2-(азетидин-1-илметил)-3,6-дифторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 1-(2-бром-3,6-дифторбензил)азетидина

В раствор 2-бром-3,6-дифторбензальдегида (8,84 г, 40,0 ммоль) в дихлорметане (160 мл) добавляли азетидин (2,28 г, 40 ммоль), после чего следовал триацетоксиборгидрид натрия (15,3 г, 72,0 ммоль), и реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч. Реакцию гасили добавлением 2 M гидроксида натрия (80 мл) и перемешивали в течение 30 минут. Смесь концентрировали при пониженном давлении и остававшийся водный слой экстрагировали этилацетатом (2×60 мл). Объединенную органическую фазу промывали солевым раствором (40 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде бесцветного масла (8,82 г, выход 84%): MS (ES+) m/z 264,1 (M+1), 262,1 (M+1).
Стадия 2. Получение 2-(азетидин-1-илметил)-3,6-дифторбензальдегида

В раствор 1-(2-бром-3,6-дифторбензил)азетидина (8,82 г, 33,8 ммоль) в безводном тетрагидрофуране (70 мл) добавляли 1,6 M раствор н-бутиллития в гексане (25,3 мл, 40,5 ммоль) при -78°C. Реакционную смесь перемешивали при -78°C в течение 10 минут и затем туда добавляли N,N-диметилформамид (4,18 мл, 54,1 ммоль). Реакционную смесь перемешивали при -78°C в течение 1 ч, оставляли нагреваться до температуры окружающей среды и перемешивали в течение 40 минут. Реакционную смесь выливали на лед (50 мл) и экстрагировали дихлорметаном (3×100 мл). Органическую фазу промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-10% метанола в дихлорметане, получали указанное в заголовке соединение в виде бесцветного масла (3,08 г, выход 42%): MS (ES+) m/z 212,2 (M+1).
Стадия 3. Получение 4-((2-(азетидин-1-илметил)-3,6-дифторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида.

К трифторуксусной кислоте (3 мл) добавляли 4-амино-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамид (0,65 г, 1,66 ммоль) при 0°C, после чего следовал триацетоксиборгидрид (0,56 г, 2,66 ммоль), и смесь перемешивали в течение 5 минут. Затем туда добавляли раствор 2-(азетидин-1-илметил)-3,6-дифторбензальдегида (0,35 г, 1,66 ммоль) в дихлорметане (5 мл) при 0°C. Реакционную смесь перемешивали в течение 1 часа при 0°C и затем выливали на холодный бикарбонат натрия (50 мл) и экстрагировали этилацетатом (3×30 мл). Объединенную органическую фазу сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-90% этилацетата в гексанах, после которого следовал градиент 0-10% метанола в дихлорметане, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,18 г, выход 22%):1H ЯМР (300 МГц, DMSO-d6) δ 11,23-11,09 (ушир. с, 1H), 8,90-8,88 (м, 1H), 7,46-7,33 (м, 3H), 6,92-6,90 (м, 1H), 6,40-6,33 (м, 2H), 4,39-4,31 (м, 4H), 3,96-3,87 (м, 4H), 2,24-2,18 (м, 2H); MS (ES+) m/z 487,2 (M+1).
ПРИМЕР 284
Синтез 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-3,6-дифторбензил)(метил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

В раствор 4-((2-(азетидин-1-илметил)-3,6-дифторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида (0,10 г, 0,20 ммоль) в трифторуксусной кислоте (2 мл) добавляли триацетоксиборгидрид натрия (0,12 г, 0,60 ммоль) при 0°C после чего следовал формальдегид (0,01 г, 0,30 ммоль) и реакционную смесь перемешивали в течение 10 минут при 0°C. Реакцию гасили добавлением воды (1 мл) и концентрировали in vacuo. Очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,02 г, выход 20%):1H ЯМР (300 МГц, DMSO-d6) δ 11,28 (ушир. с, 1H), 10,36-10,34 (ушир. с, 1H), 8,90 (д, J=2,1 Гц, 1H), 7,45-7,40 (м, 2H), 6,94 (д, J=2,2 Гц, 1H), 6,62 (дд, J=13,4, 0,3 Гц, 2H), 4,74-4,73 (м, 2H), 4,51-4,49 (м, 2H), 4,11-3,99 (м, 4H), 2,69 (с, 3H), 2,36-2,22 (м, 2H); MS (ES+) m/z 501,2 (M+1).
ПРИМЕР 285
Синтез формиата 4-((2-(азетидин-1-илметил)-6-метилбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 1-(3-хлорбензил)азетидина
В раствор 3-хлорбензальдегида (20,0 г, 142 ммоль) в дихлорметане (460 мл) добавляли азетидин (8,10 г, 142,0 ммоль), после чего следовал триацетоксиборгидрид натрия (45,1 г, 213 ммоль), и реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч. Реакцию гасили добавлением 28% водного раствора гидроксида аммония (70 мл) и перемешивали в течение 30 минут. Водный слой экстрагировали дихлорметаном (2×200 мл). Объединенную органическую фазу промывали солевым раствором (40 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-10% метанола в дихлорметане, получали указанное в заголовке соединение в виде бесцветного масла (11,5 г, выход 45%):1H-ЯМР (300 МГц, CDCl3) δ 7,30-7,28 (м, 1H), 7,25-7,16 (м, 3H), 3,55 (с, 2H), 3,23 (т, J=7,0 Гц, 4H), 2,11 (квинтет, J=7,0 Гц, 2H).
Стадия 2. Получение 2-(азетидин-1-илметил)-6-хлорбензальдегид оксима

В раствор 1-(3-хлорбензил)азетидина (10,5 г, 58,0 ммоль) в безводном тетрагидрофуране (150 мл) добавляли по каплям 1,4 M раствор втор-бутиллития в гексане (53,8 мл, 78,4 ммоль) при -78°C. Реакционную смесь перемешивали при -78°C в течение 1 ч и затем туда добавляли N,N-диметилформамид (8,00 мл, 104 ммоль). Реакционную смесь перемешивали при -78°C в течение 1 ч и затем нагревали до температуры окружающей среды. В нее добавляли раствор соли гидроксиламина и соляной кислоты (5,64 г, 81,2 ммоль) в метаноле (20 мл) и воде (20 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении и в нее добавляли воду (50 мл). Смесь экстрагировали этилацетатом (2×50 мл). Объединенную органическую фазу промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде бесцветного твердого вещества (13,0 г, количественный выход) в виде смеси E/Z изомеров 1:3:1H-ЯМР (300 МГц, CDCl3) δ 8,57 (с, 0,75H), 7,74 (с, 0,25H), 7,39-7,27 (м, 3H), 3,92 (с, 1,5H), 3,61 (с, 0,5H), 3,29 (тд, J=7,1, 3,8 Гц, 4H), 2,13 (квинтет, J=7,1 Гц, 2H), OH не наблюдали.
Стадия 3. Получение (2-(азетидин-1-илметил)-6-хлорфенил)метанамина

В раствор 2-(азетидин-1-илметил)-6-хлорбензальдегид оксима (13,0 г, 58,0 ммоль) в метаноле (35 мл) добавляли порошок цинка (7,59 г, 116,0 ммоль) и смесь нагревали с обратным холодильником. Затем в смесь добавляли по каплям 6 M соляную кислоту (67 мл, 406,0 ммоль) в течение периода 10 минут. После завершения добавления, в реакционную смесь еще добавляли порошок цинка (3,80 г, 58,0 ммоль). Реакционную смесь нагревали с обратным холодильником в течение еще 15 минут и затем оставляли остывать до температуры окружающей среды. Реакционную смесь охлаждали до 0°C и в нее добавляли 18 M гидроксид натрия (40 мл). Смесь экстрагировали этилацетатом (3×80 мл) и объединенную органическую фазу концентрировали in vacuo. Остаток растворяли в простом диэтиловом эфире (50 мл) и экстрагировали 3,5 M раствором хлорида аммония (2×30 мл). Объединенные водные слои промывали простым диэтиловым эфиром (20 мл) и затем pH корректировали гидроксидом натрия (10,0 г, 250,0 ммоль). Водную фазу экстрагировали этилацетатом (2×40 мл). Объединенную органическую этилацетатную фазу промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде масла (7,80 г, выход 58%):1H-ЯМР (300 МГц, CDCl3) δ 7,30 (дд, J=6,5, 2,8 Гц, 1H), 7,14-7,08 (м, 2H), 3,94 (с, 2H), 3,62 (с, 2H), 3,17 (т, J=7,0 Гц, 4H), 2,04 (дт, J=14,0, 7,0 Гц, 2H), NH не наблюдали.
Стадия 4. Получение трет-бутил ((4-((2-(азетидин-1-илметил)-6-хлорбензил)амино)-3-бром-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

В смесь (2-(азетидин-1-илметил)-6-хлорфенил)метанамина (2,10 г, 10,0 ммоль) и трет-бутил ((3-бром-2,4,6-трифторфенил)сульфонил)(тиазол-4-ил)карбамата (4,73 г, 10,0 ммоль), в безводном диметилсульфоксиде (50 мл) добавляли карбонат калия (2,76 г, 20,0 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч. Добавляли воду (50 мл) и смесь экстрагировали этилацетатом (3×50 мл). Объединенную органическую фазу промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и растиранием остатка в простом диэтиловом эфире (5 мл) получали указанное в заголовке соединение в виде бесцветного твердого вещества (3,10 г, выход 47%):1H-ЯМР (300 МГц, CDCl3) δ 8,82 (д, J=2,3 Гц, 1H), 7,75-7,72 (м, 1H), 7,54 (д, J=2,2 Гц, 1H), 7,40 (дд, J=7,7, 1,7 Гц, 1H), 7,27-7,18 (м, 2H), 6,66 (дд, J=13,7, 1,6 Гц, 1H), 4,62 (д, J=5,6 Гц, 2H), 3,68 (с, 2H), 3,27 (дд, J=8,8, 5,5 Гц, 4H), 2,17-2,08 (м, 2H), 1,41-1,39 (м, 9H).
Стадия 5. Получение трет-бутил ((4-((2-(азетидин-1-илметил)-6-хлорбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

В смесь трет-бутил ((4-((2-(азетидин-1-илметил)-6-хлорбензил)амино)-3-бром-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (3,0 г, 4,53 ммоль) в этаноле (20 мл) добавляли триэтиламин (2,30 мл, 18,1 ммоль) и 5% палладий на углероде (влажный, 0,24 г). Затем реакционную смесь нагревали до 60°C в течение 1 ч. После охлаждения до температуры окружающей среды, реакционную смесь фильтровали через подушку диатомовой земли и фильтровальную подушку споласкивали этанолом (2×5 мл). К объединенному фильтрату добавляли триэтиламин (2,30 мл, 18,1 ммоль) и 5% палладий на углероде (влажный, 0,24 г). Затем реакционную смесь нагревали до 60°C в течение 1 ч. После охлаждения до температуры окружающей среды, реакционную смесь фильтровали через подушку диатомовой земли и фильтровальную подушку споласкивали этанолом (2×5 мл). Объединенный фильтрат концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-40% этилацетата (содержащего 15% этанол и 5% триэтиламин) в гексанах для того, чтобы получать указанное в заголовке соединение в виде бесцветного масла (1,23 г, выход 46%): MS (ES+) m/z 587,2 (M+1), 585,2 (M+1).
Стадия 6. Получение 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-хлорбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил ((4-((2-(азетидин-1-илметил)-6-хлорбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (1,30 г, 2,22 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (5 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Затем смесь концентрировали in vacuo и растирали в этаноле (5 мл). Фильтрованием получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,91 г, выход 84%):1H ЯМР (300 МГц, DMSO-d6) δ 11,25 (с, 1H), 10,19-10,17 (ушир. с, 1H), 8,92 (д, J=2,2 Гц, 1H), 7,62 (дд, J=5,8, 3,6 Гц, 1H), 7,53-7,48 (м, 2H), 7,17-7,15 (м, 1H), 6,93 (д, J=2,2 Гц, 1H), 6,40-6,35 (м, 2H), 4,47-4,38 (м, 4H), 4,15-4,01 (м, 4H), 2,41-2,25 (м, 2H); MS (ES+) m/z 487,3 (M+1), 485,2 (M+1).
Стадия 7. Получение формиата 4-((2-(азетидин-1-илметил)-6-метилбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида
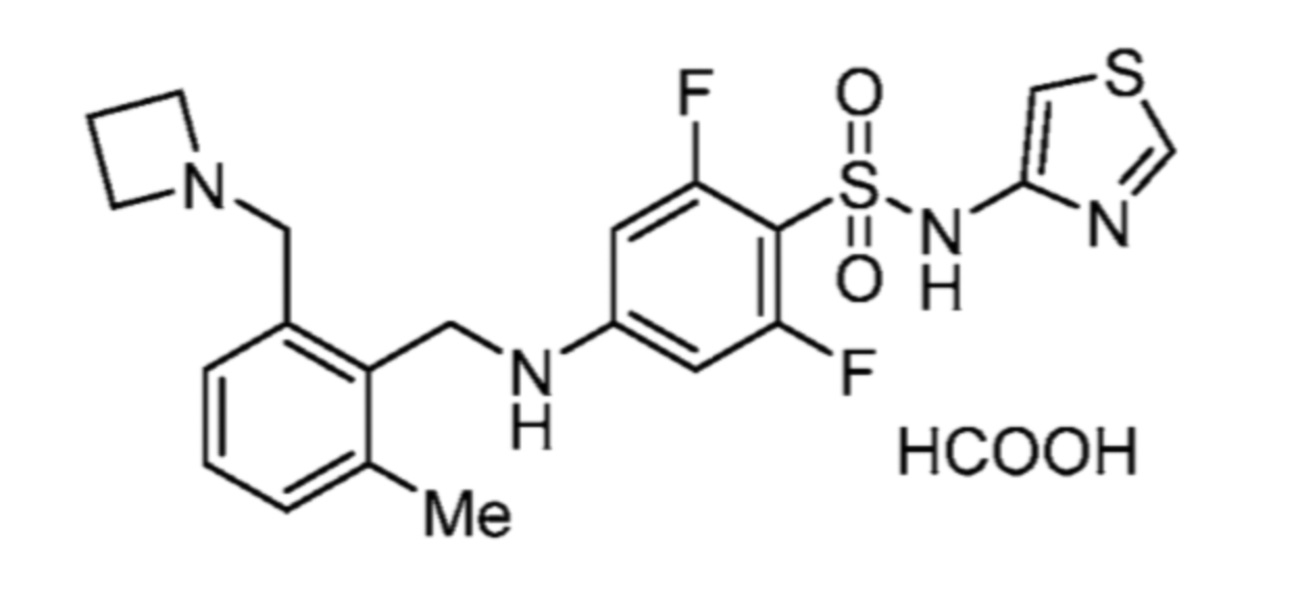
В смесь хлорида (2-дициклогексилфосфино-2′,6′-диметокси-1,1′-бифенил)[2-(2-аминоэтилфенил)]палладия(II) (0,004 г, 0,006 ммоль), 2-дициклогексилфосфино-2',6'-диметоксибифенила (0,003 г, 0,0006 ммоль), трехосновного фосфата калия (0,33 г, 1,56 ммоль) и метилбороновой кислоты (0,05 г, 0,78 ммоль) добавляли раствор 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-хлорбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида (0,06 г, 0,12 ммоль) в 1,4-диоксане (1,5 мл) и воде (0,5 мл). Затем реакционную смесь нагревали в микроволновом реакторе до 160°C в течение 45 минут. После охлаждения до температуры окружающей среды добавляли насыщенный хлорид аммония (5 мл) и смесь экстрагировали этилацетатом (2×10 мл) и дихлорметаном (10 мл). Объединенные органические слои концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-15% метанола (содержащего 0,5% муравьиной кислоты) в дихлорметане для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,03 г, выход 47%):1H ЯМР (300 МГц, DMSO-d6) δ 8,91-8,91 (м, 1H), 8,16 (с, 1H), 7,27-7,18 (м, 3H), 7,13-7,11 (м, 1H), 6,91 (дд, J=2,2, 0,6 Гц, 1H), 6,40 (д, J=12,7 Гц, 2H), 4,25 (с, 2H), 3,84 (д, J=0,3 Гц, 2H), 3,44 (т, J=7,4 Гц, 4H), 2,29 (с, 3H), 2,10-2,05 (м, 2H), NH и COOH не наблюдали; MS (ES+) m/z 467,1 (M+1).
ПРИМЕР 286
Синтез (S)-5-хлор-2-фтор-N-(тиазол-4-ил)-4-((1-(2,4,5-трифторфенил)этил)амино)бензолсульфонамида

Стадия 1. Получение (R,E)-2-метил-N-(2,4,5-трифторбензилиден)пропан-2-сульфинамида

В смесь 2,4,5-трифторбензальдегида (6,75 г, 42,2 ммоль), (R)-2-метилпропан-2-сульфинамиде (5,62 г, 46,4 ммоль) и п-толуолсульфонате пиридиния (0,53 г, 2,12 ммоль) в безводном дихлорметане (40 мл) добавляли безводный сульфат магния (25,4 г, 210 ммоль) и реакцию перемешивали при температуре окружающей среды в течение 16 ч. Реакционную смесь фильтровали и фильтрат концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с использованием 20% этилацетата в петролейном эфире, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (5,60 г, выход 50%): MS (ES+) m/z 264,1 (M+1).
Стадия 2. Получение (R)-2-метил-N-((S)-1-(2,4,5-трифторфенил)этил)пропан-2-сульфинамида

В смесь (R,E)-2-метил-N-(2,4,5-трифторбензилиден)пропан-2-сульфинамида (2,65 г, 10 ммоль) в дихлорметане (40 мл) добавляли 3 M раствор бромида метилмагния в простом диэтиловом эфире (6,60 мл, 20 ммоль) при -50°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 16 ч. Добавляли воду (60 мл) и смесь экстрагировали дихлорметаном (3×30 мл). Объединенные органические слои промывали солевым раствором (3×30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием 40% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного масла (2,73 г, выход 98%): MS (ES+) m/z 280,1 (M+1).
Стадия 3. Получение (S)-1-(2,4,5-трифторфенил)этан-1-амина

К (R)-2-метил-N-((S)-1-(2,4,5-трифторфенил)этил)пропан-2-сульфинамиду (2,70 г, 10 ммоль) добавляли 1,5 M раствор хлороводорода в метаноле (10,5 ммоль, 7,00 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Затем реакционную смесь концентрировали in vacuo. Добавляли этилацетат (20 мл) и насыщенный бикарбонат натрия (10 мл) и смесь перемешивали при температуре окружающей среды в течение 30 минут. Водную фазу экстрагировали этилацетатом (30 мл). Объединенные органические слои промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде бесцветного масла (1,43 г, выход 82%): MS (ES+) m/z 176,1 (M+1).
Стадия 4. Получение трет-бутил (S)-((5-хлор-2-фтор-4-((1-(2,4,5-трифторфенил)этил)амино)фенил)сульфонил)(тиазол-4-ил)карбамата

В смесь (S)-1-(2,4,5-трифторфенил)этан-1-амина (0,17 г, 1,00 ммоль) и трет-бутил((5-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,41 г, 1,00 ммоль) в безводном диметилсульфоксиде (6 мл) добавляли карбонат калия (0,28 г, 2,00 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч. Добавляли воду (15 мл) и смесь экстрагировали простым диэтиловым эфиром (2×20 мл). Объединенную органическую фазу промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-30% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,05 г, выход 8%): MS (ES+) m/z 566,0 (M+1), 568,0 (M+1).
Стадия 5. Получение (S)-5-хлор-2-фтор-N-(тиазол-4-ил)-4-((1-(2,4,5-трифторфенил)этил)амино)бензолсульфонамида

В раствор трет-бутил (S)-((5-хлор-2-фтор-4-((1-(2,4,5-трифторфенил)этил)амино)фенил)сульфонил)(тиазол-4-ил)карбамата (0,05 г, 0,09 ммоль) в дихлорметане (1 мл) добавляли трифторуксусную кислоту (1 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Концентрированием реакционной смеси in vacuo получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,04 г, выход 100%):1H ЯМР (300 МГц, DMSO-d6) δ 11,15 (с, 1H), 8,87 (дд, J=2,2, 0,7 Гц, 1H), 7,64-7,50 (м, 3H), 6,97 (д, J=2,1 Гц, 1H), 6,66-6,63 (м, 1H), 6,43 (д, J=13,2 Гц, 1H), 4,95-4,90 (м, 1H), 1,55-1,51 (м, 3H); MS (ES+) m/z 446,0 (M+1).
ПРИМЕР 287
Синтез (S)-5-хлор-4-((1-(2,5-дифторфенил)этил)амино)-2-фтор-N-(оксазол-2-ил)бензолсульфонамида

Стадия 1. Получение 5-хлор-2,4-дифтор-N-(оксазол-2-ил)бензолсульфонамида

В раствор трет-бутил оксазол-2-илкарбамата (1,98 г, 10,7 ммоль) в безводном тетрагидрофуране (20 мл) добавляли 1 M раствор бис(триметилсилил)амида лития в тетрагидрофуране (15,0 мл, 15,0 ммоль) при -78°C и реакционную смесь перемешивали при -78°C в течение 15 минут. Затем реакционную смесь нагревали до температуры окружающей среды, перемешивали в течение 10 минут и охлаждали до -78°C. Затем туда добавляли раствор 5-хлор-2,4-дифторбензолсульфонилхлорида (2,65 г, 10,7 ммоль) в безводном тетрагидрофуране (20 мл) и реакционную смесь перемешивали при -78°C в течение 2 ч. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 16 ч. Реакционную смесь разводили насыщенным хлоридом аммония (50 мл) и экстрагировали этилацетатом (2×40 мл). Объединенную органическую фазу промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-65% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,43 г, выход 13%):1H-ЯМР (300 МГц, CDCl3) δ 8,14-8,08 (м, 1H), 7,16 (д, J=1,7 Гц, 1H), 7,05-6,99 (м, 1H), 6,98 (д, J=1,7 Гц, 1H); MS (ES+) m/z 297,1 (M+1), 295,1 (M+1).
Стадия 2. Получение 5-хлор-2,4-дифтор-N-(4-метоксибензил)-N-(оксазол-2-ил)бензолсульфонамида

В раствор 5-хлор-2,4-дифтор-N-(оксазол-2-ил)бензолсульфонамида (0,43 г, 1,47 ммоль) в диметилсульфоксиде (5 мл) добавляли карбонат цезия (1,05 г, 3,23 ммоль) и 4-метоксибензилхлорид (0,28 мл, 2,06 ммоль) и реакционную смесь перемешивали при 40°C в течение 16 ч. Реакционную смесь разводили водой (10 мл) и экстрагировали этилацетатом (2×15 мл). Объединенную органическую фазу промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-80% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного масла (0,21 г, выход 34%): MS (ES+) m/z 417,2 (M+1), 415,2 (M+1).
Стадия 3. Получение (S)-5-хлор-4-((1-(2,5-дифторфенил)этил)амино)-2-фтор-N-(4-метоксибензил)-N-(оксазол-2-ил)бензолсульфонамида

В смесь (S)-1-(2,5-дифторфенил)этан-1-амина (0,01 г, 0,50 ммоль) и 5-хлор-2,4-дифтор-N-(4-метоксибензил)-N-(оксазол-2-ил)бензолсульфонамида (0,21 г, 0,50 ммоль) в безводном диметилсульфоксиде (4 мл) добавляли карбонат калия (0,28 г, 2,00 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч. Добавляли воду (10 мл) и смесь экстрагировали простым диэтиловым эфиром (2×20 мл). Объединенную органическую фазу промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-100% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,05 г, выход 18%): MS (ES+) m/z 554,2 (M+1), 552,2 (M+1).
Стадия 4. Получение (S)-5-хлор-4-((1-(2,5-дифторфенил)этил)амино)-2-фтор-N-(оксазол-2-ил)бензолсульфонамида

В (S)-5-хлор-4-((1-(2,5-дифторфенил)этил)амино)-2-фтор-N-(4-метоксибензил)-N-(оксазол-2-ил)бензолсульфонамид (0,05 г, 0,09 ммоль) добавляли трифторуксусную кислоту (3 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 96 ч. Затем смесь концентрировали in vacuo и остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусной кислоты в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,004 г, выход 25%):1H-ЯМР (300 МГц, DMSO-d6) δ 11,64-11,48 (ушир. с, 1H), 10,38-10,17 (ушир. с, 1H), 7,85 (к, J=8,2 Гц, 1H), 7,77 (д, J=7,4 Гц, 1H), 7,45-7,41 (м, 2H), 6,89-6,86 (м, 1H), 6,78-6,70 (м, 2H), 4,52-4,50 (м, 1H), 2,44-2,13 (м, 3H); MS (ES+) m/z 434,2 (M+1), 432,2 (M+1).
ПРИМЕР 288
Синтез гидрохлорида 4-((2-(азетидин-1-илметил)-3,6-дифторбензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида
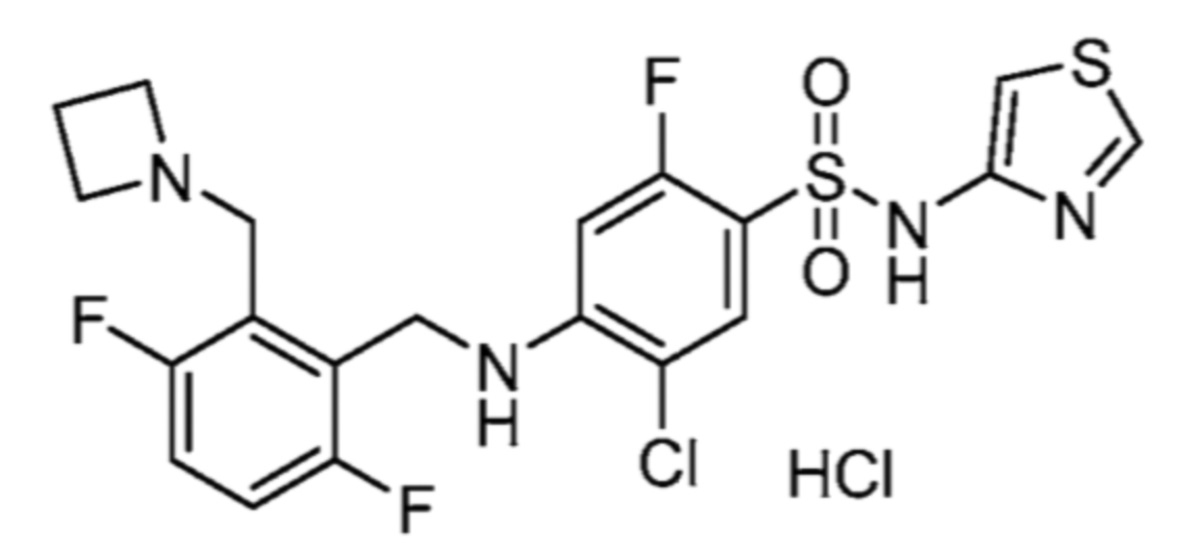
Стадия 1. Получение (2-(азетидин-1-илметил)-3,6-дифторфенил)метанола

К 3,6-дифторфталевому ангидриду (1,84 г, 10,0 ммоль) добавляли безводный тетрагидрофуран (20 мл) и азетидин (0,57 мл, 10 ммоль) и смесь перемешивали при температуре окружающей среды в течение 2 ч. Затем туда добавляли 1,0 M раствор борана в тетрагидрофуране (25 мл, 25 ммоль) и смесь перемешивали при температуре окружающей среды в течение 16 ч. Смесь медленно выливали в 2 M гидроксид натрия (30 мл) и перемешивали в течение 2 ч. Раствор экстрагировали этилацетатом (3×30 мл), промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде бесцветного масла (2,16 г, выход 99%): MS (ES+) m/z 214,1 (M+1).
Стадия 2. Получение 2-(2-(азетидин-1-илметил)-3,6-дифторбензил)изоиндолин-1,3-диона

В раствор (2-(азетидин-1-илметил)-3,6-дифторфенил)метанола (1,93 г, 8,90 ммоль), фталимида (1,43 г, 9,8 ммоль) и трифенилфосфина (3,96 г, 15,1 ммоль) в безводном тетрагидрофуране (30 мл) добавляли по каплям диизопропилазодикарбоксилат (2,70 мл, 13,3 ммоль) при 0°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды, перемешивали в течение 1 ч и затем концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-50% этилацетата в гексанах, чтобы получать указанное в заголовке соединение в виде масла (3,08 г, выход 99%): MS (ES+) m/z 343,2 (M+1).
Стадия 3. Получение (2-(азетидин-1-илметил)-3,6-дифторфенил)метанамина

В раствор 2-(2-(азетидин-1-илметил)-3,6-дифторбензил)изоиндолин-1,3-диона (3,08 г, 8,9 ммоль) в этаноле (30 мл) добавляли моногидрат гидразина (5,00 мл, 100 ммоль) и смесь нагревали с обратным холодильником в течение 1 ч. После охлаждения до температуры окружающей среды, реакционную смесь концентрировали in vacuo. К остатку добавляли воду (20 мл) и смесь экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде бесцветного масла (1,88 г, выход 99%): MS (ES+) m/z 213,1 (M+1).
Стадия 4. Получение трет-бутил ((4-((2-(азетидин-1-илметил)-3,6-дифторбензил)амино)-5-хлор-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата

В смесь (2-(азетидин-1-илметил)-3,6-дифторфенил)метанамина (2,40 г, 11,4 ммоль) и трет-бутил ((5-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (4,68 г, 11,4 ммоль) в безводном диметилсульфоксиде (30 мл) добавляли карбонат калия (3,93 г, 28,5 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч. Добавляли воду (70 мл) и смесь экстрагировали смесью простого диэтилового эфира и этилацетата 1:1 (2×50 мл). Объединенную органическую фазу промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали остаток, который очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-50% этилацетата в гексанах. Дополнительной очисткой с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-10% метанола в дихлорметане, получали указанное в заголовке соединение в виде бесцветного твердого вещества (1,08 г, выход 16%): MS (ES+) m/z 603,2 (M+1), 605,2 (M+1).
Стадия 5. Получение гидрохлорида 4-((2-(азетидин-1-илметил)-3,6-дифторбензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил ((4-((2-(азетидин-1-илметил)-3,6-дифторбензил)амино)-5-хлор-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата (1,08 г, 1,79 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (5 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Смесь концентрировали in vacuo и остаток растворяли в метаноле (5 мл). Затем туда добавляли 7 M раствор гидроксида аммония (5 мл) в метаноле при -40°C. Концентрированием in vacuo получали остаток, который растворяли в дихлорметане (10 мл) и промывали 2 M соляной кислотой (10 мл). Водный слой экстрагировали дихлорметаном (2×10 мл). Объединенную органическую фазу концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-10% метанола в дихлорметане, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,610 г, выход 63%):1H ЯМР (300 МГц, DMSO-d6) δ 11,19 (с, 1H), 10,52-10,47 (м, 1H), 8,89 (д, J=2,2 Гц, 1H), 7,62 (д, J=7,3 Гц, 1H), 7,47-7,41 (м, 2H), 7,01-7,00 (м, 1H), 6,94-6,92 (м, 1H), 6,78-6,73 (м, 1H), 4,58-4,53 (м, 4H), 4,20-4,14 (м, 2H), 4,05-3,99 (м, 2H), 2,39-2,27 (м, 2H); MS (ES+) m/z 505,2 (M+1), 503,2 (M+1).
ПРИМЕР 289
Синтез 4-(((1R,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-ил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил ((4-(((1R,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-ил)амино)-3-бром-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

В раствор трет-бутил (3-бром-2,4,6-трифторфенил)сульфонил(тиазол-4-ил)карбамата (0,30 г, 0,63 ммоль) в безводном N,N-диметилформамиде (8 мл) добавляли (1R,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-амин (0,12 г, 0,63 ммоль) и карбонат калия (0,18 г, 1,27 ммоль). Смесь перемешивали при температуре окружающей среды в течение 12 ч и затем разводили водой (10 мл) и экстрагировали этилацетатом (2×10 мл). Объединенные органические экстракты промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 100% этилацетата, получали указанное в заголовке соединение в виде твердого вещества желтого цвета (0,25 г, выход 55%):1H ЯМР (400 МГц, CDCl3) δ 8,72 (д, J=2,4 Гц, 1H), 7,44 (д, J=2,2 Гц, 1H), 7,32-7,28 (м, 2H), 7,28-7,23 (м, 2H), 6,58 (д, J=8,4 Гц, 1H), 6,54-6,44 (м, 1H), 4,87 (дт, J=7,6, 3,4 Гц, 1H), 4,05-3,94 (м, 1H), 3,46 (к, J=7,2 Гц, 2H), 3,42-3,34 (м, 2H), 2,55 (тд, J=13,4, 6,8 Гц, 1H), 2,09 (квин, J=7,2 Гц, 2H), 1,89 (т, J=3,4 Гц, 1H), 1,34 (с, 9H); MS (ES+) m/z 641,0 (M+1).
Стадия 2. Получение трет-бутил ((4-(((1R,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-ил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

В раствор трет-бутил ((4-(((1R,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-ил)амино)-3-бром-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,16 г, 0,25 ммоль) в метаноле (10 мл) добавляли палладий на древесном угле (0,020 г). Суспензию перемешивали в атмосфере водорода (50 фунтов/дюйм2) при 50°C в течение 12 ч. После охлаждения до температуры окружающей среды, реакционную смесь фильтровали и фильтрат концентрировали in vacuo. Остаток очищали с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом ацетонитрила в воде, содержащей 0,225% муравьиной кислоты, чтобы получать указанное в заголовке соединение в виде твердого вещества желтого цвета (0,13 г, выход 93%):1H ЯМР (400 МГц, CDCl3) δ 8,78 (д, J=2,2 Гц, 1H), 7,62 (д, J=8,8 Гц, 1H), 7,50 (дд, J=4,4, 2,0 Гц, 2H), 7,47-7,38 (м, 3H), 6,46 (д, J=12,0 Гц, 2H), 5,05 (т, J=8,4 Гц, 1H), 4,55 (д, J=7,6 Гц, 1H), 4,23-4,13 (м, 2H), 4,06 (д, J=7,6 Гц, 2H), 2,94-2,85 (м, 1H), 2,75-2,45 (м, 2H), 2,30-2,05 (м, 1H), 1,42 (с, 9H); MS (ES+) m/z 563,1 (M+1).
Стадия 3. Получение 4-(((1R,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-ил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано в примере 5, стадии 2 и выполняя некритические вариации, чтобы заменять трет-бутил (S)-((5-хлор-4-((1-(5-хлор-2-фторфенил)этил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамат на трет-бутил ((4-(((1R,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-ил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамат, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,080 г, выход 96%):1H ЯМР (400 МГц, CD3OD) δ 8,76 (д, J=2,4 Гц, 1H), 8,55 (с, 1H), 7,42-7,36 (м, 1H), 7,34-7,29 (м, 3H), 6,95 (д, J=2,0 Гц, 1H), 6,35 (д, J=12,4 Гц, 2H), 4,97-4,94 (м, 1H), 4,07 (т, J=6,8 Гц, 1H), 3,61-3,51 (м, 4H), 2,78 (тд, J=13,0, 7,2 Гц, 1H), 2,20 (квин, J=7,2 Гц, 2H), 1,64 (тд, J=13,2, 6,6 Гц, 1H), NH не наблюдали; MS (ES+) m/z 463,1 (M+1).
ПРИМЕР 290
Синтез 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-хлорбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 4-хлор-1,3-дигидроизобензофуран-1-ола
В смесь 4-хлоризобензофуран-1(3H)-она (3,37 г, 20,0 ммоль) в безводном дихлорметане (50 мл) добавляли 1 M раствор гидрид диизобутилалюминия в толуоле (44,0 мл, 44,0 ммоль) при -78°C. Реакционную смесь перемешивали при -78°C в течение 2 ч и затем гасили добавлением 1 M соляной кислоты (50 мл). Водный слой экстрагировали дихлорметаном (2×30 мл). Объединенные органические слои промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде бесцветного твердого вещества (2,85 г, выход 84%):1H ЯМР (300 МГц, CDCl3) δ 7,42-7,29 (м, 3H), 6,55-6,53 (м, 1H), 5,27-5,23 (м, 1H), 5,08-5,03 (м, 1H), 3,40-3,37 (м, 1H).
Стадия 2. Получение (2-(азетидин-1-илметил)-6-хлорфенил)метанола

