Тетразамещенные алкеновые соединения и их применение - RU2733741C2
Код документа: RU2733741C2
Чертежи





Описание
Перекрестные ссылки на родственные заявки
Настоящая заявка испрашивает приоритет согласно предварительной заявке США No. 62/168581, поданной 29 мая 2015 года, предварительной заявке на патент США No. 62/168529, поданной 29 мая 2015 года, и предварительной заявке США No. 62/269745, поданной 18 декабря 2015 года.
Уровень техники
Рак молочной железы является наиболее часто диагностированным злокачественным новообразованием среди женщин сегодня с почти 200000/1,7 миллиона новых случаев, диагностированных в США/мире каждый год, соответственно. Поскольку около 70% опухолей молочной железы являются положительными для рецептора эстрогена альфа (ERα) - ключевой онкогенный фактор в этом подмножестве опухолей- было разработано несколько классов терапий для противодействия функции ERα, включая 1) селективные супрессоры рецепторов эстрогена (SERD), примером которых является фулвестрант, 2) селективные модуляторы рецепторов эстрогена (SERM), примером которых является тамоксифен, и 3) ингибиторы ароматазы, которые снижают системные уровни эстрогена. Эти терапии в значительной степени эффективны в клинике, уменьшая возникновение и прогрессирование ERα+ опухоли молочной железы. Однако существуют целевые отрицательные особенности, связанные с этими различными классами соединений. Например, как было доказано, тамоксифен активирует сигнальную активность в эндометрии, что приводит к увеличению риска развития рака эндометрия в клинике (Fisher et al., (1994) J Natl Cancer Inst. Apr 6;86(7):527-37; van Leeuwen et al., (1994) Lancet Feb 19;343(8895):448-52). Напротив, поскольку фулвестрант является чистым антагонистом, он может привести к потере плотности костной ткани у женщин в постменопаузе, поскольку активность ERα имеет решающее значение для построения костной ткани. В дополнение к побочным эффектам, лекарственная устойчивость также начинает проявляться к этим классам ERα антагонистов, подчеркивая необходимость разработки соединений следующего поколения.
Было выявлено несколько механизмов устойчивости с использованием in vitro и in vivo моделей устойчивости к различным эндокринным терапиям. К ним относятся повышенные ʺперекрестные взаимодействияʺ ERα/HER2 (Shou et al., (2004) J Natl Cancer Inst. Jun 16;96(12):926-35), аберрантная экспрессия ERα коактиваторов/корепрессоров (Osborne et al., (2003) J Natl Cancer Inst. Mar 5;95(5):353-61) или потеря ERα в целом для обеспечения ER-независимого роста (Osborne CK, Schiff R (2011) Annu Rev Med 62: 233-47).
В надежде на выявление клинически значимых механизмов резистентности, в последнее время большое внимание уделено также глубокой характеристике генетики метастазов, устойчивых к эндокринной терапии, выделенных у пациентов. Несколько независимых лабораторий недавно опубликовали множество генетических повреждений, наблюдаемых в резистентных vs первичных опухолей (Li et al., (2013) Cell Rep. Sep 26;4(6):1116-30; Robinson et al., (2013) Nat Genet. Dec;45(12):1446-51; Toy et al., (2013) Nat Genet. 2013 Dec;45(12):1439-45). Среди них высоко рекуррентные мутации в лиганд-связывающем домене ESR1 (ген, который кодирует ERα белок), которые, как было установлено, значительно обогащены в примерно 20% резистентных опухолей по сравнению с опухолями, не подвергавшимися эндокринной терапии (Jeselsohn et al., (2014) Clin Cancer Res. Apr 1;20(7):1757-67; Toy et al., (2013) Nat Genet. 2013 Dec;45(12):1439-45; Robinson et al., (2013) Nat Genet. Dec;45(12):1446-51; Merenbakh-Lamin et al., (2013) Cancer Res. Dec 1;73(23):6856-64; Yu et al., (2014) Science Jul 11;345(6193):216-20; Segal and Dowsett (2014), Clin Cancer Res Apr 1;20(7):1724-6), указывающие на возможность для этих мутаций функционально управлять клинической устойчивостью. В отличие от обогащения в ESR1 мутациях, наблюдаемых в опухолях, устойчивых к терапии, мутации в других генах, связанных с раком, не показали такое сильное обогащение, что в значительной степени указывает на важность ERα мутаций в повышении резистентности (Jeselsohn et al., (2014) Clin Cancer Res. Apr 1;20(7):1757-67).
Пациенты, страдающие ER+ раком молочной железы, в среднем получают семь независимых терапий, включая химиотерапию и различные антиэстрогенные терапии, такие как тамоксифен, фульвестрант и ингибиторы ароматазы. Недавнее геномное профилирование показало, что ERα путь остается критическим фактором роста опухоли в резистентном состоянии, так как появляются активирующие мутации в ERα. Таким образом, крайне важно разработать более эффективные ER-направленные терапии, которые могут преодолеть устойчивость в клинических условиях. Следовательно, существует потребность в новых соединениях, которые могут эффективно подавлять рост дикого типа (WT) и ER α-мутант положительных опухолей.
Сущность изобретения
Описанные в настоящем документе соединения представляют собой новые соединения, используемые для лечения рака. В вариантах осуществления такие новые соединения описываются формулой II:

где: R1 выбран из группы, состоящей из метила, этила, циклобутила, циклопропила и -CH2CH2Cl, пропила, изопропила, -CH2CF3, и -CH2CH2F; R2 выбран из группы, состоящей из H и F; n равно 0-1; R3 представляет собой F; m равно 0-2; R4 являются одинаковыми или различными и независимо выбраны из группы, состоящей из F, CF3, Cl, изопропила, -OCH3, -OCHF2, -OCF3, этила и метила; p равно 0-1; R5 представляет собой F; R6 и R7 являются одинаковыми или различными и независимо выбраны из группы, состоящей из метила, этила, пропила, -CH2CH2OH и

В других вариантах осуществления такие новые соединения описываются формулой I:

где: R1 выбран из группы, состоящей из метила, этила, циклобутила, циклопропила и -CH2CH2Cl; R2 выбран из группы, состоящей из H и F; n равно 0-1; R3 представляет собой F; m равно 0-2; R4 являются одинаковыми или различными и независимо выбраны из группы, состоящей из F, CF3, Cl, изопропила, -OCH3, -OCHF2, -OCF3, этила и метила; p равно 0-1; R5 представляет собой F; R6 и R7 являются одинаковыми или различными и независимо выбраны из группы, состоящей из метила, этила, пропила, -CH2CH2OH и

В вариантах осуществления соединения формулы I могут иметь следующие предпочтительные перестановки или предпочтительную комбинацию перестановок: R1 представляет собой этил или циклобутил; R6 и R7 оба представляют собой метил; R8 представляет собой H; R2 представляет собой H или F; m равно 1 и один из R4 представляет собой F и другой R4 представляет собой Cl; m равно 2 и оба R4 представляют собой F; m равно 0; R3 представляет собой F; n равно 0; p равно 1 и R5 представляет собой F; и p равно 0.
В вариантах осуществления соединения формулы I имеют следующую перестановку: R1 представляет собой этил; R2 представляет собой H; n равно 0; m равно 0; p равно 0; R6 и R7 являются одинаковыми и представляют собой метил; R8 представляет собой H; или их фармацевтически приемлемые соли.
В вариантах осуществления соединения формулы I имеют следующую перестановку: R1 представляет собой этил; R2 представляет собой F; n равно 0; m равно 1 и один R4 представляет собой F и один R4представляет собой Cl; p равно 0; R6 и R7 являются одинаковыми и представляют собой метил; R8 представляет собой H; или их фармацевтически приемлемые соли.
В вариантах осуществления соединения формулы I выбраны из группы, состоящей из (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-1-(3-фтор-1H-индазол-5-ил)-2-(3-фтор-5-(трифторметил)фенил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-1-(4-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-2-(3,5-дифторфенил)-1-(1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-2-(3,4-дифторфенил)-1-(1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-2-(3-хлор-5-фторфенил)-1-(1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N-метил-N-(проп-2-ин-1-ил)бут-2-енамида; (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N-(бут-3-ин-1-ил)-N-метилбут-2-енамида; (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-1-(азетидин-1-ил)бут-2-ен-1-она; (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-1-(пирролидин-1-ил)бут-2-ен-1-она; (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-1-(пиперидин-1-ил)бут-2-ен-1-она; (E)-4-((2-(4-((E)-2-циклобутил-1-(1H-индазол-5-ил)-2-фенилвинил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-2-циклобутил-1-(3-фтор-1H-индазол-5-ил)-2-фенилвинил)фенокси)этил)амино)-N,N-диметилбут-2-енамид 2,2,2-трифторацетата; (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-1-морфолинобут-2-ен-1-она; (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N-этил-N-метилбут-2-енамида; (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N-метил-N-пропилбут-2-енамида; (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N-(2-гидроксиэтил)-N-метилбут-2-енамида; (E)-4-((2-(4-((E)-2-(2-хлор-4-фторфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N,3-триметилбут-2-енамида; (Z)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-2-циклобутил-1-(1H-индазол-5-ил)-2-фенилвинил)фенокси)этил)амино)-1-морфолинобут-2-ен-1-она; (E)-4-((2-(4-((E)-2-циклобутил-1-(4-фтор-1H-индазол-5-ил)-2-фенилвинил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-((5-((Z)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-(o-толил)бут-1-ен-1-ил)фенокси)этил)амино)-N-(2-гидроксиэтил)-N-метилбут-2-енамида; (E)-4-((2-(4-((E)-2-(2-хлор-4-фторфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N-(2-гидроксиэтил)-N-метилбут-2-енамида; (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-(4-изопропилфенил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-4-хлор-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-(4-изопропилфенил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-2-(2-(дифторметокси)фенил)-1-(1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-(2-(трифторметокси)фенил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-(2-изопропилфенил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-2-(2-этилфенил)-1-(1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-(o-толил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-((5-((Z)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиримидин-2-ил)окси)этил)амино)-N,N-диметилбут-2-енамида; (E)-1-(азетидин-1-ил)-4-((2-(4-((E)-2-циклобутил-1-(1H-индазол-5-ил)-2-фенилвинил)фенокси)этил)амино)бут-2-ен-1-она; (E)-1-(азетидин-1-ил)-4-((2-(4-((E)-2-циклобутил-1-(3-фтор-1H-индазол-5-ил)-2-фенилвинил)фенокси)этил)амино)бут-2-ен-1-она; (E)-4-((2-(4-((E)-2-циклобутил-1-(1H-индазол-5-ил)-2-фенилвинил)фенокси)этил)амино)-N-(2-гидроксиэтил)-N-метилбут-2-енамида; (E)-1-(азетидин-1-ил)-4-((2-(4-((E)-2-циклобутил-1-(1H-индазол-5-ил)-2-(o-толил)винил)фенокси)этил)амино)бут-2-ен-1-она; (E)-4-((2-(4-((E)-1-(3-фтор-1H-индазол-5-ил)-2-(2-фторфенил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-1-(3-фтор-1H-индазол-5-ил)-2-(3-фторфенил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-1-(3-фтор-1H-индазол-5-ил)-2-(3-фторфенил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-2-(2-хлор-4-фторфенил)-2-циклобутил-1-(3-фтор-1H-индазол-5-ил)винил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(2-фтор-4-((Z)-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-2-(2,6-дифторфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-1-(3-фтор-1H-индазол-5-ил)-2-(пиридин-3-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-1-(3-фтор-1H-индазол-5-ил)-2-(пиридин-4-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилпроп-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-2-циклопропил-1-(1H-индазол-5-ил)-2-фенилвинил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-2-(2-хлор-4-фторфенил)-1-(4-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-((5-((Z)-2-(2-хлор-4-фторфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)пиридин-2-ил)окси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-((5-((Z)-2-(2-хлор-4-фторфенил)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)пиридин-2-ил)окси)этил)амино)-N,N-диметилбут-2-енамида; (E)-N,N-диметил-4-((2-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)этил)амино)бут-2-енамида; (E)-N,N-диметил-4-((2-(4-((E)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-(3-фторпиридин-4-ил)бут-1-ен-1-ил)фенокси)этил)амино)бут-2-енамида; (E)-N,N-диметил-4-((2-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-(3-фторпиридин-4-ил)бут-1-ен-1-ил)пиридин-2-ил)окси)этил)амино)бут-2-енамида; (E)-N,N-диметил-4-((2-(4-((E)-2-фенил-1-(1H-пиразоло[4,3-b]пиридин-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)бут-2-енамида; (E)-4-((2-(3-фтор-4-((Z)-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-2-(2,4-дифторфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-1-(3,6-дифтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-1-(3-фтор-1H-индазол-5-ил)-2-(пиридин-2-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-((5-((Z)-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)этил)амино)-N,N-диметилбут-2-енамида; (E)-N,N-диметил-4-((2-(4-((E)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)бут-2-енамида; (E)-4-((2-((5-((Z)-2-циклобутил-1-(3-фтор-1H-индазол-5-ил)-2-фенилвинил)пиридин-2-ил)окси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-2-(2-хлорфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-2-(2-хлор-4-фторфенил)-1-(1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-1-(3-фтор-1H-индазол-5-ил)-2-(пиридин-4-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N-(2-гидроксиэтил)-N-метилбут-2-енамида; (E)-4-((2-(4-((E)-1-(7-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-1-(3-фтор-1H-индазол-5-ил)-2-фенилпент-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-1-(3,7-дифтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-2-(2,5-дифторфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-1-(3-фтор-1H-индазол-5-ил)-2-(3-фторпиридин-4-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-1-(3-фтор-1H-индазол-5-ил)-3-метил-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-5-((2-(4-((E)-4-фтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилпент-2-енамида; (E)-5-((2-(4-((E)-2-(2-хлор-4-фторфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилпент-2-енамида; (E)-4-((2-(4-((E)-2-(2-хлор-4-фторфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N,2-триметилбут-2-енамида; (E)-4-((2-(4-((E)-2-(2-хлор-4-фторфенил)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-(фенил-d5)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилпент-2-енамида; (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)пропил)амино)-N,N-диметилбут-2-енамида; (E)-4-((1-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)пропан-2-ил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-((6-((Z)-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-3-ил)окси)этил)амино)-N,N-диметилбут-2-енамида; (E)-4-((3-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)пропил)амино)-N,N-диметилбут-2-енамида; (E)-4-((3-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)бутан-2-ил)амино)-N,N-диметилбут-2-енамида; (E)-4-((1-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)-2-метилпропан-2-ил)амино)-N,N-диметилбут-2-енамида; (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)-2-метилпропил)амино)-N,N-диметилбут-2-енамида; и (E)-4-((2-(4-((E)-2-циклобутил-1-(3-фтор-1H-индазол-5-ил)-2-фенилвинил)фенокси)этил)амино)-N-(2-гидроксиэтил)-N-метилбут-2-енамид, и их фармацевтически приемлемых солей.
В некоторых вариантах осуществления такие новые соединения описываются формулой III:
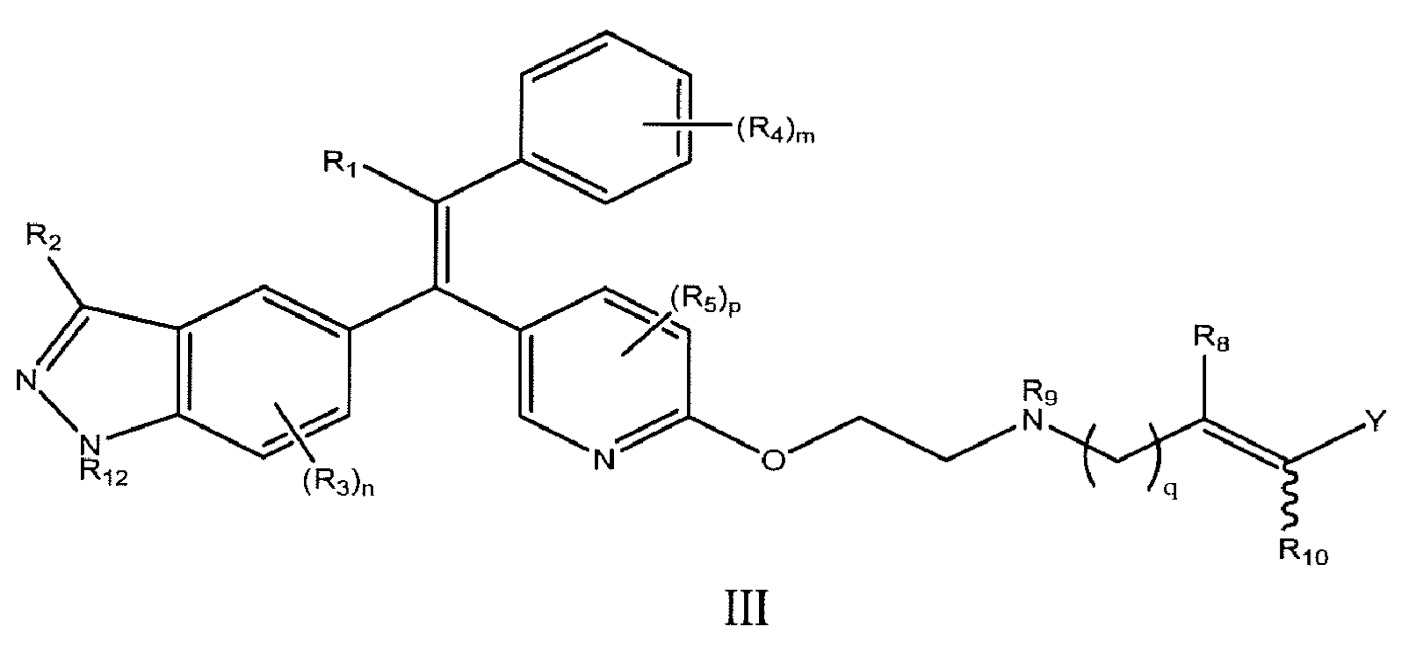
где: R1 выбран из группы, состоящей из C1-C6 алкила, C3-C6 циклоалкила и 4-6-членного гетероциклического кольца; R2 выбран из группы, состоящей из H, галогена, гидрокси, C1-C3 алкила, C3-C4 циклоалкила и C4 гетероциклического кольца; R3 являются одинаковыми или различными и независимо выбраны из группы, состоящей из H, галогена, C1-C6 алкила и C1-C3 алкокси, необязательно замещенного по меньшей мере одним галогеном; n равно 0-3; R4 являются одинаковыми или различными и независимо выбраны из группы, состоящей из H, галогена, C1-C6 алкила и OR11, где R11 выбран из группы, состоящей из C3-C6 циклоалкила, C1-C6 алкила, арила, гетероарила и 4-6-членного гетероциклического кольца; m равно 0-5; R5 являются одинаковыми или различными и независимо выбраны из группы, состоящей из H, галогена, C1-C4 алкила, C1-C4 алкокси, C3-C4 циклоалкила, C3-C6 циклоалкокси и C4 гетероцикл; p равно 0-3; q равно 1-2; R8 и R10 являются одинаковыми или различными и независимо выбраны из группы, состоящей из галогена, H и C1-C3 алкила; R9 выбран из группы, состоящей из H, C1-C6 алкила и C3-C6 циклоалкила; Y выбран из группы, состоящей из -S(O)2R6, -S(O)2NR6R7, -C(O)NR6R7, -C(O)R6, -C(O)OR6, -CN; или где Y и R10 оба представляют собой -CF3; R6 и R7 являются одинаковыми или различными и независимо выбраны из группы, состоящей из H, C1-C6 алкила, C3-C6 циклоалкила, арила, гетероарила и 4-6-членного гетероциклического кольца, где указанный алкил является насыщенным или ненасыщенным или, где R6 и R7 образуют 4-6-членное гетероциклическое кольцо с N, к которому они присоединены, необязательно также содержащее атом O; R12 выбран из группы, состоящей из H, C3-C4 циклоалкила и C1-C6 алкила; и где любой содержащий углерод фрагмент R1-R12 может быть необязательно замещен одним или несколькими атомами галогена, фторметаном, дифторметаном или трифторметаном, или -OH; или их фармацевтически приемлемые соли.
В другом варианте осуществления, R1 выбран из группы, состоящей из C1-C6 алкила, C3-C6 циклоалкила и 4-6-членного гетероциклического кольца. В другом варианте осуществления, R1 представляет собой -CH2CF3. В более конкретных вариантах осуществления такие новые соединения описываются формулой III, где: R2 выбран из группы, состоящей из H, галогена, метила и этила; R3 являются одинаковыми или различными и независимо выбраны из группы, состоящей из H, галогена, метила и этила; R4 являются одинаковыми или различными и независимо выбраны из группы, состоящей из H, галогена, C1-C6 алкила и C1-C6 алкокси; R5 являются одинаковыми или различными и независимо выбраны из группы, состоящей из H, галогена, метила и этила; R8 и R10 являются одинаковыми или различными и независимо выбраны из группы, состоящей из H и метила; R9 выбран из группы, состоящей из H, метила и этила; и R6 и R7 являются одинаковыми или различными и независимо выбраны из группы, состоящей из H и C1-C6 алкила или где R6 и R7 образуют 4-6-атомное гетероциклическое кольцо с N, к которому они присоединены, необязательно также содержащее атом O. В других вариантах осуществления R1 представляет собой циклобутил, этил, или -CH2CF3; R2 представляет собой -H или -F; n равно 0; m равно 0 или 2, и когда m равно 1, тогда один R4 представляет собой -Cl и другой R4 представляет собой -F; p равно 0; Y представляет собой -CON(CH3)2, и R8, R9, R10, и R12 все представляют собой -H.
В некоторых вариантах осуществления такие новые соединения описываются формулой IV:

где: R1 выбран из группы, состоящей из C1-C6 алкила, C3-C6 циклоалкила и 4-6-членного гетероциклического кольца; R2 выбран из группы, состоящей из H, галогена, гидрокси, C1-C3 алкила, C3-C4 циклоалкила и C4 гетероциклического кольца; R3 являются одинаковыми или различными и независимо выбраны из группы, состоящей из H, галогена, C1-C6 алкила и C1-C3 алкокси, необязательно замещенного по меньшей мере одним галогеном; n равно 0-3; R4 являются одинаковыми или различными и независимо выбраны из группы, состоящей из H, галогена, C1-C6 алкила, и OR11, где R11 выбран из группы, состоящей из C3-C6 циклоалкила, C1-C6 алкила, арила, гетероарила и 4-6-членного гетероциклического кольца; m равно 0-4; R5 являются одинаковыми или различными и независимо выбраны из группы, состоящей из H, галогена, C1-C4 алкила, C1-C4 алкокси, C3-C4 циклоалкила, C3-C6 циклоалкокси и C4 гетероцикла; p равно 0-4; q равно 1-2; R8 и R10 являются одинаковыми или различными и независимо выбраны из группы, состоящей из галогена, H и C1-C3 алкила; R9 выбран из группы, состоящей из H, C1-C6 алкила и C3-C6 циклоалкила; Y выбран из группы, состоящей из -S(O)2R6, -S(O)2NR6R7, -C(O)NR6R7, -C(O)R6, -C(O)OR6, -CN; или где Y и R10 оба представляют собой -CF3; R6 и R7 являются одинаковыми или различными и независимо выбраны из группы, состоящей из H, C1-C6 алкила, C3-C6 циклоалкила, арила, гетероарила и 4-6-членного гетероциклического кольца, где указанный алкил является насыщенным или ненасыщенным или, где R6 и R7 образуют 4-6-членное гетероциклическое кольцо с N, к которому они присоединены, необязательно также содержащее атом O; R12 выбран из группы, состоящей из H, C3-C4 циклоалкила и C1-C6 алкила; и где любой содержащий углерод фрагмент R1-R12 может быть необязательно замещен одним или несколькими атомами галогена, фторметаном, дифторметаном или трифторметаном, или -OH; или их фармацевтически приемлемые соли.
В более конкретных вариантах осуществления такие новые соединения описываются формулой IV, где: R2 выбран из группы, состоящей из H, галогена, метила и этила; R3 являются одинаковыми или различными и независимо выбраны из группы, состоящей из H, галогена, метила и этила; R4 являются одинаковыми или различными и независимо выбраны из группы, состоящей из H, галогена, C1-C6 алкила и C1-C6 алкокси; R5 являются одинаковыми или различными и независимо выбраны из группы, состоящей из H, галогена, метила и этила; R8 и R10 являются одинаковыми или различными и независимо выбраны из группы, состоящей из H и метила; R9 выбран из группы, состоящей из H, метила и этила; и R6 и R7 являются одинаковыми или различными и независимо выбраны из группы, состоящей из H и C1-C6 алкила или где R6 и R7образуют 4-6-атомное гетероциклическое кольцо с N, к которому они присоединены, необязательно также содержащее атом O.
В некоторых вариантах осуществления соединения формулы III или формулы IV имеют следующие перестановки или комбинации перестановок: Y представляет собой -C(O)NR6R7; R6 и R7 представляют собой метил; R8 и R10 оба представляют собой H; R1 представляет собой этил или циклобутил; R9 представляет собой H; R2 представляет собой F или H; m равно 2 и один из R4 представляет собой F и другой R4 представляет собой Cl; m равно 2 и оба R4 представляют собой F; m равно 0; n равно 1 и R3 представляет собой F; n равно 0; p равно 1 и R5 представляет собой F; или p равно 0.
Вариант осуществления может обеспечивать соединение, имеющее следующую формулу:

Вариант осуществления может обеспечивать соединение, имеющее следующую формулу:

Вариант осуществления может обеспечивать соединение, имеющее следующую формулу:

Еще один вариант осуществления может обеспечивать способ лечения рака молочной железы, включающий введение субъекту соединения в соответствии с любым из предшествующих абзацев. Рак молочной железы может представлять собой ER-положительный рак молочной железы. Субъект может экспрессировать мутантный ER-α белок. В одном варианте осуществления может обеспечиваться применение соединения, описанного в представленных выше абзацах, для лечения рака молочной железы. В некоторых вариантах осуществления рак молочной железы представляет собой ER-положительный рак молочной железы. В некоторых вариантах осуществления субъект экспрессирует мутантный ER-α белок. В некоторых вариантах осуществления соединение, представленное выше, используют для получения лекарственного средства для лечения рака молочной железы.
В некоторых вариантах осуществления соединения, раскрытые в настоящей заявке, являются полезными для ингибирования роста клеточных культур MCF7 ER-альфа (дикий тип) и MCF7 ER-альфа (Y537S мутант) клеток. Другие соединения (например, тамоксифен, ралоксифен и фулвестрант), известные как ингибирующие рост клеточной культуры MCF7 ER-альфа (дикий тип) клеток, в настоящее время используются для лечения рака молочной железы у пациентов людей. Следовательно, соединения, раскрытые в настоящей заявке, являются полезными для лечения ER-альфа-экспрессирующего рака молочной железы у пациентов людей и являются полезными для лечения Y537S мутант ER-альфа-экспрессирующего рака молочной железы у пациентов людей.
В вариантах осуществления соединения, раскрытые в настоящем описании, являются полезными для лечения рака молочной железы. В вариантах осуществления рак молочной железы представляет собой ER-α+. В вариантах осуществления рак молочной железы экспрессирует ER-α мутацию, которая представляет собой L536Q (Robinson et al. Nat Genet. 2013 Dec;45(12)), L536R (Toy et al. Nat Genet. 2013 Dec;45(12):1439-45), Y537S (Toy et al. Nat Genet. 2013 Dec;45(12):1439-45; Robinson et al. Nat Genet. 2013 Dec;45(12); Jeselsohn et al. Clin Cancer Res. 2014 Apr 1;20(7):1757-67), Y537N (Toy et al. Nat Genet. 2013 Dec;45(12):1439-45; Jeselsohn et al. Clin Cancer Res. 2014 Apr 1;20(7):1757-67), Y537C (Toy et al. Nat Genet. 2013 Dec;45(12):1439-45; Jeselsohn et al. Clin Cancer Res. 2014 Apr 1;20(7):1757-67) и D538G (Toy et al. Nat Genet. 2013 Dec;45(12):1439-45; Robinson et al. Nat Genet. 2013 Dec;45(12); Jeselsohn et al. Clin Cancer Res. 2014 Apr 1;20(7):1757-67; Merenbakh-Lamin et al. Cancer Res. 2013 Dec 1;73(23):6856-64); and Yu et al., (2014) Science Jul 11;345(6193):216-20, все из которых включены в полном объеме в качестве ссылки для их учения о ER-α мутациях.
Краткое описание фигур
Фиг. 1показывает in vitro эффекты пролиферации MCF7 линий дикого типа и содержащей мутантный ER при использовании клинических терапевтических средств 4-гидрокситамоксифен (4-OHT), ралоксифен и фулвестрант, где наблюдалась фенотипическая устойчивость(резистентность) в мутант-несущих линиях по сравнению с контрольными линиями к существующим клиническим соединениям, при этом MCF7 клетки, сконструированные так, чтобы чрезмерно экспрессировать различные ERαMUT, показали частичную резистентность к различным эндокринным терапиям.
Фиг. 2показывает противоопухолевый эффект и эффект на массу тела перорально вводимого Соединения 1 у самок бестимусных мышей balb/c с ксентрансплантатом MCF7.
Фиг. 3 показывает противоопухолевый эффект и эффект на массу тела перорально вводимого Соединения 1 у самок бестимусных ʺголыхʺ мышей (Crl:NU(NCr)-Foxnlnu) с ксентрансплантатом PDX-Y537S.
Фиг. 4 показывает противоопухолевый эффект и эффект на массу тела перорально вводимого Соединения 1 у самок мышей SCID-bg с ксентрансплантатом WHUM20.
Фиг. 5 показывает противоопухолевый эффект и эффект на массу тела перорально вводимого Соединения 60 у самок бестимусных мышей Balb/c с ксентрансплантатом MCF7.
Фиг. 6 показывает противоопухолевый эффект и эффект на массу тела перорально вводимого Соединения 60 у самок бестимусных ʺголыхʺ мышей (Crl:NU(NCr)-Foxnlnu) с ксентрансплантатом PDX-Y537S.
Фиг. 7 показывает противоопухолевый эффект и эффект на массу тела перорально вводимого Соединения 69 у самок бестимусных мышей Balb/c с ксентрансплантатом MCF7.
Фиг. 8 показывает противоопухолевый эффект и эффект на массу тела перорально вводимого Соединения 69 у самок бестимусных ʺголыхʺ мышей (Crl:NU(NCr)-Foxnlnu) с ксентрансплантатом PDX-Y537S.
Фиг. 9 показывает противоопухолевый эффект и эффект на массу тела Соединения 60 в модели ER+ WHIM20 PDX с гомозиготной мутацией Y537S.
Подробное описание
Все публикации и патентные документы, приведенные в настоящем описании, включены в настоящее описание в качестве ссылки, как если бы каждая такая публикация или документ была конкретно и индивидуально указана для включения в настоящее описание в качестве ссылки. В случае, когда текст настоящего раскрытия и текст одного или нескольких документов, включенных в качестве ссылки, противоречат друг другу, настоящее раскрытие является определяющим. Цитирование публикаций и патентных документов не является признанием того, что любой из них относится к предшествующему уровню техники, а также не является признанием в качестве их содержания или даты. Варианты осуществления, раскрытые в настоящем документе, имеют письменное описание, поэтому специалисту в данной области будет понятно, что описанные здесь варианты осуществления могут быть осуществлены на практике во множестве вариантов осуществления и что описание и примеры, представленные в настоящем описании, предназначены для иллюстрации, а не ограничения формулы изобретения.
Как используется в настоящем описании «алкил», «C1, C2, C3, C4, C5 или C6 алкил» или «C1-C6 алкил» предназначен для включения C1, C2, C3, C4, C5 или C6 насыщенных алифатических углеводородных групп с прямой цепью (линейных) и C3, C4, C5 или C6 разветвленных насыщенных алифатических углеводородных групп. Например, C1-C6 алкил предназначен для включения C1, C2, C3, C4, C5 и C6алкильных групп. Примеры алкила включают фрагменты, имеющие от одного до шести атомов углерода, такие как, но не ограничиваясь ими, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, н-пентил, втор-пентил или н-гексил.
В некоторых вариантах осуществления алкил с прямой цепью или разветвленной цепью имеет шесть или менее атомов углерода (например, C1-C6 для прямой цепи, C3-C6 для разветвленной цепи), и в другом варианте осуществления алкил с прямой цепью или разветвленной цепью имеет четыре или менее атомов углерода.
Как используется в настоящем описании термин «циклоалкил» относится к насыщенному или ненасыщенному неароматическому углеводородному кольцу, имеющему 3-7 атомов углерода (например, C3-C7). Примеры циклоалкила включают, но не ограничиваются ими, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклопентенил, циклогексенил и циклогептенил.
Термин «гетероциклоалкил» относится к насыщенным или ненасыщенным неароматическим 3-8-членным моноциклическим группам, 7-10-членным конденсированным бициклическим группам, имеющим один или несколько гетероатомов (таких как O, N или S), если не указано иное. Примеры гетероциклоалкильных групп включают, но не ограничиваются ими, пиперидинил, пиперазинил, пирролидинил, диоксанил, тетрагидрофуранил, изоиндолинил, индолинил, имидазолидинил, пиразолидинил, оксазолидинил, изоксазолидинил, триазолидинил, оксиранил, азетидинил, оксетанил, тиетанил, 1,2,3,6-тетрагидропиридинил, тетрагидропиранил, тетрагидротиофен, дигидропиранил, пиранил, морфолинил, 1,4-диазепанил, 1,4-оксазепанил и тому подобное.
Дополнительные примеры гетероциклоалкильных групп включают, но не ограничиваются ими, акридинил, азоцинил, бензимидазолил, бензофуранил, бензотиофуранил, бензотиофенил, бензоксазолил, бензоксазолинил, бензтиазолил, бензтриазолил, бензтетразолил, бензизоксазолил, бензизотиазолил, бензимидазолинил, карбазолил, 4аН-карбазолил, карболинил, хроманил, хроменил, циннолинил, декагидрохинолинил, 2Н,6Н-1,5,2-дитиазинил, дигидрофуро[2,3-b]тетрагидрофуран, фуранил, фуразанил, имидазолидинил, имидазолинил, имидазолил, 1Н-индазолил, индоленил, индолинил, индолизинил, индолил, 3Н-индолил, изатиноил, изобензофуранил, изохроманил, изоиндазолил, изоиндолинил, изоиндолил, изохинолинил, изотиазолил, изоксазолил, метилендиоксифенил, морфолинил, нафтиридинил, октагидроизохинолинил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, 1,2,4-оксадиазол5(4Н)-он, оксазолидинил, оксазолил, оксиндолил, пиримидинил, фенантридинил, фенантролинил, феназинил, фенотиазинил, феноксатинил, феноксазинил, фталазинил, пиперазинил, пиперидинил, пиперидонил, 4-пиперидонил, пиперонил, птеридинил, пуринил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, пиридооксазол, пиридоимидазол, пиридотиазол, пиридинил, пиридил, пиримидинил, пирролидинил, пирролинил, 2Н-пирролил, пирролил, хиназолинил, хинолинил, 4Н-хинолизинил, хиноксалинил, хинуклидинил, тетрагидрофуранил, тетрагидроизохинолинил, тетрагидрохинолинил, тетразолил, 6H-1,2,5-тиадиазинил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, тиантренил, тиазолил, тиенил, тиенотиазолил, тиенооксазолил, тиеноимидазолил, тиофенил, триазинил, 1,2,3-триазолил, 1,2,4-триазолил, 1,2,5-триазолил, 1,3,4-триазолил и ксантенил.
Термин «необязательно замещенный алкил» относится к незамещенному алкилу или алкилу, имеющему обозначенные заместители, замещающие один или несколько атомов водорода на одном или нескольких атомах углерода углеводородного скелета. Такие заместители могут включать, например, алкил, алкенил, алкинил, галоген, гидроксил, алкилкарбонилокси, арилкарбонилокси, алкоксикарбонилокси, арилоксикарбонилокси, карбоксилат, алкилкарбонил, арилкарбонил, алкоксикарбонил, аминокарбонил, алкиламинокарбонил, диалкиламинокарбонил, алкилтиокарбонил, алкоксил, фосфат, фосфонато, фосфинато, амино (включая алкиламино, диалкиламино, ариламино, диариламино и алкилариламино), ациламино (включая алкилкарбониламино, арилкарбониламино, карбамоил и уреидо), амидино, имино, сульфгидрил, алкилтио, арилтио, тиокарбоксилат, сульфаты, алкилсульфинил, сульфонато, сульфамоил, сульфонамидо, нитро, трифторметил, циано, азидо, гетероциклил, алкиларил или ароматический или гетероароматический фрагмент.
«Арилалкил» или «аралкильный» фрагмент представляет собой алкил, замещенный арилом (например, фенилметил(бензил)). «Алкиларильный» фрагмент представляет собой арил, замещенный алкилом (например, метилфенил).
«Алкенил» включает ненасыщенные алифатические группы, аналогичные по длине и возможному замещению алкилам, описанным выше, но которые содержат по меньшей мере одну двойную связь. Например, термин «алкенил» включает алкенильные группы с прямой цепью (например, этенил, пропенил, бутенил, пентенил, гексенил) и разветвленные алкенильные группы. В некоторых вариантах осуществления алкенильная группа с прямой цепью или разветвленной цепью имеет шесть или меньше атомов углерода в ее основной цепи (например, C2-C6 для прямой цепи, C3-C6 для разветвленной цепи). Термин «C2-C6» включает алкенильные группы, содержащие от двух до шести атомов углерода. Термин «C3-C6» включает алкенильные группы, содержащие от трех до шести атомов углерода.
Термин «необязательно замещенный алкенил» относится к незамещенному алкенилу или алкенилу, имеющему обозначенные заместители, замещающие один или несколько атомов водорода на одном или нескольких атомах углерода углеводородного скелета. Такие заместители могут включать, например, алкил, алкенил, алкинил, галоген, гидроксил, алкилкарбонилокси, арилкарбонилокси, алкоксикарбонилокси, арилоксикарбонилокси, карбоксилат, алкилкарбонил, арилкарбонил, алкоксикарбонил, аминокарбонил, алкиламинокарбонил, диалкиламинокарбонил, алкилтиокарбонил, алкоксил, фосфат, фосфонато, фосфинато, амино (включая алкиламино, диалкиламино, ариламино, диариламино и алкилариламино), ациламино (включая алкилкарбониламино, арилкарбониламино, карбамоил и уреидо), амидино, имино, сульфгидрил, алкилтио, арилтио, тиокарбоксилат, сульфаты, алкилсульфинил, сульфонато, сульфамоил, сульфонамидо, нитро, трифторметил, циано, гетероциклил, алкиларил или ароматический или гетероароматический фрагмент.
«Алкинил» включает ненасыщенные алифатические группы, аналогичные по длине и возможному замещению алкилам, описанным выше, но которые содержат по меньшей мере одну тройную связь. Например, «алкинил» включает алкинильные группы с прямой цепью (например, этинил, пропинил, бутинил, пентинил, гексинил) и разветвленные алкинильные группы. В некоторых вариантах осуществления алкинильная группа с прямой цепью или разветвленной цепью имеет шесть или меньше атомов углерода в ее основной цепи (например, C2-C6 для прямой цепи, C3-C6 для разветвленной цепи). Термин «C2-C6» включает алкинильные группы, содержащие от двух до шести атомов углерода. Термин «C3-C6» включает алкинильные группы, содержащие от трех до шести атомов углерода.
Термин «необязательно замещенный алкинил» относится к незамещенному алкинилу или алкинилу, имеющему обозначенные заместители, замещающие один или несколько атомов водорода на одном или нескольких атомах углерода углеводородного скелета. Такие заместители могут включать, например, алкил, алкенил, алкинил, галоген, гидроксил, алкилкарбонилокси, арилкарбонилокси, алкоксикарбонилокси, арилоксикарбонилокси, карбоксилат, алкилкарбонил, арилкарбонил, алкоксикарбонил, аминокарбонил, алкиламинокарбонил, диалкиламинокарбонил, алкилтиокарбонил, алкоксил, фосфат, фосфонато, фосфинато, амино (включая алкиламино, диалкиламино, ариламино, диариламино и алкилариламино), ациламино (включая алкилкарбониламино, арилкарбониламино, карбамоил и уреидо), амидино, имино, сульфгидрил, алкилтио, арилтио, тиокарбоксилат, сульфаты, алкилсульфинил, сульфонато, сульфамоил, сульфонамидо, нитро, трифторметил, циано, азидо, гетероциклил, алкиларил или ароматический или гетероароматический фрагмент.
Другие необязательно замещенные фрагменты (такие как необязательно замещенный циклоалкил, гетероциклоалкил, арил или гетероарил) включают как незамещенные фрагменты, так и фрагменты, имеющие один или несколько обозначенных заместителей. Например, замещенный гетероциклоалкил включает замещенные одной или несколькими алкильными группами, такие как 2,2,6,6-тетраметилпиперидинил и 2,2,6,6-тетраметил-1,2,3,6-тетрагидропиридинил.
«Арил» включает группы с ароматичностью, включая «конъюгированные» или полициклические системы с по меньшей мере одним ароматическим кольцом и не содержат какой-либо гетероатом в кольцевой структуре. Примеры включают фенил, бензил, 1,2,3,4-тетрагидронафталенил и тому подобное.
«Гетероарильные» группы представляют собой арильные группы, как определено выше, за исключением наличия от одного до четырех гетероатомов в кольцевой структуре и также могут упоминаться как «арильные гетероциклы» или «гетероароматические соединения». Как используется в настоящем описании термин «гетероарил» предназначен для включения стабильного 5-, 6- или 7-членного моноциклического или 7-, 8-, 9-, 10-, 11- или 12-членного бициклического ароматического гетероциклического кольца, которое состоит из атомов углерода и одного или нескольких гетероатомов, например, 1 или 1-2, или 1-3, или 1-4, или 1-5, или 1-6 гетероатомов, или, например, 1, 2, 3, 4, 5 или 6 гетероатомов, независимо выбранных из группы, состоящей из азота, кислорода и серы. Атом азота может быть замещенным или незамещенным (то есть N или NR', где R' представляет собой H или другие заместители, как определено). Гетероатомы азота и серы могут быть необязательно окислены (т.е., N→O и S(O)p, где p=1 или 2). Следует отметить, что общее количество атомов S и O в ароматическом гетероцикле не превышает 1.
Примеры гетероарильных групп включают пиррол, фуран, тиофен, тиазол, изотиазол, имидазол, триазол, тетразол, пиразол, оксазол, изоксазол, пиридин, пиразин, пиридазин, пиримидин и тому подобное.
Кроме того, термины «арил» и «гетероарил» включают полициклические арильные и гетероарильные группы, например, бициклические. Неограничивающий пример таких арильных групп включает, например, нафталин, бензоксазол, бензодиоксазол, бензотиазол, бензоимидазол, бензотиофен, метилендиоксифенил, хинолин, изохинолин, нафтридин, индол, бензофуран, пурин, бензофуран, деазапурин, индолизин.
В случае полициклических ароматических колец только одно из колец должно быть ароматическим (например, 2,3-дигидроиндол), хотя все кольца могут быть ароматическими (например, хинолин).
Циклоалкильное, гетероциклоалкильное, арильное или гетероарильное кольцо может быть замещено в одном или нескольких положениях кольца (например, образующем кольцо углероде или гетероатоме, таком как N) такими заместителями, как описано выше, например, алкилом, алкенилом, алкинилом, галогеном, гидроксилом, алкокси, алкилкарбонилокси, арилкарбонилокси, алкоксикарбонилокси, арилоксикарбонилокси, карбоксилатом, алкилкарбонилом, алкиламинокарбонилом, аралкиламинокарбонилом, алкениламинокарбонилом, алкилкарбонилом, арилкарбонилом, аралкилкарбонилом, алкенилкарбонилом, алкоксикарбонилом, аминокарбонилом, алкилтиокарбонилом, фосфатом, фосфонато, фосфинато, амино (включая алкиламино, диалкиламино, ариламино, диариламино и алкилариламино), ациламино (включая алкилкарбониламино, арилкарбониламино, карбамоил и уреидо), амидино, имино, сульфгидрилом, алкилтио, арилтио, тиокарбоксилатом, сульфатами, алкилсульфинилом, сульфонато, сульфамоилом, сульфонамидо, нитро, трифторметилом, циано, азидо, гетероциклилом, алкиларилом или ароматическим или гетероароматическим фрагментом. Арильные и гетероарильные группы также могут быть конденсированы с алициклическими или гетероциклическими кольцами, которые не являются ароматическими, с образованием полициклической системы (например, тетралин, метилендиоксифенил).
Когда связь к заместителю показана как пересекающая связь, соединяющая два атома в кольце (как показано ниже в примерах с заместителем R), тогда такой заместитель может быть связан с любым атомом в кольце.

Когда любая переменная (например, R1) встречается более одного раза в любой составной части или формуле соединения, ее определение в каждом случае не зависит от ее определения в каждом другом случае. Так, например, если показано, что группа замещена 0-2 группами R1, тогда группа может быть необязательно замещена до двух групп R1 и R1 в каждом случае выбирается независимо от определения R1.
Термин «гидрокси» или «гидроксил» включает группы с -OH или -O-.
Как используется в настоящем описании термин «гало» или «галоген» относится к фтору, хлору, бром и иоду. Термин «пергалогенированный» обычно относится к фрагменту, в котором все атомы водорода замещены атомами галогена. Термин «галогеналкил» или «галогеналкоксил» относится к алкилу или алкоксилу, замещенному одним или несколькими атомами галогена.
«Алкоксиалкил», «алкиламиноалкил» и «тиоалкоксиалкил» включают алкильные группы, как описано выше, где атомы кислорода, азота или серы замещают один или несколько атомов углерода углеводородного скелета.
Термин «алкокси» или «алкоксил» включает замещенные и незамещенные алкильные, алкенильные и алкинильные группы, ковалентно связанные с атомом кислорода. Примеры алкоксильных групп или алкоксильных радикалов включают, но не ограничиваются ими, метокси, этокси, изопропилокси, пропокси, бутокси и пентоксигруппы. Примеры замещенных алкоксильных групп включают галогенированные алкоксильные группы. Алкоксильные группы могут быть замещены группами, такими как алкенил, алкинил, галоген, гидроксил, алкилкарбонилокси, арилкарбонилокси, алкоксикарбонилокси, арилоксикарбонилокси, карбоксилат, алкилкарбонил, арилкарбонил, алкоксикарбонил, аминокарбонил, алкиламинокарбонил, диалкиламинокарбонил, алкилтиокарбонил, алкоксил, фосфат, фосфонато, фосфинато, амино (включая алкиламино, диалкиламино, ариламино, диариламино и алкилариламино), ациламино (включая алкилкарбониламино, арилкарбониламино, карбамоил и уреидо), амидино, имино, сульфгидрил, алкилтио, арилтио, тиокарбоксилат, сульфаты, алкилсульфинил, сульфонато, сульфамоил, сульфонамидо, нитро, трифторметил, циано, азидо, гетероциклил, алкиларил или ароматические или гетероароматические фрагменты. Примеры галогензамещенных алкоксигрупп включают, но не ограничиваются ими, фторметокси, дифторметокси, трифторметокси, хлорметокси, дихлорметокси и трихлорметокси.
«Изомеризи» означает соединения, которые имеют идентичные молекулярные формулы, но отличаются в последовательности связывания их атомов или по расположению их атомов в пространстве. Изомеры, которые различаются по расположению их атомов в пространстве, называются «стереоизомерами». Стереоизомеры, которые не являются зеркальными изображениями друг друга, называются «диастереоизомерами», и стереоизомеры, которые являются ненакладываемыми зеркальными отображениями друг друга, называются «энантиомерами» или иногда оптическими изомерами. Смесь, содержащую равные количества индивидуальных энантиомерных форм противоположной хиральности, называется «рацемической смесью».
Атом углерода, связанный с четырьмя неидентичными заместителями, называется «хиральным центром».
«Хиральный изомер» означает соединение с по меньшей мере одним хиральным центром. Соединения с более чем одним хиральным центром могут существовать или в виде отдельного диастереомера, или в виде смеси диастереомеров, называемой «диастереомерной смесью». Когда присутствует один хиральный центр, стереоизомер может быть охарактеризован по абсолютной конфигурации (R или S) этого хирального центра. Абсолютная конфигурация относится к расположению в пространстве заместителей, присоединенных к хиральному центру. Заместители, присоединенные к рассматриваемому хиральному центру, располагаются по рангу в соответствии с Правилом Последовательности Cahn, Ingold and Prelog. (Calm et al., Angew. Chem. Inter. Edit. 1966, 5, 385; errata 511; Cahn et al., Angew. Chem. 1966, 78, 413; Cahn and Ingold, J. Chem. Soc. 1951 (London), 612; Calm et al., Experientia 1956, 12, 81; Cahn, J. Chem. Educ. 1964, 41, 116).
В настоящем описании каждое число хирального центра в структурной формуле, такого как неограничивающий пример, показанный здесь:

предназначено для описания всех возможных стереоизомеров. Напротив, хиральный центр, нарисованный с помощью штриховки и клиньев, такой как неограничивающий пример, показанный здесь:

предназначен для того, чтобы изобразить стереоизомер, как указано (здесь, в этом хиральном центре с sp3 гибридизированным атомом углерода, R3 и R4 находятся в плоскости рисунка, R1 находится над плоскостью рисунка, а R2 находится за плоскостью рисунка).
«Геометрический изомер» означает диастереомеры, которые обязаны своим существованием затрудненному вращению вокруг двойных связей или циклоалкильного линкера (например, 1,3-циклобутил). Эти конфигурации дифференцированы в их наименованиях префиксами цис и транс или Z и Е, которые показывают, что группы находятся на одной или на противоположных сторонах двойной связи в молекуле, согласно правилам Cahn-Ingold-Prelog.
В настоящем описании каждая область внутри структурной формулы, включающая волнистую линию, присоединенную к двойной связи, как показано:

предназначена для отображения обоих геометрических изомеров. Напротив, такие структуры, выполненные без волнистой линии, предназначены для отображения соединения, имеющего геометрическую конфигурацию, как показано на рисунке.
«Таутомер» является одним из двух или более структурных изомеров, которые существуют в равновесии и легко превращаются из одной изомерной формы в другую. Это превращение приводит к формальной миграции атома водорода, сопровождаемой переносом соседних сопряженных двойных связей. Таутомеры существуют как смесь таутомерного набора в растворе. В растворах, где возможна таутомеризация, будет достигнуто химическое равновесие таутомеров. Точное соотношение таутомеров зависит от нескольких факторов, включая температуру, растворитель и рН. Общее понятие таутомеров, что они являются взаимопревращаемыми путем таутомеризаций, называется таутомеризмом.
Если в настоящей заявке указано соединение, подверженное таутомеризации, но только отображен один из таутомеров, подразумевается, что все таутомеры включены как часть значения представленного химического вещества. Следует понимать, что описанные здесь соединения могут быть представлены как различные таутомеры. Следует также понимать, что когда соединения имеют таутомерные формы, все таутомерные формы должны быть включены в объем изобретения, и наименование соединений не исключает какую-либо таутомерную форму.
Из различных типов таутомеризма, которые возможны, обычно наблюдаются два. В кето-енольном таутомеризме происходит одновременный сдвиг электронов и атома водорода. Кольчато-цепная таутомерия возникает как результат взаимодействия альдегидной группы (--CHO) в молекуле сахара в виде цепи с одной из гидроксигрупп (--OH) в той же молекуле с получением ее циклической (кольцеобразной) формы, как проявляется глюкозой. Обычными таутомерными парами являются: кетон-енол, амид-нитрил, лактам-лактим, амид-имидная кислота таутомерия в гетероциклических кольцах (например, в нуклеиновых основаниях, таких как гуанин, тимин и цитозин), имин-енамин и енамин-енамин.
Кроме того, структуры и другие соединения, описанные в настоящем документе, включают все атропические изомеры, причем понятно, что не все атропические изомеры могут иметь одинаковый уровень активности. «Атропические изомеры» являются типом стереоизомера, в котором атомы двух изомеров различно расположены в пространстве. Атропические изомеры обязаны своим существованием ограниченному вращению, вызванному затрудненностью вращения больших групп вокруг центральной связи. Такие атропические изомеры обычно существуют в виде смеси, однако в результате последних достижений в методах хроматографии стало возможным разделить смеси двух атропических изомеров в избранных случаях.
Термин «кристаллические полиморфы», «полиморфы» или «кристаллические формы» означает кристаллические структуры, в которых соединение (или его соль или сольват) могут кристаллизоваться в различных системах упаковки кристаллов, все из которых имеют одинаковый элементарный состав. Различные кристаллические формы обычно имеют различные рентгенограммы, инфракрасные спектральные характеристики, температуры плавления, плотность, твердости, кристаллическую форму, оптические и электрические свойства, стабильность и растворимость. Растворитель для перекристаллизации, скорость кристаллизации, температура хранения и другие факторы могут сделать одну кристаллическую форму доминирующей. Кристаллические полиморфы соединений могут быть получены кристаллизацией в различных условиях. Известно, что соединения, раскрытые в настоящем описании, могут существовать в кристаллической форме, смеси кристаллической формы или в виде его ангидрида или гидрата.
Соединения, раскрытые в настоящем описании, включают сами соединения, а также их соли и сольваты, при необходимости. Например, соль может быть образована между анионом и положительно заряженной группой (например, амино) на арил- или гетероарилзамещенном бензольном соединении. Подходящие анионы включают хлорид, бромид, йодид, сульфат, бисульфат, сульфамат, нитрат, фосфат, цитрат, метансульфонат, трифторацетат, глутамат, глюкуронат, глутарат, малат, малеат, сукцинат, фумарат, тартрат, тозилат, салицилат, лактат, нафталинсульфонат и ацетат (например, трифторацетат). Термин «фармацевтически приемлемый анион» относится к аниону, подходящему для образования фармацевтически приемлемой соли. Аналогично, соль может быть также образована между катионом и отрицательно заряженной группой (например, карбоксилатом) на арил- или гетероарилзамещенном бензольном соединении. Подходящие катионы включают ион натрия, ион калия, ион магния, ион кальция и катион аммония, такой как ион тетраметиламмония. Арил- или гетероарилзамещенные бензольные соединения также включают те соли, содержащие четвертичные атомы азота.
Дополнительно, соединения, раскрытые в настоящем описании, например, соли соединений, могут существовать как в гидратированной, так и в негидратной (безводной) форме или в виде сольватов с молекулами другого растворителя. Неограничивающие примеры гидратов включают моногидраты, дигидраты и тому подобное. Неограничивающие примеры сольватов включают сольваты с этанолом, сольваты с ацетоном и тому подобное.
Как используется в настоящем описании термин «фармацевтически приемлемые соли» относится к производным соединений, раскрытых в настоящем описании, где исходное соединение модифицировано путем получения его соли с кислотой или основанием. Примеры фармацевтически приемлемых солей включают, но не ограничиваются ими, минеральные или органические соли с кислотами основных остатков, таких как амины, щелочные или органические соли кислотных остатков, таких как карбоновые кислоты и тому подобное. Фармацевтически приемлемые соли включают обычные нетоксичные соли или соли четвертичного аммония исходного соединения, образованные, например, из нетоксичных неорганических или органических кислот. Например, такие обычные нетоксичные соли включают, но не ограничиваются ими, производные из неорганических и органических кислот, выбранных из 2-ацетоксибензойной, 2-гидроксиэтансульфоновой, уксусной, аскорбиновой, бензолсульфоновой, бензойной, бикарбоновой, угольной, лимонной, этилендиаминтетрауксусной, этандисульфоновой, 1,2-этансульфоновой, фумаровой, глюкогептоновой, глюконовой, глутаминовой, гликолевой, гликолиларсаниловой, гексилрезорциновой, гидрабаминовой, бромистоводородной, хлористоводородной, йодистоводородной, гидроксималеиновой, гидроксинафтойной, изетионовой, молочной, лактобионовой, лаурилсульфоновой, малеиновой, яблочной, миндальной, метансульфоновой, напсиловой, азотной, щавелевой, памовой, пантотеновой, фенилуксусной, фосфорной, полигалактуроновой, пропионовой, салициловой, стеариновой, субуксусной, янтарной, сульфаминовой, сульфаниловой, серной, дубильной, винной, толуолсульфоновой и общераспространенных аминокислот, например, глицина, аланина, фенилаланина, аргинина и тому подобное.
Другие примеры фармацевтически приемлемых солей включают соли гексановой (капроновой) кислоты, циклопентанпропионовой кислоты, пировиноградной кислоты, малоновой кислоты, 3-(4-гидроксибензоил)бензойной кислоты, коричной кислоты, 4-хлорбензолсульфоновой кислоты, 2-нафталинсульфоновой кислоты, 4-толуолсульфоновой кислоты, камфорсульфоновой кислоты, 4-метилбицикло-[2.2.2]-окт-2-ен-1-карбоновой кислоты, 3-фенилпропионовой кислоты, триметилуксусной кислоты, трет-бутилуксусной кислоты, муконовой кислоты и тому подобное. Настоящее изобретение также охватывает соли, образованные в случае, если в исходном соединении присутствует кислотный протон, и он либо замещен ионом металла, например, ионом щелочного металла, ионом щелочноземельного металла или ионом алюминия; либо координирован с органическим основанием, таким как этаноламин, диэтаноламин, триэтаноламин, трометамин, N-метилглюкамин и тому подобное. В солевой форме понятно, что отношение соединения к катиону или аниону соли может составлять 1:1, или любое отношение, отличное от 1:1, например, 3:1, 2:1, 1:2 или 1:3.
Следует понимать, что все ссылки на фармацевтически приемлемые соли включают формы присоединения растворителя (сольваты) или кристаллические формы (полиморфы), как определено в настоящем описании, такой же соли.
«Сольват» означает формы с добавленным растворителем, которые содержат или стехиометрическое, или не стехиометрическое количество растворителя. Некоторые соединения имеют тенденцию захватывать фиксированное молярное отношение молекул растворителя в кристаллическом твердом состоянии, образуя таким образом сольват. Если растворителем является вода, образовавшийся сольват представляет собой гидрат; и если растворителем является спирт, образовавшийся сольват представляет собой алкоголят. Гидраты образуются путем комбинации одной или нескольких молекул воды с одной молекулой вещества, в которой вода сохраняет свое молекулярное состояние как H2O.
Химические вещества, которые названы или изображены, предназначены для включения всех встречающихся в природе изотопов атомов, встречающихся в настоящих соединениях. Изотопы включают такие атомы, имеющие один и тот же атомный номер, но разные массовые числа. В качестве общего примера и без ограничения изотопы1H водорода включают тритий и дейтерий, и изотопы12C углерода включают13C и14C.
Понятно, что некоторые соединения и их изомеры, соли, сложные эфиры и сольваты, раскрытые в настоящем описании, могут проявлять большую активность in vivo или in vitro, чем другие. Также будет понятно, что некоторые виды рака можно лечить более эффективно, чем другие, и можно лечить эффективнее у некоторых видов субъектов, которые другие, используя соединения и их изомеры, соли, сложные эфиры и сольваты, соединений, раскрытых в настоящем описании.
Как используется в настоящем описании термин «лечение» означает введение субъекту фармацевтической композиции для улучшения, снижения или уменьшения симптомов заболевания. Как используется в настоящем описании термин «лечение» или «лечить» описывает лечение и уход за субъектом с целью борьбы с заболеванием, состоянием или нарушением и включает введение соединения, раскрытого в настоящем описании, или его фармацевтически приемлемой соли, полиморфа или сольвата, для облегчения симптомов или осложнений заболевания, состояния или нарушения, или устранения заболевания, состояние или нарушения. Термин «лечить» может также включать обработку клетки in vitro или животной модели.
Лечение рака может привести к уменьшению размера опухоли. Уменьшение размера опухоли также можно назвать «регрессией опухоли». Предпочтительно, после обработки размер опухоли уменьшается на 5% или более по сравнению с ее размером до обработки; более предпочтительно, размер опухоли уменьшается на 10% или более; более предпочтительно, уменьшается на 20% или более; более предпочтительно, уменьшается на 30% или более; более предпочтительно, уменьшается на 40% или более; еще более предпочтительно, уменьшается на 50% или более; и наиболее предпочтительно, уменьшается более чем на 75% или более. Размер опухоли может быть измерен с помощью любых воспроизводимых способов измерения. Размер опухоли можно измерить как диаметр опухоли.
Лечение рака может привести к уменьшению объема опухоли. Предпочтительно, после лечения объем опухоли уменьшается на 5% или более по сравнению с ее размером перед лечением, более предпочтительно, объем опухоли уменьшается на 10% или более; более предпочтительно, уменьшается на 20% или более; более предпочтительно, уменьшается на 30% или более; более предпочтительно, уменьшается на 40% или более; еще более предпочтительно, уменьшается на 50% или более; и наиболее предпочтительно, уменьшается более чем на 75% или более. Объем опухоли может быть измерен с помощью любых воспроизводимых способов измерения.
Лечение рака может привести к уменьшению числа опухолей. Предпочтительно, после лечения число опухолей уменьшается на 5% или более по сравнению с числом перед лечением; более предпочтительно, число опухолей уменьшается на 10% или более; более предпочтительно, уменьшается на 20% или более; более предпочтительно, уменьшается на 30% или более; более предпочтительно, уменьшается на 40% или более; еще более предпочтительно, уменьшается на 50% или более; и наиболее предпочтительно, уменьшается более чем на 75%. Число опухолей может быть измерено любым воспроизводимым способом измерения. Число опухолей может быть измерено путем подсчета опухолей, видимых невооруженным глазом или при определенном увеличении. Предпочтительно, определенное увеличение составляет 2×, 3×, 4×, 5×, 10× или 50×.
Лечение рака приводит к уменьшению числа метастатических поражений в других тканях или органах, расположенных в отдалении от месторасположения первичной опухоли. Предпочтительно, после лечения число метастатических поражений уменьшается на 5% или более по сравнению с числом перед лечением; более предпочтительно, число метастатических поражений уменьшается на 10% или более; более предпочтительно, уменьшается на 20% или более; более предпочтительно, уменьшается на 30% или более; более предпочтительно, уменьшается на 40% или более; еще более предпочтительно, уменьшается на 50% или более; и наиболее предпочтительно, уменьшается более чем на 75%. Число метастатических поражений может быть измерено любым воспроизводимым способом измерения. Число метастатических поражений может быть измерено путем подсчета метастатических поражений, видимых невооруженным глазом или при определенном увеличении. Предпочтительно, определенное увеличение составляет 2×, 3×, 4×, 5×, 10× или 50×.
Как используется в настоящем описании термин «субъект» или «субъекты» относится к любому животному, например, млекопитающим, включая грызунов (например, мышей или крыс), собак, приматов, лемуров или людей.
Лечение рака может привести к увеличению среднего времени продолжительности жизни совокупности субъектов, подвергшихся лечению, по сравнению с совокупностью субъектов, получавшей только носитель. Предпочтительно, среднее время продолжительности жизни увеличивается более чем на 30 дней; более предпочтительно, более чем на 60 дней; более предпочтительно, более чем на 90 дней; и наиболее предпочтительно, более чем на 120 дней. Увеличение среднего времени продолжительности жизни совокупности субъектов может быть измерено с помощью любых воспроизводимых способов измерения. Увеличение среднего времени продолжительности жизни совокупности субъектов может быть измерено, например, путем подсчета средней продолжительности жизни субъектов после начала лечения с использованием активного соединения. Увеличение среднего времени продолжительности жизни совокупности субъектов может быть также измерено, например, путем подсчета средней продолжительности жизни субъектов после завершения первого цикла лечения с использованием активного соединения.
Лечение рака может привести к увеличению среднего времени продолжительности жизни совокупности субъектов, подвергшихся лечению, по сравнению с совокупностью субъектов, не подвергавшихся лечению. Предпочтительно, среднее время продолжительности жизни увеличивается более чем на 30 дней; более предпочтительно более чем на 60 дней; более предпочтительно более чем на 90 дней; и наиболее предпочтительно более чем на 120 дней. Увеличение среднего времени продолжительности жизни совокупности субъектов может быть измерено с помощью любых воспроизводимых способов. Увеличение среднего времени продолжительности жизни совокупности субъектов может быть измерено, например, путем подсчета средней продолжительности жизни совокупности субъектов после начала лечения с использованием активного соединения. Увеличение среднего времени продолжительности жизни совокупности субъектов может быть измерено, например, путем подсчета средней продолжительности жизни совокупности субъектов после завершения первого цикла лечения с использованием активного соединения.
Лечение рака может привести к увеличению среднего времени продолжительности жизни совокупности субъектов, подвергшихся лечению, по сравнению с совокупностью субъектов, получавших монотерапию с лекарственным средством, которое не является соединением, раскрытым в настоящем описании, или его фармацевтически приемлемой солью. Предпочтительно, среднее время продолжительности жизни увеличивается более чем на 30 дней; более предпочтительно более чем на 60 дней; более предпочтительно более чем на 90 дней; и наиболее предпочтительно более чем на 120 дней. Увеличение среднего времени продолжительности жизни совокупности субъектов может быть измерено с помощью любых воспроизводимых способов. Увеличение среднего времени продолжительности жизни совокупности субъектов может быть измерено, например, путем подсчета средней продолжительности жизни совокупности субъектов после начала лечения с использованием активного соединения. Увеличение среднего времени продолжительности жизни совокупности субъектов может быть измерено, например, путем подсчета средней продолжительности жизни субъектов после завершения первого цикла лечения с использованием активного соединения.
Лечение рака может привести к снижению уровня смертности совокупности субъектов, подвергшихся лечению, по сравнению с совокупностью субъектов, получавших только носитель. Лечение рака может привести к снижению уровня смертности совокупности субъектов, подвергшихся лечению, по сравнению с совокупностью субъектов, не подвергшихся лечению. Лечение рака может привести к снижению уровня смертности совокупности субъектов, подвергшихся лечению, по сравнению с совокупностью субъектов, получавших монотерапию с лекарственным средством, которое не является соединением, раскрытым в настоящем описании, или его фармацевтически приемлемой солью, пролекарством, метаболитом, аналогом или производным. Предпочтительно уровень смертности снижается более чем на 2%; более предпочтительно более чем на 5%; более предпочтительно более чем на 10%; и наиболее предпочтительно более чем на 25%. Снижение уровня смертности совокупности субъектов, подвергшихся лечению, может быть измерено с помощью любого воспроизводимого способа. Снижение уровня смертности совокупности субъектов может быть измерено, например, путем подсчета для совокупности субъектов среднего числа смертельных случаев, связанных с заболеванием, в единицу времени после начала лечения с использованием активного соединения. Снижение уровня смертности совокупности субъектов также может быть измерено, например, путем подсчета для совокупности субъектов среднего числа смертельных случаев, связанных с заболеванием, в единицу времени после завершения первого цикла лечения с использованием активного соединения.
Лечение рака может привести к снижению скорости роста опухоли. Предпочтительно, после лечения скорость роста опухоли уменьшается по меньшей мере на 5% относительно количества перед лечением; более предпочтительно скорость роста опухоли уменьшается по меньшей мере на 10%; более предпочтительно уменьшается по меньшей мере на 20%; более предпочтительно уменьшается по меньшей мере на 30%; более предпочтительно уменьшается по меньшей мере на 40%; более предпочтительно уменьшается по меньшей мере на 50%; еще более предпочтительно уменьшается по меньшей мере на 50%; и наиболее предпочтительно уменьшается по меньшей мере на 75%. Скорость роста опухоли может быть измерена с помощью любого воспроизводимого способа измерения. Скорость роста опухоли измеряют в соответствии с изменением диаметра опухоли в единицу времени.
Лечение рака может привести к уменьшению повторного роста опухоли, например, после попыток удалить ее хирургическим путем. Предпочтительно после лечения повторный рост опухоли составляет менее 5%; более предпочтительно повторный рост опухоли составляет менее 10%; более предпочтительно менее 20%; более предпочтительно менее 30%; более предпочтительно менее 40%; более предпочтительно менее 50%; еще более предпочтительно менее 50%; и наиболее предпочтительно менее 75%. Повторный рост опухоли может быть измерен с помощью любого воспроизводимого способа измерения. Повторный рост опухоли измеряют, например, путем измерения увеличения диаметра опухоли после уменьшения размеров опухоли после лечения. Уменьшение повторного роста опухоли указывает на то, что опухоль не восстанавливается после прекращения лечения.
Лечение или предотвращение клеточного пролиферативного нарушения может приводить к уменьшению скорости клеточной пролиферации. Предпочтительно после лечения скорость клеточной пролиферации уменьшается по меньшей мере на 5%; более предпочтительно по меньшей мере на 10%; более предпочтительно по меньшей мере на 20%; более предпочтительно по меньшей мере на 30%; более предпочтительно по меньшей мере на 40%; более предпочтительно по меньшей мере на 50%; еще более предпочтительно по меньшей мере на 50%; и наиболее предпочтительно по меньшей мере на 75%. Скорость клеточной пролиферации может быть измерена с помощью любого воспроизводимого способа измерения. Скорость клеточной пролиферации измеряют, например, путем измерения числа делящихся клеток в образце ткани в единицу времени.
Лечение или предотвращение клеточного пролиферативного нарушения может приводить к уменьшению доли пролиферирующих клеток. Предпочтительно после лечения доля пролиферирующих клеток снижается по меньшей мере на 5%; более предпочтительно по меньшей мере на 10%; более предпочтительно по меньшей мере на 20%; более предпочтительно по меньшей мере на 30%; более предпочтительно по меньшей мере на 40%; более предпочтительно по меньшей мере на 50%; еще более предпочтительно по меньшей мере на 50%; и наиболее предпочтительно по меньшей мере на 75%. Доля пролиферирующих клеток может быть измерена с помощью любых воспроизводимых способов измерения. Предпочтительно, долю пролиферирующих клеток измеряют, например, путем количественной оценки числа делящихся клеток относительно числа неделящихся клеток в образце ткани. Доля пролиферирующих клеток эквивалентна митотическому индексу.
Лечение или предотвращение клеточного пролиферативного нарушения может приводить к уменьшению размера площади или зоны клеточной пролиферации. Предпочтительно после лечения размер площади или зоны клеточной пролиферации уменьшается по меньшей мере на 5% относительно его размера до лечения; более предпочтительно уменьшается по меньшей мере на 10%; более предпочтительно уменьшается по меньшей мере на 20%; более предпочтительно уменьшается по меньшей мере на 30%; более предпочтительно уменьшается по меньшей мере на 40%; более предпочтительно уменьшается по меньшей мере на 50%; еще более предпочтительно уменьшается по меньшей мере на 50%; и наиболее предпочтительно уменьшается по меньшей мере на 75%. Размер площади или зоны клеточной пролиферации может быть измерен с помощью любых воспроизводимых способов измерения. Размер площади или зоны клеточной пролиферации может быть измерен как диаметр или ширина площади или зоны клеточной пролиферации.
Лечение или предотвращение клеточного пролиферативного нарушения может приводить к уменьшению количества или доли клеток, имеющих аномальный внешний вид или морфологию. Предпочтительно после лечения количество клеток, имеющих аномальную морфологию, уменьшается по меньшей мере на 5% относительно их размера до лечения; более предпочтительно уменьшается по меньшей мере на 10%; более предпочтительно уменьшается по меньшей мере на 20%; более предпочтительно уменьшается по меньшей мере на 30%; более предпочтительно уменьшается по меньшей мере на 40%; более предпочтительно уменьшается по меньшей мере на 50%; еще более предпочтительно уменьшается по меньшей мере на 50%; и наиболее предпочтительно уменьшается по меньшей мере на 75%. Аномальный внешний вид или морфология клеток могут быть измерены с помощью любых воспроизводимых способов измерения. Аномальная клеточная морфология может быть измерена с помощью микроскопии, например, с использованием инвертированного микроскопа для культуры тканей. Аномальная клеточная морфология может иметь вид ядерного плейоморфизма.
Как используется в настоящем описании, термин «облегчение» предназначен для описания процесса, при котором уменьшается тяжесть признака или симптома нарушения. Важно отметить, что признак или симптом могут быть облегчены без устранения. В предпочтительном варианте осуществления введение фармацевтических композиций, раскрытых в настоящем описании, приводит к устранению признака или симптома, однако устранение не требуется. Эффективные дозы, как ожидается, уменьшат тяжесть признака или симптома. Например, признак или симптом нарушения, такого как рак, который может возникать в нескольких местах, облегчается, если степень тяжести рака снижается, по меньшей мере, в одном из нескольких мест.
Как используется в настоящем описании, термин «тяжесть» предназначен для описания возможности рака превращаться из предракового или доброкачественного состояния в злокачественное состояние. Альтернативно, или, в дополнение, тяжесть предназначена для описания стадии рака, например, в соответствии с системой TNM (принятой Международным Противораковым Союзом (UICC) и Американским объединенным комитетом по изучению рака (AJCC)) или другими принятыми в данной области методами. Стадия рака относится к степени или тяжести рака, основанной на таких факторах, как местоположение первичной опухоли, размер опухоли, количество опухолей и вовлечение лимфатических узлов (распространение рака в лимфатические узлы). Альтернативно, или, в дополнение, тяжесть предназначена для описания степени опухоли принятыми в данной области методами (см., National Cancer Institute, www.cancer.gov). Степень опухоли представляет собой систему, используемую для классификации раковых клеток с точки зрения того, насколько они ненормально выглядят под микроскопом и насколько быстро опухоль, вероятно, будет расти и распространяться. При определении степени опухоли учитываются многие факторы, включая структуру и характер роста клеток. Конкретные факторы, используемые для определения степени опухоли, различаются для каждого типа рака. Тяжесть также описывает гистологическую степень злокачественности, также называемую дифференцировка, которая относится к тому, насколько опухолевые клетки напоминают нормальные клетки того же типа ткани (см., National Cancer Institute, www.cancer.gov). Кроме того, тяжесть описывает степень полиморфизма ядер, который относится к размеру и форме ядра в опухолевых клетках и к проценту опухолевых клеток, которые делятся (см., National Cancer Institute, www.cancer.gov).
В другом аспекте вариантов осуществления, описанных в настоящем документе, тяжесть описывает степень, при которой опухоль секретирует факторы роста, деградирует внеклеточный матрикс, становится васкуляризированной, теряется адгезия к соседним тканям или метастазируется. Более того, тяжесть описывает количество мест, где метастазируется первичная опухоль. Наконец, тяжесть включает трудность лечения опухолей различных типов и локализаций. Например, неоперабельные опухоли, те типы рака, которые имеют больший доступ к разнообразным системам организма (гематологические и иммунологические опухоли), и те, которые наиболее устойчивы к традиционным методам лечения, считаются наиболее тяжелыми. В этих ситуациях продление ожидаемой продолжительности жизни субъекта и/или уменьшение боли, уменьшение доли раковых клеток или ограничение клеток одной системой и улучшение стадии рака/степени опухоли/гистологической степени/степени полиморфизма ядер считаются смягчающими признак или симптом рака.
Как используется в настоящем описании термин «симптом» определяется как признак заболевания, расстройства, травмы или того, что что-то не так в организме. Симптомы ощущаются или замечаются индивидуумом, испытывающим симптом, но не могут быть легко замечены специалистами, не относящимися к медицине.
«Фармацевтическая композиция» представляет собой композицию, содержащую соединение, раскрытое в настоящем описании, в форме, подходящей для введения субъекту. В одном варианте осуществления фармацевтическая композиция находится в виде объемной массы или в единичной лекарственной форме. Единичная лекарственная форма представляет собой любую из разнообразных форм, включая, например, капсулу, IV мешок, таблетку, разовый нагнетатель на аэрозольном ингаляторе или ампулу. Количество активного ингредиента (например, состав раскрытого соединения или его соли, гидрата, сольвата или изомера) в единичной дозе композиции является эффективным количеством и варьируется в зависимости от конкретного лечения. Специалист в данной области поймет, что иногда необходимо вводить обычные изменения в дозировку в зависимости от возраста и состояния пациента. Дозировка также будет зависеть от способа введения. Предполагаются различные пути, включая оральный, легочный, ректальный, парентеральный, трансдермальный, подкожный, внутривенный, внутримышечный, интраперитонеальный, ингаляционный, трансбуккальный, подъязычный, интраплевральный, интратекальный, интраназальный и тому подобное. Лекарственные формы для местного или трансдермального введения соединения, раскрытого в настоящем описании, включают порошки, спреи, мази, пасты, кремы, лосьоны, гели, растворы, патчи и ингалянты. В одном варианте осуществления активное соединение смешивают в стерильных условиях с фармацевтически приемлемым носителем и с какими-либо консервантами, буферами или пропеллентами, которые требуются.
Как используется в настоящем описании, фраза «фармацевтически приемлемый» относится к таким соединениям, анионам, катионам, материалам, композициям, носителям и/или лекарственным формам, которые в рамках здравого медицинского суждения, являются подходящими для использования в контакте с тканями людей и животных без излишней токсичности, раздражения, аллергической реакции или другой проблемы или осложнения, соразмерно с приемлемым соотношением польза/риск.
«Фармацевтически приемлемый эксципиент» означает эксципиент, который является полезным при получении фармацевтической композиции, который обычно является безопасным, нетоксичным и безвредным в биологическом или ином отношении, и включает эксципиент, который является приемлемым для ветеринарного применения, а также для фармацевтического применения у человека. «Фармацевтически приемлемый эксципиент», используемый в описании и формуле изобретения, включает как один, так и более чем один такой эксципиент.
Настоящее изобретение также относится к фармацевтическим композициям, содержащим любое соединение, раскрытое в настоящем описании, в комбинации, по меньшей мере, с одним фармацевтически приемлемым эксципиентом или носителем.
Фармацевтическую композицию, раскрытую в настоящем описании, составляют таким образом, чтобы она соответствовала предполагаемому пути введения. Примеры путей введения включают парентеральное, например, внутривенное, внутрикожное, подкожное, пероральное (например, ингаляция), трансдермальное (местно) и чреcслизистый введение. Растворы или суспензии, используемые для парентерального, внутрикожного или подкожного применения, могут включать следующие компоненты: стерильный разбавитель, такой как вода для инъекций, физиологический раствор, нелетучие масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; антибактериальные агенты, такие как бензиловый спирт или метилпарабены; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатирующие агенты, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетаты, цитраты или фосфаты, и агенты для регулирования тоничности, такие как хлорид натрия или декстроза. рH можно регулировать с помощью кислот или оснований, таких как хлористоводородная кислота или гидроксид натрия. Парентеральный препарат может быть заключен в ампулы, одноразовые шприцы или многоразовые флаконы из стекла или пластика.
Соединение или фармацевтическую композицию, раскрытую в настоящем описании, можно вводить субъекту с помощью многочисленных хорошо известных способов, используемых в настоящее время для химиотерапевтического лечения. Например, для лечения рака, соединение, раскрытое в настоящем описании, можно вводить непосредственно в опухоли, вводить в кровоток или полости тела или принимать перорально или наносить через кожу с помощью пластырей. Выбранная доза должна быть достаточной для проведения эффективного лечения, но не настолько высокой, чтобы вызвать неприемлемые побочные эффекты. Состояние заболевания (например, рака, предракового состояния и тому подобное) и здоровье пациента предпочтительно следует тщательно контролировать во время лечения и в течение разумного периода времени после лечения.
Термин «терапевтически эффективное количество», как используется в настоящем описании, относится к количеству фармацевтического агента для лечения, облегчения или предотвращения идентифицированного заболевания или состояния или для обнаружения поддающегося обнаружению терапевтического или ингибирующего действия. Такое действие может быть обнаружено с помощью любого способа анализа, известного в данной области. Точное эффективное количество для субъекта будет зависеть от массы тела, размера и здоровья субъекта; природы и степени состояния; и лекарственного препарата или комбинации лекарственных препаратов, выбранных для введения. Терапевтически эффективные количества для данной ситуации могут быть определены обычным экспериментированием, что входит в компетенцию и усмотрение лечащего врача. В предпочтительном аспекте заболевание или состояние, подлежащее лечению, представляет собой рак. В другом аспекте заболевание или состояние, подлежащее лечению, представляет собой клеточное пролиферативное нарушение.
Для любого соединения терапевтически эффективное количество может быть оценено первоначально или в анализах клеточной культуры, например, неопластических клеток, или на животных моделях, обычно крыс, мышей, кроликов, собак или свиней. Животная модель также может быть использована для определения подходящего диапазона концентраций и пути введения. Такая информация затем может быть использована для определения полезных доз и путей введения человеку. Терапевтическая/профилактическая эффективность и токсичность могут быть определены с использованием стандартных фармацевтических способов на клеточных культурах или экспериментальных животных, например, ED50 (доза, терапевтически эффективная для 50% популяции) и LD50 (доза, летальная для 50% популяции). Соотношение дозы между токсическим и терапевтическим действиями представляет собой терапевтический индекс, и он может быть выражен как соотношение LD50/ED50. Предпочтительными являются фармацевтические композиции, которые проявляют высокие терапевтические индексы. Доза может варьироваться в пределах этого диапазона в зависимости от используемой дозированной формы, чувствительности пациента и пути введения.
Дозировку и введение регулируют таким образом, чтобы обеспечить достаточные уровни активного(ых) агента(ов) или для поддержания желаемого эффекта. Факторы, которые могут быть приняты во внимание, включают тяжесть состояния заболевания, общее состояние здоровья субъекта, возраст, массу и пол субъекта, режим питания, время и частоту введения, комбинацию(и) лекарственных средств, чувствительность реакции и переносимость/ответ на терапию. Фармацевтические композиции длительного действия можно вводить каждые 3-4 дня, каждую неделю или один раз в две недели в зависимости от периода полувыведения и скорости выведения конкретного препарата.
Фармацевтические композиции, содержащие активные соединения, раскрытые в настоящем описании, могут быть получены способом, который является общеизвестным, например, с помощью обычного смешивания, растворения, гранулирования, дражирования, растирания в порошок, эмульгирования, инкапсулирования, способов получения соединений включения или лиофилизации. Фармацевтические композиции могут быть составлены обычным способом с использованием одного или нескольких фармацевтически приемлемых носителей, содержащих эксципиенты и/или вспомогательные вещества, которые облегчают переработку активных соединений в препараты, которые можно использовать в фармацевтике. Конечно, подходящая лекарственная форма зависит от выбранного пути введения.
Фармацевтические композиции, подходящие для инъекционного применения, включают стерильные водные растворы (где растворим в воде) или дисперсии и стерильные порошки для немедленного получения стерильных растворов для инъекций или дисперсии. Для внутривенного введения подходящие носители включают физиологический раствор, бактериостатическую воду, кремофор ELTM (BASF, Parsippany, N.J.) или забуференный фосфатом физиологический раствор (PBS). Во всех случаях композиция должна быть стерильной и должна быть текучей в такой степени, чтобы была возможность легкого применения с помощью шприца. Она должна быть стабильной в условиях производства и хранения и должна быть защищена от загрязняющего действия микроорганизмов, таких как бактерии и грибки. Носитель может быть растворителем или дисперсионной средой, содержащей, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль и тому подобное) и их подходящие смеси. Правильную текучесть можно поддерживать, например, с использованием оболочки, такой как лецитин, за счет поддержания требуемого размера частиц в случае дисперсии и с помощью использования поверхностно-активных веществ. Предотвращение действия микроорганизмов может быть достигнуто с помощью различных антибактериальных и противогрибковых агентов, например, парабенов, хлорбутанола, фенола, аскорбиновой кислоты, тимерозала и тому подобным. Во многих случаях предпочтительно включать в композицию изотонические агенты, например, сахара, полиспирты, такие как маннит и сорбит, и хлорид натрия. Пролонгированное всасывание инъекционных композиций можно осуществить путем включения в композицию агента, который замедляет всасывание, например, моностеарата алюминия и желатина.
Стерильные растворы для инъекций могут быть получены путем включения активного соединения в требуемом количестве в подходящий растворитель вместе с одним из перечисленных выше ингредиентов или их комбинацией, при необходимости, с последующей стерилизацией фильтрованием. Как правило, дисперсии получают путем включения активного соединения в стерильный носитель, который содержит основную дисперсионную среду и требуемые другие ингредиенты из перечисленных выше. В случае стерильных порошков для получения стерильных растворов для инъекций, способы получения представляют собой вакуумную сушку и сушку вымораживанием, что дает порошок активного ингредиента плюс любой дополнительный желаемый ингредиент из предварительно стерилизованного фильтрованием раствора.
Пероральные композиции обычно включают инертный разбавитель или пищевой фармацевтически приемлемый носитель. Они могут быть заключены в желатиновые капсулы или спрессованы в таблетки. В целях перорального терапевтического введения активное соединение может быть объединено с эксципиентами и использовано в виде таблеток, пилюль или капсул. Пероральные композиции также могут быть получены с использованием жидкого носителя для применения в виде жидкости для полоскания рта, где соединение в жидком носителе применяют перорально и полощут и выплевывают или проглатывают. Как часть композиции в нее могут быть включены фармацевтически совместимые связывающие агенты и/или вспомогательные вещества. Таблетки, пилюли, капсулы, пластинки и тому подобное могут содержать любой из следующих ингредиентов или соединений аналогичной природы: связующее вещество, такое как микрокристаллическая целлюлоза, трагакантовая камедь или желатин; эксципиент, такой как крахмал или лактоза; дезинтегрирующий агент, такой как альгиновая кислота, примогель или кукурузный крахмал; лубрикант, такой как стеарат магния или Sterotes; глидант, такой как коллоидный диоксид кремния; подсластитель, такой как сахароза или сахарин; или ароматизатор, такой как мята перечная, метилсалицилат или апельсиновый ароматизатор.
Активные соединения могут быть получены с фармацевтически приемлемыми носителями, которые будут защищать соединение от быстрого выведения из организма, такими как состав с контролируемым высвобождением, включая имплантаты и микроинкапсулированные системы доставки. Можно использовать биоразлагаемые, биосовместимые полимеры, такие как этиленвинилацетат, полиангидриды, полигликолевая кислота, коллаген, полиортоэфиры и полимолочная кислота. Способы получения таких препаратов будут очевидны для специалистов в данной области.
Особенно предпочтительно получать пероральные или парентеральные композиции в виде единичной дозированной формы для удобства введения и единообразия дозировки. Единичная дозированная форма, как используется в настоящем описании, относится к физически дискретным единицам, подходящим в качестве единичных доз для субъекта, подлежащего лечению; каждая единица содержит заранее определенное количество активного соединения, рассчитанное для получения желаемого терапевтического эффекта, в сочетании с необходимым фармацевтическим носителем. Описание единичных дозированных форм соединений, раскрытых в настоящем описании, обусловлено и непосредственно зависит от уникальных характеристик активного соединения и конкретного терапевтического эффекта, который необходимо достичь.
При терапевтическом применении дозировки фармацевтических композиций, используемых в соответствии с вариантами осуществления, раскрытыми в настоящем описании, изменяются в зависимости от агента, возраста, массы и клинического состояния пациента-реципиента, и также опыта и решения клинического или практикующего врача, проводящего лечение, наряду с другими факторами, влияющими на выбранную дозу. Обычно, доза должна быть достаточной для того, чтобы вызвать замедление и, предпочтительно, регрессию роста опухолей, и также предпочтительно вызывать полную регрессию рака. Дозы могут варьироваться примерно от 0,01 мг/кг в день до примерно 5000 мг/кг в день. В предпочтительных аспектах дозы могут составлять примерно от 1 мг/кг в день до примерно 1000 мг/кг в день. В одном из аспектов доза будет находиться в диапазоне примерно от 0,1 мг/день до примерно 50 г/день; примерно от 0,1 мг/день до примерно 25 г/день; примерно 0,1 мг/день до примерно 10 г/день; примерно 0,1 мг до примерно 3 г/день; или примерно от 0,1 мг до примерно 1 г/день в виде однократных, разделенных или непрерывных дозах (указанную дозу можно регулировать в зависимости от массы пациента в кг, площадь поверхности тела в м2 и возраста в годах). Эффективное количество фармацевтического агента является таким, которое обеспечивает объективно наблюдаемые улучшение, как отмечает клиницист или другой квалифицированный наблюдатель. Например, регрессия опухоли у пациента может быть измерена со ссылкой на диаметр опухоли. Уменьшение диаметра опухоли указывает на регрессию. На регрессию также указывает отсутствие повторного появления опухолей после прекращения лечения. Как используется в настоящем описании, термин «эффективная доза» относится к количеству активного соединения для получения желаемого биологического эффекта у субъекта или клетки.
Фармацевтические композиции могут находиться в контейнере, упаковке или дозаторе вместе с инструкциями для введения.
Способы получения и введения соединений, раскрытых в настоящем описании, могут быть найдены в Remington: the Science and Practice of Pharmacy, 19th edition, Mack Publishing Co., Easton, Pa. (1995). В одном из вариантов осуществления соединения, раскрытые в настоящем описании, и их фармацевтически приемлемые соли могут быть использованы в фармацевтических препаратах в комбинации с фармацевтически приемлемым носителем или разбавителем. Подходящие фармацевтически приемлемые носители включают инертные твердые наполнители или разбавители и стерильные водные или органические растворы. Соединения будут присутствовать в таких фармацевтических композициях в количествах, достаточных для обеспечения желаемого количества дозы в диапазоне, описанном в настоящем документе.
Примеры видов рака, которые можно лечить с использованием одного или нескольких соединений, раскрытых в настоящем описании, включают, но не ограничиваются ими, рак молочной железы, эндометрий матки, рак яичника, саркому, рак щитовидной железы, простаты, аденокарциному легкого и гепатоцеллюлярную карциному.
В вариантах осуществления соединения, раскрытые в настоящем описании, могут быть полезны для лечения рака молочной железы. В вариантах осуществления рак молочной железы представляет собой ER-α+. В вариантах осуществления, рак молочной железы экспрессирует ER-α мутацию, которая может быть L536Q (Robinson et al. Nat Genet. 2013 Dec;45(12)), L536R (Toy et al. Nat Genet. 2013 Dec;45(12):1439-45), Y537S (Toy et al. Nat Genet. 2013 Dec;45(12):1439-45; Robinson et al. Nat Genet. 2013 Dec;45(12); Jeselsohn et al. Clin Cancer Res. 2014 Apr 1;20(7):1757-67), Y537N (Toy et al. Nat Genet. 2013 Dec;45(12):1439-45; Jeselsohn et al. Clin Cancer Res. 2014 Apr 1;20(7):1757-67), Y537C (Toy et al. Nat Genet. 2013 Dec;45(12):1439-45; Jeselsohn et al. Clin Cancer Res. 2014 Apr 1;20(7):1757-67) и D538G (Toy et al. Nat Genet. 2013 Dec;45(12):1439-45; Robinson et al. Nat Genet. 2013 Dec;45(12); Jeselsohn et al. Clin Cancer Res. 2014 Apr 1;20(7):1757-67; Merenbakh-Lamin et al. Cancer Res. 2013 Dec 1;73(23):6856-64), все из которых включены в полном объеме в качестве ссылки для их изучений мутаций ER-α.
Таким образом, соединения, раскрытые в настоящем описании, также могут быть полезны для дополнительных показаний и генотипов. Мутации ESR1 (Y537C/N) были недавно обнаружены в 4 из 373 случаев рака эндометрия (Kandoth et al. Nature 2013 May 2;497(7447):67-73; Robinson et al. Nat Genet. 2013 Dec;45(12)). Поскольку было показано, что мутации ESR1 Y537C/N значительно способствуют устойчивости к доступным в настоящее время терапиям SOC, соединения, раскрытые в настоящем описании, могут быть полезны для лечения рака эндометрия ERαMUT.
Примеры клеточных пролиферативных нарушений, которые можно лечить с использованием одного или нескольких соединений, раскрытых в настоящем описании, включают, но не ограничиваются ими, рак молочной железы, предрак или предраковое состояние молочной железы, доброкачественные опухоли или поражения молочной железы, и злокачественные опухоли или поражения молочной железы, и метастатические поражения в ткани и органах в организме, отличных от молочной железы. Клеточные пролиферативные нарушения груди могут включать гиперплазию, метаплазию и дисплазию молочной железы.
Рак молочной железы, который подлежит лечению, может возникнуть у мужчины или женщины. Рак молочной железы, который подлежит лечению, может возникать у женщин в пременопаузе или у женщин в постменопаузе. Рак молочной железы, который подлежит лечению, может возникать у субъекта в возрасте 30 лет или старше или у субъекта моложе 30 лет. Рак молочной железы, который подлежит лечению, возникает у субъекта в возрасте 50 лет или старше или у субъекта моложе 50 лет. Рак молочной железы, который подлежит лечению, может возникать у субъекта в возрасте 70 лет или старше или у субъекта моложе 70 лет.
Соединение, раскрытое в настоящем описании, или его фармацевтически приемлемая соль может быть использовано для лечения или предотвращения клеточного пролиферативного нарушения молочной железы или для лечения или предотвращения рака молочной железы у субъекта, имеющего повышенный риск развития рака молочной железы относительно населения в целом, или используется для определения подходящих кандидатов для таких целей. Субъект с повышенным риском развития рака молочной железы относительно населения в целом является женщиной с семейным анамнезом или личным анамнезом рака молочной железы. Субъектом с повышенным риском развития рака молочной железы относительно населения в целом является женщина старше 30 лет, старше 40 лет, старше 50 лет, старше 60 лет, старше 70 лет, старше 80 лет или старше 90 лет.
Рак, который подлежит лечению, может включать опухоль, которая, как было определено, меньше или равна примерно 2 сантиметрам в диаметре. Рак, который подлежит лечению, может включать опухоль, которая, как было определено, составляет от примерно 2 до примерно 5 см в диаметре. Рак, который подлежит лечению, может включать опухоль, которая, как было определено, больше или равна примерно 3 см в диаметре. Рак, который подлежит лечению, может включать опухоль, которая, как было установлено, больше 5 см в диаметре. Рак, который подлежит лечению, можно классифицировать по микроскопическому виду, а также как дифференцированный, умеренно дифференцированный, слабо дифференцированный или недифференцированный. Рак, который подлежит лечению, можно классифицировать по микроскопическому виду по отношению к количеству митозов (например, количеству деления клеток) или ядерному плейоморфизму (например, изменению клеток). Рак, который подлежит лечению, может быть классифицирован по микроскопическому виду как связанный с областями некроза (например, области умирающих или дегенерирующих клеток). Рак, который подлежит лечению, может быть классифицирован как имеющий аномальный кариотип, имеющий ненормальное количество хромосом или имеющий одну или несколько хромосом, которые являются ненормальными по внешнему виду. Рак, который подлежит лечению, может быть классифицирован как анеуплоидный, триплоидный, тетраплоидный или как имеющий измененную плоидность. Рак, который подлежит лечению, может быть классифицирован как имеющий хромосомную транслокацию, или делецию или дублирование всей хромосомы, или область делеции, дублирования или амплификации части хромосомы.
Соединения или их фармацевтически приемлемые соли можно вводить перорально, назально, трансдермально, легочно, ингаляционно, буккально, сублингвально, интраперитонально, подкожно, внутримышечно, внутривенно, ректально, интраплеврально, интратекально и парентерально. В одном варианте осуществления соединение вводят перорально. Специалист в данной области поймет преимущества определенных путей введения.
Режим дозирования с использованием соединений может быть выбран в соответствии с различными факторами, включая тип, вид, возраст, массу, пол и состояние здоровья пациента; тяжесть состояния, подлежащего лечению; путь введения; функция почек и функция печени пациента; и используемое конкретное соединение или его соль. Обычно, врач или ветеринар средней квалификации может легко определить и назначить эффективное количество лекарственного средства, необходимое для предотвращения, противодействия или остановки развития состояния.
ПРИМЕРЫ
Настоящим приводятся неограничивающие примеры вариантов осуществления соединений, раскрытых в настоящем описании. Если есть какое-либо несоответствие между изображенной химической структурой соединения и ее химическим названием, изображенная химическая структура будет контролем.
Таблица 1: Результаты анализа жизнеспособности (см. Пример 101, ниже)





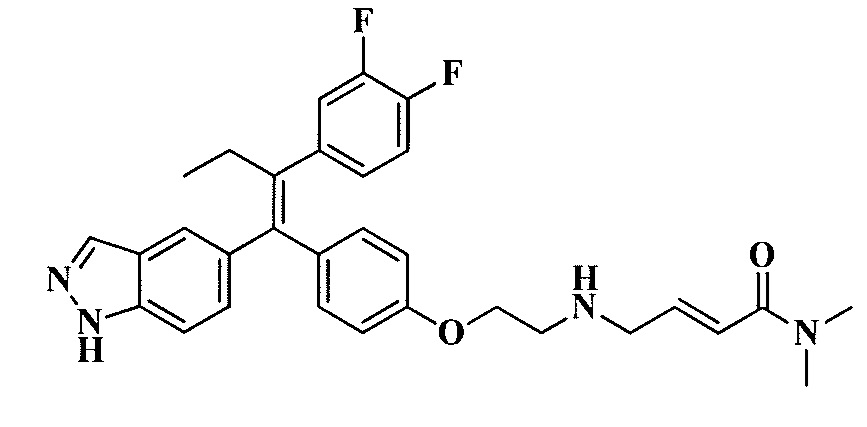


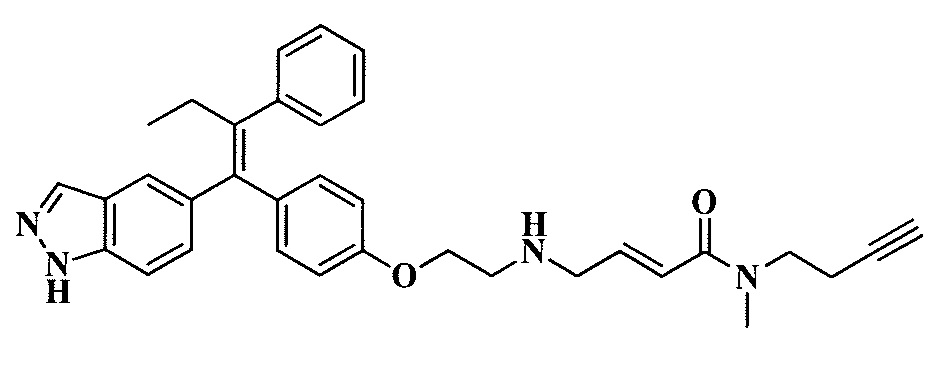



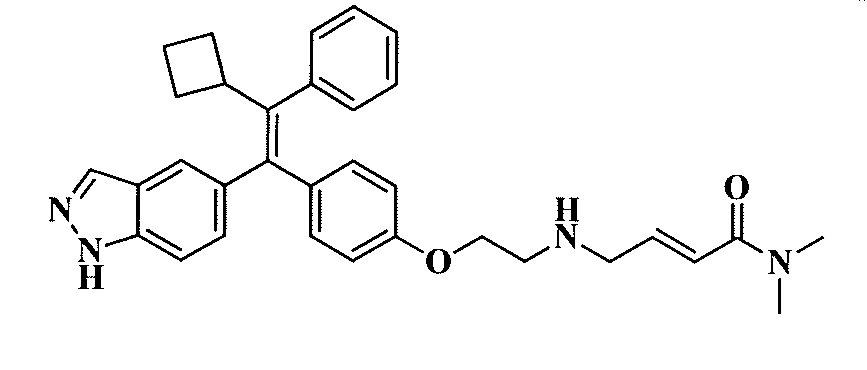








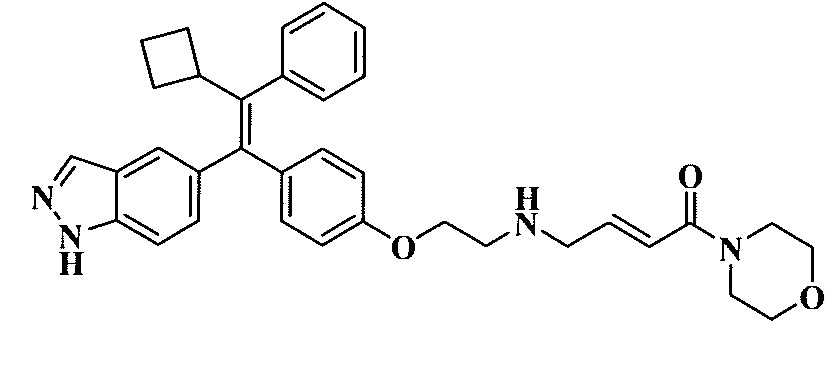



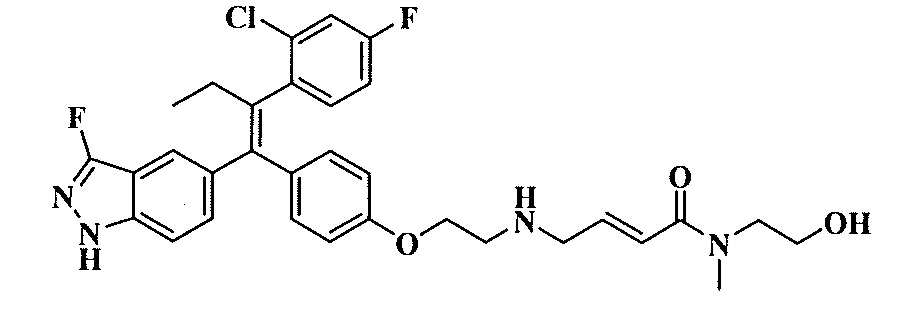








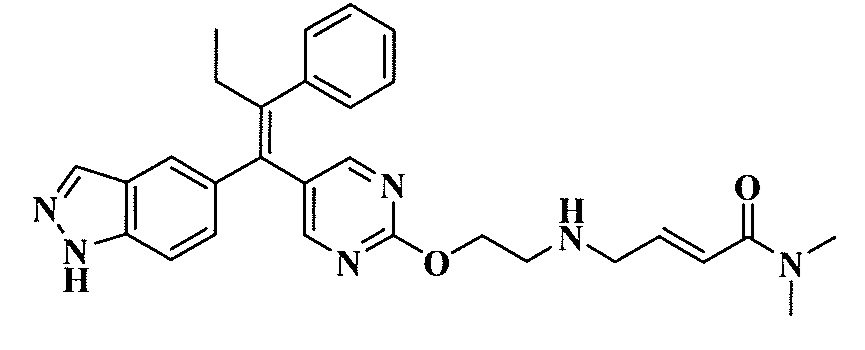

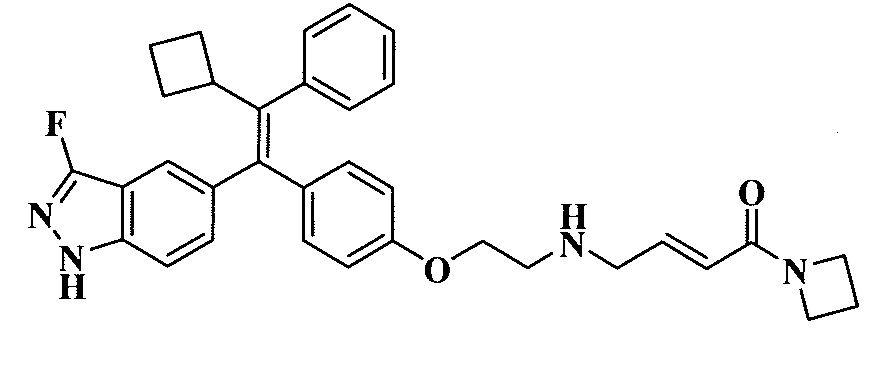

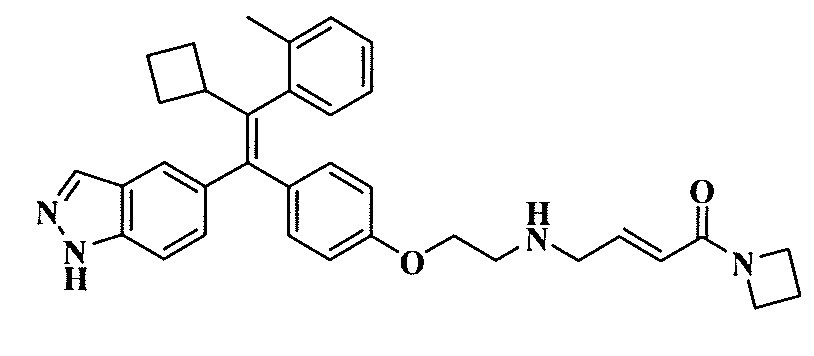


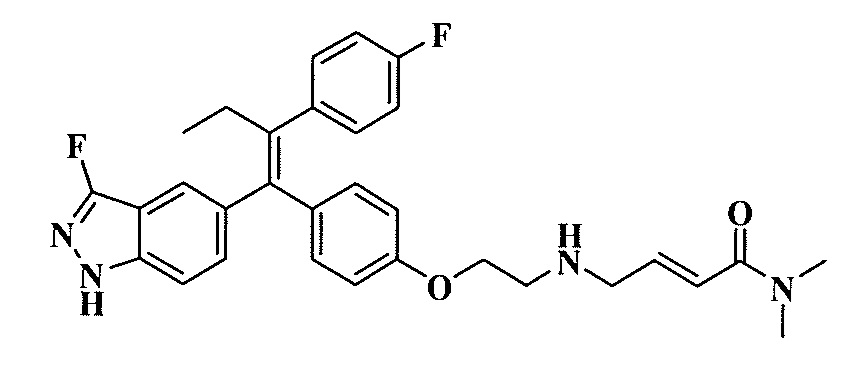



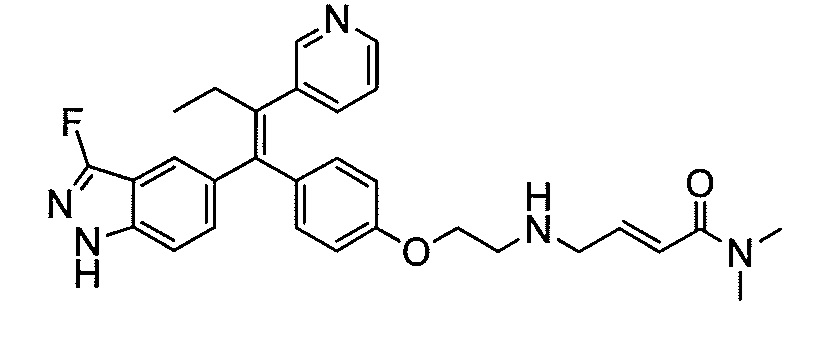
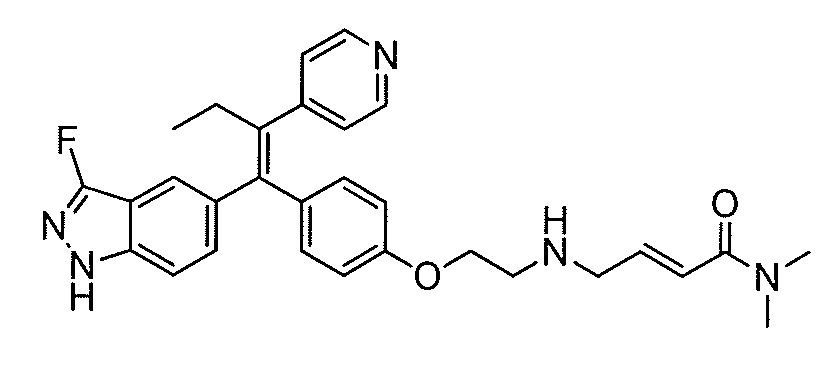



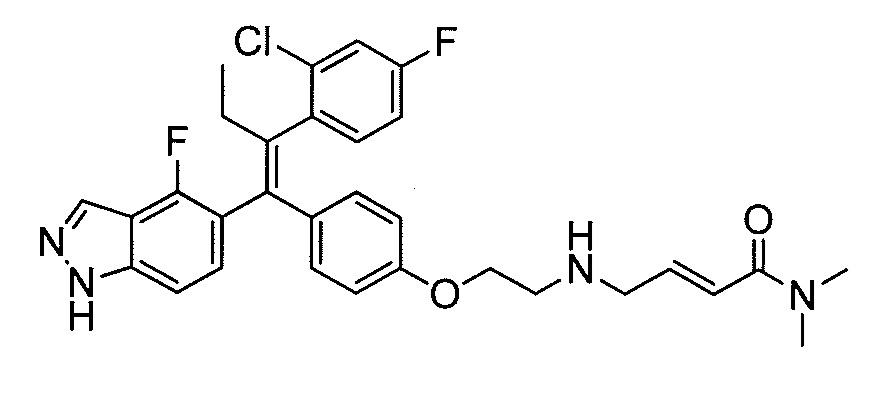
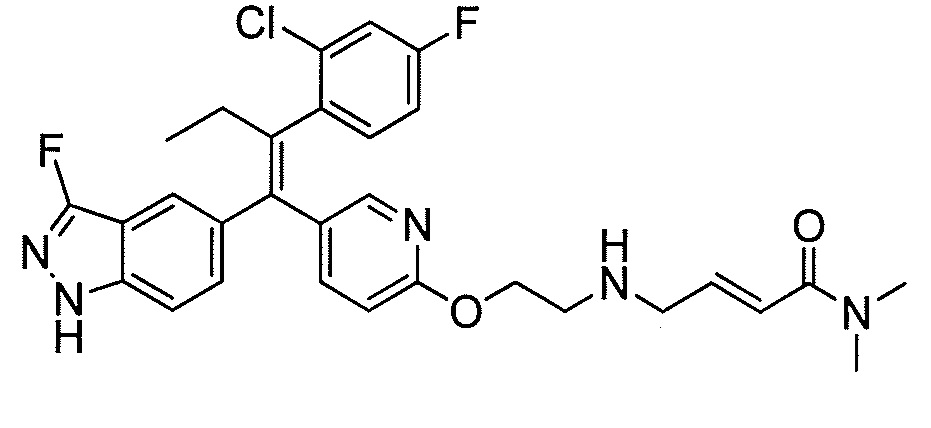






