Соединения азаазулена - RU2524202C2
Код документа: RU2524202C2
Чертежи
Описание
Перекрестные ссылки
Настоящая заявка заявляет приоритет на предварительную заявку США №61/173,883, поданную 29 апреля 2009 и озаглавленную «Новые соединения азаазулена, обладающие множественными ингибиторными активностями в отношении киназы»; ее содержание включено здесь полностью в виде ссылки.
Предпосылки настоящего изобретения
Область применения настоящего изобретения
Настоящее изобретение касается медицины, в частности соединений азаазулена, которые модулируют активность протеинкиназы (PKs) и/или лечат рак.
Описание родственной технологии
Протеинкиназы (PKs) представляют собой ферменты, которые катализируют фосфорилирование специфических остатков тирозина, серина или треонина в клеточных протеинах. Указанные протеинкиназы (PKs) являются медиаторами трансдукции (преобразования) клеточных сигналов в такие регулирующие функции клеток, как пролиферация, дифференциация, рост, клеточный цикл, клеточный метаболизм, жизнеспособность клеток, апоптоз клеток, устранение повреждений ДНК, подвижность клеток и их ответ на микросреду. Дисрегулируемая активность PKs является частой причиной таких заболеваний, как ангиогенезис, рак, рост опухолей, метастазы опухолей, артериосклероз, возрастная дегенерация желтого пятна, диабетическая ретинопатия. воспалительные заболевания и/или заболевания, вызванные паразитами.
PKs можно разделить на 2 класса - протеинтирозинкиназы (PTKs) и серин/треонинкиназы (STKs). Протеинтирозинкиназы, которые катализируют передачу гамма-фосфата АТФ к остаткам тирозина в протеиновых субстратах, являются одним из ключевых ковалентных изменений, происходящих в микроклеточных организмах как результат внеклеточной связи в процессе эмбриогенеза и защиты зрелых тканей. Фосфорилирование тирозиновых остатков модулирует ферментную активность PTKs и восстановление нижележащей сигнальной системы протеинов. В клетках представлено 2 класса PTKs: трансмембранные рецепторы PTKs и нерецепторные PTKs. PTKs представляют собой главный компонент путей передачи сигналов клеток, каталитическая активность PTKs четко регулируется. Нерегулируемая активация этих ферментов через такие механизмы, как точечные мутации и избыточная экспрессия могут привести к различным формам рака, а также к состоянию злокачественной пролиферации. Важная роль PTKs для состояний здоровья или болезни также подчеркивается наличием аберраций в сигнальной системе PTK, имеющей место при воспалительных заболеваниях и диабете. Рецепторы фактора роста, обладающие РТК-активностью, известны как рецепторы тирозинкиназы (RTKs). Они включают большое семейство трансмембранных рецепторов с многообразной биологической активностью. Внутриклеточные домены RTKs можно разделить на 2 класса: на домены, содержащие растяжку аминокислот, отделяющих домен киназы, и на такие, в которых домены киназы непрерывны. Активация киназы достигается связыванием лиганда с внеклеточным доменом, который индуцирует димеризацию или олигомеризацию рецепторов. Активируемые таким образом рецепторы способны к автофосфорилированию остатков тирозина вне каталитического домена, которое происходит путем перекрестного фосфорилирования. Результатами такого автофосфорилирования являются стабилизация конформации активного рецептора и создание фосфотирозиновых стыковочных сайтов для протеинов, преобразующих сигналы внутри клетки. Сигнальная система протеинов, которые связаны с внутриклеточным доменом рецептора тирозинкиназы зависимым от фосфотирозина образом, включают: RasGAP, PI3-киназу, фосфолипазу C, фосфотирозинфосфатазу SHP и адаптор протеинов, такой как She, Grb2 и Crk.
ERFR - рецептор эпидермального фактора роста - принадлежит к семейству рецептора тирозинкиназы млекопитающих, он состоит из 4 членов ERFR (ErB1), ErB2, ErB3 и ErB4. ERFR представляет собой 1186 аминокислотный остаток трансмембранного гликопротеина. Он содержит внеклеточный лигандсвязывающий домен, внутриклеточный домен тирозинкиназы и СООН терминальную область, содержащую сайты автофосфорилирования. Связывание специфичных лигандов (таких как EGF, трансформирующий фактор роста, бета-целлулин, гепаринсвязывающий EGF, эпирегулин или амфирегулин) приводит к фосфорилированию большого количества тирозиновых остатков на COOH-терминальном хвосте. При этом запускаются в действие клеточные сигнальные пути, которые регулируют фундаментальные клеточные процессы типа пролиферации, миграции, дифференциации и продолжительность существования. ERFR обладает избыточной экспрессией во многих типах опухолевых клеток типа клеток мочевого пузыря, легких, желудка, молочной железы, мозга, головы и шеи, шейки матки, яичника, эндометрия и т.д. Аномально высокая активность EGFR может быть характеристикой немелкоклеточного рака легких, молочной железы, яичника, мочевого пузыря, простаты, слюнных желез, поджелудочной железы, эндометрия, немелкоклеточного колоректального рака, рака почки, рака шеи и головы, а также мультиформной глиобластомы. Ингибитор тирозинкиназы, специфичный к EGFR, может быть использован для лечения раковых заболеваний, имеющих аномально высокий уровень активности киназы EGFR, а также для лечения заболеваний с нарушениями киназы EFGR.
Одно из подсемейств рецепторов тирозинкиназы (RTK) называют группой производного рецептора фактора роста тромбоцитов (PDGFR), которая включает PDGFR-альфа, PDGFR-бета, CSFIR, c-KIT и c-fms. Эти рецепторы состоят из гликозилированных внеклеточных доменов, составленных из различного количества иммуноглобулиноподобных петель, а также из внутриклеточного домена, в котором домен тирозинкиназы прерывается последовательностями неродственных аминокислот. Сигналы PDGFR индуцируют экспрессию проангиогенных сигналов в клетки эндотелия, стимулируя в дальнейшем опухолевый ангиогенезис. Сигнальный путь PDGFR может играть важную роль в пролиферации клеток, миграции клеток и ангиогенезисе, а также может опосредовать высокое давление интерстициальной жидкости в опухолях.
Другой группой, которую из-за ее схожести с подсемейством PDGFR иногда относят к последней группе, является подсемейство рецептора киназы печени эмбриона (flk). Предполагают, что эта группа построена из вкладки домена киназы - рецептора киназы-1 печени эмбриона (KDR/FLK-1, VEGF-R2), flk-1R, flk-4 и семейства, подобного тирозинкиназе (flk-1). Аномально высокая активность PDGFR может быть характеристикой рака стромы желудка, мелкоклеточного рака легкого, мультиформной глиобластомы и рака простаты. Ингибитор тирозинкиназы, специфичный к PDGFR, может быть использован для лечения раковых заболеваний, имеющих аномально высокий уровень активности киназы PDGFR, а также для лечения заболеваний с нарушениями киназы PDFGR.
FLT-3 представляет собой класс III рецепторов тирозинкиназы (RTK), структурно относящийся к PDGFR и колониестимулирующему фактору 1 (CSF). Эти рецепторы тирозинкиназы содержат во внеклеточной области 5 иммуноглобулиноподобных доменов, а также внутриклеточный домен тирозинкиназы, расщепляемый надвое путем специфического гидрофильного внедрения. Экспрессия FLT-3 была описана на CD34-позитивных клетках спинного мозга, соответствующих мультипотенциальным, миелоидным и B-лимфоидным клеткам-предшественникам, и на клетках моноцитов. Экспрессия FLT-3 ограничена клетками печени эмбриона, обладающими высокими уровнями экспрессии CD34. Функция рецептора FLT-3 может быть определена по активности его лиганда FL. FL представляет собой рано действующий фактор, он поддерживает жизнеспособность, пролиферацию и дифференциацию примитивных кроветворных клеток-предшественников. Связывание лиганда с FLT-3 способствует димеризации рецептора и последующей передаче сигнала путем фосфорилирования многочисленных протеинов цитоплазмы, включая SHC, SHP-2, SHIP, Cb1, Cb1-b, Gab1 и Gab2, а также активации нескольких путей передачи сигналов типа Ras/Raf/MARK и каскадов PI3 киназы. Внутренняя дупликация тандема (ITD) и/или вставки и (редко) делеции в FLT-3 гене в 20-25% случаев вовлечены в острую миелоидную лейкому (AML). Такая дуплицированная последовательность принадлежит экзону 11, но иногда включает интрон 11 и экзон 12. Наиболее часто используемой номенклатурой является FLT3-ITD. Более адекватным, ввиду сильной степени гетерогенности молекулярной структуры, представляется термин FLT3-LM (длинные мутации). Описано также, что он вовлечен в 5-10% стойкую анемию с миелодиспластическим синдромом (MDS) при избытке бластов (RAEB 1 и RAEB 2), а в редких случаях - в острую лимфобластную анемию (ALL).
Ингибитор тирозинкиназы, специфичный к FLT-3, может быть использован для лечения раковых заболеваний, имеющих аномально высокий уровень активности киназы FLT-3, а также для лечения заболеваний с нарушениями киназы FLT-3.
C-KIT, SCFR (рецептор фактора роста стволовых клеток, Stem Cell Factor Receptor) известен как рецептор III типа тирозинкиназы, структурно относящийся к CSF-1R, PDGFR и FLT-3, содержащим внеклеточный домен с 5 Ig-подобными петлями, трансмембранный домен с высокой степенью гидрофобности, а также внутриклеточный домен с активной тирозинкиназой, расщепленной вставкой киназы (KI) в АТФ-связывающую область и в домен фосфотрансферазы. C-KIT экспрессирован на мембране плазмы клетки в кроветворных стволовых клетках, тучных клетках, меланоцитах и на клеточных линиях зародышевых клеток. Рецептор SCF/MGF, обладающий активностью протеинтирозинкиназы (РТК), связывая лиганд SCF, индуцирует димеризацию рецептора, автофосфорилирование и преобразование сигнала посредством молекул, содержащих 8Н2-домен. При аномальной экспрессии у большинства пациентов выявляется гиперплазия тучных клеток в спинном мозге, печени, селезенке, лимфоузлах, желудочно-кишечном тракте и коже, и усиление функции мутации. Признается, что на клинические характеристики роста недоброкачественных кроветворных клеток оказывает влияние время, расположение эпизодов мутации, а также количество ассоциированных патологических изменений. Ингибитор тирозинкиназы, специфичный к c-KIT, может быть использован для лечения раковых заболеваний, имеющих аномально высокий уровень активности киназы c-KIT, а также для лечения заболеваний с нарушениями киназы c-KIT.
Другим членом семейства рецепторов фактора роста тирозинкиназы является подгруппа рецептора сосудисто-эндотелиального фактора роста (VEGRF). VEGRF представляет собой димерный гликопротеин, аналогичный PDGFR, но обладающий другими биологическими функциями и другой специфичностью к клеткам-мишеням in vivo. В частности, в настоящее время полагают, что VEGRF играет существенную роль в васкулогенезе и ангиогенезе. Ангиогенез существенен для роста и жизнеспособности опухолей. Различаются 3 рецептора VEGRF - VEGRF-1, -2 и -3. Каждый из них по отдельности вносит свой вклад в процесс ангиогенеза. Полагают, что VEGRF-1 играет роль в связывании VEGF с VEGRF-2 в процессе ангиогенеза. VEGRF-2 (KDR) стимулирует пролиферацию, миграцию и жизнеспособность клеток эндотелия в процессе ангиогенеза, и его считают рецептором, важным для ангиогенеза. VEGRF-3 стимулирует пролиферацию, миграцию и жизнеспособность клеток эндотелия в процессе лимфагиогенеза, который в свою очередь ускоряет метастазирование. Несмотря на эти кажущиеся различимыми роли, все VEGRF до некоторой степени обладают перекрывающимися функциями, приводя к существенной избыточности. Поэтому более полное подавление ангиогенеза может гарантировать ингибирование всех идентифицированных рецепторов VEGF. Ингибитор тирозинкиназы, специфичный по отношению к VEGRF, может быть использован для лечения солидных опухолей и заболеваний, связанных с сосудистыми нарушениями.
c-Met (рецептор фактора роста гепатоцитов) представляет собой обладающий высоким сродством рецептор HGF/SF, мультифункционального цитокина. При связывании с лигандом MET димеризует и трансфосфорилирует остатки тирозина в C-терминальном домене, который затем взаимодействует с элементами различных путей передачи сигнала. Они включают ассоциированное с Grb-2 связующее 1, фосфоиноситид 3' киназу и c-Src. Было показано, что в физиологичных условиях в зависимости от клетки-мишени передача сигнала MET-HGF/SF оказывает воздействие на большое количество веществ с биологической активностью. Эта активность меняется от пролиферации клеток до формирования клеток (морфогенез) и их подвижности. Координация этих разнообразных активностей составляет генетическую программу инвазивного роста, делающую возможным разветвленный морфогенез (образование эпителиальных тубулярных структур), миграцию миобластов и разветвление аксонов. MET/HGF клетки-мишени включают клетки эпителия и мезенхимальные клетки, кроветворные клетки, миобласты, спинальные двигательные нейроны. Передача сигнала MET-HGF/SF также важна для нормального развития: эмбрионы мыши, обладающие нулевой мутацией в обоих HGF аллелях, погибают в середине срока беременности и демонстрируют дефектное формирование печени. MET и его лиганд - фактор роста гепатоцитов/фактор рассеяния (HGF/SF) - экспрессированы в многочисленные ткани, хотя преимущественно - в клетки эпителия и мезенхимальные клетки соответственно. MET амплифицирован и обладает избыточной экспрессией в многие типы опухолей, включая опухоли почек, щитовидной железы, поджелудочной железы и остеосаркому. Ингибитор тирозинкиназы, специфичный к с-МЕТ, может быть использован для лечения раковых заболеваний, имеющих аномально высокий уровень активности киназы с-МЕТ, а также для лечения заболеваний с нарушениями киназы с-МЕТ.
RET представляет собой рецептор тирозинкиназы, лигандами которого являются нейротропные факторы глиальной клеточной линии, производными семейства нейротропных факторов (GDNF). Активация RET опосредована различными рецепторами гликозилфосфатидил-инозитола. У человека выявлены 3 изоформы RET - длинная изоформа (RET 51):1114 аминокислот, средняя изоформа (RET 43): 1106 аминокислот и короткая изоформа (RET 9): 1072 аминокислоты. В основном RET экспрессирована в опухоли с исходным нервным валиком - медуллярную тироидную карциному, феохромоцитому и нейробластому. У эмбриона человека RET экспрессирована в популяцию клеток мозга с нервным валиком, а также в развиваемые нервную и урогенитальную системы. Экспрессия RET обнаруживается в некоторых клеточных линиях с нервным валиком, селезенке, тимусе, лимфоузлах, слюнных железах, а в последнее время - в здоровой тироидной ткани, аденоме щитовидной железы, а также как в папиллярных, так и в фоликуллярных неопластах щитовидной железы. Ингибитор тирозинкиназы, специфичный к RET, может быть использован для лечения раковых заболеваний, имеющих аномально высокий уровень активности киназы RET, а также для лечения заболеваний с нарушениями киназы RET.
c-ABL (изменяемый гомолог вирального онкогена лейкемии мышей Абельсона, v-abi Abelson murine leukemia viral oncogene homolog) демонстрирует постоянную активность ядер и цитоплазмы, которой управляют 3 сигнала с ядерной локализацией (NLS) и 1 сигнал ядерного экспорта (NES) в непосредственной близости от C-терминальной области. BCR/ABL имеет цитоплазменную локализацию, а все слитые белки BCL-ABL, как было показано, обладают онкогенным потенциалом. Все 3 гибридных белка имеют повышенную активность протеинкиназы по сравнению с ABL:3BPI (связывающий белок), который связывает нормальный ABL на домене SH3, что предотвращает активацию SHL. Ядерный и цитоплазматический ABL может иметь различные функции. 1-Ядерный c-ABL играет важную роль в регуляции клеток после повреждения ДНК. Все агенты, индуцирующие повреждение ДНК, активируют ядерную c-ABL киназу, причем эта активация происходит в режиме, зависимом от ATM, и в присутствии р53-гомолога белка р73. Последний, после повреждения ДНК, физически ассоциирован с c-ABL посредством SH3 домена из c-ABL. Повреждение ДНК одновременно активирует также путь р53, приводя к активации Робертсоновской транслокации (Rb), которая индуцирует прекращение роста и защищает клетки от апоптоза. Точный механизм апоптоза, индуцируемого c-ABL, неизвестен. Было показано, что ядерный захват BCR-ABL также индуцирует апоптоз в лейкозных клетках. 2-Цитоплазменная c-ABL: возможная функция подачи сигнала адгезии в фибробластах как отток с-ABL из ядер в цитоплазму после адгезии. Ингибитор тирозинкиназы, специфичный к c-ABL, может быть использован для лечения раковых заболеваний, имеющих аномально высокий уровень активности киназы с-ABL, а также для лечения заболеваний с нарушениями киназы c-ABL.
TIE (тирозинкиназа с иммуноглобулиноподобным и EGF-подобным доменом) можно разделить на 2 подгруппы. TIE-1 (тирозинкиназа с Ig и EGF гомологичным доменом 1) и подсемейство TIE-2/Tek содержат рецептор тирозинкиназы с уникальными структурными характеристиками:
2 иммуноглобулиноподобных домена, фланкирующих три EGF-подобные домена эпидермального фактора роста, за ними во внеклеточной области следуют 3 повтора фибронектинового типа, а в цитоплазмической области - расщепленный домен тирозинкиназы. Эти рецепторы экспрессированы прежде всего на эндотелиальных и кроветворных клетках-предшественниках и играют существенную роль в ангиогенезе, васкулогенезе и гемопоэзе. TIE-1 кДНК человека кодирует 1124 аминокислотный остаток - предшественник белка с 18 остатками мнимых сигнальных пептидов, домен 727 внеклеточных остатков и домен 354 цитоплазмических остатков. Были идентифицированы 2 лиганда, ангиопоэтин-1 (Angi) и ангиопоэтин-2 (Ang2), которые связывают TIE-1 с высокой степенью сродства. Сообщалось, что Ang2 действует как антагонист Angi. Ингибитор тирозинкиназы, специфичный к TIE, может быть использован для лечения солидных опухолей и заболеваний, связанных с сосудистыми нарушениями.
FGFR (рецепторы фактора роста фибробластов) состоит из домена внеклеточного лиганда, содержащего 3 иммуноподобных домена, 1 трансмембранный спиральный домен, а также внутриклеточный домен, обладающий тирозинкиназной активностью. Указанные факторы роста фибробластов представляют собой самое большое семейство лигандов факторов роста, содержащее 23 члена. FGFR принимают участие в аналогичной последовательности, характеризующейся 3 внеклеточными иммуноглобулиноподобными доменами (IgI, IgII и IgIII), трансмембранным сегментом однократного прохождения сигнала, а также расщепленным доменом (TK1/TK2). Важную роль в патогенных мутациях FGFR играют бессмысленный кодон и все неудачные передачи функций мутированному белку. Некоторые мутации являются в высокой степени возвратными. Механизмом приобретения функций, выявленных для мутаций FGFR2, являются: (а) селективное усиление сродства к FGF-связыванию, (б) запрещенная специфичность FGF-связывания, (в) независимая ковалентная димеризация FGF, а также (г) эктопическая экспрессия сплайсиформ. Эти механизмы объясняют доминантную наследственность всех ассоциированных фенотипов. Ингибитор тирозинкиназы, специфичный к FGFR, может быть использован для лечения раковых заболеваний, имеющих аномально высокий уровень активности киназы FGFR, а также для лечения заболеваний с нарушениями киназы FGFR.
Инсулиноподобный фактор роста 1 (IGF1) рассматривался потенциальным кандидатом для лечения сердечной недостаточности. Однако некоторые испытания на животных и клинические опыты поставили под вопрос, действительно ли полезно хроническое повышение инсулиноподобного фактора роста. Этот нежелательный результат могут объяснить вторичные эффекты повышенного уровня сывороточного IGF1 в других тканях. Целью настоящего исследование являлось изучение роли IGF1 в миоцитах сердца при отсутствии вторичных эффектов, а также объяснение путей передачи сигналов и регулирующих транскрипционных воздействий рецептора IGFI(IGFIR). Активацией рецептора IGF1 является жизнеспособность и пролиферации в мито-компетентных клетках, а также рост (гипертрофию) в тканях, таких как мышцы скелета и сердечная мышца. Путь передачи сигнала особенно важен в период нормального развития ткани молочных желез при беременности и лактации. В этот общий процесс вовлечены несколько факторов роста и гормоны, и полагают, что IGF1R играет роль в дифференциации клеток и ключевую роль в подавлении апоптоза вплоть до того момента, пока не будет завершено отнятие ребенка от груди. IGF1 вовлечен в некоторые формы рака, наиболее заметно - в рак молочной железы. Он вовлечен также в рак молочной железы за счет увеличения потенциала метастазов исходной опухоли, препятствуя ее способности поддерживать васкулизацию. Ингибитор тирозинкиназы, специфичный к IGFR, может быть использован для лечения рака с аномально высоким уровнем активности киназы IGFR и заболеваний, связанных с повреждениями киназы IGFR.
Киназы типа c-Src, с-Abl, киназы митогенактивированного белка (MAP), фосфотидилинозитол-3 -киназы (PI3K) АКТ и рецептора эпидермального фактора роста (EGF) обычно активируются в раковых клетках, и известно, что они вносят вклад в онкогенез. Многие из них находятся на одном и том же пути передачи сигналов, например, члены семейства киназы HER1, HER3 и HER4 для того, чтобы поддерживать пролиферацию клеток, передают сигналы через киназу митогенактивированного белка и фосфотидилинозитол-3-киназу.
TrkA (тропомиозинсвязанная киназа А) является каталитическим рецептором нейротрофина, фактора роста нервов (Nerve Growth Factor, NGF), и таким образом она опосредует многочисленные действия указанного фактора, включая нейронную дифференциацию и жизнеспособность. Рецептор TrkA является частью большого семейства рецепторов тирозинкиназы.
Нарушение работы протеинтирозинкиназы (РТК) включает такие расстройства, как рак, артрит, диабетическая ретинопатия, рестеноз, цирроз печени, артериосклероз, разрастание кровеносных сосудов, гломеронефрит, диабетическая невропатия, синдромы тромбозной микроангиопатии, отторжение трансплантата, аутоиммунные заболевания и гипериммунные расстройства.
Виды рака включают, без ограничения, карциномы желчного пузыря, молочной железы, толстой кишки, почки, печени, головы и шеи, яичника, поджелудочной железы, желудка, шейки матки, щитовидной железы, простаты, кожи, гемопоэтический рак лимфоидного происхождения (например, лейкемия, острая лимфоцитная лейкемия, острая лимфобластная лейкемия, лимфома В-клеток, лимфома Т-клеток, лимфома Ходкинса, лимфома волосковых клеток и лимфома Буркетта), гемопоэтический рак миелоидного происхождения (например, миелогенная лейкемия, хроническая миелогенная лейкемия, множественная миелогенная лейкемия, миелодиспластический синдром и промиелоцитная лейкемия), рак мезенхимального происхождения (например, фибросаркома и рабдомиосаркома), рак центральной или периферической нервной системы (например, астроцитома, нейробластома, глиома и невриома), меланома, семинома, тератосаркома, остеосаркома, фолликулярный рак щитовидной железы и саркома Капоши.
Краткое содержание настоящего изобретения
Один вариант настоящего изобретения характеризует азаазуленовые соединения формулы (I):

в которой
один
каждый из A1, А2, А3, А4, А5, А6, А7, и A8 независимо представляет собой углерод или азот;
A1, A2, А3, А4, А5, А6, A7, и A8 вместе с азотом связаны с A1 и A8, образуя 6,5-слитый гетероцикл, имеющий π-электронов;
R1 представляет собой О, OR, S, SR, NH2, NHR, NRR', NH, or NR; каждый из R2, R3, R4, R5, R6, R7, R8, R9, R10, R11, R12, и R13, независимо представляет собой O, Н, галоид, C1-С10алкил, C2-С10алкенил, C2-С10алкинил, C3-С20циклоалкил, C3-С20циклоалкенил, C1-C20гетероциклоалкил, C1-C20гетероциклоалкенил, C3-C20арил, C1-C20гетероарил, NO2, NO, N3, SCN, CN, OCN, OR, OC(O)R, OC(S)R, OC(S)OR, ОС(O)SR, OC(S)SR, OC(O)NRR', OC(S)NRR”, ONRR', OS(O)R, OS(O)2R, SR, SC (O)R, SC(S)R, SC(S)OR, SC(O)SR, SC(S)SR, SC(O)NRR', SC(S)NRR', S(O)R, S(O)2R, S(O)NRR', S(O)2NRR', S(O)OR, S(O)2OR, NCO, NCS, NRR', N(R)-C(O)R”, N(R)-C(O)OR', N(R)-C(S)R', N(R)-C(S)OR', N(C(O)R)-C(O)R', N(R)-S(O)R', N(R)-S(O)OR', N(R)-S(O)2R', N(R)-S(O)2OR', N(R)-OR, N(OR)-C(O)R', N(OR)-C(O)OR', N(OR)-C(S)R', N(OR)-C(S)OR', N(OR)-C(S)SR', N(OR)-S(O)R', N(OR)-S(O)OR', N(OR)-S(O)2R', N(OR)-S(O)2OR', C(O)R, C(O)OR, C(O)NRR', C(O)SR, C(S)R, C(S)OR, C(S)NRR', C(S)SR, C(NR)-R', C(NR)-OR', C(NR)-NRR”, C(NR)-SR, C(NOR)-R', C(NOR)-OR', C(NOR)-NR'R”, или C(NOR)-SR; или R2 и R3, R3 и R4, R4 и R5, R5 и R6, R6 и R7, R8 и R9, R9 и R10, R10 и R11, R11 и R12, или R12 и R13 вместе с атомами, к которым они присоединены, представляют собой C3-C20циклоалкил, C3-C20циклоалкенил, C1-C20гетероциклоалкил, C1-C20гетероциклоалкенил, C3-C20арил, или C1-C20гетероарил; в которой каждый из R, R', и R” независимо представляет собой Н, галоид, C1-C10алкил, C2-C10алкенил, C2-C10алкинил, C3-C20циклоалкил, C3-C20циклоалкенил, C1-C20гетероциклоалкил, C1-C20гетероциклоалкенил, C3-C20арил, или C1-C20гетероарил, или R и R', R и R” или R' и R” вместе с атомами, к которым они присоединены, представляют собой C1-C20гетероциклоалкил или C1-C20гетероциклоалкенил, где каждый из A1, А3, А4, A5, А6, А7, и A8представляет собой углерод, А2 представляет собой азот,
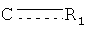

Если

Термин «арил» касается неразветвленной или разветвленной углеводородной цепи, радикальной группы, содержащей от 1 до 12 атомов углерода и присоединенной к остальной молекуле простой связью, например, метил, этил, н-пропил, 1-метилэтил (изопропил), н-бутил, н-пентил, 1,1-диметилэтил (трет-бутил) и им подобные.
Термин «алкенил» касается линейного или разветвленного углеводородного фрагмента, который содержит по меньшей мере одну двойную связь, включает от 2 до 20 атомов углерода и присоединен к остальной молекуле простой или двойной связью, например, этенил, проп-1-енил, бут-1-енил, пент-1-енил, пента-1,4-диенил и им подобные.
Термин «алкинил» касается линейного или разветвленного углеводородного фрагмента, который содержит по меньшей мере одну тройную связь, включает от 2 до 10 атомов углерода и присоединен к остальной молекуле простой или тройной связью, например, этинил, проп-1-инил, бут-1-инил, пент-1-инил, пент-3-инил и им подобные.
Термин «циклоалкил» касается насыщенного, моно-, би- или трициклического углеводородного фрагмента, который содержит от 3 до 20 атомов углерода, насыщен и присоединен к остальной молекуле простой связью, например, циклопропил, циклобутил, циклогексил, декалинил, норборнан, норборнен, адамантил, бицикло[2.2.2]октан и им подобные.
Термин «циклоалкенил» касается неароматического, моно-, би- или трициклического углеводородного фрагмента, который содержит от 3 до 20 атомов углерода и имеет по меньшей мере одну двойную связь, например циклогексенил.
Термин «гетероциклоалкил» касается насыщенного, моно-, би- или трициклического фрагмента, который содержит от 1 до 20 атомов углерода и по меньшей мере один гетероатом (например, N, О, или S), типа 4-тетрагидропиранила.
Термин «гетероциклоалкенил» касается неароматического, моно-, би- или трициклического фрагмента, который содержит от 1 до 20 атомов углерода, по меньшей мере один гетероатом (например, N, О, или S), а также по меньшей мере одну двойную связь, типа пиранила.
Термин «арил» касается углеводородного фрагмента, имеющего от 6 до 30 атомов углерода, а также содержит один и более ароматических циклов. Примеры арильных фрагментов включают фенил (Ph), фенилен, бифенил, нафталин, пиренил, антрил, азуленил и фенантрил.
Термин «гетероарил» касается фрагмента, который включает от 1 до 30 атомов углерода, а также по меньшей мере один или более ароматических циклов, содержащих по меньшей мере один гетероатом (например N, О, или S). Примеры гетероарильных фрагментов включают (но ими не ограничены): акридинил, азаазуленил, бензимидазолил, бензиндолил, бензозоксазинил, бенз[4,6]имидазо[1,2-а]пиридинил, бензофуранил, бензотиадиазолил, бензотиазолил, бензотиофенил, бензотриазолил, бензотиопиранил, бензоксазинил, бензоксазолил, бензотиазолил, бета-карбонил, карбазолил, циннолинил, дибензофуранил, фуранил, имидазолил, имидазопиридинил, имидазотиазолил, индазолил, индолизинил, индолил, изобензотиенил, изоиндолинил, изохинолинил, изотиазолидинил, изатиазолил, нафтиридинил, октагидроиндолил, октагидроизоиндолил, оксазолидинонил, оксазолил, оксиранил, перимидинил, фенантридинил, фенантролинил, фенарсазинил, феназинил, фенотиазинил, феноксазинил, фтализинил, птеридинил, пуринил, пиразинил, пиразолил, пиридазинил, пиридинил, пиридопиридинил, пиримидинил, пирролил, хиназолинил, хинолинил, хиноксалинил, тетразолил, тиадиазолил, тиазолил, тиофенил, триазинил и триазолил.
Если иного не оговорено, то упоминаемые здесь алкил, алкенил, алкинил, циклоалкил, циклоалкенил, гетероциклоалкил, гетероциклоалкенил, арил и гетероарил включают как замещенные, так и незамещенные фрагменты. Возможные заместители циклоалкила, циклоалкенила, гетероциклоалкенила, арила и гетероарила включают (но ими не ограничены): C1-C20алкил, C2-C20алкенил, C2-C20алкинил, C3-C20циклоалкил, C3-C20циклоалкенил, C1-C20гетероциклоалкил, C1-C20гетероциклоалкенил, C1-C10алкокси, C3-C30арил, C3-C30арилокси, C1-C30гетероарил, C1-C30гетероарилокси, амино, C1-C20алкиламино, C1-C20диалкиламино, C3-C20ариламино, C6-C40диариламино, C1-C10алкилсульфониламино, C3-C20арилсульфониламино, C1-C10алкилимино, C3-C20арилимино, C1-C20алкилсульфонилимино, C3-C20арил-сулофонилимино, гидроксид, галоген, тио, C1-C10алкилтио, C3-C20арилтио, C1-C10алкилсульфонил, C3-C20арилсульфонил, ациламино, аминоацил, аминотиоацил, амидино, гуанидин, уреидо, циано, нитро, нитрозо, азидо, ацил, тиоацил, ацилокси, карбоксил, и эфиры карбоновых кислот. С другой стороны, возможные заместители алкила, алкенила или алкинила включают все цитируемые выше заместители. Циклоалкил, циклоалкенил, гетероциклоалкил, гетероциклоалкенил, арил и гетероарил также могут быть слиты друг с другом.
Другой вариант настоящего изобретения характеризует способ лечения рака. Указанный способ включает введение нуждающемуся в этом субъекту эффективного количества одного или более азаазуленовых соединений с приведенной выше формулой (1). Примеры раковых заболеваний включают лейкемию (например, острая миелогенная лейкемия), рак желудочно-кишечного тракта (например, стромальная опухоль желудочно-кишечного тракта), рак почки (например, метастазирующая карцинома клеток почки), или рак легких (например, мелкоклеточный рак легких).
Термин «лечение» касается введения одного или более азаазуленовых соединений субъекту, у которого имеется описанное выше заболевание, симптомы такого заболевания или предрасположенность к нему, для того, чтобы обеспечить некоторый терапевтический эффект, например, лечение, ослабление, прекращение или предотвращение описанного выше заболевания, его симптомов или предрасположенности.
Другой вариант настоящего изобретения охватывает фармацевтический состав, который содержит эффективное количество по меньшей мере одного из упомянутых выше азаазуленовых соединений, а также фармацевтически приемлемый носитель.
Упомянутые выше азаазуленовые соединения включают как сами соединения, так и их соли, пролекарства, сольваты, комплексы или производные с радиоизотопными метками. Такая соль может быть образована, например, анионом и положительно заряженной группой (например, аминогруппой) азаазуленового соединения. Подходящие анионы включают хлорид, бромид, иодид, сульфат, нитрат, фосфат, цитрат, метансульфонат, трифторацетат, ацетат, малеат, тозилат, тартрат, фумарат, глюконат, лактат, глютарат и малеат.
Аналогично соль может быть образована также катионом и отрицательно заряженной группой (например, карбоксилат) азаазуленового соединения. Подходящие катионы включают ион натрия, ион калия, ион магния, ион кальция, а также катион аммония, типа иона тетраметиламмония. Указанные азаазуленовые соединения включают также соли, содержащие четвертичный атом азота. Примеры пролекарств включают сложные эфиры и другие фармацевтически доступные производные, которые при введении их субъекту способны обеспечить активные азаазуленовые соединения. Сольватами называют молекулы, образованные активным азаазуленовым соединением и фармацевтически приемлемым растворителем. Примеры фармацевтически приемлемых растворителей включают воду, этанол, изопропанол, этилацетат, уксусную кислоту и этаноламин. Комплекс может быть образован активным азаазуленовым соединением и комплексообразующим агентом (например, циклодекстрины или циклофаны) или активным азаазуленовым соединением и неорганическим катионом (например, катионы цинка, магния, кальция, серебра или меди).
В границы настоящего изобретения входит также состав, содержащий одно или более описанных выше азаазуленовых соединений, для их применения при лечении рака, а также использование такого состава для изготовления лекарственного средства, предназначенного для указанного выше лечения. Упоминаемые выше раковые заболевания могут включать острую миелоидную лейкемию (AML).
Настоящее изобретение обеспечивает также способ ингибирования активности протеинкиназы у субъекта. Этот способ включает введение в клетку эффективного количества одного или более азаазуленовых соединений, описанных выше формулой (I). Примеры протеинкиназы включают АМРК, BLK, CSF1R, FGFR, FGR, FLT3, KDR, KIT, LCK, LYN, МАР4К5, NTRK, PHKG1, RET, SRC, STK и YES1. Помимо этого, при способе ингибирования активности протеинкиназы у субъекта по настоящему изобретению таким субъектом может быть раковая клетка, и указанная раковая клетка может включать клетку острой миелоидной лейкемии.
В приведенных далее вариантах дано подробное описание настоящего изобретения со ссылкой на прилагаемые фигуры.
Краткое описание фигур
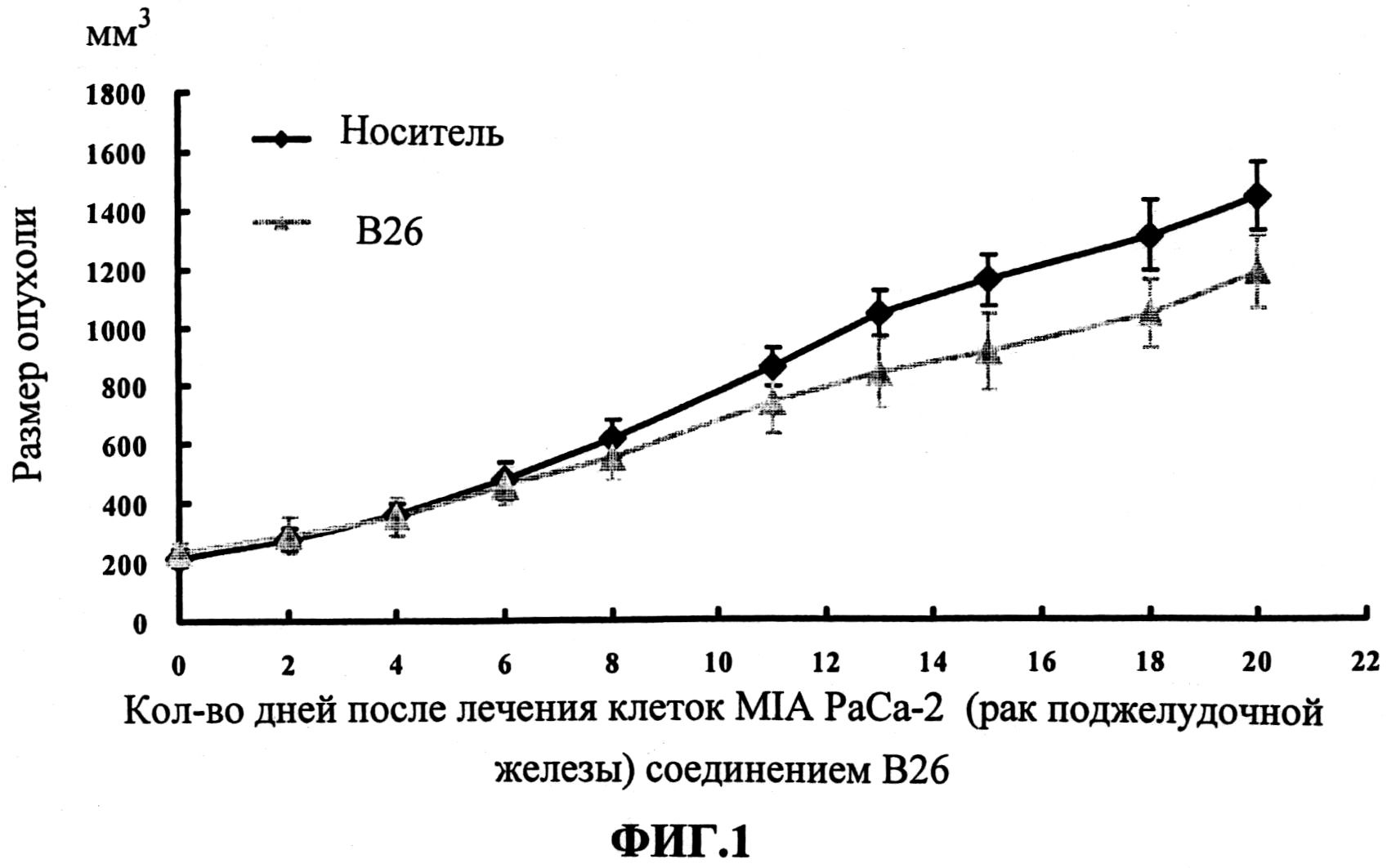
Настоящее изобретение может быть более полно понято при чтении изложенного ниже подробного описания и примеров со ссылкой на прилагаемые фигуры. На этих фигурах показано: фиг.1 демонстрирует средний объем опухолей MV4-11 подкожного рака, модель для мышей BALB/c после введения В26 или носителя.
Подробное описание изобретения
Приведенное описание представляет собой наилучший из предполагаемых способов реализации настоящего изобретения. Это описание сделано для иллюстрации основных принципов изобретения и его не следует использовать в предельном смысле. Границы настоящего изобретения наилучшим образом определены в прилагаемой формуле изобретения.
Азаазуленовые соединения по настоящему изобретению можно получить хорошо известными способами. Например, приведенная далее схема иллюстрирует типичный путь синтеза азаазуленовых соединений по настоящему изобретению.
Промежуточные соединения для конструирования азаазуленовых ядер по настоящему изобретению можно синтезировать по следующей схеме:

По этому изобретению азаазуленовые соединения, которые содержат 6,5-конденсированный гетероцикл индольного типа можно синтезировать по следующей схеме:
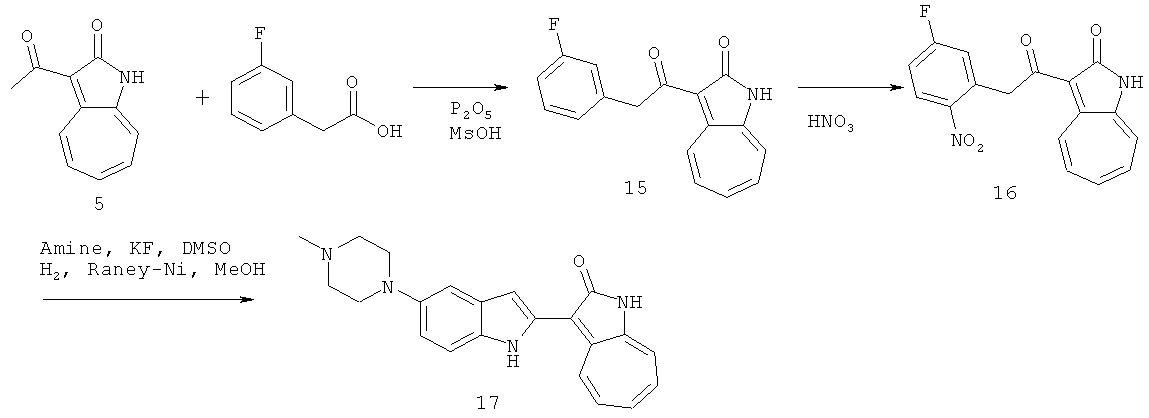
По этому изобретению азаазуленовые соединения, которые содержат 6,5-конденсированный гетероцикл бензимидазольного типа можно синтезировать по следующей схеме:
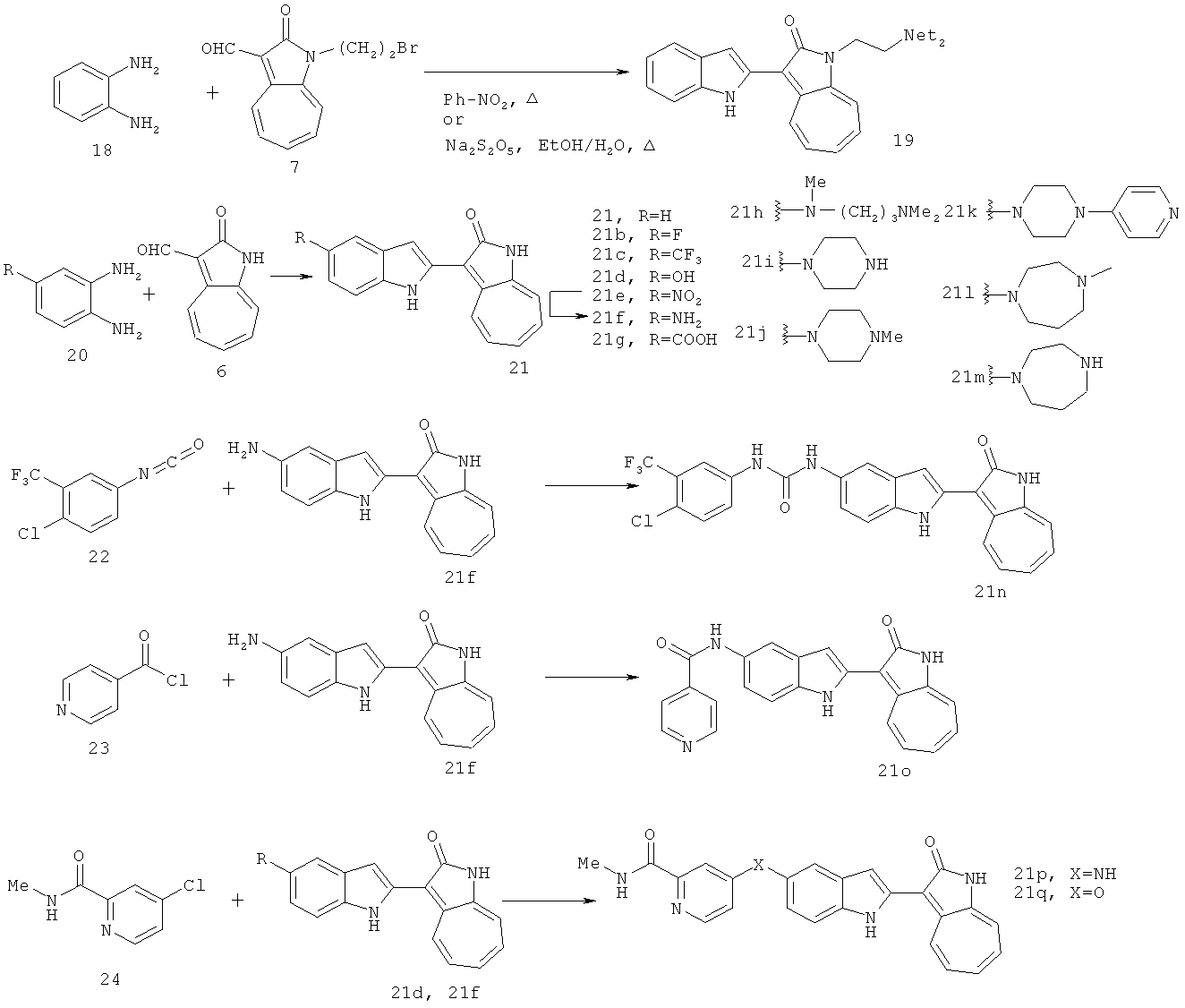
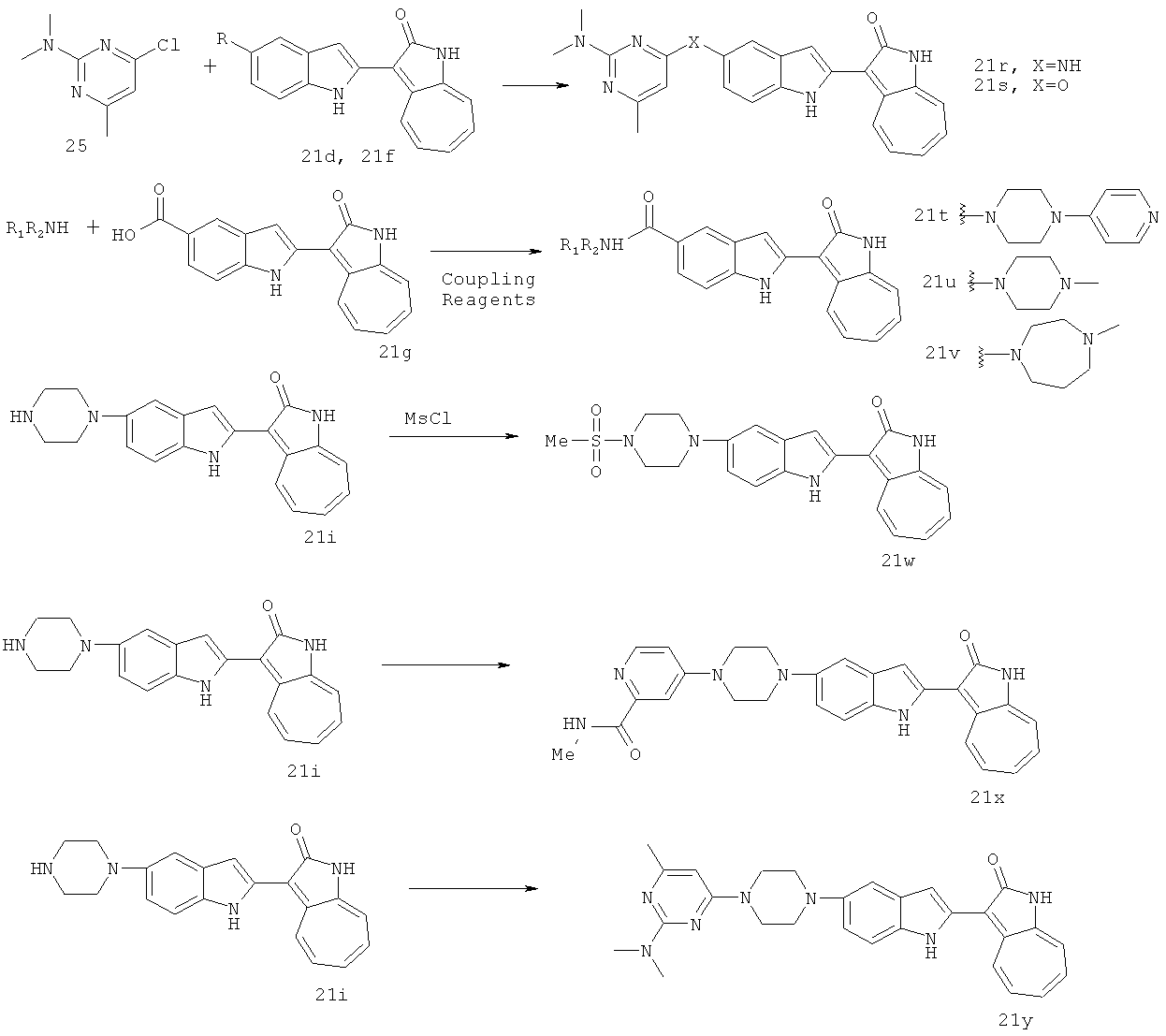
По этому изобретению азаазуленовые соединения, которые содержат 3H-имидазо[4,5-6]пиридин, 3H-имидазоо[4,5-с]пиридин, а также 6,5-конденсированный гетероцикл пуринового типа, можно синтезировать по следующей схеме:
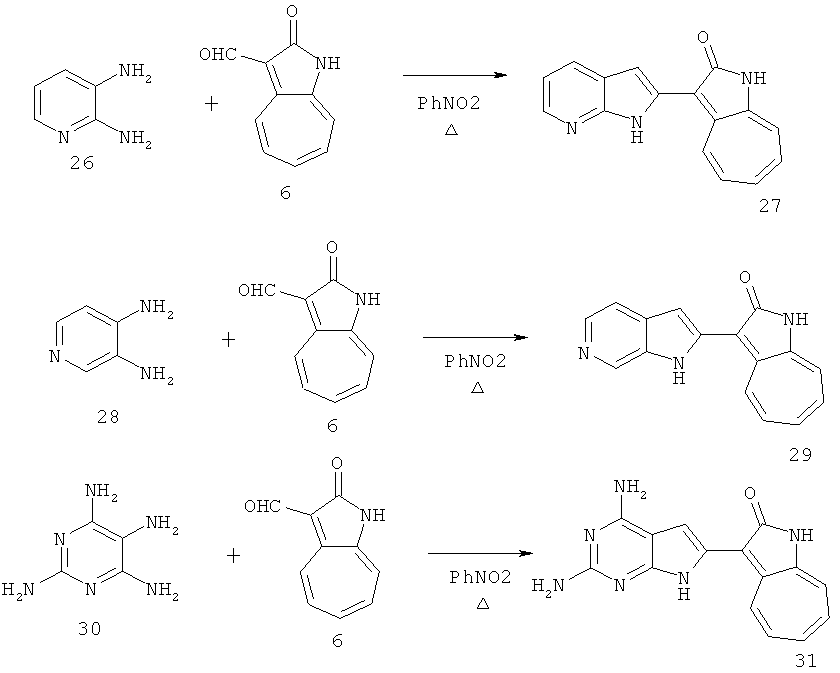
По этому изобретению азаазуленовые соединения, которые содержат 2Н-имидазол, а также 6,5-конденсированный гетероцикл 2Н-бензо[d][1,2,3]триазольного типа, можно синтезировать по следующей схеме:
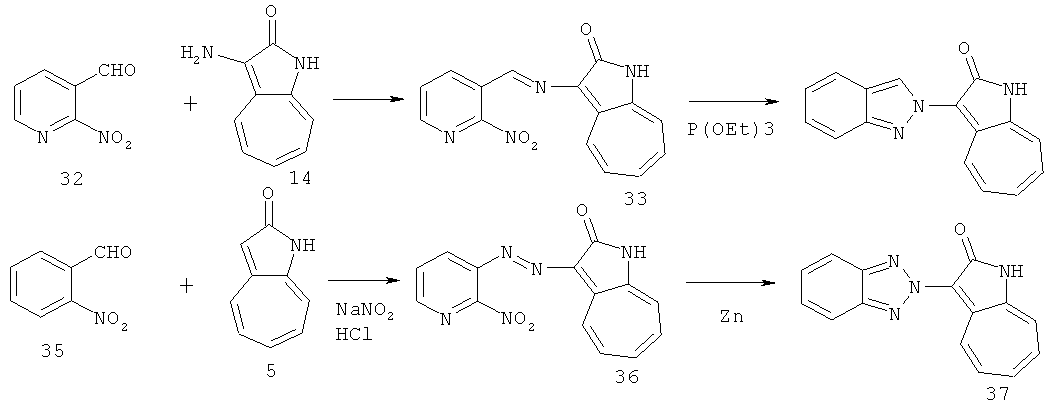
По этому изобретению азаазуленовые соединения, которые содержат имидазо[1,2-а]пиридин, имидазо[1,2-а]пиримидин, имидазо[1,2-с]пиримидин, имидазо[1,2-а]пиразин, а также 6,5-конденсированный гетероцикл имидазо[1,2-а][1,3,5]триазинового типа, можно синтезировать по следующей схеме:
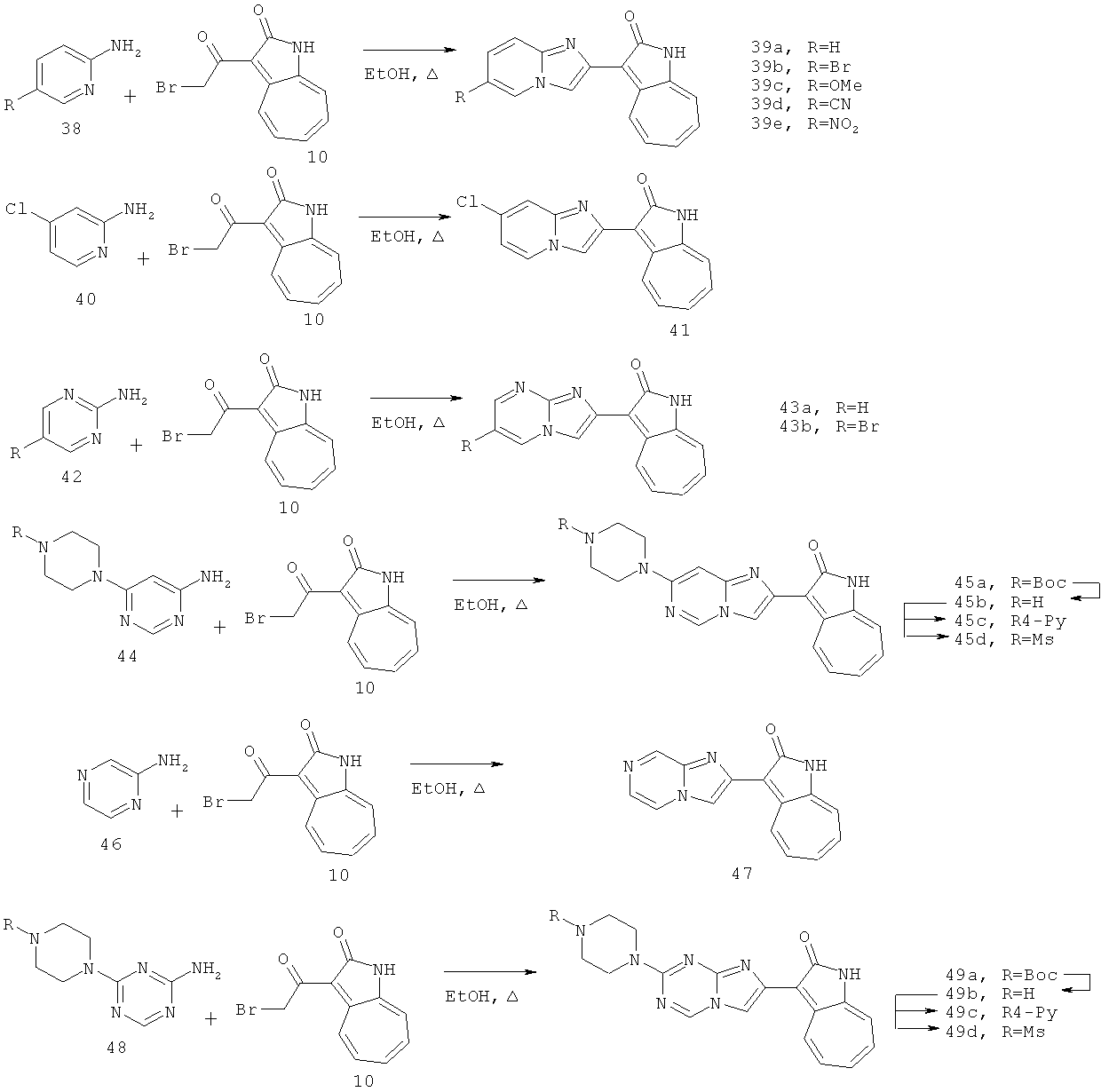
Как показано на приведенных выше схемах, в целях ускорения синтеза азаазуленовых соединений по настоящему изобретению можно использовать основание. Предпочтительно, чтобы указанное основание представляло собой соединение, содержащее атом азота (типа аммиака, метиламина, триметиламина, триэтиламина, анилина, диметиламино-пиридина, пролина, N-метиланилина, 1,8-диазобицикло[5.4.0]ундец-7-ена, диизопропилэтиламина, пирролидина, пиперидина, амида натрия, диизопропиламида лития и гексаметилдисилазанида натрия. Также можно использовать другие органические или неорганические основания, не содержащие атом азота. Они включают карбонаты, бикарбонаты, ацетаты, формиаты, алкиллитиевые соединения, ариллитиевые соединения, оксиды металла, реактивы Гриньяра, гидроксиды, фосфаты, бисульфаты, гидросульфиды и гидриды.
Как показано на приведенных выше схемах, для ускорения синтеза азаазуленовых соединений по настоящему изобретению можно использовать кислоту. Примеры органических или неорганических кислот включают уксусную, бензолсульфоновую, бензойную, камфоросульфоновую, лимонную, этенсульфоновую, дихлоруксусную, муравьиную, фумаровую, глюконовую, хлористоводородную, бромистоводородную, фтористоводородную, молочную, малеиновую, метансульфоновую, азотную, щавелевую, пантотеновую, фосфорную, янтарную, серную, винную, п-толуолсульфоновую, трифторуксусную кислоту и им подобные.
Как показано на приведенных выше схемах, для ускорения синтеза азаазуленовых соединений по настоящему изобретению можно использовать связующий реагент. Примеры связующего реагента включают ВОР, CDI, DCC, DEPBT, DIC, EDC HCl, HATU, HBTU, HCTU, PyBOP, PyBrOP, TATU, TBTU, TDBTU, TSTU и им подобные.
Как показано на приведенных выше схемах, для ускорения синтеза азаазуленовых соединений по настоящему изобретению можно использовать металлсодержащий катализатор. Примеры таких металлов включают Fe, Ni, Co, Cu, Au, Pd, Pt, Rh и Ru. Для повышения каталитической способности металла возможно присутствие лиганда.
Реакция, приведенная выше на схеме, может происходить в присутствии растворителя, который может быть или протонным, или апротонным. Примеры протонных растворителей включают гексан, толуол, бензол, метиленхлорид, хлороформ, диметилформамид, диметилсульфоксид и тетрагидрофуран. Приведенная выше на схеме реакция также может происходить и в отсутствие растворителя.
Синтезированное таким способом азаазуленовое соединение можно очистить подходящим для этого способом, таким как колоночная хроматография, высокоэффективная жидкостная хроматография (ВЭЖХ), дистилляция, сублимация и перекристаллизация.
Другие азаазуленовые соединения можно получить, используя пригодное сырье, и действуя согласно приведенной выше схеме, а также другим известным схемам. Описанные выше способы могут дополнительно включать стадии (или до, или после описанных здесь), предназначенные для присоединения или удаления защитных групп в целях оптимального проведения синтеза указанных азаазуленовых соединений. Помимо этого, возможно проведение различных стадий синтеза в меняющейся последовательности. Методики использования групп перегруппировки и защитных групп известны в химическом синтезе, для синтеза азаазуленовых соединений применимы методики, которые включают схемы, описанные, например в работах R.Larock, Comprehensive Organic Transformations, VCH Publishers (1989); T.W.Greene and P.G.Wuts, Protective Groups in Organic Synthesis, 2 sup.nd Ed., John Wiley and Sons (19991); L.Fieser and M.Fieser, Fieser and Fieser's Reagents for Organic Synthesis, John Wiley and Sons (1994); Encyclopedia of Reagents for Organic Synthesis, John Wiley and Sons (1995).
Упоминаемые здесь азаазуленовые соединения могут содержать неароматическую двойную связь, а также один или более асимметричных центра. Поэтому они могут существовать в виде рацематов, рацемических смесей, единичных энантиомеров, отдельных диастереомеров, смесей диастереомеров, а также в виде цис- или трансизомерных форм.
В границы настоящего изобретения входит также фармацевтический состав, содержащий эффективное количество по меньшей мере одного описанного азаазуленового соединения, а также фармацевтически приемлемый носитель или соль. Помимо этого, настоящее изобретение охватывает также способ введения раковому больному эффективного количества одного или более азаазуленового соединения. Термин «эффективное количество» означает такое количество активного азаазуленового соединения, которое необходимо для оказания данному субъекту требуемого терапевтического эффекта. Эффективная доза будет варьироваться в зависимости от вида заболевания, способа введения, использования наполнителей, а также возможности совместного применения вместе с другим терапевтическим лечением. Фармацевтически приемлемый носитель может включать растворитель, дисперсионную среду, покровные, антибактериальные и противогрибковые средства, изотонические средства и средства задержки действия, а также им подобные.
Азаазуленовые соединения по настоящему изобретению пригодны для выявления сайтов рестрикции азаазулена или 6,5-слитого гетероцикла. Сайтом рестрикции азаазулена или 6,5-слитого гетероцикла может быть любой энзим, рецептор, канал, переносчик, функциональный белок, РНК или ДНК сайт, который связан с азаазуленом или с 6,5-слитым гетероциклическим фрагментом азаазуленового соединения по настоящему изобретению. Поэтому соединения по настоящему изобретению могут быть использованы как диагностические средства, прогностические средства, молекулярные датчики, средства разделения и терапевтические средства, относящиеся к заболеваниям или нарушениям, или к энзимам, рецепторам, каналам, переносчикам, функциональным белкам, РНК или ДНК, связанным с этими заболеваниями или нарушениями.
Солями, пригодными для используемых компонентов согласно настоящему предмету изобретения, являются также соли с неорганическими катионами, такие как соли щелочных металлов (в частности, соли натрия, калия или аммония), соли щелочноземельных металлов (в частности, соли магния или кальция), а также соли с би- или тетравалентными катионами, например соли цинка, алюминия или циркония. Рассматриваются также соли органических оснований (такие как дициклогексиламиновые соли, метил-D-глюкамин); и соли аминокислот (таких как аргинин, лизин, гистидин, глютамин и т.д.). Основные азотсодержащие группы можно также кватернизировать с помощью таких агентов, как галогениды низших алкилов (типа метила, этила, пропила) и бутилхлоридов, бромидов и иодидов; диалкилсульфаты (такие как диметил, диэтил, дибутил и диамил сульфаты); галогениды с длинной цепью (типа децила, лаурила, миристила и стеарилхлоридов). Возможно также применение солеобразующих агентов, например низкомолекулярных алкиламинов (типа метиламина, этиламина или триэтиламина). Таким образом получают водо-, или жирорастворимые, или диспергируемые продукты.
В целях применения способа лечения по настоящему изобретению состав, содержащий одно или более азаазуленовых соединений, можно вводить субъекту (например, млекопитающему) парентерально, орально, назально, ректально, местно или буккально. Используемый здесь термин «парентеральный» касается подкожной, интрадермальной, внутривенной, внутримышечной, интраартериальной, интрасиновиальной или интра-краниальной инъекциии, а также любого пригодного инфузионного способа введения.
Стерильный инъекционный состав может представлять собой раствор или суспензию в нетоксичном парентерально приемлемом разбавителе или растворителе, типа раствора в 1,3-бутандиоле. Среди приемлемых разбавителей и растворителей, которые можно использовать, находятся: маннит, вода, раствор Рингера и изотонический раствор хлорида натрия. Помимо этого, в качестве растворителя или суспендирующей среды удобно использовать нелетучее масло (например, синтетические моно- или диглицериды). Для изготовления инъекционных форм пригодны жирные кислоты, такие как олеиновая кислота и ее глицеридные производные, поскольку (особенно в своей полиоксиэтилированной форме) они являются натуральными фармацевтически приемлемыми маслами, типа оливкового или касторового масла. Такие масляные растворы или суспензии могут также содержать спиртовой растворитель или дисперсант с длинной цепью, карбоксиметилцеллюлозу или аналогичные диспергирующие средства. Для составления рецептуры можно использовать также другие стандартно применяемые поверхностно-активные вещества, такие как Твин или Спан, или другие эмульгаторы, или биологически доступные интенсификаторы, обычно применяемые при производстве фармацевтически приемлемых твердых, жидких или иных дозировочных форм.
Составом для орального введения может быть любая орально приемлемая дозировочная форма, включая капсулы, таблетки, эмульсии и водные суспензии, дисперсии и растворы. В случае таблеток стандартно используемые носители включают лактозу и кукурузный крахмал. Также обычно вводят скользящие средства, типа стеарата магния. Пригодные для орального введения в капсульной форме разбавители включают лактозу и кукурузный крахмал. Если суспензию или эмульсию вводят орально, то активный ингредиент может быть суспендирован или растворен в масляной фазе, объединенной с эмульгатором или с суспендирующим средством. Если это требуется, то возможно дополнительное введение определенных подсластителей, ароматизаторов или красителей.
Назальный аэрозоль или ингаляционный состав можно приготовить по методике, хорошо известной в области получения фармацевтических рецептур. Например, такой состав можно получить в виде физиологического раствора, используя бензиловый спирт или другие подходящие консерваторы, промоторы абсорбции, фтороуглероды и/или другие известные способствующие солюбилизации или дисперсии агенты.
Возможно также использование состава, содержащего одно или более азаазуленовых соединений, в виде суппозитория, предназначенного для ректального введения.
Носитель в таком фармацевтическом составе может быть приемлем, если он совместим с активным ингредиентом указанного состава (и, предпочтительно, способен стабилизировать активный ингредиент), а также не вреден для проходящего лечение субъекта. В целях доставки активного азаазуленового соединения в качестве фармацевтических наполнителей возможно применение одного или более способствующего солюбилизации средства. Примеры других носителей включают коллоидный оксид кремния, стеарат магния, целлюлозу, лаурилсульфат натрия, а также D&C Yellow #10.
Описанные выше азаазуленовые соединения могут быть предварительно отобраны по их эффективности при лечении приведенных выше заболеваний, этот отбор производится путем in vitro анализа с последующим подтверждением опытами на животных и клиническими испытаниями. Квалифицированным в данной области людям очевидны также другие методы.
Приводимые далее примеры сделаны только в иллюстративных целях, они никоим образом не ограничивают остальное раскрытие. Без дальнейшего уточнения предполагается, что на основании приведенного здесь описания квалифицированный в данной области человек способен использовать настоящее изобретение в наиболее полной степени. Все цитируемые публикации включены здесь полностью в виде ссылок.
Настоящее изобретение обеспечивает также способ ингибирования активности протеинкиназы или протеинфосфатазы в клетке, используя для этого одно из описанных азаазуленовых соединений. Этот способ включает контакт клетки, экспрессирующей протеинкиназу или протеинфосфатазу, с такого рода азазуленовым соединением. Протеинкиназа и протеинфосфатаза регулируют каскады передачи сигналов. Эти каскады в свою очередь регулируют рост клеток, их миграцию, дифференциацию, экспрессию гена, сжатие мышц, метаболизм глюкозы, синтез клеточных белков, а также регулируют клеточный цикл.
Термин «Протеинкиназа» касается фермента киназы, который изменяет другие белки путем химического присоединения к ним фосфатных групп (фосфорилирования). Примеры протеинкиназы включают AMPK, BLK, CSFIR, FGFR, FOR, FLT3, KDR, KIT, LCK, LYN, MAP4K5, NTRK, PHKG1, RET, SRC, STK и YES1.
В настоящем изобретении клетки могут быть получены от раковых больных. Их здесь называют «раковыми клетками». Эти клетки выделяют из разных источников и тканей. Например, указанные клетки можно выделить из образца крови или биопсии. Такие клетки могут быть стволовыми клетками, клетками фибробласта или лимфоидными клетками. Согласно типу клеток и источнику их получения клетки можно размножить в некоторой культуре. Размножение клеток возможно без их иммортализации. Или же клетки могут быть иммортализированы, используя вирус или плазмиду, несущую антиген, или путем трансформации вирусного белка, например белка папилломы Е6 или Е7.
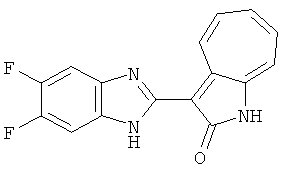
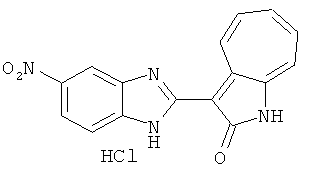
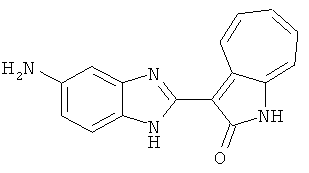
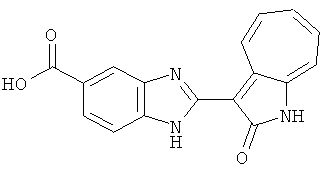
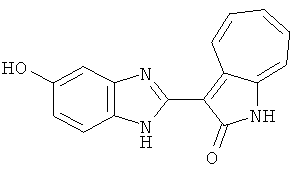

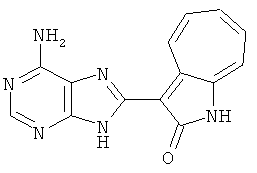

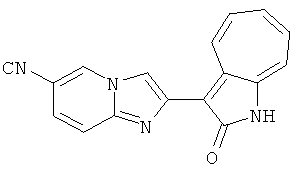
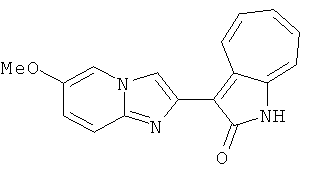
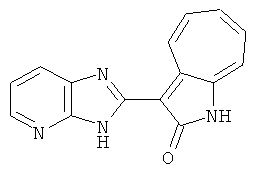
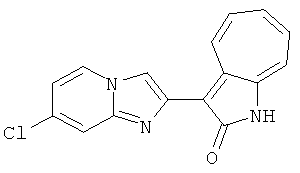
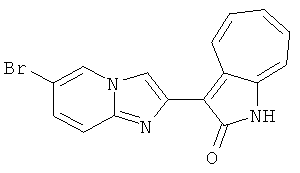
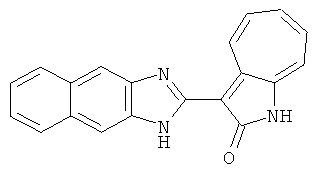
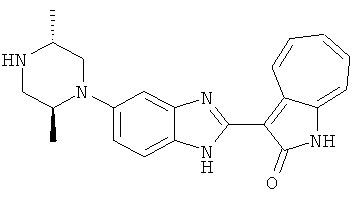

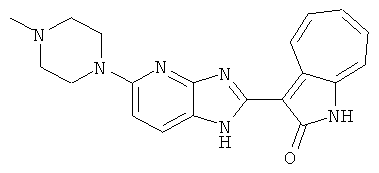
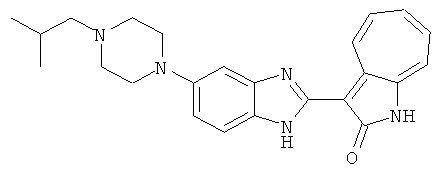
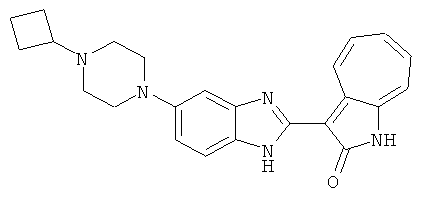
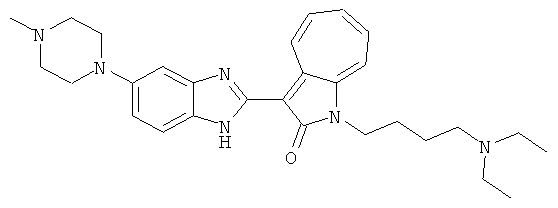
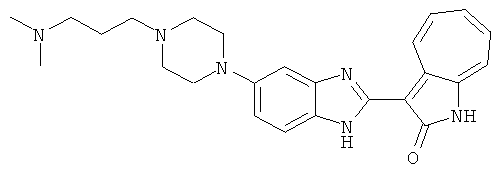
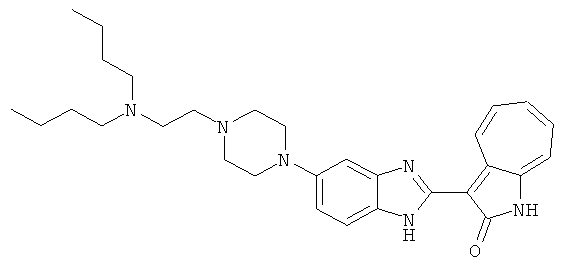
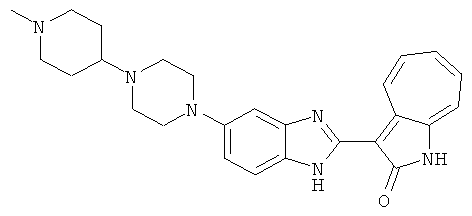

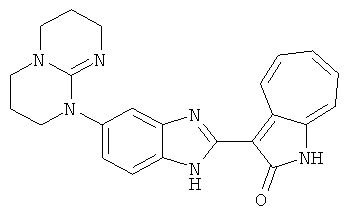

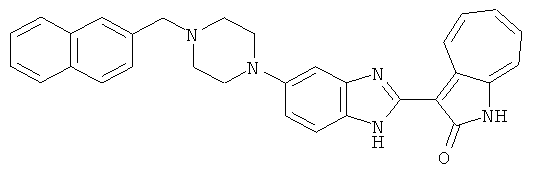
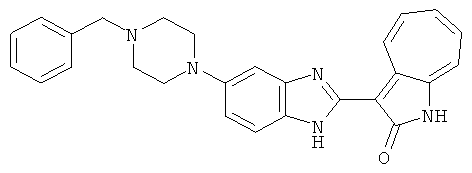
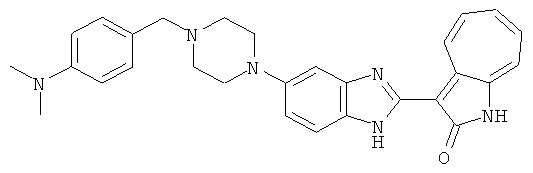
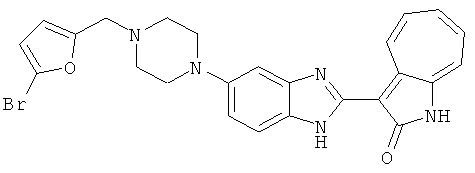
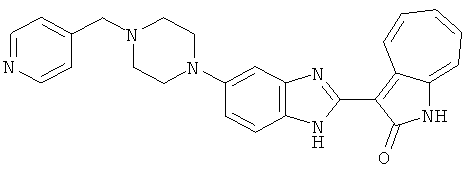
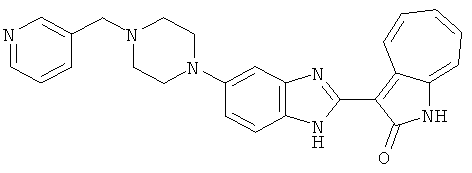
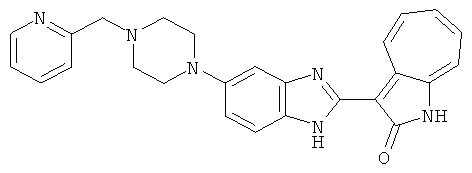

