Ингибиторы iap - RU2728789C2
Код документа: RU2728789C2
Чертежи

Описание
[0001] Настоящее изобретение относится к органическим соединениям, подходящим для лечения и/или профилактики млекопитающего, в частности к ингибиторам белков IAP, подходящим для лечения раковых заболеваний.
[0002] Апоптоз, или запрограммированная гибель клеток, представляет собой механизм, регулируемый генетически и биохимически, который играет важную роль в развитии и гомеостазе у беспозвоночных, а также у позвоночных. Нарушения апоптоза, которые приводят к преждевременной гибели клеток, связывают с рядом отклонений развития. Недостаточность апоптоза, которая приводит к снижению гибели клеток, связывают с раком и хроническими вирусными инфекциями (Thompson et al., (1995) Science 267, 1456-1462).
[0003] Одними из ключевых молекулярных эффекторов при апоптозе являются каспазы (цистеинсодержащие аспартат-спепифичные протеазы). Каспазы являются высокоактивными протеазами, которые расщепляют белки после остатков аспарагиновой кислоты и в активированной форме разрушают жизненно важные клеточные белки внутри клетки. Так как каспазы являются настолько высокоактивными протеазами, для предотвращения преждевременной гибели клеток необходим точный контроль этого семейства белков. В целом, каспазы синтезируются в виде крупных неактивных зимогенов, для активации которых требуется протеолитический процессинг. Указанный протеолитический процессинг является только одним из путей регуляции каспаз. Второй механизм опосредован семейством белков, связывающих и ингибирующих каспазы.
[0004] Семейством молекул, ингибирующих каспазы, являются ингибиторы апоптоза (IAP) (Deveraux et al., J Clin Immunol (1999), 19: 388-398). Первоначально IAP были открыты в бакуловирусе, что было обусловлено их функциональной способностью заменять белок Р35, антиапоптозный ген (Crook et al. (1993) J Virology 67, 2168-2174). Описания IAP приведены для различных организмов от Drosophila до человека. Вне зависимости от происхождения в структурах IAP содержатся от одного до трех повторяющихся доменов IAP бакуловируса (BIR), и большинство из них также содержат карбокситерминальный мотив RING finger. Домен BIR, как таковой, представляет собой цинк-связывающий домен, состоящий примерно из 70 остатков, содержащий 4 альфа-спирали и 3 бета-тяжа и остатки цистеина и гистидина, который координирует с ионом цинка (Hinds et al., (1999) Nat. Struct. Biol. 6, 648-651). Полагают, что домен BIR вызывает антиапоптотическое действие путем ингибирования каспаз и, тем самым, ингибирования апоптоза. Например, человеческий IAP, связанный с Х-хромосомой (XIAP), ингибирует каспазу 3, каспазу 7, а также опосредованную Apaf-1-цитохромом C активацию каспазы 9 (Deveraux et al., (1998) EMBO J. 17, 2215-2223). Домен BIR2 XIAP ингибирует каспазы 3 и 7, тогда как домен BIR3 XIAP отвечает за ингибирование активности каспазы 9. XIAP повсеместно экспрессируются в большинстве тканей взрослых и плода (Liston et al, Nature, 1996, 379(6563): 349), их повышенная экспрессия происходит в ряде опухолевых клеточных линий, принадлежащих к группе клеточных линий NCI 60 (Fong et al, Genomics, 2000, 70: 113; Tamm et al, Clin. Cancer Res. 2000, 6(5): 1796). Было показано, что повышенная экспрессия XIAP в опухолевых клетках обеспечивает защиту от ряда проапоптотических стимулов и промотирует резистентность к химиотерапии (LaCasse et al, Oncogene, 1998, 17(25): 3247). С этим согласуется наблюдаемая сильная корреляция между уровнем белков XIAP и выживаемостью пациентов с острой миелогенной лейкемией (Tamm et al, выше). Было показано, что понижающая регуляция экспрессии XIAP античувствительными олигонуклеотидами повышает чувствительность опухолевых клеток к гибели, вызываемой широким диапазоном проапоптотических агентов in vitro и in vivo (Sasaki et al, Cancer Res., 2000, 60(20): 5659; Lin et al, Biochem J., 2001, 353: 299; Hu et al, Clin. Cancer Res., 2003, 9(7): 2826). Также было показано, что Smac/DIABLO-производные пептиды повышают чувствительность ряда различных опухолевых клеточных линий к апоптозу, вызванному различными проапоптотическими лекарственными средствами (Arnt et al, J. Biol. Chem., 2002, 277(46): 44236; Fulda et al, Nature Med., 2002, 8(8): 808; Guo et al, Blood, 2002, 99(9): 3419; Vucic et al, J. Biol. Chem., 2002, 277(14): 12275; Yang et al, Cancer Res., 2003, 63(4): 831).
[0005] IAP меланомы (ML-IAP) представляет собой IAP, который не поддается обнаружению в большинстве нормальных тканей взрослого человека, но при меланоме происходит его сильная повышающая регуляция (Vucic et al., (2000) Current Bio 10: 1359-1366). Определение структуры белка показало значительную гомологию доменов BIR и RING finger ML-IAP с соответствующими доменами, содержащимися в человеческих XIAP, C-IAP1 и C-IAP2. Полагают, что домен BIR ML-IAP наиболее схож с BIR2 и BIR3 XIAP, С-IAP1 и C-IAP2 и отвечает за ингибирование апоптоза, что определяли при помощи делеционного анализа. Кроме того, Вучич с соавторами (Vucic et al.) показали, что ML-IAP может ингибировать апоптоз, вызванный химиотерапевтическими агентами. Агенты, такие как адриамицин и 4-третичный бутилфенол (4-ТВР), тестировали на системе клеточных культур меланомы, имеющих повышенную экспрессию ML-IAP, и эффективность химиотерапевтических агентов в отношении уничтожения клеток была значительно снижена по сравнению с нормальным контролем меланоцитов. Механизм, по которому ML-IAP осуществляет антиалоптотическую активность, отчасти опосредован ингибированием каспазы 3 и 9. ML-IAP не ингибирует эффективно каспазы 1, 2, 6 или 8.
[0006] Так как апоптоз является жестко контролируемым процессом с множеством взаимосвязанных факторов, обнаружение факта регуляции самих IAP не было неожиданным. У плодовых мушек Drosophila белки Reaper (rpr), Head Involution Defective (hid) и GRIM вступают в физическое взаимодействие и ингибируют антиалоптотическую активность семейства IAP у Drosophila. У млекопитающих действие белков SMAC/DIABLO блокирует IAP и обеспечивает прохождение апоптоза. Было показано, что при нормальном течении апоптоза происходит процессинг SMAC с переводом в активную форму и его высвобождение из митохондрий в цитоплазму, где происходит его физическое связывание с IAP, что предотвращает связывание IAP с каспазой. Указанное ингибирование IAP оставляет каспазу в активной форме и, таким образом, происходит апоптоз. Интересно отметить, что гомология последовательностей в ингибиторах IAP подтверждает наличие мотива из четырех аминокислот в N-конце процессированных активных белков. Полагают, что указанный тетрапептид связывается с гидрофобным «карманом» домена BIR и разрушает домен BIR, связывающийся с каспазами (Chai et al., (2000) Nature 406: 855-862, Liu et al., (2000) Nature 408: 1004-1008, Wu et al., (2000) Nature 408 1008-1012).
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
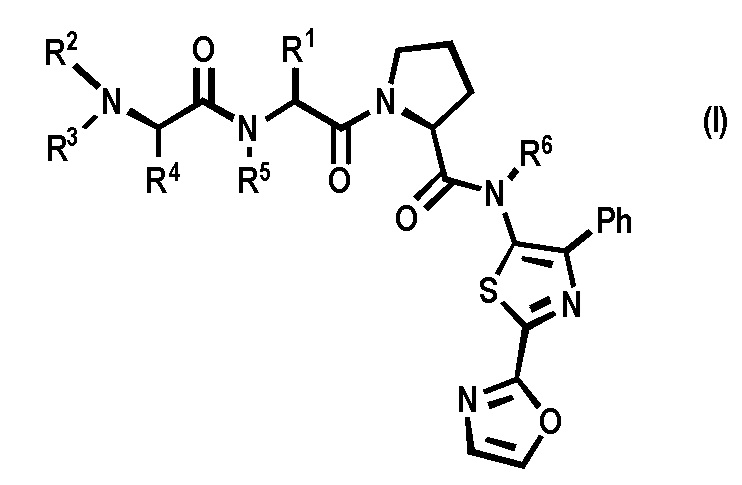
[0007] Согласно одному из аспектов настоящего изобретения предложены новые ингибиторы белков IAP, имеющие общую формулу (I)
где
[0008] R1 представляет собой C3-7 циклоалкил,
[0009] Ph представляет собой фенил,
[0010] каждый R2, R3, R4, R5 и R6 в каждом случае независимо представляет собой Н или C1-6 алкил; или
[0011] их фармацевтически приемлемые соли.
[0012] Формула I включает все стереоизомеры.
[0013] Согласно другому аспекту настоящего изобретения предложены композиции, содержащие соединения формулы I и носитель, разбавитель или вспомогательное вещество.
[0014] Согласно другому аспекту настоящего изобретения предложен способ инициирования апоптоза в клетке, включающий введение в указанную клетку соединения формулы I.
[0015] Согласно другому аспекту настоящего изобретения предложен способ увеличения чувствительности клетки к апоптотическому сигналу, включающий введение в указанную клетку соединения формулы I.
[0016] Согласно другому аспекту настоящего изобретения предложен способ ингибирования связывания белка IAP с белком каспазой, включающий приведение указанного белка IAP в контакт с соединением формулы I.
[0017] Согласно другому аспекту настоящего изобретения предложен способ лечения заболевания или состояния, связанного с повышенной экспрессией белка IAP у млекопитающего, включающий введение указанному млекопитающему эффективного количества соединения формулы I.
