Ингибиторы деметилазы lsd1 на основе арилциклопропиламина и их применение в медицине - RU2611437C2
Код документа: RU2611437C2
Описание
Настоящее изобретение относится к (гетеро)арилциклопропиламинам, предпочтительно к соединениям формулы (I), (Ia), (Ib), (II) или (III), описанным и определенным в настоящем изобретении, и к их применению в терапии, включая, например, лечение или предупреждение рака, неврологического заболевания или патологического состояния или вирусной инфекции.
Общей характеристикой многих заболеваний человека является аберрантная экспрессия генов в пораженной ткани в отличие от нормальной ткани. Это относится к раку и неврологическим заболеваниям, которые характеризуются изменениями характера экспрессии генов. Характер экспрессии генов регулируется в клетке на многих уровнях. Регулирование экспрессии генов может происходить с помощью модификаций ДНК: метилирование промотора ДНК связано с подавлением экспрессии генов. Различные ингибиторы метилирования ДНК утверждены для клинического применения, включая мощное средство видазу™. Другой класс модификаций включает модификации гистонов, которые образуют белковый каркас, с которым обычно связана ДНК (обернута вокруг) в эукариотных клетках. Гистоны играют критическую роль в организации ДНК и регулируют обертывание ДНК вокруг гистонов и ее развертывание, что критически важно для регулирования экспрессии генов - обернутая ДНК обычно не доступна для транскрипции генов. Обнаружен целый ряд модификаций гистонов, включая продукты ацетилирования гистона, метилирования лизина гистона, метилирования аргинина гистона, убихихинилиривание гистона и сумоилирование гистона, многие из которых модифицируют доступность ассоциированной ДНК с помощью механизма клеточной транскрипции. Эти гистонные метки служат для рекрутинга различных комплексов белков, участвующих в транскрипции и репрессии. Все увеличивающееся количество исследований направлено на изображение того, как различные комбинации гистонных меток регулируют экспрессию генов клеточноспецифическим образом и для описания этого подхода предложен новый термин: гистонный код.
Прототипической гистонной меткой является ацетилирование гистона. Гистонацетилтрансфераза и гистондезацетилазы являются катализаторами, участвующими в модуляции этой гистонной метки, хотя обычно эти ферменты являются частями мультипротеиновых комплексов, содержащих другие белки, участвующие в считывании и модификации гистонных меток. Компоненты этих белковых комплексов обычно являются компонентами клеточного типа и обычно представляют собой регуляторы транскрипции, репрессоры, сорепрессоры, рецепторы, связанные с модуляцией экспрессии генов (например, эстрогенный или андрогенный рецептор). Ингибиторы гистондезацетилазы меняют профиль ацетилирования хроматина гистона. В соответствии с этим показано, что ингибиторы гистондезацетилазы, такие как SAHA, TSA и многие другие, меняют экспрессию генов в различных моделях на животных in vitro и in vivo. Клинически показано, что ингибитор гистондезацетилазы обладает активностью при раке и исследовано его применение в онкологии, а также при неврологических патологических состояниях и других заболеваниях.
Другой модификацией, которая участвует в регуляции экспрессии генов, является метилирование гистона, включая метилирование лизина и аргинина. Недавно показано, что статус метилирования лизинов гистонов важен для динамической регуляции экспрессии генов.
В модификациях лизина гистона участвует группа ферментов, известных, как гистонлизинметилтрансферазы и гистонлизиндеметилазы. Недавно было установлено (Shi et al. (2004) Cell 119: 941), что одна конкретная гистонлизиндеметилаза человека под названием лизинспецифическая деметилаза-1 (LSD1) участвует в этой критически важной модификации гистона. LSD1 характеризуется значительным структурным сходством и идентичности/гомологии аминокислот с полиаминоксидазами и моноаминоксидазами, каждая из которых (т.е. MAO-A, МАО-B и LSD1) является зависимой от флавина аминоксидазой, которые катализируют окисление связей азот-водород и/или связей азот-углерод.
Несколько групп исследователей описали в литературе ингибиторы LSD1. Sharma et al. недавно описали новую группу аналогов мочевины и тиомочевины на основе предложенной ими ранее группы полиаминов, для которых показано, что они ингибируют LSD1 и модулируют метилирование гистона и экспрессию генов в клетках ((2010) J. Med. Chem. PMID: 20568780). Sharma et al. отметили, что "к настоящему времени обнаружены только несколько соединений, для которых показано, что они ингибируют LSD1." В ряде исследований получены аналоги пептида гистона, которые метилированы ферментом, другие исследования направлены на малые молекулы, такие как молекулы, основанные на известных ингибиторах МАО.
Известно, что содержащие циклопропиламин соединения ингибируют целый ряд важных для медицины мишеней, включая аминоксидазы, такие как моноаминоксидаза A (MAO-A; или МАОА), моноаминоксидаза B (MAO-В; или MAOB) и лизинспецифическая деметилаза-1 (LSD1). Известно, что транилципромин (также известный под названием 2-фенилциклопропиламин), который является активным ингредиентом препарата парнат® и одним из самых известных примеров циклопропиламина, ингибирует все эти ферменты.
Gooden et al. описали аналоги транс-2-арилциклопропиламина, которые ингибируют LSD1 и характеризуются значениями Ki, находящимися в диапазоне 188-566 мкМ (Gooden et al. ((2008) Bioorg. Med. Chem. Let. 18:3047-3051)). Большинство этих соединений более активны по отношению к МАО-A, чем по отношению к МАО-B. Ueda et al. ((2009) J. Am. Chem Soc. 131(48): 17536-17537) описали аналоги циклопропиламина, селективные по отношению к LSD1 в большей степени, чем по отношению к МАО-A и МАО-B, которые были получены на основании опубликованных рентгеноструктурных данных для аддукта этих ферментов с фенилциклопропиламин-FAD и пептидом FAD-N-пропаргиллизин. Значение IC50для фенилциклопропиламина равно примерно 32 мкМ по отношению к LSD1, а соединения 1 и 2 обладают значениями, равными 2,5 и 1,9 мкМ соответственно.
Mimasu et al. описали группу производных фенилциклопропиламина, содержащих бензоильные заместители в орто-положении (2010) Biochemistry PMID: 20568732. Орто-замещенные соединения этой группы, не содержащие бензоильную группу в орто-положении, например, содержащие фенил, алкоксигруппу или содержащие комбинацию орто- и пара-заместителей, видимо, являются менее активными ингибиторами LSD1, чем соединения, содержащие бензоильные заместители в орто-положении. Наиболее активные соединения этой группы содержат бензоильную группу в орто-положении и один или два мета-фторидных заместителя: бифенилы, такие как S1310, и соединения, содержащие большие группы в пара-положении, являются менее эффективными ингибиторами LSD1.
Фенилциклопропиламины были объектом многих исследований, посвященных изучению соотношений структура-активность для ингибирования MAO. В публикациях Kaiser et al. ((1962) J. Med. Chem. 5:1243-1265); Zirkle et al. ((1962) J. Med. Chem. 1265-1284; патентах США US №№3365458; 3471522; 3532749) описаны синтез и активность целого ряда соединений, родственных фенилциклопропиламину. Другие соединения типа фенилциклопропиламина описаны в публикации Bolesov et al. ((1974) Zhurnal Organicheskoi Khimii 10:8 1661-1669) и в патенте Росийской Федерации 230169 (19681030).
Проведены исследования соединений, родственных фенилциклопропиламину, для определения селективности по отношению к МАО-A по сравнению с МАО-B, поскольку ингибиторы MAO-A могут привести к опасным побочным эффектам (см., например, публикации Yoshida et al. (2004) Bioorg. Med Chem. 12(10): 2645-2652; Hruschka et al. (2008) Biorg Med Chem. (16): 7148-7166; Folks et al. (1983) J. Clin. Psychopharmacol. (3)249; и Youdim et al. (1983) Mod. Probl. Pharmacopsychiatry (19): 63).
Binda et al. исследовали группу производных фенилциклопропиламина в связи с их ингибирующей активностью по отношению к LSD1 и LSD2, а также исследовали стереохимические аспекты, связанные с циклопропильным кольцом (J. Am. Chem. Soc. (2010) May 19; 132(19): 6827-33). Binda et al. сообщили, что предложенным ими пара-замещенные производные фенилциклопропиламина являются неселективными и в качестве группы они, видимо, являются лучшими ингибиторами MAO-A, чем ингибиторами MAO-B. Кроме того, их ингибирующая активность по отношению к MAO-A и LSD1, была примерно одинаковой.
Замещенные циклопропиламины могут быть хиральными. Хиральные соединения часто характеризуются своей способностью вращать плоскость поляризованного света и обычно обозначаются, как (+) или (-) в зависимости он направления, в котором они вращают свет. Другими обозначениями являются d- и l-, которые являются аббревиатурами правовращающих и левовращающих. Обозначения R и S используются для указания абсолютной конфигурации, поскольку способность хиральных молекул вращать плоскость поляризованного света, например, направление вращения, не всегда коррелирует с абсолютными конфигурациями.
Транилципромин обладает двумя стереоцентрами, соответствующими атомам углерода циклопропильного кольца, которые содержат аминогруппу и фенил в качестве заместителей. Теоретически, соединение, содержащее фенилциклопропиламиновую структуру транилципромина, может обладать четырьмя стереохимическими конфигурациями: две соответствуют цис-(1S,2S или 1R,2R) и две соответствуют транс-(1S,2R или 1R,2S). Транилципромин соответствует транс-изомеру 2-фенилциклопропиламина и является рацематом (-)- и (+)-энантиомеров (т.е. 50:50 смесь 1S,2R- и 1R,2S-энантиомеров) и, таким образом, оптически неактивно.
(-)-Энантиомер транилципромина синтезировали, охарактеризовывали и определяли абсолютную конфигурацию. Riley et al. (1972) J. Med. Chem. 15(11): 1187-1188. Установлено, что при синтезе из 1R,2R-2-фенилциклопропанкарбоновой кислоты (-)-стереоизомер обладает 1R,2S абсолютной конфигурацией. Последующие исследования с использованием других методик подтвердили эту интерпретацию (Binda et al. (2010) JACS 132:6827-6833) путем получения рентгеновской структуры пара-бромпроизводного стереоизомера 2-фенилциклопропиламина и сопоставления со спектрами КД (кругового дихроизма) стереоизомеров транилципромина. Эти результаты подтверждены стереоселективным синтезом энантиомеров транилципромина.
Сообщают, по данным модели, что in vivo (Riley et al. (1972) J. Med. Chem. 15(11): 1187-1188) с помощью судорог, вызываемых триптамином (Zirkle et al. (1962) J. Med. Pharm. Chem. 5:1265) (+)-изомер (1S,2R) транс-2-фенилциклопропиламина более активен по отношению к MAO, чем (-)-изомер (1R,2S).
Binda et al. сообщили о значительном различии способности стереоизомеров транилципромина ингибировать МАО-B и по данным биохимического анализа in vitro (+)-стереоизомер является более активным. (-)-Стереоизомер (1R,2S) являлся немного более активным ингибитором LSD1, чем (+)-стереоизомер (Ki равно 168 мкМ по сравнению с Ki, равным 284 мкМ), но эти различия авторы сочли незначительными (Binda et al. ((2010) JACS 132: 6827-6833 см. стр. 6828).
Недавно группа авторов сообщила о другом исследовании стереоизомеров транилципромина применительно в LSD1 (Benelkebir et al. (2011) Bioorg Med. Chem. doi: 10.1016/j.bmc. 2011.02.017). Они установили, что стереоизомеры транилципромина примерно одинаково активны по отношению к LSD1: Ki(inact) для (+)-стереоизомера равнялось 26,6 мкМ и равнялось 28,1 мкМ для (-)-стереоизомера и 25,0 мкМ для рацемата.
Другая группа авторов исследовала влияние стереохимической конфигурации около циклопропиламиновой группы замещенных 2-фенилциклопропиламинов и сообщила, что по данным биохимических исследований in vitro и исследований ингибиторов роста клеток Hela и HEK293 стереоизомер обладающий абсолютной конфигурацией (1S,2R) (по данным ЯМР с использованием химических сдвигов хиральных реагентов), являлся более активным ингибитором LSD1, чем его энантиомер, что установлено с помощью биохимических исследований и исследований ингибирования роста клеток Hela и HEK293, тогда как энантиомеры ведут себя одинаково для клеток линии нейробластомы SH-SY5Y (Ogasawara et al. (2011) Bioorg. Med. Chem. Doi: 10, 1016/j.bmc. 2010,12,024).
С учетом различий анализов/экспериментальных методик, использованных при различных исследованиях, указанных выше, трудно сопоставить результаты этих исследований. Кроме того, из этих данных неясно, как производные или аналоги соединений, содержащих фенилциклопропиламиновое ядро, можно оптимизировать и получить активные ингибиторы LSD1 и LSD1 и MAO-B на основе стереохимической конфигурации атомов углерода циклопропильного фрагмента. Кроме того, из данных этих исследований неясно, как можно модулировать селективность N-замещенных арил- и гетероциклопропиламинов по отношению к LSD1 и MAO-B и получить соединения, которые ингибируют эти ферменты в большей степени, чем MAO-A. Предполагается, что такие соединения обладают превосходными диапазонами безопасности, когда исключается ингибирование МАОА и так называемый "эффект сыра" (гипертензия при лечении ингибиторами МАО и аналогичными соединениями).
Ввиду отсутствия адекватных средств лечения таких патологических состояний, как рак и нейродегенерация, настоятельно необходимы лекарственные средства, влияющие на заболевания, и лекарственные средства, которые воздействуют путем ингибирования новых мишеней. Необходима разработка новых селективных ингибиторов LSD1, в особенности таких, которые селективно ингибируют LSD1 или LSD1 в комбинации с MAO-B.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к выявлению соединений и их применению для лечения или предупреждения заболеваний. Настоящее изобретение относится к (гетеро)циклопропиламинам, включая соединения формулы (I), (II) или (III), описанные и определенные в настоящем изобретении. Настоящее изобретение, в частности, относится к соединению формулы (I) или его фармацевтически приемлемой соли или сольвату, фармацевтическим композициям, содержащим соединение формулы (I) или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель, и их применению для лечения заболеваний. Одним применением соединения формулы (I) является применение для лечения или предупреждения рака. Другим применением соединения формулы (I) является ингибирование LSD1. Таким образом, настоящее изобретение относится к соединению формулы (I) или его энантиомеру, диастереоизомеру или их смеси, или его фармацевтически приемлемой соли или сольвату, предназначенному для применения для лечения или предупреждения рака. Таким образом, настоящее изобретение относится к соединению формулы (I), или его фармацевтически приемлемой соли или сольвату и также относится к его применению для лечения или предупреждения заболевания человека.
В соответствии с этим, настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли или сольвату:

(A) обозначает циклильную группу, содержащую n заместителей (R3).
(B) обозначает циклильную группу или -(L1)-циклильную группу, где указанная циклильная группа или циклильный фрагмент -(L1)-циклильной группы содержит n заместителей (R2).
(L1) обозначает -O-, -NH-, -N(алкил)-, алкилен или гетероалкилен.
(D) обозначает гетероарильную группу или -(L2)-гетероарильную группу, где указанная гетероарильная группа или гетероарильный фрагмент, содержащийся в указанной -(L2)-гетероарильной группе, содержит один заместитель (R1) и, кроме того, где указанная гетероарильная группа ковалентно связана с остальной частью молекулы через кольцевой атом углерода или гетероарильный фрагмент, содержащийся в указанной -(L2)-гетероарильной группе, ковалентно связан с фрагментом (L2) через кольцевой атом углерода.
(L2) обозначает -O-, -NH-, -N(алкил)-, алкилен или гетероалкилен.
(R1) обозначает группу, связанную водородной связью.
Каждый (R2) независимо выбран из группы, включающей алкил, алкенил, алкинил, циклил, аминогруппу, амидную группу, C-амидную группу, алкиламиногруппу, гидроксигруппу, нитрогруппу, галоген, галогеналкил, галогеналкоксигруппу, цианогруппу, сульфинил, сульфонил, сульфонамидную группу, алкоксигруппу, ацил, карбоксигруппу, карбаматную группу или мочевинную группу.
Каждый (R3) независимо выбран из группы, включающей алкил, алкенил, алкинил, циклил, аминогруппу, амидную группу, C-амидную группу, алкиламиногруппу, гидроксигруппу, нитрогруппу, галоген, галогеналкил, галогеналкоксигруппу, цианогруппу, сульфинил, сульфонил, сульфонамидную группу, алкоксигруппу, ацил, карбоксигруппу, карбаматную группу или мочевинную группу;
n независимо равен 0, 1, 2, 3 или 4.
Заместители циклопропильного фрагмента, т.е. группа (A) и группа -NH-CH2-(D), предпочтительно находятся в транс-конфигурации.
Родственным объектом настоящего изобретения является фармацевтическая композиция, содержащая соединение формулы (I) или его энантиомер, диастереоизомер или их смесь, или его фармацевтически приемлемую соль или сольват, определенные выше, и фармацевтически приемлемый носитель. Предпочтительные варианты осуществления соединения формулы (I), например, предназначенные для применения в композиции, предлагаемой в настоящем изобретении, более подробно определены и описаны ниже в настоящем изобретении.
Другим объектом настоящего изобретения является способ лечения или предупреждения заболевания или патологического состояния, включающий введение пациенту (предпочтительно человеку), нуждающемуся в лечении или предупреждении, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы (I), описанное выше или описанное в его вариантах осуществления, описанных ниже, или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. Этот объект можно переформулировать, как соединение формулы (I), определенное выше в первом объекте настоящего изобретения, предназначенное для применения в качестве лекарственного средства. Родственным объектом настоящего изобретения является фармацевтическая композиция, предназначенное для применения для лечения или предупреждения заболевания или патологического состояния, где указанная композиция содержит терапевтически эффективное количество соединения формулы (I), достаточное для лечения или предупреждения указанного заболевания или патологического состояния. В более предпочтительном варианте осуществления настоящее изобретение относится к соединению формулы (I), предназначенному для применения для лечения заболевания, связанного с LSD1.
Еще одним объектом настоящего изобретения является способ ингибирования активности LSD1, включающий введение пациенту, нуждающемуся в лечении, терапевтически эффективного количества композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, достаточного для ингибирования активности LSD1. Предпочтительно, если пациентом является. Этот объект можно переформулировать, как соединение формулы (I), определенное в настоящем изобретении, предназначенное для применения в качестве ингибитора LSD1. Родственным объектом является способ лечения индивидуума, указанный способ включает выявление индивидуума, нуждающегося в лечении, и введение указанному индивидууму терапевтически эффективного количества соединения формулы (I). В предпочтительном объекте терапевтически эффективное количество соединения формулы (I) является количеством, достаточным для ингибирования LSD1. Предпочтительные варианты осуществления соединений формулы (I), предназначенные для применения в композиции и способе этого объекта настоящего изобретения, являются такими, как более подробно описанные в настоящем изобретении.
Еще одним объектом настоящего изобретения является способ лечения или предупреждения рака, включающий введение пациенту, нуждающемуся в лечении или предупреждении, терапевтически эффективного количества композиции, содержащей соединение формулы (I), определенное выше или в вариантах осуществления, более подробно описанных в настоящем изобретении, и фармацевтически приемлемый носитель. Этот объект можно переформулировать, как соединение формулы (I), определенное выше в первом объекте настоящего изобретения, предназначенное для применения для лечения или предупреждения рака. Родственным объектом настоящего изобретения является фармацевтическая композиция, предназначенная для применения для лечения или предупреждения рака, где указанная композиция содержит терапевтически эффективное количество соединения формулы (I), достаточное для лечения или предупреждения рака. Другим родственным объектом настоящего изобретения является соединение формулы (I) или фармацевтическая композиция, предназначенная для лечения или предупреждения рака, где указанный рак выбран из группы, включающей рак молочной железы, рак легких, рак предстательной железы, колоректальный рак, рак головного мозга, рак кожи, рак крови (например, лейкоз, включая, например, острый миелогенный лейкоз (ОМЛ), хронический миелогенный лейкоз (ХМЛ), хронический нейтрофильный лейкоз, хронический эозинофильный лейкоз, хронический лимфолейкоз (ХЛЛ), острый лимфобластный лейкоз (ОЛЛ) или волосатоклеточный лейкоз), лимфому и миелому. Указанная композиция предпочтительно содержит терапевтически эффективное количество соединения формулы (I), достаточное для лечения или предупреждения указанного рака. В предпочтительном объекте терапевтически эффективное количество соединения формулы (I) является количеством, достаточным для ингибирования LSD1. В другом предпочтительном объекте терапевтически эффективное количество является количеством, достаточным для модулирования степени метилирования гистона. В другом предпочтительном объекте терапевтически эффективное количество является количеством, достаточным для модулирования степени метилирования гистон-3-лизина-4. В другом предпочтительном объекте терапевтически эффективное количество является количеством, достаточным для модулирования степени метилирования гистон-3-лизина-9.
Еще одним объектом настоящего изобретения является способ лечения или предупреждения неврологического заболевания или патологического состояния, включающий введение пациенту, нуждающемуся в лечении или предупреждении, терапевтически эффективного количества композиции, содержащей соединение формулы (I), определенное выше или в вариантах осуществления, более подробно описанных в настоящем изобретении, и фармацевтически приемлемый носитель. Этот объект можно переформулировать, как соединение формулы (I), определенное выше, предназначенное для применения для лечения или предупреждения неврологического патологического состояния или заболевания. Родственным объектом настоящего изобретения является фармацевтическая композиция, предназначенная для применения для лечения или предупреждения неврологического патологического состояния или заболевания, где указанная композиция предпочтительно содержит терапевтически эффективное количество соединения формулы (I), достаточное для лечения или предупреждения указанного неврологического заболевания или патологического состояния. Другим родственным объектом настоящего изобретения является соединение формулы (I) или фармацевтическая композиция, предназначенная для лечения или предупреждения неврологического заболевания или патологического состояния, где указанное неврологическое заболевание или патологическое состояние выбрано из группы, включающей депрессию, болезнь Альцгеймера, болезнь Гентингтона, болезнь Паркинсона, боковой амиотрофический склероз, слабоумие с тельцами Леви или лобно-височное слабоумие, предпочтительно из группы, включающей депрессию, болезнь Альцгеймера, болезнь Гентингтона, болезнь Паркинсона или слабоумие с тельцами Леви. Указанная композиция предпочтительно содержит терапевтически эффективное количество соединения формулы (I), достаточное для лечения или предупреждения указанного заболевания или патологического состояния. В предпочтительном объекте терапевтически эффективное количество соединения формулы (I) является количеством, достаточным для ингибирования LSD1. В другом предпочтительном объекте терапевтически эффективное количество является количеством, достаточным для модулирования степени метилирования гистона. В другом предпочтительном объекте терапевтически эффективное количество является количеством, достаточным для модулирования степени метилирования гистон-3-лизина-4. В другом предпочтительном объекте терапевтически эффективное количество является количеством, достаточным для модулирования степени метилирования гистон-3-лизина-4. В другом предпочтительном объекте терапевтически эффективное количество является количеством, достаточным для модулирования степени метилирования гистон-3-лизина-9.
Еще одним объектом настоящего изобретения является способ выявления соединения, которое является селективным ингибитором LSD1, способ включает выбор или получение соединения формулы (I), определенного в настоящем изобретении, и определение способности соединения ингибировать LSD1 и МАО-A и/или МАО-B, где соединение, которое ингибирует LSD1 в большей степени, чем МАО-A и/или МАО-B, идентифицируют, как селективный ингибитор LSD1. Соединение этого объекта, которое является ингибитором LSD1, можно использовать для лечения заболевания, предпочтительно заболевания человека.
Еще одним объектом настоящего изобретения является способ выявления соединения, которое является двойным ингибитором LSD1 и МАО-B, способ включает выбор или получение соединения формулы (I), определенного в настоящем изобретении, и определение способности соединения ингибировать LSD1, МАО-A, и МАО-B, где соединение, которое ингибирует LSD1 и МАО-B в большей степени, чем МАО-A, идентифицируют, как двойной ингибитор LSD1/MAO-B. Соединение этого объекта, которое является двойным ингибитором LSD1/MAO-B, можно использовать для лечения заболевания, предпочтительно заболевания человека.
Таким образом, в одном варианте осуществления настоящего изобретения фармацевтическая композиция, содержащая селективный ингибитор LSD1 формулы (I) или его фармацевтически приемлемую соль или сольват, применима для лечения и/или предупреждения заболевания у индивидуума. В одном объекте терапевтически эффективное количество композиции вводят индивидууму в количестве, достаточном для предупреждения или лечения заболевания. В более предпочтительном объекте заболеванием является рак. В еще более предпочтительном объекте заболеванием является рак, выбранный из группы, включающей рак предстательной железы, головного мозга, колоректальный, легких, молочной железы, кожи и рак крови. В одном предпочтительном объекте раком является рак предстательной железы. В одном предпочтительном объекте раком является рак легких. В одном предпочтительном объекте раком является рак головного мозга. В одном предпочтительном объекте раком является рак крови (например, лейкоз, включая, например, острый миелогенный лейкоз (ОМЛ), хронический миелогенный лейкоз (ХМЛ), хронический нейтрофильный лейкоз, хронический эозинофильный лейкоз, хронический лимфолейкоз (ХЛЛ), острый лимфобластный лейкоз (ОЛЛ) или волосатоклеточный лейкоз). В одном предпочтительном объекте раком является рак молочной железы. В одном предпочтительном объекте раком является колоректальный рак. В одном предпочтительном объекте раком является лимфома. В одном предпочтительном объекте раком является миелома. В другом предпочтительном объекте терапевтически эффективное количество является количеством, достаточным для ингибирования LSD1. В другом предпочтительном объекте терапевтически эффективное количество является количеством, достаточным для модулирования степени метилирования гистона. В другом предпочтительном объекте терапевтически эффективное количество является количеством, достаточным для модулирования степени метилирования гистон-3-лизина-4. В другом предпочтительном объекте терапевтически эффективное количество является количеством, достаточным для модулирования степени метилирования гистон-3-лизина-9.
Кроме того, согласно изобретению неожиданно установлено, что стереохимическая конфигурация циклопропильных атомов углерода N-замещенных арилциклопропиламинов значительно влияет на активность ингибирования LSD1, ингибирования MAO-B и ингибирования MAO-A. Согласно изобретению установлено, что (-)-стереоизомер 5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-амина примерно в 20 раз более активен по отношению к LSD1, чем соответствующий (+)-стереоизомер. Кроме того, (-)-стереоизомер сохраняет значительную ингибирующую активность по отношению к MAO-B. Примечательно, что селективность по отношению к LSD1/MAO-A (-)/(+)-стереоизомера боле, чем в 100 раз выше, о чем свидетельствуют значения kinact/KI. Таким образом, (-)-стереоизомеры N-замещенных (гетеро)арилциклопропиламинов неожиданно оказались активными и селективными ингибиторами LSD1 в отличие от соответствующих их энантиомеров. Кроме того, соединения, предлагаемые в настоящем изобретении, обладают улучшенной селективностью по отношению к MAO-A, предпочтительно ингибируют MAO-B и LSD1. Поэтому настоящее изобретение относится к оптически активным (гетеро)арилциклопропиламинам, в частности, к оптически активным N-замещенным арил- или гетероарилциклопропиламинам и их применению для лечения или предупреждения заболевания или нарушения.
Таким образом, одним предпочтительным объектом настоящего изобретения является в основном чистый стереоизомер N-замещенного арил- или гетероарилциклопропиламина (например, соединение формулы (II) или (III), описанное и определенное ниже в настоящем изобретении), предназначенный для применения в способе лечения или предупреждения заболевания или нарушения. Предпочтительно, если заболеванием или нарушением является такое, которое можно лечить или предупреждать путем ингибирования LSD1, ингибирования LSD1 и ингибирования MAO-B или ингибирования MAO-B. В предпочтительном объекте в основном чистый стереоизомер N-замещенного арил- или гетероарилциклопропиламина означает N-замещенный арил- или гетероарилциклопропиламин, который содержит 90% или более (-)-стереоизомера и 10% или менее (+)-стереоизомера. В более предпочтительном объекте в основном чистый стереоизомер N-замещенного арил- или гетероарилциклопропиламина означает N-замещенный арил- или гетероарилциклопропиламин, который содержит 95% или более (-)-стереоизомера и 5% или менее (+)-стереоизомера. В еще более предпочтительном объекте в основном чистый стереоизомер N-замещенного арил- или гетероарилциклопропиламина означает N-замещенный арил- или гетероарилциклопропиламин, который содержит 98% или более (-)-стереоизомера и 2% или менее (+)-стереоизомера. В еще более предпочтительном объекте в основном чистый стереоизомер N-замещенного арил- или гетероарилциклопропиламина означает N-замещенный арил- или гетероарилциклопропиламин, который содержит 99% или более (-)-стереоизомера и 1% или менее (+)-стереоизомера. В еще более предпочтительном объекте в основном чистый стереоизомер N-замещенного арил- или гетероарилциклопропиламина означает N-замещенный арил- или гетероарилциклопропиламин, который содержит 99,5% или более (-)-стереоизомера и 0,5% или менее (+)-стереоизомера. В одном варианте осуществления описанные выше выраженные в процентах содержания являются молярными. В основном чистый стереоизомер N-замещенного арил- или гетероарилциклопропиламина в одном объекте предназначен для применения в способе лечения или предупреждения рака, депрессии, нейродегенеративного заболевания или нарушения или вирусной инфекции.
Кроме того, другим объектом настоящего изобретения является композиция, содержащая стереоизомер N-замещенного арил- или гетероарилциклопропиламина (например, соединение формулы (II) или (III), описанное и определенное ниже в настоящем изобретении), где указанная композиция обладает составляющим 90% или более энантиомерным избытком (-)-стереоизомера N-замещенного арил- или гетероарилциклопропиламина. В предпочтительном объекте указанная композиция обладает составляющим 95% или более энантиомерным избытком (-)-стереоизомера N-замещенного арил- или гетероарилциклопропиламина. В более предпочтительном объекте указанная композиция обладает составляющим 98% или более энантиомерным избытком (-)-стереоизомера N-замещенного арил- или гетероарилциклопропиламина. В еще более предпочтительном объекте указанная композиция обладает составляющим 99% или более энантиомерным избытком (-)-стереоизомера N-замещенного арил- или гетероарилциклопропиламина. В одном объекте настоящего изобретения композиция предназначена для применения в способе лечения или предупреждения рака, депрессии, нейродегенеративного заболевания или нарушения или вирусной инфекции.
Кроме того, другим объектом настоящего изобретения является фармацевтическая композиция, содержащая стереоизомер N-замещенного арил- или гетероарилциклопропиламина (например, соединение формулы (II) или (III), описанное и определенное ниже в настоящем изобретении) и фармацевтически приемлемый носитель, где указанная композиция обладает составляющим 90% или более энантиомерным избытком (-)-стереоизомера N-замещенного арил- или гетероарилциклопропиламина. В предпочтительном объекте указанная композиция обладает составляющим 95% или более энантиомерным избытком (-)-стереоизомера N-замещенного арил- или гетероарилциклопропиламина. В предпочтительном объекте указанная композиция обладает составляющим 99% или более энантиомерным избытком (-)-стереоизомера N-замещенного арил- или гетероарилциклопропиламина. Фармацевтическая композиция, описанная в этом абзаце, предназначена для применения в способе лечения или предупреждения рака, депрессии, нейродегенеративного заболевания или нарушения или вирусной инфекции.
В одном объекте настоящего изобретения оптически активный N-замещенный арил- или гетероарилциклопропиламин или его фармацевтически приемлемая соль или сольват, предназначенный для применения в способе лечения или предупреждения заболевания или нарушения, описанном в настоящем изобретении, описывается формулой (II):

в которой:
(AII) обозначает арильную или гетероарильную группу, содержащую 2 заместителя, R1II и R2II, и 1, 2 или 3 необязательных заместителя, где указанные необязательные заместители независимо выбраны из группы, включающей галоген, C1-C3-алкил или C1-C3-алкоксигруппу;
R1II обозначает группу ;
R3II обозначает арильную или гетероарильную группу, содержащую 1, 2, 3, 4 или 5 необязательных заместителей, независимо выбранных из группы, включающей галоген, -OH, -NHSO2RA, алкил, алкоксигруппу, цианогруппу, -CF3 или -OCF3, где RA обозначает C1-C6-алкил или фенил;
выбран из группы, включающей связь, -CH2O-, -CH2CH2O-, -OCH2-, -OCH2CH2-, -CH2CH2-, -CH2-, -CH2CH2CH2- или -O-;
R2II обозначает , где указанная циклопропильная группа содержит 2 хиральных центра, замещенных в транс-положении по отношению к атомам углерода, к которым ковалентно присоединены (A) и ;
R4II обозначает 5- или 6-членное гетероарильное кольцо, содержащее 1, 2 или 3 необязательных заместителя, выбранных из группы, включающей алкил, -NHRB, -ORB или галоген, где RB обозначает водород, C1-C3-алкил или -C(=O)CH3;
обозначает разветвленную или неразветвленную C1-C4-алкиленовую группу и где указанное соединение формулы (II) является оптически активным.
Настоящее изобретение также относится к фармацевтической композиции, содержащей оптически активный N-замещенный арил- или гетероарилциклопропиламин формулы (II), определенный выше, или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к фармацевтической композиции, содержащей оптически активный N-замещенный арил- или гетероарилциклопропиламин формулы (II), определенный выше, или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель, предназначенной для применения для лечения или предупреждения заболевания или нарушения. Предпочтительно, если заболевание или нарушение опосредуется аминоксидазой. В одном объекте аминоксидазой является LSD1 или MAO-B.
Кроме того, согласно изобретению обнаружена подгруппа оптически активных соединений формулы (II), описывающаяся формулой (III), которые являются ингибиторами LSD1 или LSD1 и MAO-B.
Таким образом, настоящее изобретение также относится к оптически активному соединению формулы (III) или его фармацевтически приемлемой соли или сольвату:

в которой:
(AIII) обозначает арильную или гетероарильную группу, содержащую 2 заместителя, R1III и R2III, и 1, 2 или 3 необязательных заместителя, независимо выбранных из группы, включающей галоген, C1-C3-алкил или C1-C3-алкоксигруппу;
R1III обозначает группу ;
R3III обозначает фенильную, пиридильную, тиазолильную или тиенильную группу, содержащую 1, 2, 3, 4 или 5 необязательных заместителей, независимо выбранных из группы, включающей галоген, -OH, -NHSO2RA, алкил, алкоксигруппу, цианогруппу, -CF3 или -OCF3, где RA обозначает C1-C6-алкил или фенил;
выбран из группы, включающей связь, -OCH2- или -CH2O-;
R2III обозначает , где указанная циклопропильная группа содержит 2 хиральных центра, замещенных в трансположении по отношению к атомам углерода, к которым ковалентно присоединены (AIII) и ;
R4III обозначает 5-членное гетероарильное кольцо, содержащее 1, 2 или 3 необязательных заместителя, где указанные необязательные заместители независимо выбраны из группы, включающей -NH2 или -NH(C1-C3)алкил;
L2III обозначает -CH2- или -CH2CH2-;
и где указанное соединение формулы (III) является оптически активным.
Кроме того, настоящее изобретение относится к фармацевтической композиции, содержащей оптически активный N-замещенный арил- или гетероарилциклопропиламин формулы (II) или (III), определенный выше, или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к фармацевтической композиции, содержащей оптически активный N-замещенный арил- или гетероарилциклопропиламин формулы (II) или (III), определенный выше, или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель, предназначенной для применения для лечения или предупреждения заболевания или нарушения.
Одним объектом настоящего изобретения является способ лечения или предупреждения заболевания или нарушения, включающий введение индивидууму, нуждающемуся в лечении, терапевтически эффективного количества оптически активного N-замещенного арил- или гетероарилциклопропиламина, предпочтительно соединения формулы (II) или (III) или его фармацевтически приемлемой соли или сольвата. В более предпочтительном объекте заболеванием или нарушением является заболевание или нарушение человека, выбранное из группы, включающей рак, депрессию, нейродегенеративное заболевание или нарушение или вирусную инфекцию. В одном объекте нейродегенеративным заболеванием или нарушением является болезнь Гентингтона, болезнь Паркинсона, болезнь Альцгеймера, боковой амиотрофический склероз, лобно-височное слабоумие или слабоумие с тельцами Леви.
Одним объектом настоящего изобретения является способ лечения или предупреждения заболевания или нарушения, включающий выявление индивидуума, нуждающегося в лечении или предупреждении, и введение указанному индивидууму терапевтически эффективного количества оптически активного N-замещенного арил- или гетероарилциклопропиламина, предпочтительно соединения формулы (II) или (III) или его фармацевтически приемлемой соли или сольвата. В более предпочтительном объекте заболеванием или нарушением является заболевание или нарушение человека, выбранное из группы, включающей рак, депрессию, нейродегенеративное заболевание или нарушение или вирусную инфекцию. В одном объекте нейродегенеративным заболеванием или нарушением является болезнь Гентингтона, болезнь Паркинсона, болезнь Альцгеймера, боковой амиотрофический склероз, лобно-височное слабоумие или слабоумие с тельцами Леви.
Одним объектом настоящего изобретения является способ обогащения энантиомера транс-N-замещенного циклопропиламина (предпочтительно соединения формулы (II) или (III), или соединения формулы (I), где заместители циклопропильного фрагмента находятся в транс-ориентации), способ включает:
Взаимодействие транс-замещенного циклопропиламина с хиральным реагентом для перекристаллизации в растворителе (предпочтительно при условиях, которые достаточны для кристаллизации соли хирального реагента для перекристаллизации и транс-замещенного циклопропиламина); и выделение закристаллизованой соли хирального реагента для перекристаллизации и транс-замещенного циклопропиламина. В одном объекте транс-циклопропиламином является N-замещенный арил- или гетероарилциклопропиламин. В одном объекте транс-циклопропиламином является 4-бензокси-2-фенилциклопропиламин или его производное, в котором аминогруппа содержит защитную группу.
Если не приведено другое определение, то все технические и научные термины, использующиеся в настоящем изобретении, обладают такими же значениями, которые обычно известны специалисту с общей подготовкой в области техники, к которой относится настоящее изобретение. Хотя при практическом осуществлении или тестировании настоящего изобретения можно использовать методики и материалы, сходные с описанными в настоящем изобретении или эквивалентные им, подходящие методики и материалы описаны ниже. В случае противоречий необходимо руководствоваться настоящим описанием, включая определения. Кроме того, материалы, методики и примеры являются лишь иллюстративными, и их не следует рассматривать в качестве ограничивающих.
Другие особенности и преимущества настоящего изобретения очевидны из приведенного ниже подробного описания и из формулы изобретения.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к выявлению соединений и их применению для лечения или предупреждения заболеваний. Настоящее изобретение относится к соединениям формулы (I), фармацевтические композиции, содержащим соединение формулы (I) или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель и их применению для лечения заболеваний. Одним применением соединений формулы (I) является применение для лечения рака. Соединения формулы (I) можно использовать в качестве селективных ингибиторов LSD1, которые ингибируют LSD1 в большей степени, чем MAO-A и MAO-B, или в качестве двойных ингибиторов LSD1/MAO-B, которые ингибируют LSD1 и MAO-B в большей степени, чем MAO-A. Соединения формулы (I), описанные в настоящем изобретении обычно являются ингибиторами LSD1, более чем в 10-20 раз или более лучшими, чем транилципромин, и обладают улучшенной селективностью по отношению к MAO-A. Таким образом, эти соединения являются селективными по отношению к LSD1 в том отношении, что они ингибируют LSD1 в большей степени, чем MAO-A и MAO-B, или являются двойными ингибиторами LSD1/MAO-B в том отношении, что они ингибируют LSD1 и MAO-B в большей степени, чем MAO-A.
Настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли или сольвату:

(A) обозначает циклильную группу, содержащую n заместителей (R3). Предпочтительно, если (A) обозначает арильную группу или гетероарильную группу, где указанная арильная группа или указанная гетероарильная группа содержит n заместителей (R3). Более предпочтительно, если (A) обозначает фенил, пиридинил, тиофенил, пирролил, фуранил или тиазолил, где (A) содержит n заместителей (R3). Еще более предпочтительно, если (А) обозначает фенил или пиридил, где указанный фенил или указанный пиридил содержит n заместителей (R3). В одном варианте осуществления (A) содержит 0 или 1 заместитель (R3). В другом варианте осуществления (A) содержит 0 заместителей (R3). В другом варианте осуществления (A) содержит 1 заместитель (R3). Следует понимать, что, если n равно 0, то циклильная группа не содержит заместителя (R3), но вместо этого может быть замещена водородом.
(B) обозначает циклильную группу или -(L1)-циклильную группу, где указанная циклильная группа или циклильный фрагмент -(L1)-циклильной группы содержит n заместителей (R2). Указанная циклильная группа или циклильный фрагмент, содержащийся в указанной -(L1)-циклильной группе, может, например, представлять собой арильную группу (например, фенил, нафтил или антраценил) или гетероарильную группу (например, пиридинил, тиофенил, пирролил, фуранил, тиазолил, оксазолил, изоксазолил, оксадиазолил, тиадиазолил, триазинил, пиридазинил, пиразинил или пиримидинил). Предпочтительно, если (B) обозначает -O-CH2-фенил или фенил, где (B) содержит n заместителей (R2). В одном варианте осуществления (B) обозначает фенил, содержащий n заместителей (R2). В другом варианте осуществления (B) обозначает -O-CH2-фенил, содержащий n заместителей (R2). В одном варианте осуществления (B) содержит 0, 1 или 2 заместителя (R2). В другом варианте осуществления (B) содержит 0 или 1 заместитель (R2). В другом варианте осуществления (B) содержит 0 заместителей (R2). В другом варианте осуществления (B) содержит 1 заместитель (R2).
(L1) обозначает -O-, -NH-, -N(алкил)-, алкилен или гетероалкилен. Указанный алкилен может, например, быть обладающим линейной цепью или разветвленной цепью алкиленом, содержащим от 1 до 6 атомов углерода. Указанный гетероалкилен может, например, быть обладающим линейной цепью или разветвленной цепью алкиленом, содержащим от 1 до 6 атомов углерода, где 1, 2 (если содержатся) или 3 (если содержатся) атома углерода все заменены гетероатомом, независимо выбранным из группы, включающей O, N или S; соответственно, указанный гетероалкилен может, например, быть обладающим линейной цепью или разветвленной цепью алкиленом, содержащим от 1 до 4 атомов углерода, где 1 или 2 несоседних атома углерода заменены на O.
(D) обозначает гетероарильную группу или -(L2)-гетероарильную группу, где указанная гетероарильная группа или гетероарильный фрагмент, содержащийся в указанной -(L2)-гетероарильной группе, содержит один заместитель (R1) и, кроме того, где указанная гетероарильная группа ковалентно связана с остальной частью молекулы через кольцевой атом углерода или гетероарильный фрагмент, содержащийся в указанной -(L2)-гетероарильной группе, ковалентно связан с фрагментом (L2) через кольцевой атом углерода. Предпочтительно, (D) обозначает тиазолил, оксадиазолил, оксазолил, изоксазолил, тиадиазолил, триазинил, пиридазинил, пиразинил, пиридинил или пиримидинил, где указанный тиазолил, оксадиазолил, оксазолил, изоксазолил, тиадиазолил, триазинил, пиридазинил, пиразинил, пиридинил или пиримидинил содержит 1 заместитель (R1). В частности, (D) может обозначать тиазолил, оксадиазолил или пиримидинил, где указанный тиазолил, указанный оксадиазолил или указанный пиримидинил содержит 1 заместитель (R1). Наиболее предпочтительно, если (D) обозначает оксадиазолил.
(L2) обозначает -O-, -NH-, -N(алкил)-, алкилен или гетероалкилен. Указанный алкилен может, например, быть обладающим линейной цепью или разветвленной цепью алкиленом, содержащим от 1 до 6 атомов углерода. Указанный гетероалкилен может, например, быть обладающим линейной цепью или разветвленной цепью алкиленом, содержащим от 1 до 6 атомов углерода, где 1, 2 (если содержатся) или 3 (если содержатся) атома углерода все заменены гетероатомом, независимо выбранным из группы, включающей O, N или S; соответственно, указанный гетероалкилен может, например, быть обладающим линейной цепью или разветвленной цепью алкиленом, содержащим от 1 до 4 атомов углерода, где 1 или 2 несоседних атома углерода заменены на O.
(R1) обозначает группу, связанную водородной связью. Например (R1) может обозначать -OH, -O(алкил), -NH2, -NH(алкил) (например, -NHCH3), -N(алкил)(алкил) (например, -N(CH3)2), амидную группу, -SO-NH2, -SO-NH(алкил), -SO-N(алкил)(алкил), -S(O)2NH2, -S(O)2NH(алкил), -S(O)2N(алкил)(алкил), -C(=O)NH2, -С(=O)N(алкил), -С(=O)N(алкил)(алкил), -алкилен-C(=O)NH2 (например, -CH2-C(=O)NH2), -алкилен-C(=O)NH(алкил) (например, -CH2-С(=O)NH(алкил)), -алкилен-C(=O)N(алкил)(алкил) (например, -CH2-С(=O)N(алкил)(алкил)), -NHC(=O)-алкил (например, -NHC(=O)CH3), -N(алкил)-С(=O)-алкил (например, -N(-CH3)-С(=O)CH3), -алкилен-NH2 (например, -CH2-NH2), -алкилен-NH(алкил) или -алкилен-N(алкил)(алкил), где предпочтительно, если указанные выше алкильные и алкиленовые группы все независимо содержат от 1 до 6 атомов углерода. Предпочтительно, если (R1) обозначает -OH, -NH2, амидную группу, -S(O)2NH2, -C(=O)NH2, -CH2-C(=O)NH2, -NHC(=O)CH3, -NHCH3, -N(CH3)2 или -CH2-NH2, предпочтительно -OH, -NH2, -NHCH3, амидную группу, -S(O)2NH2, -C(=O)NH2, -CH2-C(=O)NH2 или -CH2-NH2. Более предпочтительно, если (R1) обозначает -NH2 или -NHCH3. Еще более предпочтительно, если (R1) обозначает -NH2.
Каждый (R2) независимо выбран из группы, включающей алкил, алкенил, алкинил, циклил, аминогруппу, амидную группу, C-амидную группу, алкиламиногруппу, гидроксигруппу, нитрогруппу, галоген, галогеналкил, галогеналкоксигруппу, цианогруппу, сульфинил, сульфонил, сульфонамидную группу, алкоксигруппу, ацил, карбоксигруппу, карбаматную группу или мочевинную группу. Например, каждый (R2) может быть независимо выбран из группы, включающей гидроксигруппу, галоген (например, -Cl или -F) или галогеналкил (например, -CF3). Соответственно, каждый (R2) может, например, быть независимо выбран из группы, включающей гидроксигруппу или галогеналкил (например, -CF3). Предпочтительно, если каждый (R2) обозначает галоген, более предпочтительно -F.
Каждый (R3) независимо выбран из группы, включающей алкил, алкенил, алкинил, циклил, аминогруппу, амидную группу, C-амидную группу, алкиламиногруппу, гидроксигруппу, нитрогруппу, галоген, галогеналкил, галогеналкоксигруппу, цианогруппу, сульфинил, сульфонил, сульфонамидную группу, алкоксигруппу, ацил, карбоксигруппу, карбаматную группу или мочевинную группу. Например, каждый (R3) может быть независимо выбран из группы, включающей алкил, циклил, аминогруппу, амидную группу, алкиламиногруппу, гидроксигруппу, галоген, галогеналкил, галогеналкоксигруппу, цианогруппу, сульфонамидную группу, алкоксигруппу, ацил, карбоксигруппу, карбаматную группу или мочевинную группу.
n Независимо равен 0, 1, 2, 3 или 4. Например, каждый n может быть независимо равен 0, 1 или 2. В частности, каждый n может быть независимо равен 0 или 1.
Заместители циклопропильного фрагмента, т.е. группа (A) и -NH-CH2-(D) группа, предпочтительно находятся в транс-конфигурации.
В одном предпочтительном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы (I), где (A) обозначает арил или гетероциклил. В более предпочтительном варианте осуществления (A) обозначает фенил, пиридинил, тиофенил, пирролил, фуранил или тиазолил. В еще более предпочтительном варианте осуществления (A) обозначает фенил или пиридинил.
В одном предпочтительном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы (I), в которой (B) обозначает -L2-циклил. В более предпочтительном варианте осуществления (B) обозначает -O-фенил или -O-CH2-фенил. В еще более предпочтительном варианте осуществления (B) обозначает -O-CH2-фенил. В одном предпочтительном варианте осуществления фенильная группа указанной группы (B) содержит 1, 2, 3, или 4 необязательных заместителя (R2), независимо выбранных из группы, включающей алкил, алкенил, алкинил, циклил, аминогруппу, амидную группу, C-амидную группу, алкиламиногруппу, гидроксигруппу, нитрогруппу, галоген, галогеналкил, галогеналкоксигруппу, цианогруппу, сульфинил, сульфонил, сульфонамидную группу, алкоксигруппу, ацил, карбоксигруппу, карбаматную группу или мочевинную группу.
В одном предпочтительном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы (I), в которой (B) обозначает циклил. В более предпочтительном варианте осуществления (B) обозначает фенил. В одном конкретном варианте осуществления фенильная группа указанной группы (B) содержит 1, 2, 3, или 4 необязательных заместителя (R2), независимо выбранных из группы, включающей алкил, алкенил, алкинил, циклил, аминогруппу, амидную группу, C-амидную группу, алкиламиногруппу, гидроксигруппу, нитрогруппу, галоген, галогеналкил, галогеналкоксигруппу, цианогруппу, сульфинил, сульфонил, сульфонамидную группу, алкоксигруппу, ацил, карбоксигруппу, карбаматную группу или мочевинную группу.
В одном предпочтительном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы (I), в которой (R2) обозначает гидроксигруппу, галоген или галогеналкил. В одном предпочтительном варианте осуществления (R2) обозначает -OH или -CF3. В другом предпочтительном варианте осуществления (R2) обозначает фтор или хлор.
В одном предпочтительном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы (I), в которой (D) обозначает моноциклический гетероарил. В более предпочтительном варианте осуществления (D) обозначает тиазолил, оксадиазолил, оксазолил, изоксазолил, тиадиазолил, триазинил, пиридазинил, пиразинил, пиридинил или пиримидинил. В одном конкретном варианте осуществления указанный циклил (D) содержит 1 заместитель (R1).
В одном предпочтительном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы (I), в которой (R1) обозначает группу, связанную водородной связью. Например, (R1) может обозначать -OH, -O(алкил), -NH2, -NH(алкил) (например, -NHCH3), -N(алкил)(алкил), амидную группу, -SO-NH2, -SO-NH(алкил), -SO-N(алкил)(алкил), -S(O)2NH2, -S(O)2NH(алкил), -S(O)2N(алкил)(алкил), -C(=O)NH2, -С(=O)NH(алкил), -С(=O)N(алкил)(алкил), -алкилен-C(=O)NH2 (например, -CH2-C(=O)NH2), -алкилен-C(=O)NH(алкил) (например, -СН2-С(=O)NH(алкил)), -алкилен-C(=O)N(алкил)(алкил) (например, -СН2-С(=O)N(алкил)(алкил)), -NHC(=O)-алкил (например, -NHC(=O)CH3), -N(алкил)-С(=O)-алкил (например, -N(-CH3)-С(=O)CH3), -алкилен-NH2 (например, -СН2-NH2), -алкилен-NH(алкил) или -алкилен-N(алкил)(алкил), где предпочтительно, если указанные выше алкильные и алкиленовые группы все независимо содержат от 1 до 6 атомов углерода. В более предпочтительном варианте осуществления (R1) обозначает -NH2, -OH, амидную группу, -NHC(=O)CH3, -NHCH3 или -S(O)2NH2. В еще более предпочтительном варианте осуществления (R1) обозначает -NH2.
Соединение формулы (I), описанное и определенное в настоящем изобретении может, например, представлять собой соединение следующей формулы (Ia) или его фармацевтически приемлемую соль или сольват:

в которой (A), (B), (D), (R1), (R2), (R3) и n обладают значениями или предпочтительными значениями, указанными в настоящем изобретении для соединения формулы (I).
Предпочтительно, если соединения, предлагаемые в настоящем изобретении, включая, в частности, соединения формулы (I), (Ia) или (Ib), описанные в настоящем изобретении, применяют для лечения заболевания млекопитающего и более предпочтительно человека. Более предпочтительно, если заболевание человека выбрано из группы, включающей рак (например, рак молочной железы, рак легких, рак предстательной железы, колоректальный рак, рак головного мозга, рак кожи, рак крови (например, лейкоз, включая, например, острый миелогенный лейкоз (ОМЛ), хронический миелогенный лейкоз (ХМЛ), хронический нейтрофильный лейкоз, хронический эозинофильный лейкоз, хронический лимфолейкоз (ХЛЛ), острый лимфобластный лейкоз (ОЛЛ) или волосатоклеточный лейкоз), лимфому или миелому), неврологическое патологическое состояние или заболевание (например, депрессия, болезнь Альцгеймера, болезнь Гентингтона, болезнь Паркинсона, боковой амиотрофический склероз, лобно-височное слабоумие или слабоумие с тельцами Леви) или вирусную инфекцию.
В одном предпочтительном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы (Ia), в которой (A) обозначает арил или гетероциклил. В более предпочтительном варианте осуществления (A) обозначает фенил, пиридинил, тиофенил, пирролил, фуранил, и тиазолил. В еще более предпочтительном варианте осуществления (А) обозначает фенил или пиридинил.
В одном предпочтительном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы (Ia), в которой (B) или -L2-циклил. В более предпочтительном варианте осуществления (B) обозначает -O-фенил или -O-CH2-фенил. В еще более предпочтительном варианте осуществления (B) обозначает -O-CH2-фенил. В одном предпочтительном варианте осуществления фенильная группа указанной группы (B) содержит 1, 2, 3, или 4 необязательных заместителя (R2), независимо выбранных из группы, включающей алкил, алкенил, алкинил, циклил, аминогруппу, амидную группу, C-амидную группу, алкиламиногруппу, гидроксигруппу, нитрогруппу, галоген, галогеналкил, галогеналкоксигруппу, цианогруппу, сульфинил, сульфонил, сульфонамидную группу, алкоксигруппу, ацил, карбоксигруппу, карбаматную группу или мочевинную группу.
В одном предпочтительном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы (Ia), в которой (B) обозначает циклил. В более предпочтительном варианте осуществления (B) обозначает фенил. В одном предпочтительном варианте осуществления фенильная группа указанной группы (B) содержит 1, 2, 3, или 4 необязательных заместителя (R2), независимо выбранных из группы, включающей алкил, алкенил, алкинил, циклил, аминогруппу, амидную группу, C-амидную группу, алкиламиногруппу, гидроксигруппу, нитрогруппу, галоген, галогеналкил, галогеналкоксигруппу, цианогруппу, сульфинил, сульфонил, сульфонамидную группу, алкоксигруппу, ацил, карбоксигруппу, карбаматную группу или мочевинную группу.
В одном предпочтительном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы (Ia), в которой (R2) обозначает гидроксигруппу, галоген или галогеналкил. В одном предпочтительном варианте осуществления (R2) обозначает -OH или -CF3. В другом предпочтительном варианте осуществления (R2) обозначает фтор или хлор.
В одном предпочтительном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы (Ia), в которой (D) обозначает моноциклический гетероарил. В более предпочтительном варианте осуществления (D) обозначает тиазолил, оксадиазолил, оксазолил, изоксазолил, тиадиазолил, триазинил, пиридазинил, пиразинил, пиридинил или пиримидинил. В одном предпочтительном варианте осуществления указанный циклил (D) содержит 1 заместитель (R1).
В одном предпочтительном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы (Ia), в которой (R1) обозначает группу, связанную водородной связью. Например (R1) может обозначать -OH, -O(алкил), -NH2, -NH(алкил) (например, -NHCH3), -N(алкил)(алкил), амидную группу, -SO-NH2, -SO-NH(алкил), -SO-N(алкил)(алкил), -S(O)2NH2, -S(O)2NH(алкил), -S(O)2N(алкил)(алкил), -C(=O)NH2, -С(=O)NH(алкил), -С(=O)N(алкил)(алкил), -алкилен-C(=O)NH2 (например, -CH2-C(=O)NH2), -алкилен-C(=O)NH(алкил) (например, -СН2-С(=O)NH(алкил)), -алкилен-C(=O)N(алкил)(алкил) (например, -СН2-С(=O)N(алкил)(алкил)), -NHC(=O)-алкил (например, -NHC(=O)CH3), -N(алкил)-С(=O)-алкил (например, -N(-CH3)-С(=O)CH3), -алкилен-NH2 (например, -СН2-NH2), -алкилен-NH(алкил) или -алкилен-N(алкил)(алкил), где предпочтительно, если указанные выше алкильные и алкиленовые группы все независимо содержат от 1 до 6 атомов углерода. В более предпочтительном варианте осуществления (R1) обозначает -NH2, -OH, амидную группу, -NHC(=O)CH3, -NHCH3 или -S(O)2NH2. В более предпочтительном варианте осуществления (R1) обозначает -NH2, -OH, амидную группу, или -S(O)2NH2. В еще более предпочтительном варианте осуществления (R1) обозначает -NH2.
Таким образом, в предпочтительном объекте настоящее изобретение относится к соединению формулы (Ib) или его фармацевтически приемлемой соли или сольвату, или его применению для лечения или предупреждения заболевания или нарушения:
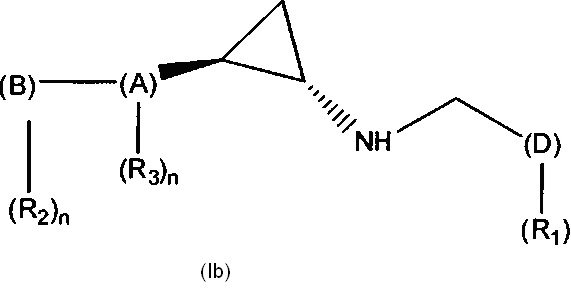
в которой:
(A) обозначает фенильную, пиридинильную, тиофенильную, пирролильную, фуранильную или тиазолильную группу, содержащую n необязательных заместителей (R3);
(B) обозначает -O-CH2-фенил или фенил, где фенильная группа содержит n необязательных заместителей (R2);
(D) обозначает тиазолил, оксадиазолил или пиримидинил, где указанный (D) содержит 1 заместитель (R1);
(R1) обозначает -NH2, -OH, амидную группу, -NHC(=O)CH3, -NHCH3 или -S(O)2NH2;
каждый (R2) независимо выбран из группы, включающей алкил, алкенил, алкинил, циклил, аминогруппу, амидную группу, C-амидную группу, алкиламиногруппу, гидроксигруппу, нитрогруппу, галоген, галогеналкил, галогеналкоксигруппу, цианогруппу, сульфинил, сульфонил, сульфонамидную группу, алкоксигруппу, ацил, карбоксигруппу, карбаматную группу или мочевинную группу;
каждый (R3) независимо выбран из группы, включающей алкил, алкенил, алкинил, циклил, аминогруппу, амидную группу, C-амидную группу, алкиламиногруппу, гидроксигруппу, нитрогруппу, галоген, галогеналкил, галогеналкоксигруппу, цианогруппу, сульфинил, сульфонил, сульфонамидную группу, алкоксигруппу, ацил, карбоксигруппу, карбаматную группу или мочевинную группу; и
n независимо равно 1, 2, 3 или 4.
В одном варианте осуществления этого объекта соединение формулы (Ib) применяют для лечения заболевания млекопитающего и более предпочтительно человека. В другом варианте осуществления заболевание или нарушение выбрано из группы, включающей рак, неврологическое патологическое состояние или заболевание или вирусную инфекцию. В одном варианте осуществления неврологическим заболеванием или нарушением является болезнь Гентингтона, болезнь Паркинсона, болезнь Альцгеймера, боковой амиотрофический склероз или лобно-височное слабоумие.
В другом варианте осуществления этого объекта заболеванием или нарушением является рак. В другом варианте осуществления раком является рак предстательной железы. В другом предпочтительном варианте осуществления этого объекта раком является рак молочной железы. В еще одном предпочтительном варианте осуществления этого объекта раком является рак легких. В еще одном предпочтительном варианте осуществления этого объекта раком является колоректальный рак. В еще одном предпочтительном варианте осуществления этого объекта раком является рак головного мозга. В еще одном предпочтительном варианте осуществления этого объекта раком является рак кожи. В еще одном предпочтительном варианте осуществления этого объекта раком является рак крови (например, лейкоз, включая, например, острый миелогенный лейкоз (ОМЛ), хронический миелогенный лейкоз (ХМЛ), хронический нейтрофильный лейкоз, хронический эозинофильный лейкоз, хронический лимфолейкоз (ХЛЛ), острый лимфобластный лейкоз (ОЛЛ) или волосатоклеточный лейкоз), лимфома или миелома.
В другом предпочтительном варианте осуществления этого объекта, настоящее изобретение относится к соединению формулы (Ib), предназначенному для применения для лечения или предупреждения заболевания или нарушения, в которой (A) обозначает арил или гетероциклил. В более предпочтительном варианте осуществления (A) обозначает фенил, пиридинил, тиофенил, пирролил, фуранил или тиазолил. В еще более предпочтительном варианте осуществления (А) обозначает фенил или пиридинил.
В другом предпочтительном варианте осуществления этого объекта, настоящее изобретение относится к соединению формулы (Ib), предназначенному для применения для лечения или предупреждения заболевания или нарушения, в которой (B) или -L2-циклил. В более предпочтительном варианте осуществления (B) обозначает -O-фенил или -O-CH2-фенил. В еще более предпочтительном варианте осуществления (B) обозначает -O-CH2-фенил. В одном предпочтительном варианте осуществления фенильная группа указанной группы (B) содержит 1, 2, 3, или 4 необязательных заместителя (R2), независимо выбранных из группы, включающей алкил, алкенил, алкинил, циклил, аминогруппу, амидную группу, C-амидную группу, алкиламиногруппу, гидроксигруппу, нитрогруппу, галоген, галогеналкил, галогеналкоксигруппу, цианогруппу, сульфинил, сульфонил, сульфонамидную группу, алкоксигруппу, ацил, карбоксигруппу, карбаматную группу или мочевинную группу.
В другом предпочтительном варианте осуществления этого объекта, настоящее изобретение относится к соединению формулы (Ib), предназначенному для применения для лечения или предупреждения заболевания или нарушения, в которой (B) обозначает циклил. В более предпочтительном варианте осуществления (B) обозначает фенил. В одном предпочтительном варианте осуществления фенильная группа указанной группы (B) содержит 1, 2, 3, или 4 необязательных заместителя (R2), независимо выбранных из группы, включающей алкил, алкенил, алкинил, циклил, аминогруппу, амидную группу, C-амидную группу, алкиламиногруппу, гидроксигруппу, нитрогруппу, галоген, галогеналкил, галогеналкоксигруппу, цианогруппу, сульфинил, сульфонил, сульфонамидную группу, алкоксигруппу, ацил, карбоксигруппу, карбаматную группу или мочевинную группу.
В другом предпочтительном варианте осуществления этого объекта, настоящее изобретение относится к соединению формулы (Ib), предназначенному для применения для лечения или предупреждения заболевания или нарушения, в которой (R2) обозначает гидроксигруппу, галоген или галогеналкил. В одном предпочтительном варианте осуществления (R2) обозначает -OH или -CF3. В другом предпочтительном варианте осуществления (R2) обозначает фтор или хлор.
В другом предпочтительном варианте осуществления этого объекта, настоящее изобретение относится к соединению формулы (Ib), предназначенному для применения для лечения или предупреждения заболевания или нарушения, в которой (D) обозначает моноциклический гетероарил. В более предпочтительном варианте осуществления (D) обозначает тиазолил, оксадиазолил, оксазолил, изоксазолил, тиадиазолил, триазинил, пиридазинил, пиразинил, пиридинил или пиримидинил. В одном предпочтительном варианте осуществления указанный циклил (D) содержит 1 заместитель (R1).
В другом предпочтительном варианте осуществления этого объекта, настоящее изобретение относится к соединению формулы (Ib), предназначенному для применения для лечения или предупреждения заболевания или нарушения, в которой (R1) обозначает группу, связанную водородной связью. Например (R1) может обозначать -OH, -O(алкил), -NH2, -NH(алкил) (например, -NHCH3), -N(алкил)(алкил), амидную группу, -SO-NH2, -SO-NH(алкил), -SO-N(алкил)(алкил), -S(O)2NH2, -S(O)2NH(алкил), -S(O)2N(алкил)(алкил), -C(=O)NH2, -С(=O)NH(алкил), -С(=O)N(алкил)(алкил), -алкилен-C(=O)NH2 (например, -CH2-C(=O)NH2), -алкилен-C(=O)NH(алкил) (например, -СН2-С(=O)NH(алкил)), -алкилен-C(=O)N(алкил)(алкил) (например, -СН2-С(=O)N(алкил)(алкил)), -NHC(=O)-алкил (например, -NHC(=O)CH3), -N(алкил)-С(=O)-алкил (например, -N(-CH3)-С(=O)CH3), -алкилен-NH2 (например, -CH2-NH2), -алкилен-NH(алкил) или -алкилен-N(алкил)(алкил), где предпочтительно, если указанные выше алкильные и алкиленовые группы все независимо содержат от 1 до 6 атомов углерода. В более предпочтительном варианте осуществления (R1) обозначает -NH2, -OH, амидную группу, -NHC(=O)CH3, -NHCH3 или -S(O)2NH2. В еще более предпочтительном варианте осуществления (R1) обозначает -NH2.
Одним объектом настоящего изобретения является стереоизомер или их смесь соединения формулы (I), (Ia) или (Ib).
Другим объектом настоящего изобретения является производное или аналог соединения формулы (I), (Ia) или (Ib).
Еще одним объектом настоящего изобретения является сольват или полиморфная форма соединения формулы (I), (Ia) или (Ib).
Еще одним объектом настоящего изобретения является пролекарство соединения формулы (I), (Ia) или (Ib).
Еще одним объектом настоящего изобретения является метаболит соединения формулы (I), (Ia) или (Ib).
Другим объектом настоящего изобретения является способ лечения или предупреждения заболевания или патологического состояния, включающий введение пациенту (предпочтительно человеку), нуждающемуся в лечении или предупреждении, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы (I), (Ia) или (Ib), определенное выше, или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель. Этот объект можно переформулировать, как соединение формулы (I), (Ia) или (Ib), предназначенное для применения в качестве лекарственного средства. Родственным объектом настоящего изобретения является фармацевтическая композиция, предназначенное для применения для лечения или предупреждения заболевания или патологического состояния, где указанная композиция содержит терапевтически эффективное количество соединения формулы (I), (Ia) или (Ib), достаточное для лечения или предупреждения указанного заболевания или патологического состояния. В более предпочтительном варианте осуществления настоящее изобретение относится к соединению формулы (I), (Ia) или (Ib), предназначенному для применения для лечения заболевания, связанного с LSD1. В другом предпочтительном объекте терапевтически эффективное количество является количеством, достаточным для модулирования степени метилирования гистона. В другом предпочтительном объекте терапевтически эффективное количество соединения формулы (I), (Ia) или (Ib) является количеством, достаточным для модулирования степени метилирования гистон-3-лизина-4. В другом предпочтительном объекте терапевтически эффективное количество является количеством, достаточным для модулирования степени метилирования гистон-3-лизина-9.
Еще одним объектом настоящего изобретения является фармацевтическая композиция, содержащая соединение формулы (I), (Ia) или (Ib) и фармацевтически приемлемый носитель. В более предпочтительном объекте фармацевтическая композиция содержит терапевтически эффективное количество соединения формулы (I), (Ia) или (Ib). В еще более предпочтительном объекте терапевтически эффективное количество соединения формулы (I), (Ia) или (Ib) является количеством, эффективным для ингибирования LSD1. В другом предпочтительном объекте терапевтически эффективное количество является количеством, достаточным для модулирования степени метилирования гистона. В другом предпочтительном объекте терапевтически эффективное количество соединения формулы (I), (Ia) или (Ib) является количеством, достаточным для модулирования степени метилирования гистон-3-лизина-4. В другом предпочтительном объекте терапевтически эффективное количество является количеством, достаточным для модулирования степени метилирования гистон-3-лизина-9.
Еще одним объектом настоящего изобретения является способ ингибирования активности LSD1, включающий введение пациенту, нуждающемуся в лечении, терапевтически эффективного количества композиции, содержащей соединение формулы (I), (Ia) или (Ib) или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель, достаточного для ингибирования активности LSD1. Этот объект можно переформулировать, как соединение формулы (I), (Ia) или (Ib), определенное в настоящем изобретении, предназначенное для применения в качестве ингибитора LSD1. Этот объект также можно переформулировать, как соединение формулы (I), (Ia) или (Ib) для приготовления лекарственного средства, предназначенного для лечения заболевания, связанного с LSD1. Родственным объектом является способ лечения индивидуума, указанный способ включает выявление индивидуума, нуждающегося в лечении, и введение указанному индивидууму терапевтически эффективного количества соединения формулы (I), (Ia) или (Ib). В предпочтительном объекте терапевтически эффективное количество соединения формулы (I), (Ia) или (Ib) является количеством, достаточным для ингибирования LSD1. В другом предпочтительном объекте терапевтически эффективное количество является количеством, достаточным для модулирования степени метилирования гистона. В другом предпочтительном объекте терапевтически эффективное количество соединения формулы (I), (Ia) или (Ib) является количеством, достаточным для модулирования степени метилирования гистон-4-лизина-3. В другом предпочтительном объекте терапевтически эффективное количество является количеством, достаточным для модулирования степени метилирования гистон-3-лизина-9.
Предпочтительные варианты осуществления соединений формулы (I), (Ia) или (Ib), предназначенные для применения в композиции и способе этого четвертого объекта настоящего изобретения являются такими, как определено выше в настоящем изобретении в первом объекте настоящего изобретения.
Еще одним объектом настоящего изобретения является способ лечения или предупреждения рака, включающий введение пациенту, нуждающемуся в лечении, терапевтически эффективного количества композиции, содержащей соединение формулы (I), (Ia) или (Ib), определенное выше в первом объекте настоящего изобретения, и фармацевтически приемлемый носитель. Этот объект можно переформулировать, как соединение формулы (I), (Ia) или (Ib), определенное выше в первом объекте настоящего изобретения, предназначенное для применения для лечения или предупреждения рака. Родственным объектом настоящего изобретения является фармацевтическая композиция, предназначенная для применения для лечения или предупреждения рака, где указанная композиция содержит терапевтически эффективное количество соединения формулы (I), (Ia) или (Ib), достаточное для лечения или предупреждения рака. Другим родственным объектом настоящего изобретения является соединение формулы (I), (Ia) или (Ib) или фармацевтическая композиция, предназначенная для лечения или предупреждения рака, где указанный рак выбран из группы, включающей тестикулярный рак, рак молочной железы, рак легких, рак предстательной железы, колоректальный рак, рак головного мозга, рак кожи, рак крови (например, лейкоз, включая, например, острый миелогенный лейкоз (ОМЛ), хронический миелогенный лейкоз (ХМЛ), хронический нейтрофильный лейкоз, хронический эозинофильный лейкоз, хронический лимфолейкоз (ХЛЛ), острый лимфобластный лейкоз (ОЛЛ) или волосатоклеточный лейкоз), лимфому и миелому, где указанная композиция содержит терапевтически эффективное количество соединения формулы (I), (Ia) или (Ib), достаточное для лечения или предупреждения указанного рака. В предпочтительном объекте терапевтически эффективное количество соединения формулы (I), (Ia) или (Ib) является количеством, достаточным для ингибирования LSD1. В другом предпочтительном объекте терапевтически эффективное количество является количеством, достаточным для модулирования степени метилирования гистона. В другом предпочтительном объекте терапевтически эффективное количество соединения формулы (I), (Ia) или (Ib) является количеством, достаточным для модулирования степени метилирования гистон-3-лизина-4. В другом предпочтительном объекте терапевтически эффективное количество является количеством, достаточным для модулирования степени метилирования гистон-3-лизина-9.
Еще одним объектом настоящего изобретения является способ для лечения или предупреждения неврологического заболевания или нарушения, включающий введение пациенту, нуждающемуся в лечении или предупреждении, терапевтически эффективного количества композиции, содержащей соединение формулы (I), (Ia) или (Ib), определенное выше в первом объекте настоящего изобретения, и фармацевтически приемлемый носитель. Этот объект можно переформулировать, как соединение формулы (I), (Ia) или (Ib), определенное выше в первом объекте настоящего изобретения, предназначенное для применения для лечения или предупреждения неврологического заболевания или нарушения. Родственным объектом настоящего изобретения является фармацевтическая композиция, предназначенная для применения для лечения или предупреждения неврологического заболевания или нарушения, где указанная композиция содержит терапевтически эффективное количество соединения формулы (I), (Ia) или (Ib), достаточное для лечения или предупреждения неврологического заболевания или нарушения. Другим родственным объектом настоящего изобретения является соединение формулы (I), (Ia) или (Ib) или фармацевтическая композиция, предназначенная для лечения или предупреждения неврологического заболевания или нарушения, где указанное неврологическое заболевание или нарушение выбрано из группы, включающей болезнь Гентингтона, болезнь Паркинсона, болезнь Альцгеймера, боковой амиотрофический склероз или лобно-височное слабоумие или слабоумие с тельцами Леви, и, кроме того, где указанная композиция предпочтительно содержит терапевтически эффективное количество соединения формулы (I), (Ia) или (Ib), достаточное для лечения или предупреждения неврологического заболевания или нарушения. В предпочтительном объекте терапевтически эффективное количество соединения формулы (I), (Ia) или (Ib) является количеством, достаточным для ингибирования LSD1. В другом предпочтительном объекте терапевтически эффективное количество соединения формулы (I), (Ia) или (Ib) является количеством, достаточным для модулирования степени метилирования гистон-3-лизина-4
Еще одним объектом настоящего изобретения является способ выявления соединения, которое является селективным ингибитором LSD1, способ включает выбор или получение соединения формулы (I) и определение способности указанного соединения ингибировать LSD1 и MAO-A и/или MAO-B, где соединение, которое ингибирует LSD1 в большей степени, чем MAO-A и/или MAO-B, идентифицируют, как селективный ингибитор LSD1. Таким образом, настоящее изобретение относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель и соединение формулы (I), (Ia) или (Ib), которое является селективным ингибитором LSD1. Селективные ингибиторы LSD1 обладают значениями Ki (IC50) для LSD1, которые меньше, чем значение Ki (IC50) для MAO-A и/или MAO-B. Предпочтительно, если значения Ki (IC50) для LSD1 в два раза меньше, чем для MAO-A и/или MAO-B. В одном воплощении этого варианта осуществления значение Ki для LSD1 по меньшей мере в 5 раз меньше, чем значение Ki (IC50) для MAO-A и/или MAO-B. В одном воплощении этого варианта осуществления значение Ki (IC50) для LSD1 по меньшей мере в 10 раз меньше, чем значение Ki (IC50) для MAO-A и/или MAO-B. В одном варианте осуществления этого объекта настоящего изобретения фармацевтическая композиция, содержащая селективный ингибитор LSD1 формулы (I), (Ia) или (Ib) или его фармацевтически приемлемую соль или сольват, применима для лечения и/или предупреждения заболевания у индивидуума. В одном конкретном варианте осуществления терапевтически эффективное количество композиции вводят индивидууму в количестве, достаточном для предупреждения или лечения заболевания или нарушения. В более предпочтительном варианте осуществления заболеванием является рак, неврологическое заболевание или патологическое состояние или вирусная инфекция. В еще более предпочтительном объекте заболеванием является рак, выбранный из группы, включающей рак предстательной железы, тестикулярный головного мозга, колоректальный, легких, молочной железы, кожи и рак крови. В другом объекте неврологическим заболеванием или нарушением является болезнь Гентингтона, болезнь Паркинсона, болезнь Альцгеймера, боковой амиотрофический склероз, лобно-височное слабоумие или слабоумие с тельцами Леви.
Еще одним объектом настоящего изобретения является способ выявления соединения, которое является двойным ингибитором LSD1 и MAO-B, способ включает выбор или получение соединения формулы (I) и определение способности указанного соединения ингибировать LSD1 и MAO-A и/или MAO-B, где соединение, которое ингибирует LSD1 и MAO-B в большей степени, чем MAO-A, идентифицируют, как двойной ингибитор LSD1/MAO-B. Таким образом, настоящее изобретение относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель и соединение формулы (I), (Ia) или (Ib), которое является двойным ингибитором LSD1/MAO-B. Двойные ингибиторы LSD1/MAO-B обладают значениями Ki (IC50) для LSD1 и MAO-B, которые меньше, чем значение Ki (IC50) для MAO-A. Предпочтительно, если значения Ki (IC50) для LSD1 и MAO-B в два раза меньше, чем для MAO-A. В одном воплощении этого варианта осуществления двойные ингибиторы LSD1/MAO-B обладают значениями Ki (IC50), которые по меньшей мере в 5 раз меньше, чем значение Ki (IC50) для MAO-A. В одном воплощении этого варианта осуществления значения Ki (IC50) для LSD1/MAO-B по меньшей мере в 10 раз меньше, чем значение Ki (IC50) для MAO-A. В одном варианте осуществления этого объекта настоящего изобретения фармацевтическая композиция, содержащая двойной ингибитор LSD1/MAO-B формулы (I), (Ia) или (Ib) или его фармацевтически приемлемую соль или сольват, применима для лечения и/или предупреждения заболевания у индивидуума. В одном конкретном варианте осуществления терапевтически эффективное количество композиции вводят индивидууму в количестве, достаточном для предупреждения или лечения заболевания или нарушения. В более предпочтительном варианте осуществления заболеванием является рак, неврологическое заболевание или патологическое состояние или вирусная инфекция. В еще более предпочтительном объекте заболеванием является рак, выбранный из группы, включающей рак предстательной железы, тестикулярный головного мозга, колоректальный, легких, молочной железы, кожи и рак крови. В другом объекте неврологическим заболеванием или нарушением является болезнь Гентингтона, болезнь Паркинсона, болезнь Альцгеймера, боковой амиотрофический склероз, лобно-височное слабоумие или слабоумие с тельцами Леви.
Недавние исследования показали, что LSD1 участвует в вирусной инфекции и реактивации вируса. В частности, показано, что фармакологические ингибиторы LSD1, такие как парнат и siRNA, лишенная LSD1, приводят к уменьшенной вирулентности и уменьшают реактивацию после латентного периода (Liang et al. (2009) Nat. Med. 15: 1312-1317). Поэтому предполагается, что соединения, предлагаемые в настоящем изобретении, можно использовать для лечения или предупреждения вирусной инфекции. Кроме того, предполагается, что соединения, предлагаемые в настоящем изобретении, могут лечить или предупреждать реактивацию вируса после латентного периода.
Таким образом, другим объектом настоящего изобретения является способ лечения или предупреждения вирусной инфекции, способ включает введение индивидууму (предпочтительно человеку) соединения формулы (I), (Ia) или (Ib), определенное выше в любом из объектов и вариантов осуществления настоящего изобретения или его фармацевтически приемлемую соль или сольват. В соответствии с этим, настоящее изобретение также относится к соединению формулы (I), (Ia) или (Ib), определенному выше в любом из объектов и вариантов осуществления настоящего изобретения или его фармацевтически приемлемой соли или сольвату, предназначенному для применения для лечения или предупреждения вирусной инфекции. В одном конкретном варианте осуществления вирусной инфекцией является инфекция, вызванная вирусом герпеса. В более предпочтительном варианте осуществления инфекция, вызванная вирусом герпеса, вызвана и/или связана с вирусом герпеса, выбранным из группы, включающей HSV-1, HSV-2 и вирус Эпштейна-Барра. В другом варианте осуществления этого седьмого объекта вирусная инфекция вызвана и/или связана с ВИЧ (вирус иммунодефицита человека). В еще более предпочтительном варианте осуществления настоящее изобретение относится к способу лечения или предупреждения реактивации вируса после латентного периода, способ включает введение индивидууму (предпочтительно человеку) соединения формулы (I), (Ia) или (Ib), определенного выше в любом из объектов и вариантов осуществления настоящего изобретения или его фармацевтически приемлемой соли или сольвата. В соответствии с этим, настоящее изобретение также относится к соединению формулы (I), (Ia) или (Ib), определенному выше в любом из объектов и вариантов осуществления настоящего изобретения или его фармацевтически приемлемой соли или сольвату, предназначенному для применения для лечения или предупреждения реактивации вируса после латентного периода. В предпочтительном варианте осуществления вирусом, который реактивируется, является вирус герпеса. В более предпочтительном варианте осуществления вирус герпеса, который реактивируется, выбран из группы, включающей HSV-1, HSV-2 и вирус Эпштейна-Барра. В еще более предпочтительном варианте осуществления вирусом, который реактивируется, является HSV.
Во время проведенного авторами изобретения исследования аминоксидаз, таких как LSD1, MAO-B и MAO-A, неожиданно было установлено, что стереохимическая конфигурация циклопропильных атомов углерода N-замещенных арилциклопропиламинов значительно влияет на активность LSD1 ингибирования. Согласно изобретению было установлено, что (-)-стереоизомер 5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-амина примерно в 20 раз более активен по отношению к LSD1, чем соответствующий (+)-стереоизомер. Кроме того, (-)-стереоизомер сохраняет значительную ингибирующую активность по отношению к MAO-B. Примечательно, что селективность по отношению к LSD1/MAO-A (-)/(+)-стереоизомера боле, чем в 100 раз выше, о чем свидетельствуют значения kinact/KI. Таким образом, (-)-стереоизомеры соединений, предлагаемых в настоящем изобретении, включая, в частности, соединения формулы (I), (Ia) и (Ib), неожиданно оказались активными ингибиторами LSD1 в отличие от соответствующих их энантиомеров. Таким образом, настоящее изобретение относится к соединению формулы (I), (Ia) или (Ib), описанному и определенному в настоящем изобретении, в которой заместители циклопропильного фрагмента (т.е. группа (А) и группа -NH-CH2-(D)) находятся в транс-конфигурации и, кроме того, соединение является оптически активным.
Одним объектом настоящего изобретения является в основном чистый оптически активный стереоизомер соединения формулы (I), (Ia) или (Ib), описанного и определенного в настоящем изобретении, в которой заместители циклопропильного фрагмента (т.е. группа (A) и группа -NH-CH2-(D)) находятся в транс-конфигурации, или его фармацевтически приемлемой соли или сольвату, а также к его применению в качестве лекарственного средства. В предпочтительном объекте в основном чистый, оптически активный стереоизомер соединения формулы (I), (Ia) или (Ib), в которой заместители циклопропильного фрагмента (т.е. группа (А) и группа -NH-CH2-(D)) находятся в транс-конфигурации, содержит 90 мол.% или более (-)-стереоизомера и 10 мол.% или менее (+)-стереоизомера. В более предпочтительном объекте в основном чистый, оптически активный стереоизомер содержит 95 мол.% или более (-)-стереоизомера и 5 мол.% или менее (+)-стереоизомера. В еще более предпочтительном объекте в основном чистый, оптически активный стереоизомер содержит 98 мол.% или более (-)-стереоизомера и 2 мол.% или менее (+)-стереоизомера. В еще более предпочтительном объекте в основном чистый, оптически активный стереоизомер содержит 99 мол.% или более (-)-стереоизомера и 1 мол.% или менее (+)-стереоизомера. В еще более предпочтительном объекте в основном чистый стереоизомер содержит 99,5 мол.% или более (-)-стереоизомера и 0,5 мол.% или менее (+)-стереоизомера. В основном чистый, оптически активный стереоизомер соединения формулы (I), (Ia) или (Ib), в которой заместители циклопропильного фрагмента (т.е. группа (А) и группа -NH-CH2-(D)) находятся в транс-конфигурации, применим для лечения или предупреждения заболевания или нарушения, предпочтительно рака, депрессии, нейродегенеративного заболевания или нарушения или вирусной инфекции.
Настоящее изобретение также относится к композиции, содержащей стереоизомер соединения формулы (I), (Ia) или (Ib), в которой заместители циклопропильного фрагмента (т.е. группа (А) и группа -NH-CH2-(D)) находятся в транс-конфигурации, или его фармацевтически приемлемую соль или сольват, где указанная композиция обладает составляющим 90% или более энантиомерным избытком (-)-стереоизомера соединения. В предпочтительном объекте указанная композиция обладает составляющим 95% или более энантиомерным избытком (-)-стереоизомера соединения. В более предпочтительном объекте указанная композиция обладает составляющим 98% или более энантиомерным избытком (-)-стереоизомера соединения. В еще более предпочтительном объекте указанная композиция обладает составляющим 99% или более энантиомерным избытком (-)-стереоизомера соединения. В одном объекте композиция предназначена для применения для лечения или предупреждения заболевания или нарушения, предпочтительно рака, депрессии, или нейродегенеративного заболевания или нарушения или вирусной инфекции.
В соответствии с этим, настоящее изобретение относится к соединению формулы (I), (Ia) или (Ib), в которой заместители циклопропильного фрагмента (т.е. группа (А) и группа -NH-CH2-(D)) находятся в транс-конфигурации, или его фармацевтически приемлемой соли или сольвату, где (-)-стереоизомер указанного соединения содержится в энантиомерном избытке, составляющем 90% или более, предпочтительно 95% или более, более предпочтительно 98% или более и еще более предпочтительно 99% или более.
Другим объектом настоящего изобретения является фармацевтическая композиция, содержащая стереоизомер соединения формулы (I), (Ia) или (Ib), в которой заместители циклопропильного фрагмента (т.е. группа (A) и группа -NH-CH2-(D)) находятся в транс-конфигурации, или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель, где указанная композиция обладает составляющим 90% или более энантиомерным избытком (-)-стереоизомера соединения. В предпочтительном объекте указанная композиция обладает составляющим 95% или более энантиомерным избытком (-)-стереоизомера соединения. В предпочтительном объекте указанная композиция обладает составляющим 99% или более энантиомерным избытком (-)-стереоизомера соединения. Фармацевтическая композиция этого объекта являются особенно подходящими для лечения или предупреждения рака, депрессии, нейродегенеративного заболевания или нарушения или вирусной инфекции.
В одном объекте настоящее изобретение относится к оптически активному соединению формулы (I), (Ia) или (Ib), в которой заместители циклопропильного фрагмента (т.е. группа (A) и группа -NH-CH2-(D)) находятся в трансконфигурации, или его фармацевтически приемлемой соли или сольвату, или композиции, содержащей любое из указанных выше соединений, где указанное соединение представляет собой (1R,2S)-энантиомер (применительно к заместителям циклопропильного кольца), в основном не содержащий (1S,2R)-энантиомера. Предпочтительно, если соединение содержит более 90 мол.% (1R,2S)-энантиомера и менее 10 мол.% (1S,2R)-энантиомера. Более предпочтительно, если соединение содержит более 95 мол.% (1R,2S)-энантиомера и менее 5 мол.% (1S,2R)-энантиомера. Еще более предпочтительно, если соединение содержит более 98 мол.% (1R,2S)-энантиомера и менее 2 мол.% (1S,2R)-энантиомера. Еще более предпочтительно, если соединение содержит более 99 мол.% (1R,2S)-энантиомера и менее 1 мол.% (1S,2R)-энантиомера. Еще более предпочтительно, если соединение содержит более 99,5 мол.% (1R,2S)-энантиомера и менее 0,5 мол.% (1S,2R)-энантиомера. Содержание энантиомера можно определить, например, с помощью хиральной ВЭЖХ (например, как это описано в примере 36).
В одном объекте настоящее изобретение относится к оптически активному соединению формулы (I), (Ia) или (Ib), в которой заместители циклопропильного фрагмента (т.е. группа (А) и группа -NH-CH2-(D)) находятся в трансконфигурации, или его фармацевтически приемлемой соли или сольвату, или композиции, содержащей любое из указанных выше соединений, где указанное соединение представляет собой (1S,2R)-энантиомер (применительно к заместителям циклопропильного кольца), в основном не содержащий (1R,2S)-энантиомера. Предпочтительно, если соединение содержит более 90 мол.% (1S,2R)-энантиомера и менее 10 мол.% (1R,2S)-энантиомера. Более предпочтительно, если соединение содержит более 95 мол.% (1S,2R)-энантиомера и менее 5 мол.% (1R,2S)-энантиомера. Еще более предпочтительно, если соединение содержит более 98 мол.% (1S,2R)-энантиомера и менее 2 мол.% (1R,2S)-энантиомера. Еще более предпочтительно, если соединение содержит более 99 мол.% (1S,2R)-энантиомера и менее 1 мол.% (1R,2S)-энантиомера. Еще более предпочтительно, если соединение содержит более 99,5 мол.% (1S,2R)-эанантиомера и менее 0,5 мол.% (1R,2S)-энантиомера. Содержание энантиомера можно определить, например, с помощью хиральной ВЭЖХ (например, как это описано в примере 36).
В одном объекте настоящее изобретение относится к оптически активному соединению формулы (I), (Ia) или (Ib), в которой заместители циклопропильного фрагмента (т.е. группа (А) и группа -NH-CH2-(D)) находятся в трансконфигурации, или его фармацевтически приемлемой соли или сольвату, в которой кольцевой атом углерода циклопропила, который связан с аминогруппой соединения, обладает (S)-конфигурацией и кольцевой атом углерода циклопропила, который связан с циклической группой (A), присоединенной к циклопропильному кольцу соединения, обладает (R)-конфигурацией. Предпочтительно, если соединение обладает энантиомерным избытком, составляющим не менее 90%. Еще более предпочтительно, если соединение обладает энантиомерным избытком, составляющим не менее 95%. Еще более предпочтительно, если соединение обладает энантиомерным избытком, составляющим не менее 98%. Еще более предпочтительно, если соединение обладает энантиомерным избытком, составляющим не менее 99%. Энантиомерный избыток можно определить, например, с помощью хиральной ВЭЖХ (например, как это описано в примере 36).
В одном объекте настоящее изобретение относится к оптически активному соединению формулы (I), (Ia) или (Ib), в которой заместители циклопропильного фрагмента (т.е. группа (А) и группа -NH-CH2-(D)) находятся в трансконфигурации, или его фармацевтически приемлемой соли или сольвату, в которой кольцевой атом углерода циклопропила, который связан с аминогруппой соединения, обладает (R)-конфигурацией и кольцевой атом углерода циклопропила, который связан с циклической группой (А), присоединенной к циклопропильному кольцу соединения, обладает (S)-конфигурацией. Предпочтительно, если соединение обладает энантиомерным избытком, составляющим не менее 90%. Еще более предпочтительно, если соединение обладает энантиомерным избытком, составляющим не менее 95%. Еще более предпочтительно, если соединение обладает энантиомерным избытком, составляющим не менее 98%. Еще более предпочтительно, если соединение обладает энантиомерным избытком, составляющим не менее 99%. Энантиомерный избыток можно определить, например, с помощью хиральной ВЭЖХ (например, как это описано в примере 36).
В одном объекте настоящее изобретение относится к оптически активному соединению формулы (I), (Ia) или (Ib), в которой заместители циклопропильного фрагмента (т.е. группа (A) и группа -NH-CH2-(D)) находятся в трансконфигурации, или его фармацевтически приемлемой соли или сольвату, предназначенному для применения для лечения или предупреждения заболевания или нарушения, такого как, например, рак, неврологическое заболевание, нарушение или патологическое состояние или вирусная инфекция.
В одном объекте неврологическим заболеванием, нарушением или патологическим состоянием является депрессия, болезнь Гентингтона, болезнь Паркинсона, болезнь Альцгеймера, боковой амиотрофический склероз, лобно-височное слабоумие или слабоумие с тельцами Леви. В одном предпочтительном объекте раком является рак предстательной железы. В другом предпочтительном объекте раком является рак молочной железы. В другом объекте раком является рак легких. В другом объекте раком является колоректальный рак. В другом предпочтительном объекте раком является рак головного мозга. В другом предпочтительном объекте раком является рак кожи. В другом предпочтительном объекте раком является рак крови (например, лейкоз или лимфома; лейкоз, который лечат или предупреждают, включает, например, острый миелогенный лейкоз (ОМЛ), хронический миелогенный лейкоз (ХМЛ), хронический нейтрофильный лейкоз, хронический эозинофильный лейкоз, хронический лимфолейкоз (ХЛЛ), острый лимфобластный лейкоз (ОЛЛ) или волосатоклеточный лейкоз). В одном объекте неврологическим заболеванием, нарушением или патологическим состоянием является депрессия, болезнь Гентингтона, болезнь Паркинсона или болезнь Альцгеймера. В одном объекте вирусной инфекцией является инфекция, вызванная HSV1 или HSV2. В одном объекте заболеванием или нарушением является депрессия. В одном объекте неврологическим заболеванием, нарушением или патологическим состоянием является нейродегенеративное заболевание, нарушение или патологическое состояние. В одном объекте нейродегенеративным заболеванием, нарушением или патологическим состоянием является болезнь Гентингтона, болезнь Паркинсона, болезнь Альцгеймера, боковой амиотрофический склероз или лобно-височное слабоумие.
Настоящее изобретение также относится к оптически активному соединению формулы (I), (Ia) или (Ib), в которой заместители циклопропильного фрагмента (т.е. группа (А) и группа -NH-CH2-(D)) находятся в трансконфигурации, или его фармацевтически приемлемой соли или сольвату, определенному в любом из приведенных выше вариантов осуществления или объектов, предназначенному для применения для лечения или предупреждения заболевания или нарушения, предпочтительно рака (например, рак молочной железы, рак легких, рак предстательной железы, колоректальный рак, рак головного мозга, рак кожи, рак крови, лейкоз (включая, например, острый миелогенный лейкоз (ОМЛ), хронический миелогенный лейкоз (ХМЛ), хронический нейтрофильный лейкоз, хронический эозинофильный лейкоз, хронический лимфолейкоз (ХЛЛ), острый лимфобластный лейкоз (ОЛЛ) или волосатоклеточный лейкоз), лимфому или миелому), неврологического заболевания или патологического состояния (например, депрессии, болезни Альцгеймера, болезни Гентингтона, болезни Паркинсона или слабоумия с тельцами Леви) или вирусной инфекции (например, вирусной инфекции, вызванной и/или связанной с ВИЧ, или инфекции, вызванной вирусом герпеса, такой как инфекция, вызванная вирусом герпеса, вызванная и/или связанная с вирусом герпеса, выбранным из группы, включающей HSV-1, HSV-2 или вирус Эпштейна-Барра) у субъекта (предпочтительно млекопитающего, более предпочтительно человека).
Настоящее изобретение также относится к оптически активному соединению формулы (I), (Ia) или (Ib), в которой заместители циклопропильного фрагмента (т.е. группа (A) и группа -NH-CH2-(D)) находятся в трансконфигурации, или его фармацевтически приемлемой соли или сольвату, определенному в любом из приведенных выше вариантов осуществления или объектов, предназначенному для применения для лечения или предупреждения заболевания или нарушения, где указанным заболеванием или нарушением является нейродегенеративное заболевание или нарушение. В одном объекте нейродегенеративным заболеванием или нарушением является болезнь Гентингтона, болезнь Паркинсона, болезнь Альцгеймера, боковой амиотрофический склероз или лобно-височное слабоумие.
Другим предпочтительным объектом настоящего изобретения является оптически активный N-замещенный арил- или гетероарилциклопропиламин или его фармацевтически приемлемая соль или сольват, предназначенный для применения в способе лечения или предупреждения заболевания или нарушения. Предпочтительно, если оптически активный N-замещенный арил- или гетероарилциклопропиламин, описанный в настоящем изобретении, представляет собой соединение формулы (II) или его фармацевтически приемлемую соль или сольват:
в которой:
(AII) обозначает арильную или гетероарильную группу, содержащую 2 заместителя, R1II и R2II, и 1, 2 или 3 необязательных заместителя, где указанные необязательные заместители независимо выбраны из группы, включающей галоген, C1-C3-алкил или C1-C3-алкоксигруппу;
R1II обозначает группу ;
R3II обозначает арильную или гетероарильную группу, содержащую 1, 2, 3, 4 или 5 необязательных заместителей, независимо выбранных из группы, включающей галоген, -OH, -NHSO2RA, алкил, алкоксигруппу, цианогруппу, -CF3 или -OCF3, где RA обозначает C1-C6-алкил или фенил;
выбран из группы, включающей связь, -CH2O-, -CH2CH2O-, -OCH2-, -OCH2CH2-, -CH2CH2-, -CH2-, -CH2CH2CH2- или -O-;
R2II обозначает , где указанная циклопропильная группа содержит 2 хиральных центра, замещенных в транс-положении по отношению к атомам углерода, к которым ковалентно присоединены (A) и ;
R4II обозначает 5- или 6-членное гетероарильное кольцо, содержащее 1, 2 или 3 необязательных заместителя, выбранных из группы, включающей алкил, -NH-RB, -ORB или галоген, где RB обозначает водород, C1-C3-алкил или -C(=O)CH3;
обозначает разветвленную или неразветвленную C1-C4-алкиленовую группу и где указанное соединение формулы (II) является оптически активным.
Оптически активные соединения, предназначенные для применения в способах, предлагаемых в настоящем изобретении, и оптически активные соединения, предлагаемые в настоящем изобретении, означают обогащенные стереоизомеры соединений, где обогащение относится к хиральным центрам, соответствующим энантиомерам транс-замещенного циклопропильного фрагмента. Другие хиральные центры могут содержаться или не содержаться в молекуле и конфигурация или оптическое вращение, приписываемые этим центрам, не входят в объем настоящего изобретения, например, их влияние на LSD1, MAO-A, или MAO-B.
Настоящее изобретение также относится к фармацевтической композиции, содержащей оптически активный N-замещенный арил- или гетероарилциклопропиламин формулы (II), определенный выше, или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к фармацевтической композиции, содержащей оптически активный N-замещенный арил- или гетероарилциклопропиламин формулы (II), определенный выше, или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель, предназначенной для применения для лечения или предупреждения заболевания или нарушения.
Предпочтительно, если оптически активный N-замещенный арил- или гетероарилциклопропиламин или его фармацевтически приемлемая соль или сольват, предназначенный для применения в способе лечения или предупреждения заболевания или нарушения, описанном в настоящем изобретении, описывается формулой (II):

в которой:
(AII) обозначает арильную или гетероарильную группу, содержащую 2 заместителя, R1II и R2II, и 1, 2 или 3 необязательных заместителя, независимо выбранных из группы, включающей галоген, C1-C3-алкил или C1-C3-алкоксигруппу;
R1II обозначает группу ;
R3II обозначает фенильную, пиридильную, тиазолильную или тиенильную группу, содержащую 1, 2 или 3 необязательных заместителя, независимо выбранных из группы, включающей галоген, -OH, -NHSO2RA, алкил, алкоксигруппу, цианогруппу, -CF3 или -OCF3, где RA обозначает C1-C6-алкил или фенил;
выбран из группы, включающей связь, -CH2O- или -CH2O-;
R2II обозначает , где указанная циклопропильная группа содержит 2 хиральных центра, замещенных в транс-положении по отношению к атомам углерода, к которым ковалентно присоединены (AII) и ;
R4II обозначает 5-членное гетероарильное кольцо, содержащее 1, 2 или 3 необязательных заместителя, независимо выбранных из группы, включающей - NH2 или -NH(C1-C3)алкил;
обозначает -CH2- или -CH2CH2-;
и где указанное соединение формулы (II) является оптически активным.
Настоящее изобретение также относится к фармацевтической композиции, содержащей оптически активный N-замещенный арил- или гетероарилциклопропиламин формулы (II), определенный выше, или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к фармацевтической композиции, содержащей оптически активный N-замещенный арил- или гетероарилциклопропиламин формулы (II), определенный выше, или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель, предназначенной для применения для лечения или предупреждения заболевания или нарушения.
Кроме того, согласно изобретению неожиданно установлено, что подгруппа оптически активных соединений формулы (II), описывающаяся формулой (III), являются активными и селективными ингибиторами LSD1 или LSD1 и MAO-B.
Поэтому настоящее изобретение относится к оптически активному соединению формулы (III) или его фармацевтически приемлемой соли или сольвату:

в которой:
(AIII) обозначает арильную или гетероарильную группу, содержащую 2 заместителя, R1III и R2III, и 1, 2 или 3 необязательных заместителя, где указанные необязательные заместители независимо выбраны из группы, включающей галоген, C1-C3-алкил или C1-C3-алкоксигруппу;
R1III обозначает группу ;
R3III обозначает фенильную, пиридильную, тиазолильную или тиенильную группу, содержащую 1, 2, 3, 4 или 5 необязательных заместителей, независимо выбранных из группы, включающей галоген, -OH, -NHSO2RA, алкил, алкоксигруппу, цианогруппу, -CF3 или -OCF3, где RA обозначает C1-C6-алкил или фенил;
выбран из группы, включающей связь, - OCH2- или -CH2O-;
R2III обозначает , где указанная циклопропильная группа содержит 2 хиральных центра, замещенных в транс-положении по отношению к атомам углерода, к которым ковалентно присоединены (AIII) и ;
R4III обозначает 5-членное гетероарильное кольцо, содержащее 1, 2 или 3 необязательных заместителя, независимо выбранных из группы, включающей -NH2 или -NH(C1-C3)алкил;
обозначает -СН2- или -СН2СН2-;
и где указанное соединение формулы (III) является оптически активным.
Предпочтительно, если оптически активное соединение формулы (III) является следующим:

в которой:
(AIII) обозначает фенильную или пиридильную группу, содержащую 2 заместителя, R1III и R2III;
R1III обозначает группу ;
R3III обозначает фенил, содержащий 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -F, -Cl, -OH, -NHSO2RA, C1-C3-алкил, C1-C3-алкоксигруппу, цианогруппу, -CF3, или -OCF3, где RA обозначает C1-C6-алкил или фенил;
выбран из группы, включающей связь, - ОСН2- или -CH2O-;
R2III обозначает , где указанная циклопропильная группа содержит 2 хиральных центра, замещенных в транс-положении по отношению к атомам углерода, к которым ковалентно присоединены (AIII) и ;
R4 обозначает 5-членное гетероарильное кольцо, где указанная цепь атомов, образующая указанное 5-членное гетероарильное кольцо, содержит 2 или 3 гетероатома, независимо выбранных из группы, включающей N, S, или O, где указанное 5-членное гетероарильное кольцо содержит 1 необязательный заместитель, где указанный необязательный заместитель представляет собой -NH2 или -NH(C1-C3)алкил;
обозначает -CH2- или -CH2CH2-;
или его фармацевтически приемлемую соль или сольват,
и где указанное соединение формулы (III) является оптически активным.
Настоящее изобретение также относится к фармацевтической композиции, содержащей оптически активный N-замещенный арил- или гетероарилциклопропиламин формулы (III), определенный выше, или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к фармацевтической композиции, содержащей оптически активный N-замещенный арил- или гетероарилциклопропиламин формулы (III), определенный выше, или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель, предназначенной для применения для лечения или предупреждения заболевания или нарушения.
Еще более предпочтительно, если оптически активное соединение формулы (III) является следующим:

в которой:
(AIII) обозначает фенильную или пиридильную группу, содержащую 2 заместителя, R1III и R2III;
R1III обозначает группу ;
R3III обозначает фенил, содержащий 1, 2 или 3 необязательных заместителя, независимо выбранных из группы, включающей -F, -Cl, -OH, -NHSO2CH3, метил, метоксигруппу, цианогруппу, -CF3 или -OCF3;
выбран из группы, включающей связь, -OCH2- или -CH2O-;
R2III обозначает , где указанная циклопропильная группа содержит 2 хиральных центра, замещенных в транс-положении по отношению к атомам углерода, к которым ковалентно присоединены (AIII) и ;
R4III обозначает оксадиазолильное, тиадиазолильное или тиазолильное кольцо, содержащее 1 необязательный заместитель, где указанный необязательный заместитель представляет собой -NH2 или -NH(C1-C3)алкил;
обозначает -CH2- или -СН2СН2-;
или его фармацевтически приемлемую соль или сольват,
и где указанное соединение формулы (III) является оптически активным.
Настоящее изобретение также относится к фармацевтической композиции, содержащей оптически активный N-замещенный арил- или гетероарилциклопропиламин формулы (III), определенный выше, или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к фармацевтической композиции, содержащей оптически активный N-замещенный арил- или гетероарилциклопропиламин формулы (III), определенный выше, или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель, предназначенной для применения для лечения или предупреждения заболевания или нарушения.
Предпочтительно, если оптически активное соединение формулы (III) является следующим:

в которой:
(AIII) обозначает фенильную или пиридильную группу, содержащую 2 заместителя, R1III и R2III;
R1III обозначает группу ;
R3III обозначает фенил, содержащий 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -F, -Cl, -OH, -NHSO2RA, C1-C3-алкил, C1-C3-алкоксигруппу, цианогруппу, -CF3 или -OCF3, где RA обозначает C1-C6-алкил или фенил;
выбран из группы, включающей связь, -ОСН2- или -CH2O-;
R2III обозначает , где указанная циклопропильная группа содержит 2 хиральных центра, замещенных в транс-положении по отношению к атомам углерода, к которым ковалентно присоединены (AIII) и ;
R4III обозначает 5-членное гетероарильное кольцо, где указанная цепь атомов, образующая указанный 5-членное гетероарильное кольцо, содержит 2 или 3 гетероатома, независимо выбранных из группы, включающей N, S, или O, содержит 1 необязательный заместитель, где указанный необязательный заместитель представляет собой -NH2 или -NH(C1-C3)алкил;
обозначает -CH2- или -СН2СН2-;
или его фармацевтически приемлемую соль или сольват,
и где указанное соединение формулы (III) является оптически активным.
Настоящее изобретение также относится к фармацевтической композиции, содержащей оптически активный N-замещенный арил- или гетероарилциклопропиламин формулы (III), определенный выше, или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к фармацевтической композиции, содержащей оптически активный N-замещенный арил- или гетероарилциклопропиламин формулы (III), определенный выше, или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель, предназначенной для применения для лечения или предупреждения заболевания или нарушения.
Еще более предпочтительно, если оптически активное соединение формулы (III) является следующим:
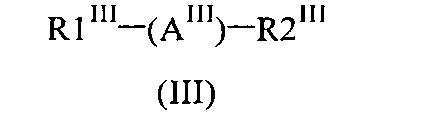
в которой:
(AIII) обозначает фенильную или пиридильную группу, содержащую 2 заместителя, R1III и R2III;
R1III обозначает группу ;
R3III обозначает фенил, содержащий 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -F, -Cl, -OH, -NHSO2CH3, метил, метоксигруппу, цианогруппу, -CF3 или -OCF3;
выбран из группы, включающей связь, -OCH2- или -CH2O-;
R2III обозначает , где указанная циклопропильная группа содержит 2 хиральных центра, замещенных в транс-положении по отношению к атомам углерода, к которым ковалентно присоединены (AIII) и ;
R4III обозначает гетероарильную группу, которая является изостерой амидной группы, содержащую 1 необязательный заместитель, где указанный необязательный заместитель представляет собой -NH2 или -NH(C1-C3)алкил;
обозначает -CH2- или -CH2CH2-;
или его фармацевтически приемлемую соль или сольват,
и где указанное соединение формулы (III) является оптически активным.
Настоящее изобретение также относится к фармацевтической композиции, содержащей оптически активный N-замещенный арил- или гетероарилциклопропиламин формулы (III), определенный выше, или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к фармацевтической композиции, содержащей оптически активный N-замещенный арил- или гетероарилциклопропиламин формулы (III), определенный выше, или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель, предназначенной для применения для лечения или предупреждения заболевания или нарушения.
Одним объектом настоящего изобретения является оптически активный N-замещенный арил- или гетероарилциклопропиламин, предназначенный для применения в способе лечения или предупреждения заболевания или нарушения. В предпочтительном объекте заболеванием или нарушением является заболевание или нарушение человека, выбранное из группы, включающей рак, депрессию, нейродегенеративное заболевание или патологическое состояние или вирусную инфекцию. В одном объекте нейродегенеративным заболеванием или нарушением является болезнь Гентингтона, болезнь Паркинсона, болезнь Альцгеймера, боковой амиотрофический склероз, лобно-височное слабоумие или слабоумие с тельцами Леви. В предпочтительном объекте оптически активный N-замещенный арил- или гетероарилциклопропиламин является таким, как определено для формулы (II) или (III).
Одним объектом настоящего изобретения является в основном чистый стереоизомер N-замещенного арил- или гетероарилциклопропиламина (например, соединение формулы (II) или (III), описанное в настоящем изобретении) или его фармацевтически приемлемая соль или сольват, предназначенный для применения в способе лечения или предупреждения заболевания или нарушения. В родственном объекте способ включает введение индивидууму терапевтически эффективного количества в основном чистого стереоизомера N-замещенного арил- или гетероарилциклопропиламина. Предпочтительно, если заболеванием или нарушением является такое, которое можно лечить или предупреждать путем ингибирования LSD1, ингибирования LSD1 и ингибирования MAO-B или ингибирования MAO-B. В предпочтительном объекте в основном чистый стереоизомер N-замещенного арил- или гетероарилциклопропиламина означает N-замещенный арил- или гетероарилциклопропиламин, который содержит 90% или более (-)-стереоизомера и 10% или менее (+)-стереоизомера. В более предпочтительном объекте в основном чистый стереоизомер N-замещенного арил- или гетероарилциклопропиламина означает N-замещенный арил- или гетероарилциклопропиламин, который содержит 95% или более (-)-стереоизомера и 5% или менее (+)-стереоизомера. В еще более предпочтительном объекте в основном чистый стереоизомер N-замещенного арил- или гетероарилциклопропиламина означает N-замещенный арил- или гетероарилциклопропиламин, который содержит 98% или более (-)-стереоизомера и 2% или менее (+)-стереоизомера. В еще более предпочтительном объекте в основном чистый стереоизомер N-замещенного арил- или гетероарилциклопропиламина означает N-замещенный арил- или гетероарилциклопропиламин, который содержит 99% или более (-)-стереоизомера и 1% или менее (+)-стереоизомера. В еще более предпочтительном объекте в основном чистый стереоизомер N-замещенного арил- или гетероарилциклопропиламина означает N-замещенный арил- или гетероарилциклопропиламин, который содержит 99,5% или более (-)-стереоизомера и 0,5% или менее (+)стереоизомера. В одном варианте осуществления описанные выше выраженные в процентах содержания являются молярными. В основном чистый стереоизомер N-замещенного арил- или гетероарилциклопропиламина этого объекта применим для лечения или предупреждения рака, депрессии, нейродегенеративного заболевания или нарушения или вирусной инфекции. В предпочтительном объекте оптически активный N-замещенный арил- или гетероарилциклопропиламин является таким, как определено для формулы (II) или (III).
Кроме того, другим объектом настоящего изобретения является композиция, содержащая стереоизомер N-замещенного арил- или гетероарилциклопропиламина (например, соединение формулы (II) или (III), описанное в настоящем изобретении), или его фармацевтически приемлемую соль или сольват, где указанная композиция обладает составляющим 90% или более энантиомерным избытком (-)-стереоизомера N-замещенного арил- или гетероарилциклопропиламина. В предпочтительном объекте указанная композиция обладает составляющим 95% или более энантиомерным избытком (-)-стереоизомера N-замещенного арил- или гетероарилциклопропиламина. В более предпочтительном объекте указанная композиция обладает составляющим 98%) или более энантиомерным избытком (-)-стереоизомера N-замещенного арил- или гетероарилциклопропиламина. В еще более предпочтительном объекте указанная композиция обладает составляющим 99% или более энантиомерным избытком (-)-стереоизомера N-замещенного арил- или гетероарилциклопропиламина. В одном объекте композиция предназначена для применения для лечения или предупреждения рака, депрессии, или нейродегенеративного заболевания или нарушения или вирусной инфекции. В предпочтительном объекте оптически активный N-замещенный арил- или гетероарилциклопропиламин является таким, как определено для формулы (II) или (III).
Кроме того, другим объектом настоящего изобретения является фармацевтическая композиция, содержащая стереоизомер N-замещенного арил- или гетероарилциклопропиламина (например, соединение формулы (II) или (III), описанное в настоящем изобретении), или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель, где указанная композиция обладает составляющим 90% или более энантиомерным избытком (-)-стереоизомера N-замещенного арил- или гетероарилциклопропиламина. В предпочтительном объекте указанная композиция обладает составляющим 95% или более энантиомерным избытком (-)-стереоизомера N-замещенного арил- или гетероарилциклопропиламина. В предпочтительном объекте указанная композиция обладает составляющим 99% или более энантиомерным избытком (-)-стереоизомера N-замещенного арил- или гетероарилциклопропиламина. Фармацевтическая композиция этого объекта применим для лечения или предупреждения рака, депрессии, нейродегенеративного заболевания или нарушения или вирусной инфекции. В предпочтительном объекте оптически активный N-замещенный арил- или гетероарилциклопропиламин является таким, как определено для формулы (II) или (III).
Одним объектом настоящего изобретения является композиция, определенная в настоящем изобретении, содержащая оптически активный N-замещенный арил- или гетероарилциклопропиламин формулы (II) или (III), описанный в любом из приведенных выше вариантов осуществления или объектов, или его сольват или фармацевтически приемлемую соль, где указанный N-замещенный арил- или гетероарилциклопропиламин представляет собой (1R,2S)-энантиомер (применительно к заместителям циклопропильного кольца), в основном не содержащий (1S,2R)-энантиомера. Предпочтительно, если N-замещенный арил- или гетероарилциклопропиламин содержит более 90% (1R,2S)-энантиомера и менее 10% (1S,2R)-энантиомера. Более предпочтительно, если N-замещенный арил- или гетероарилциклопропиламин содержит более 95% (1R,2S)-энантиомера и менее 5% (1S,2R)-энантиомера. Еще более предпочтительно, если N-замещенный арил- или гетероарилциклопропиламин содержит более 98% (1R,2S)-энантиомера и менее 2% (1S,2R)-энантиомера. Еще более предпочтительно, если N-замещенный арил- или гетероарилциклопропиламин содержит более 99% (1R,2S)-энантиомера и менее 1% (1S,2R)-энантиомера. Еще более предпочтительно, если N-замещенный арил- или гетероарилциклопропиламин содержит более 99,5% (1R,2S)-энантиомера и менее 0,5% (1S,2R)-энантиомера. Содержание энантиомера можно определить, например, с помощью хиральной ВЭЖХ например (описанной в примере 36).
Одним объектом настоящего изобретения является композиция, определенная в настоящем изобретении, содержащая оптически активный N-замещенный арил- или гетероарилциклопропиламин формулы (II) или (III), описанный в любом из приведенных выше вариантов осуществления или объектов, или его сольват или фармацевтически приемлемую соль, где указанный N-замещенный арил- или гетероарилциклопропиламин представляет собой (1S,2R)-энантиомер (применительно к заместителям циклопропильного кольца), в основном не содержащий (1R,2S)-энантиомера. Предпочтительно, если N-замещенный арил- или гетероарилциклопропиламин содержит более 90% (1S,2R)-энантиомера и менее 10% (1R,2S)-энантиомера. Более предпочтительно, если N-замещенный арил- или гетероарилциклопропиламин содержит более 95% (1S,2R)-энантиомера и менее 5% (1R,2S)-энантиомера. Еще более предпочтительно, если N-замещенный арил- или гетероарилциклопропиламин содержит более 98% (1S,2R)-энантиомера и менее 2% (1R,2S)-энантиомера. Еще более предпочтительно, если N-замещенный арил- или гетероарилциклопропиламин содержит более 99% (1S,2R)-энантиомера и менее 1% (1R,2S)-энантиомера. Еще более предпочтительно, если N-замещенный арил- или гетероарилциклопропиламин содержит более 99,5% (1S,2R)-энантиомера и менее 0,5% (1R,2S)-энантиомера. Содержание энантиомера можно определить, например, с помощью хиральной ВЭЖХ например (описанной в примере 36).
Одним объектом настоящего изобретения является оптически активный N-замещенный арил- или гетероарилциклопропиламин (например, соединение формулы (II) или (III)), определенный в любом из приведенных выше вариантов осуществления или объектов, или сольват или его фармацевтически приемлемая соль или сольват, где кольцевой атом углерода циклопропила, который связан с аминогруппой N-замещенного арил- или гетероарилциклопропиламина обладает (S)-абсолютной конфигурацией и кольцевой атом углерода циклопропила, который связан с циклической группой, соседней с циклопропильным кольцом N-замещенного арил- или гетероарилциклопропиламина, обладает (R)-абсолютной конфигурацией. Предпочтительно, если указанный N-замещенный арил- или гетероарилциклопропиламин получают с энантиомерным избытком, составляющим не менее 90%. Еще более предпочтительно, если указанный N-замещенный арил- или гетероарилциклопропиламин получают с энантиомерным избытком, составляющим не менее 95%. Еще более предпочтительно, если указанное соединение получают с энантиомерным избытком, составляющим не менее 98%. Еще более предпочтительно, если указанный N-замещенный арил- или гетероарилциклопропиламин получают с энантиомерным избытком, составляющим не менее 99%. Энантиомерный избыток можно определить, например, с помощью хиральной ВЭЖХ например (описанной в примере 36).
Одним объектом настоящего изобретения является оптически активный N-замещенный арил- или гетероарилциклопропиламин (например, соединение формулы (II) или (III)), определенный в любом из приведенных выше вариантов осуществления или объектов, или его фармацевтически приемлемая соль или сольват, где кольцевой атом углерода циклопропила, который связан с аминогруппой соединения, обладает (R)-абсолютной конфигурацией и кольцевой атом углерода циклопропила, который связан с циклической группой, соседней с циклопропильным кольцом соединения, обладает (S)-абсолютной конфигурацией. Предпочтительно, указанное соединение получают с энантиомерным избытком, составляющим не менее 90%. Еще более предпочтительно, если указанное соединение получают с энантиомерным избытком, составляющим не менее 95%. Еще более предпочтительно, если указанное соединение получают с энантиомерным избытком, составляющим не менее 98%. Еще более предпочтительно, если указанное соединение получают с энантиомерным избытком, составляющим не менее 99%. Энантиомерный избыток можно определить, например, с помощью хиральной ВЭЖХ, например (описанной в примере 36).
Одним объектом настоящего изобретения является оптически активный N-замещенный арил- или гетероарилциклопропиламин (например, соединение формулы (II) или (III), описанное в настоящем изобретении), или его фармацевтически приемлемая соль или сольват, предназначенный для применения для лечения или предупреждения заболевания или нарушения. В родственном объекте способ включает введение индивидууму, нуждающемуся в лечении, терапевтически эффективного количества оптически активного N-замещенного арил- или гетероарилциклопропиламина или его фармацевтически приемлемой соли или сольвата. В одном объекте заболеванием или нарушением является заболевание или нарушение человека, выбранное из группы, включающей рак, неврологическое заболевание или патологическое состояние или вирусную инфекцию. В одном объекте неврологическим заболеванием или нарушением является депрессия, болезнь Гентингтона, болезнь Паркинсона, болезнь Альцгеймера, боковой амиотрофический склероз, лобно-височное слабоумие или слабоумие с тельцами Леви. В одном предпочтительном объекте раком является рак предстательной железы. В другом предпочтительном объекте раком является рак молочной железы. В другом объекте раком является рак легких. В другом объекте раком является колоректальный рак. В другом предпочтительном объекте раком является рак головного мозга. В другом предпочтительном объекте раком является рак кожи. В другом предпочтительном объекте раком является рак крови (например, лейкоз (включая, например, острый миелогенный лейкоз (ОМЛ), хронический миелогенный лейкоз (ХМЛ), хронический нейтрофильный лейкоз, хронический эозинофильный лейкоз, хронический лимфолейкоз (ХЛЛ), острый лимфобластный лейкоз (ОЛЛ) или волосатоклеточный лейкоз) или лимфома). В другом предпочтительном объекте раком является миелома. В одном объекте неврологическим заболеванием или патологическим состоянием является депрессия, болезнь Гентингтона, болезнь Паркинсона или болезнь Альцгеймера. В одном объекте вирусной инфекцией является HSV1 или HSV2. В одном объекте заболеванием или патологическим состоянием является депрессия. В одном объекте неврологическим заболеванием или патологическим состоянием является нейродегенеративное заболевание или патологическое состояние. В одном объекте нейродегенеративным заболеванием или нарушением является болезнь Гентингтона, болезнь Паркинсона, болезнь Альцгеймера, боковой амиотрофический склероз или лобно-височное слабоумие. В одном объекте нейродегенеративным заболеванием или нарушением является болезнь Гентингтона. В одном объекте нейродегенеративным заболеванием или нарушением является болезнь Паркинсона. В одном объекте нейродегенеративным заболеванием или нарушением является болезнь Альцгеймера. В одном объекте нейродегенеративным заболеванием или нарушением является боковой амиотрофический склероз. В одном объекте нейродегенеративным заболеванием или нарушением является лобно-височное слабоумие.
Настоящее изобретение также относится к оптически активному N-замещенному арил- или гетероарилциклопропиламину формулы (II) или (III), или его фармацевтически приемлемой соли или сольвату, определенному в приведенных выше вариантах осуществления или объектах, предназначенному для применения для лечения или предупреждения заболевания или нарушения, предпочтительно рака (например, рак молочной железы, рак легких, рак предстательной железы, колоректальный рак, рак головного мозга, рак кожи, рак крови, лейкоз (включая, например, острый миелогенный лейкоз (ОМЛ), хронический миелогенный лейкоз (ХМЛ), хронический нейтрофильный лейкоз, хронический эозинофильный лейкоз, хронический лимфолейкоз (ХЛЛ), острый лимфобластный лейкоз (ОЛЛ) или волосатоклеточный лейкоз), лимфому или миелому), неврологического заболевания или патологического состояния (например, депрессии, болезни Альцгеймера, болезни Гентингтона, болезни Паркинсона или слабоумия с тельцами Леви) или вирусной инфекции (например, вирусной инфекции, вызванной и/или связанной с ВИЧ, или инфекции, вызванной вирусом герпеса, такой как инфекция, вызванная вирусом герпеса, вызванная и/или связанная с вирусом герпеса, выбранным из группы, включающей HSV-1, HSV-2 или вирус Эпштейна-Барра) у субъекта (предпочтительно млекопитающего, более предпочтительно человека).
Настоящее изобретение также относится к оптически активному N-замещенному арил- или гетероарилциклопропиламину формулы (II) или (III), или его фармацевтически приемлемой соли или сольвату, определенному в любом из приведенных выше вариантов осуществления, предназначенному для применения для лечения или предупреждения заболевания или нарушения, где указанным заболеванием или нарушением является нейродегенеративное заболевание или нарушение. В одном объекте нейродегенеративным заболеванием или нарушением является болезнь Гентингтона, болезнь Паркинсона, болезнь Альцгеймера, боковой амиотрофический склероз или лобно-височное слабоумие.
В одном варианте осуществления настоящее изобретение относится к соединению формулы (I), (Ia) или (Ib) или его сольвату или фармацевтически приемлемой соли, где соединение представляет собой:
5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)пиримидин-2-амин;
5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)тиазол-2-амин;
5-(((транс)-2-(6-(3-(трифторметил)фенил)пиридин-3-ил)циклопропиламино)метил)пиримидин-2-амин;
5-(((транс)-2-(6-(3-(трифторметил)фенил)пиридин-3-ил)циклопропиламино)метил)тиазол-2-амин;
3-(5-((транс)-2-((2-аминопиримидин-5-ил)метиламино)циклопропил)пиридин-2-ил)фенол;
3-(5-((транс)-2-((2-аминотиазол-5-ил)метиламино)циклопропил)пиридин-2-ил)фенол;
4'-((транс)-2-((2-аминопиримидин-5-ил)метиламино)циклопропил)бифенил-3-ол;
4'-((транс)-2-((2-аминотиазол-5-ил)метиламино)циклопропил)бифенил-3-ол;
5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,2,4-оксадиазол-3-амин;
5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-((4-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-((3-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-((3,5-дифторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-((4-хлорбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-((3-хлорбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-((2-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-N-метил-1,3,4-оксадиазол-2-амин;
N-(5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-ил)ацетамид;
4'-((транс)-2-(((5-амино-1,3,4-оксадиазол-2-ил)метил)амино)циклопропил)-[1,1'-бифенил]-3-ол;
5-((((транс)-2-(6-(3-(трифторметил)фенил)пиридин-3-ил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-тиадиазол-2-амин;
2-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)тиазол-5-амин;
4-((((транс)-2-(3'-(трифторметил)-[1,1'-бифенил]-4-ил)циклопропил)амино)метил)тиазол-2-амин;
2-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)оксазол-5-амин;
3-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)изоксазол-5-амин;
5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,2,4-оксадиазол-3-амин;
3-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,2,4-оксадиазол-5-амин;
5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,2,4-тиадиазол-3-амин;
5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)пиридин-2-амин;
6-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)пиридазин-3-амин;
5-((((транс)-2-(4-бензилокси)фенил)циклопропил)амино)метил)пиразин-2-амин;
2-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)пиримидин-5-амин;
6-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,2,4-триазин-3-амин; или
3-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,2,4-триазин-6-амин;.
В одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей фармацевтический носитель и соединение или его сольват или фармацевтически приемлемую соль, где указанное соединение представляет собой:
5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)пиримидин-2-амин;
5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)тиазол-2-амин;
5-(((транс)-2-(6-(3-(трифторметил)фенил)пиридин-3-ил)циклопропиламино)метил)пиримидин-2-амин;
5-(((транс)-2-(6-(3-(трифторметил)фенил)пиридин-3-ил)циклопропиламино)метил)тиазол-2-амин;
3-(5-((транс)-2-((2-аминопиримидин-5-ил)метиламино)циклопропил)пиридин-2-ил)фенол;
3-(5-((транс)-2-((2-аминотиазол-5-ил)метиламино)циклопропил)пиридин-2-ил)фенол;
4'-((транс)-2-((2-аминопиримидин-5-ил)метиламино)циклопропил)бифенил-3-ол;
4'-((транс)-2-((2-аминотиазол-5-ил)метиламино)циклопропил)бифенил-3-ол;
5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,2,4-оксадиазол-3-амин;
5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-((4-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-((3-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-((3,5-дифторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-((4-хлорбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-((3-хлорбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-((2-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-N-метил-1,3,4-оксадиазол-2-амин;
N-(5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-ил)ацетамид;
4'-((транс)-2-(((5-амино-1,3,4-оксадиазол-2-ил)метил)амино)циклопропил)-[1,1'-бифенил]-3-ол;
5-((((транс)-2-(6-(3-(трифторметил)фенил)пиридин-3-ил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-тиадиазол-2-амин;
2-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)тиазол-5-амин;
4-((((транс)-2-(3'-(трифторметил)-[1,1'-бифенил]-4-ил)циклопропил)амино)метил)тиазол-2-амин;
2-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)оксазол-5-амин;
3-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)изоксазол-5-амин;
5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,2,4-оксадиазол-3-амин;
3-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,2,4-оксадиазол-5-амин;
5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,2,4-тиадиазол-3-амин;
5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)пиридин-2-амин;
6-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)пиридазин-3-амин;
5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)пиразин-2-амин;
2-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)пиримидин-5-амин;
6-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,2,4-триазин-3-амин; или
3-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,2,4-триазин-6-амин;
В одном варианте осуществления настоящее изобретение относится к соединению или его сольвату или фармацевтически приемлемой соли, где указанное соединение представляет собой:
5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)пиримидин-2-амин;
5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)тиазол-2-амин;
5-(((транс)-2-(6-(3-(трифторметил)фенил)пиридин-3-ил)циклопропиламино)метил)пиримидин-2-амин;
5-(((транс)-2-(6-(3-(трифторметил)фенил)пиридин-3-ил)циклопропиламино)метил)тиазол-2-амин;
3-(5-((транс)-2-((2-аминопиримидин-5-ил)метиламино)циклопропил)пиридин-2-ил)фенол;
3-(5-((транс)-2-((2-аминотиазол-5-ил)метиламино)циклопропил)пиридин-2-ил)фенол;
4'-((транс)-2-((2-аминопиримидин-5-ил)метиламино)циклопропил)бифенил-3-ол;
4'-((транс)-2-((2-аминотиазол-5-ил)метиламино)циклопропил)бифенил-3-ол;
5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,2,4-оксадиазол-3-амин;
5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-((4-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-((3-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-((3,5-дифторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-((4-хлорбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-((3-хлорбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-((2-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-N-метил-1,3,4-оксадиазол-2-амин;
N-(5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-ил)ацетамид;
4'-((транс)-2-(((5-амино-1,3,4-оксадиазол-2-ил)метил)амино)циклопропил)-[1,1'-бифенил]-3-ол;
5-((((транс)-2-(6-(3-(трифторметил)фенил)пиридин-3-ил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-тиадиазол-2-амин;
2-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)тиазол-5-амин;
4-((((транс)-2-(3'-(трифторметил)-[1,1'-бифенил]-4-ил)циклопропил)амино)метил)тиазол-2-амин;
2-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)оксазол-5-амин;
3-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)изоксазол-5-амин;
5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,2,4-оксадиазол-3-амин;
3-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,2,4-оксадиазол-5-амин;
5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,2,4-тиадиазол-3-амин;
5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)пиридин-2-амин;
6-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)пиридазин-3-амин;
5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)пиразин-2-амин;
2-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)пиримидин-5-амин;
6-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,2,4-триазин-3-амин; или
3-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,2,4-триазин-6-амин;
предназначенному для применения в способе лечения или предупреждения неврологического заболевания или патологического состояния. В одном объекте неврологическое заболевание или патологическое состояние выбрано из группы, включающей болезнь Альцгеймера, болезнь Паркинсона, болезнь Гентингтона, боковой амиотрофический склероз, лобно-височное слабоумие или слабоумие с тельцами Леви.
В одном варианте осуществления настоящее изобретение относится к соединению или его сольвату или фармацевтически приемлемой соли, где указанное соединение представляет собой:
5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)пиримидин-2-амин;
5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)тиазол-2-амин;
5-(((транс)-2-(6-(3-(трифторметил)фенил)пиридин-3-ил)циклопропиламино)метил)пиримидин-2-амин;
5-(((транс)-2-(6-(3-(трифторметил)фенил)пиридин-3-ил)циклопропиламино)метил)тиазол-2-амин;
3-(5-((транс)-2-((2-аминопиримидин-5-ил)метиламино)циклопропил)пиридин-2-ил)фенол;
3-(5-((транс)-2-((2-аминотиазол-5-ил)метиламино)циклопропил)пиридин-2-ил)фенол;
4'-((транс)-2-((2-аминопиримидин-5-ил)метиламино)циклопропил)бифенил-3-ол;
4'-((транс)-2-((2-аминотиазол-5-ил)метиламино)циклопропил)бифенил-3-ол;
5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,2,4-оксадиазол-3-амин;
5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-((4-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-((3-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-((3,5-дифторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-((4-хлорбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-((3-хлорбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-((2-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-N-метил-1,3,4-оксадиазол-2-амин;
N-(5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-ил)ацетамид;
4'-((транс)-2-(((5-амино-1,3,4-оксадиазол-2-ил)метил)амино)циклопропил)-[1,1'-бифенил]-3-ол;
5-((((транс)-2-(6-(3-(трифторметил)фенил)пиридин-3-ил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-тиадиазол-2-амин;
2-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)тиазол-5-амин;
4-((((транс)-2-(3'-(трифторметил)-[1,1'-бифенил]-4-ил)циклопропил)амино)метил)тиазол-2-амин;
2-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)оксазол-5-амин;
3-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)изоксазол-5-амин;
5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,2,4-оксадиазол-3-амин;
3-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,2,4-оксадиазол-5-амин;
5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,2,4-тиадиазол-3-амин;
5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)пиридин-2-амин;
6-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)пиридазин-3-амин;
5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)пиразин-2-амин;
2-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)пиримидин-5-амин;
6-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,2,4-триазин-3-амин; или
3-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,2,4-триазин-6-амин;
предназначенному для применения в способе лечения или предупреждения рака. В одном объекте рак выбран из группы, включающей рак предстательной железы, тестикулярный головного мозга, колоректальный, легких, молочной железы, лимфому, рак кожи или рак крови.
В одном варианте осуществления настоящее изобретение относится к соединению или его сольвату или фармацевтически приемлемой соли, где указанное соединение представляет собой:
4'-((транс)-2-((2-аминотиазол-5-ил)метиламино)циклопропил)бифенил-3-ол;
5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,2,4-оксадиазол-3-амин;
5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-амин;
5-((((транс)-2-(4-((4-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин; или
5-((((транс)-2-(4-((3-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
предназначенному для применения в способе лечения или предупреждения неврологического заболевания или патологического состояния. В одном объекте неврологическое заболевание или патологическое состояние выбрано из группы, включающей болезнь Альцгеймера, болезнь Паркинсона, болезнь Гентингтона, боковой амиотрофической склероз, лобно-височное слабоумие или слабоумие с тельцами Леви.
В одном варианте осуществления настоящее изобретение относится к соединению или его сольвату или фармацевтически приемлемой соли, где указанное соединение представляет собой
4'-((транс)-2-((2-аминотиазол-5-ил)метиламино)циклопропил)бифенил-3-ол, предназначенный для применения в способе лечения или предупреждения неврологического заболевания или патологического состояния. В одном объекте неврологическое заболевание или патологическое состояние выбрано из группы, включающей болезнь Альцгеймера, болезнь Паркинсона, болезнь Гентингтона, боковой амиотрофический склероз, лобно-височное слабоумие или слабоумие с тельцами Леви.
В одном варианте осуществления настоящее изобретение относится к соединению или его сольвату или фармацевтически приемлемой соли, где указанное соединение представляет собой
5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,2,4-оксадиазол-3-амин, предназначенный для применения в способе лечения или предупреждения неврологического заболевания или патологического состояния. В одном объекте неврологическое заболевание или патологическое состояние выбрано из группы, включающей болезнь Альцгеймера, болезнь Паркинсона, болезнь Гентингтона, боковой амиотрофический склероз, лобно-височное слабоумие или слабоумие с тельцами Леви.
В одном варианте осуществления настоящее изобретение относится к соединению или его сольвату или фармацевтически приемлемой соли, где указанное соединение представляет собой
5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-амин, предназначенный для применения в способе лечения или предупреждения неврологического заболевания или патологического состояния. В одном объекте неврологическое заболевание или патологическое состояние выбрано из группы, включающей болезнь Альцгеймера, болезнь Паркинсона, болезнь Гентингтона, боковой амиотрофический склероз, лобно-височное слабоумие или слабоумие с тельцами Леви.
В одном варианте осуществления настоящее изобретение относится к соединению или его сольвату или фармацевтически приемлемой соли, где указанное соединение представляет собой
5-((((транс)-2-(4-((4-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин, предназначенный для применения в способе лечения или предупреждения неврологического заболевания или патологического состояния. В одном объекте неврологическое заболевание или патологическое состояние выбрано из группы, включающей болезнь Альцгеймера, болезнь Паркинсона, болезнь Гентингтона, боковой амиотрофический склероз, лобно-височное слабоумие или слабоумие с тельцами Леви.
В одном варианте осуществления настоящее изобретение относится к соединению или его сольвату или фармацевтически приемлемой соли, где указанное соединение представляет собой
5-((((транс)-2-(4-((3-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин, предназначенный для применения в способе лечения или предупреждения неврологического заболевания или патологического состояния. В одном объекте неврологическое заболевание или патологическое состояние выбрано из группы, включающей болезнь Альцгеймера, болезнь Паркинсона, болезнь Гентингтона, боковой амиотрофический склероз, лобно-височное слабоумие или слабоумие с тельцами Леви.
В одном варианте осуществления настоящее изобретение относится к оптически активному N-замещенному арил- или гетероарилциклопропиламину или его фармацевтически приемлемой соли или сольвату, где указанный оптически активный N-замещенный арил- или гетероарилциклопропиламин выбран из группы, включающей
(-)-5-((((транс)-2-(4-((3-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
(-)-5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-N-метил-1,3,4-оксадиазол-2-амин;
(-)-N-(5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-ил)ацетамид;
(-)-5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)пиримидин-2-амин;
(-)-5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-тиадиазол-2-амин; или
(-)-5-((((транс)-2-(4-((2-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин.
В родственном объекте настоящее изобретение относится к соединению, выбранному из группы, включающей:
(-)-5-((((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-амин;
(-)-5-((((транс)-2-(4-((3-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
(-)-5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-N-метил-1,3,4-оксадиазол-2-амин;
(-)-N-(5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-ил)ацетамид;
(-)-5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)пиримидин-2-амин;
(-)-5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-тиадиазол-2-амин;
(-)-5-((((транс)-2-(4-((2-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин; или
его фармацевтически приемлемой соли или сольвату;
предназначенному для применения в способе лечения или предупреждения неврологического заболевания или патологического состояния (например, депрессии). Предпочтительно, если неврологическое заболевание или патологическое состояние выбрано из группы, включающей депрессию, болезнь Альцгеймера, болезнь Гентингтона, болезнь Паркинсона, лобно-височное слабоумие, слабоумие с тельцами Леви или боковой амиотрофический склероз.
Таким образом, настоящее изобретение относится к соединению или композиции, содержащей оптически активный N-замещенный арил- или гетероарилциклопропиламин. Предпочтительно, если оптически активный N-замещенный арил- или гетероарилциклопропиламин является таким, как определено в настоящем изобретении в любых вариантах осуществления или объектах формулы (II) или (III). Более предпочтительно, если оптически активный N-замещенный арил- или гетероарилциклопропиламин обладает значением kinact/KI для LSD1, которое по меньшей мере в 50 раз больше, чем значение kinact/KI для MAO-A. Еще более предпочтительно, если значение kinact/KI для LSD1 по меньшей мере в 100 раз больше, чем значение kinact/KI для MAO-A. Еще более предпочтительно, если значение kinact/KI для LSD1 по меньшей мере в 250 раз больше, чем значение kinact/KI для MAO-A. Еще более предпочтительно, если значение kinact/KI для LSD1 по меньшей мере в 500 раз больше, чем значение kinact/KI для MAO-A.
Предпочтительно, если оптически активный N-замещенный арил- или гетероарилциклопропиламин является таким, как определено в настоящем изобретении в любых вариантах осуществления или объектах формулы (II) или (III). Более предпочтительно, если оптически активный N-замещенный арил- или гетероарилциклопропиламин обладает значениями kinact/KI для LSD1 и MAO-B, которые по меньшей мере в 50 раз больше, чем значение kinact/KI для MAO-A. Предпочтительно, если значения kinact/KI для LSD1 и MAO-B по меньшей мере в 100 раз больше, чем значение kinact/KI для MAO-A. Еще более предпочтительно, если значения kinact/KI для LSD1 и MAO-B по меньшей мере в 250 раз больше, чем значение kinact/KI для MAO-A. Еще более предпочтительно, если значения kinact/KI для LSD1 и MAO-B по меньшей мере в 500 раз больше, чем значение kinact/KI для MAO-A.
Предпочтительно, если оптически активный N-замещенный арил- или гетероарилциклопропиламин является таким, как определено в настоящем изобретении в любом из вариантов осуществления формулы (I) или (II). Более предпочтительно, если оптически активный N-замещенный арил- или гетероарилциклопропиламин обладает значением kinact/KI для MAO-B, которое по меньшей мере в 50 раз больше, чем значение kinact/KI для MAO-A. Предпочтительно, если значение kinact/KI для MAO-B по меньшей мере в 100 раз больше, чем значение kinact/KI для MAO-A. Еще более предпочтительно, если значение kinact/KI для MAO-B по меньшей мере в 500 раз больше, чем значение kinact/KI для MAO-A. Еще более предпочтительно, если значение kinact/KI для MAO-B по меньшей мере в 1000 раз больше, чем значение kinact/KI для MAO-A. Еще более предпочтительно, если значение kinact/KI для MAO-B по меньшей мере в 2000 раз больше, чем значение kinact/KI для MAO-A.
Оптически активный N-замещенный арил- или гетероарилциклопропиламин, фармацевтическая композиция, содержащая оптически активный N-замещенный арил- или гетероарилциклопропиламин или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, и способы их применения характеризуются неожиданной селективностью по отношению к LSD1 и/или МАОВ. (-)-Стереоизомеры N-замещенных арил- или гетероарилциклопропиламинов (например, соединений формулы (II) или (III), описанных в настоящем изобретении) неожиданно являются активными и селективными ингибиторами LSD1 и/или MAO-B. Исключение ингибирования других мишеней может исключить неожиданные или нежелательные побочные эффекты, такие как так называемый "эффект сыра", связанный с MAO-A.
Оптически активные соединения, предлагаемые в настоящем изобретении, можно получить с помощью хиральной ВЭЖХ, например, их рацематов, путем хирального синтеза с использованием соединений известной хиральности или путем хиральной перекристаллизации с использованием хиральных солей.
Одним объектом настоящего изобретения является способ обогащения энантиомера транс-N-замещенного циклопропиламина (например, энантиомера соединения формулы (II) или (III) или энантиомера соединения формулы (I), в которой заместители циклопропильного фрагмента, находящиеся в формуле (I) (т.е. заместитель (A) и заместитель -NH-CH2-(D)), находятся в трансконфигурации, способ включает: взаимодействие транс-замещенного циклопропиламина с хиральным реагентом для перекристаллизации в растворителе (предпочтительно при условиях, которые достаточны для кристаллизации соли хирального реагента для перекристаллизации и транс-замещенного циклопропиламина); и выделение закристаллизованной соли хирального реагента для перекристаллизации и транс-замещенного циклопропиламина. В другом предпочтительном объекте транс-циклопропиламином является транс-4-бензокси-2-фенилциклопропиламин или его содержащее защитную группу производное. В предпочтительном объекте транс-N-замещенный циклопропиламин описывается формулой (II) или (III), описанной выше, или его производное, в котором группа -L2II-R4II или группа -L2III-R4III отсутствует или содержит защитную группу. В предпочтительном объекте хиральный реагент для перекристаллизации выбран из группы, включающей (S)-(+)-миндальную кислоту, D-(-)-винную кислоту, L-(-)-ди-п-толуоилвинную кислоту или R-(-)-миндальную кислоту. В одном предпочтительном объекте хиральным реагентом для перекристаллизации является R-(-)-миндальная кислота. В одном объекте растворителем является ТГФ и H2O.
Одним объектом настоящего изобретения является способ получения энантиомера транс-N-замещенного циклопропиламина, включающий: взаимодействие транс-замещенного циклопропиламина с хиральным реагентом для перекристаллизации в растворителе (предпочтительно при условиях, которые достаточны для кристаллизации соли хирального реагента для перекристаллизации и транс-замещенного циклопропиламина); и выделение закристаллизованной соли хирального реагента для перекристаллизации и транс-замещенного циклопропиламина, таким образом, с получением энантиомера транс-N-замещенного циклопропиламина. В предпочтительном объекте транс-N-замещенный циклопропиламин описывается формулой (II) или (III), определенной выше, или его производное, в котором группа -L2II-R4II или группа -L2III-R4III отсутствует или содержит защитную группу. В другом предпочтительном объекте транс-циклопропиламин представляет собой транс-4-бензокси-2-фенилциклопропиламин или его содержащее защитную группу производное. В предпочтительном объекте хиральный реагент для перекристаллизации выбран из группы, включающей (S)-(+)-миндальную кислоту, D-(-)-винную кислоту, L-(-)-ди-п-толуоилвинную кислоту или R-(-)-миндальную кислоту. В одном предпочтительном объекте хиральным реагентом для перекристаллизации является R-(-)-миндальная кислота. В одном объекте растворителем является ТГФ и H2O.
Кроме того, настоящее изобретение относится к (+)-энантиомеру транс-N-замещенного арил- или гетероарилциклопропиламина, включая соединения формулы (II) или (III) и соединения формулы (I), в которой заместители циклопропиламинового фрагмента находятся в транс-ориентации. Например, соответствующий оптически активный (+)-энантиомер может быть выбран из группы, включающей:
(+)-5-((((транс)-2-(4-((3-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
(+)-5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-N-метил-1,3,4-оксадиазол-2-амин;
(+)-N-(5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-ил)ацетамид;
(+)-5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)пиримидин-2-амин;
(+)-5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-тиадиазол-2-амин;
(+)-5-((((транс)-2-(4-((2-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
или его фармацевтически приемлемую соль или сольват.
Кроме того, настоящее изобретение относится к следующему:
1. Оптически активный N-замещенный арил- или гетероарилциклопропиламин или его фармацевтически приемлемая соль или сольват, предназначенный для применения в способе лечения или предупреждения заболевания.
2. Оптически активный N-замещенный арил- или гетероарилциклопропиламин, соответствующий параграфу 1, где указанный N-замещенный арил- или гетероарилциклопропиламин представляет собой транс-N-замещенный арил- или гетероарилциклопропиламин, который вращает плоскость поляризованного света в (-)-направлении или является (-)-стереоизомером.
3. Оптически активный N-замещенный арил- или гетероарилциклопропиламин, соответствующий параграфу 1 или 2, где указанный N-замещенный арил- или гетероарилциклопропиламин содержит 90% или более (-)-стереоизомера и 10% или менее (+)-стереоизомера.
3. Оптически активный N-замещенный арил- или гетероарилциклопропиламин, соответствующий параграфу 1 или 2, где указанный N-замещенный арил- или гетероарилциклопропиламин содержит 95% или более (-)-стереоизомера и 5% или менее (+)-стереоизомера.
5. Оптически активный N-замещенный арил- или гетероарилциклопропиламин, соответствующий параграфу 1 или 2, где указанный N-замещенный арил- или гетероарилциклопропиламин содержит 98% или более (-)-стереоизомера и 2% или менее (+)-стереоизомера.
6. Оптически активный N-замещенный арил- или гетероарилциклопропиламин, соответствующий параграфу 1 или 2, где указанный N-замещенный арил- или гетероарилциклопропиламин содержит 99% или более (-)-стереоизомера и 1% или менее (+)-стереоизомера.
7. Оптически активный N-замещенный арил- или гетероарилциклопропиламин, соответствующий параграфу 1 или 2, где указанный N-замещенный арил- или гетероарилциклопропиламин содержит 99,5% или более (-)-стереоизомера и 0,5% или менее (+)-стереоизомера.
8. Оптически активный N-замещенный арил- или гетероарилциклопропиламин, соответствующий параграфу 1 или 2, обладающий составляющим 90% или более энантиомерным избытком (-)-стереоизомера N-замещенного арил- или гетероарилциклопропиламина.
9. Оптически активный N-замещенный арил- или гетероарилциклопропиламин, соответствующий параграфу 1 или 2, обладающий составляющим 95% или более энантиомерным избытком (-)-стереоизомера N-замещенного арил- или гетероарилциклопропиламина.
10. Оптически активный N-замещенный арил- или гетероарилциклопропиламин, соответствующий параграфу 1 или 2, обладающий составляющим 98% или более энантиомерным избытком (-)-стереоизомера N-замещенного арил- или гетероарилциклопропиламина.
11. Оптически активный N-замещенный арил- или гетероарилциклопропиламин, соответствующий параграфу 1 или 2, обладающий составляющим 99% или более энантиомерным избытком (-)-стереоизомера N-замещенного арил- или гетероарилциклопропиламина.
12. Оптически активный N-замещенный арил- или гетероарилциклопропиламин, соответствующий любому из параграфов 1-11, или его фармацевтически приемлемая соль, где указанный N-замещенный арил- или гетероарилциклопропиламин описывается формулой (II):
R1II-(AII)-R2II
в которой (AII) обозначает арильную или гетероарильную группу, содержащую 2 заместителя, R1II и R2II, и 1-3 необязательных заместителя, где указанные необязательные заместители, независимо выбранных из группы, включающей галоген, C1-C3-алкил или C1-C3-алкоксигруппу;
R1II обозначает группу -L1-R3;
R3II обозначает арильную или гетероарильную группу, содержащую 1, 2, 3, 4 или 5 необязательных заместителей, независимо выбранных из группы, включающей галоген, -OH, -NHSO2RA, алкил, алкоксигруппу, цианогруппу, -CF3 или -OCF3, где RA обозначает C1-C6-алкил или фенил;
выбран из группы, включающей связь, -CH2O-, -CH2CH2O-, -OCH2-, -OCH2CH2-, -CH2-, -CH2CH2-, -CH2CH2CH2- или -O-.
R2II обозначает , где указанная циклопропильная группа содержит 2 хиральных центра, замещенных в транс-положении по отношению к атомам углерода, к которым ковалентно присоединены (AII) и ;
R4II обозначает 5- или 6-членное гетероарильное кольцо, содержащее 1, 2 или 3 необязательных заместителя, где указанные необязательные заместители, независимо выбранных из группы, включающей алкил, NHRB, -ORB или галоген, где RB обозначает водород, C1-C3-алкил или -C(=O)CH3;
обозначает разветвленную или неразветвленную C1-C4-алкиленовую группу.
13. Оптически активный N-замещенный арил- или гетероарилциклопропиламин, соответствующий параграфу 12, где (AII) обозначает арильную или гетероарильную группу, содержащую 2 заместителя, R1II и R2II, и 1, 2 или 3 необязательных заместителя, где указанные необязательные заместители, независимо выбранных из группы, включающей галоген, C1-C3-алкил или C1-C3-алкоксигруппу.
14. Оптически активный N-замещенный арил- или гетероарилциклопропиламин, соответствующий параграфу 12, где R3II обозначает фенильную, пиридильную, тиазолильную или тиенильную группу, содержащую 1, 2 или 3 необязательных заместителя, независимо выбранных из группы, включающей галоген, -OH, -NHSO2RA, алкил, алкоксигруппу, цианогруппу, -CF3 или -OCF3, где RA обозначает C1-C6-алкил или фенил.
15. Оптически активный N-замещенный арил- или гетероарилциклопропиламин, соответствующий параграфу 12, где выбран из группы, включающей связь, -OCH2- или -CH2O-.
16. Оптически активный N-замещенный арил- или гетероарилциклопропиламин, соответствующий параграфу 12, где R4II обозначает 5-членное гетероарильное кольцо, содержащее 1, 2 или 3 необязательных заместителя, независимо выбранных из группы, включающей -NH2 или -NH(C1-C3)алкил.
17. Оптически активный N-замещенный арил- или гетероарилциклопропиламин, соответствующий параграфу 12, где обозначает -CH2- или -CH2CH2-.
18. Применение, соответствующее любому из параграфов 1-17, в котором указанный способ лечения или предупреждения представляет собой способ лечения или предупреждения рака, депрессии, нейродегенеративного заболевания или нарушения или вирусной инфекции.
19. Применение, соответствующее параграфу 17, в котором указанное нейродегенеративное заболевание или нарушение выбрано из группы, включающей болезнь Альцгеймера, болезнь Паркинсона, болезнь Гентингтона, лобно-височное слабоумие или боковой амиотрофический склероз.
20. Применение, соответствующее параграфу 17, в котором указанное нейродегенеративное заболевание или нарушение выбрано из группы, включающей болезнь Альцгеймера, болезнь Паркинсона, болезнь Гентингтона, лобно-височное слабоумие или боковой амиотрофический склероз.
21. Применение, соответствующее параграфу 17, в котором указанным нейродегенеративным заболеванием или нарушением является болезнь Альцгеймера.
22. Применение, соответствующее параграфу 17, в котором указанным нейродегенеративным заболеванием или нарушением является болезнь Паркинсона.
23. Применение, соответствующее параграфу 17, в котором указанным нейродегенеративным заболеванием или нарушением является болезнь Гентингтона.
24. Применение, соответствующее параграфу 17, в котором указанным нейродегенеративным заболеванием или нарушением является лобно-височное слабоумие.
25. Применение, соответствующее параграфу 17, в котором указанным нейродегенеративным заболеванием или нарушением является боковой амиотрофический склероз.
26. Оптически активное соединение, или его фармацевтически приемлемая соль или сольват, формулы (III):
R1III-(AIII)-R2III
в которой (AIII) обозначает арильную или гетероарильную группу, содержащую 2 заместителя, R1III и R2III, и 1-3 необязательных заместителя, где указанные необязательные заместители, независимо выбранных из группы, включающей галоген, C1-C3-алкил или C1-C3-алкоксигруппу;
R1III обозначает группу ;
R3III обозначает фенильную, пиридильную, тиазолильную или тиенильную группу, содержащую 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -F, -Cl, -OH, -NHSO2RA, C1-C3-алкил, C1-C3-алкоксигруппу, цианогруппу, -CF3 или -OCF3, где RA обозначает C1-C6-алкил или фенил;
выбран из группы, включающей связь, -CH2O- или -CH2O-,
R2III обозначает , где указанная циклопропильная группа содержит 2 хиральных центра, замещенных в транс-положении по отношению к атомам углерода, к которым ковалентно присоединены (AIII) и ;
R4III обозначает 5-членное гетероарильное кольцо, содержащее 1, 2 или 3 необязательных заместителя, где указанные необязательные заместители, независимо выбранных из группы, включающей -NH2 или -NH(C1-C3)алкил; и обозначает -CH2- или -CH2CH2-.
27. Соединение, соответствующее параграфу 26, в котором (AIII) обозначает фенильную или пиридильную группу.
28. Соединение, соответствующее параграфу 26, в котором R3III обозначает фенил, содержащий 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -F, -Cl, -OH, -NHSO2RA, C1-C3-алкил, C1-C3-алкоксигруппу, цианогруппу, -CF3 или -OCF3, где RA обозначает C1-C6-алкил или фенил.
29. Соединение, соответствующее параграфу 26, в котором выбран из группы, включающей связь, -OCH2- или -CH2O-.
30. Соединение, соответствующее параграфу 26, в котором R4III обозначает 5-членное гетероарильное кольцо, где цепь атомов, образующая указанное 5-членное гетероарильное кольцо, содержит 2 или 3 гетероатома, независимо выбранных из группы, включающей N, S, или O и указанное гетероарильное кольцо содержит 1 необязательный заместитель, где указанный необязательный заместитель, если он содержится, представляет собой -NH2 или -NH(C1-C3)алкил.
31. Соединение, соответствующее параграфу 26, в котором обозначает -CH2- или -CH2CH2-.
32. Соединение, соответствующее параграфу 26, в котором R3III обозначает фенил, содержащий 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -F, -Cl, -OH, -NHSO2CH3, метил, метоксигруппу, цианогруппу, -CF3 или -OCF3.
33. Соединение, соответствующее параграфу 26, в котором R4III обозначает оксадиазолильное, тиадиазолильное или тиазолильное кольцо, содержащее 1 необязательный заместитель, где указанный необязательный заместитель, если он содержится, представляет собой -NH2 или -NH(C1-C3)алкил.
34. Соединение, соответствующее параграфу 26, в котором R4III обозначает оксадиазолильное кольцо, содержащее 1 необязательный заместитель, выбранный из группы, включающей -NH2 или -NH(C1-C3)алкил.
35. Оптически активный N-замещенный арил- или гетероарилциклопропиламин, соответствующий любому из параграфов 26-34, где указанный N-замещенный арил- или гетероарилциклопропиламин вращает плоскость поляризованного света в (-)-направлении или представляет собой (-)-энантиомер.
36. Оптически активный N-замещенный арил- или гетероарилциклопропиламин, соответствующий любому из параграфов 26-34, где указанный N-замещенный арил- или гетероарилциклопропиламин содержит 90% или более (-)-стереоизомера и 10% или менее (+)-стереоизомера.
37. Оптически активный N-замещенный арил- или гетероарилциклопропиламин, соответствующий параграфу 1 и любому из параграфов 26-34, где указанный N-замещенный арил- или гетероарилциклопропиламин содержит 95% или более (-)-стереоизомера и 5% или менее (+)-стереоизомера.
38. Оптически активный N-замещенный арил- или гетероарилциклопропиламин, соответствующий любому из параграфов 26-34, где указанный N-замещенный арил- или гетероарилциклопропиламин 98% или более (-)-стереоизомера и 2% или менее (+)-стереоизомера.
39. Оптически активный N-замещенный арил- или гетероарилциклопропиламин, соответствующий любому из параграфов 26-34, где указанный N-замещенный арил- или гетероарилциклопропиламин 99% или более (-)-стереоизомера и 1% или менее (+)-стереоизомера.
40. Оптически активный N-замещенный арил- или гетероарилциклопропиламин, соответствующий любому из параграфов 26-34, где указанный N-замещенный арил- или гетероарилциклопропиламин 99,5% или более (-)-стереоизомера и 0,5% или менее (+)-стереоизомера.
41. Оптически активный N-замещенный арил- или гетероарилциклопропиламин, соответствующий любому из параграфов 26-34, где указанный N-замещенный арил- или гетероарилциклопропиламин обладает составляющим 90% или более энантиомерным избытком (-)-стереоизомера N-замещенного арил- или гетероарилциклопропиламина.
42. Оптически активный N-замещенный арил- или гетероарилциклопропиламин, соответствующий любому из параграфов 26-34, где указанный N-замещенный арил- или гетероарилциклопропиламин обладает составляющим 95% или более энантиомерным избытком (-)-стереоизомера N-замещенного арил- или гетероарилциклопропиламина.
43. Оптически активный N-замещенный арил- или гетероарилциклопропиламин, соответствующий любому из параграфов 26-34, где указанный N-замещенный арил- или гетероарилциклопропиламин обладает составляющим 98% или более энантиомерным избытком (-)-стереоизомера N-замещенного арил- или гетероарилциклопропиламина.
44. Оптически активный N-замещенный арил- или гетероарилциклопропиламин, соответствующий любому из параграфов 26-34, где указанный N-замещенный арил- или гетероарилциклопропиламин обладает составляющим 99% или более энантиомерным избытком (-)-стереоизомера N-замещенного арил- или гетероарилциклопропиламина.
45. Способ лечения или предупреждения заболевания или нарушения, указанный способ включает введение индивидууму, нуждающемуся в указанном лечении или предупреждении, эффективного количества оптически активного N-замещенного арил- или гетероарилциклопропиламина или его фармацевтически приемлемой соли или сольвата.
46. Способ, соответствующий параграфу 45, в котором указанный оптически активный N-замещенный арил- или гетероарилциклопропиламин является таким, как в любом из параграфов 26-44.
47. Способ, соответствующий параграфу 45 или 46, в котором указанное заболевание или нарушение выбрано из группы, включающей рак, нейродегенеративное заболевание или нарушение, вирусную инфекцию или депрессию.
48. Способ, соответствующий параграфу 45 или 46, в котором указанным заболеванием или нарушением является нейродегенеративное заболевание или нарушение, выбранное из группы, включающей болезнь Альцгеймера, болезнь Паркинсона, болезнь Гентингтона, лобно-височное слабоумие или боковой амиотрофический склероз.
49. Оптически активное соединение или его фармацевтически приемлемая соль или сольват, где указанное оптически активное соединение выбрано из группы, включающей:
(-)-5-((((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-амин;
(-)-5-((((транс)-2-(4-((3-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
(-)-5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-N-метил-1,3,4-оксадиазол-2-амин;
(-)-N-(5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-ил)ацетамид;
(-)-5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)пиримидин-2-амин; или
(-)-5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-тиадиазол-2-амин.
50. Соединение, соответствующее параграфу 49, выбранное из группы, включающей (-)-N-(5-(((2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-ил)ацетамид; (-)-5-(((2-(4-(бензилокси)фенил)циклопропил)амино)метил)-N-метил-1,3,4-оксадиазол-2-амин; или (-)-5-(((2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-амин.
51. Соединение, соответствующее параграфу 49, которое представляет собой (-)-5-(((2-(4-(бензилокси)фенил)циклопропил)амино)метил)-N-метил-1,3,4-оксадиазол-2-амин.
52. Соединение, соответствующее параграфу 49, которое представляет собой (-)-5-(((2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-амин.
53. Соединение, соответствующее параграфу 49, которое представляет собой (-)-N-(5-(((2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-ил)ацетамид.
54. Фармацевтическая композиция, содержащая оптически активное соединение, соответствующее любому из параграфов 26-44 или 49 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
55. Фармацевтическая композиция, соответствующая параграфу 54, предназначенная для применения в способе лечения или предупреждения заболевания или нарушения.
56. Фармацевтическая композиция, соответствующая параграфу 55, где указанным заболеванием или нарушением является заболевание или нарушение человека, выбранное из группы, включающей рак, неврологическое заболевание или нарушение или вирусную инфекцию.
57. Фармацевтическая композиция, соответствующая параграфу 56, где указанным неврологическим заболеванием или нарушением является депрессия или нейродегенеративное заболевание или нарушение.
58. Фармацевтическая композиция, соответствующая параграфу 57, где указанным нейродегенеративным заболеванием или нарушением является болезнь Альцгеймера, болезнь Паркинсона, болезнь Гентингтона, лобно-височное слабоумие или боковой амиотрофический склероз.
59. Оптически активное соединение, выбранное из группы, включающей:
(+)-5-((((транс)-2-(4-((3-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин;
(+)-5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-N-метил-1,3,4-оксадиазол-2-амин;
(+)-N-(5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-ил)ацетамид;
(+)-5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)пиримидин-2-амин;
или
(+)-5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-тиадиазол-2-амин; или его фармацевтически приемлемая соль или сольват.
60. Способ обогащения энантиомера транс-N-замещенного циклопропиламина, включающий: взаимодействие транс-замещенного циклопропиламина с хиральным реагентом для перекристаллизации в растворителе и при условиях, которые достаточны для кристаллизации соли хирального реагента для перекристаллизации и транс-замещенного циклопропиламина; и выделение закристаллизованой соли хирального реагента для перекристаллизации и транс-замещенного циклопропиламина.
61. Способ, соответствующий параграфу 60, в котором транс-циклопропиламин представляет собой транс-4-бензокси-2-фенилциклопропиламин или его содержащее защитную группу производное.
62. Способ, соответствующий параграфу 60, в котором хиральный реагент для перекристаллизации выбран из группы, включающей (S)-(+)-миндальную кислоту, D-(-)-винную кислоту, L-(-)-ди-п-толуоилвинную кислоту или R-(-)-миндальную кислоту.
63. Способ, соответствующий параграфу 60 или 61, в котором хиральным реагентом для перекристаллизации является R-(-)-миндальная кислота.
64. Способ, соответствующий параграфу 60, 61, 62, или 63, в котором растворителем является ТГФ и H2O.
Определения
Для описания сложной структурной группы любое определение, приведенное в настоящем изобретении, можно использовать в комбинации с любым другим определением. По определению, последним элементом любого такого определения является такой, который присоединен к исходному фрагменту. Например, сложная алкиламидная группа представляет собой алкильную группу, присоединенную к исходной молекуле через амидную группу, и термин алкоксиалкил означает алкоксигруппу, присоединенную к исходной молекуле через алкильную группу.
При использовании в настоящем изобретении термин "ацил" означает карбонил, присоединенный к алкенилу, алкилу, арилу, циклоалкилу, гетероарилу, гетероциклилу или любому другому фрагменту, в котором атомом, присоединенным к карбонилу, является атом углерода. "Ацетильная" группа означает группу -C(=O)CH3. "Алкилкарбонильная" или "алканоильная" группа означает алкильную группу, присоединенную к исходной молекуле через карбонильную группу. Примеры таких групп включают, но не ограничиваются только ими, метилкарбонил или этилкарбонил. Примеры ацильных групп включают, но не ограничиваются только ими, формил, алканоил или ароил.
При использовании в настоящем изобретении термин "алкенил" означает обладающую линейной цепью или разветвленной цепью углеводородную группу, содержащую одну или большее количество двойных связей и содержащую от 2 до 20 атомов углерода. A (C2-C6)алкенил содержит от 2 до 6 атомов углерода.
При использовании в настоящем изобретении термин "алкоксигруппа" означает простую алкильную эфирную группу, где термин алкил является таким, как определено ниже. Примеры подходящих простых алкильных эфирных групп включают, но не ограничиваются только ими, метоксигруппу, этоксигруппу, н-пропоксигруппу, изопропоксигруппу, н-бутоксигруппу, изобутоксигруппу, втор-бутоксигруппу, трет-бутоксигруппу, или n-пентоксигруппу.
При использовании в настоящем изобретении термин "алкил" означает обладающую линейной цепью или разветвленной цепью алкильную группу, содержащую от 1 до 20 атомов углерода. (C1-C10)Алкил содержит от 1 до 10 атомов углерода и (C1-C6)алкил содержит от 1 до 6 атомов углерода и (C1-C4)алкил содержит от 1 до 4 атомов углерода. Примеры алкильных групп включают, но не ограничиваются только ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил, изопентил, неопентил, изоамил, гексил, гептил, октил или нонил.
При использовании в настоящем изобретении термин "алкилен" означает алкильную группу, присоединенную в двух положениях, т.е. алкандиильную группу. Примеры включают, но не ограничиваются только ими, метилен, этилен, пропилен, бутилен, пентилен, гексилен, гептилен, октилен или нонилен. Соответственно, термин "алкилен" может, например, означать обладающую линейной цепью или разветвленной цепью алкиленовую группу, содержащую от 1 до 6 атомов углерода.
При использовании в настоящем изобретении термин "алкиламиногруппа" означает алкильную группу, присоединенную к исходному молекулярному фрагменту через аминогруппу. Подходящие алкиламиногруппы могут быть моно- или диалкилированными, образующими группы, включая, но не ограничиваясь только ими N-метиламиногруппу, N-этиламиногруппу, N,N-диметиламиногруппу, N,N-этилметиламиногруппу, N,N-диэтиламиногруппу, н-пропиламиногруппу и N,N-метилпропиламиногруппу.
При использовании в настоящем изобретении термин "алкинил" означает обладающую линейной цепью или разветвленной цепью углеводородную группу, содержащую одну или большее количество тройных связей и содержащую от 2 до 20 атомов углерода. (C2-C6)Алкинил содержит от 2 до 6 атомов углерода. (C2-C4)Алкинил содержит от 2 до 4 атомов углерода. Примеры алкинильных групп включают, но не ограничиваются только ими, этинил, пропинил, гидроксипропинил, бутин-1-ил, бутин-2-ил, пентин-1-ил, 3-метилбутин-1-ил или гексин-2-ил.
При использовании в настоящем изобретении термины "амидная группа" и "карбамоил" означает аминогруппу, описанную ниже, присоединенную к исходному молекулярному фрагменту через карбонил группу (например, -C(=O)NRR') или наоборот (-N(R)C(=O)NR'). "Амидная группа" и "карбамоил" включают "C-амидную группу", "N-амидную группу" и "ациламиногруппу", определенную в настоящем изобретении. R и R' являются такими, как определено в настоящем изобретении.
При использовании в настоящем изобретении термин "C-амидная группа" означает группу -C(=O)NRR', в которой R и R' являются такими, как определено в настоящем изобретении.
При использовании в настоящем изобретении термин "аминогруппа" означает -NRR', где R и R' независимо выбраны из группы, включающей водород, алкил, гетероалкил, арил, карбоциклил и гетероциклил. Кроме того, R и R' можно объединить с образованием гетероциклила.
При использовании в настоящем изобретении термин "арил" означает карбоциклическую ароматическую систему, содержащую одно кольцо или два кольца, или кольца, сконденсированных друг с другом, в которой все кольцевые атомы являются атомами углерода. Термин "арильные" группы включает, но не ограничивается только ими, такие группы, как фенил, нафтил или антраценил.
При использовании в настоящем изобретении термин "арилалкоксигруппа" или "арилалкоксигруппа" означает арильную группу, присоединенную к исходному молекулярному фрагменту через алкоксигруппу. Примеры арилалкоксигрупп включают, но не ограничиваются только ими, бензилоксигруппу или фенетоксигруппу.
При использовании в настоящем изобретении термин "арилалкил" или "арилалкил" означает арильную группу, присоединенную к исходному молекулярному фрагменту через алкильную группу.
При использовании в настоящем изобретении термин "арилоксигруппа" означает арильную группу, присоединенную к исходному молекулярному фрагменту через оксигруппу (-O-).
При использовании в настоящем изобретении термин "карбамат" означает O-карбамильную или N-карбамильную группу, определенную в настоящем изобретении.
При использовании в настоящем изобретении термин "карбонил", когда он содержится в виде отдельной группы, включает формил -C(=O)H и в комбинации означает -C(=O)- группу.
При использовании в настоящем изобретении термин "карбоксил" или "карбоксигруппа" означает -C(=O)OH или соответствующий "карбоксилатный" анион, такой как содержащийся в соли карбоновой кислоты. "O-Карбоксигруппа" означает группу RC(=O)O-, в которой R является таким, как определено в настоящем изобретении. "C-Карбоксигруппа" означает группы -C(=O)OR, в которых R является таким, как определено в настоящем изобретении.
При использовании в настоящем изобретении термин "цианогруппа" означает -CN.
При использовании в настоящем изобретении термин "карбоциклил" означает насыщенную или частично насыщенную моноциклическую или конденсированную бициклическую или трициклическую группу, где все кольцевые атомы циклической системы являются атомами углерода и где каждый циклический фрагмент содержит от 3 до 12 кольцевых атомов углерода. "Карбоциклил" включает бензол, сконденсированный с карбоциклильной кольцевой системой. Одна группа карбоциклилов содержит от 5 до 7 атомов углерода. Примеры карбоциклильных групп включают, но не ограничиваются только ими, циклопропил, циклобутил, циклопентил, циклогексил, и циклогептил, тетрагидронафтил, инданил, октагидронафтил, 2,3-дигидро-1H-инденил или адамантил.
При использовании в настоящем изобретении термин "циклоалкил" означает насыщенную моноциклическую, бициклическую или трициклическую группу, где все кольцевые атомы циклической системы являются атомами углерода и где каждый циклический фрагмент содержит от 3 до 12 кольцевых атомов углерода. Одна группа циклоалкилов содержит от 5 до 7 атомов углерода. Примеры циклоалкильных групп включают, но не ограничиваются только ими, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил или адамантил.
При использовании в настоящем изобретении термин "циклоалкенил" означает частично насыщенную моноциклическую, бициклическую или трициклическую группу, где все кольцевые атомы циклической системы являются атомами углерода и где каждый циклический фрагмент содержит от 3 до 12 кольцевых атомов углерода. Одна группа карбоалкенилов содержит от 5 до 7 атомов углерода. Примеры циклоалкенильных групп включают, но не ограничиваются только ими, циклобутенил, циклопентенил или циклогексенил.
При использовании в настоящем изобретении термин "циклил" означает арильную, гетероциклильную или карбоциклильную группу, определенную в настоящем изобретении.
При использовании в настоящем изобретении термин "галоген" означает хлор, фтор, бром или йод.
При использовании в настоящем изобретении термин "галогеналкоксигруппа" означает галогеналкильную группу, присоединенную к исходному молекулярному фрагменту через атом кислорода. Примеры галогеналкоксигрупп включают, но не ограничиваются только ими, трифторметоксигруппу, 2-фторэтоксигруппу, или 3-хлорпропоксигруппу.
При использовании в настоящем изобретении термин "галогеналкил" означает алкильную группу, обладает значением, определенным выше, в которой один или большее количество атомов водорода заменены галогеном. Он предпочтительно включает моногалогеналкильные, дигалогеналкильные или полигалогеналкильные группы. В одном примере моногалогеналкильная группа, может содержать атом йода, брома, хлора или фтора в группе. Дигалогенидные или полигалогеналкильные группы могут содержать два или большее количество одинаковых атомов галогенов или комбинацию разных галогенидных групп. Примеры галогеналкильных групп включают, но не ограничиваются только ими, фторметил, дифторметил, трифторметил, хлорметил, дихлорметил, трихлорметил, пентафторэтил, гептафторпропил, дифторхлорметил, дихлорфторметил, дифторэтил, дифторпропил, дихлорэтил или дихлорпропил.
При использовании в настоящем изобретении термин "гетероалкил" означает линейную или разветвленную алкильную цепь, в которой 1, 2 или 3 атома углерода, образующие алкильную цепь, заменены гетероатомом, независимо выбранным из группы, включающей O, N и S, и в которой гетероатом(ы) азота и/или серы (если содержатся) необязательно могут быть окислены и гетероатом(ы) азота (если содержатся) необязательно могут быть кватернизованы. Гетероатом(ы) О, N и S могут, например, находиться во внутреннем положении гетероалкильной группы, т.е. гетероалкил может быть связан с остальной частью молекулы через атом углерода. До двух гетероатомов могут располагаться рядом, как, например, в -CH2-NH-OCH3. Соответственно, другим примером "гетероалкильной" группы является линейная или разветвленная алкильная группа, в которой два соседних атома углерода заменены гетероатомами S и N, соответственно и гетероатом серы, кроме того, окислен с образованием таких фрагментов, как, например, -S(=O)2-NH2, -S(=O)2-NH(алкил) или -S(=O)2-N(алкил)(алкил).
При использовании в настоящем изобретении термин "гетероалкилен" означает гетероалкильную группу, присоединенную в двух положениях. Примеры включают, но не ограничиваются только ими, -СН2ОСН2-, -CH2SCH2-, и -CH2NHCH2-, -CH2S- или -CH2NHCH(CH3)CH2-. Соответственно, термин "гетероалкилен" может, например, означать линейную или разветвленную алкилен группу (т.е. линейную или разветвленную алкандиил группу), содержащую от 1 до 6 атомов углерода, где 1, 2 (если содержатся) или 3 (если содержатся) из указанных атомов углерода все заменены гетероатомом, независимо выбранным из группы, включающей О, N или S. Следует понимать, что наличие атомов водорода зависит от валентности гетероатома, заменяющего соответствующий атом углерода. Если, например, атом углерода в группе -СН2- заменен на О или S, то полученной группой будет -О- или -S- соответственно и будет -N(H)-, если атом углерода заменен на N. Аналогичным образом, если центральный атом в группе -СН2-СН(-СН3)-СН2- заменен на N, то полученной группой будет -CH2-N(-CH3)-CH2-. Примером "гетероалкиленовой" группы является линейная или разветвленная алкиленовая группа, в которой два последовательных атома углерода заменены гетероатомами S и N соответственно и гетероатом серы, кроме того окислен, что дает такие фрагменты, как, например, -S(=O)2-N(H)- или -S(=O)2-N(алкил)-. Соответственно, группы -S(=O)2-N(H)- и -S(=O)2-N(алкил)- (например, -S(=O)2-N(С1-С6-алкил)-) являются типичными "гетероалкиленовыми" группами.
При использовании в настоящем изобретении термин "гетероарил" означает 3-7-членное ненасыщенное моноциклическое кольцо, или конденсированную бициклическую или трициклическую кольцевую систему, в которой кольца являются ароматическими и в которой по меньшей мере одно кольцо содержит по меньшей мере один атом, выбранный из группы, включающей О, S и N. Одна группа гетероарилов содержит от 5 до 7 атомов углерода. Примеры гетероарильных групп включают, но не ограничиваются только ими, пиридинил, имидазолил, имидазопиридинил, пиримидинил, пиразолил, триазолил, пиразинил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксадиазолил, оксазолил, изотиазолил, пирролил, хинолинил, изохинолинил, индолил, бензимидазолил, бензофуранил, циннолинил, индазолил, индолизинил, фталазинил, пиридазинил, триазинил, изоиндолил, птеридинил, пуринил, оксадиазолил, триазолил, тиадиазолил, тиадиазолил, фуразанил, бензофуразанил, бензотиофенил, бензотиазолил, бензоксазолил, хиназолинил, хиноксалинил, нафтиридинил или фуропиридинил.
При использовании в настоящем изобретении термин "гетероциклил" или "гетероцикл" каждый означает насыщенную, частично ненасыщенную или полностью ненасыщенную моноциклическую, бициклическую, или трициклическую гетероциклическую группу, содержащую по меньшей мере один гетероатом в качестве элемента кольца, где каждый указанный гетероатом может быть независимо выбран из группы, включающей азот, кислород и серу, где атомы азота или серы могут быть окислены (например, с образованием -N=O, -S(=O)- или -S(=O)2-). Кроме того, 1, 2 или 3 атома углерода гетероциклила могут быть необязательно окислены (например, с образованием оксогруппы или =О). Одна группа гетероциклилов содержит от 1 до 4 гетероатомов в качестве элементов кольца. Другая группа гетероциклилов содержит от 1 до 2 гетероатомов в качестве элементов кольца. Одна группа гетероциклилов содержит от 3 до 8 элементов кольца в каждом кольце. Еще одна группа гетероциклилов содержит от 3 до 7 элементов кольца в каждом кольце. И другая группа гетероциклилов содержит от 5 до 6 элементов кольца в каждом кольце. "Гетероциклил" включает гетероциклильную группу, сконденсированную с карбоциклильной или бензольной кольцевыми системами. Примеры гетероциклильных групп включают, но не ограничиваются только ими, пирролидинил, тетрагидрофуранил, дигидрофуранил, тетрагидротиенил, тетрагидропиранил, дигидропиранил, тетрагидротиопиранил, пиперидиновую группу, морфолиновую группу, тиоморфолиновую группу, тиоксанил, пиперазинил, гомопиперазинил, азетидинил, оксетанил, тиетанил, гомопиперидинил, оксепанил, тиепанил, оксазепинил, диазепинил, тиазепинил, 2-пирролинил, 3-пирролинил, индолинил, 2Н-пиранил, 4Н-пиранил, диоксанил, 1,3-диоксоланил, пиразолинил, дитианил, дитиоланил, дигидропиранил, дигидротиенил, дигидрофуранил, пиразолидинилимидазолинил или имидазолидинил. Примеры гетероарилов, которые являются гетероциклилами, включают, но не ограничиваются только ими, пиридинил, имидазолил, имидазопиридинил, пиримидинил, пиразолил, триазолил, пиразинил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксадиазолил, оксазолил, изотиазолил, пирролил, хинолинил, изохинолинил, индолил, бензимидазолил, бензофуранил, циннолинил, индазолил, индолизинил, фталазинил, пиридазинил, триазинил, изоиндолил, птеридинил, пуринил, оксадиазолил, триазолил, тиадиазолил, тиадиазолил, фуразанил, бензофуразанил, бензотиофенил, бензотиазолил, бензоксазолил, хиназолинил, хиноксалинил, нафтиридинил или фуропиридинил.
При использовании в настоящем изобретении термин "гетероциклоалкил" означает гетероциклильную группу, которая не полностью ненасыщенная например, одна или большее количество кольцевых систем гетероциклоалкила не являются ароматическими. Примеры гетероциклоалкилов включают пиперазинил, морфолинил, пиперидинил или пирролидинил.
При использовании в настоящем изобретении термин "гидроксигруппа" означает -ОН.
При использовании в настоящем изобретении термин "гидроксиалкил" означает гидроксигруппу, присоединенную к исходному молекулярному фрагменту через алкильную группу.
При использовании в настоящем изобретении выражение "в главной цепи" означает самую длинную близкую или соседнюю цепь атомов углерода, начиная от положения присоединения группы к соединениям любой из формул, раскрытых в настоящем изобретении.
При использовании в настоящем изобретении выражение "линейная цепь атомов" означает самую длинную линейную цепь атомов, независимо выбранных из группы, включающей углерод, азот, кислород и серу.
При использовании в настоящем изобретении термин "низш.", если специально не приведено иного определения, означает фрагмент, содержащий от 1 включительно до 6 атомов углерода.
При использовании в настоящем изобретении термин "низш. арил" означает фенил или нафтил.
При использовании в настоящем изобретении термин "низш. гетероарил" означает 1) моноциклический гетероарил, содержащий 5 или 6 элементов кольца, из которых от 1 до 4 указанных элементов могут быть гетероатомами, выбранными из группы, включающей О, S или N.
При использовании в настоящем изобретении термин "нитрогруппа" означает -NO2.
При использовании в настоящем изобретении термины "сульфонат", "сульфоновая кислота" и "сулфоновый" означает группу -SO3H и ее анион в качестве сульфоновой кислоты использующийся для образования соли.
При использовании в настоящем изобретении термин "сульфанил" означает -S-.
При использовании в настоящем изобретении термин "сульфинил" означает -S(=O)R, где R является таким, как определено в настоящем изобретении.
При использовании в настоящем изобретении термин "сульфонил" означает -S(=O)2R, где R является таким, как определено в настоящем изобретении.
При использовании в настоящем изобретении термин "сульфонамид" означает N-сульфонамидную группу или S-сульфонамидную группу, определенную в настоящем изобретении.
При использовании в настоящем изобретении термин "N-сульфонамидная группа" означает группу RS(=O)2N(R')-, в которой R и R' являются такими, как определено в настоящем изобретении. Типичными неограничивающими N-сульфонамидными группами являются -NHSO2CH3, -NHSO2CH2CH3, NHSO2(фенил) или -NHSO2 (изопропил).
При использовании в настоящем изобретении термин "S-сульфонамидная группа" означает группу -S(=O)2NRR', в которой R и R' являются такими, как определено в настоящем изобретении.
При использовании в настоящем изобретении термин "мочевина" означает группу -N(R)C(=O)N(R), где R и R' являются такими, как определено в настоящем изобретении.
При использовании в настоящем изобретении "образующая водородную связь группа" означает замещающую группу, которая способна принимать участие в нековалентном связывании между водородом и другим атомом (обычно азотом или кислородом). Примеры включают, но не ограничиваются только ими, -NH2, -OH, амидную группу, -S(O)2NH2, -C(=O)NH2, -CH2-C(=O)NH2- и -CH2-NH2. Другие неограничивающие примеры включают NHC(=O)CH3 или -NHCH3.
При использовании в настоящем изобретении термин "изостера амидной группы" означает моноциклическую или бициклическую кольцевую систему, которая изостерна или биоозостерна с амидным фрагментом. Примеры изостер амидной группы включают, но не ограничиваются только ими, раскрытые, например, в публикации Meanwell (2011) J. Med. Chem. PMID: 21413808,
При использовании в настоящем изобретении термин "необязательно замещенная" означает предыдущую или предшествующую группу, которая может быть замещенной или незамещенной. Если она является замещенной, то заместители "необязательно замещенной" группы могут включать, без наложения ограничений, один или большее количество заместителей независимо выбранных из числа следующих групп или специально указанного набора групп, по отдельности или в комбинации: низш. алкил, низш. алкенил, низш. алкинил, низш. алканоил, низш. гетероалкил, низш. гетероциклоалкил, низш. галогеналкил, низш. циклоалкил, фенил, арил, арилоксигруппу, низш. алкоксигруппу, низш. галогеналкоксигруппу, оксогруппу, низш. ацилоксигруппу, карбонил, карбоксигруппу, низш. алкилкарбонил, низш. сложноэфирную карбоксигруппу, низш. карбоксамидную группу, цианогруппу, водород, галоген, гидроксигруппу, аминогруппу, низш. алкиламиногруппу, ариламиногруппу, аминоалкил, амидную группу, нитрогруппу, тиогруппу, низш. алкилтиогруппу, низш. галогеналкилтиогруппу, низш. пергалогеналкилтиогруппу, арилтиогруппу, сульфонатную группу, сульфоновую группу, тризамещенный силил, N3, SH, SCH3, С(O)CH3, CO2CH3, CO2H, пиридинил, тиофен, фуранил, карбаматную и мочевинную группу. Два заместителя могут быть связаны с образованием конденсированного 5-, 6- или 7-членного карбоциклического или гетероциклического кольца, содержащего от O до 3 гетероатомов, например, образующего метилендиоксигруппу или этилендиоксигруппу. Необязательно замещенная группа может быть незамещенной (например, -CH2CH3), полностью замещенной (например, -CF2CF3), монозамещенной (например, -CH2CH2F) или замещенной в диапазоне от полностью замещенной до монозамещенной (например, -CH2CF3). Если заместители указаны без определения степени замещения, то включены замещенные и незамещенные формы. Если заместитель указан, как "замещенный", то специально включена замещенная форма. Кроме того, при необходимости можно определить различные наборы необязательных заместителей конкретного фрагмента; в этих случаях необязательное замещение будет таким, как определено, часто сразу после выражения "необязательно замещенный с помощью". В одном предпочтительном определении необязательные заместители выбраны из группы, включающей гидроксигруппу, галоген, алкил, алкоксигруппу, галогеналкил, галогеналкоксигруппу, -N((C1-C3)алкил)2, -NH((C1-C3)алкил), -NHC(=O)((C1-C3)алкил), -C(=O)ОН, -C(=O)O((C1-C3)алкил), -С(=O)(C1-C3)алкил), -C(=O)NH2, -C(=O)NH(C1-C3)алкил), -С(=O)NH(циклоалкил), -С(=O)N(C1-C3)алкил)2, -S(=O)2((C1-C3)алкил), -S(=O)2NH2, -S(=O)2N((C1-C3)алкил)2, -S(=O)2NH((C1-C3)алкил), -CHF2, -OCF3, -OCHF2, -SCF3, -CF3, -CN, -NH2, -NO2 или тетразолил.
При использовании в настоящем изобретении термин "необязательный заместитель" означает, что соответствующий заместитель может содержаться или может отсутствовать. Соответственно, соединение, содержащее 1, 2 или 3 необязательных заместителя, может быть незамещенным или может содержать 1, 2 или 3 заместителя.
Термин R или термин R', указанный сам по себе и без определения количества, если не приведено другое определение, означает фрагмент, выбранный из группы, включающей водород, алкил, циклоалкил, гетероалкил, арил, гетероарил и гетероциклоалкил. Независимо от того, содержит или не содержит группа R числовое обозначение, каждая группа R, включая R, R' и Rp, в которой p=(1, 2, 3, …p), каждый заместитель, и каждый термин следует считать независящими друг от друга в отношении выбора из группы. Если любая переменная, заместитель или термин (например, арил, гетероцикл, R и т.п.) встречается в формуле или родовой структуре более одного раза, то его определение в каждом случае не зависит от определения в каждом другом случае. Специалисты в данной области техники также должны понимать, что некоторые группы могут быть присоединены к исходной молекуле или могут занимать положение в цепи элементов на любом конце, как это обозначено. Таким образом, только в качестве примера, несимметричная группа, такая как -C(=O)N(R)-, может быть присоединена к исходному фрагменту по любому атому углерода или азота.
В соединениях, раскрытых в настоящем изобретении, находятся асимметрические центры. Эти центры обозначаются символами "R" или "S" в зависимости от конфигурации заместителей вокруг хирального атома углерода. Следует понимать, что в объем настоящего изобретения входят все стереохимические изомерные формы, включая диастереоизомерные, энантиомерные и эпимерные формы, а также d-изомеры и l-изомеры и их смеси. Отдельные стереоизомеры соединений можно получить синтетически из имеющихся в продаже исходных веществ, которые содержат хиральные центры или путем получения смесей энантиомерных продуктов с последующим разделением, таким как превращение в смеси диастереоизомеров с последующим разделением или перекристаллизацией, с использованием хроматографических методик, прямое разделение энантиомеров на хиральных хроматографических колонках или по любой другой подходящей методике, известной в данной области техники. Исходные соединения, обладающие конкретной стереохимической конфигурацией, имеются в продаже или их можно получить и разделить по методикам, известным в данной области техники. Кроме того, соединения, рассмотренные в настоящем изобретении, могут существовать в виде геометрических изомеров. В объем настоящего изобретения входят все цис-, транс-, син-, анти-, (E)- и (Z)-изомеры, а также подходящие их смеси. Кроме того, соединения могут существовать в виде таутомеров; все таутомеры входят в объем настоящего изобретения. Кроме того, соединения, раскрытые в настоящем изобретении, могут находиться в несольватированной, а также сольватированной формах с фармацевтически приемлемыми растворителями, такими как вода, этанол и т.п. Обычно сольватированные формы считают эквивалентными несольватированным формам.
При использовании в настоящем изобретении термин "оптически активное" означает способность соединения вращать плоскость поляризованного света. В контексте настоящего изобретения термин означает смеси энантиомеров, которые не являются рацемическими смесями; т.е. не являются 50:50 смесью (+)-энантиомера и соответствующего (-)-энантиомера.
При использовании в настоящем изобретении термин "N-замещенный арил или гетероарилциклопропиламин" (или также "N-замещенный арил- или гетероарилциклопропиламин") означает соединение, содержащее 1,2-дизамещенное циклопропильное ядро, где положения 1 и 2 замещены замещенной аминогруппой и замещенной арильной или гетероарильной группой. Соединения формулы (II) и формулы (III), описанные в настоящем изобретении являются примерами N-замещенных арил- или гетероарилциклопропиламинов.
При использовании в настоящем изобретении термин "энантиомерный избыток" или "ЭИ" или "энантиомерный избыток в процентах" означает разность между молярной долей одного конкретного энантиомера (т.е. заданного энантиомера) и молярной долей другого энантиомера, деленную на сумму молярных долей обоих энантиомеров, выраженную в процентах, и таким образом описывает избыток одного конкретного энантиомера по сравнению с другим энантиомером. Если, например, конкретный энантиомер получен при отсутствии другого энантиомера, то энантиомерный избыток будет равен 100%, а рацемат, содержащий эквимолярные количества этих двух энантиомеров, будет характеризоваться энантиомерным избытком, составляющим 0%. Соответственно, "энантиомерный избыток" или "ЭИ" или "энантиомерный избыток в процентах" определяется следующей формулой:

При использовании в настоящем изобретении термин "предупреждение усиления симптома" означает недопущение усиления или ухудшения симптома, а также уменьшение скорости усиления симптома. Например, симптом можно охарактеризовать содержанием маркера конкретного заболевания, т.е. белка (например, биомаркера рака). В другом примере симптомом может быть ухудшение познавательной способности. Предупреждение усиления в соответствии с определением, приведенным в настоящем изобретении, означает, что степень проявления симптома (например, содержание белка или степень ухудшения познавательной способности) не усиливается, или что уменьшается скорость его усиления.
При использовании в настоящем изобретении термин "лечение заболевания или нарушения" означает замедление или обращение прогрессирования заболевания. Лечение заболевания или нарушения включает устранение симптома и/или ослабление симптома заболевания.
При использовании в настоящем изобретении термин "предупреждение заболевания или нарушения" означает замедление заболевания или начала развития заболевания или ослабление его симптомов. Предупреждение заболевания или нарушения может включать остановку развития заболевания или его симптомов. При использовании в настоящем изобретении термин "разовая дозированная форма" означает отдельные порции, такие как капсулы или таблетки, пригодные для использования в качестве разовых доз для пациентов-людей. Каждая порция содержит заранее заданное количество соединения формулы (I), (Ia), (Ib), (II) или (III), которое, как это установлено или предположено, приводит к необходимому фармакокинетическому профилю, который обеспечивает необходимый терапевтический эффект. Дозированная форма содержит соединение формулы (I), (Ia), (Ib), (II) или (III) вместе по меньшей мере с одним фармацевтически приемлемым носителем, солью, инертным наполнителем или их комбинацией.
При использовании в настоящем изобретении термин "субъект" или "пациент", или "индивидуум", такой как субъект, нуждающийся в лечении или предупреждении, может представлять собой эукариота, животного, позвоночного животного, млекопитающего, грызуна (например, морскую свинку, хомяка, крысу, мышь), представителя семейства мышиных (например, мышь), представителя семейства собачьих (например, собаку), представителя семейства кошачьих (например, кошку), представителя семейства лошадиных (например, лошадь), примата, представителя семейства обезьяньих (например, обезьяну или человекообразную обезьяну), обезьяну (например, мартышку, бабуина), человекообразную обезьяну (например, гориллу, шимпанзе, орангутана, гиббона) или человека. Значения терминов "эукариот", "животное", "млекопитающее" и т.п. хорошо известно в данной области техники и определены, например, в публикации Wehner und Gehring (1995; Thieme Verlag). В контексте настоящего изобретения особо отмечается, что следует лечить животных, которые являются экономически, агрономически важными или важными с научной точки зрения. Важные с научной точки зрения организмы включают, но не ограничиваются только ими, мышей, крыс и кроликов. Низшие организмы, такие как, например, плодовые мушки, такие как Drosophila melagonaster, и нематоды, такие как Caenorhabditis elegans, также можно использовать в научных исследованиях. Неограничивающими примерами агрономически важных животных являются овцы, крупный рогатый скот и свиньи, тогда как, например, кошек и собак можно считать экономически важными животными. Предпочтительно, если субъект/пациент/индивидуум является млекопитающим; более предпочтительно, если субъект/пациент/индивидуум является человеком или млекопитающим, не являющимся человеком (таким как, например, морская свинка, хомяк, крыса, мышь, кролик, собака, кошка, лошадь, обезьяна, человекообразная обезьяна, мартышка, бабуин, горилла, шимпанзе, орангутан, гиббон, овца, крупный рогатый скот или свинья); еще более предпочтительно, если субъект/пациент/индивидуум является человеком.
При использовании в настоящем изобретении термин "доза" или "дозировка" означает количество активного ингредиента, который индивидуум принимает или которое вводят за один раз. Например, равная 40 мг доза соединения формулы (I), (Ia), (Ib), (II) или (III) в случае режима введения два раза в сутки означает ситуацию, когда индивидуум принимает 40 мг соединения формулы (I) два раза в сутки, например, 40 мг утром и 40 мг вечером. Равную 40 мг дозу соединения формулы (I), (Ia), (Ib), (II) или (III) можно разделить на 2 или большее количество доз, например, 2 дозы по 20 мг соединения формулы (I), (Ia), (Ib), (II) или (III) в форме таблетки или 2 дозы по 20 мг соединения формулы (I), (Ia), (Ib), (II) или (III) в форме капсулы.
При использовании в настоящем изобретении "фармацевтически приемлемое пролекарство" означает соединение, которое при физиологических условиях или путем сольволиза может превратиться в заданное соединение или в фармацевтически приемлемую соль такого соединения.
При использовании в настоящем изобретении "фармацевтически активный метаболит" означает фармакологически активный продукт, образовавшийся путем происходящего в организме метаболизма из указанного соединения или его соли. Метаболиты соединения можно идентифицировать по стандартным методикам, известным в данной области техники, и их активность определяют с использованием тестов, таких как описанные в настоящем изобретении.
При использовании в настоящем изобретении "фармацевтически приемлемая соль" означает соль, которая сохраняет биологическую эффективность свободных кислот и оснований данного соединения и не является биологически или в ином отношении нежелательной. Соединение, предназначенное для применения в настоящем изобретении, может обладать достаточным количеством кислотных, достаточным количеством основных групп или обоими типами функциональных групп и соответствующим образом взаимодействовать с любым количеством неорганических или органических оснований и неорганических и органических кислот с образованием фармацевтически приемлемой соли. Типичные фармацевтически приемлемые соли включают соли, полученные по реакции соединений, предлагаемых в настоящем изобретении, с неорганической или органической кислотой или с неорганическим основанием, такие как сульфаты, пиросульфаты, бисульфаты, сульфиты, бисульфиты, фосфаты, моногидрофосфаты, дигидрофосфаты, метафосфаты, пирофосфаты, хлориды, бромиды, йодиды, ацетаты, пропионаты, деканоаты, каприлаты, акрилаты, формиаты, изобутираты, капроаты, гептаноаты, пропиолаты, оксалаты, малонаты, сукцинаты, субераты, себацинаты, фумараты, малеаты, бутин-1,4-диоаты, гексин-1,6-диоаты, бензоаты, хлорбензоаты, метилбензоаты, динитробензоаты, гидроксибензоаты, метоксибензоаты, фталаты, сульфонаты, ксилолсульфонаты, фенилацетаты, фенилпропионаты, фенилбутираты, цитраты, лактаты, гамма-гидроксибутираты, гликоляты, тартраты, метансульфонаты, пропансульфонаты, нафталин-1-сульфонаты, нафталин-2-сульфонаты и манделаты.
При использовании в настоящем изобретении "фармацевтически приемлемый носитель" означает вещества, не являющиеся АФИ (АФИ означает активный фармацевтический ингредиент), такие как разрыхлители, связующие, наполнители, и смазывающие вещества, использующиеся для приготовления фармацевтических продуктов. Они обычно являются безопасными при введении людям в соответствии с установленными правительственными стандартами, включая введенные в действие Управлением по контролю за качеством пищевых продуктов, медикаментов и косметических средств США и Европейским медицинским агентством.
Специалист в данной области техники понимает, что некоторые переменные в перечне заместителей повторяются (разные названия для одного и того же заместителя), являются родовыми для других переменных перечня и/или по значению частично перекрываются с другими терминами. Специалист в данной области техники понимает, что в соединениях, предлагаемых в настоящем изобретении, заместители могут быть присоединены к остальной части молекулы в целом ряде положений и предпочтительными являются положения, указанные в примерах.
Как описано выше в настоящем изобретении, соединение формулы (I), (Ia) или (Ib) содержат асимметрические атомы углерода и поэтому могут существовать в рацемических и оптически активных формах. Таким образом, оптические изомеры или энантиомеры, рацематы, таутомеры и диастереоизомеры также входят в число соединений формулы (I), (Ia) или (Ib). Способы, предлагаемые в настоящем изобретении, включают применение всех таких изомеров и их смесей. Методики разделения смесей энантиомеров и диастереоизомеров хорошо известны специалисту в данной области техники и, кроме того, описаны прилагаемых примерах. В объем настоящего изобретения входят любые выделенные рацемические или оптически активные формы соединения формулы (I), (Ia) или (Ib) или любые их смеси. В одном объекте соединения, предлагаемые в настоящем изобретении, транс-конфигурацией относительно циклопропильного кольца, как в транс-фенилциклопропиламине. В одном объекте соединения, предлагаемые в настоящем изобретении, обладают цис-конфигурацией относительно циклопропильного кольца, как в цис-фенилциклопропиламине. В предпочтительном объекте соединения формулы (I), (Ia) или (Ib) обладают транс-конфигурацией. В более предпочтительном объекте соединения формулы (I), (Ia) или (Ib) являются (-)-стереоизомерами, содержащими транс-конфигурации относительно циклопропильного кольца.
Обычно соединения формулы (I), (Ia), (Ib), (II) или (III) могут быть эффективны в количестве, составляющем от примерно 0,01 до примерно 100 мг/(кг сутки) в пересчете на полную массу тела. Активный ингредиент можно вводить одной порцией или можно разделить на несколько меньших доз, вводимых через заранее заданные промежутки времени. Разовая доза, подходящая для каждого введения может составлять, например, от примерно 1 мкг до примерно 2000 мг, предпочтительно от примерно 5 мкг до примерно 1000 мг. Еще более предпочтительно, если количество вводимого активного ингредиента составляет от примерно 5 мкг до примерно 100 мг в сутки. Эти дозы зависят от фармакокинетических параметров конкретного соединения и других характеристик всасывания, распределения и метаболизма, а также от эффективности соединения при конкретном состоянии заболевания.
Следует понимать, что указанные выше диапазоны доз являются лишь типичными и не ограничивают объем настоящего изобретения. Терапевтически эффективное количество каждого активного соединения может меняться от таких факторов, включая, но не ограничиваясь только ими, как активность использующегося соединения, стабильность активного соединения в организме пациента, тяжесть патологических состояний, протекание которых облегчают, полная масса подвергающегося лечению пациента, путь введения, легкость всасывания, распределения и выведения активного соединения из организма, возраст и чувствительность пациента по отношению к лечению и т.п., что должен понимать специалист в данной области техники. Вводимое количество можно регулировать, поскольку различные факторы меняются во времени.
Для пероральной доставки активные соединения можно ввести в препарат, который включает фармацевтически приемлемые носители, такие как связующие (например, желатин, целлюлоза, трагакантовая камедь), инертные наполнители (например, крахмал, лактоза), смазывающие вещества (например, стеарат магния, диоксид кремния), разрыхлители (например, альгинат, примогель и кукурузный крахмал) и подсластитель и ароматизаторы (например, глюкоза, сахароза, сахарин, метилсалицилат и перечная мята). Препарат можно доставлять перорально в виде закрытых капсул из желатина или прессованных таблеток. Капсулы и таблетки можно изготовить по любым обычным методикам. На капсулы и таблетки также можно нанести различные покрытия, известные в данной области техники, для изменения аромата, вкуса, цвета и формы капсул и таблеток. Кроме того, в капсулы можно внести жидкие носители, такие как нелетучее жидкое масло.
Препараты, подходящие для перорального введения также могут находится в форме суспензии, сиропа, жевательной резинки, облатки, эликсира и т.п. При необходимости в них также можно включить обычные агенты, изменяющие аромат, вкус, цвет и форму этих специальных форм препаратов. Кроме того, для удобства введения с помощью зонда для искусственного кормления пациентов, которые не могут глотать, активные соединения можно растворить в приемлемом липофильном растительном масле в качестве разбавителя, таком как оливковое масло, кукурузное масло и сафлоровое масло.
Активные соединения также можно вводить парентерально в виде раствора или суспензии или в лиофилизированной форме, которую перед использованием можно превратить в раствор или суспензию. В таких препаратах можно использовать разбавители или фармацевтически приемлемые носители, такие как стерильная вода и буферный физиологический раствор. Можно включить другие обычные растворители, буферы для регулирования pH, стабилизаторы, антибактериальные средства, поверхностно-активные вещества и антиоксиданты. Например, применяющиеся компоненты включают хлорид натрия, ацетатные, цитратные или фосфатные буферы, глицерин, декстрозу, нелетучие масла, метилпарабены, полиэтиленгликоль, пропиленгликоль, бисульфат натрия, бензиловый спирт, аскорбиновую кислоту и т.п. Препараты для парентерального введения можно хранить в любых обычных контейнерах, таких как флаконы и ампулы.
Пути местного введения включают назальное, трансбуккальное, проводимое через слизистую оболочку, ректальное или вагинальное введение. Для местного введения активные соединения можно приготовить в виде лосьонов, кремов, мазей, гелей, порошков, суспензий, капель и аэрозолей. В препараты можно включить один или большее количество загущающих агентов, влагоудерживающих веществ и стабилизирующих агентов. Примеры таких агентов включают, но не ограничиваются только ими, полиэтиленгликоль, сорбит, ксантановую камедь, вазелиновое масло, пчелиный воск или минеральное масло, ланолин, сквален и т.п. Специальным типом местной доставки является использование чрескожного пластыря. Методики изготовления чрескожных пластырей описаны, например, в публикации Brown, et al. (1988) Ann. Rev. Med. 39:221-229, которая включена в настоящее изобретение в качестве ссылки.
Подходящим путем введения также может быть подкожная имплантация, предназначенная для пролонгированного высвобождения активных соединений. Она включает хирургические процедуры для имплантации активного соединения в любой подходящей композиции в подкожное пространство, например, под переднюю брюшную стенку. См., например, публикацию Wilson et al. (1984) J. Clin. Psych. 45:242-247. Для пролонгированного высвобождения активных соединений в качестве носителя можно использовать гидрогели. Гидрогели в целом известны в данной области техники. Их обычно получают путем сшивки высокомолекулярных биологически совместимых полимеров в сетку, которая набухает в воде с образованием гелеобразного материала. Предпочтительно, если гидрогели являются биологически разлагающимися или биологически сорбирующимися. Для задач настоящего изобретения можно использовать гидрогели, приготовленные из полиэтиленгликолей, коллагена или сополимера гликоля с L-молочной кислотой. См., например, публикацию Phillips et al. (1984) J. Pharmaceut. Sci., 73: 1718-1720.
Активные соединения также можно конъюгировать с растворимым в воде неиммуногенным, непептидным обладающим большой молекулярной массой полимером с образованием конъюгата с полимером. Например, активное соединение ковалентно связывают с полиэтиленгликолем с образованием конъюгата. Обычно такой конъюгат обладает улучшенной растворимостью, стабильностью и пониженной токсичностью и иммуногенностью. Таким образом, при введении пациенту активное соединение в конъюгате может обладать более длительным периодом полувыведения из организма и большей эффективностью. См. публикацию Burnham (1994) Am. J. Hosp. Pharm. 15: 210-218. Пэгилированные (ПЭГ - полиэтиленгликоль) белки в настоящее время используются в белковозаместительной терапии и для других терапевтических целей. Например, пэгилированные интерферон (ПЭГ-ИНТРОН А®) используют для клинического лечения гепатита B. Пэгилированную аденозиндезаминазу (адаген®) используют для лечения тяжелого комбинированного иммунодефицита (SCIDS). Пэгилированную L-аспарагиназу (онкапспар®) используют для лечения острого лимфобластного лейкоза (ОЛЛ). Предпочтительно, если ковалентная связь между полимером и активным соединением и/или в самом полимере гидролитически разрывается при физиологических условиях. Такие конъюгаты, известные в качестве "пролекарств", могут легко высвобождать активное соединение внутри организма. Регулируемое высвобождение активного соединения также можно обеспечить путем включения активного ингредиента в микрокапсулы, нанокапсулы или гидрогели, общеизвестные в данной области техники. Другие фармацевтически приемлемые пролекарства соединений, предлагаемых в настоящем изобретении, включают, но не ограничиваются только ими, сложные эфиры, карбонаты, тиокарбонаты, N-ацилпроизводные, N-ацилоксиалкилпроизводные, четвертичные производные третичных аминов, N-основания Манниха, шиффовы основания, конъюгаты аминокислот, фосфаты, соли металлов и сульфонаты.
В качестве носителей для активных соединений, предлагаемых в настоящем изобретении, также можно использовать липосомы. Липосомы являются мицеллами, образованными из различных липидов, таких как холестерин, фосфолипиды, жирные кислоты и их производные. Также можно использовать различные модифицированные липиды. Липосомы могут уменьшать токсичность активных соединений и повышать их стабильность. Методики приготовления липосомных суспензий, содержащих активные ингредиенты, общеизвестны в данной области техники. См., например, патент U.S. №4522811; Prescott, Ed., Methods in Cell Biology, Volume XIV, Academic Press, New York, N.Y. (1976).
Активные соединения также можно вводить в комбинации с другим активным средством, которое синергетически устраняет или предупреждаем такие же симптомы или эффективно для лечения другого заболевания или устранения симптома у подвергающегося лечению пациента, если другое активное средство не мешает активному соединению или неблагоприятно не влияет на воздействие активных соединений, предлагаемых в настоящем изобретении. Такие другие активные средства включают, но не ограничиваются только ими, противовоспалительные средства, противовирусные средства, антибиотики, фунгицидные средства, антитромботические средства, сердечнососудистые средства, средства, снижающие содержание холестерина, противораковые лекарственные средства, гипотензивные лекарственные средства и т.п.
Примеры противоопухолевых средств, которые можно использовать в комбинации с соединениями и способами, предлагаемыми в настоящем изобретении, обычно включают, если это является подходящим, алкилирующие средства, антиметаболиты, эпидофиллотоксины, противоопухолевые ферменты, ингибиторы топоизомеразы, прокарбазины, митоксантроны, координационные комплексы платины, модификаторы биологического ответа и ингибиторы роста, гормональные/антигормональные терапевтические средства и гематопоэтические факторы роста. Типичные классы противоопухолевых средств включают антрациклины, алкалоиды барвинка, митомицины, блеомицины, цитотоксические нуклеозиды, эпотилоны, дискодермолиды, птеридины, диинены и подофиллотоксины. Особенно полезные представители этих классов включают, например, карминомицин, даунорубицин, аминоптерин, метотрексат, метоптерин, дихлорметотрексат, митомицин C, порфиромицин, 5-фторурацил, 6-меркаптопурин, гемцитабин, цитозин арабинозид, подофиллотоксин или производные подофиллотоксина, такие как этопозид, этопозидфосфат или тенипозид, мелфалан, винбластин, винкристин, лейрозидин, виндезин, лейрозин, паклитаксел и т.п. Другие полезные противоопухолевые средства включают эстрамустин, карбоплатин, циклофосфамид, блеомицин, гемцитабин, ифосфамид, мелфалан, гексаметилмеламин, тиотепа, цитарабин, идатрексат, триметрексат, дакарбазин, L-аспарагиназа, камптотецин, CPT-11, топотекан, ara-C, бикалутамид, флутамид, лейпролид, производные пиридобензоиндолы, интерфероны и интерлейкины.
Описание общего пути синтеза
Соединения формулы (I), (Ia), (Ib), (II) или (III) можно синтезировать в соответствии или по аналогии с общими путями, описанными на схемах 1, 2 и 3. Другие пути, известные специалисту с общей подготовкой в данной области техники, а также другие реагенты и промежуточные продукты также можно использовать для получения соединений формулы (I), (Ia), (Ib), (II) или (III).