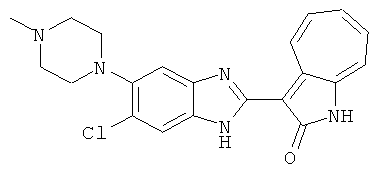
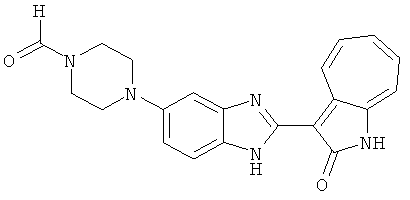
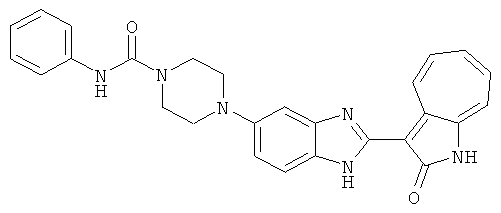
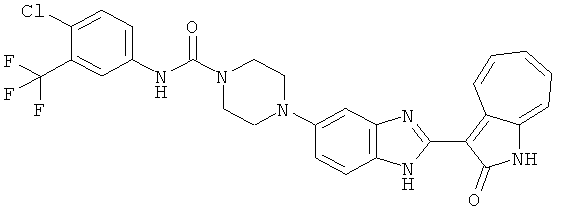
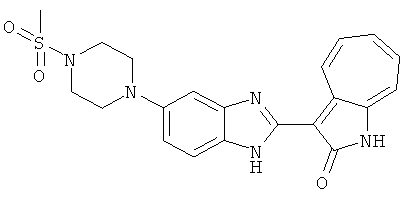
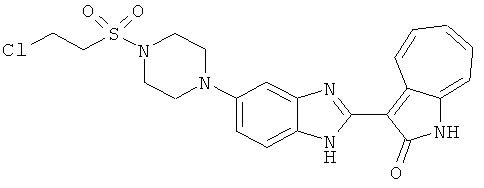
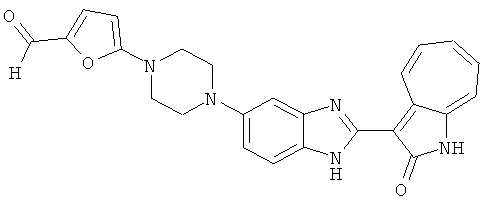
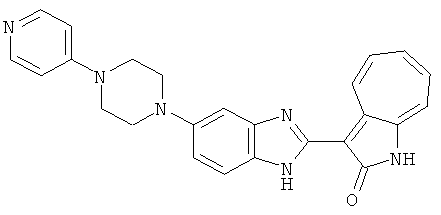
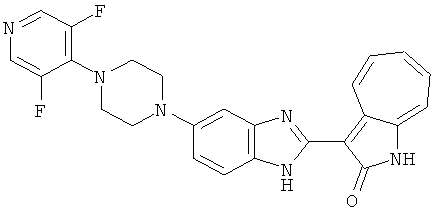
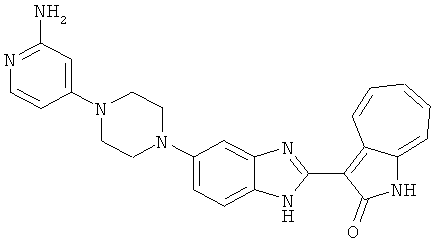
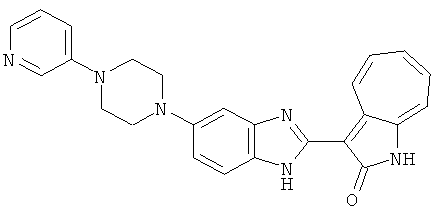
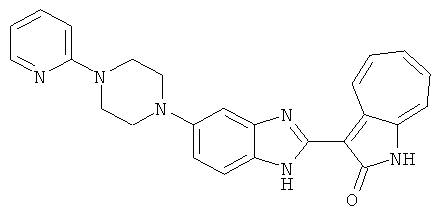
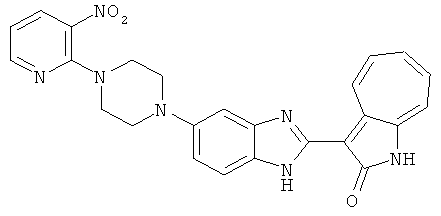
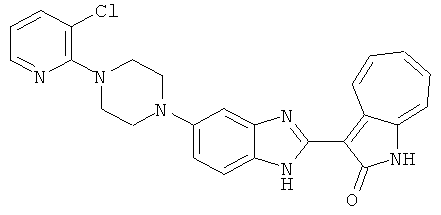
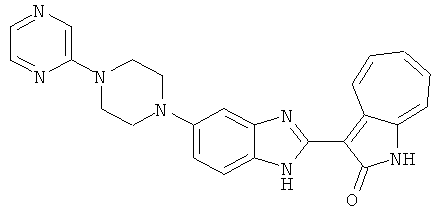


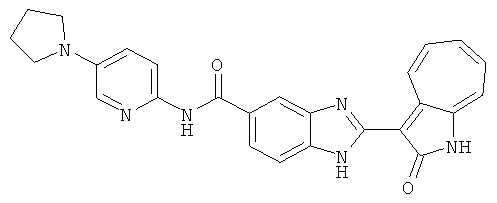



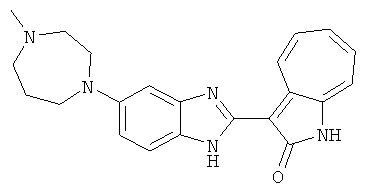
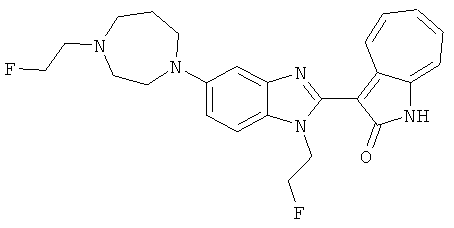
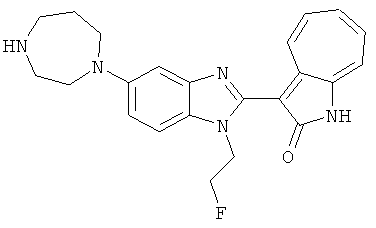
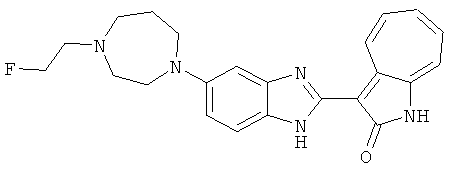

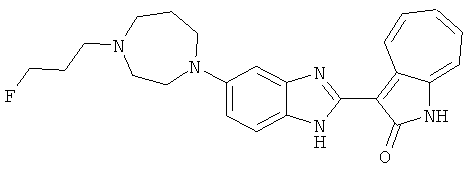
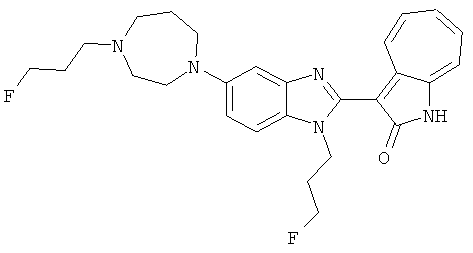
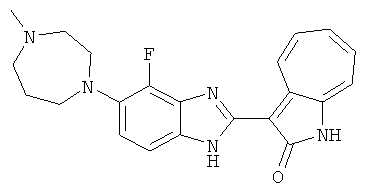
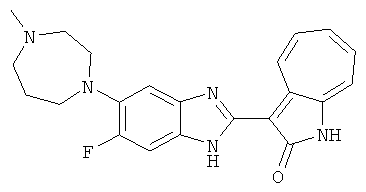
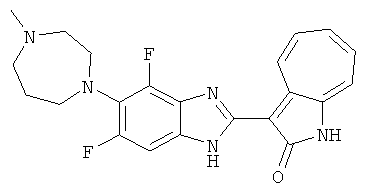

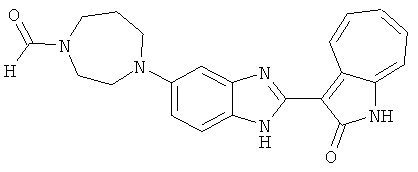
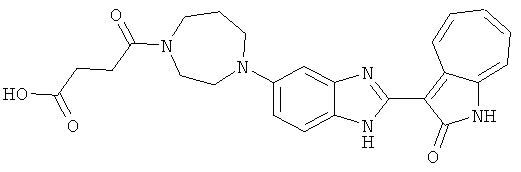
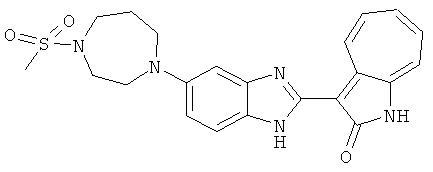
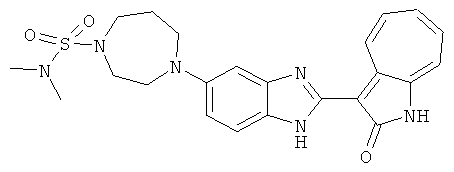
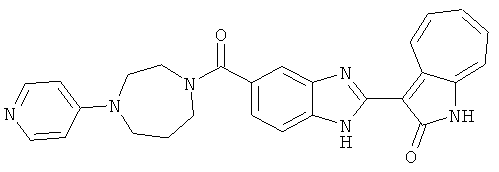
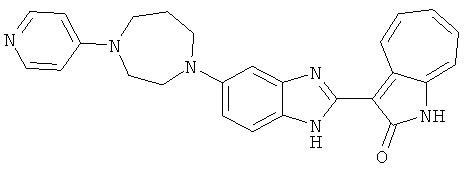
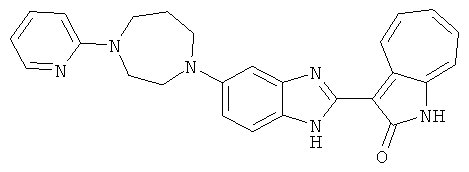
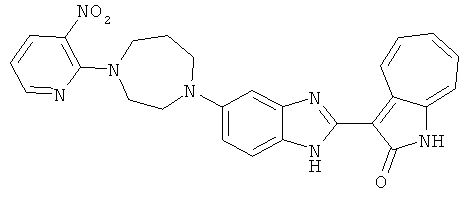

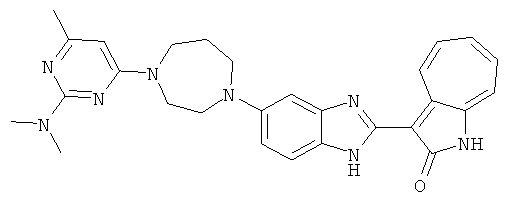
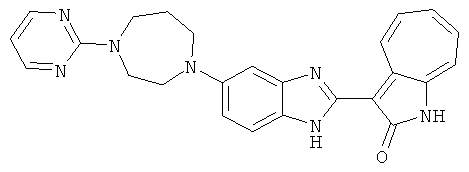

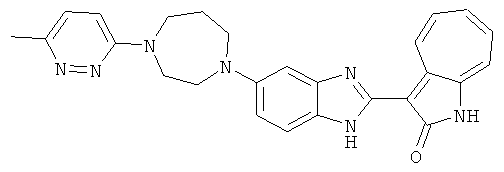
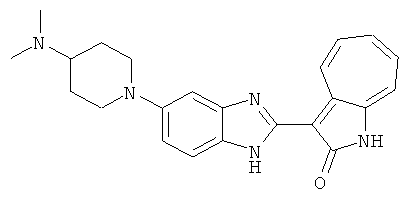
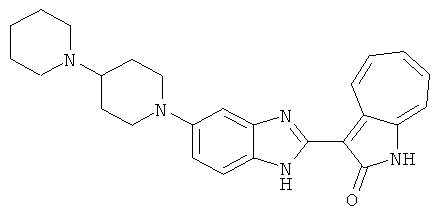

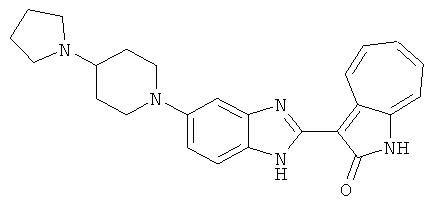
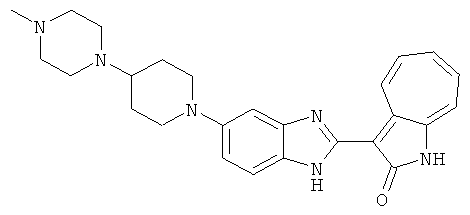
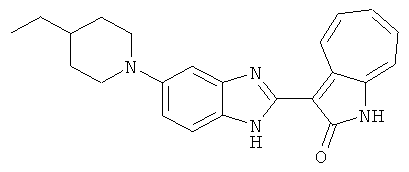
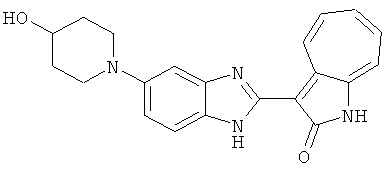
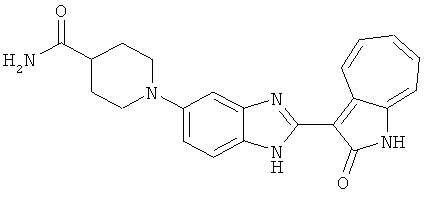

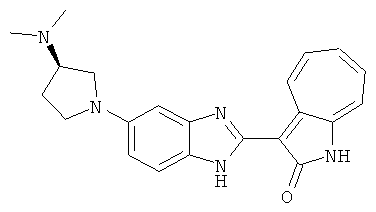
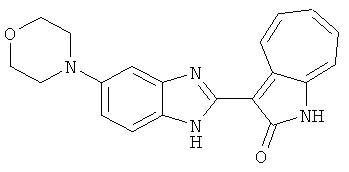


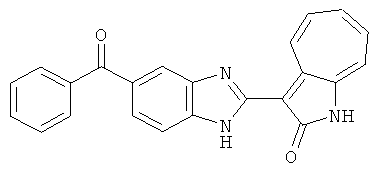

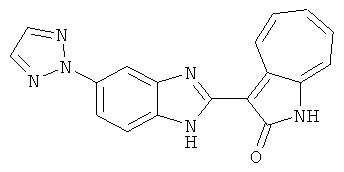

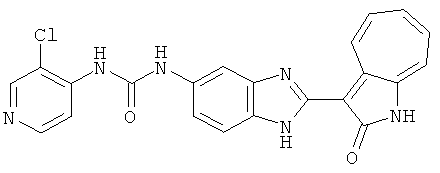

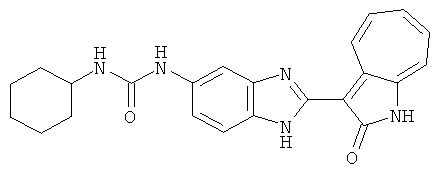
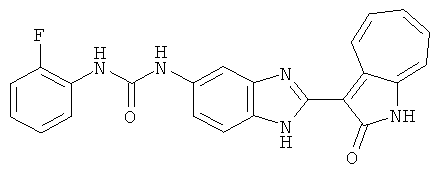
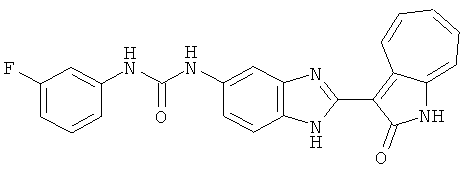
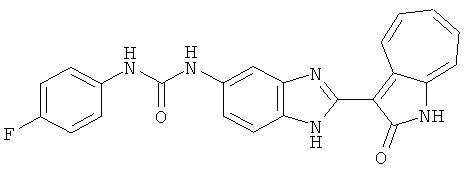
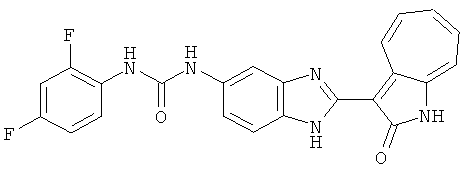
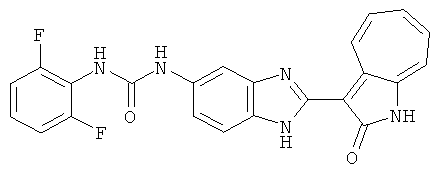

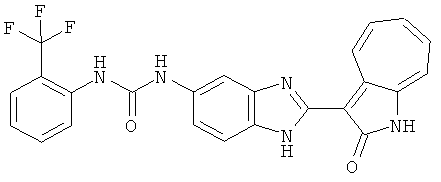

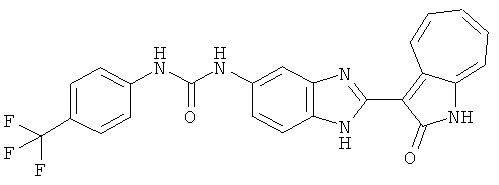
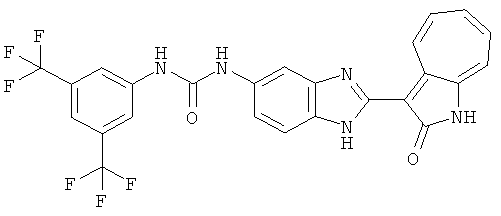
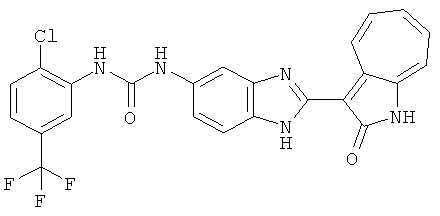
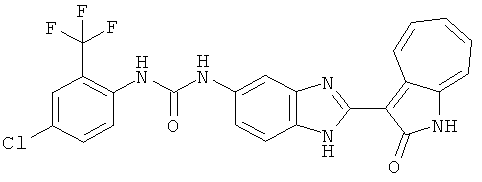
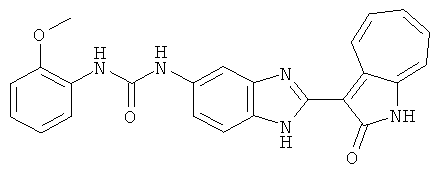
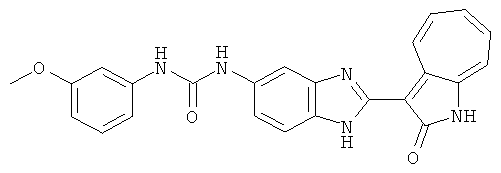
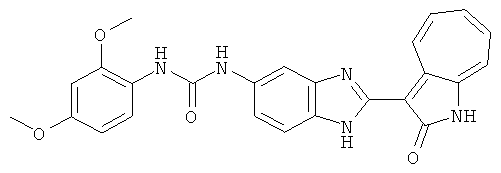
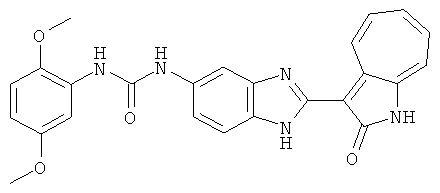