[0018] Согласно другому аспекту настоящего изобретения предложен способ лечения рака.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
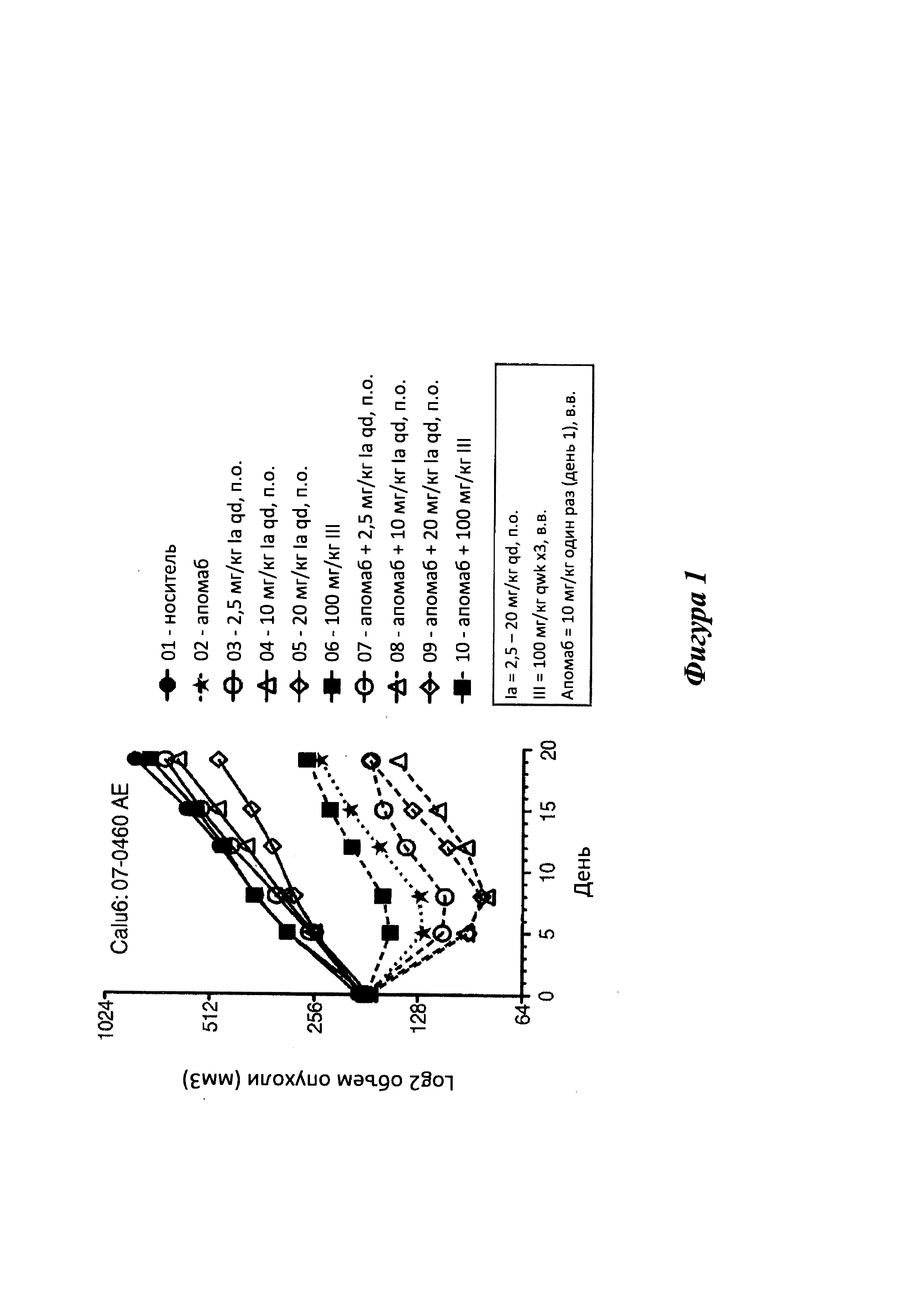
[0019] На фигуре 1 показана эффективность Ia, используемого отдельно, а также в комбинации с апомабом, совместно с данными эффективности известного в уровне техники соединения III и его комбинации с апомабом в модели ксенотрансплантата клеток аденокарциномы легких Calu-6. Ia вводили п.о. III вводили в.в. Выбранные дозы представляли собой максимальные переносимые дозы для каждого лекарственного средства.
[0020] На фигуре 2 показана эффективность Ia, используемого отдельно, а также в комбинации с апомабом, совместно с данными эффективности известного в уровне техники соединения III и его комбинации с апомабом в модели ксенотрансплантата клеток колоректальной аденокарциномы Colo205. Ia вводили п.о. III вводили в.в. Выбранные дозы представляли собой максимальные переносимые дозы для каждого лекарственного средства.
[0021] Объект, описываемый в настоящей заявке в форме единственного числа, относится к одному или более указанным объектам; например, соединение относится к одному или более соединениям или по меньшей мере к одному соединению. Таким образом, форму единственного числа, «один или более» и «по меньшей мере один» можно использовать в настоящем описании взаимозаменяемо.
[0022] Используемые в настоящем описании в качестве переходной фразы или для описания пункта формулы изобретения термины «содержать(ит)» и «содержащий» следует рассматривать как неограничивающие термины. То есть, эти термины следует рассматривать в качестве синонимов с фразами «имеющий по меньшей мере» или «включающий по меньшей мере». При использовании в контексте описания способа термин «включающий» означает, что способ включает по меньшей мере указанные стадии, но также может включать и дополнительные стадии. При использовании в контексте описания соединения или композиции термин «содержащий» означает, что соединение или композиция включает все указанные отличительные признаки или компоненты, но также может включать и дополнительные отличительные признаки или компоненты.
[0023] Термин «независимо» используют в настоящем описании для указания на то, что переменную используют в каждом случае вне зависимости от наличия или отсутствия в одном соединении переменной, имеющей такое же или отличающееся определение. Таким образом, в соединении, в котором R'' встречается дважды и определен как «независимо представляющий собой атом углерода или азота», оба R'' могут представлять собой атомы углерода, оба R'' могут представлять собой атомы азота или один R" может представлять собой атом углерода, а другой - атом азота.
[0024] Термины «возможный» или «возможно», используемые в настоящем описании, означают, что описываемые с их помощью явления или условия могут происходить, но не обязательно, и в описание включены случаи, в которых явление или условие происходит, и случаи, в которых они отсутствуют. Например, «возможно замещенный» означает, что возможно замещенный фрагмент может включать атом водорода или заместитель.
[0025] Термин «примерно» используют в настоящем описании в значении приблизительно, в районе, округленно или около. Если термин «примерно» используют совместно с числовым диапазоном, то он модифицирует указанный диапазон, расширяя его границы выше и ниже числовых значений, приведенных для указанного диапазона. В целом, термин «примерно» используют в настоящем описании для модификации числового значения в большую и меньшую сторону с погрешностью 20% от указанного значения.
[0026] При использовании в настоящем описании указание числового диапазона для переменной означает возможность реализации изобретения при использовании переменной, имеющей любое из значений внутри указанного диапазона. Таким образом, в случае переменной, которая по определению может быть исключительно дискретной, переменная может быть равна любому целочисленному значению внутри числового диапазона, включая концы диапазона. Аналогично, в случае переменной, которая по определению является непрерывной, переменная может быть равна любому фактическому значению внутри числового диапазона, включая концы диапазона. Например, переменная, описываемая как имеющая значение от 0 до 2, может составлять 0, 1 или 2 в случае переменных, которые по определению являются дискретными, и 0,0, 0,1, 0,01, 0,001 или иметь любое другое фактическое значение в случае переменных, которые по определению являются непрерывными.
[0027] Соединения формулы I проявляют таутомерию. Таутомерные соединения могут существовать в виде двух или более взаимопревращающихся молекул. Прототропные таутомеры образуются в результате миграции ковалентно связанного атома водорода между двумя атомами. Таутомеры, в целом, существуют в равновесии, и попытки выделить отдельные таутомеры обычно приводят к получению смеси, физические и химические свойства которой совпадают со свойствами смеси соединений. Положение равновесия зависит от химических особенностей молекулы. Например, во множестве алифатических альдегидов и кетонов, таких как ацетальдегид, преобладает кето-форма, тогда как в фенолах преобладает енольная форма. Распространенные прототропные таутомеры включают системы кето/енол



[0028] «Алкил» обозначает разветвленную или неразветвленную насыщенную алифатическую углеводородную группу, содержащую до 6 атомов углерода, если не указано иное, в том числе в случае использования в качестве составной части другого термина, например, «алкиламино». Примеры предпочтительных алкильных групп включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, 2-метилбутил, 2,2-диметилпропил, н-гексил, 2-метилпентил, 2,2-диметилбутил и т.д. Термины «низший алкил», «C1-C4 алкил» и «алкил, содержащий от 1 до 4 атомов углерода» являются синонимами, и их используют взаимозаменяемо для обозначения метила, этила, 1-пропила, изопропила, циклопропила, 1-бутила, втор-бутила или трет-бутила.
[0029] Циклоалкильные группы могут представлять собой моно-, би- или трициклические алифатические кольца, содержащие от 3 до 7 атомов углерода. Предпочтительные группы включают циклопропил и циклогексил, наиболее предпочтительным является циклогексил.
[0030] «Аминозащитная группа» относится к производному группы, традиционно используемому для блокировки или защиты аминогруппы, в результате чего можно проводить взаимодействия с участием других функциональных групп соединения. Примеры указанных защитных групп включают карбаматы, амиды, алкильные и арильные группы, имины, а также многочисленные N-гетероатомные производные, которые можно удалять для регенерации целевой аминогруппы. Предпочтительными аминозащитными группами являются Boc, Fmoc и Cbz. Дополнительные примеры указанных групп приведены в Т.W. Greene and P.G.M. Wuts, "Protective Groups in Organic Synthesis", 2nd ed., John Wiley & Sons, Inc., New York, NY, 1991, раздел 7; E. Haslam, "Protective Groups in Organic Chemistry", J. G.W. McOmie, Ed., Plenum Press, New York, NY, 1973, раздел 5, и T.W. Greene, "Protective Groups in Organic Synthesis", John Wiley and Sons, New York, NY, 1981. Термин «защищенный амин» относится к аминогруппе, замещенной одной или более из указанных аминозащитных групп. Указанные группы можно использовать во время синтеза.