В раствор 4-хлор-1,3-дигидроизобензофуран-1-ола (0,75 г, 4,41 ммоль) и азетидина (0,30 мл, 5,3 ммоль) в дихлорметане (10 мл) добавляли триацетоксиборгидрид натрия (2,60 г, 12,3 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч. Затем туда добавляли насыщенный бикарбонат натрия (20 мл) и смесь экстрагировали дихлорметаном (3×20 мл). Объединенную органическую фазу промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-70% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного масла (0,90 г, выход 96%): MS (ES+) m/z 212,1 (M+1), 214,1 (M+1).
Стадия 3. Получение 2-(2-(азетидин-1-илметил)-6-хлорбензил)изоиндолин-1,3-диона

Следуя процедуре как описано в примере 288, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять (2-(азетидин-1-илметил)-3,6-дифторфенил)метанол на (2-(азетидин-1-илметил)-6-хлорфенил)метанол, указанное в заголовке соединение получали в виде масла (1,38 г, выход 95%): MS (ES+) m/z 341,1 (M+1), 343,2 (M+1).
Стадия 4. Получение (2-(азетидин-1-илметил)-6-хлорфенил)метанамина

Следуя процедуре как описано в примере 288, стадии 3 и выполняя некритические вариации при необходимости, чтобы заменять 2-(2-(азетидин-1-илметил)-3,6-дифторбензил)изоиндолин-1,3-дион на 2-(2-(азетидин-1-илметил)-6-хлорбензил)изоиндолин-1,3-дион, указанное в заголовке соединение получали в виде бесцветного масла (1,88 г, выход 99%): MS (ES+) m/z 211,1 (M+1), 213,1 (M+1).
Стадия 5. Получение трет-бутил ((4-((2-(азетидин-1-илметил)-6-хлорбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

В смесь (2-(азетидин-1-илметил)-6-хлорфенил)метанамина (0,89 г, 4,24 ммоль) и трет-бутил тиазол-4-ил((2,4,6-трифторфенил)сульфонил)карбамата (1,67 г, 4,24 ммоль) в безводном N,N-диметилформамиде (10 мл) добавляли карбонат цезия (4,12 г, 12,7 ммоль) и реакционную смесь нагревали до 45°C в течение 1 ч. Реакционную смесь оставляли остывать до температуры окружающей среды и в нее добавляли воду (40 мл). Смесь экстрагировали этилацетатом (2×50 мл). Объединенную органическую фазу промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали остаток, который очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-100% этилацетата в гексанах. Дополнительной очисткой с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-10% метанола в дихлорметане, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,10 г, выход 4%): MS (ES+) m/z 587,2 (M+1), 585,2 (M+1).
Стадия 6. 2,2,2-трифторацетат 4-(((2-(азетидин-1-илметил-d2)-3,6-дифторфенил)метил-d2)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил ((4-((2-(азетидин-1-илметил)-6-хлорбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,10 г, 0,17 ммоль) в дихлорметане (3 мл) добавляли трифторуксусную кислоту (3 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Затем смесь концентрировали in vacuo для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,10 г, количественный выход):1H ЯМР (300 МГц, DMSO-d6) δ 11,25 (с, 1H), 10,19-10,17 (ушир. с, 1H), 8,92 (д, J=2,2 Гц, 1H), 7,62 (дд, J=5,8, 3,6 Гц, 1H), 7,53-7,48 (м, 2H), 7,17-7,15 (м, 1H), 6,93 (д, J=2,2 Гц, 1H), 6,40-6,35 (м, 2H), 4,47-4,38 (м, 4H), 4,15-4,01 (м, 4H), 2,41-2,25 (м, 2H); MS (ES+) m/z 487,3, 485,2 (M+1).
ПРИМЕР 291
Синтез 2,2,2-трифторацетата 4-((2-((2,6-диазаспиро[3.3]гептан-2-ил)метил)-6-хлорбензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил 6-(3-хлор-2-(гидроксиметил)бензил)-2,6-диазаспиро[3.3]гептан-2-карбоксилата
Следуя процедуре как описано в примере 290, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять азетидин на трет-бутил 2,6-диазаспиро[3.3]гептан-2-карбоксилат, получали указанное в заголовке соединение (1,51 г, выход 92%): MS (ES+) m/z 355,2 (M+1), 353,2 (M+1).
Стадия 2. Получение трет-бутил 6-(3-хлор-2-((1,3-диоксоизоиндолин-2-ил)метил)бензил)-2,6-диазаспиро[3.3]гептан-2-карбоксилата

Следуя процедуре как описано в примере 288, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять (2-(азетидин-1-илметил)-3,6-дифторфенил)метанол на трет-бутил 6-(3-хлор-2-(гидроксиметил)бензил)-2,6-диазаспиро[3.3]гептан-2-карбоксилат, указанное в заголовке соединение получали в виде масла (1,38 г, выход 95%): MS (ES+) m/z 484,2 (M+1), 482,2 (M+1).
Стадия 3. Получение трет-бутил 6-(2-(аминометил)-3-хлорбензил)-2,6-диазаспиро[3.3]гептан-2-карбоксилата

Следуя процедуре как описано в примере 288, стадии 3 и выполняя некритические вариации при необходимости, чтобы заменять 2-(2-(азетидин-1-илметил)-3,6-дифторбензил)изоиндолин-1,3-дион на трет-бутил 6-(3-хлор-2-((1,3-диоксоизоиндолин-2-ил)метил)бензил)-2,6-диазаспиро[3.3]гептан-2-карбоксилат, указанное в заголовке соединение получали в виде масла (1,12 г, выход 79%): MS (ES+) m/z 354,2 (M+1), 352,2 (M+1).
Стадия 4. Получение трет-бутил 6-(2-(((4-(N-(трет-бутоксикарбонил)-N-(тиазол-4-ил)сульфамоил)-2-хлор-5-фторфенил)амино)метил)-3-хлорбензил)-2,6-диазаспиро[3.3]гептан-2-карбоксилата

Следуя процедуре как описано в примере 288, стадии 4 и выполняя некритические вариации при необходимости, чтобы заменять (2-(азетидин-1-илметил)-3,6-дифторфенил)метанамин на трет-бутил 6-(2-(аминометил)-3-хлорбензил)-2,6-диазаспиро[3.3]гептан-2-карбоксилат, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,18 г, выход 13%): MS (ES+) m/z 728,3 (M+1), 726,3 (M+1).
Стадия 5. 2,2,2-трифторацетат 4-((2-((2,6-диазаспиро[3.3]гептан-2-ил)метил)-6-хлорбензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано в примере 290, стадии 6 и выполняя некритические вариации при необходимости, чтобы заменять трет-бутил ((4-((2-(азетидин-1-илметил)-6-хлорбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамат на трет-бутил 6-(2-(((4-(N-(трет-бутоксикарбонил)-N-(тиазол-4-ил)сульфамоил)-2-хлор-5-фторфенил)амино)метил)-3-хлорбензил)-2,6-диазаспиро[3.3]гептан-2-карбоксилат, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,18 г, выход 100%):1H ЯМР (300 МГц, DMSO-d6) δ 11,21-11,18 (ушир. с, 1H), 10,60-10,58 (м, 1H), 8,90-8,83 (м, 3H), 7,64-7,58 (м, 2H), 7,50-7,41 (м, 2H), 7,01-7,00 (м, 1H), 6,78 (д, J=13,1 Гц, 1H), 6,55-6,54 (м, 1H), 4,49-4,46 (м, 4H), 4,33-4,28 (м, 4H), 4,16-4,10 (м, 4H); MS (ES+) m/z 544,1, 542,1 (M+1).
ПРИМЕР 292
Синтез 2,2,2-трифторацетата 5-хлор-4-((2-хлор-6-((6-метил-2,6-диазаспиро[3.3]гептан-2-ил)метил)бензил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

К 2,2,2-трифторацетату 4-((2-((2,6-диазаспиро[3.3]гептан-2-ил)метил)-6-хлорбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида (0,16 г, 0,24 ммоль) добавляли тетрагидрофуран (2 мл), 37% водный формальдегид (0,1 мл, 1,2 ммоль) и цианоборогидрид натрия (0,03 г, 0,48 ммоль). Реакционную смесь перемешивали в течение 2 часов при температуре окружающей среды и затем гасили добавлением насыщенного бикарбоната натрия (10 мл). Смесь экстрагировали дихлорметаном (3×10 мл). Объединенную органическую фазу промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,04 г, выход 25%):1H ЯМР (300 МГц, DMSO-d6) δ 8,87 (д, J=2,1 Гц, 1H), 8,19 (м, 0,6H), 7,61 (д, J=7,3 Гц, 1H), 7,45-7,42 (м, 1H), 7,32 (дд, J=3,2, 2,7 Гц, 2H), 7,08-6,99 (м, 2H), 6,88-6,88 (м, 1H), 4,53-4,50 (м, 2H), 3,91-3,86 (м, 4H), 3,66 (д, J=4,9 Гц, 2H), 3,34-3,31 (м, 4H), 2,59-2,57 (с, 3H), NH не наблюдали; MS (ES+) m/z 558,2 (M+1), 556,2 (M+1).
ПРИМЕР 293
Синтез 5-хлор-4-((3,6-дифтор-2-(гидроксиметил)бензил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение (2-бром-3,6-дифторфенил)метанола

В раствор 2-бром-3,6-дифторбензальдегида (10,0 г, 45,2 ммоль) в смеси дихлорметана и метанола 1:1 (150 мл) добавляли борогидрид натрия (2,58 г, 67,8 ммоль) при 0°C частями в течение 15 минут. Реакционную смесь перемешивали при 0°C в течение 2 часов и затем гасили добавлением насыщенного хлорида аммония (50 мл). Реакционную смесь концентрировали при пониженном давлении и остававшийся водный слой экстрагировали этилацетатом (2×70 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде бесцветного твердого вещества (9,70 г, выход 96%), которое использовали на следующей стадии без дополнительной очистки.
Стадия 2. Получение ((2-бром-3,6-дифторбензил)окси)(трет-бутил)диметилсилана

В раствор (2-бром-3,6-дифторфенил)метанола (9,70 г, 43,5 ммоль) в N,N-диметилформамиде (85 мл) добавляли имидазол (3,84 г, 56,5 ммоль), после чего следовал трет-бутилдиметилсилил хлорид (6,85 г, 45,6 ммоль) при 0°C, и реакционную смесь перемешивали в течение 1 часа при 0°C. Затем туда добавляли насыщенный хлорид аммония (50 мл) и смесь экстрагировали простым диэтиловым эфиром (2×60 мл). Объединенные органические слои промывали солевым раствором (2×30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде бесцветного масла (14,0 г, выход 96%):1H-ЯМР (300 МГц, CDCl3) δ 7,08-7,04 (м, 2H), 4,85 (дд, J=2,4, 0,7 Гц, 2H), 0,92 (с, 9H), 0,13 (с, 6H).
Стадия 3. Получение 2-(((трет-бутилдиметилсилил)окси)метил)-3,6-дифторбензальдегида

В раствор ((2-бром-3,6-дифторбензил)окси)(трет-бутил)диметилсилана (14,0 г, 41,5 ммоль) в безводном тетрагидрофуране (120 мл) добавляли по каплям 1,5 M раствор н-бутиллития в гексане (36,0 мл, 54,0 ммоль) при -78°C. Реакционную смесь перемешивали в течение 10 минут при -78°C и затем туда добавляли N,N-диметилформамид (5,10 мл, 66,4 ммоль). Реакционную смесь перемешивали при -78°C в течение 15 минут, затем оставляли нагреваться до температуры окружающей среды и перемешивали в течение 40 минут. Туда добавляли насыщенный хлорид аммония (50 мл) и смесь экстрагировали простым диэтиловым эфиром (2×80 мл). Объединенные органические слои промывали солевым раствором (2×40 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде бесцветного масла (11,7 г, выход 96%):1H-ЯМР (300 МГц, CDCl3) δ 10,46 (т, J=0,4 Гц, 1H), 7,29-7,22 (м, 1H), 7,12 (дд, J=9,5, 4,1 Гц, 1H), 5,04 (д, J=1,9 Гц, 2H), 0,89 (с, 9H), 0,11 (с, 6H).
Стадия 4. Получение (E)-2-(((трет-бутилдиметилсилил)окси)метил)-3,6-дифторбензальдегид оксима

В раствор 2-(((трет-бутилдиметилсилил)окси)метил)-3,6-дифторбензальдегида (4,25 г, 14,9 ммоль) в ацетонитриле (18 мл) и воде (32 мл) добавляли бикарбонат натрия (3,70 г, 45,0 ммоль) после чего следовал гидрохлорид гидроксиламина (2,10 г, 29,7 ммоль) и тетрабутилхлорид аммония (0,21 г, 0,75 ммоль), и реакционную смесь перемешивали при температуре окружающей среды в течение 45 минут. Туда добавляли уксусную кислоту (1,2 мл) для того, чтобы корректировать pH до 6,7, после чего следовала вода (30 мл), и смесь экстрагировали простым диэтиловым эфиром (3×60 мл). Объединенные органические слои промывали солевым раствором (40 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде твердого вещества желтого цвета (4,53 г, выход 100%): MS (ES+) m/z 302,2 (M+1),.
Стадия 5. Получение (2-(((трет-бутилдиметилсилил)окси)метил)-3,6-дифторфенил)метанамина

В раствор (E)-2-(((трет-бутилдиметилсилил)окси)метил)-3,6-дифторбензальдегид оксима (4,53 г, 14,9 ммоль) в простом диэтиловом эфире (150 мл) добавляли 1,0 M раствор гидрида лития алюминия в простом диэтиловом эфире (37,7 мл, 37,7 ммоль) при 0°C и реакционную смесь перемешивали при 0°C в течение 2 ч. Затем реакцию гасили медленным добавлением воды (1,4 мл) и перемешивали в течение 5 минут. Затем туда добавляли 2 M гидроксид натрия (2,8 мл). Смесь перемешивали в течение 15 минут и в нее добавляли воду (4,2 мл), после чего следовал безводный сульфат натрия (10 г). Смесь фильтровали и фильтровальный осадок споласкивали простым диэтиловым эфиром (3×15 мл). Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде твердого вещества желтого цвета (3,57 г, выход 83%): MS (ES+) m/z 288,2 (M+1).
Стадия 6. Получение трет-бутил ((5-хлор-4-((3,6-дифтор-2-(гидроксиметил)бензил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата

Следуя процедуре как описано в примере 288, стадии 4 и выполняя некритические вариации при необходимости, чтобы заменять (2-(азетидин-1-илметил)-3,6-дифторфенил)метанамин на (2-(((трет-бутилдиметилсилил)окси)метил)-3,6-дифторфенил)метанамин, указанное в заголовке соединение получали в виде бесцветного масла (1,41 г, выход 42%): MS (ES+) m/z 566,2 (M+1), 564,2 (M+1).
Стадия 7. Получение 5-хлор-4-((3,6-дифтор-2-(гидроксиметил)бензил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил ((5-хлор-4-((3,6-дифтор-2-(гидроксиметил)бензил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата (0,2 г, 0,35 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (5 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Затем смесь концентрировали in vacuo. Остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,1% трифторуксусную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,03 г, выход 18%):1H ЯМР (300 МГц, DMSO-d6) δ 11,14 (с, 1H), 8,89 (дд, J=2,2, 0,8 Гц, 1H), 7,59 (д, J=7,4 Гц, 1H), 7,24 (т, J=6,9 Гц, 2H), 7,00-6,93 (м, 2H), 6,64-6,60 (м, 1H), 5,65-5,63 (м, 1H), 4,63-4,61 (м, 2H), 4,55-4,53 (м, 2H); MS (ES+) m/z 466,1 (M+1), 464,1 (M+1).
ПРИМЕР 294
Синтез 2,2,2-трифторацетата 2,3-дифтор-4-((2-фтор-6-(пирролидин-1-илметил)бензил)амино)-N-(изоксазол-3-ил)бензолсульфонамида

Стадия 1. Получение 2-фтор-6-(пирролидин-1-илметил)фенил)метанамина

Следуя процедуре как описано в примере 246, стадиях 1 и 2 и выполняя некритические вариации при необходимости, чтобы заменять азетидин на пирролидин, указанное в заголовке соединение получали в виде бесцветного масла (0,37 г, выход 78%):1H-ЯМР (300 МГц, DMSO-d6) δ 7,20 (тд, J=7,8, 5,9 Гц, 1H), 7,12-7,03 (м, 2H), 3,76-3,69 (м, 2H), 3,69-3,62 (м, 2H), 2,43-2,38 (м, 4H), 1,83-1,77 (м, 2H), 1,70-1,63 (м, 4H); MS (ES+) m/z 209,3 (M+1).
Стадия 2. Получение трет-бутил изоксазол-3-ил((2,3,4-трифторфенил)сульфонил)карбамата

В раствор трет-бутил изоксазол-3-илкарбамата (3,30 г, 17,9 ммоль) в безводном тетрагидрофуране (100 мл) добавляли 1 M раствор бис(триметилсилил)амида лития в тетрагидрофуране (21,5 мл, 21,5 ммоль) при -78°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 1 ч. Реакционную смесь охлаждали до -78°C, и туда добавляли раствор 2,3,4-трифторбензолсульфонилхлорида (4,10 г, 17,9 ммоль) в безводном тетрагидрофуране (20 мл). Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 3 ч. Смесь разводили этилацетатом (200 мл), промывали насыщенным хлоридом аммония (2×200 мл), солевым раствором (2×100 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 60% этилацетата в гептане, получали указанное в заголовке соединение в виде бесцветного твердого вещества (5,20 г, выход 77%): MS (ES+) m/z 379,1 (M+1).
Стадия 2. Получение трет-бутил ((2,3-дифтор-4-((2-фтор-6-(пирролидин-1-илметил)бензил)амино)фенил)сульфонил)(изоксазол-3-ил)карбамата

В смесь (2-фтор-6-(пирролидин-1-илметил)фенил)метанамина (0,21 г, 1,00 ммоль) и трет-бутил изоксазол-3-ил((2,3,4-трифторфенил)сульфонил)карбамата (0,38 г, 1,00 ммоль) в безводном N,N-диметилформамиде (4 мл) добавляли карбонат калия (0,28 г, 2,00 ммоль) и реакцию перемешивали при температуре окружающей среды в течение 16 ч. Реакционная смесь была и туда добавляли воду (10 мл). Смесь экстрагировали этилацетатом (2×15 мл). Объединенную органическую фазу промывали солевым раствором (25 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали остаток, который очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом 0-100% этилацетата в гексанах для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,03 г, выход 5%): MS (ES+) m/z 567,2 (M+1).
Стадия 3. Получение 2,2,2-трифторацетата 2,3-дифтор-4-((2-фтор-6-(пирролидин-1-илметил)бензил)амино)-N-(изоксазол-3-ил)бензолсульфонамида

Следуя процедуре как описано в примере 290, стадии 6 и выполняя некритические вариации при необходимости, чтобы заменять трет-бутил ((4-((2-(азетидин-1-илметил)-6-хлорбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамат на трет-бутил ((2,3-дифтор-4-((2-фтор-6-(пирролидин-1-илметил)бензил)амино)фенил)-сульфонил)(изоксазол-3-ил)карбамат, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,03 г, количественный выход):1H-ЯМР (300 МГц, DMSO-d6) δ 11,78-11,72 (ушир. с, 1H), 10,17-9,91 (ушир. с, 1H), 8,72 (д, J=1,8 Гц, 1H), 7,55-7,42 (м, 3H), 7,37-7,30 (м, 1H), 7,20-7,16 (м, 1H), 6,80-6,74 (м, 1H), 6,35 (д, J=1,8 Гц, 1H), 4,49 (м, 4H), 3,34-3,03 (м, 4H), 2,05-1,80 (м, 4H); MS (ES+) m/z 467,2 (M+1).
ПРИМЕР 295
Синтез формиата 4-((2-((2-окса-6-азаспиро[3.3]гептан-6-ил)метил)-6-хлорбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида
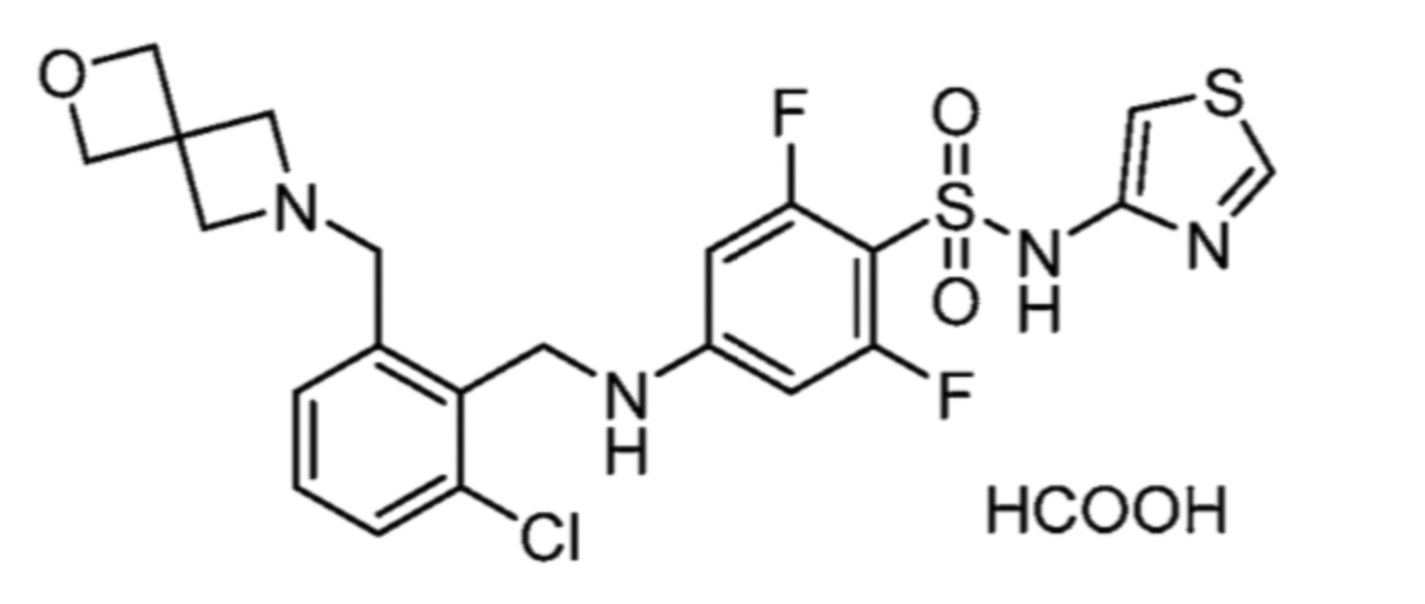
Стадия 1. Получение (2-((2-окса-6-азаспиро[3.3]гептан-6-ил)метил)-6-хлорфенил)метанола

Следуя процедуре как описано в примере 290, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять азетидин на 2-окса-6-азаспиро[3.3]гептаноксалат, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,29 г, выход 34%): MS (ES+) m/z 256,2 (M+1), 254,2 (M+1).
Стадия 2. Получение 2-(2-((2-окса-6-азаспиро[3.3]гептан-6-ил)метил)-6-хлорбензил)изоиндолин-1,3-диона

Следуя процедуре как описано в примере 288, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять (2-(азетидин-1-илметил)-3,6-дифторфенил)метанол на (2-((2-окса-6-азаспиро[3.3]гептан-6-ил)метил)-6-хлорфенил)метанол, указанное в заголовке соединение получали в виде масла (0,32 г, выход 95%): MS (ES+) m/z 385,2 (M+1), 383,2 (M+1).
Стадия 3. Получение (2-((2-окса-6-азаспиро[3.3]гептан-6-ил)метил)-6-хлорфенил)метанамина

Следуя процедуре как описано в примере 288, стадии 3 и выполняя некритические вариации при необходимости, чтобы заменять 2-(2-(азетидин-1-илметил)-3,6-дифторбензил)изоиндолин-1,3-дион на 2-(2-((2-окса-6-азаспиро[3.3]гептан-6-ил)метил)-6-хлорбензил)изоиндолин-1,3-дион, указанное в заголовке соединение получали в виде масла (0,14 г, выход 67%): MS (ES+) m/z 255,3 (M+1), 253,3 (M+1).
Стадия 4. Получение трет-бутил ((4-((2-((2-окса-6-азаспиро[3.3]гептан-6-ил)метил)-6-хлорбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

Следуя процедуре как описано в примере 290, стадии 5 и выполняя некритические вариации при необходимости, чтобы заменять (2-(азетидин-1-илметил)-6-хлорфенил)метанамин на (2-((2-окса-6-азаспиро[3.3]гептан-6-ил)метил)-6-хлорфенил)метанамин, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,12 г, выход 37%): MS (ES+) m/z 645,3 (M+1), 643,3 (M+1).
Стадия 5. Получение формиата 4-((2-((2-окса-6-азаспиро[3.3]гептан-6-ил)метил)-6-хлорбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано в примере 290, стадии 6 и выполняя некритические вариации при необходимости, чтобы заменять трет-бутил ((4-((2-(азетидин-1-илметил)-6-хлорбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамат на трет-бутил ((4-((2-((2-окса-6-азаспиро[3.3]гептан-6-ил)метил)-6-хлорбензил)амино)-5-хлор-2-фторфенил)сульфонил)(тиазол-4-ил)карбамат, очисткой с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом ацетонитрила в воде, содержащей 0,2% муравьиной кислоты, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,18 г, выход 100%):1H ЯМР (300 МГц, DMSO-d6) δ 8,91 (дд, J=1,3, 0,8 Гц, 1H), 8,14-8,13 (ушир. с, 1H), 7,44-7,31 (м, 3H), 7,12-7,10 (м, 1H), 6,91 (д, J=2,2 Гц, 1H), 6,39-6,34 (м, 2H), 4,56-4,54 (м, 2H), 4,36-4,34 (м, 2H), 3,31-3,25 (м, 8H), NH и COOH не наблюдали; MS (ES+) m/z 529,2 (M+1), 527,2 (M+1).
ПРИМЕР 296
Синтез формиата 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(5-фторпиридин-2-ил)бензолсульфонамида

Стадия 1. Получение N-(2,4-диметоксибензил)-5-фторпиридин-2-амина

В раствор 5-фторпиридин-2-амина (3,00 г, 26,8 ммоль) в безводном толуоле (80 мл) добавляли 2,4-диметоксибензальдегид (6,67 г, 40,2 ммоль) одной частью. Смесь нагревали до 140°C в течение 12 ч с использованием ловушки Дина-Старка для удаления воды. Затем смесь концентрировали при пониженном давлении. Остаток растворяли в метаноле (80 мл) и смесь охлаждали до 0°C. Затем туда добавляли борогидрид натрия (2,02 г, 53,5 ммоль) при 0°C. Смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 12 ч. Реакционную смесь разводили водой (100 мл) и экстрагировали дихлорметаном (3×100 мл). Объединенные органические слои промывали солевым раствором (3×50 мл), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 9 до 20% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (4,40 г, выход 63%):1H ЯМР (400 МГц, CDCl3) δ 7,98 (д, J=2,8 Гц, 1H), 7,24-7,15 (м, 2H), 6,51-6,34 (м, 3H), 4,89 (ушир. с, 1H), 4,39 (д, J=5,8 Гц, 2H), 3,83 (д, J=14,4 Гц, 6H); MS (ES+) m/z 263,1 (M+1).
Стадия 2. Получение N-(2,4-диметоксибензил)-2,4,6-трифтор-N-(5-фторпиридин-2-ил)бензолсульфонамида

В раствор N-(2,4-диметоксибензил)-5-фторпиридин-2-амина (2,00 г, 7,63 ммоль) в безводном тетрагидрофуране (20 мл) добавляли 1,6 M раствор метиллития в простом диэтиловом эфире (6,67 мл, 10,6 ммоль) при -78°C. Реакционную смесь нагревали до 0°C, перемешивали в течение 1 ч при 0°C и затем охлаждали до -78°C. Затем туда добавляли раствор 2,4,6-трифторбензол-1-сульфонилхлорида (2,64 г, 11,4 ммоль) в безводном тетрагидрофуране (20 мл) при -78°C. Смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 3 ч. Реакционную смесь разводили насыщенным хлоридом аммония (200 мл) и экстрагировали этилацетатом (3×200 мл). Объединенные органические слои промывали солевым раствором (3×50 мл), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 9 до 20% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (3,00 г, выход 86%):1H ЯМР (400 МГц, CDCl3) δ 8,06 (д, J=2,8 Гц, 1H), 7,31-7,18 (м, 2H), 7,12-7,07 (м, 1H), 6,66 (т, J=8,6 Гц, 2H), 6,32-6,24 (м, 2H), 4,97 (с, 2H), 3,68 (с, 3H), 3,62 (с, 3H).
Стадия 3. Получение 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-N-(2,4-диметоксибензил)-2,6-дифтор-N-(5-фторпиридин-2-ил)бензолсульфонамида

В смесь N-(2,4-диметоксибензил)-2,4,6-трифтор-N-(5-фторпиридин-2-ил)бензолсульфонамида (0,500 г, 1,10 ммоль) и (2-(азетидин-1-илметил)-6-фторфенил)метанамина (0,320 г, 1,65 ммоль) в безводном N,N-диметилформамиде (6 мл) добавляли карбонат калия (0,456 г, 3,30 ммоль) одной частью. Смесь перемешивали при температуре окружающей среды в течение 12 ч. Реакционную смесь разводили водой (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали солевым раствором (3×50 мл), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 9 до 20% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде масла желтого цвета (0,200 г, выход 29%):1H ЯМР (400 МГц, CDCl3) δ 8,07 (д, J=3,0 Гц, 1H), 7,37 (дд, J=8,8, 4,2 Гц, 1H), 7,25 (дт, J=8,4, 3,0 Гц, 1H), 7,18-7,12 (м, 2H), 7,01-6,93 (м, 2H), 6,30-6,24 (м, 2H), 6,10 (д, J=11,8 Гц, 2H), 5,00 (с, 2H), 4,25 (с, 2H), 3,67 (с, 3H), 3,62 (с, 3H), 3,52 (с, 2H), 3,14 (т, J=7,0 Гц, 4H), 2,04 (квин, J=7,0 Гц, 2H), NH не наблюдали; MS (ES+) m/z 631,2 (M+1).
Стадия 4. Получение формиата 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(5-фторпиридин-2-ил)бензолсульфонамида

В раствор 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-N-(2,4-диметоксибензил)-2,6-дифтор-N-(5-фторпиридин-2-ил)бензолсульфонамида (0,100 г, 0,159 ммоль) в дихлорметане (4 мл) добавляли трифторуксусную кислоту (3,08 г, 27,0 ммоль). После концентрирования in vacuo, остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,0623 г, выход 74%):1H ЯМР (400 МГц, CD3OD) δ 8,42 (ушир. с, 1H), 8,08 (д, J=3,0 Гц, 1H), 7,57-7,44 (м, 2H), 7,30-7,15 (м, 3H), 6,37-6,29 (м, 2H), 4,39 (с, 2H), 4,29 (с, 2H), 3,95 (т, J=7,8 Гц, 4H), 2,40 (квин, J=7,8 Гц, 2H), NH и COOH не наблюдали; MS (ES+) m/z 481,2 (M+1).
ПРИМЕР 297
Синтез (S)-4-((1-(2,5-дифторфенил)этил)амино)-5-этил-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение (S)-4-((1-(2,5-дифторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)-5-винилбензолсульфонамида

В смесь (S)-трет-бутил (5-хлор-4-((1-(2,5-дифторфенил)этил)амино)-2-фторфенил)сульфонил(тиазол-4-ил)карбамата (0,800 г, 1,46 ммоль), 4,4,5,5-тетраметил-2-винил-1,3,2-диоксаборолана (2,25 г, 14,60 ммоль, 2,47 мл) и фосфата калия (0,115 г, 0,545 ммоль) в воде (2 мл) и толуоле (8 мл) добавляли ацетат палладия(II) (0,164 г, 0,730 ммоль) и тетрафторборат трициклогексилфосфония (1,60 г, 4,36 ммоль). Реакционную смесь дегазировали и нагревали до 100°C в течение 12 ч. После охлаждения до температуры окружающей среды, смесь разводили водой (20 мл) и экстрагировали этилацетатом (3×20 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали in vacuo. Очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,060 г, выход 9%):1H ЯМР (400 МГц, CDCl3) δ 8,63 (ушир. с, 1H), 8,58 (д, J=2,0 Гц, 1H), 7,66 (д, J=8,0 Гц, 1H), 7,05 (тд, J=9,2, 4,0 Гц, 1H), 6,94 (д, J=2,2 Гц, 2H), 6,84-6,92 (м, 1H), 6,62 (дд, J=17,2, 10,8 Гц, 1H), 6,02 (д, J=12,8 Гц, 1H), 5,66 (д, J=17,6 Гц, 1H), 5,50 (д, J=11,2 Гц, 1H), 4,70-4,77 (м, 1H), 4,66 (д, J=4,8 Гц, 1H), 1,57 (д, J=6,8 Гц, 3H); MS (ES+) m/z 440,0 (M+1).
Стадия 2. Получение (S)-4-((1-(2,5-дифторфенил)этил)амино)-5-этил-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

В раствор (S)-4-((1-(2,5-дифторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)-5-винилбензолсульфонамида (0,030 г, 0,068 ммоль) в метаноле (10 мл) добавляли Pd/C (0,010 г). Смесь перемешивали в атмосфере водорода (45 фунтов/дюйм2) при температуре окружающей среды в течение 12 ч. Смесь затем фильтровали и концентрировали in vacuo. Очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,019 г, выход 63%):1H ЯМР (400 МГц, CDCl3) δ 8,69 (ушир. с, 1H), 8,60 (д, J=2,4 Гц, 1H), 7,50 (д, J=8,0 Гц, 1H), 7,06 (тд, J=9,2, 4,4 Гц, 1H), 6,92-6,99 (м, 2H), 6,89 (ддд, J=8,4, 5,6, 3,2 Гц, 1H), 6,02 (д, J=12,8 Гц, 1H), 4,72 (т, J=6,4 Гц, 1H), 4,46 (д, J=4,8 Гц, 1H), 2,51 (к, J=7,4 Гц, 2H), 1,60 (д, J=6,8 Гц, 3H), 1,30 (т, J=7,6 Гц, 3H).
ПРИМЕР 298
Синтез 5-хлор-2-фтор-4-(((6-фтор-1H-индол-7-ил)метил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 7-бром-6-фтор-1H-индола

В смесь 2-бром-1-фтор-3-нитробензола (2,30 г, 10,5 ммоль) в безводном тетрагидрофуране (30 мл) добавляли бромид винилмагния (1,0 M, 31,4 мл, 31,4 ммоль) при -78°C. Смесь перемешивали при -78°C в течение 30 минут и затем нагревали до температуры окружающей среды и перемешивали в течение 2 ч. Смесь разводили насыщенным хлоридом аммония (50 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические экстракты промывали солевым раствором (3×50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали остаток, который очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 9% этилацетата в петролейном эфире. Дополнительной очисткой с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 9% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде масла желтого цвета (0,250 г, выход 11%):1H ЯМР (400 МГц, DMSO-d6) δ 11,46 (ушир. с, 1H), 7,55 (дд, J=8,6, 5,0 Гц, 1H), 7,40 (т, J=2,8 Гц, 1H), 7,01 (дд, J=9,6, 8,6 Гц, 1H), 6,58 (дд, J=3,2, 1,8 Гц, 1H).
Стадия 2. Получение 6-фтор-1H-индол-7-карбонитрила