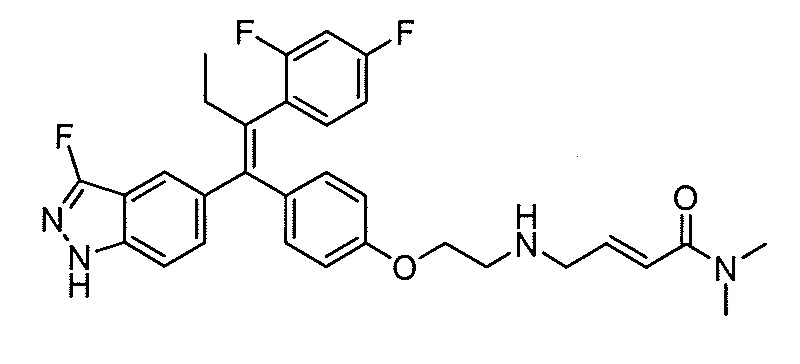














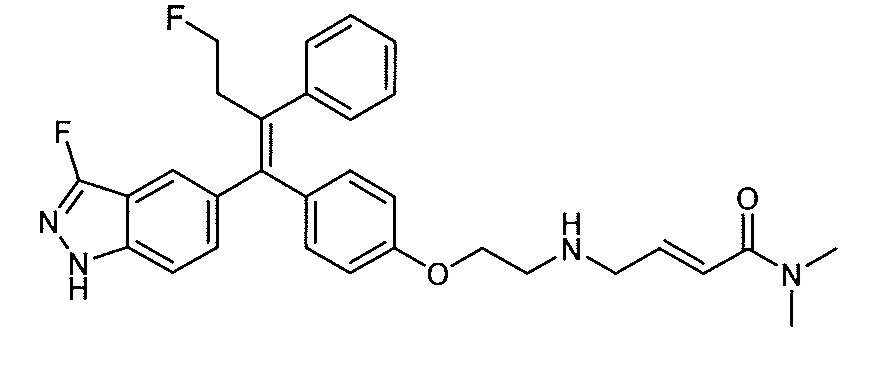








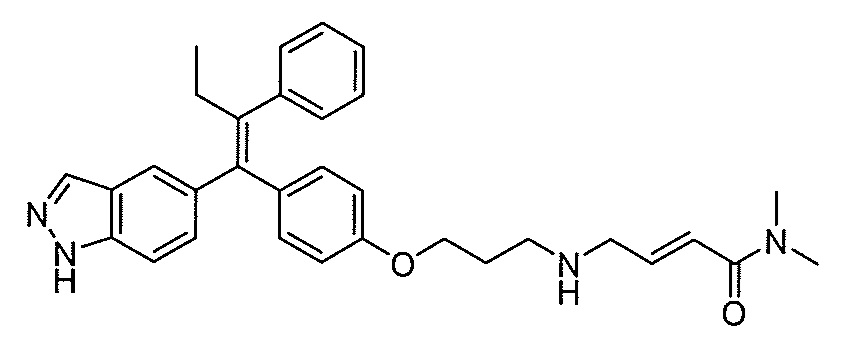



Общие способы
В настоящем описании могут использоваться следующие сокращения:
ACN: Ацетонитрил
BOC: трет-бутилоксикарбонил
CAN: нитрат аммония-церия
Конц.: концентрированный
Cs2CO3: карбонат цезия
DABCO: 1,4-диазабицикло[2.2.2]октан
DCM: дихлорметан
DHP: дигидропиран
DIPEA: N,N-диизопропилэтиламин, основание Хьюнига
DMA: диметилацетамид
DMF: диметилформамид
DMSO: диметилсульфоксид
DPEphos: (Оксиди-2,1-фенилен)бис(дифенилфосфин)
EDCI.HCl: N-(3-Диметиламинопропил)-N-этилкарбодиимид гидрохлорид
EtOH: этанол
EtOAc: Этилацетат
Et3N: триэтиламин
Пр.: Пример
ч: часы
HATU: 1-[Бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиний 3-оксид гексафторфосфат
HCl: хлористоводородная кислота
HMPA: Гексаметилфосфорамид
ВЭЖХ: высокоэффективная жидкостная хроматография
H2SO4: серная кислота
IPA: изопропиловый спирт
K2CO3: Карбонат калия
KOH: Гидроксид калия
ЖХМС: Жидкая хроматография - масс-спектрометрия
MeOH: метанол
Na2CO3: Карбонат натрия
NBS: н-бромсукцинимид
nBuLi: н-бутиллитий
NH4Cl: Хлорид аммония
NH4OH: Гидроксид аммония
ЯМР: ядерный магнитный резонанс
вн или в.н.: в течение ночи
Pd/C: Палладий (0) на углероде
Pd2(dba)3: Трис(дибензилиденацетон)дипалладий(0)
PPTS: пиридиний п-толуолсульфонат
PTSA: п-толуолсульфоновая кислота
КТ или к.т.: комнатная температура
TBAF: Тетрабутиламмоний фторид
TEA: триэтиламин
TFA: Трифторуксусная кислота
THF: Тетрагидрофуран
ТСХ: тонкослойная хроматография
Pt/C: Платина (0) на углероде
Если не указано иное, спектры 1H-ЯМР были взяты на ЯМР Varian Mercury Plus 400 МГц
Схема 1:

Стадия-1: К перемешиваемому раствору 5-бром-1H-индазола (201, 23,5 ммоль) в сухом дихлорметане (50 мл) при 23°C добавляли дигидропиран (9,9 г, 118 ммоль) с последующим добавлением п-толуолсульфоната пиридиния (0,6 г, 2,4 ммоль). Полученную смесь перемешивали при комнатной температуре 23°C в течение 16 ч. По завершении ТСХ, реакционную смесь гасили водой (50 мл) и экстрагировали дихлорметаном (2×100 мл). Объединенные органические экстракты промывали водой, насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали. Неочищенный материал очищали при помощи колоночной хроматографии на силикагеле 230-400 меш с использованием 4-5% этилацетата в гексане с получением 5-бром-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (202, 20 ммоль, 86%) в виде бледно-желтого масла.
Стадия-2a: К перемешиваемому раствору 5-бром-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (202, 3,6 ммоль) в 10 мл MeOH:DMA:H2O (1:1:1), в герметичном сосуде, добавляли йодид меди (0,068 г, 0,3 ммоль) и карбонат цезия (1,62 г, 4,9 ммоль) при 23°C. Эту смесь дегазировали с помощью трех циклов вакуум/N2, и затем добавляли бут-1-ин-1-илтриметилсилан (203, 0,899 г, 7,1 ммоль), затем Pd(PPh3)2Cl2 (0,125 г, 0,1 ммоль). Сосуд высокого давления герметизировали и нагревали при 80°C в течение 1-12 ч. По завершении ТСХ, реакционную смесь гасили водой (10 мл) и экстрагировали этилацетатом (2×50 мл). Объединенные органические экстракты промывали водой, насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали при помощи колоночной хроматографии на силикагеле 230-400 меш, с использованием 4-5% этилацетата в гексане, с получением 204 (1,4 ммоль, 55%) в виде бледно-желтого масла.
Стадия-2b: К перемешиваемому раствору 5-бром-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (202, 2,5 г, 8,3 ммоль) в 30 мл DMA, в герметичном сосуде, добавляли йодид меди (79 мг, 0,41 ммоль) и карбонат цезия (4 г, 12,4 ммоль) при комнатной температуре. реакционную смесь дегазировали тремя циклами вакуум/Н2, добавляли (циклобутилэтинил)триметилсилан (203, 1,78 г, 11,74 ммоль), затем Pd(OAc)2 (92 мг, 0,41 ммоль) и dppf (228 мг, 0,041 ммоль). Сосуд высокого давления герметизировали и нагревали при 90°C в течение 2 ч. По завершении ТСХ, реакционную смесь охлаждали до комнатной температуры и разбавляли водой (25 мл), экстрагировали этилацетатом (100 мл). Объединенные органические экстракты промывали водой, затем насыщенным солевым раствором. Органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали при помощи колоночной хроматографии на силикагеле 230-400 меш, с использованием 10% этилацетата в н-гексане в качестве элюента, с получением 5-(циклобутилэтинил)-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (204, 2,15 г, 86%).
Стадия-3: К перемешиваемому раствору 204 (39,37 ммоль) в 2-метил THF (80 мл), добавляли бис(пинаколато)диборон (10,09 г, 39,76 ммоль), тетракис(трифенилфосфин)платина(0) (372 мг, 0,299 ммоль) в атмосфере азота, реакционную смесь кипятили с обратным холодильником в течение 6 ч. После завершения реакции реакционную смесь разбавляли водой и экстрагировали EtOAc. Органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный материал использовали на следующей стадии без дополнительной очистки (206, 39 ммоль, количественный) в виде масла коричневого цвета.
Стадия-4: К перемешиваемому раствору 207 (2,34 г, 10,61 ммоль) добавляли бис(трифенилфосфин)палладий(II) дихлорид (372 мг, 0,530 ммоль), карбонат цезия (6,9 г, 21,23 ммоль) и 2-метил THF (60 мл). Эту смесь дегазировали азотом и добавляли воду (5 мл). Эту смесь перемешивали при комнатной температуре в течение 12 ч. После завершения реакции реакционную смесь разбавляли водой и экстрагировали EtOAc. Органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный материал очищали при помощи колоночной хроматографии на силикагеле 230-400 меш с использованием MeOH в дихлорметане (1,6:98,4) с получением 208 (5,5 ммоль, 43%).
Стадия-5: К перемешиваемому раствору 208 (1,8 ммоль) в 2-метил THF (30 мл), добавляли иодбензол (209, 1,8 ммоль), 4M водный KOH (5 мл) и Pd(PPh3)2Cl2 (63 мг, 0,09 ммоль) и смесь дегазировали азотом в течение 15 мин и нагревали при 80-90°C в течение 8-12 ч. По завершении реакции реакционную смесь разбавляли EtOAc. Органический слой промывали водой, затем насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали с помощью хроматографии на силикагеле (2:8 EtOAc в н-гексане) с получением желаемого продукта (211, 0,74 ммоль, 41%).
Стадия-6: К раствору 5-(бут-1-ин-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (204, 27,5 ммоль), иодбензола (209, 17 г, 82,7 ммоль), 4-гидроксифенилбороновой кислоты (210, 11,4 г, 82,7 ммоль) в N,N-диметилформамиде/воде (2:1, 50 мл) добавляли K2CO3 (11,4 г, 82,7 ммоль). Содержимое дегазировали с помощью трех циклов вакуум/N2 и затем нагревали при 45°С в течение 1 ч, пока раствор не стал однородным. Добавляли раствор Pd(PhCN)2Cl2(0,528 г, 1,4 ммоль) в N,N-диметилформамиде (1 мл) и полученную смесь перемешивали при 45°C в течение 16 ч. По завершении ТСХ, реакционную смесь гасили водой (500 мл) и экстрагировали этилацетатом (2×500 мл). Объединенные органические экстракты промывали водой, насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали. Неочищенный материал очищали при помощи колоночной хроматографии на силикагеле 230-400 меш с использованием 20% этилацетата в гексане с получением желаемого соединения (211, 11,8 ммоль, 43%) в виде бледно-желтого масла.
Схема 2:

Стадия-1: К раствору 211a (3,4 ммоль) в DMF (30 мл), при 0°C, добавляли последовательно карбонат калия (1,4 г, 10,1 ммоль) и трет-бутил (2-бромэтил)-карбамат (212, 8,5 ммоль). Реакционную смесь перемешивали при 80°C в течение 6 ч, разбавляли этилацетатом, промывали водой, насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали при пониженном давлении. Неочищенный материал очищали при помощи колоночной хроматографии на силикагеле 230-400 меш с использованием 15% этилацетата в гексане с получением желаемого продукта (211b, 1,8 ммоль, 53%) в виде светло-коричневой вязкой массы.
Стадия-2a: К перемешиваемому раствору 211b (2,5 ммоль) в этаноле (10 мл) добавляли при 0°C, 2M HCl в эфире (10 мл). Реакционную смесь перемешивали в течение 16-24 ч при 23°C. После завершения реакции, реакционную смесь подщелачивали насыщенным NaHCO3, экстрагировали 10% МеОН в дихлорметане. Органический слой концентрировали при пониженном давлении и неочищенный материал очищали при помощи колоночной хроматографии на силикагеле 230-400 меш с использованием (4-5%) MeOH в DCM с получением желаемого продукта (13, 1,25 ммоль, 50%) в виде полутвердого вещества коричневого цвета.
Стадия-2b: К перемешиваемому раствору 211 (1,5 г, 2,3 ммоль) в этаноле (3 мл) добавляли при 0°C 2M HCl в диэтиловом эфире (15 мл). Реакционную смесь перемешивали в течение 24 ч при комнатной температуре. После завершения реакции, реакционную смесь подщелачивали насыщенным NaHCO3,экстрагировали этилацетатом. Органический слой концентрировали при пониженном давлении с получением неочищенного целевого продукта (213, 1,1 г неочищенный).
Стадия-3a: К перемешиваемому раствору 213 (1,24 ммоль) в DMF (5 мл) добавляли при 0°C (E)-4-бром-N,N-диметилбут-2-енамид (214, 1,24 ммоль) и DIPEA (0,321 г, 2,49 ммоль). Реакционную смесь перемешивали в течение 12-48 ч при 23°C, разбавляли холодной водой (50 мл) и экстрагировали дихлорметаном. Органический слой промывали водой, затем насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением неочищенной смеси 215.
Стадия-3b: К перемешиваемому раствору 213 (1,1 г, 2 ммоль) в DMF (22 мл) добавляли DIPEA (0,62 г, 4 ммоль) при комнатной температуре, перемешивали в течение 15 мин при той же температуре. Раствор смеси (E)-4-бром-N,N-диметилбут-2-енамида и (E)-4-хлор-N,N-диметилбут-2-енамида (214, 0,41 г, 2 ммоль) в DMF (5 мл) добавляли по каплям, реакционную смесь перемешивали в течение 16 ч при комнатной температуре, после завершения реакции (контролируемой ТСХ), реакционную смесь разбавляли холодной водой (50 мл) и экстрагировали дихлорметаном. Органический слой промывали водой, затем насыщенным раствором NaCl, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного продукта 215 (1,8 г).
Стадия-4: Неочищенный материал, полученный на предыдущей стадии, очищали при помощи препаративной ВЭЖХ с получением чистого изомера 216 (0,06 ммоль, 5%) в виде белого твердого вещества. Собирали данные1H ЯМР, ВЭЖХ и MS.
Стадия-5: К перемешиваемому раствору 215 (1,25 ммоль) в DCM (10 мл) добавляли ди-трет-бутилдикарбонат (0,546 г, 2,5 ммоль). Реакционную смесь перемешивали в течение 1 ч при 23°C, после завершения реакции, реакционную смесь разбавляли холодной водой (50 мл) и экстрагировали дихлорметаном (100 мл). Органический слой промывали водой, затем насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенное соединение очищали при помощи колоночной хроматографии с использованием 2% МеОН в DCM с получением 211c (0,63 ммоль, 50%) в виде светло-коричневого полутвердого вещества.
Стадия-6a: К перемешиваемому раствору 211c (0,622 ммоль) в MeOH (5 мл) добавляли при 0°C 2M HCl в эфире (10 мл). Реакционную смесь перемешивали в течение 16 ч при 23°C. После завершения реакции реакционную смесь подщелачивали насыщенным NaHCO3, экстрагировали 10% МеОН в DCM. Органический слой концентрировали при пониженном давлении с получением неочищенного соединения (215, 0,07 ммоль) в виде не совсем белого твердого вещества. Неочищенное соединение очищали с помощью препаративной ВЭЖХ с получением желаемого чистого изомера (216, 0,03 ммоль, 4,1%) в виде белого твердого вещества. Собирали данные1H ЯМР, ВЭЖХ и MS.
Стадия-6b: К перемешиваемому раствору 211c (0,081 ммоль) в дихлорметане (1,2 мл) при 0°C добавляли TFA (0,3 мл). Реакционную смесь перемешивали при 23°C в течение 30 мин до 2 ч. После завершения реакции раствор подщелачивали насыщенным раствором NaHCO3 и экстрагировали дихлорметаном. Объединенный органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный материал очищали при помощи препаративной ВЭЖХ с получением желаемого чистого изомера (216, 0,12 ммоль, 33,3%) в виде белого твердого вещества. Собирали данные1H ЯМР, ВЭЖХ и MS.
Стадия-6c: К перемешиваемому раствору 211c (0,504 ммоль) в EtOH (3 мл) добавляли при 0°C 2M HCl в диэтиловом эфире (5 мл). Реакционную смесь перемешивали в течение 12 ч при комнатной температуре. После завершения реакции (контролируемой ТСХ), реакционную смесь подщелачивали насыщенным раствором NaHCO3 при 0°C,экстрагировали 10% МеОН в дихлорметане. Органический слой промывали водой, насыщенным раствором NaCl и концентрировали при пониженном давлении с получением неочищенного соединения, неочищенное соединение очищали при помощи препаративной ТСХ с получением желаемого соединения 216 (74 мг, 27%).
Схема 3:

Стадия-1: К перемешиваемому раствору 4-иодфенола (207a, 227 ммоль) в DMF (750 мл) добавляли карбонат калия (188 г, 1,363 моль) и перемешивали в течение 30 мин при 23°C, к вышеуказанной смеси добавляли трет-бутил (2-бромэтил)карбамат (212, 71,27 г, 318 ммоль). Содержимое перемешивали при 70°C в течение 12 ч. После завершения реакции, реакционную смесь выливали в ледяную воду, твердое вещество отфильтровывали и сушили при пониженном давлении с получением желаемого соединения трет-бутил (2-(4-йодфенокси)-этил)карбамата в виде не совсем белого твердого вещества (207b, 220 ммоль, 97%).
Стадия-2: К перемешиваемому раствору трет-бутил (2-(4-йодфенокси)этил)карбамата (207b, 68,6 ммоль) в этаноле (50 мл) добавляли при 0°C 2M HCl в эфире (250 мл). Реакционную смесь перемешивали в течение 12 ч при 23°C. После завершения реакции реакционную смесь подщелачивали насыщенным NaHCO3, экстрагировали 10% МеОН в DCM. Органический слой концентрировали при пониженном давлении и неочищенный материал использовали на следующей стадии без дополнительной очистки (217, 60 ммоль, 88%).
Стадия-3: К перемешиваемому раствору 2-(4-йодфенокси)этан-1-амина (217, 60,6 ммоль) в DMF (65 мл) добавляли при 0°C 4-бром-N,N-диметилбут-2-енамид (214, 42,4 ммоль) и DIPEA (11,72 г, 90,9 ммоль). Реакционную смесь перемешивали в течение 5 ч при комнатной температуре, разбавляли холодной водой (250 мл) и экстрагировали дихлорметаном. Органический слой промывали водой, затем насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный материал использовали на следующей стадии без дополнительной очистки (218, 50 ммоль, 83%, неочищенный).
Стадия-4: К перемешиваемому раствору 218 (50,26 ммоль) в сухом дихлорметане (150 мл) добавляли DIPEA (6,4 г, 50,2 ммоль) при 0°C, перемешивали в течение 15 мин при 0°C. К вышеуказанной реакционной смеси, добавляли ди-трет-бутилдикарбонат (13,1 г, 60,3 ммоль), полученную смесь перемешивали при 23°C в течение 12 ч. По окончании ТСХ реакционную смесь охлаждали до 0°С, гасили ледяной водой (500 мл) и экстрагировали дихлорметаном (500 мл). Объединенные органические экстракты промывали водой, затем насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный материал очищали при помощи колоночной хроматографии на силикагеле 230-400 меш с использованием 3% MeOH в дихлорметане в качестве элюента с получением 207c (19 ммоль, 37,8%).
Схема 4:

Стадия-1: К перемешиваемому раствору бут-2-еновой кислоты (219, 116,0 ммоль) в бензоле (150 мл) добавляли N-бромсукцинамид (31,4 г, 120,0 ммоль), затем бензоил пероксид (0,200 г, 1,4 ммоль) при 23°C. Реакционную смесь нагревали до кипения с обратным холодильником в течение 4 ч, что приводило к осаждению кристаллов сукцинамида. Кристаллы отфильтровывали и фильтрат концентрировали. Неочищенный продукт перекристаллизовывали с минимальным количеством гексана и промывали гексаном с получением 4-бромбут-2-еновой кислоты (220, 42,5 ммоль, 37%) в виде белого твердого вещества.
Стадия-2: Бромбут-2-еновую кислоту (220, 9 ммоль) брали в дихлорметане (30 мл) и охлаждали до 0°C. К этому раствору добавляли оксалилхлорид (1,6 мл, 18 ммоль), DMF (0,1 мл) и перемешивали в течение 0,5 ч при 23°C. Реакционную смесь концентрировали в атмосфере азота, остаток разбавляли THF (30 мл), охлаждали до 0°C и подщелачивали DIPEA (3,1 мл, 18 ммоль). К этой смеси медленно добавляли амин (221, 9 ммоль) в виде раствора в дихлорметане и содержимое перемешивали при 23°C в течение 1 ч. Летучие вещества удаляли концентрированием при пониженном давлении и остаток разделяли между водой и этилацетатом. Органический слой промывали водой, насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали. Неочищенный материал очищали при помощи колоночной хроматографии на силикагеле 230-400 меш с использованием 3-7% этилацетата в гексане с получением желаемого амида (214, 0,85 ммоль, 9,4%) в виде жидкости коричневого цвета.
Схема 5:
Соединения формулы III можно получить с использованием следующего соединения вместо соединения 207 на Стадии-4 Схемы 1:
Соединение, показанное здесь на схеме 5, можно получить с использованием способа, описанного на схеме 3, с использованием соответствующего пиридила вместо соединения 207a.
Схема 6:
Соединения формулы IV можно получить с использованием следующего соединения вместо соединения 209 на Стадии-5 или Стадии-6 схемы 1:

Пример 1: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 1)
Стадия-1: Синтез 5-бром-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола
Реакцию осуществляли в соответствии со Схемой 1, Стадия-1, используя 5-бром-1H-индазол (5 г, 23,5 ммоль) вместо соединения 201. Неочищенный материал очищали при помощи колоночной хроматографии на силикагеле 230-400 меш с использованием 4-5% этилацетата в гексане с получением указанного в заголовке соединения Пр.1 Стадия-1 (12,6 г, 86%) в виде бледно-желтого масла.
Стадия-2: Синтез бут-1-ин-1-илтриметилсилана:

К перемешиваемому раствору (триметилсилил)ацетилена (116 г, 1,19 моль) в сухом THF (400 мл) добавляли н-BuLi (2,5 М в THF, 500 мл) при -78°C в течение 2 ч. Полученную смесь нагревали до 0°C и перемешивали в течение 10 минут. Реакционную смесь снова охлаждали до -78°C, к вышеуказанной смеси добавляли HMPA (234 г, 1,13 моль) и перемешивали при -78°C в течение 30 минут. К вышеуказанной реакционной смеси добавляли иодэтан (200 г, 1,28 моль) и полученную смесь перемешивали при комнатной температуре в течение 16 ч. После завершения реакции, реакционную смесь гасили водой, экстрагировали этилацетатом (1000 мл). Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Продукт бут-1-ин-1-илтриметилсилан дистиллировали между 125-135°C с получением указанного в заголовке соединения Пр.1 Стадия-2 (91 г, 61%) в виде бесцветной жидкости.
Стадия-3: Синтез 5-(бут-1-ин-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола

Реакцию осуществляли в соответствии со Схемой 1, Стадия-2a, используя 5-бром-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол (1 г, 3,6 ммоль) вместо соединения 202. Неочищенный продукт очищали при помощи колоночной хроматографии на силикагеле 230-400 меш, с использованием 4-5% этилацетата в гексане, с получением указанного в заголовке соединения Пр.1 Стадия-3 (0,5 г, 55%) в виде бледно-желтого масла.
Стадия-4: Синтез (E)-4-(2-фенил-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)фенола

Реакцию осуществляли в соответствии со Схемой 1, Стадия-6, используя 5-(бут-1-ин-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол (7 г, 27,5 ммоль) вместо соединения 204, иодбензол (17 г, 82,7 ммоль) и 4-гидроксифенилбороновую кислоту (11,4 г, 82,7 ммоль) вместо соединения 210. Неочищенный материал очищали при помощи колоночной хроматографии на силикагеле 230-400 меш, используя 20% этилацетат в гексане для указанного в заголовке соединения Пр.1 Стадия-4 (5 г, 43%) в виде бледно-желтого масла.
Стадия-5: Синтез (E)-2-(2-(4-(2-фенил-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)изоиндолин-1,3-диона

Реакцию осуществляли в соответствии со Схемой 2, Стадия-1, используя (E)-4-(2-фенил-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)фенол (1 г, 2,3 ммоль) вместо соединения 211a и 2-(2-бромэтил)изоиндолин-1,3-дион (6 г, 23,5 ммоль) вместо соединения 212. Реакционную смесь перемешивали при 55°C в течение 16 ч, разбавляли этилацетатом, промывали водой, насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали при пониженном давлении. Неочищенный материал очищали при помощи хроматографии на силикагеле с использованием 30-35% этилацетата в н-гексане с получением указанного в заголовке соединения Пр.1 Стадия-5 (0,5 г, 36%) в виде светло-коричневой вязкой массы.
Стадия-6: Синтез (E)-2-(4-(2-фенил-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этан-1-амина

К раствору (E)-2-(2-(4-(2-фенил-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)изоиндолин-1,3-диона (1 г, 1,6 ммоль) в MeOH/DCM (1:2, 20 мл) добавляли гидразин гидрат (14 мл) при комнатной температуре. Реакционную смесь перемешивали при 55°C в течение 4 ч, гасили NH4OH и экстрагировали дихлорметаном. Органический слой промывали водой, насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали при пониженном давлении. Неочищенный материал очищали при помощи колоночной хроматографии на силикагеле 100-200 меш с использованием 3% метанола в DCM с получением указанного в заголовке соединения Пр.1 Стадия-6 (0,500 г, 64%) в виде вязкого соединения.
Стадия-7: Синтез (E)-4-бром-N,N-диметилбут-2-енамида

Стадия-7.1: Синтез (E)-4-бромбут-2-еновой кислоты

К перемешиваемому раствору (E)-бут-2-еновой кислоты (10,0 г, 116,0 ммоль) в бензоле (150 мл) добавляли N-бромсукцинамид (31,4 г, 120,0 ммоль), затем бензоил пероксид (0,200 г, 1,4 ммоль) при 23°C. Реакционную смесь нагревали до кипения с обратным холодильником в течение 4 ч, что приводило к осаждению кристаллов сукцинамида. Кристаллы отфильтровывали и фильтрат концентрировали. Неочищенный продукт перекристаллизовывали с минимальным количеством гексана и промывали гексаном с получением (E)-4-бромбут-2-еновой кислоты (6,97 г, 37%) в виде белого твердого вещества.
Стадия-7.2: Синтез (E)-4-бром-N,N-диметилбут-2-енамида

(E)-4-бромбут-2-еновую кислоту (2 г, 12,2 ммоль) брали в дихлорметане (20 мл) и при 0°C добавляли HATU (5,5 г, 14 ммоль), триэтиламин (2,56 мл,18,4 ммоль) и перемешивали в течение 10 мин при КТ. К этой смеси медленно добавляли N,N-диметиламин (9,2 мл, 18 ммоль) и содержимое перемешивали при комнатной температуре в течение 2 ч. Летучие вещества удаляли при пониженном давлении и остаток разделяли между водой и этилацетатом. Органический слой промывали водой, насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали. Неочищенный материал очищали при помощи колоночной хроматографии на 100-200 силикагеле, с использованием 20% этилацетата в н-гексане, с получением (E)-4-бром-N,N-диметилбут-2-енамида (0,4 г, 17%) в виде жидкости бледно-зеленого цвета.
Стадия-8: Синтез (E)-N,N-диметил-4-((2-(4-((E)-2-фенил-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)бут-2-енамида
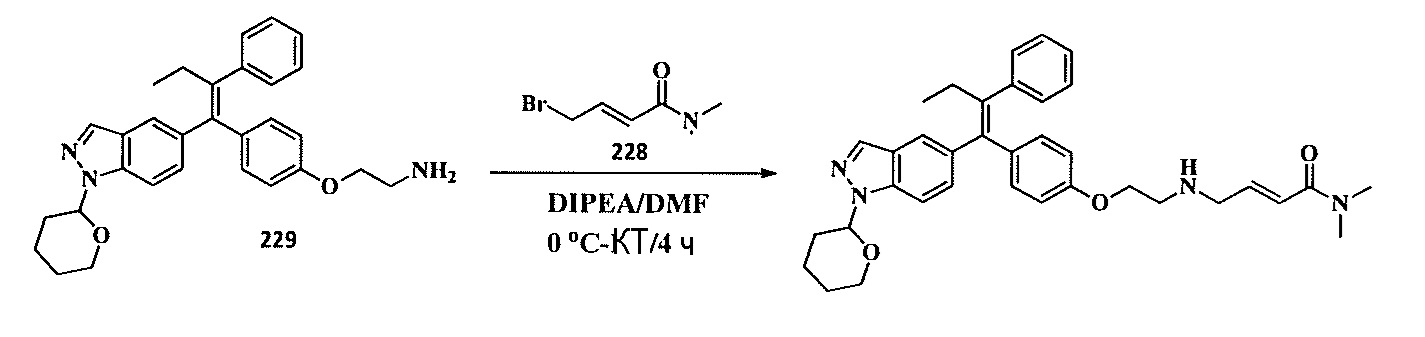
Реакцию осуществляли в соответствии со Схемой 2, Стадия-3a, используя 2-(4-(2-фенил-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этан-1-амин (0,450 г, 0,96 ммоль) вместо соединения 213. Реакционную смесь перемешивали в течение 4 ч при комнатной температуре, разбавляли холодной водой (50 мл) и экстрагировали дихлорметаном. Органический слой промывали водой, затем насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный материал очищали при помощи колоночной хроматографии на силикагеле 230-400 меш с использованием 1% метанола в дихлорметане с получением указанного в заголовке соединения Пр.1 Стадия-8 (0,200 г, 36%) в виде вязкого твердого вещества.
Стадия-9: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 1)

К перемешиваемому раствору (E)-N,N-диметил-4-((2-(4-((E)-2-фенил-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)бут-2-енамида (0,180 г, 0,31 ммоль) в этаноле (10 мл), при 0°C добавляли 2M HCl в эфире (10 мл). Реакционную смесь перемешивали в течение 3 ч при температуре кипения с обратным холодильником, разбавляли холодной водой (50 мл) и экстрагировали этилацетатом. Органический слой промывали водой, затем насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный материал очищали при помощи колоночной хроматографии на силикагеле 230-400 меш с использованием 3-4% метанола в DCM с получением соединения 1 (0,035 г, 23%) в виде бесцветного твердого вещества.
Соединение 1:1H ЯМР (400 MГц, CDCl3) δ 10,16 (шир.с, 1H), 8,07 (с, 1H), 7,66 (с, 1H), 7,5 (д, J=8,8 Гц, 1H), 7,26-7,11 (м, 6H), 6,93-6,87 (м,1H), 6,78 (д, J=8,8 Гц, 2H), 6,56 (д, J=8,8, 2H), 6,42 (д, J=15,2 Гц, 1H), 3,948 (т, J=4,9 Hz 2H), 3,45 (м, 2H), 3,06 (с, 3H), 2,99 (с, 3H), 2,50-2,45 (м, 2H), 0,95 (т, J=7,2 Гц, 3H). ЖХМС: 526,3 [M+H]+.
Пример 1A - Синтез хлористоводородная соль соединения 1
Стадия-1A: Синтез 5-бром-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола)

В чистый, сухой, промытый азотом 200 л стеклянный реактор, загружали 5-бром-индазол (11,0 кг, 0,055 моль), ацетонитрил (110 лит) и PTSA (1,0 кг г, 0,055 моль), реакционную смесь перемешивали при 28-30°C в течение 30 минут. К вышеуказанному раствору, по каплям добавляли дигидропиран (7,03 кг, 2,55 моль) в течение 20 мин. Полученную смесь перемешивали при комнатной температуре в течение 16 ч. По завершении ТСХ, реакционную смесь гасили водой (500 мл) и ацетонитрил концентрировали при пониженном давлении. Полученный продукт экстрагировали этилацетатом (25 л*3), объединенные органические экстракты промывали водой, затем насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением желаемого соединения, которое было взято, для следующей стадии, без дополнительной очистки (14 кг, выход 89%, ВЭЖХ чистота 93,93%)
1H ЯМР (300 MГц, CDCl3): δ 7,95 (с, 1H), 7,85 (с, 1H), 7,45-7,55 (д, 2H), 5,62-5,82 (д, 1H), 3,95-4,05 (д, 1H), 3,75-3,85 (д, 1H), 2,55-2,65 (т, 1H), 2,1-2,2 (м, 2H), 1,65-1,85 (м, 4H), ВЭЖХ: 93,93% при 210 нм.
Стадия-2A: Синтез (E)-4-бромбут-2-еновой кислоты

К перемешиваемому раствору (E)-бут-2-еновой кислоты (5,0 кг, 59,52 моль) в бензоле (50,0 лит) добавляли N-бромсукцинамид (10,58 кг, 59,52 моль), затем бензоил пероксид (144 г, 0,595 моль) при комнатной температуре. Реакционную смесь нагревали до кипения с обратным холодильником в течение 10 ч, что приводило к осаждению кристаллов сукцинамида. Кристаллы отфильтровывали и фильтрат концентрировали. Неочищенный продукт очищали кристаллизацией с использованием гексана с получением (E)-4-бромбут-2-еновой кислоты (2,5 кг, выход 26,5%) в виде белого твердого вещества.
1H ЯМР (300 MГц, CDCl3): δ 7,07-7,17 (м, 1H), 6,05-6,8 (д, 1H), 4,05-4,1 (д, 2H)
ВЭЖХ; 84,1% при 210 нм
Стадия-3A: Синтез (E)-4-бром-N,N-диметилбут-2-енамида

(E)-4-бромбут-2-еновую кислоту (5,0 кг г, 0,030 моль) брали в дихлорметане (600 мл) и охлаждали до 0°C. К этому раствору добавляли оксалилхлорид (5,1 кг, 0,039 моль), каталитическое количество DMF (500 мл) при той же температуре и перемешивали в течение 2 ч при комнатной температуре. Смесь охлаждали до 0°C и подщелачивали карбонатом натрия (5,79 кг, 0,054 моль). К этой смеси медленно добавляли 2М диметиламина в THF (15 л, 2 М раствор) (6-8 ч) и содержимое перемешивали при комнатной температуре в течение 1-2 ч. Летучие вещества удаляли концентрированием при пониженном давлении и остаток разделяли между водой и этилацетатом. Органический слой промывали водой, насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали. Неочищенный материал очищали при помощи колоночной хроматографии на силикагеле 230-400 меш с использованием 3-7% этилацетата в гексане с получением (E)-4-бром (хлор)-N,N-диметилбут-2-енамида (301, 55:45/Br:Cl, 2,5 кг, 73%) в виде жидкости коричневого цвета.
1H ЯМР (300 MГц, CDCl3): δ 6,84-6,94 (м, 1H), 6,4-6,5 (м, 1H), 4,15-4,2 (д, 1H), 3,95-4,0 (д, 1H), 2,9-2,95 (с, 3H), 2,95-3,0 (с, 3H), ВЭЖХ: 68,23 и 25,7% при 210 нм.
Стадия-4A: Синтез 2-Феноксиэтилхлорида

В чистый, сухой, промытый азотом 750 л стеклянный реактор (GLR), загружали толуол (150 л), 2-феноксиэтанол (15 кг, 0,108 моль) и пиридин (1,2 кг, 0,162 моль, 0,15 экв.). Реакционную массу охлаждали до 10-15°C, добавляли тионилхлорид (19,3 кг, 0,162 моль, 1,5 экв.) в течение 90 мин. Затем реакционную смесь нагревали до кипения с обратным холодильником и выдерживали в течение 10-12 часов. После завершения реакции охлаждали до 10-15°С и гасили водой (500 л). Водную фазу отделяли и отбрасывали. Процедуру промывки повторяли еще раз, прежде чем сосуд устанавливали для вакуумной перегонки и нагревали при температуре ниже 60°С до удаления толуола (500 л). Остаток затем охлаждали до 30-35°С перед тем, как его переносили в меньший сосуд(16 кг, 94%, 95,89% ВЭЖХ чистота).
1H ЯМР (400 MГц, DMSO-d6): δ 7,27-7,31 (т, 2H), 6,94-6,96 (т, 3H), 4,22-4,25(т, 2H), 3,95-4,0 (т, 2H); ВЭЖХ: 95,89% при 210 нм.
Стадия-5A: Синтез 1-[4-(2-Хлорэтокси)фенил]-2-фенил-1-бутанона

В чистый, сухой, промытый азотом 200 л стеклянный реактор загружали 2-феноксиэтил хлорид (17,0 кг, 69,94 кг при 100%, 1035 моль) и трифторуксусный ангидрид (13 лит кг), это перемешивали в течение 30 мин при 20-25°C. Добавляли 2-фенилмасляную кислоту (17,8 кг при 98,7%, 17,9 кг при 100%, 1038 моль). Реакционную смесь выдерживали при 25-30°С в течение 3 дней, после завершения реакции ее гасили водой и подщелачивали карбонатом калия. Полученный продукт экстрагировали DCM (25 л*3), промывали водой (25 л*2) затем насыщенным солевым раствором (25 л). Органический слой сушили сульфатом натрия и концентрировали при пониженном давлении. Раствор охлаждали до 20°C, и добавляли гексан (25 л). Суспензию охлаждали до 5°C и перемешивали при 0-5°C в течение 1 ч. Продукт выделяли фильтрованием и промывали холодным гексаном (5 л). Влажный продукт сушили на воздухе с получением кетона (24 кг, выход 78%, ВЭЖХ чистота 98,82%).
1H ЯМР (400 MГц, CDCl3): δ 7,95-7,97 (д, 2H), 7,286-7,28 (м, 4H), 7,18-7,22 (м, 1H), 6,86-6,88 (д, 2H), 4,38-4,40 (т, 1H), 4,22-4,25 (т, 2H), 3,78-3,85 (т, 2H), 2,18-2,22 (м, 1H), 1,85-1,90 (м, 1H), 0,95-0,98 (т, 3H), ВЭЖХ; 98,83% при 210 нм, ЖХМС: 303[M+H]+.
Стадия-6A: Синтез 1-(4-(2-хлорэтокси)фенил)-2-фенил-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бутан-1-ола

В чистый, сухой, промытый азотом 200 л стеклянный реактор загружали 1-[4-(2-хлорэтокси)фенил]-2-фенил-1-бутанон (Соединение 3) (11 кг, 0,0364 моль, 1 экв.) Соединение 300 (10,2 кг, 0,0364 моль, 1,0 экв. THF (110 л) охлаждали до -78°C. Добавляли n-BuLi (19 л 2,5 моля 13. экв.) в 6-7 ч. реакционную массу выдерживали при -78°С в течение 2 часов, после завершения реакции ее гасили насыщенным хлоридом аммония (4-5 л) в то же время в течение 2-3 ч и реакционной массе давали нагреться при комнатной температуре постепенно (8-10 ч). Добавляли 25 л воды и продукт экстрагировали 25 л этилацетата, органический слой отделяли и концентрировали в вакууме. Полученный неочищенный продукт очищали 60-120 силикагелем, используя 0-20 этилацетата в гексане, с получением чистого соединения 4 (6,7 кг, выход 36,5%, 97,83% ВЭЖХ чистота)
1H ЯМР (400 MГц, DMSO-d6): δ 8,10 (с, 1H), 7,69-7,72 (д, 1H), 7,65-7,70 (с, 1H), 7,10-7,23 (м, 6H), 6,72-6,74 (д, 2H), 6,55-6,59 (д, 2H), 5,82-5,86 (д, 1H), 3,85-3,90 (с, 1H), 3,75-3,8 (т, 3H), 2,75-2,80 (т, 2H), 1,35-1,40 (м, 3H), 1,95-2,0 (т, 2H), 1,2-2,0 (м, 7H), 0,85-,90 (т, 3H). ВЭЖХ; 97,83% при 210 нм
Стадия-7A: Синтез (E)-5-(1-(4-(2-хлорэтокси)фенил)-2-фенилбут-1-ен-1-ил)-1H-индазола

В чистый, 200 л стеклянный реактор, загружали соединение 4 (6,4 кг, 12,67 моль), метанол (64 л, 10 об.) и 11 н HCl (13 л, 2,5 об.), реакционную смесь выдерживали при 50°C в течение 14-16 ч. После завершения реакции, метанол концентрировали при пониженном давлении и полученный продукт экстрагировали этилацетатом (25 л*3), органический слой промывали водой (25 л), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный продукт суспендировали, промывали гексаном (10 л), с получение чистого соединения 5 в виде белого твердого вещества (4,4 кг, выход 86,37%, соотношение изомеров 52,92:46,5)
Стадия-8A: Синтез (E)-5-(1-(4-(2-хлорэтокси)фенил)-2-фенилбут-1-ен-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола

В чистый, сухой, промытый азотом 200 л стеклянный реактор загружали соединение 5 (5,1 кг, 12,686 моль), ацетонитрил (5 лит) и PTSA (241 г, 1,268 моль), реакционную смесь перемешивали при 28-30°C в течение 30 минут. К вышеуказанному раствору, добавляли по каплям дигидропиран 1,6 кг, 19,03 моль) в течение 20 мин. Полученную смесь перемешивали при комнатной температуре в течение 16 ч. По завершении ТСХ, реакционную смесь гасили водой (500 мл) и ацетонитрил концентрировали при пониженном давлении. Полученный продукт экстрагировали этилацетатом (25 л*3), объединенные органические экстракты промывали водой, затем насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением соединения 6 (6.кг, выход 97%,) в виде бледно-желтого вязкого материала.
1H ЯМР (400 MГц, DMSO-d6): соединение имеет оба геометрических изомера в соотношении 39,2:50,4.
Стадия-9A: Синтез (E)-2-(2-(4-(2-фенил-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)изоиндолин-1,3-диона

В чистый, сухой, промытый азотом 200 л стеклянный реактор загружали соединение 6 (5,8 кг, 11,9 моль, 1,1 экв.), DMF (6 л), фталамид калия (2,42 кг, 13,11 моль), DIPEA (4,6 кг, 35,7 моль, 3,0 экв.), реакционную массу нагревали до 90°C и выдерживали в течение 12-14 часов. После завершения реакции, массу охлаждали до 30°С, гасили 50 л воды и полученный продукт экстрагировали этилацетатом, промывали 20 л воды, органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученное твердое вещество перемешивали с 25 л 1:1 этилацетата и гексана в течение 5-6 часов, фильтровали нерастворенный Z-изомер и концентрировали фильтрат, и процесс повторяли до тех пор, пока изомерная чистота не становилась >80% при помощи ВЭЖХ (4,0 кг, 50% желаемого изомера).
Стадия-10A: Синтез (E)-2-(4-(2-фенил-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этан-1-амина

В чистый, сухой, промытый азотом 200 л стеклянный реактор, загружали соединение 7 (6,0 кг, 10,04 моль), метанол (6 л), IPA (60 л), охлаждали до 5-10°C и добавляли гидразин гидрат (10 л, 1,5 об.), После этого реакционную массу перемешивали при 30°C в течение 14-16 ч. После завершения реакции массу фильтровали для удаления сукцинамида, фильтрат гасили 10 л воды и спирт концентрировали при пониженном давлении. Полученный продукт экстрагировали этилацетатом (25 л*2), органический слой отделяли, промывали водой 25 л, сушили над безводным сульфатом натрия, концентрировали с получением соединения 8 (4,5 кг желаемой чистота изомера 59,45%. Другой изомер 15%).
Стадия-11A: Синтез (E)-2-(4-(2-фенил-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этан-1-амин сукцината

В чистый, сухой, промытый азотом 200 л стеклянный реактор добавляли янтарную кислоту (1,2 кг, 10,3 моль), этилацетат (15 л, 3 об.) и нагревали до кипения с обратным холодильником, добавляли соединение 8 (4,8 кг, 10,3 моль) растворяли в этилацетате (20 л, 4 об.) в течение 20-30 мин и продолжали кипячение с обратным холодильником еще 30 мин. реакционную массу охлаждали до 30-40°C и фильтровали твердые вещества и промывали (25 л) этилацетатом, сушили с получением 2,1 кг желаемого соединения с 93,6% чистоты ВЭЖХ (2% нежелательного геометрического изомера).
1H ЯМР (300 MГц, DMSO-d6): δ 8,10 (с, 1H), 7,65-7,72 (м, 2H), 7,13-7,21 (м, 6H), 6,75-6,78 (д, 2H), 6,62-6,64 (д, 2H), 3,87-4,01 (м, 4H), 3,69-3,73 (м, 4H), 3,04-3,05 (т, 3H), 2,27-2,38 (м, 5H), 1,98-2,0 (д, 1H), 1,6 (с, 1H), 0,85-,90 (т, 2H). ВЭЖХ; 93,6% при 210 нм
Стадия-12A: Синтез (E)-2-(4-(2-фенил-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этан-1-амина

В чистую, 20 л четырехгорлую круглодонную колбу, снабженную механической мешалкой, добавляли л воды, соединение 9 (2,1 кг, 3,58 моль) и охлаждали до 10-15°C, добавляли раствор карбоната калия до достижения значения рН 9-10, полученное основание экстрагировали 5 л*3 DCM. Объединенный органический слой промывали водой, сушили над безводным сульфатом натрия, концентрировали при пониженном давлении с получением желаемого соединения (1,5 кг, выход 89%, 90% ВЭЖХ, 2,5% другой изомер)
Стадия-13A: Синтез (E)-N,N-диметил-4-((2-(4-((E)-2-фенил-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)бут-2-енамида

В чистую, сухую, промытую азотом 10 л четырехгорлую круглодонную колбу, снабженную механической мешалкой, добавляли соединение 10 (1,5 кг, 3,211 моль) DCM (9 л), DIPEA (1,24 кг, 9,63 моль, 3,0 экв.) и реакционную смесь охлаждали до 0-5°C. Добавляли (E)-4-бром (хлор)-N,N-диметилбут-2-енамид (0,616 кг, 3,2 моль), растворенный в DCM (2 л), с использованием капельной воронки в течение 30-40 мин (E)-4-бром (хлор)-N,N-диметилбут-2-енамид, полученный на стадиях, и реакционную массу медленно нагревали до комнатной температуры и выдерживали в течение 24 часов при перемешивании. Массу охлаждали до 0-5°С, добавляли Boc-ангидрид (1,05 кг, 4,81 моль, 1,5 экв.) в DCM (2 л), и перемешивали в течение 4-5 ч. После завершения реакции, ее гасили водой, слой DCM отделяли, промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия и концентрировали при пониженном давлении до желаемого неочищенного соединения, которое очищали при помощи силикагеля 60-120 меш, с использованием 0-20 этилацетата в гексане, с получением (1,23 кг, выход 55,3%, 94,3% ВЭЖХ желаемого продукта и 2,1% другой изомер.
Стадия-14A: Синтез трет-бутил ((E)-4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-((E)-2-фенил-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)карбамата

В чистую, сухую, промытую азотом 10 л четырехгорлую круглодонную колбу, снабженную механической мешалкой и обратным холодильником, добавляли метанол (12 л) и соединение 11 (1,2 кг, 1,76 моль), реакционную смесь охлаждали до 0-5°C, добавляли концентрированную HCl (2,4 л, 2 об.) с использованием капельной воронки в течение 30 мин и реакционную смесь медленно нагревали до комнатной температуры, затем до 50°C. Это выдерживали при той же температуре в течение 4-6 ч, после завершения реакции, метанол концентрировали при пониженном давлении. Реакционную смесь подщелачивали раствором карбоната натрия при 10-15°C и полученный продукт экстрагировали 2 л*3 этилацетатом, промывали водой и сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением желаемого продукта (1,0 кг, выход 80%, 98,22% ВЭЖХ чистота)
Стадия-15A: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамид гидрохлорида

В чистую, сухую, промытую азотом 5 л четырехгорлую круглодонную колбу, снабженную механической мешалкой добавляли соединение 11 (600 г, 1,21 моль), растворенное в 9 л IPA. охлаждали до 5-10°С, добавляли IPA.HCl до достижения значения рН 2,5-3 и перемешивали при той же температуре в течение 30 мин и фильтровали образовавшееся твердое вещество и промывали 3 л холодного IPA и хорошо высушивали до 550 (выход 85%) желаемого соединения (98,07% ВЭЖХ).
ВЭЖХ: 98,07% (210 нм), ЖХМС (ESI, m/z), 494,5 [M+H]+,Температура плавления: 220-221°C
Соединение 1 (хлористоводородная соль):1H ЯМР (300 MГц, DMSO-d6): δ 13,05 (с, 1H), 9,28 (с, 2H), 8,07 (с, 1H), 7,60 (с, 1H), 7,5-7,52 (д, J=8,8 Гц, 1H), 7,1-7,22 (м, 6H), 6,78-6,84 (м, 3H), 6,52-6,66 (м, 3H), 4,11-4,13 (т, J=4,4 Гц, 2H), 3,76-3,80 (м, 2H), 3,26-3,3 (м, 2H), 3,03 (с, 3H), 2,86 (с, 3H), 2,40-2,41 (м, 2H), 0,86-,89 (т, J=7,4 Гц, 3H). ВЭЖХ; 98,07% при 210 нм.
Пример 2: Синтез (E)-4-((2-(4-((E)-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 2)
Стадия-1: Синтез 5-бром-3-фтор-1H-индазола

К перемешиваемому раствору 5-бром-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (10 г, 35,7 ммоль, полученного в Примере 1, Стадия-1) в 100 мл ацетонитрила, добавляли уксусную кислоту (4 мл) и selectfluor (25,2 г, 71,4 ммоль) при комнатной температуре. Реакционную смесь кипятили с обратным холодильником в течение 1 часа. По завершении ТСХ, реакционную смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (200 мл). Объединенные органические экстракты промывали водой, насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали при помощи колоночной хроматографии на силикагеле 230-400 меш, с использованием 1% этилацетата в н-гексане, с получением 5-бром-3-фтор-1H-индазола (6 г, 78%) в виде коричневого масла.
Стадия-2: Синтез 5-бром-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола

Реакцию осуществляли в соответствии со Схемой 1, Стадия-1 с получением неочищенного продукта, который очищали при помощи колоночной хроматографии на силикагеле 230-400 меш, используя 1% этилацетата в н-гексане с получением указанного в заголовке соединения Пр.2 Стадия-2 (5 г, 60%) в виде коричневого масла.
Стадия-3: Синтез 5-(бут-1-ин-1-ил)-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола

Реакцию осуществляли в соответствии со Схемой 1, Стадия-2a, с получением указанного в заголовке соединения Пр.2 Стадия-3 (4,2 г, 99%) в виде коричневого масла.
Стадия-4: Синтез (E)-4-(1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенола

Реакцию осуществляли в соответствии со Схемой 1, Стадия-6, с получением указанного в заголовке соединения Пр.2 Стадия-4 (1 г, 22%).
Стадия-5: Синтез трет-бутил (E)-(2-(4-(1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)карбамата

Реакцию осуществляли в соответствии со Схемой 2, Стадия-1 с получением неочищенного продукта, который очищали ппри помощи колоночной хроматографии на силикагеле 230-400 меш, используя 10% этилацетат в н-гексане, с получением указанного в заголовке соединения Пр.2 Стадия-5 (1 г, 75%) в виде светло-коричневой вязкой массы.
Стадия-6: Синтез (E)-2-(4-(1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этанамина

Реакцию осуществляли в соответствии со Схемой 2, Стадия-2a, используя (E)-(2-(4-(1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)карбамат (0,8 г, 1,61 ммоль) вместо соединения 211b. Неочищенный материал очищали при помощи хроматографии на силикагеле, используя (2:98) MeOH в DCM, с получением указанного в заголовке соединения Пр.2 Стадия-6 (0,23 г) в виде белого твердого вещества.
Стадия-7: Синтез (E)-4-((2-(4-((E)-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 2)

Реакцию осуществляли в соответствии со Схемой 2, Стадия-3a, с получением указанного в заголовке соединения Пр.2 Стадия-7 (0,13 г, неочищенный), который очищали при помощи ВЭЖХ, с получением чистого изомера (0,07 г, 37%) в виде не совсем белого твердого вещества.
Соединение 2:1H ЯМР (400 MГц, DMSO-d6) δ 12,57 (с, 1H), 7,50 (с, 1H), 7,47 (дд, J1=8,8 Гц, J2=2,0 Гц, 1H), 7,22-7,19 (м, 6H), 6,77 (с, 1H), 6,74 (с, 1H), 6,74-6,58 (м, 3H), 6,51-6,48 (м, 1H), 3,87 (т, J =5,6 Гц, 2H), 2,98 (с, 3H), 2,79 (с, 3H), 2,73 (т, J=5,6 Гц, 2H), 2,49-2,38 (м, 2H), 0,87 (т, J=7,2 Гц, 3H). ES (MS) 513,2 [M+H]+.
Пример 3: Синтез (E)-4-((2-(4-((E)-1-(3-фтор-1H-индазол-5-ил)-2-(3-фтор-5-(трифторметил)фенил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 3)
Стадия-1: Синтез (Z)-5-(1,2-бис(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола

Реакцию осуществляли в соответствии со Схемой 1, Стадия-3, используя 5-(бут-1-ин-1-ил)-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол (2,0 г, 7,29 ммоль, полученный в Примере 2, Стадия-3) вместо соединения 204. Неочищенный материал очищали при помощи колоночной хроматографии на силикагеле 230-400 меш с использованием 2% EtOAc в н-гексане с получением указанного в заголовке соединения Пр.3, Стадия-1 (2,5 г, 65%).
Стадия-2: Синтез трет-бутил (2-(4-йодфенокси)этил)карбамата

Реакцию осуществляли в соответствии со Схемой 3, Стадия-1, с получением указанного в заголовке соединения Пр.3 Стадия-2 в виде не совсем белого твердого вещества (80 г, 97%).
Стадия-3: Синтез трет-бутил (Z)-(2-(4-(1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)фенокси)этил)карбамата
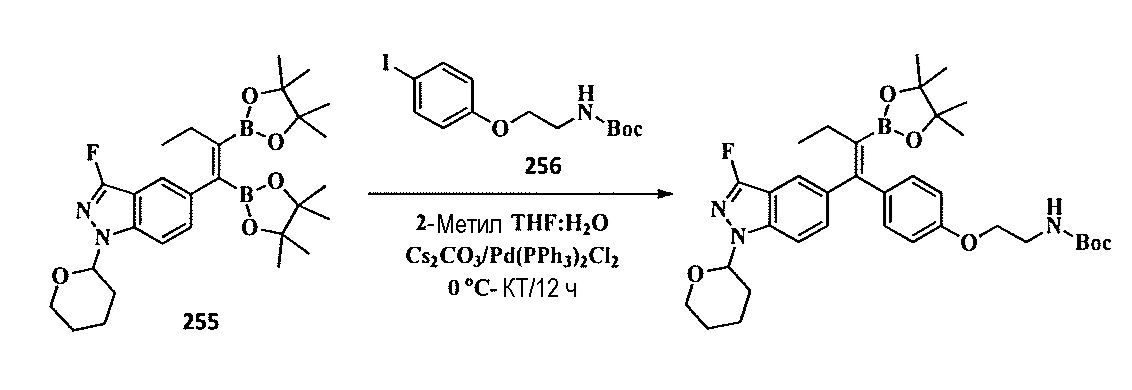
Реакцию осуществляли в соответствии со Схемой 1, Стадия-4, используя (Z)-5-(1,2-бис(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол (1,5 г, 2,85 ммоль) вместо соединения 206 и трет-бутил (2-(4-йодфенокси)этил)карбамат (1,03 г, 2,85 ммоль) вместо соединения 207. Неочищенный продукт, содержащий указанное в заголовке соединение Пр.3, Стадия-3, использовали на следующей стадии без дополнительной очистки (2 г).
Стадия-4: Синтез трет-бутил (E)-(2-(4-(1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(3-фтор-5-(трифторметил)фенил)бут-1-ен-1-ил)фенокси)этил)карбамата

Реакцию осуществляли в соответствии со Схемой 1, Стадия-5, используя трет-бутил (Z)-(2-(4-(1-(3-фтор-1-(тетрагидро-2H-пиран-2-ил)-1H-индазол-5-ил)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)-фенокси)этил)карбамат (950 мг, 1,8 ммоль) вместо соединения 208 и 1-фтор-3-иод-5-(трифторметил)бензол (523 мг, 1,8 ммоль) вместо соединения 209. Неочищенный продукт очищали с помощью хроматографии на силикагеле (2:8 EtOAc в н-гексане) с получением указанного в заголовке соединения Пр.3, Стадия-4 (0,5 г, 41%).
Стадия-5: Синтез (E)-2-(4-(1-(3-фтор-1H-индазол-5-ил)-2-(3-фтор-5-(трифторметил)фенил)бут-1-ен-1-ил)фенокси)этан-1-амина

Реакцию осуществляли в соответствии со Схемой 2, Стадия-2a, с получением указанного в заголовке соединения Пр.3, Стадия-5 (0,24 г, 66%) в виде полутвердого вещества коричневого цвета.
Стадия-6: Синтез (E)-4-((2-(4-((E)-1-(3-фтор-1H-индазол-5-ил)-2-(3-фтор-5-(трифторметил)фенил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 3).

