Соединения индола в качестве ингибиторов клеточного некроза - RU2477282C2
Код документа: RU2477282C2
Описание
Область техники
Настоящее изобретение относится к соединениям индола формулы (1), их фармацевтически приемлемым солям или изомерам и к способу и композиции для профилактики или лечения клеточного некроза и связанных с некрозом заболеваний, включающим эти соединения в качестве активного ингредиента.
Уровень техники
Большинство исследований, связанных с гибелью клеток, было сконцентрировано на апоптозе клеток, также известном как запрограммированная гибель клеток (PCD). С открытием фермента каспазы в течение последних 10 лет множество фармацевтических компаний включилось в разработку лекарственных средств, в которых использовались ингибиторы каспазы. Однако текущая ситуация состоит в том, что только некоторые из этих препаратов были одобрены FDA. Причина этого заключается в том, что апоптоз клеток представляет собой гибель клеток, которая происходит при физиологических обстоятельствах, и такая гибель клеток, вероятно, является следствием защитного механизма поддержания гомеостаза в организме. Напротив, некроз представляет собой гибель клеток, которая главным образом происходит при патологических обстоятельствах, и в большинстве случаев он характеризуется сопутствующим воспалительным ответом. Некроз был известен в течение долгого времени как неконтролируемая гибель клеток, но в недавнем исследовании (Proskurykakov S.Y. et al. 2002, Biochemistry) некроз был описан как активная/контролируемая гибель клеток. Типичные заболевания, вызванные некрозом, включают ишемические (например, инфаркт миокарда, инсульт, инфаркт почки), нейродегенеративные и воспалительные заболевания. Так как считается, что некроз является неконтролируемой, случайной гибелью клеток при патологических обстоятельствах, исследования относительно функционального механизма, молекулярных мишеней, систем трансдукции сигналов и т.д. проводились редко. Таким образом, существует серьезная потребность в открытии и разработке ингибирующих некроз веществ для лечения ишемических, нейродегенеративных и воспалительных заболеваний, которые вызываются некрозом, и в объяснении биологических, патологических причин некроза.
Производные индола согласно настоящему изобретению имеют очень полезные структуры с медицинской точки зрения, и во многих публикациях сообщалось о результатах исследований в отношении этих структур. Среди результатов исследований следующее является наиболее репрезентативным: в заявке на патент WO 2006/112549 сообщается о некоторых производных индола, имеющих активность в отношении глюкокиназы, в заявке на патент WO95/07276 сообщается о соединениях, которые могут быть использованы как противоопухолевые средства и как ингибиторы против продукции сердечно-сосудистой системы, и в заявке на патент WO 2004/018428 сообщается о соединениях, которые могут быть использованы в качестве антибиотиков.
Подробное описание изобретения
Решаемая техническая задача
Таким образом, авторы настоящего изобретения провели обширные исследования в этой области, чтобы разработать новые соединения, которые проявляли бы эффект в отношении профилактики или лечения и улучшения в случае клеточного некроза и связанных с некрозом заболеваний, особенно пригодные для профилактики или лечения заболеваний печени. В результате этого они подтвердили, что производные индола формулы (1), раскрытые ниже, проявляют превосходящий эффект в отношении профилактики и лечения клеточного некроза и связанных с некрозом заболеваний, посредством чего осуществили настоящее изобретение.
Поэтому цель настоящего изобретения состоит в том, чтобы получить новые производные индола формулы (1).
Другой целью настоящего изобретения является получение композиции для профилактики или лечения клеточного некроза и связанных с некрозом заболеваний, в частности для гепатопротекции, функционального улучшения состояния печени и профилактики или лечения острых/хронических заболеваний печени, которая содержит в качестве активного ингредиента соединения формулы (1), их фармацевтически приемлемые соли или изомеры вместе с фармацевтически приемлемым носителем или разбавителем, и способ ее получения.
Другой целью настоящего изобретения является разработка способа профилактики или лечения клеточного некроза и связанных с некрозом заболеваний, в частности гепатопротекции, функционального улучшения состояния печени и профилактики или лечения острых/хронических заболеваний печени с использованием указанной композиции.
Для решения указанных задач настоящее изобретение относится к соединениям индола следующей формулы (1):

в которой
n обозначает число от 0 до 3,
А обозначает 5-членный гетероарил или гетероцикл, каждый из которых имеет от 1 до 3 гетероатомов, выбранных из N, O и S,
R1 обозначает R5-X-B-X'-,
B обозначает прямую связь, или обозначает 3~10-членный гетероцикл или гетероарил, каждый из которых имеет от 1 до 4 гетероатомов, выбранных из N, O и S,
X и X' независимо от друг друга обозначают прямую связь, или выбраны из группы, состоящей из -NR6-, -CO-, -CONR6-, -CO2-, -OC(O)-, -S(O)m-, -O-(CH2)m-, -(CH2)m-O-, -(CH2)m-, -NR6CO-, -(R6O)2P(O)- и -NHCO2-, где m обозначает число от 0 до 3, и R6 обозначает водород, алкил или циклоалкил,
R5 обозначает водород, нитрил, гидрокси, алкил, алкокси, циклоалкил или арил, или обозначает 3~10-членный моноциклический или конденсированный циклический гетероцикл или гетероарил, каждый из которых имеет от 1 до 3 гетероатомов, выбранных из N, O и S, и может быть замещен оксо или алкилом, или
R5 и R6 могут вместе образовывать 4~8-членный цикл,
R2 обозначает -(CR8R9)p-Y-R7,
p обозначает число от 0 до 2,
R8 и R9 независимо друг от друга обозначают водород или алкил, или могут вместе образовывать 4~8-членный цикл,
Y обозначает прямую связь, или выбран из группы, состоящей из -O-, -S-, -NR6-, -NR6C(O)-, -CO2-, -C(O)-, -C(O)NR6-, -S(O)q- и -S(O)qNR6-, где q обозначает число от 0 до 2,
R7 обозначает водород, галоген, циано, гидрокси, нитро, алкил, циклоалкил или арил, или обозначает 3~10-членный гетероцикл или гетероарил, каждый из которых имеет от 1 до 3 гетероатомов, выбранных из N, S и O, и который в случае необходимости содержит оксо,
R3 обозначает водород, алкил, -(CH2)q-циклоалкил или -(CH2)q-гетероцикл,
R4 обозначает -(CH2)p-D-R10,
D обозначает прямую связь, обозначает циклоалкил, в случае необходимости содержащий оксо, обозначает арил, или обозначает 3~10-членный гетероцикл или гетероарил, каждый из которых имеет от 1 до 3 гетероатомов, выбранных из N, S и O,
R10 обозначает водород, галоген, амино, циано, нитро, гидрокси, алкил, алкилкарбонил, алкилсульфонил или -(CH2)p-NR8R9,
где алкил, алкокси, арил, циклоалкил, гетероцикл и гетероарил могут быть в случае необходимости замещены, и заместители представляют собой один или более заместителей, выбранных из группы, состоящей из гидрокси, галогена, нитрила, амино, алкиламино, диалкиламино, алкил, галогеналкил, алкилсульфонил, карбоксиалкил, алкилкарбонилокси, алкилтио, алкилоксикарбонил, алкиламинокарбонил, арилалкокси и оксо, и к их фармацевтически приемлемым солям или изомерам.
В приведенных выше определениях для соединений формулы (1), термин 'алкил' означает алифатический углеводородный радикал. Алкил может быть насыщенным алкилом, который не включает алкенильную или алкинильную группу, или ненасыщенным алкилом, который включает по меньшей мере одну алкенильную или алкинильную группу. "Алкенил" означает группу, содержащую по меньшей мере одну углерод-углеродную двойную связь, и “алкинил” означает группу, содержащую по меньшей мере одну углерод-углеродную тройную связь. Алкил может иметь разветвленную или прямую цепь, когда используется индивидуально или в сложной форме, такой как алкокси.
Алкильная группа может иметь от 1 до 20 атомов углерода, если не указано иное. Алкильная группа может быть среднеразмерным алкилом, имеющим от 1 до 10 атомов углерода. Иначе алкильная группа может быть низшим алкилом, имеющим от 1 до 6 атомов углерода. Типичные примеры включают, но не ограничены ими, метил, этил, пропил, изопропил, н-бутил, изобутил, трет-бутил, пентил, гексил, винил, пропенил, бутенил и т.д. Например, C1-C4-алкил имеет от 1 до 4 атома углерода в алкильной цепи и выбран из группы, состоящей из метила, этила, пропила, изопропила, н-бутила, изобутила, втор-бутила и трет-бутила.
Термин 'алкокси' означает алкилокси, имеющий от 1 до 10 атомов углерода, если не указано иное.
Термин 'циклоалкил' означает насыщенный алифатический 3~10-членный цикл, если не указано иное. Типичные примеры включают, но не ограничены ими, циклопропил, циклобутил, циклопентил, циклогексил и т.д.
Термин 'арил' включает по меньшей мере одно кольцо, имеющее ковалентную π электронную систему, например, моноциклические или конденсированные полициклические (т.е. циклы, имеющие общие пары смежных атомов углерода) группы. В настоящем описании «арил» обозначает ароматический 4~10-членный, предпочтительно 6~10-членный, моноциклический или полициклический цикл, включая фенил, нафтил и т.д., если не указано иное.
Термин 'гетероарил' означает ароматический 3~10-членный, предпочтительно 4~8-членный, более предпочтительно 5~6-членный цикл, который имеет от 1 до 4 гетероатомов, выбранных из N, O и S, и который может быть конденсирован с бензо или C3-C8 циклоалкилом, если не указано иное. Моноциклический гетероарил включает, но не ограничен ими, тиазол, оксазол, тиофен, фуран, пиррол, имидазол, изоксазол, изотиазол, пиразол, триазол, триазин, тиадиазол, тетразол, оксадиазол, пиридин, пиридазин, пиримидин, пиразин и т.п. Бициклический гетероарил включает, но не ограничен ими, индол, индолин, бензотиофен, бензофуран, бензимидазол, бензоксазол, бензизоксазол, бензотиазол, бензотиадиазол, бензотриазол, хинолин, изохинолин, пурин, пуропиридин и т.п.
Термин 'гетероцикл' означает 3~10-членный, предпочтительно 4~8-членный, более предпочтительно 5~6-членный цикл, который имеет от 1 до 4 гетероатомов, выбранных из N, O и S, который может быть конденсирован с бензо или C3-C8 циклоалкилом и является насыщенным или содержит 1 или 2 двойных связи, если не указано иное. Гетероцикл включает, но не ограничен ими, пирролин, пирролидин, имидазолин, имидазолидин, пиразолин, пиразолидин, пиран, пиперидин, морфолин, тиоморфолин, пиперазин, гидрофуран и т.п.
Другие термины и сокращения в настоящем описании имеют значения, обычно используемые специалистом в этой области, если не указано иное.
Предпочтительными соединениями среди соединений формулы (1), описанных выше, являются соединения, в которых
n обозначает число от 0 до 3,
А обозначает 5-членный гетероарил или гетероцикл, каждый из которых имеет от 1 до 3 гетероатомов, выбранных из N, O и S,
R1 обозначает R5-X-B-X'-,
B обозначает прямую связь, или обозначает 3~10-членный гетероцикл или гетероарил, каждый из которых имеет от 1 до 4 гетероатомов, выбранных из N, O и S,
X и X' независимо от друг друга обозначают прямую связь, или выбраны из группы, состоящей из -NR6-, -CO-, -CONR6-, -CO2-, -OC(O)-, -S(O)2-, -O-(CH2)m-, -(CH2)m-O-, -(CH2)m-, -NR6CO-, -(R6O)2P(O)- и -NHCO2-, где m обозначает число от 0 до 3, и R6 обозначает водород, C1-C6-алкил или C3-C6-циклоалкил,
R5 обозначает водород, нитрил, гидрокси, C1-C6-алкил, галоген-C1-C6-алкил, гидрокси-C1-C6-алкил, C4-C6-циклоалкил, фенил или галогенфенил, или обозначает 5~10-членный моноциклический или конденсированный циклический гетероцикл или гетероарил, каждый из которых имеет от 1 до 3 гетероатомов, выбранных из N, O и S, и может быть замещен оксо или галоген-C1-C6-алкилом, или
R5 и R6 могут вместе образовывать 4~8-членный цикл,
R2 обозначает -(CR8R9)p-Y-R7,
p обозначает число от 0 до 2,
R8 и R9 независимо друг от друга обозначают водород или C1-C6-алкил, или могут вместе образовывать 5~6-членный цикл,
Y обозначает прямую связь, или выбран из группы, состоящей из -O-, -NR6-, -NR6C(O)-, -C(O)-, -CO2-, -C(O)NR6- и -S(O)q-, где q обозначает число от 0 до 2,
R7 обозначает водород, галоген, циано, гидрокси, C1-C6-алкил, гидрокси-C1-C6-алкил или галоген-C1-C6-алкил, обозначает фенил, в случае необходимости замещенный C1-C6-алкилсульфонилом, или обозначает 5~6-членный гетероцикл или гетероарил, каждый из которых имеет от 1 до 3 гетероатомов, выбранных из N и O,
R3 обозначает водород, C1-C6-алкил, -(CH2)-C3-C6-циклоалкил или -(CH2)-гетероцикл, причем гетероцикл представляет собой 5~6-членный цикл, имеющий 1 или 2 гетероатома, выбранных из N, O и S,
R4 обозначает -(CH2)p-D-R10,
D обозначает прямую связь, обозначает C3-C6-циклоалкил, в случае необходимости содержащий оксо, или обозначает 5~6-членный гетероцикл или гетероарил, каждый из которых имеет 1 или 2 гетероатома, выбранных из N, O и S,
R10 обозначает водород, галоген, амино, C1-C6-алкил, C1-C6-алкилкарбонил, галоген-C1-C6-алкилкарбонил, C1-C6-алкилсульфонил или -(CH2)p-NR8R9.
В соединениях формулы (1) согласно настоящему изобретению А более предпочтительно обозначает цикл, который может быть представлен одной из следующих формул (i)-(viii), в которых R обозначает водород, или обозначает C1-C4-алкил, в случае необходимости замещенный гидрокси или амино.

A наиболее предпочтительно выбран из группы, состоящей из 4,5-дигидро-тиазола, тиазола, оксазолина, оксадиазола и изоксадиазола.
В формуле R5-X-B-X'- для R1 B более предпочтительно обозначает прямую связь, обозначает имидазол или оксадиазол, или обозначает 5~6-членный гетероцикл, имеющий 1 или 2 гетероатома, выбранных из N и O, и наиболее предпочтительно обозначает структуру, которая может быть представлена одной из следующих формул (ix)-(xii).