В смесь 7-бром-6-фтор-1H-индола (0,400 г, 1,87 ммоль) и цианида цинка (0,658 г, 5,61 ммоль) в безводном N,N-диметилформамиде (10 мл) добавляли тетракис[трифенилфосфин]палладий(0) (0,432 г, 0,374 ммоль). Смесь дегазировали продуванием азотом и затем нагревали до 120°C в течение 2 ч в микроволновом реакторе. После охлаждения до температуры окружающей среды, реакционную смесь разводили водой (5 мл) и экстрагировали этилацетатом (3×10 мл). Объединенные органические слои промывали солевым раствором (3×10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата при пониженном давлении и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 9% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде твердого вещества желтого цвета (0,190 г, выход 63%):1H ЯМР (400 МГц, DMSO-d6) δ 12,17 (ушир. с, 1H), 7,94 (дд, J=8,5, 5,6 Гц, 1H), 7,51 (т, J=2,6 Гц, 1H), 7,12 (т, J=9,6 Гц, 1H), 6,74-6,58 (м, 1H).
Стадия 3. Получение (6-фтор-1H-индол-7-ил)метанамина

В раствор 6-фтор-1H-индол-7-карбонитрила (0,190 г, 1,19 ммоль) в метаноле (20 мл) и гидроксиде аммония (5 мл) добавляли ренеевский никель (0,102 г, 1,19 ммоль). Реакционную смесь перемешивали в атмосфере водорода (50 фунтов/дюйм2) при температуре окружающей среды в течение 12 ч. Фильтрованием и концентрированием фильтрата при пониженном давлении получали указанное в заголовке соединение в виде твердого вещества желтого цвета (0,190 г, выход 97%):1H ЯМР (400 МГц, CDCl3) δ 9,85 (ушир. с, 1H), 7,36 (дд, J=5,0, 8,6 Гц, 1H), 7,15-7,10 (м, 1H), 6,76 (дд, J=8,6, 10,8 Гц, 1H), 6,44-6,38 (м, 1H), 4,27 (ушир. с, 2H), 1,36 (ушир. с, 2H).
Стадия 4. Получение трет-бутил ((5-хлор-2-фтор-4-(((6-фтор-1H-индол-7-ил)метил)амино)фенил)сульфонил)(тиазол-4-ил)карбамата

В смесь (6-фтор-1H-индол-7-ил)метанамина (0,190 г, 1,16 ммоль) и трет-бутил (5-хлор-2,4-дифторфенил)сульфонил(тиазол-4-ил)карбамата (0,477 г, 1,16 ммоль) в безводном N,N-диметилформамиде (5 мл) добавляли карбонат калия (0,481 г, 3,48 ммоль) и смесь перемешивали при температуре окружающей среды в течение 12 ч. Реакционную смесь разводили водой (5 мл) и экстрагировали этилацетатом (3×10 мл). Объединенные органические слои промывали солевым раствором (3×10 мл), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении. Очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 25% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде твердого вещества желтого цвета (0,250 г, выход 39%):1H ЯМР (400 МГц, CDCl3) δ 8,69 (д, J=2,2 Гц, 1H), 8,49 (ушир. д, J=2,2 Гц, 1H), 7,90 (д, J=7,2 Гц, 1H), 7,51 (дд, J=8,6, 5,2 Гц, 1H), 7,42 (д, J=2,2 Гц, 1H), 7,17 (дд, J=3,2, 2,4 Гц, 1H), 6,89 (дд, J=10,6, 8,8 Гц, 1H), 6,55 (д, J=12,2 Гц, 1H), 6,51 (дд, J=3,2, 2,2 Гц, 1H), 4,61 (д, J=4,6 Гц, 2H), 1,30 (с, 9H), NH не наблюдали; MS (ES+) m/z 554,9 (M+1).
Стадия 5. Получение 5-хлор-2-фтор-4-(((6-фтор-1H-индол-7-ил)метил)амино)-N-(тиазол-4-ил)бензолсульфонамида

В смесь трет-бутил(5-хлор-2-фтор-4-(((6-фтор-1H-индол-7-ил)метил)амино)фенил)сульфонил(тиазол-4-ил)карбамата (0,240 г, 0,432 ммоль) в трет-бутиловом спирте (3 мл) добавляли трет-бутоксид калия (0,146 г, 1,30 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Смесь фильтровали и фильтрат концентрировали при пониженном давлении. Очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,05% гидроксид аммония в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,057 г, выход 28%):1H ЯМР (400 МГц, CD3OD) δ 8,62 (д, J=1,8 Гц, 1H), 7,65 (д, J=7,4 Гц, 1H), 7,48 (дд, J=8,6, 5,2 Гц, 1H), 7,27 (д, J=3,2 Гц, 1H), 6,85 (дд, J=10,8, 8,8 Гц, 1H), 6,65 (д, J=12,6 Гц, 1H), 6,47 (д, J=3,2 Гц, 1H), 6,40 (ушир. с, 1H), 4,70 (с, 2H), способные к обмену протоны не наблюдали; MS (ES+) m/z 455,0 (M+1).
ПРИМЕР 299
Синтез (R)-2,6-дифтор-4-((2-фтор-6-(1-гидроксиэтил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил ((4-((трет-бутоксикарбонил)(2-фтор-6-(1-гидроксиэтил)бензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

В раствор трет-бутил ((4-((трет-бутоксикарбонил)(2-фтор-6-формилбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,500 г, 0,776 ммоль) в безводном тетрагидрофуране (8 мл) добавляли 3,0 M раствор бромида метилмагния в простом диэтиловом эфире (0,52 мл, 1,56 ммоль) при 0°C и смесь перемешивали при 0°C в течение 3 ч. Добавляли насыщенный хлорид аммония (2 мл) и получаемый раствор концентрировали in vacuo. Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 9 до 50% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,350 г, выход 70%):1H ЯМР (400 МГц, CDCl3) δ 8,73 (д, J=2,2 Гц, 1H), 7,45 (д, J=2,2 Гц, 1H), 7,31-7,26 (м, 1H), 7,24-7,20 (м, 1H), 6,89-6,77 (м, 3H), 5,21 (ушир. дд, J=6,2, 3,4 Гц, 1H), 5,03 (с, 2H), 1,43 (д, J=6,4 Гц, 3H), 1,37 (с, 9H), 1,28 (с, 9H), OH не наблюдали.
Стадия 2. Получение трет-бутил ((4-((2-ацетил-6-фторбензил)(трет-бутоксикарбонил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

В раствор трет-бутил ((4-((трет-бутоксикарбонил)(2-фтор-6-(1-гидроксиэтил)бензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,340 г, 0,528 ммоль) в безводном дихлорметане (6 мл) добавляли периодинан Десса-Мартина (0,269 г, 0,634 ммоль) и смесь перемешивали при температуре окружающей среды в течение 1 ч. Смесь концентрировали in vacuo. Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 5 до 33% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,260 г, выход 77%):1H ЯМР (400 МГц, CDCl3) δ 8,81 (д, J=2,2 Гц, 1H), 7,52 (д, J=2,2 Гц, 1H), 7,43-7,38 (м, 1H), 7,37-7,31 (м, 1H), 7,15 (ддд, J=10,0, 8,4, 1,2 Гц, 1H), 6,99 (д, J=10,6 Гц, 2H), 5,29 (с, 2H), 2,59 (с, 3H), 1,48 (с, 9H), 1,36 (с, 9H).
Стадия 3. Получение трет-бутил (R)-(4-(N-(трет-бутоксикарбонил)-N-(тиазол-4-ил)сульфамоил)-3,5-дифторфенил)(2-фтор-6-(1-гидроксиэтил)бензил)карбамата

В 1,0 M раствор (S)-1-метил-3,3-дифенил-тетрагидро-пирроло[1.2-c][1.3.2]оксазоборола в тетрагидрофуране (0,0312 мл, 0,031 ммоль) добавляли безводный тетрагидрофуран (2 мл) и 10,0 M раствор борандиметилсульфидного комплекса (0,020 мл, 0,20 ммоль) при -20°C. Смесь перемешивали при -20°C в течение 1 ч. Затем туда добавляли раствор трет-бутил ((4-((2-ацетил-6-фторбензил)(трет-бутоксикарбонил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,100 г, 0,156 ммоль) в безводном тетрагидрофуране (1 мл) при -20°C и реакционную смесь перемешивали при -20°C в течение 3 ч. Насыщенный хлорид аммония (1 мл) добавляли и смесь концентрировали in vacuo. Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 9 до 20% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,050 г, выход 47%):1H ЯМР (400 МГц, CDCl3) δ 8,82 (д, J=2,2 Гц, 1H), 7,54 (д, J=2,2 Гц, 1H), 7,39-7,34 (м, 1H), 7,31 (дд, J=8,0, 5,6 Гц, 1H), 6,99-6,88 (м, 3H), 5,34-5,27 (м, 1H), 5,11 (с, 2H), 1,52 (д, J=6,4 Гц, 3H), 1,46 (с, 9H), 1,37 (с, 9H), OH не наблюдали.
Стадия 4. Получение (R)-2,6-дифтор-4-((2-фтор-6-(1-гидроксиэтил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил (R)-(4-(N-(трет-бутоксикарбонил)-N-(тиазол-4-ил)сульфамоил)-3,5-дифторфенил)(2-фтор-6-(1-гидроксиэтил)бензил)карбамата (0,0400 г, 0,0621 ммоль) в дихлорметане (2 мл) добавляли трифторуксусную кислоту (1,54 г, 13,5 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч и затем концентрировали in vacuo. Очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,014 г, выход 46%):1H ЯМР (400 МГц, CD3OD) δ 8,75 (д, J=2,2 Гц, 1H), 7,47-7,34 (м, 2H), 7,16-7,02 (м, 1H), 6,97 (д, J=2,0 Гц, 1H), 6,28 (д, J=12,2 Гц, 2H), 5,08 (к, J=6,4 Гц, 1H), 4,36 (с, 2H), 1,44 (д, J=6,4 Гц, 3H), способные к обмену протоны не наблюдали; MS (ES+) m/z 444,1 (M+1).
ПРИМЕР 300
Синтез 4-((2-((2,2-диметилазетидин-1-ил)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 2-((2,2-диметилазетидин-1-ил)метил)-6-фторбензонитрила

В раствор 2,2-диметилазетидина (4,29 г, 50,38 ммоль) и N,N-диизопропилэтиламина (13,16 мл, 75,57 ммоль) в безводном N,N-диметилформамиде (100 мл) добавляли 2-(бромметил)-6-фторбензонитрил (9,80 г, 45,80 ммоль) при 0°C. Смесь перемешивали при температуре окружающей среды в течение 2 ч и затем разводили этилацетатом (170 мл). Смесь промывали насыщенным хлоридом аммония (2×100 мл), солевым раствором (100 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo для того, чтобы получать указанное в заголовке соединение в виде масла розового цвета (9,97 г, выход 99%):1H ЯМР (300 МГц, CDCl3) δ 7,52 (тд, J=8,1, 5,7 Гц, 1H), 7,40-7,36 (м, 1H), 7,10-7,04 (м, 1H), 3,73 (с, 2H), 3,18 (т, J=7,0 Гц, 2H), 1,94 (т, J=7,0 Гц, 2H), 1,26 (с, 6H); MS (ES+) m/z 219,3 (M+1).
Стадия 2. Получение (2-((2,2-диметилазетидин-1-ил)метил)-6-фторфенил)-метанамина
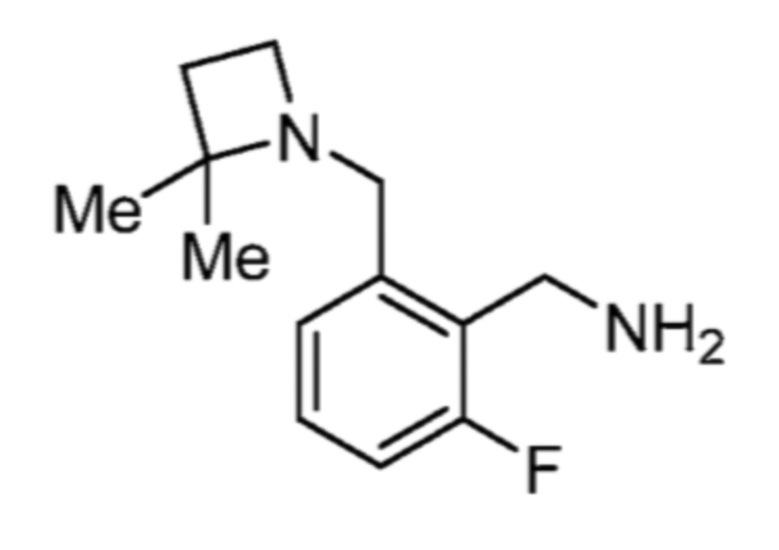
К ренеевскому никелю (50% в воде, 0,70 мл) добавляли этанол (50 мл), концентрированный водный гидроксид аммония (3 мл) и 2-((2,2-диметилазетидин-1-ил)метил)-6-фторбензонитрил (1,02 г, 4,67 ммоль). Смесь перемешивали в атмосфере водорода (1 атм.) в течение 18 ч. Смесь фильтровали через подушку из целита и концентрировали in vacuo для того, чтобы получать указанное в заголовке соединение в виде масла красного цвета (0,96 г, выход 92%):1H ЯМР (300 МГц, CDCl3) δ 7,10-6,95 (м, 3H), 3,97-3,83 (м, 2H), 3,60 (с, 2H), 3,03 (т, J=6,7 Гц, 2H), 1,83 (т, J=6,7 Гц, 2H), 1,25 (с, 6H), NH не наблюдали; MS (ES+) m/z 223,3 (M+1).
Стадия 3. Получение 4-((2-((2,2-диметилазетидин-1-ил)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида
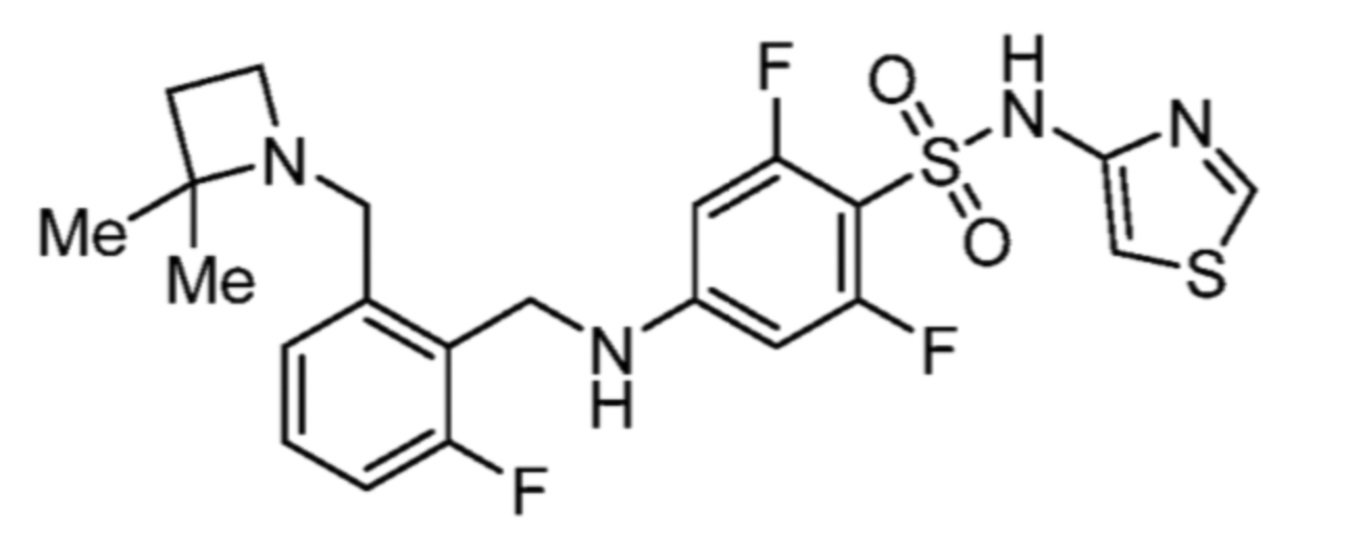
В смесь (2-((2,2-диметилазетидин-1-ил)метил)-6-фторфенил)метанамина (0,22 г, 1,00 ммоль) и карбоната цезия (0,33 г, 1,00 ммоль) в безводном N,N-диметилформамиде (12 мл) добавляли раствор трет-бутил тиазол-4-ил((2,4,6-трифторфенил)сульфонил)карбамата (0,39 г, 1,00 ммоль) в N,N-диметилформамиде (5 мл) при -42°C. Смесь перемешивали при -42°C в течение 3 ч и затем при температуре окружающей среды в течение 15 ч. Смесь разводили этилацетатом (60 мл), насыщенным хлоридлм аммония (2×50 мл), солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Остаток растворяли в дихлорметане (20 мл) и туда добавляли трифторуксусную кислоту (6 мл). Смесь перемешивали при температуре окружающей среды в течение 2 ч и затем концентрировали in vacuo. Остаток разводили 2 M гидроксидом натрия (30 мл) и солевым раствором (30 мл) и смесь экстрагировали этилацетатом (2×50 мл). Объединенную органическую фазу промывали насыщенным хлоридом аммония (50 мл), солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 10% метанола (+0,2% муравьиной кислоты) в дихлорметане для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,045 г, выход 10%):1H ЯМР (300 МГц, DMSO-d6) δ 11,16 (с, 1H), 8,89 (д, J=2,2 Гц, 1H), 7,37-7,27 (м, 2H), 7,22-7,10 (м, 2H), 6,89 (д, J=2,2 Гц, 1H), 6,47-6,39 (м, 2H), 4,41-4,35 (м, 2H), 3,56 (с, 2H), 3,02-2,97 (м, 2H), 1,84-1,74 (м, 2H), 1,13 (с, 6H); MS (ES+) m/z 497,3 (M+1).
ПРИМЕР 301
Синтез формиата 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-5-хлор-2-фтор-N-(изоксазол-3-ил)бензолсульфонамида
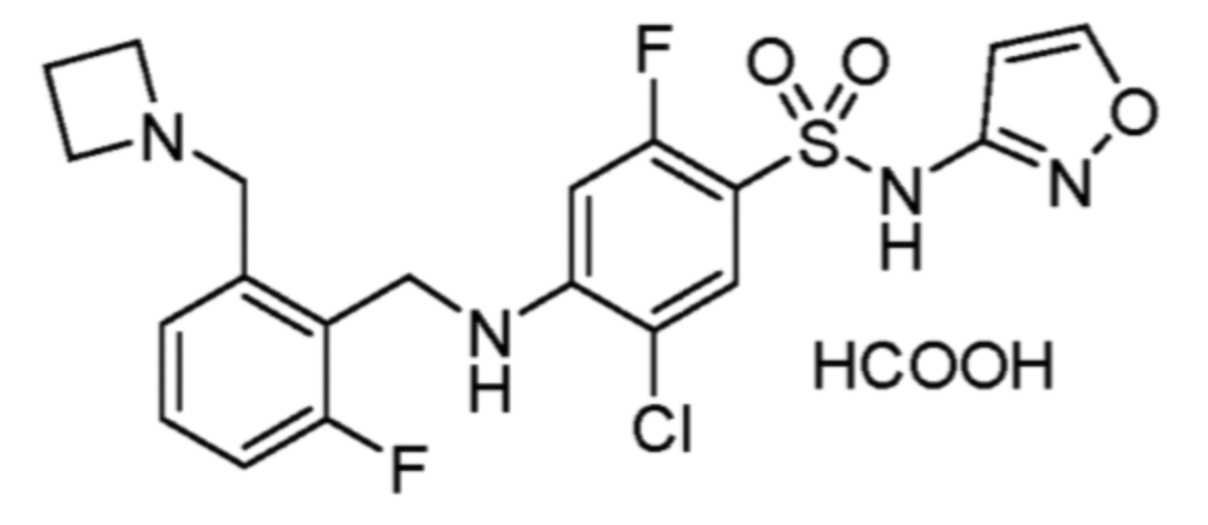
Стадия 1. Получение 5-хлор-2,4-дифтор-N-(изоксазол-3-ил)бензолсульфонамида

В раствор изоксазол-3-амина (2,16 г, 25,69 ммоль), пиридина (4,10 мл, 51,38 ммоль) и 4-(диметиламино)пиридина (0,69 г, 5,65 ммоль) в безводном дихлорметане (50 мл) добавляли раствор 5-хлор-2,4-дифторбензолсульфонилхлорида в безводном ихлорметане (20 мл) при 0°C. Смесь перемешивали при 0°C в течение 2 ч и затем температуре окружающей среды в течение 18 ч. Смесь разводили этилацетатом (120 мл), промывали 1 M соляной кислотой (3×60 мл), солевым раствором (60 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo для того, чтобы получать указанное в заголовке соединение в виде твердого вещества красного цвета (6,80 г, выход 82%):1H ЯМР (300 МГц, DMSO-d6) δ 12,23 (с, 1H), 8,77-8,75 (м, 1H), 8,10 (т, J=7,6 Гц, 1H), 7,87 (т, J=9,6 Гц, 1H), 6,44-6,39 (м, 1H); MS (ES-) m/z 293,2 (M - 1), 295,0 (M - 1).
Стадия 2. Получение 5-хлор-2,4-дифтор-N-(изоксазол-3-ил)-N-(4-метоксибензил)-бензолсульфонамида

В смесь 5-хлор-2,4-дифтор-N-(изоксазол-3-ил)бензолсульфонамида (6,80 г, 23,08 ммоль) и бикарбоната натрия (9,69 г, 115,4 ммоль) в безводном N,N-диметилформамиде (100 мл) добавляли 4-метоксибензилхлорид (6,26 мл, 46,15 ммоль). Смесь нагревали до 35°C в течение 18 ч и затем разводили этилацетатом (120 мл). Смесь промывали водой (2×150 мл), насыщенным хлоридом аммония (100 мл), солевым раствором (60 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo и остаток растирали с метанолом (50 мл) для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (4,76 г, выход 50%):1H ЯМР (300 МГц, CDCl3) δ 8,24 (д, J=1,8 Гц, 1H), 7,83 (т, J=7,3 Гц, 1H), 7,39-7,32 (м, 2H), 7,01 (т, J=8,8 Гц, 1H), 6,83-6,77 (м, 2H), 6,62 (д, J=1,8 Гц, 1H), 5,07 (с, 2H), 3,79 (с, 3H); MS (ES+) m/z 415,2 (M+1), 417,2 (M+1).
Стадия 2. Получение формиата 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-5-хлор-2-фтор-N-(изоксазол-3-ил)бензолсульфонамида

В смесь (2-(азетидин-1-илметил)-6-фторфенил)метанамина (0,31 г, 1,60 ммоль) и карбоната цезия (0,65 г, 2,00 ммоль) в безводном N,N-диметилформамиде (10 мл) добавляли 5-хлор-2,4-дифтор-N-(изоксазол-3-ил)-N-(4-метоксибензил)бензолсульфонамид (0,62 г, 1,50 ммоль) при -42°C. Смесь перемешивали при -42°C в течение 3 ч и затем при температуре окружающей среды в течение 18 ч. Смесь разводили этилацетатом (60 мл). Органическую фазу промывали насыщенным хлоридом аммония (2×40 мл), солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo и остаток растворяли в дихлорметане (40 мл). Туда добавляли трифторуксусную кислоту (10 мл) и смесь нагревали с обратным холодильником в течение 2 ч и затем концентрировали in vacuo. Остаток разводили 2 M водным гидроксидом натрия (30 мл) и солевым растворлм (30 мл). Смесь экстрагировали этилацетатом (2×50 мл) и объединяли органические фракции. Органический слой промывали насыщенным хлоридом аммония (50 мл), солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 10% метанола (содержащего 0,2% муравьиной кислоты) в дихлорметане, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,26 г, выход 34%):1H ЯМР (300 МГц, DMSO-d6) δ 8,61 (д, J=1,8 Гц, 1H), 8,15 (с, 1H), 7,64 (д, J=7,4 Гц, 1H), 7,34 (тд, J=7,9, 5,8 Гц, 1H), 7,22-7,15 (м, 2H), 6,95 (д, J=13,4 Гц, 1H), 6,31 (д, J=1,8 Гц, 1H), 4,47 (с, 2H), 3,86 (с, 2H), 3,37 (т, J=7,2 Гц, 4H), 2,12-2,04 (м, 2H), NH и COOH не наблюдали; MS (ES+) m/z 469,2 (M+1), 471,2 (M+1).
ПРИМЕР 302
Синтез 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано для примера 300, стадии 3 и выполняя некритические вариации при необходимости, чтобы заменять (2-((2,2-диметилазетидин-1-ил)метил)-6-фторфенил)метанамин на (2-(азетидин-1-илметил)-6-фторфенил)метанамин, дополнительной очисткой с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом от 10 до 55% ацетонитрила в воде, содержащей 0,1% трифторуксусной кислоты, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,040 г, выход 3%):1H ЯМР (300 МГц, DMSO-d6) δ 11,24 (с, 1H), 10,29 (с, 1H), 8,91 (д, J=2,1 Гц, 1H), 7,55-7,46 (м, 1H), 7,39-7,31 (м, 2H), 7,29-7,24 (м, 1H), 6,92 (д, J=2,2 Гц, 1H), 6,40-6,32 (м, 2H), 4,49-4,43 (м, 2H), 4,37-4,30 (м, 2H), 4,13-4,00 (м, 4H), 2,41-2,24 (м, 2H); MS (ES+) m/z 469,2 (M+1).
ПРИМЕР 303
Синтез 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-3-хлор-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил ((4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-3-хлор-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

В смесь трет-бутил ((3-хлор-2,4,6-трифторфенил)сульфонил)(тиазол-4-ил)карбамата (1,07 г, 2,50 ммоль) и N,N-диизопропилэтиламина (0,87 мл, 5,00 ммоль) в безводном диметилсульфоксиде (20 мл) добавляли (2-(азетидин-1-илметил)-6-фторфенил)метанамин (0,49 г, 2,50 ммоль). Смесь перемешивали при температуре окружающей среды в течение 3 ч и затем разводили этилацетатом (80 мл). Органическую фазу промывали насыщенным хлоридом аммония (2×50 мл), солевым раствором (40 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 10 до 70% этилацетата (содержащего 20% этанол и 0,2% гидроксид аммония) в гексанах, чтобы получать указанное в заголовке соединение в виде бесцветной пены (0,88 г, выход 58%): MS (ES+) m/z 603,4 (M+1), 605,4 (M+1).
Стадия 2. Получение 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-3-хлор-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

В смесь трет-бутил ((4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-3-хлор-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,28 г, 0,46 ммоль) в дихлорметане (10 мл) добавляли трифторуксусную кислоту (4 мл). Смесь перемешивали при температуре окружающей среды в течение 3 ч и затем концентрировали in vacuo. Очисткой остатка с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом от 10 до 55% ацетонитрила в воде, содержащей 0,1% трифторуксусной кислоты, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,050 г, выход 18%):1H ЯМР (300 МГц, DMSO-d6) δ 11,42 (с, 1H), 10,33 (с, 1H), 8,91 (д, J=2,2 Гц, 1H), 7,49 (тд, J=8,0, 5,9 Гц, 1H), 7,36-7,30 (м, 2H), 7,17-7,14 (м, 1H), 7,00 (д, J=2,2 Гц, 1H), 6,63 (дд, J=13,7, 1,3 Гц, 1H), 4,52 (с, 4H), 4,08-4,04 (м, 4H), 2,41-2,26 (м, 2H); MS (ES+) m/z 503,2 (M+1), 505,2 (M+1).
ПРИМЕР 304
Синтез 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-3-метил-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил ((4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-3-метилфенил)сульфонил)(тиазол-4-ил)карбамата

В смесь трет-бутил ((4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-3-хлор-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,60 г, 1,00 ммоль), метилбороновой кислоты (0,60 г, 10,0 ммоль) и фосфата калия (1,06 г, 5,00 ммоль) в безводном диоксане (12 мл) добавляли ацетат палладия(II) (0,045 г, 0,20 ммоль) и трициклогексилфосфинтетрафторборат (0,15 г, 0,40 ммоль). Смесь дегазировали и нагревали до 90°C в герметизированной трубке в течение 1,5 ч. После охлаждения до температуры окружающей среды, смесь разводили этилацетатом (30 мл) и фильтровали через подушку из целита. Фильтрат концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 10 до 70% этилацетата (содержащего 20% этанола и 0,2% гидроксида аммония) в гексанах, чтобы получать указанное в заголовке соединение в виде бесцветной пены (0,32 г, выход 55%): MS (ES+) m/z 583,4 (M+1).
Стадия 2. Получение 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-3-метил-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано для примера 303, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять трет-бутил ((4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-3-хлор-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамат на трет-бутил ((4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-3-метилфенил)сульфонил)(тиазол-4-ил)карбамат, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,165 г, выход 51%):1H ЯМР (300 МГц, DMSO-d6) δ 11,25 (с, 1H), 10,32 (с, 1H), 8,89 (д, J=2,2 Гц, 1H), 7,52-7,45 (м, 1H), 7,33 (т, J=9,0 Гц, 2H), 6,91 (д, J=2,2 Гц, 1H), 6,57-6,53 (м, 1H), 6,40 (д, J=13,8 Гц, 1H), 4,50 (д, J=0,3 Гц, 2H), 4,41 (д, J=3,7 Гц, 2H), 4,11-4,04 (м, 4H), 2,43-2,26 (м, 2H), 1,92 (д, J=1,8 Гц, 3H); MS (ES+) m/z 483,2 (M+1).
ПРИМЕР 305
Синтез 2,2,2-трифторацетата 4-((2-((2,2-диметилазетидин-1-ил)метил)бензил)амино)-2,6-дифтор-3-метил-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 2-((2,2-диметилазетидин-1-ил)метил)бензонитрила

Следуя процедуре как описано в примере 300, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять 2-(бромметил)-6-фторбензонитрил на 2-(бромметил)бензонитрил, получали указанное в заголовке соединение в виде масла розового цвета (1,29 г, выход 93%):1H ЯМР (300 МГц, CDCl3) δ 7,62-7,49 (м, 3H), 7,33-7,28 (м, 1H), 3,73 (с, 2H), 3,17 (т, J=7,0 Гц, 2H), 1,92 (т, J=7,0 Гц, 2H), 1,25 (с, 6H); MS (ES+) m/z 201,2 (M+1).
Стадия 2. Получение (2-((2,2-диметилазетидин-1-ил)метил)фенил)метанамина

В раствор 2-((2,2-диметилазетидин-1-ил)метил)бензонитрила (1,29 г, 6,44 ммоль) в безводном тетрагидрофуране (50 мл) добавляли 1,0 M раствор гидрида лития алюминия в тетрагидрофуране (19,30 мл, 19,30 ммоль) при 0°C. Смесь перемешивали при 0°C в течение 1 ч и затем при температуре окружающей среды в течение 4 ч. Смесь охлаждали до 0°C и гасили медленным добавлением декагидрата сульфата натрия (19,30 г, 59,90 ммоль). Смесь энергично перемешивали в течение 18 ч и затем фильтровали через подушку из целита. Фильтровальный осадок споласкивали этилацетатом (200 мл). Объединенный фильтрат концентрировали in vacuo для того, чтобы получать указанное в заголовке соединение в виде масла розового цвета (1,30 г, выход 99%):1H ЯМР (300 МГц, CDCl3) δ 7,28-7,17 (м, 4H), 3,85 (с, 2H), 3,58 (с, 2H), 3,04 (т, J=6,9 Гц, 2H), 2,64 (с, 2H), 1,84 (т, J=6,9 Гц, 2H), 1,26 (с, 6H); MS (ES+) m/z 205,3 (M+1).
Стадия 3. Получение трет-бутил ((3-бром-4-((2-((2,2-диметилазетидин-1-ил)метил)бензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

В смесь трет-бутил ((3-бром-2,4,6-трифторфенил)сульфонил)(тиазол-4-ил)карбамата (1,51 г, 3,18 ммоль) и карбоната калия (0,44 г, 3,18 ммоль) в безводном диметилсульфоксиде (10 мл) добавляли раствор (2-((2,2-диметилазетидин-1-ил)метил)фенил)метанамина (0,65 г, 3,18 ммоль) в безводном диметилсульфоксиде (15 мл). Смесь перемешивали при температуре окружающей среды в течение 18 ч и затем разводили этилацетатом (80 мл). Органическую фазу промывали водой (50 мл), насыщенным хлоридом аммония (50 мл), солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 10 до 70% этилацетата (содержащего 20% этанола и 0,2% гидроксида аммония) в гептане, чтобы получать указанное в заголовке соединение в виде бесцветной пены (1,00 г, выход 48%):1H ЯМР (300 МГц, CDCl3) δ 8,81 (д, J=2,3 Гц, 1H), 7,52 (д, J=2,3 Гц, 1H), 7,29-7,25 (м, 4H), 6,91 (с, 1H), 6,59 (д, J=12,9 Гц, 1H), 4,53 (с, 2H), 3,64 (с, 2H), 3,10 (с, 2H), 1,95 (с, 2H), 1,39 (с, 9H), 1,29 (с, 6H); MS (ES+) m/z 657,4 (M+1), 659,4 (M+1).
Стадия 4. Получение 2,2,2-трифторацетата 4-((2-((2,2-диметилазетидин-1-ил)метил)бензил)амино)-2,6-дифтор-3-метил-N-(тиазол-4-ил)бензолсульфонамида

В смесь трет-бутил ((3-бром-4-((2-((2,2-диметилазетидин-1-ил)метил)бензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (1,00 г, 1,52 ммоль), метилбороновой кислоты (0,46 г, 7,60 ммоль) и фосфата калия (1,61 г, 7,60 ммоль) в безводном диоксане (20 мл) добавляли ацетат палладия(II) (0,068 г, 0,30 ммоль) и трициклогексилфосфинтетрафторборат (0,22 г, 0,61 ммоль). Смесь дегазировали и нагревали с обратным холодильником в течение 6 ч. После охлаждения до температуры окружающей среды, смесь разводили этилацетатом (80 мл). Органическую фазу промывали насыщенным хлоридом аммония (2×60 мл), солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 10 до 70% этилацетата (содержащего 20% этанола и 0,2% гидроксида аммония) в гептане. Дополнительной очисткой с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом от 10 до 55% ацетонитрила в воде, содержащей 0,1% трифторуксусной кислоты, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,335 г, выход 36%):1H ЯМР (300 МГц, DMSO-d6) δ 11,22 (с, 1H), 9,81 (с, 1H), 8,89 (д, J=2,2 Гц, 1H), 7,50-7,45 (м, 1H), 7,40-7,26 (м, 3H), 7,02-6,98 (м, 1H), 6,88 (д, J=2,2 Гц, 1H), 6,13 (д, J=13,6 Гц, 1H), 4,54-4,49 (м, 3H), 4,31-4,17 (м, 2H), 3,93-3,87 (м, 1H), 2,42-2,31 (м, 1H), 2,20-2,11 (м, 1H), 2,04 (д, J=1,7 Гц, 3H), 1,68 (с, 3H), 1,55 (с, 3H); MS (ES+) m/z 493,3 (M+1).
ПРИМЕР 306
Синтез 4-((2-((2,2-диметилазетидин-1-ил)метил)-6-фторбензил)амино)-2-фтор-3-метил-N-(тиазол-4-ил)бензолсульфонамида

