Применение 2-имидазолов для лечения расстройств цнс - RU2440344C2
Код документа: RU2440344C2
Описание
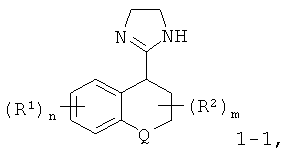
Настоящее изобретение относится к применению соединений формулы

где
R1 представляет собой атом водорода, атом трития, гидрокси, низший алкил,
низший алкокси, атом галогена, нитро, амино или низший алкил,
замещенный атомом галогена;
R2 представляет собой атом водорода, гидрокси или низший алкил;
Х представляет собой N и Y представляет собой СН, или СН2, или СН-низший алкил, или
Х представляет собой СН и Y представляет собой N;
Q представляет собой CH2, О, NH, N-алкил, или N-SO2-алкил, или
N-SO2-толуол-4-ил;
W представляет собой СН2 или связь;
m, n независимо друг от друга имеют значения 1, 2 или 3; когда m имеет значение 2 или 3, R2 могут быть одинаковыми или разными, когда n имеет значение 2 или 3, R1 могут быть одинаковыми или разными;
каждая пунктирная линия независимо от другой пунктирной линии может представлять собой связь или не является связью;
и их фармацевтически активных солей, рацемических смесей, энантиомеров, оптических изомеров и таутомерных форм соединений формулы 1 в приготовлении лекарств для лечения депрессии, тревожных расстройств, биполярного расстройства, синдрома дефицита внимания и гиперактивности, расстройств, связанных со стрессом, психотических расстройств, таких как шизофрения, неврологических заболеваний, таких как болезнь Паркинсона, нейродегенеративных расстройств, таких как болезнь Альцгеймера, эпилепсии, мигрени, гипертензии, злоупотребления веществами, вызывающими зависимость, метаболических расстройств, таких как расстройства приема пищи, диабет, диабетические осложнения, ожирение, дислипидемия, расстройства потребления и ассимиляции энергии, расстройств и нарушений температурного гомеостаза, нарушений сна и циркадного ритма и сердечно-сосудистых заболеваний
Некоторые соединения формулы I являются известными соединениями и описаны, например, в нижеприведенных ссылках или внесены в общедоступные базы данных по химическим соединениям Соединения примеров 1-14, 26-55 и 57-74 являются новыми
Установлено, что соединения формулы I обладают высокой аффинностью к рецепторам, ассоциированным со следовыми аминами (trace amine associated receptors, TAAR), особенно к TAAR1.
Классические биогенные амины (серотонин, норэпинефрин, эпинефрина, допамин, гистамин) как нейромедиаторы играют важную роль в центральной и периферической нервной системе [1]. Их синтез и хранение, а также их деградация и обратный захват после высвобождения строго регулируются Известно, что дисбаланс уровней биогенных аминов является ответственным за изменение функции мозга при многих патологических состояниях [2-5]. Соединения, образующие второй класс эндогенных аминов, так называемые следовые амины (trace amines, ТА), очень схожи с классическими биогенными аминами по своей структуре, метаболизму и субклеточной локализации. ТА включают паратирамин, β-фенилэтиламин, триптамин и октопамин, и их уровень в нервной системе млекопитающих существенно ниже уровня классических биогенных аминов [6]
Нарушение их регуляции связано с различными психическими заболеваниями, такими как шизофрения и депрессия [7], и другими состояниями, такими как синдром дефицита внимания и гиперактивности, головная боль типа мигрени, болезнь Паркинсона, злоупотребление веществами, вызывающими зависимость, и расстройства приема пищи [8, 9]
В течение долгого времени существование ТА-специфических рецепторов являлось всего лишь гипотезой, основанной на присутствии в ЦНС (центральной нервной системе) человека и других млекопитающих анатомически дискретных сайтов связывания, обладающих высокой аффинностью к ТА [10, 11]. Соответственно, считалось, что фармакологическое действие ТА опосредовано тем же известным механизмом, что и действие классических биогенных аминов, то есть либо сигналом, вызывающим их высвобождение, либо ингибированием их обратного захвата, либо "перекрестным связыванием" с их рецепторной системой [9, 12, 13]. В последнее время данная точка зрения претерпела значительные изменения в связи с идентификацией нескольких членов нового семейства GPCR (G-белок-сопряженных рецепторов), рецепторов, ассоциированных со следовыми аминами (trace amine associated receptors, TMR) [7, 14]. Обнаружено 9 TAAR-генов в геноме человека (включая 3 псевдогена) и 16 генов в геноме мыши (включая 1 псевдоген). TAAR-гены не содержат интронов (за одним исключением, TAAR2 содержит 1 интрон) и расположены рядом на одном хромосомном сегменте. Филогенетическое родство генов этих рецепторов, находящееся в соответствии с высокой степенью их сходства с GPCR-фармакофором и в соответствии с фармакологическими данными, дает возможность предположить, что эти рецепторы образуют три различных подсемейства [7, 14]. TAAR1 относится к первому подклассу, состоящему из четырех генов (TAAR1-4), которые представлены в геномах человека и грызунов высококонсервативными последовательностями. ТА активируют TAAR1 через Gα. Показано, что нарушение регуляции ТА связано с этиологией различных заболеваний, таких как депрессия, психоз, синдром дефицита внимания и гиперактивности, злоупотребление веществами, вызывающими зависимость, болезнь Паркинсона, головная боль типа мигрени, расстройства приема пищи, метаболические расстройства, и поэтому использование TAAR1-лигандов в лечении данных заболеваний может являться весьма перспективным.
Поэтому получение новых знаний о рецепторах, ассоциированных со следовыми аминами (trace amine associated receptors), весьма актуально.
Список литературы
1. Deutch, A Y. and Roth, R. H. (1999) Neurotransmitters In Fundamental
Neuroscience (2nd edn) (Zigmond, M. J., Bloom, F.E., Landis, S. С., Roberts, J.L, and Squire, L.R., eds), p.193-234, Academic Press.
2. Wong, M. L. and Licinio, J. (2001) Research and treatment approaches to depression. Nat. Rev. Neurosci. 2, 343-351.
3. Carlsson, A. et al. (2001) Interactions between monoamines, glutamate, and GABA in schizophrenia: new evidence. Anna. Rev. Pharmacol. Toxicol. 41, 237-260.
4. Tuite, P. and Riss, J. (2003) Recent developments in the pharmacological treatment of Parkinson′s disease. Expert Opin Investig Drugs 12,1335-1352.
5. Castellanos, F.X. and Tannock, R. (2002) Neuroscience of attention-
deficit/hyperactivity disorder: the search for endophenotypes. Nat. Rev. Neurosci 3, 617-628.
6. Usdin, E. and Sandier, M. eds. (1984), Trace Amines and the brain, Dekker.
7. Lindemann, L. and Hoener, M. (2005) A renaissance in trace amines inspired by a novel GPCR family. Trends in Pharmacol. Sci. 26, 274-281.
8. Branchek, T. A. and Blackburn, T.P. (2003) Trace amine receptors as targets for novel therapeutics: legend, myth and fact. Curr. Opin. Pharmacol. 3, 90-97.
9. Premont, R.T. et al. (2001) Following the trace of elusive amines. Proc. NatI Acad Sci. U. S. A. 98, 9474-9475.
10. Mousseau, D.D. and Butterworth, R.F. (1995) A high-affinity [3H] tryptamine binding site in human brain. Prog. Brain Res. 106, 285-291.
11. McCormack, J. К. et al. (1986) Autoradiographic localization of tryptamine binding sites in the rat and dog central nervous system. J. Neurosci 6, 94-101.
12. Dyck, L.E. (1989) Release of some endogenous trace amines from rat stnatal slices in the presence and absence of a monoamine oxidase inhibitor. Life Sci. 44, 1149-1156.
13. Parker, E.M. and Cubeddu, L X. (1988) Comparative effects of amphetamine, phenylethylamine and related drugs on dopamine efflux, dopamine uptake and mazindol binding. J. Pharmacol. Exp.Ther. 245,199-210.
14. Lindemann, L. et al. (2005) Trace amine associated receptors form structurally and functionally distinct subfamilies of novel G protein-coupled receptors. Genomics 85, 372-385.
Целью настоящего изобретения являются новые соединения формулы I и применение соединений формулы I и их фармацевтически приемлемых солей, рацемических смесей, энантиомеров, оптических изомеров или таутомерных форм в изготовлении лекарств для лечения заболеваний, связанных с аффинностью к рецепторам, ассоциированным со следовыми аминами (trace amine associated receptors), новые специфические соединения в объеме формулы I, их получение, лекарства на основе соединения по изобретению и их изготовление, а также применение соединений формулы I для контроля над заболеваниями, такими как депрессия, тревожные расстройства, биполярное расстройство, синдром дефицита внимания и гиперактивности, расстройства, вызванные стрессом, психотические расстройства, такие как шизофрения, неврологические заболевания, такие как болезнь Паркинсона, нейродегенеративные расстройства, такие как болезнь Альцгеймера, эпилепсия, мигрень, гипертензия, злоупотребление веществами, вызывающими зависимость, метаболические расстройства, такие как расстройства приема пищи, диабет, диабетические осложнения, ожирение, дислипидемия, расстройства потребления и ассимиляции энергии, расстройства и нарушения температурного гомеостаза, нарушения сна и циркадного ритма и сердечнососудистые заболевания, или для предупреждения перечисленных заболеваний. Другой целью настоящего изобретения является применение меченых соединений формулы 1 в качестве радиолиганда в анализе связывания с рецепторами, ассоциированными со следовыми аминами (trace amine associated receptors).
Предпочтительными показаниями к применению соединений по настоящему изобретению являются депрессия, психоз, болезнь Паркинсона, тревожность и синдром дефицита внимания и гиперактивности (ADHD).
Изобретение относится также к новым соединениям формулы 1

где
R1 представляет собой атом водорода, атом трития, гидрокси, низший алкил, низший алкокси, атом галогена, нитро, амино или низший алкил, замещенный атомом галогена,
R2 представляет собой атом водорода, гидрокси или низший алкил,
Х представляет собой N и Y представляет собой СН, или СН2, или СН-низший алкил, или
Х представляет собой СН и Y представляет собой N;
Q представляет собой СН2, О, NH, N-алкил, или N-SO2-алкил, или N-SO2-толуол-ил;
W представляет собой СН2 или связь;
m, n независимо друг от друга имеют значения 1, 2 или 3, когда m имеет значение 2 или 3, R2 могут быть одинаковыми или разными; когда n имеет значение 2 или 3, R1 могут быть одинаковыми или разными;
каждая пунктирная линия независимо от другой пунктирной линии может
представлять собой связь или не являться связью;
и к их фармацевтически активным солям, рацемическим смесям, энантиомерам,
оптическим изомерам и таутомерным формам, за исключением следующих соединений.
рац-2-(1,2,3,4-тетрагидро-1-нафтил)-2-имидазолина,
рац-2-(7-метил-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазола,
рац-2-(6-метил-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазола,
рац-2-(6-хлор-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазола,
рац-2-(5-хлор-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазола,
рац-2-(7-метокси-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазола,
рац-2-(6-метокси-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазола,
рац-2-(5-метокси-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазола,
рац-5-(4,5-дигидро-1Н-имидазол-2-ил)-5,6,7,8-тетрагидро-нафталин-2-ола,
рац-4-(1,2,3,4-тетрагидро-нафталин-1-ил)-1Н-имидазола,
рац-5-(4,5-дигидро-1Н-имидазол-2-ил)-5,6,7,8-тетрагидро-нафталин-2,3-диола или
рац-5-(4,5-дигидро-1Н-имидазол-2-ил)-5,6,7,8-тетрагидро-нафталин-1,2-диола.
Эти новые соединения формулы I также можно применять в качестве радиолиганда в анализе связывания с рецепторами, ассоциированными со следовыми аминами (trace amine associated receptors).
В контексте данного описания термин "низший алкил" означает группу с насыщенной нормальной или разветвленной цепью, содержащую от 1 до 7 атомов углерода, например, метил, этил, пропил, изопропил, н-бутил, изо-бутил, 2-бутил, трет-бутил и тому подобное. Предпочтительными алкильными группами являются группы, содержащие 1-4 атома углерода.
В контексте данного описания термин "низший алкокси" означает группу, где алкильный остаток, такой как определено выше, присоединен через атом кислорода.
В контексте данного описания термин "низший алкил, замещенный атомом галогена" означает алкильную группу, такую как определено выше, где по меньшей мере один атом водорода заменен на атом галогена, например CF3, CHF2, CH2F, СН2СF3, CH2CF2CF3 и тому подобное.
Термин "атом галогена" означает атом хлора, иода, фтор и брома.
Термин "фармацевтически приемлемые соли присоединения кислот" включает соли с неорганическими и органическими кислотами, такими как соляная кислота, азотная кислота, серная кислота, фосфорная кислота, лимонная кислота, муравьиная кислота, фумаровая кислота, малеиновая кислота, уксусная кислота, янтарная кислота, винная кислота, метан-сульфоновая кислота, пара-толуолсульфоновая кислота и тому подобное.
Одно из воплощений изобретения представляет собой применение соединений формулы

