Трициклические азотсодержащие производные имидазо[4,5-с]пиридина, обладающие ингибирующей активностью в отношении рецептора гистамина 4 (hh4r) - RU2628074C2
Код документа: RU2628074C2
Описание
Область техники
Настоящее изобретение относится к новым гетероциклическим соединениям, которые могут быть использованы в получении лекарственных средств для лечения заболеваний, связанных с различными функциями рецептора гистамина 4. Особенно указанные лекарственные средства могут быть использованы для лечения воспалительных заболеваний, аллергии, боли, носовых полипов, ринита, хронического синусита, заложенности носа, назального зуда, астмы, хронического обструктивного заболевания легких, ревматоидного артрита, атопического дерматита, псориаза, экземы, зуда, кожного зуда, крапивницы, идиопатической хронической крапивницы, склеродермии, конъюнктивита, кератоконъюнктивита, глазного воспаления, сухости глаз, кардиальной дисфункции, аритмии, атеросклероза, рассеянного склероза, воспалительного заболевания кишечника (включая колит, болезнь Крона, неспецифический язвенный колит), воспалительной боли, нейропатической боли, боли при остеоартрите, аутоиммунного заболевания щитовидной железы, иммунно-опосредованного (также известного как тип 1) диабета, волчанки, послеоперационных спаек, вестибулярных нарушений и рака.
Уровень техники
Гистамин, который является биогенным амином, играет центральную роль в иммунном и воспалительном ответе и также является нейромедиатором. Например, гистамин контролирует различные функции антигенпредставляющих клеток (дендритные клетки и макрофаги), Т-клеток, В-клеток, эпителиальных и эндотелиальных клеток и пролиферацию Т-клеток или секрецию цитокина в дендритных клетках и тучных клетках (JDDG, 2010, 8, 495-504). Существуют 4 рецептора гистамина (рецептор гистамина 1, рецептор гистамина 2, рецептор гистамина 3 и рецептор гистамина 4) (Br. J. Pharm 2006, 147, S127-S135). Острая аллергическая реакция находится под контролем рецептора гистамина 1, который распределен в теле равномерно (Br. J. Pharmac. Chemother. 1966, 27, 427-439), а секреция кислоты желудка находится под контролем рецептора гистамина 2, который также распределен в теле равномерно, как рецептор гистамина 1 (Nature 1972, 236, 385-390). Известно, что секреция нейромедиаторов в центральной нервной системе находится под контролем рецептора гистамина 3, который экспрессируется в нейронах (Nature 1983, 302, 832-837). Рецептор гистамина 4 дополнительно объясняет физиологические функции многих процессов трансдукции сигналов, которые не объясняются только рецептором гистамина 1, рецептором гистамина 2 и рецептором гистамина 3. О рецепторе гистамина 4 впервые сообщалось в 1994 году, и его клонирование было осуществлено только в 2000-х годах. Рецептор гистамина 4, который представляет собой рецептор, связанный с G-белком, состоит из 390 аминокислот и активируется при связывании с белком Gi/o, увеличивая концентрацию кальция или подавляя циклический аденозин монофосфат (цАМФ) (The Open Immunology Journal, 2009, 2, 9-41). Рецептор гистамина 4 главным образом экспрессируется в костном мозге или эозинофилах, базофилах, Т-клетках, тучных клетках, моноцитах и дендритных клетках и также наблюдается в селезенке, тимусе, легком, сердце и кишечнике (Nat. Rev. Drug Discov. 2008, 7, 41-53; Biochem. Biophys. Res. Commun. 2000, 279, 615-620). Рецептор гистамина 4 не только играет центральную роль в иммунном ответе, но также оказывает эффекты на активацию и миграцию различных иммуноцитов и продукцию цитокинов и хемокинов (J. Immunol. 2005, 174, 5224-5232; J. Pharmacol. Exp. Ther. 2003, 305, 1212-1221; J. Allergy Clin. Immnol. 2007, 120, 300-307; J. Recept. Signal Transduct. Res. 2002, 22, 431-448).
Известно, что в различных экспериментах in vivo рецептор гистамина 4 играет важную роль при воспалении и зуде (J. Allergy Clin. Immnol. 2007, 119, 176-183; J. Pharmacol. Exp. Ther. 2004, 309, 404-413). Особенно, как результаты исследований, было обнаружено в модели аллергической астмы на мышах, что антагонисты рецептора гистамина 4 облегчают воспаление легкого, контролируя реакцию Th2 (Т хелпер типа 2), и было подтверждено, что антагонисты рецептора гистамина 4 эффективно подавляют вызванный гистамином зуд. Такой двойной эффект против аллергического воспаления и зуда является основанием для того факта, что рецептор гистамина 4 может быть хорошей мишенью для лечения аллергических кожных заболеваний, таких как атопический дерматит (J. Invest. Dermatol. 2010, 130 (4), 1023-1033).
В таком иммуноците антагонизм против различных функций рецептора гистамина 4 представляет собой ключевой центр исследования воспалительных заболеваний, зуда, боли, аллергического ринита, астмы, ревматоидного артрита, атопического дерматита, идиопатической хронической крапивницы, воспалительной боли, нейропатической боли и боли при остеоартрите. Кроме того, недавно были анонсированы исследования, связанные с эффективностью рецептора гистамина 4 против рака, и таким образом, ожидается его развитие в качестве противоракового лекарственного средства.
Недавно в WO 2010/030785 сообщалось, что производные на основе хиноксалина показывают активность на рецепторе гистамина 4. Однако они не показывали достаточную фармакологическую активность in vivo на модели животных, потому что их растворимость и метаболическая стабильность являются невысокими.
Раскрытие изобретения
Техническая задача
Гетероциклические соединения согласно настоящему изобретению, включая пиридопиразин, пиридопиримидин и нафтиридин, показывают такую же или более высокую ингибирующую активность в отношении рецептора гистамина 4 по сравнению с обычными ингибиторами человеческого рецептора гистамина 4 (hH4R), такими как раскрытые в WO 2010/030785; показывают селективность для каждого из подтипов рецепторов гистамина и рецепторов, транспортеров и ионных каналов на мембране; имеют более высокую растворимость, метаболическую стабильность и, соответственно, эффективные фармакокинетические свойства для использования для лечения с более низкой дозировкой и меньшей частотой введения; показывают подавляющий эффект против вызванной гистамином инфильтрации воспалительных клеток, таких как тучные клетки и эозинофилы, и таким образом имеют выраженные противовоспалительные и противозудные эффекты в модели атопического дерматита; и имеют селективность в отношении рецептора серотонина 3, предотвращая побочные эффекты, такие как диарея или запор (Clinical and Experimental Immunology, 2010, 161, 19-27; Pharmacology & Therapeutics, 2010, 128, 146-169), из-за высокого структурного подобия между лигандами рецептора гистамина 4 (hH4R) и лигандами рецептора серотонина 3 (BMCL, 2011, 21, 5460-5464). Таким образом, цель настоящего изобретения состоит в том, чтобы получить такие новые гетероциклические соединения и содержащие их фармацевтические композиции.
Поскольку новые гетероциклические соединения согласно настоящему изобретению и содержащие их фармацевтические композиции проявляют сильную ингибирующую активность в отношении человеческого рецептора гистамина 4 (hH4R), они могут быть использованы в лечении или профилактике воспалительных заболеваний, аллергии, боли, носовых полипов, ринита, хронического синусита, заложенности носа, носового зуда, астмы, хронического обструктивного заболевания легких, ревматоидного артрита, атопического дерматита, псориаза, экземы, зуда, кожного зуда, крапивницы, идиопатической хронической крапивницы, склеродермии, конъюнктивита, кератоконъюнктивита, глазного воспаления, сухости глаз, кардиальной дисфункции, аритмии, атеросклероза, рассеянного склероза, воспалительного заболевания кишечника (включая колит, болезнь Крона, неспецифический язвенный колит), воспалительной боли, нейропатической боли, боли при остеоартрите, аутоиммунного заболевания щитовидной железы, иммуно-опосредованного (также известного как тип 1) диабета, волчанки, послеоперационных спаек, вестибулярных нарушений и рака.
Решение задачи
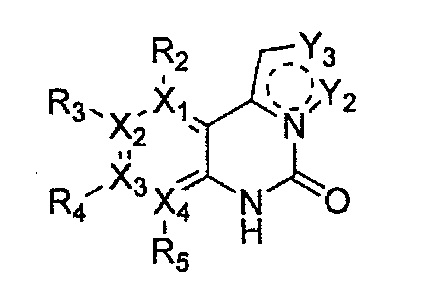
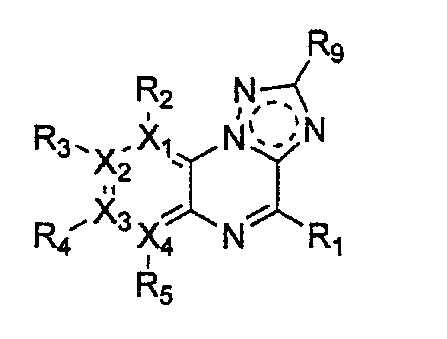
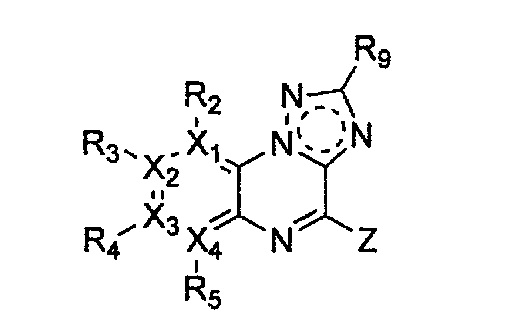
Настоящее изобретение относится к гетероциклическому соединению формулы 1
Формула 1
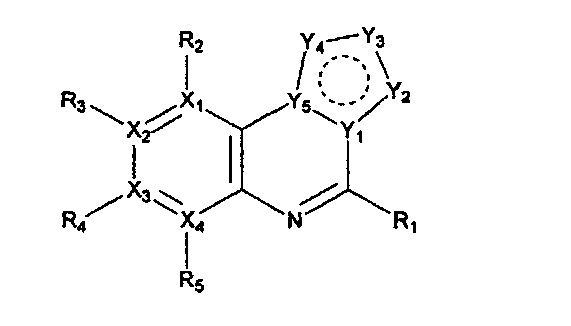
или его рацемату, изомеру или фармацевтически приемлемой соли:
в которой
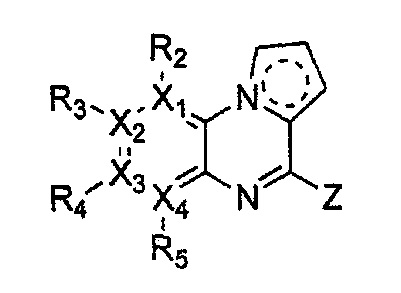
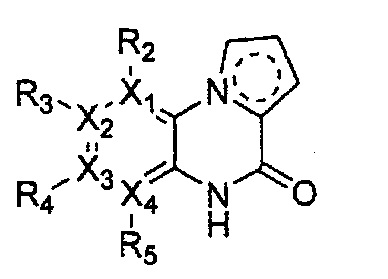
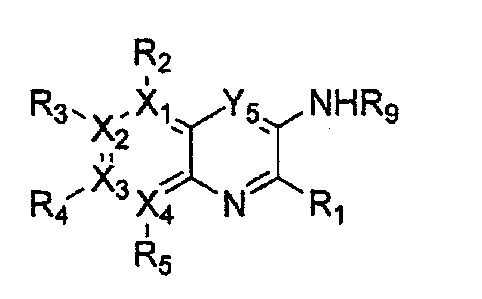
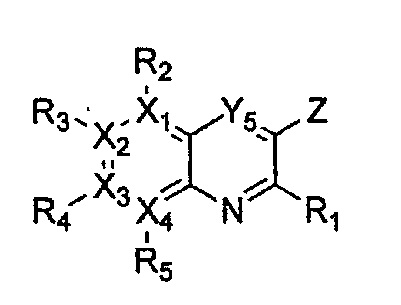
каждый из X1, X2, Х3 и X4 независимо обозначает C или N, при условии, что по меньшей мере один из X1, X2, Х3 и X4 обозначает N,
R1 обозначает насыщенный или ненасыщенный 3-12-членный моно- или поли-гетероциклил, содержащий 1-3 гетероатома (предпочтительно гетероатомы, выбранные из N, О и S), где R1 незамещен или замещен 1-3 заместителями, выбранными из -NR6R7, -C1-C6aлкил-NR6R7 и R8; или R1 выбран из -H, -NR6R7 и R8,
R2, R3, R4 и R5 могут быть одинаковыми или разными; и каждый из них независимо выбран из -H; -C1-C6алкила; -C1-C6галогеналкила; -C1-C6пергалогеналкила; -амино-C1-C6алкила; -C3-C8циклоалкила; галогена (-F, -Cl, -Br, -I); -CN; -C1-C6алкокси; -C1-C6галогеналкокси; -C1-C6пергалогеналкокси; -C2-C7алкенила; -С2-С8алкинила; -амино; -амидо; -C1-C6алкилкарбоксила; -карбоксила (-СООН); -С1-С6ацила; -OH; -нитро (-NO2); -С6-C10арила; -гетероциклила; и -O-C1-C6алкил-гетероциклила, где гетероциклил представляет собой насыщенный или ненасыщенный 3-6-членный гетероциклил, содержащий 1-3 гетероатома (предпочтительно гетероатомы, выбранные из N, О и S),
при условии, что, когда X1 обозначает N, R2 отсутствует; когда X2 обозначает N, R3 отсутствует; когда Х3 обозначает N, R4 отсутствует; и когда X4 обозначает N, R5 отсутствует,
каждый из Y1, Y2, Y3, Y4 и Y5 независимо обозначает C или гетероатом (предпочтительно гетероатом, независимо выбранный из N, О и S), при условии, что по меньшей мере один из Y1, Y2, Y3, Y4 и Y5 обозначает гетероатом, независимо выбранный из N, О и S,
каждый из Y2 и Y3 может быть независимо замещен R9,
Y4 может быть замещен -H или -C1-C6алкилом,
каждый из R6 и R7 независимо выбран из -H; -C1-C6алкила; -C3-С8циклоалкила; -гетероциклила; -амино-С1-С6моно- или диалкила; -C1-C6алкил-амино-C1-C6моно- или диалкила; -C1-C6алкил-гетероциклила; -C1-C6алкилкарбоксила; -карбоксила (-СООН); и фенила, где гетероциклил представляет собой насыщенный или ненасыщенный 3-6-членный гетероциклил, содержащий 1-3 гетероатома (предпочтительно гетероатомы, выбранные из N, О и S),
R8 обозначает -C1-C6алкил; -C1-C6алкокси; -OH; -амино; -C1-С6алкил-амино; -C3-С8циклоалкил; -S-C1-C6алкил-амино-C1-C6моно- или диалкил; -S-C1-C6алкил-гетероциклил; -О-гетероциклил; или -O-C1-C6алкил-гетероциклил, где гетероциклил представляет собой насыщенный или ненасыщенный 3-6-членный гетероциклил, содержащий 1-3 гетероатома (предпочтительно гетероатомы, выбранные из N, О и S), и
R9 выбран из -H; -OH; -C1-C6алкила; -C1-C6галогеналкила; -C1-С6пергалогеналкила; -амино-С1-С6моно- или диалкила; -C3-С7циклоалкила; -гетероциклила; -C6-C10арила; 5-12-членного гетероарила; -C1-C6алкокси; -C1-C6галогеналкокси; -галогена (-F, -Cl, -Br, -I); - амино; -амидо; -C1-C6ацила; -CN; -карбоксила (-СООН); -C1-C6алкилкарбоксила; и -нитро (-NO2), при условии, что, когда Y4 обозначает N и Y1, Y2, Y3 и Y5 обозначают С, Y3 не замещен заместителем, имеющим группу -С(=O)-,
причем каждый из алкила, циклоалкила, гетероциклила, алкокси, алкенила, алкинила, ацила и арила может быть независимо незамещенным или замещенным одним или более заместителями (например, 1-3 заместителями), выбранными из группы, состоящей из -C1-C4алкила, -галогена (-F, -Cl, -Br, -I), -CN, -C1-C4алкокси, -амино, -амидо, -карбоксила (-СООН), -C1-C6ацила, -OH, -нитро (-NO2), гетероциклила и фенила, где гетероциклил представляет собой насыщенный или ненасыщенный 3-6-членный гетероциклил, содержащий 1-3 гетероатома (предпочтительно гетероатомы, выбранные из N, О и S).
Согласно предпочтительному варианту осуществления настоящего изобретения, в приведенной выше формуле 1 каждый из X1, Х2 и Х3 независимо обозначает C или N, и X4 обозначает N.
Согласно другому предпочтительному варианту осуществления настоящего изобретения, в приведенной выше формуле 1 каждый из Y1, Y2, Y3, Y4 и Y5 независимо обозначает C или гетероатом (предпочтительно гетероатом, независимо выбранный из N, О и S), при условии, что по меньшей мере три из Yl, Y2, Y3, Y4 и Y5обозначают N.
Согласно другому предпочтительному варианту осуществления настоящего изобретения, в приведенной выше формуле 1 R1 обозначает насыщенный или ненасыщенный 3-8-членный моно- или поли-гетероциклил, содержащий 1-3 гетероатома (предпочтительно гетероатомы, выбранные из N, О и S), где R1 незамещен или замещен 1-3 заместителями, выбранными из NR6R7, C1-C6aлкил-NR6R7 и R8,
Согласно другому предпочтительному варианту осуществления настоящего изобретения, в приведенной выше формуле 1 R3 выбран из -H; -C1-C6алкила; -C1-C6галогеналкила; -C1-C6пергалогеналкила; галогена (-F, -Cl, -Br, -I); -CN; -C1-C6алкокси; -C1-C6галогеналкокси; -C1-C6реггалогеналкокси; -C2-С7алкенила; -C2-C8алкинила и -OH.
Согласно другому предпочтительному варианту осуществления настоящего изобретения, в приведенной выше формуле 1,
каждый из X1, X2, Х3 и X4независимо обозначает C или N, при условии, что по меньшей мере один из X1, X2, Х3 и X4обозначает N,
R1 обозначает насыщенный или ненасыщенный 3-12-членный моно- или поли-гетероциклил, содержащий 1-3 гетероатома (предпочтительно гетероатомы, выбранные из N, О и S), где R1 незамещен или замещен 1-3 заместителями, выбранными из -NR6R7, -C1-C6aлкил-NR6R7 и R8; или R1 выбран из -NR6R7 и R8,
R2, R3, R4 и R5 могут быть одинаковыми или разными; и каждый из них независимо выбран из -H; -C1-C6алкила; -C1-С6галогеналкила; -C1-C6пергалогеналкила; -галогена (-F, -Cl, -Br, -I); -CN; -C1-C6алкокси; -C1-C6галогеналкокси; -C1-C6пергалогеналкокси; -C2-C7алкенила; -С2-С8алкинила; и -OH,
при условии, что, когда X1 обозначает N, R2 отсутствует; когда Х2 обозначает N, R3 отсутствует; когда Х3 обозначает N, R4 отсутствует; и когда X4 обозначает N, R5 отсутствует,
каждый из Y1, Y2, Y3, Y4 и Y5 независимо обозначает C или гетероатом (предпочтительно гетероатом, независимо выбранный из N, О и S), при условии, что по меньшей мере два из Y1, Y2, Y3, Y4 и Y5 обозначают гетероатомы, независимо выбранные из N, О и S,
каждый из Y2 и Y3 может быть независимо замещен R9,
Y4 может быть замещен -H или -C1-C6алкилом,
каждый из R6 и R7 независимо выбран из -H; -C1-C6алкила; и -карбоксила (-СООН),
R8 выбран из -C1-C6алкила и -C3-С8циклоалкила и
R9 выбран из -H; -C1-C6алкила; и -C3-С7циклоалкила,
причем каждый из алкила, циклоалкила, гетероциклила, алкокси, алкенила и алкинила может быть независимо незамещенным или замещенным одним или более заместителями, выбранными из группы, состоящей из -C1-C4алкил, -OH и -C1-C4алкокси, где гетероциклил представляет собой насыщенный или ненасыщенный 3-6-членный гетероциклил, содержащий 1-3 гетероатома (предпочтительно гетероатомы, выбранные из N, O и S).
Согласно другому предпочтительному варианту осуществления настоящего изобретения, в приведенной выше формуле 1
каждый из X1, X2, Х3 и X4 независимо обозначает C или N, при условии, что по меньшей мере один из X1, X2, Х3 и X4 обозначает N,
R1 обозначает насыщенный или ненасыщенный 3-12-членный моно- или поли-гетероциклил, содержащий 1-3 гетероатома (предпочтительно гетероатомы, выбранные из N, О и S), где R1 незамещен или замещен 1-3 заместителями, выбранными из -NR6R7 и R8,
R2, R3, R4 и R5 могут быть одинаковыми или разными; и каждый из них независимо выбран из -H; -C1-C6алкила; -C1-C6галогеналкила; -галогена (-F, -Cl, -Br, -I); -CN; -C1-C6алкокси; -C1-С6галогеналкокси; -C2-C7алкенила; и -C2-C8алкинила,
при условии, что, когда X1 обозначает N, R2 отсутствует; когда Х2 обозначает N, R3 отсутствует; когда Х3 обозначает N, R4 отсутствует; и когда X4 обозначает N, R5 отсутствует,
каждый из Y1, Y2, Y3, Y4 и Y5 независимо выбран из C, N и О, при условии, что по меньшей мере два из Y1, Y2, Y3, Y4 и Y5 обозначают N или О,
каждый из Y2 и Y3 может быть независимо замещен R9,
каждый из R6 и R7 независимо выбран из -H и -C1-C6алкила,
R8 выбран из -C1-C6алкила и
R9 выбран из -H, -C1-C6алкила и -C3-С7циклоалкила,
причем каждый из алкила, циклоалкила, гетероциклила, алкокси, алкенила и алкинила может быть независимо незамещенным или замещенным одним или более заместителями, выбранными из группы, состоящей из -C1-C4алкила, -OH и -C1-C4алкокси, где гетероциклил представляет собой насыщенный или ненасыщенный 3-6-членный гетероциклил, содержащий 1-3 гетероатома (предпочтительно гетероатомы, выбранные из N, O и S).
Согласно другому предпочтительному варианту осуществления настоящего изобретения, в приведенной выше формуле 1
каждый из X1, X2, Х3 и X4 независимо обозначает C или N, при условии, что по меньшей мере один из X1, X2, Х3 и X4 обозначает N,
R1 обозначает насыщенный или ненасыщенный 3-6-членный гетероциклил, содержащий 1-3 гетероатома (предпочтительно гетероатомы, выбранные из N, О и S), где R1 незамещен или замещен -NR6R7,
R2, R3, R4 и R5 могут быть одинаковыми или разными; и каждый из них независимо выбран из -H; -C1-C6алкила; -C1-C6галогеналкила; -галогена (-F, -Cl, -Br, -I); -CN; -C1-C6алкокси; -C1-C6галогеналкокси; -C2-C7алкенила; и -С2-С8алкинила,
при условии, что, когда X1 обозначает N, R2 отсутствует; когда Х2 обозначает N, R3 отсутствует; когда Х3 обозначает N, R4 отсутствует; и когда X4 обозначает N, R5 отсутствует,
каждый из Y1, Y2, Y3, Y4 и Y5 независимо выбран из C, N и O, при условии, что по меньшей мере два из Y1, Y2, Y3, Y4 и Y5 обозначают N,
каждый из Y2 и Y3 может быть независимо замещен R9,
каждый из R6 и R7 независимо выбран из -H и -C1-C6алкила и
R9 выбран из -H, -C1-C6алкила и -C3-С7циклоалкила,
причем каждый из алкила, циклоалкила, гетероциклила, алкокси, алкенила и алкинила может быть независимо незамещенным или замещенным одним или более заместителями, выбранными из группы, состоящей из -C1-C4алкила, -OH и -C1-C4алкокси, где гетероциклил представляет собой насыщенный или ненасыщенный 3-6-членный гетероциклил, содержащий 1-3 гетероатома (предпочтительно гетероатомы, выбранные из N, О и S).
Согласно другому предпочтительному варианту осуществления настоящего изобретения, в приведенной выше формуле 1
каждый из X1, X2, Х3 и X4 независимо обозначает C или N, при условии, что по меньшей мере один из X1, X2, Х3 и X4 обозначает N,
R1 обозначает насыщенный или ненасыщенный 3-6-членный гетероциклил, содержащий 1-3 гетероатома (предпочтительно гетероатомы, выбранные из N, О и S), где R1 незамещен или замещен -NR6R7,
R2, R3, R4 и R5 могут быть одинаковыми или разными; и каждый из них независимо выбран из -H; -C1-C6алкила; -C1-C6галогеналкила; и -галогена (-F, -Cl, -Br, -I),
при условии, что, когда X1 обозначает N, R2 отсутствует; когда Х2 обозначает N, R3 отсутствует; когда Х3 обозначает N, R4 отсутствует; и когда X4 обозначает N, R5 отсутствует,
каждый из Y1, Y2, Y3, Y4 и Y5 независимо выбран из C, N и O, при условии, что по меньшей мере два из Y1, Y2, Y3, Y4 и Y5 обозначают N,
каждый из Y2 и Y3 может быть независимо замещен R9,
каждый из R6 и R7 независимо выбран из -H и -C1-C6алкила и
R9 выбран из -H и -C1-C6алкила,
причем каждый из алкила и гетероциклила может быть независимо незамещенным или замещенным одним или более заместителями, выбранными из группы, состоящей из -C1-C4алкила, -OH и -C1-C4алкокси, где гетероциклил представляет собой насыщенный или ненасыщенный 3-6-членный гетероциклил, содержащий 1-3 гетероатома (предпочтительно гетероатомы, выбранные из N, O и S).
Соединение согласно настоящему изобретению является ингибитором человеческого рецептора гистамина 4 (hH4R) и может быть использовано для лечения или профилактики воспалительных заболеваний, аллергии, боли, носовых полипов, ринита, хронического синусита, заложенности носа, носового зуда, астмы, хронического обструктивного заболевания легких, ревматоидного артрита, атопического дерматита, псориаза, экземы, зуда, кожного зуда, крапивницы, идиопатической хронической крапивницы, склеродермии, конъюнктивита, кератоконъюнктивита, глазного воспаления, сухости глаз, кардиальной дисфункции, аритмии, атеросклероза, рассеянного склероза, воспалительного заболевания кишечника (включая колит, болезнь Крона, неспецифический язвенный колит), воспалительной боли, нейропатической боли, боли при остеоартрите, аутоиммунного заболевания щитовидной железы, иммуно-опосредованного (также известного как тип 1) диабета, волчанки, послеоперационных спаек, вестибулярных нарушений и рака, и особенно пригодно в качестве средства для лечения атопического дерматита.
Если не указано иное, алкильный заместитель, как описано здесь, и алкильный остаток на других заместителях (например, алкокси), как описано здесь, может быть линейным или разветвленным. Кроме того, галоген включает фтор (F), хлор (Cl), бром (Br) и йод (I).
В качестве репрезентативных примеров соединения формулы 1 согласно настоящему изобретению могут быть упомянуты следующие соединения:
3-метил-6-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразин (соединение 1);
8-метил-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 2);
4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 3);
8-хлор-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 4);
3-хлор-6-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразин (соединение 5);
6-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразин (соединение 6);
8-хлор-4-(пиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 7);
1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-амин (соединение 8);
(R)-1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N,N-диметилпирролидин-3-амин (соединение 9);
(R)-1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилпирролидин-3-амин (соединение 10);
(R)-1-(3-хлорпиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразин-6-ил)-N-метилпирролидин-3-амин (соединение 11);
8-бром-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 12);
4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-8-карбонитрил (соединение 13);
8-хлор-1-метил-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 14);
8-хлор-1-метил-4-(пиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 15);
8-бром-4-(пиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 16);
7,8-дихлор-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 17);
1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (соединение 18);
(S)-8-хлор-4-(3-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 19);
(S)-8-хлор-4-(3,4-диметилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 20);
8-хлор-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-7-карбонитрил (соединение 21);
8-хлор-4-(3,4,5-триметилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 22);
8-хлор-7-этокси-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 23);
1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (соединение 24);
4-(3-(метиламино)азетидин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-8-карбонитрил (соединение 25);
4-(пиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-8-карбонитрил (соединение 26);
1-(7,8-дихлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (соединение 27);
8-хлор-4-(3-(метиламино)азетидин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-7-карбонитрил (соединение 28);
8-хлор-4-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 29);
(R)-1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)пирролидин-3-амин (соединение 30);
9-хлор-2-метил-5-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (соединение 31);
9-хлор-2-метил-5-(пиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (соединение 32);
1-(9-хлор-2-метилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-ил)-N-метилазетидин-3-амин (соединение 33);
9-хлор-2-циклопропил-N,N-диэтилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-амин (соединение 34);
9-хлор-5-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (соединение 35);
1-(9-хлорпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-ил)-N-метилазетидин-3-амин (соединение 36);
9-хлор-5-(пиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (соединение 37);
9-хлор-2-циклопропил-5-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (соединение 38);
9-хлор-2-циклопропил-5-(пиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (соединение 39);
9-хлор-2-(метоксиметил)-5-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (соединение 40);
9-хлор-2-этил-5-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (соединение 41);
9-хлор-5-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[4,3-c]пиримидин (соединение 42);
8-хлор-2-метил-4-(4-метилпиперазин-1-ил)-2Н-пиразоло[3,4-с][1,8]нафтилидин (соединение 43);
1-(8-хлор-2-метил-2Н-пиразоло[3,4-с][1,8]нафтилидин-4-ил)-N-метилазетидин-3-амин (соединение 44);
8-хлор-2-метил-4-(пиперазин-1-ил)-2Н-пиразоло[3,4-с][1,8]нафтилидин (соединение 45);
8-хлор-4-(4-метилпиперазин-1-ил)имидазо[1,2-а]пиридо[2,3-е]пиразин (соединение 46);
8-хлор-4-(пиперазин-1-ил)имидазо[1,2-а]пиридо[2,3-е]пиразин (соединение 47);
1-(8-йодпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (соединение 48);
8-йод-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (Соединение 49);
N-метил-1-(8-метилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-амин (соединение 50);
1-(8-(дифторметил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (соединение 51);
N-метил-1-(8-(трифторметил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-амин (соединение 52);
4-(4-метилпиперазин-1-ил)-8-(трифторметил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 53);
1-(8-этинилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (соединение 54);
N-метил-1-(8-винилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-амин (соединение 55);
1-(8-этилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (соединение 56);
4-(3-(метиламино)азетидин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-8-ол (соединение 57);
1-(8-метоксипиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (соединение 58);
1-(8-(дифторметокси)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (соединение 59);
8-хлор-7-метокси-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 60);
8-хлор-7-метокси-4-(пиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 61);
7,8-дихлор-4-(пиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 62);
8-хлор-7-этокси-4-(пиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 63);
8-хлор-4-(4-метилпиперазин-1-ил)-7-(2,2,2-трифторэтокси)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 64);
1-(8-бром-9-метилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (соединение 65);
8-бром-9-метил-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 66);
1-(8,9-дихлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (соединение 67);
1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин гидрохлорид (соединение 68);
1-(8-бромпиридо[2,3-е]тетразоло[1,5-а]пиразин-4-ил)-N-метилазетидин-3-амин гидрохлорид (соединение 69);
8-хлор-4-(5-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 70);
8-бром-4-(5-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 71);
1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-3-метилазетидин-3-амин (соединение 72);
1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N,3-диметилазетидин-3-амин (соединение 73);
8-бром-4-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 74);
4-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)-8-йодпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 75);
8-хлор-4-(4-циклопропилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 76);
4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 77);
8-хлор-4-((1S,4S)-5-метил-2,5-диазабицикло[2,2,1]гептан-2-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 78);
8-хлор-4-(1,4-диазепан-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 79);
8-хлор-4-(4-метил-1,4-диазепан-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 80);
(R)-1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилпирролидин-3-амин (соединение 81);
8-хлор-4-(гексагидро-1Н-пирроло[3,4-b]пиридин-6(2Н)-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 82);
8-хлор-4-(1-метилгексагидро-1Н-пирроло[3,4-b]пиридин-6(2Н)-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 83);
1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N,N-диметилазетидин-3-амин (соединение 84);
1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N,N-диметилазетидин-3-амин (соединение 85);
(1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)карбаминовая кислота (соединение 86);
2-((8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)амино)этанол (соединение 87);
1-(8-хлоримидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)-N-метилазетидин-3-амин (соединение 88);
1-(8-бромимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)-N-метилазетидин-3-амин (соединение 89);
(1-(8-хлор-2-метилоксазоло[4,5-с][1,8]нафтилидин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (соединение 90);
1-(8-хлор-2-метилоксазоло[4,5-с][1,8]нафтилидин-4-ил)-N-метилазетидин-3-амин (соединение 91);
8-хлор-2-метил-4-(4-метилпиперазин-1-ил)оксазоло[4,5-с][1,8]нафтилидин (соединение 92);
1-(8-хлорпиридо[2,3-е]тетразоло[1,5-а]пиразин-4-ил)-N-метилазетидин-3-амин (соединение 93);
8-хлор-4-(4-метилпиперазин-1-ил)пиридо[2,3-е]тетразоло[1,5-а]пиразин (соединение 94);
1-(8-бромпиридо[2,3-е]тетразоло[1,5-а]пиразин-4-ил)-N-метилазетидин-3-амин (соединение 95);
8-бром-4-(4-метилпиперазин-1-ил)пиридо[2,3-е]тетразоло[1,5-а]пиразин (соединение 96);
1-(8-хлор-2-метилпиридо[2,3-е][1,2,4]триазоло[1,5-а]пиразин-4-ил)-N-метилазетидин-3-амин (соединение 97);
8-хлор-2-метил-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[1,5-а]пиразин (соединение 98);
1-(8-бром-7-метилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (соединение 99);
8-бром-7-метил-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (соединение 100);
8-хлор-4-(3-(метиламино)азетидин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-7-ол соль НСl (соединение 101);
N-(1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)-N-метилгидроксиламин (соединение 102);
1-(2-бромпиридо[3,2-е]пирроло[1,2-c]пиримидин-6-ил)-N-метилазетидин-3-амин (соединение 103);
1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин 2,2,2-трифторацетат (соединение 104);
(S)-1-(8-бром-2-метил-1,2-дигидроимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)-N-метилазетидин-3-амин (соединение 105);
(R)-1-(8-бром-2-метил-1,2-дигидроимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)-N-метилазетидин-3-амин (соединение 106);
1-(8-бром-1,2-дигидроимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)-N-метилазетидин-3-амин (соединение 107);
1-(8-бром-2-метилимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)-N-метилазетидин-3-амин (соединение 108);
1-(9-бром-2-метилпиразоло[1,5-c]пиридо[3,2-е]пиримидин-5-ил)-N-метилазетидин-3-амин (соединение 109);
1-(9-бромпиразоло[1,5-c]пиридо[3,2-е]пиримидин-5-ил)-N-метилазетидин-3-амин (соединение 110);
N-метил-1-(8-нитропиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-амин (соединение 111);
4-(3-(метиламино)азетидин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-8-амин (соединение 112);
N-метил-1-(8-фенилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-амин (соединение 113);
1-(8-(фуран-2-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (соединение 114);
1-(2-бромпиридо[2,3-е]пирроло[1,2-а]пиразин-6-ил)-N-метилазетидин-3-амин (соединение 115);
1-(2-хлорпиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразин-6-ил)-N-метилазетидин-3-амин (соединение 1-16);
1-(8-хлорпиридо[3,4-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (соединение 117);
1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин соль метансульфоновой кислоты (соединение 118);
1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин соль малеиновой кислоты (соединение 119);
1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин соль 2-гидроксипропан-1,2,3-трикарбоновой кислоты (соединение 120);
1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин соль азотной кислоты (соединение 121);
1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин соль йодистоводородной кислоты (соединение 122);
1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин соль фосфорной кислоты (соединение 123);
1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин соль 4,4'-метиленбис(3-гидрокси-2-нафтойной кислоты) (соединение 124);
1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин соль бромистоводородной кислоты (соединение 125);
1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин соль серной кислоты (соединение 126);
1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин соль (2R,3R)-2,3-дигидроксиянтарной кислоты (соединение 127);
1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин соль (1S)-(+)-10-камфорсульфоновой кислоты (соединение 128);
8-бром-N-(1-метилпирролидин-3-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-амин (соединение 129);
1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин соль (S)-2-гидроксипропановой кислоты (соединение 130);
N-(азетидин-3-илметил)-8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-амин соль трифторуксусной кислоты (соединение 131); и
4-(азетидин-3-илметокси)-8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин соль хлористоводородной кислоты (соединение 132).
Вышеперечисленные названия соединений описаны в соответствии со способом номенклатуры, обеспеченным программным обеспечением ChemBioDraw Ultra (Версия 12.02.107 6) от CambridgeSoft.
В случае соединения формулы 1 согласно настоящему изобретению, являющемуся рацематом, рацемат может быть разделен на его соответствующие изомеры при использовании обычного метода разделения, например, такого как обычная хроматография на колонках, заполненных силикагелем с нормальной фазой (Isu Chemical Co., диаметр частиц: 0,040-0,063 мм и 0,063-0,200 мм), обычная хроматография на колонках, заполненных аминсиликагелем (Isu Chemical Co., диаметр частиц: 0,040-0,075 мм), или хроматография на герметичной ректификационной колонке, заполненной в обратной фазе (Yamazen, W-Prep 2XY), и с использованием соответствующего растворителя, предпочтительно смеси растворителей гексана, этилацетата, дихлорметана и метанола в нормальной фазе и смеси растворителей воды и ацетонитрила в обратной фазе.
Соединение формулы 1 согласно настоящему изобретению может также образовывать фармацевтически приемлемую соль. Репрезентативные кислоты, которые могут быть использованы в получении такой фармацевтически приемлемой соли (например, соли присоединения с кислотой) включают, но не ограничены ими, хлористоводородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту, бромистоводородную кислоту, йодистоводородную кислоту, муравьиную кислоту, лимонную кислоту, уксусную кислоту, трихлоруксусную кислоту или трифторуксусную кислоту, бензойную кислоту, фумаровую кислоту, малеиновую кислоту, метансульфоновую кислоту, бензолсульфоновую кислоту, п-толуолсульфоновую кислоту, 2,2-дихлоруксусную кислоту, ацилированные аминокислоты, адипиновую кислоту, альгиновую кислоту, аскорбиновую кислоту, L-аспарагиновую кислоту, 4-ацетамидобензойную кислоту, (+)-камфорную кислоту, камфорсульфоновую кислоту, (+)-(1S)-камфорсульфоновую кислоту, каприновую кислоту, капроновую кислоту, каприловую кислоту, коричную кислоту, цикламовую кислоту, додецилсерную кислоту, этан-1,2-дисульфоновую кислоту, этансульфоновую кислоту, 2-гидроксиэтансульфоновую кислоту, галактаровую кислоту, гентизиновую кислоту, глюкогептановую кислоту, D-глюконовую кислоту, D-глюкуроновую кислоту, L-глутаминовую кислоту, α-оксо-глутаровую кислоту, гликолевую кислоту, гиппуровую кислоту, (+)-L-молочную кислоту, (±)-DL-молочную кислоту, лактобионовую кислоту, (-)-L-яблочную кислоту, малоновую кислоту, (±)-DL-миндальную кислоту, метансульфоновую кислоту, нафталин-2-сульфоновую кислоту, нафталин-1,5-дисульфоновую кислоту, 1-гидрокси-2-нафтойную кислоту, никотиновую кислоту, олеиновую кислоту, оротовую кислоту, щавелевую кислоту, пальмитиновую кислоту, памовую кислоту, L-пироглутаминовую кислоту, салициловую кислоту, 4-амино-салициловую кислоту, себациновую кислоту, стеариновую кислоту, янтарную кислоту, дубильную кислоту, (+)-L-винную кислоту, тиоциановую кислоту, ундециленовую кислоту и т.п. Кроме того, могут быть включены соли других кислот, известных и используемых в области производных амина. Они могут быть получены известными способами.
Соединение формулы 1, как определено выше согласно настоящему изобретению, может быть получено, но не ограничиваясь ими, способами, описанными в следующих вариантах осуществления.
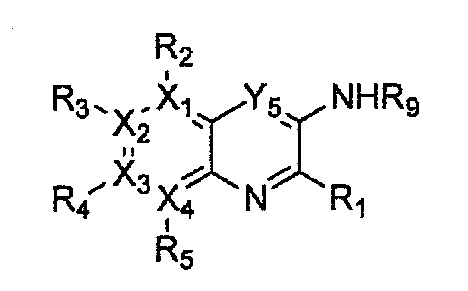
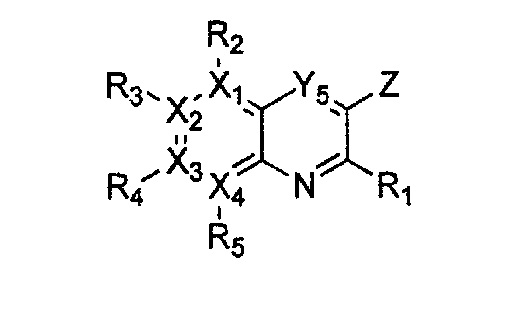
Гетероциклическое соединение, имеющее структуру формулы 1, или его рацемат, изомер или фармацевтически приемлемая соль может быть получено способом, включающим стадии:
(a) получение соединения формулы 3 через арилирование соединения формулы 4 соединением формулы R1-H (за исключением того, что R1 обозначает -H);
(b) получение соединения формулы 2 через арилирование полученного соединения формулы 3; и
(c) циклизация полученного соединения формулы 2 (или может быть включена стадия удаления защитной группы от R1):
Формула 2
Формула 3
Формула 4
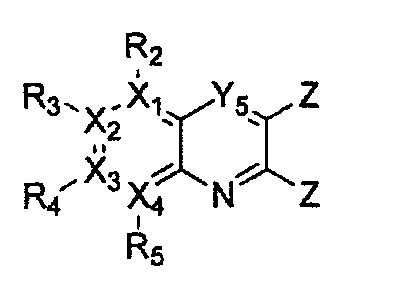
причем в формулах 2-4, X1, X2, Х3, X4, R1, R2, R3, R4, R5, R9 и Y5 имеют значения, определенные в формуле 1; и Z обозначает реактивную уходящую группу, такую как галоген (-F, -Cl, -Br).
Гетероциклическое соединение, имеющее структуру формулы 5, которая является формулой 1, как определено выше, в которой Y1, Y2 и Y4 обозначают N, или его рацемат, изомер или фармацевтически приемлемая соль могут быть получены способом, включающим стадии:
(a) получение соединения формулы 9 через галогенирование соединения формулы 10;
(b) получение соединения формулы 8 через цианирование полученного соединения формулы 9;
(c) получение соединения формулы 7 через ацилирование полученного соединения формулы 8;
(d) получение соединения формулы 6 через циклизацию полученного соединения формулы 7 с последующим галогенированием; и
(e) проведение арилирования полученного соединения формулы 6 с соединением формулы R1-H (за исключением того, что R1 обозначает -H) (или может быть включена стадия удаления защитной группы от R1):
Формула 5
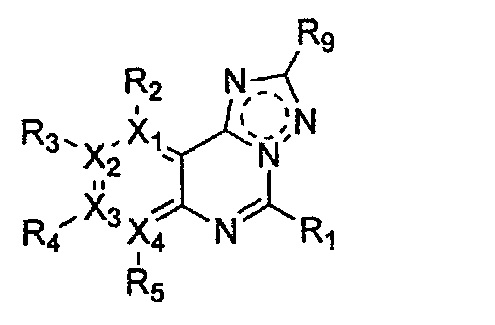
Формула 6
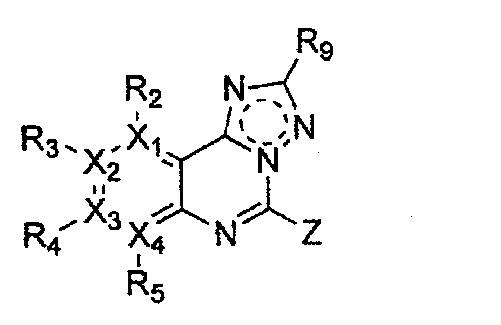
Формула 7
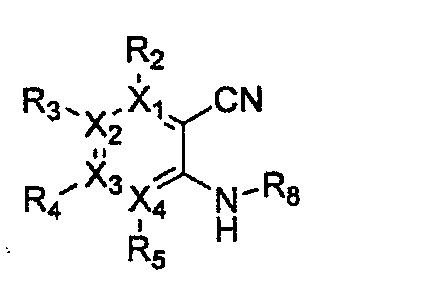
Формула 8

Формула 9
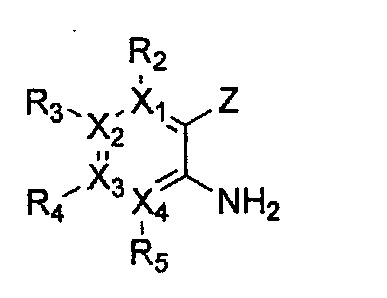
Формула 10

причем в формулах 5-10 X1, X2, Х3, X4, R1, R2, R3, R4, R5, R8 и R9 имеют значения, определенные в формуле 1; и Z обозначает реактивную уходящую группу, такую как галоген (-F, -Cl, -Br), метансульфонат, трифлат и тозилат.
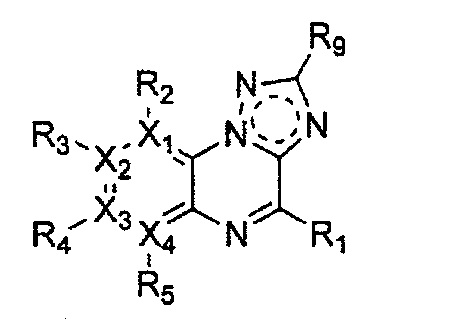
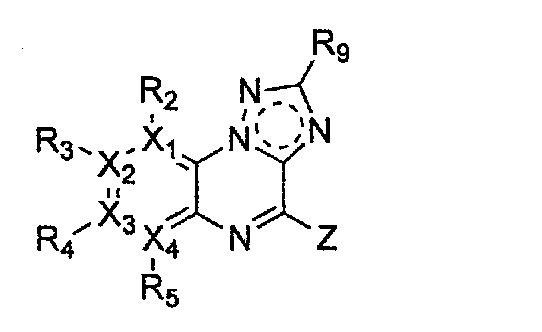
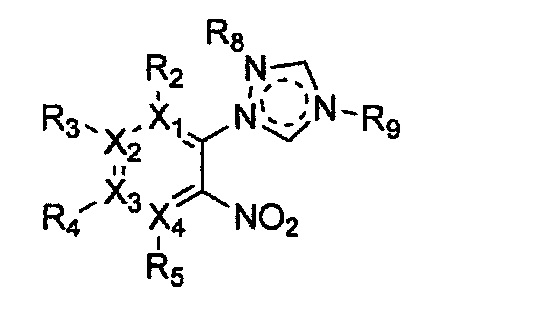
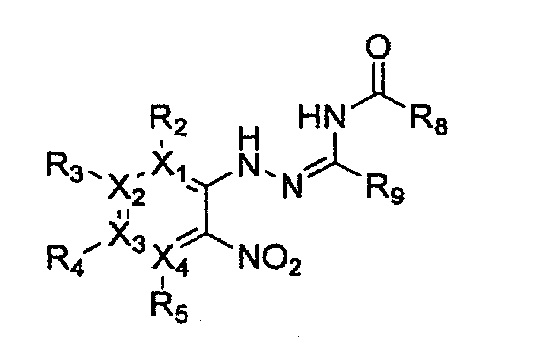
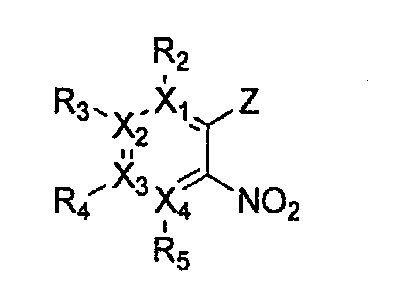
Гетероциклическое соединение, имеющее структуру формулы 11, которая является формулой 1, как определено выше, в которой Y2, Y4 и Y5 обозначают N, или его рацемат, изомер или фармацевтически приемлемая соль могут быть получены способом, включающим стадии:
(a) получение соединения формулы 16 через арилирование соединения формулы 17;
(b) получение соединения формулы 15 через имидирование полученного соединения формулы 16;
(c) получение соединения формулы 14 через ацилирование полученного соединения формулы 15;
(d) получение соединения формулы 13 через циклизацию полученного соединения формулы 14;
(e) получение соединения формулы 12 через восстановление полученного соединения формулы 13 с последующим галогенированием; и
(f) проведение арилирования полученного соединения формулы 12 с соединением формулы R1-H (за исключением того, что R1 обозначает -H) (или может быть включена стадия удаления защитной группы от R1):
Формула 11
Формула 12
Формула 13
Формула 14
Формула 15
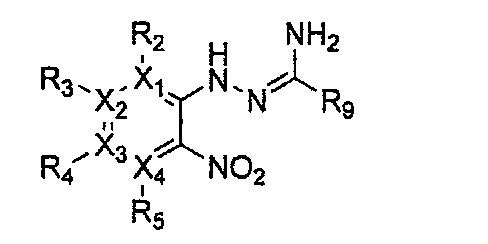
Формула 16
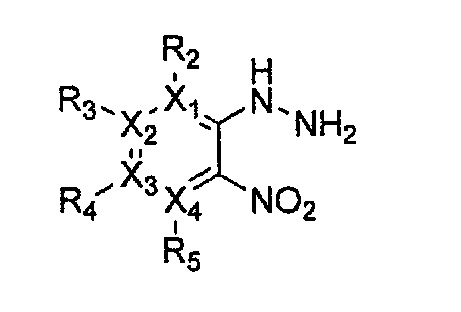
Формула 17

причем в формулах 11-17 X1, X2, Х3, X4, R1, R2, R3, R4, R5, R8 и R9 имеют значения, определенные в формуле 1; и Z обозначает реактивную уходящую группу, такую как галоген (-F, -Cl, -Br).
Гетероциклическое соединение, имеющее структуру формулы 18, которая является формулой 1, как определено выше, в которой Y2 и Y3 обозначают N, или его рацемат, изомер или фармацевтически приемлемая соль могут быть получены способом, включающим стадии:
(a) получение соединения формулы 21 через реакцию Фридель-Крафта соединения формулы 23 с соединением формулы 22;
(b) получение соединения формулы 20 через циклизацию полученного соединения формулы 21;
(c) получение соединения формулы 19 через галогенирование полученного соединения формулы 20; и
(d) проведение арилирования полученного соединения формулы 19 с соединением формулы R1-H (за исключением того, что R1 обозначает -H) (или может быть включена стадия удаления защитной группы от R1):
Формула 18
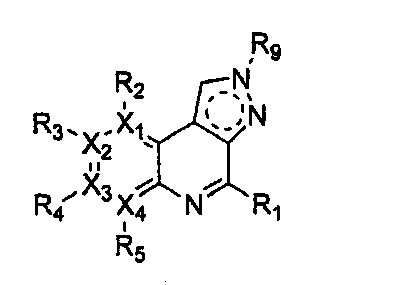
Формула 19
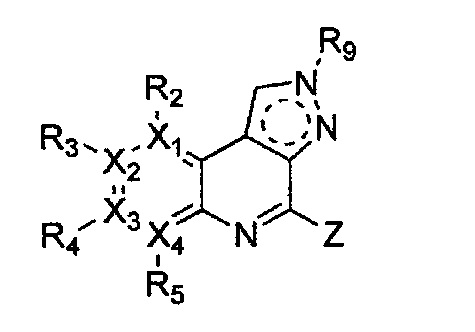
Формула 20
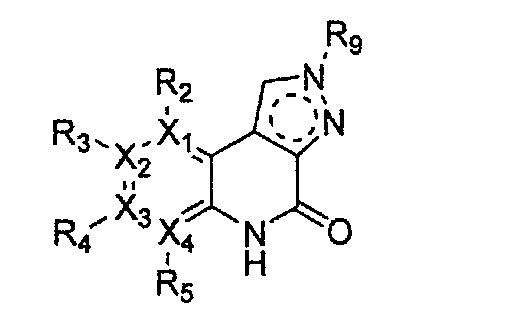
Формула 21
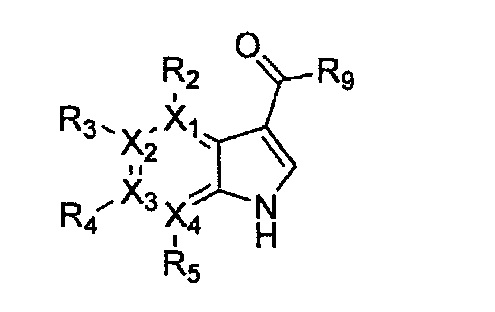
Формула 22

Формула 23
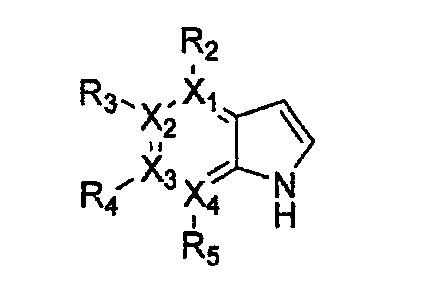
причем в формулах 18-23 X1, X2, Х3, X4, R1, R2, R3, R4, R5 и R9 имеют значения, определенные в формуле 1; и Z обозначает реактивную уходящую группу, такую как галоген (-F, -Cl, -Br).
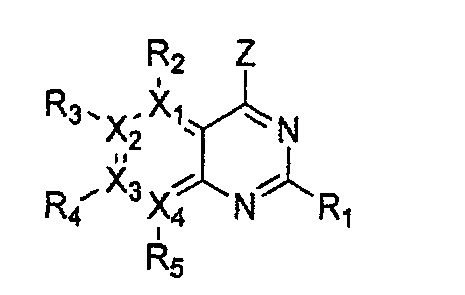
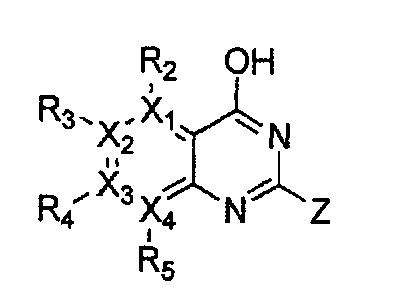
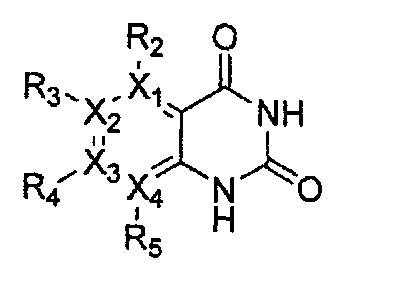
Гетероциклическое соединение, имеющее структуру формулы 24, которая является формулой 1, как определено выше, в которой Y1, Y3 и Y4 обозначают N, или его рацемат, изомер или фармацевтически приемлемая соль могут быть получены способом, включающим стадии:
(a) получение соединения формулы 28 через циклизацию соединения формулы 29;
(b) получение соединения формулы 27 через галогенирование полученного соединения формулы 28 с последующим гидроксилированием;
(c) получение соединения формулы 26 через арилирование полученного соединения формулы 27 с соединением формулы R1-H (за исключением того, что R1 обозначает -H) с последующим галогенированием;
(d) получение соединения формулы 25 через арилирование полученного соединения формулы 26; и
(e) циклизация полученного соединения формулы 25 (или может быть включена стадия удаления защитной группы от R1):
Формула 24

Формула 25
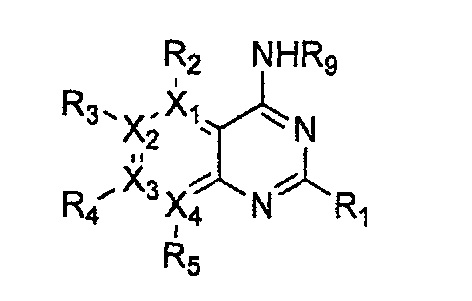
Формула 26
Формула 27
Формула 28
Формула 29
причем в формулах 24-29 X1, X2, Х3, X4, R1, R2, R3, R4, R5 и R9 имеют значения, определенные в формуле 1; и Z обозначает реактивную уходящую группу, такую как галоген (-F, -Cl, -Br).
Гетероциклическое соединение, имеющее структуру формулы 30, которая является формулой 1, как определено выше, в которой Y2 обозначает N, и Y4 обозначает O, или его рацемат, изомер или фармацевтически приемлемая соль могут быть получены способом, включающим стадии:
(a) получение соединения формулы 36 через этерификацию соединения формулы 37;
(b) получение соединения формулы 35 через арилирование полученного соединения формулы 36;
(c) получение соединения формулы 34 через циклизацию полученного соединения формулы 35;
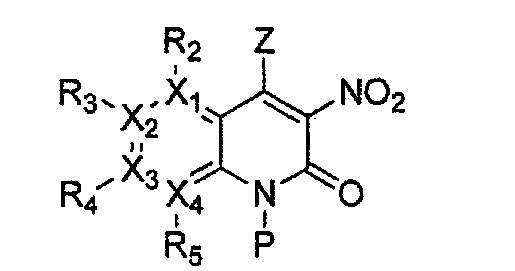
(d) получение соединения формулы 33 через реакцию присоединения енолата полученного соединения формулы 34;
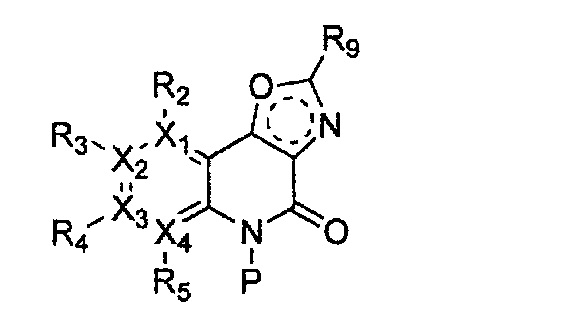
(e) получение соединения формулы 32 через циклизацию полученного соединения формулы 33;
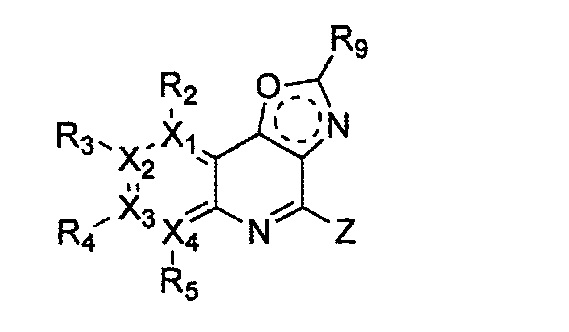
(f) получение соединения формулы 31 через галогенирование полученного соединения формулы 32; и
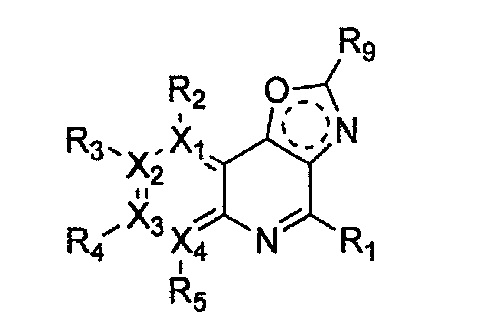
(g) проведение арилирования полученного соединения формулы 31 с соединением формулы R1-H (за исключением того, что R1 обозначает -H) (или может быть включена стадия удаления защитной группы от R1):
Формула 30
Формула 31
Формула 32
Формула 33
Формула 34
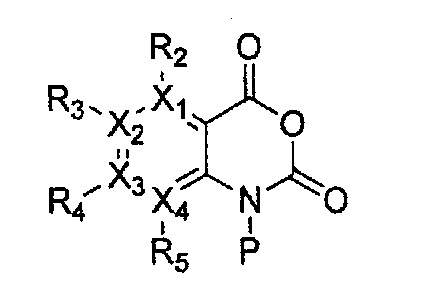
Формула 35
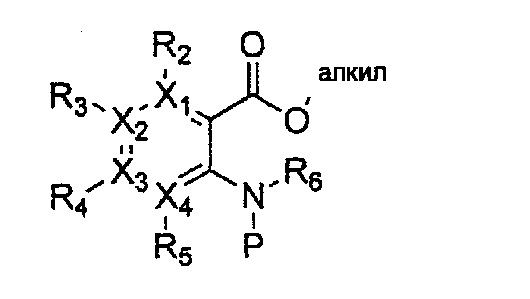
Формула 36
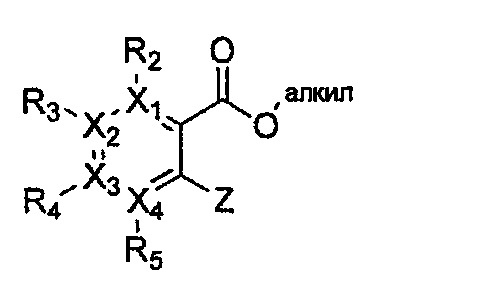
Формула 37

причем в формулах 30-37 X1, X2, Х3, X4, R1, R2, R3, R4, R5, R6 и R9 имеют значения, определенные в формуле 1; P обозначает защитную группу, такую как п-метоксибензил, 3',5'-диметоксибензил, три-метоксибензил; и Z обозначает реактивную уходящую группу, такую как галоген (-F, -Cl, -Br).
Гетероциклическое соединение, имеющее структуру формулы 38, которая является формулой 1, как определено выше, в которой Y1 обозначает N, и каждый из Y2 и Y3 независимо обозначает C или N, или его рацемат, изомер или фармацевтически приемлемая соль могут быть получены способом, включающим стадии:
(a) получение соединения формулы 40 через реакцию сочетания Сузуки соединения формулы 42 с соединением формулы 41;
(b) получение соединения формулы 39 через галогенирование полученного соединения формулы 40; и
(c) проведение арилирования полученного соединения формулы 39 с соединением формулы R1-H (за исключением того, что R1 обозначает -H) (или может быть включена стадия удаления защитной группы от R1):
Формула 38
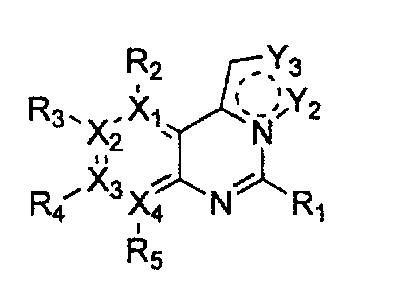
Формула 39

Формула 40
Формула 41
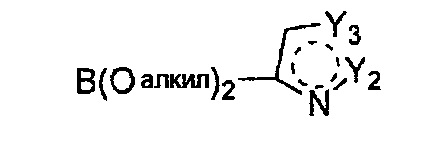
Формула 42
причем в формулах 38-42 X1, X2, Х3, X4, R1, R2, R3, R4 и R5 имеют значения, определенные в формуле 1; и Z обозначает реактивную уходящую группу, такую как галоген (-F, -Cl, -Br).
Гетероциклическое соединение, имеющее структуру формулы 43, которая является формулой 1, как определено выше, в которой Y5 обозначает N, или его рацемат, изомер или фармацевтически приемлемая соль могут быть получены способом, включающим стадии:
(a) получение соединения формулы 46 через пирролирование соединения формулы 47;
(b) получение соединения формулы 45 через циклизацию полученного соединения формулы 46;
(c) получение соединения формулы 44 через галогенирование полученного соединения формулы 45; и
(d) проведение арилирования полученного соединения формулы 44 с соединением формулы R1-H (за исключением того, что R1 обозначает -H) (или может быть включена стадия удаления защитной группы от R1):
Формула 43
Формула 44
Формула 45
Формула 46

