Бициклические арильные монобактамовые соединения и способы их применения для лечения бактериальных инфекций - RU2733402C2
Код документа: RU2733402C2
Описание
Область изобретения
Настоящее изобретение относится к новым бициклическим арильным монобактамовым соединениям, способам их получения и их применению в качестве терапевтических средств. Более конкретно, изобретение относится к бициклическим арильным монобактамовым соединениям и их применению в качестве антибиотиков для лечения бактериальных инфекций.
Уровень техники изобретения
Использование антибиотиков для лечения бактериальных инфекций является одним из величайших достижений медицины 20 века. Однако, за последние десятилетия бактерии, устойчивые к разнообразным антибиотикам, начали появляться во всем мире, угрожая эффективности антибактериальной терапии. Только в Соединенных Штатах, по меньшей мере, 23000 человек ежегодно умирают в результате инфекций, вызываемых бактериями, устойчивыми к антибиотикам, и многие другие умирают от ранее существовавших заболеваний, усугубляемых подобными инфекциями. Antibiotic Resistance Threats in the United States, 2013, Centers for Disease Control, Atlanta, Georgia. Новые антибиотики необходимы для борьбы с существующей и будущей угрозой бактерий с множественной лекарственной устойчивостью.
β-лактамы являются наиболее широко используемыми антибиотиками для лечения серьезных бактериальных инфекций. К ним относятся карбапенемы, цефалоспорины, пенициллины и монобактамы. Как было отмечено для других классов антибиотиков, появилась резистентность к β-лактамам. Для большинства грамотрицательных бактерий эта устойчивость в первую очередь обусловлена экспрессией β-лактамаз, ферментов, которые гидролизуют β-лактамные соединения. Существует 4 различных класса β-лактамаз (A, B, C и D), способных гидролизовать перекрывающиеся, но различные подмножества β-лактамов (Drawz and Bonomo, Clin. Micro. Rev., 2010, 23:160-201). В то время как β-лактамазы класса B, также известные как металло-β-лактамазы (MBL), не являются наиболее распространенными β-лактамазами, обнаруженными в клинике, частота и распределение их экспрессии возрастает и представляет значительную медицинскую угрозу, поскольку (i) MBL обладают способностью гидролизовать все β-лактамы, кроме монобактамов, и (ii) в отличие от β-лактамаз класса A и C, для MBL нет доступных ингибиторов.
Азтреонам, монобактам, был впервые одобрен в США в 1986 году для лечения аэробных грамотрицательных бактериальных инфекций и остается единственным монобактамом, который используется в настоящее время в США. Однако, азтреонам обладает слабой активностью в отношении штаммов Pseudomonas и Acinetobacter. Поскольку монобактамы по своей природе устойчивы к гидролизу MBL, несколько компаний начали разработку новых монобактамовых соединений для лечения инфекций, вызванных грамотрицательными бактериями. Монобактамовые соединения, содержащие фрагмент сидерофора, описаны в WO 2007/065288, WO2012/073138, J. Medicinal Chemistry 56: 5541-5552 (2013), и Bioorganic and Medicinal Chemstry Letters 22:5989 (2012).
В опубликованной патентной заявке США No. US 2014/0275007 раскрыты оксамазин монобактамы и их использование в качестве антибактериальных средств, и в опубликованной патентной заявке США No. US 2015/0266867 также раскрыты новые монобактамовые соединения для использования в качестве антибактериальных средств. В публикации Международной заявки на патент No. WO 2013/110643 описаны новые амидинзамещенные монобактамовые производные и их применение в качестве противомикробных средств.
Остается потребность в новых антибиотиках для преодоления множественной лекарственной устойчивости. Соединения, раскрытые в настоящем изобретении, предназначены для заполнения этой медицинской потребности посредством введения либо отдельно, либо в сочетании с одним или несколькими подходящими ингибиторами β-лактамазы.
Сущность изобретения
Изобретение относится к разработке и синтезу ряда бициклических арильных монобактамовых аналогов, нового класса высокоэффективных антибиотиков, эффективных против широкого ряда грамотрицательных бактерий. Эти соединения и их фармацевтически приемлемые соли могут быть полезны в качестве терапевтических средств для клинического лечения различных инфекций, вызванных грамотрицательными бактериями, включая штаммы, обладающие множественной лекарственной устойчивостью. Соединения могут использоваться отдельно или в комбинации с одним или несколькими подходящими ингибиторами β-лактамазы. Более конкретно, настоящее изобретение включает соединения формулы I:

или их фармацевтически приемлемую соль, где:
W представляет собой связь или O;
RХ и RZнезависимо представляют собой водород, -SC1-C3алкил, C1-C3 алкил, -(C1-C3алкилен)nOC1-C3алкил или -(C1-C3алкилен)nNC1-C3алкил, где указанный -SC1-C3алкил, C1-C3 алкил, -(C1-C3алкилен)nOC1-C3алкил и -(C1-C3алкилен)nNC1-C3алкил необязательно замещены одним-семью фторами;
или, альтернативно, Rx и Rz, вместе с углеродом, к которому они присоединены, образуют моноциклический C4-C7 циклоалкил или моноциклический C4-C7 гетероциклоалкил с 1, 2 или 3 гетероатомами в качестве кольцевых атомов, независимо выбранными из N, O и S, где указанный C4-C7 циклоалкил и C4-C7 гетероциклоалкил необязательно замещены одним-тремя заместителями, независимо выбранными из -F, -OH и -OC1-C3алкила;
X представляет собой N или CR1;
R1 представляет собой водород, C1-C3 алкил, или галоген; где указанный C1-C3 алкил необязательно замещен одним-тремя Ra;
в каждом случае Ra независимо представляет собой водород, галоген, C1-C3алкил, - NRcRd, -ORe или -C(O)NRcRd;
Z представляет собой C1-C3 алкилен, необязательно замещенный одним-тремя Rb;
в каждом случае Rb независимо представляет собой -C1-C6 алкил, -C3-C7 циклоалкил, -C(O)ORe, -C(O)NRcRd, тетразолил, оксадиазолонил, HetA, AryA, -S(O)mRe, -S(O)mNRcRd, или P(O)(Re)p, где указанный -C1-C6 алкил и -C3-C7 циклоалкил необязательно замещены одним-тремя Ra и где указанный AryA и HetA необязательно замещены одним-четырьмя R4;
AryA представляет собой 5- или 6-членное моноциклическое ароматическое кольцо с 0, 1, 2 или 3 гетероатомами в качестве кольцевых атомов, независимо выбранными из N, N в виде четвертичной соли, O и S;
HetA представляет собой 4-6-членное насыщенное или мононенасыщенное моноциклическое кольцо с 1, 2 или 3 гетероатомами в качестве кольцевых атомов, независимо выбранными из N, NH, N в виде четвертичной соли, O и S;
Y представляет собой связь, O, NR2, S или CH2;
R2 представляет собой водород, -C1-C3 алкил, -C(O)Re,-C(O)NRcRd, -S(O)mRe или -S(O)mNRcRd, где указанный -C1-C3 алкил необязательно замещен одним-тремя Ra;
A1 представляет собой 9-11-членное бициклическое ароматическое кольцо с 0, 1, 2, 3 или 4 гетероатомами в качестве кольцевых атомов, независимо выбранными из N, NH, N в виде четвертичной соли, O и S, необязательно замещенное одним-четырьмя R4;
в каждом случае R4 независимо представляет собой:
(a) -C1-C6 алкил,
(b) -C2-C6 алкенил,
(c) -C2-C6 алкинил,
(d) галоген,
(e) -ORe,
(f) -S(O)mRe,
(g) -S(O)mNRcRd,
(h) -C(O)Re,
(i) -OC(O)Re,
(j) -C(O)ORe,
(k) -CN,
(l) -C(O)NRcRd,
(m) -NRcRd,
(n) -NRcC(O)Re,
(o) -NRcC(O)ORe,
(p) -NRcC(O)NRcRd,
(q) -NRcS(O)mRe,
(r) =NH,
(s) -CF3,
(t) -OCF3,
(u) -OCHF2,
(v) -C3-C6циклоалкил,
(w) -O-C3-C6циклоалкил,
(x) -C1-C3алкилен-C3-C6циклоалкил,
(y) -O-C1-C3алкилен-C3-C6циклоалкил,
(z) HetA,
(aa) -O-HetA,
(bb) - C1-C3алкилен-HetA,
(cc) -O- C1-C3алкилен-HetA,
(dd) AryA,
(ee) -O-AryA,
(ff) -C1-C3алкилен-AryA, или
(gg) -O-C1-C3алкилен-AryA,
где указанный C1-C6 алкил, -C2-C6 алкенил, -C2-C6 алкинил, -C3-C6циклоалкил, -O-C3-C6циклоалкил, -C1-C3алкилен-C3-C6циклоалкил, -O-C1-C3алкилен-C3-C6циклоалкил, HetA, O-HetA, -C1-C3алкилен-HetA, -O-C1-C3алкилен-HetA, AryA, -O-AryA, -C1-C3алкилен-AryA и -O-C1-C3алкилен-AryA необязательно замещены одним-тремя Ra;
L представляет собой связь, -O-, -C1-C6алкилен-, -NHC(O)-, -C(O)-, -C(=NH)-, -S(O)m-, -SC1-C6алкилен-, -NR3(CH2)n-, -NHC(=NH)- или -NHS(O)m-, где -C1-C6алкилен-, -NHC(O)-, -C(=NH)-, -SC1-C6алкилен-, -NR3(CH2)n-, -NHC(=NH)- и -NHS(O)m-, необязательно замещены одним-четырьмя R7;
R3 представляет собой водород или -C1-C3 алкил;
M представляет собой -CH2OH, N(R3)2, N+(C1-C3алкил)3, C2-C6алкил, C3-C7 циклоалкил, HetA или AryA, где -CH2OH, N(R3)2, N+(C1-C3алкил)3, C2-C6алкил, C3-C7 циклоалкил, HetA и AryA необязательно замещены одним-четырьмя R6;
в каждом случае R6 независимо выбран из группы, состоящей из: галогена, -C1-C6алкила, -(CH2)nNRcRd, -(CH2)qORe, -S(O)mRe, -S(O)mNRcRd, -C(O)Re,-OC(O)Re,-C(O)ORe, -CN, -C(O)NRcRd, -C(NH)NRcRd, -NRcRd, -N(Rc)(C(O)Re), -N(Rc)(C(O)ORe), -N(Rc)(C(O)NRcRd), -N(Rc)(S(O)mRe), HetA и -C1-C3алкилен-HetA;
в каждом случае R7 независимо выбран из группы, состоящей из: галогена, -C1-C6алкила, -(CH2)nNRcRd, -(CH2)q-ORe, -S(O)mRe, -S(O)mNRcRd, -C(O)Re,-OC(O)Re,-C(O)ORe, -CN, -C(O)NRcRd, -C(NH)NRcRd, -NRcRd, -N(Rc)(C(O)Re), -N(Rc)(C(O)ORe), -N(Rc)(C(O)NRcRd), -N(Rc)(S(O)mRe), HetA и -C1-C3алкилен-HetA;
в каждом случае Rcи Rd независимо представляет собой: водород, -C1-C6алкил, -C2-C6алкенил, -C3-C6циклоалкил, -C1-C3алкилен-C3-C6 циклоалкил, HetA, -C1-C3алкилен-HetA, AryA, -C1-C3алкилен-AryA или -C1-C3алкилен-HetA, где каждый Rcи Rd необязательно замещен одним-тремя Rf;
или, альтернативно, Rcи Rd вместе с атомом азота, к которому они присоединены, взятые вместе образуют 4-7-членный циклогетероалкил, необязательно содержащий один или два дополнительных гетероатома, независимо выбранных из O, S и -NRg;
в каждом случае Reнезависимо представляет собой: водород, -C1-C6алкил, -C2-C6алкенил, -OH, -OC1-C6 алкил, -C3-C6циклоалкил, -C1-C3алкилен-C3-C6 циклоалкил, HetA, AryA, -C1-C3алкилен-AryA или -C1-C3алкилен-HetA; где каждый Re необязательно замещен одним-тремя Rh;
в каждом случае Rf независимо представляет собой: галоген, -C1-C6алкил, -OH, -OC1-C4 алкил, -S(O)mC1-C4 алкил, -CN, -CF3, -OCHF2 или -OCF3; где указанный -C1-C6алкил, -OC1-C4 алкил и -S(O)mC1-C4 алкил необязательно замещены одним-тремя заместителями, независимо выбранными из: -OH, галогена, циано и -S(O)2CH3;
в каждом случае Rgнезависимо представляет собой: водород, -C(O)Re, или -C1-C6алкил, где указанный -C1-C6алкил необязательно замещен одним-пятью фторами;
в каждом случае Rh независимо представляет собой: галоген, -C1-C6алкил, -OH, -OC1-C4 алкил, -S(O)mC1-C4 алкил, -CN, -CF3, -OCHF2 или -OCF3; где указанный -C1-C6алкил, -OC1-C4 алкил и -S(O)mC1-C4 алкил необязательно замещены одним-тремя заместителями, независимо выбранными из: -OH, галогена, циано и -S(O)2CH3;
каждый n независимо равен 0, 1, 2, 3 или 4;
каждый m независимо равен 0, 1 или 2,
каждый p равен 1 or 2; и
каждый q равен 0, 1, 2 или 3.
Настоящее изобретение также включает соединения формулы I:
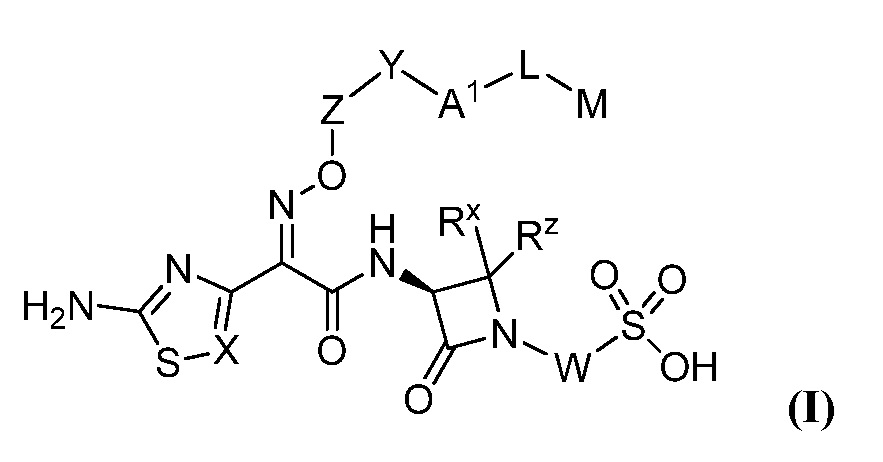
или их фармацевтически приемлемую соль, где:
W представляет собой связь или O;
RХ и RZнезависимо представляют собой водород, -SC1-C3алкил, C1-C3 алкил, -(C1-C3алкилен)nOC1-C3алкил или -(C1-C3алкилен)nNC1-C3алкил, где указанный -SC1-C3алкил, C1-C3 алкил, -(C1-C3алкилен)nOC1-C3алкил и -(C1-C3алкилен)nNC1-C3алкил необязательно замещены одним-семью фторами;
или, альтернативно, Rx и Rz, вместе с углеродом, к которому они присоединены, образуют моноциклический C4-C7 циклоалкил или моноциклический C4-C7 гетероциклоалкил с 1, 2 или 3 гетероатомами в качестве кольцевых атомов, независимо выбранными из N, O и S, где указанный C4-C7 циклоалкил и C4-C7 гетероциклоалкил необязательно замещены одним-тремя заместителями, независимо выбранными из -F, -OH и -OC1-C3алкила;
X представляет собой N или CR1;
R1 представляет собой водород, C1-C3 алкил, или галоген; где указанный C1-C3 алкил необязательно замещен одним-тремя Ra;
в каждом случае Ra независимо представляет собой водород, галоген, C1-C3алкил, -NRcRdили -ORe;
Z представляет собой C1-C3 алкилен, необязательно замещенный одним-тремя Rb;
в каждом случае Rb независимо представляет собой -C1-C6 алкил, -C3-C7 циклоалкил, -C(O)ORe, -C(O)NRcRd, тетразолил, оксадиазолонил, HetA, AryA, -S(O)mRe, -S(O)mNRcRd илиP(O)(Re)p, где указанный -C1-C6 алкил и -C3-C7 циклоалкил необязательно замещены одним-тремя Ra и, где указанный AryA и HetA необязательно замещены одним-четырьмя R4;
AryA представляет собой 5- или 6-членное моноциклическое ароматическое кольцо с 0, 1, 2 или 3 гетероатомами в качестве кольцевых атомов, независимо выбранными из N, N в виде четвертичной соли, O и S;
HetA представляет собой 4-6-членное насыщенное или мононенасыщенное моноциклическое кольцо с 1, 2 или 3 гетероатомами в качестве кольцевых атомов, независимо выбранными из, N в виде четвертичной соли, O и S;
Y представляет собой связь, O, NR2, S или CH2;
R2 представляет собой водород, -C1-C3 алкил, -C(O)Re,-C(O)NRcRd, -S(O)mRe или -S(O)mNRcRd, где указанный -C1-C3 алкил необязательно замещен одним-тремя Ra;
A1 представляет собой 9-11-членное бициклическое ароматическое кольцо с 0, 1, 2, 3 или 4 гетероатомами в качестве кольцевых атомов, независимо выбранными из N, N в виде четвертичной соли, O и S, необязательно замещенное одним-четырьмя R4;
в каждом случае R4 независимо представляет собой:
(a) -C1-C6 алкил,
(b) -C2-C6 алкенил,
(c) -C2-C6 алкинил,
(d) галоген,
(e) -ORe,
(f) -S(O)mRe,
(g) -S(O)mNRcRd,
(h) -C(O)Re,
(i) -OC(O)Re,
(j) -C(O)ORe,
(k) -CN,
(l) -C(O)NRcRd,
(m) -NRcRd,
(n) -NRcC(O)Re,
(o) -NRcC(O)ORe,
(p) -NRcC(O)NRcRd,
(q) -NRcS(O)mRe,
(r) =NH,
(s) -CF3,
(t) -OCF3,
(u) -OCHF2,
(v) -C3-C6циклоалкил,
(w) -O-C3-C6циклоалкил,
(x) -C1-C3алкилен-C3-C6циклоалкил,
(y) -O-C1-C3алкилен-C3-C6циклоалкил,
(z) HetA,
(aa) -O-HetA,
(bb) -C1-C3алкилен-HetA,
(cc) -O-C1-C3алкилен-HetA,
(dd) AryA,
(ee) -O-AryA,
(ff) -C1-C3алкилен-AryA, или
(gg) -O-C1-C3алкилен-AryA,
где указанный C1-C6 алкил, -C2-C6 алкенил, -C2-C6 алкинил, -C3-C6циклоалкил,-O-C3-C6циклоалкил, -C1-C3алкилен-C3-C6циклоалкил, -O-C1-C3алкилен-C3-C6циклоалкил, HetA, O-HetA, -C1-C3алкилен-HetA, -O-C1-C3алкилен-HetA, AryA, -O-AryA, -C1-C3алкилен-AryA и -O-C1-C3алкилен-AryA необязательно замещены одним-тремя Ra;
L представляет собой связь, -O-, -C1-C6алкилен-, -NHC(O) -, -C(O)-, -C(=NH)-, -S(O)m-, -SC1-C6алкилен-, -NR3(CH2)n-, -NHC(=NH)- или -NHS(O)m-
R3 представляет собой водород или -C1-C3 алкил;
M представляет собой N(R3)2, N+(C1-C3алкил)3, C2-C6алкил, C3-C7 циклоалкил, HetA или AryA, где указанный C2-C6алкил, C3-C7 циклоалкил, HetA и AryA необязательно замещены одним-четырьмя R6
в каждом случае R6 независимо выбран из группы, состоящей из: галогена, -C1-C6алкила, -(CH2)nNRcRd, -ORe, -S(O)mRe, -S(O)mNRcRd, -C(O)Re,-OC(O)Re,-C(O)ORe, -CN, -C(O)NRcRd, -C(NH)NRcRd, -NRcRd, -N(Rc)(C(O)Re), -N(Rc)(C(O)ORe), -N(Rc)(C(O)NRcRd) и -N(Rc)(S(O)mRe);
в каждом случае Rcи Rd независимо представляет собой: водород, -C1-C6алкил, -C2-C6алкенил, -C3-C6циклоалкил, -C1-C3алкилен-C3-C6 циклоалкил, HetA, -C1-C3алкилен-HetA, AryA, -C1-C3алкилен-AryA или - C1-C3алкилен-HetA, где каждый Rcи Rd необязательно замещен одним-тремя Rf;
или, альтернативно, Rcи Rd вместе с атомом азота, к которому они присоединены, взятые вместе образуют 4-7-членный циклогетероалкил, необязательно содержащий один или два дополнительных гетероатома, независимо выбранных из O, S и -NRg;
в каждом случае Reнезависимо представляет собой: водород, -C1-C6алкил, -C2-C6алкенил, -OH, -OC1-C6 алкил, -C3-C6циклоалкил, -C1-C3алкилен-C3-C6 циклоалкил, HetA, AryA, -C1-C3алкилен-AryA или -C1-C3алкилен-HetA; где каждый Re необязательно замещен одним-тремя Rh;
в каждом случае Rf независимо представляет собой: галоген, -C1-C6алкил, -OH, -OC1-C4 алкил, -S(O)mC1-C4 алкил, -CN, -CF3, -OCHF2 или -OCF3; где указанный -C1-C6алкил, -OC1-C4 алкил и -S(O)mC1-C4 алкил необязательно замещены одним-тремя заместителями, независимо выбранными из: -OH, галогена, циано и -S(O)2CH3;
в каждом случае Rgнезависимо представляет собой: водород, -C(O)Re или -C1-C6алкил, где указанный -C1-C6алкил необязательно замещен одним-пятью фторами;
в каждом случае Rh независимо представляет собой: галоген, -C1-C6алкил, -OH, -OC1-C4 алкил, -S(O)mC1-C4 алкил, -CN, -CF3, -OCHF2 или -OCF3; где указанный -C1-C6алкил, -OC1-C4 алкил и -S(O)mC1-C4 алкил необязательно замещены одним-тремя заместителями, независимо выбранными из: -OH, галогена, циано и -S(O)2CH3;
каждый n независимо равен 0, 1, 2, 3 или 4;
каждый m независимо равен 0, 1 или 2, и
каждый p равен 1 или 2.
Настоящее изобретение также относится к фармацевтической композиции для лечения бактериальной инфекции у субъекта, включая инфекцию грамотрицательными бактериальными штаммами с множественной лекарственной устойчивостью, содержащую бициклическое арильное монобактамовое соединение по изобретению и фармацевтически приемлемый носитель, разбавитель или эксципиент.
Соединения формулы (I) (также называемые в настоящем описании «бициклические арильные монобактамовые соединения») и их фармацевтически приемлемые соли могут быть полезны, например, для ингибирования роста грамотрицательных бактериальных штаммов, включая, но не ограничиваясь ими, штаммов Pseudomonas и Acinetobacter, и/или для лечения или профилактики их клинических проявлений у пациента.
Настоящее изобретение также относится к способам лечения грамотрицательных бактериальных инфекций у субъекта, нуждающегося в таком лечении, включающим введение субъекту эффективного количества бициклического арильного монобактамового соединения по изобретению. В конкретных вариантах осуществления изобретения способ включает введение одного или нескольких соединения(ий) ингибитора бета-лактамазы.
Варианты осуществления, подварианты осуществления и характерные черты настоящего изобретения далее либо будут описаны, либо будут очевидны из последующего описания, примеров и прилагаемых пунктов формулы изобретения.
Подробное описание изобретения
Изобретение относится к новым бициклическим арильным монобактамовым аналогам, классу высокоэффективных антибиотиков, эффективных против широкого ряда грамотрицательных бактерий. Эти соединения полезны в качестве терапевтических средств для клинического лечения различных инфекций, вызванных грамотрицательными бактериями, включая штаммы, обладающие множественной лекарственной устойчивостью, и для лечения или профилактики клинических патологий, связанных с ними.
В каждом из различных вариантов осуществления соединений по изобретению, описанных в настоящем документе, каждая переменная, включая, в том числе, формулы I, IA и IB, и различные варианты осуществления этого, каждая переменная выбирается независимо от других, если не указано иное.
Настоящее изобретение охватывает все соединения формул I, IA и IB и различные варианты осуществления, описанные в настоящем документе, например, любые сольваты, гидраты, стереоизомеры и таутомеры указанных соединений и любых их фармацевтически приемлемых солей.
Соединения формулы (I)
В одном аспекте настоящее изобретение включает соединения формулы I:
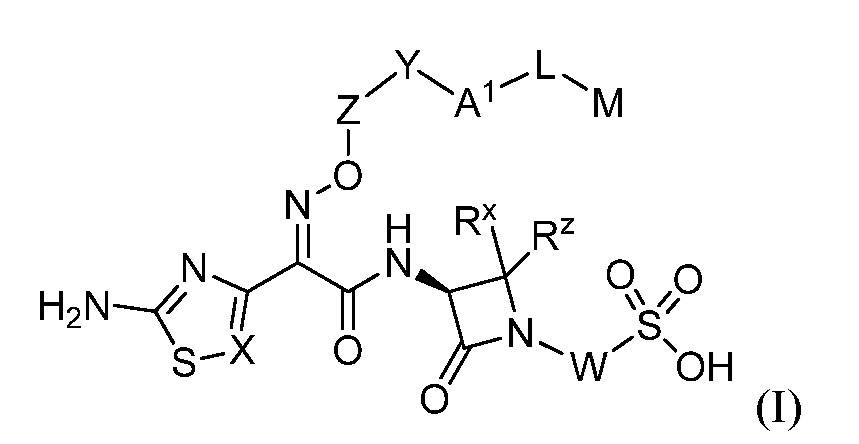
или их фармацевтически приемлемую соль, где A1, L, M, W, X, Y, Z, RX и Rzявляются такими, как определено в настоящем описании для соединений формулы (I); где соединения могут быть пригодны для использования для лечения бактериальных инфекций.
Первый вариант осуществления изобретения (вариант осуществления Е1) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где A1, L, M, W, X, Y, Z, RXи Rz являются такими, как определено в формуле I в разделе Сущность изобретения.
Второй вариант осуществления (вариант осуществления E2) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W представляет собой связь, и все другие переменные являются такими, как определено в варианте осуществления E1.
Третий вариант осуществления (вариант осуществления E3) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W представляет собой O, и все другие переменные являются такими, как определено в варианте осуществления E1.
Четвертый вариант осуществления (вариант осуществления E4) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X представляет собой N, и все другие переменные являются такими, как определено в варианте осуществления E1.
Пятый вариант осуществления (вариант осуществления E5) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X представляет собой CR1, и R1 представляет собой водород, галоген или C1-C3 алкил, необязательно замещенный одним-тремя Ra, и все другие переменные являются такими, как определено в варианте осуществления E1. В подварианте осуществления варианта осуществления E5 (вариант осуществления E5-A), R1представляет собой водород. В другом подварианте осуществления варианта осуществления E5 (вариант осуществления E5-B), R1представляет собой галоген. В дополнительном подварианте осуществления варианта осуществления E5 (вариант осуществления E5-C), R1представляет собой хлор. В еще одном подварианте осуществления варианта осуществления E5 (вариант осуществления E5-D), R1представляет собой фтор. В другом подварианте осуществления варианта осуществления E5 (вариант осуществления E5-D), R1представляет собой бром. В дополнительном подварианте осуществления варианта осуществления E5 (вариант осуществления E5-E), R1представляет собой C1-C3 алкил, необязательно замещенный одним-тремя Ra, где в каждом случае Ra независимо представляет собой водород, галоген, C1-C3алкил, -NRcRdили -ORe.
Шестой вариант осуществления (вариант осуществления E6) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z представляет собой C1-C3 алкилен, необязательно замещенный одним-тремя Rb, где в каждом случае Rb независимо представляет собой C1-C6алкил, C3-C7 циклоалкил, -C(O)ORe, -C(O)NRcRd, тетразолил, оксадиазолонил, HetA, AryA, -S(O)mRe, -S(O)mNRcRd или -P(O)(Re)p, где указанный C1-C6 алкил и указанный C3-C7 циклоалкил необязательно замещены одним-тремя Ra; и все другие переменные являются такими, как определено в варианте осуществления E1. В подварианте осуществления варианта осуществления E6, Z представляет собой C1-C3 алкилен, замещенный одним случаем Rb. В другом подварианте осуществления варианта осуществления E6, Z представляет собой C1-C3 алкилен, замещенный двумя случаями Rb.
В дополнительном подварианте осуществления варианта осуществления E6, Z представляет собой C1-C3 алкилен, замещенный тремя случаями Rb.
Седьмой вариант осуществления (вариант осуществления E7) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z представляет собой C1-C3алкилен, замещенный -C(O)ORe; и все другие переменные являются такими, как определено в варианте осуществления E1.
Восьмой вариант осуществления (вариант осуществления E8) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z представляет собой C1-C3 алкилен, замещенный -C(O)OH; и все другие переменные являются такими, как определено в варианте осуществления E1.
Девятый вариант осуществления (вариант осуществления E9) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z представляет собой C1-C3 алкилен, замещенный тетразолилом; и все другие переменные являются такими, как определено в варианте осуществления E1.
Десятый вариант осуществления (вариант осуществления E10) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z представляет собой C1-C3 алкилен, замещенный C1-C6 алкилом, необязательно замещенным одним-тремя Ra, и все другие переменные являются такими, как определено в варианте осуществления E1.
Одиннадцатый вариант осуществления (вариант осуществления E11) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z представляет собой C1-C3 алкилен, замещенный метилом; и все другие переменные являются такими, как определено в варианте осуществления E1.
Двенадцатый вариант осуществления (вариант осуществления E12) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z представляет собой C1-C3 алкилен, замещенный метилом и -C(O)OH; и все другие переменные являются такими, как определено в варианте осуществления E1.
Тринадцатый вариант осуществления (вариант осуществления E13) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z представляет собой -CH(C(O)OH)CH2- и все другие переменные являются такими, как определено в варианте осуществления E1.
Четырнадцатый вариант осуществления (вариант осуществления E14) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z представляет собой C1-C3 алкилен, замещенный оксадиазолонилом, и все другие переменные являются такими, как определено в варианте осуществления E1.
Пятнадцатый вариант осуществления (вариант осуществления E15) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y представляет собой связь, и все другие переменные являются такими, как определено в варианте осуществления E1.
Шестнадцатый вариант осуществления (вариант осуществления E16) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y представляет собой O, и все другие переменные являются такими, как определено в варианте осуществления E1.
Семнадцатый вариант осуществления (вариант осуществления E17) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y представляет собой NR2, и все другие переменные являются такими, как определено в варианте осуществления E1. В подварианте осуществления варианта осуществления E17, R2 представляет собой водород. В другом подварианте осуществления варианта осуществления E17, R2 представляет собой C1-C3 алкил, необязательно замещенный одним-тремя Ra. В дополнительном подварианте осуществления варианта осуществления E17, R2 представляет собой C(O)Re.В еще одном подварианте осуществления варианта осуществления E17, R2 представляет собой-C(O)NRcRd. В еще одном подварианте осуществления варианта осуществления E17, R2 представляет собой -S(O)mRe. В дополнительном подварианте осуществления варианта осуществления E17, R2 представляет собой -S(O)mNRcRd.
Восемнадцатый вариант осуществления (вариант осуществления E18) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y представляет собой S, и все другие переменные являются такими, как определено в варианте осуществления E1.
Девятнадцатый вариант осуществления (вариант осуществления E19) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y представляет собой CH2, и все другие переменные являются такими, как определено в варианте осуществления E1.
Двадцатый вариант осуществления (вариант осуществления E20) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 представляет собой 9-11-членное бициклическое ароматическое кольцо с 0, 1, 2, 3 или 4 гетероатомами в качестве кольцевых атомов, независимо выбранными из N, N в виде четвертичной соли, O и S, необязательно замещенное одним-четырьмя R4; и все другие переменные являются такими, как определено в варианте осуществления E1.
Двадцать первый вариант осуществления (вариант осуществления E21) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 представляет собой 9-членное бициклическое ароматическое кольцо с 0, 1, 2, 3 или 4 гетероатомами в качестве кольцевых атомов, независимо выбранными из N, N в виде четвертичной соли, O и S, необязательно замещенное одним-четырьмя R4, и все другие переменные являются такими, как определено в варианте осуществления E1.
Двадцать второй вариант осуществления (вариант осуществления E22) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 представляет собой 10-членное бициклическое ароматическое кольцо с 0, 1, 2, 3 или 4 гетероатомами в качестве кольцевых атомов, независимо выбранными из N, N в виде четвертичной соли, O и S, необязательно замещенное одним-четырьмя R4, и все другие переменные являются такими, как определено в варианте осуществления E1.
Двадцать третий вариант осуществления (вариант осуществления E23) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 представляет собой 11-членное бициклическое ароматическое кольцо с 0, 1, 2, 3 или 4 гетероатомами в качестве кольцевых атомов, независимо выбранными из N, N в виде четвертичной соли, O и S, необязательно замещенное одним-четырьмя R4, и все другие переменные являются такими, как определено в варианте осуществления E1. В подварианте осуществления варианта осуществления E21, E22 или E23, A1 имеет 1 кольцевой атом, независимо выбранный из N, N в виде четвертичной соли, O и S. В дополнительном подварианте осуществления варианта осуществления E21, E22 или E23, A1 имеет 2 кольцевых атома, независимо выбранных из N, N в виде четвертичной соли, O и S. В другом подварианте осуществления варианта осуществления E21, E22 или E23, A1 имеет 2 кольцевых атома, независимо выбранных из N, N в виде четвертичной соли, O и S. В еще одном подварианте осуществления варианта осуществления E21, E22 или E23, A1 имеет 3 кольцевых атома, независимо выбранных из N, N в виде четвертичной соли, O и S. В дополнительном подварианте осуществления варианта осуществления E21, E22 или E23, A1 имеет 4 кольцевых атома, независимо выбранных из N, N в виде четвертичной соли, O и S.
Двадцать четвертый вариант осуществления (вариант осуществления E24) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 представляет собой 9-11-членное бициклическое ароматическое кольцо, содержащее один гетероатом в качестве кольцевого атома, выбранный из N и N в виде четвертичной соли, и содержащее 0, 1, 2 или 3, дополнительных гетероатома, независимо выбранных из N, O и S, необязательно замещенное одним-четырьмя R4, и все другие переменные являются такими, как определено в варианте осуществления E1.
Двадцать пятый вариант осуществления (вариант осуществления E25) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 представляет собой:

где ** указывает место присоединения к L и * указывает место присоединения к остальной части соединения, и все другие переменные являются такими, как определено в варианте осуществления E1.
Двадцать шестой вариант осуществления (вариант осуществления E26) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 представляет собой:

и все другие переменные являются такими, как определено в варианте осуществления E1.
Двадцать седьмой вариант осуществления (вариант осуществления E27) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 представляет собой:

и все другие переменные являются такими, как определено в варианте осуществления E1.
Двадцать восьмой вариант осуществления (вариант осуществления E28) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D, или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 представляет собой:
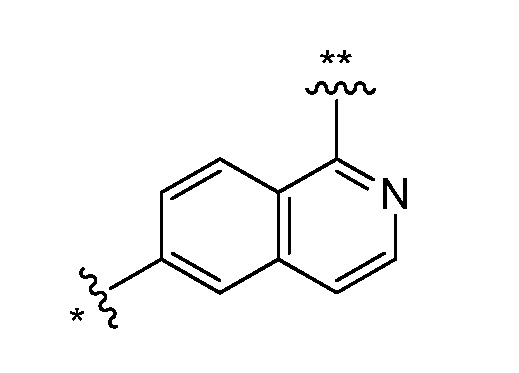
и все другие переменные являются такими, как определено в варианте осуществления E1.
Двадцать девятый вариант осуществления (вариант осуществления E29) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 представляет собой:
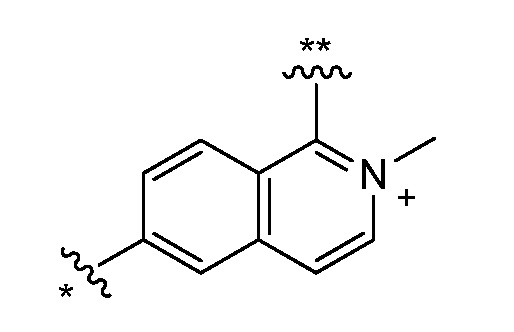
и все другие переменные являются такими, как определено в варианте осуществления E1.
Тридцатый вариант осуществления (вариант осуществления E30) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D, или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 представляет собой:
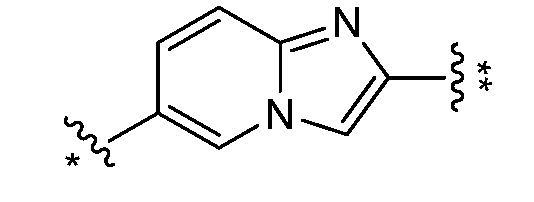
и все другие переменные являются такими, как определено в варианте осуществления E1.
Тридцать первый вариант осуществления (вариант осуществления E31) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 представляет собой:

и все другие переменные являются такими, как определено в варианте осуществления E1.
Тридцать второй вариант осуществления (вариант осуществления E32) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 представляет собой:

и все другие переменные являются такими, как определено в варианте осуществления E1.
Тридцать третий вариант осуществления (вариант осуществления E33) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 представляет собой:

и все другие переменные являются такими, как определено в варианте осуществления E1.
Тридцать четвертый вариант осуществления (вариант осуществления E34) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 представляет собой:
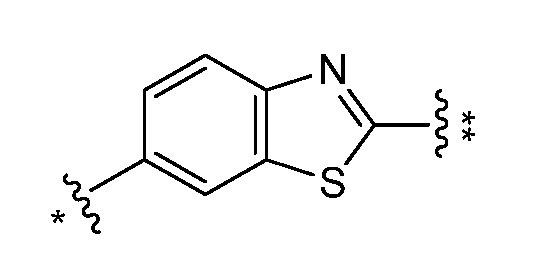
и все другие переменные являются такими, как определено в варианте осуществления E1.
Тридцать пятый вариант осуществления (вариант осуществления E35) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L представляет собой -CH2-; и все другие переменные являются такими, как определено в варианте осуществления E1.
Тридцать шестой вариант осуществления (вариант осуществления E36) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L представляет собой связь; и все другие переменные являются такими, как определено в варианте осуществления E1.
Тридцать седьмой вариант осуществления (вариант осуществления E37) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L представляет собой -O-; и все другие переменные являются такими, как определено в варианте осуществления E1.
Тридцать восьмой вариант осуществления (вариант осуществления E38) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L представляет собой -C1-C6алкилен-; и все другие переменные являются такими, как определено в варианте осуществления E1.
Тридцать девятый вариант осуществления (вариант осуществления E39) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L представляет собой -NHC(O)-; и все другие переменные являются такими, как определено в варианте осуществления E1.
Сороковой вариант осуществления (вариант осуществления E40) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L представляет собой -C(O)-; и все другие переменные являются такими, как определено в варианте осуществления E1.
Сорок первый вариант осуществления (вариант осуществления E41) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L представляет собой -C(=NH)-; и все другие переменные являются такими, как определено в варианте осуществления E1.
Сорок второй вариант осуществления (вариант осуществления E42) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L представляет собой -S(O)m-; m равен 0, 1 или 2; и все другие переменные являются такими, как определено в варианте осуществления E1.
Сорок третий вариант осуществления (вариант осуществления E43) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L представляет собой -SC1-C6алкилен-; и все другие переменные являются такими, как определено в варианте осуществления E1.
Сорок четвертый вариант осуществления (вариант осуществления E44) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L представляет собой -NR3(CH2)n-; n равен 0, 1, 2, 3 или 4; и все другие переменные являются такими, как определено в варианте осуществления E1. В подварианте осуществления варианта осуществления E44, R3 представляет собой водород. В другом подварианте осуществления варианта осуществления E44, R3 представляет собой -C1-C3 алкил. В дополнительном подварианте осуществления варианта осуществления E44, R3 представляет собой метил.
Сорок пятый вариант осуществления (вариант осуществления E45) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L представляет собой -NHC(=NH)-; и все другие переменные являются такими, как определено в варианте осуществления E1.
Сорок шестой вариант осуществления (вариант осуществления E46) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L представляет собой -NHS(O)m-; m равен 0, 1 или 2; и все другие переменные являются такими, как определено в варианте осуществления E1.
Сорок седьмой вариант осуществления (вариант осуществления E47) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M представляет собой N(R3)2; и все другие переменные являются такими, как определено в варианте осуществления E1.
Сорок восьмой вариант осуществления (вариант осуществления E48) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M представляет собой N+(C1-C3алкил)3; и все другие переменные являются такими, как определено в варианте осуществления E1.
Сорок девятый вариант осуществления (вариант осуществления E49) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M представляет собой C2-C6алкил, необязательно замещенный одним-четырьмя R6; и все другие переменные являются такими, как определено в варианте осуществления E1.
Пятидесятый вариант осуществления (вариант осуществления E50) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M представляет собой C3-C7 циклоалкил, необязательно замещенный одним-четырьмя R6; и все другие переменные являются такими, как определено в варианте осуществления E1.
Пятьдесят первый вариант осуществления (вариант осуществления E51) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M представляет собой HetA, необязательно замещенный одним-четырьмя R6; и все другие переменные являются такими, как определено в варианте осуществления E1.
Пятьдесят второй вариант осуществления (вариант осуществления E52) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M представляет собой AryA, необязательно замещенный одним-четырьмя R6; и все другие переменные являются такими, как определено в варианте осуществления E1.
Пятьдесят третий вариант осуществления (вариант осуществления E53) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M представляет собой C3-C7 циклоалкил, замещенный N(R3)2, и необязательно замещенный одним-тремя дополнительными заместителями, независимо выбранными из галогена, C1-C3алкила, -NRcRdи -ORe; и все другие переменные являются такими, как определено в варианте осуществления E1.
Пятьдесят четвертый вариант осуществления (вариант осуществления E54) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; М представляет собой 5- или 6-членное моноциклическое ароматическое кольцо, содержащее один гетероатом в качестве кольцевого атома, выбранный из N и N в виде четвертичной соли, и содержащее 0, 1 или 2 дополнительных гетероатомов в качестве кольцевых атомов, независимо выбранных из N, О и S, необязательно замещенное одним-четырьмя R6; и все другие переменные являются такими, как определено в варианте осуществления E1.
Пятьдесят пятый вариант осуществления (вариант осуществления E55) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M представляет собой 4-6-членное насыщенное или мононенасыщенное моноциклическое кольцо, содержащее один гетероатом в качестве кольцевого атома, выбранный из N и N в виде четвертичной соли, и содержащее 0, 1 или 2 дополнительных гетероатомов в качестве кольцевых атомов, независимо выбранных из N, О и S, необязательно замещенное одним-четырьмя R6; и все другие переменные являются такими, как определено в варианте осуществления E1.
Пятьдесят шестой вариант осуществления (вариант осуществления E56) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E13, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E35, L определен в любом из вариантов осуществления E36-E46; M представляет собой -NH2; и все другие переменные являются такими, как определено в варианте осуществления E1.
Пятьдесят седьмой вариант осуществления (вариант осуществления E57) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M представляет собой -NHCH3; и все другие переменные являются такими, как определено в варианте осуществления E1.
Пятьдесят восьмой вариант осуществления (вариант осуществления E58) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M представляет собой 5- или 6-членное моноциклическое ароматическое кольцо, содержащее один гетероатом в качестве кольцевого атома, выбранный из N и N в виде четвертичной соли, и содержащее 0, 1 или 2 дополнительных гетероатомов в качестве кольцевых атомов, независимо выбранных из N, О и S, необязательно замещенное одним или двумя C1-C6алкилом; и все другие переменные являются такими, как определено в варианте осуществления E1.
Пятьдесят девятый вариант осуществления (вариант осуществления E59) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M представляет собой 4-6-членное насыщенное или мононенасыщенное моноциклическое кольцо, содержащее один гетероатом в качестве кольцевого атома, выбранный из N и N в виде четвертичной соли, и содержащее 0, 1 или 2 дополнительных гетероатома в качестве кольцевых атомов, независимо выбранных из N, O и S, необязательно замещенное одним или двумя C1-C6алкилом; и все другие переменные являются такими, как определено в варианте осуществления E1.
В шестидесятом варианте осуществления (вариант осуществления E60) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M представляет собой:

и все другие переменные являются такими, как определено в варианте осуществления E1.
Шестьдесят первый вариант осуществления (вариант осуществления E61) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M представляет собой:
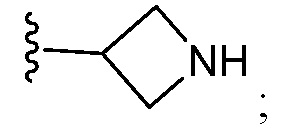
и все другие переменные являются такими, как определено в варианте осуществления E1.
Шестьдесят второй вариант осуществления (вариант осуществления E62) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M представляет собой:
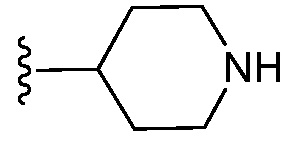
и все другие переменные являются такими, как определено в варианте осуществления E1.
Шестьдесят третий вариант осуществления (вариант осуществления E63) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M представляет собой:

и все другие переменные являются такими, как определено в варианте осуществления E1.
Шестьдесят четвертый вариант осуществления (вариант осуществления E64) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M представляет собой:
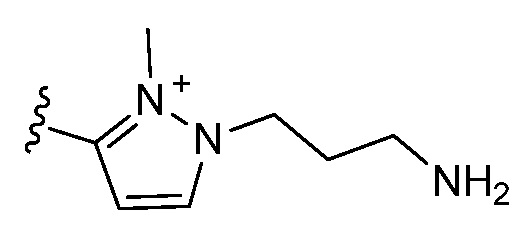
и все другие переменные являются такими, как определено в варианте осуществления E1.
Шестьдесят пятый вариант осуществления (вариант осуществления E65) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M представляет собой:
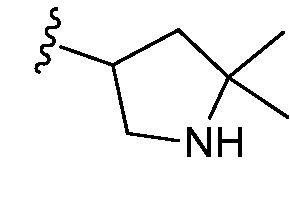
и все другие переменные являются такими, как определено в варианте осуществления E1.
Шестьдесят шестой вариант осуществления (вариант осуществления E66) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M представляет собой:
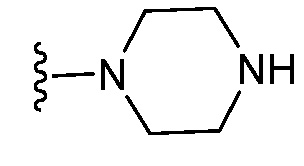
и все другие переменные являются такими, как определено в варианте осуществления E1.
Шестьдесят седьмой вариант осуществления (вариант осуществления E67) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M представляет собой:

и все другие переменные являются такими, как определено в варианте осуществления E1.
Шестьдесят восьмой вариант осуществления (вариант осуществления E68) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M представляет собой:

и все другие переменные являются такими, как определено в варианте осуществления E1.
Шестьдесят девятый вариант осуществления (вариант осуществления E69) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M представляет собой:

и все другие переменные являются такими, как определено в варианте осуществления E1.
Семидесятый вариант осуществления (вариант осуществления E70) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M представляет собой:

и все другие переменные являются такими, как определено в варианте осуществления E1.
Семьдесят первый вариант осуществления (вариант осуществления E71) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M определен в любом из вариантов осуществления E47-E70, Rx и Rzпредставляют собойметил; и все другие переменные являются такими, как определено в варианте осуществления E1.
Семьдесят второй вариант осуществления (вариант осуществления E72) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M определен в любом из вариантов осуществления E47-E70; Rx представляет собой водород и Rzпредставляет собой метил; и все другие переменные являются такими, как определено в варианте осуществления E1.
Семьдесят третий вариант осуществления (вариант осуществления E73) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M определен в любом из вариантов осуществления E47-E70; по меньшей мере один из Rx и Rzпредставляет собойSCH3,необязательно замещенный одним-тремя фторами; и все другие переменные являются такими, как определено в варианте осуществления E1.
Семьдесят четвертый вариант осуществления (вариант осуществления E74) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M определен в любом из вариантов осуществления E47-E70; по меньшей мере один из Rx и Rzпредставляет собойSC1-C3 алкил, необязательно замещенный одним-семью фторами; и все другие переменные являются такими, как определено в варианте осуществления E1.
Семьдесят пятый вариант осуществления (вариант осуществления E75) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M определен в любом из вариантов осуществления E47-E70; по меньшей мере один из Rx и Rzпредставляет собойC1-C3 алкил, необязательно замещенный одним-семью фторами; и все другие переменные являются такими, как определено в варианте осуществления E1.
Семьдесят шестой вариант осуществления (вариант осуществления E76) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M определен в любом из вариантов осуществления E47-E70; по меньшей мере один из Rx и Rzпредставляет собой(C1-C3алкилен)nOC1-C3алкил, необязательно замещенный одним-семью фторами; и все другие переменные являются такими, как определено в варианте осуществления E1.
Семьдесят седьмой вариант осуществления (вариант осуществления E77) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M определен в любом из вариантов осуществления E47-E70; по меньшей мере один из Rx и Rzпредставляет собой (C1-C3алкилен)nNC1-C3алкил, необязательно замещенный одним-семью фторами; и все другие переменные являются такими, как определено в варианте осуществления E1.
Семьдесят восьмой вариант осуществления (вариант осуществления E78) представляет собой соединение формулы I, или его фармацевтически приемлемую соль, где W определен в варианте осуществления E2 или E3, X определен в любом из вариантов осуществления E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E, Z определен в любом из вариантов осуществления E6-E14, Y определен в любом из вариантов осуществления E15-E19, A1 определен в любом из вариантов осуществления E20-E34, L определен в любом из вариантов осуществления E35-E46; M определен в любом из вариантов осуществления E47-E70; Rx и Rz, вместе с углеродом, к которому они присоединены, взятые вместе образуют моноциклический C4-C7 циклоалкил или моноциклический C4-C7 гетероциклоалкил с 1, 2 или 3 гетероатомами в качестве кольцевых атомов, независимо выбранными из N, O и S, где указанный C4-C7 циклоалкил и C4-C7 гетероциклоалкил необязательно замещены одним-тремя заместителями, независимо выбранными из -F, -OH и -OC1-C3алкила; и все другие переменные являются такими, как определено в варианте осуществления E1.
Семьдесят девятый вариант (вариант осуществления E79) представляет собой соединение формулы IA или IB или его фармацевтически приемлемую соль,
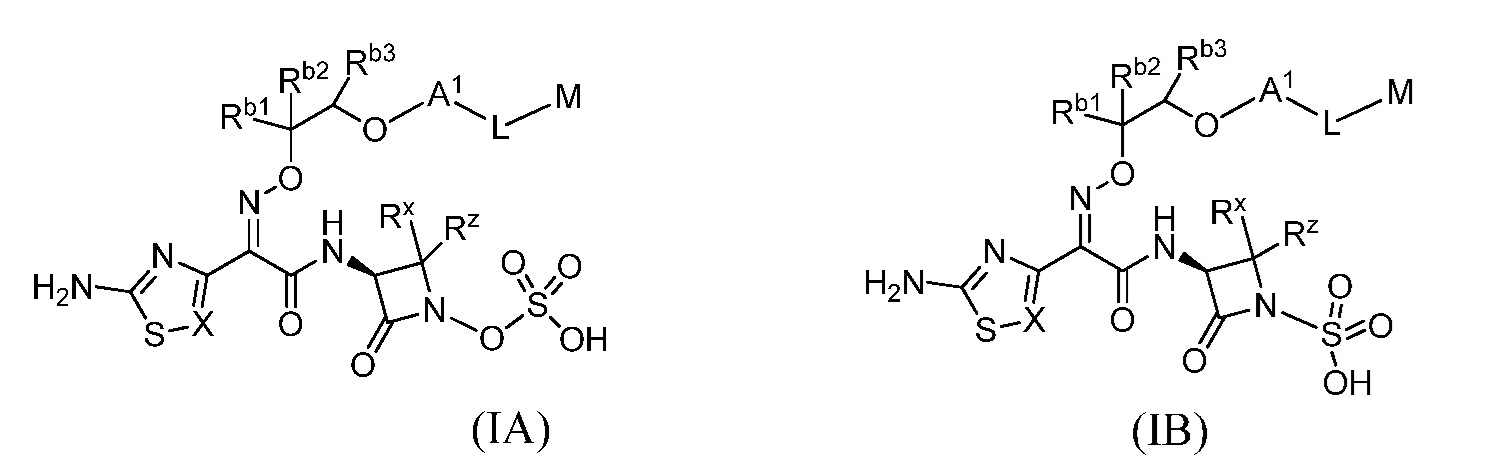
где X определен в любом из вариантов осуществления E1, E4, E5, E5-A, E5-B, E5-C, E5-D или E5-E;
RХ и RZнезависимо представляют собой водород, -SC1-C3алкил, C1-C3 алкил, -(C1-C3алкилен)nOC1-C3алкил или -(C1-C3алкилен)nNC1-C3алкил, где указанный -SC1-C3алкил, C1-C3 алкил, -(C1-C3алкилен)nOC1-C3алкил и -(C1-C3алкилен)nNC1-C3алкил необязательно замещены одним-семью фторами;
Rb1, Rb2 и Rb3независимо представляют собой водород, C1-C6 алкил, C3-C7 циклоалкил, -C(O)ORe, -C(O)NRcRd, тетразолил, оксадиазолонил, HetA, AryA, -S(O)mRe, -S(O)mNRcRd или -P(O)(Re)p, где указанный C1-C6 алкил и C3-C7 циклоалкил необязательно замещены одним-тремя Ra и где указанный AryA и HetA необязательно замещены одним-четырьмя R4;
A1 представляет собой 9-11-членное бициклическое ароматическое кольцо, содержащее один гетероатом в качестве кольцевого атома, выбранный из N и N в виде четвертичной соли, и содержащее 0, 1, 2 или 3, дополнительных гетероатома, независимо выбранных из N, O и S, необязательно замещенное одним-четырьмя R4;
M выбран из группы, состоящей из:
(a) N(R3)2,
(b) N+(C1-C3 алкил)3,
(c) C3-C7 циклоалкила, замещенного N(R3)2, и необязательно замещенного одним-тремя дополнительными заместителями, независимо выбранными из галогена, C1-C3алкила, - NRcRd и - ORe,
(d) 5- или 6-членного моноциклического ароматического кольца, содержащего один гетероатом в качестве кольцевого атома, выбранный из N и N в виде четвертичной соли, и содержащего 0, 1 или 2 дополнительных гетероатомов в качестве кольцевых атомов, независимо выбранных из N, О и S, необязательно замещенного одним-четырьмя R6; и
(e) 4-6-членного насыщенного или мононенасыщенного моноциклического кольца, содержащего один гетероатом в качестве кольцевого атома, выбранный из N и N в виде четвертичной соли, и содержащего 0, 1 или 2 дополнительных гетероатома в качестве кольцевых атомов, независимо выбранных из N, O и S, необязательно замещенного одним-четырьмя R6;
L определен в любом из вариантов осуществления E35-E46; и все другие переменные являются такими, как определено в варианте осуществления E1.
Восьмидесятый вариант осуществления (вариант осуществления E80) представляет собой соединение формулы IA или IB или его фармацевтически приемлемую соль, где Rb1 и Rb2 независимо представляют собой водород, C1-C3 алкил, тетразолил, оксадиазолонил или -C(O)ORe; и Rb3 представляет собой водород, и все другие переменные являются такими, как определено в варианте осуществления E79.
Восемьдесят первый вариант осуществления (вариант осуществления E81) представляет собой соединение формулы IA или IB или его фармацевтически приемлемую соль, где Rb1 представляет собой -C(O)OH, Rb2 представляет собой водород, и
Rb3 представляет собой водород, и все другие переменные являются такими, как определено в варианте осуществления E79.
Восемьдесят второй вариант осуществления (вариант осуществления E82) представляет собой соединение формулы IA или IB или его фармацевтически приемлемую соль, где Rb1 представляет собой тетразолил, Rb2 представляет собой водород, и Rb3 представляет собой водород, и все другие переменные являются такими, как определено в варианте осуществления E79.
Восемьдесят третий вариант осуществления (вариант осуществления E83) представляет собой соединение формулы IA или IB или его фармацевтически приемлемую соль, где Rb1 представляет собой оксадиазолонил, Rb2 представляет собой водород, и Rb3 представляет собой водород, и все другие переменные являются такими, как определено в варианте осуществления E79.
Восемьдесят четвертый вариант осуществления (вариант осуществления E84) представляет собой соединение, имеющее структуру:




