Новые лекарственные средства для ингибирования агрегации белков, вовлеченных в заболевания, связанные с агрегацией белков, и нейродегенеративные заболевания - RU2531915C2
Код документа: RU2531915C2
Чертежи


























Описание
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединению, представленному формулой (Е). Настоящее изобретение относится также к соединению, представленному формулой (Е), для применения при лечении или предотвращения заболеваний, связанных с агрегацией белков, и/или нейродегенеративных заболеваний. Кроме того, настоящее изобретение относится к фармацевтическим и диагностическим композициям, включающим соединение согласно настоящему изобретению, а также к набору, включающему соединение согласно изобретению. Изобретение относится также к способу визуализации скоплений агрегированных белков. Также раскрыт набор для получения соединения с обнаруживаемой меткой согласно настоящему изобретению.
В тексте настоящего изобретения упоминаются некоторые документы. Содержание документов, цитированных в данном изобретении (включая любые описания, инструкции производителей и т.д.), введено в данное изобретение в виде ссылки.
УРОВЕНЬ ТЕХНИКИ
M. Ono et al. (Bioorganic & Medicinal Chemistry 16 (2008) 6867-6872) описывают некоторые бета-амилоидные зонды на основе 3,5-дифенил-1,2,4-оксадиазолов.
В US 2007/02760034 описываются некоторые бис- и трис-дигидроксиарильные соединения и их метилендиокси-аналоги и фармацевтически приемлемые сложные эфиры, которые, как утверждается, подходят для лечения синуклеопатий.
В WO 2008/131148 описываются специфические дифенилгетероарильные производные и их применение для связывания и визуализации амилоидных бляшек.
Гетероциклические соединения, которые применимы в качестве NURR-1 активаторов, описаны в WO 2004/072050.
Этиленгликоль или пропиленгликоль с радиоактивной меткой применяются в качестве группы-метки в соединениях, которые могут применяться для визуализации тканей, как описано в WO 2007/002540.
В WO 98/17652 описываются некоторые производные оксадиазола, которые, как заявлено, подходят для лечения нейродегенеративных расстройств и церебральной ишемии.
Известно большое количество неврологических и нейродегенеративных заболеваний, многие из которых в настоящее время не поддаются лечению. Эти заболевания включают такие болезненные состояния, как болезнь Паркинсона, хорея Хантингтона, болезнь Галлервордена-Шпатца, болезнь Альцгеймера, старческое слабоумие, болезнь Крейтцфельдта-Якоба, артериосклеротическое слабоумие, облитерирующий церебральный тромбангиит, деменция с тельцами Леви (DLB), множественная системная атрофия (MSA) и множество других болезненных состояний.
Прионные болезни, которые включают такие заболевания, как болезнь Крейтцфельдта-Якоба (CJD), скрейпи и коровий губчатый энцефалит (BSE), патологически характеризуются спонгиформной дегенерацией мозга. Прионные болезни вызываются особым возбудителем инфекции, который состоит главным образом из неправильно сложенной в виде бэта-листов агрегированной изоформы PrPSc мембранного бета-гликопротеида PrPC.
Прионные болезни вызывали большой интерес в общественном здравоохранении вследствие появления BSE. Научные данные подтверждают, что BSE передавалась людям, вызывая новый вариант болезни Крейтцфельда-Якоба (nvCJD) (Will et al. 1996, Bruce et al. 1997). Не известно, сколько людей инфицировано и сколько еще подвергнется инфицированию vCJD в будущем. Имеющиеся на сегодняшний день данные не исключают возможность распространение эпидемии, затрагивающей большое количество пациентов (Andrews et al., 2000). Это повышает необходимость разработки эффективной терапии в дополнение к уже введенным в действие мерам, предупреждающим дальнейшее распространения заболевания. Кроме того, недавно полученные данные подтверждают, что может иметь место и вторичная передача болезни посредством переливания крови (LLewelyn et al., 2004).
Главным событием в патогенезе прионных болезней является превращение клеточного прионного белка PrPC в патологическую изоформу PrPSc, которая агрегируется в большие скопления белка. Такое формирование скоплений PrPSc является признаком патогенеза прионных болезней. Имеющиеся на сегодняшний день доказательства свидетельствуют о том, что PrPSc действует и как матрица этого превращения и как нейротоксическое средство, вызывающее нейронную дисфункцию и смерть клетки (Prusiner 1998, Giese и Kretzschmar 2001). Поэтому наиболее перспективным терапевтическим подходом к лечению прионных болезней является подавление PrPSc амплификации. Данные, полученные из исследований на клеточной культуре и живых организмах, говорят о том, что при ингибировании образования PrPSc может иметь место клиренс PrPSc (Mallucci 2003). Таким образом, данная терапевтическая стратегия также могла бы быть эффективной в конце инкубационноого периода и даже после проявления клинических признаков болезни и имеет большое значение при адресном применении для борьбы с прионной болезнью человека.
Было показано, что некоторые соединения эффективны в подавлении PrPSc амплификации in vitro, такие как, например, производные акридина, Congo Red, порфирины/фталоцианины, Ср-60, пептиды, разрушающие бета-слой, и разновидности PrP (Caughey etal. 1998, Chabry et al. 1998, Demaimay et al. 2000, Horiuchi etal. 2000, Perrier et al. 2000, Rudyk et al. 2000, Soto et al. 2000). Однако до настоящего времени ни одно из этих соединений не нашло применения в лечении болезни или в качестве исходного соединения для получения соединений с повышенной терапевтической эффективностью и фармакологическими свойствами.
Соединения, идентифицированные в качестве потенциальных терапевтических средств, были обнаружены, главным образом, случайно. Существует несколько методик испытаний in vitro, подходящих для высокопроизводительного скрининга больших библиотек соединений с целью выявления потенциальных антиприонных лекарственных средств. В недавно опубликованных исследованиях были предложены два различных подхода к системному скринингу: один основан на дрожжах (Bach et al. 2003), а в другом используются инфицированные ScN2a клеточные культуры (Kocisko et al. 2004, Kocisko et al. 2003). Однако эти подходы позволяли осуществлять скрининг в библиотеках, ограниченных 2500 и 2000 соединениями, соответственно, и оказались трудоемкими.
Помимо применения низкомолекулярных соединений в настоящее время испытано еще три возможных подхода. В первом подходе для подавления образования PrPSc используются антитела против PrP. Этот метод успешно использовался в испытании на клеточной культуре, а также в испытании на мышах при интраперитонеальных инъекциях (Enari et al., 2001; White et al., 2003). Другим подходом является применение CpG олигонуклеотидов, которые, как установлено, увеличивают продолжительность инкубационного периода у скрейпи-инфицированных мышей (Sethi et al., 2002). Однако механизм действия этого метода пока не объяснен. И наконец, изучается подавление экспрессии PrPC в нейронах инфицированных животных или людей с помощью siRNA. Было показано, что данный метод приводит к ингибированию образования PrPSc в клеточных культурах (Daude et al., 2003). Все три указанных метода имеют один и тот же недостаток, а именно - прохождение молекул через гематоэнцефалический барьер. Вследствие данного недостатка указанные подходы применимы только для профилактики установленной экспозиции в периферийных органах, но не подходят для терапевтического лечения заболевания центральной нервной системы.
Другой класс нейродегенеративных заболеваний, так называемые синуклеинопатии, характеризуется внутриклеточным аккумулированием белковых скоплений, олигомеров, протофиблилл и фибрилл, содержащих, главным образом, α-синуклеин. Считается, что в случаях синуклеинопатий патологическое воздействие на нервные клетки вызвано образованием олигомерных агрегатов α-синуклеина и последующим образованием мембранных пор. Примерами синуклеинопатий являются болезнь Паркинсона (PD), деменция с тельцами Леви (DLB) и множественная системная атрофия (MSA). До настоящего времени нет приемлемых терапевтических стратегий для подавления агрегации α-синуклеина.
Таким образом, существует потребность в идентификации новых соединений, подходящих для лечения заболеваний, связанных с агрегацией белков, таких как прионные болезни и синуклеинопатии.
Поэтому техническая задача, составляющая основу настоящего изобретения, заключается в создании соединений для лечения прионных болезней, синуклеинопатий и других болезней, характеризующихся агрегацией белков, в частности, болезни Паркинсона. Кроме того, существует необходимость создания соединений, которые являются подходящими зондами для визуализации скоплений агрегированных белков в упомянутых выше расстройствах.
ОПИСАНИЕ ФИГУР
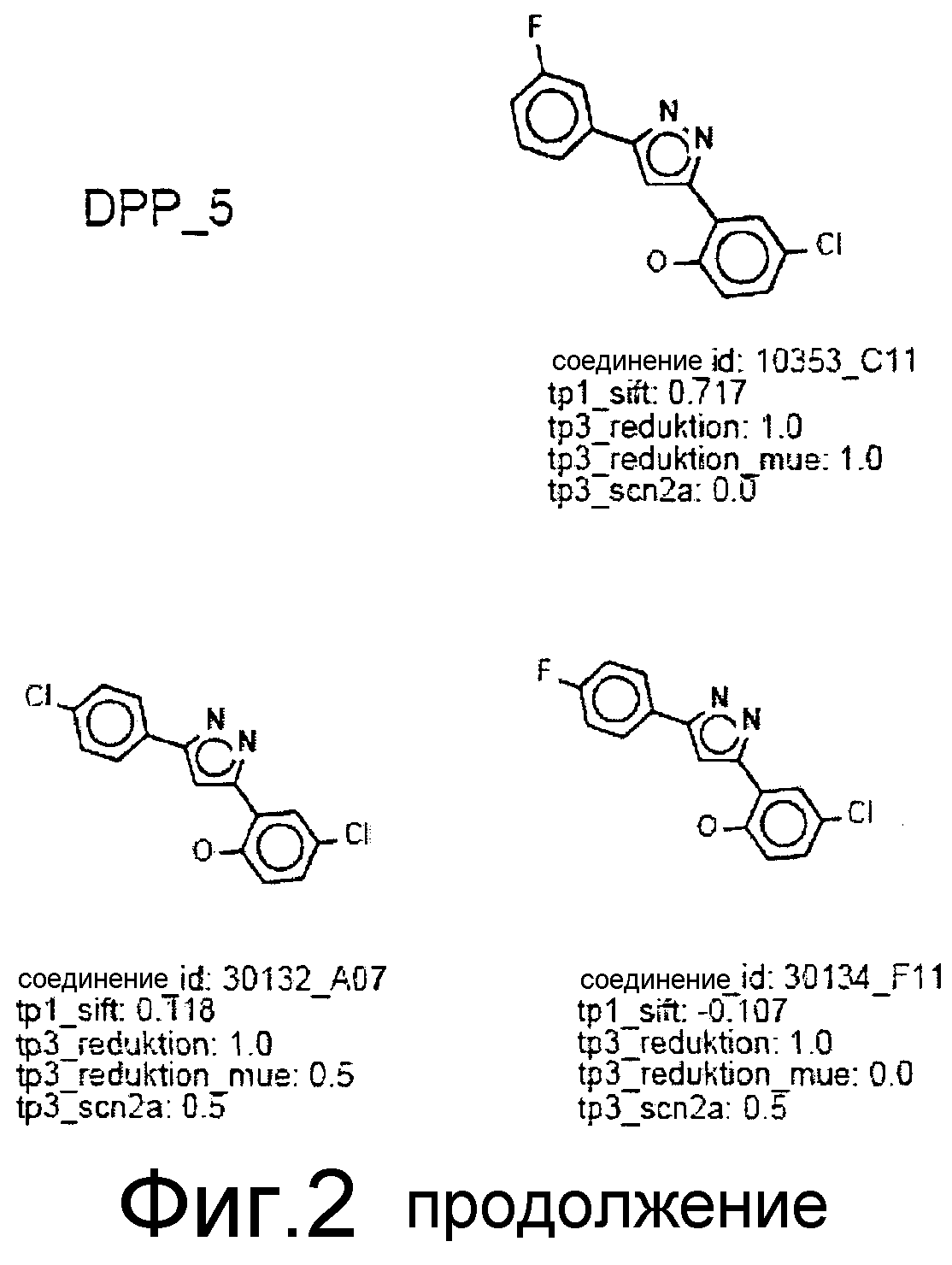
Фиг.1: SAR-карта, составленная для DIVERSet 1 и 2, скрининг которых проводился методом 2D-SIFT и тестированием в клеточной культуре. На карте показаны кластеры структурно подобных соединений (обозначенные звездочками или квадратами), сформированные из 837 лучших соединений, выявленных первичным скринингом в клеточной культуре из DIVERSet 1 и 2. Кластеры, в свою очередь, расположены таким образом, что аналогичные кластеры находятся в непосредственной близости друг к другу. Символами, представляющими кластеры, являются масштаб, форма и окраска в соответствии с их размером и количественными соотношениями между активностями в SIFT-анализе и в клеточных структурах, соответственно, как указано в пояснениях. Таким образом, большие кластеры, содержащие большие соотношения SIFT-активностей и активностей в клеточных культурах, обозначены большими красными звездами. Выбрано пять кластеров, названных DPP_1-DPP_5, и показаны соединения-прототипы, представляющие эти кластеры.
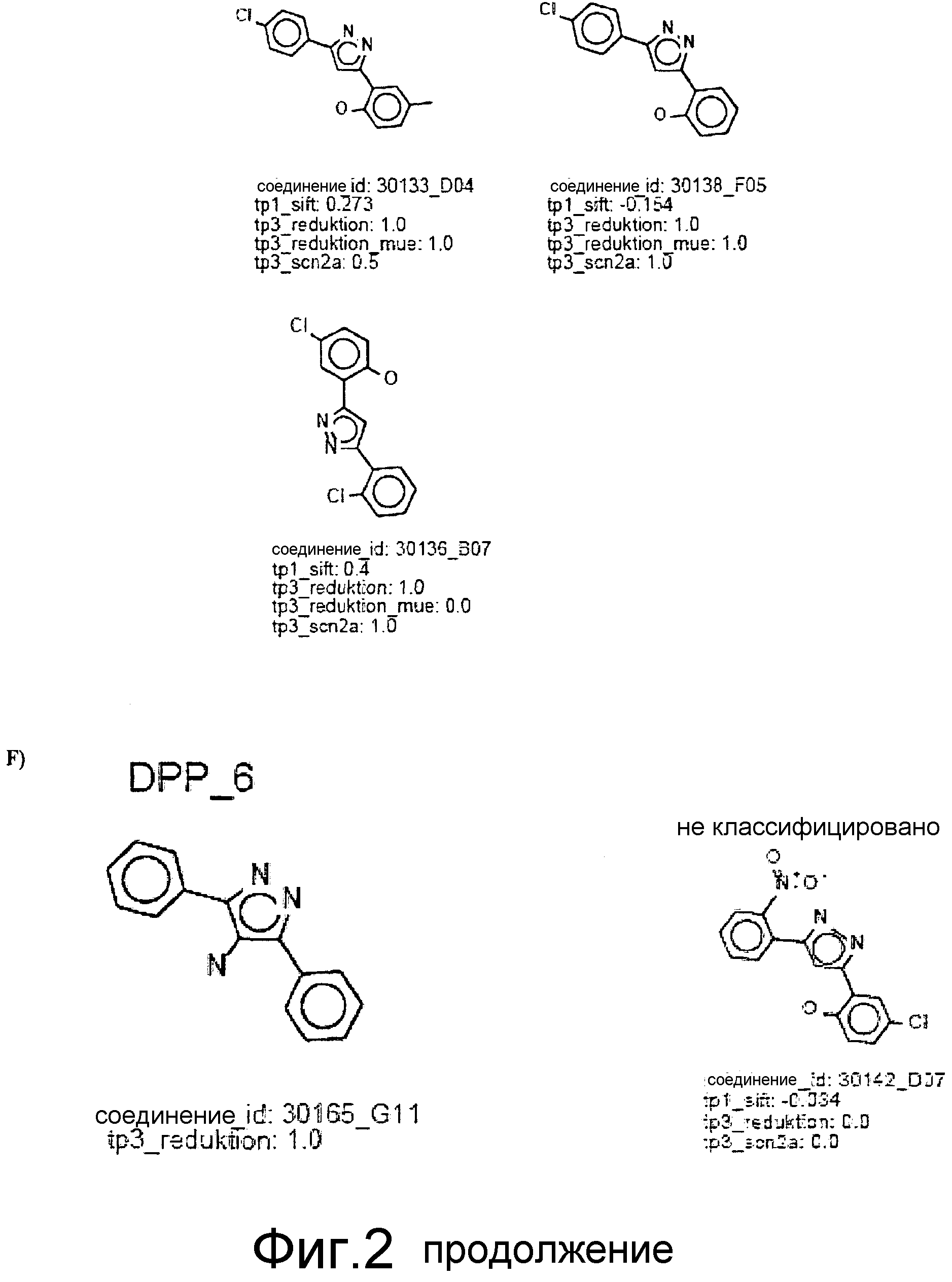
Фиг.2: Все соединения, распределенные в кластеры, которые идентифицированы выше (фиг.1), представлены на фиг.2 (А-F) с их активностями в различных тестах. Все они относятся к химическому классу производных 3,5-дифенилпиразола (DPP) (структура DPP показана на Фиг.1). На фиг.2F показан дополнительный кластер DPP_6, который не содержится в SAR-карте, представленной на фиг.1, и включает единственное соединение, активное в отношении клеточной культуры, с атомом N, присоединенным к пиразольному циклу. На фиг.2F дополнительно показаны 4 соединения из 33 DPP из DIVERSet 1 и 2, которые, как установлено, не активны в клеточной культуре и которые, как определено DM программой, не сходны с шестью соединениями класса DPP. Заявителями изобретения идентифицированы эти соединения с помощью поиска в библиотеке структур DPP.
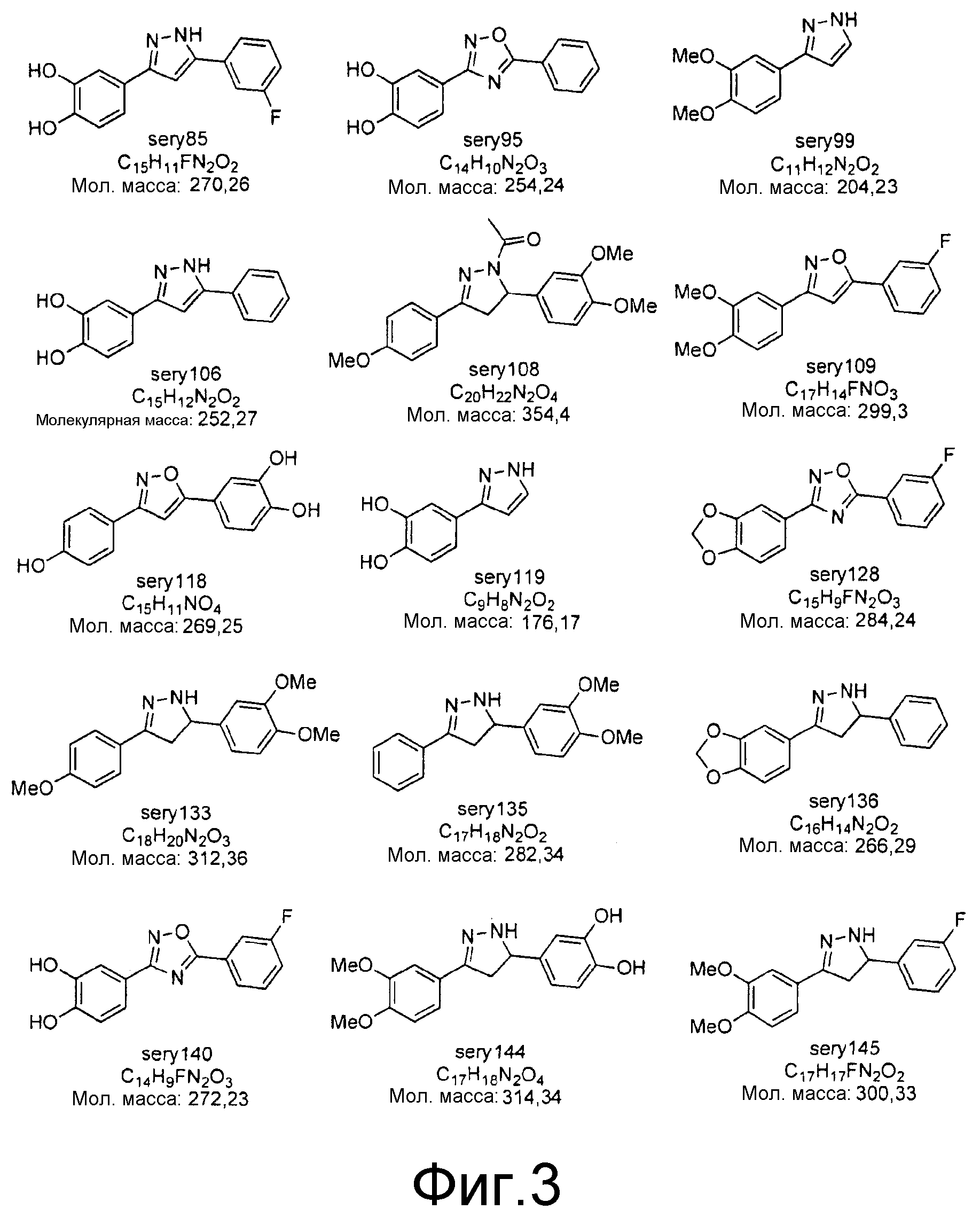
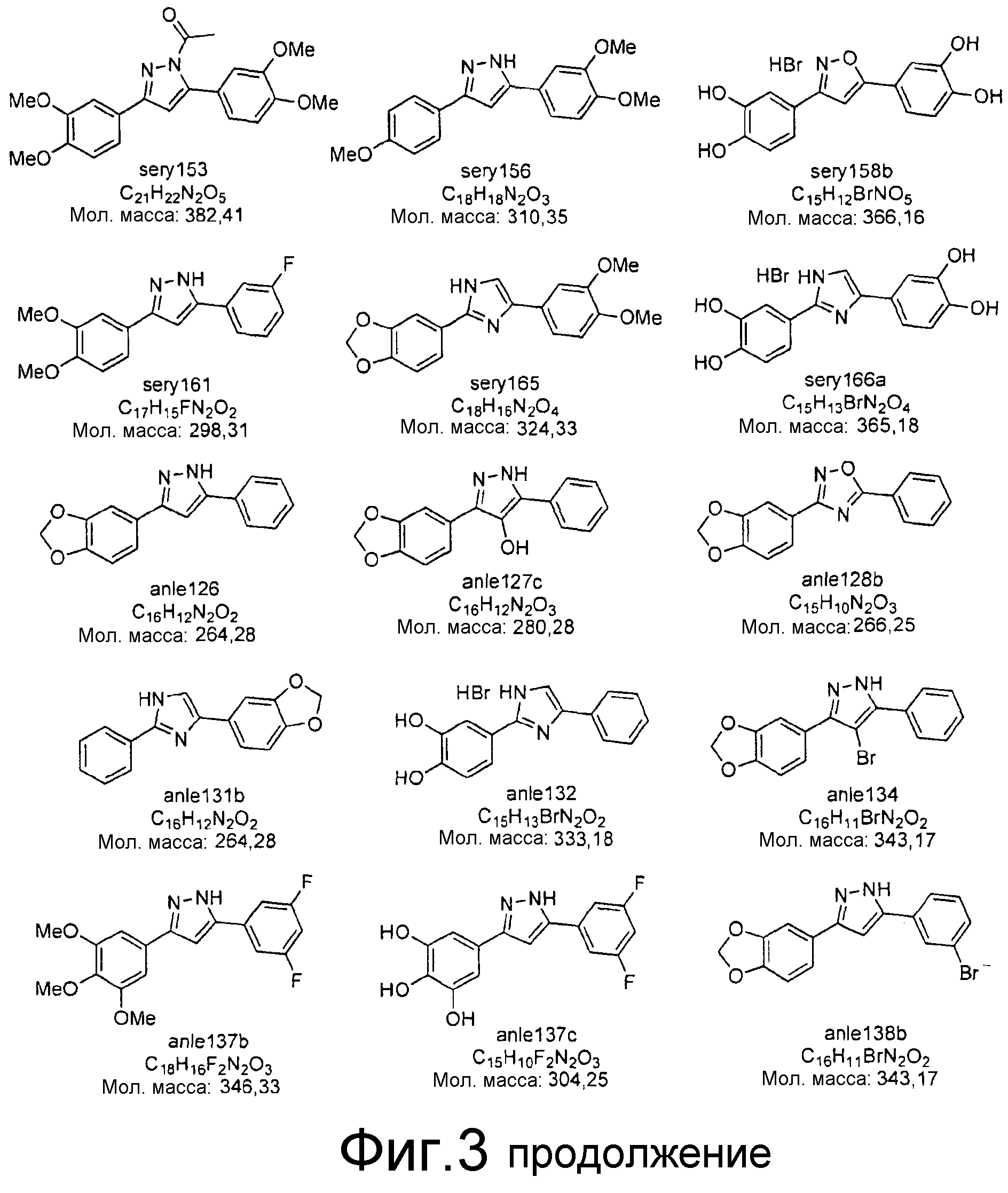
Фиг.3: Перечень соединений, которые были синтезированы согласно способам, описанным в примере 2, на основании результатов первичного скрининга и медицинских химических соображений. Эти соединения были испытаны с использованием различных способов анализа (т.е. SIFT, анализы в моделях клеточных культур прионных болезней, тесты in vivo на животных или биохимические анализы агрегации α-синуклеина).
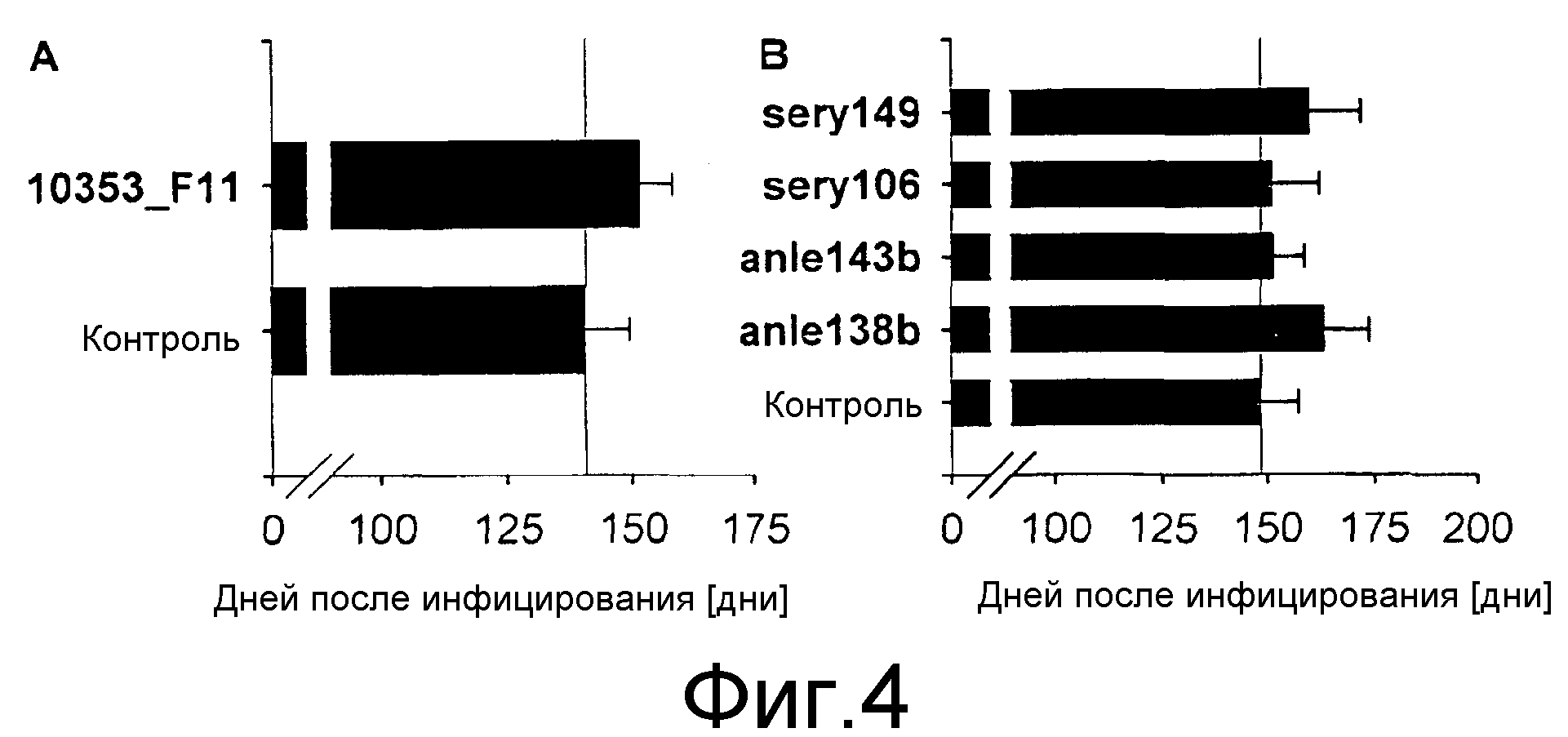
Фиг.4: Влияние лечения на период выживания мыши после интрацеребрального инфицирования RML скрейпи. Соединения вводились ежедневно в течение 14 дней, начиная с 80 дня после инфицирования (50 мкл 10 мМ соединения). На схеме (А) показано, что лечение соединением 10353_F11 увеличивает продолжительность периода выживания интрацеребрально инфицированных мышей (р<0,05). На схеме (В) показаны средние продолжительности периодов выживания после лечения различными соединениями. Ежедневная интраперитонеальная инъекция соединений anle138b и sery149 значительно увеличивает продолжительность периодов выживания после заражения RML скрейпи (anle138b: p<0,01; sery149: p<0,05). Средние значения времени выживания выражены в днях ± среднеквадратическое отклонение.
Фиг.5: Влияние лечения на уровень содержания PrPSc в селезенке мышей, интраперитонеально инфицированных RML скрейпи. (А) После инокуляции скрейпи-прионами мышей лечили один раз в день соединением (25 мкл 100 мМ соединения для интраперитонеального введения и 50 мг/кг для перорального введения). (В) Денситометрический анализ уровня содержания PrPSc в селезенке в дот-блот анализе. Лечение с помощью anle138b вызывает стойкое снижение уровней содержания PrPSc по сравнению с контролем (р=0,001). (С) Иммуногистологические анализы селезенок скрейпи-инфицированных мышей. После лечения anle138b процент селезенок с низкими PrPSc скоплениями возрастает (качество +) и прочные PrPSc скопления отсутствуют (качество +++). На (D) показаны два образца окрашенных PrPSc скоплений (стрелки) в селезенках. На фигуре слева показано сильно окрашенное PrPSc скопление (качество +++), в то время как справа показано PrPSc скопление с незначительным окрашиванием (качество +). Планки погрешностей в блок-схемах показывают среднее значение PrPSc ± стандартное отклонение.
Фиг.6: Иммуноблот и гистологический анализ мозга мышей. (А) Лечение мышей после интрацеребральной инокуляции RML скрейпи начинали через 80 дней после инфицирования. Соединения вводили в указанных временных точках (25 мкл 100 мМ соединения для i.p. введения и 50 мг/кг для перорального введения). (В) Количественное определение уровней содержания PrPSc в гомогенатах мозга прион-инокулированных мышей в различных временных точках. Лечение с помощью anle138b полностью блокирует аккумулирование PrPSc в мозге. Количество PrPSc в 106 день еще находится на уровне количества, выявленного в день 80. Лечение с помощью anle186b приводит к снижению аккумулирования PrPSc в мозге мышей. (С) Изменение относительных уровней содержания PrPSc после лечения соединениями по сравнению с необработанным контролем на день 80. (D) Гистологическое исследование апоптотических клеток (стрелка в (Е)) в Н&Е окрашенных тонких срезах мозга в указанных временных точках. На графике показаны средние количества апоптотических клеток ± среднеквадратическая ошибка. На (Е) показана окрашенная область с апоптотической клеткой (стрелка).
Фиг.7: Увеличение продолжительности периода выживания при ежедневном введении соединений согласно настоящему изобретению. Мыши, которые были интрацеребрально инфицированы штаммом RML скрейпи, показывают более продолжительный период выживания до достижения конечной стадии скрейпи-инфекции.
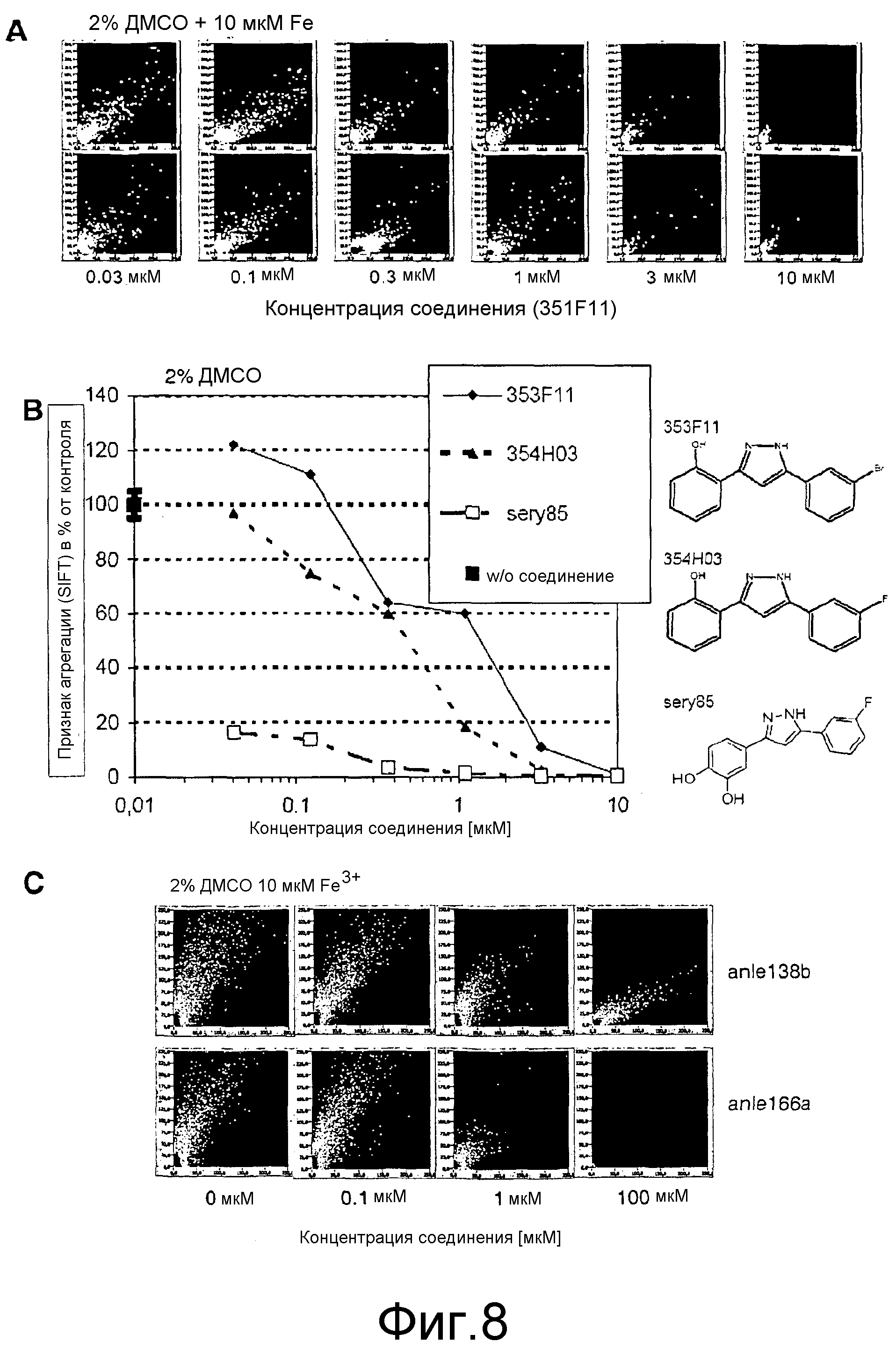
Фиг.8: Подавление агрегации α-синуклеина соединениями согласно настоящему изобретению. (А) DPP-соединение 351F11 способно ингибировать образование мультимерных комплексов α-синуклеина зависимым от дозы образом. (В)(С) Зависимое от дозы ингибиторное действие на агрегацию α-синуклеина, выявленное для других DPP-родственных соединений.
Фиг.9: Культуры клеток, инфицированных прионами, обрабатывают DPP-родственными соединениями согласно настоящему изобретению. DPP-родственные соединения проявили высокоэффективное снижение PrPSc в клеточной культуре при низких микромолярных и даже субмикромолярных концентрациях.
Фиг.10: Влияние ежедневного лечения соединением anle138b на аккумуляцию PrPSc и прионную патологию у мышей, инфицированных RML скрейпи. (А) Срезы мозга, подкрашенные для PrPSc (верхний ряд: кора и гиппокапм, нижний ряд: мозжечок), показывают, что лечение соединением 138b аккумулированием PrPSc по сравнению с животными, обработанными ДМСО. (В) Квантификация содержания PrPSc в мозговых гомогенатах прион-инокулированных мышей в указанные моменты времени показывает, что аккумулирование PrPSc в anle138b-обработанных мышах значительно снижается даже после начала лечения на последней стадии болезни (120 дней после инфицирования). (С) Гистологическое определение количества апоптотических клеток в мозжечке в Н&Е окрашенных срезах показывает, что ингибирование PrPSc аккумуляции приводит к ингибированию гибели нейронных клеток. (D) Контрольные мыши, обработанные смесью ДМСО+арахисовое масло без соединения, показывают прогрессирующую потерю массы. Лечение с помощью anle138b, начиная с 80 дня после инфицирования, предотвращает потерю массы на ~100 дней. Лечение, начиная со 120 дня после инфицирования, ингибирует потерю массы на ~70 дней. Планки погрешностей на В и С показывают стандартную ошибку (n=4 мыши). Подрисуночные подписи к фиг.В также применимы к фиг.С и D.
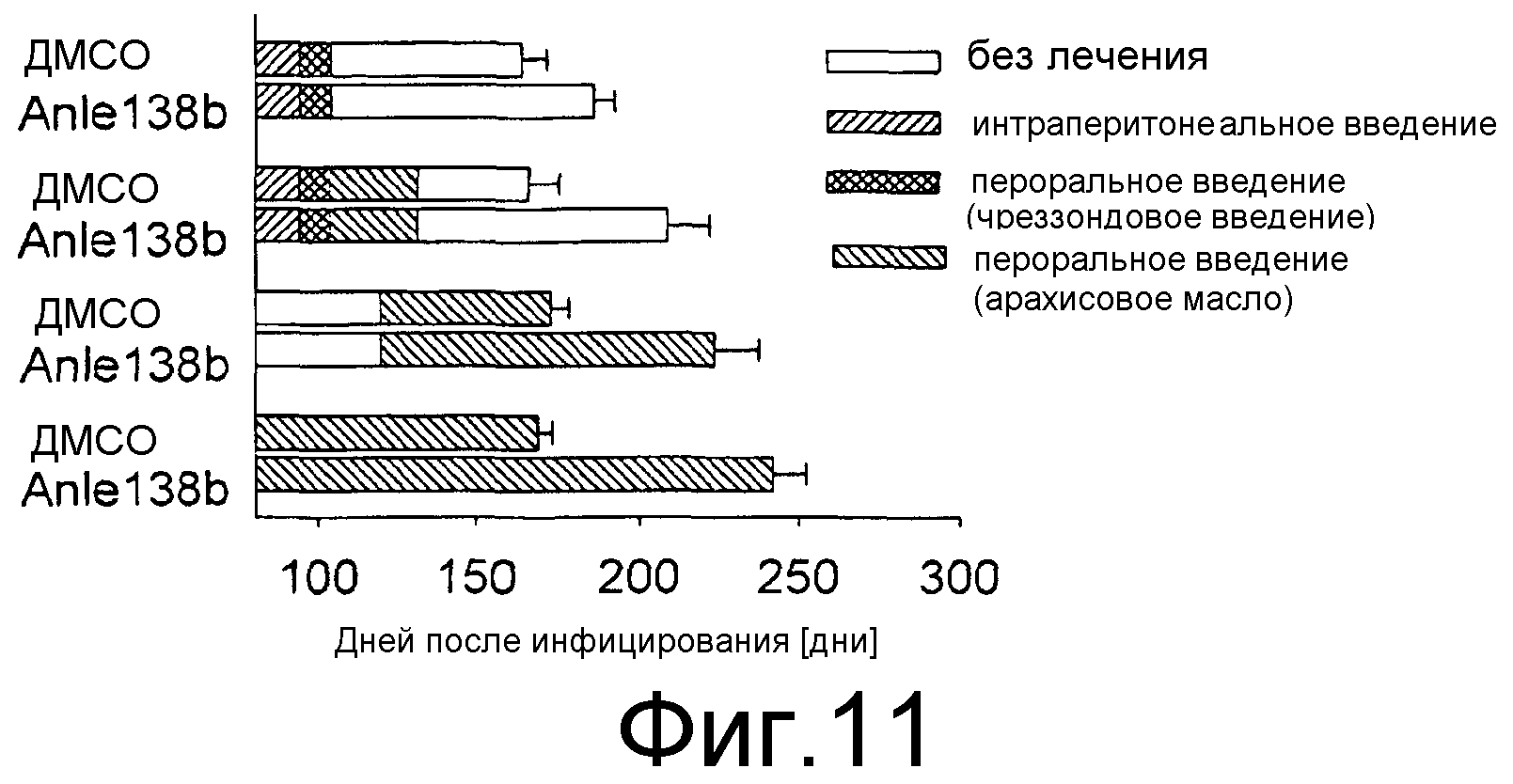
Фиг.11: Сравнение различных методик лечения. Лечение с помощью anle138b в разное время и по различным схемам, как показано на данной фигуре, значительно увеличивает продолжительность периодов выживания после введения RML скрейпи (р<0,01). Средний период выживания представлен в днях ± среднеквадратическое отклонение.
Фиг.12: Зависимое от дозы действие введения anle138b на уровни содержания PrPSc в мозге. C57BL/6 мышей инокулировали интрацеребрально (i.c.) 30 мкл 1% мозгового гомогената (RNL скрейпи). Лечение начинали на дни 80 после инфицирования различными количествами anle138b (как показано на фигуре), вводимыми перорально в смеси с буфером ДМСО + арахисовое масло. На дни 120 животных забивали и определяли количество PrPSc в мозге в сравнении с животными, забитыми на день 80 после инфицирования. Планки погрешностей показывают среднеквадратическую ошибку (n=4 мыши).
Фиг.13: Количественное определение содержания PrPSc иммуноблоттингом ткани мозга от неинфицированных мышей, обрабатываемых 1 мг в день соединения anle138b в смеси с буфером ДМСО + арахисовое масло в течение 1 недели. Каждая планка погрешности показывает погрешность для группы из четырех мышей.
Фиг.14: Фармакокинетический анализ anle138b. Разовую дозу anle138b вводили неинфицированным C57BL/6 мышам, как было показано. В различные моменты времени после введения соединения количество соединения определяли в мозге и сыворотке крови у 2 животных для каждой временной точки и экспериментальной группы с помощью ЖХ-МС.
Фиг.15: Ингибирование образования α-синуклеиновых скоплений с помощью различных соединений. Представлены структуры соединений, результаты испытаний которых представлены в таблице 2.
Фиг.16: Количественное определение потери нейронов у МРТР-обработанных мышей по сравнению с МРТР-необработанными мышами с помощью тирозингидроксилаза-(ТН)-положительных клеток substantia nigra pars compacta (SNpc) в 50 мкм срезах, иммуноокрашенных антителом против ТН. Каждый второй срез SNPc анализировали с использованием программного обеспечения Stereo investigator (MicroBrightfield, Colchester, VT, USA). Иммуноокрашенные клетки подсчитывались с помощью фракционного метода с использованием 20× объектива. Стереологические подсчеты проводились «вслепую» двумя независимыми исследователями.
Фиг.17: Влияние anle138c на Abeta агрегацию. Abeta агрегацию анализировали с помощью динамического рассеяния света. Мономерная и олигометрическая Abeta в отсутствие (верхний график) и в присутствии anle138C (средний график). Нижний график показывает распределение по размерам амилоидного фибриллярного состояния Abeta40, количественно определенное после центрифугирования образца.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Изобретение описано с помощью вариантов осуществления, представленных в формуле изобретения. Следует представлять, что сочетания всех предпочтительных вариантов осуществления изобретения, приведенных далее и в формуле изобретения, должны рассматриваться как часть области настоящего изобретения.
Настоящее изобретение относится к соединению, представленному общей формулой (Е)