X более предпочтительно обозначает прямую связь или выбран из группы, состоящей из -CO-, -CONR6-, -CO2-, -SO2-, -(CH2)m- и -O-(CH2)m-, где m обозначает число от 0 до 2, и R6 обозначает водород, C1-C6-алкил или C3-C6-циклоалкил. Наиболее предпочтительно X выбран из группы, состоящей из -CO-, -CONH-, -CO2-, -SO2-, -(CH2)2-, -O- и -O-CH2-.
X' более предпочтительно обозначает прямую связь, или выбран из группы, состоящей из -(CH2)2-, -NH-, -CO-, -CO2-, -CONH-, -S(O)2-, -(R6O)2P(O)-, -NHC(O)- и -NHCO2-.
R5 более предпочтительно обозначает водород, нитрил, гидрокси, C1-C6-алкил, галоген-C1-C6-алкил, гидрокси-C1-C6-алкил, C4-C6-циклоалкил, фенил или галогенфенил, или обозначает моноциклический или конденсированный циклический 5~9-членный гетероцикл или 5~6-членный гетероарил, каждый из которых имеет от 1 до 3 гетероатомов, выбранных из N, O и S, и может быть замещен оксо или трифторметилом. Наиболее предпочтительно, R5 выбран из группы, состоящей из водорода, нитрила, гидрокси, метила, этила, изопропила, изобутила, гидроксиметила, трифторметила, циклобутила, циклопентила, циклогексила, пирролидина, пиперидина, 2-оксопиперазина, 2-оксопирролидина, тетрагидрофурана, тетрагидропирана, тетрагидротиопирана, морфолина, фурана, пиридина, 1,3-пиразина, 1,1-диоксо-тиоморфолина, тетразола, имидазола, пиразола и 3-трифторметил-5,6,7,8-тетрагидро-2H-[1,2,4]триазоло[4,3-a]пиразина.
В формуле -(CR8R9)p-Y-R7 для R2, R8 и R9, каждый, более предпочтительно обозначает водород.
Y более предпочтительно выбран из группы, состоящей из -O-, -NR6-, -NR6C(O)-, -C(O)-, -C(O)NR6- и -S(O)2-, где R6 имеет значения, определенные выше в предпочтительных значениях. Наиболее предпочтительно, Y выбран из группы, состоящей из -O-, -NH-, -NHC(O)-, -SO2- и -C(O)-.
R7 более предпочтительно обозначает водород, галоген, гидрокси, C1-C6-алкил, гидроксиметил или галоген-C1-C6-алкил, обозначает фенил, в случае необходимости замещенный C1-C6-алкилсульфонилом, или обозначает 5~6-членный гетероцикл или гетероарил, каждый из которых имеет 1 или 2 гетероатома, выбранных из N и O. Наиболее предпочтительно, R7 выбран из группы, состоящей из водорода, брома, фтора, хлора, метила, этила, пропила, гидроксиметила, трифторметила, фенила, 4-метилсульфонил-фенила, пиперидина, пирролидина, фурана, пиррола, пиразола и пиридина.
R3 более предпочтительно обозначает водород, метил или изобутил.
R4 более предпочтительно обозначает -R10, -D-R10, или -CH2-D-R10, причем D обозначает C3-C6-циклоалкил, в случае необходимости содержащий оксо, обозначает 5~6-членный гетероцикл, имеющий 1 или 2 гетероатома, выбранных из N, O и S, или обозначает 5~6-членный гетероарил, имеющий 1 или 2 гетероатома, выбранных из N и S, и R10 обозначает водород, галоген, амино, C1-C6-алкил, C1-C3-алкилкарбонил, галоген-C1-C3-алкилкарбонил, C1-C3-алкилсульфонил или -(CH2)p-NR8R9, где p, R8 и R9 имеют значения, определенные выше в предпочтительных значениях. Наиболее предпочтительно, R4 выбран из группы, состоящей из водорода, изопропила, изобутила, циклопропилметила, циклопентилметила, циклобутила, циклопентила, циклогексила, 4-метил-циклогексила, 4,4-дифторциклогексила, 4-оксо-циклогексила, тетрагидропиран-4-ила, (тетрагидропиран-4-ил)метила, (тетрагидропиран-2-ил)метила, тетрагидрофуран-3-ила, пиперидин-4-ила, метансульфонила, 1-ацетил-пиперидин-4-ила, 1-метансульфонил-пиперидин-4-ила, 1-трифторацетил-пиперидин-4-ила, 1-ацетил-пирролидин-3-ила, тетрагидротиопиран-4-ила, тиофен-3-ила и 5-амино-пиридин-2-ила.
Соединения формулы (1) согласно настоящему изобретению могут также образовывать фармацевтически приемлемую соль. Такая “фармацевтически приемлемая соль” включает нетоксичную соль присоединения с кислотой, содержащую фармацевтически приемлемый анион, например, соль с неорганическими кислотами, такими как серная кислота, соляная кислота, азотная кислота, фосфорная кислота, бромистоводородная кислота, йодистоводородная кислота и т.д.; соль с органическими карбоновыми кислотами, такими как винная кислота, муравьиная кислота, лимонная кислота, уксусная кислота, трихлоруксусная кислота, трифторуксусная кислота, глюконовая кислота, бензойная кислота, молочная кислота, фумаровая кислота, малеиновая кислота, салициловая кислота и т.д.; или соль с сульфоновыми кислотами, такими как метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота, нафталинсульфоновая кислота и т.д. Соединения формулы (I) могут также образовывать фармацевтически приемлемую соль присоединения с основанием, например, соль со щелочными металлами или щелочноземельными металлами, такими как литий, натрий, калий, кальций, магний и т.д.; соль с аминокислотами, такими как лизин, аргинин, гуанидин и т.д.; или органическую соль с дициклогексиламином, N-метил-D-глюкамином, трис(гидроксиметил)метиламином, диэтаноламином, холином, триэтиламином и т.д. Соединения формулы (I) согласно настоящему изобретению могут быть преобразованы в их соли согласно любому из обычных способов, и образование соли может быть легко выполнено специалистом на основании структуры формулы (1) без дополнительных объяснений.
Термин 'изомер' в настоящем описании означает такие, которые имеют ту же самую химическую или молекулярную формулу, как и соединение формулы (1), но оптически или пространственно отличаются от соединения формулы (1) или его солей. Соединения формулы (1) согласно настоящему изобретению могут иметь асимметрический углеродный центр (центры) в структуре, и, таким образом, могут существовать в форме оптического изомера (изомера R или S), рацемата, смеси диастереомеров, или индивидуального диастереомера и т.д. Когда соединения имеют двойную связь, они могут существовать в форме геометрического изомера (транс- или цис-изомер). Все изомеры и их смеси также охвачены настоящим изобретением.
В дальнейшем описании соединения формулы (1) включают их фармацевтически приемлемые соли и изомеры, если не указано иное. Следует понимать, что соли и изомеры охвачены настоящим изобретением. Для удобства они указаны в настоящем описании как соединения формулы (1).
Типичные соединения среди соединений формулы (1) выбраны из следующих соединений:
Циклопентил-[2-(4,5-дигидро-1,3-тиазол-2-ил)-1Н-индол-7-ил]амин;
[(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-1,3-тиазол-4-ил]метанол;
метиловый эфир [(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты;
[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этанол;
(R)-2-[7-циклопентиламино-5-(гидроксиметил)-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-илметанол;
[2-(4,5-Дигидро-тиазол-2-ил)-1Н-индол-7-ил]пиперидин-4-иламин;
[(R)-2-(5-метил-7-(тетрагидропиран-4-ил)метиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(S)-2-(5-метил-7-(1-трифторацетилпиперидин-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
2-[(S)-2-(7-(тетрагидропиран-2-ил)метиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(S)-2-(7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(S)-2-(7-(тетрагидропиран-4-ил)метиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(S)-2-(5-метил-7-(1-ацетилпирролидин-3-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(S)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(R)-2-(5-фенокси-7-(тетрагидропиран-4-ил)метиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(S)-2-(5-фенокси-7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(S)-2-(5-метил-7-(4,4-дифторциклогексан-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(S)-2-(5-хлор-7-(тетрагидропиран-4-ил)метиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(S)-2-(5-метил-7-циклобутиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(S)-2-(5-метил-7-(тетрагидрофуран-3-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(S)-2-(5-метил-7-циклопропилметиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(S)-2-(5-метил-7-(тетрагидропиран-4-ил)метиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(S)-2-(5-метил-7-циклопентилметиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(S)-2-(5-метил-7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этанол;
[(S)-2-(5-метил-7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(S)-2-(5-метил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
(тиофен-3-ил)метил-[2-(4,5-дигидро-1,3-тиазол-2-ил)-1Н-индол-7-ил]амин;
(3-тетрагидрофуран)-[2-(4,5-дигидро-1,3-тиазол-2-ил)-1Н-индол-7-ил]амин;
[(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-1,3-тиазол-4-ил]метанол;
3-[(R)-2-(7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовая кислота;
[(R)-2-(5-хлор-7-изопропиламино-1Н-индол-2-ил)-4,5-дигидро-1,3-тиазол-4-ил]метанол;
(тетрагидропиран-4-ил)-[2-(4,5-дигидро-4-метил-тиазол-2-ил)-1Н-индол-7-ил]амин;
[(R)-2-(5-(морфолин-4-ил)метил-7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-(диметиламино)метил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-(пиррол-3-ил)метил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-(1,3-имидазол-1-ил)метил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-(пиразол-1-ил)метил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-ацетиламино-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-феноксиметил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
метиловый эфир [(R)-2-(5-(пирролидин-1-ил)метил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты;
Циклопентил-[5-хлор-2-((R)-4-изобутил-4,5-дигидро-тиазол-2-ил)-1Н-индол-7-ил)амин;
[(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовой кислоты этиловый эфир;
{(R)-2-[7-((3R)-1-ацетилпирролидин-3-ил)амино-1Н-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил}метанол;
Циклопентил-[5-фтор-2-((R)-4-этил-4,5-дигидро-тиазол-2-ил)-1Н-индол-7-ил]амин;
{(R)-2-[7-(метил-циклопентил)амино-5-фтор-1Н-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил}метанол;
метиловый эфир [(S)-2-(5-этокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты;
[(S)-2-(5-этокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(S)-2-(5-этокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этанол;
этиловый эфир [(R)-2-(5-метил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]карбоновой кислоты;
[(S)-2-(5-фенокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(R)-2-(7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(7-(тетрагидрофуран-3-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(7-(1-(метансульфонил)пирролидин-3-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-фтор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-фтор-7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этанол;
[(R)-2-(5-хлор-7-(тетрагидротиопиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-бром-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-бром-7-(тетрагидропиран-4-ил)-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-метокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-(пиридин-3-ил)окси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-(пиридин-3-ил)окси-7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-метансульфонилметил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-(изоиндол-1,3-дион-2-ил)метил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
метиловый эфир [(R)-2-(5-хлор-7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты;
[(R)-2-(5-хлор-7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(R)-2-(5-хлор-7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этанол;
[(R)-2-(5-бром-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(R)-2-(5-бром-7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этанол;
[(R)-2-(5-бром-7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(R)-2-(5-фтор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
этиловый эфир [(R)-2-(5-фтор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты;
[(R)-2-(5-фтор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этанол;
[(R)-2-(5-фтор-7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
этиловый эфир [(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты;
[(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этанол;
[(R)-2-(7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]карбоновая кислота;
[(R)-2-(7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этанол;
метиловый эфир [(R)-2-(5-метокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты;
[(R)-2-(5-метокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(R)-2-(5-метокси-7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(R)-2-(5-этокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(R)-2-(5-пропилокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(R)-2-(5-фенокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(R)-2-(5-фенокси-7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
метиловый эфир [(R)-2-(5-(пиридин-3-ил)окси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты;
[(R)-2-(5-(пиридин-3-ил)окси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
метиловый эфир [(R)-2-(5-(пиридин-3-ил)окси-7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты;
[(R)-2-(5-(пиридин-3-ил)окси-7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
метиловый эфир [(R)-2-(5-метил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты;
[(R)-2-(5-метил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(R)-2-(5-метил-7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(R)-2-(5-(4-(метансульфонил)фенокси)-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
метиловый эфир [(R)-2-(5-феноксиметил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты;
метиловый эфир [(R)-2-(5-фениламинометил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты;
метиловый эфир [(R)-2-(5-метансульфонилметил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты;
[(R)-2-(5-метансульфонилметил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(R)-2-(5-метансульфонилметил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этанол;
[(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этилацетамид;
3-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовая кислота;
[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропанол;
3-[(R)-2-(5-хлор-7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовая кислота;
3-[(R)-2-(5-хлор-7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропанол;
этиловый эфир 3-[(R)-2-(5-метил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовой кислоты;
3-[(R)-2-(5-метил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовая кислота;
3-[(R)-2-(5-фенокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовая кислота;
этиловый эфир 3-[(R)-2-(5-фтор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовой кислоты;
3-[(R)-2-(5-фтор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовая кислота;
этиловый эфир 3-[(R)-2-(5-бром-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовой кислоты;
3-[(R)-2-(5-бром-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовая кислота;
3-[(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовая кислота;
3-[(R)-2-(5-метокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовая кислота;
этиловый эфир 3-[(R)-2-(5-этокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовой кислоты;
3-[(R)-2-(5-этокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовая кислота;
этиловый эфир 3-[(R)-2-(5-трифторметокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовой кислоты;
3-[(R)-2-(5-трифторметокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовая кислота;
[(R)-2-(5-метил-7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-оксазол-4-ил]этанол;
[(S)-2-(5-метил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-оксазол-4-ил]уксусная кислота;
[(S)-2-(5-метил-7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил)-4,5-дигидро-оксазол-4-ил]уксусная кислота;
[(S)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-оксазол-4-ил]уксусная кислота;
[(S)-2-(5-хлор-7-(тетрагидропиран-4-ил)метиламино-1Н-индол-2-ил)-4,5-дигидро-оксазол-4-ил]уксусная кислота;
[2-((4S,5R)-5-аминометил-4-бензил-дигидро-оксазол-2-ил)-5-хлор-1Н-индол-7-ил]циклопентил-амин;
{2-[(R)-5-((S)-1-амино-2-фенил-этил)-4,5-дигидро-оксазол-2-ил]-5-хлор-1Н-индол-7-ил}циклопентил-амин;
(тетрагидропиран-4-ил)-[2-(4,5-дигидро-оксазол-2-ил)-1Н-индол-7-ил]амин;
[2-(7-(тетрагидропиран-4-ил)метиламино-1Н-индол-2-ил)-4,5-дигидро-изоксадиазол-4-ил]уксусная кислота;
[2-(7-(тетрагидропиран-4-ил)метиламино-1Н-индол-2-ил)-4,5-дигидро-изоксадиазол-4-ил]этанол;
Циклопентил-[2-(4,5-дигидро-оксадиазол-2-ил)-1Н-индол-7-ил]амин;
[2-(5-метил-7-циклопентиламино-1Н-индол-2-ил)тиазол-4-ил]метанол;
[2-(5-метил-7-циклопентиламино-1Н-индол-2-ил)тиазол-5-ил]метанол;
этиловый эфир [2-(5-метил-7-циклопентиламино-1Н-индол-2-ил)тиазол-4-ил]карбоновой кислоты;
[2-(5-метил-7-циклопентиламино-1Н-индол-2-ил)тиазол-4-ил]карбоновая кислота;
[2-(7-Циклопентиламино-1Н-индол-2-ил)тиазол-4-ил]метанол;
метиловый эфир [2-(7-Циклопентиламино-1Н-индол-2-ил)тиазол-4-ил]карбоновой кислоты;
{(R)-2-[5-метил-7-(4-оксо-циклогексиламино)-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-ил}уксусная кислота;
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-морфолин-4-илэтанон;
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-(морфолин-4-ил)этиламино-этанон;
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-(морфолин-4-ил)пропиламино-этанон;
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-метиламино-этанон;
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-диметиламино-этанон;
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-[4-(метил)пиперазин-1-ил]этанон;
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-(3-диметиламинопирролидин-1-ил)этанон;
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-(пиперидин-4-ил)этанон;
2-[(R)-2-(5-хлор-7-(тетрагидропиран-4-ил)метиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-метиламино-этанон;
2-[(R)-2-(5-хлор-7-(тетрагидропиран-4-ил)метиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-(морфолин-4-ил)этанон;
2-[(R)-2-(5-фтор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-(4-(метил)пиперазин-1-ил)этанон;
2-[(R)-2-(5-фтор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-(морфолин-4-ил)этиламино-этанон;
2-[(R)-2-(5-метокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-(метиламино)-4-илэтанон;
2-[(R)-2-(5-метокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-(морфолин-4-ил)этанон;
2-[(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-метиламино-этанон;
2-[(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-(морфолин-4-ил)этанон;
2-[(R)-2-(7-(тетрагидропиран-4-ил)метиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-метиламино-этанон;
2-[(R)-2-(7-(тетрагидропиран-4-ил)метиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-(морфолин-4-ил)этанон;
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-(морфолин-4-ил)этиламино-этанон;
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-(4-(метил)пиперазин-1-ил)этанон;
Циклопентил-{5-метансульфонилметил-2-[(R)-4-(2-морфолин-4-ил-этил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}амин;
1-(4-{2-[(R)-2-(7-циклопентиламино-5-метансульфонилметил-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-ил)этанон;
Циклопентил-[2-((R)-4-пирролидин-1-илметил-4,5-дигидро-тиазол-2-ил)-1Н-индол-7-ил]амин;
{5-хлор-2-[(R)-4-(2-диметиламино-этил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}циклопентил-амин;
{5-хлор-2-[(R)-4-(2-пиперазин-1-илэтил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}циклопентил-амин;
(5-хлор-2-{(R)-4-[2-(4-этансульфонил-пиперазин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
1-(4-{2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-ил)-2-гидрокси-этанон;
{5-хлор-2-[(R)-4-(2-пиразол-1-ил-этил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}циклопентил-амин;
(S)-1-{2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пирролидин-2-карбоновая кислота;
{5-хлор-2-[(R)-4-(2-метансульфонил-этил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}циклопентил-амин;
этиловый эфир 3-{2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}-5-метил-3H-имидазол-4-карбоновой кислоты;
3-{2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}-5-метил-3H-имидазол-4-карбоновая кислота;
1-(2-{(R)-2-[5-хлор-7-(тетрагидро-пиран-4-иламино)-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-ил}этил)пиперидин-3-карбоновая кислота;
трет-бутиловый эфир [(S)-1-(2-{(R)-2-[5-хлор-7-(тетрагидро-пиран-4-иламино)-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-ил}этил)пирролидин-3-ил]карбаминовой кислоты;
(2-{(R)-4-[2-((S)-3-амино-пирролидин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-5-хлор-1Н-индол-7-ил)-(тетрагидро-пиран-4-ил)амин;
N-[(S)-1-(2-{(R)-2-[5-хлор-7-(тетрагидро-пиран-4-иламино)-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-ил}этил)пирролидин-3-ил]ацетамид;
Циклопентил-{2-[(R)-4-(2-метокси-этил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}амин;
[2-((R)-4-аминометил-4,5-дигидро-тиазол-2-ил)-1Н-индол-7-ил]циклопентил-амин;
{2-[(R)-4-((R)-3-амино-пирролидин-1-илэтил)-4,5-дигидро-тиазол-2-ил]-5-хлор-1Н-индол-7-ил}циклопентил-амин;
4-[(R)-2-(7-циклопентиламино-5-этокси-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-илэтил]пиперазин-2-он;
{2-[(R)-4-((S)-3-амино-пирролидин-1-илэтил)-4,5-дигидро-тиазол-2-ил]-5-хлор-1Н-индол-7-ил}циклопентил-амин;
(5-хлор-2-{(S)-4-[2-(3-диметиламино-фенил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
1-(4-{2-[(S)-2-(7-циклопентиламино-5-хлор-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-ил)этанон;
1-(4-{2-[(S)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-ил)этанон;
(5-метокси-2-{(R)-4-[2-(пирролидин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(2-{(R)-4-[(пирролидин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-(тетрагидро-пиран-4-ил)амин;
(2-{(S)-4-[(2-оксопиперазин-4-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-(тетрагидро-пиран-4-ил)амин;
(5-метансульфонилметил-2-{(S)-4-[(2-оксопиперазин-4-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-метил-2-{(S)-4-[(морфолин-4-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-(тетрагидро-пиран-4-ил)амин;
1-(4-{2-[(R)-2-(7-циклопентиламино-5-хлор-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-ил)этанон;
(5-хлор-2-{(R)-4-[4-метил-пиперазин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-хлор-2-{(R)-4-[4-(гидрокси)пиперидин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-хлор-2-{(R)-4-[2-оксопиперазин-4-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-хлор-2-{(R)-4-[(пиперидин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-хлор-2-{(R)-4-[(1,1-диоксо-тиоморфолин-4-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-хлор-2-{(R)-4-[(2-оксопирролидин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-хлор-2-{(R)-4-[((3S)-3-(диметиламинокарбокси)пиперидин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-(тетрагидро-пиран-4-ил)амин;
(5-хлор-2-{(R)-4-[(пиперазин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-(тетрагидро-пиран-4-ил)амин;
1-(4-{2-[(R)-2-(5-хлор-7-(тетрагидро-пиран-4-иламино)-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-ил)-2-гидрокси-этанон;
(5-хлор-2-{(R)-4-[(1-(трифторацетил)пиперазин-4-ил)этил]4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-(тетрагидро-пиран-4-ил)амин;
(5-хлор-2-{(R)-4-[(1-[(фуран-2-ил)карбонил]пиперазин-4-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-(тетрагидро-пиран-4-ил)амин;
(5-хлор-2-{(R)-4-[(1,4-пиразин-2-ил)пиперазин-4-ил-этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-(тетрагидро-пиран-4-ил)амин;
(5-хлор-2-{(R)-4-[(1,3-пиразин-2-ил)пиперазин-4-илэтил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-(тетрагидро-пиран-4-ил)амин;
(5-Фтор-2-{(R)-4-(2-аминоэтил)-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
1-(4-{2-[(R)-2-(5-фтор-7-циклопентиламино)-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-ил)-2-гидрокси-этанон;
(5-Фтор-2-{(R)-4-[(морфолин-4-ил)-этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-Фтор-2-{(R)-4-(2-диметиламино-этил)-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-Фтор-2-{(R)-4-[(пирролидин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-Фтор-2-{(R)-4-[(1,1-диоксо-тиоморфолин-4-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-Фтор-2-{(R)-4-[(2-оксопирролидин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
1-(4-{2-[(5-фтор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-ил)-2-гидрокси-этанон;
(5-Фтор-2-{(R)-4-[метансульфонил-этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-Фтор-2-{(R)-4-(2-диметиламино-этил)-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-(тетрагидро-пиран-4-ил)амин;
(5-Фтор-2-{(R)-4-[(пирролидин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-(тетрагидро-пиран-4-ил)амин;
(5-Фтор-2-{(R)-4-[(морфолин-4-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-(тетрагидро-пиран-4-ил)амин;
1-(4-{2-[(R)-2-(7-(тетрагидро-пиран-4-иламино)-5-фтор-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-ил)этанон;
(5-Фтор-2-{(R)-4-[(1,1-диоксо-тиоморфолин-4-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-(тетрагидро-пиран-4-ил)амин;
(5-Фтор-2-{(R)-4-[2-метансульфонил-этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-(тетрагидро-пиран-4-ил)амин;
(5-Фтор-2-{(R)-4-[2-оксопиперазин-4-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-(тетрагидро-пиран-4-ил)амин;
1-(4-{2-[(R)-2-(5-фтор-7-(тетрагидро-пиран-4-иламино)-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-ил)-2-гидрокси-этанон;
(2-{(R)-4-[2-диметиламино-этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(2-{(R)-4-[(пиперидин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
2-{(R)-4-[2-метансульфонил-этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-(тетрагидро-пиран-4-ил)амин;
1-(4-{2-[(R)-2-(5-метокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-ил)-2-гидрокси-этанон;
1-(4-{2-[(R)-2-(7-циклопентиламино)-5-хлор-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропил}пиперазин-1-ил)этанон;
2-{(R)-4-[(морфолин-4-ил)метил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-хлор-2-{(R)-4-[(морфолин-4-ил)пропил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(2-{(R)-4-[2-диметиламино-метил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-хлор-2-{(S)-4-[(морфолин-4-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
{5-метил-2-[(R)-4-(2-морфолин-4-илэтил)-4,5-дигидро-оксазол-2-ил]-1Н-индол-7-ил}-(тетрагидропиран-4-ил)амин;
{5-метил-2-[(S)-4-(2-морфолин-4-ил-этил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}-(тетрагидро-пиран-4-илметил)амин;
1-(4-{2-[(S)-2-(5-фенокси-7,7-диизобутиламино)-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-ил)этанон;
(5-Фенокси-2-{(S)-4-[(пирролидин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)диизобутил-амин;
1-(4-{2-[(S)-2-(5-фенокси-7-циклопентиламино)-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-ил)-2-гидрокси-этанон;
(5-Фенокси-2-{(S)-4-[(пиперазин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
трет-бутил-(4-{2-[(S)-2-(5-фенокси-7-циклопентиламино)-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-карбоксилат;
Циклопентил-(5-фенокси-2-{(S)-4-[2-(3-фторметил-5,6-дигидро-8H-[1,2,4]триазоло[4,3-a]пиразин-7-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)амин;
(5-Фенокси-2-{(S)-4-[2-оксопиперазин-4-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(4-{2-[(S)-2-(5-фенокси-7-циклопентиламино)-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-ил)-оксоран-2-ил-метанон;
(5-Фенокси-2-{(S)-4-[(пиридин-2-ил)пиперазин-этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-Фенокси-2-{(S)-4-[(2-фторфенил)пиперазин-этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-хлор-2-{(S)-4-[2-оксопиперазин-4-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-Фенокси-2-{(S)-4-[(3S)-3-(амино)пирролидин-1-илэтил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
1-(4-{2-[(S)-2-(5-фенокси-7-циклопентиламино)-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-ил)этанон;
(5-метил-2-{(S)-4-[2-(аминокарбонил)пирролидин-1-илэтил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-(тетрагидропиран-4-ил)метиламин;
(5-метил-[(S)-2-(7-(тетрагидропиран-4-ил)метиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил])пирролидин-2-илметанол;
(5-хлор-[(S)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил])пирролидин-3-илацетамид;
(5-Фенокси-2-{(S)-4-[4-(бензил)пиперазин-1-илэтил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-метил-2-{(S)-4-[2-диэтоксифосфорил-этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-(тетрагидропиран-4-ил)метиламин;
(5-метил-2-{(S)-4-[морфолин-4-илэтил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-(тетрагидропиран-4-ил)метиламин;
(5-Фенокси-2-{(R)-4-[пирролидин-1-илэтил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-Фенокси-2-{(S)-4-[морфолин-4-илэтил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-(тетрагидропиран-4-ил)метиламин;
(5-Фенокси-2-{(S)-4-[2-оксопиперазин-4-илэтил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-(тетрагидропиран-4-ил)метиламин;
(5-Фенокси-2-{(S)-4-[пирролидин-1-илэтил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-метил-2-{(S)-4-[2-оксопиперазин-4-илэтил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-4,4-дифторциклогексил-амин;
(5-метил-2-{(S)-4-[морфолин-4-илэтил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-4,4-дифторциклогексил-амин;
(тетрагидропиран-4-ил)-(5-метил-2-{(S)-4-[2-(3-фторметил-5,6-дигидро-8H-[1,2,4]триазоло[4,3-a]пиразин-7-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)амин;
(5-метил-2-{(S)-4-[2-оксопиперазин-4-илэтил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-(тетрагидропиран-4-ил)метиламин;
(5-хлор-2-{(S)-4-[1-(пиридин-2-ил)пиперазин-4-илэтил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-(тетрагидропиран-4-ил)амин;
(4-{2-[(S)-2-(5-хлор-7-(тетрагидропиран-4-ил)амино)-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-ил)оксоран-2-илметанон;
(5-метокси-2-{(R)-4-[2-оксопиперазин-4-илэтил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-(тетрагидропиран-4-ил)амин;
1-(4-{2-[(R)-2-(5-метокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-ил)-2-гидрокси-этанон;
[(R)-2-(5-аминометил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[7-циклопентиламино-2-((R)-4-гидроксиметил-4,5-дигидро-тиазол-2-ил)-1Н-индол-5-илметил]амид фуран-2-карбоновой кислоты;
метиловый эфир [(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-илметокси]уксусной кислоты;
[(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-илметокси]уксусная кислота;
Циклопентил-{2-[(R)-4-(3-циклопентил-[1,2,4]оксадиазол-5-илметил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}амин; и
Циклопентил-{2-[(R)-4-(3-пиперидин-1-ил-[1,2,4]оксадиазол-5-илметил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}амин.
Настоящее изобретение также относится к способам получения соединений формулы (1). Далее способы получения соединений формулы (1) проиллюстрированы для лучшего понимания схемами примеров реакции. Однако специалист в области, к которой относится настоящее изобретение, может получить соединения формулы (1) различными путями в зависимости от их структур, и такие способы должны рассматриваться как входящие в объем настоящего изобретения. Другими словами, соединения формулы (1) можно получить, в случае необходимости комбинируя различные способы синтеза, которые описаны в настоящем описании или раскрыты в уровне техники. Способы получения соединений формулы (1) охватывают даже такие процессы и не ограничены объясненными ниже.
Прежде всего, соединения формулы (1) могут быть получены реакцией амидного сочетания или реакцией алкилирования из соединений (2) и (3) согласно следующей реакционной схеме (1).
Реакционная схема 1

в приведенной выше реакционной схеме (1)
A, R1, R2, R3, R4 и n имеют значения, определенные выше, и
W обозначает карбоновую кислоту, используемую в реакции амидного сочетания, или галоген, алкилсульфонат и т.д. используемые в реакции сочетания.
В частности, R1 в приведенной выше Реакционной схеме (1) предпочтительно обозначает группу, содержащую амин или нуклеофильный углерод.
Реакцию амидирования можно осуществлять, используя агент сочетания, такой как, например, дициклогексилкарбодиимид (DCC), EDC, N-[диметиламино-1Н-1,2,3-триазол[4,5-b]-пиридин-1-илметилен]-N-метилметанаминий (HATU) и т.д. вместе с HOBT. Реакцию проводят в DMF или DCM в присутствии основания Et3N, DIPEA и т.д., в течение 4-12 часов и при температуре окружающей среды. В случае нуклеофила, содержащего атом азота, реакцию алкилирования можно проводить, используя различные основания, такие как Et3N, K2CO3, NMPA, DBU и т.д., в растворителе, таком как ацетонитрил, THF или DMF, при температуре от 25 до 80°C и в течение 4-24 часов. Большинство соединений (3) коммерчески доступно.
В следующей реакционной схеме (2) соединения (2-1) и (2-2), в которых A обозначает 4,5-дигидро-тиазол, могут быть получены гидролизом сложных (4,5-дигидро-тиазол-4-ил)-овых эфиров соединения (4), или могут быть получены синтезом спиртов путем восстановления и введением галогена или сульфонильной группы в качестве удаляемой группы.
Реакционная схема 2

в приведенной выше реакционной схеме (2)
n, R2, R3 и R4 имеют значения, определенные выше,
Q обозначает удаляемую группу, предпочтительно галоген или алкилсульфонат, и
R' обозначает алкил, предпочтительно метил, этил, изопропил и т.д.
В частности, соединение карбоновой кислоты (2-1) может быть получено гидролизом эфирного соединения (4), где от 2 до 10 экв. NaOH, LiOH, KOH и т.д. используют в качестве основания, и используют один или более растворителей, выбранных из воды, метанола, THF и диоксана. Эту реакцию гидролиза проводят в течение от 30 минут до 12 часов при температуре от температуры окружающей среды до 100°C.
Спиртовое соединение (5) может также быть получено восстановлением эфирного соединения (4), где NaBH4, LiBH4, LAH и т.д. используют в качестве восстановителя, и спирт, такой как метанол, THF, диоксан и т.д., используют в качестве растворителя. Эту реакцию восстановления осуществляют в течение от 30 минут до 24 часов при температуре от температуры окружающей среды до 100°C. Восстановитель обычно используется в количестве от 3 до 5 экв., но может использоваться в избытке приблизительно 10 экв., если необходимо.
Реакция галогенирования спиртового соединения (5) может быть осуществлена с использованием агента, выбранного из йода, брома, N-йодсукцинимида (NIS), N-бромсукцинимида (NBS), тетрахлорида углерода (CCl4), тетрабромида углерода (CBr4) и т.д., в присутствии основания, такого как имидазол, диметиламинопиридин (DMAP) и т.д. и фосфина, такого как трифенилфосфин (Ph3P), трибутилфосфин (Bu3P) и т.д. Каждый из галогенирующего реагента, основания и фосфина обычно используется в количестве от 1 до 10 экв. относительно соединения (5). Реакция может протекать в растворителе, выбранном из простых эфиров, таких как тетрагидрофуран, простой диэтиловый эфир и т.д., дихлорметан, хлороформ и т.д., при температуре от 0 до 50°C и в течение от 10 минут до 12 часов.
Реакцию сульфонилирования спиртового соединения (5) можно проводить, используя агент, выбранный из метансульфонилхлорида, п-толуолсульфонилхлорида и т.д. в количестве от 1 до 10 экв. В присутствии органического основания, такого как пиридин, триэтиламин и т.д. Эта реакция может протекать в растворителе, выбранном из дихлорметана, дихлорэтана и т.д., при температуре от 0 до 50°C и в течение от 10 минут до 12 часов.
Соединение индол-4,5-дигидро-тиазола (4') может быть получено, как показано в следующей реакционной схеме (3), то есть введением 4,5-дигидро-тиазола в исходное соединение сложного эфира 7-нитроиндола, восстановлением нитрогруппы и реакцией восстановительного аминирования с введением R3 и R4.
Реакционная схема 3

в приведенной выше реакционной схеме (3)
R2 имеет значения, определенные выше,
R” обозначает п-метоксибензил (p-MeOBn) или трифенилметил (Ph3C), и
R''' обозначает R1 или защищенный R1, и обычно обозначает алкилоксикарбонил (алкил-OC(O)-) или алкил карбоксилат (алкил-CO2-).
Реакцию гидролиза в реакционной схеме (3) проводят тем же самым образом, как объяснено для реакционной схемы (2), и реакцию амидного сочетания проводят, как объяснено для Реакционной схемы (1). Реакция циклизации может быть проведена с использованием пентахлорида фосфора (PCl5) в дихлорметане в качестве растворителя, когда R” является п-метоксибензилом, или с использованием ангидрида трифторметансульфоновой кислоты (Tf2O) и трифенилфосфиноксида (Ph3PO) в дихлорметане в качестве растворителя, когда R” является трифенилметилом.
Восстановление нитрогруппы в 7-нитроиндольном соединении (10) может быть осуществлено с использованием кислотного катализатора и металла или с использованием металлического катализатора в атмосфере газообразного водорода. В реакции с использованием кислотного катализатора в качестве металлов могут использоваться железо, цинк, литий, натрий или олово (обычно, хлорид олова), и в качестве кислотного катализатора могут использоваться неорганические кислоты, такие как соляная кислота, серная кислота, азотная кислота, фосфорная кислота и т.д.; органические карбоновые кислоты, такие как уксусная кислота, трифторуксусная кислота и т.д.; кислые соли амина, такие как хлорид аммония и т.д., предпочтительно соляная кислота, уксусная кислота или хлорид аммония. Кроме того, в реакции восстановления с использованием металлического катализатора в атмосфере газообразного водорода, в качестве металлического катализатора, который может использоваться, можно назвать палладий, никель, платину, рутений, родий и т.д., предпочтительно палладий или никель.
Реакцию восстановительного аминирования осуществляют, используя соединение, содержащее карбонильную группу (кетон или альдегид). В качестве восстановителя, который может использоваться, может быть назван боргидрид натрия, цианоборгидрид натрия, триацетоксиборгидрид натрия и т.д. Реакция может быть облегчена использованием кислоты в качестве катализатора. В качестве кислотного катализатора, который может использоваться, могут быть названы неорганические кислоты, такие как соляная кислота, серная кислота, азотная кислота, фосфорная кислота и т.д.; органические карбоновые кислоты, такие как уксусная кислота, трифторуксусная кислота и т.д.; кислые соли амина, такие как хлорид аммония и т.д., более предпочтительно соляная кислота или уксусная кислота.
В следующей реакционной схеме (4) соединение (11) и соединение индола (7) подвергают реакции сочетания, получая соединение (9-1), которое затем циклизуют в кислой среде и гидролизуют, получая соединение (2-3), в котором R”' является спиртом. Кроме того, соединение (2-4), которое получают, замещая спиртовую группу соединения (2-3) удаляемой группой, может быть введено в реакцию с соединением амина (14) с получением соединения амина (2-5).
Реакционная схема 4

в приведенной выше реакционной схеме (4)
Q, R2, R5, R6 и R” имеют значения, определенные выше.
Реакции амидного сочетания и циклизации могут быть проведены в тех же самых условиях, как объяснено для реакционных схем (1) и (3).
Реакцию гидролиза для получения соединения (2-3) осуществляют в смеси воды и органического растворителя и в присутствии основания, где в качестве органического растворителя может использоваться THF, метанол, диоксан и т.д., и в качестве основания может использоваться LiOH, KOH, NaOH и т.д.
Реакция введения удаляемой группы Q может быть осуществлена в тех же самых условиях, как объяснено для реакционной схемы (2). Синтез соединения (2-5) вследствие введения аминогруппы осуществляют путем введения в реакцию соединения амина (14) с соединением (2-4) в присутствии основания при 25-80°C и в течение от 20 минут до 24 часов. В качестве основания можно назвать Et3N, DIPEA, DMAP и т.д., но реакция может также быть выполнена в отсутствие основания, если необходимо.
7-нитроиндольное соединение (6) коммерчески доступно или может быть получено согласно следующей реакционной схеме (5).
Реакционная схема 5

в приведенной выше реакционной схеме (5) R2 имеет значения, определенные выше.
Соединение нитра-фениламина (15) коммерчески доступно или может быть получено согласно способу, известному из Heterocycles, 68(11), 2285-99, 2006, или Bioorganic & Medicinal Chemistry Letters, 14(19), 4903-4906, 2004.
Гидразиновое соединение (16) также коммерчески доступно или может быть получено модификацией аминогруппы соединения (15) в гидразиновую группу согласно способу, известному из Journal of the America Chemical Society, 198(48), 15374-75, 2006.
Соединение гидразина (18) может быть получено комбинацией соединения кетона (17) с гидразиновым соединением (16). Основание не используется, когда гидразиновое соединение (16) представляет собой нейтральную форму, но должно использоваться, когда соединение представляет собой форму кислой соли, чтобы перевести его в нейтральную форму. В качестве основания могут быть названы гидроксиды металлов, такие как гидроксид натрия, гидроксид лития и т.д., карбонаты металлов, такие как бикарбонат натрия, карбонат калия, и т.д., ацетаты металлов, такие как ацетат натрия, и т.д., органические основания, такие как триэтиламин, пиридин и т.д., предпочтительно ацетат натрия, бикарбонат натрия и т.д.
Соединение гидразина (18) может быть получено реакцией соли диазония с соединением кетона (19) в присутствии основания согласно способу перегруппировки Japp-Klingemann, известному из Organic Process Research & Development, 2, 1988, 214-220.
Реакция циклизации соединения (18) может быть осуществлена согласно способу, известному из Journal of Organic Chemistry, 68(24), 2003, 9506~9509, Tetrahedron, 55(34), 1999, 10271-10282, и т.д. Кислотой, которая может использоваться в этой реакции, может быть полифосфорная кислота, соляная кислота, п-толуолсульфоновая кислота, серная кислота, уксусная кислота и т.д. В случае полифосфорной кислоты она может использоваться индивидуально или вместе с ароматическим углеводородом, выбранным из бензола, толуола и т.д.
Соединение, которое модифицировано в положении 5 индольного кольца, может быть получено из соединения (6'), как показано на следующей реакционной схеме (6).
Реакционная схема 6

в приведенной выше реакционной схеме (6)
R”, R''' и R7 имеют значения, определенные выше.
Соединение (20) может быть получено присоединением защитной группы к аминогруппе метилового эфира (5-метил-7-нитро-1Н-индол-2-ил)карбоновой кислоты (6') с использованием Boc2O в присутствии основания и превращением метильной группы в положении 5 в бромметильную группу с использованием бромирующего агента.
Последующее ацетилирование на соединении (20) с использованием ацетата натрия, удаление защитной группы от группы ВОС и реакция гидролиза могут дать спиртовое соединение (22).
Реакция циклизации может быть осуществлена с использованием пентахлорида фосфор на соединении (9-2), которое получают реакцией амидного сочетания между соединениями (22) и (8), как объяснено для реакционной схемы (3), с получением соединения (2-6), в котором спирт заменен хлоридом, и соединение (2-6) может быть преобразовано в соединение (2-7) введением группы R7.
Соединение (1-2), имеющее аминогруппу в положении 5 индольного кольца, может быть получено путем получения соединения (24) из фталимидного соединения (23) и ацилирования аминогруппы соединения (1-1), которое получают из соединения (24), как изображено на следующей реакционной схеме (7).
Реакционная схема 7