ПРИМЕРЫ
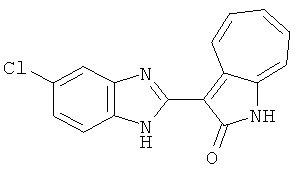
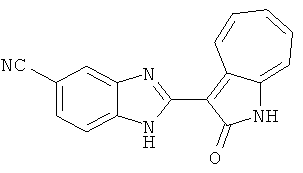
Сравнительный пример (Соединение A1)
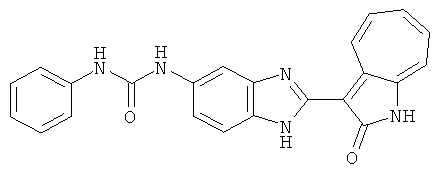
Получение 3-(бензимидазол-2-ил)-1-азаазулен-2-он (A1): 3-формил-1-азаазулен-2-он (1 ммол, согласно методике, описанной в Chem. Pharm. Bull., 1994, vol.42, #12, pp.2491-2499 с небольшими изменениями) был растворен в насыщенном растворе гидросульфита натрия (3 мл) и перемешан в течение 3 часов. После этого добавили о-фенилендиамин (1.2 ммол) и этанол (10 мл) и нагревали вплоть до дефлегмации в течение 3 часов. Этанол выпарили и далее в остаток добавили воду (50 мл). Осадок отфильтровали, промыв водой (50 мл, 3 раза), и извлекли для сушки. Масса продукта составила 197.3 мг, выход 75%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.28 (s, 1H), 9.41 (d, 1H), 7.70-7.68 (m, 2H), 7.63 (t, 1H), 7.55 (t, 1H), 7.45 (d, 1H), 7.32 (t, 1H), 7.20-7.18 (m, 2H). LC-MS (m/z) 262 [M+1].
Пример 1
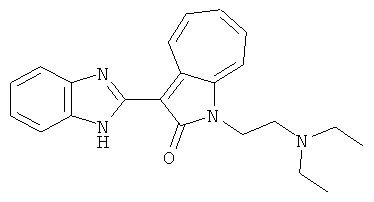
Получение B1: 3-Формил-1-азаазулен-2-онs (1 ммол) и калий т-бутоксид (2 ммол) были растворены в DMF (10 мл) и перемешаны до образования прозарачного раствора. Далее, в раствор добавили диэтиламиноэтил хлорид гидрохлорид (1 ммол) и перемешивали в течение ночи, при комнатной температуре. С утра остаток был очищен колоночной хроматографией, используя дихлорметан/этилацетат/триэтиламин (9:1:0.1 to 5:5:0.1) как элюент, до получения 76% 1-диэтиламинэтил-3-формил-1-азаазулен-2-он. 1-диэтиламинэтил-3-формил-1-азаазулен-2-он (0.1 ммол) растворили в смеси этанола (10 мл) и воды (5 мл). Далее добавили о-финилинидиамин (0.15 ммол) и бисульфат натрия (0.2 ммол) и нагревали вплоть до дефлегмации в течение 1 дня. С утра остаток очистили колоночной хроматографией используя дихлорметан/этилацетат/триэтиламина (9:1:0.1 to 3:7:0.1) в качестве элюента, до получения 54% целевого соединения B1.1H-ЯМР (500 МГц, CDCl3) δ (ppm) 11.53 (s, 1H), 9.56 (d, 1H), 7.81 (d, 1H), 7.53-7.149 (m, 7H), 4.26 (t, 2H), 2.78 (t, 2H), 2.62 (q, 4H), 1.02 (t, 6H). LC-MS (m/z) 361 [M+1].
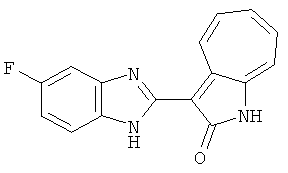
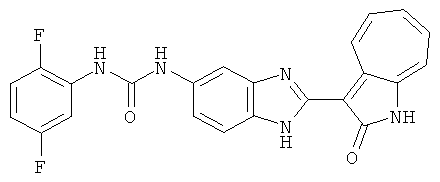
Получение B2: 3-формил-1-азаазулен-2-он (1 ммол) растворили в насыщенном растворе бисульфита натрия (3 мл) и перемешали в течение 3 часов. Далее добавили 4-фтор-1,2-фенилендиамин (1.2 ммол) и этанол (10 мл), нагрели смесь вплоть до дефлегмации в течение 3 часов. Этанол выпарили и далее в остаток добавили воду (50 мл). Осадок отфильтровали, промыв водой (50 мл, 3 раза), и извлекли для сушки. Масса целевого продукта составила 210.5 мг, выход 75%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.31 (s, 1H), 9.36 (d, 1H), 7.68 (t, 1H), 7.65 (t, 1H), 7.57 (t, 1H), 7.48-7.45 (m, 2H), 7.34 (t, 1H), 7.04 (dt, 1H). LC-MS (m/z) 280 [M+1].
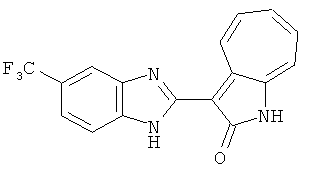
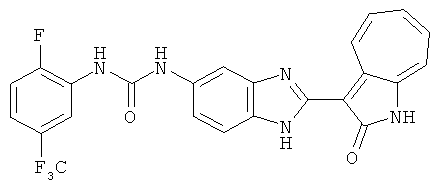
Получение B4: 3-Формил-1-азаазулен-2-он (1 ммол) растворили в насыщенном растворе бисульфита натрия (3 мл), перемешали в течение 3 часов. Далее добавили 3,4-диаминобензотрифторид (1.2 ммол) и этанол (10 мл), нагрели смесь вплоть до дефлегмации в течение 3 часов. Этанол выпарили и далее добавили воду (50 мл). Осадок отфильтровали, промыв водой (50 мл, 3 раза), и извлекли для сушки. Масса целевого продукта составила 245.5 мг, выход 74%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.58 (s, 1H), 12.39 & 12.37 (s, 1H), 9.44 & 9.42 (d, 1H), 8.07 & 8.01 (s, 1H), 7.87 & 7.86 (t, 1H), 7.73 & 7.14 (t, 1H), 7.63 & 7.62 (t, 1H), 7.54-7.49 (m, 2H), 7.41 & 7.39 (t, 1H). LC-MS (m/z) 330 [M+1].
Получение B5: 3-Формил-1-азаазулен-2-он (5 ммол) растворили в насыщенном растворе бисульфита натрия (10 мл) и перемешали в течение 30 минут. Далее добавили 4-нитро-1,2-фенилендиамин (6 ммол) и этанол (150 мл) и нагревали смесь вплоть до дефлегмации в течение 3 часов. Этанол выпарили и далее в остаток добавили воду (50 мл). Осадок отфильтровали, промыв водой (50 мл, 3 раза) и 1N HCl в этаноле (50 мл, 3 раза), и в конце извлекли для сушки. Масса целевого продукта составила 1.06 г, выход 69%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.55 (s, 1H), 9.38 (d, 1H), 8.64 (s, 1H), 8.58 (s, 1H), 8.15 (d, 1H), 7.85 (d, 1H), 7.80 (t, 1H), 7.71 (t, 1H), 7.63 (d, 1H), 7.48 (t, 1H).
Получение В6: Соединение B5 (2.8 ммол) растворили в метаноле (350 мл) и далее добавили 10% Pd/C (100 мг). Смесь гидрировали, используя водород при давлении 1 атм при комнатной температуре. Смесь для удаления катализатора отфильтровали и далее выпарили. Остаток очистили колоночной хроматографией с помощью метанола в качестве элюента. После выпаривания смеси масса целевого продукта составила 230 мг, выход 26%.1Н-ЯМР (500 МГц, DMSO-d6) 5 (ppm) 12.74 (s, 1H), 9.11 (d, 1H), 7.73-7.81 (m, 5H), 7.56 (t, 3H), 7.27 (s, 2H).
Получение B7: 3-Формил-1-азаазулен-2-онs (4 ммол) добавили в насыщенный раствор бисульфита натрия (10 мл) и этанола (40 мл). Смесь перемешали в течение 30 минут при комнатной температуре и далее добавили 3,4-диаминобензойную кислоту (4.5 ммол) и в течение ночи нагревали вплоть до дефлегмации. Этанол выпарили. В остаток добавили воду (100 мл) и затем окислили с 6 N хлористоводородой кислотой (3 мл). Продукт отфильтровали с водой (2 мл, 4 раза) и высушили при 80°C. Масса целевого продукта составила 981.7 мг, выход 80%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.39 (s, 1H), 8.31 (s, 1H), 7.84 (d, 1H), 7.63 (t, 1H), 7.53 (d, 1H), 7.40 (t, 1H), 7.36 (t, 2H), 3.97 (d, 1H), 3.40(s, 1H).
Получение B8: 4-амино-3-нитрофенол (3 ммол) растворили в метаноле (100 мл), добавили 5% Pd/C (100 мг) и смесь гидрировали. Катализатор отфильтровали и затем в фильтрат добавили 3-формил-1-азаазулен-2-онs (3 ммол) и насыщенный раствор бисульфита натрия (3 мл) и нагрели вплоть до дефлегмации. Осадок отфильтровали, промыли метанолом (20 мл, 5 раз) и водой (20 мл, 5 раз). Продукт высушили вакуумом до получения 411.2 мг целевого продукта, выход 49%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.18 (s, 1H), 11.90,11.95 (s, 1H), 9.30, 9.35 (d, 1H), 8.95, 9.12 (s, 1H), 7.44-7.60 (m, 3H), 7.36, 7.39 (d, 1H), 7.22-7.29 (m, 1H), 7.00, 7.07 (d, 1H), 6.69 (dt, 1H).
Получение B15: 3-Формил-1-азаазулен-2-он (0.5 ммол) и насыщенный раствор гидросульфита натрия (0.5 мл) добавили в этанол (20 мл) и перемешали в течение 30 минут. Добавили 2,3-диаминпиридин (0.5 ммол) и нагрели вплоть до дефлегмации в течение 8 часов. Этанол выпарили вакуумом. Остаток распределили на этилацетат и воду.
Органический слой отделили, высушили с ангидридмагнийсульфатом, отфильтровали и затем концентрировали до 20 мл. Раствор подвергли колоночной хроматографии с силикагелем и элютированию, сначала с этилацетатом и затем с ацетоном до получения 58.6 мг целевого продукта, выход 45%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.37 (s, 1H), 9.44 (broad, 1H), 8.36 (s, 1H), 8.02 (d, J=7.0 Hz, 1H), 7.73 (t, J=10 Hz, 1H), 7.63 (t, J=10.0 Hz, 1H), 7.54 (d, J=9.0 Hz, 1H), 7.41 (t, J=9.5 Hz, 1H), 7.22 (broad, 1H). LC-MS (m/z) 263 [M+1].
Получение B18: 2,3-диамононафталин (1 ммол), 3-формил-1-азаазулен-2-онs (1 ммол) и аммоний метавандат (0.05 ммол) добавили в метанол (50 мл) и перемешали при температуре 50°C в течение 24 часов. Этилацетат (200 мл) добавили в реакционную смесь и затем пропустили через планшет силикагеля. Собранный фильтрат выпарили под вакуумом. Твердый осадок обработали с помощью EtOAc/MeOH (10/1, 40 мл), отфильтровали и повторно промыли смесью растворителей (10 мл). Масса целевого продукта составила 65.4 мг, выход 20.9%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.38 (s, 1H), 12.19 (s, 2H), 9.30 (d, 2H), 8.16 (s, 1H), 8.09 (s, 1H), 7.98 (d, 1H), 7.93 (d, 1H), 7.73 (t, 1H), 7.61 (t, 1H), 7.53 (d, 1H), 7.32-7.40 (m, 3H).
Получение B19: 3-Формил-1-азаазулен-2-онs (4 ммол) добавили в насыщенный раствор бисульфита натрия (10 мл) и этанола (40 мл). Смесь перемешали в течение 30 минут при комнатной температуре и далее добавили 3,4-диаминобензойная кислоту (4.5 ммол) и в течение ночи нагревали вплоть до дефлегмации. Этанол выпарили. В остаток добавили воду (100 мл). Продукт отфильтровали с водой (5 мл, 4 раза) и высушили при температуре 80°C. Масса целевого продукта составила 761.6 мг, выход 80%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.59 (d, 1H), 12.36 (d, 1H), 9.35-9.40 (m, 1H), 8.07, 8.13 (s, 1H), 7.38-7.83 (m, 6H).
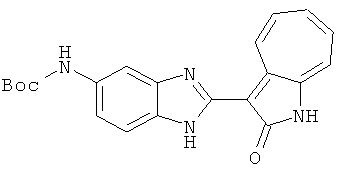
Получение B20: дикарбонат ди-трет-бутила (38.41 г, 0.176 ммол) добавили в раствор 2-нитро-п-фенилендиамин (24.50 г, 0.16 ммол) безводный дихлорметан (1.6 Л) при комнатной температуре. После перемешивания в течение 2 дней реакционную смесь пропустили через короткий планшет активной глины и концентрировали под вакуумом. Высушили под вакуумом при температуре 50°C до получения твердого продукта ярко-оранжевого цвета. Масса целевого продукта составила 40.52 г, выход 100%. Нитрофенилендиамин (15.20 г, 60 ммол) растворили в метаноле (500 мл) и затем добавили никель Ренея (5.0 г). Смесь гидрировали, используя водород при давлении 1 атм в течение 48 часов.
Смесь для удаления катализатора отфильтровали и далее промыли с 200 мл метанола два раза. Далее, 3-формил-1-азаазулен-2-он (10.39 г, 60 ммол) аммоний метавандат (0.35 г, 3 ммол) добавили в фильтрат и перемешали при температуре 50°C в течение 18 часов. Метанол выпарили и остаток растворили в смеси EtOAc/MeOH (10/1, 500 мл). Смесь отфильтровали через короткую колонку силикагеля и выпарили под вакуумом. Твердый остаток обработали с EtOAc/DCM (1/5, 10 мл) и отфильтровали до получения 2.42 г целевого продукта, выход 10.7%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.16, 12.19 (s, 1H), 12.02 (s, 1H), 9.30, 9.35 (d, 1H), 9.16, 9.24 (s, 1H), 7.75, 7.89 (s, 1H), 7.45-7.60 (m, 3H), 7.37 (t, 1H), 7.16-7.29 (m, 2H), 1.50 (s, 9H).
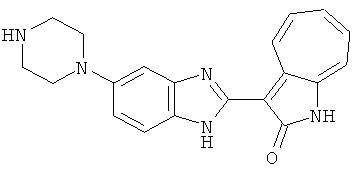
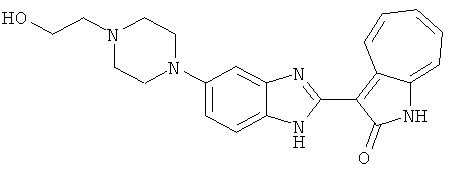
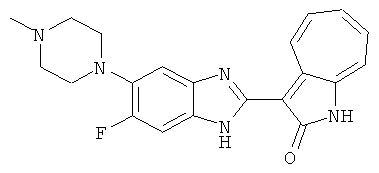
Получение B22: 2-нитро-5-пиперазин-1-ил-анилин (6.12 ммол, согласно методике описанной в Pharmazie, 1984, vol. 39, #11 p.747-749) растворили в метаноле (100 мл). Добавили никель Ренея (1.0 г) и гидрировали, используя водород при давлении 1 атм в течение 1 дня. Смесь отфильтровали, затем фильтрат добавили в колбу, содержащую 3-формил-1-азаазулен-2-он (1.01 г, 5.8 ммол) и насыщеную смесь гидросульфата натрия (7 мл). Смесь дефлегмировали в течение 1 дня. Сырой продукт очистили колоночной хроматографией с помощью дихлорметанметанола в качестве элюента (20:1 to 2.5:1). Масса продукта 1.46 g, выход 72%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.28 (s, 1H), 12.00, 12.05 (s, 1H), 9.32, 9.37 (d, 1H), 7.41-7.60 (m, 4H), 7.27 (m, 2H), 6.99 (d, 1H), 3.28-3.35 (m, 8H).
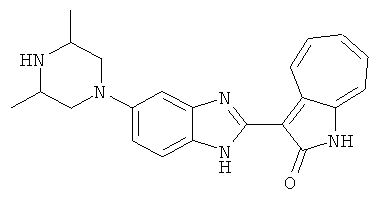
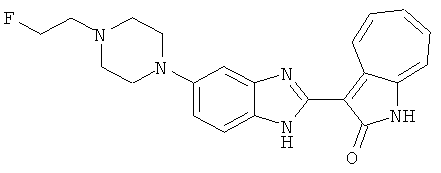
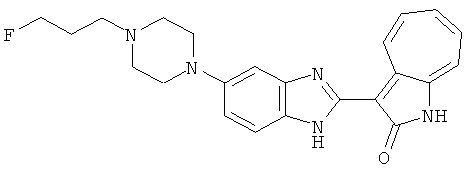
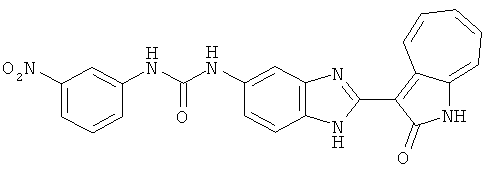
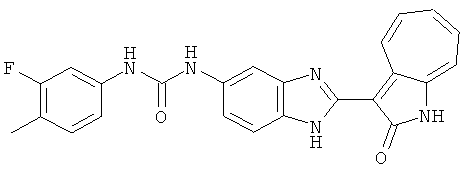
Получение B24: 2,6-диметилпиперазин (12.56 г, 0.11 ммол), 5-фтор-2-нитроанилин (15.61 г, 0.1 ммол) и триэтиламин (11.13 г, 0.11 ммол) добавили в ацетонитрил (200 мл) и нагревали до дефлегмации в течение 12 часов. Растворитель выпарили, добавили воду (100 мл) в остаток и перемешали в течение 30 минут. Отфильтровали твердый продукт желтого цвета, промыли с водой (50 мл, 2 раза) и в конце высушили под вакуумом до получения 23.15 г продукта, выход 92%. Используя такой же способ как при получении B22, соединение 24 было получено с выходом 78%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 11.10 (d, 1H), 9.40 (m, 1H), 7.73 (d, 1H), 7.44 (m, 3H), 7.16 (m, 1H), 7.04 (m 3H), 3.60 (d, 2H), 3.20 (b, 1H), 2.41 (q, 2H), 1.24 (d, 6H).
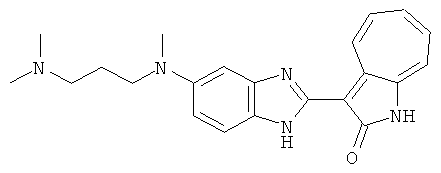
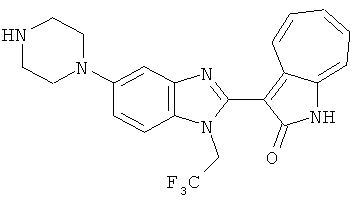
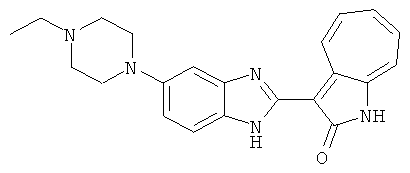
Получение B26: Используя такой же способ, как при получении B24, 5-(4-метилпепиразин-1-ил)-2-нитроанилин был получен с выходом 90%. Далее были проведены реакции восстановления и конденсации аналогично, как при получении соединения B22, целевое соединение B26 было получено с выходом 72%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.20 (s, 1H), 11.97 & 11.91 (s, 1H), 9.38 & 9.31 (d, 1H), 7.58-6.94 (m, 7H), 3.34 (s, 4H), 3.14 (s, 4H), 2.24 (s, 3H). LC-MS (m/z) 360 [M+1].
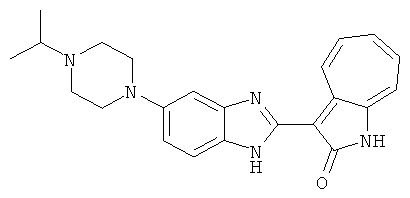
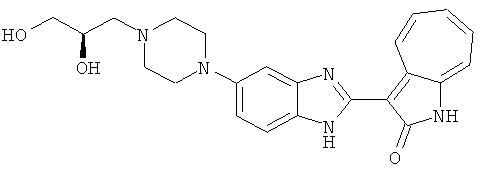
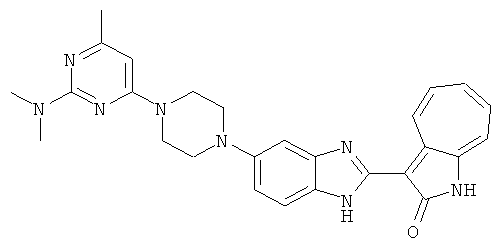
Получение B33: Используя такой же способ, как при получении B24, 5-(4-(3-(диметиламино)пропил)пиперазин-1-ил)-2-нитроанилин был получен с выходом 81%. Далее реакции восстановления и конденсации были проведены аналогично, как при получении соединения B22, целевое соединение B33 было получено с выходом 65%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.82 (s, 1H), 11.47 (s, 1H), 10.80 (s, 1H), 8.94 (d, 1H), 7.74-7.84 (m, 3H), 7.58 (t, 1H), 7.32 (s, 1H), 7.28 (d, 1H), 3.85 (d, 2H), 3.67 (d, 2H), 2.80 (s, 6H), 2.27 (dd, 2H), 2.11 (s, 2H).
Получение B34: Используя такой же способ, как при получении B24, 5-(4-(3-(дипропиламино)пропил)пиперазин-1-ил)-2-нитроанилин был получен с выходом 91%. Далее реакции восстановления и конденсации были проведены аналогично, как при получении соединения B22, целевое соединение B34 было получено с выходом 75%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 8.85 (d, 1H), 7.31 (d, 1H), 7.21 (t, 1H), 7.08 (m, 1H), 7.04 (m, 4H), 6.94 (m, 1H), 6.75 (d, 1H), 3.50 (t, 4H), 2.97 (d, 4H), 2.46 (s, 2H), 2.34 (m, 2H), 2.27 (m, 4H), 1.41 (m, 4H), 0.76 (m, 6H).
Получение B35: Используя такой же способ, как при получении B24, 5-(4-(1-метилпиперидин-4-ил)пиперазин-1-ил)-2-нитроанилин был получен с выходом 93%. Далее реакции восстановления и конденсации были проведены аналогично, как при получении соединения В22, целевое соединение B35 было получено с выходом 77%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 11.19 (d, 1H), 9.93 (m, 1H), 7.56 (m, 3H), 7.36 (t, 1H), 7.24 (q, 1H), 7.15 (d, 1H), 6.91 (t, 1H), 3.25 (m, 2H), 3.17 (s, 4H), 2.80 (d, 2H), 2.65 (s, 4H), 2.14 (s, 3H), 1.98 (m, 1H), 1.77 (d, 2H), 1.47 (q, 2H).
Получение B36: Используя такой же способ, как при получении B24, 3-(4-(3-амино-4-нитрофенил)пиперазин-1-ил)пропан-1-ол был получен с выходом 85%. Далее реакции восстановления и конденсации были проведены аналогично, как при получении соединения В22, целевое соединение B36 было получено с выходом 67%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 9.20 (d, 1H), 7.62 (m, 2H), 7.56 (m, 1H), 7.38 (m, 4H), 7.17 (m, 1H), 7.10 (d, 1H), 3.80 (m, 2H), 3.69 (m, 4H), 2.83 (m, 4H), 2.68 (m, 2H), 2.07 (s, 1H).
Получение B50: Используя такой же способ, как при получении B24, 4-фтор-5-(4-метилпиперазин-1-ил)-2-нитроанилин был получен с выходом 95%. Далее реакции восстановления и конденсации были проведены аналогично, как при получении соединения B22, целевое соединение B50 было получено с выходом 87%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.23 (s, 1H), 12.10, 12.12 (s, 1H), 9.31, 9.34 (d, 1H), 7.59 (t, 1H), 7.51 (t, 1H), 7.38-7.44 (m, 2H), 7.27-7.33 (m, 1H), 3.03 (s, 4H), 2.53 (s, 4H), 2.27 (s, 3H).
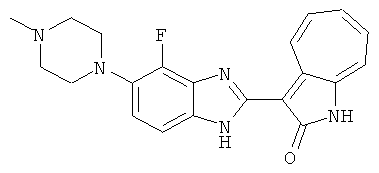
Получение B51: Используя такой же способ, как при получении B24, 2-фтор-3-(4-метилпиперазин-1-ил)-6-нитроанилин был получен с выходом 94%. Далее реакции восстановления и конденсации были проведены аналогично, как при получении соединения B22, целевое соединение B51 было получено с выходом 89%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.26 (s, 1H), 12.19 (s, 1H), 9.35 (d, 1H), 7.64 (t, 1H), 7.53 (t, 1H), 7.44 (d, 1H), 7.38 (d, 1H), 7.31 (t, 1H), 6.96 (t, 1H), 3.03 (s, 4H), 2.53 (s, 4H), 2.26 (s, 3H).
Получение B52: Используя такой же способ, как при получении B24, 2,4-дифтор-3-(4-метилпиперазин-1-ил)-6-нитроанилин было получено с выходом 91%. Далее реакции восстановления и конденсации были проведены аналогично, как при получении соединения B22, целевое соединение B52 было получено с выходом 85%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.38 (s, 1H), 9.30 (d, 1H), 7.67 (t, 1H), 7.57 (t, 1H), 7.48 (d, 1H), 7.29-7.36 (m, 2H), 3.54 (s, 3H), 3.16 (s, 2H), 2.48 (s, 2H), 2.27 (s, 2H), 1.26 (s, 2H).
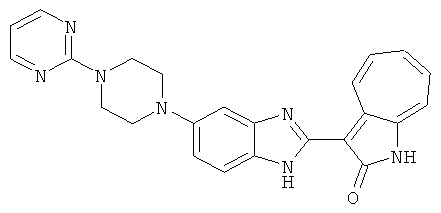
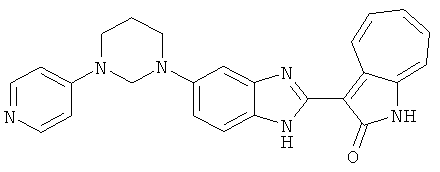
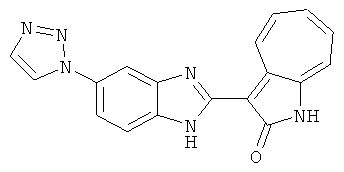
Получение B60: Используя такой же способ, как при получении B24, 2-нитро-5-(4-(пиредин-4-ил)пиперазин-1-ил)анилин был получен с выходом 81%. Далее реакции восстановления и конденсации были проведены аналогично, как при получении соединения B22, целевое соединение B60 было получено с выходом 82%.1H-ЯМР (500 МГц, DMSO-d6+TFA-d) δ (ppm) 13.50 (s, 1H), 12.81 (s, 1H), 8.57 (d, 1H), 8.32 (d, 2H), 7.88 (m, 1H), 7.77 & 7.73 (d, 1H), 7.63 (t, 1H), 7.33 (t, 1H), 7.30 (d, 2H), 3.96 (s, 4H), 3.46 (s, 4H). LC-MS (m/z) 422 [M+1].
Получение B76: Используя такой же способ, как при получении B24, 5-(1,4-диазепан-1-ил)-2-нитроанилин был получен с выходом 80%. Далее реакции восстановления и конденсации были проведены аналогично, как при получении соединения B22, целевое соединение B76 было получено с выходом 80%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 11.67, 11.79 (s, 1H), 9.24, 9.34 (d, 1H), 7.39-7.50 (m, 3H), 7.31 (t, 1H), 7.19 (t, 1H), 6.89, 6.95 (s, 1H), 6.70 (d, 1H), 3.50-3.57 (m, 5H), 2.91 (s, 2H), 2.65 (s, 2H), 1.84 (s, 2H).
Получение B77: Используя такой же способ, как при получении B24, 5-(4-метил-1,4-диазепан-1-ил)-2-нитроанилин был получен с выходом 85%. Далее реакции восстановления и конденсации были проведены аналогично, как при получении соединения В22, целевое соединение B77 было получено с выходом 89%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.13 (s, 1H), 11.69, 11.81 (s, 1H), 9.25, 9.35 (d, 1H), 7.17-7.53 (m, 5H), 6.89, 6.94 (s, 1H), 6.69 (d, 1H), 3.47-4.07 (m, 6H), 3.17 (s, 2H), 2.27 (s, 3H), 1.93 (d, 2H).
Получение B85: Используя такой же способ, как при получении B24, 4-фтор-5-(4-метил-1,4-диазепан-1-ил)-2-нитроанилин был получен с выходом 89%. Далее реакции восстановления и конденсации были проведены аналогично, как при получении соединения B22, целевое соединение B85 было получено с выходом 88%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.18 (s, 1H), 11.96, 12.02 (s, 1H), 9.26, 9.30 (d, 1H), 7.51-7.57 (m, 2H), 7.44-7.49 (m, 2H), 7.33-7.38 (m, 2H), 7.21-7.30 (m, 2H), 2.72 (s, 2H), 2.63 (s, 2H), 2.32 (s, 2H), 1.94 (s, 2H), 1.25 (s, 2H).
Получение B86: Используя такой же способ, как при получении B24, 2,4-дифтор-3-(4-метил-1,4-диазепан-1-ил)-6-нитроанилин был получен с выходом 96%. Далее реакции восстановления и конденсации были проведены аналогично, как при получении соединения B22, целевое соединение B86 было получено с выходом 88%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.32 (s, 1H), 9.29 (d, 1H), 7.64 (t, 1H), 7.54 (t, 1H), 7.44 (d, 1H), 7.32 (t, 1H), 7.27 (d, 1H), 3.28 (s, 3H), 2.68 (t, 2H), 2.64 (t, 2H), 2.33(s, 4H), 1.87-1.91 (m, 2H).
Получение B101: Используя такой же способ, как при получении B24, 1-(3-амино-4-нитрофенил)-N,N-диметилпиперадин-4-амин был получен с выходом 86%. Далее реакции восстановления и конденсации были проведены аналогично, как при получении соединения В22, целевое соединение B101 было получено с выходом 78%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 11.90 (d, 1H), 9.30 (m, 1H), 7.55 (m, 3H), 7.46 (t, 1H), 7.25 (m, 3H), 6.93 (m 1H), 3.67 (d, 2H), 2.68 (m, 2H), 2.29 (s, 6H), 1.90 (d, 2H), 1.58 (m, 2H).
Получение B102: Используя такой же способ, как при получении B24, 5-([1,4′-бипиперадин]-1′-ил)-2-нитроанилин был получен с выходом 76%. Далее реакции восстановления и конденсации были проведены аналогично, как при получении соединения B22, соединение B102 было получено с выходом 79%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.20 (b, 1H), 11.90 (d, 1H), 8.30 (s, 1H), 9.30 (m, 1H), 7.54 (m, 3H), 7.40 (s 1H), 7.38 (m, 2H), 7.20 (m, 1H), 3.73 (d, 2H), 2.82 (m, 2H), 2.03 (m, 2H), 1.70 (m, 2H), 1.21 (m, 2H).
Получение B103: Используя такой же способ, как при получении B24, 5-(4-морфолинпиперадин-1-ил)-2-нитроанилин был получен с выходом 76%. Далее реакции восстановления и конденсации были проведены аналогично, как при получении соединения В22, целевое соединение В 103 было получено с выходом 81%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 11.90 (d, 1H), 9.28 (m, 1H), 7.50 (m, 3H), 7.35 (t, 1H), 7.19 (m, 3H), 6.91 (t 1H), 3.63 (d, 2H), 3.58 (s, 1H), 2.66 (m, 2H), 2.26 (s, 6H), 1.88 (d, 2H), 1.55 (b, 2H).
Получение B104: Используя такой же способ, как при получении B24, 2-нитро-5-(4-(пирролидин-1-ил)пиперидин-1-ил) анилин был получен с выходом 77%. Далее реакции восстановления и конденсации были проведены аналогично, как при получении соединения B22, целевое соединение B104 было получено с выходом 78%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 11.0 (d, 1H), 9.46 (m, 1H), 7.6 (d, 1H), 7.49 (m, 2H), 7.2-7.51 (m, 3H), 7.01 (b, 2H), 3.69 (m, 4H), 2.79 (q, 1H), 2.71 (b, 4H), 2.05 (b, 2H), 1.84 (b, 4H), 1.25 (s, 2H).
Получение B105: Используя такой же способ, как при получении B24, 5-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)-2-нитроанилин был получен с выходом 79%. Далее реакции восстановления и конденсации были проведены аналогично, как при получении соединения B22, целевое соединение B105 было получено с выходом 72%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 11.96 (d, 1H), 9.37 (m, 1H), 7.56 (m, 3H), 7.40 (t, 1H), 7.29 (m, 3H), 6.92 (m, 1H), 3.67 (m, 1H), 3.3 (s, 4H), 3.12 (m, 2H), 2.69 (m, 2H), 2.53 (s, 4H), 2.17 (s, 3H), 1.92 (m, 2H), 1.62 (m, 2H). B106,1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 9.16 (d, 1H), 7.59 (t, 1H), 7.48 (t, 1H), 7.35 (m, 5H), 7.251 (1H, s), 7.08 (d, 1H), 3.37 (s, 2H), 3.29 (t, 1H), 2.75 (d, 4H), 2.58 (m, 4H), 1.22 (t, 3H).
Получение B107: Используя такой же способ, как при получении B24, 1-(3-амино-4-нитрофенил)пиперидин-4-ол был получен с выходом 79%. Далее реакции восстановления и конденсации были проведены аналогично, как при получении соединения B22, целевое соединение B107 было получено с выходом 72%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.19 (b, 1H), 11.88 (d, 1H), 9.28 (m, 1H), 9.31 (m, 1H), 7.55 (m, 3H), 7.41 (t, 1H), 7.21 (m, 3H), 6.85 (t, 1H), 3.66 (m, 1H), 3.53 (b, 2H), 2.85 (q, 2H), 1.89 (b, 2H), 1.58 (m, 2H).
Получение B108: Используя такой же способ, как при получении B24, 1-(3-амино-4-нитрофенил)пиперидин-4-карбоксамид был получен с выходом 85%. Далее реакции восстановления и конденсации были проведены аналогично, как при получении соединения B22, соединение B108 было получено с выходом 67%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 9.18 (d, 1H), 7.60 (m, 2H), 7.52 (t, 1H), 7.37 (m, 4H), 7.27 (t, 1H), 7.10 (d, 1H), 3.74 (d, 2H), 2.41 (t,1H), 2.30 (m, 4H), 1.94 (m, 4H).
Получение B109: Используя такой же способ, как при получении B24, 5-(4-(этоксиметил)пиперидин-1-ил)-2-нитроанилин был получен с выходом 82%. Далее реакции восстановления и конденсации были проведены аналогично, как при получении соединения B22, целевое соединение B109 было получено с выходом 72%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 9.05 (d, 1H), 7.51 (m, 2H), 7.37 (m, 1H), 7.24 (m, 4H), 7.07 (m, 1H), 6.98 (t, 1H), 3.61 (d, 2H), 3.5 (m, 2H), 2.70 (t, 4H), 1.87 (d, 4H), 1.46 (m, 1H), 1.23 (m, 3H).
Получение B110: Используя такой же способ, как при получении B24, (R)-1-(3-амино-4-нитрофенил)-N,N-диметилпиролидин-3-амин был получен с выходом 68%. Далее реакции восстановления и конденсации были проведены аналогично, как при получении соединения B22, целевое соединение B110 было получено с выходом 67%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.1 (b, 1H), 11.77 (m, 1H), 9.32 (m, 1H), 7.42-7.50 (m, 3Н), 7.34 (t, 1H), 7.22 (t, 1H), 6.79 (s, 1H), 6.59 (m, 1H), 3.48 (m, 2H), 3.20 (m, 1H), 3.11 (m, 1H), 2.88 (m, 1H), 2.25 (s, 6H), 2.21 (m, 1H), 1.87(m, 1H).
Получение B111: Используя такой же способ, как при получении B24, 5-морфолино-2-нитроанилин был получен с выходом 77%. Далее реакции восстановления и конденсации были проведены аналогично, как при получении соединения B22, целевое соединение B111 было получено с выходом 79%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 9.30 (d, 1H), 7.56 (m, 2H), 7.38 (d, 1H), 7.26 (m, 4H), 7.17 (s, 1H), 6.95 (d, 1H), 3.04 (s, 4H), 1.98 (s, 4H).
Получение B112: К раствору этилацетата (10 мл) 1-метилпиперазине (400.6 мг, 4 ммол) и триэтиламине (404.8 мг, 4 ммол) добавили в раствор этилацетата (40 мл) 4-хлор-3-нитробензинсульфонил хлорид (1.02 г, 4 ммол). После перемешивания при комнатной температуре в течение 30 минут раствор промыли водой (50 мл), 1 М раствором бикарбоната натрия (20 мл) и насыщенным солевым раствором. Органический слой отделили, высушили с ангидридмагнийсульфатом, отфильтровали и затем концентрировали под вакуумом до получения качественного продукта. Вышеупомянутый сульфамид (639.5 мг, 2 ммол) растворили в растворе метанольного амония (7М, 20 мл) размещенного в герметичном стальном баллоне, и затем раствор нагрели до температуры 100°C в течение 18 часов. Реакционную смесь концентрировали. Остаток обработали раствором 1М карбоната натрия (50 мл), этилацетат (100 мл) и изопропанола (15 мл) и перемешали в течение 30 минут. Раствор этилацетата разделили, высушили с ангидридмагнийсульфатом, отфильтровали и затем концентрировали под пониженным давлением до получения 422.3 мг продукта, выход 70.3%. Нитроанилин (157.2 мг, 0.52 ммол) растворили в метаноле (30 мл) и затем добавили никель Ренея (0.5 г). Смесь гидрировали при давлении 1 атм в течение 1 дня. Никель Ренея отфильтровали и промыли с метанолом (30 мл, 2 раза). Далее, 3-формил-1-азаазулен-2-он (86.6 мг, 0.5 ммол) и аммонийметавандат (2.9 мг, 0.025 ммол) добавили в фильтрат и перемешали при температуре 50°C в течение 48 часов. Метанол выпарили, и далее остаток пропустили через колонну с силикагелем и элютировали с DCM/MeOH (10/1) до получения 100 мг продукта, выход 47.2%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.58, 12.62 (s, 1H), 12.34 (s, 1H), 9.39, 9.41 (d, 1H), 7.97, 8.11 (s, 1H), 7.83, 7.87 (d, 1H), 7.70 (dd, 1H), 7.58-7.63 (dt, 1H), 7.49-7.53 (m, 2H), 7.36-7.40 (dt, 1H), 2.90 (s, 4H), 2.36 (d, 4H), 2.12 (d, 3H).
Получение B113: Используя такой же способ, как при получении B112, как сульфамид 1-((4-хлор-3-нитрофенил)сульфонил)-4-метил-1,4-диазепан, так и нитроанилин 4-((4-метил-1,4-диазепан-1-ил)сульфонил)-2-нитроанлин были получены с качественным выходом. Целевое соединение В113 было получено с выходом 21%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.54, 12.58 (s, 1H), 12.33 (s, 1H), 9.38, 9.40 (d, 1H), 8.00, 8.14 (s, 1H), 7.80, 7.84 (d, 1H), 7.69 (dd, 2H), 7.56-7.62 (m, 2H), 7.49-7.54 (m, 1H), 7.35-7.39 (m, 1H), 3.32 (s, 3H), 2.55 (s, 2H), 2.22 (s, 3H), 1.72 (d, 2H).
Получение B114: Используя такой же способ, как при получении B18, целевое соединение B114 было получено с выходом 40%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.49, 12.53 (s, 1H), 12.31 (s, 1H), 9.38, 9.43 (d, 1H), 7.98, 8.17 (s, 1H), 7.75-7.82 (m, 3H), 7.63-7.72 (m, 3H), 7.55-7.61 (m, 3H), 7.49 (t, 1H), 7.35 (dd, 1H).
Получение B115: Имидазол (7.49 г, 0.11 ммол), 5-фтор-2-нитроанилин (15.61 г, 0.1 ммол) и триэтиламин (11.13 г, 0.11 мл) были добавлены в ацетонитрил (200 мл) и нагреты вплоть до дефлегмации в течение 12 часов. Растворитель выпарили, добавили воду (50 мл) к остатку и перемешали в течение 30 минут. Твердый продукт желтого цвета отфильтровали, промыли с водой (25 мл, 2 раза) и в конце высушили в вакууме до получения 18.71 г 5-(1H-имидазод-1-ил)-2-нитроанилина, выход 92%. Используя такой же способ, как при получении B22, целевое соединение B115 было получено с выходом 88%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.30 (b, 1H), 9.38 (d, 1H), 8.6 (b, 1H), 7.90-7.88 (m, 2H), 7.79 (d, 1H), 7.66 (t 1H), 7.56 (t, 1H), 7.47 (m, 2H), 7.36 (m, 2H).
Получение B116: Используя такой же способ, как при получении B115, 2-нитро-5-(2H-1,2,3-триазол-2-ил)анилин был получен с выходом 68%. Используя такой же способ, как при получении B22, целевое соединение B116 было получено с выходом 75%.1H-ЯМР (DMSO-d6) δ (ppm) 12.00 (s, 1Н), 9.18 (d, 1Н), 8.69 (s, 1H), 8.05 (s, 2H), 7.56-7.66 (m, 2H), 7.45-7.48 (m, 2H), 7.34 (t, 1H), 7.25 (d, 1H), 7.16 (d, 1H).
Пример 2
Получение B23: К раствору B22 (345.4 мг, 1 ммол) с ацетонитрилом (10 мл) добавили диизопропилэтиламин (142.2 мг, 1.1 ммол) и затем трифторэтилйодид (314.9 мг, 1.5 ммол), реакционную смесь перемешали до дефлегмации в течение 16 часов. Полученной смеси дали охладится при комнатной температуре, концентрировали под вакуумом и далее очистили хроматографией на силикагеле с помощью этилацетатметанола (10:1-2:1), в качестве элюента, до получения 210.4 мг целевого соединения B23, выход 49.2%.1Н NMR (500 МГц, DMSO-d6) δ (ppm) 11.9 (b, 1H), 9.27 (d, 1H), 7.56-7.50 (m, 2H), 7.46 (t, 1H), 7.39 (d, 1H), 7.25 (t, 1H), 7.17, (s, 1H), 6.93 (d, 1H), 3.16 (s, 2H), 3.13 (t, 4H), 3.00 (t, 4H).
Получение B28: Используя такой же способ, как при получении B23 и этилйодид, в качестве алкилирующего агента, целевое соединение B28 было получено с выходом 75%.1Н NMR (500 МГц, DMSO-d6) δ (ppm) 12.18 (s, 1H), 11.87, 11.93 (2s, 1H), 9.28, 9.34 (2d, 1H), 7.49-7.55 (m, 2H), 7.45 (t, 1H), 7.36 (t, 1H), 7.24 (t, 1H), 7.13, 7.18 (2s, 1H), 6.92 (t, 1H), 3.11 (s, 4H), 2.54 (s, 4H), 2.39 (q, 2H), 1.04 (t, 3H).
Получение B38: Используя такой же способ, как при получении B23, и фторэтилтозилат, в качестве алкилирующего агента, целевое соединение B38 было получено с выходом 70%.1H NMR (500 МГц, DMSO-d6) δ (ppm) 11.88, 11.94 (2s, 1H), 9.28, 9.34 (2d, 1H), 7.43-7.56 (m, 3H), 7.37 (t, 1H), 7.18-7.26 (m, 2H), 6.93 (t, 1H), 4.63 (t, 1H), 4.54 (t, 1H), 3.12 (s, 4H), 2.72 (t, 1H), 2.64 (s, 4H).
Получение B39: Используя такой же способ, как при получении B38, и фторпропилтозилат, в качестве алкилирующего агента, целевое соединение B38 было получено с выходом 65%.1H NMR (500 МГц, DMSO-d6) δ (ppm) 11.88, 11.94 (2s, 1H), 9.29, 9.35 (d, 1H), 7.43-7.55 (m, 3H), 7.37 (t, 1H), 7.23 (q, 1H), 7.15 (d, 1H), 6.92 (t, 1H), 4.55 (t, 1H), 4.45 (t, 1H), 3.11 (s, 4H), 2.55 (t, 4H), 2.43 (t, 2H),1.88 (t, 1H), 1.82 (t, 1H).
Получение B78, B79 и B80: К раствору B76 (359.4 мг, 1 ммол) в ацетонитриле (10 мл) добавили диизопропилэтиламин (142.2 мг, 1.1 ммол) затем фторэтилтозилат (327.4 мг, 1.5 ммол), реакционную смесь перемешали до дефлегмации в течение 16 часов. Полученной смеси дали охладится при комнатной температуре, концентрировали под вакуумом и далее очистили хроматографией на силикагеле с помощью этилацетатметанола (10:1-2:1), в качестве элюента, до получения 94.8 мг целевого соединения B78, выход 21%; 141.9 мг целевого соединения B79, выход 35%; и 89.2 мг целевого соединения B80, выход 22%.
B78,1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 11.74, 11.88 (d, 1H), 9.40, 9.50 (d, 1H), 7.49-7.67 (m, 4H), 7.25-7.33 (m, 1H), 6.87-7.00 (m, 1H), 6.70-6.76 (m, 1H), 1.87-4.81 (m, 18H).
B79,1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 11.72, 11.85 (s, 1H), 9.39, 9.49 (d, 1H), 7.44-7.67 (m, 4H), 7.25-7.33 (m, 1H), 6.90, 6.94 (s, 1H), 6.72 (t, 1H), 4.76 (d, 2H), 4.55 (d, 2H), 3.50-3.58 (m, 4H), 2.92 (d, 2H), 2.67 (d, 2H), 1.84-1.86 (m, 2H).
B80,1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.13 (s, 1H), 11.68, 11.71, 11.28 (s, 1H), 9.24, 9.34 (d, 1H), 7.40-7.53 (m, 3H), 7.31-7.35 (m, 1H), 7.18-7.25 (m, 1H), 6.95-7.00 (m, 1H), 6.73-6.75 (m, 1H), 1.86-3.93 (m, 14H).
Получение B81, B82 и B83: Используя такой же способ, как при получении B78, и фторпропилтозилат, в качестве алкилирующего агента, целевые соединения B81, B82 и B83 были получены с выходом 19%, 31% и 21%.
B81,1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 11.31, 11.73, 11.87 (s, 1H), 9.38, 9.48, 9.75, 9.82 (d, 1H), 8.33-8.34, 7.81-7.84, 7.40-7.61 (m, 4H), 7.24-7.32 (m, 1H), 6.84-6.99 (m, 1H), 6.72 (t, 1H), 1.75-4.92 (m, 17H), В83,1H-ЯМР (CDCl3, 500 МГц) δ 1.81-4.55 (m, 22H), 11.19, 11.27 (s, 1H), 9.44, 9.50 (d, 1H), 6.68-7.62 (m, 6H).
В82,1Н-ЯМР (CDCl3, 500 МГц) δ 1.17-4.54 (m, 16H), 10.85, 11.11 (s, 2H), 9.37-9.40 (m, 1H), 7.58 (s, 1H), 7.11-7.43 (m, 5H), 6.71-6.76 (m, 1H).
Пример 3
Получение B43: К раствору соединения B22 (345.4 мг, 1 ммол) в этаноле (10 мл) добавили триэтиламин (253.0 мг, 2.5 ммол) и 2-нафталингид (553.6 мг, 3.5 ммол). После одного часа в смесь добавили цианоборгидрид натрия (351.9 мг, 5.6 ммол), которую перемешали при комнатной температуре в течение 1 дня. Смесь концентрировали под пониженным давлением. Сырой продукт очистили хроматографией на силикагеле с помощью этилацетатметанола (10:1-2:1), в качестве элюента, до получения 317.4 мг B43, выход 65%.1Н NMR (500 МГц, DMSO-d6) δ (ppm) 12.18 (s, 1H), 11.87, 11.93 (2s, 1H), 9.27, 9.33 (2d, 1H), 7.84-7.90 (m, 4H), 7.45-7.60 (m, 6H), 7.38 (t, 1H), 7.18-7.26 (m, 2H), 6.93 (t, 1H), 3.71 (s, 2H), 3.14 (s, 4H), 2.62 (s, 4H).
Получение B44: Используя такой же способ, как при получении B43, и бензальдегид, в качестве алкилирующего агента, целевое соединение B44 было получено с выходом 79%.1H NMR (500 МГц, DMSO-d6) δ (ppm) 12.14 (s, 1H), 11.85, 11.91 (2s, 1H), 9.31, 9.36 (2d, 1H), 7.80 (d, 2H), 7.58 (d, 2H), 7.50-7.55 (m, 2H), 7.44 (t, 1H), 7.38 (t, 1H), 7.24 (q, 1H), 7.17 (d, 1H), 6.93 (t, 1H), 3.66 (s, 2H), 3.20 (s, 4H), 2.57 (s, 4H).
Получение B45: Используя такой же способ, как при получении B43, и п-диметиламинобензальдегид, в качестве алкилирующего агента, целевое соединение B45 было получено с выходом 77%.1H NMR (500 МГц, DMSO-d6) δ (ppm) 12.20 (s, 1H), 11.85, 11.91 (2s, 1H), 9.25, 9.31 (2d, 1H), 7.40-7.52 (m, 3H), 7.33 (t, 1H), 7.21 (q, 1H), 7.13 (t, 1H), 7.10 (d, 2H), 6.89 (t, 1H), 6.66 (d, 1H), 3.39 (s, 2H), 3.08 (s, 4H), 2.83 (s, 6H), 2.50 (s, 4H).
Получение B46: Используя такой же способ, как при получении B43, и 5-бромфуран-2-карбальдегид, в качестве алкилирующего агента, целевое соединение B46 было получено с выходом 87%.1Н NMR (500 МГц, DMSO-d6) δ (ppm) 12.18 (s, 1H), 11.87, 11.93 (2s, 1H), 9.28, 9.35 (2d, 1H), 8.60 (s, 1H), 7.43-7.57 (m, 3H), 7.38 (t, 1H), 7.13-7.27 (m, 2H), 6.92 (t, 1H), 6.51 (d, 1H), 6.41 (d, 1H), 3.66 (s, 2H), 3.27 (s, 4H), 2.75 (s, 4H).5.
Получение B47: Используя такой же способ, как при получении B43 и пиридин-4-карбальдегид, в качестве алкилирующего агента, целевое соединение B47 было получено с выходом 82%.1H NMR (500 МГц, DMSO-d6) δ (ppm) 11.88, 11.94 (2s, 1H), 9.27, 9.30 (2d, 1H), 8.53 (d, 2H), 7.43-7.55 (m, 3H), 7.37 (d, 2H), 7.34 (t, 1H), 7.14-7.25 (m, 3H), 6.92 (t, 1H), 3.59 (s, 2H), 3.15 (s, 4H), 2.58 (s, 4H).
Получение B48: Используя такой же способ, как при получении B43 и пиридин-3-карбальдегид, в качестве алкилирующего агента, целевое соединение B48 было получено с выходом 91%.1Н NMR (500 МГц, DMSO-d6) δ (ppm) 11.87, 11.94 (2s, 1H), 9.27, 9.33 (2d, 1H), 8.54 (s, 1H), 8.42 (d, 2H), 7.75 (d, 1H), 7.43-7.56 (m, 3H), 7.36-7.39 (m, 2H), 7.23 (q, 1H), 7.15 (d, 2H), 6.92 (t, 1H), 3.58 (s, 2H), 3.12 (s, 4H), 2.57 (s, 4H).
Получение B49: Используя такой же способ, как при получении B43, пиридин-2-карбальдегид, в качестве алкилирующего агента, целевое соединение B49 было получено с выходом 81%.1H NMR (500 МГц, DMSO-d6) δ (ppm) 11.88, 11.94 (2s, 1H), 9.28, 9.34 (2d, 1H), 8.42, 8.52 (2d, 1H), 7.78 (t, 1H), 7.43-7.57 (m, 3H), 7.38 (t, 1H), 7.18-7.35 (m, 3Н), 6.93 (t, 1H), 6.50 (s, 1H), 3.67 (s, 2H), 3.14 (s, 4H), 2.62 (s, 4H).
Получение B87: Используя такой же способ, как при получении B48, и соединения B76 в качестве исходного сырья, целевое соединение B87 было получено с выходом 90%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.11, 12.16 (s, 1H), 11.68, 11.80 (s, 1H), 9.25, 9.34 (d, 1H), 8.49 (s, 1H), 8.44 (d, 1H), 7.69 (d, 1H), 7.17-7.51 (m, 5H), 6.89, 6.95 (s, 1H), 6.70 (d, 1H), 3.66 (s, 2H), 3.56 (s, 2H), 3.52 (s, 2H), 2.75 (s, 2H), 2.56 (s, 2H), 1.98 (s, 2H).
Пример 4
Получение B59: 5-бомофуран -2-карбальдегид (262.5 мг, 1.5 ммол), соединение BN22 (345.4 мг, 1 ммол) и диизопропилэтиламин (258.5, 2 ммол) в н-бутаноле (10 мл) перемешали при температуре 140°C в течение 1 дня. Смесь охладили при комнатной температуре, разбавили водой (50 мл), и выделили с этилацетатом (20 мл, 2 раза). Растворы этилацетатата объединили, высушили с ангидридмагнийсульфатом и далее выпарили под пониженным давлением. Остаток очистили хроматографией на силикагеле с помощью этилацетатметанола (10:1-2:1), в качестве элюента, до получения 258.6 мг целевого соединения B59, выход 59%.1H NMR (500 МГц, DMSO-d6) δ (ppm) 11.94, 11.99 (2s, 1H), 9.28, 9.34 (2d, 1H), 9.01 (s, 1H), 7.51-7.59 (m, 3H), 7.46 (t, 1H), 7.38 (t, 1H), 7.24-7.27 (m, 2H), 6.99 (t, 1H), 5.71 (d, 1H), 3.57 (t, 4H), 3.23 (t, 4H).
Получение B61: Используя такой же способ, как при получении B59, и 3,4,5-трифторпиридин, в качестве исходного сырья, целевое соединение B61 было получено с выходом 80%.1H NMR (500 МГц, DMSO-d6) δ (ppm) 11.30, 11.33 (2s, 1H), 10.44, 10.53 (2s, 1H), 9.46, 9.49 (2d, 1H), 8.15 (s, 2H), 7.50 (q, 1H), 7.30-7.47 (m, 3H), 7.21 (t, 1H), 7.01-7.05 (m, 2H), 3.73 (s, 4H), 3.31 (s, 4H).
Получение B63: Используя такой же способ, как при получении B59 и 3-фторпиридин, в качестве исходного сырья, целевое соединение B63 было получено с выходом 65%.1H NMR (500 МГц, DMSO-d6) δ (ppm) 12.00, 12.06 (2s, 1H), 9.42, 9.48 (2d, 1H), 7.51-7.70 (m, 6H), 7.33 (q, 1H), 7.20 (d, 1H), 6.97 (t, 1H), 3.57 (t, 4H), 3.13 (t, 2H), 3.07 (t, 2H).
Получение B64: Используя такой же способ, как при получении B59, и 2-хлорпиридин, в качестве исходного сырья, целевое соединение B64 было получено с выходом 85%.1H NMR (500 МГц, DMSO-d6) δ (ppm) 11.97, 12.02 (2s, 1H), 9.30, 9.36 (2d, 1H), 8.18 (d, 1H), 7.57-7.61 (m, 3H), 7.48 (t, 1H), 7.40 (t, 1H), 7.24-7.28 (m, 2H), 7.03 (t, 1H), 6.93 (d, 1H), 6.70 (t, 1H), 3.68 (t, 4H), 3.22 (t, 4H).
Получение B65: Используя такой же способ, как при получении B59, и 2-хлор-3-нитропиридин, в качестве исходного сырья, целевое соединение B65 было получено с выходом 81%.1Н NMR (500 МГц, CDCl3) δ (ppm), 11.20, 11.26 (2s, 1H), 9.45 (d, 1H), 8.35 (t, 1H), 8.15 (d, 1H), 7.17-7.50 (m, 7H), 7.01 (d, 1H), 6.67 (m, 1H), 3.67 (s, 4H), 3.32 (s, 4H).
Получение B67: Используя такой же способ, как при получении B59, и 4-хлор-N,N,6-триметилпиримидин-2-амин, в качестве исходного сырья, целевое соединение В67 было получено с выходом 81%.1H NMR (500 МГц, CDCl3) δ (ppm) 11.30, 11.36 (2s, 1H), 9.45 (m, 1H), 7.69-7.71 (m, 1H), 7.29-7.51 (m, 6H), 7.04 (m, 2H), 3.78 (t, 4H), 3.20 (t, 4H), 3.15 (s, 6H), 2.33 (s, 3Н).
Получение B68: Используя такой же способ, как при получении B59, и 2-хлорпиримидин, в качестве исходного сырья, целевое соединение B68 было получено с выходом 80%.1H NMR (500МГц, DMSO-d6) δ (ppm) 11.93, 11.98 (2s, 1H), 9.29, 9.30 (2d, 1H), 8.39, 8.40 (d, 2H), 7.53-7.58 (m, 2H), 7.42-7.48 (m, 1H), 7.39 (t, 1H), 7.21-7.28 (m, 2H), 7.00 (t, 1H), 6.65 (t, 1H), 3.93 (t, 4H), 3.18 (t, 4H).
Получение B69: Используя такой же способ, как при получении B59, и 2-хлорпиразин, в качестве исходного сырья, целевое соединение B69 было получено с выходом 75%.1H NMR (500 МГц, DMSO-d6) δ (ppm) 11.94, 12.00 (2s, 1H), 9.29, 9.35 (2d, 1H), 8.40 (s, 1H), 8.12 (t, 1H), 7.87 (d, 1H), 7.52-7.56 (m, 2H), 7.46 (t, 1H), 7.37 (t, 1H), 7.22-7.27 (m, 2H), 6.71-7.00 (m, 1H),3.76(t, 4H), 3.17 (t, 4H).
Получение B93: 4-хлорпиридигидрохлорид (205.5 мг, 1.5 ммол), соединение B76 (359.4 мг, 1 ммол) и диизопропилэтиламин (258.5, 2 ммол) в н-бутаноле (10 мл) перемешали при температуре 140°C в течение 1 дня. Смесь охладили при комнатной температуре, разбавили водой (50 мл), и разделили с этилацетатом (20 мл, 2 раза). Растворы этилацетатата объединили, высушили с ангидридмагнийсульфатом и далее выпарили под пониженным давлением. Остаток очистили хроматографией на силикагеле с помощью этилацетатметанола (10:1-2:1), в качестве элюента, до получения 315.6 мг целевого соединения B93, выход 72%.1H NMR (500 МГц, DMSO-d6) δ (ppm) 12.17 (s, 1H), 11.74 (s, 1H), 8.11 (m, 2H), 7.49-7.53 (m, 1H), 7.46 (d, 2H), 7.35 (m, 1H), 7.23 (m, 1Н), 7.03 (m, 1H), 6.93 (d, 2H), 6.77 (d, 1H), 3.82 (s, 1H), 3.70 (s, 1H), 3.56 (s, 1H), 3.51 (s, 1H), 3.17 (s, 1H), 2.09 (s, 2H), 1.76 (s, 1H), 1.24 (m, 2H).
Получение B94: Используя такой же способ, как при получении B93, и 2-хлорпиридин, в качестве исходного сырья, целевое соединение B94 было получено с выходом 75%.1Н-ЯМР (DMSO-d6, 500 МГц) δ 2.04-4.04 (m, 10Н), 12.12, 12.16 (s, 1H), 11.69, 11.81 (s, 1H), 9.24, 9.35 (d, 1H), 7.40-8.05 (m, 4H), 7.30-7.35 (m, 1H), 7.18-7.25 (m, 1H), 6.97, 7.03 (s, 1H), 6.75-6.85 (m, 1H), 6.67 (t, 1H), 6.50 (t, 1H).
Получение B95: Используя такой же способ, как при получении B93, и 2-хлор-3-нитропиридин, в качестве исходного сырья, целевое соединение B95 было получено с выходом 85%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.12, 12.15 (s, 1H), 11.66, 11.79 (s, 1H), 9.22, 9.32 (d, 1H), 8.30 (d, 1H), 8.05 (d, 1H), 7.18-7.54 (т, 5Н), 6.92, 6.96 (s, 1H), 6.68-6.72 (т, 2Н), 2.03-3.82 (d, 10H).
Получение B96: Используя такой же способ, как при получении B93, и 2,6-дихлорпиридин, в качестве исходного сырья, целевое соединение B96 было получено с выходом 85%.1H-ЯМР (DMSO-d6, 500 МГц) δ.1.93-3.65 (m, 10Н), 12.14 (s, 1H), 11.71, 11.83 (s, 1H), 9.26 (d, 1H), 7.42-7.95 (m, 5Н), 7.33 (d, 1H), 7.22 (t, 1H), 7.00 (s, 1H), 6.75 (d, 1H).
Получение B97: Используя такой же способ, как при получении B93, и 4-хлор-N,N,6-триметилпиримидин-2-амин, в качестве исходного сырья, целевое соединение B97 было получено с выходом 81%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.13 (s, 1H), 11.69, 11.81 (s, 1H), 9.26 (s, 1H), 7.41-7.52 (m, 3H), 7.32 (d, 1H), 7.23 (t, 1H), 7.01 (s, 1H), 6.76 (d, 1H), 5.89 (s, 1H), 3.68 (s, 2H), 3.48 (s, 2H), 3.31 (s, 4H), 3.04 (s, 6H), 2.11 (s, 3H), 2.01 (t, 2H).
Получение B98: Используя такой же способ, как при получении B93, и 2-хлопиримидин, в качестве исходного сырья, целевое соединение B97 было получено с выходом 87%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.13 (s, 1H), 11.69, 11.80 (s, 1H), 9.23, 9.33 (d, 1H), 7.32-7.55 (m, 4H), 7.19-7.26 (m, 1H), 6.96, 7.01 (s, 1H), 6.77 (t, 1H), 6.55 (t, 1H), 3.98 (s, 2H), 3.66 (s, 2H), 3.62 (d, 2H), 2.55 (s, 2H), 2.00 (d, 2H).
Получение B99: Используя такой же способ, как при получении B93, и 2-хлопиразин, в качестве исходного сырья, целевое соединение B99 было получено с выходом 80%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.12 (s, 1H), 11.69, 11.81 (s, 1H), 9.25 (s, 1H), 8.18 (s, 1H), 8.00 (s, 1H), 7.71 (s, 1H), 7.41-7.52 (m, 3H), 7.33 (d, 1H), 7.21 (t, 1H), 7.02 (s, 1H), 6.77 (d, 1H), 3.90 (s, 2H), 3.68 (s, 2H), 3.57 (s, 2H), 3.50 (s, 2H), 2.04 (t, 2H).
Получение B100: Используя такой же способ, как при получении B93, и 2-хлор-6-метилпиразин, в качестве исходного сырья, целевое соединение В 100 было получено с выходом 78%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.11 (s, 1H), 11.70, 11.82 (s, 1H), 9.23, 9.34 (d, 1H), 7.30-7.53 (m, 4H), 7.16 7.24 (m, 2H), 6.97-7.07 (m, 2H), 6.75(d, 1H), 3.93 (s, 2H), 3.67 (s, 2H), 3.54 (s, 2H), 3.46 (s, 2H), 2.38 (s, 3H), 2.03 (t, 2H).
Пример 5
Получение B57: К смеси соединения B22 (345.4 мг, 1 ммол) и диизопропилэтиламин (193.8 мг, 1.5 ммол) в дихлорметане (10 мл) добавили метансульфонил хлорид (137.5 мг, 1.2 ммол) и перемешали в течение ночи при комнатной температуре. Смесь промыли насыщенным раствором бикарбоната натрия (10 мл). Раствор дихлорметана разделили, высушили с ангидридмагнийсульфатом и далее выпарили под пониженным давлением. Остаток очистили хроматографией на силикагеле с помощью этилацетатметанола (10:1-10:3), в качестве элюента, до получения 385.4 мг целевого соединения B57, выход 91%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.20 (s, 1H), 11.97, 12.02 (s, 1H), 9.32, 9.38 (d, 1H), 7.47-7.61 (m, 3H), 7.40 (t, 1H), 7.23-7.31 (m, 2H), 6.97-7.00 (m, 1H), 3.19-3.25 (m, 8H), 2.97 (s, 3H).
Получение B58: Используя такой же способ, как при получении B57, и 2-хлорэтанолсульфонил хлорид, в качестве сульфирующего агента, целевое соединение B58 было получено с выходом 78%.1H NMR (500 МГц, DMSO-d6) δ (ppm) 12.7 (s, 1H), 9.27 (s, 1H), 8.92 (d, 1H), 7.68-7.78 (m, 3H), 7.52 (t, 1H), 7.30 (s, 1H), 7.20 (t, 1H), 3.29-3.41 (m, 12H).
Получение B89: К смеси соединения B79 (359.4 мг, 1 ммол) и диизопропилэтиламин (193.8 мг, 1.5 ммол) в дихлорметане (10 мл) добавили янтарный ангидрид (120.1 мг, 1.2 ммол) и перемешали при комнатной температуре в течение ночи. Затем смесь промыли 0.1 М раствором лимонной кислоты (10 мл). Раствор дихлорметана разделили, высушили с ангидридмагнийсульфатом, отфильтровали и далее выпарили под пониженным давлением. Полученный остаток очистили хроматографией на силикагеле с помощью этилацетатметанола (10:1-2:1), в качестве элюента, до получения 425.1 мг целевого соединения B57, выход 92%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 11.71, 11.83 (s, 1H), 9.24, 9.34 (d, 1H), 7.33-8.09 (m, 5H), 7.18-7.22 (m, 1H), 6.96, 7.01 (s, 1H), 6.58, 6.75 (d, 1H), 3.66 (m, 4H), 2.42 (t, 2H), 2.34 (t, 2H), 2.25 (t, 2H), 1.98 (t, 2H), 1.88 (t, 2H).
Получение B90: Используя такой же способ, как при получении B57, и B75, в качестве исходного сырья, целевое соединение B90 было получено с выходом 88%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.13, 12.17 (s, 1H), 11.71, 11.84 (s, 1H), 9.25, 9.35 (d, 1H), 7.41-7.54 (m, 3H), 7.33 (t, 1H), 7.19-7.26 (m, 1H), 6.97, 7.02 (s, 1H), 6.76 (t, 1H), 1.95-3.67 (m, 13H).
Получение B91: Используя такой же способ, как при получении B57, и диметилсульфомоил хлорид, в качестве сульфирующего агента, целевое соединение B91 было получено с выходом 87%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.13, 12.17 (s, 1H), 11.71, 11.83 (s, 1H), 9.25, 9.34 (d, 1H), 7.39-7.52 (m, 3H), 7.33 (t, 1H), 7.22 (dd, 1H), 6.75 (d, 1H), 6.67, 7.01 (s, 1H), 1.39-3.65 (m, 16H).
Пример 6
Получение B55: Соединение B22 (345.4 мг, 1 ммол) и фенилизоцинат (178.7 мг, 1.5 ммол) были добавлены в дихлорметан (10 мл) и вступили реакцию при комнатной температуре в течение 1 дня. Смесь добавили в колонку силикагеля и далее вымыли смесью этилацетатметанола (10:1-2:1). После выпаривания 401.2 мг целевого соединения В55 было получено с выходом 86%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.21 (s, 1H), 11.92, 11.98 (2s, 1H), 9.28, 9.35 (2d, 1H), 8.60 (s, 1H), 7.46-7.58 (m, 5H), 7.39 (t, 1H), 7.23-7.28 (m, 4H), 6.99 (t, 1H), 6.94 (t, 1H), 3.65 (s, 4H), 3.15 (s, 4H).