В смесь (2-((2,2-диметилазетидин-1-ил)метил)-6-фторфенил)метанамина (0,96 г, 4,32 ммоль) и карбоната калия (0,60 г, 4,32 ммоль) в безводном диметилсульфоксиде (25 мл) добавляли трет-бутил ((3-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамат (1,77 г, 4,32 ммоль). Смесь перемешивали при температуре окружающей среды в течение 3 ч и затем разводили этилацетатом (80 мл). Органическую фазу промывали водой (50 мл), насыщенным хлоридом аммония (50 мл), солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo и остаток растворяли в безводном диоксане (45 мл). Смесь дегазировали продуванием аргоном. Затем в эту смесь добавляли метилбороновую кислоту (1,29 г, 21,60 ммоль), фосфат калия (4,59 г, 21,60 ммоль), ацетат палладия (0,19 г, 0,86 ммоль) и трициклогексилфосфинтетрафторборат (0,64 г, 1,73 ммоль). Смесь нагревали с обратным холодильником в течение 6 ч. После охлаждения до температуры окружающей среды, смесь разводили этилацетатом (80 мл). Органическую фазу промывали насыщенным хлоридом аммония (2×60 мл), солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 10 до 70% этилацетата (содержащего 20% этанола и 0,2% гидроксида аммония) в гептане. Дополнительной очисткой с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом от 10 до 55% ацетонитрила в воде, содержащей 0,1% трифторуксусной кислоты, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,30 г, выход 14%):1H ЯМР (300 МГц, DMSO-d6) δ 8,85 (д, J=2,2 Гц, 1H), 7,52 (т, J=8,6 Гц, 1H), 7,27 (тд, J=7,8, 5,8 Гц, 1H), 7,19-7,07 (м, 2H), 6,84 (д, J=2,2 Гц, 1H), 6,73 (д, J=9,0 Гц, 1H), 6,43 (с, 1H), 4,45 (с, 2H), 3,62 (с, 2H), 3,00 (т, J=6,8 Гц, 2H), 1,99 (д, J=1,9 Гц, 3H), 1,79 (т, J=6,9 Гц, 2H), 1,13 (с, 6H), NH не наблюдали; MS (ES+) m/z 493,3 (M+1).
ПРИМЕР 307
Синтез 5-хлор-4-((2,5-дифторбензил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

В смесь трет-бутил ((5-хлор-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,41 г, 1,00 ммоль) и N,N-диизопропилэтиламина (0,35 мл) в безводном N,N-диметилформамиде (5 мл) добавляли (2,5-дифторфенил)-метанамин (0,14 мл), 1,20 ммоль). Смесь перемешивали при температуре окружающей среды в течение 18 ч и затем разводили этилацетатом (100 мл). Органическую фазу промывали 1 M соляной кислотой (2×10 мл), солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 40% этилацетата в гексане. Затем очищенный остаток разводили дихлорметаном (10 мл) и туда добавляли трифторуксусную кислоту (0,5 мл). Смесь перемешивали при температуре окружающей среды в течение 2 ч и затем концентрировали in vacuo. Остаток очищали растиранием с простым диэтиловым эфиром (10 мл) для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,22 г, выход 51%):1H ЯМР (300 МГц, DMSO-d6) δ 11,13 (с, 1H), 8,88 (д, J=2,0 Гц, 1H), 7,62 (д, J=7,4 Гц, 1H), 7,33-6,94 (м, 5H), 6,57 (д, J=13,2 Гц, 1H), 4,49 (д, J=5,9 Гц, 2H); MS (ES+) m/z 434,1 (M+1), 436,1 (M+1).
ПРИМЕР 308
Синтез 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,5-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил тиазол-4-ил((2,4,5-трифторфенил)сульфонил)карбамата

В раствор трет-бутил тиазол-4-илкарбамата (3,46 г, 17,30 ммоль) в безводном тетрагидрофуране (150 мл) добавляли 1,0 M раствор бис(триметилсилил)амида лития в тетрагидрофуране (20,80 мл, 20,80 ммоль) при -78°C. Смесь перемешивали при -78°C в течение 2 ч и затем при температуре окружающей среды в течение 1 ч. Смесь охлаждали до -78°C и туда добавляли раствор 2,4,5-трифторбензолсульфонилхлорида (3,99 г, 17,30 ммоль) в безводном тетрагидрофуране (30 мл). Смесь перемешивали при -78°C в течение 2 ч и затем при температуре окружающей среды в течение 18 ч. Смесь разводили этилацетатом (250 мл), промывали насыщенным хлоридом аммония (2×100 мл), солевым раствором (2×100 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 10 до 70% этилацетата в гексанах, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (4,23 г, выход 62%):1H ЯМР (300 МГц, CDCl3) δ 8,81-8,80 (м, 1H), 8,09-8,01 (м, 1H), 7,54 (дд, J=1,6, 0,6 Гц, 1H), 7,17-7,09 (м, 1H), 1,38 (д, J=0,9 Гц, 9H); MS (ES+) m/z 394,7 (M+1).
Стадия 2. Получение 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,5-дифтор-N-(тиазол-4-ил)бензолсульфонамида

В смесь трет-бутил тиазол-4-ил((2,4,5-трифторфенил)сульфонил)карбамата (0,43 г, 1,09 ммоль) и N,N-диизопропилэтиламина (0,38 мл, 2,20 ммоль) в безводном N,N-диметилформамиде (5 мл) добавляли (2-(азетидин-1-илметил)-6-фторфенил)метанамина (0,23 г, 1,20 ммоль). Смесь перемешивали при температуре окружающей среды в течение 18 ч и затем разводили этилацетатом (100 мл). Органическую фазу промывали водой (2×10 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 80% этилацетата (содержащего 10% изопропанола и 10% триэтиламина) в гексанах. Дополнительной очисткой растиранием с простым диэтиловым эфиром (10 мл) получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,052 г, выход 10%):1H ЯМР (300 МГц, DMSO-d6) δ 11,14-10,93 (м, 1H), 8,88 (с, 1H), 8,02-7,93 (м, 1H), 7,38 (дд, J=11,1, 6,2 Гц, 1H), 7,34-7,25 (м, 1H), 7,21-7,10 (м, 2H), 7,01-6,92 (м, 2H), 4,42 (с, 2H), 3,67 (с, 2H), 3,15 (т, J=7,0 Гц, 4H), 2,06-1,91 (м, 2H); MS (ES+) m/z 469,1 (M+1).
ПРИМЕР 309
Синтез 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-хлор-3-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 1-(5-хлор-2-фторбензил)азетидина

В смесь 5-хлор-2-фторбензальдегида (3,95 г, 24,9 ммоль) и азетидина (1,71 г, 29,9 ммоль) в безводном 1,2-дихлорэтане (50 мл) добавляли триацетоксиборгидрид натрия (10,6 г, 50,0 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч. Смесь разводили этилацетатом (100 мл), промывали насыщенным хлоридом аммония (80 мл) и солевым раствором (50 мл) и затем концентрировали in vacuo. Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 10% метанола в дихлорметане, получали указанное в заголовке соединение в виде бесцветного масла (2,0 г, выход 40%):1H ЯМР (300 МГц, CDCl3) δ 7,38 (дд, J=6,3, 2,7 Гц, 1H), 7,28-7,20 (м, 1H), 6,99 (м, 1H), 3,82 (с, 2H), 3,55 (т, J=7,5 Гц, 4H), 2,27-2,17 (м, 2H); MS (ES+) m/z 200,2 (M+1), 202,0 (M+1).
Стадия 2. Получение (E)-2-(азетидин-1-илметил)-6-хлор-3-фторбензальдегид оксима
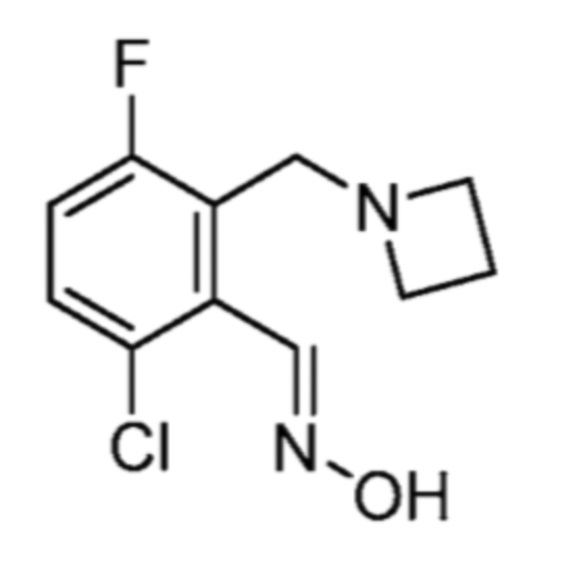
В раствор 1-(5-хлор-2-фторбензил)азетидина (2,53 г, 12,7 ммоль) в безводном тетрагидрофуране (36 мл) добавляли 0,64 M раствор втор-бутиллития в циклогексане (25,7 мл, 16,4 ммоль) при -78°C. Смесь перемешивали при -78°C в течение 80 минут и затем в нее добавляли безводный N,N-диметилформамид (1,77 мл, 22,9 ммоль) при -78°C. Смесь перемешивали при -78°C в течение 50 минут и затем добавляли раствор гидрохлорида гидроксиламина (1,32 г, 19,0 ммоль) в смеси метанола и воды 1:1 (6 мл) при -78°C. Смесь перемешивали при температуре окружающей среды в течение 2 ч и затем концентрировали in vacuo. К остатку добавляли воду (50 мл) и смесь экстрагировали смесью дихлорметана и метанола 10:1 (6×40 мл). Объединенные органические экстракты сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде масла желтого цвета (2,64 г, выход 86%), которое использовали без дополнительной очистки: MS (ES+) m/z 243,2 (M+1).
Стадия 3. Получение (2-(азетидин-1-илметил)-6-хлор-3-фторфенил)метанамина

В раствор (E)-2-(азетидин-1-илметил)-6-хлор-3-фторбензальдегид оксима (2,64 г, 10,87 ммоль) в метаноле (3 мл) добавляли цинковую пыль (2,12 г, 32,4 ммоль). Смесь охлаждали до 0°C, к ней медленно добавляли 6 M соляную кислоту (18 мл, 108 ммоль) и смесь нагревали с обратным холодильником в течение 1 ч. Смесь охлаждали до 0°C и в нее медленно добавляли 3,5 M гидроксид натрия (30 мл, 105 ммоль), после чего следовал концентрированный гидроксид аммония (50 мл) и дихлорметан (10 мл). Смесь перемешивали в течение 16 ч. Слои разделяли и водный слой экстрагировали дихлорметаном (2×25 мл). Объединенные органические слои сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде масла желтого цвета (2,03 г, выход 82%), которое использовали без дополнительной очистки: MS (ES+) m/z 229,2 (M+1).
Стадия 4. Получение трет-бутил ((4-((2-(азетидин-1-илметил)-6-хлор-3-фторбензил)амино)-3-бром-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

В смесь трет-бутил ((3-бром-2,4,6-трифторфенил)сульфонил)-(тиазол-4-ил)карбамата (1,62 г, 3,42 ммоль) и карбоната калия (0,944 г, 6,83 ммоль) в безводном диметилсульфоксиде (17 мл) добавляли раствор (2-(азетидин-1-илметил)-6-хлор-3-фторфенил)метанамина (2,03 г, 8,88 ммоль) в безводном диметилсульфоксиде (2 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 2,5 ч. Смесь охлаждали до 0°C и в нее добавляли воду (40 мл), после чего следовал этилацетат (60 мл). Органический слой промывали солевым раствором (5×20 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 20 до 35% этилацетата (содержащего 10% изопропанола и 10% триэтиламина) в гептане, получали указанное в заголовке соединение в виде пены светло-желтого цвета (0,503 г, выход 22%):1H ЯМР (300 МГц, CDCl3) δ 8,81 (д, J=2,3 Гц, 1H), 7,92-7,86 (м, 1H), 7,52 (д, J=2,3 Гц, 1H), 7,34 (дд, J=8,9, 5,1 Гц, 1H), 7,06-7,00 (м, 1H), 6,63 (дд, J=13,5, 1,7 Гц, 1H), 4,58 (д, J=5,3 Гц, 2H), 3,74 (д, J=2,6 Гц, 2H), 3,33 (т, J=7,1 Гц, 4H), 2,16-2,06 (м, 2H), 1,41 (с, 9H); MS (ES+) m/z 681,1 (M+1), 683,2 (M+1).
Стадия 5. Получение трет-бутил ((4-((2-(азетидин-1-илметил)-6-хлор-3-фторбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

В смесь трет-бутил ((4-((2-(азетидин-1-илметил)-6-хлор-3-фторбензил)амино)-3-бром-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,491 г, 0,720 ммоль), тетракис(трифенилфосфин)палладия(0) (0,083 г, 0,072 ммоль), муравьиной кислоты (0,14 мл, 3,7 ммоль) в безводном 1,4-диоксане (7,2 мл) добавляли триэтиламин (1,0 мл, 7,2 ммоль) и смесь дегазировали продуванием азотом в течение 10 минут. Затем реакционную смесь нагревали с обратным холодильником в течение 4 ч. Смесь оставляли остывать до температуры окружающей среды и разводили этилацетатом (60 мл). Смесь промывали насыщенным бикарбонатом натрия (2×25 мл), солевым раствор (20 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 40 до 80% этилацетата (содержащего 10% изопропанола и 10% триэтиламина) в гептане, получали указанное в заголовке соединение в виде пены желтого цвета (0,146 г, выход 34%):1H ЯМР (300 МГц, CDCl3) δ 8,79 (д, J=2,3 Гц, 1H), 8,18-8,12 (м, 1H), 7,50 (д, J=2,3 Гц, 1H), 7,34 (дд, J=8,9, 5,1 Гц, 1H), 7,05-6,99 (м, 1H), 6,30-6,25 (м, 2H), 4,52-4,51 (м, 2H), 3,68 (д, J=2,7 Гц, 2H), 3,30 (т, J=7,1 Гц, 4H), 2,17-2,07 (м, 2H), 1,40 (с, 9H); MS (ES+) m/z 603,3 (M+1), 605,3 (M+1).
Стадия 6. Получение 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-хлор-3-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил ((4-((2-(азетидин-1-илметил)-6-хлор-3-фторбензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,050 г, 0,083 ммоль) в дихлорметане (3 мл) добавляли трифторуксусную кислоту (1,5 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч и затем концентрировали in vacuo. Растиранием остатка в простом диэтиловом эфире (8 мл) получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,044 г, выход 86%):1H ЯМР (300 МГц, DMSO-d6) δ 11,25 (ушир. с, 1H), 10,09 (ушир. с, 1H), 8,91 (д, J=2,2 Гц, 1H), 7,73 (дд, J=9,0, 5,2 Гц, 1H), 7,47 (м, 1H), 7,14 (м, 1H), 6,93 (д, J=2,2 Гц, 1H), 6,37 (д, J=12,4 Гц, 2H), 4,48 (ушир. с, 2H), 4,39 (д, J=3,9 Гц, 2H), 4,20-4,00 (м, 4H), 2,39-2,17 (м, 2H); MS (ES+) m/z 503,2 (M+1), 505,2 (M+1).
ПРИМЕР 310
Синтез 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)бензил)амино)-2,6-дифтор-3-метил-N-(пиримидин-4-ил)бензолсульфонамида

Стадия 1. Получение 3-хлор-N-(2,4-диметоксибензил)-2,4,6-трифтор-N-(пиримидин-4-ил)бензолсульфонамида

В раствор N-(2,4-диметоксибензил)пиримидин-4-амина (2,67 г, 10,9 ммоль) в безводном тетрагидрофуране (40 мл) добавляли бис(триметилсилил)амид лития (1,0 M в тетрагидрофуране, 13,1 мл, 13,1 ммоль) при -78°C. Смесь нагревали до 0°C, перемешивали в течение 1 ч и затем охлаждали до -78°C. Туда добавляли раствор 3-хлор-2,4,6-трифторбензолсульфонилхлорида (3,46 г, 13,1 ммоль) в безводном тетрагидрофуране (14 мл) при -78°C. Смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 16 ч. Смесь концентрировали in vacuo, и остаток распределялся между этилацетатом (150 мл) и водой (30 мл). Органический слой промывали солевым раствором (30 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 20 до 25% этилацетата (содержащего 10% изопропанола и 10% триэтиламина) в гептане, получали указанное в заголовке соединение в виде пены светло-желтого цвета (0,619 г, выход 12%):1H ЯМР (300 МГц, CDCl3) δ 8,78 (д, J=0,9 Гц, 1H), 8,48 (д, J=5,9 Гц, 1H), 7,25-7,22 (м, 1H), 7,02 (дд, J=5,9, 1,3 Гц, 1H), 6,94-6,87 (м, 1H), 6,46-6,42 (м, 2H), 5,25 (с, 2H), 3,83 (с, 3H), 3,78 (с, 3H); MS (ES+) m/z 474,2 (M+1), 476,2 (M+1).
Стадия 3. Получение 4-((2-(азетидин-1-илметил)бензил)амино)-3-хлор-N-(2,4-диметоксибензил)-2,6-дифтор-N-(пиримидин-4-ил)бензолсульфонамида

В смесь 3-хлор-N-(2,4-диметоксибензил)-2,4,6-трифтор-N-(пиримидин-4-ил)бензолсульфонамида (0,593 г, 1,25 ммоль) и карбоната калия (0,346 г, 2,50 ммоль) в безводном диметилсульфоксиде (4 мл) добавляли раствор (2-(азетидин-1-илметил)фенил)метанамина (0,221 г, 1,25 ммоль) в безводном диметилсульфоксиде (2 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 4 ч. Смесь распределялась между этилацетатом (25 мл) и водой (20 мл), и водную фазу экстрагировали этилацетатом (25 мл). Объединенные органические слои промывали солевым раствором (5×15 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 25 до 30% этилацетата (содержащего 10% изопропанола и 10% триэтиламина) в гептане, получали указанное в заголовке соединение в виде бледной пены (0,532 г, выход 67%):1H ЯМР (300 МГц, CDCl3) δ 8,80 (д, J=1,0 Гц, 1H), 8,44 (д, J=6,0 Гц, 1H), 8,21 (ушир. с, 1H), 7,29-7,21 (м, 6H), 6,44-6,36 (м, 3H), 5,31 (с, 2H), 4,33 (с, 2H), 3,82 (с, 3H), 3,76 (с, 3H), 3,59 (с, 2H), 3,24-3,19 (м, 4H), 2,11-2,02 (м, 2H); MS (ES+) m/z 630,3 (M+1), 632,3 (M+1).
Стадия 4. Получение 4-((2-(азетидин-1-илметил)бензил)амино)-N-(2,4-диметоксибензил)-2,6-дифтор-3-метил-N-(пиримидин-4-ил)бензолсульфонамида
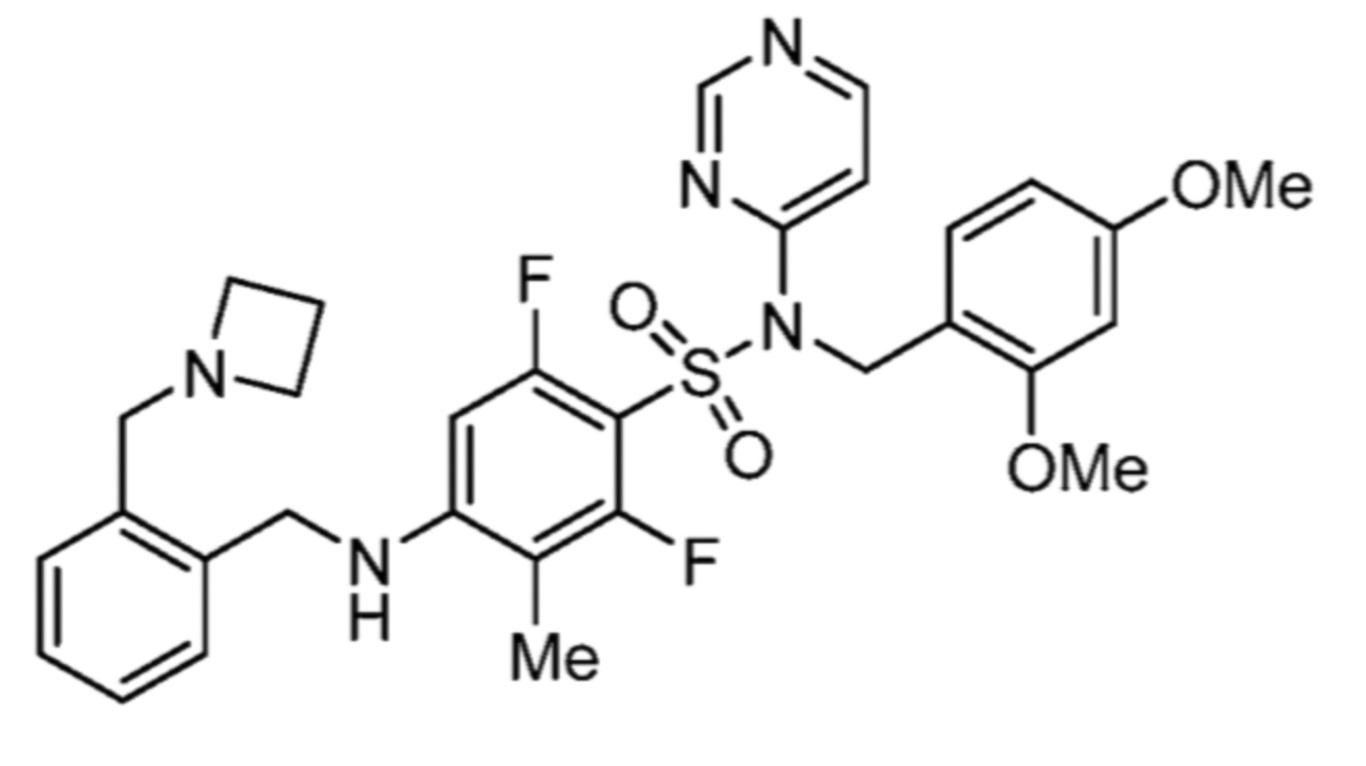
В смесь 4-((2-(азетидин-1-илметил)бензил)амино)-3-хлор-N-(2,4-диметоксибензил)-2,6-дифтор-N-(пиримидин-4-ил)бензолсульфонамида (0,250 г, 0,397 ммоль), метилбороновой кислоты (0,190 г, 3,17 ммоль), трициклогексилфосфинтетрафторбората (0,029 г, 0,079 ммоль) и ацетата палладия(II) (0,0089 г, 0,040 ммоль) в безводном 1,4-диоксане (7,9 мл) добавляли трехосновный фосфат калия (0,253 г, 1,19 ммоль). Смесь дегазировали посредством барботирования азотом в течение 20 минут и затем нагревали до 100°C в течение 2,5 ч. Смесь оставляли остывать до температуры окружающей среды и распределяли между этилацетатом (50 мл) и водой (10 мл). Смесь фильтровали через подушку из целита, и органическую фазу фильтрата промывали солевым раствором (20 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 30 до 40% этилацетата (содержащего 10% изопропанола и 10% триэтиламина) в гептане, получали указанное в заголовке соединение в виде бледной пены (0,194 г, выход 80%):1H ЯМР (300 МГц, CDCl3) δ 8,78 (д, J=1,2 Гц, 1H), 8,42 (д, J=6,0 Гц, 1H), 7,46 (ушир. с, 1H), 7,36 (дд, J=6,0, 1,2 Гц, 1H), 7,31-7,21 (м, 5H), 6,44 (д, J=2,3 Гц, 1H), 6,40 (дд, J=8,3, 2,4 Гц, 1H), 6,30-6,25 (м, 1H), 5,35 (с, 2H), 4,28 (с, 2H), 3,83 (с, 3H), 3,76 (с, 3H), 3,58 (с, 2H), 3,21 (т, J=7,1 Гц, 4H), 2,10-2,00 (м, 5H); MS (ES+) m/z 610,3 (M+1).
Стадия 5. Получение 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)бензил)амино)-2,6-дифтор-3-метил-N-(пиримидин-4-ил)бензолсульфонамида

В раствор 4-((2-(азетидин-1-илметил)бензил)амино)-N-(2,4-диметоксибензил)-2,6-дифтор-3-метил-N-(пиримидин-4-ил)бензолсульфонамида (0,184 г, 0,302 ммоль) в дихлорметане (2,8 мл) добавляли трифторуксусную кислоту (2,8 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 3 ч и затем концентрировали in vacuo. к остатку добавляли метанол (10 мл) и смесь фильтровали через подушку из целита. Фильтрат концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с использованием 15% метанола в дихлорметане, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,116 г, выход 67%):1H ЯМР (300 МГц, DMSO-d6) δ 12,47 (ушир. с, 1H), 10,30 (ушир. с, 1H), 8,61 (с, 1H), 8,42-8,33 (м, 1H), 7,44-7,25 (м, 4H), 6,98-6,97 (м, 2H), 6,16 (д, J=14,0 Гц, 1H), 4,51-4,50 (м, 4H), 4,12 (т, J=8,0 Гц, 4H), 2,44-2,33 (м, 2H), 2,03 (д, J=1,6 Гц, 3H);19F ЯМР (282 МГц, DMSO-d6) δ -73,5 (с, 3F), -111,4 (с, 1F), -112,4 (с, 1F); MS (ES+) m/z 460,3 (M+1).
ПРИМЕР 311
Синтез формиата 4-((2-(азетидин-1-илметил)бензил)амино)-2,6-дифтор-N-(пиримидин-4-ил)бензолсульфонамида

В раствор 4-((2-(азетидин-1-илметил)бензил)амино)-3-хлор-N-(2,4-диметоксибензил)-2,6-дифтор-N-(пиримидин-4-ил)бензолсульфонамида (0,261 г, 0,414 ммоль) в этаноле (8 мл) добавляли 10% палладий на углероде (0,047 г), после чего следовал триэтиламин (0,23 мл, 1,6 ммоль). Смесь нагревали до 60°C в атмосфере водорода в течение 23 ч. После охлаждения до температуры окружающей среды, смесь фильтровали через подушку из целита. Фильтрат концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 20 до 50% этилацетата (содержащего 10% изопропанола и 10% триэтиламина) в гептане, чтобы получать пленку желтого цвета (0,029 г). К этому остатку добавляли дихлорметан (2,4 мл) и трифторуксусную кислоту (2,4 мл) и раствор перемешивали при температуре окружающей среды в течение 2 ч. Концентрированием in vacuo получали остаток. К этому остатку добавляли смесь метанола и дихлорметана 1:1 (10 мл) и смесь фильтровали через подушку из целита. Фильтрат концентрировали in vacuo и остаток очищали с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом от 10 до 40% ацетонитрила в воде, содержащей 0,5% муравьиной кислоты, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,010 г, выход 5% на 2 стадии):1H ЯМР (300 МГц, DMSO-d6) δ 8,38 (с, 1H), 8,15 (с, 1H), 8,06 (д, J=5,7 Гц, 1H), 7,31-7,19 (м, 4H), 6,70 (д, J=5,9 Гц, 1H), 6,26 (д, J=12,3 Гц, 2H), 4,35 (с, 2H), 3,68 (с, 2H), 3,25 (т, J=7,1 Гц, 4H), 2,08-1,99 (м, 2H), NH и COOH не наблюдали;19F ЯМР (282 МГц, DMSO-d6) δ -108,6 (с, 2F); MS (ES+) m/z 446,3 (M+1).
ПРИМЕР 312
Синтез 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)бензил)амино)-2,3-дифтор-6-метил-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил ((4-((2-(азетидин-1-илметил)бензил)амино)-6-бром-2,3-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

В смесь трет-бутил ((6-бром-2,3,4-трифторфенил)сульфонил)(тиазол-4-ил)карбамата (2,11 г, 4,46 ммоль) и карбоната калия (1,23 г, 8,90 ммоль) в диметилсульфоксиде (15 мл) добавляли раствор (2-(азетидин-1-илметил)фенил)метанамина (0,786 г, 4,46 ммоль) в диметилсульфоксиде (7 мл) и смесь перемешивали при температуре окружающей среды в течение 18 ч. Смесь распределялась между этилацетатом (50 мл) и водой со льдом (70 мл). Органический слой промывали солевым раствором (5×25 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 20 до 30% этилацетата (содержащего 10% изопропанола и 10% триэтиламина) в гептане, получали указанное в заголовке соединение в виде бледной пены (1,55 г, выход 55%):1H ЯМР (300 МГц, CDCl3) δ 8,93 (ушир. с, 1H), 8,79 (д, J=2,3 Гц, 1H), 7,54 (м, 1H), 7,35-7,24 (м, 4H), 6,96 (дд, J=7,2, 1,8 Гц, 1H), 4,34 (с, 2H), 3,60 (с, 2H), 3,19 (т, J=7,0 Гц, 4H), 2,15-2,06 (м, 2H), 1,37 (с, 9H); MS (ES+) m/z 629,2 (M+1), 631,2 (M+1).
Стадия 2. Получение трет-бутил ((4-((2-(азетидин-1-илметил)бензил)амино)-2,3-дифтор-6-метилфенил)сульфонил)(тиазол-4-ил)карбамата
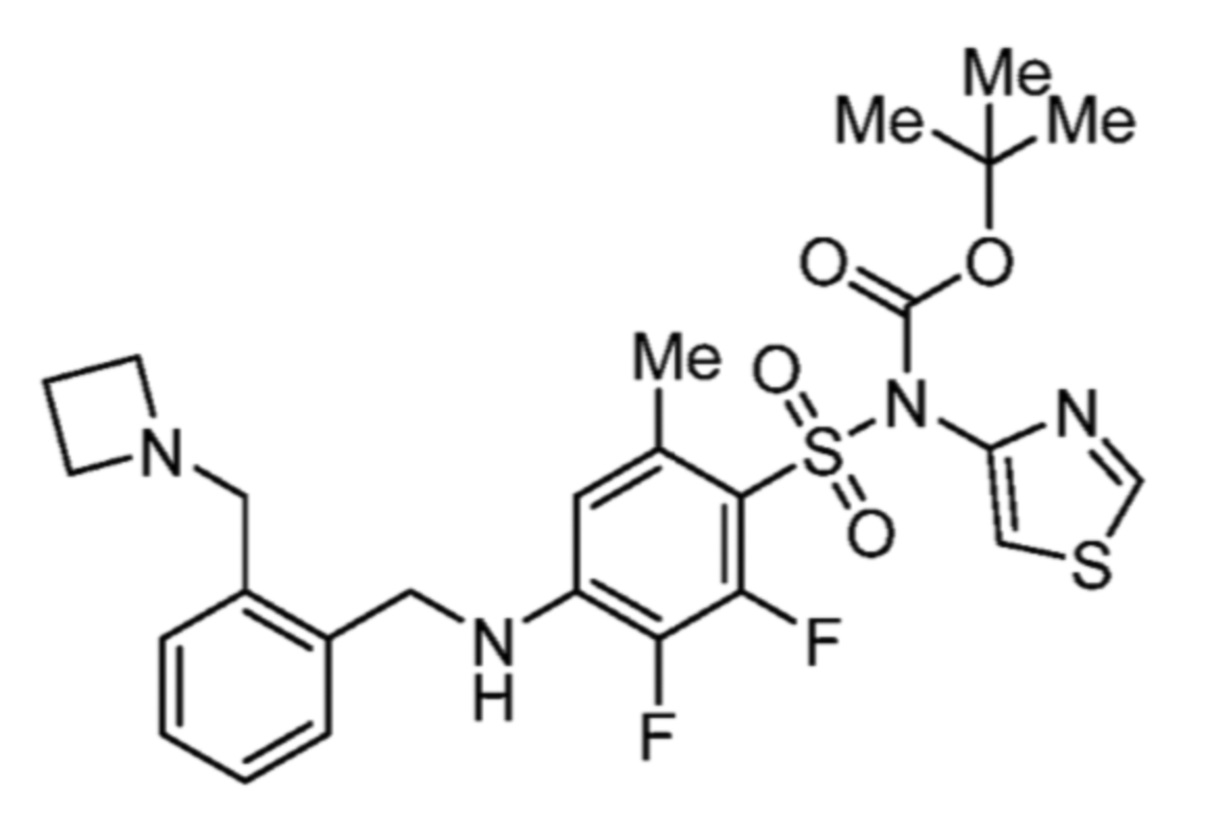
В смесь трет-бутил ((4-((2-(азетидин-1-илметил)бензил)амино)-6-бром-2,3-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,504 г, 0,801 ммоль), метилбороновой кислоты (0,384 г, 6,41 ммоль) и тетракис(трифенилфосфин)палладия(0) (0,093 г, 0,080 ммоль) в безводном 1,4-диоксане (16 мл) добавляли трехосновный фосфат калия (0,510 г, 2,40 ммоль). Смесь дегазировали посредством барботирования азотом в течение 15 минут и затем нагревали до 90°C в течение 3 ч. Смесь оставляли остывать до температуры окружающей среды, и она распределялась между этилацетатом (60 мл) и водой (20 мл). Смесь фильтровали через подушку из целита и органический слой фильтрата промывали солевым раствором (20 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 10 до 30% этилацетата (содержащего 10% изопропанола и 10% триэтиламина) в гептане, получали указанное в заголовке соединение в виде бледного масла (0,312 г, выход 69%):1H ЯМР (300 МГц, CDCl3) δ 8,78 (д, J=2,3 Гц, 1H), 7,50 (дд, J=2,2, 0,8 Гц, 1H), 7,38-7,26 (м, 4H), 6,46-6,43 (м, 1H), 4,38 (с, 2H), 3,62 (с, 2H), 3,27-3,18 (м, 4H), 2,66 (с, 3H), 2,16-2,07 (м, 2H), 1,33 (с, 9H), NH не наблюдали; MS (ES+) m/z 565,4 (M+1).
Стадия 3. Получение 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)бензил)амино)-2,3-дифтор-6-метил-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил ((4-((2-(азетидин-1-илметил)бензил)амино)-2,3-дифтор-6-метилфенил)сульфонил)(тиазол-4-ил)карбамата (0,295 г, 0,522 ммоль) в дихлорметане (4 мл) добавляли трифторуксусную кислоту (2 мл) и раствор перемешивали при температуре окружающей среды в течение 1 ч. Концентрированием in vacuo получали остаток, который растворяли в метаноле (1 мл) и растирали с простым диэтиловым эфиром (80 мл) для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,254 г, выход 84%):1H ЯМР (300 МГц, DMSO-d6) δ 11,08 (ушир. с, 1H), 10,13 (ушир. с, 1H), 8,88 (д, J=2,2 Гц, 1H), 7,43-7,32 (м, 4H), 7,24-7,19 (м, 1H), 6,92 (д, J=2,2 Гц, 1H), 6,35 (д, J=7,8 Гц, 1H), 4,50-4,48 (м, 4H), 4,14-4,04 (м, 4H), 2,44-2,27 (м, 5H);19F ЯМР (282 МГц, DMSO-d6) δ -73,4 (с, 3F), -135,1 (д, J=21,7 Гц, 1F), -161,9 (д, J=21,7 Гц, 1F); MS (ES+) m/z 465,4 (M+1).
ПРИМЕР 313
Синтез 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-хлор-3-фторбензил)амино)-2,6-дифтор-3-метил-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил ((4-((2-(азетидин-1-илметил)-6-хлор-3-фторбензил)амино)-2,6-дифтор-3-метилфенил)сульфонил)(тиазол-4-ил)карбамата