X, Y и L в цикле D независимо ненаправленно выбраны из -C(R1)(R2)-, -C(R3)=, -N(R4)-, -N=, -N+(R5)=, -O- и -S-;
М и Z независимо ненаправленно выбраны из


- - - - означает необязательную двойную связь,
Разумеется, что X, Y, Z, L и M будут выбираться в соответствии с подходящей валентностью и стабильностью соединений.
R1, R2, R3, R4, R5, R6 и R7 независимо выбраны из водорода, C1-4 алкила; группы -C1-4 алкилен-галоген; группы -C1-4 алкилен-OH; группы -C1-4 алкилен-C1-4 алкокси; -C(O)-C1-4 алкила; и C6-10 арила, где арильный цикл может быть необязательно замещен C1-4 алкилом или галогеном. C6-10 арильная группа конкретно ограничена и может быть выбрана, например, из фенила и нафтила. Атом галогена может представлять собой F, Cl, Br или I и обычно представляет собой F или Cl.
Предпочтительно R1, R2, R3, R4, R5, R6 и R7 независимо выбраны из водорода; C1-4алкила; группы -C1-4алкилен-галоген; группы -C1-4 алкилен-OH; группы -C1-4 алкилен-C1-4алкокси; и -C(O)-C1-4 алкила. Более предпочтительно R1, R2, R3, R4, R5, R6 и R7 выбраны из водорода; C1-4 алкила; и группы -C1-4 алкилен-галоген.
Выбор заместителя может зависеть от предполагаемого применения соединений формулы (E). В одном предпочтительном варианте осуществления изобретения, по меньшей мере, один из R1, R2, R3, R4, R5, R6 и R7 (более предпочтительно, по меньшей мере, один из R4, R5 и R7) представляет собой группу -C1-4 алкилен-галоген. Это в особенности применимо, если соединения должны применяться в качестве зонда для визуализации скоплений агрегированных белков, поскольку в эту группу можно быстро и рационально ввести обнаруживаемую метку, такую как обнаруживаемый изотоп галогена. Примеры различимых изотопов галогена включают18F,125I,123I,131I,77Br и76Br, в частности18F. Разумеется, можно использовать обнаруживаемый изотоп галогена в качестве любых других атомов галогена, присутствующих в соединениях согласно настоящему изобретению, таких как атомы галогенов, присоединенные к фенильному циклу.
Альтернативно,11C может использоваться в качестве обнаруживаемой метки в соединениях согласно настоящему изобретению.11C может присутствовать, по меньшей мере, в одном из R1, R2, R3, R4, R5, R6 и R7 (более предпочтительно, по меньшей мере, в одном из R4, R5 и R7) или в любой другой части соединения согласно настоящему изобретению.
В альтернативном предпочтительном варианте осуществления изобретения R1, R2, R3, R4, R5, R6 и R7 независимо выбраны из водорода и C1-4 алкила, предпочтительно водорода.
Цикл D конкретно не ограничен. Типичные примеры цикла D включают

Особенно предпочтительными примерами цикла D являются

В приведенных выше формулах R8 выбран из водорода; C1-4 алкила; группы -C1-4 алкилен-галоген; группы -C1-4 алкилен-OH; группы -C1-4 алкилен-C1-4 алкокси; -C(O)-C1-4 алкила; и C6-10 арила, где арильный цикл может быть необязательно замещен C1-4 алкилом или галогеном. Предпочтительно R8 выбран из водорода; C1-4 алкила; группы -C1-4 алкилен-галоген; группы -C1-4 алкилен-OH; группы -C1-4 алкилен-C1-4 алкокси; и -C(O)-C1-4 алкила. Более предпочтительно, R8 выбран из водорода; C1-4 алкила; группы -C1-4 алкилен-галоген. В одном варианте осуществления изобретения R8 выбран из водорода; и C1-4 алкила, более предпочтительно представляет собой водород. В альтернативном варианте осуществления изобретения R8 представляет собой группу -C1-4 алкилен-галоген. Как указано выше, R8 может быть помечен обнаруживаемой меткой, если это необходимо.
В приведенных выше формулах R9 выбран из водорода; C1-4 алкила; группы -C1-4 алкилен-галоген; группы -C1-4 алкилен-OH; группы -C1-4 алкилен-C1-4 алкокси; -C(O)-C1-4 алкила; и C6-10 арила, где арильный цикл может быть необязательно замещен C1-4 алкилом или галогеном. Предпочтительно R9 выбран из водорода; C1-4 алкила; группы -C1-4 алкилен-галоген; группы -C1-4 алкилен-OH; группы -C1-4 алкилен-C1-4 алкокси и -C(O)-C1-4 алкила. Более предпочтительно, R9 выбран из водорода; C1-4 алкила; группы -C1-4 алкилен-галоген. В одном варианте осуществления изобретения R9 выбран из водорода; и C1-4 алкила, более предпочтительно представляет собой водород. В альтернативном варианте осуществления изобретения R9 представляет собой -C1-4 алкилен-галоген. Как указано выше, R9 может быть помечен различимой обнаруживаемой меткой, если это необходимо.
В дополнительном варианте осуществления изобретения R8 и R9 представляют собой водород. В еще одном варианте осуществления изобретения R8 представляет собой группу -C1-4 алкилен-галоген и R9 представляет собой водород.
Hal выбран из F, Cl, Br и I и предпочтительно представляет собой F, Cl или Br, более предпочтительно Cl или Br, более предпочтительно Br.
RE1выбран из гидроксильной группы, C1-6 алкокси и -NRE5RE6.
RE2 выбран из водорода, галогена, гидроксильной группы, C1-6 алкокси и -NRE5RE6, предпочтительно RE2 выбран из водорода, гидроксильной группы, C1-6 алкокси и -NRE5RE6.
В альтернативном варианте осуществления изобретения RE1и RE2 вместе могут ненаправленно образовывать структуру -T-(CRE7RE8)n-V-, а также соответствующие структуры, в которых присутствует двойная связь, если они присоединены к соседним атомам углерода. В данной структуре Т выбран из CRE9RE10, NH и О, V выбран из CRE9RE10, NH и О. Предпочтительно по меньшей мере один из Т и V представляет собой NH или О. Примеры таких структур включают -O-(CH2)n-O-, -O-(CF2)n-O-, -O-(CH2)n-CH2-, -NH-(CH2)n-NH-, -NH-(CF2)n-NH-, -NH-(CH2)n-CH2- или соответствующую структуру, в которой присутствует двойная связь. Например, если n=1, то -N=CH-NH- представляет собой структуру, в которой присутствует двойная связь и которая соответствует -NH-CH2-NH-. Предпочтительно RE1 и RE2 вместе образуют структуру -O-(CH2)n-O-. Подразумевается, что данная группа также может подвергаться гидролизу in vivo до соответствующих гидроксильных групп.
n принимает значения от 1 до 3; предпочтительно n равно 1 или 2, более предпочтительно n равно 1.
RE5 и RE6 независимо выбраны из водорода и С1-6 алкила; предпочтительно RE5 и RE6 независимо выбраны из водорода и С1-4 алкила.
RE7и RE8 независимо представляют собой Н или F, предпочтительно представляют собой Н.
RE9 и RE10 независимо представляют собой Н или F, предпочтительно представляют собой Н.
Положение, в котором RE1 и RE2 присоединены к фенильному циклу, может изменяться.
В одном варианте осуществления изобретения RE1 и RE2 независимо представляют собой гидроксильную группу или алкоксигруппу и присоединены в мета- или пара-положение относительно атома углерода, который связывает фенильное кольцо с циклом D.
В другом варианте осуществления изобретения RE1 и RE2представляют собой структуру -T-(CRE7RE8)n-V- или соответствующую структуру, в которой присутствует двойная связь, и присоединены в мета- и пара-положении относительно атома углерода, который связывает фенильный цикл с циклом D. Приведенные выше предпочтительные определения структуры -T-(CRE7RE8)n-V- аналогично применимы к данному варианту осуществления.
В третьем варианте осуществления изобретения RE1 представляет собой -NRE5RE6 и присоединен в пара-положении относительно атома углерода, который связывает фенильный цикл с циклом D.
Помимо RE1 и RE2 на фенильном цикле могут необязательно присутствовать дополнительные заместители RE3. RE3 может представлять собой С1-6 алкильную группу или С5-10 арильную группу (такую как фенильная или нафтильная группа), предпочтительно С1-6 алкильную группу, более предпочтительно С1-4алкильную группу. Число заместителей m конкретно не ограничено и обычно находится в интервале от 0 до 2, предпочтительно от 0 до 1, обычно равно 0.
Могут также присутствовать дополнительные заместители RE4. Обычно они представляют собой атом галогена, С1-6 алкильную группу или С5-10 арильную группу (такую как фенильная или нафтильная группа), предпочтительно атом галогена или С1-6 алкильную группу, более предпочтительно С1-6 алкильную группу, наиболее предпочтительно С1-4алкильную группу. Число заместителей р конкретно не ограничено и обычно находится в интервале от 0 до 2, предпочтительно равно 0 или 1, обычно равно 0.
В некоторые варианты осуществления не включены следующие соединения:
3(5)-(2-гидрокси-5-метилфенил)-5(3)-(4-хлорфенил)пиразол (DE 4126543: соединение 26 в таблице 1);
орто-гидроксифенил-5 дихлор-3'-4'-фенил-3 метил-2 пиразол (FR 2.104.932: Пример IV);
орто-гидроксифенил-5 дихлор-3'-4'-фенил-3-фенил-2 пиразол (FR 2.104.932: Пример IV);