Формула 47

причем в формулах 43-47 X1, X2, Х3, X4, R1, R2, R3, R4 и R5 имеют значения, определенные в формуле 1; и Z обозначает реактивную уходящую группу, такую как галоген (-F, -Cl, -Br).
Соединение формулы 1 согласно настоящему изобретению имеет превосходную ингибирующую активность в отношении человеческого рецептора гистамина 4 (hH4R). Поэтому настоящее изобретение также относится к фармацевтической композиции, включающей эффективное количество соединения формулы 1 или его рацемата, изомера или фармацевтически приемлемой соли; и фармацевтически приемлемый носитель.
Фармацевтическая композиция согласно настоящему изобретению может быть получена путем смешивания эффективного количества соединения формулы 1, или его рацемата, изомера или фармацевтически приемлемой соли, с фармацевтически приемлемым носителем, связующим, стабилизатором и/или разбавителем. Кроме того, когда фармацевтическая композиция согласно настоящему изобретению получена в форме жидкости для инъекции, фармацевтически приемлемый буфер, адъювант растворения и/или изотонический агент может быть смешан с соединением формулы 1, или его рацематом, изомером или фармацевтически приемлемой солью.
Поскольку фармацевтическая композиция согласно настоящему изобретению демонстрирует сильную ингибирующую активность в отношении человеческого рецептора гистамина 4 (hH4R), она может быть использована для лечения или профилактики воспалительных заболеваний, аллергии, боли, носовых полипов, ринита, хронического синусита, заложенности носа, носового зуда, астмы, хронического обструктивного заболевания легких, ревматоидного артрита, атопического дерматита, псориаза, экземы, зуда, кожного зуда, крапивницы, идиопатической хронической крапивницы, склеродермии, конъюнктивита, кератоконъюнктивита, глазного воспаления, сухости глаз, кардиальной дисфункции, аритмии, атеросклероза, рассеянного склероза, воспалительного заболевания кишечника (включая колит, болезнь Крона, неспецифический язвенный колит), воспалительной боли, нейропатической боли, боли при остеоартрите, аутоиммунного заболевания щитовидной железы, иммуно-опосредованного (также известного как тип 1) диабета, волчанки, послеоперационных спаек, вестибулярных нарушений и рака.
Фармацевтическая композиция согласно настоящему изобретению может быть получена в форме доставки фармацевтической композиции, включающей одну или более единиц дозировки фармацевтического средства, при использовании методики получения, известной или доступной специалисту, и подходящий фармацевтический эксципиент. В способе согласно настоящему изобретению композиция может вводиться через подходящий путь доставки, например, такой как пероральное или парентеральное, чрескожное, ректальное, топическое или глазное введение, или введение ингаляцией. Фармацевтический состав может быть в форме таблетки, капсулы, саше, засахаренной пилюли, порошка, гранулы, таблетки, порошка для восстановления, жидкого препарата или суппозитория. Например, композиция может быть составлена в форме для внутривенной инъекции, спрея, топического или перорального введения.
В случае получения состава в пероральной лекарственной форме, могут использоваться любые обычные фармацевтические носители. Например, вода, гликоли, масла, спирты и т.п. могут использоваться в качестве носителя в случае пероральных жидких составов, таких как суспензии, сиропы, эликсиры и растворы; и крахмалы, сахара, каолин, лубриканты, связующие, разрыхлители и т.п. могут использоваться в качестве носителя в случае твердых составов, таких как порошки, пилюли, капсулы и таблетки. В силу легкости введения, таблетки и капсулы представляют собой самые удобные лекарственные формы, и таблетки и пилюли предпочтительно получают в форме составов, имеющих энтеросолюбильное покрытие.
В случае парентеральных составов, обычно используется стерилизованная вода, и другой ингредиент(ы), такой как адъювант растворения, может также быть включен. Составы для инъекции, например, суспензия на основе стерилизованной воды или масла для инъекции, могут быть получены согласно известным методикам при использовании подходящего диспергирующего агента, смачивающего вещества или суспендирующего агента. Растворители, пригодные для использования с этой целью, включают воду, раствор Рингера и изотонический раствор NaCl, и стерилизованные, иммобилизированные масла также обычно используются как растворитель или суспендирующая среда. Любые нераздражающие иммобилизированные масла, включая моно- и диглицериды, могут использоваться с этой целью, и жирные кислоты, такие как олеиновая кислота, могут использоваться для получения состава для инъекции.
В случае чрескожных составов, усиливающее проникновение средство и/или подходящее смачивающее вещество могут использоваться как носитель, в случае необходимости, в комбинации с подходящей нераздражающей по отношению к коже добавкой(ами). В качестве таких добавок могут быть выбраны полезные для усиления введения через кожу и/или получения желаемой композиции. Чрескожный состав может вводиться различными способами, например, такими как трансдермальный пластырь, средство для точечной обработки или мазь.
Время введения и дозировка фармацевтической композиции согласно настоящему изобретению могут быть соответственно определены в зависимости от заболевания, состояния, возраста, массы тела пациента и формы введения. В случае взрослых, фармацевтическая композиция может вводиться в количестве 0,1-2,000 мг, предпочтительно 1-200 мг в сутки, в единственной дозе или в множественных дозах, но не ограничиваясь этим.
Поскольку фармацевтическая композиция согласно настоящему изобретению демонстрирует сильную ингибирующую активность в отношении человеческого рецептора гистамина 4 (hH4R), она может быть использована для лечения или профилактики воспалительных заболеваний, аллергии, боли, носовых полипов, ринита, хронического синусита, заложенности носа, носового зуда, астмы, хронического обструктивного заболевания легких, ревматоидного артрита, атопического дерматита, псориаза, экземы, зуда, кожного зуда, крапивницы, идиопатической хронической крапивницы, склеродермии, конъюнктивита, кератоконъюнктивита, глазного воспаления, сухости глаз, кардиальной дисфункции, аритмии, атеросклероза, рассеянного склероза, воспалительного заболевания кишечника (включая колит, болезнь Крона, неспецифический язвенный колит), воспалительной боли, нейропатической боли, боли при остеоартрите, аутоиммунного заболевания щитовидной железы, иммуно-опосредованного (также известного как тип 1) диабета, волчанки, послеоперационных спаек, вестибулярных нарушений и рака.
Если не указано иное, алкильная группа и алкильная часть других групп (например, алкоксигруппы), описанные в настоящем изобретении, могут иметь линейные или разветвленные разновидности. Кроме того, галоген включает фтор, хлор, бром и йод.
Соединение формулы 1 согласно настоящему изобретению может быть рацематом. Рацемат может быть разделен на его соответствующие изомеры при использовании обычного метода разделения, например, нормальной хроматографии на колонках, заполненных силикагелем с нормальной фазой (Merck, 0,040-0,063 мм и 0,063-0,200 мм), нормальной хроматографии на колонках, заполненных аминосиликагелем (chromatorex, 100-200 меш) или препаративной хроматографии под давлением на колонках с обратной фазой (Yonglin, SDV 30 plus), с использованием соответствующего растворителя, предпочтительно смеси растворителей гексана, этилацетата, дихлорметана и метанола в нормальной фазе и смеси растворителей воды и ацетонитрила в обратной фазе.
Соединение формулы 1 согласно настоящему изобретению может также образовывать фармацевтически приемлемую соль. Такая фармацевтически приемлемая соль включает соли присоединения с кислотой, полученные из кислот, которые образуют нетоксичные соли присоединения, содержащие фармацевтически приемлемые анионы, например, неорганических кислот, таких как хлористоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, бромистоводородная кислота, йодистоводородная кислота, органических кислот, таких как винная кислота, муравьиная кислота, лимонная кислота, уксусная кислота, трихлоруксусная кислота или трифторуксусная кислота, глюконовая кислота, бензойная кислота, молочная кислота, фумаровая кислота, малеиновая кислота, сульфоновых кислот, таких как метансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота, нафталинсульфоновая кислота; -и соли щелочного металла, такого как натрий, калий и т.д. Кроме того, могут быть включены соли с кислотой или щелочным металлом, известные и используемые в области ароматических производных амидина и производных лактама. Для получения солей могут использоваться обычные способы.
Соединение формулы 1 согласно настоящему изобретению может быть получено следующим способом. Соответственно, настоящее изобретение также относится к способу получения соединения формулы 1.
Более конкретно, соединение формулы 1 может быть получено, но не ограничено ими, каждым из способов 1-8, описанных далее.
Способ 1
Соединение, имеющее структуру следующей формулы 2, которая является формулой 1 как определено выше, в которой Y2 и Y5 обозначают N, может быть получено способом, включающим стадии: получение соединения формулы 3 через арилирование соединения формулы 4 соединением формулы R1-H (за исключением того, что R1 обозначает -H); получение соединения формулы 2 через арилирование полученного соединения формулы 3; и циклизация полученного соединения формулы 2:
Формула 2
Формула 3
Формула 4
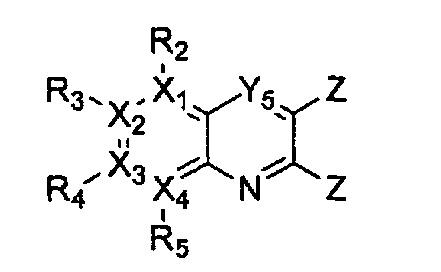
причем в формулах 2-4 X1, X2, Х3, X4, R1, R2, R3, R4, R5, R6, R7, R8 и R9 имеют значения, определенные в формуле 1; и Z обозначает реактивную уходящую группу, предпочтительно атом галогена (-F, -Cl, -Br).
Способ 1 более специфично описан далее.
Соединения формул 2, 3 и 4, используемые как исходный материал, могут быть получены согласно известным способам в данной области техники (например, J. Med. Chem. 1990, 33, 2240-2254, J. Med. Chem. 1997, 40, 2053-2063).
Получение соединения формулы 3 через арилирование соединения формулы 4 соединением формулы R1-H (за исключением того, что R1 обозначает -H) осуществляют согласно обычному способу в присутствии подходящего растворителя и основания.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является дихлорметан.
Следующую стадию получения соединения формулы 2 через арилирование полученного соединения формулы 3 осуществляют согласно обычному способу в присутствии подходящего растворителя.
Вообще, обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является этанол.
Следующую стадию получения соединения формулы 1 через циклизацию полученного соединения формулы 2 осуществляют согласно обычному способу в присутствии подходящего растворителя.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является этанол.
Реакцию циклизации осуществляют при 50-200°C в течение от 0,1 до 24 часов, предпочтительно в условиях 80°C и 1 часа с триметилортоформиатом или триэтил ортоформиатом.
Вышеописанный способ получения соединения формулы 2 более специфично описан в следующем примере.
Способ 2
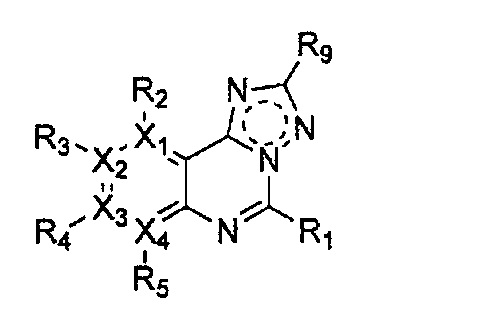
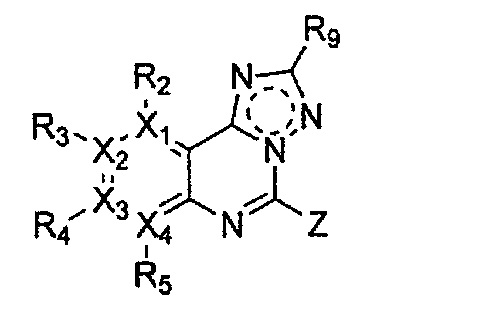
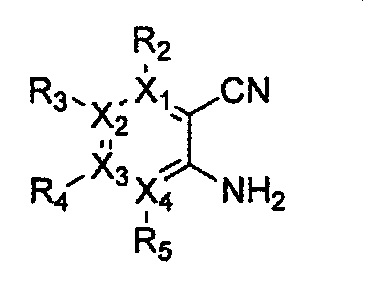
Соединение, имеющее структуру следующей формулы 5, которая является формулой 1 как определено выше, в которой Y1, Y2 и Y4 обозначают N, может быть получено способом, включающим стадии: получение соединения формулы 9 через галогенирование соединения следующей формулы 10; получение соединения формулы 8 через цианирование полученного соединения формулы 9; получение соединения формулы 7 через ацилирование полученного соединения формулы 8; получение соединения формулы 6 через циклизацию полученного соединения формулы 7 с последующим галогенированием; и проведение арилирования полученного соединения формулы 6 соединением формулы R1-H (за исключением того, что R1 обозначает -H):
Формула 5
Формула 6
Формула 7

Формула 8
Формула 9
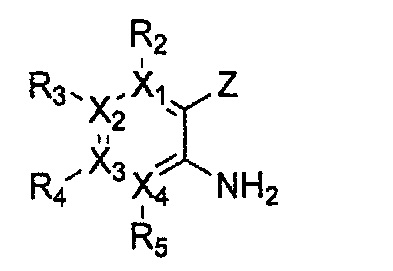
Формула 10
причем в формулах 5-10 X1, X2, Х3, X4, R1, R2, R3, R4, R5, R6, R7, R8, и R9 имеют значения, определенные в формуле 1; и Z обозначает реактивную уходящую группу, предпочтительно атом галогена (-F, -Cl, -Br), метансульфонат, трифлат и тозилат.
Способ 2 более специфично описан далее.
Соединения формул 5, 6, 7, 8, 9 и 10, используемые как исходный материал, могут быть получены согласно известным способам в данной области техники.
Получение соединения формулы 9 через галогенирование соединения формулы 10 осуществляют согласно обычному способу в присутствии подходящего растворителя.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является хлороформ.
Примеры галогенирующих реагентов для реакции включают бром, трихлорид фосфора (3), фосфорил хлорид, оксибромид фосфора, фенилфосфонил хлорид и пентахлорид фосфора. Предпочтительным является бром. Реакция может быть проведена при, но не ограничиваясь этим, 50-200°C в течение приблизительно от 0,1 до 24 часов.
Следующую стадию получения соединения формулы 8 через цианирование полученного соединения формулы 9 осуществляют согласно обычному способу в присутствии подходящего растворителя и цианирующего агента.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является N-метилпирролидон.
Примеры цианирующих агентов для этой реакции включают KCN, NaCN, Zn(CN)2, CuCN, (СН3)2C(ОН)CN и TMSCN. Реакция может быть проведена при, но не ограничиваясь этим, 50-200°C в течение приблизительно от 0,1 до 24 часов.
Следующую стадию получения соединения формулы 7 через ацилирование полученного соединения формулы 8 осуществляют согласно обычному способу в присутствии подходящего растворителя.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительный растворитель для реакции 2-бутанон.
Следующую стадию получения соединения формулы 6 через циклизацию полученного соединения формулы 7 с последующим галогенированием осуществляют согласно обычному способу в присутствии подходящего растворителя.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является простой дифениловый эфир.
Примеры галогенирующих реагентов для этой реакции включают трихлорид фосфора (3), фосфорил хлорид, оксибромид фосфора, фенилфосфонил хлорид и пентахлорид фосфора. Предпочтительным является фосфорил хлорид. Реакция может быть проведена при, но не ограничиваясь этим, 50-200°C в течение приблизительно от 0,1 до 24 часов.
Следующую стадию получения соединения формулы 5 через арилирование полученного соединения формулы 6 соединением формулы R1-H (за исключением того, что R1 обозначает -H) осуществляют согласно обычному способу в присутствии подходящего растворителя и основания.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является N,N-диметилформамид.
Вышеописанный способ получения соединения формулы 5 более специфично описан в следующем примере.
Способ 3
Соединение, имеющее структуру следующей формулы 11, которая является формулой 1 как определено выше, в которой Y2, Y4 и Y5 обозначают N, может быть получен способом, включающим стадии: получение соединения формулы 16 через арилирование соединения формулы 17; получение соединения формулы 15 через имидирование полученного соединения формулы 16; получение соединения формулы 14 через ацилирование полученного соединения формулы 15; получение соединения формулы 13 через циклизацию полученного соединения формулы 14; получение соединения формулы 12 через восстановление полученного соединения формулы 13 с последующим галогенированием; и проведение арилирования полученного соединения формулы 12 соединением формулы R1-H (за исключением того, что R1 обозначает -H):
Формула 11
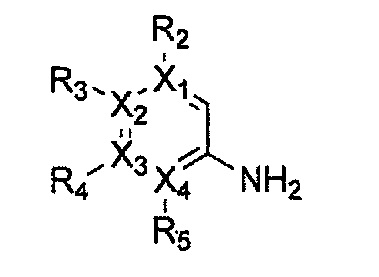
Формула 12
Формула 13
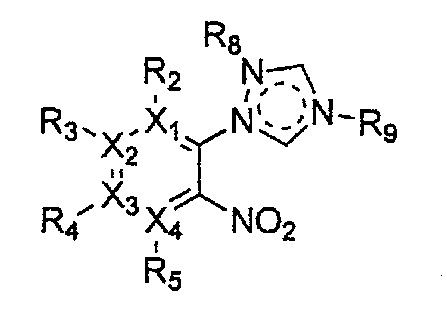
Формула 14
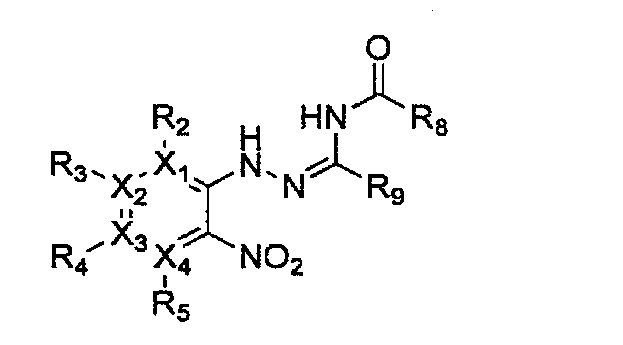
Формула 15
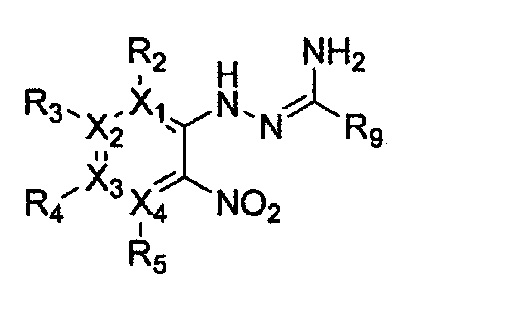
Формула 16
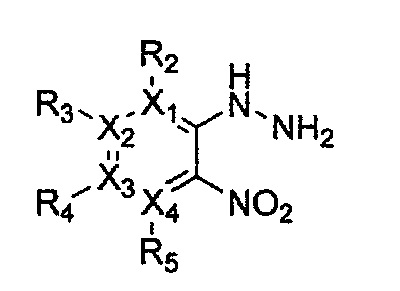
Формула 17
причем в формулах 11-17 X1, X2, Х3, X4, R1, R2, R3, R4, R5, R6, R7, R8 и R9 имеют значения, определенные в формуле 1; и Z обозначает реактивную уходящую группу, предпочтительно атом галогена (-F, -Cl, -Br).
Способ 3 более специфично описан далее.
Соединения формул 11, 12, 13, 14, 15, 16 и 17, используемые как исходный материал, могут быть получены способом, известным в данной области техники.
Получение соединения формулы 16 через арилирование соединения формулы 17 осуществляют согласно обычному способу в присутствии подходящего растворителя.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является этанол.
Следующую стадию получения соединения формулы 15 через имидирование полученного соединения формулы 16 осуществляют согласно обычному способу в присутствии подходящего растворителя.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является пиридин.
Следующую стадию получения соединения формулы 14 через ацилирование полученного соединения формулы 15 осуществляют согласно обычному способу в присутствии подходящего растворителя.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является простой диэтиловый эфир.
Следующую стадию получения соединения формулы 13 через циклизацию полученного соединения формулы 14 осуществляют согласно обычному способу в присутствии подходящего растворителя.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является толуол.
Реакция может быть проведена при, но не ограничиваясь этим, 50-200°C в течение приблизительно от 0,1 до 24 часов.
Следующую стадию получения соединения формулы 12 через восстановление полученного соединения формулы 13 с последующим галогенированием осуществляют согласно обычному способу в присутствии подходящего растворителя, восстановителя и галогенирующего реагента.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является уксусная кислота.
Примеры восстановителей для этой реакции включают катализатор на основе палладия на углероде (5% вес./вес.), катализатор на основе палладия на углероде (10% вес./вес.), никель Ренея, цинк и железо. Особенно предпочтительным является железо. Реакция может быть проведена при, но не ограничиваясь этим, 50-200°C в течение приблизительно от 0,1 до 24 часов.
Кроме того, примеры галогенирующих реагентов для этой реакции включают трихлорид фосфора (3), фосфорил хлорид, оксибромид фосфора, фенилфосфонил хлорид и пентахлорид фосфора. Предпочтительным является фосфорил хлорид. Реакция может быть проведена при, но не ограничиваясь этим, 50-200°C в течение приблизительно от 0,1 до 24 часов.
Следующую стадию получения соединения формулы 11 через арилирование полученного соединения формулы 12 соединением формулы R1-H (за исключением того, что R1 обозначает -H) осуществляют согласно обычному способу в присутствии подходящего растворителя и основания.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является N,N-диметилформамид.
Вышеописанный способ получения соединения формулы 11 более специфично описан в следующем примере.
Способ 4
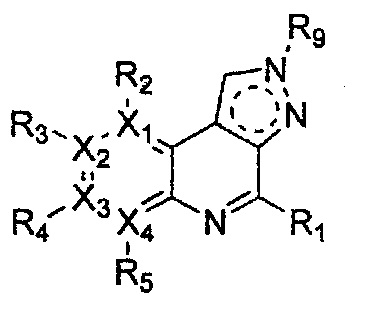
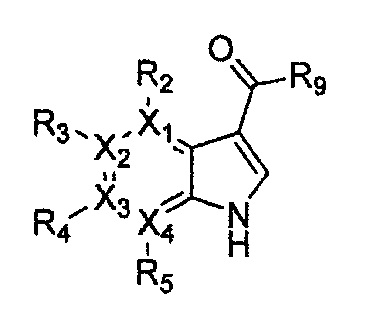
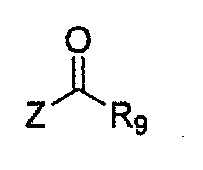
Соединение, имеющее структуру следующей формулы 18, которая является формулой 1 как определено выше, в которой Y2 и Y3 обозначают N, может быть получено способом, включающим стадии: получение соединения следующей формулы 21 через реакцию Фриделя-Крафта соединения следующей формулы 23 с соединением следующей формулы 22; получение соединения формулы 20 через циклизацию полученного соединения формулы 21; получение соединения формулы 19 через галогенирование полученного соединения формулы 20; и проведение арилирования полученного соединения формулы 19 соединением формулы R1-H (за исключением того, что R1 обозначает -H):
Формула 18
Формула 19

Формула 20

Формула 21
Формула 22
Формула 23
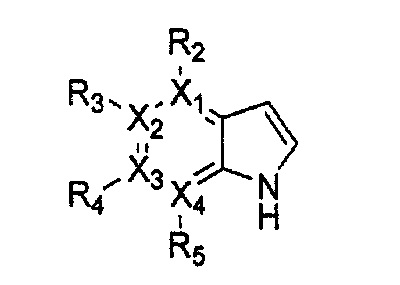
причем в формулах 18-23 X1, X2, Х3, X4, R1, R2, R3, R4, R5, R6, R7, R8 и R9 имеют значения, определенные в формуле 1; и Z обозначает реактивную уходящую группу, предпочтительно атом галогена (-F, -Cl, -Br).
Способ 4 более специфично описан далее.
Соединения формул 18, 19, 20, 21, 22 и 23, используемые как исходный материал, могут быть получены способом, известным в данной области техники.
Получение соединения формулы 21 через реакцию Фриделя-Крафта соединения формулы 23 с соединением формулы 22 осуществляют согласно обычному способу в присутствии подходящего растворителя.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является дихлорметан.
Следующую стадию получения соединения формулы 20 через циклизацию полученного соединения формулы 21 осуществляют согласно обычному способу в присутствии подходящего растворителя.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является этанол.
Реакция может быть проведена при, но не ограничиваясь этим, 50-200°C в течение приблизительно от 0,1 до 24 часов.
Следующую стадию получения соединения формулы 19 через галогенирование полученного соединения формулы 20 осуществляют согласно обычному способу в присутствии подходящего растворителя и галогенирующего реагента.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является толуол. Растворитель может не использоваться в настоящей реакции.
Примеры галогенирующих реагентов для этой реакции включают трихлорид фосфора (3), фосфорил хлорид, оксибромид фосфора, фенилфосфонил хлорид и пентахлорид фосфора. Предпочтительным является фосфорил хлорид. Реакция может быть проведена при, но не ограничиваясь этим, 50-200°C в течение приблизительно от 0,1 до 24 часов.
Следующую стадию получения соединения формулы 18 через арилирование полученного соединения формулы 19 соединением формулы R1-H (за исключением того, что R1 обозначает -H) осуществляют согласно обычному способу в присутствии подходящего растворителя и основания.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является N,N-диметилформамид.
Вышеописанный способ получения соединения формулы 18 более специфично описан в следующем примере.
Способ 5
Соединение, имеющее структуру следующей формулы 24, которая является формулой 1 как определено выше, в которой Y1, Y3 и Y4 обозначают N, может быть получен способом, включающим стадии: получение соединения следующей формулы 28 через циклизацию соединения следующей формулы 29; получение соединения формулы 27 через галогенирование полученного соединения формулы 28 с последующим гидроксилированием; получение соединения формулы 26 через арилирование полученного соединения формулы 27 соединением формулы R1-H (за исключением того, что R1 обозначает -H) с последующим галогенированием; получение соединения формулы 25 через арилирование полученного соединения формулы 26; и циклизация полученного соединения формулы 25:
Формула 24
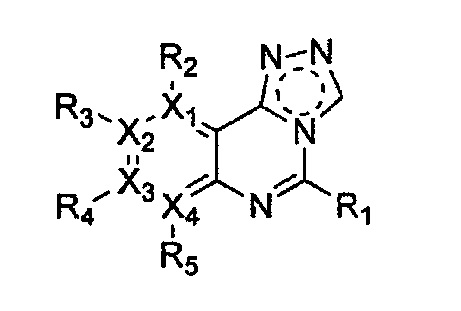
Формула 25
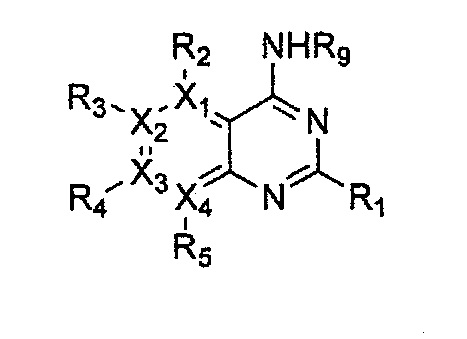
Формула 26
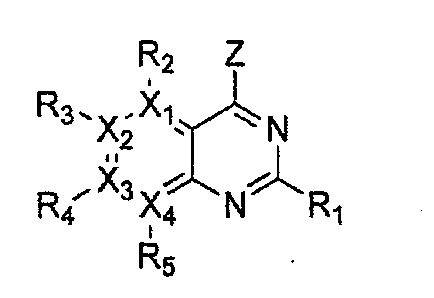
Формула 27

Формула 28
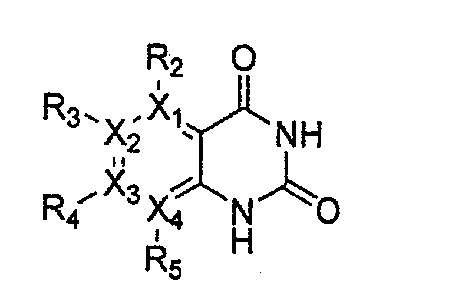
Формула 29
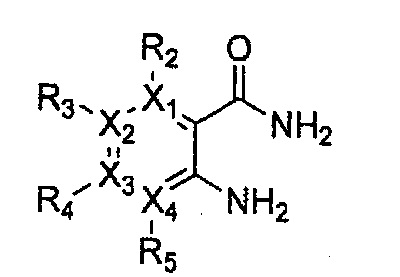
причем в формулах 24-29 X1, X2, Х3, X4, R1, R2, R3, R4, R5, R6, R7, R8 и R9 имеют значения, определенные в формуле 1; и Z обозначает реактивную уходящую группу, предпочтительно атом галогена (-F, -Cl, -Br).
Способ 5 более специфично описан далее.
Соединения формул 24, 25, 26, 27, 28 и 29, используемые как исходный материал, могут быть получены способом, известным в данной области техники.
Получение соединения формулы 28 через циклизацию соединения формулы 29 осуществляют согласно обычному способу в присутствии подходящего растворителя и агента циклизации.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является диоксан.
Примеры агентов циклизации для этой реакции включают дифосген и трифосген. Предпочтительным является дифосген. Реакция может быть проведена при, но не ограничиваясь этим, 50-200°C в течение приблизительно от 0,1 до 24 часов.
Следующую стадию получения соединения формулы 27 через галогенирование полученного соединения формулы 28 с последующим гидроксилированием осуществляют согласно обычному способу в присутствии подходящего растворителя, галогенирующего реагента и гидроксилирующего агента.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является н-бутанол.
Примеры галогенирующих реагентов для этой реакции включают трихлорид фосфора (3), фосфорил хлорид, оксибромид фосфора, фенилфосфонил хлорид и пентахлорид фосфора. Предпочтительным является фосфорил хлорид. Реакция может быть проведена при, но не ограничиваясь этим, 50-200°C в течение приблизительно от 0,1 до 24 часов.
Примеры гидроксилирующих агентов для этой реакции включают гидроксид лития, гидроксид натрия, гидроксид калия, гидроксид кальция и гидроксид магния. Предпочтительным является гидроксид натрия. Реакция может быть проведена, но не ограничиваясь этим, при комнатной температуре в течение приблизительно от 0,1 до 24 часов.
Следующую стадию получения соединения формулы 26 через арилирование полученного соединения формулы 27 соединением формулы R1-H (за исключением того, что R1 обозначает -H) с последующим галогенированием осуществляют согласно обычному способу в присутствии подходящего растворителя и галогенирующего реагента.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться в арилирование реакции. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является этанол.
Примеры галогенирующих реагентов для этой реакции включают трихлорид фосфора (3), фосфорил хлорид, оксибромид фосфора, фенилфосфонил хлорид и пентахлорид фосфора предпочтительным является фосфорил хлорид. Реакция может быть проведена при, но не ограничиваясь этим, 50-200°C в течение приблизительно от 0,1 до 24 часов.
Следующую стадию получения соединения формулы 25 через арилирование полученного соединения формулы 26 осуществляют согласно обычному способу в присутствии подходящего растворителя.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является этанол.
Следующую стадию получения соединения формулы 24 через циклизацию полученного соединения формулы 25 осуществляют согласно обычному способу в присутствии подходящего растворителя и агента циклизации.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является этанол.
Примеры предпочтительных агентов циклизации для этой реакции включают триметилортоформиат и триэтоксиметан. Реакция может быть проведена при, но не ограничиваясь этим, 50-200°C в течение приблизительно от 0,1 до 24 часов.
Вышеописанный способ получения соединения формулы 24 более специфично описан в следующем примере.
Способ 6
Соединение, имеющее структуру следующей формулы 30, которая является формулой 1 как определено выше, в которой Y2 обозначает N, и Y4 обозначает O, может быть получено способом, включающим стадии: получение соединения следующей формулы 36 через этерификацию соединения следующей формулы 37; получение соединения формулы 35 через арилирование полученного соединения формулы 36; получение соединения формулы 34 через циклизацию полученного соединения формулы 35; получение соединения формулы 33 через реакцию присоединения енолата полученного соединения формулы 34; получение соединения формулы 32 через циклизацию полученного соединения формулы 33; получение соединения формулы 31 через галогенирование полученного соединения формулы 32; и проведение арилирования полученного соединения формулы 31 соединением формулы R1-H (за исключением того, что R1 обозначает -H):
Формула 30
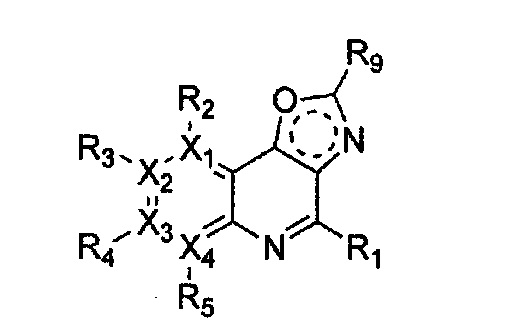
Формула 31
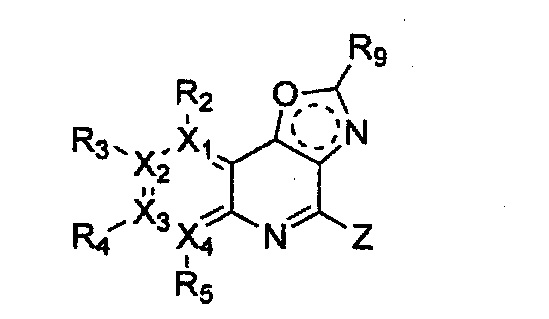
Формула 32
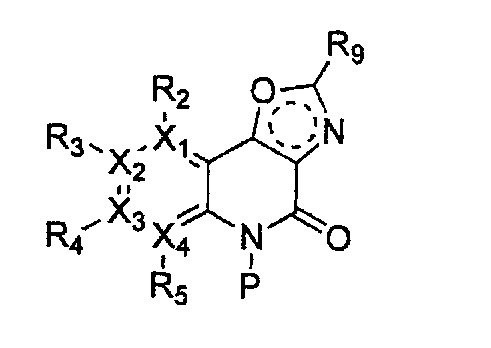
Формула 33
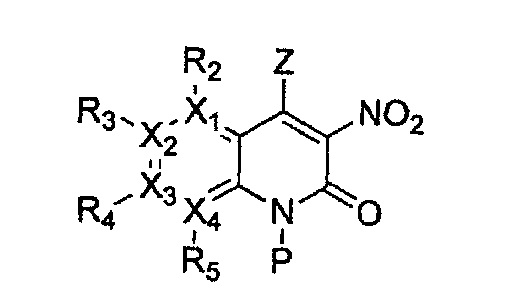
Формула 34
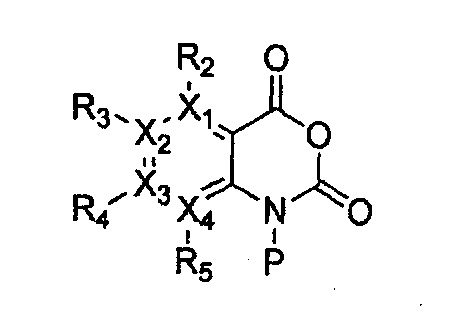
Формула 35
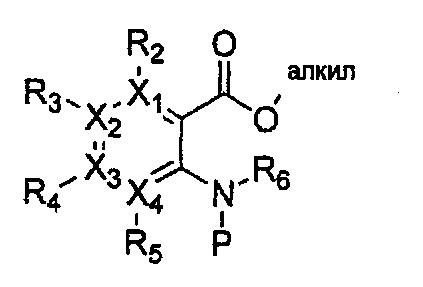
Формула 36

Формула 37

причем в формулах 30-37 X1, X2, Х3, X4, R1, R2, R3, R4, R5, R6, R7, R8 и R9 имеют значения, определенные в формуле 1; Z обозначает реактивную уходящую группу, предпочтительно атом галогена (-F, -Cl, -Br); и P обозначает защитную группу, предпочтительно п-метоксибензил, 3',5'-диметоксибензил и три-метоксибензил.
Способ 6 более специфично описан далее.
Соединения формул 30, 31, 32, 33, 34, 35, 36 и 37, используемые как исходный материал, могут быть получены способом, известным в данной области техники.
Получение соединения следующей формулы 36 через этерификацию соединения следующей формулы 37 осуществляют согласно обычному способу в присутствии подходящего растворителя и этерифицирующего агента.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительными растворителями для этой реакции являются дихлорметан и метанол.
Следующую стадию получения соединения формулы 35 через арилирование полученного соединения формулы 36 осуществляют согласно обычному способу в присутствии подходящего растворителя.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является этанол.
Следующую стадию получения соединения формулы 34 через циклизацию полученного соединения формулы 35 осуществляют согласно обычному способу в присутствии подходящего растворителя и агента циклизации.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является диоксан.
Примеры агентов циклизации для этой реакции включают дифосген и трифосген. Предпочтительным является дифосген. Реакция может быть проведена при, но не ограничиваясь этим, 50-200°C в течение приблизительно от 0,1 до 24 часов.
Следующую стадию получения соединения формулы 33 через реакцию присоединения енолата полученного соединения формулы 34 осуществляют согласно обычному способу в присутствии подходящего растворителя.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является тетрагидрофуран.
Реакция может быть проведена при, но не ограничиваясь этим, 50-200°C в течение приблизительно от 0,1 до 24 часов.
Следующую стадию получения соединения формулы 32 через циклизацию полученного соединения формулы 33 осуществляют согласно обычному способу в присутствии подходящего растворителя.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является уксусная кислота.
Реакция может быть проведена при, но не ограничиваясь этим, 50-200°C в течение приблизительно от 0,1 до 24 часов.
Следующую стадию получения соединения формулы 31 через галогенирование полученного соединения формулы 32 осуществляют согласно обычному способу в присутствии подходящего галогенирующего реагента.
Примеры галогенирующих реагентов для этой реакции включают трихлорид фосфора (3), фосфорил хлорид, оксибромид фосфора, фенилфосфонил хлорид и пентахлорид фосфора, предпочтительным является фосфорил хлорид. Реакция может быть проведена при, но не ограничиваясь этим, 50-200°C в течение приблизительно от 0,1 до 24 часов.
Следующую стадию получения соединения формулы 30 через арилирование полученного соединения формулы 31 соединением формулы R1-H (за исключением того, что R1 обозначает -H) осуществляют согласно обычному способу в присутствии подходящего растворителя и основания.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является N,N-диметилформамид.
Вышеописанный способ получения соединения формулы 30 более специфично описан в следующем примере.
Способ 7
Соединение, имеющее структуру следующей формулы 38, которая является формулой 1 как определено выше, в которой Y1 обозначает N, каждый из Y2 и Y3 независимо обозначает C или N, и по меньшей мере один из Y1, Y2 и Y3 обозначает N, может быть получено способом, включающим стадии: получение соединения следующей формулы 40 через реакцию сочетания Сузуки соединения следующей формулы 42 с соединением следующей формулы 41; получение соединения формулы 39 через галогенирование полученного соединения формулы 40; и проведение арилирования полученного соединения формулы 39 соединением формулы R1-H (за исключением того, что R1 обозначает -H):
Формула 38

Формула 39
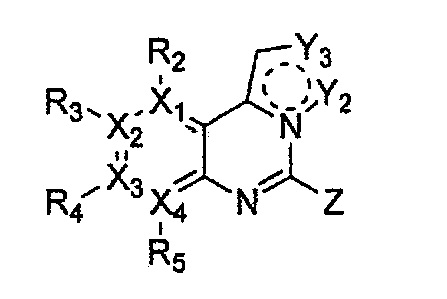
Формула 40

Формула 41
Формула 42
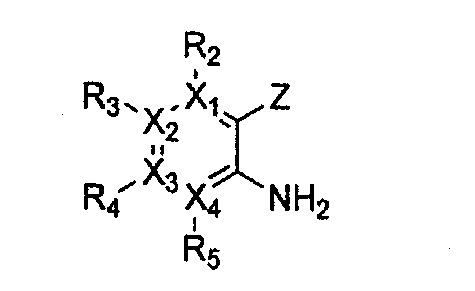
причем в формулах 38-42 X1, X2, Х3, X4, R1, R2, R3, R4, R5, R6, R7, R8 и R9 имеют значения, определенные в формуле 1; и Z обозначает реактивную уходящую группу, предпочтительно атом галогена (-F, -Cl, -Br).
Способ 7 более специфично описан далее.
Соединения формул 38, 39, 40, 41 и 42, используемые как исходный материал, могут быть получены способом, известным в данной области техники.
Получение соединения формулы 40 через реакцию сочетания Сузуки соединения формулы 42 с соединением формулы 41 осуществляют согласно обычному способу в присутствии подходящего растворителя и катализатора.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительными растворителями для этой реакции являются N,N-диметилформамид и вода.
Примеры катализаторов для этой реакции включают тетракис (трифенилфосфин) палладий (Pd(PPh3)4), ацетат палладия (II) (Pd(ОАс)2), бис(трифенилфосфин)палладий (II) дихлорид (PdCl2(PPh3)2), [1,1-бис(дифенилфосфино)ферроцен]дихлор-палладий (II) (PdCl2(dppf)), (дибензилиденацетон) дипалладий (0) (Pd(dba)2) и хлорид палладия (II) (PdCl2). Предпочтительным является тетракис(трифенилфосфин)палладий (Pd(PPh3)4). Реакция может быть проведена при, но не ограничиваясь этим, 50-200°C в течение приблизительно от 0,1 до 24 часов.
Следующую стадию получения соединения формулы 39 через галогенирование полученного соединения формулы 40 осуществляют согласно обычному способу в присутствии подходящего галогенирующего реагента.
Примеры галогенирующих реагентов для этой реакции включают трихлорид фосфора (3), фосфорил хлорид, оксибромид фосфора, фенилфосфонил хлорид и пентахлорид фосфора. Предпочтительным является фосфорил хлорид. Реакция может быть проведена при, но не ограничиваясь этим, 50-200°C в течение приблизительно от 0,1 до 24 часов.
Следующую стадию получения соединения формулы 38 через арилирование полученного соединения формулы 39 соединением формулы R1-H (за исключением того, что R1 обозначает -H) осуществляют согласно обычному способу в присутствии подходящего растворителя и основания.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является дихлорметан.
Вышеописанный способ получения соединения формулы 5 более специфично описан в следующем примере.
Способ 8
Соединение, имеющее структуру следующей формулы 43, которая является формулой 1 как определено выше, в которой Y5 обозначает N, может быть получено способом, включающим стадии: получение соединения следующей формулы 46 через пирролирование соединения следующей формулы 47; получение соединения формулы 45 через циклизацию полученного соединения формулы 46; получение соединения формулы 44 через галогенирование полученного соединения формулы 45; и проведение арилирования полученного соединения формулы 44 соединением формулы R1-H (за исключением того, что R1 обозначает -H):
Формула 43
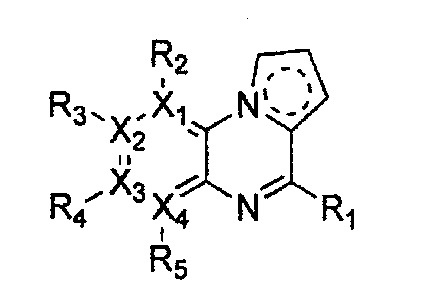
Формула 44

Формула 45
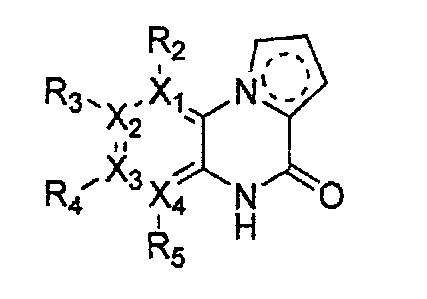
Формула 46
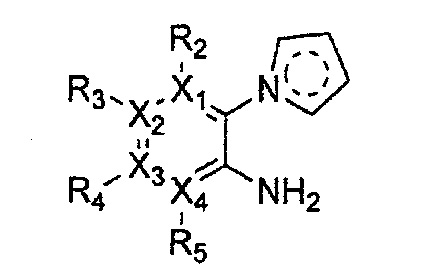
Формула 47
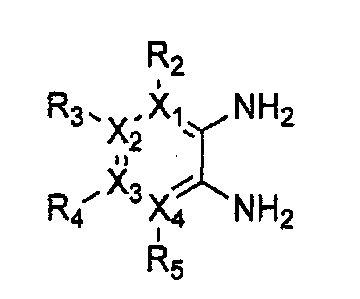
причем в формулах 43-47 X1, X2, Х3, X4, R1, R2, R3, R4, R5, R6, R7, R8 и R9 имеют значения, определенные в формуле 1; и Z обозначает реактивную уходящую группу, предпочтительно атом галогена (-F, -Cl, -Br).
Способ 8 более специфично описан далее.
Соединения формул 43, 44, 45, 46 и 47, используемые как исходный материал, могут быть получены способом, известным в данной области техники.
Получение соединения формулы 46 через пирролирование соединения формулы 47 осуществляют согласно обычному способу в присутствии подходящего растворителя.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является уксусная кислота. Реакция может быть проведена при, но не ограничиваясь этим, 50-200°C в течение приблизительно от 0,1 до 24 часов.
Следующую стадию получения соединения формулы 45 через циклизацию полученного соединения формулы 46 осуществляют согласно обычному способу в присутствии подходящего растворителя и агента циклизации.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является толуол.
Примеры агентов циклизации для этой реакции включают дифосген и трифосген. Предпочтительным является трифосген. Реакция может быть проведена при, но не ограничиваясь этим, 50-200°C в течение приблизительно от 0,1 до 24 часов.
Следующую стадию получения соединения формулы 44 через галогенирование полученного соединения формулы 45 осуществляют согласно обычному способу в присутствии подходящего галогенирующего реагента.
Примеры галогенирующих реагентов для этой реакции включают трихлорид фосфора (3), фосфорил хлорид, оксибромид фосфора, фенилфосфонил хлорид и пентахлорид фосфора. Предпочтительным является фосфорил хлорид. Реакция может быть проведена при, но не ограничиваясь этим, 50-200°C в течение приблизительно от 0,1 до 24 часов.
Следующую стадию получения соединения формулы 43 через арилирование полученного соединения формулы 44 соединением формулы R1-H (за исключением того, что R1 обозначает -H) осуществляют согласно обычному способу в присутствии подходящего растворителя и основания.
Обычные растворители, которые не оказывают никаких неблагоприятных эффектов на реакцию, могут использоваться на этой стадии. Предпочтительные примеры растворителя включают растворители на основе простых эфиров, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, 2-бутанон, диглим и т.д.; растворители на основе углеводородов, такие как бензол, пиридин, толуол, гексан, ксилол и т.д.; растворители на основе галогензамещенных водородов, такие как дихлорметан, хлороформ, тетрахлорметан, 1,2-дихлорэтан и т.д.; растворители на основе спиртов, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.д.; растворители на основе сложных эфиров, такие как этилацетат, метилацетат, бутилацетат и т.д.; полярные растворители, такие как ацетон, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, диметил сульфоксид, ацетонитрил и т.д.; и смеси этих растворителей. Особенно предпочтительным растворителем для этой реакции является N,N-диметилацетамид.
Выгодные эффекты изобретения
Новые гетероциклические соединения согласно настоящему изобретению показывают такую же или более сильную ингибирующую активность в отношении рецептора гистамина 4 по сравнению с обычными ингибиторами человеческого рецептора гистамина 4 (hH4R); показывают селективность для каждого из подтипов рецепторов гистамина и рецепторов, транспортеров и ионных каналов на мембране; и имеют более высокую растворимость, метаболическую стабильность, и соответственно, как результат фармакокинетического анализа и сравнения с соединением, раскрытым в WO 2010/030785, при использовании модели животных, такой как крыса SD, соединения по изобретению показали в 7-8 раз более высокие эффекты в отношении фармакокинетиках профилей, таких как AUCinf и максимальная концентрация в крови, чем сравнительное соединение. Относительно симптомов, обнаружено что зуд, вызванный гистамином, веществом P и соединением 48/80 и т.д., эффективно подавляется, и что гетероциклическое соединение согласно настоящему изобретению имеет в 3 раза более сильный эффект, чем соединение, раскрытое в WO 2010/030785, в отношении супрессии вызванной гистамином инфильтрации воспалительных клеток, таких как тучные клетки и эозинофилы. В модели оксазолон-индуцированного атопического дерматита гетероциклическое соединение согласно настоящему изобретению показало намного более сильный противовоспалительный эффект, чем соединение, раскрытое в WO 2010/030785. Особенно в мышиной NC/Nga модели Dermatophagoides farina-индуцированного атопического дерматита обнаружено, что гетероциклическое соединение согласно настоящему изобретению демонстрирует такой же противовоспалительный эффект, как Tacrolimus, который является иммуносупрессивным лекарственным средством, и обнаружено соединение, имеющее селективность в отношении рецептора серотонина 3.
Поэтому новые гетероциклические соединения согласно настоящему изобретению и содержащие их фармацевтические композиции могут быть очень эффективными в лечении или профилактике воспалительных заболеваний, аллергии, боли, носовых полипов, ринита, хронического синусита, заложенности носа, носового зуда, астмы, хронического обструктивного заболевания легких, ревматоидного артрита, атопического дерматита, псориаза, экземы, зуда, кожного зуда, крапивницы, идиопатической хронической крапивницы, склеродермии, конъюнктивита, кератоконъюнктивита, глазного воспаления, сухости глаз, кардиальной дисфункции, аритмии, атеросклероза, рассеянного склероза, воспалительного заболевания кишечника (включая колит, болезнь Крона, неспецифический язвенный колит), воспалительной боли, нейропатической боли, боли при остеоартрите, аутоиммунного заболевания щитовидной железы, иммуно-опосредованного (также известного как тип 1) диабета, волчанки, послеоперационных спаек, вестибулярных нарушений и рака.
Способ осуществления изобретения
Настоящее изобретение будет объяснено в дальнейших деталях со ссылкой на следующие примеры и эксперименты. Однако эти примеры и эксперименты предназначены только для иллюстрации настоящего изобретения, и настоящее изобретение ни в коем случае ими не ограничено.
Аббревиатуры, используемые в следующих примерах, определены следующим образом:



Пример 1
Синтез 3-метил-6-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразина
(a) Синтез 2-хлор-3-гидразинил-7-метилпиридо[2,3-b]пиразина
2,3-дихлор-7-метилпиридо[2,3-b]пиразин (500,0 мг, 2,33 ммоль) и моногидрат гидразина (234,0 мг, 4,66 ммоль) растворяли в EtOH (10,0 мл), перемешивали в течение 12 часов при комнатной температуре и затем перегоняли при пониженном давлении. Добавляли Et2O, получая твердое вещество. Образовавшееся твердое вещество затем отфильтровывали и высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 2-хлор-3-гидразинил-7-метилпиридо[2,3-b]пиразин.
ЖХ/МС ESI (+): 210 (М+1), 212 (М+3)
(b) Синтез 6-хлор-3-метилпиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразина
Смесь неочищенного 2-хлор-3-гидразинил-7-метилпиридо[2,3-b]пиразина и триметилортоформиата (5,0 мл) перемешивали при 100°C в течение одного часа и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество. Образовавшееся твердое вещество затем отфильтровывали и высушивали при пониженном давлении, получая твердое соединение кремового цвета, представляющее собой 6-хлор-3-метилпиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразин (340,0 мг, 66% за 2 стадии).
ЖХ/МС ESI (+): 220 (М+1), 222 (М+3)
(c) Синтез 3-метил-6-(4-метилпиперазин-1-ил)пиридо[3,2-e][1,2,4]триазоло[4,3-а]пиразина
6-хлор-3-метилпиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразин (50,0 мг, 0,23 ммоль) растворяли в DMF (2,0 мл), и N-метилпиперазин (48,0 мг, 0,48 ммоль) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение одного часа и перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (от DCM:MeOH=100:0 до DCM:MeOH=98:2) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение белого цвета, представляющее собой 3-метил-6-(4-метил-1-ил)пиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразин (25,0 мг, 38%).
ЖХ/МС ESI (+): 284 (М+1), 286 (М+3)
1H-ЯМР (300 МГц, ДMCO-d6); δ: 9,83 (с, 1Н), 8,20 (м, 1Н), 7,82 (м, 1Н), 4,34 (шир. с, 4Н), 2,50 (м, 4Н), 2,42 (с, 3Н), 2,24 (с, 3H).
Пример 2
Синтез 8-метил-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
(а) Синтез 2-хлор-7-метил-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразина
2,3-дихлор-7-метилпиридо[2,3-b]пиразин (500,0 мг, 2,30 ммоль) растворяли в DCM (8,0 мл), и N-метилпиперазин (468,0 мг, 2,60 ммоль) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов и выливали в воду, и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:МеОН от 100:0 до 99:1) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 2-хлор-7-метил-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин.
ЖХ/МС ESI (+): 278 (М+1), 280 (М+3)
(b) Синтез 8-метил-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
Неочищенный 2-хлор-7-метил-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин (300,0 мг, 1,08 ммоль) и моногидрат гидразина (108,0 мг, 2,16 ммоль) растворяли в EtOH (10,0 мл), перемешивали в течение 12 часов при 50°C и затем перегоняли при пониженном давлении. Реакционную смесь выливали в воду и экстрагировали DCM. Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем упаривали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 2-гидразинил-7-метил-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин. Смесь неочищенного 2-гидразинил-7-метил-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразина и триметилортоформиата (2,0 мл) перемешивали при 100°C в течение одного часа и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали. Остаток очищали хроматографией на колонках (DCM:MeOH=99:1) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 8-метил-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (94,0 мг, 31%).
ЖХ/МС ESI (+): 284 (М+1)
1Н-ЯМР (300 МГц, ДMCO-d6); δ: 9,99 (с, 1Н), 8,45 (м, 2Н), 4,37 (шир. с, 4Н), 2,50 (м, 4Н), 2,43 (с, 3H), 2,24 (с, 3H).
Пример 3
Синтез 4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
(а) Синтез 2-хлор-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразина
2,3-дихлорпиридо[2,3-b]пиразин (300,0 мг, 1,50 ммоль) растворяли в DMF (5,0 мл), и N-метилпиперазин (0,3 мл, 2,97 ммоль) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем выливали в насыщенный водный раствор NH4Cl, и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=100:0) на аминосиликагеле. Фракции, содержащие продукты, собирали и упаривали, получая твердое соединение светло-желтого цвета, представляющее собой 2-хлор-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин (265,0 мг, 67%).
ЖХ/МС ESI (+): 264 (М+1), 266 (М+3)
(b) Синтез 4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин
2-хлор-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин (260,0 мг, 0,99 ммоль) и моногидрат гидразина (63,0 мг, 1,97 ммоль) растворяли в EtOH (10,0 мл), перемешивали при комнатной температуре в течение 2 часов и затем перегоняли при пониженном давлении. Реакционную смесь выливали в насыщенный водный раствор NH4Cl и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем упаривали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 2-гидразинил-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин. Смесь неочищенного 2-гидразинил-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразина и триметилортоформиата (2,0 мл) перемешивали при 80°C в течение одного часа и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=100:0) на аминосиликагеле. Фракции, содержащие продукт, собирали, получая твердое соединение светло-желтого цвета, представляющее собой 4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (186,0 мг, 70%).
ЖХ/МС ESI (+): 270 (М+1)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 10,04 (с, 1Н), 8,61-8,54 (м, 2Н), 7,39-7, 32 (м, 1Н), 4,60-4,21 (м, 4Н), 2, 55-2, 44 (м, 4Н), 2,25 (с, 3H).
Пример 4
Синтез 8-хлор-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
(а) Синтез 2,3,7-трихлорпиридо[2,3-b]пиразина
5-хлорпиридин-2,3-диамин (10000,0 мг, 69,65 ммоль) добавляли к диэтилоксалату (30,0 мл), и реакционную смесь перемешивали при 100°C в течение 12 часов и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество. Образовавшееся твердое вещество затем отфильтровывали и высушивали при пониженном давлении, получая твердое соединение ярко-коричневого цвета, представляющее собой 7-хлорпиридо[2,3-b]пиразин-2,3-диол. Смесь неочищенного 7-хлорпиридо[2,3-b]пиразин-2,3-диола и POCl3 (30,0 мл) перемешивали при 130°C в течение 12 часов и затем охлаждали до комнатной температуры. Реакционную смесь выливали в воду со льдом. Сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая твердое соединение коричневого цвета, представляющее, собой 2,3,7-трихлорпиридо[2,3-b]пиразин (13700,0 мг, 84% за 2 стадии).
ЖХ/МС ESI (+): 234 (М+1), 236 (М+3)
1H-ЯМР (300 МГц, ДMCO-d6); δ: 9,23 (д, 1Н, J=2,6 Гц), 8,86 (д, 1Н, J=2,6 Гц).
(b) Синтез 2,7-дихлор-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразина
2,3,7-трихлорпиридо[2,3-b]пиразин (428,0 мг, 1,83 ммоль) и TEA (2,5 мл, 18,30 ммоль) растворяли в DCM (10,0 мл), и N-метилпиперазин (0,2 мл, 2,01 ммоль), разбавленный в DCM (5,0 мл), медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем вливали в насыщенный водный раствор NH4Cl и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=100:0) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 2,7-дихлор-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин (290,0 мг, 53%).
ЖХ/МС ESI (+): 298 (М+1), 300 (М+3)
(с) Синтез 8-хлор-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
2,7-дихлор-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин (290,0 мг, 0,97 ммоль) и моногидрат гидразина (98,0 мг, 1,96 ммоль) растворяли в EtOH (10,0 мл), перемешивали в течение 2 часов при комнатной температуре и затем перегоняли при пониженном давлении. Реакционную смесь выливали в насыщенный водный раствор NH4Cl и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем упаривали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 7-хлор-2-гидразинил-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин. Смесь неочищенного 7-хлор-2-гидразинил-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразина и триметилортоформиата (5,0 мл) перемешивали при 80°C в течение одного часа и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=100:0) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 8-хлор-4-(4-метилпиперазин-1-ил)пиридо[2,3-е] [1,2,4]триазоло[4,3-а]пиразин (215,0 мг, 73% за 2 стадии).
ЖХ/МС ESI (+): 304 (М+1), 306 (М+3)
1H-ЯМР (300 МГц, ДМСО-d6); δ: 10,01 (с, 1Н), 8,85 (д, 1Н, J=2,4 Гц), 8,58 (д, 1Н, J=2,4 Гц), 4,40 (шир. с, 4Н), 2,53 (м, 4Н), 2,24 (с, 3H).
Пример 5
Синтез 3-хлор-6-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразина
(а) Синтез 2,7-дихлор-3-гидразинилпиридо[2,3-b]пиразина 2,3,7-трихлорпиридо[2,3-b]пиразин (150,0 мг, 0,64 ммоль), TEA (129,0 мг, 1,28 ммоль), моногидрат гидразина (35,0 мг, 0,74 ммоль) растворяли в EtOH (5,0 мл), перемешивали при комнатной температуре в течение 12 часов и затем перегоняли при пониженном давлении. Добавляли Et2O, получая твердое вещество. Сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 2,7-дихлор-3-гидразинилпиридо [2,3-b]пиразин.
ЖХ/МС ESI (+): 230 (М+1), 232 (М+3)
(b) Синтез 3-хлор-6-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразина
Смесь неочищенного 2,7-дихлор-3-гидразинилпиридо[2,3-b]пиразина и триметилортоформиата (2,0 мл) перемешивали при 100°C в течение 2 часов и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество. Сформированное твердое вещество отфильтровывали, получая твердое соединение желтого цвета, представляющее собой 3,6-дихлорпиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразин. Неочищенный 3,6-дихлорпиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразин растворяли в DMF (2,0 мл), и затем N-метилпиперазин (0,1 мл, 0,90 ммоль), разбавленный в DMF (1,0 мл), медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и выливали в воду, и экстрагировали EtOAc (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=99:1) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 3-хлор-6-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразин (24,0 мг, 12% за 3 стадии).
ЖХ/МС ESI (+): 304 (М+1), 306 (М+3)
1H-ЯМР (300 МГц, CDCl3); δ: 9,46 (с, 1Н), 8,22 (д, 1Н, J=2,4 Гц), 7,93 (д, 1Н, J=2,4 Гц), 4,54 (шир. с, 4Н), 2,61 (м, 4Н), 2,37 (с, 3H).
Пример 6
Синтез 6-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразина
2,3-дихлорпиридо[2,3-b]пиразин (300,0 мг, 1,50 ммоль) и моногидрат гидразина (48,1 мг, 1,50 ммоль) растворяли в EtOH (7,0 мл), перемешивали при комнатной температуре в течение 2 часов и затем перегоняли при пониженном давлении. Реакционную смесь выливали в насыщенный водный раствор NH4Cl и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем упаривали при пониженном давлении, получая 2-хлор-3-гидразинилпиридо[2,3-b]пиразин. Смесь неочищенного 2-хлор-3-гидразинилпиридо[2,3-b]пиразина и триметилортоформиата (2,0 мл) перемешивали при 80°C в течение одного часа и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество. Сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая 6-хлорпиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразин. Неочищенный 6-хлорпиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразин растворяли в DMF (5,0 мл), и затем N-метилпиперазин (0,3 мл, 2,97 ммоль) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем вливали в насыщенный водный раствор NH4Cl и экстрагировали DCM. Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=100:0) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение светло-желтого цвета, представляющее собой 6-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразин (70,0 мг, 17% за 3 стадии).
ЖХ/МС ESI (+): 270 (М+1)
1Н-ЯМР (300 МГц, CDCl3); δ: 9,51 (с, 1Н), 8,30 (дд, 1Н, J=1,5 Гц, J=4,6 Гц), 7,94 (дд, 1Н, J=1,5 Гц, J=8,0 Гц), 7,44 (дд, 1Н, J=4,6 Гц, J=8,0 Гц), 4,70-4,34 (м, 4Н), 2,58-2,56 (м, 4Н), 2,38 (с, 3H).
Пример 7
Синтез 8-хлор-4-(пиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
(a) Синтез 4-(2,7-дихлорпиридо[2,3-b]пираэин-3-ил)пиперазин-1-трет-бутилкарбоксилата
2,3,7-трихлорпиридо[2,3-b]пиразин (800,0 мг, 3,41 ммоль) и TEA (2,4 мл, 17,05 ммоль) растворяли в DCM (20,0 мл), и затем пиперазин-1-трет-бутилкарбоксилат (667,0 мг, 3,58 ммоль), разбавленный в DCM (10,0 мл), медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов и затем вливали в насыщенный водный раствор NH4Cl и экстрагировали DCM (50,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (n-Hex:EtOAc от 90:10 до 80:20) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 4-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)пиперазин-1-трет-бутилкарбоксилат (892,0 мг, 68%).
ЖХ/МС ESI (+): 384 (М+1), 389 (М+3)
(b) Синтез 4-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)пиперазин-1-трет-бутилкарбоксилата
4-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)пиперазин-1-трет-бутилкарбоксилат (892,0 мг, 2,32 ммоль) и моногидрат гидразина (244,0 мг, 4,87 ммоль) растворяли в EtOH (50,0 мл), перемешивали при комнатной температуре в течение 2 часов и затем перегоняли при пониженном давлении. Реакционную смесь выливали в насыщенный водный раствор NH4Cl и экстрагировали DCM (50,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 4-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)пиперазин-1-трет-бутилкарбоксилат.
ЖХ/МС ESI (+): 380 (М+1), 382 (М+3)
(с) Синтез 4-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)пиперазин-1-трет-бутилкарбоксилата
Смесь неочищенного 4-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)пиперазин-1-трет-бутилкарбоксилата и триметилортоформиата (20,0 мл) перемешивали при 70°C в течение одного часа и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество. Сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=100:0) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 4-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)пиперазин-1-трет-бутилкарбоксилат (814,0 мг, 90%).
ЖХ/МС ESI (+): 390 (М+1), 392 (М+3)
(d) Синтез 8-хлор-4-(пиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
4-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)пиперазин-1-трет-бутилкарбоксилат (814,0 мг, 2,09 ммоль) растворяли в DCM (20,0 мл), и TFA (5,0 мл) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем перегоняли при пониженном давлении. Остаток нейтрализовали водным раствором NaHCO3 (рН=7) и затем экстрагировали DCM (50,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH от 90:10 до 80:20) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 8-хлор-4-(пиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (503,0 мг, 83%).
ЖХ/МС ESI (+): 290 (М+1), 292 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 10,00 (с, 1Н), 8,84 (д, 1Н, J=2,4 Гц), 8,56 (д, 1Н, J=2,4 Гц), 4,40 (шир. с, 4Н), 2,88 (м, 4Н).
Пример 8
Синтез 1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-амина
(а) Синтез (1-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)-трет-бутилкарбамата
2,3,7-трихлорпиридо[2,3-b]пиразин (200,0 мг, 0,85 ммоль) и TEA (1,2 мл, 8,50 ммоль) растворяли в DCM (10,0 мл), и азетидин-3-ил-трет-бутилкарбамат (162,0 мг, 0,94 ммоль), разбавленный в DCM (5,0 мл), медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов и выливали в насыщенный водный раствор NH4Cl и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH от 100:0 до 99:1) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение коричневого цвета, представляющее собой (1-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)-трет-бутилкарбамат (224,0 мг, 71%).
ЖХ/МС ESI (+): 370 (М+1), 372 (М+3)
(b) Синтез (1-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил) азетидин-3-ил)-трет-бутилкарбамата
(1-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)-трет-бутилкарбамат (224,0 мг, 0,61 ммоль) и моногидрат гидразина (76,0 мг, 1,53 ммоль) растворяли в EtOH (10,0 мл), перемешивали при комнатной температуре в течение 2 часов и затем перегоняли при пониженном давлении. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой (1-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)-трет-бутилкарбамат (100,0 мг, 41%).
ЖХ/МС ESI (+): 366 (М+1), 368 (М+3)
с) Синтез (1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)-трет-бутилкарбамата
Смесь (1-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил) азетидин-3-ил)-трет-бутилкарбамата (100,0 мг, 0,27 ммоль) и триметилортоформиата (5,0 мл) перемешивали при 70°C в течение одного часа и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая твердое соединение кремового цвета, представляющее собой (1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)-трет-бутилкарбамат (70,0 мг, 68%).
ЖХ/МС ESI (+): 376 (М+1), 378 (М+3)
(d) Синтез 1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-амина
(1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)-трет-бутилкарбамат (70,0 мг, 0,19 ммоль) растворяли в DCM (5,0 мл), и TFA (2,0 мл) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем перегоняли при пониженном давлении. Остаток нейтрализовали водным раствором NaНCO3 (рН=7) и затем экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH от 90:10 до 50:50) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-амин (6,3 мг, 12%).
ЖХ/МС ESI (+): 276 (М+1), 278 (М+3)
1Н-ЯМР (300 МГц, ДMCO-d6); δ: 9,95 (с, 1Н), 8,80 (д, 1Н, J=2,3 Гц), 8,53 (д, 1Н, J=2,4 Гц), 4,93 (м, 1Н), 4,47 (м, 1Н), 4,35 (м, 1Н), 3,93 (м, 2Н).
Пример 9
Синтез (R)-1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N,N-диметилпирролидин-3-амина
(R)-1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)пирролидин-3-амин (20,0 мг, 0,07 ммоль) и формиат натрия (20,0 мг, 0,29 ммоль) растворяли в смеси муравьиная кислота/формамид (1,0 мл/1,0 мл), перемешивали при 100°C в течение одного часа и затем перегоняли при пониженном давлении. Реакционную смесь нейтрализовали водным раствором NaHCO3 (рН=7) и затем экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH от 90:10 до 80:20) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой (R)-1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N,N-диметилпирролидин-3-амин (13,0 мг, 59%).
ЖХ/МС ESI (+): 318 (М+1), 320 (М+3)
1Н-ЯМР (300 МГц, CDCl3); δ: 9,15 (с, 1Н), 8,59 (д, 1Н, J=2,2 Гц), 8,01 (д, 1Н, J=2,3 Гц), 4,90 (м, 1Н), 4,40-3, 60 (м, 3H), 2,90 (м, 1Н), 2,39 (с, 3H), 2,37 (с, 3H), 2,30 (м, 1Н), 2,00 (м, 1Н).
Пример 10
Синтез (R)-1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилпирролидин-3-амина
(а) Синтез (R)-(1-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)пирролидин-3-ил)(метил)-трет-бутилкарбамата
2,3,7-трихлорпиридо[2,3-b]пиразин (100,0 мг, 0,43 ммоль) и TEA (0,6 мл, 4,30 ммоль) растворяли в DCM (10,0 мл), (R)-пирролидин-3-ил-трет-бутилкарбамат (94,0 мг, 0,47 ммоль), разбавленный в DCM (5,0 мл), медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов и затем выливали в насыщенный водный раствор NH4Cl и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=100:0) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой (R)-(1-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)пирролидин-3-ил)(метил)-трет-бутилкарбамат (169,0 мг, 99%).
ЖХ/МС ESI (+): 398 (М+1), 400 (М+3)
(b) Синтез (R)-(1-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)пирролидин-3-ил)(метил)-трет-бутилкарбамата
(R)-(1-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)пирролидин-3-ил)(метил)-трет-бутилкарбамат (169,0 мг, 0,42 ммоль) и моногидрат гидразина (53,0 мг, 1,05 ммоль) растворяли в EtOH (5,0 мл), перемешивали при комнатной температуре в течение 2 часов и затем перегоняли при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой (R)-(1-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)пирролидин-3-ил)(метил)-трет-бутилкарбамат.
ЖХ/МС ESI (+): 394 (М+1), 396 (М+3)
(c) Синтез (R)-(1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)пирролидин-3-ил)(метил)-трет-бутилкарбамата
Смесь неочищенного (R)-(1-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)пирролидин-3-ил)(метил)-трет-бутилкарбамата и триметилортоформиата (5,0 мл) перемешивали при 80°C в течение одного часа и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая твердое соединение кремового цвета, представляющее собой (R)-(1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)пирролидин-3-ил)(метил)-трет-бутилкарбамат.
ЖХ/МС ESI (+): 404 (М+1), 406 (М+3)
(d) Синтез (R)-1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилпирролидин-3-амина
Неочищенный (R)-(1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)пирролидин-3-ил)(метил)-трет-бутилкарбамат растворяли в DCM (3,0 мл), и TFA (1,0 мл) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем перегоняли при пониженном давлении. Остаток нейтрализовали водным раствором NaHCO3 (рН=7) и затем экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH от 90:10 до 50:50) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой (R)-1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилпирролидин-3-амин (58,6 мг, 45% за 3 стадии).
ЖХ/МС ESI (+): 304 (М+1), 306 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 9,97 (с, 1Н), 8,80 (д, 1Н, J=2,4 Гц), 8,53 (д, 1Н, J=2,4 Гц), 4,50-4,20 (м, 2Н), 3,90-3,40 (м, 2Н), 3,30 (м, 1Н), 2,32 (с, 3H), 2,20-1,80 (м, 2Н).
Пример 11
Синтез (R)-1-(3-хлорпиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразин-6-ил)-N-метилпирролидин-3-амина
Твердое соединение кремового цвета, представляющее собой (R)-1-(3-хлорпиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразин-6-ил)-N-метилпирролидин-3-амин (18,0 мг, 14%) получали как описано в примере 10.
ЖХ/МС ESI (+): 304 (М+1), 306 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 9,84 (с, 1Н), 8,31 (д, 1Н, J=2,2 Гц), 8,04 (д, 1Н, J=2,2 Гц), 4,50-4,20 (м, 2Н), 3,90-3,50 (м, 2Н), 3,30 (м, 1Н), 2,33 (с, 3H), 2,20-1,80 (м, 2Н).
Пример 12
Синтез 8-бром-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
(а) Синтез 7-бром-2,3-дихлорпиридо[2,3-b]пиразина
5-бромпиридин-2,3-диамин (5000,0 мг, 2,66 ммоль) добавляли к диэтилоксалату (20,0 мл), и смесь перемешивали при 120°C в течение 12 часов и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество. Образовавшееся твердое вещество затем отфильтровывали и высушивали при пониженном давлении, получая твердое соединение светло-коричневого цвета, представляющее собой 7-бромпиридо[2,3-b]пиразин-2,3-диол. Смесь неочищенного 7-бромпиридо[2,3-b]пиразин-2,3-диола и POCl3 (20,0 мл) перемешивали при 130°C в течение 12 часов и затем охлаждали до комнатной температуры. Реакционную смесь выливали в воду со льдом, получая твердое вещество. Сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая твердое соединение коричневого цвета, представляющее собой 7-бром-2,3-дихлорпиридо[2,3-b]пиразин (6500,0 мг, 72% за 2 стадии).
ЖХ/МС ESI (+): 278 (М+1), 280 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 9,28 (д, 1Н, J=2,4 Гц), 8,99 (д, 1Н, J=2,4 Гц).
(b) Синтез 7-бром-2-хлор-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразина
7-бром-2,3-дихлорпиридо[2,3-b]пиразин (1000,0 мг, 3,59 ммоль) и TEA (5,0 мл, 35,90 ммоль) растворяли в DCM (20,0 мл), и N-метилпиперазин (0,2 мл, 2,01 ммоль), разбавленный в DCM (1,0 мл), медленно добавляли при -10°C. Реакционную смесь перемешивали в течение 12 часов и затем вливали в насыщенный водный раствор NH4Cl и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=100:0) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 7-бром-2-хлор-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин (1000,0 мг, 81%).
ЖХ/МС ESI (+): 342 (М+1), 344 (М+3)
(с) Синтез 8-бром-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
7-бром-2-хлор-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин (413,0 мг, 0,12 ммоль) и моногидрат гидразина (150,0 мг, 0,30 ммоль) растворяли в EtOH (10,0 мл), перемешивали при комнатной температуре в течение 4 часов и затем перегоняли при пониженном давлении. Добавляли Et2O, получая твердое вещество. Образовавшееся твердое вещество затем отфильтровывали и высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 7-бром-2-гидразинил-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин. Смесь неочищенного 7-бром-2-гидразинил-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразина и триметилортоформиата (10,0 мл) перемешивали при 80°C в течение 2 часов и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество. Образовавшееся твердое вещество затем отфильтровывали и высушивали при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH от 100:0 до 95:5) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 8-бром-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (368,0 мг, 29% за 3 стадии).
ЖХ/МС ESI (+): 348 (М+1), 350 (М+3)
1H-ЯМР (300 МГц, ДMCO-d6); δ: 10,01 (с, 1Н), 8,95 (д, 1Н, J=2,3 Гц), 8,63 (д, 1Н, J=2,2 Гц), 4,40 (шир. с, 4Н), 2,53 (м, 4Н), 2,24 (с, 3H).
Пример 13
Синтез 4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-8-карбонитрила
8-бром-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (50,0 мг, 0,14 ммоль), Zn(CN)2 (17,0 мг, 0,14 ммоль) и Pd(PPh3)4 (33,0 мг, 0,01 ммоль) растворяли в DMF (1,0 мл) и давали взаимодействовать в микроволновом реакторе в условиях 60 Вт, 90°C, в течение 2 часов и затем охлаждали до комнатной температуры. Реакционную смесь затем выливали в воду и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH от 100:0 до 95:5) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-8-карбонитрил (6,0 мг, 13%).
ЖХ/МС ESI (+): 295 (М+1)
1H-ЯМР (300 МГц, ДMCO-d6); δ: 10,01 (с, 1Н), 9,13 (д, 1Н, J=2,0 Гц), 8,93 (д, 1Н, J=2,0 Гц), 4,88 (шир. с, 2Н), 4,14 (шир. с, 2Н), 2,51 (м, 4Н), 2,26 (с, 3H).
Пример 14
Синтез 8-хлор-1-метил-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
(а) Синтез 2,7-дихлор-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразина
2,3,7-трихлорпиридо[2,3-b]пиразин (200,0 мг, 0,85 ммоль) и TEA (1180,0 мкл, 8,53 ммоль) растворяли в DCM (8,5 мл), и N-метилпиперазин (220,0 мкл, 0,94 ммоль), растворенный в DCM (0,5 мл), медленно добавляли при -20°C. Реакционную смесь перемешивали при -20°C в течение 12 часов и затем вливали в насыщенный водный раствор NH4Cl и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонке (DCM:MeOH=99:1) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое вещество желтого цвета, представляющее собой 2,7-дихлор-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин (110,0 мг, 43%).
ЖХ/МС ESI (+): 298 (М+1), 300 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6) δ: 8,94 (д, 1Н, J=2,7 Гц), 8,51 (д, 1Н, J=2,7 Гц), 3,61 (м, 4Н), 2,52 (м, 4Н), 2,25 (с, 3H).
(b) Синтез 7-хлор-2-гидразинил-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразина
2,7-дихлор-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин (100,0 мг, 0,36 ммоль) и моногидрат гидразина (46,0 мкл, 0,84 ммоль) растворяли в EtOH (3,0 мл), перемешивали при комнатной температуре в течение 12 часов и затем упаривали при пониженном давлении, получая твердое вещество желтого цвета, представляющее собой 7-хлор-2-гидразинил-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин.
ЖХ/МС ESI (+): 294 (М+1), 296 (М+3)
(с) Синтез 8-хлор-1-метил-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
Смесь неочищенного 7-хлор-2-гидразинил-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразина (50,0 мг, 0,17 ммоль) и триметилортоацетата (1,0 мл) перемешивали при 80°C в течение 2 часов и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=95:5) на аминосиликагеле. Фракции, содержащие продукт, собирали и испаряли твердое соединение кремового цвета, представляющее собой 8-хлор-1-метил-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (30,0 мг, 56%).
ЖХ/МС ESI (+): 318 (М+1), 320 (М+3).
1H-ЯМР (300 МГц, ДМСО-d6); δ: 8,56 (д, 1Н, J=2,4 Гц), 8,42 (д, 1Н, J=2,4 Гц), 4,36 (м, 4Н), 3,02 (с, 3H), 2,49 (м, 4Н), 2,25 (с, 3H).
Пример 15
Синтез 8-хлор-1-метил-4-(пиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
(а) Синтез 4-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)пиперазин-1-трет-бутилкарбоксилата
2,3,7-трихлорпиридо[2,3-b]пиразин (200,0 мг, 0,85 ммоль) и TEA (1,2 мл, 8,53 ммоль) растворяли в DCM (8,5 мл), и пиперазин-1-трет-бутилкарбоксилат (174,7 мг, 0,94 ммоль), разбавленный в DCM (0,5 мл), медленно добавляли при -20°C. Реакционную смесь перемешивали при -20°C в. течение 12 часов и затем выливали в насыщенный водный раствор NH4Cl и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=100:0) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 4-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)пиперазин-1-трет-бутилкарбоксилат (110,0 мг, 64%).
ЖХ/МС ESI (+): 384 (М+1), 386 (М+3)
1Н-ЯМР (300 МГц, ДМСO-d6); δ: 8,96 (д, 1Н, J=2,4 Гц), 8,54 (д, 1Н, J=2,4 Гц), 3,61 (м, 4Н), 3,54 (м, 4Н), 1,43 (с, 9Н).
(b) Синтез 4-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)пиперазин-1-трет-бутилкарбоксилата
4-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)пиперазин-1-трет-бутилкарбоксилат (110,0 мг, 0,29 ммоль) и моногидрат гидразина (35,0 мкл, 0,72 ммоль) растворяли в EtOH (2,0 мл), перемешивали при комнатной температуре в течение 12 часов и затем упаривали при пониженном давлении, получая твердое вещество желтого цвета, представляющее собой 4-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)пиперазин-1-трет-бутилкарбоксилат (55,0 мг, 51%).
ЖХ/МС ESI (+): 380 (М+1), 382 (М+3)
(с) Синтез 4-(8-хлор-1-метилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)пиперазин-1-трет-бутилкарбоксилата
Смесь неочищенного 4-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)пиперазин-1-трет-бутилкарбоксилата (55,0 мг, 0,15 ммоль) и триметилортоацетата (0,7 мл) перемешивали при 80°C в течение 2 часов и затем охлаждали до комнатной температуры. Добавляли Et2O и N-Hex (30,0 мл), получая твердое вещество, и сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая 4-(8-хлор-1-метилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)пиперазин-1-трет-бутилкарбоксилат (35,0 мг, 60%).
(d) Синтез 8-хлор-1-метил-4-(пиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
Неочищенный 4-(8-хлор-1-метилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)пиперазин-1-трет-бутилкарбоксилат (35,0 мг, 0,09 ммоль) растворяли в DCM (2,0 мл), и TFA (0,5 мл) медленно добавляли при комнатной температуре, и затем смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=80:20) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 8-хлор-1-метил-4-(пиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (9,0 мг, 35%).
ЖХ/МС ESI (+): 304 (М+1), 306 (М+3)
1H-ЯМР (300 МГц, ДMCO-d6); δ: 8,55 (м, 1Н), 8,42 (м, 1Н), 4,34 (м, 4Н), 3,04 (с, 3H), 2,87 (м, 4Н).
Пример 16
Синтез 8-бром-4-(пиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
(а) Синтез 4-(7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил) пиперазин-1-трет-бутилкарбоксилата
7-бром-2,3-дихлорпиридо[2,3-b]пиразин (1000,0 мг, 2,33 ммоль) и TEA (3,3 мл, 23,30 ммоль) растворяли в DCM (20,0 мл), и пиперазин-1-трет-бутилкарбоксилат (478,0 мг, 2,56 ммоль), разбавленный в DCM (10,0 мл), медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов и затем выливали в насыщенный водный раствор NH4Cl и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=100:0) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 4-(7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил)пиперазин-1-трет-бутилкарбоксилат.
ЖХ/МС ESI (+): 428 (М+1), 430 (М+3)
(b) Синтез 4-(7-бром-2-гидразинилпиридо[2,3-b]пиразин-3-ил)пиперазин-1-трет-бутилкарбоксилата
Неочищенный 4-(7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил)пиперазин-1-трет-бутилкарбоксилат и моногидрат гидразина (292,0 мг, 5,83 ммоль) растворяли в EtOH (50,0 мл), перемешивали при комнатной температуре в течение 12 часов и затем перегоняли при пониженном давлении. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 4-(7-бром-2-гидразинилпиридо[2,3-b]пиразин-3-ил)пиперазин-1-трет-бутилкарбоксилат.
ЖХ/МС ESI (+): 424 (М+1), 426 (М+3)
(с) Синтез 4-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)пиперазин-1-трет-бутилкарбоксилата
Смесь неочищенного 4-(7-бром-2-гидразинилпиридо[2,3-b]пиразин-3-ил)пиперазин-1-трет-бутилкарбоксилата и триметилортоформиата (10,0 мл) перемешивали при 80°C в течение одного часа и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 4-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)пиперазин-1-трет-бутилкарбоксилат.
ЖХ/МС ESI (+): 434 (М+1), 436 (М+3)
(d) Синтез 8-бром-4-(пиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
Неочищенный 4-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)пиперазин-1-трет-бутилкарбоксилат растворяли в DCM (8,0 мл), и TFA (2,0 мл) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем перегоняли при пониженном давлении. Остаток нейтрализовали водным раствором NaHCO3 (рН=7) и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=99:1) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 8-бром-4-(пиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (270,0 мг, 35% за 3 стадии).
ЖХ/МС ESI (+): 334 (М+1), 336 (М+3)
1H-ЯМР (300 МГц, ДMCO-d6); δ: 10,01 (с, 1Н), 8,94 (д, 1Н, J=2,3 Гц), 8,63 (д, 1Н, J=2,2 Гц), 4,36 (шир. с, 4Н), 2,89 (м, 4Н).
Пример 17
Синтез 7,8-дихлор-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
(а) Синтез 5,6-дихлорпиридин-2,3-диамина
5-хлор-3-нитропиридин-2-амин (2000,0 мг, 11,52 ммоль) и SnCl2 (8740,0 мг, 46,09 ммоль) добавляли к концентрированной HCl (20,0 мл) и затем перемешивали при 80-100°C в течение 0,5 часа. Реакционную смесь нейтрализовали насыщенным 1 н. водным раствором NaOH (рН=7) и затем экстрагировали EtOAc (200,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (n-Hех:EtOAc=90:10) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 5,6-дихлорпиридин-2,3-диамин (1000,0 мг, 49%).
1Н-ЯМР (300 МГц, ДMCO-d6); δ: 6,80 (с, 1Н), 6,04 (с, 2Н), 5,11 (с, 2Н).
(b) Синтез 2,3,6,7-тетрахлорпиридо[2,3-b]пиразина
5,6-дихлорпиридин-2,3-диамин (1000, 0 мг, 5,62 ммоль) добавляли к диэтилоксалату (20,0 мл), и смесь перемешивали при 120°C в течение 12 часов и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении, получая твердое соединение коричневого цвета, представляющее собой 6,7-дихлорпиридо[2,3-b]пиразин-2,3(1Н,4Н)-дион. Смесь неочищенного 6,7-дихлорпиридо[2,3-b]пиразин-2,3(1Н,4Н)-диона и РОСl3 (20,0 мл) перемешивали при 130°C в течение 48 часов и затем охлаждали до комнатной температуры. Реакционную смесь выливали в воду со льдом, получая твердое вещество, и сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая твердое соединение коричневого цвета, представляющее собой 2,3,6,7-тетрахлорпиридо[2,3-b]пиразин (1200,0 мг, 72% за 2 стадии).
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 9,09 (с, 1Н).
(с) Синтез 2,6,7-трихлор-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразина
2,3,6,7-тетрахлорпиридо[2,3-b]пиразин (190,0 мг, 0,71 ммоль) и TEA (1,0 мл, 7,10 ммоль) растворяли в DCM (8,0 мл), N-метилпиперазин (85,0 мг, 0,85 ммоль), разбавленный в DCM (2,0 мл), медленно добавляли при -20°C и перемешивали в течение 12 часов. Реакционную смесь выливали в насыщенный водный раствор NH4Cl и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 2,6,7-трихлор-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин.
ЖХ/МС ESI (+): 332 (М+1), 334 (М+3)
(d) Синтез 7,8-дихлор-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
Неочищенный 2,6,7-трихлор-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин и моногидрат гидразина (89,0 мг, 1,78 ммоль) растворяли в EtOH (10,0 мл), перемешивали при комнатной температуре в течение 12 часов и затем перегоняли при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 6,7-дихлор-2-гидразинил-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин. Смесь неочищенного 6,7-дихлор-2-гидразинил-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразина и триметилортоформиата (5,0 мл) перемешивали при 80°C в течение одного часа и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:МеОН=100:0) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 7,8-дихлор-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (107,0 мг, 45% за 3 стадии).
ЖХ/МС ESI (+): 338 (М+1), 340 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 9,99 (с, 1Н), 9,01 (с, 1Н), 4,80-3,80 (м, 4Н), 2,51 (м, 4Н), 2,24 (с, 3H).
Пример 18
Синтез 1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина
(a) Синтез (1-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
2,3,7-трихлорпиридо[2,3-b]пиразин (100,0 мг, 0,43 ммоль) растворяли в DCM (4,2 мл), азетидин-3-ил(метил)-трет-бутилкарбамат (88,0 мг, 0,47 ммоль) и TEA (0,2 мл, 1,28 ммоль) добавляли при 0°C и затем перемешивали в течение 1 часа. Растворитель удаляли из реакционной смеси при пониженном давлении. Остаток очищали хроматографией на колонке (EtOAc:n-Hех=1:5) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое вещество желтого цвета, представляющее собой (1-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (118,0 мг, 72%).
ЖХ/МС ESI (+): 384 (М+1), 386 (М+3)
1H-ЯМР (300 МГц, ДMCO-d6); δ: 8,83 (д, 1Н, J=2,4 Гц), 8,40 (д, 1Н, J=2,4 Гц), 4,86 (м, 1Н), 4,62 (м, 2Н), 4,48 (м, 2Н), 2,90 (с, 3H), 1,41 (с, 9Н).
(b) Синтез (1-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил) азетидин-3-ил)(метил)-трет-бутилкарбамата
(1-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил) (метил)-трет-бутилкарбамат (116,0 мг, 0,30 ммоль) растворяли в EtOH (4,2 мл) и затем добавляли моногидрат гидразина (24,0 мл, 0,76 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение одного часа. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой (1-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (114,0 мг, 100%).
ЖХ/МС ESI (+): 380 (М+1), 382 (М+3)
1H-ЯMP (300 МГц, ДМСО-d6); δ: 7,77 (шир. с, 1Н), 7,25 (шир. с, 1Н), 7,05 (м, 3H), 4,65 (м, 2Н), 4,46 (м, 1Н), 4,24 (м, 2Н), 2,86 (с, 3H), 1,40 (с, 9Н).
(с) Синтез (1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
(1-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (119,0 мг, 0,31 ммоль) растворяли в триметилортоформиате (1,5 мл) и затем перемешивали при 75°C в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры и затем высушивали при пониженном давлении, получая твердое вещество желтого цвета, представляющее собой (1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (120,0 мг, 98%).
ЖХ/МС ESI (+): 390 (М+1), 392 (М+3)
(d) Синтез 1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина
(1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (120,0 мг, 0,31 ммоль) растворяли в DCM (0,6 мл) и затем добавляли TFA (0,4 мл). Реакционную смесь перемешивали при комнатной температуре в течение одного часа. Растворитель затем удаляли из реакционной смеси при пониженном давлении. Остаток очищали хроматографией на колонках (МеОН:DCM=1:60) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (50,0 мг, 57%).
ЖХ/МС ESI (+): 290 (М+1), 292 (М+3)
1H-ЯMP (300 МГц, ДMCO-d6); δ: 9,95 (с, 1Н), 8,81 (д, 1Н, J=2,4 Гц), 8,54 (д, 1Н, J=2,4 Гц), 4,89 (м, 1Н), 4,43 (м, 2Н), 3,99 (м, 1Н), 3,71 (м, 1Н), 2,29 (с, 3H).
Пример 19
Синтез (S)-8-хлор-4-(3-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
(а) Синтез (S)-4-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)-2-метилпиперазин-1-трет-бутилкарбоксилата
2,3,7-трихлорпиридо[2,3-b]пиразин (200,0 мг, 0,85 ммоль) и TEA (1180,0 мкл, 8,53 ммоль) растворяли в DCM (8,5 мл), и (S)-2-метилпиперазин-1-трет-бутилкарбоксилат (187,8 мг, 0,94 ммоль), растворенный в DCM (0,5 мл), медленно добавляли при -20°C.Реакционную смесь перемешивали при -20°C в течение 12 часов и вливали в насыщенный водный раствор NH4Cl и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонке (EtOAc:n-Hех=30:70) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое вещество желтого цвета, представляющее собой (S)-4-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)-2-метилпиперазин-1-трет-бутилкарбоксилат (233,0 мг, 67%).
ЖХ/МС ESI (+): 398 (М+1), 400 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 8,96 (д, 1Н, J=2,7 Гц), 8,54 (д, 1Н, J=2,7 Гц), 4,30 (м, 1Н), 4,09 (м, 2Н), 3,88 (м, 1Н), 3,20 (м, 2Н), 3,04 (м, 1Н), 1,43 (с, 9Н), 1,24 (д, 3H, J=6,6 Гц).
(b) Синтез (S)-4-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)-2-метилпиперазин-1-трет-бутилкарбоксилата
(S)-4-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)-2-метилпиперазин-1-трет-бутилкарбоксилат (230,0 мг, 0,58 ммоль) и моногидрат гидразина (70,0 мкл, 1,44 ммоль) растворяли в EtOH (2,0 мл). Смесь затем перемешивали при комнатной температуре в течение 12 часов при пониженном давлении, получая твердое вещество желтого цвета, представляющее собой (S)-4-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)-2-метилпиперазин-1-трет-бутилкарбоксилат (220,0 мг, 97%).
ЖХ/МС ESI (+): 394 (М+1), 396 (М+3)
(c) Синтез (S)-4-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-2-метилпиперазин-1-трет-бутилкарбоксилата
Смесь неочищенного (S)-4-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)-2-метилпиперазин-1-трет-бутилкарбоксилата (150,0 мг, 0,38 ммоль) и триметилортоформиата (2,0 мл) перемешивали при 80°C в течение 2 часов и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая (S)-4-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-2-метилпиперазин-1-трет-бутилкарбоксилат (150,0 мг, 81%).
(d) Синтез (S)-8-хлор-4-(3-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
Неочищенный (S)-4-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-2-метилпиперазин-1-трет-бутилкарбоксилат (150,0 мг, 0,37 ммоль) растворяли в DCM (1,0 мл). TFA (0,2 мл) медленно добавляли при комнатной температуре и затем смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=95:5) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой (S)-8-хлор-4-(3-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (70,0 мг, 63%).
ЖХ/МС ESI (+): 304 (М+1), 306 (М+3)
1H-ЯМР (300 МГц, CDCl3); δ: 9,15 (с, 1Н), 8,61 (д, 1Н, J=2,4 Гц), 8,04 (д, 1Н, J=2,4 Гц), 6,24 (м, 1Н), 5,21 (м, 1Н), 3,02 (м, 5Н), 1,21 (д, 3H, J=6 Гц).
Пример 20
Синтез (S)-8-хлор-4-(3,4-диметилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
(S)-8-хлор-4-(3-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (50,0 мг, 0,16 ммоль), полученный на стадии (d) примера 19, и 37%-й формальдегид (73,5 мкл, 0,99 ммоль) растворяли в МеОН (0,5 мл). Медленно добавляли NaBH4 (37,0 мг, 0,99 ммоль), и смесь затем перемешивали при комнатной температуре в течение 12 часов. В реакционную смесь выливали солевой раствор, и смесь экстрагировали DCM (30,0 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонке (DCM:MeOH=95:5) на силикагеле, получая твердое вещество желтого цвета, представляющее собой (S)-8-хлор-4-(3,4-диметилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (70,0 мг, 48%).
ЖХ/МС ESI (+): 318 (М+1), 320 (М+3)
1Н-ЯМР (300 МГц, CDCl3); δ: 9,15 (с, 1Н), 8,62 (д, 1Н, J=2,4 Гц), 8,03 (д, 1Н, J=2,1 Гц), 6,06 (м, 1Н), 5,12 (м, 1Н), 3,56 (м, 2Н), 2,99 (м, 1Н), 2,37 (м, 5Н), 1,22 (д, 3H, J=6,3 Гц).
Пример 21
Синтез 8-хлор-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-7-карбонитрила
7,8-дихлор-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (10,0 мг, 0,03 ммоль), Zn(CN)2 (4,0 мг, 0,03 ммоль) и Pd(PPh3)4 (3,0 мг, 0,003 ммоль) растворяли в DMF (1,0 мл). Смеси давали взаимодействовать в микроволновом реакторе в условиях 60 Вт, 100°C, в течение одного часа и затем охлаждали до комнатной температуры. Реакционную смесь выливали в воду и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=100:0) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 8-хлор-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-7-карбонитрил (2,0 мг, 20%).
ЖХ/МС ESI (+): 329 (М+1), 331 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 10,04 (с, 1Н), 9,07 (с, 1Н), 4,80 (м, 2Н), 4,09 (м, 2Н), 2,54 (м, 4Н), 2,25 (с, 3H).
Пример 22
Синтез 8-хлор-4-(3,4,5-триметилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
(a) Синтез 3,5-диметилпиперазин-1-трет-бутилкарбоксилата
2,6-диметилпиперазин (200,0 мг, 1,75 ммоль) и TEA (0,6 мл, 4,37 ммоль) растворяли в DCM (6,0 мл), и (Boc)2O (458,7 мг, 2,10 ммоль) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=95:5) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая желтое жидкое соединение 3,5-диметилпиперазин-1-трет-бутилкарбоксилат (210,0 мг, 56%).
1Н-ЯМР (300 МГц, CDCl3); δ: 3,95 (м, 2Н), 2,79 (м, 2Н), 2,33 (м, 2Н), 1,46 (с, 9Н), 1,07 (д, 6Н, J=6,3 Гц).
(b) Синтез 3,4,5-триметилпиперазин-1-трет-бутилкарбоксилата
3,5-диметилпиперазин-1-трет-бутилкарбоксилат (200,0 мг, 0,93 ммоль) и 37%-й формальдегид (440,0 мкл, 5,56 ммоль) растворяли в МеОН (5,0 мл) и NaBH4 (172,6 мг, 5,56 ммоль) медленно добавляли и перемешивали при комнатной температуре в течение 12 часов. В реакционную смесь выливали солевой раствор, и смесь экстрагировали DCM (30,0 мл). Органический слой высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонке (DCM:MeOH=95:5) на силикагеле, получая желтое жидкое соединение 3,4,5-триметилпиперазин-1-трет-бутилкарбоксилат (57,0 мг, 27%). (с) Синтез 1,2,6-триметилпиперазина 3,4,5-триметилпиперазин-1-трет-бутилкарбоксилат (57,0 мг, 0,25 ммоль) растворяли в DCM (1,0 мл), и TFA (0,2 мл) медленно добавляли при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем перегоняли при пониженном давлении и очищали хроматографией на колонках (DCM:MeOH=95:5) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая желтое жидкое соединение 1,2,6-триметилпиперазин (32,0 мг, 100%).
1Н-ЯМР (300 МГц, CDCl3); δ: 2,88 (м, 2Н), 2,58 (м, 2Н), 2,28 (с, 3H), 2,14 (м, 2Н), 1,07 (д, 6Н, J=6 Гц).
(d) Синтез 2,7-дихлор-3-(3,4,5-триметилпиперазин-1-ил)пиридо[2,3-b]пиразина
2,3,7-трихлорпиридо[2,3-b]пиразин (58,4 мг, 0,25 ммоль) и TEA (347,0 мкл, 2,49 ммоль) растворяли в DCM (1,0 мл), и 1,2,6-триметилпиперазин (32,0 мг, 0,25 ммоль) в DCM (0,5 мл) медленно добавляли при -20°C. Реакционную смесь перемешивали при -20°C в течение 12 часов и затем вливали в насыщенный водный раствор NH4Cl и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем упаривали при пониженном давлении, получая твердое вещество желтого цвета, представляющее собой 2,7-дихлор-3-(3,4,5-триметилпиперазин-1-ил)пиридо[2,3-b]пиразин (23,0 мг, 28%).
ЖХ/МС ESI (+): 326 (М+1), 328 (М+3)
(е) Синтез 7-хлор-2-гидразинил-3-(3,4,5-триметилпиперазин-1-ил)пиридо[2,3-b]пиразина
2,7-дихлор-3-(3,4,5-триметилпиперазин-1-ил)пиридо[2,3-b]пиразин (23,0 мг, 0,07 ммоль) и моногидрат гидразина (8,0 мкл, 0,18 ммоль) растворяли в EtOH (0,3 мл) и перемешивали при комнатной температуре в течение 12 часов, и затем упаривали при пониженном давлении, получая твердое вещество желтого цвета, представляющее собой 7-хлор-2-гидразинил-3-(3,4,5-триметилпиперазин-1-ил)пиридо[2,3-b]пиразин (23,0 мг, 100%).
ЖХ/МС ESI (+): 322 (М+1), 324 (М+3)
(f) Синтез 8-хлор-4-(3,4,5-триметилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
Смесь неочищенного 7-хлор-2-гидразинил-3-(3,4,5-триметилпиперазин-1-ил)пиридо[2,3-b]пиразина (23,0 мг, 0,07 ммоль) и триметилортоформиата (2,0 мл) перемешивали при 80°C в течение 2 часов и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая 8-хлор-4-(3,4,5-триметилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (5,0 мг, 21%).
ЖХ/МС ESI (+): 332 (М+1), 334 (М+3)
1Н-ЯМР (300 МГц, CDCl3); δ: 9,15 (с, 1Н), 8,62 (д, 1Н, J=2,4 Гц), 8,04 (д, 1Н, J=2,4 Гц), 6,17 (м, 1Н), 5,18 (м, 1Н), 3,28 (м, 1Н), 2,93 (м, 1Н), 2,39 (м, 2Н), 2,34 (с, 3H) 1,26 (с, 6Н).
Пример 23
Синтез 8-хлор-7-этокси-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
7,8-дихлор-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (20,0 мг, 0,06 ммоль) и NaOEt (5,0 мг, 0,07 ммоль) растворяли в EtOH (1,0 мл) и давали взаимодействовать в микроволновом реакторе в условиях 60 Вт, 90°C, в течение 2 часов, и затем охлаждали до комнатной температуры. Реакционную смесь очищали хроматографией на колонках (DCM:MeOH=98:2) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 8-хлор-7-этокси-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (3,0 мг, 14%).
ЖХ/МС ESI (+): 348 (М+1), 350 (М+3)
1Н-ЯМР (300 МГц, ДMCO-d6); δ: 9,91 (с, 1Н), 8,84 (с, 1Н), 4,44 (кв., 2Н, J=7,0 Гц), 4,40 (м, 4Н), 2,50 (м, 4Н), 2,24 (с, 3H), 2,24 (т, 3H, J=7,0 Гц).
Пример 24
Синтез 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина
(а) Синтез (1-(7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
7-бром-2,3-дихлорпиридо[2,3-b]пиразин (200,0 мг, 0,72 ммоль) и TEA (1,0 мл, 7,20 ммоль) растворяли в DCM (8,0 мл), и азетидин-3-ил(метил)-трет-бутилкарбамат (147,0 мг, 0,79 ммоль), разбавленный в DCM (2,0 мл), медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов и затем выливали в насыщенный водный раствор NH4Cl и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=100:0) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение коричневого цвета, представляющее собой (1-(7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат.
ЖХ/МС ESI (+): 428 (М+1), 430 (М+3)
(b) Синтез (1-(7-бром-2-гидразинилпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
Неочищенный (1-(7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (224,0 мг, 0,61 ммоль) и моногидрат гидразина (72,0 мг, 3,78 ммоль) растворяли в EtOH (10,0 мл), перемешивали при комнатной температуре в течение 12 часов и затем перегоняли при пониженном давлении. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой (1-(7-бром-2-гидразинилпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат.
ЖХ/МС ESI (+): 424 (М+1), 426 (М+3)
(с) Синтез (1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
Смесь неочищенного (1-(7-бром-2-гидразинилпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата и триметилортоформиата (10,0 мл) перемешивали при 80°C в течение одного часа и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая твердое соединение кремового цвета, представляющее собой (1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (137,0 мг, 4 5% за 3 стадии).
ЖХ/МС ESI (+): 434 (М+1), 436 (М+3)
(d) Синтез 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина
(1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (87,0 мг, 0,20 ммоль) растворяли в DCM (4,0 мл), и TFA (1,0 мл) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов и перегоняли при пониженном давлении. Остаток нейтрализовали водным раствором NaHCO3 (рН=7) и затем экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=98:2) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (16,0 мг, 23%).
ЖХ/МС ESI (+): 334 (М+1), 336 (М+3)
1Н-ЯМР (300 МГц, ДMCO-d6); δ: 9,95 (с, 1Н), 8,90 (д, 1Н, J=2,1 Гц), 8,59 (д, 1Н, J=2,1 Гц), 4,89 (м, 1Н), 4,42 (м, 2Н), 4,00 (м, 1Н), 3,73 (м, 1Н), 2,29 (с, 3H).
Пример 25
Синтез 4-(3-(метиламино)азетидин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-8-карбонитрила
(1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (50,0 мг, 0,12 ммоль), Zn(CN)2 (14,0 мг, 0,12 ммоль) и Pd(PPh3)4 (13,0 мг, 0,01 ммоль) растворяли в DMF (1,0 мл) и давали взаимодействовать в микроволновом реакторе в условиях 60 Вт, 90°C, в течение 2 часов, и затем охлаждали до комнатной температуры. Реакционную смесь затем очищали хроматографией на колонках (DCM:MeOH=98:2) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой (1-(8-цианопиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат. (1-(8-цианопиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат растворяли в DCM (4,0 мл), и TFA (1,0 мл) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов и затем перегоняли при пониженном давлении. Остаток нейтрализовали водным раствором NaHCO3 (рН=7) и затем экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=98:2) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 4-(3-(метиламино)азетидин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-8-карбонитрил (10,0 мг, 22%).
ЖХ/МС ESI (+): 281 (М+1)
1H-ЯМР (300 МГц, ДМСО-d6); δ: 9,93 (с, 1Н), 9,06 (д, 1Н, J=1,9 Гц), 8,88 (д, 1Н, J=1,9 Гц), 4,95 (м, 1Н), 4,49 (м, 2Н), 4,04 (м, 1Н), 3,73 (м, 1Н), 2,30 (с, 3H).
Пример 26
Синтез 4-(пиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-8-карбонитрила
8-бром-4-(пиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (50,0 мг, 0,15 ммоль), Zn(CN)2 (18,0 мг, 0,15 ммоль) и Pd(PPh3)4 (3,0 мг, 0,02 ммоль) растворяли в DMF (1,0 мл) и давали взаимодействовать в микроволновом реакторе в условиях 60 Вт, 90°C, в течение одного часа, и затем охлаждали до комнатной температуры. Реакционную смесь очищали хроматографией на колонках (DCM:МеОН=98:2) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 4-(пиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-8-карбонитрил (4,4 мг, 10%).
ЖХ/МС ESI (+): 281 (М+1)
1Н-ЯМР (300 МГц, ДMCO-d6); δ: 9,98 (с, 1Н), 9,09 (д, 1Н, J=2,0 Гц), 8,90 (д, 1Н, J=2,1 Гц), 4,80 (м, 2Н), 4,05 (м, 2Н), 2,90 (м, 4Н).
Пример 27
Синтез 1-(7,8-дихлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина
(а) Синтез (1-(2,6,7-трихлорпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)-трет-бутилметилкарбамата
2,3,6,7-тетрахлорпиридо[2,3-b]пиразин (200,0 мг, 0,74 ммоль) и TEA (1,0 мл, 7,40 ммоль) растворяли в DCM (8,0 мл), и азетидин-3-ил(метил)-трет-бутилкарбамат (162,0 мг, 0,94 ммоль), разбавленный в DCM (2,0 мл), медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов, вливали в насыщенный водный раствор NH4Cl и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=100:0) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение коричневого цвета, представляющее собой (1-(2,6,7-трихлорпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)-трет-бутилметилкарбамат.
ЖХ/МС ESI (+): 418 (М+1), 420 (М+3)
(b) Синтез (1-(6,7-дихлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
Неочищенный (1-(2,6,7-трихлорпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)-трет-бутилметилкарбамат и моногидрат гидразина (37,0 мг, 1,85 ммоль) растворяли в EtOH (10,0 мл) и затем перемешивали при комнатной температуре в течение 12 часов, и затем перегоняли при пониженном давлении. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой (1-(6,7-дихлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат.
ЖХ/МС ESI (+): 414 (М+1), 416 (М+3)
(c) Синтез (1-(7,8-дихлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
Смесь неочищенного (1-(6,7-дихлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата и триметилортоформиата (20,0 мл) перемешивали при 70°C в течение одного часа и затем охлаждали до комнатной температуры. Медленно добавляли Et2O, и сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая твердое соединение кремового цвета, представляющее собой (1-(7,8-дихлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (173,0 мг, 55% за 3 стадии).
ЖХ/МС ESI (+): 424 (М.++1), 426 (М+3)
(d) Синтез 1-(7,8-дихлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина
(1-(7,8-дихлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (123,0 мг, 0,29 ммоль) растворяли в DCM (4,0 мл), и TFA (1,0 мл) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и перегоняли при пониженном давлении. Остаток нейтрализовали водным раствором NaHCO3 (рН=7) и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=98:2) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 1-(7,8-дихлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (19,0 мг, 20%).
ЖХ/МС ESI (+): 324 (М+1), 326 (М+3)
1Н-ЯМР (300 МГц, ДMCO-d6); δ: 9,95 (с, 1Н), 8,98 (с, 1Н), 4,94 (м, 1Н), 4,46 (м, 2Н), 4,00 (м, 1Н), 3,73 (м, 1Н), 2,30 (с, 3H).
Пример 28
Синтез 8-хлор-4-(3-(метиламино)азетидин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-7-карбонитрила
(1-(7,8-дихлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (50,0 мг, 0,15 ммоль), Zn(CN)2 (18,0 мг, 0,15 ммоль) и Pd(PPh3)4 (18,0 мг, 0,02 ммоль) растворяли в DMF (1,0 мл) и давали взаимодействовать в микроволновом реакторе в условиях 60 Вт, 90°C, в течение одного часа и затем охлаждали до комнатной температуры. Реакционную смесь очищали хроматографией на колонках (DCM:MeOH=98:2) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой (1-(8-хлор-7-цианопиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат. Неочищенный (1-(8-хлор-7-цианопиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат растворяли в DCM (2,0 мл), и TFA (0,5 мл) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов и затем перегоняли при пониженном давлении. Остаток нейтрализовали водным раствором NaHCO3 (рН=7) и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=98:2) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 8-хлор-4-(3-(метиламино)азетидин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-7-карбонитрил (20,0 мг, 41%).
ЖХ/МС ESI (+): 315 (М+1), 317 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 9,98 (с, 1Н), 9,03 (с, 1Н), 4,93 (м, 1Н), 4,48 (м, 2Н), 4,00 (м, 1Н), 3,73 (м, 1Н), 2,30 (с, 3H).
Пример 29
Синтез 8-хлор-4-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
(а) Синтез 2,7-дихлор-3-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)пиридо[2,3-b]пиразина
2,3,7-трихлорпиридо[2,3-b]пиразин (100,0 мг, 0,43 ммоль) и октагидропирроло[1,2-а]пиразин (60,0 мг, 0,47 ммоль) вводили в реакцию, получая твердое соединение желтого цвета, представляющее собой 2,7-дихлор-3-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)пиридо[2,3-b]пиразин (83,0 мг, 60%), таким же образом, как в примере 18 (а).
ЖХ/МС ESI (+): 324 (М+1), 326 (М+3)
1H-ЯМР (300 МГц, CDCl3); δ: 8,86 (д, 1Н, J=2,7 Гц), 8,17 (д, 1Н, J=2,4 Гц), 4,45 (м, 2Н), 3,32 (м, 1Н), 3,19 (м, 2Н), 2,97 (м, 1Н), 2,49 (м, 1Н), 2,26 (м, 2Н), 1,91 (м, 2Н), 1,81 (м, 1Н), 1,51 (м, 1Н).
(b) Синтез 7-хлор-3-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)-2-гидразинилпиридо[2,3-b]пиразина
2,7-дихлор-3-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)пиридо[2,3-b]пиразин (80,0 мг, 0,25 ммоль) и моногидрат гидразина (19,0 мкл, 0,62 ммоль) вводили в реакцию таким же образом, как в примере 18 (b), получая твердое соединение оранжевого цвета, представляющее собой 7-хлор-3-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)-2-гидразинилпиридо[2,3-b]пиразин (50,0 мг, 63%).
ЖХ/МС ESI (+): 320 (М+1), 322 (М+3)
1H-ЯМР (300 МГц, CDCl3); δ: 8,60 (д, 1Н, J=3,0 Гц), 8,01 (д, 1Н, J=3,0 Гц), 6,49 (шир. с, 1Н), 4,20 (д, 2Н, J=3,0 Гц), 3,94 (м, 2Н), 3,17 (м, 3H), 2,87 (м, 1Н), 2,46 (м, 1Н), 2,22 (м, 2Н), 1,89 (м, 2Н), 1,78 (м, 1Н), 1,50 (м, 1Н).
(c) Синтез 8-хлор-4-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
7-хлор-3-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)-2-гидразинилпиридо[2,3-b]пиразин (45,0 мг, 0,14 ммоль) и триметилортоформиат (1,0 мл) вводили в реакцию таким же образом, как в примере 18 (с), при 90°C в течение 5 часов, получая твердое соединение оранжевого цвета, представляющее собой 8-хлор-4-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин.
ЖХ/МС ESI (+): 330 (М+1), 332 (М+3)
1Н-ЯМР (300 МГц, CDCl3); δ: 9,16 (с, 1Н), 8,61 (д, 1Н, J=2,1 Гц), 8,04 (д, 1Н, J=2,4 Гц), 6,39 (м, 1Н), 5,50 (м, 1Н), 3,55 (м, 0,5Н), 3,20 (м, 3H), 2,90 (м, 0,5Н), 2,42 (м, 1Н), 2,20 (м, 2Н), 1,88 (м, 3H), 1,52 (м, 1Н).
Пример 30
Синтез (R)-1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)пирролидин-3-амина
(а) Синтез (R)-(1-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)пирролидин-3-ил)-трет-бутилкарбамата
2,3,7-трихлорпиридо[2,3-b]пиразин (200,0 мг, 0,85 ммоль) и TEA (1,2 мл, 8,50 ммоль) растворяли в DCM (10,0 мл), и (R)-пирролидин-3-ил-трет-бутилкарбамат (175,0 мг, 0,94 ммоль), разбавленный в DCM (5,0 мл), медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов, вливали в насыщенный водный раствор NH4Cl и затем экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH от 100:0 до 98:2) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой (R)-(1-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)пирролидин-3-ил)-трет-бутилкарбамат (284,0 мг, 86%).
ЖХ/МС ESI (+): 384 (М+1), 386 (М+3)
(b) Синтез (R)-(1-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)пирролидин-3-ил)-трет-бутилкарбамата
(R)-(1-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)пирролидин-3-ил)-трет-бутилкарбамат (284,0 мг, 0,74 ммоль) и моногидрат гидразина (92,0 мг, 1,85 ммоль) растворяли в EtOH (10,0 мл), перемешивали при комнатной температуре в течение 2 часов и затем перегоняли при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой (R)-(1-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)пирролидин-3-ил)-трет-бутилкарбамат.
ЖХ/МС ESI (+): 380 (М+1), 382 (М+3)
(c) Синтез (R)-(1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)пирролидин-3-ил)-трет-бутилкарбамата
Смесь неочищенного (R)-(1-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)пирролидин-3-ил)-трет-бутилкарбамата и триметилортоформиата (5,0 мл) перемешивали при 70°C в течение одного часа и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении, получая твердое соединение кремового цвета, представляющее собой (R)-(1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)пирролидин-3-ил)-трет-бутилкарбамат (272,0 мг, 94% за 2 стадии).
ЖХ/МС ESI (+): 390 (М+1), 392 (М+3)
(d) Синтез (R)-1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)пирролидин-3-амина
(R)-(1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)пирролидин-3-ил)-трет-бутилкарбамата (272,0 мг, 0,70 ммоль) растворяли в DCM (20,0 мл), и затем TFA (4,0 мл) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и перегоняли при пониженном давлении. Остаток нейтрализовали водным раствором NaHCO3 (рН=7) и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH от 90:10 до 50:50) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой (R)-1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)пирролидин-3-амин (79,0 мг, 39%).
ЖХ/МС ESI (+): 290 (М+1), 292 (М+3)
1Н-ЯМР (300 МГц, ДMCO-d6); δ: 9,99 (с, 1Н), 8,81 (д, 1Н, J=2,4 Гц), 8,53 (д, 1Н, J=2,4 Гц), 4,6-3,4 (м, 5Н), 2,4-1,6 (м, 2Н).
Пример 31
Синтез 9-хлор-2-метил-5-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидина
(а) Синтез 3-бром-5-хлорпиридин-2-амина
5-хлорпиридин-2-амин (5000,0 мг, 38,90 ммоль) растворяли в СНСl3 (78,0 мл) и затем добавляли Br2 (2,0 мл, 38,90 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение одного часа, и растворитель удаляли при пониженном давлении. Остаток растворяли в EtOAc, и смесь затем промывали насыщенным водным раствором NaHCO3 и солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (EtOAc:n-Hex=1:9) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 3-бром-5-хлорпиридин-2-амин (7500,0 мг, 93%).
ЖХ/МС ESI (+): 207 (М+1), 209 (М+3)
1H-ЯМР (300 МГц, CDCl3); δ: 7,98 (д, 1Н, J=2,1 Гц), 7,66 (д, 1Н, J=2,1 Гц), 4,93 (шир. с, 2Н).
(b) Синтез 2-амино-5-хлорникотинонитрила
3-бром-5-хлорпиридин-2-амин (2700,0 мг, 13,00 ммоль) растворяли в NMP (60,0 мл) и затем добавляли Zn(CN)2 (2300,0 мг, 19,50 ммоль) и Pd(PPh3)4 (1500,0 мг, 1,30 ммоль). Реакционную смесь перемешивали при 110°C в течение 5 часов и затем охлаждали до комнатной температуры. К реакционной смеси добавляли воду и EtOAc, и смесь перемешивали в течение 10 минут и затем фильтровали через целит. Фильтрат экстрагировали EtOAc. Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонке (EtOAc:n-Hех=1:9) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение белого цвета, представляющее собой 2-амино-5-хлорникотинонитрил (1900,0 мг, 100%).
ЖХ/МС ESI (+): 154 (М+1), 156 (М+3)
1H-ЯМР (300 МГц, ДМСО-d6); δ: 8,22 (д, 1Н, J=2,7 Гц), 8,07 (д, 1Н, J=2,7 Гц), 7,14 (с, 2Н).
(с) Синтез 9-хлор-2-метилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-ола
2-амино-5-хлорникотинонитрил (300,0 мг, 1,95 ммоль) растворяли в МЕК (2,0 мл) и добавляли NaHCO3 (492, 0 мг, 5,86 ммоль) и хлорэтилформиат (3,0 мл). Реакционную смесь нагревали с обратным холодильником и затем охлаждали до комнатной температуры, и смесь фильтровали и затем высушивали при пониженном давлении, получая нетипичное соединение желтого цвета, представляющее собой (5-хлор-3-цианопиридин-2-ил)этилкарбамат. Неочищенный (5-хлор-3-цианопиридин-2-ил)этилкарбамат растворяли в простом дифениловом эфире (2,0 мл) и добавляли ацетогидразид (144,0 мг, 1,95 ммоль). Реакционную смесь перемешивали при 180°C в течение 30 минут и затем охлаждали до комнатной температуры, и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонке (EtOAc:n-Hех=1:4) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 9-хлор-2-метилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-ол (91,0 мг, 20% за 2 стадии)
ЖХ/МС ESI (+): 236 (М+1), 238 (М+3)
1Н-ЯМР (300 МГц, ДMCO-d6); δ: 12,9 (с, 1Н), 8,73 (д, 1Н, J=2,4 Гц), 8,58 (д, 1Н, J=2,4 Гц).
(d) Синтез 5,9-дихлор-2-метилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидина
9-хлор-2-метилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-ол (80,0 мг, 0,34 ммоль) растворяли в РОСl3 (1,5 мл) и добавляли DIPEA (120,0 мкл, 0,68 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 12 часов и затем охлаждали до комнатной температуры. Реакционную смесь выливали в воду со льдом, нейтрализовали насыщенным водным раствором NaHCO3, и затем смесь экстрагировали EtOAc. Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (EtOAc:n-Hех=1:5) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение белого цвета, представляющее собой 5,9-дихлор-2-метилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (30,0 мг, 35%).
ЖХ/МС ESI (+): 254 (М+1), 256 (М+3)
(е) Синтез 9-хлор-2-метил-5-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидина
5,9-дихлор-2-метилпиридо[3,2-е][1,2,4]триазоло[1,5-с] пиримидин (28,0 мг, 0,11 ммоль) растворяли в DMF (1,1 мл) и добавляли N-метилпиперазин (24,0 мкл, 0,22 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение одного часа, и остаток очищали хроматографией на колонках (МеОН:DCM=1:40) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 9-хлор-2-метил-5-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (18,0 мг, 51%).
ЖХ/МС ESI (+): 318 (М+1), 320 (М+3)
1H-ЯMP (300 МГц, ДМСО-d6); δ: 8,84 (д, 1Н, J=2,4 Гц), 8,63 (д, 1Н, J=2,4 Гц), 4,12 (м, 4Н), 2,56 (с, 3H), 2,54 (м, 4Н), 2,24 (с, 3H).
Пример 32
Синтез 9-хлор-2-метил-5-(пиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидина
(а) Синтез 4-(9-хлор-2-метилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-ил)пиперазин-1-трет-бутилкарбоксилата
5,9-дихлор-2-метилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (20,0 мг, 0,08 ммоль), полученный в примере 31 (d), растворяли в DMF (1,0 мл) и добавляли пиперазин-1-трет-бутилкарбоксилат (29,0 мг, 0,16 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение одного часа и очищали хроматографией на колонках (EtOAc:n-Hех=1:5) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 4-(9-хлор-2-метилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-ил)пиперазин-1-трет-бутилкарбоксилат (32,0 мг, 100%).
ЖХ/МС ESI (+): 404 (М+1), 406 (М+3)
(b) Синтез 9-хлор-2-метил-5-(пиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидина
4-(9-хлор-2-метилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-ил)пиперазин-1-трет-бутилкарбоксилат (30,0 мг, 0,07 ммоль) растворяли в DCM (0,6 мл) и добавляли TFA (0,4 мл). Реакционную смесь перемешивали при комнатной температуре в течение одного часа и очищали хроматографией на колонках (МеОН:DCM=1:40) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 9-хлор-2-метил-5-(пиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (22,5 мг, 100%).
ЖХ/МС ESI (+): 304 (М+1), 306 (М+3)
1Н-ЯМР (300 МГц, CDCl3); δ: 8,79 (д, 1Н, J=2,7 Гц), 8,58 (д, 1Н, J=2,7 Гц), 4,26 (м, 4Н), 3,11 (м, 4Н), 2,64 (с, 3H).
Пример 33
Синтез 1-(9-хлор-2-метилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-ил)-N-метилазетидин-3-амина
(а) Синтез (1-(9-хлор-2-метилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
5,9-дихлор-2-метилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (12,0 мг, 0,05 ммоль), полученный в примере 31 (d), растворяли в DMF (1,0 мл) и добавляли азетидин-3-ил(метил)-трет-бутилкарбамат (17,6 мг, 0,09 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение одного часа, и растворитель удаляли при пониженном давлении. Остаток очищали хроматографией на колонках (МеОН:DCM=1:40) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой (1-(9-хлор-2-метилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (19,0 мг, 100%).
ЖХ/МС ESI (+): 404 (М+1), 406 (М+3)
(b) Синтез 1-(9-хлор-2-метилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-ил)-N-метилазетидин-3-амина
(1-(9-хлор-2-метилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (18,0 мг, 0,04 ммоль) растворяли в DCM (0,6 мл) и добавляли TFA (0,4 мл). Реакционную смесь перемешивали при комнатной температуре в течение одного часа и очищали хроматографией на колонках (МеОН:DCM=1:40) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 1-(9-хлор-2-метилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-ил)-N-метилазетидин-3-амин (7,0 мг, 55%).
ЖХ/МС ESI (+): 304 (М+1), 306 (М+3)
1H-ЯМР (300 МГц, CDCl3); δ: 8,73 (д, 1Н, J=2,7 Гц), 8,51 (д, 1Н, J=2,7 Гц), 4,84 (м, 2Н), 4,38 (м, 2Н), 3,80 (м, 1Н), 2,60 (с, 3H), 2,49 (с, 3H).
Пример 34
Синтез 9-хлор-2-циклопропил-N,N-диэтилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-амина
(а) Синтез 9-хлор-2-циклопропилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-ола
(5-хлор-3-цианопиридин-2-ил)этилкарбамат синтезировали из 2-амино-5-никотинонитрила (500,0 мг, 3,26 ммоль) таким же образом, как в примере 31 (с), и вводили в реакцию с циклопропанкарбогидразидом (326,0 мг, 3,26 ммоль), получая твердое соединение коричневого цвета, представляющее собой 9-хлор-2-циклопропилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-ол (37,0 мг, 4% за 2 стадии).
ЖХ/МС ESI (+): 262 (М+1), 264 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 12,86 (шир. с, 1Н), 8,71 (д, 1Н, J=2,7 Гц), 8,53 (д, 1Н, J=2,7 Гц), 2,23 (м, 1Н), 1,09 (м, 2Н), 0,99 (м, 2Н).
(b) Синтез 9-хлор-2-циклопропил-N,N-диэтилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-амина
9-хлор-2-циклопропилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-ол (35,0 мг, 0,13 ммоль) растворяли в РОСl3 (1,5 мл) и добавляли TEA (37,0 мкл, 0,27 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 12 часов и затем охлаждали до комнатной температуры. Реакционную смесь выливали в воду со льдом и нейтрализовали насыщенным водным раствором NaHCO3, и смесь экстрагировали EtOAc. Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (EtOAc:n-Hех=1:4) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 9-хлор-2-циклопропил-N,N-диэтилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-амин (32,0 мг, 74%).
ЖХ/МС ESI (+): 317 (М+1), 319 (М+3)
1Н-ЯМР (300 МГц, CDCl3); δ: 8,71 (д, 1Н, J=2,7 Гц), 8,51 (д, 1Н, J=2,7 Гц), 4,04 (кв., 4Н, J=6,9 Гц), 2,23 (м, 1Н), 1,39 (т, 6Н, J=6,9 Гц), 1,13 (м, 4Н).
Пример 35
Синтез 9-хлор-5-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидина
(а) Синтез 9-хлорпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5(6Н)-она
2-амино-5-никотинонитрил (500,0 мг, 3,26 ммоль) растворяли в МЕК (3,5 мл) и добавляли NaHCO3 (820,4 мг, 9,77 ммоль) и хлорзтилформиат (5,0 мл). Реакционную смесь нагревали с обратным холодильником в течение 24 часов и затем охлаждали до комнатной температуры и фильтровали и затем высушивали при пониженном давлении, получая нетипичное соединение желтого цвета, представляющее собой, представляющее собой (5-хлор-3-цианопиридин-2-ил)этилкарбамат. Неочищенный (5-хлор-3-цианопиридин-2-ил)этилкарбамат растворяли в простом дифениловом эфире (3,5 мл) и затем добавляли формгидразид (195,5 мг, 3,26 ммоль). Реакционную смесь перемешивали при 180°C в течение одного часа и затем охлаждали до комнатной температуры и очищали хроматографией на колонках (EtOAc:n-Hex=1:1) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 9-хлорпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5(6Н)-он (63,0 мг, 9%).
ЖХ/МС ESI (+): 221 (М+1), 223 (М+3)
1H-ЯMP (300 МГц, ДМСО-d6); δ: 12,95 (с, 1Н), 8,74 (д, 1Н, J=2,1 Гц), 8,63 (д, 1Н, J=2,4 Гц), 8,59 (с, 1Н).
(b) Синтез 5,9-дихлорпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидина
9-хлорпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5(6Н)-он (63,0 мг, 0,29 ммоль) растворяли в РОСl3 (2,0 мл) и добавляли DIPEA (150,0 мкл, 0,86 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 12 часов и затем охлаждали до комнатной температуры. Реакционную смесь выливали в воду со льдом и нейтрализовали насыщенным водным раствором NaHCO3 и экстрагировали EtOAc. Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (EtOAc:n-Hex=1:1) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение белого цвета, представляющее собой 5,9-дихлорпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (50,4 мг, 72%).
ЖХ/МС ESI (+): 240 (М+1), 241 (М+3)
(с) Синтез 9-хлор-5-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидина
5,9-дихлорпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (15,0 мг, 0,06 ммоль) растворяли в DMF (0,5 мл), N-метилпиперазин (10,3 мкл, 0,09 ммоль) и добавляли TEA (25,0 мкл, 0,19 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение одного часа и затем очищали хроматографией на колонках (МеОН:DCM=1:19) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 9-хлор-5-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (6,0 мг, 33%).
ЖХ/МС ESI (+): 304 (М+1), 306 (М+3)
1Н-ЯМР (300 МГц, CDCl3); δ: 8,82 (д, 1Н, J=3,0 Гц), 8,63 (д, 1Н, J=2,7 Гц), 8,37 (с, 1Н), 4,33 (м, 4Н), 2,65 (м, 4Н), 2,38 (с, 3H).
Пример 36
Синтез 1-(9-хлорпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-ил)-N-метилазетидин-3-амина
5,9-дихлорпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (15,0 мг, 0,06 ммоль), полученный в примере 35 (b), растворяли в DMF (0,5 мл) и добавляли трет-бутилазетидин-3-ил(метил)карбамат (17,3 мг, 0,09 ммоль) и TEA (25,0 мкл, 0,19 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение одного часа и перегоняли при пониженном давлении, и затем растворяли в DCM (1,0 мл) без очистки, и затем добавляли TFA (0,2 мл). Реакционную смесь перемешивали при комнатной температуре в течение одного часа и очищали хроматографией на колонках (МеОН:DCM=1:19) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 1-(9-хлорпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-ил)-N-метилазетидин-3-амин (2,6 мг, 15%).
ЖХ/МС ESI (+): 290 (М+1), 292 (М+3)
1H-ЯМР (300 МГц, CDCl3+MeOH-d4); δ: 8,70 (д, 1Н, J=2,7 Гц), 8,53 (д, 1Н, J=2,7 Гц), 8,28 (д, 1Н, J=2,7 Гц), 4,84 (м, 2Н), 4,45 (м, 2Н), 3,82 (м, 1Н), 2,46 (с, 3H).
Пример 37
Синтез 9-хлор-5-(пиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидина
5,9-дихлорпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (15,0 мг, 0,06 ммоль), полученный в примере 35 (b), растворяли в DMF (0,5 мл), и затем добавляли пиперазин-1-трет-бутилкарбоксилат (16,7 мг, 0,09 ммоль) и TEA (25,0 мкл, 0,19 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение одного часа и концентрировали при пониженном давлении, и затем смесь растворяли в DCM (1,0 мл) без очистки, и добавляли TFA (0,2 мл). Реакционную смесь перемешивали при комнатной температуре в течение одного часа и очищали хроматографией на колонках (МеОН:DCM=1:19) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение белого цвета, представляющее собой 9-хлор-5-(пиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (1,7 мг, 10%).
ЖХ/МС ESI (+): 290 (М+1), 292 (М+3)
1H-ЯMP (300 МГц, CDCl3+MeOH-d4); δ: 8,82 (д, 1Н, J=2,4 Гц), 8,68 (д, 1Н, J=2,7 Гц), 8,38 (с, 1Н), 4,51 (м, 4Н), 3,39 (м, 4Н).
Пример 38
Синтез 9-хлор-2-циклопропил-5-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидина
(а) Синтез 5,9-дихлор-2-циклопропилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидина
9-хлор-2-циклопропилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-ол (125,0 мг, 0,48 ммоль) вводили в реакцию с РOСl3 (2,5 мл) и DIPEA (0,2 мл, 0,96 ммоль) таким же образом, как в примере 31 (d), получая твердое соединение кремового цвета, представляющее собой 5,9-дихлор-2-циклопропилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (90,0 мг, 67%).
ЖХ/МС ESI (+): 280 (М+1), 282 (М+3)
1H-ЯMP (300 МГц, CDCl3); δ: 9,01 (д, 1Н, J=2,7 Гц), 8,76 (д, 1Н, J=2,7 Гц), 2,34 (м, 1Н), 1,22 (м, 4Н).
(b) Синтез 9-хлор-2-циклопропил-5-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидина
5,9-дихлор-2-циклопропилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (25,0 мг, 0,09 ммоль) и N-метилпиперазин (20,0 мкл, 0,18 ммоль) вводили в реакцию таким же образом, как в примере 31 (е), получая твердое соединение кремового цвета, представляющее собой 9-хлор-2-циклопропил-5-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (22,0 мг, 72%).
ЖХ/МС ESI (+): 344 (М+1), 346 (М+3)
1H-ЯMP (300 МГц, CDCl3); δ: 8,77 (д, 1Н, J=2,7 Гц), 8,56 (д, 1Н, J=2,7 Гц), 4,31 (т, 4Н, J=5,1 Гц), 2,64 (т, 4Н, J=5,1 Гц), 2,38 (с, 3H), 2,25 (м, 1Н), 1,15 (м, 4Н).
Пример 39
Синтез 9-хлор-2-циклопропил-5-(пиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидина
5,9-дихлор-2-циклопропилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (30,0 мг, 0,11 ммоль), полученный таким же путем, как в примере 38 (а), растворяли в DCM (1,0 мл) и добавляли пиперазин-1-трет-бутилкарбоксилат (40,0 мг, 0,21 ммоль). Реакционную смесь перемешивали в течение одного часа и затем добавляли TFA (0,8 мл), и смесь вводили в реакцию в течение другого часа. Реакционную смесь очищали хроматографией на колонках (МеОН:DCM=1:40) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали. Добавляли Et2O и n-Hех, и полученное твердое вещество отфильтровывали, получая твердое соединение кремового цвета, представляющее собой 9-хлор-2-циклопропил-5-(пиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (31,0 мг, 89%).
ЖХ/МС ESI (+): 330 (М+1), 332 (М+3)
1Н-ЯМР (300 МГц, CDCl3); δ: 8,77 (д, 1Н, J=2,7 Гц), 8,56 (д, 1Н, J=2,7 Гц), 4,25 (т, 4Н, J=5,1 Гц), 3,10 (т, 4Н, J=5,1 Гц), 2,25 (м, 1Н), 1,15 (м, 4Н).
Пример 40
Синтез 9-хлор-2-(метоксиметил)-5-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидина
(а) Синтез 9-хлор-2-(метоксиметил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-ола
2-амино-5-никотинонитрил (500,0 мг, 3,26 ммоль) растворяли в МЕК (4,0 мл) и добавляли К2СO3 (1350, 0 мг, 9,78 ммоль), MgSO4 (250,0 мг, 1,25 ммоль) и хлорэтилформиат (6,0 мл). Реакционную смесь нагревали с обратным холодильником в течение 24 часов, затем охлаждали до комнатной температуры, фильтровали и затем высушивали при пониженном давлении, получая нетипичное соединение желтого цвета, представляющее собой, представляющее собой (5-хлор-3-цианопиридин-2-ил)этилкарбамат. Неочищенный (5-хлор-3-цианопиридин-2-ил)этилкарбамат растворяли в простом дифениловом эфире (2,0 мл) и добавляли 2-метоксиацетогидразид (271,0 мг, 2,61 ммоль). Реакционную смесь перемешивали при 150°C в течение 3 часов и затем охлаждали до комнатной температуры, и затем очищали хроматографией на колонке (EtOAc:n-Hex=1:4) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 9-хлор-2-(метоксиметил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-ол (150,0 мг, 17% за 2 стадии).
ЖХ/МС ESI (+): 266 (М+1), 268 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 12,99 (с, 1Н), 8,74 (д, 1Н, J=2,4 Гц), 8,64 (д, 1Н, J=2,7 Гц), 4,63 (с, 2Н), 3,38 (с, 3H).
(b) Синтез 5,9-дихлор-2-(метоксиметил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидина
9-хлор-2-(метоксиметил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-ол (146,0 мг, 0,51 ммоль) вводили в реакцию с РОСl3 (1,5 мл) и DIPEA (0,2 мл, 1,03 ммоль) таким же образом, как в примере 31 (d), получая твердое соединение кремового цвета, представляющее собой 5,9-дихлор-2-(метоксиметил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (89,0 мг, 61%).
ЖХ/МС ESI (+): 284 (М+1), 286 (М+3)
1Н-ЯМР (300 МГц, CDCl3); δ: 9,06 (д, 1Н, J=2,4 Гц), 8,87 (д, 1Н, J=2,7 Гц), 4,85 (с, 2Н), 3,59 (с, 3H).
(с) Синтез 9-хлор-2-(метоксиметил)-5-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидина
5,9-дихлор-2-(метоксиметил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (25,0 мг, 0,09 ммоль) и N-метилпиперазин (20,0 мкл, 0,18 ммоль) вводили в реакцию в DCM (1,0 мл) таким же образом, как в примере 31 (е), получая твердое соединение кремового цвета, представляющее собой 9-хлор-2-(метоксиметил)-5-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (18,0 мг, 58%).
ЖХ/МС ESI (+): 348 (М+1), 350 (М+3)
1H-ЯMP (300 МГц, CDCl3); δ: 8,81 (д, 1Н, J=2,7 Гц), 8,67 (д, 1Н, J=2,7 Гц), 4,76 (с, 2Н), 4,33 (т, 4Н, J=4,8 Гц), 3,57 (с, 3H), 2,64 (т, 4Н, J=4,8 Гц), 2,38 (с, 3H).
Пример 41
Синтез 9-хлор-2-этил-5-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидина
(a) Синтез 9-хлор-2-этилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-ола
(5-хлор-3-цианопиридин-2-ил)этилкарбамат синтезировали из 2-амино-5-никотинонитрила (500,0 мг, 3,26 ммоль) таким же образом, как в примере 31 (с), и вводили в реакцию с этилкарбогидразидом (150,0 мг, 1,79 ммоль), получая твердое соединение желтого цвета, представляющее собой 9-хлор-2-этилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-ол (95,0 мг, 12% за 2 стадии).
ЖХ/МС ESI (+): 250 (М+1), 252 (М+3)
1H-ЯМР (300 МГц, ДMCO-d6); δ: 12,90 (с, 1Н), 8,72 (д, 1Н, J=2,7 Гц), 8,59 (д, 1Н, J=2,7 Гц), 2,86 (кв., 2Н, J=7,5 Гц), 1,35 (т, 3H, J=7,5 Гц).
(b) Синтез 5,9-дихлор-2-этилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидина
9-хлор-2-этилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин-5-ол (92,0 мг, 0,37 ммоль) растворяли в POCl3 (1,5 мл) и добавляли DIPEA (130,0 мкл, 0,74 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 12 часов и затем охлаждали до комнатной температуры. К реакционной смеси добавляли триэтилбензиламмоний хлорид (92,0 мг, 0,42 ммоль), перемешивали при 130°C в течение 2 часов и затем охлаждали до комнатной температуры. Реакционную смесь вливали в смесь воды со льдом, нейтрализовали насыщенным водным раствором NaHCO3 и затем экстрагировали EtOAc. Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонке (DCM) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение бледно-желтого цвета, представляющее собой 5,9-дихлор-2-этилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (21,0 мг, 21%)
ЖХ/МС ESI (+): 268 (М+1), 270 (М+3)
1Н-ЯМР (300, МГц, CDCl3); δ: 9,12 (д, 1Н, J=2,7 Гц), 8,96 (д, 1Н, J=2,7 Гц), 2,99 (кв., 2Н, J=7,5 Гц), 1,35 (т, 3H, J=7,5 Гц).
(с) Синтез 9-хлор-2-этил-5-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидина
5,9-дихлор-2-этилпиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (10,0 мг, 0,04 ммоль) и N-метилпиперазин (8,3 мкл, 0,08 ммоль) в DCM вводили в реакцию таким же образом, как в примере 31 (е), получая твердое соединение белого цвета, представляющее собой 9-хлор-2-этил-5-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[1,5-с]пиримидин (5,2 мг, 42%).
ЖХ/МС ESI (+): 332 (М+1), 334 (М+3)
1Н-ЯМР (300 МГц, CDCl3); δ: 8,84 (д, 1Н, J=2,7 Гц), 8,64 (д, 1Н, J=2,7 Гц), 4,13 (м, 4Н), 2,91 (кв., 2Н, J=7,5 Гц), 2,53 (м, 4Н), 2,24 (с, 3H), 1,36 (т, 3H, J=7,5 Гц).
Пример 42
Синтез 9-хлор-5-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[4,3-c]пиримидина
(a) Синтез 6-хлорпиридо[2,3-d]пиримидин-2,4(1Н,3H)-диона
Смесь 2-амино-5-хлорникотинамида (169,0 мг, 0,98 ммоль) и дифосгена (646,0 мг, 3,14 ммоль), растворенного в 1,4-диоксане (10,0 мл), перемешивали при 120°C в течение 12 часов и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и. затем высушивали при пониженном давлении, получая твердое соединение кремового цвета, представляющее собой 6-хлорпиридо[2,3-d]пиримидин-2,4(1Н,3H)-дион (163,0 мг, 84%).
1H-ЯМР (300 МГц, ДMCO-d6); δ: 11,59 (с, 2Н), 8,60 (д, 1Н, J=2,8 Гц), 8,19 (д, 1Н, J=2,6 Гц).
(b) Синтез 2,4,6-трихлорпиридо[2,3-d]пиримидина
Смесь 6-хлорпиридо[2,3-d]пиримидин-2,4(1Н,3H)-диона (163,0 мг, 0,82 ммоль) и BPOD (2,0 мл) перемешивали при 180°C в течение 6 часов и затем охлаждали до комнатной температуры. Реакционную смесь выливали в воду и нейтрализовали насыщенным водным раствором NaHCO3 (рН=7) и затем экстрагировали EtOAc (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении, получая твердое соединение белого цвета, представляющее собой 2,4,6-трихлорпиридо[2,3-d]пиримидин (169,0 мг, 88%).
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 8,97 (д, 1Н, J=2,8 Гц), 8,49 (д, 1Н, J=2,8 Гц).
(с) Синтез 2,6-дихлорпиридо[2,3-d]пиримидин-4-ола
2,4,6-трихлорпиридо[2,3-d]пиримидин (90,0 мг, 0,38 ммоль) растворяли в 1 н. NaOH/BuOH (0,8 мл/0,8 мл), перемешивали при комнатной температуре в течение 2 часов, затем перегоняли при пониженном давлении, нейтрализовали 1 н. водным раствором HCl (рН=7) и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4 и затем фильтровали при пониженном давлении, получая твердое соединение кремового цвета, представляющее собой 2,6-дихлорпиридо[2,3-d]пиримидин-4-ол (63,0 мг, 76%).
ЖХ/МС ESI (+): 216 (М+1), 218 (М+3)
(d) Синтез 6-хлор-2-(4-метилпиперазин-1-ил)пиридо[2,3-d]пиримидин-4(3H)-она
Смесь 2,6-дихлорпиридо[2,3-d]пиримидин-4-ола (63,0 мг, 0,29 ммоль) и N-метилпиперазина (0,5 мл), растворенных в EtOH (5,0 мл), перемешивали при 80°C в течение одного часа, затем охлаждали до комнатной температуры и перегоняли при пониженном давлении. Реакционную смесь выливали в воду и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении, получая твердое соединение кремового цвета, представляющее собой 6-хлор-2-(4-метилпиперазин-1-ил)пиридо[2,3-d]пиримидин-4(3H)-он (63,0 мг, 77%).
ЖХ/МС ESI (+): 280 (М+1), 282 (М+3)
(е) Синтез 4,6-дихлор-2-(4-метилпиперазин-1-ил)пиридо[2,3-d]пиримидина
Смесь 6-хлор-2-(4-метилпиперазин-1-ил)пиридо[2,3-d]пиримидин-4(3H)-она (63,0 мг, 0,23 ммоль) и РОСl3 (1,0 мл) перемешивали при 90°C в течение 12 часов и затем перегоняли при пониженном давлении, получая твердое соединение коричневого цвета, представляющее собой 4,6-дихлор-2-(4-метилпиперазин-1-ил)пиридо[2,3-d]пиримидин.
ЖХ/МС ESI (+): 298 (М+1), 300 (М+3)
(f) Синтез 6-хлор-4-гидразинил-2-(4-метилпиперазин-1-ил)пиридо[2,3-d]пиримидина
Неочищенный 4,6-дихлор-2-(4-метилпиперазин-1-ил)пиридо[2,3-d]пиримидин, TEA (233,0 мг, 2,30 ммоль) и моногидрат гидразина (115,0 мг, 2,30 ммоль) растворяли в EtOH (10,0 мл). Смесь перемешивали при -20°C в течение одного часа и затем перегоняли при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 6-хлор-4-гидразинил-2-(4-метилпиперазин-1-ил)пиридо[2,3-d]пиримидин.
ЖХ/МС ESI (+): 294 (М+1), 296 (М+3)
(g) Синтез 9-хлор-5-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[4,3-c]пиримидина
Смесь неочищенного 6-хлор-4-гидразинил-2-(4-метилпиперазин-1-ил)пиридо[2,3-d]пиримидина и триметилортоформиата (2,0 мл) перемешивали при 100°C в течение 2 часов и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали, получая твердое соединение фиолетового цвета, представляющее собой 9-хлор-5-(4-метилпиперазин-1-ил)пиридо[3,2-е][1,2,4]триазоло[4,3-с]пиримидин (0,1 мг, 0,1%).
ЖХ/МС ESI (+): 304 (М+1), 306 (М+3)
1H-ЯМР (300 МГц, ДМСО-d6); δ: 9,54 (с, 1Н), 8,82 (с, 1Н), 8,76 (с, 1Н), 3,66 (шир. с, 4Н), 2,55 (м, 4Н), 2,26 (с, 3H).
Пример 43
Синтез 8-хлор-2-метил-4-(4-метилпиперазин-1-ил)-2Н-пиразоло[3,4-с][1,8]нафтилидина
(а) Синтез 2-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-ил)-2-оксоэтилацетата
5-хлор-1Н-пирроло[2,3-b]пиридин (500,0 мг, 3,28 ммоль) и AlCl3 (2180,0 мг, 16,38 ммоль) растворяли в DCM (10,0 мл), и 2-хлор-2-оксоэтилацетат (44,0 мкл, 3,93 ммоль) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. К реакционной смеси при 0°C добавляли EtOH и лед и затем экстрагировали DCM. Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 2-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-ил)-2-оксоэтилацетат (200,0 мг, 23%).
ЖХ/МС ESI (+): 253 (М+1), 255 (М+3)
1Н-ЯМР (300 МГц, CDCl3); δ: 10,39 (с, 1Н), 8,72 (д, 1Н, J=2,4 Гц), 8,68 (д, 1Н, J=3 Гц), 8,39 (д, 1Н, J=2,4 Гц), 4,44 (кв., 2Н, J=7,2 Гц), 1,45 (т, 3H, J=6,9 Гц).
(b) Синтез 8-хлор-2-метил-2Н-пиразоло[3,4-с][1,8]нафтилидин-4(5Н)-она
2-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-ил)-2-оксоэтилацетат (200,0 мг, 0,79 ммоль), метилгидразин сульфат (228,0 мг, 1,58 ммоль) и уксусную кислоту (68,0 мкл, 11,87 ммоль) растворяли в EtOH (2,0 мл) и давали взаимодействовать в микроволновом реакторе в условиях 100 Вт, 120°C, в течение 30 минут, и затем охлаждали до комнатной температуры. Затем добавляли водный раствор NH4Cl и экстрагировали DCM. Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали, затем перегоняли при пониженном давлении и очищали хроматографией на колонках (MeOH:DCM=5:95) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 8-хлор-2-метил-2Н-пиразоло[3,4-с][1,8]нафтилидин-4(5Н)-он (30,0 мг, 16%).
ЖХ/МС ESI (+): 235 (М+1), 237 (М+3)
1Н-ЯMP (300 МГц, CDCl3); δ: 11,88 (с, 1Н), 8,71 (с, 1Н), 8,52 (д, 1Н, J=2,7 Гц), 8,38 (д, 1Н, J=2,4 Гц), 4,14 (с, 3H).
(c) Синтез 4,8-дихлор-2-метил-2Н-пиразоло[3,4-с][1,8]нафтилидина
8-хлор-2-метил-2Н-пиразоло[3,4-с][1,8]нафтилидин-4(5Н)-он (30,0 мг, 0,13 ммоль) растворяли в POCl3 (1,5 мл) и затем добавляли DIPEA (66,0 мкл, 0,38 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 12 часов и затем охлаждали до комнатной температуры. Реакционную смесь выливали в воду со льдом, нейтрализовали насыщенным водным раствором NaHCO3 (рН=7) и затем экстрагировали EtOAc. Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем концентрировали при пониженном давлении, получая твердое соединение, представляющее собой 4,8-дихлор-2-метил-2Н-пиразоло[3,4-с][1,8]нафтилидин (30,0 мг, 90%).
ЖХ/МС ESI (+): 253 (М+1), 255 (М+3)
(d) Синтез 8-хлор-2-метил-4-(4-метилпиперазин-1-ил)-2Н-пиразоло[3,4-с][1,8]нафтилидина
4,8-дихлор-2-метил-2Н-пиразоло[3,4-с][1,8]нафтилидин (10,0 мг, 0,04 ммоль) растворяли в DMF (0,3 мл) и добавляли N-метилпиперазин (23,3 мкл, 0,21 ммоль) и TEA (58,0 мкл, 0,42 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение одного часа и очищали хроматографией на колонках (МеОН:DCM=1:9) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 8-хлор-2-метил-4-(4-метилпиперазин-1-ил)-2Н-пиразоло[3,4-с][1,8]нафтилидин (3,7 мг, 30%).
ЖХ/МС ESI (+): 317 (М+1), 319 (М+3)
1Н-ЯМР (300 МГц, CDCl3); δ: 8,55 (с, 1Н), 8,11 (с, 1Н), 7,99 (д, 1Н, J=2,4 Гц), 4,46 (м, 4Н), 4,22 (с, 3H), 2,60 (м, 4Н), 2,36 (с, 3H).
Пример 44
Синтез 1-(8-хлор-2-метил-2Н-пиразоло[3,4-с][1,8]нафтилидин-4-ил)-N-метилазетидин-3-амина
4,8-дихлор-2-метил-2Н-пиразоло[3,4-с][1,8]нафтилидин (10,0 мг, 0,04 ммоль) растворяли в DMF (0,3 мл) и добавляли азетидин-3-ил(метил)-трет-бутилкарбамат (39,1 мг, 0,21 ммоль) и TEA (58,0 мкл, 0,42 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение одного часа, концентрировали при пониженном давлении, растворяли в DCM (1,0 мл) без очистки и затем добавляли TFA (0,2 мл). Реакционную смесь перемешивали при комнатной температуре в течение одного часа и очищали хроматографией на колонках (МеОН:DCM=5:95) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 1-(8-хлор-2-метил-2Н-пиразоло[3,4-с][1,8]нафтилидин-4-ил)-N-метилазетидин-3-амин (3,2 мг, 25%).
ЖХ/МС ESI (+): 303 (М+1), 305 (М+3)
1H-ЯMP (300 МГц, CDCl3+MeOH-d4); δ: 8,47 (с, 1Н), 8,27 (с, 1Н), 8,13 (д, 1Н, J=2,4 Гц), 4,89 (м, 2Н), 4,71 (м, 2Н), 4,23 (с, 3H), 3,99 (м, 1Н), 2,76 (с, 3H).
Пример 45
Синтез 8-хлор-2-метил-4-(пиперазин-1-ил)-2Н-пиразоло[3,4-с][1,8]нафтилидина
4,8-дихлор-2-метил-2Н-пиразоло[3,4-с][1,8]нафтилидин (10,0 мг, 0,04 ммоль) растворяли в DMF (0,3 мл) и добавляли пиперазин-1-трет-бутилкарбоксилат (39,1 мг, 0,21 ммоль) и TEA (58,0 мкл, 0,42 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение одного часа, концентрировали при пониженном давлении, растворяли в DCM (1,0 мл) без очистки и затем добавляли TFA (0,2 мл). Реакционную смесь перемешивали при комнатной температуре в течение одного часа и очищали хроматографией на колонках (МеОН:DCM=5:95) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 8-хлор-2-метил-4-(пиперазин-1-ил)-2Н-пиразоло[3,4-с][1,8]нафтилидин (2,0 мг, 16%).
ЖХ/МС ESI (+): 303 (М+1), 305 (М+3)
1Н-ЯМР (300 МГц, CDCl3); δ: 8,50 (д, 1Н, J=2,7 Гц), 8,16 (с, 1Н), 8,04 (д, 1Н, J=2,7 Гц), 4,52 (м, 4Н), 4,20 (с, 3H), 3,17 (м, 4Н).
Пример 46
Синтез 8-хлор-4-(4-метилпиперазин-1-ил)имидазо[1,2-а]пиридо[2,3-е]пиразина
(а) Синтез 2,7-дихлор-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразина
2,3,7-трихлорпиридо[2,3-b]пиразин (500,0 мг, 2,13 ммоль) и TEA (3,1 мл, 21,30 ммоль) растворяли в DCM (15,0 мл), и N-метилпиперазин (0,3 мл, 2,34 ммоль), разбавленный в DCM (5,0 мл), медленно добавляли при 0°C.Реакционную смесь перемешивали при комнатной температуре в течение 12 часов, вливали в насыщенный водный раствор NH4Cl и затем экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=20:1) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 2,7-дихлор-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин (331,0 мг, 52%).
ЖХ/МС ESI (+): 298 (М+1), 300 (М+3)
(b) Синтез 7-хлор-N-(2,2-диэтоксиэтил)-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин-2-амина
Смесь 2,7-дихлор-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразина (100,0 мг, 0,34 ммоль) и 2,2-диэтоксиэтиламина (2,0 мл) перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь выливали в воду и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 7-хлор-N-(2,2-диэтоксиэтил)-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин-2-амин.
ЖХ/МС ESI (+): 395 (М+1), 397 (М+3)
(с) Синтез 8-хлор-4-(4-метилпиперазин-1-ил)имидазо[1,2-а]пиридо[2,3-е]пиразина
Смесь неочищенного 7-хлор-N-(2,2-диэтоксиэтил)-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин-2-амина и TsOH (128,0 мг, 0,68 ммоль), растворенных в IPA (5,0 мл), перемешивали при 100°C в течение 2 часов, затем охлаждали до комнатной температуры и перегоняли при пониженном давлении. Реакционную смесь вливали в и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH от 20:1 до 90:10) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 8-хлор-4-(4-метилпиперазин-1-ил)имидазо[1,2-а]пиридо[2,3-е]пиразин (56,0 мг, 55% за 2 стадии).
ЖХ/МС ESI (+): 303 (М+1), 305 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 8,81 (д, 1Н, J=2,4 Гц), 8,74 (д, 1Н, J=1,2 Гц), 8,53 (д, 1Н, J=2,4 Гц), 7,72 (д, 1Н, J=1,4 Гц), 4,42 (шир. с, 4Н), 2,49 (м, 4Н), 2,23 (с, 3H).
Пример 47
Синтез 8-хлор-4-(пиперазин-1-ил)имидазо[1,2-а]пиридо[2,3-е]пиразина
(а) Синтез 4-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)пиперазин-1-трет-бутилкарбоксилата
2,3,7-трихлорпиридо[2,3-b]пиразин (400,0 мг, 1,71 ммоль) и TEA (2,4 мл, 17,10 ммоль) растворяли в DCM (8,0 мл), и пиперазин-1-трет-бутилкарбоксилат (350,0 мг, 1,88 ммоль), разбавленный в DCM (2,0 мл), медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, вливали в насыщенный водный раствор NH4Cl и затем экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=19:1) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 4-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)пиперазин-1-трет-бутилкарбоксилат.
ЖХ/МС ESI (+): 384 (М+1), 386 (М+3)
(b) Синтез 4-(7-хлор-2-((2,2-диэтоксиэтил)амино)пиридо[2,3-b]пиразин-3-ил)пиперазин-1-трет-бутилкарбоксилата
Смесь неочищенного 4-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил) пиперазин-1-трет-бутилкарбоксилата и 2,2-диэтоксиэтиламина (2,0 мл) перемешивали при комнатной температуре в течение 12 часов, вливали в воду и затем экстрагировали EtOAc (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 4-(7-хлор-2-((2,2-диэтоксиэтил)амино)пиридо[2,3-b]пиразин-3-ил)пиперазин-1-трет-бутилкарбоксилат.
ЖХ/МС ESI (+): 481 (М+1), 483 (М+3)
(с) Синтез 8-хлор-4-(пиперазин-1-ил)имидазо[1,2-а]пиридо[2,3-е]пиразина
Смесь неочищенного 4-(7-хлор-2-((2,2-диэтоксиэтил)амино)пиридо[2,3-b]пиразин-3-ил)пиперазин-1-трет-бутилкарбоксилата и TsOH (662,0 мг, 3,48 ммоль), растворенных в IPA (10,0 мл) перемешивали при 100°C в течение одного часа, затем охлаждали до комнатной температуры и перегоняли при пониженном давлении. Реакционную смесь выливали в воду, нейтрализовали водным раствором Na2SО4 (рН=7) и затем экстрагировали DCM (50,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH от 20:1 до 90:10) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 8-хлор-4-(пиперазин-1-ил)имидазо[1,2-а]пиридо[2,3-е]пиразин (155,0 мг, 31% за 2 стадии).
ЖХ/МС ESI (+): 289 (М+1), 291 (М+3)
1H-ЯМР (300 МГц, ДМСО-d6); δ: 8,79 (д, 1Н, J=2,5 Гц), 8,74 (м, 1Н), 8,52 (д, 1Н, J=2,4 Гц), 7,72 (м, 1Н), 4,35 (шир. с, 4Н), 2,84 (м, 4Н).
Пример 48
Синтез 1-(8-йодпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина
(а) Синтез (1-(8-йодпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
(1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (50,0 мг, 0,11 ммоль), N,N'-диметилэтан-1,2-диамин (1,0 мг, 0,01 ммоль), CuI (1,1 мг, 5,50 ммоль) и KI (38,0 мг, 0,22 ммоль) добавляли к 1-бутанолу (1,0 мл), перемешивали при 100°C в течение 18 часов, перегоняли при пониженном давлении и затем очищали хроматографией на колонке (DCM:MeOH от 98:2 до 95:5) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое вещество белого цвета, представляющее собой (1-(8-йодпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (25,0 мг, 45%).
ЖХ/МС ESI (+): 482 (М+1)
(b) Синтез 1-(8-йодпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина
(1-(8-йодпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (25,0 мг, 0,05 ммоль) растворяли в DCM (3,0 мл), и TFA (0,5 мл) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение одного часа и перегоняли при пониженном давлении. Остаток нейтрализовали водным раствором NaHCO3 (рН=7) и экстрагировали смесью DCM/MeOH (9/1, 30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали, перегоняли при пониженном давлении и затем очищали хроматографией на колонке (DCM:МеОН=96:4) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 1-(8-йодпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (15,0 мг, 75%).
ЖХ/МС ESI (+): 382 (М+1)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 9,96 (с, 1Н), 8,97 (д, 1Н, J=2,1 Гц), 8,68 (д, 1Н, J=2,1 Гц), 4,89 (м, 1Н), 4,42 (м, 2Н), 4,00 (м, 1Н), 3,70 (м, 1Н), 2,29 (с, 3H).
Пример 49
Синтез 8-йод-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
(а) Синтез 5-йодпиридин-2,3-диамина
5-йод-3-нитропиридин-2-амин (2000,0 мг, 7,55 ммоль), Fe (4800,0 мг, 85,90 ммоль) и концентрированную HCl (0,1 мл) добавляли к смеси EtOH (7,0 мл) и воды (2,0 мл), и суспензию перемешивали при 100°C в течение 30. Реакционную смесь охлаждали до комнатной температуры, фильтровали через целит и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонке (МеОН:DCM=1:40) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение коричневого цвета, представляющее собой 5-йодпиридин-2,3-диамин (1710,0 мг, 97%).
ЖХ/МС ESI (+): 236 (М+1)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 7,37 (д, 1Н, J=2,1 Гц), 6,91 (д, 1Н, J=2,1 Гц), 5,59 (с, 2Н), 4,91 (с, 2Н).
(b) Синтез 2,3-дихлор-7-йодпиридо[2,3-b]пиразин
Смесь 5-йодпиридин-2,3-диамина (1400,0 мг, 5,96 ммоль) и диэтилоксалата (10,0 мл) перемешивали при 100°C в течение 12 часов и затем охлаждали до комнатной температуры. К реакционной смеси добавляли Et2O, получая твердое вещество, и затем сформированное твердое вещество фильтровали и высушивали при пониженном давлении, получая твердое соединение коричневого цвета, представляющее собой 7-йодпиридо[2,3-b]пиразин-2,3-диол. Смесь неочищенного 7-йодпиридо[2,3-b]пиразин-2,3-диола и РОСl3 (15,0 мл) перемешивали при 150°C в течение 12 часов и затем охлаждали до комнатной температуры. Реакционную смесь вливали в смесь воды со льдом, и образовавшееся твердое вещество фильтровали и затем высушивали при пониженном давлении, получая твердое соединение коричневого цвета, представляющее собой 2,3-дихлор-7-йодпиридо[2,3-b]пиразин (1080,0 мг, 56%).
ЖХ/МС ESI (+): 326 (М+1), 328 (М+3)
(c) Синтез 2-хлор-7-йод-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразина
2,3-дихлор-7-йодпиридо[2,3-b]пиразин (50,0 мг, 0,15 ммоль) и TEA (213,0 мкл, 1,53 ммоль) растворяли в DCM (2,0 мл) и N-метилпиперазин (16,0 мкл, 0,15 ммоль) медленно добавляли при 0°C. Реакционную смесь перемешивали при 0°C в течение одного часа, концентрировали и затем очищали хроматографией на колонках (МеОН:DCM=5:95) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 2-хлор-7-йод-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин (40,0 мг, 67%).
ЖХ/МС ESI (+): 390 (М+1), 392 (М+3)
(d) Синтез 2-гидразинил-7-йод-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразина
2-хлор-7-йод-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин (40,0 мг, 0,10 ммоль) и моногидрат гидразина (7,5 мкл, 0,15 ммоль) растворяли в EtOH (1,0 мл), перемешивали при комнатной температуре в течение 3 часов, получая твердое вещество. Сформированное твердое вещество отфильтровывали, получая твердое соединение желтого цвета, представляющее собой 2-гидразинил-7-йод-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин (39,4 мг, 100%).
ЖХ/МС ESI (+): 386 (М+1)
(е) Синтез 8-йод-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
Смесь 2-гидразинил-7-йод-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразина (39,4 мг, 0,10 ммоль) и триметилортоформиата (2,0 мл) перемешивали при 80°C в течение 3 часов и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении. Остаток очищали хроматографией на колонках (МеОН:DCM=5:95) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 8-йод-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (13,5 мг, 34%).
ЖХ/МС ESI (+): 396 (М+1)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 10,00 (с, 1Н), 9,00 (д, 1Н, J=2,1 Гц), 8,71 (д, 1Н, J=2,1 Гц), 4,57-4,17 (м, 4Н), 2,52 (м, 4Н), 2,24 (с, 3H).
Пример 50
Синтез N-метил-1-(8-метилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-амина
(a) Синтез 5-метилпиридин-2,3-диамина
5-метил-3-нитропиридин-2-амин (50000,0 мг, 0,33 моль) растворяли в смеси MeOH/EtOAc (1000,0 мл) и добавляли 10% Pd/C (5000,0 мг). Колбу замещали водородом. Реакционную смесь вводили в реакцию при комнатной температуре в течение 8 часов, затем фильтровали через целит и упаривали при пониженном давлении, получая твердое соединение коричневого цвета, представляющее собой 5-метилпиридин-2,3-диамин (98000,0 мг, 82%).
1Н-ЯМР (300 МГц, CDCl3); δ: 7,48 (с, 1Н), 6,74 (с, 1Н), 4,06 (шир. с, 2Н), 3,27 (шир. с, 2Н), 2,16 (с, 3H).
(b) Синтез 7-метилпиридо[2,3-b]пиразин-2,3-диола 5-метилпиридин-2,3-диамин (98000,0 мг, 0,80 моль) и щавелевую кислоту (76900,0 мг, 0,85 моль) добавляли к 3 н. HCl (784,0 мл). Смесь перемешивали при 120°C в течение 24 часов и затем охлаждали до комнатной температуры. Сформированное твердое вещество фильтровали и высушивали при пониженном давлении, получая твердое соединение коричневого цвета, представляющее собой 7-метилпиридо[2,3-b]пиразин-2,3-диол (140000,0 мг, 100%).
1Н-ЯМР (300 МГц, ДMCO-d6); δ: 12,26 (шир. с, 1Н), 11,97 (шир. с, 1Н), 7,90 (с, 1Н), 7,25 (с, 1Н), 2,25 (с, 3H).
(с) Синтез 2,3-дихлор-7-метилпиридо[2,3-b]пиразина
Смесь 7-метилпиридо[2,3-b]пиразин-2,3-диола (140000,0 мг, 0,79 моль) и POCl3 (900,0 мл) перемешивали при 130°C в течение 24 часов и затем охлаждали до комнатной температуры. Реакционную смесь вливали в смесь воды со льдом, получая твердое вещество. Сформированное твердое вещество фильтровали, высушивали при пониженном давлении и затем очищали хроматографией на колонке (DCM) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение коричневого цвета, представляющее собой 2,3-дихлор-7-метилпиридо[2,3-b]пиразин (55000,0 мг, 33%).
(d) Синтез (1-(2-хлор-7-метилпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
2,3-дихлор-7-метилпиридо[2,3-b]пиразин (750,0 мг, 3,47 ммоль) и азетидин-3-ил(метил)-трет-бутилкарбамат гидрохлорид (850,0 мг, 3,82 ммоль) вводили в реакцию таким же образом, как в примере 2 (с), получая твердое соединение светло-желтого цвета, представляющее собой (1-(2-хлор-7-метилпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (720,0 мг, 57%).
ЖХ/МС ESI (+): 364 (М+1)
1H-ЯМР (300 МГц, CDCl3); δ: 8,74 (д, 1Н, J=2,1 Гц), 7,91 (д, 1Н, J=2,1 Гц), 5,05 (м, 1Н), 4,71 (м, 2Н), 4,53 (м, 2Н), 2,99 (с, 3H), 2,51 (с, 3H), 1,50 (с, 9Н).
(e) Синтез (1-(8-метилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)-трет-бутилметилкарбамата
(1-(2-хлор-7-метилпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (320,0 мг, 0,88 ммоль) и моногидрат гидразина (0,1 мл, 3,50 ммоль) растворяли в EtOH (8,0 мл) и перемешивали при 40°C в течение 12 часов. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении, получая соединение темно-коричневого цвета, представляющее собой (1-(2-гидразинил-7-метилпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат. Неочищенное соединение (1-(2-гидразинил-7-метилпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат и триметилортоформиат (4,0 мл) вводили в реакцию, получая твердое соединение коричневого цвета, представляющее собой (1-(8-метилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)-трет-бутилметилкарбамат (92,0 мг, 29% за 2 стадии).
ЖХ/МС ESI (+): 370 (М+1)1H
1H-ЯМР (300 МГц, CDCl3); δ: 9,14 (с, 1Н), 8,52 (д, 1Н, J=2,1 Гц), 7,84 (д, 1Н, J=2,1 Гц), 4,60 (м, 5Н), 2,99 (с, 3H), 2,46 (с, 3H), 1,47 (с, 9Н).
(f) Синтез N-метил-1-(8-метилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-амина
(1-(8-метилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)-трет-бутилметилкарбамат (90,0 мг, 0,24 ммоль) растворяли в смеси DCM (2,0 мл) и TFA (3,0 мл). Реакционную смесь перемешивали при 0°C в течение одного часа и затем очищали хроматографией на колонках (DCM:MeOH=50:1) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой N-метил-1-(8-метилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-амин (39,0 мг, 59%).
ЖХ/МС ESI (+): 270 (М+1)
1Н-ЯМР (300 МГц, CDCl3); δ: 9,13 (с, 1Н), 8,51 (д, 1Н, J=2,1 Гц), 7,83 (д, 1Н, J=2,1 Гц), 5,09 (м, 1Н), 4,75-4, 55 (м, 2Н), 4,20 (м, 1Н), 3,89 (м, 1Н), 2,50 (с, 6Н).
Пример 51
Синтез 1-(8-(дифторметил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина
(а) Синтез (1-(8-формилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
2,5 М n-BuLi в растворе гексана медленно добавляли к суспензии (1-(8-йодпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата (270,0 мг, 0,56 ммоль) и THF (5,6 мл) при -78°C. Реакционную смесь перемешивали при -78°C в течение 10 минут и затем добавляли DMF (0,5 мл) и THF (2,0 мл). Реакционную смесь перемешивали в течение одного часа и добавляли водный раствор NH4Cl, и экстрагировали EtOAc. Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонке (МеОН:DCM=1:60) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение белого цвета, представляющее собой (1-(8-формилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (18,0 мг, 8%).
ЖХ/МС ESI (+): 384 (М+1)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 10,14 (с, 1Н), 10,10 (с, 1Н), 9,04 (д, 1Н, J=2,1 Гц), 8,97 (д, 1Н, J=2,1 Гц), 5,09-5,00 (м, 2Н), 4,89 (м, 1Н), 4,60 (м, 1Н), 4,46 (м, 1Н), 2,94 (с, 3H), 1,42 (с, 9Н).
(b) Синтез (1-(8-(дифторметил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
Суспензии (1-(8-формилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата (11,0 мг, 0,03 ммоль) и дезоксофтора (0,8 мл) давали взаимодействовать в микроволновом реакторе в условиях 50 Вт, 70°C, в течение 1,5 часа и затем охлаждали до комнатной температуры. Реакционную смесь выливали в воду со льдом и добавляли водный раствор NH4Cl, и смесь экстрагировали раствором МеОН:DCM=1:20, Органический слой высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (МеОН:DCM=1:60) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой (1-(8-(дифторметил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (5,0 мг, 45%).
1Н-ЯМР (300 МГц, CDCl3); δ: 9,23 (с, 1Н), 8,76 (с, 1Н), 8,19 (с, 1Н), 7,04-6, 66 (м, 1Н), 5,18 (м, 1Н), 4,99 (м, 1Н), 4,74 (м, 1Н), 4,56 (м, 1H), 4, 35-4,25 (м, 1H), 3,01 (с, 3H), 1,49 (с, 9Н).
(с) Синтез 1-(8-(дифторметил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина
TFA (0,4 мл) добавляли к смеси (1-(8-(дифторметил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата (5,0 мг, 0,01 ммоль) и DCM (0,6 мл), и затем смесь перемешивали при комнатной температуре в течение одного часа. Реакционную смесь очищали хроматографией на колонках (МеОН:DCM=1:40) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали. К остатку добавляли Et2O, получая твердое вещество. Сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении, получая твердое соединение кремового цвета, представляющее собой 1-(8-(дифторметил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (1,5 мг, 41%).
ЖХ/МС ESI (+): 306 (М+1)
1Н-ЯМР (300 МГц, ДMCO-d6); δ: 10,07 (с, 1Н), 8,83 (с, 1Н), 8,69 (с, 1Н), 7,42-7,05 (м, 1Н), 4,93 (м, 1Н), 4,52-4,40 (м, 2Н), 4,01 (м, 1Н), 3,72 (м, 1Н), 2,30 (с, 3H).
Пример 52
Синтез N-метил-1-(8-(трифторметил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-амина
(а) Синтез 3-нитро-5-(трифторметил)пиридин-2-амина
5-(трифторметил)пиридин-2-амин (1700,0 мг, 10,49 ммоль) растворяли в концентрированной H2SO4 (10,0 мл) и затем медленно добавляли концентрированную HNO3 (1,7 мл, 26,22 ммоль). Реакционную смесь перемешивали при 80°C в течение 4 8 часов, затем добавляли в смесь воды со льдом, подщелачивали 1 н. водным раствором NaOH (рН=9) и затем экстрагировали EtOAc (200,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонке (n-Hех:EtOAc=50:50) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 3-нитро-5-(трифторметил)пиридин-2-амин (538,0 мг, 25%).
ЖХ/МС ESI (+): 208 (М+1)
1Н-ЯМР (300 МГц, CDCl3); δ: 8,67 (д, 1Н, J=1,7 Гц), 8,59 (д, 1Н, J=1,7 Гц), 7,92 (шир. с, 1Н), 6,10 (шир. с, 1Н).
(b) Синтез 5-(трифторметил)пиридин-2,3-диамина
3-Hитро-5-(трифторметил)пиридин-2-амин (538,0 мг, 2,60 ммоль) и SnCl2⋅2H2O (2340,0 мг, 10,39 ммоль) добавляли к DMF (5,0 мл), и смесь перемешивали при 60°C в течение 12 часов. Реакционную смесь вливали в насыщенный водный раствор NaHCO3 и нейтрализовали (рН=7), и затем экстрагировали EtOAc (200,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем упаривали при пониженном давлении, получая твердое соединение коричневого цвета, представляющее собой 5-(трифторметил)пиридин-2,3-диамин.
ЖХ/МС ESI (+): 178 (М+1)
(c) Синтез 2,3-дихлор-7-(трифторметил)пиридо[2,3-b]пиразина
Неочищенный 5-(трифторметил)пиридин-2,3-диамин добавляли к диэтилоксалату (10,0 мл). Смесь перемешивали при 120°C в течение 12 часов и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество фильтровали при пониженном давлении, получая твердое соединение коричневого цвета, представляющее собой 7-(трифторметил)пиридо[2,3-b]пиразин-2,3-диол. Смесь неочищенного 7-(трифторметил)пиридо[2,3-b]пиразин-2,3-диола и POCl3 (10,0 мл) перемешивали при 130°C в течение 12 часов и затем охлаждали до комнатной температуры. Реакционную смесь вливали в воду со льдом, получая твердое вещество. Сформированное твердое вещество фильтровали и затем высушивали при пониженном давлении, получая твердое соединение коричневого цвета, представляющее собой 2,3-дихлор-7-(трифторметил)пиридо[2,3-b]пиразин (370,0 мг, 53% за 3 стадии).
ЖХ/МС ESI (+): 268 (М+1), 270 (М+3), 272 (М+5)
(d) Синтез (1-(2-хлор-7-(трифторметил)пиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
2,3-дихлор-7-(трифторметил)пиридо[2,3-b]пиразин (100,0 мг, 0,37 ммоль) и TEA (0,26 мл, 1,85 ммоль) растворяли в DCM (10,0 мл), и азетидин-3-ил(метил)-трет-бутилкарбамат гидрохлорид (83,0 мг, 0,37 ммоль), разбавленный в DCM (5,0 мл), и TEA (0,26 мл, 1,85 ммоль) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение одного часа. Реакционную смесь вливали в насыщенный водный раствор NH4Cl и экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонке (DCM:MeOH=100:0) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение коричневого цвета, представляющее собой (1-(2-хлор-7-(трифторметил)пиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат.
ЖХ/МС ESI (+): 418 (М+1)
(е) Синтез (1-(2-гидразинил-7-(трифторметил)пиридо[2,3-Ь]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
Неочищенный (1-(2-хлор-7-(трифторметил)пиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат и моногидрат гидразина (47,0 мг, 0,93 ммоль) растворяли в EtOH (5,0 мл), перемешивали при комнатной температуре в течение 12 часов и затем перегоняли при пониженном давлении. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой (1-(2-гидразинил-7-(трифторметил)пиридо[2,3-b]пиразин-3-ил) азетидин-3-ил)(метил)-трет-бутилкарбамат.
ЖХ/МС ESI (+): 414 (М+1)
(f) Синтез (1-(8-(трифторметил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)-трет-бутилметилкарбамата
Смесь неочищенного (1-(2-гидразинил-7-(трифторметил)пиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат и триметилортоформиат (10,0 мл) перемешивали при 80°C в течение одного часа и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество фильтровали и затем высушивали при пониженном давлении, получая твердое соединение кремового цвета, представляющее собой (1-(8-(трифторметил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)-трет-бутилметилкарбамат.
ЖХ/МС ESI (+): 424 (М+1)
(g) Синтез N-метил-1-(8-(трифторметил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-амина
Неочищенный (1-(8-(трифторметил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)-трет-бутилметилкарбамат растворяли в DCM (4,0 мл), и затем TFA (0,5 мл) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение одного часа. Реакционную смесь перегоняли при пониженном давлении. Остаток нейтрализовали водным раствором NaHCO3 (рН=7) и затем экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонке (DCM:MeOH=98:2) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой N-метил-1-(8-(трифторметил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-амин (26,7 мг, 20% за 4 стадии).
ЖХ/МС ESI (+): 324 (М+1)
1H-ЯМР (300 МГц, ДMCO-d6); δ: 10,1 (с, 1Н), 9,01 (шир. с, 1Н), 8,83 (шир. с, 1Н), 4,95 (м, 1Н), 4,48 (м, 2Н), 4,03 (м, 1Н), 3,74 (м, 1Н), 2,30 (с, 3H).
Пример 53
Синтез 4-(4-метилпиперазин-1-ил)-8-(трифторметил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
(a) Синтез 2-хлор-3-(4-метилпиперазин-1-ил)-7-(трифторметил)пиридо[2,3-b]пиразина
2,3-дихлор-7-(трифторметил)пиридо[2,3-b]пиразин (50,0 мг, 0,19 ммоль) и TEA (0,3 мл, 1,90 ммоль) растворяли в DCM (5,0 мл), и N-метилпиперазин (21,0 мкл, 0,19 ммоль), разбавленный в DCM (5,0 мл), медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и вливали в насыщенный водный раствор NH4Cl, и затем экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонке (DCM:MeOH=100:0) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 2-хлор-3-(4-метилпиперазин-1-ил)-7-(трифторметил)пиридо[2,3-b]пиразин (53,0 мг, 85%).
ЖХ/МС ESI (+): 332 (М+1), 334 (М+3)
(b) Синтез 4-(4-метилпиперазин-1-ил)-8-(трифторметил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
2-хлор-3-(4-метилпиперазин-1-ил)-7-(трифторметил)пиридо[2,3-b]пиразин (53,0 мг, 0,16 ммоль) и моногидрат гидразина (0,1 мл, 2,04 ммоль) растворяли в EtOH (2,0 мл), перемешивали при комнатной температуре в течение одного часа и затем перегоняли при пониженном давлении. Реакционную смесь выливали в насыщенный водный раствор NH4Cl и затем экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 2-гидразинил-3-(4-метилпиперазин-1-ил)-7-(трифторметил)пиридо[2,3-b]пиразин. Смесь неочищенного 2-гидразинил-3-(4-метилпиперазин-1-ил)-7-(трифторметил)пиридо[2,3-b]пиразина и триметилортоформиата (2,0 мл) перемешивали при 80°C в течение одного часа и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество фильтровали и затем высушивали при пониженном давлении. Остаток очищали хроматографией на колонке (DCM:MeOH=100:0) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 4-(4-метилпиперазин-1-ил)-8-(трифторметил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (14,3 мг, 23% за 3 стадии).
ЖХ/МС ESI (+): 338 (М+1)
1Н-ЯMP (300 МГц, ДМСО-d6); δ: 10,13 (с, 1Н), 9,05 (д, 1Н, J=2,1 Гц), 8,87 (д, 1Н, J=2,1 Гц), 5,00-4,00 (м, 4Н), 2,53 (м, 4Н), 2,26 (с, 3H).
Пример 54
Синтез 1-(8-этинилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина
(а) Синтез (1-(8-((триметилсилил)этинил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)-трет-бутилметилкарбамата
(1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (50,0 мг, 0,12 ммоль), TMS-ацетилен (81,0 мкл, 0,58 ммоль), PdCl2(PPh3)2 (8,1 мг, 0,01 ммоль), CuI (4,4 мг, 0,02 ммоль) и TEA (0,5 мл) добавляли к DMF (0,5 мл), и смеси давали взаимодействовать в микроволновом реакторе в условиях 50 Вт, 70°C, в течение 1,5 часа, затем охлаждали до комнатной температуры. К реакционной смеси добавляли DCM и воду, получая твердое вещество, и сформированное твердое вещество отфильтровывали, получая твердое вещество серого цвета. Полученное твердое вещество серого цвета перемешивали в растворе MeOH:DCM=1:9 и затем отфильтровывали. Фильтрат перегоняли при пониженном давлении, получая твердое соединение серого цвета, представляющее собой (1-(8-((триметилсилил)этинил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)-трет-бутилметилкарбамат (50,0 мг, 96%).
ЖХ/МС ESI (+): 452 (М+1)
1Н-ЯМР (300 МГц, ДМСО-d6) δ: 10,00 (с, 1Н), 8,75 (д, 1Н, J=2,1 Гц), 8,57 (д, 1Н, J=2,1 Гц), 5,25-4,85 (м, 2Н), 4,82 (м, 1Н), 4,51 (м, 1Н), 4,41 (м, 1Н), 2,93 (с, 3H), 1,42 (с, 9Н), 0,27 (с, 9Н).
(b) Синтез 1-(8-этинилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина
(1-(8-((триметилсилил)этинил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)-трет-бутилметилкарбамат (50,0 мг, 0,11 ммоль) растворяли в 4 н. растворе диоксана HCl (1,0 мл). Реакционную смесь перемешивали при комнатной температуре в течение одного часа, получая твердое вещество, и сформированное твердое вещество отфильтровывали. Полученное твердое вещество растворяли в THF (1,0 мл) и добавляли TBAF (0,2 мл, 0,22 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение одного часа и добавляли МеОН, получая твердое вещество. Сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая твердое соединение кремового цвета, представляющее собой 1-(8-этинилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (6,0 мг, 19%).
ЖХ/МС ESI (+): 280 (М+1)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 9,99 (с, 1Н), 8,76 (д, 1Н, J=1,8 Гц), 8,60 (д, 1Н, J=1,8 Гц), 4,94 (м, 1Н), 4, 60-4, 40 (м, 3H), 4,10 (м, 1Н), 3,81 (м, 1Н), 3,16 (м, 1Н), 2,36 (с, 3H).
Пример 55
Синтез N-метил-1-(8-винилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-амина
(а) Синтез (1-(8-винилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)-трет-бутилметилкарбамата
(1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (150,0 мг, 0,35 ммоль), трибутилвинилолово (1,0 мл, 3,45 ммоль), Pd(OAc)2 (15,0 мг, 0,07 ммоль), CuI (39,4 мг, 0,21 ммоль), Р(о-Tol)3 (42,0 мг, 0,14 ммоль) и TEA (240,0 мкл, 1,73 ммоль) добавляли к CH3CN (2,5 мл). Смеси давали взаимодействовать в микроволновом реакторе в условиях 100 Вт, 100°C, в течение 3 часов и затем охлаждали до комнатной температуры. Реакционную смесь очищали хроматографией на колонках (МеОН:DCM=1:60) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой (1-(8-винилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)-трет-бутилметилкарбамат (15,0 мг, 11%).
ЖХ/МС ESI (+): 382 (М+1)
1H-ЯМР (300 МГц, ДМСО-d6); δ: 10,00 (с, 1Н), 8,78 (с, 1Н), 8,62 (шир. с, 1Н), 6,86 (м, 1Н), 6,06 (м, 1Н), 5,45 (м, 1Н), 5,20-4,65 (м, 3H), 4,65-4,20 (м, 2Н), 2,93 (с, 3H), 1,42 (с, 9Н).
(b) Синтез N-метил-1-(8-винилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-амина
(1-(8-винилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)-трет-бутилметилкарбамат (15,0 мг, 0,02 ммоль) растворяли в DCM (0,6 мл) и затем добавляли TFA (0,4 мл). Реакционную смесь перемешивали при комнатной температуре в течение одного часа и затем очищали хроматографией на колонках (МеОН:DCM=1:40) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой N-метил-1-(8-винилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-амин (1,5 мг, 29%).
ЖХ/МС ESI (+): 282 (М+1)
1Н-ЯМР (300 МГц, ДMCO-d6); δ: 9,99 (с, 1Н), 8,76 (с, 1Н), 8,60 (с, 1Н), 6,86 (м, 1Н), 6,06 (м, 1Н), 5,45 (м, 1Н), 4,90 (м, 1Н), 4,55-4,25 (м, 2Н), 3,99 (м, 1Н), 3,71 (м, 1Н), 2,29 (с, 3H).
Пример 56
Синтез 1-(8-этилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина
(1-(8-винилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)-трет-бутилметилкарбамат (12,0 мг, 0,03 ммоль) растворяли в МеОН (1,0 мл) и добавляли 10% Pd/C (4,8 мг). Колбу замещали водородом и затем давали взаимодействовать в течение 12 часов. Реакционную смесь фильтровали через целит и затем перегоняли при пониженном давлении. Остаток растворяли в DCM (0,6 мл) и добавляли TFA (0,4 мл). Реакционную смесь перемешивали в течение одного часа и затем очищали хроматографией на колонках (МеОН:DCM=1:40) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 1-(8-этилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (3,5 мг, 39%).
ЖХ/МС ESI (+): 284 (М+1)
1Н-ЯМР (300 МГц, CDCl3); δ: 9,15 (с, 1Н), 8,52 (д, 1Н, J=2,1 Гц), 7,82 (д, 1Н, J=2,1 Гц), 5,09 (м, 1Н), 4,78-4,50 (м, 2Н), 4,22 (м, 1Н), 3,85 (м, 1Н), 2,80 (кв., 2Н, J=7,5 Гц), 2,49 (с, 3H), 1,35 (т, 3H, J=7,5 Гц).
Пример 57
Синтез 4-(3-(метиламино)азетидин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-8-ола
1-(8-Метоксипиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (38,0 мг, 0,13 ммоль) растворяли в DCM (3,0 мл) и затем охлаждали до -78°C. Добавляли 1 М трибромборан (3,0 мл), растворенный в DCM в концентрации 1 М. Температуру постепенно повышали до 40°C. Реакционную смесь перемешивали в течение 12 часов и затем охлаждали до -78°C и добавляли МеОН (3,0 мл). Температуре реакционной смеси давали увеличиться до комнатной температуры. Затем реакционную смесь концентрировали и очищали хроматографией на колонках (DCM:MeOH от 95:5 до 90:10) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение белого цвета, представляющее собой 4-(3-(метиламино)азетидин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-8-ол (18,0 мг, 50%).
ЖХ/МС ESI (+): 272 (М+1)
1H-ЯМР (300 МГц, MeOH-d4); δ: 9,73 (с, 1Н), 8,20 (м, 1Н), 7,90 (м, 1Н), 4,81 (м, 2Н), 4,45 (м, 2Н), 4,01 (м, 1Н), 2,59 (с, 3H).
Пример 58
Синтез 1-(8-метоксипиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина
(а) Синтез (1-(8-метоксипиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
Смесь, содержащую МеОН (4,5 мл) и NaH (450, 0 мг), перемешивали при комнатной температуре в течение 30 минут и затем добавляли к раствору (1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата (300,0 мг, 0,69 ммоль) и CuI (262, 8 мг, 1,38 ммоль), растворенных в DMF (4,5 мл). Реакционной смеси давали взаимодействовать в микроволновом реакторе в условиях 60 Вт, 90°C, в течение 30 минут, охлаждали до комнатной температуры, затем концентрировали и очищали хроматографией на колонках (МеОН:DCM=5:95) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение белого цвета, представляющее собой (1-(8-метоксипиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (49,0 мг, 55%).
ЖХ/МС ESI (+): 386 (М+1)
(b) Синтез 1-(8-метоксипиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина
(1-(8-метоксипиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (49,0 мг, 0,13 ммоль) растворяли в DCM (0,6 мл), и TFA (0,4 мл) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение одного часа, затем концентрировали и очищали хроматографией на колонках (МеОН:DCM=5:95) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение белого цвета, представляющее собой 1-(8-метоксипиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (25,0 мг, 33%).
ЖХ/МС ESI (+): 286 (М+1)
1Н-ЯМР (300 МГц, МеОН-d4); δ: 9,82 (с, 1Н), 8,30 (д, 1Н, J=2,7 Гц), 8,15 (д, 1Н, J=2,7 Гц), 4,76-4,19 (м, 4Н), 4,00 (с, 3H), 3,83 (м, 1Н), 2,43 (с, 3H).
Пример 59
Синтез 1-(8-(дифторметокси)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина
(a) Синтез (1-(8-(бензилокси)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
(1-(8-(бензилокси)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата (200,0 мг, 0,60 ммоль), 1,10-фенантролина (21,6 мг, 0,12 ммоль), карбоната цезия (391,0 мг, 1,20 ммоль) и CuI (114,3 мг, 0,60 ммоль) растворяли в бензиловом спирте (4,0 мл). Смеси давали взаимодействовать в микроволновом реакторе в условиях 100 Вт, 100°C, в течение 2 часов и затем охлаждали до комнатной температуры и добавляли 36,0 мл воды. Реакционную смесь концентрировали при пониженном давлении и очищали хроматографией на колонках (МеОН:DCM=5:95) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение белого цвета, представляющее собой (1-(8-(бензилокси)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (120,0 мг, 43%).
ЖХ/МС ESI (+): 462 (М+1)
(b) Синтез (1-(8-гидроксипиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
(1-(8-(бензилокси)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (120,0 мг, 0,32 ммоль) и 10% Pd/C (30,0 мг) растворяли в МеОН (10,0 мл). Колбу замещали водородом и затем перемешивали при комнатной температуре в течение 3 дней. Реакционную смесь фильтровали через целит, перегоняли при пониженном давлении и затем очищали хроматографией на колонках (МеОН:DCM=5:95). Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение белого цвета, представляющее собой (1-(8-гидроксипиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (40,0 мг, 34%).
ЖХ/МС ESI (+): 372 (М+1)
(с) Синтез 1-(8-(дифторметокси)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина
(1-(8-гидроксипиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (20,0 мг, 0,05 ммоль) растворяли в CH3CN (0,7 мл) и добавляли КОН (60,0 мг, 1,08 ммоль), растворенный в воде (0,7 мл), и затем добавляли диэтил(бромдифторметил)фосфонат (60,0 мкл, 0,32 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении и растворяли в DCM (1,0 мл) без дальнейшей очистки, и затем добавляли TFA (0,2 мл). Реакционную смесь перемешивали при комнатной температуре в течение одного часа и очищали хроматографией на колонках (МеОН:DCM=5:95) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение белого цвета, представляющее собой 1-(8-(дифторметокси)пиридо[2,3-е] [1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (10,0 мг, 17%).
ЖХ/МС ESI (+): 322 (М+1)
1Н-ЯМР (300 МГц, МеОН-d4); δ: 9,79 (с, 1Н), 8,41 (м, 2Н), 7,24-6,75 (м, 1Н), 5,03 (м, 1Н), 4,57 (м, 2Н), 4,15 (м, 1Н), 3,82 (м, 1Н), 2,43 (с, 3H).
Пример 60
Синтез 8-хлор-7-метокси-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
7,8-дихлор-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (10,0 мг, 0,03 ммоль) и NaH (4,0 мг: в 60%-ом масле, 0,12 ммоль) растворяли в МеОН (1,0 мл). Смеси давали взаимодействовать в микроволновом реакторе в условиях 60 Вт, 90°C в течение одного часа. Реакционную смесь перегоняли при пониженном давлении и затем очищали хроматографией на клонке (DCM:MeOH=99:1) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 8-хлор-7-метокси-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (3,7 мг, 37%).
ЖХ/МС ESI (+): 334 (М+1), 336 (М+3)
1H-ЯMP (300 МГц, ДМСО-d6); δ: 9,91 (с, 1Н), 8,85 (с, 1Н), 4,41 (м, 4Н), 4,01 (м, 3H), 2,50 (м, 4Н), 2,26 (с, 3H).
Пример 61
Синтез 8-хлор-7-метокси-4-(пиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
4-(7,8-дихлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)пиперазин-1-трет-бутилкарбоксилат (20,0 мг, 0,05 ммоль), полученный из 2,3,6,7-тетрахлорпиридо[2,3-b]пиразина таким же образом, как (с)-(e) Примера 2, и NaH (9,0 мг: в 60%-ом масле, 0,25 ммоль) растворяли в МеОН (1,0 мл). Смесь перемешивали в микроволновом реакторе в условиях 60 Вт, 90°C, нейтрализовали TEA (рН=7) и затем очищали хроматографией на колонках (DCM:MeOH=99:1) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 8-хлор-7-метокси-4-(пиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (9,0 мг, 60% за 2 стадии).
ЖХ/МС ESI (+): 320 (М+1), 322 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 9,98 (с, 1Н), 8,92 (с, 1Н), 4,40 (м, 4Н), 4,08 (с, 3H), 2,94 (м, 4Н).
Пример 62
Синтез 7,8-дихлор-4-(пиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
4-(7,8-дихлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)пиперазин-1-трет-бутилкарбоксилат (20,0 мг, 0,05 ммоль) растворяли в DCM (2,0 мл), и TFA (0,5 мл) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и перегоняли при пониженном давлении. Остаток нейтрализовали TEA (рН=7) и очищали хроматографией на колонке (DCM:MeOH=99:1) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 7,8-дихлор-4-(пиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (14,0 мг, 92%).
ЖХ/МС ESI (+): 324 (М+1), 326 (М+3)
1Н-ЯМР (300 МГц, ДMCO-d6); δ: 10,01 (с, 1Н), 9,08 (с, 1Н), 5,00-4,00 (м, 4Н), 2,95 (м, 4Н).
Пример 63
Синтез 8-хлор-7-этокси-4-(пиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
4-(7,8-дихлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)пиперазин-1-трет-бутилкарбоксилат (20,0 мг, 0,05 ммоль) и NaOEt (16,0 мг, 0,25 ммоль) растворяли в EtOH (1,0 мл). Смеси давали взаимодействовать в микроволновом реакторе в условиях 60 Вт, 90°C, фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 8-хлор-7-этокси-4-(пиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (4,0 мг, 25% за 2 стадии).
ЖХ/МС ESI (+): 334 (М+1), 336 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 9,98 (м, 1Н), 8,91 (м, 1Н), 4,53 (кв., 2Н, J=7,1 Гц), 4,40 (м, 4Н), 2,95 (м, 4Н), 1,46 (т, 3H, J=7,1 Гц).
Пример 64
Синтез 8-хлор-4-(4-метилпиперазин-1-ил)-7-(2,2,2-трифторэтокси)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
7,8-дихлор-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (10,0 мг, 0,03 ммоль) и NaH (12,0 мг: в 60%-ом масле, 0,30 ммоль), растворяли в CF3CH2OH (1,0 мл). Смесь затем перемешивали в микроволновом реакторе в условиях 60 Вт, 120°C, в течение 1 часа, перегоняли при пониженном давлении и затем очищали хроматографией на колонке (DCM:MeOH=99:1) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 8-хлор-4-(4-метилпиперазин-1-ил)-7-(2,2,2-трифторэтокси)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (3,51 мг, 29%).
ЖХ/МС ESI (+): 402 (М+1), 404 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 9,93 (м, 1Н), 8,93 (м, 1Н), 5,14 (кв., 2Н, J=8,9 Гц), 4,43 (м, 4Н), 2,50 (м, 4Н), 2,25 (с, 3H).
Пример 65
Синтез 1-(8-бром-9-метилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина
(a) Синтез 5-бром-4-метилпиридин-2,3-диамина
5-бром-4-метил-3-нитропиридин-2-амин (700,0 мг, 3,02 ммоль), Fe (1680,0 мг, 30,20 ммоль) и концентрированную HCl (50,0 мкл) добавляли к EtOH (2,8 мл) и воде (0,7 мл). Суспензия перемешивали при 100°C в течение 30 минут. Реакционную смесь охлаждали до комнатной температуры, фильтровали через целит и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонке (МеОН:DCM=1:40) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение коричневого цвета, представляющее собой 5-бром-4-метилпиридин-2,3-диамин (550,0 мг, 90%).
ЖХ/МС ESI (+): 202 (М+1), 204 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 7,37 (с, 1Н), 5,49 (с, 2Н), 4,74 (с, 2Н), 2,12 (с, 3H).
(b) Синтез 7-бром-8-метилпиридо[2,3-b]пиразин-2,3-диола
Смесь 5-бром-4-метилпиридин-2,3-диамина (550,0 мг, 2,72 ммоль) и диэтилоксалата (10,0 мл) перемешивали при 100°C в течение 12 часов и затем охлаждали до комнатной температуры. К реакционной смеси добавляли Et2O, получая твердое вещество, и сформированное твердое вещество фильтровали и высушивали при пониженном давлении, получая твердое соединение коричневого цвета, представляющее собой 7-бром-8-метилпиридо[2,3-b]пиразин-2,3-диол (595,0 мг, 85%).
ЖХ/МС ESI (+): 256 (М+1), 258 (М+3)
1H-ЯMP (300 МГц, ДМСО-d6); δ: 12,40 (с, 1Н), 11,57 (с, 1Н), 8,19 (с, 1Н), 2,45 (с, 3H).
(c) Синтез 7-бром-2,3-дихлор-8-метилпиридо[2,3-b]пиразина
Смесь 7-бром-8-метилпиридо[2,3-b]пиразин-2,3-диола (560,0 мг, 2,19 ммоль) и POCl3 (5,0 мл) перемешивали при 100°C в течение 12 часов и затем охлаждали до комнатной температуры. Реакционную смесь вливали в смесь воды со льдом, получая твердое вещество. Сформированное твердое вещество фильтровали и высушивали при пониженном давлении. Полученное твердое вещество растворяли в DCM и очищали хроматографией на колонке (EtOAc:n-Hех=1:9) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение красного цвета, представляющее собой 7-бром-2,3-дихлор-8-метилпиридо[2,3-b]пиразин (365,0 мг, 54%).
ЖХ/МС ESI (+): 292 (М+1), 294 (М+3), 296 (М+5)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 9,25 (с, 1Н), 2,78 (с, 3H).
(d) Синтез (1-(7-бром-2-хлор-8-метилпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
TEA (72,0 мкл, 0,51 ммоль) добавляли к смеси 7-бром-2,3-дихлор-8-метилпиридо[2,3-b]пиразина (50,0 мг, 0,17 ммоль), азетидин-3-ил(метил)-трет-бутилкарбамат гидрохлорида (40,0 мг, 0,18 ммоль) и DCM (1,7 мл) при 0°C и перемешивали в течение одного часа. Реакционную смесь очищали хроматографией на колонке (EtOAc:n-Hex=1:5) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой (1-(7-бром-2-хлор-8-метилпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (58,0 мг, 76%).
ЖХ/МС ESI (+): 442 (М+1), 444 (М+3)
1H-ЯМР (300 МГц, ДМСО-d6); δ: 8,85 (с, 1Н), 4,86 (м, 1Н), 4, 65-4, 55 (м, 2Н), 4, 49-4, 40 (м, 2Н), 2,90 (с, 3H), 2,66 (с, 3H), 1,41 (с, 9Н).
(е) Синтез (1-(8-бром-9-метилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
Моногидрат гидразина (20,0 мкл, 0,63 ммоль) добавляли к суспензии (1-(7-бром-2-хлор-8-метилпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата (56,0 мг, 0,13 ммоль) и EtOH (1,0 мл). Реакционную смесь перемешивали при комнатной температуре в течение одного часа и затем перегоняли при пониженном давлении. К остатку добавляли Et2O, получая твердое вещество. Сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении, получая твердое соединение коричневого цвета, представляющее собой (1-(7-бром-2-гидразинил-8-метилпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат. Неочищенный (1-(7-бром-2-гидразинил-8-метилпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат растворяли в триметилортоформиате (2,0 мл), и смесь перемешивали при 85°C в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонке (МеОН:DCM=1:40) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение светло-коричневого цвета, представляющее собой (1-(8-бром-9-метилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (25,0 мг, 45%).
ЖХ/МС ESI (+): 448 (М+1), 450 (М+3)
(f) Синтез 1-(8-бром-9-метилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина
TFA (0,4 мл) добавляли к смеси (1-(8-бром-9-метилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата (25,0 мг, 0,06 ммоль) и DCM (0,6 мл), и смесь перемешивали при комнатной температуре в течение одного часа. Реакционную смесь очищали хроматографией на колонках (МеОН:DCM=1:40) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали. Et2O к остатку добавляли и получали твердое вещество, и сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении, получая твердое соединение кремового цвета, представляющее собой 1-(8-бром-9-метилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (13,0 мг, 68%).
ЖХ/МС ESI (+): 348 (М+1), 350 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 9,82 (с, 1Н), 8,63 (с, 1Н), 4,90 (м, 1Н), 4,45-4,35 (м, 2Н), 3,99 (м, 1Н), 3,71 (м, 1Н), 2,90 (с, 3H), 2,30 (с, 3H).
Пример 66
Синтез 8-бром-9-метил-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
(а) Синтез 7-бром-2-хлор-8-метил-3-(4-метилпиперазин-1-ил)пиридо [2,3-b]пиразина
TEA (72,0 мкл, 0,51 ммоль) добавляли к смеси 7-бром-2,3-дихлор-8-метилпиридо[2,3-b]пиразина (50,0 мг, 0,17 ммоль), N-метилпиперазина (20,0 мкл, 0,18 ммоль) и DCM (1,7 мл) при 0°C, и смесь перемешивали в течение одного часа. Реакционную смесь очищали хроматографией на колонке (EtOAc:n-Hех=1:5) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 7-бром-2-хлор-8-метил-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин (50,0 мг, 82%).
ЖХ/МС ESI (+): 356 (М+1), 358 (М+3), 360 (М+5)
1H-ЯМР (300 МГц, ДМСO-d6); δ=8,96 (с, 1Н), 3, 64-3, 58 (м, 4Н), 2,69 (с, 3H), 2,55-2,50 (м, 4Н), 2,25 (с, 3H).
(b) Синтез 8-бром-9-метил-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
Моногидрат гидразина (22,0 мкл, 0,70 ммоль) добавляли к суспензии 7-бром-2-хлор-8-метил-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразина (50,0 мг, 0,14 ммоль) и EtOH (1,0 мл). Реакционную смесь перемешивали при комнатной температуре в течение одного часа и затем перегоняли при пониженном давлении. К остатку добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 7-бром-2-гидразинил-8-метил-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин. Неочищенный 7-бром-2-гидразинил-8-метил-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин растворяли в триметилортоформиат (2,0 мл) и затем перемешивали при 85°C в течение 3 часов. Затем реакционную смесь охлаждали до комнатной температуры и перегоняли при пониженном давлении. Остаток очищали хроматографией на колонке (МеОН:DCM=1:40) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 8-бром-9-метил-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (24,0 мг, 47%).
ЖХ/МС ESI (+): 362 (М+1), 364 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 9,85 (с, 1Н), 8,66 (с, 1Н), 4, 60-4,30 (м, 4Н), 2,92 (с, 3H), 2, 54-2, 50 (м, 4Н), 2,25 (с, 3H).
Пример 67
Синтез 1-(8,9-дихлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина
(а) Синтез N-(4-хлорпиридин-2-ил)пиваламида
4-хлорпиридин-2-амин (1500,0 мг, 11,70 ммоль) растворяли в пиридине (6,0 мл), и пивалоил хлорид (2,2 мл, 17,50 ммоль) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов, вливали в воду и экстрагировали EtOAc (100,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонке (n-Hex:EtOAc от 90:10 до 80:20) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение белого цвета, представляющее собой N-(4-хлорпиридин-2-ил)пиваламид (2460,0 мг, 99%)
ЖХ/МС ESI (+): 213 (М+1), 215 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 10,12 (с, 1Н), 8,32 (д, 1Н, J=5,3 Гц), 8,16 (с, 1Н), 7,25 (д, 1Н, J=5,3 Гц), 1,23 (с, 9Н).
(b) Синтез N-(4,5-дихлорпиридин-2-ил)пиваламида
N-(4-хлорпиридин-2-ил)пиваламид (2160,0 мг, 10,16 ммоль) и N-хлорсукцинимид (6781,0 мг, 50,78 ммоль) растворяли в безводном CH3CN (100,0 мл). Смесь перемешивали при 70°C в течение 2 часов и затем охлаждали до комнатной температуры. Реакционную смесь вливали в воду и экстрагировали EtOAc (100,0 мл). Органический слой промывали 1 н. NaOH и солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонке (n-Hex:EtOAc=90:10) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой N-(4,5-дихлорпиридин-2-ил)пиваламид (1990,0 мг, 79%).
ЖХ/МС ESI (+): 247 (М+1), 249 (М+3), 251 (М+5)
1Н-ЯМР (300 МГц, ДMCO-d6); δ: 10,29 (с, 1Н), 8,54 (с, 1Н), 8,34 (с, 1Н), 1,23 (с, 9Н).
(c) Синтез 4,5-дихлор-3-нитропиридин-2-амина
N-(4,5-дихлорпиридин-2-ил)пиваламид (1990,0 мг, 8,05 ммоль) медленно добавляли и растворяли в концентрированной H2SO4 (11,0 мл) при 10°C, и затем медленно добавляли смесь концентрированная HNO3/концентрированная H2SO4 (332,0 мкл/415,0 мкл). Реакционную смесь перемешивали при комнатной температуре в течение 2,5 часа, вливали в смесь воды со льдом, подщелачивали 1 н. водным раствором NaOH (рН=9) и затем экстрагировали EtOAc (200,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонке (n-Hex:EtOAc от 90:10 до 80:20) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 4,5-дихлор-3-нитропиридин-2-амин (548,0 мг, 32%).
ЖХ/МС ESI (+): 208 (М+1), 210 (М+3), 212 (М+5)
(d) Синтез 4,5-дихлорпиридин-2,3-диамина
4,5-дихлор-3-нитропиридин-2-амин (548,0 мг, 2,63 ммоль), порошок цинка (1274,0 мг, 19,50 ммоль) и безводный CaCl2 (1578,0 мг, 14,20 ммоль) добавляли к 95% EtOH (20,0 мл), и смесь перемешивали при 100°C в течение одного часа. Реакционную смесь фильтровали через целит и упаривали при пониженном давлении, получая твердое соединение коричневого цвета, представляющее собой 4,5-дихлорпиридин-2,3-диамин.
ЖХ/МС ESI (+): 178 (М+1), 180 (М+3), 182 (М+5)
(e) Синтез 2,3,7,8-тетрахлорпиридо[2,3-b]пиразина
Неочищенный 4,5-дихлорпиридин-2,3-диамин добавляли к диэтилоксалату (10,0 мл). Смесь перемешивали при 120°C в течение 12 часов и затем охлаждали до комнатной температуры. Et2O добавляли через фильтр, получая твердое вещество, и сформированное твердое вещество фильтровали и высушивали при пониженном давлении, получая твердое соединение светло-коричневого цвета, представляющее собой 7,8-дихлорпиридо[2,3-b]пиразин-2,3(1Н,4Н)-дион. Смесь неочищенного 7,8-дихлорпиридо[2,3-b]пиразин-2,3(1Н,4Н)-диона и POCl3 (10,0 мл) перемешивали при 130°C в течение 48 часов и затем охлаждали до комнатной температуры. Реакционную смесь вливали в смесь воды со льдом, получая твердое вещество, и сформированное твердое вещество фильтровали и затем высушивали при пониженном давлении, получая твердое соединение коричневого цвета, представляющее собой 2,3,7,8-тетрахлорпиридо[2,3-b]пиразин (290,0 мг, 41% за 3 стадии).
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 9,35 (с, 1Н).
(f) Синтез (1-(2,7,8-трихлорпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)-трет-бутилметилкарбамата
2,3,7,8-тетрахлорпиридо[2,3-b]пиразин (100,0 мг, 0,37 ммоль) и TEA (0,3 мл, 1,85 ммоль) растворяли в DCM (8,0 мл), и азетидин-3-ил(метил)-трет-бутилкарбамат гидрохлорид (83,0 мг, 0,37 ммоль), разбавленный в DCM (2,0 мл), и TEA (0,3 мл, 1,85 ммоль) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение одного часа и вливали в насыщенный водный раствор NH4Cl, и затем экстрагировали DCM (30,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и перегоняли при пониженном давлении. Остаток очищали хроматографией на колонке (DCM:MeOH=100:0) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение коричневого цвета, представляющее собой (1-(2,7,8-трихлорпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)-трет-бутилметилкарбамат.
ЖХ/МС ESI (+): 418 (М+1), 420 (М+3)
(g) Синтез (1-(7,8-дихлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
Неочищенный (1-(2,7,8-трихлорпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)-трет-бутилметилкарбамат и моногидрат гидразина (46,0 мг, 0,93 ммоль) растворяли в EtOH (3,0 мл). Смесь перемешивали при комнатной температуре в течение 20 минут и перегоняли при пониженном давлении, получая (1-(7,8-дихлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат.
ЖХ/МС ESI (+): 414 (М+1), 416 (М+3)
(h) Синтез (1-(8,9-дихлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
Смесь неочищенного (1-(7,8-дихлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата и триметилортоформиата (2,0 мл) перемешивали при 80°C в течение одного часа и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество фильтровали и затем высушивали при пониженном давлении, получая (1-(8,9-дихлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил) (метил)-трет-бутилкарбамат.
ЖХ/МС ESI (+): 424 (М+1), 426 (М+3)
(i) Синтез 1-(8,9-дихлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина
Неочищенный (1-(8,9-дихлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат растворяли в DCM (2,0 мл), и TFA (0,5 мл) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение одного часа и перегоняли при пониженном давлении. Остаток нейтрализовали TEA и очищали хроматографией на колонке (DCM:MeOH=98:2) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 1-(8,9-дихлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (3,5 мг, 3% за 4 стадии).
ЖХ/МС ESI (+): 324 (М+1), 326 (М+3)
1H-ЯMP(300 МГц, ДMCO-d6); δ: 10,30 (с, 1Н), 8,68 (с, 1Н), 4,94 (м, 1Н), 4,46 (м, 2Н), 4,03 (м, 1Н), 3,73 (м, 1Н), 2,30 (с, 3H).
Пример 68
Синтез 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин гидрохлорида
Смесь 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина (200,0 мг, 0,60 ммоль) и 4 н. раствора диоксана в HCl (10,0 мл) перемешивали при комнатной температуре в течение 12 часов. Сформированное твердое вещество отфильтровывали и затем высушивали, получая твердое соединение белого цвета, представляющее собой 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин гидрохлорид (200,0 мг, 90%).
ЖХ/МС ESI (+): 334 (М+1), 336 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 10,0 (с, 1Н), 9,69 (шир. с, 2Н), 9,00 (д, 1Н, J=2,2 Гц), 8,66 (д, 1Н, J=2,2 Гц), 5,20-4,80 (м, 2Н), 4,70-4,40 (м, 2Н), 4,20 (м, 1Н), 2,64 (м, 3H).
Пример 69
Синтез 1-(8-бромпиридо[2,3-е]тетразоло[1,5-а]пиразин-4-ил)-N-метилазетидин-3-амин гидрохлорида
Суспензию 1-(8-бромпиридо[2,3-е]тетразоло[1,5-а]пиразин-4-ил)-N-метилазетидин-3-амина (190,0 мг, 0,57 ммоль) и 4 н. раствора диоксана в HCl (5,0 мл) перемешивали при комнатной температуре в течение 8 часов. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 1-(8-бромпиридо[2,3-е]тетразоло[1,5-а]пиразин-4-ил)-N-метилазетидин-3-амин гидрохлорид (210,0 мг, 99%).
ЖХ/МС ESI (+): 335 (М+1), 337 (М+3)
1H-ЯМР (300 МГц, ДМСО-d6); δ: 9,77 (шир. с, 2Н), 9,00 (д, 1Н, J=2,4 Гц), 8,88 (д, 1Н, J=2,4 Гц), 5,10-4,90 (м, 2Н), 4,75-4,45 (м, 2Н), 4,31 (м, 1Н), 2,66 (с, 3H).
Пример 70
Синтез 8-хлор-4-(5-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
(а) Синтез 2,7-дихлор-3-(5-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиридо[2,3-b]пиразина
2,3,7-трихлорпиридо[2,3-b]пиразин (100,0 мг, 0,43 ммоль) и TEA (0,6 мл, 4,26 ммоль) растворяли в DCM (3,0 мл), и 2-метилоктагидропирроло[3,4-с]пиррол (59,1 мг, 0,47 ммоль), разбавленный в DCM (1,0 мл), медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов, затем концентрировали и очищали хроматографией на колонках (МеОН:DCM=5:95) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 2,7-дихлор-3-(5-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиридо[2,3-b]пиразин (72,0 мг, 52%).
ЖХ/МС ESI (+): 324 (М+1), 326 (М+3)
(b) Синтез 7-хлор-2-гидразинил-3-(5-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиридо[2,3-b]пиразина
2,7-дихлор-3-(5-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиридо[2,3-b]пиразин (72,0 мг, 0,22 ммоль) и моногидрат гидразина (32,0 мкл, 0,67 ммоль) растворяли в EtOH (3,0 мл). Смесь перемешивали при комнатной температуре в течение 12 часов, получая твердое вещество. Сформированное твердое вещество отфильтровывали, получая твердое соединение желтого цвета, представляющее собой 7-хлор-2-гидразинил-3-(5-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиридо[2,3-b]пиразин (71,0 мг, 100%).
ЖХ/МС ESI (+): 320 (М+1), 322 (М+3)
(c) Синтез 8-хлор-4-(5-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
Смесь 7-хлор-2-гидразинил-3-(5-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиридо[2,3-b]пиразина (71,0 мг, 0,22 ммоль) и триметилортоформиата (3,0 мл) перемешивали при 80°C в течение 3 часов и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении. Остаток очищали хроматографией на колонках (МеОН:DCM=5:95) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 8-хлор-4-(5-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (20,0 мг, 27%).
ЖХ/МС ESI (+): 330 (М+1), 332 (М+3)
1Н-ЯМР (300 МГц, CDCl3); δ: 9,14 (с, 1Н), 8,58 (д, 1Н, J=2,4 Гц), 8,01 (д, 1Н, J=2,4 Гц), 4,70-4, 58 (м, 2Н), 4,23-4,02 (м, 2Н), 3,14-3,03 (м, 2Н), 2,71-2,65 (м, 4Н), 2,34 (с, 3H).
Пример 71
Синтез 8-бром-4-(5-метилгексапядропирроло[3,4-с]пиррол-2(1Н)-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
(а) Синтез 7-бром-2-хлор-3-(5-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиридо[2,3-b]пиразина
7-бром-2,3-дихлорпиридо[2,3-b]пиразин (100,0 мг, 0,36 ммоль) и TEA (0,5 мл, 3,58 ммоль) растворяли в DCM (3,0 мл), и 2-метилоктагидропирроло[3,4-с]пиррол (49,6 мг, 0,39 ммоль), разбавленный в DCM (1,0 мл), медленно добавляли при 0°C.Реакционную смесь перемешивали при комнатной температуре в течение 12 часов, затем концентрировали и очищали хроматографией на колонках (МеОН:DCM=5:95) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 7-бром-2-хлор-3-(5-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиридо[2,3-b]пиразин (63,0 мг, 48%).
ЖХ/МС ESI (+): 368 (М+1), 370 (М+3)
(b) Синтез 7-бром-2-гидразинил-3-(5-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиридо[2,3-b]пиразина
7-бром-2-хлор-3-(5-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиридо[2,3-b]пиразин (63,0 мг, 0,17 ммоль) и моногидрат гидразина (25,0 мкл, 0,51 ммоль) растворяли в EtOH (3,0 мл). Смесь перемешивали при комнатной температуре в течение 12 часов, получая твердое вещество. Сформированное твердое вещество отфильтровывали, получая твердое соединение желтого цвета, представляющее собой 7-бром-2-гидразинил-3-(5-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиридо[2,3-b]пиразин (62,0 мг, 100%).
ЖХ/МС ESI (+): 364 (М+1), 366 (М+3)
(c) Синтез 8-бром-4-(5-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
Смесь 7-бром-2-гидразинил-3-(5-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиридо[2,3-b]пиразина (62,0 мг, 0,17 ммоль) и триметилортоформиат (3,0 мл) перемешивали при 90°C в течение 6 часов и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении. Остаток очищали хроматографией на колонках (EtOAc:n-Hех:МеОН=4:4:1) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 8-бром-4-(5-метилгексагидропирроло[3,4-с]пиррол-2(1Н)-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (11,0 мг, 16%).
ЖХ/МС ESI (+): 374 (М+1), 376 (М+3)
1Н-ЯМР (300 МГц, CDCl3); δ: 9,17 (с, 1Н), 8,64 (д, 1Н, J=2,1 Гц), 8,14 (д, 1Н, J=2,1 Гц), 4,73-4,55 (м, 2Н), 4,22-3, 98 (м, 2Н), 3,12-3,02 (м, 2Н), 2,63 (м, 4Н), 2,32 (с, 3H).
Пример 72
Синтез 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-3-метилазетидин-3-амина
(a) Синтез (1-(7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил)-3-метилазетидин-3-ил)-трет-бутилкарбамата
(3-метилазетидин-3-ил)-трет-бутилкарбамат гидрохлорид (43,0 мг, 0,23 ммоль) и TEA (0,8 мл, 0,54 ммоль) добавляли к смеси 7-бром-2,3-дихлорпиридо[2,3-b]пиразина (50,0 мг, 0,18 ммоль) и DCM (1,8 мл) при 0°C и затем перемешивали в течение одного часа. Реакционную смесь концентрировали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой (1-(7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил)-3-метилазетидин-3-ил)-трет-бутилкарбамат.
ЖХ/МС ESI (+): 428 (М+1), 430 (М+3)
(b) Синтез (1-(7-бром-2-гидразинилпиридо[2,3-b]пиразин-3-ил)-3-метилазетидин-3-ил)-трет-бутилкарбамата
Моногидрат гидразина (28,0 мкл, 0,90 ммоль) добавляли к суспензии неочищенного (1-(7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил)-3-метилазетидин-3-ил)-трет-бутилкарбамата и EtOH (1,0 мл). Реакционную смесь перемешивали при комнатной температуре в течение одного часа и концентрировали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой (1-(7-бром-2-гидразинилпиридо[2,3-b]пиразин-3-ил)-3-метилазетидин-3-ил)-трет-бутилкарбамат.
ЖХ/МС ESI (+): 424 (М+1), 426 (М+3)
(с) Синтез (1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-3-метилазетидин-3-ил)-трет-бутилкарбамата
Суспензию неочищенного (1-(7-бром-2-гидразинилпиридо[2,3-b]пиразин-3-ил)-3-метилазетидин-3-ил)-трет-бутилкарбамата и триметилортоформиата (3,0 мл) перемешивали при 90°C в течение 3 часов и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество фильтровали и давление понижали, получая твердое вещество бледно-коричневого цвета, представляющее собой (1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-3-метилазетидин-3-ил)-трет-бутилкарбамат (25,0 мг, 32% за 3 стадии).
ЖХ/МС ESI (+): 434 (М+1), 436 (М+3)
(d) Синтез 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-3-метилазетидин-3-амина
(1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-3-метилазетидин-3-ил)-трет-бутилкарбамат (25,0 мг, 0,06 ммоль) растворяли в DCM (0,6 мл) и добавляли TFA (0,4 мл). Реакционную смесь перемешивали при комнатной температуре в течение одного часа и затем очищали хроматографией на колонках (МеОН:DCM=1:40) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение коричневого цвета, представляющее собой 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-3-метилазетидин-3-амин (9,0 мг, 47%).
ЖХ/МС ESI (+): 334 (М+1), 336 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 9,95 (с, 1Н), 8,90 (д, 1Н, J=2,4 Гц), 8,59 (д, 1Н, J=2,4 Гц), 4,58 (м, 1Н), 4,47 (м, 1Н), 4,15-3,95 (м, 2Н), 2,40 (шир. с, 2Н), 1,43 (с, 3H).
Пример 73
Синтез 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N,3-диметилазетидин-3-амина
(а) Синтез 3-((трет-бутоксикарбонил)амино)-3-метилазетидин-1-бензилкарбоксилата
(3-метилазетидин-3-ил)-трет-бутилкарбамат гидрохлорид (400,0 мг, 1,80 ммоль) растворяли в DCM (9,0 мл), и бензилхлорформиат (0,8 мл, 5,39 ммоль) и TEA (1,3 мл, 9,00 ммоль) добавляли при 0°C. Реакционной смеси давали взаимодействовать при комнатной температуре в течение 12 часов и затем очищали хроматографией на колонке (EtOAc:n-Hех=1:4) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение белого цвета, представляющее собой 3-((трет-бутоксикарбонил)амино)-3-метилазетидин-1-бензилкарбоксилат (680,0 мг, 99%).
ЖХ/МС ESI (+): 321 (М+1)
1Н-ЯМР (300 МГц, CDCl3); δ: 7,38-7,30 (м, 5Н), 5,10 (с, 2Н), 4,76 (шир. с, 1Н), 4,20-4,10 (м, 2Н), 3,90-3,75 (м, 2Н), 1,53 (с, 3H), 1,44 (м, 9Н).
(b) Синтез 3-((трет-бутоксикарбонил)амино(метил))-3-метилазетидин-1-бензилкарбоксилата
3-((трет-бутоксикарбонил)амино)-3-метилазетидин-1-бензилкарбоксилат (676,0 мг, 2,11 ммоль) растворяли в THF (21,0 мл) и добавляли NaH (110,0 мг, 2,74 ммоль). Реакционную смесь перемешивали в течение 10 минут и затем добавляли MeI (0,2 мл, 3,17 ммоль) и перемешивали при комнатной температуре в течение 12 часов, и затем давали взаимодействовать при 50°C в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры и очищали хроматографией на колонке (EtOAc:n-Hех=1:7) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая липкое бесцветное жидкое соединение, представляющее собой 3-((трет-бутоксикарбонил)амино(метил))-3-метилазетидин-1-бензилкарбоксилат (180,0 мг, 26%).
ЖХ/МС ESI (+): 335 (М+1)
1Н-ЯМР (300 МГц, CDCl3); δ: 7,40-7,30 (м, 5Н), 5,11 (с, 2Н), 4,12-4,05 (м, 2Н), 3,75-3,60 (м, 2Н), 2,69 (с, 3H), 1,48 (с, 3H), 1,45 (м, 9Н).
(c) Синтез (3-метилазетидин-3-ил)-трет-бутилметилкарбамата
3-((трет-бутоксикарбонил)амино(метил))-3-метилазетидин-1-бензилкарбоксилат (180,0 мг, 0,54 ммоль) растворяли в МеОН (2,0 мл) и добавляли 10% Pd/C (18,0 мг). Колбу замещали водородом и перемешивали в течение 12 часов. Реакционную смесь фильтровали через целит и перегоняли при пониженном давлении, получая липкое бесцветное жидкое соединение, представляющее собой (3-метилазетидин-3-ил)-трет-бутилметилкарбамат (50,0 мг, 46%).
1H-ЯMP (300 МГц, ДМСО-d6); δ: 3,85-3,75 (м, 2Н), 3,23-3,15 (м, 2Н), 2,64 (с, 3H), 1,52 (с, 3H), 1,44 (м, 9Н).
(d) Синтез (1-(7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил)-3-метилазетидин-3-ил)(метил)-трет-бутилкарбамата
TEA (90,0 мкл, 0,65 ммоль) добавляли при 0°C к смеси (3-метилазетидин-3-ил)-трет-бутилметилкарбамата (47,0 мг, 0,24 ммоль), 7-бром-2,3-дихлорпиридо[2,3-b]пиразина (60,0 мг, 0,22 ммоль) и DCM (2,2 мл), и смесь перемешивали в течение одного часа. Реакционную смесь очищали хроматографией на колонке (EtOAc:n-Hех=1:5) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой (1-(7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил)-3-метилазетидин-3-ил)(метил)-трет-бутилкарбамат (10,0 мг, 11%).
ЖХ/МС ESI (+): 442 (М+1), 444 (М+3)
(е) Синтез (1-(7-бром-2-гидразинилпиридо[2,3-b]пиразин-3-ил)-3-метилазетидин-3-ил)(метил)-трет-бутилкарбамата
(1-(7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил)-3-метилазетидин-3-ил)(метил)-трет-бутилкарбамат (10,0 мг, 0,02 ммоль) растворяли в EtOH (1,0 мл) и добавляли моногидрат гидразина (3,5 мкл, 0,11 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение одного часа и концентрировали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой (1-(7-бром-2-гидразинилпиридо[2,3-b]пиразин-3-ил)-3-метилазетидин-3-ил)(метил)-трет-бутилкарбамат.
ЖХ/МС ESI (+): 438 (М+1), 440 (М+3)
(f) Синтез (1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-3-метилазетидин-3-ил)(метил)-трет-бутилкарбамата
Неочищенный (1-(7-бром-2-гидразинилпиридо[2,3-b]пиразин-3-ил)-3-метилазетидин-3-ил)(метил)-трет-бутилкарбамат растворяли в триметилортоформиате (1,0 мл), и смесь перемешивали при 80°C в течение одного часа. Реакционную смесь затем охлаждали до комнатной температуры и перегоняли при пониженном давлении, получая атипичное соединение желтого цвета, представляющее собой (1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-3-метилазетидин-3-ил)(метил)-трет-бутилкарбамат.
ЖХ/МС ESI (+): 448 (М+1), 450 (М+3)
(g) Синтез 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N,3-диметилазетидин-3-амина
Неочищенный (1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-3-метилазетидин-3-ил)(метил)-трет-бутилкарбамат растворяли в 4 н. растворе диоксана HCl, и реакционную смесь перемешивали при комнатной температуре в течение одного часа и перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (MeOH:DCM=1:60) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N,3-диметилазетидин-3-амин (3,0 мг, 38% за 3 стадии).
ЖХ/МС ESI (+): 348 (М+1), 350 (М+3)
1Н-ЯМР (300 МГц, ДMCO-d6); δ: 9,95 (с, 1Н), 8,90 (д, 1Н, J=2,4 Гц), 8,59 (д, 1Н, J=2,4 Гц), 4,60-4,40 (м, 2Н), 4,18-3,96 (м, 2Н), 2,27 (с, 3H), 1,41 (с, 3H).
Пример 74
Синтез 8-бром-4-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
(а) Синтез 7-бром-2-хлор-3-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)пиридо[2,3-b]пиразина
7-бром-2,3-дихлорпиридо[2,3-b]пиразин (206,0 мг, 0,74 ммоль) и октагидропирроло[1,2-а]пиразин (102,0 мг, 0,81 ммоль) вводили в реакцию таким же образом, как в примере 24 (а), получая твердое соединение желтого цвета, представляющее собой 7-бром-2-хлор-3-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)пиридо[2,3-b]пиразин (180,0 мг, 66%).
ЖХ/МС ESI (+): 370 (М+1), 372 (М+3)
1Н-ЯМР (300 МГц, CDCl3); δ: 8,93 (д, 1Н, J=2,4 Гц), 8,32 (д, 1Н, J=2,4 Гц), 4,46 (м, 2Н), 3,15 (м, 3H), 2,92 (м, 1Н), 2,48 (м, 1Н), 2,22 (м, 2Н), 1,89 (м, 3H), 1,57 (м, 1Н).
(b) Синтез 8-бром-4-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
7-бром-2-хлор-3-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)пиридо[2,3-b]пиразин (100,0 мг, 0,36 ммоль) и моногидрат гидразина (0, 028 мл, 0,90 ммоль) растворяли в EtOH (8,0 мл), и смесь перемешивали при 40°C в течение 12 часов. Добавляли Et2O/EtOH, получая твердое вещество, и сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении, получая соединение желтого цвета, представляющее собой 7-бром-3-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)-2-гидразинилпиридо[2,3-b]пиразин. Смесь неочищенного 7-бром-3-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)-2-гидразинилпиридо[2,3-b]пиразина (60,0 мг, 0,17 ммоль) и триметилортоформиат (1,0 мл) перемешивали при 80°C в течение 1,5 часа и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении, получая твердое соединение коричневого цвета, представляющее собой 8-бром-4-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (35,0 мг, 26% за 2 стадии).
ЖХ/МС ESI (+): 374 (М+1), 376 (М+3)
1Н-ЯМР (300 МГц, CDCl3); δ: 9,16 (с, 1Н), 8,70 (д, 1Н, J=2,1 Гц), 8,17 (д, 1Н, J=2,1 Гц), 6,40 (м, 1Н), 5,52 (м, 1Н), 3,57 (м, 1Н), 3,20 (м, 2Н), 2,92 (м, 1Н), 2,44 (м, 1Н), 2,20 (м, 2Н), 1,79 (м, 3H), 1,55 (м, 1Н).
Пример 75
Синтез 4-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)-8-йодпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
(a) Синтез 2-хлор-3-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)-7-йодпиридо[2,3-b]пиразина
2,3-дихлор-7-йодпиридо[2,3-b]пиразин (50,0 мг, 0,15 ммоль) и TEA (213,0 мкл, 1,53 ммоль) растворяли в DCM (2,0 мл), и октагидропирроло[1,2-а]пиразин (19,0 мг, 0,15 ммоль) добавляли при 0°C. Реакционную смесь перемешивали при 0°C в течение одного часа, затем концентрировали и очищали хроматографией на колонках (МеОН:DCM=3:97) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 2-хлор-3-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)-7-йодпиридо[2,3-b]пиразин (38,0 мг, 58%).
ЖХ/МС ESI (+): 416 (М+1), 418 (М+3)
(b) Синтез 3-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)-2-гидразинил-7-йодпиридо[2,3-b]пиразина
2-хлор-3-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)-7-йодпиридо[2,3-b]пиразин (38,0 мг, 0,09 ммоль) и моногидрат гидразина (6,7 мкл, 0,14 ммоль) растворяли в EtOH (1,0 мл). Смесь перемешивали при комнатной температуре в течение 3 часов, получая твердое вещество, и сформированное твердое вещество отфильтровывали, получая твердое соединение желтого цвета, представляющее собой 3-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)-2-гидразинил-7-йодпиридо[2,3-b]пиразин (37,5 мг, 100%).
ЖХ/МС ESI (+): 412 (М+1)
(с) Синтез 4-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)-8-йодпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
Смесь 3-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)-2-гидразинил-7-йодпиридо[2,3-b]пиразина (39,4 мг, 0,10 ммоль) и триметилортоформиата (2,0 мл) перемешивали при 80°C в течение 3 часов и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении. Остаток очищали хроматографией на колонках (МеОН:DCM=5:95) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 4-(гексагидропирроло[1,2-а]пиразин-2(1Н)-ил)-8-йодпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (9,0 мг, 26%).
ЖХ/МС ESI (+): 422 (М+1)
1H-ЯМР (300 МГц, ДМСО-d6); δ: 10,01 (с, 1Н), 9,01 (д, 1Н, J=2,1 Гц), 8,71 (д, 1Н, J=2,1 Гц), 6,18-5,25 (м, 1Н), 3,42-3,35 (м, 1Н), 3,28-3,24 (м, 1Н), 3,20-3,15 (м, 1Н), 3,08-2, 87 (м, 2Н), 2,25 (м, 1Н), 2,15-2,03 (м, 2Н), 1,88 (м, 1Н), 1,81-1,68 (м, 2Н), 1,45 (м, 1Н).
Пример 76
Синтез 8-хлор-4-(4-циклопропилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
(а) Синтез 2,7-дихлор-3-(4-циклопропилпиперазин-1-ил)пиридо[2,3-b]пиразина
2,3,7-трихлорпиридо[2,3-b]пиразину (70,0 мг, 0,30 ммоль) и 1-циклопропилпиперазину (59,7 мг, 0,30 ммоль) давали взаимодействовать таким же образом, как в примере 2 (с), получая твердое соединение желтого цвета, представляющее собой 2,7-дихлор-3-(4-циклопропилпиперазин-1-ил)пиридо[2,3-b]пиразин (59,5 мг, 61%).
ЖХ/МС ESI (+): 324 (М+1), 326 (М+3)
1Н-ЯМР (300 МГц, CDCl3); δ: 8,93 (д, 1Н, J=2,7 Гц), 8,50 (д, 1Н, J=2,7 Гц), 3,59 (м, 4Н), 2,74 (м, 4Н), 1,71 (м, 1Н), 0,45 (м, 2Н), 0,39 (м, 2Н).
(b) Синтез 7-хлор-3-(4-циклопропилпиперазин-1-ил)-2-гидразинилпиридо[2,3-b]пиразина
2,7-дихлор-3-(4-циклопропилпиперазин-1-ил)пиридо[2,3-b]пиразин (56,7 мг, 0,17 ммоль) и моногидрат гидразина (14,0 мкл, 0,43 ммоль) вводили в реакцию таким же образом, как в примере 2 (d), получая твердое соединение оранжевого цвета, представляющее собой 7-хлор-3-(4-циклопропилпиперазин-1-ил)-2-гидразинилпиридо[2,3-b]пиразин (50,0 мг, 100%).
ЖХ/МС ESI (+): 320 (М+1), 322 (М+3)
(с) Синтез 8-хлор-4-(4-циклопропилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
7-хлор-3-(4-циклопропилпиперазин-1-ил)-2-гидразинилпиридо[2,3-b]пиразину (64,0 мг, 0,14 ммоль) и триметилортоформиату (2,0 мл) давали взаимодействовать при 85°C в течение 2 часов таким же образом, как в примере 2 (е), получая твердое соединение кремового цвета, представляющее собой 8-хлор-4-(4-циклопропилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (40,6 мг, 62%).
ЖХ/МС ESI (+): 330 (М+1), 332 (М+3)
1Н-ЯМР (300 МГц, CDCl3); δ: 10,01 (с, 1Н), 8,85 (д, 1Н, J=2,7 Гц), 8,57 (д, 1Н, J=2,7 Гц), 4,80-3,93 (м, АН), 2,73 (м, 4Н), 1,68 (м, 1Н), 0,46 (м, 2Н), 0,40 (м, 2Н).
Пример 77
Синтез 4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
(а) Синтез (1S,4S)-5-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)-2,5-диазабицикло [2,2,1]гептан-2-трет-бутилкарбоксилата
При 0°C TEA (0,2 мл, 1,28 ммоль) добавляли к смеси 2,3,7-трихлорпиридо[2,3-b]пиразина (100,0 мг, 0,43 ммоль), (1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-трет-бутилкарбоксилата (89,0 мг, 0,45 ммоль) в DCM (4,3 мл) и затем перемешивали в течение 3 часов. Реакционную смесь очищали хроматографией на колонке (EtOAc:n-Hех=1:5) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой (1S,4S)-5-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)-2,5-диазабицикло[2,2,1]гептан-2-трет-бутилкарбоксилат (120, 0 мг, 71%).
ЖХ/МС ESI (+): 396 (М+1), 398 (М+3)
1Н-ЯМР (300 МГц, CDCl3); δ: 8,79 (д, 1Н, J=2,7 Гц), 8,11 (д, 1Н, J=2,7 Гц), 5,30 (м, 1H), 4,78-4,55 (м, 1H), 4,20 (м, 1H), 3,95-3,63 (м, 2H), 3,53 (м, 1Н), 1,99 (м, 2Н), 1,56 (м, 9Н).
(b) Синтез (1S,4S)-5-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-2,5-диазабицикло[2,2,1]гептан-2-трет-бутилкарбоксилата
Моногидрат гидразина (48,0 мкл, 1,51 ммоль) добавляли к суспензии (1S,4S)-5-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)-2,5-диазабицикло[2,2,1]гептан-2-трет-бутилкарбоксилата (120,0 мг, 0,30 ммоль) и EtOH (1,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение одного часа. Затем добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении. К сформированному твердому веществу добавляли триметилортоформиат (3,0 мл). Смесь перемешивали при 85°C в течение одного часа и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество фильтровали и затем высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой (1S,4S)-5-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-2,5-диазабицикло[2,2,1]гептан-2-трет-бутилкарбоксилат (91,0 мг, 83%).
ЖХ/МС ESI (+): 402 (М+1), 404 (М+3)
1Н-ЯМР (300 МГц, ДMCO-d6); δ: 9,98 (м, 1Н), 8,83 (м, 1Н), 8,56 (м, 1Н), 6,27 (с, 0,7Н), 5,27 (с, 0,3H), 4,57 (м, 1Н), 4,24 (м, 0,7Н), 3,80 (м, 1Н), 3,50 (м, 1Н), 3,40 (м, 1,3H), 2,20-1,98 (м, 2Н), 1,38-1,49 (м, 9Н).
(с) Синтез 4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
TFA (0,8 мл) добавляли к смеси (1S,4S)-5-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-2,5-диазабицикло[2,2,1]гептан-2-трет-бутилкарбоксилата (90,0 мг, 0,22 ммоль) и DCM (1,2 мл), и затем смесь перемешивали при комнатной температуре в течение одного часа. Реакционную смесь очищали хроматографией на колонках (МеОН:DCM=1:60) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали. К остатку добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении, получая твердое соединение кремового цвета, представляющее собой 4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (58,0 мг, 85%).
ЖХ/МС ESI (+): 302 (М+1), 304 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 9,96 (м, 1Н), 8,79 (м, 1Н), 8,52 (с, 1Н), 6,19 (с, 0,7Н), 5,17 (с, 0,3H), 4,16 (с, 0,7Н), 3,78 (с, 1Н), 3,70 (м, 1,3H), 3,12-2,80 (м, 2Н), 1,95-1,70 (м, 2Н).
Пример 78
Синтез 8-хлор-4-((1S,4S)-5-метил-2,5-диазабицикло [2,2,1]гептан-2-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
37% формальдегида (28,0 мкл, 0,38 ммоль) и NaBH4 (14,0 мг, 0,38 ммоль) добавляли к суспензии 4-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина (38,0 мг, 0,13 ммоль) и МеОН (1,3 мл). Реакционную смесь перемешивали при комнатной температуре в течение одного часа и затем перегоняли при пониженном давлении. К остатку добавляли EtOAc и воду и экстрагировали EtOAc. Органический слой высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (МеОН:DCM=1:60) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, и затем добавляли DCM/Et2O, получая твердое вещество. Сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая твердое соединение кремового цвета, представляющее собой 8-хлор-4-((1S,4S)-5-метил-2,5-диазабицикло[2,2,1]гептан-2-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (20,0 мг, 50%).
ЖХ/МС ESI (+): 316 (М+1), 318 (М+3)
1H-ЯMP (300 МГц, ДMCO-d6); δ: 9,96 (м, 1Н), 8,79 (м, 1Н), 8,53 (м, 1Н), 6,13 (с, 0,7Н), 5,13 (с, 0,3H), 4,40 (м, 0,3H), 4,04 (м, 0,3H), 3,88 (м, 0,7Н), 3,60 (м, 1,7Н), 2,97 (м, 1Н), 2,64 (м, 1Н), 2,36 (с, 3H), 2,08-1,80 (м, 2Н).
Пример 79
Синтез 8-хлор-4-(1,4-диазепан-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
(а) Синтез 4-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)-1,4-диазепан-1-трет-бутилкарбоксилата
2,3,7-трихлорпиридо[2,3-b]пиразин (70,0 мг, 0,30 ммоль) растворяли в DCM (3,0 мл), и затем 1,4-диазепан-1-трет-бутилкарбоксилат (60,0 мкл, 0,30 ммоль) и TEA (135,0 мкл, 0,90 ммоль) добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем разбавляли DCM. Органический слой промывали водой и солевым раствором, высушивали над безводным MgSO4, фильтровали и затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке (n-Hex:EtOAc=3:1) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая жидкое соединение желтого цвета, представляющее собой 4-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)-1,4-диазепан-1-трет-бутилкарбоксилат (85,0 мг, 71%)
ЖХ/МС ESI (+): 398 (М+1), 400 (М+3)
1Н-ЯМР (300 МГц, CDCl3); δ: 8,82 (д, 1Н, J=2,67 Гц), 8,12 (д, 1Н, J=2,67 Гц), 4,15-3,92 (м, 4Н), 3,78-3,70 (м, 2Н), 3,57-3,38 (м, 2Н), 2,18-2,06 (м, 2Н), 1,41 (с, 9Н).
(b) Синтез 4-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)-1,4-диазепан-1-трет-бутилкарбоксилата
Моногидрат гидразина (6,5 мкл, 0,60 ммоль) добавляли к смеси 4-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)-1,4-диазепан-1-трет-бутилкарбоксилата (82,0 мг, 0,20 ммоль) и EtOH (2,0 мл) и затем перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь разбавляли в Et2O и затем перемешивали при комнатной температуре в течение одного часа. Сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 4-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)-1,4-диазепан-1-трет-бутилкарбоксилат.
ЖХ/МС ESI (+): 394 (М+1), 396 (М+3)
(с) Синтез 8-хлор-4-(1,4-диазепан-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
Смесь неочищенного 4-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)-1,4-диазепан-1-трет-бутилкарбоксилата и триметилортоформиата (1,1 мл) перемешивали при 80°C в течение 2 часов и затем охлаждали до комнатной температуры. Реакционную смесь концентрировали при пониженном давлении. Неочищенный 4-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-1,4-диазепан-1-трет-бутилкарбоксилат растворяли в DCM (1,0 мл), и TFA (0,5 мл) медленно добавляли при комнатной температуре и затем перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке (DCM:MeOH=10:1) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 8-хлор-4-(1,4-диазепан-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (28,0 мг, 35% за 3 стадии).
ЖХ/МС ESI (+): 304 (М+1), 306 (М+3)
1Н-ЯМР (300 МГц, CDCl3); δ: 9,14 (с, 1Н), 8,60 (д, 1Н, J=2,3 Гц), 8,01 (д, 1Н, J=2,3 Гц), 4,84-4,72 (м, 2Н), 4,28-4,17 (м, 2Н), 3,21 (т, 2Н, J=5,7 Гц), 2,94 (т, 2Н, J=5,7 Гц), 2,12-2,02 (м, 2Н).
Пример 80
Синтез 8-хлор-4-(4-метил-1,4-диазепан-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
(а) Синтез 2,7-дихлор-3-(4-метил-1,4-диазепан-1-ил)пиридо[2,3-b]пиразина
2,3,7-трихлорпиридо[2,3-b]пиразин (50,0 мг, 0,21 ммоль) растворяли в DCM (2,0 мл), и 1-метил-1,4-диазепане (26,1 мкл, 0,21 ммоль) и TEA (94,7 мкл, 0,63 ммоль) добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем разбавляли в DCM. Органический слой промывали водой и солевым раствором, высушивали над безводным MgSO4, фильтровали и затем концентрировали при пониженном давлении. «Остаток очищали хроматографией на колонке (DCM:MeOH=10:1) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение коричневого цвета, представляющее собой 2,7-дихлор-3-(4-метил-1,4-диазепан-1-ил)пиридо[2,3-b]пиразин (42,0 мг, 64%).
ЖХ/МС ESI (+): 312 (М+1), 314 (М+3)
1H-ЯMP (300 МГц, CDCl3); δ: 8,80 (д, 1Н, J=2,6 Гц), 8,11 (д, 1Н, J=2,6 Гц), 4,09-4,03 (м, 2Н), 4,01-3,95 (м, 2Н), 3,02-2,93 (м, 2Н), 2,78-2,69 (м, 2Н), 2,46 (с, 3H), 2,25-2,13 (м, 2Н).
(b) Синтез 7-хлор-2-гидразинил-3-(4-метил-1,4-диазепан-1-ил)пиридо[2,3-b]пиразина
Моногидрат гидразина (3,9 мкл, 0,36 ммоль) добавляли к смеси 2,7-дихлор-3-(4-метил-1,4-диазепан-1-ил)пиридо[2,3-b]пиразина (38,5 мг, 0,12 ммоль) и EtOH (1,2 мл) и затем перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли в Et2O, и смесь перемешивали при комнатной температуре в течение одного часа. Сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении, получая неочищенное твердое соединение желтого цвета, представляющее собой 7-хлор-2-гидразинил-3-(4-метил-1,4-диазепан-1-ил)пиридо[2,3-b]пиразин.
ЖХ/МС ESI (+): 308 (М+1), 310 (М+3)
(с) Синтез 8-хлор-4-(4-метил-1,4-диазепан-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
Смесь неочищенного 7-хлор-2-гидразинил-3-(4-метил-1,4-диазепан-1-ил)пиридо[2,3-b]пиразина и триметилортоформиата (0,7 мл) перемешивали при 80°C в течение 2 часов и затем охлаждали до комнатной температуры. Реакционную смесь концентрировали при пониженном давлении и затем очищали хроматографией на колонке (DCM:MeOH=20:1) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 8-хлор-4-(4-метил-1,4-диазепан-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (16,0 мг, 42% за 2 стадии).
ЖХ/МС ESI (+): 318 (М+1), 320 (М+3)
1H-ЯМР (300 МГц, CDCl3); δ: 9,14 (с, 1Н), 8,59 (д, 1Н, J=2,3 Гц), 8,01 (д, 1Н, J=2,3 Гц), 4,87-4,74 (м, 2Н), 4,18-4,14 (м, 2Н), 2,92-2,86 (м, 2Н), 2,67-2,60 (м, 2Н), 2,39 (с, 3H), 2,21-2,09 (м, 2Н).
Пример 81
Синтез (R)-1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилпирролидин-3-амина
(а) Синтез (R)-(1-(7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил)пирролидин-3-ил)(метил)-трет-бутилкарбамата
7-бром-2,3-дихлорпиридо[2,3-b]пиразин (70,0 мг, 0,25 ммоль) и TEA (175,0 мкл, 1,26 ммоль) растворяли в DCM (1,0 мл), и (R)-(пирролидин-3-ил)-трет-бутилметилкарбамат (55,2 мг, 0,28 ммоль), растворенный в DCM (0,5 мл), добавляли при 0°C. Реакционную смесь перемешивали при 0°C в течение 12 часов и затем концентрировали, получая (R)-(1-(7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил)пирролидин-3-ил)(метил)-трет-бутилкарбамат (111,1 мг, 100%).
ЖХ/МС ESI (+): 442 (М+1), 444 (М+3)
(b) Синтез (R)-(1-(7-бром-2-гидразинилпиридо[2,3-b]пиразин-3-ил)пирролидин-3-ил)(метил)-трет-бутилкарбамата
(R)-(1-(7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил)пирролидин-3-ил)(метил)-трет-бутилкарбамат (111,1 мг, 0,25 ммоль) и моногидрат гидразина (25,0 мкл, 0,50 ммоль) растворяли в EtOH (1,5 мл). Смесь перемешивали при комнатной температуре в течение 3 часов, получая твердое вещество, и сформированное твердое вещество отфильтровывали, получая твердое соединение желтого цвета, представляющее собой (R)-(1-(7-бром-2-гидразинилпиридо[2,3-b]пиразин-3-ил)пирролидин-3-ил)(метил)-трет-бутилкарбамат (110,0 мг, 100%).
ЖХ/МС ESI (+): 438 (M+1), 440 (M+3)
(c) Синтез (R)-(1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)пирролидин-3-ил)(метил)-трет-бутилкарбамата
Смесь (R)-(1-(7-бром-2-гидразинилпиридо[2,3-b]пиразин-3-ил)пирролидин-3-ил)(метил)-трет-бутилкарбамата (110,0 мг, 0,25 ммоль) и триметилортоформиата (1,0 мл) перемешивали при 80°C в течение 2 часов и затем охлаждали до комнатной температуры. Реакционную смесь концентрировали и затем сначала очищали хроматографией на колонках (МеОН:DCM=5:95) на силикагеле, и затем очищали хроматографией на колонках (EtOAc:n-Hех=50:50) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой (R)-(1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)пирролидин-3-ил)(метил)-трет-бутилкарбамат (40,0 мг, 36%).
ЖХ/МС ESI (+): 448 (М+1), 450 (М+3)
(d) Синтез (R)-1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилпирролидин-3-амина
(R)-(1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)пирролидин-3-ил)(метил)-трет-бутилкарбамат (40,0 мг, 0,09 ммоль) растворяли в DCM (1,0 мл) и добавляли TFA (0,4 мл). Реакционную смесь перемешивали при комнатной температуре в течение одного часа и затем очищали хроматографией на колонках (МеОН:DCM=5:95) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой (R)-1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилпирролидин-3-амин (20,7 мг, 67%).
ЖХ/МС ESI (+): 348 (М+1), 350 (М+3)
1Н-ЯМР (300 МГц, ДMCO-d6); δ: 9,98 (с, 1Н), 8,89 (д, 1Н, J=2,1 Гц), 8,57 (д, 1Н, J=2,1 Гц), 4,36-4,20 (м, 2Н), 3,81-3,61 (м, 2Н), 3,30 (м, 1Н), 2,32 (с, 3H), 2,11-1,86 (м, 3H).
Пример 82
Синтез 8-хлор-4-(гексагидро-1Н-пирроло[3,4-b]пиридин-6(2Н)-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
(а) Синтез 6-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)октагидро-1Н-пирроло[3,4-b]пиридин-1-трет-бутилкарбоксилата
2,3,7-трихлорпиридо[2,3-b]пиразин (150,0 мг, 0,64 ммоль) растворяли в DCM (6,0 мл). Октагидро-1Н-пирроло[3,4-b]пиридин-1-трет-бутилкарбоксилат (151,0 мг, 0,67 ммоль) и TEA (288,0 мкл, 1,92 ммоль) затем добавляли к смеси при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем разбавляли в DCM. Органический слой промывали водой и солевым раствором, высушивали над безводным MgSO4, фильтровали и затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке (n-Hex:EtOAc=3:1) на аминосиликагеле. Фракции, содержащие продукт, собирали и концентрировали, получая твердое соединение кремового цвета, представляющее собой 6-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)октагидро-1Н-пирроло[3,4-b]пиридин-1-трет-бутилкарбоксилат (173,0 мг, 61%).
ЖХ/МС ESI (+): 424 (М+1), 426 (М+3)
1Н-ЯМР (300 МГц, ДMCO-d6); δ: 8,81 (д, 1Н, J=2,7 Гц), 8,38 (д, 1Н, J=2,7 Гц), 4,69 (м, 1Н), 4,00-3,80 (м, 4Н), 3,70 (м, 1Н), 2,85 (м, 1Н), 2,26 (м, 1Н), 1,77-1,61 (м, 2Н), 1,42 (с, 9Н), 1,38-1,26 (м, 2Н).
(b) Синтез 6-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)октагидро-1Н-пирроло[3,4-b]пиридин-1-трет-бутилкарбоксилата
Моногидрат гидразина (37,7 мкл, 1,20 ммоль) добавляли к смеси 6-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)октагидро-1Н-пирроло[3,4-b]пиридин-1-трет-бутилкарбоксилата (170,0 мг, 0,40 ммоль) и EtOH (4,0 мл), и эту смесь затем перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь разбавляли в Et2O и перемешивали при комнатной температуре в течение одного часа, получая твердое вещество. Сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 6-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)октагидро-1Н-пирроло[3,4-b]пиридин-1-трет-бутилкарбоксилат.
ЖХ/МС ESI (+): 420 (М+1), 422 (М+3)
(с) Синтез 6-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)октагидро-1Н-пирроло[3,4-b]пиридин-1-трет-бутилкарбоксилата
Смесь неочищенного 6-(7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)октагидро-1Н-пирроло[3,4-b]пиридин-1-трет-бутилкарбоксилата и триметилортоформиата (4,0 мл) перемешивали при 80°C в течение 2 часов и затем охлаждали до комнатной температуры. Реакционную смесь концентрировали при пониженном давлении и очищали хроматографией на колонке (n-Hex:EtOAc=3:1) на аминосиликагеле. Фракции, содержащие продукт, собирали и концентрировали, получая твердое соединение желтого цвета, представляющее собой 6-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)октагидро-1Н-пирроло[3,4-b]пиридин-1-трет-бутилкарбоксилат (79,0 мг, 46% за 2 стадии).
ЖХ/МС ESI (+): 430 (М+1), 432 (М+3)
1Н-ЯМР (300 МГц, CDCl3); δ: 9,16 (с, 1Н), 8,59 (д, 1Н, J=2,3 Гц), 8,03 (д, 1Н, J=2,3 Гц), 4,91 (м, 1Н), 4,75 (м, 1Н), 4,35-4,00 (м, 3H), 3,94-3,71 (м, 1Н), 2,81 (м, 1Н), 2,37 (м, 1Н), 1,91-1,68 (м, 2Н), 1,62-1,31 (м, 11Н).
(d) Синтез 8-хлор-4-(гексагидро-1Н-пирроло[3,4-b]пиридин-6(2Н)-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
6-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)октагидро-1Н-пирроло[3,4-b]пиридин-1-трет-бутилкарбоксилат (76,0 мг, 0,17 ммоль) растворяли в DCM (2,0 мл), и TFA (126,0 мкл, 1,70 ммоль) медленно добавляли при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 8 часов. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=10:1) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 8-хлор-4-(гексагидро-1Н-пирроло[3,4-b]пиридин-6(2Н)-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (51,0 мг, 91%).
ЖХ/МС ESI (+): 330 (М+1), 332 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 9,95 (с, 1Н), 8,77 (д, 1Н, J=2,7 Гц), 8,50 (д, 1Н, J=2,7 Гц), 4,53-4,41 (м, 1Н), 4,27-4,10 (м, 1Н), 3,84-3,64 (м, 2Н), 3,42-3,31 (м, 1Н), 2,89-2 79 (м, 1Н), 2,58-2,51 (м, 1Н), 2,44-2,26 (м, 2Н), 1,84-1,35 (м, 4Н).
Пример 83
Синтез 8-хлор-4-(1-метилгексагидро-1Н-пирроло[3,4-b]пиридин-6(2Н)-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
8-хлор-4-(гексагидро-1Н-пирроло[3,4-b]пиридин-6(2Н)-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (35,0 мг, 0,10 ммоль) растворяли в МеОН (0,7 мл), и 37% формальдегида (40,5 мг, 0,50 ммоль), растворенного в МеОН (0,3 мл), добавляли при 0°C, и затем добавляли NaBH4 (18,9 мг, 0,50 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь разбавляли в EtOAc. Затем органический слой промывали водой и солевым раствором, высушивали над безводным MgSO4, фильтровали и затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке (DCM:MeOH=20:1) на аминосиликагеле. Фракции, содержащие продукт, собирали и концентрировали, получая твердое соединение коричневого цвета, представляющее собой 8-хлор-4-(1-метилгексагидро-1Н-пирроло[3,4-b]пиридин-6(2Н)-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (7,4 мг, 22%).
ЖХ/МС ESI (+): 344 (М+1), 346 (М+3)
1Н-ЯМР (300 МГц, CDCl3); δ: 9,15 (с, 1Н), 8,58 (д, 1Н, J=2,3 Гц), 8,02 (д, 1Н, J=2,3 Гц), 5,06 (м, 0,4Н), 4,69-4,60 (м, 0,6Н), 4,51-4,39 (м, 1Н), 4,19 (м, 0,4Н), 4,03 (м, 1Н), 3,80 (м, 0,6Н), 2,89-2,75 (м, 2Н), 2,67-2,47 (м, 1Н), 2,34 (с, 3H), 2,18 (м, 1Н), 1,95-1,55 (м, 4Н).
Пример 84
Синтез 1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N,N-диметилазетидин-3-амина
NaBH4 (29,0 мг, 0,77 ммоль) добавляли к суспензии 1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина (74,0 мг, 0,26 ммоль), 37% формальдегида (29,0 мкл, 0,38 ммоль) и МеОН (2,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 5 часов и добавляли EtOAc и воду, и экстрагировали EtOAc. Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (МеОН:DCM=1:100) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 1-(8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N,N-диметилазетидин-3-амин (12,0 мг, 16%).
ЖХ/МС ESI (+): 304 (М+1), 306 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 9,95 (с, 1Н), 8,81 (д, 1Н, J=2,4 Гц), 8,54 (д, 1Н, J=2,4 Гц), 4,81 (м, 1Н), 4,53 (м, 1Н), 4,34, (м, 1Н), 4,11 (м, 1Н), 3,35 (м, 1Н), 2,18 (с, 6Н).
Пример 85
Синтез 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N,N-диметилазетидин-3-амина
NaBH4 (11,0 мг, 0,29 ммоль) добавляли к суспензии 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина (32,0 мг, 0,10 ммоль), 37% формальдегида (11,0 мкл, 0,14 ммоль) и МеОН (1,0 мл). Реакционную смесь перемешивали при комнатной температуре в течение 5 часов и затем добавляли EtOAc и воду, и экстрагировали EtOAc. Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (МеОН:DCM=1:100) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N,N-диметилазетидин-3-амин (3,0 мг, 9%).
ЖХ/МС ESI (+): 348 (М+1), 350 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 9,95 (с, 1Н), 8,90 (д, 1Н, J=2,4 Гц), 8,60 (д, 1Н, J=2,4 Гц), 4,81 (м, 1Н), 4,51 (м, 1Н), 4,31 (м, 1Н), 4,10 (м, 1Н), 3,40 (м, 1Н), 2,18 (с, 6Н).
Пример 86
Синтез (1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)карбаминовой кислоты
1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин, полученный из (1-(7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата (13000,0 мг, 30,3 ммоль) таким же образом, как в примере 3 (е)-(f), очищали хроматографией на колонках (DCM:MeOH=90:10) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой (1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)карбаминовую кислоту (56,0 мг, 0,5%).
ЖХ/МС ESI (+): 378 (М+1), 380 (М+3)
1H-ЯМР (300 МГц, ДМСО-d6); δ: 9,97 (с, 1Н), 9,03 (шир. с, 1Н), 8,95 (д, 1Н, J=2,2 Гц), 8,65 (д, 1Н, J=2,2 Гц), 4,40-4,30 (м, 2Н), 4,10-4,00 (м, 1Н), 3,90-3,80 (м, 2Н), 2,89 (м, 3H).
Пример 87
Синтез 2-((8-хлорпиридо[2,3-е][1,2,4] триазоло[4,3-а]пиразин-4-ил)амино)этанола
(а) Синтез 2-((2,7-дихлорпиридо[2,3-b]пиразин-3-ил) амино)этанола
2,3,7-трихлорпиридо[2,3-b]пиразин (500,0 мг, 2,14 ммоль) растворяли в DCM (20,0 мл), и этаноламин (135,0 мкл, 2,25 ммоль) и TEA (965,0 мкл, 6,42 ммоль) добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, разбавляли в DCM и затем перемешивали при комнатной температуре в течение одного часа. Сформированное твердое вещество фильтровали и высушивали при пониженном давлении, получая неочищенное твердое вещество коричневого цвета, представляющее собой 2-((2,7-дихлорпиридо[2,3-b]пиразин-3-ил)амино)этанол.
ЖХ/МС ESI (+): 259 (М+1), 261 (М+3)
(b) Синтез 2-((7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)амино)этанола
Моногидрат гидразина (27 6,0 мкл, 8,7 9 ммоль) добавляли к смеси неочищенного 2-((2,7-дихлорпиридо[2,3-b]пиразин-3-ил)амино)этанола и EtOH (30,0 мл), и эту смесь затем перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь разбавляли в Et2O и затем перемешивали при комнатной температуре в течение одного часа. Сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая неочищенное твердое соединение коричневого цвета, представляющее собой 2-((7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)амино)этанол.
ЖХ/МС ESI (+): 255 (М+1), 257 (М+3)
(с) Синтез 2-((8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)амино)этанола
Смесь неочищенного 2-((7-хлор-2-гидразинилпиридо[2,3-b]пиразин-3-ил)амино)этанола и триметилортоформиата (16,0 мл) перемешивали при 80°C в течение 4 часов и затем охлаждали до комнатной температуры. Реакционную смесь концентрировали при пониженном давлении и затем растворяли в МеОН. К смеси добавляли EtOAc, и смесь разбавляли. Органический слой промывали водой и солевым раствором, высушивали над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. К остатку добавляли EtOAc (20,0 мл), и смесь перемешивали в течение одного часа. Сформированное твердое вещество фильтровали и высушивали при пониженном давлении, получая твердое соединение белого цвета, представляющее собой 2-((8-хлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)амино)этанол (82,0 мг, 14% за 3 стадии).
ЖХ/МС ESI (+): 265 (М+1), 267 (М+3)
1H-ЯMP (300 МГц, ДМСО-d6); δ: 9,93 (с, 1Н), 8,82 (д, 1Н, J=2,7 Гц), 8,64-8,58 (шир. с, 1Н), 8,55 (д, 1Н, J=2,7 Гц), 4,84 (м, 1Н), 3,70-3,64 (м, 4Н).
Пример 88
Синтез 1-(8-хлоримидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)-N-метилазетидин-3-амина
(а) Синтез (1-(7-хлор-2-((2,2-диэтоксиэтил)амино)пиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
(1-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (180,0 мг, 0,47 ммоль) растворяли в 2,2-диэтоксиэтанамине (2,0 мл), перемешивали при комнатной температуре в течение 12 часов и добавляли растворитель EtOAc. Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой (1-(7-хлор-2-((2,2-диэтоксиэтил)амино)пиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат.
(b) Синтез 1-(8-хлоримидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)-N-метилазетидин-3-амина
Неочищенный (1-(7-хлор-2-((2,2-диэтоксиэтил)амино)пиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (225,1 мг, 0,47 ммоль) и 4-метилбензолсульфоновую кислоту (1160,0 мг, 6,08 ммоль) растворяли в IPA (3,0 мл), перемешивали при 100°C в течение 2 часов и затем охлаждали до комнатной температуры. К реакционной смеси добавляли воду. Полученный раствор подщелачивали насыщенным водным раствором NaHCO3 (рН=9) и затем экстрагировали DCM. Органический слой высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (МеОН:DCM=5:95) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 1-(8-хлоримидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)-N-метилазетидин-3-амин (70,0 мг, 52%).
ЖХ/МС ESI (+): 289 (М+1), 291 (М+3)
1H-ЯMP (300 МГц, CDCl3); δ: 8,53 (м, 1Н), 7,94-7,89 (м, 2Н), 7,66 (м, 1Н), 5,12-4,13 (м, 4Н), 3,83 (м, 1Н), 2,49 (с, 3H).
Пример 89
Синтез 1-(8-бромимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)-N-метилазетидин-3-амина
(a) Синтез (1-(7-бром-2-((2,2-диэтоксиэтил)амино)пиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
(1-(7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (177,0 мг, 0,41 ммоль) растворяли в 2,2-диэтоксиэтанамине (2,0 мл), и смесь перемешивали при комнатной температуре в течение 12 часов. Затем к реакционной смеси добавляли растворитель EtOAc, и смесь промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой (1-(7-бром-2-((2,2-диэтоксиэтил)амино)пиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат.
(b) Синтез 1-(8-бромимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)-N-метилазетидин-3-амина
Неочищенный (1-(7-бром-2-((2,2-диэтоксиэтил)амино)пиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (215,0 мг, 0,41 ммоль) и 4-метилбензолсульфоновую кислоту (1020,0 мг, 5,36 ммоль) растворяли в IPA (3,0 мл), и смесь перемешивали при 100°C в течение 2 часов и затем охлаждали до комнатной температуры. К реакционной смеси добавляли воду, и смесь подщелачивали насыщенным водным раствором NaHCO3 (рН=9) и затем экстрагировали DCM. Органический слой высушивали над безводным Na2SO4, фильтровали и перегоняли при пониженном давлении, и затем очищали хроматографией на колонках (МеОН:DCM=5:95) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой 1-(8-бромимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)-N-метилазетидин-3-амин (65,0 мг, 47%).
ЖХ/МС ESI (+): 333 (М+1), 335 (М+3)
1Н-ЯМР (300 МГц, CDCl3); δ: 8,60 (д, 1Н, J=2,1 Гц), 8,06 (д, 1Н, J=2,1 Гц), 7,89 (д, 1Н, J=1,5 Гц), 7,65 (д, 1Н, J=1,5 Гц), 5,08-4,20 (м, 4Н), 3,82 (м, 1Н), 2,48 (с, 3H).
Пример 90
Синтез (1-(8-хлор-2-метилоксазоло[4,5-с][1,8]нафтилидин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
(а) Синтез 2,5-дихлорметилникотината
2,5-дихлорникотиновую кислоту (2150,0 мг, 11,20 ммоль) растворяли в растворе смеси DCM (10,0 мл) и МеОН (5,0 мл), и триметилсилилдиазометан (2,0 М в гексане) (11,2 мл, 22,40 ммоль) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение одного часа и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (EtOAc:n-Hех=1:9) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение белого цвета, представляющее собой 2,5-дихлорметилникотинат (2020,0 мг, 87%).
ЖХ/МС ESI (+): 206 (М+1)
1Н-ЯМР (300 МГц, CDCl3); δ: 8,48 (д, 1Н, J=2,6 Гц), 8,16 (д, 1Н, J=2,6 Гц), 3,98 (с, 3H).
(b) Синтез 5-хлор-2-((4-метоксибензил)амино)метилникотината
EtOH (30,0 мл) добавляли к 2,5-дихлорметилникотинату (2020,0 мг, 9,79 ммоль) и затем добавляли 4-метоксибензиламин (2,6 мл, 19,60 ммоль). Смесь перемешивали при 65°C в течение 12 часов и затем охлаждали до комнатной температуры. Реакционную смесь перегоняли при пониженном давлении, и остаток очищали хроматографией на колонках (EtOAc:n-Hех=1:9) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая бесцветное жидкое соединение, представляющее собой 5-хлор-2-((4-метоксибензил)амино)метилникотинат (840,0 мг, 28%).
ЖХ/МС ESI (+): 307 (М+1)
(с) Синтез 6-хлор-1-(4-метоксибензил)-1Н-пиридо[2,3-d][1,3]оксазин-2,4-диона
5-хлор-2-((4-метоксибензил)амино)метилникотинат (205,0 мг, 0,67 ммоль) растворяли в 1,4-диоксане (3,0 мл), и дифосген (120,0 мкл, 1,01 ммоль) медленно добавляли при комнатной температуре. Реакционную смесь перемешивали при 110°C в течение 12 часов. Реакционную смесь затем перегоняли при пониженном давлении. Остаток растворяли в толуоле (5,0 мл), снова перегоняли при пониженном давлении и затем высушивали, получая твердое соединение светло-желтого цвета, представляющее собой 6-хлор-1-(4-метоксибензил)-1Н-пиридо[2,3-d][1,3]оксазин-2,4-дион.
ЖХ/МС ESI (+): 319 (М+1)
(d) Синтез 6-хлор-4-гидрокси-1-(4-метоксибензил)-3-нитро-1,8-нафтилидин-2(1Н)-она
DMA (3,0 мл) добавляли к NaH (40,4 мг: в 60%-ом масле, 1,01 ммоль) и затем добавляли этилнитроацетат (110,0 мкл, 1,01 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут. Раствор 6-хлор-1-(4-метоксибензил)-1Н-пиридо[2,3-d][1,3]оксазин-2,4-диона, растворенный в DMA (1,0 мл), медленно добавляли к реакционной смеси при комнатной температуре. Реакционную смесь перемешивали при 110°C в течение 12 часов, охлаждали до комнатной температуры и затем фильтровали через целит. Фильтрат перегоняли при пониженном давлении. Остаток растворяли в толуоле (5,0 мл) и перегоняли при пониженном давлении. Сформированное твердое вещество перемешивали в смеси DCM и Et2O и затем фильтровали, получая твердое соединение темно-желтого цвета, представляющее собой 6-хлор-4-гидрокси-1-(4-метоксибензил)-3-нитро-1,8-нафтилидин-2(1Н)-он (232,0 мг, 95% за 2 стадии).
ЖХ/МС ESI (+): 362 (М+1)
(е) Синтез 8-хлор-5-(4-метоксибензил)-2-метилоксазоло[4,5-с][1,8]нафтилидин-4(5Н)-она
Смесь 6-хлор-4-гидрокси-1-(4-метоксибензил)-3-нитро-1,8-нафтилидин-2(1Н)-она (200,0 мг, 0,55 ммоль), Zn (220,0 мг, 3,36 ммоль), ангидрид уксусной кислоты (2,7 мл, 28,30 ммоль) и уксусную кислоту (4,0 мл) перемешивали при 120°C в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры и затем фильтровали через целит. Фильтрат концентрировали при пониженном давлении и добавляли TEA (0,5 мл), и смесь очищали хроматографией на колонках (EtOAc:n-Hex=l:1) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение белого цвета, представляющее собой 8-хлор-5-(4-метоксибензил)-2-метилоксазоло[4,5-с][1,8]нафтилидин-4(5Н)-он (108,0 мг, 52%).
ЖХ/МС ESI (+): 356 (М+1)
(f) Синтез 4,8-дихлор-2-метилоксазоло[4,5-с][1,8]нафтилидина
8-хлор-5-(4-метоксибензил)-2-метилоксазоло[4,5-с][1,8]нафтилидин-4(5Н)-он (108,0 мг, 0,30 ммоль) растворяли в POCl3 (2,0 мл), и смесь перемешивали при 140°C в течение 12 часов. Реакционную смесь охлаждали до 0°C, вливали в насыщенный водный раствор Na2CО3 с кусочками льда и затем экстрагировали DCM. Органический слой высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (EtOAc:n-Hех=1:3) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение светло-желтого цвета, представляющее собой 4,8-дихлор-2-метилоксазоло[4,5-с][1,8]нафтилидин (42,0 мг, 54%).
ЖХ/МС ESI (+): 254 (М+1)
1Н-ЯМР (300 МГц, CDCl3); δ: 9,04 (д, 1Н, J=2,7 Гц), 8,49 (д, 1Н, J=2,7 Гц), 2,85 (с, 3H).
(g) Синтез (1-(8-хлор-2-метилоксазоло[4,5-с][1,8]нафтилидин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
DMF (2,0 мл) добавляли к 4,8-дихлор-2-метилоксазоло[4,5-с][1,8]нафтилидину (19,4 мг, 0,08 ммоль) и затем добавляли азетидин-3-ил(метил)-трет-бутилкарбамат гидрохлорид (26,0 мг, 0,12 ммоль) и TEA (30,0 мкл, 0,23 ммоль). Реакционную смесь перемешивали в течение 6 часов и перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (EtOAc:n-Hех=1:2) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение светло-желтого цвета, представляющее собой (1-(8-хлор-2-метилоксазоло[4,5-с][1,8]нафтилидин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (30,0 мг, 97%).
ЖХ/МС ESI (+): 404 (М+1)
1Н-ЯМР (300 МГц, CDCl3); δ: 8,73 (д, 1Н, J=2,7 Гц), 8,19 (д, 1Н, J=2,7 Гц), 5,41-4,91 (м, 1Н), 4,82-4,68 (м, 2Н), 4,59-4,47 (м, 2Н), 2,99 (с, 3H), 2,71 (с, 3H), 1,48 (с, 9Н).
Пример 91
Синтез 1-(8-хлор-2-метилоксазоло[4,5-с][1,8]нафтилидин-4-ил)-N-метилазетидин-3-амина
(1-(8-хлор-2-метилоксазоло[4,5-с][1,8]нафтилидин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (30,0 мг, 0,07 ммоль) растворяли в DCM (2,0 мл) и добавляли TFA (1,0 мл, 13,50 ммоль). Смесь перемешивали в течение 6 часов. Реакционную смесь перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (МеОН:DCM=1:50) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение светло-желтого цвета, представляющее собой 1-(8-хлор-2-метилоксазоло[4,5-с][1,8]нафтилидин-4-ил)-N-метилазетидин-3-амин (20,3 мг, 87%).
ЖХ/МС ESI (+): 304 (М+1)
1Н-ЯМР (300 МГц, CDCl3); δ: 8,71 (д, 1Н, J=2,7 Гц), 8,16 (д, 1Н, J=2,7 Гц), 4,78-4,68 (м, 2Н), 4,29-4,19 (м, 2Н), 3,87-3,76 (м, 1Н), 2,71 (с, 3H), 2,48 (с, 3H).
Пример 92
Синтез 8-хлор-2-метил-4-(4-метилпиперазин-1-ил)оксазоло[4,5-с][1,8]нафтилидина
4,8-дихлор-2-метилоксазоло[4,5-с][1,8]нафтилидин (10,0 мг, 0,04 ммоль) добавляли к DMF (1,0 мл) и добавляли N-метилпиперазин (26,0 мг, 0,12 ммоль) и TEA (30,0 мкл, 0,23 ммоль). Реакционную смесь перемешивали в течение 12 часов и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (МеОН:DCM=1:10) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение светло-желтого цвета, представляющее собой 8-хлор-2-метил-4-(4-метилпиперазин-1-ил)оксазоло[4,5-с][1,8]нафтилидин (7,9 мг, 83%).
ЖХ/МС ESI (+): 318 (М+1)
1Н-ЯМР (300 МГц, CDCl3); δ: 8,72 (д, 1Н, J=2,7 Гц), 8,17 (д, 1Н, J=2,7 Гц), 4,51-4,28 (м, 4Н), 2,72 (с, 3H), 2,62-2,53 (м, 4Н), 2,36 (с, 3H).
Пример 93
Синтез 1-(8-хлорпиридо[2,3-е]тетразоло[1,5-а]пиразин-4-ил)-N-метилазетидин-3-амина
(а) Синтез (1-(8-хлорпиридо[2,3-е]тетразоло[1,5-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
NaN3 (122,0 мг, 1,87 ммоль) добавляли к суспензии (1-(2,7-дихлорпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата (80,0 мг, 0,21 ммоль) и EtOH (2,1 мл). Реакционную смесь перемешивали при 70°C в течение 12 часов, охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Добавляли воду, получая твердое вещество, и сформированное твердое вещество высушивали при пониженном давлении, получая твердое соединение белого цвета, представляющее собой (1-(8-хлорпиридо[2,3-е]тетразоло[1,5-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (75,0 мг, 93%).
ЖХ/МС ESI (+): 391 (М+1), 393 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 8,88 (д, 1Н, J=2,4 Гц), 8,78 (д, 1Н, J=2,4 Гц), 5,21-4,85 (м, 2Н), 4,82 (м, 1Н), 4,46 (м, 1Н), 4,45 (м, 1Н), 2,93 (с, 3H), 1,42 (с, 9Н).
(b) Синтез 1-(8-хлорпиридо[2,3-е]тетразоло[1,5-а]пиразин-4-ил)-N-метилазетидин-3-амина
TFA (0,4 мл) добавляли к смеси (1-(8-хлорпиридо[2,3-е]тетразоло[1,5-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата (75,0 мг, 0,19 ммоль) и DCM (0,6 мл), и эту смесь перемешивали при комнатной температуре в течение одного часа. Реакционную смесь очищали хроматографией на колонках (МеОН:DCM=1:60) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали. К остатку добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая твердое соединение белого цвета, представляющее собой 1-(8-хлорпиридо[2,3-е]тетразоло[1,5-а]пиразин-4-ил)-N-метилазетидин-3-амин (42,0 мг, 75%).
ЖХ/МС ESI (+): 291 (М+1), 293 (М+3)
1H-ЯMP (300 МГц, ДМСО-d6); δ: 8,83 (д, 1Н, J=2,4 Гц), 8,76 (д, 1Н, J=2,4 Гц), 4,90 (м, 1Н), 4,60-4,39 (м, 2Н), 4,05 (м, 1Н), 3,75 (м, 1H), 2,46 (м, 1H), 2,31 (с, 3H).
Пример 94
Синтез 8-хлор-4-(4-метилпиперазин-1-ил)пиридо[2,3-е]тетразоло[1,5-а]пиразина
NaN3 (55,0 мг, 0,85 ммоль) добавляли к суспензии 2,7-дихлор-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразина (28,0 мг, 0,09 ммоль) и EtOH (1,0 мл). Смесь перемешивали при 70°C в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры и перегоняли при пониженном давлении. Затем добавляли воду, получая твердое вещество, и сформированное твердое вещество фильтровали и высушивали при пониженном давлении, получая твердое соединение оранжевого цвета, представляющее собой 8-хлор-4-(4-метилпиперазин-1-ил)пиридо[2,3-е]тетразоло[1,5-а]пиразин (21,0 мг, 75%).
ЖХ/МС ESI (+): 305 (М+1), 307 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 8,87 (д, 1Н, J=2,1 Гц), 8,79 (д, 1Н, J=2,4 Гц), 4,80-3,90 (м, 4Н), 2,58-2, 50 (м, 4Н), 2,26 (с, 3H).
Пример 95
Синтез 1-(8-бромпиридо[2,3-е]тетразоло[1,5-а]пиразин-4-ил)-N-метилазетидин-3-амина
(а) Синтез (1-(8-бромпиридо[2,3-е]тетразоло[1,5-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
NaN3 (71,0 мг 1,09 ммоль) добавляли к суспензии (1-(7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата (52,0 мг, 0,12 ммоль) и EtOH (1,2 мл). Смесь перемешивали при 70°C в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры и перегоняли при пониженном давлении. Затем добавляли воду, получая твердое вещество, и сформированное твердое вещество фильтровали и высушивали при пониженном давлении, получая твердое соединение белого цвета, представляющее собой (1-(8-бромпиридо[2,3-е]тетразоло[1,5-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (45,0 мг, 85%).
ЖХ/МС ESI (+): 435 (М+1), 437 (М+3)
1Н-ЯМР (300 МГц, ДMCO-d6); δ: 8,94 (д, 1Н, J=2,4 Гц), 8,84 (д, 1Н, J=2,4 Гц), 5,25-4,90 (м, 2Н), 4,82 (м, 1Н), 4,65-4,30 (м, 2Н), 2,94 (с, 3H), 1,43 (с, 9Н).
(b) Синтез 1-(8-бромпиридо[2,3-е]тетразоло[1,5-а]пиразин-4-ил)-N-метилазетидин-3-амина
TFA (0,4 мл) добавляли к смеси (1-(8-бромпиридо[2,3-е]тетразоло[1,5-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата (45,0 мг, 0,10 ммоль) и DCM (0,6 мл). Реакционную смесь перемешивали при комнатной температуре в течение одного часа и очищали хроматографией на колонках (МеОН:DCM=1:60) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали. Et2O к остатку добавляли, получая твердое вещество. Сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая твердое соединение белого цвета, представляющее собой 1-(8-бромпиридо[2,3-е]тетразоло[1,5-а]пиразин-4-ил)-N-метилазетидин-3-амин (28,0 мг, 80%).
ЖХ/МС ESI (+): 335 (М+1), 337 (М+3)
1H-ЯМР (300 МГц, ДМСО-d6); δ: 8,92 (шир. с, 1Н), 8,81 (шир. с, 1Н), 4,90 (м, 1Н), 4, 55-4, 38 (м, 2Н), 4,04 (м, 1Н), 3,76 (м, 1Н), 2,31 (с, 3H).
Пример 96
Синтез 8-бром-4-(4-метилпиперазин-1-ил)пиридо[2,3-е]тетразоло[1,5-а]пиразина
NaN3 (50,0 мг, 0,76 ммоль) добавляли к суспензии 7-бром-2-хлор-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразина (29,0 мг, 0,08 ммоль) и EtOH (1,0 мл). Реакционную смесь перемешивали при 70°C в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры и перегоняли при пониженном давлении. Затем добавляли воду, получая твердое вещество, и сформированное твердое вещество фильтровали и высушивали при пониженном давлении, получая твердое соединение оранжевого цвета, представляющее собой 8-бром-4-(4-метилпиперазин-1-ил)пиридо[2,3-е]тетразоло[1,5-а]пиразин (24,0 мг, 80%).
ЖХ/МС ESI (+): 349 (М+1), 351 (М+3)
1H-ЯМР (300 МГц, ДMCO-d6); δ: 8,95 (д, 1Н, J=2,4 Гц), 8,85 (д, 1Н, J=2,4 Гц), 4,65-4,05 (м, 4Н), 2,60-2,50 (м, 4Н), 2,26 (с, 3H).
Пример 97
Синтез 1-(8-хлор-2-метилпиридо[2,3-е][1,2,4]триазоло[1,5-а]пиразин-4-ил)-N-метилазетидин-3-амина
(а) Синтез 5-хлор-3-фтор-2-нитропиридина
5-хлор-3-фторпиридин-2-амин (500,0 мг, 3,41 ммоль) растворяли в H2SO4 (1,5 мл) и добавляли Na2S2O8 (406,1 мг, 1,71 ммоль). Смесь перемешивали при комнатной температуре в течение 12 часов, и затем в реакционную смесь выливали воду. Полученный раствор подщелачивали насыщенным водным раствором NaHCO3 (рН=9) и затем экстрагировали EtOAc. Органический слой высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (EtOAc:n-Hех=10:90) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение белого цвета, представляющее собой 5-хлор-3-фтор-2-нитропиридин (200,0 мг, 33%).
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 8,21 (д, 1Н, J=1,8 Гц), 8,17 (м, 1Н).
(b) Синтез 5-хлор-3-гидразинил-2-нитропиридина
5-хлор-3-фтор-2-нитропиридин (190,0 мг, 1,08 ммоль) и моногидрат гидразина (80,0 мкл, 1,61 ммоль) растворяли в EtOH (3,0 мл), и смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали, получая твердое соединение оранжевого цвета, представляющее собой 5-хлор-3-гидразинил-2-нитропиридине (170,0 мг, 84%).
1H-ЯМР (300 МГц, ДМСО-d6); δ: 9,06 (шир. с, 1Н), 8,10 (м, 1Н), 7,76 (д, 1Н, J=2,4 Гц).
(c) Синтез (Z)-N'-(5-хлор-2-нитропиридин-3-ил)ацетогидразонамида
5-хлор-3-гидразинил-2-нитропиридин (100,0 мг, 0,53 ммоль) и этилацетимидат гидрохлорид (102,2 мг, 0,80 ммоль) добавляли к пиридину (1,8 мл), и эту смесь перемешивали при комнатной температуре в течение 3 часов. К реакционной смеси добавляли насыщенный водный раствор Na2CO3, и эту смесь затем экстрагировали EtOAc. Органический слой высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (EtOAc:n-Hех=50:50) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение красного цвета, представляющее собой (Z)-N'-(5-хлор-2-нитропиридин-3-ил)ацетогидразонамид (77,0 мг, 63%).
1H-ЯМР (300 МГц, ДMCO-d6); δ: 9,53 (шир. с, 1Н), 7,88 (д, 1Н, J=2,4 Гц), 7,78 (д, 1Н, J=2,4 Гц), 6,62 (шир. с, 2Н), 1,92 (с, 3H).
(d) Синтез 1-(5-хлор-2-нитропиридин-3-ил)-3-метил-1Н-1,2,4-триазол-5-этилкарбоксилата
(Z)-N'-(5-хлор-2-нитропиридин-3-ил)ацетогидразонамид (60,0 мг, 0,26 ммоль) и 2-хлор-2-оксоэтилацетат (54,0 мкл, 0,52 ммоль) растворяли в Et2O (0,6 мл), перемешивали при комнатной температуре в течение одного часа и затем добавляли толуол (6,0 мл). Смесь перемешивали при 80°C в течение одного часа, перемешивали при 180°C в течение 2 часов и затем охлаждали до комнатной температуры, и добавляли воду и EtOAc, и затем смесь подщелачивали водным раствором КОН (рН=12) и экстрагировали EtOAc. Органический слой высушивали над безводным Na2SO4, фильтровали и перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (EtOAc:n-Hех=50:50) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение красного цвета, представляющее собой 1-(5-хлор-2-нитропиридин-3-ил)-3-метил-1Н-1,2,4-триазол-5-этилкарбоксилат (78,0 мг, 56%).
ЖХ/МС ESI (+): 312 (М+1), 314 (М+3)
1Н-ЯМР (300 МГц, ДМСО-d6); δ: 8,95 (д, 1Н, J=2,4 Гц), 8,83 (д, 1Н, J=2,4 Гц), 4,28-4,21 (м, 2Н), 2,40 (с, 3H), 1,20-1,15 (м, 3H).
(e) Синтез 8-хлор-2-метилпиридо[2,3-е][1,2,4]триазоло[1,5-а]пиразин-4(5Н)-она
1-(5-хлор-2-нитропиридин-3-ил)-3-метил-1Н-1,2,4-триазол-5-этилкарбоксилат (62,0 мг, 0,20 ммоль) и Fe (166,6 мг, 2,98 ммоль) добавляли к уксусной кислоте (13,0 мл), перемешивали при 90°C в течение 2 часов и затем охлаждали до комнатной температуры. К реакционной смеси добавляли 1 н. водный раствор HCl для полного растворения и затем экстрагировали EtOAc. Органический слой высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках (МеОН:DCM=5:95) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение белого цвета, представляющее собой 8-хлор-2-метилпиридо[2,3-е][1,2,4]триазоло[1,5-а]пиразин-4(5Н)-он (40,0 мг, 86%).
ЖХ/МС ESI (+): 236 (М+1), 238 (М+3)
1Н-ЯМР (300 МГц, ДMCO-d6); δ: 12,89 (шир. с, 1Н), 8,52 (д, 1Н, J=2,4 Гц), 8,44 (д, 1Н, J=2,4 Гц), 2,53 (с, 3H).
(f) Синтез 4,8-дихлор-2-метилпиридо[2,3-е][1,2,4]триазоло[1,5-а]пиразина
8-хлор-2-метилпиридо[2,3-е][1,2,4]триазоло[1,5-а]пиразин-4(5Н)-он (40,0 мг, 0,17 ммоль) растворяли в POCl3 (1,0 мл) и добавляли DIPEA (60,0 мкл, 0,34 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 5 часов и затем охлаждали до комнатной температуры. Реакционную смесь выливали в воду со льдом, нейтрализовали насыщенным водным раствором NaHCO3 и затем экстрагировали EtOAc. Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем перегоняли при пониженном давлении, получая твердое соединение кремового цвета, представляющее собой 4,8-дихлор-2-метилпиридо[2,3-е][1,2,4]триазоло[1,5-а]пиразин (43,0 мг, 100%).
ЖХ/МС ESI (+): 254 (М+1), 256 (М+3)
1H-ЯМР (300 МГц, ДМСО-d6); δ: 9,03 (м, 1Н), 8,94 (м, 1Н), 2,67 (с, 3H).
(q) Синтез 1-(8-хлор-2-метилпиридо[2,3-е][1,2,4]триазоло[1,5-а]пиразин-4-ил)-N-метилазетидин-3-амина
4,8-дихлор-2-метилпиридо[2,3-е][1,2,4]триазоло[1,5-а]пиразин (10,0 мг, 0,04 ммоль) и TFA (16,5 мкл, 0,12 ммоль) растворяли в DMF (0,2 мл) и добавляли азетидин-3-ил(метил)-трет-бутилкарбамат гидрохлорид (13,0 мг, 0,06 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем концентрировали и очищали хроматографией на колонках (МеОН:DCM=5:95) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение белого цвета, представляющее собой (1-(8-хлор-2-метилпиридо[2,3-е][1,2,4]триазоло[1,5-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат. Неочищенный (1-(8-хлор-2-метилпиридо[2,3-е][1,2,4]триазоло[1,5-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат растворяли в DCM (1,0 мл) и добавляли TFA (0,4 мл). Реакционную смесь перемешивали при комнатной температуре в течение одного часа и затем очищали хроматографией на колонках (МеОН:DCM=5:95) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение белого цвета, представляющее собой 1-(8-хлор-2-метилпиридо[2,3-е][1,2,4]триазоло[1,5-а]пиразин-4-ил)-N-метилазетидин-3-амин (7,0 мг, 45%).
ЖХ/МС ESI (+): 304 (М+1), 306 (М+3)
1H-ЯМР (300 МГц, ДMCO-d6); δ: 8,60 (д, 1Н, J=2,4 Гц), 8,45 (д, 1Н, J=2,4 Гц), 4,83 (м, 1Н), 4,50-4,30 (м, 2Н), 4,00 (м, 1Н), 3,69 (м, 1Н), 2,57 (с, 3H), 2,30 (шир. с, 3H).
Пример 98
Синтез 8-хлор-2-метил-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[1,5-а]пиразина
4,8-дихлор-2-метилпиридо[2,3-е][1,2,4]триазоло[1,5-а]пиразин (10,0 мг, 0,04 ммоль) и TFA (16,5 мкл, 0,12 ммоль) растворяли в DMF (0,2 мл) и добавляли N-метилпиперазин (6,5 мкл, 0,06 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем концентрировали и очищали хроматографией на колонках (МеОН:DCM=5:95) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение белого цвета, представляющее собой 8-хлор-2-метил-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[1,5-а] пиразин (8,0 мг, 66%).
ЖХ/МС ESI (+): 318 (М+1), 320 (М+3)
1H-ЯМР (300 МГц, ДМСО-d6); δ: 8,65 (д, 1Н, J=2,4 Гц), 8,49 (д, 1Н, J=2,4 Гц), 4,45-4,18 (м, 4Н), 2,59 (с, 3H), 2,52 (м, 4Н), 2,25 (с, 3H).
Пример 99
Синтез 1-(8-бром-7-метилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина
(a) Синтез 7-бром-6-метилпиридо[2,3-b]пиразин-2,3-диола
Смесь 5-бром-6-метилпиридин-2,3-диамина (500,0 мг, 2,47 ммоль) и диэтилоксалата (3,0 мл) перемешивали при 100°C в течение 12 часов и затем охлаждали до комнатной температуры. К реакционной смеси добавляли Et2O, получая твердое вещество. Затем сформированное твердое вещество фильтровали и высушивали при пониженном давлении, получая твердое соединение светло-коричневого цвета, представляющее собой 7-бром-6-метилпиридо[2,3-b]пиразин-2,3-диол (611,0 мг, 97%)
ЖХ/МС ESI (+): 256 (М+1), 258 (М+3)
1Н-ЯМР (400 МГц, ДМСО-d6); δ: 12,40 (с, 1Н), 11,94 (с, 1Н), 7,53 (с, 1Н), 2,48 (с, 3H).
(b) Синтез 7-бром-2,3-дихлор-6-метилпиридо[2,3-b]пиразина
Смесь 7-бром-6-метилпиридо[2,3-b]пиразин-2,3-диола (300,0 мг, 1,17 ммоль) и POCl3 (6,0 мл) перемешивали при 95°C в течение 12 часов и затем охлаждали до комнатной температуры. Реакционную смесь вливали в смесь воды со льдом, получая твердое вещество. Сформированное твердое вещество фильтровали и высушивали при пониженном давлении, получая твердое соединение черного цвета, представляющее собой 7-бром-2,3-дихлор-6-метилпиридо[2,3-b]пиразин (340,0 мг, 99%).
ЖХ/МС ESI (+): 292 (М+1), 294 (М+3), 296 (М+5)
1H-ЯМР (400 МГц, CDCl3); δ: 8,53 (с, 1Н), 2,95 (с, 3H).
(c) Синтез (1-(7-бром-2-хлор-6-метилпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
TEA (140,0 мкл, 1,02 ммоль) добавляли к смеси 7-бром-2,3-дихлор-6-метилпиридо[2,3-b]пиразина (100,0 мг, 0,34 ммоль), азетидин-3-ил(метил)-трет-бутилкарбамат гидрохлорида (76,0 мг, 0,34 ммоль) и DCM (3,4 мл) при 0°C, и смесь перемешивали в течение 1 часа. Реакционную смесь очищали хроматографией на колонке (EtOAc:n-Hех=1:4) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение красного цвета, представляющее собой (1-(7-бром-2-хлор-6-метилпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (43,0 мг, 28%).
ЖХ/МС ESI (+): 442 (М+1), 444 (М+3)
1H-ЯМР (400 МГц, CDCl3); δ: 8,23 (с, 1Н), 5,02-4,90 (м, 1Н), 4,78-4,68 (м, 2Н), 4,50-4,49 (м, 2Н), 2,96 (с, 3H), 2,836 (с, 3H), 1,48 (с, 9Н).
(d) Синтез (1-(8-бром-7-метилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
Моногидрат гидразина (15,0 мкл, 0,49 ммоль) добавляли к суспензии (1-(7-бром-2-хлор-6-метилпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата (43,0 мг, 0,10 ммоль) и EtOH (1,0 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и затем концентрировали при пониженном давлении. Остаток растворяли в триметилортоформиат (1,0 мл) и перемешивали при 85°C в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке (МеОН:DCM=1:40) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение светло-коричневого цвета, представляющее собой (1-(8-бром-7-метилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (35,0 мг, 80%).
ЖХ/МС ESI (+): 448 (М+1), 450 (М+3)
(е) Синтез 1-(8-бром-7-метилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина
TFA (0,4 мл) добавляли к смеси (1-(8-бром-7-метилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата (35,0 мг, 0,08 ммоль) и DCM (0,6 мл) и затем перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь очищали хроматографией на колонках (МеОН:DCM=1:40) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали. К остатку добавляли Et2O, получая твердое вещество. Сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 1-(8-бром-7-метилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (12,0 мг, 43%).
ЖХ/МС ESI (+): 348 (М+1), 350 (М+3)
1H-ЯMP (400 МГц, ДМСО-d6); δ: 9,93 (с, 1Н), 8,63 (с, 1Н), 4,90 (м, 1Н), 4,45-4,35 (м, 2Н), 3,97 (м, 1Н), 3,71 (м, 1Н), 2,60 (с, 3H), 2,30 (с, 3H).
Пример 100
Синтез 8-бром-7-метил-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
(а) Синтез 7-бром-2-хлор-6-метил-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразина
TEA (140,0 мкл, 1,02 ммоль) добавляли к смеси 7-бром-2,3-дихлор-6-метилпиридо[2,3-b]пиразина (100,0 мг, 0,34 ммоль), полученного со стадии (b) Примера 99, N-метилпиперазина (38,0 мкл, 0,34 ммоль) и DCM (3,4 мл) при 0°C и затем перемешивали в течение 1 часа. Реакционную смесь очищали хроматографией на колонке (EtOAc:n-Hех=1:4) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение красного цвета, представляющее собой 7-бром-2-хлор-6-метил-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразин (23,0 мг, 19%).
ЖХ/МС ESI (+): 356 (М+1), 358 (М+3), 360 (М+5)
1Н-ЯМР (400 МГц, ДМСО-d6); δ: 8,32 (с, 1Н), 3,79-3,70 (м, 4Н), 2,87 (с, 3H), 2,65-2,62 (м, 4Н), 2,38 (с, 3H).
(b) Синтез 8-бром-7-метил-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина
Моногидрат гидразина (10,0 мкл, 0,32 ммоль) добавляли к суспензии 7-бром-2-хлор-6-метил-3-(4-метилпиперазин-1-ил)пиридо[2,3-b]пиразина (23,0 мг, 0,06 ммоль) и EtOH (1,0 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и затем концентрировали при пониженном давлении. Остаток растворяли в триметилортоформиате (1,0 мл) и перемешивали при 85°C в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке (МеОН:DCM=1:40) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение красного цвета, представляющее собой 8-бром-7-метил-4-(4-метилпиперазин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин (1,1 мг, 4%).
ЖХ/МС ESI (+): 362 (М+1), 364 (М+3)
1Н-ЯМР (400 МГц, ДМСО-d6); δ: 9,99 (с, 1Н), 8,93 (с, 1Н), 4,46-4,30 (м, 4Н), 3,32 (с, 3H), 2,63 (с, 3H) 2,54-2,50 (м, 4Н).
Пример 101
Синтез 8-хлор-4-(3-(метиламино)азетидин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-7-ола соль HCl
(а) Синтез (1-(8-хлор-7-гидроксипиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
CsF (32,2 мг, 0,21 ммоль) добавляли к смеси (1-(7,8-дихлорпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата (30,0 мг, 0,07 ммоль), полученного со стадии (b) Примера 27, и NMP (1,0 мл). Реакционную смесь перемешивали при 135°C в течение 12 часов и затем охлаждали до комнатной температуры. Затем добавляли CsF (161,0 мг, 1,06 ммоль) и перемешивали при 145°C в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры и очищали хроматографией на колонке (МеОН:DCM=1:40) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой (1-(8-хлор-7-гидроксипиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (10,0 мг, 35%).
ЖХ/МС ESI (+): 406 (М+1), 408 (М+3)
(b) Синтез 8-хлор-4-(3-(метиламино)азетидин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-7-ола соль HCl
Суспензию (1-(8-хлор-7-гидроксипиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата (9,0 мг, 0,02 ммоль) и 4 н. HCl в 1,4-диоксане (0,5 мл) перемешивали при комнатной температуре в течение 1 часа. К реакционной смеси добавляли Et2O, получая твердое вещество. Сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 8-хлор-4-(3-(метиламино)азетидин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-7-ол соль HCl (3,2 мг, 43%).
ЖХ/МС ESI (+): 306 (М+1), 308 (М+3)
1H-ЯМР (400 МГц, ДMCO-d6); δ: 13,00-12,30 (м, 1Н), 9,80 (с, 1Н), 9,75-9,65 (м, 2Н), 8,76 (с, 1Н), 5,02 (м, 1Н), 4,89 (м, 1Н), 4,53 (м, 2Н), 4,23 (м, 1Н), 2,63 (с, 3H).
Пример 102
Синтез N-(1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)-N-метилгидроксиламина
(а) Синтез О-бензоил-N-(1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)-N-метилгидроксиламина
CHCl3 (5,0 мл) добавляли к смеси 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина (120,0 мг, 0,36 ммоль), бензоилпероксида (116,0 мг, 0,36 ммоль) и К2CO3 (174,0 мг, 1,26 ммоль) и перемешивали при 65°C в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры, фильтровали через целит и затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке (DCM:MeOH от 98:2 до 93:7) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение серого цвета, представляющее собой О-бензоил-N-(1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)-N-метилгидроксиламин (150,0 мг, 92%).
ЖХ/МС ESI (+): 454 (М+1), 456 (М+3)
(b) Синтез N-(1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)-N-метилгидроксиламина
NaOH (24,0 мг, 0,60 ммоль) растворяли в МеОН (8,0 мл) и добавляли О-бензоил-N-(1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)-N-метилгидроксиламин (120,0 мг, 0,26 ммоль). Смесь перемешивали в течение 12 часов и очищали хроматографией на колонках (DCM:MeOH=91:9) на силикагеле. Фракции, содержащие продукт, собирали и упаривали. Остаток растворяли в ДМСО и затем очищали высокоэффективной хроматографией с обратной фазой (ACN:Н2O=65:35). Фракции, содержащие продукт, собирали и упаривали, получая твердое вещество бежевого цвета. Полученное твердое вещество перемешивали в Et2O в течение 30 минут и затем фильтровали, получая твердое соединение бежевого цвета, представляющее собой N-(1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)-N-метилгидроксиламин (71,0 мг, 77%).
ЖХ/МС ESI (+): 350 (М+1), 352 (М+3)
1Н-ЯМР (400 МГц, ДМСО-d6); δ: 9,95 (с, 1Н), 8,90 (д, 1Н, J=2,2 Гц), 8,59 (д, 1Н, J=2,2 Гц), 4,76 (м, 2Н), 4,30 (м, 2Н), 3,77 (м, 1Н), 2,51 (с, 3H).
Пример 103
Синтез 1-(2-бромпиридо[3,2-е]пирроло[1,2-с]пиримидин-6-ил)-N-метилазетидин-3-амина
(a) Синтез 2-бромпиридо[3,2-е]пирроло[1,2-c]пиримидин-6-ола
Pd(PPh3)4 (193,0 мг, 0,17 ммоль) добавляли к суспензии 5-бром-3-йодпиридин-2-амина (500,0 мг, 1,67 ммоль), (1-(трет-бутоксикарбонил)-1Н-пиррол-2-ил) бороновой кислоты (423,0 мг, 2,00 ммоль), Cs2CO3 (1600, 0 мг, 5,01 ммоль), H2O (4,0 мл) и DME (16,0 мл). Смеси давали взаимодействовать в микроволновом реакторе в условиях 100 Вт, 110°C, в течение 2 часов и затем охлаждали до комнатной температуры. К реакционной смеси добавляли Et2O, получая твердое вещество. Сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая твердое соединение белого цвета, представляющее собой 2-бромпиридо[3,2-е]пирроло[1,2-c]пиримидин-6-ол (180,0 мг, 41%).
ЖХ/МС ESI (+): 264 (М+1), 266 (М+3)
1Н-ЯМР (400 МГц, ДMCO-d6); δ: 12,11 (с, 1Н), 8,66 (д, 1Н, J=1,5 Гц), 8,39 (д, 1Н, J=1,5 Гц), 7,66 (м, 1Н), 7,23 (м, 1Н), 6,72 (м, 1Н).
(b) Синтез (1-(2-бромпиридо[3,2-е]пирроло[1,2-c]пиримидин-6-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
DIPEA (24,0 мкл, 0,14 ммоль) добавляли к суспензии 2-бромпиридо[3,2-е]пирроло[1,2-c]пиримидин-6-ола (30,0 мг, 0,11 ммоль) и POCl3 (1,1 мл), и эту смесь затем перемешивали при 120°C в течение 20 часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Остаток растворяли в DCM (1,1 мл) и добавляли азетидин-3-ил(метил)-трет-бутилкарбамат соль HCl (50,0 мг, 0,23 ммоль) и DIPEA (200,0 мкл, 1,13 ммоль). Смесь перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь очищали хроматографией на колонках (МеОН:DCM=1:50) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой (1-(2-бромпиридо[3,2-е]пирроло[1,2-c]пиримидин-6-ил)азетидин-3-ил) (метил)-трет-бутилкарбамат (28,0 мг, 57%).
ЖХ/МС ESI (+): 433 (М+1), 435 (М+3)
1H-ЯMP (400 МГц, ДМСО-d6); δ: 8,65 (д, 1Н, J=1,8 Гц), 8,46 (д, 1Н, J=1,8 Гц), 7,60 (м, 1Н), 7,25 (м, 1Н), 6,84 (м, 1Н), 5,01-4,90 (м, 1Н), 4,68 (м, 2Н), 4,54 (м, 2Н), 2,93 (с, 3H), 1,41 (с, 9Н).
(с) Синтез 1-(2-бромпиридо[3,2-е]пирроло[1,2-c]пиримидин-6-ил)-N-метилазетидин-3-амина
TFA (0,4 мл) добавляли к смеси (1-(2-бромпиридо[3,2-е]пирроло[1,2-c]пиримидин-6-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата (28,0 мг, 0,06 ммоль) и DCM (0,6 мл) и перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь очищали хроматографией на колонках (МеОН:DCM=1:40) на аминосиликагеле, и фракции, содержащие продукт, собирали и упаривали. К остатку добавляли Et2O, получая твердое вещество. Сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 1-(2-бромпиридо[3,2-е]пирроло[1,2-c]пиримидин-6-ил)-N-метилазетидин-3-амин (14,0 мг, 64%).
ЖХ/МС ESI (+): 332 (М+1), 334 (М+3)
1Н-ЯМР (400 МГц, ДМСО-d6); δ: 8,62 (д, 1Н, J=1,8 Гц), 8,44 (д, 1Н, J=1,8 Гц), 7,60 (м, 1Н), 7,25 (м, 1Н), 6,82 (м, 1Н), 4,61 (м, 2Н), 4,17 (м, 2Н), 3,60 (м, 1Н), 2,38 (м, 1Н), 2,37 (с, 3H).
Пример 104
Синтез 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин 2,2,2-трифторацетата
Синтез 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин 2,2,2-трифторацетата
(1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (300,0 мг, 0,69 ммоль) растворяли в DCM (1,8 мл), и TFA (0,9 мл) медленно добавляли при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 30 минут и затем концентрировали при пониженном давлении. Добавляли МеОН (1,84 мл), получая твердое вещество. Сформированное твердое вещество отфильтровывали и высушивали, получая твердое соединение белого цвета, представляющее собой 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин 2,2,2-трифторацетат (270,0 мг, 90%).
ЖХ/МС ESI (+): 334 (M+1)
1H-ЯМР (400 МГц, ДMCO-d6); δ: 10,03 (с, 1Н), 9,31 (с, 1Н), 8,99 (д, 1Н, J=1,5 Гц), 8,67 (д, 1Н, J=1,5 Гц), 5,04 (м, 1Н), 4,85 (м, 1Н), 4,59 (м, 1Н), 4,42 (м, 1Н), 4,27 (м, 1Н), 2,69 (с, 3H).
Пример 105
Синтез (S)-1-(8-бром-2-метил-1,2-дигидроимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)-N-метилазетидин-3-амина
(а) Синтез (S)-(1-(7-бром-2-((1-гидроксипропан-2-ил)амино)пиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
Смесь (1-(7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата (500,0 мг, 1,17 ммоль) и (S)-2-аминопропан-1-ола (193,0 мг, 2,57 ммоль) в EtOH (20,0 мл) перемешивали при 80°C в течение 8 часов. Реакционную смесь затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH от 98:2 до 95:5) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой (S)-(1-(7-бром-2-((1-гидроксипропан-2-ил)амино)пиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (349,0 мг, 64%).
ЖХ/МС ESI (+): 467 (М+1), 469 (М+3)
1Н-ЯМР (400 МГц, ДМСО-d6); δ: 8,40 (д, 1Н, J=2,4 Гц), 7,97 (д, 1Н, J=2,4 Гц), 6,18 (д, 1Н, J=7,8 Гц), 4,85 (шир. с, 1Н), 4,80 (т, 1Н, J=5,6 Гц), 4,48 (кв., 2Н, J=8,4 Гц), 4,20-4,35 (м, 3H), 3,50 (м, 2Н), 2,88 (с, 3H), 1,40 (с, 9Н), 1,19 (д, 3H, J=6,6 Гц).
(b) Синтез (S)-(1-(8-бром-2-метил-1,2-дигидроимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат
Смесь (S)-(1-(7-бром-2-((1-гидроксипропан-2-ил)амино)пиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата (199,0 мг, 0,43 ммоль), MsCl (63,0 мг, 0,56 ммоль) и TEA (129,0 мг, 1,29 ммоль) в DCM (10,0 мл) перемешивали при комнатной температуре в течение 2 часов и затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH от 98:2 до 95:5) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой (S)-(1-(8-бром-2-метил-1,2-дигидроимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (211,0 мг, колич.).
ЖХ/МС ESI (+): 449 (М+1), 451 (М+3)
(c) Синтез (S)-1-(8-бром-2-метил-1,2-дигидроимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)-N-метилазетидин-3-амина
(S)-(1-(8-бром-2-метил-1,2-дигидроимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (100,0 мг, 0,22 ммоль) растворяли в DCM (1,0 мл), и TFA (1,0 мл) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении. Остаток нейтрализовали TEA и очищали хроматографией на колонке (DCM:МеОН=90:10) на аминосиликагеле и силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой (S)-1-(8-бром-2-метил-1,2-дигидроимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)-N-метилазетидин-3-амин (42,0 мг, 54%).
ЖХ/МС ESI (+): 349 (М+1), 351 (М+3)
1H-ЯМР (400 МГц, ДМСО-d6); δ: 7,95 (д, 1Н, J=2,2 Гц), 7,27 (д, 1Н, J=2,2 Гц), 4,68 (м, 1Н), 4,35 (м, 1Н), 4,23 (м, 1Н), 4,04 (т, 2Н, J=10,6 Гц), 3,80 (м, 1Н), 3,45-3,57 (м, 2Н), 2,23 (с, 3H), 1,25 (д, 3H, J=6,7 Гц).
Пример 106
Синтез (R)-1-(8-бром-2-метил-1,2-дигидроимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)-N-метилазетидин-3-амина
(а) Синтез (R)-(1-(7-бром-2-((1-гидроксипропан-2-ил)амино)пиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
Смесь (1-(7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата (500,0 мг, 1,17 ммоль) и (R)-2-аминопропан-1-ола (193,0 мг, 2,57 ммоль) в EtOH (20,0 мл) перемешивали при 80°C в течение 8 часов. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH от 98:2 до 95:5) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой (R)-(1-(7-бром-2-((1-гидроксипропан-2-ил)амино)пиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (37 8,0 мг, 69%).
ЖХ/МС ESI (+): 467 (M+1), 469 (M+3)
(b) Синтез (R)-(1-(8-бром-2-метил-1,2-дигидроимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
Смесь (R)-(1-(7-бром-2-((1-гидроксипропан-2-ил)амино)пиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата (378, 0 мг, 0,81 ммоль), MsCl (120,0 мг, 1,05 ммоль) и TEA (246,0 мг, 2,43 ммоль) в DCM (10,0 мл) перемешивали при комнатной температуре в течение 12 часов и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:МеОН от 98:2 до 95:5) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой (R)-(1-(8-бром-2-метил-1,2-дигидроимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (320,0 мг, 88%).
ЖХ/МС ESI (+): 449 (М+1), 451 (М+3)
(c) Синтез (R)-1-(8-бром-2-метил-1,2-дигидроимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)-N-метилазетидин-3-амина
(R)-(1-(8-бром-2-метил-1,2-дигидроимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (150,0 мг, 0,33 ммоль) растворяли в DCM (1,5 мл), и TFA (1,5 мл) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов и затем концентрировали при пониженном давлении. Остаток нейтрализовали TEA и очищали хроматографией на колонке (DCM:MeOH=90:10) на аминосиликагеле и силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой (R)-1-(8-бром-2-метил-1,2-дигидроимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)-N-метилазетидин-3-амин (75,0 мг, 64%).
ЖХ/МС ESI (+): 349 (М+1), 351 (М+3)
1Н-ЯМР (400 МГц, ДMCO-d6); δ: 7,95 (д, 1Н, J=2,2 Гц), 7,27 (д, 1Н, J=2,2 Гц), 4,68 (м, 1Н), 4,35 (м, 1Н), 4,22 (м, 1Н), 4,03 (т, 2Н, J=10,6 Гц), 3,80 (м, 1Н), 3,44-3,58 (м, 2Н), 2,24 (с, 3H), 1,25 (д, 3H, J=6,7 Гц).
Пример 107
Синтез 1-(8-бром-1,2-дигидроимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)-N-метилазетидин-3-амина
(a) Синтез (1-(7-бром-2-((2-гидроксиэтил)амино)пиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
Смесь (1-(7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата (500,0 мг, 1,17 ммоль) и 2-аминоэтанола (157,0 мг, 2,57 ммоль) в EtOH (20,0 мл) перемешивали при 90°C в течение 12 часов. Реакционную смесь затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH от 98:2 до 95:5) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой (1-(7-бром-2-((2-гидроксиэтил)амино)пиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (481,0 мг, 91%).
ЖХ/МС ESI (+): 453 (М+1), 455 (М+3)
(b) Синтез (1-(8-бром-1,2-дигидроимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
Смесь (1-(7-бром-2-((2-гидроксиэтил)амино)пиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата (481,0 мг, 1,06 ммоль), MsCl (158,0 мг, 1,38 ммоль) и TEA (322,0 мг, 3,18 ммоль) в DCM (10,0 мл) перемешивали при комнатной температуре в течение 6 часов и затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=98:2) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой (1-(8-бром-1,2-дигидроимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (435,0 мг, 94%).
ЖХ/МС ESI (+): 435 (М+1), 437 (М+3)
1H-ЯМР (400 МГц, ДМСО-d6); δ: 7,96 (д, 1Н, J=2,2 Гц), 7,31 (д, 1Н, J=2,2 Гц), 4,83 (шир. с, 1Н), 4,74 (м, 1Н), 4,57 (м, 1Н), 4,31 (м, 1Н), 4,20 (м, 1Н), 4,04 (м, 2Н), 3,92 (м, 2Н), 2,87 (с, 3H), 1,41 (с, 9Н).
(с) Синтез 1-(8-бром-1,2-дигидроимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)-N-метилазетидин-3-амина
(1-(8-бром-1,2-дигидроимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (435,0 мг, 1,00 ммоль) растворяли в DCM (1,0 мл), и TFA (2,0 мл) медленно добавляли при 0°C.Реакционную смесь перемешивали при комнатной температуре в течение 3 часов и затем концентрировали при пониженном давлении. Остаток нейтрализовали TEA и очищали хроматографией на колонке (DCM:MeOH=90:10) на аминосиликагеле и силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 1-(8-бром-1,2-дигидроимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)-N-метилазетидин-3-амин (186,0 мг, 56%).
ЖХ/МС ESI (+): 335 (М+1), 337 (М+3)
1Н-ЯМР (400 МГц, ДМСО-d6); δ: 7,95 (д, 1Н, J=2,2 Гц), 7,28 (д, 1Н, J=2,2 Гц), 4,67 (м, 1Н), 4,23 (м, 2Н), 4,03 (м, 2Н), 3,91 (м, 2Н), 3,81 (м, 1Н), 3,53 (м, 1Н), 2,32 (шир. с, 1Н), 2,23 (с, 3H).
Пример 108
Синтез 1-(8-бром-2-метилимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)-N-метилазетидин-3-амина
(а) Синтез (1-(8-бром-2-метилимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
Смесь (S)-(1-(8-бром-2-метил-1,2-дигидроимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата (139,0 мг, 0,31 ммоль) и DDQ (140,0 мг, 0,62 ммоль) в ксилоле (5,0 мл) перемешивали при 90°C в течение 8 часов, и реакционную смесь концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=98:2) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой смесь (S)-(1-(8-бром-2-метил-1,2-дигидроимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата и (1-(8-бром-2-метилимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат.
ЖХ/МС ESI (+): 447 (М+1), 449 (М+3)
(b) Синтез 1-(8-бром-2-метилимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)-N-метилазетидин-3-амина
Смесь (S)-(1-(8-бром-2-метил-1,2-дигидроимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата и (1-(8-бром-2-метилимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата растворяли в DCM (1,0 мл), и TFA (2,0 мл) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали при пониженном давлении. Остаток нейтрализовали TEA (рН=7) и очищали хроматографией на колонке (DCM:MeOH от 98:2 до 90:10) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 1-(8-бром-2-метилимидазо[1,2-а]пиридо[2,3-е]пиразин-4-ил)-N-метилазетидин-3-амин (14,0 мг, 13% за две стадии).
ЖХ/МС ESI (+): 347 (М+1), 349 (М+3)
1H-ЯMP (4 00 МГц, ДМСО-d6); δ: 8,75 (д, 1Н, J=2,1 Гц), 8,53 (д, 1Н, J=2,1 Гц), 8,43 (с, 1Н), 4,85 (м, 1Н), 4,38 (м, 2Н), 3,96 (м, 1Н), 3,63 (м, 1Н), 2,38 (с, 3H), 2,30 (с, 3H).
Пример 109
Синтез 1-(9-бром-2-метилпиразоло[1,5-с]пиридо[3,2-е]пиримидин-5-ил)-N-метилазетидин-3-амина
(а) Синтез 5-бром-3-(3-метил-1Н-пиразол-5-ил)пиридин-2-амина
Pd(PPh3)4 (190,0 мг, 0,17 ммоль) добавляли к суспензии 5-бром-3-йодпиридин-2-амина (500,0 мг, 1,67 ммоль), (1-(трет-бутоксикарбонил)-3-метил-1Н-пиразол-5-ил)бороновой кислоты (755, 0 мг, 3,34 ммоль), Cs2CO3 (1600,0 мг, 5,01 ммоль), H2O (3,0 мл) и DME (12,0 мл). Реакционной смеси давали взаимодействовать в микроволновом реакторе в условиях 100 Вт и 130°C в течение 2 часов и 30 минут, затем 100 Вт и 150°C в течение 2 часов и затем охлаждали до комнатной температуры. К реакционной смеси добавляли воду и экстрагировали 2 раза EtOAc. Органический слой высушивали над безводным Na2SO4, фильтровали и затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках (МеОН:DCM=1:40) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение коричневого цвета, представляющее собой смесь 5-бром-3-(3-метил-1Н-пиразол-5-ил)пиридин-2-амина и 9-бром-2-метилпиразоло[1,5-с]пиридо[3,2-е]пиримидин-5-ола (78,0 мг).
(b) Синтез 9-бром-2-метилпиразоло[1,5-с]пиридо[3,2-е]пиримидин-5-ола
Дифосген (0,05 мл) добавляли к суспензии смеси (78,0 мг) 5-бром-3-(3-метил-1Н-пиразол-5-ил)пиридин-2-амина и 9-бром-2-метилпиразоло[1,5-с]пиридо[3,2-е]пиримидин-5-ола и 1,4-диоксана (1,0 мл). Реакционную смесь перемешивали при 110°C в течение 1 часа и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество. Сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении, получая соединение кремового цвета, представляющее собой 9-бром-2-метилпиразоло[1,5-с]пиридо[3,2-е]пиримидин-5-ол (57,0 мг, 12% за 2 стадии).
ЖХ/МС ESI (+): 279 (М+1), 281 (М+3)
1H-ЯMP (400 МГц, ДМСО-d6); δ: 12,34 (с, 1Н), 8,76 (м, 1Н), 8,57 (м, 1Н), 7,17 (с, 1Н), 2,38 (с, 3H).
(с) Синтез (1-(9-бром-2-метилпиразоло[1,5-с]пиридо[3,2-е]пиримидин-5-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
DIPEA (22,0 мкл, 0,13 ммоль) добавляли к суспензии 9-бром-2-метилпиразоло[1,5-с]пиридо[3,2-е]пиримидин-5-ола (30,0 мг, 0,11 ммоль) и POCl3 (2,0 мл), и эту смесь перемешивали при 120°C в течение 20 часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Остаток растворяли в DCM (1,0 мл). Добавляли азетидин-3-ил(метил)-трет-бутилкарбамат соль НС1 (119,0 мг, 0,54 ммоль) и DIPEA (400,0 мкл, 2,14 ммоль). Смесь затем перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь очищали хроматографией на колонках (МеОН:DCM=1:50) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой (1-(9-бром-2-метилпиразоло[1,5-с]пиридо[3,2-е]пиримидин-5-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (32,0 мг, 67%).
ЖХ/МС ESI (+): 448 (М+1), 450 (М+3)
1Н-ЯМР (400 МГц, CDCl3); δ: 8,65 (м, 1Н), 8,17 (м, 1Н), 6,67 (с, 1Н), 5,20-5,10 (м, 1Н), 4,86 (м, 2Н), 4,65 (м, 2Н), 2,99 (с, 3H), 2,46 (м, 3H), 1,48 (с, 9Н).
(d) Синтез 1-(9-бром-2-метилпиразоло[1,5-с]пиридо[3,2-е]пиримидин-5-ил)-N-метилазетидин-3-амина
TFA (0,4 мл) добавляли к смеси (1-(9-бром-2-метилпиразоло[1,5-с]пиридо[3,2-е]пиримидин-5-ил)азетидин-3-ил) (метил)-трет-бутилкарбамата (32,0 мг, 0,07 ммоль) и DCM (0,6 мл), и эту смесь перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь очищали хроматографией на колонках (МеОН:DCM=1:40) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали. К остатку добавляли Et2O и получали твердое вещество. Сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении, получая соединение кремового цвета, представляющее собой 1-(9-бром-2-метилпиразоло[1,5-с]пиридо[3,2-е]пиримидин-5-ил)-N-метилазетидин-3-амин (15,0 мг, 60%).
ЖХ/МС ESI (+): 347 (М+1), 349 (М+3)
1Н-ЯМР (400 МГц, CDCl3); δ: 8,64 (д, 1Н, J=1,8 Гц), 8,15 (д, 1Н, J=1,8 Гц), 6,65 (с, 1Н), 4,82 (м, 2Н), 4,37 (м, 2Н), 3,75 (м, 1Н), 2,48 (с, 3H), 2,47 (с, 3H).
Пример 110
Синтез 1-(9-бромпиразоло[1,5-с]пиридо[3,2-е]пиримидин-5-ил)-N-метилазетидин-3-амина
(а) Синтез 5-бром-3-(1Н-пиразол-5-ил)пиридин-2-амина
Pd(PPh3)4 (39,0 мг, 0,03 ммоль) добавляли к суспензии 5-бром-3-йодпиридин-2-амина (100,0 мг, 0,34 ммоль), (1-(трет-бутоксикарбонил)-1Н-пиразол-5-ил)бороновой кислоты (99,0 мг, 0,47 ммоль), Cs2CO3 (327,0 мг, 1,01 ммоль), H2O (0,5 мл) и DME (2,0 мл). Реакционной смеси давали взаимодействовать в микроволновом реакторе в условиях 100 Вт и 120°C в течение 1 часа и 30 минут, затем охлаждали до комнатной температуры и добавляли Pd(PPh3)4 (20,0 мг, 0,02 ммоль) и (1-(трет-бутоксикарбонил)-1Н-пиразол-5-ил)бороновую кислоту (50,0 мг, 0,24 ммоль). Реакционной смеси давали взаимодействовать в микроволновом реакторе в условиях 100 Вт и 130°C в течение 2 часов и затем охлаждали до комнатной температуры. К реакционной смеси добавляли воду и экстрагировали 2 раза DCM. Органический слой высушивали над безводным Na2SO4, фильтровали и затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках (МеОН:DCM=1:50) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение коричневого цвета, представляющее собой смесь 5-бром-3-(1Н-пиразол-5-ил)пиридин-2-амина и 9-бромпиразоло[1,5-с]пиридо[3,2-е]пиримидин-5-ола (39,0 мг).
(b) Синтез 9-бромпиразоло[1,5-с]пиридо[3,2-е]пиримидин-5-ола
Дифосген (0,05 мл) добавляли к суспензии смеси 5-бром-3-(1Н-пиразол-5-ил)пиридин-2-амина и 9-бромпиразоло[1,5-с]пиридо[3,2-е]пиримидин-5-ола и 1,4-диоксана (1,0 мл). Реакционную смесь перемешивали при 110°C в течение 1 часа и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество. Сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 9-бромпиразоло[1,5-с]пиридо[3,2-е]пиримидин-5-ол (32,0 мг, 36% за 2 стадии).
ЖХ/МС ESI (+): 265 (М+1), 267 (М+3)
1Н-ЯМР (400 МГц, CDCl3+ДMCO-d6); δ: 8,51 (д, 1Н, J=1,5 Гц), 8,43 (д, 1Н, J=1,5 Гц), 8,04 (м, 1Н), 7,11 (м, 1Н).
(c) Синтез (1-(9-бромпиразоло[1,5-с]пиридо[3,2-е]пиримидин-5-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
DIPEA (25,0 мкл, 0,15 ммоль) добавляли к суспензии 9-бромпиразоло[1,5-с]пиридо[3,2-е]пиримидин-5-ола (32,0 мг, 0,12 ммоль) и POCl3 (2,0 мл), и эту смесь перемешивали при 120°C в течение 24 часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Остаток растворяли в DCM (1,2 мл) и добавляли азетидин-3-ил(метил)-трет-бутилкарбамат соль HCl (135,0 мг, 0,61 ммоль) и DIPEA (420,0 мкл, 2,42 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов и затем очищали хроматографией на колонках (EtOAc:n-Hех=1:5) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой (1-(9-бромпиразоло[1,5-с]пиридо[3,2-е]пиримидин-5-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (32,0 мг, 62%).
ЖХ/МС ESI (+): 433 (М+1), 435 (М+3)
1H-ЯMP (400 МГц, CDCl3); δ: 8,68 (м, 1Н), 8,24 (м, 1Н), 7,97 (м, 1Н), 6,89 (м, 1Н), 5,20-5,10 (м, 1Н), 4,89 (м, 2Н), 4,68 (м, 2Н), 2,99 (с, 3H), 1,48 (с, 9Н).
(d) Синтез 1-(9-бромпиразоло[1,5-с]пиридо[3,2-е]пиримидин-5-ил)-N-метилазетидин-3-амина
TFA (0,4 мл) добавляли к смеси (1-(9-бромпиразоло[1,5-с]пиридо[3,2-е]пиримидин-5-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата (32,0 мг, 0,07 ммоль) и DCM (0,6 мл), и эту смесь перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь очищали хроматографией на колонках (МеОН:DCM=1:40) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали. К остатку добавляли Et2O и получали твердое вещество. Сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении, получая твердое соединение кремового цвета, представляющее собой 1-(9-бромпиразоло[1,5-с]пиридо[3,2-е]пиримидин-5-ил)-N-метилазетидин-3-амин (16,0 мг, 64%).
ЖХ/МС ESI (+): 333 (М+1), 335 (М+3)
1Н-ЯМР (400 МГц, CDCl3); δ: 8,67 (д, 1Н, J=1,8 Гц), 8,22 (д, 1Н, J=1,8 Гц), 7,97 (д, 1H, J=1,5 Гц), 6,88 (д, 1H, J=1,5 Гц), 4,86 (м, 2H), 4,40 (м, 2Н), 3,76 (м, 1Н), 2,48 (с, 3H).
Пример 111
Синтез N-метил-1-(8-нитропиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-амина
(a) Синтез 5-нитропиридин-2,3-диамина
3,5-динитропиридин-2-амин (300,0 мг, 1,63 ммоль) и раствор сульфида аммония (2,4 мл, 8,15 ммоль) растворяли в МеОН (11,3 мл) и перемешивали при 75°C в течение 30 минут, и затем охлаждали до комнатной температуры. Сформированное твердое вещество отфильтровывали и затем высушивали при пониженном давлении, получая твердое соединение красного цвета, представляющее собой 5-нитропиридин-2,3-диамин (250,0 мг, 99%).
ЖХ/МС ESI (+): 155 (М+1)
1H-ЯМР (400 МГц, ДMCO-d6); δ: 8,29 (д, 1Н, J=1,8 Гц), 7,36 (д, 1Н, J=1,8 Гц), 6,99 (с, 2Н), 5,32 (с, 2Н).
(b) Синтез 7-нитропиридо[2,3-b]пиразин-2,3-диола
5-Hитропиридин-2,3-диамин (25,0 мг, 1,63 ммоль) добавляли к диэтилоксалату (5,0 мл), и реакционную смесь перемешивали при 180°C в течение 72 часов и затем охлаждали до комнатной температуры. К реакционной смеси добавляли Et2O, получая твердое вещество. Сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении, получая твердое соединение светло-коричневого цвета, представляющее собой 7-нитропиридо[2,3-b]пиразин-2,3-диол (167,0 мг, 49%).
1Н-ЯМР (400 МГц, ДMCO-d6); δ: 12,95 (с, 1Н), 12,23 (с, 1Н), 8,93 (д, 1Н, J=1,8 Гц), 8,11 (д, 1Н, J=1,8 Гц).
(с) Синтез 2,3-дихлор-7-нитропиридо[2,3-b]пиразина
Смесь 7-нитропиридо[2,3-b]пиразин-2,3-диола (167,0 мг, 0,80 ммоль) и POCl3 (2,6 мл) перемешивали при 150°C в течение 48 часов и затем охлаждали до комнатной температуры. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках (n-Hex:EtOAc=80:20) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение белого цвета, представляющее собой 2,3-дихлор-7-нитропиридо[2,3-b]пиразин (68,0 мг, 35%).
ЖХ/МС ESI (+): 245 (М+1), 247 (М+3)
(d) Синтез (1-(2-хлор-7-нитропиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
2,3-дихлор-7-нитропиридо[2,3-b]пиразин (68,0 мг, 0,28 ммоль) и азетидин-3-ил(метил)-трет-бутилкарбамат соль HCl (61,8 мг, 0,28 ммоль) добавляли к DCM (6,0 мл), и TEA (0,12 мл, 0,84 ммоль) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке (n-Hex:EtOAc=70:30) на силикагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение белого цвета, представляющее собой (1-(2-хлор-7-нитропиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (95,0 мг, 87%).
ЖХ/МС ESI (+): 395 (М+1)
(е) Синтез (1-(2-гидразинил-7-нитропиридо[2,3-b]пиразин-3-ил)азетидин-3-ил) (метил)-трет-бутилкарбамата
(1-(2-хлор-7-нитропиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (90,0 мг, 0,23 ммоль) и гидразин гидрат (24,0 мкл, 0,68 ммоль) растворяли в EtOH (2,3 мл) и перемешивали при комнатной температуре в течение 1 часа, и затем концентрировали при пониженном давлении. К реакционной смеси добавляли Et2O и получали твердое вещество. Сформированное твердое вещество фильтровали и высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой (1-(2-гидразинил-7-нитропиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат. Смесь неочищенного (1-(2-гидразинил-7-нитропиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата и триметилортоформиата (2,5 мл) перемешивали при 75°C в течение 5 часов и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество. Сформированное твердое вещество фильтровали и очищали хроматографией на колонке (DCM:MeOH=95:5) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение кремового цвета, представляющее собой (1-(2-гидразинил-7-нитропиридо[2,3-b]пиразин-3-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (64,0 мг, 66%).
ЖХ/МС ESI (+): 401 (М+1)
1Н-ЯMР (400 МГц, ДМСO-d6); δ: 10,23 (с, 1Н), 9,47 (д, 1Н, J=1,8 Гц), 9,32 (д, 1Н, J=1,8 Гц), 5,00 (м, 3H), 4,52 (м, 1Н), 2,95 (с, 3H), 1,44 (с, 9Н).
(f) Синтез N-метил-1-(8-нитропиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-амина
(1-(8-нитропиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)-трет-бутилметилкарбамат (30,0 мг, 0,08 ммоль) растворяли в DCM (1,0 мл), и TFA (0,5 мл) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке (DCM:MeOH=95:5) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой N-метил-1-(8-нитропиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-амин (20,0 мг, 89%).
ЖХ/МС ESI (+): 301 (М+1)
1H-ЯМР (400 МГц, ДМСО-d6); δ: 10,22 (с, 1Н), 9,44 (д, 1Н, J=1,8 Гц), 9,30 (д, 1Н, J=1,8 Гц), 4,97 (м, 1Н), 4,53 (м, 2Н), 4,09 (м, 1Н), 3,77 (м, 1Н), 2,32 (с, 3H).
Пример 112
Синтез 4-(3-(метиламино)азетидин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-8-амина
(а) Синтез (1-(8-аминопиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
(1-(8-нитропиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)-трет-бутилметилкарбамат (35,0 мг, 0,09 ммоль) и 10% Pd/C (9,0 мг) растворяли в МеОН (3,0 мл). Колбу замещали водородом и затем перемешивали при комнатной температуре в течение 4 0 минут. Реакционную смесь фильтровали через целит и затем концентрировали при пониженном давлении, получая (1-(8-аминопиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (32,4 мг).
ЖХ/МС ESI (+): 371 (М+1)
(b) Синтез 4-(3-(метиламино)азетидин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-8-амина
Неочищенный (1-(8-аминопиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (32,4 мг, 0,09 ммоль) растворяли в DCM (1,5 мл), и TFA (0,5 мл) медленно добавляли при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках (n-Hex:EtOA:MeOH=40:40:20) на аминосиликагеле. Фракции, содержащие продукт, собирали и упаривали, получая твердое соединение желтого цвета, представляющее собой 4-(3-(метиламино)азетидин-1-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-8-амин (5,0 мг, 21% за 2 стадии).
ЖХ/МС ESI (+): 271 (М+1)
1H-ЯМР (400 МГц, ДМСО-d6); δ: 9,83 (с, 1Н), 8,05 (д, 1Н, J=1,8 Гц), 7,56 (д, 1Н, J=1,8 Гц), 5,63 (с, 2Н), 4,55 (м, 2Н), 4,11 (м, 2Н), 3,68 (м, 1Н), 2,29 (с, 3H).
Пример 113
Синтез N-метил-1-(8-фенилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-амина
Pd(PPh3)4 (17,0 мг, 0,02 ммоль) добавляли к суспензии 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина (50,0 мг, 0,15 ммоль), фенилбороновой кислоты (27,0 мг, 0,22 ммоль), CS2CO3 (122,0 мг, 0,38 ммоль), H2O (0,3 мл) и DME (1,2 мл). Реакционную смесь вводили в реакцию в микроволновом реакторе в условиях 50 Вт и 80°C в течение 30 минут и затем охлаждали до комнатной температуры. Реакционную смесь очищали хроматографией на колонках (МеОН:DCM=1:40) на аминосиликагеле. Фракции, содержащие продукт, собирали и концентрировали. К остатку добавляли Et2O и получали твердое вещество. Сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой N-метил-1-(8-фенилпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-амин (42,0 мг, 85%).
ЖХ/МС ESI (+): 332 (М+1)
1H-ЯМР (400 МГц, ДМСО-d6); δ: 10,08 (с, 1Н), 8,91 (м, 2Н), 7,87 (м, 2Н), 7,55 (м, 2Н), 7,44 (м, 1Н), 4,91 (м, 1Н), 4,45 (м, 2Н), 4,02 (м, 1Н), 3,72 (м, 1Н), 2,45 (м, 1Н), 2,31 (с, 1Н).
Пример 114
Синтез 1-(8-(фуран-2-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина
Pd(PPh3)4 (17,0 мг, 0,02 ммоль) добавляли к суспензии 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина (50,0 мг, 0,15 ммоль), фуран-2-илбороновой кислоты (34,0 мг, 0,30 ммоль), Cs2CO3 (122, 0 мг, 0,38 ммоль), H2O (0,3 мл) и DME (1,2 мл). Реакционную смесь вводили в реакцию в микроволновом реакторе в условиях 50 Вт и 80°C в течение 30 минут и затем охлаждали до комнатной температуры. Реакционную смесь очищали хроматографией на колонках (МеОН:DCM=1:40) на аминосиликагеле. Фракции, содержащие продукт, собирали и концентрировали. К остатку добавляли Et2O и получали твердое вещество. Сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 1-(8-(фуран-2-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (31,0 мг, 65%).
ЖХ/МС ESI (+): 322 (М+1)
1Н-ЯМР (400 МГц, ДМСО-d6); δ: 10,07 (с, 1Н), 8,91 (с, 1Н), 8,82 (с, 1Н), 7,87 (с, 1Н), 7,12 (м, 1Н), 6,70 (м, 1Н), 4,91 (м, 1Н), 4,44 (м, 2Н), 4,02 (м, 1Н), 3,72 (м, 1Н), 2,31 (с, 3H).
Пример 115
Синтез 1-(2-бромпиридо[2,3-е]пирроло[1,2-а]пиразин-6-ил)-N-метилазетидин-3-амина
(а) Синтез 5-бром-3-(1Н-пиррол-1-ил)пиридин-2-амина
Смесь 5-бромпиридин-2,3-диамина (2000,0 мг, 10,6 ммоль) и 2,5-диметокситетрагидрофурана (1,5 мл, 11,7 ммоль) в АсОН (10,0 мл) перемешивали при 90°C в течение 4 часов. Реакционную смесь выливали в воду, нейтрализовали 10%-ым водным раствором NaOH (рН=7) и затем экстрагировали EtOAc (200,0 мл). Органический слой промывали солевым раствором, высушивали над безводным Na2SO4, фильтровали и затем концентрировали. Остаток очищали хроматографией на колонках (EtOAc:n-Hех=1:4) на силикагеле. Фракции, содержащие продукт, собирали и концентрировали, получая твердое соединение желтого цвета, представляющее собой 5-бром-3-(1Н-пиррол-1-ил)пиридин-2-амин (614,0 мг, 24%).
ЖХ/МС ESI (+): 238 (М+1), 240 (М+3)
1H-ЯМР (400 МГц, ДМСО-d6); δ: 8,13 (д, 1Н, J=2,1 Гц), 7,53 (д, 1Н, J=2,1 Гц), 6,83 (м, 2Н), 6,37 (м, 2Н), 4,63 (шир. с, 2Н).
(b) Синтез 2-бромпиридо[2,3-е]пирроло[1,2-а]пиразин-6(5Н)-она
Смесь 5-бром-3-(1Н-пиррол-1-ил)пиридин-2-амина (200,0 мг, 0,84 ммоль) и трифосгена (374,0 мг, 1,26 ммоль) в безводном толуоле (8,0 мл) перемешивали при 110°C в течение 3 часов, охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:MeOH=30:1) на силикагеле. Фракции, содержащие продукт, собирали и концентрировали, получая твердое соединение темно-зеленого цвета, представляющее собой 2-бромпиридо[2,3-е]пирроло[1,2-а]пиразин-6(5Н)-он (178,0 мг, 80%).
ЖХ/МС ESI (+): 264 (М+1), 266 (М+3)
1H-ЯМР (400 МГц, ДМСО-d6); δ: 11,86 (с, 1Н), 8,81 (м, 1Н), 8,37 (м, 1Н), 8,28 (м, 1Н), 7,08 (м, 1Н), 6,74 (м, 1Н).
(c) Синтез (1-(2-бромпиридо[2,3-е]пирроло[1,2-а]пиразин-6-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата
2-бромпиридо[2,3-е]пирроло[1,2-а]пиразин-6(5Н)-он (17 8,0 мг, 0,67 ммоль) растворяли в POCl3 (3,0 мл) и добавляли затем DIPEA (141,0 мкл, 0,81 ммоль). Реакционную смесь перемешивали при 120°C в течение 12 часов, охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Неочищенный 2-бром-6-хлорпиридо[2,3-е]пирроло[1,2-а]пиразин и TEA (0,5 мл, 3,35 ммоль) растворяли в DMA (7,0 мл) и добавляли азетидин-3-ил(метил)-трет-бутилкарбамат гидрохлорид (164,0 мг, 0,74 ммоль). Реакционную смесь перемешивали при 110°C в течение 12 часов, охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках (DCM:МеОН=97:3) на аминосиликагеле. Фракции, содержащие продукт, собирали и концентрировали, получая твердое соединение коричневого цвета, представляющее собой (1-(2-бромпиридо[2,3-е]пирроло[1,2-а]пиразин-6-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (179,0 мг, 62% за 2 стадии).
ЖХ/МС ESI (+): 432 (М+1), 434 (М+3)
(d) Синтез 1-(2-бромпиридо[2,3-е]пирроло[1,2-а]пиразин-6-ил)-N-метилазетидин-3-амина
(1-(2-бромпиридо[2,3-е]пирроло[1,2-а]пиразин-6-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат (17 9,0 мг, 0,41 ммоль) растворяли в DCM (2,0 мл), и TFA (2,0 мл) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем концентрировали при пониженном давлении. Остаток нейтрализовали NEt3 и очищали хроматографией на колонке (DCM:MeOH от 100:0 до 90:10) на аминосиликагеле. Фракции, содержащие продукт, собирали и концентрировали, получая твердое соединение коричневого цвета, представляющее собой 1-(2-бромпиридо[2,3-е]пирроло[1,2-а]пиразин-6-ил)-N-метилазетидин-3-амин (91,0 мг, 66%).
ЖХ/МС ESI (+): 332 (М+1), 334 (М+3)
1Н-ЯМР (400 МГц, ДМСО-d6); δ: 8,75 (м, 1Н), 8,39 (м, 2Н), 6,90 (м, 1Н), 6,82 (м, 1Н), 4,54 (м, 2Н), 4,09 (м, 2Н), 3,65 (м, 1Н), 2,28 (с, 3H).
Пример 116
Синтез 1-(2-хлорпиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразин-6-ил)-N-метилазетидин-3-амина
(a) Синтез 6-бром-3-нитропиридин-2-амина
2,6-дибром-3-нитропиридин (700,0 мг, 2,48 ммоль) добавляли к 2 М раствору гидроксида аммония в EtOH (25,0 мл, 49,66 ммоль). Смесь перемешивали при комнатной температуре в течение 12 часов и концентрировали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 6-бром-3-нитропиридин-2-амин (526,0 мг, 97%).
1Н-ЯМР (400 МГц, ДMCO-d6); δ: 8,26 (м, 3H), 6,91 (д, 1Н, J=8,4 Гц).
(b) Синтез 6-бромпиридин-2,3-диамина
6-бром-3-нитропиридин-2-амин (520,0 мг, 2,39 ммоль) и SnCl22H2O (2140,0 мг, 9,54 ммоль) добавляли к DMF (8,6 мл), и реакционную смесь перемешивали при 80°C в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, подщелачивали 1 н. NaOH (рН=9) и затем экстрагировали EtOAc. Органический слой высушивали над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении, получая твердое соединение коричневого цвета, представляющее собой 6-бромпиридин-2,3-диамин (238,0 мг, 44%).
1H-ЯМР (400 МГц, ДМСО-d6); δ: 6,62 (д, 1Н, J=8,4 Гц), 6,48 (д, 1Н, J=8,4 Гц), 5,81 (шир. с, 2Н), 4,79 (шир. с, 2Н).
(c) Синтез 6-бромпиридо[2,3-b]пиразин-2,3-диола
6-бромпиридин-2,3-диамин (238,0 мг, 1,26 ммоль) добавляли к диэтилоксалату (4,0 мл). Смесь перемешивали при 150°C в течение 4 часов и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество. Сформированное твердое вещество фильтровали и затем высушивали при пониженном давлении, получая твердое соединение коричневого цвета, представляющее собой 6-бромпиридо[2,3-b]пиразин-2,3-диол (250,0 мг, 82%).
1Н-ЯМР (400 МГц, ДМСО-d6); δ: 12,50 (шир. с, 1Н), 12,06 (шир. с, 1Н), 7,36 (м, 2Н).
(d) Синтез 2,3,6-трихлорпиридо[2,3-b]пиразина
Смесь 6-бромпиридо[2,3-b]пиразин-2,3-диола (250,0 мг, 1,03 ммоль) и РОСl3 (4,0 мл) перемешивали при 150°C в течение 10 часов и затем охлаждали до комнатной температуры. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках (n-Hex:EtOAc=70:30) на силикагеле. Фракции, содержащие продукт, собирали и концентрировали, получая твердое соединение белого цвета, представляющее собой 2,3,6-трихлорпиридо[2,3-b]пиразин (180,0 мг, 63%).
ЖХ/МС ESI (+): 234 (М+1), 236 (М+3)
1Н-ЯМР (400 МГц, ДMCO-d6); δ: 8,65 (д, 1Н, J=8,8 Гц), 8,08 (д, 1Н, J=8,8 Гц).
(е) Синтез 2,6-дихлорпиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразина
Моногидрат гидразина (18,0 мкл, 0,51 ммоль) добавляли к суспензии 2,3,6-трихлорпиридо[2,3-b]пиразина (80,0 мг, 0,34 ммоль) и EtOH (1,4 мл). Реакционную смесь перемешивали при комнатной температуре в течение 90 минут. Добавляли Et2O, получая твердое вещество. Сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 2,6-дихлор-3-гидразинилпиридо[2,3-b]пиразин. Неочищенный 2,6-дихлор-3-гидразинилпиридо[2,3-b]пиразин (60,0 мг, 0,26 ммоль) растворяли в триметилортоформиат (3,0 мл) и затем перемешивали при 85°C в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке (n-Hех:EtOAc=70:30) на силикагеле. Фракции, содержащие продукт, собирали и концентрировали, получая твердое соединение бежевого цвета, представляющее собой 2,6-дихлорпиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразин (35,0 мг, 56% за 2 стадии).
1Н-ЯМР (400 МГц, ДMCO-d6); δ: 10,14 (с, 1Н), 8,60 (д, 1Н, J=8,4 Гц), 7,96 (д, 1Н, J=8,4 Гц).
(f) Синтез 1-(2-хлорпиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразин-6-ил)-N-метилазетидин-3-амина
2,6-дихлорпиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразин (35,0 мг, 0,12 ммоль) и азетидин-3-ил(метил)-трет-бутилкарбамат гидрохлорид (41,0 мг, 0,18 ммоль) добавляли к DCM (1,0 мл), и TEA (51,0 мкл, 0,37 ммоль) медленно добавляли при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 30 минут и затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках (n-Hex:EtOAc=70:30) на силикагеле. Фракции, содержащие продукт, собирали и концентрировали, получая твердое соединение желтого цвета, представляющее собой (1-(2-хлорпиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразин-6-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат. TFA (0,4 мл) добавляли к смеси (1-(2-хлорпиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразин-6-ил)азетидин-3-ил) (метил)-трет-бутилкарбамата и DCM (1,0 мл), и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках (МеОН:DCM=5:95) на аминосиликагеле. Фракции, содержащие продукт, собирали и концентрировали, получая твердое вещество бежевого цвета, представляющее собой 1-(2-хлорпиридо[3,2-е][1,2,4]триазоло[4,3-а]пиразин-6-ил)-N-метилазетидин-3-амин (30,0 мг, 86% за 2 стадии).
ЖХ/МС ESI (+): 290 (М+1)
1Н-ЯМР (400 МГц, ДМСО-d6); δ: 9,31 (с, 1Н), 8,02 (д, 1Н, J=8,4 Гц), 7,61 (д, 1Н, J=8,4 Гц), 4,90 (м, 1Н), 4,43 (м, 2Н), 4,00 (м, 1Н), 3,73 (м, 1Н), 2,30 (с, 3H).
Пример 117
Синтез 1-(8-хлорпиридо[3,4-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина
(а) Синтез 6-бромпиридин-3,4-диамина
2-бром-5-нитропиридин-4-амин (300,0 мг, 2,48 ммоль) и Fe (300,0 мг, 5,37 ммоль) добавляли к АсОН, и реакционную смесь перемешивали при 75°C в течение 4 часов, и затем охлаждали до комнатной температуры. Реакционную смесь фильтровали через целит и затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке (n-Hex:EtOAc=50:50) на аминосиликагеле. Фракции, содержащие продукт, собирали и концентрировали, получая твердое вещество коричневого цвета, представляющее собой 6-бромпиридин-3,4-диамин (200,0 мг, 78%).
1Н-ЯМР (400 МГц, ДMCO-d6); δ: 7,39 (с, 1Н), 6,42 (с, 1Н), 5,74 (шир. с, 2Н), 4,66 (шир. с, 2Н).
(b) Синтез 7-бромпиридо[3,4-b]пиразин-2,3-диола
6-бромпиридин-3,4-диамин (200,0 мг, 1,06 ммоль) добавляли к диэтилоксалату (3,0 мл). Смесь перемешивали при 130°C в течение 12 часов и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество. Сформированное твердое вещество фильтровали и высушивали при пониженном давлении, получая твердое вещество коричневого цвета, представляющее собой 7-бромпиридо[3,4-b]пиразин-2,3-диол (195,0 мг, 76%).
1Н-ЯМР (400 МГц, ДMCO-d6); δ: 12,23 (шир. с, 1Н), 12,11 (шир. с, 1Н), 8,09 (с, 1Н), 7,05 (с, 1Н).
(c) Синтез 2,3,7-трихлорпиридо[3,4-b]пиразина
Смесь 7-бромпиридо[3,4-b]пиразин-2,3-диола (190,0 мг, 0,79 ммоль) и РОСl3 (3,0 мл) перемешивали при 150°C в течение 12 часов и затем охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество, и сформированное твердое вещество отфильтровывали. Полученное твердое вещество очищали хроматографией на колонках (n-Hex:EtOAc=50:50) на силикагеле. Фракции, содержащие продукт, собирали и концентрировали, получая твердое соединение белого цвета, представляющее собой 2,3,7-трихлорпиридо[3,4-b]пиразина (1,0 мг, 5%).
ЖХ/МС ESI (+): 234 (М+1), 236 (М+3)
1H-ЯMP (400 МГц, ДMCO-d6); δ: 9,39 (с, 1Н), 8,31 (с, 1Н).
(d) Синтез 4,8-дихлорпиридо[3,4-е][1,2,4]триазоло[4,3-а]пиразина
Моногидрат гидразина (1,9 мкл, 0,05 ммоль) добавляли к суспензии 2,3,7-трихлорпиридо[3,4-b]пиразина (10,0 мг, 0,03 ммоль) и EtOH (0,5 мл) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли Et2O, получая твердое вещество. Сформированное твердое вещество фильтровали и высушивали при пониженном давлении, получая твердое вещество красного цвета, представляющее собой 3,7-дихлор-2-гидразинилпиридо[3,4-b]пиразин. Неочищенный 3,7-дихлор-2-гидразинилпиридо[3,4-b]пиразин растворяли в триметилортоформиате (0,3 мл), и реакционную смесь перемешивали при 80°C в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры. Добавляли Et2O, получая твердое вещество. Сформированное твердое вещество фильтровали и высушивали при пониженном давлении, получая твердое вещество оранжевого цвета, представляющее собой 4,8-дихлорпиридо[3,4-е][1,2,4]триазоло[4,3-а]пиразин (3,5 мг, 42% за 2 стадии).
ЖХ/МС ESI (+): 240 (М+1)
1H-ЯМР (400 МГц, CDCl3); δ: 9,35 (с, 1Н), 9,19 (с, 1Н), 7,90 (с, 1Н).
(е) Синтез 1-(8-хлорпиридо[3,4-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина
4,8-дихлорпиридо[3,4-е][1,2,4]триазоло[4,3-а]пиразин (3,5 мг, 0,01 ммоль) и азетидин-3-ил(метил)-трет-бутилкарбамат гидрохлорид (4,1 мг, 0,02 ммоль) добавляли к DCM (0,2 мл), и TEA (5,0 мкл, 0,04 ммоль) добавляли при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и концентрировали при пониженном давлении, получая (1-(8-хлорпиридо[3,4-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамат. TFA (0,2 мл) добавляли к смеси неочищенного (1-(8-хлорпиридо[3,4-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)азетидин-3-ил)(метил)-трет-бутилкарбамата и DCM (1,0 мл), и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали хроматографией на колонках (МеОН:DCM=5:95) на аминосиликагеле. Фракции, содержащие продукт, собирали и концентрировали, получая твердое вещество бежевого цвета, представляющее собой 1-(8-хлорпиридо[3,4-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин (2,0 мг, 57% за 2 стадии).
ЖХ/МС ESI (+): 290 (М+1)
1H-ЯМР (400 МГц, CDCl3); δ: 9,12 (с, 1Н), 8,77 (с, 1Н), 7,62 (с, 1Н), 5,07 (м, 1Н), 4,59 (м, 2Н), 4,18 (м, 1Н), 3,88 (м, 1Н), 2,50 (с, 3H).
Пример 118
Синтез 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина соли метансульфоновой кислоты
Метансульфоновую кислоту (38,9 мкл, 0,60 ммоль) медленно добавляли к суспензии 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина (200,0 мг, 0,60 ммоль) и этанола (3,0 мл). Реакционную смесь перемешивали при комнатной температуре в течение 20 часов, получая твердое вещество. Сформированное твердое вещество фильтровали при пониженном давлении, промывали этанолом и затем высушивали при пониженном давлении, получая твердое вещество кремового цвета, представляющее собой 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин соль метансульфоновой кислоты (226,0 мг, 88%).
ЖХ/МС ESI (+): 334 (М+1), 336 (М+3)
1H-ЯМР (400 МГц, ДМСО-d6); δ: 10,02 (с, 1Н), 9,14 (шир. с, 2Н), 8,99 (д, 1Н, J=2,3 Гц), 8,67 (д, 1Н, J=2,3 Гц), 5,03 (м, 1Н), 4,84 (м, 1Н), 4,58 (м, 1Н), 4,40 (м, 1Н), 4,27 (м, 1Н), 2,68 (с, 3H), 2,30 (с, 3H).
Пример 119
Синтез 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина соли малеиновой кислоты
Малеиновую кислоту (139,0 мг, 1,20 ммоль) медленно добавляли к суспензии 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина (200,0 мг, 0,60 ммоль) и этанола (3,0 мл). Реакционную смесь перемешивали при комнатной температуре в течение 20 часов. Сформированное твердое вещество отфильтровывали при пониженном давлении, промывали этанолом и затем высушивали при пониженном давлении, получая твердое соединение белого цвета, представляющее собой 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин соль малеиновой кислоты (242,0 мг, 90%).
ЖХ/МС ESI (+): 334 (М+1), 336 (М+3)
1Н-ЯМР (400 МГц, ДMCO-d6); δ: 10,02 (с, 1Н), 8,99 (шир. с, 2Н), 8,98 (д, 1Н, J=2,3 Гц), 8,66 (д, 1Н, J=2,3 Гц), 6,02 (с, 2Н), 5,03 (м, 1Н), 4,82 (м, 1Н), 4,58 (м, 1Н), 4,38 (м, 1Н), 4,23 (м, 1Н), 2, 66 (с, 3H).
Пример 120
Синтез 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина соли 2-гидроксипропан-1,2,3-трикарбоновой кислоты
2-Гидроксипропан-1,2,3-трикарбоновую кислоту (230,0 мг, 1,20 ммоль) медленно добавляли к суспензии 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина (200,0 мг, 0,60 ммоль) и THF (6,0 мл). Реакционную смесь перемешивали при комнатной температуре в течение 20 часов. Сформированное твердое вещество отфильтровывали при пониженном давлении, промывали этанолом и затем высушивали при пониженном давлении, получая твердое соединение белого цвета, представляющее собой 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин соль 2-гидроксипропан-1,2,3-трикарбоновой кислоты (246,0 мг, 78%).
ЖХ/МС ESI (+): 334 (М+1), 336 (М+3)
1H-ЯMP(400 МГц, ДМСО-d6); δ: 9,98 (с, 1Н), 8,94 (д, 1Н, J=2,3 Гц), 8,63 (д, 1Н, J=2,3 Гц), 4,96 (м, 1Н), 4,62 (м, 1Н), 4,50 (м, 1Н), 4,18 (м, 1Н), 3,95 (м, 1Н), 2,55 (м, 4Н), 2,47 (с, 3H).
Пример 121
Синтез 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина соли азотной кислоты
70%-ую азотную кислоту (13,0 мкл, 0,30 ммоль) добавляли к суспензии 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина (50,0 мг, 0,15 ммоль) и этанола (1,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Сформированное твердое вещество фильтровали при пониженном давлении, промывали этанолом и затем высушивали при пониженном давлении, получая твердое соединение белого цвета, представляющее собой 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин соль азотной кислоты (55,8 мг, 94%).
ЖХ/МС ESI (+): 334 (М+1), 336 (М+3)
1H-ЯМР (400 МГц, ДМСО-d6); δ: 10,02 (с, 1Н), 9,15 (шир. с, 2Н), 8,98 (д, 1Н, J=2,4 Гц), 8,66 (д, 1Н, J=2,4 Гц), 5,03 (м, 1Н), 4,83 (м, 1Н), 4,58 (м, 1Н), 4,40 (м, 1Н), 4,27 (м, 1Н), 2,69 (с, 3H).
Пример 122
Синтез 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина соли йодистоводородной кислоты
55% HI (41,0 мкл, 0,30 ммоль) добавляли к суспензии 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина (50,0 мг, 0,15 ммоль) и этанола (1,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Сформированное твердое вещество фильтровали при пониженном давлении, промывали этанолом и затем высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин соль йодистоводородной кислоты (60,0 мг, 87%).
ЖХ/МС ESI (+): 334 (М+1), 336 (М+3)
1Н-ЯМР (400 МГц, ДМСО-d6); δ: 10,02 (с, 1Н), 9,11 (шир. с, 2Н), 8,99 (д, 1Н, J=2,0 Гц), 8,67 (д, 1Н, J=2,0 Гц), 5,03 (м, 1Н), 4,83 (м, 1Н), 4,58 (м, 1Н), 4,40 (м, 1Н), 4,26 (м, 1Н), 2,69 (с, 3H).
Пример 123
Синтез 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина соли фосфорной кислоты
85%-ую фосфорную кислоту (25,9 мкл, 0,22 ммоль) медленно добавляли к суспензии 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина (50,0 мг, 0,15 ммоль) и этанола (2,0 мл). Реакционную смесь перемешивали при комнатной температуре в течение 20 часов. Сформированное твердое вещество фильтровали при пониженном давлении, промывали этанолом и затем высушивали при пониженном давлении, получая твердое соединение белого цвета, представляющее собой 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин соль фосфорной кислоты (59,0 мг, 91%).
ЖХ/МС ESI (+): 334 (М+1), 336 (М+3)
1H-ЯМР (400 МГц, ДМСО-d6); δ: 9,97 (с, 1Н), 8,93 (д, 1Н, J=2,3 Гц), 8,61 (д, 1Н, J=2,3 Гц), 4,94 (м, 1Н), 4,59 (м, 1Н), 4,48 (м, 1Н), 4,15 (м, 1Н), 3,90 (м, 1Н), 2,42 (с, 3H).
Пример 124
Синтез 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилаэетидин-3-амина соли 4,4'-метиленбис(3-гидрокси-2-нафтойной кислоты)
Памовую кислоту (87,2 мг, 0,22 ммоль) добавляли к суспензии 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина (50,0 мг, 0,15 ммоль) и этанола (2,0 мл). Реакционную смесь перемешивали при комнатной температуре в течение 20 часов. Сформированное твердое вещество отфильтровывали при пониженном давлении, промывали этанолом и затем высушивали при пониженном давлении, получая твердое соединение зеленого цвета, представляющее собой 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин соль 4,4'-метиленбис(3-гидрокси-2-нафтойной кислоты) (95,0 мг, 88%).
ЖХ/МС ESI (+): 334 (М+1), 336 (М+3)
1H-ЯMP (400 МГц, ДМСО-d6); δ: 9,98 (с, 1Н), 8,91 (д, 1Н, J=2,3 Гц), 8,63 (д, 1Н, J=2,3 Гц), 8,35 (с, 2Н), 8,14 (м, 2Н), 7,79 (м, 2Н), 7,30 (м, 2Н), 7,15 (м, 2Н), 5,04 (м, 1Н), 4,91 (м, 1Н), 4,74 (с, 2Н), 4,59 (м, 1Н), 4,47 (м, 1Н), 4,29 (м, 1Н), 2,72 (с, 3H).
Пример 125
Синтез 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина соли бромистоводородной кислоты
33%-ый бромид водорода в уксусной кислоте (41,3 мкл, 0,23 ммоль) медленно добавляли к суспензии 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина (50,0 мг, 0,15 ммоль) и этанола (1,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 20 часов. Сформированное твердое вещество фильтровали при пониженном давлении, промывали этанолом и затем высушивали при пониженном давлении, получая твердое соединение белого цвета, представляющее собой 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин соль бромистоводородной кислоты (54,3 мг, 87%).
ЖХ/МС ESI (+): 334 (М+1), 336 (М+3)
1H-ЯMP (400 МГц, ДМСО-d6); δ: 10,04 (с, 1Н), 9,23 (шир. с, 2Н), 9,00 (д, 1Н, J=2,2 Гц), 8,67 (д, 1Н, J=2,2 Гц), 5,05 (м, 1Н), 4,87 (м, 1Н), 4,59 (м, 1Н), 4,43 (м, 1Н), 4,29 (м, 1Н), 2,69 (с, 3H).
Пример 126
Синтез 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина соли серной кислоты
Раствор серной кислоты (22,5 мг, 0,23 ммоль) в этаноле (0,5 мл) медленно добавляли к суспензии 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина (50,0 мг, 0,15 ммоль) и этанола (1,0 мл). Реакционную смесь перемешивали при комнатной температуре в течение 20 часов. Сформированное твердое вещество отфильтровывали при пониженном давлении, промывали этанолом и высушивали - при пониженном давлении, получая твердое соединение белого цвета, представляющее собой 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин соль серной кислоты (64,6 мг, 99%).
ЖХ/МС ESI (+): 334 (М+1), 336 (М+3)
1H-ЯMP (400 МГц, ДМСО-d6); δ: 10,03 (с, 1Н), 9,20 (шир. с, 2Н), 9,01 (д, 1Н, J=2,2 Гц), 8,67 (д, 1Н, J=2,2 Гц), 5,05 (м, 1Н), 4,86 (м, 1Н), 4,60 (м, 1Н), 4,43 (м, 1Н), 4,29 (м, 1Н), 2,69 (т, 3H, J=5,0 Гц).
Пример 127
Синтез 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина соли (2R,3R)-2,3-дигидроксиянтарной кислоты
Раствор (2R,3R)-2,3-дигидроксиянтарной кислоты (45 мг, 0,30 ммоль) в этаноле (0,5 мл) медленно добавляли к суспензии 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина (50,0 мг, 0,15 ммоль) и этанола (1,0 мл). Реакционную смесь перемешивали при комнатной температуре в течение 48 часов. Сформированное твердое вещество отфильтровывали при пониженном давлении, промывали этанолом и высушивали при пониженном давлении, получая твердое соединение белого цвета, представляющее собой 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин соль (2R,3R)-2,3-дигидроксиянтарной кислоты (62,9 мг, 86%).
ЖХ/МС ESI (+): 334 (М+1), 336 (М+3)
1H-ЯМР (400 МГц, ДMCO-d6); δ: 9,97 (с, 1Н), 8,92 (д, 1Н, J=2,2 Гц), 8,60 (д, 1Н, J=2,2 Гц), 4,94 (м, 1Н), 4,57 (м, 1Н), 4,47 (м, 1Н), 4,14 (м, 1Н), 4,13 (с, 2Н), 3,90 (м, 1Н), 2,42 (с, 3H).
Пример 128
Синтез 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазешидин-3-амина соли (1S)-(+)-10-камфорсульфоновой кислоты
(1S)-(+)-10-камфорсульфоновую кислоту (69,5 мг, 0,30 ммоль) добавляли к суспензии 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина (50,0 мг, 0,15 ммоль) и этанола (1,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Сформированное твердое вещество отфильтровывали при пониженном давлении, промывали этанолом и высушивали при пониженном давлении, получая твердое соединение белого цвета, представляющее собой 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин соль (1S)-(+)-10-камфорсульфоновой кислоты (39,0 мг, 46%).
ЖХ/МС ESI (+): 334 (М+1), 336 (М+3)
1Н-ЯМР (400 МГц, ДМСО-d6); δ: 10,03 (с, 1Н), 9,03 (шир. с, 2Н), 8,99 (д, 1Н, J=2,0 Гц), 8,67 (д, 1Н, J=2,0 Гц), 5,04 (м, 1Н), 4,84 (м, 1Н), 4,59 (м, 1Н), 4,40 (м, 1Н), 4,25 (м, 1Н), 2,86 (д, 1Н, J=14,8 Гц), 2,71 (м, 1Н), 2,68 (с, 3H), 2,36 (д, 1Н, J=14,8 Гц), 2,23 (м, 1Н), 1,93 (м, 1Н), 1,85 (1Н, м), 1,80 (д, 1Н, J=18,4 Гц), 1,27 (м, 2Н), 1,05 (с, 3H), 0,75 (с, 3H).
Пример 129
Синтез 8-бром-N-(1-метилпирролидин-3-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-амина
(а) Синтез 7-бром-2-хлор-N-(1-метилпирролидин-3-ил)пиридо[2,3-b]пиразин-3-амина
TEA (150,0 мкл, 1,07 ммоль) добавляли к смеси 7-бром-2,3-дихлорпиридо[2,3-b]пиразина (100,0 мг, 0,36 ммоль), 1-метилпирролидин-3-амина (43,0 мг, 0,43 ммоль) и DCM (1,8 мл), и реакционную смесь перемешивали при комнатной температуре в течение 5 часов. Реакционную смесь очищали хроматографией на колонках (МеОН:DCM=1:40) на аминосиликагеле. Фракции, содержащие продукт, собирали и концентрировали, получая твердое соединение желтого цвета, представляющее собой 7-бром-2-хлор-N-(1-метилпирролидин-3-ил)пиридо[2,3-b]пиразин-3-амин (30,0 мг, 24%).
ЖХ/МС ESI (+): 342 (М+1), 344 (М+3)
(b) Синтез 8-бром-N-(1-метилпирролидин-3-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-амина
Моногидрат гидразина (5,5 мкл, 0,18 ммоль) добавляли к суспензии 7-бром-2-хлор-N-(1-метилпирролидин-3-ил)пиридо[2,3-b] пиразин-3-амина (26,0 мг, 0,06 ммоль) и EtOH (1,0 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и затем концентрировали при пониженном давлении. Остаток растворяли в триметилортоформиате (1,0 мл), и реакционную смесь перемешивали при 100°C в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке (МеОН:DCM=1:40) на аминосиликагеле. Фракции, содержащие продукт, собирали и концентрировали. К остатку добавляли Et2O, получая твердое вещество. Сформированное твердое вещество. фильтровали и высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 8-бром-N-(1-метилпирролидин-3-ил)пиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-амин (6,0 мг, 30%).
ЖХ/МС ESI (+): 348 (М+1), 350 (М+3)
1H-ЯМР (400 МГц, ДМСО-d6); δ: 9,95 (с, 1Н), 8,92 (д, 1Н, J=2,0 Гц), 8,78 (м, 1Н), 8,61 (д, 1Н, J=2,0 Гц), 4,72 (м, 1Н), 2,86 (м, 1Н), 2,68-2,50 (м, 3H), 2,33-2,10 (м, 4Н), 2,01 (м, 1Н).
Пример 130
Синтез 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина соли (S)-2-гидроксипропановой кислоты
Раствор (S)-2-Гидроксипропановой кислоты (135,0 мг, 1,50 ммоль) в воде (0,8 мл) добавляли к суспензии 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амина (50,0 мг, 0,15 ммоль) и этанола (0,8 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов и затем концентрировали при пониженном давлении. К остатку добавляли EtOH и Et2O, получая твердое вещество. Полученное твердое вещество отфильтровывали, промывали Et2O и высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 1-(8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)-N-метилазетидин-3-амин соль (S)-2-гидроксипропановой кислоты (34,0 мг, 53%).
ЖХ/МС ESI (+): 334 (М+1), 336 (М+3)
1Н-ЯМР (400 МГц, ДМСО-d6); δ: 9,96 (с, 1Н), 8,91 (д, 1Н, J=2,0 Гц), 8,60 (д, 1Н, J=2,0 Гц), 4,91 (м, 1Н), 4,45 (м, 2Н), 4,02 (м, 2Н), 3,75 (м, 1Н), 2,33 (с, 3H), 1,22 (м, 3H).
Пример 131
Синтез N-(азетидин-3-илметил)-8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-амина соли трифторуксусной кислоты
(а) Синтез 3-(((7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил)амино)метил)азетидин-1-трет-бутилкарбоксилата
7-бром-2,3-дихлорпиридо[2,3-b]пиразин (100,0 мг, 0,56 ммоль) растворяли в DCM (5,6 мл), и 3-(аминометил)азетидин-1-трет-бутилкарбоксилат (104,1 мг, 0,56 ммоль) и TEA (0,23 мл, 1,68 ммоль) медленно добавляли при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 48 часов и затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках (n-Hex:EtOAc=70:30) на силикагеле. Фракции, содержащие продукт, собирали и концентрировали, получая твердое соединение желтого цвета, представляющее собой 3-(((7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил)амино)метил)азетидин-1-трет-бутилкарбоксилат (72,0 мг, 30%).
ЖХ/МС ESI (+): 428 (М+1)
1H-ЯMP (400 МГц, ДMCO-d6); δ: 8,85 (д, 1Н, J=2,4 Гц), 8,48 (д, 1Н, J=2,4 Гц), 8,20 (т, 1Н, J=5,6 Гц), 3,90 (м, 2Н), 3,70 (м, 4Н), 2,92 (м, 1Н), 1,38 (с, 9Н).
(b) Синтез 3-(((7-бром-2-гидразинилпиридо[2,3-b]пиразин-3-ил)амино)метил)азетидин-1-трет-бутилкарбоксилата
3-(((7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил)амино)метил)азетидин-1-трет-бутилкарбоксилат (50,0 мг, 0,12 ммоль) и моногидрат гидразина (12,0 мкл, 0,35 ммоль) растворяли в EtOH (0,6 мл), и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли Et2O, получая твердое вещество. Сформированное твердое вещество отфильтровывали и высушивали при пониженном давлении, получая твердое соединение желтого цвета, представляющее собой 3-(((7-бром-2-гидразинилпиридо[2,3-b]пиразин-3-ил)амино)метил)азетидин-1-трет-бутилкарбоксилат (48,0 мг, 98%).
ЖХ/МС ESI (+): 424 (М+1)
(с) Синтез N-(азетидин-3-илметил)-8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-амина соли трифторуксусной кислоты
Реакционную смесь 3-(((7-бром-2-гидразинилпиридо[2,3-b]пиразин-3-ил)амино)метил)азетидин-1-трет-бутилкарбоксилата (50,0 мг, 0,12 ммоль) и триметилортоформиата (1,5 мл), перемешивали при 70°C в течение 12 часов, охлаждали до комнатной температуры и затем высушивали при пониженном давлении, получая 3-(((8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)амино)метил)азетидин-1-трет-бутилкарбоксилат. TFA (0,3 мл) добавляли к смеси неочищенного 3-(((8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)амино)метил)азетидин-1-трет-бутилкарбоксилата и DCM (1,0 мл), и реакционную смесь затем перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении, и остаток очищали хроматографией на колонках (МеОН:DCM=5:95) на аминосиликагеле. Фракции, содержащие продукт, собирали и концентрировали, получая твердое вещество бежевого цвета, представляющее собой N-(азетидин-3-илметил)-8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-амин соль трифторуксусной кислоты (1,5 мг, 3% за 2 стадии).
ЖХ/МС ESI (+): 334 (М+1)
1H-ЯMP (400 МГц, МеОН-d4); δ: 9,77 (с, 1Н), 8,80 (д, 1Н, J=2,0 Гц), 8,64 (м, 1Н), 3,94 (м, 2Н), 3,76 (м, 2Н), 3,63 (м, 2Н), 3,35 (с, 1Н).
Пример 132
Синтез 4-(азетидин-3-илметокси)-8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразина соли хлористоводородной кислоты
(а) Синтез 3-(((7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил)окси)метил)азетидин-1-трет-бутмлкарбоксилата
60%-ый NaH (10 мг, 0,25 ммоль) добавляли к раствору 3-(гидроксиметил)азетидин-1-трет-бутилкарбоксилата (47,9 мг, 0,25 ммоль), растворенного в THF (2,5 мл), под азотом при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 30 минут и затем добавляли при комнатной температуре 7-бром-2,3-дихлорпиридо[2,3-b]пиразин (60,0 мг, 0,25 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и разбавляли EtQAc. Органический слой промывали водой и солевым раствором, высушивали над безводным MgSO4, фильтровали и затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках (n-Hex:EtOAc=2:1) на силикагеле. Фракции, содержащие продукт, собирали и концентрировали, получая твердое соединение белого цвета, представляющее собой 3-(((7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил)окси)метил)азетидин-1-трет-бутилкарбоксилат (65,8 мг, 61%).
ЖХ/МС ESI (+): 429 (М+1), 431 (М+3), 451 (М+23), 453 (М+25)
1H-ЯМР (400 МГц, CDCl3); δ: 9,00 (д, 1Н, J=2,3 Гц), 8,45 (д, 1Н, J=2,3 Гц), 4,79 (д, 2Н, J=6,6 Гц), 4,13 (т, 2Н, J=8,5 Гц), 3,85 (дд, 2Н, J=5,2, 3,6 Гц), 3,13 (м, 1Н), 1,45 (с, 9Н).
(b) Синтез 3-(((8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)окси)метил)азетидин-1-трет-бутилкарбоксилата
Моногидрат гидразина (14,1 мкл, 0,45 ммоль) добавляли к смеси 3-(((7-бром-2-хлорпиридо[2,3-b]пиразин-3-ил)окси)метил)азетидин-1-трет-бутилкарбоксилата (63,8 мг, 0,15 ммоль) и EtOH (1,5 мл), и реакционную смесь перемешивали при комнатной температур в течение 4 часов. Реакционную смесь концентрировали при пониженном давлении, и неочищенный 3-(((7-бром-2-гидразинилпиридо[2,3-b]пиразин-3-ил)окси)метил)азетидин-1-трет-бутилкарбоксилат растворяли в триметилортоформиате (1,5 мл). Реакционную смесь перемешивали при 80°C в течение 4 часов и концентрировали при пониженном давлении, получая неочищенное твердое соединение коричневого цвета, представляющее собой 3-(((8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)окси)метил)азетидин-1-трет-бутилкарбоксилат.
ЖХ/МС ESI (+): 435 (М+1), 437 (М+3), 457 (М+23), 459 (М+25)
(с) Синтез 4-(азетидин-3-илметокси)-8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин гидрохлорида
Неочищенный 3-(((8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин-4-ил)окси)метил)азетидин-1-трет-бутилкарбоксилат растворяли в DCM (1,5 мл), и затем 4 н. раствор 1,4-диоксана в НСl (75,0 мкл, 0,30 ммоль) медленно добавляли при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 2 часов и затем концентрировали при пониженном давлении. Остаток растворяли в ДМСО (0,5 мл) и затем очищали препаративной ВЭЖХ (YL9110S YoungLin, ацетонитрил:вода=8:92). Фракции, содержащие продукт, собирали и высушивали замораживанием, получая твердое соединение белого цвета, представляющее собой 4-(азетидин-3-илметокси)-8-бромпиридо[2,3-е][1,2,4]триазоло[4,3-а]пиразин гидрохлорид (6,4 мг, 11% за 3 стадии).
ЖХ/МС ESI (+): 335 (М+1), 337 (М+3)
1H-ЯМР (400 МГц, ДМСО-d6); δ: 10,05 (с, 1Н), 9,15 (д, 1Н, J=2,0 Гц), 8,97 (шир. с, 2Н), 8,78 (д, 1Н, J=2,0 Гц), 4,79 (д, 2Н, J=6,64 Гц), 4,04 (м, 2Н), 3,90 (м, 2Н), 3,33 (м, 1Н).
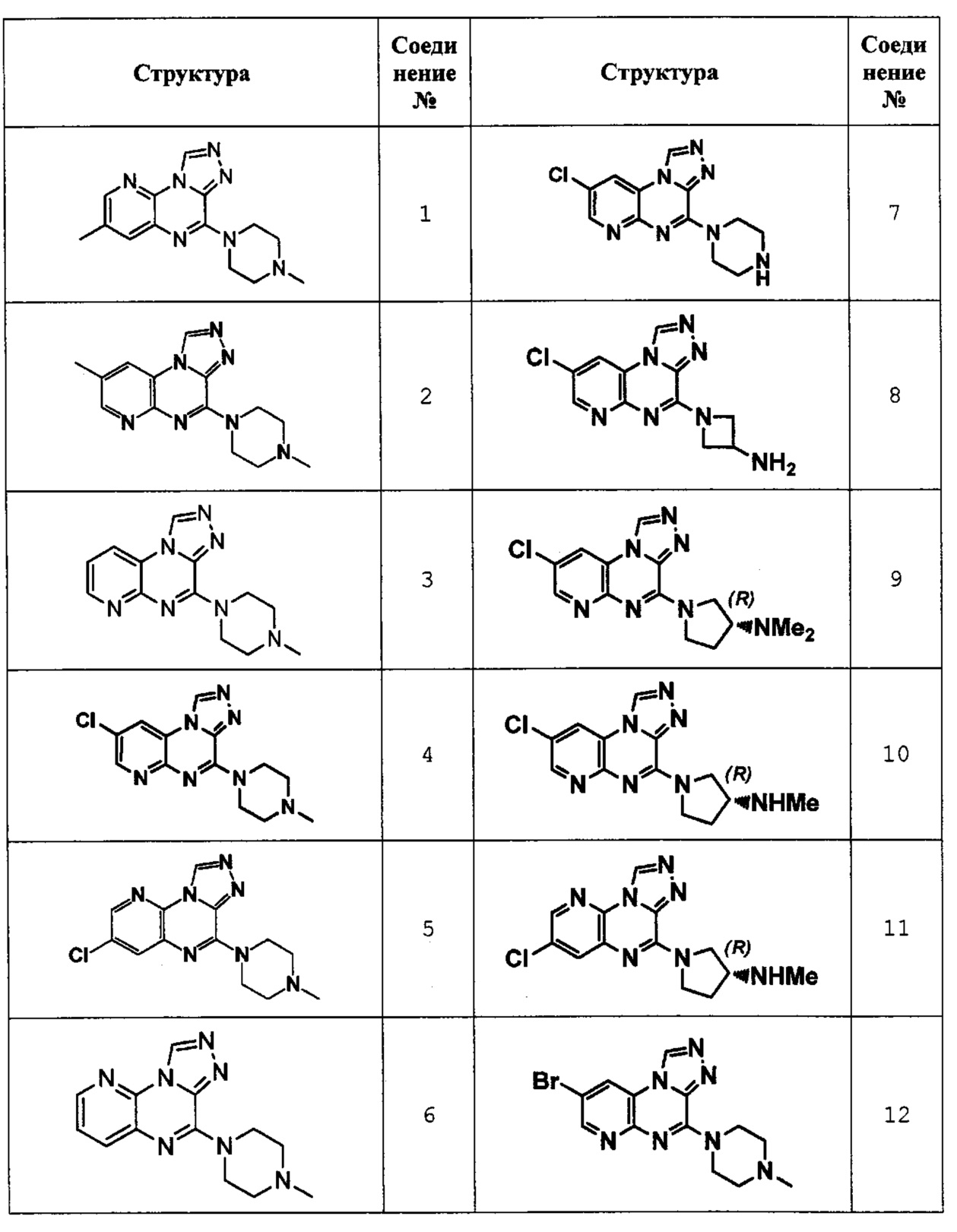
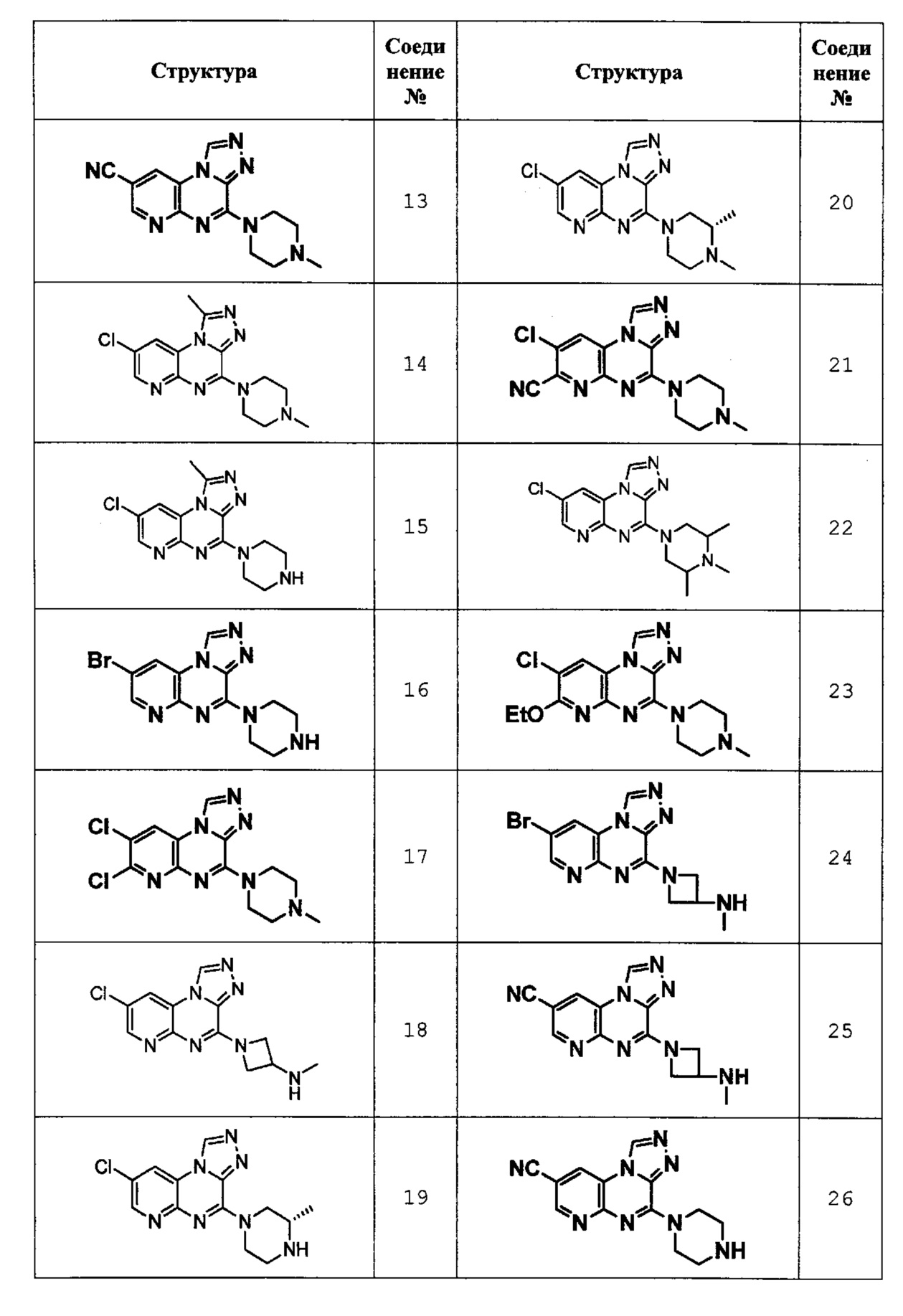
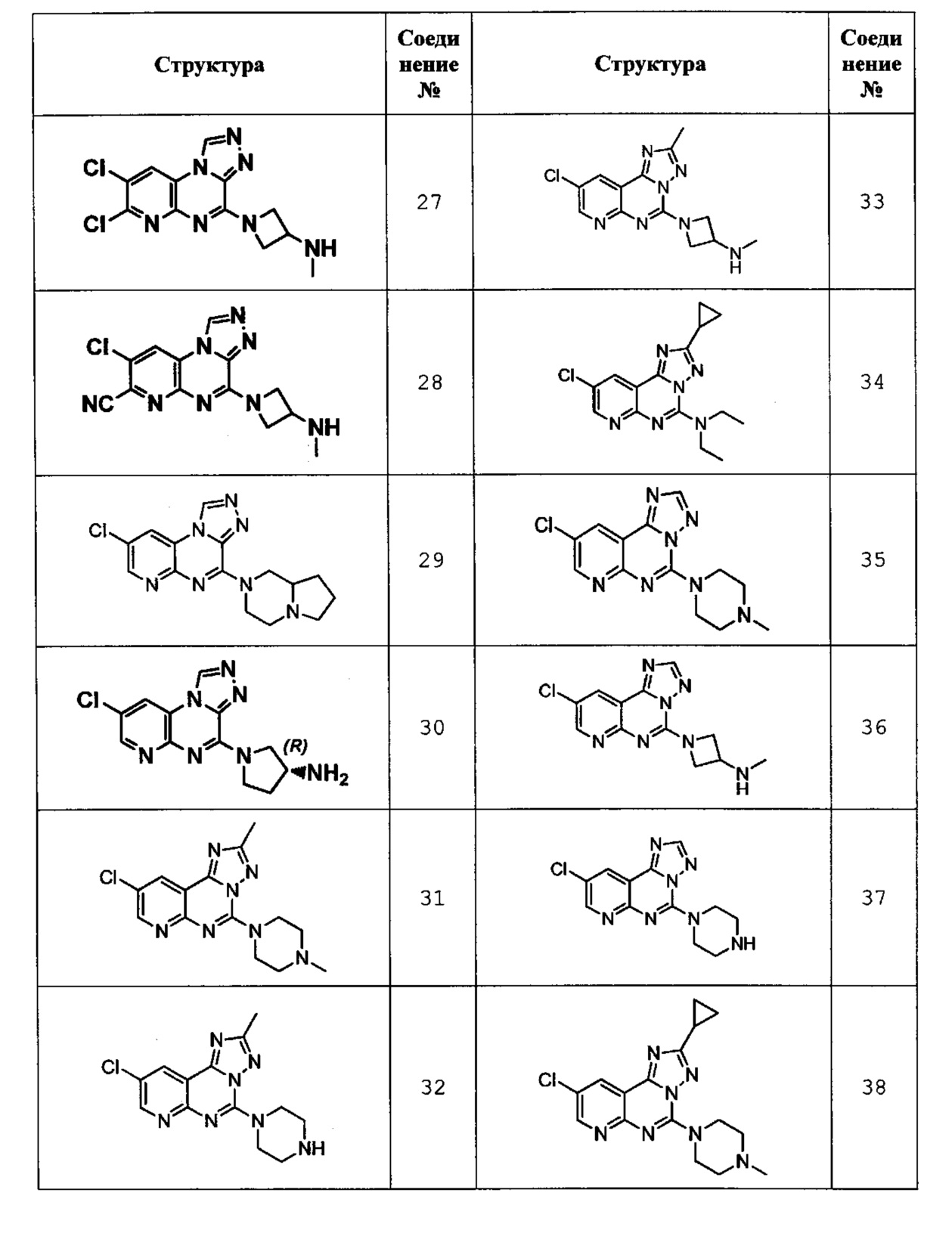
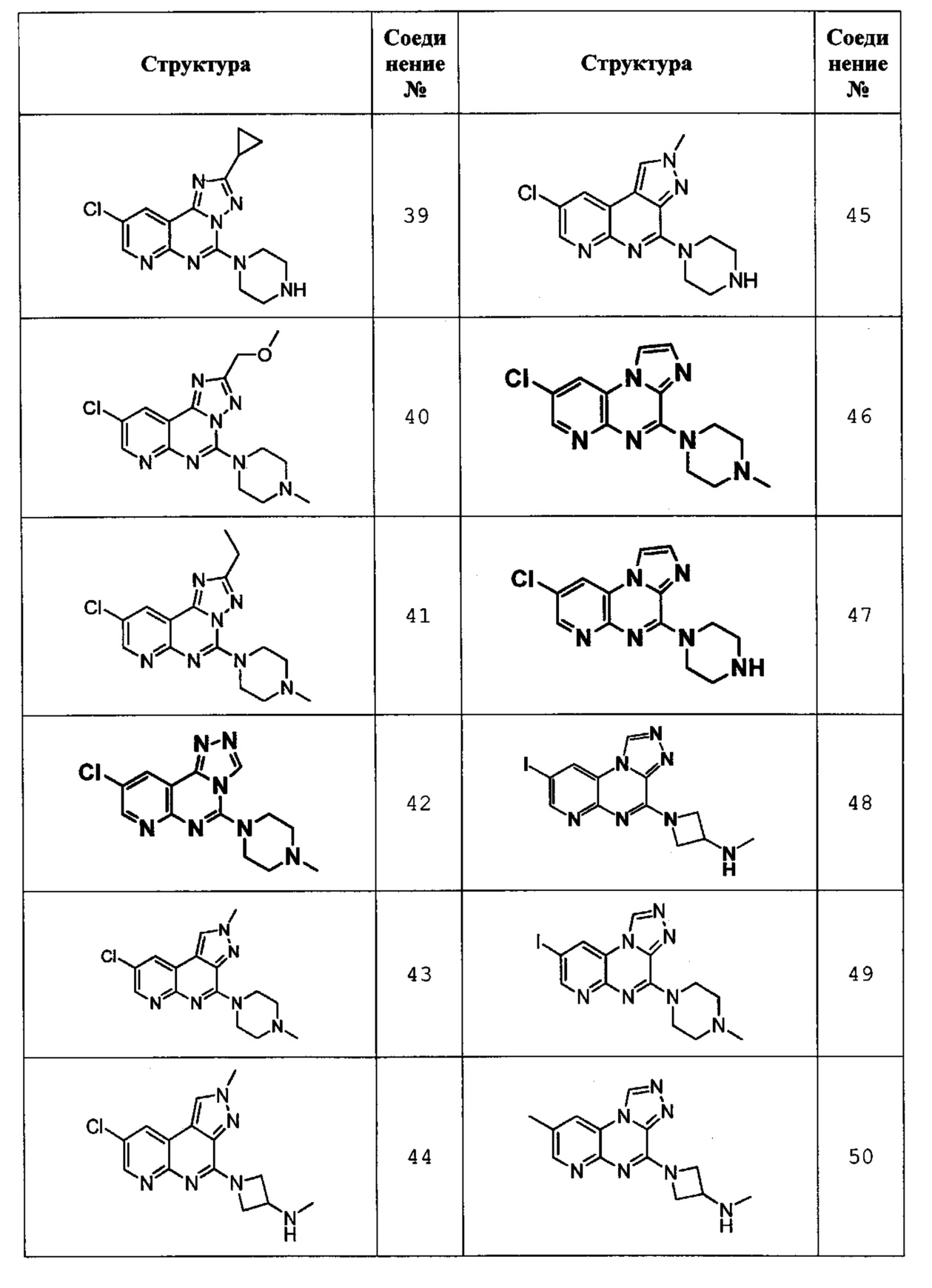