Реакцию осуществляли в соответствии со Схемой 2, Стадия-3a с получением указанного в заголовке соединения Пр.3, Стадия-6 (0,1 г, 29%) в виде белого твердого вещества.
Соединение 3:1H ЯМР (400 MГц, DMSO-d6): δ 12,60 (с, 1H), 7,57 (с, 1H), 7,48 (дд, J1=8,8 Гц, J2=6,8 Гц, 1H), 7,41 (т, J=8,8 Гц, 2H), 7,27 (с, 1H), 7,23 (дд, J1=8,4 Гц, J2=0,8 Гц, 1H), 6,80 (д, J=8,8 Гц, 2H), 6,68-6,47 (м, 4H), 3,89 (т, J=5,2 Гц, 2H), 2,99 (с, 3H), 2,84 (с, 3H), 2,77 (т, J=5,6 Гц, 2H), 2,49-2,42 (м, 2H), 0,89 (т, J=7,2 Гц, 3H). ES (MS): 599,3 [M+H]+.
Пример 4: Синтез (E)-4-((2-(4-((E)-1-(4-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида. (Соединение 4)
Соединение синтезировали в соответствии с подходом, описанным в примере 2, с использованием на стадии-2 5-бром-4-фтор-1H-индазола (получение показано ниже в Пр.4 Стадии 1-2) вместо соединения 225 с получением соединения 4 (0,03 г, 5%) в виде белого твердого вещества.
Стадия-1: Синтез 4-бром-3-фтор-2-метиланилина

К перемешиваемому раствору 3-фтор-2-метиланилина (15 г, 0,1199 моль) в ацетонитриле (300 мл) добавляли N-бромсукцинамид (23,5 г, 0,131 моль) при 0°C. Реакционную смесь перемешивали в течение 30 мин при 10°C, после завершения реакции (контролируемой ТСХ), реакционную смесь разбавляли EtOAc. Органический слой промывали водой, затем насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт использовали на следующей стадии без дополнительной очистки с получением 4-бром-3-фтор-2-метиланилина (25 г, неочищенный) в виде белого твердого вещества.
Стадия-2: Синтез 5-бром-4-фтор-1H-индазола

К перемешиваемому раствору 4-бром-3-фтор-2-метиланилина (20 г, 98 ммоль) в уксусной кислоте добавляли нитрит натрия (8,1 г, 117 ммоль) при 10°C, и содержимое перемешивали при комнатной температуре в течение 5 ч. После завершения реакции (контролируемой ТСХ), реакционную смесь охлаждали до 0°C, 50% раствор NaOH добавляли по каплям до тех пор, пока рН раствора не становился равен 9. Реакционную смесь разбавляли этилацетатом и органический слой промывали водой, затем насыщенным солевым раствором. Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное соединение очищали при помощи колоночной хроматографии на силикагеле толщиной 230-400 меш с использованием (10-15%) этилацетата в н-гексане в качестве элюента с получением указанного в заголовке соединения Пр.4 Стадия-2 (6,1 г, 29%) в виде желтого твердого вещества.
Соединение 4:1H ЯМР (400 MГц, DMSO-d6): δ 13,39 (с, 1H), 8,17 (с, 1H), 7,39 (д, J=8,4 Гц, 1H), 7,24-7,2 (м, 2H), 7,18-7,15 (м, 4H), 6,77 (д, J=8,4 Гц, 2H), 6,62-6,57 (м, 3H), 6,51-6,47 (м, 1H), 3,86 (т, J=5,6 Гц, 2H), 3,31 (м, 2H), 2,98 (с, 3H), 2,83 (с, 3H), 2,77 (т, J=5,4 Гц, 2H), 2,33-2,30 (м, 2H), 0,89 (т, J=7,4 Гц, 3H). ES (MS): 513,3 [M+H]+.
Пример 5: Синтез (E)-4-((2-(4-((E)-2-(3,5-дифторфенил)-1-(1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 5)
Стадия-1: Синтез трет-бутил (E)-(2-(4-(2-(3,5-дифторфенил)-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)карбамата

К перемешиваемому раствору 5-(бут-1-ин-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (0,4 г, 1,574 ммоль, Пример 1, Стадия-3) в 2-метил THF (5 мл), добавляли бис(пинаколато)диборон (0,44 г, 1,732 ммоль), тетракис(трифенилфосфин)платина(0) (14,6 мг, 0,0118 ммоль) в атмосфере азота, реакционную смесь нагревали при 90°C в течение 5 ч. После завершения реакции (контролируемой ТСХ), реакционной смеси давали остыть до 4°C и добавляли трет-бутил (2-(4-йодфенокси)этил)карбамат (571 мг, 1,574 ммоль, Пример 3, Стадия-2), бис(трифенилфосфин)палладий(II) дихлорид (55 мг, 0,078 ммоль), карбонат цезия (1,023 г, 3,140 ммоль) и 2-метил THF (5 мл). Эту смесь дегазировали азотом и добавляли воду (1,2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. После завершения реакции (контролируемой ТСХ), добавляли 1,3-дифтор-5-иодбензол (528 мг, 2,20 ммоль) и 4M водный KOH (3 мл) и смесь дегазировали азотом и нагревали при 70°C в течение 5 ч. По завершении реакционную смесь фильтровали через слой целита/силикагеля и промывали EtOAc. Фильтрат промывали водой, насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью хроматографии на силикагеле (2:8 EtOAc в н-гексане) с получением указанного в заголовке соединения Пр. 5 Стадия-1 (500 мг, 54%).
Стадия-2: Синтез (E)-2-(4-(2-(3,5-дифторфенил)-1-(1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этан-1-амина
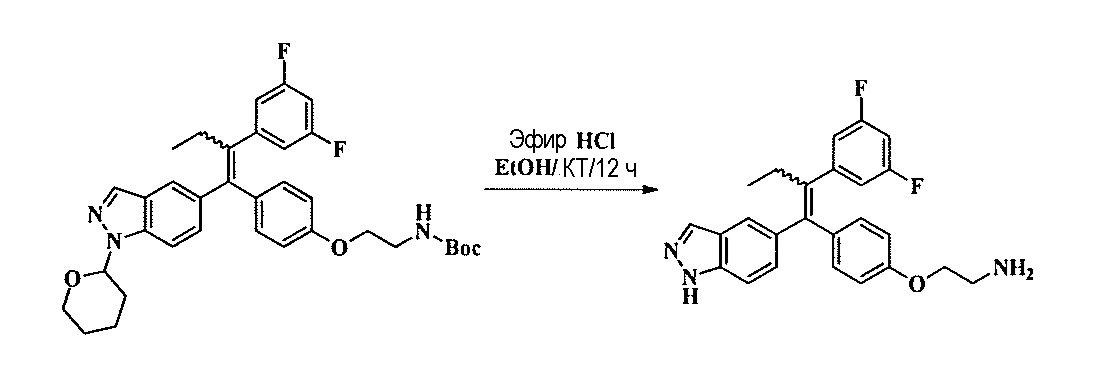
Реакцию осуществляли в соответствии со Схемой 2, Стадия-2a, с использованием трет-бутил (E)-(2-(4-(2-(3,5-дифторфенил)-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)карбамата вместо соединения 211b с получением указанного в заголовке соединения Пр.5 Стадия-2 (0,340 г, 97%) в виде белого твердого вещества.
Стадия-3: Синтез (E)-4-((2-(4-((E)-2-(3,5-дифторфенил)-1-(1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 5)

Соединение получали согласно схеме 2, Стадия-3а, с использованием (E)-2-(4-(2-(3,5-дифторфенил)-1-(1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этан-1-амина вместо соединения 213 с получением соединения 5 (60 мг, 14%) в виде белого твердого вещества.
Соединение 5:1H ЯМР (400 MГц, DMSO-d6): δ 13,08 (с, 1H), 8,07 (с, 1H), 7,61 (с, 1H), 7,52 (д, J=8,8 Гц, 1H), 7,12 (д, J=8,4 Гц, 1H), 6,99 (м, 1H), 6,868 (м, 1H), 6,795 (д, J=8,8 Гц, 2H), 6,67 (д, J=8,8 Гц, 2H), 6,63-6,58 (м, 1H), 3,9 (т, J=5,6 Гц, 2H), 3,31 (м, 2H), 2,99 (с, 3H), 2,84 (с, 3H), 2,79 (т, J=5,6 Гц, 2H), 2,39 (кв, J=7,2 Гц, 2H), 0,89 (т, J=7,4 Гц, 3H). ЖХМС: 531,3 [M+H]+.
Пример 6: Синтез (E)-4-((2-(4-((E)-2-(3,4-дифторфенил)-1-(1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 6)
Соединение синтезировали в соответствии с подходом, описанным в примере 3, используя на стадии-4 трет-бутил (E)-(2-(4-(2-(3,4-дифторфенил)-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)карбамат вместо соединения 226 (получение показано ниже в примере 6, Стадия-1) и 1,2-дифтор-5-иодбензол вместо соединения 227 с получением соединения 6 (50 мг, 8%) в виде белого твердого вещества.
Стадия-1: Синтез трет-бутил (E)-(2-(4-(2-(3,4-дифторфенил)-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)карбамата

К перемешиваемому раствору 5-(бут-1-ин-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (2,5 г, 9,84 ммоль, полученного в Примере 1, Стадия-3) в 2-метил THF (20 мл), добавляли бис(пинаколато)диборон (2,52 г, 9,94 ммоль), тетракис(трифенилфосфин)платина(0) (93 мг, 0,0748 ммоль) в атмосфере азота, реакционную смесь нагревали при 80°C в течение 6 ч. Раствору давали охладиться до 4°C, и добавляли трет-бутил (2-(4-йодфенокси)этил)карбамат (3,57 г, 9,84 ммоль, полученный в Примере 3, Стадия-2), бис(трифенилфосфин)палладий(II) дихлорид (345 мг, 0,492 ммоль), карбонат цезия (6,4 г, 19,68 ммоль) и 2-метил THF (10 мл). Эту смесь дегазировали азотом и добавляли воду (6 мл). Эту смесь перемешивали при комнатной температуре в течение 12 ч. После завершения реакции реакционную смесь разбавляли водой и экстрагировали EtOAc. Органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный материал очищали при помощи колоночной хроматографии на силикагеле 230-400 меш с использованием EtOAc в н-гексане (3:7) с получением указанного в заголовке соединения Пр.6 Стадия-1 (3,85 г, 64%) в виде липкого твердого вещества.
Соединение 6:1H ЯМР (400 MГц, DMSO-d6): δ 13,09 (с, 1H), 8,07 (с, 1H), 7,60 (с, 1H), 7,51 (д, J=8,0 Гц, 1H), 7,29-7,20 (м, 2H), 7,11 (д, J=8,8 Гц, 1H), 6,94 (шир.с, 1H), 6,77 (д, J=8,80 Гц, 2H), 6,65 (д, J=8,8 Гц, 2H), 6,63-6,48 (м, 2H), 3,71 (т, J=5,6 Гц, 2H), 2,99 (с, 3H), 2,84 (с, 3H), 2,78 (т, J=5,2 Гц, 2H), 2,43-2,32 (м, 2H), 0,89 (т, J=7,2 Гц, 3H). ЖХМС: 531,3 [M+H]+.
Пример 7: Синтез (E)-4-((2-(4-((E)-2-(3-хлор-5-фторфенил)-1-(1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида
Соединение синтезировали в соответствии с подходом, описанным в примере 3, с использованием на стадии-4 трет-бутил (Z)-(2-(4-(1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)фенокси)-этил)карбамата (1,5 г, 2,43 ммоль, полученного в Примере 6, Стадия-1) вместо соединения 226 и 1-хлор-3-фтор-5-иодбензола вместо соединения 227 на стадии-4 с получением указанного в заголовке соединения (50 мг, 9%) в виде белого твердого вещества.
Соединение 7:1H ЯМР (400 MГц, DMSO-d6): δ 13,10 (с, 1H), 8,08 (с, 1H), 7,61 (с, 1H), 7,52 (д, J=8,8 Гц, 1H), 7,19 (д, J=8,8 Гц, 1H), 7,12 (д, J=8,6 Гц, 1H), 7,05 (с, 2H), 7,00 (д, J=8,8 Гц, 2H), 6,68 (д, J=8,8 Гц, 2H), 6,63-6,58 (м, 1H), 6,52-6,48 (м, 1H), 3,9 (т, J=5,6 Гц, 2H), 3,33-3,31 (м, 2H), 2,99 (с, 3H), 2,84 (с, 3H), 2,79 (т, J=5,6 Гц, 2H), 2,44-2,32 (м, 2H), 0,89 (т, J=7,4 Гц, 3H). ESMS: 547,3 [M+H]+.
Пример 8: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N-метил-N-(проп-2-ин-1-ил)бут-2-енамида (Соединение 8)
Соединение синтезировали в соответствии с подходом, описанным в примере 1, с использованием на стадии-8 (E)-4-бром-N-метил-N-(проп-2-ин-1-ил)бут-2-енамида (получение показано ниже в Пр.8 Стадия-1) вместо соединения 228 и 2-(4-(1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этан-1-амина (получение показано ниже в Пр.9, Стадия 4-5) вместо соединения 229 с получением соединения 8 (4 мг, выход: 5%) в виде светло-зеленого твердого вещества.
Стадия-1: Синтез (E)-4-бром-N-метил-N-(проп-2-ин-1-ил)бут-2-енамида
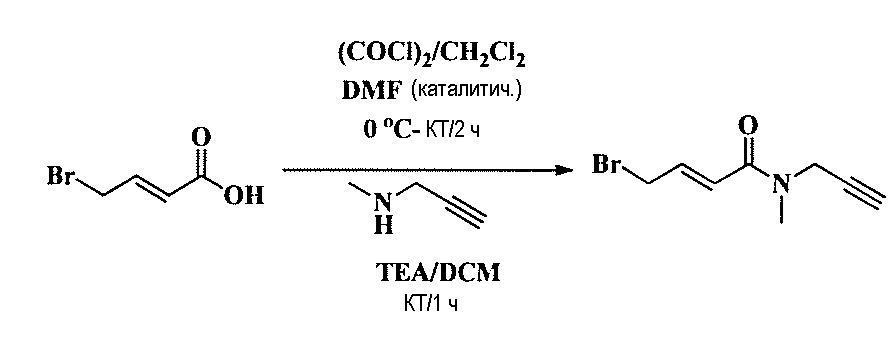
Реакцию осуществляли в соответствии со Схемой 4, Стадия-2, используя N-метилпроп-2-ин-1-амин вместо соединения 221 c получением смеси (E)-4-бром-N-метил-N-(проп-2-ин-1-ил)бут-2-енамида (0,5 г, 19%) в виде жидкости коричневого цвета.
Соединение 8:1H ЯМР (400 MГц, DMSO-d6): δ 13,06 (с, 1H), 8,06 (с, 1H), 7,61 (с, 1H), 7,51 (д, J=7,2 Гц, 1H), 7,22-7,10 (м, 6H), 6,74 (д, J=8,8 Гц, 2H), 6,60-6,52 (м, 4H), 4,22-4,16 (м, 2H), 3,87 (т, J=5,2 Гц, 2H), 3,02 (с, 2H), 2,87 (шир.с, 1H), 2,78 (т, J=5,6 Гц, 2H), 2,43-2,37 (м, 2H), 0,87 (т, J=7,6 Гц, 3H). ESMS: 519,2 [M+H]+.
Пример 9: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N-(бут-3-ин-1-ил)-N-метилбут-2-енамида (Соединение 9)
Стадия-1: Синтез бут-3-ин-1-ил 4-метилбензолсульфоната

К перемешиваемому раствору бут-3-ин-1-ола (5 г, 0,0714 ммоль) в дихлорметане (50 мл) при 0°C добавляли триэтиламин (10,82 г, 0,107 ммоль) и тозилхлорид (13,61 г, 0,0714 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 12 ч, после завершения реакции (контролируемой ТСХ), реакционную смесь разбавляли дихлорметаном. Органический слой промывали водой, насыщенным солевым раствором и сушили над безводным Na2SO4и концентрировали при пониженном давлении с получением бут-3-ин-1-ил 4-метилбензолсульфоната (13 г, 81%).
Стадия-2: Синтез N-метилбут-3-ин-1-амина

Бут-3-ин-1-ил 4-метилбензолсульфонат (10 г, 44,6 ммоль) добавляли к 40% водному метиламину (30 мл). Содержимое нагревали при 70°C в атмосфере азота в течение 2 часов. После завершения реакции реакционную смесь охлаждали до комнатной температуры, летучие вещества удаляли в вакууме и экстрагировали дихлорметаном (250 мл). Органический слой промывали водой, насыщенным солевым раствором и сушили над безводным сульфатом натрия. Органический слой сушили при пониженном давлении с получением N-метилбут-3-ин-1-амина (3,7 г, неочищенный).
Стадия-3: Синтез (E)-4-бром-N-(бут-3-ин-1-ил)-N-метилбут-2-енамида

Реакцию осуществляли в соответствии со Схемой 4, Стадия-2, используя N-метилбут-3-ин-1-амин вместо соединения 221, с получением смеси (E)-4-бром-N-(бут-3-ин-1-ил)-N-метилбут-2-енамида (1,8 г, неочищенный) в виде жидкости коричневого цвета.
Стадия-4: Синтез трет-бутил (2-(4-(2-фенил-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)карбамата

К раствору (E)-4-(2-фенил-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)фенола (3 г, 7,0 ммоль, полученного в Примере 1, Стадия-4) в DMF (30 мл) добавляли последовательно карбонат калия (4,8 г, 35,3 ммоль) и трет-бутил (2-бромэтил)карбамат (3,16 г, 14,1 ммоль). Реакционную смесь перемешивали при 80°C в течение 12 ч, после завершения реакции, реакционную смесь выливали в ледяную воду. Отделенное твердое вещество фильтровали и сушили при пониженном давлении с получением указанного в заголовке соединения Пр.9 Стадия-4 (2,5 г, 64%) в виде не совсем белого твердого вещества.
Стадия-5: Синтез 2-(4-(1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этан-1-амина

Реакцию осуществляли в соответствии со Схемой 2, Стадия-2a, используя трет-бутил (2-(4-(2-фенил-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)карбамат (2,5 г, 4,4 ммоль) вместо соединения 211b с получением указанного в заголовке соединения Пр.9 Стадия-5 (0,8 г, 47%) в виде не совсем белого твердого вещества.
Соединение 9:1H ЯМР (400 MГц, DMSO-d6): δ 13,07 (с, 1H), 8,06 (с, 1H), 7,61 (с, 1H), 7,51 (д, J=8,8 Гц, 1H), 7,20-7,11 (м, 6H), 6,74 (д, J=8,8 Гц, 2H), 6,60-6,57 (м, 4H), 3,86 (т, J=4,8 Гц, 2H), 3,49-3,38 (м, 3H), 3,03 (с, 2H), 2,86-2,84 (м, 3H), 2,77 (т, J=6,40 Гц, 2H), 2,43-2,32 (м, 4H), 0,87 (т, J=7,2 Гц, 3H). ЖХМС: 533,4 [M+H]+.
Пример 10: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-1-(азетидин-1-ил)бут-2-ен-1-она (Соединение 10)
Соединение синтезировали в соответствии с подходом, описанным в примере 9, с использованием на Стадии-4 (E)-1-(азетидин-1-ил)-4-бромобут-2-ен-1-она (получение показано ниже в Пр.10 Стадия-1) вместо соединения 230 с получением соединения 10 (22 мг, 4,1%) в виде белого твердого вещества.
Стадия-1: Синтез (E)-1-(азетидин-1-ил)-4-бромобут-2-ен-1-она

Реакцию осуществляли в соответствии со Схемой 4, Стадия-2, используя азиридин вместо соединения 221, с получением (E)-1-(азетидин-1-ил)-4-бромобут-2-ен-1-она (1 г, 27%) в виде жидкости коричневого цвета.
Соединение 10:1H ЯМР (400 MГц, DMSO-d6): δ 13,08 (с, 1H), 8,07 (с, 1H), 7,6 (с, 1H), 7,51 (д, J=8,4 Гц, 1H), 7,2 (д, J=7,6 Гц, 2H), 7,15-7,11 (м, 4H), 6,75 (д, J=8,8 Гц, 2H), 6,61-6,57 (м, 3H), 6,04 (д, J=16 Гц, 1H), 4,12 (т, J=7,6 Гц, 2H), 3,88-3,84 (м, 4H), 3,32-3,3 (м, 2H), 2,77 (т, J=5,6 Гц, 2H), 2,42-2,4 (м, 2H), 2,19-2,15 (м, 2H), 0,88 (т, J=7,6 Гц, 3H). ЖХМС: 507,1 [M+H]+.
Пример 11: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-1-(пирролидин-1-ил)бут-2-ен-1-она (Соединение 11)
Соединение синтезировали в соответствии с подходом, описанным в примере 9, с использованием на стадии-4 (E)-4-бром-1-(пирролидин-1-ил)бут-2-ен-1-она (получение показано ниже в Пр.9 Стадия-1) вместо соединения 230 с получением указанного в заголовке соединения 11 (7,5 мг, 6,6%) в виде белого твердого вещества.
Стадия-1: Синтез (E)-4-бром-1-(пирролидин-1-ил)бут-2-ен-1-она

Реакцию осуществляли в соответствии со Схемой 4, Стадия-2, с использованием пирролидина, с получением (E)-4-бром-1-(пирролидин-1-ил)бут-2-ен-1-она (0,31 г, 6%) в виде жидкости коричневого цвета.
Соединение 11:1H ЯМР (400 MГц, DMSO-d6): δ 13,06 (с, 1H), 8,07 (с, 1H), 7,61 (с, 1H), 7,51 (д, J=8,8 Гц, 1H), 7,22-7,11 (м, 6H), 6,75 (д, J=8,8 Гц, 2H), 6,65-6,58 (м, 3H), 6,31 (д, J=15,2 Гц, 1H), 3,86 (т, J=5,6 Гц, 2H), 3,45 (т, J=6,8 Гц, 2H), 2,77 (т, J=5,6 Гц, 2H), 2,43-2,38 (м, 2H), 1,9-1,73 (м, 6H), 0,88 (т, J=7,2 Гц, 5H). ЖХМС: 521,3 [M+H]+.
Пример 12: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-1-(пиперидин-1-ил)бут-2-ен-1-она (Соединение 12)
Стадия-1: Синтез (E)-4-бром-1-(пиперидин-1-ил)бут-2-ен-1-она

Реакцию осуществляли в соответствии со Схемой 4, Стадия-2, используя пиперидин вместо соединения 221, с получением (E)-4-бром-1-(пиперидин-1-ил)бут-2-ен-1-она (3,9 г, 55%) в виде жидкости коричневого цвета.
Стадия-2: Синтез (2E)-4-((2-(4-(1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-1-(пиперидин-1-ил)бут-2-ен-1-она

Реакцию осуществляли в соответствии со Схемой 2, Стадия-3a, используя 2-(4-(1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)-этан-1-амин (полученный в Примере 9, Стадия 4-5) вместо соединения 213, с получением неочищенного материала, который использовали на следующей стадии без дополнительной очистки (716 мг, неочищенный).
Стадия-3: Синтез трет-бутил (2-(4-(1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)((E)-4-оксо-4-(пиперидин-1-ил)бут-2-ен-1-ил)карбамата

Реакцию осуществляли в соответствии со Схемой 2, Стадия-5, используя (2E)-4-((2-(4-(1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-1-(пиперидин-1-ил)бут-2-ен-1-он (0,716 г, 1,34 ммоль) вместо соединения 215. Неочищенное соединение очищали при помощи колоночной хроматографии с использованием 4% МеОН в дихлорметане с получением указанного в заголовке соединения Пр.12 Стадия-3 (0,32 г, 37%).
Стадия-4: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-1-(пиперидин-1-ил)бут-2-ен-1-она (Соединение 12)

Реакцию осуществляли в соответствии со Схемой 2, Стадия-6c, используя трет-бутил (2-(4-(1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)((E)-4-оксо-4-(пиперидин-1-ил)бут-2-ен-1-ил)карбамат (0,32 г, 0,504 ммоль) вместо соединения 211с с получением неочищенного соединения, которое очищали при помощи препаративной ТСХ с получением соединения 12 (74 мг, 27%).
Соединение 12:1H ЯМР (400 MГц, DMSO-d6): δ 13,06 (с, 1H), 8,06 (с, 1H), 7,61 (с, 1H), 7,51 (д, J=8,8 Гц, 1H), 7,2 (д, J=7,6 Гц, 2H), 7,15-7,1 (м, 4H), 6,75 (д, J=8,4 Гц, 2H), 6,6-6,5 (м, 4H), 3,86 (т, J=6 Гц, 2H), 3,42 (т, J=5,6 Гц, 4H), 2,77 (т, J=5,6 Гц, 2H), 2,5 (м, 3H), 2,43-2,38 (м, 2H), 1,58-1,53 (м, 2H), 1,42 (д, J=3,6 Гц, 4H), 0,87 (т, J=7,2 Гц, 3H). ЖХМС: 535,3 [M+H]+.
Пример 13: Синтез (E)-4-((2-(4-((E)-2-циклобутил-1-(1H-индазол-5-ил)-2-фенилвинил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 13)
Стадия-1: Синтез (6-хлоргекс-1-ин-1-ил)триметилсилана

Раствор триметилсилилацетилена (10 г, 101 ммоль) в сухом тетрагидрофуране (90 мл) перемешивали при 0°С в атмосфере азота; n-BuLi (44,5 мл 2,6 М раствора в гексане, 111,2 ммоль) добавляли по каплям. Раствор перемешивали в течение 30 минут, добавляли раствор 1-хлор-4-йодбутана (13,6 мл, 111,2 ммоль) в сухом тетрагидрофуране (30 мл). Реакционной смеси давали нагреться до 20°C и перемешивали в течение 12 ч. Реакционную смесь выливали в воду, водную смесь экстрагировали диэтиловым эфиром. Эфирные экстракты промывали водой и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении с получением 6-хлор-1-триметилсилилгекс-1-ина (18 г) в виде желтой жидкости и использовали на следующей стадии без дополнительной очистки.
Стадия-2: Синтез (циклобутилэтинил)триметилсилана

К раствору диизопропиламина (17 мл, 95,7 ммоль) в безводном THF (50 мл) при 0°С добавляли по каплям n-BuLi (2,6 М в гексане, 36,8 мл, 95,7 ммоль). Смесь перемешивали в течение 20 минут при 0°C и затем охлаждали до -78°C. К этой смеси добавляли по каплям раствор (6-хлоргекс-1-ин-1-ил)триметилсилана (9 г, 47 ммоль) в безводном THF (50 мл). Полученной смеси давали нагреться до комнатной температуры и перемешивали в течение 12 ч. Реакционную смесь осторожно гасили при комнатной температуре насыщенным водным NH4Cl (100 мл), экстрагировали пентаном (200 мл). Объединенные органические слои промывали насыщенным солевым раствором и сушили над безводным Na2SO4. Растворитель выпаривали при пониженном давлении. Остаток перегоняли при 160-162°C/760 мм. рт.ст. с получением указанного в заголовке соединения в виде бесцветной жидкости (4 г, 55%).
Стадия-3: Синтез 5-(циклобутилэтинил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола

Реакцию осуществляли в соответствии со Схемой 1, Стадия-2, используя циклобутилэтинил)триметилсилан вместо соединения 203, с получением указанного в заголовке соединения Пр.13 Стадия-3 (450 мг, 84%) в виде коричневого масла.
Стадия-4: Синтез (Z)-5-(2-циклобутил-1,2-бис(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)винил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола

Реакцию осуществляли в соответствии со Схемой 1, Стадия-3, с получением указанного в заголовке соединения Пр.13 Стадия-4 (0,5 г, 66%).
Стадия-5: Синтез трет-бутил (Z)-(2-(4-(2-циклобутил-1-(1-(тетрагидро-2H-пиран-2-ил)-1H-индазол-5-ил)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)винил)фенокси)-этил)карбамата

Реакцию осуществляли в соответствии со Схемой 1, Стадия-4, с использованием трет-бутил (2-(4-йодфенокси)этил)карбамата (полученного в Примере 3, Стадия-2) вместо соединения 207 с получением указанного в заголовке соединения Пр.13 Стадия-5 (0,36 г, 50%).
Стадия-6: Синтез трет-бутил (E)-(2-(4-(2-циклобутил-2-фенил-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)винил)фенокси)этил)карбамата

Реакцию осуществляли в соответствии со Схемой 1, Стадия-5, используя йодбензол вместо соединения 209 с получением указанного в заголовке соединения Пр.13 Стадия-6 (0,17 г, 51%).
Стадия-7: Синтез (E)-2-(4-(2-циклобутил-1-(1H-индазол-5-ил)-2-фенилвинил)фенокси)этан-1-амина

Реакцию осуществляли в соответствии со Схемой 2, Стадия-2a, с получением указанного в заголовке соединения Пр.13 Стадия-7 (0,04 г, 34%) в виде полутвердого вещества коричневого цвета.
Стадия-8: Синтез (E)-4-((2-(4-((E)-2-циклобутил-1-(1H-индазол-5-ил)-2-фенилвинил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 13).

Реакцию осуществляли в соответствии со Схемой 2, Стадия-3a, используя (E)-4-бром-N,N-диметилбут-2-енамид (полученный в Примере 1, Стадия-7) вместо соединения 214 с получением соединения 13 (0,2 г, 40%) в виде белого твердого вещества.
Соединение 13:1H ЯМР (400 MГц, DMSO-d6): δ 13,06 (с, 1H), 8,07 (с, 1H), 7,58 (с, 1H), 7,51 (д, J=6,8 Гц, 1H), 7,24 (д, J=5,2 Гц, 2H), 7,17-7,09 (м, 4H), 6,78 (д, J=6,8 Гц, 2H), 6,60-6,46 (м, 4H), 3,83 (т, J=4,4 Гц, 2H), 3,50-3,39 (м, 2H), 2,97 (с, 3H), 2,83 (с, 3H), 2,75 (т, J=4,4 Гц, 2H), 1,8-1,77 (м, 4H), 1,57-1,55 (м, 1H), 1,36-1,24 (м, 2H). ЖХ (МС) 521,3 [M+H]+.
Пример 14: Синтез (E)-4-((2-(4-((E)-2-циклобутил-1-(3-фтор-1H-индазол-5-ил)-2-фенилвинил)фенокси)этил)амино)-N,N-диметилбут-2-енамид 2,2,2-трифторацетата (Соединение 14)
Стадия-1: Синтез 5-(циклобутилэтинил)-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола

Реакцию осуществляли в соответствии со Схемой 1, Стадия-2b, используя 5-бром-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол (полученный в Примере 2, Стадия-2) вместо соединения 202 и циклобутилэтинил)триметилсилан (полученный в Примере 13, Стадия-3) вместо соединения 203 с получением указанного в заголовке соединения Пр.14 Стадия-1 (2,15 г, 86%).
Стадия-2: Синтез (Z)-5-(2-циклобутил-1,2-бис(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)винил)-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола

Реакцию осуществляли в соответствии со Схемой 1, Стадия-3, с получением указанного в заголовке соединения Пр.14 Стадия-2 (4,3 г, 51,8%).
Стадия-3: Синтез трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)карбамата

Стадия-3.1: Синтез 2-(4-йодфенокси)этан-1-амина

К перемешиваемому раствору трет-бутил (2-(4-йодфенокси)этил)карбамата (25 г, 68,6 ммоль, Пример 3, Стадия-2) в этаноле (50 мл) добавляли при 0°C, 2M HCl в эфире (250 мл). Реакционную смесь перемешивали в течение 12 ч при комнатной температуре. После завершения реакции реакционную смесь подщелачивали насыщенным NaHCO3, экстрагировали 10% МеОН в DCM. Органический слой концентрировали при пониженном давлении и неочищенный материал использовали на следующей стадии без дополнительной очистки (16 г, 88%).
Стадия-3.2: Синтез (E)-4-((2-(4-йодфенокси)этил)амино)-N,N-диметилбут-2-енамида

Реакцию осуществляли в соответствии со Схемой 2, Стадия-3a, используя (E)-4-бром-N,N-диметилбут-2-енамид (Пример 1, Стадия-7) вместо соединения 214 с получением неочищенного продукта, который использовали на следующей стадии без дополнительной очистки (18,8 г, неочищенный).
Стадия-3.3: Синтез трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)карбамата

К перемешиваемому раствору (E)-4-((2-(4-йодфенокси)этил)амино)-N,N-диметилбут-2-енамида (18,8 г, 50,26 ммоль) в сухом дихлорметане (150 мл) добавляли DIPEA (6,4g, 50,2 ммоль) при 0°C, перемешивали в течение 15 мин при 0°C. К вышеуказанной реакционной смеси добавляли boc-ангидрид (13,1 г, 60,3 ммоль), полученную смесь перемешивали при комнатной температуре в течение 12 ч. По завершении ТСХ, реакционную смесь охлаждали до 0°C, гасили ледяной водой (500 мл) и экстрагировали дихлорметаном (500 мл). Объединенные органические экстракты промывали водой, затем насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный материал очищали при помощи колоночной хроматографии на силикагеле 230-400 меш с использованием 3% MeOH в дихлорметане в качестве элюента с получением трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)карбамата (9 г, 37,8%).
Стадия-4: Синтез трет-бутил (2-(4-((E)-2-циклобутил-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-фенилвинил)фенокси)этил)((E)-4-(диметиламино)-4-оксобут-2-ен-1-ил)карбамата

Реакцию осуществляли в соответствии со Схемой 1, Стадия-4 и Стадия-5, с получением неочищенного продукта. Неочищенный продукт, содержащий указанное в заголовке соединение Пр.14, Стадия-4, использовали на следующей стадии без дополнительной очистки (340 мг, неочищенный).
Стадия-5: Синтез (E)-4-((2-(4-((E)-2-циклобутил-1-(3-фтор-1H-индазол-5-ил)-2-фенилвинил)фенокси)этил)амино)-N,N-диметилбут-2-енамид 2,2,2-трифторацетата (Соединение 14)
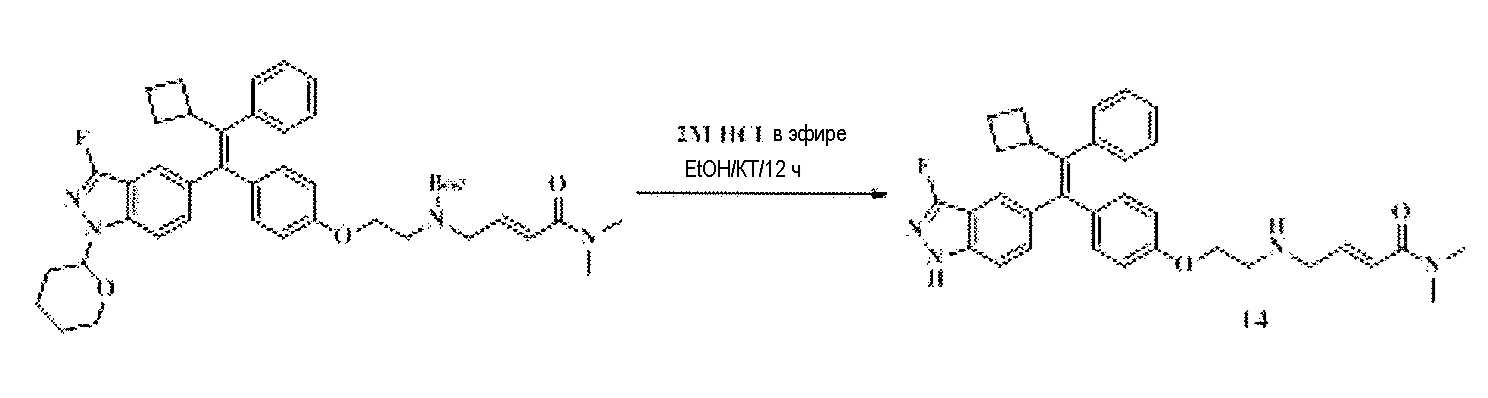
Реакцию осуществляли в соответствии со Схемой 2, Стадия-6c, с получением неочищенного соединения, которое очищали препаративной ВЭЖХ с получением соединения 14 (15 мг, 7%) в виде не совсем белого твердого вещества.
Соединение 14:1H ЯМР (400 MГц, DMSO-d6): δ 12,59 (с, 1H), 8,08 (с, 1H), 7,49-7,27 (м, 2H), 7,26-7,21 (м, 3H), 7,2-7,16 (м, 1H), 7,1 (д, J=6,8 Гц, 2H), 6,78 (д, J=14,8 Гц, 1H), 6,63 (д, J=8,8 Гц, 2H), 6,57-6,52 (м, 2H), 4,05 (т, J=4,8 Гц, 2H), 3,78 (д, J=6,0 Гц, 2H), 3,38-3,36 (м, 1H), 3,24 (с, 2H), 3,02 (с, 3H), 2,87 (с, 3H), 1,8-1,76 (м, 4H), 1,6-1,5 (м, 1H), 1,35 (м, 1H). ЖХМС: 539,3 [M+H]+.
Пример 15: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-1-морфолинобут-2-ен-1-она (Соединение 15)
Соединение синтезировали в соответствии с подходом, описанным в примере 9, с использованием на стадии-4 (E)-4-бром-1-морфолинобут-2-ен-1-она (получение показано ниже в Пр.15 Стадия-1) вместо соединения 230 с получением соединения 15 (15 мг, 3,2%) в виде белого твердого вещества.
Стадия-1: Синтез (E)-4-бром-1-морфолинобут-2-ен-1-она

Реакцию осуществляли в соответствии со Схемой 4, Стадия-2, используя морфолин вместо соединения 221, с получением (E)-4-бром-1-(пиперидин-1-ил)бут-2-ен-1-она (2,1 г, 29,6%) в виде жидкости коричневого цвета.
Соединение 15:1H ЯМР (400 MГц, DMSO-d6) δ 13,08 (с, 1H), 8,07 (с, 1H), 7,61 (с, 1H), 7,51 (д, J=6,8 Гц, 1H), 7,21-7,11 (м, 6H), 6,74 (д, J=8,8 Гц, 2H), 6,7-6,62 (м, 1H), 6,59 (д, J=8,4 Гц, 2H), 6,51 (д, J=15,2 Гц, 1H), 3,86 (т, J=5,2 Гц, 2H), 3,58-3,4 (м, 10H), 2,77 (т, J=5,2 Гц, 2H), 2,41-2,39 (м, 2H), 0,88 (т, J=7,2 Гц, 3H). ЖХМС: 537,1 [M+H]+.
Пример 16: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N-этил-N-метилбут-2-енамида (Соединение 16)
Соединение синтезировали в соответствии с подходом, описанным в примере 12, с использованием на стадии-2 (E)-4-бром-N-этил-N-метилбут-2-енамида (получение показано ниже в Пр.16 Стадия-1) вместо соединения 232 с получением соединения 16 (0,147 г, 96%).
Стадия-1: Синтез (E)-4-бром-N-этил-N-метилбут-2-енамида

Реакцию осуществляли в соответствии со Схемой 4, Стадия-2, используя (E)-4-бромбут-2-еновую кислоту (Пример 1, Стадия-7) и N-метилэтиламин, с получением неочищенного материала, который очищали колоночной хроматографией на силикагеле 230-400 меш, используя 15% этилацетат в н-гексане, с получением (E)-4-бром-N-этил-N-метилбут-2-енамида (2,8 г, 44%).
Соединение 16: 1H ЯМР (400 MГц, DMSO-d6): δ 13,06 (с, 1H), 8,06 (с, 1H), 7,61 (с, 1H), 7,51 (д, J=8,8 Гц, 1H), 7,22-7,1 (м, 6H), 6,74 (д, J=8,8 Гц, 2H), 6,6-6,65 (м, 3H), 6,46 (д, J=15,2 Гц, 1H), 3,87 (т, J=5,6 Гц, 2H), 3,36-3,35 (м, 3H), 2,96 (с, 2H), 2,82 (с, 2H), 2,79-2,76 (т, J=5,6 Гц, 2H), 2,43-2,38 (м, 2H), 1,06-0,97 (м, 3H), 0,88 (т, J=7,2 Гц, 3H). ЖХМС: 509,4 [M+H]+.
Пример 17: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N-метил-N-пропилбут-2-енамида (Соединение 17)
Соединение синтезировали в соответствии с подходом, описанным в примере 9, с использованием на стадии-4 (E)-4-бром-N-метил-N-пропилбут-2-енамида (получение показано ниже в Пр.17 Стадия-2) вместо соединения 230 с получением соединения 17 (0,015 г, 3,1%).
Стадия-1: Синтез (Z)-5-(1,2-бис(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола

Реакцию осуществляли в соответствии со Схемой 1, Стадия-3, используя 5-(бут-1-ин-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол (полученный в Пр.1, Стадия-3) вместо соединения 204. Неочищенный материал, содержащий указанное в заголовке соединение Пр.17 Стадия-1, использовали на следующей стадии без дополнительной очистки (20 г) в виде масло коричневого цвета.
Стадия-2: Синтез (E)-4-бром-N-метил-N-пропилбут-2-енамида

Реакцию осуществляли в соответствии со Схемой 4, Стадия-2, используя (E)-4-бромбут-2-еновую кислоту (полученную в Примере 1, Стадия-7.1) вместо соединения 204 и N-метилпропиламин, с получением неочищенного материала, который очищали колоночной хроматографией на силикагеле 230-400 меш, используя 15% этилацетата в н-гексане, с получением (E)-4-бром-N-метил-N-пропилбут-2-енамида (0,536 г, 23%).
Соединение 17:1H ЯМР (400 MГц, DMSO-d6): δ 13,08 (с, 1H), 8,07 (с, 1H), 7,61 (с, 1H), 7,51 (д, J=8,4 Гц, 1H), 7,22-7,19 (м, 2H), 7,18-7,11 (м, 4H), 6,75 (д, J=8,8 Гц, 2H), 6,59 (д, J=8,8 Гц, 3H), 7,5-7,44 (д, J=15,2 Гц, 1H), 3,86 (т, J=5,6 Гц, 2H), 3,32-3,30 (м, 4H), 2,96 (с, 1H), 2,83 (с, 2H), 2,67 (д, J=2,0 Гц, 2H), 2,44-2,38 (м, 2H), 1,49-1,45 (м, 2H), 0,88 (т, J=7,2 Гц, 3H), 0,80 (т, 7,2 Гц, 3H). ЖХМС: 523,2 [M+H]+.
Пример 18: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N-(2-гидроксиэтил)-N-метилбут-2-енамид (Соединение 18)
Соединение синтезировали в соответствии с подходом, описанным в примере 9, с использованием на стадии-4 (E)-4-бром-N-(2-гидроксиэтил)-N-метилбут-2-енамида (получение показано ниже в Пр.18 Стадия-1) вместо соединения 230 с получением соединения 18 (0,017 г, 3,1%).
Стадия-1: Синтез (E)-4-бром-N-(2-гидроксиэтил)-N-метилбут-2-енамида

Реакцию осуществляли в соответствии со Схемой 4, Стадия-2, используя (E)-4-бромбут-2-еновую кислоту (Пример 1, Стадия-7) вместо соединения 220 и 2-(метиламино)этан-1-ол вместо соединения 221, с получением неочищенного материала, который очищали колоночной хроматографией на силикагеле 230-400 меш, используя 30% этилацетата в н-гексане, с получением (E)-4-бром-N-(2-гидроксиэтил)-N-метилбут-2-енамида (1 г, 25%).
Соединение 18:1H ЯМР (400 MГц, DMSO-d6): δ 13,08 (с, 1H), 8,06 (с, 1H), 7,61 (с, 1H), 7,51 (д, J=8,3 Гц, 1H), 7,21 (д, J=7,3 Гц, 2H), 7,19-7,11 (м, 4H), 6,74 (д, J=8,8 Гц, 2H), 6,59 (д, J=8,8 Гц, 3H), 6,53-6,51 (м, 1H), 4,79-4,66 (м, 1H), 3,86 (т, J=5,1 Гц, 2H), 3,49 (д, J =8,8 Гц, 2H), 3,39-3,35 (м, 2H), 3,03 (с, 1H), 2,86 (с, 2H), 2,77 (с, 2H), 2,40 (д, J=7,4 Гц, 2H), 0,87 (т, J=7,3 Гц, 3H). ЖХМС: 525,2 [M+H]+.
Пример 19: Синтез (E)-4-((2-(4-((E)-2-(2-хлор-4-фторфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Пример 19)
Соединение синтезировали в соответствии с подходом, описанным в примере 14, с использованием на стадии-4 (Z)-5-(1,2-бис(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (полученного в Примере 3, Стадия-1) вместо соединения 233 и 2-хлор-4-фтор-1-иодбензола с получением соединения 19 (0,11 г, 12%) в виде не совсем белого твердого вещества.
Соединение 19:1H ЯМР (400 MГц, DMSO-d6): δ 12,6 (с, 1H), 7,6 (с, 1H), 7,5 (д, J=6,0 Гц, 1H), 7,35-7,31 (м, 2H), 7,25 (д,, J=1,2 Гц, 1H), 7,14-7,10 (м, 1H), 6,83 (д,, J=6,8 Гц, 2H), 6,63 (д,, J=7,4 Гц, 2H), 6,59 (т, J=4 Гц, 1H), 6,5 (д, J=8,8 Гц, 1H), 3,87 (т, J=4,4 Гц, 2H), 2,99 (с, 3H), 2,84 (с, 3H), 2,77 (т, J=4,4 Гц, 2H), 3,3 (м, 2H), 2,4 (кв, J=6 Гц, 2H), 0,88 (т, J=6,0 Гц, 3H). ЖХМС: 565,3 [M+H]+.
Пример 20: Примера 20 нет.
Пример 21: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N,3-триметилбут-2-енамида (Соединение 21)
Соединение синтезировали в соответствии с подходом, описанным в примере 14, с использованием на стадии-4 (Z)-5-(1,2-бис(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (полученного в Примере 17, Стадия-1) вместо соединения 233 и трет-бутил (E)-(4-(диметиламино)-2-метил-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)карбамата (получение показано ниже на стадиях 1-6) вместо соединения 234 с получением соединения 21 (35 мг, 10%).
Стадия-1: Синтез этил (E)-4-гидрокси-3-метилбут-2-еноата

К перемешиваемому раствору 1-гидроксипропан-2-она (10 г, 134 ммоль) в ацетонитриле (300 мл), добавляли этил 2- (трифенилфосфоранилиден)ацетат (56 г, 161 ммоль) и реакционную смесь нагревали до 80°C в течение 16 ч. Реакционную смесь выпаривали при пониженном давлении, разбавляли диэтиловым эфиром (200 мл), затем фильтровали с использованием воронки бюхнера, твердое вещество промывали диэтиловым эфиром (2×100 мл), фильтрат выпаривали с получением неочищенного продукта, который очищали при помощи колоночной хроматографии на силикагеле и колонку элюировали 15% EtoAc в н-гексане в качестве элюента, с получением указанного в заголовке соединения Пр.21 Стадия-1 в виде бесцветной воскообразной жидкости (13,8 г, 71%).
Стадия-2: Синтез этил (E)-4-бром-3-метилбут-2-еноата

К охлажденному льдом раствору этил (E)-4-гидрокси-3-метилбут-2-еноата (10 г, 6,9 ммоль) в ацетонитриле (20 мл), добавляли трифенилфосфин (18,2 г, 6,9 ммоль) и тетрабромид углерода (23 г, 6,9 ммоль) и реакционную смесь оставляли при комнатной температуре в течение 5 ч. Реакционную смесь концентрировали для получения неочищенного продукта, который очищали при помощи колоночной хроматографии на силикагеле и колонки, элюируя 10% EtoAc в н-гексане в качестве элюента, с получением указанного в заголовке соединения Пр.21 Стадия-2 в виде бесцветного масла (8,5 г, 59%).
Стадия-3: Синтез этил (E)-4-((2-(4-йодфенокси)этил)амино)-3-метилбут-2-еноата

Реакцию осуществляли в соответствии со Схемой 3, Стадия-3, с получением неочищенного продукта, который очищали при помощи колоночной хроматографии на силикагеле и колонку элюировали 30% EtoAc в н-гексане в качестве элюента, с получением указанного в заголовке соединения Пр.21 Стадия-3 (3,1 г, 42%).
Стадия-4: Синтез этил (E)-4-((трет-бутоксикарбонил)(2-(4-йодфенокси)-этил)амино)бут-2-еноата

Реакцию осуществляли в соответствии со Схемой 3, Стадия-4, с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле и колонке, элюируя 20% EtoAc в н-гексане в качестве элюента, с получением указанного в заголовке соединения Пр.21 Стадия-4 в виде бесцветного масла (3,8 г, 97%).
Стадия-5: Синтез (E)-4-((трет-бутоксикарбонил)(2-(4-йодфенокси)этил)амино)-3-метилбут-2-еновой кислоты
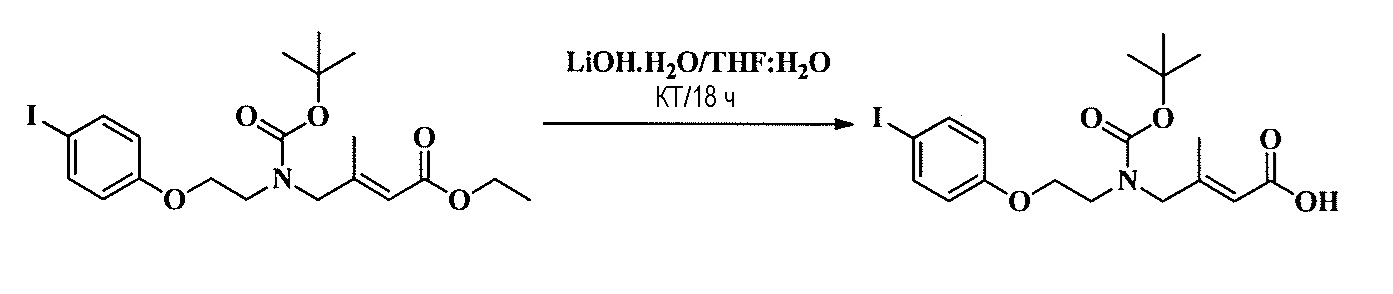
К перемешиваемому раствору этил (E)-4-((трет-бутоксикарбонил)(2-(4-йодфенокси)этил)амино)бут-2-еноата (3,8 г, 7,7 ммоль) в THF:H2O (40:10 мл), добавляли моногидрат гидроксида лития (0,65 г, 15,5 ммоль) и реакционную смесь перемешивали в течение 18 ч при комнатной температуре. К реакционной смеси добавляли воду, промывали диэтиловым эфиром (100 мл), водный слой доводили до рН 2-3 с использованием 10% лимонной кислоты, затем экстрагировали дихлорметаном (2×100 мл), органический слой сушили над безводным сульфатом натрия, концентрировали с получением указанного в заголовке соединения Пр.21 Стадия-5 (1,7 г, 48%).
Стадия-6: Синтез трет-бутил (E)-(4-(диметиламино)-2-метил-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)карбамата

Реакцию проводили, как описано в примере 1, с использованием на стадии-7.2 (E)-4-((трет-бутоксикарбонил)(2-(4-йодфенокси)этил)амино)-3-метилбут-2-еновой кислоты вместо соединения 235 и использованием DMA вместо CH2Cl2 в качестве растворителя с получением указанного в заголовке соединения Пр.21 Стадия-6 (2,5 г, неочищенный).
Соединение 21:1H ЯМР (400 MГц, DMSO-d6): δ 13,08 (с, 1H), 8,07 (с, 1H), 7,61 (с, 1H), 7,51 (д, J=8,8 Гц, 1H), 7,22-7,21 (м, 2H), 7,19-7,11 (м, 4H), 6,74 (д, J=8,4 Гц, 2H), 6,59 (д, J=8,8 Гц, 2H), 6,04 (с, 1H), 3,87 (т, J =5,2 Гц, 2H), 3,14 (с, 2H), 2,9 (с, 3H), 2,8(с, 3H), 2,74 (т, J=5,2 Гц, 2H), 2,43-2,38 (м, 2H), 1,75 (с, 3H), 0,88 (т, J=6,8 Гц, 3H). ЖХМС: 509,3[M+H]+.
Пример 22: Синтез (Z)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 22)
Соединение синтезировали в соответствии с подходом, описанным в примере 14, с использованием на стадии-4 (Z)-5-(1,2-бис(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (полученного в Примере 17, Стадия-1) вместо соединения 233 и трет-бутил (Z)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)карбамата (получение показано ниже в Пр.22 Стадии 1-7) вместо соединения 234 с получением соединения 22 (0,27 г, 38%).
Стадия-1: Синтез 1-(2-бромэтокси)-4-иодбензола

К перемешиваемому раствору 4-иодфенола (20 г, 90 ммоль) добавляли карбонат калия (9,4 г, 136 ммоль) и перемешивали в течение 10 мин при 0°C, к вышеуказанной смеси добавляли 1,2-дибромэтан (145 г, 772 ммоль). Содержимое перемешивали при 130°C в течение 22 ч. После завершения реакции, реакционную смесь выливали в ледяную воду, и экстрагировали этилацетатом. Объединенные органические слои промывали водой, затем насыщенным раствором NaCl, сушили над безводным Na2SO4и концентрировали при пониженном давлении. Неочищенный материал очищали при помощи колоночной хроматографии на силикагеле 230-400 меш с использованием 3% EtOAc в н-гексане в качестве элюента с получением желаемого соединения 1-(2-бромэтокси)-4-иодбензола (20,1 г, 67,6%).
Стадия-2: Синтез 2-((2-(4-йодфенокси)этил)амино)этан-1-ола

К перемешиваемому раствору 1-(2-бромэтокси)-4-иодбензола (20 г, 61 ммоль) в DMF (100 мл) добавляли 2-аминоэтан-1-ол (37,36 г, 610 ммоль). Реакционную смесь перемешивали в течение 1 ч при 60°C, после завершения реакции (контролируемой ТСХ), реакционную смесь разбавляли холодной водой (50 мл) и экстрагировали этилацетатом. Объединенные органические слои промывали водой, затем насыщенным раствором NaCl, сушили над безводным Na2SO4и концентрировали при пониженном давлении. Неочищенный материал очищали при помощи колоночной хроматографии на силикагеле 230-400 меш с использованием 20% MeOH в дихлорметане в качестве элюента с получением желаемого соединения 2-((2-(4-йодфенокси)этил)амино)этан-1-ол (13,2 г, 70,58%).
Стадия-3: Синтез трет-бутил (2-гидроксиэтил)(2-(4-йодфенокси)этил)карбамата

Реакцию осуществляли в соответствии со Схемой 4, Стадия-4 с получением неочищенного материала, который очищали колоночной хроматографией на силикагеле 230-400 меш, используя МеОН в дихлорметане в качестве элюента, с получением желаемого соединения трет-бутил (2-гидроксиэтил)(2-(4-йодфенокси)этил)карбамата (16,7 г, 95,9%).
Стадия-4: Синтез трет-бутил (2-(4-йодфенокси)этил)(2-оксоэтил)карбамата

К перемешиваемому раствору трет-бутил (2-гидроксиэтил)(2-(4-йодфенокси)этил)карбамата (6,7 г, 16,4 ммоль) в дихлорметане (150 мл) добавляли периодинан Десса-Мартина (10,46 г, 24,6 ммоль). Реакционную смесь перемешивали в течение 1 ч при комнатной температуре, после завершения реакции (контролируемой ТСХ), реакционную смесь разбавляли смесью 1:1 hypo и раствора NaHCO3и экстрагировали дихлорметаном (100 мл). Органический слой промывали водой, затем насыщенным раствором NaCl, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт использовали на следующей стадии без дополнительной очистки (5,9 г, 89,9%).
Стадия-5: Синтез этил этил (Z)-4-((трет-бутоксикарбонил)(2-(4-йодфенокси)этил)амино)бут-2-еноата

