Соединения мочевины и их применение в качестве ингибиторов ферментов - RU2685425C2
Код документа: RU2685425C2
Описание
Область изобретения
Настоящее изобретение относится к соединениям и их применению, а также, в частности, к соединениям и их терапевтическому применению для лечения или предупреждения заболеваний, связанных с субстратами, такими как нейротрансмиттер анандамид, которые расщепляются ферментом гидролазы амидов жирных кислот (FAAH).
Исходные данные изобретения
Фермент FAAH расщепляется такими амидами жирных кислот, как анандамид (N-арахидоноилэтаноламин), N-олеоилэтаноламин, N-пальмитоилэтаноламин и олеамид. Анандамид, также известный как N-арахидоноилэтаноламин или АЕА, является эндогенным нейротрансмиттером каннабиноида, который встречается в органах животных и человека, особенно в мозге. Также было установлено, что анандамид вступает в связь с ванилоидным рецептором. Анандамид разлагается под действием фермента гидролазы амидов жирных кислот (FAAH) до этаноламиновой и арахидоновой кислоты. Соответственно, ингибиторы FAAH обеспечивают повышение уровня содержания анандамида.
Анандамид является нейротрансмиттером эндоканнабиноидной системы, который стимулирует деятельность каннабиноидных рецепторов. Каннабиноидные рецепторы, такие как СВ1 и СВ2, являются рецепторами, сопряженными с G-белком. СВ1 встречается главным образом в центральной нервной системе, тогда как СВ2 присутствует, в основном, в периферических тканях. Установлено, что эндоканнабиноидная система участвует в выполнении большого количества физиологических функций, как в центральной, так и в периферической нервных системах, а также в периферических органах. Доказано, что изменение активности эндоканнабиноидной системы потенциально может обеспечить терапевтическое воздействие на большое количество разнообразных заболеваний и патологических состояний. Поэтому эндоканнабиноидная система и, в частности, фермент FAAH стали мишенью для терапевтического воздействия с целью разработки возможных средств лечения многих заболеваний. Установлено, что эндоканнабиноидная система связана с контролем аппетита, ожирением, метаболическими нарушениями, кахексией, анорексией, болями, воспалениями, нейротоксичностями, травмами нервной системы, инсультами, множественным склерозом, повреждениями спинного мозга, болезнью Паркинсона, дискинезией, вызванной леводопой, болезнью Хантингтона, синдромом Туретта, поздней дискинезией, дистонией, боковым амиотрофическим склерозом, болезнью Альцгеймера, эпилепсией, шизофренией, тревожным расстройством, депрессией, бессонницей, тошнотой, рвотой, алкогольными психозами, привыканием к чрезмерному употреблению опиата, никотина, кокаина, алкоголя и психостимуляторов, гипертензией, злокачественной гипертензией, реперфузионным повреждением миокарда, атеросклерозом, астмой, глаукомой, ретинопатией, раком, воспалительными заболеваниями кишечника, острой и хронической печеночной недостаточностью, такой как гепатит и цирроз печени, артритом и остеопорозом. Эндоканнабиноидная система и заболевания, с которыми она связана, подробно рассматриваются в статье Pacher et al. (2006) Pharmacol. Rev. (Фармакологическое обозрение) 58:389-462.
Чтобы регулировать уровень эндогенных субстратов FAAH, таких как анандамид, которые, в свою очередь, регулируют эндоканнабиноидную систему, были разработаны ингибиторы ферментов FAAH. Это обеспечивает возможность хотя бы частичного лечения или предупреждения состояний и заболеваний, связанных с эндоканнабиноидной системой.
Поскольку субстраты FAAH связываются с другими рецепторами, например, с ванилоидным рецептором, и/или участвуют в иных сигнальных путях, ингибиторы FAAH могут также обеспечить возможность хотя бы частичного лечения или предупреждения заболеваний и болезней, связанных с другими путями или системами, например с ванилоидной системой.
В WO 2010/074588 представлены соединения, являющиеся ингибиторами FAAH. В работе Käsnänen et al. (Heikki Käsnänen, Mikko J. Myllymäki, Anna Minkkilä, Antti O. Kataja, Susanna M. Saario, Tapio Nevalainen, Ari M.P. Koskinen, and Antti Poso. (Хейкки Кененен, Микко Й. Милимяки, Анна Минкиля, Анти О. Катая, Сузанна М. Саарио, Тапио Невалайнен, Ари М.П. Коскинен и Анти Посо.) Chem Med Chem. 2010, 5(2), 213-231) раскрыты соединения карбамата, являющиеся ингибиторами FAAH. В частности, соединение 6b является ингибитором FAAH, в котором содержится имидазоловая структура. Однако при этом данное соединение является слабым ингибитором FAAH по сравнению с многими другими соединениями карбамата, описание которых приводится в настоящей работе, и которые не содержат имидазоловой структуры.
Краткое изложение сущности изобретения
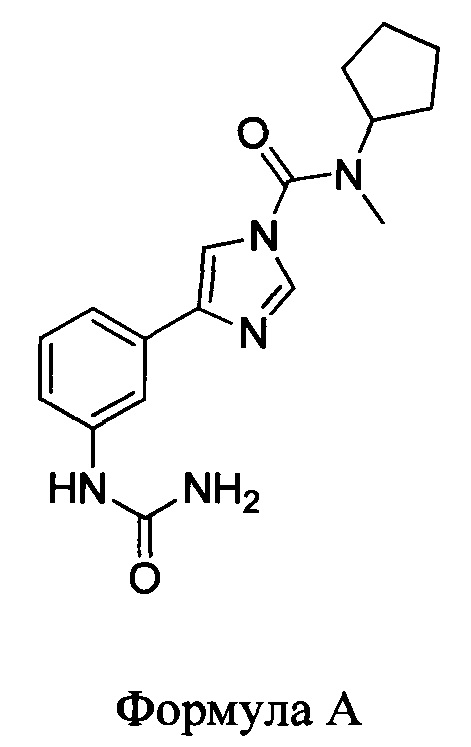
В первом аспекте настоящего изобретения представлено соединение, имеющее следующую структуру:
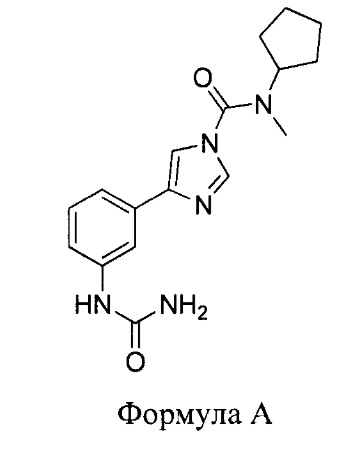
или его фармацевтически приемлемая соль или производное.
Установлено, что соединение согласно изобретению регулирует активность фермента гидролазы амидов жирных кислот (FAAH). Кроме того, доказано, что оно имеет относительно сильное действие, относительно высокую периферическую селективность (т.е. оно замедляет FAAH гораздо сильнее в периферических тканях по сравнению с тканями центральной нервной системы), а также является относительно метаболически стабильным. В частности, было доказано, что соединение согласно изобретению обеспечивает более высокие результаты в отношении одного или нескольких из этих свойств по сравнению с соединениями, представленными в WO 2010/074588.
«Фармацевтически приемлемые соли» соединений, рассматриваемых в настоящем изобретении, включают соли с неорганическими основаниями, соли с органическими основаниями, соли с неорганическими кислотами, соли с органическими кислотами, а также соли с основными или кислыми аминокислотами. Соли с кислотами могут, в частности, применяться в некоторых случаях. Среди солей, которые можно привести в качестве примера, гидрохлорид, ацетат, трифторацетат, метансульфонат, 2-гидроксипропан-1,2,3-трикарбоксилат, (2R,3R)-2,3-дигидроксисукцинат, фосфат, сульфат, бензоат, 2-гидроксибензоат, S-(+)-манделат, S-(-)-малат, S-(-)пироглутамат, пируват, p-толуенсульфонат, 1-R-(-)-камфорсульфонат, фумарат и оксалат. Соединение, рассматриваемое в настоящем изобретении, может быть представлено в виде сольвата (например, гидрат) или несольвата (например, негидрат). Когда оно имеет форму сольвата, в качестве дополнительных растворителей могут выступать спирты, такие как пропан-2-ол.
«Фармацевтически приемлемые производные» соединения согласно изобретению являются производными, в которых одна или несколько групп соединения изменены в результате реакции с другой молекулой. Например, производные включают изменение группы NH2 до формы NHR или NR2, в которой R может быть представлен алкилом С1-18 (например, алкил C1-6), арилом, гетероарилом, циклоарилом С3-8 или их сочетаниями. Такие производные могут образовываться в соответствии со следующей Схемой.