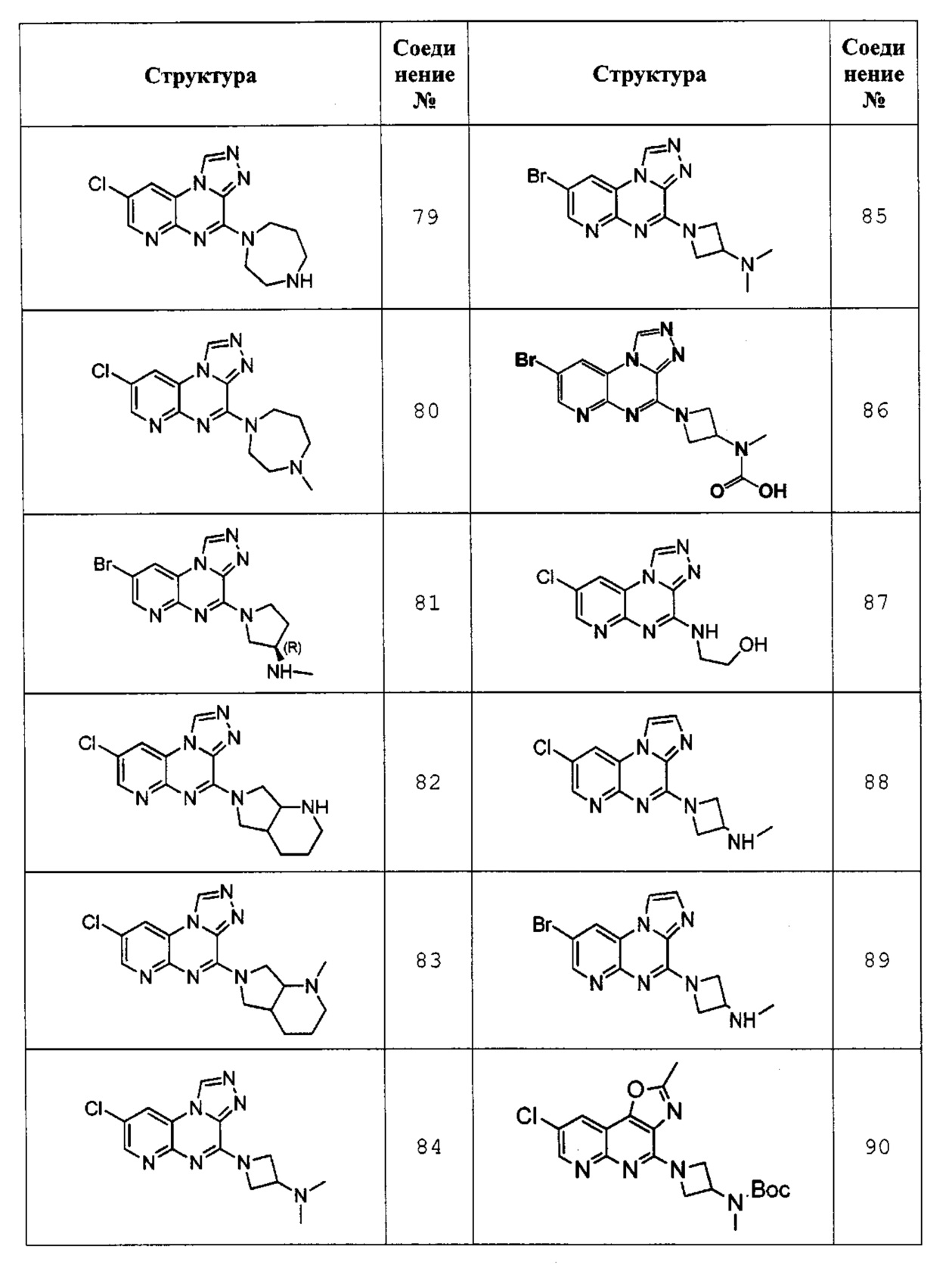
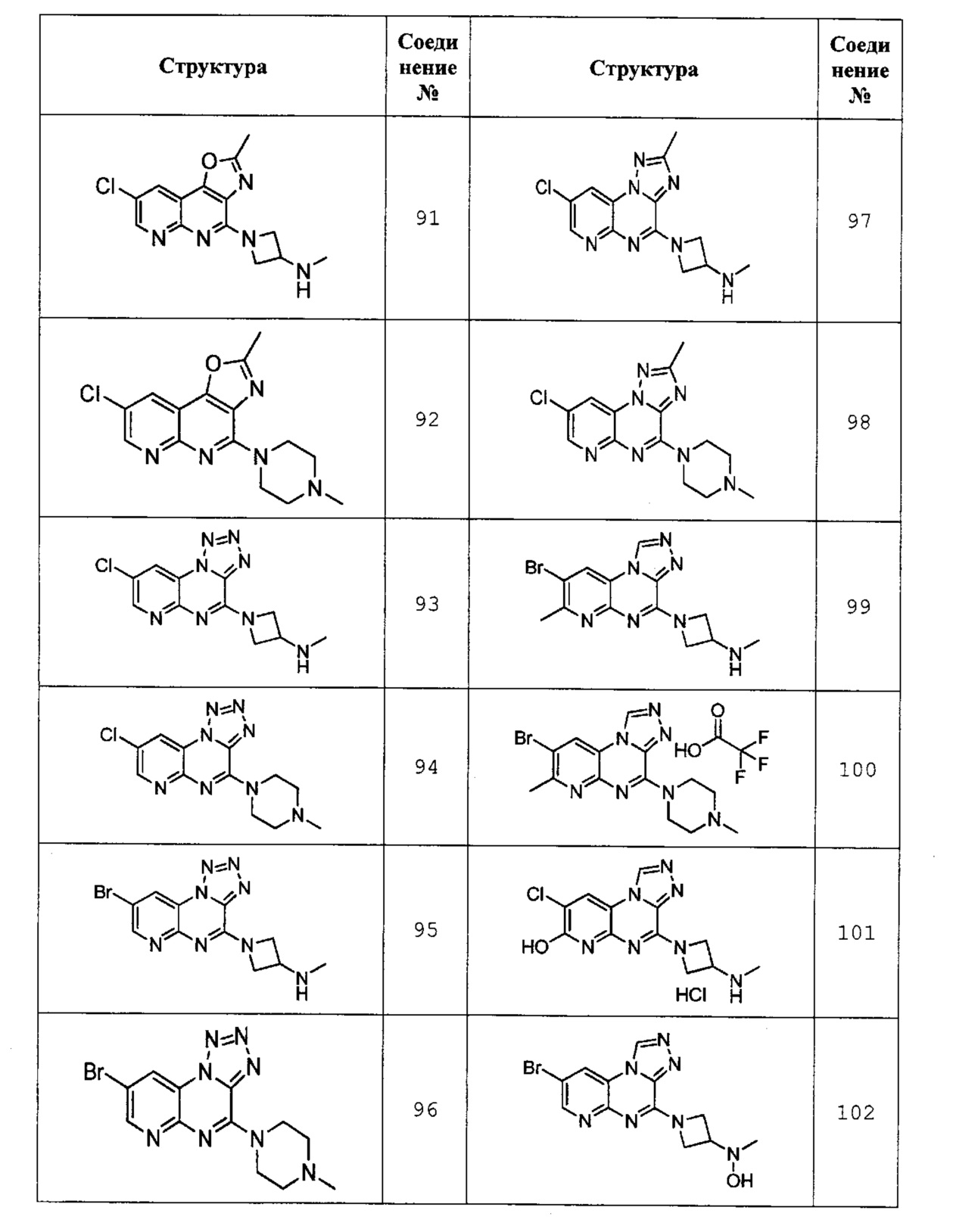
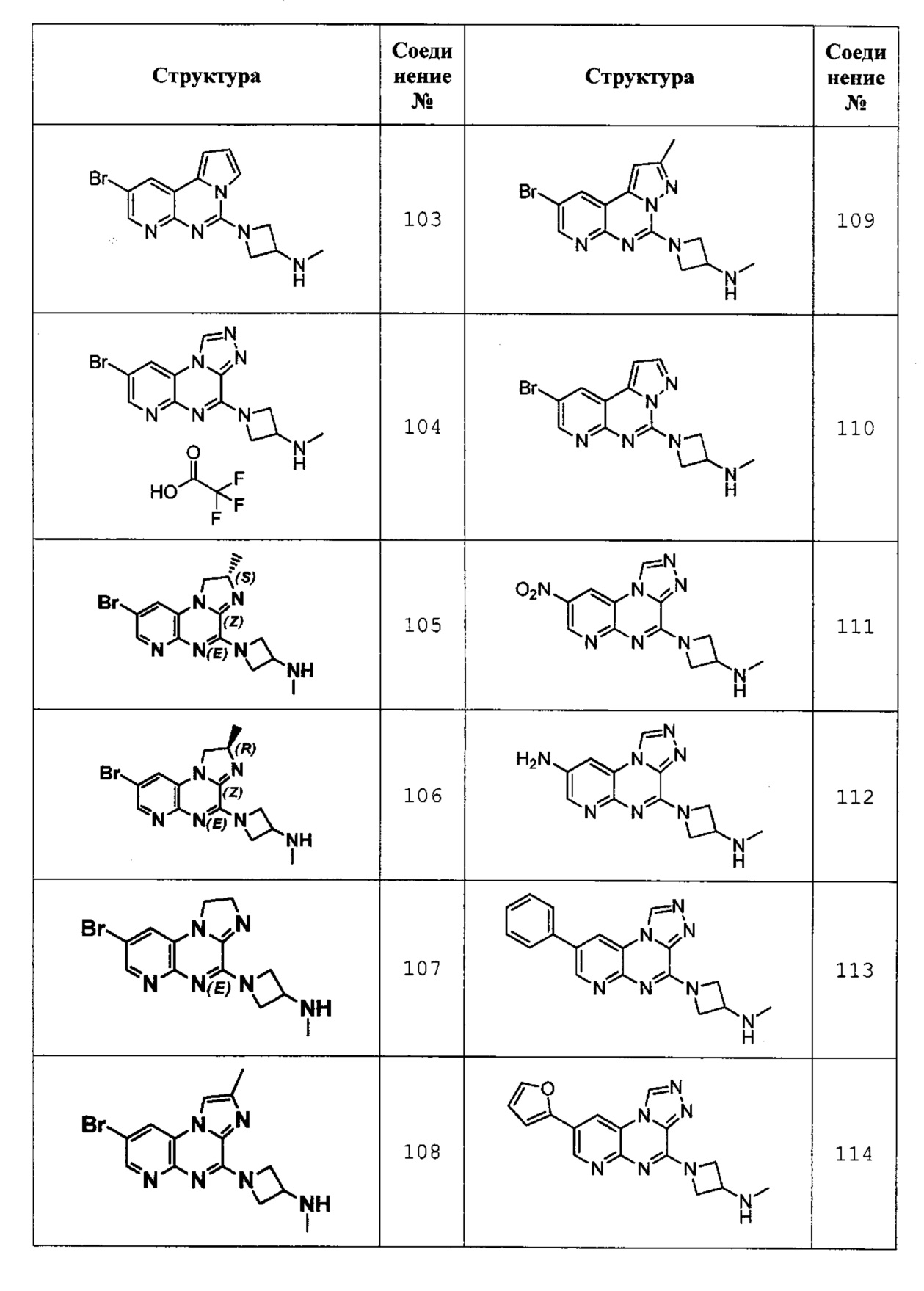
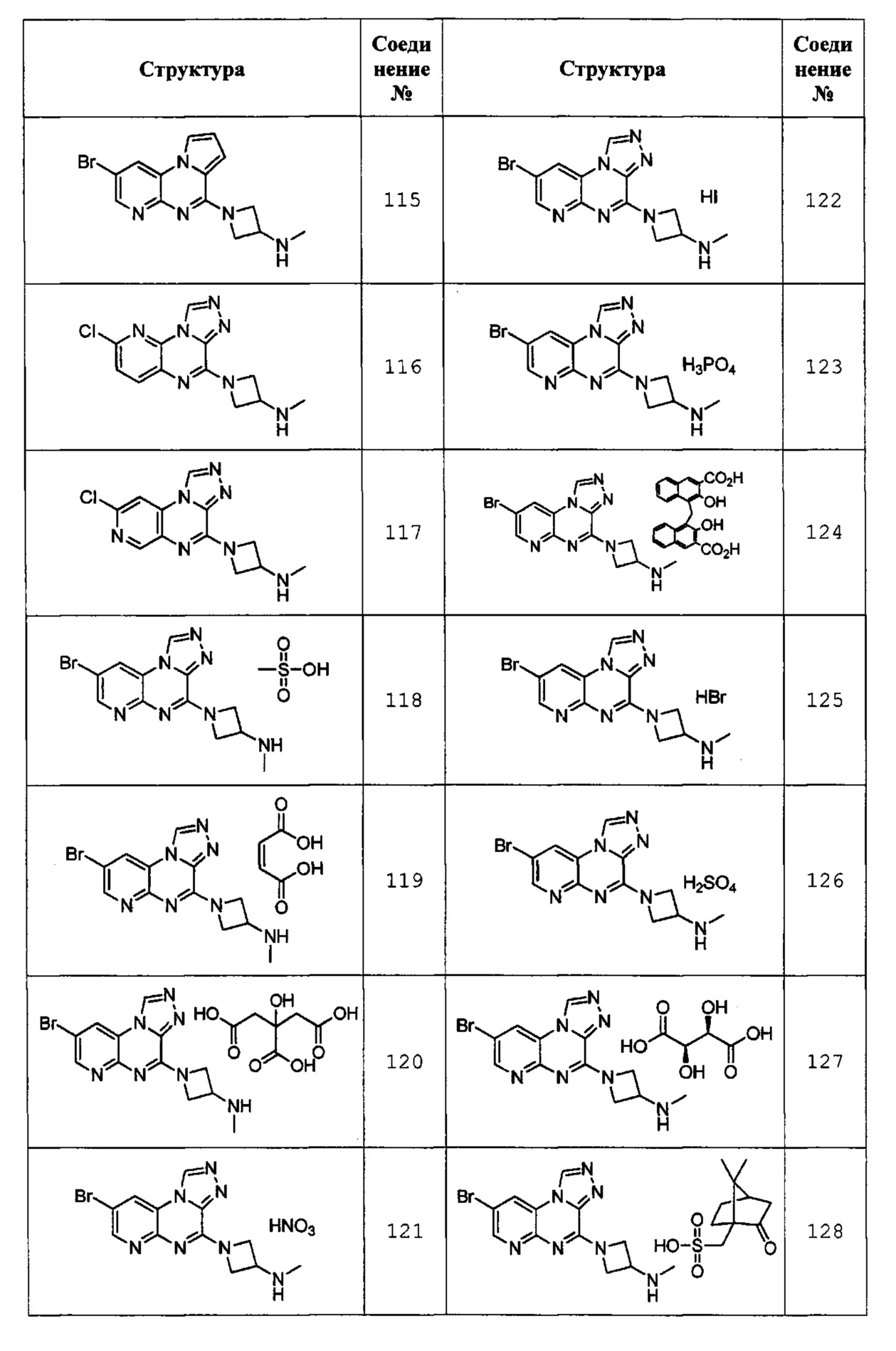
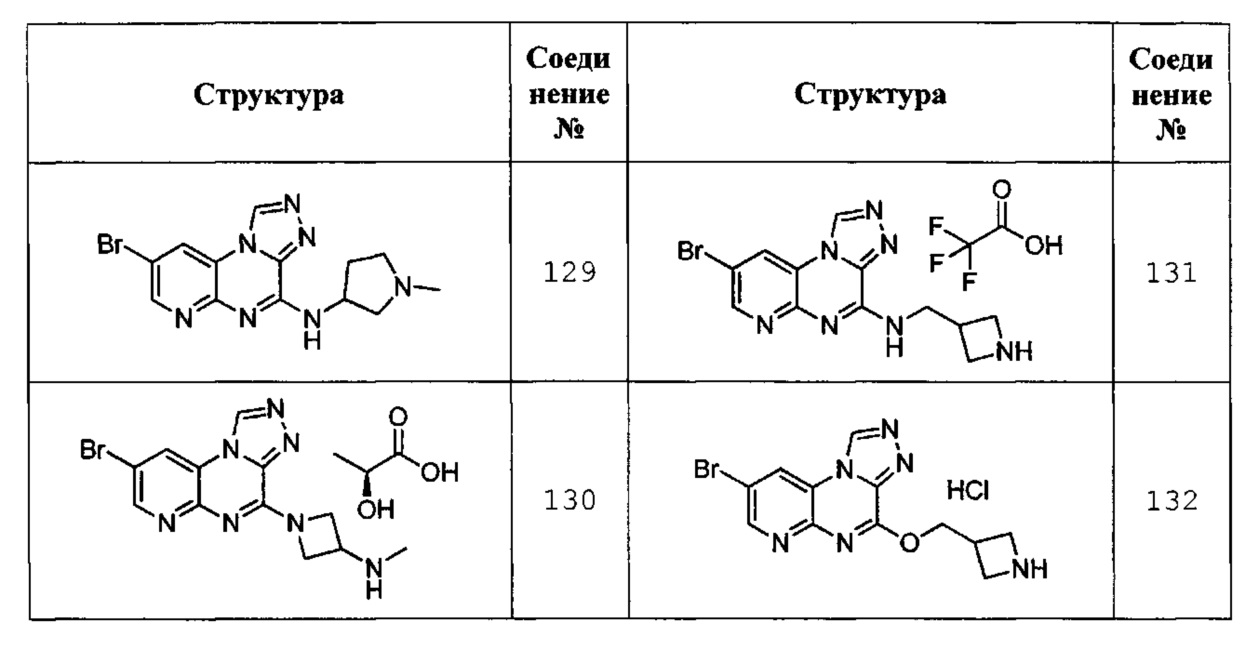
Экспериментальный пример 1: анализ сродства связывания человеческого рецептора гистамина 4 (hH4R)
Каждое соединение согласно настоящему изобретению, полученное в примерах, разбавляли в 1000 раз (об./вес.) ДМСО, и затем 1 мкл разбавленного раствора соединения смешивали с 99 мкл буферного раствора для анализа (50 мМ tris-HCl pH 7,4, 5 мМ EDTA), получая концентрации 0,1, 0,3, 1, 3, 10 и 100 мкМ. 20 мкл полученного раствора соединения переносили в каждую лунку 96-луночного планшета, и затем 20 мкл 100 мкМ гистамина, разбавленного буферным раствором для анализа и 1% ДМСО, переносили в каждую лунку, чтобы рассчитать неспецифическое связывание и степень общего связывания. 25 мкг мембран клеток, суперэкспрессирующих человеческий рецептор гистамина 4 (PerkinElmer), разбавляли в 160 мкл буферного раствора для анализа и переносили в каждую лунку. [3H]-меченный гистамин (PerkinElmer) разбавляли в концентрации 1 мкМ, 20 мкл помещали в каждую лунку и затем помещали в инкубатор при 27°C на 30 минут. После реакции 200 мкл смеси переносили на планшет из стекловолокна, предварительно смоченный 0,5% полиэтиленамина, и затем несвязанный [3H]-меченный гистамин удаляли в вакууме. После 4-кратной промывки 200 мкл промывочного буферного раствора (50 мМ tris-HCl, pH 7,4) планшет высушивали в печи при 37°C в течение 18 часов. 100 мкл раствора бета-сцинтилляционного коктейля добавляли в каждую лунку и через 1 час значение СРМ (число импульсов в минуту) для [3H]-меченного гистамина измеряли с использованием счетчика Wallac beta-counter TriLux. Сродство связывания (IС50) человеческого рецептора гистамина 4 согласно настоящему изобретению анализировали с помощью программы Excel, и результаты анализа показаны в таблице 1.
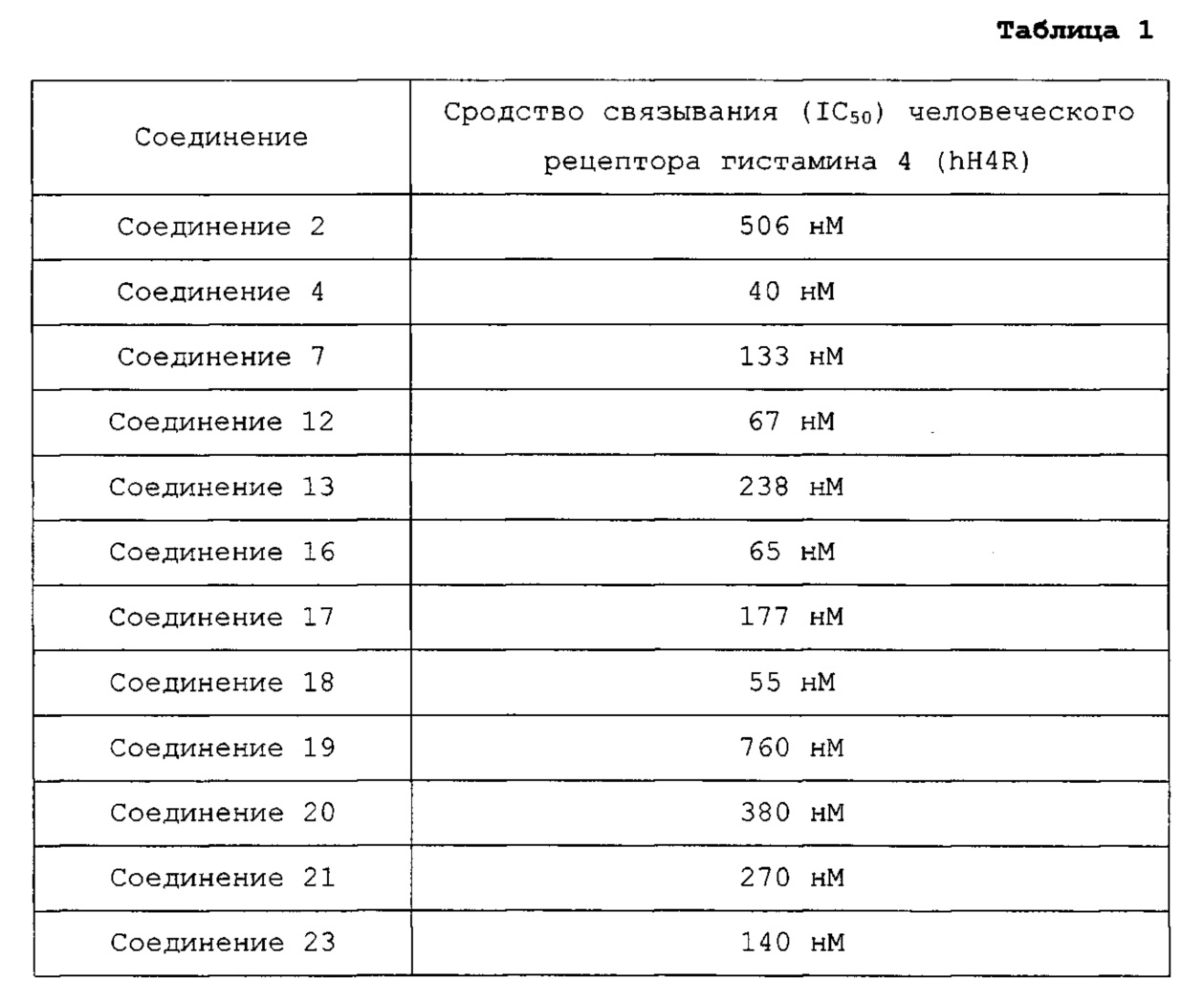