К перемешиваемому раствору этил 2-(дифеноксифосфорил)ацетата (4,71 г, 14,7 ммоль) и тритон-B (9,39 мл, 17,7 ммоль) в THF (70 мл) при -78°C, добавляли по каплям трет-бутил (2-(4-йодфенокси)этил)(2-оксоэтил)карбамат (5,97 г, 14,7 ммоль) в THF (30 мл). Реакционную смесь перемешивали в течение 1 ч при 10°C, после завершения реакции (контролируемой ТСХ), реакционную смесь разбавляли раствором хлорида аммония и экстрагировали EtOAc (100 мл). Органический слой промывали водой, затем насыщенным раствором NaCl, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле 230-400 меш, элюируя 9% EtOAc в н-гексане, с получением указанного в заголовке соединения Пр.22 Стадия-5 (0,82 г, 11,7%).
Стадия-6: Синтез (Z)-4-((трет-бутоксикарбонил)(2-(4-йодфенокси)этил)амино)бут-2-еновой кислоты

К перемешиваемому раствору этил (Z)-4-((трет-бутоксикарбонил)(2-(4-йодфенокси)этил)амино)бут-2-еноата (0,8 г, 1,6 ммоль) в THF (10 мл) добавляли LiOH.H2O (0,353 г, 8,4 ммоль) и воду (5 мл). Реакционную смесь перемешивали в течение 72 ч при комнатной температуре, после завершения реакции (контролируемой ТСХ), реакционную смесь разбавляли водой и экстрагировали дихлорметаном (100 мл). Органический слой промывали водой, затем насыщенным раствором NaCl, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт использовали на следующей стадии без дополнительной очистки (0,705 г, 93,7%).
Стадия-7: Синтез трет-бутил (Z)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)карбамата

Реакцию осуществляли, как описано в примере 1, Стадия-7.2, с использованием (Z)-4-((трет-бутоксикарбонил)(2-(4-йодфенокси)этил)амино)бут-2-еновой кислоты вместо соединения 235 и используя DMF вместо CH2Cl2 в качестве растворителя, с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле на 230-400 меш, элюируя 40-70% EtOAc в н-гексане, с получением указанного в заголовке соединения Пр. 22 Стадия-7 (0,97 г, 70,8%).
Соединение 22:1H ЯМР (400 MГц, DMSO-d6): δ 13,06 (с, 1H), 8,06 (с, 1H), 7,61 (с, 1H), 7,51 (д, J=8,8 Гц, 2H), 7,12-7,10 (м, 4H), 674 (д, J=8,8 Гц, 2H), 6,58 (д, J=8,8 Гц, 2H), 6,15 (д, J=11,6 Гц, 2H), 5,93-5,86 (м, 1H), 3,84 (т, J=5,4 Гц, 2H), 3,40 (дд, J1= 6,4 Гц, J2= 1,6 Гц, 2H), 2,93 (с, 3H), 2,80 (с, 3H), 2,77 (т, J=5,6 Гц, 2H), 2,40 (кв, J=7,3 Гц, 2H), 0,88 (т, J=7,2 Гц, 3H). ЖХМС: 495,3 [M+H]+.
Пример 23: Синтез (E)-4-((2-(4-((E)-2-циклобутил-1-(1H-индазол-5-ил)-2-фенилвинил)фенокси)этил)амино)-1-морфолинобут-2-ен-1-она (Соединение 23)
Реакцию осуществляли в соответствии с подходом, описанным в примере 2, Стадия-7, с использованием в Пр.2, Стадия-7, (E)-2-(4-(2-циклобутил-1-(1H-индазол-5-ил)-2-фенилвинил)фенокси)этанамина (полученного в Примере 13, Стадия-7) вместо соединения 237 и (E)-4-бром-1-морфолинобут-2-ен-1-она (полученного в Примере 15, Стадия-1) вместо соединения 238 с получением соединения 23 (0,045 г, 5%).
Соединение 23:1H ЯМР (400 MГц, DMSO-d6): δ 13,06 (с, 1H), 8,07 (с, 1H), 7,58 (с, 1H), 7,51 (д, J=8,8 Гц, 1H), 7,27-7,23 (м, 2H), 7,17-7,13 (м, 2H), 7,11-7,08 (м, 2H), 6,77 (д, J=8,8 Гц, 2H), 6,66-6,61 (м, 1H), 6,53 (д, J=8,8 Гц, 3H), 3,82 (т, J =5,6 Гц, 2H), 3,51-3,49 (м, 8H), 3,40 (т, J=8,5 Гц, 1H), 3,29 (с, 2H), 2,74 (т, J=5,7 Гц, 2H), 1,82-1,76 (м, 4H), 1,57-1,54 (м, 1H), 1,23 (м, 1H). ЖХМС: 563,3 [M+H]+.
Пример 24: Синтез (E)-4-((2-(4-((E)-2-циклобутил-1-(4-фтор-1H-индазол-5-ил)-2-фенилвинил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 24)
Соединение синтезировали в соответствии с подходом, описанным в примере 14, с использованием на стадии-1 5-бром-4-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (получение показано ниже в Пр.24 Стадия-1) вместо соединения 240 с получением соединения 24 (0,03 г, 5%).
Стадия-1: Синтез 5-бром-4-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола

Реакцию осуществляли в соответствии со Схемой 1, Стадия-1, с получением неочищенного материала, который очищали при помощи колоночной хроматографии на силикагеле 230-400 меш, используя 3-4% этилацетата в н-гексане, с получением 5-бром-4-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (5 г, 72%) в виде коричневого масла.
Соединение 24:1H ЯМР (400 MГц, DMSO-d6): δ 13,4 (с, 1H), 8,2 (с, 1H), 7,39 (д, J=8,8 Гц, 1H), 7,29-2,26 (м 2H), 7,2-7,16 (м, 2H), 7,11 (д, J=7,2 Гц, 2H), 6,8 (д, J=8,8 Гц, 2H), 6,6-6,54 (м, 3H), 6,52 (д, J=16,0 Гц, 1H), 3,84 (т, J=5,6 Гц, 2H), 3,3-3,26 (м, 3H), 2,97 (с, 3H), 2,83 (с, 3H), 2,76 (т, J=5,6 Гц, 2H), 1,8-1,7 (м, 4H), 1,56-1,5 (м, 1H), 1,34-1,3 (м, 1H). ЖХМС: 539,3 [M+H]+.
Пример 25: Синтез (E)-4-((2-((5-((Z)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)этил)амино)-N,N-диметилбут-2-енамида
Соединение синтезировали в соответствии с подходом, описанным в примере 14, с использованием на стадии-4 (Z)-5-(1,2-бис(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (полученного в Примере 17, Стадия-1) вместо соединения 233 и трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-((5-йодпиридин-2-ил)окси)этил)карбамата (получение показано ниже в Пр.25 Стадии 1-2) вместо соединения 234, с получением соединения 25 (80 мг, 18%) в виде не совсем белого твердого вещества.
Стадия-1: Синтез трет-бутил (2-((5-йодпиридин-2-ил)окси)этил)карбамата

К перемешиваемому раствору 2-фтор-5-йодпиридина (5 г, 22,4 ммоль) в DMF (25 мл) добавляли гидрид натрия (0,7 г, 33,5 ммоль) и перемешивали в течение 10 мин при 0°C, к вышеуказанной смеси добавляли трет-бутил (2-гидроксиэтил)карбамат (1,8 г, 11,2 ммоль). Содержимое перемешивали при 0°C в течение 30 мин. После завершения реакции, реакционную смесь выливали в ледяную воду, и экстрагировали этилацетатом. Объединенные органические слои промывали водой, затем насыщенным раствором NaCl, сушили над безводным Na2SO4и концентрировали при пониженном давлении. Неочищенный материал очищали при помощи колоночной хроматографии на силикагеле 230-400 меш, с использованием 15% EtOAc в н-гексане в качестве элюента, с получением желаемого соединения трет-бутил (2-((5-йодпиридин-2-ил)окси)этил)карбамата в виде не совсем белого твердого вещества (3,5 г, 43%).
Стадия-2: Синтез трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-((5-йодпиридин-2-ил)окси)этил)карбамата

Реакцию осуществляли в соответствии с подходом, описанным в примере 14, Стадия-3.1, с использованием в Пр.14, Стадия-3.1, трет-бутил (2-((5-йодпиридин-2-ил)окси)этил)карбамата вместо соединения 241 с получением указанного в заголовке соединения Пр.25 Стадия-2 (3,6 г, 47%).
Соединение 25:1H ЯМР (400 MГц, DMSO-d6): δ 13,1 (с, 1H), 8,1 (с, 1H), 7,65 (с, 1H), 7,58 (д, J=2,0 Гц, 1H), 7,54 (д, J=8,0 Гц, 1H), 7,23 (д, J=6,8 Гц, 2H), 7,17-7,12 (м, 5H), 6,6-6,5 (м, 1H), 6,5-6,47 (м, 2H), 4,1 (т, J=5,6 Гц, 2H), 3,3 (м, 2H), 2,98 (с, 3H), 2,84 (с, 3H), 2,75 (т, J=5,6 Гц, 2H), 2,46-2,40 (м, 2H), 0,89 (т, J=7,2 Гц, 3H). ЖХМС: 496,3 [M+H]+.
Пример 26: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-(o-толил)бут-1-ен-1-ил)фенокси)этил)амино)-N-(2-гидроксиэтил)-N-метилбут-2-енамида (Соединение 26)
Стадия-1: Синтез (E)-5-(1-(4-(2-хлорэтокси)фенил)-2-(o-толил)бут-1-ен-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола

Те же способы, как описано в примере 5, с использованием на стадии-1 (i) 5-(бут-1-ин-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (полученного в Примере 1, Стадия-3) вместо соединения 242, (ii) 1-(2-хлорэтокси)-4-иодбензола вместо соединения 243, и (iii) 1-иод-2-метилбензола вместо соединения 244, с получением неочищенного материала, который очищали при помощи колоночной хроматографии на силикагеле 230-400 меш с использованием 5% EtOAc в н-гексане с получением указанного в заголовке соединения Пр.26 Стадия-1 (2,9 г, 29,8%) в виде не совсем белого твердого вещества.
Стадия-2: Синтез (E)-2-((2-(4-(1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(o-толил)бут-1-ен-1-ил)фенокси)этил)амино)этан-1-ола

К перемешиваемому раствору (E)-5-(1-(4-(2-хлорэтокси)фенил)-2-(o-толил)бут-1-ен-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (2,8 г, 5,6 ммоль) в 2-метоксиэтаноле (28 мл) добавляли 2-аминоэтан-1-ол (3,3 мл, 56 ммоль). Реакционную смесь перемешивали в течение 16 ч при 135°C, после завершения реакции (контролируемой ТСХ), реакционную смесь разбавляли холодной водой (50 мл) и экстрагировали этилацетатом. Объединенные органические слои промывали водой, затем насыщенным раствором NaCl, сушили над безводным Na2SO4и концентрировали при пониженном давлении. Неочищенный продукт использовали на следующей стадии без дополнительной очистки (3,2 г, неочищенный).
Стадия-3: Синтез трет-бутил (E)-(2-гидроксиэтил)(2-(4-(1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(o-толил)бут-1-ен-1-ил)фенокси)этил)карбамата

Реакцию осуществляли в соответствии со Схемой 3, Стадия-4, с получением неочищенного продукта, который использовали на следующей стадии без дополнительной очистки (3,8 г, неочищенный).
Стадия-4: Синтез трет-бутил (E)-(2-оксоэтил)(2-(4-(1-(1-(тетрагидро-2H-пиран-2-ил)-1H-индазол-5-ил)-2-(o-толил)бут-1-ен-1-ил)фенокси)этил)карбамата

Реакцию осуществляли, как описано в примере 22, Стадия-4, используя соединение 306 вместо соединения 245, с получением неочищенного продукта, который очищали при помощи колоночной хроматографии на силикагеле на 100-200 меш, элюируя 30% EtOAc в н-гексане, с получением указанного в заголовке соединения Пр.26 Стадия-4 (1,5 г, 40%).
Стадия-5: Синтез этил (E)-4-((трет-бутоксикарбонил)(2-(4-((E)-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(o-толил)бут-1-ен-1-ил)фенокси)этил)амино)бут-2-еноата

Реакцию осуществляли, как описано в примере 22, Стадия-5, используя соединение 248 вместо соединения 246 и используя соединение 249 вместо соединения 247, с получением неочищенного продукта, который очищали при помощи колоночной хроматографии на силикагеле 230-400 меш, элюируя 20% EtOAc в н-гексане, с получением указанного в заголовке соединения Пр.26 Стадия-5 (1,37 г, 82%).
Стадия-6: Синтез (E)-4-((трет-бутоксикарбонил)(2-(4-((E)-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(o-толил)бут-1-ен-1-ил)фенокси)этил)амино)бут-2-еновой кислоты
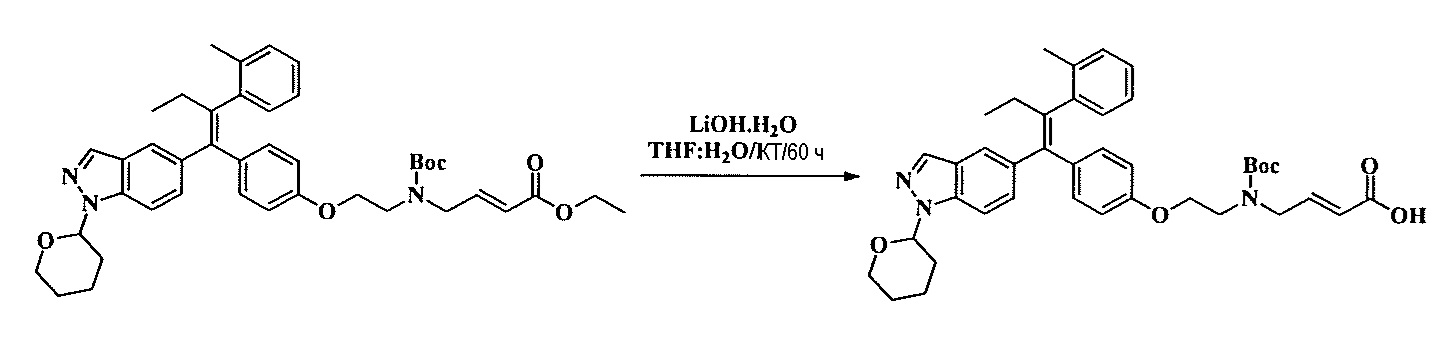
К перемешиваемому раствору этил (E)-4-((трет-бутоксикарбонил)(2-(4-((E)-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(o-толил)бут-1-ен-1-ил)фенокси)этил)амино)бут-2-еноата (0,38 г, 0,54 ммоль) в THF (16 мл) добавляли LiOH.H2O (0,15 г, 3,78 ммоль) и воду (4 мл). Реакционную смесь перемешивали в течение 60 ч при комнатной температуре, после завершения реакции (контролируемой ТСХ), реакционную смесь разбавляли водой и экстрагировали дихлорметаном (100 мл). Органический слой промывали водой, затем насыщенным раствором NaCl, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью combi-flash, с получением указанного в заголовке соединения Пр. 26 Стадия-7 (0,33 г, 91%).
Стадия-8: Синтез трет-бутил ((E)-4-((2-гидроксиэтил)(метил)амино)-4-оксобут-2-ен-1-ил)(2-(4-((E)-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(o-толил)бут-1-ен-1-ил)фенокси)этил)карбамата

К перемешиваемому раствору (E)-4-((трет-бутоксикарбонил)(2-(4-((E)-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(o-толил)бут-1-ен-1-ил)фенокси)этил)амино)бут-2-еновой кислоты (0,33 г, 0,49 ммоль) в DMA (14 мл) добавляли DIPEA (0,25 мл, 1,47 ммоль), HATU (0,18 г, 0,49 ммоль) и 2-(метиламино)этан-1-ол (0,036 г, 0,49 ммоль). Реакционную смесь перемешивали в течение 3 ч при комнатной температуре, после завершения реакции (контролируемой ТСХ), реакционную смесь разбавляли водой и экстрагировали дихлорметаном (100 мл). Органический слой промывали водой, затем насыщенным раствором NaCl, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле 230-400 меш, элюируя 3% MeOH в дихлорметане, с получением указанного в заголовке соединения Пр.26 Стадия-8 (0,4 г, неочищенный).
Стадия-9: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-(o-толил)бут-1-ен-1-ил)фенокси)этил)амино)-N-(2-гидроксиэтил)-N-метилбут-2-енамида (Соединение 26)

Реакцию осуществляли в соответствии со Схемой 2, Стадия-6c, используя соединение 250 вместо соединения 211c, с получением неочищенного соединения, которое очищали препаративной ВЭЖХ с получением соединения 26 (20 мг).
Соединение 26:1H ЯМР (400 MГц, DMSO-d6): δ 13,07 (с, 1H), 8,08 (с, 1H), 7,65 (с, 1H), 7,53 (д, J=8,4 Гц, 1H), 7,23 (д, J=8,8 Гц, 1H), 7,16-7,12 (м, 2H), 7,09-7,03 (м, 3H), 6,74 (д, J=8,8 Гц, 2H), 6,63-6,46 (м, 4H), 4,76-4,64 (м, 1H), 3,84 (т, J=5,2 Гц, 2H), 3,50-3,47 (м, 2H), 3,39-3,36 (м, 2H), 3,02 (с, 4H), 2,86 (с, 1H), 2,75-2,67(м, 1H), 2,33-2,30 (м, 2H), 2,10 (с, 3H), 2,0 (шир.с, 1H), 0,86 (т, J =7,4 Гц, 3H). ЖХМС: 539,4 [M+H]+.
Пример 27: Примера 27 нет.
Пример 28: Синтез (E)-4-((2-(4-((E)-2-(2-хлор-4-фторфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N-(2-гидроксиэтил)-N-метилбут-2-енамида (Соединение 28)
Стадия-1: Синтез (E)-4-((2-(4-((E)-2-(2-хлор-4-фторфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N-(2-гидроксиэтил)-N-метилбут-2-енамида

Соединение синтезировали, следуя подходу, описанному в примере 3, (i) с использованием на стадии-4 2-хлор-4-фтор-1-иодбензола вместо соединения 227 и (ii) с использованием на Стадии-6 Пр.3 (E)-4-бром-N-(2-гидроксиэтил)-N-метилбут-2-енамида (полученного в Примере 18, Стадия-1) вместо соединения 251, с получением указанного в заголовке соединения в виде неочищенного (0,5 г) и без дополнительной очистки.
Стадия-2: Синтез трет-бутил (2-(4-((E)-2-(2-хлор-4-фторфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)((E)-4-((2-гидроксиэтил)(метил)амино)-4-оксобут-2-ен-1-ил)карбамата

Реакцию осуществляли в соответствии со Схемой 2, Стадия-5, с получением неочищенного соединения, которое очищали препаративной ВЭЖХ с получением очищенного указанного в заголовке соединения Стадии-2 Пр.28 (0,28 г, 48%).
Стадия-3: Синтез (E)-4-((2-(4-((E)-2-(2-хлор-4-фторфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N-(2-гидроксиэтил)-N-метилбут-2-енамида (Соединение 28)

Реакцию осуществляли в соответствии со Схемой 2, Стадия-6b, с получением неочищенного продукта, который очищали препаративной ВЭЖХ с получением соединения 28 (0,025 г, 11%).
Соединение 28:1H ЯМР (400 MГц, DMSO-d6): δ 12,59 (с, 1H), 7,54 (с, 1H), 7,49 (д, J=8,8 Гц, 1H), 7,34-7,31 (м, 2H), 7,24 (д, J=8,8 Гц, 1H), 7,14-7,11 (м, 1H), 6,83 (д, J=8,4 Гц, 2H), 6,63 (д, J=8,3 Гц, 2H), 6,59-6,52 (м, 2H), 4,76-4,64 (м, 1H), 3,87 (т, J=4,7 Гц, 2H), 3,50-3,46 (м, 2H), 3,40-3,38 (м, 4H), 3,03 (с, 1H), 2,86- 2,81 (м, 4H), 2,36-2,33 (м, 2H), 0,88 (т, J=7,3 Гц, 3H). ЖХМС: 595,2 [M+H]+.
Пример 29: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-(4-изопропилфенил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 29)
Стадия-1: Синтез трет-бутил (2-(4-((E)-1-(1H-индазол-5-ил)-2-(4-изопропилфенил)бут-1-ен-1-ил)фенокси)этил)((E)-4-(диметиламино)-4-оксобут-2-ен-1-ил)карбамата

Соединение синтезировали в соответствии с подходом, описанным в примере 3, с использованием (i) на стадии-1 5-(бут-1-ин-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (полученного в примере 1, Стадия-3) вместо соединения 252 и (ii) на стадии-4 4-изопропилйодбензола вместо соединения 227. Полученное промежуточное соединение подвергали условиям реакции, описанным на схеме 2, стадия-5, с получением неочищенного продукта, который очищали при помощи колоночной хроматографии на силикагеле 230-400 меш, колонку элюировали 2% MeOH в дихлорметане в качестве элюента с получением указанного в заголовке соединения Пр.29 Стадии-1 (0,096 г, 29%).
Стадия-2: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-(4-изопропилфенил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 29)

Реакцию осуществляли в соответствии со Схемой 2, Стадия-6c, с получением неочищенного соединения, которое очищали при помощи препаративной ВЭЖХ с получением соединения 29 (0,010 г, 12%).
Соединение 29:1H ЯМР (400 MГц, DMSO-d6): δ 13,05 (с, 1H), 8,06 (с, 1H), 7,6 (с, 1H), 7,5 (д, J=8,8 Гц, 1H), 7,1-7,05 (м, 5H), 6,74 (д, J=8,8 Гц, 2H), 6,63-6,58 (м, 3H), 6,5 (д, J=15,2 Гц, 1H), 3,66 (т, J=5,6 Гц, 2H), 3,31 (м, 2H), 2,98 (с, 3H), 2,84 (с, 3H), 2,82-2,76 (м, 3H), 2,4-2,35 (м, 2H), 1,16 (д, J=6,8 Гц, 6H), 0,88 (т, J=7,3 Гц, 3H). ЖХМС: 537,4 [M+H]+.
Пример 30: Синтез (E)-4-((2-(4-((E)-4-хлор-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 30)
Соединение синтезировали в соответствии с подходом, описанным в примере 5, с использованием на стадии-1 5-(4-хлорбут-1-ин-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (получение показано ниже в Пр.30 Стадии 1-2) вместо соединения 242, трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)карбамата (полученного в Примере 14, Стадия-3) вместо соединения 243, и иодбензола вместо соединения 244 и продолжая Пр.5 Стадия-2, с получением соединения 30 (0,04 г, 6%).
Стадия-1: Синтез 4-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-3-ин-1-ола

Реакцию осуществляли в соответствии со Схемой 1, Стадия-2, используя 5-бром-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол (Пример 1, Стадия-3) вместо соединения 202 и бут-3-ин-1-ол вместо соединения 203, с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле, используя 15% этилацетата в н-гексане, с получением указанного в заголовке соединения Пр.30 Стадия-1 (1,3 г, 54%).
Стадия-2: Синтез 5-(4-хлорбут-1-ин-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола

К ледяному раствору 4-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-3-ин-1-ола (2,6 г, 9,6 ммоль) в пиридине (36 мл), добавляли POCl3. Реакционную смесь доводили до комнатной температуры в течение 16 ч. По завершении реакции избыток POCl3 удаляли в вакууме, подщелачивали раствором бикарбоната натрия, экстрагировали этилацетатом (2×100 мл), объединенные органические слои промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия. Органический слой концентрировали досуха с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле, колонку элюировали 10% EtOAc в н-гексане с получением указанного в заголовке соединения Пр.30 Стадия-2 (1,5 г, 54%).
Соединение 30:1H ЯМР(400 MГц, DMSO-d6): δ 13,1 (с, 1H), 8,08 (с, 1H), 7,7(с, 1H), 7,53 (д, J=8,8 Гц, 1H), 7,26-7,16 (м, 6H), 6,77 (д, J=8,8 Гц, 2H), 6,6 (д, J=8,0 Гц, 3H), 6,5 (т, J=5,2 Гц, 1H), 3,87 (д, J=5,6 Гц, 2H), 3,46 (т, J=6,8 Гц, 2H), 3,3 (м, 2H), 2,98 (с, 3H), 2,89 (т, J=7,2 Гц, 2H), 2,84 (с, 3H), 2,77 (т, J=5,2 Гц, 2H). ЖХМС: 529,3[M+H]+.
Пример 31: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-(4-изопропилфенил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 31)
Соединение синтезировали в соответствии с подходом, описанным в примере 3, с использованием на стадии-4 (Z)-трет-бутил (2-(4-(1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)фенокси)этил)карбамата (полученного в Примере 6, Стадия-1) вместо соединения 226 и 2-иод-1-метоксибензола вместо соединения 227, с получением соединения 31 (0,014 г, 3%).
Соединение 31:1H ЯМР (400 MГц, DMSO-d6): δ 13,05 (с, 1H), 8,07 (с, 1H), 7,6 (с, 1H), 7,5 (д, J=8,4 Гц, 1H), 7,15-7,11 (м, 2H), 6,94 (д, J=8,4 Гц, 1H), 6,87 (д, J=7,2 Гц, 1H), 6,78-6,72 (м, 3H), 6,58-6,54 (м, 3H), 6,5 (д, J=15,2 Гц, 1H), 3,85 (т, J=5,6 Гц, 2H), 3,77 (с, 3H), 3,31 (м, 2H), 2,98 (с, 3H), 2,84 (с, 3H), 2,76 (т, J=5,6 Гц, 2H), 2,33 (с, 2H), 0,88 (т, J=7,2 Гц, 3H). ЖХМС: 525,3 [M+H]+.
Пример 32: Синтез (E)-4-((2-(4-((E)-2-(2-(дифторметокси)фенил)-1-(1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 32)
Соединение синтезировали в соответствии с подходом, описанным в примере 3, с использованием на стадии-4 (Z)-трет-бутил (2-(4-(1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)фенокси)этил)карбамата (полученного в Примере 6, Стадия-1) вместо соединения 226 и 2-иод-1-дифторметоксибензола вместо соединения 227, с получением соединения 32 (0,022 г, 4%).
Соединение 32:1H-ЯМР(400 MГц, DMSO-d6): δ 13,08 (с, 1H), 8,09 (с, 1H), 7,62 (с, 1H), 7,53 (д, J=8,8 Гц, 1H), 7,25-7,23 (м, 1H), 7,22-7,08 (м, 5H), 6,78 (д, J=8,8 Гц, 2H), 6,62-6,57 (м, 3H), 6,5 (д, J=15,2 Гц, 1H), 3,85 (т, J=5,2 Гц, 2H), 3,32-3,3 (м, 2H), 2,98 (с, 3H), 2,84 (с, 3H), 2,77 (т, J=5,2 Гц, 2H), 2,33 (м, 2H), 0,87 (т, J=7,2 Гц, 3H), ЖХ-МС: 561,3[M+H]+.
Пример 33: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-(2-(трифтор-метокси)фенил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 33)
Соединение синтезировали в соответствии с подходом, описанным в примере 3, используя на стадии-4 трет-бутил ((E)-4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-((Z)-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)фенокси)этил)карбамат (получение показано ниже в Пр.33 Стадия-1) вместо соединения 226 и 2-иод-1-трифторметоксибензол вместо соединения 227 и продолжая Пр.3 Стадию-5, с получением соединения 33 (0,030 г, 26%).
Стадия-1: Синтез трет-бутил ((E)-4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-((Z)-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)фенокси)этил)карбамата

Реакцию осуществляли в соответствии со Схемой 1, Стадия-4, используя (Z)-5-(1,2-бис(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол (полученный в Примере 17, Стадия-1) вместо соединения 206 и трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)карбамат (полученный в Примере 14, Стадия-3) вместо соединения 207, с получением неочищенного материала, который очищали колоночной хроматографией на силикагеле 230-400 меш, используя MeOH в дихлорметане (1,6:98,4), с получением указанного в заголовке соединения Пр.33 Стадия-1 (4,04 г, 43%).
Соединение 33:1H-ЯМР(400 MГц, DMSO-d6): δ 13,10 (с, 1H), 8,1 (с, 1H), 7,6 (с, 1H), 7,5 (д, J=8,8 Гц, 1H), 7,42-7,39 (м, 1H), 7,38-7,26 (м, 2H), 7,22-7,18 (м, 1H), 7,13-7,11 (м, 1H), 6,74 (д, J=8,80 Гц, 2H), 6,62-6,58 (м, 3H), 6,51-6,48 (д, J=15,2 Гц, 1H), 3,86 (т, J=5,6 Гц, 2H), 3,31-3,3 (м, 2H), 2,98 (с, 3H), 2,84 (с, 3H), 2,76 (т, J=5,6 Гц, 2H), 2,43-2,42 (м, 2H), 0,86 (т, J=7,60 Гц, 3H). ЖХМС: 579,0[M+H]+.
Пример 34: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-(2-изопропилфенил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 34)
Соединение синтезировали в соответствии с подходом, описанным в примере 12, с использованием на стадии-2 (E)-2-(4-(1-(1H-индазол-5-ил)-2-(2-изопропилфенил)бут-1-ен-1-ил)фенокси)этан-1-амина (получение показано ниже в Пр.34 Стадия-1)вместо соединения 253 и (E)-4-бром-N,N-диметилбут-2-енамида (полученного в Примере 1, Стадия-7) вместо соединения 232, с получением соединения 34 (0,010 г, 6%).
Стадия-1: Синтез трет-бутил (E)-(2-(4-(2-(2-изопропилфенил)-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)карбамата

Соединение синтезировали в соответствии с подходом, описанным в примере 7, используя 2-иод-1-изопропилбензол на стадии-1, продолжая стадию-2, и непосредственно переходя на стадию 4, с получением указанного в заголовке соединения (0,5 г, 98%).
Соединение 34:1H-ЯМР(400 MГц,DMSO-d6): δ 13,07 (с, 1H), 8,10 (с, 1H), 7,64 (с, 1H), 7,53 (д, J=8,8 Гц, 1H), 7,24 (д, J=6,8 Гц, 1H), 7,19-7,14 (м, 4H), 6,72 (д, J=8,8 Гц, 2H), 6,6-6,46 (м, 4H), 3,84 (т, J=5,6 Гц, 2H), 3,30 (м, 2H), 3,17 (т, J=6,80 Гц, 1H), 2,98 (с, 3H), 2,84 (с, 3H), 2,75 (т, J=5,6 Гц, 1H), 2,67 (т, J=6,0 Гц, 1H), 2,36-2,30 (м, 2H), 1,13 (д, J=6,8 Гц, 3H), 0,86 (т, J=7,60 Гц, 3H), 0,61 (д, J=6,80 Гц, 3H), LC-MS: 537,3[M+H]+.
Пример 35: Синтез (E)-4-((2-(4-((E)-2-(2-этилфенил)-1-(1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 35)
Соединение синтезировали в соответствии с подходом, описанным в примере 5, используя на стадии-1 трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)карбамат (полученный в Примере 14, Стадия-3) вместо соединения 243 и 2-иод-1-метилбензол вместо соединения 244 и продолжая Стадию-2, с получением соединения 35 (0,079 г, 10%).
Соединение 35:1H ЯМР(400 MГц, DMSO-d6): δ 13,06 (с, 1H), 8,08 (с, 1H), 7,64 (с, 1H), 7,53 (д, J=8,4 Гц, 1H), 7,2-7,13 (м, 5H), 6,73 (д, J=8,8 Гц, 2H), 6,6-6,47 (м, 4H), 3,84 (т, J=5,6 Гц, 2H), 3,3-3,29 (м, 2H), 2,98 (с, 3H), 2,84 (с, 3H), 2,75 (т, J=5,6 Гц, 2H), 2,67-2,63 (м, 1H), 2,41-2,26 (м, 3H), 1,02 (т, J=7,6 Гц, 3H), 0,86 (т, J=7,6 Гц, 3H) ЖХМС: 523,3[M+H]+.
Пример 36: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-(o-толил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 36)
Стадия-1: Синтез трет-бутил (2-(4-((E)-1-(1H-индазол-5-ил)-2-(o-толил)бут-1-ен-1-ил)фенокси)этил)((E)-4-(диметиламино)-4-оксобут-2-ен-1-ил)карбамата

Соединение синтезировали в соответствии с подходом, описанным в примере 3, с использованием на стадии-4 (Z)-трет-бутил (2-(4-(1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)фенокси)этил)карбамата (полученного в Примере 6, Стадия-1) вместо соединения 226 и 2-иод-1-метилбензола вместо соединения 227. Полученное промежуточное соединение подвергали условиям реакции, описанным в схеме 2, Стадия-5, с получением неочищенного продукта, который очищали при помощи хроматографии на силикагеле с использованием 2% MeOH в дихлорметане в качестве элюента, с получением указанного в заголовке соединения Пр.36 Стадия-1 (0,12 г, 27%).
Стадия-2: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-(o-толил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 36)

Реакцию осуществляли в соответствии со Схемой 2, Стадия-6c, с получением неочищенного продукта, который очищали при помощи препаративной ТСХ, с получением указанного в заголовке соединения Пр.36 Стадия-2 (0,015 г, 15%).
Соединение 36:1H ЯМР(400 MГц, DMSO-d6): δ 13,08 (с, 1H), 8,08 (с, 1H), 7,65 (с, 1H), 7,53 (д, J=8,8 Гц, 1H), 7,4 (д, J=7,2 Гц, 1H), 7,16-7,13 (м, 2H), 7,1-7,03 (м, 2H), 6,74 (д, J=8,8 Гц, 2H), 6,6-6,47 (м, 4H), 3,84 (т, J=5,6 Гц, 2H), 3,29-3,1 (м, 2H), 2,98 (с, 3H), 2,84 (с, 3H), 2,76 (т, J=5,6 Гц, 2H), 2,33-2,31 (м, 2H), 2,1 (с, 3H), 0,86 (т, J=7,6 Гц, 3H). ЖХМС: 509,3[M+H]+.
Пример 37: Примера 37 нет.
Пример 38: Синтез (E)-4-((2-((5-((Z)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиримидин-2-ил)окси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 38)
Соединение синтезировали в соответствии с подходом, описанным в примере 12, с использованием на стадии-2 (E)-4-((2-((5-((Z)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиримидин-2-ил)окси)этил)амино)-N,N-диметилбут-2-енамида (получение показано ниже в Пр.38 Стадии 1-4) вместо соединения 253 и (E)-4-бром-N,N-диметилбут-2-енамида (полученного в Примере 1, Стадия-7) вместо соединения 232, с получением соединения 38 (0,11 г, 13%).
Стадия-1: Синтез трет-бутил (2-((5-бромпиримидин-2-ил)окси)этил)карбамата

К перемешиваемому раствору трет-бутил (2-гидроксиэтил)карбамата (3,25 г, 20,2 ммоль) в DMF (90 мл), добавляли карбонат цезия (7,57 г, 23,3 ммоль). Реакционную смесь перемешивали в течение приблизительно 15 мин при комнатной температуре, к реакционной смеси добавляли 5-бром-2-хлорпиримидин (3,0 г, 15,5 ммоль) и перемешивали в течение 14 ч. После завершения, реакционную смесь разбавляли водой, экстрагировали этилацетатом, объединенные органические слои промывали водой, затем насыщенным солевым раствором. Органический слой сушили над безводным сульфатом натрия, концентрировали досуха при пониженном давлении. Неочищенный продукт очищали при помощи колоночной хроматографии на силикагеле, элюируя 15-20% EtoAc в н-гексане, с получением указанного в заголовке соединения (4,5 г, 55%).
Стадия-2: Синтез трет-бутил (2-((5-йодпиримидин-2-ил)окси)этил)карбамата

К перемешиваемому раствору трет-бутил (2-((5-бромпиримидин-2-ил)окси)этил)карбамата (4,0 г, 12,6 ммоль) в 1,4-диоксане (80 мл), добавляли NaI (3,78 г, 25,2 ммоль), CuI (2,4 г, 12,6 ммоль) и N,N′-диметилэтилендиамин (2,2 г, 25,2 ммоль). Реакционную смесь нагревали до 100°C в течение 6 ч. По завершении, реакционную смесь охлаждали до комнатной температуры, фильтровали через слой целита и слой целита промывали этилацетатом (200 мл). Полученный фильтрат промывали водой, насыщенным солевым раствором и сушили над безводным сульфатом натрия и концентрировали досуха. Неочищенный продукт очищали колоночной хроматографией на силикагеле, элюируя 15% EtOAc в н-гексане, с получением указанного в заголовке соединения Пр.38 Стадия-1 (3,2 г, 69%).
Стадия-3: Синтез трет-бутил (Z)-(2-((5-(2-фенил-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)пиримидин-2-ил)окси)этил)карбамата

Реакции проводили, как описано в примере 5, Стадия-1, с использованием 5-(бут-1-ин-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (Пример 1, Стадия-3) и использованием йодбензола вместо соединения 244, с получением неочищенного продукта, который очищали при помощи хроматографии на силикагеле, элюируя 25-30% EtOAc в н-гексане, с получением указанного в заголовке соединения Пр.38 Стадия-3 (2,9 г, неочищенный).
Стадия-4: Синтез (Z)-2-((5-(1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиримидин-2-ил)окси)этан-1-амина

Реакцию осуществляли в соответствии со Схемой 2, Стадия-2b, с получением указанного в заголовке соединения Пр.38 Стадия-4 (1,6 г, 85%).
Соединение 38:1H ЯМР (400 MГц, DMSO-d6): δ 13,12 (с, 1H), 8,09 (с, 1H), 7,99 (с, 2H), 7,69 (с, 1H), 7,56 (д, J=8,4 Гц, 1H), 7,3-7,17 (м, 6H), 6,62-6,47 (м, 2H), 4,18 (т, J=6 Гц, 2H), 3,30-3,28 (м, 2H), 2,98 (с, 3H), 2,83 (с, 3H), 2,68 (т, J=6 Гц, 2H), 2,46-2,42 (м, 2H), 0,90 (т, J =7,2 Гц, 3H). ЖХМС: 497,3 [M+H]+.
Пример 39: Синтез (E)-1-(азетидин-1-ил)-4-((2-(4-((E)-2-циклобутил-1-(1H-индазол-5-ил)-2-фенилвинил)фенокси)этил)амино)бут-2-ен-1-она (Соединение 39)
Соединение синтезировали в соответствии с подходом, описанным в примере 12, с использованием на стадии-2 (E)-2-(4-(2-циклобутил-1-(1H-индазол-5-ил)-2-фенилвинил)фенокси)этан-1-амина (полученного в Примере 13, Стадия-7) вместо соединения 253 и (E)-1-(азетидин-1-ил)-4-бромобут-2-ен-1-она (полученного в Примере 10, Стадия-1) вместо соединения 232, с получением соединения 39 (0,030 г, 4%).
Соединение 39:1H ЯМР(400 MГц, DMSO-d6): δ 13,06 (с, 1H), 8,07 (с, 1H), 7,58 (с, 1H), 7,51 (д, J=8,8 Гц, 1H), 7,27-7,23 (м, 2H), 7,17-7,08 (м, 5H), 6,79-6,76 (м, 2H), 6,59 (т, J=5,2 Гц, 1H), 6,57-6,53 (м,2H), 6,04-5,75 (м, 1H), 4,11 (т, J=7,6 Гц, 2H), 3,87-3,80 (м, 4H), 3,41 (т, J=8,8 Гц, 1H), 3,29-3,27 (м, 2H), 2,74 (т, J=5,6 Гц, 2H), 2,17 (т, J=7,6 Гц, 2H), 1,82-1,76 (м, 4H), 1,59-1,55 (м, 1H), 1,36-1,32 (м, 1H). ЖХМС: 533,3[M+H]+.
Пример 40: Синтез (E)-1-(азетидин-1-ил)-4-((2-(4-((E)-2-циклобутил-1-(3-фтор-1H-индазол-5-ил)-2-фенилвинил)фенокси)этил)амино)бут-2-ен-1-она (Соединение 40)
Соединение синтезировали в соответствии с подходом, описанным в примере 3, (i) с использованием на стадии-4 (Z)-5-(1,2-бис(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (полученного в Примере 3, Стадия-1) вместо соединения 226 и иодбензола вместо соединения 227 и (ii) с использованием на стадии-6 (E)-1-(азетидин-1-ил)-4-бромобут-2-ен-1-она (полученного в Примере 10, Стадия-1) вместо соединения 251, с получением указанного в заголовке соединения (0,007 г, 3%).
Соединение 40:1H ЯМР(400 MГц, DMSO-d6): δ 12,57 (с, 1H), 7,49 (с, 1H), 7,46 (д, J=8,0 Гц, 1H), 7,27-7,1 (м, 6H), 6,8 (д, J=8,8 Гц, 2H), 6,6 (т, J=5,2 Гц, 1H), 6,55 (д, J=8,8 Гц, 2H), 6,02 (д, J=15,6 Гц, 1H), 4,12 (т, J=7,4 Гц, 2H), 3,87-3,83 (м, 4H), 3,4-3,37 (м, 1H), 3,29-3,27 (м, 2H), 2,74 (т, J=5,6 Гц, 2H), 2,2-2,1 (м, 2H), 1,83-1,75 (м, 4H), 1,6-1,56 (м, 1H), 1,34-1,24 (м, 1H). ЖХМС: 551,3[M+H]+.
Пример 41: Синтез (E)-4-((2-(4-((E)-2-циклобутил-1-(1H-индазол-5-ил)-2-фенилвинил)фенокси)этил)амино)-N-(2-гидроксиэтил)-N-метилбут-2-енамида (Соединение 41)
Соединение синтезировали в соответствии с подходом, описанным в примере 12, с использованием на стадии-2 (E)-2-(4-(2-циклобутил-1-(1H-индазол-5-ил)-2-фенилвинил)фенокси)этан-1-амина (полученного в Примере 13, Стадия-7) вместо соединения 253 и (E)-4-бром-N-(2-гидроксиэтил)-N-метилбут-2-енамида (полученного в Примере 18, Стадия-1) вместо соединения 232, с получением соединения 41 (0,012 г, 9%).
Соединение 41:1H ЯМР(400 MГц, DMSO-d6): δ 13,06 (с, 1H), 8,08 (с, 1H), 7,58 (с, 1H), 7,51 (д, J=8,0 Гц, 1H), 7,27-7,24 (м, 2H), 7,17-7,09 (м, 4H), 7,78 (д, J=7,6 Гц, 2H), 6,58-6,52 (м, 4H), 4,77-4,64 (м, 1H), 3,85 (т, J=5,2 Гц, 2H), 3,51-3,41 (м, 2H), 3,39-3,34 (м, 5H), 3,03 (с, 1H), 2,86 (с, 2H), 2,80-2,79 (м, 2H), 1,82-1,76 (м, 4H), 1,57-1,55 (м, 1H), 1,35-1,32 (м, 1H), ЖХМС: 551,3[M+H]+.
Пример 42: Синтез (E)-1-(азетидин-1-ил)-4-((2-(4-((E)-2-циклобутил-1-(1H-индазол-5-ил)-2-(o-толил)винил)фенокси)этил)амино)бут-2-ен-1-она (Соединение 42)
Соединение синтезировали в соответствии с подходом, описанным в примере 3, (i) с использованием на стадии-4 (Z)-трет-бутил (2-(4-(2-циклобутил-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)винил)фенокси)этил)карбамата (полученного в Примере 13, Стадия-5) вместо соединения 226 и 1-иод-2-метилбензола вместо соединения 227 и (ii) с использованием на стадии-6 (E)-1-(азетидин-1-ил)-4-бромобут-2-ен-1-она (полученного в Примере 10, Стадия-1) вместо соединения 251, с получением соединения 42 (0,05 г, 4,5%).
Соединение 42:1H ЯМР(400 MГц, DMSO-d6): δ 13,07 (с, 1H), 8,09 (с, 1H), 7,60 (с, 1H), 7,53 (д, J=2,0 Гц, 1H), 7,16-7,08 (м, 5H), 6,77 (д, J=8,8 Гц, 2H), 6,61-6,57 (м, 1H), 6,54 (д, J=8,8 Гц, 2H), 6,03 (д, J=15,6 Гц, 1H), 4,11 (т, J=7,6 Гц, 2H), 3,87-3,82 (м, 4H), 3,43-3,39 (м, 1H), 3,31 (с, 2H), 2,77 (т, J=5,6 Гц, 2H), 2,19-2,15 (м, 2H), 2,12 (с, 3H), 1,89-1,73 (м, 3H), 1,61-1,53 (м, 2H), 1,36-1,32 (м, 1H). ЖХМС: 547,3[M+H]+.
Пример 43: Синтез (E)-4-((2-(4-((E)-1-(3-фтор-1H-индазол-5-ил)-2-(2-фторфенил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 43)
Соединение синтезировали в соответствии с подходом, описанным в примере 3, с использованием на стадии-4 1-иод-2-фторбензола вместо соединения 227, с получением соединения 43 (0,025 г, 10%).
Соединение 43:1H ЯМР(400 MГц, DMSO-d6): δ 12,59 (с, 1H), 7,52 (с, 1H), 7,48 (дд, J1=8,8 Гц, J2=2,0 Гц, 1H), 7,24-7,18 (м, 3H), 7,08-7,03 (м, 2H), 6,80 (д, J=8,4 Гц, 2H), 6,63-6,57 (м, 3H), 6,49 (д, J=5,2 Гц, 1H), 3,86 (т, J=5,6 Гц, 2H), 3,33-3,31 (м, 2H), 2,98 (с, 3H), 2,84 (с, 3H), 2,77 (т, J=5,6 Гц, 2H), 2,37-2,32 (м, 2H), 0,88 (т, J=7,2 Гц, 3H). ЖХМС: 531,3[M+H]+.
Пример 44: Синтез (E)-4-((2-(4-((E)-1-(3-фтор-1H-индазол-5-ил)-2-(3-фторфенил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 44)
Соединение синтезировали в соответствии с подходом, описанным в примере 3, с использованием на стадии-4 1-иод-3-фторбензола вместо соединения 227, с получением соединения 44 (0,010 г, 4%).
Соединение 44:1H ЯМР(400 MГц, DMSO-d6): δ 12,60 (с, 1H), 7,52 (с, 1H), 7,47 (д, J=8,8 Гц, 1H), 7,25-7,19 (м, 2H), 6,99-6,94 (м, 3H), 6,78 (д, J=8,8 Гц, 2H), 6,64 (д, J=8,8 Гц, 2H), 6,59 (т, J=5,2 Гц, 1H), 6,50 (д, J=15,2 Гц, 1H), 3,88 (д, J=5,6 Гц, 2H), 3,33-3,31 (м, 2H), 2,99 (с, 3H), 2,84 (с, 3H), 2,78 (т, J=5,2Гц,2H), 2,42-2,38 (м, 2H), 0,88 (т, J=7,6 Гц, 3H). ЖХМС: 531,3[M+H]+.
Пример 45: Синтез (E)-4-((2-(4-((E)-1-(3-фтор-1H-индазол-5-ил)-2-(3-фторфенил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 45)
Соединение синтезировали в соответствии с подходом, описанным в примере 3, с использованием на стадии-4 1-иод-4-фторбензола вместо соединения 227, с получением соединения 45 (0,008 г, 3,8%).
Соединение 45:1H ЯМР(400 MГц, DMSO-d6): δ 12,60 (с, 1H), 7,51 (с, 1H), 7,47 (дд, J1=8,8 Гц, J2=1,6 Гц, 1H), 7,21-7,19 (м, 3H), 7,18-7,02 (м, 2H), 6,75 (д, J=8,8 Гц, 2H), 6,64-6,58 (м, 3H), 6,51 (д, J=15,2 Гц, 1H), 3,88 (т, J=5,6 Гц, 2H), 3,31 (д, J=4,4 Гц, 2H), 2,99 (с, 3H), 2,84 (с, 3H), 2,78 (т, J=5,2 Гц, 2H), 2,39-2,37 (м, 2H), 0,87 (т, J=7,2 Гц, 3H). ЖХМС: 531,3[M+H]+.
Пример 46: Синтез (E)-4-((2-(4-((E)-2-(2-хлор-4-фторфенил)-2-циклобутил-1-(3-фтор-1H-индазол-5-ил)винил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 46)
Соединение синтезировали в соответствии с подходом, описанным в примере 3, (i) с использованием на стадии-3 (Z)-5-(2-циклобутил-1,2-бис(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)винил)-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (полученного в Примере 14, Стадия-2) вместо соединения 255 и (E)-трет-бутил (4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)-карбамата (полученного в Примере 14, Стадия-3) вместо соединения 256 и (ii) с использованием на стадии-4 2-хлор-4-фтор-1-иодбензола для соединения 227 и (iii) продолжая Стадию-5, с получением соединения 46 (0,02 г, 10%).
Соединение 46:1H ЯМР (400 MГц, DMSO-d6): δ 12,6 (с, 1H), 7,49 (д, J=5,2 Hz 2H), 7,37-7,31 (м, 2H), 7,23 (д, J=8,8 Гц, 1H), 7,18-7,15 (м, 1H), 6,87 (д, J=8,8 Гц, 2H), 6,63-6,57 (м, 3H), 6,5 (д, J=15,2 Гц, 1H), 3,87 (т, J=5,6 Гц, 2H), 3,51 (с, 1H), 3,39-3,33 (м, 2H), 2,98 (с, 3H), 2,84 (с, 3H), 2,79 (т, J=5,4 Гц, 2H), 1,87-1,78 (м, 3H), 1,68-1,58 (м, 2H), 1,38-1,36 (м, 1H). ЖХМС: 591,2 [M+H]+.
Пример 47: Примера 47 нет.
Пример 48: Примера 48 нет.
Пример 49: Синтез (E)-4-((2-(2-фтор-4-((Z)-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 49)
Соединение синтезировали в соответствии с подходом, описанным в примере 12, с использованием на стадии-2 (Z)-2-(2-фтор-4-(1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этан-1-амина (получение показано ниже в Пр.49 Стадии 1-3) вместо соединения 253 и смеси (E)-4-бром-N,N-диметилбут-2-енамида и (E)-4-хлор-N,N-диметилбут-2-енамида (получение показано ниже в Пр.49 стадия 4) вместо соединения 232, с получением соединения 49 (0,03 г, 7%).
Стадия-1: Синтез 2-фтор-4-иодфенола

К смеси 2-фторфенола (5 г, 44 ммоль) и иодида калия (8,14 г, 49 ммоль) в метаноле (150 мл) добавляли трет-бутил гидропероксид (6,43 мл, 66,9 ммоль) и перемешивали при комнатной температуре в течение 12 ч. После завершения реакции, реакционную смесь гасили тиосульфатом натрия и экстрагировали EtOAc. Органический слой промывали водой, затем насыщенным солевым раствором, концентрировали при пониженном давлении с получением неочищенного соединения. Неочищенный материал очищали при помощи колоночной хроматографии на силикагеле 230-400 меш с использованием 15% EtOAc в н-гексане с получением указанного в заголовке соединения (5,7 г).
Стадия-2: Синтез трет-бутил (2-(2-фтор-4-йодфенокси)этил)карбамата

К раствору 2-фтор-4-иодфенола (4,6 г, 21 ммоль) в DMF (40 мл) добавляли карбонат калия (16 г, 116 ммоль) и трет-бутил (2-бромэтил)карбамат (8,62 г, 38,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 12 ч. Реакционную смесь гасили водой и экстрагировали этилацетатом (250 мл×2). Объединенные органические слои промывали водой, насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный материал очищали при помощи колоночной хроматографии на силикагеле 230-400 меш с использованием 30% этилацетата в н-гексане с получением трет-бутил (2-(2-фтор-4-йодфенокси)этил)карбамата (4,53 г, 61%).
Стадия-3: Синтез (Z)-2-(2-фтор-4-(1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этан-1-амина

Соединение синтезировали в соответствии с подходом, описанным в примере 3, с использованием трет-бутил (2-(2-фтор-4-йодфенокси)этил)карбамата вместо соединения 256 на Стадии-3, и с использованием йодбензола вместо соединения 227 на Стадии-4. Неочищенный материал использовали на следующей стадии без дополнительной очистки (2,1 г).
Стадия-4: Синтез смеси (E)-4-бром-N,N-диметилбут-2-енамида и (E)-4-хлор-N,N-диметилбут-2-енамида:

Реакцию осуществляли в соответствии со Схемой 4, Стадия-2, с использованием (E)-4-бромбут-2-еновой кислоты (Пример 1, Стадия-7.1) и диметиламина с получением неочищенного материала, который очищали колоночной хроматографией на силикагеле, с получением смеси (E)-4-бром-N,N-диметилбут-2-енамида и (E)-4-хлор-N,N-диметилбут-2-енамида (~60:40/Br:Cl, 18 г, 51%) в виде жидкости коричневого цвета.1H ЯМР(400 MГц, DMSO-d6): δ 6,95-6,86 (м, 1H), 6,57-6,46 (м, 1H), 4,19-4,18 (м, 1H), 4,05-4,03 (м, 1H), 3,09-3,05 (м, 3H), 3,0-2,98 (м, 3H). ЖХМС: 194,0 и 148,1 [M+H]+
Соединение 49:1H ЯМР(400 MГц, DMSO-d6): 12,59 (с, 1H), 7,54 (с, 1H), 7,48 (д, J=7,2 Гц, 1H), 7,25-7,22 (м, 3H), 7,17-7,15 (м, 3H), 6,84 (т, J=8,6 Гц, 1H), 6,64-6,47 (м, 4H), 3,94 (т, J=5,4 Гц, 2H), 3,31-3,20 (м, 2H), 2,98 (с, 3H), 2,84 (с, 3H), 2,79 (т, J=5,8 Гц, 2H), 2,41 (кв, J=6,8 Гц, 2H), 0,86 (т, J=7,6 Гц, 3H). ЖХМС: 531,3[M+H]+
Пример 50: Синтез (E)-4-((2-(4-((E)-2-(2,6-дифторфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 50)
Стадия-1: Синтез трет-бутил (E)-(2-(4-(2-(2,6-дифторфенил)-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)карбамата

Реакцию осуществляли в соответствии со Схемой 1, Стадия-5, используя трет-бутил (Z)-(2-(4-(1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)фенокси)этил)карбамат (2,79 г, 4,39 ммоль, полученный в Примере 3, Стадия-3) вместо соединения 208. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя 5-15% EtOAc в н-гексане, с получением указанного в заголовке соединения Пр.50 Стадия-1 (2,1 г, 77,2%).
Стадия-2: Синтез (E)-2-(4-(2-(2,6-дифторфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этан-1-амина

Реакцию осуществляли в соответствии со Схемой 2, Стадия-2b, с использованием трет-бутил (E)-(2-(4-(2-(2,6-дифторфенил)-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)карбамата (2,1 г, 3,38 ммоль, полученного в Примере 50, Стадия 1). Неочищенный материал использовали на следующей стадии без дополнительной очистки (1,2 г, неочищенный).
Стадия-3: Синтез (E)-4-((2-(4-((E)-2-(2,6-дифторфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида

Реакцию осуществляли в соответствии со Схемой 2, Стадия-3b, используя (E)-2-(4-(2-(2,6-дифторфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этан-1-амин (1,2 г, 2,7 ммоль) и (E)-4-бром-N,N-диметилбут-2-енамид вместо соединения 213 и (E)-4-хлор-N,N-диметилбут-2-енамид смесь (полученную в Примере 49, Стадия-4) вместо соединения 214, с получением неочищенного продукта, который использовали на следующей стадии без дополнительной очистки (1,15 г, неочищенный).
Стадия-4: Синтез трет-бутил (2-(4-((E)-2-(2,6-дифторфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)((E)-4-(диметиламино)-4-оксобут-2-ен-1-ил)карбамата