Данные соединения описаны как соединения IA-44, IA-47, IA-81, IA-106 и IA-115 в WO 2004/080972.
В других вариантах осуществления изобретения данные соединения не исключаются.
Предпочтительные примеры соединения, представленного формулой (Е), включают соединения, представленные формулой (А)

Определения Х, Y, Z, M, L, цикла D, m, p и Hal, приведенные выше для формулы (Е), аналогично применимы к формуле (А).
RA1 и RA2, каждый независимо, выбран из водорода, галогена, гидроксильной группы, С1-6 алкоксигруппы и -NRA5RA6 при условии, что по меньшей мере один из RA1и RA2представляет собой гидроксильную группу, С1-6 алкоксигруппу или -NRA5RA6. Предпочтительно, RA1и RA2 независимо выбраны из водорода, гидроксильной группы, С1-6 алкоксигруппы и -NRA5RA6.
Альтернативно, RA1 и RA2 могут вместе ненаправленно образовывать структуру -T-(CRE7RE8)n-V-. Определения, приведенные для RE1 и RE2, входящих в состав такой структуры, и в частности, приведенные выше определения RE7, RE8, T, n и V, аналогично применимы к RA1 и RA2, образующим такую структуру.
В одном варианте осуществления изобретения RA1 и RA2независимо представляют собой гидроксильную или алкоксигруппу.
Во втором варианте осуществления изобретения RA1 и RA2 представляют собой структуру -T-(CRE7RE8)n-V- или соответствующую структуру, в которой присутствует двойная связь. Приведенные выше предпочтительные определения для структуры -T-(CRE7RE8)n-V- аналогично применимы к данному варианту осуществления изобретения.
В третьем варианте осуществления изобретения RA1 представляет собой -NRA5RA6 и RA2 представляет собой водород.
Помимо RA1 и RA2 на фенильном цикле могут необязательно присутствовать дополнительные заместители RA3. RA3 может представлять собойС1-6 алкильную группу или С5-10 арильную группу (такую как фенильная или нафтильная группа), предпочтительно С1-6 алкильную группу, более предпочтительно С1-4 алкильную группу.
Также могут присутствовать дополнительные заместители RA4. Обычно они представляют собой атом галогена, С1-6 алкильную группу или С5-10 арильную группу (такую как фенильная или нафтильная группа), предпочтительно атом галогена или С1-6 алкильную группу, более предпочтительно С1-6 алкильную группу, наиболее предпочтительно С1-4 алкильную группу.
RA5 и RA6 независимо выбраны из водорода и С1-6 алкила; предпочтительно RA5 и RA6 независимо выбраны из водорода и C1-4 алкила.
Предпочтительные примеры соединения, представленного формулой (Е), включают соединения, представленные формулой (В)

Определения Х, Y, Z, M, L, цикла D, m, p и Hal, приведенные выше для формулы (Е), аналогично применимы к формуле (В).
RB1 выбран из гидроксильной группы, С1-6 алкоксигруппы и -NRB5RB6. Предпочтительно, RB1 представляет собой гидроксильную группу или С1-6 алкоксигруппу.
RB2 выбран из водорода, галогена, гидроксильной группы, С1-6 алкоксигруппы и -NRA5RA6, предпочтительно, RB2 выбран из водорода, гидроксильной группы, С1-6 алкоксигруппы и -NRA5RA6.
В одном варианте осуществления изобретения RB1 представляет собой гидроксильную или С1-6 алкоксигруппу и RB2 представляет собой водород.
RB5и RB6 независимо выбраны из водорода и С1-6 алкила, предпочтительно RB5и RB6 независимо выбраны из водорода и С1-4 алкила.
На фенильном цикле помимо RB1 и RB2 могут присутствовать дополнительные заместители RB3, RB3 могут представлять собой С1-6 алкильную группу или С5-10 арильную группу (такую как фенильная или нафтильная группа), предпочтительно С1-6 алкильную группу, более предпочтительно С1-4 алкильную группу.
Также могут присутствовать дополнительные заместители RB4. Обычно они могут представлять собой атом галогена, С1-6 алкильную группу или С5-10 арильную группу (такую как фенильная или нафтильная группа), предпочтительно атом галогена или С1-6 алкильную группу, более предпочтительно С1-6 алкильную группу, наиболее предпочтительно С1-4 алкильную группу.
Предпочтительные соединения согласно настоящему изобретению включают
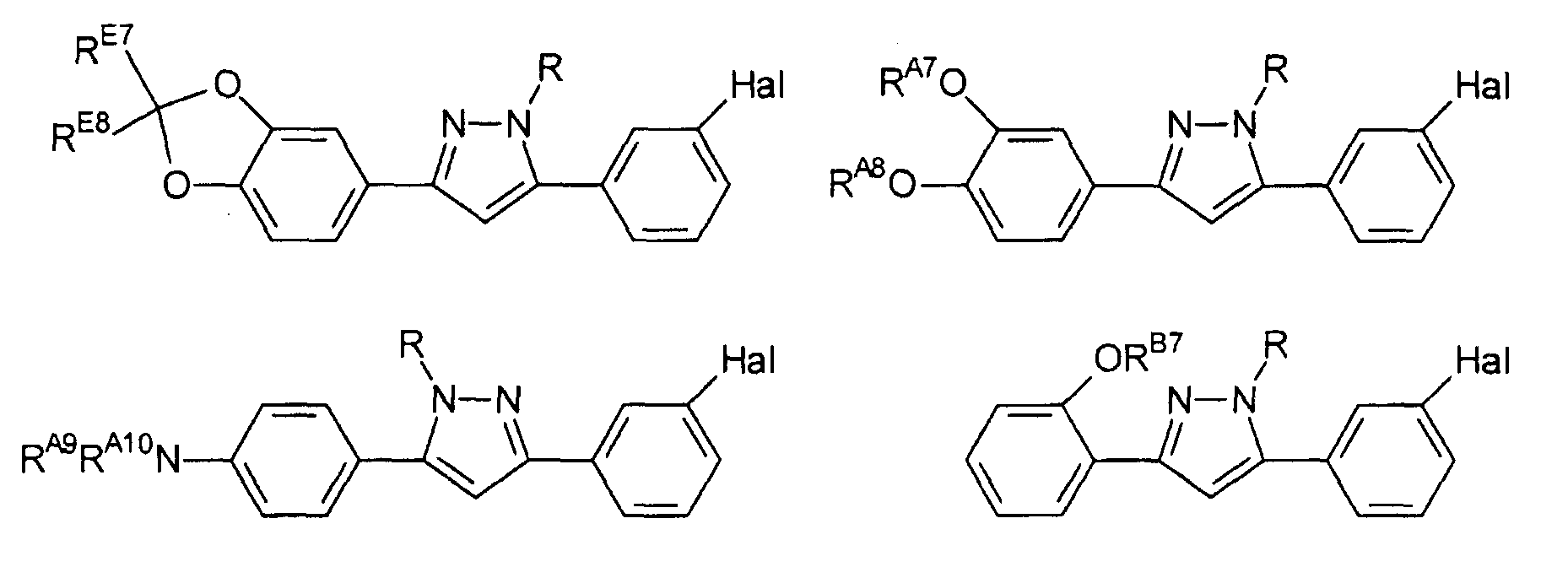
Определения, приведенные выше для RE7, RE8 и Hal, аналогично применимы и к данным соединениям.
R выбран из водорода; С1-4 алкила; группы -С1-4 алкилен-галоген; и С6-10 арила (такого как фенил и нафтил), где арильный цикл может быть необязательно замещен С1-4 алкилом или галогеном. Предпочтительно R выбран из водорода, С1-4 алкила; группы -С1-4 алкилен-галоген. В одном варианте осуществления изобретения R выбран из водорода; и С1-4 алкила, более предпочтительно представляет собой водород. В альтернативном варианте осуществления изобретения R представляет собой группу -С1-4 алкилен-галоген. Как было пояснено выше, R может содержать обнаруживаемую метку, если это необходимо.
RA7 представляет собой H или С1-6 алкил, предпочтительно H или С1-4 алкил.
RA8 представляет собой H или С1-6 алкил, предпочтительно H или C1-4 алкил.
RA9 представляет собой H или С1-6 алкил, предпочтительно H или С1-4 алкил.
RA10 представляет собой H или С1-6 алкил, предпочтительно H или С1-4 алкил.
RB7 представляет собой H или С1-6 алкил, предпочтительно H или C1-4 алкил.
Приведенные далее соединения особенно предпочтительны, поскольку, как установлено, обладают высокой эффективностью ингибирования агрегации белков или визуализации агрегированных белков:

где Hal представляет собой Cl или Br, предпочтительно Hal представляет собой Br.
Наиболее предпочтительны следующие соединения