Экспериментальный пример 2: анализ сродства связывания человеческого рецептора гистамина 3 (hH3R)
Каждое соединение согласно настоящему изобретению, полученное в примерах, было получено в ДМСО в концентрации 0,02, 0,06, 0,3 и 2 мМ. 10 мкл полученного раствора соединения распределяли в каждую лунку планшета с 96 лунками, и затем 10 мкл 200 мкМ (R)(-)-α-метилгистамина (RaMH), разбавленного буфером для анализа и 1% ДМСО, переносили в каждую лунку, чтобы рассчитать неспецифическое связывание и степень общего связывания. 15 мкг мембран клеток, суперэкспрессирующих человеческий рецептор гистамина 3 (PerkinElmer), разбавляли в 18 0 мкл буферного раствора для анализа (50 мМ tris-HCl pH 7,4, 5 мМ MgCl2) и переносили в каждую лунку. [3H]-меченный N-α-метилгистамин (PerkinElmer) разбавляли в концентрации 20 мкМ, 10 мкл добавляли в каждую лунку и затем помещали в инкубатор при 27°C на 30 минут. После реакции 200 мкл смеси переносили на планшет из стекловолокна, предварительно смоченный 0,5% полиэтиленамина, и затем несвязанный [3H]-меченный N-α-метилгистамин удаляли в вакууме. После 5-кратной промывки 200 мкл промывочного буферного раствора (50 мМ tris-HCl, pH 7,4) планшет высушивали в печи при 37°C в течение 18 часов. 100 мкл раствора бета-сцинтилляционного коктейля добавляли в каждую лунку и через 1 час значение СРМ для [3H]-меченного N-α-метилгистамина измеряли с использованием счетчика Wallac beta-counter TriLux. Сродство связывания (IС50) человеческого рецептора гистамина 3 согласно настоящему изобретению анализировали с помощью программы Excel, и результаты анализа показаны в таблице 2.
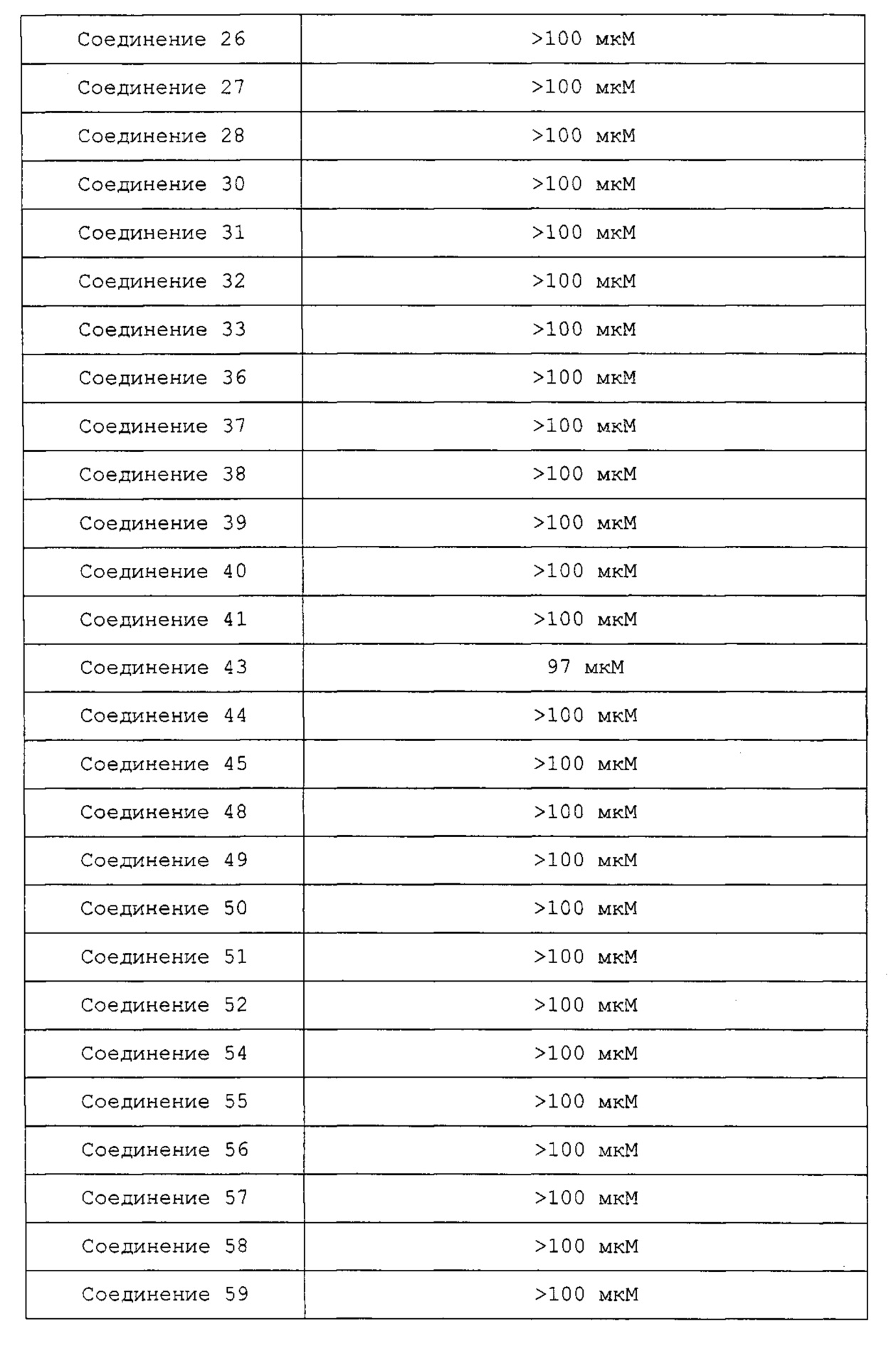
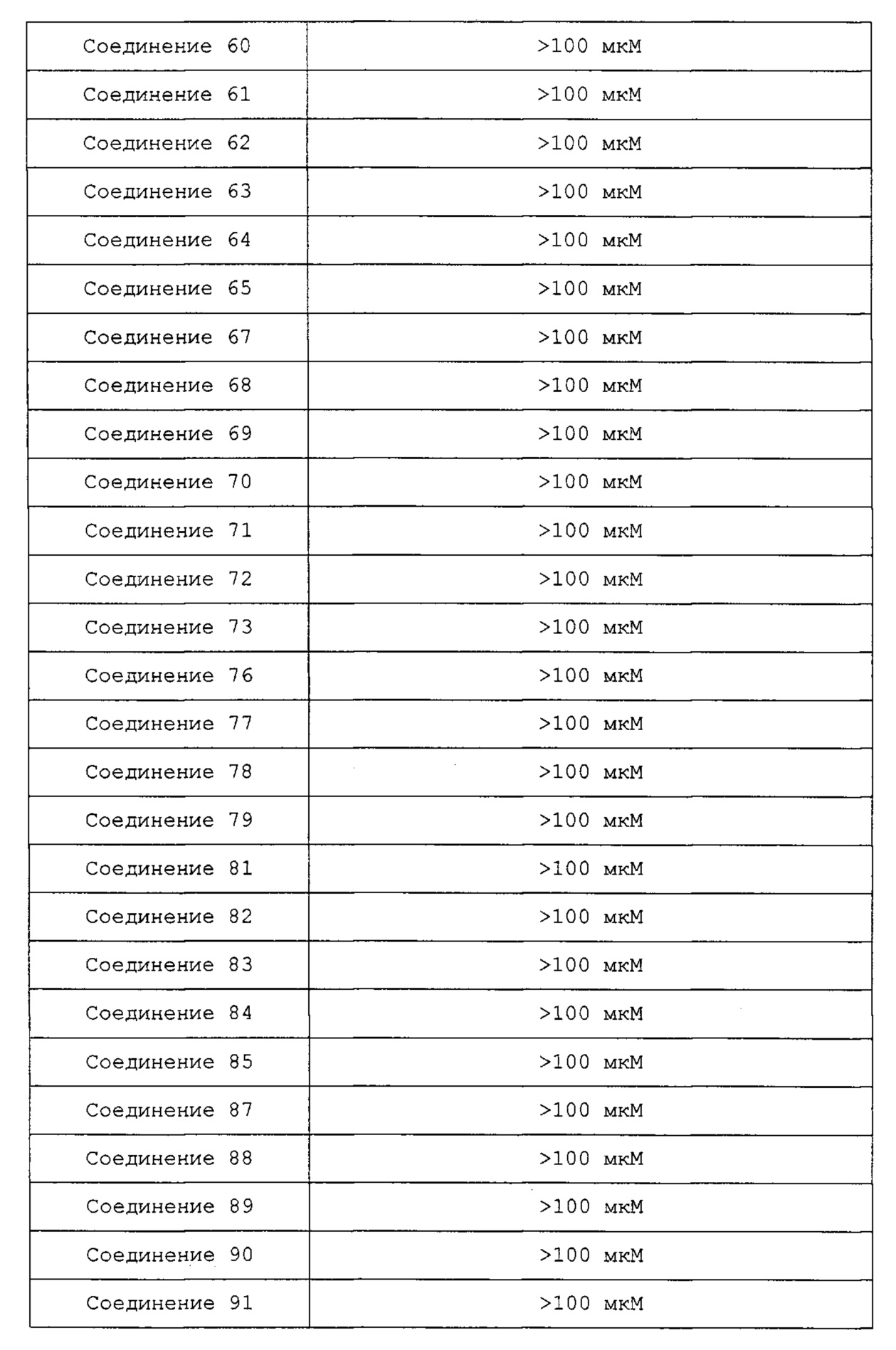
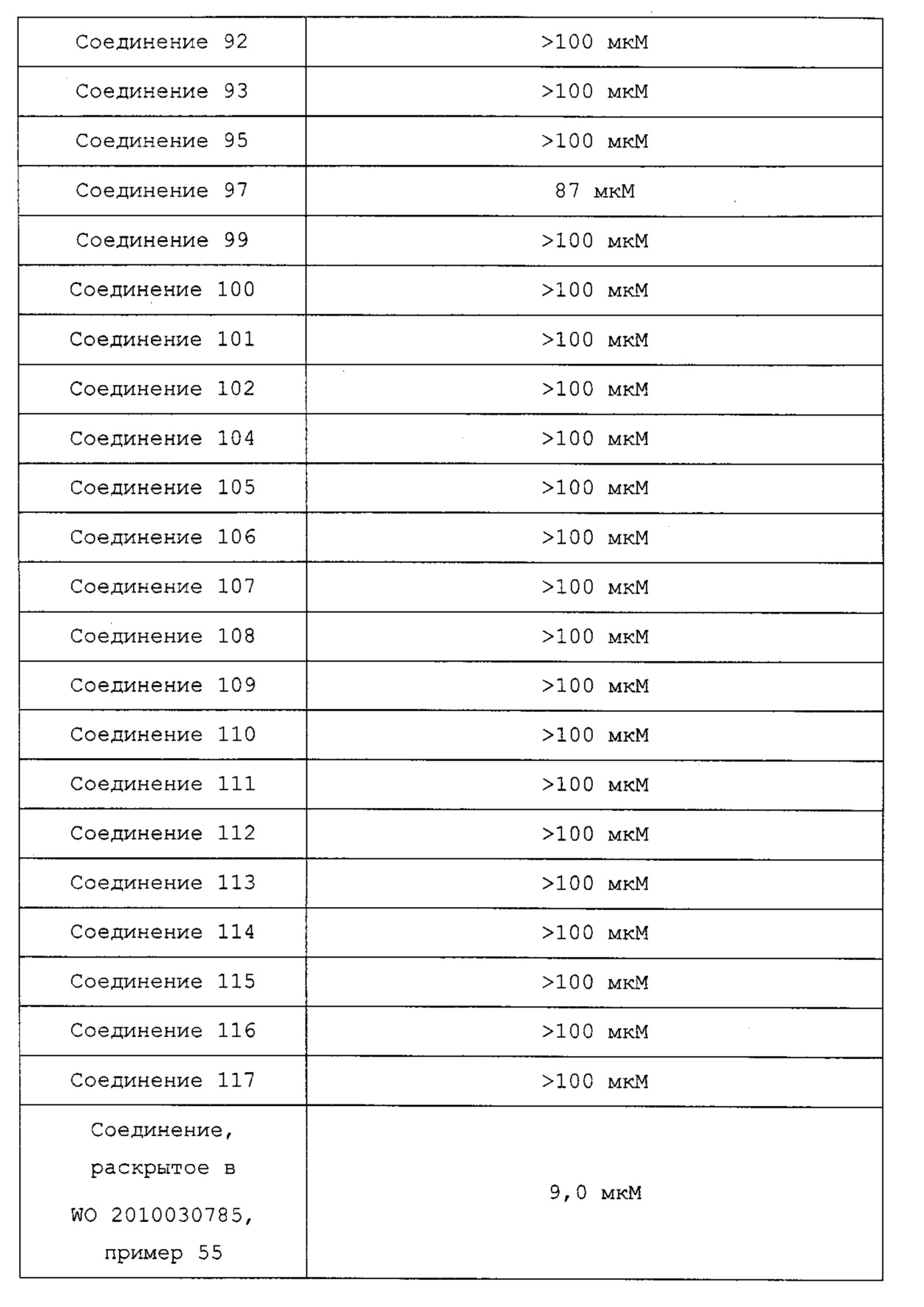
Экспериментальный пример 3: анализ сродства связывания человеческого рецептора серотонина 3 (человеческий 5-HТ3 рецептор)
Тесты связывания человеческого рецептора серотонина 3 согласно настоящему изобретению были осуществлены в Ceper (Пуатье, Франция), и результаты анализа показаны в таблице 3.
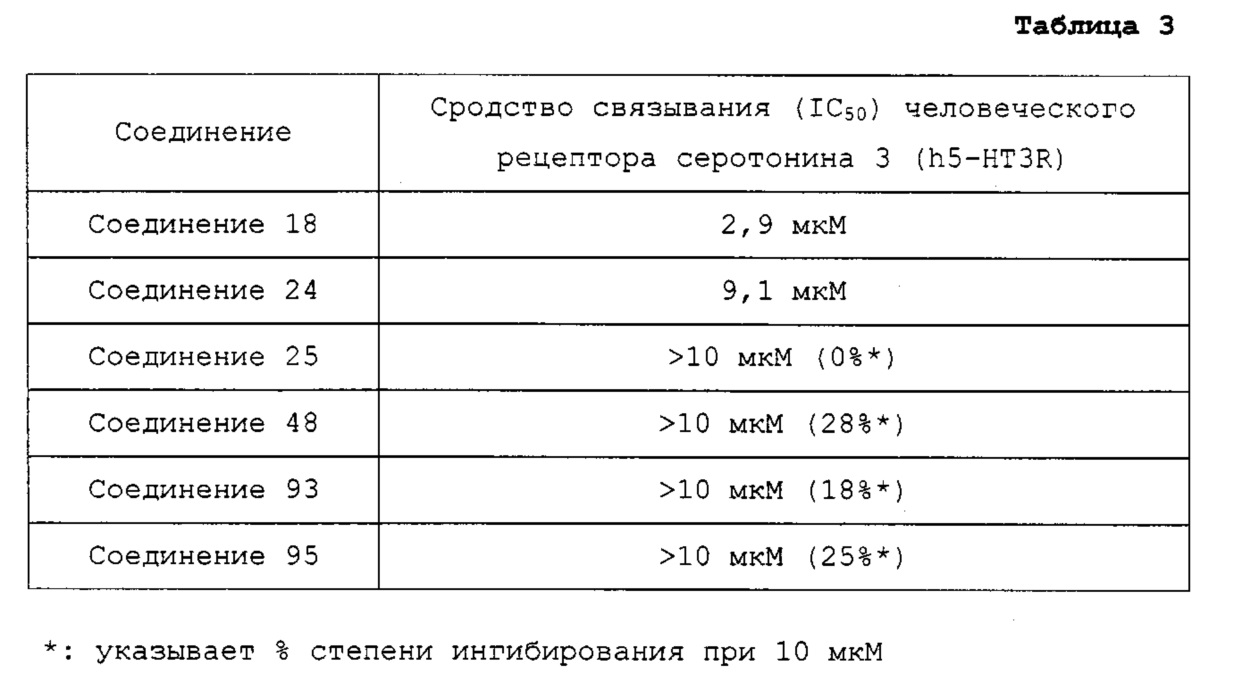
Экспериментальный пример 4: тест на растворимость в искусственном растворе желудочно-кишечного тракта
Первый раствор
2,0 г хлорида натрия добавляли в 7,0 мл 37% хлористоводородной кислоты и воды, получая полный объем 1 л. pH доводили до значения ровно 1,2 с помощью 1 н. гидроксида натрия или 1 н. хлористоводородной кислоты. Небольшое количество (20 мл) этого раствора брали и добавляли в 25 мкл Тритона Х-100, чтобы получить полный объем 25 мл.
Второй раствор
0, 348 г гидроксида натрия, 3, 438 г NaH2O4, 6,186 г хлорида натрия и воду добавляли, чтобы получить полный объем 1 л. pH доводили до значения ровно 6,5 с помощью 1 н. гидроксида натрия или 1 н. хлористоводородной кислоты. Небольшое количество (20 мл) этого раствора брали и добавляли 41,25 мг гидрата таурохолата натрия и 25 мкл 750 мМ лецитина, растворенного в этаноле, чтобы получить полный объем 25 мл.
Эксперимент
Для соединения согласно настоящему изобретению, полученного в примерах, образцы для испытаний получали в каждом из первого и второго растворов с концентрацией 2 мг/мл, и затем их перемешивали в течение 1 минуты, обрабатывали ультразвуком в течение 1 минуты и нагревали при 37°C в течение 1 часа для первого раствора и 2 часов для второго раствора. Нагретые растворы тестируемого образца фильтровали, чтобы удалить нерастворенные соединения. Наконец, отбирали 100 мкл раствора полностью растворенного соединения и добавляли 100 мкл CH3CN, получая раствор тестируемого образца. Для каждого анализа раствора тестируемого образца растворимость измеряли, используя жидкостную хроматографию. Результаты теста на растворимость в первом и втором растворах показаны в таблице 4.