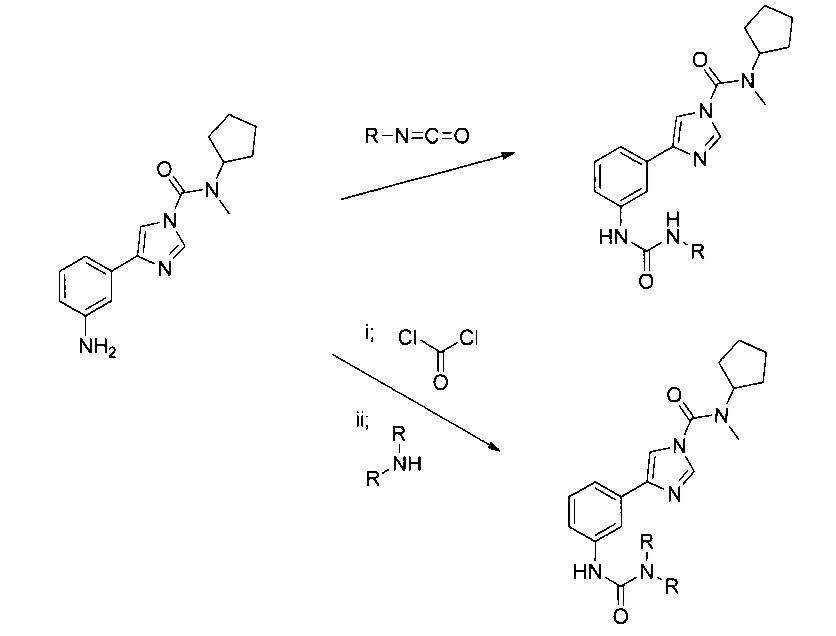
Например, производные включают продукты, образовавшиеся в результате реакции NH2 группы 4-(3-аминофенил)-N-циклопентил-N-метил-1H-имидазол-1-карбоксамида с изоцианатом R-N=C=O (см. R.G. Arnold, J.A. Nelson, J.J. Verbanc: Recent Advances in Isocyanate Chemistry Chemical Reviews (Последние достижения в области химии изоцианатов Химическое обозрение), 57(1), 47-76, 1957 и список литературы в данной работе) до формы производного NH-(C=O)-NHR, или с Сl-(С=O)-Сl и NHR2 (см. H. Babad, A.G. Zeiler: Chemistry of Phosgene Chemical Reviews (Химия фосгенов Химическое обозрение), 73(1), 75-91, 1973 список литературы в данной работе) до формы NH-(CO)-NR2, в которой R может быть представлен алкилом C1-18 (например, алкил С1-6), арилом, гетероарилом, циклоарилом C3-8 или их сочетаниями. Фармацевтически приемлемые производные могут изготавливаться любым соответствующим способом, при этом способы их изготовления являются очевидными для любого специалиста в данной области на основании общеизвестных принципов органической и медицинской химии (например, соответствующие методы представлены в Vogel's Textbook of Practical Organic Chemistry (Учебник Фогеля по практической органической химии), издание 5-е, из-во Longman, 1989). Очевидно, что данные производные должны иметь способность ингибировать FAAH и должны замедлять периферическую селективность, т.е. они должны обладать теми же свойствами, что и указанная выше структура. Подходящие способы проверки данных свойств хорошо известны специалистам в данной области, и их описание приводится в настоящем документе.
Используемое в данном документе понятие «алкил Сх-у» относится к линейной или разветвленной насыщенной углеводородной группе, содержащей от х до у атомов углерода. Например, «алкил C1-6» относится к линейной или разветвленной насыщенной углеводородной группе, содержащей от 1 до 6 атомов углерода. В качестве примера групп алкилов C1-6 можно привести метил, этил, н-пропил, изопропил, 2-бутил, 3-бутил, н-пентил, изопентил, неопентил и гексил. Предпочтительно, углеводородная группа является линейной.
При использовании в данном документе термин «алкил» относится к моноциклическому или бициклическойму углеводородному кольцу С6-12, причем, по меньшей мере, одно кольцо является ароматическим. В качестве примера таких групп можно привести фенил, нафталенил и тетрагидронафталенил.
При использовании в данном документе термин «гетероарил» относится к 5-6-членному моноциклическому ароматическому или к сочлененному 8-10-членному бициклическому ароматическому кольцу, при этом такое моноциклическое или бициклическое кольцо включает от 1 до 4 гетероатомов, выбранных из кислорода, азота или серы. В качестве примера таких моноциклических ароматических колец можно привести тиенил, фурил, фуразанил, пирролил, триазолил, тетразолил, имидазолил, имидазолил, оксазолил, тиазолил, оксадиазолил, изотиазолил, изоксазолил, тиадиазолил, пиранил, пиразолил, пиримидил, пиридазинил, пиразинил, пиридил, триазинил, тетразинил и аналогичные вещества. В качестве примера таких бициклических ароматических колец можно привести хинолинил, изохинолинил, хиназолинил, хиноксалинил, птеридинил, циннолинил, фталазинил, нафтиридинил, индолил, изоиндолил, азаиндолил, индолизинил, индазолил, пуринил, пирролопиридил, фуропиридил, бензофуранил, изобензофуранил, бензотиенил, бензоизотиазолил, бензоксазолил, бензоизоксазолил, бензотиазолил, бензоизотиазолил, бензоксадиазолил, бензотиадиазолил и имидазопиридил.
Термины «бициклическое кольцо» и «конденсированное» применительно к бициклическому кольцу относятся к двум кольцам, которые соединены вместе по связи между двумя атомами (например, нафталин), по последовательности атомов с формированием моста (например, хинуклидин) или в одном атоме с формированием спиро-соединения (например, 1,4-диокса-8-аза-спиро[4.5]декан и N,3,3-диметил-1,5-диоксаспирол[5.5]ундекан-9-ил).
Используемое в данном документе понятие «циклоалкил Сх-у» относится к насыщенному углеводородному кольцу, состоящему из от x до у атомов углерода, которые могут быть моно-, би- или трициклическими. Например, циклоалкил С3-8 относится к насыщенному моно-, би- или трициклическому углеводородному кольцу, состоящему из от 3 до 8 атомов углерода. Примеры групп циклоалкилов С3-8 включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил.
Специалистам в данной области хорошо известны общие способы получения солей и производных. Фармацевтическая приемлемость солей и производных будет зависеть от разнообразных факторов, включая характеристики обработки препаративной формы и поведение in vivo, при этом специалист сразу сможет оценить факторы, имеющие отношение к информации, приводимой в настоящем документе.
В случае если соединения согласно изобретению присутствуют в форме таутомеров (например, кето/энол, амид/имидокислота), изобретение относится к отдельным изолированным таутомерам, а также к смесям таутомеров во всех возможных пропорциях.
В соответствии со вторым аспектом изобретения предусматривается фармацевтическая композиция, содержащая соединение по первому аспекту изобретения вместе с одним или несколькими фармацевтически приемлемыми вспомогательными веществами.
Фармацевтические композиции данного изобретения включают любые соединения по первому аспекту настоящего изобретения с любым фармацевтически приемлемым носителем, адъювантом или веществом-основой. Фармацевтически приемлемые носители, адъюванты или вещества-основы, которые могут использоваться при изготовлении фармацевтических композиций согласно настоящему изобретению, являются веществами, которые обычно используют для изготовления фармацевтических препаративных форм, и которые включают, но не ограничиваются ими, сахара, сахарные спирты, крахмалы, ионообменные вещества, оксид алюминия, стеарат алюминия, лецитин, сывороточные белки, такие как человеческий сывороточный альбумин, буферные вещества, такие как фосфаты, глицерин, сорбиновую кислоту, сорбат калия, частичные смеси глицерида с насыщенными растительными жирными кислотами, воду, соли или электролиты, такие как протаминсульфат, динатрия гидрофосфат, гидрофосфат калия, хлорид натрия, соли цинка, коллоидную окись кремния, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, натрий-карбоксилметилцеллюлозу, полиакрилаты, воски, блок-сополимеры полиэтилена и полиоксипропилена, полиэтиленгликоль и шерстяной жир.
Фармацевтические композиции согласно настоящему изобретению могут вводиться орально, парентерально, посредством вдыхания при распылении, ректально, через нос, трансбуккально, вагинально или через встроенный резервуар. Предпочтительным является оральное введение. Фармацевтические композиции согласно настоящему изобретению могут содержать любые традиционные нетоксичные фармацевтически приемлемые носители, адъюванты или вещества-основы. При использовании в настоящем документе термин «парентеральный» включает подкожные, внутрикожные, внутривенные, внутримышечные, внутрисуставные, надчревные, внутриоболочечные, внутриочаговые и внутричерепные методы введения инъекций и вливаний.
Фармацевтические композиции могут быть в форме стерильных инъекционных препаратов, таких как стерильная инъекционная водная или маслянистая суспензия. Данная суспензия может быть составлена в соответствии с общеизвестными в данной области методами с использованием подходящих дисперсионных или увлажняющих реагентов (таких как, например, Tween 80) и суспендирующих агентов. Стерильным инъекционным препаратом может также быть стерильный инъекционный раствор или суспензия в нетоксичном разбавителе или растворителе, подходящем для парентерального введения, как, например, раствор в 1,3-бутандиоле. Среди приемлемых веществ-основ и растворителей могут быть маннитол, вода, раствор Рингера, изотонические растворы хлористого натрия. Кроме того, в качестве растворителя или суспендирующей среды традиционно используют стерильные нелетучие масла. Для этого можно использовать любое нейтральное нелетучее масло, включая синтетические моно- или диглицериды. Жирные кислоты, такие как олеиновая кислота и ее глицеридные производные, могут использоваться для изготовления инъекционных препаратов, таких как натуральные фармацевтически приемлемые масла, такие как оливковое масло и касторовое масло, особенно в их полиоксиэтилированных вариантах. Эти масляные растворы и суспензии также могут содержать длинноцепочечные спиртовые растворители и дисперсанты, такие как те, описание которых приводится в работе Ph. Helv (Φ. Хелв), или аналогичный спирт.
Фармацевтическая композиция согласно настоящему изобретению может приниматься внутрь в виде любой приемлемой лекарственной формы, включая, но не ограничиваясь ими, капсулы, таблетки, порошки, гранулы, а также водные суспензии и растворы. Приготовление данных лекарственных форм осуществляют в соответствии с методами, которые являются общеизвестными в области приготовления лекарственных средств. В случае с таблетками, которые принимают внутрь, широко применяют такие носители, как лактоза и кукурузный крахмал. Также привычной практикой является добавление замасливателей, таких как стеарат магния. При принятии внутрь в капсулах удобно использовать такие растворители, как лактоза и высушенный кукурузный крахмал. При приеме внутрь водных суспензий активный фармацевтический ингредиент объединяют с эмульгирующими и суспендирующими средствами. При необходимости, могут быть добавлены подсластители и/или ароматизирующие и/или окрашивающие вещества.
Фармацевтическая композиция согласно изобретению может также применяться в форме суппозиториев для ректального введения. Эти композиции можно получать смешиванием соединения по данному изобретению со вспомогательным веществом противораздражающего действия, твердым при комнатной температуре и жидким при ректальной температуре, которое соответственно будет размягчаться в прямой кишке и, таким образом, высвобождать активные компоненты. Такие материалы включают, но не ограничиваются ими, масло какао, пчелиный воск и полиэтиленгликоли.