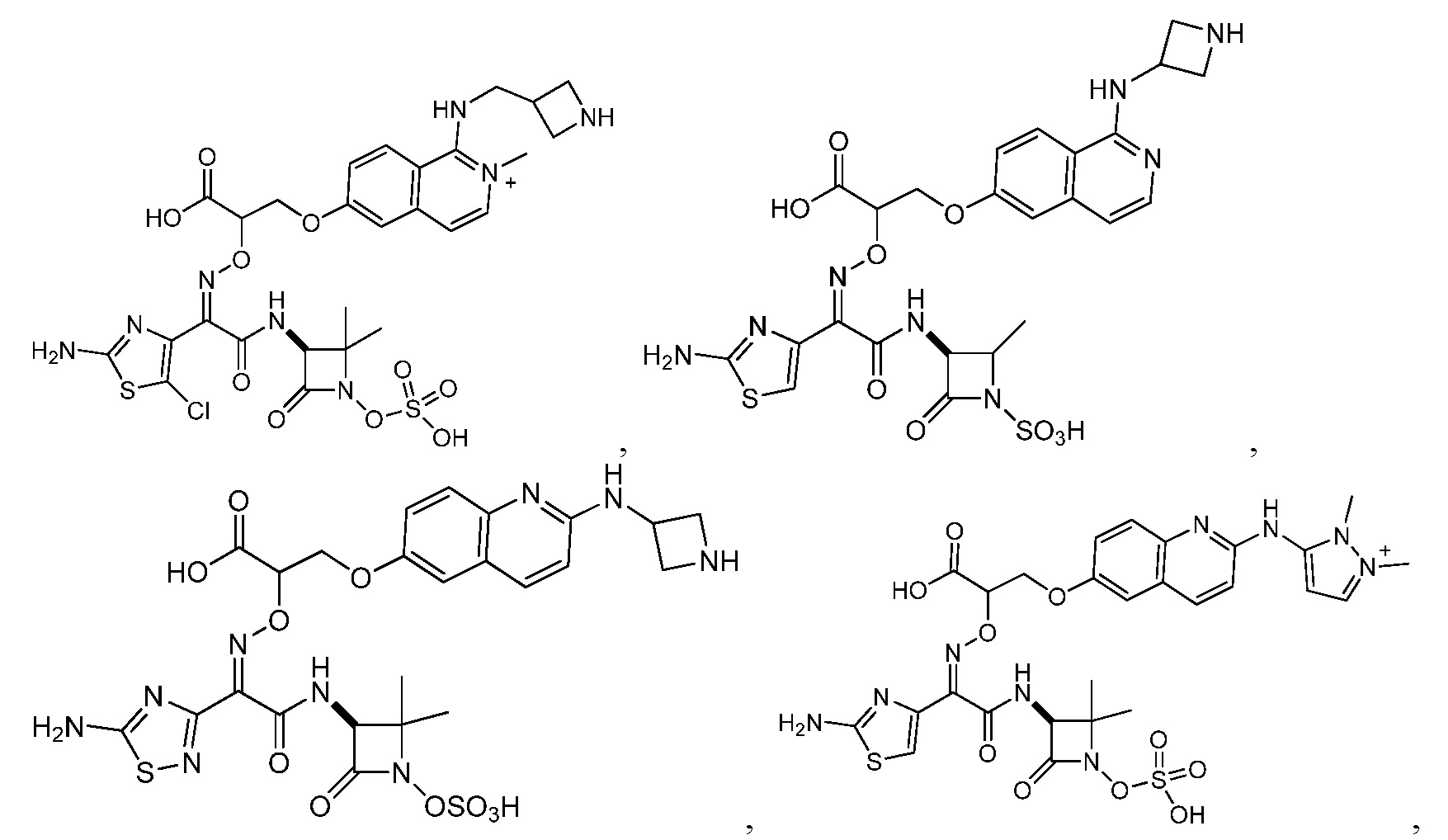
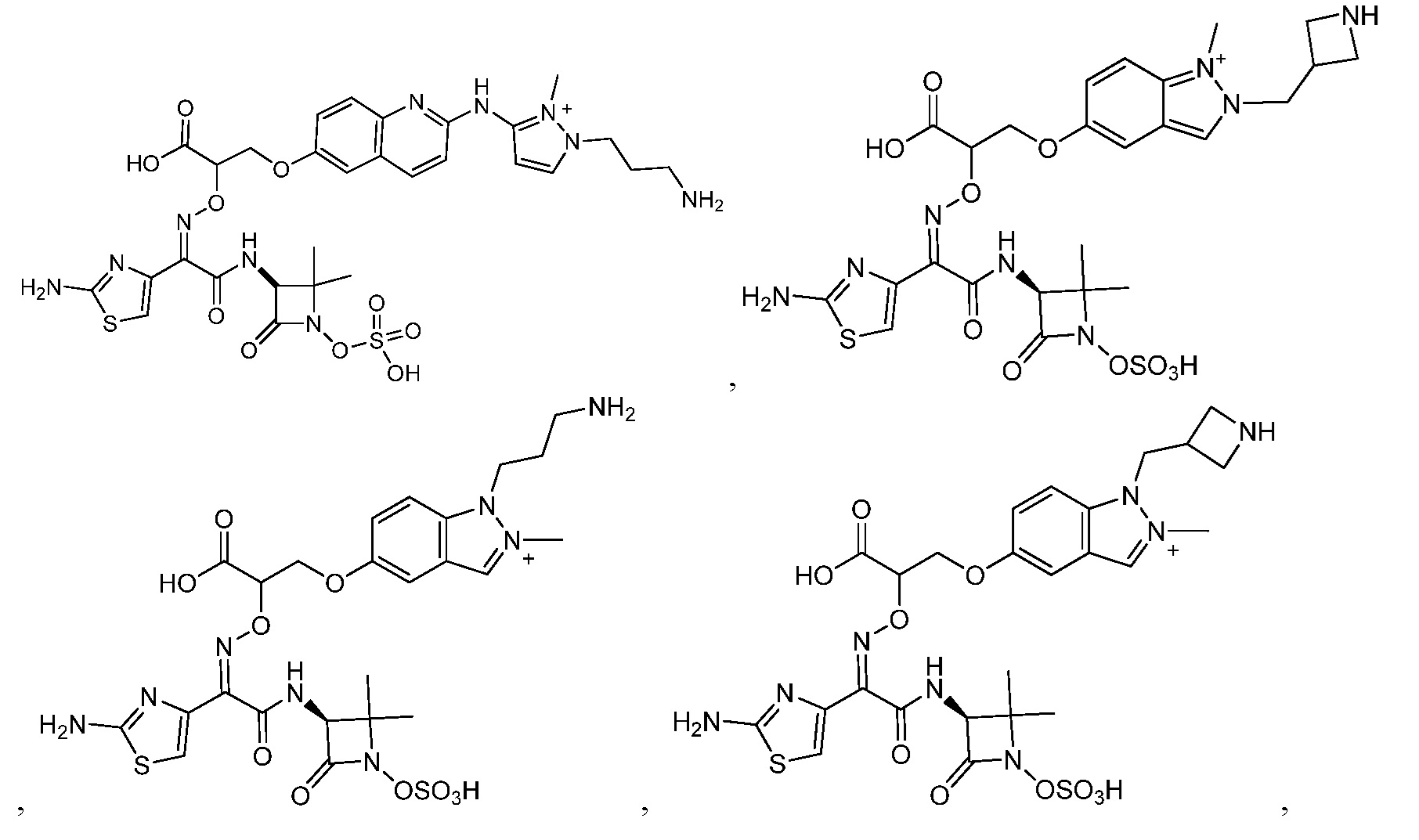
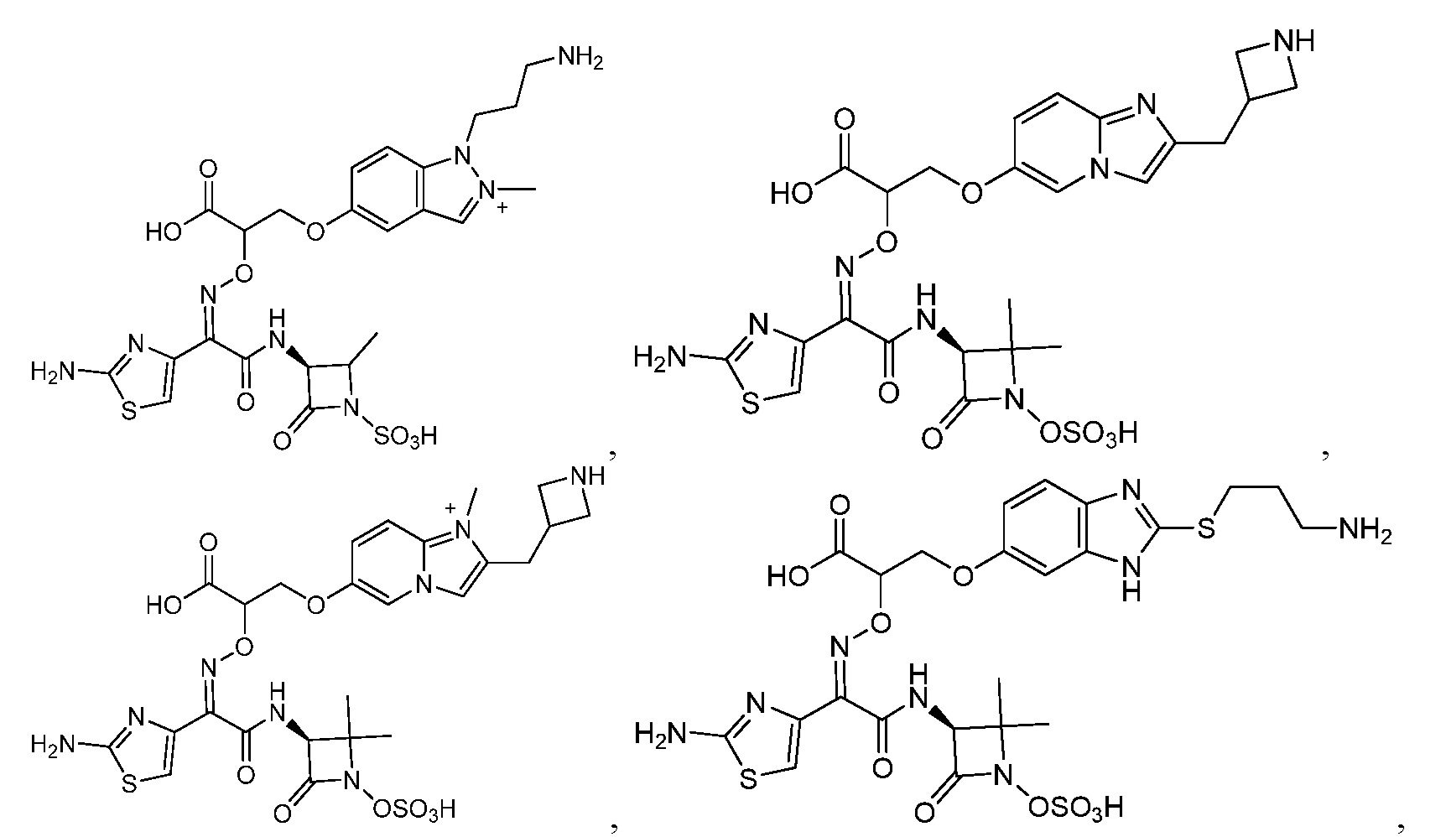
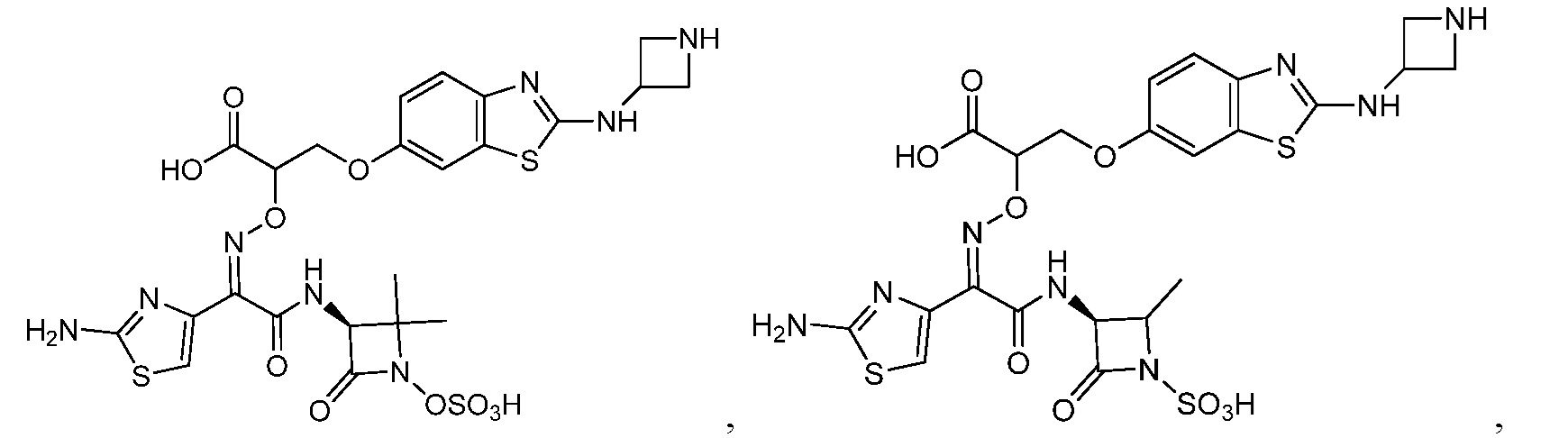
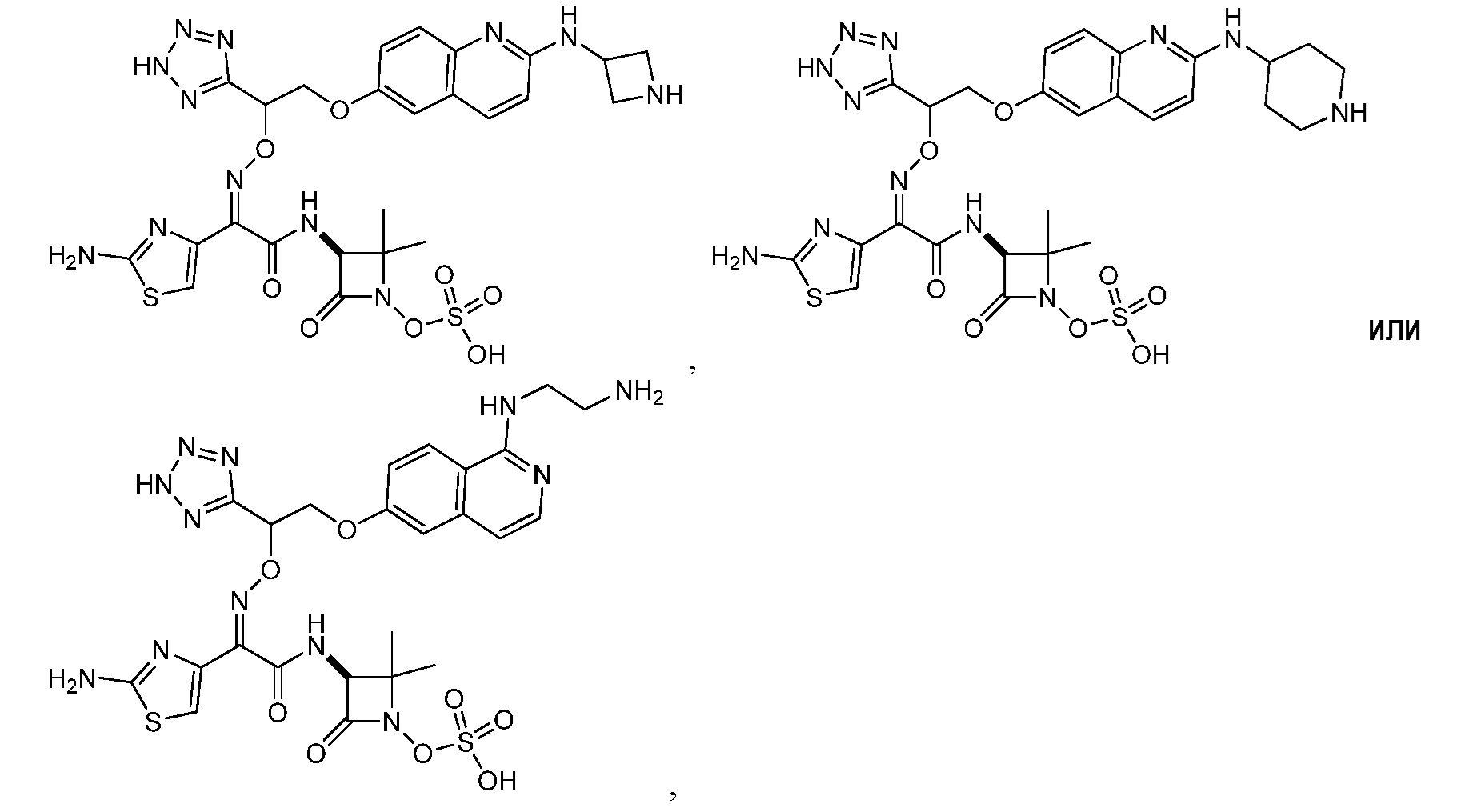
или его фармацевтически приемлемую соль.
Восемьдесят пятый вариант осуществления (вариант осуществления E85) представляет собой соединение, имеющее структуру:




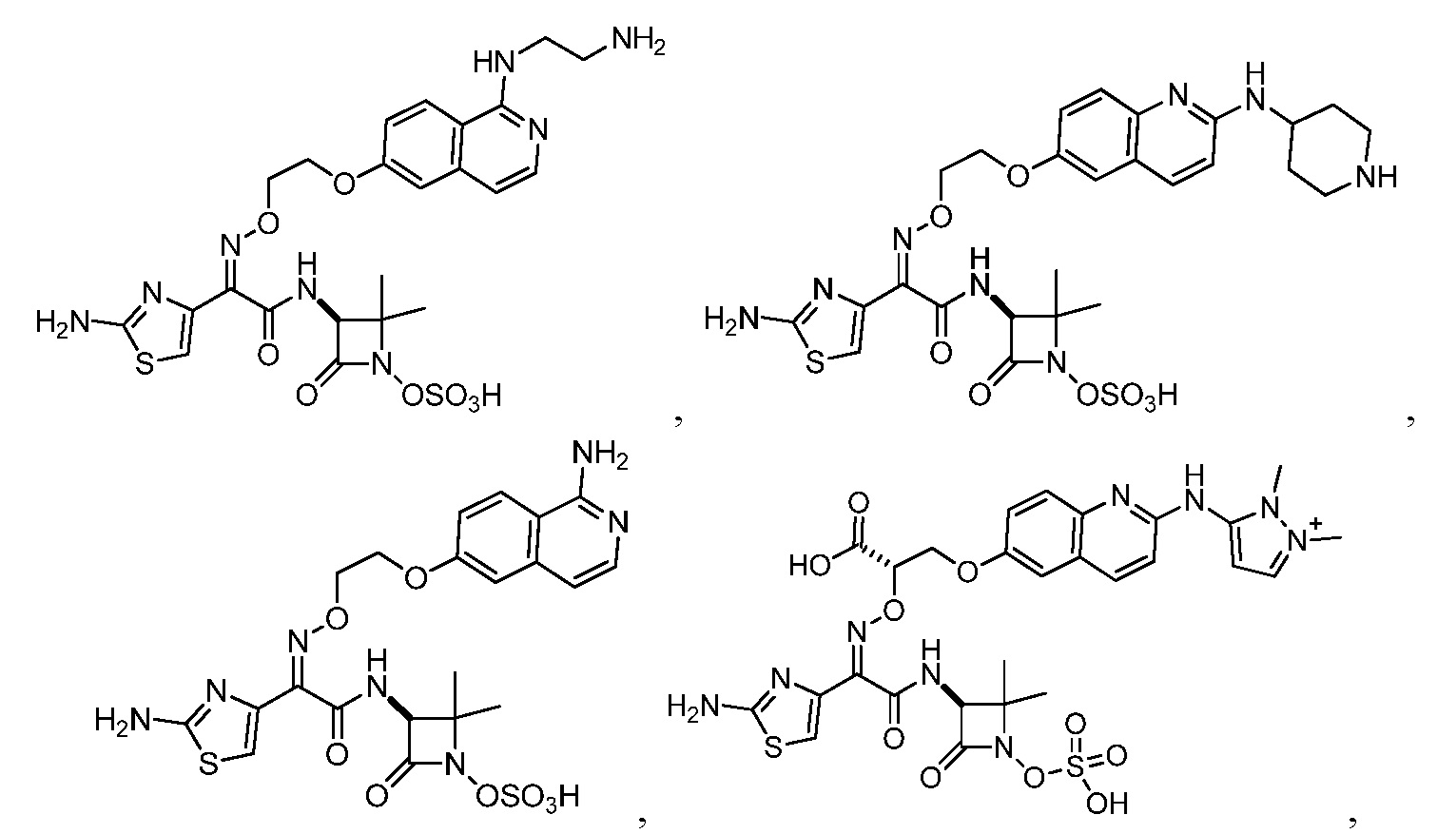
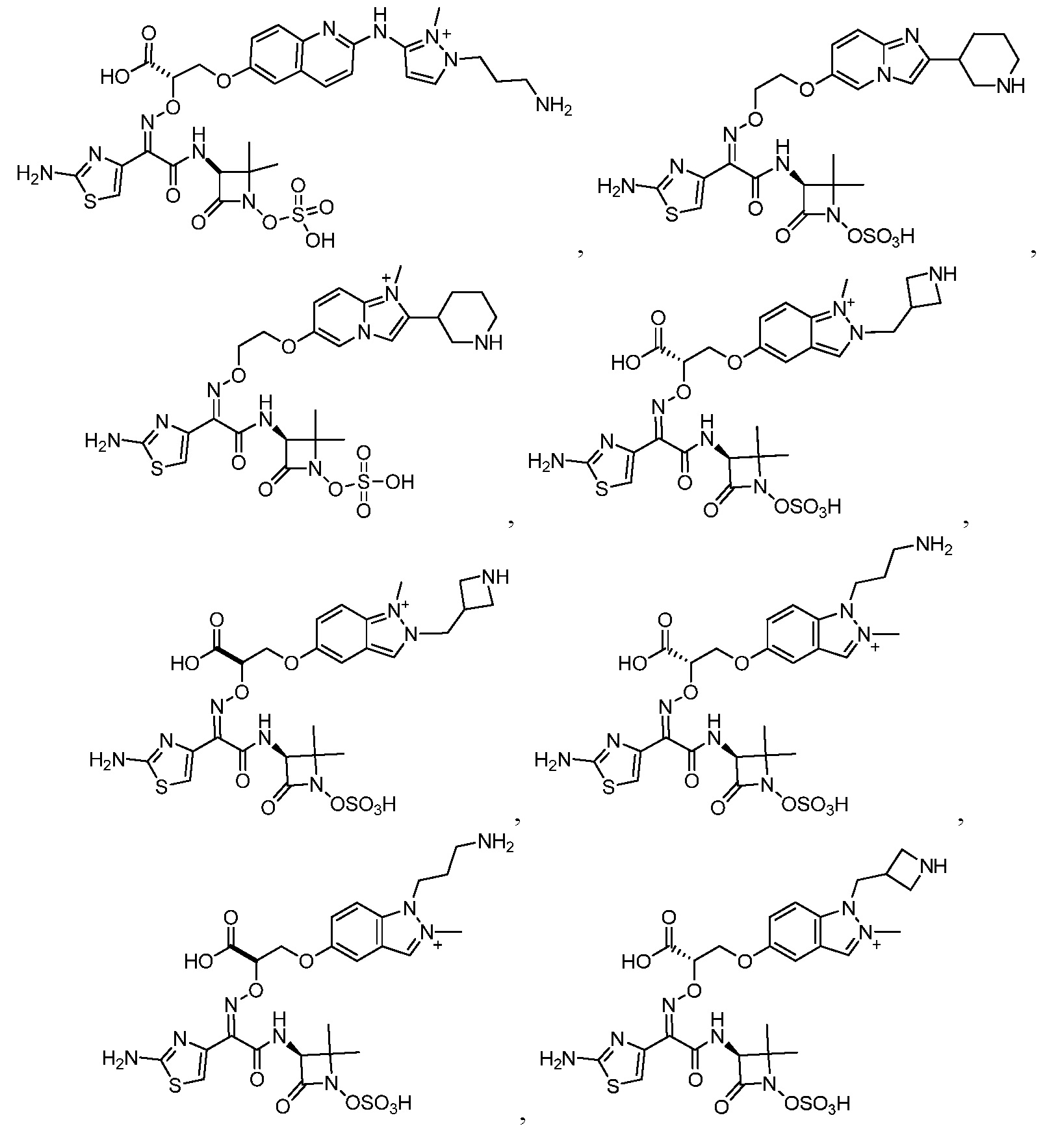
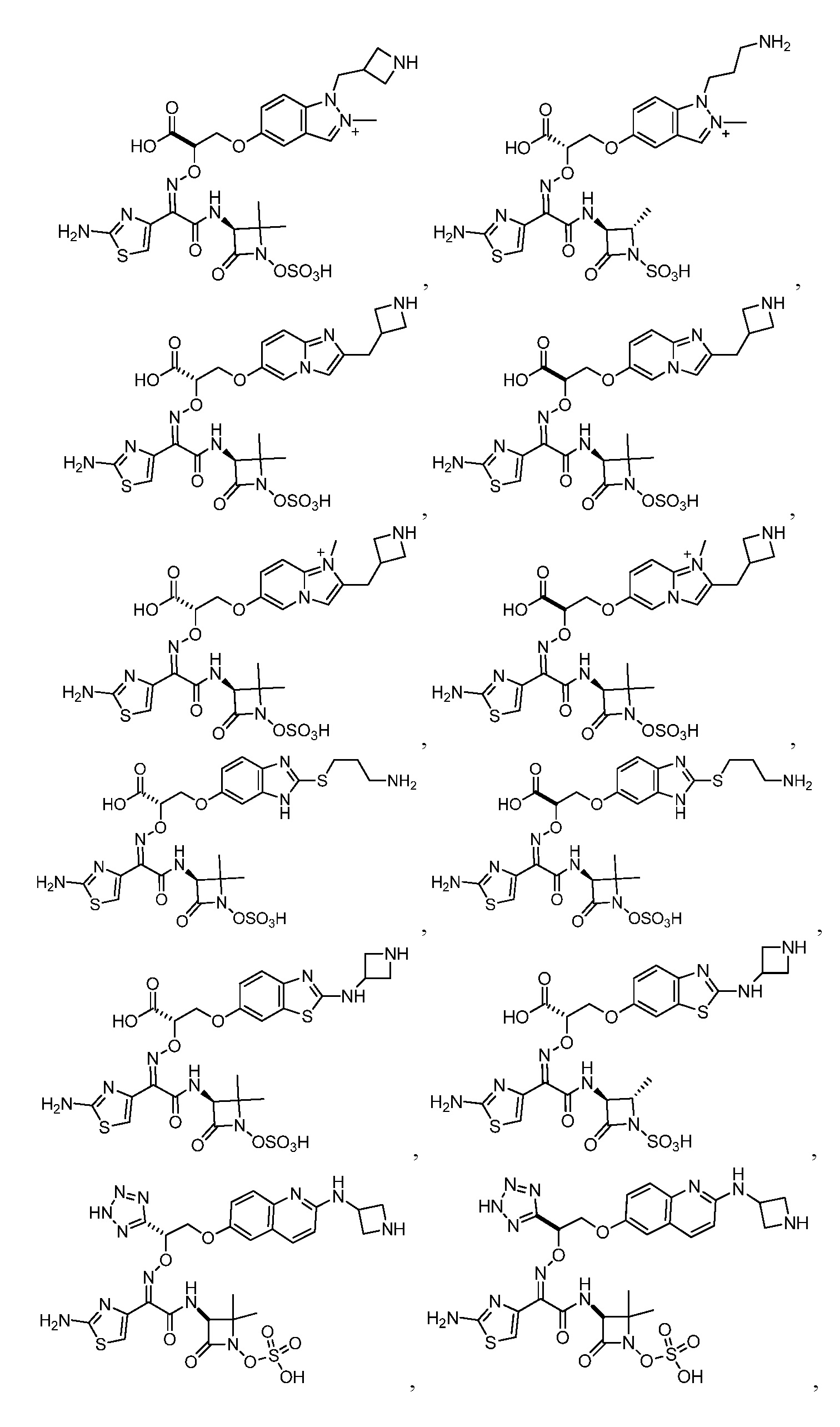
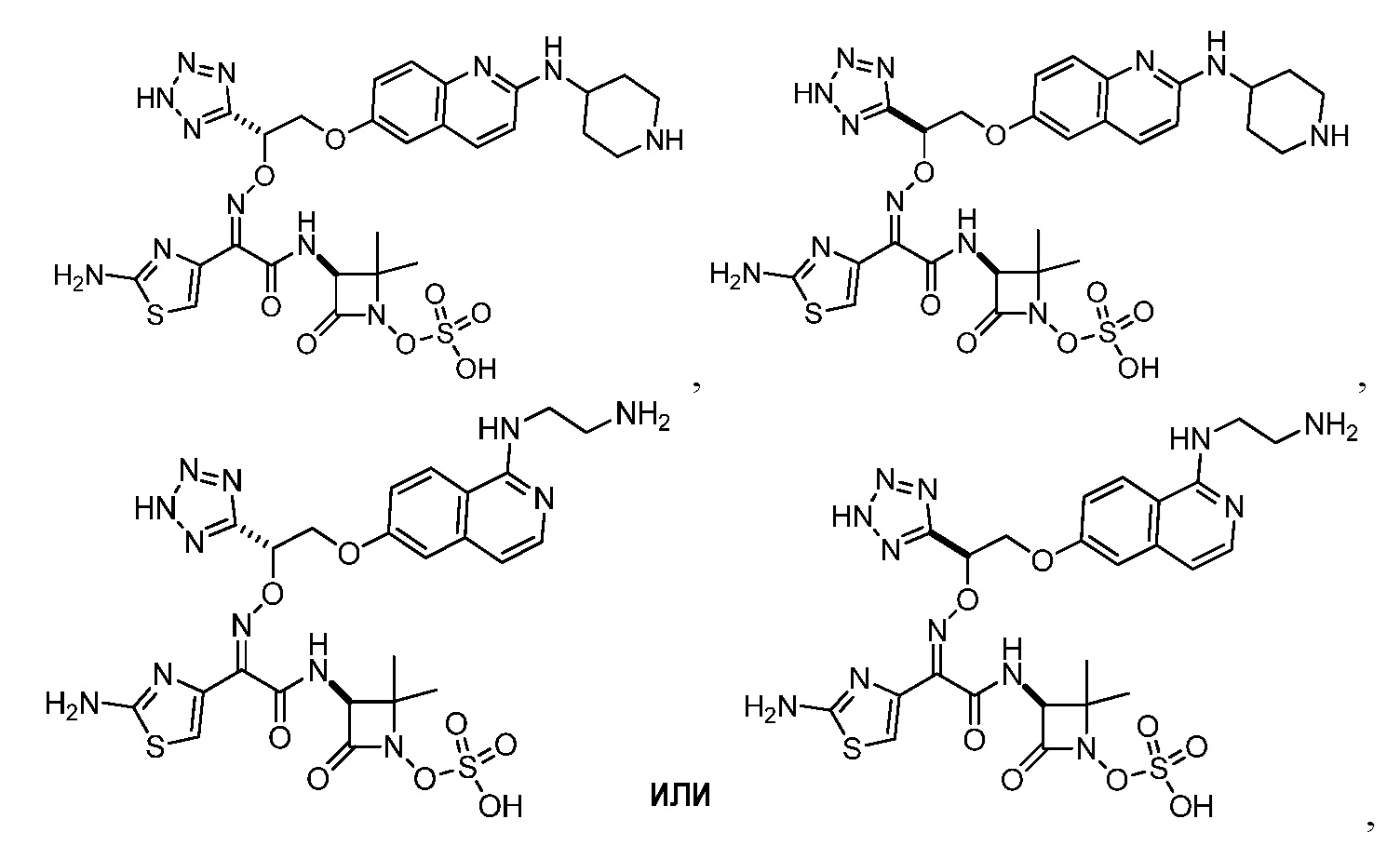
или его фармацевтически приемлемую соль.
В другом варианте осуществления настоящего изобретения, W представляет собой O.
В другом варианте осуществления настоящего изобретения, RХ и RZнезависимо представляют собой водород и C1-C3 алкил, где C1-C3 алкил необязательно замещен одним-семью фторами. В другом варианте осуществления настоящего изобретения, Rx и Rzнезависимо представляют собой C1-C3 алкил, где C1-C3 алкил необязательно замещен одним-семью фторами. В другом варианте осуществления настоящего изобретения, Rx и Rzпредставляют собой CH3.
В другом варианте осуществления настоящего изобретения, X представляет собой CH.
В другом варианте осуществления настоящего изобретения, R1 представляет собой водород.
В другом варианте осуществления настоящего изобретения, в каждом случае Ra независимо представляет собой -ORe или -C(O)NRcRd. В другом варианте осуществления настоящего изобретения, в каждом случае Ra независимо представляет собой -OH или C(O)NH2.
В другом варианте осуществления настоящего изобретения, Z представляет собой CH2CHRb или CH. В другом варианте осуществления настоящего изобретения, Z представляет собой CH2CHRb. В другом варианте осуществления настоящего изобретения, Z представляет собой CH2CHRb.
В другом варианте осуществления настоящего изобретения, в каждом случае Rb независимо представляет собой -C(O)ORe или тетразолил. В другом варианте осуществления настоящего изобретения, в каждом случае Rb независимо представляет собой -CO2H или тетразолил. В другом варианте осуществления настоящего изобретения, в каждом случае Rb представляет собой -CO2H. В другом варианте осуществления настоящего изобретения, в каждом случае Rb представляет собой тетразолил.
В другом варианте осуществления настоящего изобретения, Y представляет собой O.
В другом варианте осуществления настоящего изобретения, в каждом случае R4 независимо представляет собой: -C1-C6 алкил, галоген или -NRcRd, где указанный C1-C6 алкил необязательно замещен одним-тремя Ra. В другом варианте осуществления настоящего изобретения, в каждом случае R4 независимо представляет собой: -CH3, галоген или -NH2.
В другом варианте осуществления настоящего изобретения, A1 представляет собой хинолин, изохинолин, имидазо[1,2-а]пиридин, индазол, бензо[d]имидазол, бензо[d]тиазол или нафталин, необязательно замещенный одним-четырьмя R4.
В другом варианте осуществления настоящего изобретения, A1 представляет собой:
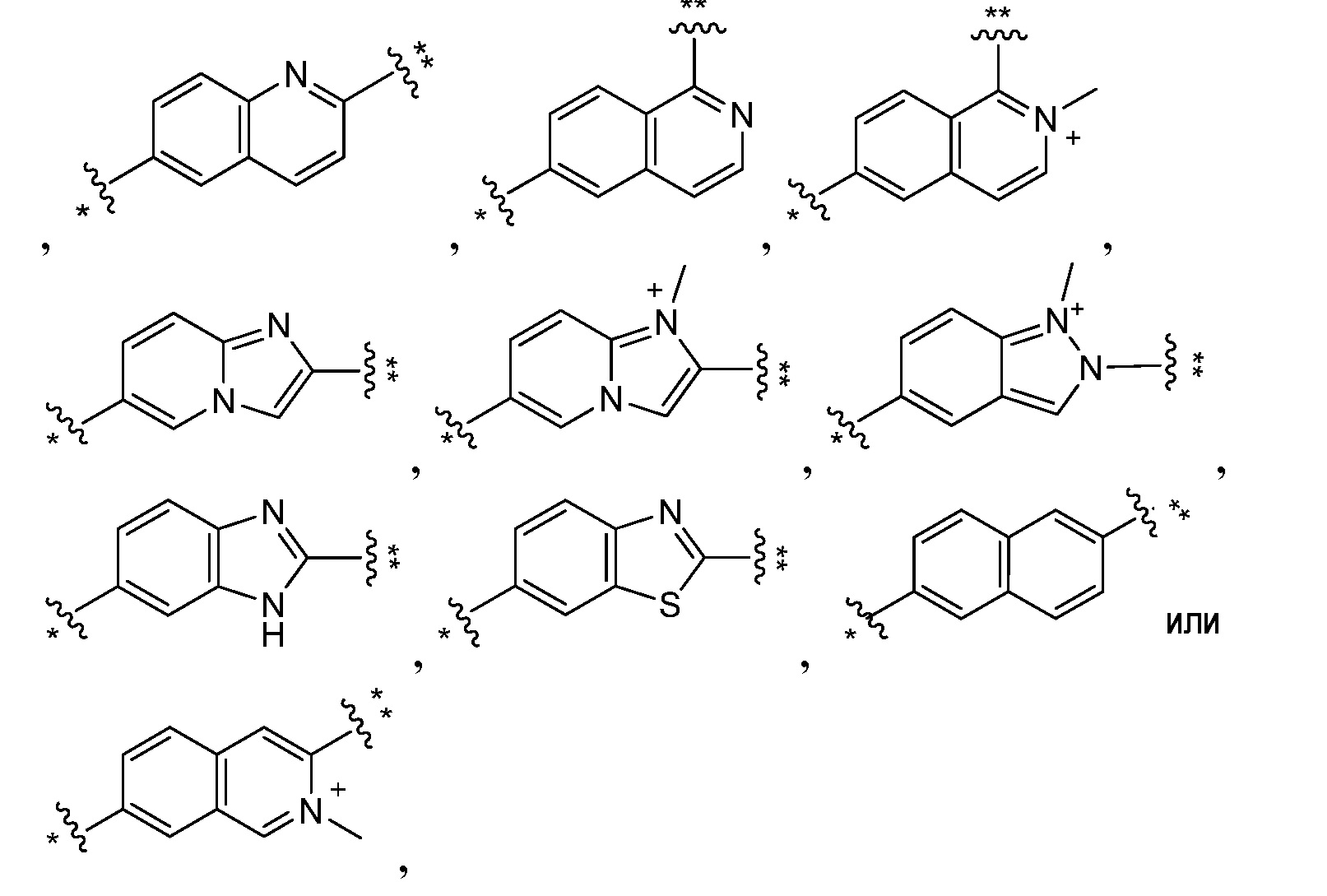
где ** указывает место присоединения к L и * указывает место присоединения к остальной части соединения.
В другом варианте осуществления, L представляет собой связь, -O-, -C1-C6алкилен-, -NHC(O) -, -C(O)-, -C(=NH)-, -S(O)m-, -SC1-C6алкилен-, -NR3(CH2)n-, -NHC(=NH)- или -NHS(O)m-, где -C1-C6алкилен-, -NHC(O) -, -C(=NH)-, -SC1-C6алкилен-, -NR3(CH2)n-, -NHC(=NH)- и -NHS(O)m- необязательно замещены одним-четырьмя R7.
В другом варианте осуществления, L представляет собой связь.
В другом варианте осуществления, L представляет собой -C1-C6алкилен- или -NR3(CH2)n-, где -C1-C6алкилен- и -NR3(CH2)n- необязательно замещены одним-четырьмя R7.
В другом варианте осуществления, L представляет собой -C1-C6алкилен-, где -C1-C6алкилен- необязательно замещен одним-четырьмя R7.
В другом варианте осуществления, L представляет собой -NR3(CH2)n-, где -NR3(CH2)n- необязательно замещен одним-четырьмя R7.
В другом варианте осуществления, R3 представляет собой водород. В другом варианте осуществления, R3 представляет собой -C1-C3 алкил.
В другом варианте осуществления, M представляет собой: -CH2OH, -NH2, -NHCH3 или -N+(CH3)3, где M необязательно замещен одним-двумя R6.
В другом варианте осуществления, M представляет собой -CH2OH, -NH2, -NHCH3 или -N+(CH3)3.
В другом варианте осуществления, M представляет собой -CH2OH.
В другом варианте осуществления, M представляет собой N(R3)2, N+(C1-C3алкил)3, C2-C6алкил, C3-C7 циклоалкил, HetA или AryA, где указанный C2-C6алкил, C3-C7 циклоалкил, HetA и AryA необязательно замещены одним-четырьмя R6.
В другом варианте осуществления, M представляет собой N(R3)2, N+(C1-C3алкил)3, C2-C6алкил, C3-C7 циклоалкил, HetA или AryA, где указанный C2-C6алкил, C3-C7 циклоалкил, HetA и AryA необязательно замещены одним-четырьмя R6.
В другом варианте осуществления, M представляет собой N(R3)2, N+(C1-C3алкил)3, C2-C6алкил, C3-C7 циклоалкил или HetA, где указанный C2-C6алкил, C3-C7 циклоалкил и HetA необязательно замещены одним-четырьмя R6.
В другом варианте осуществления настоящего изобретения, M представляет собой:
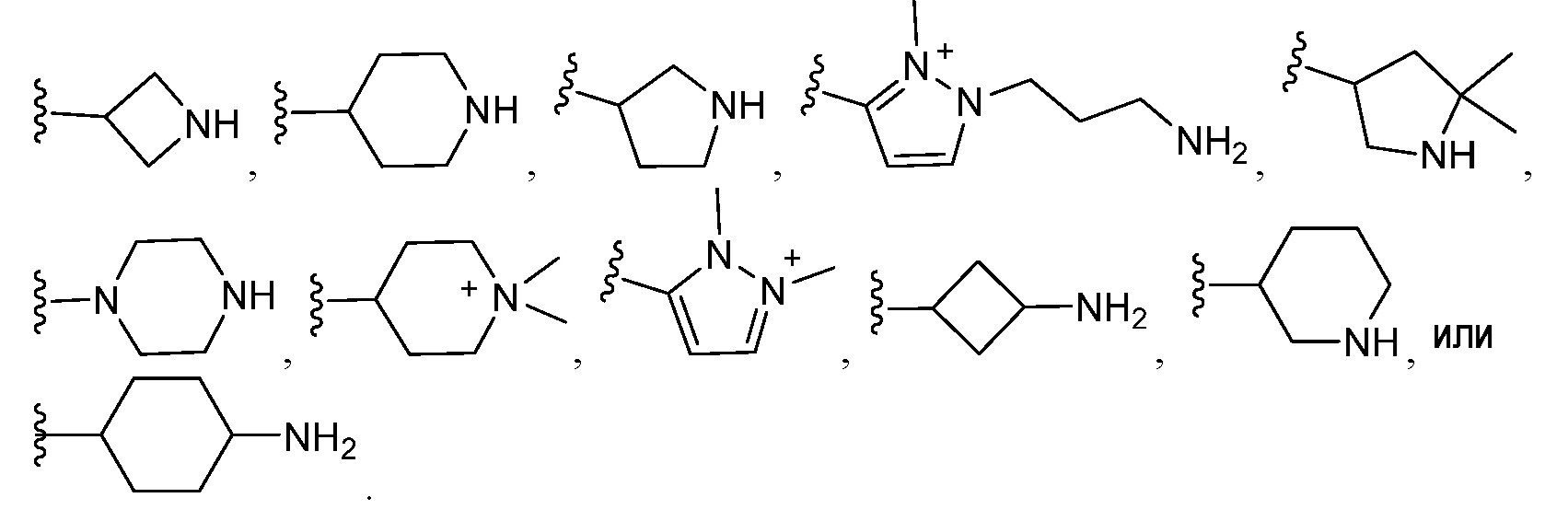
В другом варианте осуществления, в каждом случае R6 независимо выбран из группы, состоящей из: галогена, -C1-C6алкила, -(CH2)nNRcRd, -(CH2)qORe, -S(O)mRe, -S(O)mNRcRd, -C(O)Re,-OC(O)Re,-C(O)ORe, -CN, -C(O)NRcRd, -C(NH)NRcRd, -NRcRd, -N(Rc)(C(O)Re), -N(Rc)(C(O)ORe), -N(Rc)(C(O)NRcRd), -N(Rc)(S(O)mRe), и -C1-C3алкилен-HetA. В другом варианте осуществления, в каждом случае R6 независимо выбран из группы, состоящей из: -(CH2)qORe и -C1-C3алкилен-HetA.В другом варианте осуществления, в каждом случае R6 представляет собой -C1-C3алкилен-HetA.
В другом варианте осуществления, в каждом случае R7 независимо выбран из группы, состоящей из: галогена, -C1-C6алкила, -(CH2)nNRcRd, -(CH2)q-ORe, -S(O)mRe, -S(O)mNRcRd, -C(O)Re,-OC(O)Re,-C(O)ORe, -CN, -C(O)NRcRd, -C(NH)NRcRd, -NRcRd, -N(Rc)(C(O)Re), -N(Rc)(C(O)ORe), -N(Rc)(C(O)NRcRd), -N(Rc)(S(O)mRe)и -C1-C3алкилен-HetA.
В другом варианте осуществления, в каждом случае R7 независимо выбран из группы, состоящей из: галогена, -C1-C6алкила, -(CH2)nNRcRd, -(CH2)q-ORe, -C(O)NRcRd и -C1-C3алкилен-HetA.
В другом варианте осуществления, в каждом случае Rcи Rd независимо представляет собой: водород, -C1-C6алкил, -C2-C6алкенил, -C3-C6циклоалкил, -C1-C3алкилен-C3-C6 циклоалкил, HetA, -C1-C3алкилен-HetA, AryA, -C1-C3алкилен-AryA или -C1-C3алкилен-HetA, где каждый Rcи Rd необязательно замещен одним-тремя Rf.
В другом варианте осуществления, в каждом случае Rc независимо представляет собой: водород или -C1-C6алкил.
В другом варианте осуществления, в каждом случае Rd независимо представляет собой: водород или -C1-C6алкил.
В другом варианте осуществления, в каждом случае Re независимо представляет собой: водород, -C1-C6алкил, -C2-C6алкенил, -OH или -OC1-C6 алкил. В другом варианте осуществления, в каждом случае Reнезависимо представляет собой: водород или -C1-C6алкил. В другом варианте осуществления, Reпредставляет собой водород. В другом варианте осуществления, Reпредставляет собой -C1-C6алкил.
В другом варианте осуществления настоящего изобретения соединение формулы I выбрано из:
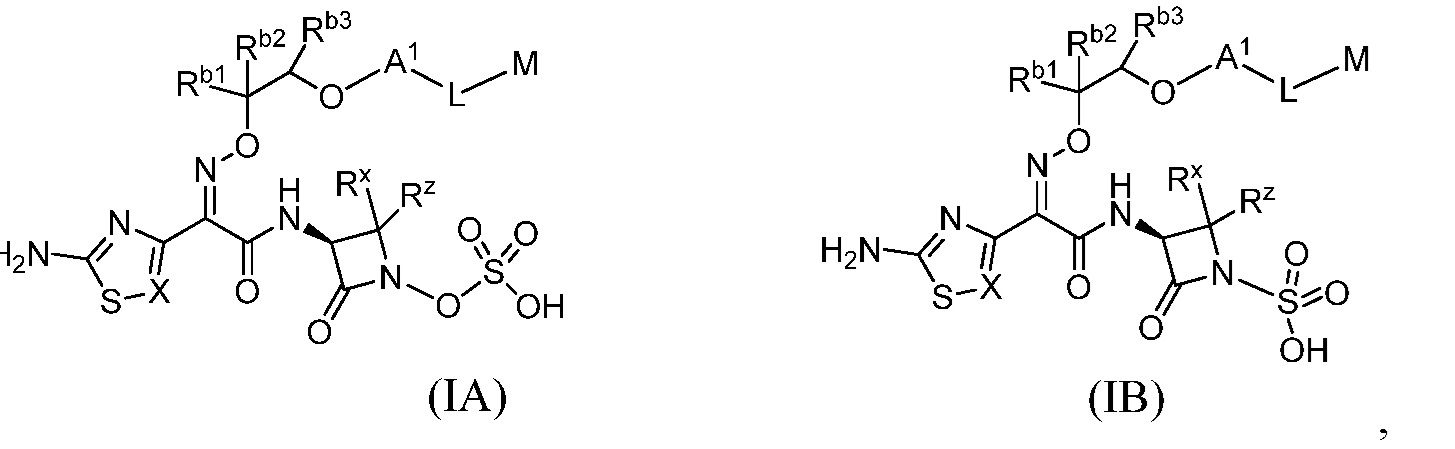
или его фармацевтически приемлемая соль.
В другом варианте осуществления настоящего изобретения соединение формулы I выбрано из:
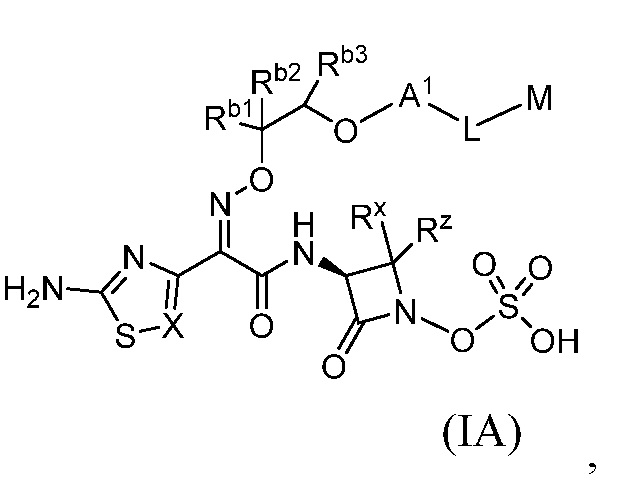
или его фармацевтически приемлемая соль.
В другом варианте осуществления настоящего изобретения соединение формулы I выбрано из:
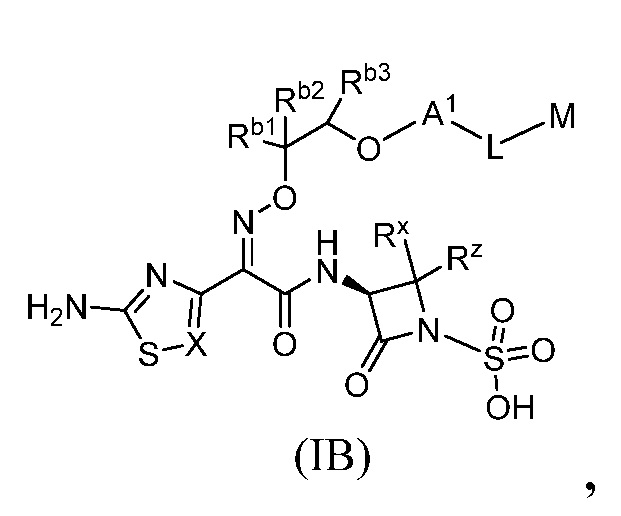
или его фармацевтически приемлемая соль.
В другом варианте осуществления настоящего изобретения соединение формулы (I) выбрано из:



или его фармацевтически приемлемая соль.
Другие варианты осуществления настоящего изобретения включают следующее:
(a) Фармацевтическая композиция, содержащая эффективное количество соединения формулы I, IA или IB, как определено в настоящем описании, или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
(b) Фармацевтическая композиция (а), дополнительно содержащая одно или несколько соединений ингибиторов бета-лактамазы.
(c) Фармацевтическая композиция (b), где по меньшей мере одно из одного или нескольких соединений ингибиторов бета-лактамазы, выбраны из группы, включающей: релебактам, тазобактам, клавулановую кислоту, сульбактам и авибактам.
(d) Фармацевтическая композиция, содержащая (i) соединение формулы I, IA или IB или его фармацевтически приемлемую соль и (ii) одно или несколько дополнительных соединений, где одно или несколько дополнительных соединений являются соединениями ингибиторами бета-лактамазы, где соединение формулы I, IA или IB или его фармацевтически приемлемая соль и одно или несколько дополнительных соединений, каждое используют в количестве, которое делает комбинацию эффективной для лечения или профилактики бактериальной инфекции.
(e) Комбинация (d), где одно или несколько дополнительных соединений выбраны из группы, состоящей из: релебактама, тазобактама, клавулановой кислоты, сульбактама и авибактама.
(f) Способ лечения бактериальной инфекции у субъекта, который включает введение субъекту, нуждающемуся в таком лечении, эффективного количества соединения формулы I, IA или IB или его фармацевтически приемлемой соли.
(g) Способ профилактики и/или лечения бактериальной инфекции, который включает введение субъекту, нуждающемуся в таком лечении, фармацевтической композиции, содержащей фармацевтически эффективное количество соединения формулы I, IA или IB или его фармацевтически приемлемой соли, и фармацевтически приемлемый носитель.
(h) Способ лечения бактериальной инфекции, который включает введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества композиции (a), (b), (c), (d) или (e).
(i) Способ лечения бактериальной инфекции, как указано в (f), (g) или (h), где бактериальная инфекция обусловлена грамотрицательными бактериями.
(j) Способ лечения бактериальной инфекции, как указано в (f), (g), (h) или (i), где бактериальная инфекция обусловлена Pseudomonas aeruginosa или Acinetobacter baumannii.
Настоящее изобретение также включает соединение формулы I, IA или IB или его фармацевтически приемлемую соль, (i) для использования в, (ii) для использования в качестве лекарственного средства для, или (iii) для использования при получении (или производстве) лекарственного средства для, в медицине или для лечения бактериальной инфекции, включая инфекцию бактериальным штаммом с множественной лекарственной устойчивостью. В этих применениях соединения по настоящему изобретению могут быть необязательно использованы в комбинации с одним или несколькими вторыми терапевтическими средствами, включая релебактам, тазобактам, клавулановую кислоту, сульбактам и авибактам.
Дополнительные варианты осуществления изобретения включают фармацевтические композиции, комбинации и способы, изложенные в (а)-(j) выше, и применения, изложенные в предыдущем абзаце, где соединение по настоящему изобретению, используемое в нем, представляет собой соединение одного из вариантов осуществления, подвариантов осуществления, классов или подклассов, описанных выше. В этих вариантах осуществления соединение может быть необязательно использовано в форме фармацевтически приемлемой соли.
В вариантах осуществления соединений и солей, представленных выше, следует понимать, что каждый вариант осуществления может быть объединен с одним или несколькими другими вариантами осуществления в той степени, в которой такая комбинация обеспечивает стабильное соединение или соль и соответствует описанию вариантов осуществления. Кроме того, следует понимать, что варианты осуществления композиций и способов, представленные в (а)-(j) выше, подразумевают включение всех вариантов осуществления соединений и/или солей, включая такие варианты осуществления, которые являются результатом комбинаций вариантов осуществления.
Дополнительные варианты осуществления настоящего изобретения включают каждые из фармацевтических композиций, комбинаций, способов и применений, изложенных в предыдущих параграфах, где используемое здесь соединение по настоящему изобретению или его соль являются по существу чистыми. Что касается фармацевтической композиции, содержащей соединение формулы I, IA или IB или его соль и фармацевтически приемлемый носитель и необязательно один или несколько эксципиентов, понятно, что термин «по существу чистое» относится к соединению формулы I, IA или IB или его соли как таковой; т.е. чистоте этого активного ингредиента в композиции.
Определения и аббревиатуры
Термины, используемые в настоящем описании, имеют свое обычное значение и значение таких терминов является независимым в каждом его случае. Несмотря на это и за исключением случаев, когда указано иное, в описании и формуле изобретения используются следующие определения. Химические названия, общие названия и химические структуры могут использоваться взаимозаменяемо для описания одной и той же структуры. Если химическое соединение определено с использованием как химической структуры, так и химического названия, и существует расхождение между структурой и названием, структура преобладает. Эти определения применяются независимо от того, используется ли термин сам по себе или в сочетании с другими терминами, если не указано иное. Таким образом, определение «алкил» относится к «алкилу», а также «алкильным» фрагментам «гидроксиалкила», «галогеналкила», «-O-алкила» и т.д.
Как используется здесь и во всем настоящем описании, следующие термины, если не указано иное, понимаются как имеющие следующие значения:
Термин «ингибитор β-лактамазы» относится к соединению, которое способно ингибировать активность фермента из β-лактамаз. Как используется в настоящем описании, ингибирование активности β-лактамазы означает ингибирование активности β-лактамазы класса A, C и/или D. В случае противомикробных применений предпочтительно, чтобы концентрация, при которой достигается 50% ингибирование, составляла примерно 100 микрограмм/мл или ниже, или примерно 50 микрограмм/мл или ниже, или примерно 25 микрограмм/мл. Термины β-лактамазы «класса А», «класса В», «класса С» и «класса D» понятны специалистам в данной области и описаны в S. G. Waley, β-lactamase: mechanisms of action, in The Chemistry of β-Lactams, M. I. Page, Ed.; Chapman and Hall, London, (1992) 198-228.
Термин «металло-β-лактамаза» обозначает металлопротеин, способный инактивировать β-лактамный антибиотик. β-Лактамаза может являться ферментом, который катализирует гидролиз β-лактамного кольца β-лактамного антибиотика. Особый интерес в настоящем описании представляют микробные металло-β-лактамазы. Металло-β-лактамаза может являться, например, металло-β-лактамазой, содержащей цинк. β-Лактамазы, представляющие интерес, включают те, которые описаны, например, в S. G. Waley, β-lactamase: mechanisms of action, in The Chemistry of β-Lactams, M. I. Page, Ed.; Chapman and Hall, London, (1992) 198-228. Представляющие в настоящем описании особый интерес β-лактамазы включают металло-β-лактамазы Escherichia coli (такие как металло-β-лактомаза из Нью-Дели (New Delhi Metallo-β-lactamase), NDM), Serratia marcescens (такие как IMP) и Klebsiella spp. (такие как интегронокодируемая металло-β-лактамаза из Вероны (Verona integron-encoded metallo-β-lactamase), VIM).). Дополнительные металло-β-лактамазы, представляющие интерес, включают ферменты SPM-, GIM-, SIM-, KHM-, AIM-, DIM-, SMB-, TMB- и FIM-типа.
Термин «антибиотик» относится к соединению или композиции, которые снижают жизнеспособность микроорганизма, или которые ингибируют рост или пролиферацию микроорганизма. Фраза «ингибирует рост или пролиферацию" означает увеличение времени генерации (то есть времени, требующегося для деления бактериальной клетки или для удвоения популяции), по меньшей мере, примерно в 2 раза. Предпочтительными антибиотиками являются антибиотики, которые могут увеличивать время генерации, по меньшей мере, примерно в 10 раз или более (например, по меньшей мере, примерно в 100 раз или даже до бесконечности, как в случае смерти всех клеток). Как используется в настоящем описании, дополнительно предполагается, что антибиотик включает противомикробное, бактериостатическое, или бактерицидное средство. Примеры антибиотиков включают пенициллины, цефалоспорины и карбапенемы.
Термин «β-лактамный антибиотик» относится к соединению с антибиотическими свойствами, которое содержит β-лактамную функциональность. Неограничивающие примеры β-лактамных антибиотиков включают пенициллины, цефалоспорины, пенемы, карбапенемы и монобактамы.
Термин «примерно» при изменении количества (например, кг, л или эквивалентов) вещества или композиции или величины физического свойства или величины параметра, характеризующего стадию процесса (например, температура при который проводят стадию процесса) или тому подобное, относится к отклонению численной величины, которое может происходить, например, при обычных методиках измерения, манипулирования и отбора проб, связанных с получением, характеристикой и/или использованием вещества или композиции; вследствие случайной ошибки в этих методиках; вследствие различий в производстве, источнике, или чистоте ингредиентов, используемых для получения или применения композиций, или в осуществлении методик; и тому подобное. В некоторых вариантах осуществления «примерно» может означать колебание ± 0,1, 0,2, 0,3, 0,4, 0,5, 1,0, 2,0, 3,0, 4,0 или 5,0 соответствующей единицы. В некоторых вариантах осуществления «примерно» может означать колебание ± 1%, 2%, 3%, 4%, 5%, 10% или 20%.
Другим вариантом осуществления настоящего изобретения является соединение формулы I, IA или IB или его фармацевтически приемлемая соль, как определено выше или как определено в любом из вышеприведенных вариантов осуществления, подвариантах осуществления, аспектах, классах или подклассах, где соединение или его соль находится в по существу чистой форме. Как используется в настоящем описании термин «по существу чистый» означает, соответственно, по меньшей мере примерно 60% мас., обычно, по меньшей мере примерно 70% мас., предпочтительно по меньшей мере примерно 80% мас., более предпочтительно, по меньшей мере примерно 90% мас. (например, от примерно 90% мас. до примерно 99% мас.), еще более предпочтительно по меньшей мере примерно 95% мас. (например, от примерно 95% мас. до примерно 99% мас. или от примерно 98% мас. до 100% мас.) и наиболее предпочтительно по меньшей мере примерно 99% мас. (например, 100% мас.) продукта, содержащего соединение формулы I, IA или IB или их соли (например, продукт, выделенный из реакционной смеси, обеспечивающий соединение или соль), состоит из соединения или соли. Степень чистоты соединений и солей может быть определена с использованием стандартного метода анализа, такого как тонкослойная хроматография, гель-электрофорез, высокоэффективная жидкостная хроматография и/или масс-спектрометрия. Если используют более чем один метод анализа, и методы обеспечивают в условиях эксперимента значительные различия в степени чистоты, то тогда метод, обеспечивающий самую высокую степень чистоты, имеет приоритетное значение. Соединение или соль 100% чистоты является соединением или солью, которые не содержат поддающиеся обнаружению примеси, определяемые стандартным методом анализа.
Что касается соединения по изобретению, которое имеет один или несколько центров асимметрии и может существовать в виде смесей стереоизомеров, по существу чистое соединение может быть или по существу чистой смесью стереоизомеров, или по существу чистым индивидуальным диастереомером или энантиомером, если явно не указано иное. Настоящее изобретение охватывает все стереоизомерные формы соединений формулы I, IA и IB. Если не указана конкретная стереохимия, настоящее изобретение предназначено для охвата всех таких изомерных форм этих соединений. Центры асимметрии, которые присутствуют в соединениях формулы I, IA и IB, могут независимо друг от друга иметь (R) или (S) конфигурацию. Когда связи с хиральным атомом углерода изображены в виде прямых линий в структурных формулах изобретения, понятно, что как (R), так и (S) конфигурации хирального атома углерода и, следовательно, как энантиомеры, так и их смеси включены в формулу. Аналогично, когда название соединения приведено без хирального обозначения для хирального углерода, подразумевается, что название включает как (R), так и (S) конфигурации хирального атома углерода и, следовательно, отдельные энантиомеры и их смеси. Получение конкретных стереоизомеров или их смесей может быть определено в примерах, где были получены такие стереоизомеры или смеси, но это никоим образом не ограничивает включение всех стереоизомеров и их смесей в объем настоящего изобретения.
Изобретение включает все возможные энантиомеры и диастереомеры и смеси двух или более стереоизомеров, например, смеси энантиомеров и/или диастереомеров, во всех соотношениях. Таким образом, энантиомеры являются предметом изобретения в энантиомерно чистой форме, в форме как левовращающих, так и правовращающих антиподов, в форме рацематов и в форме смесей двух энантиомеров во всех соотношениях. В случае цис/транс-изомерии изобретение включает как цис-форму, так и транс-форму, а также смеси этих форм во всех соотношениях. Получение индивидуальных стереоизомеров может быть осуществлено, при необходимости, путем разделения смеси обычными методами, например, хроматографией или кристаллизацией, с использованием стереохимически однородных исходных материалов для синтеза или стереоселективным синтезом. Необязательно дериватизация может быть проведена до разделения стереоизомеров. Разделение смеси стереоизомеров может проводиться на промежуточной стадии во время синтеза соединения формулы I, IA и IB, или это можно сделать после получения конечного рацемического продукта. Абсолютная стереохимия может быть определена с помощью рентгеновской кристаллографии кристаллических продуктов или кристаллических промежуточных продуктов, которые при необходимости дериватизируются реагентом, содержащим стереогенный центр известной конфигурации. Когда соединения по настоящему изобретению способны к таутомеризации, все индивидуальные таутомеры, а также их смеси включены в объем настоящего изобретения. Если не указан конкретный изомер, соль, сольват (включая гидраты) или сольватированная соль такого рацемата, энантиомера, диастереомера или таутомера, настоящее изобретение включает все такие изомеры, а также соли, сольваты (включая гидраты) и сольватированные соли таких рацематов, энантиомеров, диастереомеров и таутомеров и их смесей.
ʺAcʺ представляет собой ацетил, который представляет собой CH3C(=O)-.
«Алкил» означает насыщенные углеродные цепи, которые могут быть линейными или разветвленными или их комбинациями, если углеродная цепь не определена иначе. Другие группы, имеющие префикс «алк», такие как алкокси и алканоил, также могут быть линейными или разветвленными или их комбинациями, если углеродная цепь не определена иначе. Примеры алкильных групп включают метил, этил, пропил, изопропил, бутил, втор- и трет-бутил, пентил, гексил, гептил, октил, нонил и тому подобное.
ʺАлкилен,ʺ как используется в настоящем описании, относится к алкильной группе, как определено выше, в которой один из атомов водорода алкильной группы заменен на связь. Неограничивающие примеры алкиленовых групп включают -CH2-, -CH2CH2-, -CH2CH2CH2-, -CH2CH2CH2CH2-, -CH(CH3)CH2CH2-, -CH(CH3)- и -CH2CH(CH3)CH2-. В одном варианте осуществления алкиленовая группа имеет от 1 до приблизительно 6 атомов углерода. В другом варианте осуществления алкиленовая группа имеет от 1 до 3 атомов углерода. В другом варианте осуществления, алкиленовая группа является разветвленной. В другом варианте осуществления, алкиленовая группа является линейной. В одном варианте осуществления алкиленовая группа представляет собой -CH2-. Термин ʺC1-C6 алкиленʺ относится к алкиленовой группе, имеющей от 1 до 6 атомов углерода.
«Алкенил» означает углеродные цепи, которые содержат по меньшей мере одну углерод-углеродную двойную связь и которые могут быть линейными или разветвленными, или их комбинации, если не указано иное. Примеры алкенила включают винил, аллил, изопропенил, пентенил, гексенил, гептенил, 1-пропенил, 2-бутенил, 2-метил-2-бутенил и тому подобное.
"Алкинил" означает углеродные цепи, которые содержат по меньшей мере одну углерод-углеродную тройную связь, и которые могут быть линейными или разветвленными или их комбинации, если не указано иное. Примеры алкинила включают этинил, пропаргил, 3-метил-1-пентинил, 2-гептинил и тому подобное.
«Ароматическая кольцевая система» или «ароматическое» по отношению к кольцу означает моноциклическое, бициклическое или трициклическое ароматическое кольцо или кольцевую систему, содержащую 5-14 кольцевых атомов, причем по меньшей мере одно из колец является ароматическим. Термин может использоваться для описания насыщенного или мононенасыщенного карбоциклического кольца, конденсированного с арильной группой. Например, 5-7-членный циклоалкил может быть конденсирован через два соседних кольцевых атома с 5-6-членным гетероарилом, содержащим 1, 2 или 3 гетероатома в качестве кольцевых атомов, выбранных из N, О и S. В другом примере гетеромоноциклическое кольцо конденсировано через два кольцевых атома с фенилом или 5-6-членным гетероарилом, содержащим 1, 2 или 3 гетероатома, выбранных из N, О и S. В случае гетеромоноциклического кольца, содержащего один или несколько атомов N, N может быть в форме четвертичного амина. В некоторых вариантах осуществления кольцевой атом N может быть в форме N-оксида.
«9-11-членное бициклическое ароматическое кольцо» означает бициклическую кольцевую систему, в которой по меньшей мере одно из колец является ароматическим. Термин может быть использован для описания циклоалкильного кольца или циклоалкенильного кольца, конденсированного с арилом или гетероарильным кольцом. Этот термин также может быть использован для описания гетероциклоалкильного кольца или гетероциклоалкенильного кольца, конденсированного с арилом или гетероарильным кольцом. Например, 5-7-членный циклоалкил может быть конденсирован через два соседних кольцевых атома с 5-6-членным гетероарилом, содержащим 1, 2 или 3 гетероатома в качестве кольцевых атомов, выбранных из N, NH, O и S, или арилом. В другом примере гетероциклоалкильное кольцо конденсировано через два кольцевых атома с арильным или 5-6-членным гетероарильным кольцом, содержащим 1, 2 или 3 гетероатома, выбранных из N, NH, O и S. В случае гетероциклоалкильного кольца, содержащего один или несколько атомов N, N может быть в форме четвертичного амина. В некоторых вариантах осуществления кольцевой атом N может быть в форме N-оксида. Примеры 9-11-членного бициклического ароматического кольца включают, но не ограничиваются ими, хинолин, изохинолин, имидазо[1,2-а]пиридин, индазол, бензо[d]имидазол, бензо[d]тиазол или нафталин.
«Арил» означает моноциклическое, бициклическое или трициклическое карбоциклическое ароматическое кольцо или кольцевую систему, содержащую 6-14 атомов углерода, где по меньшей мере одно из колец является ароматическим. Примеры арила включают фенил и нафтил. В одном варианте осуществления настоящего изобретения, арил представляет собой фенил.
«Циклоалкил» означает насыщенное моноциклическое, бициклическое или мостиковое карбоциклическое кольцо, имеющее определенное количество атомов углерода. Примеры циклоалкила включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, инданил и тому подобное. В одном варианте осуществления настоящего изобретения, циклоалкил выбран из: циклопропана, циклобутана, циклопентана и циклогексана.
«Циклоалкенил» означает неароматическое моноциклическое или бициклическое карбоциклическое кольцо, содержащее по меньшей мере одну двойную связь. Примеры циклоалкенила включают циклопропенил, циклобутенил, циклопентенил, циклогексенил, циклогептенил, циклооксенил и тому подобное.
«Гетероциклоалкил», как используется в настоящем описании, относится к неароматической насыщенной моноциклической или полициклической кольцевой системе, содержащей 3-11 кольцевых атомов, где от 1 до 4 кольцевых атомов независимо представляют собой N, NH, S (включая SO и SO2) и O, и остальная часть кольцевых атомов представляют собой атомы углерода. Гетероциклоалкильная группа может быть присоединена через кольцевой атом углерода или кольцевой атом азота (если присутствует). Когда кольцо или кольцевая система содержит один или несколько атомов N, N может быть в форме четвертичного амина. В одном варианте осуществления гетероциклоалкильная группа является моноциклической и имеет от примерно 3 до примерно 7 кольцевых атомов. В другом варианте осуществления, гетероциклоалкильная группа является моноциклической, имеет от примерно 4 до примерно 7 кольцевых атомов. В другом варианте осуществления, гетероциклоалкильная группа является бициклической и имеет от примерно 7 до примерно 11 кольцевых атомов. Когда гетероциклоалкил содержит два кольца, кольца могут быть конденсированными или спироциклическими. В еще одном варианте осуществления гетероциклоалкильная группа является моноциклической и имеет 5 или 6 кольцевых атомов. В одном варианте осуществления гетероциклоалкильная группа является моноциклической. В другом варианте осуществления, a гетероциклоалкильная группа является бициклической. В кольцевой системе отсутствуют смежные атомы кислорода и/или серы. Любая -NH-группа в гетероциклоалкильном кольце может быть защищена такой как, например, -N(BOC), -N(Cbz), -N(Tos) группа и тому подобное; такие защищенные гетероциклоалкильные группы считаются частью настоящего изобретения. Атом азота или серы гетероциклоалкила (если присутствует) может быть необязательно окислен до соответствующего N-оксида, S-оксида или S,S-диоксида. Неограничивающие примеры моноциклических гетероциклоалкильных колец включают оксетанил, пиперидил, пирролидин, пиперазинил, морфолинил, тиоморфолинил, тиазолидинил, 1,4-диоксанил, тетрагидрофуранил, дельта-лактам, дельта-лактон, силациклопентан, силапирролидин и тому подобное, и все их изомеры.
«Гетероциклоалкенил» означает неароматическое моноциклическое, бициклическое или мостиковое карбоциклическое кольцо или кольцевую систему, содержащую по меньшей мере одну двойную связь, и содержащую по меньшей мере один гетероатом, выбранный из N, NH, S и O. В одном варианте осуществления гетероциклоалкенил относится к неароматической насыщенной моноциклической или полициклической кольцевой системе, содержащей 3-11 кольцевых атомов, содержащей по меньшей мере одну двойную связь, где от 1 до 4 кольцевых атомов независимо представляют собой N, NH, S (включая SO и SO2) и O, а остальная часть кольцевых атомов представляют собой атомы углерода.
«Гетероарил» означает моноциклическое, бициклическое или трициклическое кольцо или кольцевую систему, содержащую 5-14 атомов углерода, и содержащую по меньшей мере один кольцевой гетероатом, выбранный из N, NH, S (включая SO и SO2) и O, где по меньшей мере одно из колец, содержащих гетероатом, является ароматическим. В случае гетероарильной кольцевой системы, где одно или несколько колец являются насыщенными и содержат один или несколько атомов N, N может быть в форме четвертичного амина. В одном варианте осуществления гетероарильная группа имеет 5-10 кольцевых атомов. В другом варианте осуществления, гетероарильная группа является моноциклической и имеет 5 или 6 кольцевых атомов. В другом варианте осуществления, гетероарильная группа является бициклической. Любой атом азота гетероарила может быть необязательно окислен до соответствующего N-оксида. Термин «гетероарил» также охватывает гетероарильную группу, как определено выше, которая конденсирована с бензольным кольцом. Примеры гетероарила включают пирролил, изоксазолил, изотиазолил, пиразолил, пиридил, оксазолил, оксадиазолил, тиадиазолил, тиазолил, имидазолил, триазолил, тетразолил, фуранил, триазинил, тиенил, пиримидил, пиридазинил, пиразинил, бензизоксазолил, бензоксазолил, бензотиазолил, бензимидазолил, бензопиразолил, бензотиофуранил, бензотиофенил (включая S-оксид и диоксид), бензотриазолил, фуро(2,3-b)пиридил, хинолил, индолил, изохинолил, хиназолинил, дибензофуранил и тому подобное. В одном варианте осуществления настоящего изобретения гетероарил выбран из: пиридина, пиримидина, тиазола, бензимидазола, бензтиазола, бензоксазола и бензизоксазола. В другом варианте осуществления настоящего изобретения, гетероарил представляет собой пиридин. Примеры бициклических колец включают:

