Водорастворимые производные 3,5-дифенилдиазольных соединений - RU2764131C2
Код документа: RU2764131C2
Чертежи




Описание
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретения относится к водорастворимым производным 3,5-дифенилдиазольных соединений, которые являются эффективными терапевтическими агентами при применении в лечении заболеваний, связанных с агрегацией белка, и/или нейродегенеративных заболеваний, таких как болезнь Альцгеймера (AD), болезнь Паркинсона (PD) и трансмиссивная губчатая энцефалопатия (TSEs), такая как болезнь Крейтцфельда-Якоба (CJD).
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Известно большое число неврологических и нейродегенеративных заболеваний, многие из которых в настоящее время неизлечимы. Все распространенные нейродегенеративные заболевания характеризуются неправильным сворачиванием, агрегацией и/или осаждением определенных белков в мозге. Эти заболевания включают медицинские состояния, такие как болезнь Паркинсона (PD), болезнь Альцгеймера (AD), трансмиссивная губчатая энцефалопатия (TSEs), такая как болезнь Крейтцфельда-Якоба (CJD), старческое слабоумие, АА-амилоидоз, артериосклеротическая деменция, болезнь Хантингтона (HD), облитерирующий церебральный тромбангиит, деменция с тельцами Леви (DLB), мультисистемная атрофия (MSA) и многие другие. Диабет 2 типа представляет собой еще одно заболевание, патогенез которого включает упорядоченную агрегацию белка.
В случае TSE неправильно свернутый белок, который образует агрегаты, называется «прион», что происходит от «белковый (proteinaceous)» и «инфекционный (infectious)». Поэтому TSE также называют прионными заболеваниями. Основным событием в патогенезе TSE является конверсия клеточного прионного белка PrPС в патологическую изоформу PrPSc, которая скапливается в крупные белковые агрегаты. Прионы распространяются при передаче неправильно свернутого белка. Если прион попадает в здоровый организм, то он вызывает превращение существующих правильно сложенных белков в прионную форму, связанную с заболеванием. Эти вновь образованные прионы могут затем сами продолжать превращать больше белков; это запускает цепную реакцию, которая продуцирует большое количество белков прионной формы. Все известные прионы индуцируют образование амилоидной складчатости, при которой белок полимеризуется в агрегат, состоящий из плотно упакованных бета-листов. Амилоидные агрегаты представляют собой фибриллы, растущие на своих концах, а репликация при разрыве приводит к тому, что растущие два конца становятся четырьмя концами. Эта измененная структура чрезвычайно устойчива и накапливается в инфицированной ткани. Теория распространения, описанная для прионного белка, может также относится к образованию амилоидов при других заболеваниях с некорректным сворачиванием белков (PMD).
Еще один класс нейродегенеративных заболеваний, так называемых синуклеинопатий, характеризуется внутриклеточным накоплением белковых агрегатов, олигомеров, протофибрилл и фибрилл, содержащих в основном α-синуклеин. В случае синуклеопатий считается, что патологические эффекты на нервные клетки индуцируются путем образования олигомерных агрегатов α-синуклеина и последующего образования мембранных пор. Примерами синуклеопатий являются болезнь Паркинсона, деменция с тельцами Леви (DLB) и мультисистемная атрофия.
Фактически, конформационные изменения белка, связанные с патогенезом большинства PMD, приводят к образованию патологических белков, которые богаты структурой β-листа, частично устойчивы к протеолизу и в высокой степени склонны к образованию агрегатов более крупного порядка, что сходно с прионами. Образование амилоида зависит от медленного взаимодействия неправильно свернутых белковых мономеров с образованием олигомерных ядер, вокруг которых происходит более быстрая фаза удлинения. Способность олигомерных структур инициализировать собственный рост аналогична самораспространяющейся активности прионов.
Эти олигомеры, которые возникают в процессе агрегации, описаны в литературе как основной токсический агент, приводящий к дисфункции клеток и клеточной гибели. Одним из возможных механизмов, приводящих к гибели клеток, является перфорация мембран, вызванная белковыми агрегатами. Для лечения заболевания, вызванного агрегацией белка в ткани пациента, необходимо предотвратить, уменьшить агрегацию белка или, в лучшем случае, удалить ее из ткани.
В случае прионных заболеваний это может быть достигнуто с помощью терапевтического подхода, направленного на вмешательство в формирование и амплификацию инфекционного белка (PrPSc). Доказательства, полученные на клеточной культуре, и in vivo исследования позволяют предполагать, что после ингибирования образования PrPSc может происходить удаление PrPSc. Таким образом, этот терапевтический подход также может быть эффективным в конце инкубационного периода и даже после проявления клинических признаков заболевания, что важно для его применения в лечении прионного заболевания человека.
Существует ряд соединений, которые, как было показано, эффективно влияют на амплификацию PrPcS in vitro, такие как производные акридина, конго-красный, порфирины/фталоцианины, Cp-60, пептиды, разрушающие бета-листы, и варианты PrP. Однако ни одно из этих соединений до сих пор не имело эффективного применения при лечении заболеваний или в качестве соединений-прототипов для разработки соединений с повышенной терапевтической активностью и фармакологическими свойствами.
В WO02010/00372 описаны соединения, которые, как было показано, эффективны для ингибирования агрегации белков. Широкий скрининг основывался на сочетании метода сканирования интенсивно флуоресцирующих мишеней (SIFT) и клеточных анализов по измерению числа агрегаций α-синуклеина (PD) и прионного белка (CJD). В этом скрининге выяснилось, что 3,5-дифенилпиразольные (DPP) соединения являются активным каркасом, который может быть легко модифицирован путем органического синтеза. Была синтезирована матрица из примерно 250 соединений в этом классе и были оценены соединения для пероральной доступности и эффективности на моделях животных, имитирующих различные указанные заболевания (AD, CJD, PD). Соединение, обозначенное как «anle138b», имеющее следующую структуру:

5-(3-Бромфенил)-3-(3,4-метилендиоксифенил)-1H-пиразол
было идентифицирован как эффективный модулятор образования олигомеров на животных моделях, имитирующих болезнь Паркинсона (PD) (1,2), болезнь Альцгеймера (AD) (3) и болезнь Крейтцфельда-Якоба (CJD) (1,2). Было показано, что перфорация мембран, которая может быть механизмом нейротоксичности, вызванной олигомером, ингибируется anle138b (1).
Поскольку агрегация белка происходит непрерывно и приводит к нейродисфункции и потере нейронов, нейропротективное лечение потребует постоянного применения терапевтического агента. Такая терапия должна быть нетоксичной при постоянном дозировании и с действенными концентрациями соединений. Нетоксичность anle138b при лечении в течение более одного года была показана на некоторых мышиных моделях, как описано выше. Чтобы свести к минимуму необходимую пероральную дозу и при этом достигать эффективных уровней соединения, терапевтическое соединение также должно наиболее эффективно поглощаться после перорального применения. Было показано, что для того, чтобы быть эффективным, терапевтический агент должен введен per os и затем перемещен в кишечник, где он будет поглощен и транспортирован через кровоток в ткань, подверженную агрегации белка. Следовательно, соединение должно обладать способностью проходить гематоэнцефалический барьер. Чтобы быть эффективными, концентрация соединения в кишечнике перед поглощением в кровоток должна быть достаточно высокой.
Однако, хотя, как было показано, производные 3,5-дифенилпиразола (DPP) эффективны при ингибировании агрегации белков, было обнаружено, что производные DPP характеризуются также плохой растворимостью в водном растворе. Например, anle138b имеет растворимость 0,2 мкМ в воде.
Эта проблема была решена в случае anle138b путем разбавления соединения ДМСО и диспергирования разбавленного соединения в масле или арахисовом масле. В ДМСО 1 часть anle138b может быть растворена в 2 частях ДМСО. Было обнаружено, что резорбция из таких суспензий оливковое масло/ДМСО является высокоэффективной. Эта композиция оказалась хорошо переносимой у мышей и крыс. Однако ДМСО неприемлем для перорального применения у людей. Поэтому необходимо было исследовать альтернативные применения.
Еще один подход заключался в том, чтобы измельчить сухое соединение и смешать его с сухой едой для мыши/крысы. Однако это привело к приемлемым уровням веществ только у мышей, но не у крыс. Фармакокинетические исследования показали, что крысы не поглощают соединение из сухого корма. Этот способ применения также не подходит для длительной терапии пациента.
Еще один подход заключался в том, чтобы модифицировать терапевтический агент для повышения его водорастворимости. В предшествующем уровне техники обсуждалось использование фосфатов, сложных эфиров или соединений ПЭГ для повышения растворимости соединения. Было обнаружено, что путем объединения anle138b с эксципиентами на основе ПЭГ/кремофора у крыс может быть достигнуто воздействие 25% anle138b, достигаемое при введении sery433. Однако этот подход имеет несколько недостатков. Во-первых, необходимо использовать объемы порядка 20:1 эксципиент/anle138b, и, во-вторых, эксципиенты плохо переносятся некоторыми видами животных, включая людей. Например, кремофор вызывает аллергические реакции у людей. Кроме того, эксципиенты являются дорогостоящими, а объемы капсул будут в несколько раз больше объемов, требуемых для применения с водорастворимым порошком. Кроме того, в клинической практике соблюдение пациентом инструкций по приему исследуемого препарата может быть поставлено под угрозу, если пациенты, страдающие нейродегенерацией и поэтому часто имеющие проблемы с глотанием, должны принимать большое количество больших капсул.
Было обнаружено, что повышение водорастворимости anle138b особенно сложно, потому что растворимость в воде anle138b составляет всего 0,2 мкМ (т.е. 70 нг/мл), что даже ниже, чем у эталонного соединения «кирпичная пыль» DP-TAT-59 (4). Поэтому разработка эффективного водорастворимого производного терапевтического агента является ключевой проблемой, которую невозможно преодолеть просто путем применения подходов, предложенных в уровне техники.
Чтобы разработать удачное эффективное производное терапевтического агента, необходимо поддерживать терапевтические характеристики агента, повышая водорастворимость и биодоступность. Например, модификация терапевтического соединения должна приводить к тому, что терапевтическое соединение доставляется в кишечник большей частью в монодисперсной форме, так чтобы оно могло поглощаться в кровоток. Другие положительные характеристики немодифицированного терапевтического соединения также должны быть сохранены или даже улучшены. Например, модифицированное соединение должно быть стабильным во время хранения и в процессе прохождения от рта до кишечника. Оно также должен оставаться нетоксичным, чтобы быть пригодным для терапевтического использования.
Были исследованы некоторые обычные модификации этих соединений, но большинство из них были либо нестабильны, либо не доходили до полезных уровней в плазме, либо то и другое.
Таким образом, задача, решаемая настоящим изобретением, состоит в том, чтобы предложить 3,5-дифенилдиазольное соединение, подходящее для использования при лечении заболеваний, связанных с агрегацией белка, в стабильной форме, которое может применяться в терапевтически полезной концентрации и предпочтительно обеспечивает монодисперсный раствор в кишечнике. Предпочтительно 3,5-дифенилдиазольное соединение должно быть представлено в форме, которая не требует смешивания с большим количеством эксципиентов, так что можно достичь небольшого объема на дозу. В частности, целью настоящего изобретения является разработка водорастворимой формы 3,5-дифенилдиазольного соединения, которая может быть использована для перорального введения. Кроме того, поскольку вышеуказанные заболевания влияют на мозг, также необходимо, чтобы эффективная часть соединения могла пересекать гематоэнцефалический барьер и могла переноситься в мозг, то есть оставаться стабильной после того, как соединение высвобождается в кишечнике и достигает уровней в плазме крови, достаточных для лечения.
ОПИСАНИЕ ЧЕРТЕЖЕЙ
Изобретение также поясняется с отсылкой к следующим чертежам.
На фигуре 1 показана хроматограмма анализа ВЭЖХ sery433, пролекарства anle138b.
На фигуре 2 показана хроматограмма анализа ВЭЖХ anle423b, пролекарства anle253b.
На фигуре 3 показана концентрация anle138b в тканях мышей после применения дозы 1 мг пролекарства sery433.
На фигуре 4 показана концентрация во времени anle138b в образцах плазмы крыс после применения пролекарства sery433. В эксперименте взрослым крысам Sprague Dawley вводили пероральную дозу anle138b в PEG/кремофор («фаза 1») и sery433 в водном растворе («фаза 2») при номинальной дозе 10 мг/кг. Эксперименты проводились на трех крысах, которые были идентифицированы как «1M», «2M» и «3M».
ОПРЕДЕЛЕНИЯ
В данном описании и в формуле изобретения дается ссылка на ряд терминов, которые должны быть определены как имеющие следующие значения:
«3,5-Дифенилдиазольное соединение» в контексте настоящего изобретения относится к соединению, имеющему диазольное ядро, которое замещено двумя заместителями или незамещенными фенильными группами в положениях 3 и 5, или в положениях 2 и 4, или в положениях 2 и 5. Диазольное ядро является производным либо пиразола, либо имидазола. Другими словами, 3,5-дифенилдиазольное соединение по настоящему изобретению, в частности, представляет собой соединение 3,5-дифенил-пиразола (DPP) или соединение 2,4-дифенил-имидазола (DPI), или 2,5-дифенилимидазола (DPI), где фенильные группы могут быть замещенными или незамещенными. Всякий раз, когда описывается DPP соединение, понятно, что описание также относится к соответствующему соединению DPI, если позволяет контекст.
Диазольное ядро соединений по настоящему изобретению может существовать в двух таутомерных формах. Как каждая таутомерная форма сама по себе, так и смесь их обеих охватываются термином «диазол».
Термин «anle138b» используется для описания терапевтического агента, также известного как 5-(3-бромфенил)-3-(3,4-метилендиоксифенил)-1H-пиразол. Пиразольное кольцо соединения существует в двух таутомерных формах:

Поэтому «anle138b» можно описать следующими структурными формулами:

Всякий раз, когда в описании указывается одна из вышеупомянутых структур, предполагается, что должна быть охвачена и другая структура, а также смеси обеих структур. То же самое относится к другим структурным формулам, описывающим производные диазола по настоящему изобретению, например, DPP производные по настоящему изобретению такие как sery335b и anle253b.
Термин «заболевание, связанное с агрегацией белка» используется для всех состояний, расстройств или заболеваний, характеризующихся наличием агрегированной формы по меньшей мере одного белка или его фрагмента или производного, где белок, предпочтительно, выбран из группы, включающей прионный белок, белок-предшественник амилоида (APP), альфа-синуклеин, супероксиддисмутазу, тау, иммуноглобулин, амилоид-А, транстиретин, бета-2-микроглобулин, цистатин С, аполипопротеин A1, TDP-43, островковый амилоидный полипептид, ANF, гельсолин, инсулин, лизоцим, фибриноген, хантингин и атаксин и другие белки с растяжением Poly-Q.
Термин «прионное заболевание» или TSE используется для всех состояний, расстройств или заболеваний, вызванных образованием прионов, то есть белков, которые ошибочно используют и индуцируют неправильное сворачивание других белковых молекул. Примеры прионных заболеваний включают спорадическую и генетическую болезнь Крейтцфельдта-Якоба, вариант болезни Крейтцфельдта-Якоба (vCJD), расстройство у человека, вызванное инфекционным агентом губчатой энцефалопатии крупного рогатого скота (BSE), синдром Герстмана-Штрауслера-Шейнкера, фатальную семейную бессонницу и куру. TSE у других млекопитающих включают BSE крупного рогатого скота, скрейпи овец и коз, губчатую энцефалопатию кошек (FSE), трансмиссивную энцефалопатию норок (TME), энцефалопатию экзотических копытных (EUE) антилоп ньяла и большой куду, а также хроническую изнуряющую болезнь (CWD) оленей и лосей.
Термин «нейродегенеративное заболевание» охватывает болезни или состояния, такие как болезнь Паркинсона (PD), болезнь Альцгеймера (AD), трансмиссивная губчатая энцефалопатия (TSEs), такая как болезнь Крейтцфельда-Якоба (CJD), старческое слабоумие, АА-амилоидоз, артериосклеротическая деменция, болезнь Хантингтона (HD), облитерирующий церебральный тромбангиит, деменция с тельцами Леви (DLB), фронтотемпоральная деменция, боковой амиотрофический склероз, спинально-церебеллярная атаксия и другие болезни Poly-Q, мультисистемная атрофия (MSA), наследственная церебральная амилоидная ангиопатия, семейная амилоидная полинейропатия, первичный системный амилоидоз (AL амилоидоз), реактивный системный амилоидоз (АА-амилоидоз), диабет типа II, инъекционно-локализованный амилоидоз, бета-2-микроглобулиновый амилоидоз, наследственный не-нейропатический амилоидоз и финский наследственный системный амилоидоз.
Наиболее распространенной является болезнь Альцгеймера, которая поражает в настоящее время около 7 миллионов пациентов в Европе, Японии и США, и признаками которой являются агрегация белка Aβ с образованием бляшек Альцгеймера и тау-белка с образованием нейрофибриллярных клубков. Агрегация тау также несет ответственность за прогрессирующий супрануклеарный паралич (PSP), кортико-базальную дегенерацию (CBD) и фронтотемпоральную деменцию, включая болезнь Пика.
Болезнь Паркинсона поражает в настоящее время около 3,5 миллионов пациентов в вышеперечисленных странах и характеризуется агрегацией α-синуклеина, образующих тельца Леви. Мультисистемная атрофия (MSA) и деменция с тельцами Леви (LBD) также связаны с агрегацией α-синуклеина.
Болезнь Крейтцфельда-Якоба в виде спорадической формы в Европе менее распространена при примерно 500 новых пациентов в году. Новые правила забоя скота более или менее искоренили появление CJD. Следующими нейродегенеративными заболеваниями, связанными с агрегацией белков, являются болезнь Хантингтона (HD), а также боковой амиотрофический склероз (ALS).
Термины «расстройства, обусловленные неправильно свернутыми белками» (PMD), или протеопатия используются для описания заболеваний, расстройств или состояний, при которых определенные белки становятся структурно аномальными и тем самым нарушают функцию клеток, тканей и органов организма. Помимо вышеупомянутых прионных заболеваний и нейрогенеративных расстройств, где нейроны являются пораженными клетками, другие типы клеток также могут быть поражены неправильно свернутыми белками. Одним из примеров является диабет 2 типа, где белок амилин, также называемый «островковый амилоидный полипептид» (IAPP), является основным компонентом связанных с диабетом островковых амилоидных отложений.
Термин «пролекарство» используется для описания соединения, вводимого пациенту в фармакологически неактивной форме, которая затем превращается в активную форму посредством нормального метаболического процесса. Следовательно, «пролекарство» представляет собой химическое соединение-предшественник лекарственного средства. Производные по настоящему изобретению используются в качестве пролекарств.
ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к производному терапевтического агента для использования при лечении заболевания, связанного с неправильным сворачиванием белка/агрегацией. Производное диазольного соединения по настоящему изобретению, также называемое пролекарством, которое представляет собой производное терапевтического агента, является водорастворимым и может вводиться перорально в достаточно высокой концентрации в кишечник пациента. Пролекарство стабильно при нормальных условиях хранения. Пролекарство по настоящему изобретению нетоксично в терапевтических концентрациях, поэтому оно пригодно для использования при лечении пациентов даже в течение длительного периода времени. Пролекарство, то есть соединение, заявленное в пункте 1 формулы изобретения, является терапевтически неактивным, но будет превращено в биологически активное лекарственное средство в организме пациента.
Соединения, заявленные в данной заявке, представляют собой водорастворимые производные 3,5-дифенилдиазольных соединений, которые могут быть получены способом по настоящему изобретению. Предпочтительно, соединение выбрано из группы, включающей 3,5-дифенил-пиразольные соединения anle138b, sery335b, sery345 и anle253b.
Как указано выше, диазольные соединения по настоящему изобретению могут существовать в виде таутомеров, и каждый из двух таутомеров для всех соединений специалист может представить себе по названию или формуле соединения. Таким образом, включены каждый таутомер, а также смеси обоих таутомеров/изомеров в любом соотношении. Кроме того, далее показаны структуры одного таутомера/изомера каждого из трех вышеупомянутых соединений, но включенным является и другой изомер, который специалист может представить себе для всех соединений, и включены также смеси обоих таутомеров/изомеров в любом соотношении.
Anle138b (5-(3-Бромфенил)-3-(3,4-метилендиоксифенил)-1H-пиразол) представлен следующей структурой:

5-(3-Бромфенил)-3-(3,4-метилендиоксифенил)-1H-пиразол
Sery335b (5-(3-Хлорфенил)-3-(3,4-метилендиоксифенил)-1Н-пиразол) представлен следующей структурой:

5-(3-Хлорфенил)-3-(3,4-метилендиоксифенил)-1H-пиразол
Anle253b, которое даже менее растворим, чем anle138b, представлен следующей структурой:
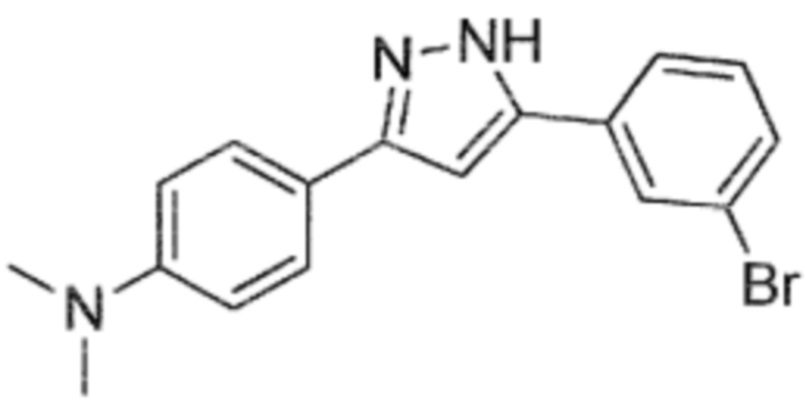
5-(3-Бромфенил)-3-(4-диметиламинофенил)-1H-пиразол
Sery345 представлен следующей структурой:

2-(1,3-бензодиоксол-5-ил)-5-(3-бромфенил)-1H-имидазол
Эти соединения известны своей эффективностью при лечении заболеваний, включающих агрегацию белка. Например, anle138b обладает способностью восстанавливать функциональность нейронов (1,2). Однако эти соединения также характеризуются своей очень низкой растворимостью в воде, что является менее желательным в медицинских препаратах для перорального введения. В настоящем изобретении теперь предоставлены производные, которые являются стабильными и подходящими пролекарствами для перорального применения у людей.
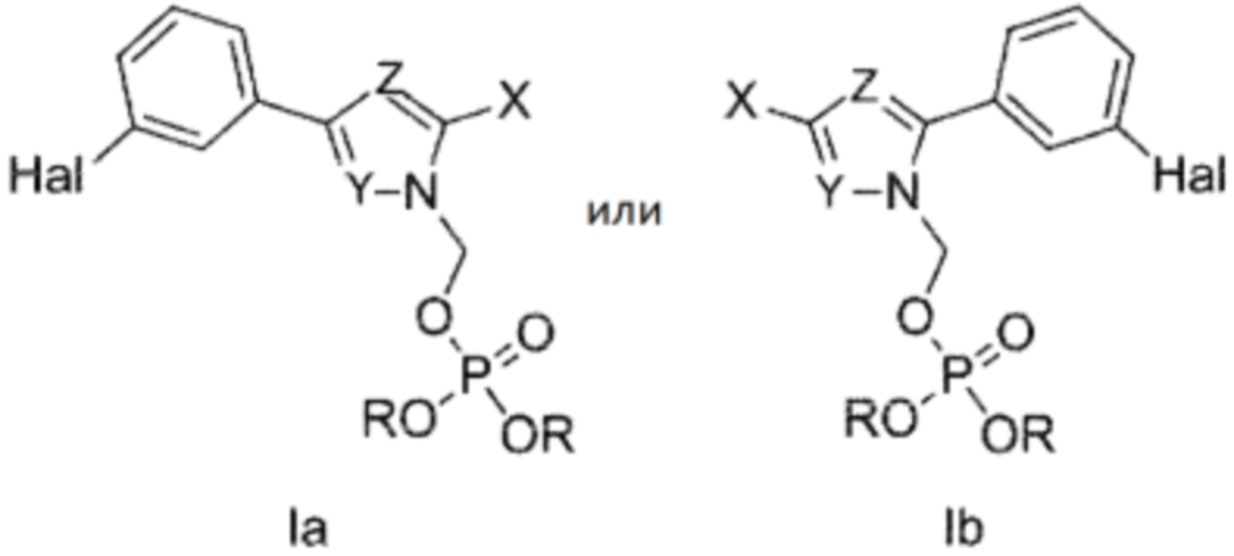
Авторы изобретения неожиданно обнаружили, что замещение метил фосфатом на одном азоте в диазольном кольце 3,5-дифенил-пиразольного или 3,5-дифенил-имидазольного соединения улучшает растворимость в воде без ухудшения стабильности. Эта химическая модификация согласно настоящему изобретению приводит к получению пролекарства этого ценного терапевтического агента, представленного следующими изомерными структурами:

где один из Y и Z представляет собой N, и другой представляет собой CR2;
где R2 выбран из водорода, C1-4 алкила, C1-4 алкила, замещенного по меньшей мере одним галогеном; и C6-10 арила, где арильное кольцо необязательно может быть замещено C1-4 алкилом или галогеном;
где либо X представляет собой

X представляет собой
где каждый R независимо представляет собой водород или катион.
В предпочтительном варианте осуществления соединения по настоящему изобретению представлены следующими изомерными структурами:

где Hal представляет собой галоген, выбранный из хлора или брома, и где каждый R независимо выбран из водорода или катиона.
В следующем предпочтительном варианте осуществления производное по настоящему изобретению представляет собой пролекарство anle253b, имеющее следующие изомерные структуры:

где каждый R независимо выбран из водорода или катиона.
Катионом может быть любой катион, который является фармацевтически приемлемым для такого типа соединения. Предпочтительно, катион представляет собой одновалентный катион. Примерами являются катион натрия, лития, калия, аммония, особенно, натрия. Другие примеры групп, совместимых с фосфатной группой соединений по настоящему изобретению, включают группы структуры RR'R''N, такие как этаноламин, холин, лизин, меглумин, пиперазин и трометамин в их протонированной форме RR'R''NH+. В соединениях по настоящему изобретению оба R могут быть как водородом, так и оба R могут быть катионами (одинаковыми или разными катионами), или один R может быть водородом, а другой может быть катионом. Двухвалентные катионы, такие как Ca2+, Mg2+ и Zn2+, или трехвалентные катионы, такие как Al3+, не являются предпочтительными, так как полученные соли менее водорастворимые.
Кроме того, форма свободной кислоты (R=H) метилфосфатного производного по настоящему изобретению также подходит в качестве пролекарства.
Настоящее изобретение также относится к фармацевтической композиции, которая содержит по меньшей мере одно из соединений, определенных в пункте 1 формулы изобретения. В соответствии с настоящим изобретением термин «фармацевтическая композиция» относится к композиции для введения пациенту, предпочтительно, млекопитающему, более предпочтительно, человеку. Фармацевтическая композиция по настоящему изобретению содержит вышеуказанные соединения и, необязательно, дополнительные вещества, способные изменять характеристики соединений по изобретению, тем самым, например, стабилизируя, модулируя и/или активируя их действие. Композиция может быть в твердой, жидкой или газообразной форме.
Фармацевтические композиции по изобретению могут вводиться перорально, ректально, парентерально, интрацистернально, интравагинально, внутрибрюшинно, местно (в виде порошков, мазей, капель или трансдермального пластыря), трансдермально, путем внутриартериальной инъекции, внутривенно, внутрибрюшинно, внутримышечно, подкожно или с помощью орального или назального спрея. Предпочтительно, фармацевтическая композиция должна вводиться перорально.
Что касается перорального введения, фармацевтические композиции по настоящему изобретению могут быть предоставлены, например, в виде таблетки, пилюли, порошка (включая порошок для растворения), пастилки, саше, каше, эликсира, суспензии, эмульсии, раствора, сиропа, аэрозоля или капсулы, предпочтительно, в виде таблетки, пилюли или порошка (включая порошок для растворения). Если желательно, фармацевтические композиции и, в частности, пероральные композиции, упомянутые выше, могут быть составлены таким образом, чтобы они обеспечивали быстрое, продолжительное или замедленное высвобождение и/или высвобождение на определенной стадии прохождения через организм (например, с энтеросолюбильным покрытием).
Фармацевтические композиции, описанные в настоящем документе, могут быть составлены с использованием одной или нескольких фармацевтически приемлемых добавок (веществ), обычно используемых в технологии рецептуры, например, таких как, среди прочего, упомянутые в 5-м издании Fiedler's «Lexikon der Hilfstoffe» 5th Edition, Editio Cantor Verlag Aulendorf 2002, «The Handbook of Pharmaceutical Excipients», 4th Edition, American Pharmaceuticals Association, 2003. Фармацевтически приемлемые добавки могут быть выбраны из носителей, разбавителей или наполнителей, связующих агентов/связующих, разрыхлителей, смазывающих веществ, придающих скольжение веществ, стабилизирующих агентов, поверхностно-активных веществ, смягчителей, смачивающих агентов, подсластителей, пигментов/красителей, антиоксидантов, консервантов и тому подобное. Подходящими носителями, связывающими агентами, разрыхлителями, смазывающими веществами и придающими скольжение веществами могут быть, например, те, которые более подробно описаны ниже в качестве фармацевтически приемлемых добавок. Композиции, содержащие такие добавки, могут быть получены обычными способами.
Подходящие носители включают, без ограничения, полиолы, такие как маннит, сорбит, ксилит; дисахариды, такие как лактоза, сахароза, декстроза и мальтоза; полисахариды, такие как мальтодекстрин и декстраны; крахмалы, такие как кукурузный крахмал; целлюлозы, такие как микрокристаллическая целлюлоза, натрий карбоксиметилцеллюлоза, гидроксипропилцеллюлоза, гидроксилэтилцеллюлоза, гидроксипропилцеллюлоза или их смеси; циклодекстрины и неорганические агенты, такие как дикальций фосфат, кальций фосфат; гидроксиапатит, трикальцийфосфат, тальк и диоксид кремния.
Подходящие связывающие агенты включают, без ограничения, гидроксипропилметилцеллюлозу (НРМС), гидроксипропилцеллюлозу (HPC), предварительно желатинизированный крахмал и их комбинации.
Подходящие разрыхляющие вещества включают, без ограничения, кальций карбоксиметилцеллюлозу (CMC-Ca), натрий карбоксиметилцеллюлозу (CMC-Na), сшитые PVP (например, кросповидон, полипласдон® или коллидон® XL), альгиновую кислоту, натрий альгинат, гуаровую камедь, (натрий кроскармеллозу, например, Ac-Di-Sol®), карбоксиметилкрахмал-Na (натрий-крахмалгликолят) (например, Primojel® или Explotab®).
Подходящие смазывающие вещества включают, без ограничения, стеарат магния, силикат алюминия или кальция, стеариновую кислоту, модифицированное касторовое масло, тальк, глицерилбегенат, натрий стеарат фумарат и их комбинации.
Подходящие скользящие вещества включают, без ограничения, коллоидный SiO2 (например, Aerosil® 200), трисиликат магния, порошкообразную целлюлозу, тальк и их комбинации.
Таким образом, таблетки могут содержать эксципиенты, такие как микрокристаллическая целлюлоза, лактоза, цитрат натрия, карбонат кальция, двухосновный фосфат кальция и глицин, разрыхлители, такие как крахмал (например, кукурузный, картофельный крахмал или крахмал тапиоки), натрийгликолят крахмал, натрий кроскармеллозу и некоторые сложные силикаты и связующие вещества для грануляции, такие как поливинилпирролидон, гидроксипропилметилцеллюлоза (HPMC), гидроксипропилцеллюлоза (HPC), сахароза, желатин и аравийская камедь. Кроме того, могут быть включены смазывающие агенты, такие как стеарат магния, стеариновая кислота, глицерилбегенат и тальк.
Твердые композиции аналогичного типа могут также использоваться в качестве наполнителей в желатиновых капсулах. Предпочтительные наполнители в этом отношении включают лактозу, крахмал, целлюлозу или высокомолекулярные полиэтиленгликоли.
Кроме того, фармацевтические композиции/лекарственные формы, как описано в настоящем документе, могут быть покрыты с использованием пленочных покрытий или покрытий с модифицированным высвобождением, применяя способы покрытия, хорошо известные специалисту в данной области, с использованием коммерчески доступных материалов для покрытия, таких как смесь пленкообразующих полимеров, непрозрачных материалов, красителей и пластификаторов.
Перечень других подходящих эксципиентов также можно найти в учебниках, таких как Remington′s Pharmaceutical Sciences, 18th Ed. (Alfonso R. Gennaro, ed.; Mack Publishing Company, Easton, PA, 1990); Remington: the Science and Practice of Pharmacy 19th Ed.(Lippincott, Williams & Wilkins, 1995); Handbook of Pharmaceutical Excipients, 3rd Ed. (Arthur H. Kibbe, ed.; Amer. Pharmaceutical Assoc, 1999); the Pharmaceutical Codex: Principles and Practice of Pharmaceutics 12th Ed. (Walter Lund ed.; Pharmaceutical Press, London, 1994); The United States Pharmacopeia: The National Formulary (United States Pharmacopeial Convention); and Goodman and Gilman's: the Pharmacological Basis of Therapeutics (Louis S. Goodman and Lee E. Limbird, eds.; McGraw Hill, 1992), описание которых включено в настоящий документ посредством ссылки.
Дозированные формы, как описано в настоящем документе, могут быть составлены в соответствии со способами, хорошо известными специалисту в данной области, например, как описано в «Pharmazeutische Technologie», 11th Edition, Deutscher Apotheker Verlag 2010, or «Pharmazeutische Techologie», 9th Edition, Wissenschaftliche Verlagsgesellschaft Stuttgart, 2012.
Фармацевтическая композиция будет составлена и дозироваться в соответствии с надлежащей медицинской практикой, принимая во внимание клиническое состояние отдельного пациента, область доставки фармацевтической композиции, способ введения, режим введения и другие факторы, известные практикующим специалистам. Таким образом, «терапевтически эффективное количество» фармацевтической композиции для целей по настоящему изобретению определяется такими соображениями.
Как правило, врач будет определять фактическую дозу, которая будет наиболее подходящей для отдельного субъекта. Конкретный уровень дозы и частота дозировки для любого конкретного индивидуума могут варьироваться и будут зависеть от множества факторов, включая конкретное соединение, режим и время введения, тяжесть конкретного состояния и пациента, подвергающегося терапии (возраст, масса тела, общее состояние здоровья, пол и т. д.).
Фармацевтические композиции, предпочтительно, представлены в виде стандартных лекарственных форм. Соединения по настоящему изобретению обычно вводят в терапевтически эффективном количестве. Это количество особо не ограничено, и предлагаемая доза соединений в соответствии с настоящим изобретением для введения человеку (приблизительно 70 кг массы тела) составляет от около 10 до около 1000 мг, предпочтительно, от около 50 до около 500 мг активного ингредиента на единичную дозу. Единичную дозу можно вводить, например, 1-4 раза в день. Доза будет зависеть от пути введения. Понятно, что может потребоваться внести обычные изменения в дозировку в зависимости от возраста и веса пациента, а также от тяжести состояния, подлежащего лечению. Точная доза и путь введения будут в конечном счете на усмотрении лечащего врача.
Композиция может содержать одну изомерную форму соединения по настоящему изобретению или обе изомерные формы. Композиция может также содержать более одного соединения, и любое из соединений, используемых в композиции, может быть либо одним из обоих изомеров, либо их смесью. Таким образом, фармацевтическая композиция для использования при лечении вышеупомянутых заболеваний может включать один из изомеров или смесь обоих изомеров. Обе изомерные формы превращаются в биологически активное лекарственное средство внутри организма.
В одном варианте осуществления представленные соединения находятся в растворенной форме в водном растворе. В настоящем изобретении «растворенная форма» означает, что настоящее соединение полностью растворимо при выбранной концентрации при комнатной температуре (например, 25°С) в растворителе, так что невооруженным глазом нерастворенное соединение не видно.
Установлено, что динатрий фосфатное производное является стабильным и дает высокие уровни активного соединения в головном мозге и крови.
В предпочтительном варианте осуществления соединение представляет собой динатрий фосфат одной из следующих структур или их смесь:

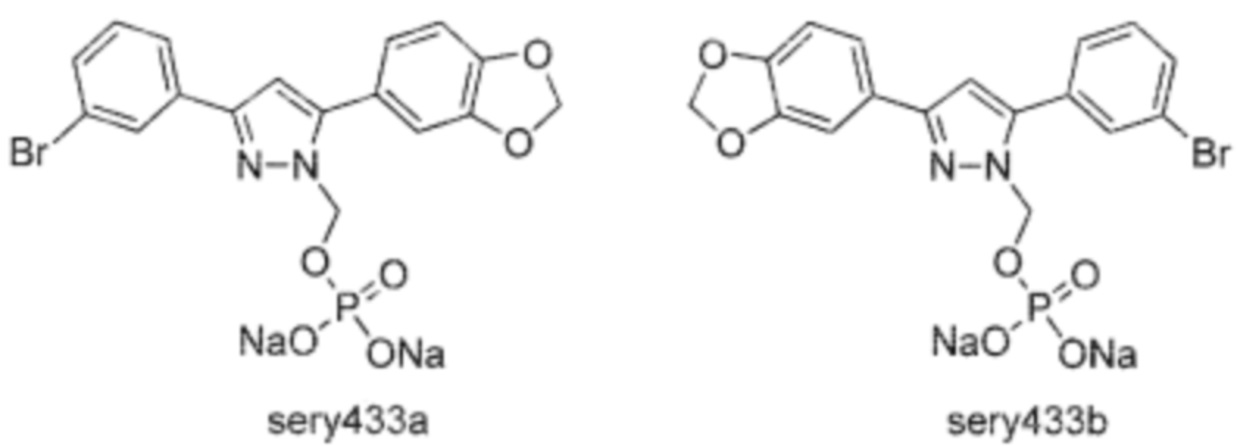
В предпочтительном варианте осуществления производное по настоящему изобретению представляет собой динатрий метилфосфат терапевтического агента anle138b, то есть соединение, как показано выше, где Hal представляет собой хлор или бром. Первое из указанных производных называется sery335b, последнее производное называется sery433. Оба они могут быть в форме одного из изомеров или могут быть смесью обоих изомеров. Изомеры sery433a или sery433b показаны ниже:

В предпочтительном варианте осуществления sery433 представляет собой изомерную смесь sery433a и sery433b, например, при соотношении sery433b:sery433a=2:3 в соответствии с экспериментальными данными, полученными с помощью ЯМР 2D-NOESY. Обе изомерные формы превращаются в биологически активное лекарственное средство внутри организма.
Неожиданно было обнаружено, что химическая модификация согласно настоящему изобретению дает терапевтическое соединение со следующими благоприятными характеристиками:
эффективное превращение в активную форму посредством нормальных метаболических процессов,
стабильность при нормальных условиях хранения,
улучшенная растворимость в воде,
улучшенная биодоступность активного соединения,
нетоксичность и
стабильность в процессе прохождения от рта до кишечника.
Ввиду улучшенной растворимости в воде, а также повышенной биодоступности настоящие соединения можно предоставить в значительно меньших объемах фармацевтически приемлемой добавки. Это улучшает соблюдение больным режима и схемы лечения. Кроме того, можно использовать обычные фармацевтически приемлемые добавки. В отличие от этого, при применении anle138b для введения этого высоко липофильного соединения необходимы специальные добавки (такие как кремофор). Таким образом, соединения по настоящему изобретению могут быть успешно использованы для длительного применения. Наконец, ввиду использования стандартных добавок настоящие соединения могут быть легко предоставлены в стандартных дозированных формах, таких как таблетка, пилюля или порошок (включая порошок для растворения), и нет необходимости предоставлять их в более сложных лекарственных формах, таких как капсулы.
Как можно видеть из примеров, представленных ниже, некоторые другие подходы с использованием пролекарств оказались неудачными, и, таким образом, является совершенно неожиданным, что вышеупомянутые преимущества могут быть достигнуты с помощью заявленных теперь соединений.
Пролекарства по изобретению представляют собой модифицированные 3,5-дифенилдиазольные производные, которые характеризуются улучшенной растворимостью в водных растворах по сравнению с исходными соединениями. С предпочтительным пролекарством sery433 достигается 4-кратное увеличение воздействие anle138b по сравнению с использованием anle138b/эксципиент. Таким образом, устраняются недостатки, связанные с непереносимостью высокой дозы добавок, и может быть реализован благоприятный протокол введения, который требует меньше и/или меньших капсул/таблеток.
Пролекарства по настоящему изобретению характеризуются очень высокой растворимостью в воде (см. пример 8). Пролекарства по настоящему изобретению также характеризуются своей устойчивостью в воде (см. пример 9).
При использовании производных по настоящему изобретению терапевтически активное соединение находится в мозге и крови в терапевтически полезных количествах. Предполагается, не будучи связанным теорией, что активное терапевтическое соединение высвобождается из пролекарства по настоящему изобретению с помощью мембраносвязанной кишечной щелочной фосфатазы (IAP) и, таким образом, близко к эпителию кишечника, где оно прямо передается в кровоток. Возможно, соединение высвобождается в основном в монодисперсной форме, которая затем может быть захвачена из кишечника в кровь. Фармакокинетические характеристики пролекарств по настоящему изобретению приводят к терапевтически эффективным уровням (см. примеры 10 и 11) активного соединения в головном мозге и крови у мышей и крыс. Самое главное, что фармакокинетическая оценка пролекарства sery433 по настоящему изобретению демонстрируют значительное улучшение воздействия, как показывают значения AUC и Cmax. В частности, введение пролекарства sery433 по сравнению с применением anle138b в эксципиенте PEG/кремофор приводит к системному воздействию anle138b, либо в терминах Cmax и AUC, значительно увеличиваясь примерно в 4 раза выше в случае пролекарства sery433 (см. фиг.4).
Например, активное соединение anle138b высвобождается из пролекарства sery433 на стенке кишечника путем ферментативного отщепления фосфатной группы из соединения с помощью мембраносвязанной кишечной щелочной фосфатазы (IAP). Не ограничиваясь теорией, предполагается, что возможны высокие уровни активного соединения в крови и головном мозге, поскольку концентрация активного соединения на стенке кишечника ниже концентрации, приводящей к осаждению активного соединения. Кроме того, предполагается, что IAP не высвобождает активное терапевтическое соединение в просвет кишечника при расщеплении, но удерживает соединение до тех пор, пока оно не пройдет на и через мембрану кишечника для пассивного переноса в кровь.
ПРИМЕРЫ
Предпочтительные варианты осуществления изобретения изложены в следующих примерах, которые не следует интерпретировать как ограничивающие объем или дух изобретения.
Существуют различные концепции солюбилизации липофильного соединения, чтобы сделать соединения водорастворимыми, то есть придать им заряд. Недавний обзор по таким подходам дан Müller, Chem. Biodiversity (7). В примерах были исследованы различные концепции, большинство из которых не привели к полезным продуктам.
В следующих примерах проиллюстрировано получение производных по настоящему изобретению и для сравнения, кроме того, подходы к созданию пролекарств.
МАТЕРИАЛЫ И МЕТОДЫ
Все исходные материалы и растворители были приобретены у компаний ABCR, Acros, Alfa Aesar, Fluorochem, Sigma-Aldrich или Merck и использованы как таковые, если не указано иное. Точки плавления определяли на аппарате плавления Stuart Scientific (BIBBY, UK) с использованием открытых стеклянных капилляров и не корректировали. Тонкослойная хроматография (ТСХ): пластинки с предварительно нанесенным покрытием Macherey-Nagel, 0,25 мм пластины ALUGRAM® SIL G/UV254, обнаружение с помощью УФ и/или путем обработки 10 мас.% раствором реагента фосфомолибденовой кислоты в этаноле с последующим нагреванием при 200°С. Колоночную флэш-хроматографию проводили с использованием силикагеля Merck 60 (0,063-0,100 мм). Аналитическую и препаративную высокоэффективную жидкостную хроматографию (ВЭЖХ) проводили с использованием системы Waters ВЭЖХ с детектором длин волн Photodiode Waters 996. Все разделения включают подвижную фазу 0,1% трифторуксусной кислоты (ТФУ) (об./об.) в воде (растворитель А) и 0,1% ТФУ в ацетонитриле (растворитель В). ВЭЖХ проводили с использованием колонки с обращенной фазой (ОФ) Eurospher RP 18, 100Ǻ, 5 мкм, 250×4,6 мм (аналитическая) и 250×16 мм (препаративная) при скорости потока 1 мл⋅мин-1 (аналитическая) и 7 мл⋅мин-1 (препаративная). Анализ электрораспылительной ионизационной масс-спектрометрии (ESI-MS) и жидкостной хроматографии/масс-спектрометрии (ЖХ/МС) осуществляли с использованием масс-спектрометра Waters Micromass ZQ 4000 в сочетании с описанным выше аппаратом Waters ВЭЖХ. Масс-спектры высокого разрешения (HRMS) регистрировали с использованием гибридного масс-спектрометра Thermo Scientific LTQ Orbitrap XL, и они были представлены в m/z. Спектры ЯМР регистрировали при температуре 298°K с использованием спектрометра Bruker Avance с частотой 400 МГц (Bruker AG, Rheinstetten, Germany), оснащенного зондом TXI HCN z-gradient. Все спектры обрабатывали с использованием TOPSPIN2 (Bruker AG, Karlsruhe, Germany). Химические сдвиги1H-ЯМР (δ) приведены в миллионных долях (м. д.) относительно CHCl3 и [D5]ДМСО в качестве внутренних стандартов. Данные представлены следующим образом: химический сдвиг, мультиплетность (с=синглет, д=дублет, т=триплет, шир.=широкий, м=мультиплет), константы связывания (J, заданные в Гц), интегральная интенсивность. Химические сдвиги13C ЯМР (δ) приведены в миллионных долях (м. д.) относительно CHCl3 и [D6]ДМСО в качестве внутренних стандартов. Для регистрации резонансов соединений использовались следующие эксперименты:1H-1D,13C-1D ЯМР-спектры и13C-APT (attached proton test с одним временем J-эволюции 1/145 сек, спектры обрабатываются таким образом, что четвертичные и метиленовые группы имеют положительный знак, а метильные и метиновые группы отрицательный знак). Чтобы разрешить перекрытие резонансных сигналов и восстановить необнаруживаемые резонансные сигналы в спектрах1H и APT, для некоторых соединений регистрировали 2D-[13C,1H]-HSQC (гетероядерная одноквантовая когерентность), 2D-[13C,1H]-HMBC (гетероядерная многоквантовая когерентность) и 2D-NOESY. 1,3-Диарилпропан-1,3-дионы представлены на рисунках как дикетоны, несмотря на то, что в спектрах преобладает форма енола.
Пример 1: Синтез sery433, пролекарство anle138b, расщепляемое с помощью IAP