Фармацевтические композиции согласно изобретению могут приниматься посредством разбрызгивания или вдыхания через нос. Приготовление таких композиций осуществляют в соответствии с методами, которые являются общеизвестными в области приготовления лекарственных средств. Они могут приготавливаться в виде соляных растворов с применением бензилового спирта и других соответствующих предохраняющих средств, стимуляторов абсорбции для увеличения биодоступности, фторуглеродов и/или иных известных повышающих растворимость диспергаторов.
Соединения согласно настоящему изобретению могут приниматься в дозировке от около 1 до около 20000 мкг/кг на дозу, например, от около 1 до около 10000 мкг/кг, от около 1 до около 5000 мкг/кг, от около 1 до около 3000 мкг/кг, от около 1 до около 2000 мкг/кг, от около 1 до около 1500 мкг/кг, от около 1 до около 1000 мкг/кг, от около 1 до около 500 мкг/кг, от около 1 до около 250 мкг/кг, от около 1 до около 100 мкг/кг, от около 1 до около 50 мкг/кг или от около 1 до около 25 мкг/кг на дозу, в зависимости от состояния, которое необходимо излечить или предотвратить, а также характеристик пациента, принимающего соединение. Во многих случаях дозировка может составлять от около 1 до около 10 мкг/кг на дозу. В определенных вариантах осуществления изобретения дозировка может составлять около 250 мкг/кг на дозу, около 100 мкг/кг или около 10 мкг/кг на дозу. Режим дозировки для конкретного соединения может быть легко определен специалистом, получившим доступ к данному документу.
В одном из частных случаев осуществления изобретения фармацевтическая композиция согласно изобретению может также содержать один или несколько дополнительных активных фармацевтических ингредиентов. Соединение согласно изобретению может приниматься с одним или несколькими дополнительными активными фармацевтическими ингредиентами, такими как анандамид, олеоилэтаноламид или пальмитоилэтаноламид. Оно может иметь форму отдельной композиции, включающей соединение согласно изобретению, а также один или несколько дополнительных активных фармацевтических ингредиентов. В качестве альтернативы, оно может быть представлено в виде двух или более отдельных композиций, в которых содержится соединение согласно изобретению, при этом в одной или в нескольких отдельных композициях будет содержаться один или несколько дополнительных активных фармацевтических ингредиентов.
Поэтому введение соединений согласно настоящему изобретению может осуществляться одновременно или поочередно с одним или несколькими дополнительными активными фармацевтическими ингредиентами.
В третьем аспекте настоящее изобретение предусматривает по первому аспекту изобретения или композицию по второму аспекту для применения в терапии.
В третьем аспекте настоящее изобретение предусматривает соединение по первому аспекту изобретения или композицию по второму аспекту для применения при лечении или предупреждении состояния, развитие и симптомы которого связаны с субстратом фермента FAAH.
Изобретение также предусматривает применение соединения по первому аспекту изобретения или композиции по второму аспекту при изготовлении медикамента для лечения или предупреждения состояния, развитие и симптомы которого связаны с субстратом фермента FAAH.
Разнообразные заболевания, развитие или симптомы которых связаны с субстратом фермента FAAH, хорошо известны специалистам. Некоторые из них были рассмотрены выше.
В пятом аспекте изобретение также предусматривает способ лечения или предупреждения состояния, развитие или симптомы которого связаны с субстратом фермента FAAH, включающий введение пациенту, нуждающемуся в таком лечении или предупреждении, терапевтически эффективного количества соединения по первому аспекту изобретения или композиции по второму аспекту.
Соединение по четвертому аспекту или способ по пятому аспекту, отличающиеся тем, что указанное состояние представляет собой заболевание, связанное с эндоканнабиноидной системой.
В некоторых вариантах осуществления изобретения такое подлежащее лечению состояние может быть выбрано из следующих:
(i) боль, в частности, острая или хроническая нейрогенная боль, такая как мигрень и невропатическая боль (например, диабетическая невропатическая боль, постгерпетическая невралгия, тригеминальная невралгия); мигрень, острая или хроническая воспалительная боль, такая как боль, связанная с воспалительными заболеваниями, как артрит, ревматоидный артрит, остеоартрит, спондилит, подагра, васкулит, болезнь крона, а также синдром раздраженной толстой кишки; острая или хроническая периферическая боль; боль, связанная с раковым заболеванием;
(ii) головокружение, тошнота и рвота, в частности, после химиотерапии;
(iii) расстройства, связанные с приемом пищи, в частности, нарушение аппетита, нарушение обмена веществ, различные виды анорексии и кахексии;
(iv) неврологические и психиатрические патологии, такие как тремор, дискинезия, дистония, тошнота, рвота, аддиктивные заболевания (такие как алкогольная или наркотическая зависимость), мышечная спастичность, навязчивый невроз, синдром Туретта, все формы депрессии и тревоги любого характера и происхождения, бессонница, расстройство настроения, а также психозы, такие как шизофрения;
(v) острые и хронические нейродегенеративные заболевания, такие как болезнь Паркинсона, болезнь Альцгеймера, сенильная деменция, хорея Гентингтона, патологические изменения, связанные с церебральной ишемией, а также с черепно-мозговыми травмами;
(vi) эпилепсия;
(vii) нарушение сна, включая апноэ во сне;
(viii) сердечнососудистые заболевания, такие как сердечная недостаточность, гипертензия, циркуляторный шок, реперфузионное повреждение миокарда, аритмия сердца, артериосклероз/атеросклероз, сердечный приступ, сердечная ишемия, васкулит и почечная ишемия;
(ix) раковые заболевания, например, доброкачественные опухоли кожи, опухоли простаты, опухоли мозга (глиобластома, медуллоэпителиома, медуллобластома, нейробластома, опухоли эмбрионального происхождения, астроцитома, астробластома, эпендимальная глиома, олигодендроглиома, опухоли сплетений, эпендимоглиома, опухоль эпифиза, эпендимобластома, злокачественная менингиома, саркоматоз, злокачественная меланома, шванома);
(x) заболевания иммунной системы, в частности, аутоиммунные заболевания, такие как псориаз, красная волчанка, заболевания соединительных тканей или коллагенозы, синдром Шегрена, анкилозирующий спондилоартрит, недифференцированный спондилит, синдром Бехчета, аутоиммунная гемолитическая анемия, рассеянный склероз, боковой амиотрофический склероз, амилоидоз, отторжение трансплантата, заболевания, воздействующие на плазмоциты, аллергические заболевания; аллергическая реакция немедленного или замедленного типа, аллергический ринит и конъюнктивит, контактный дерматит;
(xi) паразитические, вирусные и бактериальные инфекционные заболевания, такие как СПИД и менингит;
(xii) воспалительные заболевания, в частности, заболевания суставов, такие как артрит, ревматоидный артрит, остеоартрит, спондилит, подагра, васкулит, болезнь крона, синдром раздраженной/воспаленной толстой кишки, астма;
(xiii) остеопороз;
(xiv) заболевания глаза, такие как гипертензия глаза, ретинопатия и глаукома;
(xv) заболевания легких, включая болезни дыхательных путей, бронхоспазм, кашель, астма, хронический бронхит, хроническая непроходимость дыхательных путей, эмфизема;
(xvi) желудочно-кишечный заболевания, такие как синдром раздраженной/воспаленной толстой кишки, воспалительные кишечные расстройства, язвы, диарея, недержание мочи и воспаление мочевого пузыря;
(xvii) острые и хронические заболевания печени, такие как гепатит и цирроз;
(xviii) неврологические заболевания, такие как механическое повреждение нерва, припадок, рассеянный склероз, повреждение спинного мозга, болезнь Паркинсона, дискинезия, вызванная леводопой, болезнь/хорея Гантингтона, синдром Туретта, поздняя дискинезия, дистония, боковой амиотрофический склероз, болезнь Альцгеймера, эпилепсия.
В шестом аспекте изобретения предусматривается способ получения кислой соли N-метилциклопентиламина, включающий реакцию циклопентиламина с хлорформиатом или ди-трет-бутил карбонатом, с образованием циклопентилкарбамата, с последующим восстановлением циклопентилкарбамата и окислением до кислой соли N-метилциклопентиламина.
Способ, предусмотренный шестым аспектом, может использоваться для эффективного получения кислой соли N-метилциклопентиламина, основного промежуточного продукта при приготовлении соединения по Формуле А. Данный способ предусматривает большой выход и высокое качество продукта. Предпочтительной кислой солью является соль НСl. Однако также целесообразно использовать и другие неорганические и органические кислые соли.
В вариантах осуществления изобретения образование циклопентилкарбамата выполняется при основных условиях, например, неорганические основания, такие как NaOH (например, 3М), NаНСО3, Nа2СО3 или К2СО3 или органические основания, такие как триэтил амин, ди-изопропил этил амин, в органическом растворителе, таком как ТГФ, метил ТГФ, диоксан, метил-трет-бутил эфир или дихлорметан. Хлорформиат, используемый на данном этапе, может быть представлен, например, в виде хлороформиата С1-4, такого как этил. Этап восстановления может выполняться с использованием алюмогидрида лития (АГЛ) в органическом растворителе, таком как ТГФ или метил ТГФ. Диапазон температур составляет от 30°С до рефлюкса, предпочтительно при 60°С. Для изоляции продукта удобно добавить неорганическую кислоту, такую как НСl, HBr, HI (например, концентрированную) или органическую кислоту, такую как уксусная кислота, чтобы образовать соответствующую кислую соль N-метилциклопентиламина.
В седьмом аспекте изобретение также предусматривает способ получения кислой соли N-метилциклопентиламина, включающий восстановительное аминирование циклопентанона в присутствии кислой соли метиламина.