[0031] «Карбоксизащитная группа» относится к одному из сложноэфирных производных карбоксильной группы, традиционно применяемых для блокировки или защиты карбоксильной группы, в результате чего можно проводить взаимодействия с участием других функциональных групп соединения. Примеры указанных защитных групп карбоновых кислот включают 4-нитробензил, 4-метоксибензил, 3,4-диметоксибензил, 2,4-диметоксибензил, 2,4,6-триметоксибензил, 2,4,6-триметилбензил, пентаметилбензил, 3,4-метилендиоксибензил, бензгидрил, 4,4'-диметоксибензгидрил, 2,2',4,4'-тетраметоксибензгидрид, алкил, такой как трет-бутил или трет-амил, тритил, 4-метокситритил, 4,4'-диметокситритил, 4,4',4''-триметокситритил, 2-фенилпроп-2-ил, триметилсилил, трет-бутилдиметилсилил, фенацил, 2,2,2-трихлорэтил, бета-(триметилсилил)этил, бета-(ди(н-бутил)метилсилил)этил, п-толуолсульфонилэтил, 4-нитробензилсульфонилэтил, аллил, циннамил, 1-(триметилсилилметил)проп-1-ен-3-ил и схожие фрагменты. Вид применяемой карбоксизащитной группы не важен, так как производные карбоновых кислот являются стабильными в условиях, в которых проводят последующее(ие) взаимодействие(я) по другим положениям молекулы, и их можно удалять в подходящий момент, не разрушая остаток молекулы. В частности, важно не подвергать молекулу с защищенными карбоксигруппами воздействию сильных нуклеофильных оснований, таких как гидроксид лития или NaOH, или воздействию в условиях восстановления с использованием высокоактивных гидридов металлов, таких как LiAlH4. (Следует избегать использования таких жестких условий и при удалении аминозащитных групп и гидроксизащитных групп). Предпочтительными защитными группами карбоновых кислот являются алкил (например, метил, этил, трет-бутил), аллил, бензил и п-нитробензильные группы. Для защиты карбокси-заместителей также можно использовать карбоксизащитные группы, схожие с тем, что применяют в области цефалоспоринов, пенициллинов и пептидов. Дополнительные примеры указанных групп приведены в Т.W. Greene and P.G.M. Wuts, "Protective Groups in Organic Synthesis", 2nd ed., John Wiley & Sons, Inc., New York, N.Y., 1991, раздел 5; E. Haslam, "Protective Groups in Organic Chemistry", J. G.W. McOmie, Ed., Plenum Press, New York, N.Y., 1973, раздел 5, и T.W. Greene, "Protective Groups in Organic Synthesis", John Wiley and Sons, New York, NY, 1981, раздел 5. Термин «защищенный карбокси» относится к карбоксигруппе, замещенной одной из указанных выше карбоксизащитных групп. Указанные группы можно использовать во время синтеза.
[0032] «Гидроксизащитная группа» относится к производному гидроксигруппы, традиционно используемому для блокировки или защиты гидроксигрупп, в результате чего можно проводить взаимодействия с использованием других групп соединения. Примеры указанных защитных групп включают тетрагидропиранилокси, бензоил, ацетокси, карбамоилокси, бензил и простые силильные эфиры (например, TBS, TBDPS). Дополнительные примеры указанных групп приведены в Т.W. Greene and P.G.M. Wuts, "Protective Groups in Organic Synthesis", 2nd ed., John Wiley & Sons, Inc., New York, NY, 1991, разделы 2-3; E. Haslam, "Protective Groups in Organic Chemistry", J.G.W. McOmie, Ed., Plenum Press, New York, NY, 1973, раздел 5, и T.W. Greene, "Protective Groups in Organic Synthesis", John Wiley and Sons, New York, NY, 1981. Термин «защищенный гидрокси» относится к гидроксигруппе, замещенной одной из указанных выше гидроксизащитных групп. Указанные группы можно использовать во время синтеза.
[0033] «Ингибитор» обозначает соединение, которое снижает или предотвращает связывание белков IAP с белками каспазами или снижает или предотвращает ингибирование апоптоза белком IAP (например, с-IAP1, C-IAP2, X-IAP или ML-IAP). В качестве альтернативы «ингибитор» обозначает соединение, которое предотвращает связывание X-IAP с каспазами или связывание ML-IAP с SMAC.
«Фармацевтически приемлемые соли» включают соли присоединения кислот и оснований. «Фармацевтически приемлемая соль присоединения кислоты» относится к солям, которые сохраняют биологическую эффективность и свойства свободных оснований, не являются по биологическим или иным причинам нежелательными, полученным из неорганических кислот, таких как хлороводородная кислота, бромоводородная кислота, серная кислота, азотная кислота, угольная кислота, фосфорная кислота и т.д., и органических кислот, которые могут быть выбраны из классов алифатических, циклоалифатических, ароматических, аралифатических, гетероциклических, карбоновых и сульфо-органических кислот, таких как муравьиная кислота, уксусная кислота, пропановая кислота, гликолевая кислота, глюконовая кислота, молочная кислота, пировиноградная кислота, щавелевая кислота, яблочная кислота, малеиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, аспарагиновая кислота, аскорбиновая кислота, глутаминовая кислота, антраниловая кислота, бензойная кислота, коричная кислота, миндальная кислота, эмбоновая кислота, фенилуксусная кислота, метансульфокислота, этансульфокислота, п-толуолсульфокислота, салициловая кислота и т.д. «Фармацевтически приемлемые соли присоединения оснований» включают соли, полученные из неорганических оснований, такие как соли натрия, калия, лития, аммония, кальция, магния, железа, цинка, меди, марганца, алюминия и т.д. Особенно предпочтительными являются соли аммония, калия, натрия, кальция и магния. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, замещенных аминов, включая природные замещенные амины, циклических аминов и основных ионообменных смол, такие как соли изопропил амина, триметиламина, диэтиламина, триэтил амина, трипропил амина, этаноламина, 2-диэтиламиноэтанола, триметамина, дициклогексиламина, лизина, аргинина, гистидина, кофеина, прокаина, гидрабамина, холина, бетаина, этилендиамина, глюкозамина, метилглюкамина, теобромина, пуринов, пиперизина, пиперидина, N-этилпиперидина, полиаминовых смол и т.д. Особенно предпочтительными органическими нетоксичными основаниями являются изопропиламин, диэтиламин, этаноламин, триметамин, дициклогексиламин, холин и кофеин. Предполагается, что формула I охватывает гидраты и сольваты соединений.
[0034] В настоящем изобретении предложены новые соединения, имеющие общую формулу I
[0035] В конкретном варианте реализации R1 представляет собой циклогексил. В другом конкретном варианте реализации R1 представляет собой циклопентил. В конкретном варианте реализации R1 ориентирован таким образом, что аминокислота или аналог аминокислоты, составной частью которого он является, имеет L-конфигурацию.
[0036] R2 и R3 независимо представляют собой Н или C1-6 алкил. В одном из вариантов реализации R2 и R3 оба представляют собой H. В другом варианте реализации R2 представляет собой метил, а R3 представляет собой H.
[0037] R4 представляет собой Н или C1-6 алкил. В конкретном варианте реализации R4 представляет собой Н или метил. В другом варианте реализации R4 представляет собой метил. В другом варианте реализации R4 ориентирован таким образом, что аминокислота или аналог аминокислоты, составной частью которого он является, имеет L-конфигурацию.
[0038] Каждый R5 и R6 независимо представляет собой Н или C1-6 алкил. В одном из вариантов реализации R5 и R6 представляют собой Н или метил. В одном из вариантов реализации R5 представляет собой Н, а R6 представляет собой метил. В другом варианте реализации R5 представляет собой метил, а R6 представляет собой Н. В другом варианте реализации R5 и R6 оба представляют собой метил. В другом варианте реализации R5 и R6 оба представляют собой Н.
[0039] Согласно другому аспекту настоящего изобретения соединение формулы I представляет собой (2-оксазол-2-ил-4-фенилтиазол-5-ил)амид (S)-1-[(S)-2-циклогексил-2-((S)-2-метиламинопропиониламино)ацетил]пирролидин-2-карбоновой кислоты (Ia).

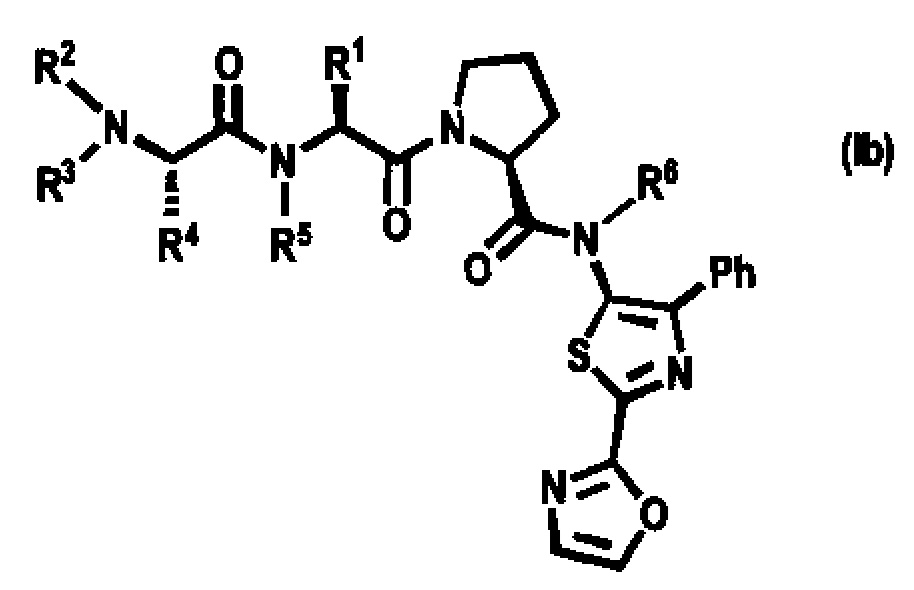
[0040] Соединения согласно настоящему изобретению содержат один или более асимметрических атомов углерода. Соответственно, соединение может существовать в виде стереоизомеров, включая, диастереомеры, энантиомеры или их смеси. В способах синтеза соединений в качестве исходных или промежуточных веществ можно применять рацематы, диастереомеры или энантиомеры. Диастереомерные соединения можно разделять при помощи способов хроматографии или кристаллизации. По аналогии, энантиомерные смеси можно разделять при помощи таких же или других способов, известных в данной области техники. Каждый из асимметрических атомов углерода может иметь R- или S-конфигурацию, и обе указанные конфигурации включены в объем настоящего изобретения. Предпочтительно соединения согласно настоящему изобретению имеют следующую стереохимическую конфигурацию формулы Ib, где R1, R2, R3, R4, R5 и R6 такие, как описано в настоящей заявке.