Ди-трет-бутилхлорметилфосфат (sery430)
Соединение sery433 было получено в соответствии с опубликованном протоколом (7). К смеси ди-трет-бутилфосфата калия (35 г, 141 ммоль), K2HPO4⋅3H2O (127 г, 557 ммоль), n-Bu4NHSO4 (4 г, 11 ммоль), H2O (125 мл) и tBuOMe (170 мл) при непрерывном интенсивном перемешивании в течение 25 минут при 0°C добавляли по каплям раствор хлорметилхлорсульфата (35 г, 212 ммоль) в tBuOMe (35 мл). После завершения добавления перемешивание продолжали в течение 2 часов при комнатной температуре (реакционную смесь охлаждали, если внутренняя температура превышала 30°С). Реакционную смесь гасили H2O (350 мл) и tBuOMe (200 мл), органическую фазу отделяли, промывали 1M водным раствором K2HPO4(200 мл), водой (200 мл), насыщенным солевым раствором (50 мл) и сушили над Na2SO4. После фильтрации сульфата натрия к раствору добавляли n-Bu3N (3 мл), и раствор концентрировали при пониженном давлении с получением продукта в виде масла (34,4 г, 94%). К продукту добавляли дополнительную порцию n-Bu3N (3 мл), чтобы повысить стабильность при хранении в морозильной камере (-21°C).
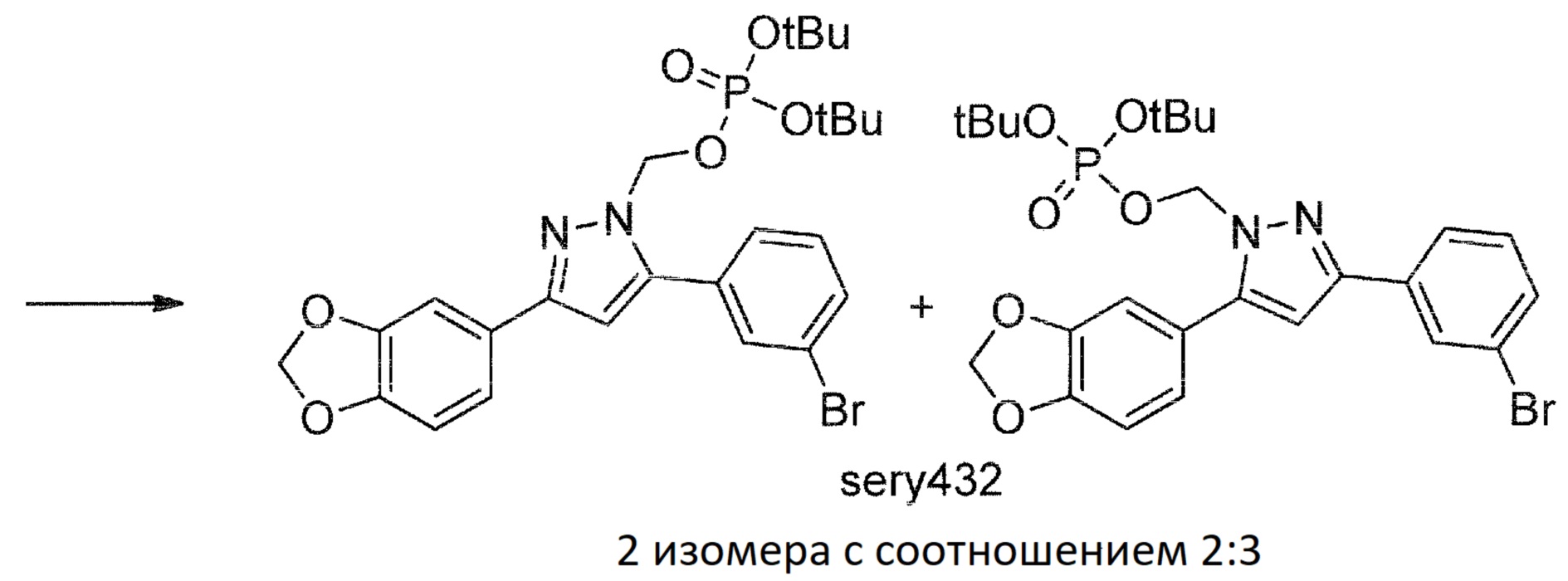
Sery432
К смеси anle138b (1) (25 г, 72,8 ммоль), Cs2CO3 (35,6 г, 109 ммоль) в ДМСО (200 мл) одной порцией добавляли ди-трет-бутилхлорметилфосфат (28,1 г, 109 ммоль). После перемешивания при комнатной температуре в течение 5 часов реакционную смесь разбавляли водой (1200 мл) и экстрагировали диэтиловым эфиром (500+200+100 мл). Объединенные органические фракции промывали водой (500 мл), насыщенным солевым раствором (100 мл) и сушили над Na2SO4. После фильтрации сульфата натрия раствор концентрировали при пониженном давлении с получением продукта в виде масла (всего 49,5 г, 40,8 г sery432, 82%, смесь двух изомеров с соотношением 2:3). Полученный продукт также содержал 10,5% sery430 и 7,1% Bu3N, согласно1H ЯМР спектру. Продукт использовали на следующей стадии без дополнительной очистки. Sery432 представлял собой смесь изомеров в соотношении 2:3 (1H-ЯМР).
Sery433
К охлажденному раствору sery432 (34,4 г, 60,9 ммоль) в DCM (400 мл) в течение 5 мин при непрерывном интенсивном перемешивании при 0°C добавляли ТФУ (23,5 мл). Через 2 часа добавляли дополнительную порцию ТФУ (23 мл), и реакционную смесь перемешивали при 0°С в течение 8 часов. После разбавления холодным толуолом (300 мл) реакционную смесь концентрировали при пониженном давлении при температуре 0°C, остаток смешивали с холодным толуолом (300 мл) и снова концентрировали при температуре 0°C. (DCM выпаривали при 50-100 мбар, ТФУ и толуол выпаривали с помощью высоковакуумного роторного испарителя). Полученную смесь разбавляли холодным ацетонитрилом (300 мл) и перемешивали в течение одного часа при 0°C. Белый осадок отфильтровывали и сушили при пониженном давлении, получая sery433 в виде кислоты (31,0 г всего, 29,0 г sery433 в виде кислоты и 2 г ацетонитрила). К сырому sery433 в виде кислоты добавляли 1М водный раствор NaOH (129 мл, 129 ммоль, 2 экв.) и воду (80 мл), полученный раствор отфильтровывали (фильтр Millipore Express Plus), и фильтрат лиофилизировали с получением динатриевой соли sery433 (30,0 г, 99%, смесь двух изомеров с соотношением 2:3) в виде порошка белого цвета.
Два изомера sery433, производного по настоящему изобретению, показаны ниже.
Были получены следующие пролекарства:
Стабильность соединений в водном растворе определяли с помощью ЯМР. Образец, содержащий раствор соединения (5 мг) в D2O (0,5 мл), инкубировали при комнатной температуре в течение 1 дня; 1H-спектры регистрировали и анализировали каждые 12 часов, а именно во временных точках 0 ч, 12 ч, 24 ч. Результаты приведены в таблице ниже.
Пример 2: Синтез anle423b (пролекарство anle253b)

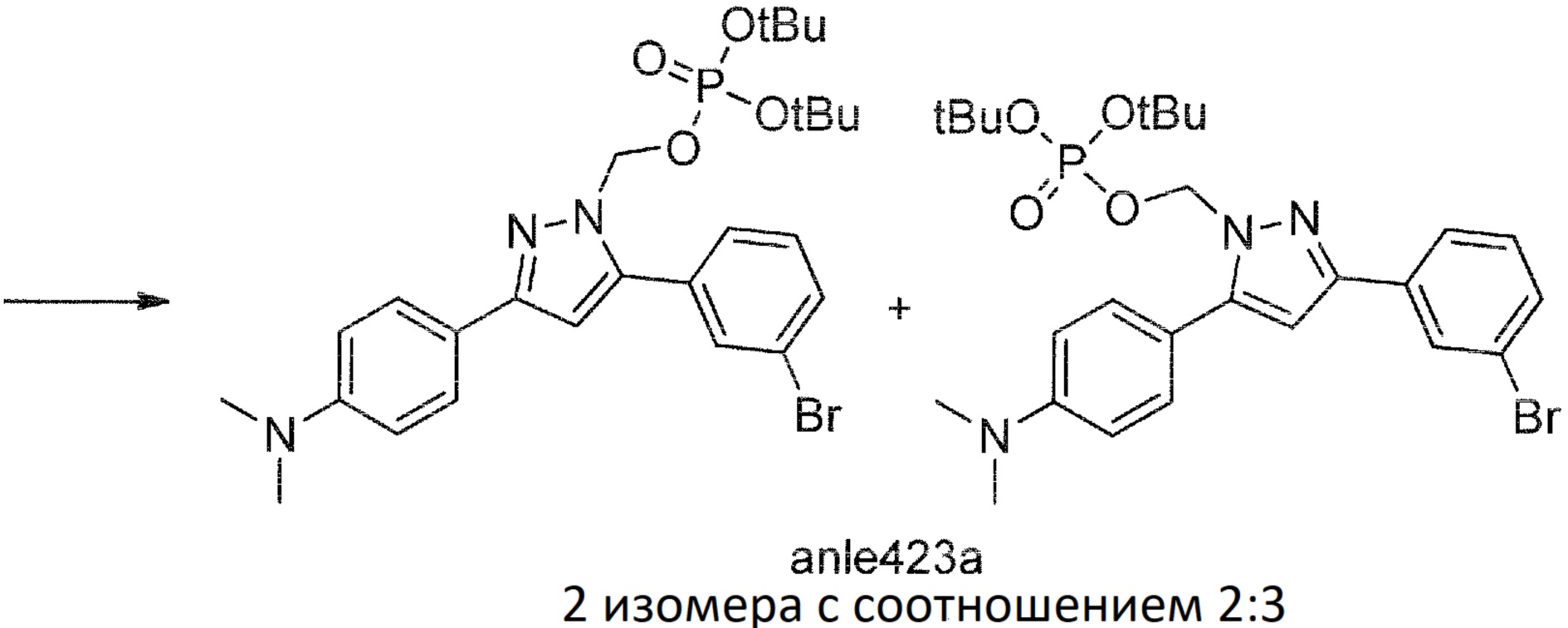

Anle423a
К смеси anle253b (500 мг, 1,46 ммоль), Cs2CO3 (620 мг, 1,9 ммоль) в ДМСО (5 мл) одной порцией добавляли ди-трет-бутил хлорметил фосфат (525 мг, 1,9 ммоль). После 15 часов перемешивания при комнатной температуре завершение реакции было показано с помощью тонкослойной хроматографии (ТСХ) (SiO2, гексан:EtOAc=2:1, Rf эдукта 0,33, Rf продукта 0,18). Реакционную смесь разбавляли водой (30 мл) и экстрагировали этилацетатом (2×15 мл). Объединенные экстракты промывали водой (10 мл), насыщенным солевым раствором (10 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением продукта в виде масла (1,08 г). Продукт использовали на следующей стадии без дополнительной очистки.
Anle423b (диаммониевая соль)
К охлажденной суспензии anle423a (1,08 г) в DCM (10 мл) в течение 1 минуты при непрерывном интенсивном перемешивании при температуре 0°С добавляли ТФУ (2 мл) и реакционную смесь перемешивали при температуре 0°С в течение 8 часов. Смесь фильтровали (GHP 0,45 мкм), разбавляли толуолом (10 мл) и концентрировали при пониженном давлении при температуре 20°C, остаток смешивали с толуолом (10 мл) и снова концентрировали при температуре 20°C. (DCM выпаривали при 50-100 мбар, ТФУ и толуол выпаривали с помощью высоковакуумного роторного испарителя). Полученный вязкий стекловидный остаток растирали в холодном ацетоне (30 мл), перемешивали в течение одного часа при 0°С. Белый осадок отфильтровывали, промывали ацетоном (10 мл) и сушили при пониженном давлении, получая anle423b в виде кислоты (448 мг, 0,99 ммоль, 68%, смесь двух изомеров с соотношением anle423ba:anle423bb=3:2 в соответствии с данными ЯМР 2D-NOESY) в виде порошка желтоватого цвета. К anle423b (дикислота, 156 мг, 0,345 ммоль) добавляли воду (3 мл) и 25% NH4OH (12,6 М, 150 мкл, 1,89 ммоль). Смесь перемешивали до полного растворения, полученный раствор замораживали и лиофилизировали с получением диаммониевой соли anle423b (161 мг, 331 мкмоль, 96%, смесь двух изомеров с соотношением anle423ba:anle423bb=3:2) в виде порошка желтоватого цвета.