«Гетероцикл» означает моноциклическую или бициклическую насыщенную, частично ненасыщенную или ненасыщенную кольцевую систему, содержащую 5-10 атомов и содержащую по меньшей мере один кольцевой гетероатом, выбранный из N, S и O. В некоторых вариантах осуществления кольцевая система содержит 1-4 гетероатома, выбранных из N, S и O. Когда гетероцикл содержит два кольца, кольца могут быть конденсированными, мостиковыми или спироциклическими. Примеры моноциклических гетероциклических колец включают пиперазин, пиперидин и морфолин. Примеры бициклических гетероциклических колец включают 1,4-диазабицикло[2,2,2]октан и 2,6-диазаспирогептан.
«Галоген» включает фтор, хлор, бром и йод.
«Оксо» означает атом кислорода, связанный с другим атомом двойной связью, и может быть представлен «= О».
Когда любая переменная (например, R1, Rа, и т.п.) появляется более одного раза в любой составляющей или в формуле I, IA и IB, ее определение для каждого случая не зависит от ее определения для каждого другого случая. Кроме того, комбинации заместителей и/или переменных являются допустимыми, только если такие комбинации приводят к образованию стабильных соединений. Волнистая линия по связи в переменной заместителя представляет собой точку присоединения.
«Устойчивый к лекарственным средствам» означает по отношению к грамотрицательным бактериальным штаммом штамм, который больше не чувствителен по меньшей мере к одному ранее эффективному лекарственному средству; который развил способность противостоять воздействию антибиотика по меньшей мере одного ранее эффективного лекарственного средства. ʺС множественной лекарственной устойчивостьюʺ означает штамм, который больше не чувствителен к двум или более ранее эффективным лекарственным средствам; который развил способность противостоять воздействию антибиотика двумя или более ранее эффективными препаратами. Штамм, устойчивый к лекарственным средствам, может передавать эту способность переносить действие лекарственного средства своему потомству. Упомянутая устойчивость может быть результатом случайных генетических мутаций в бактериальных клетках, которая изменяет чувствительность к одному лекарственному средству или различным лекарственным средствам.
«Стабильным» соединением является соединение, которое может быть получено и выделено, и чья структура и свойства остаются или могут в результате принятых мер оставаться практически неизменными в течение периода времени, достаточного для использования соединения для целей, описанных в настоящем документе (например, введение субъекту в терапевтических целях). Соединения по настоящему изобретению ограничиваются стабильными соединениями, охватываемыми формулой I, IA и IB.
В соответствии со стандартной номенклатурой, используемой на протяжении описании, концевая часть обозначенной боковой цепи описывается последней, которой предшествует соседняя функциональность по направлению к точке присоединения.
При выборе соединений по настоящему изобретению специалисту в данной области будет понятно, что различные заместители, то есть R1, R2 и т.п., должны выбираться в соответствии с известными принципами связности и стабильности химической структуры.
Считается, что термин «замещенный» включает множественные степени замещения названным заместителем. Когда описано или заявлено несколько частей-заместителей, соединение может быть независимо замещено одним или несколькими описанными или заявленными частями-заместителями однократно или многократно. Под независимо замещенным подразумевается, что заместители (два или несколько) могут быть одинаковыми или различными. Когда группа, например, C1-C6алкил, обозначена как замещенная, такие замены могут также происходить, если такая группа является частью более крупного заместителя, например, -C1-C6алкил-C3-C7циклоалкил и -C1-C6алкил-арил.
В соединениях формулы I, IA и IB атомы могут иметь свои природные изотопные составы, или один или несколько атомов могут быть искусственно обогащены конкретным изотопом, имеющим один и тот же атомный номер, но атомную массу или массовое число, отличное от атомной массы или массового числа, которое преимущественно встречается в природе. Подразумевается, что настоящее изобретение включает все подходящие изотопные вариации соединений формулы I, IA и IB. Например, различные изотопные формы водорода (H) включают протий (1H) и дейтерий (2H или D). Протий является преобладающим изотопом водорода, обнаруженным в природе. Обогащение для дейтерия может дать определенные терапевтические преимущества, такие как увеличение периода полувыведения in vivo или снижение необходимой дозировки, или может обеспечить соединение, применимое в качестве стандарта для характеристики биологических образцов. Изотопно-обогащенные соединения формул I, IA и IB могут быть получены без дополнительного эксперимента с использованием общепринятых спобов, хорошо известных специалистам в данной области, или способами, аналогичными описанным на схемах и примерах, представленных в настоящем описании, с использованием подходящих изотопно-обогащенных реагентов и/или промежуточных соединений.
Если прямо не указано иное в конкретном контексте, то любое из различных циклических колец и кольцевых систем, описанных в настоящем описании, могут быть присоединены к остальной части соединения по любому кольцевому атому (то есть любому углеродному атому или любому гетероатому) при условии, что это приводит к образованию стабильного соединения.
Если прямо не указано иное, то все приводимые здесь интервалы являются включающими в себя интервалами. Например, гетероароматическое кольцо, описываемое как содержащее от "1 до 4 гетероатомов", означает, что кольцо может содержать 1, 2, 3 или 4 гетероатома. Кроме того, следует иметь в виду, что любой приводимый здесь интервал включает внутри себя область всех подинтервалов внутри этого интервала. Так, например, предполагается, что гетероциклическое кольцо, описываемое как содержащее от "1 до 4 гетероатомов", включает в качестве его аспектов гетероциклические кольца, содержащие от 2 до 4 гетероатомов, 3 или 4 гетероатома, от 1 до 3 гетероатомов, 2 или 3 гетероатома, 1 или 2 гетероатома, 1 гетероатом, 2 гетероатома, 3 гетероатома, и 4 гетероатома. Аналогично, C1-C6 при использовании с цепью, например, алкильной цепью, означает, что цепь может содержать 1, 2, 3, 4, 5 или 6 атомов углерода. Он также включает все диапазоны, содержащиеся в нем, включая C1-C5, C1-C4, C1-C3, C1-C2, C2-C6, C3-C6, C4-C6, C5-C6, и все другие возможные комбинации.
Следует также отметить, что любой углерод, а также гетероатом с неудовлетворенными валентностями в тексте, схемах, примерах и таблицах, здесь, как предполагается, имеют достаточное количество атома(атомов) водорода для удовлетворения валентностей.
Соединения по настоящему изобретению имеют по меньшей мере один центр асимметрии и могут иметь один или несколько дополнительных центров в результате присутствия определенных заместителей и/или структур заместителя. Соответственно, соединения по изобретению могут встречаться в виде смесей стереоизомеров, или в виде отдельных диастереомеров, или энантиомеров. Все изомерные формы этих соединений, независимо друг от друга или в смесях, входят в объем настоящего изобретения.
Термин «соединение» относится к свободной форме соединения и любому его гидрату или сольвату, при условии, что они являются стабильными. Гидрат представляет собой соединение, образовавшее комплекс с водой, и сольват представляет собой соединение, образовавшее комплекс с органическим растворителем.
Как указано выше, соединения по настоящему изобретению могут быть использованы в форме фармацевтически приемлемых солей. Следует понимать, что, как используется в настоящем описании, соединения по настоящему изобретению могут также включать фармацевтически приемлемые соли, а также соли, которые не являются фармацевтически приемлемыми, когда они используются в качестве предшественников для свободных соединений или их фармацевтически приемлемых солей или при других операциях синтезирования.
Термин «фармацевтически приемлемая соль» относится к соли, которая обладает эффективностью исходного соединения и которая не является биологически или иным образом нежелательной (например, не является токсичной ни иным образом опасной для реципиента). Термин «фармацевтически приемлемая соль» относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований или кислот, включая неорганические или органические основания и неорганические или органические кислоты. Соли основных соединений, охваченные термином "фармацевтически приемлемая соль", относятся к нетоксичным солям соединений по настоящему изобретению, которые обычно получают при взаимодействии свободного основания с подходящей органической или неорганической кислотой. Характерные соли основных соединений по настоящему изобретению включают, без ограничения, следующие: ацетат, аскорбат, адипат, альгинат, аспират, бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, бромид, бутират, камфорат, камфорсульфонат, камзилат, карбонат, хлорид, клавуланат, цитрат, циклопентанпропионат, диэтилацетат, диглюконат, дигидрохлорид, додецилсульфанат, эдетат, эдисилат, эстолат, эзилат, этансульфонат, формиат, фумарат, глюцептат, глюкогептаноат, глюконат, глутамат, глицерофосфат, гликоллиларсанилат, гемисульфат, гептаноат, гексаноат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, 2-гидроксиэтансульфонат, гидроксинафтоат, иодид, изоникотинат, изотионат, лактат, лактобионат, лаурат, малат, малеат, манделат, мезилат, метилбромид, метилнитрат, метилсульфат, метансульфонат, мукат, 2-нафталинсульфонат, напсилат, никотинат, нитрат, соль N-метилглюкамин аммония, олеат, оксалат, памоат (эмбонат), пальмитат, пантотенат, пектинат, персульфат, фосфат/дифосфат, пимелиновой кислоты, фенилпропионат, полигалактуронат, пропионат, салицилат, стеарат, сульфат, субацетат, сукцинат, таннат, тартрат, теоклат, тиоцианат, тозилат, триэтиодид, трифторацетат, ундеконат, валерат и тому подобное. Кроме того, если соединения по изобретению несут кислотный фрагмент, их подходящие фармацевтически приемлемые соли включают, но не ограничиваются ими, соли, полученные из неорганических оснований, включая алюминий, аммоний, кальций, медь, железо, двухвалентное железо, литий, магний, трехвалентный марганец, двухвалентный марганец, калий, натрий, цинк и тому подобное. Особенно предпочтительными являются аммониевые, кальциевые, магниевые, калиевые и натриевые соли. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, циклических аминов, дициклогексиламинов и основные ионообменные смолы, такие как аргинин, бетаин, кофеин, холин, N,N-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этиламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и тому подобное. Также, основные азотсодержащие группы могут быть кватернизованы такими агентами, как низшие алкилгалогениды, такие как метил-, этил-, пропил- и бутилхлорид, бромиды и иодиды; диалкилсульфатами, такими как диметил, диэтил, дибутил; и диамилсульфаты, галогенидами с длинной цепью, такими как децил-, лаурил-, миристил- и стеарилхлориды, бромиды и иодиды, аралкилгалогенидами, такими как бензил- и фенетилбромиды и другие.
Эти соли могут быть получены известными способами, например, путем смешивания соединения по настоящему изобретению с эквивалентным количеством и раствором, содержащим нужную кислоту, основание или тому подобное, и затем собирание желаемой соли путем фильтрации соли или отгонки растворителя. Соединения по настоящему изобретению и их соли могут образовывать сольваты с растворителем, таким как вода, этанол или глицерин. Соединения по настоящему изобретению могут образовывать кислотно-аддитивную соль и соль с основанием в то же время в зависимости от типа заместителя боковой цепи.
Соединение по изобретению можно также использовать в форме пролекарства. Любое пролекарство предшественник, известное в данной области, может быть использовано для образования пролекарства по изобретению. В некоторых аспектах настоящего варианта осуществления водород в -COOH в формуле I может быть заменен на любые следующие группы: C1-6 алкил, C3-6 циклоалкил, -C1-6 алкилен-C3-6 циклоалкил, C3-7 циклогетероалкил,-C1-6 алкилен-C3-7 циклогетероалкил, арил, -C1-10 алкилен-арил, гетероарил, и -C1-10 алкилен-гетероарил. В некоторых аспектах этого варианта осуществления, C1-6алкил, C3-6 циклоалкил или C3-7 циклогетероалкил может быть замещены. В других аспектах этого варианта осуществления каждый арил и гетероарил могут быть замещены.
Как указано выше, настоящее изобретение включает фармацевтические композиции, содержащие соединение формулы I по настоящему изобретению, необязательно включающие один или несколько других активных компонентов (например, ингибитор β-лактамазы) и фармацевтически приемлемый носитель. Характеристики носителя будут зависеть от пути введения. Под понятием "фармацевтически приемлемый" подразумевается то, что ингредиенты фармацевтической композиции должны быть совместимы друг с другом, не препятствуют эффективности активного ингредиента (ингредиентов), и не являются опасными (например, токсичными) для реципиента. Таким образом, композиции по изобретению могут, помимо ингибитора, содержать разбавители, наполнители, соли, буферы, стабилизаторы, солюбилизаторы и другие материалы, хорошо известные в данной области.
Также, как указано выше, настоящее изобретение включает способ лечения бактериальной инфекции, который включает введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы I, IA и IB или его фармацевтически приемлемой соли, необязательно в комбинации с одним или несколькими ингибиторами β-лактамазы. Термин "субъект" (или, альтернативно, "пациент"), как используется в настоящем описании, относится к животному, предпочтительно, млекопитающему, наиболее предпочтительно человеку, который являлся объектом лечения, исследования или эксперимента. Термин «введение» и его варианты (например, «введение» соединения) по отношению к соединению формулы I, IA и IB означает предоставление соединения или его фармацевтически приемлемой соли человеку, нуждающемуся в лечении. Когда соединение или его соль предоставляют в комбинации с одним или несколькими другими активными средствами (например, ингибитором β-лактамазы), «введение» и его варианты, как понимается, каждый включают предоставление соединения или его соли и других средств одновременно или в разное время. Когда средства комбинации вводят одновременно, они могут быть введены вместе в одной композиции или они могут быть введены раздельно. Следует иметь в виду, что «комбинацией» активных средств может быть одна композицией, содержащей все активные средства, или множество композиций, каждая из которых содержит один или несколько активных средств. В случае двух активных средств комбинация может представлять собой или одну композицию, содержащую оба средства, или две отдельные композиции, каждая из которых содержит одно из средств; в случае трех активных средств комбинация может представлять собой или одну композицию, содержащую все три средства, три отдельные композиции, каждая из которых содержит один из средств, или две композиции, одна из которых содержит два средства, и другая содержит третье средство; и так далее.
Композиции и комбинации настоящего изобретения вводят соответствующим образом в эффективных количествах. Термин «эффективное количество», как используется в настоящем описании, означает количество активного соединения, достаточное для ингибирования роста бактерий, и, таким образом, вызывающего желаемый ответ (то есть «ингибирующее эффективное количество») в клетке, ткани, системе, животном или человеке. В одном варианте осуществления эффективное количество представляет собой «терапевтически эффективное количество» для облегчения симптомов заболевания или состояния, подвергаемых лечению (например, лечение состояний, связанных с бактериальной инфекцией и/или устойчивостью бактерий к действию лекарственного средства). В другом варианте осуществления, эффективным количеством является «профилактически эффективное количество» для профилактики симптомов заболевания или состояния, которые хотят предотвратить. Когда активное соединение (то есть активный ингредиент) вводят в виде соли, ссылки на количество активного ингредиента относятся к форме свободной кислоты или форме свободного основания соединения.
Введение композиции по настоящему изобретению осуществляется соответствующим образом парентерально, перорально, сублингвально, трансдермально, местно, интраназально, интратрахеально, интраокулярно или интраректально, где композицию соответствующим образом готовят для введения путем выбранного способа с использованием способов получения, хорошо известных в данной области, включая, например, способы получения и введения лекарственных форм, описанных в главах 39, 41, 42, 44 и 45 в Remington - The Science and Practice of Pharmacy, 21st edition, 2006. В одном варианте осуществления соединения по изобретению вводят внутривенно в условиях стационара. В другом варианте осуществления, введением является пероральное введение в форме таблетки или капсулы или другой подобной форме. При системном введении, терапевтическую композицию, например, вводят подходящим образом в достаточной дозировке для достижения концентрации ингибитора в крови по меньшей мере около 1 микрограмм/мл и в дополнительном варианте осуществления составляет по меньшей мере около 10 микрограмм/мл и по меньшей мере около 25 микрограмм/мл. Для локализованного введения эффективными могут являться значительно более низкие концентрации, чем эти, и допустимыми могут являться значительно более высокие концентрации.
Внутривенное введение соединения по изобретению может быть осуществлено путем восстановления порошкообразной формы соединения с приемлемым растворителем. Подходящие растворители включают, например, физиологические растворы (например, 0,9% раствор хлорида натрия для инъекций) и стерильную воду (например, стерильную воду для инъекций, бактериостатическую воду для инъекций с метилпарабеном и пролпилпарабеном, или бактериостатическую воду для инъекций с 0,9% бензилового спирта). Порошкообразная форма соединения может быть получена путем облучение соединения гамма-лучами или путем лиофилизации раствора соединения, после чего порошок можно хранить (например, в герметичной ампуле) при комнатной температуре или ниже, пока он не будет восстановлен. Концентрация соединения в восстановленном растворе для внутривенного вливания может находиться, например, в интервале от примерно 0,1 мг/мл до примерно 20 мг/мл.
Настоящее изобретение также включает способ ингибирования бактериального роста, который включает введение в бактериальную клеточную культуру, или в бактериально инфицированную клеточную культуру, ткань, или организм, ингибирующего эффективного количества соединения формулы I, IA и IB. Дополнительные варианты осуществления изобретения включают включают только что описанный способ ингибирования бактериального роста, где используемым в нем соединением по настоящему изобретению является соединение одного из вариантов осуществления, подвариантов или классов, описанных выше. В этих вариантах осуществления соединение может необязательно быть использовано в форме фармацевтически приемлемой соли. Способ может включать введение соединения формулы I, IA и IB в экспериментальную клеточную культуру in vitro для предотвращения роста бактерий, устойчивых к действию β-лактама. Способ может, в качестве альтернативы, включать введение соединения формулы I животному, включая человека, для предотвращения роста бактерий, устойчивых к β-лактаму, in vivo. В этих случаях соединение формулы I, IA и IB обычно вводят совместно с одним или несколькими соединениями ингибиторами β-лактамазы.
Способы раскрытого в настоящее момент предмета изобретения являются полезными для лечения этих состояний тем, что они ингибируют начало, рост или распространение состояния, вызывают регрессию состояния, вылечивают состояние или иным образом улучшают общее самочувствие субъекта, страдающего или подверженного риску заражения состоянием. Таким образом, в соответствии с раскрытым предметом настоящего изобретения термины «лечить», «лечение» и их грамматические вариации, а также фраза «способ лечения» предназначены для охвата любого желаемого терапевтического вмешательства, включая, но не ограничиваясь этим, способ лечения существующей инфекции у субъекта и способ профилактики (т.е. предотвращения) инфекции, например, у субъекта, который был подвергнут воздействию микроорганизма, как раскрыто в настоящем документе, или который имеет вероятность подвергнуться воздействию микроорганизма, как раскрыто в настоящем документе.
Соединения изобретения могут применяться для лечения, профилактики или ингибирования бактериального роста или инфекций, вызванных бактериями, которые устойчивы к действию β-лактамовых антибиотиков. Более конкретно, бактериями могут являться металло-β-лактамазовые положительные штаммы, которые обладают высокой устойчивостью к действию β-лактамовых антибиотиков. Термины "малоустойчивые" и "высокоустойчивые" широко используются обычными специалистами в данной области (см., например, Payne et al., Antimicrobial Agents and Chemotherapy 38:767-772 (1994); Hanaki et al., Antimicrobial Agents and Chemotherapy 30:11.20-11.26 (1995)). Для целей настоящего изобретения штаммами бактерий, которые обладают высокой устойчивостью к имипенему, являются штаммы, против которых MIC (минимальная ингибирующая концентрация) для имипенема составляет >16 мкг/мл, и штаммами бактерий, которые имеют малую устойчивость к действию имипенема, являются штаммы, против которых MIC для имипенема составляет >4 мкг/мл.
Соединения по изобретению могут использоваться в комбинации с одним или несколькими ингибиторами β-лактамазы для лечения инфекций, вызванных штаммами, продуцирующими β-лактамазу, в дополнение к тем инфекциям, которые включены в антибактериальный спектр антибиотического средства. Примерами бактерий, продуцирующих β-лактамазу, являются Pseudomonas aeruginosa, Pseudomonas putida, Enterobacter cloacae, Klebsiella pneumoniae, Klebsiella oxytoca, Escherichia coli, Serratia marcescens, Enterobacter aerogenes, Enterobacter asburiae, Citrobacter freundii, Proteus mirabilis, Morganella morganii, Providencia rettgeri, Stenotrophomonas maltophilia и Acinetobacter baumannii.
Обычно полезно использовать соединение формулы I, IA и IB в смеси или в сочетании с одним или несколькими ингибиторами β-лактамазы, или его пролекарством. Полезно использовать соединение формулы I в комбинации с ингибитором β-лактамазы класса A и C из-за свойств устойчивости к β-лактамазе класса B соединений. Также полезно использовать соединение формулы I в комбинации с одним или несколькими ингибиторами β-лактамазы класса А, С или D для дальнейшего ограничения восприимчивости β-лактама. Как уже отмечалось, соединение формулы I и один или несколько ингибиторов β-лактамазы можно вводить отдельно (одновременно или в разное время) или в форме одной композиции, содержащей оба активных ингредиента.
Релебактам, тазобактам, клавулановую кислоту, сульбактам, авибактам и другие ингибиторы β-лактамазы и металло-β-лактамазы, подходящие для использования в настоящем изобретении, включают те, которые, как известно, проявляют ингибирующую активность к β-лактамазам.
Используемые в настоящем описании аббревиатуры включают следующие: водн.=водный; ACN=ацетонитрил; BLI=ингибитор β-лактамазы; Bn=бензил; BOC (или Boc)=трет-бутилоксикарбонил; BOC2O= ди-трет-бутилдикарбонат; CAN= церий аммония нитрат; CELITE=диатомовая земля; CBZ (или Cbz)=карбобензокси (в качестве варианта, бензилоксикарбонил); CDCl3= дейтерированный хлороформ; CH3CN=ацетонитрил; DBU=1,8-диазабицикло-[5.4.0]ундец-7-ен; DCC=дициклогексилкарбодиимид; DCE= 1,2-дихлорэтан; DCM=дихлорметан; DEAD= диэтилазодикарбоксилат; DIAD= диизопропил азодикарбоксилат; DIBAL-H= диизобутилалюминий-гидрид; DIEA= диизопропилэтиламин; DMA=диметилацетамид; DMAP=4-диметиламинопиридин или N,N-диметиламинопиридин; DMF=N,N-диметилформамид; DMSO=диметилсульфоксид; EA представляет собой этилацетат; EDC=1-этил-3-(3-диметиламинопропил) карбодиимид; эк. или эквив.=эквивалент(ы); Et=этил; Et2O= этиленоксид; EtOAc=этилацетат; EtOH=этанол; HOBT=1-гидроксибензотриазол; ВЭЖХ=высокоэффективная жидкостная хроматография; IPA=изопропиловый спирт; LC/MS или LC-MS=жидкостная хроматография/масс-спектрометрия; LDA=диизопропиламид лития; m-CPBA=м-хлорпероксибензойная кислота; MBL=металло-β-лактамаза; Me=метил; MeOH=метанол; MeI= метилиодид; MITC=минимальная ингибирующая пороговая концентрация; MPLC=жидкостная хроматография среднего давления; MTBE=метил трет-бутиловый эфир; NBS=N-бромсукцинимид; NCS=N-хлорсукцинимид; ЯМР=ядерный магнитный резонанс; MS=масс-спектрометрия; MW=молекулярная масса; PE представляет собой петролейный эфир; Pd/c=палладий на угле; PdCl2(dppf)=[1,2'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II); PG=защитная группа; Ph=фенил; RP-HPLC= обращенно-фазовая высокоэффективная жидкостная хроматография; комн.темп.=комнатная температура; насыщ.=насыщенный; предкатализатор RuPhos второго поколения представляет собой хлор(2-дициклогексилфосфино-2′,6′-диизопропокси-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладий(II) (или RuPhos-Pd-G2); SEM-Cl=2-(триметилсилил)этоксиметил хлорид; tBu=трет-бутил; TBAF=фторид тетрабутиламмония; TBME= трет-бутил метиловый эфир; TBS= трет-бутилдиметилсилил; t-BuOH=трет-бутанол; TBSO=трет-бутилдиметилсилил; TEA=триэтиламин; TFA=трифторуксусная кислота; THF=тетрагидрофуран; TLC=тонкослойная хроматография; и TMS= триметилсилил.
Способы получения соединений формулы (I):
Соединения, раскрытые в настоящем описании, могут быть получены и протестированы в соответствии со следующими схемами реакции и примерами или их модификациями с использованием легко доступных исходных материалов, реагентов и общепринятых способов синтеза. В этих реакциях также можно использовать варианты, которые сами по себе известны специалистам в данной области, но не упоминаются здесь более подробно. Кроме того, другие способы получения соединений, описанных в настоящем документе, будут легко понятны специалисту в данной области с помощью следующей схемы реакции и примеров. Если не указано иное, все переменные определены выше.
Схема 1
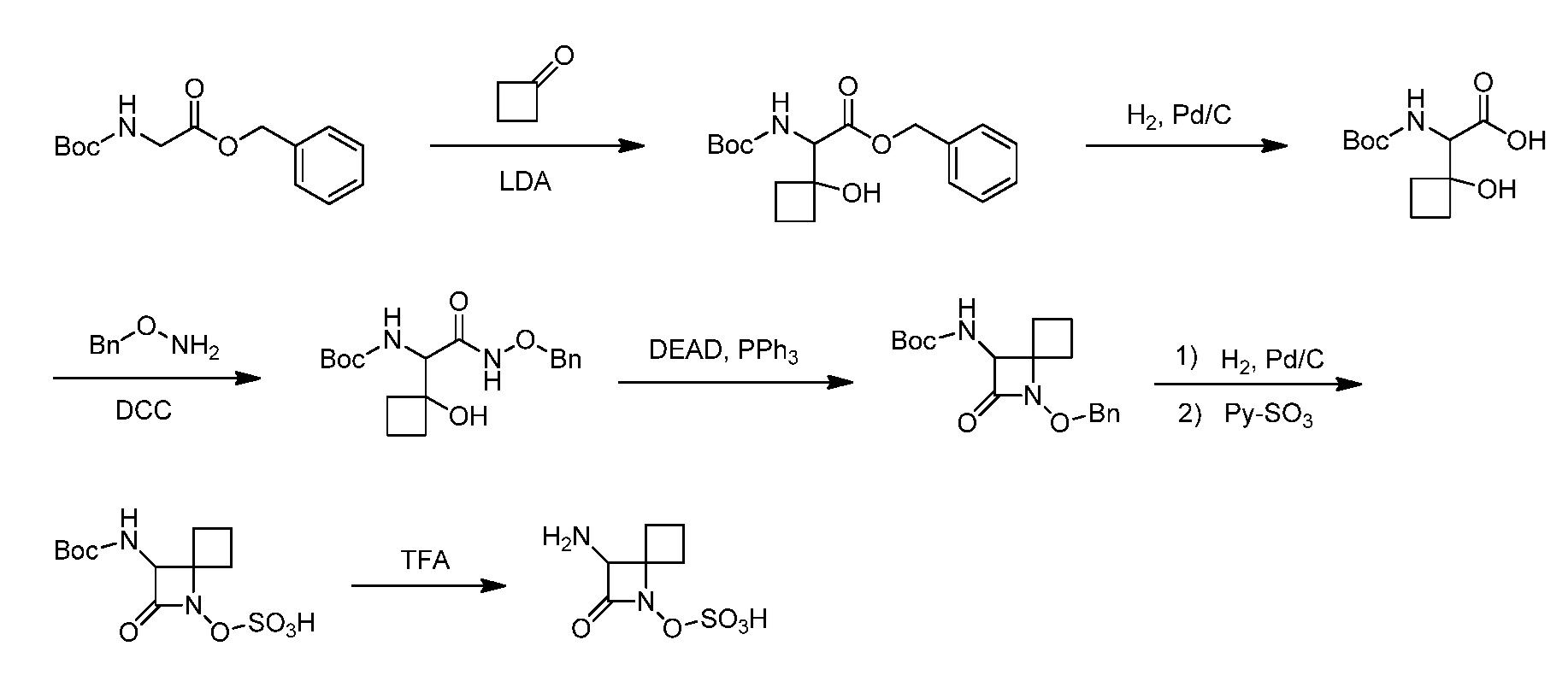
Монобактамовые соединения, содержащие спироцикл при Rx и Rx, могут быть синтезированы по вышеприведенной схеме, которая показывает синтез β-лактамового промежуточного соединения, где Rx и Rz взятые вместе образуют 4-членное спироциклическое кольцо. Конечное β-лактамовое промежуточное соединение, показанное на схеме, альтернативно можно приобрести из коммерческих источников. Схема синтеза также подробно обсуждалась в литературе. (см. EP 0229012). Этот амин можно превратить в конечные монобактамовые соединения, используя процедуры, аналогичные тем, которые показаны в следующих примерах.
Промежуточное соединение 1
трет-бутил оксиран-2-карбоксилат

В 1-литровую 2-горлую круглодонную колбу, снабженную конденсатором, m-CPBA добавляли частями к раствору трет-бутил акрилата (20 г, 156 ммоль) в DCM (200 мл) (48,5 г, 281 ммоль). Полученный раствор нагревали до 58-60°С с масляной баней и кипятили с обратным холодильником в течение 2 ½ дней. Смесь проверяли с помощью ЯМР, чтобы убедиться, что реакция завершена. Смесь затем охлаждали до комнатной температуры и добавляли небольшие порции насыщенного раствора тиосульфата натрия (примерно 40 мл, экзотермия, добавляли небольшими порциями, пока не было больше тепла). После перемешивания смеси в течение примерно 1 часа появлялось большое количество осадка. Для рассеивания эмульсии и получения двухфазной системы добавляли примерно 60-100 мл воды и 100-200 мл DCM. Водный слой отделяли и органическую фазу промывали насыщенным NaHCO3 (2×200 мл) и насыщенным солевым раствором (100 мл), сушили над MgSO2 и концентрировали досуха (температура водяной бани при 35°C). Остаток суспендировали в 150 мл гексана и оставляли стоять при комнатной температуре в течение 1 часа. Смесь затем фильтровали, и фильтрат концентрировали для удаления гексана в роторном испарителе (BÜCHI Labortechnik AG, Flawil, Switzerland) (< 35°C), что приводит к желаемому продукту.1Н ЯМР (500 МГц, CDCl3) δ 3,35 (м, 1H), 2,86 (м, 2H), 1,52 (с, 9H).
Промежуточное соединение 2
(R, R)-Co катализатор

Ссылка: J. Am. Chem. Soc. 1999, 121, 6086-6087. К раствору перфтор-трет-бутанола (1,96 г, 8,28 ммоль) в DCM (97 мл) добавляли (R,R)-(-)-N,N'-бис(3,5-ди-трет-бутилсалицилиден)-1,2-циклогександиаминокобальт(II) (5 г, 8,28 ммоль). Затем смесь перемешивали при 30°C в течение 45 минут на открытом воздухе. Затем реакционную смесь концентрировали и сушили с помощью HiVac с получением твердого вещества.
Промежуточное соединение 3
2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусная кислота

Стадия A: Получение этил 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоацетат К раствору этил 2-(2-аминотиазол-4-ил)-2-оксоацетата (10 г, 49,9 ммоль) в ацетонитриле (250 мл) добавляли BOC-ангидрид (23,2 мл, 100 ммоль), затем N,N,N',N'-тетраметилэтилендиамин (9,80 мл, 64,9 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов. Растворитель удаляли, и остаток разделяли между EtOAc и 1 н HCl. Органический слой промывали NaHCO3(насыщенный водный раствор) и насыщенным солевым раствором, и сушили над Na2SO4. Растворитель удаляли под вакуумом. Остаток очищали с помощью колоночной хроматографии на силикагеле (redi flash 220g), и элюируя смесью EtOAc/гексан (0-30%, 5cv; 30%, 10cv) с получением целевого продукта в виде твердого вещества. LC-MS [M+H]: m/z 301.
Стадия B: Получение 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусной кислоты
Этил 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоацетат (10,2 г, 34,1 ммоль) растворяли в THF (140 мл)/MeOH (50 мл), и добавляли гидроксид натрия (68,3 мл, 68,3 ммоль, 1M). Раствор перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь выливали в воду (1 л) и экстрагировали EtOAc (3×200 мл). Водный слой подкисляли раствором HCl (1н) и повторно экстрагировали EtOAc (3×200 мл). Органический слой промывали насыщенным солевым раствором и сушили над Na2SO4, и концентрировали с получением целевого продукта в виде твердого вещества. LC-MS [M+H]: m/z 273.1Н ЯМР (500 МГц, CDCl3) δ 8,35 (с, 1H), 1,55 (с, 9H).
Промежуточное соединение 4
2-(2-((трет-бутоксикарбонил)амино)-5-хлортиазол-4-ил)-2-оксоуксусная кислота
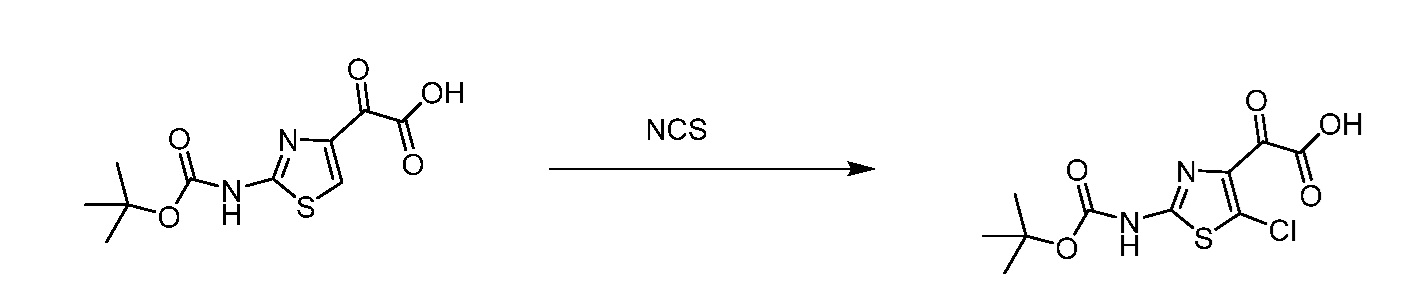
NCS (0,589 г, 4,41 ммоль) добавляли к суспензии 2-(2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)-2-оксоуксусной кислоты (Промежуточное соединение 3) (1 г, 3,67 ммоль) в DMF (10,0 мл). Смесь нагревали до 50°C в течение ночи. Затем его разбавляли EtOAc (100 мл), и промывали водой (3×30 мл) и насыщенным солевым раствором. Органический слой сушили над Na2SO4. Растворитель удаляли под вакуумом с получением целевого продукта в виде смолы. LC-MS [M+H]: m/z 307.
Промежуточное соединение 5
2-(5-((трет-бутоксикарбонил)амино)-1,2,4-тиадиазол-3-ил)-2-оксоуксусная кислота
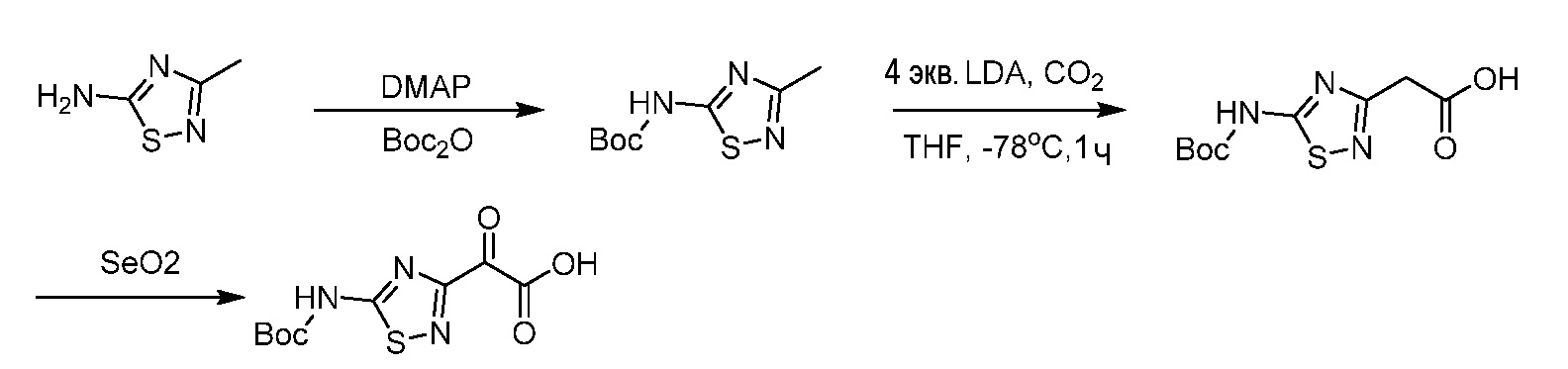
Стадия A: Получение трет-бутил (3-метил-1,2,4-тиадиазол-5-ил)карбамата В 5-литровую четырехгорлую круглодонную колбу, продутую и поддерживаемую инертной атмосферой азота, помещали 3-метил-1,2,4-тиадиазол-5-амин (167 г, 1,45 mol, 1,00 эквив.), 4-диметиламино-пиридин (17,7 г, 144,88 ммоль, 0,10 эквив.), ди-трет-бутилдикарбонат (348 г, 1,59 mol, 1,10 эквив.), и бутан-1-ол (1670 мл). Полученный раствор перемешивали в течение 1 часа при 40oC. Полученную смесь концентрировали в вакууме. Остаток промывали гексаном. Это привело к получению трет-бутил N-(3-метил-1,2,4-тиадиазол-5-ил)карбамата в виде твердого вещества.
Стадия B: Получение 2-(5-((трет-бутоксикарбонил)амино)-1,2,4-тиадиазол-3-ил)уксусной кислоты В 5000-мл четырехгорлую круглодонную колбу, продутую и поддерживаемую инертной атмосферой азота, помещали раствор трет-бутил N-(3-метил-1,2,4-тиадиазол-5-ил)карбамата (128 г, 595 ммоль, 1,00 эквив.) в тетрагидрофуране (640 мл). Полученный раствор перемешивали при -78°C и добавляли LDA (1190,69 мл, 4,00 эквив.). Через 30 минут в раствор вводили CO2 (g) в течение 30 минут при -30°C. Затем реакцию гасили добавлением 1280 мл воды. Полученный раствор экстрагировали 640 мл этилацетата и водные слои объединяли. Значение рН раствора доводили до 2 с помощью HCl (2М моль/л). Полученный раствор экстрагировали 2,5 л этилацетата и органические слои объединяли. Полученную смесь промывали 2000 мл насыщенного солевого раствора. Смесь сушили над безводным сульфатом натрия и концентрировали под вакуумом. Это привело к получению 2-(5-[[(трет-бутокси)карбонил]амино]-1,2,4-тиадиазол-3-ил)уксусной кислоты в виде твердого вещества.
Стадия C: Получение 2-(5-((трет-бутоксикарбонил)амино)-1,2,4-тиадиазол-3-ил)-2-оксоуксусной кислоты В 2000-мл четырехгорлую круглодонную колбу, продутую и поддерживаемую инертной атмосферой азота, помещали раствор 2-(5-[[(трет-бутокси)карбонил]амино]-1,2,4-тиадиазол-3-ил)уксусной кислоты (76 г, 293,12 ммоль, 1,00 эквив.) в диоксане (1520 мл) и SeO2 (65,14 г, 587 ммоль, 2,00 эквив.). Полученный раствор перемешивали в течение 3 часов при 80°C на масляной бане и затем концентрировали. Неочищенный продукт очищали Flash-Prep-HPLC со следующими условиями (IntelFlash-1): колонка, силикагель; подвижная фаза, ACN/H2O (0,5% HCl)=10/90-30/70, увеличивающаяся до ACN/H2O (0,5% HCl)=90/10-70/30 в течение 20 мин; Детектор, УФ 254 нм. Это привело к получению 2-(5-[[(трет-бутокси)карбонил]амино]-1,2,4-тиадиазол-3-ил)-2-оксоуксусной кислоты в виде твердого вещества. LC-M: (ES, m/z): (M+H)=274. H-ЯМР (300 МГц, DMSO, ppm): δ 1,523-1,502 (с, 9H), 12,806 (с, 1H).
Промежуточное соединение 6
трет-бутил 3-((6-бромимидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилат

Стадия A: 2-(1-(трет-бутоксикарбонил)азетидин-3-ил)уксусный (изобутил угольный) ангидрид
Изобутил хлорформиат (3,2 г, 23 ммоль) добавляли к 0°C охлажденной смеси 2-(1-(трет-бутоксикарбонил)азетидин-3-ил)уксусной кислоты (5 г, 23 ммоль) и 4-метилморфолина (2,6 г, 26 ммоль) в THF (80 мл) в атмосфере азота. Смесь перемешивиал при 26°C в течение 30 минут. LCMS указала, что реакция завершена. Смесь быстро фильтровали через слой целита (толщиной около 5 мм), который непосредственно использовали на следующей стадии без очистки. LCMS m/z [M+Na]+: 338,1
Стадия B: трет-бутил 3-(3-диазо-2-оксопропил)азетидин-1-карбоксилат К раствору свежеполученного диазометана (9,8 г, 230 ммоль) в Et2O (540 мл) добавляли по каплям 2-(1-(трет-бутоксикарбонил)азетидин-3-ил)уксусный (изобутил угольный) ангидрид (8,8 г, 28 ммоль) в THF (100 мл) при 0°C. Реакционную смесь перемешивали при 0°C в течение 0,5 часа и затем нагревали до 25°C в течение 1 часа. Через смесь в течение 5 минут барботировали медленный поток азота для удаления избытка диазометана. Затем растворитель осторожно удаляли на роторном испарителе (<35°C) и сушили на вакуумном насосе с получением трет-бутил 3-(3-диазо-2-оксопропил)азетидин-1-карбоксилата, который непосредственно использовали на следующей стадии без очистки.
Стадия C: трет-бутил 3-(3-бром-2-оксопропил)азетидин-1-карбоксилат К раствору трет-бутил 3-(3-диазо-2-оксопропил)азетидин-1-карбоксилата (6,7 г, 28 ммоль) в THF (120 мл) добавляли водн. 48% HBr (3,2 мл, 28 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 60 минут. Полученную смесь гасили насыщенным водн. NaHCO3 (60 мл), и растворитель удаляли в вакууме. Затем реакционную смесь разбавляли EtOAc (50 мл) и водой (100 мл). Водный слой экстрагировали EtOAc (3×60 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением трет-бутил 3-(3-бром-2-оксопропил)азетидин-1-карбоксилата, который использовали непосредственно для следующей стадии без очистки. LCMS m/z [M-55+41]+: 279
Стадия D: трет-бутил 3-((6-бромимидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилат
К смеси трет-бутил 3-(3-бром-2-оксопропил)азетидин-1-карбоксилата (6 г, 12 ммоль) и 5-бромпиридин-2-амина (2,7 г, 15 ммоль) в EtOH (90 мл) добавляли бикарбонат натрия (2,0 г, 24 ммоль). Реакционную смесь перемешивали при 80°C в течение 12 часов. Этанол удаляли в вакууме и полученный материал смешивали с водой (50 мл) и этилацетатом (50 мл). Водный слой экстрагировали EtOAc (3×30 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии (SiO2, PE:EtOAc=20:1-1:1) с получением трет-бутил 3-((6-бромимидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата. LCMS m/z [M+H]+: 3661H ЯМР (400 МГц, CHCl3-d) δ 7,44-7,39 (м, 1H), 7,33-7,31 (м, 1H), 7,23-7,18 (м, 1H), 4,07-4,01 (м, 2H), 3,71-3,66 (м, 2H), 3,06-2,99 (м, 3H), 1,43 (с, 9H).
Промежуточное соединение 7
трет-бутил (R)-3-(3-формил-4-нитрофенокси)-2-гидроксипропаноат

Смесь трет-бутил оксиран-2-карбоксилата (4,42 г, 30,7 ммоль), 5-гидрокси-2-нитробензальдегида (2,33 г, 13,94 ммоль), молекулярных сит (1 г) и кобальтового катализатора (R,R) (0,585 г, 0,697 ммоль, Ссылка: J. Am. Chem. Soc. 1999, 121, 6086-6087) в t-BuOMe (20 мл) перемешивали при комнатной температуре в течение 3 дней. После фильтрации через целитTM, фильтрат концентрировали и остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением указанного в заголовке соединения. LC/MS: [M+H]+: 311,2; (2M+23)+: 645,2.1H ЯМР (500 МГц, CDCl3): δ 10,49 (с, 1H), 8,19-8,17 (д, J=9,0 Гц, 1H), 7,38-7,37 (д, J=4,1 Гц, 1H), 7,22-7,20 (дд, J=9,8 и 3,3 Гц, 1H), 4,46-4,37 (м, 3H), 3,29 (с, 1H), 1,51 (с, 9H).
Промежуточное соединение 8
трет-бутил (S)-(3-амино-2-((трет-бутилдиметилсилил)окси)пропил)карбамат
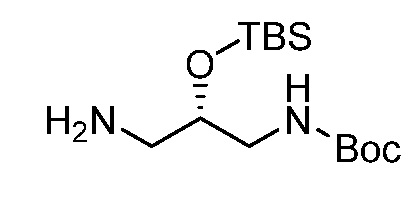
Стадия A: (S)-бензил трет-бутил (2-гидроксипропан-1,3-диил)дикарбамат К раствору (S)-трет-бутил (3-амино-2-гидроксипропил)карбамата (2,38 г, 12,5 ммоль) в DCM (50 мл) при 0°C добавляли TEA (4,53 мл, 32,5 ммоль) и CBZ-Cl (2,32 мл, 16,3 ммоль). Полученный раствор перемешивали от 0°C до комнатной температуры в течение ночи. Смесь концентрировали и остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением указанного в заголовке соединения. LC/MS: [M+1]+= 325,2.
Стадия B: (S)-бензил трет-бутил (2-((трет-бутилдиметилсилил)окси)пропан-1,3-диил)дикарбамат К раствору (S)-бензил трет-бутил (2-гидроксипропан-1,3-диил)дикарбамата (2,94 г, 9,06 ммоль) в DMF (20 мл) добавляли имидазол (3,09 г, 45,3 ммоль), TBS-Cl (2,73 г, 18,1 ммоль), и DMAP (0,111 г, 0,906 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 2 ч. Затем смесь разделяли между EtOAc (300 мл) и водой (200 мл). Органическую фазу промывали водой (200 мл×2), и насыщенным солевым раствором (200 мл), сушили над Na2SO4 и концентрировали. Полученный остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением указанного в заголовке соединения. LC/MS: [M+1]+= 439,4.
Стадия C: (S)-трет-бутил (3-амино-2-((трет-бутилдиметилсилил)окси)пропил)карбамат К раствору (S)-бензил трет-бутил (2-((трет-бутилдиметилсилил)окси)пропан-1,3-диил)дикарбамата (3,99 г, 9,10 ммоль) в MeOH (100 мл) добавляли 10% Pd/C (0,968 г, 0,910 ммоль). Полученную смесь гидрировали с помощью баллонного H2 при комнатной температуре в течение 1 часа. Затем смесь фильтровали через целитTM, и фильтрат концентрировали с получением указанного в заголовке соединения. LC/MS: [M+1]+= 305,3.
Промежуточное соединение 9
ди-трет-бутил (2-(аминометил)пропан-1,3-диил)дикарбамат
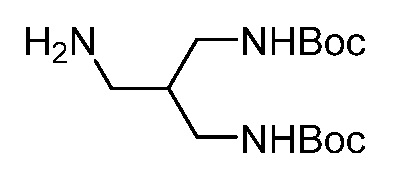
Стадия A: метил 3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)амино)метил)пропаноат К раствору метил 3-амино-2-(аминометил)пропаноат дигидрохлорида (1,01 г, 4,92 ммоль) в диоксане (50 мл), воде (10 мл) и THF (20 мл) при 0°C добавляли ди-трет-бутилдикарбонат (2,36 г, 10,8 ммоль) и ди-изопропилэтиламин (1,89 мл, 10,8 ммоль). Полученный раствор перемешивали при комнатной температуре в течение ночи. Летучие вещества удаляли. Полученный остаток растворяли в EtOAc (200 мл), и промывали ледяной HCl (0,5 н, 3×100 мл), затем насыщенным NaHCO3 (2×100 мл), затем сушили над Na2SO4, и концентрировали с получением указанного в заголовке соединения. LC/MS: [M+1]+=333,3.
Стадия B: ди-трет-бутил (2-(гидроксиметил)пропан-1,3-диил)дикарбамат К смеси метил 3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)амино)метил)пропаноата (1,66 г, 4,99 ммоль), литий боргидрида (0,544 г, 25,0 ммоль), и хлорида лития (1,06 г, 25,0 ммоль) в THF (безводный, 40 мл) при 0°C добавляли MeOH (6 мл) по каплям в течение 10 мин. Ледяную баню удалили и реакционную смесь перемешивали при комнатной температуре в течение прибл. 50 мин. Затем реакционную смесь охлаждали до 0°C и и гасили 16 мл насыщенным NaCl (16 мл); добавляли дополнительное количество твердого NaCl для обеспечения насыщения, и затем разбавляли EtOAc (200 мл). Органический слой собирали. Водный слой снова экстрагировали DCM (100 мл×3). Органические слои объединяли, сушили над Na2SO4, и концентрировали в вакууме с получением указанного в заголовке соединения. LC/MS: [M+1]+: 305,3.
Стадия C: 3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)амино)метил)пропил метансульфонат К раствору ди-трет-бутил (2-(гидроксиметил)пропан-1,3-диил)дикарбамата (283 мг, 0,93 ммоль) в DCM (безводный, 8 мл) при 0°C добавляли TEA (0,259 мл, 1,86 ммоль) и MsCl (0,087 мл, 1,12 ммоль). Полученный раствор перемешивали при 0°C в течение 1 часа, затем реакционный раствор разделяли между DCM (100 мл) и 0,2 M KHSO4. Органическую фазу отделяли, промывали 0,2 M KHSO4, сушили над Na2SO4, и концентрировали с получением указанного в заголовке соединения. LC/MS: [M+1]+: 383,3.
Стадия D: ди-трет-бутил (2-(азидометил)пропан-1,3-диил)дикарбамат К раствору 3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)амино)метил)пропил метансульфоната (1,57 г, 4,10 ммоль) в DMSO (6 мл) добавляли азид натрия (0,801 г, 12,3 ммоль). Полученную смесь нагревали при 90°C в течение ночи. Затем смесь разбавляли EtOAc (200 мл) и промывали водой (3×100 мл), сушили над Na2SO4, и концентрировали. Полученный остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением указанного в заголовке соединения. LC/MS: [M+1]+= 330,4.
Стадия E: ди-трет-бутил (2-(аминометил)пропан-1,3-диил)дикарбамат К раствору ди-трет-бутил (2-(азидометил)пропан-1,3-диил)дикарбамата (1,4 г, 4,25 ммоль) в MeOH (50 мл) добавляли Pd/C (4,52 г, 4,25 ммоль). Полученную смесь гидрировали с помощью баллонного H2 при комнатной температуре в течение 3 часов. Затем смесь фильтровали через целитTM, и фильтрат концентрировали с получением указанного в заголовке соединения. LC/MS: [M+1]+: 304,3.
Промежуточное соединение 10
трет-бутил (R)-2-((трет-бутилдиметилсилил)окси)-3-((3-хлоризохинолин-7-ил)окси)пропаноат

Стадия A: (Z)-2-(гидроксиимино)-6-метокси-2,3-дигидро-1Н-инден-1-он 6-Метокси-2,3-дигидро-1Н-инден-1-он (30 г, 185 ммоль) растворяли в MeOH (400 мл). Смесь нагревали до 40°C, затем добавляли изоамилнитрит (52,3 мл, 388 ммоль) и концентрировали HCl (30,4 мл, 370 ммоль). Реакционную смесь перемешивали при 40°C в течение 2 ч, и затем охлаждали до комнатной температуры. Полученный осадок собирали фильтрованием и сушили в высоком вакууме с получением указанного в заголовке соединения, которое использовали непосредственно без дополнительной очистки. LCMS (ESI) вычислено для C10H9NO3[M+Na]+: 192,0, найдено: 192,0
Стадия B: 1,3-дихлор-7-метоксиизохинолин К суспензии (Z)-2-(гидроксиимино)-6-метокси-2,3-дигидро-1Н-инден-1-она (10 г, 52,3 ммоль) в POCl3 (91 мл, 978 ммоль), и PCl5 (17,10 г, 82 ммоль) добавляли при 0°C, затем через раствор барботировали газообразный HCl (избыток) до тех пор, пока раствор не будет насыщен HCl. Реакционную смесь перемешивали при 30°C в течение 18 ч, затем растворитель удаляли в вакууме и воду со льдом (50 мл) добавляли к полученному остатку. Полученный осадок собирали фильтрованием, промывали водой (5 мл), и сушили под высоким вакуумом с получением указанного в заголовке соединения, которое использовали непосредственно без дополнительной очистки. LCMS (ESI) вычислено для C10H7Cl2NO[M+H]+: 228,0, 230,0, найдено: 228,0.
Стадия C: 3-хлор-7-метоксиизохинолин 1,3-дихлор-7-метоксиизохинолин (11 г, 48,2 ммоль) суспендировали в уксусной кислоте (90 мл) и концентрировали HCl (30 мл), и затем обрабатывали Sn (17,18 г, 144,7 ммоль), и затем перемешивали при 60°C в течение 24 ч. Полученную смесь подщелачивали до pH=9 концентрированной NH4OH и затем экстрагировали этилацетатом (150 мл×3). Объединенные органические слои промывали насыщенным раствором NaHCO3 (400 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Полученный остаток очищали с помощью колонки с силикагелем (SiO2; EtOAc/PE=1:20-2:1) с получением указанного в заголовке соединения. LC-MS (ESI) вычислено для C10H8ClNO[M+H]+: 194,0, найдено: 194,0.
Стадия D: 3-хлоризохинолин-7-ол 3-хлор-7-метоксиизохинолин (4,7 г, 24,3 ммоль) растворяли в DCM (80 мл). Затем BBr3 (6,20 мл, 65,5 ммоль) медленно добавляли при 25°C и раствор перемешивали при 25°C в течение 18 ч. После охлаждения до 0°C, метанол (40 мл) медленно добавляли для гашения реакции. Раствор перемешивали в течение дополнительных 10 минут, затем концентрировали при пониженном давлении. Полученный остаток обрабатывали метанолом (40 мл) и концентрировали при пониженном давлении. Полученное масло медленно обрабатывали насыщенным водным раствором бикарбоната натрия при перемешивании до достижения рН ~ 7-8. Полученное твердое вещество собирали с использованием вакуумной фильтрации и промывали водой (10 мл) и метиленхлоридом (10 мл) с получением указанного в заголовке соединения, которое использовали непосредственно без дополнительной очистки. LCMS (ESI) вычислено для C9H6ClNO[M+H]+: 180,0, найдено: 180,0.
Стадия E: (R)-трет-бутил 3-((3-хлоризохинолин-7-ил)окси)-2-гидроксипропаноат К смеси Co-катализатора (1,31 г, 1,56 ммоль), 3-хлоризохинолин-7-ола (3,5 г, 19,5 ммоль) и молекулярных сит (200 мг) в TBME (10 мл) добавляли трет-бутил оксиран-2-карбоксилат (10 г, 55,5 ммоль). Полученную суспензию перемешивали при 25°C в атмосфере N2 в течение 72 ч. Затем реакционную смесь фильтровали и очищали с помощью хроматографии на силикагеле (SiO2, EA:PE=0%-40%) с получением указанного в заголовке соединения. LC-MS (ESI) вычислено для C16H18ClNO4 [M+H]+: 324,0,найдено: 324,1.
Стадия F: (R)-трет-бутил 2-((трет-бутилдиметилсилил)окси)-3-((3-хлоризохинолин-7-ил)окси)пропаноат К смеси (R)-трет-бутил 3-((3-хлоризохинолин-7-ил)окси)-2-гидроксипропаноата (1,9 г, 5,87 ммоль), имидазола (1,2 г, 17,6 ммоль) в DMF (20 мл) добавляли TBS-Cl (1,77 г, 11,7 ммоль), полученную суспензию перемешивали при 25°C в течение 18 ч. LCMS показала сформировавшийся желаемый продукт. Реакционную смесь фильтровали и разбавляли EtOAc (200 мл), промывали насыщенным солевым раствором (3*180 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали. Смесь остатков очищали с помощью хроматографии на силикагеле (SiO2, EA:PE=0%-30%) с получением указанного в заголовке соединения. LCMS (ESI) вычислено для C22H32ClNO4Si [M+H]+: 438,1, найдено: 438,2.1H ЯМР (400 МГц, хлороформ-d) δ=8,92-8,88 (м, 1H), 8,84 (с, 1H), 7,60-7,51 (м, 2H), 7,27 (дд, J=2,3, 9,0 Гц, 1H), 7,13 (д, J=2,3 Гц, 1H), 4,45-4,40 (м, 1H), 4,24 (шир. д, J=3,3 Гц, 1H), 4,15 (шир. д, J=7,0 Гц, 1H), 1,40 (с, 9H), 0,81 (с, 9H), 0,05 (с, 3H), 0,00 (с, 3H).
Промежуточное соединение 11
трет-бутил 3-(((3-((трет-бутоксикарбонил)амино)пропил)амино)метил)азетидин-1-карбоксилат
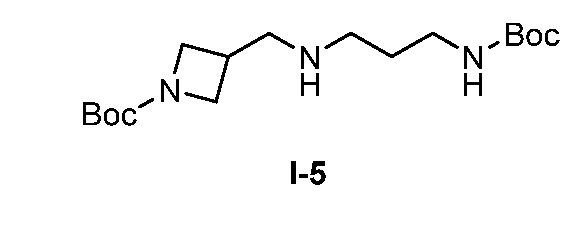
К раствору трет-бутил (3-аминопропил)карбамата (1,6 г, 9,18 ммоль) в MeOH (50 мл) добавляли трет-бутил 3-формилазетидин-1-карбоксилат (1,70 г, 9,18 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли триацетоксиборгидрид натрия (3,89 г, 18,4 ммоль) и AcOH (1,05 мл, 18,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 5 часов и гасили водой. MeOH удаляли под вакуумом. Раствор разбавляли EtOAc, промывали водн. раствором NaHCO3, водой и насыщенным солевым раствором. Органический раствор сушили с Na2SO4 и концентрировали с получением указанного в заголовке соединения. LC-MS [M+H]: m/z 344,41.
Промежуточное соединение 12
трет-бутил (R)-3-((4-(N,N-бис(трет-бутоксикарбонил)амино)-2-хлорхинолин-6-ил)окси)-2-((трет-бутилдиметилсилил)окси)пропаноат

Стадия A: 2,4-дихлор-6-метоксихинолин POCl3 (80 мл) добавляли через конденсатор в 250-мл круглодонную колбу, содержащую малоновую кислоту (25,3 г, 244 ммоль) при 20°C. При перемешивании небольшими порциями в течение 15 минут добавляли 4-метоксианилин (20 г, 162 ммоль). Реакционную смесь нагревали и перемешивали при 105°C в течение 3 часов. Затем реакционную смесь охлаждали до 20°C и концентрировали в вакууме для удаления POCl3. Полученный остаток растворяли в DCM (200 мл). Затем смесь выливали в концентрированный гидроксид аммония, и конечный рН водного слоя составлял около 10. Водный слой экстрагировали DCM (3×100 мл). Объединенные органические слои сушили над Na2SO4, фильтровалии концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии (SiO2, PE:EtOAc =100:1-10:1) с получением указанного в заголовке соединения.
Стадия B: 2-хлор-6-метокси-N-(4-метоксибензил)хинолин-4-амин К смеси 2,4-дихлор-6-метоксихинолина (6,0 г, 26,3 ммоль) и (2,4-диметоксифенил)метанамина (6,60 г, 39,5 ммоль) в DMSO (80 мл) добавляли Et3N (11,00 мл, 79 ммоль). Реакционную смесь перемешивали при 90°C в течение 48 ч. Затем реакционную смесь охлаждали до 20°C, и разбавляли EtOAc (100 мл) и водой (100 мл). Водный слой экстрагировалиEtOAc (3×100 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии (SiO2, PE:EtOAc =10:1-1:1) с получением указанного в заголовке соединения.
Стадия C: 4-амино-2-хлорхинолин-6-ол К смеси 2-хлор-N-(2,4-диметоксибензил)-6-метоксихинолин-4-амина (5 г, 13,9 ммоль) в DCM (150 мл) добавляли BBr3 (6,59 мл, 69,7 ммоль). Реакционную смесь перемешивали при 25°C в течение 16 ч. Затем МеОН (10 мл) добавляли по каплям к смеси для гашения реакции. Полученную смесь концентрировали в вакууме. Полученный остаток промывали PE/EtOAc (1:1; 20 мл×3), и затем сушили под вакуумом с получением указанного в заголовке соединения, которое использовали на следующей стадии без дополнительной очистки. LCMS (ESI) вычислено для C9H7ClN2O [M+H]+: 195, найдено: 195,0.
Стадия D: трет-бутил 3-((4-амино-2-хлорхинолин-6-ил)окси)-2-гидроксипропаноат К смеси 4-амино-2-хлорхинолин-6-ола (1,5 г, 7,71 ммоль) и трет-бутил оксиран-2-карбоксилата (4,44 г, 30,8 ммоль) в N,N-диметилформамиде (5 мл) добавляли Cs2CO3 (5,02 г, 15,41 ммоль). Полученную суспензию перемешивали при 40°C в атмосфере N2 в течение 16 ч. Затем смесь фильтровали и фильтрат очищали с помощью колоночной хроматографии на силикагеле (SiO2, EtOAc:PE=0~50%), затем препаративной ВЭЖХ (Waters Xbridge Prep OBD C18 150*30 5u; подвижная фаза A: вода (0,05% гидроксид аммиака об/об) подвижная фаза B: ацетонитрил; градиент: 24-54% B, 10,0 мин; 100% B, 2 мин; скорость потока: 25 мл/мин) с получением указанного в заголовке соединения. LCMS (ESI) вычислено для C16H19ClN2O4 [M+H]+: 339, найдено: 339,0.1H ЯМР (400 МГц, хлороформ-d) δ 7,85 (д, J=9,2 Гц, 1H), 7,33 (шир. дд, J=2,6, 9,1 Гц, 1H), 7,00 (д, J=2,5 Гц, 1H), 6,61 (с, 1H), 4,71 (шир. с, 2H), 4,45 (шир. с, 1H), 1,50 (с, 9H)
Стадия E: трет-бутил 3-((4-амино-2-хлорхинолин-6-ил)окси)-2-((трет-бутилдиметилсилил)окси)пропаноат К смеси трет-бутил 3-((4-амино-2-хлорхинолин-6-ил)окси)-2-гидроксипропаноата (450 мг, 1,33 ммоль) и имидазола (136 мг, 1,99 ммоль) в N,N-диметилформамиде (8 мл) добавляли TBS-Cl (240 мг, 1,59 ммоль). Полученную суспензию перемешивали при 25°C в атмосфере N2 в течение 16 ч. Затем смесь разбавляли EtOAc (30 мл), промывали насыщенным солевым раствором (10 мл×3), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (SiO2, EtOAc/Pentane=0~30%) с получением указанного в заголовке соединения. LCMS (ESI) вычислено для C22H33ClN2O4Si [M+H]+: 453, найдено: 453,2.
Стадия F: трет-бутил (R)-3-((4-(N,N-бис(трет-бутоксикарбонил)амино)-2-хлорхинолин-6-ил)окси)-2-((трет-бутилдиметилсилил)окси)пропаноат К смеси трет-бутил 3-((4-амино-2-хлорхинолин-6-ил)окси)-2-((трет-бутилдиметилсилил)окси)пропаноата (750 мг, 1,655 ммоль) и TEA (0,346 мл, 2,483 ммоль) в дихлорметане (10 мл) добавляли BOC2O (0,461 мл, 1,987 ммоль) и DMAP (20,22 мг, 0,166 ммоль). Полученную суспензию перемешивали при 20°C в атмосфере N2 в течение 16 ч. Затем смесь выпаривали под вакуумом, полученный остаток очищали с помощью колоночной хроматографии на силикагеле (SiO2, EtOAc:PE=0~20) с получением указанного в заголовке соединения. LC-MS (ESI) вычислено для C32H49ClN2O8Si [M+H]+: 653, найдено: 653,3.1H ЯМР (400 МГц, хлороформ-d) δ 7,96 (д, J=9,2 Гц, 1H), 7,45-7,37 (м, 1H), 7,23 (с, 1H), 6,98 (д, J=2,7 Гц, 1H), 4,52 (дд, J=3,5, 6,7 Гц, 1H), 4,32-4,11 (м, 2H), 1,53-1,49 (м, 9H), 1,36 (с, 18H), 0,93 (с, 9H), 0,20-0,10 (м, 6H).
Нижеследующие примеры описывают синтез соединений по изобретению в форме конкретных солей. Форма свободного основания этих солей может быть получена путем очистки конечного продукта с помощью ВЭЖХ с обращенной фазой с использованием муравьиной кислоты в качестве модификатора. Муравьиную кислоту можно удалить из образца лиофилизацией, которую можно повторить один или несколько раз, если желательно, для удаления остаточной соли TFA и улучшения содержание свободной основы.
ПРИМЕР 1
3-((6-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)хинолин-2-ил)амино)азетидин-1-ий 2,2,2-трифторацетат (C1)

Стадия A. Получение трет-бутил (R)-3-((2-хлорхинолин-6-ил)окси)-2-гидроксипропаноата
Смесь 2-хлорхинолин-6-ола (1,0 г, 5,6 ммоль), кобальтового катализатора (Промеж.соед. 2) (0,93 г, 1,1 ммоль), молекулярных сит (1 г, порошок), и трет-бутил оксиран-2-карбоксилата (1,8 г, 12 ммоль) в TBME (5 мл) перемешивали при комнатной температуре а атмосфере N2 в течение выходных. Добавляли снова кобальтовый катализатор (0,5 г) и трет-бутил оксиран-2-карбоксилат (0,8 мл). Смесь перемешивали при комнатной температуре еще 2 дня. Реакционную смесь разбавляли EtOAc для растворения большинства реагентов и продукта. Твердое вещество отфильтровывали и растворитель удаляли. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью EtOAc/гексан (35%) с получением целевого продукта в виде твердого вещества. LC-MS [M+H]+: m/z 324,18
Стадия B. Получение трет-бутил (R)-2-((трет-бутилдиметилсилил)окси)-3-((2-хлорхинолин-6-ил)окси)пропаноата К раствору трет-бутил 3-((2-хлорхинолин-6-ил)окси)-2-гидроксипропаноата (0,5 г, 1,5 ммоль), имидазола (0,53 г, 7,7 ммоль) и TBS-Cl (3,9 мл, 3,9 ммоль) в ацетонитриле (10 мл) добавляли DMAP (0,019 г, 0,154 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 3 часов. После концентрации, остаток растворяли в EtOAc, промывали насыщенным NaHCO3, водой и насыщенным солевым раствором. Растворитель удаляли. Остаток очищали на колонке с силикагелем (24 г), используя смесь 0-20% EtOAc/гексан в качестве градиента с получением трет-бутил 2-((трет-бутилдиметилсилил)окси)-3-((2-хлорхинолин-6-ил)окси)пропаноата в виде твердого вещества. LC-MS [M+H]+: m/z 440,24
Стадия C. Получение трет-бутил (R)-3-((6-(3-(трет-бутокси)-2-((трет-бутилдиметилсилил)окси)-3-оксопропокси)хинолин-2-ил)амино)азетидин-1-карбоксилата К раствору трет-бутил (R)-2-((трет-бутилдиметилсилил)окси)-3-((2-хлорхинолин-6-ил)окси)пропаноата (150 мг, 0,34 ммоль) в диоксане (2 мл) добавляли трет-бутил 3-аминоазетидин-1-карбоксилат, предкатализатор RuPhos второго поколения (39,9 мг, 0,051 ммоль) и Cs2CO3 (223 мг, 0,68 ммоль). Затем раствор дегазировали и наполняли N2, смесь нагревали при 70°C в течение ночи. Смесь разбавляли EtOAc, промывали NH4Cl, водой и насыщенным солевым раствором. Растворитель удаляли. Остаток очищали с помощью колоночной хроматографии на силикагеле (24 г), элюируя смесью EtOAc/гексан (30%), с получением целевого продукта в виде масла. LC-MS [M+H]+: m/z 574,52
Стадия D. Получение трет-бутил (R)-3-((6-(3-(трет-бутокси)-2-гидрокси-3-оксопропокси)-хинолин-2-ил)амино)азетидин-1-карбоксилата К раствору трет-бутил (R)-3-((6-(3-(трет-бутокси)-2-((трет-бутилдиметилсилил)окси)-3-оксопропокси)хинолин-2-ил)амино)азетидин-1-карбоксилата (0,129 г, 0,225 ммоль) в THF (3 мл) добавляли TBAF (0,225 мл, 0,225 ммоль) при комнатной температуре. Раствор перемешивали в течение часа и растворитель удаляли. Остаток очищали с помощью колоночной хроматографии на силикагеле (24 г), элюируя смесью EtOAc/гексан (80%, 15cv), с получением целевого продукта. LC-MS [M+H]: m/z 460,43
Стадия E. Получение трет-бутил (S)-3-((6-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)хинолин-2-ил)амино)азетидин-1-карбоксилата 2-Гидроксиизоиндолин-1,3-дион (0,039 г, 0,24 ммоль) и трифенилфосфин (0,068 г, 0,26 ммоль) добавляли к раствору трет-бутил (R)-3-((6-(3-(трет-бутокси)-2-гидрокси-3-оксопропокси)хинолин-2-ил)амино)азетидин-1-карбоксилата (0,1 г, 0,22 ммоль) в THF (3 мл), затем DIAD (0,051 мл, 0,26 ммоль) при комнатной температуре. Раствор перемешивали в течение ночи. Растворитель удаляли. Остаток очищали с помощью колоночной хроматографии на силикагеле (12 г), элюируя смесью EtOAc/гексан (70% 10cv), с получением целевого продукта в виде масла. LC-MS [M+H]+: m/z 605,55
Стадия F. Получение трет-бутил (S)-3-((6-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)-хинолин-2-ил)амино)азетидин-1-карбоксилата К раствору трет-бутил (S)-3-((6-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)хинолин-2-ил)амино)азетидин-1-карбоксилата в EtOH (2 мл) добавляли гидразин (6,75 мкл, 0,215 ммоль). Раствор перемешивали при комнатной температуре в течение 1 часа. Растворитель удаляли. DCM (3 мл) добавляли к остатку и перемешивали при комнатной температуре в течение 1 часа. Затем твердое вещество отфильтровывали. Раствор концентрировали с получением целевого продукта в виде пленки. LC-MS [M+H]+: m/z 475,38
Стадия G. Получение (S,Z)-2-(((1-(трет-бутокси)-3-((2-((1-(трет-бутоксикарбонил)азетидин-3-ил)амино)хинолин-6-ил)окси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)уксусной кислоты Раствор 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусной кислоты (Промежуточное соединение 3) (0,048 г, 0,18 ммоль) и трет-бутил (S)-3-((6-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)хинолин-2-ил)амино)азетидин-1-карбоксилата (0,1 г, 0,166 ммоль) в EtOH (2 мл) и DCE (1 мл) перемешивали при комнатной температуре в течение ночи. Смесь концентрировали и использовали на следующей стадии. LC-MS [M+H]+: m/z 729,63
Стадия H. Получение трет-бутил 3-((6-((S)-3-(трет-бутокси)-2-((((Z)-1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)-амино)окси)-3-оксопропокси)хинолин-2-ил)амино)азетидин-1-карбоксилата К раствору (S,Z)-2-(((1-(трет-бутокси)-3-((2-((1-(трет-бутоксикарбонил)азетидин-3-ил)амино)хинолин-6-ил)окси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)уксусной кислоты (0,14 г, 0,164 ммоль) в DMF (1 мл) добавляли DCC (0,084 г, 0,41 ммоль) и HOBT (0,063 г, 0,41 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 30 минут перед добавлением (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфата (0,069 г, 0,33 ммоль) и бикарбоната натрия (0,069 г, 0,82 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Смесь фильтровали. Раствор очищали с помощью RP-HPLC (Gilson) (колонка C-18), элюируя 20-100% ACN/вода с 0,05% TFA. Фракцию продукта лиофилизировали с получением целевого продукта в виде твердого вещества. LC-MS [M+H]+: m/z 921,67
Стадия I. Получение соединения (S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-((2-(азетидин-3-иламино)хинолин-6-ил)окси)пропановой кислоты с 2,2,2-трифторуксусной кислотой (1:1)
К раствору трет-бутил 3-((6-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-хинолин-2-ил)амино)азетидин-1-карбоксилата, TFA (86 мг, 0,083 ммоль) в CH2Cl2 (1 мл) добавляли TFA (2 мл, 26,0 ммоль). Раствор перемешивали при комнатной температуре в течение 0,5 часа. Растворитель удаляли под вакуумом. Остаток дважды промывали Et2O. Твердый неочищенный продукт собирали и сушили. Остаток растворяли в DMSO и очищали с помощью RP-HPLC (Gilson) (колонка C-18), элюируя 2-40% ACN/вода с 0,05% TFA. Фракцию продукта лиофилизировали с получением целевого продукта в виде твердого вещества. LC-MS [M+H]+: m/z 665,34.1Н ЯМР (500 МГц, CDCl3) δ: 8,33 (1H, д, J=9,8 Гц), 7,81 (1H, д, J=9,5 Гц), 7,44-7,51 (2H, м), 7,13 (2H, т, J=8,4 Гц), 5,28 (1H, с), 5,15 (1H, д, J=8,0 Гц), 4,55-4,70 (5H, м), 4,31 (2H, д, J=9,4 Гц), 1,46-1,50 (3H, с), 1,16 (3H, с).
Таблица 1. Используя те же общие процедуры, описанные в примере 1, заменяя соответствующие реактивы и реагенты, следующие соединения были синтезированы и охарактеризованы с помощью LC/MS.