R4, R”, R''' и R10 имеют значения, определенные выше.
Фталимид калия, используемый в реакции алкилирования для введения фталимидной группы, является коммерчески доступным, и эта реакция может быть осуществлена в присутствии тетрагидрофурана, N,N-диметилформамида, N-метилпирролидинона и т.д. Реакция гидролиза может быть осуществлена тем же самым образом, как способ получения соединения (2-3) на реакционной схеме (4). Амидное сочетание соединения (23), циклизация и восстановление и восстановительное аминирование соединения (24) могут также быть осуществлены тем же самым образом, как объяснено выше. Реакция удаления фталимидной группы соединения (24) может быть осуществлена с использованием гидразина. Соединение (1-2) может быть получено ацилированием соединения амина (1-1) с использованием соединения хлорангидрида кислоты (25), где основание, выбранное из Et3N, DIPEA, DMAP, пиридина и т.д., используется обычно в количестве 2 экв. или больше относительно соединения амина.
В следующей реакционной схеме (8) соединения (8-1) и (8-2) получают, защищая тиоловую группу аминокислот, таких как цистеин, или вводя тиоловую группу в производные аминокислот, полученные из глутаминовой кислоты, аспарагиновой кислоты и т.д.
Реакционная схема 8

в приведенной выше реакционной схеме (8)
R” имеет значения, определенные выше, и
R”” обозначает алкил, предпочтительно метил, этил, изопропил или циклогексил.
Соединение (8-1) может быть получено реакцией защищенного по амину соединения аминокислоты (28) с диазометаном с получением азосоединения, удлинением на один атом углерода с использованием иона серебра, такого как бензоат серебра, этерификацией кислотной группы и удалением защитной группы аминогруппы. В частности, реакция удлинения на один атом углерода может быть осуществлена путем введения в реакцию соединения (28) с этилхлорформиатом (EtOCOCl) или изобутилхлорформиатом (iBuOCOCl) в присутствии основания [например, N-метилморфолина (NMM), триэтиламина и т.д.] в растворителе, представляющем собой тетрагидрофуран, при температуре окружающей среды согласно способу, известному из Helvetica Chimica Acta, 87, 2004, 3131~3159, с получением ангидрида, введением полученного ангидрида в реакцию с водным раствором гидроксида калийдиазометана в простом диэтиловом эфире в качестве растворителя при 0°C и затем с ионом Ag [например, трифторацетатом серебра (CF3CO2Ag), бензоатом серебра и т.д.] и алкиловым спиртом (например, метанолом, этанолом и т.д.) в темноте с получением соединения сложного алкилового эфира.
В вышеописанной реакции диазометан может быть получен реакцией Diazald, N-метил-N-нитрозогуанидина или N-метил-N-нитрозомочевины в присутствии основания KOH согласно известному из уровня техники обычному способу. Эфирное соединение, имеющее один дополнительный атом углерода, может быть получено из диазосоединения введением его в реакцию с бензоатом серебра в спиртовом растворителе, где подходящая температура реакции составляет приблизительно -15°C. Для завершения реакции реакционную смесь после добавления бензоата серебра нагревают до температуры окружающей среды. В качестве растворителя могут использоваться метанол или этанол. Группа ВОС может быть удалена с использованием трифторуксусной кислоты или 4 н. раствора соляная кислота/простой эфир или соляная кислота/диоксан.
Соединение аминокислоты (28), в котором аминогруппа защищена группой ВОС, может быть получено защитой тиоловой группы цистеина в основных условиях и защитой аминогруппы группой ВОС. В частности, защита тиоловой группы может быть осуществлена с использованием п-метоксибензилхлорида (PMBCl) или трифенилметилхлорида (TrCl) в присутствии основания, выбранного из NaOH, NaH и т.д. Защита группой ВОС аминогруппы может быть осуществлена с использованием (ВОС)2O в основных условиях, где основание может включать NaOH, Et3N, NaHCO3 и т.д., и может использоваться растворитель, выбранный из DCM, диоксана, воды и т.д.
Соединение (8-2) может быть получено введением тиоловой группы в соединение (31) и удалением из него группы ВОС. В частности, добавление тиоловой группы можно осуществить, используя PMB-SH (а-метоксибензилтиол) в присутствии основания, выбранного из NaH, CeCO3, K2CO3 и т.д. С другой стороны, соединение (31) может быть получено защитой спиртового соединения (29) с использованием метансульфонилхлорида в присутствии основания Et3N или DIPEA.
Соединение (29) может синтезироваться из исходной глутаминовой кислоты соединения или аспарагиновой кислоты согласно способу, известному из Synlett, 15, 2005, 2397~2399 или Journal of Organic Chemistry, 66(5), 2001,1919~1923, и т.д.
В следующей Реакционной схеме (9) группу карбоновой кислоты производного цистеина (28) модифицируют, получая соединение (11).
Реакционная схема 9

в приведенной выше реакционной схеме (9) R” имеет значения, определенные выше.
Защиту аминогруппы соединения (28) можно осуществить, используя (ВОС)2O, ацилирование можно осуществить, используя хлорангидрид изомасляной кислоты, хлорангидрид трет-масляной кислоты и т.д. в присутствии основания, и восстановление можно осуществить, используя NaBH4.
Ацилирование соединения (32) осуществляют, используя пивалоилхлорид в присутствии основания, и удаление ВОС осуществляют, как объяснено выше.
Соединения, способы получения которых специфически не объясняются в настоящем описании, известны per se, или могут быть получены из известного соединения согласно известному способу или подобному ему способу.
В способах согласно настоящему изобретению смеси обычно разделяют хроматографией на колонках. В случае конечного продукта, он может быть отделен после завершения реакции перекристаллизацией или нормальной ВЭЖХ или ВЭЖХ с обратной фазой (Waters, Delta Pack, 300×50 мм внутренний диаметр, C18 5 мкм, 100A). Когда продукт очищают перекристаллизацией или ВЭЖХ, соединение может быть получено в форме соли с трифторуксусной кислотой. Когда желаема соль с соляной кислотой, может быть использована ионообменная смола.
Как объяснено выше, соединения согласно настоящему изобретению, исходные материалы, промежуточные соединения и т.д. для их получения могут быть получены различными способами, и такие способы получения соединения формулы (1) должны быть рассмотрены как входящие в объем настоящего изобретения.
Эффект
Настоящее изобретение также относится к композиции для профилактики или лечения некроза и связанных с ним заболеваний, которая содержит терапевтически эффективное количество соединений формулы (1), их фармацевтически приемлемых солей или изомеров в качестве активного ингредиента вместе с фармацевтически приемлемыми носителями или разбавителями.
Настоящее изобретение также относится к способу профилактики или лечения некроза и связанных с ним заболеваний с использованием вышеописанной композиции.
Некроз и связанные с ним заболевания, которые могут быть подвергнуты лечению и/или профилактике согласно настоящему изобретению, включают острое/хроническое заболевание печени (например, гепатит, фиброз печени, цирроз печени), нейродегенеративное заболевание (например, деменция, болезнь Паркинсона, заболевание Гентингтона), ишемическое заболевание сердца, реперфузионное повреждение, ишемический инсульт или ишемическое повреждение, панкреатит, бактериальный/вирусный сепсис, сахарный диабет или диабетические осложнения, диабетическое заболевание сосудов [в частности, эти виды диабета вызываются веществами, разрушающими панкреатические клетки, и опосредуются вирусом, гипергликемией, жирной кислотой, диетой, токсином, стрептозотоцином и т.п.], некротический проколит, муковисцидоз, ревматоидный артрит, дегенеративный артрит, нефропатию, бактериальную инфекцию, вирусную инфекцию (например, ВИЧ), рассеянный склероз, лейкоз, лимфому, респираторный дистресс-синдром новорожденных, асфиксию, туберкулез, эндометриоз, ангиастению, псориаз, обморожение, осложнения лечения стероидами, гангрену, пролежни, гемоглобинурию, ожоги, гипертермию, болезнь Крона, глютеиновую болезнь, синдром сдавления, повреждение спинного мозга, гломерулонефрит, мышечную дистрофию, наследственное метаболическое заболевание, микоплазматическое заболевание, сибирскую язву, болезнь Андерсена, врожденное митохондриальное заболевание, фенилкетонурию, инфаркт плаценты, сифилис, асептический некроз и т.д. Кроме того, некроз и связанные с ним заболевания, вызванные лекарственными и токсичными веществами, выбраны из группы, состоящей из некроза, связанного с хроническим алкоголизмом, контакта с, и/или введения и/или самовведения, кокаина, лекарственных средств (например, парацетаола), антибиотиков, противоракового средства, адриамицина, пуромицина, блеомицина, NSAID, циклоспорина, химических токсинов (например, тетрахлорида углерода, цианида, метанола, этиленгликоля), ядовитого газа, агрохимикатов, тяжелых металлов (например, свинца, ртути, кадмия), или повреждения вследствие контакта с радиацией/УФ и связанного с этим некроза.
В частности, композиция согласно настоящему изобретению демонстрирует не только эффекты в отношении гепатопротекции и функционального улучшения состояния печени, но также и профилактические и терапевтические эффекты в отношении хронического заболевания печени, такого как ожирение печени, фиброз печени, цирроз печени и т.д. и острого/хронического заболевания печени, такого как гепатит и т.д., вызванных вирусами или лекарственными средствами. Следовательно, осложнения заболевания печени, включая, но не ограничиваясь ей, портальную гипертензию, также могут быть подвергнуты профилактике или лечению. Более конкретно, медицинская композиция согласно настоящему изобретению также эффективна для лечения или профилактики заболевания печени, выбранного из трансплантации печени, алкогольного или безалкогольного ожирения печени, фиброза печени, цирроза печени и гепатита, вызванного вирусом или лекарственными средствами, и эффективна в отношении алкогольного острого/хронического заболевания печени.
Далее, композиция согласно настоящему изобретению эффективна для лечения или профилактики вызванного жирной кислотой ожирения печени или острого/хронического заболевания печени, являющегося следствием ожирения печени.
В рамках изобретения "лечение" означает прерывание или отсрочивание развития заболевания применительно к пациенту, у которого показано возникновение симптомов заболевания, и "профилактика" означает прерывание или отсрочивание появления признаков начала заболевания применительно к пациенту, у которого не показано, но существует риск возникновения симптомов заболевания.
Указанная “фармацевтическая композиция” может включать фармацевтически приемлемые носители, разбавители, эксципиенты или их комбинации, если необходимо, вместе с соединениями согласно настоящему изобретению. Фармацевтическая композиция облегчает введение соединения в живой организм. Существует множество методик введения соединения, и они включают, но не ограничены ими, пероральное, путем инъекции, аэрозольное, парентеральное и топическое введение.
В рамках изобретения "носитель" означает вещество, которое облегчает включение соединения в клетки или ткани. Например, диметилсульфоксид (ДМСО) является типичным носителем, который используется для облегчения введения различных органических соединений в клетки или ткани живых организмов.
В рамках изобретения "разбавитель" определяют как вещество, которое разбавлено в воде, которое растворяет соединение, а также стабилизирует биологически активную форму используемого соединения. Соли, растворенные в буферном растворе, используют в данной области техники в качестве разбавителей. Обычно используемым буферным раствором является фосфатный буферизованный солевой раствор, который имитирует солевую форму среды человеческого организма. Буферные разбавители редко изменяют биологические активности соединения, поскольку буферные соли могут контролировать рН раствора в низкой концентрации.
В рамках изобретения “фармацевтически приемлемый” означает свойство, которое не ухудшает биологические активности и физические свойства соединения.
Соединения согласно настоящему изобретению могут быть составлены как различные фармацевтические лекарственные формы в зависимости от желаемой цели. Для получения фармацевтической композиции согласно настоящему изобретению, активный ингредиент, в частности, соединения формулы (1), их фармацевтически приемлемые соли или изомеры смешивают вместе с различными фармацевтически приемлемыми носителями, которые могут быть выбраны в зависимости от получаемого состава. Например, фармацевтическая композиция согласно настоящему изобретению может быть составлена как препарат для инъекции, пероральный препарат и т.д., в зависимости от желаемой цели.
Соединения согласно настоящему изобретению могут быть составлены способами, известными из уровня техники, в которых используют фармацевтические носители и эксципиенты, известные из уровня техники, и могут быть заключены в контейнеры для лекарственной формы в виде разовой дозы или формы для множества доз. Форма препарата может представлять собой растворы, суспензии или эмульсии в масляных или водных средах и может содержать обычные диспергирующие агенты, суспендирующие агенты или стабилизаторы. Далее, например, это может быть форма сухого порошка, которая предназначена для восстановления путем растворения в стерильной, апирогенной воде до использования. Соединения согласно настоящему изобретению также могут быть составлены в формы суппозитория с использованием обычной основы суппозитория, такой как масло какао или другие глицериды. В качестве твердых лекарственных форм для перорального введения могут быть получены капсулы, таблетки, пилюли, порошок и гранулы, и особенно предпочтительны капсулы и таблетки. Предпочтительно, таблетки и пилюли получают в формах, имеющих энтеросолюбильное покрытие. Твердые лекарственные формы можно получить, смешивая соединения согласно настоящему изобретению вместе с носителями, такими как один или более инертных разбавителей, таких как сахароза, лактоза, крахмал и т.д., лубрикантами, такими как стеарат магния, дезинтеграторами, связующими и т.д.
Если необходимо, соединения согласно настоящему изобретению или содержащие их фармацевтические композиции могут также вводиться в комбинации с другими активными средствами, включая цитопротекторные средства с различными механизмами действия различных типов, особенно существующие средства, используемые для гепатопротекции, функционального улучшения состояния печени и профилактики или лечения заболевания печени - промоторами регенерации гепатоцитов, функциональными печеночными адъювантами, противовирусными средствами, иммуносупрессорами, ингибиторами фиброза и т.д.
Соединения согласно настоящему изобретению или содержащие их фармацевтические композиции можно использовать совместно с профилактическими или терапевтическими средствами для профилактики или лечения любого вызванного лекарственным средством некроза и связанных с ним заболеваний. Эти лекарственные средства включают лекарственные средства для любой группы заболеваний, такие как антибиотики, противораковые средства, противовирусные средства, противоинфекционные средства, противовоспалительные средства, антикоагулирующие средства, средства, улучшающие липидный профиль, ингибиторы гибели клеток, антигипертензивные средства, антидиабетические/против ожирения средства, терапевтические средства для лечения сердечно-сосудистого заболевания, терапевтические средства для лечения нейродегенеративного заболевания, средства, замедляющие старение, терапевтические средства для лечения метаболических заболеваний и т.д.
Соединения согласно настоящему изобретению или содержащие их фармацевтические композиции могут использоваться для профилактики повреждения клеток и последующего некроза и связанных с ним заболеваний, вызванных различными причинами, такими как токсины, и эти причины включают реакционноспособные формы кислорода (ROS), тяжелые металлы, алкоголь, пищу, добавки, радиацию, диету и т.д.
Дозировка соединений формулы (1) зависит от предписания врача, принимая во внимание такие факторы, как масса тела, пол, возраст, состояние здоровья и режим питания пациента, конкретную природу заболевания, время введения средства, способ введения, соотношение средств в смеси и серьезность заболевания и т.д. Однако доза, необходимая для лечения взрослого, обычно составляет приблизительно от 1,0 мг до 2000 мг в сутки, в зависимости от интенсивности и частоты введения. При введении взрослому внутримышечным или внутривенным путями, полная доза, составляющая приблизительно от 1,0 мг до 300 мг в сутки, обычно будет достаточной при отдельном введении в виде единственной дозы, но для некоторых пациентов может быть желательной более высокая ежедневная доза.
Настоящее изобретение также относится к способу получения композиции для профилактики или лечения некроза и связанных с ним заболеваний, который включает стадию смешивания соединений формулы (1), их фармацевтически приемлемых солей или изомеров в качестве активного ингредиента вместе с фармацевтически приемлемыми носителями или разбавителями.
Наилучший способ осуществления изобретения
Настоящее изобретение будет более подробно объяснено следующими примерами получения и примерами. Однако следует понимать, что они предназначены для иллюстрации настоящего изобретения, но никоим образом не для ограничения объема настоящего изобретения. В следующих примерах получения и примерах М обозначает молярную концентрацию, и н. означает нормальную концентрацию.
Следующие Примеры получения объясняют более подробно получение промежуточных соединений, которые требуются для синтеза соединений из примеров. Сокращения, используемые в следующих примерах получения и примерах, являются следующими.
Ac: ацетил
AIBN: 2,2'-азобис(2-метилпропионитрил)
BOC: трет-бутоксикарбонил
Bu: бутил
Bn: бензил
c-Pen: циклопентил
c-Hex: циклогексил
CBZ(Cbz): бензилоксикарбонил
DME: диметоксиэтан
DCM: дихлорметан
DIPEA: диизопропилэтиламин
DMAP: 4-диметиламинопиридин
DMF: N,N-диметилформамид
EDC: 1-(3-диметиламинопропил)-3-этилкарбодиимид, гидрохлорид
Et: этил
EtOAC: этил ацетат
Hex: н-гексан
HOBT: гидроксибензотриазол
HBTU: 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилуроний гексафторфосфат
i-Pr: изопропил
i-Pen: изопентил
Me: метил
Ph: фенил
Pid: пиперидин
Piz: пиперазин
Pyd: пирролидин
PMB: параметоксибензил
TEA: триэтиламин
TFA: трифторуксусная кислота
THF: тетрагидрофуран
THP: тетрагидропиран
t-Bu: трет-бутил
Пример получения 1
4-(Пиридин-3-илокси)фениламин
Стадия A: 4-(Пиридин-3-ил)окси-1-нитробензол
1-хлор-4-нитробензол (40 г, 0,25 моль) и 3-гидроксипиридин (36 г, 0,38 моль) растворяли в N,N-диметилформамид (100 мл). Добавляли карбонат калия (52,6 г, 0,38 моль), и смесь перемешивали в течение 20 часов при 100°C. После завершения реакции добавляли воду. Реакционную смесь экстрагировали этилацетатом, промывали насыщенным раствором хлорида натрия, и высушивали над безводным сульфатом магния, получая целевое соединение.
Стадия B: 4-(Пиридин-3-илокси)фениламин
4-(Пиридин-3-ил)окси-1-нитробензол, полученный на стадии A, растворяли в смеси воды (100 мл), тетрагидрофурана (100 мл) и метанола (100 мл). Добавляли железный порошок (103 г, 1,84 моль) и хлорид аммония (99 г, 1,84 моль), и смесь перемешивали в течение 3 часов при 80°C, используя приводную мешалку. После завершения реакции реакционную смесь фильтровали через целит, промывали метанолом и концентрировали при пониженном давлении. Полученное таким образом твердое вещество отфильтровывали, промывали простым эфиром и высушивали, получая целевое соединение (17 г, выход 36%).
Масса [M+H]: 186 (M+1).
Пример получения 2
4-(4-Метансульфонил-фенокси)фениламин
Стадия A: 1-(4-метилсульфанилфенокси)-4-нитробензол
1-хлор-4-нитробензол (15 г, 95 ммоль) и 4-(метилмеркапто)фенол (13,3 г, 95 ммоль) растворяли в диметилсульфоксиде (100 мл). Добавляли карбонат калия (15,8 г, 134 ммоль), и смесь перемешивали в течение 12 часов при 100°C. После завершения реакции, добавляли избыток воды, чтобы осадить твердое вещество, которое затем отфильтровывали и высушивали, получая целевое соединение.
Стадия B: 4-(4-метансульфонил-фенокси)нитробензол
1-(4-Метилсульфанилфенокси)-4-нитробензол (86 г, 330 ммоль), полученный на стадии A, растворяли в дихлорметане (500 мл). Добавляли mCPBA (3-хлорпербензойная кислота) (83 г, 330 ммоль), и смесь перемешивали в течение 2 часов при температуре от 0°C до температуры окружающей среды. После завершения реакции добавляли избыток 6 н. водного раствора гидроксида натрия. Реакционную смесь экстрагировали этилацетатом и дихлорметаном, промывали насыщенным раствором хлорида натрия и высушивали над безводным сульфатом магния, получая целевое соединение (28 г, Выход 100%).
Стадия C: 4-(4-метансульфонил-фенокси)фениламин
4-(4-Метансульфонил-фенокси)нитробензол (28 г, 95 ммоль), полученный на стадии B, растворяли в метаноле (500 мл) и добавляли этилацетат (500 мл). 10%-ный Pd/C (1,0 г), и смесь перемешивали в атмосфере газообразного водорода при атмосферном давлении в течение 3 часов. После завершения реакции реакционную смесь фильтровали через целит, промывали метанолом, концентрировали при пониженном давлении, и высушивали над безводным сульфатом магния, получая целевое соединение (25 г, выход 100%).
Масса [M+H]: 263 (M+1).
Пример получения 3
4-Этокси-2-нитро-фениламин
Стадия A: 4-Этокси-1-ацетиламинобензол
4-Этоксианилин (40 г, 0,29 моль) и триэтиламин (61 мл, 0,44 моль) растворяли в дихлорметане (200 мл). Добавляли по каплям уксусный ангидрид (30 мл, 0,32 моль), и смесь перемешивали в течение 1 часа при температуре от 0°C до температуры окружающей среды. Добавляли 1 н. раствор соляной кислоты, и реакционную смесь экстрагировали этилацетатом, промывали раствором хлорида натрия и высушивали над безводным сульфатом магния, получая целевое соединение.
Стадия B: 4-Этокси-2-нитро-1-фениламин
4-Этокси-1-ацетиламинобензол (51 г, 0,29 моль), полученный на стадии A, растворяли в дихлорметане (200 мл). Дымящую азотную кислоту (13 мл, 0,29 моль) добавляли по каплям при 0°C, и смесь перемешивали в течение 1 часа при температуре от 0°C до температуры окружающей среды. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия, затем экстрагировали этилацетатом, промывали насыщенным раствором хлорида натрия и высушивали над безводным сульфатом магния. Полученное нитросоединение растворяли в метаноле (100 мл) и тетрагидрофуране (100 мл). 6 н. гидрид натрия добавляли по каплям, и смесь перемешивали в течение 6 часов при температуре окружающей среды. После завершения реакции реакционную смесь нейтрализовали до приблизительно рН 7 с использованием 6 н. раствора соляной кислоты, экстрагировали этилацетатом, промывали насыщенным раствором хлорида натрия и высушивали над безводным сульфатом магния, получая целевое соединение (44 г, выход 83%).
Масса [M+H]: 182 (M+1).
Примеры получения 4-13:
Соединения фениламина, полученные в примерах получения 1 и 2, и коммерчески доступные анилины вводили в реакцию согласно той же самой процедуре, как в примере получения 3, для синтеза соединений из примеров получения, показанных в следующей таблице.
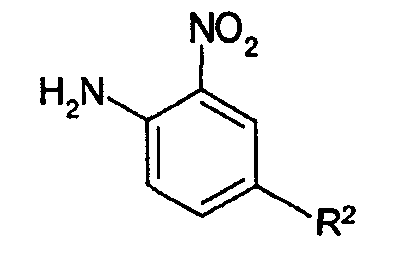
Пример получения 14
Метиловый эфир 5-хлор-7-нитро-1Н-индол-2-карбоновой кислоты
<Способ A>
Стадия A: (4-хлор-2-нитро-фенил)гидразин хлорид
4-Хлор-2-нитроанилин (40 г, 0,23 моль), полученный в примере получения 13, растворяли в 12 н. соляной кислоте (100 мл). Нитрит натрия (16 г, 0,23 моль), растворенный в воде (50 мл), медленно добавляли по каплям при 0°C, и смесь перемешивали в течение 30 минут при температуре от 0°C до температуры окружающей среды. Реакционную смесь охлаждали до 0°C, медленно добавляли по каплям хлорид олова (II) (132 г, 0,70 моль), растворенный в 12 н. соляной кислоте (100 мл), и смесь перемешивали в течение 3 часов при температуре от 0°C до температуры окружающей среды. Полученное твердое вещество желтого цвета отфильтровывали, промывали небольшим количеством 6 н. HCl, и высушивали, получая целевое соединение (30 г, выход 63%).
1H-ЯМР (400 МГц, ДМСО-d6); δ 9,21 (с, 1H), 7,98 (д, J=2,4 Гц, 1H), 7,66 (д, J=9,6 Гц, 1H), 7,55 (дд, J=2,4, 9,6 Гц, 1H), 4,74 (ушир.с, 2H).
Стадия B: метиловый эфир 2-[(4-хлор-2-нитро-фенил)гидразоно]пропионовой кислоты
(4-Хлор-2-нитро-фенил)гидразин хлорид (30 г, 0,14 моль), полученный на стадии A, и метилпируват (14,4 мл, 0,16 моль) растворяли в метаноле (300 мл) и добавляли ацетат натрия (14,2 г, 0,17 моль). Реакционный раствор перемешивали в течение 8 часов при температуре окружающей среды, и полученное твердое вещество желтого цвета отфильтровывали, промывали водой и метанолом и высушивали, получая целевое соединение (30 г, выход 82%).
1H-ЯМР (400 МГц, CDCl3); δ 10,88 (с, 1H), 8,21 (д, J=2,4 Гц, 1H), 8,01 (д, J=9,2 Гц, 1H), 7,56 (дд, J=2,4, 9,2 Гц, 1H), 3,90 (с, 3H), 2,23 (c, 3H).
Стадия C: метиловый эфир 5-хлор-7-нитро-1Н-индол-2-карбоновой кислоты
К метиловому эфиру 2-[(4-хлор-2-нитро-фенил)гидразоно]пропионовой кислоты (13 г, 46 ммоль), полученному на стадии B, добавляли полифосфорную кислоту (100 мл), и смесь нагревали в течение 4 часов при 100°C. После завершения реакции к реакционной смеси при 0°C добавляли воду. Полученную смесь перемешивали в течение 2 часов, и фильтровали, чтобы собрать твердое вещество. Это твердое вещество промывали водой и высушивали, получая целевое соединение (6,0 г, выход 49%).
1H-ЯМР (400 МГц, CDCl3); δ 10,32 (ушир.с, 1H), 8,29 (д, 1H), 8,03 (д, J=2,4 Гц, 1H), 7,31 (д, J=2,0 Гц, 1H), 4,01 (с, 3H).
<Способ B>
Стадия A: метиловый эфир 2-[(4-хлор-2-нитро-фенил)гидразоно]пропионовой кислоты
4-Хлор-2-нитро-фениламин (11,0 г, 64,05 ммоль), полученный в примере получения 13, растворяли в концентрированной соляной кислоте (32 мл) в колбе A и охлаждали до -10°C. Добавляли лед (90 г), медленно добавляли нитрит натрия (4,42 г, 64,05 ммоль), растворенный в воде (50 мл), и смесь перемешивали, пока она не стала прозрачной.
Метиловый эфир 2-метил-3-оксо-масляной кислоты (8,32 г, 64,05 ммоль) растворяли в этаноле (76 мл) в колбе B и охлаждали до -10°C. Добавляли гидроксид калия (19,05 мл), растворенный в воде (19 мл), к которому при -10°C добавляли раствор, полученный в колбе. Смесь перемешивали в течение 1 часа. Полученное твердое вещество красного цвета отфильтровывали, получая целевое соединение (7,54 г, выход 49%).
Стадия B: Метиловый эфир 5-хлор-7-нитро-1Н-индол-2-карбоновой кислоты
Метиловый эфир 2-[(4-хлор-2-нитро-фенил)гидразоно]пропионовой кислоты, полученный на стадии A, вводили в реакцию согласно той же самой процедуре, как на стадии C <Способа A> примера получения 14, получая целевое соединение.
1H-ЯМР (400 МГц, CDCl3); δ 10,32 (ушир.с, 1H), 8,29 (д, 1H), 8,03 (д, J=2,4 Гц, 1H), 7,31 (д, J=2,0 Гц, 1H), 4,01 (с, 3H).
Примеры получения 15-26
Соединения из примеров получения вводили в реакцию с метилпируватом, этилпируватом, метиловым эфиром 2-метил-3-оксо-масляной кислоты или этиловым эфиром 2-метил-3-оксо-масляной кислоты согласно способу A или B примера получения 14, чтобы синтезировать соединения примеров получения, показанные в следующей таблице.