Сравнительный пример 3: Синтез sery447, расщепляемого эстеразами
Одним из подходов, который был исследован, было введение группы, которая может быть расщеплена эстеразами и должна обеспечивать стабильность через катион.
Таким образом, было синтезировано еще одно потенциальное пролекарство anle138b, называемое sery447:


Хлорметиловый эфир никотиновой кислоты
Соединение sery447 получали в соответствии с опубликованным протоколом (8), исходя из никотиновой кислоты, и очищали флэш-хроматографией на колонке с силикагелем (CH2Cl2) с получением хлорметилового эфира никотиновой кислоты (выход 55%) в виде масла желтого цвета. ТСХ (CH2Cl2): RF=0,2.
Sery445
К суспензии гидрида натрия (220 мг, 5,5 ммоль, 60%-ная суспензия в минеральном масле) в безводном ДМФА (10 мл) в течение 10 минут при непрерывном интенсивном перемешивании при комнатной температуре добавляли раствор anle138b (1,71 г, 5 ммоль) в безводном ДМФА (5 мл). Смесь перемешивали в течение 30 мин при комнатной температуре и затем по каплям добавляли раствор хлорметилового эфира никотиновой кислоты (0,94 г, 5,5 ммоль) в безводном ДМФА (5 мл). После инкубации в течение 24 часов при комнатной температуре с последующим выпариванием ДМФА при пониженном давлении остаток растворяли в EtOAc (50 мл) и раствор промывали водой (50 мл), насыщенным солевым раствором (25 мл) и концентрировали при пониженном давлении. Сырой продукт очищали с помощью колоночной хроматографии на колонке с силикагелем с градиентным элюированием (EtOAc:гексан, от 1:3 об./об. до 1:1 об./об.), получая sery445 (2,05 г, 86%) в виде твердого вещества. Sery445 представлял собой смесь изомеров с соотношением 1:2 (1H-ЯМР). ТСХ (EtOAc:гексан, 1:3 по объему): RF=0,13.
Sery447
Раствор sery445 (1 г, 2,09 ммоль) и MeI (1,48 г, 10,4 ммоль) в ацетонитриле (15 мл) перемешивали в течение 22 часов при комнатной температуре с последующим добавлением дополнительной порции MeI (0,45 г, 3,2 ммоль). После перемешивания в течение еще 24 ч при комнатной температуре смесь концентрировали при пониженном давлении и остаток вновь суспендировали в EtOAc (30 мл). Образовавшийся осадок собирали фильтрованием и сушили с получением sery447 (1,17 г, 90%) в виде твердого вещества желтого цвета. Sery447 представлял собой смесь изомеров с соотношением 1:1,2 (1H ЯМР).
Экспериментальные данные, однако, показали, что у sery447 была низкая растворимость в воде (<1 мМ). Поэтому sery447 не был классифицирован как подходящее пролекарство для решения упомянутых выше проблем.
Сравнительный пример 4: Синтез sery435, расщепляемого пептидазами
В другом подходе было исследовано введение группы, которая должна расщепляться пептидазами. Стабильность должна быть обеспечена через катион. Было синтезировано еще одно потенциальное пролекарство anle138b, называемое sery435:


Sery434b
К суспензии anle138b (1,03 г, 3,0 ммоль) и Et3N (0,81 г, 8 ммоль) в DCM (25 мл) небольшими порциями при непрерывном интенсивном перемешивании при комнатной температуре добавляли гидрохлорид хлорангидрида никотиновой кислоты (0,62 г, 3,5 ммоль). После перемешивания при комнатной температуре в течение 4 дней добавляли дополнительные порции гидрохлорида хлорангидрида никотиновой кислоты (0,20 г, 1,9 ммоль) и Et3N (0,22 г, 2,2 ммоль) и перемешивание продолжали в течение еще 3 дней. Смесь гасили DCM (25 мл), промывали 1М водным фосфатным буфером (25 мл, рН 7,0), водой (25 мл), насыщенным солевым раствором (10 мл) и сушили над Na2SO4. Растворитель выпаривали при пониженном давлении и остаток очищали флэш-хроматографией на колонке с силикагелем (EtOAc:гексан, 1:1 об./об.), с получением sery434b (1,2 г, 89%) в виде твердого вещества. Sery434b представляет собой смесь изомеров с соотношением 1:1,2 (1H ЯМР). ТСХ (EtOAc:гексан, 1:1 об./об.): RF=0,64.
Sery435
Раствор sery434b (1 г, 2,23 ммоль) и MeI (1,5 г, 10,56 ммоль) в ацетонитриле (12 мл) перемешивали в течение 24 часов при комнатной температуре. Образовавшийся осадок собирали фильтрованием и сушили с получением sery435 (1 г, 76%) в виде твердого вещества желтого цвета. Sery435 представлял собой смесь изомеров с соотношением 1:1,2 (1H ЯМР).
Экспериментальные данные, однако, показали, что у sery435 была низкая растворимость в воде (<1 мМ). Поэтому sery435 не был классифицирован как подходящее пролекарство для решения упомянутых выше проблем.
Сравнительный пример 5: Синтез sery453, расщепляемого гидролизом
В другом подходе было исследовано, может ли гидролизуемая группа обеспечить водорастворимое и стабильное пролекарство, когда высвобождение терапевтического агента anle138b происходило бы посредством гидролиза.
Было синтезировано еще одно потенциальное пролекарство anle138b, называемое sery453:
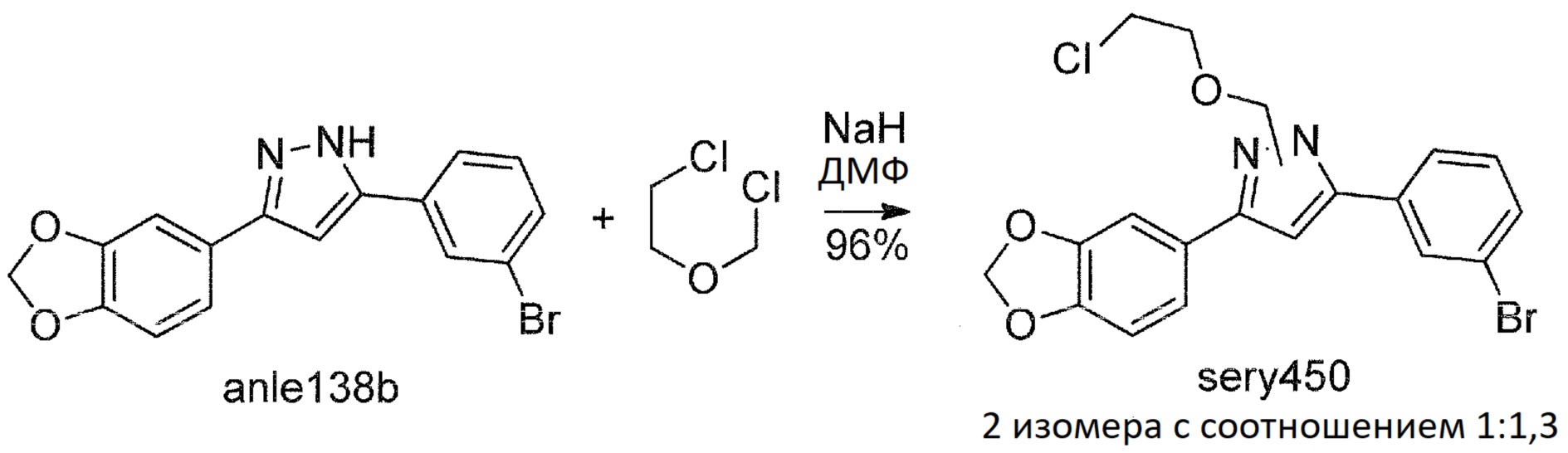


Sery450
Sery450
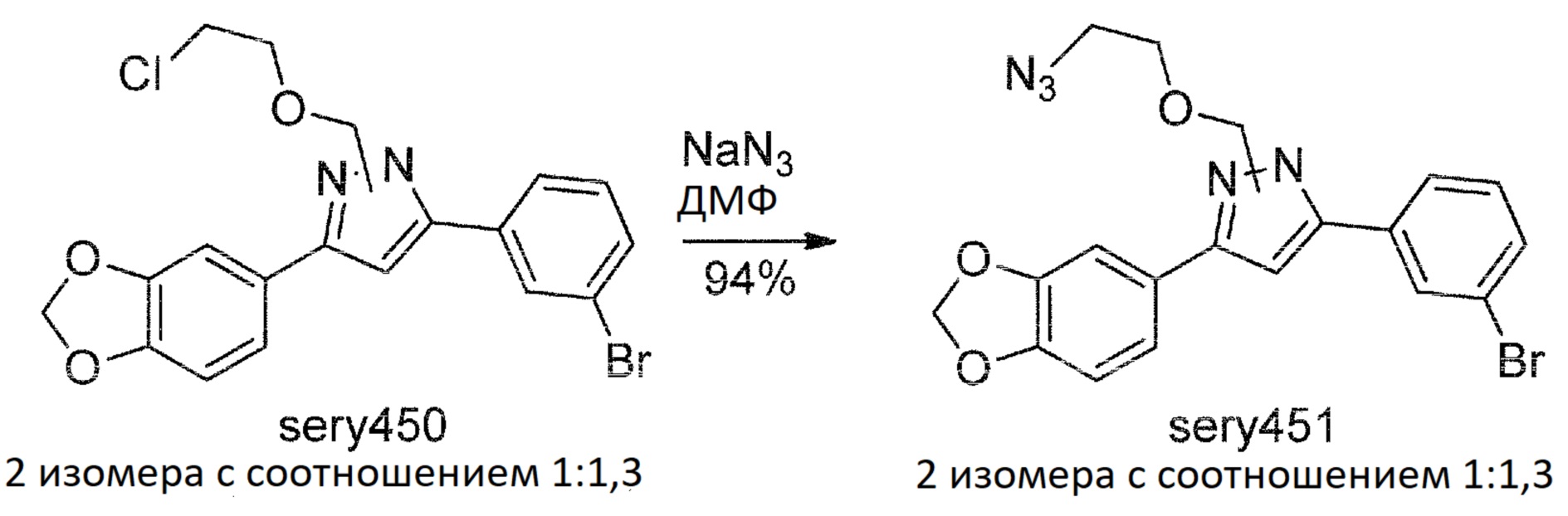
К суспензии гидрида натрия (220 мг, 5,5 ммоль, 60%-ная суспензия в минеральном масле) в безводном ДМФА (10 мл) в течение 10 минут при непрерывном интенсивном перемешивании при комнатной температуре добавляли раствор anle138b (1,71 г, 5 ммоль) в безводном ДМФА (5 мл). Смесь перемешивали в течение 30 мин при комнатной температуре и затем по каплям добавляли 2-хлорэтилхлорметиловый эфир (0,7 г, 5,5 ммоль). После инкубации в течение 30 мин при комнатной температуре с последующим выпариванием ДМФА при пониженном давлении остаток растворяли в EtOAc (60 мл) и раствор промывали водой (50 мл), насыщенным солевым раствором (25 мл), и концентрировали при пониженном давлении. Сырой продукт очищали хроматографией на колонке с силикагелем (EtOAc:гексан, 1:3 об./об.), с получением sery450 (2,1 г, 96%). Sery450 представлял собой смесь изомеров с соотношением 1:1,3 (1H ЯМР). ТСХ (EtOAc:гексан, 1:3 об./об.): RF=0,58.
Sery451
Смесь sery450 (1,87 г, 4,3 ммоль), NaN3 (2,79 г, 43 ммоль), NaI (0,1 г) в ДМФА (25 мл) перемешивали в течение 15 ч при температуре 80°С и затем концентрировали при пониженном давлении. Остаток растворяли в EtOAc (90 мл), органическую фазу промывали водой (2×50 мл), насыщенным солевым раствором и сушили над Na2SO4. После фильтрования от сульфата натрия раствор концентрировали при пониженном давлении и полученную смесь очищали хроматографией на колонке с силикагелем (EtOAc:гексан, 1:4 об./об.), с получением sery451 (1,81 г, 95%). Sery451 представлял собой смесь изомеров с соотношением 1:1,3 (1H ЯМР). ТСХ (EtOAc:гексан, 1:5 об./об.): RF=0,36.
Sery452
Смесь sery451 (1,67 г, 3,78 ммоль), Ph3P (1,49 г, 5,67 ммоль), воды (2 мл) в ТГФ (25 мл) перемешивали в течение 18 часов при комнатной температуре и затем концентрировали при пониженном давлении. Полученную смесь очищали хроматографией на колонке с силикагелем с градиентным элюированием CHCl3:MeOH, от 30:1 об./об. до 9:1 об./об.), с получением sery452 (0,38 г, 96%). Sery452 представлял собой смесь изомеров с соотношением 1:1,3 (1H ЯМР). ТСХ (CHCl3: MeOH, 9:1 об./об.): RF=0,19.
Sery453
Смесь sery452 (1,45 г, 3,48 ммоль), MeI (2,97 г, 20,9 ммоль), KHCO3 (2,1 г, 21 ммоль) в ацетонитриле (25 мл) перемешивали в течение 24 часов при комнатной температуре. Нерастворимое вещество отфильтровывали и фильтрат концентрировали при пониженном давлении с получением sery453 с йодом в качестве противоиона. Используя анионообменную смолу, противоион йода был заменен противоионом хлора, получением sery453 (0,88 мг, 51%) в виде твердого вещества белого цвета. Sery453 представлял собой смесь изомеров с соотношением 1:1,3 (1H ЯМР).
Хотя экспериментальные данные показали, что у sery453 была хорошая растворимость в воде, это соединение также не решало вышеупомянутых проблем, так как концентрация anle138b в мозге и крови мышей была относительно низкой (см. пример 10). Sery453 достигал уровня в 8-10 раз ниже (временные точки 2 ч и 4 ч) по сравнению с sery433 и поэтому не рассматривался далее.
Сравнительный пример 6: Синтез sery474, подлежащего расщеплению с помощью IAP
Дальнейший подход был использован при попытке обеспечить водорастворимое стабильное пролекарство: была введена группа, которая могла быть отщеплена внутренней щелочной фосфатазой.
Было синтезировано еще одно потенциальное пролекарство anle138b, называемое sery474:

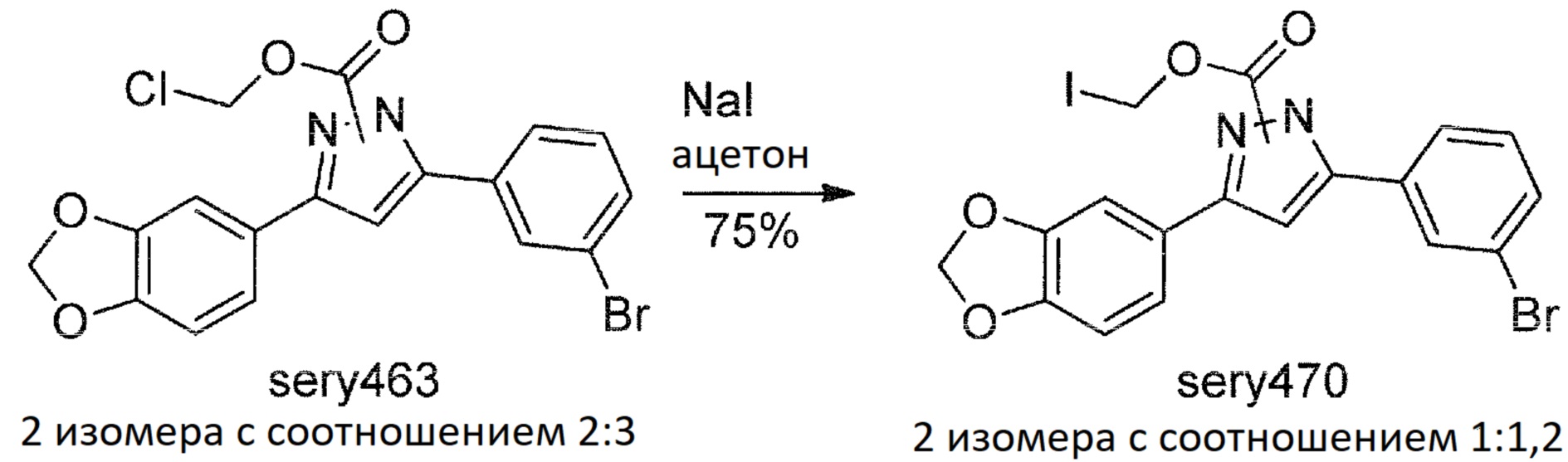
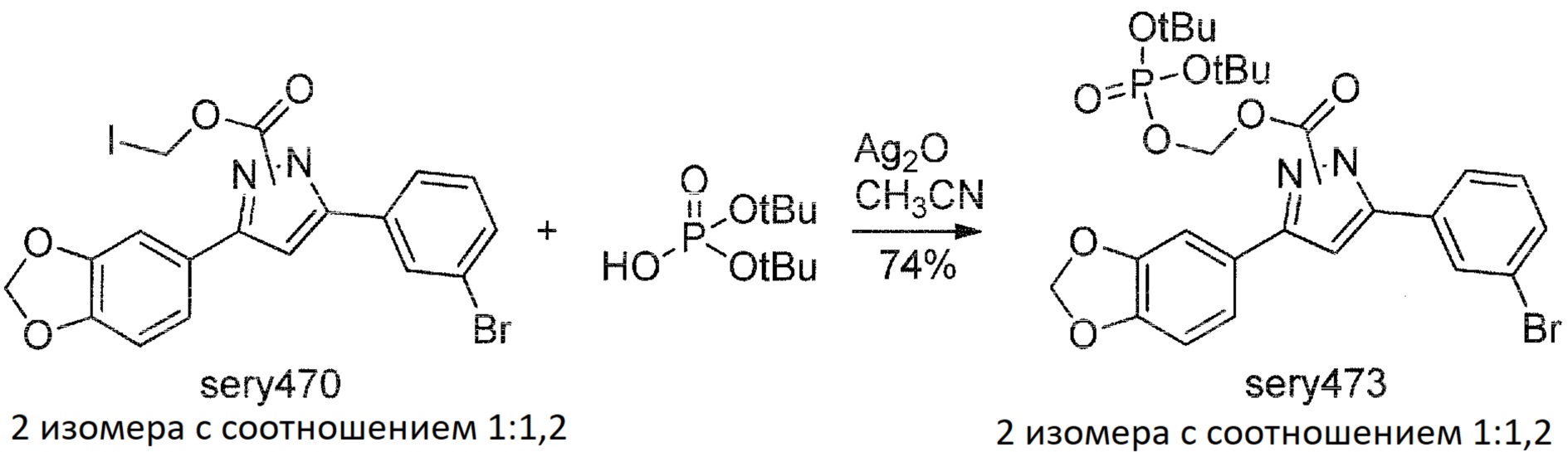
Sery463
К смеси anle138b (1 г, 2,9 ммоль) и пиридина (0,276 г, 3,5 ммоль) в DCM (10 мл) в течение 10 минут при непрерывном интенсивном перемешивании при комнатной температуре добавляли раствор хлорметилхлорформиата (0,452 г, 3,5 ммоль) в DCM (5 мл). После перемешивания при комнатной температуре в течение 2 часов реакционную смесь разбавляли DCM (20 мл), органическую фазу промывали водой (2×30 мл), насыщенным солевым раствором и концентрировали при пониженном давлении. Полученный остаток перекристаллизовывали из ацетонитрила, получая sery463 (0,85 г, 67%) в виде твердого вещества белого цвета. Sery463 представлял собой смесь изомеров с соотношением 2:3 (1H-ЯМР).
Sery470
Смесь sery463 (0,8 г, 1,84 ммоль), NaI (0,413 г, 2,76 ммоль) в ацетоне (15 мл) перемешивали в течение 3 дней при температуре 40°С и затем концентрировали при пониженном давлении. Остаток растворяли в DCM (30 мл), органическую фазу промывали водой (20 мл), 1М водным раствором Na2S2O3 (20 мл), насыщенным солевым раствором и сушили над Na2SO4. После фильтрования от сульфата натрия раствор концентрировали при пониженном давлении и полученную смесь очищали хроматографией на колонке с силикагелем (EtOAc:гексан, 1:3 об./об.), с получением sery470 (0,73 г, 75%) в виде твердого вещества. Sery470 представлял собой смесь изомеров с соотношением 1:1,2 (1H ЯМР). ТСХ (EtOAc:гексан, 1:3 об./об.): RF=0,55.
Sery473
Смесь sery470 (53 мг, 0,1 ммоль), калиевой соли ди-трет-бутилфосфата (42 мг, 0,2 ммоль) и Ag2O (24 мг, 0,1 ммоль) в ацетонитриле (3 мл) перемешивали в течение 2 часов при комнатной температуре. Затем нерастворимый материал отфильтровывали, и твердое вещество промывали на фильтре ацетонитрилом (2 мл). Объединенный органический раствор концентрировали при пониженном давлении, и полученное масло очищали хроматографией на колонке с силикагелем (EtOAc:гексан, 1:2 об./об.) с получением sery473 (45 мг, 74%) в виде масла. Sery473 представлял собой смесь изомеров с соотношением 1:1,2 (1H ЯМР). ТСХ (EtOAc:гексан, 1:2 об./об.): RF=0,24.
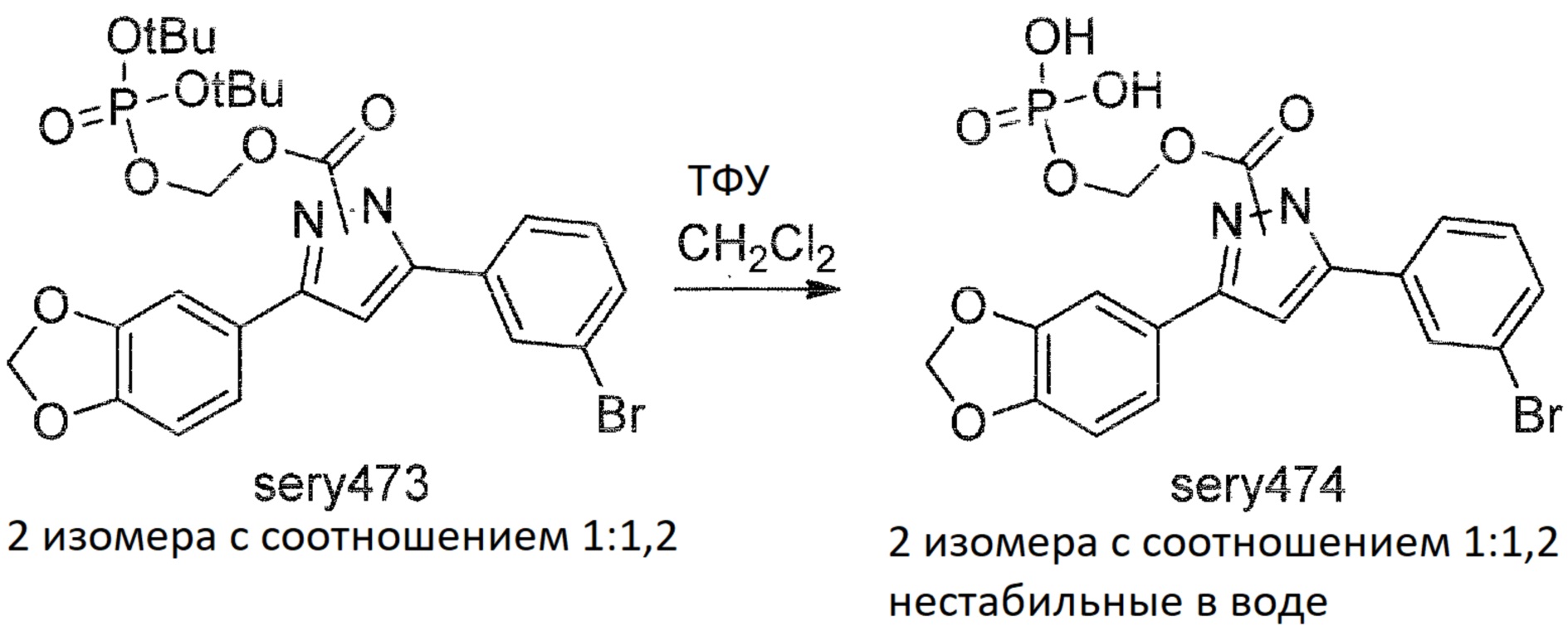
Sery474
Смесь sery473 (10 мг), ТФУ (20 мг) в дейтерированном хлороформе (0,7 мл) помещали в пробирку для ЯМР анализа, и протекание реакции контролировали с помощью ЯМР. После 6 ч инкубации при комнатной температуре1H спектр демонстрировал завершение реакции, обработка водой в нейтральных условиях приводила к разложению sery474, приводящему к образованию anle138b.
Экспериментальные данные показали, что, хотя пролекарство sery474 растворяется в воде, оно не может решить проблему, поскольку оно не обладает достаточной стабильностью. Предполагается, что это связано с самогидролизом ацетальной функциональной группы. Поэтому sery474 не был классифицирован как подходящее пролекарство. Из-за низкой стабильности sery474 в водном растворе дальнейшая разработка была прекращена.
Сравнительный пример 7: Синтез anle380 на основе PEG600, расщепляемого пептидазами
Общим подходом при разработке водорастворимого и стабильного пролекарства является получение производных с помощью дикарбоновой кислоты PEG600, которая сама по себе смешивается с водой во всех соотношениях. Таким образом, было синтезировано еще одно потенциальное пролекарство anle138b, называемое anle380:

Anle380
Смесь бисхлорангидрида на основе PEG (полученного взаимодействием соответствующего (поли(этиленгликоль)бис(карбоксиметил)эфира дикислоты, среднее значение Mn 600) (3 г, 5 ммоль) и SOCl2), anle138b (3,43 г, 10 ммоль) и Et3N (1,01 г, 10 ммоль) в EtOAc (150 мл) перемешивали в течение 24 часов при комнатной температуре. Нерастворимое вещество отфильтровывали; фильтрат промывали 5%-ным водным раствором лимонной кислоты (50 мл) и насыщенным солевым раствором. Полученный раствор концентрировали при пониженном давлении до объема примерно 20 мл и затем медленно выливали в гексан (150 мл). Осадок белого цвета собирали фильтрованием и сушили с получением anle380 (5,8 г) в виде твердого вещества белого цвета. Согласно анализу ЖХ-МС этой смеси, 4 изомера желаемого продукта являются основными компонентами, тогда как anle138b и соединения с одним гетероциклическим фрагментом были обнаружены в качестве основных примесей.
Экспериментальные данные, однако, показали, что у anle380 была низкая растворимость в воде (<1 мМ). Неожиданно было то, что производное дикарбоновой кислоты PEG600 не достигало растворимости с присоединенными двумя остатками anle138b. Поэтому anle380 не может использоваться в качестве подходящего пролекарства.
Пример 8: Исследование растворимости в воде
Растворимость каждого соединения в воде оценивали по следующему протоколу. В стеклянную трубку, содержащую 10 мг соединения добавляли деионизированную воду (1 мл). После встряхивания в течение 10 минут образец визуально исследовали, и регистрировали образование раствора или суспензии. Результаты приведены в таблице ниже.
Результаты показывают, что sery433 и sery452 приблизительно в 2,19 млн. раз более растворимы в воде, чем anle138b.
Пример 9: Исследование стабильности
Стабильность соединений в водном растворе определяли с помощью ЯМР. Образец, содержащий раствор соединения (5 мг) в D2O (0,5 мл), инкубировали при комнатной температуре в течение 1 дня;1H спектры регистрировали и анализировали каждые 12 часов, а именно: во временных точках 0 ч, 12 ч, 24 ч. Прекрасные результаты приведены в таблице ниже.
Таким образом, в отличие от приведенных выше сравнительных примеров, производные по настоящему изобретению являются стабильными. Активное соединение высвобождается широко распространенным ферментом, участвующим в расщеплении, и, как ни удивительно, фермент не выделяет гидрофобное соединение в просвет кишечника, а «держится» в нем до тех пор, пока не будет передан на мембрану кишечника для пассивного переноса в кровь.
Пример 10: Фармакокинетический анализ sery433 и sery453 на мышах
Растворы sery433 и sery453 в виде динатриевой соли (7,25 мг, экв. 5 мг anle138b) в стерильной воде Millipore (50 мкл) вводили через зонд мышам (C57/BL6, возраст 77 дней). Животных умерщвляли смещением шейных позвонков во временных точках 1 ч, 2 ч, 4 ч и 8 ч после введения (по два животных в каждый момент времени). Мозги извлекали, промывали 50 мМ Трис-буфером с рН 7,0 и сразу замораживали в жидком азоте. Образцы хранили при -80°С.
Перед использованием ткани оттаивали при 4°C. Ткань дважды гомогенизировали в 5 мл ацетонитрила при максимальной скорости в течение 3 минут, используя гомогенизатор (рабочая станция IKA ULTRA-TURRAX Tube Drive, Германия). Гомогенат подвергали ультразвуковой обработке при 30°С в течение 5 минут и центрифугировали при 5000×g в течение 10 минут. Аликвоту (100 мкл) супернатанта вводили в систему ВЭЖХ. Образцы количественно оценивали, используя соотношение площадей пиков соединений и внешних стандартов (см. фиг.3).
Пример 11: Фармакокинетический анализ sery433 на крысах
Раствор sery433 в виде динатриевой соли (уровень дозы 10 мг/кг, объем дозы 10 мл/кг) в 25 мМ фосфатном буфере с pH 7 вводили крысам CD с помощью желудочного зонда. Образцы плазмы собирали в соответствующие моменты времени, замораживали в жидком азоте и хранили при -80°C. Перед тем, как использовать, ткани оттаивали при 4°C. Их гомогенизировали дважды в 5 мл ацетонитрила с максимальной скоростью в течение 3 минут, используя гомогенизатор (IKA ULTRA-TURRAX Tube drive workstation, Germany). Гомогенат подвергали ультразвуковой обработке при 30°С в течение 5 минут и центрифугировали при 5000×g в течение 10 минут. Аликвоту (100 мкл) супернатанта вводили в систему ВЭЖХ. Образцы оценивали количественно, используя соотношение площадей пиков соединений и внешних стандартов (см. фиг.3).
Пример 12: Фармакокинетический анализ sery433 на крысах
Данное исследование было разработано для оценки фармакокинетики anle138b и sery433 на самцах крыс Sprague Dawley после однократного перорального введения anle138b (фаза 1) и sery433 (фаза 2). Во время фазы 1 и фазы 2 животным вводили каждое исследуемое соединение, соответствующим образом подготовленное, в виде одной из двух отдельных доз при перекрестном дизайне. Уровень дозы составлял 10 мг/кг для обоих соединений. Количество sery433 не было откорректировано до количества anle138b, которое представлено в пролекарстве, так что из-за более высокой молекулярной массы sery433 фактически вводилось меньшее количество активного агента.
Исследование проводили в две фазы.
Фаза 1: трем ранее не подверженным экспериментам самцам крыс CD перорально вводили дозу anle138b при целевой дозы 10 мг/кг.
Фаза 2: через неделю (период промывания) тем же животным перорально вводили sery433 при целевой дозе 10 мг/кг.
На каждой фазе выстраивали индивидуальный серийный профиль плазмы для каждого животного в течение 24 часов после введения дозы.
Получение состава для пероральной дозы (фаза 1)
Несущую среду получали, взвешивая кремофор RH40 (45% от конечного объема); PEG400 (35% от конечного объема) и Capryol 90 (20% от конечного объема). Смесь перемешивали и плавили при приблизительно 50°С (например, термостатированная ванна) прибл. 15 минут (мин) до получения прозрачной жидкости. Затем исследуемое соединение взвешивали и добавляли к несущей среде при перемешивании при 50°С. Смесь перемешивали еще 15 мин и обрабатывали ультразвуком в течение 10 мин до получения прозрачного (при визуальном осмотре) раствора.
Получение состава для пероральной дозы (фаза 2)
Несущую среду готовили, взвешивая Na2HPO4 (3,0 г/л) и NaCl (2,64 г/л); затем добавляли 1н HCl (1,9 мл до рН 7) вместе с 1/2 от общего объема колбы H2O, и смесь обрабатывали ультразвуком в течение 10 минут. Затем исследуемое соединение взвешивали и добавляли к несущей среде при перемешивании магнитной мешалкой в течение приблизительно 15 мин. Затем смесь обрабатывали ультразвуком прибл. 5 мин и снова перемешивали в течение 15 мин до получения прозрачного (визуальный осмотр) раствора.
Номинальная концентрация фосфатного буфера составляла 20 мМ.
После перорального введения образцы крови собирали из хвостовой вены каждой крысы в следующие временные точки: перед введением дозы, 0,5, 1, 2, 4, 6, 8 и 24 часа после введения дозы.
Результаты показаны на фигуре 4. Как видно, концентрация в плазме anle138b после введения sery433 была намного выше, чем соответствующая концентрация в плазме anle138b после введения только anle138b.
ССЫЛКИ
[1] J. Wagner et al. «Anle138b: a novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson′s disease», Acta Neuropathol. 125, 795-813 (2013).
[2] J. Levin et al. «The oligomer modulator anle138b inhibits disease progression in a Parkinson mouse model even with treatment started after disease onset» Act. Neuropath. 127, 779-780 (2014).
[3] J. Wagner, et al. «Reducing tau aggregates with anle138b delays disease progression in a mouse model of tauopathies» Act. Neuropathol (2015), in print (doi: 10.1007/s0041-015-1483-3).
[4] J. Rautio et al. «Prodrugs: design and clinical applications» Nat. Rew. Drug Disc. 7, 255-270 (2008).
[5] A. H. Burstein, D. Cox, B. Mistry, N. Eddington «Phenytoin pharmacokinetics following oral administration of phenytoin suspension and fosphenytoin solution to rats» Epilepsy Res. 34:129-133 (1999).
[6] T. Heimbach et al. «Enzyme-mediated precipitation of parent drugs from their phosphate prodrugs» Int. J. Pharm. 261, 81-92 (2003).
[7] C. E. Müller, «Prodrug approaches for enhancing the bioavailability of drugs with low solubility» Chem. Biodiversity 6,2071-2083(2009)ato, K. et al. Composition containing chloromethyl phosphate derivative with improved stability and method for producing the same. EP2133355A1 (2009).
[8] Bodor, N. S. Redox systems for brain-targeted drug delivery. EP0327766A2 (1990).
Реферат
Настоящее изобретение относится к соединению, представленному одной из следующих изомерных структур Ia и Ib, которое используется для лечения или профилактики заболеваний, связанных с агрегацией белка, к фармацевтической композиции на его основе, к способу лечения или профилактики заболеваний, связанных с агрегацией белка, с использованием заявленного соединения, а также к применению заявленного соединения или композиции на его основе для производства лекарственного средства:



Формула



Документы, цитированные в отчёте о поиске
Новые лекарственные средства для ингибирования агрегации белков, вовлеченных в заболевания связанные с агрегацией белков, и нейродегенеративные заболевания
Комментарии