Получение B118: Соединение B6 (276.3 мг, 1 ммол) и 4-хлор-3-(трифторметил)фенилизоцинат (265.9 мг, 1.2 ммол) были смешаны с N-метилпрролидином (10 мл) и вступили в реакцию при комнатной температуре в течение 1 дня. Смесь выпарили в вакууме для удаления большинства NMP. Остаток обработали дихлорметаном (5 мл), наполнили им колонку силикагеля и далее вымыли смесью этилацетатметанола (10:1-2:1). После выпаривания, 412.5 мг целевое соединение B118, было получено с выходом выходом 83%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.25 (s, 1H), 12.13 & 12.11 (s, 1H), 9.41 & 9.36 (d, 1H), 9.15 & 9.11 (s, 1H), 8.87 & 8.76 (s, 1H) 8.18-8.17 (m, 1H), 7.92 & 7.86 (s, 1H), 7.70-7.23 (m, 8H). LC-MS (m/z) 498 [M+1].
Получение B119: Используя такой же способ, как при получении В 118, и 2-фтор-5-(трифторметил)фенилизоцинат, в качестве уреидного агента, целевое соединение B119 было получено с выходом 81%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.21 (s, 1H), 12.09 (s, 1H), 9.35 (s, 1H), 9.17 (s, 1H), 8.84 (s, 1H), 8.68 (d, 1H), 7.89 (s, 1H), 7.58 (dd, 2H), 7.49 (t, 2H), 7.40 (d, 2H), 7.28 (t, 1H), 7.20 (d, 1H).
Получение B120: Используя такой же способ, как при получении B118,, и 2,5-дифторфенилизоцинат, в качестве уреидного агента, целевое соединение B120 было получено с выходом 89%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.09, 12.20 (s, 1H), 12.05, 12.08 (s, 1H), 9.32, 9.37 (d, 1H), 8.09-9.16 (m, 2H), 7.87 (t, 1H), 7.55-7.62 (m, 2H), 7.46-7.52 (m, 1H), 7.36-7.41 (m, 1H), 6.80-7.30 (m, 3Н).
Получение B121: Используя такой же способ, как при получении B118,, и фенилизоцинат, в качестве уреидного агента, целевое соединение B121 было получено с выходом 83%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.04, 12.06 (s, 1H), 9.31, 9.36 (d, 1H), 8.66, 8.70 (s, 1H), 8.60, 8.63 (s, 1H), 7.84, 7.87 (s, 1H), 7.54-7.61 (m, 2H), 7.44-7.51 (m, 3Н), 7.38 (t, 1H), 7.16-7.30 (m, 4H), 6.96 (t, 1H).
Получение B123: Используя такой же способ, как при получении B118,, и 2-этил-6-метилфенилизоцинат, в качестве уреидного агента, целевое соединение B123 было получено с выходом 78%.1Н-ЯМР (DMSO-d6, 500 МГц) δ 12.02 (s, 1H), 9.30, 9.34 (d, 1H), 8.62, 8.74 (s, 1H), 7.85 (s, 1H), 7.64 (s, 1H), 7.50-7.59 (m, 2H), 7.46 (t, 1H), 7.35-7.40 (m, 1H), 7.20-7.28 (m, 2H), 7.10-7.15 (m, 3Н), 2.62 (dd, 2H), 2.24 (s, 3H),1.15 (t, 3H).
Получение B124: Используя такой же способ, как при получении B118,, и циклогексил изоцинат, в качестве уреидного агента, целевое соединение B124 было получено с выходом 71%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.17 (s, 1H), 11.96, 11.99 (s, 1H), 9.29, 9.34 (d, 1H), 8.16, 8.27 (s, 1H), 7.76 (s, 1H), 7.36 (t, 1H), 7.34-7.59 (m, 3Н), 7.24 (dd, 1H), 7.06, 7.12 (d, 1H).
Получение B125: Используя такой же способ, как при получении B118,, и 2-фторфенилизоцинат, в качестве уреидного агента, целевое соединение B125 было получено с выходом 85%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.20 (s, 1H), 12.06, 12.08 (s, 1H), 9.32, 9.37 (d, 1H), 8.99, 9.08 (s, 1H), 8.49 (d, 1H), 8.21 (t, 1H), 7.87 (s, 1H), 7.52 (m, 2H), 7.48 (t, 1H), 7.39 (t, 1H), 7.13-7.30 (m, 4H), 6.99 (dd, 1H).
Получение B126: Используя такой же способ, как при получении B118, и 3-фторфенилизоцинат, в качестве уреидного агента, целевое соединение B126 было получено с выходом 88%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.05 (s, 1H), 9.31, 9.36 (d, 1H), 9.30, 9.32 (s, 1H), 9.07, 9.16 (s, 1H), 7.84, 7.89 (s, 1H), 7.52-7.60 (m, 3Н), 7.47 (t, 1H), 7.38 (t, 1H), 7.15-7.31 (m, 4H), 6.74 (t, 1H).
Получение B127: Используя такой же способ, как при получении B118, и 4-фторфенилизоцинат, в качестве уреидного агента, целевое соединение B127 было получено с выходом 80%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.04, 12.06 (s, 1H), 9.30, 9.35 (d, 1H), 8.91, 8.93 (s, 1H), 8.82, 8.91 (s, 1H), 7.83, 7.87 (s, 1H), 7.45-7.60 (m, 5H), 7.38 (t, 1H), 7.17-7.28 (m, 2H), 7.11 (t, 2H).
Получение B128: Используя такой же способ, как при получении B118,, и 2,4-дифторфенилизоцинат, в качестве уреидного агента, целевое соединение B128 было получено с выходом 77%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.20 (s, 1H), 12.07, 12.08 (s, 1H), 9.32, 9.36 (d, 1H), 8.94, 9.04 (s, 1H), 8.46, 8.47 (s, 1H), 8.15 (dd, 1H), 7.86 (s, 1H), 7.51-7.61 (m, 2H), 7.48 (t, 1H), 7.39 (t, 1H), 7.13-7.32 (m, 3H), 7.05 (t, 1H).
Получение В 129: Используя такой же способ, как при получении B118, и 2,6-дифторфенилизоцинат, в качестве уреидного агента, целевое соединение B129 было получено с выходом 75%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.19 (s, 1H), 12.06 (s, 1H), 9.32 (d, 1H), 8.79, 8.88 (s, 1H), 8.00 (1H, s), 7.84 (1H, s), 7.56 (t, 2H), 7.48 (t, 1H), 7.38 (d, 1H), 7.24-7.33 (m, 2H), 7.13-7.20 (m, 3H).
Получение B130: Используя такой же способ, как при получении B118,, и 3-хлор-4-фторфенилизоцинат, в качестве уреидного агента, целевое соединение B130 было получено с выходом 82%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.19 (s, 1H), 12.06 (s, 1H), 9.32, 9.37 (d, 1H), 8.87 (d, 1H), 8.69, 8.79 (s, 1H), 7.78-7.85 (m, 2H), 7.46-7.60 (m, 3H), 7.39 (t, 1H), 7.17-7.33 (m, 4H).
Получение B131: Используя такой же способ, как при получении B118, и 2-(трифторметил)фенилизоцинат, в качестве уреидного агента, целевое соединение В 131 было получено с выходом 82%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.19 (s, 1H), 12.08 (s, 1H), 9.32, 9.36 (d, 1H), 9.31, 9.40 (s, 1H), 8.01, 8.03 (s, 2H), 7.85, 7.88 (s, 1H), 7.56-7.68 (m, 4H), 7.48 (t, 1H), 7.39 (t, 1H), 7.13-7.30 (m, 3Н).
Получение B132: Используя такой же способ, как при получении B118, и 3-(трифторметил)фенилизоцинат, в качестве уреидного агента, целевое соединение В 13 2 было получено с выходом 82%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.20 (s, 1H), 12.06, 12.08 (s, 1H), 9.32, 9.37 (d, 1H), 8.98, 9.01 (s, 1H), 8.69, 8.79 (s, 1H), 8.05 (s, 1H), 7.83, 7.89 (s, 1H), 7.46-7.61 (m, 5H), 7.39 (t, 1H), 7.20-7.30 (m, 3H).
Получение В133: Используя такой же способ, как при получении B118, и 4-(трифторметил)фенилизоцинат, в качестве уреидного агента, целевое соединение В 133 было получено с выходом 83%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.20 (s, 1H), 12.08 (s, 1H), 9.32, 9.37 (d, 1H), 9.02, 9.05 (s, 1H), 8.69, 8.79 (s, 1H), 7.84, 7.88 (s, 1H), 7.58-7.69 (m, 6H), 7.48 (t, 1H), 7.39 (t, 1H), 7.18-7.28 (m, 2H).
Получение B134: Используя такой же способ, как при получении B118, и 2,5-бис(трифторметил)фенилизоцинат, в качестве уреидного агента, целевое соединение B134 было получено с выходом 88%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 10.02-12.20 (m, 1H), 8.90-9.39 (m, 2H), 6.52-8.18 (m, 12H).
Получение B135: Используя такой же способ, как при получении B118, и 2-хлор-5-(трифторметил)фенилизоцинат, в качестве уреидного агента, целевое соединение B135 было получено с выходом 80%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.08, 12.10 (s, 1H), 9.49, 9.58 (s, 1H), 9.32, 9.38 (d, 1H), 7.88, 7.91 (d, 1H), 7.71 (d, 1H), 7.52-7.61 (m, 2H), 7.49 (t, 1H), 7.40 (t, 1H), 7.35 (dd, 1H), 7.16-7.31 (m, 2H).
Получение B136: Используя такой же способ, как при получении B118, и 4-хлор-2-(трифторметил)фенилизоцинат, в качестве уреидного агента, целевое соединение B136 было получено с выходом 82%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.09 (s, 1H), 9.39, 9.47 (s, 1H), 9.32, 9.35 (d, 1H), 8.14 (s, 1H), 8.08 (dd, 1H), 7.85-7.88 (m, 1H), 7.71-7.75 (m, 3H), 7.52-7.61 (m, 2H), 7.48 (t, 1H), 7.40 (t, 1H), 7.13-7.30 (m, 2H).
Получение B137: Используя такой же способ, как при получении B118, и 2-метоксифенилизоцинат, в качестве уреидного агента, целевое соединение B137 было получено с выходом 89%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.04, 12.06 (s, 1H), 9.31, 9.36 (d, 1H), 9.26 (d, 1H), 8.18 (t, 2H), 7.88 (d, 1H), 7.51-7.61 (m, 2H), 7.47 (t, 1H), 7.39 (t, 1H), 7.13-7.30 (m, 2H,), 7.02 (d, 1H), 6.891-6.954 (m, 2H), 3.894 (s, 3H).
Получение B138: Используя такой же способ, как при получении B118, и 3-метоксифенилизоцинат, в качестве уреидного агента, целевое соединение B138 было получено с выходом 80%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.04, 12.06 (s, 1H), 9.31, 9.35 (d, 1H), 8.62, 8.65 (d, 2H), 7.83, 7.87 (s, 1H), 7.54-7.61 (m, 2H), 7.48 (t, 1H), 7.39 (t, 1H), 7.15-7.30 (m, 4H), 6.95 (t, 1H), 6.54 (d, 1H), 3.74 (s, 3H).
Пример 7
Получение B71: Соединение B7 (305.3 мг, 1 ммол) и HBTU (568.9 мг, 1.5 ммол) были растворены дихлорметане (10 мл) при комнатной температуре. После 10 минут добавили 1-метилпиперазин (300.5 мг, 3 ммол). Реакционную смесь перемешивали всю ночь при комнатной температуре с азотом. Смесь разбавили этилацетатом (50 мл) и промыли раствором карбоната натрия (1 M, 20 мл, 2 раза). Органический слой отделили, высушили с ангидридмагнийсульфатом, отфильтровали и затем выпаривали под вакуумом. Сырой продукт очистили испарительной колонной с помощью дихлорметанэтилацетата (5:1), в качестве элюента, до получения 325 мг целевого продукта, выход 84%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.42 (d, 1H), 10.10 (s, 1H), 9.42 (d, 1H), 7.73-7.82 (m, 3H), 7.63 (t, 1H), 7.52 (d, 1H), 7.40 (t, 1H), 7.30 (d, 1H), 3.55 (t, 4H), 2.94 (t, 4H), 2.66 (s, 3H).
B72,1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 9.39 (d, 2H), 8.51 (s, 2H), 8.24 (s, 1H), 7.82 (d, 1H), 7.59-7.67 (m, 3H), 7.55 (d, 1H), 7.50 (d, 1H), 7.32 (t, 2H), 6.69 (s, 1H), 3.50 (t, 4H), 3.29 (t, 4H).
Получение B73: Это соединение было получено с использованием сходной методики при получении целевого соединения B71 с выходом 78%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.42 (s, 1H), 12.28 (br, 1H), 10.25 (d, 1H), 9.43 (dd, 1H), 8.02 (dd, 1H), 7.90 (t, 1H), 7.76 (d, 1H), 7.72 (m, 2H), 7.62 (m, 1H), 7.50 (m, 1H), 7.35 (m, 1H), 7.05 (d, 1H), 3.30 (t, 4H), 2.01 (t, 4H).
Получение B74: Это соединение было получено с использованием сходной методики при получении целевого соединения B71 с выходом 88%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.36 (s, 1H), 9.41 (s, 1H), 7.74 (s, 1H), 7.67-7.69 (m, 2H), 7.56 (d, 1H), 7.47 (d, 1H), 7.36 (t, 1H), 7.22 (s, 1H), 4.11 (s, 1H), 3.67 (d, 1H), 3.51 (s, 1H), 3.20 (s, 1H), 2.64 (m, 1H), 2.53 (s, 3Н), 2.32 (d, 2H), 1.83 (d, 2H), 1.79 (s, 1H), 1.26 (s, 1H).
Получение B92: Это соединение было получено с использованием сходной методики при получении целевого соединения B71 с выходом 75%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 12.35 (s, 1H), 9.40 (t, 1H), 8.03 (m, 2H), 7.66 (m, 2H), 7.60 (m, 2H), 7.47 (d, 1H), 7.37 (t, 1H), 7.09 (m, 1H), 6.86 (m, 1H), 6.65 (m, 1H), 3.59-3.84 (m, 5H), 3.20 (s, 1H), 1.99 (s, 2H), 1.83 (d, 2H), 1.61-1.82 (m, 2H), 1.26 (s, 1H).
Пример 8
Получение B9: 1-Азаазулен-2-он (8.71 г, 60 ммол) был растворен в дихлорметане (250 мл), далее хлорид алюминия (120 ммол) и бромацетилхлорид (10 мл, 90 ммол, 1.5 экв) были добавлены и перемешаны при комнатной температуре в течение ночи. В смесь был добавлен лед (100 г), и далее соляная кислота (6N, 20 мл) и вода (150 мл), все перемешали в течение 30 минут. Твердый продукт отфильтровали и промыли водой (100 мл, 4 раза). Продукт 3-(бромацетил)-1-азаазулен-2-он массой 6.36 г, выход 40%. 3-(бромацетил)-1-азаазулен-2-он (1 ммол) и 2-аминопиридин (1 ммол) были добавлены в этанол (5 мл) и нагреты вплоть до дефлегмации в течение 1 дня. Растворитель выпарили в вакууме, остаток разделили на этилацетат и раствор карбоната натрия.
Слой этилацетата был отделен и выпарен. Остаток очистили колоночной хроматографией с помощью этилацетатметанола (10:1-3:1), в качестве элюента, до получения 175 мг целевых соединений, выход 67%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 11.84 (s, 1H), 9.33 (d, 1H), 8.67 (d, 1H), 8.66 (s, 1H), 7.64 (d, 1H), 7.29 (t, 1H), 7.27 (t, 1H), 7.25 (t, 1H), 7.13 (d, 1H), 7.04 (t, 1H), 6.92 (t, 1H). LC-MS (m/z) 262 [M+1].
Получение B13: 3-(бромацетил)-1-азаазулен-2-он (1 ммол) и 2-амино-5-цианопиридин (1 ммол) были добавлены в этанол (5 мл) и нагреты вплоть до дефлегмации в течение 1 дня. Слой этилацетата был отделен и выпарен. Остаток очистили колоночной хроматографией с помощью этилацетатметанола (10:1-2:1), в качестве элюента, до получения 115 мг целевых соединений, выход 40%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 11.95 (s, 1H), 9.44 (s, 1H), 9.30 (d, 1H), 8.76 (s, 1H), 7.79 (d, 1H), 7.53 (d, 1H), 7.31-7.40 (m, 2H), 7.22 (d, 1H), 7.11 (t, 1H).
Получение B14: 3-(бромацетил)-1-азаазулен-2-он (1 ммол) и 2-амино-5-метоксипиридин (1 ммол) были добавлены в этанол(5 мл) и нагреты вплоть до дефлегмации в течение 1 дня. Слой этилацетата был отделен и выпарен. Остаток очистили колоночной хроматографией с помощью этилацетатметанола (10:1-3:1), в качестве элюента, до получения 254 мг целевых соединений, выход 87%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 9.03 (d, 1H), 8.43 (s, 1H), 8.19 (s, 1H), 7.53 (d, 1H), 7.30-7.7.36 (m, 2H), 7.13(t, 1H), 3.90 (s, 3H).
Получение B16: 3-(бромацетил)-1-азаазулен-2-он (1 ммол) и 2-амино-4-хлорпиридин (1 ммол) были добавлены в этанол (5 мл) и нагреты вплоть до дефлегмации в течение 1 дня. Слой этилацетата был отделен и выпарен. Остаток очистили колоночной хроматографией с помощью этилацетатметанола (10:1-4:1), в качестве элюента, до получения 213 мг целевых соединений, выход 72%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 11.84 (s, 1H), 9.25 (d, 1H), 8.68 (d, 1H), 8.67 (s, 1H), 7.77 (s, 1H), 7.22-7.30 (m, 2H), 7.13 (d, 1H), 6.68-7.04 (m, 2H).
B17,1H-ЯМР (DMSO-d6, 500 МГц) δ 11.85 (s, 1H), 9.26 (d, 1H), 9.00 (s, 1H), 8.65 (s, 1H), 7.60 (d, 1H), 7.23-7.37 (m, 3H), 7.14 (d, 1H), 7.04 (t, 1H).
Пример 9
Получение B12: 3-(бромацетил)-1-азаазулен-2-он (1 ммол) и 2-аминопирмидин (1 ммол) были добавлены в этанол (5 мл) и нагреты вплоть до дефлегмации в течение 1 дня. Слой этилацетата был отделен и выпарен. Остаток очистили колоночной хроматографией с помощью этилацетатметанола (10:1-1:1), в качестве элюента, до получения 198 мг целевых соединений, выход 76%.1H-ЯМР (500 МГц, DMSO-d6) δ (ppm) 11.91 (broad, 1H), 9.36 (d, 1H), 9.07 (d, 1H), 8.64 (s, 1H), 8.55 (s, 1H), 7.37 (t, 1H), 7.33 (t, 1H), 7.20 (d, 1H), 7.12-7.08 (m, 2H). LC-MS (m/z) 263 [M+1].
Пример 10
Получение B143: 2-(3-фторфенил) уксусная кислота (1 ммол) и 1-азаазулен-2-он (1 ммол) были добавлены в реактив Итона (2 мл) и нагреты при температуре 80 градусов в течение 1 дня. Смесь пересыпали в воду (50 мл) и перемешали в течение 30 минут. Продукт отфильтровали, промыли с водой и высушили до получения 201 мг целевого продукта, выход 72%. 3-(2-(3-фторфенилацетил)-1-азаазул -2-он (201 мг, 0.72 ммол) был растворен в концентрированной серной кислоте (2 мл), охлажденной в ледяной ванне и далее добавили концентрированную азотную кислоту (65 мг) и перемешали в течение 1 часа. Реакционную смесь разбавили водой (5 мл). Твердый продукт отфильтровали, промыли с водой (5 мл) и метанолом (5 мл) и далее высушили до получения 57 мг нитрированного продукта, выход 24%. 3-(2-(5-фтор-2-нитрофенил)ацетил)-1-азаазул-2-он (57 мг, 0.17 ммол), KF (41 мг) and N-метилпиперазине (109 мг) были растворены в DMSO (3 мл) и нагреты при температуре 120 градусов в течение 3 часов. Реакционную смесь разбавили водой (10 мл). Твердый продукт отфильтровали, промыли с водой (5 мл) и метанолом (5 мл), и далее высушили до получения 58 мг целевого продукта, выход 84%. 3-(2-(5-(4-метилпиперазин-1-ил)-2-нитрофенил)ацетил)-1-азаазул-2-он (10 мг, 0.025 ммол) растворили в метаноле (5 мл), гидрировали при давлении 1 атм водорода при наличии никеля Ренея в течение 14 часов. Смесь отфильтровали, и фильтрат концентрировали в вакууме до получения 5.2 мг целевого соединения 3-(5-(4-метилпиперазин-1-ил)-1H-индол -2-ил)-1-азаазул-2-он, выход 58%.1Н-ЯМР (500 МГц, DMSO-d6) δ (ppm) 10.04 (s, 1H), 11.01 (s, 1H), 8.07 (d, 1H), 7.49 (d, 1H), 7.29(t, 2H), 7.16 (d, 1H), 7.11 (s, 1H), 7.06 (t, 1H), 6.94 (d, 1H), 6.90 (s, 1H), 3.49 (b, 4H), 2.82 (b, 4H), 2.53 (s, 3H).
Ингибирующая активность соединений по настоящей заявке.
Пример 11: Образцы для анализа киназы
Азаазуленовые соединения по настоящему изобретению исследовались на их эффективность по ингибированию активностей FLT-3, C-KIT и KDR-киназ, исследования проводили биохимическими анализам DELFIA по описанной ниже методике. Анализы осуществлялись в Division of Cell Engineering, Biomedical Engineering Research Laboratories, Industrial Technology Research Institute, Bidg. 53, 195, sec.4, Chung Hsing Rd. Chutung, Hsinchu, Taiwan 310, R.O.C.
Анализ FLT-3 проводился по протоколу, описанному в руководстве «Protocol for HTScang FLT-3 Kinase Assay Kit (Cell Signaling TechnologyRTM)». Условия анализа были следующими: источник FLT-3: слитый белок GST-киназы получали при использовании системы экспрессии бакуловируса с элементом конструкции, экспрессирующей FLT-3 человека (Arg571-Ser993) (GenBank серийный № NM.sub.-004119) с аминотерминальным хвостом GST, субстрат: 1,5 мкМ биотинированный пептидный предшественник гастрина (с Tyr87 в качестве сайта фосфорилирования), носитель: 1% диметилсульфоксид (ДМСО), время до инкубирования/температура: 5 мин при комнатной температуре, время инкубирования/температура: 30 мин при комнатной температуре, буфер для инкубирования: 60 мМ HEPES pH 7.5, 5 мМ MgCl2, 5 мМ MnCl2, 3 мкМ Na3VO4, 1,25 мМ DTT, 20 мкМ АТР. Использовался также и количественный анализ: DELFLRTM Assay.
В приведенной далее таблице 2 сведены ингибирующие действия азаазуленовых соединений по настоящему изобретению в отношении FLT-3 киназы.
Анализ c-KIT проводился по протоколу, описанному в руководстве «Protocol for HTScarRTM c-KIT Kinase Assay Kit (Cell Signaling TechnologyRTM)». Условия анализа были следующими: источник C-KIT: слитый белок GST-c-KIT получали при использовании системы экспрессии бакуловируса с элементом конструкции, экспрессирующей c-KIT человека (Thr544-Val976) с аминотерминальным хвостом GST, субстрат: 1,5 мкМ биотинированного пептида, содержащего остатки, окружающие Tyr-996 из KDR, носитель: 1% диметилсульфоксид (ДМСО), время до инкубирования/температура: 5 мин при комнатной температуре, время инкубирования/температура: 30 мин при комнатной температуре, буфер для инкубирования: 60 мМ HEPES рН 7.5, 5 мМ MgCl2, 5 мМ MnCl2, 3 мкМ Na3VO4, 1,25 мМ DTT, 20 мкМ АТР. Использовался также и количественный анализ: DELFIARTM Assay.
[00232] В приведенной далее таблице 3 сведены ингибирующие действия азаазуленовых соединений по настоящему изобретению в отношении c-KIT киназы.
Анализ KDR проводился по протоколу, описанному в руководстве «Protocol for HTScanR™ VEGFR-2 Kinase Assay Kit (Cell Signaling Technology.RTM)». Условия анализа были следующими: источник KDR: слитый белок GST-киназы получали при использовании системы экспрессии бакуловируса с элементом конструкции, экспрессирующей VEGFR-2 человека (Val789-Vall356) (GenBank Accession No. NM.sub.-002253) с аминотерминальным хвостом GST, субстрат: 1,5 мкМ биотинированный пептидный предшественник гастрина (с Tyr87 в качестве сайта фосфорилирования), носитель: 1% диметилсульфоксид (ДМСО), время до инкубирования/температура: 5 мин при комнатной температуре, время инкубирования/температура: 30 мин при комнатной температуре, буфер для инкубирования: 60 мМ HEPES Ph 7.5, 5 мМ MgCl2, 5 мМ MnCl2, 3 мкМ Na3VO4, 1,25 мМ DTT, 20 мкМ АТР. Использовался также и количественный анализ: DELFIARTM Assay.
В приведенной далее таблице 4 сведены ингибирующие действия азаазуленовых соединений по настоящему изобретению в отношении KDR киназы.
Пример 12: Анализы активности киназы
Анализ активности киназы в соединении B26 по настоящему изобретению осуществлялся с помощью SelectScreen® Kinase Profiling Services of Invitrogen. Ингибирующее действие 1000 нМ соединения B26 в отношении различных киназ, у которых ингибирующее действие было >50%, приведены далее в таблице 5.
Таблица 5.
Ингибирующая активность киназ из B26 в отношении различных киназ
Пример 13: Анализ клеточной активности in vitro Клеточная линия человека MV4-11 (FLT3-ITD) была получена из Американской Коллекции клеточных культур (American Tissue Culture Collection, ATCC номер CRL-9591). Эту клеточную линию культивировали с RPMI 1640, содержащей 10% бычьей сыворотки, 1 ммол/л пирувата натрия и 10 ммол/л HEPES (рН 7.4). Клетки росли и выдерживались во влажной атмосфере при 37°C и 5% содержании диоксида углерода.
Клетку MV4-11 поместили на микротитровальные пластины с 96 ячейками (10,000 клеток на каждую ячейку) и добавили последовательные разведения исследуемых соединений. В конце инкубационного периода (72 ч при 37°C) с помощью красящего тетразолия, МТТ (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолий-бромид) (Promega, Madison, WI) определили жизнеспособность клетки. Кристаллы формазана растворили в диметилсульфоксиде (ДМСО) и с помощью считывающего устройства ELISA зарегистрировали абсорбцию при длине волны, составляющей 600 нм. Величины IC50 вычисляли, используя нелинейную регрессию, и определили их как такую концентрацию, которая необходима для 50% снижения абсорбции обработанных клеток по сравнению с необработанными контрольными.
Ингибирующее действие азаазуленовых соединений по настоящему изобретению в отношении клеток MV4-11 сведено в приведенной ниже таблице 6.
Пример 14: Анализ клеточной активности in vitro
Методика разработки ксенотрансплантата опухоли, а также дозировка B26 осуществлялись в соответствии с правилами ухода и использования животных ITRI, IACUC. Женские особи мышей BALB/c, лишенные шерстного покрова, в возрасте 6-8 недель получены от BioLASCO Co., Ltd, (Taipei, Taiwan). Этим мышам подкожно в правый бок вводили клетки MV4-11 (FLT3-ITD) в количестве 1×107 клеток на каждую особь. Обработка началась, когда размер опухоли достиг 150-200 мм3. Мыши были случайным образом распределены на когорты (обычно по 4 мыши в каждой группе, из соображений эффективности исследования). Начиная с 16 дня после инокуляции мышам орально давали B26 (50 и 150 мг/кг) и носитель, процедуру проводили в течение 14 дней. Размеры опухолей оценивали ежедневно, а массы тел - два раза в неделю. Сделанные кронциркулем измерения были преобразованы в формулу, позволяющую узнать средний объем опухоли: 0,5 × [длина × (ширина)2].
В модели подкожного введения опухолевых клеток MV4-11 (FLT3-ITD) в виде ксенотрансплантата BALB/c мышам при оральном применении B26 в дозе 50 или 150 мг/кг в течение 14 дней был показано сильное и значительное противопухолевое действие, зависящее от дозы препарата. Доза B26, составляющая 50 и 150 мг/кг, продемонстрировала полную регрессию опухоли у всех мышей на 9 и 7 день соответственно. Обратимся к фиг.1, на ней показано, что в процессе эксперимента у группы, обрабатываемой B26, не было никакой существенной потери веса и не наблюдалось смертности. По настоящему изобретению соединение B26 представляет собой новый и потенциальный ингибитор FLT3, обещающий противораковУЮ активность и активность против лейкемии.
Пример A: Анализ клеточной активности In vitro для соединения В39 в отношении различных клеточных линий AML
Клеточная линия человека MV4-11 (FLT3-ITD) была получена из Американской Коллекции клеточных культур (American Tissue Culture Collection, ATCC номер CRL-9591). Эту клеточную линию культивировали с RPMI 1640, содержащей 10% бычьей сыворотки, 1 ммол/л пирувата натрия и 10 ммол/л HEPES (pH 7.4). Клетки росли и выдерживались во влажной атмосфере при 37°C и 5% содержании диоксида углерода.
Клетку MV4-11 поместили на микротитровальные пластины с 96 ячейками (10,000 клеток на каждую ячейку) и добавили последовательные разведения исследуемых соединений. В конце инкубационного периода (72 ч при 37°C) с помощью красящего тетразолия, МТТ (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолий-бромид) (Sigma) определили жизнеспособность клетки. Кристаллы формазана растворили в диметилсульфоксиде (ДМСО) и с помощью считывающего устройства ELISA зарегистрировали абсорбцию при длине волны, составляющей 600 нм. Величины IC50 вычисляли, используя нелинейную регрессию, и определили их как такую концентрацию, которая необходима для 50% снижения абсорбции обработанных клеток по сравнению с необработанными контрольными.
Клеточная линия человека MOLM13 (FLT3-ITD) была получена из Американской Коллекции клеточных культур. Эту клеточную линию культивировали с RPMI 1640, содержащей 10% бычьей сыворотки, 1 ммол/л пирувата натрия и 10 ммол/л HEPES (pH 7.4). Клетки росли и выдерживались во влажной атмосфере при 37°C и 5% содержании диоксида углерода.
Клетку MOLM13 (FLT3-ITD) поместили на микротитровальные пластины с 96 ячейками (10,000 клеток на каждую ячейку) и добавили последовательные разведения исследуемых соединений. В конце инкубационного периода (72 ч при 37°C) с помощью красящего тетразолия, МТТ (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолий-бромид) (Sigma) определили жизнеспособность клетки. Кристаллы формазана растворили в диметилсульфоксиде (ДМСО) и с помощью считывающего устройства ELISA зарегистрировали абсорбцию при длине волны, составляющей 600 нм. Величины IC50 вычисляли, используя нелинейную регрессию, и определили их как такую концентрацию, которая необходима для 50% снижения абсорбции обработанных клеток по сравнению с необработанными контрольными.
Клеточная линия человека RS4;11 (FLT3wt) была получена из Американской Коллекции клеточных культур (American Tissue Culture Collection, ATCC номер CRL-1873). Эту клеточную линию культивировали с RPMI 1640, содержащей 10% бычьей сыворотки, 1 ммол/л пирувата натрия и 10 ммол/л HEPES (pH 7.4). Клетки росли и выдерживались во влажной атмосфере при 37°C и 5% содержании диоксида углерода.
Клетку RS4; 11 поместили на микротитровальные пластины с 96 ячейками (10,000 клеток на каждую ячейку) и добавили последовательные разведения исследуемых соединений. В конце инкубационного периода (72 ч при 37°C) с помощью красящего тетразолия, МТТ (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолий-бромид) (Sigma) определили жизнеспособность клетки. Кристаллы формазана растворили в диметилсульфоксиде (ДМСО) и с помощью считывающего устройства ELISA зарегистрировали абсорбцию при длине волны, составляющей 600 нм. Величины IC50 вычисляли, используя нелинейную регрессию, и определили их как такую концентрацию, которая необходима для 50% снижения абсорбции обработанных клеток по сравнению с необработанными контрольными.
Клеточная линия человека ТНР-1 (FLT3wt) была получена из Американской Коллекции клеточных культур. Эту клеточную линию культивировали с RPMI 1640, содержащей 10% бычьей сыворотки, 1 ммол/л пирувата натрия и 10 ммол/л HEPES (pH 7.4). Клетки росли и выдерживались во влажной атмосфере при 37°C и 5% содержании диоксида углерода.
Клетку ТНР-1 поместили на микротитровальные пластины с 96 ячейками (10,000 клеток на каждую ячейку) и добавили последовательные разведения исследуемых соединений. В конце инкубационного периода (72 ч при 37°C) с помощью красящего тетразолия, МТТ (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолий-бромид) (Sigma) определили жизнеспособность клетки. Кристаллы формазана растворили в диметилсульфоксиде (ДМСО) и с помощью считывающего устройства ELISA зарегистрировали абсорбцию при длине волны, составляющей 600 нм. Величины IC50 вычисляли, используя нелинейную регрессию, и определили их как такую концентрацию, которая необходима для 50% снижения абсорбции обработанных клеток по сравнению с необработанными контрольными.
Клеточная линия человека Kasumi-1 была получена из Американской Коллекции клеточных культур (American Tissue Culture Collection, ATCC номер CRL-2724). Эту клеточную линию культивировали с RPMI 1640, содержащей 20% бычьей сыворотки, 1 ммол/л пирувата натрия и 10 ммол/л HEPES (pH 7.4). Клетки росли и выдерживались во влажной атмосфере при 37°C и 5% содержании диоксида углерода.
Клетку Kasumi-1 поместили на микротитровальные пластины с 96 ячейками (10,000 клеток на каждую ячейку) и добавили последовательные разведения исследуемых соединений. В конце инкубационного периода (72 ч при 37°C) с помощью красящего тетразолия, МТТ (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолий-бромид) (Sigma) определили жизнеспособность клетки. Кристаллы формазана растворили в диметилсульфоксиде (ДМСО) и с помощью считывающего устройства ELISA зарегистрировали абсорбцию при длине волны, составляющей 570 нм. Величины IC50 вычисляли, используя нелинейную регрессию, и определили их как такую концентрацию, которая необходима для 50% снижения абсорбции обработанных клеток по сравнению с необработанными контрольными.
Пример В: Анализ клеточной активности in vitro для соединения B26 в отношении различных твердоопухолевых клеточных линий.
Клеточная линия рака прямой кишки.
Клеточная линия человека НСТ-15 была получена из Центра Сбора и Исследования Биологических источников (Bioresource Collection & Research Center) (BCRC номер: 60539). Эту клеточную линию культивировали в среде RPMI 1640, содержащей 10% бычьей сыворотки, 2 ммол/л L-глутамина и 10 ммол/л HEPES (pH 7.4). Клетки росли и выдерживались во влажной атмосфере при 37°C и 5% содержании диоксида углерода.
Клетку НСТ-15 поместили на микротитровальные пластины с 96 ячейками (10,000 клеток на каждую ячейку) и добавили последовательные разведения исследуемых соединений. В конце инкубационного периода (48 ч при 37°C) с помощью красящего тетразолия, МТТ (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолий-бромид) (Sigma) определили жизнеспособность клетки. Кристаллы формазана растворили в диметилсульфоксиде (ДМСО) и с помощью считывающего устройства ELISA зарегистрировали абсорбцию при длине волны, составляющей 570 нм. Величины IC50 вычисляли, используя нелинейную регрессию, и определили их как такую концентрацию, которая необходима для 50% снижения абсорбции обработанных клеток по сравнению с необработанными контрольными.
Клеточная линия человека НТ-29 была получена из Американской Коллекции клеточных культур (American Tissue Culture Collection, ATCC номер НТВ-39). Эту клеточную линию культивировали в среде RPMI 1640, содержащей 10% бычьей сыворотки, 1 ммол/л пирувата натрия и 10 ммол/л HEPES (pH 7.4). Клетки росли и выдерживались во влажной атмосфере при 37°C и 5% содержании диоксида углерода.
Клетку НТ-29 поместили на микротитровальные пластины с 96 ячейками (10,000 клеток на каждую ячейку) и добавили последовательные разведения исследуемых соединений. В конце инкубационного периода (48 ч при 37°C) с помощью красящего тетразолия, МТТ (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолий-бромид) (Sigma) определили жизнеспособность клетки. Кристаллы формазана растворили в диметилсульфоксиде (ДМСО) и с помощью считывающего устройства ELISA зарегистрировали абсорбцию при длине волны, составляющей 570 нм. Величины IC50 вычисляли, используя нелинейную регрессию, и определили их как такую концентрацию, которая необходима для 50% снижения абсорбции обработанных клеток по сравнению с необработанными контрольными.
Клеточная линия рака груди
Клеточная линия человека MCF-7 была получена из Центра Сбора и Исследования Биологических источников (Bioresource Collection & Research Center) (BCRC номер: 60539). Эту клеточную линию культивировали в среде RPMI 1640, содержащей 10% бычьей сыворотки, 2 ммол/л L-глутамина и 10 ммол/л HEPES (pH 7.4). Клетки росли и выдерживались во влажной атмосфере при 37°C и 5% содержании диоксида углерода.
Клетку MCF-7 поместили на микротитровальные пластины с 96 ячейками (10,000 клеток на каждую ячейку) и добавили последовательные разведения исследуемых соединений. В конце инкубационного периода (48 ч при 37°C) с помощью красящего тетразолия, МТТ (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолий-бромид) (Sigma) определили жизнеспособность клетки. Кристаллы формазана растворили в диметилсульфоксиде (ДМСО) и с помощью считывающего устройства ELISA зарегистрировали абсорбцию при длине волны, составляющей 570 нм. Величины IC50 вычисляли, используя нелинейную регрессию, и определили их как такую концентрацию, которая необходима для 50% снижения абсорбции обработанных клеток по сравнению с необработанными контрольными.
Клеточная линия человека MDA-MB-231 была получена из Центра Сбора и Исследования Биологических источников (Bioresource Collection & Research Center) (BCRC номер: 60539). Эту клеточную линию культивировали в среде Лейбовица (Leibovitz's) содержащей 10% бычьей сыворотки (pH 7.4). Клетки росли и выдерживались во влажной атмосфере при 37°C и 5% содержании диоксида углерода.
Клетку MDA-MB-231 поместили на микротитровальные пластины с 96 ячейками (10,000 клеток на каждую ячейку) и добавили последовательные разведения исследуемых соединений. В конце инкубационного периода (48 ч при 37°C) с помощью красящего тетразолия, МТТ (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолий-бромид) (Sigma) определили жизнеспособность клетки. Кристаллы формазана растворили в диметилсульфоксиде (ДМСО) и с помощью считывающего устройства ELISA зарегистрировали абсорбцию при длине волны, составляющей 570 нм. Величины IC50 вычисляли, используя нелинейную регрессию, и определили их как такую концентрацию, которая необходима для 50% снижения абсорбции обработанных клеток по сравнению с необработанными контрольными.
Клеточная линия гепатоаденомы.
Клеточная линия человека Huh7 была получена из Японского Центра Сбора и Исследования Биологических источников (Bioresource Collection & Research Center) (JCRB номер: CRB0403). Эту клеточную линию культивировали в среде DMEM содержащей 10% бычьей сыворотки, 0.1 ммол/л второстепенных аминокислот (pH 7.4). Клетки росли и выдерживались во влажной атмосфере при 37°C и 5% содержании диоксида углерода.
Клетку Huh7 231 поместили на микротитровальные пластины с 96 ячейками (10,000 клеток на каждую ячейку) и добавили последовательные разведения исследуемых соединений. В конце инкубационного периода (48 ч при 37°C) с помощью красящего тетразолия, МТТ (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолий-бромид) (Sigma) определили жизнеспособность клетки. Кристаллы формазана растворили в диметилсульфоксиде (ДМСО) и с помощью считывающего устройства ELISA зарегистрировали абсорбцию при длине волны, составляющей 570 нм. Величины IC50 вычисляли, используя нелинейную регрессию, и определили их как такую концентрацию, которая необходима для 50% снижения абсорбции обработанных клеток по сравнению с необработанными контрольными.
Клеточная линия человека HepG2 была получена из Центра Сбора и Исследования Биологических источников (Bioresource Collection & Research Center) (BCRC номер: 60539). Эту клеточную линию культивировали в среде DMEM содержащей 10% бычьей сыворотки, 0.1 ммол/л второстепенных аминокислот (pH 7.4). Клетки росли и выдерживались во влажной атмосфере при 37°C и 5% содержании диоксида углерода.
Клетку HepG2 поместили на микротитровальные пластины с 96 ячейками (10,000 клеток на каждую ячейку) и добавили последовательные разведения исследуемых соединений. В конце инкубационного периода (48 ч при 37°C) с помощью красящего тетразолия, МТТ (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолий-бромид) (Sigma) определили жизнеспособность клетки. Кристаллы формазана растворили в диметилсульфоксиде (ДМСО) и с помощью считывающего устройства ELISA зарегистрировали абсорбцию при длине волны, составляющей 570 нм. Величины IC50 вычисляли, используя нелинейную регрессию, и определили их как такую концентрацию, которая необходима для 50% снижения абсорбции обработанных клеток по сравнению с необработанными контрольными.
Клеточная линия человека Hep3B из Центра Сбора и Исследования Биологических источников (Bioresource Collection & Research Center) (BCRC номер: 60434). Эту клеточную линию культивировали в среде альфа MEM содержащей 10% бычьей сыворотки (pH 7.4). Клетки росли и выдерживались во влажной атмосфере при 37°C и 5% содержании диоксида углерода.
Клетку Hep3B поместили на микротитровальные пластины с 96 ячейками (10,000 клеток на каждую ячейку) и добавили последовательные разведения исследуемых соединений. В конце инкубационного периода (48 ч при 37°C) с помощью красящего тетразолия, МТТ (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолий-бромид) (Sigma) определили жизнеспособность клетки. Кристаллы формазана растворили в диметилсульфоксиде (ДМСО) и с помощью считывающего устройства ELISA зарегистрировали абсорбцию при длине волны, составляющей 570 нм. Величины IC50 вычисляли, используя нелинейную регрессию, и определили их как такую концентрацию, которая необходима для 50% снижения абсорбции обработанных клеток по сравнению с необработанными контрольными.
Клеточная линия человека SK-Hep1 была получена из Американской Коллекции клеточных культур (American Tissue Culture Collection) (ATCC номер: НТВ-52). Эту клеточную линию культивировали в среде DMEM, содержащей 10% бычьей сыворотки, 0.1 ммол/л второстепенных аминокислот (pH 7.4). Клетки росли и выдерживались во влажной атмосфере при 37°C и 5% содержании диоксида углерода.
Клетку SK-Hep1 поместили на микротитровальные пластины с 96 ячейками (10,000 клеток на каждую ячейку) и добавили последовательные разведения исследуемых соединений. В конце инкубационного периода (48 ч при 37°C) с помощью красящего тетразолия, МТТ (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолий-бромид) (Sigma) определили жизнеспособность клетки. Кристаллы формазана растворили в диметилсульфоксиде (ДМСО) и с помощью считывающего устройства ELISA зарегистрировали абсорбцию при длине волны, составляющей 570 нм. Величины IC50 вычисляли, используя нелинейную регрессию, и определили их как такую концентрацию, которая необходима для 50% снижения абсорбции обработанных клеток по сравнению с необработанными контрольными.
Клеточная линия рака поджелудочной железы
Клеточная линия человека MIA-PaCa-2 была получена из Центра Сбора и Исследования Биологических источников (Bioresource Collection & Research Center) (BCRC номер: 60139). Эту клеточную линию культивировали в среде DMEM, содержащей 10% бычьей сыворотки, 4 ммол/л L-глутамина. Клетки росли и выдерживались во влажной атмосфере при 37°C и 5% содержании диоксида углерода.
Клетку MIA-PaCa-2 поместили на микротитровальные пластины с 96 ячейками (10,000 клеток на каждую ячейку) и добавили последовательные разведения исследуемых соединений. В конце инкубационного периода (48 ч при 37°C) с помощью красящего тетразолия, МТТ (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолий-бромид) (Sigma) определили жизнеспособность клетки. Кристаллы формазана растворили в диметилсульфоксиде (ДМСО) и с помощью считывающего устройства ELISA зарегистрировали абсорбцию при длине волны, составляющей 570 нм. Величины IC50 вычисляли, используя нелинейную регрессию, и определили их как такую концентрацию, которая необходима для 50% снижения абсорбции обработанных клеток по сравнению с необработанными контрольными.
Клеточная линия человека ВхРС-3 была получена из Центра Сбора и Исследования Биологических источников (Bioresource Collection & Research Center) (BCRC номер: 60283). Эту клеточную линию культивировали в среде RPMI 1640, содержащей 10% бычьей сыворотки, 1 ммол/л HERPES (pH 7.4). Клетки росли и выдерживались во влажной атмосфере при 37°C и 5% содержании диоксида углерода.
Клетку ВхРС-3 поместили на микротитровальные пластины с 96 ячейками (10,000 клеток на каждую ячейку) и добавили последовательные разведения исследуемых соединений. В конце инкубационного периода (48 ч при 37°C) с помощью красящего тетразолия, МТТ (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолий-бромид) (Sigma) определили жизнеспособность клетки. Кристаллы формазана растворили в диметилсульфоксиде (ДМСО) и с помощью считывающего устройства ELISA зарегистрировали абсорбцию при длине волны, составляющей 570 нм. Величины IC50 вычисляли, используя нелинейную регрессию, и определили их как такую концентрацию, которая необходима для 50% снижения абсорбции обработанных клеток по сравнению с необработанными контрольными.
Клеточная линия рака легких
Клеточная линия человека NCI-H460 была получена из Центра Сбора и Исследования Биологических источников (Bioresource Collection & Research Center) (BCRC номер: 60373). Эту клеточную линию культивировали в среде RPMI 1640, содержащей 10% бычьей сыворотки, 1 ммол/л пирувата натрия 10 и ммол/л HEPES (pH 7.4). Клетки росли и выдерживались во влажной атмосфере при 37°C и 5% содержании диоксида углерода.
Клетку NCI-H460 поместили на микротитровальные пластины с 96 ячейками (10,000 клеток на каждую ячейку) и добавили последовательные разведения исследуемых соединений. В конце инкубационного периода (48 ч при 37°C) с помощью красящего тетразолия, МТТ (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолий-бромид) (Sigma) определили жизнеспособность клетки. Кристаллы формазана растворили в диметилсульфоксиде (ДМСО) и с помощью считывающего устройства ELISA зарегистрировали абсорбцию при длине волны, составляющей 570 нм. Величины IC50 вычисляли, используя нелинейную регрессию, и определили их как такую концентрацию, которая необходима для 50% снижения абсорбции обработанных клеток по сравнению с необработанными контрольными.
Пример C: Анализ активности ксенотрансплантата опухоли при раке поджелудочной железы in vivo
Клеточная линия человека для рака поджелудочной железы MIA РаСа-2 была получена из Американской Коллекции клеточных культур (American Tissue Culture Collection, ATCC номер CRL-1420™).
Методика разработки ксенотрансплантата опухоли, а также дозировка азаазуленового соединения В26, осуществлялись в соответствии с правилами ухода и использования животных ITRI, IACUC.
Женские особи мышей BALB/c, лишенные шерстного покрова, в возрасте 6-8 недель получены от BioLASCO Co., Ltd, (Taipei, Taiwan). Этим мышам подкожно в правый бок вводили клетки 5×106 MIA РаСа-2 в количестве 5×106 клеток на каждую особь.
Обработка началась, когда размер опухоли достиг 200 мм3. Мыши были случайным образом распределены на когорты (обычно по 4 мыши в каждой группе, из соображений эффективности исследования).
Начиная с 9 дня после инокуляции мышам путем интраперитонеальной инъекции давали азаазуленовые соединения B26 (25 мг/кг/день) и носитель, процедуру проводили в течение 14 дней и увеличили дозу до 100 мг/кг/день в течение 7 дней. Размеры опухолей и массы тела оценивали три раза в неделю. Сделанные кронциркулем измерения были преобразованы в формулу, позволяющую узнать средний объем опухоли: 0,5 × [длина × (ширина)2].
Коэффициент (TGI; %) роста опухоли вычислили как [1-(конечный размер опухоли - первоначальный размер опухоли для леченной группы) / (конечный размер опухоли - первоначальный размер опухоли для группы с носителем)] × 100.
В модели подкожного введения опухолевых клеток MIA РаСа-2 в виде ксенотрансплантата BALB/c мышам размер опухоли достиг 1437±117 мм3 на 20 день после лечения. Интраперитонеальное введение азаазуленовых соединения B26 проявило противоопухолевую активность; средний размер опухоли составил 1177±123 мм3; и коэффициент ингибирования роста опухоли на 20 день лечения составил 22±9% (p=0.1616).
Ниже приведен результат исследований азаазуленового соединения B26 как отдельного средства, проявляющего активность в отношении клеток MIA РаСа-2 ксенотрансплантата опухоли при раке поджелудочной железы.
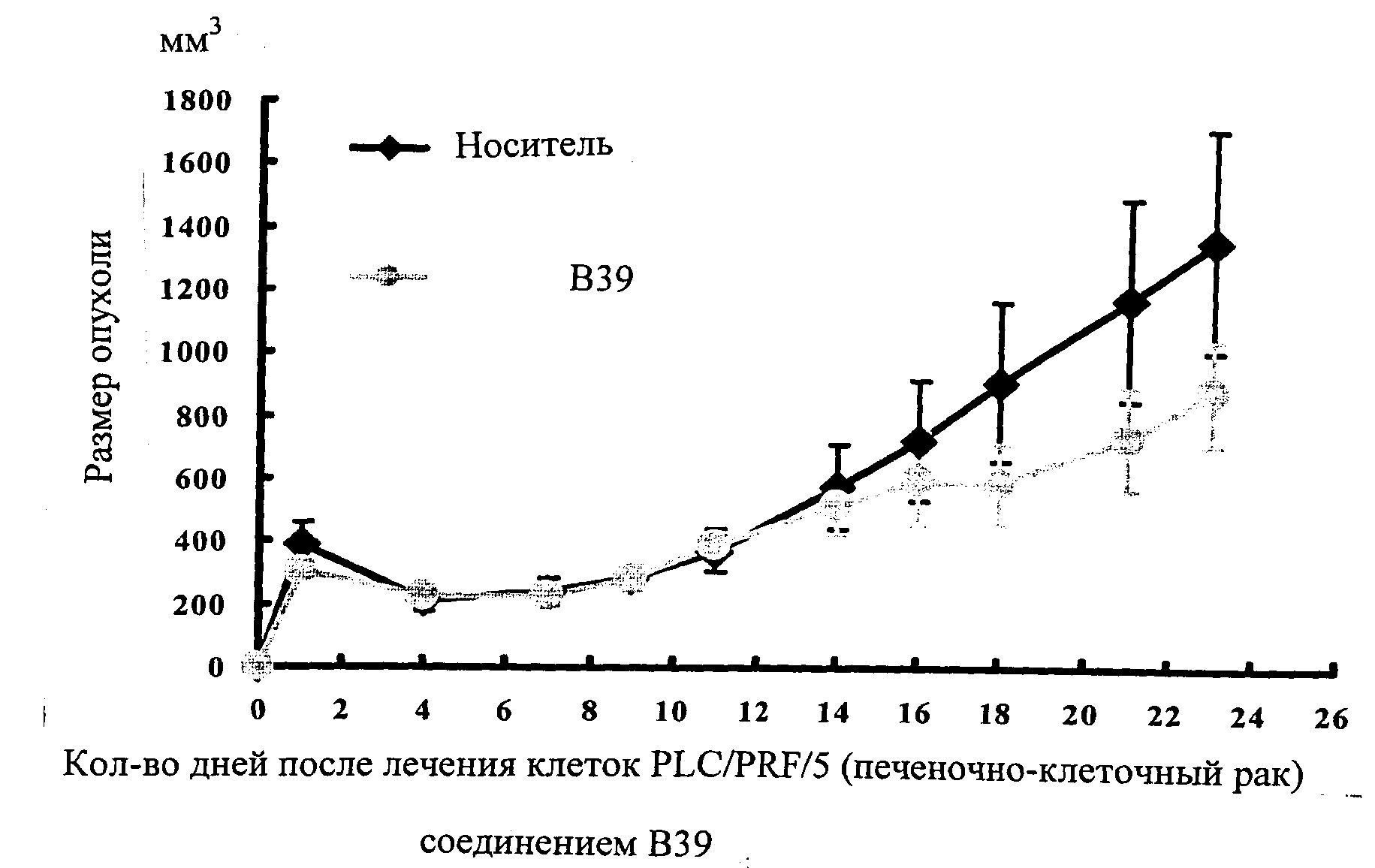
Пример D: Анализ активности ксенотрансплантата опухоли при печеночно-клеточном раке in vivo
Клеточная линия человека для печеночно-клеточного рака PLC/PRF/5 была получена из Американской Коллекции клеточных культур (American Tissue Culture Collection, ATCC номер CRL-8024™). Методика разработки ксенотрансплантата опухоли, а также дозировка ITRI-260 осуществлялись в соответствии с правилами ухода и использования животных ITRI, IACUC. Женские особи мышей BALB/c, лишенные шерстного покрова, в возрасте 6-8 недель получены от BioLASCO Co., Ltd, (Taipei, Taiwan). Этим мышам подкожно в правый бок вводили клетки 5×106 PLC/PRF/5 в количестве 5×106 клеток на каждую особь. Обработка началась, когда размер опухоли достиг 200 мм3. Мыши были случайным образом распределены на когорты (обычно по 4 мыши в каждой группе, из соображений эффективности исследования). Начиная с 9 дня после инокуляции мышам орально давали азаазуленовые соединения B26 (300 мг/кг/день), B39 (150 мг/кг/день) и носитель, процедуру проводили в течение 14 дней.
Размеры опухолей оценивали ежедневно, а массы тел - два раза в неделю. Сделанные кронциркулем измерения были преобразованы в формулу, позволяющую узнать средний объем опухоли: 0,5 × [длина × (ширина)2].
Коэффициент (TGI; %) роста опухоли вычислили как [1-(конечный размер опухоли - первоначальный размер опухоли для леченной группы) / (конечный размер опухоли - первоначальный размер опухоли для группы с носителем)] × 100.
В модели подкожного введения опухолевых клеток PLC/PRF/5 в виде ксенотрансплантата SCID мышам с носителем размер опухоли достиг 1070±345 мм3 на 23 день после инакуляции клеток. Введение азаазуленовых соединений B26 (300 мг/кг/день) и B39 (150 мг/кг/день) проявило противоопухолевую активность; средние размеры опухоли составили 972±96 мм3 и 873±167 мм3; и коэффициенты ингибирования роста опухоли на 23 день составили 35±8% (p=0.3622) и 45±13% (P=0.3622). Ниже приведен результат исследований азаазуленовых соединений B26 и B39 как отдельного средства, проявляющего активность в отношении клеток PLC/PRF/5 ксенотрансплантата опухоли при печеночно-клеточном раке.
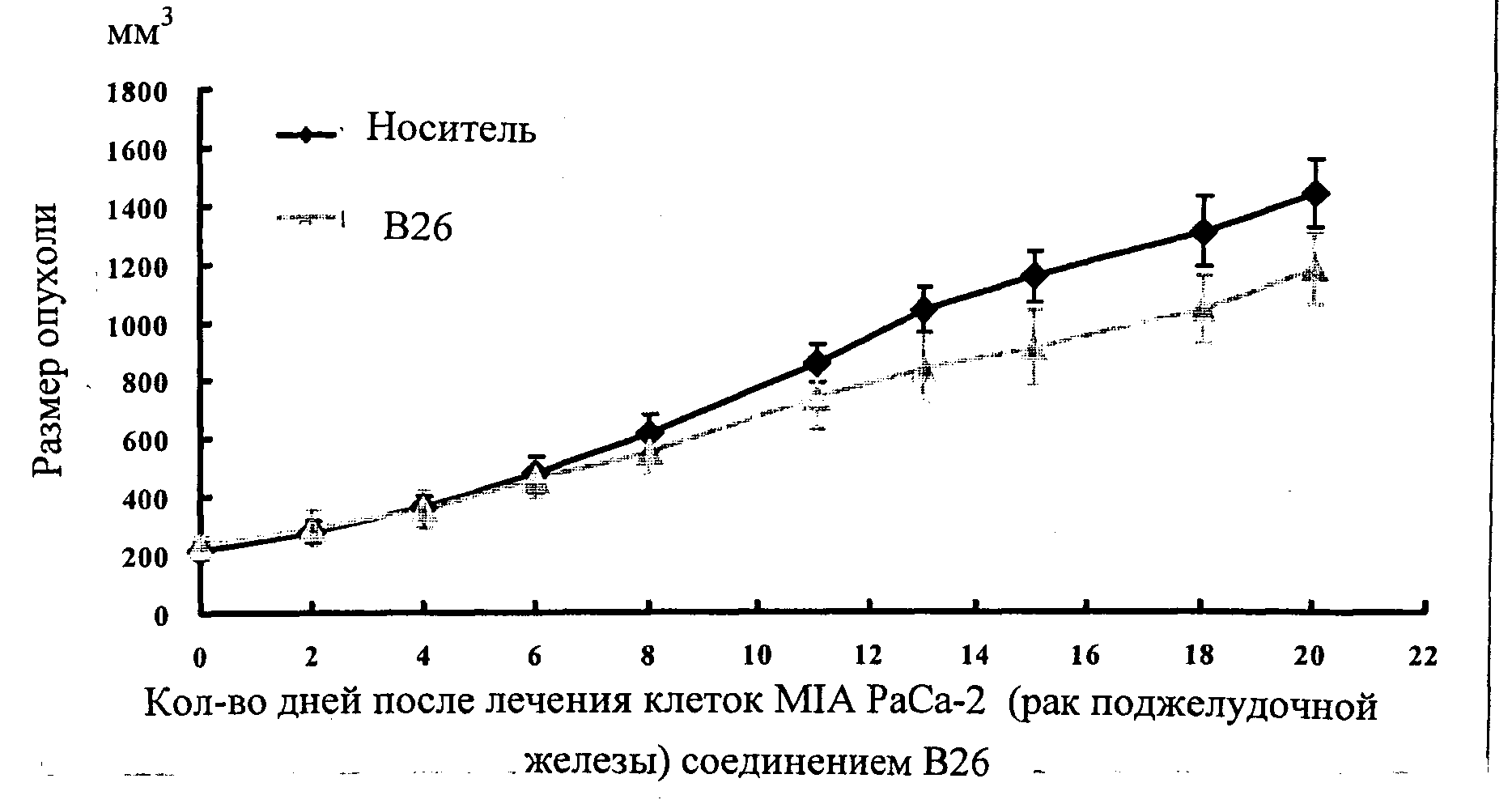
Несмотря на то что настоящее изобретение было описано с помощью примеров и в терминах предпочтительных вариантов, необходимо понимать, что раскрытыми вариантами оно не ограничивается. Напротив, оно предназначено для охвата различных модификаций и сходных процессов. Следовательно, прилагаемая формула изобретений должна соответствовать самым широким интерпретациям всех таких модификаций и сходных процессов.
Реферат
Описывается конкретный перечень разнообразных новых азаазуленовых соединений, содержащих 6,5-конденсированный гетероцикл индольного типа, бензимидазольного типа, пуринового типа, 3Н-имидазо[4,5-b]пиридин, 3Н-имидазо[4,5-с] пиридин т.д., которые могут быть изображены общей формулой (I).где R- =О; R- Н или диэтиламиноСалкил; R-R- Н; другие переменные в этой формуле (I) отражены в конкретных структурных формулах описываемых соединений. Также описывается фармацевтическая композиция, их содержащая. Соединения обладают противоопухолевой активностью и могут быть использованы для лечения рака, такого как рак груди, рак легких, рак поджелудочной железы, рак толстой кишки острая миелоидная лейкемия. 2 н.и 3 з.п. ф-лы, 2 ил., 6 табл., 14 пр.
Формула
Таблица А