Имеющиеся в продаже нитростиролы формулы (1) вводили в реакцию циклопропанирования с использованием триметилсульфоксониййодида и трет-бутилата калия. Затем нитрогруппу полученных транс-нитроциклопропилпроизводных формулы (2) (они являются транс-((1S,2R), (1R,2S))-смесью, хотя можно использовать отдельные диастереоизомеры, соответствующие (1S,2R) и (1R,2S)) восстанавливали цинком в хлористоводородной кислоте и получали циклопропиламинопроизводные формулы (3). Затем восстановительное алкилирование с помощью имеющихся в продаже альдегидов формулы (4) с использованием триацетоксиборогидрида натрия в качестве восстановительного реагента приводило к образованию циклопропиламинопроизводных формулы (5), которые являются объектами настоящего изобретения.

Имеющиеся в продаже альдегиды формулы (6) вводили в реакцию Хорнера-Уодсворта-Эммонса с использованием триэтилфосфоноацетата и трет-бутоксида калия в тетрагидрофуране при 0°C и получали этилакрилатные производные формулы (7), которые вводили в реакцию циклопропанирования с использованием триметилсульфоксониййодида и гидрида натрия в диметилсульфоксиде в качестве растворителя и получали (транс)-этилциклопропанкарбоксилатные производные формулы (8) (они являются транс-((1S,2R), (1R,2S))-смесью, хотя можно использовать отдельные диастереоизомеры, соответствующие (1S,2R) и (1R,2S)). Гидролиз с получением соответствующих производных (транс)-циклопропанкарбоновой кислоты формулы (9) проводили с помощью NaOH в MeOH. Реакция, сначала с этилхлорформиатом и триэтиламином в ацетоне и затем с азидом натрия в воде приводила к образованию (транс)-циклопропанкарбонилазидных производных формулы (10). Реакция с трет-бутанолом приводила к образованию трет-бутил-(транс)-циклопропилкарбаматных производных формулы (11). Реакция с имеющейся в продаже бороновой кислотой или ее эфирами формулы (12) с использованием ацетонитрила и воды в качестве растворителя, карбоната калия в качестве основания и тетракис(трифенилфосфин)палладия(0) в качестве катализатора приводила к образованию трет-бутил-(транс)-циклопропилкарбаматных производных формулы (13).
Удаление защитной группы Boc с помощью 2M HCl в диэтиловом эфире с использованием диэтилового эфира в качестве растворителя приводило к образованию соответствующих гидрохлоридов (транс)-циклопропанаминовых производных формулы (9). Восстановительное алкилирование с помощью имеющихся в продаже альдегидов формулы (4) с использованием триацетоксиборогидрида натрия в качестве восстановительного реагента приводило к образованию циклопропиламинопроизводных формулы (5), которые являются объектами настоящего изобретения.
Алкилирование трет-бутил-(транс)-циклопропилкарбаматных производных формулы (13) с помощью имеющихся в продаже алкилгалогенидов формулы (14) с использованием гидрида натрия в качестве основания и ДМФ в качестве растворителя приводило к образованию трет-бутил-(транс)-циклопропилкарбаматных производных формулы (15). Удаление защитной группы Boc с помощью 2М HCl в диэтиловом эфире с использованием диэтилового эфира в качестве растворителя приводило к образованию соответствующих гидрохлоридов транс)-циклопропанаминовых производных формулы (5), которые являются объектом настоящего изобретения, как это определено выше.
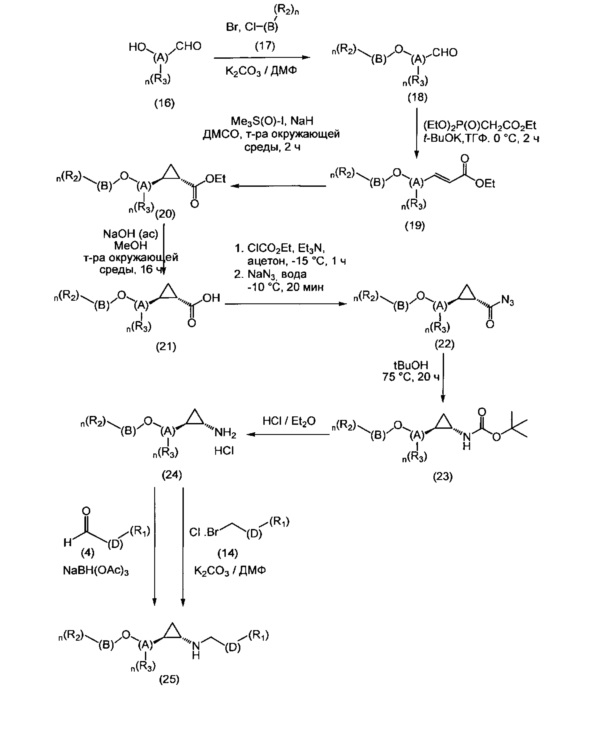

Алкилирование имеющихся в продаже альдегидов формулы (16) с использованием имеющихся в продаже алкилгалогенидов формулы (17), карбоната калия в N,N-диметилформамиде приводило к образованию производных альдегидов формулы (18). Реакция Хорнера-Уодсворта-Эммонса с использованием триэтилфосфоноацетата и трет-бутоксида калия в тетрагидрофуране при 0°C давала этилакрилатные производные формулы (19), которые вводили в реакцию циклопропанирования с использованием триметилсульфоксониййодида и гидрида натрия в диметилсульфоксиде в качестве растворителя и получали (транс)-этилциклопропанкарбоксилатные производные формулы (20). Гидролиз с получением соответствующих производных (транс)-циклопропанкарбоновой кислоты формулы (21) проводили с помощью NaOH в МеОН. Реакция, сначала с этилхлорформиатом и триэтиламином в ацетоне и затем с азидом натрия в воде приводила к образованию (транс)-циклопропанкарбонилазидных производных формулы (22). Реакция с трет-бутанолом приводила к образованию трет-бутил-(транс)-циклопропилкарбаматных производных формулы (23). Удаление защитной группы Boc с помощью 2М HCl в диэтиловом эфире с использованием диэтилового эфира в качестве растворителя приводило к образованию соответствующих гидрохлоридов (транс)-циклопропанаминовых производных формулы (24).
Восстановительное алкилирование с помощью имеющихся в продаже альдегидов формулы (4) с использованием триацетоксиборогидрида натрия в качестве восстановительного реагента приводило к образованию (транс)-циклопропиламинопроизводных формулы (25), которые также являются объектами настоящего изобретения.
Альтернативно, алкилирование (транс)-циклопропанаминовых производных формулы (24) с использованием имеющихся в продаже алкилгалогенидов формулы (14) и карбоната калия в качестве основания и N,N-диметилформамида в качестве растворителя также приводило к образованию (транс)-циклопропиламинопроизводных формулы (25), которые являются объектом настоящего изобретения, как это определено выше.
Как известно специалистам в данной области техники, (транс)-циклопропиламинопроизводные формулы (5) и (25) также можно получить из (транс)-циклопропанаминовых производных формулы (3) и (24) соответственно по хорошо известным реакциям (т.е. с помощью циклизации).
Оптически чистые или энантиомерно обогащенные соединения можно выделить на различных стадиях процедуры синтеза и можно использовать на следующих стадиях.
Примеры
Для образования названий, соответствующих структурам, приведенным ниже в примерах соединений, использовали программу ChemBioDraw Ultra 11.0.1. Эта программа образует названия молекул в конфигурации (1S,2R), что обусловлено конфигурацией вводимой в программу структуры, и вместо термина (1S,2R), заданного в программе, используют термин "транс". Структуры, приведенные ниже в примерах соединений представлены, как обладающие одной определенной стереохимической конфигурацией атомов углерода циклопропильного фрагмента фенилциклопропиламинового ядра, (1S,2R). Если не указано иное, соединения, синтезированные в примерах, являются смесями, содержащими обе конфигурации (1R,2S) и (1S,2R), т.е. они обладают "транс"-конфигурацией применительно к заместителям циклопропильной кольцевой системы. Это обусловлено тем, что циклопропильные производные, использующиеся в качестве исходного вещества, обладают "транс"-конфигурацией. Предполагается, что в качестве исходного вещества можно использовать соединение в цис-конфигурации или отдельные диастереоизомеры и все они имеются в продаже или их можно синтезировать. Таким образом, настоящее изобретение относится к соединениям формулы (I), (Ia), (Ib), (II) или (III), включая соединения примеров, которые обладают указанными стереохимическими конфигурациями циклопропильного кольца, например, транс-((1R,2S) и (1S,2R)) и цис-((1R,2R) и (1S,2S)). Предпочтительной стереохимической конфигурацией циклопропильного кольца является трансконфигурация.
Соединения, указанные в примерах, также можно синтезировать или они поставляются в виде солей. Специалист в данной области техники знает, как получить и может получить солевые формы и/или подвергнуть превращению солевые формы соединений, предлагаемых в настоящем изобретении, например, соединений формулы (I), (Ia), (Ib), (II) или (III) и соединения примеров. В некоторых случаях соединения формулы (I), (Ia), (Ib), (II) или (III) и соединения примеров в форме солей могут быть стабильнее, чем свободные основания.
Промежуточные продукты (и аналоги промежуточных продуктов или их производные), указанные на схемах синтеза, представленных в настоящем изобретении, можно получить по приведенным ниже методикам.
Промежуточный продукт A: 1-(Бензилокси)-4-[(транс)-2-нитроциклопропил]бензол