[0041] Соединения формулы II, где A представляет собой возможно замещенный 5-членный гетероцикл, содержащий от 1 до 4 гетероатомов, был предложен в опубликованной заявке на патент США №20060014700. В некоторых соединениях, предложенных в указанной опубликованной заявке, А представляет собой N-(4-фенилтиазол-5-ил).

[0042] Авторы настоящей заявки обнаружили, что соединения согласно настоящему изобретению, в которых A представляет собой 2-(оксазол-2-ил)-4-фенилтиазол-5-ил, обладают неожиданно увеличенной активностью и пероральной биодоступностью. Кроме того, соединения согласно настоящему изобретению, в целом, имеют пониженные побочные эффекты, включая улучшенную токсичность в легких, например, по сравнению с соединением III, предложенным в опубликованной заявке на патент США №20060014700. На фигурах 1 и 2 приведено сравнение активности в моделях ксенотрансплантатов опухолей после в.в. введения соединения III и перорального введения соединения Ia согласно настоящему изобретению.
СИНТЕЗ
[0043] Соединения согласно настоящему изобретению получали при помощи стандартных способов органического синтеза из коммерчески доступных исходных веществ и реактивов. Общие способы синтеза описаны в международной заявке на патент WO 98/46576 и в патенте США №7244851, и в настоящую заявку посредством ссылок включены описания способов получения, предложенных в указанных документах. Следует понимать, что выбор способов синтеза, применяемых для получения соединений согласно настоящему изобретения, зависит от конкретных заместителей, содержащихся в соединении, а также что в соответствии со стандартной практикой органического синтеза могут требоваться различные стадии введения и снятия защиты. Согласно общей схеме синтеза соединения согласно настоящему изобретению можно получать при помощи традиционных способов химии пептидов путем сочетания аналогов остатков аминокислот с использованием типовых процедур амидного сочетания. На схеме 1 проводят сочетание аминозащищенных остатков аминокислот и последующее снятие защиты с получением конечных соединений согласно протоколам синтеза пептидов.
Схема 1

[0044] Следует понимать, что сочетание аналогов аминокислот можно проводить в любом порядке, и их можно получать с использованием твердофазной подложки, что является традиционным в данной области техники.
[0045] Если соединения согласно настоящему изобретению содержат заместители R2 или R3, отличные от Н, их также можно получать путем замещения подходящей промежуточной кислоты, содержащей уходящую группу, с использованием целевого амина. Например, Br-CH(R4)-C(O)-OH замещают с использованием амина R2-NH2 или R2-NH-R3 согласно схеме 2.
Схема 2

[0046] В качестве альтернативы реакцию замещения для введения заместителей R2 или R3 можно проводить в качестве конечной стадии способа получения соединения, как проиллюстрировано на схеме 3.
Схема 3
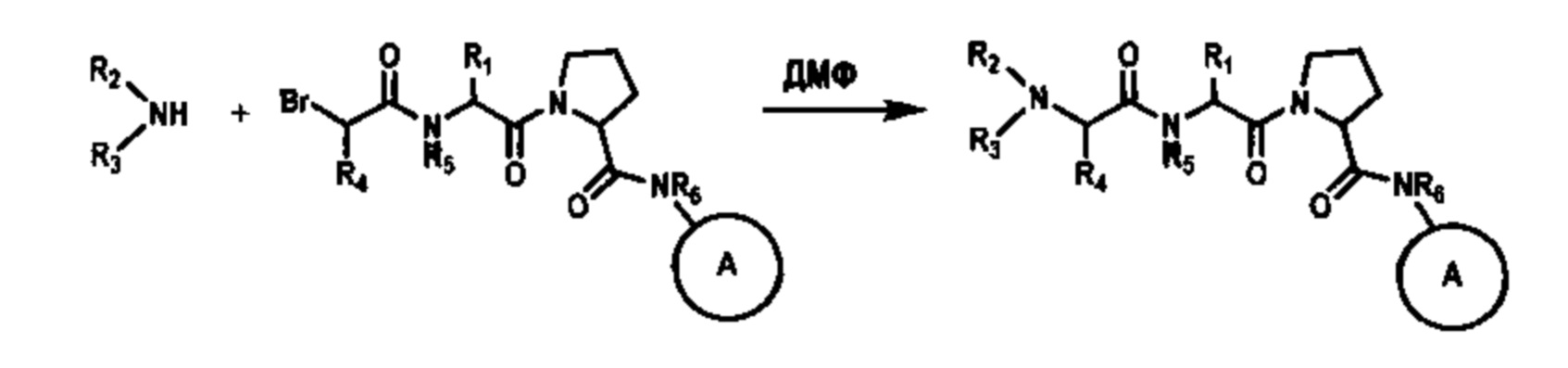
[0047] В конкретном варианте реализации проводят взаимодействие 2-бромпропановой кислоты с соответствующими аминами, растворенными в ДМФ, при кипячении до полного замещения с образованием N-замещенного остатка аланина.
[0048] Соединения с аминозамещенным кольцом A, которые выступают в качестве промежуточных веществ при получении соединений согласно настоящему изобретению, являются коммерчески доступными или в ином случае их можно получать из коммерчески доступных реактивов при помощи стандартных способов органической химии. 2-(оксазол-2-ил)-4-фенилтиазол-5-амин можно получать путем конденсации гидрохлорида α-аминофенилацетонитрила и оксазол-2-карбальдегида в присутствии серы и ТЭА (схема 4).

ПРИМЕНЕНИЕ
[0049] Соединения согласно настоящему изобретению ингибируют связывание белков IAP с каспазами, в частности связывание X-IAP с каспазами 3 и 7. Соединения также ингибируют связывание ML-IAP с белком Smac. Соответственно, соединения согласно настоящему изобретению подходят для инициирования апоптоза в клетках или для увеличения чувствительности клеток, в частности раковых клеток, к апоптотическим сигналам. Соединения согласно настоящему изобретению подходят для инициирования апоптоза в клетках, в которых происходит повышенная экспрессия белков IAP (например, C-IAP1, C-IAP2, X-IAP или ML-IAP). В качестве альтернативы соединения согласно настоящему изобретению подходят для инициирования апоптоза в клетках, в которых в результате нарушения митохондриального апоптотического пути происходит ингибирование высвобождения Smac из белков ML-IAP, например, за счет повышающей регуляции Bcl-2 или понижающей регуляции Bax/Bak. В более широком смысле соединения можно применять для лечения рака.
[0050] Они особенно подходят для лечения всех типов рака, при которых не происходит апоптоз. Примеры указанных типов рака включают нейробластому, карциному кишечника, такую как карцинома прямой кишки, карцинома толстой кишки, семейный аденоматозный полипоз и врожденный неполипозный колоректальный рак, карциному пищевода, карциному губы, карциному гортани, карциному гипофаринкса, карциному языка, карциному слюнных желез, карциному желудка, аденокарциному, медуллярную карциному щитовидной железы, папиллярную карциному щитовидной железы, карциному почки, карциному паренхимы почек, карциному яичников, карциному шейки матки, карциному тела матки, карциному эндометрия, хорионкарциному, карциному поджелудочной железы, карциному простаты, карциному яичек, карциному груди, карциному мочевыводящих путей, меланому, опухоли мозга, такие как глиобластома, астроцитома, менингиома, медуллобластома, и периферические нейроэктодермальные опухоли, лимфому Ходжкина, неходжкинскую лимфому, лимфому Беркитта, острую лимфатическую лейкемию (ОЛЛ), хроническую лимфатическую лейкемию (ХЛЛ), острую миелоидную лейкемию (ОМЛ), хроническую лимфатическую лейкемию (ХМЛ), T-клеточную лейкемию/лимфому взрослых, печеночноклеточную карциному, карциному желчного пузыря, бронхиальную карциному, мелкоклеточную карциному легких, немелкоклеточную карциному легких, множественную миелому, базалиому, тератому, ретинобластому, хориоидальную меланому, семиному, рабдомиосаркому, краниофарингеому, остеосаркому, хондросаркому, миосаркому, липосаркому, фибросаркому, саркому Юинга и плазмоцитому. Возможно применение для лечения солидных опухолей. Также возможно применение для лечения рака груди, аденокарциномы поджелудочной железы или злокачественной меланомы.
[0051] Соединения согласно настоящему изобретению подходят для увеличения чувствительности клеток к апоптотическим сигналам. Соответственно, соединения можно вводить до, во время или после проведения радиационной терапии или цитостатической или антинеопластической химиотерапии. Подходящие соединения для цитостатической химиотерапии включают, но не ограничиваются ими, (i) антиметаболиты, такие как цитарабин, флударабин, 5-фтор-2'-деоксиуридин, гемцитабин, гидроксимочевина или метотрексат; (ii) агенты, фрагментирующие ДНК, такие как блеомицин, (iii) агенты, обеспечивающие перекрестную сшивку ДНК, такие как хлорамбуцил, цисплатин, циклофосфамид или азотистый иприт; (iv) интеркалирующие агенты, такие как адриамицин (доксорубицин) или митоксантрон; (v) ингибиторы синтеза белков, такие как L-аспарагиназа, циклогексимид, пуромицин или токсин дифтерии; (vi) «яды» топоизомеразы I, такие как камптотецин или топотекан; (vii) «яды» топоизомеразы II, такие как этопозид (VP-16) или тенипозид; (viii) агенты направленного действия в микротрубочках, такие как колцемид, колхицин, паклитаксел, винбластин или винкристин; (ix) ингибиторы киназ, такие как флавопиридол, стауроспорин, STI571 (CPG 57148 В) или UCN-01 (7-гидроксистауроспорин); (x) вспомогательные агенты для исследований, такие как тиоплатин, PS-341, фенилбутират, ET-18-OCH3 или ингибиторы фарнезилтрансферазы (L-739749, L-744832); полифенолы, такие как кверцетин, ресвератрол, пицеатаннол, галлат эпигаллокатехина, теафлавины, флаванолы, процианидины, бетулиновую кислоту и ее производные; (xi) гормоны, такие как глюкокортикоиды или фенретинид; (xii) антагонисты гормонов, такие как тамоксифен, финастерид или антагонисты РФЛГ. В предпочтительном варианте реализации соединения согласно настоящему изобретению вводят совместно с цитостатическим соединением, выбранным из группы, состоящей из цисплатина, доксорубицина, паклитаксела, доцетаксела и митомицина С. Наиболее предпочтительным цитостатическим соединением является доксорубицин. Можно применять комбинации с 5-FU, гемцитабином, капецитабином, винорелбином, бевацизумабом или таксанами.