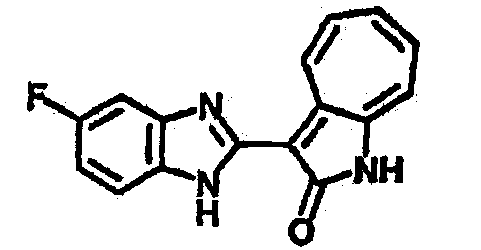

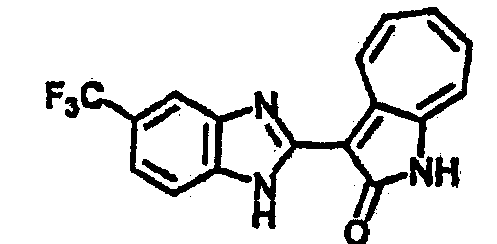
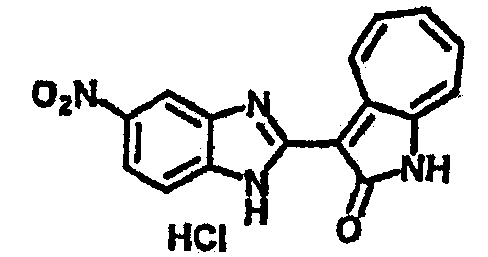
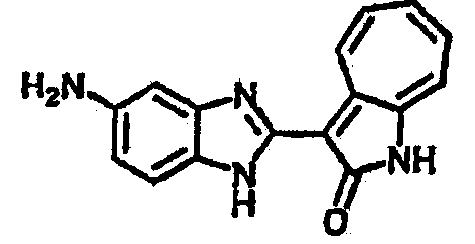
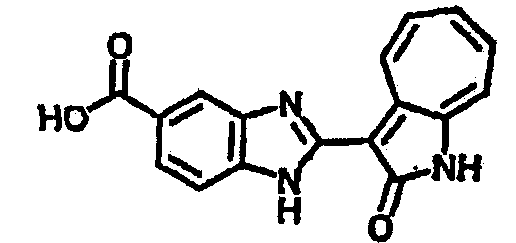
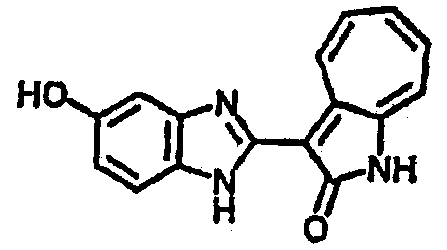
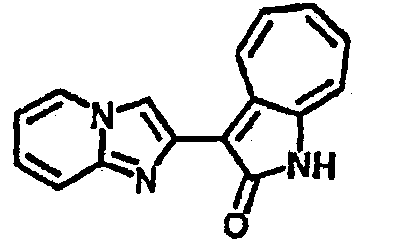
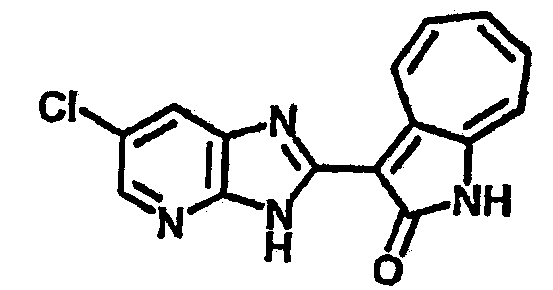
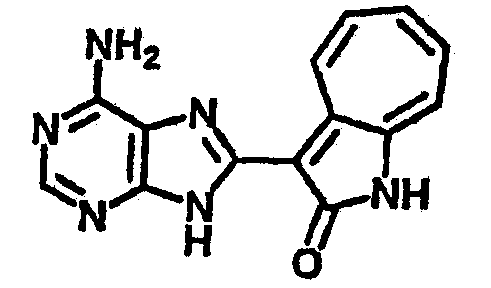
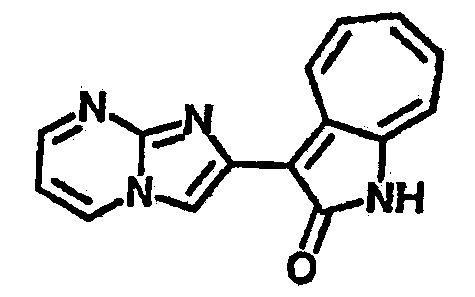
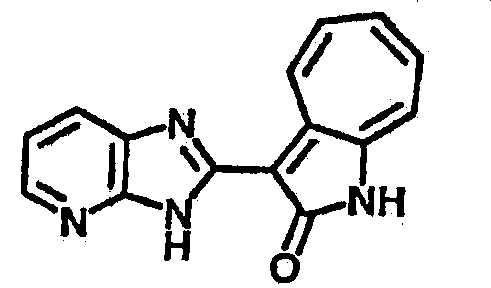