Следуя процедуре как описано в примере 312, стадии 2 и выполняя некритические модификации при необходимости, чтобы заменять трет-бутил ((4-((2-(азетидин-1-илметил)бензил)амино)-6-бром-2,3-дифторфенил)сульфонил)(тиазол-4-ил)карбамат на трет-бутил ((4-((2-(азетидин-1-илметил)-6-хлор-3-фторбензил)амино)-3-бром-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамат, указанное в заголовке соединение получали в виде бледного масла (0,118 г, выход 33%):1H ЯМР (300 МГц, CDCl3) δ 8,80 (д, J=2,3 Гц, 1H), 7,51 (д, J=2,4 Гц, 1H), 7,35 (дд, J=8,9, 5,1 Гц, 1H), 7,05-6,99 (м, 1H), 6,53-6,48 (м, 1H), 4,55 (д, J=5,1 Гц, 2H), 3,72 (д, J=2,8 Гц, 2H), 3,31 (т, J=7,1 Гц, 4H), 2,12-2,03 (м, 5H), 1,39 (с, 9H), NH не наблюдали; MS (ES+) m/z 617,4 (M+1), 619,4 (M+1).
Стадия 2. Получение 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-хлор-3-фторбензил)амино)-2,6-дифтор-3-метил-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил ((4-((2-(азетидин-1-илметил)-6-хлор-3-фторбензил)амино)-2,6-дифтор-3-метилфенил)сульфонил)(тиазол-4-ил)карбамата (0,113 г, 0,183 ммоль) в дихлорметане (4 мл) добавляли трифторуксусную кислоту (2 мл) и раствор перемешивали при температуре окружающей среды в течение 3 ч. Концентрированием in vacuo и растиранием остатка в простом диэтиловом эфире (20 мл) получали указанное в заголовке соединение в виде бледного твердого вещества (0,093 г, выход 80%):1H ЯМР (300 МГц, DMSO-d6) δ 11,27 (с, 1H), 10,01 (ушир. с, 1H), 8,90 (д, J=2,2 Гц, 1H), 7,70 (дд, J=9,1, 5,4 Гц, 1H), 7,48-7,42 (м, 1H), 6,92 (д, J=2,1 Гц, 1H), 6,48 (д, J=13,6 Гц, 1H), 6,31-6,24 (м, 1H), 4,54-4,44 (м, 4H), 4,23-3,97 (м, 4H), 2,41-2,15 (м, 2H), 1,89 (д, J=1,1 Гц, 3H); MS (ES+) m/z 517,2 (M+1), 519,2 (M+1).
ПРИМЕР 314
Синтез 2,2,2-трифторацетата 4-((2-((3,3-диметилазетидин-1-ил)метил)-6-фторбензил)амино)-2,3-дифтор-6-метил-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 2-((3,3-диметилазетидин-1-ил)метил)-6-фторбензонитрила
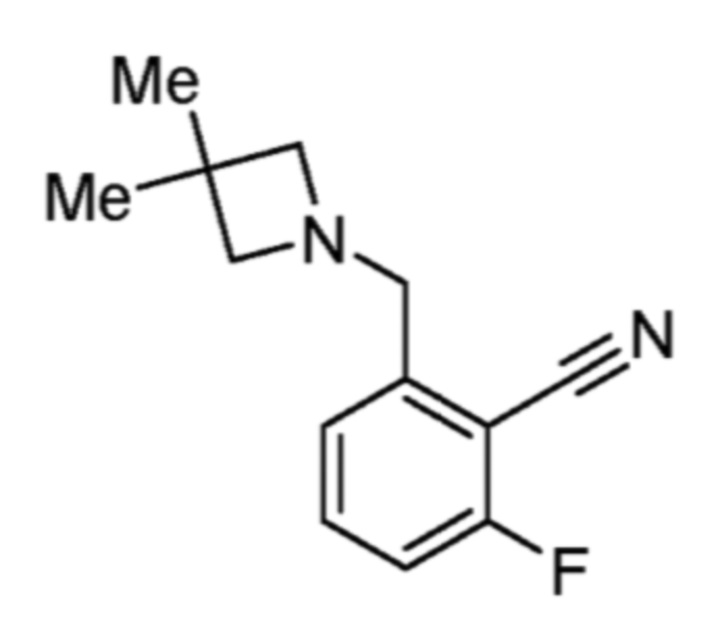
Смесь 2-циано-3-фторбензилбромида (0,500 г, 2,34 ммоль), гидрохлорида 3,3-диметилазетидина (0,305 г, 2,51 ммоль) и N,N-диизопропилэтиламина (0,52 мл, 3,0 ммоль) в дихлорметане (16 мл) перемешивали при температуре окружающей среды в течение 4 ч. Затем туда добавляли еще N,N-диизопропилэтиламин (0,52 мл, 2,99 ммоль) и реакционную смесь перемешивали при температур окружающей среды в течение 16 ч. Реакционную смесь промывали насыщенным хлоридом аммония (10 мл). Водный слой экстрагировали дихлорметаном (2×10 мл) и объединенные органические слои промывали солевым раствором (10 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 30% этилацетата (содержащего 10% изопропанола и 10% триэтиламина) в гексанах, получали указанное в заголовке соединение в виде масла оранжевого цвета (0,390 г, выход 76%): MS (ES+) m/z 219,4 (M+1).
Стадия 2. Получение (2-((3,3-диметилазетидин-1-ил)метил)-6-фторфенил)-метанамина

В смесь 2-((3,3-диметилазетидин-1-ил)метил)-6-фторбензонитрила (0,390 г, 1,79 ммоль) в безводном тетрагидрофуране (12 мл) добавляли 1,0 M раствор гидрида лития алюминия в тетрагидрофуране (2,7 мл, 2,7 ммоль) при 0°C и смесь оставляли нагреваться до температуры окружающей среды в течение 2 ч. Затем туда добавляли декагидрат сульфата натрия (4,0 г, 12 ммоль) и смесь перемешивали при температуре окружающей среды в течение 16 ч. Смесь фильтровали и фильтрат сушили над безводным сульфатом магния. Фильтрованием и концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде масла желтого цвета (0,378 г, выход 95%), которое использовали без дополнительной очистки:1H ЯМР (300 МГц, DMSO-d6) δ 7,19 (тд, J=7,8, 5,9 Гц, 1H), 7,09-7,01 (м, 2H), 3,69 (м, 2H), 3,63 (с, 2H), 2,86 (с, 4H), 1,87 (ушир. с, 2H), 1,15 (с, 6H); MS (ES+) m/z 223,3 (M+1).
Стадия 3. Получение трет-бутил ((6-бром-4-((2-((3,3-диметилазетидин-1-ил)метил)-6-фторбензил)амино)-2,3-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

В смесь трет-бутил ((6-бром-2,3,4-трифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,554 г, 1,17 ммоль) и карбоната калия (0,323 г, 2,34 ммоль) в безводном диметилсульфоксиде (4 мл) добавляли раствор (2-((3,3-диметилазетидин-1-ил)метил)-6-фторфенил)метанамина (0,260 г, 1,17 ммоль) в безводном диметилсульфоксиде (2 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 2,5 ч. Смесь распределялась между этилацетатом (50 мл) и водой со льдом (50 мл). Органический слой промывали солевым раствором (5×15 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 10 до 30% этилацетата (содержащего 10% изопропанола и 10% триэтиламина) в гептане, получали указанное в заголовке соединение в виде пены желтого цвета (0,371 г, выход 47%):1H ЯМР (300 МГц, CDCl3) δ 8,79 (д, J=2,3 Гц, 1H), 7,67 (ушир. с, 1H), 7,54 (д, J=2,3 Гц, 1H), 7,28-7,21 (м, 1H), 7,11-7,03 (м, 3H), 4,45 (с, 2H), 3,65 (с, 2H), 3,02 (с, 4H), 1,38 (с, 9H), 1,24 (с, 6H); MS (ES+) m/z 675,3 (M+1), 677,3 (M+1).
Стадия 4. Получение трет-бутил ((4-((2-((3,3-диметилазетидин-1-ил)метил)-6-фторбензил)амино)-2,3-дифтор-6-метилфенил)сульфонил)(тиазол-4-ил)карбамата

Следуя процедуре как описано в примере 312, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять трет-бутил ((4-((2-(азетидин-1-илметил)бензил)амино)-6-бром-2,3-дифторфенил)сульфонил)(тиазол-4-ил)карбамат на трет-бутил ((6-бром-4-((2-((3,3-диметилазетидин-1-ил)метил)-6-фторбензил)амино)-2,3-дифторфенил)сульфонил)(тиазол-4-ил)карбамат, указанное в заголовке соединение получали в виде пены желтого цвета (0,209 г, выход 64%):1H ЯМР (300 МГц, CDCl3) δ 8,79 (д, J=2,3 Гц, 1H), 7,50 (дд, J=2,3, 0,9 Гц, 1H), 7,27-7,20 (м, 1H), 7,07-7,01 (м, 2H), 6,59-6,56 (м, 1H), 4,46 (с, 2H), 3,65 (с, 2H), 3,02 (с, 4H), 2,68 (с, 3H), 1,33 (с, 9H), 1,24 (с, 6H), NH не наблюдали; MS (ES+) m/z 611,4 (M+1).
Стадия 5. Получение 2,2,2-трифторацетата 4-((2-((3,3-диметилазетидин-1-ил)метил)-6-фторбензил)амино)-2,3-дифтор-6-метил-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил ((4-((2-((3,3-диметилазетидин-1-ил)метил)-6-фторбензил)амино)-2,3-дифтор-6-метилфенил)сульфонил)(тиазол-4-ил)карбамата (0,193 г, 0,316 ммоль) в дихлорметане (4 мл) добавляли трифторуксусную кислоту (2 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч и затем концентрировали in vacuo. Очисткой остатка с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом от 15 до 80% ацетонитрила в воде, содержащей 0,1% трифторуксусной кислоты, получали указанное в заголовке соединение в виде бледного твердого вещества (0,038 г, выход 19%):1H ЯМР (300 МГц, DMSO-d6) δ 11,12 (с, 1H), 10,25-10,15 (м, 1H), 8,88 (д, J=2,2 Гц, 1H), 7,51-7,44 (м, 1H), 7,35-7,29 (м, 2H), 6,95-6,92 (м, 2H), 6,57 (д, J=8,1 Гц, 1H), 4,53-4,42 (м, 4H), 3,96-3,79 (м, 4H), 2,48 (с, 3H), 1,30 (с, 3H), 1,23 (м, 3H);19F ЯМР (282 МГц, DMSO-d6) δ -74,0 (с, 3F), -115,4 (с, 1F), -135,2 (д, J=21,7 Гц, 1F), -161,2 (д, J=21,7 Гц, 1F); MS (ES+) m/z 511,4 (M+1).
ПРИМЕР 315
Синтез 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2-фтор-N-(изоксазол-3-ил)-3-метилбензолсульфонамида

Стадия 1. Получение трет-бутил (2-(азетидин-1-илметил)-6-фторбензил)(2-бром-4-(N-(трет-бутоксикарбонил)-N-(изоксазол-3-ил)сульфамоил)-3-фторфенил)карбамата

В смесь трет-бутил (2-(азетидин-1-илметил)-6-фторбензил)карбамата (0,336 г, 1,14 ммоль) в безводном N,N-диметилформамиде (5,5 мл) добавляли 60% дисперсию гидрида натрия в минеральном масле (0,069 г, 1,7 ммоль) при 0°C. Смесь перемешивали при 0°C в течение 30 минут и затем добавляли по каплям в раствор трет-бутил ((3-бром-2,4-дифторфенил)сульфонил)(изоксазол-3-ил)карбамата (0,502 г, 1,14 ммоль) в безводном N,N-диметилформамиде (11 мл) при 0°C. Смесь перемешивали при 0°C в течение 4 ч и затем разводили этилацетатом (100 мл). Органическую фазу промывали смесью воды и солевого раствора 5:1 (5×50 мл), солевым раствором (15 мл) и сушили над безводным сульфатом магния. Фильтрованием и концентрированием фильтрата in vacuo получали остаток. Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 0 до 30% этилацетата (содержащего 10% изопропанола и 10% триэтиламина) в гептане, получали указанное в заголовке соединение в виде бледной пены (0,113 г, выход 14%): MS (ES) m/z 713,4 (M+1), 715,4 (M+1).
Стадия 2. Получение 2,2,2-трифторацетата 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2-фтор-N-(изоксазол-3-ил)-3-метилбензолсульфонамида

В смесь трет-бутил (2-(азетидин-1-илметил)-6-фторбензил)(2-бром-4-(N-(трет-бутоксикарбонил)-N-(изоксазол-3-ил)сульфамоил)-3-фторфенил)карбамата (0,199 г, 0,279 ммоль), метилбороновой кислоты (0,100 г, 1,67 ммоль), тетракис(трифенилфосфин)палладия(0) (0,032 г, 0,028 ммоль) в безводном 1,4-диоксане (6 мл) добавляли трехосновный фосфат калия (0,178 г, 0,839 ммоль). Смесь дегазировали и затем нагревали до 100°C в течение 21 ч. Смесь оставляли остывать до температуры окружающей среды, и она распределялась между этилацетатом (30 мл) и водой (10 мл). Смесь фильтровали через подушку из целита, и органическую фазу фильтрата промывали солевым раствором (2×10 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo получали пену коричневого цвета (0,224 г). К раствору этого остатка в дихлорметане (4 мл) добавляли трифторуксусную кислоту (4 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 3 ч и затем концентрировали in vacuo. Очисткой с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом от 15 до 50% ацетонитрила в воде, содержащей 0,1% трифторуксусной кислоты, получали указанное в заголовке соединение в виде твердого вещества желтого цвета (0,042 г, выход 27% на 2 стадии):1H ЯМР (300 МГц, DMSO-d6) δ 11,51 (с, 1H), 10,25 (ушир. с, 1H), 8,67 (д, J=1,8 Гц, 1H), 7,59-7,44 (м, 2H), 7,35-7,29 (м, 2H), 6,57 (д, J=8,8 Гц, 1H), 6,36-6,32 (м, 2H), 4,54-4,45 (м, 4H), 4,14-4,03 (м, 4H), 2,45-2,25 (м, 2H), 1,98 (с, 3H);19F ЯМР (282 МГц, DMSO-d6) δ -74,04 (с, 3F), -113,94 (с, 1F), -115,28 (с, 1F); MS (ES+) m/z 449,0 (M+1).
ПРИМЕР 316
Синтез (S)-5-хлор-2-фтор-4-((1-(2-фторфенил)пропил)амино)-N-(6-фторпиридин-2-ил)бензолсульфонамида
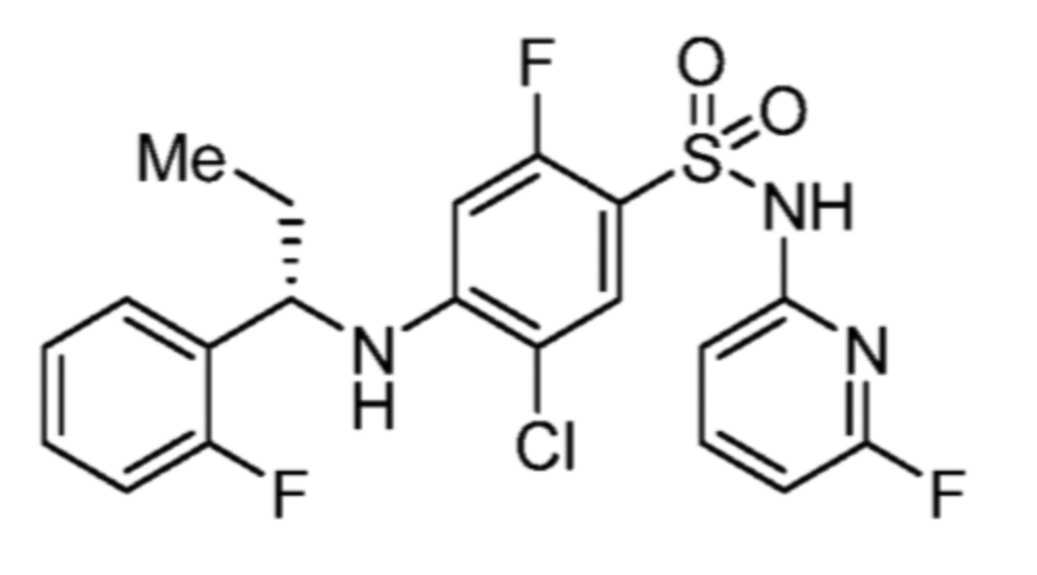
Стадия 1. Получение (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(2-фторфенил)пропил)амино)-N-(6-фторпиридин-2-ил)бензолсульфонамида

Смесь 5-хлор-N-(2,4-диметоксибензил)-2,4-дифтор-N-(6-фторпиридин-2-ил)бензолсульфонамида (0,500 г, 1,06 ммоль), гидрохлорида (S)-1-(2-фторфенил)пропан-1-амина (0,200 г, 1,06 ммоль) и карбоната калия (0,497 г, 3,60 ммоль) в безводном диметилсульфоксиде (5 мл) перемешивали при 75°C в течение 18 часов. Смесь разводили этилацетатом (10 мл) и водой (10 мл). Слои разделяли и водную фазу экстрагировали этилацетатом (3×10 мл). Объединенные органические фазы промывали солевым раствором (5 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 5 до 70% этилацетата в гексанах, получали указанное в заголовке соединение в виде бесцветного масла (0,519 г, выход 81%): MS (ES+) m/z 606,0 (M+1), 608,0 (M+1).
Стадия 2. Получение (S)-5-хлор-2-фтор-4-((1-(2-фторфенил)пропил)амино)-N-(6-фторпиридин-2-ил)бензолсульфонамида

Следуя процедуре как описано для примера 222, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять трет-бутил (S)-((5-хлор-4-((1-(2-хлорфенил)пропил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамат на (S)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-4-((1-(2-фторфенил)пропил)амино)-N-(6-фторпиридин-2-ил)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,135 г, выход 35%):1H-ЯМР (300 МГц, DMSO-d6) δ 11,50 (с, 1H), 7,82 (к, J=8,2 Гц, 1H), 7,75 (д, J=7,5 Гц, 1H), 7,46 (тд, J=7,8, 1,7 Гц, 1H), 7,32-7,25 (м, 1H), 7,21-7,18 (м, 1H), 7,16-7,13 (м, 1H), 6,84 (дд, J=7,9, 2,1 Гц, 1H), 6,70 (дд, J=8,0, 2,4 Гц, 2H), 6,37 (д, J=13,4 Гц, 1H), 4,72-4,64 (м, 1H), 2,09-2,00 (м, 1H), 1,87-1,78 (м, 1H), 0,89 (т, J=7,3 Гц, 3H); MS (ES+) m/z 455,9 (M+1), 457,9 (M+1).
ПРИМЕР 317
Синтез формиата 2,6-дифтор-4-((2-фтор-6-(2-метилпиридин-4-ил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил ((4-((трет-бутоксикарбонил)(2-фтор-6-(2-метилпиридин-4-ил)бензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

Смесь трет-бутил ((4-((2-бром-6-фторбензил)(трет-бутоксикарбонил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,20 г, 0,29 ммоль), (2-метил-4-пиридил)бороновой кислоты (0,048 г, 0,35 ммоль), карбоната натрия (0,062 г, 0,59 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) (0,043 г, 0,058 ммоль) в безводном 1,4-диоксане (5 мл) и воде (1,2 мл) дегазировали и затем нагревали до 90°C в течение 2 ч. Добавляли воду (10 мл), и получаемую смесь экстрагировали этилацетатом (3×20 мл). Объединенные органические экстракты промывали солевым раствором (3×20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 33% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде твердого вещества желтого цвета (0,10 г, выход 49%): MS (ES+) m/z 691,4 (M+1).
Стадия 2. Получение формиата 2,6-дифтор-4-((2-фтор-6-(2-метилпиридин-4-ил)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил ((4-((трет-бутоксикарбонил)(2-фтор-6-(2-метилпиридин-4-ил)бензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата (0,10 г, 0,15 ммоль) в дихлорметане (2 мл) добавляли трифторуксусную кислоту (1,50 г, 13,50 ммоль) и смесь перемешивали при температуре окружающей среды в течение 12 ч. Реакционную смесь концентрировали in vacuo и остаток очищали с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом ацетонитрила в воде, содержащей 0,225% муравьиную кислоту, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,057 г, выход 72%):1H ЯМР (400 МГц, CD3OD) δ 8,75 (д, J=2,2 Гц, 1H), 8,42 (д, J=5,2 Гц, 1H), 8,25 (ушир. с, 1H), 7,48 (дт, J=8,0, 5,6 Гц, 1H), 7,31-7,24 (м, 2H), 7,24-7,21 (м, 1H), 7,17 (д, J=7,6 Гц, 1H), 6,96 (д, J=2,2 Гц, 1H), 6,07-6,00 (м, 2H), 4,20 (с, 2H), 2,47 (с, 3H), NH не наблюдали; MS (ES+) m/z 491,3 (M+1).
ПРИМЕР 318
Синтез 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(изотиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 2,4,6-трифтор-N-(изотиазол-4-ил)бензолсульфонамида

В раствор изотиазол-4-амина (0,40 г, 4,00 ммоль) в дихлорметане (2 мл) добавляли пиридин (0,63 г, 8,00 ммоль) и 4,4-диметиламинопиридин (0,049 г, 0,40 ммоль). Затем туда добавляли раствор 2,4,6-трифторбензол-1-сульфонилхлорида (1,11 г, 4,80 ммоль) в дихлорметане (1 мл) и смесь перемешивали при температуре окружающей среды в течение 12 ч. Смесь концентрировали in vacuo и остаток очищали с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом ацетонитрила в воде (содержащей 0,225% муравьиную кислоту), чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,70 г, выход 59%):1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 8,32 (с, 1H), 7,12-7,01 (м, 2H), NH не наблюдали; MS (ES+) m/z 295,1 (M+1).
Стадия 2. Получение 2,4,6-трифтор-N-(изотиазол-4-ил)-N-((2-(триметилсилил)этокси)метил)-бензолсульфонамида

В смесь 2,4,6-трифтор-N-(изотиазол-4-ил)бензолсульфонамида (0,60 г, 2,00 ммоль) в безводном N,N-диметилформамиде (3 мл) добавляли карбонат калия (0,84 г, 6,10 ммоль) и смесь перемешивали при температуре окружающей среды в течение 30 минут. Затем смесь охлаждали до 0°C и туда добавляли 2-(триметилсилил)этоксиметилхлорид (0,68 г, 4,08 ммоль). Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 12 ч. Реакционную смесь разводили насыщенным хлоридом аммония (20 мл) и экстрагировали этилацетатом (3×20 мл). Объединенные органические экстракты промывали солевым раствором (3×20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 10 до 50% этилацетата/петролейного эфира, чтобы получать указанное в заголовке соединение в виде масла желтого цвета (0,60 г, выход 69%):1H ЯМР (400 МГц, CDCl3) δ 8,59 (с, 1H), 8,46 (с, 1H), 6,80-6,71 (м, 2H), 5,15 (с, 2H), 3,70-3,61 (м, 2H), 0,98-0,86 (м, 2H), 0,01 (с, 9H).
Стадия 3. Получение 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(изотиазол-4-ил)-N-((2-(триметилсилил)этокси)метил)бензолсульфонамида

В раствор 2,4,6-трифтор-N-(изотиазол-4-ил)-N-((2-триметилсилил)этокси)метил)бензолсульфонамида (0,30 г, 0,71 ммоль) в безводном N,N-диметилформамиде (2 мл) добавляли (2-(азетидин-1-илметил)-6-фторфенил)метанамин (0,14 г, 0,71 ммоль) и карбонат калия (0,20 г, 1,41 ммоль) и смесь нагревали до 40°C в течение 5 ч. После охлаждения до температуры окружающей среды, реакционную смесь разводили насыщенным хлоридом аммония (20 мл) и экстрагировали этилацетатом (3×20 мл). Объединенные органические экстракты промывали солевым раствором (3×20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 10 до 50% петролейного эфира в этилацетате, получали указанное в заголовке соединение в виде масла желтого цвета (0,20 г, выход 47%): MS (ES+) m/z 599,4 (M+1).
Стадия 4. Получение 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(изотиазол-4-ил)бензолсульфонамида

К 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(изотиазол-4-ил)-N-((2-(триметилсилил)этокси)метил)бензолсульфонамиду (0,20 г, 0,33 ммоль) добавляли 4 M раствор хлороводорода в 1,4-диоксане (2,00 мл, 8,00 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 30 минут. Смесь корректировали до pH 7 триэтиламином и затем смесь концентрировали in vacuo. Остаток очищали с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом ацетонитрила в воде, содержащей 0,225% муравьиную кислоту, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,044 г, выход 27%):1H ЯМР (400 МГц, CD3OD) δ 8,40 (с, 1H), 8,32 (с, 1H), 7,28-7,24 (м, 1H), 7,12-7,04 (м, 2H), 6,21 (д, J=12,2 Гц, 2H), 4,35 (с, 2H), 3,68 (с, 2H), 3,32 (т, J=7,2 Гц, 4H), 2,18 (квин, J=7,1 Гц, 3H), NH не наблюдали; MS (ES+) m/z 469,3 (M+1)
ПРИМЕР 319
Синтез 4-((2-(2-(3,3-дифторазетидин-1-ил)этил)бензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 2-(2-(3,3-дифторазетидин-1-ил)этил)бензонитрила

Следуя процедуре как описано для примера 261, стадии 3 и выполняя некритические вариации при необходимости, чтобы заменять гидрохлорид азетидина на 3,3-дифторазетидин, указанное в заголовке соединение получали в виде масла желтого цвета (0,25 г, выход 36%):1H ЯМР (400 МГц, CDCl3) δ 7,65 (д, J=7,6 Гц, 1H), 7,58-7,52 (м, 1H), 7,39-7,32 (м, 2H), 3,63 (т, J=12,0 Гц, 4H), 2,97-2,85 (м, 4H); MS (ES+) m/z 223,0 (M+1).
Стадия 2. Получение (2-(2-(3,3-дифторазетидин-1-ил)этил)фенил)метанамина

Следуя процедуре как описано для примера 261, стадии 4 и выполняя некритические вариации при необходимости, чтобы заменять 2-(2-(азетидин-1-ил)этил)бензонитрил на 2-(2-(3,3-дифторазетидин-1-ил)этил)бензонитрил, указанное в заголовке соединение получали в виде масла желтого цвета (0,21 г, выход 83%):1H ЯМР (400 МГц, CDCl3) δ 7,29-7,23 (м, 1H), 7,17-7,06 (м, 3H), 3,82 (с, 2H), 3,49 (т, J=12,0 Гц, 4H), 2,78-2,66 (м, 4H), NH не наблюдали.
Стадия 3. Получение трет-бутил ((4-((2-(2-(3,3-дифторазетидин-1-ил)этил)бензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

Следуя процедуре как описано для примера 261, стадии 5 и выполняя некритические вариации при необходимости, чтобы заменять (2-(2-(азетидин-1-ил)этил)фенил)метанамин на (2-(2-(3,3-дифторазетидин-1-ил)этил)фенил)метанамин, указанное в заголовке соединение получали в виде бесцветного масла (0,23 г, выход 43%):1H ЯМР (400 МГц, CDCl3) δ 8,80 (д, J=2,4 Гц, 1H), 7,51 (д, J=2,4 Гц, 1H), 7,38-7,29 (м, 2H), 7,28-7,23 (м, 2H), 6,53 (ушир. с, 1H), 6,24-6,17 (м, 2H), 4,29 (с, 2H), 3,42 (т, J=12,0 Гц, 4H), 3,01-2,92 (м, 2H), 2,87-2,80 (м, 2H), 1,42 (с, 9H); MS (ES+) m/z 601,3 (M+1).
Стадия 4. Получение 4-((2-(2-(3,3-дифторазетидин-1-ил)этил)бензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано для примера 261, стадии 6 и выполняя некритические вариации при необходимости, чтобы заменять трет-бутил(4-((2-(2-(азетидин-1-ил)этил)бензил)амино)-5-хлор-2-фторфенил)сульфонил(тиазол-4-ил)карбамат на трет-бутил ((4-((2-(2-(3,3-дифторазетидин-1-ил)этил)бензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамат, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,0235 г, выход 14%):1H ЯМР (400 МГц, CDCl3) δ 8,71 (д, J=2,0 Гц, 1H), 7,35-7,30 (м, 1H), 7,28-7,21 (м, 3H), 7,05 (д, J=2,0 Гц, 1H), 6,39-6,29 (м, 1H), 6,11 (д, J=11,6 Гц, 2H), 4,21 (д, J=4,4 Гц, 2H), 3,37 (т, J=12,0 Гц, 4H), 2,97-2,89 (м, 2H), 2,83-2,75 (м, 2H), NH не наблюдали; MS (ES+) m/z 501,3 (M+1).
ПРИМЕР 320
Синтез формиата 4-((2-(2-(3-фторазетидин-1-ил)этил)бензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 2-(2-(3-фторазетидин-1-ил)этил)фенил)метанамина

Следуя процедуре как описано для примера 261, стадии 3 и выполняя некритические вариации при необходимости, чтобы заменять гидрохлорид азетидина на 3-фторазетидин, указанное в заголовке соединение получали в виде масла желтого цвета (0,20 г, 35%):1H ЯМР (400 МГц, CDCl3) δ 7,64 (д, J=7,6 Гц, 1H), 7,54 (тд, J=7,6, 1,2 Гц, 1H), 7,39-7,31 (м, 2H), 5,25-5,01 (м, 1H), 3,79-3,65 (м, 2H), 3,28-3,13 (м, 2H), 2,94-2,87 (м, 2H), 2,86-2,79 (м, 2H); MS (ES+) m/z 205,3 (M+1)
Стадия 2. Получение 2-(2-(3-фторазетидин-1-ил)этил)фенил)метанамина

Следуя процедуре как описано для примера 261, стадии 4 и выполняя некритические вариации при необходимости, чтобы заменять 2-(2-азетидин-1-ил)этил)фенил)-метанамин на 2-(2-(3-фторазетидин-1-ил)этил)фенил)метанамин, указанное в заголовке соединение получали в виде масла желтого цвета (0,12 г, 59%):1H ЯМР (400 МГц, CDCl3) δ 7,37-7,32 (м, 1H), 7,26-7,17 (м, 3H), 5,24-5,03 (м, 1H), 3,91 (ушир. с, 2H), 3,78-3,64 (м, 2H), 3,23-3,16 (м, 1H), 3,15-3,10 (м, 1H), 2,75 (с, 4H), NH не наблюдали.
Стадия 3. Получение трет-бутил ((4-((2-(2-(3-фторазетидин-1-ил)этил)бензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамата
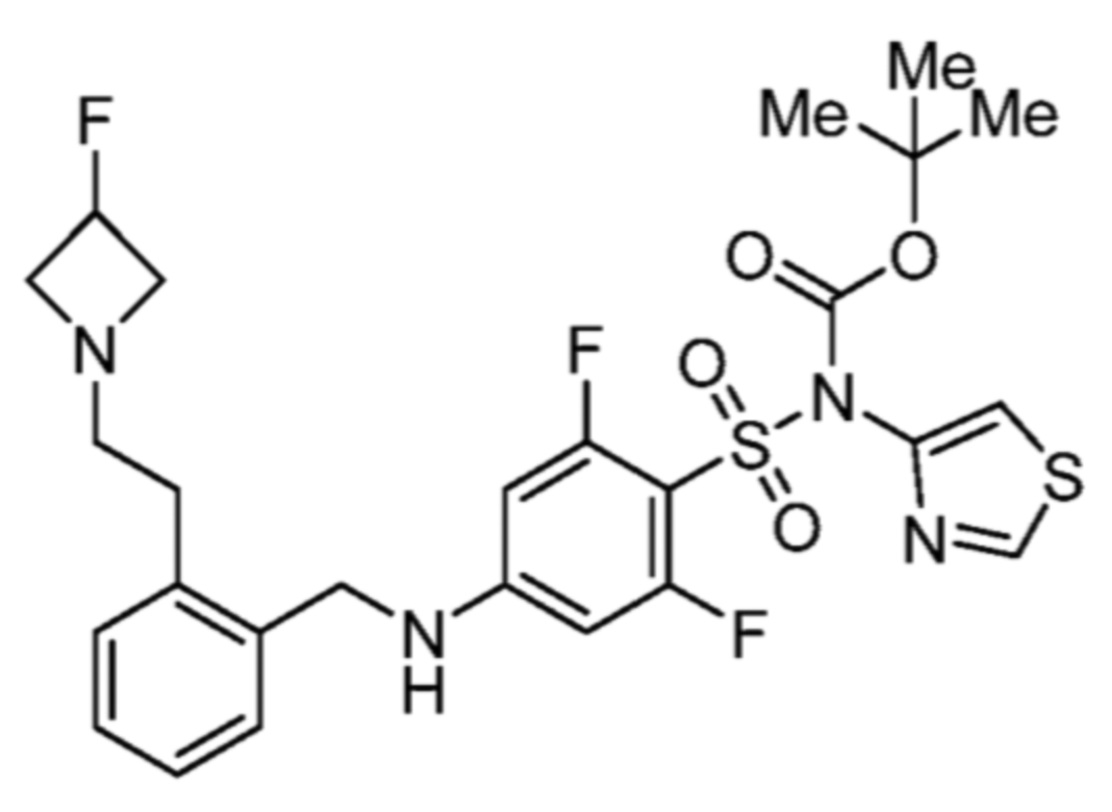
Следуя процедуре как описано для примера 261, стадии 5 и выполняя некритические вариации при необходимости, чтобы заменять (2-(2-(азетидин-1-ил)этил)фенил)метанамин на (2-(2-(3-фторазетидин-1-ил)этил)фенил)метанамин, указанное в заголовке соединение получали в виде бесцветного масла (0,080 г, выход 19%):1H ЯМР (400 МГц, CDCl3) δ 8,72 (д, J=2,2 Гц, 1H), 7,43 (д, J=2,0 Гц, 1H), 7,27-7,21 (м, 2H), 7,18-7,13 (м, 2H), 6,87 (ушир. с, 1H), 6,12 (д, J=11,6 Гц, 2H), 5,07-4,83 (м, 1H), 4,19 (д, J=4,4 Гц, 2H), 3,36-3,24 (м, 2H), 3,05-2,93 (м, 2H), 2,81-2,77 (м, 2H), 2,73-2,68 (м, 2H), 1,34 (с, 9H); MS (ES+) m/z 583,4 (M+1).
Стадия 2. Получение формиата 4-((2-(2-(3-фторазетидин-1-ил)этил)бензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано для примера 261, стадии 6 и выполняя некритические вариации при необходимости, чтобы заменять трет-бутил(4-((2-(2-(азетидин-1-ил)этил)бензил)амино)-5-хлор-2-фторфенил)сульфонил(тиазол-4-ил)карбамат на трет-бутил ((4-((2-(2-(3-фторазетидин-1-ил)этил)бензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамат, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,0376 г, выход 51%):1H ЯМР (400 МГц, CD3OD) δ 8,75 (д, J=2,2 Гц, 1H), 8,39 (ушир. с, 1H), 7,37-7,21 (м, 4H), 6,96 (д, J=2,2 Гц, 1H), 6,30-6,19 (м, 2H), 5,38-5,13 (м, 1H), 4,33 (с, 2H), 4,13-3,98 (м, 2H), 3,84-3,68 (м, 2H), 3,17-3,08 (м, 2H), 2,90-2,80 (м, 2H), NH и COOH не наблюдали; MS (ES+) m/z 483,3 (M+1).
ПРИМЕР 321
Синтез формиата 4-(((1R,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-ил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение (R)-3-фенил-3-(2,2,2-трифторацетамидо)пропановой кислоты

Следуя процедуре как описано для примера 264, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять (S)-3-амино-3-фенилпропановую кислоту на (R)-3-амино-3-фенилпропановую кислоту, указанное в заголовке соединение получали в виде бесцветного твердого вещества (7,40 г, неочищенное):1H ЯМР (400 МГц, DMSO-d6) δ 13,31-11,70 (м, 1H), 9,96 (д, J=8,2 Гц, 1H), 7,43-7,33 (м, 5H), 5,29-5,19 (м, 1H), 2,96-2,86 (м, 1H), 2,84-2,74 (м, 1H); MS (ES+) m/z 262,1 (M+1).
Стадия 2. Получение (R)-3-фенил-3-(2,2,2-трифторацетамидо)пропанoилхлорида