где
R1 представляет собой атом водорода, атом трития, гидрокси, низший алкил, низший алкокси, атом галогена или низший алкил, замещенный атомом галогена;
Q представляет собой СН2 или О;
n имеет значение 1, 2 или 3; когда n имеет значение 2 или 3, R1 могут быть одинаковыми или разными,
пунктирная линия может представлять собой связь или не являться связью;
и их фармацевтически активных солей, рацемических смесей, энантиомеров, оптических изомеров и таутомерных форм соединений формулы 1А в приготовлении лекарств для лечения депрессии, тревожных расстройств, биполярного расстройства, синдрома дефицита внимания и гиперактивности, расстройств, связанных со стрессом, психотических расстройств, таких как шизофрения, неврологических заболеваний, таких как болезнь Паркинсона, нейродегенеративных расстройств, таких как болезнь Альцгеймера, эпилепсии, мигрени, гипертензии, злоупотребления веществами, вызывающими зависимость, метаболических расстройств, таких как расстройства приема пищи, диабет, диабетические осложнения, ожирение, дислипидемия, расстройства потребления и ассимиляции энергии, расстройств и нарушений температурного гомеостаза, нарушений сна и циркадного ритма и сердечно-сосудистых заболеваний.
Предпочтительными соединениями формулы I в соответствии с вышеописанным применением являются те соединения формулы I, где Х представляет собой N.
Предпочтительными соединениями в данной группе являются те соединения, где Q представляет собой CH2 и R1 представляет собой атом галогена, например следующие соединения:
рац-2-(5-бром-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол,
рац-2-(7-хлор-5-фтор-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол,
рац-2-(6-хлор-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол или
рац-2-(5-хлор-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол.
Предпочтительными соединениями формулы I в соответствии с вышеописанным применением являются те соединения формулы I, где Q представляет собой СН; и R1 представляет собой низший алкил, например следующие соединения:
рац-2-(5,7-диметил-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол
или
рац-2-(5,7-диметил-1,2,3,4-тетрагидро-нафталин-1-ил)-1Н-имидазол.
Предпочтительными соединениями формулы I в соответствии с вышеописанным применением являются те соединения формулы I, где Q представляет собой СН2 и R1 представляет собой низший алкокси, например следующие соединения:
рац-2-(7-метокси-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол,
рац-2-(6-метокси-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол или
рац-2-(5-метокси-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол.
Предпочтительными соединениями формулы I в соответствии с вышеописанным применением являются те соединения формулы I, где Q представляет собой О или NH и R1 представляет собой атом водорода или атом галогена, например
рац-2-(6,8-дихлор-хроман-4-ил)-1Н-имидазол или
рац-4-(1 Н-имидазол-2-ил)-1,2,3,4-тетрагидро-хинолин.
Предпочтительными соединениями формулы I в соответствии с вышеописанным применением являются те соединения формулы I, где Х представляет собой СН.
Предпочтительными соединениями в данной группе являются те соединения, где Q представляет собой СН2 и R1 представляет собой атом водорода, например следующие соединения:
(4-(3,4-дигидро-нафталин-1-ил)-1Н-имидазол или
рац-4-(1,2,3,4-тетрагидро-нафталин-1-ил)-1Н-имидазол.
Предпочтительными соединениями в данной группе являются те соединения, где Q представляет собой О и R1 представляет собой атом водорода, например следующее соединение:
рац-5-хроман-4-ил-1Н-имидазол гидрохлорид, или таутомер.
Другими предпочтительными соединениями в данной группе являются те соединения, где Q представляет собой О и R1 представляет собой низший алкил, например следующие соединения:
рац-5-(7-метил-хроман-4-ил)-1Н-имидазол, или таутомер, или
рац-5-(5-метил-хроман-4-ил)-1Н-имидазол, или таутомер.
Другими предпочтительными соединениями в данной группе являются те соединения, где Q представляет собой О и R1 представляет собой атом галогена, например следующие соединения:
рац-5-(6-фтор-хроман-4-ил)-1Н-имидазол, или таутомер;
5-(8-хлор-2Н-хромен-4-ил)-1Н-имидазол, или таутомер;
5-(6-хлор-2Н-хромен-4-ил)-1Н-имидазол, или таутомер;
рац-5-(7-фтор-хроман-4-ил)-1Н-имидазол, или таутомер, или
рац-5-(5-фтор-хроман-4-ил)-1Н-имидазол, или таутомер.
Предпочтительными новыми соединениями являются следующие:
- Соединения формулы I, где Х представляет собой N, Q представляет собой СН2 и R1 представляет собой атом галогена, например следующие соединения:
рац-2-(5-бром-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол или
рац-2-(7-хлор-5-фтор-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол.
- Соединения формулы I, где Х представляет собой N, Q представляет собой СН2 и R представляет собой атом трития, например
рац-2-(7-тритио-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол.
- Соединения формулы I, где Х представляет собой N и Q представляет собой -О-, например следующие соединения:
рац-2-хроман-4-ил-4,5-дигидро-1Н-имидазол,
рац-2-хроман-4-ил-1Н-имидазол или
рац-2-(6-фтор-хроман-4-ил)-1Н-имидазол.
- Соединения формулы I, где Х представляет собой N, Q представляет собой О или NH и R1 представляет собой атом водорода или атом галогена, например
рац-2-(6,8-дихлор-хроман-4-ил)-1Н-имидазол или
рац-4-(1Н-имидазол-2-ил)-1,2,3,4-тетрагидро-хинолин.
- Соединения формулы I, где Х представляет собой СН, Q представляет собой СН2 и R1 представляет собой атом водорода, например следующее соединение:
(4-(3,4-дигидро-нафталин-1-ил)-1Н-имидазол.
- Соединения формулы I, где Х представляет собой СН, Q представляет собой О и R1 представляет собой атом водорода, например следующее соединение:
рац-5-хроман-4-ил-1Н-имидазол гидрохлорид, или таутомер.
- Соединения формулы I, где Х представляет собой СН, Q представляет собой О и R1 представляет собой низший алкил, например следующие соединения
рац-5-(7-метил-хроман-4-ил)-1Н-имидазол, или таутомер, или рац-5-(5-метил-хроман-4-ил)-1Н-имидазол, или таутомер.
- Соединения формулы I, где Х представляет собой СН, Q представляет собой О и R1 представляет собой атом галогена, например следующие соединения:
рац-5-(6-фтор-хроман-4-ил)-1Н-имидазол, или таутомер;
5-(8-хлор-2Н-хромен-4-ил)-1Н-имидазол, или таутомер;
5-(6-хлор-2Н-хромен-4-ил)-1Н-имидазол, или таутомер;
рац-5-(7-фтор-хроман-4-ил)-1Н-имидазол, или таутомер, или
рац-5-(5-фтор-хроман-4-ил)-1 Н-имидазол, или таутомер.
Соединения по настоящему изобретению формулы I и их фармацевтически приемлемые соли могут быть получены с помощью методик, известных в данной области техники, например, с использованием нижеописанных методик, которые включают
а) взаимодействие соединения формулы

с этилендиамином формулы
Н2NСН2СН2NН2 III
с получением соединения формулы
где R1, R2, Q, m и n являются такими, как определено выше, или
б) восстановление соединения формулы

путем каталитической гидрогенизации в присутствии Pd/C или действием комплексного гидрида
с получением соединения формулы

где R1, R2, Q, m и n являются такими, как определено выше, или
в) восстановление соединения формулы

путем каталитической гидрогенизации в присутствии Pd/C или действием комплексного гидрида
с получением соединения формулы
где R1, R2, Q, m и n являются такими, как определено выше, или 10
г) деблокирование соединения формулы

действием муравьиной кислоты
с получением соединения формулы

где R1, R2, Q, m и n являются такими, как определено выше, или
д) взаимодействие соединения формулы

с ДМСО и оксалилхлоридом в дихлорметане или перманганате, абсорбированном на силикагеле, в ацетонитриле или с Pd/C в толуоле
с получением соединения формулы
где R1, R2, Q, m и n являются такими, как определено выше, или
е) взаимодействие соединения формулы
с NaOH и гидратом гидразина
с получением соединения формулы
где R1, R2, m и n являются такими, как определено выше, или
ж) взаимодействие соединения формулы

с HBr, уксусной кислотой и анизолом
с получением соединения формулы

где R1, R2, m и n являются такими, как определено выше, или
з) взаимодействие соединения формулы

с NaOH и гидратом гидразина
с получением соединени формулы

где R1, R2, m и n являются такими, как определено выше, и Q представляет собой О или СН2, и,
при желании, превращение полученных соединений в фармацевтически приемлемые соли присоединения кислот.
Бициклические замещенные 2-имидазолины, 2-имидазолы и 2-имидазольные соединения, описанные в данной заявке, получали в соответствии с методиками, аналогичными методикам, описанными в литературе, с использованием реакций, приведенных на Схемах 1-6.
Данные методики описаны в следующих ссылках:
[1] J. Med. Chem. 1986, 29, 1413.
[2] Bull. Korean Chem. Soc. 2003, 24,1354.
[3] J. Med. Chem. 1987, 30, 1482.
[4] Chem. Pharm Bull. 1987, 35,1058 и Synthesis 1990, 78.
[5] J. Med. Chem. 1997, 40, 3014.
[6] Tempahedron 2004, 60, 9857.
[7] Synth. Common. 1990, 20, 2483.
[8] Org. Left. 2002,4, 3051.
Все исходные вещества или имеются в продаже, или же описаны в химической литературе, или могут быть получены с помощью методик, хорошо известных в данной области техники.
МЕТОДИКА А
Синтез бициклических замещенных имидазолинов
Схема 1
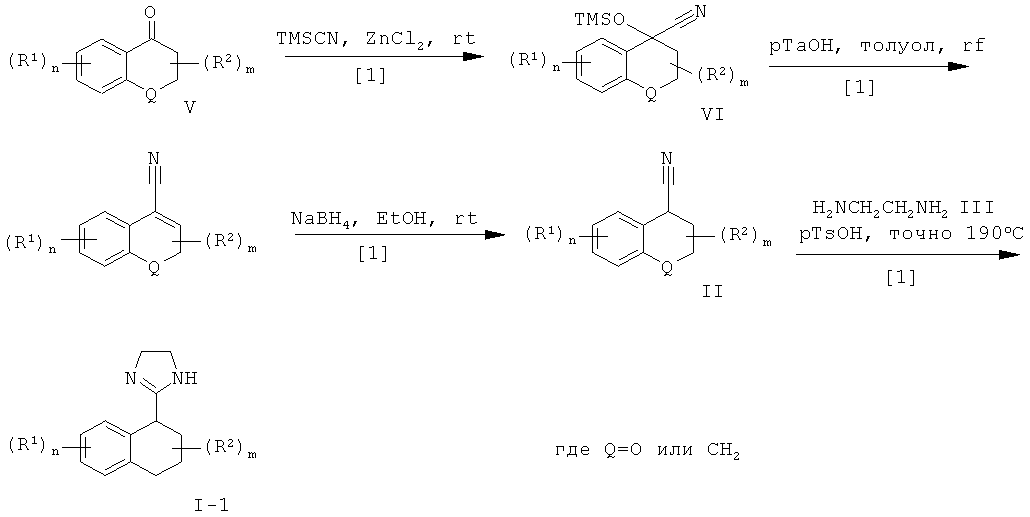
TMSN - триметилсилилцианид
pTsOH - пара-толуолсульфоновая кислота
rt - комнатная температура
rf - температура дефлегмации.
2-Имидазолины формулы I-1 могут быть получены путем взаимодействия нитрила формулы II с этилендиамином формулы III. Циклизация с диамином может быть выполнена путем нагревания диаминовой моносоли пара-толуолсульфоновой кислоты с нитрилом строго в диапазоне температур от 100°С до 250°С, предпочтительно от 140°С до 240°С, в течение нескольких часов, предпочтительно в течение 2-6 часов, или путем нагревания раствора нитрила в этилендиамине или его производном, взятых в избытке, в присутствии каталитического количества серы, предпочтительно от 10 мол.% до 50 мол.%, в герметически закрытой пробирке в микроволноовой печи при температуре до 200°С в течение 10-60 минут, предпочтительно в течение 15-30 минут [2], или путем взаимодействия комплекса, образованного из триметилалюминия и этилендиамина, или его производного, в толуоле при температуре ниже температуры окружающей среды, предпочтительно при температуре от 0°С до 10°С, с нитрилом в толуоле при температуре дефлегмации в течение 4-24 часов, предпочтительно в течение 16-20 часов [3]. В последней методике указанный нитрил может быть заменен соответствующим сложным низшим алкиловым эфиром.
Нитрилы формулы II, являющиеся производными циклических кетонов формулы V, могут быть получены путем трехстадийного синтеза в соответствии с методиками, описанными в литературе. Синтез начинают с добавления синтетического эквивалента цианистого водорода, например триметилсилилцианида, что приводит к образованию О-защищенного циангидрина формулы VI, например триметилсилил-О. Это добавление выполняют в присутствии катализатора, например иодида цинка, строго при температуре окружающей среды с интенсивным перемешиванием в течение 18-часов. Отщепление триметилсиланола в присутствии каталитического количества кислоты, предпочтительно пара-толуолсульфоновой кислоты, в органическом растворителе, таком как бензол, толуол, ксилол и тому подобное, предпочтительно в толуоле, при температуре дефлегмации в течение 1-6 часов, предпочтительно в течение 2-3 часов, приводит к образованию α,β-ненасыщенного нитрила формулы VII. Восстановление двойной связи в данном нитриле комплексным гидридом, предпочтительно боргидридом натрия, в низшем спирте, таком как метанол, этанол, изопропанол, предпочтительно в этаноле, при температуре дефлегмации в течение 0,5-2 часов, предпочтительно в течение 0,5-1 часа, приводит к образованию нитрила формулы II.
МЕТОДИКА Б
Синтез бициклических замещенных имидазолов
Схема 2а: 2-имидазолы. где Q представляет собой О, СН2, N-алкил и N-SO2-арил

где Q=О, СН2, N-алкил, N-SO2-толуол-4-ил
ТГФ - тетрагидрофуран.
Прямое введение 2-имидазольного остатка выполняют путем взаимодействия арилкетона V с металлированным N-защищенным имидазолом, который сначала получают in situ путем депротонирования N-защищенного имидазола сильным основанием, таким как алкиллитий или ариллитий, предпочтительно н-бутиллитием, в инертном органическом растворителе, например в тетрагидрофуране или диэтиловом эфире, при температуре ниже температуры окружающей среды, предпочтительно при -78°С. Выделяемый основной продукт представляет собой третичный спирт формулы VIII.
α,β-Ненасыщенные 2-имидазолы формулы IV получают из соответствующих третичных спиртов путем отщепления воды в присутствии кислотного катализатора. Предпочтительным катализатором является пара-толуолсульфоновая кислота, реакцию проводят в растворителе, образующем азеотропную смесь, таком как бензол или толуол, предпочтительно в толуоле, при температуре дефлегмации в течение 1-4 часов, предпочтительно в течение 2-3 часов. Данная реакция может быть также выполнена путем добавления соответствующих третичных спиртов к концентрированной серной кислоте, в диапазоне температур от 0°С до температуры окружающей среды, предпочтительно при температуре от 0°С до 10°С, и последующего перемешивания смеси при температуре окружающей среды в течение 5-30 минут, предпочтительно в течение 10-15 минут.
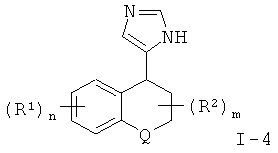
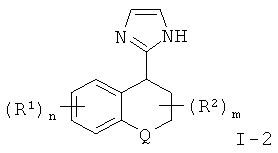
2-Имидазолы формулы 1-2 получают из соответствующих α,β-ненасыщенных 2-имидазолов формулы IV путем восстановления двойной связи либо путем каталитической гидрогенизации в присутствии Pd/C (палладия на углероде) в полярном растворителе, предпочтительно в низшем спирте, либо действием комплексного гидрида, такого как алюмогидрид лития, в апротонном растворителе, таком как тетрагидрофуран или диэтиловый эфир, при температуре окружающей среды или при повышенной температуре в течение 2-12 часов, предпочтительно в течение 4-8 часов. В том случае, когда Q представляет собой N-SO2-арил, восстановление с использованием алюмогидрида лития при повышенной температуре приводит к образованию смеси соответствующих продуктов формулы I-2, где Q представляет собой N-SO2-арил и Q представляет собой NH. Образованию последнего соединения способствует увеличение времени реакции или повышение температуры реакции.