Соединения согласно настоящему изобретению также могут быть представлены в форме пролекарств, сложных эфиров, сольватов или солей.
Соединения согласно настоящему изобретению образуют соли, которые также относятся к области данного изобретения. Следует представлять, что ссылка в описание на соединение согласно настоящему изобретению включает ссылку на его соли, если не указано иного. Термин «соль(и)», когда применяется в данном описании, означает кислотные и/или основные соли, образованные с неорганическими и/или органическими кислотами и основаниями. Кроме того, когда соединение содержит основный и кислотный фрагмент, могут образовываться цвиттерионы («внутренние соли»), которые также включены в область значений термина «соль(и)», используемого в данном описании. Фармацевтически приемлемые (т.е. нетоксичные, физиологически приемлемые) соли являются предпочтительными, хотя также могут использоваться и другие соли, например, на стадиях выделения или очистки в процессе получения. Соли соединений согласно настоящему изобретению могут быть получены, например, взаимодействием соединения с количеством кислоты или основания, таким как эквивалентное количество, в среде, такой как среда, в которой соль выпадает в осадок, или в водной среде с последующей лиофилизацией.
Соединения, которые содержат основный фрагмент, могут образовывать соли с различными органическими и неорганическими кислотами. Примеры кислотно-аддитивных солей включают ацетаты (такие как ацетаты, образованные с уксусной или тригалогенуксусной кислотой, например, трифторуксусной кислотой), адипаты, альгинаты, аскорбаты, аспартаты, бензоаты, бензолсульфонаты, бисульфаты, бораты, бутираты, цитраты, камфораты, камфорсульфонаты, циклопентанпропионаты, диглюконаты, додецилсульфаты, этансульфонаты, фумараты, глюкогептаноаты, глицерофосфаты, гемисульфаты, гептаноаты, гексаноаты, гидрохлориды, гидробромиды, гидройодиды, гидроксиэтансульфонаты (например, 2-гидроксиэтансульфонаты), лактаты, малеаты, метансульфонаты, нафталинсульфонаты (например, 2-нафталинсульфонаты), никотинаты, нитраты, оксалаты, пектинаты, персульфаты, фенилпропионаты (например, 3-фенилпропионаты), фосфаты, пикраты, пивалаты, пропионаты, салицилаты, сукцинаты, сульфаты (например, соли, образованные с серной кислотой), сульфонаты (например, соли, указанные в описании), тартраты, тиоцианаты, толуолсульфонаты, такие как тозилаты, ундеканоаты и т.п.
Соединения, которые содержат кислотный фрагмент, могут образовывать соли с различными органическими и неорганическими основаниями. Типичные основные соли включают аммониевые соли, соли щелочных металлов, такие как натриевые, литиевые и калиевые соли, соли щелочноземельных металлов, такие как кальциевые и магниевые соли, соли с органическими основаниями (например, органическими аминами), такими как бензатины, дициклогексиламины, гидрабамины (образованные с N,N-бис(дегидроабиетил)этилендиамином), N-метил-D-глюкамины, N-метил-D-гликамиды, трет-бутиламины, и соли с аминокислотами, такие как аргинин, лизин и т.п. Основные азотсодержащие группы, могут быть кватернизованными с помощью таких соединений, как низшие алкилгалогениды (например, метил-, этил-, пропил- и бутилхлориды, -бромиды и йодиды), диалкилсульфаты (например, диметил-, диэтил-, дибутил- и диамилсульфаты), галогениды с длинной углеродной цепью (например, децил-, лаурил-, миристил- и стеарилхлориды, -бромиды и йодиды), аралкилгалогениды (например, бензил- и фенетилбромиды) и т.д.
Пролекарства и сольваты соединений согласно настоящему изобретению также включены в область настоящего изобретения. Термин «пролекарство», когда используется в данном описании, означает соединение, которое после введения субъекту подвергается химическому превращению посредством метаболических и химических процессов с получением соединения согласно настоящему изобретению или его соли и/или сольвата.
Сольваты соединений согласно настоящему изобретению включают, например, гидраты.
Сложные эфиры соединений согласно настоящему изобретению включают сложные С1-6 алкиловые, предпочтительно сложные С1-4 алкиловые эфиры.
Соединения согласно настоящему изобретению могут существовать в их таутомерной форме (например, в форме амида или простого иминоэфира). Подразумевается, что все такие таутомерные формы составляют часть настоящего изобретения.
Подразумевается, что все стереоизомеры соединений согласно настоящему изобретению (например, соединения, которые могут существовать вследствие наличия асимметричных атомов углерода на различных заместителях), в том числе энантиомерные и диастереомерные формы, включены в область настоящего изобретения. Отдельные стереоизомеры соединений согласно изобретению могут быть, например, по существу свободными от других изомеров (например, чистый оптический изомер или по существу чистый оптический изомер, обладающий специфической активностью) или могут быть смешанными, например, в виде рацематов или со всеми другими или иначе выбранными стереоизомерами. Хиральные центры соединений согласно настоящему изобретению могут иметь S- или R-конфигурацию, как определено в IUPAC 1974 Recommendations.
Рацемические формы могут разделяться физическими методами, такими как фракционная кристаллизация, разделение или кристаллизация диастереомерных производных или разделение с помощью хиральной колоночной хроматографии. Отдельные оптические изомеры могут быть получены из рацематов любым подходящим способом, включая, но без ограничения, получение соли с оптически активной кислотой с последующей кристаллизацией.
Все пространственные изомеры соединений согласно настоящему изобретению подразумеваются либо в виде смеси, либо в чистой или в по существу чистой форме. Определение соединений согласно настоящему изобретению включает цис (Z) и транс (Е) алкиленовые изомеры, а также цис- и транс-изомеры углеводородных циклов или гетероциклов.
В данном описании группы и их заместители могут выбираться при условии получения стабильных фрагментов и соединений.
Все соединения формулы (Е) могут быть предоставлены в форме фармацевтической или диагностической композиции, которая необязательно включает фармацевтически приемлемый носитель.
Применяя системы биохимического анализа, основанные на методе «сканирования интенсивно флуоресцирующих мишеней» (SIFT), в сочетании с клеточным анализом в моделях клеточной культуры прионных болезней заявителями данного изобретения in vitro был произведен скрининг больших библиотек синтетических соединений в качестве ингибиторов агрегации процессов, сопровождающих нейродегенеративные болезни, и в частности, прионные болезни и синуклеопатии, на молекулярном уровне. Такие ингибиторы потенциально могут быть новыми терапевтическими лекарственными средствами для лечения этих болезней.
Данная аналитическая система несомненно превосходит все аналитические системы, которые применяются для выявления новых лекарственных средств для ингибирования агрегации белков, по степени автоматизации, скорости измерения (75 секунд на образец), количествам химических соединений (только 200 пикомолей на первичных анализ), а также необходимому количеству используемого образца (например, только эквивалент 0,2 мг мозга от CJD-пациента на анализ). Эти относительно невысокие потребности в образцах и временных затратах позволяют эффективно оценить и анализировать такое большое количество (то есть 20000) соединений. Кроме того, картирование всех данных скрининга в централизованную базу данных и их автоматизированный анализ позволяет эффективно оценивать и анализировать взаимосвязь «структура-активность». Сочетание с методиками скрининга в клеточной культуре, которые были включены в настоящее изобретение, позволяет идентифицировать соединения, активные как в биохимическом анализе, так и в испытании на клеточном уровне. Таким образом, идентифицированы соединения, которые активны не только в условиях in vitro, но и на клеточном уровне, что, например, гарантирует подходящую стабильность и реакционную способность идентифицированных соединений для дальнейшей разработки в применениях in vivo.
Таким образом, заявителями настоящего изобретения в данном первичном скрининге был идентифицирован ряд соединений, активность которых впоследствии была подтверждена в серии разбавлений для идентификации соединений, активных в очень низких концентрациях. Соединения, охарактеризованные как «активные» в первичном скрининге, были подвергнуты к кластер-анализу, в результате которого выявлена группа пяти близких кластеров (от DPP_1 до DPP_5; фиг.1), включая высоко активные соединения класса производных 3,5-дифенилпиразола (DPP) (сравнить фрагмент DPP на фиг.1).
Авторы настоящего изобретения вводили различные заместители идентифицированного класса соединений для идентификации родственных соединений, подходящих в качестве ингибиторов процессов агрегации, сопровождающих нейродегенеративные заболевания, в частности прионные болезни, и синуклеинопатий на молекулярном уровне. При использовании такого медико-химического подхода был синтезирован ряд дополнительных соединений. Указанные соединения вместе с соединениями, выбранными в результате первичного скрининга, были подвергнуты дополнительному тестированию, включая SIFT анализ, анализы на основе клеточной культуры, эксперименты на мышах в условиях in vivo, а также биохимическим анализам, направленным на агрегацию α-синуклеина (см. примеры). Таким образом, была верифицирована активность данных соединений как in vitro, так и in vivo. Обнаружение того, что данные соединения способны также эффективно ингибировать мультимерное образование α-синуклеина, является точным подтверждением того, что идентифицированные соединения могут функционировать не только как антиприонные соединения, но обладают также терапевтическим потенциалом для лечения синуклиенопатий, таких как болезнь Паркинсона, DLB и MSA, направленным воздействием на патологический механизм на молекулярном уровне. Более того, ингибиторная активность указанных соединений в отношении агрегации прионных белков и α-синуклеинов in vitro может свидетельствовать об их общей противоагрегаторной активности в отношении более широкого спектра заболеваний, связанных с агрегации белков, где белок, неправильно складывающийся в преимущественно бета-листовые конформации, образует базис для последующей агрегации белков в амилоидные фибриллы. Таким образом, данные соединения и соединения, относящиеся к соединениям класса производных DPP, обладают потенциалом применения в качестве терапевтических средств для казуативного лечения всего перечня (нейродегенеративных) заболеваний, связанных с агрегацией белков, включая, но без ограничения, болезнь Паркинсона, прионнную болезнь, болезнь Альцгеймера, множественную системную атрофию, болезнь диффузных телец Леви, лобно-височную деменцию, боковой амиотрофический склероз, болезнь Хантингтона, атаксию, связанную с заболеваниями спинного мозга и мозжечка, и другие поли-Q-заболевания, наследственную церебральную амилоидную ангиопатию, наследственную амилоидную полиневропатию, первичный системный амилоидоз (AL амилоидоз), реактивный системный амилоидоз (AA амилоидоз), диабет II типа, инъекция-локализованный амилоидоз бета-2-микроглобулиновый амилоидоз, наследственный не невропатический амилоидоз, наследственный финский системный амилоидоз.
Настоящее изобретение относится также к соединению согласно настоящему изобретению, а также к его пролекарству, сложному эфиру, сольвату или соли для применения в лечении или предотвращении заболевания, связанного с агрегацией белков, и/или нейродегенеративного заболевания. Дополнительными вариантами осуществления настоящего изобретения являются применение соединения согласно настоящему изобретению для получения фармацевтической композиции для лечения или предотвращения заболевания, связанного с агрегацией белков, и/или нейродегенеративного заболевания, а также способ лечения или предотвращения заболевания, связанного с агрегацией белков, и/или нейродегенеративного заболевания, которые включают введение терапевтически эффективного количества соединения согласно настоящему изобретению пациенту, нуждающемуся в таком введении.
Термин «агрегация», согласно настоящему изобретению, относится к образованию олигомерных или мультимерных комплексов, обычно одного или несколько типов белков, которое может сопровождаться интеграцией в комплекс дополнительных биомолекул, таких как углеводы, нуклеиновые кислоты и липиды.
Термин «белок, вовлеченный в заболевание, связанное с агрегацией белков, и/или нейродегенеративное заболевание», когда используется в данном описании, относится к заболеваниям, которые характеризуются наличием агрегированных белков. Такие агрегированные белки могут образовывать скопления в специфических тканях, более предпочтительно, в нервной ткани или ткани мозга. Степень агрегации зависит от конкретного заболевания.
Настоящее изобретение относится также к применению соединения согласно настоящему изобретению, как определено выше, для получения фармацевтической композиции для лечения или предотвращения заболевания, связанного с агрегацией белка, и/или нейродегеративного заболевания.
Согласно настоящему изобретению, термин «фармацевтическая композиция» относится к композиции для введения пациенту, предпочтительно человеку. Фармацевтическая композиция согласно настоящему изобретению включает соединения, определенные выше, и, необязательно, дополнительные молекулы, способные изменять характеристики соединений согласно изобретению, например, стабилизируя, модулируя и/или активируя их функцию. Соединение может быть в твердой, жидкой или газообразной форме и может быть, inter alia, в форме порошка(ов), таблетки(ок), раствора(ов) или аэрозоля(ей). Фармацевтическая композиция согласно настоящему изобретению может, необязательно и дополнительно, включать фармацевтически приемлемый носитель. Примеры подходящих фармацевтических носителей хорошо известны в данной области техники и включают фосфатно-буферные растворы соли, воду, эмульсии, такие как эмульсии масло-в-воде, различные типы смачивающих агентов, стерильные растворы, органические растворители, включая ДМСО и т.д. Композиции, включающие такие носители, могут быть получены стандартными способами, хорошо известными в данной области техники.
Фармацевтическая композиция будет получена и дозирована способом, согласующимся с традиционной медицинской практикой, принимая во внимание клиническое состояние отдельного пациента, место доставки фармацевтической композиции, способ введения, схему введения и другие факторы, известные практикующим врачам. Таким образом, термин «эффективное количество» в отношении фармацевтической композиции согласно настоящему изобретению, соответствует указанным соображениям. Квалифицированный специалист данной области техники знает, что эффективное количество фармацевтических композиций, вводимых индивидууму, будет, inter alia, зависеть от природы вводимого соединения.
Фармацевтические композиции согласно изобретению могут вводиться перорально, ректально, парентерально, интрацистернально, интравагинально, интраперитонеально, местно (в виде порошков, мазей, капель или чрескожных заплат), буккально или в виде перорального или назального спрея. Термин «фармацевтически приемлемый носитель» означает нетоксический твердый, полутвердый или жидкий наполнитель, разбавитель, инкапсулирующий материал или вспомогательную добавку любого типа. Термин «парентеральный», когда используется в данном описании, относится к способам введения, которые включают внутривенные, внутримышечные, интраперитонеальные, интрастернальные (в грудину), подкожные и внутрисуставные инъекции и вливание.
Фармацевтическая композиция также подходящим образом вводится с помощью систем постепенного высвобождения действующего вещества. Подходящие примеры композиций с постепенным высвобождением действующего вещества включают полупроницаемые полимерные матрицы в виде формованных частиц, например, пленок или микрокапсул. Матрицы с постепенным высвобождением действующего вещества включают полиактиды (патент США № 3773919, ЕР 58481), сополимеры L-глутаминовой кислоты и гамма-этил-L-глутамата (Sidman, U. et al., Biopolymers 22:547-556 (1983)), поли(2-гидроксиэтилметакрилат) (R. Langer et al., J. Biomed. Mater. Res. 15:167-277 (1981), R. Langer, Chem. Tech. 12:98-105 (1982)), этиленвинилацетат (R. Langer et al., Id.) или поли-D-(-)-3-гидроксимасляную кислоту (ЕР 133988). Фармацевтическая композиция с постепенным высвобождением действующего вещества включает также липосомно захваченное соединение. Липосомы, содержащие фармацевтическую композицию, получают способами, известными в данной области техники: DE 3218121; Epstein et al., Proc. Natl. Acad. Sci. (USA) 82:3688-3692 (1985); Hwang et al., Proc. Natl. Acad. Sci. (USA) 77:4030-4034 (1980); EP 52322; EP 36676; EP 88046; EP 143949; EP 142641; заявка на патент Японии 83-118008; патенты США № 4485045 и 4544545; и EP 102324. Обычно липосомы представляют собой однослойные частицы небольшого размера (примерно 200-800 ангстрем), в которых липидное содержание составляет более примерно 30% мол., причем выбранная доля корректируется для достижения оптимального терапевтического действия.
Фармацевтическую композицию для парентерального введения обычно получают смешением соединения желательной степени чистоты в форме стандартной дозы для инъекции (раствор, суспензия или эмульсия) с фармацевтически приемлемым носителем, т.е. носителем, который является нетоксичным для реципиентов в применяемых дозах и концентрациях и совместим с другими ингредиентами препарата.
Обычно препараты получают однородным тщательным смешиванием компонентов фармацевтической композиции с жидкими носителями или тонко измельченными твердыми носителями, либо с теми и другими. Затем, если это необходимо, полученное изделие формуют в желательный препарат. Предпочтительно носитель представляет собой носитель для парентерального введения, более предпочтительно раствор, который является изотоническим с кровью реципиента.
Примеры таких несущих разбавителей включают воду, раствор соли, раствор Рингера и раствор декстрозы. Применимы также неводные разбавители, такие как нелетучие масла и этилолеат, а также липосомы. Носитель подходящим образом содержит незначительные количества добавок, такие как вещества, повышающие изотоничность и химическую стабильность. Такие добавки являются нетоксичными для реципиентов в применяемых дозах и концентрациях и включают буферные добавки, такие как фосфаты, цитраты, сукцинаты, уксусная кислота и другие органические кислоты и их соли; антиоксиданты, такие как аскорбиновая кислота; низкомолекулярные (менее примерно десяти остатков) (поли)пептиды, например, полиаргинин или трипептиды; белки, такие как альбумин сыворотки крови, желатин или иммуноглобулин; гидрофильные полимеры, такие как поливинилпирролидон; аминокислоты, такие как глицин, глутаминовая кислота, аспаргиновая кислота или аргинин; моносахариды, дисахариды и другие углеводы, включая целлюлозу или ее производные, глюкозу, маннозу или декстрины; хелатообразующие агенты, такие как EDTA; сахарные спирты, такие как маннит или сорбит; противоионы, такие как натрий; и/или неионогенные поверхностно-активные вещества, такие как полисорбаты, полоксамеры или ПЭГ.
Компоненты фармацевтической композиции, предназначенной для терапевтического введения, должны быть стерильными. Стерильность легко достигается фильтрацией через стерильные фильтрационные мембраны (например, 0,2 мкм мебраны). Терапевтические компоненты фармацевтической композиции обычно помещаются в контейнер со стерильным входным отверстием, например, мешок для внутривенного раствора или пузырек с пробкой, которая может прокалываться иглой для подкожной инъекции.
Компоненты фармацевтической композиции обычно будут храниться в контейнерах единичной дозы или множественных доз, например, в герметично запаянных ампулах или пузырьках, в виде водного раствора или в виде лиофилизированного препарата для восстановления. В качестве примера получения лиофилизированного препарата пузырьки объемом 10 мл заполняют 5 мл стерильно отфильтрованного 1% (масс./об.) водного раствора и полученную смесь лиофилизируют. Раствор для вливания получают восстановлением лиофилизированного(ых) соединения(й) с использованием бактериостатической воды для инъекции.
Настоящее изобретение относится также к способу лечения или предотвращения заболевания, связанного с агрегацией белков, и/или нейродегенеративного заболевания, указанный способ включает введение пациенту, нуждающемуся в таком введении, терапевтически эффективного количества соединения согласно настоящему изобретению.
Термин «терапевтически эффективное количество», когда используется в данном описании, относится к количеству, достаточному для достижения желательного биологического ответа. В настоящем изобретении желательным биологическим ответом является ингибирование агрегации белка.
Настоящее изобретение относится также к способу идентификации соединения, обладающего повышенной эффективностью ингибирования агрегации белка, вовлеченного в заболевание, связанное с агрегацией белков, и/или нейродегенеративное заболевание, указанный способ включает стадии (а) контактирования меченого мономерного белка и иным образом меченного агрегата указанного белка в присутствии (1) и/или в отсутствие (2) потенциального ингибитора агрегации, который представляет собой производное соединения, которое определено выше; (b) определение количества расположенных вблизи меток, отображающее степень связывания мономерного белка с агрегатом указанного белка; и (с) сравнение результата, полученного в присутствии и в отсутствие указанного соединения, где уменьшение расположенных вблизи меток в присутствии указанного соединения является показателем способности соединения ингибировать агрегацию указанного белка.
Термин «мономерный белок» относится к молекулярной единице, состоящей из единственной (поли)пептидной цепи с трехмерной конформацией, специфической для каждого конкретного белка, который предпочтительно является растворимым в водных растворах обычно до наномольных, микромольных или миллимольных концентраций и может модифицироваться посредством ковалентного связывания одного или нескольких углеводов, углеводных производных, липидов, фосфата, сульфата, жирных кислот и нуклеотидов с отдельными аминокислотами в цепи. Предпочтительно, указанная модификация представляет собой фосфорилирование, гликозилирование, протеолитический процессинг, гликацию, окисление и нитрование. Термин «(поли)пептид», когда используется в данном изобретении, относится к группе молекул, которая включает группу пептидов, состоящих из до 30 аминокислот, а также группу полипептидов, состоящих из более 30 аминокислот. Термин «белок», когда используется в данном изобретении, относится также к (поли)пептидам.
Термин «агрегированный белок» означает нековалентно связанные олигомеры или мультимеры одного или нескольких типов «мономерного(ых) белка(ов) или полипептида(ов)», как определено выше, которые характеризуются измененной трехмерной конформацией комплексных единиц белка относительно мономерных единиц белка и обычно низкой растворимостью комплексов в водных растворах.
Термин «соединение для ингибирования агрегации белка» относится к соединению, которое способно предупреждать образование скоплений белка и/или которое способно дезинтегрировать или разрушать существующие скопления белка, где указанные соединения получены из соединений согласно изобретению дериватизацией. Предпочтительно, такие соединения разрабатываются компьютерным моделированием, где термин «компьютерное моделирование» означает применение средств виртуального скрининга для поиска соединений, которые связываются с мономерной или агрегированной формой белка или с обеими формами. Обычно эти способы основаны на трехмерной структуре белков, предпочтительно белков, кристаллизованных вместе с субстратом. Более предпочтительно, субстрат замещают потенциальным модулятором или ингибитором.
Термин «меченый … белок» относится к белку, к которому присоединена метка. Указанная метка может присоединяться непосредственно или опосредованно. Опосредованное мечение, в частности, относится к меченым (поли)пептидам, точнее к меченым антителам. Присоединение метки может осуществляться рядом методов, известных специалисту данной области техники, и описано в стандартных учебниках (см., например, Harlow and Lane "Antibodies, A Laboratory Manual", CSH Press, Cold Spring Harbor, 1998).
Термин «различным образом меченный белок» означает, что к агрегированной и мономерной изоформе белка присоединены разные метки. Типичным примером является присоединение «FITC» к агрегированному белку и «Texas red» к мономерному белку. Поскольку эти метки обнаруживаемы при различных длинах волн света, можно определить количество и/или расположение изоформ белка. Точнее, применение различных меток позволяет количественно определять наличие близко расположенных меток, т.е. меток, которые обнаружены в непосредственной близости друг от друга.
«Определение количества близко расположенных меток» может осуществляться, например, раздельным измерением (т.е. специфическим для данной длины волны) числа одиночных фотонов, по меньшей мере, двух различных длин волн, приходящих от одного и того же малого элемента объема, обычно менее 1 фемтолитра, образца в течение очень короткого промежутка времени, обычно менее 100 мкс, с последующим компьютеризированным сравнением соответствующего числа фотонов, которое может быть представлено графически на многомерной гистограмме с одной осью для числа фотонов одной конкретной длины волны. Таким образом, в случае двух длин волн число фотонов в конкретный период времени может быть представлено в виде одиночных точек в двухмерной гистограмме интенсивности флуоресценции.
Термин «сравнение результата, полученного в присутствии и в отсутствие указанного соединения» означает оценку влияния соединения на образование и/или количество скоплений белков. В данном описании снижение количества расположенных в тесной близости меток более чем на 10%, более предпочтительно более чем на 25%, еще более предпочтительно более чем на 50% и наиболее предпочтительно более чем на 95%, в присутствии соединения, которое является потенциальным ингибитором агрегации, является показателем способности соединений ингибировать агрегацию белка. Термин «отсутствие указанного соединения» означает, что ингибитор или потенциальный ингибитор не был добавлен в агрегирующийся белок. В особых случаях он может применяться для добавления отрицательного контроля, т.е. соединений, которые не обладают способностью влиять на агрегацию белка. Термин «отсутствие указанного соединения» относится также к таким случаям. Аналогично, любое из соединений настоящего изобретения, которые ингибируют агрегацию белка, может применяться в качестве положительного контроля в анализах для идентификации новых ингибиторных соединений. Очевидно, что термин «присутствие» также относится к качеству. По понятным причинам, соединения, упомянутые в настоящем изобретении, имеют различные эффективные концентрации. Предпочтительно, эффективные концентрации составляют менее 100 мкМ, более предпочтительно менее 10 мкМ, еще более предпочтительно менее 1 мкМ.
Способ согласно настоящему изобретению особенно применим для идентификации новых соединений, способных с высокой эффективностью препятствовать агрегации белка. Это позволяет осуществлять скрининг больших библиотек производных соединений и дает возможность с высокой точностью идентифицировать соединения, обладающие ингибиторной активностью. В соответствии с одним аспектом настоящего изобретения способ основан на флуоресцентной корреляционной спектроскопии. В последние годы флуоресцентная корреляционная спектроскопия (FCS) была признана в качестве метода, который дает возможность осуществлять высокочувствительный анализ агрегации белка на молекулярном уровне при нейродегенеративных заболеваниях, таких как прионные болезни (Bieschke and Schwille 1997, Bieschke et al. 2000, Giese et al. 2000, Post et al. 1998). Кроме того, FCS все более миниатюризируется и автоматизируется и становится методом, адаптированным для высокопроизводительного скрининга в фармацевтической промышленности (Koltermann et al. 1998). Флуоресцентная корреляционная спектроскопия в ее конфокальной форме анализирует флуктуации сигнала, вызываемые диффузией одиночных флуоресцентно меченных молекул через открытый элемент объема, определенный лучом лазера возбуждения, фокусируемого через светосильный объектив микроскопа и конфокально отображаемого на счетном детекторе единичных фотонов (Schwille et al. 1997). В наиболее предпочтительном варианте осуществления изобретения способ согласно настоящему изобретению основан на этой технологии. Данный способ подходит для высокопроизводительного скрининга, основанного на ингибировании, например, связывания PrPC в скопления PrPSc или образования олигомеров или протофибрилл либо фибрилл α-синуклеина.
Данная система испытания для выявления ингибиторов агрегации белков может применяться для поиска новых терапевтических средств для лечения нейродегенеративного заболевания, которое связано с агрегации специфических белков, такого как болезнь Альцгеймера и болезнь Паркинсона. Кроме того, это должно позволить осуществлять скрининг потенциальных терапевтических средств для лечения всех заболеваний, в патогенезе которых важную роль играет образование мультимера безотносительно природы его компонентов.
В предпочтительном варианте осуществления настоящего изобретения указанными метками являются флуоресцентные метки.
Предпочтительно, метка выбрана из группы, включающей флуорохром, например изотиоцианат флуоресцеина (FITC), родамин, Texas Red, Alexa 488, Alexa 647, фикоэритрин, аллофикоцианин, 6-карбоксифлуоресцеин (6-FAM), 2',7'-диметокси-4',5'-дихлор-6-карбоксифлуоресцеин (JOE), 6-карбокси-Х-родамин (ROX), 6-карбокси-2',4',7',4,7-гексахлорфлуоресцеин (НЕХ), 5-карбоксифлуоресцеин (5-FAM) или N,N,N',N'-тетраметил-6-карбоксиродамин (TAMRA), радиоактивные метки, например32P,35S,3H; и т.д. Метка также может представлять собой систему, состоящую из двух частей, где, например, белок или (поли)пептид либо соединение согласно настоящему изобретению конъюгирован(о) с биотином, гептенами и т.д., обладающими высоким сродством к связывающему партнеру, например, адивину, специфическим антителам и т.д., где связывающий партнер конъюгирован с обнаруживаемой меткой.
В другом предпочтительном варианте осуществления настоящего изобретения указанные метки присоединены к антителу или фрагменту антитела, специфически связанному с указанным белком.
Термин «специфическое связывание» в применении к антителам может быть описан, например, исходя из их перекрестной реактивности. Предпочтительно, термин «антитело, специфически связанное с …», относится к антителам, которые не связывают (поли)пептиды, кодированные посредством агрегации белка, с идентичностью менее 98%, менее 95%, менее 90%, менее 85%, менее 80%, менее 75%, менее 70% и менее 65% (в соответствии с расчетами по методикам, известным в данной области техники). Однако антитела также могут быть описаны или специфически определены по их сродству к связыванию. Предпочтительно, сродство к связыванию включает сродство к связыванию, константа диссоциации которого или Kd составляет менее 5×10-6M, 10-6M, 5×10-7M, 10-7M, 5×10-8M, 10-8M, 5×10-9M, 10-9M, 5×10-10M, 10-10M, 5×10-11M, 10-11M, 5×10-12M, 10-12M, 5×10-13M, 10-13M, 5×10-14M, 10-14M, 5×10-15M и 10-15M.
Термин «антитело» относится к поликлональным, моноклональным, химерным, «гуманизированным» антителам, антителу с одиночной цепью, антителу с одиночной Fv цепью, антителу или фрагментам антител, подобным, inter alia, Fab фрагментам. Фрагменты антител или их производные включают также фрагменты F(ab')2, Fv или scFv (см., например, Harlow and Lane (1988), (1999), loc. cit.). В данной области техники известны различные методики, которые могут применяться для получения таких антител и/или фрагментов. Таким образом, производные (антител) могут быть получены с помощью пептидомиметиков. Далее, методики, описанные для получения антител с одиночной цепью (см., inter alia, патент США № 4946778), также могут адаптироваться для получения антител с одиночной цепью, специфических для полипептида(ов) и гибридных белков данного изобретения. Кроме того, для экспрессии гуманизированных антител, специфических для полипептидов и гибридных белков согласно настоящему изобретению, могут использоваться трансгенные животные. Наиболее предпочтительно, антитело согласно данному изобретению представляет собой моноклональное антитело. Для получения моноклональных антител может использоваться любая методика, которая предоставляет антитела, полученные с помощью культур стабильной клеточной линии. Примеры таких методик включают метод гибридомы (Köhler and Milstein Nature 256 (1975), 495-497), метод триомы, метод гуманизированной В-клеточной гибридомы (Kozbor, Immunology Today 4 (1983), 72) и метод EBV-гибридомы для получения моноклональных антител человека (Cole et al., Monoclonal Antibodies and Cancer Therapy, Alan R. Liss, Inc. (1985), 77-96). Поверхностный плазменный резонанс, который используется в BIAcore системе, может применяться для повышения эффективности антител против бактериального вируса, которые связывают эпитоп полипептида согласно изобретению (Schier, Human Antibodies Hybridomas 7 (1996), 97-105; Malmborg, J. Immunol. Methods 183 (1995), 7-13). Контекстом данного изобретения также предусмотрено, что термин «антитело» включает генно-инженерный конструкт антитела, который может быть экспрессирован в клетках, например, конструкт антитела, который может быть трансфектирован и/или преобразован через, помимо прочих, вирусы или плазмидные векторы. Квалифицированному специалисту известно, что во многих случаях антитела могут быть заменены другими специфически связывающими соединениями, такими как пептиды, экспозированные на поверхности бактериального вируса (визуальное изображение бактериального вируса), или изолированными (поли)пептидами. Антитело или (поли)пептиды могут быть немечеными или меченными любой из меток, описанных в данном изобретении. Предпочтительно, антитела могут быть получены из человека, мыши, крысы, козы или кролика.
В более предпочтительном варианте осуществления настоящего изобретения указанное антитело способно различать агрегированный и мономерный белок.
Термин «способное различать» относится к антителу, которое специфично к мономерной или агрегированной изоформе белка. Предпочтительно, значение Kd указанного антитела в 5 раз превышает значение Kd изоформы белка, более предпочтительно в 10 раз. Следовательно, указанное антитело способно связываться с одной изоформой белка, в то время как по существу не связывается с другой изоформой.
В другом предпочтительном варианте осуществления настоящего изобретения количество метки, находящейся в непосредственной близости, определяется с использованием способа «сканирования интенсивно флуоресциирующей метки (SIFT)» (Bieschke et al. 2000), FRET или конфокальная визуализация высокого разрешения.
Предпочтительно, указанная конфокальная визуализация высокого разрешения осуществляется с помощью лазерного сканирующего конфокального микроскопа или с помощью микроскопа, в котором используется технология спиннинг-диска.
В еще одном предпочтительном варианте осуществления настоящего изобретения указанные мономерные и агрегирующиеся белки выбраны из группы, включающей прионный белок, амилоидный белок-предшественник (АРР), альфа-синуклеин, супероксиддисмутазу, тау-белок, иммуноглобулин, амилоид-А, транстиретин, бета-2-микроглобулин, цистатин С, аполипопротеин А1, TDP-43, островковый амилоидный полипептид, ANF, гельзолин, инсулин, лисозим, фибриноген, хантингтин, атаксин и другие белки с поли-Q-последовательностью, а также фрагменты или производные указанных белков. В предпочтительном варианте осуществления изобретения мономерные и агрегирующиеся белки выбраны из группы, включающей амилоидный белок-предшественник (АРР) и альфа-синуклеин. В более предпочтительном варианте осуществления изобретения мономерные и агрегирующиеся белки представляют собой альфа-синуклеин.
Предпочтительно, указанные белки с поли-Q-последовательностью представляют собой белки, содержащие по меньшей мере 36 консекутивных глутаминовых остатков. Более предпочтительно, указанные белки c с поли-Q-последовательностью выбраны из группы, включающей хантингтин и атаксин.
Предпочтительно, указанные фрагменты или производные выбраны из группы, модифицированной фосфорилированием, гликозилированием, протеолитическим процессингом, гликацией, окислением и нитрованием. (Поли)пептиды, упомянутые в данном изобретении, могут содержать один или несколько углеводов, производных углеводов, липидов, фосфат, сульфат, жирные кислоты и нуклеотиды, присоединенные к отдельным аминокислотам в цепи. Предпочтительно, указанная модификация представляет собой фосфорилирование, гликозилирование, протеолитический процессинг, гликацию, окисление и нитрование.
Белок может представлять собой белок позвоночного или беспозвоночного животного. Предпочтительно, белок представляет собой белок млекопитающего или птичий белок. Более предпочтительно, белок млекопитающего выбран из белка примата, человека, мыши, крысы, крупного рогатого скота, свиньи или овцы. В особых случаях может быть предпочтительно применение смешанных изоформ, т.е., например, агрегированной формы PrPSc, полученной от человека, и мономерной формы, полученной от мыши.
Белки могут быть выделены из животного или из культуры ткани либо могут быть получены рекомбинантно. Заявителями настоящего изобретения предусмотрено, что белки могут быть химически модифицированы или обработаны ферментами, такими как протеазы или гликозидазы, для облегчения работы с ними в аналитической системе.
В другом более предпочтительном варианте осуществления настоящего изобретения мономерный белок представляет собой прионный белок и агрегированный белок представляет собой PrPSc (Prusiner, 1998).
В еще одном более предпочтительном варианте осуществления настоящего изобретения мономерный белок представляет собой альфа-синуклеин и указанный агрегированный белок выбран из группы, включающей олигомеры, протофибриллы или фибриллы альфа-синуклеина.
Настоящее изобретение относится также к способу выявления соединений с улучшенной in vivo эффективностью в лечении заболеваний, связанных с агрегацией белков, и/или нейродегенеративных заболеваний, указанный способ включает (а) введение исследуемого соединения, которое представляет собой производное соединения, как определено в данном изобретении, в клеточную культуру или животному с изоформой белка, которая может подвергаться агрегации, как определено в данном изобретении; (b) определение количества обнануживаемых скоплений; и (с) идентификацию соединения, которое способно уменьшить скопления или образование скоплений указанного белка.
Данный способ согласно настоящему изобретению позволяет тестировать соединения-кандидаты in vivo, т.е. в клетках внутри живого организма, или вне живого организма. Соединения-кандидаты, тестируемые in vivo, представляют собой, например, соединения, представленные в примерах (см. ниже). Тестирование соединений-кандидатов in vivo дает важную дополнительную информацию, включая данные, касающиеся токсичности, стабильности в присутствии сложной химической окружающей среды и способности достигать месторасположения, где достигается желательное молекулярное действие.
Предпочтительно, соединение вводится в различных концентрациях для определения концентрации, при которой может быть достигнуто действие на агрегацию белка, и для вычисления значения ЕС50, которая представляет собой (молярную) концентрацию соединения, которая обеспечивает 50% от максимально возможного ответа для данного соединения.
Настоящее изобретение относится также к применению соединения, как определено выше, для ингибирования агрегации белка в организме животного, in vitro или ex vivo.
В предпочтительном варианте осуществления изобретения животное не является человеком.
Настоящее изобретение относится также к фармацевтической или диагностической композиции, включающей соединение согласно изобретению и, необязательно, фармацевтически приемлемые носитель или наполнители.
В соответствии с настоящим изобретением термин «диагностическая композиция» относится к композициям для диагностики отдельных пациентов на их возможный ответ на фармацевтически приемлемую композицию или возможность лечения с помощью фармацевтических композиций согласно настоящему изобретению. Термин «диагностическая композиция» относится также к композициям для определения наличия агрегированных белков, которые составляют основу заболеваний, указанных выше. Диагностическая композиция согласно изобретению включает соединения, указанные выше. Диагностическая композиция может дополнительно включать подходящие буферные добавки, ферменты, такие как обратная транскриптаза, термостабильные полимеразы и т.д. Диагностические композиции могут расфасовываться в контейнер или множество контейнеров.
В более предпочтительном варианте осуществления изобретения эффективность указанного соединения дополнительно повышена дериватизацией.
Термин «дериватизация» в соответствии с настоящим изобретением относится к получению химически родственных соединений, которые содержат модификации по меньшей мере в одной части молекулы.
В предпочтительном варианте осуществления настоящего изобретения указанное соединение является обнаруживаемым или помечено обнаруживаемой меткой. В соответствии с настоящим изобретением понятно, что соединение является обнаруживаемым или помечено обнаруживаемой меткой, если его присутствие может контролироваться стандартными методами, такими как ЯМР спектроскопия, оптическое детектирование, позитрон-эмиссионная томография (РЕТ), электронная микроскопия, магнитно-резонансная интроскопия (MRI), спектрометрия, хроматография, ELISA анализ, обнаружение радиоактивного излучения, предпочтительно сцинтилляционным подсчетом или гамма-подсчетом, предпочтительно РЕТ.
Когда соединения согласно настоящему изобретению предназначены для применения в качестве зондов для визуализации агрегированных белков, в частности амилоидных скоплений, они должны быть мечеными. Специфическая природа метки будет зависеть от способа, который должен использоваться для визуализации. Обычно могут применяться радиоактивные метки, которые испускают позитроны (PET) и имеют короткий период полураспада, такие как18F111C,125I,123I,131I,77Br и76Br, в особенности,18F и11C. Ввиду короткого периода полураспада меченые соединения согласно настоящему изобретению должны быть получены непосредственно перед их применением для тестирования. Следовательно, диагностическая композиция согласно настоящему изобретению может также предоставляться в форме набора, включающего предшественники соединений согласно настоящему изобретению, которые взаимодействуют с получением целевого соединения. Такой набор особенно подходит, если соединение согласно настоящему изобретению содержит по меньшей мере один фрагмент, представляющий собой Х, Y или L, который, в свою очередь, представляет собой -N(R4)-, где R4включает обнаруживаемую метку.
В предпочтительном варианте осуществления настоящего изобретения соединение, которое используется для визуализации, содержит фрагмент -N(R4)- в качестве X, Y или L, где R4 представляет собой группу -С1-4 алкилен-галоген, где атом галогена является радиоактивным. В другом предпочтительном варианте осуществления настоящего изобретения соединение, которое используется для визуализации, содержит фрагмент -N(R4)- в качестве X, Y или L, где R4 представляет собой -С1-4 алкил, содержащий, по меньшей мере, один11С изотоп.
Квалифицированный специалист сможет разработать способы, с помощью которых обнаруживаемая метка может присоединяться к соединениям согласно настоящему изобретению. Приведенные далее схемы могут служить в качестве иллюстративных примеров.
2-[18F]Фторэтилтозилат 2 применим в качестве предшественника для введения18F (период полураспада 109,8 мин.) через фторэтилирование соединений, включающих кислород-, серо- и азотсодержащий нуклеофилы, или через реакции метилирования, опосредуемые различными металлами (R. Schirrmacher et al., J. Label. Compd. Radiopharm. 2002, 45, 763-774). 2-[18F]фторэтилтозилат может синтезироваться посредством двухстадийного синтеза прямым нуклеофильным замещением этиленгликоль-1,2-дитозилата 1 с добавлением K[18F]/Kryptofix 2.2.2 для получения агента18F-фторэтилирования 2. Нерадиоактивный реагент 2 может применяться для синтеза нерадиоактивных соединений sery363A, sery363B и sery388B в соответствии со схемой А. Такие же условия могут применяться для синтеза радиоактивных аналогов указанных соединений.
Схема А