Триметилсульфоксониййодид (0,62 г, 2,82 ммоля) порциями добавляли к раствору t-BuOK (0,32 г, 2,82 ммоля) в сухом ДМСО (5 мл). Через 10 мин с помощью канюли добавляли раствор 1-(бензилокси)-4-[(E)-2-нитровинил]бензола (0,60 г, 2,35 ммоля) в ДМСО (5 мл) и смесь перемешивали при комнатной температуре в течение 6 ч. Реакционную смесь выливали в воду (10 мл) и экстрагировали с помощью Et2O (3×10 мл); органические слои промывали рассолом (2×15 мл), сушили над безводным Na2SO4 и фильтровали. После удаления растворителя оставшееся оранжевое масло очищали с помощью колоночной хроматографии на силикагеле (5% EtOAc/гексаны) и получали 0,16 г 1-(бензилокси)-4-[(транс)-2-нитроциклопропил]бензола [Rf=0,5 (20% EtOAc/гексаны), белое твердое вещество, выход 26%].
Промежуточный продукт B: Транс-2-[4-(бензилокси)фенил]циклопропанамин

Цинковую пыль (1,97 г, 30 моля) небольшими порциями в течение 30 мин при энергичном перемешивании добавляли к раствору 1-(бензилокси)-4-[(транс)-2-нитроциклопропил]бензола (промежуточный продукт A, 0,81 г, 3,0 ммоля) в i-PrOH (25 мл) и HCl (11 мл 2,7 н. водного раствора, 30 ммолей). Через 17 ч смесь фильтровали через слой целита, который промывали с помощью 10 мл метанола. Фильтрат концентрировали и добавляли 10 мл воды, промывали с помощью CH2Cl2 (3×15 мл). Органические слои сушили над безводным Na2SO4 и фильтровали. После удаления растворителя неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (10% МеОН/CH2Cl2) и получали 0,50 г (транс)-2-[4-(бензилокси)фенил]циклопропанамина [Rf=0,2 (10% МеОН/CH2Cl2), белое твердое вещество, выход 70%].1H-ЯМР (МеОН, 250 МГц, δ): 7,45-7,27 (m, 5Н, ArH); 6,96 (d, J=8,5 Гц, 2Н, ArH); 6,86 (d, J=8,5 Гц, 2Н, ArH); 5,03 (s, 2Н, CH2); 2,41-2,34 (m, 1Н, CH); 1,86-1,76 (m, 1Н, CH); 0,98-0,85 (m, 2Н, CH2).
Промежуточный продукт C: (E)-Этил-3-(6-бромпиридин-3-ил)акрилат
Триэтилфосфоноацетат (26,6 г, 118,8 ммоля) по каплям медленно добавляли к смеси трет-бутоксида калия (14,5 г, 129,6 ммоля) с сухим ТГФ (200 мл) при -5°C, перемешивали в течение 20 мин и затем по каплям при -5°C медленно добавляли раствор 6-бромпиридин-3-карбоксальдегида (20 г, 108 ммоля) в сухом ТГФ (100 мл) и перемешивали в течение 30 мин. После завершения реакции реакционную смесь выливали на лед с водой (350 мл) и экстрагировали с помощью EtOAc (2×300 мл). Объединенные органические экстракты промывали насыщенным раствором NAHCO3 (250 мл), водой (250 мл) и рассолом (250 мл) и сушили над безводным Na2SO4, фильтровали и выпаривали и получали (E)-этил-3-(6-бромпиридин-3-ил)акрилат (20 г, 72,9%) в виде коричневой жидкости. Его использовали на следующей стадии без дополнительной очистки.
Промежуточный продукт D: (Транс)-этил-2-(6-бромпиридин-3-ил)циклопропанкарбоксилат

Триметилсульфоксониййодид (20,8 г, 94,7 ммоля) небольшими порциями добавляли к суспензии гидрида натрия (4 г, 170,6 ммоля) в сухом ДМСО (400 мл) при КТ, перемешивали в течение 1 ч до образования прозрачного раствора. Добавляли раствор (E)-этил-3-(6-бромпиридин-3-ил)акрилата (промежуточный продукт C, 20 г, 78,7 ммоля) в сухом ДМСО (20 мл) и перемешивали в течение 4 ч. После завершения реакции реакционную смесь выливали на лед с водой (700 мл), экстрагировали с помощью ЕtOАс (2×350 мл). Объединенные органические экстракты промывали водой (250 мл), рассолом (250 мл) и сушили над безводным Na2SO4, фильтровали и выпаривали и получали (транс)-этил-2-(6-бромпиридин-3-ил)циклопропанкарбоксилат (10 г, 47%) в виде коричневой жидкости.
Промежуточный продукт E: Гидрохлорид(транс)-2-(6-бромпиридин-3-ил)циклопропанкарбоновой кислоты

4 н. Раствор NaOH (60 мл) добавляли к раствору (транс)-этил-2-(6-бромпиридин-3-ил)циклопропанкарбоксилата (промежуточный продукт D, 10 г, 37,1 ммоля) в метаноле (100 мл) и реакционную смесь перемешивали при КТ в течение 4 ч. После завершения реакции растворитель выпаривали и остаток разбавляли водой со льдом (250 мл) и подкисляли 4 н. раствором HCl, водный слой экстрагировали с помощью EtOAc (2×350 мл). Объединенные органические экстракты промывали водой (250 мл), рассолом (250 мл) и сушили над безводным Na2SO4, фильтровали и выпаривали и получали гидрохлорид (транс)-2-(6-бромпиридин-3-ил)циклопропанкарбоновой кислоты (5 г, 55,8%) в виде светло-коричневого твердого вещества.
Промежуточный продукт F: (Транс)-2-(6-бромпиридин-3-ил)циклопропанкарбонилазид
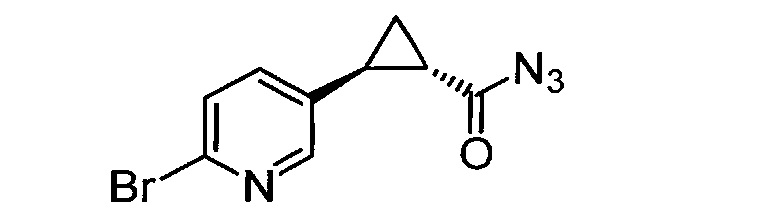
Этилхлорформиат (5,8 мл, 62 ммоля) добавляли к раствору гидрохлорида (транс)-2-(6-бромпиридин-3-ил)циклопропанкарбоновой кислоты (промежуточный продукт E, 5 г, 20,7 ммоля) и Et3N (14,2 мл, 103,7 ммоля) в ацетоне (100 мл) при -5°C, затем реакционную смесь перемешивали при -5°C в течение 1 ч, затем раствор NaN3 (2,7 г, 41,4 ммоля) в воде (10 мл) добавляли и перемешивали в течение 30 мин при КТ. После завершения реакции растворитель выпаривали в вакууме. Неочищенный остаток растворяли в этилацетате (200 мл), промывали водой (80 мл), рассолом (80 мл), сушили над безводным Na2SO4, фильтровали и выпаривали и получали (транс)-2-(6-бромпиридин-3-ил)циклопропанкарбонилазид (2,5 г, 45,5%) в виде коричневой вязкой жидкости.
Промежуточный продукт G: трет-Бутил-(транс)-2-(6-бромпиридин-3-ил)циклопропилкарбамат
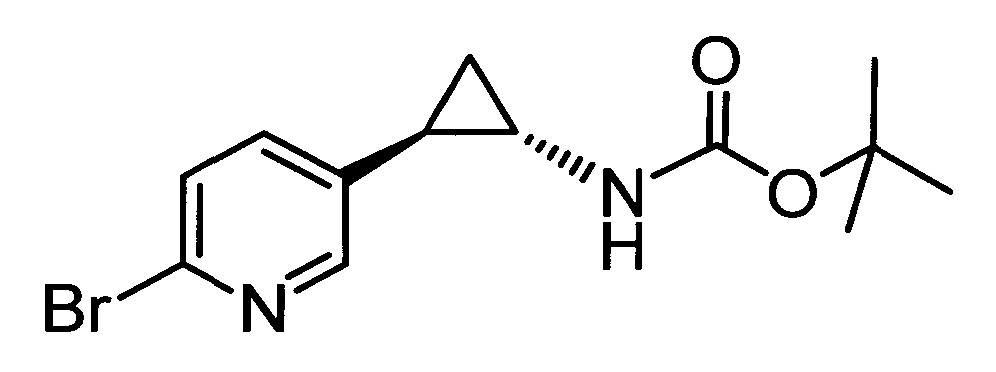
Раствор (транс)-2-(6-бромпиридин-3-ил)циклопропанкарбонилазида (промежуточный продукт F, 2,5 г, 9,36 ммоля) в трет-бутаноле (80 мл) нагревали при 90°C в течение 16 ч. После завершения реакции растворитель выпаривали в вакууме и остаток переносили в воду (100 мл) и экстрагировали с помощью EtOAc (2×100 мл). Объединенные органические экстракты промывали водой (100 мл), рассолом (100 мл) и сушили над безводным Na2SO4, фильтровали и выпаривали. Неочищенный остаток очищали с помощью колоночной флэш-хроматографии (SiO2) при элюировании смесью EtOAc: гексан (2:8) и получали трет-бутил-(транс)-2-(6-бромпиридин-3-ил)циклопропилкарбамат (1,1 г, 37,5%) в виде светло-желтого твердого вещества.1H-ЯМР (CDCl3) δ (част./млн): 1,16 (q, 1Н), 1,23 (квинтет, 1H), 1,45 (s, 9H), 2,01 (m, 1H), 2,69 (m, 1H), 4,88 (br, 1H), 7,36 (s, 2H), 8,20 (s, 1H).
Промежуточный продукт H: (E)-Этил-3-(4-бромфенил)акрилат
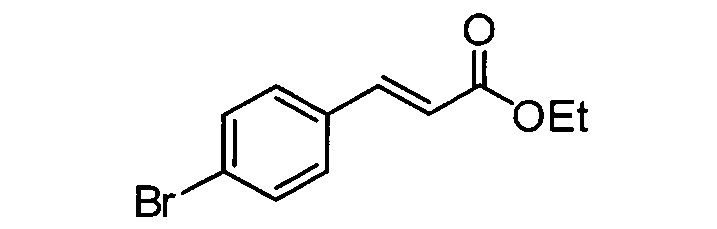
Раствор триэтилфосфоноацетата (13,1 г, 0,0589 моля) медленно добавляли (по каплям) к раствору трет-бутоксида калия (6,59 г, 0,0589 моля) в сухом ТГФ (150 мл) при -5°C, перемешивали в течение 30-45 мин при такой же температуре, затем при -5°C в течение 15 мин медленно по каплям добавляли раствор 4-бромбензальдегид (10 г, 0,054 моля), в сухом ТГФ (50 мл), реакционную смесь перемешивали в течение 30 мин при такой же температуре. После завершения реакции по данным ТСХ (тонкослойная хроматография), реакционную смесь выливали на лед с водой (300 мл), экстрагировали с помощью EtOAc (2×200 мл). Объединенные органические экстракты промывали насыщенным раствором NaHCO3 (200 мл), водой (200 мл), рассолом (200 мл) и сушили над безводным Na2SO4, фильтровали и выпаривали и получали неочищенный (E)-этил-3-(4-бромфенил)акрилат (10 г, 72%) в виде бледно-зеленой жидкости. Его использовали на следующей стадии без дополнительной очистки.
Промежуточный продукт I: (Транс)-этил-2-(4-бромфенил)циклопропанкарбоксилат

Триметилсульфоксониййодид (5,19 г, 0,0236 моля) небольшими порциями в течение 20 мин при КТ медленно добавляли к суспензии гидрида натрия (0,44 г, 0,0236 моля) в сухом ДМСО (80 мл), перемешивали в течение 1 ч до образования прозрачного раствора. Затем медленно по каплям добавляли раствор (E)-этил-3-(4-бромфенил)акрилата (промежуточный продукт H, 5 г, 0,01968) в сухом ДМСО (20 мл), перемешивали при КТ в течение 30 мин. После завершения реакции по данным ТСХ реакционную смесь выливали на лед с водой (200 мл), экстрагировали с помощью EtOAc (2×150 мл). Объединенные органические экстракты промывали водой со льдом (2×150 мл), рассолом (150 мл), сушили над безводным Na2SO4, фильтровали и выпаривали и получали (транс)-этил-2-(4-бромфенил)циклопропанкарбоксилат (4 г, 75,9%) в виде зеленой жидкости. Неочищенное вещество использовали на следующей стадии без дополнительной очистки.
Промежуточный продукт J: (Транс)-2-(4-бромфенил)циклопропанкарбоновая кислота
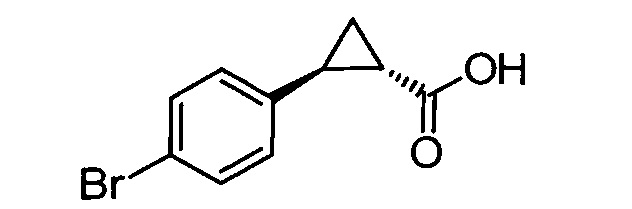
NaOH 4 н. (20 мл) добавляли к раствору (транс)-этил-2-(4-бромфенил)циклопропанкарбоксилата (промежуточный продукт I, 4 г, 0,0149 моля) в метаноле (40 мл) и перемешивали при КТ в течение 2 ч. После завершения реакции по данным ТСХ растворитель выпаривали и остаток разбавляли водой (50 мл), подкисляли 4 н. раствором HCl, образовавшееся твердое вещество отфильтровывали и сушили и получали (транс)-2-(4-бромфенил)циклопропанкарбоновую кислоту (2,59 г, 72%) в виде белого твердого вещества.
Промежуточный продукт K: (Транс)-2-(4-бромфенил)циклопропанкарбонилазид
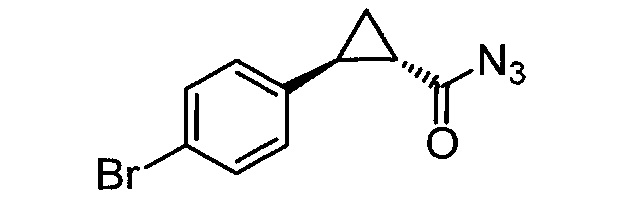
Этилхлорформиат (1,9 мл) добавляли к раствору (транс)-2-(4-бромфенил)циклопропанкарбоновой кислоты (промежуточный продукт J, 4 г, 0,0165 моля) и Et3N (2,51 мл, 0,0199 моля) в ацетоне (60 мл) при -20°C, перемешивали при такой же температуре в течение 1 ч, затем добавляли раствор NaN3 (1,3 г, 0,0199 моля) в воде (5 мл) и перемешивали в течение 30 мин при КТ. После завершения реакции по данным ТСХ растворитель выпаривали и неочищенный остаток растворяли в этилацетате (100 мл), промывали водой (40 мл), сушили над безводным Na2SO4, фильтровали и выпаривали и получали (транс)-2-(4-бромфенил)циклопропанкарбонилазид (4 г). Неочищенный остаток использовали на следующей стадии без дополнительной очистки.
Промежуточный продукт L: трет-бутил-(транс)-2-(4-бромфенил)циклопропилкарбамат
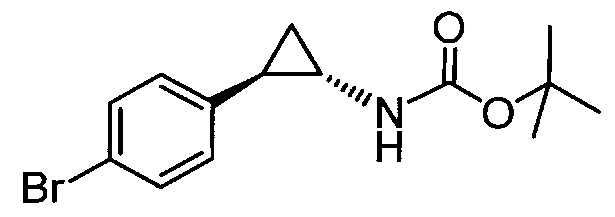
Раствор (транс)-2-(4-бромфенил)циклопропанкарбонилазида (промежуточный продукт K, 4 г) в трет-бутаноле (40 мл) нагревали при 90°C в течение 16 ч. После завершения реакции по данным ТСХ растворитель выпаривали, остаток выливали в воду (50 мл), экстрагировали с помощью EtOAc (2×50 мл). Объединенные органические экстракты промывали водой (50 мл), рассолом (50 мл), сушили над безводным Na2SO4, фильтровали и выпаривали. Неочищенный остаток очищали с помощью колоночной хроматографии (SiO2) при элюировании смесью EtOAc: петролейный эфир (2:98) и получали трет-бутил-(транс)-2-(4-бромфенил)циклопропилкарбамат (2,5 г, 48%, суммарный за 2 стадии) в виде белого твердого вещества.1H-ЯМР (CDCl3, 250 МГц) δ (част./млн): 1,07-1,19 (m, 2Н), 1,44 (s, 9Н); 2,05-1,94 (m, 1Н); 2,72-2,62 (m, 1Н); 4,85 (br, 1H); 7,09-6,96 (m, 2Н); 7,44-7,33 (m, 2Н).
Промежуточный продукт M: Этил-5-((трет-бутоксикарбонил)амино)-1,3,4-оксадиазол-2-карбоксилат

Гидрид натрия (280 мг, 0,007 моля) в ДМФ (10 мл) при 0°C добавляли к суспензии этил-5-амино-1,3,4-оксадиазол-2-карбоксилата (1 г, 0,006 моля) в ДМФ (2 мл), перемешивали в течение 10 мин, затем добавляли ди-трет-бутилдикарбонат (1,65 г, 0,0076 моля) и перемешивали при КТ в течение 16 ч. После завершения реакции реакционную смесь выливали на лед с водой (25 мл) и экстрагировали с помощью EtOAc (3×25 мл). Объединенные экстракты промывали холодной водой (2×25 мл), рассолом (25 мл), сушили над безводным Na2SO4, фильтровали и выпаривали. Неочищенный остаток очищали с помощью колоночной хроматографии (SiO2) с использованием смеси EtOAc: петролейный эфир (1:3) в качестве элюента и получали этил-5-((трет-бутоксикарбонил)амино)-1,3,4-оксадиазол-2-карбоксилат (900 мг, 56,2%) в виде белого твердого вещества.
Промежуточный продукт N: трет-Бутил-(5-(гидроксиметил)-1,3,4-оксадиазол-2-ил)карбамат

NaBH4 (330 мг, 0,0087 моля) при 0°C добавляли к раствору этил-5-((трет-бутоксикарбонил)амино)-1,3,4-оксадиазол-2-карбоксилата (промежуточный продукт M, 900 мг, 0,0035 моля) в ТГФ (18 мл) и перемешивали при КТ в течение 16 ч. После завершения реакции растворитель выпаривали и остаток переносили в воду (15 мл) и экстрагировали с помощью EtOAc (3×20 мл). Объединенные экстракты промывали водой (20 мл), рассолом (20 мл), сушили над безводным Na2SO4, фильтровали и выпаривали. Неочищенный остаток очищали с помощью колоночной хроматографии (SiO2) с использованием смеси EtOAc: петролейный эфир (8:2) в качестве элюента и получали трет-бутил-(5-(гидроксиметил)-1,3,4-оксадиазол-2-ил)карбамат (450 мг, 54,2%) в виде белого твердого вещества.
Промежуточный продукт O: трет-Бутил-(5-формил-1,3,4-оксадиазол-2-ил)карбамат
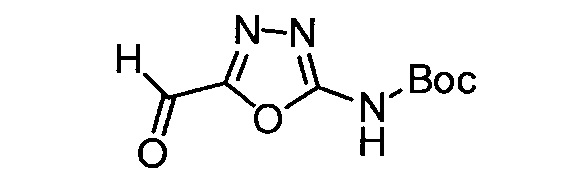
MnO2 (500 мг) при КТ добавляли к раствору трет-бутил-(5-(гидроксиметил)-1,3,4-оксадиазол-2-ил)карбамата (промежуточный продукт N, 450 мг, 0,0021 моля) в ТГФ (9 мл) и перемешивали в течение 16 ч. После завершения реакции реакционную смесь фильтровали через слой целита и фильтрат выпаривали и получали неочищенный трет-бутил-(5-формил-1,3,4-оксадиазол-2-ил)карбамат (250 мг). Это неочищенное вещество использовали на следующей стадии без дополнительной очистки.
Промежуточный продукт P: 4-(Бензилокси)бензальдегид

Карбонат калия (678 г, 4,91 моля) добавляли к раствору 4-гидроксибензальдегида (200 г, 1,63 моля) в ДМФ (2 л), затем добавляли бензилбромид (214 мл, 1,80 моля) при 0°C и перемешивали в течение 18 ч при КТ. После завершения реакции реакционную смесь выливали на лед с водой (3 л), твердое вещество отфильтровывали и сушили и получали 4-(бензилокси)бензальдегид (230 г, 66%).
Промежуточный продукт Q: (E)-Этил-3-(4-(бензилокси)фенил)акрилат

Триэтилфосфоноацетат (259 мл, 1,3 моля) по каплям медленно добавляли к раствору трет-бутоксида калия (145 г, 1,29 моля) в сухом ТГФ (2 л) при -5°C и перемешивали в течение 30-45 мин. Затем при -10°C в течение 15 мин по каплям медленно добавляли раствор 4-(бензилокси)бензальдегида (промежуточный продукт P, 230 г, 1,08 моля) в сухом ТГФ (1,5 л) и перемешивали в течение 30 мин. После завершения реакции реакционную смесь выливали на лед с водой (1 л) и экстрагировали с помощью EtOAc (2×1,5 л). Объединенные органические экстракты промывали насыщенным раствором NaHCO3 (1 л), водой (1 л), рассолом (1 л), сушили над безводным Na2SO4, фильтровали и выпаривали и получали неочищенный (E)-этил-3-(4-(бензилокси)фенил)акрилат (290 г, 95%). Неочищенное вещество использовали на следующей стадии без дополнительной очистки.
Промежуточный продукт R: (Транс)-этил-2-(4-(бензилокси)фенил)циклопропанкарбоксилат

Триметилсульфоксониййодид (224 г, 1,02 моля) порциями добавляли к суспензии NaH (40,8 г, 1,02 моля) в сухом ДМСО (2 л) при КТ в течение 20 мин и перемешивали в течение 1 ч до образования прозрачного раствора. По каплям добавляли раствор (E)-этил-3-(4-(бензилокси)фенил)акрилата (промежуточный продукт Q, 240 г, 0,85 моля) в сухом ДМСО (2 л) и перемешивали при КТ в течение 30 мин. После завершения реакции реакционную смесь выливали на лед с водой (2 л), экстрагировали с помощью EtOAc (2×1 л). Объединенные органические экстракты промывали водой со льдом (1 л), рассолом (1 л), сушили над безводным Na2SO4, фильтровали и выпаривали и получали (транс)-этил-2-(4-(бензилокси)фенил)циклопропанкарбоксилат (142 г, 58,6%) в виде почти белого твердого вещества. Неочищенное вещество использовали на следующей стадии без дополнительной очистки.
Промежуточный продукт S: (Транс)-2-(4-(бензилокси)фенил)циклопропанкарбоновая кислота

4 н. Раствор NaOH (4 л) при 0°C добавляли к раствору (транс)-этил-2-(4-(бензилокси)фенил)циклопропанкарбоксилата (промежуточный продукт R, 250 г, 0,844 моля) в метаноле (1,2 л) и перемешивали при КТ в течение 4 ч. После завершения реакции растворитель выпаривали, остаток разбавляли водой (1 л), подкисляли 4 н. раствором HCl, экстрагировали с помощью EtOAc (2×2 л). Объединенные органические экстракты промывали водой (1 л), рассолом (1 л), сушили над безводным Na2SO4, фильтровали и выпаривали и получали (транс)-2-(4-(бензилокси)фенил)циклопропанкарбоновую кислоту (190 г, 84%) в виде почти белого твердого вещества. Неочищенное вещество использовали на следующей стадии без дополнительной очистки.
Промежуточный продукт T: (Транс)-2-(4-(бензилокси)фенил)циклопропанкарбонилазид
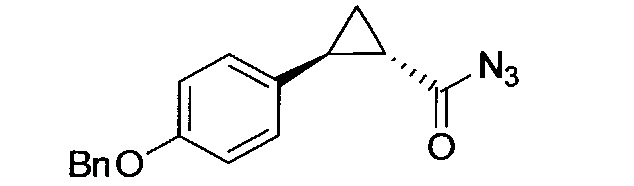
Этилхлорформиат (143 мл, 1,48 моля) при -20°C добавляли к раствору (транс)-2-(4-(бензилокси)фенил)циклопропанкарбоновой кислоты (промежуточный продукт S, 190 г, 0,70 моля), триэтиламина (229 мл, 1,63 моля) в ацетоне (2,8 л) и перемешивали в течение 1 ч, затем добавляли раствор NaN3 (138 г, 2,1 моля) в воде (200 мл) и перемешивали при КТ в течение 30 мин. После завершения реакции растворитель выпаривали, остаток растворяли в EtOAc (2 л), промывали водой (2 л), рассолом (1 л), сушили над безводным Na2SO4, фильтровали и выпаривали и получали (транс)-2-(4-(бензилокси)фенил)циклопропанкарбонилазид (178 г, 85,9%).
Промежуточный продукт U: Трет-бутил-((транс)-2-(4-(бензилокси)фенил)циклопропил)карбамат

Раствор (транс)-2-(4-(бензилокси)фенил)циклопропанкарбонилазида (промежуточный продукт T, 178 г, 0,64 моля) в трет-бутаноле (2,6 л) нагревали при 90°C в течение 16 ч. После завершения реакции растворитель выпаривали и неочищенный остаток очищали с помощью колоночной хроматографии с использованием (SiO2) смеси EtOAc: петролейный эфир (4:96) и получали трет-бутил-((транс)-2-(4-(бензилокси)фенил)циклопропил)карбамат (78 г, 37,8%) в виде почти белого твердого вещества.
Промежуточный продукт V: (Транс)-2-(4-(бензилокси)фенил)циклопропанамингидрохлорид
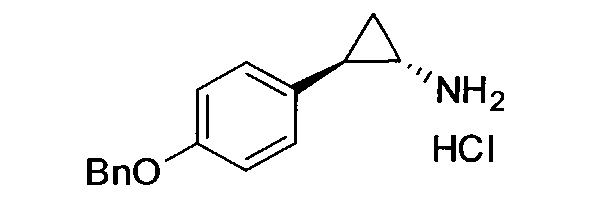
HCl в диоксане (390 мл) при 0°C добавляли к раствору трет-бутил-((транс)-2-(4-(бензилокси)фенил)циклопропил)карбамата (промежуточный продукт U, 78 г, 0,23 моля) в 1,4-диоксане (780 мл) и перемешивали при КТ в течение 12 ч. После завершения реакции растворитель выпаривали и остаток растирали с диэтиловым эфиром (1 л) затем с гексаном (1 л) и получали (транс)-2-(4-(бензилокси)фенил)циклопропанамингидрохлорид (55 г, 87%) в виде почти белого твердого вещества.
Промежуточный продукт W: Этил-2-амино-2-тиооксоацетат

P2S5 (28,5 г, 128 ммоля) в течение 30 мин порциями добавляли к раствору этил-2-амино-2-оксоацетата (30 г, 25,6 ммоля) в пиридине (300 мл) и перемешивали при 90°C в течение 3 ч. После завершения реакции растворитель выпаривали, остаток разбавляли водой (300 мл) и экстрагировали с помощью EtOAc (2×300 мл). Объединенные экстракты промывали водой (2×200 мл), рассолом (200 мл) и сушили над безводным Na2SO4, фильтровали и выпаривали. Неочищенное вещество очищали с помощью колоночной хроматографии (SiO2) при элюировании смесью (1:9) EtOAc: гексан и получали этил-2-амино-2-тиооксоацетат (18 г, 52,9%) в виде белого твердого вещества.
Промежуточный продукт X: 2-(Этоксикарбонил)тиазол-5-карбоновая кислота

Бромпировиноградную кислоту (22,7 г, 135,33 ммоля) добавляли к раствору этил-2-амино-2-тиооксоацетата (промежуточный продукт W, 18 г, 135,3 ммоля) в диоксане (200 мл) и кипятили с обратным холодильником в течение 5 ч. После завершения реакционную смесь выливали в воду (200 мл), остаток подщелачивали насыщенным раствором NaHCO3 и экстрагировали с помощью EtOAc (2×250 мл). Водный слой подкисляли с помощью 2 н. HCl и экстрагировали с помощью EtOAc (2×250 мл). Объединенные экстракты промывали водой (250 мл), рассолом (250 мл), сушили над безводным Na2SO4, фильтровали и выпаривали и получали 2-(этоксикарбонил)тиазол-5-карбоновую кислоту (13 г неочищенного вещества). Неочищенное вещество использовали на следующей стадии без дополнительной очистки.
Промежуточный продукт Y: Этил-5-(азидокарбонил)тиазол-2-карбоксилат

Этилхлорформиат (9,8 г, 83,6 ммоля) при -20°C добавляли к раствору 2-(этоксикарбонил)тиазол-5-карбоновой кислоты (промежуточный продукт X, 13 г, 64,67 ммоля), ТЭА (триэтиламин) (9,79 г, 97,01 ммоля) в ацетоне (130 мл), перемешивали в течение 1 ч, затем добавляли раствор NaN3 (5,4 г, 83,6 ммоля) в воде (15 мл) и перемешивали при КТ в течение 30 мин. После завершения реакции растворитель выпаривали, неочищенный остаток разбавляли водой (150 мл) и экстрагировали с помощью EtOAc (2×150 мл). Объединенные экстракты промывали водой (100 мл), рассолом (100 мл), сушили над безводным Na2SO4, фильтровали и выпаривали и получали Этил-5-(азидокарбонил)тиазол-2-карбоксилат (11 г неочищенного вещества) в виде коричневой жидкости. Неочищенное вещество использовали на следующей стадии без дополнительной очистки.
Промежуточный продукт Z: Этил-5-((трет-бутоксикарбонил)амино)тиазол-2-карбоксилат

Раствор этил-5-(азидокарбонил)тиазол-2-карбоксилата (промежуточный продукт Y, 11 г, 48,6 ммоля) в трет-бутаноле (150 мл) кипятили с обратным холодильником при 90°C в течение 16 ч. После завершения реакции растворитель выпаривали. Неочищенный остаток очищали с помощью колоночной хроматографии с использованием (SiO2), при элюировании смесью EtOAc: петролейный эфир (2:98) и получали этил-5-((трет-бутоксикарбонил)амино)тиазол-2-карбоксилат (4 г, 30,23%) в виде белого твердого вещества.
Промежуточный продукт AA: Трет-бутил-(2-(гидроксиметил)тиазол-5-ил)карбамат
NaBH4 (1,1 г, 29,2 ммоля) при 0°C порциями добавляли к раствору этил-5-(трет-бутоксикарбониламино)тиазол-2-карбоксилата (промежуточный продукт Z, 4 г, 14,6 ммоля) в МеОН (40 мл) в течение 30 мин и перемешивали при КТ в течение 16 ч. После завершения реакции растворитель выпаривали, твердый остаток растворяли в воде со льдом (50 мл) и экстрагировали с помощью EtOAc (2×50 мл). Объединенные экстракты промывали водой (50 мл), рассолом (50 мл), сушили над безводным Na2SO4, фильтровали и выпаривали. Неочищенное вещество очищали с помощью колоночной хроматографии с использованием SiO2, при элюировании смесью EtOAc: петролейный эфир (2:8) и получали трет-бутил-(2-(гидроксиметил)тиазол-5-ил)карбамат (2,9 г, 84,8%) в виде белого твердого вещества.
Промежуточный продукт AB: трет-Бутил-(2-формилтиазол-5-ил)карбамат
MnO2 (1,5 г, 18,2 ммоля) добавляли к раствору трет-бутил-(2-(гидроксиметил)тиазол-5-ил)карбамата (промежуточный продукт AA, 700 мг, 3,04 ммоля) в ДХМ (дихлорметан) (15 мл) и перемешивали при КТ в течение 16 ч. После завершения реакционную смесь разбавляли с помощью ДХМ, фильтровали через целит. Фильтрат концентрировали в вакууме и получали трет-бутил-(2-формилтиазол-5-ил)карбамат (500 мг неочищенного вещества). Неочищенное вещество использовали на следующей стадии без дополнительной очистки.
Соединения, описанные в примерах 1-24, являются рацемическими, т.е. представляют собой 50:50 смесь энантиомеров, соответствующую трансрацемату.
Пример 1: 5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)пиримидин-2-амин

Триацетоксиборогидрид натрия (883 мг, 4,166 ммоля) при 0°C медленно добавляли к раствору (транс)-2-(4-(бензилокси)фенил)циклопропанамина (промежуточный продукт B, 500 мг, 2,083 ммоля), 2-аминопиримидин-5-карбальдегида (256 мг, 2,083 ммоля) в ДХЭ (дихлорэтан) (10 мл) и перемешивали в течение 20 ч. После завершения реакции растворитель выпаривали. Остаток растворяли в метаноле (15 мл), при 0°C медленно добавляли NaBH4 (237 мг, 6,249 ммоля) и перемешивали в течение 3 ч. После завершения реакции растворитель выпаривали, остаток растворяли в воде со льдом (20 мл) и экстрагировали с помощью EtOAc (2×20 мл). Объединенные органические слои промывали рассолом (20 мл) и сушили над безводным Na2SO4, фильтровали и выпаривали. Неочищенный остаток очищали с помощью препаративной ВЭЖХ и получали 5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)пиримидин-2-амин (180 мг, 25%) в виде белого твердого вещества, 1H-ЯМР (400 МГц, ДМСО-d6) δ (част./млн): 0,85 (q, 1H), 0,90 (квинтет, 1Н), 1,73 (m, 1Н), 2,07 (m, 1H), 2,75 (brs, 1H), 3,53 (s, 2Н), 5,04 (s, 2Н), 6,46 (s, 2Н), 6,85 (d, 2Н), 6,92 (d, 2Н), 7,33 (m, 1H), 7,42 (m, 4Н), 8,11 (s, 2Н). Масса (M+H): 347,3
Соединение следующего примера синтезировали по методике, описанной в примере 1, и с использованием соответствующих исходных веществ.
Пример 2: 5-(((Транс)-2-4-(бензилокси)фенил)циклопропиламино)тиазол-2-амингидрохлорид

1H-ЯМР (400 МГц, ДМСО-d6) δ (част./млн): 1,22 (q, 1H), 1,48 (квинтет, 1Н), 2,46 (m, 1Н), 2,80 (br, 1Н), 4,35 (s, 2Н), 5,08 (s, 2Н), 6,93 (d, 2Н), 7,06 (d, 2Н), 7,32 (m, 2Н), 7,40 (m, 4Н), 8,98 (br, 1Н), 9,90 (br, 2Н). Масса (M+H): 351,9
Указанные ниже соединения можно синтезировать по методикам, описанным на схеме 1 и 2, или по другим путям синтеза, известным специалисту с общей подготовкой в данной области техники.
Пример 3: 5-(((Транс)-2-(6-(3-(трифторметил)фенил)пиридин-3-ил)циклопропиламино)метил)пиримидин-2-амин

Пример 4: 5-(((Транс)-2-(6-(3-(трифторметил)фенил)пиридин-3-ил)циклопропиламино)метил)тиазол-2-амин

Пример 5: 3-(5-((Транс)-2-((2-аминопиримидин-5-ил)метиламино)циклопропил)пиридин-2-ил)фенол

Пример 6: 3-(5-((Транс)-2-((2-аминотиазол-5-ил)метиламино)циклопропил)пиридин-2-ил)фенол

Пример 7: 4'-((Транс)-2-((2-аминопиримидин-5-ил)метиламино)циклопропил)бифенил-3-ол