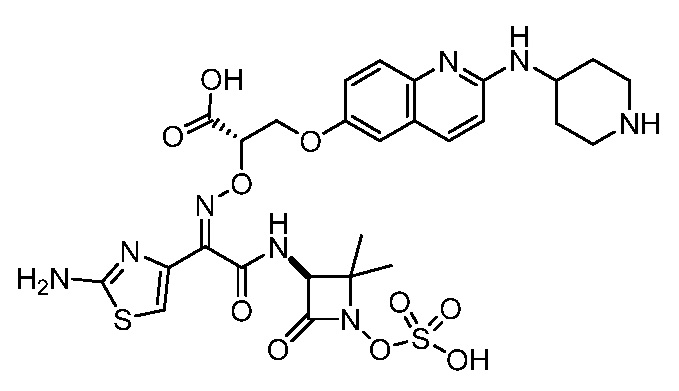
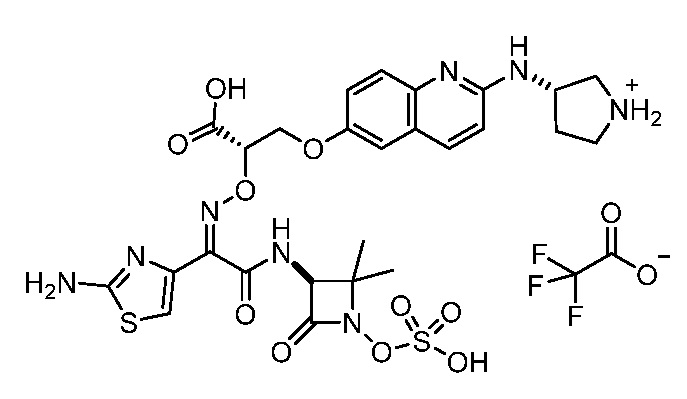

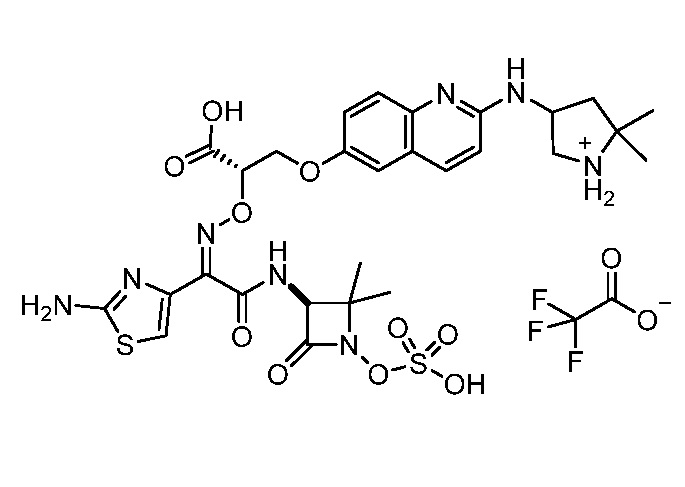
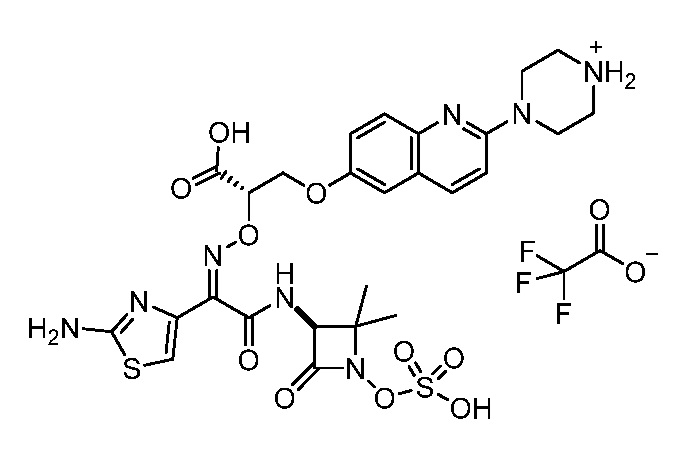
ПРИМЕР 8
(S)-3-((2-((2-аминоэтил)амино)хинолин-6-ил)окси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота (C8)

Соединение 8 получали с использованием тех же процедур, что и в примере 1, за исключением того, что использовали условия, описанные ниже для стадии С. К раствору трет-бутил (R)-2-((трет-бутилдиметилсилил)окси)-3-((2-хлорхинолин-6-ил)окси)пропаноата (0,25 г, 0,57 ммоль) в диоксане (1 мл) добавляли трет-бутил (2-аминоэтил)-карбамат (0,18 г, 1,14 ммоль), MorDalphos - G3-palladacycle (0,095 г, 0,114 ммоль) и Cs2CO3 (0,46 г, 1,43 ммоль). Смесь дегазировали и заполняли N2. Смесь нагревали при 75°C в течение ночи. Твердое вещество отфильтровывали и растворитель удаляли в ваккуме. Остаток очищали с помощью колоночной хроматографии на силикагеле (24 г), элюируя EtOAc/гексаном с получением целевого продукта в виде смолы.
Остальные стадии были такими же, как в примере 1. Указанное в заголовке соединение 8 охарактеризовывали с помощью LC/MS и ЯМР. LC-MS [M+H]+: m/z 653,47.1Н ЯМР (500 МГц, D2O) δ 8,10-8,12 (1H, м), 7,62 (1H, д, J=9,4 Гц), 7,25-7,37 (2H, м), 6,96-6,99 (1H, м), 6,87 (1H, с), 4,90-4,93 (1H, м), 4,35-4,46 (2H, м), 3,80 (2H, дд, J=6,5, 5,7 Гц), 3,25-3,31 (2H, м), 1,31 (3H, с), 0,92 (3H, с).
Таблица 2. Используя те же общие процедуры, которые описаны в примере 8, заменяя соответствующие реактивы и реагенты, следующие соединения были синтезированы и охарактеризованы с помощью LC/MS.
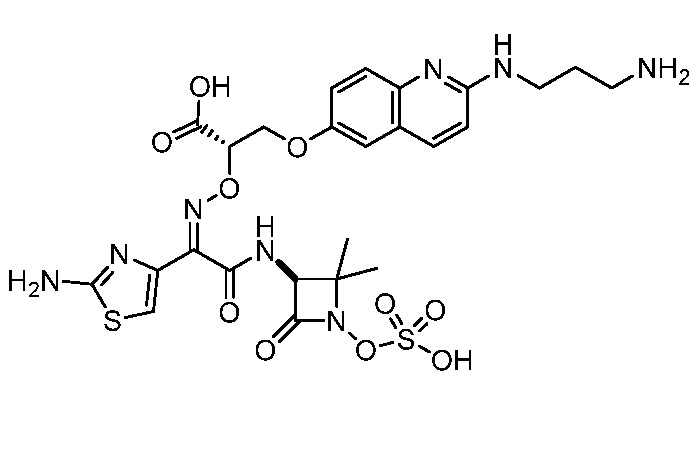

Пример 11
(S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-((1-((1-(трет-бутоксикарбонил)азетидин-3-ил)амино)изохинолин-6-ил)окси)пропановая кислота (C11)
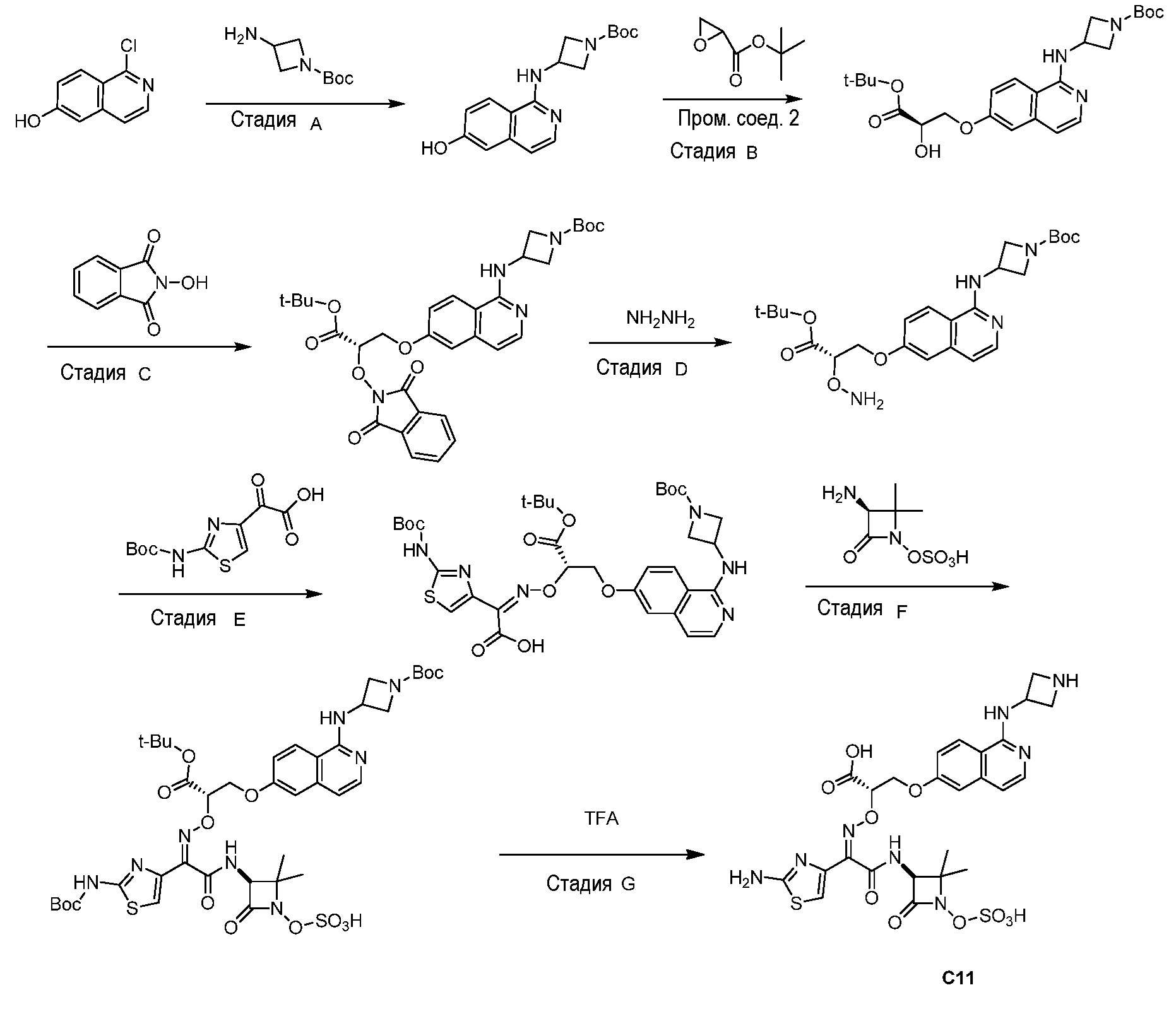
Стадия A. Получение трет-бутил 3-((6-гидроксиизохинолин-1-ил)амино)азетидин-1-карбоксилата К раствору 1-хлоризохинолин-6-ола (0,2 г, 1,1 ммоль) в диоксане (3 мл) добавляли трет-бутил 3-аминоазетидин-1-карбоксилат (0,29 г, 1,7 ммоль), предкатализатор RuPhos второго поколения (0,13 г, 0,17 ммоль) и Cs2CO3 (1,1 г, 3,3 ммоль). Герметично закупоренный флакон дегазировали и заполняли N2 и нагревали при 80°C в течение ночи. Твердое вещество отфильтровывали и растворитель удаляли. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью EtOAc/гексан с получением целевого продукта в виде смолы. LC-MS [M+H]+: m/z 316,33.
Стадия B. Получение трет-бутил (R)-3-((6-(3-(трет-бутокси)-2-гидрокси-3-оксопропокси)изохинолин-1-ил)амино)азетидин-1-карбоксилата К раствору трет-бутил 3-((6-гидрокси-изохинолин-1-ил)амино)азетидин-1-карбоксилата (0,21 г, 0,67 ммоль) в MTBE (1 мл) добавляли трет-бутил оксиран-2-карбоксилат (0,21 г, 1,5 ммоль) и кобальтовый катализатор (Промеж.соед. 2) (0,11 г, 0,13 ммоль). Смесь дегазировали и заполняли N2 и перемешивали при комнатной температуре в течение выходных. Твердое вещество отфильтровывали и растворитель удаляли. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью EtOAc/гексан с получением целевого продукта в виде смолы. LC-MS [M+H]+: m/z 460,40.
Стадия C. Получение трет-бутил (S)-3-((6-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)изохинолин-1-ил)амино)азетидин-1-карбоксилата 2-гидрокси-изоиндолин-1,3-дион (0,070 г, 0,43 ммоль) и трифенилфосфин (0,12 г, 0,47 ммоль) добавляли к раствору трет-бутил (R)-3-((6-(3-(трет-бутокси)-2-гидрокси-3-оксопропокси)-изохинолин-1-ил)амино)азетидин-1-карбоксилата (0,18 г, 0,39 ммоль) в THF (4 мл), затем DIAD (0,091 мл, 0,47 ммоль) при комнатной температуре. Смесь перемешивали в течение 1 часа и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью EtOAc/гексан с получением целевого продукта в виде масла. LC-MS [M+H]+: m/z 605,45.
Остальная часть процедуры от стадии D до стадии G была такой же, как описано на стадии F до стадии I в примере 1 с соответствующими изохинолиновыми промежуточными соединениями. Указанное в заголовке соединение 11, (S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-((1-((1-(трет-бутоксикарбонил)азетидин-3-ил)амино)изохинолин-6-ил)окси)пропановую кислоту, охарактеризовывали с помощью LC/MS и ЯМР. LC-MS [M+H]+: m/z 665,29.1Н ЯМР (500 МГц, D2O) δ 8,17 (1H, д), 7,32 (1H, д), 7,20 (1H, д), 7,16 (1H, с), 7,08 (1H, д), 7,02 (1H, с), 5,12 (1H, с), 5,00 (1H, м), 4,40-4,54 (5H, м), 4,26-4,32 (2H, м), 1,23 (3H, с), 0,90 (3H, с).
Таблица 3. Используя те же общие процедуры, которые описаны в примере 11, заменяя соответствующие реактивы и реагенты, следующие соединения были синтезированы и охарактеризованы с помощью LC/MS.

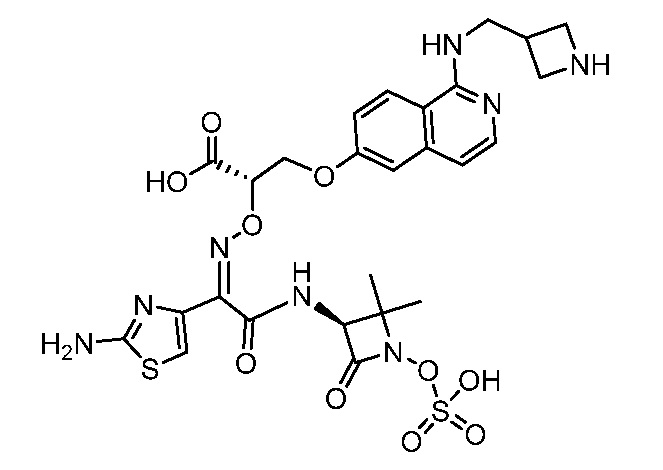

ПРИМЕР 15
6-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-1-(азетидин-3-иламино)-2-метилизохинолин-2-ий 2,2,2-трифторацетат (C15)


Стадия A. Получение (S)-6-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-1-((1-(трет-бутоксикарбонил)азетидин-3-ил)амино)-2-метилизохинолин-2-ия MeI (0,052 мл, 0,83 ммоль) добавляли к промежуточному соединению из стадии C в Примере 11 трет-бутил (S)-3-((6-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-изохинолин-1-ил)амино)азетидин-1-карбоксилату (50 мг, 0,083 ммоль) в ацетонитриле (1 мл) в герметичном флаконе для микроволновой печи. Раствор нагревали при 90°C в течение ночи. Растворитель удаляли под вакуумом и неочищенный продукт использовали на следующей стадии. LC-MS [M+H]: m/z 619,41. Остальную часть процедуры от стадии В до стадии Е выполняли по той же процедуре, как описано на стадии F до стадии I в примере 1, с соответствующими промежуточными соединениями. Указанное в заголовке соединение 15, 6-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-1-(азетидин-3-иламино)-2-метилизохинолин-2-ий 2,2,2-трифторацетат, охарактеризовывали с помощью LC/MS и ЯМР. LC-MS [M+H]+: m/z 679,66.1H ЯМР (CH3OH-d4, 500 MГц): δ 7,99 (1H, д, J=9,3 Гц), 7,85 (1H, д, J=7,1 Гц), 7,44-7,50 (3H, м), 7,10 (1H, с), 5,30-5,37 (2H, м), 4,84 (1H, дд, J=11,5, 2,3 Гц), 4,61-4,73 (3H, м), 4,54-4,54 (1H, м), 4,49 (2H, т, J=9,3 Гц), 4,06 (3H, с), 1,51 (3H, с), 1,19 (3H, с).
ПРИМЕР 16
(S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-2-((1-((азетидин-3-илметил)амино)-2-метилизохинолин-2-ий-6-ил)окси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат (C16)

Способ получения соединения 16 был таким же, как описанный в примере 15, с исходным промежуточным соединением трет-бутил (S)-3-(((6-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)изохинолин-1-ил)амино)метил)азетидин-1-карбоксилатом (промежуточное соединение, используемое для получения соединения 13, полученное на стадиях А-С примера 11 с использованием 1-хлоризохинолин-6-ола и трет-бутил 3-(аминометил)азетидин-1-карбоксилата). Указанное в заголовке соединение 16 охарактеризовывали с помощью LC/MS и ЯМР. LC-MS [M+H]+: m/z 693,29.1Н ЯМР (500 МГц, D2O) δH 8,33 (1H, м), 7,62 (1H, д, J=7,2 Гц), 7,37-7,29 (3H, м), 6,99 (1H, с), 5,07 (1H, с), 4,26 (2H, т, J=10,0 Гц), 4,10 (2H, м), 4,06-3,98 (3H, м), 3,92 (3H, с), 1,40 (3H, с), 1,05 (3H, с).
ПРИМЕР 17
2-((6-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)хинолин-2-ил)амино)-N,N,N-триметилэтан-1-аминий 2,2,2-трифторацетат (C17)


Стадия A. Получение трет-бутил (R)-2-((трет-бутилдиметилсилил)окси)-3-((2-((2-(диметиламино)-этил)амино)хинолин-6-ил)окси)пропаноата К раствору промежуточного соединения трет-бутил (R)-2-((трет-бутилдиметилсилил)окси)-3-((2-хлорхинолин-6-ил)окси)пропаноата (со стадии В примера 1) (0,3 г, 0,68 ммоль) в диоксане (1 мл) добавляли N1,N1-диметилэтан-1,2-диамин (0,12 г, 1,4 ммоль), MorDalphos-G3-palladacycle (0,057 г, 0,068 ммоль) и Cs2CO3 (0,56 г, 1,7 ммоль). Смесь дегазировали и заполняли N2. Затем смесь нагревали при 75°C в течение ночи и концентрировали. Смесь очищали при помощи колоночной хроматографии на силикагеле, элюируя EtOAc/10% 7н NH3 в MeOH с получением целевого продукта в виде масла. LC-MS [M+H]: m/z 490,44.
Стадия B. Получение трет-бутил (R)-3-((2-((2-(диметиламино)этил)амино)хинолин-6-ил)окси)-2-гидроксипропаноата К раствору трет-бутил (R)-2-((трет-бутилдиметилсилил)окси)-3-((2-((2-(диметиламино)этил)амино)хинолин-6-ил)окси)пропаноата (0,25 г, 0,51 ммоль) в THF (10 мл) добавляли TBAF (0,51 мл, 0,51 ммоль) при комнатной температуре. Раствор перемешивали в течение 1 часа и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя EtOAc и 7н NH3 в MeOH с получением целевого продукта в виде масла. LC-MS [M+H]: m/z 376,34.
Стадия C. Получение трет-бутил (S)-3-((2-((2-(диметиламино)этил)амино)хинолин-6-ил)окси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноата 2-Гидроксиизоиндолин-1,3-дион (0,081 г, 0,50 ммоль) и трифенилфосфин (0,14 г, 0,54 ммоль) добавляли к раствору трет-бутил (R)-3-((2-((2-(диметиламино)этил)амино)хинолин-6-ил)окси)-2-гидрокси-пропаноата (0,17 г, 0,45 ммоль) в THF (3 мл), затем DIAD (0,11 мл, 0,54 ммоль) при комнатной температуре. Раствор перемешивали в течение 4 часов и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя EtOAc для 10 объемов колонки и затем Et3N/ацетон 5%/95% с получением целевого продукта в виде масла. LC-MS [M+H]: m/z 521,39.
Стадия D. Получение (S)-2-((6-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)хинолин-2-ил)амино)-N,N,N-триметилэтан-1-аминия К раствору трет-бутил (S)-3-((2-((2-(диметиламино)этил)амино)хинолин-6-ил)окси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-пропаноата (75 мг, 0,14 ммоль) в ACN (1 мл) добавляли MeI (0,036 мл, 0,58 ммоль) в герметичном флаконе. Смесь перемешивали при комнатной температуре в течение 3 часов и концентрировали в высоком вакууме с получением неочищенного продукта, который использовали как есть на следующей стадии. LC-MS [M+H]: m/z 535,49.
Остальная часть процедуры от стадии Е до стадии Н была такой же, как описано на стадии F до стадии I примера 1 с соответствующими промежуточными соединениями. Указанное в заголовке соединение 17, 2-((6-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)хинолин-2-ил)амино)-N,N,N-триметилэтан-1-аминий 2,2,2-трифторацетат, охарактеризовывали с помощью LC/MS и ЯМР. LC-MS [M+H]: m/z 695,34.1H ЯМР (CH3OH-d4, 500 MГц): δH 8,27 (1H, д, J=9,5 Гц), 7,81 (1H, д, J=9,2 Гц), 7,49 (1H, д, J=9,4 Гц), 7,42 (1H, с), 7,11 (1H, д, J=9,7 Гц), 7,02 (1H, с), 5,23 (1H, д, J=5,5 Гц), 4,58 (1H, дд, J=11,8, 5,8 Гц), 4,47 (1H, с), 4,13 (2H, д, J=6,9 Гц), 3,82 (2H, т, J=6,8 Гц), 3,30 (9H, с), 1,48 (3H, с), 1,11 (3H, с).
Таблица 4. Используя, в общем, ту же процедуру, что и в примере 17, заменяя соответствующие реактивы и реагенты, следующие соединения были синтезированы и охарактеризованы с помощью LC/MS.

ПРИМЕР 19
(S)-3-((Z)-2-(2-амино-5-хлортиазол-4-ил)-2-(((S)-2-((1-((азетидин-3-илметил)амино)-2-метилизохинолин-2-ий-6-ил)окси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат, 2,2,2-трифторацетатная соль (C19)
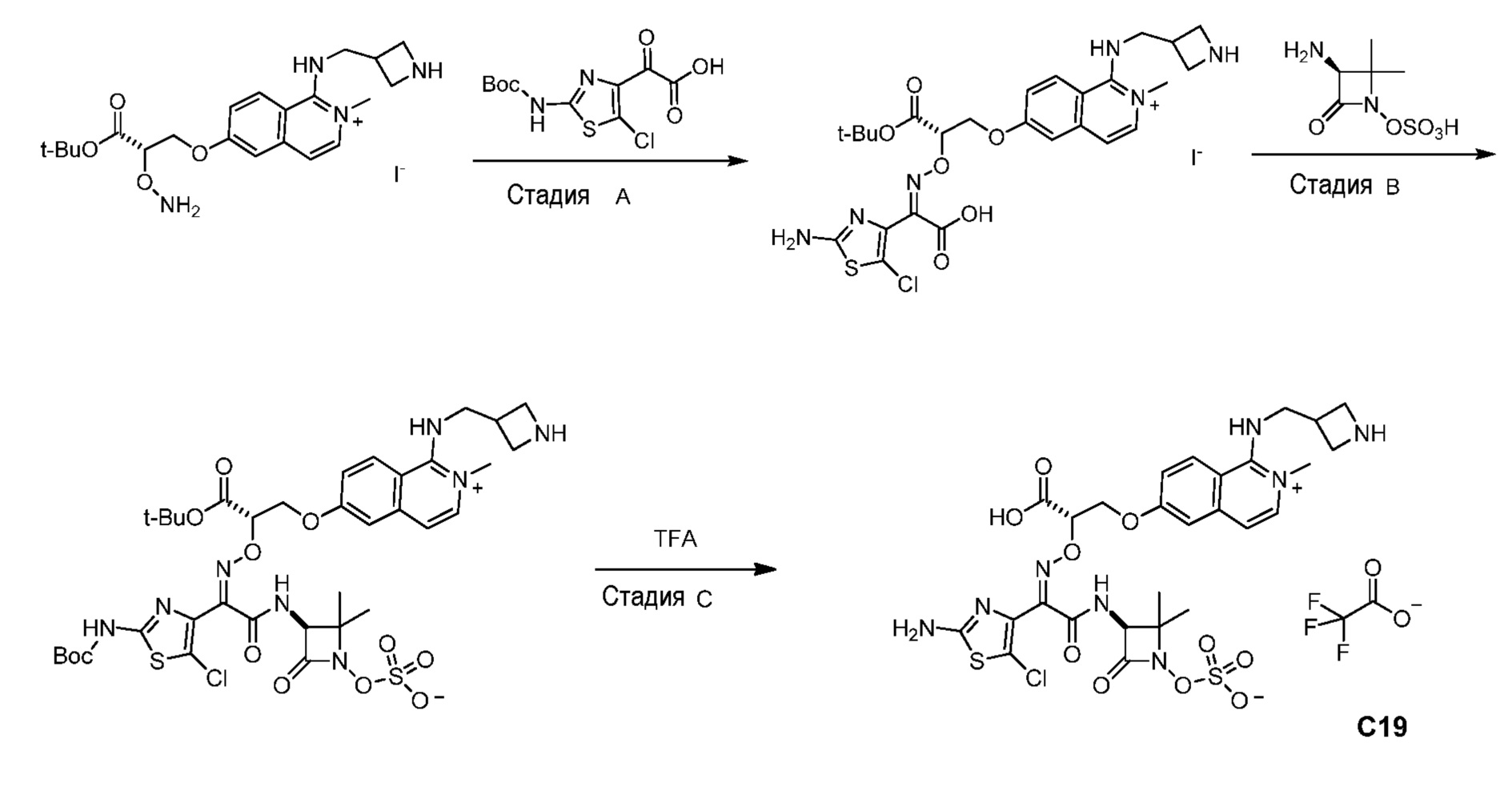
Стадия A. Получение (S,Z)-6-(3-(трет-бутокси)-2-(((1-(2-((трет-бутоксикарбонил)амино)-5-хлортиазол-4-ил)-2-карбокси-2-оксоэтилиден)амино)окси)-3-оксопропокси)-1-(((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)амино)-2-метилизохинолин-2-ия Раствор (S)-6-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)-1-(((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)амино)-2-метилизохинолин-2-ий, йодида (промежуточное соединение из стадии B в примере 16) (40 мг, 0,063 ммоль) и 2-(2-((трет-бутоксикарбонил)амино)-5-хлортиазол-4-ил)-2-оксоуксусной кислоты (промеж. соед. 4) (21,4 мг, 0,070 ммоль) в EtOH (2 мл) и DCE (1 мл) перемешивали при комнатной температуре в течение ночи и затем концентрировали под вакуумом. Полученную смесь использовали как неочищенную на следующей стадии. LC-MS [M+H]: m/z 791,37.
Стадию B и стадию C выполняли по той же процедуре, что и на стадии H и на стадии I в примере 1. Указанное в заголовке соединение 19, (S)-3-((Z)-2-(2-амино-5-хлортиазол-4-ил)-2-(((S)-2-((1-((азетидин-3-илметил)амино)-2-метилизохинолин-2-ий-6-ил)окси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат, 2,2,2-трифторацетатная соль, охарактеризовывали с помощью LC/MS и ЯМР. LC-MS [M+H]: m/z 727,30.1H ЯМР (H2O-d2, 500 MГц): δΗ 8,24-8,26 (1H, м), 7,59-7,61 (1H, м), 7,25-7,37 (3H, м), 5,15-5,20 (2H, м), 4,19-4,24 (2H, м), 4,16 (2H, д, J=7,5 Гц), 3,98 (2H, дд, J=11,0, 7,4 Гц), 3,89 (3H, с), 3,84-3,88 (1H, м), 3,44 (1H, т, J=7,9 Гц), 3,16-3,19 (1H, м), 1,39 (3H, с), 1,00 (3H, с).
ПРИМЕР 20
3-((6-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((2S,3S)-2-метил-4-оксо-1-сульфоазетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)изохинолин-1-ил)амино)азетидин-1-ий (C20)
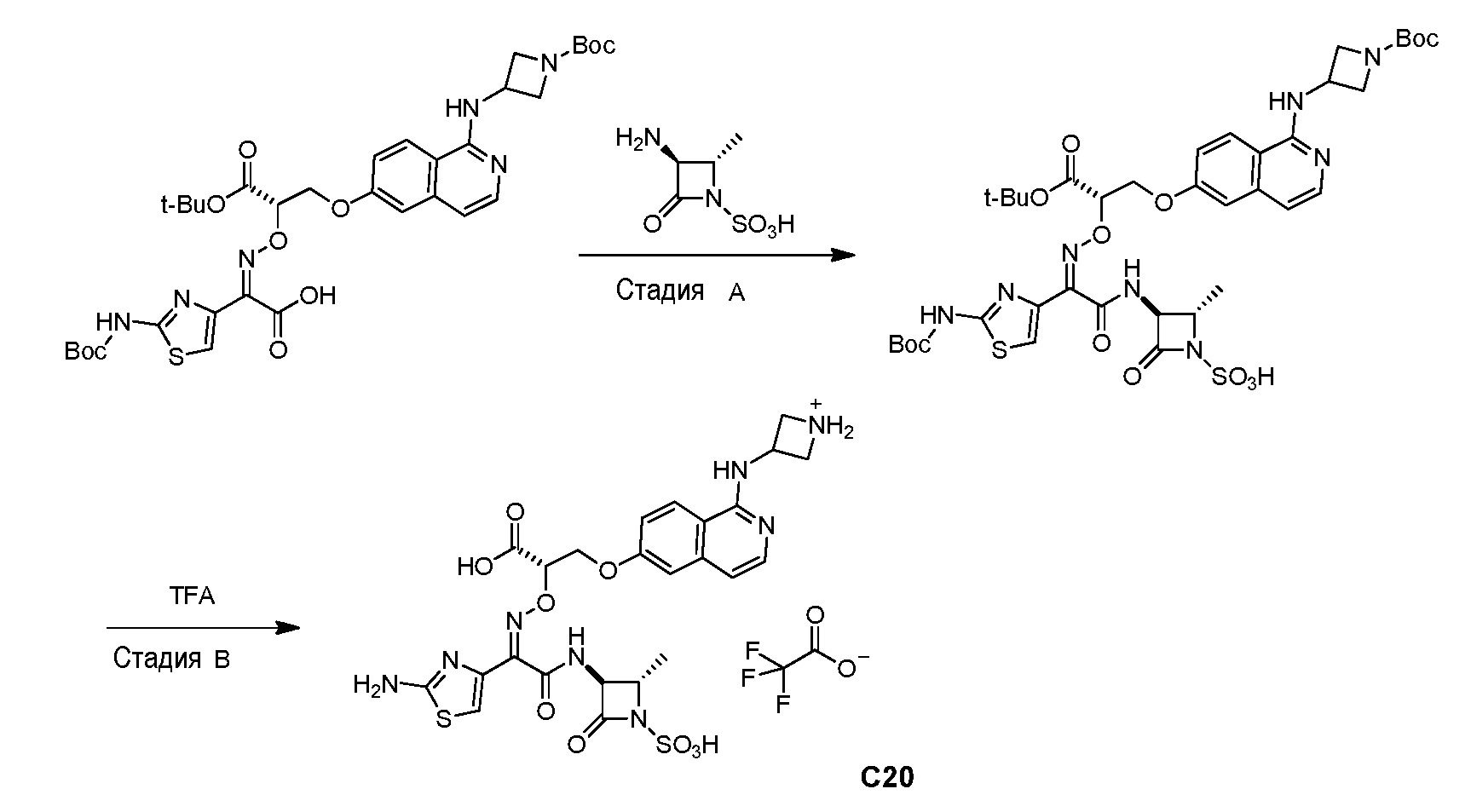
Стадия A. Получение (2S,3S)-3-((Z)-2-((((S)-1-(трет-бутокси)-3-((1-((1-(трет-бутокси-карбонил)азетидин-3-ил)амино)изохинолин-6-ил)окси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)ацетамидо)-2-метил-4-оксоазетидин-1-сульфоновой кислоты К раствору (S,Z)-2-(((1-(трет-бутокси)-3-((1-((1-(трет-бутоксикарбонил)азетидин-3-ил)амино)изохинолин-6-ил)окси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутокси-карбонил)-амино)тиазол-4-ил)уксусной кислоты (со стадии E в примере 11) (60 мг, 0,082 ммоль) в DMF (3 мл) добавляли DCC (42 мг, 0,21 ммоль) и HOBT (32 мг, 0,21 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 30 минут перед добавлением (2S,3S)-3-амино-2-метил-4-оксоазетидин-1-сульфоновой кислоты (29,7 мг, 0,165 ммоль) и бикарбоната натрия (34,6 мг, 0,412 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи и фильтровали. Раствор очищали с помощью RP-HPLC (Gilson) (колонка C-18), элюируя 20-100% ACN/вода с 0,05% TFA. Фракцию продукта лиофилизировали с получением целевого продукта в виде твердого вещества. LC-MS [M+H]: m/z 891,75.
Стадия B. Получение (S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((2S,3S)-2-метил-4-оксо-1-сульфоазетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-((1-(азетидин-3-иламино)-изохинолин-6-ил)окси)пропановой кислоты соединения с 2,2,2-трифторуксусной кислотой (1:1) К раствору (2S,3S)-3-((Z)-2-((((S)-1-(трет-бутокси)-3-((1-((1-(трет-бутоксикарбонил)-азетидин-3-ил)амино)изохинолин-6-ил)окси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)ацетамидо)-2-метил-4-оксоазетидин-1-сульфоновой кислоты (41 мг, 0,046 ммоль) в CH2Cl2 (0,5 мл) добавляли TFA (0,5 мл, 6,5 ммоль). Смесь перемешивали при комнатной температуре в течение 0,5 часа и концентрировали. Остаток дважды промывали Et2O и сушили на воздухе с получением неочищенного твердого вещества. Остаток очищали с помощью RP-HPLC (Gilson колонка C-18), элюируя 0-40% ACN/вода с 0,05% TFA. Фракцию продукта лиофилизировали с получением целевого продукта в виде твердого вещества. LC-MS [M+H]: m/z 635,29.1H ЯМР (H2O-d2, 500 MГц): δH 8,25 (1H, д, J=9,2 Гц), 7,40 (1H, д, J=7,1 Гц), 7,31 (1H, д, J=9,5 Гц), 7,26 (1H, д, J=2,7 Гц), 7,08-7,10 (1H, м), 6,98 (1H, с), 5,10 (1H, д, J=5,0 Гц), 5,00 (1H, т, J=7,5 Гц), 4,60-4,64 (2H, м), 4,51 (2H, т, J=9,7 Гц), 4,36-4,41 (2H, м), 3,36 (1H, дд, J=6,4, 2,9 Гц), 1,05 (3H, д, J=6,2 Гц).
ПРИМЕР 21
3-((6-((S)-2-((((Z)-1-(5-амино-1,2,4-тиадиазол-3-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)хинолин-2-ил)амино)азетидин-1-ий 2,2,2-трифторацетат (C21)
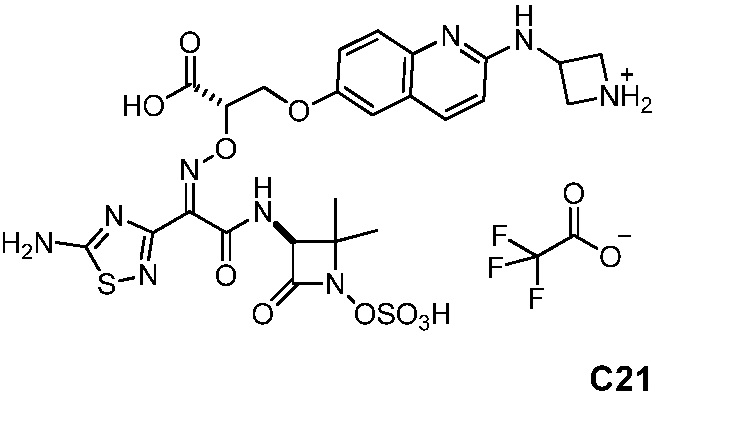
Соединение 21 получали согласно процедуре, как и в примере 1, за исключением того, что на стадии G использовали реагент 2-(5-((трет-бутоксикарбонил)амино)-1,2,4-тиадиазол-3-ил)-2-оксоуксусную кислоту. LC-MS [M+H]: m/z 666,20
ПРИМЕР 22
(S,Z)-4-((6-(2-(((1-(2-аминотиазол-4-ил)-2-((2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)этокси)изохинолин-1-ил)амино)пиперидин-1-ий (C22)
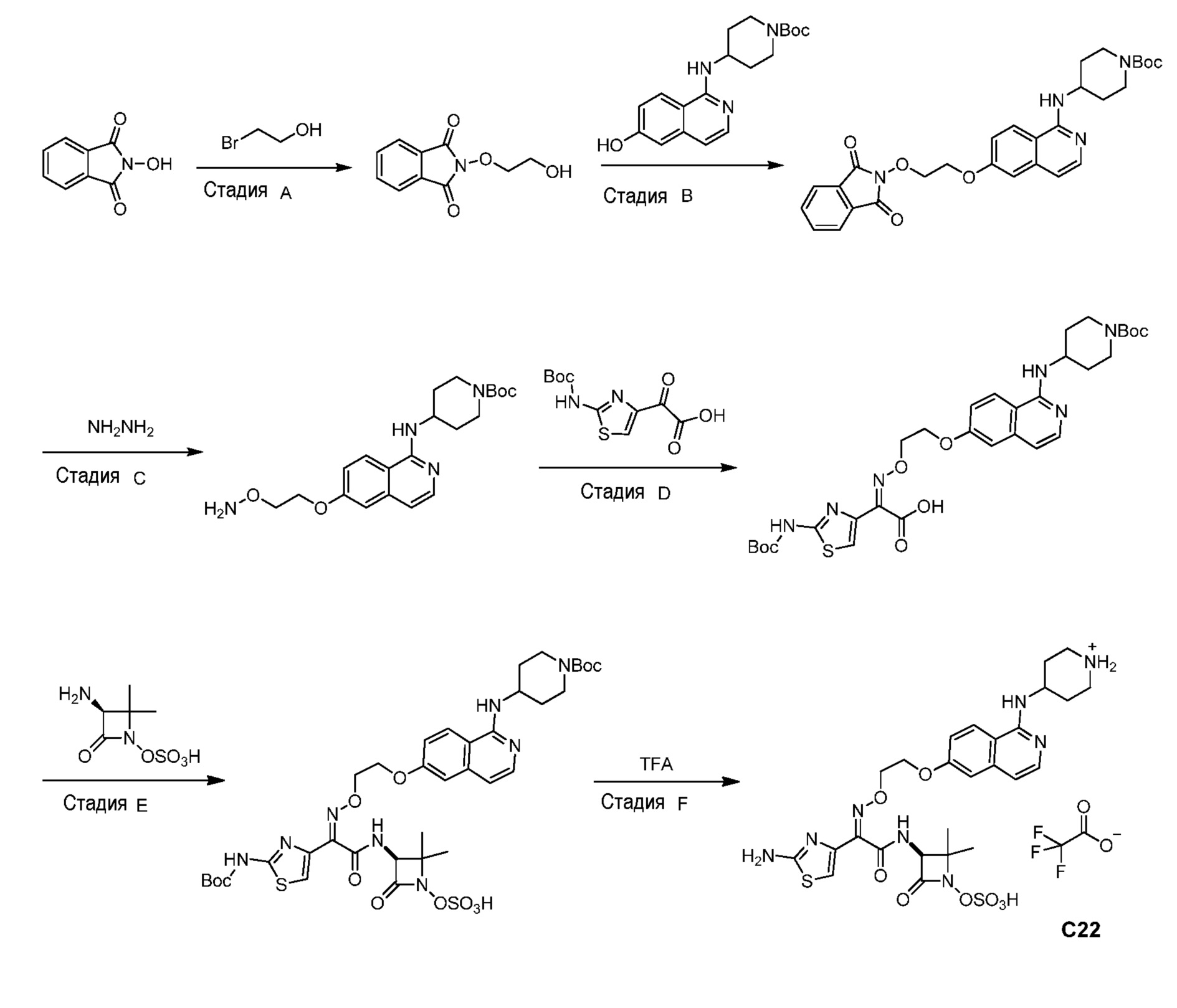
Стадия A. Получение 2-(2-гидроксиэтокси)изоиндолин-1,3-диона Раствор N-гидроксифталимида (5,0 г, 31 ммоль), бромэтанола (6,5 мл, 92 ммоль) и DBU (4,6 мл, 31 ммоль) в DMF (60 мл) нагревали до 50°C и перемешивали в течение ночи. Смесь разбавляли водой и экстрагировали EtOAc. Органический слой промывали водой и насыщенным солевым раствором, и сушили над Na2SO4. Раствор концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью EtOAc/гексан с получением целевого продукта в виде твердого вещества. LC-MS [M+H]: m/z 208,48.
Стадия B. Получение трет-бутил 4-((6-(2-((1,3-диоксоизоиндолин-2-ил)окси)этокси)изохинолин-1-ил)амино)пиперидин-1-карбоксилата К раствору трет-бутил 4-((6-гидроксиизохинолин-1-ил)амино)пиперидин-1-карбоксилата (Общий промежуточный продукт из получения примера 14, полученный на стадии А примера 11 с использованием 1-хлоризохинолин-6-ола и трет-бутил 4-аминопиперидин-1-карбоксилата) (50 мг, 0,15 ммоль) в DCM (2 мл) добавляли 2-(2-гидроксиэтокси)изоиндолин-1,3-дион (30 мг, 0,15 ммоль) и трифенилфосфин (42 мг, 0,16 ммоль), затем DIAD (0,084 мл, 0,16 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов и растворитель удаляли. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью EtOAc/гексан с получением целевого продукта в виде смолы. LC-MS [M+H]: m/z 533,38.
Стадия C. Получение трет-бутил 4-((6-(2-(аминоокси)этокси)изохинолин-1-ил)амино)-пиперидин-1-карбоксилата К раствору трет-бутил 4-((6-(2-((1,3-диоксоизоиндолин-2-ил)окси)этокси)изохинолин-1-ил)амино)пиперидин-1-карбоксилата (40 мг, 0,075 ммоль) в этаноле (5 мл) добавляли гидразин (2,8 мкл, 0,090 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут. Растворитель удаляли. Остаток растворяли в 5 мл DCM и перемешивали при комнатной температуре в течение 15 минут. Твердое вещество отфильтровывали. Растворитель удаляли с получением неочищенного продукта и использовали на следующей стадии. LC-MS [M+H]: m/z 403,30.
Процедуры от стадии D до стадии F были такими же, как от стадии G до стадии I в примере 1. Указанное в заголовке соединение 22 охарактеризовывали с помощью LC/MS и ЯМР. LC-MS [M+H]: m/z 649,26.1H ЯМР (CH3OH-d4, 500 MГц): δΗ 8,48 (1H, д, J=9,3 Гц), 7,57 (1H, д, J=7,0 Гц), 7,44 (1H, дд, J=9,3, 2,5 Гц), 7,38 (1H, д, J=2,6 Гц), 7,21 (1H, д, J=7,1 Гц), 7,01 (1H, с), 4,64-4,67 (2H, м), 4,58-4,62 (1H, м), 4,48-4,51 (1H, м), 4,13-4,17 (1H, м), 3,62 (2H, д, J=13,1 Гц), 3,20 (2H, т, J=12,8 Гц), 2,37 (2H, д, J=13,9 Гц), 2,06-2,14 (2H, м), 1,51 (3H, с), 1,27 (3H, с).
Таблица 5. Используя те же общие процедуры, которые описаны в примере 22, заменяя соответствующие реактивы и реагенты, следующие соединения были синтезированы и охарактеризованы с помощью LC/MS.
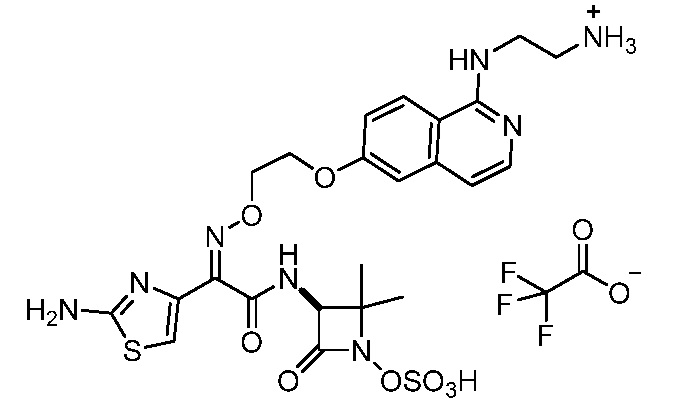
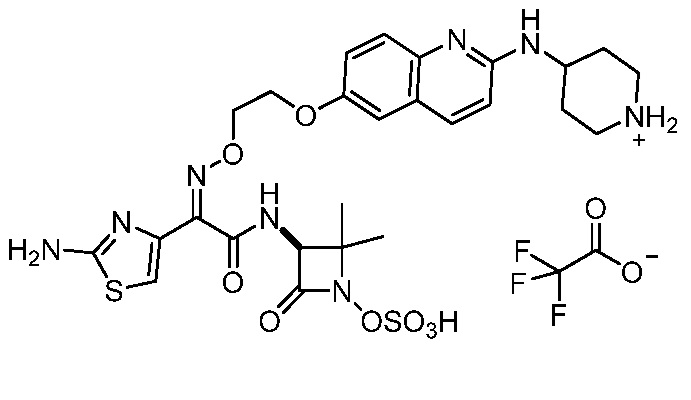
ПРИМЕР 25
(S,Z)-1-амино-6-(2-(((1-(2-аминотиазол-4-ил)-2-((2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)этокси)изохинолин-2-ий 2,2,2-трифторацетат (C25)

Соединение 25 получали путем использования в основном такой же процедуры в примере 22, с реагентом 1-аминоизохинолин-6-олом на стадии В. Указанное в заголовке соединение 25 охарактеризовывали с помощью LC/MS. LC-MS [M+H]: m/z 566,36.
ПРИМЕР 26
5-((6-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)хинолин-2-ил)амино)-1,2-диметил-1H-пиразол-2-ий 2,2,2-трифторацетат (C26)
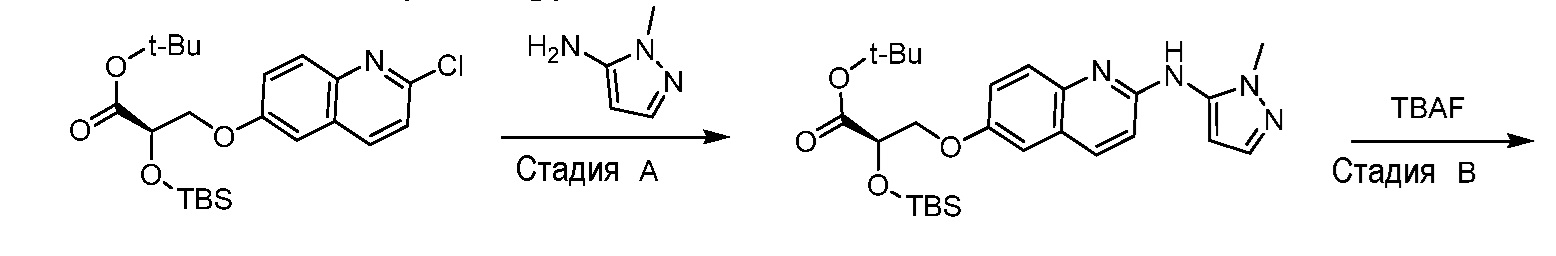
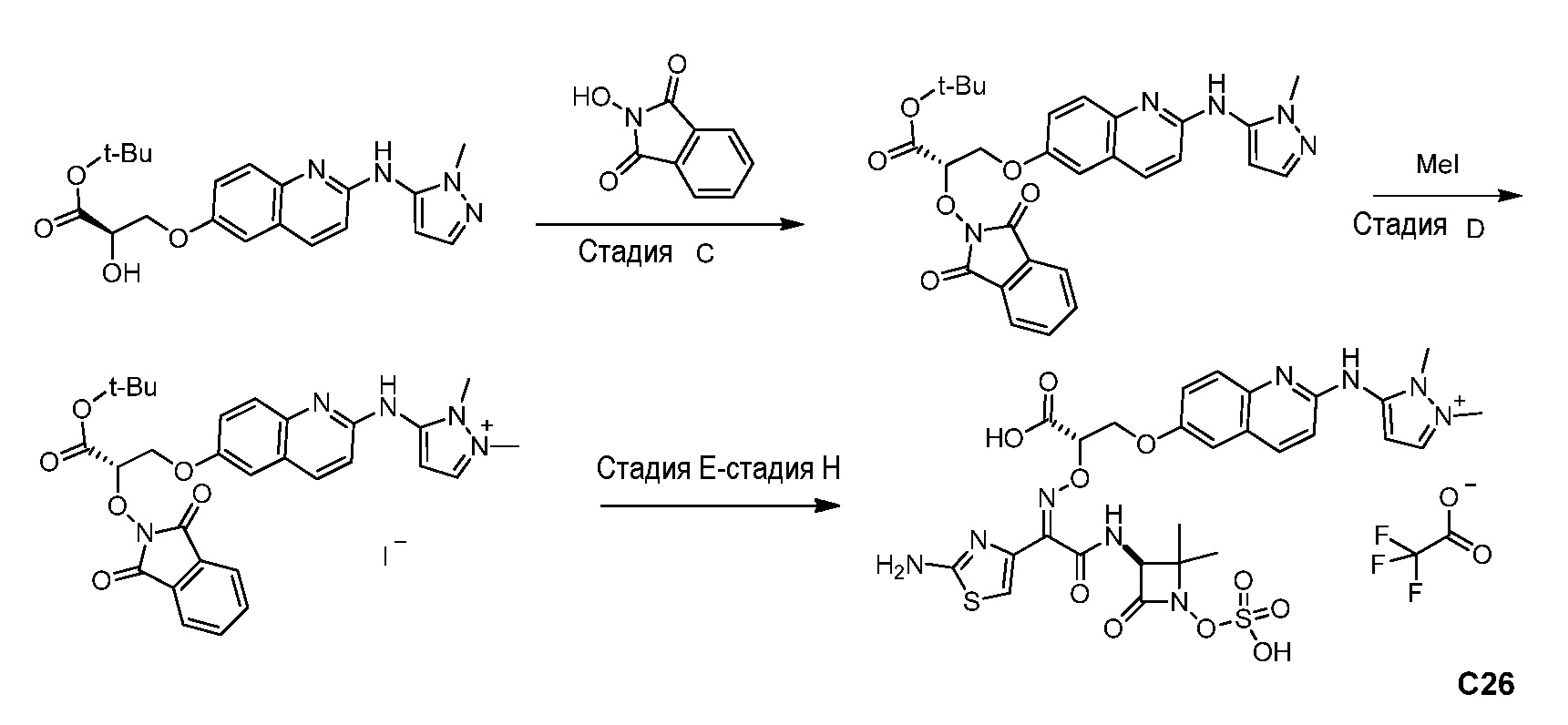
Процедуры от стадии А до стадии С соединения 26 были, в общем, такими же процедурами, как от стадии С до стадии Е в примере 1.
Стадия D: Получение (S)-5-((6-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)хинолин-2-ил)амино)-1,2-диметил-1H-пиразол-2-ий йодида К раствору (S)-трет-бутил 2-((1,3-диоксоизоиндолин-2-ил)окси)-3-((2-((1-метил-1H-пиразол-5-ил)амино)хинолин-6-ил)окси)пропаноата (80 мг, 0,15 ммоль) в ACN (1 мл) добавляли MeI (0,094 мл, 1,511 ммоль). Реакционную смесь нагревали при 60°C в течение 3 часов и концентрировали с получением неочищенного продукта, который использовался как есть на следующей стадии. LC-MS [M+H]: m/z 544,23.
Стадии E-H, в общем, были такими же, как процедуры от стадии B до стадии E в примере 15. Указанное в заголовке соединение 26 охарактеризовывали с помощью LC/MS и ЯМР. LC-MS [M+H]: m/z 704,43.1H ЯМР (CH3OH-d4, 500 MГц): δH 8,19 (1H, д, J=8,9 Гц), 8,14 (1H, с), 7,75 (1H, д, J=9,1 Гц), 7,39 (1H, д, J=9,3 Гц), 7,32 (1H, с), 7,27 (2H, д, J=9,1 Гц), 7,17 (1H, с), 5,31 (1H, с), 4,72 (1H, с), 4,63 (2H, м), 4,07 (3H, с), 4,01 (3H, с), 1,49 (3H, с), 1,28 (3H, с).
ПРИМЕР 27
(S)-3-((Z)-2-(((S)-2-((2-((1-(3-аминопропил)-2-метил-1H-пиразол-2-ий-3-ил)амино)хинолин-6-ил)окси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат (C27)
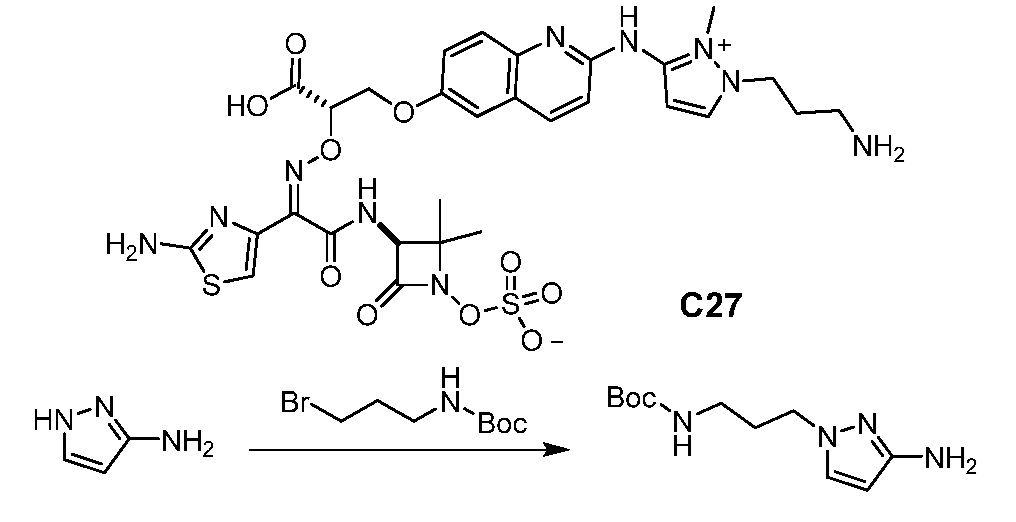
Стадия A. Получение трет-бутил (3-(3-амино-1H-пиразол-1-ил)пропил)карбамата
К раствору 1H-пиразол-3-амина (0,5 г, 6,0 ммоль) в DMF (5 мл) добавляли NaH (0,26 г, 6,6 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 10 минут. Добавляли трет-бутил (3-бромпропил)карбамат (1,6 г, 6,6 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь гасили и разбавляли водой, экстрагировали EtOAc. Объединенный органический слой промывали водой и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя EtOAc с получением целевого продукта в виде масла. LC-MS [M+H]: m/z 241,22. Используя, в общем, ту же самую процедуру в примере 26, соединение 27 синтезировали с вышеуказанным промежуточным соединением и охарактеризовывали с помощью LC/MS. LC-MS [M+H]: m/z 747,39.
ПРИМЕР 28
3-(6-(2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)этокси)имидазо[1,2-а]пиридин-2-ил)пиперидин-1-ий 2,2,2-трифторацетат (C28)