Реакцию осуществляли в соответствии со Схемой 2, Стадия-5, используя (E)-4-((2-(4-((E)-2-(2,6-дифторфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамид (1,15 г, 2,13 ммоль) вместо соединения 215. Неочищенный материал очищали колоночной хроматографией на силикагеле 230-400 меш, используя (2,5%) MeOH в дихлорметане, с получением указанного в заголовке соединения Пр.50 Стадия-4 (0,3 г, 22,2%).
Стадия-5: Синтез (E)-4-((2-(4-((E)-2-(2,6-дифторфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида

Реакцию осуществляли в соответствии со Схемой 2, Стадия-6b, используя трет-бутил (2-(4-((E)-2-(2,6-дифторфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)((E)-4-(диметиламино)-4-оксобут-2-ен-1-ил)карбамат (0,3 г, 0,462 ммоль) вместо соединения 211c. Неочищенное соединение очищали при помощи препаративной ВЭЖХ с получением соединения 50 (0,065 г, 25,6%).
Соединение 50:1H ЯМР(400 MГц, DMSO-d6): δ 12,60 (с, 1H), 7,52 (с, 1H), 7,49 (дд, J1=8,8 Гц, J2=1,2 Гц, 1H), 7,23-7,12 (м, 1H), 7,11-7,02 (м, 3H), 6,83 (д, J=8,4 Гц, 2H), 6,65 (д, J=8,4 Гц, 2H), 6,62-6,48 (м, 2H), 3,88 (т, J=5,6 Гц, 2H), 3,32-3,31 (м, 2H), 2,99 (с, 3H), 2,84 (с, 3H), 2,79 (т, J=5,4 Гц, 2H), 2,35 (кв, J=7,0 Гц, 2H), 0,91 (т, J=7,4 Гц, 3H). ЖХМС: 549,3[M+H]+
Пример 51: Синтез (E)-4-((2-(4-((E)-1-(3-фтор-1H-индазол-5-ил)-2-(пиридин-3-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 51)
Соединение синтезировали в соответствии с подходом, описанным в примере 50, с использованием на стадии-1 3- йодпиридина вместо соединения 257, с получением соединения 51 (0,02 г, 10%).
Соединение 51:1H ЯМР(400 MГц, DMSO-d6): δ 12,59 (с, 1H), 8,31-8,30 (м, 1H), 8,26 (д, J=2 Гц, 1H), 7,62 (дд, J1=6,0 Гц, J2=2,0 Гц, 1H), 7,54 (с, 1H), 7,48 (дд, J1=8,0 Гц, J2=2,0 Гц, 1H), 7,28-7,24 (м, 1H), 7,22 (дд, J1=8,8 Гц, J2=1,2 Гц, 1H), 6,79-6,77 (м, 2H), 6,65-6,60 (м, 3H), 6,58-6,48 (м, 1H), 3,89 (т, J=5,4 Гц, 2H), 3,32-3,31 (м, 2H), 2,99 (с, 3H), 2,84 (с, 3H), 2,79 (т, J=5,4 Гц, 2H), 2,42 (кв, J=7,4 Гц, 2H), 0,88 (т, J=7,4 Гц, 3H). ЖХМС: 514,3 [M+H]+
Пример 52: Синтез (E)-4-((2-(4-((E)-1-(3-фтор-1H-индазол-5-ил)-2-(пиридин-4-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 52)
Соединение синтезировали в соответствии с подходом, описанным в примере 50, с использованием на стадии-1 4-йодпиридина вместо соединения 257, с получением указанного в заголовке соединения 52 (0,05 г, 19,9%).
Соединение 52:1H ЯМР(400 MГц, DMS-d6): δ 12,60 (с, 1H), 8,39-8,37 (м, 2H), 7,53 (с, 1H), 7,50-7,47 (м, 1H), 7,21 (дд, J1=8,6 Гц, J2=1,4 Гц, 1H), 7,14-7,13 (м, 2H), 6,80-6,78 (м, 2H), 6,66-6,60 (м, 2H), 6,59-6,48 (м, 2H), 3,89 (т, J=5,4 Гц, 2H), 3,33-3,31 (м, 2H), 2,99 (с, 3H), 2,84 (с, 3H), 2,79 (т, J=5,8 Гц, 2H), 2,42 (кв, J=7,4 Гц, 2H), 0,87 (т, J=7,2 Гц, 3H). ЖХМС: 514,3[M+H]+
Пример 53: Синтез (E)-4-((2-(4-((E)-2-циклобутил-1-(3-фтор-1H-индазол-5-ил)-2-фенилвинил)фенокси)этил)амино)-N-(2-гидроксиэтил)-N-метилбут-2-енамида (Соединение 53)
Соединение синтезировали в соответствии с подходом, описанным в примере 50, с использованием на стадии-3 (E)-2-(4-(2-циклобутил-1-(3-фтор-1H-индазол-5-ил)-2-фенилвинил)фенокси)этан-1-амина (получение показано ниже в Пр.53 Стадия-1) вместо соединения 258 и (E)-4-бром-N-(2-гидроксиэтил)-N-метилбут-2-енамида (полученного в Примере 18, Стадия-1) вместо соединения 259, с получением соединения 53 (0,011 г, 5%).
Стадия-1: Синтез (E)-2-(4-(2-циклобутил-1-(3-фтор-1H-индазол-5-ил)-2-фенилвинил)фенокси)этан-1-амина

Соединение синтезировали в соответствии с подходом, описанным в примере 3, с использованием трет-бутил (Z)-(2-(4-(2-циклобутил-1-(1-(тетрагидро-2H-пиран-2-ил)-1H-индазол-5-ил)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)винил)фенокси)-этил)карбамата (полученного в Примере 13, Стадия-5) вместо соединения 226 и иодбензола вместо соединения 227 на стадии-4 и после Стадии-5, получая указанное в заголовке соединение (0,85, неочищенный).
Соединение 53:1H ЯМР(400 MГц, DMSO-d6): δ 12,56 (с, 1H), 7,48 (с, 2H), 7,46-7,23 (м, 4H), 7,21 (дд, J1=8,8 Гц, J2=1,6 Гц, 2H), 6,80-6,78 (м, 2H), 6,60-6,47 (м, 4H), 4,77-4,64 (м, 1H), 3,85 (т, J=5,2 Гц, 2H), 3,51-3,41 (м, 2H), 3,39-3,34 (м, 4H), 3,03 (с, 1H), 2,86 (с, 2H), 2,80-2,79 (м, 2H), 1,82-1,76 (м, 4H), 1,57-1,55 (м, 1H), 1,35-1,32 (м, 1H). ЖХМС: 569,3[M+H]+
Пример 54: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилпроп-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 54)
Указанное в заголовке соединение синтезировали в соответствии со способом, описанным в примере 1. Не желая связывать себя теорией, считается, что соединение 54 было получено за счет включения метилиодида в этилиодид, используемый на стадии-2 примера 1.
Соединение 54:1H ЯМР (400 MГц, DMSO-d6): δ 13,05 (с, 1H), 8,05 (с, 1H), 7,6 (с, 1H), 7,50 (д, J=8,4 Гц, 1H), 7,21-7,09 (м, 6H), 6,75 (д, J=8,8 Гц, 2H), 6,62-6,48 (м, 4H), 3,88 (т, J=5,2 Гц, 2H), 3,31 (д, J=4,8 Гц, 2H), 2,99 (с, 3H), 2,84 (с, 3H), 2,79-2,77 (м, 2H), 2,07 (с, 3H). ЖХМС: 481,3 [M+H]+
Пример 55: Примера 55 нет.
Пример 56: Синтез (E)-4-((2-(4-((E)-2-циклопропил-1-(1H-индазол-5-ил)-2-фенилвинил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 56)
Соединение синтезировали в соответствии с подходом, описанным в примере 50, с использованием на стадии-1 трет-бутил (Z)-(2-(4-(2-циклопропил-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)винил)фенокси)этил)карбамата (получение показано ниже на стадии 1-2) вместо соединения 260 и иодбензола вместо соединения 257 с получением соединения 56 (0,12 г).
Стадия-1: Синтез 5-(циклопропилэтинил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола

Реакцию осуществляли в соответствии со Схемой 1, Стадия-2, заменяя 5-бром-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол (5,5 г, 19,57 ммоль) в 88 мл триэтиламине и этинилциклопропан (2,5 г, 39,14 ммоль) и нагревали при 90°C в течение 12 ч. По завершении ТСХ, реакционную смесь гасили водой (10 мл) и экстрагировали этилацетатом (2×50 мл). Объединенные органические экстракты промывали водой, насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали при помощи колоночной хроматографии на силикагеле 230-400 меш, с использованием 5% этилацетата в н-гексане, с получением 5-(циклопропилэтинил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (4,5 г, 86%).
Стадия-2: Синтез трет-бутил (Z)-(2-(4-(2-циклопропил-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)винил)фенокси)этил)карбамата

Соединение синтезировали в соответствии с подходом, описанным в примере 3, используя на стадии-1 5-(циклопропилэтинил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол вместо соединения 252, и продолжая стадии 2-3, с получением указанного в заголовке соединения Пр.56 Стадия-2, которое использовали на следующей стадии без дополнительной очистки (6 г, неочищенный).
Соединение 56:1H ЯМР(400 MГц, DMSO-d6): δ 13,04 (с, 1H), 8,06 (с, 1H), 7,73 (с, 1H), 7,51 (д, J=8,8 Гц, 1H), 7,27 (д, J=8,4 Гц, 1H), 7,21-7,04 (м, 5H), 6,72 (д, J=8,4 Гц, 2H), 6,61-6,46 (м, 4H), 3,83 (т, J=5,6 Гц, 2H), 3,31-3,29 (м, 2H), 2,98 (с, 3H), 2,83 (с, 3H), 2,75 (т, J=5,2 Гц, 2H), 2,0-2,30 (м, 1H), 1,74-1,71 (м, 1H), 0,58-0,56 (м, 2H), 0,21-0,20 (м, 2H). ЖХМС: 507,4[M+H]+
Пример 57: Синтез (E)-4-((2-(4-((E)-2-(2-хлор-4-фторфенил)-1-(4-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 57)
Соединение синтезировали в соответствии с подходом, описанным в примере 50, с использованием на стадии-2 трет-бутил (E)-(2-(4-(2-(2-хлор-4-фторфенил)-1-(4-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)карбамата (получение показано ниже на стадии-1) вместо соединения 261, с получением соединения 57 (0,04 г, 15,7%).
Стадия-1: Синтез трет-бутил (E)-(2-(4-(2-(2-хлор-4-фторфенил)-1-(4-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)карбамата

Соединение синтезировали в соответствии с подходом, описанным в примере 3, используя 5-(бут-1-ин-1-ил)-4-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол (Пример 4, Стадия-4) вместо соединения 252 на стадии-1, непосредственно переходя на стадию-3, и используя 2-хлор-4-фтор-1-иодбензол вместо соединения 227 на стадии-4, с получением указанного в заголовке соединения этой стадии. Неочищенный продукт очищали колоночной хроматографией на силикагеле, элюируя 2% MeOH в дихлорметане, с получением указанного в заголовке соединения (1,3 г, 16%).
Соединение 57:1H ЯМР (400 MГц, DMSO-d6): δ 13,42 (с, 1H), 8,18 (с, 1H), 7,42 (д, J=6,4 Гц, 1H), 7,36 (дд, J1=7,2 Гц, J2=2,0 Гц, 1H), 7,31 (дд, J1=6,8 Гц, J2=5,2 Гц, 1H), 7,25-7,22 (м, 1H), 7,14 (dt, J1=6,8 Гц, J2=5,2 Гц, 1H), 6,84 (д, J=7,2 Гц, 2H), 6,63 (д, J=7,2 Гц, 2H), 6,61-6,57 (м, 1H), 6,49 (д, J=12,4 Гц, 1H), 3,87 (т, J=4,4 Гц, 2H), 3,33-3,31 (м, 2H), 2,97 (с, 3H), 2,84 (с, 3H), 2,78 (т, J=4,4 Гц, 2H), 2,30-2,25 (м, 2H), 0,82 (т, J=6,2 Гц, 3H). ЖХМС: 565,3 [M+H]+
Пример 58: Синтез (E)-4-((2-((5-((Z)-2-(2-хлор-4-фторфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)пиридин-2-ил)окси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 58)
Соединение синтезировали в соответствии с подходом, описанным в примере 14, с использованием на стадии-4 (i) (Z)-5-(1,2-бис(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (полученного в Примере 3, Стадия-1) вместо соединения 233, (ii) трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-((5-йодпиридин-2-ил)окси)этил)карбамата (полученного в Примере 25, Стадия-2) вместо соединения 234, и (iii) 2-хлор-4-фтор-1-иодбензола вместо соединения 262, с получением соединения 58 (0,115 г, 10%).
Соединение 58:1H ЯМР (400 MГц, DMSO-d6): δ 12,63 (с, 1H), 7,69 (д, J=2,0 Гц, 1H), 7,59 (с, 1H), 7,51 (дд, J1=8,6 Гц, J2=1,8 Гц, 1H), 7,43-7,35 (м, 2H), 7,29 (д, J=1,4 Гц, 1H), 7,27-7,15 (м, 2H), 6,62-6,47 (м, 3H), 4,14 (т, J=5,9 Гц, 2H), 2,93 (с, 3H), 2,84 (с, 3H), 2,78 (т, J=5,8 Гц, 2H), 2,37 (кв, J=7,3 Гц, 2H), 0,89 (т, J=7,3 Гц, 3H). ЖХМС: 566,3 [M+H]+.
Пример 59: Синтез (E)-4-((2-((5-((Z)-2-(2-хлор-4-фторфенил)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)пиридин-2-ил)окси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 59)
Стадия-1: Синтез 3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-5-((триметилсилил)этинил)-1H-индазола

К перемешиваемому раствору 5-бром-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (4,5 г, 15 ммоль, полученного в Примере 2, Стадия-2) в 35 мл THF:Et3N (5:1) в герметичном сосуде, добавляли йодид меди (0,288 г, 1,5 ммоль) при комнатной температуре. Эту смесь дегазировали с помощью трех циклов вакуум/N2, и добавляли этинилтриметилсилан (2,22 г, 22 ммоль), затем Pd(PPh3)2Cl2 (0,5 г, 0,7 ммоль). Сосуд высокого давления герметизировали и нагревали при 80°C в течение 48 ч. По завершении ТСХ, реакционную смесь разбавляли водой (250 мл) и экстрагировали EtOAc (2×100 мл). Объединенные органические экстракты промывали водой, насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали при помощи combo-flash, используя 5% EtOAc в н-гексане, с получением 3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-5-((триметилсилил)этинил)-1H-индазола (3,2 г, 72%).
Стадия-1A: Альтернативный синтез 3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-5-((триметилсилил)этинил)-1H-индазола

Стадия-1А представляет предлагаемую альтернативу Стадии-1 примера 59 для возможного получения 3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-5-((триметилсилил)этинил)-1H-индазола, который может быть использован в качестве промежуточного соединения для дальнейших путей синтеза, которые включают это соединение. К перемешиваемому раствору 5-бром-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (5 г, 16,71 ммоль) в 30 мл 2-метил THF(6V) в 100 мл круглодонную колбу, добавляли триэтиламин (5,07 г, 50 ммоль), йодид меди (0,16 г, 0,84 ммоль) при комнатной температуре. Эту смесь дегазировали с помощью трех циклов вакуум/N2, и добавляли этинилтриметилсилан (4,10 г, 41,74 ммоль), затем Pd-134 (0,31 г, 0,41 ммоль) или Pd-117. Реакционную смесь нагревали при 40°C-75°C в течение 3-8 ч. По завершении ТСХ, реакционную смесь фильтровали через слой целита и промывали 30 мл 2-Метил-THF. Фильтрат промывали насыщенным солевым раствором и концентрировали при пониженном давлении с получением неочищенного 3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-5-((триметилсилил)этинил)-1H-индазола (6,1 г) и использовали для следующей стадии с учетом 100% выхода.
Стадия-2: Синтез 5-этинил-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола

К перемешиваемому раствору 3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-5-((триметилсилил)этинил)-1H-индазола (3,2 г, 10 ммоль) в метаноле 32 мл добавляли карбонат калия (0,151 г, ммоль), реакционную смесь перемешивали в течение 16 ч при комнатной температуре. После завершения реакции, реакционную смесь разбавляли этилацетатом и органический слой промывали водой, затем насыщенным солевым раствором. Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением указанного в заголовке соединения Пр.59 Стадии-2 (2,8 г, неочищенный).
Стадия-3: Синтез 3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-5-(4,4,4-трифторбут-1-ин-1-ил)-1H-индазола

К перемешиваемому раствору 5-этинил-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (2,6 г, 10,6 ммоль) в 20 мл толуола, добавляли 1,1,1-трифтор-2-иодэтан (4,47 г, 21,3 ммоль) при комнатной температуре. Эту смесь дегазировали с помощью трех циклов вакуум/N2, и добавляли Pd2(dba)3 (0,487 г, 0,5 ммоль), затем DPEphos (1,14 г, 2,1 ммоль) и DABCO (2,39 г, 21,3 ммоль). Реакционную смесь нагревали при 80°C в течение 24 ч. По завершении ТСХ, реакционную смесь разбавляли водой (250 мл) и экстрагировали EtOAc (2×100 мл). Объединенные органические экстракты промывали водой, насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью колоночной хроматографии, используя 5% EtOAc в н-гексане, с получением 3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-5-(4,4,4-трифторбут-1-ин-1-ил)-1H-индазола (1,6 г, 46%).
Стадия-4: Синтезтрет-бутил ((E)-4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-((5-((E)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)пиридин-2-ил)окси)этил)карбамата

К перемешиваемому раствору 3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-5-(4,4,4-трифторбут-1-ин-1-ил)-1H-индазола (1 г, 3,06 ммоль) в 2-метил THF (20 мл), добавляли бис(пинаколато)диборон (0,934 г, 3,68 ммоль), тетракис(трифенилфосфин)платина(0) (0,026 г, 0,02 ммоль) в атмосфере азота, реакционную смесь перемешивали при 80°C в течение 12 ч. Раствору давали остыть до комнатной температуры и добавляли трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-((5-йодпиридин-2-ил)окси)этил)карбамат (1,12 г, 2,37 ммоль), бис(трифенилфосфин)палладий(II) дихлорид (0,092 г, 0,13 ммоль), карбонат цезия (1,7 г, 5,26 ммоль) и 2-метил THF (20 мл). Эту смесь дегазировали азотом и добавляли воду (0,3 мл). Эту смесь перемешивали при комнатной температуре в течение 12 ч. После завершения реакции реакционную смесь разбавляли водой с последующим экстрагированием этилацетатом, органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного соединения. Неочищенное соединение очищали при помощи combi-flash, с использованием 3% MeOH в дихлорметане, с получением указанного в заголовке соединения Пр.59 Стадия-4 (1,2 г).
Стадия-5: Синтез трет-бутил (2-((5-((Z)-2-(2-хлор-4-фторфенил)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)пиридин-2-ил)окси)этил)((E)-4-(диметиламино)-4-оксобут-2-ен-1-ил)карбамата

Реакцию осуществляли в соответствии со Схемой 1, Стадия-5, используя трет-бутил ((E)-4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-((5-((E)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)пиридин-2-ил)окси)этил)карбамат (1,2 г, 1,49 ммоль) вместо соединения 208 в 2-m и 2-хлор-4-фтор-1-иодбензол (0,383 г, 1,49 ммоль) вместо соединения 209. Реакционную смесь перемешивали при 80°C в течение 8 ч. Неочищенный материал очищали с помощью combi-flash, с использованием 3% MeOH в дихлорметане, с получением указанного в заголовке соединения Пр.59 Стадия-5 (0,92 г, 76%).
Стадия-6: Синтез (E)-4-((2-((5-((Z)-2-(2-хлор-4-фторфенил)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)пиридин-2-ил)окси)этил)амино)-N,N-диметилбут-2-енамида

Реакцию осуществляли в соответствии со Схемой 2, Стадия-6b, используя трет-бутил (2-((5-((Z)-2-(2-хлор-4-фторфенил)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-1-ен-1-ил)пиридин-2-ил)окси)этил)((E)-4-(диметиламино)-4-оксобут-2-ен-1-ил)карбамат (0,92 г, 1,14 ммоль) вместо соединения 211c, с получением неочищенного соединения, которое очищали препаративной ВЭЖХ с получением соединения 59 (0,27 г, 38%) в виде свободного основания.
Compound 59:1H ЯМР (400 MГц, DMSO-d6): δ 12,71 (с, 1H), 7,71 (д, J=2 Гц, 2H), 7,65 (м, 2H), 7,57-7,52 (м, 2H), 7,37 (дд, J1=8,8 Гц, J2=2,4 Гц, 1H), 7,28-7,19 (м, 3H), 6,62-6,46 (м, 3H), 4,15 (т, J=4,1 Гц, 2H), 3,54-3,40 (м, 2H), 3,37-3,28 (м, 2H), 2,98 (с, 3H), 2,84 (с, 3H), 2,76 (т, J=4,1 Гц, 2H). ЖХМС: 620,2 [M+H]+.
Пример 60: Синтез (E)-N,N-диметил-4-((2-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)этил)амино)бут-2-енамида (Соединение 60)
Стадия-1: Синтезтрет-бутил ((E)-4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)этил)карбамата

К перемешиваемому раствору 3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-5-(4,4,4-трифторбут-1-ин-1-ил)-1H-индазола (0,66 г, 2,02 ммоль, полученного, как указано в примере 59, Стадия-1-Стадия-3) в 2-метил THF (10 мл), добавляли бис(пинаколато)диборон (0,566 г, 2,22 ммоль), тетракис(трифенилфосфин)платину(0) (0,025 г, 0,02 ммоль) в атмосфере азота, реакционную смесь перемешивали при 85°C в течение 5 ч. Раствору давали остыть до комнатной температуры и добавляли трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-((5-йодпиридин-2-ил)окси)этил)карбамат (0,72 г, 1,51 ммоль,полученный в Примере 25, Стадия-2), бис(трифенилфосфин)палладий(II) дихлорид (0,071 г, 0,1 ммоль), карбонат цезия (1,3 г, 4,04 ммоль) и 2-метил THF (10 мл). Эту смесь дегазировали азотом и добавляли воду (0,12 мл). Эту смесь перемешивали при комнатной температуре в течение 16 ч. После завершения реакции, к вышеуказанной реакционной смеси добавляли 4M KOH (2,78 мл, 11,13 ммоль) и иодбензол (0,33 г, 1,61 ммоль). Реакционную смесь перемешивали при 85°C в течение 7 ч. После завершения реакции, реакционную смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали EtOAc. Органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный материал очищали с помощью combi-flash, с использованием 100% этилацетата в качестве элюента, с получением трет-бутил ((E)-4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)этил)карбамата (0,35 г, 23%).
Стадия-2: Синтез (E)-N,N-диметил-4-((2-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)этил)амино)бут-2-енамида

К перемешиваемому раствору трет-бутил ((E)-4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)этил)карбамата (0,35 г, 0,46 ммоль) в дихлорметане (10 мл) добавляли при 0°C, TFA (1 мл). Реакционную смесь перемешивали в течение 16 ч при комнатной температуре. После завершения реакции реакционную смесь подщелачивали насыщенным NaHCO3,экстрагировали этилацетатом. Органический слой концентрировали при пониженном давлении с получением неочищенного соединения, которое очищали препаративной ВЭЖХ с получением желаемого соединения (E)-N,N-диметил-4-((2-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)этил)амино)бут-2-енамида (0,03 г, 11%).
Соединение 60:1H ЯМР (400 MГц, DMSO-d6): δ 12,71 (с, 1H), 7,63 (с, 2H), 7,54 (м, 1H), 7,26-7,18 (м, 7H), 6,62-6,46 (м, 3H), 4,13 (т, J=5,8 Гц, 2H), 3,51-3,43 (м, 2H), 3,34-3,28 (м, 2H), 2,98 (с, 3H), 2,83 (с, 3H), 2,75 (т, J=5,8 Гц, 2H). ЖХМС: 568,2 [M+H]+.
Пример 60A: Синтез хлористоводородной соли соединения 60
Стадия-1: Синтез (E)-N,N-диметил-4-((2-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)этил)амино)бут-2-енамид гидрохлорида

К перемешиваемому раствору (E)-N,N-диметил-4-((2-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)этил)амино)бут-2-енамида (4,1 г, 7,23 ммоль) в этаноле (24 мл) добавляли 2M HCl в диэтиловом эфире (7,5 мл) при 0°C. Белое твердое вещество наблюдали в реакционной смеси после перемешивания в течение 30 мин при комнатной температуре. Реакционную смесь концентрировали в вакууме при 35°С и полученное твердое вещество подвергали совместной перегонке с дихлорметаном в вакууме при 45°C. Полученное твердое вещество промывали н-пентаном и сушили в вакууме при 50°С в течение 4 ч с получением указанного в заголовке соединения (E)-N,N-диметил-4-((2-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)этил)амино)бут-2-енамид гидрохлорида (4,3 г, 98%).1H ЯМР(400 MГц, DMSO-d6): δ 12,74 (с, 1H), 9,26 (шир.с, 2H),7,69-7,34 (м, 3H), 7,28-7,17 (м, 7H), 6,81 (д, J=15,2 Гц, 1H), 6,62-6,53 (м, 2H), 4,37 (т, J=4,4 Гц, 2H), 3,77-3,76 (м, 2H), 3,51-3,43 (м, 2H), 3,23 (шир.с, 2H), 3,03 (с, 3H), 2,86 (с, 3H). ЖХМС: 568,3[M+H]+
Пример 60B: Синтез хлористоводородной соли соединения 60
К перемешиваемому раствору (E)-N,N-диметил-4-((2-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)этил)амино)бут-2-енамида (0,200 г, 0,35 ммоль) в этаноле (0,6 мл) добавляли 2M HCl в диэтиловом эфире (0,88 мл) при 0°C. Белое твердое вещество наблюдали в реакционной смеси после перемешивания в течение 30 мин при комнатной температуре. Реакционную смесь концентрировали в вакууме при 35°С и полученное твердое вещество подвергали совместной перегонке с дихлорметаном в вакууме при 45°C. Полученное твердое вещество промывали н-пентаном и сушили в вакууме при 50°С в течение 4 ч с получением указанного в заголовке соединения (E)-N,N-диметил-4-((2-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)этил)амино)бут-2-енамид гидрохлорида (,150 г, 67%).
1H ЯМР (400 MГц, DMSO-d6): δ 12,71 (с, 1H), 9,07 (шир.с, 2H), 7,69-7,54 (м, 3H), 7,26-7,18 (м, 7H), 6,80 (д, J=15,2 Гц, 1H), 6,62-6,46 (м, 2H), 4,35 (м, 2H), 3,75 (м, 2H), 3,51-3,43 (м, 2H), 3,34-3,28 (м, 2H), 2,98 (с, 3H), 2,83 (с, 3H). ЖХМС: 568,2 [M+H]+.
Пример 61: Синтез (E)-N,N-диметил-4-((2-(4-((E)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-(3-фторпиридин-4-ил)бут-1-ен-1-ил)фенокси)этил)амино)бут-2-енамида (Соединение 61)
Соединение синтезировали в соответствии с подходом, описанным в примере 60, с использованием на стадии-1 трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)карбамата (полученного в Примере 14, Стадия-3) вместо соединения 263 и 3-фтор-4-йодпиридина вместо соединения 264, с получением соединения 61 (0,02 г, 5,8%).
Соединение 61:1H ЯМР (400 MГц, DMSO-d6): δ 12,71 (с, 1H), 8,39 (с, 1H), 8,34 (д, J=2 Гц, 1H), 7,59 (с, 1H), 7,56-7,49 (м, 2H), 7,20 (д, J=2,4 Гц, 1H), 6,85 (д, J=8,9 Гц, 2H), 6,85 (д, 1H), 6,68 (д, J=8,9 Гц, 2H), 6,63-6,47 (м, 1H), 4,15 (м, 2H), 3,79 (т, J=5,7 Гц, 2H), 3,45 (кв, J=10,8 Гц, 2H), 3,25 (м, 2H), 2,98 (с, 3H), 2,84 (с, 3H). ЖХМС: 586,2 [M+H]+.
Пример 62: Синтез (E)-N,N-диметил-4-((2-((5-((Z)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-(3-фторпиридин-4-ил)бут-1-ен-1-ил)пиридин-2-ил)окси)этил)амино)бут-2-енамида (Соединение 62)
Соединение синтезировали в соответствии с подходом, описанным в примере 59, с использованием на стадии-5 3-фтор-4-йодпиридина вместо соединения 265, с получением соединения 62 (0,07 г, 12%).
Соединение 62:1H ЯМР (400 MГц, DMSO-d6): δ 12,76 (с, 1H), 8,43-8,39 (м, 2H), 7,72-7,56 (м, 4H), 7,28-7,24 (м, 2H), 6,62-6,48 (м, 3H), 4,15 (т, J=5,8 Гц, 2H), 4,15 (м, 2H), 3,51-3,46 (м, 2H), 3,38-3,31 (м, 2H), 2,98 (с, 3H), 2,84 (с, 3H). ЖХМС: 587,2 [M+H]+
Пример 63: Синтез (E)-N,N-диметил-4-((2-(4-((E)-2-фенил-1-(1H-пиразоло[4,3-b]пиридин-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)бут-2-енамида (Соединение 63)
Соединение синтезировали в соответствии с подходом, описанным в примере 50, с использованием на стадии-2 трет-бутил (E)-(2-(4-(2-фенил-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-пиразоло[4,3-b]пиридин-5-ил)бут-1-ен-1-ил)фенокси)этил)карбамата (получение показано ниже в Пр.63 Стадия 1-6) вместо соединения 261, с получением соединения 63 (0,013 г, 5,2%).
Стадия-1: Синтез 6-бром-2-метилпиридин-3-амина

К перемешиваемому раствору 6-бром-2-метил-3-нитропиридина (15 г, 69,12 ммоль) в этаноле (280 мл) добавляли железный порошок (57,9 г, 1036 ммоль), а затем конц. HCl (30 мл). Реакционную смесь перемешивали в течение 6 ч при 100°C, после завершения реакции (контролируемой ТСХ), реакционную смесь охлаждали до комнатной температуры, фильтровали через целит. Фильтрат подщелачивали насыщенным раствором NaHCO3, экстрагировали EtOAc. Органический слой промывали водой, затем насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт использовали на следующей стадии без дополнительной очистки с получением желаемого соединения (5,6 г, 65%) в виде не совсем белого твердого вещества.
Стадия-2: Синтез 5-бром-1H-пиразоло[4,3-b]пиридина

К перемешиваемому раствору 6-бром-2-метил-3-нитропиридина (5 г, 26,73 ммоль) в хлороформе добавляли ацетат калия (3,14 г, 32,08 ммоль) и уксусный ангидрид (10,9 г, 106 ммоль) при 10°C, содержимое перемешивали при температуре 65°C в течение 2 ч. После завершения реакции (контролируемой ТСХ), реакционную смесь охлаждали до комнатной температуры, добавляли по каплям в течение 15 мин изоамилнитрит (3,75 г, 32,06 ммоль), затем добавляли 18-краун-6 (0,7 г, 2,67 ммоль), реакционную смесь перемешивали при 65°C в течение 16 ч. После завершения реакции (контролируемой ТСХ), реакционную смесь охлаждали до комнатной температуры, смесь метанола и воды 120 мл (1:1) добавляли к реакционной смеси, затем карбоната калия (34,48 г, 24,99 ммоль) и KOH (4,2 г, 74,97 ммоль), реакционную смесь перемешивали в течение 3 ч при комнатной температуре. После завершения реакции реакционную смесь добавляли, разбавляли этилацетатом и органический слой промывали водой, затем насыщенным солевым раствором. Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении, получая указанное в заголовке соединение (5 г) в виде не совсем белого твердого вещества.
Стадия-3: Синтез 5-бром-1-(тетрагидро-2Н-пиран-2-ил)-1H-пиразоло[4,3-b]пиридина

Реакцию осуществляли в соответствии со Схемой 1, Стадия-1, с получением неочищенного материала, который очищали колоночной хроматографией на силикагеле 230-400 меш, используя 10-15% этилацетата в н-гексане, с получением 5-бром-1-(тетрагидро-2Н-пиран-2-ил)-1H-пиразоло[4,3-b]пиридина (5 г, 78%).
Стадия-4: Синтез 5-(бут-1-ин-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-пиразоло[4,3-b]пиридина

Реакцию осуществляли в соответствии со Схемой 1, Стадия-2, с получением неочищенного материала, который очищали с помощью колоночной хроматографии на силикагеле 230-400 меш, используя 10-15% этилацетат в н-гексане, с получением указанного в заголовке соединения Пр.63 Стадия-4 (3,5 г, 77,7%) в виде коричневого масла.
Стадия-5: Синтез трет-бутил (2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)этил)карбамата

К раствору трет-бутил (2-(4-йодфенокси)этил)карбамата (25 г, 68,87 ммоль) в DMSO (200 мл) добавляли ацетат калия (20,24 г, 206,6 ммоль) ибис(пинаколато)диборон (34,98 г, 137,74 ммоль). Содержимое дегазировали с помощью трех циклов вакуум/N2, добавляли Pd(dppf)Cl2.CH2Cl2(2,8 г, 3,44 ммоль) и полученную смесь перемешивали при 75°C в течение 16 ч. По завершении ТСХ, реакционную смесь гасили водой (500 мл) и экстрагировали этилацетатом (2×250 мл). Объединенные органические экстракты промывали водой, насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный материал очищали при помощи колоночной хроматографии, элюировали 10-15% этилацетатом в н-гексане с получением указанного в заголовке соединения Пр. 63 Стадия-5 (50 г, смесь с бис(пинаколато)дибороном).
Стадия-6: Синтез трет-бутил (E)-(2-(4-(2-фенил-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-пиразоло[4,3-b]пиридин-5-ил)бут-1-ен-1-ил)фенокси)этил)карбамата

Реакцию проводили согласно схеме 1, Стадия-6, с использованием 5-(бут-1-ин-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-пиразоло[4,3-b]пиридина для соединения 204, иодбензола для соединения 209, трет-бутил (2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)этил)карбамата для соединения 210, с получением неочищенного материала, который очищали колоночной хроматографией на силикагеле с получением указанного в заголовке соединения Пр. 63 Стадия-6 (4 г, неочищенный).
Соединение 63:1H ЯМР (400 MГц, DMSO-d6): δ 13,29 (с, 1H), 8,26 (с, 1H), 7,97 (с, J=8,8 Гц, 1H), 7,24-7,13 (м, 6H), 6,75 (д, J=8,8 Гц, 2H), 6,62-6,57 (м, 3H), 6,50 (д, J=15,2 Гц, 1H), 3,87 (т, J=5,6 Гц, 2H), 3,3-3,29 (м, 2H), 2,98 (с, 3H), 2,84 (с, 3H), 2,78 (т, J=5,4 Гц, 2H), 2,37-2,30 (м, 2H), 0,86 (т, J=7,6 Гц, 3H). ЖХМС: 496,4 [M+H]+.
Пример 64: Синтез (E)-4-((2-(3-фтор-4-((Z)-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 64)
Соединение синтезировали в соответствии с подходом, описанным в примере 50, с использованием на стадии-2 трет-бутил (Z)-(2-(3-фтор-4-(1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)карбамата (получение показано ниже на стадии 1-4) вместо соединения 261, с получением соединения 64 (0,16 г, 17,4%).
Стадия-1: Синтез 4-амино-3-фторфенола

Смесь 3-фтор-4-нитрофенола (6 г, 38,4 ммоль) и 10% Pd/C (1,8 г) в EtOH (60 мл) и THF (36 мл) перемешивали в атмосфере H2 (1 атм) в течение 16 часов. После фильтрации фильтрат концентрировали с получением 4,68 г (96,5%) 4-амино-3-фторфенола.
Стадия-2: Синтез 3-фтор-4-иодфенола
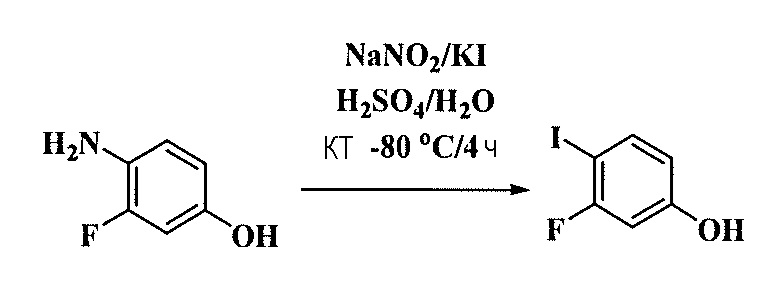
4-Амино-3-фторфенол (5 г, 39,4 ммоль) суспендировали в 15 мл воды и H2SO4 (4,62 г), смесь охлаждали на бане с ледяной солью до -5°C. К этой смеси добавляли по каплям раствор нитрита натрия (2,71 г, 39,4 ммоль) в 15 мл воды. Внутренняя температура была ниже +2°C. Полученный коричневый раствор перемешивали еще 15 мин при -5°C, затем по каплям медленно добавляли раствор иодида калия (7,87 г, 47,2 ммоль) в 30 мл воды. После завершения добавления, реакционную смесь перемешивали при комнатной температуре в течение 4 ч. По завершении реакции реакционную смесь разбавляли EtOAc. Органический слой промывали водой, затем насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения Пр.64 Стадия-2 (5 г, 53,3%).
Стадия-3: Синтез трет-бутил (2-(3-фтор-4-йодфенокси)этил)карбамата

Реакцию осуществляли в соответствии со Схемой 3, Стадия-1, с использованием 3-фтор-4-иодфенола и трет-бутил (2-бромэтил)карбамата, с получения неочищенного материала, который очищали колоночной хроматографией на силикагеле, с получением трет-бутил (2-(3-фтор-4-йодфенокси)этил)карбамата (5 г).
Стадия-4: Синтез трет-бутил (Z)-(2-(3-фтор-4-(1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)карбамата

Реакцию осуществляли в соответствии с подходом, описанным в примере 50, Стадия-1, с использованием (Z)-5-(1,2-бис(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (8 г, 15,19 ммоль) вместо соединения 260 и трет-бутил (2-(3-фтор-4-йодфенокси)этил)карбамата (5,4 г, 14,43 ммоль) вместо соединения 257 с получением неочищенного материала. Материал очищали хроматографией на силикагеле, используя 15% EtOAc в н-гексане, с получением указанного в заголовке соединения Пр.64 Стадия-4 (3,5 г, 38%).
Соединение 64:1H ЯМР (400 MГц, DMSO-d6): δ 12,57 (с, 1H), 7,49 (с, 1H), 7,46 (д, J=2,0 Гц, 1H), 7,27 (дд, J1=8,5 Гц, J2=1,2 Гц, 1H), 7,21-7,09 (м, 3H), 6,92 (т, J=8,6 Гц, 1H), 6,62-6,47 (м, 4H), 3,88 (т, J=5,6 Гц, 2H), 3,30 (с, 2H), 2,98 (с, 3H), 2,83 (с, 3H), 2,76 (т, J=5,6 Гц, 2H), 2,44 (кв, J=7,8 Гц, 2H), 2,10 (шир.с, 1H), 0,89 (т, J=7,3 Гц, 3H). ЖХМС: 531,3 [M+H]+.
Пример 65: Синтез (E)-4-((2-(4-((E)-2-(2,4-дифторфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 65)
Соединение синтезировали в соответствии с подходом, описанным в примере 50, с использованием на стадии-1 2,4-дифтор-1-иодбензола, с получением соединения 65 (0,06 г, 23,7%).
Соединение 65:1H ЯМР(400 MГц, DMSO-d6): δ 12,59 (с, 1H), 7,52 (с, 1H), 7,49 (дд, J1=8,8 Гц, J2=1,2 Гц, 1H), 7,28-7,21 (м, 2H), 7,09 (дд, J1=10 Гц, J2=2,4 Гц, 1H), 6,97 (дд, J1=8,4 Гц, J2=2,4 Гц, 1H), 6,80 (д, J=8,4 Гц, 2H), 6,64 (д, J=8,8 Гц, 2H), 6,63-6,58 (м, 1H), 6,50 (д, J=15,2 Гц, 1H), 3,88 (т, J=5,6 Гц, 2H), 3,32-3,31 (м, 2H), 2,98 (с, 3H), 2,84 (с, 3H), 2,78 (т, J=5,4 Гц, 2H) 2,34 (кв, J=7,6 Гц, 2H), 0,89 (т, J=7,6 Гц, 3H). ЖХМС: 549,3[M+H]+
Пример 66: Синтез (E)-4-((2-(4-((E)-1-(3,6-дифтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 66)
Соединение синтезировали в соответствии с подходом, описанным в примере 50, посредством (i) использования на стадии-1 трет-бутил (Z)-(2-(4-(1-(3,6-дифтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)фенокси)этил)карбамата (получение показано ниже в Пр.66 Стадии 1-7) вместо соединения 260 и иодбензола вместо соединения 251, и (ii) проведения стадии 5 пр.50, следуя условиям реакции, описанным на схеме 2, стадия-6b, с получением соединения 66 (0,012 г).
Стадия-1: Синтез N-(5-фтор-2-метилфенил)ацетамида

К перемешиваемому раствору 5-фтор-2-метиланилина (25 г, 200 ммоль) в толуоле добавляли уксусный ангидрид (25 мл) при 10°C, содержимое перемешивали при температуре кипения с обратным холодильником в течение 2 ч. После завершения реакции (контролируемой ТСХ), реакционную смесь охлаждали до комнатной температуры, полученный твердый продукт отфильтровывали и промывали диэтиловым эфиром и гексаном с получением желаемого соединения (28 г, 84%).
Стадия-2: Синтез N-(4-бром-5-фтор-2-метилфенил)ацетамида

К перемешиваемому раствору N-(5-фтор-2-метилфенил)ацетамида (28 г, 167 ммоль) в уксусной кислоте (150 мл) добавляли бром (9,6 мл, 186 ммоль) при 10°C. Реакционную смесь перемешивали в течение 12 ч при комнатной температуре, после завершения реакции (контролируемой ТСХ), твердое вещество отфильтровывали и сушили при пониженном давлении. Неочищенный продукт использовали на следующей стадии без дополнительной очистки с получением N-(4-бром-5-фтор-2-метилфенил)ацетамида (40 г, 97%).
Стадия-3: Синтез 1-(5-бром-6-фтор-1H-индазол-1-ил)этан-1-она

К перемешиваемому раствору 4-бром-5-фтор-2-метиланилина (40 г, 163 ммоль) в хлороформе (400 мл) добавляли ацетат калия (32 г, 326 ммоль) и уксусный ангидрид (45 мл) при 10°C, содержимое перемешивали при температуре 65°C в течение 2 ч. После завершения реакции (контролируемой ТСХ), реакционную смесь охлаждали до комнатной температуры, добавляли по каплям в течение 15 мин изоамилнитрит (50 мл, 371 ммоль), затем добавляли 18-краун-6 (2,16 г, 8,18 ммоль), реакционную смесь перемешивали при 65°C в течение 12 ч. После завершения реакции (контролируемой ТСХ), реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом. Органический слой промывали водой, затем насыщенным солевым раствором. Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением указанного в заголовке соединения Пр.66 Стадии-3 (38,3 г, 44%).
Стадия-4: Синтез 5-бром-6-фтор-1H-индазола

К перемешиваемому раствору 1-(5-бром-6-фтор-1H-индазол-1-ил)этан-1-она (20 г, 78,43 ммоль) в 3M HCl (400 мл) добавляли метанол (80 мл), реакционную смесь перемешивали при 90°C в течение 3 ч. После завершения реакции (контролируемой ТСХ), реакционную смесь охлаждали до комнатной температуры и подщелачивали 1M NaOH, твердое вещество отфильтровывали и сушили при пониженном давлении с получением указанного в заголовке соединения Пр.66 Стадия-4 (7,6 г, 45%).
Стадия-5: Синтез 5-бром-6-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола

Реакцию проводили в соответствии со Схемой 1, Стадия-1, с использованием 5-бром-6-фтор-1H-индазола (7,6 г, 35,34 ммоль) вместо соединения 201. Неочищенный материал, полученный в результате реакции, очищали хроматографией на силикагеле, используя 4% этилацетат в н-гексане, с получением 5-бром-6-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (8,83 г, 84%).
Стадия-6: Синтез 5-(бут-1-ин-1-ил)-3,6-дифтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола

Соединение синтезировали в соответствии с подходом, описанным в примере 2, (i) с использованием на стадии-1 5-бром-6-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола вместо соединения 270 и (ii) следуя условиям реакции стадий 2 и 3 Пр. 2, с получением указанного в заголовке соединения Пр.66 Стадия-6 (12,19 г, 87%).
Стадия-7: Синтез трет-бутил (Z)-(2-(4-(1-(3,6-дифтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)фенокси)этил)карбамата

Соединение синтезировали в соответствии с подходом, описанным в примере 14 (i) с использованием на стадии-2 5-(бут-1-ин-1-ил)-3,6-дифтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола вместо соединения 271 и (ii) с использованием на стадии-4 трет-бутил (2-(4-йодфенокси)этил)карбамата (полученного в Примере 3, Стадия-2) вместо соединения 234, с получением указанного в заголовке соединения в Пр.66 Стадия-7.
Соединение 66:1H ЯМР (400 MГц, DMSO-d6): δ 12,65 (с, 1H), 7,69 (д, J=6,8 Гц, 1H), 7,34 (дд, J1=6,0 Гц, J2=2,0 Гц, 1H), 7,25-7,21 (м, 2H), 7,17-7,14 (м, 3H), 6,79 (д, J=8,8 Гц, 2H), 6,64-6,57 (м, 3H), 6,49 (д, J=15,2 Гц, 1H), 3,87 (т, J=5,6 Гц, 2H), 2,98 (с, 3H), 2,84 (с, 3H), 2,77 (т, J=5,6 Гц, 2H), 2,33-2,31(м, 2H), 0,81 (т, J =7,6 Гц, 3H). ЖХМС: 531,3 [M+H]+.
Пример 67: Синтез (E)-4-((2-(4-((E)-1-(3-фтор-1H-индазол-5-ил)-2-(пиридин-2-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 67)
Соединение синтезировали в соответствии с подходом, описанным в примере 14, с использованием на стадии-4 (Z)-5-(1,2-бис(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (полученного в Примере 3, Стадия-1) вместо соединения 233 и 2-йодпиридина вместо соединения 262, с получением соединения 67 (0,010 г) в виде не совсем белого твердого вещества.
Соединение 67:1H ЯМР (400 MГц, DMSO-d6): δ 12,6 (с, 1H), 8,56 (д, J=4,4 Гц, 1H), 7,53 (с, 1H), 7,50-7,45 (м, 2H), 7,21 (дд, J1=8,8 Гц, J2=1,4 Гц, 1H), 7,13 (дд, J1=4,9 Гц, J2=6,9 Гц, 1H), 6,87 (д, J=7,8 Гц, 1H), 6,72 (д, J=8,8 Гц, 2H), 6,64-6,57 (м, 3H), 6,50 (д, J=15,1 Гц, 1H), 3,88 (т, J=5,7 Гц, 2H), 3,32-3,21 (м, 2H), 299 (с, 3H), 2,84 (с, 3H), 2,78 (т, J=5,6 Гц, 2H), 2,54-2,52 (м, 2H), 0,86 (т, J=7,3 Гц, 3H). ЖХМС: 514,3 [M+H]+.
Пример 68: Синтез (E)-4-((2-((5-((Z)-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-2-ил)окси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 68)
Соединение синтезировали в соответствии с подходом, описанным в примере 14, с использованием на стадии-4 (Z)-5-(1,2-бис(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (полученного в Примере 3, Стадия-1) вместо соединения 233 и трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-((5-йодпиридин-2-ил)окси)этил)карбамата (полученного в Примере 25, Стадия-2) вместо соединения 234, с получением соединения 68 (0,165 г, 11%).
Соединение 68:1H ЯМР (400 MГц, DMSO-d6): δ 12,6 (с, 1H), 7,59 (д, J=2,0 Гц, 1H), 7,56 (с, 1H), 7,50 (дд, J1=8,8 Гц, J2=2,0 Гц, 1H), 7,26-7,22 (м, 3H), 7,18-7,14 (м, 4H), 6,61-6,56 (м, 1H), 6,51-6,46 (м, 2H), 4,13 (т, J=5,6 Гц, 2H), 3,30-3,29 (м, 2H), 3,29 (с, 3H), 2,84 (с, 3H), 2,76 (т, J=5,9 Гц, 2H), 2,42 (кв, J=7,4 Гц, 2H), 2,1-2,0 (шир.с, 1H), 0,88 (т, J=7,3 Гц, 3H). ЖХМС: 514,3 [M+H]+.
Пример 69: Синтез (E)-N,N-диметил-4-((2-(4-((E)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)бут-2-енамида (Соединение 69)
Стадия-1: Синтезтрет-бутил (E)-(2-(4-(4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)карбамата

К перемешиваемому раствору 3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-5-(4,4,4-трифторбут-1-ин-1-ил)-1H-индазола (1,6 г, 4 ммоль, Пример 59, Стадия-3) в 2-метил THF (30 мл), добавляли бис(пинаколато)диборон (1,24 г, 4 ммоль), тетракис(трифенилфосфин)платина(0) (0,06 г, 0,04 ммоль) в атмосфере азота, реакционную смесь перемешивали при 90°C в течение 12 ч. Раствору давали остыть до комнатной температуры и добавляли трет-бутил (2-(4-йодфенокси)этил)карбамат (1,78 г, 4 ммоль, Пример 3, Стадия-2), бис(трифенилфосфин)палладий(II) дихлорид (0,172 г, 0,2 ммоль), карбонат цезия (3,19 г, 9 ммоль) и 2-метил THF (30 мл). Эту смесь дегазировали азотом и добавляли воду (0,3 мл). Эту смесь перемешивали при комнатной температуре в течение 16 ч. После завершения реакции, к вышеуказанной реакционной смеси добавляли 4M KOH (1,47 г, 26 ммоль) и иодбензол (1,95 г, 9 ммоль). Реакционную смесь перемешивали при 90°C в течение 12 ч. После завершения реакции, реакционную смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали EtOAc. Органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный материал очищали при помощи колоночной хроматографии на силикагеле 230-400 меш с использованием 15% EtOAc в н-гексане, с получением трет-бутил (E)-(2-(4-(4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)карбамата (1,5 г, 50%).
Стадия-2: Синтез (E)-2-(4-(4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этан-1-амина

Реакцию осуществляли в соответствии со Схемой 2, Стадия-2, с использованием трет-бутил (E)-(2-(4-(4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)карбамата (1,5 г, 2,3 ммоль) с получением неочищенного указанного в заголовке соединения (1,1 г неочищенный).
Стадия-3: Синтез (E)-N,N-диметил-4-((2-(4-((E)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)бут-2-енамида

Реакцию осуществляли в соответствии со Схемой 2, Стадия-3, используя (E)-2-(4-(4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этан-1-амин (1,1 г, 2 ммоль) и смесь (E)-4-бром-N,N-диметилбут-2-енамида и (E)-4-хлор-N,N-диметилбут-2-енамида (0,41 г, 2 ммоль, Пример 63, Стадия-8). Неочищенный материал использовали на следующей стадии без дополнительной очистки (1,8 г, неочищенный).
Стадия-4: Синтез трет-бутил ((E)-4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-((E)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)карбамата

Процедуру, описанную на схеме 2, стадия-5, осуществляли с (E)-N,N-диметил-4-((2-(4-((E)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)бут-2-енамидом (1,8 г, 3,18 ммоль), с получением неочищенного материала, который очищали хроматографией на силикагеле с использованием (2%) MeOH в дихлорметане, с получением трет-бутил ((E)-4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-((E)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)карбамата (0,2 г).
Стадия-5: Синтез (E)-N,N-диметил-4-((2-(4-((E)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)бут-2-енамида

Реакцию осуществляли в соответствии со Схемой 2, Стадия-6b, с использованием трет-бутил ((E)-4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-((E)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)карбамата (0,2 г, 0,3 ммоль), с получением неочищенного соединения, которое очищали препаративной ТСХ с получением желаемого соединения (E)-N,N-диметил-4-((2-(4-((E)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)бут-2-енамида (0,05 г, 29%).
Соединение 69:1H ЯМР (400 MГц, CDCl3): δ 9,40 (с, 1H), 7,66 (с, 1H), 7,39-7,15 (м, 7H), 6,91-6,86 (м, 1H), 6,79 (д, J=8,8 Гц, 2H), 6,56 (д, J=8,8 Гц, 2H), 6,44 (д, J=15,1 Гц, 1H), 3,95 (т, J=5,7 Гц, 2H), 3,46 (кв, J=10,8 Гц, 2H), 3,32-3,29 (м, 2H), 3,06 (с, 3H), 3,00 (с, 3H), 2,95 (т, J=5,4 Гц, 2H). ЖХМС: 567,3 [M+H]+.
Пример 69A: Синтез (E)-N,N-диметил-4-((2-(4-((E)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)бут-2-енамид гидрохлорида
Стадия-1A: Синтез 3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-5-((триметилсилил)этинил)-1H-индазола

К перемешиваемому раствору 5-бром-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (1,5 кг, 5,014 моль) в Et3N (10 л) в герметичном сосуде, добавляли йодид меди (95,5 г, 0,501 моль) при комнатной температуре. Эту смесь дегазировали с помощью трех циклов вакуум/N2, добавляли этинилтриметилсилан (0,73 кг, 7,52 моль), затем Pd(PPh3)2Cl2 (175 г, 0,25 моль). Сосуд высокого давления герметизировали и нагревали при 80°C в течение 16 ч. По завершении ТСХ, реакционную смесь разбавляли EtOAc (10 л), промывали водой. Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного продукта 3-фтор--(тетрагидро-2Н-пиран-2-ил)-5-((триметилсилил)этинил)-1H-индазола в виде темно-коричневого масла (1,71 кг, неочищенный) 65,7% чистота ВЭЖХ.
Стадия-2A: Синтез 5-этинил-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола

К перемешиваемому раствору 3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-5-((триметилсилил)этинил)-1H-индазола (1,7 кг, 5,37 моль) в метаноле 12 л добавляли карбонат калия (74,6 г, 0,53 моль), реакционную смесь перемешивали в течение 16 ч при комнатной температуре. После завершения реакции метанол выпаривали, остаток разбавляли этилацетатом (6 л) и органический слой промывали водой, затем насыщенным солевым раствором. Объединенный органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением указанного в заголовке соединения (1,5 кг неочищенный) 72,3% чистота ВЭЖХ.
Стадия-3A: Синтез 3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-5-(4,4,4-трифторбут-1-ин-1-ил)-1H-индазола

Смесь Pd2(dba)3 (0,187 кг, 0,204 моль), DPEphos (0,44 кг, 0,818 моль) и DABCO (0,91 кг, 0,818 моль) в атмосфере азота в толуоле (5 л) добавили раствор 5-этинил-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (1 кг, 4,093 моль) в толуоле (5 л). Реакционную смесь дегазировали азотом в течение 10 мин, к вышеуказанной смеси добавляли 1,1,1-трифтор-2-иодэтан (1,71 кг, 8,187 моль). Эту смесь снова дегазировали с помощью трех циклов вакуум/N2, нагревали при 80°C в течение 16 ч. По завершении ТСХ, реакционную смесь охлаждали до комнатной температуры, фильтровали через целит. Фильтрат разбавляли водой и экстрагировали EtOAc (2×3 л). Органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный материал очищали при помощи колоночной хроматографии на силикагеле 60-120 меш, используя 6% этилацетат в н-гексане в качестве элюента, с получением 3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-5-(4,4,4-трифторбут-1-ин-1-ил)-1H-индазола (1,1 кг, выход 82%, 78% чистота ВЭЖХ).
Стадия-4A: Синтезтрет-бутил ((E)-4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)фенокси)этил)карбамата