Пример 8: 4'-((Транс)-2-((2-аминотиазол-5-ил)метиламино)циклопропил)бифенил-3-ол

Пример 9: 5-((Транс)-2-(4-бензилокси)фенил)циклопропиламино)метил)-1,2,4-оксадиазол-3-амин
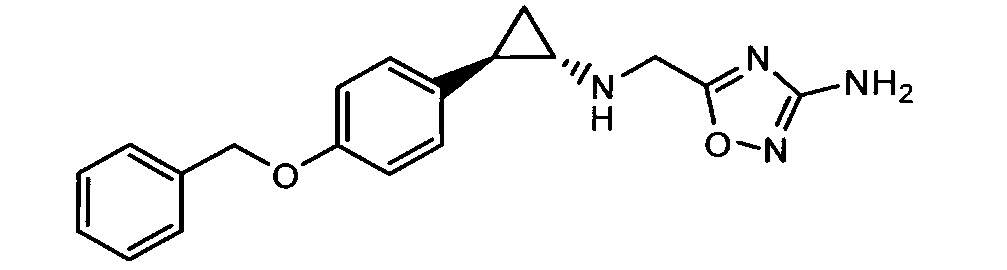
Пример 10: 5-(((Транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-амин

Это соединение можно синтезировать по схеме 1 или схеме 3, или по другим путям синтеза, известным специалисту с общей подготовкой в данной области техники.
Схема 1, методика
Стадия 1:
трет-Бутил-(5-формил-1,3,4-оксадиазол-2-ил)карбамат (промежуточный продукт O, 220 мг, 1,041 ммоля) и триацетоксиборогидрид натрия (441 мг, 2,08 ммоля) при 0°C добавляли к раствору транс-2-[4-(бензилокси)фенил]циклопропанамина (промежуточный продукт B, 250 мг, 1,041 ммоля) в сухом дихлорэтане (2,5 мл) и перемешивали при КТ в течение 24 ч, затем растворитель выпаривали. Остаток переносили в MeOH (2,5 мл) и при 0°C добавляли NaBH4 (116 мг, 3,138 ммоля) и перемешивали в течение 2 ч при КТ. После завершения реакции растворитель выпаривали, остаток переносили в воду (10 мл) и экстрагировали с помощью EtOAc (4×10 мл). Объединенные экстракты промывали водой (10 мл), рассолом (10 мл), сушили над безводным Na2SO4, фильтровали и выпаривали. Остаток очищали с помощью колоночной хроматографии (SiO2) с использованием MeOH:CHCl3 (1:99) и получали трет-бутил-(5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-ил)карбамат (70 мг, 15,3%) в виде бледно-зеленой жидкости.
Стадия 2:
HCl в 1,4-диоксане (1 мл) добавляли к раствору трет-бутил-(5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-ил)карбамат (100 мг) в 1,4-диоксане (1 мл) при 0°C и перемешивали в течение 18 ч. После завершения реакции растворитель выпаривали и остаток растворяли в воде (10 мл), подщелачивали раствором Na2CO3, экстрагировали с помощью EtOAc (3×5 мл). Объединенные экстракты промывали водой (5 мл), рассолом (5 мл), сушили над безводным Na2SO4, фильтровали и выпаривали. Неочищенный остаток очищали с помощью колоночной хроматографии с использованием смеси МеОН:CHCl3 (5:95) в качестве элюента и получали 5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-амин (40 мг, 52%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, ДМСО-d6) δ (част./млн): 0,85 (m, 2Н), 1,72 (m, 1Н), 2,2 (m, 1H), 3,0 (m, 1H), 3,75 (s, 2Н), 5,08 (s, 2Н), 6,8-7,0 (m, 6Н), 7,4 (m, 5Н); Масса (M+H): 337,1
Схема 3, методика
Это соединение можно синтезировать по такой же методике, как описанная на схеме 1, но на стадии 1 используют промежуточный продукт V вместо промежуточного продукта B.
Указанные ниже соединения можно синтезировать по методике, описанной в примере 10 с использованием методики, представленной на схеме 3, и соответствующих имеющихся в продаже алкилгалогенидов и получить подходящие производные промежуточного продукта P.
Пример 11: 5-((((Транс)-2-(4-((4-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин

1H ЯМР (400 МГц, ДМСО d6) δ (част./млн): 0,84 (m, 2Н), 1,79 (m, 1Н), 2,20 (m, 1H), 3,12 (m, 1H), 3,78 (s, 1Н), 5,02 (s, 2Н), 6,85-7,00 (m, 6Н), 7,2 (t, 2Н), 7,46 (t, 2Н); Масса (M-H): 353,3
Пример 12: 5-((((Транс)-2-(4-((3-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин

1H ЯМР (400 МГц, ДМСО d6) δ (част./млн): 0,85 (m, 2Н), 1,73 (m, 1Н), 2,19 (m, 1Н), 3,00 (m, 1Н), 3,75 (s, 2Н), 5,07 (s, 2Н), 6,85-7,00 (m, 6Н), 7,14 (t, 1Н), 7,25 (t, 2Н), 7,41 (m, 1Н); Масса (M-H): 353,3
Пример 13: 5-((((Транс)-2-(4-((3,5-дифторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин

1H ЯМР (400 МГц, ДМСО d6) δ (част./млн): 0,87 (m, 2Н), 1,75 (m, 1H), 2,20 (m, 1Н), 3,04 (m, 1Н), 3,75 (s, 2Н), 5,13 (s, 2Н), 6,80-7,05 (m, 6Н), 7,16 (m, 3H); Масса (M+H): 373,0
Пример 14: 5-((((Транс)-2-(4-((4-хлорбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин

1H ЯМР (400 МГц, ДМСО d6) δ (част./млн): 0,86 (m, 2Н), 1,73 (m, 1Н), 2,18 (m, 1Н), 2,98 (m, 1Н), 3,75 (s, 2Н), 5,05 (s, 2Н), 6,82-6,95 (m, 6Н), 7,44 (m, 4Н); Масса (M-H): 369,0
Пример 15: 5-((((Транс)-2-(4-((3-хлорбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин

1H ЯМР (400 МГц, ДМСО d6) δ (част./млн): 0,85 (m, 2Н), 1,73 (m, 1Н), 2,19 (m, 1Н), 3,00 (m, 1Н), 3,75 (s, 2Н), 5,07 (s, 2Н), 6,84-7,02 (m, 6Н), 7,39-7,54 (m, 4Н); Масса (M+H): 371,0
Указанные ниже соединения можно синтезировать по методикам, описанным на схеме 1, 2 и 3. Альтернативно, как это известно специалистам в данной области техники, указанные ниже соединения также можно получить из производных (транс)-циклопропанамина формулы (3) и (24) соответственно по хорошо известным реакциям (т.е. образования гетероцикла или циклизации)
Пример 16: 5-((((Транс)-2-(4-((2-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин

Пример 17: 5-((((Транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-N-метил-1,3,4-оксадиазол-2-амин

Пример 18: N-(5-((((Транс)-2-(4-бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-ил)ацетамид

Пример 19: 4'-((Транс)-2-(((5-амино-1,3,4-оксадиазол-2-ил)метил)амино)циклопропил)-[1,1'-бифенил]-3-ол

Пример 20: 5-((((Транс)-2-(6-(3-(трифторметил)фенил)пиридин-3-ил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин

Пример 21: 5-((((Транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-тиадиазол-2-амин
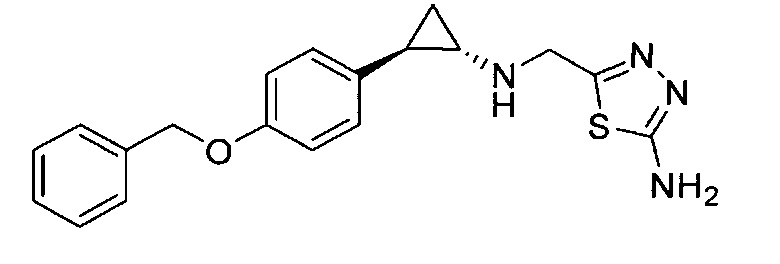
Пример 22: 2-((((Транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)тиазол-5-амин

Пример 23: 4-((((Транс)-2-(3'-(трифторметил)-[1,1'-бифенил]-4-ил)циклопропил)амино)метил)тиазол-2-амин
Пример 24: 2-((((Транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)оксазол-5-амин

Пример 25: 3-((((Транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)изоксазол-5-амин
Пример 26: 5-((((Транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-N,N-диметил-1,3,4-оксадиазол-2-амин
Пример 27: 3-((((Транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,2,4-оксадиазол-5-амин

Пример 28: 5-((((Транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,2,4-тиадиазол-3-амин

Пример 29: 5-((((Транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)пиридин-2-амин

Пример 30: 6-((((Транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)пиридазин-3-амин

Пример 31: 5-((((Транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)пиразин-2-амин

Пример 32: 2-((((Транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-пиримидин-5-амин

Пример 33: 6-((((Транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,2,4-триазин-3-амин

Пример 34: 3-((((Транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,2,4-триазин-6-амин

Пример 35: Получение энантиомерно обогащенных или оптически активных соединений
5-(((Транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-амин можно синтезировать по методике, описанной в примере 10. Альтернативно, энантиомерно обогащенные или чистые промежуточные продукты можно получить и затем использовать в последующих реакциях для синтеза соответствующего (-)- или (+)-энантиомера, например, 5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-амина.
Стадия 1
R-(-)-Миндальную кислоту (22,2 г, 0,14 моля) добавляли к раствору (транс)-2-(4-(бензилокси)фенил)циклопропанамингидрохлорида (промежуточный продукт V) (35 г, 0,14 моля) в смеси ТГФ и H2O (6:4) (650 мл) и кипятили с обратным холодильником в течение 1 ч. После образования прозрачного раствора реакционную смесь охлаждали до КТ. Образовавшееся осажденное твердое вещество отфильтровывали, подщелачивали насыщенным раствором NaHCO3 и экстрагировали этилацетатом (3×500 мл). Объединенные органические экстракты промывали водой (500 мл), рассолом (500 мл), сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме и получали транс-2-(4-(бензилокси)фенил)циклопропанамин (энантиомер-(-)) (14 г, 46,6%) в виде почти белого твердого вещества.
Стадия 2
трет-Бутил-(5-(хлорметил)-1,3,4-оксадиазол-2-ил)карбамат (141 мг, 0,606 ммоля) добавляли к раствору 2-(4-(бензилокси)фенил)циклопропанамина (энантиомер-(-)) (145 мг, 0,606 ммоля) и K2CO3 (166 мг, 1,213 ммоля) в сухом ДМФ (1,5 мл) и перемешивали при КТ в течение 2 ч. После завершения реакции реакционную смесь выливали на лед с водой (10 мл) и экстрагировали с помощью EtOAc (4×10 мл). Объединенные органические экстракты промывали водой (3×10 мл), рассолом (10 мл), сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии (SiO2) с использованием смеси MeOH:CHCl3 (1:99) в качестве элюента и получали трет-бутил-(5-(((2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-ил)карбамат (энантиомер-(-)) (100 мг, 37,7%) в виде бледно-зеленой жидкости.
Стадия 3
К раствору (-)-трет-бутил-5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-илкарбамата (100 мг, 0,229 ммоля) в 1,4-диоксане (1 мл) при 0°C добавляли HCl в 1,4-диоксане (1 мл) и перемешивали в течение 18 ч. После завершения реакции растворитель выпаривали и остаток растворяли в воде (10 мл), подщелачивали раствором Na2CO3, экстрагировали с помощью EtOAc (3×5 мл). Объединенные экстракты промывали водой (5 мл), рассолом (5 мл), сушили над безводным Na2SO4, фильтровали и выпаривали. Неочищенный остаток очищали с помощью колоночной хроматографии с использованием МеОН:CHCl3 (5:95) в качестве элюента и получали (-)-5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-амин (40 мг, 52%) в виде белого твердого вещества.
1H ЯМР (400 МГц, ДМСО d6) δ: 7,46 (t, 2Н), 7,2 (t, 2Н), 6,98 (q, 6Н), 5,0 (s, 2Н), 3,78 (s, 1H), 3,1 (brs, 1H), 2,2 (brs, 1H), 1,79 (brs, 1H), 0,92 (m, 2H)
Масса (M+H): 337,1
Чистота по данным ВЭЖХ (высокоэффективная жидкостная хроматография): 96,02%
Хиральная чистота по данным ВЭЖХ: 95,12%
Удельное оптическое вращение (c=0,5% в ДМСО): -37,76°
Соответствующий энантиомер-(+)- можно синтезировать по такой же методике, но с использованием S-(+)-миндальной кислоты на стадии 1.
1H-ЯМР (400 МГц, ДМСО d6) δ: 7,46 (t, 2Н), 7,2 (t, 2Н), 6,98 (q, 6Н), 5,0 (s, 2Н), 3,78 (s, 1H), 3,1 (brs, 1H), 2,2 (brs, 1H), 1,79 (brs, 1H), 0,92 (m, 2H);
Масса (M+H): 337,1
Чистота по данным ВЭЖХ: 98,16%
Хиральная чистота по данным ВЭЖХ: 98,34%
Удельное оптическое вращение (C=0,5% в ДМСО): +37,76°
Соли для хиральной перекристаллизации включают (S)-(+)-миндальную кислоту,
D-(-)-винную кислоту,
L-(-)-ди-п-толуоилвинную кислоту, или
R-(-)-миндальную кислоту.
Указанные ниже соединения можно получить в соответствии с описанием синтеза, приведенным в настоящем изобретении, и опытом специалиста с общей подготовкой в данной области техники, где абсолютная конфигурация также указана в изображенной структуре:
5-((((Транс)-2-(4-((3-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин

5-((((Транс)-2-(4-((3-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин

5-((((Транс)-2-(4-((2-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин
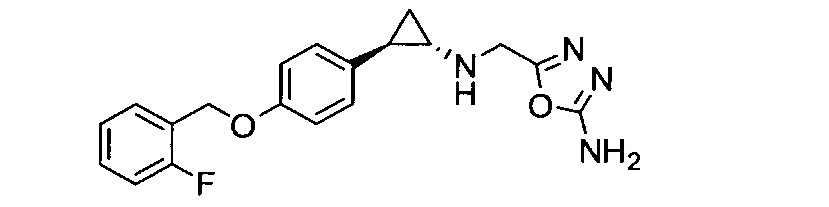
5-((((Транс)-2-(4-((2-фторбензил)окси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин

5-((((Транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-N-метил-1,3,4-оксадиазол-2-амин

5-((((Транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-N-метил-1,3,4-оксадиазол-2-амин

N-(5-((((Транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-ил)ацетамид

N-(5-((((Транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-ил)ацетамид

5-((((Транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)пиримидин-2-амин

5-((((Транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)пиримидин-2-амин
5-((((Транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-тиадиазол-2-амин
5-((((Транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-тиадиазол-2-амин
5-((((Транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-N,N-диметил-1,3,4-оксадиазол-2-амин

5-((((Транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-N,N-диметил-1,3,4-оксадиазол-2-амин

Пример 36: Выделение отдельных энантиомеров (транс)-рацемических N-замещенных арил- или гетероарилциклопропиламинов
Хиральная ВЭЖХ: Условия для проведения хирального разделения соединений или промежуточных продуктов, предлагаемых в настоящем изобретении, могут быть аналогичны следующим:
Разделение с помощью хиральной препаративной ВЭЖХ: При каждом инжектировании вводят, например, примерно 15 мг N-замещенного арил- или гетероарил-транс-циклопропиламина, растворенного в смеси EtOH, н-пентана и ГФИПС (1,1,1,3,3,3-гексафтор-2-пропанол). Оптически активный N-замещенный арил- или гетероарил-транс-циклопропиламин (например (-)-5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-амин) можно разделить, например, на ChiralPak-IA (250×20 мм внутренний диаметр) 5 мкм при температуре окружающей среды при элюировании с помощью 0,1% ДЭА (диэтаноламин) в смеси 70/30 гексан/EtOH со скоростью, равной 18 мл/мин. Растворы после этого хирального разделения можно сконцентрировать в вакууме (15 фунт-сила/дюйм2, 35°C) и получить разделенные энантиомеры.
Аналитическое определение энантиомерного избытка (ЭИ): ChiralPak IA 250×4,6 мм внутренний диаметр, 5 мкм, 0,1% ДЭА в смеси 80/20 гексан/EtOH при скорости, равной 1 мл/мин, при температуре окружающей среды, по данным УФ-анализа при 230 нм. Энантиомеры элюируются при 11,35 и 16,51 мин, каждый с энантиомерным избытком >90%.
Аналитическая чистота: Acquity UPLC BEH C18 100×2,1 мм внутренний диаметр, 1,7 мкм, 0,025% ТФК (трифторуксусная кислота) в градиентном режиме H2O:АЦН (Т/% B, 0/30, 4/80, 6/80, 6,1/30) при скорости, равной 0,4 мл/мин, при температуре окружающей среды, по данным УФ-анализа при 229 нм. Элюируются при 1,64 мин, каждый с чистотой >95,0%. Если не ограничиваться теоретическими соображениями, то можно полагать, что смеси, например, рацематов соответствующие соединению формулы (I), (Ia), (Ib), (II) или (III), можно разделить на отдельные энантиомеры или энантиомер, в основном не содержащий другого энантиомера. Таким образом, специалист в данной области техники с учетом раскрытия, описанного в настоящем изобретении, может выделить или очистить энантиомеров из рацематов или смеси энантиомеров с учетом раскрытия, приведенного в настоящем изобретении, с использованием стандартных для органической химии методик разделения энантиомеров.
Энантиомер 1, (-)-оптический стереоизомер 5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-амина, охарактеризован следующим образом:1H ЯМР (400 МГц, ДМСО d6) δ: 7,46 (t, 2Н), 7,2 (t, 2Н), 6,98 (q, 6Н), 5,0 (s, 2Н), 3,78 (s, 1Н), 3,1 (brs, 1H), 2,2 (brs, 1H), 1,79 (brs, 1H), 0,92 (m, 2H); Масса (M+H): 337,1; чистота по данным ВЭЖХ: 96,02%; Хиральная чистота по данным ВЭЖХ: 95,18%; Удельное оптическое вращение (c=0,5% в ДМСО): -37,76°.
Энантиомер 2, (+)-оптический стереоизомер 5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-амина охарактеризован следующим образом:1H ЯМР (400 МГц, ДМСО d6) δ: 7,46 (t, 2Н), 7,2 (t, 2Н), 6,98 (q, 6Н), 5,0 (s, 2Н), 3,78 (s, 1Н), 3,1 (brs, 1H), 2,2 (brs, 1H), 1,79 (brs, 1H), 0,92 (m, 2H); Масса (M+H): 337,1; чистота по данным ВЭЖХ: 98,16%; Хиральная чистота по данным ВЭЖХ: 98,34%; Удельное оптическое вращение (c=0,5% в ДМСО): +37,76°.
Эксперимент по определению оптической активности проводили с помощью поляриметра Jasco-P-1030 при температуре, равной примерно 26,9 и 27,2°C и указанной концентрации соединения (0,5%) и в выбранном растворителе, например ДМСО.
Пример 37: Определение кинетических параметров для оптически активных соединений, предлагаемых в настоящем изобретении,
Кинетические параметры для ингибирования LSD1 деметилазы получали по методике реакции связанной пероксидазы. В этом исследовании реакцию деметилазы инициировали путем одновременного смешивания белка LSD1 (31 нМ или 10 нМ) (BPS), пептида 31,25 мкМ H3-K4me2 (Millipore), увеличивающихся концентраций исследуемого соединения (например, оптически активного стереоизомера 5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-амина или оптически активного N-замещенного арил- или гетероарилциклопропиламина) и 50 мкМ Amplex® Red и 0,1 Ед/мл пероксидазы хрена (HPR) (Invitrogen) в буфере, содержащем 50 мМ натрийфосфатного буфера pH=7,4. Конечная концентрация ДМСО равнялась 0,7% и при исследовании была одинаковой во всех лунках.
За превращением реагента Amplex® Red в резоруфин вследствие генерации H2O2 непрерывно следили по флуоресценции (возбуждение при 540 нм, испускание при 590 нм) с использованием считывающего устройства для микропланшетов (Infinite 200, Tecan). 1 мкМ Раствор H2O2 использовали для калибровки сигнала флуоресценции и температуру постоянно поддерживали равной 25°C.
Кинетические параметры получали по методике, описанной в публикации Szewczuk et al. ((2007) Biochemistry, 46, 6892-6902).
Вкратце, методика заключалась в следующем: зависимости, полученные в присутствии исследуемого соединения, аппроксимировали для получения наблюдаемых значений kobs (k) с помощью следующего уравнения:

Затем значения kobs использовали для получения кинетических констант с помощью следующих уравнений (анализ Kitz и Wilson):

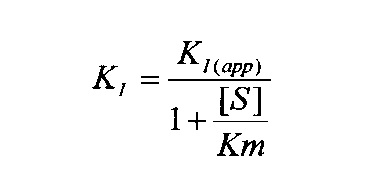
где Km=24 мкМ.
Определение кинетических констант для ингибирования обоих MAO проводили по той же методике, что и для LSD1, со следующими изменениями:
Для MAO-A концентрацию белка поддерживали равной 1,8 нг/мкл (Sigma M7316) и в качестве субстрата использовали 64 мкМ кинурамина (Sigma). В этом случае Km равнялось 64 мкМ.
Для MAO-B концентрацию белка поддерживали равной от 1,8 до 3,6 нг/мкл (Sigma М7441) и в качестве субстрата использовали 50 мкМ кинурамина (Sigma). В этом случае Km равнялось 32 мкМ.
Для обоих исследований МАО конечная концентрация ДМСО равнялась 0,54%.
Селегингидрохлорид и разагилинмезилат получали от фирм Sigma-Aldrich и Carbone Scientific Co. Ltd соответственно.
Эти исследования использовали для расчета значений, приведенных в таблице 1.
Результаты, описанные в настоящем изобретении, показывают, что (-)-стереоизомер 5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-амина является активным и высокоселективным ингибитором LSD1 и MAOB. Селективность (-)-стереоизомера 5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-амина по отношению к LSD1, MAO-A и MAO-B, характеризующаяся индексом селективности kinact/KI, показывает, что соединение высокоселективно по отношению к LSD1 и MAO-B. В частности, индекс селективности (-)-стереоизомера 5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-амина по отношению к MAO-B/MAO-A равен примерно 2253 и, таким образом, он лучше, чем соответствующие значения для розагилина и селегилина, которые равны 120 и <325 соответственно. Индекс селективности (+)-стереоизомера 5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-амина по отношению к MAO-B/MAO-A равен примерно 192. Кроме того, другие необратимые ингибиторы моноаминоксидазы, такие как разагилин и селегилин, по данным этих исследований неактивны по отношению к LSD1. Примечательно, что отношение kinact/KI для LSD1/MAO-A было для (-)-стереоизомера более, чем в 100 раз больше, чем для (+)-стереоизомера 5-(((транс)-2-(4-(бензилокси)фенил)циклопропиламино)метил)-1,3,4-оксадиазол-2-амина. Таким образом, согласно изобретению неожиданно было установлено, что оптически активные N-замещенные арил- или гетероарил-транс-циклопропиламины, включая соединения формулы (I), в которой заместители циклопропильного фрагмента находятся в транс-ориентации, а также соединения формулы (II) или (III), обладают неожиданной селективностью ингибировании LSD1 и ингибировании LSD1 и MAO-B.
Пример 38: Биологические исследования
Можно исследовать способность соединений, предлагаемых в настоящем изобретении, ингибировать LSD1. Способность соединений, предлагаемых в настоящем изобретении, ингибировать LSD1 можно изучить следующим образом. Рекомбинантный белок LSD1 человека приобретали у фирмы BPS Bioscience Inc. Для изучения ферментативной активности LSD1 и/или степени его ингибирования рассматриваемым ингибитором (ингибиторами), предлагаемым в настоящем изобретении, в качестве субстрата выбран диметилированный пептид H3-K4 (Millipore). Активность деметилазы оценивали при анаэробных условиях путем определения количества H2O2, образовавшегося при каталитической реакции с использованием набора для исследования Amplex® Red пероксид/пероксидаза (Invitrogen, Carlsbad, СА).
Вкратце, методика заключалась в следующем: определенное количество LSD1 инкубировали на льду в течение 15 мин при отсутствии и/или в присутствии ингибитора в разных концентрациях (например, от 0 до 75 мкМ, в зависимости от активности ингибитора). В качестве контроля для ингибирования использовали транилципромин (Biomol International). При проведении эксперимента каждую концентрацию ингибитора исследовали трижды. После введения фермента в реакцию с ингибитором в каждую реакционную смесь добавляли 12,5 мкМ диметилированного пептида H3-K4 и реакционную смесь выдерживали в течение 1 ч при 37°C в темноте. Реакции ферментов проводили в буфере, содержащем 50 мМ фосфат натрия, pH 7,4. После завершения инкубации к реакционной смеси добавляли реагент Amplex® Red и раствор пероксидазы хрена (ПРХ) в соответствии с инструкциями поставщика (Invitrogen) и инкубацию проводили в течение еще 30 мин при комнатной температуре в темноте. Для проверки эффективности набора использовали 1 мкМ раствор H2O2. За превращением реагента Amplex® Red в резоруфин вследствие наличия H2O2 в реакционной смеси следили по флуоресценции (возбуждение при 540 нм, испускание при 590 нм) с использованием считывающего устройства для микропланшетов (Infinite 200, Tecan). Для определения содержания H2O2, образовавшегося при отсутствии и/или в присутствии ингибитора использовали произвольные единицы.
Максимальная активность деметилазы по отношению к LSD1 наблюдалась при отсутствии ингибитора и вносили поправку на фоновую флуоресценцию при отсутствии LSD1. Значение Ki (IC50) для каждого ингибитора определяли при половине максимальной активности.
В приведенной ниже таблице 2 представлены данные исследования по ингибированию LSD1 целым рядом соединений примеров. Обнаружено, что парнат (2-транс фенилциклопропиламин) характеризуется значением Ki (IC50), равным примерно от 15 до 35 мкМ в зависимости от препарата фермента. Это исследование показывают, что соединения, предлагаемые в настоящем изобретении, неожиданно активно ингибируют LSD1.
Пример 39: Биологические исследования - использование моноаминоксидазы для определения селективности соединений, предлагаемых в настоящем изобретении, по отношению к LSD1
Рекомбинантные белки моноаминоксидазы человека MAO-A и MAO-B приобретали у фирмы Sigma Aldrich. МАО катализируют окислительное дезаминирование первичных, вторичных и третичных аминов. Для определения ферментативной активности МАО и/или степени их ингибирования рассматриваемым ингибитором (ингибиторами) использовали скрининг ингибиторов на основе флуоресценции. В качестве субстрата выбрано нефлуоресцирующее соединение 3-(2-аминофенил)-3-оксопропанамин (кинураминдигидробромид, Sigma Aldrich). Кинурамин является неспецифическим субстратом для определения активности обоих МАО. При изучении окислительного дезаминирования с помощью МАО кинурамин превращается во флуоресцирующий продукт, 4-гидроксихинолин (4-HQ).
Активность моноаминоксидазы оценивали путем определения степени превращения кинурамина в 4-гидроксихинолин. Исследования проводили в черных 96-луночных планшетах с прозрачным дном (Corning) при конечном объеме, равном 100 мкл. Буфером для исследования являлся 100 мМ HEPES (N-2-гидроксиэтилпиперазин-N-2-этансульфоновая кислота), pH 7,5. Каждый эксперимент в каждой серии проводили трижды.
Вкратце, методика заключалась в следующем: определенное количество МАО (0,25 мкг MAO-A и 0,5 мкг MAO-B) инкубировали на льду в течение 15 мин в буфере для проведения реакции при отсутствии и/или в присутствии ингибитора в разных концентрациях (например, от 0 до 50 мкМ, в зависимости от активности ингибитора). В качестве контроля для ингибирования использовали транилципромин (Biomol International).
После введения фермента (ферментов) в реакцию с ингибитором в каждую реакционную смесь добавляли от 60 до 90 мкМ кинурамина для исследования MAO-B и MAO-A соответственно и реакционную смесь выдерживали в течение 1 ч при 37°C в темноте. Окислительное дезаминирование субстрата останавливали путем добавления 50 мкл (об./об.) 2 н. NaOH. За превращением кинурамина в 4-гидроксихинолин следили по флуоресценции (возбуждение при 320 нм, испускание при 360 нм) с использованием считывающего устройства для микропланшетов (Infinite 200, Tecan). Для определения интенсивности флуоресценции при отсутствии и/или в присутствии ингибитора использовали произвольные единицы.
Максимальная активность дезаминирования наблюдалась при определении содержания 4-гидроксихинолина, образовавшегося из кинурамина при отсутствии ингибитора с поправкой на фоновую флуоресценцию при отсутствии ферментов MAO. Значение Ki (IC50) для каждого ингибитора определяли при Vmax/2.
Диапазоны значений Ki, приведенные в таблице 2, составляют для MAO-A, MAO-B и LSD1 - I = от 1 мкМ до 40 мкМ; II = от 0,1 мкМ до 1 мкМ; III от 0,001 мкМ до 0,1 мкМ.
В целом установление, что соединения примеров обладают значениями Ki (IC50) для MAO-A и MAO-B, превышающими значения Ki для LSD1, тогда как значения Ki для LSD1 были меньше чем 0,6 мкМ.
Таким образом, соединения, предлагаемые в настоящем изобретении, неожиданно являются активными LSD1 ингибиторами и неожиданно селективны по отношению к LSD1 в отличие от MAO-A и MAO-B или соединений, являющихся двойными ингибиторами LSD1 и MAO-B.
Для некоторых соединений примеров исследована антипролиферативная/цитотоксическая активность и установлено, что по отношению к линиям раковых клеток, включая HCT-116, они обладают активностью, находящейся в диапазоне значений от микромолей до нескольких миллимолей.
В предшествующих публикациях об LSD1 установлено, что она участвует в пролиферации и росте клеток. В некоторых исследованиях LSD1 рассматривалась в качестве мишени терапевтического воздействия при раке. В публикации Huang et al. (2007) PNAS 104: 8023-8028 установлено, что полиаминовые ингибиторы LSD1 в раковых клетках приводят к умеренной реэкспресии генов, которые в результате аберрации стали молчащими, в особенности при колоректальном раке (Huang et al. Clin Cancer Res. (2009) Dec 1; 15(23): 7217-28. Epub 2009 Nov 24. PMID: 19934284). В публикации Scoumanne Scoumanne et al. ((2007) J. Biol. Chem. May 25; 282(21): 15471-5) установлено, что недостаток LSD1 приводит к частичной остановке клеточного цикла в G2/M и сенсибилизированных клетках и усилению супрессии, вызванной повреждением ДНК. В публикации Kahl et al. ((2006) Cancer Res. 66(23): 11341-7) установлено, что экспрессия LSD1 коррелирует с агрессивностью рака предстательной железы. В публикации Metzger et al. сообщают, что модулирование LSD1 с помощью siRNA и паргилина регулирует андрогенный рецептор (АР) и, вероятно, может использоваться для лечения типов рака, при которых играет роль АР, таких как рак предстательной железы, яичников и головного мозга. В публикации Lee et al. ((2006) Chem. Biol. 13: 563-567) сообщают, что в некоторых линиях раковых клеток транилципромин дерепрессирует ген Egr-1. Имеется много данных о том, что во многих случаях Egr-1 является геном-супрессором опухолей. Например, в публикации Calogero et al. (2004) Cancer Cell International 4:1) экзогенная экспрессия Egr-1 приводит к остановке роста и в конечном счете к гибели первичных линий раковых клеток; в публикации Lucerna et al. (2006) Cancer Research 66, 6708-6713) показано, что длительная экспрессия Egr-1 в некоторых моделях приводит к антиангиогенному эффекту и подавляет рост опухолей; в публикации Ferraro et al. ((2005) J. Clin. Oncol. Mar 20; 23(9): 1921-6) сообщают, что Egr-1 подвергается понижающей регуляции у пациентов, страдающих раком легких с высоким риском рецидива, и может быть более устойчивым к терапии. Таким образом, усиление экспрессии Egr-1 путем ингибирования LSD1 является методикой лечения некоторых типов рака. В недавних исследованиях показано, что LSD1 участвует в раке головного мозга (Schulte et al. (2009) Cancer Res. Mar 1; 69(5): 2065-71). В других исследованиях показано, что LSD1 участвует в раке молочной железы (Lims et al. Carcinogenesis. 2009 dec 30. [Epub ahead of print] PMID: 20042638).
Таким образом, большое количество данных показывает, что LSD1 участвует в целом ряде раковых заболеваний, что указывает на то, что LSD1 является мишенью терапевтического воздействия при раке. Авторы настоящего изобретения обнаружили класс ингибиторов LSD1, которые можно использовать для лечения заболеваний, в которых мишенью для лечения является LSD1, таких как рак. В соответствии с этим фенилциклопропиламины, предлагаемые в настоящем изобретении, можно использовать для лечения таких заболеваний.
Недавние исследования показали, что LSD1 участвует в вирусной инфекции и реактивации вируса. В частности, показано, что фармакологические ингибиторы LSD1, такие как парнат и siRNA, лишенная LSD1, приводят к уменьшенной вирулентности и уменьшают реактивацию после латентного периода (Liang et al. (2009) Nat. Med. 15: 1312-1317). Поэтому предполагается, что соединения, предлагаемые в настоящем изобретении, можно использовать для лечения или предупреждения вирусной инфекции. Кроме того, предполагается, что соединения, предлагаемые в настоящем изобретении, могут лечить или предупреждать реактивацию вируса после латентного периода.
Таким образом, если не ограничиваться теоретическими соображениями, то можно сделать вывод о том, согласно изобретению выявлен новый класс производных циклопропанамина, содержащий ингибиторы LSD1, обладающие неожиданной активностью и селективностью по отношению к LSD1, биологически важной мишени при онкологических и других заболеваниях и/или LSD1/MAO-B.
Все публикации и заявки на патенты, отмеченные в описании, указывают на уровень специалистов в области техники, к которой относится настоящее изобретение. Все публикации и заявки на патенты включены в настоящее изобретение в качестве ссылки в такой же степени, как, если бы для каждой отдельной публикации или заявки на патент было специально и по отдельности указано, что она включена в настоящее изобретение в качестве ссылки. Простое указание на публикации и заявки на патенты необязательно является признанием того, что они являются предшествующим уровнем техники для настоящей заявки.
Хотя выше настоящее изобретение подробно описано в качестве иллюстрации и примера для облегчения понимания, очевидно, что в объеме прилагаемой формулы изобретения в него можно внести некоторые изменения и модификации.
Реферат
Изобретение относится к новым соединениям формулы (I) или его фармацевтически приемлемой соли или сольвату, которые обладают активностью ингибитора лизинспецифической деметилазы-1 (LSD1). Соединения могут найти применение для лечения или предупреждения рака, выбранного из рака предстательной железы, рака молочной железы, рака легких, колоректального рака, рака головного мозга, рака крови или лейкоза; неврологического заболевания, включающего депрессию, болезнь Альцгеймера, болезнь Гентингтона, болезнь Паркинсона, боковой амиотрофический склероз, лобно-височное слабоумие или слабоумие с тельцами Леви; или вирусной инфекции вызванной вирусом герпеса, или выбранной из группы HSV-1, HSV-2, и вируса Эпштейна-Барра. В соединении формулы (I)(A) обозначает фенил или пиридил, где указанный фенил или указанный пиридил содержит n заместителей (R3); (B) обозначает -О-СН-фенил или фенил, где указанный фенил или фенильный фрагмент, содержащийся в указанном -О-СН-фениле, содержит n заместителей (R2); (D) обозначает моноциклическую гетероарильную группу, содержащую 5 или 6 кольцевых членов, где от одного до трех указанных кольцевых членов являются гетероатомами, выбранными из О, S и N, где указанная гетероарильная группа содержит один заместитель (R1) и, кроме того, где указанная гетероарильная группа ковалентно связана с остальной частью молекулы через кольцевой атом углерода; (R1) обозначает -NH; каждый (R2) независимо выбран из группы, включающей C-Салкил, гидроксигруппу, галоген, C-Сгалогеналкил, C-Сгалогеналкоксигруппу или С-Салкоксигруппу; каждый (R3) независимо выбран из группы, включающей C-Салкил, гидроксигруппу, галоген, C-Сгалогеналкил, C-Сгалогеналкоксигруппу или C-Салкоксигруппу; и n независимо
Формула

Комментарии