Следуя процедуре как описано для примера 264, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять (R)-3-амино-3-фенилпропановую кислоту на трет-бутил ((4-((2-(2-(3-фторазетидин-1-ил)этил)бензил)амино)-2,6-дифторфенил)сульфонил)(тиазол-4-ил)карбамат, указанное в заголовке соединение получали в виде твердого вещества желтого цвета (7,90 г, неочищенное).
Стадия 3. Получение (R)-2,2,2-трифтор-N-(3-оксо-2,3-дигидро-1H-инден-1-ил)ацетамида

Следуя процедуре как описано для примера 264, стадии 3 и выполняя некритические вариации при необходимости, чтобы заменять (S)-3-фенил-3-(2,2,2-трифторацетамидо)пропанoилхлорид на (R)-3-фенил-3-(2,2,2-трифторацетамидо)пропанoилхлорид, указанное в заголовке соединение получали в виде твердого вещества желтого цвета (5,60 г, выход 82%):1H ЯМР (400 МГц, CDCl3) δ 7,77-7,70 (м, 2H), 7,64 (д, J=7,6 Гц, 1H), 7,54 (т, J=7,2 Гц, 1H), 7,05 (с, 1H), 5,73 (тд, J=8,0, 3,2 Гц, 1H), 3,27 (дд, J=19,2, 7,6 Гц, 1H), 2,60 (дд, J=19,2, 3,2 Гц, 1H).
Стадия 4. Получение N-((1R,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-ил)-2,2,2-трифторацетамида

В раствор (R)-2,2,2-трифтор-N-(3-оксо-2,3-дигидро-1H-инден-1-ил)ацетамида (5,60 г, 23,03 ммоль) и гидрохлорида азетидина (4,31 г, 46,06 ммоль) в безводном тетрагидрофуране (100 мл) по каплям добавляли изопропоксид титана(IV) (26,18 г, 92,12 ммоль). Реакционную смесь перемешивали при 60°C в течение 1 ч. Затем туда добавляли цианоборогидрид натрия (5,79 г, 92,12 ммоль) и смесь перемешивали при температуре окружающей среды в течение 11 ч. Смесь разводили дихлорметаном (50 мл) и насыщенным хлоридом аммония (50 мл). Смесь фильтровали и органический слой промывали солевым раствором (100 мл), сушили над безводным сульфатом магния и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,05% гидроксид аммония в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (1,00 г, выход 15%):1H ЯМР (400 МГц, CDCl3) δ 7,53-7,37 (м, 2H), 7,36-7,32 (м, 1H), 7,31-7,29 (м, 1H), 5,44 (ушир. д, J=5,8 Гц, 1H), 4,21-4,03 (м, 1H), 3,74 (д, J=5,4 Гц, 1H), 3,40-3,36 (м, 2H), 3,21 (к, J=6,8 Гц, 2H), 2,45-2,36 (м, 1H), 2,06-2,02 (м, 2H), 1,87 (дд, J=1,2, 13,8 Гц, 1H); MS (ES+) m/z 285,2 (M+1).
Стадия 5. Получение (1R,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-амина

Следуя процедуре как описано для примера 264, стадии 5 и выполняя некритические вариации при необходимости, чтобы заменять N-((1S,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-ил)-2,2,2-трифторацетамид на N-((1R,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-ил)-2,2,2-трифторацетамид, указанное в заголовке соединение получали в виде твердого вещества желтого цвета (0,200 г, выход 50%): MS (ES+) m/z 189,3 (M+1).
Стадия 6. Получение трет-бутил (4-(((1R,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-ил)амино)-5-хлор-2-фторфенил)сульфонил(тиазол-4-ил)карбамата
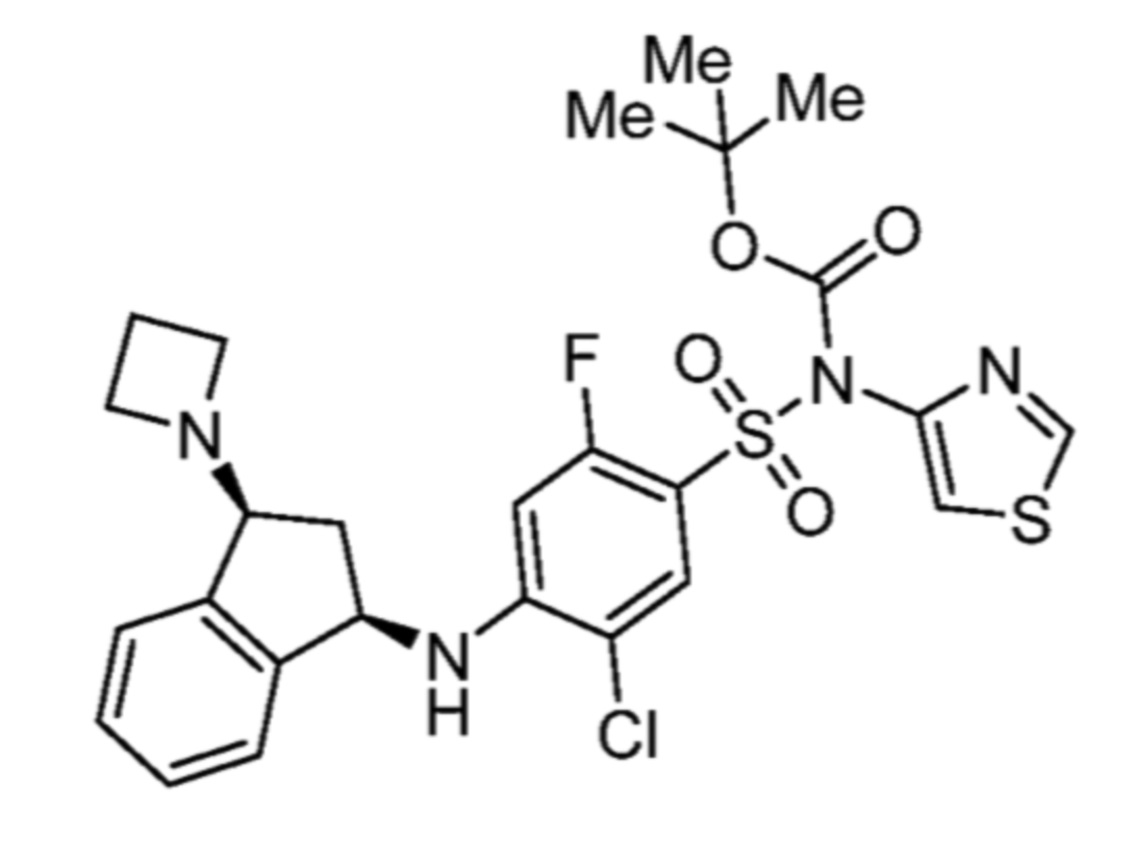
Следуя процедуре как описано для примера 264, стадии 6 и выполняя некритические вариации при необходимости, чтобы заменять (1S,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-амин на (1R,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-амин, указанное в заголовке соединение получали в виде масла желтого цвета (0,120 г, выход 43%): MS (ES+) m/z 579,2 (M+1), 581,2 (M+1).
Стадия 7. Синтез формиата 4-(((1R,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-ил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано для примера 264, стадии 7 и выполняя некритические вариации при необходимости, чтобы заменять трет-бутил (4-(((1S,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-ил)амино)-5-хлор-2-фторфенил)сульфонил(тиазол-4-ил)карбамат на (4-(((1R,3S)-3-(азетидин-1-ил)-2,3-дигидро-1H-инден-1-ил)амино)-5-хлор-2-фторфенил)сульфонил(тиазол-4-ил)карбамат, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,022 г, выход 20%):1H ЯМР (400 МГц, CDCl3) δ 8,63 (д, J=2,2 Гц, 1H), 8,22 (с, 1H), 7,67 (д, J=7,2 Гц, 1H), 7,35-7,27 (м, 4H), 6,90 (д, J=2,2 Гц, 1H), 6,45 (д, J=12,4 Гц, 1H), 6,17 (ушир. д, J=8,6 Гц, 1H), 4,84-4,78 (м, 1H), 4,20 (дд, J=4,4, 7,2 Гц, 1H), 3,72-3,60 (м, 4H), 2,75-2,66 (м, 1H), 2,24 (квин, J=7,6 Гц, 2H), 1,88 (тд, J=13,8, 4,2 Гц, 1H), NH и COOH не наблюдали; MS (ES+) m/z 479,2 (M+1), 481,2 (M+1).
ПРИМЕР 322
Синтез формиата 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(6-фторпиридин-2-ил)бензолсульфонамида
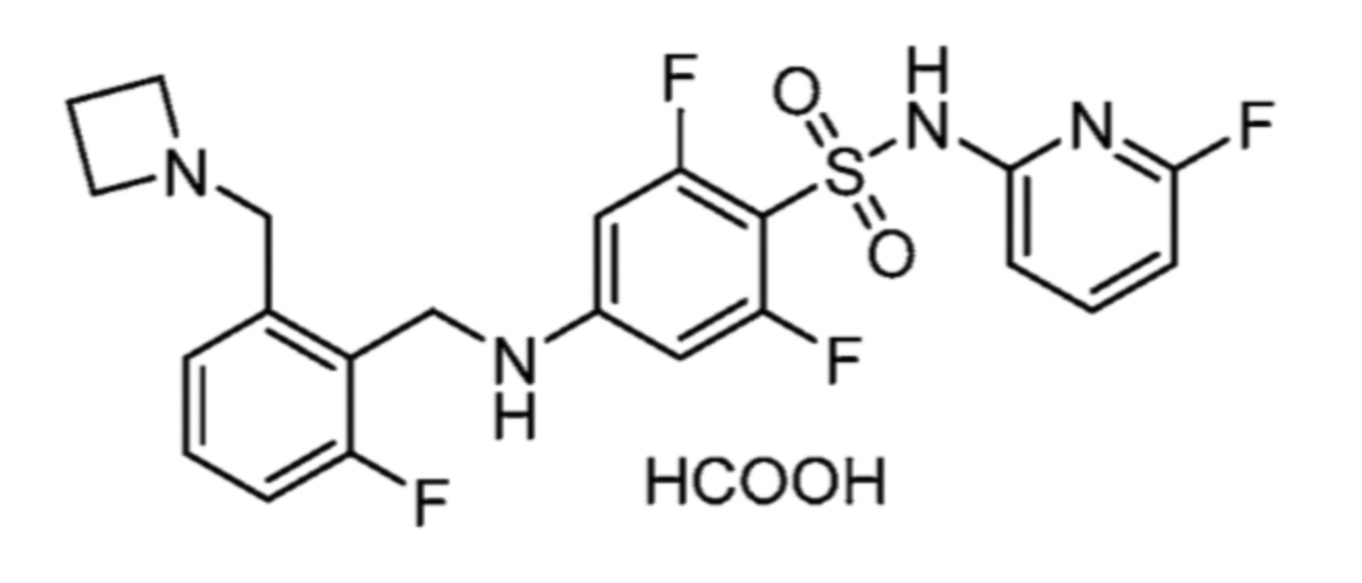
Стадия 1. Синтез 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-N-(2,4-диметоксибензил)-2,6-дифтор-N-(6-фторпиридин-2-ил)бензолсульфонамида

В раствор N-(2,4-диметоксибензил)-2,4,6-трифтор-N-(6-фторпиридин-2-ил)бензолсульфонамида (0,25 г, 0,55 ммоль) и (2-(азетидин-1-илметил)-6-фторфенил)метанамина (0,138 г, 0,712 ммоль) в N,N-диметилформамиде (5 мл) добавляли карбонат калия (0,151 г, 1,10 ммоль). Смесь перемешивали при температуре окружающей среды в течение 12 ч. Затем смесь разводили водой (50 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические экстракты промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 33% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде твердого вещества желтого цвета (0,12 г, выход 35%):1H ЯМР (400 МГц, CDCl3) δ 7,67 (к, J=8,2 Гц, 1H), 7,37-7,30 (м, 2H), 7,24 (дд, J=7,6, 5,6 Гц, 1H), 7,10-7,03 (м, 2H), 6,62 (дд, J=8,0, 2,8 Гц, 1H), 6,43-6,37 (м, 2H), 6,19 (д, J=12,0 Гц, 2H), 5,16 (с, 2H), 4,34 (с, 2H), 3,77 (д, J=2,8 Гц, 6H), 3,61 (с, 2H), 3,23 (т, J=6,8 Гц, 4H), 2,13 (квин, J=7,2 Гц, 2H), NH не наблюдали.
Стадия 2. Синтез формиата 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(6-фторпиридин-2-ил)бензолсульфонамида

К 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-N-(2,4-диметоксибензил)-2,6-дифтор-N-(6-фторпиридин-2-ил)бензолсульфонамиду (0,120 г, 0,190 ммоль) добавляли раствор 4 M хлороводорода в этилацетате (4 мл) и смесь перемешивали при температуре окружающей среды в течение 1 ч. Смесь концентрировали in vacuo и остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,0307 г, выход 30%):1H ЯМР (400 МГц, CDCl3) δ 8,31 (с, 1H), 7,73 (к, J=8,0 Гц, 1H), 7,33-7,29 (м, 1H), 7,28-7,26 (м, 1H), 7,15-7,06 (м, 2H), 6,61 (дд, J=8,0, 2,4 Гц, 1H), 6,22 (д, J=12,0 Гц, 2H), 4,36 (с, 2H), 3,81 (с, 2H), 3,46 (ушир. т, J=7,2 Гц, 4H), 2,24 (квин, J=7,2 Гц, 2H), NH и COOH не наблюдали; MS (ES+) m/z 481,1 (M+1).
ПРИМЕР 323
Синтез формиата 4-((2-(азетидин-1-илметил)бензил)амино)-2-фтор-N-(тиазол-4-ил)-5-винилбензолсульфонамида

Стадия 1. Получение трет-бутил ((5-бром-2,4-дифторфенил)сульфонил)(тиазол-4-ил)карбамата

В смесь трет-бутил тиазол-4-илкарбамата (2,00 г, 9,99 ммоль) в безводном N,N-диметилформамиде (20 мл) добавляли гидрид натрия (60% дисперсия в минеральном масле, 0,799 г, 20,0 ммоль) при 0°C. Смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 30 минут. После охлаждения до 0°C медленно добавляли раствор 5-бром-2,4-дифторбензол-1-сульфонилхлорида (3,79 г, 13,0 ммоль) в безводном N,N-диметилформамиде (5 мл). Смесь перемешивали при 0°C в течение 30 минут и температуре окружающей среды в течение 1 ч. Затем остаток выливали в воду со льдом (50 мл) и водную фазу экстрагировали этилацетатом (3×50 мл). Объединенные органические экстракты промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 9 до 13% этилацетата в гексанах, чтобы получать указанное в заголовке соединение в виде твердого вещества желтого цвета (3,00 г, выход 66%):1H ЯМР (400 МГц, CDCl3) δ 8,81 (д, J=2,0 Гц, 1H), 8,38 (т, J=7,2 Гц, 1H), 7,54 (д, J=2,0 Гц, 1H), 7,08 (дд, J=9,2, 8,0 Гц, 1H), 1,39 (с, 9H).
Стадия 2. Получение 4-((2-(азетидин-1-илметил)бензил)амино)-5-бром-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

В раствор трет-бутил (5-бром-2,4-дифторфенил)сульфонил(тиазол-4-ил)карбамата (0,60 г, 1,32 ммоль) и карбоната калия (0,36 г, 2,6 ммоль) в безводном N,N-диметилформамиде (5 мл) по каплям добавляли (2-(азетидин-1-илметил)фенил)метанамин (0,348 г, 1,98 ммоль). Смесь перемешивали при температуре окружающей среды в течение 12 ч. Затем смесь разводили водой (60 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические экстракты промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,20 г, выход 30%):1H ЯМР (400 МГц, CDCl3) δ 8,66 (д, J=2,0 Гц, 1H), 7,90 (д, J=7,6 Гц, 1H), 7,28-7,25 (м, 4H), 6,97 (д, J=2,0 Гц, 1H), 6,53 (д, J=13,2 Гц, 1H), 4,36 (с, 2H), 3,61 (с, 2H), 3,23 (т, J=7,2 Гц, 4H), 2,12-2,08 (м, 2H), NH не наблюдали; MS (ES+) m/z 511,0 (M+1), 513,0 (M+1).
Стадия 3. Получение формиата 4-((2-(азетидин-1-илметил)бензил)амино)-2-фтор-N-(тиазол-4-ил)-5-винилбензолсульфонамида

В раствор 4-((2-(азетидин-1-илметил)бензил)амино)-5-бром-2-фтор-N-(тиазол-4-ил)бензолсульфонамида (0,20 г, 0,39 ммоль), 4,4,5,5-тетраметил-2-винил-1,3,2-диоксаборолана (0,12 г, 0,78 ммоль) и карбоната натрия (0,0829 г, 0,782 ммоль) в безводном 1,4-диоксане (3 мл) одной частью добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (0,0572 г, 0,0782 ммоль). Смесь перемешивали при 100°C в течение 12 ч. После охлаждения до температуры окружающей среды, смесь разводили водой (30 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические экстракты промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,057 г, выход 31%):1H ЯМР (400 МГц, CDCl3) δ 8,66 (д, J=2,0 Гц, 1H), 8,39 (с, 1H), 7,73 (д, J=8,2 Гц, 1H), 7,40-7,31 (м, 4H), 6,96 (д, J=2,0 Гц, 1H), 6,72 (дд, J=17,2, 10,8 Гц, 1H), 6,36 (д, J=13,2 Гц, 1H), 5,61 (дд, J=17,2, 0,8 Гц, 1H), 5,40-5,31 (м, 1H), 4,38 (с, 2H), 3,94 (с, 2H), 3,62 (т, J=7,6 Гц, 4H), 2,29 (квин, J=7,6 Гц, 2H), NH и COOH не наблюдали; MS (ES+) m/z 459,2 (M+1).
ПРИМЕР 324
Синтез 4-((2-(азетидин-1-илметил)бензил)амино)-2,6-дифтор-N-(6-фторпиридин-2-ил)бензолсульфонамида

Стадия 1. Получение N-(2,4-диметоксибензил)-2,4,6-трифтор-N-(6-фторпиридин-2-ил)бензолсульфонамида
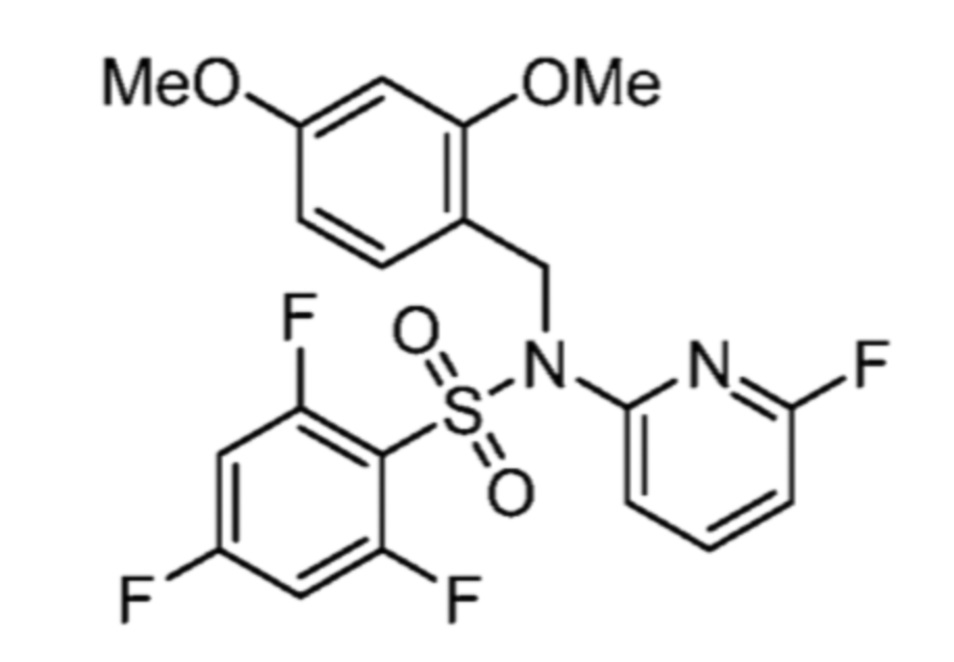
В раствор N-(2,4-диметоксибензил)-6-фторпиридин-2-амина (0,20 г, 0,76 ммоль) в безводном тетрагидрофуране (10 мл) по каплям добавляли метиллитий (1,6 M, 0,67 мл) при -78°C. Смесь нагревали до 0°C, перемешивали в течение 30 минут и затем охлаждали до -78°C. Затем туда добавляли раствор 2,4,6-трифторбензол-1-сульфонилхлорида (0,281 г, 1,22 ммоль) в безводном тетрагидрофуране (3 мл) при -78°C. Смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 2 ч. Затем смесь разводили водой (50 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические экстракты промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 9% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде масла желтого цвета (0,14 г, выход 40%):1H ЯМР (400 МГц, CDCl3) δ 7,69 (к, J=8,0 Гц, 1H), 7,27 (д, J=9,2 Гц, 1H), 7,15 (дд, J=7,6, 1,6 Гц, 1H), 6,81-6,73 (м, 2H), 6,69 (дд, J=8,0, 2,4 Гц, 1H), 6,44-6,39 (м, 2H), 5,13 (с, 2H), 3,79 (с, 3H), 3,76 (с, 3H); MS (ES+) m/z 479,1 (M+23).
Стадия 2. Получение 4-((2-(азетидин-1-илметил)бензил)амино)-N-(2,4-диметоксибензил)-2,6-дифтор-N-(6-фторпиридин-2-ил)бензолсульфонамида

В раствор N-(2,4-диметоксибензил)-2,4,6-трифтор-N-(6-фторпиридин-2-ил)бензолсульфонамида (0,11 г, 0,24 ммоль) и (2-(азетидин-1-илметил)фенил)метанамина (0,0637 г, 0,362 ммоль) в безводном N,N-диметилформамиде (4 мл) добавляли карбонат калия (0,0666 г, 0,482 ммоль). Смесь перемешивали при температуре окружающей среды в течение 12 ч и затем разводили водой (50 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические экстракты промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде масла желтого цвета (0,14 г, выход 95%): MS (ES+) m/z 613,2 (M+1).
Стадия 3. Получение 4-((2-(азетидин-1-илметил)бензил)амино)-2,6-дифтор-N-(6-фторпиридин-2-ил)бензолсульфонамида

К 4-((2-(азетидин-1-илметил)бензил)амино)-N-(2,4-диметоксибензил)-2,6-дифтор-N-(6-фторпиридин-2-ил)бензолсульфонамиду (0,10 г, 0,16 ммоль) добавляли 4 M раствор хлороводорода в этилацетате (5 мл) и смесь перемешивали при температуре окружающей среды в течение 1 ч. Концентрированием in vacuo и очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,05% гидроксид аммония, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,0269 г, выход 35%):1H ЯМР (400 МГц, DMSO-d6) δ 7,72 (к, J=8,0 Гц, 1H), 7,52 (ушир. с, 1H), 7,34-7,18 (м, 4H), 6,78 (дд, J=8,0, 2,0 Гц, 1H), 6,54 (ушир. д, J=6,0 Гц, 1H), 6,35 (д, J=12,4 Гц, 2H), 4,38 (с, 2H), 3,60 (с, 2H), 3,16 (т, J=6,8 Гц, 4H), 2,00 (квин, J=6,8 Гц, 2H), NH не наблюдали;MS (ES+) m/z 463,1 (M+1), 464,1 (M+1).
ПРИМЕР 325
Синтез (S)-5-хлор-4-((1-(2,5-дифторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил (S)-((5-хлор-4-((1-(2,5-дифторфенил)этил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата

Следуя процедуре как описано для примера 5, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять гидрохлорид (S)-1-(5-хлор-2-фторфенил)этан-1-амина на гидрохлорид (S)-1-(2,5-дифторфенил)этан-1-амина, указанное в заголовке соединение получали в виде твердого вещества желтого цвета (0,800 г, выход 50%): MS (ES+) m/z 447,9 (M - 99).
Стадия 2. Получение (S)-5-хлор-4-((1-(2,5-дифторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано для примера 5, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять трет-бутил (S)-((5-хлор-4-((1-(5-хлор-2-фторфенил)этил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамат на трет-бутил (S)-((5-хлор-4-((1-(2,5-дифторфенил)этил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамат, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,153 г, выход 61%):1H ЯМР (400 МГц, DMSO-d6) δ 8,84 (д, J=2,0 Гц, 1H), 7,62 (д, J=7,4 Гц, 1H), 7,32-7,22 (м, 2H), 7,18-7,10 (м, 1H), 6,90 (ушир. с, 1H), 6,61 (д, J=7,8 Гц, 1H), 6,39 (д, J=13,0 Гц, 1H), 4,93 (квин, J=6,8 Гц, 1H), 1,55 (д, J=6,8 Гц, 3H), NH не наблюдали; MS (ES+) m/z 447,9 (M+1).
ПРИМЕР 326
Синтез формиата 2,6-дифтор-4-((2-фтор-6-((1-метилазетидин-3-ил)окси)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил 3-(2-циано-3-фторфенокси)азетидин-1-карбоксилата

Смесь 2-фтор-6-гидроксибензонитрила (1,50 г, 10,9 ммоль), трет-бутил 3-(2-циано-3-фторфенокси)азетидин-1-карбоксилата (3,10 г, 10,9 ммоль) и карбоната калия (3,78 г, 27,3 ммоль) в безводном N,N-диметилформамиде (3 мл) перемешивали при 90°C в течение 12 ч. Реакционную смесь разводили водой (60 мл) и затем экстрагировали этилацетатом (3×30 мл). Объединенную органическую фазу промывали солевым раствором (40 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата при пониженном давлении и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом 18-25% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (2,00 г, выход 63%):1H ЯМР (400 МГц, CDCl3) δ 7,49 (тд, J=8,6, 6,6 Гц, 1H), 6,85 (т, J=8,4 Гц, 1H), 6,42 (д, J=8,6 Гц, 1H), 5,02-4,95 (м, 1H), 4,34 (ддд, J=9,8, 6,4, 1,0 Гц, 2H), 4,07-4,14 (м, 2 H), 1,46 (с, 9H); MS (ES+) m/z 315,2 (M+23).
Стадия 2. Получение трет-бутил 3-(2-(аминометил)-3-фторфенокси)азетидин-1-карбоксилата

В смесь трет-бутил 3-(2-циано-3-фторфенокси)азетидин-1-карбоксилата (2,00 г, 6,84 ммоль) в метаноле (100 мл) и концентрированном гидроксиде аммония (25 мл) добавляли ренеевский никель (0,58 г, 6,84 ммоль) и смесь перемешивали в атмосфере водорода (50 фунтов/дюйм2) при температуре окружающей среды в течение 12 ч. Затем реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении, чтобы получать указанное в заголовке соединение в виде бесцветного масла (1,00 г, выход 49%):1H ЯМР (400 МГц, CDCl3) δ 7,10 (ушир. с, 1H), 6,76 (ушир. с, 1H), 6,31 (ушир. д, J=6,1 Гц, 1H), 4,92 (ушир. с, 1H), 4,33 (дд, J=9,4, 6,2 Гц, 2H), 4,17-3,81 (м, 4H), 1,45 (с, 9H), NH не наблюдали; MS (ES+) m/z 297,1 (M+1)
Стадия 3. Получение трет-бутил 3-(2-(((4-(N-(трет-бутоксикарбонил)-N-(тиазол-4-ил)сульфамоил)-3,5-дифторфенил)амино)метил)-3-фторфенокси)азетидин-1-карбоксилата

В раствор трет-бутил 3-(2-(аминометил)-3-фторфенокси)азетидин-1-карбоксилата (0,20 г, 0,67 ммоль), трет-бутил тиазол-4-ил((2,4,6-трифторфенил)сульфонил)карбамата (0,28 г, 0,70 ммоль) в безводном N,N-диметилформамиде (15 мл) добавляли карбонат калия (0,18 г, 1,3 ммоль). Смесь перемешивали при температуре окружающей среды в течение 12 ч и затем добавляли воду (100 мл) и смесь экстрагировали этилацетатом (3×300 мл). Объединенные органические слои промывали солевым раствором (40 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата при пониженном давлении и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 20 до 50% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного масла (0,23 г, выход 51%):1H ЯМР (400 МГц, CDCl3) δ 8,79 (д, J=2,3 Гц, 1H), 7,49 (д, J=2,3 Гц, 1H), 7,19-7,26 (м, 1H), 6,78 (т, J=8,6 Гц, 1H), 6,35 (д, J=8,2 Гц, 1H), 6,29 (д, J=11,6 Гц, 2H), 5,03-5,01 (м, 1H), 5,01-4,95 (м, 1H), 4,45 (ушир. д, J=5,8 Гц, 2H), 4,37 (дд, J=9,8, 6,26 Гц, 2H), 4,03 (дд, J=10,4, 3,8 Гц, 2H), 1,47 (с, 9H), 1,37 (с, 9H).
Стадия 4. Получение 4-((2-(азетидин-3-илокси)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида
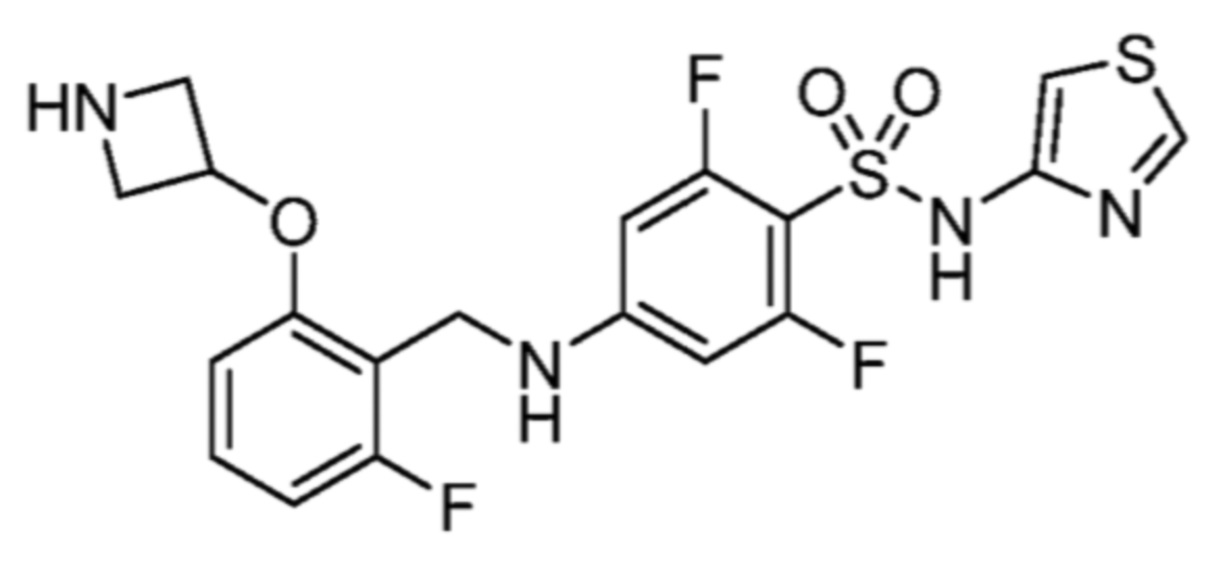
К трет-бутил 3-(2-(((4-(N-(трет-бутоксикарбонил)-N-(тиазол-4-ил)сульфамоил)-3,5-дифторфенил)амино)метил)-3-фторфенокси)азетидин-1-карбоксилату (0,23 г, 0,34 ммоль) добавляли 4 M раствор хлороводорода в (20 мл) и смесь перемешивали при температуре окружающей среды в течение 2 ч. Концентрированием in vacuo и очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,05% гидроксида аммония, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,070 г, выход 43%): MS (ES+) m/z 471,1 (M+1).
Стадия 5. Получение формиата 2,6-дифтор-4-((2-фтор-6-((1-метилазетидин-3-ил)окси)бензил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Раствор 4-((2-(азетидин-3-илокси)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида (0,06 г, 0,1 ммоль), параформальдегида (0,023 г, 0,26 ммоль) и уксусной кислоты (0,00076 г, 0,013 ммоль, 0,73 мкл) в метаноле (2 мл) перемешивали при температуре окружающей среды в течение 30 минут. Затем туда добавляли цианоборогидрид натрия (0,016 г, 0,26 ммоль) и смесь перемешивали при температуре окружающей среды в течение 2,5 ч. Реакционную смесь разводили водой (50 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические экстракты промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 10% метанола в дихлорметане, после которой следовала препаративная HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,0055 г, выход 9%):1H ЯМР (400 МГц, CD3OD) δ 8,72 (д, J=2,2 Гц, 1H), 8,46 (ушир. с, 1H), 7,28 (дт, J=8,4, 6,8 Гц, 1H), 6,94 (д, J=2,4 Гц, 1H), 6,81 (т, J=8,8 Гц, 1H), 6,55 (д, J=8,4 Гц, 1H), 6,34-6,23 (м, 2H), 5,05-4,98 (м, 1H), 4,37 (с, 2H), 4,24 (дд, J=11,2, 6,2 Гц, 2H), 3,76 (ушир. дд, J=11,0, 4,5 Гц, 2H), 2,71 (с, 3H), NH и COOH не наблюдали.
ПРИМЕР 327
Синтез формиата 4-((2-(азетидин-1-илметил)бензил)амино)-3-хлор-2,6-дифтор-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида

Стадия 1. Получение 4-((2-(азетидин-1-илметил)бензил)амино)-3-хлор-N-(2,4-диметоксибензил)-2,6-дифтор-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида

В раствор 3-хлор-N-(2,4-диметоксибензил)-2,4,6-трифтор-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида (0,600 г, 1,25 ммоль) в безводном N,N-диметилформамиде (3 мл) добавляли карбонат цезия (0,815 г, 2,50 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 12 ч. Реакционную смесь разводили этилацетатом (50 мл) и водой (20 мл) и водную фазу экстрагировали этилацетатом (2×50 мл). Объединенные органические фазы промывали солевым раствором (3×20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 5 до 50% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде твердого вещества светло-коричневого цвета (0,288 г, выход 36%):1H ЯМР (400 МГц, CDCl3) δ 8,12 (с, 1H), 7,25-7,18 (м, 4H), 7,12 (д, J=8,4 Гц, 1H), 6,29-6,24 (м, 1H), 6,20 (дд, J=13,5, 1,2 Гц, 1H), 6,14 (д, J=2,2 Гц, 1H), 5,28 (с, 2H), 4,24 (с, 2H), 3,61 (с, 6H), 3,53 (с, 2H), 3,15 (т, J=7,2 Гц, 4H), 2,05-1,97 (м, 2H), NH не наблюдали.
Стадия 2. Получение формиата 4-((2-(азетидин-1-илметил)бензил)амино)-3-хлор-2,6-дифтор-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида

В смесь 4-((2-(азетидин-1-илметил)бензил)амино)-3-хлор-N-(2,4-диметоксибензил)-2,6-дифтор-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида (0,050 г, 0,079 ммоль) в дихлорметане (10 мл) добавляли трифторуксусную кислоту (0,090 г, 0,786 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Концентрированием in vacuo и очисткой остатка с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,0190 г, выход 46%):1H ЯМР(400 МГц, CDCl3) δ 7,94 (с, 1H), 7,50-7,32 (м, 4H), 6,11 (д, J=11,6 Гц, 1H), 4,57 (д, J=3,4 Гц, 4H), 4,26 (т, J=8,2 Гц, 4H), 2,53 (квин, J=8,1 Гц, 2H), NH и COOH не наблюдали;19F ЯМР (376,5 МГц, CDCl3) δ -109,1 (с, 1F), -109,2 (с, 1F); MS (ES+) m/z 486,2 (M+1), 488,2 (M+1).
ПРИМЕР 328
Синтез 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(пиридин-2-ил)бензолсульфонамида