К перемешиваемому раствору 3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-5-(4,4,4-трифторбут-1-ин-1-ил)-1H-индазола (500 г, 1,532 моль) в 2-метил THF (2,5 л), добавляли бис(пинаколато)диборон (0,39 кг, 1,532 моль), тетракис(трифенилфосфин)платину(0) (19 г, 0,0153 моль) в атмосфере азота, реакционную смесь перемешивали при 85°C в течение 7 ч. Реакционной смеси давали остыть до комнатной температуры, добавляли трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)карбамат (0,62 кг, 1,072 моль), бис(трифенилфосфин)палладий(II) дихлорид (53,7 г, 0,076 моль), карбонат цезия (0,6 кг, 3,064 моль), 2-метил THF (5 л) и воду (250 мл). Эту смесь дегазировали азотом в течение 15 мин, перемешивали при комнатной температуре в течение 36 ч. После завершения реакции реакционную смесь охлаждали до комнатной температуры, фильтровали через целит. Фильтрат разбавляли водой и экстрагировали EtOAc (2×3 л). Органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный материал очищали колоночной хроматографией на силикагеле 60-120 меш, используя 5% MeOH в дихлорметане в качестве элюента, для получения трет-бутил ((E)-4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)фенокси)этил)карбамата (600 г, 49%), чистота ВЭЖХ 58%.
Стадия-5A: Синтез трет-бутил ((E)-4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-((E)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)карбамата

К перемешиваемому раствору трет-бутил ((E)-4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-((Z)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)фенокси)этил)карбамата (600 г, 0,74 моль) в 2-метил THF (6 л), добавляли к вышеуказанной реакционной смеси 4M KOH (1,2 л, 2 об.), добавляли тетракис(трифенилфосфин)палладий(0) (43 г, 0,037 моль) и иодбензол (128 г, 0,62 моль). Реакционную смесь перемешивали при 90°C в течение 5 ч. После завершения реакции реакционную смесь охлаждали до комнатной температуры, а затем фильтровали через целит. Фильтрат разбавляли водой и экстрагировали EtOAc (2×2 л). Органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением указанного в заголовке соединения трет-бутил ((E)-4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-((E)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)карбамата (700 г, неочищенный) 48% чистота ВЭЖХ.
Стадия-6A: Синтез (E)-N,N-диметил-4-((2-(4-((E)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)бут-2-енамида

К перемешиваемому раствору трет-бутил ((E)-4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-((E)-4,4,4-трифтор-1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)карбамата (700 г, 0,93 моль) в метаноле (3,5 л) добавляли при 0°C конц.HCl (2,1 л, 3 об.). Реакционную смесь перемешивали в течение 16 ч при комнатной температуре. После завершения реакции, реакционную смесь концентрировали при пониженном давлении. Вязкую реакционную массу подщелачивали насыщенным Na2CO3, экстрагировали дихлорметаном (3×2 л). Объединенные органические слои сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного соединения, которое очищали при помощи колоночной хроматографии на силикагеле 60-120 меш с использованием 5-7% MeOH в дихлорметане с получением желаемого соединения (E)-N,N-диметил-4-((2-(4-((E)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)бут-2-енамида (260 г,) 65,3% чистота ВЭЖХ.
Полученное соединение дополнительно очищали с помощью колоночной хроматографии combi flash с получением указанного в заголовке соединения с >92% ВЭЖХ.
Стадия-7A: Синтез (E)-N,N-диметил-4-((2-(4-((E)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)бут-2-енамид гидрохлорида

К перемешиваемому раствору (E)-N,N-диметил-4-((2-(4-((E)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)бут-2-енамида (5,6 г, 9,88 ммоль) в этаноле (6,6 мл) добавляли 2M HCl в диэтиловом эфире (56 мл) при 0°C. Белое твердое вещество наблюдали после перемешивания в течение 1 ч при комнатной температуре. Реакционную смесь концентрировали в вакууме при 30°C для удаления избытка эфира HCl и этанола. Полученное твердое вещество сушили при 50°С в течение 1 ч, остаток растирали с диэтиловым эфиром и эфирный слой декантировали (500 мл×3). Полученное твердое вещество подвергали совместной перегонке с этилацетатом (300 мл), затем растирали с этилацетатом (500 мл), фильтровали и сушили при пониженном давлении с получением указанного в заголовке соединения (E)-N,N-диметил-4-((2-(4-((E)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)бут-2-енамид гидрохлорида (5,4 г, 90%).
Соединение 69 (хлористоводородная соль):1H ЯМР (400 MГц, DMSO-d6): δ 12,70 (с, 1H), 9,15 (с, 2H), 7,57 (с, 1H),7,54 (м, 2H), 7,25-7,14 (м, 6H), 6,86-6,79 (м, 3H), 6,69 (д, J=5,8 Гц, 2H), 6,61-6,54 (м, 1H), 4,13 (т, J=4,4 Гц, 2H), 3,79 (м, 2H), 3,48-3,37 (м, 2H), 3,26 (м, 2H) 3,03 (с, 3H), 2,87 (с, 3H). ЖХМС: 567,2 [M+H]+.
На стадиях 8А-12А представлен синтез промежуточного соединения трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)карбамата
Стадия 8A: Синтез трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)карбамата

Стадия-9A: Синтез трет-бутил (2-бромэтил)карбамата

Бромэтиламин гидробромид (1 кг, 4,88 моль) добавляли к перемешиваемому раствору карбоната натрия (1,55 кг, 14,6 моль), ди-трет-бутилдикарбоната (1,6 кг, 7,32 моль) в смеси 1,4-диоксан-вода (2:1, 3 л) при 0°С и смесь перемешивали при комнатной температуре в течение 12 ч. После завершения реакции, избыток 1,4-диоксана удаляли при пониженном давлении. Реакционную смесь экстрагировали этилацетатом (2×4 л), органический слой промывали водой, затем насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт использовали на следующей стадии без дополнительной очистки с получением (1,1 кг, %).1H ЯМР(400 MГц, DMSO-d6): δ 7,07 (шир.с, 1H), 3,42 (т, 2H), 3,31 (т, 2H), 1,38 (с, 9H).
Стадия-10A: Синтез трет-бутил (2-(4-йодфенокси)этил)карбамата

К перемешиваемому раствору 4-иодфенола (1 кг, 4,54 моль) в DMF (15 л) добавляли карбонат калия (1,8 кг, 13,6 моль) и перемешивали в течение 30 мин при комнатной температуре, к вышеуказанной смеси добавляли трет-бутил (2-бромэтил)карбамат (1,48 кг, 6,36 моль). Содержимое перемешивали при 70°C в течение 12 ч. После завершения реакции, реакционную смесь выливали в ледяную воду, твердое вещество отфильтровывали и сушили при пониженном давлении с получением желаемого соединения трет-бутил (2-(4-йодфенокси)этил)карбамата в виде не совсем белого твердого вещества (1,4 кг, 84%).1H ЯМР(400 MГц, DMSO-d6): δ 7,58 (д, J=8,8 Гц, 2H), 7,01 (т, J=5,2 Гц, 1H), 7,78 (д, J=8,8 Гц, 2H), 3,92 (т, J=5,6 Гц, 2H), 1,37 (с, 9H).
Стадия-11A: Синтез 2-(4-йодфенокси)этан-1-амина

К перемешиваемому раствору трет-бутил (2-(4-йодфенокси)этил)карбамата (1,4 кг, 3,85 моль) в метаноле (10 л) добавляли при 0°C конц.HCl (2,8 л). Реакционную смесь перемешивали в течение 12 ч при комнатной температуре. После завершения реакции метанол выпаривали, остаток подщелачивали насыщенным Na2CO3, экстрагировали 10% МеОН в DCM. Органический слой концентрировали при пониженном давлении и неочищенный материал использовали на следующей стадии без дополнительной очистки (700 г, выход 70%, 98% чистота ВЭЖХ).1H ЯМР(400 MГц, DMSO-d6): δ 7,59-7,55 (м, 2H), 6,80-6,76 (м, 2H), 3,88 (т, J=5,8 Гц, 2H), 3,85 (т, J=6,0 Гц, 2H), 1,61 (шир.с, 2H).
Стадия-12A: Синтез (E)-4-((2-(4-йодфенокси)этил)амино)-N,N-диметилбут-2-енамида

К перемешиваемому раствору 2-(4-йодфенокси)этан-1-амина (700 г, 2,66 моль) в DCM (4 л) добавляли DIPEA (1 л, 7,98 моль) и охлаждали до 0°C. Затем, (E)-4-бром-(хлор)-N,N-диметилбут-2-енамид (505 г, 2,66 моль) в DCM (2 л) добавляли по каплям при 0°C. Реакционную смесь перемешивали в течение 12 ч при комнатной температуре. После этого, реакционную смесь охлаждали до 0°C и добавляли по каплям Boc ангидрид (870 г, 3,9 моль) в DCM (2 л) и перемешивали при комнатной температуре в течение 12 ч. По завершении ТСХ, реакционную смесь охлаждали до 0°C, гасили ледяной водой (5 л) и экстрагировали дихлорметаном (2 л). Объединенные органические экстракты промывали водой, затем насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный материал очищали колоночной хроматографией на силикагеле 60-120 меш, используя 20% EtOAc в гексане в качестве элюента, с получением трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)карбамата (480 г, выход 37%). 69% чистота ВЭЖХ.1H ЯМР(400 MГц, DMSO-d6): δ 7,58 (д, J=8,8 Гц, 2H), 6,79 (д, J=6,8 Гц, 2H), 6,51-6,39 (м, 2H), 4,0-3,99 (м, 2H), 3,95 (т, J=5,6 Гц, 2H), 3,38 (д, J=5,6 Гц, 2H), 2,96 (с, 3H), 2,84 (с,3H), 1,37 (с, 9H). ЖХМС: 595,4[M+H]+.
Пример 70: Примера 70 нет.
Пример 71: Примера 71 нет.
Пример 72: Синтез (E)-4-((2-((5-((Z)-2-циклобутил-1-(3-фтор-1H-индазол-5-ил)-2-фенилвинил)пиридин-2-ил)окси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 72)
Соединение синтезировали в соответствии с подходом, описанным в примере 14, с использованием на стадии-4 трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-((5-йодпиридин-2-ил)окси)этил)карбамата (полученного в Примере 25, Стадия-2) вместо соединения 234 и заменяя Стадию-5 на условия реакции, описанные на схеме 2, Стадия-6b, с получением соединения 72 (0,09 г, 11%).
Соединение 72:1H ЯМР (400 MГц, DMSO-d6): δ 12,59 (с, 1H), 7,67 (д, J=2,0 Гц, 1H), 7,54 (с, 1H), 7,49 (дд, J1=8,8 Гц, J2=1,6 Гц, 1H), 7,28-7,12 (м, 7H), 6,61-6,44 (м, 3H), 4,09 (т, J=6,0 Гц, 2H), 3,42-3,38 (м, 1H), 3,28-3,27 (м, 2H), 2,97 (с, 3H), 2,83 (с, 3H), 2,73 (т, J=5,8 Гц, 2H), 1,83-1,76 (м, 4H), 1,63-1,58 (м, 1H), 1,35-1,33 (м, 1H). ЖХМС: 540,3[M+H]+.
Пример 73: Синтез (E)-4-((2-(4-((E)-2-(2-хлорфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 73)
Соединение синтезировали в соответствии с подходом, описанным в примере 60, с использованием на стадии-1 (i) 5-(бут-1-ин-1-ил)-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (полученного в Примере 2, Стадия-3) вместо соединения 275, (ii) трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)карбамата (полученного в Примере 14, Стадия-3) вместо соединения 263, и (iii) 1-хлор-2-иодбензола вместо соединения 264, с получением соединения 73 (0,23 г, 5,6%) в виде не совсем белого твердого вещества.
Соединение 73:1H ЯМР(400 MГц, DMSO-d6): δ 12,59 (с, 1H), 7,54 (с, 1H), 7,49 (дд, J1=8,8 Гц, J2=2,0 Гц, 1H), 7,35 (дд, J1=7,4 Гц, J2=1,4 Гц, 1H), 7,27-7,16 (м, 4H), 6,83 (д, J=8,8 Гц, 2H), 6,60 (д, J=8,8 Гц, 2H), 6,57-6,47 (м, 2H), 3,86 (т, J=5,6 Гц, 2H), 2,98 (с, 3H), 2,84 (с, 3H), 2,77 (т, J=5,4 Гц, 2H), 2,43-2,32 (м, 4H), 0,88 (т, J=7,4 Гц, 3H). ЖХМС: 547,2[M+H]+
Пример 74: Синтез (E)-4-((2-(4-((E)-2-(2-хлор-4-фторфенил)-1-(1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 74)
Соединение синтезировали в соответствии с подходом, описанным в примере 60, с использованием на стадии-1 (i) 5-(бут-1-ин-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (полученного в Примере 1, Стадия-3) вместо соединения 275, (ii) трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)карбамата (полученного в Примере 14, Стадия-3) вместо соединения 263 и (iii) 2-хлор-4-фтор-1-иодбензола вместо соединения 264 с выделением соединения 74 (0,26 г, 31%).
Соединение 74:1H ЯМР (400 MГц, DMSO-d6): δ 13,08 (с, 1H), 8,08 (с, 1H), 7,65 (с, 1H), 7,53 (д, J=8,8 Гц, 1H), 7,34-7,17 (м, 2H), 7,16-7,10 (м, 2H), 6,81 (д, J=8,8 Гц, 2H), 6,62-6,47 (м, 4H), 3,86 (т, J=5,6 Гц, 2H), 2,98 (с, 3H), 2,83 (с, 3H), 2,76 (т, J=5,4 Гц, 2H), 2,38-2,33 (м, 3H), 0,88 (т, J=7,4 Гц, 3H). ЖХМС: 547,3 [M+H]+.
Пример 75: Синтез (E)-4-((2-(4-((E)-1-(3-фтор-1H-индазол-5-ил)-2-(пиридин-4-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N-(2-гидроксиэтил)-N-метилбут-2-енамида (Соединение 75)
Соединение синтезировали в соответствии с подходом, описанным в примере 14, (а) с использованием на стадии-4 (i) (Z)-5-(1,2-бис(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (полученного в Примере 3, Стадия-1) вместо соединения 233, (ii) трет-бутил (E)-(4-((2-гидроксиэтил)(метил)амино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)карбамата (получение показано ниже в Пр.75 Стадия 1-2) вместо соединения 234, (iii) 4-йодпиридина вместо соединения 262, и (b) используя условия реакции, описанные на схеме 2, стадия-6b, в Пр.14 Стадия-5, с получением соединения 75 (0,1 г, 20,6%).
Стадия-1: Синтез (E)-N-(2-гидроксиэтил)-4-((2-(4-йодфенокси)этил)амино)-N-метилбут-2-енамида

Реакцию осуществляли в соответствии со Схемой 3, Стадия-3, используя 2-(4-йодфенокси)этан-1-амин (7,3 г, 27 ммоль, Пример 14, Стадия-3) вместо соединения 217 и (E)-4-бром-N-(2-гидроксиэтил)-N-метилбут-2-енамид вместо соединения 214 и (E)-4-хлор-N-(2-гидроксиэтил)-N-метилбут-2-енамид (5 г, 27 ммоль, Пример 50, Стадия-1) вместо соединения 214. Неочищенный материал использовали на следующей стадии без дополнительной очистки (9,5 г, неочищенный).
Стадия-2: Синтез трет-бутил (E)-(4-((2-гидроксиэтил)(метил)амино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)карбамата

Реакцию осуществляли в соответствии со Схемой 3, Стадия-4, используя (E)-N-(2-гидроксиэтил)-4-((2-(4-йодфенокси)этил)амино)-N-метилбут-2-енамид (9,5 г, 23,5 ммоль) вместо соединения 218. Неочищенный материал очищали при помощи колоночной хроматографии на силикагеле 230-400 меш, используя 3% MeOH в дихлорметане в качестве элюента, с получением указанного в заголовке соединения Пр.75 Стадия-2 (2,5 г, 21%).
Соединение 75:1H ЯМР(400 MГц, DMSO-d6): δ 12,60 (с, 1H), 8,38 (дд, J1=4,4 Гц, J2=1,7 Гц, 1H), 7,53 (с, 1H), 7,49 (д, J=1,6 Гц, 1H), 7,47 (д, J=2,0 Гц, 1H), 7,22 (д, J=1,6 Гц, 1H), 7,20 (д, J=1,6 Гц, 1H), 7,13 (дд, J1=4,2 Гц, J2=1,4 Гц, 2H), 6,79 (д, J=8,8 Гц, 2H), 6,65 (д, J=8,8 Гц, 2H), 6,60-6,51 (м, 2H), 3,88 (т, J=5,4 Гц, 2H), 3,50-3,48 (м, 2H), 3,47-3,34 (м, 2H), 3,03-2,85 (м, 4H), 2,86-2,78 (м, 2H), 2,42 (кв, J=7,4 Гц, 2H), 0,87 (т, J=7,2 Гц, 3H). ЖХМС: 544,3[M+H]+.
Пример 76: Синтез (E)-4-((2-(4-((E)-1-(7-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 76)
Соединение синтезировали в соответствии с подходом, описанным в примере 14, с использованием на стадии-2 5-(бут-1-ин-1-ил)-7-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (получение показано ниже в Пр. 76 Стадия 1-5) вместо соединения 271 и заменяя стадию 5 условиями реакции, описанными на схеме 2, стадия-6с, с получением соединения 76 (0,195 г, 17,7%) в виде не совсем белого твердого вещества.
Стадия-1: Синтез 5-бром-2,3-дифторбензальдегида

К перемешиваемому раствору 2,3-дифторбензальдегида (30 г, 211 ммоль) в H2SO4 (120 мл) добавляли N-бромсукцинамид (45 г, 253 ммоль) при 0°C. Реакционную смесь перемешивали в течение 16 ч при 60°C, после завершения реакции (контролируемой ТСХ), реакционную смесь разбавляли EtOAc. Органический слой промывали водой, затем насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали при помощи колоночной хроматографией, используя 0,8% EtOAc в н-гексане, с получением 5-бром-2,3-дифторбензальдегида (11,7 г, 23,5%) в виде белого твердого вещества.
Стадия-2: Синтез (E)-5-бром-2,3-дифторбензальдегид O-метилоксима

К перемешиваемому раствору 5-бром-2,3-дифторбензальдегида (2 г, 9 ммоль) в DMF (20 мл) добавляли O-метилгидроксиламин гидрохлорид (1,51 г, 18 ммоль) и K2CO3 (2,75 г, 19,9 ммоль), содержимое перемешивали при 40°C в течение 16 ч. После завершения реакции (контролируемой ТСХ), реакционную смесь охлаждали до 0°C, разбавляли этилацетатом и органический слой промывали водой, затем насыщенным солевым раствором. Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное соединение очищали с помощью combi-flash, с использованием 2% этилацетата в н-гексане в качестве элюента, с получением указанного в заголовке соединения Пр.76 Стадия-2 (1 г, 44%).
Стадия-3: Синтез 5-бром-7-фтор-1H-индазола

К перемешиваемому раствору (E)-5-бром-2,3-дифторбензальдегид O-метилоксима (1 г, 4 ммоль) в THF (10 мл) добавляли гидразин гидрат (4 мл), содержимое перемешивали при 90°C в течение 12 ч. После завершения реакции (контролируемой ТСХ), реакционную смесь охлаждали до 0°C, разбавляли этилацетатом и органический слой промывали водой, затем насыщенным солевым раствором. Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное соединение очищали с помощью combi-flash, с использованием 10% этилацетата в н-гексане в качестве элюента, с получением указанного в заголовке соединения Пр.76 Стадия-3 (0,2 г, 21%).
Стадия-4: Синтез 5-бром-7-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола
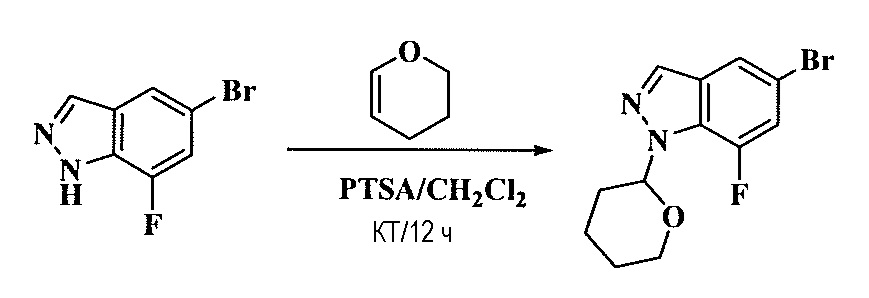
Реакцию осуществляли в соответствии со Схемой 1, Стадия-1, с использованием 5-бром-7-фтор-1H-индазола (1,82 г, 8,46 ммоль). Неочищенное соединение очищали с помощью combi-flash, с использованием 5% этилацетата в н-гексане в качестве элюента, с получением указанного в заголовке соединения Пр.76 Стадия-4 (2,39 г, 94%).
Стадия-5: Синтез 5-(бут-1-ин-1-ил)-7-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола

Реакцию осуществляли в соответствии со Схемой 1, Стадия-2, используя 5-бром-7-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол (4,49 г, 15 ммоль) и бут-1-ин-1-илтриметилсилан (3,8 г, 30 ммоль, Пример 1, Стадия-2). Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле 230-400 меш, используя 20% этилацетат в н-гексане, с получением указанного в заголовке соединения Пр.76 Стадия-5 (3,2 г).
Соединение 76:1H ЯМР(400 MГц, DMSO-d6): δ 13,66 (с, 1H), 8,19 (д, J=2,4 Гц, 1H), 7,48 (с, 1H), 7,22-7,21 (м, 2H), 7,19-7,11 (м, 3H), 6,90 (д, J=12,0 Гц, 1H), 6,76 (д, J=8,4 Гц, 2H), 6,63-6,59 (м, 3H), 6,57-6,47 (м, 1H), 3,87 (т, J=5,4 Гц, 2H), 3,37-3,30 (м, 2H), 2,98 (с, 3H), 2,84 (с, 3H), 2,77 (т, J=5,6 Гц, 2H), 2,41 (кв, J=7,2 Гц, 2H), 2,32-2,10 (м, 1H), 0,87 (т, J=7,2 Гц, 3H). ЖХМС: 513,3[M+H]+.
Пример 77: Синтез (E)-4-((2-(4-((E)-1-(3-фтор-1H-индазол-5-ил)-2-фенилпент-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 77)
Соединение синтезировали в соответствии с подходом, описанным в примере 59, (i) с использованием на стадии-4 3-фтор-5-(пент-1-ин-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (получение показано ниже в Пр. 77 Стадия 1-2) вместо соединения 276 и трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)карбамата (полученного в Примере 14, Стадия-3) вместо соединения 277 и (ii) с использованием на стадии-5 иодбензола вместо соединения 265, с получением соединения 77 (0,06 г, 19,3%) в виде не совсем белого твердого вещества.
Стадия-1: Синтез триметил(пент-1-ин-1-ил)силана

К перемешиваемому раствору (триметилсилил)ацетилена (29 г, 295 ммоль) в сухом THF (100 мл) добавляли н-BuLi (2,5 М в THF, 125 мл) при -78°C в течение 2 ч. Полученную смесь нагревали до 0°C и перемешивали в течение 10 минут. Реакционную смесь снова охлаждали до -78°C, к вышеуказанной смеси добавляли HMPA (58 г, 324 ммоль) и перемешивали при -78°C в течение 30 минут. К вышеуказанной реакционной смеси добавляли 1-йодпропан (53 г, 315 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 12 ч. После завершения реакции, реакционную смесь гасили водой, экстрагировали этилацетатом (500 мл). Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Продукт бут-1-ин-1-илтриметилсилан перегоняли между 110-160°C с получением желаемого продукта (38 г).
Стадия-2: Синтез 3-фтор-5-(пент-1-ин-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола

Реакцию осуществляли в соответствии со Схемой 1, Стадия-2, используя 5-бром-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол (4 г, 13,4 ммоль, Пример 2, Стадия-2) вместо соединения 202 и триметил(пент-1-ин-1-ил)силан (2,8 г, 20,1 ммоль) вместо соединения 203. Неочищенный продукт очищали при помощи хроматографии на 230-силикагеле, используя 5% этилацетата в н-гексане, с получением 3-фтор-5-(пент-1-ин-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (1,18 г, 30,8%).
Соединение 77:1H ЯМР(400 MГц, DMSO-d6): δ 12,56 (с, 1H), 7,49 (с, 1H), 7,47 (д, J=6,8 Гц, 1H), 7,46-7,11 (м, 6H), 6,77 (д, J=8,8 Гц, 2H), 6,62-6,48 (м, 4H), 3,87 (т, J=5,6 Гц, 2H), 3,33-3,31 (м, 2H), 2,98 (с, 3H), 2,84 (с, 3H), 2,78 (т, J=5,6 Гц, 2H), 2,36-2,32 (м, 2H), 1,29-1,23 (м, 2H), 0,74 (т, J=7,4 Гц, 3H). ЖХМС: 527,3 [M+H]+.
Пример 78: Примера 78 нет.
Пример 79: Синтез (E)-4-((2-(4-((E)-1-(3,7-дифтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 79)
Соединение синтезировали в соответствии с подходом, описанным в примере 14, с использованием на стадии-2 5-(бут-1-ин-1-ил)-3,7-дифтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (получение показано ниже в Пр.79 Стадия-1) вместо соединения 271 и заменяя стадию-5 Пр.14 условиями реакции, описанными в схеме 2, Стадия-6b, с выделением соединения 79 (0,046 г) в виде не совсем белого твердого вещества.
Стадия-1: Синтез 5-(бут-1-ин-1-ил)-3,7-дифтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола

Соединение синтезировали в соответствии с подходом, описанным в примере 2, с использованием на стадии-1 5-бром-7-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола вместо соединения 270 и продолжая стадии 2-3 Пр.2, с получением указанного в заголовке соединения Пр.79 Стадия-1 (0,78 г, 76%).
Соединение 79:1H ЯМР(400 MГц, DMSO-d6): δ 13,20 (с, 1H), 7,40 (с, 1H), 7,22-7,21 (м, 2H), 7,19-7,11 (м, 3H), 6,90 (д, J=12,0 Гц, 1H), 6,8 (д, J=8,4 Гц, 2H), 6,63-6,59 (м, 3H), 6,57-6,47 (м, 1H), 3,87 (т, J=5,4 Гц, 2H), 3,37-3,30 (м, 2H), 2,98 (с, 3H), 2,84 (с, 3H), 2,77 (т, J=5,6 Гц, 2H), 2,41 (кв, J=7,2 Гц, 2H), 0,87 (т, J=7,2 Гц, 3H) ЖХМС: 531,3[M+H]+.
Пример 80: Синтез (E)-4-((2-(4-((E)-2-(2,5-дифторфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 80)
Соединение синтезировали в соответствии с подходом, описанным в примере 14, с использованием на стадии-4 (i) (Z)-5-(1,2-бис(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (полученного в Примере 3, Стадия-1) вместо соединения 233, (ii) трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)карбамата (полученного в Примере 14, Стадия-3) вместо соединения 234, (iii) 1,4-дифтор-2-иодбензола вместо соединения 262 и (iv) заменяя Пр.14 Стадию-5 условиями реакции, описанными на схеме 2, Стадия-6b, с выделением соединения 80 (0,16 г, 23,7%) в виде не совсем белого твердого вещества.
Соединение 80:1H ЯМР(400 MГц, DMSO-d6): δ 12,60 (с, 1H), 7,52 (с, 1H), 7,49 (дд, J1=8,8 Гц, J2=1,2 Гц, 1H), 7,23-7,12 (м, 1H), 7,11-7,02 (м, 3H), 6,83 (д, J=8,4 Гц, 2H), 6,65 (д, J=8,4 Гц, 2H), 6,62-6,48 (м, 2H), 3,88 (т, J=5,6 Гц, 2H), 3,32-3,31 (м, 2H), 2,99 (с, 3H), 2,84 (с, 3H), 2,79 (т, J=5,4 Гц, 2H), 2,35 (кв, J=7,0 Гц, 2H), 0,91 (т, J=7,4 Гц, 3H). ЖХМС: 549,3[M+H]+.
Пример 81: Синтез (E)-4-((2-(4-((E)-1-(3-фтор-1H-индазол-5-ил)-2-(3-фторпиридин-4-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 81)
Соединение синтезировали в соответствии с подходом, описанным в примере 14, с использованием на стадии-4 (i) (Z)-5-(1,2-бис(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (полученного в Примере 3, Стадия-1) вместо соединения 233, (ii) трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)карбамата (Пример 14, Стадия-3) вместо соединения 234, и (iii) 3-фтор-4-йодпиридина вместо соединения 262, и (iv) заменяя Пр.14 Стадия-5 условиями реакции, описанными на схеме 2, Стадия-6b, с выделением соединения 81 (0,24 г, 34%) в виде не совсем белого твердого вещества.
Соединение 81:1H ЯМР(400 MГц, DMSO-d6): δ 12,60 (с, 1H), 8,4 (с, 1H), 8,30 (с, 1H), 7,6 (м, 2H), 7,49 (д, J=1,6 Гц, 1H), 7,22 (д, J=1,6 Гц, 1H), 6,8 (д, J=8,8 Гц, 2H), 6,65 (д, J=8,8 Гц, 3H), 6,40 (м, 1H), 3,88 (т, J=5,4 Гц, 2H), 3,03 (с, 3H), 2,80 (с, 3H), 2,78 (т, J=5,4 Гц, 2H), 2,42 (кв, J=7,4 Гц, 2H), 0,87 (т, J=7,2 Гц, 3H). ЖХМС: 532,3[M+H]+.
Пример 82: Синтез (E)-4-((2-(4-((E)-1-(3-фтор-1H-индазол-5-ил)-3-метил-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 82)
Соединение синтезировали в соответствии с подходом, описанным в примере 50, (i) с использованием на стадии-3 (E)-2-(4-(1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-3-метил-2-фенилбут-1-ен-1-ил)фенокси)этан-1-амина (получение показано ниже в Пр. 82 Стадия 1-3) вместо соединения 258 и (ii) заменяя Пр.50 Стадия-5 условиями реакции, описанными на схеме 2, Стадия-6c, с выделением соединения 82 (0,07 г).
Стадия-1: Синтез (E)-2-(4-(1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-3-метил-2-фенилбут-1-ен-1-ил)фенокси)этан-1-амина

Соединение синтезировали в соответствии с подходом, описанным в примере 2, (i) с использованием на стадии-3 3-метилбут-1-ина вместо соединения 278, (ii) с использованием на стадии-4 трет-бутил (2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси)этил)карбамата (полученного в Примере 63, Стадия-5) вместо соединения 274, и (iii) продолжая Пр.2 Стадию 6, с выделением указанного в заголовке соединения Пр.82 Стадия-1.
Стадия-2: Синтез (E)-2-(4-(1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-3-метил-2-фенилбут-1-ен-1-ил)фенокси)этан-1-амина

К перемешиваемому раствору (E)-2-(4-(1-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-3-метил-2-фенилбут-1-ен-1-ил)фенокси)этан-1-амина (1,5 г, 3 ммоль) в дихлорметане (20 мл) добавляли при 0°C TFA (3,4 мл, 45 ммоль). Реакционную смесь перемешивали в течение 12 ч при комнатной температуре. После завершения реакции реакционную смесь подщелачивали насыщенным NaHCO3,экстрагировали 10% MeOH в дихлорметане. Органический слой концентрировали при пониженном давлении и неочищенный материал очищали с помощью combi-flash, с использованием 10% MeOH в дихлорметане, с получением указанного в заголовке соединения Пр.82 Стадия-2 (1,1 г).
Соединение 82:1H ЯМР (400 MГц, DMSO-d6): δ 12,55 (с, 1H), 7,56 (с, 1H), 7,48 (д, J=8,8 Гц, 1H), 7,30 (д, J=8,8 Гц, 1H), 7,23-7,19 (м, 2H), 7,14-7,10 (м, 3H), 6,84 (д, J=8,8 Гц, 2H), 6,61-6,46 (м, 4H), 3,83 (т, J=5,6 Гц, 2H), 3,31 (шир.с, 2H), 2,97 (с, 3H), 2,83 (м, 4H), 2,75 (т, J=5,4 Гц, 2H), 0,88 (м, 6H). ЖХМС: 527,3 [M+H]+.
Пример 83: Примера 83 нет.
Пример 84: Синтез (E)-5-((2-(4-((E)-4-фтор-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилпент-2-енамида (Соединение 84)
Соединение синтезировали в соответствии с подходом, описанным в примере 60, с использованием на стадии-1 3-фтор-5-(4-фторбут-1-ин-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (получение показано ниже в Пр.84 Стадия 1-2) вместо соединения 275 и трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)карбамата (полученного в Примере 14, Стадия-3) вместо соединения 263, с выделением соединения 84 (0,075 г, 10%).
Стадия-1: Синтез 4-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-3-ин-1-ола

Реакцию осуществляли в соответствии со Схемой 1, Стадия-2, используя 5-бром-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол (10 г, 33 ммоль, полученный в Примере 2, Стадия-2) и бут-3-ин-1-ол (3,52 г, 50 ммоль) вместо соединения 202. Неочищенный продукт очищали колоночной хроматографией на силикагеле, используя 30% этилацетат в н-гексане, с получением указанного в заголовке соединения Пр.84 Стадия-1 (5,7 г, 59%).
Стадия-2: Синтез 3-фтор-5-(4-фторбут-1-ин-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола

К перемешиваемому раствору 4-(3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)бут-3-ин-1-ола (4 г, 13 ммоль) в 40 мл дихлорметана, добавляли DAST (3,35 г) при -78°C, реакционную смесь перемешивали в течение 2 ч. По завершении ТСХ, реакционную смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (250 мл). Объединенные органические экстракты промывали водой, насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле 230-400 меш, используя 6% этилацетат в н-гексане, с получением указанного в заголовке соединения Пр.84 Стадия-2 (1,2 г, 30%).
Соединение 84:1H ЯМР (400 MГц, DMSO-d6): δ 12,61 (с, 1H), 7,60 (с, 1H), 7,47 (дд, J1=8,6 Гц, J2=1,8 Гц, 1H), 7,25-7,13 (м, 6H), 6,9-6,75 (м, 2H), 6,64-6,57 (м, 2H), 6,52-6,48 (м, 2H), 4,36 (т, J=6 Гц, 1H), 4,25 (т, J=6,2 Гц, 1H), 3,87 (т, J=5,6 Гц, 2H), 3,37-3,31 (м, 2H), 2,99 (с, 3H), 2,84 (с, 3H), 2,82-2,70 (м, 4H). ЖХМС: 531,3 [M+H]+.
Пример 85: Синтез (E)-5-((2-(4-((E)-2-(2-хлор-4-фторфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилпент-2-енамида (Соединение 85)
Соединение синтезировали в соответствии с подходом, описанным в примере 60, с использованием на стадии-1 (i) трет-бутил (E)-(5-(диметиламино)-5-оксопент-3-ен-1-ил)(2-(4-йодфенокси)этил)карбамата (получение показано ниже в Пр.85 Стадия 1-7) вместо соединения 263, 5-(бут-1-ин-1-ил)-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (полученного в Примере 2, Стадия-3) вместо соединения 275, и 2-хлор-4-фтор-1-иодбензола вместо соединения 264, с выделением соединения 85 (0,046 г).
Стадия-1: Синтез 1-(2-хлорэтокси)-4-иодбензола

К перемешиваемому раствору 4-иодфенола (15 г, 68 ммоль) добавляли 5M NaOH (90 мл) и перемешивали в течение 10 мин при 0°C, к вышеуказанной смеси добавляли 1,2-дихлорэтан (225 мл), затем TBAB (0,43 г, 1,36 ммоль). Содержимое перемешивали при кипении с обратным холодильником в течение 16 ч. После завершения реакции, реакционную смесь выливали в ледяную воду, и экстрагировали этилацетатом. Объединенные органические слои промывали водой, затем насыщенным раствором NaCl, сушили над безводным Na2SO4и концентрировали при пониженном давлении с получением желаемого соединения 1-(2-хлорэтокси)-4-иодбензола (20 г, неочищенный).
Стадия-2: Синтез 3-((2-(4-йодфенокси)этил)амино)пропан-1-ола

К перемешиваемому раствору 1-(2-хлорэтокси)-4-иодбензола (10 г, 35 ммоль) в DMF (25 мл) добавляли карбонат калия (19,5 г, 141 ммоль) и 3-аминопропан-1-ол (26,6 г, 350 ммоль). Реакционную смесь перемешивали в течение 24 ч при 50°C, после завершения реакции (контролируемой ТСХ), реакционную смесь разбавляли холодной водой (50 мл), твердое вещество отфильтровывали и сушили при пониженном давлении. Неочищенный продукт использовали на следующей стадии без дополнительной очистки (8,6 г, неочищенный).
Стадия-3: Синтез трет-бутил (3-гидроксипропил)(2-(4-йодфенокси)этил)карбамата

К перемешиваемому раствору 3-((2-(4-йодфенокси)этил)амино)пропан-1-ола (8,6 г, 26 ммоль) в дихлорметане (86 мл) добавляли boc-ангидрид (6,4 г, 29 ммоль). Реакционную смесь перемешивали в течение 2 ч при комнатной температуре, после завершения реакции (контролируемой ТСХ), реакционную смесь разбавляли холодной водой (100 мл) и экстрагировали дихлорметаном (250 мл). Органический слой промывали водой, затем насыщенным раствором NaCl, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью combi-flash, с использованием 40% EtOAc в н-гексане, с получением указанного в заголовке соединения Пр.85 Стадия-3 (9,5 г).
Стадия-4: Синтез трет-бутил (2-(4-йодфенокси)этил)(3-оксопропил)карбамата

К перемешиваемому раствору трет-бутил (3-гидроксипропил)(2-(4-йодфенокси)этил)карбамата (9 г, 21 ммоль) в дихлорметане (90 мл) добавляли периодинан Десса-Мартина (11,78 г, 27 ммоль). Реакционную смесь перемешивали в течение 1 ч при комнатной температуре, после завершения реакции (контролируемой ТСХ), реакционную смесь разбавляли смесью 1:1 hypo и раствора NaHCO3и экстрагировали дихлорметаном (250 мл). Органический слой промывали водой, затем насыщенным раствором NaCl, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением указанного в заголовке соединения Пр.85 Стадия-4 (9 г).
Стадия-5: Синтез этил (E)-5-((трет-бутоксикарбонил)(2-(4-йодфенокси)этил)амино)пент-2-еноата

К перемешиваемому раствору трет-бутил (2-(4-йодфенокси)этил)(3-оксопропил)карбамата (9 г, 28 ммоль) в дихлорметане (90 мл) добавляли этил 2-(трифенил-l5-фосфанилиден)ацетат (9,8 г, 28 ммоль). Реакционную смесь перемешивали в течение 16 ч при комнатной температуре, после завершения реакции (контролируемой ТСХ), реакционную смесь разбавляли водой и экстрагировали дихлорметаном (250 мл). Органический слой промывали водой, затем насыщенным раствором NaCl, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали при помощи combi-flash, элюируя 9% EtOAc в н-гексане, с получением указанного в заголовке соединения Пр.85 Стадия-5 (6,7 г).
Стадия-6: Синтез (E)-5-((трет-бутоксикарбонил)(2-(4-йодфенокси)этил)амино)пент-2-еновой кислоты

К перемешиваемому раствору этил (E)-5-((трет-бутоксикарбонил)(2-(4-йодфенокси)этил)амино)пент-2-еноата (6,7 г, 13 ммоль) в THF (100 мл) добавляли LiOH.H2O (2,87 г, 68 ммоль) и воду (33 мл). Реакционную смесь перемешивали в течение 32 ч при комнатной температуре, после завершения реакции (контролируемой ТСХ), реакционную смесь разбавляли водой и нейтрализовали лимонной кислотой, экстрагировали EtOAc (250 мл). Органический слой промывали водой, затем насыщенным раствором NaCl, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт использовали на следующей стадии без дополнительной очистки (5,4 г).
Стадия-7: Синтез трет-бутил (E)-(5-(диметиламино)-5-оксопент-3-ен-1-ил)(2-(4-йодфенокси)этил)карбамата

К перемешиваемому раствору (E)-5-((трет-бутоксикарбонил)(2-(4-йодфенокси)этил)амино)пент-2-еновой кислоты (2,5 г, 5,3 ммоль) в DMF (25 мл) добавляли DIPEA (2,32 мл, 13 ммоль), HATU (2,45 г, 6,4 ммоль) и диметиламин гидрохлорид (0,526 г, 6,4 ммоль). Реакционную смесь перемешивали в течение 16 ч при комнатной температуре, после завершения реакции (контролируемой ТСХ), реакционную смесь разбавляли водой и экстрагировали EtOAc (100 мл). Органический слой промывали водой, затем насыщенным раствором NaCl, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением указанного в заголовке соединения Пр.85 Стадия-7 (2 г, 71%).
Соединение 85:1H ЯМР (400 MГц, DMSO-d6): δ 12,62 (с, 1H), 7,54 (с, 1H), 7,49 (дд, J1=8,8 Гц, J2=1,2 Гц, 1H), 7,36-7,35 (м, 2H), 7,34-7,31 (м, 1H), 7,15-7,11 (м, 1H), 6,82 (д, J=8,8 Гц, 2H), 6,65-6,58 (м, 3H), 6,43-6,39 (м, 1H), 3,85 (т, J=5,7 Гц, 2H), 2,98 (с, 3H), 2,83 (с, 3H), 2,79-2,78 (м, 2H), 2,62 (т, J=6,8 Гц, 2H), 2,33-2,27 (м, 4H), 0,88 (т, J=7,4 Гц, 3H). ЖХМС: 579,3 [M+H]+.
Пример 86: Синтез (E)-4-((2-(4-((E)-2-(2-хлор-4-фторфенил)-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N,2-триметилбут-2-енамид (Соединение 86)
Соединение синтезировали в соответствии с подходом, описанным в примере 59, с использованием на стадии-4 5-(бут-1-ин-1-ил)-3-фтор-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (полученного в Примере 2, Стадия-3) вместо соединения 276 и трет-бутил (E)-(4-(диметиламино)-3-метил-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)карбамата (получение показано ниже в Пр.86 Стадия 1-3) вместо соединения 277, с выделением соединения 86 (0,11 г, 7,6%).
Стадия-1: Синтез метил (E)-4-((трет-бутоксикарбонил)(2-(4-йодфенокси)этил)амино)-2-метилбут-2-еноата

К перемешиваемому раствору (1-метокси-1-оксопропан-2-ил)трифенилфосфоний бромида (10,8 г, 25 ммоль) в дихлорметане (50 мл) добавляли триэтиламин (6,35 г, 62 ммоль) при 0°C, перемешивали в течение 30 мин. К вышеуказанному раствору, добавляли по каплям трет-бутил (2-(4-йодфенокси)этил)(2-оксоэтил)карбамат (8,5 г, 20 ммоль, Пример 22, Стадия-4) в дихлорметане (35 мл). Реакционную смесь перемешивали в течение 3,5 ч при комнатной температуре, после завершения реакции (контролируемой ТСХ), реакционную смесь разбавляли раствором хлорида аммония и экстрагировали EtOAc (100 мл). Органический слой промывали водой, затем насыщенным раствором NaCl, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле 230-400 меш, элюируя 5% EtOAc в н-гексане, с получением указанного в заголовке соединения Пр.86 Стадия-1 (7,1 г, 71,7%).
Стадия-2: Синтез (E)-4-((трет-бутоксикарбонил)(2-(4-йодфенокси)этил)амино)-2-метилбут-2-еновой кислоты

К перемешиваемому раствору метил (E)-4-((трет-бутоксикарбонил)(2-(4-йодфенокси)этил)амино)-2-метилбут-2-еноата (7,1 г, 14,9 ммоль) в смеси MeOH:THF:H2O (1:1:1) (70 мл) добавляли LiOH.H2O (3,13 г, 74 ммоль). Реакционную смесь перемешивали в течение 16 ч при комнатной температуре, после завершения реакции (контролируемой ТСХ), реакционную смесь разбавляли водой, с последующим подкислением 0,5 M HCl и экстрагировали EtOAc (250 мл). Органический слой промывали водой, затем насыщенным раствором NaCl, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт использовали на следующей стадии без дополнительной очистки (6,5 г, 94%).
Стадия-3: Синтез трет-бутил (E)-(4-(диметиламино)-3-метил-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)карбамата

К перемешиваемому раствору (E)-4-((трет-бутоксикарбонил)(2-(4-йодфенокси)этил)амино)-2-метилбут-2-еновой кислоты (3 г, 6,5 ммоль) в DMF (30 мл) добавляли DIPEA (3,1 г, 16,25 ммоль), HATU (2,96 г, 7,8 ммоль) и диметиламин гидрохлорид (0,63 г, 7,8 ммоль). Реакционную смесь перемешивали в течение 16 ч при комнатной температуре, после завершения реакции (контролируемой ТСХ), реакционную смесь разбавляли водой и экстрагировали дихлорметаном (100 мл). Органический слой промывали водой, затем насыщенным раствором NaCl, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле 230-400 меш, элюируя 70% EtOAc в н-гексане, с получением указанного в заголовке соединения Пр.86 Стадия-3 (2,6 г, 82%).
Соединение 86:1H ЯМР (400 MГц, DMSO-d6): δ 12,63 (с, 1H), 7,54 (с, 1H), 7,49 (дд, J1=8,6 Гц, J2=1,8 Гц, 1H), 7,36-7,31 (м, 2H), 7,24 (дд, J1=8,8 Гц, J2=1,2 Гц, 1H), 7,14 (дд, J1=8,0 Гц, J2=2,4 Гц, 1H), 6,82 (д, J=8,8 Гц, 2H), 6,62 (д, J=8,8 Гц, 2H), 5,43-5,40 (м, 1H), 3,86 (т, J=5,6 Гц, 2H), 3,21 (д, J=6,4 Гц, 2H), 2,87-2,75 (м, 8H), 2,33 (кв, J=7,2 Гц, 2H), 2,08-1,85 (м, 1H), 1,70 (с, 3H), 0,88 (т, J=7,6 Гц, 3H). ЖХМС: 579,3 [M+H]+.
Пример 87: Синтез (E)-4-((2-(4-((E)-2-(2-хлор-4-фторфенил)-4,4,4-трифтор-1-(3-фтор-1H-индазол-5-ил)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 87)
Соединение синтезировали в соответствии с подходом, описанным в примере 59, с использованием на стадии-4 трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)этил)карбамата (полученного в Примере 14, Стадия-3) вместо соединения 277, с выделением соединения 87 (0,17 г, 24%).
Соединение 87:1H ЯМР (400 MГц, DMSO-d6): δ 12,69 (с, 1H), 7,59 (с, 1H), 7,55 (дд, J1=8,6 Гц, J2=1,8 Гц, 1H), 7,53 (дд, J1=8,6 Гц, J2=1,8 Гц, 1H), 7,43 (дд, J1=8,6 Гц, J2=1,8 Гц, 1H), 7,36-7,21 (м, 2H), 6,85 (д, J=8,9 Гц, 2H), 6,67-6,57 (м, 3H), 6,51-6,47 (м, 1H), 3,88 (т, J=5,7 Гц, 2H), 3,45 (кв, J=10,8 Гц, 2H), 2,98 (с, 3H), 2,84 (с, 3H), 2,78 (т, J=5,6 Гц, 2H). ЖХМС: 619,3 [M+H]+.
Пример 88: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-(фенил-d5)бут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 88)
Соединение синтезировали в соответствии с подходом, описанным в примере 59, с использованием на стадии-5 трет-бутил ((E)-4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-((Z)-1-(1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол-5-ил)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бут-1-ен-1-ил)фенокси)этил)карбамата (полученного в Примере 33, Стадия-1) вместо соединения 278 и иодбензол-d5 вместо соединения 265, с выделением соединения 88 (0,023 мг) в виде не совсем белого твердого вещества.
Соединение 88:1H ЯМР (400 MГц, DMSO-d6) δ 13,05 (с, 1H), 8,06 (с, 1H), 7,60 (с, 1H), 7,50 (д, J=8,8 Гц, 1H), 7,11 (д, J=8,4 Гц, 1H), 6,73 (д, J=8,8, 2H), 6,59-6,51 (м, 3H), 6,47 (д, J=8,8 Гц, 1H), 3,86 (т, J=5,2 Гц, 2H), 3,31 (д, J=4,8 Гц, 2H), 2,98 (с, 3H), 2,83 (с, 3H), 2,78-2,75 (м, 2H), 2,40 (кв, J=7,6 Гц, 2H), 0,89 (т, J=7,6 Гц, 3H). ЖХМС: 500,4 [M+H]+.
Пример 89: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)этил)амино)-N,N-диметилпент-2-енамида (Соединение 89)
Соединение синтезировали в соответствии с подходом, описанным в примере 59, с использованием на стадии-4 5-(бут-1-ин-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (Пример 1, Стадия-3) вместо соединения 276 и трет-бутил (E)-(5-(диметиламино)-5-оксопент-3-ен-2-ил)(2-(4-йодфенокси)этил)карбамата (получение показано ниже в Пр.89 Стадия 1-4) вместо соединения 277 и с использованием на стадии-5 иодбензола вместо соединения 265, с выделением соединения 89 (0,08 г, 5,5%).
Стадия-1: Синтез (E)-4-бромпент-2-еновой кислоты

К перемешиваемому раствору (E)-бут-2-еновой кислоты (5 г, 50 ммоль) в четыреххлористом углероде (50 мл) добавляли N-бромсукцинамид (8,6 г, 65 ммоль). Реакционную смесь нагревали до кипения с обратным холодильником в течение 24 часов, что приводило к осаждению кристаллов сукцинамида. Кристаллы отфильтровывали и фильтрат концентрировали. Неочищенный продукт перекристаллизовывали с минимальным количеством гексана и промывали гексаном с получением (E)-4-бромпент-2-еновой кислоты (4 г, 45%) в виде белого твердого вещества.
Стадия-2: Синтез (E)-4-бром-N,N-диметилпент-2-енамида

(E)-4-бромпент-2-еновую кислоту (4 г, 24,2 ммоль) брали в дихлорметане (40 мл) и охлаждали до 0°C. К этому раствору добавляли оксалилхлорид (3,6 г, 29 ммоль), DMF (0,5 мл) и перемешивали в течение 3 ч при комнатной температуре. Реакционную смесь концентрировали в атмосфере азота, остаток разбавляли дихлорметаном (40 мл), охлаждали до -30°C и подщелачивали DIPEA (7,7 мл, 43 ммоль). К этой смеси медленно добавляли 2М диметиламина в THF (14,3 мл, 28,6 ммоль) и содержимое перемешивали при 0°C в течение 30 мин. Летучие вещества удаляли концентрированием при пониженном давлении и остаток разделяли между водой и этилацетатом. Органический слой промывали водой, насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали. Неочищенный материал очищали при помощи combi-flash, с получением (E)-4-бром-N,N-диметилпент-2-енамида (2,3 г, 50%).
Стадия-3: Синтез (E)-4-((2-(4-йодфенокси)этил)амино)-N,N-диметилпент-2-енамида

Реакцию осуществляли в соответствии со Схемой 3, Стадия-3, используя 2-(4-йодфенокси)этан-1-амин (1,5 г, 5,86 ммоль, Пример 14, Стадия-3.1) вместо соединения 217 и (E)-4-бром-N,N-диметилпент-2-енамид (1,8 г, 5,86 ммоль) вместо соединения 214. Неочищенный материал использовали на следующей стадии (3 г, неочищенный).
Стадия-4: Синтез трет-бутил (E)-(5-(диметиламино)-5-оксопент-3-ен-2-ил)(2-(4-йодфенокси)этил)карбамата

Реакцию осуществляли в соответствии со Схемой 3, Стадия-4, используя (E)-4-((2-(4-йодфенокси)этил)амино)-N,N-диметилпент-2-енамид (4,5 г, 12 ммоль) вместо соединения 218, с получением неочищенного продукта, который очищали при помощи combi-flash, элюируя 70% этилацетата в н-гексане, с получением указанного в заголовке соединения в Пр.90 Стадия-4 (2,8 г, 50%).
Соединение 89:1H ЯМР (400 MГц, DMSO-d6): δ 13,05 (с, 1H), 8,06 (с, 1H), 7,60 (с, 1H), 7,51 (д, J=8,8 Гц, 1H), 7,22-7,10 (м, 6H), 6,74 (д, J=8,8 Гц, 2H), 6,57 (д, J=8,8 Гц, 2H), 6,47-6,41 (м, 2H), 3,84 (т, J=5,6 Гц, 2H), 3,30 (шир.с, 2H), 2,98 (с, 3H), 2,83 (с, 3H), 2,75-2,66 (м, 2H), 2,43-2,32 (м, 2H), 1,07 (д, J=6,8 Гц, 3H), 0,87 (т, J=7,2 Гц, 3H). ЖХМС: 509,3 [M+H]+.
Пример 90: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)пропил)амино)-N,N-диметилбут-2-енамида (Соединение 90)
Соединение синтезировали в соответствии с подходом, описанным в примере 59, с использованием на стадии-4 5-(бут-1-ин-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (Пример 1, Стадия-3) вместо соединения 276 и трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)пропил)карбамата (получение показано ниже Пр.90 Стадия 1-5) вместо соединения 277 и с использованием на стадии-5 иодбензола вместо соединения 265 с получением соединения 90 (0,06 г, 10%).
Стадия-1: Синтез трет-бутил (2-гидроксипропил)карбамата
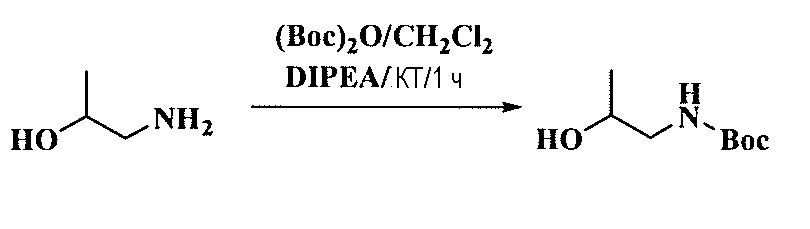
К перемешиваемому раствору 1-аминопропан-2-ола (5 г, 66,6 ммоль) в дихлорметане (50 мл) добавляли boc-ангидрид (17 мл, 79,92 ммоль) и DIPEA (18 мл, 99,9 ммоль), реакционную смесь перемешивали при комнатной температуре в течение примерно 1 часа. После завершения реакции, рекционную смесь концентрировали при пониженном давлении и неочищенный продукт очищали при помощи combi-flash, элюируя 50% этилацетата в н-гексане, с получением указанного в заголовке соединения в Пр.90 Стадия-1 (10 г, 86%).
Стадия-2: Синтез трет-бутил (2-(4-йодфенокси)пропил)карбамата

К смеси 4-иодфенола (4 г, 18,1 ммоль), трет-бутил (2-гидроксипропил)карбамата (2,6 г, 21,8 ммоль), трифенилфосфина (5,7 г, 21,8 ммоль) в THF (40 мл) добавляли по каплям DEAD (3,8 г, 21,8 ммоль), затем реакционную смесь перемешивали при комнатной температуре в течение 1 ч. После завершения реакции, реакционную смесь гасили водой, экстрагировали этилацетатом (2×100 мл), объединенный органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия. Органический слой концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле 100-200 меш, элюируя 10% EtoAc в н-гексане, с получением указанного в заголовке соединения в Пр.90 Стадия-2 (2,1 г, 32%).
Стадия-3: Синтез 2-(4-йодфенокси)пропан-1-амина

Реакцию осуществляли в соответствии со Схемой 3, Стадия-2, используя трет-бутил (2-(4-йодфенокси)пропил)карбамат (4,8 г, 12,76 ммоль) вместо соединения 207b, с получением неочищенного материала, который использовали на следующей стадии без дополнительной очистки (2,8 г).
Стадия-4: Синтез (E)-4-((2-(4-йодфенокси)пропил)амино)-N,N-диметилбут-2-енамида