где Q=CH2 или O, Tr=тратил
ТФУ - трифторуксусная кислота
Прямое введение 4-имидазольного остатка выполняют путем взаимодействия арилкетона формулы V с металлированным N-защищенным имидазолом, который сначала получают in situ из N-защищенного 4-иод-имидазола путем обработки магнийорганическим реагентом, предпочтительно бромидом этилмагния, в инертном органическом растворителе, предпочтительно в смеси дихлорметана и тетрагидрофурана, при температуре окружающей среды. Выделяемый основной продукт представляет собой третичный спирт формулы IX.
α,β-Ненасыщенные и N-деблокированные 4-имидазолы формулы I-3 получают из соответствующих третичных спиртов путем отщепления воды в присутствии кислотного катализатора в соответствии с тем, как описано для 2-имидазолов. Тритильную группу на имидазоле также отщепляют в этих же условиях. Для получения α,β-ненасыщенных и детритилированных 4-имидазолов формулы I-3 наряду с упомянутыми методиками получения α,β-ненасыщенных 2-имидазолов также может быть использовано взаимодействие с 30-80% трифторуксусной кислотой в воде, предпочтительно с 60%, при температуре окружающей среды в течение 12-24 часов, предпочтительн в течение 14-18 часов.
N-деблокированные 4-имидазолы формулы I-5, все еще являющиеся третичными спиртами, получают путем деблокирования соответствующего N-тритил-имидазола в присутствии кислотного катализатора, а имено смесью муравьиная кислота/ТГФ/вода (1:1:0,1).
По аналогии с 2-имидазолами 4-имидазолы формулы I-4 получают из соответствующих α,β-ненасыщенных 4-имидазолов формулы I-3 путем восстановления двойной связи либо путем каталитической гидрогенизации в присутствии Pd/C в полярном растворителе, таком как метанол, этанол, пропанол, изопропанол или этилацетат, предпочтительно в низшем спирте, таком как метанол или этанол, либо путем восстановления с использованием комплексного гидрида, такого как алюмогидрид лития, в апротонном растворителе, таком как тетрагидрофуран или диэтиловый эфир, при температуре окружающей среды в течение 2-12 часов, предпочтительно в течение 4-8 часов
МЕТОДИКА В (В1 и В2)
Дегидрогенизация имидазолинов до имидазолов
Схема 3

2-Имидазолы формулы I-2 могут быть также получены путем дегидрогенизации соответствующих 2-имидазолинов. Для данного превращения использованы две методики, описанные в литературе, окисление по Сверну и каталитическая дегидрогенизация.
МЕТОДИКА Г

Прямое введение 4-имидазольного остатка также может быть выполнено в соответствии с методикой, аналогичной методике, опубликованной S. Ohta et al. (Synthesis 1990, 78), путем взаимодействия арилкетон формулы V с металлированным N(1)-, С(2)-дизащищенным имидазолом, предпочтительно с 2-(трет-бутил-диметил-силанил)-имидазол-1-сульфоновой кислоты диметиламидом, который депротонируют in situ сильным основанием, таким как алкиллитий или ариллитий, предпочтительно н-бутиллитием, в инертном органическом растворителе, например в тетрагидрофуране или диэтиловом эфире, при температуре ниже температуры окружающей среды, предпочтительно при -78°С. Выделяемый основной продукт представляет собой третичный спирт формулы X.
Нагревание раствора третичного спирта Х в разбавленной неорганической кислоте, предпочтительно в 1-4 н. HCI, при температуре дефлегмации в течение 2-6 часов приводит к образованию α,β-ненасыщенного бициклического продукта формулы 1-3, содержащего деблокированный 4-имидазолильный остаток.
4-Имидазолы формулы I-4 получают из соответствующих α,β-ненасыщенных 2-имидазолов формулы I-3 путем восстановления двойной связи либо путем каталитической гидрогенизации водородом по давлением 50-150 бар (0,5-1,5·107 Па), предпочтительно под давлением 100 бар (107 Па), в присутствии Pd/C в полярном растворителе, таком как метанол, этанол, пропанол, изопропанол или этилацетат, предпочтительно в этилацетате, в диапазоне температур от температуры окружающей среды до 150°С, предпочтительно при температуре 50°С, в течение 12-24 часов, предпочтительно в течение 16-20 часов, или путем восстановления с использованием комплексного гидрида, такого как алюмогидрид лития, в апротонном растворителе, таком как тетрагидрофуран или диэтиловый эфир, при температуре окружающей среды в течение 2-12 часов, предпочтительно в течение 4-8 часов.
МЕТОДИКА Д

Ar представляет собой толуол-4-ил.
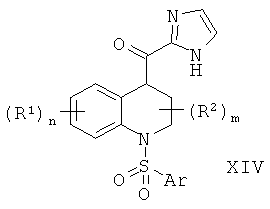
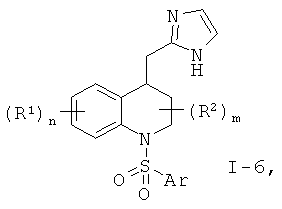
2-Имидазольные соединения формулы 1-7, где W представляет собой СН2 и Q представляет собой NH, N-алкил, N-SO2-алкил или N-SO2-толуол-4-ил, могут быть получены в соответствии со Схемой 5. Исходными веществами являются производные 1,2,3,4-тетрагидро-хинолин-4-карбоновой кислоты формулы XI, которые могут быть получены с помощью методик, уже описанных в литературе, например путем восстановления в присутствии никеля Ренея соответствующих производных хинолин-4-карбоновой кислоты, как описано в Khimiya Geterotsiklicheskikh Soedinemi 1988, 77-9. Производные карбоновой кислоты формулы XI превращают в соответствующие производные амида Вайнреба формулы XII путем обработки гидрохлоридом N,O-диметилгидроксиламина и сшивающим реагентом, таким как 1-этил-3-(3′-диметиламинопропил)карбодиимида гидрохлорид (EDCI), в присутствии третичного амина в качестве основания, такого как триэтиламин или N-метилморфолин. Реакцию проводят в галогенированном органическом растворителе, таком как дихлорметан.
После получения производных амида Вайнреба формулы XII атом азота 1,2,3,4-тетрагидро-хинолиновой кольцевой системы защищают, например в виде соответствующего арилсульфонамида, путем обработки арилсульфохлоридом в присутствии третичного амина в качестве основания, такого как триэтиламин, в галогенированном органическом растворителе, таком как дихлорметан или 1,2-дихлорэтан. Данную реакцию можно проводить при комнатной температуре или при температуре дефлегмации используемого растворителя.
Затем амидная группировка Вайнреба, присутствующая в соединениях формулы XIII, может быть подвергнута взаимодействию с металлированным N-защищенным имидазолом, например с 2-(1-диэтоксиметил-1Н-имидазол-2-ил)-литием, который сначала получают in situ путем депротонирования соответствующего N-защищенного имидазола сильным основанием, таким как алкиллитий или ариллитий, предпочтительно н-бутиллитием, в инертном эфирном растворителе, например в тетрагидрофуране или диэтиловом эфире, при температуре ниже температуры окружающей среды, предпочтительно при -78°С. Взаимодействие производного амида Вайнреба формулы XIII и металлированного N-защищенного имидазола проводят в инертном эфирном растворителе, например в тетрагидрофуране или диэтиловом эфире, при температуре ниже температуры окружающей среды, предпочтительно при температуре от -78°С до 0°С. Выделяемый основной продукт представляет собой кетон формулы XIV.
Затем кетон формулы XIV может быть подвергнут восстановлению Вольфа-Кижнера с получением соединения формулы I-6, например с использованием методики, описанной в Arch. Pharm. 1989, 322, 363-367, которая включает обработку гидроксидом натрия и гидратом гидразина в органическом растворителе, имеющем высокую точку кипения, таком как триэтиленгликоль, при повышенной температуре, предпочтительно при температуре от 110°С до 200°С.
В конце защитная группа в соединении формулы I-6 может быть удалена, например, в результате взаимодействия с протонной кислотой, такой как HBr, в уксусной кислоте в присутствии анизола, с получением желаемого соединения формулы I-7.
МЕТОДИКА Е

Исходное вещество, амид Вайнреба формулы XVI, получают из соответствующей карбоновой кислоты формулы XV в соответствии с методиками, известными в данной области техники (сравни со Схемой 5). Прямое введение 2-имидазольного остатка выполняют путем взаимодействия данного амида Вайнреба с металлированным N-защищенным имидазолом, который получают in situ из N-защищенного имидазола действием сильного основания, такого как алкиллитий или ариллитий, предпочтительно действием н-бутиллития, в инертном органическом растворителе, например в тетрагидрофуране или диэтиловом эфире, при температуре ниже температуры окружающей среды, предпочтительно при -78°С. Выделяемый основной продукт представляет собой кетон формулы XVII.
Путем восстановления данного кетона в соответствии с методиками, известными в данной области техники, например путем восстановления по Вольфу-Кижнеру (сравни со Схемой 5), получают конечный продукт формулы I-8
Соответствующие 4-имидазолы могут быть получены в соответствии с методикой синтеза, приведенной на Схеме 4, с использованием 1,2-дизащищенных имидазолов, показанных на Схеме 4.
Соединения формулы I, где R представляет собой тритий, могут быть получены из соответствующих галогенизированных (хлором, бромом или иодом) соединений, которые предпочтительно представляют собой бром-замещенные соединения, путем каталитической гидрогенизации газообразным тритием.
Выделение и очистка соединений
Выделение и очистка соединений и промежуточных соединений, описанных в данном изобретении, могут быть выполнены, при желании, с использованием любой подходящей методики разделения или очистки, такой как, например, фильтрация, экстракция, кристаллизация, колоночная хроматография, тонкослойная хроматография, хроматография в толстом слое, препаративная жидкостная хроматография низкого или высокого давления, или с использованием комбинации данных методик. Для конкретной иллюстрации подходящих методик разделения или выделения можно сослаться на препараты и примеры, приведенные в данном описании ниже. Однако могли бы быть использованы и другие эквивалентные методики разделения или выделения. Рацемические смеси хиральных соединений формулы I могут быть разделены с использованием хиральной ЖХВД (жидкостной хроматографии высокого давления).
Соли соединений формулы I
Соединения формулы I являются основаниями и могут быть превращены в соответствующую соль присоединения кислоты. Такое превращение осуществляют путем обработки по меньшей мере стехиометрическим количеством подходящей кислоты, такой как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и тому подобное и такие органические кислоты, как уксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, яблочная кислота, малоновая кислота, янтарная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, пара-толуолсульфоновая кислота, салициловая кислот и тому подобное. Обычно свободное основание растворяют в инертном органическом растворителе, таком как диэтиловый эфир, этилацетат, хлороформ, этанол или метанол и тому подобное, и добавляют кислоту в похожем растворителе. Температуру поддерживают в диапазоне от 0°С до 50°С. Получающаяся соль или спонтанно выпадает в осадок, или может быть осаждена из раствора с использованием менее полярного растворителя.
Соли присоединения кислот основных соединений формулы I могут быть превращены в соответствующие свободные основания путем их обработки по меньшей мере стехиометрическим эквивалентом подходящего основания, такого как гидроксид натрия или калия, карбонат калия, бикарбонат натрия, аммиак и тому подобное.
Соединения формулы I и их фармацевтически приемлемые соли присоединения обладают полезными фармакологическими свойствами. Конкретно, найдено, что соединения по настоящему изобретению обладают высокой аффинностью к рецепторам, ассоциированным со следовыми аминами (trace amine associated receptors, TAAR), особенно к TAAR1.
Данные соединения были исследованы в соответствии с тестом, приведенным в данном описании ниже.
Материалы и методики
Конструирование TAAR-экспрессирующих плазмид и стабильно трансфицированных клеточных линий
Для конструирования экспрессирующих плазмид амплифицировали кодирующие последовательности TAAR1 из геномной ДНК человека, крысы и мыши по существу таким способом, как описано Lindemann et al. [14]. Использовали Expand High Fidelity PCR System (Roche Diagnostics) с 1,5 мМ Mg2+, и очищенные ПЦР-продукты клонировали в клонирующий вектор pCR2.1-TOPO (Invitrogen) в соответствии с инструкцией производителя. ПЦР-продукты субклонировали в вектор plRESneo2 (BD Clontech, Palo Alto, California), и до введения в клеточные линии последовательности полученных экспрессирующих плазмид подтверждали путем секвенирования.
Клетки НЕК293 (АТСС (Американская коллекция типовых культур) #CRL-1573) культивировали по существу так, как описано Lindemann et al. (2005). Для получения стабильно трансфицированных клеточных линий клетки НЕК293 трансфицировали экспрессирующими плазмидами plRESneo2, содержащими кодирующие последовательности TAAR (описанные выше) с использованием реагента для трансфекции Lipofectamine 2000 (Invitrogen) в соответствии с инструкцией производителя, и через 24 часа после трансфекции в культуральную среду добавляли G418 (Sigma, Buchs, Switzerland) в концентрации 1 мг/мл. После культивирования в течение приблизительно 10 суток клоны выделяли, рассевали и исследовали их отвечаемость на следовые амины (все соединения приобретены в Sigma) с использованием cAMP Biotrak Enzyme immunoassay (EIA) System (Amersham) в соответствии с протоколом EIA (иммуноферментного анализа) без ацетилирования, предоставленным производителем Для всех последующих исследований использовали моноклональные клеточные линии, которые показывали стабильную EC50 (концентрацию, требуемую для достижения 50% эффекта) в течение периода культивации, составляющего 15 пассажей.
Приготовление мембран и связывание радиолиганда
Клетки из монослоя клеточной культуры смывали охлажденным до 0°С фосфатно-солевым буфером без Са2+ и Мg2+, содержащим 10 мМ ЭДТА (этилендиаминтетраацетат), и осаждали путем центрифугирования при 1000 об/мин в течение 5 мин при 4°С. Затем осадок дважды промывали охлажденным до 0°С фосфатно-солевым буфером и клеточный осадок сразу замораживали путем погружения в жидкий азот и хранили до использования при -80°С. Затем клеточный осадок суспендировали в 20 мл буфера HEPES-NaOH (20 мМ), рН 7,4, содержащего 10 мМ ЭДТА, и гомогенизировали на Polytron (РТ 3000, Kinematica) при 10000 об/мин в течение 10 с. Данный гомогенат центрифугировали при 48000xg в течение 30 мин при 4°С и полученный осадок ресуспендировали в 20 мл буфера HEPES-NaOH (20 мМ), рН 7,4, содержащего 0,1 мМ ЭДТА, (буфер А) и гомогенизировали на Polytron при 10000 об/мин в течение 10 с. Затем гомогенат центрифугировали при 48000хg в течение 30 мин при 4°С, и полученный осадок ресуспендировали в 20 мл буфера А и гомогенизировали на Polytron при 10000 об/мин в течение 10 с. Концентрацию белка определяли согласно методике Pierce (Rockford, IL). Затем гомогенат центрифугировали при 48000xg в течение 10 мин при 4°С, осадок ресуспендировали в буфере HEPES-NaOH (20 мМ), рН 7,0, содержащем MgCl2 (10 мМ) и CaCl2 (2 мМ), (буфер В) до концентрации белка 200 г на мл и гомогенизировали на Polytron при 10000 об/мин в течение 10 с.
Анализ связывания выполняли при 4°С в конечном объеме 1 мл, время инкубации составляло 30 мин. Радиолиганд [3H]-рац-2-(1,2,3,4-тетрагидро-1-нафтил)-2-имидазолин использовали в концентрации, равной рассчитанному значению Кл 60 нМ, при которой связывается приблизительно 0,1% всего добавленного радиолиганда, и специфическое связывание составляет приблизительно 70-80% от общего связывания. Неспецифическое связывание определяли как количество [3H]-рац-2-(1,2,3,4-тетрагидро-1-нафтил)-2-имидазолина, связанного в присутствии соответствующего немеченого лиганда (10 мкМ). Конкурирующие лиганды исследовали в широком диапазоне концентраций (10 пМ-30 мкМ). Конечная концентрация диметилсульфоксида в пробах составляла 2%, и это не оказывало влияния на связывание радиолиганда. Каждый эксперимент выполняли с двойной повторностью. Инкубацию останавливали путем быстрой фильтрации через планшеты UmFilter-96 (Packard Instrument Company) и стеклянный фильтр GF/C, который предварительно вымачивали в течение по меньшей мере 2 ч в 0,3% полиэтиленимине, с использованием харвестера Filtermate 96 Cell Harvester (Packard Instrument Company). Затем пробирки и фильтры 3 раза промывали аликвотами холодного буфера В объемом 1 мл. Фильтры без предварительной сушки вымачивали в Ultima gold (45 мкл/лунку, Packard Instrument Company) и связанную радиоактивность подсчитывали на счетчике TopCount Microplate Scintillation Counter (Packard Instrument Company).
Ниже в таблице приведены предпочтительные соединения, которые в анализе с использованием мышиных TAAR1 показывали значение Ki в диапазоне 0,009-0,060 мкМ.
Соединения формулы I и фармацевтически приемлемые соли соединений формулы 1 можно применять в качестве лекарств, например, в форме фармацевтических препаратов. Данные фармацевтические препараты можно вводить перорально, например в форме таблеток, таблеток, покрытых оболочкой, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий. Однако они могут быть ведены также ректально, например в форме суппозиториев, парентерально, например в форме инъекционных растворов.
Для получения фармацевтических препаратов соединения формулы I могут быть приготовлены вместе фармацевтически инертными, неорганическими или органическими носителями. Например, в качестве таких носителей для таблеток, таблеток, покрытых оболочкой, драже и твердых желатиновых капсул может быть использована лактоза, кукурузный крахмал или его производные, тальк, стеариновые кислоты или их соли и тому подобное. Подходящими носителями для мягких желатиновых капсул являются, например, растительные масла, воски, жиры, полутвердые и жидкие полиолы и тому подобное. В случае мягких желатиновых капсул носители обычно не требуются, однако это зависит от природы активного вещества. Подходящими носителями для приготовления растворов и сиропов является, например, вода, полиол, глицерин, растительное масло и тому подобное. Подходящими носителями для суппозиториев являются, например, natural или отвержденные масла, воски, жиры, полужидкие или жидкие полиолы и тому подобное.
Кроме того, данные фармацевтические препараты могут содержать консерванты, солюбилизаторы, стабилизаторы, увлажняющие агенты, эмульгаторы, подсластители, красящие вещества, корригенты, соли для регулирования осмотического давления, буферные агенты, маскирующие агенты или антиоксиданты. Они также могут дополнительно содержать другие терапевтически полезные вещества.
Лекарства, содержащие соединение формулы I, или его фармацевтически приемлемую соль, и терапевтически инертный носитель, также являются предметом настоящего изобретения, как и способ их изготовления, который включает использование одного или более чем одного соединения формулы I, и/или фармацевтически приемлемых солей присоединения кислот, и, при желании, одного или более чем одного другого терапевтически полезного вещества в форме галенова препарата вместе с одним или более чем с одним терапевтически инертным носителем.
Наиболее предпочтительными показаниями согласно настоящему изобретению являются показания, которые включают расстройства центральной нервной системы, например лечение или предупреждение депрессии, психоза, болезни Паркинсона, тревожности и синдрома дефицита внимания и гиперактивности (ADHD).
Конечно, дозировку можно варьировать в широких пределах, и обычно в каждом конкретном случае она должна быть подобрана в соответствии с индивидуальными потребностями. В случае перорального введения доза для взрослых может изменяться в диапазоне от приблизительно 0,01 мг до приблизительно 1000 мг в сутки для соединения общей формулы I или в соответствующем диапазоне для его фармацевтически приемлемой соли. Суточную дозу можно вводить в виде однократной дозы или в виде дробных доз, и, кроме того, также может быть превышен верхний предел, когда для этого имеются показания.