В вариантах осуществления изобретения по седьмому аспекту восстановление происходит в присутствии каталитического количества ценного металла на углероде, такого как Pd/C (например, 5% или 10%), например, в присутствии органического основания, такого как триэтиламин или ди-изопропил этил амин, и спиртового растворителя, такого как метанол, этанол, пропанол или бутанол под водородом. Давление может быть в диапазоне от атмосферного до 10 бар. Может использоваться температура примерно 50-80 (или 60-70)°С. Предпочтительной кислой солью по седьмому аспекту является соль НСl, отличающаяся тем, что в данном способе используют метиламин НСl. Однако также целесообразно использовать и другие неорганические и органические кислые соли. Таким образом, неорганические кислые соли, такие как HBr, HI или органические кислые соли, такие как соли уксусной кислоты, могут быть приготовлены с использованием аналогичного способа.
Соль N-метилциклопентиламина (такая как соль НСl), получаемая в соответствии с шестым или седьмым аспектами, может впоследствии использоваться при получении соединения по Формуле А с применением способов приготовления данного соединения, описание которых приводится в настоящем документе. Таким образом, изобретение предусматривает способ получения соединения по Формуле А, в который включены способы, предусмотренные по шестому или седьмому аспектам. Также предусматривается соединение по Формуле А, полученное или которое можно получить при помощи такого способа.
Подробное описание изобретения
Далее изобретение будет описано более подробно исключительно на примерах:
1. Синтетические методики
Методы, которые используют для получения соединений согласно изобретению, наглядно представлены на общих схемах ниже. Характеристика всем соединениям и промежуточным продуктам была дана по результатам ядерно-магнитного резонанса (ЯМР). Исходные материалы и реагенты, которые используют при получении данных соединений, имеются в продаже у поставщиков или могут быть получены с использованием методов, являющихся очевидными для специалистов в данной области. Данные общие схемы используют исключительно для иллюстрации методов, при помощи которых можно получить соединения согласно настоящему изобретению, при этом схемы можно как угодно изменять, что и предлагается выполнить специалистам в данной области, которые будут ознакомлены с данной работой.
Комнатная температура на схемах означает температуру в диапазоне от 20°С до 25°С.
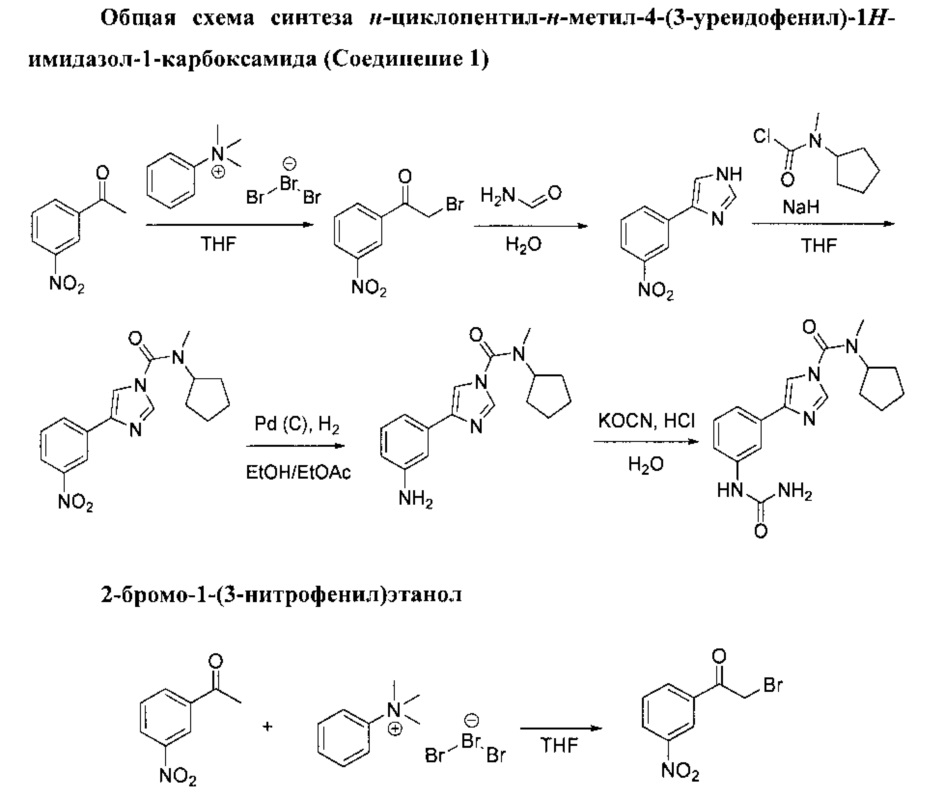
Раствор трибромида фенилтриметиламмония (50,1 г, 133 ммоль) в ТГФ (200 мл) добавили по каплям в перемешиваемый раствор 1-(3-нитрофенил)этанола (20 г, 121 ммоль) в ТГФ (200 мл) при комнатной температуре. Реакционную смесь оставили перемешиваться при комнатной температуре в течение 1 ч. Белую суспензию отфильтровали, после чего осадок на фильтре промыли при помощи ТГФ, а фильтрат выпарили в вакууме для получения желтого масла. Затем осадок растворили в этилацетате и промыли водой. Органический слой осушили (MgSO4) и выпарили в вакууме для получения желтого масла, которое застыло до твердого желтого вещества. Твердое вещество рекристаллизовали из пропан-2-ола, и конечный продукт изолировали в виде серовато-белого твердого вещества. 2-бромо-1-(3-нитрофенил)этанол (20,5 г, выход 70%).
(1Н, 600 MHz, 20°С, CDCl3) δ: 8.83 (1Н, t, J=2Hz), 8.49 (1Н, ddd, J=1.0, 2.3, 8.2 Hz), 8.34 (1H, ddd, J=1.0, 1.7, 7.8 Hz), 7.75 (1H, t, J=8.1 Hz), 4.49 (2H, s).
(13С, 150 MHz, 20°С, CDCl3) δ: 189.3, 148.5, 135.1, 134.4, 130.2, 128.1, 123.8, 29.9
Температура плавления (mp): 90-91°С.
Ниже представлен альтернативный путь протекания реакции бромирования:
К раствору 3-нитроацетофенона (1 мас., 1 экв.) в уксусной кислоте (10 об.) на протяжении не менее чем 2 часов при поддержании температуры ниже 30°С добавляют раствор брома (0,34 об., 1,08 экв.). После перемешивания на протяжении 1 часа при температуре от 25°С до 30°С реакцию проверяют на предмет завершения. После завершения реакции добавляют холодную воду (12 об.), в результате чего образуется белый осадок. Суспензию перемешивают еще на протяжении часа при 15°С, после чего ее фильтруют. Осадок промывают водой (4,5 об.). Продукт осушают в вакууме при температуре не выше 45°С, пока не будут зафиксированы потери при высушивании <1,0%. Изолированный выход продукта реакции бромирования составил примерно 66%. Данный альтернативный подход может лучше подойти для пропорционального увеличения.
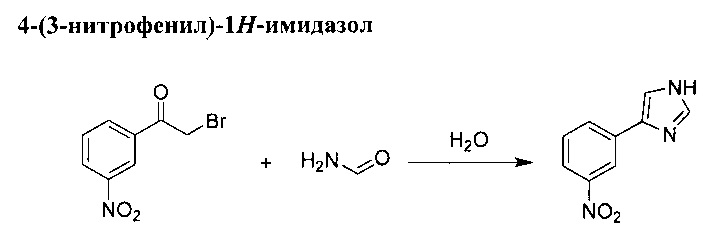
В перемешиваемую суспензию 2-бромо-1-(3-нитрофенил)этанола (57,1 г, 234 ммоль) и формамида (116 мл, 1,9 моль) добавили воду (8 мл). Смесь оставили перемешиваться при температуре 140°С в течение 5 ч. Коричневый остаток вылили в 300 мл воды, а полученный осадок отделили при помощи фильтрации, после чего промыли в растворе 1М НСl. Фильтрат ощелочили в 50% растворе NaOH, а полученный желтый осадок отделили при помощи фильтрации, после чего промыли его водой. Твердое вещество высушили, после чего его рекристаллизовали из пропан-2-ола. 4-(3-нитрофенил)-1N-имидазол (7,05 г, выход 44%).
(1Н, 600 MHz, 20°С, DMSO) δ: 12.37 (1Н, s, br), 8.58 (1H, mt, J=2.0 Hz), 8.21 (1H, ddd, J=1.0, 1.6, 7.8 Hz), 8.02 (1H, ddd, J=1.0, 2.5, 8.2 Hz), 7.88 (1H, dd, J=1.2 Hz), 7.79 (1H, dd, J=1.1 Hz), 7.64 (1H, t, J=8.1 Hz)
(13C, 150 MHz, 20°C, DMSO) δ: 148.4, 137.9, 136.8, 136.6, 130.5, 130.0, 120.5, 118.3, 114.6
Температура плавления: 221°C (dec.)
Что касается улучшений по данному этапу способа, было установлено, что самостоятельное использование формамида (т.е. без воды) в качестве суспензионной среды приводит к повышению выхода, так же как и увеличение температуры от 140 до 170°С (до 80%). Таким образом, улучшенный протокол выглядит следующим образом:
Раствор 2-бромо-1-(3-нитрофенил)этанола (1 мас., 1 экв.) в формамиде (10 об.) подогревают до 170°С и перемешивают в течение не более чем 4 часов. После перемешивания на протяжении 4 часов реакцию проверяют на предмет завершения. После завершения реакции смесь охлаждают до комнатной температуры и добавляют в нее воду (15 об.). Суспензию фильтруют, а осадок фильтра промывают при помощи 3N НСl (2 об.), после чего исходный раствор фильтруют снова. Показатель рН раствора доводят до 14 посредством добавления 50% NaOH (2 об.) с поддержанием температуры смеси в диапазоне от 0°С до 5°С. После перемешивания суспензии при 0/5°С на протяжении не менее 30 минут осадок необходимо отфильтровать и промыть водой (5 об.). Продукт осушают в вакууме при температуре не выше 45°С, пока не будут зафиксированы потери при высушивании <1,0%.