Реакцию осуществляли в соответствии со Схемой 3, Стадия-3, используя 2-(4-йодфенокси)пропан-1-амин (1 г, 3,6 ммоль) вместо соединения 217 в DMF (10 мл) и (E)-4-хлор-N,N-диметилбут-2-енамид вместо соединения 214 и (E)-4-бром-N,N-диметилбут-2-енамида смесь (0,7 г, 3,6 ммоль, Пример 63, Стадия-7) вместо соединения 214. Неочищенный материал использовали на следующей стадии (0,5 г, неочищенный).
Стадия-5: Синтез трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)пропил)карбамата

Реакцию осуществляли в соответствии со Схемой 3, Стадия-4, используя (E)-4-((2-(4-йодфенокси)пропил)амино)-N,N-диметилбут-2-енамид (2 г, 5,3 ммоль) вместо соединения 218, с получением неочищенного продукта, который очищали с помощью combi-flash, элюируя 70% этилацеатом в н-гексане, с получением указанного в заголовке соединения в Пр.90 Стадия-5 (0,5 г, 20%).
Соединение 90:1H ЯМР (400 MГц, DMSO-d6): δ 13,07 (с, 1H), 8,07 (с, 1H), 7,61 (с, 1H), 7,52 (д, J=11,2 Гц, 1H), 7,22-7,11 (м, 6H), 6,78 (д, J=8,8 Гц, 2H), 6,71-6,52 (м, 4H), 4,52-4,31 (м, 1H), 3,61 (д, J=4,4 Гц, 2H), 3,96-3,04 (м, 5H), 2,86 (с, 3H), 2,42-2,32 (м, 2H), 1,14 (д, J=6,0 Гц, 3H), 0,87 (т, J=7,6 Гц, 3H). ЖХМС: 509,3 [M+H]+.
Пример 91: Примера 91 нет.
Пример 92: Синтез (E)-4-((1-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)пропан-2-ил)амино)-N,N-диметилбут-2-енамида (Соединение 92)
Соединение синтезировали в соответствии с подходом, описанным в примере 59, (i) с использованием на стадии-4 5-(бут-1-ин-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (полученного в Примере 1, Стадия-3) вместо соединения 276 и трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(1-(4-йодфенокси)пропан-2-ил)карбамата (получение показано ниже в Пр.92 Стадия 1-4) вместо соединения 277 и (ii) с использованием на стадии-5 иодбензола вместо соединения 265 с выделением соединения 92 (0,015 г).
Стадия-1: Синтез 2-(3-(4-иодфенил)пропил)изоиндолин-1,3-диона

К смеси 4-иодфенола (5 г, 22 ммоль), трет-бутил (1-гидроксипропан-2-ил)карбамата (4,66 г, 26 ммоль), трифенилфосфина (7,12 г, 26 ммоль) в THF (100 мл) добавляли по каплям DEAD (4,66 г, 26 ммоль), затем реакционную смесь перемешивали при комнатной температуре в течение 1 ч. После завершения реакции, реакционную смесь гасили водой, экстрагировали этилацетатом (2×100 мл), объединенный органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия. Органический слой концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле 100-200 меш, элюируя 10% EtoAc в н-гексане, с получением указанного в заголовке соединения в Пр.92 Стадия-1 (2 г).
Стадия-2: Синтез 1-(4-йодфенокси)пропан-2-амина

Реакцию осуществляли в соответствии со Схемой 3, Стадия-2, используя трет-бутил (1-(4-йодфенокси)пропан-2-ил)карбамат (4 г, 10,6 ммоль) вместо соединения 207b, с получением неочищенного материала, который использовали на следующей стадии без дополнительной очистки (2 г).
Стадия-3: Синтез (E)-4-((2-(4-йодфенокси)этил)амино)-N,N-диметилпент-2-енамида

Реакцию осуществляли в соответствии со Схемой 3, Стадия-3, используя 1-(4-йодфенокси)пропан-2-амин (4 г, 14 ммоль) в DMF (35 мл) вместо соединения 217 и смесь (E)-4-хлор-N,N-диметилбут-2-енамида и (E)-4-бром-N,N-диметилбут-2-енамида (2,75 г, 14 ммоль, Пример 63, Стадия-7) вместо соединения 214. Неочищенный материал использовали на следующей стадии (5,6 г, неочищенный).
Стадия-4: Синтез трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(1-(4-йодфенокси)пропан-2-ил)карбамата
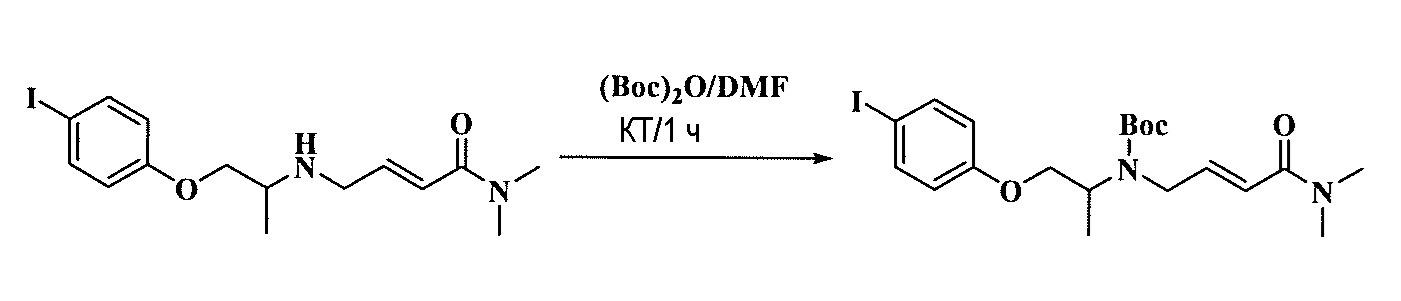
Реакцию осуществляли в соответствии с (E)-4-((1-(4-йодфенокси)пропан-2-ил)амино)-N,N-диметилбут-2-енамидом (5,6 г, 14 ммоль) с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле с 40% этилацетатом в н-гексане с получением указанного в заголовке соединения в Пр.92 Стадия-4 (2,5 г).
Соединение 92:1H ЯМР (400 MГц, DMSO-d6): δ 13,05 (с, 1H), 8,06 (с, 1H), 7,60 (с, 1H), 7,51 (д, J=8,0 Гц, 1H), 7,22-7,11 (м, 6H), 6,75 (д, J=8,4 Гц, 2H), 6,6-6,47 (м, 4H), 3,74-3,71 (м, 1H), 3,65-3,61 (м, 1H), 3,39-3,32 (м, 2H), 2,97 (с, 3H), 2,91-2,83 (м, 4H), 2,43-2,23 (м, 2H), 1,07 (д, J=6,4 Гц, 3H), 0,87 (т, J=7,2 Гц, 3H). ЖХМС: 509,3 [M+H]+.
Пример 93: Синтез (E)-4-((2-((6-((Z)-1-(3-фтор-1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)пиридин-3-ил)окси)этил)амино)-N,N-диметилбут-2-енамида (Соединение 93)
Соединение синтезировали в соответствии с подходом, описанным в примере 3, (i) с использованием на стадии-3 Пр.3 трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-((6-йодпиридин-3-ил)окси)этил)карбамата (получение показано ниже в Пр.93 Стадия 1-5) вместо соединения 256, (ii) с использованием на стадии-4 Пр.3 иодбензола вместо соединения 227, и (iii) заменяя Стадию-5 Пр.3 условиями реакции, описанными в схеме 2, Стадия-6b, с получением соединения 93 (0,12 г).
Стадия-1: Синтез трет-бутил (2-((6-бромпиридин-3-ил)окси)этил)карбамата

К перемешиваемому раствору 6-бромпиридин-3-ола (5 г, 28,9 ммоль) в 50 мл DMF, добавляли карбонат цезия (28,2 г, 86 ммоль) и трет-бутил (2-бромэтил)карбамат (12,89 г, 57,8 ммоль). Затем смесь перемешивали в течение 16 ч при 40°C. После завершения реакции, реакционную смесь добавляли в ледяную воду. Отделенное твердое вещество фильтровали и сушили с получением трет-бутил (2-((6-бромпиридин-3-ил)окси)этил)карбамата (8,2 г).
Стадия-2: Синтез трет-бутил (2-((6-йодпиридин-3-ил)окси)этил)карбамата

К перемешиваемому раствору трет-бутил (2-((6-бромпиридин-3-ил)окси)этил)карбамата (8,1 г, 25 ммоль) в 1,4-диоксане (80 мл) добавляли йодид натрия (19,2 г, 128 ммоль), йодид меди (0,244 г, 1,28 ммоль) и N,N'-диметилэтилендиамин (0,28 мл, 2,5 ммоль), перемешивали в течение 16 ч при 110°C. После завершения реакции, реакционную смесь разбавляли этилацетатом, органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия, и затем концентрировали при пониженном давлении. Полученный неочищенный продукт использовали на следующей стадии без дополнительной очистки (8,7 г, неочищенный).
Стадия-3: Синтез 2-((6-йодпиридин-3-ил)окси)этан-1-амина

К перемешиваемому раствору трет-бутил (2-((6-йодпиридин-3-ил)окси)этил)карбамата (8 г, 21,97 ммоль) в этаноле (2 мл) добавляли при 0°C конц. HCl (20 мл). Реакционную смесь перемешивали в течение 16 ч при комнатной температуре. После завершения реакции, реакционную смесь подщелачивали насыщенным NaHCO3,экстрагировали этилацетатом. Органический слой концентрировали при пониженном давлении и неочищенный материал использовали на следующей стадии без дополнительной очистки (5 г, неочищенный).
Стадия-4: Синтез (E)-4-((2-((6-йодпиридин-3-ил)окси)этил)амино)-N,N-диметилбут-2-енамида
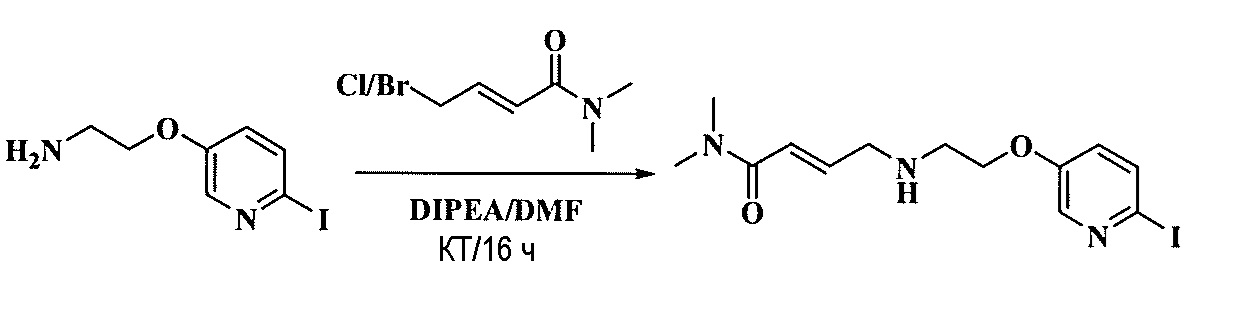
Реакцию осуществляли в соответствии со Схемой 3, Стадия-3, используя 2-((6-йодпиридин-3-ил)окси)этан-1-амин (5 г, 18,9 ммоль) в DMF (50 мл) вместо соединения 217 и смесь (E)-4-бром-N,N-диметилбут-2-енамида и (E)-4-хлор-N,N-диметилбут-2-енамида (2,63 г, 15 ммоль, Пример 63, Стадия-7) вместо соединения 214. Неочищенный материал использовали на следующей стадии без дополнительной очистки (7,08 г, неочищенный).
Стадия-5: Синтез трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-((6-йодпиридин-3-ил)окси)этил)карбамата

Реакцию осуществляли в соответствии со Схемой 3, Стадия-4, используя (E)-4-((2-((6-йодпиридин-3-ил)окси)этил)амино)-N,N-диметилбут-2-енамид (7,08 г, 18,9 ммоль) вместо соединения 218. Неочищенный материал очищали с помощью combi-flash, элюируя 1,5% MeOH в дихлорметане в качестве элюента, с получением указанного в заголовке соединения Пр.93 Стадия-5 (2,7 г, 30%).
Соединение 93:1H ЯМР (400 MГц, DMSO-d6): δ 12,5 (с, 1H), 8,03 (с, 1H), 7,48-7,44 (м, 2H), 7,25 (дд, J1=8,8 Гц, J2=1,6 Гц, 2H), 7,21-7,10 (м, 4H), 7,02-6,99 (м, 1H), 6,78 (д, J=8,0 Гц, 1H), 6,62-6,47 (м, 2H), 3,94 (т, J=5,6 Гц, 2H), 2,98 (с, 3H), 2,84-2,78 (м, 5H), 2,47-2,42 (м, 2H), 0,87 (т, J=7,2 Гц, 3H). ЖХМС: 514,3 [M+H]+.
Пример 94: Примера 94 нет.
Пример 95: Синтез (E)-4-((3-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)пропил)амино)-N,N-диметилбут-2-енамида (Соединение 95)
Соединение синтезировали в соответствии с подходом, описанным в примере 59, (i) с использованием на стадии-4 Пр.59 5-(бут-1-ин-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (полученного в Примере 1, Стадия-3) вместо соединения 276 и трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(3-(4-йодфенокси)пропил)карбамата (получение показано ниже в Пр.95 Стадия 1-5) вместо соединения 277 и (ii) с использованием на стадии-5 Пр.59 иодбензола вместо соединения 265, с выделением соединения 95 (0,035 г).
Стадия-1: Синтез 1-(3-хлорпропокси)-4-иодбензола

К перемешиваемому раствору 4-иодфенола (10 г, 45,5 ммоль) в ацетоне (150 мл) добавляли карбонат калия (12,6 г, 91 ммоль) и перемешивали в течение 30 мин при комнатной температуре, к вышеуказанной смеси добавляли 1-бром-3-хлорпропан (10,75 г, 68,2 ммоль). Содержимое перемешивали при 70°C в течение 18 ч. После завершения реакции, реакционную смесь фильтровали через слой целита, фильтрат концентрировали при пониженном давлении с получением желаемого соединения 1-(3-хлорпропокси)-4-иодбензола в виде стеклообразного твердого вещества (13,2 г, 98%).
Стадия-2: Синтез 2-(3-(4-йодфенокси)пропил)изоиндолин-1,3-диона

К перемешиваемому раствору 1-(3-хлорпропокси)-4-иодбензола (12 г, 40,5 ммоль) в DMF (100 мл) добавляли фталимид калия (9 г, 48,6 ммоль) в одной порции при комнатной температуре и реакционную смесь перемешивали при 100°C в течение 5 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой, твердое вещество отфильтровывали и сушили при пониженном давлении, получая указанное в заголовке соединение Пр.95 Стадия-2 (15,2 г, 92%).
Стадия-3: Синтез 3-(4-йодфенокси)пропан-1-амина

К раствору 2-(3-(4-йодфенокси)пропил)изоиндолин-1,3-диона (16 г, 39 ммоль) в MeOH/CH2Cl2 (160 мл/1:2) добавляли гидразин гидрат (28 мл) при комнатной температуре. Реакционную смесь перемешивали в течение 2 ч и гасили водой и экстрагировали дихлорметаном. Органический слой промывали водой, насыщенным раствором NaCl, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 3-(4-йодфенокси)пропан-1-амина (10,6 г, 97%) в виде светло-коричневой жидкости.
Стадия-4: Синтез (E)-4-((3-(4-йодфенокси)пропил)амино)-N,N-диметилбут-2-енамида

Реакцию осуществляли в соответствии со Схемой 3, Стадия-3, используя 3-(4-йодфенокси)пропан-1-амин (10,6 г, 38 ммоль) вместо соединения 217 и смесь (E)-4-хлор-N,N-диметилбут-2-енамида и (E)-4-бром-N,N-диметилбут-2-енамида (5,2 г, 30,6 ммоль, Пример 63, Стадия-7) вместо соединения 214. Неочищенный материал использовали на следующей стадии (14,7 г, неочищенный).
Стадия-5: Синтез трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(3-(4-йодфенокси)пропил)карбамата

Реакцию осуществляли в соответствии со Схемой 3, Стадия-4, используя (E)-4-((3-(4-йодфенокси)пропил)амино)-N,N-диметилбут-2-енамид (14,7 г, 37,8 ммоль) вместо соединения 218, с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле 60-120 меш, элюируя 60-80% этилацетата в н-гексане, с получением указанного в заголовке соединения Пр.95 Стадия-5 (7,1 г, 39%).
Соединение 95:1H ЯМР (400 MГц, DMSO-d6): δ 13,05 (с, 1H), 8,06 (с, 1H), 7,60 (с, 1H), 7,52 (д, J=8,4 Гц, 1H), 7,20 (д, J=7,6 Гц, 2H), 7,15-7,11 (м, 4H), 6,74 (д, J=8,4 Гц, 2H), 6,58-6,48 (м, 4H), 3,86 (т, J=6,0 Гц, 2H), 3,25 (д, J=4,8 Гц, 2H), 2,96 (с, 3H), 2,83 (с, 3H), 2,56 (т, J=6,8 Гц, 2H), 2,41 (м, 2H), 1,90 (шир.с, 1H), 1,76 (т, J=6,0 Гц, 2H), 0,89 (т, J=7,6 Гц, 3H). ЖХМС: 509,4 [M+H]+.
Пример 96: Синтез (E)-4-((3-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)бутан-2-ил)амино)-N,N-диметилбут-2-енамида (Соединение 96)
Соединение синтезировали в соответствии с подходом, описанным в примере 59, с использованием на стадии-4 Пр.59 5-(бут-1-ин-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (полученного в Примере 1, Стадия-3) вместо соединения 276 и трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(3-(4-йодфенокси)бутан-2-ил)карбамата (получение показано ниже в Пр.96 Стадия 1-4) вместо соединения 277 и с использованием на стадии-5 Пр.59 иодбензола вместо соединения 265 с выделением соединения 96 (0,165 г).
Стадия-1: Синтез 3-(4-йодфенокси)бутан-2-она

К перемешиваемому раствору 4-иодфенола (5 г, 22,7 ммоль) в бутаноне (100 мл) добавляли карбонат калия (4,7 г, 34 ммоль) и иодид калия (0,376 г, 2,27 ммоль). Реакционную смесь перемешивали в течение 30 мин при комнатной температуре, к вышеуказанной смеси добавляли 3-хлорбутан-2-он (2,4 г, 22,7 ммоль). Содержимое перемешивали при комнатной температуре в течение 12 ч. После завершения реакции, реакционную смесь выливали в ледяную воду и экстрагировали этилацетатом (2×250 мл). Объединенные органические экстракты промывали водой, насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением желаемого соединения 3-(4-йодфенокси)бутан-2-она (6 г, 86%).
Стадия-2: Синтез 3-(4-йодфенокси)бутан-2-амина

К перемешиваемому раствору 3-(4-йодфенокси)бутан-2-она (3 г, 10,3 ммоль) в метаноле (50 мл) добавляли при 0°C цианоборгидрид натрия (0,98 г, 15,5 ммоль) и ацетат аммония (11,8 г, 154,5 ммоль). Реакционную смесь перемешивали в течение 12 ч при комнатной температуре. После завершения реакции, реакционную смесь разбавляли конц. HCl и водой, экстрагировали диэтиловым эфиром. Водный слой подщелачивали 5% раствором NaOH, экстрагировали этилацетатом. Органический слой концентрировали при пониженном давлении и неочищенный материал использовали на следующей стадии без дополнительной очистки (2,2 г, 70%).
Стадия-3: Синтез (E)-4-((3-(4-йодфенокси)бутан-2-ил)амино)-N,N-диметилбут-2-енамида

Реакцию осуществляли в соответствии со Схемой 3, Стадия-3, используя 3-(4-йодфенокси)бутан-2-амин (2,9 г, 10,03 ммоль) и смесь (E)-4-хлор-N,N-диметилбут-2-енамида и (E)-4-бром-N,N-диметилбут-2-енамида (1,73 г, 9,03 ммоль, Пример 63, Стадия-7). Неочищенный материал использовали на следующей стадии (4 г, неочищенный).
Стадия-4: Синтез трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(3-(4-йодфенокси)бутан-2-ил)карбамата

Реакцию осуществляли в соответствии со Схемой 3, Стадия-4, используя (E)-4-((3-(4-йодфенокси)бутан-2-ил)амино)-N,N-диметилбут-2-енамид (4 г, 9,9 ммоль) вместо соединения 218. Неочищенный продукт очищали с помощью combi-flash, элюируя 70% этилацетата в н-гексане, с получением указанного в заголовке соединения Пр.96 Стадия-4 (2,9 г, 60%).
Соединение 96:1H ЯМР (400 MГц, DMSO-d6): δ 13,05 (с, 1H), 8,06 (с, 1H), 7,61 (с, 1H), 7,51 (д, J=11,2 Гц, 1H), 7,22-7,10 (м, 6H), 6,74-6,71 (м, 2H), 6,61-6,43 (м, 4H), 4,23-4,20 (м, 1H), 3,35-3,28 (м, 2H), 2,94-2,81 (м, 6H), 2,71-2,67 (м, 1H), 2,42-2,32 (м, 2H), 1,09-0,84 (м, 9H). ЖХМС: 523,3 [M+H]+.
Пример 97: Примера 97 нет.
Пример 98: Синтез (E)-4-((1-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)-2-метилпропан-2-ил)амино)-N,N-диметилбут-2-енамида (Соединение 98)
Соединение синтезировали в соответствии с подходом, описанным в примере 3, (i) используя на стадии-1 Пр.3 5-(бут-1-ин-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазол (полученный в Примере 1, Стадия-3) вместо соединения 252, (ii) непосредственно переходя от Стадии-1 к Стадии-3 и используя на Стадии-3 Пр.3 трет-бутил (1-(4-йодфенокси)-2-метилпропан-2-ил)карбамат (получение показано ниже в Пр.98 Стадия 1-4) вместо соединения 256, (iii) используя на Стадии-4 Пр.3 иодбензол вместо соединения 227, (iv) заменяя Стадию-5 Пр.3 условиями реакции, описанными на схеме 2, Стадия-6b, и (v) используя на стадии-6 Пр.3 смесь (E)-4-хлор-N,N-диметилбут-2-енамида и (E)-4-бром-N,N-диметилбут-2-енамида (полученную в Примере 49, Стадия-4) вместо соединения 251, с получением соединения 98 (0,035 г).
Стадия-1: Синтез 2-метил-1-(4-нитрофенокси)пропан-2-амина

К перемешиваемому раствору 2-амино-2-метилпропан-1-ола (4,5 г) в THF (25 мл) добавляли раствор трет-бутоксида калия (5,7 г, 51 ммоль) в THF (25 мл) при 0°C, перемешивали в течение 1 ч при той же температуре. 1-фтор-4-нитробензол (5 г, 35,4 ммоль) добавляли к вышеуказанной реакционной смеси и реакционную смесь перемешивали при комнатной температуре в течение 4 ч. После завершения реакции, реакционную смесь разбавляли ледяной водой, экстрагировали EtOAc. Органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением указанного в заголовке соединения Пр.98 Стадия-1 (6 г, 80%).
Стадия-2: Синтез трет-бутил (2-метил-1-(4-нитрофенокси)пропан-2-ил)карбамата

К перемешиваемому раствору 2-метил-1-(4-нитрофенокси)пропан-2-амина (5 г, 23,5 ммоль) в DCM (50 мл) добавляли TEA (6,3 мл, 35,3 ммоль) при комнатной температуре, перемешивали в течение 15 мин. Добавляли Boc ангидрид (6,1 мл, 28,2 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. После завершения реакции, реакционную смесь разбавляли ледяной водой, экстрагировали EtOAc. Органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением указанного в заголовке соединения Пр.98 Стадия-2 (7,3 г).
Стадия-3: Синтез трет-бутил (1-(4-аминофенокси)-2-метилпропан-2-ил)карбамата

К раствору трет-бутил (2-метил-1-(4-нитрофенокси)пропан-2-ил)карбамата (6 г, 19,35 ммоль) в метаноле (70 мл) добавляли 10% Pd/C (1,5 г). Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода в течение 3 ч. После завершения реакции, реакционную смесь фильтровали через целит, фильтрат концентрировали при пониженном давлении с получением неочищенного продукта, неочищенное соединение использовали на следующей стадии без дополнительной очистки (6 г, неочищенный).
Стадия-4: Синтез трет-бутил (1-(4-йодфенокси)-2-метилпропан-2-ил)карбамата
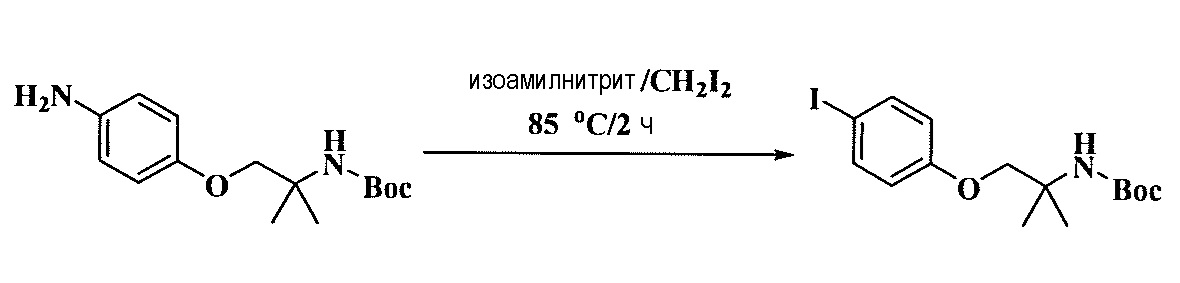
Смесь трет-бутил (1-(4-аминофенокси)-2-метилпропан-2-ил)карбамата (4 г, 14,2 ммоль), изоамилнитрита (19 мл, 142 ммоль) и дийодметана (11,4 мл, 142 ммоль) перемешивали при 85°C в течение 2 ч. После завершения реакции, реакционную смесь разбавляли ледяной водой, экстрагировали EtOAc. Органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного продукта, неочищенное соединение с помощью combi-flash, элюируя 5% этилацетата в н-гексане, с получением указанного в заголовке соединения в Пр.98 Стадия-4 (0,9 г, 37%).
Соединение 98:1H ЯМР (400 MГц, DMSO-d6): δ 13,05 (с, 1H), 8,06 (с, 1H), 7,60 (с, 1H), 7,51 (д, J=8,8 Гц, 1H), 7,20 (д, J=7,2 Гц, 1H), 7,15-7,10 (м, 5H), 6,74 (д, J=8 Гц, 2H), 6,65-6,58 (м, 3H), 6,49 (д, J=15,2 Гц, 1H), 3,60 (с, 2H), 3,28-3,06 (м, 2H), 2,98 (с, 3H), 2,89 (с, 3H), 2,40 (д, J=8 Гц, 2H), 1,04 (с, 6H), 0,87 (т, J=7,2 Гц, 3H). ЖХМС: 523,4 [M+H]+.
Пример 99: Синтез (E)-4-((2-(4-((E)-1-(1H-индазол-5-ил)-2-фенилбут-1-ен-1-ил)фенокси)-2-метилпропил)амино)-N,N-диметилбут-2-енамида (Соединение 99)
Соединение синтезировали в соответствии с подходом, описанным в примере 59, (i) с использованием на стадии-4 Пр.59 5-(бут-1-ин-1-ил)-1-(тетрагидро-2Н-пиран-2-ил)-1H-индазола (полученный в примере 1, Стадия-3) вместо соединения 276 и трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)-2-метилпропил)карбамата (получение показано ниже Пр.99 стадия 1-5) вместо соединения 277, и (ii) с использованием на стадии-5 Пр.59 иодбензола вместо соединения 265 с получением соединения 99 (0,19 г).
Стадия-1: Синтез этил 2-(4-йодфенокси)-2-метилпропаноата

К перемешиваемому раствору 4-иодфенола (10 г, 45,45 ммоль) в ацетонитриле (100 мл) добавляли карбонат цезия (29,5 г, 90 ммоль) и 2-бром-2-метилпентан-3-он (8,86 г, 45,45 ммоль). Содержимое перемешивали при 70°C в течение 12 ч. После завершения реакции, реакционную смесь выливали в ледяную воду и экстрагировали этилацетатом (2×250 мл). Объединенные органические экстракты промывали водой, насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением желаемого соединения этил 2-(4-йодфенокси)-2-метилпропаноата (15 г).
Стадия-2: Синтез 2-(4-йодфенокси)-2-метилпропановой кислоты

К перемешиваемому раствору этил 2-(4-йодфенокси)-2-метилпропаноата (15 г, 44,91 ммоль) в MeOH:H2O (60 мл, 3:1) добавляли LiOH.H2O (3,77 г, 89,82 ммоль). Реакционную смесь перемешивали в течение 12 ч при комнатной температуре, после завершения реакции (контролируемой ТСХ), реакционную смесь разбавляли водой и подкисляли 2M HCl, экстрагировали EtOAc (300 мл). Органический слой промывали водой, затем насыщенным раствором NaCl, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт использовали на следующей стадии без дополнительной очистки (12 г).
Стадия-3: Синтез 2-(4-йодфенокси)-2-метилпропанамида
К перемешиваемому раствору 2-(4-йодфенокси)-2-метилпропановой кислоты (13 г, 42,48 ммоль) в DMF (25 мл) добавляли DIPEA (14,6 мл, 84,96 ммоль), EDCI.HCl (12,21 г, 63,72 ммоль), HOBT (8,6 г, 63,72 ммоль) и хлорид аммония (4,58 г, 84,96 ммоль). Реакционную смесь перемешивали в течение 12 ч при комнатной температуре, после завершения реакции (контролируемой ТСХ), реакционную смесь разбавляли водой и экстрагировали EtOAc (500 мл). Органический слой промывали водой, затем насыщенным раствором NaCl, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением неочищенного соединения, неочищенное соединение очищали с помощью combi-flash, с использованием 2% MeOH в дихлорметане, с получением указанного в заголовке соединения Пр.99 Стадия-3 (12 г, 97%).
Стадия-4: Синтез 2-(4-йодфенокси)-2-метилпропан-1-амина

Раствор 2-(4-йодфенокси)-2-метилпропанамида (11,2 г, 36,72 ммоль), в THF (110 мл) добавляли к раствору комплекса боран-метилсульфида (36,7 мл, 73,44 ммоль) добавляли по каплям и содержимое перемешивали в течение 6 ч при 70°C. Реакционную смесь гасили метанолом, к реакционной смеси добавляли воду, дважды экстрагировали этилацетатом. Объединенные органические слои промывали водой, затем насыщенным солевым раствором и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении с получением указанного в заголовке соединения в Пр.99 Стадия-4 (8,5 г, 80%).
Стадия-5: Синтез трет-бутил (E)-(4-(диметиламино)-4-оксобут-2-ен-1-ил)(2-(4-йодфенокси)-2-метилпропил)карбамата

Реакцию осуществляли в соответствии со Схемой 3, Стадия-3, с использованием 2-(4-йодфенокси)-2-метилпропан-1-амина (5 г, 17,18 ммоль) в DMF (50 мл) и смеси (E)-4-хлор-N,N-диметилбут-2-енамида и (E)-4-бром-N,N-диметилбут-2-енамида (2,39 г, 13,74 ммоль, Пример 63, Стадия-7). Неочищенный продукт очищали с помощью combi-flash, элюируя 2% MeOH в дихлорметане, с получением указанного в заголовке продукта Пр.99 Стадия5 (5 г, 58%).
Соединение 99:1H ЯМР (400 MГц, DMSO-d6): δ 13,06 (с, 1H), 8,07 (с, 1H), 7,63 (с, 1H), 7,52 (д, J=8,8 Гц, 1H), 7,11-7,10 (м, 5H), 7,09 (д, J=2 Гц, 1H), 6,75 (д, J=8,4 Гц, 2H), 6,63-6,60 (м, 2H), 6,51 (т, J=5,4 Гц, 1H), 6,50 (с, 1H), 2,98 (с, 3H), 2,84 (с, 3H), 2,41 (кв, J=7,2 Гц, 2H), 1,12 (с, 6H), 0,87 (т, J=7,2 Гц, 3H). ЖХМС: 523,3 [M+H]+.
Пример 100: Примера 100 нет.
ПРИМЕРЫ
Пример 101 - Соединения, которые ингибируют активность ERαWT/MUT in vitro
Культура клеток
MCF7 BUS клетки (Coser, et al., (2003) PNAS 100(24): 13994-13999) поддерживали в модифицированной по способу Дульбекко среде Игла, дополненной 10% FBS, 4 мМ L-глутамином и 1x незаменимыми аминокислотами. Lenti-X 293T клетки (Clontech, Cat # 632180) обычно культивировали в модифицированной по способу Дульбекко среде Игла, дополненной 10% FBS.
Сайт-направленный мутагенез и конструирование клеточной линии
Набор для сайт-направленного мутагенеза QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies, Cat # 200523) использовали для генерации мутаций Y537S, Y537C, Y537N и D538G в экзоне 8 ERα. кДНК ESR1 дикого типа (GeneCopoeia Inc., Cat# GC-A0322, номер доступа NM 000125) использовали в качестве матрицы со следующими мутагенезными праймера (где подчеркнутые нуклеотиды представляют участок мутаций); Y537S: F-AAG AAC GTG GTG CCC CTC TCT GAC CTG CTG CTG GAG ATG (SEQ ID NO:1), R-CAT CTC CAG CAG CAG GTC AGA GAG GGG CAC CAC GTT CTT (SEQ ID NO:2); Y537N: F-AAG AAC GTG GTG CCC CTC AAT GAC CTG CTG CTG GAG ATG (SEQ ID NO:3), R-CAT CTC CAG CAG CAG GTC ATT GAG GGG CAC CAC GTT CTT (SEQ ID NO: 4); Y537C: F-AAG AAC GTG GTG CCC CTC TGT GAC CTG CTG CTG GAG ATG (SEQ ID NO:5), R-CAT CTC CAG CAG CAG GTC ACA GAG GGG CAC CAC GTT CTT (SEQ ID NO:6); D538G: F-AAC GTG GTG CCC CTC TAT GGC CTG CTG CTG GAG ATG CTG (SEQ ID NO:7), R-CAG CAT CTC CAG CAG CAG GCC ATA GAG GGG CAC CAC GTT (SEQ ID NO:8). кДНК ESR1 дикого типа и мутантных ESR1 клонировали в лентивирусный вектор, обозначенный pLenti6.3/V5-Dest (Invitrogen, Cat #V533-06). Для создания лентивируса ДНК (ESR1 дикого типа и мутант ESR1) совместно трансфицировали упаковочными плазмидами в Lenti-X 293T клетки с использованием TransIT (Mirus, Cat #MIR 2700). Через 48 ч после трансфекции среду, содержащую вирус, фильтровали и добавляли к клеткам MCF7 в присутствии 8 мкг/мл полибрена в течение ночи. Через два дня после заражения клетки помещали для селекции с 10 мкг/мл бластицидина в течение 2 недель для стабильной экспрессии.
Анализы пролиферации in vitro
MCF7-WT и -Y537S клетки высевали при 1500 клеток/лунку в 96-луночные планшеты с черной стенкой (аналитические планшеты, Costar, Cat # 3904). Параллельно клетки также высевали в отдельный 96-луночный планшет (8 лунок/клеточная линия, контрольный планшет), для которого на следующий день измеряли CTG (CellTiter-Glo® Luminescent Viability Assay, Promega, Cat #G7572) (показания день 0). Показания дня 0 использовали для расчета GI50 при завершении эксперимента. На следующий день после посева соединения добавляли в планшеты для анализа. Кратко, серийное разведение 1:4 готовили в DMSO в 200x конечной концентрации для общего количества концентраций 10 (9 разведений, содержащих соединение, и одно содержит только DMSO). Серийно разведенные соединения пипетировали в среду для получения смеси соединение-среда с 10x конечной концентрацией. 10 мкл смеси соединение-среда добавляли к MCF7-WT и -Y537S клеткам при 3 лунки/концентрация (в трех повторностях для каждой концентрации). На 3 день среду/соединение удаляли и заменяли свежей средой/соединением, как описано выше. На 6-й день измеряли CTG и сравнивали с показаниями дня 0 с контрольной пластины для оценки GI50.
Результаты
Фиг. 1 показывает, что эктопическая экспрессия ERαY537S/N/C, D538G в клетках MCF7 придает фенотипическую устойчивость к применяемым в настоящее время методам лечения тамоксифеном (SERM), ралоксифеном (SERM) и фулвестрантом (SERD). Аналогичные наблюдения были недавно опубликованы несколькими независимыми лабораториями (Jeselsohn et al., (2014) Clin Cancer Res. Apr 1;20(7):1757-67; Toy et al., (2013) Nat Genet. 2013 Dec;45(12):1439-45; Robinson et al., (2013) Nat Genet. Dec;45(12):1446-51; Merenbakh-Lamin et al., (2013) Cancer Res. Dec 1;73(23):6856-64; Yu et al., (2014) Science Jul 11;345(6193):216-20). Подтвердив, что ERαMUT управляет устойчивость к существующим эндокринным терапиям, поиск новых соединений, которые уменьшали бы пролиферацию ERαMUT-несущих MCF7 клеток более эффективно, чем соответствующее клиническое соединение 4-гидрокситамоксифен, является востребованным. Используя анализ жизнеспособности WT (дикого типа) и мутанта в качестве формы для скрининга, были идентифицированы соединения, которые были более эффективными по отношению к линии YF37S с MCF7, относительно 4-гидрокситамоксифена. Результаты теста анализа жизнеспособности показаны в таблице 1 выше.
Способы in vivo c ксенотрансплантатом
Методы и материалы
В следующих примерах (примеры 102-109) и на фигурах, которые упоминаются в этих примерах, ссылки на соединение 1, соединение 60 и соединение 69 относятся к хлористоводородной соли соответственно пронумерованных соединений. Эти хлористоводородной соли получают, как описано выше в примерах 1А, 60А и 69А.
Исследование ксенотрансплантата MCF7
Линию клеток MCF7 (АТСС) рака молочной железы человека ER+ с ESR1 дикого типа культивировали в среде DMEM с добавлением 10% FBS при 37°С в атмосфере 5% СО2 и хранили в экспоненциальной фазе роста. Клетки собирали в трипсине и повторно суспендировали в смеси 1:1 матригеля и HBSS при конечной концентрации 5×107 клеток/мл. 0,2 мл аликвот клеток вводили подкожно в 3-ю жировую ткань молочной железы 6-8-недельных самок ʺголыхʺ мышей Balb/c, вводя 1×107 клеток/мышь. Когда средний объем опухоли достигал приблизительно 155 мм3, 92 животных рандомизировали до лечения.
Противоопухолевую активность в модели ксенотрансплантата MCF7 исследовали с использованием соединения 1, соединения 60 и соединения 69. Все соединения вводили перорально каждый день в дозах от 1 до 30 мг/кг. Каждое лечение было начато в День 0, и график приема препарата продолжался в течение 17 дней. Объем введения рассчитывали по индивидуальным массам тела мыши до введения дозы. Массы тела измеряли ежедневно, а объемы опухоли измеряли два раза в неделю. Объемы опухоли (TV) рассчитывали по формуле:
TV=длина×ширина2×0,5
длина: наибольший диаметр опухоли (мм)
ширина: диаметр перпендикулярно длине (мм)
% Ингибирования роста опухоли (TGI) рассчитывали по следующей формуле:
% Ингибирования роста опухоли (TGI)=((средний объем опухоли в контрольной группе в День Х-объем опухоли в обработанной группе в День Х)/(средний объем опухоли в контрольной группе в День Х))×100
Где День X является конечной точкой измерения.
Исследование PDx ксенотрансплантата Y537S положительного
Модель опухоли ксенотрансплантата, полученного из тканей пациента (PDX), представляющая собой ESR1-Y537S мутантный ER+ рак молочной железы человека, обозначенный как PDX-Y537S, выращивали подкожно у иммунокомпрометированных мышей. Опухоли вырезали через 60 дней после имплантации и обрабатывали в смешанные фрагменты опухоли. Ткани солидной опухоли истощали некротическими компонентами, разрезали на фрагменты 70 мг, смешивали с матригелем и подкожно имплантировали в правый бок 6-12-недельных самок бестимусных ʺголыхʺ мышей (Crl:NU(NCr)-Foxn1nu). Точное количество фрагментов и объем матригеля определяли в каждом конкретном случае. Когда средний объем опухоли достиг приблизительно 200 мм3, животных рандомизировали до лечения. Все первичные опухоли человека, используемые в этом исследовании, прошли 7 пассирований in vivo.
Противоопухолевую активность в модели PDX-Y537S исследовали с использованием соединения 1, соединения 60 и соединения 69. Эстроген не был добавлен в исследованиях. Все соединения вводили перорально каждый день при дозах от 3 до 200 мг/кг. Каждое лечение начинали в День 0, и график приема препарата продолжался в течение 35 дней. Объем введения рассчитывали по индивидуальным массам тела мыши до введения дозы. Массы тела измеряли ежедневно, а объемы опухоли измеряли два раза в неделю. Объемы опухолей рассчитывали на основе ранее описанной формулы.
Исследование ксенотрансплантата WHIM20
Модель опухоли ксенотрансплантата, полученного из тканей пациента (PDX), WHIM20, представляющая собой ESR1-Y537S мутантный ER+ рак молочной железы человека, выращивали у мышей. Опухоли вырезали и обрабатывали в смешанные фрагменты опухоли, и фрагменты повторно имплантировали подкожно новым мышам-реципиентам. Для текущей работы ткани солидной опухоли истощали некротическими компонентами, разрезали на фрагменты, смешивали с матригелем и подкожно имплантированы в правый бок 6-8-недельных самок мышей SCID-bg. Точное количество фрагментов и объем матригеля определяли в каждом конкретном случае. Когда средний объем опухоли достиг приблизительно 370 мм3, животных рандомизировали до лечения. Все первичные опухоли человека, используемые в этом исследовании, прошли 4 пассирования in vivo.
Противоопухолевую активность в модели ксенотрансплантата WHIM20, полученной от пациента, исследовали с использованием соединения 1 и соединения 60 в отдельных исследованиях. Эстроген не был добавлен в исследованиях WHIM20. Соединения 1 и 60 вводили перорально каждый день, в указанных дозах. Каждое лечение начинали в День 0, и график приема препарата продолжался в течение указанных дней. Объем введения рассчитывали по индивидуальным массам тела мыши до введения дозы. Массы тела измеряли ежедневно, а объемы опухоли измеряли два раза в неделю. Объемы опухолей рассчитывали на основе ранее описанной формулы.
Статистический анализ
Данные выражали в виде среднее±SEM для объема опухоли и среднее±SEM для массы тела. Различия в объеме опухоли в течение периода исследования между группой, обработанной носителем, и группой, обработанной соединением, анализировали с помощью двухфакторного дисперсионного анализа (ANOVA), а затем с помощью апостериорного критерия множественного сравнения Даннетта. Статистический анализ проводили с использованием GraphPad Prism® version 5.04 (GraphPad Software, La Jolla, CA).
Пример 102
Фиг. 2 показывает противоопухолевый эффект и эффект на массу тела соединения 1 в модели ксенотрансплантата MCF7, несущей дикий тип ER, выращенного в иммунокомпрометированных мышах. Соединение 1 ингибировало рост ксенотрансплантата дозозависимым образом с 10 мг/кг один раз в день и 30 мг/кг один раз в день, существенно ингибируя рост на 17-й день по сравнению с контролем (TGI 68% и 83% и р<0,0001 для обеих доз, соответственно). Обработка соединения 1 1 мг/кг один раз в день и 3 мг/кг один раз в день статистически не отличалась от группы, получавшей контроль (TGI 19% и 41%, соответственно). Все дозы и схемы лечения хорошо переносились без существенной потери массы тела.
Соединение 1 давали перорально один раз в день в течение всего периода исследования. Данные на фиг.2 представляют среднее±SEM (объем опухоли) или среднее±SEM (масса тела) (N=6 для групп лечения, N=8 для контрольной группы с носителем). *p<0,0001 в сравнении с контролем носителем на день 17 (двухфакторный дисперсионный анализ (ANOVA), затем критерий множественного сравнения Даннетта).
Пример 103
Фиг.3 показывает противоопухолевый эффект и эффект на массу тела Соединения 1 в модели PDX-Y537S с гетерозиготной мутацией Y537S. Соединение 1, вводимое ежедневно, ингибировало рост ксенотрансплантата дозозависимым образом с обработкой 10 мг/кг, 30 мг/кг, 100 мг/кг и 200 мг/кг, существенно ингибируя рост на 35-й день (TGI 35%, 63%, 76% и 81% и p<0,01, p<0,0001, p<0,0001 и p<0,0001, соответственно). Обработанные ежедневно соединением 1 при дозе 3 мг/кг статистически не отличалось от группы, обработанной носителем (TGI 3%). Все дозы и схемы лечения хорошо переносились без существенной потери массы тела.
Соединение 1 давали перорально один раз в день в течение всего периода исследования. Данные представляют среднее±SEM (объем опухоли) или среднее±SEM (масса тела) (N=8 для всех групп). *p<0,01, **p<0,0001 в сравнении с контролем носителем на день 35 (двухфакторный дисперсионный анализ (ANOVA), затем апостериорный критерий множественного сравнения Даннетта).
Пример 104
Фиг.4 показывает противоопухолевый эффект и эффект на массу тела Соединения 1 в ER+ WHIM20 PDX модели с гомозиготной мутацией Y537S. Ежедневная обработка соединением 1 при 100 мг/кг существенно уменьшала рост опухоли по сравнению с контролем носителем (TGI 45%; p<0,05). Эта доза соединения 1 переносилась в соответствии с внутренними рекомендациями комитета по уходу за животными и использованию.
Соединение 1 давали перорально один раз в день в течение всего периода исследования. Данные представляют среднее±SEM (объем опухоли) или среднее±SEM (масса тела) (N=9 для всех групп). *p<0,05 в сравнении с контролем носителем на день 27 (двухфакторный дисперсионный анализ (ANOVA), затем апостериорный критерий множественного сравнения Даннетта).
Пример 105
Фиг. 5 показывает противоопухолевый эффект и эффект на массу тела соединения 60 в модели подкожного ксенотрансплантата MCF7, несущей дикий тип ER, выращенной у иммунокомпрометированных мышей. Соединение 60 ингибировало рост ксенотрансплантата дозозависимым образом с обработкой 3 мг/кг один раз в день, 10 мг/кг один раз в день и 30 мг/кг один раз в день, ингибируя рост на день 17 (TGI 75%, 80% и 85% и p<0,0001 для всех доз, соответственно). Обработка соединением 60 1 мг/кг один раз в день×18 не показало статистически значимого отличия от группы, получавшей контроль (TGI 36%, p>0,05). Все дозы и схемы лечения хорошо переносились без существенной потери массы тела или клинических признаков.
Соединение 60 давали перорально один раз в день в течение всего периода исследования. Данные представляют среднее±SEM (объем опухоли) или среднее±SEM (масса тела) (N=6 для групп лечения, N=8 для контрольной группы с носителем). *p<0,0001 в сравнении с контролем носителем на день 17 (двухфакторный дисперсионный анализ (ANOVA), затем критерий множественного сравнения Даннетта).
Пример 106
Фиг.6 показывает противоопухолевый эффект и эффект на массу тела соединения 60 в повторном исследовании в модели ER+ PDX-Y537S с гетерозиготной мутацией Y537S. Соединение 60 ингибировало рост ксенотрансплантата дозозависимым образом с обработкой 3 мг/кг один раз в день, 10 мг/кг один раз в день, 30 мг/кг один раз в день и 100 мг/кг один раз в день, существенно ингибируя рост на 28 день (TGI 61%, 85%, 81% и 84% и p<0,001, p<0,0001, p<0,0001 и p<0,0001, соответственно). Все дозы хорошо переносились без значительного снижения массы тела или клинических признаков.
Соединение 60 давали перорально один раз в день в течение всего периода исследования. Данные представляют среднее ± SEM (объем опухоли) или среднее ± SEM (масса тела) (N=6 для соединения 60 и N=8 для носителя). *p<0,001, **p<0,0001 в сравнении с контролем носителем на день 28 (двухфакторный дисперсионный анализ (ANOVA), затем критерий множественного сравнения Даннетта).
Пример 107
Фиг. 7 показывает противоопухолевый эффект и эффект на массу тела соединения 69 в модели подкожного ксенотрансплантата MCF7, несущей дикий тип ER, выращенной иммунокомпрометированных мышей. Соединение 69 ингибировало рост ксенотрансплантата дозозависимым образом с обработкой 3 мг/кг один раз в день, 10 мг/кг один раз в день и 30 мг/кг один раз в день, ингибируя рост на день 17 (TGI 72%, 80% и 83% и p<0,0001 для всех доз, соответственно). Обработка соединением 69 1 мг/кг один раз в день не показала статистически значимого отличия от группы, получавшей контроль (TGI 42%). Все дозы хорошо переносились без значительного снижения массы тела или клинических признаков.
Соединение 69 давали перорально один раз в день в течение всего периода исследования. Данные представляют среднее ± SEM (объем опухоли) или среднее ± SEM (масса тела) (N=6 для групп лечения, N=8 для контрольной группы с носителем). *p<0,0001 в сравнении с контролем носителем на день 17 (двухфакторный дисперсионный анализ (ANOVA), затем критерий множественного сравнения Даннетта).
Пример 108
Фиг.8 показывает противоопухолевый эффект и эффект на массу тела соединения 69 в модели ER+ PDX-Y537S с гетерозиготной мутацией Y537S. Соединение 69 показало значительную эффективность с обработкой 3 мг/кг один раз в день, 10 мг/кг один раз в день, 30 мг/кг один раз в день и 100 мг/кг один раз в день, ингибируя рост на день 28 по отношению к группе, обработанной носителем (TGI 62%, 72%, 67% и 76% и p<0,001, p<0,0001, p<0,001 и p<0,0001, соответственно). Все дозы хорошо переносились без значительного снижения массы тела или клинических признаков.
Соединение 69 давали перорально один раз в день в течение всего периода исследования. Данные представляют среднее±SEM (объем опухоли) или среднее±SEM (масса тела) (N=8). *p<0,001, **p<0,0001 в сравнении с контролем носителем на день 28 (двухфакторный дисперсионный анализ (ANOVA), затем критерий множественного сравнения Даннетта).
Пример 109
Фиг.9 показывает противоопухолевый эффект и эффект на массу тела соединения 60 в ER+ WHIM20 PDX модели с гомозиготной мутацией Y537S. Соединение 60 ингибировало рост ксенотрансплантата дозозависимым образом с обработкой 10 мг/кг один раз в день, 30 мг/кг один раз в день и 100 мг/кг один раз в день, существенно ингибируя рост на 22 день (TGI 26%, 36%, и 48% и p<0,05, p<0,01 и p<0,0001, соответственно). Эта доза соединения 60 переносилась в соответствии с внутренними рекомендациями комитета по уходу за животными и использованию.
Соединение 60 давали перорально один раз в день в течение всего периода исследования. Данные представляют среднее±SEM (объем опухоли) или среднее±SEM (масса тела) (N=8 для всех групп). *p<0,05, **p<0,01 и ***p<0,0001, соответственно, в сравнении с контролем носителем на день 22 (двухфакторный дисперсионный анализ (ANOVA), затем критерий множественного сравнения Даннетта).
Для специалистов в данной области будет очевидно, что в данном описании представлены новые, улучшенные и неочевидные композиции с достаточной подробностью. Кроме того, для специалистов в данной области будет очевидно, что существуют модификации, вариации, замены и эквиваленты для признаков композиций, которые существенно не отходят от сущности и объема вариантов осуществления, раскрываемых в настоящем описании. Таким образом, предполагается, что все такие модификации, изменения, замены и эквиваленты, находятся в пределах сущности и объема настоящего изобретения, как определено прилагаемой формулой изобретения, должны быть охвачены прилагаемой формулой изобретения.
--->
СПИСОК ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> Eisai R&D Management Co., Ltd.
Bock, Mark
Korpal, Manav
Hao, Ming-Hong
Nyavanandi, Vijay K
Puyang, Xiaoling
Samajdar, Susanta
Smith, Peter G
Wang, John
Zheng, Guo Zhu
Zhu, Ping
<120> ТЕТРАЗАМЕЩЕННЫЕ АЛКЕНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ
<130> 0080171-000246
<160> 8
<170> PatentIn version 3.5
<210> 1
<211> 39
<212> ДНК
<213> искусственная последовательность
<220>
<223> мутагенезный праймер
<400> 1
aagaacgtgg tgcccctctc tgacctgctg ctggagatg 39
<210> 2
<211> 39
<212> ДНК
<213> искусственная последовательность
<220>
<223> мутагенезный праймер
<400> 2
catctccagc agcaggtcag agaggggcac cacgttctt 39
<210> 3
<211> 39
<212> ДНК
<213> искусственная последовательность
<220>
<223> мутагенезный праймер
<400> 3
aagaacgtgg tgcccctcaa tgacctgctg ctggagatg 39
<210> 4
<211> 39
<212> ДНК
<213> искусственная последовательность
<220>
<223> мутагенезный праймер
<400> 4
catctccagc agcaggtcat tgaggggcac cacgttctt 39
<210> 5
<211> 39
<212> ДНК
<213> искусственная последовательность
<220>
<223> мутагенезный праймер
<400> 5
aagaacgtgg tgcccctctg tgacctgctg ctggagatg 39
<210> 6
<211> 39
<212> ДНК
<213> искусственная последовательность
<220>
<223> мутагенезный праймер
<400> 6
catctccagc agcaggtcac agaggggcac cacgttctt 39
<210> 7
<211> 39
<212> ДНК
<213> искусственная последовательность
<220>
<223> мутагенезный праймер
<400> 7
aacgtggtgc ccctctatgg cctgctgctg gagatgctg 39
<210> 8
<211> 39
<212> ДНК
<213> искусственная последовательность
<220>
<223> мутагенезный праймер
<400> 8
cagcatctcc agcagcaggc catagagggg caccacgtt 39
<---
Реферат
В настоящем изобретении описываются соединения формул II и III с радикалами, раскрытыми в материалах заявки, или их фармацевтически приемлемые соли, а также способы применения соединений для лечения рака молочной железы путем введения субъекту, нуждающемуся в этом, терапевтически эффективного количества соединений или их фармацевтически приемлемых солей. Рак молочной железы может быть ER-положительным раком молочной железы и/или субъект, нуждающийся в лечении, может экспрессировать мутантный ER-α белок. 8 н. и 35 з.п. ф-лы, 9 ил., 1 табл., 109 пр.
Формула




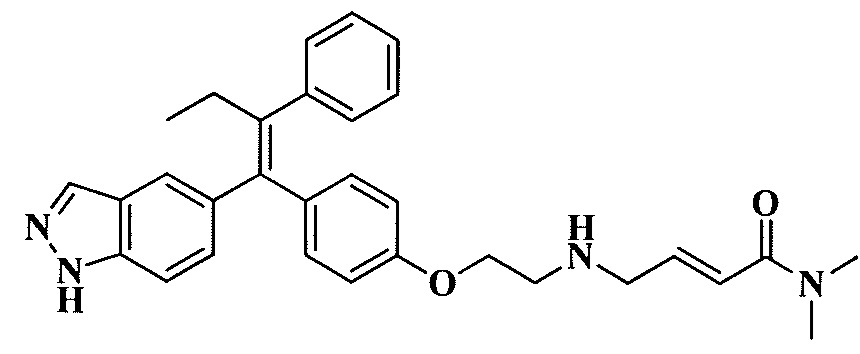



Документы, цитированные в отчёте о поиске
Соединение, фармацевтическая композиция, способ лечения или предупреждения эндометриоза и промежуточное соединение
Комментарии