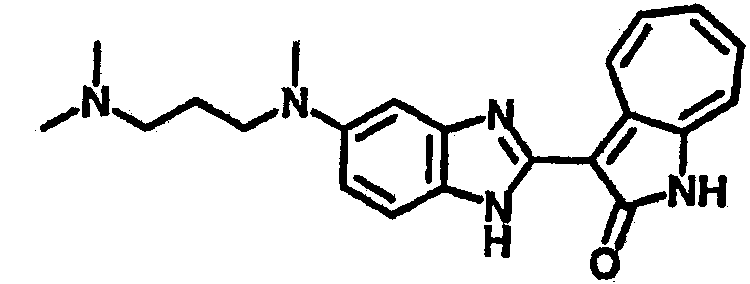
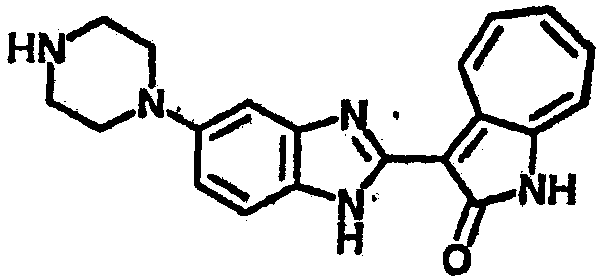
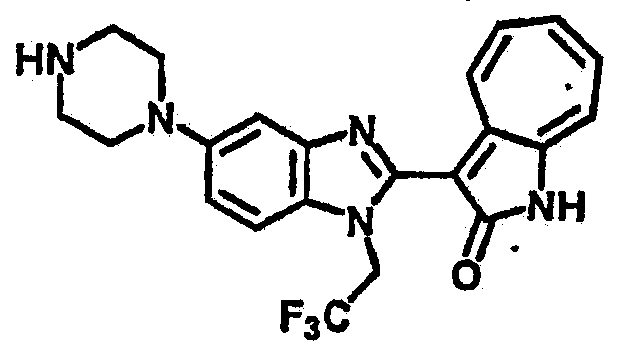
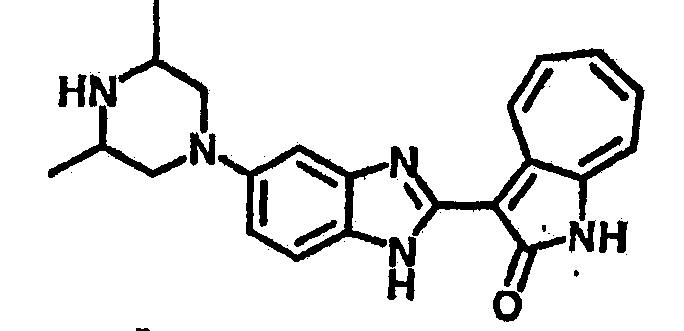
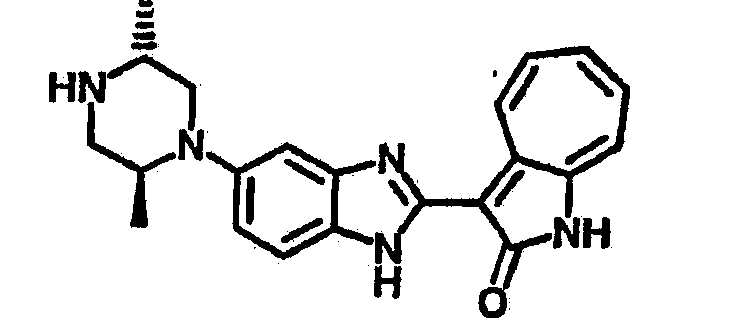
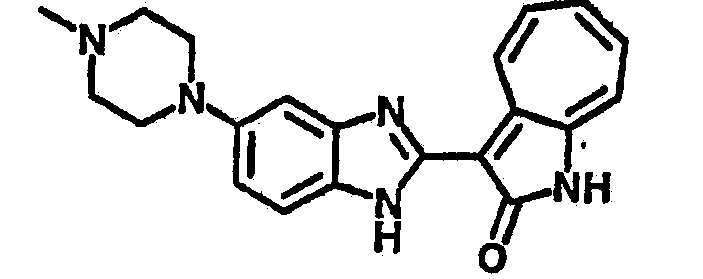

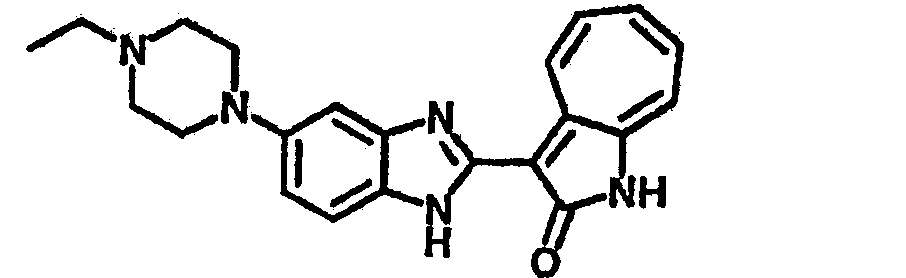

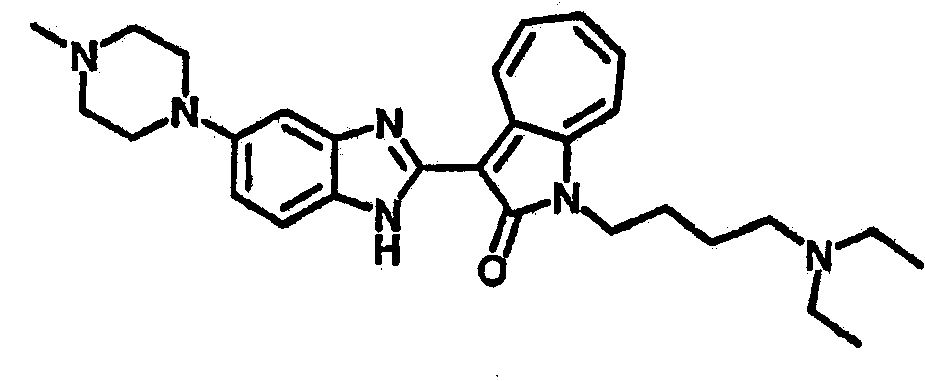
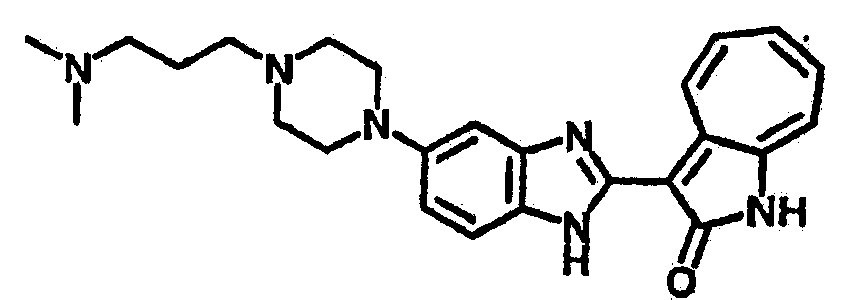


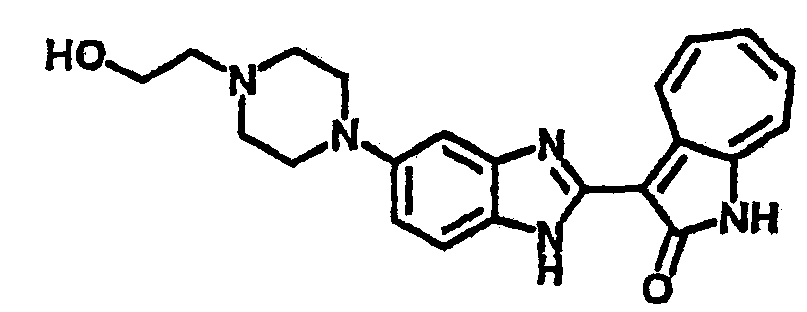
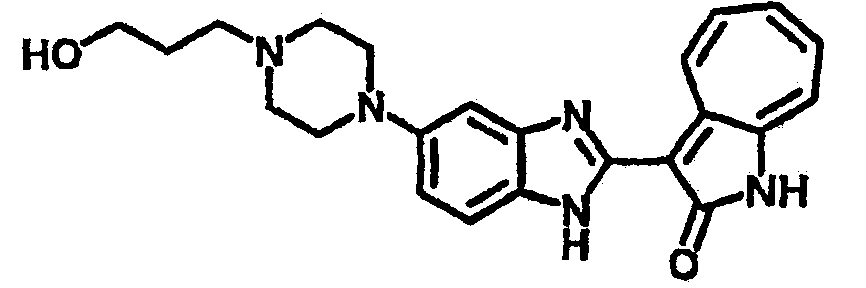
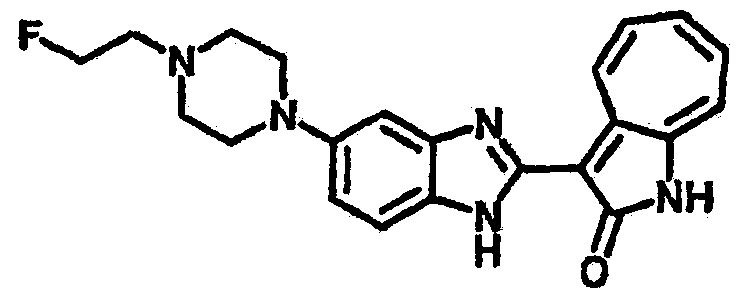

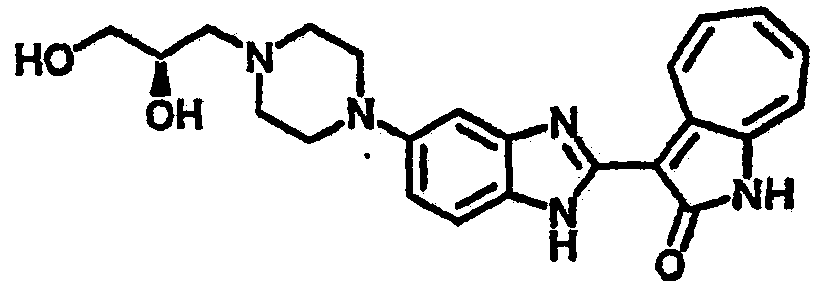
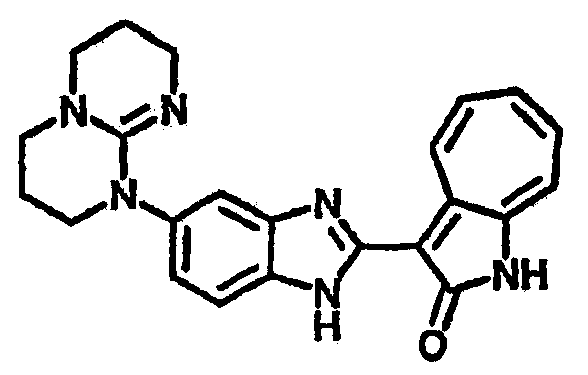
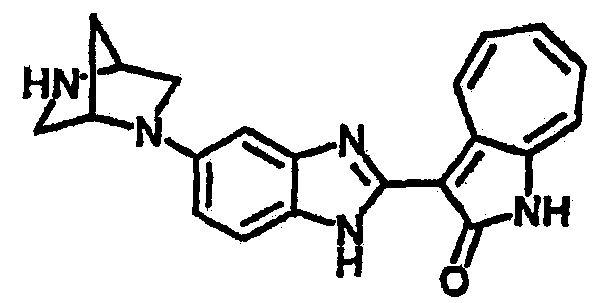

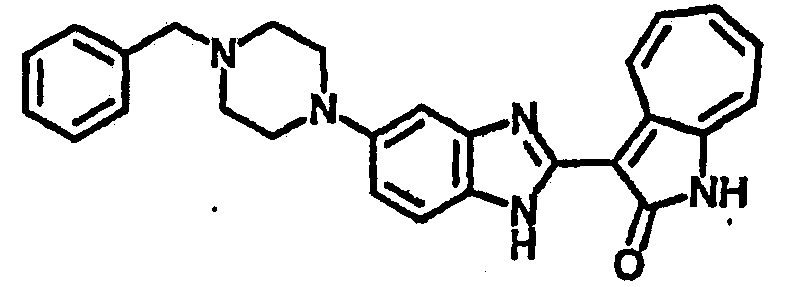
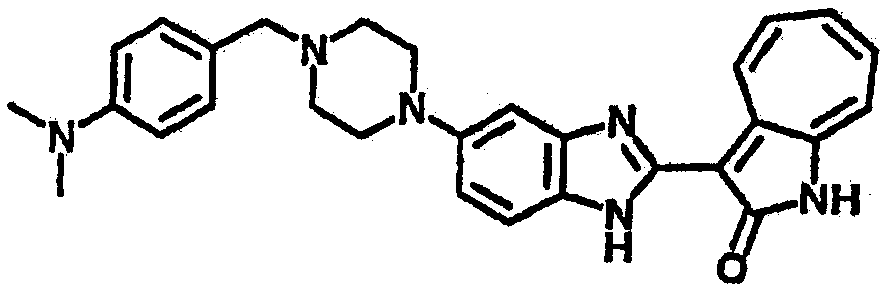
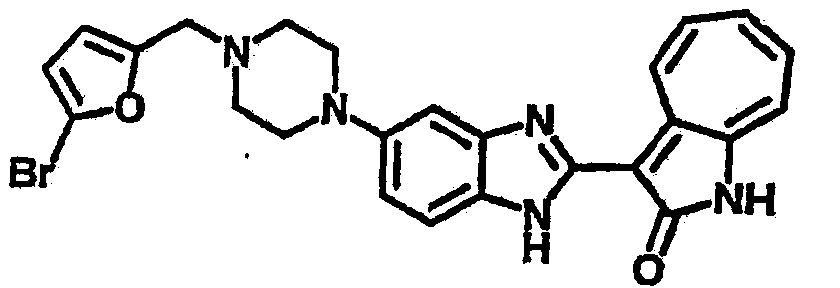


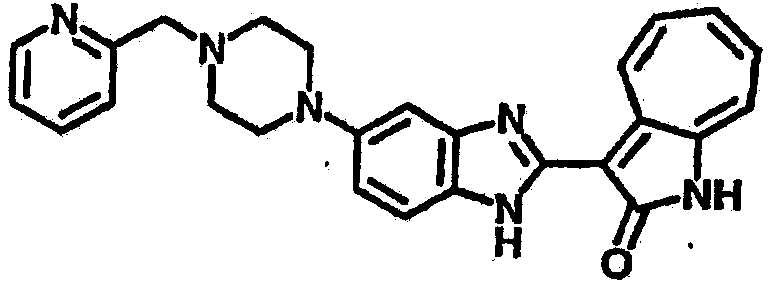
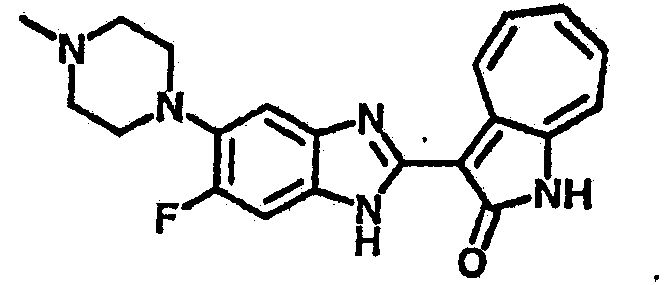

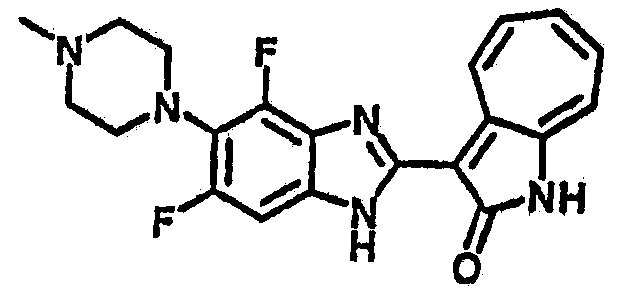
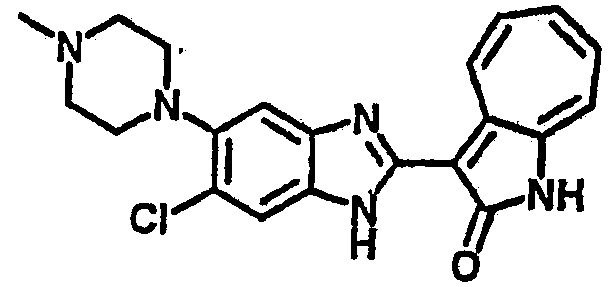
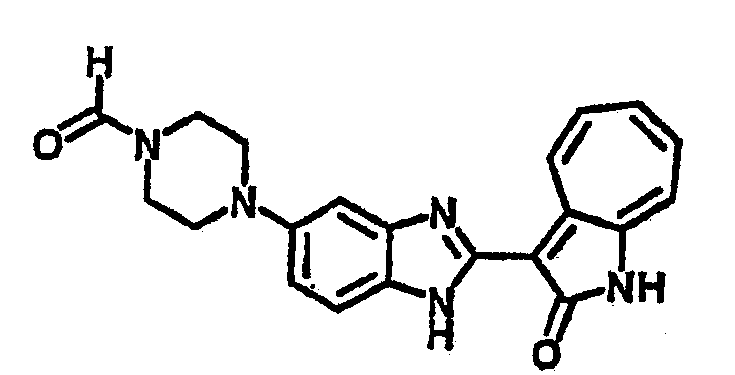
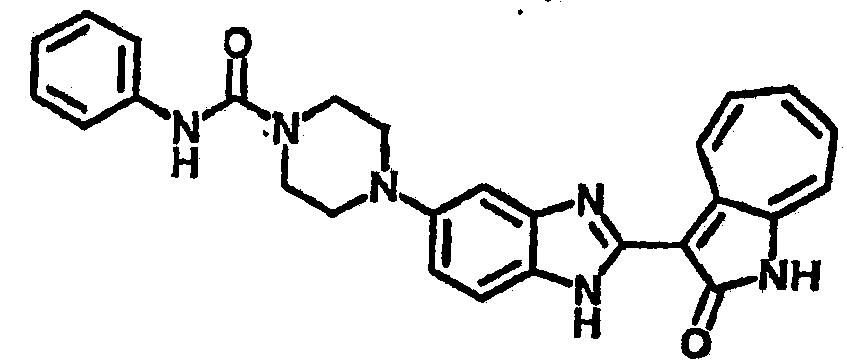
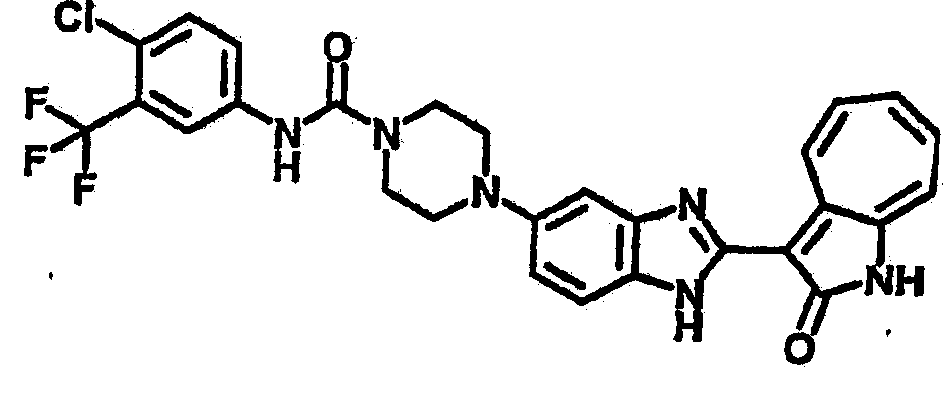

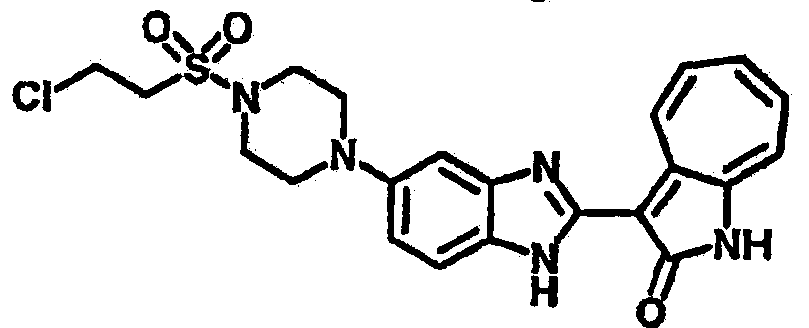
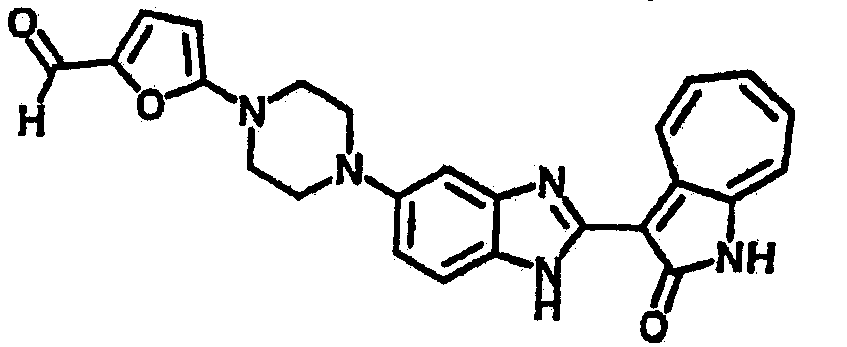

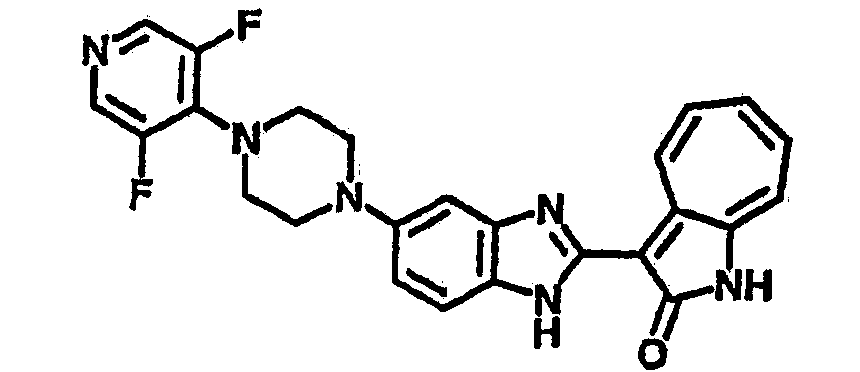
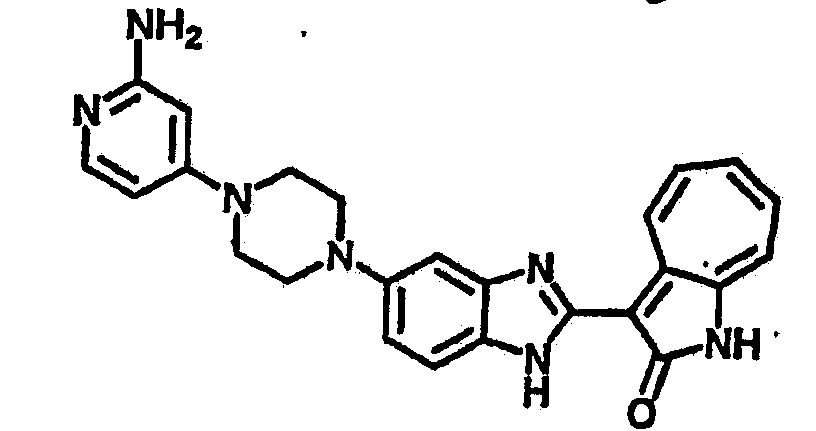
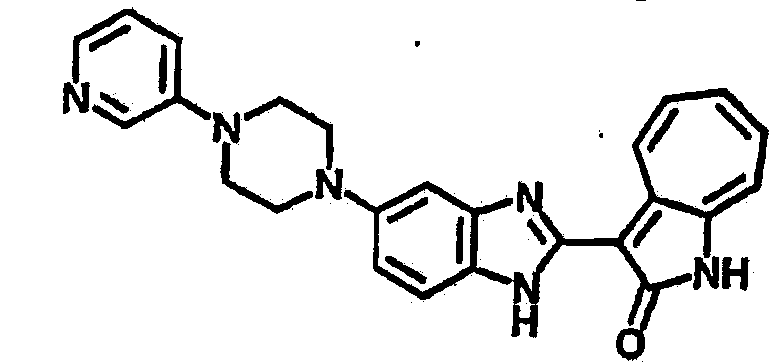
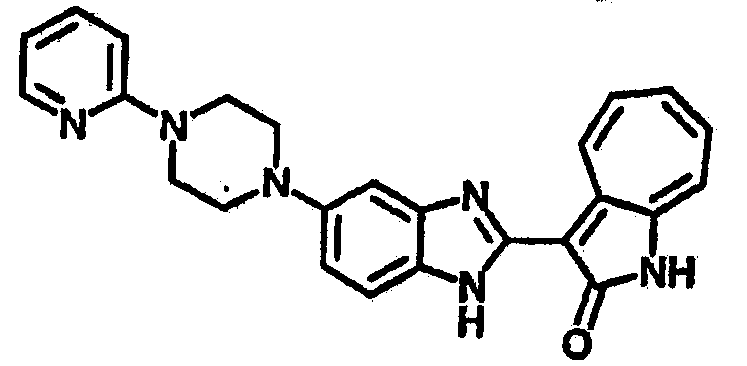
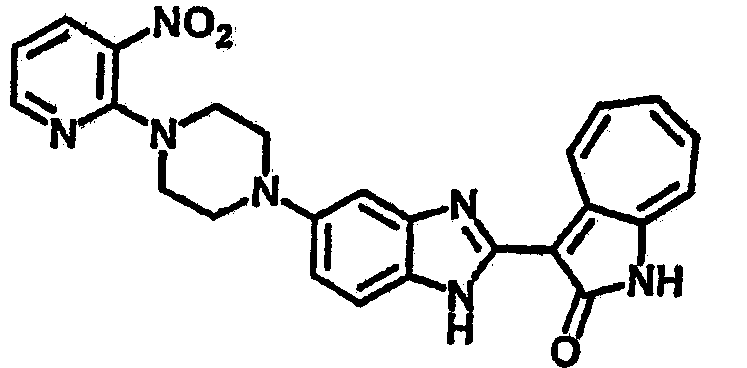
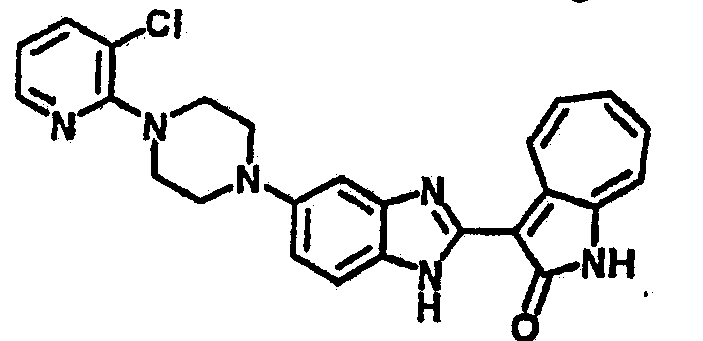

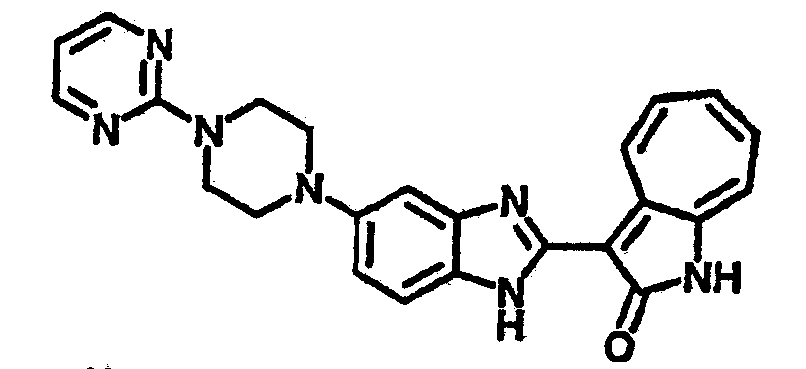
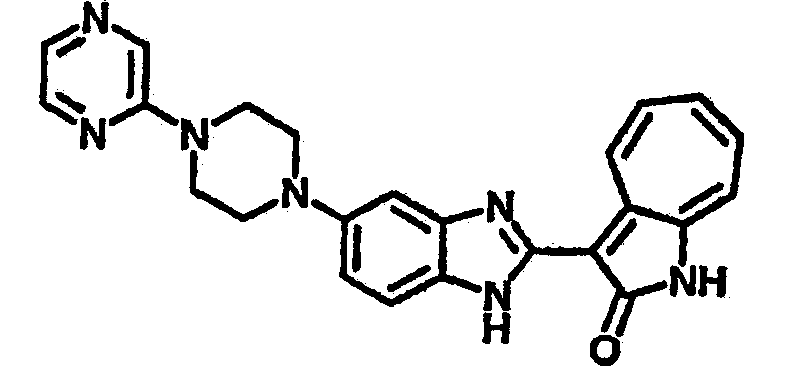
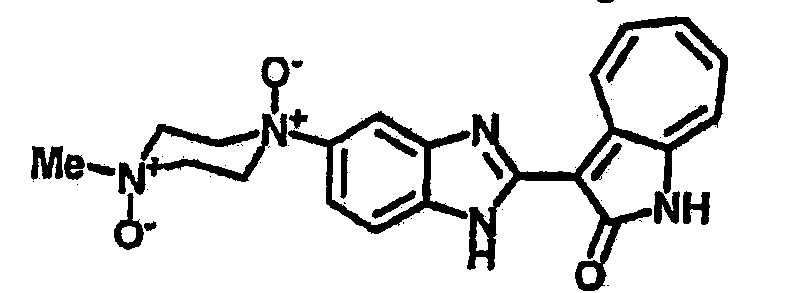
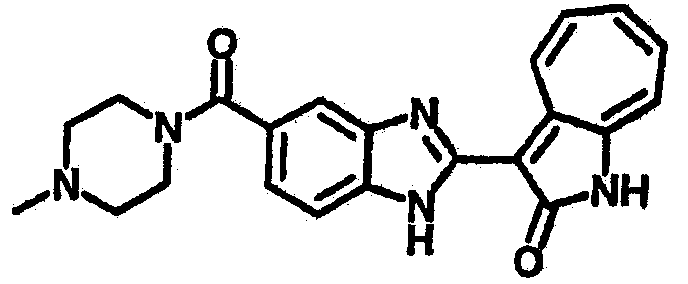

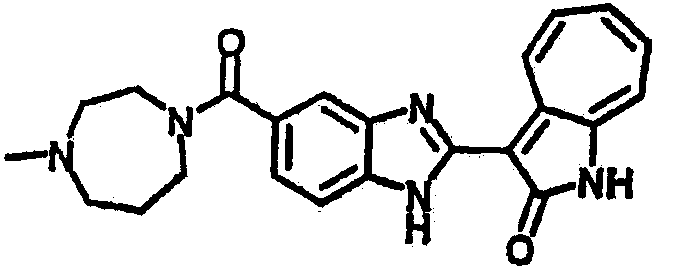
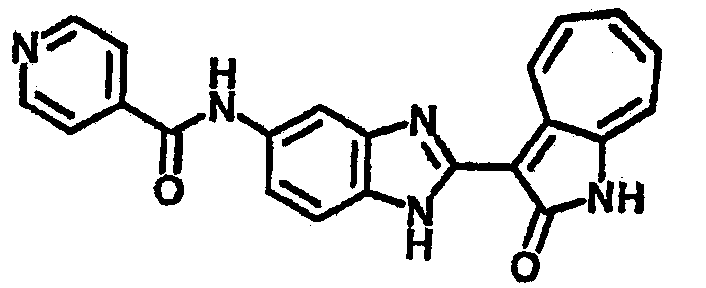
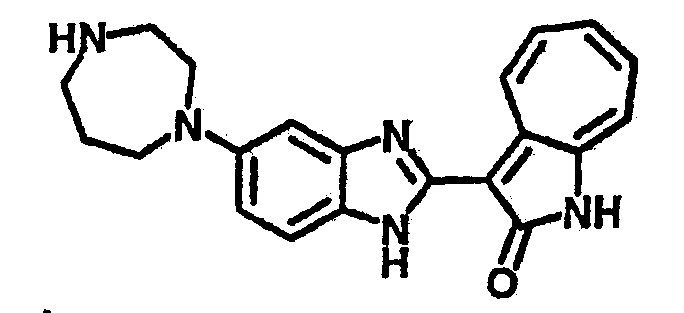

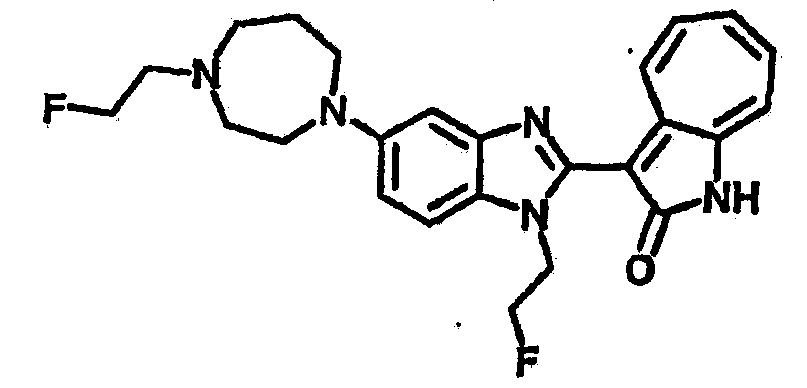

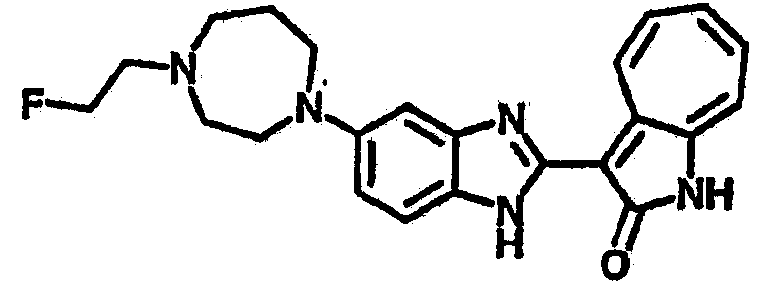
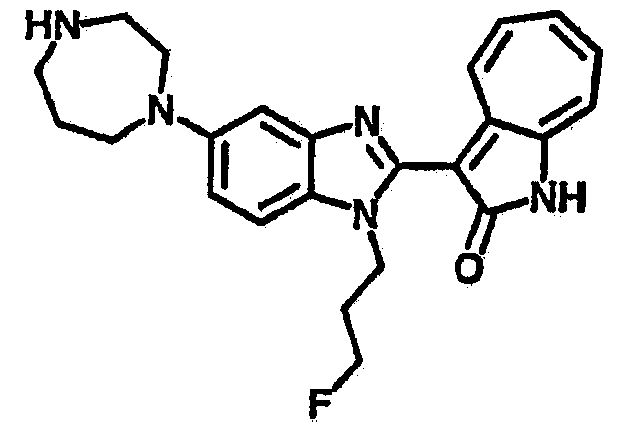
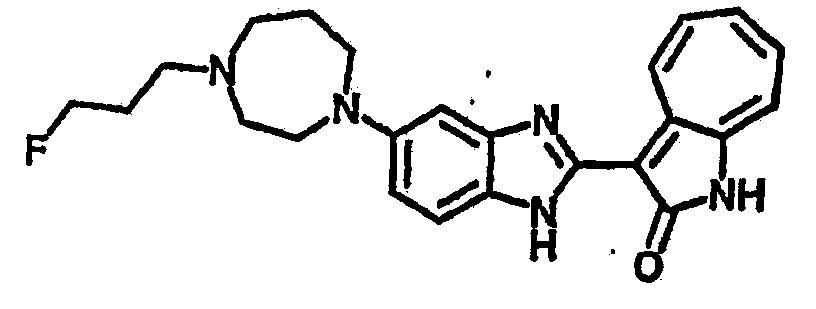
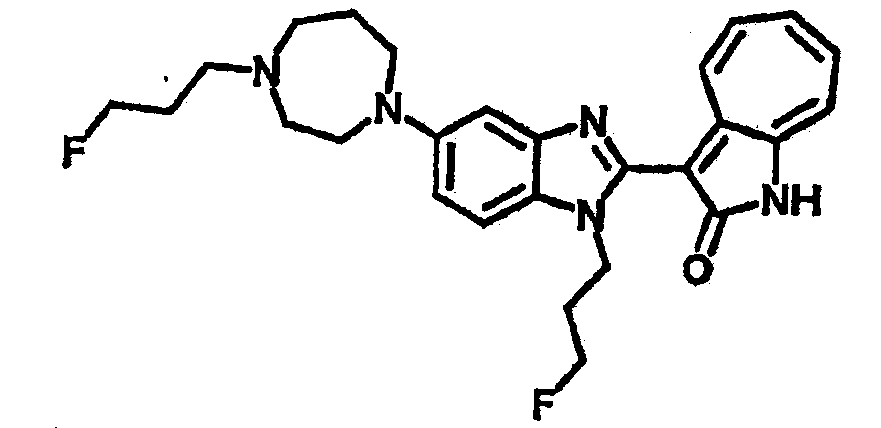
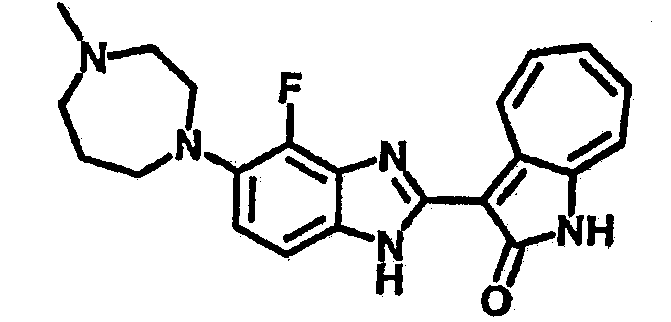
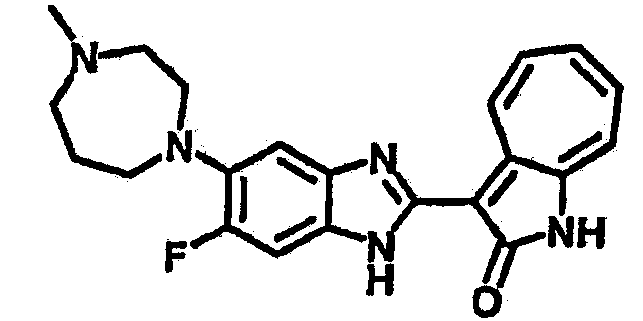
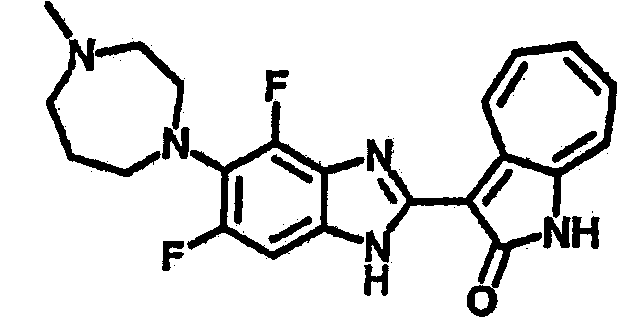
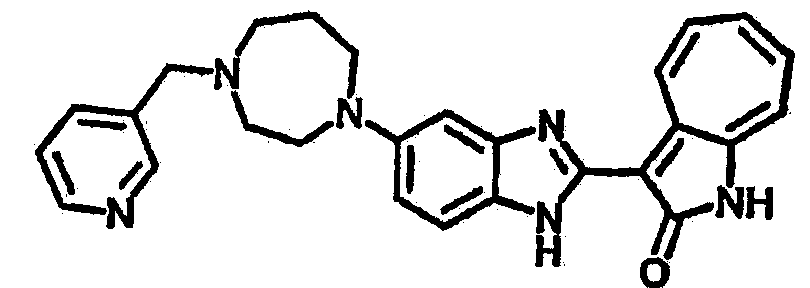