Может также применяться излучатель позитронов11С (период полураспада 20,38 мин.), который может вводиться через [11C]метилйодид (J. Eriksson et al., J. Label. Compd. Radiopharm. 2006, 49, 1177-1186) с помощью нуклеофильного замещения такого же типа. Нерадиоактивный метилйодид использовался для синтеза нерадиоактивного sery392A и sery392B в соответствии со схемой В. Такие же условия могут применяться для синтеза радиоактивных аналогов указанных соединений.
Схема В

Несмотря на то, что предпочтительно присоединять обнаруживаемый фрагмент к циклу D, поскольку такие соединения особенно легко синтезировать, это не является необходимым условием. В равной степени можно получить соединения согласно настоящему изобретению, в которых обнаруживаемый фрагмент находится в другом положении в молекуле.
Настоящее изобретение предоставляет способ визуализации агрегированного белка, который включает следующие стадии:
(i) введение субъекту обнаруживаемого количества композиции, включающей обнаруживаемо меченное соединение согласно настоящему изобретению;
(ii) предоставление периода времени, достаточного для ассоциирования соединения с агрегированным белком;
(iii) обнаружение соединения, связанного с агрегированным белком.
В предпочтительном варианте осуществления способа визуализации агрегированный белок выбран из группы, включающей амилоидный белок-предшественник (АРР) и альфа-синуклеин. В более предпочтительном варианте осуществления агрегированный белок представляет собой альфа-синуклеин.
Композиция, включающая обнаруживаемо меченное соединение, может вводиться субъекту посредством любых способов введения, описанных выше, таких как, например, пероральное или парентеральное введение. Меченое соединение может вводиться пациенту и, после истечения промежутка времени, достаточного для того, чтобы соединение стало связанным с агрегированным белком, меченое соединение обнаруживается внутри пациента атравматичным способом. Альтернативно, меченое соединение может вводиться пациенту, далее предоставляется промежуток времени, достаточный для того, чтобы соединение стало связанным с агрегированным белком, затем у пациента берется образец ткани и меченое соединение обнаруживается в ткани отдельно от пациента. Образец ткани может также удаляться из организма пациента перед введением меченого соединения в образец ткани. По истечении времени, достаточного для того, чтобы соединение стало связанным с агрегированным белком, соединение может быть обнаружено.
Способы обнаружения меченого соединения, связанного с агрегированным белком, хорошо известны в данной области техники и включают, но без ограничения, магнитно-резонансную интроскопию (MRI), позитрон-эмиссионную томографию (РЕТ) или компьютерную томографию эмиссии одиночного позитрона (SPECT) для обнаружения радиоактивно меченных соединений. Метка, которая вводится в соединение, зависит от способа ее обнаружения, который должен применяться. Таким образом, например, если в качестве способа обнаружения выбрана PET, соединение должно содержать позитрон-испускающий атом, такой как11С или18F.
Визуализация агрегированных белков может также осуществляться таким образом, что может быть определено и количество агрегированного белка.
В предпочтительном варианте осуществления изобретения указанное заболевание, связанное с агрегацией белков, характеризуется присутствием агрегированных форм по меньшей мере одного белка, его фрагмента или его производного, где белок выбран из группы, включающей прионный белок, амилоидный белок-предшественник (АРР), альфа-синуклеин, супероксиддисмутазу, тау-белок, иммуноглобулин, амилоид-А, транстиретин, бета-2-микроглобулин, цистатин С, аполипопротеин А1, TDP-43, островковый амилоидный полипептид, ANF, гельзолин, инсулин, лизозим, фибриноген, хантингтин, атаксин и другие белки с поли-Q-последовательностью. В предпочтительном варианте осуществления изобретения белок выбран из группы, включающей амилоидный белок-предшественник (АРР) и альфа-синуклеин. В более предпочтительном варианте осуществления изобретения белок представляет собой альфа-синуклеин.
Предпочтительно, указанные белки с поли-Q-последовательностью представляют собой белки, содержащие, по меньшей мере, 36 консекутивных глутаминовых остатков. Более предпочтительно, указанные белки с поли-Q-последовательностью выбраны из группы, включающей хантингтин и атаксин.
Специалисту данной области техники известно, что указанные белки могут существовать в различных изоформах, включая белки, модифицированные фосфорилированием, гликозилированием, протеолитическим процессингом и т.д. Термин «по меньшей мере, один …» относится к тому факту, известному специалисту данной области, что заболевания могут быть связаны с присутствием более чем одного белка в агрегированной форме. Например, при болезни Альцгеймера обычно могут быть обнаружены скопления фрагментов амилоидного белка-предшественника (АРР) и скопления тау-белка.
В данном описании термин «нейродегенеративные болезни» включает такие болезни, как болезнь Паркинсона, прионная болезнь, болезнь Альцгеймера, множественная системная атрофия, болезнь диффузных телец Леви, лобно-височное слабоумие, боковой амиотрофический склероз, болезнь Хантингтона, атаксия, связанная с заболеваниями спинного мозга и мозжечка, и другие поли-Q-заболевания, наследственная церебральная амилоидная ангиопатия, наследственная амилоидная полиневропатия. Кроме того, термин «заболевание, связанное с агрегацией белка» относится к заболеваниям, проявляющимся, главным образом, вне нервной системы, и включает такие заболевания как первичный системный амилоидоз (AL амилоидоз), реактивный системный амилоидоз (AA амилоидоз), диабет II типа, инъекция-локализованный амилоидоз, бета-2-микроглобулиновый амилоидоз, наследственный не невропатический амилоидоз и наследственный финский системный амилоидоз.
В другом предпочтительном варианте осуществления изобретения заболевание выбрано из группы, включающей болезнь Паркинсона, прионную болезнь, болезнь Альцгеймера, множественную системную атрофию, болезнь диффузных телец Леви, лобно-височное слабоумие, боковой амиотрофический склероз, болезнь Хантингтона, атаксию, связанную с заболеваниями спинного мозга и мозжечка, и другие поли-Q-заболевания, наследственную церебральную амилоидную ангиопатию, наследственную амилоидную полиневропатию, первичный системный амилоидоз (AL амилоидоз), реактивный системный амилоидоз (AA амилоидоз), диабет II типа, инъекция-локализованный амилоидоз, бета-2-микроглобулиновый амилоидоз, наследственный не невропатический амилоидоз и наследственный финский системный амилоидоз. В более предпочтительном варианте осуществления изобретения заболевание представляет собой болезнь Паркинсона.
В предпочтительном варианте осуществления изобретения указанная прионная болезнь выбрана из болезни Крейтцфельдта-Якоба, варианта болезни Крейтцфельдта-Якоба, наследственной прионной болезни человека, коровьей губчатой энцефалопатии (BSE) и скрейпи.
И наконец, настоящее изобретение относится к набору, включающему соединение, как определено в данном изобретении, и, дополнительно антитело или фрагмент антитела, специфически связывающиеся с указанным соединением; и/или мономерный или агрегированный белок, как определено в настоящем изобретении; и/или мономерный или агрегированный белок, как определено в настоящем изобретении, необязательно связанный в комплекс с указанным соединением, и инструкции для применения - в одном или нескольких контейнерах.
ОБЩИЕ МЕТОДИКИ ЭКСПЕРИМЕНТОВ
Соединения согласно настоящему изобретению могут быть получены с помощью любой из подходящих методик органического синтеза. Многие из таких методик подробно описаны в L.F. Tietze, Th. Eicher "Reaktionen und Synthesen", 2. Auflage (Georg Thieme Verlag, Stuttgart, NY, 1991), T.W. Greene, P.G.M. Wuts "Protective Groups in Organic Synthesis", Third Edition (John Wiley & Sons, NY, 1999), а также в J. March "Advanced Organic Chemistry", Third Edition (John Wiley & Sons, NY, 1985).
Ряд типичных примеров способов получения соединений согласно изобретению представлен ниже. Данные способы предназначены только для иллюстрации сущности таких методик получения и не предназначены для ограничения области подходящих способов.
Обычно условия реакций, такие как температура, продолжительность реакции, растворители, способы обработки и т.п., являются стандартными в данной области техники для конкретной реакции, которая должна осуществляться. Указанные ссылки вместе с их содержанием включают подробные описания таких условий. Обычно температура будет находиться в интервале от -80°C до 150°C, растворители будут апротонными и протонными и продолжительность реакций будет находиться в интервале от 10 секунд до 10 дней. Обработка обычно включает гашение любых непрореагировавших реагентов с последующим распределением между системой вода/органический слой (экстракция) и отделением слоя, содержащего продукт.
Стандартные методики синтеза, такие как применение безводных условий реакции (например, среда инертного газа), являются традиционными для данной области техники и будут применяться, когда это возможно.
Модификация каждой из представленной ниже схем приводит к различным аналогам специфических примеров соединений, полученных ниже. Пояснения, приведенные ниже и описывающие подходящие способы органического синтеза, применимы к таким модификациям.
В каждом из приведенных ниже типичных примеров схем может быть полезно выделять продукты реакции и/или отделять их от исходных веществ. Целевые продукты каждой стадии отделяются и/или очищаются (далее - выделяются) до желательной степени однородности с помощью методик, традиционных для данной области техники. Обычно такие способы выделения включают многофазные экстракции, кристаллизацию из растворителя или смеси растворителей, отгонку, возгонку или хроматографию. Хроматография может включать любое число методов, включая, например, эксклюзионную хроматографию или ионообменную хроматографию, хроматографию высокого, среднего или низкого давления, хроматографию малых образцов или препаративную тонкослойную хроматографию, а также методы тонкослойной хроматографии малых образцов и флэш-хроматографии.
Другой класс способов выделения включает обработку смеси реагентом, выбранным для связывания или для придания целевому продукту, непрореагировавшему исходному веществу или побочному продукту и т.д. других свойств, позволяющих его выделить. Такие реагенты включают адсорбенты, такие как активированный уголь, молекулярные сита, ионообменную среду и т.п. Альтернативно, реагенты могут представлять собой кислоты в случае основного материала, основания в случае кислотного материала, связывающие реагенты, такие как антитела, связывающие белки, селективные хелатообразующие вещества, такие как простые краун-эфиры, жидкостная/липидная ионная экстракция и т.п.
Выбор подходящих способов выделения зависит от природы задействованных материалов, например, температуры кипения и молекулярной массы при отгонке и возгонке, наличия или отсутствия полярных функциональных групп при хроматографии, стабильности материалов в кислотной или основной средах при многоступенчатой экстракции и т.п. Специалист данной области сможет применить методики, наиболее подходящие для достижения желательного выделения.
В частности, соединения согласно настоящему изобретению могут быть получены способом, аналогичным методикам, которые описаны, например, в M. Ono et al. (Bioorganic & Medicinal Chemistry 16 (2008) 6867-6872), WO 2008/131148, WO 2004/080972, WO 2004/072050, WO 98/17652. Альтернативные пути также рассмотрены в разделе Примеры согласно настоящему изобретению.
Приведенные далее примеры предназначены для иллюстрации изобретения. Однако они не должны рассматриваться как примеры, ограничивающие область настоящего изобретения.
ПРИМЕРЫ
Пример 1: Идентификация нового класса соединений для ингибирования агрегации белков
Для выявления ингибиторов распространения прионного заболевания тестируют две подгруппы коммерческой библиотеки соединений DIVERSet (ChemBridge Corp., San Diego, CA, USA), каждая из которых содержит 10000 соединений, названные заявителями изобретения DIVERSet 1 и 2, тестирование проводят с использованием антиприонного анализа 2D-SIFT (Bertsch et al. 2005) и модели прионной болезни в клеточной культуре. В обоих тестах первичные ингибиторы выявляют тестированием соединений в единой концентрации с последующей проверкой активности в серии разбавлений. Дополнительно, выявленные первичные ингибиторы клеточной культуры испытывают с использованием другой клеточной линии.
2D-SIFT скрининг
Для проведения оценки ингибиторного действия лекарственных средств на скопление PrPC и PrPSC в высокопроизводительном и высокосодержательном скрининговом тесте заявители используют методику «сканирования интенсивно флуоресцентных мишеней» (SIFT), в которой применяется установка инвертированного двойного цветного конфокального микроскопа с детекторами одиночных протонов для двух цветов люминесцентного света. Образцы получают в 96- или 384-луночных титрационных микропланшетах со сдвижными стеклами-донышками. Аналитическая смесь состоит из рекомбинантного мышиного PrP (rPrP), моноклонального антитела (mAb) L42, которое не распознает мышиный PrP, но распознает человеческий PrP, и скоплений PrPSc, полученных из CJD мозга человека. Молекулы rPrP и mAb мечены зелеными и красными флуорофорами, соответственно. Связывание некоторых молекул rPrP и mAb с PrPSc скоплениями будет приводить к образованию трехчастных комплексов, проявляющих большое количество красных и зеленых присоединенных флуорофоров. Такие скопления можно идентифицировать и анализировать с помощью SIFT метода, поскольку одновременно вызывают высокие интенсивности в обоих флуоресцентных каналах. Распределение интенсивностей красной и зеленой флуоресценции можно оценивать с помощью двухмерной гистограммы интенсивности флуоресценции. В любом случае, когда в аналитической системе присутствует ингибитор ассоциирования rPrP и PrPSc, интенсивность трехчастных комплексов должна снижаться. Цветовое распределение скоплений в 2D-гистограмме после этого будет сдвигаться в направлении «красных» секторов данной гистограммы.
Первичный SIFT-скрининг 20000 соединений
Описанную выше аналитическую систему применяют к двум библиотекам, каждая из библиотек содержит 10000 различных соединений со свойствами, подобными свойствам лекарственных средств (ChemBridge; “DIVERSet1” и “DIVERSet2”) в 96-луночных титровальных микропланшетах для первичного скрининга, включающего отрицательные контроли любых дополнительных соединений и положительные контроли, содержащие 17 мкМ DOSPA, а также контроли без CJD палочек и соединений (служащие для проверки отсутствия агрегации в смеси антитела и rPrP).
Образцы, содержащие DOSPA, показывают снижение SIFT сигнала в тех секторах, которые контролируют сигналы агрегации, преимущественно меченные зеленым rPrP. Это указывает на то, что в этих контролях меньше rPrP связывается с DJD прионными палочками. Поскольку прионные палочки маркированы красными метками антитела, их флуоресценция все еще генерирует SIFT сигнал в «красных» секторах. Большинство из соединений не влияет на распределение SIFT сигнала. Но некоторые из DIVERSet соединений снижали число скоплений, обнаруженных в «зеленых» секторах. SIFT кривые таких образов сдвигаются в направлении DOSPA контролей. Таким образом, соответствующие соединения могут рассматриваться как первичные соединения-кандидаты в потенциальные антиприонные лекарственные средства. Время от времени некоторые технические проблемы приводят к артефактам, которые делают непонятным результат, полученный на всем титровальном микропланшете. Кроме того, только примерно 7% измерений, в которых не было зафиксировано изменение распределения, должны были обрабатываться как «выбросы» и не подходили для автоматизированного SIFT анализа вследствие, главным образом, внутренней флуоресценции испытываемых соединений. Такой довольно низкий процент соединений, не подлежащих обработке SIFT анализом, является неожиданным и подчеркивает универсальность и надежность данного анализа. Идентификация проблематичных измерений и соединений облегчается высокосодержательной природой данных, полученных в данном SIFT анализе. Для каждого образца одновременно записываются несколько флуоресцентных параметров, таких как, например, средние значения интенсивности флуоресценции каждого канала цветности. Они отображаются вместе с суммарными значениями событий высокой интенсивности в каждом секторе гистограммы распределения цветности. Поэтому образцы с высокой внутренней флуоресценцией могут легко исключаться из анализа.
Первичные кандидаты, полученные в SIFT анализе, и проверка их активности посредством серии разбавлений
Для соединения, которое должно классифицироваться как первичные соединения-кандидаты, заявители изобретения анализируют сумму столбцов в «зеленых» секторах от 1 до 5 и определяют значение приблизительно 50% ингибирования влияния DOSPA в сравнении с необработанным контролем как минимального необходимого эффекта. Исходя из результатов SIFT-скрининга, DIVERSet соединениям присваивают скалярные значения SIFT активности (tp1_sift), характеризующие их ингибиторное действие на прионную агрегацию, как описано в публикации Bertsch et al. 2005. Значения активности менее нуля свидетельствуют об отсутствии ингибиторного эффекта, в то время как значения больше нуля свидетельствуют об ингибиторном действии. В данном случае значения, близкие к единице, указывают на значительное ингибиторное действие, равное ингибиторному действию положительного контроля на данном микропланшете.
Зависимость ингибиторного действия от дозы каждого из соединений, выявленных в SIFT анализе, в отношении связывания PrPC-PrPSc проверяют с использованием ряда разбавлений из шести точек (0,1-100 мкМ). Кривые «доза-эффект» данных соединений подтверждают зависимость ингибиторной активности от концентрации. По сравнению в эффектом 17 мкМ DOSPA, пятидесяти процентное от максимального ингибирования связывание rPrP с прионными палочками наблюдается при значениях EC50 в интервале от 0,3 до 60 мкМ.
Первичные соединения-кандидаты в скрининге на клеточной культуре и проверка из активности посредством серий разбавлений
Помимо SIFT анализа, DIVERSet библиотеки подвергают скринингу в моделях клеточных культур прионных болезней. В указанных анализах антиприонную активность DIVERSet соединений вначале испытывают в концентрации 20 мкМ. Результаты первичного скрининга в клеточной культуре (на SMB клетках при 20 мкМ) кодируют двоичной переменной (tp_3 reduktion), где значение «0,1» кодирует «активный» образец, а значение «0,0» кодирует «неактивный». Активность соединений, идентифицированных в первичном скрининге, проверяют с целью идентифицировать соединения, активные даже при очень низких концентрациях. Верификацию первичной клеточной культуры проводят в серии разбавлении при четырех концентрациях (1 мкМ, 4 мкМ, 10 мкМ, 40 мкМ) для DIVERSet 1 и только при двух концентрациях (0,2 мкМ, 2 мкМ) для DIVERSet 2. Результаты (tp3_reduktion_mue) обозначены «1,0» для активных при 1 мкМ (DS1) или при 0,2 мкМ (DS2) и «0,5» для активных при 2 мкМ (DS2). Соединения, неактивные при концентрациях более 20 мкМ, обозначают «0,0» (DS1 + 2). Кроме того, активность первичных кандидатов, выявленных в SMB клеточной культуре, проверяют при единых концентрациях на ScN2a клетках (tp3_scn2a), где снова «0,0» и «1,0» означают неактивные и активные соединения, соответственно.
Дифенилпиразолы (DPP) и родственные соединения в качестве новых соединений с антиприонной активностью
Создание SAR карт
DIVERSet соединения, характеризованные как «активные» в первичном скрининге в клеточной культуре, подвергают кластерному анализу с использованием пакета программного обеспечения Benchware HTS DataMiner (DM; Tripos Inc., St. Louis, MO, USA) с получением SAR-карты, представленной на фиг.1. Поскольку большое число соединений, включенных в две библиотеки (20000), является слишком большим в качестве исходной точки для кластерного анализа с использованием DataMiner, анализ сначала ограничили первичными кандидатами (837 соединений) скрининга в клеточной культуре DIVERSet 1 и 2. Таким образом, кластеры определяют только для активных соединений. В данном случае DM программа группирует в кластеры структурно аналогичные и активные соединения и, следовательно, подходит для идентификации потенциальных релевантных новых ведущих структур. На второй стадии определенную таким образом классификацию применяют к остальным соединениям библиотеки, неактивным в клеточной культуре. В этом случае DM программа добавляет к гетенированным кластерам (неактивные) соединения, если применяемое измерение указывает на высокое структурное сходство.
Результаты кластерного анализа графически представляют с помощью DateMiner в форме SAR-карты, на которой кластеры соединений S обозначены символами. DM располагает символ таким образом, что они находятся в непосредственной близости друг к другу, если представленные кластеры являются структурно схожими. Размер, форма и цвет символов назначается на основании кластер-специфических свойств, которые могут быть выбраны пользователем.
Таким образом, определяют, что размер символов пропорционален размеру кластера /S/, т.е. числу соединений С, которые содержатся в нем. Форму символов определяют в соответствии с формулой