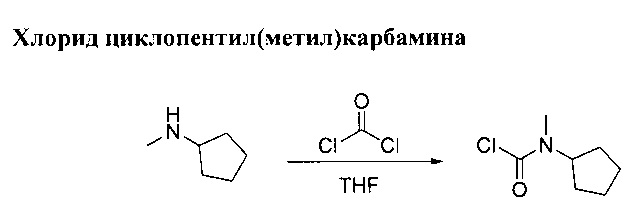
Раствор N-метилциклопентанамина (10 г, 101 ммоль) в ТГФ (126 мл) добавили по каплям в раствор фосгена (63.7 г, 121 ммоль, 20% в толуоле) при 0°С для получения белой суспензии. Реакционную смесь оставили перемешиваться при комнатной температуре в течение 1 ч. Раствор вылили на лед. Органический слой разбавили этилацетатом, выпарили и промыли в 1М НСl, затем осушили (MgSO4) и выпарили в вакууме до получения прозрачного дорожного масла. Хлорид циклопентил(метил)карбамина (13,1 г, выход 80%).
(1Н, 600 MHz, 20°С, CDCl3) δ: 4.65 (1H, m), 3.0, 2.93 (3Н, 2 синглеты), 1.92 (2Н, m), 1.73 (2Н, m), 1.59 (4Н, m)
(13С, 150 MHz, 20°С, CDCl3) δ: 149.7, 149.3, 61.1, 59.5, 33.1, 31.1, 28.8, 28.5, 24.0
Этап карбамоилирования также может выполняться с использованием трифосгена/дихлорметана и карбоната натрия в следующем порядке:
Раствор трифосгена (1,2 мас., 0,4 экв.) в дихлорметане (10 об.) охлаждают до 0/5°С и перемешивают в течение не более чем 10 минут. Раствор N-метилциклопентиламина (1 мас., 1 экв.) в дихлорметане (5 об.) загружают при поддержании температуры реакции ниже 10°С. После добавления раствора амина необходимо загрузить Na2CO3 (2,14 мас., 2 экв.) и дать ему нагреться до комнатной температуры. После перемешивания на протяжении 2 часов реакционную смесь фильтруют, а осадок промывают в дихлорметане (1 об.). После концентрации до уровня сухости получают желтое масло, которое используют в том виде, в котором оно есть, без дальнейшей обработки.
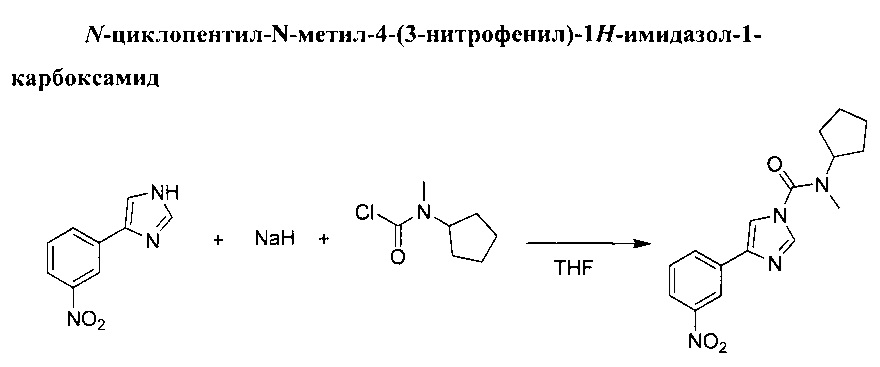
Гидрид натрия (5,1 г, 127 ммоль, 60% дисперсия в минеральном масле) добавили по порциям в перемешиваемую суспензию 4-(3-нитрофенил)-1Н-имидазола (20 г, 106 ммоль) в ТГФ (500 мл) при 0°С. Желтая суспензия превратилась в темно-красную. Смеси дали перемешаться при комнатной температуре на протяжении 30 минут, после чего добавили раствор хлорида циклопентил(метил)карбамина (25,6 г, 159 ммоль) в ТГФ (26 мл). Затем суспензию оставили перемешиваться при комнатной температуре в течение 2 ч. При температуре 0°С добавили воду, и ТГФ выпарили. Органический остаток извлекли при помощи дихлорметана, органический слой отделили, затем осушили (MgSO4) и выпарили в вакууме до получения твердого бежевого вещества. Данное твердое вещество измельчили в порошок при помощи пропан-2-ола. N-циклопентил-N-метил-4-(3-нитрофенил)-1H-имидазол-1-карбоксамид (25,18 г, выход 76%).
(1Н, 600 MHz, 20°С, CDCl3) δ: 8.63 (1Н, mt, J=2.0 Hz), 8.16 (1H, ddd, J=1.0, 1.6, 7.8 Hz), 8.14 (1H, ddd, J=1.0, 2.3, 8.2 Hz), 7.96 (1H, d, J=1.3 Hz), 7.65 (1H, dd, J=1.3 Hz), 7.58 (1H, t, J=8.1 Hz), 4.45 (1H, m), 3.03 (3H, s), 1.98 (2H, m), 1.80 (2H, m), 1.73 (2H, m), 1.66 (2H, m)
(13C, 150 MHz, 20°C, CDCl3) δ: 151.3, 148.7, 140.1, 137.3, 134.9, 130.9, 129.7, 122.1, 119.9, 114.6, 59.4,31.3,28.9, 24.4
Температура плавления: 121-122°C
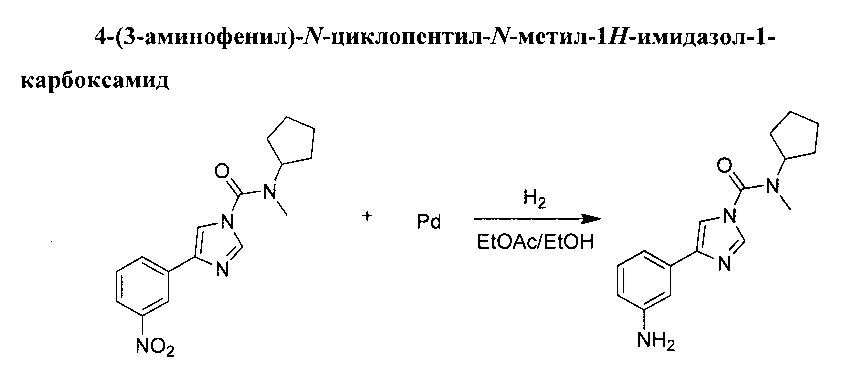
Смесь этилацетата (160 мл) и этилового спирта (160 мл) добавили в сырой Pd/C (0,846 г, 0,795 ммоль, 10%) при атмосферном давлении аргона. К этому добавили N-циклопентил-N-метил-4-(3-нитрофенил)-1Н-имидазол-1-карбоксамид (5 н, 15,91 ммоль) по порциям, а затем суспензии дали перемешаться при комнатной температуре в течение ночи под атмосферным давлением водорода. Смесь промыли струей аргона и отфильтровали через целит, а целит промыли в дихлорметане. Фильтрат выпарили в вакууме до получения прозрачного масла, которое застыло и превратилось в твердое бесцветное вещество. Твердое вещество рекристаллизовали из пропан-2-ола. 4-(3-аминофенил)-N-циклопентил-N-метил-1H-имидазол-1-карбоксамид (3,62 г, выход 80%).
(1Н, 600 MHz, 20°С, DMSO) δ: 8.06 (1H, d, J=1.3 Hz), 7.77 (1H, d, J=1.1 Hz), 7.08 (1H, t, J=1.9 Hz), 7.0 (1H, t, J=7.8 Hz), 6.98 (1H, md, J=7.7 Hz), 6.45 (1H, ddd, J=1.2, 2.3, 7.7 Hz), 5.07 (2H, s), 4.37 (1H, m), 2.92 (3H, s), 1.87 (2H, m), 1.68 (4H, m), 1.53 (2H, m)
(13C, 150 MHz, 20°C, DMSO) δ: 151.2, 148.8, 141.4, 137.3, 133.8, 129.0, 113.7, 112.9, 112.8, 110.4,58.4,31.2,28.2, 24.0
Температура плавления: 108-109°C
В альтернативном варианте осуществления изобретения производный продукт анилина по данному этапу может быть использован при последующих этапах без очистки, т.е. так, что данный и последующий этап можно выполнять с накладкой друг на друга.
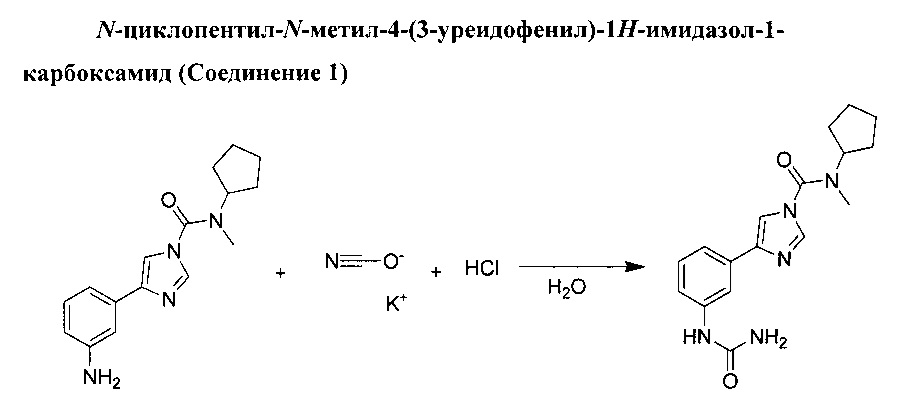
Цианат калия (0,445 г, 5,49 ммоль) добавили по порциям в перемешиваемый раствор 4-(3-аминофенил)-N-циклопентил-N-метил-1Н-имидазол-1-карбоксамида (1,3 г, 4,57 ммоль) в смеси 2N хлорида водорода (2,286 мл, 4,57 ммоль) в воде (4 мл) при температуре 0°С. Данной смеси дали перемешаться при комнатной температуре на протяжении 24 ч. Добавили цианат калия (0,220 г, 7,77 ммоль) и полученной смеси дали перемешаться при комнатной температуре на протяжении еще одной ночи. Добавили воду, и органический слой разбавили со смесью дихлорметана/пропан-2-ола в соотношении 7:3. Органический слой отделили и промыли в водном растворе 1N НСl. Органический слой отделили, затем осушили (MgSO4) и выпарили в вакууме до получения бесцветной пены. Данный продукт очистили с применением колоночной хроматографии (кварц, дихлорметан/МеОН 5%, 10%), а затем изолировали в виде бесцветного твердого вещества. Данное твердое вещество рекристаллизовали из этилового спирта при температуре 0°С. N-циклопентил-N-метил-4-(3-уреидофенил)-1H-имидазол-1-карбоксамид (0,403 г, выход 26%).