Стадия 1. Получение N-(2,4-диметоксибензил)пиридин-2-амина

Раствор 2-фторпиридина (1,00 г, 10,3 ммоль) в (2,4-диметоксифенил)метанамине (1,72 г, 10,3 ммоль) перемешивали при 110°C в течение 12 ч. После охлаждения до температуры окружающей среды смесь очищали с помощью колоночной хроматографии с обращенной фазой, осуществляя элюирование с градиентом 5-60% ацетонитрила в воде, чтобы получать указанное в заголовке соединение в виде твердого вещества желтого цвета (1,35 г, выход 52%):1H ЯМР (400 МГц, CDCl3) δ 8,10 (дд, J=5,2, 1,0 Гц, 1H), 7,40 (ддд, J=8,6, 7,0, 1,8 Гц, 1H), 7,23 (д, J=8,2 Гц, 1H), 6,57 (ддд, J=7,2, 5,0, 0,8 Гц, 1H), 6,49 (д, J=2,4 Гц, 1H), 6,46-6,40 (м, 2H), 5,11 (ушир. с, 1H), 4,42 (ушир. д, J=3,4 Гц, 2H), 3,85 (с, 3H), 3,81 (с, 3H); MS (ES+) m/z 245,3 (M+1).
Стадия 2. Получение N-(2,4-диметоксибензил)-2,4,6-трифтор-N-(пиридин-2-ил)бензолсульфонамида

В раствор N-(2,4-диметоксибензил)пиридин-2-амина (0,80 г, 3,3 ммоль) в безводном тетрагидрофуране (10 мл) добавляли 1,6 M раствор метиллития (3,07 мл, 4,9 ммоль) при -78°C. Реакционную смесь перемешивали в течение 30 минут при 0°C и затем добавляли раствор 2,4,6-трифторбензолсульфонилхлорида (1,06 г, 4,58 ммоль) в безводном тетрагидрофуране (2 мл) при -78°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 3 ч. Реакционную смесь концентрировали in vacuo и остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 9 до 11% этилацетата в петролейном эфире, чтобы получать указанное в заголовке соединение в виде масла желтого цвета (1,00 г, выход 48%):1H ЯМР (400 МГц, CDCl3) δ 8,28 (дд, J=4,8, 1,2 Гц, 1H), 7,61 (дт, J=7,8, 1,8 Гц, 1H), 7,26-7,19 (м, 2H), 7,07 (ддд, J=7,4, 4,8, 0,8 Гц, 1H), 6,73 (т, J=8,8 Гц, 2H), 6,41-6,36 (м, 2H), 5,13 (с, 2H), 3,76 (с, 3H), 3,72 (с, 3H); MS (ES+) m/z 461,0 (M+23).
Стадия 3. Получение 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-N-(2,4-диметоксибензил)-2,6-дифтор-N-(пиридин-2-ил)бензолсульфонамида

Смесь (2-(азетидин-1-илметил)-6-фторфенил)метанамина (0,31 г, 1,6 ммоль), N-(2,4-диметоксибензил)-2,4,6-трифтор-N-(пиридин-2-ил)бензолсульфонамида (0,70 г, 1,6 ммоль) и карбоната калия (0,442 г, 3,20 ммоль) в безводном N,N-диметилформамиде (15 мл) перемешивали при температуре окружающей среды в течение 12 ч. Реакционную смесь разводили этилацетатом (50 мл) и водой (20 мл) и водную фазу экстрагировали этилацетатом (2×50 мл). Объединенные органические фазы промывали солевым раствором (3×20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии с обращенной фазой, осуществляя элюирование с использованием от 5 до 60% ацетонитрила в воде, получали указанное в заголовке соединение в виде бесцветного масла (0,10 г, выход 10%):1H ЯМР (400 МГц, CDCl3) δ 8,30 (дд, J=4,8, 1,4 Гц, 1H), 7,63-7,56 (м, 1H), 7,44 (д, J=8,2 Гц, 1H), 7,27 (с, 2H), 7,25-7,20 (м, 1H), 7,09-7,00 (м, 3H), 6,37 (кд, J=4,4, 2,3 Гц, 2H), 6,18 (д, J=11,9 Гц, 2H), 5,17 (с, 2H), 4,33 (д, J=1,0 Гц, 2H), 3,75 (с, 3H), 3,72 (с, 3H), 3,62 (с, 2H), 3,25 (ушир. с, 4H), 2,13 (квин, J=6,9 Гц, 2H); MS (ES+) m/z 613,2 (M+1).
Стадия 4. Получение 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(пиридин-2-ил)бензолсульфонамида

В раствор 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-N-(2,4-диметоксибензил)-2,6-дифтор-N-(пиридин-2-ил)бензолсульфонамида (0,10 г, 0,16 ммоль) в дихлорметане (10 мл) добавляли трифторуксусную кислоту (0,04 г, 0,3 ммоль). Смесь перемешивали при температуре окружающей среды в течение 1 ч и затем концентрировали in vacuo. Остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,0638 г, выход 80%):1H ЯМР (400 МГц, CDCl3) δ 8,31 (ушир. д, J=5,8 Гц, 1H), 7,75-7,66 (м, 1H), 7,55 (д, J=8,8 Гц, 1H), 7,27-7,21 (м, 1H), 7,11-7,01 (м, 2H), 6,78 (т, J=6,4 Гц, 1H), 6,19 (ушир. д, J=12,0 Гц, 2H), 4,32 (с, 2H), 3,78 (ушир. с, 2H), 3,42 (ушир. с, 4H), 2,28-2,14 (м, 2H), NH не наблюдали; MS (ES+) m/z 463,0 (M+1).
ПРИМЕР 329
Синтез 4-((2-(азетидин-1-илметил)бензил)амино)-2-фтор-N-(6-фторпиридин-2-ил)-5-метилбензолсульфонамида

Стадия 1. Получение 4-((2-(азетидин-1-илметил)бензил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(6-фторпиридин-2-ил)-5-метилбензолсульфонамида

В смесь 4-бром-N-(2,4-диметоксибензил)-2-фтор-N-(6-фторпиридин-2-ил)-5-метилбензолсульфонамида (0,300 г, 0,584 ммоль) в безводном 1,4-диоксане (2 мл) добавляли (2-(азетидин-1-илметил)фенил)метанамин (0,206 г, 1,17 ммоль), карбонат цезия (0,571 г, 1,75 ммоль) и хлор(2-дициклогексилфосфино-2′,6′-диметокси-1,1′-бифенил)[2-(2′-амино-1,1′-бифенил)]палладий(II) (0,042 г, 0,058 ммоль). Реакционную смесь нагревали до 90°C в течение 12 ч. После охлаждения до температуры окружающей среды, реакционную смесь разводили водой (10 мл) и водную фазу экстрагировали этилацетатом (3×10 мл). Объединенные органические экстракты промывали солевым раствором (3×10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 50% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде масла желтого цвета (0,200 г, выход 56%):1H ЯМР (400 МГц, CDCl3) δ 7,57 (к, J=8,2 Гц, 1H), 7,41 (д, J=8,0 Гц, 1H), 7,28 (дд, J=7,8, 1,8 Гц, 1H), 7,25-7,19 (м, 4H), 6,71 (ушир. с, 1H), 6,52 (дд, J=7,8, 3,0 Гц, 1H), 6,34-6,28 (м, 3H), 5,00 (с, 2H), 4,21 (с, 2H), 3,67 (д, J=7,2 Гц, 6H), 3,52 (с, 2H), 3,14 (т, J=7,0 Гц, 4H), 2,09 (с, 3H), 2,02-1,98 (м, 2H), NH не наблюдали; MS (ES+) m/z 609,4 (M+1).
Стадия 2. Получение 4-((2-(азетидин-1-илметил)бензил)амино)-2-фтор-N-(6-фторпиридин-2-ил)-5-метилбензолсульфонамида

В раствор 4-((2-(азетидин-1-илметил)бензил)амино)-N-(2,4-диметоксибензил)-2-фтор-N-(6-фторпиридин-2-ил)-5-метилбензолсульфонамида (0,200 г, 0,329 ммоль) в дихлорметане (2 мл) добавляли трифторуксусную кислоту (1,54 г, 13,5 ммоль) и смесь перемешивали при температуре окружающей среды в течение 1 ч. Реакционную смесь концентрировали in vacuo. Остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,05% гидроксид аммония, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,045 г, выход 29%):1H ЯМР (400 МГц, CDCl3) δ 7,58 (к, J=8,0 Гц, 1H), 7,45 (д, J=8,2 Гц, 1H), 7,21 (дд, J=3,8, 1,4 Гц, 3H), 7,10 (дд, J=7,8, 1,8 Гц, 1H), 7,02-6,63 (м, 1H), 6,48 (дд, J=2,4, 7,8 Гц, 1H), 6,35 (д, J=13,4 Гц, 1H), 4,22 (с, 2H), 3,54 (с, 2H), 3,17 (ушир. с, 4H), 2,10 (с, 3H), 2,00 (тд, J=14,0, 6,8 Гц, 2H), NH не наблюдали;19F ЯМР (376,5 МГц, CDCl3) -69,2 (с, 1F), -109,8 (с, 1F); MS (ES+) m/z 459,2 (M+1).
ПРИМЕР 330
Синтез 4-((2-(азетидин-1-илметил)бензил)амино)-2,6-дифтор-3-метил-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида

Стадия 1. Получение 4-((2-(азетидин-1-илметил)бензил)амино)-3-бром-N-(2,4-диметоксибензил)-2,6-дифтор-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида

В раствор 3-бром-N-(2,4-диметоксибензил)-2,4,6-трифтор-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида (1,00 г, 1,91 ммоль) в безводном N,N-диметилформамиде (20 мл) добавляли карбонат цезия (1,87 г, 5,73 ммоль) и (2-(азетидин-1-илметил)фенил)метанамин (0,673 г, 3,82 ммоль). Смесь перемешивали при температуре окружающей среды в течение 12 ч. Реакционную смесь разводили водой (20 мл) и водную фазу экстрагировали этилацетатом (2×20 мл). Объединенные органические экстракты промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 9 до 33% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде масла желтого цвета (0,600 г, выход 46%):1H ЯМР (400 МГц, CDCl3) δ 8,21 (с, 1H), 7,33-7,29 (м, 3H), 7,22 (д, J=8,4 Гц, 1H), 6,39-6,30 (м, 2H), 6,30-6,22 (м, 1H), 6,22-6,19 (м, 1H), 4,37 (с, 2H), 3,75 (д, J=7,8 Гц, 2H), 3,71 (с, 3H), 3,67 (с, 3H), 3,65 (с, 2H), 3,28 (т, J=7,2 Гц, 4H), 2,12 (квин, J=7,2 Гц, 2H), NH не наблюдали.
Стадия 2. Получение 4-((2-(азетидин-1-илметил)бензил)амино)-N-(2,4-диметоксибензил)-2,6-дифтор-3-метил-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида

В смесь 4-((2-(азетидин-1-илметил)бензил)амино)-3-бром-N-(2,4-диметоксибензил)-2,6-дифтор-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида (0,500 г, 0,734 ммоль) в безводном толуоле (8 мл) добавляли метилбороновую кислоту (0,087 г, 1,5 ммоль), 2-дициклогексилфосфино-2',6'-диметоксибифенил (0,060 г, 0,15 ммоль), фосфат калия (0,467 г, 2,20 ммоль) и ацетат палладия(II) (0,033 г, 0,147 ммоль). Смесь дегазировали и нагревали до 110°C в течение 12 ч. После охлаждения до температуры окружающей среды, реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 9 до 33% этилацетата в петролейном эфире, чтобы получать указанное в заголовке соединение в виде твердого вещества желтого цвета (0,300 г, выход 66%):1H ЯМР (400 МГц, CDCl3) δ 8,18 (с, 1H), 7,35-7,30 (м, 4H), 7,20 (д, J=8,4 Гц, 1H), 6,36 (дд, J=2,4, 8,4 Гц, 1H), 6,30 (д, J=2,4 Гц, 1H), 6,21 (ушир. д, J=13,6 Гц, 1H), 5,33 (с, 2H), 4,40-4,28 (м, 2H), 3,76-3,74 (м, 3H), 3,73 (с, 3H), 3,70 (с, 2H), 3,34 (ушир. с, 4H), 2,14 (ушир. с, 2H), 2,00 (д, J=1,8 Гц, 3H), NH не наблюдали; MS (ES+) m/z 616,4 (M+1).
Стадия 3. Получение 4-((2-(азетидин-1-илметил)бензил)амино)-2,6-дифтор-3-метил-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида

В смесь 4-((2-(азетидин-1-илметил)бензил)амино)-N-(2,4-диметоксибензил)-2,6-дифтор-3-метил-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамида (0,280 г, 0,454 ммоль) в дихлорметане (10 мл) добавляли трифторуксусную кислоту (2 мл) и смесь перемешивали при температуре окружающей среды в течение 30 минут. Реакционную смесь концентрировали in vacuo и остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,065 г, выход 31%):1H ЯМР (400 МГц, DMSO-d6) δ 7,87 (с, 1H), 7,41-7,25 (м, 4H), 6,55 (ушир. с, 1H), 6,04 (д, J=13,4 Гц, 1H), 4,46 (ушир. с, 2H), 4,41 (с, 2H), 4,02 (ушир. т, J=7,8 Гц, 4H), 2,34 (квин, J=7,8 Гц, 2H), 2,00 (д, J=0,8 Гц, 3H), сигнал NH не наблюдали; MS (ES+) m/z 466,3 (M+1).
ПРИМЕР 331
Синтез (S)-5-хлор-2-фтор-4-((1-(2-фтор-5-метоксифенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил (S)-((5-хлор-2-фтор-4-((1-(2-фтор-5-метоксифенил)этил)амино)фенил)сульфонил)(тиазол-4-ил)карбамата

В раствор трет-бутил (5-хлор-2,4-дифторфенил)сульфонил(тиазол-4-ил)карбамата (0,250 г, 0,609 ммоль) и (S)-1-(2-фтор-5-метоксифенил)этанамина (0,124 г, 0,730 ммоль) в безводном диметилсульфоксиде (2 мл) добавляли карбонат цезия (0,397 г, 1,22 ммоль) одной частью. Смесь перемешивали при температуре окружающей среды в течение 12 ч. Смесь разводили водой (30 мл) и водную фазу экстрагировали этилацетатом (3×20 мл). Объединенные органические экстракты промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 33% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного масла (0,150 г, выход 44%):1H ЯМР (400 МГц, CDCl3) δ 8,79 (д, J=2,0 Гц, 1H), 7,97 (д, J=7,2 Гц, 1H), 7,49 (д, J=2,0 Гц, 1H), 7,04 (т, J=8,0 Гц, 1H), 6,81-6,75 (м, 1H), 6,75-6,71 (м, 1H), 6,20 (д, J=12,0 Гц, 1H), 5,32-5,26 (м, 1H), 4,87-4,77 (м, 1H), 3,75 (с, 3H), 1,67 (д, J=6,4 Гц, 3H), 1,37 (с, 9H); MS (ES+) m/z 460,0 (M - 99).
Стадия 2. Получение (S)-5-хлор-2-фтор-4-((1-(2-фтор-5-метоксифенил)этил)амино)-N-(тиазол-4-ил)бензолсульфонамида

К трет-бутил (S)-((5-хлор-2-фтор-4-((1-(2-фтор-5-метоксифенил)этил)амино)фенил)-сульфонил)(тиазол-4-ил)карбамату (0,150 г, 0,268 ммоль) добавляли 4 M раствор хлороводорода в этилацетате (5 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 30 минут. Смесь концентрировали in vacuo. Остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,088 г, выход 66%:1H ЯМР (400 МГц, CDCl3) δ 10,00 (с, 1H), 8,68 (д, J=2,0 Гц, 1H), 7,76 (д, J=7,2 Гц, 1H), 7,02 (т, J=9,2 Гц, 1H), 6,92 (д, J=2,0 Гц, 1H), 6,78-6,73 (м, 1H), 6,71 (дд, J=6,2, 3,2 Гц, 1H), 6,12 (д, J=12,0 Гц, 1H), 5,18 (д, J=5,2 Гц, 1H), 4,74 (к, J=6,4 Гц, 1H), 3,74 (с, 3H), 1,62 (д, J=6,8 Гц, 3H); MS (ES+) m/z 460,1 (M+1).
ПРИМЕР 332
Синтез (R)-5-хлор-2-фтор-N-(тиазол-2-ил)-4-((2,2,2-трифтор-1-(2-фторфенил)этил)амино)бензолсульфонамида

Стадия 1. Получение (R)-2-метил-N-(2,2,2-трифтор-1-(2-фторфенил)этилиден)пропан-2-сульфинамида

В раствор 2,2,2-трифтор-1-(2-фторфенил)этанона (8,0 г, 42 ммоль) и (R)-2-метилпропан-2-сульфинамида (6,3 г, 52 ммоль) в безводном простом диэтиловом эфире (300 мл) добавляли изопропоксид титана(IV) (29,5 г, 104 ммоль, 30,8 мл). Смесь перемешивали при 50°C в течение 24 ч и затем охлаждали до температуры окружающей среды. Неочищенное указанное в заголовке соединение использовали без дополнительной очистки.
Стадия 2. Получение (R)-2-метил-N-((R)-2,2,2-трифтор-1-(2-фторфенил)этил)пропан-2-сульфинамида

В раствор (R)-2-метил-N-(2,2,2-трифтор-1-(2-фторфенил)этилиден)пропан-2-сульфинамида (10,0 г, 33,8 ммоль) в безводном простом диэтиловом эфире (200 мл) частями добавляли борогидрид натрия (3,8 г, 101,5 ммоль) при -78°C. Смесь перемешивали при этой температуре в течение 3 ч и затем разводили солевым раствором (200 мл) при -78°C. После нагревания до температуры окружающей среды водную фазу экстрагировали этилацетатом (3×100 мл). Объединенные органические экстракты концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии, осуществляя элюирование с использованием 25% этилацетата в петролейном эфире, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (6,0 г, выход 60%, 94% de). Диастереомерный избыток определяли с помощью1H ЯМР:1H ЯМР (400 МГц, CDCl3) δ 7,35-7,50 (м, 2H), 7,18-7,25 (м, 1H), 7,14 (тд, J=9,2, 0,8 Гц, 1H), 5,25 (квин, J=6,8 Гц, 1H), 4,00 (д, J=5,2 Гц, 1H), 1,22 (с, 9H); MS (ES+) m/z 298,1 (M+1).
Стадия 3. Получение (R)-2,2,2-трифтор-1-(2-фторфенил)этан-1-амина

К (R)-2-метил-N-((R)-2,2,2-трифтор-1-(2-фторфенил)этил)пропан-2-сульфинамиду (1,5 г, 5,0 ммоль) добавляли в 4 M раствор хлороводорода в метаноле (10 мл) и получаемую смесь перемешивали при температуре окружающей среды в течение 4 ч. Реакционную смесь концентрировали in vacuo. Остаток разводили метанолом (1 мл) и очищали посредством перекристаллизации из простого трет-бутилметилового эфира (30 мл) для того, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,920 г, выход 94%):1H ЯМР (400 МГц, CD3OD) δ 7,59-7,77 (м, 2H), 7,28-7,50 (м, 2H), 5,63 (к, J=7,6 Гц, 1H), NH не наблюдали.
Стадия 4. Получение (R)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)-4-((2,2,2-трифтор-1-(2-фторфенил)этил)амино)бензолсульфонамида

В смесь (R)-2,2,2-трифтор-1-(2-фторфенил)этанамина (0,170 г, 0,880 ммоль) и 4-бром-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)бензолсульфонамида (0,4592 г, 0,8801 ммоль) в безводном толуоле (2,5 мл) добавляли трет-бутоксид калия (0,395 г, 3,51 ммоль) и аддукт хлорида (2-дициклогексилфосфино-2′,6′-диметокси-1,1′-бифенил)[2-(2-аминоэтилфенил)]палладия(II) и простого трет-бутилметилового эфира (0,067 г, 0,088 ммоль). Реакционную смесь дегазировали нагреванием до 80°C в течение 12 ч. Смесь разводили этилацетатом (40 мл) и водой (30 мл) и водную фазу промывали этилацетатом (3×40 мл). Объединенные органические фазы концентрировали in vacuo. Остаток очищали с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 25% этилацетата в петролейном эфире, чтобы получать указанное в заголовке соединение в виде масла желтого цвета (0,110 г, выход 20%): MS (ES+) m/z 634,1 (M+1), 636,1 (M+1).
Стадия 5. Получение (R)-5-хлор-2-фтор-N-(тиазол-2-ил)-4-((2,2,2-трифтор-1-(2-фторфенил)этил)амино)бензолсульфонамида

Следуя процедуре как описано для примера 327, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять 4-((2-(азетидин-1-илметил)бензил)амино)-3-хлор-N-(2,4-диметоксибензил)-2,6-дифтор-N-(1,2,4-тиадиазол-5-ил)бензолсульфонамид на (R)-5-хлор-N-(2,4-диметоксибензил)-2-фтор-N-(тиазол-2-ил)-4-((2,2,2-трифтор-1-(2-фторфенил)этил)амино)бензолсульфонамид, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,0369 г, выход 48%):1H ЯМР (400 МГц, CDCl3) δ 12,93 (ушир. с, 1H), 7,94 (д, J=7,2 Гц, 1H), 7,37-7,50 (м, 2H), 7,13-7,27 (м, 3H), 6,54 (д, J=4,4 Гц, 1H), 6,38 (д, J=11,6 Гц, 1H), 5,57 (д, J=8,3 Гц, 1H), 5,24-5,40 (м, 1H); MS (ES+) m/z 484,1 (M+1), 486,1 (M+1).
ПРИМЕР 333
Синтез формиата 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(пиридазин-3-ил)бензолсульфонамида

Стадия 1. Получение 2,4,6-трифтор-N-(пиридазин-3-ил)бензолсульфонамида

В раствор пиридазин-3-амина (10,0 г, 105 ммоль), пиридина (16,6 г, 210 ммоль, 17 мл) и 4-диметиламинопиридина (1,28 г, 10,5 ммоль) в дихлорметане (150 мл) добавляли 2,4,6-трифторбензол-1-сульфонилхлорид (29,1 г, 126 ммоль) по каплям. Смесь перемешивали при температуре окружающей среды в течение 12 ч. Смесь концентрировали in vacuo и остаток разводили водой (200 мл). Слои разделяли и водную фазу экстрагировали этилацетатом (3×200 мл). Объединенные органические экстракты промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 17 до 50% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде твердого вещества желтого цвета (1,50 г, выход 5%):1H ЯМР (400 МГц, CDCl3) δ 12,99 (ушир. с, 1H), 8,21 (дд, J=4,0, 1,6 Гц, 1H), 7,50-7,45 (м, 1H), 7,43-7,37 (м, 1H), 6,82-6,73 (м, 2H).
Стадия 2. Получение 2,4,6-трифтор-N-(пиридазин-3-ил)-N-((2-(триметилсилил)этокси)метил)бензолсульфонамида

В раствор 2,4,6-трифтор-N-(пиридазин-3-ил)бензолсульфонамида (1,50 г, 5,19 ммоль) и карбоната калия (1,43 г, 10,4 ммоль) в безводном N,N-диметилформамиде (20 мл) по каплям добавляли 2-(триметилсилил)этоксиметилхлорид (1,04 г, 6,22 ммоль) при 0°C. Смесь перемешивали при температуре окружающей среды в течение 1 ч. Смесь разводили водой (50 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические экстракты промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 17 до 25% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде твердого вещества желтого цвета (1,10 г, выход 51%):1H ЯМР (400 МГц, CDCl3) δ 8,42 (дд, J=9,6, 1,6 Гц, 1H), 8,14 (дд, J=4,0, 1,6 Гц, 1H), 7,43 (дд, J=9,6, 4,0 Гц, 1H), 6,81-6,72 (м, 2H), 5,69 (с, 2H), 3,78-3,68 (м, 2H), 0,98-0,90 (м, 2H), 0,00 (с, 9H); MS (ES+) m/z 420,3 (M+1).
Стадия 3. Получение 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(пиридазин-3-ил)-N-((2-(триметилсилил)этокси)метил)бензолсульфонамида

В раствор 2,4,6-трифтор-N-(пиридазин-3-ил)-N-((2-(триметилсилил)этокси)метил)бензолсульфонамида (1,00 г, 2,38 ммоль) и (2-(азетидин-1-илметил)-6-фторфенил)метанамина (0,74 г, 3,8 ммоль) в безводном N,N-диметилформамиде (15 мл) одной частью добавляли карбонат калия (0,66 г, 4,8 ммоль). Смесь перемешивали при температуре окружающей среды в течение 12 ч. Смесь разводили водой (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические экстракты промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали in vacuo. Очисткой с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,05% гидроксид аммония, в качестве элюента, получали указанное в заголовке соединение в виде масла желтого цвета (0,13 г, выход 9%):1H ЯМР (400 МГц, CDCl3) δ 8,39 (дд, J=9,6, 1,6 Гц, 1H), 8,05 (дд, J=4,0, 1,6 Гц, 1H), 7,92-7,53 (м, 1H), 7,36-7,31 (м, 1H), 7,29-7,22 (м, 1H), 7,11-7,04 (м, 2H), 6,25 (д, J=11,6 Гц, 2H), 5,70 (с, 2H), 4,36 (с, 2H), 3,81-3,73 (м, 2H), 3,63 (ушир. с, 2H), 3,32-3,19 (м, 4H), 2,22-2,11 (м, 2H), 1,02-0,93 (м, 2H), 0,00 (с, 9H); MS (ES+) m/z 594,4 (M+1).
Стадия 4. Получение формиата 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(пиридазин-3-ил)бензолсульфонамида

Твердый 4-((2-(азетидин-1-илметил)-6-фторбензил)амино)-2,6-дифтор-N-(пиридазин-3-ил)-N-((2-(триметилсилил)этокси)метил)бензолсульфонамид (0,12 г, 0,20 ммоль) растворяли в 4 M растворе хлороводорода в метаноле (12 мл) и перемешивали при температуре окружающей среды в течение 1 ч. После концентрирования in vacuo, очисткой с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,0924 г, выход 90%):1H ЯМР (400 МГц, CDCl3) δ 8,39 (ушир. с, 1H), 8,18 (дд, J=3,6, 1,6 Гц, 1H), 7,42-7,34 (м, 2H), 7,32-7,28 (м, 1H), 7,14-7,07 (м, 2H), 6,22 (д, J=12,0 Гц, 2H), 4,37 (с, 2H), 3,88 (с, 2H), 3,52 (т, J=7,2 Гц, 4H), 2,26 (квин, J=7,2 Гц, 2H), отсутствуют 1 сигнал COOH и 2 сигнала N-H;19F ЯМР (376,5 МГц, CDCl3) δ -108,0, -115,1; MS (ES+) m/z 464,1 (M+1).
ПРИМЕР 334
Синтез формиата 5-хлор-2-фтор-4-(((6-фтор-1H-индазол-7-ил)метил)амино)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 7-бром-6-фтор-1H-индазола
В раствор 3-бром-2,4-дифторбензальдегида (3,00 г, 13,6 ммоль) в 1,2-диметоксиэтане (20 мл) по каплям добавляли моногидрат гидразина (10,3 г, 206 ммоль). Смесь перемешивали при 100°C в течение 12 ч. После охлаждения до температуры окружающей среды, смесь концентрировали in vacuo, разводили водой (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические экстракты промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 9 до 25% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде твердого вещества желтого цвета (1,50 г, выход 51%):1H ЯМР (400 МГц, CDCl3) δ 10,14 (ушир. с, 1H), 8,08 (ушир. с, 1H), 7,59 (дд, J=8,8, 4,4 Гц, 1H), 6,96 (т, J=8,8 Гц, 1H); MS (ES+) m/z 215,1 (M+1), 217,1 (M+1).
Стадия 2. Получение 6-фтор-1H-индазол-7-карбонитрила
Смесь 7-бром-6-фтор-1H-индазола (1,50 г, 6,98 ммоль), тетракис[трифенилфосфин]палладия(0) (0,40 г, 0,349 ммоль) и цианида цинка (2,46 г, 20,9 ммоль, 1,33 мл) в безводном N,N-диметилформамиде (10 мл) нагревали до 120°C в течение 1 ч в микроволновом реакторе. После охлаждения до температуры окружающей среды, смесь разводили водой (100 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические экстракты промывали солевым раствором (100 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии с обращенной фазой, осуществляя элюирование с градиентом ацетонитрила в воде, содержащей 0,1% гидроксида аммония, получали указанное в заголовке соединение в виде твердого вещества желтого цвета (0,37 г, выход 33%):1H ЯМР (400 МГц, CDCl3) δ 8,26 (с, 1H), 8,01 (дд, J=8,8, 4,8 Гц, 1H), 7,09 (т, J=9,2 Гц, 1H), NH не наблюдали; MS (ES+) m/z 162,2 (M+1).
Стадия 3. Получение 6-фтор-1-((2-(триметилсилил)этокси)метил)-1H-индазол-7-карбонитрила

В раствор 6-фтор-1H-индазол-7-карбонитрила (0,37 г, 2,3 ммоль) и карбоната калия (0,636 г, 4,60 ммоль) в безводном N,N-диметилформамиде (8 мл) по каплям добавляли 2-(триметилсилил)этоксиметилхлорид (0,575 г, 3,45 ммоль) при 0°C. Смесь перемешивали при 0°C в течение 1 ч и затем разводили водой (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические экстракты промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 33% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного твердого вещества (0,27 г, выход 40%):1H ЯМР (400 МГц, CDCl3) δ 8,12 (с, 1H), 7,97 (дд, J=8,8, 4,8 Гц, 1H), 7,11 (т, J=9,2 Гц, 1H), 5,99 (с, 2H), 3,69-3,61 (м, 2H), 0,99-0,92 (м, 2H), 0,03 (с, 9H).
Стадия 4. Получение (6-фтор-1-((2-(триметилсилил)этокси)метил)-1H-индазол-7-ил)метанамина

В раствор 6-фтор-1-((2-(триметилсилил)этокси)метил)-1H-индазол-7-карбонитрила (0,13 г, 0,45 ммоль) в метаноле (8 мл) и гидроксиде аммония (2 мл) одной частью добавляли ренеевский никель (0,0038 г, 0,045 ммоль). Смесь перемешивали при температуре окружающей среды в атмосфере водорода (50 фунтов/дюйм2) в течение 12 ч. Смесь фильтровали и фильтрат концентрировали in vacuo, чтобы получать указанное в заголовке соединение в виде твердого вещества желтого цвета (0,13 г, выход 98%): MS (ES+) m/z 296,3 (M+1).
Стадия 5. Получение трет-бутил ((5-хлор-2-фтор-4-(((6-фтор-1-((2-(триметилсилил)этокси)метил)-1H-индазол-7-ил)метил)амино)фенил)сульфонил)(тиазол-4-ил)карбамата

В раствор (6-фтор-1-((2-(триметилсилил)этокси)метил)-1H-индазол-7-ил)метанамина (0,11 г, 0,37 ммоль) и трет-бутил (5-хлор-2,4-дифторфенил)сульфонил(тиазол-4-ил)карбамата (0,153 г, 0,372 ммоль) в безводном N,N-диметилформамиде (5 мл) добавляли карбонат калия (0,103 г, 0,745 ммоль) одной частью. Смесь перемешивали при температуре окружающей среды в течение 12 ч. Смесь разводили водой (50 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические экстракты промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали in vacuo. Очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 33% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного масла (0,10 г, выход 39%):1H ЯМР (400 МГц, CDCl3) δ 8,85 (д, J=2,0 Гц, 1H), 8,06 (с, 1H), 8,02 (д, J=7,2 Гц, 1H), 7,78 (дд, J=8,8, 5,2 Гц, 1H), 7,57 (д, J=2,0 Гц, 1H), 7,16-7,08 (м, 1H), 6,81 (д, J=12,0 Гц, 1H), 5,79 (с, 2H), 5,42 (ушир. с, 1H), 4,91 (ушир. д, J=4,0 Гц, 2H), 3,66-3,60 (м, 2H), 1,44 (с, 9H), 0,99-0,93 (м, 2H), 0,00 (с, 9H); MS (ES+) m/z 686,2 (M+1).
Стадия 6. Получение формиата 5-хлор-2-фтор-4-(((6-фтор-1H-индазол-7-ил)метил)амино)-N-(тиазол-4-ил)бензолсульфонамида

К трет-бутил ((5-хлор-2-фтор-4-(((6-фтор-1-((2-(триметилсилил)этокси)метил)-1H-индазол-7-ил)метил)амино)фенил)сульфонил)(тиазол-4-ил)карбамату (0,090 г, 0,13 ммоль) добавляли 4 M раствор хлороводорода в метаноле (9 мл), и смесь перемешивали при температуре окружающей среды в течение 12 ч. После концентрирования in vacuo, остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,0326 г, выход 49%):1H ЯМР (400 МГц, DMSO-d6) δ 13,34 (ушир. с, 1H), 8,76 (с, 1H), 8,30 (ушир. с, 0,5H), 8,11 (с, 1H), 7,74 (дд, J=8,8, 5,2 Гц, 1H), 7,55 (д, J=7,2 Гц, 1H), 7,02 (дд, J=10,0, 8,8 Гц, 1H), 6,97 (ушир. с, 1H), 6,68 (ушир. с, 1H), 6,62 (ушир. д, J=12,8 Гц, 1H), 4,71 (ушир. д, J=6,0 Гц, 2H); MS (ES+) m/z 456,0 (M+1).
ПРИМЕР 335
Синтез (R)-5-хлор-4-((1-(2,5-дифторфенил)-2,2-дифторэтил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение 1-(2,5-дифторфенил)-2,2-дифторэтан-1-она
В раствор N,N-диизопропиламина (5,85 г, 57,9 ммоль, 8,13 мл) в безводном тетрагидрофуране (100 мл) добавляли н-бутиллитий (2,5 M, 23,14 мл, 57,9 ммоль) при -70°C. После перемешивания в течение 30 минут, в смесь по каплям добавляли 1,4-дифторбензол (6,00 г, 52,6 ммоль, 5,13 мл) при -70°C. После перемешивания при -70°C в течение еще 30 минут, по каплям добавляли этил 2,2-дифторацетат (7,83 г, 63,11 ммоль). Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 1 ч. Смесь разводили водой (100 мл) и экстрагировали этилацетатом (2×100 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали, и фильтрат концентрировали in vacuo. Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 1 до 5% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного масла (5,00 г, выход 49%):1H ЯМР (400 МГц, CDCl3) δ 7,56 (ддд, J=8,2, 5,2, 3,2 Гц, 1H), 7,29 (тдд, J=8,8, 7,2, 3,6 Гц, 1H), 7,18-7,08 (м, 1H), 6,35 (тд, J=53,2, 2,8 Гц, 1H).
Стадия 2. Получение (R)-N-((R)-1-(2,5-дифторфенил)-2,2-дифторэтил)-2-метилпропансульфинамида