из тех соединений С в соответствующих кластерах S, первичная активность которых а(С), как определено SIFT скринингом, выше выбранного порога amin=0,25. Таким образом, кластеры, у которых PSIFT выше 50%, обозначают звездочками, в то время как остальные, главным образом неактивные кластеры, обозначают квадратом. Аналогично, цвет символов кодируется в соответствии с формулами кластеров
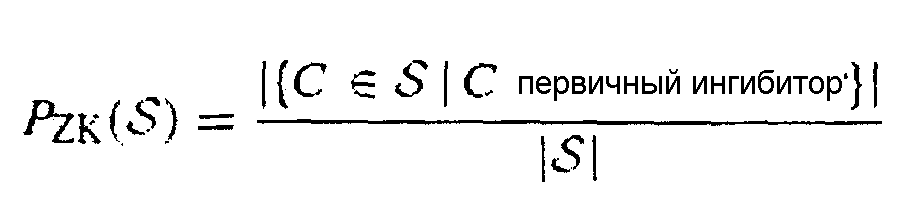
соответствующих первичным ингибиторам, выявленным в испытании в клеточной культуре, где символы кластеров с более чем 50% активных веществ являются красными, а остальные кластеры являются серыми. На фиг.1 представлена SAR-карта, полученная в результате описанного выше кластерного анализа. Большие звездочки красного цвета символизируют кластеры с высокой долей соединений, проявивших положительное действие в SIFT-анализе и анализе в культуре клеток. Эти кластеры представляют потенциально ведущие структуры. С использованием DataMiner кластеры, представляющие интерес, подвергают дополнительному анализу и идентифицируют группу, состоящую из пяти ближайших кластеров (DPP_1-DPP_5), которые показаны на фиг.1 полужирным шрифтом. Все соединения, выделенные в эти кластеры, представлены на фиг.2 вместе с их активностями, выявленными в различных анализах. Тот факт, что эти кластеры расположены в непосредственной близости друг от друга, означает, что они содержат соединения, которые являются структурно подобными, т.е. все они принадлежат к химическому классу производных 3,5-дифенилпиразола (DPP) (сравнить фрагмент DPP, представленный на фиг.1).
Пример 2. Синтез новых лекарственных средств для ингибирования агрегации с точки зрения медико-химических аспектов
Исходя из открытия новой лидирующей структуры, как описано выше, селективным замещением различных заместителей синтезируют ряд дополнительных соединений, как представлено ниже.
Схема 1: Синтез изоксазолов
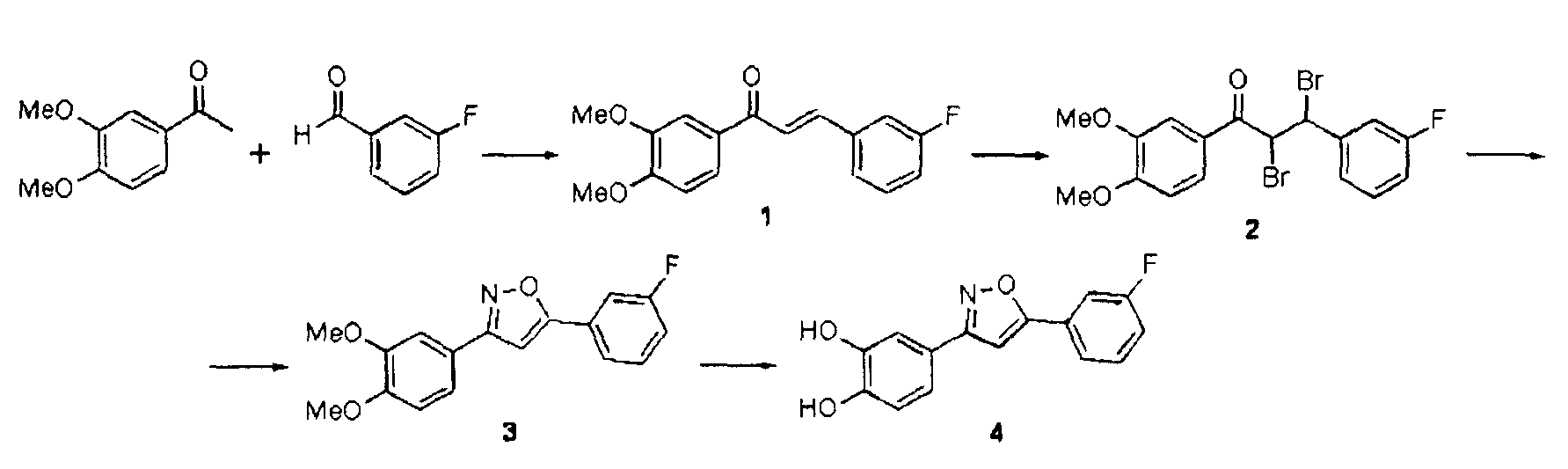
(E)-1-(3,4-диметоксифенил)-3-(3-фторфенил)-2-пропен-1-он(1) [Nam et al., 2004]
Раствор 3,4-диметоксиацетофенона (1,8 г, 10 ммоль), 3-фторбензальдегида (1,24 г, 10 ммоль), NaOH (50 мг, 1,25 ммоль) и Ba(OH)2·8H2O (100 мг, 0,32 ммоль) в метаноле (10 мл) перемешивают при комнатной температуре в течение 24 часов. Реакционную смесь охлаждают до +4°C, полученный осадок собирают фильтрацией, перекристаллизовывают из метанола и сушат, получая 1 (1,65 г, 58%) в виде порошка желтого цвета.
2,3-дибром-1-(3,4-диметоксифенил)-3-(3-фторфенил)пропан-1-он(2) [Harris et al., 1977]
К раствору 1 (715 мг, 2,5 ммоль) в хлороформе (11 мл) по каплям добавляют раствор брома (400 мг, 2,5 ммоль) в хлороформе (4 мл) при 0°C. Смесь перемешивают в течение 2 часов при 0°C, затем разбавляют петролейным эфиром (20 мл), полученную смесь замораживают (-24°C) и выдерживают при данной температуре в течение 10 часов, полученный осадок собирают фильтрацией, промывают н-гексаном (10 мл) и сушат, получая 2 (780 мг, 70%) в виде белого порошка.
3-(3,4-диметоксифенил)-5-(3-фторфенил)изоксазол(3) [Harris et al., 1977]
Раствор 2 (450 мг, 1 ммоль) в этаноле (6 мл) обрабатывают гидрохлоридом гидроксиламина (306 мг, 4,4 ммоль) и затем раствором NaOH (460 мг, 11,5 ммоль) в воде (1,5 мл). Смесь кипятят с обратным холодильником в течение 2 часов, охлаждают и обрабатывают водой (3 мл). Смесь охлаждают (4°C), оставляют на ночь при указанной температуре, продукт собирают фильтрацией, промывают водой (5 мл) и сушат, получая 3 (180 мг, 60%) в виде белого порошка.
3-(3,4-Дигидроксифенил)-5-(3-фторфенил)изоксазол (4) [Vanelle et al., 2000]
Раствор 3 (100 мг, 0,33 ммоль) в дихлорметане (5 мл) охлаждают до -78°C, обрабатывают трибромидом бора (0,16 мл, 1,7 ммоль), перемешивают при -78°C в течение 3 часов и затем в течение ночи при комнатной температуре. Смесь охлаждают до -78°C и гасят метанолом (5 мл). Смесь перемешивают в течение 3 часов при комнатной температуре, после этого растворители удаляют при пониженном давлении, остаток подвергают четыре раза совместному выпариванию с метанолом (10 мл). Полученный остаток кипятят с обратным холодильником в 5 мл хлороформа, после охлаждения продукт собирают фильтрацией и сушат, получая 4 (60 мг, 67%) в виде белого порошка.
Схема 2: Синтез пиразолов

1,3-Бис(3,4-диметоксифенил)пропан-1,3-дион (5) [Anselme, 1967]
60% суспензию гидрида натрия в минеральном масле (0,4 г, 10 ммоль) дважды промывают петролейным эфиром (20 мл), добавляют безводный ДМСО (10 мл). Смесь перемешивают в течение 30 минут при комнатной температуре в атмосфере аргона, добавляют ТГФ (5 мл), колбу охлаждают до 15°C и добавляют этил-3,4-диметоксибензоат (2,1 г, 10 ммоль). Температуре смеси дают возможность понизиться до 10°C и к смеси добавляют раствор 3,4-диметоксиацетофенона (1,08 г, 6 ммоль) в ДМСО (4 мл) с такой скоростью, чтобы температура смеси не поднималась выше 15°C. После завершения реакции реакционную смесь перемешивают в течение 72 часов при комнатной температуре, затем медленно выливают в измельченный лед (250 г), содержащий 85% фосфорную кислоту (1 мл). Полученный осадок собирают фильтрацией, промывают водой (50 мл) и сушат, получая 5 (2,1 г, 99%) в виде порошка желтого цвета.
3,5-Бис(3,4-диметоксифенил)пиразол (6) [Hauser et al., 1957]
Раствор 5 (1,0 г, 2,9 ммоль) и гидразингидрата (218 мг, 4,4 ммоль) в этаноле (15 мл) кипятят с обратным холодильником при перемешивании в течение 3 часов. Прозрачный раствор желтого цвета упаривают при пониженном давлении, добавляют воду, полученный осадок собирают фильтрацией, промывают водой и сушат, получая 6 (960 мг, 97%) в виде порошка желтого цвета.
Гидробромид 3,5-бис(3,4-дигидроксифенил)пиразола (7) [Vanelle et al., 2000]
Раствор 6 (120 мг, 0,35 ммоль) в дихлорметане (5 мл) охлаждают до -78°C, обрабатывают трибромидом бора (0,34 мл, 3,5 ммоль), перемешивают при -78°C в течение 3 часов и затем в течение ночи при комнатной температуре. Смесь охлаждают до -78°C и гасят метанолом (5 мл). Смесь перемешивают в течение 3 часов при комнатной температуре, после этого растворители удаляют при пониженном давлении, остаток подвергают четыре раза совместному выпариванию с метанолом (10 мл). Полученный остаток кипятят с обратным холодильником в 5 мл хлороформа, после охлаждения продукт собирают фильтрацией и сушат, получая 7 (108 мг, 85%) в виде порошка желтого цвета.

1-(1,3-Бензодиоксол-5-ил)-3-(3-бромфенил)пропан-1,3-дион(21) [Anselme, 1967]
1. Методика
Сухую трехгорлую колбу объемом 500 мл снабжают магнитной мешалкой с тефлоновым покрытием (Teflon®), резиновой мембраной, термометром и обратным холодильником, к которому присоединена T-образная трубка, соединенная с источником чистого азота. Оставшуюся точку присоединения T-образной трубки соединяют с барботирующим устройством таким образом, чтобы скорость потока азота могла регулироваться в процессе реакции.
Аппарат располагают таким образом, чтобы колба могла периодически охлаждаться на водяной бане. После продувки колбы азотом в колбе поддерживают постоянную атмосферу азота для реакции. В колбу загружают 60% дисперсию гидрида натрия (5 г, 0,125 моль) в минеральном масле (примечание 1). Минеральное масло вымывают из гидрида с помощью петролейного эфира 40/60 (3×40 мл) (примечание 2). Верхний слой петролейного эфира удаляют с помощью гиподермического шприца с наконечником Люэра и иглой из нержавеющей стадии, вводимой через резиновую мембрану. Остаточный гидрид натрия смешивают с 80 мл диметилсульфоксида (примечание 3) и резиновую мембрану замещают капельной воронкой, компенсирующей давление. Раствор 16,4 г (0,1 моль) 1-(1,3-бензодиоксол-5-ил)этанона (примечание 4) и 26,9 г (0,125 моль) метил-3-бромбензоата (примечание 5) в 60 мл диметилсульфоксида помещают в капельную воронку. Воронку закрывают, перемешивание начинают и содержимое колбы охлаждают на водяной бане до 15°C. К содержимому колбы медленно добавляют 1-(1,3-бензодиоксол-5-ил)этанон и метил-3-бромбензоат таким образом, чтобы выделение водорода происходило с контролируемой скоростью и температура не превышала 20°C в течение 60 минут (примечание 6). После завершения добавления баню удаляют и реакционную смесь перемешивают при комнатной температуре (23°C) в течение 15 часов. Оставшуюся гомогенную реакционную смесь красновато-коричневого цвета медленно выливают в смесь 500 мл льда и воды, содержащую 5 мл 85% ортофосфорной кислоты (примечание 7) при перемешивании. Смесь перемешивают в течение 1 часа, после чего продукт удаляют фильтрацией (примечание 8), промывают водой с использованием вакуум-фильтра (2×100 мл) и сушат в вакууме до постоянной массы при 40°C в течение 6 часов, получая 34,4 г сырого (примечание 9) продукта. Перекристаллизация из 200 мл 99,9% этанола и 200 мл этилацетата (примечание 10) и сушка в вакууме до постоянной массы при 40°C в течение 6 часов приводят к получению 28,5 г (82% выход) чистого (примечание 11) 1-(1,3-бензодиоксол-5-ил)-3-(3-бромфенил)пропан-1,3-диона, т.пл. 136-137°C, фильтрат концентрируют при пониженном давлении до объема приблизительно 30 мл, твердые кристаллы, которые выпадают в осадок, собирают фильтрацией, промывают с использованием вакуум-фильтра (2×10 мл) и сушат в вакууме до постоянной массы при 40°C в течение 6 часов, получая дополнительный выход сырого продукта. Сырой продукт дважды перекристаллизовывают из 20 мл 99,9% этанола и 20 мл этилацетата и сушат в вакууме до постоянной массы 40°C в течение 6 часов, получая дополнительные 1,8 г (5% выход) чистого 1-(1,3-бензодиоксол-5-ил)-3-(3-бромфенил)пропан-1,3-диона (примечание 12). Общий выход продукта 30,3 г (87%).
2. Примечание
1. Используют гидрид натрия в виде 57-63% масляной дисперсии, доступна под порядковым номером 13431 от Alfa Aesar GmbH & Co KG, Karlsruhe.
2. Используют петролейный эфир GR для анализа (интервал температуры кипения 40-60°C), доступен под порядковым номером 101775 от Merck KGaA, Darmstadt.
3. Используют без дополнительной очистки диметилсульфоксид GR для анализа, порядковый номер 102952 от Merck KGaA, Darmstadt.
4. Используют 1-(1,3-бензодиоксол-5-ил)этанон, 98%, порядковый номер A13597, доступен от Alfa Aesar GmbH & Co KG, Karlsruhe.
5. Используют метил-3-бромбензоат, 98%+, порядковый номер A16174, доступен от Alfa Aesar GmbH & Co KG, Karlsruhe.
6. В процессе добавления наблюдают образование пены. Применение механической мешалки и антипенного агента, такого как полиэтиленгликольдиметилэфир, может быть необходимо для повышения выхода.
7. Используют ортофосфорную кислоту, 85% масс./масс. водный раствор, GR для анализа, порядковый номер 100573 от Merck KGaA, Darmstadt.
8. Значение pH реакционной смеси равно 7. При подкислении до рН=2 добавлением 15 мл ортофосфорной кислоты может быть получено 1,3 г 3-бромбензойной кислоты.
9. Чистота продукта, согласно ВЭЖХ, равна 96,3%.
10. Используют абсолютный этанол 99,9% GR для анализа, порядковый номер 100983, и этилацетат GR для анализа, порядковый номер 109623 от Merck KGaA, Darmstadt.
11. Чистота продукта, согласно ВЭЖХ, равна 99,3%. Аналитическую ВЭЖХ проводят с использованием системы ВЭЖХ Waters с фотодиодным детектором Waters 996 Photodiode Array Detector. Все разделения включают применение подвижной фазы 0,1% об./об. трифторуксусной кислоты (ТФУК) в воде (растворитель A) и 0,1% об./об. ТФУК в ацетонитриле (растворитель B) с применением колонки с обращенной фазой (ОФ) Eurospher RP 18, 100 Å, 5 мкм, 250×4,6 мм при скорости истечения 1 мл/мин. Соединение растворяют в ацетонитриле GR для ВЭЖХ при концентрации 1 мг/мл. Пики со значениями времени удерживания (RT) 20,9 и 10,3 мин соответствуют енольной и кето-формам, соответственно, ANLE 138A. Повторный впрыск отдельно собранных пиков 20,9 или 10,3 мин снова дает такие же два пика с идентичными значениями времени удерживания (RT).
12. Чистота продукта, согласно ВЭЖХ, равна 98,4%.
3-(1,3-Бензодиоксол-5-ил)-5-(3-бромфенил)-1H-пиразол (22) [Hauser et al., 1957]
1. Методика
Смесь 28,4 г (81,8 ммоль) 1-(1,3-бензодиоксол-5-ил)-3-(3-бромфенил)пропан-1,3-диона (примечание 1) и 200 мл н-бутилового спирта (примечание 2) помещают в круглодонную колбу объемом 500 мл, снабженную магнитной мешалкой с тефлоновым покрытием (Teflon®), обратным холодильником и нагревательной рубашкой с электрическим подогревом. Начинают перемешивание и нагрев, добавляют 6 мл (6,2 г, 123,4 ммоль) гидразина моногидрата (примечание 3) с растворением твердых веществ и реакционную смесь кипятят с обратным холодильником при перемешивании в течение 4 часов. Реакционную смесь охлаждают до 20°C, выдерживают в течение 1 часа при 0°C, выпавший в осадок продукт собирают фильтрацией с вакуум-насосом. Промывка водой (100 мл) и сушка в вакууме до постоянной массы при 40°C в течение 36 часов приводит к получению 26,8 г (95% выход) 3-(1,3-бензодиоксол-5-ил-5-(3-бромфенил)-1H-пиразола (примечание 4) т.пл. 195-197°C.
2. Примечания
1. 1-(1,3-Бензодиоксол-5-ил)-3-(3-бромфенил)пропан-1,3-дион получают в соответствии с методикой получения ANLE 138A.
2. Используют н-бутиловый спирт (99,4% GR "Baker analyzed"), порядковый номер 8017, доступен от J.T. Baker B. V., Deventer, Holland.
3. Используют гидразинмоногидрат GR purum, порядковый номер 53850, доступен от Sigma-Aldrich Chemie GmbH, Taufkirchen.
4. Чистота продукта, согласно ВЭЖХ, равна 99,3%. Аналитическую ВЭЖХ проводят с использованием системы ВЭЖХ Waters с фотодиодным детектором Waters 996 Photodiode Array Detector. Все разделения включают применение подвижной фазы 0,1% об./об. трифторуксусной кислоты (ТФУК) в воде (растворитель A) и 0,1% об./об. ТФУК в ацетонитриле (растворитель B) с применением колонки с обращенной фазой (ОФ) Eurospher RP 18, 100 Å, 5 мкм, 250×4,6 мм при скорости истечения 1 мл/мин. Соединение растворяют в ацетонитриле GR для ВЭЖХ при концентрации 1 мг/мл.
Схема 3: Синтез имидазолов

4-(3,4-Диметоксифенил)-2-фенилимидазол (8) [Li et al., 2000]
Смесь гидрохлорида бензамидина (313 мг, 2 ммоль) и бикарбоната натрия (672 мг, 8 ммоль) в ТГФ (6 мл) и воде (1,5 мл) кипятят с обратным холодильником. К смеси в течение 30 минут добавляют раствор α-бром-3,4-диметоксиацетофенона (518 мг, 2 ммоль) в ТГФ (1,5 мл) при кипячении с обратным холодильником. После завершения добавления реакционную смесь кипятят с обратным холодильником в течение 2 часов и ТГФ выпаривают при пониженном давлении. К смеси добавляют этилацетат (20 мл), органический слой отделяют, промывают насыщенным раствором соли (5 мл), сушат над сульфатом натрия и смесь упаривают при пониженном давлении. Полученный сырой продукт очищают колоночной хроматографией на силикагеле (хлороформ/метанол 100:1), получая 8 (470 мг, 84%) в виде твердого продукта.
Гидробромид 4-(3,4-дигидроксифенил)-2-фенилимидазола (9) [Vanelle et al., 2000]
Раствор 8 (190 мг, 0,68 ммоль) в дихлорметане (5 мл) охлаждают до -78°C, обрабатывают трибромидом бора (0,32 мл, 3,4 ммоль), перемешивают при -78°C в течение 3 часов и затем в течение ночи при комнатной температуре. Смесь охлаждают до -78°C и гасят метанолом (5 мл). Смесь перемешивают в течение 3 часов при комнатной температуре, растворители выпаривают при пониженном давлении, остаток подвергают четыре раза совместному выпариванию с метанолом (10 мл). Полученный остаток кипятят с обратным холодильником в 5 мл хлороформа, после охлаждения продукт собирают фильтрацией и сушат, получая 9 (192 мг, 85%) в виде порошка.
Схема 4: Синтез пирролов

(Е)-1-Фенил-3-(3,4-диметоксифенил)-2-пропен-1-он (10)
Соединение 10 получают в соответствии с методикой получения 1. Выход 90%.
3-(3,4-диметоксифенил)-4-нитро-1-фенилбутан-1-он (11) [Hall et al., 2005]
Раствор 10 (774 мг, 2,9 ммоль) в MeOH (30 мл) обрабатывают диэтиламином (1,55 мл, 15 ммоль) и нитрометаном (0,81 мл, 15 ммоль) и полученную смесь кипятят с обратным холодильником в течение 24 часов. Раствор охлаждают, распределяют между дихлорметаном (60 мл) и водой (50 мл) и подкисляют 1M соляной кислотой. Органический слой отделяют, водный слой экстрагируют дихлорметаном (20 мл). Объединенные органические слои промывают водой (50 мл) и раствором соли (50 мл) и сушат над сульфатом натрия. Растворитель удаляют при пониженном давлении и полученное масло очищают колоночной хроматографией на силикагеле (н-гексан/этилацетат 3:2), получая 11 (780 мг, 82%) в виде твердого вещества.
4-(3,4-диметоксифенил)-1-фенилпиррол(12) [Hall et al., 2005]
Раствор 11 (400 мг, 1,22 ммоль) в метаноле (13 мл) и ТГФ (26 мл) при комнатной температуре с перемешиванием обрабатывают гидроксидом калия (343 мг, 6,1 ммоль). Спустя 1 час реакционную смесь по каплям добавляют к раствору серной кислоты (2,44 мл) в метаноле (13 мл) при 0°C и полученную смесь перемешивают в течение 1 часа при комнатной температуре. К смеси добавляют воду (20 мл) и лед (20 мл), смесь нейтрализуют 1M водным раствором гидроксидом натрия и экстрагируют дихлорметаном (2×50 мл). Объединенные органические фракции промывают раствором соли (25 мл), сушат над сульфатом натрия и упаривают при пониженном давлении. Полученное масло обрабатывают уксусной кислотой (8 мл) и хлоридом аммония (470 мг), раствор нагревают до 100°C и выдерживают при данной температуре в течение 1 часа. Реакционную смесь охлаждают, добавляют лед (50 мл) и смесь нейтрализуют 1M водным раствором гидроксида натрия. Раствор экстрагируют дихлорметаном (2×50 мл). Объединенные органические фракции промывают раствором соли (25 мл), сушат над сульфатом натрия и упаривают при пониженном давлении. Сырой продукт очищают колоночной хроматографией на силикагеле, (н-гексан/этилацетат 3:1) и затем перекристаллизовывают из смеси н-гексан/этилацетат (2:1), получая 12 (150 мг, 44%) в виде твердого вещества.
4-(3,4-Дигидроксифенил)-1-фенилпиррол (13) [Vanelle et al., 2000]
Раствор 12 (80 мг, 0,29 ммоль) в дихлорметане (5 мл) охлаждают до -78°C, обрабатывают трибромидом бора (0,13 мл, 1,4 ммоль), перемешивают при -78°C в течение 3 часов и затем в течение ночи при комнатной температуре. Смесь охлаждают до -78°C и гасят метанолом (5 мл). Смесь перемешивают в течение 3 часов при комнатной температуре, растворители выпаривают при пониженном давлении, остаток подвергают четыре раза совместному выпариванию с метанолом (10 мл). Полученный сырой продукт очищают колоночной хроматографией на силикагеле (хлороформ/метанол 95:5) и затем перекристаллизовывают из хлороформа с несколькими каплями ацетонитрила, получая 13 (36 мг, 50%) в виде твердого вещества.
Схема 5: Синтез пиразолинов

3-(3,4-Диметоксифенил)-5-(3-фторфенил)-4,5-дигидро-1H-пиразол (14)
Суспензию 1 (57 мг, 0,2 ммоль) и гидразингидрата (0,5 мл, 10 ммоль) в воде (0,14 мл) нагревают до 100°C и выдерживают при данной температуре в течение 1,5 часа при перемешивании. Реакционную смесь охлаждают, добавляют воду (0,2 мл), полученный осадок собирают фильтрацией, промывают водой и сушат, получая 14 (37 мг, 62%) в виде твердого белого вещества.
Схема 6: Синтез N-Ac-пиразолинов
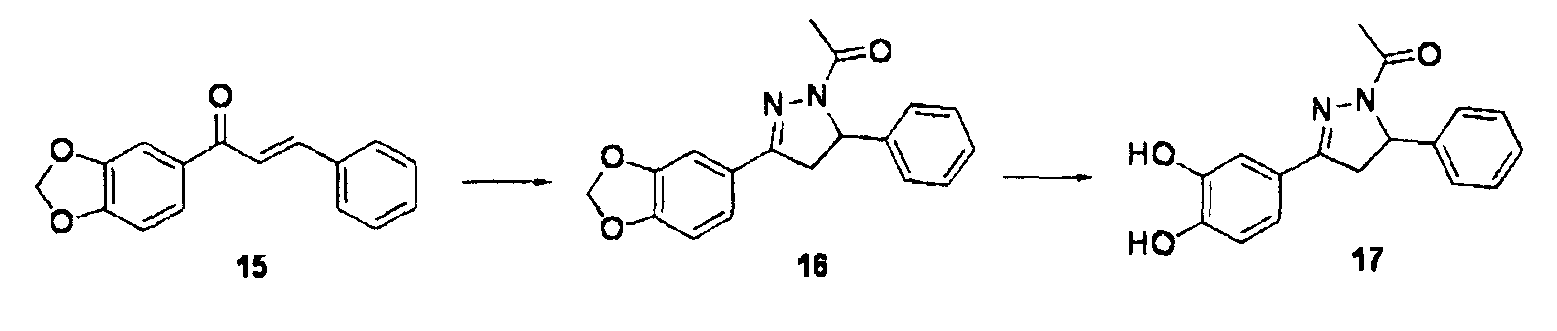
(Е)-1-(3,4-метилендиоксифенил)-3-фенил-2-пропен-1-он (15)
Соединение 15 получают в соответствии с методикой получения 1. Выход 64%.
1-Ацетил-3-(3,4-метилендиоксифенил)-5-фенил-4,5-дигидропиразол (16) [Chimenti et al., 2004]
Раствор 15 (504 мг, 2 ммоль) и гидразингидрата (250 мг, 5 ммоль) в уксусной кислоте (12 мл) нагревают до 120°C и выдерживают при данной температуре в течение 24 часов при перемешивании. Реакционную смесь охлаждают, добавляют охлажденную воду (40 мл), полученный осадок собирают фильтрацией, перекристаллизовывают из этанола и сушат, получая 16 (458 мг, 74%) в виде твердого белого вещества.
1-Ацетил-3-(3,4-дигидроксифенил)-5-фенил-4,5-дигидропиразол(17) [Vanelle et al., 2000]
Раствор 17 (70 мг, 0,23 ммоль) в дихлорметане (3 мл) охлаждают до -78°C, обрабатывают трибромидом бора (0,11 мл, 1,16 ммоль), перемешивают при -78°C в течение 3 часов и затем в течение ночи при комнатной температуре. Смесь охлаждают до -78°C и гасят метанолом (5 мл). Смесь перемешивают в течение 3 часов при комнатной температуре, растворители выпаривают при пониженном давлении, остаток подвергают четыре раза совместному выпариванию с метанолом (10 мл). Полученный сырой продукт очищают колоночной хроматографией на силикагеле (н-гексан/этилацетат 1:1), получая 17 (25 мг, 37%) в виде твердого белого вещества.
Схема 7: Синтез 1,2,4-оксадиазолов