Методика приготовления
1. Смешивают вещества 1, 2, 3 и 4 и гранулируют с очищенной водой.
2. Сушат гранулы при 50°С.
3. Пропускают гранулы через подходящее помольное оборудование
4. Добавляют вещество 5 и перемешивают в течение трех минут; прессуют на подходящем прессе.

Методика приготовления
1. Смешивают вещества 1, 2 и 3 в подходящем смесителе в течение 30 минут.
2. Добавляют вещества 4 и 5 и смешивают в течение 3 минут.
3. Заполняют подходящую капсулу.
Эксперименты
Следующие примеры иллюстрируют изобретение, но предполагается, что они не ограничивают его объем.
МЕТОДИКА А
Пример 1
рац-2-(5-Бром-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол

Смесь рац-5-бром-1,2,3,4-тетрагидро-нафталин-1-карбонитрила (400 мг, 1,7 ммоль) и этилендиаминовой моносоли пара-толуолсульфоновой кислоты (511 мг, 2,2 ммоль) нагревали строго до 150°С и полученную жидкость перемешивали в течение 6 часов при этой же температуре. Затем охлажденную реакционную смесь разбавляли водой и насыщенным водным раствором карбоната калия. Данный раствор экстрагировали этилацетатом, объединенные экстракты промывали рассолом, сушили над Na2SO4, фильтровали и упаривали. Неочищенный продукт очищали путем флэш-хроматографии на силикагеле, используя в качестве элюента метанол/концентрированный раствор аммиака (98:2), с получением чистого рац-2-(5-бром-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазола в виде бесцветного твердого вещества; МС (ISP (ионораспыление в режиме положительных ионов)): 281,0 и 279,0 ((М+Н)+).
Пример 2
рац-2-(5,7-Диметил-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол

рац-2-(5,7-Диметил-1,2,3,4-тетрагидро-нафтапин-1-ил)-4,5-дигидро-1Н-имидазол получали аналогично соединению Примера 1, но с использованием рац-5,7-диметил-1,2,3,4-тетрагидро-нафталин-1-карбонитрила, с выходом бесцветного твердого вещества, МС (ЭИ (электронная ионизация)): 228,3 (М+).
Пример 3
рац-2-(7-Хлор-5-фтор-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол
а) рац-7-Хлор-5-фтор-1-триметилсиланилокси-1,2,3,4-тетрагидро-нафталин-1-карбонитрил

К 7-хлор-5-фтор-3,4-дигидро-2Н-нафталин-1-ону (2,00 г, 11,3 ммоль) добавляли йодистый цинк (0,11 г, 0,35 ммоль) и при интенсивном перемешиваниии триметилсилилцианид (3,72 г (4,69 мл, 37,4 ммоль), по каплям в течение 15 мин. Данную смесь перемешивали при температуре окружающей среды в течение ночи и затем разбавляли этилацетатом. Органическую фазу дважды промывали насыщенным водным раствором бикарбоната натрия, рассолом, сушили над Na2SO4, фильтровали и упаривали. Неочищенный продукт фильтровали через подушку силикагеля, используя в качестве элюента гептан/этилацетат (4:1), с получением рац-7-хлор-5-фтор-1-триметилсиланилокси-1,2,3,4-тетрагидро-нафталин-1-карбонитрила в виде светло-желтой жидкости: МС (ЭИ): 297,2 (M+), 282,2 ((М-СН3)+), 271,2 ((М-СN)+), 255,1 (((М-(СН3+HCN))+, 100%), 207,1 ((M-(CH3)3SiOH)+).
б)7-Хлор-5-фтор-3.4-дигидро-нафталин-1-карбонитрил

К охлажденной до 0°С концентрированной (96%) серной кислоте (4,5 мл) при интенсивном перемешивании добавляли рац-7-хлор-5-фтор-1-триметилсиланилокси-1,2,3,4-тетрагидро-нафталин-1-карбонитрил (1,00 г, 3,4 ммоль), по каплям в течение 5 мин. Затем охлаждающую баню удаляли и смесь перемешивали в течение 10 мин. Затем добавляли лед и данную смесь подщелачивали путем добавления концентрированного водного раствора гидроксида натрия. Полученный водный раствор экстрагировали дихлорметаном, объединенные органические экстракты промывали рассолом, сушили над Na2SO4, фильтровали и упаривали. Неочищенный продукт фильтровали через силикагель, используя в качестве элюента гептан/этилацетат (1:1), с получением 7-хлор-5-фтор-3,4-дигидро-нафталин-1-карбонитрила (0,63 г, 90%)
в) рац-7-Хлор-5-фтор-1,2.3.4-тетрагидро-нафталин-1-карбонитрил
К раствору 7-хлор-5-фтор-3,4-дигидро-нафталин-1-карбонитрила (300 мг, 1,44 ммоль) в этаноле (4 мл) добавляли боргидрид натрия (328 мг, 8,67 ммоль) и данную смесь нагревали до температуры дефлегмации в течение 30 мин. Реакционную смесь охлаждали и концентрировали. Остаток распределяли между водой и дихлорметаном. Объединенные органические экстракты промывали рассолом, сушили над Na2SO4, фильтровали и упаривали. Неочищенный продукт очищали путем флэш-хроматографии, используя в качестве элюента градиент гептан/этилацетат. рац-7-Хлор-5-фтор-1,2,3,4-тетрагидро-нафталин-1-карбонитрил получали в виде бесцветного масла: МС (ЭИ): 209,2 (М+), 182,1 ((М-НСN)+), 156,1 ((M-CH2=CHCN)+), 147,2 (((M-(Cl+HCN))+), 100%)
г) рац-2-(7-Хлор-5-фтор-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол

рац-2-(7-Хлор-5-фтор-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол получали аналогично соединению Примера 1, но с использованием рац-7-хлор-5-фтор-1,2,3,4-тетрагидро-нафталин-1-карбонитрила и нагреванием до 240°С в течение 2 часов, с выходом бесцветного кристаллического твердого вещества; МС (ISP): 253,1 ((М+Н)+).
Пример 4
рац-2-(7-Фтор-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол
а) рац-7-фтор-1-триметилсиланилокси-1,2,3,4-тетрагидро-нафталин-1-карбонитрил
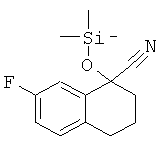
рац-7-Фтор-1-триметилсиланилокси-1,2,3,4-тетрагидро-нафталин-1-карбонитрил получали аналогично соединению Примера 3 а), но с использованием 7-фтор-3,4-дигидро-2Н-нафталин-1-она, с выходом светло-желтой жидкости; МС (ЭИ): 263,2 (М+), 248,2 ((М-СН3)+), 237,2 ((M-CN)+), 221,2 ((М-(СН3+HCN))+), 173,2 (((M-(CH3)3SiOH)+), 100%).
б) 7-Фтор-3,4-дигидро-нафталин-1-карбонитрил

7-Фтор-3,4-дигидро-нафталин-1-карбонитрил получали из рац-7-фтор-1-триметил-силанилокси-1,2,3,4-тетрагидро-нафталин-1-карбонитрила по аналогии с соединением Примера 3 (б).
в) рац-7-Фтор-1,2,3,4-тетрагидро-нафталин-1-карбонитрил
рац-7-Фтор-1,2,3,4-тетрагидро-нафталин-1-карбонитрил получали аналогично соединению Примера 3 (в), но с использованием 7-фтор-3,4-дигидро-нафталин-1-карбонитрила, с выходом светло-желтой жидкости; МС (ЭИ): 175,2 (М+), 148,2(((М-HCN)+), 100%), 122,1 ((M-CH2CHCN)+).
г) рац-2-(7-Фтор-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол

рац-2-(7-Фтор-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол получали аналогично соединению Примера 3 (г), но с использованием рац-7-фтор-1,2,3,4-тетрагидро-нафталин-1-карбонитрила, с выходом светло-желтого твердого вещества; МС (ЭИ): 218,2 (М+).
Пример 5
рац-2-(8-Метокси-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол

рац-2-(8-Метокси-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол получали аналогично соединению Примера 3 (г), но с использованием рац-8-метокси-1,2,3,4-тетрагидро-нафталин-1-карбонитрила, с выходом светло-желтой камеди; МС (ISP): 231,2 ((М+Н)+).
Пример 6
рац-2-(7-Бром-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол
а) рац-7-Бром-1-триметилсиланилокси-1,2,3,4-тетрагидро-наФталин-1-карбонитрил

рац-7-Бром-1-триметилсиланилокси-1,2,3,4-тетрагидро-нафталин-1-карбонитрил получали аналогично соединению Примера 3 (а), но с использованием 7-бром-3,4-дигидро-2Н-нафталин-1-она, с выходом бесцветного твердого вещества, т.пл. (температура плавления) 45-47°С; МС (ЭИ): 325,1 и 323,1 (М+), 310,1 и 308,1 ((М-СН3)+), 283,1 и 281,0 ((М-(СН3+НСN))+), 235,1 и 233,1 (((М-(CH3)3SiOH)+), 100%), 202,2 ((М-(СН3+HCN+Br))+).
б) 7-Бром-3,4-дигидро-нафталин-1-карбонитрил

7-Бром-3,4-дигидро-нафталин-1-карбонитрил получали аналогично соединению Примера 3 (б), но с использованием рац-7-бром-1-триметилсиланилокси-1,2,3,4-тетрагидро-нафталин-1-карбонитрила, с выходом бесцветного твердого вещества, т. пл. 113-115°С;1H-ЯМР (CDCl3): 2.48-2.55 m, 2H(=СН-СН2), 2.81 t, J=8.1 Гц, 2H (CH2-арил), 6.94 t, J=4.2 Гц, 1Н(=СН), 7.03 d, J=7.8 Гц, 1Н и 7.38 dd, J=7.8 и 1.8 Гц, 1Н, и 7.59 d, J=1.8 Гц, 1Н (арил-Н).
в) рац-7-Бром-1,2,3,4-тетрагидро-нафталин-1-карбонитрил

рац-7-Бром-1,2,3,4-тетрагидро-нафталин-1-карбонитрил получали аналогично соединению Примера 3 (в), но с использованием 7-бром-3,4-дигидро-нафталин-1-карбонитрила, с выходом бесцветного масла; МС (ЭИ): 236,0 и 234,9 (M+), 210,0 и 207,9 ((M-HCN)+), 129,0 (((M-(HCN+Br))+), 100%).
г) рац-2-(7-Бром-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол

рац-2-(7-Бром-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол получали аналогично соединению Примера 1, но с использованием рац-7-бром-1,2,3,4-тетрагидро-нафталин-1-карбонитрила и нагреванием до 210°С в течение 2 часов, в виде бесцветного твердого вещества, т.пл. 156-158°С; МС (ISP): 281,1 и 279,0 ((М+Н)+).
Пример 7
рац-2-(5,7-Дибром-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол
а) рац-5,7-Дибром-1-триметилсиланилокси-1,2,3,4-тетрагидро-нафталин-1-карбонитрил

рац-5,7-Дибром-1-триметилсиланилокси-1,2,3,4-тетрагидро-нафталин-1-карбонитрил получали аналогично соединению Примера 3 (а), но с использованием 5,7-дибром-3,4-дигидро-2Н-нафталин-1-она, с выходом серого твердого вещества.
б) 5,7-Дибром-3,4-дигидро-нафталин-1-карбонитрил
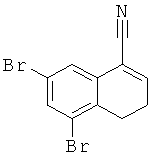
5,7-Дибром-3,4-дигидро-нафталин-1-карбонитрил получали аналогично соединению Примера 3 (б), но с использованием рац-5,7-дибром-1-триметилсиланилокси-1,2,3,4-тетрагидро-нафталин-1-карбонитрила, с выходом желтоватого твердого вещества.
в) рац-5.7-Дибром-1,2,3,4-тетрагидро-нафталин-1-карбонитрил
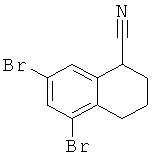
рац-5,7-Дибром-1,2,3,4-тетрагидро-нафталин-1-карбонитрил получали аналогично соединению Примера 3 (в), но с использованием 5,7-дибром-3,4-дигидро-нафталин-1-карбонитрила, с выходом бесцветной жидкости.
г) рац-2-(5,7-Дибром-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол рац-2-(5,7-Дибром-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол получали аналогично соединению Примера 1, но с использованием рац-5,7-дибром-1,2,3,4-тетрагидро-нафталин-1-карбонитрила и нагреванием до 210°С в течение 2 часов, с выходом светло-коричневого твердого вещества; МС (ЭИ): 360,0 и 358,0 (100%) и 356,0 (М+).
Пример 8
рац-4-Метил-2-(1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол или таутомер