[0052] Другим классом соединений, которые можно применять в настоящем изобретении, являются соединения, которые могут увеличивать чувствительность или инициировать апоптоз путем связывания с рецепторами смерти («агонисты рецептора смерти»). Указанные агонисты рецепторов смерти включают лиганды рецепторов смерти, такие как фактор некроза опухоли а (ФНО-α), фактор некроза опухоли β (ФНО-β, лимфотоксин-α), LT-β (лимфотоксин-β), TRAIL (Apo2L, лиганд DR4), лиганд CD95 (Fas, APO-1), лиганд TRAMP (DR3, Apo-3), лиганд DR6, а также фрагменты и производные любого из указанных лигандов. Предпочтительно лигандом рецептора смерти является ФНО-α. Более предпочтительно лигандом рецептора смерти является Apo2L/TRAIL. Кроме того, агонисты рецепторов смерти содержат агонистические антитела к рецепторам смерти, такие как антитело к CD95, антитело к TRAIL-R1 (DR4), антитело к TRAIL-R2 (DR5), антитело к TRAIL-R3, антитело к TRA1L-R4, антитело к DR6, антитело к ФНО-α и антитело к TRAMP (DR3), а также фрагменты и производные любого из указанных антител.
[0053] Для увеличения чувствительности клеток к апоптозу соединения согласно настоящему изобретению также можно применять в комбинации с радиационной терапией. Фраза «радиационная терапия» относится к использованию электромагнитного или корпускулярного излучения для лечения новообразований. Радиационная терапия основана на том принципе, что высокодозное облучение, доставляемое к целевому участку, приводит к гибели репродуктивных клеток опухоли и нормальных тканей. Схему дозирования радиационного излучения, в целом, определяют с учетом поглощаемой дозы излучения (рад), времени и фракционирования, и ее точный выбор должен осуществляться онкологом. Количество излучения, получаемого пациентом, зависит от различных факторов, но двумя наиболее важными являются расположение опухоли относительно других жизненно важных структур или органов организма и степень распространения опухоли. Примеры радиотерапевтических агентов предложены, но не ограничиваются ими, в работах по радиационной терапии и известны в данной области техники (Hellman, Principles of Radiation Therapy, Cancer, in Principles I and Practice of Oncology, 24875 (Devita et al., 4th ed., vol 1, 1993)). Последние достижения в области радиационной терапии включают согласованное по трем измерениям облучение внешним лучом, радиационную терапию с модулированной интенсивностью (РТМИ), стереотаксическую радиохирургию и брахитерапию (интерстициальную радиационную терапию), в последнем способе источник излучения размещают непосредственно в опухоль в виде имплантируемых «зерен». При помощи указанных современных режимов лечения происходит доставка более высоких доз излучения к опухоли, что обуславливает повышенную эффективность по сравнению со стандартной терапией с использованием внешнего луча.
[0054] Ионизирующее излучение с использованием радионуклидов, испускающих бета-частицы, считают наиболее подходящим для применения в радиационной терапии вследствие умеренной линейной передачи энергии (ЛПЭ) ионизирующей частицы (электрона) и умеренного диапазона воздействия (как правило, на несколько миллиметров ткани). При использовании гамма-лучей происходит доставка меньших дозировок на значительно более высокие расстояния. Альфа-частицы являются другим крайним случаем, они доставляют очень высокую дозировку ЛПЭ, но имеют сильно ограниченный диапазон и, таким образом, должны находиться в непосредственном контакте с клетками обрабатываемой ткани. Кроме того, источники альфа-излучения, в целом, представляют собой тяжелые металлы, что ограничивает возможные химические условия и представляет повышенную угрозу, вызванную возможностью утечки радионуклида из подвергающегося лечению участка. В зависимости от опухоли, подвергающейся лечению, все виды источников излучения рассматривают в рамках объема настоящего изобретения.
[0055] Кроме того, настоящее изобретение охватывает различные типы неионизирующего излучения, например, ультрафиолетовое (УФ) излучение, высокоэнергетическое видимое излучение, микроволновое излучение (при гипертермической терапии), инфракрасное (ИК) излучение и лазеры. В конкретном варианте реализации настоящего изобретения применяют УФ излучение.
[0056] Более конкретно, соединения согласно настоящему изобретению можно использовать в комбинированной терапии. «Комбинированная терапия» включает введение субъекту соединений в комбинации с другими биологически активными ингредиентами (включая, но не ограничиваясь им, второй и отличающийся антинеопластический агент) и с проведением нелекарственной терапии (включая, но не ограничиваясь ими, хирургию или радиационное лечение). Например, соединения согласно настоящему изобретению можно применять в комбинации с другими фармацевтически активными соединениями, предпочтительно с соединениями, которые способны усиливать действие соединений согласно настоящему изобретению. Соединения согласно настоящему изобретению можно вводить одновременно (в составе одного препарата или различных препаратов) или последовательно с другой лекарственной терапией. В целом, комбинированная терапия подразумевает введение двух или более лекарственных средств во время одного цикла или курса терапии.
Таким образом, согласно одному из аспектов настоящего изобретения предложенные соединения можно вводить в комбинации с одним или более отдельными агентами, которые модулируют протеинкиназы, задействованные в различных болезненных состояниях или мишени в нисходящих сигналах. Примеры указанных киназ могут включать, но не ограничиваются ими, серин/треонин-специфические киназы, рецепторные тирозин-специфические киназы и нерецепторные тирозин-специфические киназы. Серин/треонин киназы включают митоген-активируемые протеинкиназы (МАРК), мейоз-специфическую киназу (МЕК), RAF и киназу Aurora. Примеры семейств рецепторных киназ включают рецептор эпидермального фактора роста (РЭФР) (например, HER2/neu, HER3, HER4, ErbB, ErbB2, ErB3, ErbB4, Xmrk, DER, Let23); рецептор фактора роста фибробластов (FGF) (например, FGF-R1,GFF-R2/BEK/CEK3, FGF-R3/CEK2, FGF-R4/TKF, KGF-R); рецептор фактора роста/рассеяния гепатоцитов (HGFR) (например, MET, RON, SEA, SEX); инсулиновый рецептор (например, IGFI-R, PI3K, АКТ, mTor); Eph (например, СЕК5, СЕК8, ЕВК, ЕСК, ЕЕК, ЕНК-1, ЕНК-2, ELK, EPH, ERK, НЕК, MDK2, MDK5, SEK); Axl (например, Mer/Nyk, Rse); RET; и рецептор фактора роста, выделенного из тромбоцитов (PDGFR) (например, PDGFα-R, PDGβ-R, CSF1-R/FMS, SCF-R/C-KIT, VEGF-R/FLT, NEK/FLK1, FLT3/FLK2/STK-1). Семейства нерецепторных протеинкиназ включают, но не ограничиваются ими, BCR-ABL (например, p43abl, ARG); ВТК (например, ITK/EMT, TEC); CSK, FAK, FPS, JAK, SRC, ВМХ, FER, CDK и SYK.
Согласно другому аспекту изобретения предложенные соединения можно вводить в комбинации с одним или более отдельными агентами, которые модулируют некиназные биологические мишени или процессы. Указанные мишени включают деацетилазы гистонов (HDAC), метилтрансферазу ДНК (DNMT), белки теплового шока (например, HSP90), ингибиторы Hedgehog и протеосомы.
В предпочтительном варианте реализации предложенные соединения можно объединять с антинеопластическим агентами (например, с небольшими молекулами, моноклональными антителами, античувствительной РНК и гибридными белками), которые ингибируют одну или более биологических мишеней, такими как эриведж, золинза, тарцева, пресса, тайкерб, гливек, сутент, сприцел, нексавар, CNF2024, RG108, BMS387032, аффинитак, авастин, герцептин, эрбитукс, AG24322, PD325901, ZD6474, PD184322, обатодакс, ABT737 и AEE788. Также включены моноклональные антитела, специфические к киназам и/или рецепторам, например, указанные в настоящей заявке или иные. Указанные комбинации могут повышать терапевтическую эффективность по сравнению с эффективностью, достигаемой при использовании любого из агентов по отдельности, и могут предотвращать или отсрочивать появление резистентных мутантных изменений.
В конкретных предпочтительных вариантах реализации соединения согласно настоящему изобретению вводят в комбинации с химиотерапевтическим агентом. Химиотерапевтические агенты охватывают широкий диапазон способов лечения онкологии. Указанные агенты вводят на различных стадиях заболевания для уменьшения размера опухоли, уничтожения раковых клеток, оставшихся после хирургии, инициирования ремиссии, поддержания ремиссии и/или ослабления симптомов, относящихся к раку или к способу его лечения. Примеры указанных агентов (некоторые из которых также обсуждались выше) включают, но не ограничиваются ими, алкилирующие агенты, такие как производные газообразного иприта (мехлорэтамин, циклофосфамид, хлорамбуцил, мелфалан, ифосфамид), этиленимины (тиотепа, гексаметилмеланин), алкилсульфонаты (бусульфан), гидразины и триазины (алтретамин, прокарбазин, дакарбазин и темозоломид), нитрозомочевины (кармустин, ломустин и стрептозоцин), ифосфамид и соли металлов (карбоплатин, цисплатин и оксалиплатин); растительные алкалоиды, такие как подофиллотоксины (этопозид и тенипозид), таксаны (паклитаксел и доцетаксел), алкалоиды винка (винкристин, винбластин, виндесин и винорелбин) и аналоги камптотекана (иринотекан и топотекан); противоопухолевые антибиотики, такие как хромомицины (дактиномицин и пликамицин), антрациклины (доксорубицин, даунорубицин, эпирубицин, митоксантрон, валрубицин и идарубицин) и вспомогательные антибиотики, такие как митомицин, актиномицин и блеомицин; антиметаболиты, такие как антагонисты фолиевой кислоты (метотрексат, пеметрексед, ралтитрексед, аминоптерин), пиримидиновые антагонисты (5-фторурацил, флоксуридин, цитарабин, капецитабин и гемцитабин), пуриновые антагонисты (6-меркаптопурин и 6-тиогуанин) и ингибиторы аденозиндезаминазы (кладрибин, флударабин, меркаптопурин, клофарабин, тиогуанин, неларабин и пентостатин); ингибиторы топоизомеразы, такие как ингибиторы топоизомеразы I (иринотекан, топотекан) и ингибиторы топоизомеразы II (амсакрин, этопозид, этопозида фосфат, тенипозид); моноклональные антитела (алемтузумаб, гемтузумаб озогамицин, ритуксимаб, трастузумаб, ибритумомаб тиуксетан, цетоксимаб, панитумумаб, тоситумумаб, бевацизумаб); и вспомогательные антинеопластические средства, такие как ингибиторы рибонуклеотидредуктазы (гидроксимочевина); ингибиторы адренокортикальных стероидов (митотан); ферменты (аспарагиназа и пегаспаргаза); антимикротрубочковые агенты (эстрамустин); и ретиноиды (бексаротен, изотретиноин, третиноин (ATRA)) и т.д.