Пример получения 27
Метиловый эфир (R)-3-амино-4-(4-метокси-бензилсульфанил)масляной кислоты гидрохлорид
Стадия A: (R)-2-амино-3-(4-метокси-бензилсульфанил)пропионовая кислота
4-Метоксибензиловый спирт (280 г, 1780 ммоль), растворенный в простом диэтиловом эфире (400 мл), добавляли по каплям к смеси простого диэтилового эфира (400 мл) и концентрированной соляной кислоты (400 мл) за 2 часа, и смесь перемешивали в течение 1 часа. Органический слой отделяли и добавляли к раствору, полученному растворением L-цистеина (197 г, 1625 ммоль) и 2 н. водного раствора гидроксида натрия (980 мл) в этаноле (1890 мл). Смесь перемешивали в течение 2 часов при температуре окружающей среды. После завершения реакции реакционную смесь охлаждали до 0°C и нейтрализовали до рН 7 с использованием 3 н. водного раствора соляной кислоты. Полученное твердое вещество отфильтровывали и высушивали, получая целевое соединение (250 г, выход 64%).
Стадия B: (R)-2-BOC-амино-3-(4-метокси-бензилсульфанил)пропионовая кислота
(R)-2-Амино-3-(4-метокси-бензилсульфанил)пропионовую кислоту (30,7 г, 127,3 ммоль), полученную на стадии A, растворяли в тетрагидрофуране (150 мл) и воде (150 мл). Добавляли карбонат калия (26,4 г, 190 ммоль) и (ВОС)2O (27,7 г, 127,3 ммоль), и смесь перемешивали в течение 2 часов при температуре окружающей среды. После завершения реакции реакционную смесь перегоняли при пониженном давлении, чтобы удалить тетрагидрофуран. Остаток охлаждали до 0°C и подкисляли до рН 3, используя 3 н. водный раствор соляной кислоты. Полученное твердое вещество промывали водой и высушивали, получая целевое соединение (43 г, выход 99%).
Стадия C: трет-бутиловый эфир [(R)-3-диазо-1-(4-метокси-бензилсульфанилметил)-2-оксо-пропил]карбаминовой кислоты
(R)-2-BOC-амино-3-(4-метокси-бензилсульфанил)пропионовую кислоту (43 г, 132 ммоль), полученную на стадии B, 1-метилморфолин (14,5 мл, 132 ммоль) и этилхлорформиат (14,1 мл, 132 ммоль) растворяли в тетрагидрофуране (500 мл), и смесь перемешивали в течение 1 часа при -25°C. В то же самое время гидроксид калия (75 г, 1336 ммоль) растворяли в воде (75 мл) и простом диэтиловом эфире (750 мл), за 2 часа при 0°C добавляли по каплям N-метил-нитрозомочевину (26 г, 252 ммоль), и смесь перемешивали в течение 30 мин. Эти два таким образом полученных раствора смешивали вместе и перемешивали в течение 3 часов при температуры от -25°C до температуры окружающей среды. После завершения реакции к реакционной смеси добавляли воду, промывали насыщенным водным раствором бикарбоната натрия и насыщенным водным раствором хлорида аммония в этом порядке. Органический слой концентрировали, получая целевое соединение (46,0 г, выход 95%).
1H-ЯМР (400 МГц, CDCl3); δ 7,25 (д, J=8,8 Гц, 2H), 6,86 (д, J=8,8 Гц, 2H), 5,48 (ушир.с, 1H), 5,29 (м, 1H), 4,31 (м, 1H), 3,79 (с, 3H), 3,69 (с, 2H), 2,76 (д, J=6,0 Гц, 2H), 1,45 (с, 9H).
Стадия D: (R)-3-трет-бутоксикарбониламино-4-(4-метокси-бензилсульфанил)масляной кислоты метиловый эфир
Трет-бутиловый эфир [(R)-3-диазо-1-(4-метокси-бензилсульфанилметил)-2-оксо-пропил]карбаминовой кислоты (40 г, 109 ммоль), полученный на стадии C, растворяли в метаноле (600 мл), и смесь охлаждали до -25°C. Добавляли трифторацетат серебра, и смесь медленно нагревали. После завершения реакции твердую часть удаляли фильтрацией через целит. Добавляли насыщенный водный раствор NH4Cl, и смесь экстрагировали EtOAc и высушивали над MgSO4. Растворитель удаляли при пониженном давлении, и остаток очищали хроматографией на колонках (элюент: EtOAc/n-Hex=1/3), получая целевое соединение (30,6 г, выход 76%).
1H-ЯМР (500 МГц, CDCl3); δ 7,24 (д, J=8,6 Гц, 2H), 6,83 (д, J=8,6 Гц, 2H), 5,09 (м, 1H), 4,08 (м, 1H), 3,79 (c, 3H), 3,68 (c, 2H), 3,66 (c, 3H), 2,70-2,52 (м, 4H), 1,44 (c, 9H).
Стадия E: метиловый эфир (R)-3-амино-4-(4-метокси-бензилсульфанил)масляной кислоты
Метиловый эфир (R)-3-трет-бутоксикарбониламино-4-(4-метокси-бензилсульфанил)-масляной кислоты (30 г, 81,3 ммоль), полученный на стадии D, растворяли в дихлорметане (70 мл). Добавляли раствор 4 н. соляная кислота/1,4-диоксан (71 мл), и смесь перемешивали в течение 2 часов при температуре окружающей среды. После завершения реакции реакционный раствор концентрировали при пониженном давлении. К остатку добавляли дихлорметан (30 мл) и простой диэтиловый эфир (150 мл). Полученное твердое вещество отфильтровывали и высушивали, получая целевое соединение (19,2 г, выход 87%).
1H ЯМР (400 МГц, ДМСО-d6); δ 8,21 (ушир.с, 3H), 7,25 (д, 2H), 6,83 (д, 2H), 3,78 (с, 3H), 3,68 (с, 2H), 3,65 (с, 3H), 3,29 (м, 1H), 2,51-2,48 (м, 2H), 2,35-2,31 (м, 2H).
Пример получения 28
(R)-3-Амино-4-(4-метокси-бензилсульфанил)этилбутирилат гидрохлорид
Трет-бутиловый эфир [(R)-3-диазо-1-(4-метокси-бензилсульфанилметил)-2-оксо-пропил]карбаминовой кислоты, полученный на стадии C примера получения 27, и этанол вводили в реакцию согласно тем же самым процедурам, как на стадиях D и E примера получения 27, последовательно, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3); δ 8,37 (ушир.с, 3H), 7,28 (д, J=8,0 Гц, 2H), 6,87 (д, J=8,0 Гц, 2H), 4,11 (м, 2H), 3,73 (с, 3H), 3,70 (с, 2H), 2,81-2,67 (м, 4H), 1,18 (т, 3H).
Пример получения 29
Этиловый эфир (R)-4-амино-5-(4-метокси-бензилсульфанил)валериановой кислоты гидрохлорид
Стадия A: этиловый эфир (R)-4-BOC-амино-5-гидрокси-пентановой кислоты
Коммерчески доступный 5-этил-1-метиловый эфир (R)-2-BOC-аминопентановой кислоты (57,8 г, 200 ммоль) растворяли в метаноле (200 мл). Добавляли LiBH4 (1н. раствор в THF, 400 мл), и смесь перемешивали в течение 2 часов, поддерживая температуру 10°C или ниже. После завершения реакции реакционную смесь охлаждали до 0°C и медленно добавляли воду, чтобы завершить реакцию. Метанол удаляли при пониженном давлении, и остаток разбавляли насыщенным водным раствором NaHCO3. Смесь экстрагировали EtOAc и высушивали над MgSO4. Растворитель удаляли при пониженном давлении, и остаток очищали хроматографией на колонках, получая целевое соединение (39 г, выход 75%).
Стадия B: этиловый эфир (R)-4-BOC-амино-5-метансульфонилокси-пентановой кислоты
Этиловый эфир (R)-4-BOC-амино-5-гидрокси-пентановой кислоты (36 г, 137,8 ммоль), полученный на стадии A, и триэтиламин (38,4 мл, 275,5 ммоль) растворяли в дихлорметане (200 мл). Метансульфонилхлорид (11,7 мл, 151,5 ммоль) добавляли по каплям, и смесь перемешивали в течение 1 часа при температуре от 0°C до температуры окружающей среды. После завершения реакции добавляли 1 н. раствор соляной кислоты, затем экстрагировали этилацетатом, промывали насыщенным водным раствором хлорида натрия и высушивали над безводным сульфатом магния, получая целевое соединение.
Стадия C: этиловый эфир (R)-4-BOC-амино-5-(4-метокси-бензилсульфанил)валериановой кислоты
Гидрид натрия (5,5 г, 137,8 ммоль) и 4-метоксибензилмеркаптан (15,4 мл, 110,2 ммоль) растворяли в N,N-диметилформамиде (150 мл), и смесь перемешивали в течение 10 минут при 0°C. К полученному раствору добавляли по каплям этиловый эфир (R)-4-BOC-амино-5-метансульфонилокси-пентановой кислоты (46,7 г, 137,8 ммоль), полученный на стадии B, и смесь перемешивали в течение 4 часов при 0°C. Добавляли воду, чтобы завершить реакцию, и реакционную смесь экстрагировали этилацетатом, промывали насыщенным водным раствором хлорида натрия и высушивали над безводным сульфатом магния. Растворитель удаляли при пониженном давлении, и остаток очищали хроматографией на колонках, получая целевое соединение.
1H-ЯМР (400 МГц, CDCl3); δ 7,25 (д, J=8,8 Гц, 2H), 6,85 (д, J=8,8 Гц, 2H), 4,56 (м, 1H), 4,12 (м, 2H), 3,79 (c, 3H), 3,69 (c, 2H), 2,53 (м, 2H), 2,33 (т, 2H), 1,93 (м, 1H), 1,70 (м, 1H), 1,44 (c, 9H), 1,25 (т, 3H).
Стадия D: этиловый эфир (R)-4-амино-5-(4-метокси-бензилсульфанил)валериановой кислоты гидрохлорид
Этиловый эфир (R)-4-BOC-амино-5-(4-метокси-бензилсульфанил)валериановой кислоты (11 г, 62,7 ммоль), полученный на стадии C, растворяли в дихлорметане (200 мл). Добавляли раствор 4 н. соляной кислоты/этилацетат (20 мл), и смесь перемешивали в течение 2 часов при температуре окружающей среды. После завершения реакции растворитель полностью удаляли при пониженном давлении. Остаток перекристаллизовывали из простого диэтилового эфира (150 мл) и высушивали, получая целевое соединение (20 г, выход 96%).
1H ЯМР (400 МГц, ДМСО-d6); δ 8,69 (ушир.с, 3H), 7,29 (д, J=8,0 Гц, 2H), 6,89 (д, J=8,0 Гц, 2H), 4,08 (м, 2H), 3,74 (м, 5H), 3,26 (м, 1H), 2,76~2,63 (м, 2H), 2,49~2,40 (м, 2H), 1,89 (м, 2H), 1,20 (т, 3H).
Пример получения 30
Изопропиловый эфир (S)-3-амино-4-(метокси-бензилсульфанил)масляной кислоты
Стадия A: 4-изопропил-1-метиловый эфир (S)-2-BOC-амино-янтарной кислоты
Коммерчески доступный 1-метиловый эфир (S)-2-BOC-амино-янтарной кислоты (2,4 г, 10 ммоль) растворяли в DCM (30 мл) и добавляли триэтиламин (2,8 мл, 20 ммоль). К смеси добавляли изопропанол (660 мг, 11 ммоль), EDC (2,5 г, 26 ммоль) и HOBt (2,3 г, 30 ммоль), и смесь перемешивали в течение 4 часов при температуре окружающей среды. Реакционную смесь гасили насыщенным водным раствором NaHCO3. Реакционную смесь экстрагировали EtOAc, промывали насыщенным водным раствором хлорида натрия и высушивали над безводным сульфатом магния. Растворитель удаляли при пониженном давлении, и остаток очищали хроматографией на колонках, получая целевое соединение (2,5 г, выход 87%).
Стадия B: изопропиловый эфир (S)-3-амино-4-(метокси-бензилсульфанил)масляной кислоты
4-Изопропил-1-метиловый эфир (S)-2-BOC-амино-янтарной кислоты, полученный на стадии A, вводили в реакцию согласно той же самой процедуре, как в примере получения 29, получая целевое соединение.
Масса [M+H]=397.
Пример получения 31
(R)-2-амино-3-(4-метокси-бензилсульфанил)пропиловый эфир 2,2-диметилпропионовой кислоты
Стадия A: метиловый эфир (R)-2-амино-3-(4-метокси-бензилсульфанил)пропионовой кислоты
(R)-2-BOC-амино-3-(4-метокси-бензилсульфанил)пропионовую кислоту, полученную на стадии B примера получения 27, и метанол вводили в реакцию согласно той же самой процедуре, как в примере получения 30, получая целевое соединение.
1H ЯМР (400 МГц, ДМСО-d6, соль HCl); δ 8,81 (ушир.с, 3H), 7,29 (д, J=8,4 Гц, 2H), 6,91 (д, J=8,4 Гц, 2H), 4,28 (м, 1H), 3,18 (ушир.с, 8H), 2,95 (м, 2H).
Стадия B: метиловый эфир (R)-2-BOC-амино-3-(4-метокси-бензилсульфанил)пропионовой кислоты
Метиловый эфир (R)-2-амино-3-(4-метокси-бензилсульфанил)пропионовой кислоты (30,7 г, 127,3 ммоль), полученный на стадии A, растворяли в DCM. Добавляли Et3N (26,4 г, 190 ммоль) и (ВОС)2O (27,7 г, 127,3 ммоль), и смесь перемешивали в течение 2 часов при температуре окружающей среды. После завершения реакции реакционную смесь перегоняли при пониженном давлении, чтобы удалить DCM, и использовали в следующей реакции без дальнейшей очистки.
Стадия C: трет-бутиловый эфир [(R)-2-гидрокси-1-(4-метокси-бензилсульфанилметил)этил]карбаминовой кислоты
Метиловый эфир (R)-2-BOC-амино-3-(4-метокси-бензилсульфанил)пропионовой кислоты, полученный на стадии B, вводили в реакцию согласно той же самой процедуре, как на стадии А примера получения 29, получая целевое соединение.
1H ЯМР (500 МГц, ДМСО-d6); δ 7,24 (д, J=8,6 Гц, 2H), 6,84 (д, J=8,6 Гц, 2H), 4,96 (ушир.с, 1H), 3,78 (с, 3H), 3,76 (ушир.с, 1H), 3,70 (с, 2H), 3,7~3,66 (м, 3H), 2,58 (м, 2H), 1,44 (с, 9H).
Стадия D: (R)-2-трет-бутоксикарбониламино-3-(4-метокси-бензилсульфанил)пропиловый эфир 2,2-диметилпропионовой кислоты
Трет-бутиловый эфир [(R)-2-гидрокси-1-(4-метокси-бензилсульфанилметил)этил]карбаминовой кислоты (71,3 г, 227,9 ммоль), полученный на стадии C, растворяли в дихлорметане (300 мл). Добавляли триэтиламин (58 мл, 414,4 ммоль) и хлорид триметилуксусной кислоты (28 мл, 227,9 ммоль), и смесь перемешивали в течение 6 часов при 0°C. Добавляли воду, чтобы завершить реакцию. Реакционную смесь экстрагировали этилацетатом, промывали насыщенным водным раствором хлорида натрия и высушивали над безводным сульфатом магния. Растворитель удаляли при пониженном давлении, и остаток очищали хроматографией на колонках, получая целевое соединение (81,0 г, выход 95%).
1H ЯМР (400 МГц, CDCl3); δ 7,25 (д, J=8,8 Гц, 2H), 6,85 (д, J=8,8 Гц, 2H), 4,71 (м, 1H), 4,11 (м, 2H), 3,79 (c, 3H), 3,70 (c, 2H), 2,55 (д, J=6,4 Гц, 2H), 1,52 (c, H), 1,27 (c, 9H).
Стадия E: (R)-2-амино-3-(4-метокси-бензилсульфанил)пропиловый эфир 2,2-диметилпропионовой кислоты
(R)-2-трет-бутоксикарбониламино-3-(4-метокси-бензилсульфанил)пропиловый эфир 2,2-диметилпропионовой кислоты (81 г, 196 ммоль), полученный на стадии D, растворяли в дихлорметане (300 мл). Добавляли раствор 4 н. соляной кислоты/1,4-диоксан (100 мл), и смесь перемешивали в течение 8 часов при температуре окружающей среды. После завершения реакции растворитель полностью удаляли при пониженном давлении. Остаток перекристаллизовывали из простого диэтилового эфира и высушивали, получая целевое соединение (68 г, выход 95%).
1H ЯМР (400 МГц, ДМСО-d6, свободная форма); δ 7,24 (д, J=12,0 Гц, 2H), 6,85 (дд, J=4,0, 8,0 Гц, 2H), 4,04 (м, 1H), 3,95 (м, 1H), 3,80 (c, 3H), 3,68 (c, 2H), 3,10 (м, 1H), 2,60 (м, 1H), 2,36 (м, 1H), 1,18 (c, 9H).
Пример получения 32
(S)-2-амино-3-(4-метокси-бензилсульфанил)пропиловый эфир 2,2-диметилпропионовой кислоты
Стадия A: метиловый эфир (S)-2-BOC-амино-3-метилсульфонилокси-пропионовой кислоты
Коммерчески доступный метиловый эфир (S)-2-BOC-амино-3-гидрокси-пропионовой кислотный и метансульфонилхлорид вводили в реакцию согласно той же самой процедуре, как на стадии B примера получения 29, получая целевое соединение.
Стадия B: метиловый эфир (S)-2-BOC-амино-3-(4-метокси-бензилсульфанил)пропионовой кислоты
Метиловый эфир (S)-2-BOC-амино-3-метилсульфонилокси-пропионовой кислоты, полученный на стадии A, и 4-метокси-бензил-тиол вводили в реакцию согласно той же самой процедуре, как на стадии C примера получения 29, получая целевое соединение.
Стадия C: (S)-2-трет-бутоксикарбониламино-3-(4-метокси-бензилсульфанил)пропанол
Метиловый эфир (S)-2-BOC-амино-3-(4-метокси-бензилсульфанил)пропионовой кислоты, полученный на стадии B, и LiBH4 вводили в реакцию согласно той же самой процедуре, как на стадии А примера получения 29, получая целевое соединение.
Стадия D: (R)-2-амино-3-(4-метокси-бензилсульфанил)пропиловый эфир 2,2-диметилпропионовой кислоты
(S)-2-трет-бутоксикарбониламино-3-(4-метокси-бензилсульфанил)пропанол, полученный на стадии C, триметилацетилхлорид и HCl (4 н. раствор в диоксане) вводили в реакцию согласно тем же самым процедурам, как на стадиях D и E примера получения 31, последовательно получая целевое соединение.
Масса [M+H]=397.
Пример получения 33
Метиловый эфир 2-амино-3-(4-метокси-бензилсульфанил)пропионовой кислоты
Коммерчески доступный BOC-Ser-OMe вводили в реакцию согласно тем же самым процедурам, как на стадиях B, C и D примера получения 29, последовательно получая целевое соединение.
Масса [M+H]=255 (M+1).
Пример получения 34
2-[(4-Метокси-бензилсульфанил)этиламин
Коммерчески доступный BOC-амино-этанол вводили в реакцию согласно тем же самым процедурам, как на стадиях B и C примера получения 29 и на стадии E примера получения 31, последовательно получая целевое соединение.
Масса [M+H]=197 (M+1).
Пример получения 35
(R)-1-[(4-Метокси-бензилсульфанил)метил]пропиламин
Коммерчески доступный (R)-2-BOC-амино-1-бутанол вводили в реакцию согласно тем же самым процедурам, как на стадиях B и C примера получения 29 и на стадии E примера получения 31, последовательно получая целевое соединение.
Масса [M+H]=225 (M+1).
Пример получения 36
(R)-1-[(4-Метокси-бензилсульфанил)метил]-2-метил-1-пропиламин
Коммерчески доступный метиловый эфир 2-BOC-амино-3-метил-масляной кислоты вводили в реакцию согласно тем же самым процедурам, как на стадиях A, B и C примера получения 29 и на стадии E примера получения 31, последовательно получая целевое соединение.
Масса [M+H]=239 (M+1).
Пример получения 37
Метиловый эфир 5-бромметил-7-нитро-индол-2-карбоновой кислоты
Стадия A: метиловый эфир 1-BOC-5-метил-7-нитро-индол-2-карбоновой кислоты
Метиловый эфир 5-метил-7-нитро-1Н-индол-2-карбоновой кислоты (24,0 г, 100 ммоль), полученный в примере получения 15, растворяли в дихлорметане (500 мл), добавляли триэтиламин (84 мл, 601 ммоль) и 4-(диметиламино)пиридин (600 мг, 5 ммоль). Добавляли по каплям (ВОС)2O (43,7 г, 200 ммоль), растворенный в дихлорметане (100 мл), и смесь перемешивали в течение 8 часов при температуре окружающей среды. После завершения реакции добавляли воду. Реакционную смесь экстрагировали этилацетатом, промывали насыщенным водным раствором хлорида натрия, высушивали над безводным сульфатом магния и концентрировали при пониженном давлении, получая целевое соединение (34,0 г, выход 100%).
1H-ЯМР (500 МГц, CDCl3); δ 7,80 (с, 1H), 7,67 (с, 1H), 7,15 (с, 1H), 3,93 (с, 3H), 2,51 (с, 3H), 1,62 (с, 9H).
Стадия B: метиловый эфир 1-BOC-5-бромметил-7-нитро-индол-2-карбоновой кислоты
Метиловый эфир 1-BOC-5-метил-7-нитро-индол-2-карбоновой кислоты (34 г, 101,7 ммоль), полученный на стадии A, растворяли в тетрахлорметане (100 мл). Добавляли N-бромсукцинимид (27,2 г, 152,6 ммоль) и AIBN (1,7 г, 10,2 ммоль), и смесь перемешивали в течение 5 часов при 80°C. После завершения реакции реакционную смесь перегоняли при пониженном давлении и очищали хроматографией на колонках, получая целевое соединение (48,0 г, выход 100%).
1H-ЯМР (500 МГц, CDCl3); δ 8,01 (с, 1H), 7,90 (с, 1H), 7,21 (с, 1H), 4,60 (с, 2H), 3,93 (с, 3H), 1,62 (с, 9H).
Стадия C: метиловый эфир 5-Бромметил-7-нитро-индол-2-карбоновой кислоты
Метиловый эфир 1-BOC-5-бромметил-7-нитро-индол-2-карбоновой кислоты, полученный на стадии B, вводили в реакцию согласно той же самой процедуре, как на стадии E примера получения 31, получая целевое соединение.
Масса [M+H]=313 (M+1).
Пример получения 38
5-Гидроксиметил-7-нитро-1Н-индол-2-карбоновая кислота
Стадия A: метиловый эфир 1-BOC-5-ацетоксиметил-7-нитро-индол-2-карбоновой кислоты
Метиловый эфир 1-BOC-5-бромметил-7-нитро-индол-2-карбоновой кислоты (10,0 г, 24,2 ммоль), полученный на стадии B примера получения 37, растворяли в N,N-диметилформамиде (50 мл). Добавляли ацетат натрия (2,4 г, 29,0 ммоль), и смесь перемешивали в течение 4 часов при температуре окружающей среды. После завершения реакции растворитель удаляли перегонкой при пониженном давлении. Остаток экстрагировали этилацетатом, промывали насыщенным водным раствором хлорида натрия и высушивали над безводным сульфатом магния. Остаток очищали хроматографией на колонках, получая целевое соединение (4,7 г, выход 50%).
1H-ЯМР (500 МГц, CDCl3); δ 7,99 (с, 1H), 7,90 (с, 1H), 7,21 (с, 1H), 5,22 (с, 2H), 3,94 (с, 3H), 2,12 (с, 3H), 1,63 (с, 9H).
Стадия B: метиловый эфир 5-ацетоксиметил-7-нитро-1Н-индол-2-карбоновой кислоты
Метиловый эфир 1-BOC-5-ацетоксиметил-7-нитро-индол-2-карбоновой кислоты (4,7 г, 12,0 ммоль), полученный на стадии A, растворяли в дихлорметане (50 мл). Добавляли 2 н. раствор соляной кислоты (30 мл, 60 ммоль), и смесь перемешивали в течение 12 часов при температуре окружающей среды. Реакционную смесь перегоняли при пониженном давлении, получая твердое целевое соединение (3,5 г, выход 100%).
1H-ЯМР (500 МГц, CDCl3); δ 10,33 (ушир.с, 1H), 8,32 (с, 1H), 8,06 (с, 1H), 7,34 (с, 1H), 5,24 (с, 2H), 3,99 (с, 3H), 2,12 (с, 3H).
Стадия C: 5-Гидроксиметил-7-нитро-1Н-индол-2-карбоновая кислота
Метиловый эфир 5-ацетоксиметил-7-нитро-1Н-индол-2-карбоновой кислоты (3,5 г, 12,0 ммоль), полученный на стадии B, растворяли в смеси тетрагидрофурана, метанола и воды (1:1:1, 100 мл). Добавляли гидрат гидроксида лития (1,5 г, 35,9 ммоль), и смесь перемешивали в течение 3 часов при температуре окружающей среды. После завершения реакции метанол и тетрагидрофуран удаляли перегонкой при пониженном давлении. К остатку добавляли 1 н. соляную кислоту. Реакционную смесь экстрагировали этилацетатом, высушивали над безводным сульфатом магния и перегоняли при пониженном давлении, чтобы удалить растворитель. Остаток очищали хроматографией на колонках, получая целевое соединение (2,3 г, выход 81%).
1H-ЯМР (500 МГц, ДМСО-d6); δ 11,02 (ушир.c, 1H), 8,21 (c, 1H), 8,10 (c, 1H), 7,34 (c, 1H), 5,43 (ушир.c, 1H), 4,64 (c, 2H).
Пример получения 39
Метиловый эфир 5-(1,3-Диоксо-1,3-дигидро-изоиндол-2-илметил)-7-нитро-1Н-индол-2-карбоновой кислоты
Стадия A: метиловый эфир 1-BOC-5-(1,3-диоксо-1,3-дигидро-изоиндол-2-илметил)-7-нитро-1Н-индол-2-карбоновой кислоты
Метиловый эфир 1-BOC-5-бромметил-7-нитро-индол-2-карбоновой кислоты (4,9 г, 11,4 ммоль), полученный на стадии B примера получения 37, растворяли в N,N-диметилформамиде (50 мл). Добавляли фталимид калия (2,7 г, 14,8 ммоль), и смесь перемешивали в течение 4 часов при температуре окружающей среды. Добавляли воду, чтобы завершить реакцию. Реакционную смесь экстрагировали этилацетатом, промывали насыщенным водным раствором хлорида натрия и высушивали над безводным сульфатом магния. Растворитель удаляли перегонкой при пониженном давлении, и остаток очищали хроматографией на колонках, получая целевое соединение (3,6 г, выход 66%).
1H-ЯМР (500 МГц, CDCl3); δ 8,04 (c, 1H), 7,98 (c, 1H), 7,85 (м, 2H), 7,71 (м, 2H), 7,17 (c, 1H), 4,96 (c, 2H), 4,37 (кв, 2H), 1,59 (c, 9H), 1,39 (т, 3H).
Стадия B: метиловый эфир 5-(1,3-Диоксо-1,3-дигидро-изоиндол-2-илметил)-7-нитро-1Н-индол-2-карбоновой кислоты
Метиловый эфир 1-BOC-5-(1,3-диоксо-1,3-дигидро-изоиндол-2-илметил)-7-нитро-1Н-индол-2-карбоновой кислоты, полученный на стадии A, вводили в реакцию согласно той же самой процедуре, как на стадии B примера получения 38, получая целевое соединение.
Масса [M+H]=379 (М+1).
Пример получения 40
(R)-2-(7-нитро-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-илметиловый эфир 2,2-диметилпропионовой кислоты
Стадия A: 7-Нитро-1Н-индол-2-карбоновая кислота
Метиловый эфир 7-Нитро-1Н-индол-2-карбоновой кислоты (13 г, 59 ммоль), полученный в примере получения 17, растворяли в смеси тетрагидрофурана и воды (1:1, 300 мл), добавляли 1 н. водный раствор гидроксида натрия (180 мл, 177 ммоль). Смесь перемешивали в течение 3 часов при температуре окружающей среды, и добавляли избыток 6 н. раствор соляной кислоты. Реакционную смесь экстрагировали этилацетатом. Экстракт промывали насыщенным водным раствором хлорида натрия, высушивали над безводным сульфатом магния и фильтровали. Растворитель удаляли перегонкой при пониженном давлении, и остаток высушивали, получая целевое соединение (12 г, выход 99%).
Стадия B: (R)-3-(4-метокси-бензилсульфанил)-2-[(7-нитро-1Н-индол-2-карбонил)амино]пропиловый эфир 2,2-диметилпропионовой кислоты
7-Нитро-1Н-индол-2-карбоновую кислоту (8,2 г, 22,7 ммоль), полученную на стадии A, и (R)-2-амино-3-(4-метокси-бензилсульфанил)пропиловый эфир 2,2-диметилпропионовой кислоты (13,2 г, 27,2 ммоль), полученный в примере получения 31, растворяли в N,N-диметилформамиде (100 мл). Добавляли EDC (6,6 г, 25,0 ммоль) и HOBT (4,6 г, 25,0 ммоль), и смесь перемешивали в течение 8 часов при температуре окружающей среды. К смеси добавляли насыщенный водный раствор бикарбоната натрия, чтобы завершить реакцию. Реакционную смесь экстрагировали этилацетатом, высушивали над безводным сульфатом магния, фильтровали и перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках, получая целевое соединение (8,1 г, выход 71%).
1H-ЯМР (400 МГц, CDCl3); δ 10,47 (ушир.с, 1H), 8,27 (д, J=8,0 Гц, 1H), 8,01 (д, J=8,0 Гц, 1H), 7,26 (м, 2H), 6,93 (д, J=4,0 Гц, 1H), 6,83 (м, 2H), 6,74 (д, J=8,0 Гц, 1H), 4,56 (м, 1H), 4,44 (м, 1H), 4,24 (м, 1H), 3,74 (м, 5H), 2,77 (м, 1H), 2,62 (м, 1H), 1,18 (с, 9H).
Стадия C: [(R)-2-(7-нитро-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метиловый эфир 2,2-диметилпропионовой кислоты
(R)-3-(4-метокси-бензилсульфанил)-2-[(7-нитро-1Н-индол-2-карбонил)амино]пропиловый эфир 2,2-диметилпропионовой кислоты (1,6 г, 3,2 ммоль), полученный на стадии B, растворяли в дихлорметане (50 мл). Добавляли пентахлорид фосфора (1,3 г, 6,4 ммоль), и смесь перемешивали в течение 5 часов при температуре окружающей среды. К смеси добавляли насыщенный водный раствор бикарбоната натрия, чтобы завершить реакцию. Реакционную смесь экстрагировали этилацетатом, высушивали над безводным сульфатом магния, фильтровали и перегоняли при пониженном давлении. Остаток очищали хроматографией на колонках, получая целевое соединение (0.8 г, выход 69%).
1H-ЯМР (400 МГц, CDCl3); δ 10,53 (ушир.с, 1H), 8,26 (д, J=8,0 Гц, 1H), 7,99 (д, J=8,0 Гц, 1H), 7,04 (д, J=2,0 Гц, 1H), 6,90 (д, J=7,6 Гц, 1H), 4,78 (м, 1H), 4,46 (м, 1H), 4,30 (м, 1H), 3,59 (м, 1H), 3,36 (м, 1H), 1,20 (с, 9H).
Пример получения 41
(R)-2-[(5-хлорметил-7-нитро-1Н-индол-2-ил)-(4,5-дигидро-тиазол-4-ил)метиловый эфир 2,2-диметилпропионовой кислоты
Стадия A: (R)-2-[(5-гидроксиметил-7-нитро-1Н-индол-2-карбонил)амино]-3-(4-метокси-бензилсульфанил)пропиловый эфир 2,2-диметилпропионовой кислоты
5-Гидроксиметил-7-нитро-1Н-индол-2-карбоновую кислоту, полученную в примере получения 38, и (R)-2-амино-3-(4-метокси-бензилсульфанил)пропиловый эфир 2,2-диметилпропионовой кислоты, полученный в примере получения 31, вводили в реакцию согласно той же самой процедуре, как на стадии B примера получения 40, получая целевое соединение.
Стадия B: (R)-2-[(5-хлорметил-7-нитро-1Н-индол-2-ил)-(4,5-дигидро-тиазол-4-ил)метиловый эфир 2,2-диметилпропионовой кислоты
(R)-2-[(5-гидроксиметил-7-нитро-1Н-индол-2-карбонил)амино]-3-(4-метокси-бензилсульфанил)пропиловый эфир 2,2-диметилпропионовой кислоты, полученный на стадии A, вводили в реакцию согласно той же самой процедуре, как на стадии C примера получения 40, получая целевое соединение.
Масса [M+H]=395 (M+1).
Пример получения 42
Метиловый эфир (R)-2-[(5-хлорметил-7-нитро-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты
5-Гидроксиметил-7-нитро-1Н-индол-2-карбоновую кислоту, полученную в примере получения 38, и гидрохлорид метилового эфира (R)-3-амино-4-(4-метокси-бензилсульфанил)масляной кислоты, полученный в примере получения 27, вводили в реакцию согласно тем же самым процедурам, как на стадиях B и C примера получения 40, получая целевое соединение.
Масса [M+H]=353 (M+1).
Пример получения 43
(R)-2-[5-(2,2-диметил-пропионилоксиметил)-7-нитро-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-илметиловый эфир 2,2-диметилпропионовой кислоты
Стадия A: (R)-2-{[5-(2,2-диметил-пропионилоксиметил)-7-нитро-1Н-индол-2-карбонил]амино}-3-(4-метокси-бензилсульфанил)пропиловый эфир 2,2-диметилпропионовой кислоты
(R)-2-[(5-гидроксиметил-7-нитро-1Н-индол-2-карбонил)амино]-3-(4-метокси-бензилсульфанил)пропиловый эфир 2,2-диметилпропионовой кислоты (3,1 г, 6,01 ммоль), полученный на стадии А примера получения 41, растворяли в DCM, добавляли Et3N (1,68 мл, 12,02 ммоль). Добавляли пивалоилхлорид (0,92 мл, 6,61 ммоль), и смесь перемешивали в течение 12 часов при температуре окружающей среды. После завершения реакции растворитель удаляли при пониженном давлении, и остаток очищали хроматографией на колонках (элюент: EtOAc/n-Hex=1/1), получая целевое соединение.
Стадия B: (R)-2-{[5-(2,2-диметил-пропионилоксиметил)-7-нитро-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-илметиловый эфир 2,2-диметилпропионовой кислоты
(R)-2-{[5-(2,2-диметил-пропионилоксиметил)-7-нитро-1Н-индол-2-карбонил]амино}-3-(4-метокси-бензилсульфанил)пропиловый эфир 2,2-диметилпропионовой кислоты, полученный на стадии A, вводили в реакцию согласно той же самой процедуре, как на стадии C примера получения 40, получая целевое соединение.
Масса [M+H]=475 (M+1).
Пример получения 44
(R)-2-{[5-метансульфонилметил-7-нитро-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-илметиловый эфир 2,2-диметилпропионовой кислоты
Стадия A: метиловый эфир (5-метансульфонилметил-7-нитро-1Н-индол-2-ил)карбоновой кислоты
Метиловый эфир 5-Бромметил-7-нитро-индол-2-карбоновой кислоты (251 мг, 0,805 ммоль), полученный в примере получения 37, добавляли к DMF (8 мл). Добавляли NaSO2Me (290 мг, 2,415 ммоль), и смесь перемешивали в течение 2 часов при температуре окружающей среды. Реакционную смесь гасили насыщенным водным раствором NaHCO3. Реакционную смесь экстрагировали EtOAc и высушивали над безводным сульфатом магния. Растворитель удаляли при пониженном давлении, и остаток использовали в следующей реакции без дальнейшей очистки.
Стадия B: (R)-2-{[5-метансульфонилметил-7-нитро-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-илметиловый эфир 2,2-диметилпропионовой кислоты
Метиловый эфир (5-метансульфонилметил-7-нитро-1Н-индол-2-ил)карбоновой кислоты, полученный на стадии A, и (R)-2-амино-3-(4-метокси-бензилсульфанил)пропиловый эфир 2,2-диметилпропионовой кислоты, полученный в примере получения 31, вводили в реакцию согласно той же самой процедуре, как в примере получения 40, получая целевое соединение.
Масса [M+H]=453 (M+1).
Пример получения 45
Метиловый эфир (R)-2-[5-метансульфонилметил-7-нитро-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-илуксусной кислоты
Метиловый эфир (5-метансульфонилметил-7-нитро-1Н-индол-2-ил)карбоновой кислоты, полученный на стадии А примера получения 44, и гидрохлорид метилового эфира (R)-3-амино-4-(4-метокси-бензилсульфанил)масляной кислоты, полученный в примере получения 27, вводили в реакцию согласно той же самой процедуре, как в примере получения 40, получая целевое соединение.
Масса [M+H]=411 (M+1).
Примеры получения 46-91
Соединения из примеров получения 14-26, 38 и 39 и соединения из примеров получения 27-36 выборочно использовали для синтеза соединений из примеров получения, показанных в следующей таблице, согласно способу, выбранному из примеров получения 40-45.