3,4-Диметоксибензамидоксим (18) [Chalquest, 2001]
Раствор 3,4-диметоксибензонитрила (4,0 г, 24,5 ммоль), гидрохлорида гидроксиламина (2,0 г, 28,8 ммоль), N,N-диизопропилэтиламина (5,0 мл, 29,2 ммоль) в этаноле (70 мл) перемешивают при комнатной температуре в течение 48 часов. Этанол выпаривают при пониженном давлении, добавляют охлажденную воду (60 мл), полученный осадок собирают фильтрацией и сушат, получая 18 (3,4 г, 71%) в виде белого порошка.
3,5-Бис(3,4-диметоксифенил)-1,2,4-оксадиазол (19) [Korbonits, 1982]
К раствору 18 (700 мг, 3,57 ммоль) и этил-3,4-диметоксибензоата (834 мг, 3,97 ммоль) в этаноле (12 мл) добавляют трет-бутоксид калия (425 мг, 3,79 ммоль) и полученную смесь кипятят с обратным холодильником в течение 12 часов. Смесь охлаждают, осадок собирают фильтрацией, промывают теплым этанолом и сушат, получая 19 (540 мг, 44%) в виде белого порошка.
Гидробромид 3,5-бис(3,4-дигидроксифенил)-1,2,4-оксадиазола(20) [Vanelle et al., 2000]
Раствор 19 (220 мг, 0,64 ммоль) в дихлорметане (6 мл) охлаждают до -78°C, обрабатывают трибромидом бора (0,59 мл, 6,1 ммоль), перемешивают при -78°C в течение 3 часов и затем в течение ночи при комнатной температуре. Смесь охлаждают до -78°C и гасят метанолом (5 мл). Смесь перемешивают в течение 3 часов при комнатной температуре, растворители выпаривают при пониженном давлении, остаток подвергают четыре раза совместному выпариванию с метанолом (10 мл). Полученный остаток кипятят с обратным холодильником в 5 мл хлороформа, после охлаждения смеси продукт собирают фильтрацией и сушат, получая 20 (190 мг, 81%) в виде порошка.
Приведенные выше примеры представляют собой примеры синтеза целевых соединений и их производных. Остальные соединения, показанные на фиг.3, синтезируют в соответствии с данными примерами. Указанные химически синтезированные соединения вместе с выбранными соединениями первичного скрининга, подвергают дополнительным испытаниям, включая SIFT анализ, анализы в клеточной культуре, in vivo эксперименты на мышах, а также биохимические анализы по агрегации α-синуклеина (см. ниже). Перечень испытанных соединений представлен на фиг.3.
Пример 3: Используемый материал и методы
Библиотеки соединений
Библиотеки, подвергнутые скринингу, содержат 10000 соединений каждая и названы заявителями изобретения DIVERSet1 и DIVERSet2, поскольку они включают только часть большей DIVERSet библиотеки (ChemBridge Corp., San Diego, CA). DIVERSet представляет собой коллекцию рационально подобранных разнообразных небольших молекул со свойствами, подобными свойствам лекарственных средств. Соединения поставляются в виде раствора в диметилсульфоксиде (ДМСО) и на 96-луночных титрационных микропланшетах. База данных, содержащая молекулярные структуры, и некоторые физико-химические данные для каждого из соединений доступны на http://www..
Получение рекомбинантной мыши PrP 23-231
Рекомбинантный PrP получают и очищают по существу как описано в публикации Liemann et al. (1998) с тем отличием, что клетки бактериальной экспрессии BL21DE3 RIL E.coli (Novagen) трансформируют с плазмидой pET17b-MmPrP23-231WT31 мыши PrP23-231. Кроме того, бактерии выращивают до оптической плотности 0,5 перед продуцированием белка, которое индуцируют добавлением 1 мМ IPTG, и клетки собирают спустя два часа. Бактерии лизируют добавлением 0,5% Triton X-100 к лизисному буферу и инкубированием в течение 30 минут при 37°C вместо применения френч-пресса (French press). Далее, фильтрацию геля заменяют стадией хроматографией по сродству к хелату никеля. Конечную стадию катионообменной хроматографии после рефолдинга также пропускают.
В частности, PrP, предварительно очищенный ионообменной хроматографией, подвергают окислению, как описано, окисление заканчивают добавлением 0,1 мМ EDTA и доведением значения рН до примерно 6. После добавления 0,1 мМ NiCl2 до 50 мг PrP применяют к 2 мл хелатообразующей сефарозы (Pharmacia), предварительно заряженной с помощью NiCl2 в соответствии с рекомендациями производителей и предварительно уравновешенной с помощью буфера А (8 М мочевины, 10 мМ MOPS рН 7,0). Связывание PrP с Ni-хелатной матрицей проводят в течение по меньшей мере 3 часов при комнатной температуре, непрерывно переворачивая смесь. Матрицу переносят в хроматографическую колонку (poly-prep, BioRad) и вымывают с потоком, проходящим через колонку. Колонку дважды промывают 5 мл буфера В (8М мочевины, 10 мМ MOPS pH 7,0 500 мМ NaCl) и затем последовательно элюируют 6 раз 5 мл буфера D (7,2М мочевины, 10 мМ MOPS рН 7,0, 150 мМ NaCl, 50 мМ имидазола). Фракции, содержащие очищенный PrP, объединяют, концентрируют в центрифуге (centriprep device) и наконец разбавляют 1:50 для рефолдинга в 10 мМ MES (pH 6,0).
Введение флуоресцентной метки в антитела и рекомбинантный PrP
В моноклональное антитело L42 (r-biopharm, Darmstadt, Germany) вводят метку Alexa Fluor 647 (Alexa-647; Invitrogen, Eugene) в соответствии с инструкцией производителя. В рекомбинантную PrP 23,231 мышь вводят метку Alexa Fluor 488 (Alexa-488; Invitrogen, Eugene) в 20 мМ фосфатно-калиевом буфере (рН 6, 0,1% Nonidet P40), 40 мМ бикарбонатном буфере бикарбоната натрия (рН 8,3). Несвязанные флуорофоры отделяют гельфильтрацией на PD10 колонках (GE Healthcare, Freiburg, Germany), уравновешенных 20 мМ фосфатнокалиевым буфером (рН 6, 0,1% Nonidet P40). Контроль качества реакции введения метки и коэффициент введения метки проводят с помощью флуоресцентной корреляционной спектроскопии (FCS) на датчике считывания Insight Reader (Evotec Technologies, Humburg, Germany). Коэффициент введения метки составляет приблизительно 1,3 флуорофора на молекулу rPrP.
Анализ ассоциации PrPC-PrPSC
PrPSc получают из мозга CJD пациентов в соответствии с публикацией (Safar et al. (1998)), аликвоты конечного пеллета, ресуспендированные в 1×PBS+0,1% саркосильного раствора, разбавляют в пять раз в буфере А (20 мМ фосфатнокалиевый буфер при рН 6,0, 0,1% Nonidet P40) и обрабатывают ультразвуком в устройстве для обработки ультразвуком на водяной бане в течение 60 секунд. Обработанную смесь центрифугируют при 1000 об/мин в течение 1 минуты и полученный супернатант разбавляют в 100 раз буфером А для анализа.
Смесь меченого мышиного rPrP и меченого L42 моноклонального антитела приготавливают в 20 мМ фосфатнокалиевом буфере (рН 6, 0,1% Nonidet P40) таким образом, что меченые молекулы составляют приблизительно равный избыток при 2-6 мМ. В аналитическом объеме 20 мкл смешивают объем 8 мкл смеси rPrP/антитело, 2 мкл соединения и 10 мкл разбавленного препарата PrPSc. Образцы загружают в 96-луночные микропланшеты со сдвижными стеклами-донышками (Evotec-Technologies, Humburg, Germany) и проводят измерения на Insight Reader.
Определение количества одиночных частиц и анализ
Измерения FIDA проводят при энергиях возбуждения 200 мкВт для 488 нм лазера и 300 мкВт для 633 нм лазера. Параметры сканирования: длина пути сканирования 100 мкм, частота сканирующего луча 50 Гц, перемещение координатного стола 2000 мкм. Время измерения 10 секунд. Флуоресценцию от двух флуорофоров считывают отдельно детекторами одиночных фотонов и число обнаруженных фотонов в течение интервалов времени постоянной продолжительности (бины) суммируют, используя продолжительность бина 40 микросекунд. Определяют число считанных красных и зеленых флуоресцентных фотонов и анализируют его в двухмерной гистограмме распределения интенсивностей, как описано выше (Bieschke et al., 2000).
Данные распределения интенсивностей оценивают с использованием модуля программного обеспечения 2D-SIFT (Evotec-Technologies, Hamburg, Germany) суммированием бинов высокой интенсивности в секторах. Значения выключения для биновых интенсивностей для каждой серии измерений регулируют вручную в соответствии с контрольными измерениями.
Пример 4: Терапевтическое применение новых потенциальных антиприонных соединений в скрейпи-инфицированных мышах спустя 80 дней после инфицирования
Для доказательства эффективности указанных новых потенциальных антиприонных соединений в подавлении инфекционных губчатых энцефалопатий (TSE) или прионных болезней проводят эксперимент на животных, в котором соединения, обладающие антиприонной активностью в соответствии с SIFT анализом и/или анализом на культуре скрейпи клеток, используют для лечения мыши, инфицированной RML штаммом скрейпи, на поздней стадии периода инкубации. Заявители выбирают интраперитонеальное применение соединений с интервалом 14 дней спустя 80 дней после инфицирования, поскольку обычно это время, когда у животных, инфицированных данным прионным штаммом, проявляются первые субклинические симптомы. Это должно соответствовать самому раннему времени, когда человек, зараженный TSE и проявивший первые симптомы заболевания, должен получить реальное терапевтическое лечение. Выбор таких жестких условий для терапевтического лечения заявители делают для оценки функциональности испытываемых соединений в качестве терапевтических средств в реальных условиях. До сих пор, как правило, терапевтические средства для лечения TSE должны были тестировать только на профилактическое действие после экспозиции в экспериментах на животных, где они применяются приблизительно в одно время с инфицированием прионами. В реальной жизни только в очень редких случаях выполним такой терапевтический режим для пациентов, инфицированных TSE. Для большинства пациентов, инфицированных TSE, время инфицирования или начало инкубационного времени для наследственных и спорадических случаев (которые составляют подавляющее большинство) неизвестно и не может быть выявлено. Поэтому большинство пациентов, инфицированных TSE, могут получить лечение только после проявления первых симптомов TSE.
Методики экспериментов:
Самок мышей C57BI6 возраста 6-7 недель инокулируют RML скрейпи интрацеребральной инъекцией 30 мкл 1% стерильного мозгового гомогената в фосфатно-буферном растворе (PBS) от мыши, смертельно больной RML скрейпи-штаммом. Спустя 80 дней после инфицирования мышей лечат выбранными новыми потенциальными антиприонными соединениями или разбавителем (водным диметилсульфоксидом - ДМСО). Для лечения выбирают пять потенциальных антиприонных соединений в соответствии с их антиприонной активностью в модели культуры клеток, которые были обозначены как 10353F11 в соответствии с плейт-позицией (plate-position) в Diverset библиотеке химических соединений (Chembridge Corp., San Diego, USA) и anle138b, anle143b, sery106 и sery149. Лечение указанными соединениями проводят в течение последующих 14 дней интраперитонеальной инъекцией 50 мкл в день соединения 10353F11 при разбавлении с 25 мкл других соединений в разбавителе (ДМСО). Соединение 10353F11 используют в концентрации 10 мМ, а соединения anle138b, anle143b, sery106 и sery149 вводят в концентрации 100 мМ инъекцией в течение всего периода. Животных обследуют ежедневно на выявление признаков болезни обученные смотрители за животными спустя 80 дней после инфицирования. Животных забивают, когда они достигают конечной стадии болезни, характеризующейся определенными клиническими симптомами (атаксия, тремор, затруднение выхода из положения лежа на спине в нормальное положение и окоченение хвоста) и агонизируют. Обычно развитие болезни, прошедшее через конечную стадию, будет приводить к смерти животного в пределах одного или двух дней. Из забитых животных одно полушарие и половину селезенки замораживают в свежем виде при -80°C для вестерн-блоттинга, а второе полушарие и вторую часть селезенки, а также все внутренние органы фиксируют в 4% растворе формальдегида для (иммунной) гистологии.
Результаты
Как показано на фиг.4, лечение мыши, которая была интрацеребрально инфицирована штаммом RML скрейпи, на последней стадии инкубационного периода (80 дней p.i.) посредством ежедневного интраперитонеального введения выбранных потенциальных антиприонных лекарственных средств приводит к увеличению продолжительности инкубационного периода до тех пор, пока не достигается последняя стадия скрейпи инфекции для 10353F11 (фиг.4А) и anle138b и sery149 (фиг.4В). Средняя продолжительность периода выживания, определенная для соединений anle138b и sery149 при испытании на группах из семи и восьми животных каждая, увеличивается на 14,9 и 11,5 дней, соответственно, по сравнению с группой из двенадцати животных, которые принимают только разбавитель 100% ДМСО. Для соединений sery106 и anle143b увеличение продолжительности периода выживания в группе из восьми обрабатываемых животных по сравнению с такой же контрольной группой ниже статистически значимого значения. При лечении соединением 10353F11 период выживания, определенный для группы из восьми животных каждая, увеличивается на 11 дней по сравнению с контрольной группой. Наблюдаемое увеличение продолжительности периода выживания в экспериментах приблизительно соответствует продолжительности лечения. Это может означать, что лечение данными лекарственными средствами приостанавливает развитие болезни, пока вводятся лекарственные средства. В этом случае лечение данными лекарственными средствами могло бы привести к увеличению продолжительности жизней TSE-инфицированных пациентов и к стабилизации их состояния здоровья, защищая его от дальнейшего ухудшения посредством остановки болезни.
Пример 5. Терапевтическое применение потенциального антиприонного соединения в скрейпи-инфицированной мыши после интраперитонеального инфицирования
Для доказательства эффективности указанного нового антиприонного соединения в подавлении инфекционных губчатых энцефалопатий (TSE) или прионных болезней проводят дополнительный эксперимент на животных. В данном эксперименте мышей инфицируют интраперитонельно RML скрейпи и лечат соединением anle138b, которое обладает доказанной антиприонной активностью в опыте на животных на последней стадии развития прионной болезни (пример 4). Заявители выбирают сочетание интраперитонеального и перорального введения соединения и начинают лечение сразу после инфицирования.
Методики эксперимента:
Скрейп-инфицирование и лечение мышей
Самок мышей C57BI6 возраста 6-7 недель инокулируют RML скрейпи интраперитонеальной инокуляцией 100 мкл 1% стерильного мозгового гомогената в фосфатно-буферном растворе (PBS) от мыши, смертельно больной RML скрейпи-штаммом.Лечение этих мышей соединением anle138b или разбавителем, в качестве которого используется водный диметилсульфоксид (ДМСО), начинают сразу после инфицирования. Лечение данным соединением проводят в течение последующих 14 дней интраперитонеальной инъекцией 25 мкл в день соединения, разбавленного разбавителем (ДМСО) и последующим пероральным введением в течение 4 и затем 5 дней 50 мкл соединения в день в смеси растительное масло/ДМСО c помощью зонда для кормления (фиг.5А). Соединение anle138b используют в концентрации 100 мМ при интраперитонеальном применении и в дозе 50 мг/кг при пероральномвведении. Животных забивают на 35 день после инфицирования, когда PrPSc может четко обнаруживаться в селезенке интраперитонеально инфицированных мышей. Одну часть селезенки забитых животных замораживают в свежем виде при -80°C для вестерн-блоттинга, в то время как вторую часть селезенки и все внутренние органы фиксируют в 4% растворе формальдегида для иммуногистологии.
РЕТ блоттинг
Формалин-фиксированную ткань мозга разрезают на блоки толщиной 2 мм, дезинфицируют в концентрированной муравьиной кислоте в течение 1 часа, постфиксируют в 4% фосфатно-буферном растворе формалина в течение 48 часов в соответствии с методикой, описанной в публикации Brown et al. (1990) и заливают в парафин. Секции (5-7 мкм) разрезают на микротоме, помещают в водяную баню (55°C), собирают на предварительно увлажненной нитроцеллюлозной мембране с порами размером 0,45 мкм (Bio-Rad, Richmond, CA) и сушат в течение, по меньшей мере, 30 минут при 55°C. Нитроцеллюлозную мембрану депарафинируют ксилолом. Ксилол заменяют изопропанолом и затем постадийно регидрируют. На последней стадии регидратации в дистиллированную воду добавляют Tween 20 в конечной концентрации 0,1%. Мембраны сушат и хранят при комнатной температуре в течение месяцев без потери качества последующего PrPSC окрашивания.
После предварительного смачивания TBST (10 ммоль/л Tris-HCl, pH 7,8; 100 ммоль/л NaCl; 0,05% Tween 20) проводят биологическую переработку с помощью 250 мкг/мл протеиназы К (Boehringer) в фосфатно-калиевой буфере (10 ммоль/л Tris-HCl, pH 7,8; 100 ммоль/л NaCl; 0,1% Brij 35) в течение 8 часов при 55°C. На этой стадии мембран-присоединенные белки фиксируются на мембране. После троекратной промывки TBST белки на мембране денатурируют 3 моль/л гуанидинизоцианата в 10 ммоль Tris-HCl (pH 7,8) в течение 10 минут. Гуанидин промывают три раза с помощью TBST. Иммунологический анализ проводят после предварительной инкубации в блокирующем растворе (0,2% казеина в TBST) в течение 30 минут. В качестве первичного антитела использую поликлональное антитело кролика против рекомбинантного мышиного PrP, которое обозначено как CDC1, при 1:500 разбавлении в растворе антитело-разбавитель (Ventana). Смесь инкубируют в течение по меньшей мере 1 часа. После промывки три раза TBST инкубирование проводят в течение по меньшей мере 1 часа с алкалинфосфатаза-связанным антителом кролика против антитела мыши (Dako, Hamburg) при разбавлении 1:500. После пяти промывок в TBST в течение 10 минут значение рН мембран доводят до щелочного значения посредством инкубирования два раза в течение 5 минут в NTM (100 ммоль/л Tris-HCl, pH 9,5; 100 ммоль/л NaCl; 50 ммоль/л MgCl2). Визуализацию реакции антитела проводят реакцией формазана с использованием NBT/BCIP. Блоты оценивают с помощью препаровальной лупы Olympus.
Результаты
Ткань селезенки, экстрагированную после гибели мышей, получавших лечение лекарственных средством, и мышей, получавших разбавитель, иммуногистохимически окрашивают для обнаружения присутствия скоплений PrPSc и анализируют на уровень содержания селезеночного PrPSc с помощью иммуноблоттинга. Как показано на фиг.5В, уровень содержания PrPSc в селезенке животных, получавших лечение, значительно снижается по сравнению с мышами, получавшими разбавитель. Исследование селезеночной ткани от инфицированных мышей показывает, что скопления PrPSc уменьшаются после лечения соединением anle138b. Процентное содержание селезенок с низким содержанием скоплений PrPSc возрастает, а содержание прочных PrPSc скоплений снижается (фиг.5С). На фиг.5D представлены примеры двух РЕТ блотов PrPSc скоплений. Результаты четко показывают антиприонную эффективность данного соединения в периферийных тканях при подборе плана эксперимента и схемы терапевтического лечения.
Пример 6: Терапевтическое применение новых антиприонных соединений для лечения скрейпи-инфицированных мышей спустя 80 дней после инфицирования
Для подтверждения результата анализа, полученного в примере 4, что увеличение времени выживания соответствует продолжительности лечения животного проводят эксперимент на животных, в котором соединения вводятся в более высокой дозировке и в течение более длительного времени лечения мышей, инфицированных RML штаммом скрейпи, на поздней стадии инкубационного периода. Заявители выбирают сочетанию интраперитонеального и перорального применение соединений спустя 80 дней после инфицирования, поскольку обычно этот период времени соответствует времени появления первых субклинических симптомов у животных, инфицированных данным прионным штаммом.
Методики эксперимента:
Самок мышей C57BI6 возраста 6-7 недель инокулируют RML скрейпи интрацеребральной инъекцией 30 мкл 1% стерильного мозгового гомогената в фосфатно-буферном растворе (PBS) от мыши, смертельной больной RML скрейпи-штаммом. Спустя 80 дней после инфицирования этих мышей лечат новыми антиприонными соединениями или разбавителем ДМСО. Используют два потенциальных антиприонных соединения: anle138b и anle186b. Лечение этими соединениями проводят в течение последующих 14 дней интраперитонеальной инъекцией 25 мкл в день соединения, разбавленного в ДМСО, и последующими двумя пероральными введениями в течение 5 дней 50 мкл в день соединения в смеси растительное масло/ДМСО посредством зонда для кормления (фиг.6А). 3 контрольным мышам и 2 мышам, получающим лечение соединением 138b, дополнительно вводят соединение перорально, начиная со 109 дня по 136 день, предоставляя гранулы пищевого арахисового масла в смеси с основными растворами ДМСО/соединение. Указанные соединения используют в концентрации 100 мм для интраперитонеального введения и в дозе 50 мг/кг для перорального введения. Из каждой группы в указанные моменты времени забивают четырех животных (фиг.6A). Восемь животных из каждой группы обученный персонал ежедневно проверяет на наличие признаков болезни, начиная с 80 дня после инфицирования. Животных забивают, когда они достигают конечной стадии болезни, которая характеризуется определенными клиническими симптомами (атаксия, тремор, затруднение выхода из положения лежа на спине в нормальное положение и окоченение хвоста) и агонизируют. Обычно развитие болезни, прошедшее через конечную стадию, будет приводить к смерти животного в пределах одного или двух дней. Из забитых животных одно полушарие и половину селезенки подвергают замораживанию в свежем виде при -80°C для вестерн-блоттинга, а второе полушарие и вторую часть селезенки, а также все внутренние органы фиксируют в 4% растворе формальдегида для гистологии.
Результаты
Уровень содержания PrPSc в мозговых гомогенатах и гибель апоптотических клеток после инфицирования
Мозговые гомогенаты мышей, принимающих лечение, и мышей, принимающих разбавитель, анализируют на уровни содержания PrPSc посредством иммуноблот-анализа. Как показано на фиг.6В, уровень содержания PrPSc в мозге всех животных, проверенных в указанные моменты времени, из группы, принимающей лечение соединением anle138b, может удерживаться на уровне содержания PrPSc в организме необработанных мышей на 80 день, в то время как уровень содержания PrPSc в контрольной группе возрастает. Результаты, полученные для группы животных, получавших соединение anle186b, находится между результатами контрольной группы и группы, получающей anle138b. Увеличение PrPSc могло быть замедлено (фиг.6B). На фиг.6С показано изменение относительных уровней содержания PrPSc после лечения соединениями по сравнению с необработанным контролем на 80 день. Уровень содержания PrPSc в мозге животных медленно снижается после лечения соединением anle138b. Гистологический анализ H&E окрашенных срезов мозговой ткани мышей, принимавших лечение соединениями anle138b и anle186b в указанные моменты времени, показал снижение патологических изменений. Число апоптотических клеток в слое клеток-зерен мозжечка инфицированных мышей из групп, принимавших лечение, снижается по сравнению с контрольной группой (фиг.6D). Эти результаты показывают, что оба соединения могут преодолевать гематоэнцефалический барьер. Следовательно, терапевтическое лечение способно предупреждать дальнейшее отложение PrPSc и развитие болезни в мозге животных в процессе лечения. Эти результаты означают, что лечение соединением anle138b приостанавливает развитие болезни, пока вводятся лекарственные средства. Эти результаты также показывают, что можно влиять на развитие болезни при терапевтическом лечении с использованием данного соединения, воздействуя на образование PrPSc. В этом случае лечение должно привести к увеличению продолжительности жизни TSE-инфицированных индивидуумов и стабилизацию или, возможно, улучшению состояния их здоровья, защищая от дальнейшего ухудшения состояния здоровья посредством остановки развития болезни.
Увеличение продолжительности периода инкубации
Как показано на фиг.7, лечение мышей, которые были интрацеребрально инфицированы штаммом RML скрейпи, на последней стадии инкубационного периода (80 день p.i.) посредством ежедневного введения соединения приводит к увеличению продолжительности периода выживания до тех пор, пока не достигается конечная стадия скрейпи-инфицирования (фиг.7). Кроме того, более продолжительные периоды выживания двух мышей, которые принимали дополнительное лечение, начиная со 109 дня по 136 день, введением соединения anle138b в смеси с арахисовым маслом показали, что i) выживание коррелирует с продолжительностью лечения, ii) соединение является эффективным при пероральном введении и iii) соединение преодолевает гематоэнцефалический барьер.
Пример 7: Подавление агрегации α-синуклеина в условиях invitro
Синуклеинопатии представляют собой (нейродегенеративные) заболевания, характеризующиеся внутриклеточной аккумуляцией скоплений и фибрилл, состоящих, главным образом, из белка α-синуклеина (см. обзор: Goedert, 2001). Наиболее известными нейродегенеративными синуклеинопатиями являются болезнь Паркинсона (PD), деменция с тельцами Леви (DLB), множественная системная атрофия (MSA).
Было показано, что агрегация α-синуклеина in vitro имеет место в присутствии соединений, таких как органические растворители, которые имитируют диэлектрические условия, которые имеют место в естественных условиях в непосредственной близости от биологических мембран (Munishkina et al., 2003). Были разработаны испытания агрегации в условиях in vitro, которые обеспечивают модельные системы для исследования основных аспектов неправильного сложения и агрегации α-синуклеина и образования токсических разновидностей скоплений в процессе течения болезни (Kostka et al., 2008).
Для тестирования выбранных соединений на их способность подавлять агрегацию α-синуклеина заявители изобретения используют систему in vitro, где используется органический растворитель диметилсульфоксид (ДМСО) в низких концентрациях (<3%) и, в некоторых экспериментах, трехвалентное железо для индуцирования агрегации α-синуклеина in vitro. Мультимерное образование контролируют с использованием установки флуоресцентной корреляции одиночных частиц посредством кросс-корреляционного анализа и SIFT-анализа, применяемых к смесям мономеров α-синуклеина, меченного зеленым Alexa488- или красным Alexa647 флуорофорами, соответственно. Такие α-синуклеиновые смеси агрегируют в параллели к образцам, где соединение добавляют в реакцию.
Методики эксперимента:
Введение флуоресцентной метки в α-синуклеин
Введение метки в α-синуклеин осуществляют с помощью флуоресцентных красителей, способных взаимодействовать по аминогруппе Alexa Fluor-488-О-сукцинимидиловый сложный эфир) или Alexa Fluor-647-О-сукцинимидиловый сложный эфир (Molecular Probes, USA), соответственно. После завершения реакции несвязанные молекулы красителя отделяют эксклюзионной хроматографией реакционных смесей, пропуская их через две последовательно соединенные колонки PD10 (Amersham Bioscience, Germany) в соответствии с инструкциями производителя. Эффективность введения метки и удаления несвязанного красителя определяют посредством измерений прямого светорассеяния (FSC) с подходящими разбавлениями фракций, содержащих меченые мономеры α-синуклеина.
Определение показателей флуоресцентной корреляционной спектроскопии одиночных частиц
В объеме 20 мкл в лунках специального титрационного микропланшета со сдвижным стеклом-донышком для проведения измерений с помощью флуоресцентной корреляционной спектроскопии (Evotec-Technologies, Germany) проводят агрегацию α-синуклеина в буфере, содержащем 50 мМ Tris при рН 7,0 и смесь мономеров α-синуклеина, меченных Alexa488- или Alexa647-флуорофорами, соответственно, в конечной концентрации приблизительно 5-10 нМ каждого вида α-синуклеина. Измерения проводят на датчике Insight (Evotec Technologies, Germany) с использованием 40× объектива микроскопа 1,2 NA (Olympus, Japan) c FIDA оптическими настройками, диаметром перфорированного отверстия 70 мкм и энергией возбуждения 200 мкВт для 488 нм лазера и энергией возбуждения 300 мкВт для 633 нм лазера. Время измерения равно 10 секундам, в процессе которого фокус лазера перемещают по лунке посредством устройства сканирования луча (длина пути сканирования 100 мкм при частоте сканирования 50 Гц, частота сканирующего луча 50 Гц, перемещение координатного стола 2000 мкм). Это эквивалентно скорости сканирования приблизительно 10 мм/с. В результате получают двумерные гистограммы распределения интенсивностей, которые анализируют с использованием 2-D SIFT программного обеспечения (Evotec OAI, Germany).
Результаты: Подавление агрегации α-синуклеина соединениями
Агрегация α-синуклеина, вызываемая ДМСО и ДМСО/Fe3+, проявляется в образовании мультимерных комплексов α-синуклеина, которые содержат и зеленые, и красные меченые единицы α-синуклеина в больших количествах. Таким образом, контрольная реакция без добавления ингибиторных соединений показывает присутствие большого числа комплексов, которые испускают большие количества фотонов. Добавление DPP-соединения 351F11 к опытному раствору может в значительной степени ингибировать образование мультимерных комплексов α-синуклеинов зависимым от дозы образом, как показано на фиг.8А. Следовательно, соединение 351F11 способно эффективно ингибировать образование мультимерных комплексов α-синуклеина при низкой микромолярной концентрации в данной in vitro модели патологической агрегации белка, обнаруженной в синуклеопатиях. Это является четким показателем того, что соединение 351F11 может функционировать не только как антиприонное соединение, но также обладает потенциалом терапевтического лекарственного средства для лечения синуклеинопатий, таких как болезнь Паркинсона, DLB и MSA, которое воздействует на патологический механизм на молекулярном уровне. Ингибиторное воздействие зависимым от дозы образом на агрегацию α-синуклеина также может обнаруживаться и у других исследуемых DPP-производных (фиг.8-В, С). Следовательно, данные соединения представляют собой новую группу соединений с доказанной способностью ингибировать агрегацию α-синуклеина in vitro, что позволит разработать каузальную терапию против болезни Паркинсона и других синуклеинопатий.
Кроме того, ингибиторная активность данных соединений в отношении агрегации in vitro прионных белков и α-синуклеина может свидетельствовать об их общей противоагрегаторной активности в отношении более широкого спектра заболеваний, связанных с агрегацией белков, в которых неправильное сложение белков преимущественно в β-листовые конформации составляет дает возможность их агрегации в амилоидные фибриллы. Следовательно, данные соединение и другие представители класса DPP-производных обладают потенциалом применения в качестве терапевтических средств для каузального лечения полного перечня (нейродегенеративных) заболеваний, связанных с агрегацией белков, включая болезнь Альцгеймера, прионную болезнь, болезнь Паркинсона, множественную системную атрофию, болезнь диффузных телец Леви, лобно-височную деменцию, боковой амиотрофический склероз, болезнь Хантингтона, атаксию, связанную с заболеваниями спинного мозга и мозжечка, и другие поли-Q-заболевания, наследственную церебральную амилоидную ангиопатию, наследственную амилоидную полиневропатию, первичный системный амилоидоз (AL амилоидоз), реактивный системный амилоидоз (AA амилоидоз), диабет II типа, инъекция-локализованный амилоидоз, бета-2 микроглобулиновый амилоидоз, наследственный не невропатический амилоидоз и наследственный финский системный амилоидоз.
Пример 8: Ингибирование PrPSc в культуре клеток
Методики эксперимента:
Прион-инфицированные культуры клеток обрабатывают вновь синтезированными соединениями, как описано выше для первичного скрининга. Соединения добавляют в концентрациях, указанных на фиг.9. Структуры соединений показаны ниже, а также на фиг.3.