[0057] Изобретение также включает фармацевтические композиции или лекарственные средства, содержащие соединения согласно настоящему изобретению и терапевтически инертный носитель, разбавитель или вспомогательное вещество, а также способы применения соединений согласно настоящему изобретению для получения указанных композиций и лекарственных средств. Как правило, соединения формулы I, применяемые в способах согласно настоящему изобретению, вводят в состав путем перемешивания при температуре окружающей среды и соответствующем pH и при желаемой степени чистоты с физиологически приемлемыми носителями, т.е. с носителями, которые являются нетоксичными для потребителя в дозировках и концентрациях, применяемых в галеновых лекарственных формах. pH состава зависит, главным образом, от конкретного применения и концентрации соединения, но предпочтительно находится в диапазоне от примерно 3 до примерно 8. Подходящим вариантом реализации является состав в ацетатном буфере с pH 5.
[0058] Ингибирующее соединение для применения согласно настоящему изобретению предпочтительно является стабильным. Как правило, соединение хранят в виде твердой композиции, хотя лиофилизированные составы или водные растворы также являются приемлемыми.
[0059] Композиции согласно настоящему изобретению получают, дозируют и вводят в соответствии с надлежащей медицинской практикой. Важные в этом контексте факторы включают конкретное нарушение, подвергающееся лечению, конкретное млекопитающее, подвергающееся лечению, клиническое состояние конкретного пациента, причину нарушения, участок доставки агента, способ введения, схему введения и другие факторы, известные практикующим врачам. «Эффективное количество» вводимого соединения зависит от указанных факторов и представляет собой минимальное количество, необходимое для лечения указанных заболеваний, например, для ингибирования взаимодействия IAP с каспазами, инициирования апоптоза или увеличения чувствительности злокачественных клеток к апоптотическому сигналу. Указанное количество предпочтительно является более низким по сравнению с количеством, которое является токсичными для нормальных клеток или для млекопитающего в целом.
[0060] В целом, соединения согласно настоящему изобретению можно вводить один или несколько раз в день. Также их можно вводить непрерывно в течение дня без перерывов во введении. Таким образом, их также можно вводить и с перерывами. Указанные варианты также являются доступными и в случае применения совместно с другими агентами или согласно другой схеме. Первоначальное фармацевтически эффективное количество соединения согласно настоящему изобретению во вводимой парентерально дозе находится в диапазоне примерно 0,01-100 мг/кг, предпочтительно примерно от 0,1 до 20 мг/кг массы тела пациента в день, при этом обычно первоначальный диапазон количества вводимого соединения составляет от 0,3 до 15 мг/кг/день. Пероральные стандартные лекарственные формы, такие как таблетки и капсулы, предпочтительно содержат от примерно 25 до примерно 1000 мг соединения согласно настоящему изобретению. Пероральное введение является предпочтительным. Можно использовать схемы введения с интервалами (режим on-off), которые являются традиционными для лечения рака, например, ежедневное пероральное введение в течение 1, 2, 3, 4 и т.д. недель, последующий перерыв в лечении в течение 1, 2 и т.д. недель, и снова ежедневное пероральное введение в течение 1, 2, 3, 4 и т.д. недель и т.д. В предпочтительном варианте реализации соединения согласно настоящему изобретению вводят ежедневно в диапазоне от примерно 25 до примерно 3000 мг в день, более предпочтительно от примерно 300 до примерно 1500 мг соединения в день.
[0061] Соединение согласно настоящему изобретению можно вводить при помощи любых подходящих способов, включая пероральное, местное, трансдермальное, парентеральное, подкожное, интраперитонеальное, внутрилегочное и интраназальное, а также при желании в случае местного лечения внутриочаговое введение. Парентеральные инфузии включают внутримышечное, внутривенное, внутриартериальное, интраперитонеальное или подкожное введение. Примером подходящей пероральной лекарственной формы является таблетка, содержащая примерно 25 мг, 50 мг, 100 мг, 250 мг или 500 мг соединения согласно настоящему изобретению совместно с примерно 90-30 мг безводной лактозы, примерно 5-40 мг кроскармеллозы натрия, примерно 5-30 мг поливинилпирролидона (ПВП) K30 и примерно 1-10 мг стеарата магния. Сначала перемешивают друг с другом порошковые ингредиенты, а затем их перемешивают с раствором ПВП. Полученную композицию можно сушить, гранулировать, смешивать со стеаратом магния и прессовать в таблетку при помощи стандартного оборудования. Состав в виде аэрозоля можно получать путем растворения, например, 5-400 мг соединения согласно настоящему изобретению в подходящем буферном растворе, например, в фосфатном буфере, и добавления агента, изменяющего тоничность, например, соли, такой как хлорид натрия, при желании. Для удаления примесей и загрязняющих веществ раствор, как правило, фильтруют, например, на фильтре 0,2 микрон.
[0062] Более конкретно, соединения согласно настоящему изобретению можно применять согласно описанию, приведенному в международной заявке на патент WO 98/46576 и патенте США №7244851, и приведенные в них общие рекомендации по применению соединений включены в настоящую заявку посредством ссылок.
ПРИМЕРЫ
[0063] Изобретение станет более понятным при рассмотрении следующих примеров. Тем не менее, их не следует рассматривать как ограничивающие объем изобретения. Далее приведены сокращения, используемые в настоящем описании:
[0064] ACN: ацетонитрил;
[0065] Chg: циклогексилглицин;
[0066] ДХМ: дихлорметан;
[0067] DLPEA: диизопропилэтиламин;
[0068] DMAP: 4-диметиламинопиридин;
[0069] ДМЭ: 1,2-диметоксиэтан;
[0070] ДМФ: диметилформамид;
[0071] ДМСО: диметилсульфоксид;
[0072] EDC: 1-этил-3-(3-диметиламинопропил)карбодиимид;
[0073] EEDQ: 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин;
[0074] ЖХМС: жидкостная хроматография-масс-спектрометрия;
[0075] HATU: гексафторфосфат O-(7-азобензотриазол-1-ил)-1,1,3,3-тетраметилурония;
[0076] HOBt: N-гидроксибензотриазол;
[0077] HBTU: гексафторфосфат 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония;
[0078] ВЭЖХ: высокоэффективная жидкостная хроматография;
[0079] NBS: N-бромсукцинимид;
[0080] TASF: дифтортриметилсиликат трис(диметиламино)сульфония;
[0081] ТЭА: триэтиламин;
[0082] ТФА: трифторацетат;
[0083] ТТФ: тетрагидрофуран;
[0084] Пример сравнения 1
[0085] (2-фенил-2Н-пиразол-3-ил)амид 1-[2-циклогексил-2-(2-метиламинопропиониламино)ацетил]пирролидин-2-карбоновой кислоты

[0086] Раствор Boc-MeAla-Chg-Pro-OH (47,0 мг, 0,107 ммоль) и пиридина (26 мкл, 0,32 ммоль) в безводном дихлорметане (300 мкл) охлаждали до 0°C и по каплям добавляли раствор оксалилхлорида в дихлорметане (54 мкл, 2,0 М, 0,11 ммоль) в течение 10 минут. Смесь перемешивали при 0°C в течение 15 минут, затем при температуре окружающей среды в течение 45 минут и добавляли раствор 5-амино-1-фенилпиразола (15,9 мг, 0,100 ммоль; № в каталоге TCI America А0174) и пиридина (15,5 мкл, 0,191 ммоль) в дихлорметане (0,5 мл). Полученную смесь перемешивали при температуре окружающей среды в течение 16 часов, разбавляли дихлорметаном до объема 20 мл и промывали 0,2н. водным гидроксидом натрия (20 мл). Органическую фазу сушили (MgSO4) и концентрировали при пониженном давлении. Неочищенный продукт очищали путем колоночной хроматографии (силикагель, 60% этилацетат в гексанах, затем 100% этилацетат) с получением желтой маслянистой жидкости: m/z 581 (M+H+). Маслянистую жидкость обрабатывали 5% трифторуксусной кислотой в дихлорметане (2 мл), через 18 часов растворитель удаляли в вакууме. Полученную маслянистую жидкость (29,3 мг, выход 57% за 2 стадии) дополнительно очищали путем обращенно-фазовой ВЭЖХ с получением продукта (ТФА соль, 9,6 мг, выход 15%): m/z 481 (M+H+), 503 (M+Na+).
Пример сравнения 2
[0087] Способ сочетания с фторангидридом

[0088] Раствор Boc-MeAla-Chg-Pro-OH (2,3 ммоль) и пиридина (6,9 мкмоль) в безводном ДХМ (23 мл) охлаждали до 0°C и по каплям добавляли фторангидрид циануровой кислоты (2,3 ммоль) в течение 30 секунд. Смесь перемешивали при 0°C в течение 15 минут, при КТ в течение 5 часов, а затем реакцию гасили водой. Смесь трижды экстрагировали ДХМ (общий объем 100 мл), объединенные органические фазы промывали солевым раствором и сушили (Na2O4). Фильтрование и концентрирование в вакууме приводили к получению фторангидрида пептида в виде прозрачной бесцветной маслянистой жидкости, которую использовали непосредственно без дополнительной очистки.