Стадия A: Получение трет-бутил 3-(6-гидроксиимидазо[1,2-а]пиридин-2-ил)пиперидин-1-карбоксилата В 250-мл круглодонной колбе 6-аминопиридин-3-ол и соль HCl (2,0 г, 13 ммоль) суспендировали в EtOH (40 мл), и затем добавляли бикарбонат натрия (1100 мг, 13 ммоль) и перемешивали в течение 5 минут. После образования раствора, добавляли трет-бутил 3-(2-хлорацетил)пиперидин-1-карбоксилат (5 г, 19 ммоль) и смесь нагревали до 90°C в течение 16 часов. Реакционную смесь концентрировали и остаток очищали с помощью MPLC с обращенной фазой с ACN и водой, забуференной 0,05% TFA, с получением твердой соли TFA продукта. Соль растворяли в воде (15 мл), рН доводили до ~7-8 с помощью насыщенного водного раствора NaHCO3, и затем полученный продукт экстрагировали смесью IPA:CHCl3 (1:3). Органический слой промывали насыщенным солевым раствором (1x), сушили (Na2SO4), фильтровали и концентрировали. Остаток перекристаллизовывали из THF с получением трет-бутил 3-(6-гидроксиимидазо[1,2-а]пиридин-2-ил)пиперидин-1-карбоксилата. LC-MS m/z [M+H]+: 318,08.
Стадия B: Трет-бутил 3-(6-(2-((1,3-диоксоизоиндолин-2-ил)окси)этокси)имидазо[1,2-а]пиридин-2-ил)пиперидин-1-карбоксилат 2-(2-гидроксиэтокси)изоиндолин-1,3-дион (118 мг, 0,57 ммоль), трет-бутил 3-(6-гидроксиимидазо[1,2-а]пиридин-2-ил)пиперидин-1-карбоксилат (120 мг, 0,38 ммоль), и трифенилфосфин (150 мг, 0,57 ммоль) растворяли в THF (4 мл) и охлаждали до -70°C и затем добавляли DEAD (0,093 мл, 0,57 ммоль). Реакционную смесь нагревали до комнатной температуры в течение 1 часа. Реакционную смесь концентрировали и остаток очищали с помощью MPLC с 0-50% EtOAc/этанол 1:3 с получением трет-бутил 3-(6-(2-((1,3-диоксоизоиндолин-2-ил)окси)этокси)имидазо[1,2-а]пиридин-2-ил)пиперидин-1-карбоксилата. LC-MS m/z [M+H]+: 507,78.
Стадия C: Трет-бутил 3-(6-(2-(аминоокси)этокси)имидазо[1,2-а]пиридин-2-ил)пиперидин-1-карбоксилат Трет-бутил 3-(6-(2-((1,3-диоксоизоиндолин-2-ил)окси)этокси)имидазо[1,2-а]пиридин-2-ил)пиперидин-1-карбоксилат (112 мг, 0,22 ммоль) растворяли в этаноле (7 мл) и охлаждали до 0°C, затем добавляли гидразин (8,5 мкл, 0,26 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали и перемешивали в DCM (20 мл) в течение 15 минут. Суспензию DCM фильтровали и концентрировали с получением трет-бутил 3-(6-(2-(аминоокси)этокси)имидазо[1,2-а]пиридин-2-ил)пиперидин-1-карбоксилата. LC-MS m/z [M+H]+: 377,12
Стадия D: (Z)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-((2-((2-(1-(трет-бутоксикарбонил)пиперидин-3-ил)имидазо[1,2-а]пиридин-6-ил)окси)этокси)имино)уксусная кислота
Трет-бутил 3-(6-(2-(аминоокси)этокси)имидазо[1,2-а]пиридин-2-ил)пиперидин-1-карбоксилат (85 мг, 0,23 ммоль) растворяли в смеси этанола (5,6 мл) и хлороформа (1,9 мл), и затем добавляли 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусную кислоту (Промежуточное соединение 3) (62 мг, 0,226 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали с получением (Z)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-((2-((2-(1-(трет-бутоксикарбонил)пиперидин-3-ил)имидазо[1,2-а]пиридин-6-ил)окси)этокси)имино)уксусной кислоты. LC-MS m/z [M+H]+: 631,51.
Стадия E: Получение трет-бутил 3-(6-(2-((((Z)-1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)-окси)этокси)имидазо[1,2-а]пиридин-2-ил)пиперидин-1-карбоксилата (Z)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-((2-((2-(1-(трет-бутоксикарбонил)пиперидин-3-ил)имидазо[1,2-а]пиридин-6-ил)окси-)этокси)-имино)уксусную кислоту (0,14 г, 0,22 ммоль) растворяли в DMF (4 мл), добавляли 1H-бензо[d][1,2,3]триазол-1-ол гидрат (0,071 г, 0,450 ммоль) и N,N'-метандиилидендициклогексанамин (0,094 г, 0,450 ммоль), и затем смесь перемешивали при комнатной температуре в течение 0,5 часа. Добавляли (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфат (0,095 г, 0,45 ммоль), затем гидрокарбонат натрия (0,076 г, 0,90 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь фильтровали и очищали с помощью ВЭЖХ с обращенной фазой с ACN и водой, забуференной 0,05% TFA, с получением трет-бутил 3-(6-(2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-этокси)имидазо[1,2-а]пиридин-2-ил)пиперидин-1-карбоксилата. LC-MS m/z [M+H]+: 823,59
Стадия F: Получение 3-(6-(2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)этокси)имидазо[1,2-а]пиридин-2-ил)пиперидин-1-ий 2,2,2-трифторацетата. Трет-бутил 3-(6-(2-(((Z)-(1-(2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксо-этилиден)амино)окси)этокси)имидазо[1,2-а]пиридин-2-ил)пиперидин-1-карбоксилат (5 мг, 6,1 мкмоль) растворяли в DCM (1 мл), добавляли TFA (0,47 мкл, 6,1 мкмоль) и затем смесь перемешивали при комнатной температуре в течение 0,5 часа. Реакционную смесь концентрировали без подогрева. Остаток очищали с помощью ВЭЖХ с обращенной фазой с ACN и водой, забуференной 0,05% TFA, с получением продукта 3-(6-(2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-этокси)имидазо[1,2-а]пиридин-2-ил)пиперидин-1-ий 2,2,2-трифторацетата. LC-MS m/z [M+H]+: 623,32.1H ЯМР (CD3OD-d1, 500 MГц): 1,29 (3H, с), 1,50 (3H, с), 1,94-1,90 (2H, м), 2,13-2,08 (1H, м), 2,31-2,28 (1H, м), 3,14-3,06 (1H, м), 3,47-3,39 (2H, м), 3,73-3,71 (1H, м), 4,42-4,39 (1H, м), 4,49-4,46 (1H, м), 4,60 (4H, м), 6,94 (1H, с), 7,74 (2H, с), 8,05 (1H, с), 8,45 (1H, с).
ПРИМЕР 29
(3S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-((2-((1-метил-2-(пиперидин-1-ий-3-ил)имидазо[1,2-а]пиридин-1-ий-6-ил)окси)этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат 2,2,2-трифторацетат (C29)
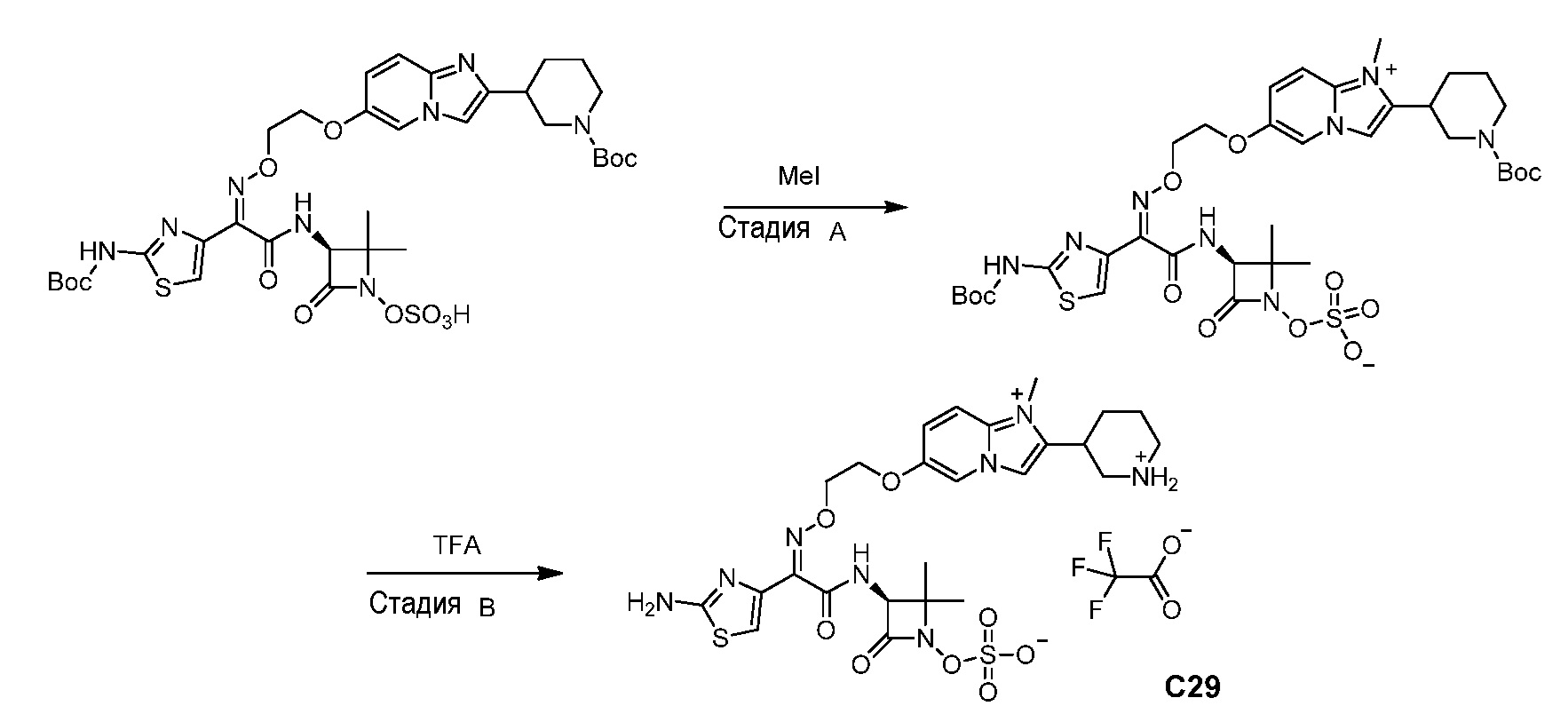
Стадия A: Получение (3S)-3-((Z)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-((2-((2-(1-(трет-бутоксикарбонил)пиперидин-3-ил)-1-метилимидазо[1,2-a]пиридин-1-ий-6-ил)окси)-этокси)имино)-ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата Трет-бутил 3-(6-(2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)этокси)имидазо[1,2-а]пиридин-2-ил)пиперидин-1-карбоксилат (Стадия E, Пример 28) (10 мг, 0,012 ммоль) растворяли в ацетонитриле (1 мл) в пробирке для реакций под воздействием микроволнового излучения, затем добавляли иодметан (7,6 мкл, 0,122 ммоль) и пробирку закупоривали. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. К реакционной смеси добавляли бикарбонат натрия (2,04 мг, 0,024 ммоль), которую нагревали при 43°C в течение 16 часов. Реакционную смесь фильтровали и остаток очищали с помощью ВЭЖХ с обращенной фазой с ACN и водой, забуференной 0,05% TFA, с получением продукта. LC-MS m/z [M+H]+: 838
Стадия B: Получение (3S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-((2-((1-метил-2-(пиперидин-1-ий-3-ил)имидазо[1,2-а]пиридин-1-ий-6-ил)окси)этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат 2,2,2-трифторацетат (3S)-3-((Z)-2-(2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)-2-((2-((2-(1-(трет-бутоксикарбонил)пиперидин-3-ил)-1-метилимидазо[1,2-a]пиридин-1-ий-6-ил)окси)-этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат (8 мг, 9,6 мкмоль) растворяли в DCM (1 мл), и затем добавляли TFA (0,74 мкл, 9,6 мкмоль). Реакционную смесь перемешивали при комнатной температуре в течение 0,5 часа. Реакционную смесь концентрировали и остаток очищали с помощью ВЭЖХ с обращенной фазой с ACN и водой, забуференной 0,05% TFA, с получением (3S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-((2-((1-метил-2-(пиперидин-3-ил)имидазо[1,2-а]пиридин-1-ий-6-ил)окси)этокси)-имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат 2,2,2-трифторацетата. LC-MS m/z [M+H]+: 637,41.1H ЯМР (CH3OH-d4, 500 MГц): 1,31-1,23 (3H, м), 1,50 (3H, д, J=5,4 Гц), 1,97-1,90 (2H, м), 2,14-2,11 (1H, м), 2,30-2,28 (1H, м), 3,17-3,05 (1H, м), 3,30-3,28( 1H, м), 3,51-3,38(1H, м), 3,76-3,66 (1H, м), 4,0 (3H, с), 4,45-4,37 (1H, м), 4,51-4,47 (1H, м), 4,63-4,59 (3H, м), 6,98-6,97 (1H, м), 7,75 (1H, с), 7,79 (1H, д дд, J=9,9, 5,3, 2,2 Гц), 7,94 (1H, д, J=9,9 Гц), 8,05( 1H, с), 8,12 (1H, с), 8,45-8,44 (1H, м).
Примеры 30 и 31
5-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-2-(азетидин-3-илметил)-1-метил-2H-индазол-1-ий 2,2,2-трифторацетат (C30) и 5-((R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-2-(азетидин-3-илметил)-1-метил-2H-индазол-1-ий 2,2,2-трифторацетат (C31)
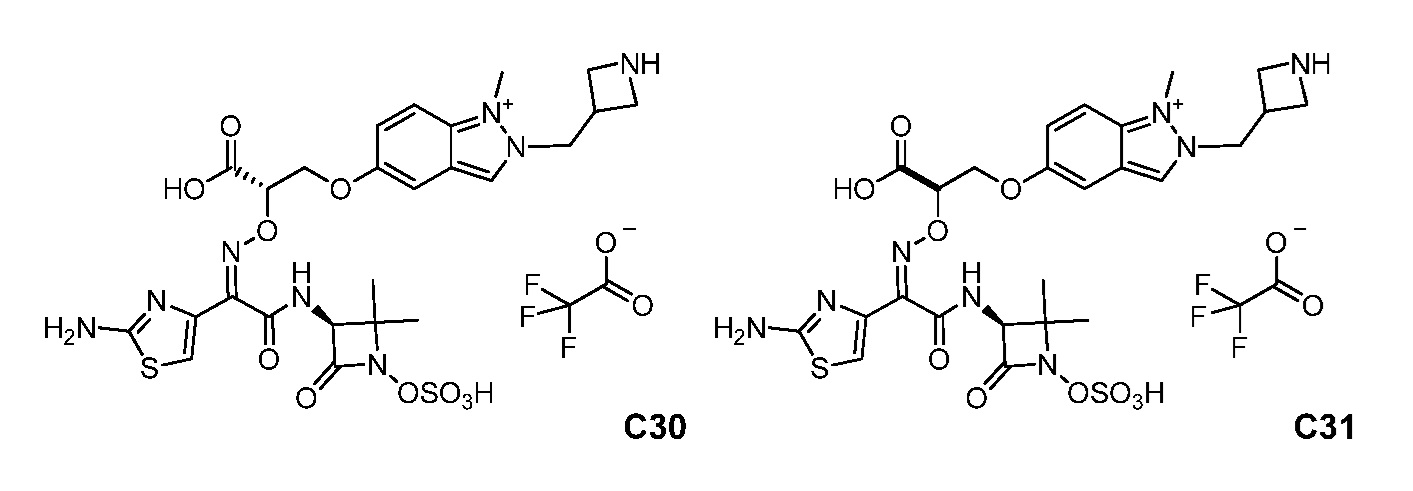
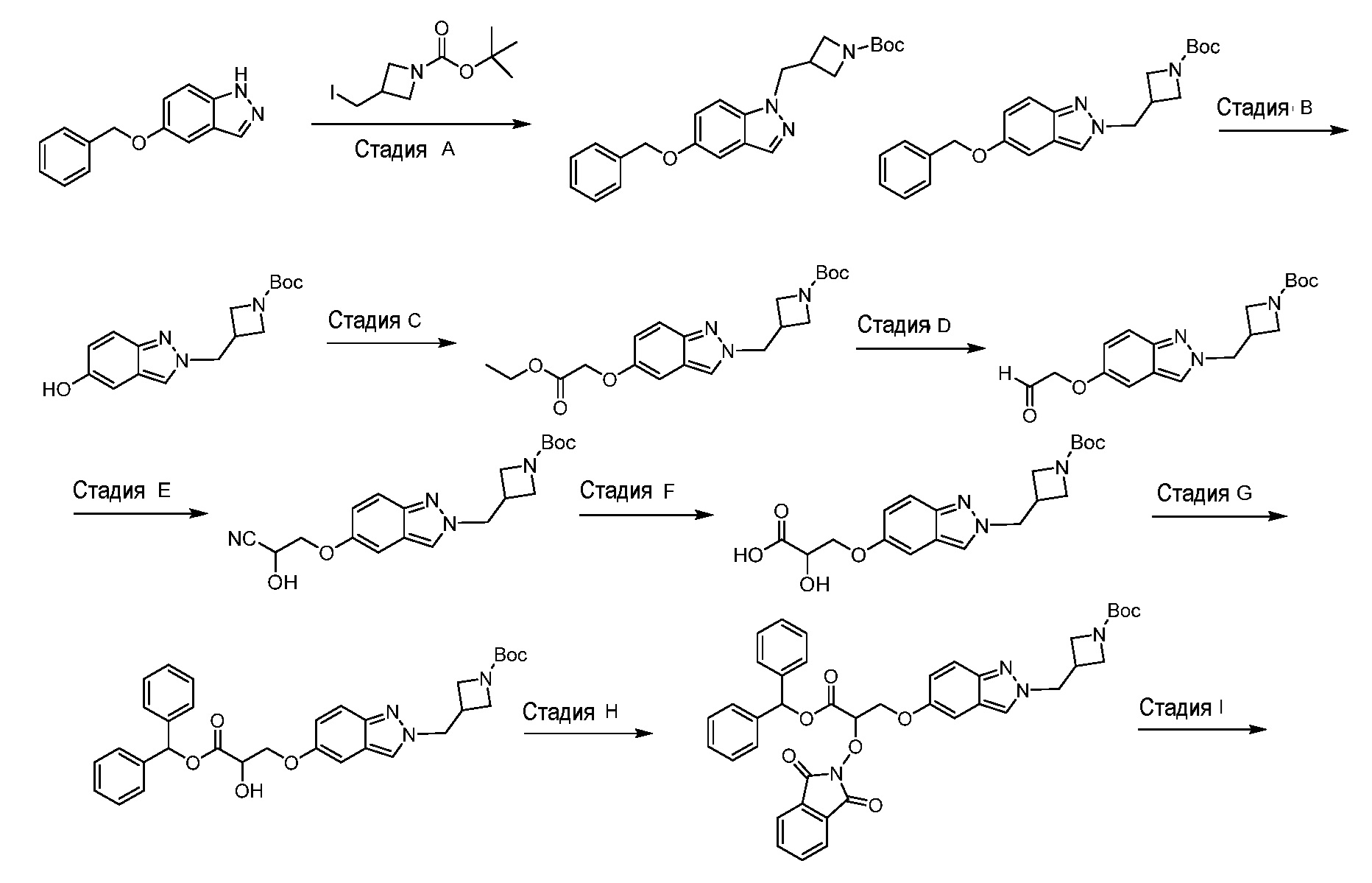

Стадия A: Получение трет-бутил 3-((5-(бензилокси)-2H-индазол-2-ил)метил)азетидин-1-карбоксилата и трет-бутил 3-((5-(бензилокси)-1H-индазол-1-ил)метил)азетидин-1-карбоксилата К раствору 5-(бензилокси)-1H-индазола (1 г, 4,5 ммоль) в DMF (16 мл) при 0°C добавляли NaH (60%, 0,27 г, 6,7 ммоль). Полученный раствор перемешивали при 0°C в течение 30 минут перед добавлением трет-бутил 3-(иодметил)азетидин-1-карбоксилата (1,3 г, 4,5 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 5 часов. После гашения путем добавления воды смесь разделяли между EtOAc (200 мл) и водой (100 мл), и органическую фазу промывали насыщенным NaHCO3 (3×100 мл), сушили над Na2SO4, концентрировали и остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением трет-бутил 3-((5-(бензилокси)-2H-индазол-2-ил)метил)-азетидин-1-карбоксилата. LC/MS: (M+1)+=394,4.1H ЯМР (CDCl3, 500 MГц): δ 7,79 (с, 1H), 7,64-7,62 (д, J=9,5 Гц, 1H), 7,50-7,48 (д, J=7,2 Гц, 2H), 7,44-7,41 (т, J=7,2 Гц, 2H), 7,37-7,36 (д, J=7,2 Гц, 1H), 7,12-7,10 (дд, J=2,1 Гц и 9,3 Гц, 1H), 6,96-6,95 (д, J=2,1 Гц, 1H), 5,10 (с, 2H), 4,59-4,58 (д, J=6,1 Гц, 2H), 4,09-4,05 (т, J=8,2 Гц, 2H), 3,81-3,77 (м, 2H), 3,25-3,20 (м, 1H), 1,45 (с, 9H); и трет-бутил 3-((5-(бензилокси)-1H-индазол-1-ил)-метил)азетидин-1-карбоксилата. LC/MS: (M+1)+=394,4,1H ЯМР (CDCl3, 500 MГц): δ 7,9 (с, 1H), 7,50-7,49 (д, J=7,5 Гц, 2H), 7,44-7,41 (т, J=8,0 Гц, 2H), 7,38-7,35 (м, 2H), 5,13 (с, 2H), 4,55-4,53 (д, J=7,6 Гц, 2H), 4,06-4,03 (т, J=8,6 Гц, 2H), 3,83-3,80 (м, 2H), 3,20-3,15 (м, 1H), 1,46 (с, 9H).
Стадия B: Получение трет-бутил 3-((5-гидрокси-2H-индазол-2-ил)метил)азетидин-1-карбоксилата К раствору трет-бутил 3-((5-(бензилокси)-2H-индазол-2-ил)метил)-азетидин-1-карбоксилата (1,03 г, 2,6 ммоль) в MeOH (50 мл) добавляли 10% Pd/C (0,28 г, 0,26 ммоль). Полученную смесь гидрировали при 50 фунт/кв.дюйм в течение 40 часов. Смесь фильтровали через целит, и фильтрат концентрировали с получением трет-бутил 3-((5-гидрокси-2H-индазол-2-ил)метил)азетидин-1-карбоксилата. LC/MS: (M+1)+=304,2
Стадия C: Получение трет-бутил 3-((5-(2-этокси-2-оксоэтокси)-2H-индазол-2-ил)метил)-азетидин-1-карбоксилата К раствору трет-бутил 3-((5-гидрокси-2H-индазол-2-ил)метил)азетидин-1-карбоксилата (0,89 г, 2,9 ммоль) в этилацетате (100 мл) добавляли K2CO3 (0,81 г, 5,9 ммоль) и этил-2-бромацетат (0,39 мл, 3,5 ммоль). Полученную смесь нагревали с обратным холодильником в течение ночи. После фильтрации через целит, фильтрат концентрировали и остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением трет-бутил 3-((5-(2-этокси-2-оксоэтокси)-2H-индазол-2-ил)метил)-азетидин-1-карбоксилата. LC/MS: (M+1)+=390,3
Стадия D: Получение трет-бутил 3-((5-(2-оксоэтокси)-2H-индазол-2-ил)метил)азетидин-1-карбоксилата К раствору трет-бутил 3-((5-(2-этокси-2-оксоэтокси)-2H-индазол-2-ил)метил)азетидин-1-карбоксилата (0,31 г, 0,80 ммоль) в CH2Cl2 (14 мл) при -78°C добавляли по каплям DIBAL-H (1,6 мл, 1,6 ммоль). Полученный раствор перемешивали при -78°C в течение 4 часов. Реакцию гасили добавлением MeOH (3 мл) с последующим добавлением насыщенного тартрата калия (100 мл). Полученную смесь перемешивали при комнатной температуре в течение 8 часов, и смесь экстрагировали DCM (3×100 мл). Объединенную органическую фазу сушили над Na2SO4, и концентрировали с получением трет-бутил 3-((5-(2-оксоэтокси)-2H-индазол-2-ил)метил)азетидин-1-карбоксилата. LC/MS: (M+1+18)+=364,3
Стадия E: Получение трет-бутил 3-((5-(2-циано-2-гидроксиэтокси)-2H-индазол-2-ил)метил)азетидин-1-карбоксилата К смеси трет-бутил 3-((5-(2-оксоэтокси)-2H-индазол-2-ил)метил)азетидин-1-карбоксилата (0,47 г, 1,4 ммоль) в t-BuOMe (10 мл) и воды (2 мл) добавляли по каплям уксусную кислоту (3 мл) и цианид натрия (0,10 г, 2,04 ммоль). Полученный раствор перемешивали при комнатной температуре в течение ночи. Раствор добавляли к насыщенному Na2CO3 (100 мл) при 0°C. Смесь экстрагировали EtOAc (2×150 мл), и объединенную органическую фазу промывали насыщенным NaHCO3(2×100 мл), сушили над Na2SO4, и концентрировали. Остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением трет-бутил 3-((5-(2-циано-2-гидроксиэтокси)-2H-индазол-2-ил)метил)азетидин-1-карбоксилата. LC/MS: (M+1)+=373,3
Стадия F: Получение 3-((2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-2H-индазол-5-ил)окси)-2-гидроксипропановой кислоты В раствор трет-бутил 3-((5-(2-циано-2-гидроксиэтокси)-2H-индазол-2-ил)метил)азетидин-1-карбоксилата (0,21 г, 0,56 ммоль) в MeOH (20 мл) при 0°C барботировали HCl (g) в течение 20 минут, и затем полученный раствор перемешивали от 0°C до комнатной температуры в течение ночи. После концентрации, остаток растворяли в диоксане (10 мл) и воде (2 мл). К полученному раствору добавляли NaOH (5 мл, 5,00 ммоль) и Boc2O (0,16 мл, 0,68 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 часа. После удаления летучих веществ, водную фазу экстрагировали DCM (2×10 мл), и затем подкисляли до pH 3, и осадок экстрагировали DCM (3×50 мл). Объединенную органическую фазу сушили над Na2SO4, и концентрировали с получением 3-((2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-2H-индазол-5-ил)окси)-2-гидроксипропановой кислоты. LC/MS: (M+1)+=392,3
Стадия G: Получение трет-бутил 3-((5-(3-(бензгидрилокси)-2-гидрокси-3-оксопропокси)-2H-индазол-2-ил)метил)азетидин-1-карбоксилата К раствору 3-((2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-2H-индазол-5-ил)окси)-2-гидроксипропановой кислоты (190 мг, 0,48 ммоль) в MeOH (10 мл) добавляли дифенилдиазометан (470 мг, 2,4 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 часа. После концентрации, остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением трет-бутил 3-((5-(3-(бензгидрилокси)-2-гидрокси-3-оксопропокси)-2H-индазол-2-ил)метил)азетидин-1-карбоксилата. LC/MS: (M+1)+=558,4
Стадия H: Получение трет-бутил 3-((5-(3-(бензгидрилокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2H-индазол-2-ил)метил)азетидин-1-карбоксилата К раствору трет-бутил 3-((5-(3-(бензгидрилокси)-2-гидрокси-3-оксопропокси)-2H-индазол-2-ил)метил)азетидин-1-карбоксилата (200 мг, 0,36 ммоль) в THF (4 мл) добавляли 2-гидроксиизоиндолин-1,3-дион (70 мг, 0,43 ммоль), трифенилфосфин (141 мг, 0,54 ммоль) и DEAD (0,085 мл, 0,54 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 2 часов. После концентрации, остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением трет-бутил 3-((5-(3-(бензгидрилокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2H-индазол-2-ил)метил)азетидин-1-карбоксилата. LC/MS: (M+1)+=703,5
Стадия I: Получение 5-(3-(бензгидрилокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1-метил-2H-индазол-1-ий йодида К раствору трет-бутил 3-((5-(3-(бензгидрилокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2H-индазол-2-ил)метил)азетидин-1-карбоксилата (230 мг, 0,33 ммоль) в ацетонитриле (5 мл) добавляли MeI (0,20 мл, 3,3 ммоль). Полученный раствор нагревали при 70°C в течение 4 дней. Затем добавляли дополнительное количество MeI (0,20 мл, 3,3 ммоль) и полученный раствор нагревали при 80°C в течение 24 часов. Раствор затем концентрировали с получением 5-(3-(бензгидрилокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1-метил-2H-индазол-1-ий йодида. LC/MS: M+=717,6
Стадия J: Получение (Z)-5-(3-(бензгидрилокси)-2-((((2-((трет-бутоксикарбонил)амино)-тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)-азетидин-3-ил)метил)-1-метил-2H-индазол-1-ия К раствору 5-(3-(бензгидрилокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1-метил-2H-индазол-1-ий йодида (240 мг, 0,33 ммоль) в этаноле (5 мл) и CH2Cl2 (5,00 мл) при 0°C добавляли гидразин (10 мкл, 0,33 ммоль). Полученный раствор перемешивали при 0°C в течение 1 часа. Затем 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусную кислоту (Промежуточное соединение 3) (134 мг, 0,49 ммоль) добавляли к вышеуказанному раствору, и полученный раствор перемешивали при 0°C до комнатной температуры в течение 2 часов. Реакционную смесь концентрировали и остаток очищали с помощью колонки MPLC с обращенной фазой с использованием ацетонитрила (0,05% TFA)/воды (0,05% TFA) в качестве элюирующих растворителей с получением (Z)-5-(3-(бензгидрилокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1-метил-2H-индазол-1-ия. LC/MS: M+=841,5
Стадия K: Получение 5-(3-(бензгидрилокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)-тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксо-этилиден)амино)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1-метил-2H-индазол-1-ия К раствору (Z)-5-(3-(бензгидрилокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1-метил-2H-индазол-1-ия (64 мг, 0,076 ммоль) в DMF (2 мл) добавляли DCC (125 мг, 0,61 ммоль) и HOBT (47 мг, 0,30 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 30 минут перед добавлением (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфата (64 мг, 0,30 ммоль) и бикарбоната натрия (77 мг, 0,91 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. После фильтрации, фильтрат очищали с помощью MPLC с обращенной фазой с использованием ацетонитрила (0,05% TFA)/воды (0,05% TFA) в качестве элюирующих растворителей с получением 5-(3-(бензгидрилокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1-метил-2H-индазол-1-ия. LC/MS: M+=1033,6
Стадия L: Получение 5-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-2-(азетидин-3-илметил)-1-метил-2H-индазол-1-ий 2,2,2-трифторацетата (C30) и 5-((R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-2-(азетидин-3-илметил)-1-метил-2H-индазол-1-ий 2,2,2-трифторацетата (C31) К раствору 5-(3-(бензгидрилокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)-азетидин-3-ил)метил)-1-метил-2H-индазол-1-ия (70 мг, 0,068 ммоль) в CH2Cl2 (2 мл) добавляли TFA (2 мл, 26 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 40 минут, концентрировали и затем остаток обрабатывали Et2O и концентрировали снова. Остаток очищали с помощью ВЭЖХ с обращенной фазой с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05%TFA) с получением 5-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-2-(азетидин-3-илметил)-1-метил-2H-индазол-1-ий 2,2,2-трифторацетата (C30, быстро элюируемый диастереоизомер). LC/MS: (M+1)+=667,1,1H ЯМР (500 МГц, D2O): δ 8,68(с, 1H), 7,62-7,60(д, J=8,0 Гц, 1H), 7,45-7,43(д, J=8,6 Гц, 1H), 7,23(с, 1H), 7,03(с, 1H), 5,07(с, 1H), 4,97-4,95(д, J=7,5 Гц, 2H), 4,65(с,1H), 4,49-4,44(м, 2H), 4,24-4,20(т, J=9,8 Гц, 2H), 4,13(с, 3H), 4,10-4,06(т, J=9,5 Гц, 2H), 3,68-3,61(м,1H), 1,32(с, 3H), 0,89(с, 3H); и 5-((R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)-азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-2-(азетидин-3-илметил)-1-метил-2H-индазол-1-ий 2,2,2-трифторацетата (C31, медленно элюируемый диастереоизомер). LC/MS: (M+1)+=667,1,1H ЯМР (500 МГц, D2O): δ 8,68 (с, 1H), 7,63-7,61 (д, J=10,0 Гц, 1H), 7,45-7,43 (дд, J=10,0 Гц и 1,5 Гц, 1H), 7,24 (д, J=1,5 Гц, 1H), 7,02 (с, 1H), 5,02-4,99 (м, 1H), 4,97-4,95 (д, J=7,8 Гц, 2H), 4,65 (с, 1H), 4,46-4,41 (м, 2H), 4,24-4,20 (т, J=10,9 Гц, 2H), 4,13 (с, 3H), 4,10-4,06 (т, J=9,8 Гц, 2H), 3,68-3,61 (м, 1H), 1,32 (с, 3H), 0,95 (с, 3H).
Примеры 32 и 33
1-(3-аминопропил)-5-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-2-метил-1H-индазол-2-ий 2,2,2-трифторацетат (C32) и 1-(3-аминопропил)-5-((R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-2-метил-1H-индазол-2-ий 2,2,2-трифторацетат (C33)
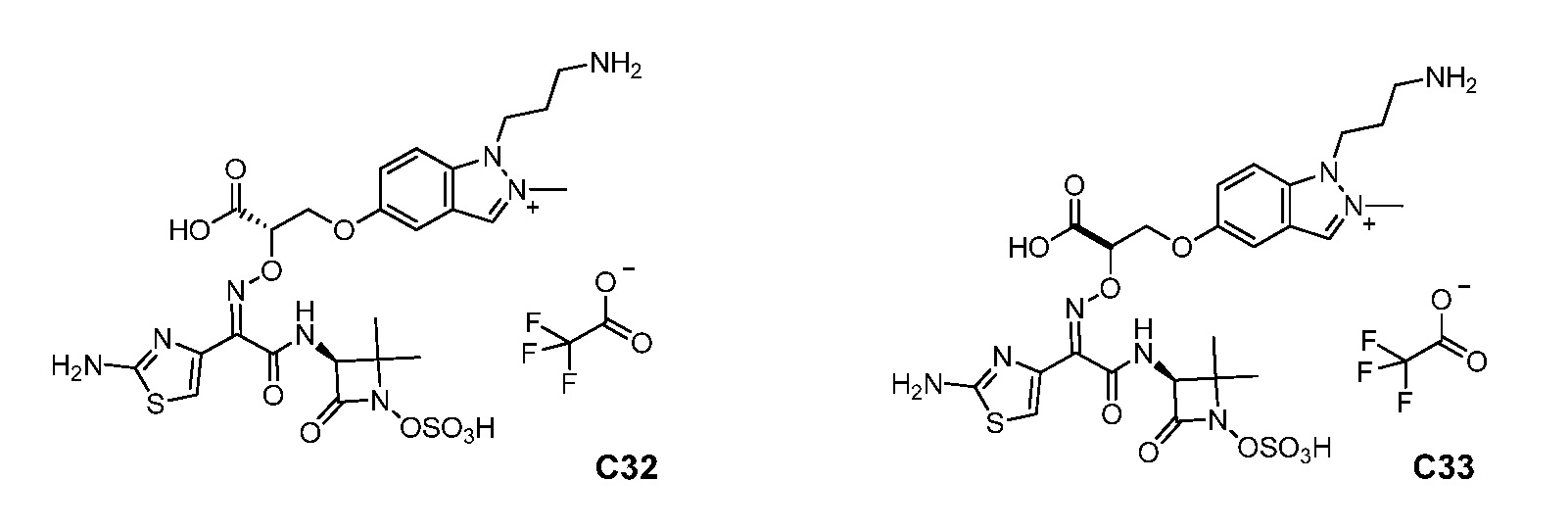
Соединения 32 и 33 получали по той же общей методике, что и в примере 30 и 31.
Соединение 32: 1-(3-аминопропил)-5-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-2-метил-1H-индазол-2-ий 2,2,2-трифторацетат. LC/MS: (M+1)+= 655,4
1Н ЯМР (500 МГц, D2O): δ 8,66 (с, 1H), 7,65-7,63 (д, J=9,2 Гц, 1H), 7,47-7,44 (дд, J=1,6 Гц и 9,4 Гц, 1H), 7,47-7,44 (д, J=2,2 Гц, 1H), 7,03 (с, 1H), 5,06-5,04 (м, 1H), 4,67 (с, 1H), 4,46-4,39 (м, 2H), 4,28 (с, 3H), 3,05-3,02 (м, 2H), 2,21-2,17 (м, 2H), 1,34 (с, 3H), 0,95 (с, 3H).
Соединение 33: 1-(3-аминопропил)-5-((R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-2-метил-1H-индазол-2-ий 2,2,2-трифторацетат. LC/MS: (M+1)+= 655,4,1Н ЯМР (500 МГц, D2O): δ 8,65 (с, 1H), 7,65-7,63 (д, J=9,6 Гц, 1H), 7,46-7,44 (дд, J=9,9 Гц и 2,4 Гц, 1H), 7,27 (д, J=2,4 Гц, 1H), 7,04 (с, 1H), 5,02-5,00 (м, 1H), 4,69 (с, 1H), 4,47-4,41 (м, 2H), 4,28 (с, 3H), 3,05-3,02 (т, J=7,6 Гц, 2H), 2,2-2,16 (м, 2H), 1,32 (с, 3H), 0,97 (с, 3H).
Примеры 34 и 35
5-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-1-(азетидин-3-илметил)-2-метил-1H-индазол-2-ий 2,2,2-трифторацетат (C34) и 5-((R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-1-(азетидин-3-илметил)-2-метил-1H-индазол-2-ий 2,2,2-трифторацетат (C35)
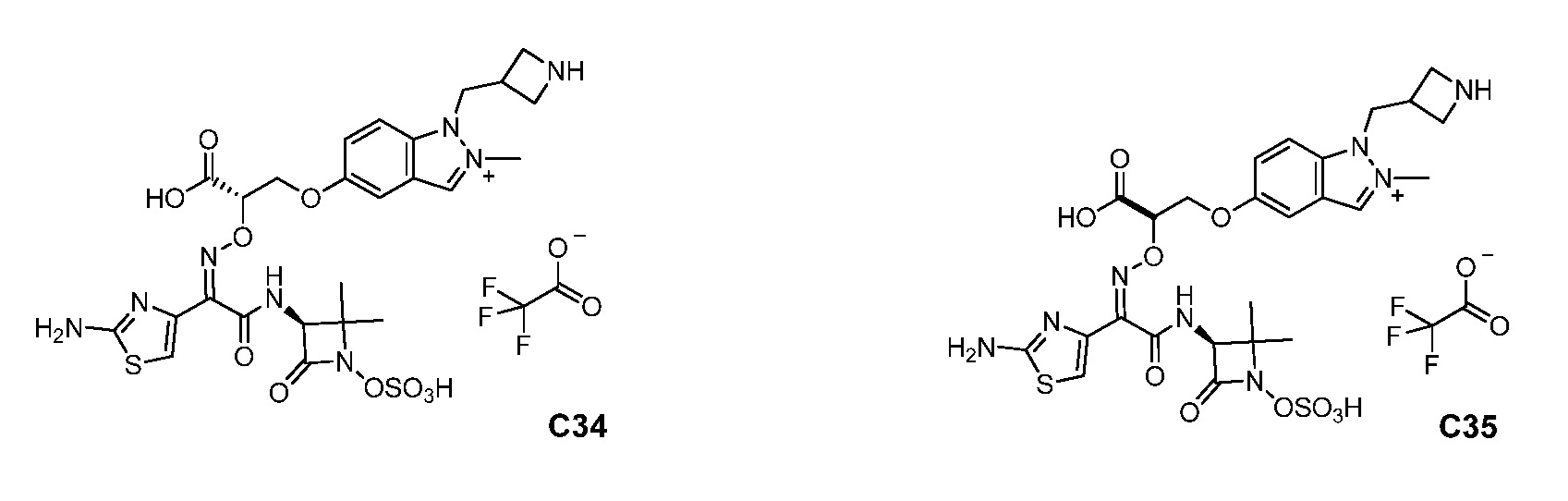
Соединения 34 и 35 получали по той же общей методике, что и в примере 30 и 31.
Соединение 34: 5-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-1-(азетидин-3-илметил)-2-метил-1H-индазол-2-ий 2,2,2-трифторацетат. LC/MS: (M+1)+= 667,4.1H ЯМР (500 МГц, D2O): δ 8,68 (с, 1H), 7,76-7,74 (д, J=9,1 Гц, 1H), 7,48-7,46 (м, 1H), 7,28-7,27 (м, 1H), 7,05 (с, 1H), 5,11-5,10 (м, 1H), 4,99-4,97 (д, J=6,7 Гц, 2H), 4,68 (с, 1H), 4,48-4,40 (м, 2H), 4,26 (с, 3H), 4,12-4,02 (м, 4H), 3,56-3,50 (м, 1H), 1,34 (с, 3H), 0,93 (с, 3H).
Соединение 35: 5-((R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-1-(азетидин-3-илметил)-2-метил-1H-индазол-2-ий 2,2,2-трифторацетат. LC/MS: (M+1)+= 667,1,1H ЯМР (500 МГц, D2O): δ 8,68(с, 1H), 7,76-7,74(д, J=9,0 Гц, 1H), 7,48-7,46(дд, J=9,8Гц и 2,3 Гц, 1H), 7,27-7,26(д, J=2,3 Гц, 1H), 7,04(с, 1H), 5,05(м, 1H), 4,99-4,97(д, J=9,2 Гц, 2H), 4,49-4,42(м, 2H), 4,26(с, 3H), 4,12-4,01(м, 4H), 3,56-3,49(м, 1H), 1,32(с, 3H), 0,96(с, 3H).
ПРИМЕР 36
1-(3-аминопропил)-5-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((2S,3S)-2-метил-4-оксо-1-сульфоазетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-2-метил-1H-индазол-2-ий 2,2,2-трифторацетат

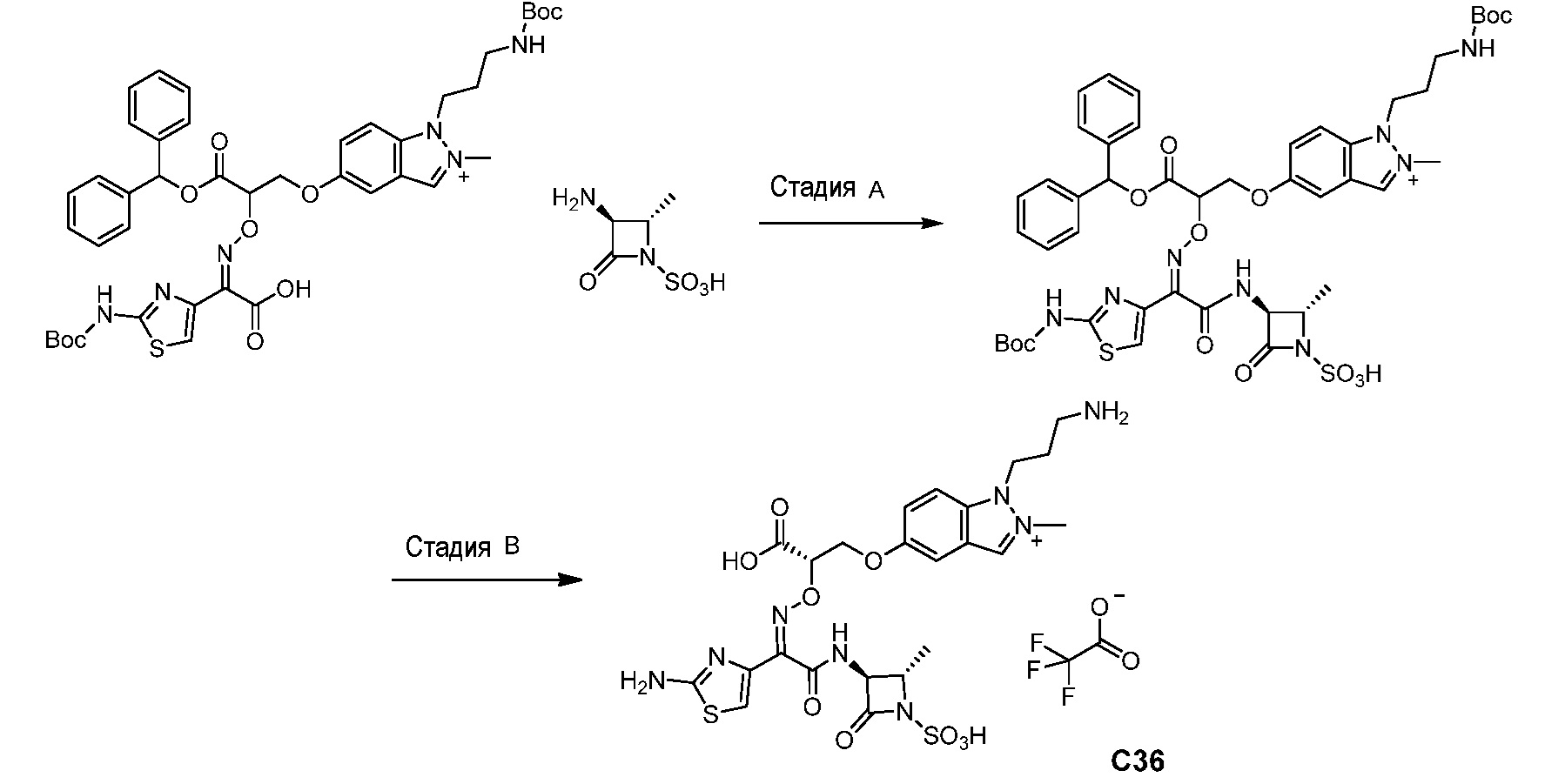
Стадия A: Получение 5-(3-(бензгидрилокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)-тиазол-4-ил)-2-(((2S,3S)-2-метил-4-оксо-1-сульфоазетидин-3-ил)амино)-2-оксо-этилиден)амино)окси)-3-оксопропокси)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-индазол-2-ия (Z)-5-(3-(бензгидрилокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)-амино)окси)-3-оксопропокси)-1-(3-((трет-бутоксикарбонил)амино)-пропил)-2-метил-1H-индазол-2-ий получали из 5-(бензилокси)-1H-индазола и трет-бутил (3-иодопропил)карбамата, следуя той же процедуре из стадий A-J в примерах 30 и 31. К раствору (Z)-5-(3-(бензгидрилокси)-2-((((2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)-1-(3-((трет-бутокси-карбонил)амино)пропил)-2-метил-1H-индазол-2-ия (54 мг, 0,065 ммоль) в DMF (2 мл) добавляли DCC (67 мг, 0,32 ммоль) и HOBT (40 мг, 0,26 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут перед добавлением (2S,3S)-3-амино-2-метил-4-оксоазетидин-1-сульфоновой кислоты (47 мг, 0,26 ммоль) и бикарбоната натрия (55 мг, 0,65 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 5 часов. Смесь фильтровали и фильтрат очищали с помощью ВЭЖХ с обращенной фазой с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05%TFA) в качестве подвижной фазы с получением 5-(3-(бензгидрилокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((2S,3S)-2-метил-4-оксо-1-сульфоазетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-1-(3-((трет-бутоксикарбонил)-амино)пропил)-2-метил-1H-индазол-2-ия. LC/MS: M+= 991,6
Стадия B: Получение 1-(3-аминопропил)-5-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((2S,3S)-2-метил-4-оксо-1-сульфоазетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-2-метил-1H-индазол-2-ий 2,2,2-трифторацетата К раствору 5-(3-(бензгидрилокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((2S,3S)-2-метил-4-оксо-1-сульфоазетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-1-(3-((трет-бутоксикарбонил)амино)пропил)-2-метил-1H-индазол-2-ия (42 мг, 0,042 ммоль) в DCM (2 мл) добавляли TFA (2 мл, 26 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 30 минут. Раствор концентрировали и остаток очищали с помощью ВЭЖХ с обращенной фазой с использованием смеси ацетонитрил (0,05% TFA)/вода (0,0-5%TFA) с получением 1-(3-аминопропил)-5-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((2S,3S)-2-метил-4-оксо-1-сульфоазетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-2-метил-1H-индазол-2-ий 2,2,2-трифторацетата (более полярный диастереомер). LC/MS: (M+1)+= 625,4,1Н ЯМР (500 МГц, CD3OD): δ 8,86 (с, 1H),7,82-7,80(д, J=9,8 Гц, 1H), 7,57-7,55(д, J=9,8 Гц, 1H), 7,48(с, 1H), 6,90(с, 1H), 5,20(с, 1H), 4,81-4,66(м, 2H), 4,42(с, 3H), 4,16-4,15(д, J=1,7 Гц, 1H), 3,81-3,77(м, 1H), 3,13-3,10(т, J=7,6 Гц, 2H), 2,32-2,22(м, 2H), 1,43(д, J=6,9 Гц, 3H).
Примеры 37 и 38
(S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-((2-(азетидин-3-илметил)имидазо[1,2-а]пиридин-6-ил)окси)пропановая кислота, 2,2,2-трифторацетатная соль (C37) и (R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-((2-(азетидин-3-илметил)имидазо[1,2-а]пиридин-6-ил)окси)пропановая кислота, 2,2,2-трифторацетатная соль (C38)