Соединения согласно настоящему изобретению охарактеризованы при помощи теста на температуру плавления и ЯМР, описание которых более подробно представлено ниже. Спектры ЯМР фиксировали при помощи спектрометра Bruker 600 МГц Avance III с применением растворителя в качестве внутреннего эталона. Спектры 13С были зафиксированы на частоте 150 МГц, а спектры 1Н - на частоте 600 МГц. Данные представлены в отчете в следующем порядке: ориентировочный химический сдвиг (ч./млн.), количество протонов, мультиплетность (br, широкая; d, дублет; m, мальтиплет; s, синглет, t; триплет) и константа взаимодействия (Гц).
Соединение №1 (температура плавления: 204°С).
(13С, 150 MHz, 20°С, DMSO) δ: 156, 151.1, 140.9, 140.8, 137.5, 133.7, 128.9, 117.9, 116.6, 114.2, 114.2, 58.4, 31.2, 28.2, 24.
(1H, 600 MHz, 20°С, DMSO) δ: 8.55 (1Η, s), 8.09 (1H, d, J=1.2 Hz), 7.86 (1H, d, J=1.2 Hz), 7.85 (1H, t, J=1.8 Hz), 7.35 (1H, md), 7.34 (1H, md), 7.22 (1H, t, J=7.8 Hz), 5.84 (2H, s), 4.36 (1H, m), 2.93 (3H, s), 1.87 (2H, m), 1.69 (4H, m), 1.54 (2H, m).
Улучшение данного окончательного этапа (образование мочевины на фенильном кольце) состоит в использовании уксусной кислоты вместо воды в качестве растворителя 4-(3-аминофенил)-N-циклопентил-N-метил-1Н-имидазол-1-карбоксамида. Это обеспечивает увеличение выхода (около 78%) и улучшение протокола изоляции. Описание улучшенного этапа можно изложить следующим образом: 4-(3-аминофенил)-N-циклопентил-N-метил-1H-имидазол-1-карбоксамид растворяют в АсОН (8,8 об.) при комнатной температуре. В получившийся раствор при комнатной температуре добавляют раствор цианата калия (0,65 мас., 2,5 экв.) в воде (0,9 об.). Полученный раствор перемешивают при комнатной температуре до завершения реакции (исходные материал <0,1%). В течение 1 ч произошло осаждение продукта мочевины. В полученный раствор добавляют воду (5 об.), при этом выводят дополнительные объемы твердого вещества. Затем бежевую суспензию выдерживают в течение 1 ч при комнатной температуре, после чего ее фильтруют. Твердое бежевое вещество промывают водой (10 об.), и осушают в вакуумной печи до потерь при высушивании <1,5%.
Если в результате данного улучшенного этапа (с использованием уксусной кислоты) образуются N-ацетилированные анилиновые примеси, то можно провести рекристаллизацию. Это можно выполнить в следующем порядке:
На протяжении 30 минут в раствор продукта мочевины (1 мас.) в уксусной кислоте (5 об.) при комнатной температуре по каплям добавляли воду. После затравки добавили воду (2 об.), и дали раствору отстояться в течение 1 ч. при комнатной температуре. Раствор охлаждают до 10°С, перемешивают при 10°С на протяжении минимум 1 ч., а затем фильтруют. Серовато-белое твердое вещество промывают смесью воды/уксусной кислоты в соотношении 9:1 (2 об.), затем водой (10 об.), а затем осушают в вакуумной печи при 55°С. Далее серовато-белое твердое вещество (0,82 мас.) растворяют в уксусной кислоте (3,9 об.) при комнатной температуре, а потом по каплям в течение 30 минут добавляют воду (4,1 об.). Затем в раствор добавляют затравку, а потом воду (1,6 об.). Полученный раствор перемешивали при комнатной температуре в течение, по меньшей мере, 1 часа, а затем охлаждали при 10°С. После отстаивания раствора при 10°С в течение, по меньшей мере, 1 часа, твердое вещество фильтруют, промывают в смеси воды/уксусной кислоты в соотношении 9:1 (1,6 об.) и в воде (10 об.), затем осушают в вакуумной печи при температуре 55°С до потерь при высушивании <1,5%.
2. Биологическая эффективность
Испытания in vivo выполняли в соответствии с протоколом, описание которого приводится ниже. BRh (гомогенат мозга) указывает на поглощение в тканях центральной нервной системы, и в этом случае, в мозге, a LVh (гомогенат печени) указывает на поглощение в периферических тканях, в данном случае, в печени. В качестве контрольных групп использовался показатель сокращения количества испытуемых соединений в реакционной смеси. Поэтому низкое значение количества испытуемых соединений указывает на наличие мощного ингибитора. Значение 100 указывает на то, не имело место какое-либо измеримое поглощение.
Протокол испытаний in vivo
Лечение животных
В экспериментах в качестве животных использовали самцов мышей линии NMRI (весом 27-44 г), предоставленные Interfauna Ibérica (Испания). Мышей содержали по 5 особей в клетке и контролировали в условиях окружающей среды (цикл света/темноты 12 часов и комнатная температура 22±1°С). Пища и водопроводная вода были в их свободном распоряжении, а все эксперименты выполняли в светлое время суток.
Животным давали дозу в 30 мг/кг или 3 мг/кг соединения согласно изобретению или сравнительного соединения с приемом внутрь через рот (8 мл/кг; соединение, осажденное в 0,5% растворе карбоксиметилцеллюлозы (КМЦ) или растворенное в воде) или вещества-основы (контрольные группы) с использованием искривленных иголок для кормления животных из нержавеющей стали (Perfectum, США). За пятнадцать минут до убийства подопытного животного в рамках эксперимента им через брюшную полость в качестве анестезии вводили пентобарбитал. Из мышей извлекали фрагмент печени, левую долю легкого и мозг без мозжечка, после чего все это укладывали в пластиковые контейнеры с мембранной перегородкой (3 ммоль MgCl2, 1 ммоль EDTA, 50 ммоль Tris НСl рН 7.4). Ткани до момента проведения анализов хранили при температуре -30°С.
Перед каждым вводом соединений на протяжении ночи животных не кормили за исключением временных точек >18 ч., когда пищу извлекали утром в день ввода соединений, при этом соединения вводили в полдень того же дня. Затем животным не давали ничего кроме воды.
Все процедуры работы с животными выполняли строго в соответствии с Европейской директивой по защите позвоночных животных, используемых в экспериментах и для других научных целей (86/609СЕЕ), а также в соответствии с законодательством Португалии (Законодательный декрет 129/92, декреты 1005/92 и 1131/97). Животных использовали в минимально возможном количестве в соответствии с действующими правилами и научными принципами.
Реагенты и растворы
Анандамид [этаноламин-1-3Н-] (40-60 Ки/ммоль) был предоставлен компанией American Radiochemicals. Все другие реагенты были предоставлены Sigma-Aldrich. Optiphase Supermix был предоставлен компанией Perkin Elmer, а активированный уголь - компанией Sigma-Aldrich.
Подготовка ткани
Ткани отогрели ото льда и гомогенизировали в 10 объемах мембранного буфера (3 ммоль MgCl2, 1 ммоль EDTA, 50 ммоль Трис-HCl рН 7,4) при помощи гомогенизатора Поттера-Эльвейхема или Heidolph Diax (печень - при 2 движениях в положении 5 по 20 секунд с 30-секундными паузами).
Общее содержание белка в тканях определяли при помощи белкового маркера BioRad (BioRad) с использованием стандартной кривой BSA (50-250 мкг/мл).
Ферментный анализ
Реакционная смесь (общий объем 200 мкг) содержала: 2 мкМ АЕА (2 мкМ АЕА+5 нМ3Н-АЕА), 0,1% BSA, который не содержит жирных кислот, 15 мкг (мозг), 5 мкг (печень) или 50 мкг (легкое) белок, в 1 ммоль EDTA, 10 ммоль Трис рН 7.6. После прединкубационного периода продолжительностью 15 минут при температуре 37°С, реакция началась после добавления субстратного раствора (холодный АЕА + меченный АЕА + BSA). Реакция проходила на протяжении 10 минут (мозг и легкое) или 7 минут (печень) до тех пор, пока не была прервана добавлением 400 мкл суспензии активированного угля (8 г угля в 32 мл 0,5 м НСl при непрерывном перемешивании). После инкубационного периода продолжительностью 30 минут при комнатной температуре с перемешиванием, выполняли осаждение угля за счет центрифугирования в микроцентрифуге (10 минут при 13000 оборотах в минуту). К сцинтиляционному коктейлю Optiphase Supermix в объеме 800 мкл, предварительно помещенному на 24-луночные планшеты, добавили супернатант в объеме 200 мкл. Число импульсов в минуту (с/мин) определили при помощи сцинтилляционного счетчика MicrobetaTriLux.
При каждом анализе осуществляли подготовку холостых проб (без белка).
Процент остаточной энзимной активности рассчитывали применительно к контрольным группам и после холостой субтракции.
Определение ED50
Испытываемые соединения давали животным все в больших дозировках (10, 3, 1, 0.3, 0,03 и 0.01 мг/кг) и в соответствии с указанным выше протоколом регистрации активности FAAH in-vivo через 8 часов после приема определяли активность, после чего производили расчет значений ED50 при помощи программного обеспечения «Prisma» с доверительными интервалами на уровне 95%.
Анализ метаболической стабильности цитохром
Стабильность тестируемых соединений определяли в ММП (микросомы мышиной печени) или МЧП (микросомы человеческой печени) в присутствии и при отсутствии NADP.