рац-4-Метил-2-(1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол получали из 1,2,3,4-тетрагидро-нафталин-1-карбоновой кислоты метилового эфира и комплекса триметилалюминия с 1,2-диаминопропаном в толуоле при температуре дефлегмации в течение 1 часа в соответствии с методикой, аналогичной методике, приведенной в [3], с выходом оранжевой камеди, МС (ISP): 215,1 ((М+Н)+)
МЕТОДИКА В1
Пример 9
рац-2-(1,2,3,4-Тетрагидро-нафталин-1-ил)-1Н-имидазол

К охлажденному до -78°С раствору диметилсульфоксида (390 мг, 0,354 мл, 5 ммоль) в дихлорметане (20 мл) добавляли раствор оксалилхлорида (634 мг, 0,422 мл, 5 ммоль) в дихлорметане (20 мл). Данную смесь перемешивали в течение 30 минут при -78°С и затем добавляли раствор рац-2-(1,2,3,4-тетрагидро-1-нафтил)-2-имидазолина (200 мг, 2 ммоль) в дихлорметане (20 мл), и перемешивание продолжали при -78°С в течение 1 часа. Затем добавляли триэтиламин (1,01 г, 1,4 мл) и данную реакционную смесь нагревали до температуры окружающей среды и перемешивание продолжали в течение 20 минут. Добавляли концентрированный раствор аммиака и реакционную смесь экстрагировали дихлорметаном, объединенные экстракты промывали рассолом, сушили над Na2SО4, фильтровали и упаривали. После очистки на силикагеле путем флэш-хроматографии с использованием градиента гептан/этилацетат получали рац-2-(1,2,3,4-тетрагидро-нафталин-1-ил)-1Н-имидазол (71 мг) в виде бесцветного твердого вещества: МС (ЭИ): 198,1 (М+).
МЕТОДИКА В2
Пример 10
рац-2-(5,7-Диметил-1,2,3,4-тетрагидро-нафталин-1-ил)-1Н-имидазол

Смесь рац-2-(5,7-диметил-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазола (57 мг, 0,25 ммоль) и 10% Pd на угле (57 мг) в толуоле (10 мл) нагревали до температуры дефлегмации в течение 40 часов. Затем добавляли дополнительную порцию 10% Pd на угле (30 мг) и продолжали нагревание до температуры дефлегмации в течение еще 24 часов. Эту процедуру повторяли еще два раза, через 8 и 16 часов. Через 88 часов (полное время, в течение которого реакционная смесь находилась при температуре дефлегмации) реакционную смесь охлаждали, фильтровали через подушку силикагеля с получением коричневого масла (28 мг), которое очищали путем флэш-хроматографии на силикагеле, используя в качестве элюента этилацетат. рац-2-(5,7-Диметил-1,2,3,4-тетрагидро-нафталин-1-ил)-1Н-имидазол получали в виде бесцветного кристаллического твердого вещества, т. пл. 161-163°С; МС (ЭИ): 226,3 (М+).
Пример 11
МЕТОДИКА А
рац-2-Хроман-4-ил-4,5-дигидро-1Н-имидазол

рац-2-Хроман-4-ил-4,5-дигидро-1Н-имидазол получали аналогично соединению Примера 1, но с использованием рац-хроман-4-карбонитрила и нагреванием до 210°С в течение 2 часов, с выходом бесцветного твердого вещества; МС (ISP): 202,8 ((М+Н)+).
МЕТОДИКА Б
Пример 12
рац-2-Хроман-4-ил-1Н-имидазол
а) рац-4-(1Н-Имидазол-2-ил)-хроман-4-ол

рац-4-(1Н-Имидазол-2-ил)-хроман-4-ол получали из 4-хроманона и 2-(1-диэтоксиметил-1Н-имидазол-2-ил)-лития (полученного in situ из 1-(диэтокси-метил)имидазола путем обработки бутиллитием в тетрагидрофуране при -78°С) с использованием методики Ohta (Synthesis 1990, 78) в виде бесцветного твердого вещества; МС (ЭИ): 216,2 (М+), 95,1 (((О=C-2-имидазол)+), 100%).
б)2-(-2Н-Хромен-4-ил)-1Н-имидазол

2-(2Н-Хромен-4-ил)-1Н-имидазол получали аналогично соединению Примера 3 (б), но с использованием рац-4-(1Н-имидазол-2-ил)-хроман-4-ола и поддерживанием температуры 0°С, с выходом светло-зеленого твердого вещества: MC(ISP): 199,1 ((M+H)+).
в) рац-2-Хроман-4-ил-1Н-имидазол

К раствору 2-(2Н-хромен-4-ил)-1Н-имидазола (100 мг, 0,50 ммоль) в тетрагидрофуране (5 мл) добавляли 2,02 мл 1 М раствора алюмогидрида лития в тетрагидрофуране и данную смесь нагревали до температуры дефлегмации в течение 2 часов. Затем реакционную смесь охлаждали до температуры окружающей среды и реакцию гасили путем медленного добавления изопропанола. Добавляли воду и данную смесь экстрагировали трет-бутил метиловым эфиром, объединенные органические фазы промывали рассолом, сушили над Na2SO4, фильтровали и упаривали. После очистки неочищенного продукта путем флэш-хроматографии на Si-NH2 колонке, используя в качестве элюента этилацетат, получали рац-2-хроман-4-ил-1Н-имидазол в виде бесцветного твердого вещества; МС (ЭИ): 200,1 (М+), 185,1 (((М-СН3)+), 100%).
Соединение Примера 13 получали аналогично соединению Примера 12.
Пример 13
рац-2-(6-Фтор-хроман-4-ил)-1Н-имидазол
а) рац-6-Фтор-4-(1Н-имидазол-2-ил)-хроман-4-ол

рац-6-Фтор-4-(1Н-имидазол-2-ил)-хроман-4-ол получали аналогично соединению Примера 12 (а), но с использованием 6-фтор-хроман-4-она, с выходом бесцветного твердого вещества; МС (ISP): 234,9 ((М+Н)+).
б) 2-(6-Фтор-2Н-хромен-4-ил)-1Н-имидазол

2-(6-Фтор-2Н-хромен-4-ил)-1Н-имидазол получали аналогично соединению Примера 12 (б), но с использованием рац-6-фтор-4-(1Н-имидазол-2-ил)-хроман-4-ола, с выходом бесцветного твердого вещества. МС (ЭИ): 216,2 (М+).
в) рац-2-(6-Фтор-хроман-4-ил)-1Н-имидазол

рац-2-(6-Фтор-хроман-4-ил)-1Н-имидазол получали аналогично соединению Примера 12 (в), но с использованием 2-(6-фтор-2Н-хромен-4-ил)-1Н-имидазола, с выходом бесцветного твердого вещества: МС (ЭИ): 218,2 (М+), 203,2 (((М-СН3)+), 100%).
Соединения, меченые тритием
Синтез [3Н]-меченых соединений с использованием Pd-катализируемой реакции тритий-дегалогенирования бромированных предшедственников газообразным тритием:
Общая методика.
Реакционный сосуд объемом 2 мл, содержащий раствор 25-50 мкМ бромированного предшественника, 15-20 мг Pd/C (10%) и 6-10 мкл триэтиламина в
1 мл метанола, присоединяли к тритиевому манифольду (RC TRITEC AG, Teufen, Switzerland). Реакционный сосуд и его содержимое дегазировали, выполняя несколько циклов замораживания-оттаивания и откачивания, и затем наполняли свободным от носителя газообразным тритием (10-18 Ки). Перемешивание продолжали в течение 2-5 ч при комнатной температуре.
Раствор упаривали под вакуумом и обменный тритий удаляли путем повторных лиофилизаций (3х) из 1 мл метанола. Остаток растворяли в этаноле (1-2 мл) и фильтровали через шприцевой фильтр PTFE (0,2 мкм) для удаления катализатора. После промывания фильтра этанолом (4-8 мл) растворитель выпаривали, остаток растворяли в метаноле и затем очищали путем ЖХВД на стандартной колонке С-18 или С-8.
Пример 14
рац-2-(7-Тритио-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол

рац-2-(7-Тритио-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол получали из рац-2-(7-бром-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазола путем каталитической гидрогенизации газообразным тритием. Радиохимическая чистота >98%, удельная радиоактивность 32 Ки/ммоль.
Известные соединения:











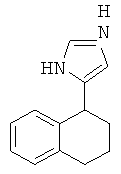
Пример 26
рац-2-(5-Нитро-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазола гидрохлорид

рац-2-(5-Нитро-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол получали аналогично соединению Примера 3, но с использованием 5-нитро-3,4-дигидро-2Н-нафталин-1-она, с выходом бесцветного твердого вещества; МС (ISP): 246,1 ((М+Н)+).
Пример 27
рац-8-(4,5-Дигидро-1Н-имидазол-2-ил)-5,6,7,8-тетрагидро-нафталин-2-иламин
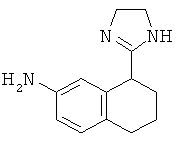
рац-8-(4,5-Дигидро-1Н-имидазол-2-ил)-5,6,7,8-тетрагидро-нафталин-2-иламин получали аналогично соединению Примера 3, но с использованием 7-нитро-3,4-дигидро-2Н-нафталин-1-она и с получением, соответственно, рац-2-(7-нитро-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1 Н-имидазола, который с помощью методик, известных в данной области техники, восстанавливали до указанного в заголовке соединения, бесцветного твердого вещества; МС (ISP): 216,4((M+H)+).
Пример 28
2-(2Н-Хромен-4-ил)-1Н-имидазол

2-(2Н-Хромен-4-ил)-1 Н-имидазол получали аналогично соединению Примера 3 (б), но с использованием рац-4-(1Н-имидазол-2-ил)-хроман-4-ола и поддерживанием температуры 0°С, с выходом светло-зеленого твердого вещества: МС (ISP): 199,1 ((М+Н)+).
Пример 29
2-(6,8-Дихлор-2Н-хромен-4-ил)-1Н-имидазол
а) рац-6,8-Пихлоо-4-(1Н-имидазол-2-ил)-хроман-4-ол

рац-6,8-Дихлор-4-(1Н-имидазол-2-ил)-хроман-4-ол получали аналогично соединению Примера 12 (а), но с использованием 6,8-дихлор-хроман-4-она, с выходом бесцветного твердого вещества; МС (ISP): 284,8 ((М+Н)+).
б) 2-(6,8-Дихлор-2Н-хромен-4-ил)-1Н-имидазол

2-(6,8-Дихлор-2Н-хромен-4-ил)-1Н-имидазол получали аналогично соединению Примера 3 (б), но с использованием рац-6,8-дихлор-4-(1Н-имидазол-2-ил)-хроман-4-ола и поддерживанием температуры 0°С, с выходом бесцветного твердого вещества: МС (ЭИ): 266,1 ((M+), 100%).
Пример 30
2-(8-Хлор-2Н-хромен-4-ил)-1Н-имидазол
а) рац-8-Хлор-4-(1Н-имидазол-2-ил)-хроман-4-ол

рац-8-Хлор-4-(1Н-имидазол-2-ил)-хроман-4-ол получали аналогично соединению Примера 12 (а), но с использованием 8-хлор-хроман-4-она, с выходом бесцветного твердого вещества; МС (ISP): 250,9 (((М+Н)+), 100%).
б) 2-(8-Хлор-2Н-хромен-4-ил)-1Н-имидазол

2-(8-Хлор-2Н-хромен-4-ил)-1Н-имидазол получали аналогично соединению Примера 3 (б), но с использованием рац-8-хлор-4-(1Н-имидазол-2-ил)-хроман-4-ола и поддерживанием температуры 0°С: в виде желтоватого твердого вещества МС (ЭИ): 232,1 ((M+), 100%).
Пример 31
2-(6-Хлор-2Н-хромен-4-ил)-1Н-имидазол
а) рац-6-Хлор-4-(1Н-имидазол-2-ил)-хроман-4-ол
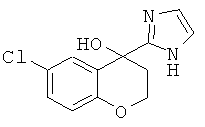
рац-6-Хлор-4-(1Н-имидазол-2-ил)-хроман-4-ол получали аналогично соединению Примера 12 (а), но с использованием 6-хлор-хроман-4-она, с выходом желтоватого твердого вещества; МС (ISP): 250,9 (((М+Н)+), 100%).
б) 2-(6-Хлор-2Н-хромен-4-ил)-1Н-имидазол

2-(6-Хлор-2Н-хромен-4-ил)-1Н-имидазол получали аналогично соединению Примера 3 (б), но с использованием рац-6-хлор-4-(1Н-имидазол-2-ил)-хроман-4-ола и поддерживанием температуры 0°С, с выходом светло-коричневого твердого вещества МС (ЭИ): 232,1 ((М+), 100%).
Пример 32
рац-2-(8-Хлор-хроман-4-ил)-1Н-имидазол

рац-2-(8-Хлор-хроман-4-ил)-1Н-имидазол получали аналогично соединению Примера 12 (в), но с использованием 2-(8-хлор-2Н-хромен-4-ил)-1Н-имидазола, с выходом бесцветного твердого вещества. МС (ЭИ): 234,2 (М+), 219,1 (((М-СН3)+), 100%).
Пример 33
рац-2-(6-Хлор-хроман-4-ил)-1Н-имидазол

рац-2-(6-Хлор-хроман-4-ил)-1Н-имидазол получали аналогично соединению Примера 12 (в), но с использованием 2-(6-хлор-2Н-хромен-4-ил)-1Н-имидазола, с выходом бесцветного твердого вещества: МС (ISP): 235,1 (((М+Н)+), 100%). Пример 34
рац-2-(6-Метокси-хроман-4-ил)-1Н-имидазол
а) рац-4-(1Н-Имидазол-2-ил)-6-метокси-хроман-4-ол

рац-4-(1Н-Имидазол-2-ил)-6-метокси-хроман-4-ол получали аналогично соединению Примера 12 (а), но с использованием 6-метокси-хроман-4-она, с выходом желтоватого твердого вещества; МС (ISP): 247,0 ((М+Н)+).
б) 2-(6-Метокси-2Н-хромен-4-ил)-1Н-имидазол
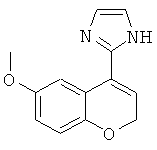
2-(6-Метокси-2Н-хромен-4-ил)-1Н-имидазол получали аналогично соединению Примера 12 (б), но с использованием рац-6-фтор-4-(1Н-имидазол-2-ил)-хроман-4-ола, с выходом желтоватого твердого вещества: МС (ЭИ): 228,2 ((М+), 100%), 213,1((M-CH3)+).
в) рац-2-(6-Метокси-хроман-4-ил)-1Н-имидазол

рац-2-(6-Метокси-хроман-4-ил)-1Н-имидазол получали аналогично соединению Примера 12 (в), но с использованием 2-(6-метокси-2Н-хромен-4-ил)-1Н-имидазола, с выходом желтоватого твердого вещества: МС (ЭИ): 230,2 ((М+), 100%) 215,2 ((M-CH3)+).
Пример 35
рац-2-(8-Метокси-хроман-4-ил)-1Н-имидазол
a) рац-4-(1Н-Имидазол-2-ил)-8-метокси-хроман-4-ол