Экспериментальный пример 5: тест на метаболическую стабильность
Получение раствора микросомы
Микросому с концентрацией белка 20 мг/мл (человек, мышь, крыса) разбавляли 0,1 М фосфатного буфера (рН 7,4) и получали в концентрации 1,316 мг/мл. Соединение согласно настоящему изобретению, полученное в примерах, растворяли в ДМСО, получая 2,5 мМ раствор, и разбавляли дистиллированной водой, получая 125 мкМ раствор. Разбавленный раствор микросом, полученный, как описано выше, и разбавленный раствор соединения смешивали, получая раствор микросом с концентрацией белка 1,2 5 мг/мл и концентрацией соединения 6,25 мкМ.
Получение раствора NADPH
NADPH растворяли в 0,1 М фосфатном буфере, получая 5 мМ раствор.
Эксперимент
Раствор микросом в концентрации 6,25 мкМ соединения согласно настоящему изобретению, полученный в примерах, распределяли в каждые 80 мкл, помещали на баню с температурой 37°C и держали в течение 5 минут, и затем инициировали реакцию, добавляя 20 мкл 5 мМ NADPH, получая конечные концентрации 1 мг/мл микросомального белка, 5 мкМ соединения и 1 мМ NADPH. Реакцию завершали добавлением 100 мкл ацетонитрила в 0, 10, 20 и 30 минут, центрифугировали, и затем супернатант анализировали, используя жидкостную хроматографию. Период полужизни определяли, используя пиковую область оставшихся соединений в 0, 10, 20 и 30 минут, и высчитывали параметр CLh.int, представляющий метаболическую стабильность. Результаты теста на метаболическую стабильность показаны в таблице 5.
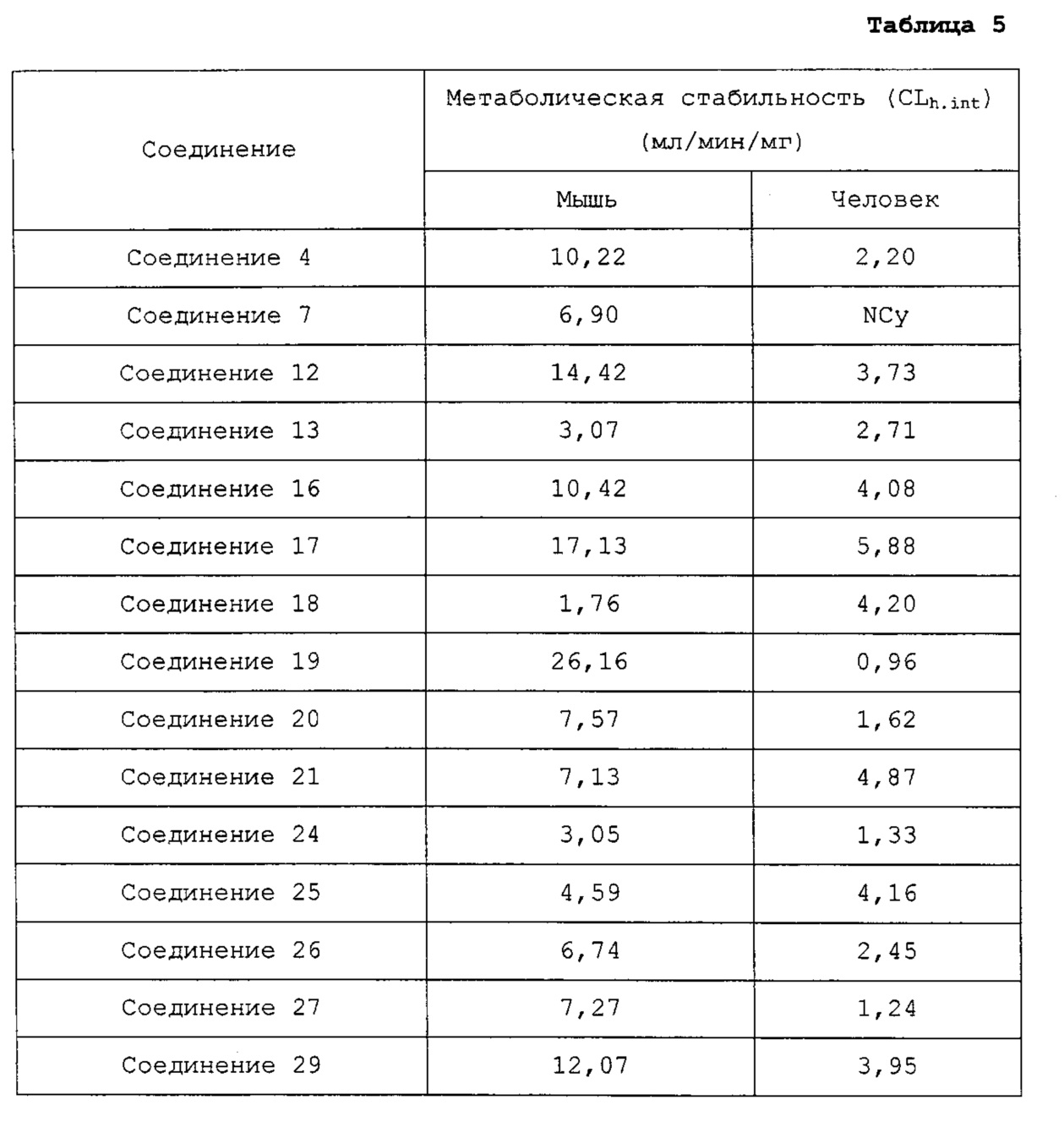
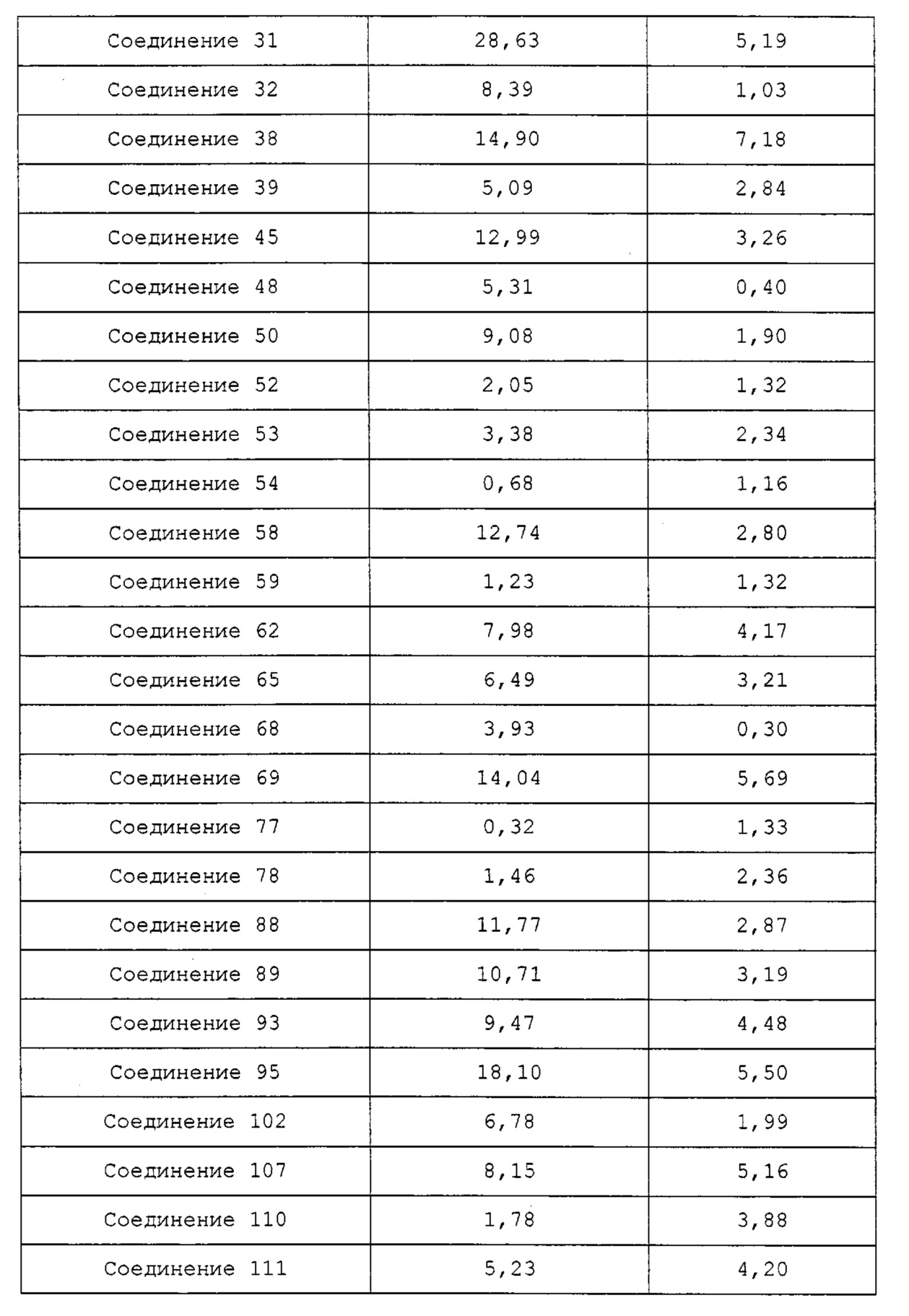