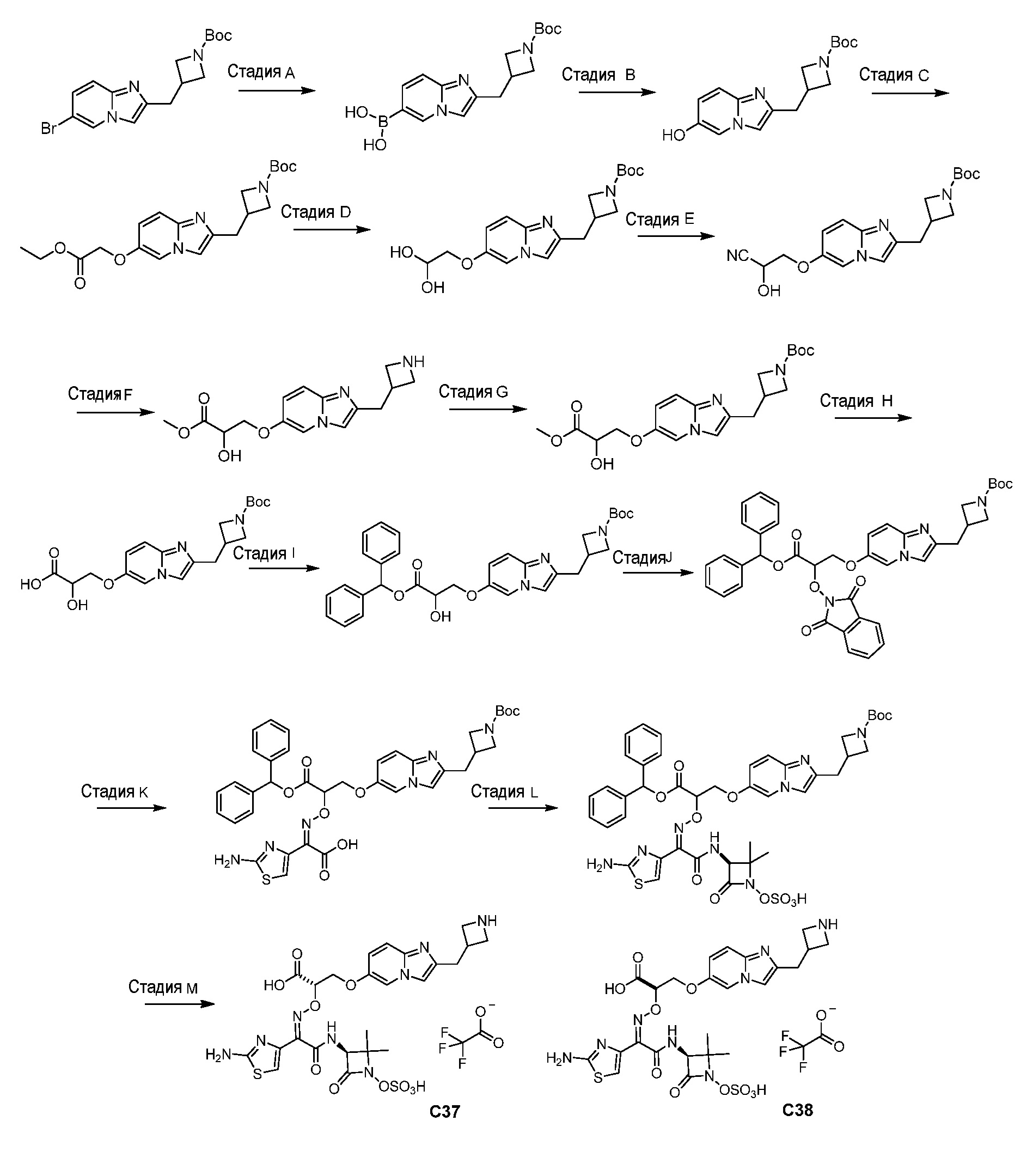
Стадия A: Получение (2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)имидазо[1,2-а]пиридин-6-ил)бороновой кислоты Смесь ацетата калия (178 мг, 1,8 ммоль), PdCl2(dppf)-CH2Cl2аддукта (49 мг, 0,060 ммоль), бис(пинаколато)диборона (184 мг, 0,72 ммоль), и трет-бутил 3-((6-бромимидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата (Промеж.соед. 6) (221 мг, 0,60 ммоль) дегазировали с помощью вакуума/заполняли N2 три раза в сосуде для микроволнового излучения, с последующим добавлением диоксана (4 мл). Полученную смесь дополнительно дегазировали с помощью вакуума/заполняли N2 три раза, и затем нагревали при 100°C в течение ночи. После фильтрации смеси через целит, фильтрат концентрировали и остаток очищали с помощью MPLC с обращенной фазой (150 г колонка) с использованием ацетонитрила (0,05% TFA)/воды (0,05% TFA) в качестве подвижной фазы с получением (2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-имидазо[1,2-а]пиридин-6-ил)бороновой кислоты. LC/MS: (M+1)+=332,2
Стадия B: Получение трет-бутил 3-((6-гидроксиимидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата К раствору (2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)имидазо-[1,2-a]пиридин-6-ил)бороновой кислоты (950 мг, 2,9 ммоль) в THF (40 мл) и этаноле (20 мл) при 0°C добавляли бикарбонат натрия (1400 мг, 17 ммоль) и пероксид водорода (1,2 мл, 11,5 ммоль). Полученную смесь перемешивали при 0°C до комнатной температуры в течение ночи. Реакцию гасили добавлением насыщенного тиосульфата (100 мл), и затем летучие вещества выпаривали и водную фазу экстрагировали DCM (3×100 мл). Объединенную органическую фазу сушили над Na2SO4, концентрировали и остаток очищали на колонке с силикагелем с использованием смеси MeOH/DCM в качестве элюирующих растворителей с получением трет-бутил 3-((6-гидроксиимидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата. LC/MS: (M+1)+=304,2
Стадия C: Получение трет-бутил 3-((6-(2-этокси-2-оксоэтокси)имидазо[1,2-а]пиридин-2-ил)-метил)азетидин-1-карбоксилата К раствору трет-бутил 3-((6-гидроксиимидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата (0,5 г, 1,6 ммоль) в этилацетате (20 мл) добавляли карбонат калия (0,46 г, 3,3 ммоль) и этилбромацетат (0,18 мл, 1,6 ммоль). Полученную смесь нагревали при 60°C в течение ночи. После фильтрации через целит, остаток очищали на колонке с силикагелем с использованием MeOH/DCM с получением трет-бутил 3-((6-(2-этокси-2-оксоэтокси)имидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата. LC/MS: (M+1)+=390,3
Стадия D: Получение трет-бутил 3-((6-(2,2-дигидроксиэтокси)имидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата К раствору трет-бутил 3-((6-(2-этокси-2-оксо-этокси)имидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата (405 мг, 1,04 ммоль) в DCM (12 мл) при -78°C добавляли DIBAL-H (1M в DCM) (2,1 мл, 2,1 ммоль). Полученный раствор перемешивали при -78°C в течение 5 часов. Реакцию гасили добавлением MeOH (4 мл) и насыщенного тартрата калия (50 мл). Смесь перемешивали при комнатной температуре в течение ночи. Твердое вещество собирали фильтрованием и фильтрат экстрагировали 30% IPA в DCM (3×150 мл). Объединенную органическую фазу сушили над Na2SO4, концентрировали с получением трет-бутил 3-((6-(2,2-дигидроксиэтокси)имидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата. LC/MS: (M+1)+=364,3
Стадия E: Получение трет-бутил 3-((6-(2-циано-2-гидроксиэтокси)имидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата К раствору трет-бутил 3-((6-(2,2-дигидрокси-этокси)имидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата (0,52 г, 1,4 ммоль) в метил трет-бутиловом эфире (10 мл), уксусной кислоты (2 мл), и воды (2 мл) добавляли цианид натрия (0,10 г, 2,1 ммоль). Полученный раствор перемешивали при комнатной температуре в течение ночи. Раствор добавляли ко льду, охлаждали насыщенным Na2CO3 (150 мл), и смесь экстрагировали EtOAc (100 мл) и 30% IPA в DCM (2×100 мл). Объединенную органическую фазу сушили над Na2SO4, концентрировали и остаток очищали на колонке с силикагелем, используя MeOH/DCM в качестве элюирующих растворителей с получением трет-бутил 3-((6-(2-циано-2-гидрокси-этокси)имидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата. LC/MS: (M+1)+=373,3
Стадия F: Получение метил 3-((2-(азетидин-3-илметил)имидазо[1,2-а]пиридин-6-ил)окси)-2-гидроксипропаноата Раствор трет-бутил 3-((6-(2-циано-2-гидроксиэтокси)имидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата (0,17 г, 0,46 ммоль) в MeOH (10 мл) при 0°C барботировали HCl (газ) в течение 10 минут. Полученный раствор перемешивали от 0°C до комнатной температуры в течение ночи. Раствор концентрировали с получением метил 3-((2-(азетидин-3-илметил)имидазо[1,2-а]пиридин-6-ил)окси)-2-гидроксипропаноата. LC/MS: (M+1)+=306,2
Стадия G: Получение трет-бутил 3-((6-(2-гидрокси-3-метокси-3-оксопропокси)имидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата К смеси метил 3-((2-(азетидин-3-илметил)имидазо[1,2-а]пиридин-6-ил)окси)-2-гидроксипропаноата (140 мг, 0,46 ммоль) в CH2Cl2 (12 мл) добавляли Boc2O (0,14 мл, 0,59 ммоль), воду (24 мл), диоксан (12,00 мл), и триэтиламин (0,317 мл,2,276 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Смесь концентрировали с получением трет-бутил 3-((6-(2-гидрокси-3-метокси-3-оксопропокси)имидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата. LC/MS: (M+1)+=406,3
Стадия H: 3-((2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)имидазо[1,2-а]пиридин-6-ил)окси)-2-гидроксипропановая кислота К раствору трет-бутил 3-((6-(2-гидрокси-3-метокси-3-оксопропокси)имидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата (185 мг, 0,46 ммоль) в THF (20 мл), MeOH (20 мл), и воды (10 мл) добавляли NaOH (6 мл, 6,00 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 2 часов. После испарения летучих веществ, водную фазу подкисляли до pH 3 с помощью 1н HCl. Смесь лиофилизировали, и остаток затем очищали с помощью MPLC с обращенной фазой с использованием ацетонитрила (0,05% TFA)/воды (0,05% TFA) в качестве подвижной фазы с получением 3-((2-((1-(трет-бутоксикарбонил)-азетидин-3-ил)метил)имидазо[1,2-а]пиридин-6-ил)окси)-2-гидроксипропановой кислоты. LC/MS: (M+1)+=392,3
Стадия I: Получение трет-бутил 3-((6-(3-(бензгидрилокси)-2-гидрокси-3-оксопропокси)-имидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата К раствору 3-((2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)имидазо[1,2-а]пиридин-6-ил)окси)-2-гидроксипропановой кислоты (82 мг, 0,21 ммоль) в MeOH (5 мл) добавляли дифенилдиазометан (205 мг, 1,05 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Смесь концентрировали и остаток очищали на силикагеле, используя MeOH/DCM в качестве элюирующих растворителей, с получением трет-бутил 3-((6-(3-(бензгидрилокси)-2-гидрокси-3-оксопропокси)имидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата. LC/MS: (M+1)+=558,4
Стадия J: Получение трет-бутил 3-((6-(3-(бензгидрилокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)имидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата К раствору трет-бутил 3-((6-(3-(бензгидрилокси)-2-гидрокси-3-оксопропокси)имидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата (47 мг, 0,084 ммоль) в THF (2 мл) добавляли 2-гидроксиизоиндолин-1,3-дион (16,50 мг, 0,101 ммоль), трифенилфосфин (33,2 мг, 0,126 ммоль) и DEAD (0,020 мл, 0,126 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 2 часов. Раствор концентрировали и остаток очищали на колонке с силикагелем с использованием смеси MeOH/DCM в качестве элюирующих растворителей с получением трет-бутил 3-((6-(3-(бензгидрилокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)имидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата. LC/MS: (M+1)+=703,4
Стадия K: Получение (Z)-2-(((1-(бензгидрилокси)-3-((2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)имидазо[1,2-а]пиридин-6-ил)окси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)уксусной кислоты К раствору трет-бутил 3-((6-(3-(бензгидрилокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)имидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата (29 мг, 0,041 ммоль) в CH2Cl2 (2 мл) и EtOH (2 мл) добавляли гидразин (2,6 мкл, 0,081 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 5 часов. Затем 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусную кислоту (Промежуточное соединение 3) (33 мг, 0,12 ммоль) добавляли к вышеуказанному раствору и полученный раствор перемешивали при комнатной температуре в течение 1 часа. Раствор затем концентрировали и остаток очищали с помощью MPLC с обращенной фазой с использованием ацетонитрила (0,05% TFA)/воды (0,05% TFA) с получением (Z)-2-(((1-(бензгидрилокси)-3-((2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)имидазо[1,2-а]пиридин-6-ил)окси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)уксусной кислоты. LC/MS: (M+1)+=827,4
Стадия L: Получение трет-бутил 3-((6-(3-(бензгидрилокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксо-этилиден)амино)окси)-3-оксопропокси)имидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата К раствору (Z)-2-(((1-(бензгидрилокси)-3-((2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-имидазо[1,2-а]пиридин-6-ил)окси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)уксусной кислоты (36 мг, 0,043 ммоль) в DMF (2 мл) добавляли DCC (71 мг, 0,34 ммоль) и HOBt (26 мг, 0,17 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут, и затем добавляли (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфат (36 мг, 0,17 ммоль) и бикарбонат натрия (72 мг, 0,86 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 6 часов. После фильтрации, фильтрат очищали с помощью ВЭЖХ с обращенной фазой с использованием смеси ацетонитрил (0,05% TFA)/вода (0,0%TFA) в качестве подвижной фазы с получением трет-бутил 3-((6-(3-(бензгидрилокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)имидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата. LC/MS: (M+1)+=1019,8
Стадия M: Получение (S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-((2-(азетидин-3-ил-метил)-имидазо[1,2-а]пиридин-6-ил)окси)пропановой кислоты, 2,2,2-трифторацетатной соли и (R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-((2-(азетидин-3-илметил)имидазо[1,2-а]пиридин-6-ил)окси)-пропановой кислоты, 2,2,2-трифторацетатной соли К раствору трет-бутил 3-((6-(3-(бензгидрилокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)имидазо-[1,2-a]пиридин-2-ил)метил)азетидин-1-карбоксилата (16 мг, 0,016 ммоль) в CH2Cl2 (2 мл) добавляли TFA (2 мл, 26 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 часа. После концентрации, остаток очищали с помощью ВЭЖХ с обращенной фазой с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05%TFA) с получением:
Соединения 37: (S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-((2-(азетидин-3-илметил)имидазо[1,2-а]пиридин-6-ил)окси)пропановая кислота, 2,2,2-трифторацетатная соль (быстро элюируемый диастереоизомер). LC/MS:: (M+1)+=653,16,1ЯМР (D2O, 500 MГц): δ 8,56 (с, 1H), 7,68 (с, 1H), 7,62-7,60 (д, J=10,0 Гц, 1H), 7,55-7,53(д, J=10,0 Гц, 1H), 6,99 (с, 1H), 5,00 (с, 1H), 4,67 (с, 1H), 4,45-4,40 (м, 2H), 4,16-4,13 (т, J=10,3 Гц, 2H), 3,91-3,87 (т, J=9,3 Гц, 2H), 3,36-3,29 (м, 1H), 3,16-3,14(д, J=7,9 Гц, 2H), 1,37(с, 3H), 1,04(с, 3H); и
Соединения 38: (R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-((2-(азетидин-3-илметил)имидазо[1,2-а]пиридин-6-ил)окси)пропановая кислота, 2,2,2-трифторацетатная соль (медленно элюируемый диастереоизомер). LC/MS:: (M+1)+=653,16.1ЯМР (D2O, 500MГц): δ 8,19(д, J=1,2 Гц, 1H), 7,72(с, 1H), 7,63-7,61(д, J=9,1 Гц, 1H), 7,57-7,54(дд, J=9,1 Гц и 2,4 Гц, 1H), 6,98(с, 1H), 4,96(с, 1H), 4,68(с, 1H), 4,43-4,42(м, 2H), 4,16-4,13(т, J=9,5 Гц, 2H), 3,91-3,87(т, J=8,8 Гц, 2H), 3,36-3,14(д, J=8,0 Гц, 2H), 1,36(с, 3H), 1,07(с, 3H).
Примеры 39 и 40
6-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-2-(азетидин-3-илметил)-1-метилимидазо[1,2-a]пиридин-1-ий 2,2,2-трифторацетат (C39) и 6-((R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-2-(азетидин-3-илметил)-1-метилимидазо[1,2-a]пиридин-1-ий 2,2,2-трифторацетат (C40)
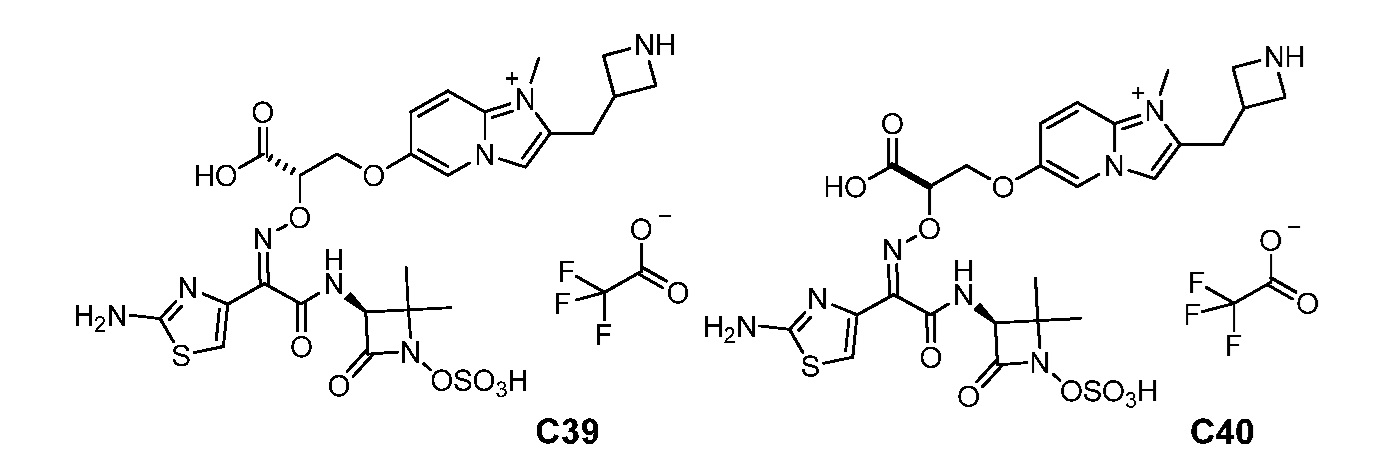
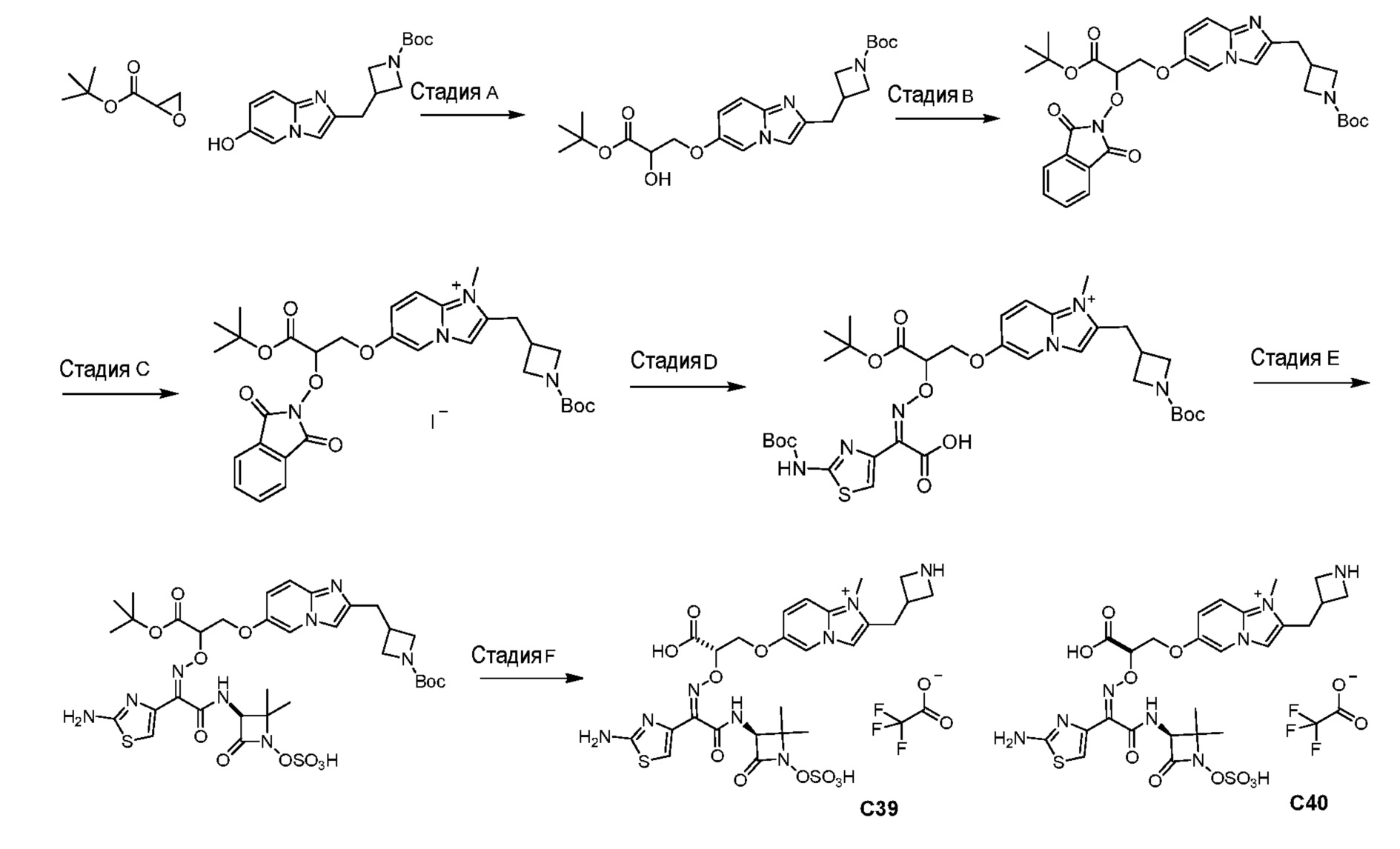
Стадия A: Получение трет-бутил 3-((6-(3-(трет-бутокси)-2-гидрокси-3-оксопропокси)имидазо-[1,2-a]пиридин-2-ил)метил)азетидин-1-карбоксилата К суспензии трет-бутил 3-((6-гидроксиимидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата (стадия B примеров 37 и 38) (1,1 г, 3,6 ммоль) и трет-бутил оксиран-2-карбоксилата (2,1 г, 14,5 ммоль) в t-BuOMe (6 мл) добавляли хлорид лития (0,15 г, 3,6 ммоль) и NaH (0,29 г, 7,2 ммоль, 60%). Полученную смесь нагревали при 50°C в течение ночи, и затем добавляли дополнительное количество гидрида натрия (0,145 г) и трет-бутил оксиран-2-карбоксилата (1,04 г) и полученную смесь нагревали при 50°C в течение 3 дней. Реакционную смесь гасили добавлением по каплям насыщенного NaHCO3 (100 мл). Смесь затем экстрагировали DCM (3×70 мл), и объединенную органическую фазу сушили над Na2SO4 и концентрировали. Остаток очищали на колонке с силикагелем с использованием смеси MeOH/DCM в качестве элюирующих растворителей с получением трет-бутил 3-((6-(3-(трет-бутокси)-2-гидрокси-3-оксопропокси)имидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата. LC/MS: (M+1)+=448,3
Стадия B: Получение трет-бутил 3-((6-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)имидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата К раствору трет-бутил 3-((6-(3-(трет-бутокси)-2-гидрокси-3-оксопропокси)имидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата (310 мг, 0,69 ммоль) в THF (4 мл) добавляли 2-гидроксиизоиндолин-1,3-дион (136 мг, 0,83 ммоль), трифенилфосфин (273 мг, 1,039 ммоль) и DEAD (0,165 мл, 1,04 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 2 часов. После концентрации, остаток очищали на колонке с силикагелем с использованием смеси MeOH/DCM в качестве элюирующих растворителей с получением трет-бутил 3-((6-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)имидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата. LC/MS: (M+1)+=593,1
Стадия C: Получение 6-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1-метилимидазо[1,2-a]пиридин-1-ий йодида К раствору трет-бутил 3-((6-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)имидазо[1,2-а]пиридин-2-ил)метил)азетидин-1-карбоксилата (0,37 г, 0,62 ммоль) в ацетонитриле (8 мл) добавляли MeI (0,12 мл, 1,9 ммоль). Полученный раствор нагревали при 60°C в течение 20 часов. Раствор концентрировали с получением 6-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1-метилимидазо[1,2-a]пиридин-1-ий йодида. LC/MS: M+=607,4
Стадия D: Получение (Z)-6-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1-метилимидазо[1,2-a]пиридин-1-ий К раствору 6-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1-метилимидазо[1,2-a]пиридин-1-ий йодида (0,47 г, 0,64 ммоль) в этаноле (8 мл) и CH2Cl2 (8 мл) добавляли гидразин (0,030 мл, 0,96 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 2 часов. Затем 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусную кислоту (Промежуточное соединение 3) (0,26 г, 0,96 ммоль) добавляли к вышеуказанной смеси.
Полученную смесь перемешивали при комнатной температуре в течение 2 часов, и раствор концентрировали и остаток очищали с помощью MPLC с обращенной фазой с использованием ацетонитрила (0,05% TFA)/воды (0,05% TFA) в качестве подвижной фазы с получением (Z)-6-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1-метилимидазо[1,2-a]пиридин-1-ия. LC/MS: M+=731,3
Стадия E: Получение 6-(3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)-окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1-метилимидазо[1,2-a]пиридин-1-ия К раствору (Z)-6-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1-метилимидазо[1,2-a]пиридин-1-ия (120 мг, 0,16 ммоль) в DMF (4 мл) добавляли DCC (270 мг, 1,3 ммоль) и HOBt (100 мг, 0,66 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут перед добавлением (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфата (138 мг, 0,66 ммоль) и бикарбоната натрия (275 мг, 3,3 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. После фильтрации, фильтрат очищали с помощью MPLC с обращенной фазой с использованием ацетонитрила (0,05% TFA)/воды (0,05% TFA) в качестве подвижной фазы с получением 6-(3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-2-((1-(трет-бутокси-карбонил)-азетидин-3-ил)метил)-1-метилимидазо[1,2-a]пиридин-1-ия. LC/MS: (M+1)+/2=462,4
Стадия F: Получение 6-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-2-(азетидин-3-илметил)-1-метилимидазо[1,2-a]пиридин-1-ий 2,2,2-трифторацетата и 6-((R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-2-(азетидин-3-илметил)-1-метилимидазо[1,2-a]пиридин-1-ий 2,2,2-трифторацетата К раствору 6-(3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)-азетидин-3-ил)метил)-1-метилимидазо[1,2-a]пиридин-1-ия (106 мг, 0,115 ммоль) в CH2Cl2 (2 мл) добавляли TFA (4 мл, 51,9 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 50 минут. Раствор концентрировали и остаток обрабатывали Et2O. Твердое вещество собирали и сушили в высоком вакууме. Остаток затем растворяли в DMSO (2 мл) и очищали с помощью ВЭЖХ с обращенной фазой с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05%TFA) в качестве подвижной фазы с получением:
Соединения 39: 6-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-2-(азетидин-3-илметил)-1-метилимидазо[1,2-a]пиридин-1-ий 2,2,2-трифторацетата (быстро элюируемый), LC/MS: (M+1)+=667,1,1H-ЯМР (500 МГц, D2O), δ 8,19-8,18 (д, J=2,1 Гц, 1H), 7,73-7,71 (д, J=11,0 Гц, 2H), 7,62-7,59 (дд, J=9,8 Гц и 3,0 Гц, 1H), 7,05 (с, 1H), 5,09-5,07 (м, 1H), 4,68 (с, 1H), 4,50-4,43 (м, 2H), 4,24-4,20 (т, J=11,6 Гц, 2H), 3,95-3,90 (м, 2H), 3,79 (с, 3H), 3,41-3,34 (м, 1H), 3,21-3,19 (д, J=6,2 Гц, 2H), 1,38 (с, 3H), 1,02 (с, 3H).
Соединения 40: 6-((R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-2-(азетидин-3-илметил)-1-метилимидазо[1,2-a]пиридин-1-ий 2,2,2-трифторацетат (медленно элюируемый), LC/MS: (M+1)+=667,0,1H-ЯМР (500 МГц, D2O), δ 8,21-8,20(д, J=2,8 Гц, 1H), 7,75-7,73(т, J=4,9 Гц, 2H), 7,64-7,61(дд, J=10,3 Гц и 2,8 Гц, 2H), 7,05(с, 1H), 5,04-5,02(м, 1H), 4,48-4,44(м, 2H), 4,25-4,20(м, 2H), 3,95-3,91(м, 2H), 3,80(с, 3H), 3,41-3,34(м, 1H), 3,21-3,19(д, J=7,7 Гц, 2H), 1,38(с, 3H), 1,06(с, 3H).
Примеры 41 и 42
3-((6-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-1H-бензо[d]имидазол-2-ил)тио)пропан-1-аминий 2,2,2-трифторацетат (C41) и 3-((6-((R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-1H-бензо[d]имидазол-2-ил)тио)пропан-1-аминий 2,2,2-трифторацетат (C42)
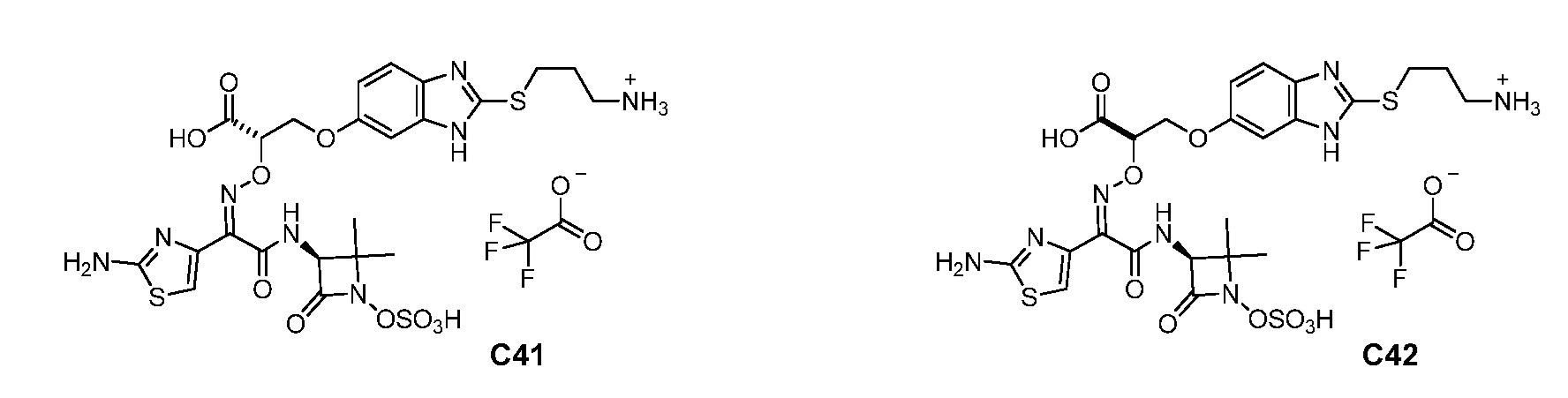
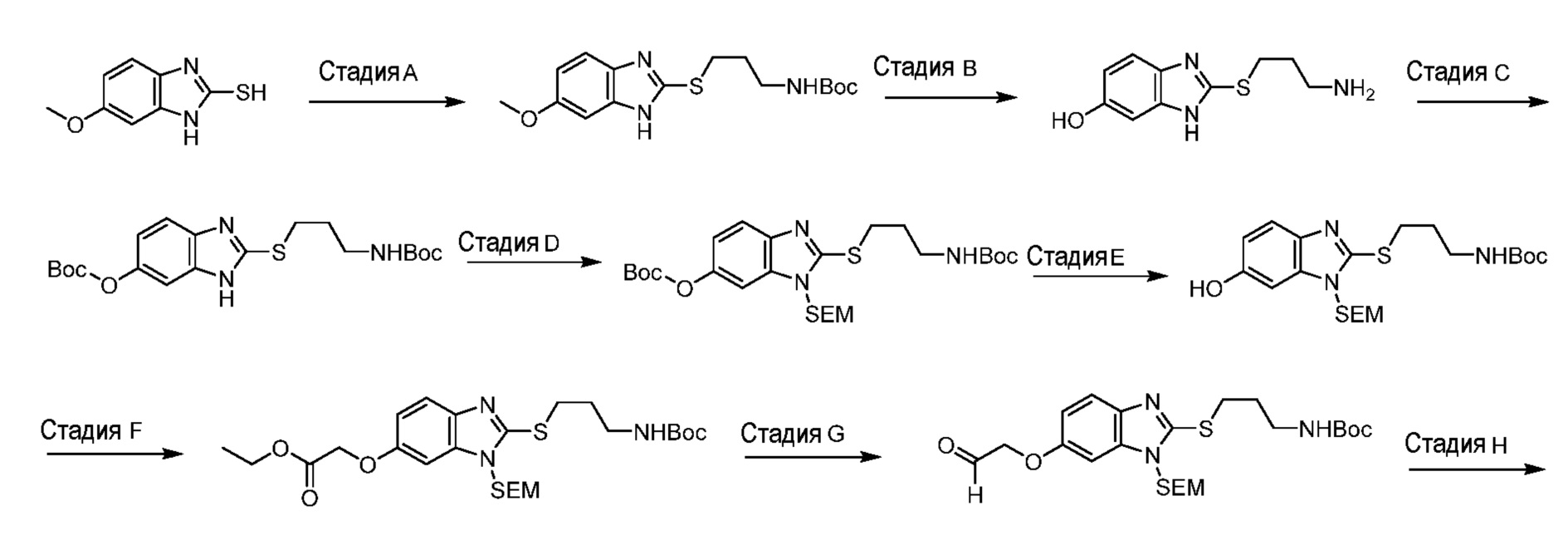

Стадия A: Получение трет-бутил (3-((5-метокси-1H-бензо[d]имидазол-2-ил)тио)пропил)-карбамата К раствору 5-метокси-1H-бензо[d]имидазол-2-тиола (1,06 г, 5,9 ммоль) в DMF (8 мл) при 0°C добавляли бикарбонат натрия (0,49 г, 5,9 ммоль), и раствор трет-бутил (3-иодопропил)-карбамата (1,7 г, 5,9 ммоль) в DMF (6 мл) по каплям. Полученный раствор перемешивали при 0°C в течение ночи. Смесь разделяли между EtOAc и насыщенным NaHCO3. Органическую фазу промывали насыщенным NaHCO3 три раза, сушили над Na2SO4, концентрировали и остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением трет-бутил (3-((5-метокси-1H-бензо[d]имидазол-2-ил)тио)пропил)карбамата. LC/MS: (M+1)+: 338,6
Стадия B: Получение 2-((3-аминопропил)тио)-1H-бензо[d]имидазол-5-ола К раствору трет-бутил (3-((5-метокси-1H-бензо[d]имидазол-2-ил)тио)пропил)карбамата (2,5 г, 7,3 ммоль) в CH2Cl2 (30 мл) при 0°C добавляли BBr3 (22 мл, 22 ммоль). Полученный раствор перемешивали при 0°C в течение 4 часов, и затем реакцию гасили добавлением MeOH. Смесь концентрировали с получением 2-((3-аминопропил)тио)-1H-бензо[d]имидазол-5-ола. LC /MS: (M+1)+: 224,2
Стадия C: Получение трет-бутил (3-((5-((трет-бутоксикарбонил)окси)-1H-бензо[d]имидазол-2-ил)тио)пропил)карбамата К 2-((3-аминопропил)тио)-1H-бензо[d]имидазол-5-олу (1,6 г, 7,3 ммоль) в диоксане (50 мл) и воде (50,0 мл) добавляли K2CO3 (5,1 г, 37 ммоль) и Boc2O (5,6 мл, 24 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Добавляли NaOH (20 мл, 20 ммоль) и дополнительное количество BOC2O (5,61 мл, 24,16 ммоль) и полученную смесь перемешивали при комнатной температуре в течение ночи. После удаления летучих веществ, к водной фазе добавляли MeOH (40 мл) и THF (80 мл) и NaOH (1 н, 80 мл). Полученный раствор перемешивали при комнатной температуре в течение 0,5 часа. После удаления летучих веществ, водную фазу экстрагировали DCM три раза, и объединенную органическую фазу сушили над Na2SO4 и концентрировали. Остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением трет-бутил (3-((5-((трет-бутокси-карбонил)окси)-1H-бензо[d]имидазол-2-ил)тио)пропил)карбамата. LC/MS: (M+1)+: 424,3
Стадия D: Получение трет-бутил (3-((5-((трет-бутоксикарбонил)окси)-1-((2-(триметилсилил)-этокси)метил)-1H-бензо[d]имидазол-2-ил)тио)пропил)карбамата К раствору трет-бутил (3-((5-((трет-бутоксикарбонил)окси)-1H-бензо[d]имидазол-2-ил)тио)пропил)карбамата (0,73 г, 1,7 ммоль) в DCM (50 мл) при 0°C добавляли DIEA (0,60 мл, 3,4 ммоль), и SEM-Cl (0,37 мл, 2,1 ммоль). Полученный раствор перемешивали от 0°C до комнатной температуры в течение 8 часов. Раствор разделяли между DCM и насыщенным NaHCO3, и водную фазу экстрагировали DCM. Объединенную органическую фазу сушили над Na2SO4 и концентрировали. Остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением трет-бутил (3-((5-((трет-бутоксикарбонил)окси)-1-((2-(триметилсилил)этокси)метил)-1H-бензо[d]имидазол-2-ил)тио)пропил)карбамата. LC /MS: (M+1)+: 554,5
Стадия E: Получение трет-бутил (3-((5-гидрокси-1-((2-(триметилсилил)этокси)метил)-1H-бензо[d]имидазол-2-ил)тио)пропил)карбамата К раствору трет-бутил (3-((5-((трет-бутоксикарбонил)окси)-1-((2-(триметилсилил)этокси)метил)-1H-бензо[d]имидазол-2-ил)тио)пропил)карбамата (0,91 г, 1,6 ммоль) в THF (10 мл) и MeOH (20 мл) добавляли NaOH (6,6 мл, 6,6 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 4 часов. После концентрации, водную фазу подкисляли до pH 8, и затем добавляли насыщенный NaHCO3 (100 мл). Смесь экстрагировали DCM три раза. Объединенную органическую фазу сушили над Na2SO4 и концентрировали с получением трет-бутил (3-((5-гидрокси-1-((2-(триметилсилил)этокси)метил)-1H-бензо[d]имидазол-2-ил)тио)пропил)-карбамата. LC/MS: (M+1)+: 454,8
Стадия F: Получение этил 2-((2-((3-((трет-бутоксикарбонил)амино)пропил)тио)-1-((2-(триметилсилил)этокси)метил)-1H-бензо[d]имидазол-5-ил)окси)ацетата Смесь K2CO3 (0,43 г, 3,1 ммоль), этилбромацетата (0,21 мл, 1,8 ммоль), и трет-бутил (3-((5-гидрокси-1-((2-(триметилсилил)этокси)метил)-1H-бензо[d]имидазол-2-ил)тио)пропил)карбамата (0,7 г, 1,5 ммоль) нагревали при 80°C в течение ночи. После фильтрации, фильтрат концентрировали и остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением этил 2-((2-((3-((трет-бутоксикарбонил)амино)пропил)тио)-1-((2-(триметилсилил)этокси)-метил)-1H-бензо[d]имидазол-5-ил)окси)ацетата. LC /MS: (M+1)+: 540,5

Стадия H: Получение трет-бутил (3-((5-(2-циано-2-гидроксиэтокси)-1-((2-(триметилсилил)-этокси)метил)-1H-бензо[d]имидазол-2-ил)тио)пропил)карбамата К раствору трет-бутил (3-((5-(2-оксоэтокси)-1-((2-(триметилсилил)этокси)метил)-1H-бензо[d]имидазол-2-ил)тио)-пропил)карбамата (0,96 г, 1,9 ммоль) в t-BuOMe (16 мл) и воде (3 мл) при 0°C добавляли уксусную кислоту (4 мл) и NaCN (0,19 г, 3,9 ммоль). Полученный раствор перемешивали от 0°C до комнатной температуре в течение ночи. Раствор добавляли к насыщенному Na2CO3 (200 мл) при 0°C, и смесь экстрагировали EtOAc. Объединенную органическую фазу промывали насыщенным NaHCO3, сушили над Na2SO4, и затем концентрировали с получением трет-бутил (3-((5-(2-циано-2-гидроксиэтокси)-1-((2-(триметилсилил)-этокси)метил)-1H-бензо[d]имидазол-2-ил)тио)пропил)карбамата. LC /MS: (M+1)+: 523,4
Стадия I: Получение 3-((2-((3-((трет-бутоксикарбонил)амино)пропил)тио)-1H-бензо[d]имидазол-5-ил)окси)-2-гидроксипропановой кислоты Раствор трет-бутил (3-((5-(2-циано-2-гидроксиэтокси)-1-((2-(триметилсилил)этокси)метил)-1H-бензо[d]имидазол-2-ил)тио)пропил)карбамата (0,83 г, 1,6 ммоль) в MeOH (20 мл) при 0°C барботировали HCl в течение 15 минут. Затем полученный раствор перемешивали от 0°C до комнатной температуры в течение ночи. Добавляли по каплям воду (5 мл) и полученный раствор перемешивали при комнатной температуре в течение 0,5 часа, и затем нагревали с обратным холодильником в течение 0,5 часа. После удаления летучих веществ, водную фазу подщелачивали до рН 10 путем добавления насыщенного NaOH при 0°C. Затем добавляли диоксан (20 мл) и Boc2O (0,92 мл, 4,0 ммоль) и полученный раствор перемешивали при комнатной температуре в течение 2 часов. После удаления летучих веществ, водную фазу экстрагировали DCM, и затем подкисляли до pH 4. Раствор затем очищали с помощью MPLC с обращенной фазой с использованием ацетонитрила (0,05% TFA)/воды (0,05% TFA) с получением 3-((2-((3-((трет-бутоксикарбонил)амино)пропил)тио)-1H-бензо[d]имидазол-5-ил)окси)-2-гидроксипропановой кислоты. LC/MS: (M+1)+: 412,3
Соединения 41/42 получали из 3-((2-((3-((трет-бутоксикарбонил)амино)пропил)тио)-1H-бензо[d]имидазол-5-ил)окси)-2-гидроксипропановой кислоты в соответствии со стадиями I-M, как описано для соединений 37/38.
Соединение 41: 3-((6-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-1H-бензо[d]имидазол-2-ил)тио)пропан-1-аминий 2,2,2-трифторацетат (быстро элюируемый диастереоизомер). LC/MS: (M+1)+=673,6,1H-ЯМР (500 МГц, D2O): δ 7,47-7,46 (д, J=8,8 Гц, 1H), 7,10-7,09 (д, J=2,5 Гц, 1H), 7,05-7,03 (дд, J=2,3Гц и 9,2 Гц, 1H), 7,02 (с, 1H), 8,08-8,07 (м, 1H), 4,61 (с, 1H), 4,50-4,41 (м, 2H), 3,38-3,33 (т, J=7,9 Гц, 2H), 3,10-3,07 (т, J=7,7 Гц, 2H), 2,07-2,04 (м, 2H), 1,32 (с, 3H), 0,99 (с, 3H).
Соединение 42: 3-((6-((R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-1H-бензо[d]имидазол-2-ил)тио)пропан-1-аминий 2,2,2-трифторацетат (медленно элюируемый диастереоизомер). LC/MS: (M+1)+=673,6,1H-ЯМР(500 МГц, D2O): δ 7,47-7,46 (д, J=9,1 Гц, 1H), 7,11-7,10 (д, J=3,1 Гц, 1H), 7,05-7,03 (дд, J=8,4Гц и 2,6 Гц, 1H), 7,02 (с, 1H), 5,02-5,00 (м,1H), 4,62 (с, 1H), 4,47-4,40 (м, 2H), 3,36-3,33 (т, J=8,0 Гц, 2H), 3,10-3,07 (т, J=8,0 Гц, 2H), 2,08-2,02(м, 2H), 1,31 (с, 3H), 0,93 (с, 3H).
ПРИМЕР 43
3-((6-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)бензо[d]тиазол-2-ил)амино)азетидин-1-ий 2,2,2-трифторацетат (C43)
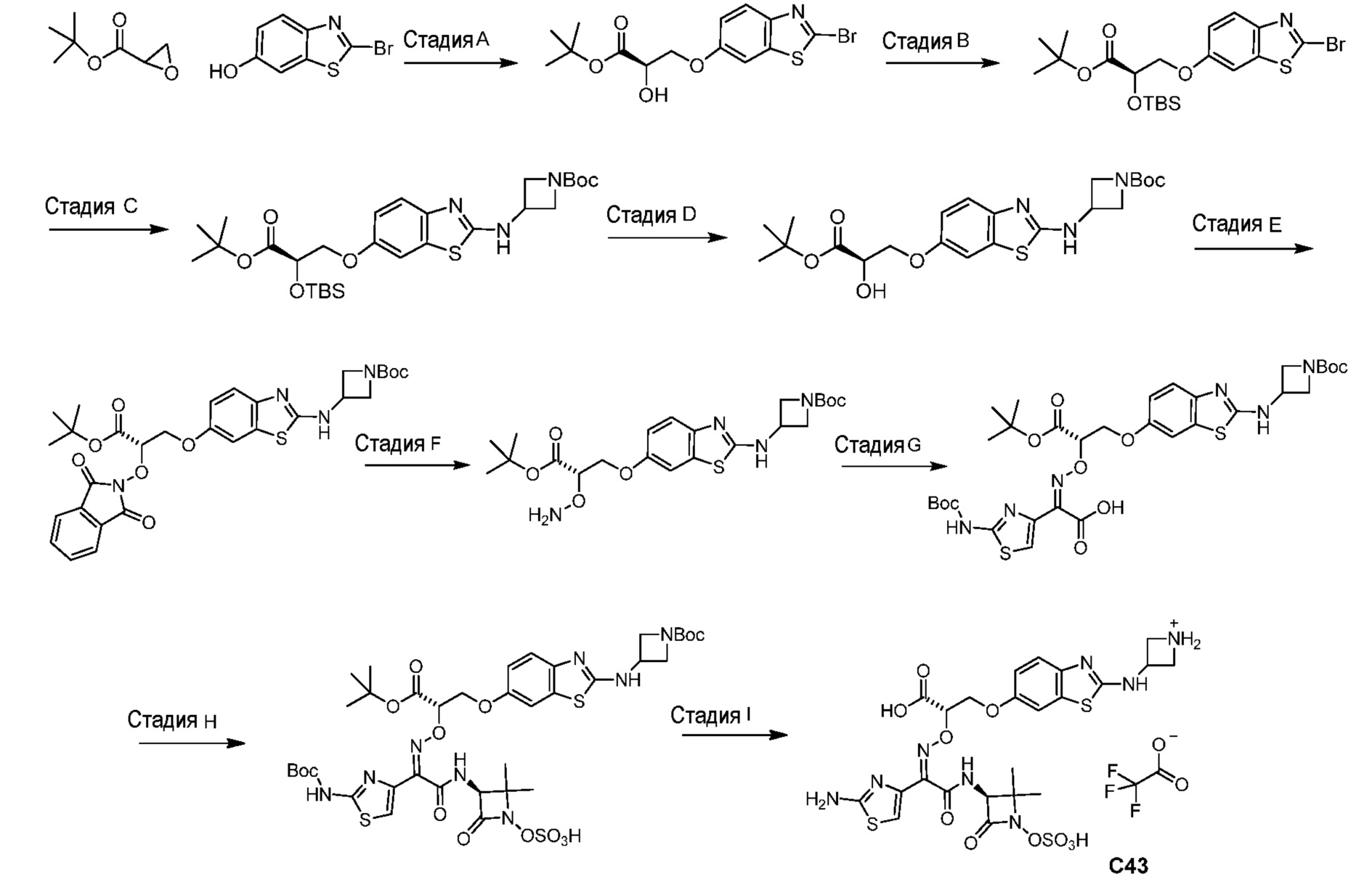
Стадия A: Получение (R)-трет-бутил 3-((2-бромбензо[d]тиазол-6-ил)окси)-2-гидроксипропаноата Смесь трет-бутил оксиран-2-карбоксилата (1,44 г, 10,0 ммоль), 2-бромбензо[d]тиазол-6-ола (0,92 г, 4,0 ммоль), и молекулярные сита (4Å) (1000 мг) и катализатор (R,R)-Cobal Jacobsen (промеж.соед. 2) (0,17 г, 0,20 ммоль) (Ref. J. Am. Chem. Soc., 1999, 121, 6086-6087) в t-BuOMe (8 мл) перемешивали при комнатной температуре в течение 2 дней. Смесь фильтровали, фильтрат концентрировали, и остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением (R)-трет-бутил 3-((2-бромбензо[d]тиазол-6-ил)окси)-2-гидроксипропаноата. LC/MS: (M+1)+=374,1, 376,1
Стадия B: Получение (R)-трет-бутил 3-((2-бромбензо[d]тиазол-6-ил)окси)-2-((трет-бутилдиметилсилил)окси)пропаноата К раствору (R)-трет-бутил 3-((2-бромбензо[d]тиазол-6-ил)окси)-2-гидроксипропаноата (0,34 г, 0,91 ммоль) в ацетонитриле (4 мл) добавляли имидазол (0,31 г, 4,5 ммоль), TBS-Cl (0,20 г, 1,4 ммоль) и DMAP (5,6 мг, 0,045 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 2 часов. После концентрации, остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением (R)-трет-бутил 3-((2-бромбензо[d]тиазол-6-ил)окси)-2-((трет-бутилдиметилсилил)окси)пропаноата. LC/MS: (M+1)+=488,3, 490,2
Стадия C: Получение (R)-трет-бутил 3-((6-(3-(трет-бутокси)-2-((трет-бутилдиметилсилил)окси)-3-оксопропокси)бензо[d]тиазол-2-ил)амино)азетидин-1-карбоксилата Смесь трет-бутил 3-аминоазетидин-1-карбоксилата (123 мг, 0,71 ммоль), трет-бутил (R)-3-((2-бромбензо[d]тиазол-6-ил)окси)-2-((трет-бутилдиметилсилил)окси)пропаноата (290 мг, 0,59 ммоль) и карбоната цезия (580 мг, 1,8 ммоль) в диоксане (10 мл) дегазировали с помощью вакуума/наполняли N2 в герметичной пробирке три раза, с последующим добавлением предкатализатора Ruthphos G2 (92 мг, 0,12 ммоль). Полученную смесь дополнительно дегазировали с помощью вакуума/заполняли N2 три раза, и затем нагревали при 80°C в течение 16 часов. Смесь фильтровали через целит. Фильтрат концентрировали и остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением (R)-трет-бутил 3-((6-(3-(трет-бутокси)-2-((трет-бутилдиметилсилил)окси)-3-оксопропокси)бензо[d]тиазол-2-ил)амино)азетидин-1-карбоксилата. LC/MS: (M+1)+=580,5
Стадия D: Получение (R)-трет-бутил 3-((6-(3-(трет-бутокси)-2-гидрокси-3-оксопропокси)-бензо[d]-тиазол-2-ил)амино)азетидин-1-карбоксилата К раствору (R)-трет-бутил 3-((6-(3-(трет-бутокси)-2-((трет-бутилдиметилсилил)окси)-3-оксопропокси)бензо[d]тиазол-2-ил)амино)азетидин-1-карбоксилата (240 мг, 0,40 ммоль) в THF (5 мл) при 0°C добавляли TBAF (0,40 мл, 0,40 ммоль). Полученный раствор перемешивали при 0°C в течение 30 минут. После концентрации, остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением (R)-трет-бутил 3-((6-(3-(трет-бутокси)-2-гидрокси-3-оксопропокси)-бензо[d]тиазол-2-ил)амино)азетидин-1-карбоксилата. LC/MS: (M+1)+=466,3
Стадия E: Получение (S)-трет-бутил 3-((6-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)бензо[d]тиазол-2-ил)амино)азетидин-1-карбоксилата К раствору (R)-трет-бутил 3-((6-(3-(трет-бутокси)-2-гидрокси-3-оксопропокси)бензо[d]тиазол-2-ил)амино)-азетидин-1-карбоксилата (48 мг, 0,10 ммоль) в THF (2 мл) добавляли 2-гидроксиизоиндолин-1,3-дион (18 мг, 0,11 ммоль), трифенилфосфин (38 мг, 0,14 ммоль) и DEAD (0,023 мл, 0,14 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1,5 часов. После концентрации, остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением (S)-трет-бутил 3-((6-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)бензо[d]тиазол-2-ил)амино)азетидин-1-карбоксилата. LC/MS: (M+1)+=611,4
Стадия F: Получение (S)-трет-бутил 3-((6-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)-бензо[d]тиазол-2-ил)амино)азетидин-1-карбоксилата К раствору (S)-трет-бутил 3-((6-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)бензо[d]тиазол-2-ил)амино)-азетидин-1-карбоксилата (53 мг, 0,087 ммоль) в EtOH (4 мл) и DCM (2 мл) при 0°C добавляли гидразин (3,3 мкл, 0,10 ммоль). Полученный раствор перемешивали при 0°C в течение 1 часа. Раствор концентрировали с получением (S)-трет-бутил 3-((6-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)бензо[d]тиазол-2-ил)амино)азетидин-1-карбоксилата. LC/MS: (M+1)+=481,4
Стадия G: Получение (S,Z)-2-(((1-(трет-бутокси)-3-((2-((1-(трет-бутоксикарбонил)азетидин-3-ил)амино)бензо[d]тиазол-6-ил)окси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутокси-карбонил)амино)тиазол-4-ил)уксусной кислоты К раствору (S)-трет-бутил 3-((6-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)бензо[d]тиазол-2-ил)амино)азетидин-1-карбоксилата (42 мг, 0,087 ммоль) в EtOH (6 мл) и DCM (6 мл) добавляли 2-(2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)-2-оксоуксусную кислоту (Промежуточное соединение 3) (36 мг, 0,13 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 часа. После концентрации, остаток очищали с помощью ВЭЖХ с обращенной фазой с использованием ацетонитрила и воды (0,05%TFA) с получением (S,Z)-2-(((1-(трет-бутокси)-3-((2-((1-(трет-бутокси-карбонил)азетидин-3-ил)амино)бензо[d]тиазол-6-ил)окси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)уксусной кислоты. LC/MS: (M+1)+=735,4
Стадия H: Получение трет-бутил 3-((6-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)бензо[d]тиазол-2-ил)амино)азетидин-1-карбоксилата К раствору (S,Z)-2-(((1-(трет-бутокси)-3-((2-((1-(трет-бутоксикарбонил)-азетидин-3-ил)амино)бензо[d]тиазол-6-ил)окси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутокси-карбонил)амино)тиазол-4-ил)уксусной кислоты (24 мг, 0,033 ммоль) в DMF (2 мл) добавляли DCC (34 мг, 0,16 ммоль) и HOBt (20 мг, 0,13 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут перед добавлением (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфата (27 мг, 0,13 ммоль) и бикарбоната натрия (27 мг, 0,33 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли дополнительное количество DCC (33,7 мг, 0,163 ммоль) и полученную смесь непрерывно перемешивали при комнатной температуре в течение ночи. После фильтрации, фильтрат очищали с помощью ВЭЖХ с обращенной фазой с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05%TFA) в качестве подвижной фазы с получением трет-бутил 3-((6-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)-амино)окси)-3-оксопропокси)-бензо[d]тиазол-2-ил)амино)азетидин-1-карбоксилата. LC/MS: (M+1)+=927,6
Стадия I: Получение 3-((6-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)-бензо[d]тиазол-2-ил)амино)азетидин-1-ий 2,2,2-трифторацетата К раствору трет-бутил 3-((6-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)бензо[d]тиазол-2-ил)амино)азетидин-1-карбоксилата (17 мг, 0,018 ммоль) в DCM (1 мл) добавляли TFA (2 мл, 26,0 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 часа. После концентрации, остаток очищали с помощью ВЭЖХ с обращенной фазой с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05%TFA) в качестве подвижной фазы с получением целевого продукта. LC/MS: (M+1)+=671,1,1H-ЯМР (CD3OD, 500 MГц): δ 7,46-7,44 (д, J=8,6 Гц, 1H), 7,35-7,34 (д, J=2,5 Гц, 1H), 7,14 (с, 1H), 7,00-6,98 (дд, J=8,8Hz и 2,2 Гц, 1H), 5,22-5,20 (м, 1H), 4,82-4,76(м, 1H), 4,54-4,29(м, 2H), 4,47-4,43 (т, J=9,9 Гц, 2H), 4,33-4,29 (м, 2H), 3,53-3,49 (м, 1H), 1,54 (с, 3H), 1,31 (с, 3H).
ПРИМЕР 44
3-((6-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((2S,3S)-2-метил-4-оксо-1-сульфоазетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)бензо[d]тиазол-2-ил)амино)азетидин-1-ий 2,2,2-трифторацетат (C44)

Стадия A: (2S,3S)-3-((Z)-2-((((S)-1-(трет-бутокси)-3-((2-((1-(трет-бутоксикарбонил)азетидин-3-ил)-амино)бензо[d]тиазол-6-ил)окси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)ацетамидо)-2-метил-4-оксоазетидин-1-сульфоновая кислота К раствору (S,Z)-2-(((1-(трет-бутокси)-3-((2-((1-(трет-бутоксикарбонил)азетидин-3-ил)амино)бензо[d]-тиазол-6-ил)окси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутокси-карбонил)амино)тиазол-4-ил)уксусной кислоты (Стадия G примера 43) (24 мг, 0,033 ммоль) в DMF (2 мл) добавляли DCC (34 мг, 0,16 ммоль) и HOBt (20,01 мг, 0,131 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут перед добавлением (2S,3S)-3-амино-2-метил-4-оксоазетидин-1-сульфоновой кислоты (23 мг, 0,13 ммоль) и бикарбоната натрия (27 мг, 0,33 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли дополнительно DCC (33,7 мг, 0,163 ммоль) и полученную смесь продолжали перемешивать при комнатной температуре в течение ночи. После фильтрации, фильтрат очищали с помощью ВЭЖХ с обращенной фазой с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05%TFA) в качестве подвижной фазы с получением (2S,3S)-3-((Z)-2-((((S)-1-(трет-бутокси)-3-((2-((1-(трет-бутокси-карбонил)азетидин-3-ил)амино)бензо[d]тиазол-6-ил)окси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутокси-карбонил)амино)тиазол-4-ил)ацетамидо)-2-метил-4-оксоазетидин-1-сульфоновой кислоты. LC/MS: (M+1)+=897,5
Стадия B: Получение 3-((6-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((2S,3S)-2-метил-4-оксо-1-сульфоазетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-карбоксиэтокси)бензо-[d]тиазол-2-ил)амино)азетидин-1-ий 2,2,2-трифторацетата К раствору (2S,3S)-3-((Z)-2-((((S)-1-(трет-бутокси)-3-((2-((1-(трет-бутоксикарбонил)азетидин-3-ил)амино)-бензо[d]тиазол-6-ил)окси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)ацетамидо)-2-метил-4-оксоазетидин-1-сульфоновой кислоты (21 мг, 0,023 ммоль) в CH2Cl2 (1 мл) добавляли TFA (2 мл, 26 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 40 минут. Реакционную смесь концентрировали и остаток очищали с помощью ВЭЖХ с обращенной фазой с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05%TFA) в качестве подвижной фазы с получением целевого продукта. LC/MS: (M+1)+=641,4,1H-ЯМР(500 МГц, CD3OD): δ 7,45-7,43(д, J=9,1 Гц, 1H), 7,36-7,35 (д, J=2,5 Гц, 1H), 7,13(с, 1H), 7,02-7,00 (дд, J=8,6 Гц, 2,5 Гц, 1H), 5,20-5,18(м, 1H), 4,77-4,74 (м, 1H), 4,58-4,55(м, 2H), 4,47-4,43(т, J=9,8 Гц, 2H), 4,42-4,41 (д, J=3,2 Гц, 1H), 4,34-4,30(м, 2H), 4,00-3,99 (м, 1H), 1,44-1,43 (д, J=6,3 Гц, 3H).
Примеры 45 и 46
3-((6-((R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-(2H-тетразол-5-ил)этокси)хинолин-2-ил)амино)азетидин-1-ий 2,2,2-трифторацетат (C45) и 3-((6-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-(2H-тетразол-5-ил)этокси)хинолин-2-ил)амино)азетидин-1-ий 2,2,2-трифторацетат (C46)
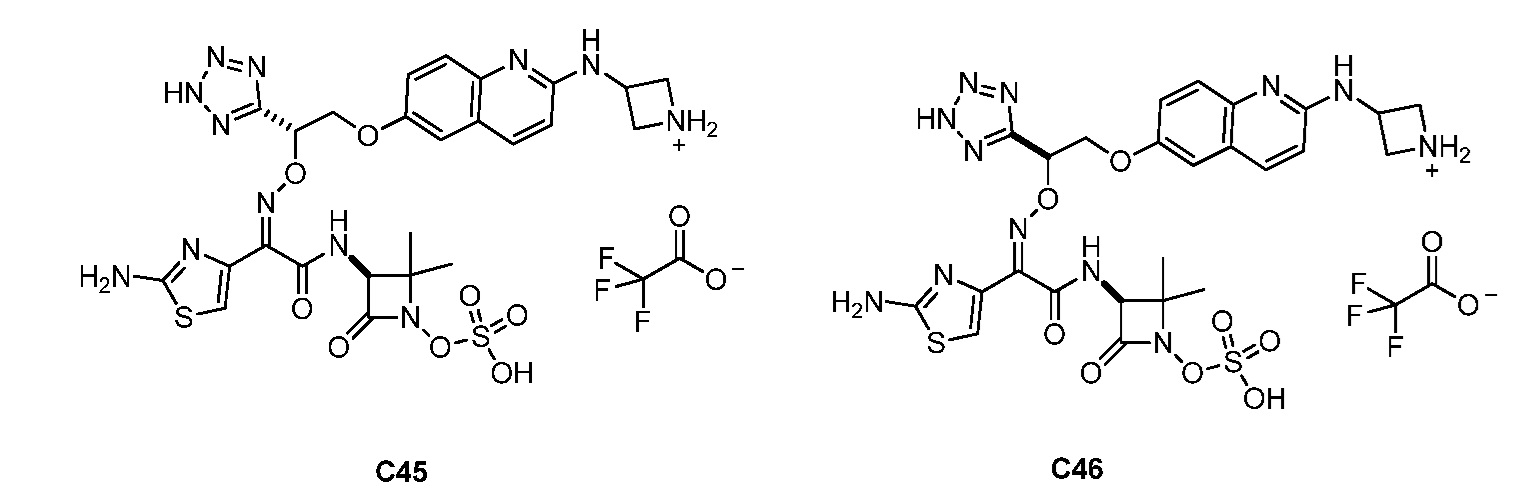

Стадия A: Получение этил 2-((2-хлорхинолин-6-ил)окси)ацетата К раствору 2-хлорхинолин-6-ола (5,3 г, 30 ммоль) в EtOAc (100 мл) добавляли K2CO3 (6,1 г, 44 ммоль) и этилбромацетат (3,9 мл, 35 ммоль). Полученную смесь нагревали при 60°C в течение ночи. После охлаждения до комнатной температуры, смесь фильтровали через целит и фильтрат концентрировали. Остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением этил 2-((2-хлорхинолин-6-ил)окси)ацетата. LC/MS: (M+1)+= 266,7
Стадия B: Получение 2-((2-хлорхинолин-6-ил)окси)этан-1,1-диола К раствору этил 2-((2-хлорхинолин-6-ил)окси)ацетата (5,9 г, 22 ммоль) в DCM (100 мл) при -78°C добавляли по каплям DIBAL-H (25 мл, 25 ммоль). Полученный раствор перемешивали при -78°C в течение 3 часов. Реакцию гасили добавлением метанола (5 мл). Смесь затем перемешивали при -78°C в течение 10 минут. Добавляли насыщенный тартрат калия (500 мл) и DCM (200 мл) и полученную смесь перемешивали при комнатной температуре в течение ночи. Смесь фильтровали, и твердое вещество промывали водой, и сушили под вакуумом с получением 2-((2-хлорхинолин-6-ил)окси)этан-1,1-диола. LC/MS: (M+1)+=239,9
Стадия C: Получение 3-((2-хлорхинолин-6-ил)окси)-2-гидроксипропаннитрила К смеси 2-((2-хлорхинолин-6-ил)окси)этан-1,1-диола (4,2 г, 18 ммоль) в THF (60 мл), уксусной кислоты (20 мл) и воды (5 мл) добавляли цианид натрия (1,7 г, 35 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Затем реакционный раствор добавляли к раствору карбоната натрия (60 г) в 300 мл воды при 0°C по каплям. Летучие вещества выпаривали и водный раствор дважды экстрагировали EtOAc. Объединенную органическую фазу промывали водой два раза, сушили над Na2SO4, и концентрировали с получением 3-((2-хлорхинолин-6-ил)окси)-2-гидроксипропаннитрила. LC/MS: (M+1)+= 248,8
Стадия D: Получение 2-((трет-бутилдиметилсилил)окси)-3-((2-хлорхинолин-6-ил)окси)пропаннитрила К раствору 3-((2-хлорхинолин-6-ил)окси)-2-гидрокси-пропаннитрила (4,3 г, 17 ммоль), имидазола (5,9 г, 87 ммоль), и TBS-Cl (6,6 г, 43 ммоль) в ацетонитриле (20 мл) добавляли DMAP (0,212 г, 1,737 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 часа. После концентрации, остаток разбавляли в DCM (50 мл). Твердое вещество отфильтровывали и фильтрат очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением 2-((трет-бутилдиметилсилил)окси)-3-((2-хлорхинолин-6-ил)окси)пропаннитрила. LC/MS: (M+1)+= 363,1
Стадия E: Получение 6-(2-((трет-бутилдиметилсилил)окси)-2-(2H-тетразол-5-ил)этокси)-2-хлорхинолина К смеси оксида дибутилолова (0,83 г, 3,4 ммоль) и 2-((трет-бутилдиметилсилил)окси)-3-((2-хлорхинолин-6-ил)окси)пропаннитрила (6,1 г, 17 ммоль) в толуоле (100 мл) добавляли TMS-N3 (6,7 мл, 50 ммоль). Полученную смесь нагревали при 110°C в течение 1 часа. Смесь концентрировали и остаток разделяли между EtOAc (200 мл) и насыщенным NH4Cl (100 мл). Органическую фазу промывали насыщенным NH4Cl дважды, сушили над Na2SO4, и концентрировали и остаток очищали на колонке с силикагелем с использованием смеси MeOH/DCM в качестве элюирующих растворителей с получением 6-(2-((трет-бутилдиметилсилил)окси)-2-(2H-тетразол-5-ил)этокси)-2-хлорхинолина. LC/MS: (M+1)+=406,1
Стадия F: Получение 6-(2-((трет-бутилдиметилсилил)окси)-2-(2-(4-метоксибензил)-2H-тетразол-5-ил)этокси)-2-хлорхинолина и 6-(2-((трет-бутилдиметилсилил)окси)-2-(1-(4-метоксибензил)-1H-тетразол-5-ил)этокси)-2-хлорхинолина К смеси 6-(2-((трет-бутилдиметилсилил)окси)-2-(2H-тетразол-5-ил)этокси)-2-хлорхинолина (5,4 г, 11 ммоль), тетрабутиламмоний хлорид гидрата (0,33 г, 1,1 ммоль), K2CO3 (4,6 г, 33 ммоль) в DCE (100 мл), воды (50 мл) и ацетонитрила (50 мл) добавляли 4-метоксибензил хлорид (1,7 мл, 12,2 ммоль). Полученную смесь нагревали при 40°C в течение ночи. После испарения летучих веществ, смесь суспендировали в DCM (200 мл) и затем фильтровали. Фильтрат экстрагировали DCM три раза. Объединенную органическую фазу сушили над Na2SO4, концентрировали и остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением двух региоизомеров 6-(2-((трет-бутилдиметилсилил)окси)-2-(2-(4-метоксибензил)-2H-тетразол-5-ил)этокси)-2-хлорхинолина и 6-(2-((трет-бутилдиметилсилил)окси)-2-(1-(4-метоксибензил)-1H-тетразол-5-ил)этокси)-2-хлорхинолина. LC/MS: (M+1)+=527,7
Стадия G: Получение трет-бутил 3-((6-(2-((трет-бутилдиметилсилил)окси)-2-(2-(4-метоксибензил)-2H-тетразол-5-ил)этокси)хинолин-2-ил)амино)азетидин-1-карбоксилата Смесь трет-бутил 3-аминоазетидин-1-карбоксилата (290 мг, 1,7 ммоль), 6-(2-((трет-бутилдиметилсилил)окси)-2-(2-(4-метоксибензил)-2H-тетразол-5-ил)этокси)-2-хлорхинолина (810 мг, 1,54 ммоль), карбоната цезия (1500 мг, 4,6 ммоль) в диоксане (10 мл) дегазировали с помощью вакуума/заполняли N2 три раза, с последующим добавлением предкатализатора Ruthphos G2 (240 мг, 0,31 ммоль). Полученную смесь дополнительно дегазировали с помощью вакуума /заполняли N2 три раза, и затем нагревали при 80°C в течение 16 часов. Смесь фильтровали через целит и фильтрат концентрировали. Остаток очищали на колонке с силикагелем с использованием EtOAc/гексана с получением трет-бутил 3-((6-(2-((трет-бутилдиметилсилил)окси)-2-(2-(4-метоксибензил)-2H-тетразол-5-ил)этокси)хинолин-2-ил)амино)азетидин-1-карбоксилата. LC/MS: (M+1)+=662,6
Стадия H: Получение трет-бутил 3-((6-(2-гидрокси-2-(2-(4-метоксибензил)-2H-тетразол-5-ил)этокси)хинолин-2-ил)амино)азетидин-1-карбоксилата К раствору трет-бутил 3-((6-(2-((трет-бутилдиметилсилил)окси)-2-(2-(4-метоксибензил)-2H-тетразол-5-ил)этокси)хинолин-2-ил)амино)азетидин-1-карбоксилата (0,53 г, 0,80 ммоль) в THF (10 мл) при 0°C добавляли TBAF (0,80 мл, 0,80 ммоль). Полученный раствор перемешивали при 0°C в течение 1 часа. Раствор концентрировали и остаток очищали на колонке с силикагелем с использованием смеси MeOH/DCM в качестве элюирующих растворителей с получением трет-бутил 3-((6-(2-гидрокси-2-(2-(4-метокси-бензил)-2H-тетразол-5-ил)этокси)хинолин-2-ил)амино)азетидин-1-карбоксилата. LC/MS: (M+1)+=548,2
Стадия I: Получение трет-бутил 3-((6-(2-((1,3-диоксоизоиндолин-2-ил)окси)-2-(2-(4-метокси-бензил)-2H-тетразол-5-ил)этокси)хинолин-2-ил)амино)азетидин-1-карбоксилата К раствору трифенилфосфина (0,16 г, 0,59 ммоль), трет-бутил 3-((6-(2-гидрокси-2-(2-(4-метокси-бензил)-2H-тетразол-5-ил)этокси)хинолин-2-ил)амино)азетидин-1-карбоксилата (0,25 г, 0,46 ммоль) и 2-гидроксиизоиндолин-1,3-диона (0,082 г, 0,50 ммоль) в THF (20 мл) добавляли DEAD (0,097 мл, 0,59 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 3 часов. Раствор затем концентрировали и остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением трет-бутил 3-((6-(2-((1,3-диоксоизоиндолин-2-ил)окси)-2-(2-(4-метоксибензил)-2H-тетразол-5-ил)этокси)хинолин-2-ил)амино)азетидин-1-карбоксилата. LC/MS: (M+1)+=693,9
Стадия J: Получение трет-бутил 3-((6-(2-(аминоокси)-2-(2-(4-метоксибензил)-2H-тетразол-5-ил)этокси)хинолин-2-ил)амино)азетидин-1-карбоксилата К раствору трет-бутил 3-((6-(2-((1,3-диоксоизоиндолин-2-ил)окси)-2-(2-(4-метоксибензил)-2H-тетразол-5-ил)этокси)хинолин-2-ил)амино)азетидин-1-карбоксилата (0,36 г, 0,52 ммоль) в смеси растворителей этанола (10 мл) и DCE (20 мл) при 0°C добавляли гидразин (0,018 мл, 0,57 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 2 часов, раствор концентрировали и остаток суспендировали в DCM (10 мл). Полученную смесь перемешивали при комнатной температуре в течение 10 минут. Осадок отфильтровывали и фильтрат концентрировали с получением трет-бутил 3-((6-(2-(аминоокси)-2-(2-(4-метоксибензил)-2H-тетразол-5-ил)этокси)хинолин-2-ил)амино)азетидин-1-карбоксилата. LC/MS: (M+1)+=563,4
Стадия K: Получение (Z)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-((2-((2-((1-(трет-бутоксикарбонил)азетидин-3-ил)амино)хинолин-6-ил)окси)-1-(2-(4-метоксибензил)-2H-тетразол-5-ил)этокси)имино)уксусной кислоты К раствору трет-бутил 3-((6-(2-(аминоокси)-2-(2-(4-метоксибензил)-2H-тетразол-5-ил)этокси)хинолин-2-ил)амино)азетидин-1-карбоксилата (240 мг, 0,43 ммоль) в этаноле (10 мл) и DCE (10 мл) добавляли 2-(2-((трет-бутокси-карбонил)амино)тиазол-4-ил)-2-оксоуксусную кислоту (Промежуточное соединение 3) (139 мг, 0,51 ммоль). Полученный раствор перемешивали при комнатной температуре в течение ночи. Затем раствор концентрировали с получением (Z)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-((2-((2-((1-(трет-бутоксикарбонил)азетидин-3-ил)амино)хинолин-6-ил)окси)-1-(2-(4-метоксибензил)-2H-тетразол-5-ил)этокси)имино)уксусной кислоты. LC/MS: (M+1)+=817,4
Стадия L: Получение (Z)-2-(2-аминотиазол-4-ил)-2-((2-((2-(азетидин-3-иламино)хинолин-6-ил)окси)-1-(2H-тетразол-5-ил)этокси)имино)уксусной кислоты Во флакон, содержащий (Z)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-((2-((2-((1-(трет-бутоксикарбонил)азетидин-3-ил)амино)хинолин-6-ил)окси)-1-(2-(4-метоксибензил)-2H-тетразол-5-ил)этокси)имино)уксусную кислоту (350 мг, 0,43 ммоль) добавляли TFA (4 мл). Полученный раствор перемешивали при комнатной температуре в течение 30 минут, и затем концентрировали. Остаток повторно растворяли в TFA (4 мл) и нагревали при 70°C в течение 1 часа. Раствор концентрировали с получением (Z)-2-(2-аминотиазол-4-ил)-2-((2-((2-(азетидин-3-иламино)хинолин-6-ил)окси)-1-(2H-тетразол-5-ил)этокси)имино)уксусной кислоты. LC/MS: (M+1)+=497,3
Стадия M: Получение (Z)-2-(2-аминотиазол-4-ил)-2-((2-((2-((1-(трет-бутоксикарбонил)-азетидин-3-ил)амино)хинолин-6-ил)окси)-1-(2H-тетразол-5-ил)этокси)имино)уксусной кислоты К раствору (Z)-2-(2-аминотиазол-4-ил)-2-((2-((2-(азетидин-3-иламино)хинолин-6-ил)окси)-1-(2H-тетразол-5-ил)этокси)имино)уксусной кислоты (210 мг, 0,43 ммоль) в DMF (4 мл) при 0°C добавляли TEA (0,476 мл, 3,42 ммоль) и Boc2O (0,12 мл, 0,51 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 часа. После удаления летучих веществ, раствор очищали с помощью MPLC с обращенной фазой с использованием ацетонитрила (0,05% TFA)/воды (0,05% TFA) с получением (Z)-2-(2-аминотиазол-4-ил)-2-((2-((2-((1-(трет-бутоксикарбонил)азетидин-3-ил)амино)-хинолин-6-ил)окси)-1-(2H-тетразол-5-ил)этокси)имино)уксусной кислоты. LC/MS: (M+1)+=597,4
Стадия N: трет-бутил 3-((6-(2-(((Z)-(1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-(2H-тетразол-5-ил)этокси)хинолин-2-ил)амино)азетидин-1-карбоксилат К раствору соединения (Z)-2-(2-аминотиазол-4-ил)-2-((2-((2-((1-(трет-бутоксикарбонил)азетидин-3-ил)амино)хинолин-6-ил)окси)-1-(2H-тетразол-5-ил)этокси)имино)уксусной кислоты (142 мг, 0,20 ммоль) в DMF (6 мл) добавляли DCC (270 мг, 1,3 ммоль) и HOBt (200 мг, 1,3 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 30 минут перед добавлением (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфата (168 мг, 0,80 ммоль) и бикарбоната натрия (235 мг, 2,8 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 4 часов. Раствор фильтровали и фильтрат очищали с помощью ВЭЖХ с обращенной фазой с использованием ацетонитрила (0,05%TFA)/воды (0,05%TFA) с получением трет-бутил 3-((6-(2-(((Z)-(1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-(2H-тетразол-5-ил)этокси)хинолин-2-ил)амино)азетидин-1-карбоксилата. LC/MS: (M+1)+=789,4
Стадия O: Получение (S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((R)-2-((2-(азетидин-3-иламино)хинолин-6-ил)окси)-1-(2H-тетразол-5-ил)этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил гидросульфата и (S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-2-((2-(азетидин-3-иламино)хинолин-6-ил)окси)-1-(2H-тетразол-5-ил)этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил гидросульфата К раствору трет-бутил 3-((6-(2-(((Z)-(1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-(2H-тетразол-5-ил)этокси)хинолин-2-ил)амино)азетидин-1-карбоксилата (135 мг, 0,150 ммоль) в CH2Cl2 (5 мл) добавляли TFA (5 мл, 64,9 ммоль), и полученный раствор перемешивали при комнатной температуре в течение 30 минут. После концентрации, остаток очищали с помощью ВЭЖХ с обращенной фазой с использованием смеси ацетонитрил (0,05% TFA)/вода (0,05%TFA) в качестве подвижной фазы с получением (S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((R)-2-((2-(азетидин-3-иламино)хинолин-6-ил)окси)-1-(2H-тетразол-5-ил)этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил гидросульфата в виде соли TFA (быстро элюируемый), LC/MS:(M+1)+: 689,2.1Н ЯМР (500 МГц, CD3OD): δ 8,36-8,34(д, J=9,8 Гц, 1H), 7,83-7,81(д, J=8,7 Гц, 1H), 7,53-7,48(м, 2H), 7,12-7,11(д, J=9,6 Гц, 1H), 7,04(с, 1H), 6,12-6,10(м, 1H), 5,18-5,12(м, 1H), 4,75-4,72(м, 2H), 4,58-4,55(м,H), 4,54(с, 1H), 4,34-4,29(м, 2H), 1,46(с, 3H), 1,04(с, 3H), и (S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-2-((2-(азетидин-3-иламино)хинолин-6-ил)окси)-1-(2H-тетразол-5-ил)этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил гидросульфата в виде соли TFA (медленно элюируемый). LC/MS: (M+1)+: 689,2.1Н ЯМР (500 МГц, CD3OD): δ 8,35-8,33(д, J=10,6 Гц, 1H), 7,81-7,79(д, J=8,8 Гц, 1H), 7,53-7,51(м, 1H), 7,49-7,48(м, 1H), 7,12-7,10(д, J=9,3 Гц, 1H), 7,02(с, 1H), 6,08-6,06(т, J=4,8 Гц, 1H), 5,17-5,11(м, 1H), 4,83-4,81(м, 2H), 4,56(с, 1H), 4,58-4,53(м, 2H), 4,34-4,30(м, 2H), 1,5(с, 3H),1,28(с, 3H).
Примеры 47 и 48
4-((6-((R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-(2H-тетразол-5-ил)этокси)хинолин-2-ил)амино)пиперидин-1-ий 2,2,2-трифторацетат (C47) и 4-((6-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-(2H-тетразол-5-ил)этокси)хинолин-2-ил)амино)пиперидин-1-ий 2,2,2-трифторацетат (C48)