рац-4-(1Н-Имидазол-2-ил)-8-метокси-хроман-4-ол получали аналогично соединению Примера 12 (а), но с использованием 8-метокси-хроман-4-она, с выходом бесцветного твердого вещества, МС (ЭИ): 246,2 (М+), 228,2 ((М-Н2О)+), 95,2 (((С(=О)-2-имидазолил)+), 100%).
б) 2-(8-Метокси-2Н-хромен-4-ил)-1Н-имидазол

2-(8-Метокси-2Н-хромен-4-ил)-1Н-имидазол получали аналогично соединению Примера 12 (б), но с использованием рац-4-(1Н-имидазол-2-ил)-8-метокси-хроман-4-ола, с выходом желтоватого твердого вещества МС (ЭИ) 228,1 ((М+), 100%).
в) рац-2-(8-Метокси-хроман-4-ил)-1Н-имидазол

рац-2-(8-Метокси-хроман-4-ил)-1Н-имидазол получали аналогично соединению Примера 12 (в), но с использованием 2-(8-метокси-2Н-хромен-4-ил)-1Н-имидазола, с выходом бесцветного твердого вещества: МС (ISP): 231,1 ((М+Н)+).
Пример 36
рац-2-(6,8-Дихлор-хроман-4-ил)-1Н-имидазол

рац-2-(6,8-Дихлор-хроман-4-ил)-1Н-имидазол получали аналогично соединению Примера 3 (б), но с использованием 2-(6,8-дихлор-2Н-хромен-4-ил)-1Н-имидазола и поддерживанием в течение 2 часов температуры на уровне температуры окружающей среды, с выходом бесцветного твердого вещества: МС (ЭИ): 268,1 (М+), 253,1 (((М-СН3)+), 100%).
Пример 37
рац-2-(7-Метил-хроман-4-ил)-1Н-имидазол
а) рац-4-(1Н-Имидазол-2-ил)-7-метил-хроман-4-ол

рац-4-(1Н-Имидазол-2-ил)-7-метил-хроман-4-ол получали аналогично соединению Примера 12 (а), но с использованием 7-метил-хроман-4-она, с выходом бесцветного твердого вещества; МС (ЭИ): 230,2 (М+), 212,2 ((M-H2О)+), 183,2 ((M-(H2О+H+СО))+), 95,2 (((C(=О)-2-имидазолил)+), 100%).
б) 2-(7-Метил-2Н-хромен-4-ил)-1Н-имидазол

2-(7-Метил-2Н-хромен-4-ил)-1Н-имидазол получали аналогично соединению Примера 12 (б), но с использованием рац-4-(1Н-имидазол-2-ил)-7-метил-хроман-4-ола, с выходом светло-желтого твердого вещества: МС (ЭИ): 212,2 ((М+), 100%)
в) рац-2-(7-Метил-хроман-4-ил)-1Н-имидазол

рац-2-(7-Метил-хроман-4-ил)-1Н-имидазол получали аналогично соединению Примера 3 (б), но с использованием 2-(7-метил-2Н-хромен-4-ил)-1Н-имидазола и поддерживанием в течение 18 часов температуры на уровне температуры окружающей среды, с выходом бесцветного твердого вещества: МС (ЭИ): 214,2 (M+), 199,2 (((М-СН3)+), 100%).
Пример 38
2-(5-Метил-2Н-хромен-4-ил)-1Н-имидазол
а) рац-4-(1Н-Имидазол-2-ил)-5-метил-хроман-4-ол

рац-4-(1Н-Имидазол-2-ил)-5-метил-хроман-4-ол получали аналогично соединению Примера 12 (а), но с использованием 5-метил-хроман-4-она, с выходом бесцветного твердого вещества; МС (ЭИ): 230,2 ((М+), 100%), 95,2 ((С(=О)-2-имидазолил)+).
б) 2-(5-Метил-2Н-хромен-4-ил)-1Н-имидазол

2-(5-Метил-2Н-хромен-4-ил)-1Н-имидазол получали путем нагревания раствора рац-4-(1Н-имидазол-2-ил)-5-метил-хроман-4-ола в 4 н. водном растворе HCl в течение 16 часов. Реакционную смесь охлаждали до температуры окружающей среды, рН доводили до 10 путем добавления аммиака и экстрагировали /трет-бутилметиловым эфиром. Собранные органические фазы промывали рассолом, сушили над Na2SО4, фильтровали и упаривали с получением бесцветного твердого вещества: МС (ISP): 213,1 ((М+Н)+).
Пример 39
рац-2-(7-Фтор-хроман-4-ил)-1Н-имидазол

рац-2-(7-Фтор-хроман-4-ил)-1Н-имидазол получали аналогично соединению Примера 13, но с использованием 7-фтор-хроман-4-она, с выходом бесцветного твердого вещества, МС (ЭИ): 218,1 (М+), 203,1 (((М-СН3)+), 100%).
Пример 40
рац-4-(1Н-Имидазол-2-ил)-1-(толуол-4-сульфонил)-1,2,3,4-тетрагидро-хинолин

а) 1-Фенил-азетидин-2-он

1-Фенил-азетидин-2-он получали из азетидин-2-она и иодбензола путем обработки транс-1,2-диаминоциклогексаном, иодидом меди (I) и карбонатом калия в соответствии с методикой, описанной в J. Am. Chem. Soc. 2001, 123, 7727-7729, в виде желтоватого кристаллического твердого вещества; МС (ISP): 148,4 ([М+Н]+ 100%).
б) 2,3-Дигидро-4(1H)-хинолинон

2,3-Дигидро-4(1Н)-хинолинон получали из 1-фенил-азетидин-2-она путем обработки трифторметансульфоновой кислотой в 1,2-дихлорэтане в соответствии с методикой, описанной в Tempahedron 2002, 58, 8475-8481, с выходом желтого масла, МС (ISP): 148,3 ([М+Н]+, 100%).
в) 1-(Толуол-4-сульфонил)-2,3-дигидро-1Н-хинолин-4-он

К раствору 2,3-дигидро-4(1Н)-хинолинона (2,57 г, 17,5 ммоль) в дихлорметане (20 мл) при 0°С добавляли по каплям триэтиламин (9,09 мл, 65,6 ммоль). Затем добавляли пара-толуолсульфокислоты хлорангидрид (5,25 г, 27,5 ммоль) и данную реакционную смесь нагревали при температуре дефлегмации в течение 16 часов. После охлаждения до комнатной температуры смесь разбавляли дихлорметаном и промывали последовательно 1 М водным раствором соляной кислоты, насыщенным водным раствором бикарбоната натрия и насыщенным рассолом. Фазы разделяли, и органическую фазу сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Остаток очищали путем хроматографии (силикагель, этилацетат/гептан) с получением указанного в заголовке соединения (1,55 г, 29%) в виде белого кристаллического твердого вещества. МС (ISP): 302,4 ([M+H]+, 100%).
г) рац-4-(1Н-Имидазол-2-ил)-1-(толуол-4-сульфонил)-1,2,3,4-тетрагидро-хинолин-4-ол

рац-4-(1Н-Имидазол-2-ил)-1-(толуол-4-сульфонил)-1,2,3,4-тетрагидро-хинолин-4-ол получали аналогично соединению Примера 12 (а), но с использованием 1-(толуол-4-сульфонил)-2,3-дигидро-1Н-хинолин-4-она и 2-(1-диэтоксиметил-1Н-имидазол-2-ил)-лития, с выходом желтоватой пены, МС (ISP): 370,1 ([М+Н]+, 100%).
д) 4-(1Н-Имидазол-2-ил)-1-(толуол-4-сульфонил)-1,2-дигидро-хинолин

4-(1Н-Имидазол-2-ил)-1-(толуол-4-сульфонил)-1,2-дигидро-хинолин получали аналогично соединению Примера 12 (б), но с использованием рац-4-(1Н-имидазол-2-ил)-1-(толуол-4-сульфонил)-1,2,3,4-тетрагидро-хинолин-4-ола и серной кислоты, в этаноле при 0-20°С, с выходом белого кристаллического твердого вещества: МС (ISP): 352,3 ([М+Н]+, 100%).
е) рац-4-(1Н-Имидазол-2-ил)-1-(толуол-4-сульфонил)-1,2,3,4-тетрагидро-хинолин
рац-4-(1Н-Имидазол-2-ил)-1-(толуол-4-сульфонил)-1,2,3,4-тетрагидро-хинолин получали аналогично соединению Примера 12 (в), но с использованием 4-(1Н-имидазол-2-ил)-1-(толуол-4-сульфонил)-1,2-дигидро-хинолина и алюмогидрида лития, в тетрагидрофуране при температуре дефлегмации, с выходом желтоватой пены; МС (ISP): 354,3 ([М+Н]+, 100%).
Пример 41
рац-4-(1Н-Имидазол-2-ил)-1,2,3,4-тетрагидро-хинолин

рац-4-(1Н-Имидазол-2-ил)-1,2,3,4-тетрагидро-хинолин получали в качестве побочного продукта в процессе получения рац-4-(1Н-имидазол-2-ил)-1-(толуол-4-сульфонил)-1,2,3,4-тетрагидро-хинолина в соответствии с тем, как описано в Примере 40 (е), с выходом желтоватого аморфного твердого вещества; МС (ISP). 200,4 ([М+Н]+, 100%).
Пример 42
рац-2-(5-Метил-хроман-4-ил)-1Н-имидазол

рац-2-(5-Метил-хроман-4-ил)-1Н-имидазол получали из 2-(5-метил-2Н-хромен-4-ил)-1Н-имидазола путем гидрогенизации под давлением 100 бар (107 Па) в присутствии 10% Pd/C, в качестве катализатора, в этилацетате при 50°С в течение 18 часов. После обычной обработки остаток очищали путем флэш-хроматографии на силикагеле, используя в качестве элюента градиент этилацетат/метанол 5-30%, с получением указанного в заголовке соединения в виде бесцветного твердого вещества; МС (ISP): 215,2 ((М+Н)+).
Пример 43
рац-2-(5-Фтор-хроман-4-ил)-1Н-имидазол
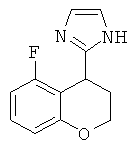
рац-2-(5-Фтор-хроман-4-ил)-1 Н-имидазол получали аналогично соединению Примера 13, но с использованием 5-фтор-хроман-4-она, с выходом бесцветного твердого вещества; МС (ISP): 219,1 ((М+Н)+.
Пример 44
рац-4-(1Н-Имидазол-2-ил)-1-метил-1,2,3,4-тетрагидро-хинолин

а) 1-Метил-2,3-дигидро-1Н-хинолин-4-он

Данное соединение получали в соответствии с методикой, описанной в J. Med. Chem. 2003, 46, 1962-1979. К раствору 2,3-дигидро-4(1Н)-хинолинона (220 мг, 1,49 ммоль) в ацетоне (3 мл), находящемуся в сосуде, выдерживающем повышенное давление, добавляли карбоната калия (620 мг, 4,48 ммоль). Затем добавляли по каплям иодметан (0,38 мл, 5,98 ммоль), пробирку герметично закрывали и реакционную смесь нагревали при 80°С в течение 16 часов. После охлаждения до комнатной температуры смесь разбавляли этилацетатом и промывали насыщенным рассолом. Фазы разделяли и органическую фазу сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Остаток очищали путем хроматографии (силикагель, этилацетат/гептан) с получением указанного в заголовке соединения (147 мг, 61%) в виде светло-желтого масла. МС (ISP): 162,1 ([М+Н]+ 100%).
б) рац-4-(1Н-Имидазол-2-ил)-1-метил-1,2,3,4-тетрагидро-хинолин-4-ол

рац-4-(1Н-Имидазол-2-ил)-1-метил-1,2,3,4-тетрагидро-хинолин-4-ол получали аналогично соединению Примера 12 (а), но с использованием 1-метил-2,3-дигидро-1Н-хинолин-4-она и 2-(1-диэтоксиметил-1Н-имидазол-2-ил)-лития, с выходом желтоватого кристаллического твердого вещества; МС (ISP): 230,4 ([М+Н]+, 54%), 212,1 ([М+Н-H2O]+, 100%).
в) 4-(1Н-Имидазол-2-ил)-1-метил-1,2-дигидро-хинолин

4-(1Н-Имидазол-2-ил)-1-метил-1,2-дигидро-хинолин получали аналогично соединению Примера 12 (б), но с использованием рац-4-(1Н-имидазол-2-ил)-1-метил-1,2,3,4-тетрагидро-хинолин-4-ола и серной кислоты, в этаноле при 50°С, с выходом оранжевого кристаллического твердого вещества; МС (ISP): 212,3 ([M+H]+, 100%).
г) рац-4-(1Н-Имидазол-2-ил)-1-метил-1,2,3,4-тетрагидро-хинолин
рац-4-(1Н-Имидазол-2-ил)-1-метил-1,2,3,4-тетрагидро-хинолин получали аналогично соединению Примера 12 (в), но с использованием 4-(1Н-имидазол-2-ил)-1-метил-1,2-дигидро-хинолина и алюмогидрида лития, в тетрагидрофуране при температуре дефлегмации, с выходом желтоватого кристаллического твердого вещества; МС (ISP): 214,1 ([M+H]+, 100%).
Пример 45
2-(3-Метил-2Н-хромен-4-ил)-1Н-имидазол
а) рац-4-1Н-Имидазол-2-ил)-3-метил-хроман-4-ол

рац-4-(1Н-Имидазол-2-ил)-3-метил-хроман-4-ол получали аналогично соединению Примера 12 (а), но с использованием 3-метил-хроман-4-она, с выходом бесцветного твердого вещества, МС (ISP): 231,1 ((М+Н)+).
б) 2-(3-Метил-2Н-хромен-4-ил)-1Н-имидазол

2-(3-Метил-2Н-хромен-4-ил)-1Н-имидазол получали аналогично соединению Примера 38 (б), но с использованием рац-4-(1Н-имидазол-2-ил)-3-метил-хроман-4-ола, с выходом светло-коричневого твердого вещества, МС (ISP): 213,0 ((М+Н)+.
Пример 46
2-(2,2-Диметил-2Н-хромен-4-ил)-1Н-имидазол

2-(2,2-Диметил-2Н-хромен-4-ил)-1Н-имидазол получали аналогично 10 соединению Примера 38, но с использованием 2,2-диметил-хроман-4-она, с выходом желтого твердого вещества; МС (ISP): 227,0 ((М+Н)+).
Пример 47
рац-2-(2,2-Диметил-хроман-4-ил)-1Н-имидазол

рац-2-(2,2-Диметил-хроман-4-ил)-1Н-имидазол получали аналогично соединению Примера 42, но с использованием 2-(2,2-диметил-2Н-хромен-4-ил)-1Н-имидазола, с выходом бесцветного твердого вещества; МС (ISP): 229,2 ((М+Н)+).
Пример 48
рац-2-(2-Метил-2Н-хромен-4-ил)-1Н-имидазол

рац-2-(2-Метил-2Н-хромен-4-ил)-1Н-имидазол получали аналогично соединению Примера 38, но с использованием рац-2-метил-хроман-4-она, с выходом светло-коричневого твердого вещества; МС (ISP): 213,0 ((М+Н)+).
Пример 49
(3R,4S или 3S,4R)-2-(3-Метил-хроман-4-ил)-1Н-имидазол