Стабильность определяли при помощи инкубационной смеси (общий объем 100 мкл), содержащейся в 1 мг/мл общего белка, MgCl2 5 ммоль и 50 ммоль К-фосфатного буфера. Инкубирование проб осуществляли в присутствии и при отсутствии NADP объемом 1 ммоль. Прединкубационный период при реакциях составлял 5 минут, и реакция начиналась с тестируемым соединением (5 мкМ для МЧП и 50 мкМ для ММП). Инкубирование образцов выполняли на протяжении 60 минут в водяной бане-шейкере при температуре 37°С. Реакцию останавливали путем добавления 100 мкл ацетонитрила. Образцы подвергали обработке центрифугированием, фильтровали и в ВЭЖХ-МСД нагнетали супернатант. Тестовые соединения также растворялись в ДМСО, при этом окончательная концентрация ДМСО при реакции составила менее 0,5% (объемное содержание). Перед добавлением соединения при температуре 0 добавили ацетонитрил. Все эксперименты выполняли с дубликатами образцов.
Также протестировали соединение 1 (N-циклопентил-N-метил-4-(3-урейдофенил)-1H-имидазол-1-карбоксамид; также именуемое соединением вышеприведенной Формулы А). Кроме того, протестировали два сравнительных соединения, описанных в документе WO 2010/074588. К таким соединениям относятся:
Сравнительное соединение 1 - N-циклогексил-4-(3-гуанидинофенил)-N-метил-1N-имидазол-1-карбоксамид.
Сравнительное соединение 2 - N-циклопентил-4-(4-флюоро-3-гидроксифенил)-N-метил-1H-имидазол-1-карбоксамид.
Сравнительное соединение 1 сходно по структуре с соединением 1, несмотря на наличие совершенно четких различий между указанными соединениями. Сравнительное соединение 2 также сходно по структуре с соединением 1, но опять же, существуют совершенно четкие различия между этими двумя соединениями.

Как видно из приведенной ниже таблицы, соединение 1 является наиболее действенным соединением с точки зрения ингибирования FAAH в печени. В частности, соединение 1 намного более эффективно, чем сравнительное соединение 1.
Расчет переферийной селективности может быть выполнен путем деления активности FAAH в печени на активность FAAH в мозгу. При выполнении указанного расчета меньшее значение указывает на соединение, которое обладает более высокой переферийной селективностью. Результаты приведены в таблице ниже:
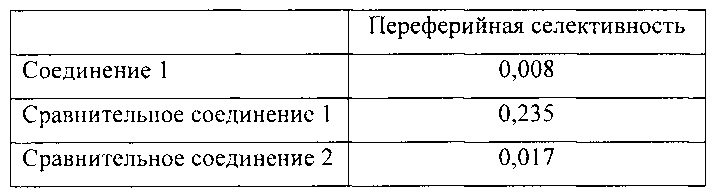
Данные результаты показывают, что соединение 1 является соединением с наиболее высокой переферийной селективностью, значение которой выше более чем в два раза. Кроме того, соединение 1 обладает намного более высокой переферийной селективностью, чем сравнительное соединение 1.
Дополнительные данные по активности FAAH при различных концентрациях соединения 1 и сравнительного соединения 2 приведены в таблице ниже:
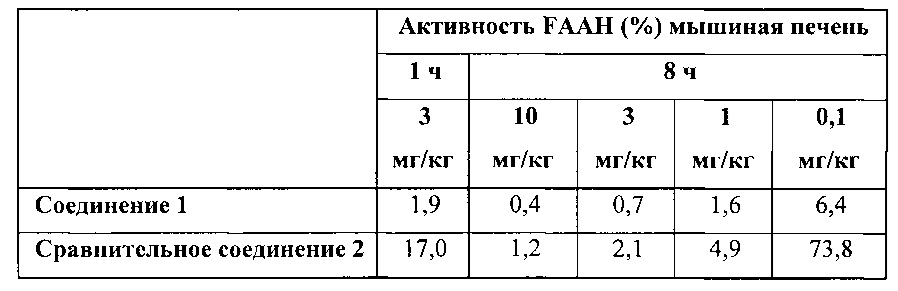
Как видно при любых дозах активность FAAH значительно ниже после введения соединения 1 по сравнению с сравнительным соединением 2. В частности, при 0,1 мг/кг по прошествии 8 часов после приема активность FAAH значительно ниже в случае с соединением 1 по сравнению с сравнительным соединением 2. Это свидетельствует о том, что соединение 1 намного более эффективно, чем сравнительное соединение 2. При концентрации 0,1 мг/кг соединение 1 в 10 раз более эффективно, чем сравнительное соединение 2. Это необычно большое различие в плане эффекта. Указанные данные также свидетельствуют о том, что соединение 1 является метаболически стабильным, поскольку при проведении экспериментов по ингибированию in vivo, метаболическая стабильность соединения также будет влиять на уровень ингибирования и продолжительность процесса ингибирования.
В приведенной ниже таблице показаны результаты ингибирования FAAH по ED50 (средняя эффективная дозировка, дозировка соединения, необходимая для 50%-ного ингибирования FAAH в печени) для соединений, вводимых мышам перорально. Учитывали доверительные интервалы (95%).
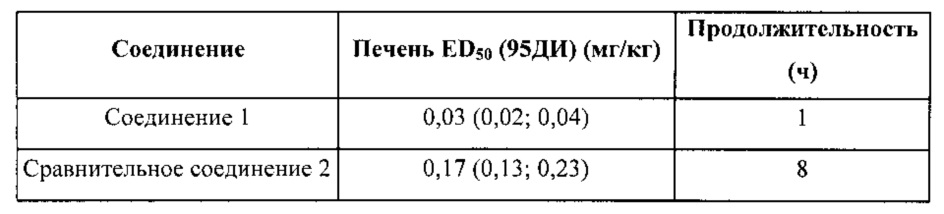
В таблице ниже представлена информация по метаболической стабильности соединения 1 и сравнительного соединения 2. Данные по стабильности приведены в виде % от остатка соединения после 1 часа воздействия на ММП или МЧП. 100% означает полное отсутствие какой-либо метаболической реакции, а 0% соответствует полному распаду под действием ферментов. «CYP-» указывает на отсутствие кофактора (NADPH), который является важным для метаболических реакций цитохром. Таким образом «CYP-» может рассматриваться в качестве контрольного значения. «CYP+» указывает на присутствие кофактора, а распад под действием ферментов может происходить в зависимости от стабильности тестируемого соединения. Как видно, соединение 1 является более стабильным, чем сравнительное соединение 2 как в случае с ММП, так и в случае с МЧП.
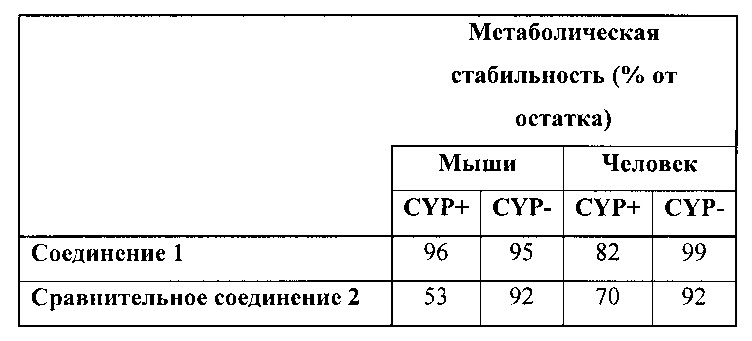
3. Определение значения IC50 для соединения 1
3.1 Материалы и методы
а) Реагенты и растворы
Анандамид [этаноламин-1-3Н-] был предоставлен компанией American Radiochemicals - для него характерна специфическая активность на уровне 60 Ки/ммоль. Все другие реагенты были предоставлены компанией Sigma-Aldrich. Optiphase Supermix был предоставлен компанией Perkin Elmer, а активированный уголь - компанией Sigma.
b) Подготовка ткани
Замороженные мозги 4 крыс линии Вистар гомогенизировали в 20 мл 1 ммоль MgCl2, 20 ммоль ГЭПЭ (4-(2-гидроксиэтил)-1-пиперазин этансерной кислоты) рН 7,0 при помощи гомогенизатора Поттера-Эльвейхема (8 движений при 500 об./мин.). Гомогенаты подвергали центрифугированию на протяжении 20 минут при 36000 г при 4°С (Beckman, ротор 70Ti). Гранулы повторно поместили в 15 мл аналогичного буфера и подвергли центрифугированию в аналогичных условиях. Гранулы повторно поместили в 15 мл аналогичного буфера и на протяжении 15 минут подвергали инкубации при температуре 37°С, после чего их подвергали центрифугированию на протяжении 20 минут при 36000 г при 4°С. Каждую гранулу затем повторно поместили в 10 мл 3 ммоль MgCl2, 1 ммоль EDTA (этилендиаминтетрауксусная кислота), 50 ммоль Tris (2-амино-2-гидроксиметил-пропан-1,3-диол) рН 7,4, а белок определили при помощи белкового анализа BioRad (BioRad) с использованием стандартной кривой BSA (бычий сывороточный альбумин), (50-250 мкг/мл).
Мембранные суспензии поделили на аликвоты и хранили при температуре -30°С.
c) Ферментный анализ
Реакционная смесь (общий объем 200 мкг) содержала: 2 мкМ АЕА (2 мкМ АЕА+5 нМ3Н-АЕА), 0,1% BSA, кот. не содержит жирных кислот, 5 мкг или 10 мкг белка, в 1 ммоль EDTA, 10 ммоль Трис рН 7,6, и соединение 1 при различных концентрациях. Исходный раствор (10 ммоль) подготовили в 100% ДМСО (диметил сульфоксид), а концентрация ДМСО при анализе составит 0,1%. После прединкубационного периода продолжительностью 15 минут при температуре 37°С реакция началась после добавления субстратного раствора (холодный АЕА + меченный АЕА + ВSA). Реакция проходила на протяжении 10 минут до тех пор, пока не была прервана добавлением 400 мкл суспензии активированного угля (8 г угля в 32 мл 0,5 м НСl при непрерывном перемешивании). После инкубационного периода продолжительностью 30 минут при комнатной температуре с перемешиванием, выполняют осаждение угля за счет центрифугирования в микроцентрифуге (10 минут при 15000 г). К сцинтиляционному коктейлю Optiphase Supermix в объеме 800 мкл, предварительно помещенному на 24-луночные планшеты, добавили супернатант в объеме 200 мкл. Число импульсов в минуту (с/мин) или распадов в минуту (р/мин) определили при помощи сцинтилляционного счетчика MicrobetaTriLux. При каждом анализе присутствовали холостые образцы (отсутствие белка) и контрольные группы (отсутствие соединений).
d) Системы тестирования
Сцинтилляционный счетчик Wallac 1450 MicrobetaTriLux.
e) Метод тестирования
Использовали следующие условия подсчета:

f) Другое оборудование
Spectramax Plus - SOFTmax® PRO, версия 3.0
g) Сбор и анализ данных
Сбор необработанных данных выполнили с использованием программного обеспечения Microbeta TriLux Windows workstation, версия 4.01.