Пример получения 92
[(R)-2-(5-диметиламинометил-7-нитро-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метиловый эфир 2,2-диметилпропионовой кислоты
[(R)-2-(5-хлорметил-7-нитро-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил] метиловый эфир 2,2-диметилпропионовой кислоты (1,0 г, 2,4 ммоль), полученный в примере получения 41, растворяли в диметилсульфоксиде (15 мл). Добавляли диизопропилэтиламин (0,6 г, 4,9 ммоль) и диметиламин (0,2 г, 4,9 ммоль), и смесь перемешивали в течение 6 часов при температуре окружающей среды. После завершения реакции добавляли воду. Реакционную смесь экстрагировали этилацетатом, высушивали над безводным сульфатом магния и фильтровали. Фильтрат перегоняли при пониженном давлении и очищали хроматографией на колонках, получая целевое соединение (0,7 г, выход 71%).
Масса [M+H]=418 (M+1).
Пример получения 93
{(R)-2-[5-(морфолин-4-ил)метил-7-нитро-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-ил}метиловый эфир 2,2-диметилпропионовой кислоты
[(R)-2-(5-хлорметил-7-нитро-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метиловый эфир 2,2-диметилпропионовой кислоты, полученный в примере получения 41, и морфолин вводили в реакцию согласно той же самой процедуре, как в примере получения 92, за исключением того, что DMF использовали как растворитель вместо ДМСО, получая целевое соединение.
Масса [M+H]=460 (M+1).
Пример получения 94
{(R)-2-[5-(пиразол-1-ил)метил-7-нитро-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-ил}метиловый эфир 2,2-диметилпропионовой кислоты
[(R)-2-(5-хлорметил-7-нитро-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метиловый эфир 2,2-диметилпропионовой кислоты, полученный в примере получения 41, и пиразол вводили в реакцию согласно той же самой процедуре, как в примере получения 92, за исключением того, что DMF использовали как растворитель вместо ДМСО, получая целевое соединение.
Масса [M+H]=441 (M+1).
Пример получения 95
{(R)-2-[5-(1,3-имидазол-1-ил)метил-7-нитро-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-ил}метиловый эфир 2,2-диметилпропионовой кислоты
[(R)-2-(5-хлорметил-7-нитро-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метиловый эфир 2,2-диметилпропионовой кислоты, полученный в примере получения 41, и 1,3-имидазол вводили в реакцию согласно той же самой процедуре, как в примере получения 92, за исключением того, что DMF использовали как растворитель вместо ДМСО, получая целевое соединение.
Масса [M+H]=441 (M+1).
Пример получения 96
{(R)-2-[5-(1,2,4-триазол-1-ил)метил-7-нитро-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-ил}метиловый эфир 2,2-диметилпропионовой кислоты
[(R)-2-(5-хлорметил-7-нитро-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метиловый эфир 2,2-диметилпропионовой кислоты, полученный в примере получения 41, и 1,2,4-триазол вводили в реакцию согласно той же самой процедуре, как в примере получения 92, за исключением того, что DMF использовали как растворитель вместо ДМСО, получая целевое соединение.
Масса [M+H]=442 (M+1).
Пример получения 97
{(R)-2-[5-(пиррол-1-ил)метил-7-нитро-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-ил}метиловый эфир 2,2-диметилпропионовой кислоты
[(R)-2-(5-хлорметил-7-нитро-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метиловый эфир 2,2-диметилпропионовой кислоты, полученный в примере получения 41, и пиррол вводили в реакцию согласно той же самой процедуре, как в примере получения 92, за исключением того, что DMF использовали как растворитель вместо ДМСО, получая целевое соединение.
Масса [M+H]=440 (M+1).
Пример получения 98
Метиловый эфир [(R)-2-(7-нитро-5-феноксиметил-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты
Фенол (192 мг, 2,04 ммоль) растворяли в DMF. Добавляли NaH (60% в минеральном масле, 82 мг, 2,04 ммоль), и реакционный раствор охлаждали до 0°C. Медленно добавляли метиловый эфир (R)-2-(5-хлорметил-7-нитро-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-илуксусной кислоты (500 мг, 1,36 ммоль), полученный в примере получения 42, который растворяли в DMF. Реакционный раствор нагревали до температуры окружающей среды и перемешивали в течение 4 часов. После добавления насыщенного водного раствора NH4Cl для завершения реакции реакционную смесь экстрагировали EtOAc, высушивали над MgSO4 и фильтровали. Растворитель удаляли при пониженном давлении, и остаток очищали хроматографией на колонках (элюент: EtOAc/n-Hex=1/2), получая целевое соединение.
Масса [M+H]=425 (M+1).
Пример получения 99
{(R)-2-[7-нитро-5-(фениламино)метил-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-ил}метиловый эфир 2,2-диметилпропионовой кислоты
Метиловый эфир (R)-2-(5-хлорметил-7-нитро-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-илуксусной кислоты, полученный в примере получения 42, и фениламин вводили в реакцию согласно той же самой процедуре, как в примере получения 98, получая целевое соединение.
Масса [M+H]=466 (M+1).
Пример получения 100
{(R)-2-[7-нитро-5-(пирролидин-1-ил)метил-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-ил}метиловый эфир 2,2-диметилпропионовой кислоты
(R)-2-[(5-хлорметил-7-нитро-1Н-индол-2-ил)-(4,5-дигидро-тиазол-4-ил)метиловый эфир 2,2-диметилпропионовой кислоты, полученный в примере получения 41, и пирролидин вводили в реакцию согласно той же самой процедуре, как в примере получения 92, за исключением того, что DMF использовали как растворитель вместо ДМСО, получая целевое соединение.
Масса [M+H]=444 (M+1).
Пример получения 101
2-(4,5-Дигидро-тиазол-2-ил)-7-нитро-1Н-индол
Стадия A: 7-Нитро-1Н-индол-2-карбоновая кислота
Коммерчески доступный 7-нитроиндол-2-этилкарбоксилат (500 мг, 2,14 ммоль) растворяли в смеси тетрагидрофурана и воды (1:1, 20 мл). Добавляли гидрат гидроксида лития (448 мг, 10,7 ммоль), и смесь перемешивали в течение 8 часов при температуре окружающей среды. Добавляли 1 н. раствор соляной кислоты, и реакционную смесь экстрагировали этилацетатом. Экстракт высушивали над безводным сульфатом магния и фильтровали. Фильтрат перегоняли при пониженном давлении, получая целевое соединение.
Стадия B: (2-хлорэтил)-амид 7-Нитро-1Н-индол-2-карбоновой кислоты
7-Нитро-1Н-индол-2-карбоновую кислоту (371 мг, 3,2 ммоль), полученную на стадии A, растворяли в N,N-диметилформамиде (10 мл). Добавляли триэтиламин (0,6 мл, 4,3 ммоль), EDC (614 мг, 3,2 ммоль) и HOBT (433 мг, 3,2 ммоль), добавляли 2-хлорэтиламин (252,8 мг, 3,2 ммоль), и смесь перемешивали в течение 8 часов при температуре окружающей среды. Добавляли 1 н. раствор соляной кислоты, и реакционную смесь экстрагировали этилацетатом, промывали насыщенным раствором бикарбоната натрия, высушивали над безводным сульфатом магния и фильтровали. Фильтрат перегоняли при пониженном давлении, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3); δ 10,51 (ушир.с, 1H), 8,28 (д, J=6,4 Гц, 1H), 8,02 (д, J=6,4 Гц, 1H), 7,27 (т, 1H), 7,03 (с, 1H), 6,62 (ушир.с, 1H), 3,86 (м, 2H), 3,77 (м, 2H).
Стадия C: 2-(4,5-Дигидро-тиазол-2-ил)-7-нитро-1Н-индол
(2-Хлорэтил)-амид 7-Нитро-1Н-индол-2-карбоновой кислоты (267 мг, 1 ммоль), полученный на стадии B, растворяли в дихлорэтане (10 мл) и толуоле (10 мл) и добавляли реактив Лоуссона (1,29 г, 3,2 ммоль). Смесь нагревали с обратным холодильником в течение 4 часов и перегоняли при пониженном давлении. Добавляли воду, и реакционную смесь экстрагировали этилацетатом, высушивали над безводным сульфатом магния и фильтровали. Фильтрат перегоняли при пониженном давлении, и концентрат очищали хроматографией на колонках, получая целевое соединение как продукт реакции циклизации (148 мг, выход 60%).
1H-ЯМР (400 МГц, CDCl3); δ 10,49 (ушир.с, 1H), 8,24 (д, J=8,0 Гц, 1H), 7,98 (д, J=7,6 Гц, 1H), 7,23 (т, 1H), 7,02 (с, 1H), 4,47 (т, 2H), 3,51 (т, 2H).
Пример получения 102
Изопропиловый эфир [(S)-2-(5-хлор-7-нитро-1Н-индол-2-ил)-4,5-дигидрооксазол-4-ил]-карбоновой кислоты
Стадия A: изопропиловый эфир (S)-3-амино-4-гидрокси-масляной кислоты
4-Изопропил-1-метиловый эфир (S)-2-BOC-амино-янтарной кислоты, полученный на стадии А примера получения 30, вводили в реакцию согласно тем же самым процедурам, как на стадиях А-D примера получения 29, последовательно, получая целевое соединение.
Стадия B: изопропиловый эфир [(S)-3-(5-хлор-7-нитро-1Н-индол-2-карбонил)амино]-4-гидроксимасляной кислоты
5-Хлор-7-нитро-1Н-индол-2-карбоновую кислоту, полученную гидролизом метилового эфира 5-хлор-7-нитро-1Н-индол-2-карбоновой кислоты, полученного в примере получения 14 согласно стадии А примера получения 40, и изопропиловый эфир (S)-3-амино-4-гидрокси-масляной кислоты, полученный на стадии A, вводили в реакцию согласно той же самой процедуре, как на стадии B примера получения 40, получая целевое соединение.
Стадия C: изопропиловый эфир [(S)-3-(5-хлор-7-нитро-1Н-индол-2-карбонил)амино]-4-метансульфонилокси-масляной кислоты
Изопропиловый эфир [(S)-3-(5-хлор-7-нитро-1Н-индол-2-карбонил)амино]-4-гидроксимасляной кислоты, полученный на стадии B, вводили в реакцию согласно той же самой процедуре, как на стадии B примера получения 29, получая целевое соединение.
Стадия D: изопропиловый эфир [(S)-2-(5-хлор-7-нитро-1Н-индол-2-ил)-4,5-дигидрооксазол-4-ил]карбоновой кислоты
Изопропиловый эфир [(S)-3-(5-хлор-7-нитро-1Н-индол-2-карбонил)амино]-4-метансульфонилокси-масляной кислоты (930 мг, 2 ммоль), полученный на стадии C, добавляли к THF (10 мл). Добавляли K2CO3 (330 мг, 10 ммоль), и смесь перемешивали в течение 2 часов при 80°C. Добавляли воду, чтобы завершить реакцию. Реакционную смесь экстрагировали EtOAc и высушивали над MgSO4. Растворитель удаляли при пониженном давлении, и остаток очищали хроматографией на колонках (элюент: EtOAc/n-Hex/DMC=1/4/1), получая целевое соединение (445 мг, выход 61%).
Масса [M+H]=365 (M+1).
Пример получения 103
трет-Бутиловый эфир (1S,2R)-(3-амино-1-бензил-2-гидрокси-пропил)карбаминовой кислоты
Стадия A: трет-бутиловый эфир (1S,2R)-(3-азидо-1-бензил-2-гидрокси-пропил)карбаминовой кислоты
Коммерчески доступный трет-бутиловый эфир (1-оксиран-2-фенил-этил)карбаминовой кислоты (2,6 г, 10 ммоль) растворяли в DMF (30 мл). Добавляли азид натрия (655 мг, 10 ммоль), и смесь перемешивали в течение 12 часов при 80°C. Добавляли воду, чтобы завершить реакцию. Реакционную смесь экстрагировали EtOAc и высушивали над MgSO4. Растворитель удаляли при пониженном давлении, и остаток очищали хроматографией на колонках, получая целевое соединение (2,75 г, выход 90%).
Стадия B: трет-бутиловый эфир (1S,2R)-(3-амино-1-бензил-2-гидрокси-пропил)карбаминовой кислоты
Трет-бутиловый эфир (1S,2R)-(3-азидо-1-бензил-2-гидрокси-пропил)карбаминовой кислоты (2,5 г, 8,17 ммоль), полученный на Стадии A, растворяли в метаноле (15 мл), добавляли Pd/C (100 мг). Смесь вводили в реакцию в течение 12 часов в водородном реакторе (50 psi), и фильтровали через Целит. Растворитель удаляли при пониженном давлении, и остаток использовали в следующей реакции без дальнейшей очистки.
Масса [M+H]=280 (M+1).
Пример получения 104
трет-Бутиловый эфир [(1S,2R)-1-бензил-3-(1,3-диоксо-1,3-дигидро-изоиндол-2-ил)-2-гидроксипропил]карбаминовой кислоты
Трет-бутиловый эфир (1R,2S)-(3-амино-1-бензил-2-гидрокси-пропил)карбаминовой кислоты (1,4 г, 5 ммоль), полученный в Примере получения 103, растворяли в DMF (20 мл), добавляли фталевый ангидрид (735 мг, 5 ммоль). Добавляли Et3N (1,4 мл, 10 ммоль), и смесь перемешивали в течение 24 часов при 80°C. Реакционную смесь гасили водой, и реакционную смесь экстрагировали EtOAc и высушивали над MgSO4. Растворитель удаляли при пониженном давлении, и остаток растворяли в DCM. Добавляли TFA, и смесь перемешивали в течение 2 часов при температуре окружающей среды. После завершения реакции растворитель удаляли при пониженном давлении. Остаток перекристаллизовывали из DCM/Hex, получая целевое соединение (1,18 г, выход 71%).
Масса [M+H]=410 (M+1).
Примеры получения 105-109
Эфиры индол-2-карбоновой кислоты, полученные в примерах получения 14, 15 и 17, и производные аминоэтанола, которые являются коммерчески доступными или могут быть получены в примерах получения 103 и 104, вводили в реакцию согласно той же самой процедуре, как в примере получения 101, чтобы синтезировать соединения примера получения, приведенные в следующей таблице.