Соединения 47 и 48 получали в соответствии с аналогичной процедурой, описанной в примерах 45 и 46.
Соединение 47: 4-((6-((R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-(2H-тетразол-5-ил)этокси)хинолин-2-ил)амино)пиперидин-1-ий 2,2,2-трифторацетат. LC/MS: (M+1)+=717,1,1H-ЯМР (500 МГц, CD3OD): δ 8,30-8,28(д, J=10,1 Гц, 1H), 7,86-7,84(д, J=8,9 Гц, 1H), 7,53-7,51(м, 1H), 7,47-7,46(д, J=2,8 Гц, 1H), 7,10-7,08(м, 1H), 7,04(с, 1H), 6,12-6,10(м, 1H), 4,87-4,84(м, 1H), 4,77-4,73(м, 1H), 4,55(с, 1H), 4,25-4,21(м, 1H), 3,60-3,57(д, J=13,2 Гц, 2H), 3,25-3,20(т, J=12,9 Гц, 2H), 2,41-2,38(д, J=15. Гц, 2H), 1,94-1,88(м, 2H), 1,47(с, 3H), 1,09(с, 3H);
Соединение 48: 4-((6-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-(2H-тетразол-5-ил)этокси)хинолин-2-ил)амино)пиперидин-1-ий 2,2,2-трифторацетат. LC/MS: (M+1)+= 717,0,1H-ЯМР (500 МГц, CD3OD): δ 8,30-8,28(д, J=9,4 Гц, 1H), 7,85-7,83(д, J=9,4 Гц, 1H), 7,54-7,51(м, 1H), 7,48-7,47(д, J=2,7 Гц, 1H), 7,11-7,08(м, 1H), 7,03(с, 1H), 6,08-6,06(т, J=4,8 Гц, 1H), 4,83-4,82(д, J=6,8 Гц, 2H), 4,58(с, 1H), 4,26-4,21(м, 1H), 3,60-3,57(д, J=13,2 Гц, 2H), 3,26-3,21(т, J=13,0 Гц, 2H), 2,40-2,37(д, J=14,2 Гц, 2H), 1,95-1,87(м, 2H), 1,50(с, 3H), 1,30(с, 3H).
Примеры 49 и 50
2-((6-((R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-(2H-тетразол-5-ил)этокси)изохинолин-1-ил)амино)этан-1-аминий 2,2,2-трифторацетат (C49) и 2-((6-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-(2H-тетразол-5-ил)этокси)изохинолин-1-ил)амино)этан-1-аминий 2,2,2-трифторацетат (C50)

Соединения 49 и 50 получали, как правило, в соответствии с процедурой, описанной в примерах 45 и 46.
Соединение 49: 2-((6-((R)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-(2H-тетразол-5-ил)этокси)изохинолин-1-ил)амино)этан-1-аминий 2,2,2-трифторацетат. LC/MS: (M+1)+=677,1,1H-ЯМР (500 МГц, CD3OD): δ 8,41-8,40(д, J=8,9 Гц, 1H), 7,60-7,59(д, J=6,1 Гц, 1H), 7,49-7,47(д, J=7,1 Гц, 2H), 7,28-7,27(д, J=7,1 Гц, 1H), 7,04(с, 1H), 6,17-6,15(м, 1H), 5,00-4,97(м, 1H), 4,87-4,82(м, 2H), 4,53(с, 1H), 3,99-3,88(м, 2H), 3,50-3,43(м, 2H), 1,46(с, 3H), 1,00(с, 3H).
Соединение 50: 2-((6-((S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-2-(2H-тетразол-5-ил)этокси)изохинолин-1-ил)амино)этан-1-аминий 2,2,2-трифторацетат. LC/MS: (M+1)+= 677,3,1H-ЯМР (500 МГц, CD3OD): δ 4,82-4,80 (д, J=8,5 Гц, 1H), 7,60-7,59 (д, J=6,5 Гц, 1H), 7,51-7,47 (м, 2H), 7,26-7,24 (д, J=6,9 Гц, 1H), 7,01 (с, 1H), 6,13-6,11 (м, 1H), 5,03-4,98 (м, 1H), 4,55 (с, 1H), 3,96-3,92 (м, 2H), 3,47-3,45 (т, J=7,6 Гц, 2H), 1,50 (с, 3H), 1,27 (с, 3H).
ПРИМЕР 51
(S)-3-((Z)-2-(((S)-2-((2-((R)-3-амино-2-гидроксипропил)-1-метил-2H-индазол-1-ий-5-ил)окси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
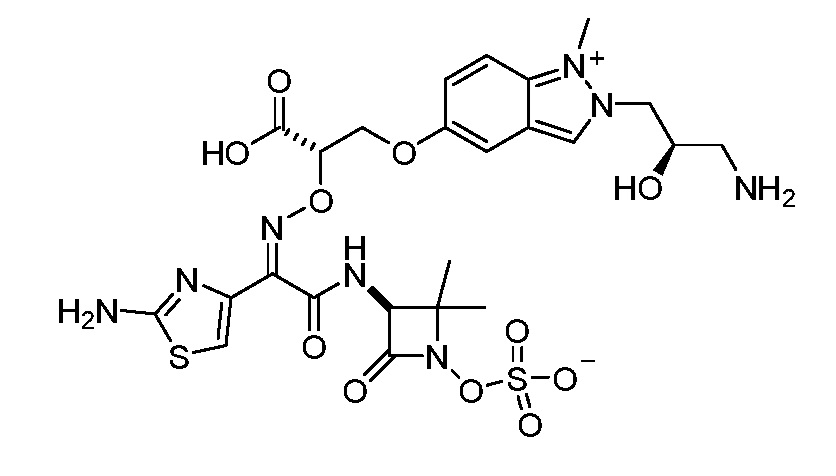
Стадия A: (R)-трет-бутил 3-((2-((R)-3-((трет-бутоксикарбонил)амино)-2-((трет-бутил-диметилсилил)окси)пропил)-2H-индазол-5-ил)окси)-2-гидроксипропаноат Раствор (R)-трет-бутил 3-(3-формил-4-нитрофенокси)-2-гидроксипропаноата (2,35 г, 7,55 ммоль) и (S)-трет-бутил (3-амино-2-((трет-бутилдиметилсилил)окси)пропил)карбамата (2,53 г, 8,30 ммоль) в 2-пропаноле (20 мл) нагревали при 80°C в течение 4 ч. Затем раствор охлаждали до комнатной температуры, и добавляли три-н-бутилфосфин (5,71 мл, 22,6 ммоль). Полученный раствор нагревали при 80°C в течение 16 ч и затем концентрировали. Полученный остаток очищали на колонке с силикагелем, элюируя смесью EtOAc/гексан с получением указанного в заголовке соединения. LC/MS: [M+1]+= 566,5.
Стадия B: (S)-трет-бутил 3-((2-((R)-3-((трет-бутоксикарбонил)амино)-2-((трет-бутилдиметилсилил)окси)пропил)-2H-индазол-5-ил)окси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноат К раствору (R)-трет-бутил 3-((2-((R)-3-((трет-бутоксикарбонил)амино)-2-((трет-бутилдиметилсилил)окси)пропил)-2H-индазол-5-ил)окси)-2-гидроксипропаноата (1,31 г, 2,32 ммоль) в THF (20 мл) добавляли 2-гидроксиизоиндолин-1,3-дион (0,453 г, 2,78 ммоль), трифенилфосфин (0,911 г, 3,47 ммоль) и DEAD (0,567 мл, 3,47 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч и затем концентрировали. Полученный остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением указанного в заголовке соединения. LC/MS: [M+1]+= 711,6.
Стадия C: 5-((S)-3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2-((R)-3-((трет-бутоксикарбонил)амино)-2-((трет-бутилдиметилсилил)окси)пропил)-1-метил-2H-индазол-1-ий К раствору (S)-трет-бутил 3-((2-((R)-3-((трет-бутоксикарбонил)амино)-2-((трет-бутилдиметилсилил)окси)пропил)-2H-индазол-5-ил)окси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноата (2,06 г, 2,32 ммоль) в ацетонитриле (30 мл) при 0°C добавляли метил трифторметансульфонат (0,306 мл, 2,78 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1,5 ч, и затем концентрировали с получением указанного в заголовке соединения. LC/MS: [M]+= 725,6.
Стадия D: 5-((S)-2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)-2-((R)-3-((трет-бутоксикарбонил)амино)-2-((трет-бутилдиметилсилил)окси)пропил)-1-метил-2H-индазол-1-ий К раствору 5-((S)-3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2-((R)-3-((трет-бутоксикарбонил)амино)-2-((трет-бутилдиметилсилил)окси)пропил)-1-метил-2H-индазол-1-ия (1,68 г, 2,32 ммоль) в CH2Cl2 (15 мл) и EtOH (15 мл) при 0°C добавляли гидразин (0,087 мл, 2,78 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, затем концентрировали с получением указанного в заголовке соединения. LC/MS: [M]+= 595,6.
Стадия E: 5-((S)-3-(трет-бутокси)-2-(((Z)-((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)-метилен)амино)окси)-3-оксопропокси)-2-((R)-3-((трет-бутоксикарбонил)амино)-2-((трет-бутилдиметилсилил)окси)пропил)-1-метил-2H-индазол-1-ий К раствору 5-((S)-2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)-2-((R)-3-((трет-бутоксикарбонил)амино)-2-((трет-бутилдиметилсилил)окси)пропил)-1-метил-2H-индазол-1-ия (1,25 г, 2,10 ммоль) в MeOH (12 мл) и CH2Cl2 (12 мл) добавляли 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусную кислоту (0,743 г, 2,73 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 2 ч, затем концентрировали с получением указанного в заголовке соединения. LC-MS: [M]+= 849,6.
Стадия F: 5-((S)-3-(трет-бутокси)-2-(((Z)-((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)-метилен)амино)окси)-3-оксопропокси)-2-((R)-3-((трет-бутоксикарбонил)амино)-2-гидроксипропил)-1-метил-2H-индазол-1-ий К раствору 5-((S)-3-(трет-бутокси)-2-(((Z)-((2-((трет-бутокси-карбонил)-амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)-2-((R)-3-((трет-бутоксикарбонил)-амино)-2-((трет-бутилдиметилсилил)окси)-пропил)-1-метил-2H-индазол-1-ия (1,78 г, 2,10 ммоль) в THF (20 мл) при 0°C добавляли TBAF (5,24 мл, 5,24 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч и затем концентрировали. Полученный остаток очищали с помощью MPLC с обращенной фазой (C18) с использованием ацетонитрила (0,05% TFA)/воды (0,05% TFA) в качестве элюирующих растворителей с получением указанного в заголовке соединения. LC/MS: [M]+= 735,5.
Стадия G: 5-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-2-((R)-3-((трет-бутоксикарбонил)амино)-2-гидроксипропил)-1-метил-2H-индазол-1-ий К раствору 5-((S)-3-(трет-бутокси)-2-(((Z)-((2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)-2-((R)-3-((трет-бутоксикарбонил)амино)-2-гидроксипропил)-1-метил-2H-индазол-1-ия (1,0 г, 1,36 ммоль) в DMF (10 мл) добавляли DCC (1,68 г, 8,15 ммоль) и HOBT (0,832 г, 5,44 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин, затем добавляли (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфат (0,857 г, 4,08 ммоль) и бикарбонат натрия (1,14 г, 13,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем смесь фильтровали и фильтрат очищали с помощью MPLC с обращенной фазой (C18) с использованием ацетонитрила (0,05% TFA)/воды (0,05% TFA) в качестве подвижной фазы с получением указанного в заголовке соединения. LC-MS: [M]+= 927,8.
Стадия H: (S)-3-((Z)-2-(((S)-2-((2-((R)-3-амино-2-гидроксипропил)-1-метил-2H-индазол-1-ий-5-ил)окси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат К раствору 5-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-2-((R)-3-((трет-бутоксикарбонил)-амино)-2-гидроксипропил)-1-метил-2H-индазол-1-ия (711 мг, 0,766 ммоль) в CH2Cl2 (4 мл) добавляли TFA (8 мл, 104 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 50 мин, затем концентрировали. Полученный остаток осаждали из Et2O. Фазу Et2O переносили в колбу с помощью пипетки и процесс повторяли три раза. Твердое вещество далее сушили под вакуумом, и затем растворяли в DMSO (5,5 мл) и очищали с помощью ВЭЖХ с обращенной фазой, используя ацетонитрил/воду (20 мM NH4OAc), с получением указанного в заголовке соединения. LCMS: [M+1]+= 671,5.1H ЯМР (500 МГц, D2O): δ 8,74(с, 1H), 7,64-7,62(д, J=8,2 Гц, 1H), 7,48-7,46(д, J=8,2 Гц, 1H), 7,27(с, 1H), 6,93(с, 1H), 4,97-4,90(м, 3H), 4,44-4,35(м, 3H), 4,20(с, 3H), 3,38-3,35(м, 1H), 3,10-3,06(м, 1H), 1,40(с, 3H), 1,04(с, 3H).
ПРИМЕР 52
(S)-3-((Z)-2-(((S)-2-((2-(3-аминопропил)-1-метил-2H-индазол-1-ий-5-ил)окси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
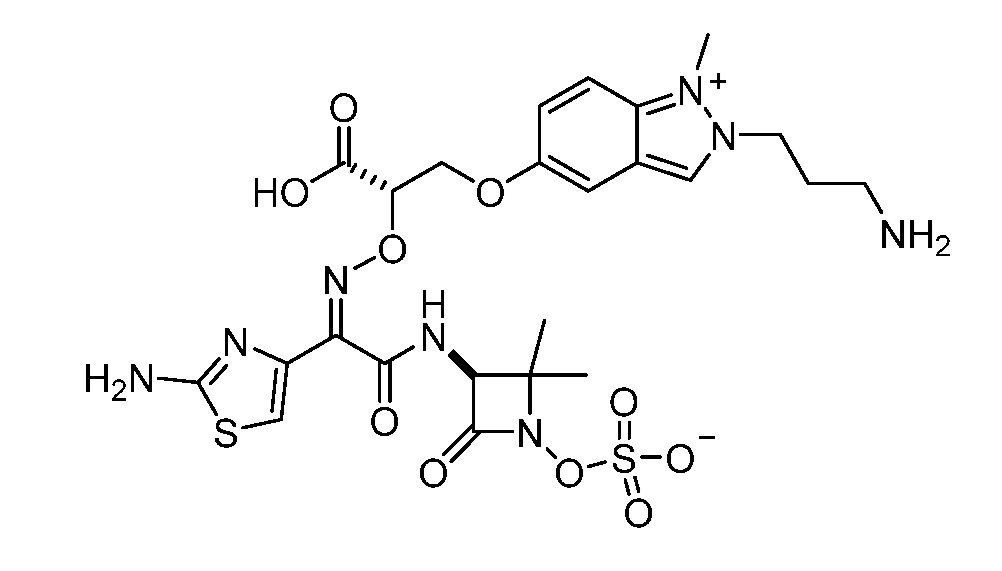
Стадия A: (R)-трет-бутил 3-((2-(3-((трет-бутоксикарбонил)амино)пропил)-2H-индазол-5-ил)окси)-2-гидроксипропаноат Раствор (R)-трет-бутил 3-(3-формил-4-нитрофенокси)-2-гидроксипропаноата (0,5 г, 1,61 ммоль) и трет-бутил (3-аминопропил)карбамата (0,308 г, 1,77 ммоль) в 2-пропаноле (4 мл) нагревали при 80°C в течение 4 ч. Затем реакционную смесь охлаждали до комнатной температуры и добавляли три-н-бутилфосфин (1,22 мл, 4,82 ммоль). Реакционную смесь нагревали при 80°C в течение 16 ч, затем концентрировали. Полученный остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением указанного в заголовке соединения. LC-MS: [M+1]+: 436,3.
Стадия B: (S)-трет-бутил 3-((2-(3-((трет-бутоксикарбонил)амино)пропил)-2H-индазол-5-ил)окси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноат К раствору (R)-трет-бутил 3-((2-(3-((трет-бутоксикарбонил)-амино)пропил)-2H-индазол-5-ил)окси)-2-гидроксипропаноата (0,302 г, 0,693 ммоль) в THF (10 мл) добавляли 2-гидрокси-изоиндолин-1,3-дион (0,136 г, 0,832 ммоль), трифенилфосфин (0,273 г, 1,04 ммоль) и DEAD (0,170 мл, 1,04 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч и затем концентрировали. Полученный остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением указанного в заголовке соединения. LC/MS: [M+1]+: 581,4.
Стадия C: (S)-5-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2-(3-((трет-бутоксикарбонил)амино)пропил)-1-метил-2H-индазол-1-ий К раствору (S)-трет-бутил 3-((2-(3-((трет-бутоксикарбонил)амино)пропил)-2H-индазол-5-ил)окси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноата (379 мг, 0,653 ммоль) в ацетонитриле (10 мл) при 0°C добавляли метил трифторметансульфонат (0,079 мл, 0,718 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, затем концентрировали с получением указанного в заголовке соединения. LC/MS: M+: 595,4.
Стадия D: (S)-5-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)-2-(3-((трет-бутоксикарбонил)амино)пропил)-1-метил-2H-индазол-1-ий К раствору (S)-5-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2-(3-((трет-бутоксикарбонил)амино)пропил)-1-метил-2H-индазол-1-ия (389 мг, 0,653 ммоль) в этаноле (5 мл) и CH2Cl2 (5 мл) добавляли гидразин (0,029 мл, 0,914 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч и затем концентрировали с получением указанного в заголовке соединения. LC/MS: M+: 465,3
Стадия E: (S)-3-(2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоацетамидо)-2,2-диметил-4-оксоазетидин-1-ил гидросульфат К смеси 2-(2-((трет-бутокси-карбонил)амино)тиазол-4-ил)-2-оксоуксусной кислоты гидрохлорида (4,02 г, 13,02 ммоль) в ацетонитриле (60 мл) при 0°C добавляли пиридин (3,16 мл, 39,1 ммоль), (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфат (5,47 г, 26,0 ммоль) и EDC (6,24 г, 32,6 ммоль). Реакционную смесь перемешивали при 0°C в течение 3 ч, затем концентрировали. Полученный остаток разделяли между 30% IPA/DCM (300 мл) и насыщенным солевым раствором (200 мл). Органическую фазу отделяли, промывали насыщенным солевым раствором, сушили над Na2SO4, и концентрировали. Полученный остаток очищали с помощью MPLC с обращенной фазой с использованием смеси ацетонитрил (0,1% муравьиной кислоты)/вода (0,1% муравьиной кислоты) в качестве элюирующих растворителей с получением указанного в заголовке соединения. LC/MS: [M+1]+: 465,1.
Стадия F: 5-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-2-(3-((трет-бутоксикарбонил)амино)пропил)-1-метил-2H-индазол-1-ий К раствору (S)-5-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)-2-(3-((трет-бутоксикарбонил)-амино)пропил)-1-метил-2H-индазол-1-ия (304 мг, 0,653 ммоль) в MeOH (5 мл) и CH2Cl2 (5,00 мл) добавляли (S)-3-(2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоацетамидо)-2,2-диметил-4-оксоазетидин-1-ил гидросульфат (425 мг, 0,914 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем концентрировали. Полученный остаток очищали с помощью MPLC с обращенной фазой с использованием смеси ацетонитрил (0,1% муравьиной кислоты)/вода (0,1% муравьиной кислоты) с получением указанного в заголовке соединения. LC/MS: M+: 911,6.
Стадия G: (S)-3-((Z)-2-(((S)-2-((2-(3-аминопропил)-1-метил-2H-индазол-1-ий-5-ил)окси)-1-карбокси-этокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат К раствору 5-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил-)амино)-тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)-амино)окси)-3-оксопропокси)-2-(3-((трет-бутоксикарбонил)амино)пропил)-1-метил-2H-индазол-1-ия (306 мг, 0,336 ммоль) в CH2Cl2 (2 мл) добавляли TFA (4 мл, 51,9 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 45 мин, и затем концентрировали. Полученный остаток обрабатывали Et2O три раза. Полученное твердое вещество сушили в высоком вакууме, затем растворяли в DMSO и очищали с помощью ВЭЖХ с обращенной фазой с использованием ацетонитрила (0,1% муравьиной кислоты)/воды (0,1% муравьиной кислоты) в качестве элюирующих растворителей с получением указанного в заголовке соединения. LC/MS: [M+1]+: 655,3.1H ЯМР (500 МГц, D2O): δ 8,73(с, 1H), 7,67-7,65(д, J=9,9 Гц, 1H), 7,49-7,47(м, 1H), 7,29(с, 1H), 7,07(с, 1H), 5,10(с, 1H), 4,79-4,75(д, J=8,8 Гц, 2H), 4,53-4,44(м, 3H), 4,19(с, 3H), 3,13-3,10(т, J=7,8 Гц, 2H), 2,40-2,35(м, 2H), 1,37(с, 3H), 0,94 (с,1H).
ПРИМЕР 53
(S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-1-карбокси-2-((2-((3-гидроксиазетидин-3-ил)метил)-1-метил-2H-индазол-1-ий-5-ил)окси)этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат

Стадия A: (R)-трет-бутил 3-((5-(3-(трет-бутокси)-2-гидрокси-3-оксопропокси)-2H-индазол-2-ил)метил)-3-гидроксиазетидин-1-карбоксилат Раствор (R)-трет-бутил 3-(3-формил-4-нитрофенокси)-2-гидрокси-пропаноата (1,4 г, 4,50 ммоль) и трет-бутил 3-(аминометил)-3-гидроксиазетидин-1-карбоксилата (1,00 г, 4,95 ммоль) в 2-пропаноле (7 мл) нагревали при 80°C в течение 4 ч. Затем раствор охлаждали до комнатной температуры и добавляли три-н-бутилфосфин (3,40 мл, 13,49 ммоль). Реакционную смесь нагревали при 80°C в течение 16 ч и затем концентрировали. Полученный остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением указанного в заголовке соединения. LC/MS: [M+1]+= 464,3.
Стадия B: (S)-трет-бутил 3-((5-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксо-пропокси)-2H-индазол-2-ил)метил)-3-гидроксиазетидин-1-карбоксилат К раствору (R)-трет-бутил 3-((5-(3-(трет-бутокси)-2-гидрокси-3-оксопропокси)-2H-индазол-2-ил)метил)-3-гидроксиазетидин-1-карбоксилата (1,17 г, 2,52 ммоль) в THF (20 мл) добавляли по каплям 2-гидроксиизоиндолин-1,3-дион (0,453 г, 2,78 ммоль), трифенилфосфин (0,993 г, 3,79 ммоль и DEAD (0,599 мл, 3,79 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч и затем концентрировали. Полученный остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением указанного в заголовке соединения. LC/MS: [M+1]+= 609,4.
Стадия C: (S)-5-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2-((1-(трет-бутокси-карбонил)-3-гидроксиазетидин-3-ил)метил)-1-метил-2H-индазол-1-ий К раствору (S)-трет-бутил 3-((5-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2H-индазол-2-ил)метил)-3-гидроксиазетидин-1-карбоксилата (500 мг, 0,822 ммоль) в ацетонитриле (4 мл) при 0°C добавляли метил трифторметансульфонат (0,109 мл, 0,986 ммоль). Полученный раствор перемешивали при 0°C и затем перемешивали при комнатной температуре в течение 1 часа. Затем реакционную смесь концентрировали с получением указанного в заголовке соединения. LC/MS: [M]+= 623,4.
Стадия D: (S)-5-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)-3-гидроксиазетидин-3-ил)метил)-1-метил-2H-индазол-1-ий К раствору (S)-5-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)-3-гидроксиазетидин-3-ил)метил)-1-метил-2H-индазол-1-ия (512 мг, 0,821 ммоль) в этаноле (5 мл) и CH2Cl2 (5 мл) добавляли гидразин (0,028 мл, 0,903 ммоль) при °C. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч и затем концентрировали. Полученный остаток обрабатывали DCM (5 мл) и смесь перемешивали при комнатной температуре в течение 15 мин, и затем фильтровали. Фильтрат концентрировали с получением указанного в заголовке соединения. LC/MS: [M]+= 493,3.
Стадия E: (S,Z)-5-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)-метилен)амино)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)-3-гидроксиазетидин-3-ил)метил)-1-метил-2H-индазол-1-ий К раствору (S)-5-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)-2-((1-(трет-бутокси-карбонил)-3-гидроксиазетидин-3-ил)метил)-1-метил-2H-индазол-1-ия (405 мг, 0,821 ммоль) в MeOH (2 мл) и CH2Cl2 (2 мл) добавляли 2-(2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)-2-оксоуксусную кислоту (268 мг, 0,985 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, затем концентрировали с получением указанного в заголовке соединения. LC/MS: [M]+= 747,4.
Стадия F: 5-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)-3-гидроксиазетидин-3-ил)метил)-1-метил-2H-индазол-1-ий К раствору (S,Z)-5-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)(карбокси)метилен)амино)-окси)-3-оксопропокси)-2-((1-(трет-бутокси-карбонил)-3-гидроксиазетидин-3-ил)метил)-1-метил-2H-индазол-1-ия (614 мг, 0,821 ммоль) в DMF (8 мл) добавляли DCC (678 мг, 3,28 ммоль) и HOBT (377 мг, 2,463 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 0,5 ч, затем добавляли (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфат (518 мг, 2,46 ммоль) и бикарбонат натрия (690 мг, 8,21 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем смесь фильтровали и фильтрат очищали с помощью MPLC с обращенной фазой с использованием ацетонитрила (0,05% TFA)/воды(0,05% TFA) в качестве элюирующих растворителей с получением указанного в заголовке соединения. LC/MS: [M]+= 939,5.
Стадия G: (S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-1-карбокси-2-((2-((3-гидроксиазетидин-3-ил)метил)-1-метил-2H-индазол-1-ий-5-ил)окси)этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат К раствору 5-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутокси-карбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)-3-гидроксиазетидин-3-ил)метил)-1-метил-2H-индазол-1-ия (287 мг, 0,305 ммоль) в CH2Cl2 (2 мл) добавляли TFA (4 мл, 51,9 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 45 мин и затем концентрировали. Полученный остаток обрабатывали Et2O. Полученное твердое вещество сушили в высоком вакууме, растворяли в DMSO (3 мл) и очищали с помощью ВЭЖХ с обращенной фазой с использованием ацетонитрила (0,1% муравьиной кислоты)/воды (0,1% муравьиной кислоты). Полученный продукт повторно очищали с помощью ВЭЖХ с обращенной фазой с использованием ацетонитрил/ацетат аммония (20 мМ) буфера с получением указанного в заголовке соединения. LC/MS: [M+1]+= 683,4.1H ЯМР (500 МГц, D2O): δ 8,79 (с, 1H), 7,62-7,60 (д, J=9 Гц, 1H), 7,47-7,45 (дд, J=2,3 Гц и 9,0 Гц, 1H), 7,24 (д, J=2,3 Гц, 1H), 6,90 (с, 1H), 5,08 (с, 2H), 4,96-4,94 (м, 1H), 4,41-4,36 (м, 4H), 4,17 (с, 3H), 4,11-4,06 (т, J=8,8 Гц, 2H), 1,35 (с, 3H), 0,98 (с, 3H).
ПРИМЕР 54
(S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-2-((2-((3-карбамоилазетидин-3-ил)метил)-1-метил-2H-индазол-1-ий-5-ил)окси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
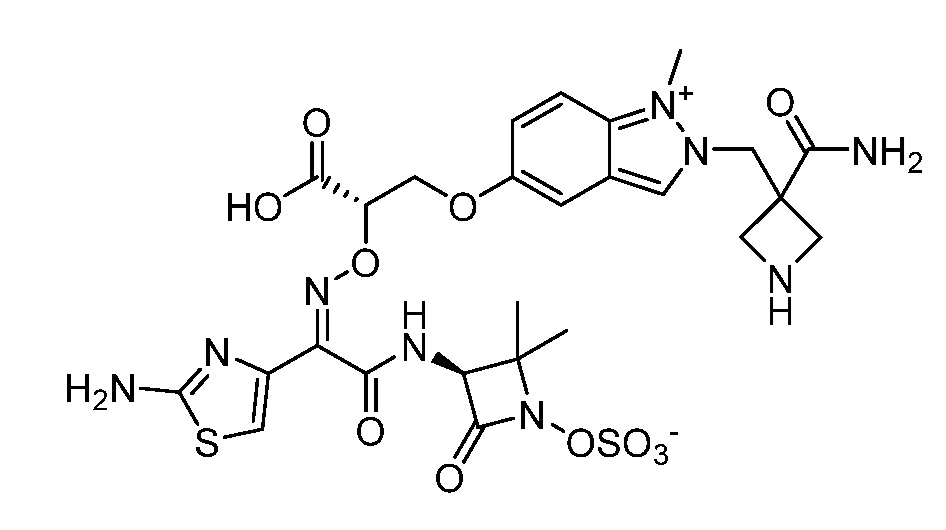
Стадия A: (R)-трет-бутил 3-((5-(3-(трет-бутокси)-2-гидрокси-3-оксопропокси)-2H-индазол-2-ил)метил)-3-карбамоилазетидин-1-карбоксилат Раствор трет-бутил 3-(аминометил)-3-карбамоилазетидин-1-карбоксилата (1 г, 4,36 ммоль) и (R)-трет-бутил 3-(3-формил-4-нитро-фенокси)-2-гидроксипропаноата (1,36 г, 4,36 ммоль) в 2-пропаноле (8 мл) нагревали при 80°C в течение 4 ч. После охлаждения до комнатной температуры добавляли трибутилфосфин (3,30 мл, 13,1 ммоль), и реакционную смесь нагревали при 80°C в течение 16 ч. Затем реакционную смесь концентрировали и полученный остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением указанного в заголовке соединения. LC/MS: [M+1]+= 491,4.
Стадия B: (S)-трет-бутил 3-((5-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксо-пропокси)-2H-индазол-2-ил)метил)-3-карбамоилазетидин-1-карбоксилат К раствору (R)-трет-бутил 3-((5-(3-(трет-бутокси)-2-гидрокси-3-оксопропокси)-2H-индазол-2-ил)метил)-3-карбамоилазетидин-1-карбоксилата (325 мг, 0,663 ммоль) в THF (5 мл) добавляли 2-гидроксиизоиндолин-1,3-дион (130 мг, 0,795 ммоль), трифенилфосфин (261 мг, 0,994 ммоль) и DEAD (0,157 мл, 0,994 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч, затем концентрировали. Полученный остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением указанного в заголовке соединения. LC/MS: [M+1]+= 636,5.
Стадия C: (S)-трет-бутил 3-((5-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)-2H-индазол-2-ил)метил)-3-карбамоилазетидин-1-карбоксилат К раствору (S)-трет-бутил 3-((5-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2H-индазол-2-ил)метил)-3-карбамоилазетидин-1-карбоксилата (421 мг, 0,662 ммоль) в CH2Cl2 (8 мл) и EtOH (8 мл) добавляли гидразин (0,025 мл, 0,795 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, затем концентрировали. Полученный остаток перемешивали в DCM (20 мл) в течение 10 мин. Образовавшийся осадок отфильтровывали и фильтрат концентрировали с получением указанного в заголовке соединения. LC/MS: [M+1]+= 506,5.
Стадия D: (S,Z)-2-(((1-(трет-бутокси)-3-((2-((1-(трет-бутоксикарбонил)-3-карбамоилазетидин-3-ил)метил)-2H-индазол-5-ил)окси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)уксусная кислота К раствору (S)-трет-бутил 3-((5-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)-2H-индазол-2-ил)метил)-3-карбамоилазетидин-1-карбоксилата (335 мг, 0,663 ммоль) в CH2Cl2 (10 мл) и MeOH (10 мл) добавляли 2-(2-((трет-бутокси-карбонил)амино)тиазол-4-ил)-2-оксоуксусную кислоту (180 мг, 0,663 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, затем концентрировали. Полученный остаток очищали с помощью MPLC с обращенной фазой (C18) с использованием ацетонитрила (0,05% TFA)/воды (0,05% TFA) в качестве подвижной фазы с получением указанного в заголовке соединения. LC/MS: [M+1]+= 760,5.
Стадия E: трет-бутил 3-((5-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)-тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксо-этилиден)амино)окси)-3-оксопропокси)-2H-индазол-2-ил)метил)-3-карбамоилазетидин-1-карбоксилат К раствору (S,Z)-2-(((1-(трет-бутокси)-3-((2-((1-(трет-бутоксикарбонил)-3-карбамоилазетидин-3-ил)метил)-2H-индазол-5-ил)окси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)уксусной кислоты (313 мг, 0,412 ммоль) в ацетонитриле (10 мл) при 0°C добавляли EDC (237 мг, 1,24 ммоль), (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфат (260 мг, 1,24 ммоль) и пиридин (0,133 мл, 1,65 ммоль). Реакционную смесь перемешивали при 0°C в течение 2 ч, затем концентрировали. Полученный остаток очищали с помощью MPLC с обращенной фазой (C18) с использованием ацетонитрила (0,05% TFA)/воды (0,05% TFA) с получением указанного в заголовке соединения. LC/MS: [M+1]+= 952,6.
Стадия F: 5-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)-3-карбамоилазетидин-3-ил)метил)-1-метил-2H-индазол-1-ий К раствору трет-бутил 3-((5-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-2H-индазол-2-ил)метил)-3-карбамоилазетидин-1-карбоксилата (203 мг, 0,213 ммоль) в ацетонитриле (5 мл) при 0°C добавляли бикарбонат натрия (35,8 мг, 0,426 ммоль) и метил трифторметансульфонат (0,028 мл, 0,256 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, и фильтровали. Фильтрат концентрировали, и полученный остаток очищали с помощью MPLC с обращенной фазой (C18) с использованием ацетонитрила (0,05% TFA)/воды (0,05% TFA) в качестве подвижной фазы с получением указанного в заголовке соединения. LC/MS: M+= 966,6.
Стадия G: (S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-2-((2-((3-карбамоилазетидин-3-ил)-метил)-1-метил-2H-индазол-1-ий-5-ил)окси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат К раствору 5-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)-3-карбамоилазетидин-3-ил)метил)-1-метил-2H-индазол-1-ия(80 мг, 0,083 ммоль) в CH2Cl2 (2 мл) добавляли TFA (4 мл, 51,9 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 45 мин и затем концентрировали под вакуумом. Полученный остаток обрабатывали Et2O, и полученное твердое вещество сушили под вакуумом, затем очищали с помощью ВЭЖХ с обращенной фазой (C18) с использованием ацетонитрила (0,05% TFA)/воды (0,05% TFA) в качестве подвижной фазы с получением указанного в заголовке соединения. LC/MS: [M+1])+= 710,5.1H ЯМР (500 МГц, D2O): δ 8,77 (с, 1H), 7,65-7,65 (д, J=10,7 Гц, 1), 7,51-7,49 (д, J=10,7 Гц, 1H), 7,26 (с, 1H), 7,04 (с, 1H), 5,32 (с, 1H), 5,08-5,06 (м, 1H), 4,50-4,45 (м, 4H), 4,40-4,36 (м, 2H), 4,13 (с, 3H), 1,35 (с, 3H), 0,95 (с, 3H).
ПРИМЕР 55
(S)-3-((Z)-2-(((S)-2-((2-(3-амино-2-(аминометил)пропил)-1-метил-2H-индазол-1-ий-5-ил)окси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
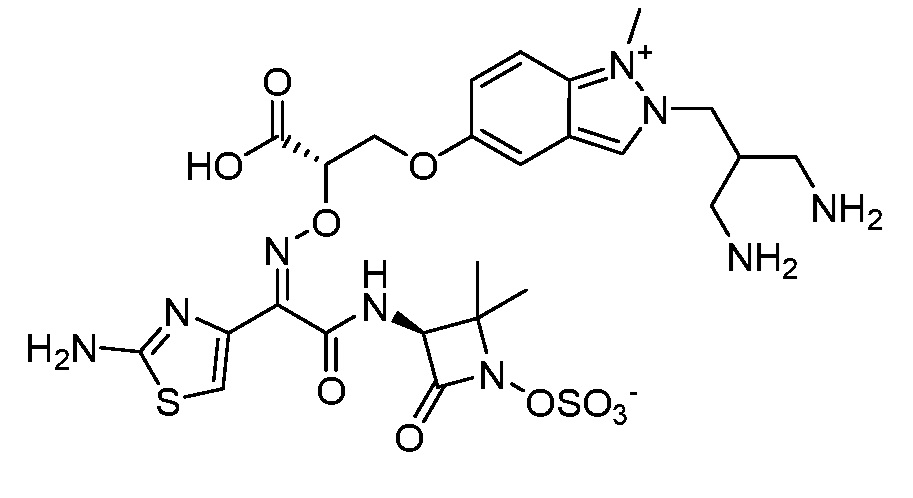
Стадия A: (R)-трет-бутил 3-((2-(3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)-амино)метил)-пропил)-2H-индазол-5-ил)окси)-2-гидроксипропаноат Раствор (R)-трет-бутил 3-(3-формил-4-нитрофенокси)-2-гидроксипропаноата (1,32 г, 4,25 ммоль) и ди-трет-бутил (2-(аминометил)пропан-1,3-диил)дикарбамата (1,29 г, 4,25 ммоль) в 2-пропаноле (10 мл) нагревали при 80°C в течение 4 ч. Затем реакционную смесь охлаждали до комнатной температуры, и добавляли три-н-бутилфосфин (3,22 мл, 12,8 ммоль). Реакционную смесь нагревали при 80°C в течение 16 ч, затем концентрировали. Полученный остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением указанного в заголовке соединения. LC/MS: [M+1]+: 565,5.
Стадия B: (S)-трет-бутил 3-((2-(3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)-амино)-метил)пропил)-2H-индазол-5-ил)окси)-2-((1,3-диоксоизоиндолин-2-ил)окси)пропаноат К раствору (R)-трет-бутил 3-((2-(3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)амино)метил)пропил)-2H-индазол-5-ил)окси)-2-гидроксипропаноата (1,1 г, 1,95 ммоль) в THF (20 мл) добавляли 2-гидрокси-изоиндолин-1,3-дион (0,381 г, 2,34 ммоль), трифенилфосфин (60,8 г, 2,92 ммоль) и DEAD (0,463 мл, 2,92 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, затем концентрировали. Полученный остаток очищали на колонке с силикагелем с использованием смеси EtOAc/гексан в качестве элюирующих растворителей с получением указанного в заголовке соединения. LC/MS: [M+1]+: 710,6
Стадия C: (S)-5-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2-(3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)амино)метил)пропил)-1-метил-2H-индазол-1-ий К раствору (S)-трет-бутил 3-((2-(3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)амино)-метил)пропил)-2H-индазол-5-ил)окси)-2-((1,3-диоксо-изоиндолин-2-ил)окси)пропаноата (1,29 г, 1,82 ммоль) в ацетонитриле (20 мл) при 0°C добавляли метил трифторметансульфонат (0,240 мл, 2,18 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, затем концентрировали с получением указанного в заголовке соединения. LC/MS: M+: 724,6.
Стадия D: (S)-5-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)-2-(3-((трет-бутоксикарбонил)-амино)-2-(((трет-бутоксикарбонил)амино)метил)пропил)-1-метил-2H-индазол-1-ий К раствору (S)-5-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2-(3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)амино)метил)пропил)-1-метил-2H-индазол-1-ия (1,32 г, 1,82 ммоль) в CH2Cl2 (10 мл) и EtOH (10,00 мл) при 0°C добавляли гидразин (0,068 мл, 2,180 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, затем концентрировали с получением указанного в заголовке соединения. LC/MS: M+: 594,6.
Стадия E: (S,Z)-5-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)-метилен)амино)окси)-3-оксопропокси)-2-(3-((трет-бутоксикарбонил)амино)-2-(((трет-бутокси-карбонил)-амино)метил)пропил)-1-метил-2H-индазол-1-ий К раствору (S)-5-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)-2-(3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)амино)метил)пропил)-1-метил-2H-индазол-1-ия (1,08 г, 1,82 ммоль) в CH2Cl2 (10 мл) и MeOH (10 мл) добавляли 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусную кислоту (0,643 г, 2,36 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, затем концентрировали с получением указанного в заголовке соединения. LC/MS: M+: 848,7.
Стадия F: 5-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-2-(3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)амино)метил)-пропил)-1-метил-2H-индазол-1-ий К раствору (S,Z)-5-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)-2-(3-((трет-бутоксикарбонил)амино)-2-(((трет-бутокси-карбонил)амино)метил)пропил)-1-метил-2H-индазол-1-ия (1,52 г, 1,79 ммоль) в DMF (10 мл) добавляли DCC (1,48 г, 7,16 ммоль) и HOBT (0,823 г, 5,37 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин, затем добавляли (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфат (1,13 г, 5,37 ммоль) и бикарбонат натрия (1,50 г, 17,9 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем фильтровали. Фильтрат очищали с помощью MPLC с обращенной фазой (колонка C18) с использованием ацетонитрила (0,05% TFA)/воды (0,05% TFA) в качестве подвижной фазы с получением указанного в заголовке соединения. LC/MS: M+: 1041,4.
Стадия G: (S)-3-((Z)-2-(((S)-2-((2-(3-амино-2-(аминометил)пропил)-1-метил-2H-индазол-1-ий-5-ил)окси)-1-карбоксиэтокси)имино)-2-(2-аминотиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат К раствору 5-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутокси-карбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-2-(3-((трет-бутоксикарбонил)амино)-2-(((трет-бутоксикарбонил)амино)метил)пропил)-1-метил-2H-индазол-1-ия (780 мг, 0,749 ммоль) в CH2Cl2 (4 мл) добавляли TFA (8 мл, 104 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 70 мин и затем концентрировали при комнатной температуре. Полученный остаток обрабатывали Et2O (10 мл). Полученное твердое вещество собирали, и раствор Et2O концентрировали и объединяли с твердым веществом. Твердое вещество растворяли в DMSO (6,5 мл) и очищали с помощью ВЭЖХ с обращенной фазой (колонка С18) с использованием ацетонитрила (0,1% муравьиной кислоты)/воды (0,1% муравьиной кислоты) с получением указанного в заголовке соединения. LC/MS: [M+1]+: 684,5.1H ЯМР (500 МГц, D2O): δ 8,80 (с, 1H), 7,64-7,62 (д, J=10,1 Гц, 1H), 7,49-7,47 (д, J=10,1 Гц, 1H), 7,25 (с, 1H), 6,94 (с, 1H), 4,98 (с, 1H), 4,88-4,87 (д, J=7,2 Гц, 2H), 4,46-4,43 (м, 2H), 4,21 (с, 3H), 3,53-3,48 (м,1H), 3,27-3,22 (м, 2H), 3,10-3,06 (м, 2H), 2,86-2,80 (м, 1H), 1,40 (с, 3H), 1,00 (с, 3H).
ПРИМЕР 56
(S)-3-((Z)-2-(2-Аминотиазол-4-ил)-2-(((S)-1-карбокси-2-((1-метил-2-((R)-пирролидин-3-ил)-2H-индазол-1-ий-5-ил)окси)этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
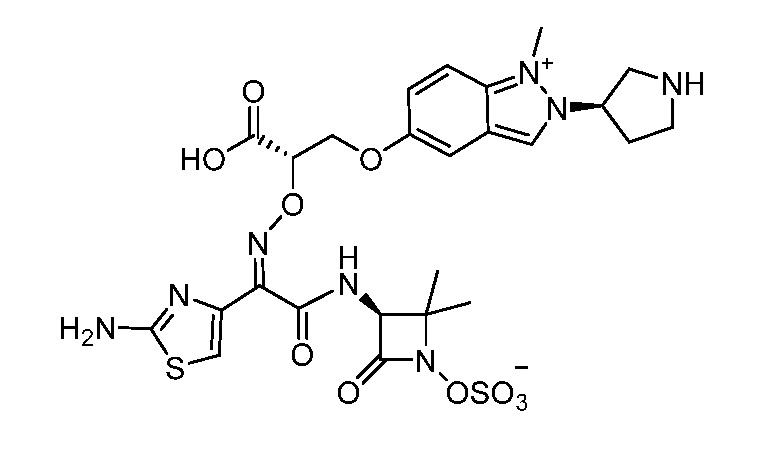
Стадия A: трет-бутил (R)-3-(5-((R)-3-(трет-бутокси)-2-гидрокси-3-оксопропокси)-2H-индазол-2-ил)пирролидин-1-карбоксилат Раствор (R)-трет-бутил 3-(3-формил-4-нитрофенокси)-2-гидроксипропаноата (300 мг, 0,964 ммоль) и (R)-(+)-1-Boc-3-аминопирролидина в 2-пропаноле (2 мл) нагревали при 80°C в течение 4 ч. Затем реакционную смесь охлаждали до комнатной температуры и добавляли три-н-бутилфосфин (0,729 мл, 2,89 ммоль). Реакционную смесь нагревали при 80°C в течение 16 ч, затем концентрировали. Полученный остаток очищали на колонке с силикагелем (40 г) с использованием 0-60% EtOAc/гексана с получением указанного в заголовке соединения. LC/MS: m/e 448,21 (M+H)+.
Стадия B: трет-бутил (R)-3-(5-((S)-3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксо-пропокси)-2H-индазол-2-ил)пирролидин-1-карбоксилат К раствору (R)-трет-бутил 3-(5-((R)-3-(трет-бутокси)-2-гидрокси-3-оксопропокси)-2H-индазол-2-ил)пирролидин-1-карбоксилата (288 мг, 0,644 ммоль) в THF (4,29 мл) при комнатной температуре добавляли по каплям 2-гидроксиизоиндолин-1,3-дион (126 мг, 0,772 ммоль), трифенилфосфин (253 мг, 0,965 ммоль) и диизопропил азодикарбоксилат (190 мкл, 0,965 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем концентрировали. Полученный остаток очищали на колонке с силикагелем (40 г) с использованием 0-70% EtOAc/гексана с получением указанного в заголовке соединения. LC/MS: m/e 593,17 (M+H)+.
Стадия C: 5-((S)-3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2-((R)-1-(трет-бутоксикарбонил)пирролидин-3-ил)-1-метил-2H-индазол-1-ий К раствору (R)-трет-бутил 3-(5-((S)-3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2H-индазол-2-ил)пирролидин-1-карбоксилата (360 мг, 0,607 ммоль) в ацетонитриле (6 мл) при 0°C добавляли метил трифторметансульфонат (73,6 мкл, 0,668 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, затем концентрировали с получением указанного в заголовке соединения, которое использовали на следующей стадии без дополнительной очистки. LC/MS: m/e 607,23 (M)+.
Стадия D: 5-((S)-2-(Аминоокси)-3-(трет-бутокси)-3-оксопропокси)-2-((R)-1-(трет-бутоксикарбонил)пирролидин-3-ил)-1-метил-2H-индазол-1-ий К раствору 5-((S)-3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2-((R)-1-(трет-бутоксикарбонил)-пирролидин-3-ил)-1-метил-2H-индазол-1-ия (0,369 г, 0,607 ммоль) в этаноле (4,1 мл) и CH2Cl2 (4,1 мл) добавляли гидразин (0,023 мл, 0,728 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, затем концентрировали. Полученный остаток обрабатывали CH2Cl2 (10 мл), и смесь перемешивали при комнатной температуре в течение 30 мин. Смесь фильтровали и фильтрат концентрировали с получением указанного в заголовке соединения. LC/MS: m/e 477,21 (M)+.
Стадия E: 5-((S)-3-(трет-бутокси)-2-((((Z)-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-(карбокси)-метилен)амино)окси)-3-оксопропокси)-2-((R)-1-(трет-бутоксикарбонил)пирролидин-3-ил)-1-метил-2H-индазол-1-ий К раствору 5-((S)-2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)-2-((R)-1-(трет-бутоксикарбонил)пирролидин-3-ил)-1-метил-2H-индазол-1-ия (0,290 г, 0,607 ммоль) в MeOH (3,04 мл) и CH2Cl2 (3,04 мл) добавляли 2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-оксоуксусную кислоту (0,198 г, 0,728 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, затем концентрировали с получением указанного в заголовке соединения. LC/MS: m/e 731,24 (M)+.
Стадия F: 5-((S)-3-(трет-бутокси)-2-((((Z)-1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-2-((R)-1-(трет-бутоксикарбонил)пирролидин-3-ил)-1-метил-2H-индазол-1-ий К раствору 5-((S)-3-(трет-бутокси)-2-(((Z)-((2-((трет-бутоксикарбонил)амино)-тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)-2-((R)-1-(трет-бутоксикарбонил)-пирролидин-3-ил)-1-метил-2H-индазол-1-ия (444 мг, 0,607 ммоль) в DMF (4047 мкл) при комнатной температуре добавляли DCC (376 мг, 1,82 ммоль) и HOBT (279 мг, 1,82 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 0,5 ч, затем добавляли (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфат (383 мг, 1,82 ммоль) и NaHCO3 (510 мг, 6,07 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Затем смесь фильтровали и фильтрат очищали с помощью MPLC с обращенной фазой (C18, колонка 100 г) с использованием 10-100% ацетонитрил (0,05% TFA)/вода (0,05% TFA) с получением указанного в заголовке соединения. LC/MS: m/e 923,22 (M+H)+.
Стадия G: (S)-3-((Z)-2-(2-Аминотиазол-4-ил)-2-(((S)-1-карбокси-2-((1-метил-2-((R)-пирролидин-3-ил)-2H-индазол-1-ий-5-ил)окси)этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат К раствору 5-((S)-3-(трет-бутокси)-2-(((Z)-(1-(2-((трет-бутокси-карбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-2-((R)-1-(трет-бутоксикарбонил)-пирролидин-3-ил)-1-метил-2H-индазол-1-ия (70 мг, 0,076 ммоль) в CH2Cl2 (2 мл) добавляли TFA (4 мл, 51,9 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 40 мин, затем концентрировали. дважды промывали Et2O и сушили под вакуумом. Полученный неочищенный продукт очищали с помощью RP-HPLC (Gilson) (Sunfire, prep C18, OBD, 10 мкм, 30×150 мм, 5-25% MeCN/H2O с 0,05%TFA, 12 мин) с получением указанного в заголовке соединения. LC/MS: m/e 667,06 (M+H)+.1H ЯМР (500 МГц, D2O): δ (ppm) 0,86 (с, 3 H); 1,30 (с, 3 H); 2,67-2,59 (м, 1 H); 2,83 (дт, J=14,7, 7,4 Гц, 1 H); 3,68-3,54 (м, 2 H); 3,79 (дд, J=13,7, 4,9 Гц, 1 H); 4,07 (дд, J=13,7, 7,9 Гц, 1 H); 4,17 (с, 3 H); 4,40 (дд, J=11,2, 5,8 Гц, 1 H); 4,48-4,46 (м, 1 H); 4,64 (с, 1H); 5,07 (дд, J=5,6, 2,1 Гц, 1 H); 5,76 (т, J=6,7 Гц, 1 H); 7,02 (с, 1 H); 7,24 (д, J=2,3 Гц, 1 H); 7,44 (дд, J=9,5, 2,3 Гц, 1 H); 7,61 (д, J=9,5 Гц, 1 H); 8,88 (с, 1 H).
Таблица 6. Соединения в примерах 57-63 получали в соответствии с процедурой примера 56 с использованием соответствующих исходных материалов и реагентов.
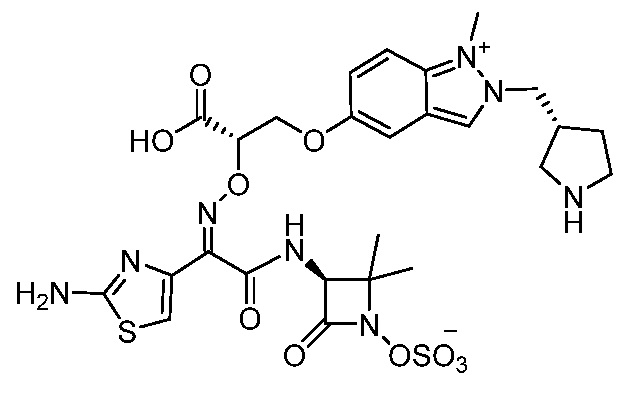
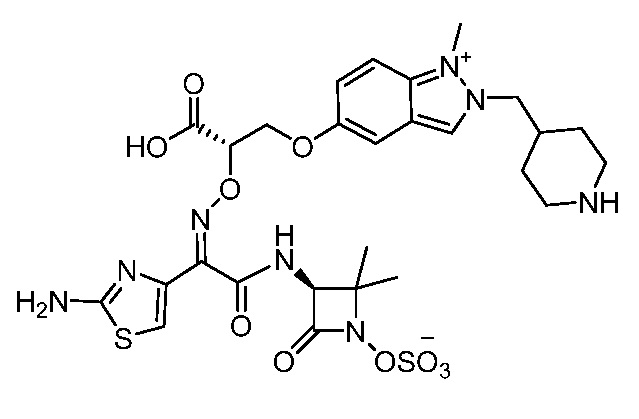
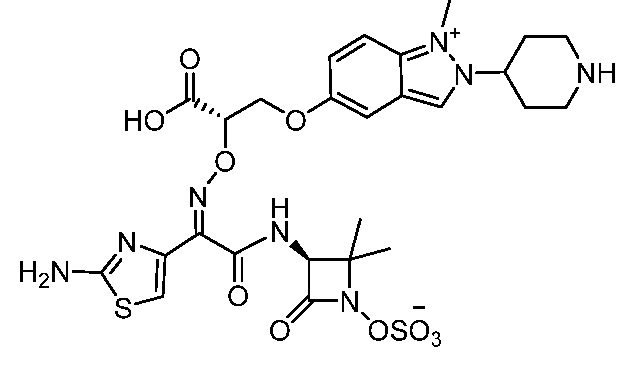
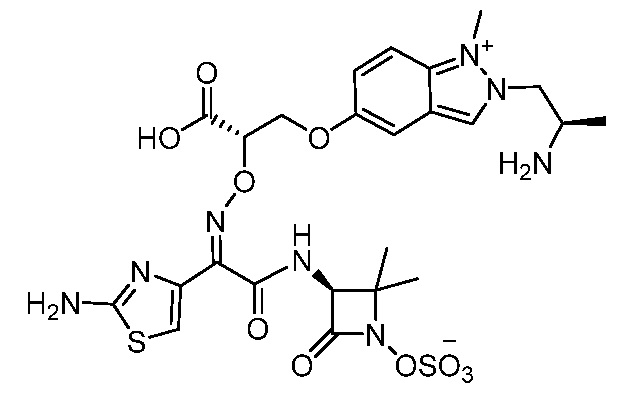



ПРИМЕР 64
(S)-3-((Z)-2-(2-Аминотиазол-4-ил)-2-(((S)-1-карбокси-2-((2-((3R,4S)-4-гидроксипирролидин-3-ил)-1-метил-2H-индазол-1-ий-5-ил)окси)этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
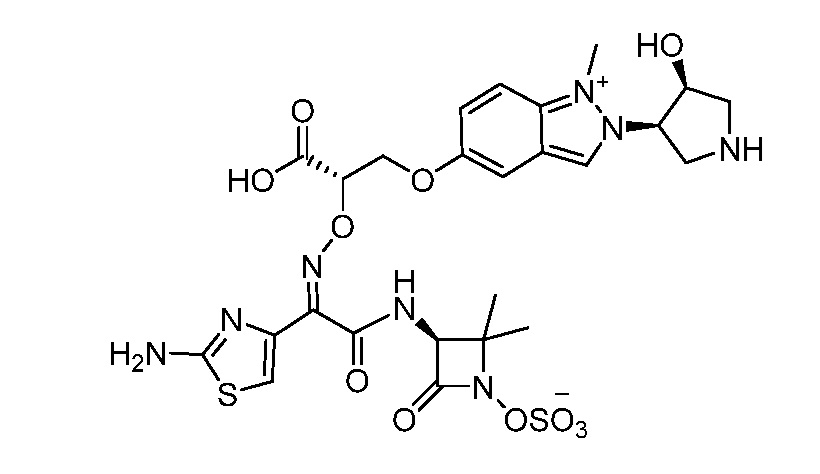
Стадия A: трет-бутил (3R,4S)-3-амино-4-((трет-бутилдиметилсилил)окси)пирролидин-1-карбоксилат К раствору (3R,4S)-трет-бутил 3-амино-4-гидроксипирролидин-1-карбоксилата (250 мг, 1,24 ммоль) и имидазола (210 мг, 3,09 ммоль) в DCM (12 мл) добавляли TBDMS-Cl (224 мг, 1,48 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем разделяли между EtOAc и водой. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали, концентрировали и очищали на колонке с силикагелем (40 г) с использованием 10-10% MeOH/DCM с получением указанного в заголовке соединения.
Стадия B: трет-бутил (3R,4S)-3-(5-((R)-3-(трет-бутокси)-2-гидрокси-3-оксопропокси)-2H-индазол-2-ил)-4-((трет-бутилдиметилсилил)окси)пирролидин-1-карбоксилат Указанное в заголовке соединение получали в соответствии с процедурой, описанной в примере 56, с использованием трет-бутил (3R,4S)-3-амино-4-((трет-бутилдиметилсилил)окси)пирролидин-1-карбоксилата. LC/MS: m/e 578,23 (M+H)+.
Стадия C: трет-бутил (3R, 4S)-3-(5-((S)-3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2H-индазол-2-ил)-4-((трет-бутилдиметилсилил)окси)пирролидин-1-карбоксилат Указанное в заголовке соединение получали в соответствии с процедурой, описанной в примере 56, с использованием трет-бутил (3R,4S)-3-(5-((R)-3-(трет-бутокси)-2-гидрокси-3-оксопропокси)-2H-индазол-2-ил)-4-((трет-бутилдиметилсилил)окси)пирролидин-1-карбоксилата. LC/MS: m/e 723,24 (M+H)+.
Стадия D: 5-((S)-3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2-((3R,4S)-1-(трет-бутоксикарбонил)-4-((трет-бутилдиметилсилил)окси)пирролидин-3-ил)-1-метил-2H-индазол-1-ий Указанное в заголовке соединение получали в соответствии с процедурой, описанной в примере 56, с использованием трет-бутил (3R,4S)-3-(5-((S)-3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2H-индазол-2-ил)-4-((трет-бутилдиметилсилил)окси)пирролидин-1-карбоксилата. LC/MS: m/e 737,22 (M)+.
Стадия E: 5-((S)-2-(Аминоокси)-3-(трет-бутокси)-3-оксопропокси)-2-((3R,4S)-1-(трет-бутоксикарбонил)-4-((трет-бутилдиметилсилил)окси)пирролидин-3-ил)-1-метил-2H-индазол-1-ий Указанное в заголовке соединение получали в соответствии с процедурой, описанной в примере 56, с использованием 5-((S)-3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2-((3R,4S)-1-(трет-бутоксикарбонил)-4-((трет-бутилдиметилсилил)окси)пирролидин-3-ил)-1-метил-2H-индазол-1-ия. LC/MS: m/e 607,28 (M)+.
Стадия F: 5-((S)-3-(трет-бутокси)-2-((((Z)-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)-2-((3R,4S)-1-(трет-бутоксикарбонил)-4-((трет-бутилдиметилсилил)окси)пирролидин-3-ил)-1-метил-2H-индазол-1-ий Указанное в заголовке соединение получали в соответствии с процедурой, описанной в примере 56, с использованием 5-((S)-2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)-2-((3R,4S)-1-(трет-бутоксикарбонил)-4-((трет-бутилдиметилсилил)окси)пирролидин-3-ил)-1-метил-2H-индазол-1-ия. LC/MS: m/e 861,47 (M)+.
Стадия G: 5-((S)-3-(трет-бутокси)-2-((((Z)-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)-2-((3R,4S)-1-(трет-бутоксикарбонил)-4-гидроксипирролидин-3-ил)-1-метил-2H-индазол-1-ий К раствору 5-((S)-3-(трет-бутокси)-2-(((Z)-((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)-2-((3R,4S)-1-(трет-бутоксикарбонил)-4-((трет-бутилдиметилсилил)окси)пирролидин-3-ил)-1-метил-2H-индазол-1-ия (274 мг, 0,318 ммоль) в THF (3 мл) при комнатной температуре добавляли TBAF (382 мкл, 0,382 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, затем концентрировали досуха и очищали с помощью колонки C18 (150 г, 10-100% MeCN (0,05%TFA)/H2O (0,05%TFA)) с получением указанного в заголовке соединения. LC/MS: m/e 747,22 (M)+.
Стадия H: 5-((S)-3-(трет-бутокси)-2-((((Z)-1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-2-((3R,4S)-1-(трет-бутоксикарбонил)-4-гидроксипирролидин-3-ил)-1-метил-2H-индазол-1-ий Указанное в заголовке соединение получали в соответствии с процедурой, описанной в примере 56, с использованием 5-((S)-3-(трет-бутокси)-2-((((Z)-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)-2-((3R,4S)-1-(трет-бутоксикарбонил)-4-гидроксипирролидин-3-ил)-1-метил-2H-индазол-1-ия. LC/MS: m/e 939,31 (M+H)+.
Стадия I: (S)-3-((Z)-2-(2-Аминотиазол-4-ил)-2-(((S)-1-карбокси-2-((2-((3R,4S)-4-гидрокси-пирролидин-3-ил)-1-метил-2H-индазол-1-ий-5-ил)окси)этокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат Указанное в заголовке соединение получали в соответствии с процедурой, описанной в примере 56, с использованием 5-((S)-3-(трет-бутокси)-2-((((Z)-1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-2-((3R,4S)-1-(трет-бутоксикарбонил)-4-гидроксипирролидин-3-ил)-1-метил-2H-индазол-1-ия.1H ЯМР (500 МГц, D2O): δ (ppm) 0,92 (с, 3 H); 1,37 (с, 3 H); 3,68 (м, 2 H); 4,06 (д, J=10,8 Гц, 1 H); 4,15 (д, J=10,4 Гц, 1 H); 4,26 (с, 3 H); 4,50 (м, 1 H); 4,55 (м, 1 H); 4,82 (с, 1 H); 5,15 (с, 1 H); 5,80 (м, 1 H); 7,10 (с, 1 H); 7,33 (с, 1 H); 7,53 (д, J=9,4 Гц, 1 H); 7,70 (д, J=9,5 Гц, 1 H); 9,02 (с, 1 H). LC/MS: m/e 683,67 (M+H)+.
Таблица 7. Соединения в примерах 65 и 66 получали в соответствии с процедурой примера 64 с использованием соответствующих исходных материалов и реагентов.