Экспериментальный пример 6: тест на фармакокинетические свойства
Получение лекарственного средства
Соединение согласно настоящему изобретению, полученное в примерах, растворяли в 20% растворе гидроксипропил-(3-циклодекстрина, получая раствор для введения с концентрацией 5 мг/мл для мышей, и последовательно добавляли ДМСО (5%), 5% раствор декстрозы (94,75%) и 2 н. хлористоводородную кислоту (0,25%), получая раствор для введения с концентрацией 2 мг/мл для крыс.
Эксперимент
Масса тела каждой мыши ICR и крысы SD составляла, соответственно, 20-30 г и 200-300 г, и дозировка соединения согласно настоящему изобретению составляла 10 мл/кг для мышей и 5 мл/кг для крыс, и ее вводили перорально с использованием Zonde. Забор крови осуществляли в 0,25, 0,5, 1, 2, 4, 6, 8 и 24 часа из орбитальной вены, используя капиллярную трубку, покрытую антикоагулирующим средством, и затем плазму изолировали, используя центрифугу, и сохраняли в морозильнике.
Анализ
Плазму, забранную у животных, и материал в стандартной концентрации предварительно обрабатывали, используя твердофазную экстракцию, и концентрацию соединения по изобретению определяли, используя жидкостную хроматографию с масс-спектрометрией (ВЭЖХ Agilent, API-3000).
Согласно полученному значению концентрации, фармакокинетические параметры находили, используя WinNonlin (версия 6,2), и период полужизни (t1/2), максимальная концентрация в крови (Сmax) и бесконечная область под кривой (AUCinf) показаны ниже в таблицах 6 и 7.