[0089] Раствор неочищенного фторангидрида (0,50 ммоль) и пиридина (1,5 ммоль) в ДХМ (2,5 мл) добавляли в твердый амин (14,0,50 ммоль), полученную смесь перемешивали при КТ или 50°C (в герметичном сосуде). Смесь выливали в водный NaHCO3, а затем трижды экстрагировали дихлорметаном (общий объем 100 мл). Объединенные органические фазы промывали солевым раствором, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Неочищенный амид пептида использовали непосредственно без дополнительной очистки.
[0090] Пример сравнения 3
[0091] (4-фенил-[1,2,3]тиадиазол-5-ил)амид 1-[2-циклогексил-2-(2-метиламинопропиониламино)ацетил]пирролидин-2-карбоновой кислоты

[0092] Стадия 1: В раствор Boc-L-Pro-OH (2 экв.), HOBt (1,9 экв.), EDC-HCl (1,9 экв.) и DLPEA (5 экв.) в ДМФ (10-15 об.) добавляли 4-фенил-1,2,3-тиадиазол-5-амин (16). Реакция сначала проходила с умеренным выделением тепла, смесь нагревали до 75°C и перемешивали в течение ночи, охлаждали до КТ, ДМФ частично удаляли в вакууме. Раствор разбавляли EtOAc (10-15 об.), после чего промывали 1М HCl (2х), NaHCO3 (1x) и солевым раствором (1x) (1:1 водн./орг.). Органический слой концентрировали в вакууме и полученное твердое вещество суспендировали в кипящем MeCN (в минимальном объеме, необходимом для простоты перемешивания) в течение 30 минут, а затем охлаждали до КТ. Вакуумное фильтрование приводило к получению Boc-защищенного сопряженного продукта в виде беловатого кристаллического твердого вещества с примерно 77% выходом. Boc-защищенный продукт суспендировали в 4М растворе HCl/диоксан (4-5 экв. кислоты) и MeCN (1 об. экв. относительно раствора в диоксане) и перемешивали при КТ до тех пор, пока при помощи анализа ЖХМС не подтверждали полное снятие защиты (примерно 1 час). Реакционную смесь концентрировали в вакууме и полученное твердое вещество интенсивно суспендировали в кипящем MeCN (в минимальном объеме, необходимом для простоты перемешивания), охлаждали до КТ, твердое вещество собирали путем вакуумного фильтрования и промывали холодным MeCN до исчезновения окраски осадка с получением соли НС1 и (S)-N-(4-фенил-1,2,3-тиадиазолил-5-ил)пирролидин-2-карбоксамида (17) в виде беловатого твердого вещества примерно с количественным выходом.
[0093] Стадия 2: В раствор 17 и DLPEA (5 экв.) в ДМФ (10-15 об.) добавляли Boc-L-Chg (1,5 экв.), HOBt (1,4 экв.) и EDC-HCl (1,4 экв.). Реакционную смесь перемешивали в течение примерно 2 часов, затем разбавляли EtOAc (15 об.) и промывали 1М HCl (2х), NaHCO3 (1х) и солевым раствором (1х) (1:1 водн/орг). Органический экстракт сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученное твердое вещество суспендировали в смеси EtOH/гексан (20:80) (в минимальном объеме, необходимом для простоты перемешивания) и фильтровали с получением Boc-защищенного сопряженного продукта в виде белого рыхлого твердого вещества с примерно 80% выходом. Boc-защищенное вещество растворяли в 4М растворе HCl/диоксан (4-5 экв. кислоты) и MeCN (0,25 об. экв. относительно раствора в диоксане) и перемешивали при КТ в течение примерно 1 часа. Реакционную смесь концентрировали с толуолом (2х) досуха (в таком же объеме, что и раствор, который применяли для снятия защиты) с получением соли HCl и (S)-1-((S)-2-амино-2-циклогексил ацетил)-N-(4-фенил-1,2,3-тиадиазол-5-ил)пирролидин-2-карбоксамида (18) в виде белого кристаллического твердого вещества примерно с количественным выходом.
[0094] Стадия 3: В раствор 18 и DLPEA (5 экв.) в ДМФ (10-15 об.) добавляли Boc-L-N-метил-Ala (1,5 экв.), HOBt (1,4 экв.) и EDC-HCl (1,4 экв.). Реакционную смесь перемешивали в течение 1 часа, разбавляли EtOAc (15 об.) и промывали 1М HCl (2х), NaHCO3 (1х) и солевым раствором (1х) (1:1 водн/орг). Органический экстракт сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением Boc-защищенного сопряженного продукта в виде бежевой твердой пены примерно с 85% выходом. Boc-защищенный продукт растворяли в 4М растворе HCl/диоксан (4-5 экв. кислоты) и MeCN (0,25 об. экв. относительно раствора в диоксане) с перемешивали при КТ в течение примерно 1 часа. Реакционную смесь концентрировали с толуолом (2х) досуха (в таком же объеме, что и раствор, который применяли для снятия защиты), полученное твердое вещество суспендировали в растворе МТБЭ/EtOAc (70:30) (в минимальном объеме, необходимом для простоты перемешивания), фильтровали и собирали с получением неочищенного (S)-1-((S)-2-циклогексил-2-((S)-2-(метиламино)пропанамидо)ацетил)-N-(4-фенил-1,2,3-тиадиазол-5-ил)ггарролидин-2-карбоксамида (d) в виде беловатого свободно текучего твердого вещества. Неочищенную соль HCl суспендировали в MeOH (4 об., минимальное количество) и растворяли при перемешивании при 65°C. Теплый изопропилацетат (6-8 об.) добавляли двумя частями, поддерживая температуру примерно 60°C, затем раствор оставляли охлаждаться при перемешивании. Быстро происходила кристаллизация, суспензию перемешивали при КТ в течение нескольких часов, затем перемешивали при 0°C в течение еще часа, после чего твердое вещество собирали путем вакуумного фильтрования. Неочищенный продукт промывали MeOH/iPrOAc (1:4, 2 об.) и сушили с получением 19 в виде белого/беловатого кристаллического твердого вещества примерно с 80% выходом.
[0095] Пример сравнения 4
[0096] 5-амино-2-(оксазол-2-ил)-4-фенилтиазол
[0097] Суспензию гидрохлорида а-аминофенилацетонитрила (1,52 г, 8,99 ммоль), порошковой серы (289 мг, 9,01 ммоль) и оксазол-2-карбальдегида (873 мг, 8,99 ммоль) в ЕЮН (18 мл) обрабатывали ТЭА (1,88 мл, 13,5 ммоль) и смесь перемешивали при 50°C в течение 60 минут. Охлажденную смесь обрабатывали водным гидроксиламином (1,00 мл, 50 масс. %, 15 ммоль) при КТ в течение ночи, фильтровали и концентрировали в вакууме. Остаток разделяли в EtOAc и водном NaHCO3, выделенную органическую фазу промывали солевым раствором, сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением темно-коричневой маслянистой жидкости. Неочищенную маслянистую жидкость предварительно наносили в SiO2 и очищали путем автоматизированной флэш-хроматографии, элюируя с градиентом 5-70% смесями этилацетата в гексанах, с получением 5-амино-2-(оксазол-2-ил)-4-фенилтиазола (20, 159 мг, 7,3%).
Пример 1
[0098] (2-оксазол-2-ил-4-фенилтиазол-5-ил)амид (S)-1-[(S)-2-циклогексил-2-((S)-2-метиламинопропиониламино)ацетил]пирролидин-2-карбоновой кислоты (Ia)
[0099] Стадия 1: В раствор Boc-L-пролина (4,5 г, 0,02 ммоль) и пиридина (8,45 мл, 0,104 ммоль) в ДХМ (20 мл), охлажденный на ледяной бане, по каплям добавляли фторангидрид циануровой кислоты (5,35 мл, 0,0627 ммоль). После завершения добавления реакционная смесь приобретала молочно-белый цвет. Раствор перемешивали при 0°C в течение 10 минут, затем нагревали до КТ и перемешивали в течение 4 часов. Реакцию гасили водой и смесь трижды экстрагировали ДХМ. Объединенные органические экстракты промывали солевым раствором, сушили и концентрировали в вакууме с получением трет-бутил-2-(фторкарбонил)пирролидин-1-карбоксилата. Неочищенный фторангидрид немедленно использовали в следующей реакции сочетания.
[00100] Свежеполученный фторангидрид кислоты (4,55 г, 0,02 ммоль) растворяли в MeCN (20 мл) и обрабатывали 20 (1,7 г, 0,007 ммоль) и пиридином (2,82 мл, 0,035 ммоль). Реакционную смесь нагревали до 50°C в течение ночи. Реакцию гасили нас. NaHCO3 и трижды экстрагировали EtOAc. Объединенные органические слои промывали солевым раствором, сушили и концентрировали. Неочищенный продукт очищали путем хроматографии на SiO2, элюируя с градиентом смесями EtOAc/гексан (от 50 до 80% EtOAc), с получением (S)-трет-бутил-2-(2-(оксазол-2-ил)-4-фенилтиазол-5-илкарбамоил)-пирролидин-1-карбоксилата (21).
[00101] Удаление Boc-защитной группы и последующее сочетание с BocNH-Chg-OH и H(Me)N-Ala-OH проводили согласно способам, описанным для стадий 2 и 3 в примере сравнения 3. Очистку продукта сочетания Chg проводили путем хроматографии SiO2, элюируя с градиентом смесями EtOAc/гексан (от 50 до 80% EtOAc). Неочищенный Boc-защищенный трипептидный продукт очищали путем ISCO (50-80% EtOAc/гексан). После удаления Boc-группы конечный продукт очищали путем препаративной ВЭЖХ с получением чистого Ia: (М+Н)+ = мс 565,3.