Пример 67
(3S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((1S)-2-((2-(азетидин-3-илметил)-1-(2,3-дигидроксипропил)-2H-индазол-1-ий-5-ил)окси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат

Стадия A: (S)-1-аллил-5-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-2H-индазол-1-ий 3-иодпроп-1-ен (569 мкл, 6,22 ммоль) добавляли к раствору (S)-трет-бутил 3-((5-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2H-индазол-2-ил)метил)азетидин-1-карбоксилата (737 мг, 1,244 ммоль) в CH3CN при комнатной температуре. Реакционную смесь перемешивали при 60°C в течение 72 ч, затем охлаждали до комнатной температуры и концентрировали. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (80 г), элюируя MeOH/DCM (0-15%), повторно очищали с помощью колонки С18 (150 г, 0-100% MeCN/H2O (0,05%TFA) с получением указанного в заголовке соединения. LC/MS: m/e 633,45 (M)+.
Стадия B: 5-((S)-3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2-((1-(трет-бутокси-карбонил)азетидин-3-ил)метил)-1-(2,3-дигидроксипропил)-2H-индазол-1-ий К раствору (S)-1-аллил-5-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)-азетидин-3-ил)метил)-2H-индазол-1-ия (294 мг, 0,464 ммоль) в CH2Cl2 (9 мл) при комнатной температуре добавляли 4-метилморфолин N-оксид (163 мг, 1,392 ммоль) и тетроксид осмия (2,36 мг, 9,28 мкмоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем гасили насыщенным Na2S2O3, экстрагировали IPA/CHCl3(1:3) (3x), сушили над MgSO4, фильтровали, и концентрировали. Полученный остаток очищали с помощью колонки С18 (150 г, 0-100% MeCN/H2O w/ 0,05% TFA) с получением указанного в заголовке соединения.
Стадия C: 5-((S)-2-(Аминоокси)-3-(трет-бутокси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)-азетидин-3-ил)метил)-1-(2,3-дигидроксипропил)-2H-индазол-1-ий Указанное в заголовке соединение получали в соответствии с процедурой, описанной в примере 56, с использованием 5-((S)-3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1-(2,3-дигидроксипропил)-2H-индазол-1-ия. LC/MS: m/e 537,67 (M)+.
Стадия D: 5-((S)-3-(трет-бутокси)-2-((((Z)-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-(карбокси)метилен)-амино)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1-(2,3-дигидроксипропил)-2H-индазол-1-ий Указанное в заголовке соединение получали в соответствии с процедурой, описанной в примере 56, с использованием 5-((S)-2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1-(2,3-дигидроксипропил)-2H-индазол-1-ия. LC/MS: m/e 791,00 (M)+.
Стадия E: 5-((S)-3-(трет-бутокси)-2-((((Z)-1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1-(2,3-дигидроксипропил)-2H-индазол-1-ий Указанное в заголовке соединение получали в соответствии с процедурой, описанной в примере 56, с использованием 5-((S)-3-(трет-бутокси)-2-((((Z)-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1-(2,3-дигидроксипропил)-2H-индазол-1-ия. LC/MS: m/e 983,92 (M+H)+.
Стадия F: (3S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((1S)-2-((2-(азетидин-3-илметил)-1-(2,3-дигидроксипропил)-2H-индазол-1-ий-5-ил)окси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат Вышеуказанное соединение получали в соответствии с процедурой, описанной в примере 56, с использованием 5-((S)-3-(трет-бутокси)-2-((((Z)-1-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-оксопропокси)-2-((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)-1-(2,3-дигидроксипропил)-2H-индазол-1-ия.1H ЯМР (500 МГц, D2O): δ (ppm) 0,89 (д, J=7,3 Гц, 3 H); 1,30 (с, 3 H); 3,58-3,55 (м, 3 H); 4,03 (м, 3 H); 4,18 (м, 2 H); 4,45-4,40 (м, 2 H); 4,64-4,67 (м, 2H): 4,75-4,79 (м, 1H); 5,04-5,00 (м, 3 H); 7,01 (с, 1 H); 7,24 (с, 1 H); 7,44 (д, J=9,6 Гц, 1 H); 7,64 (д, J=9,5 Гц, 1 H); 8,72 (с, 1 H). LC/MS: m/e 727,22 (M+H)+.
Пример 68
(S)-3-((Z)-2-(2-Аминотиазол-4-ил)-2-(((S)-2-((3-((азетидин-3-илметил)амино)-2-метилизохинолин-2-ий-7-ил)окси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфат
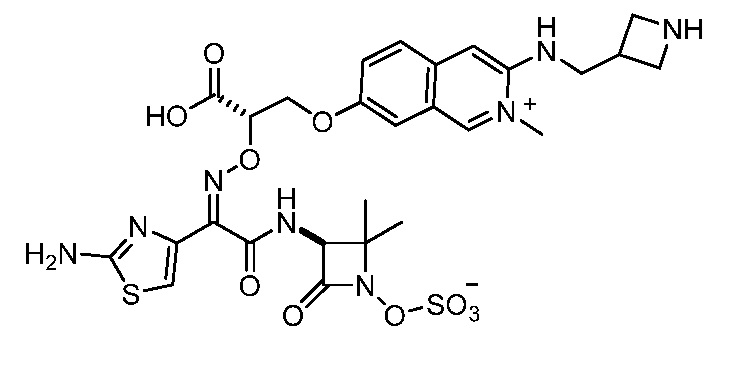
Стадия A. Получение трет-бутил (R)-3-(((7-(3-(трет-бутокси)-2-((трет-бутилдиметилсилил)окси)-3-оксопропокси)изохинолин-3-ил)амино)метил)азетидин-1-карбоксилат К раствору (R)-трет-бутил 2-((трет-бутилдиметилсилил)окси)-3-((3-хлоризохинолин-7-ил)окси)пропаноата (I-4, 300 мг, 0,685 ммоль) в диоксане (5 мл) добавляли трет-бутил 3-(аминометил)азетидин-1-карбоксилат (191 мг, 1,03 ммоль), предкатализатор RuPhos второго поколения (106 мг, 0,137 ммоль) и Cs2CO3 (558 мг, 1,71 ммоль). После дегазации и заполнения N2, реакционную смесь нагревали при 70°C в течение ночи. Затем реакционную смесь разбавляли EtOAc, промывали NH4Cl, водой и насыщенным солевым раствором. Растворитель удаляли, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (40 г), элюируя смесью EtOAc/гексан (40%) с получением указанного в заголовке соединения. LC-MS [M+H]+: m/z 589,52.
Стадия B. Получение трет-бутил (R)-3-(((7-(3-(трет-бутокси)-2-гидрокси-3-оксопропокси)изохинолин-3-ил)амино)метил)азетидин-1-карбоксилата К раствору (R)-трет-бутил 3-(((7-(3-(трет-бутокси)-2-((трет-бутилдиметилсилил)окси)-3-оксопропокси)изохинолин-3-ил)амино)метил)азетидин-1-карбоксилата (0,13 г, 0,221 ммоль) в THF (3 мл) добавляли TBAF в THF (0,221 мл, 0,221 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, затем растворитель удаляли. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (12 г), элюируя смесью EtOAc/гексан (30%, 15cv) с получением указанного в заголовке соединения. LC-MS [M+H]: m/z 474,42.
Стадия C. Получение трет-бутил (S)-3-(((7-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)изохинолин-3-ил)амино)метил)азетидин-1-карбоксилат Раствор (R)-трет-бутил 3-(((7-(3-(трет-бутокси)-2-гидрокси-3-оксопропокси)изохинолин-3-ил)амино)метил)азетидин-1-карбоксилата (0,12 г, 0,253 ммоль) в THF (1 мл) добавляли 2-гидроксиизоиндолин-1,3-дион (0,050 г, 0,304 ммоль), и трифенилфосфин (0,080 г, 0,304 ммоль), затем DIAD (0,059 мл, 0,304 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение ночи, затем растворитель удаляли. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (24 г), элюируя смесью EtOAc/гексан (70% 15cv) с получением указанного в заголовке соединения. LC-MS [M+H]: m/z 619,36
Стадия D. Получение (S)-7-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-3-(((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)амино)-2-метилизохинолин-2-ий йодида К раствору (S)-трет-бутил 3-(((7-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)изохинолин-3-ил)амино)метил)азетидин-1-карбоксилата (80 мг, 0,129 ммоль) в ACN (0,5 мл) добавляли MeI (0,081 мл, 1,29 ммоль). Реакционную смесь нагревали при 75°C в течение ночи, затем растворитель удаляли с получением указанного в заголовке соединения. LC-MS [M+H]: m/z 633,44
Стадия E. Получение (S)-7-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)-3-(((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)амино)-2-метилизохинолин-2-ий йодида К раствору (S)-7-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксопропокси)-3-(((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)амино)-2-метилизохинолин-2-ий йодида в ACN (1 мл) добавляли гидразин (3,96 мкл, 0,126 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и затем концентрировали. К полученному остатку добавляли DCM (3 мл), и смесь перемешивали при комнатной температуре в течение 1 часа. Затем твердое вещество отфильтровывали и растворитель удаляли с получением указанного в заголовке соединения. LC-MS [M+H]: m/z 503,28
Стадия F. Получение (S,Z)-7-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)амино)окси)-3-оксопропокси)-3-(((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)амино)-2-метилизохинолин-2-ий,йодида Раствор (S)-7-(2-(аминоокси)-3-(трет-бутокси)-3-оксопропокси)-3-(((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)амино)-2-метилизохинолин-2-ий, йодида (60 мг, 0,095 ммоль) и 2-(2-((трет-бутоксикарбонил)-амино)тиазол-4-ил)-2-оксоуксусной кислоты (33 мг, 0,12 ммоль) в EtOH (1,5 мл) и CH2ClCH2Cl (0,5 мл) перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь концентрировали с получением указанного в заголовке соединения. LC-MS [M+H]: m/z 757,49
Стадия G. Получение (S)-3-((Z)-2-((((S)-1-(трет-бутокси)-3-((3-(((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)амино)-2-метилизохинолин-2-ий-7-ил)окси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата К раствору (S,Z)-7-(3-(трет-бутокси)-2-((((2-((трет-бутоксикарбонил)амино)тиазол-4-ил)(карбокси)метилен)-амино)окси)-3-оксопропокси)-3-(((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)амино)-2-метил-изохинолин-2-ия (84 мг, 0,095 ммоль) в DMF (2 мл) добавляли DCC (58,8 мг, 0,285 ммоль), и HOBt (43,6 мг, 0,285 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин, затем добавляли (S)-3-амино-2,2-диметил-4-оксоазетидин-1-ил гидросульфат (49,9 мг, 0,238 ммоль) и бикарбонат натрия (39,9 мг, 0,475 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем твердое вещество отфильтровывали. Фильтрат очищали с помощью RP (C-18 колонка) (130 г), элюируя 20-100% ACN/вода, содержащая 0,05% TFA (10 CV), с получением указанного в заголовке соединения. LC-MS [M+H]: m/z 949,83
Стадия H: Получение (S)-3-((Z)-2-(2-аминотиазол-4-ил)-2-(((S)-2-((3-((азетидин-3-илметил)амино)-2-метилизохинолин-2-ий-7-ил)окси)-1-карбоксиэтокси)имино)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата К раствору (S)-3-((Z)-2-((((S)-1-(трет-бутокси)-3-((3-(((1-(трет-бутоксикарбонил)азетидин-3-ил)метил)амино)-2-метилизохинолин-2-ий-7-ил)окси)-1-оксопропан-2-ил)окси)имино)-2-(2-((трет-бутоксикарбонил)амино)тиазол-4-ил)ацетамидо)-2,2-диметил-4-оксоазетидин-1-ил сульфата (30 мг, 0,032 ммоль) в CH2Cl2 (1 мл) добавляли TFA (2 мл, 51,9 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 40 мин, затем растворитель удаляли под вакуумом. Полученный остаток дважды промывали Et2O, затем растворяли в DMSO (0,5 мл) и очищали с помощью RP-HPLC (Gilson) (колонка C-18), элюируя 2-40% ACN/вода, содержащая 0,05% TFA (12 мин), с получением указанного в заголовке соединения в виде соли TFA. LC-MS [M+H]: m/z 692,23.1Н ЯМР (500 МГц, CDCl3) δ 8,84 (1H, с), 7,73 (1H, д), 7,45 (1H, д), 7,35 (1H, с), 7,26 (1H, с), 7,09 (1H, с), 5,18 (1H, с), 4,62-4,54 (4H, м), 4,21 (2H, т), 4,05 (3H, с), 3,98(2H, т), 3,71 (2H, д), 3,39 (1H, м), 3,15 (1H, м), 1,37 (3H, с), 0,94 (3H, с).
Пример 69
(S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-((2-((1-(азетидин-3-илметил)пиперидин-4-ил)амино)хинолин-6-ил)окси)пропановая кислота
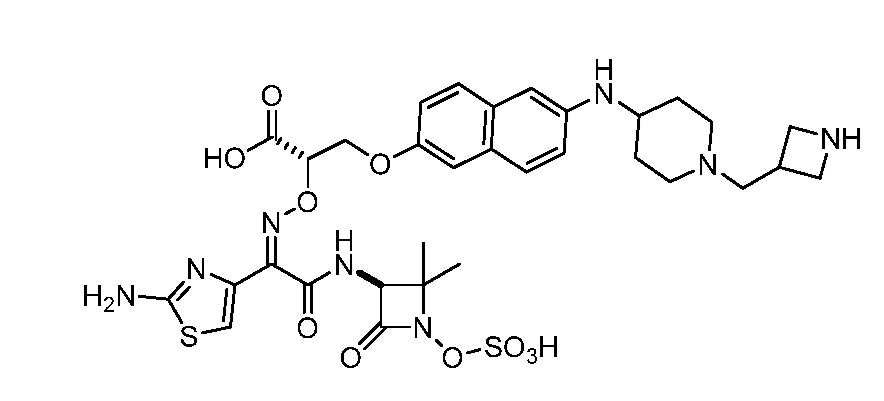
Стадия A: (S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)-азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-((2-((1-((1-(трет-бутоксикарбонил)-азетидин-3-ил)метил)пиперидин-4-ил)амино)хинолин-6-ил)окси)пропановая кислота К раствору (S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-((2-(пиперидин-4-иламино)хинолин-6-ил)окси)-пропановой кислоты (Пример 3, 15 мг, 0,019 ммоль) в DMSO (0,5 мл) добавляли трет-бутил 3-формилазетидин-1-карбоксилат (6,89 мг, 0,037 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Затем добавляли триацетоксиборгидрид натрия (7,88 мг, 0,037 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь непосредственно загружали на RP-HPLC (колонка Gilson C-18 (элюируя 20-100% ACN/вода, содержащая 0,05% TFA (12 мин)) с получением указанного в заголовке соединения. LC-MS [M+H]: m/z 862,79.
Стадия B: (S)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)-азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)-3-((2-((1-(азетидин-3-илметил)пиперидин-4-ил)амино)хинолин-6-ил)окси)пропановая кислота К раствору (S)-2-((((Z)-1-(2-амино-тиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)-амино)окси)-3-((2-((1-((1-(трет-бутоксикарбонил)-азетидин-3-ил)метил)-пиперидин-4-ил)амино)-хинолин-6-ил)окси)пропановой кислоты (5 мг, 5,12 мкмоль) в CH2Cl2 (0,25 мл) добавляли TFA (0,5 мл, 6,49 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 0,5 ч, затем растворитель удаляли. Полученный остаток дважды промывали Et2O и сушили под вакуумом, и затем очищали с помощью RP-HPLC (Gilson) (колонка C-18), элюируя 0-40% ACN/вода, содержащая 0,05% TFA (12 мин) с получением указанного в заголовке соединения в виде соли TFA. LC-MS [M+H]: m/z 760,57.1Н ЯМР (500 МГц, CDCl3) δ 8,08 (1H, с), 7,68 (1H, д), 7,32 (1H, д), 7,25 (1H, с), 6,99 (1H, с), 6,94 (1H, с), 5,04 (1H, д), 4,64-4,42 (4H, м), 4,21 (2H, т), 4,12 (1H, м), 4,02 (2H, т), 3,58 (2H, д), 3,49 (3H, м), 3,16 (2H, м), 2,33 (2H, м), 1,85 (2H, м), 1,31 (3H, с), 0,91 (3H, с).
Таблица 8. Соединения в примерах 70 и 71 получали в соответствии с процедурой примера 69, начиная с примера 9, и с использованием соответствующих реагентов

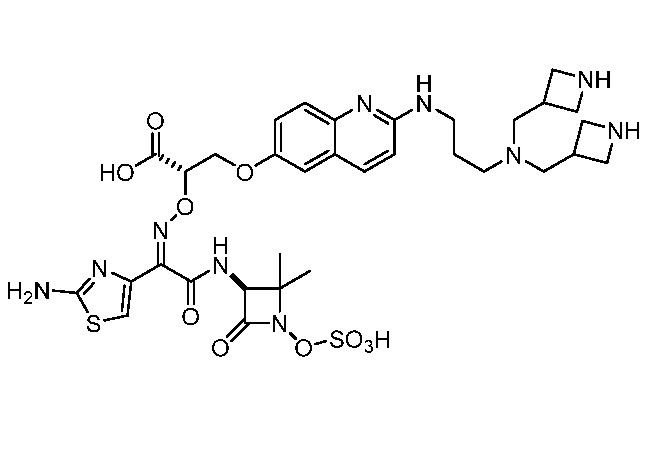
Пример 72
(S)-3-((2-((3-аминопропил)(азетидин-3-илметил)амино)хинолин-6-ил)окси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота

Стадия A. Получение трет-бутил (R)-3-(((6-(3-(трет-бутокси)-2-((трет-бутилдиметилсилил)окси)-3-оксо-пропокси)хинолин-2-ил)(3-((трет-бутоксикарбонил)амино)пропил)амино)метил)-азетидин-1-карбоксилат К раствору (R)-трет-бутил 2-((трет-бутилдиметилсилил)окси)-3-((2-хлорхинолин-6-ил)окси)пропаноата (I-4, 1 г, 2,28 ммоль) в диоксане (18 мл) добавляли трет-бутил 3-(((3-((трет-бутоксикарбонил)амино)пропил)-амино)метил)азетидин-1-карбоксилат (1,18 г, 3,42 ммоль), предкатализатор RuPhos второго поколения (0,266 г, 0,342 ммоль) и Cs2CO3 (1,49 г, 4,57 ммоль). Реакционный флакон дегазировали и заполняли N2. Реакционную смесь нагревали при 75°C в течение ночи, затем твердое вещество отфильтровывали и растворитель удаляли. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле Redi 40g gold, элюируя смесью EtOAc/гексан 30% с получением указанного в заголовке соединения. LC-MS [M+H]: m/z 745,87.
Стадия B. Получение трет-бутил (S)-3-(((6-(3-(трет-бутокси)-2-гидрокси-3-оксопропокси)-хинолин-2-ил)(3-((трет-бутоксикарбонил)амино)пропил)амино)метил)азетидин-1-карбоксилата К раствору (R)-трет-бутил 3-(((6-(3-(трет-бутокси)-2-((трет-бутилдиметилсилил)окси)-3-оксопропокси)хинолин-2-ил)(3-((трет-бутоксикарбонил)амино)пропил)амино)метил)азетидин-1-карбоксилату (0,3 г, 0,403 ммоль) в THF (5 мл) добавляли TBAF в THF (0,403 мл, 0,403 ммоль, 1M). Раствор перемешивали при комнатной температуре в течение 2 ч, затем растворитель удаляли. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (Redi 12g gold), элюируя смесью EtOAc/гексан (0-70%, 6cv; 70%, 10cv) с получением указанного в заголовке соединения.
LC-MS [M+H]: m/z 631,59.
Стадия C. Получение трет-бутил (S)-3-(((6-(3-(трет-бутокси)-2-((1,3-диоксоизоиндолин-2-ил)окси)-3-оксо-пропокси)хинолин-2-ил)(3-((трет-бутоксикарбонил)амино)пропил)амино)-метил)азетидин-1-карбоксилата К раствору (R)-трет-бутил 3-(((6-(3-(трет-бутокси)-2-гидрокси-3-оксопропокси)хинолин-2-ил)(3-((трет-бутоксикарбонил)амино)-пропил)амино)-метил)азетидин-1-карбоксилата (0,22 г, 0,349 ммоль) в THF (2 мл) добавляли 2-гидроксиизоиндолин-1,3-дион (0,068 г, 0,419 ммоль) и трифенилфосфин (0,110 г, 0,419 ммоль), затем DIAD (0,081 мл, 0,419 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение ночи и затем концентрировали. Полученный остаток очищали колоночной хроматографией на силикагеле (Redi 24g gold), элюируя смесью EtOAc/гексан (70%, 15cv) с получением указанного в заголовке соединения. LC-MS [M+H]: m/z 776,82.
Стадии D-G проводили по такой же методике, как стадии Е-Н примера 1, с получением указанного в заголовке соединения. LC-MS [M+H]+: m/z 736,89.1Н ЯМР (500 МГц, CDCl3) δ 8,22 (1H, д), 7,78 (1H, д), 7,46 (1H, д), 7,38 (1H, с), 7,31 (1H, д), 7,09 (1H, с), 5,17 (1H, д), 4,58-4,50 (2H, м), 4,19 (1H, м), 3,89-3,80 (2H, м), 3,59 (1H, м), 3,25 (2H, д), 3,10 (2H, м), 2,82 (1H, м), 2,11 (2H, м), 1,35 (3H, с), 0,86 (3H, с).
Пример 73
(S)-3-((4-амино-2-((азетидин-3-илметил)амино)хинолин-6-ил)окси)-2-((((Z)-1-(2-аминотиазол-4-ил)-2-(((S)-2,2-диметил-4-оксо-1-(сульфоокси)азетидин-3-ил)амино)-2-оксоэтилиден)амино)окси)пропановая кислота
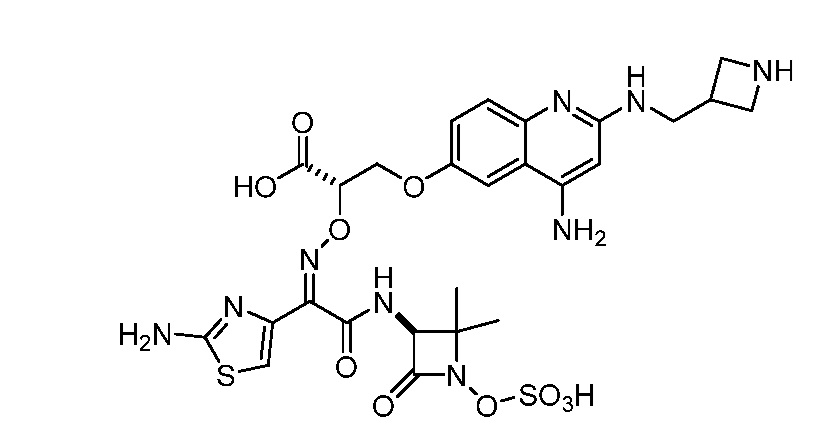
Соединение примера 73 синтезировали в соответствии с процедурой примера 72 с использованием промежуточного соединения 12 и трет-бутил 3-(аминометил)азетидин-1-карбоксилата в качестве исходных материалов. LC-MS [M+H]+: m/z 692,5.1Н ЯМР (500 МГц, CDCl3) δ 8,18 (1H, с), 7,53 (1H, с), 7,15 (1H, с), 6,85 (1H, с), 6,55 (1H, с), 5,82 (1H, с), 4,77 (1H, д), 4,73 (1H, д), 4,51-4,41(2H, м), 4,02 (2H, м), 3,82 (2H, м), 3,50 (1H, м), 3,06 (2H, м), 1,47 (3H, с), 1,30 (3H, с)
Биологический анализ
Антибиотическая активность: Определение ингибирующей концентрации роста
Концентрации соединений, необходимых для ингибирования роста различных штаммов бактерий, определяли в анализе, который оценивал рост бактерий путем измерения оптической плотности при 600 нм (ОП600). Испытуемые бактериальные штаммы включали клинические штаммы Escherichia coli, экспрессирующие NDM-1 (CLB30016), Klebsiella pneumoniae, экспрессирующие KPC-1 (CL6569), Acinetobacter baumannii, экспрессирующие TEM-1, AmpC, и Oxa-24/40 (CL6188) и Pseudomonas aeruginosa, экспрессирующие AmpC (CL5701). Все соединения тестировали в присутствии ингибитора β-лактамазы (BLi, Релебактам) в 384-луночных микропланшетах. Клинические штаммы хранили в виде замороженных материалов для однократного применения, оттаивали и разводили в 1,1X катионном бульоне Мюллера-Хинтона II для достижения примерно 2×105 КОЕ/мл. Испытуемые соединения растворяли в DMSO и разбавляли 1:50 в анализе, в результате чего конечный диапазон концентраций составлял от 100 мкМ до 0,098 мкМ. В день анализа в планшет добавляли 1 мкл испытуемого соединения, а затем 4 мкл 50 мкг/мл BLi в буфере MOPS и 45 мкл разбавленных бактерий. Планшеты центрифугировали при 1000 об/мин в течение 30 секунд, встряхивали при приблизительно 800 об/мин в течение 1 минуты, и инкубировали при 35±2°C в течение 22 часов. Концентрация BLi, используемая в анализе, составляла 4 мкг/мл. В конце инкубации определяли поглощение при 600 нм с использованием спектрофотометра. Ингибирование количественно определяли путем определения самой низкой концентрации тестируемого соединения, которая требовалась для ингибирования 95% роста бактерий. Результаты для примеров 1-73 представлены в таблице I, выраженной как концентрация соединения, которое ингибирует 95% роста бактерий (минимальная ингибирующая пороговая концентрация; MITC95).
Типичные соединения по настоящему изобретению проявляют ингибирующее рост действие. Например, были определены типичные соединения примеров 1-73 для ингибирования роста в концентрациях 100 мкМ или менее.
Таблица I. Антибактериальная активность соединений 1-73
.
Реферат
Настоящее изобретение относится к бициклическим арильным монобактамовым соединениям, перечисленным в формуле изобретения, и их фармацевтически приемлемым солям, в частности трифторуксусным солям, обладающим антибактериальной активностью. 4 н.п. ф-лы, 9 табл., 73 пр.
Формула


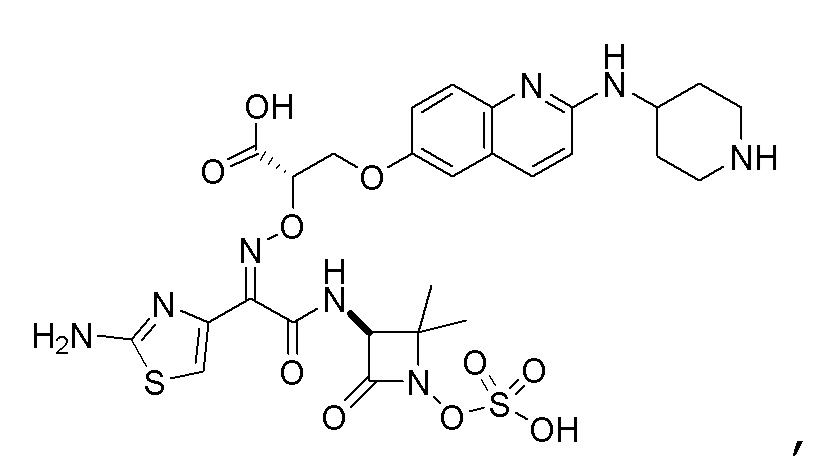

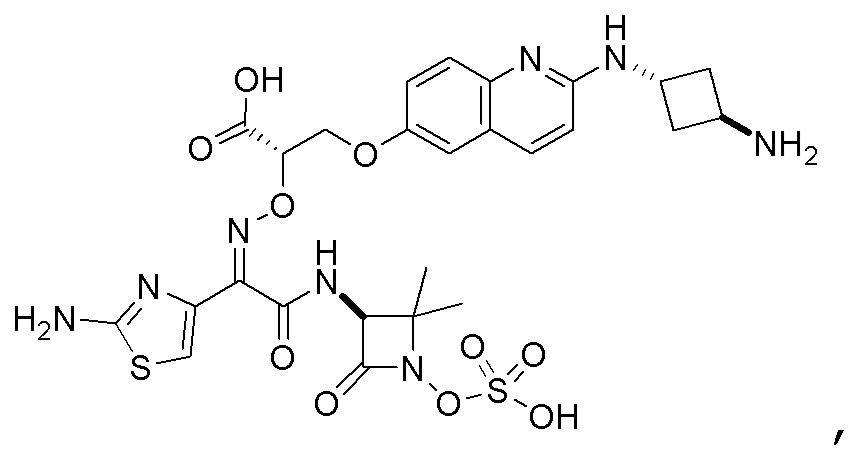
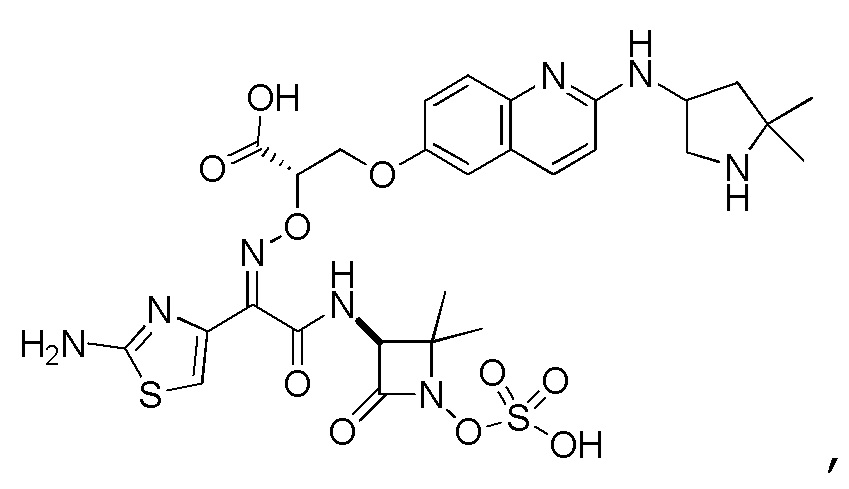
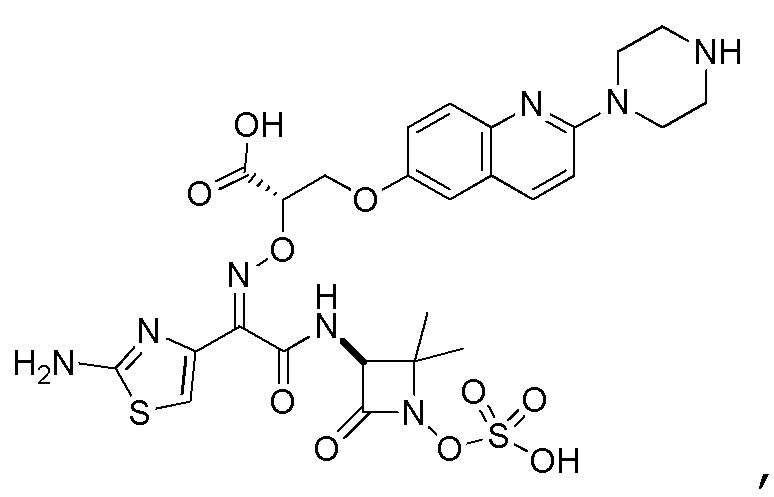

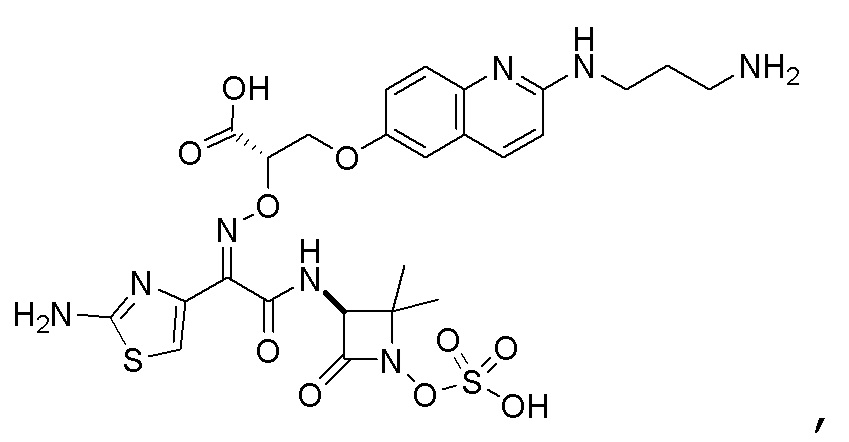
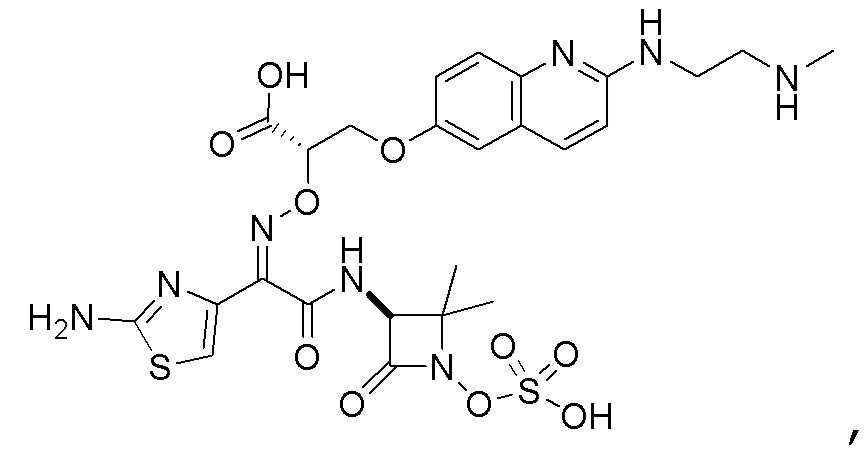


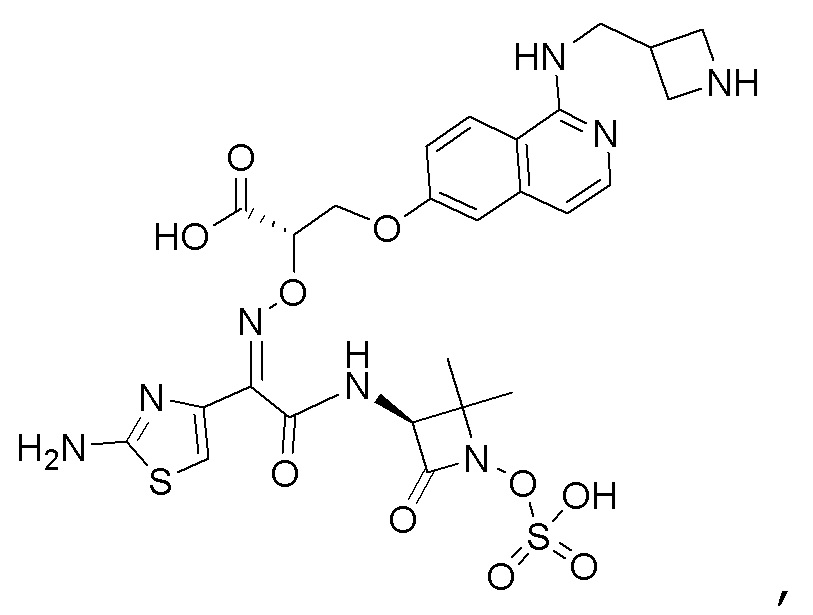

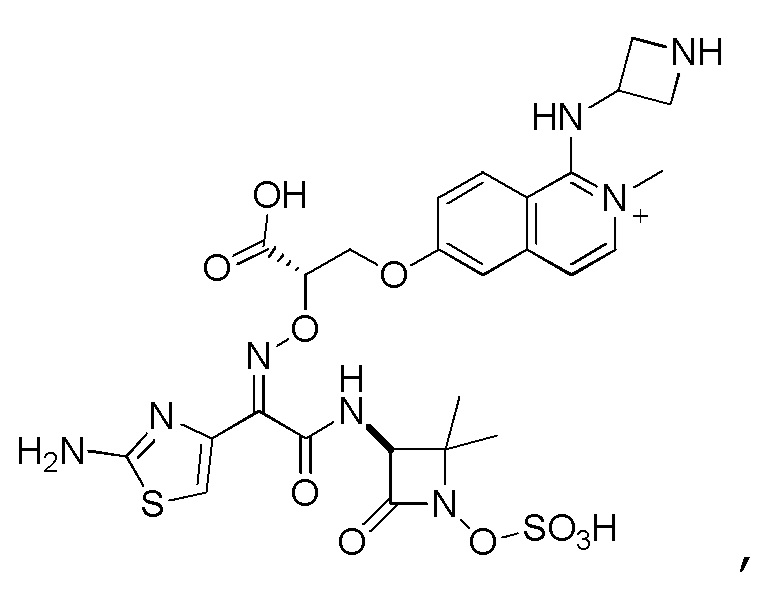
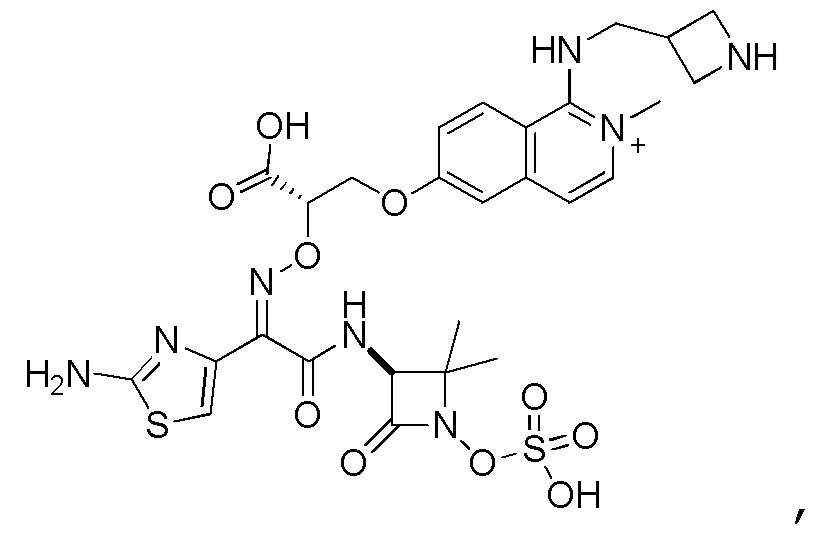

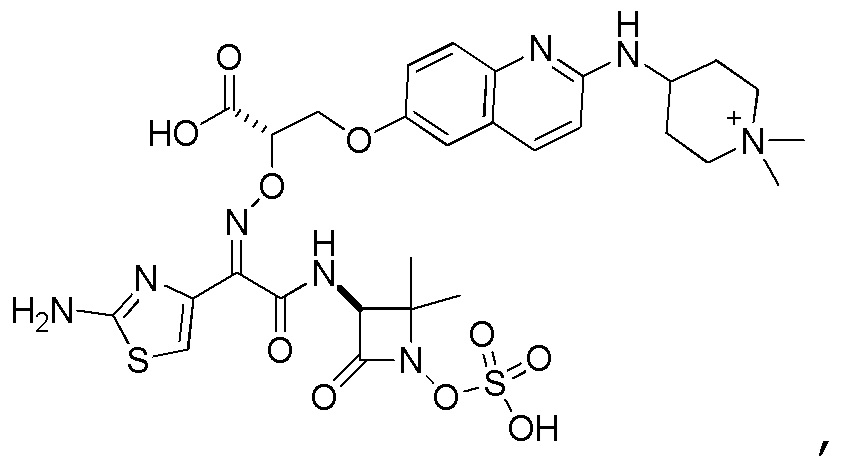

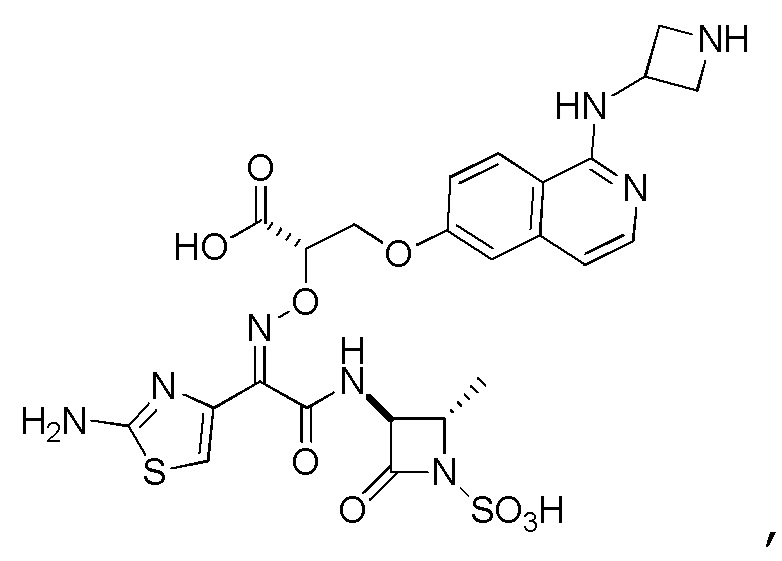
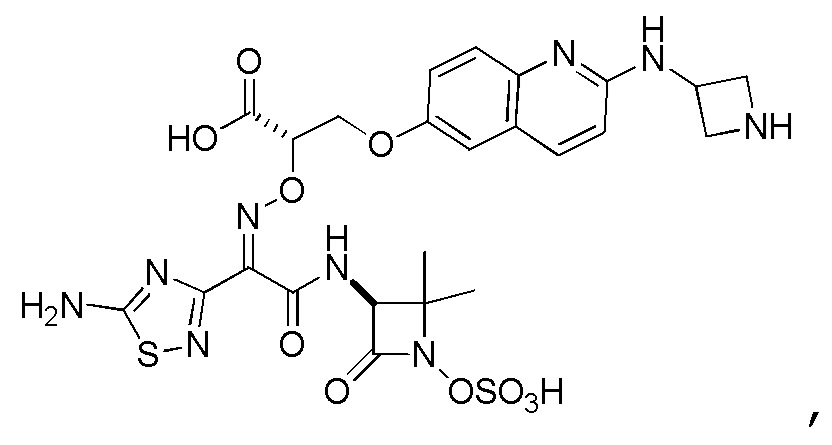


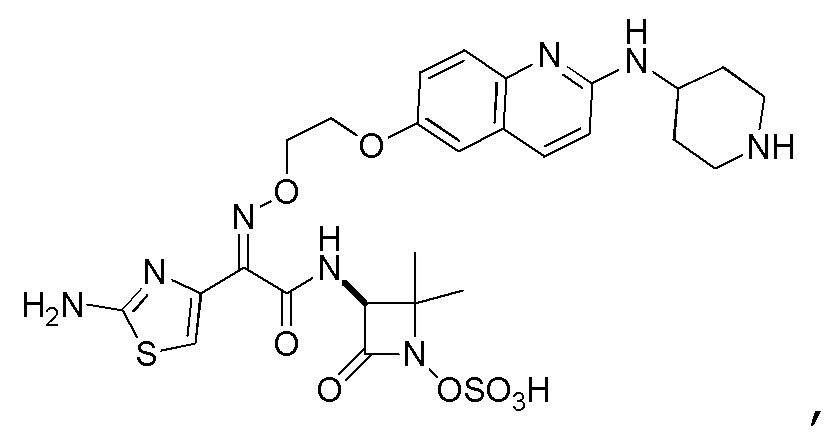


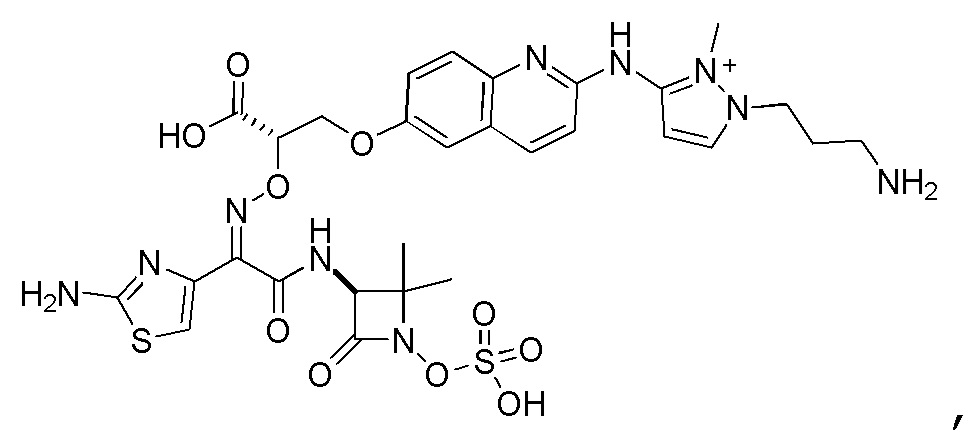

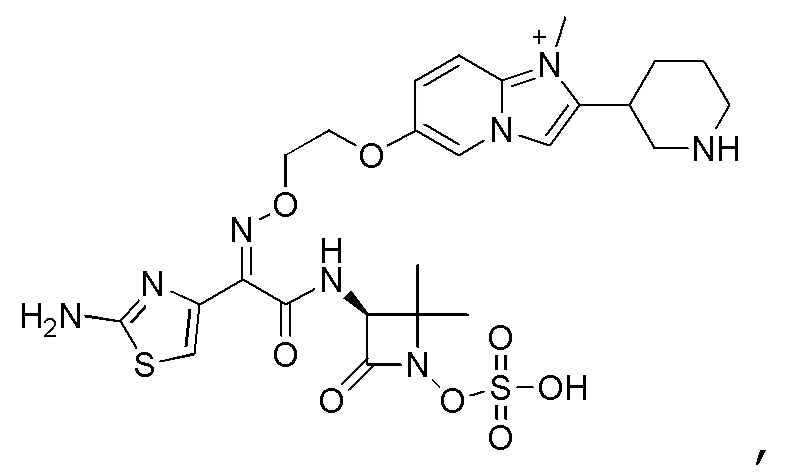

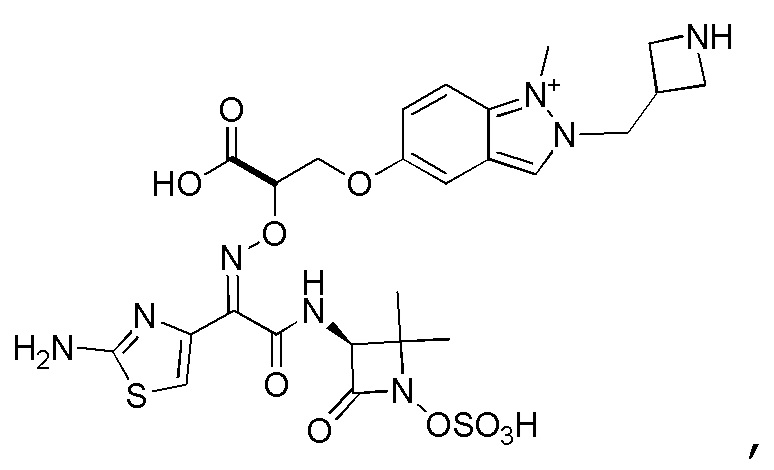
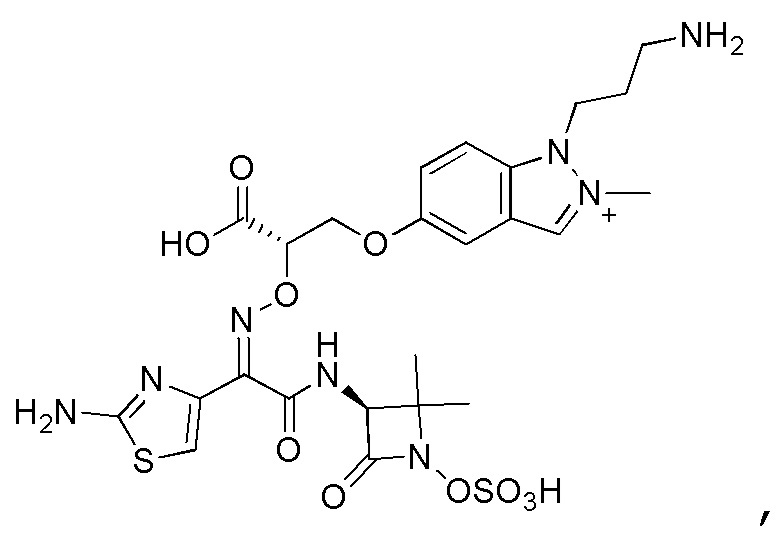
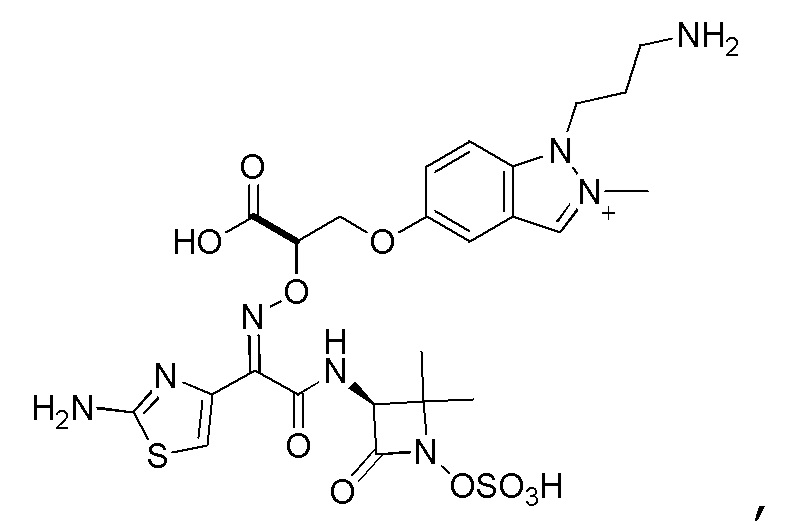




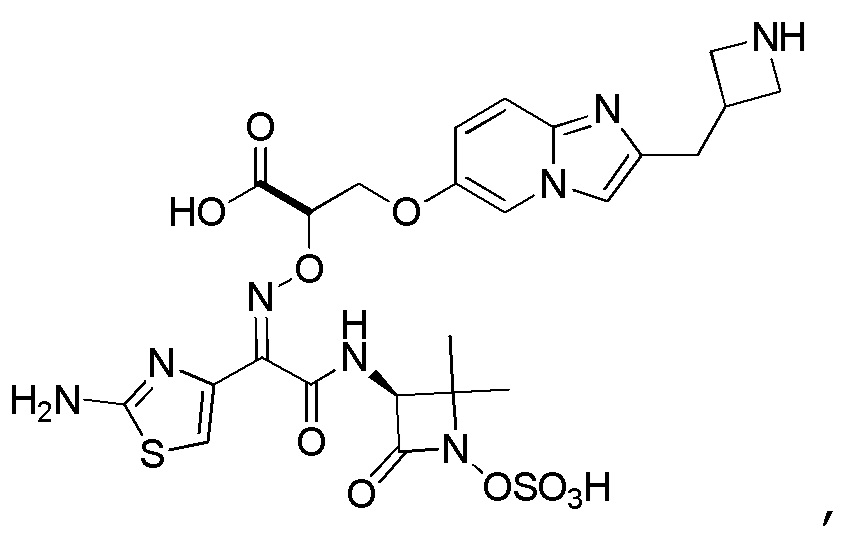

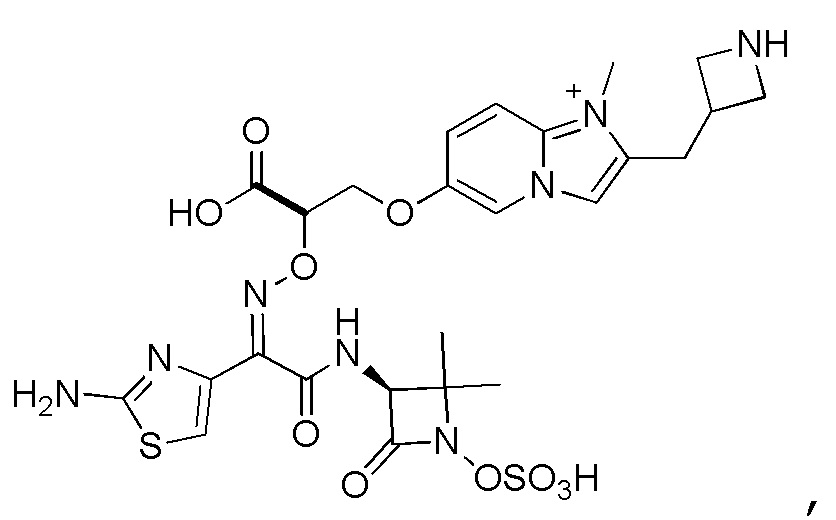

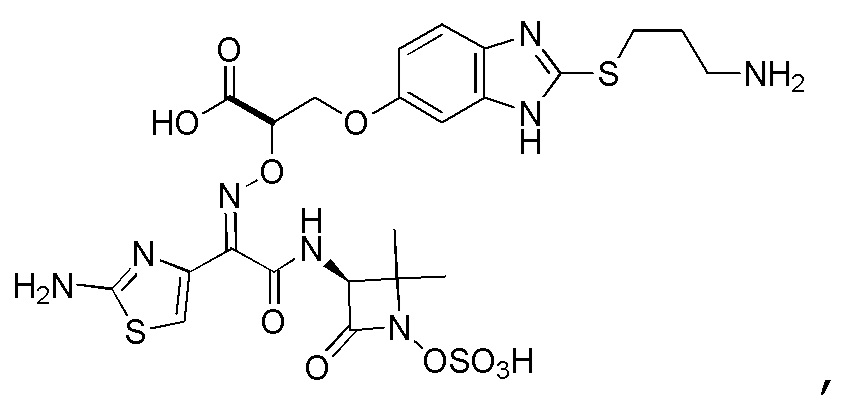



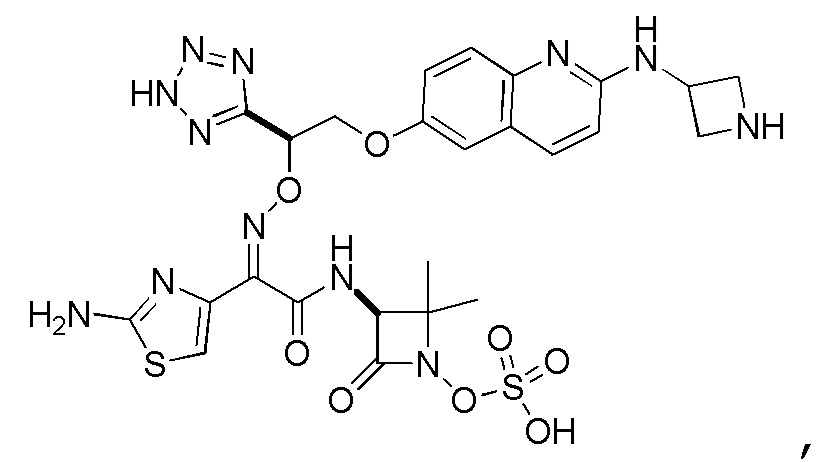
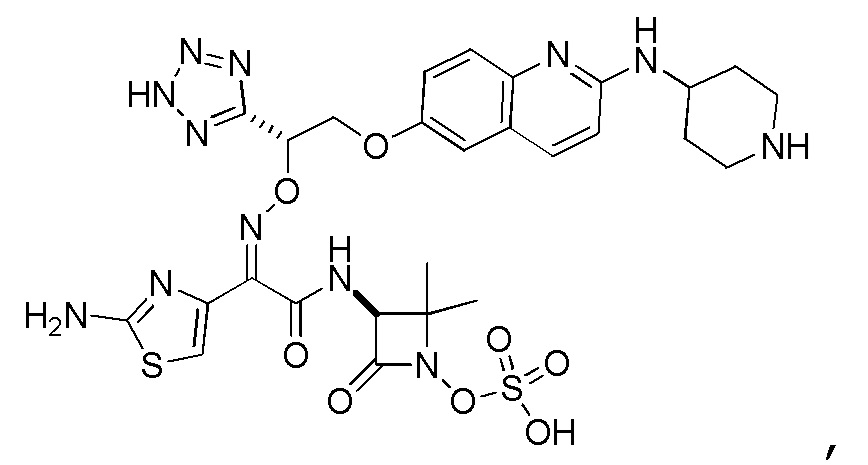

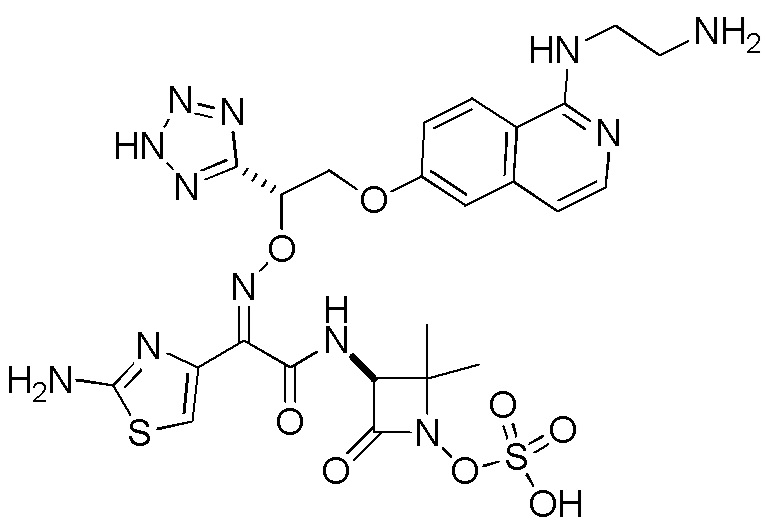
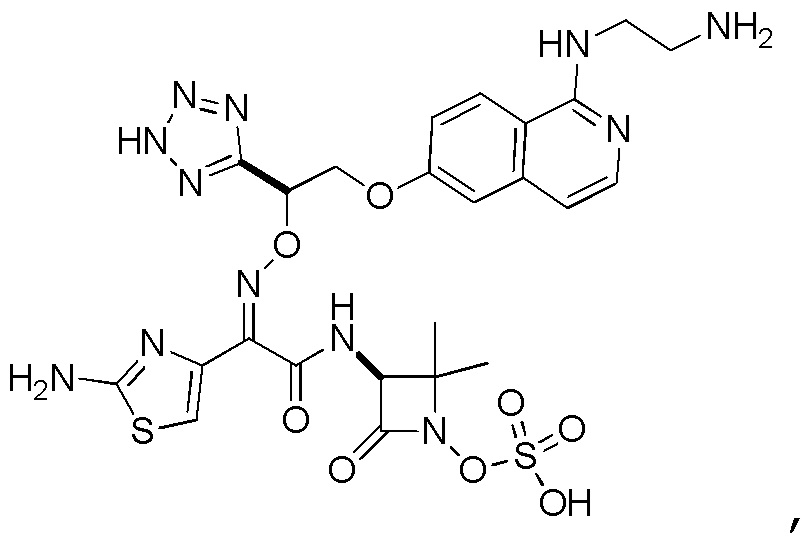



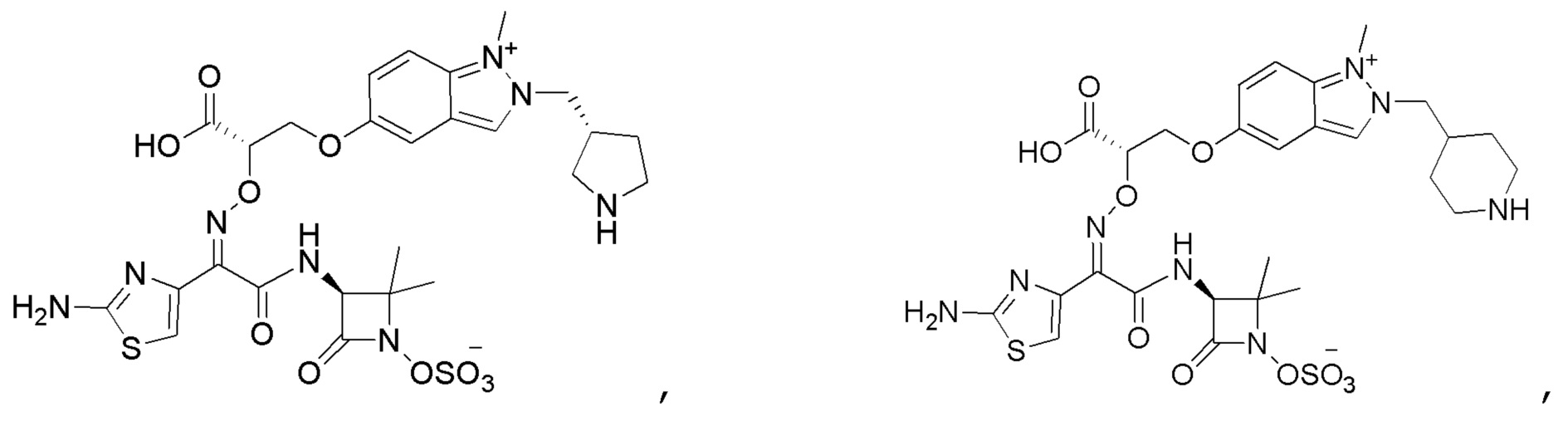



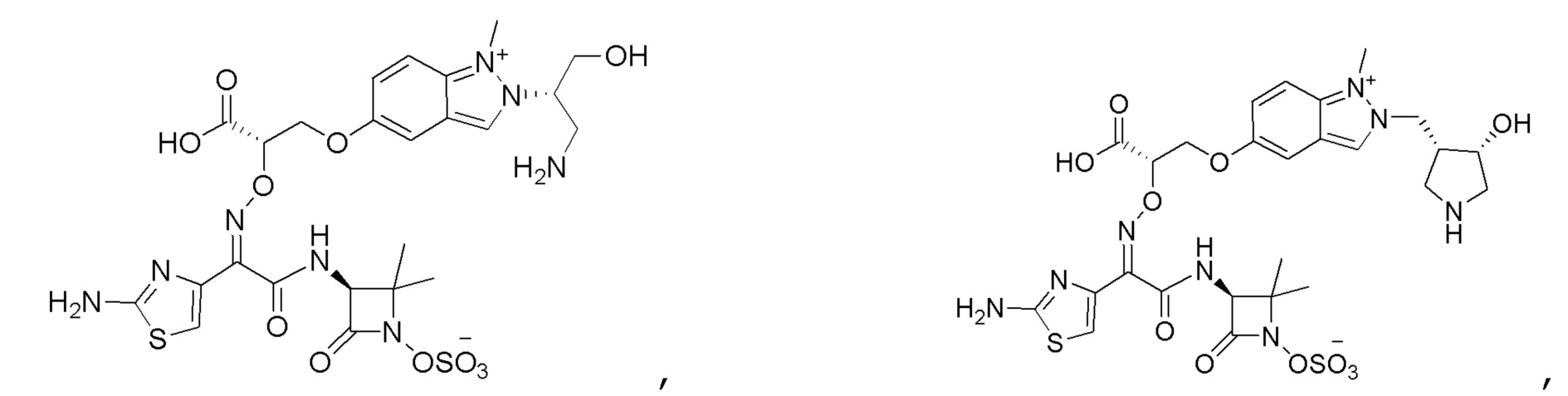
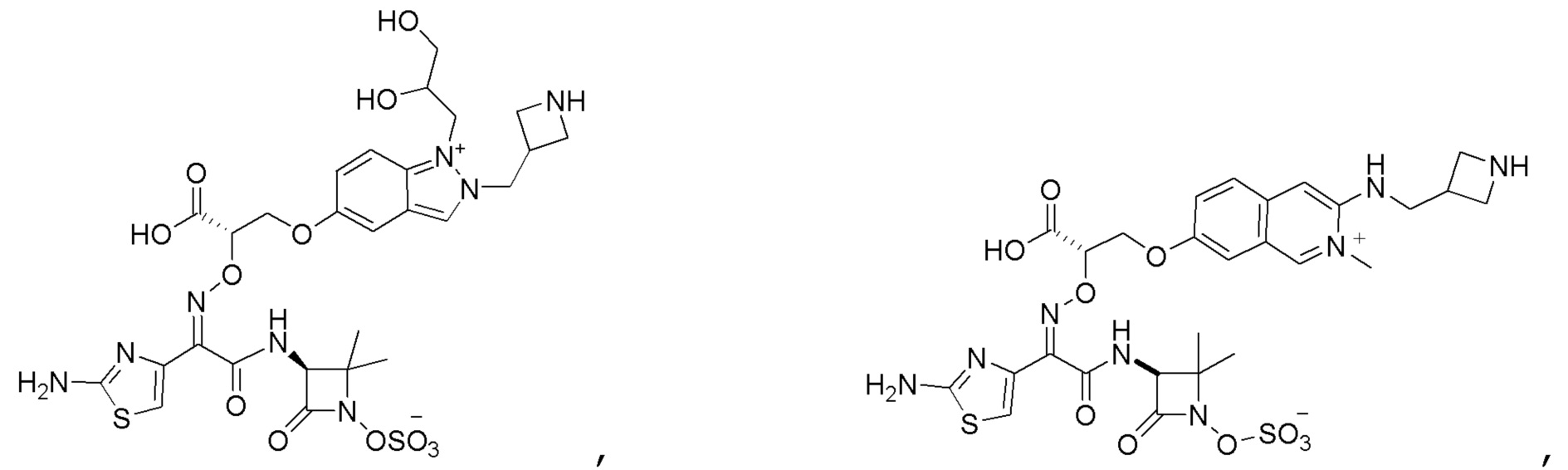



Комментарии