Пример получения 110
2-(Тиазол-2-ил)-7-нитро-1Н-индол
Стадия A: амид 7-Нитро-1Н-индол-2-тиокарбоновой кислоты
7-Нитро-1Н-индол-2-карбоновую кислоту, полученную на стадии А примера получения 101, и хлорид аммония вводили в реакцию согласно тем же самым процедурам, как на стадиях B и C примера получения 101, получая целевое соединение.
Стадия B: 2-(Тиазол-2-ил)-7-нитро-1Н-индол
Амид 7-Нитро-1Н-индол-2-тиокарбоновой кислоты (1,15 г, 5 ммоль), полученный на стадии A, растворяли в DMF (15 мл). Добавляли 2-бром-1,1-диоксиэтан (985 мг, 5 ммоль), и смесь перемешивали с обратным холодильником в течение 2 часов при 100°C. Реакционную смесь гасили водой, и реакционную смесь экстрагировали EtOAc и высушивали над MgSO4. Растворитель удаляли при пониженном давлении, и остаток очищали хроматографией на колонках, получая целевое соединение (612 мг, выход 65%).
Масса [M+H]=245 (M+1).
Пример получения 111
Метиловый эфир 2-(7-нитро-1Н-индол-2-ил)тиазол-4-карбоновой кислоты
Метиловый эфир 3-хлор-2-оксо-пропионовой кислоты и амид 7-нитро-1Н-индол-2-тиокарбоновой кислоты, полученный на стадии А примера получения 110, вводили в реакцию согласно той же самой процедуре, как в примере получения 110, получая целевое соединение.
Масса [M+H]=303 (M+1).
Пример получения 112
[2-(7-нитро-1Н-индол-2-ил)тиазол-4-ил]метанол
Метиловый эфир 2-(7-нитро-1Н-индол-2-ил)тиазол-4-карбоновой кислоты, полученный в примере получения 111, вводили в реакцию согласно тем же самым процедурам, как на стадии А примера получения 29, получая целевое соединение.
Масса [M+H]=275 (M+1).
Пример получения 113
Этиловый эфир 2-(5-метил-7-нитро-1Н-индол-2-ил)тиазол-4-карбоновой кислоты
Стадия A: амид (5-метил-7-нитро-1Н-индол-2-ил)карботиокислоты
(5-Метил-7-нитро-1Н-индол-2-ил)карбоновую кислоту вводили в реакцию согласно той же самой процедуре, как на стадии А примера получения 110, получая целевое соединение.
Стадия B: этиловый эфир 2-(5-метил-7-нитро-1Н-индол-2-ил)-тиазол-4-карбоновой кислоты
Амид (5-метил-7-нитро-1Н-индол-2-ил)карботиокислоты, полученный на стадии A, и этиловый эфир 3-хлор-2-оксо-пропионовой кислоты вводили в реакцию согласно той же самой процедуре, как на стадии B примера получения 110, получая целевое соединение.
Масса [M+H]=331 (M+1).
Пример получения 114
[2-(5-метил-7-нитро-1Н-индол-2-ил)тиазол-4-ил]метанол
Метиловый эфир 2-(5-метил-7-нитро-1Н-индол-2-ил)тиазол-4-карбоновой кислоты, полученный в примере получения 113, вводили в реакцию согласно той же самой процедуре, как на стадии А примера получения 29, получая целевое соединение.
Масса [M+H]=289 (M+1).
Пример получения 115
Метиловый эфир [5-(7-нитро-1Н-индол-2-ил)-[1,2,4]оксадиазол-3-ил]уксусной кислоты
Стадия A: метиловый эфир 2-(N-гидроксикарбамимидоил)уксусной кислоты
Метиловый эфир 2-цианоуксусной кислоты (990 мг, 10 ммоль) растворяли в THF (30 мл). Добавляли гидроксиамин (690 мг, соль HCl, 10 ммоль), и смесь перемешивали в течение 2 часов при 80°C. После завершения реакции добавляли 1н. HCl. Реакционную смесь экстрагировали EtOAc и высушивали над MgSO4. Растворитель удаляли при пониженном давлении, и остаток использовали в следующей реакции без дальнейшей очистки.
Стадия B: метиловый эфир 3-(Гидроксиимино)-3-[(7-нитро-1Н-индол-2-карбонил)амино]пропионовой кислоты
Метиловый эфир 2-(N-гидроксикарбамимидоил)уксусной кислоты, полученный на стадии A, и 7-нитро-1Н-индол-2-карбоновую кислоту, полученную на стадии А примера получения 101, вводили в реакцию согласно той же самой процедуре, как на стадии B примера получения 101, получая целевое соединение.
Стадия C: метиловый эфир 2-[5-(7-нитро-1Н-индол-2-ил)-[1,2,4]оксадиазол-3-ил]уксусной кислоты
Метиловый эфир 3-(гидроксиимино)-3-[(7-нитро-1Н-индол-2-карбонил)амино]пропионовой кислоты (960 мг, 1 ммоль), полученный на стадии B, растворяли в DMF (10 мл). Добавляли пиридин (1 мл), и смесь перемешивали в течение 4 часов при 80°C. Реакционную смесь гасили водным раствором NH4Cl. Реакционную смесь экстрагировали EtOAc и высушивали над MgSO4. Растворитель удаляли при пониженном давлении, и остаток очищали хроматографией на колонках, получая целевое соединение (830 мг, выход 90%).
Масса [M+H]=302 (M+1).
Пример получения 116
2-[5-(7-нитро-1Н-индол-2-ил)-[1,2,4]оксадиазол-3-ил]этанол
Метиловый эфир 2-[5-(7-нитро-1Н-индол-2-ил)-[1,2,4]оксадиазол-3-ил]уксусной кислоты, полученный в примере получения 115, вводили в реакцию согласно той же самой процедуре, как на стадии А примера получения 29, получая целевое соединение.
Масса [M+H]=274 (M+1).
Пример получения 117
5-метил-7-нитро-2-[1,3,4]оксадиазол-2-ил-1Н-индол
Метиловый эфир 5-метил-7-нитро-1Н-индол-2-карбоновой кислоты (234 мг, 1 ммоль), полученный в примере получения 15, растворяли в метаноле (10 мл), добавляли гидразин (3 мл). Реакционный раствор нагревали с обратным холодильником в течение 3 часов и концентрировали при пониженном давлении. К концентрату добавляли триметоксиметан (10 мл), и смесь нагревали с обратным холодильником в течение 8 часов. Реакционную смесь перегоняли при пониженном давлении, и полученное твердое вещество промывали этилацетатом, получая целевое соединение (49 мг, выход 20%).
Пример 1
Циклопентил-[2-(4,5-дигидро-1,3-тиазол-2-ил)-1Н-индол-7-ил]амин
Стадия A: [2-(4,5-Дигидро-1,3-тиазол-2-ил)-1Н-индол-7-ил]амин
2-(4,5-Дигидро-тиазол-2-ил)-7-нитро-1Н-индол, полученный в примере получения 101, вводили в реакцию согласно той же самой процедуре, как на стадии B примера получения 1, получая целевое соединение.
Стадия B: Циклопентил-[2-(4,5-дигидро-1,3-тиазол-2-ил)-1Н-индол-7-ил]амин
[2-(4,5-Дигидро-1,3-тиазол-2-ил)-1Н-индол-7-ил]амин (15 мг, 0,07 ммоль), полученный на стадии A, растворяли в 1,2-дихлорэтане (10 мл). Добавляли циклопентанон (12 мг, 0,14 ммоль) и триацетоксиборгидрид натрия (29 мг, 0,14 ммоль), и смесь перемешивали в течение 3 часов при температуре окружающей среды. Реакционную смесь гасили водой, и реакционную смесь экстрагировали этилацетатом, высушивали над безводным сульфатом магния и фильтровали. Растворитель удаляли при пониженном давлении, и остаток очищали хроматографией на колонках, получая целевое соединение (6,7 мг, выход 34%).
1H-ЯМР (400 МГц, CDCl3); δ 10,27 (c, 1H), 7,06 (д, J=8,0 Гц, 1H), 7,00 (т, J=7,6 Гц, 1H), 6,92 (c, 1H), 6,52 (д, J=7,2 Гц, 1H), 4,42 (м, 2H), 4,38 (м, 1H), 4,35 (м, 2H), 2,00 (м, 2H), 1,64 (м, 4H), 1,46 (м, 2H).
Пример 2
[(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-1,3-тиазол-4-ил]метанол
Стадия A: [(R)-2-(7-циклопентиламино)-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метиловый эфир 2,2-диметилпропионовой кислоты
(R)-2-(7-амино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-илметиловый эфир 2,2-диметилпропионовой кислоты, полученный в примере получения 40, вводили в реакцию согласно той же самой процедуре, как на стадии B примера 1, получая целевое соединение.
Стадия B: [(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-1,3-тиазол-4-ил]метанол
[(R)-2-(7-циклопентиламино)-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метиловый эфир 2,2-диметилпропионовой кислоты, полученный на стадии A, вводили в реакцию согласно той же самой процедуре, как на стадии А примера получения 29, получая целевое соединение.
1H-ЯМР (500 МГц, CDCl3); δ 11,17~11,08 (м, 1H), 7,09 (м, 1H), 6,99 (т, 1H), 6,96 (c, 1H), 6,52 (м, 1H), 4,72 (м, 1H), 4,04 (м, 1H), 3,75 (м, 1H), 3,65 (м, 1H), 3,51 (м, 1H), 3,40 (м, 1H), 1,90 (м, 2H), 1,60~1,49 (м, 4H), 1,41~1,24 (м, 2H).
Пример 3
Метиловый эфир [(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты
Метиловый эфир [(R)-2-(5-хлор-7-нитро-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты, полученный в примере получения 67, и циклопентанон вводили в реакцию согласно тем же самым процедурам, как на стадиях A и B примера 1, получая целевое соединение.
1H ЯМР (ДМСО-d6, м.д.); δ 11,51 (c, 1H), 6,79 (c, 1H), 6,79 (c, 1H), 6,16 (c, 1H), 6,13 (д, 1H), 4,85 (м, 1H), 3,80 (м, 1H), 3,62 (м, 1H), 3,58 (c, 3H), 3,19 (м, 1H), 2,71 (м, 1H), 2,63 (м, 1H), 1,93 (м, 2H), 1,69 (м, 2H), 1,56 (м, 4H).
Пример 4
[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота
Метиловый эфир [(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты, полученный в примере 3, вводили в реакцию согласно той же самой процедуре, как на стадии А примера получения 29, получая целевое соединение.
1H ЯМР (ДМСО-d6, м.д.); δ 12,51 (ушир.c, 1H), 11,51 (c, 1H), 6,79 (c, 1H), 6,79 (c, 1H), 6,16 (c, 1H), 6,14 (д, 1H), 4,87 (м, 1H), 3,80 (м, 1H), 3,61 (м, 1H), 3,19 (м, 1H), 2,72 (м, 1H), 2,64 (м, 1H), 1,93 (м, 2H), 1,69 (м, 2H), 1,56 (м, 4H).
Пример 5
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этанол
[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-уксусную кислоту (391 мг, 1 ммоль), полученную в примере 4, растворяли в DCM (10 мл). Добавляли триэтиламин (280 мкл, 2 ммоль) и хлорангидрид изомасляной кислоты (106 мг, 1 ммоль), и смесь перемешивали в течение 30 минут при 0°C. После завершения реакции растворитель удаляли при пониженном давлении. Остаток разбавляли THF. Добавляли NaBH4 (74 мг, 2 ммоль), и смесь перемешивали в течение 12 часов. Реакционную смесь гасили небольшим количеством воды. К реакционной смеси опять добавляли избыток воды, затем смесь перемешивали в течение 30 минут, экстрагировали EtOAc, высушивали над MgSO4 и фильтровали. Растворитель удаляли при пониженном давлении, и остаток очищали хроматографией на колонках, получая целевое соединение (272 мг, выход 72%).
1H ЯМР (400 МГц, ДМСО-d6); δ 11,47 (с, 1H), 6,79 (с, 1H), 6,67 (с, 1H), 6,11 (с, 1H), 6,09 (м, 1H), 4,65 (т, 1H), 4,54 (м, 1H), 3,80 (м, 2H), 3,61 (м, 2H), 3,52 (м, 1H), 3,15 (м, 1H), 2,47 (м, 1H), 1,97 (м, 2H), 1,68 (м, 2H), 1,54 (м, 4H).
Пример 6
(R)-2-[7-циклопентиламино-5-(гидроксиметил)-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-илметанол
Стадия A: (R)-2-[7-циклопентиламино-5-(2,2-диметил-пропионилоксиметил)-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-илметиловый эфир 2,2-диметилпропионовой кислоты
(R)-2-[5-(2,2-диметил-пропионилоксиметил)-7-нитро-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-илметиловый эфир 2,2-диметилпропионовой кислоты, полученный в примере получения 43, и циклопентанон вводили в реакцию согласно той же самой процедуре, как в примере 1, получая целевое соединение.
Стадия B: (R)-2-[7-циклопентиламино-5-(гидроксиметил)-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-илметанол
(R)-2-[7-циклопентиламино-5-(2,2-диметил-пропионилоксиметил)-1Н-индол-2-ил]амино}-4,5-дигидро-тиазол-4-илметиловый эфир 2,2-диметилпропионовой кислоты, полученный на стадии A, вводили в реакцию согласно той же самой процедуре, как на стадии А примера получения 29, получая целевое соединение.
1H-ЯМР (400 МГц, CDCl3); δ 9,63 (ушир.c, 1H), 7,40 (c, 1H), 7,18 (c, 1H), 6,90 (c, 1H), 4,80 (м, 1H), 4,73 (c, 2H), 4,06 (м, 1H), 3,84 (м, 1H), 3,66 (м, 2H), 3,48 (м, 1H), 3,31 (м, 1H), 1,79 (м, 2H), 1,43 (м, 4H), 1,26 (м, 2H).
Пример 7
[2-(4,5-Дигидро-тиазол-2-ил)-1Н-индол-7-ил]пиперидин-4-иламин
Стадия A: [2-(4,5-Дигидро-тиазол-2-ил)-1Н-индол-7-ил]-(1-BOC-пиперидин-4-ил)амин
2-(4,5-Дигидро-тиазол-2-ил)-7-нитро-1Н-индол, полученный в примере получения 101, и 1-BOC-4-пиперидон вводили в реакцию согласно той же самой процедуре, как в примере 1, получая целевое соединение.
Стадия B: [2-(4,5-Дигидро-тиазол-2-ил)-1Н-индол-7-ил]пиперидин-4-иламин
[2-(4,5-Дигидро-тиазол-2-ил)-1Н-индол-7-ил]-(1-BOC-пиперидин-4-ил)амин, полученный на стадии A, вводили в реакцию согласно той же самой процедуре, как на стадии D примера получения 29, получая целевое соединение.
1H-ЯМР (400 МГц, CDCl3, MeOH-d4); δ 7,39 (c, 1H), 7,07 (д, J=8,0 Гц, 1H), 6,99 (т, J=8,0 Гц, 1H), 6,93 (c, 1H), 6,47 (д, J=7,6 Гц, 1H), 4,41 (м, 2H), 3,77 (м, 1H), 3,48 (м, 4H), 3,11 (м, 2H), 2,29 (м, 2H), 1,87 (м, 2H).
Примеры 8-117
Соединения, полученные в примерах получения 40, 48-100, вводили в реакцию, чтобы синтезировать соединения из примеров, показанных в следующей таблице, согласно способу, выбранному из примеров 1-7.


Примеры 118-123
Соединения, полученные в примерах получения 102 и 105-109, вводили в реакцию, чтобы синтезировать соединения из примеров, показанных в следующей таблице, согласно способу, выбранному из примеров 1-7.

Пример 124
[2-((4S,5R)-5-аминометил-4-бензил-дигидро-оксазол-2-ил)-5-хлор-1Н-индол-7-ил]циклопентил-амин
Стадия A: 2-[(4S,5R)-бензил-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-оксазол-5-илметил]изоиндол-1,3-дион
2-[(4S,5R)-бензил-2-(5-хлор-7-нитро-1Н-индол-2-ил)-4,5-дигидро-оксазол-5-илметил]изоиндол-1,3-дион, полученный в примере получения 108, и циклопентанон вводили в реакцию согласно той же самой процедуре, как в примере 1, получая целевое соединение.
Стадия B: [2-((4S,5R)-5-аминометил-4-бензил-дигидро-оксазол-2-ил)-5-хлор-1Н-индол-7-ил]циклопентил-амин
2-[(4S,5R)-бензил-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-оксазол-5-илметил]изоиндол-1,3-дион (50 мг, 1 ммоль), полученный на стадии A, растворяли в этаноле (10 мл). Добавляли гидразин хлорид (1,8 мл, 0,33 ммоль), и смесь перемешивали в течение 4 часов при 80°C. После завершения реакции растворитель удаляли при пониженном давлении, и остаток очищали хроматографией на колонках, получая целевое соединение (16 мг, выход 39%).
1H-ЯМР (400 МГц, MeOH-d4); δ 7,36-7,18 (6H, м), 6,95 (1H, c), 6,75 (1H, c), 6,27 (1H, д, J=1,2 Гц), 3,84 (1H, ушир.c), 3,77 (1H, ушир.c), 3,66-3,56 (1H, м), 3,34 (1H, ушир.c), 2,77 (2H, ушир.c), 1,93 (2H, ушир.c), 1,54-1,48 (6H, м).
Пример 125
{2-[(R)-5-((S)-1-амино-2-фенил-этил)-4,5-дигидро-оксазол-2-ил]-5-хлор-1Н-индол-7-ил}циклопентил-амин
Трет-бутиловый эфир {(S)-1-[(R)-2-(5-хлор-7-нитро-1Н-индол-2-ил)-4,5-дигидро-оксазол-5-ил]-2-фенилэтил}карбаминовой кислоты, полученный в примере получения 107, вводили в реакцию согласно той же самой процедуре, как в примере 1, и полученный таким образом продукт (50 мг, 1 ммоль) растворяли в DCM (2 мл). Добавляли TFA (2 мл), и смесь перемешивали в течение 2 часов. Растворитель удаляли при пониженном давлении, и остаток очищали хроматографией на колонках, получая целевое соединение (38 мг, выход 92%).
1H-ЯМР (400 МГц, MeOH-d4); δ 10,4 (1H, ушир.c), 7,34-7,31 (2H, м), 7,27-7,20 (3H, м), 7,03 (1H, д, J=1,6 Гц), 6,95 (1H, c), 6,46 (1H, д, J=1,6 Гц), 4,65-4,59 (1H, м), 4,25-4,04 (2H, м), 3,92-3,83 (2H, м), 3,15-3,07 (1H, м), 2,90-2,85 (1H, м), 2,72-2,64 (1H, м), 2,05-2,00 (2H, м), 1,58 (2H, ушир.c), 1,68-1,63 (4H, м), 1,49-1,47 (2H, м).
Примеры 126-134
Соединения, полученные в примерах получения 110-117, вводили в реакцию, чтобы синтезировать соединения из примеров, показанные в следующей таблице, согласно способу, выбранному из примеров 1-7.

Пример 135
{(R)-2-[5-метил-7-(4-оксо-циклогексиламино)-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-ил}уксусная кислота
Стадия A: этиловый эфир {(R)-2-[7-(1,4-диокса-спиро[4,5]дец-8-иламино)-5-метил-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-ил}уксусной кислоты
Этиловый эфир [(R)-2-(5-метил-7-нитро-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты, полученный в примере получения 54, и 1,4-диокса-спиро[4,5]декан-8-он вводили в реакцию согласно той же самой процедуре, как в примере 1, получая целевое соединение.
Стадия B: {(R)-2-[5-метил-7-(4-оксо-циклогексиламино)-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-ил}уксусная кислота
Этиловый эфир {(R)-2-[7-(1,4-диокса-спиро[4,5]дец-8-ил)амино-5-метил-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-ил}уксусной кислоты (177 мг, 4 ммоль), полученный на стадии A, растворяли в метаноле (10 мл). Добавляли HCl (конц. 2 мл), и смесь вводили в реакцию в течение 6 часов при 60°C. Растворитель удаляли при пониженном давлении, и к остатку добавляли воду. Реакционную смесь экстрагировали EtOAc, высушивали, концентрировали и очищали ВЭЖХ, получая целевое соединение (76 мг, выход 50%).
1H-ЯМР (500 МГц, CDCl3); δ 11,99 (ушир.с, 1H), 7,00 (c, 1H), 6,79 (c, 1H), 6,30 (c, 1H), 5,34 (м, 1H), 3,89 (м, 1H), 3,71 (м, 1H), 3,21 (м, 1H), 2,66 (м, 2H), 2,59 (м, 2H), 2,43-2,35 (м, 5H), 2,26 (м, 2H), 1,97 (м, 2H).
Пример 136
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-морфолин-4-илэтанон
[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-уксусную кислоту, полученную в примере 4, и морфолин вводили в реакцию согласно той же самой процедуре, как на стадии B примера получения 101, получая целевое соединение.
1H-ЯМР (500 МГц, ДМСО-d6); δ 11,52 (ушир.c, 1H), 6,80 (c, 1H), 6,69 (c, 1H), 6,16 (c, 1H), 6,12 (м, 1H), 4,95 (м, 1H), 3,81 (м, 1H), 3,63 (м, 1H), 3,41 (м, 8H), 3,12 (м, 1H), 2,85 (м, 1H), 2,69 (м, 1H), 1,93 (м, 2H), 1,68 (м, 2H), 1,56 (м, 4H).
Примеры 137-155
Соединения, полученные в примерах 4, 11, 66, 71, 75, 81 и 101, и коммерчески доступные амины вводили в реакцию, чтобы синтезировать соединения из примеров, показанные в следующей таблице, согласно той же самой процедуре, как на стадии B примера получения 101.