Анализ данных выполнили с использованием программного обеспечения для Windows Prism 5, версия 5.02 (GraphPad Software Inc., Сан Диего, Калифорния). Значение IC50 для соединения определили путем подстановки экспериментальных данных в логарифм зависимость (ингибитора) от нормальной реакции - уравнение изменяемого наклона:
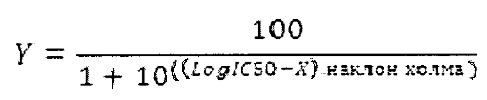
3.2 Результаты
С использованием данного протокола установили, что IC50 для соединения 1 составляет 27 нм.
Как видно из приведенных выше результатов, соединение 1 обладает значительно большим эффектом, является переферийно более селективным и/или метаболически стабильным, чем любое из сравнительных соединений 1 или 2.
4. Синтез соли НСl N-метилциклопентиламина
4.1 Метод уменьшения количества карбаматов
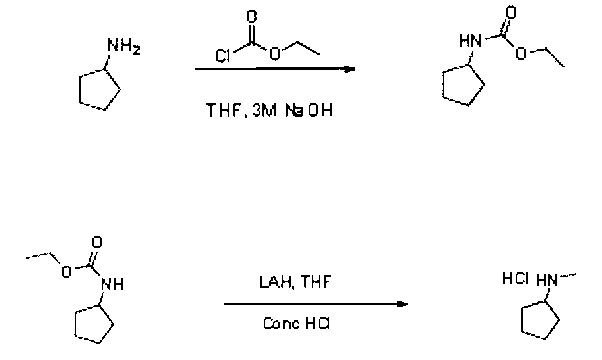
Этап 1: Образование этилкарбамата
В раствор циклопентиламина (3 мл, 30,3 ммоль) в ТГФ (20 мл) при температуре 0°С добавили соответственно 3М гидроксид натрия (15,15 мл, 45,5 ммоль) и этил хлороформат (3,47 мл, 36,4 ммоль). Полученную двухфазную смесь перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь разбавили МТБЭ (30 мл) и гидроксидом аммония (5 мл). Полученную смесь перемешивали при комнатной температуре в течение 10 минут, после чего ей дали отделиться. Органический слой промыли водой, 0,5 м НСl, высушили над Na2SO4, профильтровали. При пониженном давлении получили концентрат фильтрата. Этил циклопентилкарбамат (4,35 г) получили в виде бесцветной маслянистой жидкости в 95% выходе и использовали на следующем этапе без дальнейшей очистки.
Данная реакция протекает очень хорошо. Выход и качество продукта были высокими.
Этап 2: Уменьшение количества этилкарбамата
Уменьшение количества карбаматов под действием метиламина, в целом, хорошо известно. Такое уменьшение, как правило, предполагает необходимость использования излишков литийалюминийгидрида (ЛАГ) в ТГФ при обратном потоке. Тем не менее, использование литийалюминийгидрида в больших масштабах может потребовать более детальной проработки. Таким образом, использовали метод Физера (для x г ЛАГ, используют x мл воды, x мл 15-25% NaOH, после чего применяют 3х мл воды), который безопаснее и проще в реализации. Первую попытку успешно провели на трет-бутилкарбамате с использованием ЛАГ, после применения метода Физера образовалась соль гидрохлорида, получившаяся в результате добавления концентрированной НСl. Гидрохлорид н-метил циклопентиламина получен в 63% после выделения. Качество выхода по результатам данной первой попытки привело к повторному использованию этилкарбамата, что дало молярный выход на уровне 83% и диапазон качества >98% по ЯМР.
В суспензию ЛАГ (2414 г, 63,6 ммоль, 5 эк.) в ТГФ (20 мл) при комнатной температуре под азотом добавляли раствор этил-циклопентил-карбамата (2 г, 12,72 ммоль) в ТГФ (8 мл) на протяжении более чем 20 минут. Примечание: выделение газа. Капельную воронку промыли ТГФ (2 мл). Реакционную смесь нагревали до 65°С (внутренняя температура, обратный поток) в течение 6 часов. Суспензию охладили до 0°С (водяная баня со льдом). Суспензию растворили при помощи МТБЭ (30 мл). В суспензию по каплям добавили 2,4 мл воды (наблюдалось сильное газовыделение и экзотермическая реакция), 3,6 мл 10% NaO (необходимо хорошее помешивание) и, наконец, 7,2 литра воды. Полученный раствор подогрели до комнатной температуры и перемешивали при комнатной температуре в течение 30 минут. В белую суспензию добавили MgSO4 (10 г). Полученный раствор помешивали в течение 10 минут, затем отфильтровали. Твердую фазу промыли МТБЭ (20 мл).
В совокупный фильтрат добавили концентрированную НСl (1,272 мл, 15,27 ммоль, 1,2 эк.). Полученную смесь перемешивали при комнатной температуре в течение ночи, после чего получили сухой концентрат. Остаток растворили в пропан-2-оле (20 мл), после чего концентрацию довели до 2 объемов (4 мл). В полученный раствор затем добавили МТБЭ (12 мл), и выдавили белую твердую кристаллическую фазу. Раствор перемешивали при комнатной температуре в течение 1 часа, после чего произвели сбор твердой фазы, промыли ее при помощи МТБЭ (4 мл) и высушивали в вакуумной печи при температуре 50°С на протяжении 4 часов. Получили первую культуру белых игл (884 мг). Получили сухой общий концентрат исходного раствора и промывок. К остатку добавили изопропил ацетат (iPrOAc), после чего начали появляться белые кристаллы. Добавили дополнительное количество iPrOAc, однако часть твердой фазы осталась в виде корки на стенках колбы. Добавили некоторое количество дихлорметана и получили чистую твердую фракцию. Дихлорметан удалили, белую твердую фазу выдавили, профильтровали и промыли iPrOAc. Белую кристаллическую твердую фазу просушивали в вакуумной печи при температуре 50°С на протяжении 4 часов. Получили вторую культуру белых игл (547 мг). Гидрохлорид н-метил-циклопентиламина получили в виде белых игл в 83% молярного выхода.
4.2 Метод восстановительного аминирования
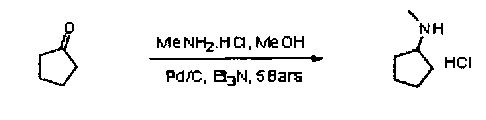
Выяснили, что наилучшие результаты дает использование циклопентанона и гидрохлорида N-метиламитна в присутствии каталитического количества триэтиламина и Pd/C при давлении водорода в метаноле при температуре 65°С. В указанных условиях гидрохлорид N-метилциклопенталамина был выделен в качестве белой твердой фазы в 49% выхода.
Источник палладия и эквиваленты реагента протестировали с целью улучшения выхода и качества продукта (удаление метиламина и гидрохлорида). При помощи Pd/C (JM, паста 5R39) с незначительными излишками гидрохлорида метиламина (1,1 эк.) выход улучшили до 69%.
Обратите внимание на то, что гидрохлорид метиламина можно удалить за счет суспендирования гидрохлорида N-метилциклопентиламина в дихлорментане в присутствии карбоната натрия и последующей дистилляции. В конечном продукте метиламин обнаружен не был.
Описание протокола
В 5% палладиевую чернь, пасту 5R 39 (0,75 г, 0,176 ммоль, 0,001 эк.) последовательно добавили МеОН (105 мл), гидрохлорид метиламина (13,24 г, 196 ммоль), циклопентанон (15,77 мл, 178 ммоль) и, наконец, триэтиламин (0,621 мл, 4.46 ммоль). Полученный раствор поместили в автоклав с подачей водорода при 5 бар. Температуру в автоклаве довели до 65°С, а перемешивание выполняли на протяжении всей ночи. Реакционную смесь медленно охладили, при этом по результатам ТСХ (растворяющий ПЭ/ацетат этила 8:2, перманганат) исходный материал выявлен не был. Черный раствор отфильтровали через целит и промыли МеОН (10 мл). Метанол удалили и заместили изопропанолом (60 мл). Концентрацию раствора довели до 2 объемов, а также добавили изопропиловый эфир (60 мл). Полученную смесь перемешивали при комнатной температуре. Наблюдалось появление белой твердой фазы, затем раствор перемешивали при температуре 0°С на протяжении 1 часа, после чего его профильтровали. Твердую фазу промыли изопропиловым эфиром/пропан-2-олом 9:1 (30 мл) и просушивали в вакуумной печи на протяжении ночи. Получили белую кристаллическую твердую фазу N-метилциклопентиламина НСl (16,9 г, выход на уровне 69,5%).
Реферат
Изобретение относится к области органической химии, а именно к гетероциклическому соединению формулы А или его фармацевтически приемлемой соли. Также изобретение относится к фармацевтической композиции на его основе и способу лечения или предупреждения указанных состояний. Технический результат: получено новое гетероциклическое соединение, полезное при лечении или предупреждении состояния, связанного с субстратом фермента FAAH. 3 н. и 5 з.п. ф-лы.
Формула
Документы, цитированные в отчёте о поиске
Производные имидазола и фармацевтическая композиция на их основе
Комментарии