[3R,4S или 3S,4R)-2-(3-Метил-хроман-4-ил)-1Н-имидазол получали путем хроматографического разделения диастереоизомерной смеси рац-2-(3-метил-хроман-4-ил)-1Н-имидазола (Пример 53) на колонке Chiralpak AD, используя в качестве элюента гептан/этанол (93:7), с выходом бесцветного твердого вещества;
MC (ISP): 215,1 ((М+Н)+).
Пример 50
рац-2-(2-Метил-хроман-4-ил)-1Н-имидазол

рац-2-(2-Метил-хроман-4-ил)-1Н-имидазол получали аналогично соединению Примера 42, но с использованием рац-2-(2-метил-2Н-хромен-4-ил)-1 Н-имидазола, с выходом светло-зеленого твердого вещества, МС (ISP): 215,1 ((М+Н)+).
Пример 51
(2R,4S или 2S,4R)-2-(2-Метил-хроман-4-ил)-1Н-имидазол

(2R,4S или 2S,4R)-2-(2-Метил-хроман-4-ил)-1Н-имидазол получали аналогично соединению Примера 49, но с использованием рац-2-(2-метил-хроман-4-ил)-1Н-имидазола, с выходом бесцветного твердого вещества; МС (ISP): 215,1 ((М+Н)+).
Пример 52
(2S,4R или 2R,4S)-2-(2-Метил-хроман-4-ил)-1Н-имидазол

(2S,4R или 2Р,45)-2-(2-Метил-хроман-4-ил)-1Н-имидазол получали аналогично соединению Примера 49, но с использованием рац-2-(2-метил-хроман-4-ил)-1Н-имидазола, с выходом бесцветного твердого вещества; МС (ISP): 215,1 ((М+Н)+).
Пример 53
рац-2-(3-Метил-хроман-4-ил)-1Н-имидазол

рац-2-(3-Метил-хроман-4-ил)-1Н-имидазол получали из рац-4-(1Н-имидазол-2-ил)-3-метил-хроман-4-ола путем восстановления литием в жидком аммиаке в течение 30 мин. Голубую реакционную смесь гасили путем добавления хлорида аммония в виде твердого вещества, аммиак выпаривали и остаток распределяли между водой и трет-бутилметиловым эфиром. Органическую фазу промывали рассолом, сушили над сульфатом натрия, фильтровали и упаривали. рац-2-(3-Метил-хроман-4-ил)-1Н-имидазол получали в виде бесцветного твердого вещества; МС (ISP): 215,1 ((М+Н)+.
Пример 54
(3S,4S и 3R,4R)-2-(3-Метил-хроман-4-ил)-1Н-имидазол
Рацемическую смесь (3S.4S и 3R,4R)-2-(3-метил-хроман-4-ил)-1Н-имидазола получали (вместе с обоими разделяемыми цис-изомерами) по аналогии с Примером 49, но с использованием рац-2-(3-метил-хроман-4-ил)-1Н-имидазола, с выходом бесцветного твердого вещества, МС (ISP): 215,1 ((М+Н)+).
Пример 55
(4-(3,4-Дигидро-нафталин-1-ил)-1Н-имидазол
а) 1-(1-Тритил-1Н-имидазол-4-ил)-1,2,3,4-тетрагидро-нафталин-1-ол

1-(1-Тритил-1Н-имидазол-4-ил)-1,2,3,4-тетрагидро-нафталин-1-ол получали из 4-иод-1-тритил-1Н-имидазола и альфа-тетралона в соответствии с методикой, описанной X. Zhang et at., J. Med. Chem. 40, 3014 (1997), с выходом бесцветного твердого вещества; МС (ISP): 457,5 ((М+Н)+).
б) (4-(3,4-Дигидро-нафталин-1-ил)-1Н-имидазол
4-(3,4-Дигидро-нафталин-1-ил)-1Н-имидазол получали путем взаимодействия 1-(1-тритил-1Н-имидазол-4-ил)-1,2,3,4-тетрагидро-нафталин-1-ола с раствором трифторуксусной кислоты в воде (6:4) в соответствии с методикой, описанной X. Zhang et al., J. Med. Chem. 40, 3014 (1997), с выходом бесцветного твердого вещества; МС (ISP): 197,3 ((М+Н)+).
Пример 56
рац-4-(1,2,3,4-Тетрагидро-нафталин-1-ил)-1Н-имидазол

рац-4-(1,2,3,4-Тетрагидро-нафталин-1-ил)-1Н-имидазол получали аналогично соединению Примера 42, но использовали 4-(3,4-дигидро-нафталин-1-ил)-1Н-имидазол, давление водорода поддерживали на уровне 3,5 бар (3,5·105 Па) и реакцию проводили при температуре окружающей среды в течение 5 часов, указанное в заголовке соединение получали в виде бесцветного твердого вещества, МС (ЭИ): 198,2 ((М+), 100%).
Пример 57
(4-(3,4-Дигидро-нафталин-1-ил)-1Н-имидазол
а) 2-(трет-Бутил-диметил-силанил)-4-1-гидрокси-1,2,3,4-тетрагидро-нафталин-1-ил)-имидазол-1-сульфоновой кислоты диметиламид

2-(трет-Бутил-диметил-силанил)-4-(1-гидрокси-1,2,3,4-тетрагидро-нафталин-1-ил)-имидазол-1-сульфоновой кислоты диметиламид получали из 2-(трет-бутил-диметил-силанил)-имидазол-1-сульфоновой кислоты диметиламида и хроман-4-она в соответствии с методикой, аналогичной методике, описанной S. Ohta et al., Synthesis 1990, 78, с выходом светло-коричневого вязкого масла; МС (ISP): 438,5 ((М+Н)+).
б) 5-(2Н-Хромен-4-ил)-1Н-имидазол или таутомер

5-(2Н-Хромен-4-ил)-1Н-имидазол получали аналогично соединению Примера 38 (б), но использовали 2-(трет-бутил-диметил-силанил)-4-(1-гидрокси-1,2,3,4-тетрагидро-нафталин-1-ил)-имидазол-1-сульфоновой кислоты диметиламид и реакцию проводили в 2 н. водном растворе HCl при температуре дефлегмации в течение 2 часов, с выходом бесцветного твердого вещества; МС (ЭИ): 198,2 ((М+), 100%).
Пример 58
5-(6-Фтор-2Н-хромен-4-ил)-1Н-имидазол или таутомер

5-(6-Фтор-2Н-хромен-4-ил)-1Н-имидазол получали аналогично соединению Примера 57, но с использованием 2-(трет-бутил-диметил-силанил)-имидазол-1- сульфоновой кислоты диметиламида и 6-фтор-хроман-4-она, с выходом желтоватого твердого вещества; МС (ЭИ): 216,1 ((М+), 100%).
Пример 59
5-(7-Метил-2Н-хромен-4-ил)-1Н-имидазол или таутомер

5-(7-Метил-2Н-хромен-4-ил)-1Н-имидазол получали аналогично соединению Примера 57, но с использованием 2-(трет-бутил-диметил-силанил)-имидазол-1-сульфоновой кислоты диметиламида и 7-метил-хроман-4-она, с выходом светло-коричневого твердого вещества; МС (ЭИ): 212,2 ((М+), 100%).
Пример 60
рац-5-(7-Метил-хроман-4-ил)-1Н-имидазол или таутомер

рац-5-(7-Метил-хроман-4-ил)-1Н-имидазол получали аналогично соединению Примера 42, но с использованием 5-(7-метил-2Н-хромен-4-ил)-1Н-имидазола, с выходом бесцветного твердого вещества; МС (ЭИ): 214,2 ((М+), 100%).
Пример 61
5-(5-Фтор-2Н-хромен-4-ил)-1Н-имидазол или таутомер

5-(5-Фтор-2Н-хромен-4-ил)-1Н-имидазол получали аналогично соединению Примера 57, но с использованием 2-(трет-бутил-диметил-силанил)-имидазол-1-сульфоновой кислоты диметиламида и 5-фтор-хроман-4-она, с выходом бесцветного твердого вещества; МС (ЭИ): 216,2 ((М+), 100%).
Пример 62
рац-5-(6-Фтор-хроман-4-ил)-1Н-имидазол или таутомер

рац-5-(6-Фтор-хроман-4-ил)-1Н-имидазол получали аналогично соединению Примера 12 (в), но с использованием 5-(6-фтор-2Н-хромен-4-ил)-1Н-имидазола, с выходом бесцветного твердого вещества; МС (ЭИ): 218,2 ((М+), 100%).
Пример 63
5-(8-Хлор-2Н-хромен-4-ил)-1Н-имидазол или таутомер

5-(8-Хлор-2Н-хромен-4-ил)-1Н-имидазол получали аналогично соединению Примера 57, но с использованием 2-(трет-бутил-диметил-силанил)-имидазол-1-сульфоновой кислоты диметиламида и 8-хлор-хроман-4-она, с выходом желтоватого твердого вещества; МС (ЭИ): 232,1 ((M+), 100%).
Пример 64
5-(6-Хлор-2Н-хромен-4-ил)-1Н-имидазол или таутомер
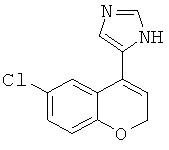
5-(6-Хлор-2Н-хромен-4-ил)-1Н-имидазол получали аналогично соединению Примера 57, но с использованием 2-(трет-бутил-диметил-силанил)-имидазол-1-сульфоновой кислоты диметиламида и 6-хлор-хроман-4-она, с выходом желтоватого твердого вещества, МС (ЭИ): 232,1 ((М+), 100%).
Пример 65
рац-5-(7-Фтор-хроман-4-ил)-1Н-имидазол или таутомер

рац-5-(7-Фтор-хроман-4-ил)-1Н-имидазол получали аналогично соединению Примера 42, но с использованием 5-(7-фтор-2Н-хромен-4-ил)-1Н-имидазола, с выходом бесцветного твердого вещества; МС (ISP): 219,1 ((М+Н)+.
Пример 66
рац-5-(5-Метил-хроман-4-ил)-1Н-имидазол или таутомер
а) 5-(5-Метил-2Н-хромен-4-ил)-1Н-имидазол или таутомер
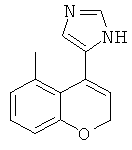
5-(5-Метил-2Н-хромен-4-ил)-1Н-имидазол получали аналогично соединению Примера 57, но с использованием 2-(трет-бутил-диметил-силанил)-имидазол-1-сульфоновой кислоты диметиламида и 5-метил-хроман-4-она, с выходом бесцветного твердого вещества; МС (ЭИ): 212,2 ((M+), 100%).
б) рац-5-(5-Метил-хроман-4-ил)-1Н-имидазол или таутомер

рац-5-(5-Метил-хроман-4-ил)-1Н-имидазол получали аналогично соединению Примера 42, но с использованием 5-(5-метил-2Н-хромен-4-ил)-1Н-имидазола, с выходом бесцветного твердого вещества, МС (ISP): 215,2 ((М+Н)+).
Пример 67
рац-5-(5-Фтор-хроман-4-ил)-1Н-имидазол или таутомер

рац-5-(5-Фтор-хроман-4-ил)-1Н-имидазол получали аналогично соединению Примера 42, но с использованием 5-(5-фтор-2Н-хромен-4-ил)-1Н-имидазола, с выходом бесцветного твердого вещества, МС (ISP): 219,1 ((М+Н)+).
Пример 68
5-(7-Фтор-2Н-хромен-4-ил)-1Н-имидазол или таутомер

5-(7-Фтор-2Н-хромен-4-ил)-1Н-имидазол получали аналогично соединению Примера 57, но с использованием 2-(трет-бутил-диметил-силанил)-имидазол-1-сульфоновой кислоты диметиламида и 7-фтор-хроман-4-она, с выходом светло-коричневого твердого вещества, МС (ISP): 217,1 ((М+Н)+).
Пример 69
рац-5-Хроман-4-ил-1Н-имидазола гидрохлорид или таутомер

рац-5-Хроман-4-ил-1Н-имидазол получали аналогично соединению Примера 12 (в), но с использованием 5-(2Н-хромен-4-ил)-1Н-имидазола, с выходом желтоватого твердого вещества. Соединение выделяли в виде гидрохлорида; МС (ISP): 201,1 ((M+H)+).
Пример 70
5-(3-Метил-2Н-хромен-4-ил)-1Н-имидазол или таутомер

5-(3-Метил-2Н-хромен-4-ил)-1Н-имидазол получали аналогично соединению 25 Примера 57, но с использованием 2-(трет-бутил-диметил-силанил)-имидазол-1-сульфоновой кислоты диметиламида и 3-метил-хроман-4-она, с выходом светло-коричневого твердого вещества, МС (ISP): 213,0 ((М+Н)+.
Пример 71
рац-1-(1Н-Имидазол-4-ил)-1,2,3,4-тетрагидро-нафталин-1-ол или таутомер

рац-1-(1Н-Имидазол-4-ил)-1,2,3,4-тетрагидро-нафталин-1-ол получали из рац-1-(1-тритил-1 Н-имидазол-4-ил)-1,2,3,4-тетрагидро-нафталин-1-ола (Пример 55 (а)) путем деблокирования смесью муравьиная кислота/тетрагидрофуран/вода (1:1:0,1) в соответствии с методикой, аналогичной методике, описанной A. Ojima et al., Org Lett. 4, 3051 (2002), с выходом бесцветного твердого вещества; МС (ISP): 215,3 ((М+Н)+).
Пример 72
рац-2-Chroman-4-илметил-1Н-имидазол
а) рац-Хроман-4-карбоновой кислоты метокси-метил-амид

К раствору хроман-4-карбоновой кислоты (500 мг, 2,8 ммоль) в дихлорметане (10 мл) добавляли N,О-диметилгидроксиламина гидрохлорид (330 мг, 3,2 ммоль) и N-(3-диметиламинопропил)-N′-этил-карбодиимида гидрохлорид (993 мг, 3,2 ммоль) и данную смесь перемешивали при температуре окружающей среды в течение 5 мин. Затем добавляли по каплям триэтиламин (710 мг, 0,98 мл, 10 ммоль) и полученную смесь перемешивали при температуре окружающей среды в течение 4 часов. Обрабатывали путем добавления 2 М раствора HCl, органический растворитель выпаривали и остаток экстрагировали трет-бутилметиловым эфиром, объединенную органическую фазу промывали рассолом, сушили над Na2SO4, фильтровали и упаривали с получением 503 мг рац-хроман-4-карбоновой кислоты метокси-метил-амида в виде светло-коричневого масла; МС (ЭИ): 221,2 (М+), 133,1 (((М-C(=O)N(CH3)OCH3)+), 100%).
б) рац-Хроман-4-ил-(1Н-имидазол-2-ил)-метанон
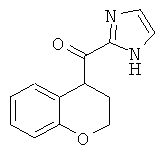
рац-Хроман-4-ил-(1Н-имидазол-2-ил)-метанон получали аналогично соединению Примера 12 (а), но с использованием рац-хроман-4-карбоновой кислоты метокси-метил-амида, с выходом бесцветной камеди; МС (ISP): 228,9 ((М+Н)+).
в) рац-2-Хроман-4-илметил-1Н-имидазол

рац-2-Хроман-4-илметил-1Н-имидазол получали из рац-хроман-4-ил-(1Н-имидазол-2-ил)-метанона путем восстановления по Вольфу-Кижнеру в соответствии с методикой, описанной Е. Reimann et al., Arch Pharm. (Weinheim) 322, 363 (1989), с выходом желтой камеди; МС (ISP): 215,1 М+Н)+).
Пример 73
рац-4-(1Н-Имидазол-2-илметил)-1,2,3,4-тетрагидро-хинолин