В раствор (R)-2-метилпропан-2-сульфинамида (4,73 г, 39,1 ммоль) и изопропоксида титана(IV) (14,79 г, 52,06 ммоль, 15,41 мл) в безводном простом диэтиловом эфире (100 мл) добавляли 1-(2,5-дифторфенил)-2,2-дифторэтанон (5,00 г, 26,0 ммоль) при температуре окружающей среды. Реакционную смесь нагревали с обратным холодильником в течение 1 ч. Затем смесь охлаждали до -70°C и частями добавляли борогидрид натрия (2,36 г, 62,5 ммоль). Получаемую смесь перемешивали при -70°C в течение 3 ч, разводили насыщенным солевым раствором (20 мл) и оставляли нагреваться до температуры окружающей среды. Получаемую суспензию фильтровали через пробку целита и фильтровальный осадок промывали этилацетатом (20 мл). Объединенный фильтрат промывали солевым раствором (20 мл). Водный слой экстрагировали этилацетатом (2×100 мл). Объединенные органические экстракты сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали in vacuo. Очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 9 до 33% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде бесцветного масла (3,00 г, выход 39%):1H ЯМР (400 МГц, CDCl3) δ 7,14-7,08 (м, 3H), 6,08 (т, J=55,2 Гц, 1H), 4,99 (ддт, J=12,0, 6,0, 3,6 Гц, 1H), 3,94 (ушир. д, J=5,6 Гц, 1H), 1,27 (с, 9H).
Стадия3. Получение (R)-1-(2,5-дифторфенил)-2,2-дифторэтан-1-амина

К (R)-N-((R)-1-(2,5-дифторфенил)-2,2-дифторэтил)-2-метилпропан-2-сульфинамиду (1,20 г, 4,04 ммоль) добавляли 4 M раствор хлороводорода в метаноле (12,01 мл) и смесь перемешивали при температуре окружающей среды в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,600 г, выход 77%):1H ЯМР (400 МГц, DMSO-d6) δ 9,52 (ушир. с, 2H), 7,86-7,70 (м, 1H), 7,53-7,34 (м, 2H), 6,61 (т, J=54,0 Гц, 1H), 5,18-4,96 (м, 1H); MS (ES+) m/z 194,0 (M+1).
Стадия 4. Получение трет-бутил (R)-((5-хлор-4-((1-(2,5-дифторфенил)-2,2-дифторэтил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата

В раствор трет-бутил (5-хлор-2,4-дифторфенил)сульфонил(тиазол-4-ил)карбамата (0,300 г, 0,730 ммоль) в безводном диметилсульфоксиде (5 мл) добавляли N,N-диизопропилэтиламин (0,319 мл, 1,83 ммоль) и (R)-1-(2,5-дифторфенил)-2,2-дифторэтанамин (0,423 г, 2,19 ммоль). Затем смесь нагревали до 70°C в течение 24 ч. Реакционную смесь разводили водой (10 мл) и экстрагировали этилацетатом (2×10 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 20% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде масла желтого цвета (0,080 г, выход 19%): MS (ES+) m/z 483,9 (M - 99), 485,9 (M - 99).
Стадия 5. Получение (R)-5-хлор-4-((1-(2,5-дифторфенил)-2,2-дифторэтил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида
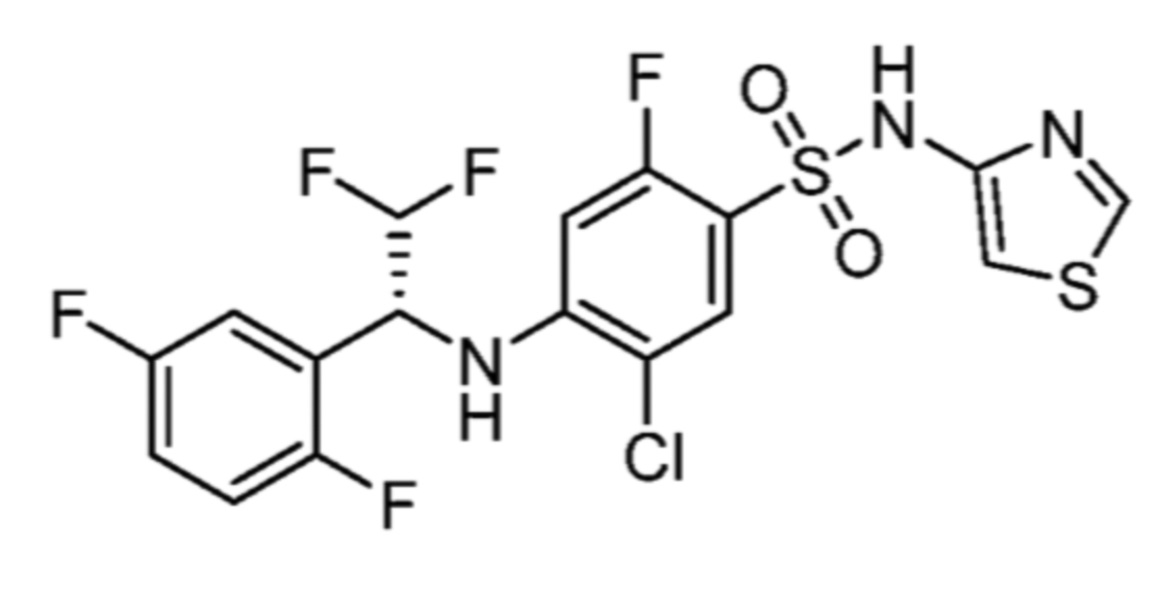
К трет-бутил (R)-((5-хлор-4-((1-(2,5-дифторфенил)-2,2-дифторэтил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамату (0,080 г, 0,137 ммоль) добавляли 4 M раствор хлороводорода в метаноле (5 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 30 минут. После концентрирования in vacuo, остаток очищали с помощью препаративной HPLC с обращенной фазой, используя ацетонитрил в воде, содержащей 0,225% муравьиную кислоту, в качестве элюента, чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,017 г, выход 26%):1H ЯМР (400 МГц, CD3OD) δ 8,70 (д, J=2,0 Гц, 1H), 8,51 (с, 1H), 7,75 (д, J=7,2 Гц, 1H), 7,31-7,22 (м, 2H), 7,22-7,13 (м, 1H), 6,92 (д, J=1,2 Гц, 1H), 6,55 (д, J=12,4 Гц, 1H), 6,33 (дт, J=55,2, 4,0 Гц, 1H), 5,31 (дт, J=11,8, 4,0 Гц, 1H), NH не наблюдали. MS (ES+) m/z 483,9 (M+1), 485,9 (M+1).
ПРИМЕР 336
Синтез (S)-5-хлор-4-((1-(2,5-дифторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил (S)-((5-хлор-4-((1-(2,5-дифторфенил)этил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамата
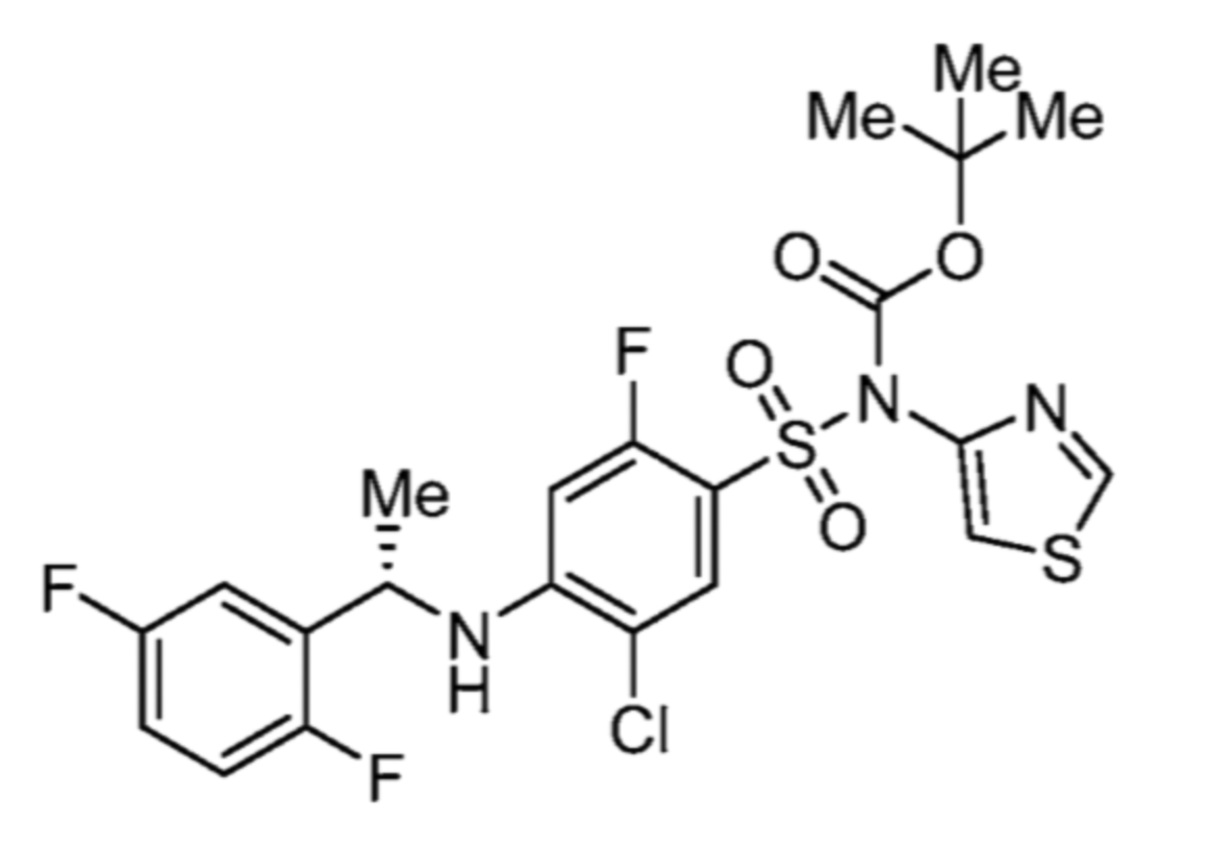
Следуя процедуре как описано для примера 5, стадии 1 и выполняя некритические вариации при необходимости, чтобы заменять гидрохлорид (S)-1-(5-хлор-2-фторфенил)этан-1-амина на гидрохлорид (S)-1-(2,5-дифторфенил)этан-1-амина, указанное в заголовке соединение получали в виде твердого вещества желтого цвета (0,800 мг, 50%): MS (ES+) m/z 447,9 (M - 99).
Стадия 2. Получение (S)-5-хлор-4-((1-(2,5-дифторфенил)этил)амино)-2-фтор-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано для примера 5, стадии 2 и выполняя некритические вариации при необходимости, чтобы заменять трет-бутил (S)-((5-хлор-4-((1-(5-хлор-2-фторфенил)этил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамат на трет-бутил (S)-((5-хлор-4-((1-(2,5-дифторфенил)этил)амино)-2-фторфенил)сульфонил)(тиазол-4-ил)карбамат, указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,153 мг, выход 61%):1H ЯМР (400 МГц, DMSO-d6) 8,84 (д, J=2,0 Гц, 1H), 7,62 (д, J=7,4 Гц, 1H), 7,32-7,22 (м, 2H), 7,18-7,10 (м, 1H), 6,90 (ушир. с, 1H), 6,61 (д, J=7,8 Гц, 1H), 6,39 (д, J=13,0 Гц, 1H), 4,93 (квин, J=6,8 Гц, 1H), 1,55 (д, J=6,8 Гц, 3H), NH не наблюдали; MS (ES+) m/z 447,9 (M+1).
ПРИМЕР 337
Синтез формиата 2,6-дифтор-4-((2-фтор-6-((3-фтор-3-метилазетидин-1-ил)метил)бензил)амино)-N-(изоксазол-3-ил)бензолсульфонамида

Стадия 1. Получение 2-фтор-6-((3-фтор-3-метилазетидин-1-ил)метил)бензонитрила

В раствор 2-(бромметил)-6-фторбензонитрила (0,35 г, 1,64 ммоль) и гидрохлорида 3-фтор-3-метилазетидина (0,27 г, 2,13 ммоль) в безводном N,N-диметилформамиде (10 мл) добавляли карбонат калия (0,90 г, 6,54 ммоль). Смесь перемешивали при температуре окружающей среды в течение 12 ч и затем разводили водой (30 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические экстракты промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с градиентом от 20 до 35% этилацетата в петролейном эфире, получали 2-фтор-6-((3-фтор-3-метилазетидин-1-ил)метил)бензонитрил в виде бесцветного масла (0,36 г, выход 99%):1H ЯМР (400 МГц, CDCl3) δ 7,48 (тд, J=8,2, 5,6 Гц, 1H), 7,24 (д, J=7,6 Гц, 1H), 7,03 (т, J=8,4 Гц, 1H), 3,80 (с, 2H), 3,40-3,23 (м, 4H), 1,64-1,54 (м, 3H); MS (ES+) m/z 223,3 (M+1).
Стадия 2. Получение (2-фтор-6-((3-фтор-3-метилазетидин-1-ил)метил)фенил)метанамина

В раствор 2-фтор-6-((3-фтор-3-метилазетидин-1-ил)метил)бензонитрила (0,31 г, 1,39 ммоль) в метаноле (20 мл) и гидроксиде аммония (4 мл) добавляли ренеевский никель (0,024 г, 0,28 ммоль). Смесь перемешивали в атмосфере водорода (50 фунтов/дюйм2) при температуре окружающей среды в течение 12 ч. Смесь фильтровали и фильтрат концентрировали in vacuo. Остаток очищали с помощью препаративной HPLC с обращенной фазой, используя градиент ацетонитрила в воде (содержащей карбонат аммония, 0,010 M), чтобы получать указанное в заголовке соединение в виде бесцветного масла (0,11 г, выход 35%):1H ЯМР (400 МГц, CDCl3) δ 7,14-7,07 (м, 1H), 6,98-6,91 (м, 2H), 3,84 (д, J=1,6 Гц, 2H), 3,66 (с, 2H), 3,31-3,13 (м, 4H), 1,58-1,48 (м, 3H), NH не наблюдали; MS (ES+) m/z 227,3 (M+1).
Стадия 3. Получение 2,4,6-трифтор-N-(изоксазол-3-ил)бензолсульфонамида

В смесь изоксазол-3-амина (10,00 г, 119 ммоль), пиридина (18,80 г, 238 ммоль) и N,N-диметилпиридин-4-амина (1,45 г, 11,9 ммоль) в безводном дихлорметане (100 мл) добавляли 2,4,6-трифторбензолсульфонилхлорид (32,90 г, 142,70 ммоль) и смесь перемешивали при температуре окружающей среды в течение 3 ч. Реакционную смесь гасили 1 M соляной кислотой (500 мл) и экстрагировали дихлорметаном (3×800 мл). Объединенные органические слои промывали солевым раствором (3×100 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью колоночной хроматографии, осуществляя элюирование с использованием от 10 до 100% этилацетата в петролейном эфире, получали указанное в заголовке соединение в виде твердого вещества желтого цвета (13,8 г, выход 42%):1H ЯМР (400 МГц, CDCl3) δ 8,18 (д, J=1,8 Гц, 1H), 6,71 (т, J=8,4 Гц, 2H), 6,53 (д, J=1,8 Гц, 1H), NH не наблюдали; MS (ES+) m/z 279,0 (M+1).
Стадия 4. Получение 2,4,6-трифтор-N-(изоксазол-3-ил)-N-(4-метоксибензил)-бензолсульфонамида

В раствор 2,4,6-трифтор-N-(изоксазол-3-ил)бензолсульфонамида (9,00 г, 32,40 ммоль) в безводном N,N-диметилформамиде (100 мл) добавляли бикарбонат натрия (13,06 г, 155,48 ммоль) и 4-метоксибензилхлорид (6,59 г, 42,10 ммоль). Смесь нагревали до 40°C в течение 4 ч и затем разводили водой (500 мл) и экстрагировали этилацетатом (3×500 мл). Объединенные органические слои промывали солевым раствором (3×100 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата под давлением получали остаток, который очищали с помощью колоночной хроматографии, осуществляя элюирование с использованием от 10 до 33% этилацетата в петролейном эфире. Дополнительной очисткой растиранием с метанолом (30 мл) получали указанное в заголовке соединение в виде бесцветного твердого вещества (10,0 г, выход 77%):1H ЯМР (400 МГц, CDCl3) δ 8,13 (д, J=1,8 Гц, 1H), 7,31 (д, J=8,8 Гц, 2H), 6,81-6,63 (м, 4H), 6,54 (д, J=1,8 Гц, 1H), 5,02 (с, 2H), 3,69 (с, 3H); MS (ES+) m/z 421,0 (M+23).
Стадия 5. Получение 2,6-дифтор-4-((2-фтор-6-((3-фтор-3-метилазетидин-1-ил)метил)бензил)амино)-N-(изоксазол-3-ил)-N-(4-метоксибензил)бензолсульфонамида

В раствор (2-фтор-6-((3-фтор-3-метилазетидин-1-ил) метил)фенил)метанамина (0,090 г, 0,40 ммоль) и 2,4,6-трифтор-N-(изоксазол-3-ил)-N-(4-метоксибензил)бензолсульфонамида (0,16 г, 0,40 ммоль) в безводном N,N-диметилформамиде (5 мл) добавляли карбонат калия (0,11 г, 0,80 ммоль). Смесь перемешивали при температуре окружающей среды в течение 12 ч, затем разводили водой (50 мл) и экстрагировали этилацетатом (3×40 мл). Объединенные органические экстракты промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo и очисткой остатка с помощью препаративной тонкослойной хроматографии, осуществляя элюирование с использованием 33% петролейного эфира в этилацетате, получали указанное в заголовке соединение в виде бесцветного масла (0,11 г, выход 46%):1H ЯМР (400 МГц, CDCl3) δ 8,15 (д, J=1,6 Гц, 1H), 7,38 (д, J=8,4 Гц, 2H), 7,26-7,18 (м, 1H), 7,11-7,01 (м, 2H), 6,80 (д, J=8,4 Гц, 2H), 6,66 (д, J=1,6 Гц, 1H), 6,18 (д, J=12,2 Гц, 2H), 5,05 (с, 2H), 4,34 (ушир. с, 2H), 3,75 (с, 3H), 3,70 (с, 2H), 3,37-3,17 (м, 4H), 1,63-1,52 (м, 3H), NH не наблюдали; MS (ES+) m/z 605,3 (M+1).
Стадия 6. Получение формиата 2,6-дифтор-4-((2-фтор-6-((3-фтор-3-метилазетидин-1-ил)метил)бензил)амино)-N-(изоксазол-3-ил)бензолсульфонамида

В раствор 2,6-дифтор-4-((2-фтор-6-((3-фтор-3-метилазетидин-1-ил)метил)бензил)амино)-N-(изоксазол-3-ил)-N-(4-метоксибензил)бензолсульфонамида (0,11 г, 0,18 ммоль) в дихлорметане (3 мл) добавляли трифторуксусную кислоту (3 мл). Смесь перемешивали при температуре окружающей среды в течение 12 ч и затем концентрировали in vacuo. Остаток очищали с помощью препаративной HPLC с обращенной фазой, осуществляя элюирование с градиентом ацетонитрила в воде (содержащей 0,22% муравьиную кислоту), чтобы получать указанное в заголовке соединение в виде бесцветного твердого вещества (0,045 г, выход 47%):1H ЯМР (400 МГц, CD3OD) δ 8,45 (д, J=1,6 Гц, 1H), 8,14 (ушир. с, 1H), 7,34 (тд, J=8,0, 5,6 Гц, 1H), 7,19 (д, J=7,2 Гц, 1H), 7,11 (т, J=9,2 Гц, 1H), 6,43 (д, J=1,6 Гц, 1H), 6,37-6,30 (м, 2H), 4,42 (д, J=1,2 Гц, 2H), 3,83 (с, 2H), 3,40 (с, 2H), 3,37-3,35 (м, 2H), 1,60-1,53 (м, 3H), способные к обмену протоны не наблюдали; MS (ES+) m/z 485,0 (M+1).
ПРИМЕР 338
Синтез (S)-5-хлор-2-фтор-4-(1-(2-фторфенил)пропокси)-N-(тиазол-4-ил)бензолсульфонамида

Стадия 1. Получение трет-бутил (S)-((5-хлор-2-фтор-4-(1-(2-фторфенил)пропокси)фенил)сульфонил)(тиазол-4-ил)карбамата

В смесь (S)-1-(2-фторфенил)пропан-1-ола (0,070 г, 0,45 ммоль) и трет-бутил (5-хлор-2,4-дифторфенил)сульфонил(тиазол-4-ил)карбамата (0,22 г, 0,54 ммоль) в безводном диметилсульфоксиде (3 мл) добавляли карбонат цезия (0,30 г, 0,91 ммоль). Смесь перемешивали при температуре окружающей среды в течение 4 ч и затем выливали на воду со льдом (30 мл). Смесь экстрагировали этилацетатом (3×30 мл). Объединенные органические экстракты промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия и фильтровали. Концентрированием фильтрата in vacuo получали указанное в заголовке соединение в виде масла желтого цвета (0,30 г, количественный выход): MS (ES+) m/z 545,2 (M+1).
Стадия 2. Получение (S)-5-хлор-2-фтор-4-(1-(2-фторфенил)пропокси)-N-(тиазол-4-ил)бензолсульфонамида

Следуя процедуре как описано в примере 145, стадии 5 и выполняя некритические вариации, чтобы заменять трет-бутил (2-(азетидин-1-илметил)-6-фторбензил)(4-(N-(2,4-диметоксибензил)-N-(пиримидин-2-ил)сульфамоил)-3,5-дифторфенил)карбамат на трет-бутил (S)-((5-хлор-2-фтор-4-(1-(2-фторфенил)пропокси)фенил)сульфонил)(тиазол-4-ил)карбамат, очисткой с помощью препаративной HPLC с обращенной фазой, используя градиент ацетонитрила в воде (содержащей 0,225% муравьиную кислоту), указанное в заголовке соединение получали в виде бесцветного твердого вещества (0,069 г, выход 30%):1H ЯМР (400 МГц, CDCl3) δ 9,33 (с, 1H), 8,63 (д, J=2,4 Гц, 1H), 7,87 (д, J=7,6 Гц, 1H), 7,34-7,30 (м, 2H), 7,14 (д, J=7,6 Гц, 2H), 6,98 (д, J=2,0 Гц, 1H), 6,54 (д, J=11,6 Гц, 1H), 5,46 (т, J=6,4 Гц, 1H), 2,16 (к, J=7,2 Гц, 1H), 2,01 (к, J=7,2 Гц, 1H), 1,06 (т, J=7,2 Гц, 3H); MS (ES+) m/z 445,1 (M+1).
БИОЛОГИЧЕСКИЕ АНАЛИЗЫ
В данной области известны различные приемы тестирования активности соединения по изобретению или определения его растворимости в известных фармацевтически приемлемых эксципиентах. Для того чтобы изобретение, описанное в настоящем описании, можно было понять более полно, изложены следующие биологические анализы. Следует понимать, что эти примеры служат лишь иллюстративным целям, и их не следует толковать в качестве ограничения этого изобретения каким-либо образом.
БИОЛОГИЧЕСКИЙ ПРИМЕР 1
Электрофизиологический анализ (анализ in vitro)
Электрофизиология с локальной фиксацией потенциала допускает прямое измерение и количественное определение блокирования потенциалзависимых натриевых каналов (NaV) и допускает определение зависимости блокирования от времени и потенциала, которую интерпретируют как дифференциальное связывание с покоящимся, открытым и неактивированным состояниями натриевого канала (Hille, B., Journal of General Physiology (1977), 69: 497-515).
Следующие электрофизиологические исследования с локальной фиксацией потенциала осуществляли на репрезентативных соединениях по изобретению, используя эмбриональные клетки почки человека (HEK), перманентно трансфицированные экспрессирующим вектором, содержащим полноразмерную кДНК, кодирующую желаемую α-субъединицу натриевого канала человека, которые растили в культуральных средах, содержащих 10% FBS, 1% PSG и 0,5 мг/мл G418 при 37°C с 5% CO2. Клетки HEK, используемые для электрофизиологической (EP) регистрации, имели количество пассажей меньше 40 для всех исследований, и их использовали в пределах трех суток от высевания. Клетки HEK-293 стабильно экспрессировали кДНК NaV1.1, NaV1.5 и NaV1.6 (NM_001165964 (SCN1A), NM_000335 (SCN5A) и NM_014191 (SCN8A), соответственно).
Натриевые токи измеряли с использованием приема локальной фиксации потенциала в конфигурации целой клетки, используя или автоматизированную фиксацию потенциала PatchXpress или вручную с использованием усилителя Axopatch 200B (Axon Instruments) или Model 2400 (A-M Systems). Ручной протокол фиксации потенциала представлял собой следующее: микропипетки из боросиликатного стекла полировали огнем до диаметра кончика, дающего сопротивление 2-4 МОм в рабочих растворах. Пипетку заполняли раствором, состоявшим из: 5 мМ NaCl, 10 мМ CsCl, 120 мМ CsF, 0,1 мМ CaCl2, 2 мМ MgCl2, 10 мМ HEPES, 10 мМ EGTA; и корректировали до pH 7,2 с использованием CsOH. Внешний раствор имел следующую композицию: 140 мМ NaCl, 5 мМ KCl, 2 мМ CaCl2, 1 мМ MgCl2, 10 мМ HEPES; и корректировали до pH 7,4 с использованием NaOH. В некоторых исследованиях внешний натрий снижали посредством эквимолярной замены на холин. Осмолярность во внутреннем CsF и внешнем NaCl растворах корректировали до 300 мОсм/кг и 310 мОсм/кг с использованием глюкозы, соответственно. Всю регистрацию осуществляли при температуре окружающей среды в камере с ванной объемом 150 мкл. Контрольные натриевые токи измеряли в 0,5% DMSO. Контроли и репрезентативные соединения по изобретению вносили в ристрационную камеру через систему клапанов с 4 или 8 зажимамии для перфузии ванны производства ALA Scientific Instruments.
Токи регистрировали на частоте дискретизации40 кГц, фильтровали на 5 Гц и хранили с использованием Digidata-1322A аналогово-цифрового интерфейса с использованием программного обеспечения pClamp (Axon Instruments). Применяли компенсацию последовательного сопротивления (60-80%). Клетки не использовали, если токи демонстрировали недостаточный контроль напряжения (как судили по IV зависимости в ходе ступенчатой активации). Всю статистику в этом исследовании приводили в виде среднего±SD.
Мембранный потенциал поддерживали при напряжении, при котором инактивация канала является полной. Затем напряжение ступенчато меняли обратно к очень отрицательному напряжению (Vудерж.= 150 мВ) на 20 мс и затем применяли тестовый импульс для того, чтобы количественно определять блокирование соединением. 20 мс короткая реполяризация была достаточно длинной для каналов без соединений, чтобы полностью восстанавливаться от быстрой инактивации, но каналы, связанные с соединениями, восстанавливались более медленно, так что в течение этого интервала могло возникать незначительное восстановление. Процент снижения натриевого тока после намыва соединения брали в качестве процента блокирования натриевых каналов.
Репрезентативные соединения по изобретению, когда их тестировали в этом анализе, демонстрировали IC50 как изложено далее в таблице 1.
БИОЛОГИЧЕСКИЙ ПРИМЕР 2
Анализ входящего натриевого тока (анализ in vitro)
В этом анализе входящего натриевого тока используют проникающий в клетки натрий-чувствительный краситель ANG2 для того, чтобы количественно определять входящий ток ионов натрия через натриевые каналы, которые поддерживают в открытом состоянии, используя модуляторы натриевых каналов. Этот анализ входящего натриевого тока высокой пропускной способности делает возможным быстрое профилирование и определение характеристик блокаторов натриевых каналов.
В целом, клетки Trex HEK293 стабильно трансфицировали индуцибельным экспрессирующим вектором, содержащим полноразмерную кДНК, кодирующую желаемую α-субъединицу натриевого канала человека, и экспрессирующим вектором, содержащим полноразмерную кДНК, кодирующую β1-субъединицу. Клеточные линии, экспрессирующие натриевые каналы, индуцировали тетрациклином (1 мкг/мл) и высевали на 384-луночные покрытые PDL планшеты с плотностью 25 30 тыс. клеток/лунка в культуральных средах (DMEM, содержащая 10% FBS и 1% L глутамин). После инкубации в течение ночи (37°C, 5% CO2), среды для культивирования удаляли и клетки нагружали 5 мкМ красителя ANG2 в течение 1-1,5 ч в буфере 1 (155 мМ NMDG, 5 мМ KCl, 2 мМ CaCl2, 1 мМ MgCl2, 10 мМ HEPES, 10 мМ глюкоза, pH корректировали до 7,4 с использованием Tris). Доступный краситель удаляли, а клетки инкубировали с тестируемыми соединениями в течение 1 ч в буфере 1, содержащем модулятор(ы) натриевых каналов, при комнатной температуре. Hamamatsu FDSS μCell использовали для того, чтобы осуществлять 1:1 добавление Na/K стимулирующий буфер (140 мМ NaCl, 20 мМ HEPES, 1 мМ CaCl2, 15 мМ KCl, 1 мМ MgCl2, 10 мМ глюкоза, pH корректировали до 7,4 с использованием Tris) и одновременно считывали планшеты на длине волны возбуждения 530 нм и длине волны испускания 558 нм. Процент ингибирования входящего тока ионов натрия вычисляли для каждого тестируемого соединения в каждой тестируемой концентрации для того, чтобы определять значения IC50.
Репрезентативные соединения по изобретению при тестировании в этой модели демонстрировали аффинности для инактивированного состояния NaV1.6, NaV1.5 и NaV1.1, как изложено далее в таблице 1.
Номера примеров, приведенные в таблице 1, соответствуют примерам в настоящем описании, «Ток» относится к анализу входящего натриевого тока и «EP» относится к электрофизиологическому анализу:
Таблица 1: ингибирование NaV1.1, NaV1.5 и NaV1.6
БИОЛОГИЧЕСКИЙ ПРИМЕР 3
Анализ пароксизмов при электрической стимуляции
Для того чтобы идентифицировать соединения с антиконвульсивной активностью, т. е. те, которые повышают порог пароксизмов, использовали многие пароксизмальные тесты с электрической стимуляцией. Два примера анализов пароксизмов при электрической стимуляции, часто исползуемых в данной области, представляют собой 6 Гц психомоторный пароксизмальный анализ (6 Гц) и максимальные электрошоковые пароксизмы (MES). 6 Гц анализ считают моделью частичных пароксизмов, наблюдаемых у человека (Löscher, W. and Schmidt, D., Epilepsy Res. (1988), том 2, стр. 145-81; Barton, M.E. et al., Epilepsy Res. (2001), том 47, стр. 217-27). Анализ MES является моделью генерализованных тонико-клонических пароксизмов у человека и дает указание на способность соединения предотвращать распространение пароксизмов, когда все нейронные цепи в головном мозге максимально активны. Эти пароксизмы обладат высокой воспроизводимостью и электрофизиологически соответствуют пароксизмам человека (Toman et al., 1946; Piredda et al., 1984; White et al., 1995). Эксперименты можно осуществлять на здоровых животных или на животных, подверженных пароксизмам, которых генетически модифицировали для того, чтобы моделировать генетические эпилептические синдромы (Piredda, S. G. et al., J. Pharmacol. Exp. Ther. (1985), том 232, стр. 741-5; Toman, J.E. et al., J. Neurophysiol. (1946), том 9, стр. 231-9; и White, H.S. et al., Ital. J. Neurol. Sci. (1995), том 16 (1-2), стр. 73-7).
Для того чтобы содействовать тестированию, мышей можно предварительно лечить тестируемым соединением или подходящим носителем перед применением электрошока. Каждую группу лечения (n=4-8 мышей/группа) исследуют на противосудорожные эффекты в различные моменты времени после введения соединения и носителя. Сначала глаза мышей анестезируют топическим нанесением алкаина (гидрохлорид пропаракаина) 0,5%, по одной капле в каждый глаз за 30 мин до стимуляции. Затем пароксизмы индуцируют, помещая электроды на глаза, которые доставляют ток через роговицу.
6 Гц психомоторный пароксизмальный тест:
После предварительного лечения, каждую мышь стимулируют низкочастотной (6 Гц, ширина импульса 0,3 мс) стимуляцией в течение 3 с, доставляемой через электроды на роговице с несколькими интенсивностями (12-44 мА). Животных удерживают вручную и выпускают сразу после стимуляции и наблюдают на присутствие или отсутствие пароксизмальной активности. Обычно, 6 Гц стимуляция ведет к пароксизму, который отличает минимальная клоническая фаза, за которой следует стереотипное автоматическое поведение, включая судорожные сокращения вибрисс и хвост Штрауба, или генерализованный тонико-клонический пароксизм. Осуществляют мониторинг присутствия, типа и латентности пароксизма (в секундах) после подачи тока. Животных, которые не проявляют клонический или генерализованный тонико-клонический пароксизм, считают «защищенными». Всех животных умерщвляют в конце анализа. Берут образцы плазмы и головного мозга.
Максимальный электрошоковый тест (MES):
После предовартельного лечения, каждую мышь стимулирут переменным током (60 Гц, ширина импульса 0,4-0,6 мс) в течение 0,2-0,5 с, который доставляют через электроды на роговице с интенсивностями (44-55 мА).
Обычно MES стимуляция ведет к генерализовнному тоническому пароксизму, за которым может следовать клонический пароксизм, автоматическое поведение и Straub-tail. Осуществляют мониторинг присутствия, типа и задержки до пароксизма (в секундах) после подачи тока. Животное считают «защищенным» от MES-индуцированных пароксизмов при избавлении от тонического компонента пароксизма в разгибателях задних конечностей. После пароксизма ожидают, что мыши восстанавливают нормальное исследующее поведение в пределах от 1 до 4 минут. Задержку до пароксизма регистрируют с порогом 1 минута, после чего всех животных умерщвляют.
* * * * *
Все патенты США, публикации патентных заявок США, патентные заявки США, иностранные патенты, иностранные патентные заявки и непатентные публикации, упоминаемые в этом описании, включены в настоящее описание посредством ссылки во всей их полноте.
Несмотря на то, что вышеуказанное изобретение описано в некоторых подробностях, чтобы содействовать пониманию, очевидно, что на практике можно осуществлять определенные изменения и модификации в пределах объема приложенной формулы изобретения. Соответственно, описанные варианты осуществления следует рассматривать как иллюстративные и не ограничивающие, и изобретение не подлежит ограничению подробностями, приведенными в настоящем описании, и его можно модифицировать в пределах объема и эквивалентов приложенной формулы изобретения.
Реферат
Изобретение относится к соединениям, выбранным из: 4-((2-(азетидин-1-илметил)бензил)амино)-5-хлор-2-фтор-N-(тиазол-4-ил)бензолсульфонамида; 4-((2-((2-окса-6-азаспиро[3.3]гептан-6-ил)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида; 4-((2-((3-азабицикло[3.1.0]гексан-3-ил)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида; 4-((2-((трет-бутил(метил)амино)метил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида; 4-((2-(азетидин-1-илметил)бензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида; (S)-4-((2-(1-(азетидин-1-ил)этил)-6-фторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида; 4-((2-(азетидин-1-илметил)-3,6-дифторбензил)амино)-2,6-дифтор-N-(тиазол-4-ил)бензолсульфонамида; 2,3-дифтор-4-((2-фтор-6-(пирролидин-1-илметил)бензил)амино)-N-(изоксазол-3-ил)бензолсульфонамида; 4-((2-((2,2-диметил азетидин-1-ил)метил)бензил)амино)-2,6-дифтор-3-метил-N-(тиазол-4-ил)бензолсульфонамида и 2,6-дифтор-4-((2-фтор-6-((3-фтор-3-метилазетидин-1-ил)метил)бензил)амино)-N-(изоксазол-3-ил)бензолсульфонамида или их фармацевтически приемлемым солям или сольватам, а также к фармацевтическим композициям и применению соединений для лечения заболевания или состояния, ассоциированного с активностью Nav1.6. Технический результат: получены новые соединения, которые могут быть применимы для лечения заболевания или состояния, ассоциированного с активностью Nav1.6. 3 н. и 10 з.п. ф-лы, 1 табл., 338 пр.
Формула
Документы, цитированные в отчёте о поиске
Биарильные простоэфирные сульфонамиды и их применение в качестве терапевтических средств
Комментарии