Пример 156
Циклопентил-{5-метансульфонилметил-2-[(R)-4-(2-морфолин-4-илэтил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}амин
Стадия A: Циклопентил-{2-[(R)-4-(2-йодэтил)-4,5-дигидро-тиазол-2-ил]-5-метансульфонилметил-1Н-индол-7-ил}амин
2-[(R)-2-(7-циклопентиламино-5-метансульфонилметил-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этанол (178 мг, 0,42 ммоль), полученный в примере 99, растворяли в тетрагидрофуране (10 мл). Добавляли йод (161 мг, 0,63 ммоль), трифенилфосфин (166 мг, 0,63 ммоль) и имидазол (86 мг, 1,23 ммоль), и смесь перемешивали в течение 8 часов при температуре окружающей среды. Реакционную смесь гасили водой, и реакционную смесь экстрагировали этилацетатом. Экстракт промывали насыщенным водным раствором хлорида натрия, высушивали над безводным сульфатом магния и фильтровали. Растворитель удаляли при пониженном давлении, и остаток очищали хроматографией на колонках, получая целевое соединение (120 мг, выход 54%).
Стадия B: Циклопентил-{5-метансульфонилметил-2-[(R)-4-(2-морфолин-4-илэтил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}амин
Циклопентил-{2-[(R)-4-(2-йодэтил)-4,5-дигидро-тиазол-2-ил]-5-метансульфонилметил-1Н-индол-7-ил}амин (116 мг, 0,22 ммоль), полученный на стадии A, растворяли в N,N-диметилформамиде (4 мл). Добавляли морфолин (57 мг, 0,66 ммоль), и смесь перемешивали в течение 8 часов при температуре окружающей среды. Реакционную смесь гасили водой, и реакционную смесь экстрагировали этилацетатом. Экстракт высушивали над безводным сульфатом магния. Растворитель удаляли при пониженном давлении, и остаток очищали хроматографией на колонках, получая целевое соединение (68 мг, выход 64%).
1H-ЯМР (500 МГц, CDCl3); δ 10,60 (ушир.c, 1H), 6,99 (c, 1H), 6,89 (c, 1H), 6,49 (c, 1H), 4,79 (м, 1H), 4,26 (c, 2H), 3,86 (м, 1H), 3,57 (м, 5H), 3,19 (м, 1H), 2,72 (c, 3H), 2,45 (м, 2H), 2,32 (м, 2H), 2,26 (м, 2H), 2,04 (м, 2H), 1,80 (м, 2H), 1,66 (м, 4H), 1,41 (м, 2H).
Пример 157
1-(4-{2-[(R)-2-(7-циклопентиламино-5-метансульфонилметил-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-ил)этанон
Циклопентил-{2-[(R)-4-(2-йодэтил)-4,5-дигидро-тиазол-2-ил]-5-метансульфонилметил-1Н-индол-7-ил}амин, полученный на стадии А примера 156, и ацетилпиперазин вводили в реакцию согласно той же самой процедуре, как на стадии B примера 156, получая целевое соединение.
1H-ЯМР (500 МГц, CDCl3); δ 10,62 (ушир.c, 1H), 6,99 (c, 1H), 6,89 (c, 1H), 6,46 (c, 1H), 4,77 (м, 1H), 4,26 (c, 2H), 3,87 (м, 1H), 3,57 (м, 1H), 3,30 (м, 2H), 3,16 (м, 1H), 2,72 (c, 3H), 2,46 (м, 2H), 2,31 (м, 2H), 2,21 (м, 2H), 2,04 (c, 3H), 2,03 (м, 2H), 1,79 (м, 2H), 1,64 (м, 4H), 1,45 (м, 2H).
Пример 158
Циклопентил-[2-((R)-4-пирролидин-1-илметил-4,5-дигидро-тиазол-2-ил)-1Н-индол-7-ил]амин
[(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол, полученный в примере 2, и пирролидин вводили в реакцию согласно той же самой процедуре, как в примере 156, получая целевое соединение.
1H-ЯМР (400 МГц, ДМСО-d6); δ 11,37 (ушир.с, 1H), 6,83 (м, 1H), 6,75 (д, J=2,0 Гц, 1H), 6,29 (д, J=8,0 Гц, 1H), 5,86 (д, J=8,0 Гц, 1H), 4,80 (м, 1H), 3,87 (м, 1H), 3,52 (м, 1H), 3,43 (м, 1H), 3,33 (м, 2H), 2,78 (м, 2H), 2,61 (м, 2H), 1,99 (м, 2H), 1,72 (м, 6H), 1,60 (м, 4H).
Пример 159
{5-хлор-2-[(R)-4-(2-диметиламино-этил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}циклопентил-амин
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этанол, полученный в примере 5, и диметиламин вводили в реакцию согласно той же самой процедуре, как в примере 156, получая целевое соединение.
1H-ЯМР (500 МГц, CDCl3); δ 10,07 (ушир.с, 1H), 6,99 (с, 1H), 6,80 (с, 1H), 6,42 (с, 1H), 4,67 (м, 1H), 3,54 (м, 1H), 3,16 (м, 1H), 2,46 (м, 1H), 2,37 (м, 1H), 2,19 (с, 6H), 2,02 (м, 3H), 1,81 (м, 4H), 1,69 (м, 4H).
Пример 160
{5-Хлор-2-[(R)-4-(2-пиперазин-1-илэтил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}циклопентил-амин
Стадия A: {5-хлор-2-[(R)-4-(2-1-BOC-пиперазин-4-илэтил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}циклопентил-амин
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этанол, полученный в примере 5, и 1-ВОС-пиперазин вводили в реакцию согласно той же самой процедуре, как в примере 156, получая целевое соединение.
Стадия B: {5-хлор-2-[(R)-4-(2-пиперазин-1-илэтил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}циклопентил-амин
{5-хлор-2-[(R)-4-(2-1-BOC-пиперазин-4-илэтил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}циклопентил-амин, полученный на стадии A, растворяли в дихлорметане (50 мл). Добавляли 4 н. раствор соляной кислоты в этилацетате (1,3 мл, 5,28 ммоль), и смесь перемешивали в течение 4 часов при температуре окружающей среды. После завершения реакции растворитель удаляли при пониженном давлении. Остаток перекристаллизовывали из DCM и простого этилового эфира, получая целевое соединение (125 мг, выход 55%).
1H ЯМР (ДМСО-d6, м.д.); δ 11,48 (1H, с), 6,79 (1H, с), 6,67 (1H, с), 6,11 (1H, с), 6,10 (1H, д), 4,61 (1H, м), 3,80 (1H, м), 3,54 (1H, м), 3,15 (1H, м), 2,93 (2H, м), 2,50-2,41 (2H, м), 2,31 (3H, м), 1,95 (4H, м), 1,79 (1H, м), 1,68 (3H, м), 1,57-1,50 (4H, м), 1,20 (1H, м).
FAB MC (m/e)=432.
Пример 161
(5-хлор-2-{(R)-4-[2-(4-этансульфонил-пиперазин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин
{5-хлор-2-[(R)-4-(2-пиперазин-1-илэтил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}циклопентил-амин, полученный в примере 160, и этилсульфонилхлорид вводили в реакцию согласно той же самой процедуре, как на стадии B примера получения 29, получая целевое соединение.
1H-ЯМР (500 МГц, CDCl3); δ 11,29 (ушир.с, 1H), 6,97 (с, 1H), 6,86 (с, 1H), 6,37 (с, 1H), 4,93 (м, 1H), 3,92 (ушир.с, 1H), 3,77 (м, 1H), 3,57 (м, 1H), 3,16 (м, 1H), 2,95 (м, 2H), 2,80 (м, 4H), 2,42-2,28 (м, 4H), 2,03 (м, 4H), 1,74 (м, 3H), 1,63 (м, 4H), 1,43 (м, 1H), 1,32 (т, 3H).
Пример 162
1-(4-{2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-ил)-2-гидрокси-этанон
{5-хлор-2-[(R)-4-(2-пиперазин-1-илэтил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}циклопентил-амин, полученный в примере 160, и гликолевую кислоту вводили в реакцию согласно той же самой процедуре, как на стадии B примера получения 101, получая целевое соединение.
1H-ЯМР (500 МГц, ДМСО-d6); δ 11,47 (ушир.с, 1H), 6,79 (с, 1H), 6,68 (с, 1H), 6,16 (с, 1H), 6,10 (м, 1H), 4,63 (м, 1H), 4,50 (м, 1H), 4,04 (м, 2H), 3,81 (м, 1H), 3,55 (м, 1H), 3,43 (м, 2H), 3,16 (м, 1H), 2,52 (м, 2H), 2,35 (м, 4H), 1,95 (м, 3H), 1,81 (м, 1H), 1,68 (м, 2H), 1,53 (м, 4H).
Пример 163
{5-хлор-2-[(R)-4-(2-пиразол-1-илэтил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}циклопентил-амин
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этанол (81 мг, 0,11 ммоль), полученный в примере 5, растворяли в тетрагидрофуране (4 мл). Добавляли йод (13,2 мг, 0,11 ммоль) и имидазол (9,7 мг, 0,14 ммоль), и смесь перемешивали в течение 2 часов при температуре окружающей среды. После завершения реакции реакционную смесь фильтровали, чтобы удалить твердую часть. Растворитель удаляли при пониженном давлении, и к остатку добавляли тетрагидрофуран (4 мл). Добавляли пиразол (58 мг, 0,85 ммоль) и гидрид натрия (60% в минеральном масле, 21 мг, 0,85 ммоль), и смесь перемешивали в течение 8 часов при температуре окружающей среды. Реакционную смесь гасили водой, и реакционную смесь экстрагировали этилацетатом, высушивали над безводным сульфатом магния и фильтровали. Растворитель удаляли при пониженном давлении, и остаток очищали хроматографией на колонках, получая целевое соединение (24 мг, выход 34%).
1H ЯМР (ДМСО-d6, м.д.); δ 11,50 (1H, с), 7,76 (1H, с), 7,42 (1H, с), 6,80 (1H, с), 6,70 (1H, с), 6,22 (1H, с), 6,17 (1H, с), 6,11 (1H, д), 4,49 (1H, кв), 4,32 (2H, м), 3,80 (1H, м), 3,53 (1H, т), 3,12 (1H, т), 2,38 (1H, м), 2,14 (1H, м), 1,92 (2H, м), 1,68 (2H, м), 1,59-1,50 (4H, м).
Пример 164
(S)-1-{2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пирролидин-2-карбоновая кислота
Стадия A: метиловый эфир 2-{2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пирролидин-2-карбоновой кислоты
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этанол, полученный в примере 5, и метиловый эфир (S)-пирролидин-2-илкарбоновой кислоты вводили в реакцию согласно той же самой процедуре, как в примере 156, получая целевое соединение.
Стадия B: (S)-1-{2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пирролидин-2-карбоновая кислота
Метиловый эфир 2-{2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пирролидин-2-карбоновой кислоты, полученный на стадии A, гидролизовали согласно той же самой процедуре, как на стадии А примера получения 101, получая целевое соединение.
1H ЯМР (CDCl3, м.д.); δ 12,04 (1H, с), 11,02 (1H, с), 6,85 (1H, с), 6,69 (1H, с), 6,31 (1H, с), 6,24 (1H, м), 4,37 (1H, м), 4,10 (1H, м), 3,86 (1H, м), 3,79 (1H, м), 3,59 (1H, м), 3,28 (1H, м), 3,17 (1H, м), 2,88 (2H, м), 2,59 (1H, м), 2,21 (1H, м), 2,06-1,59 (11H, м), 1,23 (1H, м).
Пример 165
{5-хлор-2-[(R)-4-(2-метансульфонил-этил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}циклопентил-амин
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этанол (50 мг, 0,11 ммоль), полученный в примере 5, растворяли в N,N-диметилформамиде (2 мл). Добавляли метансульфонат натрия (54 мг, 0,55 ммоль), и смесь перемешивали в течение 8 часов при температуре окружающей среды. Реакционную смесь гасили водой. Реакционную смесь экстрагировали этилацетатом, высушивали над безводным сульфатом магния и фильтровали. Растворитель удаляли при пониженном давлении, и остаток очищали хроматографией на колонках, получая целевое соединение (19 мг, выход 45%).
1H-ЯМР (400 МГц, CDCl3); δ 10,39 (ушир.c, 1H), 7,03 (c, 1H), 6,89 (c, 1H), 6,48 (c, 1H), 6,17 (c, 1H), 4,77 (м, 1H), 3,87 (м, 1H), 3,59 (м, 1H), 3,29 (м, 1H), 3,17 (м, 2H), 2,86 (c, 3H), 2,26 (м, 2H), 2,10 (м, 2H), 1,70 (м, 4H), 1,51 (м, 2H).
Пример 166
Этиловый эфир 3-{2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}-5-метил-3H-имидазол-4-карбоновой кислоты
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этанол, полученный в примере 5, и этиловый эфир 5-метил-3H-имидазол-4-карбоновой кислоты вводили в реакцию согласно той же самой процедуре, как в примере 156, получая целевое соединение.
1H-ЯМР (500 МГц, ДМСО-d6); δ 11,49 (ушир.c, 1H), 7,71 (c, 1H), 6,80 (c, 1H), 6,72 (c, 1H), 6,17 (c, 1H), 6,08 (м, 1H), 4,56 (м, 1H), 4,16 (м, 4H), 3,81 (м, 1H), 3,58 (м, 1H), 3,18 (м, 1H), 2,46 (c, 3H), 2,11 (м, 2H), 1,95 (м, 2H), 1,68 (м, 2H), 1,53 (м, 4H), 1,22 (м, 3H).
Пример 167
3-{2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}-5-метил-3H-имидазол-4-карбоновая кислота
Этиловый эфир 3-{2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}-5-метил-3H-имидазол-4-карбоновой кислоты, полученный в примере 166, вводили в реакцию согласно той же самой процедуре, как на стадии А примера получения 101, получая целевое соединение.
1H ЯМР (ДМСО-d6, м.д.); δ 11,50 (1H, с), 7,71 (1H, с), 6,80 (1H, с), 6,72 (1H, с), 6,17 (1H, с), 6,08 (1H, м), 4,55 (1H, м), 4,13 (2H, м), 3,80 (1H, м), 3,55 (2H, м), 2,19-2,15 (2H, м), 1,95 (3H, м), 1,68 (3H, м), 1,51 (5H, м).
Пример 168
1-(2-{(R)-2-[5-хлор-7-(тетрагидро-пиран-4-иламино)-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-ил}этил)пиперидин-3-карбоновая кислота
Стадия A: этиловый эфир 1-(2-{(R)-2-[5-хлор-7-(тетрагидро-пиран-4-иламино)-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-ил}этил)пиперидин-3-карбоновой кислоты
2-{2-[(R)-5-хлор-7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-ил]}этанол, полученный в примере 67, и этиловый эфир пиперидин-3-карбоновой кислоты вводили в реакцию согласно той же самой процедуре, как в примере 156, получая целевое соединение.
1H ЯМР (ДМСО-d6, м.д.); δ 11,48 (1H, с), 6,82 (1H, с), 6,67 (1H, с), 6,29 (1H, с), 6,04 (1H, д), 4,61 (1H, квин), 4,47 (1H, м), 3,87 (2H, м), 3,62 (2H, кв), 3,56 (2H, м), 3,44-3,39 (4H, м), 3,14 (2H, м), 2,52 (1H, м), 2,37-2,30 (6H, м), 1,96-1,92 (3H, м), 1,81 (1H, м), 1,42 (2H, м), 1,28 (3H, т).
Стадия B: 1-(2-{(R)-2-[5-хлор-7-(тетрагидро-пиран-4-иламино)-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-ил}этил)пиперидин-3-карбоновая кислота
Этиловый эфир 1-(2-{(R)-2-[5-хлор-7-(тетрагидро-пиран-4-иламино)-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-ил}этил)пиперидин-3-карбоновой кислоты, полученный на стадии A, вводили в реакцию согласно той же самой процедуре, как на стадии А примера получения 101, получая целевое соединение.
1H ЯМР (ДМСО-d6, м.д.); δ 13,17 (1H, c), 11,94 (1H, c), 6,80 (1H, c), 6,68 (1H, c), 6,28 (1H, c), 6,04 (1H, д), 4,62 (1H, квин), 4,47 (1H, м), 3,87 (2H, м), 3,56 (2H, м), 3,44-3,39 (4H, м), 3,14 (2H, м), 2,52 (1H, м), 2,37-2,30 (6H, м), 1,96-1,92 (3H, м), 1,80 (1H, м), 1,40 (2H, м).
Пример 169
трет-Бутиловый эфир [(S)-1-(2-{(R)-2-[5-хлор-7-(тетрагидро-пиран-4-иламино)-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-ил}этил)пирролидин-3-ил]карбаминовой кислоты
2-{2-[(R)-5-хлор-7-(тетрагидропиран-4-ил)амино-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-ил]}этанол, полученный в примере 67, и (S)-3-BOC-амино-пирролидин вводили в реакцию согласно той же самой процедуре, как в примере 156, получая целевое соединение.
1H-ЯМР (500 МГц, ДМСО-d6); δ 11,48 (ушир.c, 1H), 6,92 (м, 1H), 6,81 (c, 1H), 6,68 (c, 1H), 6,28 (м, 1H), 6,05 (м, 1H), 4,63 (м, 1H), 3,86 (м, 3H), 3,59 (м, 1H), 3,54 (м, 1H), 3,44 (м, 2H), 3,14 (м, 1H), 2,71-2,58 (м, 2H), 2,25 (м, 1H), 1,95 (м, 4H), 1,75 (м, 1H), 1,52 (м, 1H), 1,39 (м, 2H), 1,37-1,32 (м, 11H).
Пример 170
(2-{(R)-4-[2-((S)-3-амино-пирролидин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-5-хлор-1Н-индол-7-ил)-(тетрагидро-пиран-4-ил)амин
Трет-бутиловый эфир [(S)-1-(2-{(R)-2-[5-хлор-7-(тетрагидро-пиран-4-иламино)-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-ил}этил)пирролидин-3-ил]карбаминовой кислоты (150 мг, 0,27 ммоль), полученный в примере 169, растворяли в дихлорметане (30 мл). Добавляли 4 н. раствор соляной кислоты в диоксане (0,34 мл, 1,35 ммоль), и смесь перемешивали в течение 4 часов при температуре окружающей среды. После завершения реакции растворитель удаляли при пониженном давлении. Остаток перекристаллизовывали из смеси DCM/простой этиловый эфир, получая целевое соединение (92 мг, выход 75%).
1H ЯМР (ДМСО-d6, м.д.); δ 10,92 (1H, с), 8,63 (2H, с, ушир.), 6,86 (1H, с), 6,83 (1H, с), 6,43 (1H, с), 6,11 (1H, м), 4,72 (1H, м), 3,65 (5H, м), 3,45 (5H, м), 3,22 (3H, м), 2,37 (2H, м), 2,19 (3H, м), 1,90 (2H, м), 1,49 (2H, м).
Пример 171
N-[(S)-1-(2-{(R)-2-[5-хлор-7-(тетрагидро-пиран-4-иламино)-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-ил}этил)пирролидин-3-ил]ацетамид
(2-{(R)-4-[2-((S)-3-амино-пирролидин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-5-хлор-1Н-индол-7-ил)-(тетрагидро-пиран-4-ил)амин (85 мг, 0,19 ммоль), полученный в примере 170, растворяли в дихлорметане (10 мл). Добавляли диизопропилэтиламин (0,13 мл, 0,75 ммоль) и ацетилхлорид (0,013 мл, 0,19 ммоль), и смесь перемешивали в течение 30 минут при температуре окружающей среды. Реакционную смесь гасили водой, и реакционную смесь экстрагировали этилацетатом, высушивали над безводным сульфатом магния и фильтровали. Растворитель удаляли при пониженном давлении, и остаток очищали хроматографией на колонках, получая целевое соединение (39 мг, выход 42%).
1H ЯМР (ДМСО-d6, м.д.); δ 11,49 (1H, с), 7,97 (1H, с), 6,81 (1H, с), 6,69 (1H, с), 6,28 (1H, с), 6,05 (1H, д), 4,64 (1H, квинт), 4,12 (1H, м), 3,85 (2H, м), 3,53 (2H, м), 3,44 (2H, т), 3,34 (2H, м), 3,15 (1H, т), 2,72-2,60 (3H, м), 2,39 (1H, м), 2,05-1,87 (4H, м), 1,80-1,72 (4H, м), 1,53 (1H, м), 1,37 (2H, м).
Пример 172
Циклопентил-{2-[(R)-4-(2-метокси-этил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}амин
2-[2-(7-Циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этанол, полученный в примере 77, и метилат натрия вводили в реакцию согласно той же самой процедуре, как в примере 165, получая целевое соединение.
1H-ЯМР (500 МГц, ДМСО-d6); δ 10,62 (ушир.с, 1H), 7,03 (д, J=7,95 Гц, 1H), 6,99 (т, 1H), 6,93 (с, 1H), 6,48 (д, J=7,35 Гц, 1H), 4,83 (м, 1H), 3,83 (м, 1H), 3,56 (м, 1H), 3,46 (м, 2H), 3,20 (м, 4H), 2,05~1,87 (м, 4H), 1,70~1,38 (м, 6H).
Примеры 173-224
Соединения, полученные в примерах 2, 5, 24, 56, 67, 73, 77, 79, 99 и 102, и коммерчески доступные амины или метансульфонат натрия вводили в реакцию, чтобы синтезировать соединения из примеров, показанные в следующей таблице, согласно способу, выбранному из примеров 156-172.
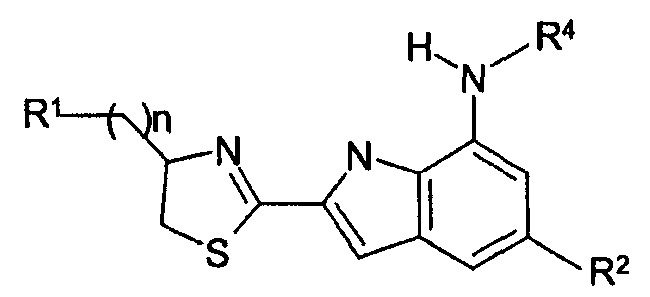
Пример 225
{5-метил-2-[(R)-4-(2-морфолин-4-илэтил)-4,5-дигидро-оксазол-2-ил]-1Н-индол-7-ил}-(тетрагидропиран-4-ил)амин
2-[(R)-2-(5-метил-7-нитро-1Н-индол-2-ил)-4,5-дигидро-оксазол-4-ил]уксусную кислоту, полученную в примере 104, тетрагидропиран-4-он и морфолин последовательно вводили в реакцию согласно тем же самым процедурам как в примере 1, на стадии А примера получения 29 и в примере 156, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3); δ 10,11 (ушир.c, 1H), 6,91 (c, 1H), 6,86 (c, 1H), 6,34 (c, 1H), 4,60 (т, 1H), 4,48 (м, 1H), 4,10~3,93 (м, 5H), 3,63~3,52 (м, 3H), 2,39 (c, 3H), 2,07 (д, 2H), 1,94 (м, 2H), 1,58 (м, 2H).
Пример 226
{5-метил-2-[(S)-4-(2-морфолин-4-илэтил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}-(тетрагидро-пиран-4-илметил)амин
Стадия A: изопропиловый эфир ((S)-2-{5-метил-7-[(тетрагидро-пиран-4-илметил)амино]-1Н-индол-2-ил}-4,5-дигидро-тиазол-4-ил)уксусной кислоты
Изопропиловый эфир [(5-метил-7-нитро-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты, полученный в примере получения 52, и тетрагидропиран-4-карбоксиальдегид вводили в реакцию согласно той же самой процедуре, как в примере 1, получая целевое соединение.
Стадия B: 2-((R)-2-{5-метил-7-[(тетрагидро-пиран-4-илметил)амино]-1Н-индол-2-ил}-4,5-дигидро-тиазол-4-ил)этанол
Изопропиловый эфир ((S)-2-{5-метил-7-[(тетрагидро-пиран-4-илметил)амино]-1Н-индол-2-ил}-4,5-дигидро-тиазол-4-ил)уксусной кислоты, полученный на стадии A, вводили в реакцию согласно той же самой процедуре, как в примере 5, получая целевое соединение.
Стадия C: {5-метил-2-[(S)-4-(2-морфолин-4-илэтил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}-(тетрагидро-пиран-4-илметил)амин
2-((R)-2-{5-метил-7-[(тетрагидро-пиран-4-илметил)амино]-1Н-индол-2-ил}-4,5-дигидро-тиазол-4-ил)этанол, полученный на стадии B, и морфолин вводили в реакцию согласно той же самой процедуре, как в примере 156, получая целевое соединение.
1H ЯМР (500 МГц, CDCl3); δ 11,13 (c, 1H), 6,82 (д, 2H), 6,24 (c, 1H), 4,81-4,78 (м, 1H), 3,88-3,81 (м, 2H), 3,60-3,46 (м, 5H), 3,35-3,30 (м, 2H), 3,19-3,17 (м, 1H), 3,01 (ушир, 2H), 2,38-2,26 (м, 7H), 2,14 (c, 2H), 1,91-1,88 (м, 1H), 1,75-1,71 (м, 2H), 1,53~1,47 (м, 2H), 1,28-1,16 (м, 2H).
Примеры 227-257
Соединения из примеров 10, 22, 51, 66 и 82 вводили в реакцию согласно той же самой процедуре, как в примере 226, или соединения из примеров получения 52 и 71 и коммерчески доступные альдегиды или кетоны и амины выборочно вводили в реакцию согласно той же самой процедуре, как в примере 226, чтобы синтезировать соединения из примеров, показанные в следующей таблице.


Пример 258
[(R)-2-(5-аминометил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-метанол
2-[7-Циклопентиламино-2-((R)-4-гидроксиметил-4,5-дигидро-тиазол-2-ил)-1Н-индол-5-илметил]изоиндол-1,3-дион (27 мг, 0,07 ммоль), полученный в примере 64, растворяли в этаноле (3 мл). Добавляли гидрат гидразина (0,6 мл, 0,11 ммоль), и смесь перемешивали в течение 3 часов при 80°C. После завершения реакции реакционную смесь перегоняли при пониженном давлении и очищали хроматографией на колонках, получая целевое соединение (7 мг, выход 37%).
1H-ЯМР (500 МГц, CDCl3); δ 10,50 (ушир.с, 1H), 6,98 (с, 1H), 6,88 (с, 1H), 6,46 (с, 1H), 4,72 (м, 1H), 4,40 (м, 1H), 3,86 (с, 2H), 3,81 (м, 1H), 3,70 (м, 1H), 3,44 (м, 2H), 1,97 (м, 2H), 1,59 (м, 4H), 1,41 (м, 2H).
Пример 259
[7-циклопентиламино-2-((R)-4-гидроксиметил-4,5-дигидро-тиазол-2-ил)-1Н-индол-5-илметил]амид фуран-2-карбоновой кислоты
[(R)-2-(5-аминометил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-метанол, полученный в примере 258, и фуран-2-карбоновую кислоту вводили в реакцию согласно той же самой процедуре, как на стадии B примера получения 101, получая целевое соединение.
1H-ЯМР (CDCl3); δ 11,01 (1H, ушир), 7,38 (1H, с), 7,12 (1H, д, J=3,7 Гц), 7,03 (1H, с), 6,90 (1H, с), 6,58 (1H, ушир), 6,49~6,45 (2H, м), 4,76~4,67 (1H, м), 4,60 (2H, д, J=5,5 Гц), 4,06~4,01 (1H, м), 3,80~3,73 (1H, м), 3,70~3,64 (1H, м), 3,52~3,46 (1H, м), 3,45~3,38 (1H, м), 1,99~1,86 (2H, м), 1,62~1,46 (4H, м), 1,41~1,32 (1H, м), 1,32~1,24 (1H, м).
Пример 260
Метиловый эфир [(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-илметокси]уксусной кислоты
[(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-1,3-тиазол-4-ил]-метанол, полученный в примере 2, и бромэтилацетат вводили в реакцию согласно той же самой процедуре, как на стадии B примера получения 29, получая целевое соединение.
1H-ЯМР (400 МГц, CDCl3); δ 10,16 (ушир.c, 1H), 6,87 (c, 1H), 6,66 (дд, J=2,4, 9,2 Гц, 1H), 6,30 (дд, J=2,4, 11,8 Гц, 1H), 4,94 (м, 1H), 4,25 (кв, 2H), 4,13 (д, J=5,6 Гц, 2H), 3,87 (м, 1H), 3,76 (д, J=6,4 Гц, 2H), 3,56 (м, 1H), 3,44 (м, 1H), 2,07 (м, 2H), 1,67 (м, 4H), 1,51 (м, 2H), 1,30 (т, 3H).
Пример 261
[(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-илметокси]уксусная кислота
Метиловый эфир [(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-илметокси]уксусной кислоты, полученный в примере 260, вводили в реакцию согласно той же самой процедуре, как на стадии А примера получения 101, получая целевое соединение.
1H-ЯМР (400 МГц, ДМСО-d6); δ 12,70 (ушир.c, 1H), 7,07 (с, 1H), 6,57 (д, J=8,8 Гц, 1H), 6,23 (д, J=12 Гц, 1H), 5,13 (м, 1H), 4,34 (м, 1H), 4,07 (м, 2H), 3,89 (м, 1H), 3,63 (м, 3H), 2,03 (м, 2H), 1,58 (м, 6H).
Пример 262
Циклопентил-{2-[(R)-4-(3-циклопентил-[1,2,4]оксадиазол-5-илметил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}амин
[(7-Циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусную кислоту (140 мг, 0,41 ммоль), полученную в примере 75, растворяли в N,N-диметилформамиде (5 мл). Добавляли 1,1'-дикарбонилдиимидазол (73 мг, 0,45 ммоль), и смесь перемешивали в течение 30 минут при температуре окружающей среды. Добавляли N-гидрокси-циклопентанкарбоксамидин (260 мг, 2,03 ммоль), и смесь перемешивали в течение 5 часов при 80°C. После завершения реакции добавляли воду. Реакционную смесь экстрагировали этилацетатом, высушивали над безводным сульфатом магния и фильтровали. Фильтрат перегоняли при пониженном давлении, и остаток очищали хроматографией на колонках, получая целевое соединение (100 мг, выход 56%).
1H-ЯМР (400 МГц, CDCl3); δ 10,62 (ушир.с, 1H), 7,04 (д, 1H), 6,97 (т, 1H), 6,92 (д, 1H), 6,49 (д, 1H), 5,20 (м, 1H), 3,83 (м, 2H), 3,64 (м, 1H), 3,39 (м, 1H), 3,31 (м, 1H), 3,17 (м, 1H), 3,01 (м, 1H), 1,97 (м, 4H), 1,73 (м, 4H), 1,60 (м, 6H), 1,46 (м, 2H), 1,34 (м, 2H).
Пример 263
Циклопентил-{2-[(R)-4-(3-пиперидин-1-ил-[1,2,4]оксадиазол-5-илметил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}амин
[(7-Циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусную кислоту, полученную в примере 75, и N-гидроксипиперидинкарбоксамидин вводили в реакцию согласно той же самой процедуре, как в примере 262, получая целевое соединение.
1H-ЯМР (400 МГц, CDCl3); δ 10,56 (ушир.с, 1H), 7,01 (д, 1H), 6,96 (т, 1H), 6,90 (д, 1H), 6,46 (д, 1H), 5,23 (м, 1H), 3,83 (м, 2H), 3,64 (м, 1H), 3,36 (м, 1H), 3,31 (м, 1H), 3,17 (м, 2H), 3,01 (м, 1H), 1,95 (м, 2H), 1,68-1,43 (м, 11H), 1,35 (м, 1H).
Экспериментальный пример 1. Измерения и анализ соединений из примеров в отношении эффекта защиты гепатоцитов против токсичности для гепатоцитов, вызванной веществами.
Различные эндогенные/экзогенные атаки на клетки запускают механизмы смерти клеток, которые обычно подразделяют на два типа, то есть апоптоз или некроз. Используя эти механизмы смерти клеток, в настоящем экспериментальном примере, первичные гепатоциты, выделенные от крыс, обрабатывали лекарственными средствами, которые, как было клинически показано, приводят к серьезным побочным эффектам в отношении токсичности для гепатоцитов, или различными химическими соединениями, которые приводят к гибели клеток, и соединения, синтезированные в Примерах, оценивали на их защитные эффекты в отношении гепатоцита, после 24-48 часов. Вещества, используемые для того, чтобы вызвать гибель гепатоцитов, включают CCl4, ActD, H2O2, доксорубицин, анти-Fas Ab/Актиномицин D, ацетаминофен, EtOH, CdCl2, пальмитат, стеарат, циклофосфамид, терфенадин, дикловенак, симвастатин и адефовир. Первичные гепатоциты выделяли, используя способ Seglen PO (Experimental Cell Research 74(1972) pp.450-454). Кратко, гепатоциты выделяли согласно двухступенчатому способу перфузии коллагеназы, и мертвые клетки удаляли низкоскоростным центрифугированием (500 об/мин) в течение 10 минут, используя градиент Перколла (Kreamer B.L. etc, In Vitro Cellular & Developmental Biology 22(1986) pp.201-211). В ходе этой стадии жизнеспособность клеток поддерживали на уровне 90% или выше. Клетки суспендировали в среде HepatoZYME (Gibco BRL), и подсчитывали число клеток. 1,5×104 клеток в 100 мл помещали в покрытый коллагеном 96-луночный планшет (BD biocoat), и оставляли на 3-4 ч для их адгезии ко дну.
Чтобы оценивать защитный эффект в отношении гепатоцитов, вышеописанные адгезивные клетки предварительно обрабатывали в течение 30 мин соединениями согласно примерам. В это время концентрацию соединений из примеров последовательно разбавляли в 2 или 3 раза за 5 стадий, начиная с 30 мкМ, 10 мкМ или 1 мкМ в зависимости от эксперимента, и конечную концентрацию ДМСО устанавливали равной 0,2%. Через 30 минут после обработки соединениями клетки обрабатывали веществами, вызывающими гибель гепатоцитов, или гепатотоксичными лекарственными средствами в концентрациях, показанных в таблице 1. Через 24-48 часов определяли жизнеспособность клеток для оценки защитных эффектов в отношении гепатоцитов. Жизнеспособность клеток определяли, используя способ WST-1 (MK-400, Takeda) на основании поглощения света при 440 нм. Защитные эффекты соединений согласно Примерам в отношении гепатоцитов представлены как “EC50”, которую вычисляли на основании измеренных значений. “EC50” здесь означает концентрацию соединения, при которой в эксперименте наблюдали 50% максимального защитного эффекта.
Предпочтительно, EC50 соединений согласно примерам составляет 30 мкМ или меньше, более предпочтительно, 10 мкМ или меньше, и особенно предпочтительно, 1,0 мкМ или меньше.
В таблице 1 показаны концентрации различных использованных веществ, вызывающих гепатотоксичность, и защитный эффект соединения из примера 4 в отношении гепатоцитов. В таблице 2 показаны защитные эффекты соединений согласно примерам в отношении гепатоцита против доксорубицина в качестве вещества, которое приводит к гепатотоксичности.
Экспериментальный пример 2: защитные эффекты при обработке гепатоцитов и других клеток, полученных из различных тканей, tBHP (трет-бутилгидроксипероксидом; t-BuOOH)
1) Защитный эффект при обработке первичных гепатоцитов tBHP
Гепатоциты выделяли согласно той же самой процедуре, как в экспериментальном примере 1, суспендировали в среде DMEM (Gibco + 10% FBS + 1X антибиотики) и распределяли на планшете. Через 24 часа после распределения гепатоцитов соединения последовательно 3-кратно разбавляли до конечных концентраций 30, 10, 3, 1, 0,3, 0,1 мкМ, которыми клетки предварительно обрабатывали в течение 30 мин. Клетки обрабатывали tBHP в конечной концентрации 300 мкМ, и защитные эффекты определяли через 1 час. Как в экспериментальном примере 1, после обработки WST-1 (Takeda, 10 мкл) в течение 1,5 часов, значения EC50 вычисляли путем измерения поглощения света при 440 нм, используя SpectraMax (Molecular Device).
Предпочтительно, EC50 соединений согласно примерам составляет 30 мкМ или меньше, более предпочтительно, 10 мкМ или меньше, и особенно предпочтительно, 1,0 мкМ или меньше.
2) Защитный эффект при обработке панкреатических клеток (Linm5F) tBHP
Для определения защитного эффекта на панкреатических клетках, клетки Linm5F, разновидность бета-клеток, распределяли в планшет с 96 лунками в количестве 2×104 клеток/лунка и инкубировали в течение 24 часов. Соединения согласно Примерам последовательно 3-кратно разбавляли до конечных концентраций 30, 10, 3, 1, 0,3, 0,1 мкМ, которыми каждую лунку обрабатывали в течение 1 часа. Клетки обрабатывали tBHP в конечной концентрации 400 мкМ, и далее инкубировали в течение 5 часов. Защитные эффекты определяли, используя способ SRB (сульфородамин B-белок), в котором окрашивается общее количество клеточного белка. Кратко, клетки инкубировали в течение 5 часов, к каждой лунке добавляли 50 мкл 4%-ного раствора формальдегида, чтобы зафиксировать клетки, и сохраняли в течение приблизительно 30 минут при температуре окружающей среды. После отбрасывания среды каждую лунку промывали дистиллированной водой 2-3 раза, и планшет высушивали в сушильном шкафу при 50°C. К каждой лунке добавляли 50 мкл раствора SRB и оставляли приблизительно на 30 минут при температуре окружающей среды. После удаления раствора SRB, планшет промывали 2-3 раза 1%-ным раствором уксусной кислоты. После высушивания планшета в сушильном шкафу при 50°C добавляли 100 мкл 10 мМ Tris, чтобы элюировать SRB, который окрашивал внутриклеточный белок. Поглощение света измеряли при 590 нм и 650 нм, используя SpectrMax, и поглощение при 650 нм вычитали из поглощения при 590 нм, чтобы вычислить значение EC50.
Предпочтительно, EC50 соединений согласно примерам составляет 30 мкМ или меньше, более предпочтительно, 10 мкМ или меньше, и особенно предпочтительно, 1,0 мкМ или меньше. Так, EC50 соединения из Примера 14 составляла 0,15 мкМ, и EC50соединения из примера 22 составляла 0,20 мкМ.
3) Защитный эффект при обработке клеткок сердца (H9C2, кардиомиоциты белой крысы) tBHP
Чтобы оценить защитный эффект на клетках сердца, клетки H9C2 распределяли в количестве 1,5×104 клеток/лунка и инкубировали в течение 24 часов. Соединения согласно Примерам последовательно 3-кратно разбавляли до конечных концентраций 30, 10, 3, 1, 0,3, 0,1 мкМ, которыми каждую лунку обрабатывали в течение 45 мин. Клетки обрабатывали tBHP в конечной концентрации 400 мкМ и инкубировали в течение 2 часов. Защитный эффект каждого соединения определяли, используя тот же самый способ SRB, как в случае Linm5F в описанном выше тесте 2.
Предпочтительно, EC50 соединений согласно Примерам составляет 30 мкМ или меньше, более предпочтительно, 10 мкМ или меньше, и особенно предпочтительно, 1,0 мкМ или меньше. Значения EC50 репрезентативных соединений согласно примерам были следующими: пример 22: 0,17 мкМ и пример 85: 0,7 мкМ.
4) Защитный эффект при обработке клеток почек (LLC-PK1) tBHP
Чтобы определить защитный эффект на клетках почек, 4×104 клеток распределяли в каждую лунку и инкубировали в течение 24 часов. Клетки обрабатывали соединениями согласно примерам в конечной концентрации 30, 10, 3, 1, 0,3, 0,1 мкМ и инкубировали в течение 30 мин. Клетки обрабатывали 400 мкМ tBHP и далее инкубировали в течение 6 часов. Защитный эффект каждого соединения определяли, используя тот же самый способ SRB, как в случае Linm5F в описанном выше тесте 2.
Предпочтительно, EC50 соединений согласно примерам составляет 30 мкМ или меньше, более предпочтительно, 10 мкМ или меньше, и особенно предпочтительно, 1,0 мкМ или меньше.
5) Защитный эффект, при обработке хондроцитов tBHP
Чтобы определить защитный эффект на хондроцитах, хондроциты изолировали от 2 задних конечностей 16 крыс SD недельного возраста (масса тела: 450-460 г). Способ изоляции был следующим. Хрящ, выделенный из коленных областей задних конечностей крыс, переносили на планшет 100 pi, содержащий PBS (+1X антибиотики). PBS поддерживали при 4°C в ванне со льдом. PBS заменяли свежим и центрифугировали при 1000 об/мин. После удаления PBS добавляли 3 мл 1X трипсина (Gibco) при температуре 37°C и обрабатывали в течение 15 мин. Супернатант после центрифугирования отбрасывали и снова промывали PBS. Супернатант после центрифугирования отбрасывали. После добавления 0,2% коллагеназы (Worthington, тип II) клетки изолировали инкубацией в течение ночи во вращающемся инкубаторе, в котором поддерживали температуру 37°C. Отфильтрованный раствор клеток центрифугировали, и супернатант отбрасывали. После промывки PBS клетки суспендировали в 10 мл DMEM/F-12 (Gibco, 10% FBS). 2×104 клеток распределяли в каждую лунку и инкубировали в течение 24 часов. Соединения согласно Примерам последовательно 3-кратно разбавляли до конечных концентраций 30, 10, 3, 1, 0,3, 0,1 мкМ, которыми каждую лунку обрабатывали в течение 1 часа. Клетки обрабатывали tBHP в конечной концентрации 500 мкМ и инкубировали в течение 3 часов. Защитный эффект каждого соединения определяли, используя тот же самый способ окрашивания SRB, как в случае Linm5F в описанном выше тесте 2.
Предпочтительно, EC50 соединений согласно примерам составляет 30 мкМ или меньше, более предпочтительно, 10 мкМ или меньше, и особенно предпочтительно, 1,0 мкМ или меньше.
6) Защитный эффект при обработке клеток головного мозга (SK-N-MC) tBHP
Чтобы оценить защитный эффект на клетках головного мозга, 2×104 клеток головного мозга распределяли в планшет с 96 лунками, используя среду DMEM (Gibco, 10% FBS), и инкубировали в течение 24 часов. Соединения согласно Примерам последовательно 3-кратно разбавляли до конечных концентраций 30, 10, 3, 1, 0,3, 0,1 мкМ, которыми каждую лунку обрабатывали в течение 1 часа. Клетки обрабатывали tBHP в конечной концентрации 400 мкМ и инкубировали в течение 6 часов. 50 мкл среды забирали из каждой лунки, чтобы провести тест LDH (Promega). В тесте LDH 50 мкл среды смешивали с 50 мкл тестового раствора. После реакции в течение 30 минут при температуре окружающей среды измеряли поглощение света при 490 нм, используя SpectraMax (Molecular Device).
Предпочтительно, EC50 соединений согласно примерам составляет 30 мкМ или меньше, более предпочтительно, 10 мкМ или меньше, и особенно предпочтительно, 1,0 мкМ или меньше. Так, соединение из примера 4, например, показало превосходную активность в настоящем эксперименте, и его значение EC50 составляло 0,1 мкМ или меньше.
Промышленная применимость
Как демонстрируется приведенными выше результатами, новые соединения согласно настоящему изобретению не только показывают эффекты гепатопротекции и функционального улучшения состояния печени, но также могут быть использованы для профилактики и лечения хронических заболеваний печени, таких как ожирение печени, фиброз печени, цирроз печени и т.д., и острых/хронических заболеваний печени, таких как гепатит и т.д., вызванных вирусом или лекарственными средствами. Соединения согласно настоящему изобретению также показывают некроз-подавляющую эффективность в клетках поджелудочной железы, почек, мозга, хряща и сердца.
Таким образом, соединения согласно настоящему изобретению могут быть использованы в профилактике и лечении некроза и связанных с ним заболеваний.
Специалист в состоянии осуществить различные применения и модификации, не отступая от объема настоящего изобретения.
Реферат
Изобретение относится к новым соединениям индола формулы (1):в которой А обозначает 5-членный гетероарил или гетероцикл, каждый из которых имеет от 1 до 3 гетероатомов, выбранных из N, О и S, Rобозначает R-X-B-X'-, Rобозначает -(CRR)p-Y-R, Rобозначает водород, C-С-алкил или -(СН)С-С-циклоалкил, Rобозначает С-С-циклоалкил (остальные значения радикалов представлены в п.1 формулы изобретения), их фармацевтически приемлемым солям или изомерам, которые могут быть использованы для профилактики или лечения клеточного некроза и связанных с некрозом заболеваний. 3 н. и 31 з.п. ф-лы, 2 табл., 263 пр.
Формула