Пример 2
[00102] Другие соединения согласно настоящему изобретению, например,
[00103] 1. (2-оксазол-2-ил-4-фенилтиазол-5-ил)амид (S)-1-[(S)-2-циклопропил-2-((S)-2-метиламинопропиониламино)ацетил]пирролидин-2-карбоновой кислоты;
[00104] 2. (2-оксазол-2-ил-4-фенилтиазол-5-ил)амид (S)-1-[(S)-2-циклопентил-2-((S)-2-метиламинопропиониламино)ацетил]пирролидин-2-карбоновой кислоты;
[00105] 3. (2-оксазол-2-ил-4-фенилтиазол-5-ил)амид (S)-1-[(S)-2-циклогептил-2-((S)-2-метиламинопропиониламино)ацетил]пирролидин-2-карбоновой кислоты;
[00106] 4. (2-оксазол-2-ил-4-фенилтиазол-5-ил)амид (S)-1-[(S)-2-циклогексил-2-((S)-2-этиламинопропиониламино)ацетил]пирролидин-2-карбоновой кислоты;
[00107] 5. (2-оксазол-2-ил-4-фенилтиазол-5-ил)-N-метиламид (S)-1-[(S)-2-циклогексил-2-((S)-2-метиламинопропиониламино)ацетил]пирролидин-2-карбоновой кислоты; и
[00108] 6. (2-оксазол-2-ил-4-фенилтиазол-5-ил)амид (S)-1-[(S)-2-циклогексил-2-((S)-2-метиламинопропиониламино)-2-метилацетил]пирролидин-2-карбоновой кислоты,
[00109] можно получать аналогично соединению согласно примеру 1 с использованием соответствующих аналогичных исходных веществ в соответствии со способом согласно примеру 1.
Пример 3
[00110] Исследования ингибирования IAP
[00111] В следующих экспериментах использовали химерный домен BIR, называемый MLXBIR3SG, в котором 11 из 110 остатков соответствовали тем, что содержатся в XIAP-BIR3, а остаток соответствовал ML-IAP-BIR. Было показано, что химерный белок MLXBIR3SG значительно лучше связывает и ингибирует каспазу 9 по сравнению с естественными доменами BIR, при этом аффинность связывания с пептидами на основе Smac и «созревшим» Smax схожа с показателем для естественного ML-IAP-BIR. Улучшенное ингибирование каспазы-9 химерным доменом BIR в MLXBIR3SG связывают с улучшенным ингибированием апоптоза, вызванного доксорубицином, при трансфицировании клеток MCF7.
[00112] Последовательность MLXBIR3SG:
[00113]



[00114] Исследование пептидного связывания РПЭФ-ВР
[00115] Проводили конкурентные эксперименты в рамках исследования резонансного переноса энергии флуоресценции с временным разрешением на многоцелевом анализаторе Wallac VictoR2 (Perkin Elmer Life and Analytical Sciences, Inc.) согласно способам, описанным в Kolb et al (Journal of Biomolecular Screening, 1996, 1(4): 203). Готовили коктейль реагентов, содержащий 300 нМ his-меченый MLXBIR3SG; 200 нМ биотинилированный пептид SMAC (AVPI); 5 мкг/мл анти-his аллофикоцианин (XL665) (CISBio International); и 200 нг/мл стрептавидин-европий (Perkin Elmer), в буфере для реагентов (50 мМ Tris [pH 7,2], 120 нМ NaCl, 0,1% бычьих глобулинов, 5 мМ DTT и 0,05% октилглюкозид). (В качестве альтернативы указанный коктейль можно получали с использованием меченого европием анти-His (Perkin Elmer) и стрептавидин-аллофикоцианина (Perkin Elmer) в концентрациях 6,5 нМ и 25 нМ, соответственно). Коктейль реагентов инкубировали при комнатной температуре в течение 30 минут. После завершения инкубации коктейль добавляли к соединению-антагонисту в концентрации, полученной путем последовательных разбавлений 1:3 (от начальной концентрации 50 мкМ), в 384-луночные черные планшеты FIA (Greiner Bio-One, Inc.). После 90-минутной инкубации при комнатной температуре анализировали флуоресценцию с использованием фильтров для длин волн возбуждения европия (340 нм) и испускания европия (615 нм) и аллофикоцианина (665 нм). Данные антагонистов рассчитывали как отношение сигнала испускания аллофикоцианина при 665 нм к сигналу испускания европия при 615 нм (для упрощения обработки данных значение этого отношения умножали на 10000). Строили графики зависимости полученных значений от концентрации антагониста, и полученные значения подставляли в 4-параметровое уравнение с использованием программного обеспечения Kaleidograph (Synergy Software, Reading, PA). Данные активности антагонистов определяли по значениям IC50. Было обнаружено, что соединения согласно настоящему изобретению имели ингибирующую активность в отношении IPA, что продемонстрировали в указанном исследовании.
[00116] Поляризационное флуоресцентное исследование связывания пептидов
[00117] Эксперименты с использованием поляризации проводили на Analyst НТ 96-384 (Molecular Devices Corp.) согласно способу, приведенному в Keating, S.M., Marsters, J, Beresini, M., Ladner, С., Zioncheck, К., Clark, К., Arellano, F., and Bodary., S.(2000), Proceedings of SPIE: In Vitro Diagnostic mstrumentation (Conn, G.E., Ed.) pp 128-137, Bellingham, WA. Образцы для определения аффинности по поляризации флуоресценции получали путем добавления MLXBIR3SG в конечной концентрации, полученной путем последовательного разбавления 1:2, начиная с 5 мкМ, в поляризационном буфере (50 мМ Tris [pH 7,2], 120 мМ NaCl, 1% бычьих глобулинов, 5 мМ DTT и 0,05% октилглюкозид) в 5-карбоксифлуоресцеин-сопряженный AVPdi-Phe-NH2 (AVP-diPhe-FAM) в конечной концентрации 5 нМ.

[00118] Реакционные смеси анализировали после 10-минутной инкубации при комнатной температуре с использованием стандартных ограничивающих фильтров для фторофора флуоресцеина (λвозб=485 НМ; λисп=530 нм) в 96-луночных черных планшетах НЕ96 (Molecular Devices Corp.). Строили графики зависимости значений флуоресценции от концентрации белка, значения IC50 получали путем подстановки данных в 4-параметровое уравнение с использованием программного обеспечения Kaleidograph (Synergy Software, Reading, PA). Проводили конкурентные эксперименты путем добавления 30 нМ MLXBIR3SG в лунки, содержащие 5 нМ зонд AVP-diPhe-FAM, а также соединения-антагонисты в концентрации, полученной путем последовательного разбавления 1:3, начиная с 300 мкМ концентрации, в поляризационном буфере. Образцы анализировали после 10-минутной инкубации. Строили графики зависимости значений поляризации флуоресценции от концентрации антагониста, значения IC50 получали путем подстановки данных в 4-параметровое уравнение с использованием программного обеспечения Kaleidograph (Synergy Software, Reading, PA). Константы ингибирования (Ki) антагонистов определяли по значениям IC50. Было обнаружено, что соединения согласно настоящему изобретению имеют ингибирующую активность в отношении IAP, что продемонстрировали в указанном исследовании. Например, значение Ki для соединения Ia составляло 0,014 (ML-IAP-BIR).
[00119] Пример 4
[00120] Исследования ксенотрансплантатов опухолей (фиг. 1 и 2)
[00121] Все процедуры с применением животных проводили согласно рекомендациям институционального комитета по содержанию и использованию животных компании Genentech. Раковые клетки, такие как клетки рака груди человека MDA-MB-231, колоректального рака Colo205, или НМРЛ Calu6, получали в Американской коллекции типовых культур (Manassas, VA). Клетки повторно суспендировали в HBSS (Colo205), или же клеточную суспензию перемешивали в отношении 1:1 с матригелем (BD Biosciences; MDA-MB-231, Calu-6). Затем клетки (1,5×107 в случае MDA-MB-231; 5,0×107 в случае Colo205, Calu6) подкожно имплантировали в правый бок самок бестимусных мышей (Charles River Laboratories, Hollister, CA) возрастом 6-8 недель. Рассчитывали объем опухолей с использованием значений среднего диаметра, измеренных при помощи штангенциркуля, по формуле ν=0,5×a×b2, где a и b представляют собой наибольший и наименьший взаимно перпендикулярные диаметры опухоли, соответственно. По десять мышей с соответствующим средним объемом опухоли случайным образом распределяли в каждую из шести групп. На момент начала исследования (день 0) средний объем опухоли ± стандартная ошибка среднего (COC) для всех шести групп составлял 168±3 мм3. Мышей наблюдали каждый день во время исследования, объем опухолей и массу тела измеряли два раза в неделю. Подавление роста опухоли в процентах рассчитывали по формуле %TGI = 100 × (1 - объем опухолидоза/объем опухолиноситель).
[00122] В этом исследовании с использованием клеток рака груди человека

[00123] Отличительные признаки, описанные в приведенном выше описании или в последующей формуле изобретения, выраженные конкретно или в виде способов реализации предложенной функции или способа или процесса достижения предложенного результата, можно по отдельности или в качестве какой-либо комбинации указанных признаков применять для реализации настоящего изобретения в его разнообразных формах.
[00124] Для ясности и простоты понимания предложенное выше изобретение было подробно описано при помощи иллюстраций и примеров. Специалистам в данной области техники должно быть очевидным, что в рамках объема прилагаемой формулы изобретения можно реализовывать изменения и модификации. Таким образом, следует понимать, что приведенное выше описание является иллюстративным и неограничивающим. Таким образом, объем изобретения должен определяться не приведенным выше описанием, но последующей прилагаемой формулой изобретения, а также полным объемом эквивалентов, возможных для указанной формулы изобретения.
[00125] Патенты, опубликованные заявки на патент и научная литература, ссылки на которые приведены в настоящем описании, определяют уровень знаний специалистов в данной области техники и включены в настоящую заявку во всей полноте посредством ссылок в той же степени, как и в случае если бы для каждой отдельной работы было конкретно указано, что она включена посредством ссылки. Любые противоречия между какой-либо ссылкой, приведенной в настоящем описании, и конкретными сведениями, приведенными в настоящем описании, следует разрешать в пользу последних. Аналогично, любые противоречия между определением слова или фразы, подразумеваемым в уровне техники, и конкретным определением слова или фразы, приведенным в настоящем описании, следует разрешать в пользу последнего.
Реферат
Изобретение относится к способу лечения заболевания или состояния, связанного с избыточной экспрессией IAP, путем инициирования апоптоза и/или увеличения чувствительности указанной раковой клетки к апоптозу, введением млекопитающему ингибитора IAP общей формулы (I). 5 н. и 13 з.п. ф-лы, 2 ил., 4 пр.
Формула

Документы, цитированные в отчёте о поиске
Ингибиторы iap
Комментарии