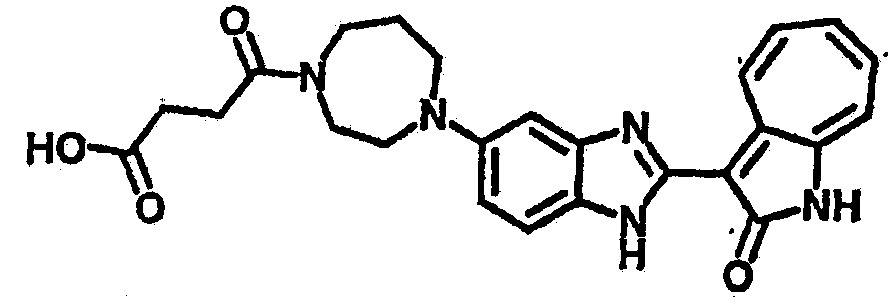
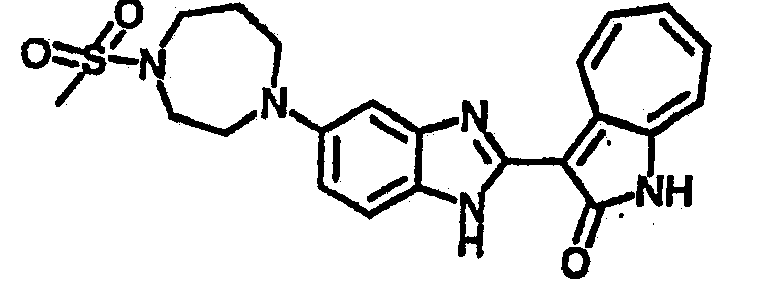

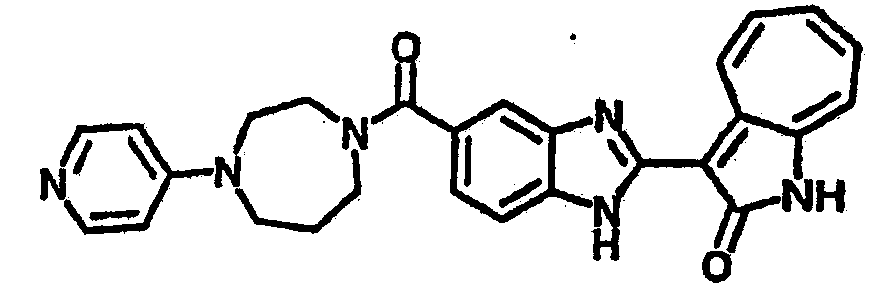

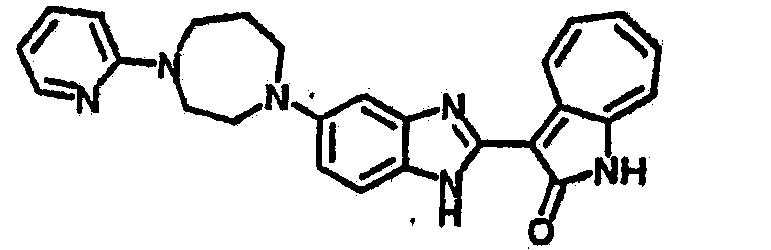
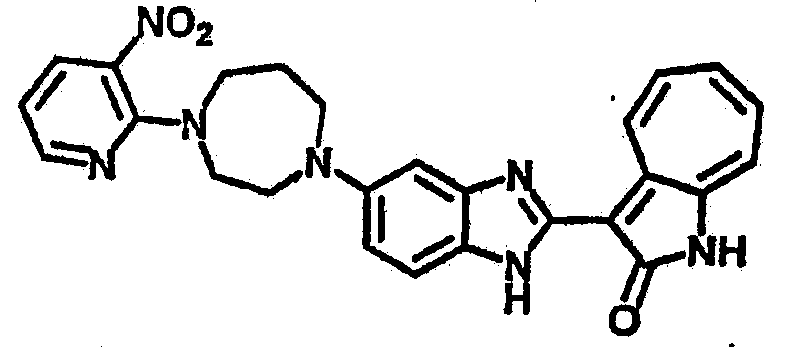
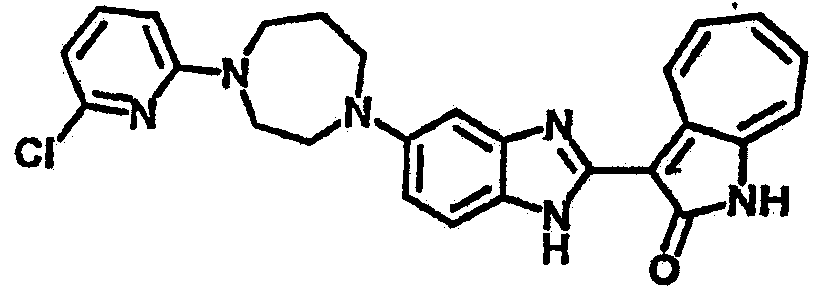
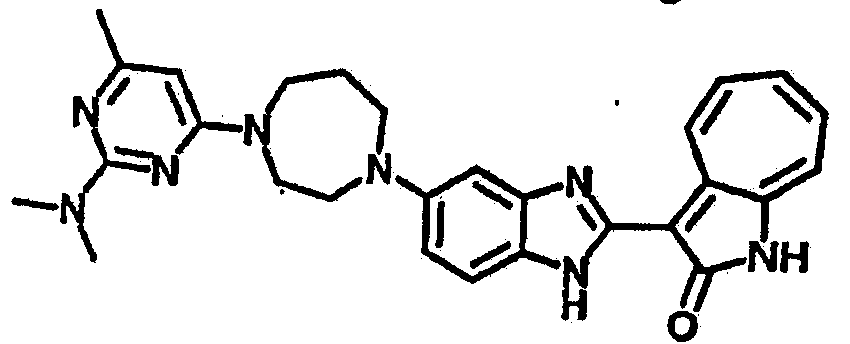
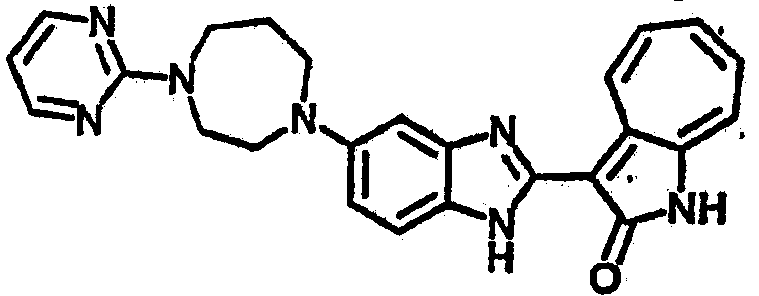
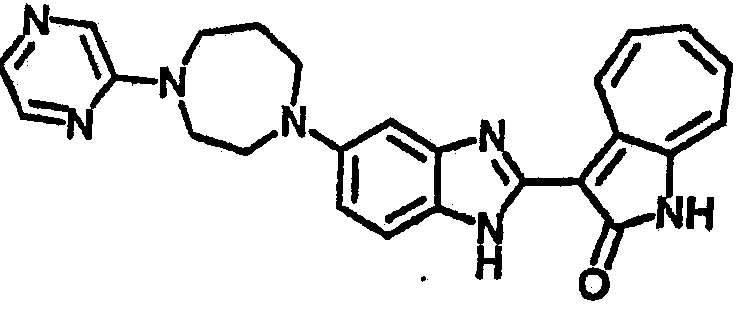
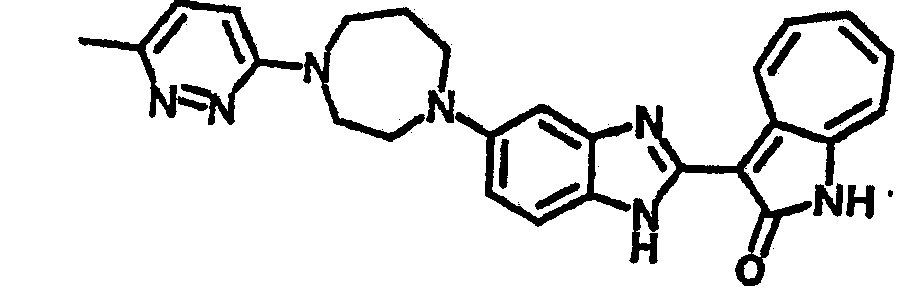
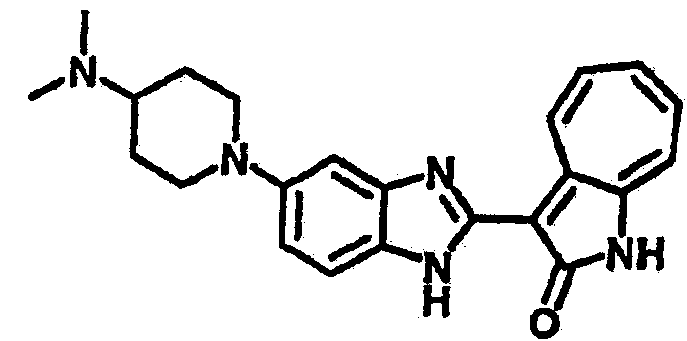
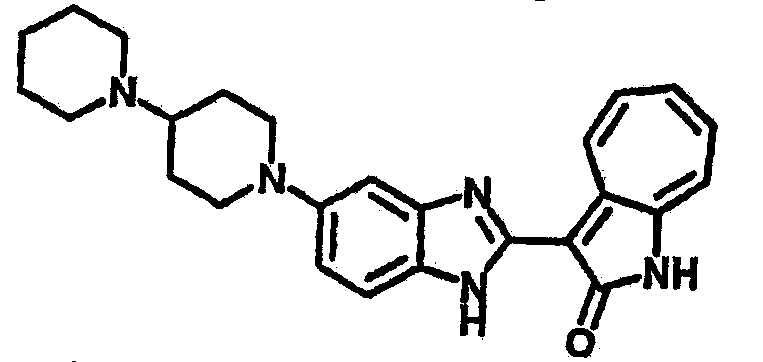
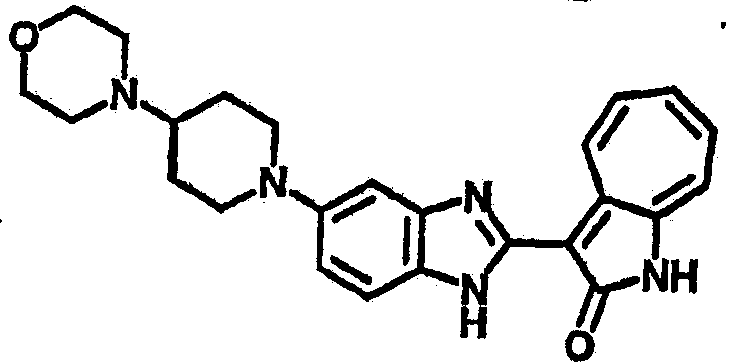
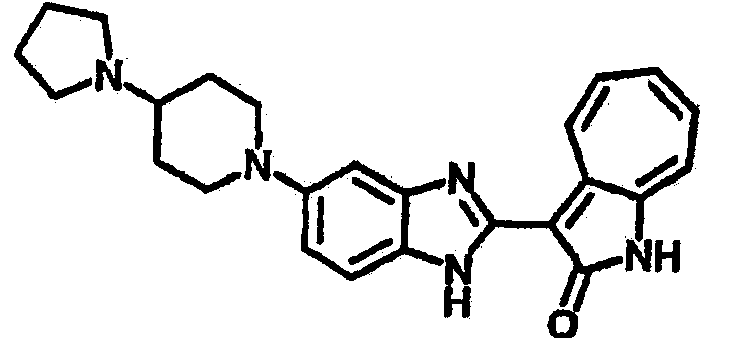
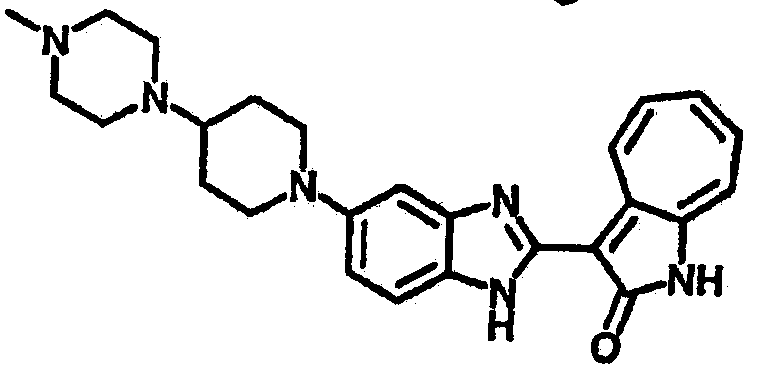
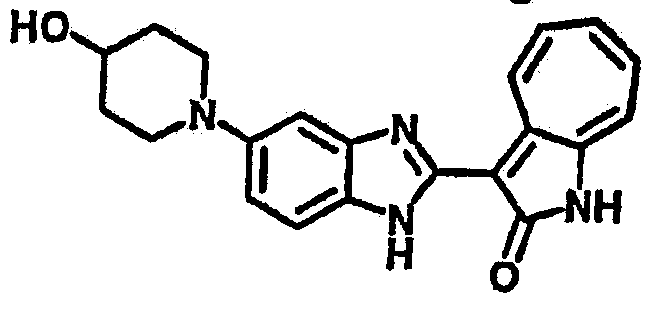


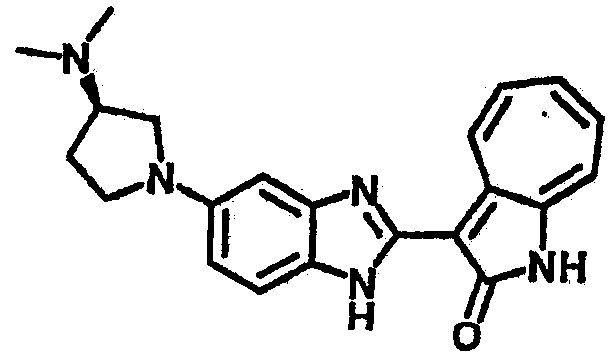
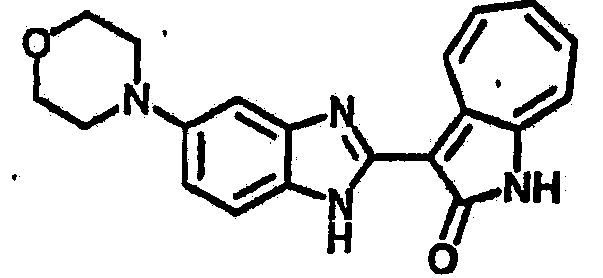
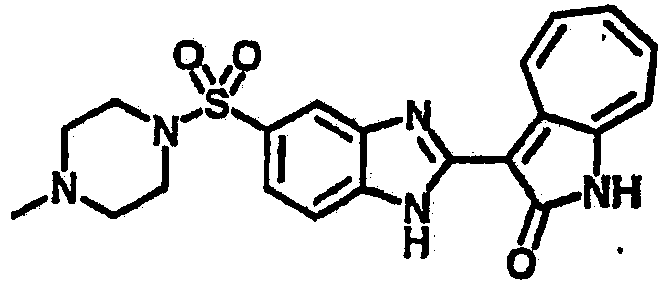
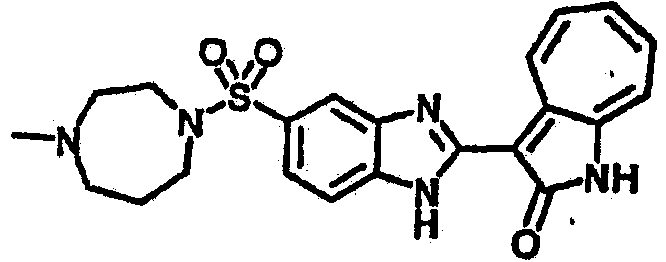
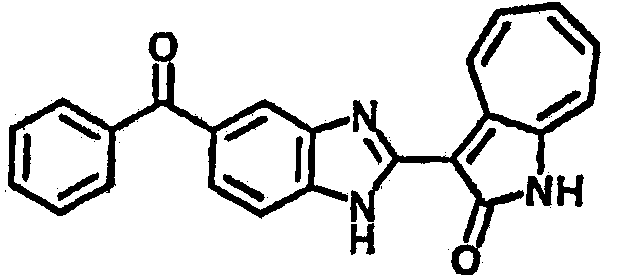
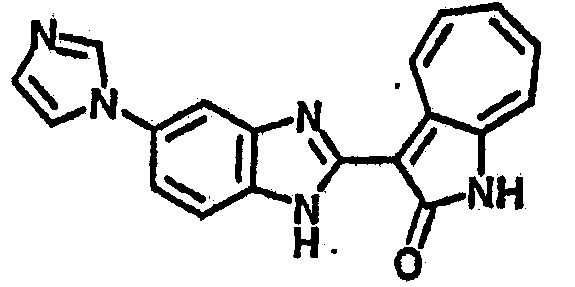
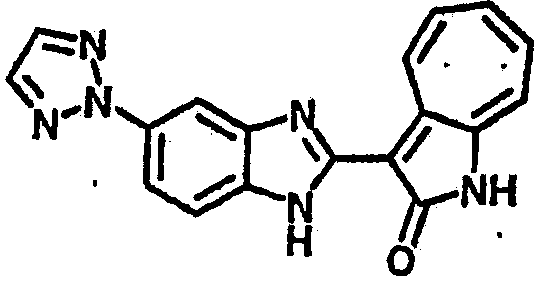
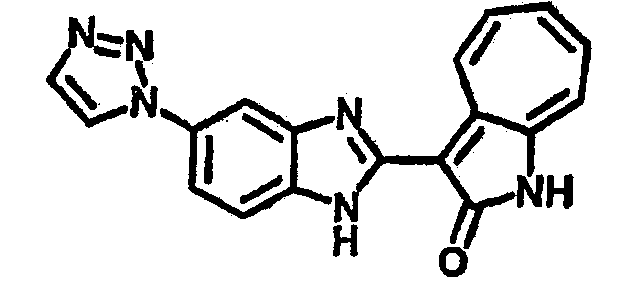
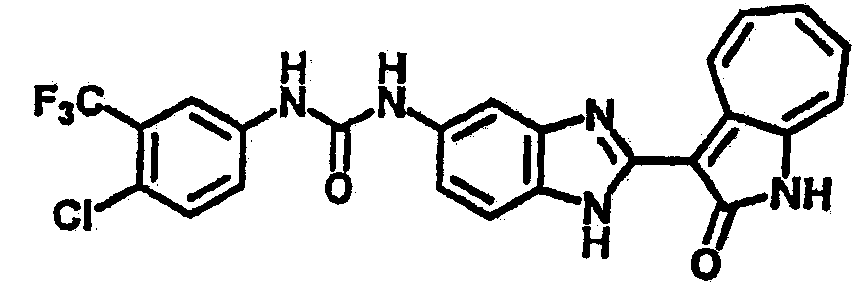

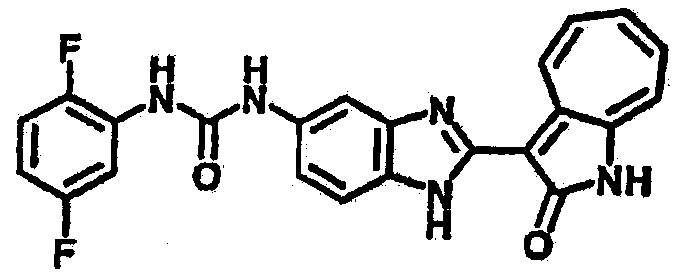
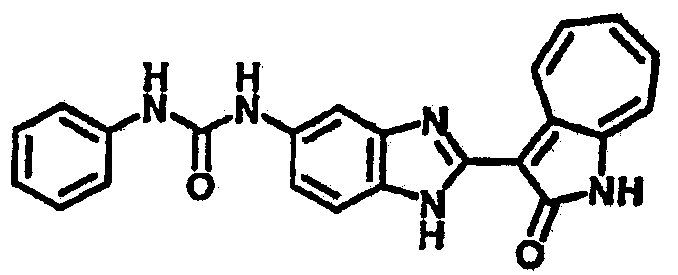
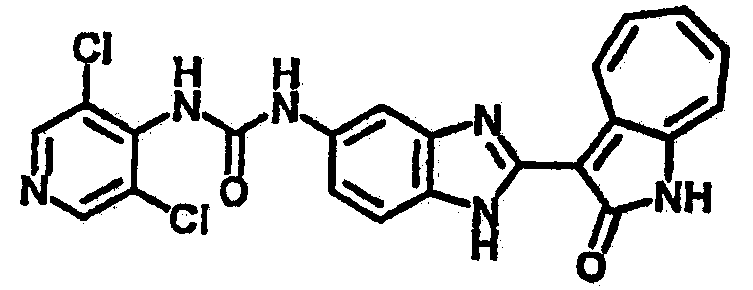
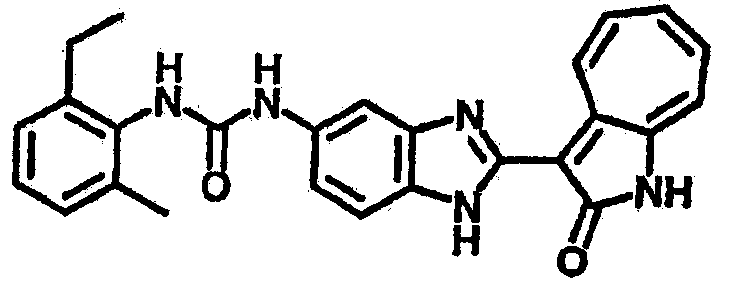
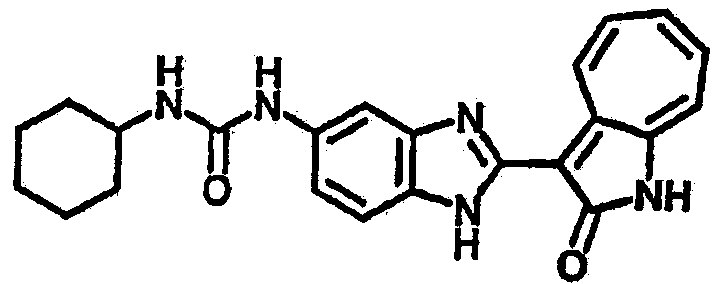
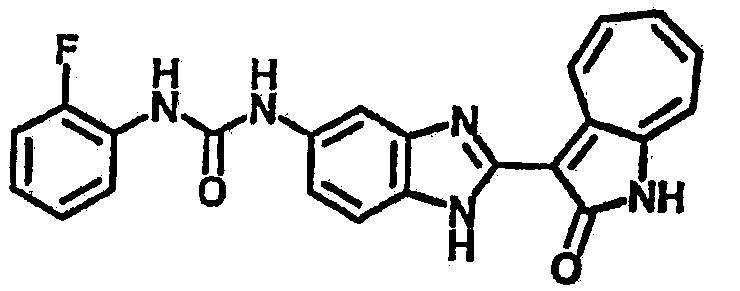
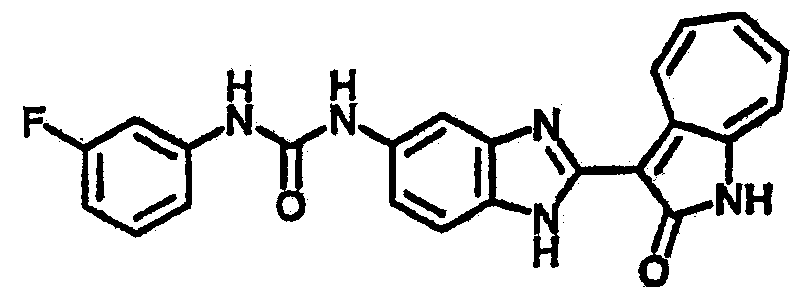
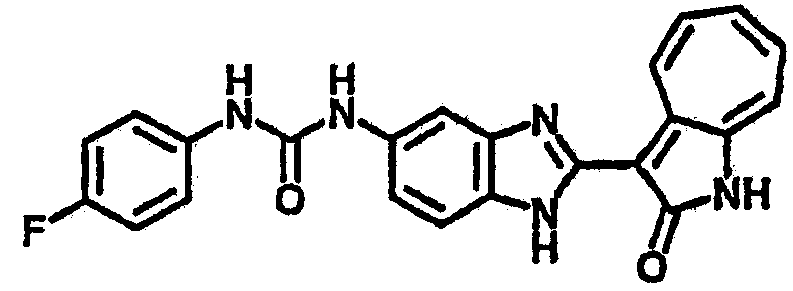
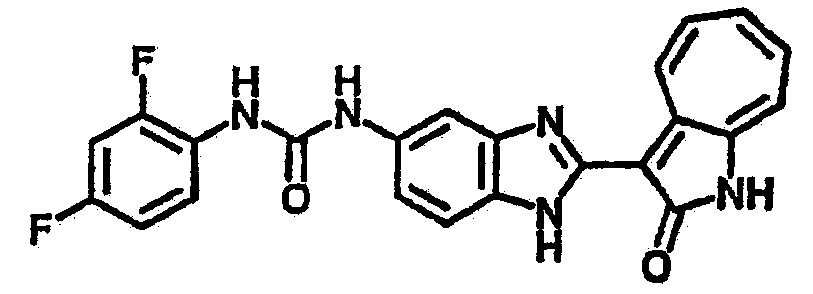
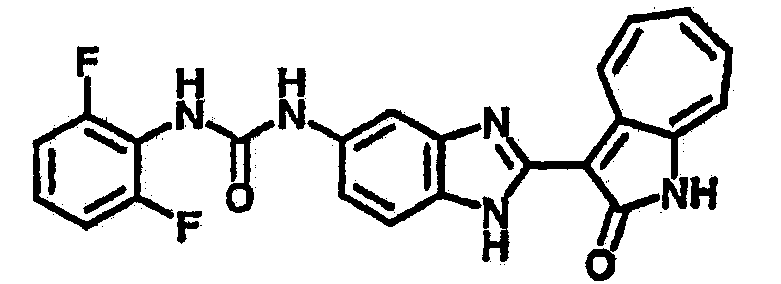
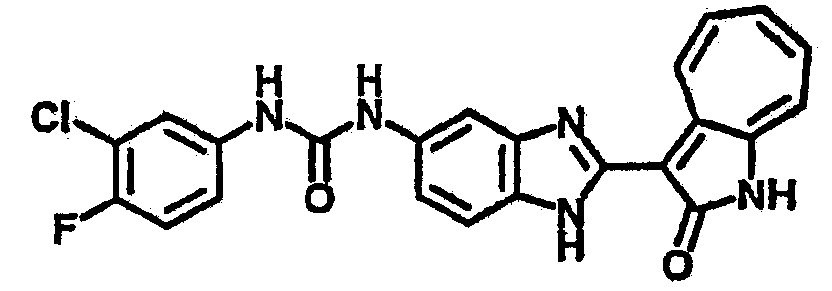

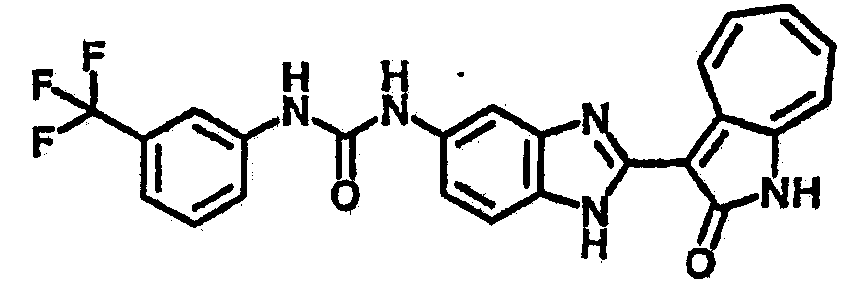
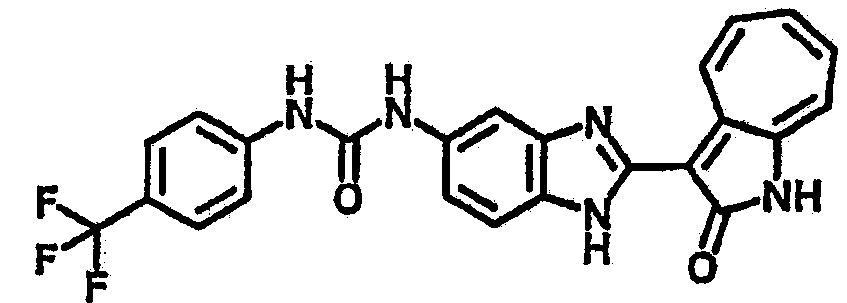
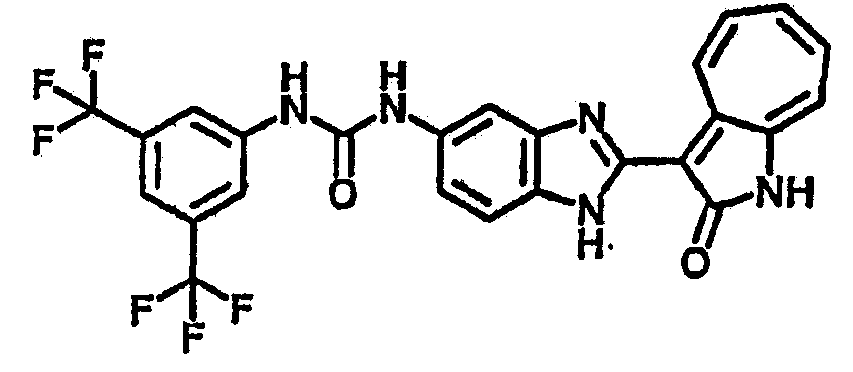
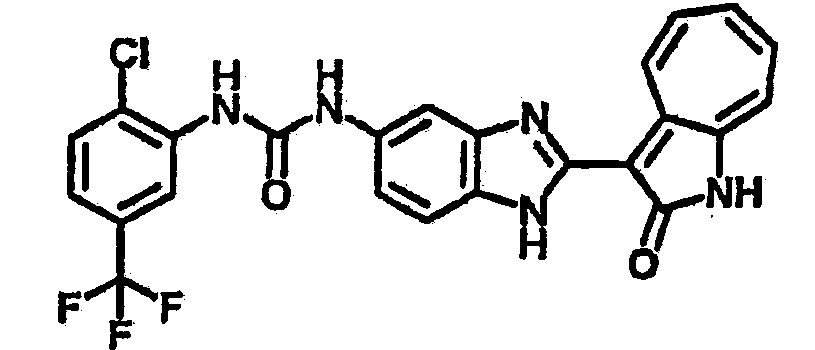
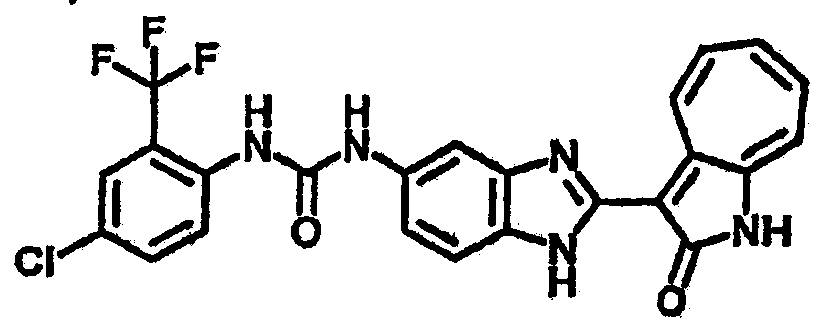
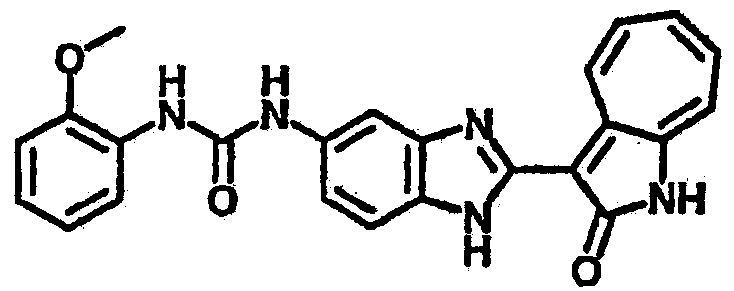
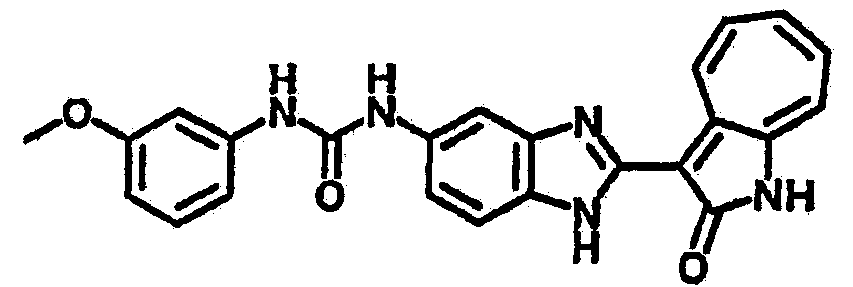
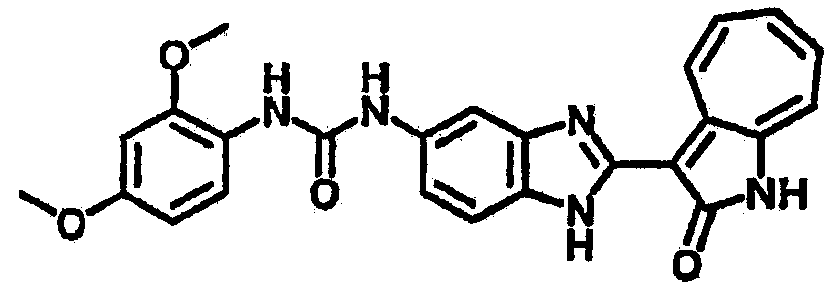
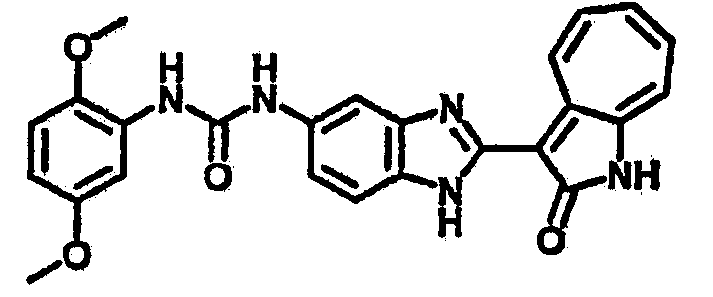
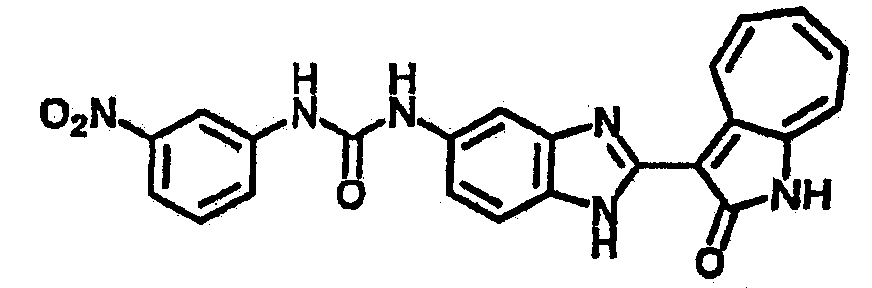
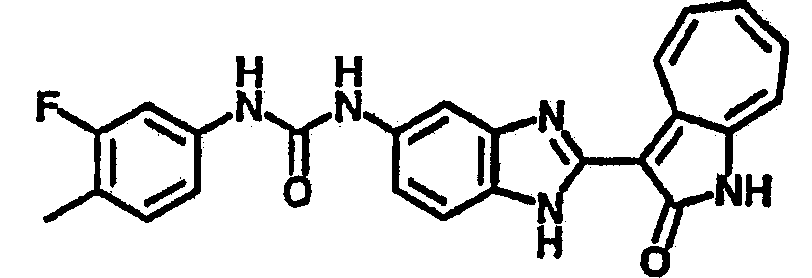
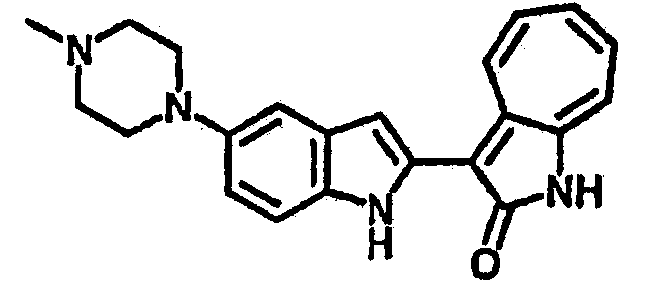
Комментарии