в которой
n обозначает число от 0 до 3,
А обозначает 5-членный гетероарил или гетероцикл, каждый из которых имеет от 1 до 3 гетероатомов, выбранных из N, О и S,
R1 обозначает R5-X-B-X'-,
В обозначает прямую связь или обозначает 3~10-членный гетероцикл или гетероарил, каждый из которых имеет от 1 до 4 гетероатомов, выбранных из N, О и S,
Х и X' независимо от друг друга обозначают прямую связь, или выбраны из группы, состоящей из -NR6-, -СО-, -CONR6-, -СО2-, -ОС(О)-, -S(O)m-, -O-(CH2)m-, -(СН2)m-O-, -(CH2)m-, -NR6CO-, -(R6O)2P(О)- и -NHCO2-, где m обозначает число от 0 до 3 и R6обозначает водород или C1-C6-алкил,
R5 обозначает водород, гидрокси, C1-C6-алкил, C1-С10-алкокси, С4-С6-циклоалкил или фенил или обозначает 3~10-членный моноциклический или конденсированный циклический гетероцикл или гетероарил, каждый из которых имеет от 1 до 3 гетероатомов, выбранных из N, О и S, и может быть замещен оксо или C1-C6-алкилом,
R2 обозначает -(CR8R9)p-Y-R7,
p обозначает число от 0 до 2,
R8 и R9 независимо от друг друга обозначают водород или C1-С6-алкил,
Y обозначает прямую связь, или выбран из группы, состоящей из -О-, -S-, -NR6-, -NR6C(O)-, -C(O)NR6- и -S(O)q-, где q обозначает число от 0 до 2,
R7 обозначает водород, галоген, гидрокси, C1-С6-алкил или фенил или обозначает 3~10-членный гетероцикл или гетероарил, каждый из которых имеет от 1 до 3 гетероатомов, выбранных из N, S и О и который в случае необходимости содержит оксо,
R3 обозначает водород, C1-C6-алкил или -(СН2)q-С3-С6-циклоалкил,
R4 обозначает С3-С6-циклоалкил,
где алкил, алкокси, фенил, циклоалкил, гетероцикл и гетероарил могут быть в случае необходимости замещены и заместители представляют собой один или более заместителей, выбранных из группы, состоящей из гидрокси, галогена, амино, аминометила С1-С6-алкила, галоген-С1-С6-алкила, С1-С6-алкилсульфонила, карбоксиалкила и оксо, и их фармацевтически приемлемые соли или изомеры.
n обозначает число от 0 до 3,
А обозначает 5-членный гетероарил или гетероцикл, каждый из которых имеет от 1 до 3 гетероатомов, выбранных из N, О и S,
R1 обозначает R5-X-B-X'-,
В обозначает прямую связь или обозначает 3~10-членный гетероцикл или гетероарил, каждый из которых имеет от 1 до 4 гетероатомов, выбранных из N, О и S,
Х и X' независимо от друг друга обозначают прямую связь, или выбраны из группы, состоящей из -NR6-, -CO-, -CONR6-, -CO2-, -ОС(О)-, -S(O)2-, -O-(СН2)m-, -(СН2)m-O-, -(СН2)m-, -NR6CO-, -(R6O)2P(O)- и -NHCO2-, где m обозначает число от 0 до 3 и R6обозначает водород или C1-C6-алкил,
R5 обозначает водород, гидрокси, C1-C6-алкил, галоген-С1-С6-алкил, гидрокси-C1-C6-алкил, С4-С6-циклоалкил, фенил или галогенфенил или обозначает 5~10-членный моноциклический или конденсированный циклический гетероцикл или гетероарил, каждый из которых имеет от 1 до 3 гетероатомов, выбранных из N, О и S, и может быть замещен оксо или галоген-C1-С6-алкилом,
R2 обозначает -(CR8R9)p-Y-R7,
p обозначает число от 0 до 2,
R8 и R9 независимо от друг друга обозначают водород или C1-С6-алкил,
Y обозначает прямую связь, или выбран из группы, состоящей из -О-, -NR6-, -NR6C(O)-, -C(O)NR6- и -S(O)q-, где q обозначает число от 0 до 2,
R7 обозначает водород, галоген, гидрокси, C1-C6-алкил, гидрокси-C1-С6-алкил или галоген-С1-С6-алкил, обозначает фенил, в случае необходимости замещенный C1-C6-алкилсульфонилом, или обозначает 5~6-членный гетероцикл или гетероарил, каждый из которых имеет от 1 до 3 гетероатомов, выбранных из N и О,
R3 обозначает водород, C1-C6-алкил или -(CH2)-С3-С6-циклоалкил,
R4 обозначает С3-С6-циклоалкил, который может содержать оксогруппу.

в которых
n и R1 имеют значения, определенные в п.2, и
R обозначает водород или аминометил.

в которых R5 имеет значения, определенные в п.2.
циклопентил-[2-(4,5-дигидро-1,3-тиазол-2-ил)-1Н-индол-7-ил]амин;
[(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-!,3-тиазол-4-ил]метанол;
метиловый эфир [(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты;
[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этанол;
(R)-2-[7-циклопентиламино-5-(гидроксиметил)-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-илметанол;
[(S)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(S)-2-(5-метил-7-(4,4-дифторциклогексан-4-ил)амино-1H-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(S)-2-(5-метил-7-циклобутиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(S)-2-(5-метил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-1,3-тиазол-4-ил]метанол;
[(R)-2-(5-(диметиламино)метил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-(пиррол-3-ил)метил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-(1,3-имидазол-1-ил)метил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-(пиразол-1-ил)метил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-ацетиламино-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-феноксиметил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
метиловый эфир [(R)-2-(5-(пирролидин-1-ил)метил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты;
циклопентил-[5-хлор-2-((R)-4-изобутил-4,5-дигидро-тиазол-2-ил)-1Н-индол-7-ил)амин;
этиловый эфир [(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовой кислоты;
циклопентил-[5-фтор-2-((R)-4-этил-4,5-дигидро-тиазол-2-ил)-1Н-индол-7-ил]амин;
{(R)-2-[7-(метил-циклопентил)амино-5-фтор-1Н-индол-2-ил]-4,5-дигидро-1,3-тиазол-4-ил}метанол;
метиловый эфир [(S)-2-(5-этокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты;
[(S)-2-(5-этокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(S)-2-(5-этокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазоп-4-ил]этанол;
этиловый эфир [(R)-2-(5-метил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]карбоновой кислоты;
[(S)-2-(5-фенокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазоп-4-ил]уксусная кислота;
[(R)-2-(5-фтор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этанол;
[(R)-2-(5-бром-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-метокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-(пиридин-3-ил)окси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-метансульфонилметил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-(изоиндол-1,3-дион-2-ил)метил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[(R)-2-(5-бром-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(R)-2-(5-фтор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазо,и-4-ил]уксусная кислота;
этиловый эфир [(R)-2-(5-фтор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты;
[(R)-2-(5-фтор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазои-4-ил]этанол;
[(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
этиловый эфир [(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты;
метиловый эфир [(R)-2-(5-метокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты;
[(R)-2-(5-метокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазоп-4-ил]уксусная кислота;
[(R)-2-(5-этокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазоп-4-ил]уксусная кислота;
[(R)-2-(5-пропилокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(R)-2-(5-фенокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазои-4-ил]уксусная кислота;
метиловый эфир [(R)-2-(5-(пиридин-3-ил)окси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты;
[(R)-2-(5-(пиридин-3-ил)окси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
метиловый эфир [(R)-2-(5-метил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты;
[(R)-2-(5-метил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазои-4-ил]уксусная кислота;
[(R)-2-(5-(4-(метансульфонил)фенокси)-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
метиловый эфир [(R)-2-(5-феноксиметил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты;
метиловый эфир [(R)-2-(5-фениламинометил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты;
метиловый эфир [(R)-2-(5-метансульфонилметил-7-циклопентиламлно-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусной кислоты;
[(R)-2-(5-метансульфонилметил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]уксусная кислота;
[(R)-2-(5-метансульфонилметил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этанол;
[(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этилацетамид;
3-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовая кислота;
[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазоп-4-ил]пропанол;
этиловый эфир 3-[(R)-2-(5-метил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовой кислоты;
3-[(R)-2-(5-метил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовая кислота;
3-[(R)-2-(5-фенокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовая кислота;
этиловый эфир 3-[(R)-2-(5-фтор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовой кислоты;
3-[(R)-2-(5-фтор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовая кислота;
этиловый эфир 3-[(R)-2-(5-бром-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовой кислоты;
3-[(R)-2-(5-бром-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовая кислота;
3-[(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовая кислота;
3-[(R)-2-(5-метокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил] пропионовая кислота;
этиловый эфир 3-[(R)-2-(5-этокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовой кислоты;
3-[(R)-2-(5-этокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовая кислота;
этиловый эфир 3-[(R)-2-(5-трифторметокси-7-циклопентиламлно-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовой кислоты;
3-[(R)-2-(5-трифторметокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропионовая кислота;
[(S)-2-(5-метил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-оксазол-4-ил]уксусная кислота;
[(S)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-оксазол-4-ил]уксусная кислота;
[2-((4S,5R)-5-аминометил-4-бензил-дигидро-оксазол-2-ил)-5-хлор-1Н-индол-7-ил]циклопентил-амин;
{2-[(R)-5-((S)-1-амино-2-фенил-этил)-4,5-дигидро-оксазол-2-ил]-5-хлор-1Н-индол-7-ил}циклопентил-амин;
циклопентил-[2-(4,5-дигидро-оксадиазол-2-ил)-1Н-индол-7-ил]амин;
[2-(5-метил-7-циклопентиламино-1Н-индол-2-ил)тиазол-4-ил]метанол;
[2-(5-метил-7-циклопентиламино-1Н-индол-2-ил)тиазол-5-ил]метанол;
этиловый эфир [2-(5-метил-7-циклопентиламино-1Н-индол-2-ил)тиазол-4-ил]карбоновой кислоты;
[2-(5-метил-7-циклопентиламино-1Н-индол-2-ил)тиазол-4-ил]карбоновая кислота;
[2-(7-циклопентиламино-1Н-индол-2-ил)тиазол-4-ил]метанол;
метиловый эфир [2-(7-циклопентиламино-1Н-индол-2-ил)тиазол-4-ил]карбоновой кислоты;
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-морфолин-4-илэтанон;
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазои-4-ил]-1-(морфолин-4-ил)этиламино-этанон;
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-морфолин-4-ил)пропиламино-этанон;
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазоп-4-ил]-1-метиламино-этанон;
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-диметиламино-этанон;
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазои-4-ил]-1-[4-(метил)пиперазин-1-ил]этанон;
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазои-4-ил]-1-(3-диметиламинопирролидин-1-ил)этанон;
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазои-4-ил]-1-(пиперидин-4-ил)этанон;
2-[(R)-2-(5-фтор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-(4-(метил)пиперазин-1-ил)этанон;
2-[(R)-2-(5-фтор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазоп-4-ил]-1-(морфолин-4-ил)этиламиноэтанон;
2-[(R)-2-(5-метокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-(метиламино)-4-илэтанон;
2-[(R)-2-(5-метокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-(морфолин-4-ил)этанон;
2-[(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-метиламино-этанон;
2-[(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-(морфолин-4-ил)этанон;
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазоч-4-ил]-1-(морфолин-4-ил)этиламино-этанон;
2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]-1-(4-(метил)пиперазин-1-ил)этанон;
циклопентил-{5-метансульфонилметил-2-[(R)-4-(2-морфолин-4-ил-этил)-4,5-цигидро-тиазол-2-ил]-1Н-индол-7-ил}амин;
1-(4-{2-[(R)-2-(7-циклопентиламино-5-метансульфонилметил-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-ил)этанон;
циклопентил-[2-((R)-4-пирролидин-1-илметил-4,5-дигидро-тиазол-2-ил)-1-Н-индол-7-ил]амин;
{5-хлор-2-[(R)-4-(2-диметиламино-этил)-4,5-дигидро-тиазол-2-ил]-1H-индол-7-ил}циклопентил-амин;
{5-хлор-2-[(R)-4-(2-пиперазин-1-ил-этил)-4,5-дигидро-тиазол-2-ил]-1-Н-индол-7-ил}циклопентил-амин;
(5-хлор-2-{(R)-4-[2-(4-этансульфонил-пиперазин-1-ил)этил]-4,5-дигидро-тиазол-1Н-индол-7-ил)циклопентил-амин;
1-(4-{2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиaзол-4-ил]этил}пиперазин-1-ил)-2-гидрокси-этанон;
{5-хлор-2-[(R)-4-(2-пиразол-1-ил-этил)-4,5-дигидро-тиазол-2-ил]-1H-индол-7-ил}циклопентил-амин;
(S)-1-{2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидpo-тазол-4-ил]-этил}-пирролидин-2ил)карбоновая кислота;
{5-хлор-2-[(R)-4-(2-метансульфонил-этил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}циклопентил-амин;
этиловый эфир 3-{2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}-5-метил-ЗН-имидазол-4-карбоновой кислоты;
3-{2-[(R)-2-(5-хлор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}-5-метил-3Н-имидазол-4-карбоновая кислота;
циклопентил-{2-[(R)-4-(2-метокси-этил)-4,5-дигидро-тиазол-2-ил]-1H-индол-7-ил}амин;
[2-((R)-1-аминометил-4,5-дигидро-тиазол-2-ил)-1Н-индол-7-ил]циклопентил-амин;
{2-[(R)-1-((R)-3-амино-пирролидин-1-илэтил)-4,5-дигидро-тиазол-2-ил]-5-хлор-1Н-индол-7-ил}циклопентил-амин;
4-[(R)-2-(7-циклопентиламино-5-этокси-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-илэтил]пиперазин-2-он;
{2-[(R)-1-((S)-3-амино-пирролидин-1-илэтил)-4,5-дигидро-тиазол-2-ил]-3-хлор-1Н-индол-7-ил}циклопентил-амин;
(5-хлор-2-{(S)-4-[2-(3-диметиламино-фенил)-этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
1-(4-{2-[(S)-2-(7-циклопентиламино-5-хлор-1Н-индол-2-ил)-4,5-дигидро-тазол-4-ил]этил}пиперазин-1-ил)этанон;
1-(4-{2-[(S)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-ил)этанон;
(5-метокси-2-{(R)-4-[2-(пирролидин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Ниндол-7-ил)циклопентил-амин;
(5-метансульфонилметил-2-{(S)-4-[(2-оксопиперазин-4-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
1-(4-{2-[(R)-2-(7-циклопентиламино-5-хлор-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-ил)этанон;
(5-хлор-2-{(R)-4-[4-метил-пиперазин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-хлор-2-{(R)-4-[4-(гидрокси)пиперидин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-хлор-2-{(R)-4-[2-оксопиперазин-4-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-хлор-2-{(R)-4-[(пиперидин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-1H-индол-7-ил)циклопентил-амин;
(5-хлор-2-{(R)-4-[(1,1-диоксо-тиоморфолин-4-ил)этил]-4,5-дигидро-тиазоп-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-хлор-2-{(R)-4-[(2-оксопирролидин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-фтор-2-{(R)-4-(2-аминоэтил)-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
1-(4-{2-[(R)-2-(5-фтор-7-циклопентиламино)-1Н-индол-2-ил)-4,5-дигидро-тлазол-4-ил]этил}пиперазин-1-ил)-2-гидрокси-этанон;
(5-фтор-2-{(R)-4-[(морфолин-4-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-фтор-2-{(R)-4-(2-диметиламино-этил)-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-фтор-2-{(R)-4-[(пирролидин-1-ил)-этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-фтор-2-{(R)-4-[(1,1-диоксо-тиоморфолин-4-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-фтор-2-{(R)-4-[(2-оксопирролидин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-1H-индол-7-ил)циклопентил-амин;
1-(4-{2-[(5-фтор-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-ил)-2-гидрокси-этанон;
(5-фтор-2-{(R)-4-[метансульфонил-этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(2-{(R)-1-[2-диметиламино-этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(2-{(R)-1-[(пиперидин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
1-(4-{2-[(R)-2-(5-метокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигид))-тиазол-4-ил]этил}пиперазин-1-ил)-2-гидрокси-этанон;
1-(4-{2-[(R)-2-(7-циклопентиламино)-5-хлор-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]пропил}пиперазин-1-ил)этанон;
2-{(R)-4-[(морфолин-4-ил)-метил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-хлоp-2-{(R)-4-[(морфолин-4-ил)пропил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(2-{(R)-1-[2-диметиламино-метил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-хлор-2-{(S)-4-[(морфолин-4-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
1-(4-{2-[(S)-2-(5-фенокси-7-циклопентиламино)-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}-пиперазин-1-ил)-2-гидрокси-этанон;
(5-феноски-2-{(S)-4-[(пиперазин-1-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
трет-бутил-[-(4-{2-[(S)-2-(5-фенокси-7-циклопентиламино)-1Н-индол-2-ил]-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-карбоксила;
циклопентил-(5-фенокси-2-{(S)-4-[2-(3-фторметил-5,6-дигидро-8H-[1,2,4]триазоло[4,3-а]пиразин-7-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)амин;
(5-фенокси-2-{(S)-4-[2-оксопиперазин-4-ил)этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(4-{2[(S)-2-(5-фенокси-7-циклопентиламино)-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-ил)-оксоран-2-ил-метанон;
(5-фенокси-2-{(S)-4-[(пиридин-2-ил)пиперазин-этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-фенокси-2-{(S)-4-[(2-фторфенил)пиперазин-этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-хлор-2-{(S)-4-[2-оксопиперазин-4-ил)этил]-4,5-дигидро-тиазол-2-ил}-1-индол-7-ил)циклопентил-амин;
(5-фенокси-2-{(S)-4-[(3S)-3-(амино)пирролидин-1-илэтил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
1-(4-2-(S)-2-(5-фенокси-7-циклопентиламино)-1Н-индол-2-ил)-4,5-дигидро)-тиазол-4-ил]этил}пиперазин-1-ил)этанон;
(5-хлор-{(S)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил])пирролидин-3-илацетамид;
(5-фенокси-2-{(S)-4-[4-(бензил)пиперазин-1-ил-этил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)циклопентил-амин;
(5-фенокси-2-{(R)-4-[пирролидин-1-илэтил]-4,5-дигидро-тиазол-2-ил}-1-индол-7-ил)циклопентил-амин;
(5-фенокси-2-{(S)-4-[пирролидин-1-илэтил]-4,5-дигидро-тиазол-2-ил-2ил}-1-индол-7-ил)циклопентил-амин;
(5-метил-2-{(S)-4-[2-оксопиперазин-4-илэтил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-4,4-дифторциклогексил-амин;
(5-метил-2-{(S)-4-[морфолин-4-илэтил]-4,5-дигидро-тиазол-2-ил}-1Н-индол-7-ил)-4,4-дифторциклогексил-амин;
1-(4-{2-[(R)-2-(5-метокси-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]этил}пиперазин-1-ил)-2-гидрокси-этанон;
[(R)-2-(5-аминометил-7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-ил]метанол;
[7-циклопентиламино-2-((R)-4-гидроксиметил-4,5-дигидро-тиазол-2-ил)-1Н-индол-5-илметил]амид фуран-2-карбоновой кислоты;
метиловый эфир [(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-илметокси]уксусной кислоты;
[(R)-2-(7-циклопентиламино-1Н-индол-2-ил)-4,5-дигидро-тиазол-4-илметокси]уксусная кислота;
циклопентил-{2-[(R)-4-(3-циклопентил-[1,2,4]оксадиазол-5-илметил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}амин; и
циклопентил-{2-[(R)-4-(3-пиперидин-1-ил-[1,2,4]оксадиазол-5-илметил)-4,5-дигидро-тиазол-2-ил]-1Н-индол-7-ил}амин.
Комментарии