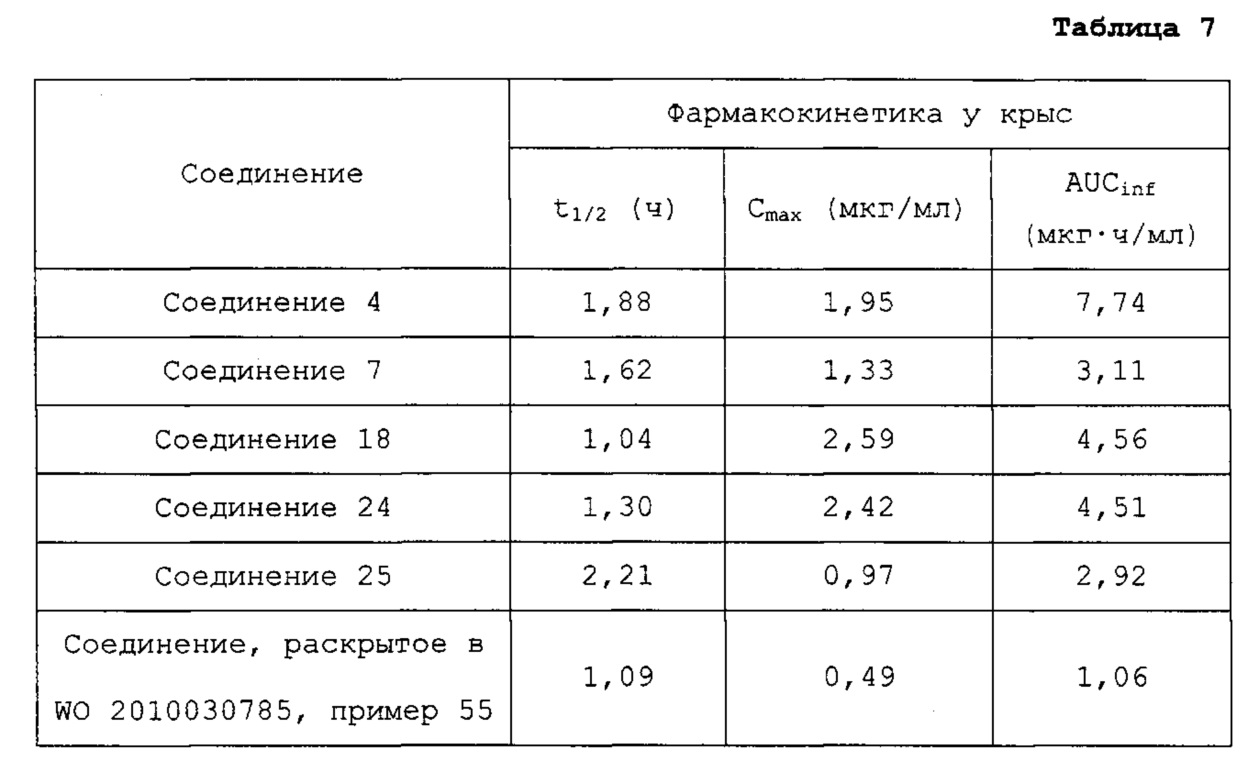
Экспериментальный пример 7: хтостамин-индуцированная миграция тучных клеток
Животное
Самок мышей Balb/c (7 недель, 20±3 г) получали от OrientBio Co., Ltd. Животных содержали в условиях контролируемой температуры (23±3°C), влажности (50±5%) и освещенности с пищей и водой, доступными ad libitum.
Эксперимент
Мышам вводили либо носитель (20%-ый циклодекстрин во второй дистиллированной воде), либо соединение путем перорального введения. Соединение (20 мг/кг) растворяли в носителе и вводили перорально в объеме 10 мл/кг. Через 15 минут мышам делали провокацию путем 20-минутной аэрозольной ингаляции PBS (забуференный фосфатом солевой раствор) или 0,1 М гистамина (растворяющегося в PBS). Этот процесс повторяли в течение 2 дней. После заключительной провокации трахеи освобождали от крови перфузией PBS, и трахеи извлекали, трахеи фиксировали в 10% (вес./об.) формальдегида для последующего получения препаратов поперечных срезов в парафине и окрашивания толуидином синим. Общее количество тучных клеток вычисляли и сообщали среднее значение. (The Journal of Pharmacology and Experimental Therapeutics, 2004, 309, 404-413). Ингибирующие эффекты соединения показаны в таблице 8. Статистический анализ проводили, используя односторонний ANOVA с тестом Dunnett. Значения p<0,05 считали статистически значимыми.


Экспериментальный пример 8: модель гистамин-индуцированного зуда на мышах ICR
Животное
Самок мышей ICR (8 недель) получали от OrientBio, Ltd. Животных содержали в условиях контролируемой температуры (23±3°C), влажности (50±5%) и освещенности с пищей и водой, доступными ad libitum.
Эксперимент: стимул зуда и его измерение
Волоски срезали, используя ножницы (8000AD, THRIVE), на ростральной части спины за 24 часа перед экспериментом под анастезией изофлюраном. Гистамин (300 нмоль) растворяли в солевом растворе, и объем 4 0 мкл вводили внутрикожно в ростральную часть спины. Мышам вводили либо носитель (20%-ый циклодекстрин во второй дистиллированной воде), либо соединение путем перорального введения. Соединение (50 мг/кг) растворяли в носителе (20%-ый циклодекстрин во второй дистиллированной воде) и вводили перорально в объеме 10 мл/кг.
Мышей (n=10 на группу) разделяли на 3 группы (нормальная группа, контрольная группа и экспериментальная группа). Животных рандомизировали для равномерного распределения массы тела. Носитель или соединение вводили за 20 минут до инъекции гистамина или солевого раствора. Немедленно после внутрикожной инъекции животных возвращали в акриловую клетку (приблизительно 30×30×30 см, доступная продукция), составленную из четырех клеток, к которым они были акклиматизированы в течение по меньшей мере 1 часа перед экспериментом, для наблюдения реакций зуда. Камера (Samsung, VLUUNV30) была установлена выше мышей, чтобы записывать их реакции. Зуд измеряли слепым подсчетом числа почесываний за период 20 минут немедленно после внутрикожной инъекции. Число почесываний определяли как 3 или более индивидуальных быстрых движений почесывания задними лапами вокруг участка инъекции (J. Allergy Clin. Immunol. 2007, 19 (1), 176-183). Все данные анализировали при использовании Excel и Prism, и все значения выражали как среднее ± S.E.M. Ингибирующий эффект соединений показан как процент от максимальной реакции на гистамин (100% контрольная группа).
Статистический анализ проводили, используя односторонний ANOVA с тестом Dunnett. Значения p<0,05 считали статистически значимыми. Ингибирующие эффекты соединения показаны в таблице 9.

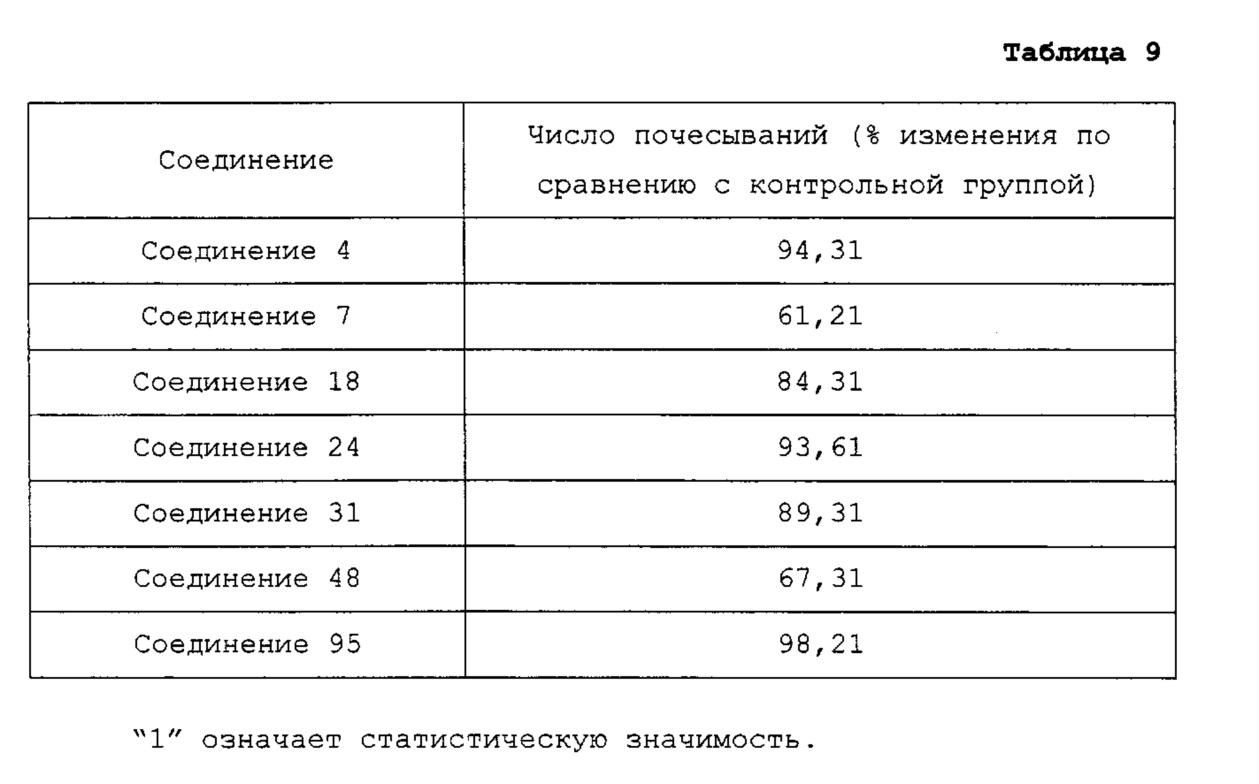
Экспериментальный пример 9: модель индуцированного веществом P зуда на мышах ICR
Животное
Самок мышей ICR (8 недель) получали от OrientBio, Ltd. Животных содержали в условиях контролируемой температуры (23±3°C), влажности (50±5%) и освещенности с пищей и водой, доступными ad libitum.
Эксперимент: индукция зуда и его измерение
Волоски срезали, используя ножницы (8000AD, THRIVE), на ростральной части спины за 24 часа перед экспериментом под анастезией изофлюраном. Вещество P (100 нмоль) растворяли в солевом растворе, и объем 40 мкл вводили внутрикожно в ростральную часть спины. Мышам вводили либо носитель (20%-ый циклодекстрин во второй дистиллированной воде), либо соединение путем перорального введения. Соединение (50 мг/кг) растворяли в носителе (20%-ый циклодекстрин во второй дистиллированной воде) и вводили перорально в объеме 10 мл/кг.
Мышей (n=10 на группу) разделяли на 3 группы (нормальная группа, контрольная группа и экспериментальная группа). Животных рандомизировали для равномерного распределения массы тела. Носитель или соединение вводили за 20 минут до инъекции вещества P или солевого раствора. Немедленно после внутрикожной инъекции животных возвращали в акриловую клетку (приблизительно 30×30×30 см, доступная продукция), составленную из четырех клеток, к которым они были акклиматизированы в течение по меньшей мере 1 часа перед экспериментом, для наблюдения реакций зуда. Камера (Samsung, VLUUNV30) была установлена выше мышей, чтобы записывать их реакции. Зуд измеряли слепым подсчетом числа почесываний за период 20 минут немедленно после внутрикожной инъекции. Число почесываний определяли как 3 или более индивидуальных быстрых движений почесывания задними лапами вокруг участка инъекции (J. Toxicol. Sci. 2009, 34 (4), 427-431; J. Pharmacol. Sci. 2006, 100, 285-288). Все данные анализировали при использовании Excel и Prism, и все значения выражали как среднее ± S.E.M. Ингибирующий эффект соединений показан как процент от максимальной реакции на гистамин (100% контрольная группа). Статистический анализ проводили, используя односторонний ANOVA с тестом Dunnett. Значения p<0,05 считали статистически значимыми. Ингибирующие эффекты соединения показаны в таблице 10.


Экспериментальный пример 10: модель индуцированного соединением 48/80 зуда на мышах ICR
Животное
Самок мышей ICR (8 недель) получали от OrientBio, Ltd. Животных содержали в условиях контролируемой температуры (23±3°C), влажности (50±5%) и освещенности с пищей и водой, доступными ad libitum.
Эксперимент: индукция зуда и его измерение
Волоски срезали, используя ножницы (8000AD, THRIVE), на ростральной части спины за 24 часа перед экспериментом под анастезией изофлюраном. Соединение 48/80 (100 мкг) растворяли в солевом растворе, и объем 4 0 мкл вводили внутрикожно в ростральную часть спины. Мышам вводили либо носитель (20%-ый циклодекстрин во второй дистиллированной воде), либо соединение путем перорального введения. Соединение (50 мг/кг) растворяли в носителе (20%-ый циклодекстрин во второй дистиллированной воде) и вводили перорально в объеме 10 мл/кг.
Мышей (n=8-10 на группу) разделяли на 3 группы (нормальная группа, контрольная группа и экспериментальная группа). Животных рандомизировали для равномерного распределения массы тела. Носитель или соединение вводили за 20 минут до инъекции соединения 48/80 или солевого раствора. Немедленно после внутрикожной инъекции животных возвращали в акриловую клетку (приблизительно 30×30×30 см, доступная продукция), составленную из четырех клеток, к которым они были акклиматизированы в течение по меньшей мере 1 часа перед экспериментом, для наблюдения реакций зуда. Камера (Samsung, VLUUNV30) была установлена выше мышей, чтобы записывать их реакции. Зуд измеряли слепым подсчетом числа почесываний за период 20 минут немедленно после внутрикожной инъекции. Число почесываний определяли как 3 или более индивидуальных быстрых движений почесывания задними лапами вокруг участка инъекции (J. Pharmacol. Sci. 2006, 100, 285-288; European J. of Pharmacol. 2002, 448, 175-183). Все данные анализировали при использовании Excel и Prism, и все значения выражали как среднее ± S.E.M. Ингибирующий эффект соединений показан как процент от максимальной реакции на гистамин (100% контрольная группа). Статистический анализ проводили, используя односторонний ANOVA с тестом Dunnett. Значения p<0,05 считали статистически значимыми. Ингибирующие эффекты соединения показаны в таблице 11.

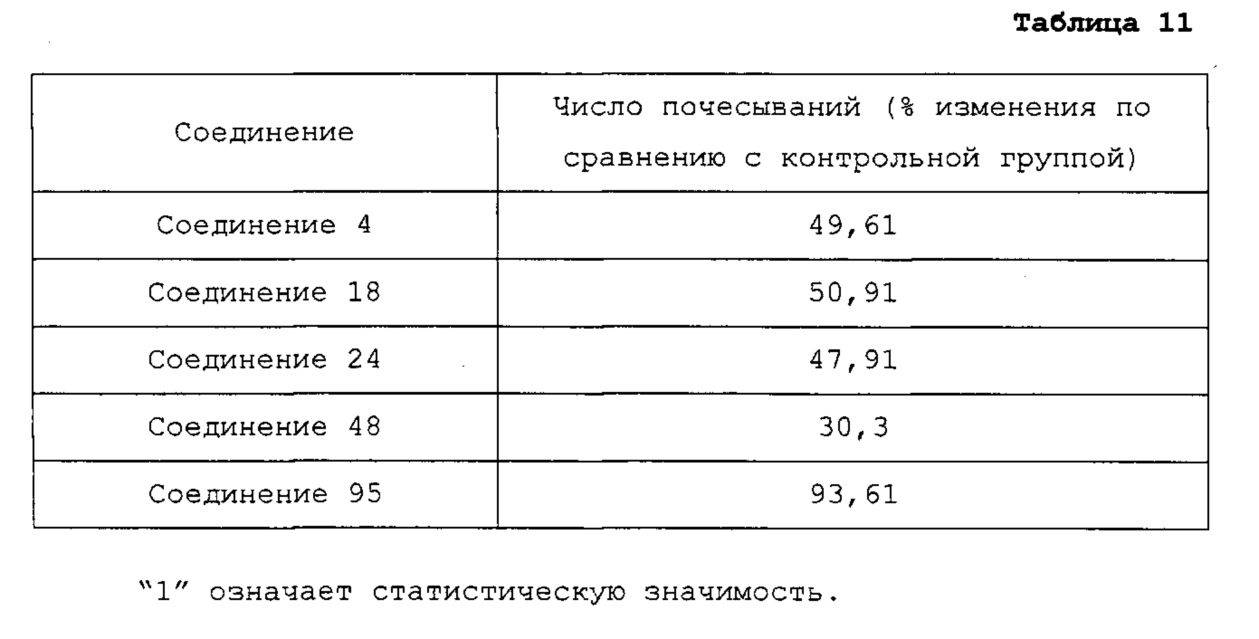
Экспериментальный пример 11: модель оксазолон-индуцированного атопического дерматита на мышах Balb/c
Животное
Самок мышей Balb/c (6 недель, 20±3 г) получали от OrientBio Co., Ltd. Животных содержали в условиях контролируемой температуры (23±3°C), влажности (50±5%) и освещенности с пищей и водой, доступных ad libitum.
Эксперимент
В начальный день (день 1) мышей сенсибилизировали топическим нанесением 50 мкл 1% оксазолона в ацетоне и оливковом масле (отношение 4:1) на кожу брюшной стороны. В день 8, 10, 12, 15, 17, 19, 22, 24 и 26 25 мкл 0,2% оксазолона наносили топически на внутреннюю поверхность правого уха каждой мыши. Между днями 8 и 28 соединение по изобретению вводили перорально (10 мл/кг) два раза в день утром и днем, и в дни 12, 19 и 27 измеряли толщину правого уха мыши. В день 28 ушную ткань мыши извлекали, фиксировали в 10%-ом водном растворе формальдегида, и затем срезы ткани получали, окрашивали Н&Е (гематоксилин и эозин), и затем наблюдали толщину уха (Journal of Investigative Dermatology, 2008, 128, 79-86). Статистический анализ проводили, используя односторонний ANOVA с тестом Dunnett. Значения p<0,05 считали статистически значимыми. Ингибирующие эффекты соединения показаны в таблице 12.

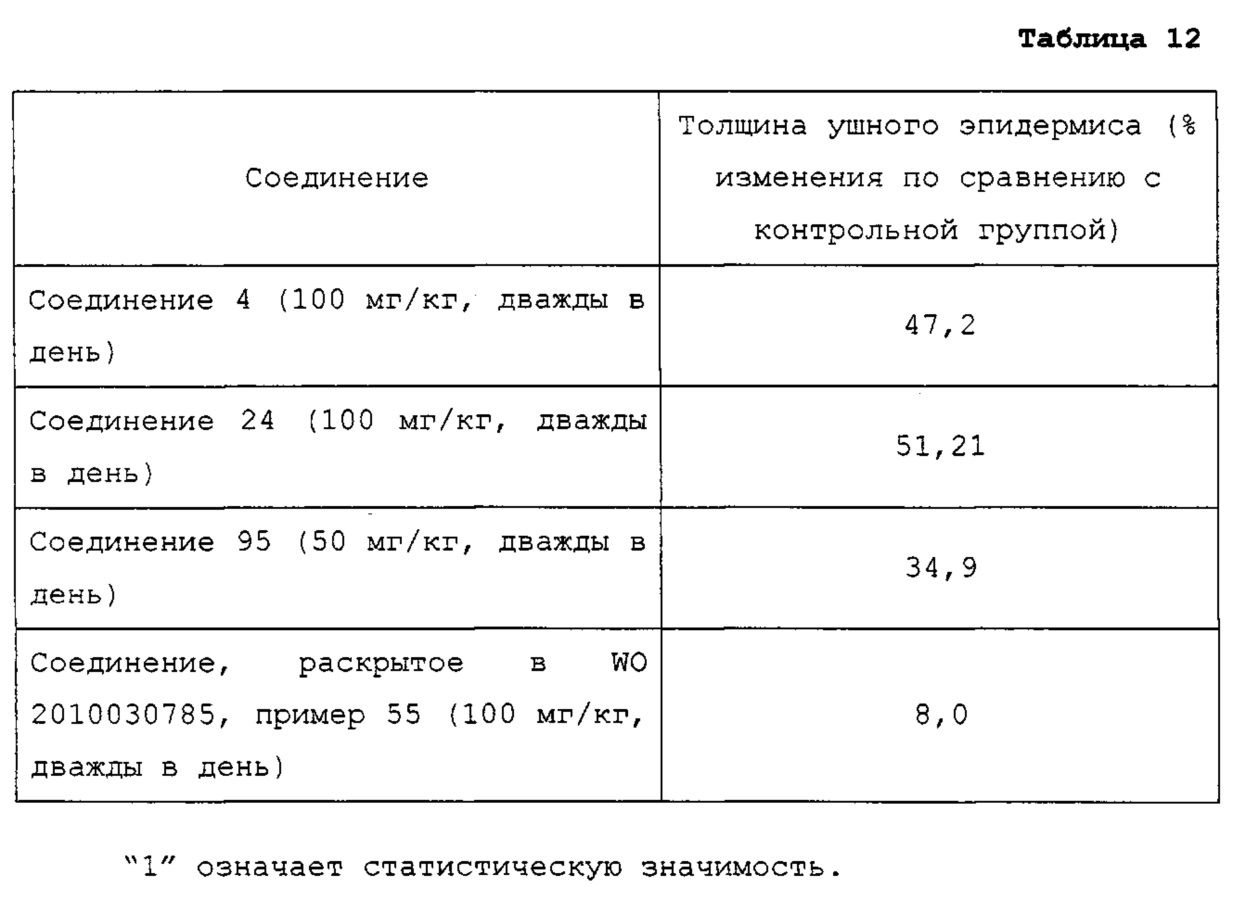
Экспериментальный пример 12: определение фармакологического эффекта в случае индуцированного наружным нанесением Dermatophagoides farina атопического дерматита на мышах NC/Nga
Животное
Самок мышей NC/Nga (3 недели, 15-17 г) получали от Central Lab. Animal Inc. Животных содержали в условиях контролируемой температуры (23±3°C), влажности (50±5%) и освещенности с пищей и водой, доступными ad libitum.
Волосы со спины мышей сбривали ножницами, и оставшиеся волоски удаляли полностью, используя крем для бритья. Для разрушения барьера 150 мкл 4%-ного додецилсульфата натрия (SDS) наносили на бритую кожу спины, мазь Df (100 мг/мышь) наносили два раза в неделю в течение 3 недель. Соединение вводили перорально два раза в день утром и днем в течение 3 недель, и затем серьезность дерматита оценивали в дни 2, 7, 10, 14 и 21, Развитие (1) эритемы/кровотечения, (2) заживления/сухости, (3) отека, (4) экскориации/эрозии оценивали как 0 (отсутствие), 1 (слабое), 2 (умеренное) и 3 (тяжелое). Полную оценку кожи определяли как сумму индивидуальных оценок. (Scandinavian Journal of Immunol. 2011, 73, 536-545). Статистический анализ проводили, используя односторонний ANOVA с тестом Dunnett. Значения p<0,05 считали статистически значимыми. Ингибирующие эффекты соединения показаны в таблице 13.

Как указано выше, соединение согласно настоящему изобретению было существенно улучшено в отношении растворимости и метаболической стабильности по сравнению с обычными лекарственными средствами. Как следует из сравнительного анализа фармакокинетики с использованием моделей животных на мышах ICR и крысах SD по сравнению с соединением, раскрытым в WO 2010/030785, соединение согласно настоящему изобретению показало превосходные эффекты в фармакокинетическом профиле, такие как AUC и максимальная концентрация в крови, в 7-8 раз превосходящие соединение, используемое для сравнения. Было обнаружено, что гистамин-индуцированная миграция воспалительных клеток, таких как тучные клетки и эозинофилы, опосредуется рецептором гистамина 4 (The Journal of Pharmacology and Experimental Therapeutics, 2003, 305, 1212-1221), и антагонисты рецептора гистамина 4 могут оказывать противовоспалительный эффект за счет супрессии увеличения воспалительных клеток. В соответствии с улучшением фармакокинетического профиля, соединение согласно настоящему изобретению показало превосходные фармакологические эффекты, в 3 раза превосходящие таковые соединения, раскрытого в WO 2010/030785, как следует из эксперимента по супрессии миграции в трахею тучных клеток, индуцируемой гистамином. Соединение согласно настоящему изобретению показало сильные снимающие зуд эффекты в модели гистамин-индуцируемого зуда и т.п., и сильные противовоспалительные эффекты в атопической модели, индуцируемой оксазолоном, который является одним из человеческих миметиков (J. Invest. Dermatol. 2008, 128, 79-86), по сравнению с соединением, раскрытым в WO 2010/030785.
Кроме того, соединение согласно настоящему изобретению показало такие же или более сильные эффекты в отношении улучшения внешнего вида кожи в атопической модели на мышах NC/Nga, которые являются человеческими миметиками (J. Clin. Invest. 1999, 104, 1097-1105), по сравнению с Такролимусом, который в настоящее время используется в качестве лекарственного средства для лечения атопии. Соответственно, можно ожидать, что это соединение будет очень эффективным для лечения пациентов, страдающих атопией.
Реферат
Изобретение относится к области органической химии, а именно к гетероциклическому соединению формулы 1, или к его рацемату, изомеру, или фармацевтически приемлемой соли, где Xи Хобозначают С; каждый из Хи Хнезависимо обозначает С или N, при условии, что один из Хи Хобозначает N; Rобозначает насыщенный 4-9-членный моно- или би-гетероциклил, содержащий 1-2 гетероатома (причем гетероатомы представляют собой N), где Rнезамещен или замещен 1-3 заместителями, выбранными из -NRRи R; или Rвыбран из -NRRи R; R, R, Rи Rмогут быть одинаковыми или разными; и каждый из них независимо выбран из -Н; -C-Салкила; -C-Сгалогеналкила; -C-Спергалогеналкила; -галогена (-F, -Cl, -Br, -I); -CN; -C-Салкокси; -C-Сгалогеналкокси; -C-Спергалогеналкокси; Салкенила; -С-Салкинила; -амино; -ОН; -нитро (-NO); -С-Сарила; и фурана; при условии, что, когда Хобозначает N, Rотсутствует; и когда Хобозначает N, Rотсутствует, каждый из Y, Y, Y, Yи Yнезависимо обозначает С или гетероатом (предпочтительно гетероатом, независимо выбранный из N, О и S), при условии, что по меньшей мере два из Y, Y, Y, Yи Yобозначают гетероатомы, независимо выбранные из N и О; каждый из Yи Yможет быть независимо замещен R; Yможет быть замещен -Н или -C-Салкилом; каждый из Rи Rнезависимо выбран из -Н; -C-Салкила; и -карбоксила (-СООН); Rобозначает -C-Салкил или -Сциклоалкил; и Rвыбран из -Н; -C-Салкила; и -Сциклоалкила; причем алкил и гетероциклил может быть независимо незамещенным или замещенным одним или более заместителями (например, 1-3 заместителями), выбранными из группы, состоящей из -С-Салкила, -С-Салкокси и -ОН. Также изобретение относится к конкретным соединениям и фармацевтической композиции на основе указанных соединений. Технический результат: получены новые гетероциклические соединения, полезные для ингибирования человеческого рецептора гистамина 4 (hH4R). 3 н. и 10 з.п. ф-лы, 13 табл., 144 пр.формула 1
Формула
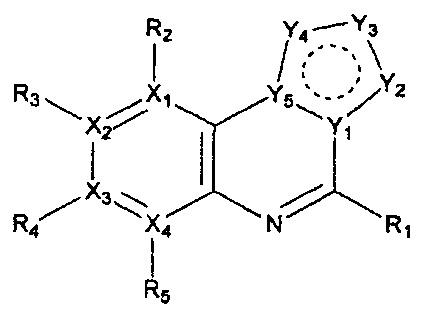
Комментарии