а) рац-1,2,3,4-Тетрагидро-хинолин-4-карбоновая кислота

рац-1,2,3,4-Тетрагидро-хинолин-4-карбоновую кислоту получали из хинолин-4-карбоновой кислоты путем обработки никелем Ренея в водном растворе гидроксида натрия в соответствии с методикой, описанной в Khimiya Geterotsiklicheskikh Soedinenii 1988, 77-9, с выходом коричневых кристаллов, 1Н-ЯМР(CDCl3): 2.04 (1H, m), 2.29 (1H, m), 3.24-3.46 (br m, 3 Н, СН2н. и NH), 3.77 (1H, t, CHCO2), 6.55 (1H, d, АrН), 6.67 (1H, dd, ArH), 7.04 (1H, dd, ArH), 7.15 (1H, d, ArH).
б) рац-1,2,3,4-Тетрагидро-хинолин-4-карбоновой кислоты метокси-метил-амид

К раствору рац-1,2,3,4-тетрагидро-хинолин-4-карбоновой кислоты (0,50 г, 2,82 ммоль) в дихлорметане (12 мл) добавляли N,O-диметилгидроксиламина гидрохлорид (0,36 г, 3,67 ммоль) и N-метилморфолин (0,40 мл 3,67 ммоль). Данную смесь охлаждали до 0°С, затем добавляли 1-этил-3-(3′-диметиламинопропил)карбодиимида гидрохлорид (EDCl) (0,70 г, 3,67 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Затем смесь концентрировали под вакуумом и остаток очищали путем хроматографии (силикагель, градиент этилацетат/гептан) с получением указанного в заголовке соединения (0,29 г, 47%) в виде желтого кристаллического твердого вещества MC(ISP): 221,4 ([М+Н]+, 100%).
в) рац-1-(Толуол-4-сульфонил)-1,2,3,4-тетрагидро-хинолин-4-карбоновой кислоты метокси-метил-амид

К раствору рац-1,2,3,4-тетрагидро-хинолин-4-карбоновой кислоты метокси-метил-амида (0,29 г, 1,32 ммоль) в 1,2-дихлорэтане (5 мл) добавляли по каплям триэтиламин (0,46 мл, 3,29 ммоль). Затем добавляли хлорангидрид пара-толуолсульфокислоты (0,33 г, 1,71 ммоль) и данную реакционную смесь нагревали при 70°С в течение 4 часов. После охлаждения до комнатной температуры смесь концентрировали под вакуумом и остаток очищали путем хроматографии (силикагель, градиент метанол/дихлорметан) с получением указанного в заголовке соединения (0,44 г, 90%) в виде коричневого кристаллического твердого вещества. MC(ISP): 375,1 ([М+Н]+, 100%).
г) рац-(1Н-Имидазол-2-ил)-[1 -(толуол-4-сульфонил)-1,2,3,4-тетрагидро-хинолин-4-ил]-метанон

К раствору 1-(диэтоксиметил)имидазола (0,21 мл, 1,29 ммоль) в тетрагидрофуране (2 мл) при -78°С добавляли по каплям 1,6 М раствор н-бутиллития (0,88 мл, 1,41 ммоль) в гексане. Полученный раствор 2-(1-диэтоксиметил-1Н-имидазол-2-ил)-лития перемешивали при -78°С и затем добавляли по каплям к раствору рац-1-(толуол-4-сульфонил)-1,2,3,4-тетрагидро-хинолин-4-карбоновой кислоты метокси-метил-амида (0,44 г, 1,18 ммоль) в тетрагидрофуране (4 мл) при 0°С Затем данную реакционную смесь перемешивали при 0°С в течение 1 ч и гасили путем добавления по каплям 2 М водного раствора соляной кислоты. Смесь подщелачивали путем добавления водного раствора бикарбоната натрия и разбавляли этилацетатом. Фазы разделяли и органическую фазу промывали насыщенным рассолом, сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Остаток очищали путем хроматографии (силикагель, градиент этилацетат/гептан) с получением указанного в заголовке соединения (0,23 г, 52%) в виде светло-желтого кристаллического твердого вещества. МС (ISP): 382,3 ([М+Н]+, 100%).
д) рац-4-(1Н-Имидазол-2-илметил)-1-(толуол-4-сульфонил)-1,2,3,4-тетрагидро-хинолин
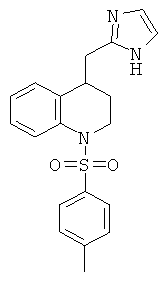
Данное соединение получали в соответствии с методикой, описанной в Arch. Pharm. 1989, 322, 363-367. К раствору рац-(1Н-имидазол-2-ил)-[1-(толуол-4-сульфонил)-1,2,3,4-тетрагидро-хинолин-4-ил]-метанона (0,23 г, 0,60 ммоль) в триэтиленгликоле (3 мл) последовательно добавляли порошок гидроксида натрия (96 мг, 2,41 ммоль) и гидрат гидразина (0,10 мл, 1,99 ммоль). Данную реакционную смесь перемешивали при 110°С в течение 1 ч и затем при 200°С в течение 2 ч. После охлаждения до комнатной температуры смесь разбавляли этилацетатом и промывали последовательно 2 н. водным раствором соляной кислоты, водным раствором бикарбоната натрия, водой и насыщенным рассолом. Фазы разделяли и органическую фазу сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Остаток очищали путем хроматографии (силикагель, градиент метанол/дихлорметан) с получением указанного в заголовке соединения (41 мг, 19%) в виде желтого кристаллического твердого вещества. МС (ISP): 368,1 ([М+Н]+, 100%).
е) рац-4-(1Н-Имидазол-2-илметил)-1,2,3,4-тетрагидро-хинолин
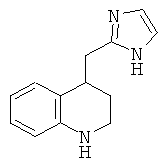
Данное соединение получали в соответствии с методикой, описанной в J. Med. Chem. 1997, 40, 105-111. К рац-4-(1Н-имидазол-2-илметил)-1-(толуол-4-сульфонил)-1,2,3,4-тетрагидро-хинолину (40 мг, 0,11 ммоль) последовательно добавляли 0,57 мл (3,27 ммоль) 33% раствора НВr в уксусной кислоте и анизол (0,06 мл, 0,54 ммоль). Данную реакционную смесь перемешивали при комнатной температуре в течение 3 ч и затем вливали в 2 н. водный раствор гидроксида натрия. Эту смесь разбавляли этилацетатом, фазы разделяли и органическую фазу промывали насыщенным рассолом. Фазы разделяли и органическую фазу сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Остаток очищали путем хроматографии (силикагель, градиент метанол/дихлорметан) с получением указанного в заголовке соединения (16 мг, 69%) в виде белой пены. МС (ISP): 214,3 ([М+Н]+, 100%).
Пример 74
рац-2-(1,2,3,4-Тетрагидро-нафталин-1-илметил)-1Н-имидазол

рац-2-(1,2,3,4-Тетрагидро-нафталин-1-илметил)-1Н-имидазол получали аналогично соединению Примера 72 (б) и (в), но с использованием рац-1,2,3,4-тетрагидро-нафталин-1-карбоновой кислоты метокси-метил-амида, с выходом бесцветного твердого вещества; МС (ISP): 213,0 М+Н)+).
Реферат
Настоящее изобретение относится к применению соединений формулы I, R1 представляет собой атом водорода, атом трития, гидрокси, низший алкил, низший алкокси, атом галогена, нитро, амино или низший алкил, замещенный атомом галогена; R представляет собой атом водорода, гидрокси или низший алкил; X представляет собой N и Y представляет собой СН, или СН2, или СН-низший алкил, или X представляет собой СН и Y представляет собой N; Q представляет собой СН2, О, NH, N-алкил, N-SO2-алкил или N-SO2-толуол-4-ил; W представляет собой СН2 или связь; m, n независимо друг от друга имеют значения 1, 2 или 3; когда m имеет значение 2 или 3, R могут быть одинаковыми или разными; когда n имеет значение 2 или 3, R1 могут быть одинаковыми или разными; каждая пунктирная линия независимо от другой пунктирной линии может представлять собой связь или не являться связью; и их фармацевтически активных солей, рацемических смесей, энантиомеров, оптических изомеров и таутомерных форм соединений формулы I в приготовлении лекарств для лечения биполярного расстройства, шизофрении, болезни Паркинсона, болезни Альцгеймера, эпилепсии, расстройств приема пищи, диабетических осложнений, дислипидемии, расстройств потребления и ассимиляции энергии, расстройств и нарушений температурного гомеостаза, нарушений сна и циркадного ритма. Также изобретение относится к применению соединения формулы IA, к соединению формулы I и лекарству на основе соединения формулы I. Технический результат: получены новые производные 2-имидазолов. обладающие полезными биологическими свойствами. 2 табл., 5 н. и 30 з.п. ф-лы.
Формула

где R1 представляет собой атом водорода, атом трития, гидрокси, низший алкил, низший алкокси, атом галогена, нитро, амино или низший алкил, замещенный атомом галогена;
R2 представляет собой атом водорода, гидрокси или низший алкил;
X представляет собой N, и Y представляет собой СН, или СН2, или СН-низший алкил, или
X представляет собой СН, и Y представляет собой N;
Q представляет собой СН2, О, NH, N-алкил, N-SO2-алкил или N-SO2-толуол-4-ил;
W представляет собой СН2 или связь;
m, n независимо друг от друга имеют значения 1, 2 или 3; когда m имеет значение 2 или 3, R2 могут быть одинаковыми или разными; когда n имеет значение 2 или 3, R1 могут быть одинаковыми или разными;
каждая пунктирная линия независимо от другой пунктирной линии может представлять собой связь или не являться связью;
и их фармацевтически активных солей, рацемических смесей, энантиомеров, оптических изомеров и таутомерных форм соединений формулы I в приготовлении лекарств для лечения биполярного расстройства, шизофрении, болезни Паркинсона, болезни Альцгеймера, эпилепсии, расстройств приема пищи, диабетических осложнений, дислипидемии, расстройств потребления и ассимиляции энергии, расстройств и нарушений температурного гомеостаза, нарушений сна и циркадного ритма.
где R1 представляет собой атом водорода, атом трития, гидрокси, низший алкил, низший алкокси, атом галогена или низший алкил, замещенный атомом галогена;
Q представляет собой СН2 или О;
n имеет значение 1, 2 или 3; когда n имеет значение 2 или 3, R1 могут быть одинаковыми или разными;
пунктирная линия может представлять собой связь или не являться связью;
и их фармацевтически активных солей, рацемических смесей, энантиомеров, оптических изомеров и таутомерных форм соединений формулы IA в приготовлении лекарств для лечения биполярного расстройства, шизофрении, болезни Паркинсона, болезни Альцгеймера, эпилепсии, расстройств приема пищи, диабетических осложнений, дислипидемии, расстройств потребления и ассимиляции энергии, расстройств и нарушений температурного гомеостаза, нарушений сна и циркадного ритма.
рац-2-(7-хлор-5-фтор-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол,
рац-2-(6-хлор-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол или
рац-2-(5-хлор-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-Н-имидазол.
рац-2-(5,7-диметил-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол или
рац-2-(5,7-диметил-1,2,3,4-тетрагидро-нафталин-1-ил)-1Н-имидазол.
рац-2-(6-метокси-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол или
рац-2-(5-метокси-1,2,3,4-тетрагидро-нафталин-1-ил)-4,5-дигидро-1Н-имидазол.
рац-2-(6,8-дихлор-хроман-4-ил)-1Н-имидазол или
рац-4-(1Н-имидазол-2-ил)-1,2,3,4-тетрагидро-хинолин.
рац-4-(1,2,3,4-тетрагидро-нафталин-1-ил)-1Н-имидазол.
рац-5-(7-метил-хроман-4-ил)-1Н-имидазол, или таутомер, или
рац-5-(5-метил-хроман-4-ил)-1Н-имидазол, или таутомер.
рац-5-(6-фтор-хроман-4-ил)-1Н-имидазол, или таутомер,
5-(8-хлор-2Н-хромен-4-ил)-1Н-имидазол, или таутомер,
5-(6-хлор-2Н-хромен-4-ил)-1Н-имидазол, или таутомер,
рац-5-(7-фтор-хроман-4-ил)-1Н-имидазол, или таутомер,
или рац-5-(5-фтор-хроман-4-ил)-1Н-имидазол, или таутомер.

где R1 представляет собой атом водорода, атом трития, низший алкил, низший алкокси, атом галогена, нитро или амино;
R2 представляет собой атом водорода или низший алкил;
X представляет собой N, и Y представляет собой СН, или СН2, или СН-низший алкил, или
X представляет собой СН, и Y представляет собой N;
Q представляет собой О, NH, N-алкил, или N-SO2-алкил, или N-SO2-толуол-ил;
W представляет собой СН2 или связь;
m, n независимо друг от друга имеют значения 1, 2;
каждая пунктирная линия независимо от другой пунктирной линии может представлять собой связь или не являться связью;
и их фармацевтически активные соли, рацемические смеси, энантиомеры, оптические изомеры и таутомерные формы, за исключением следующих соединений:
2-(7,8-диметокси-хроман-4-ил)-4,5-дигидро-1Н-имидазола,
5-хроман-4-илметил-1 Н-имидазола.
рац-2-хроман-4-ил-4,5-дигидро-1Н-имидазол,
рац-2-хроман-4-ил-1Н-имидазол или
рац-2-(6-фтор-хроман-4-ил)-1Н-имидазол.
рац-2-(6,8-дихлор-хроман-4-ил)-1Н-имидазол или
рац-4-(1Н-имидазол-2-ил)-1,2,3,4-тетрагидро-хинолин.
рац-5-(7-метил-хроман-4-ил)-1Н-имидазол, или таутомер, или
рац-5-(5-метил-хромен-4-ил)-1Н-имидазол, или таутомер.
рац-5-(6-фтор-хроман-4-ил)-1Н-имидазол, или таутомер,
5-(8-хлор-2Н-хромен-4-ил)-1Н-имидазол, или таутомер,
5-(6-хлор-2Н-хромен-4-ил)-1Н-имидазол, или таутомер,
рац-5-(7-фтор-хроман-4-ил)-1Н-имидазол, или таутомер, или
рац-5-(5-фтор-хроман-4-ил)-1Н-имидазол, или таутомер.

где R1 представляет собой атом трития;
R2 представляет собой атом водорода, гидрокси или низший алкил;
X представляет собой N, и Y представляет собой СН, или СН2, или СН-низший алкил, или
X представляет собой СН, и Y представляет собой N;
Q представляет собой СН2, О, NH, N-алкил, N-SO2-алкил или N-SO2-толуол-4-ил;
W представляет собой СН2 или связь;
m, n независимо друг от друга имеют значения 1, 2 или 3; когда m имеет значение 2 или 3, R2 могут быть одинаковыми или разными; когда n имеет значение 2 или 3, R1 могут быть одинаковыми или разными;
каждая пунктирная линия независимо от другой пунктирной линии может представлять собой связь или не являться связью;
и их фармацевтически активных солей, рацемических смесей, энантиомеров, оптических изомеров и таутомерных форм соединений формулы I в качестве радиолиганда в анализе связывания с рецепторами, ассоциированными со следовыми аминами.
Комментарии