5-(3,5-дибромфенил)-3-(3,4,5-тригидроксифенил)пиразол (anle145d)

5-(3-бромфенил)-3-(3,4-диметоксифенил)пиразол (sery255b)
Результаты:
Очень высокая часть представителей класса DPP-производных проявляет эффективное снижение PrPSc в культуре клеток при низких микромолярных и даже при субмикромолярных концентрациях. Это указывает на то, что данные соединения представляют группу производных с антиприонной активностью.
Пример 9: Ингибиторное действие различных DPP-производных в мозге и селезенке
Соединения тестируют на их ингибиторное действие в отношении аккумулирования PrPScin vivo в соответствии со следующими тремя методиками:
a) C57BL/6 мышей инокулируют интрацеребрально (i.c.) 30 мкл 1% мозгового гомогената (RML скрейпи). Лечение начинают спустя 80 дней после инфицирования пероральным введением 1 мг соединения в день в смеси с ДМСО + кокосовое масло. Уровень содержания PrPSc в мозге определяют иммуноблот-анализом на 120 день после инфицирования;
b) C57BL/6 мышей инокулируют интрацеребрально (i.c.) 30 мкл 1% мозгового гомогената (RML скрейпи). Лечение начинают спустя 80 дней после инфицирования интраперитонеальной инъекцией 0,84 мг соединения (в ДМСО) в день в течение 14 дней с последующим лечением 2×5 дней (с перерывом между этим лечением 2 дня) 1 мг соединения (в ДМСО + арахисовое масло), вводимого перорально с помощью зонда для кормления. Уровень содержания PrPSc определяют на 106 день после инфицирования;
с) C57BL/6 мышей инокулируют интраперитонеально (i.р.) 100 мкл 1% мозгового гомогената (RML скрейпи). Уровень содержания PrPSc в селезенке определяют на 35 день после инфицирования после 34 дней лечения 1 мг в день соединения в ДМСО + арахисовое масло буфере.
Относительное ингибирование аккумуляции PrPSc по сравнению с группой, получавшей ДМСО, показано в таблице 1 (среднее значение ингибирования у животных, получающих ДМСО, определяют как 0% ингибирование, среднее значение для контрольных животных в начале периода лечение определяют как 100% ингибирование).

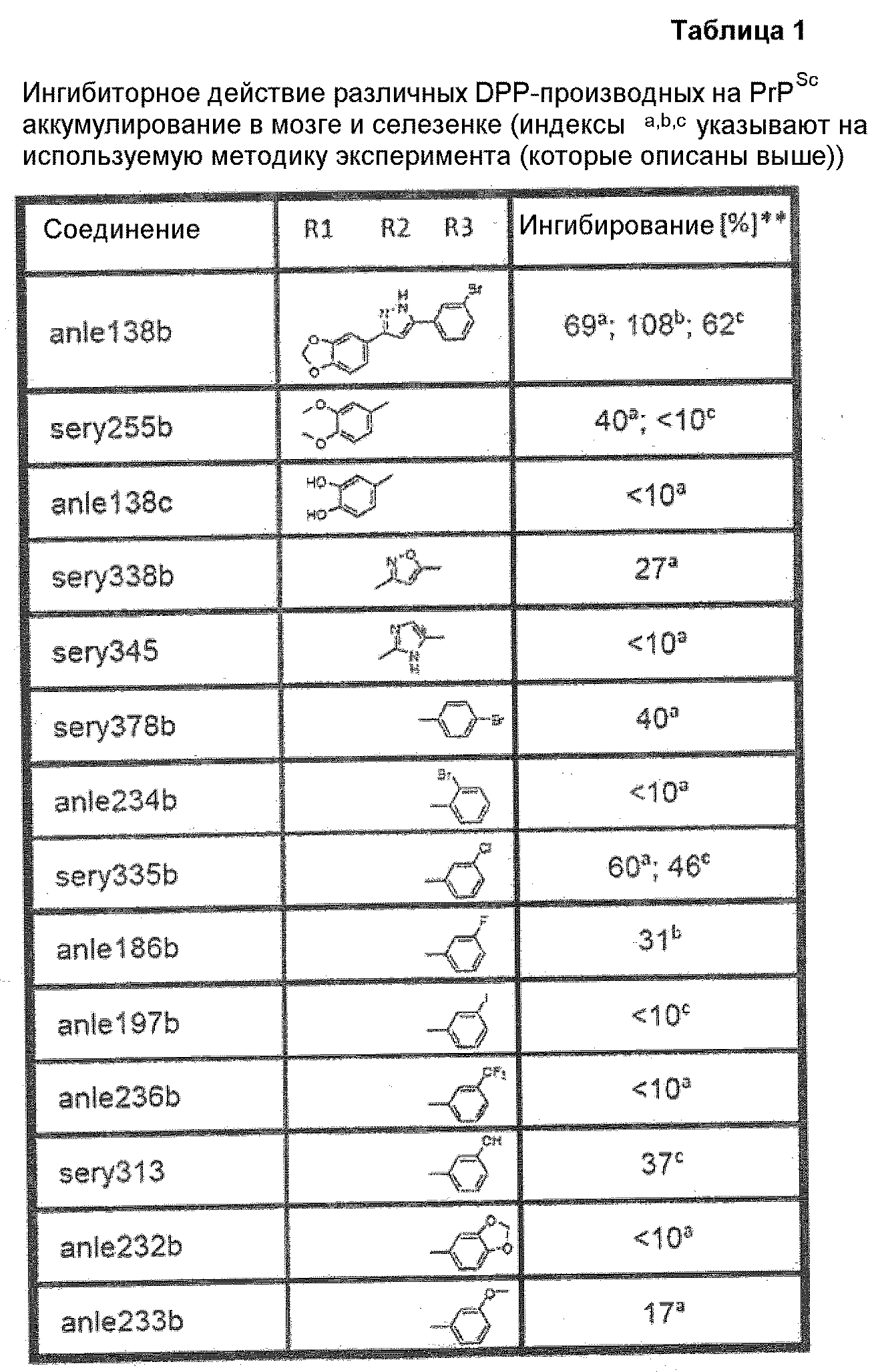
Результаты экспериментов показывают, что в данных экспериментальных условиях i) соединение anle138b и химически сходные соединения обеспечивают высокоэффективное ингибирование амплификации прионов in vivo; ii) существует зависимость активности от структуры, которая показывает, что для данного применения в данных экспериментальных условиях соединение anle138b предоставляет относительный оптимум активности.
Пример 10: Возможность прохождения гематоэнцефалического барьера и наблюдаемое взаимодействие с патологическими скоплениями белков
C57BL/6 мышей инокулируют интрацеребрально (i.c.) 30 мкл 1% мозгового гомогената (RML скрейпи). Лечение начинают спустя 80 дней после инфицирования пероральным введением 1 мг соединения (sery383) или 3 мг соединения (sery363a) в смеси с буфером ДМСО + арахисовое масло. Уровень содержания PrPSc в мозге определяют на 120 день иммуноблоттингом.

По сравнению с контрольными группами, получающими ДМСО, введение sery363a приводит к снижению аккумуляции PrPSc на 35%, введение sery383 приводит к снижению аккумуляции PrPSc на 30%.
Sery363a синтезируют и испытывают в данных экспериментах, поскольку оно представляет собой модификацию anle138b и хорошо подходит для введения изотопной метки, которая необходима для применения в качестве изотопного индикатора в РЕТ визуализации.
Sery383 тестируют в данном анализе, поскольку было установлено, что данное соединение, а также его структурные аналоги, содержащие -NH2 группу и атом галогена, являются высокоэффективными для ингибирования агрегации α-синуклеина (см. пример 16 «Ингибирование образования скоплений α-синуклеина различными соединениями»).
Результаты данных экспериментов показывают, что оба соединения могут проходить гематоэнцефалический барьер и взаимодействовать с патологическими скоплениями белков, и свидетельствует о том, что данные соединения обладают свойствами, которые могут эксплуатироваться при их применении в качестве терапевтического соединения, а также в качестве диагностического соединения.
Пример 11: Влияние ежедневного лечения соединением anle138b на аккумуляцию PrPSc и прионную патологию у мышей, инфицированных RML скрейпи
C57BL/6 мышей инокулируют интрацеребрально (i.c.) 30 мкл 1% мозгового гомогената (RML скрейпи). Лечение начинают спустя 80 или 120 дней после инфицирования пероральным введением 5 мг соединения в день в смеси с буфером ДМСО + арахисовое масло (фиг.10).
Секции мозга, окрашенные для выявления PrPSc (фиг.10А), показывают, что лечение соединением anle138b снижает аккумуляцию PrPSc по сравнению с животными, принимающими ДМСО. Количественное определение уровней содержания в мозговых гомогената прион-инокулированных мышей в различные моменты времени показывает, что аккумуляция PrPSc в организмах мышей, получавших лечение соединением anle138b, значительно снижается, даже когда лечение начинают на поздней стадии развития болезни (120 дней после инокулирования, фиг.10В). Гистологическое количественное определение апоптотических клеток в тонком срезе H&E окрашенной мозговой ткани, показывает, что ингибирование аккумуляции PrPSc приводит к ингибированию гибели нейронных клеток (фиг.10С). Контрольные мыши, обработанные буфером ДМСО + арахисовое масло без соединения, показывают прогрессирующую потерю массы тела (фиг.10D). Лечение соединением anle138b, начиная с дня 80 после инокулирования, предотвращает дальнейшую потерю массы в течение ~100 дней. Лечение со 120 дня после инокулирования ингибирует потерю массы в течение ~70 дней.
Полученные экспериментальные данные показывают, что лечение соединением ингибирует аккумуляцию PrPSc, гибель нейронных клеток и развитие клинических признаков заболевания даже в том случае, когда лечение начинают на поздней стадии развития болезни после проявления очевидных признаков болезни.
Пример 12: Сравнение различных методик лечения
C57BL/6 мышей инокулируют интрацеребрально (i.c.) 30 мкл 1% мозгового гомогената (RML скрейпи). Лечение соединением anle138b в разное время и в соответствии с разными графиками (как указано в подрисуночной подписи к фиг.11) значительно продлевает время выживания после введения RML скрейпи (р<0,01). Средняя продолжительность периодов выживания представлена в днях ± среднеквадратичное отклонение.
Как показано на фиг.11, представленные результаты экспериментов показывают, что i) лечение соединением также эффективно при пероральном введения, ii) лечение также эффективно, когда его начинают на поздней клинической стадии развития болезни, и iii) более продолжительное лечение приводит к более продолжительному периоду выживания.
Пример 13: Зависимое от дозы действие соединения anle138b на уровни содержания PrPSc в мозге
C57BL/6 мышей инокулируют интрацеребрально (i.c.) 30 мкл 1% мозгового гомогената (RML скрейпи). Лечение начинают спустя 80 дней после инфицирования пероральным введением различных количеств соединения anle138b (как показано на Фиг.12) в смеси с буфером ДМСО + арахисовое масло. На день 120 после инфицирования животных забивают и определяют количество PrPSc в мозге в сравнении с животными, забитыми на 80 день после инфицирования.
Данные, представленные на фиг.12, показывают, что соединение anle138b снижает PrPSc аккумуляцию в мозге зависимым от дозы образом.
Пример 14: Количественное определение PrPC посредством иммуноблоттинга ткани мозга от неинфицированных мышей, принимавших по 1 мг в день соединения anle138b в смеси с буфером ДМСО + арахисовое масло в течение 1 недели
Как показано на фиг.13, не наблюдается снижения уровня содержания PrPC у мышей, принимающих соединение anle138b по сравнению с контрольными мышами.
Данные результаты экспериментов показывают, что терапевтическое действие в скрейпи-инфицированных мышах не обусловлено снижением экспрессии PrPC, но является результатом ингибирования образования патологически агрегированных разновидностей белков.
Пример 15: Фармакокинетический анализ соединения anle138b
Разовую дозу соединения anle138b вводят неинфицированным C57BL/6 мышам, как показано на фиг.14. В различные моменты времени после введения определяют количество соединения в мозге и сыворотке для 2 животных в каждую временную точку и для каждой экспериментальной группы с помощью ЖХ-МС.
Результаты данного эксперимента показывают, что имеет место хорошая биологическая доступность при перорального введении и хорошее проникновение в мозг. Соединение anle138c обнаруживают в крови мышей, так что допускается, что оно является метаболитом соединения 138b.
Пример 16: Ингибирование образования скоплений α-синуклеина различными соединениями
Агрегацию α-синуклеина индуцируют ДМСО и 10 мкМ FeCl3 и анализируют конфокальной спектроскопией одиночных молекул, как описано в публикации Kostka et al. (J Biol Chem (2008) 283: 10992-11003). Влияние различных соединений, добавленных в концентрации 10 мкМ, на эффект образования промежуточных II олигомеров исследуют в сравнении с контролем (без соединения), как показано в таблице 2. Структуры соответствующих соединений представлены на фиг.15.
Результаты данного эксперимента показывают, что указанные соединения ингибируют образование токсичных скоплений α-синуклеина и что указанных структурно схожие соединения обладают потенциалом применения для лечения заболеваний, связанных с агрегацией белков, и, в частности, для лечения заболеваний, при которых может наблюдаться агрегация α-синуклеина.
Пример 16: Действие соединений in vivo в модели болезни Паркинсона на мышах
Экспериментальные данные подтверждают, что в экпериментальных моделях болезни Паркинсона с применением митохондриальных токсинов, таких как MPTP и ротенон, в подходящих концентрациях, может наблюдаться образование агрегированного α-синуклеина, что способствует гибели нейронных клеток. Мышам вводят MPTP (ежедневно, 30 мг/кг массы тела) интраперитонеальной инъекцией для индуцирования дегенерации допаминергических нейронов в substantia nigra. Животных (3-10 на экспериментальную группу) лечат различными соединениями или разбавителем (ежедневно 250 мг/кг массы тела, пероральное введение (зондовое питание соединением в 12,5 мкл ДМСО в смеси с 487,5 мкл оливкового масла) в 0-12 дни). Количественное определение потери нейронов в сравнении с мышами, не обработанными MPTP (контроль, определен как 0% гибели клеток), и мышами, обработанными MPTP, которых лечат только разбавителем (ДМСО, определены как 100% гибели клеток), проводят на 12 день. Для количественного определения тирозингидроксилазы (TH)-положительных клеток (substantia nigra parts compacta - SNpc) секции толщиной 50 мкм окрашивают с использованием иммунной метки анти-ТН-антитело. Каждую вторую секцию внутри SNpc анализируют с использованием программного обеспечения Stereo investigator (MicroBrightfield, Colchester, VT, USA). Клетки, окрашенные с использованием иммунной метки, подсчитывают с применением оптической фракционирующей колонки с 20× объективом. Стереологические подсчеты проводят вслепую двумя независимыми исследователями.
Экспериментальные данные, представленные на фиг.16, показывают, что испытываемые соединения снижают гибель клеток в модели болезни Паркинсона в условиях in vivo.
Пример 17: Влияние соединения anle138c на агрегацию Abeta
Abeta40 в концентрации 50 мкМ инкубируют в течение 30 часов в следующих условиях: 50 мМ фосфата натрия, 50 мМ хлорида натрия, 0,01% азида натрия, рН 7,4, 37°C; - перемешивают магнитными мешалками с добавлением или без добавления 50 мкМ соединения anle138c. К контрольному образцу добавляют ДМСО в такой же концентрации, что и в опытный образец. Концентрация ДМСО равна 2% (об./об.), концентрация соединения anle138c с исходным раствором составляет 3 мМ. Пептидный раствор центрифугируют при 16000 г в течение 15 минут перед DLS экспериментами.
В то время как самый большой пик в мономерном Abeta40 соответствует гидродинамическому радиусу примерно 1,5 нм, пик олигомера находится при приблизительно 30 нм (верхняя панель), агрегатное состояние Abeta40 в присутствии anle138C (средняя панель) показало пик олигомера вблизи 20 нм в дополнение к пику мономера. Нижняя панель показывает распределение частиц по размерам для амилоидного фибриллярного состояния Abeta40, измеренного после центрифугирования образца. Агрегацию ABeta анализируют динамическим рассеянием света. DLS измерения проводят дважды при 25°C на аппарате DynaPro Titan (Wyatt Technology Corp., CA) с лазером 827,08 нм. Угол рассеивания составляет 90°. DLS измерение состоит из двадцати измерений продолжительностью 10 секунд. Показатель преломления (RI) раствора устанавливают на множество в 1,333 при 589 нм и 20°C, и RI при длине волне исследования получают в соответствии с уравнением Коши (Cauchy) с коэффициентом 3119 нм2. Вязкость равна 1,019 сп при 20°C, и температура-зависимые изменения рассчитывают с помощью водной модели. Распределение частиц по размерам определяют методом ограниченной регуляризации.
Полученные экспериментальные данные, которые представлены на фиг.17, показывают, что соединение anle138c ингибирует образование больших Abeta40 олигомеров, соединение anle138c и соединения близкого химического строения также могут воздействовать на агрегацию Abeta и могут применяться для терапевтических и диагностических целей при таких заболеваниях, как болезнь Альцгеймера, которые нейропатологически характеризуются скоплением агрегированного Abeta.
Альтернативный вариант соединений согласно настоящему изобретению суммирован в следующих пунктах. Данные соединения могут применяться способом, аналогичным способу применения описанных выше соединений согласно изобретению.
1. Соединение, представленное формулой (I)

где
X, Y и L независимо ненаправленно выбраны из -C(R11)(R12)-, -C(R13)=, -N(R14)-, -N=, -N+(R17)=, -O- и -S-;
M и Z независимо ненаправленно выбраны из
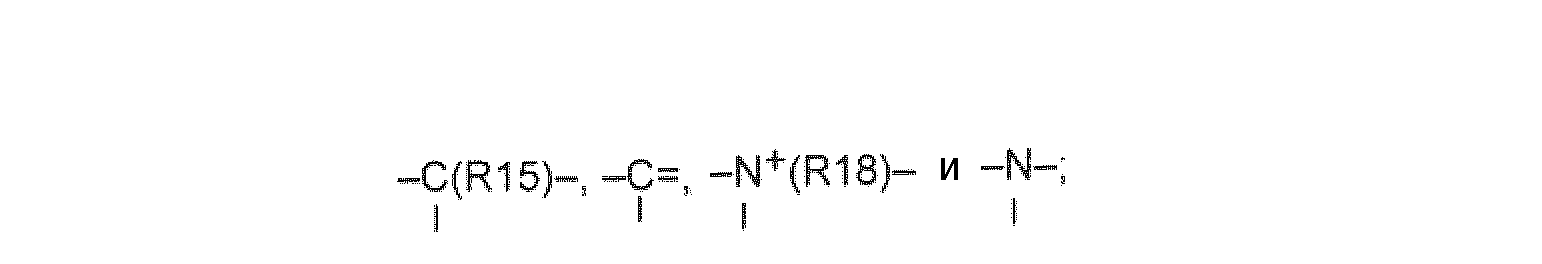
----- представляет необязательную двойную связь;
R1-R15, или R17, или R18 независимо выбраны из водорода, галогена, циано, гидроксильной группы, нитро, амино, азидо, сульфонила, тио, фосфонила, карбоксильной группы, карбониламидо, алкила, алкенила, алкинила, алкокси, ацила, ацилокси, ациламино, карбоциклической группы, карбоциклоокси, карбоциклоалкила, карбоциклоалкенила, арила, арилалкила, арилалкенила, арилалкинила, арилокси, арилалкокси, гетероциклической группы, гетероциклоокси, гетероциклоалкила, гетероарильной группы, гетероарилокси, гетероарилалкила, гетероарилалкенила и гетероарилалкокси, или две соседние группы могут соединяться с образованием мостиковой группы, содержащей от 1 до 6 атомов углерода, где один или два атома углерода могут замещаться -O-, -S- или -N(R')-, где R' выбран из H и C1-4 алкила; каждый из которых является необязательно замещенным; а также его пролекарство, сложный эфир, сольват или соль;
при условии, что соединение не является одним из следующих соединений (а), (b) или (c)

2. Соединение по п.1, где цикл A ненаправленно выбран из следующих структур:

3. Соединение по п.1 или 2, где R7 представляет собой галоген, циано, гидроксильную группу, нитро, азидо, алкокси, тио, алкилтио, амино, галогеналкокси, алкил или галогеналкил.
4. Соединение по любому из пп.1-3, где R2 и R3, каждый независимо, выбран из гидроксильной группы и C1-6 алкокси; или R2 и R3 вместе образуют структуру -O-(CH2)n-O-, где n принимает значения от 1 до 3, предпочтительно n равно 1.
5. Соединение, представленное формулой (I)
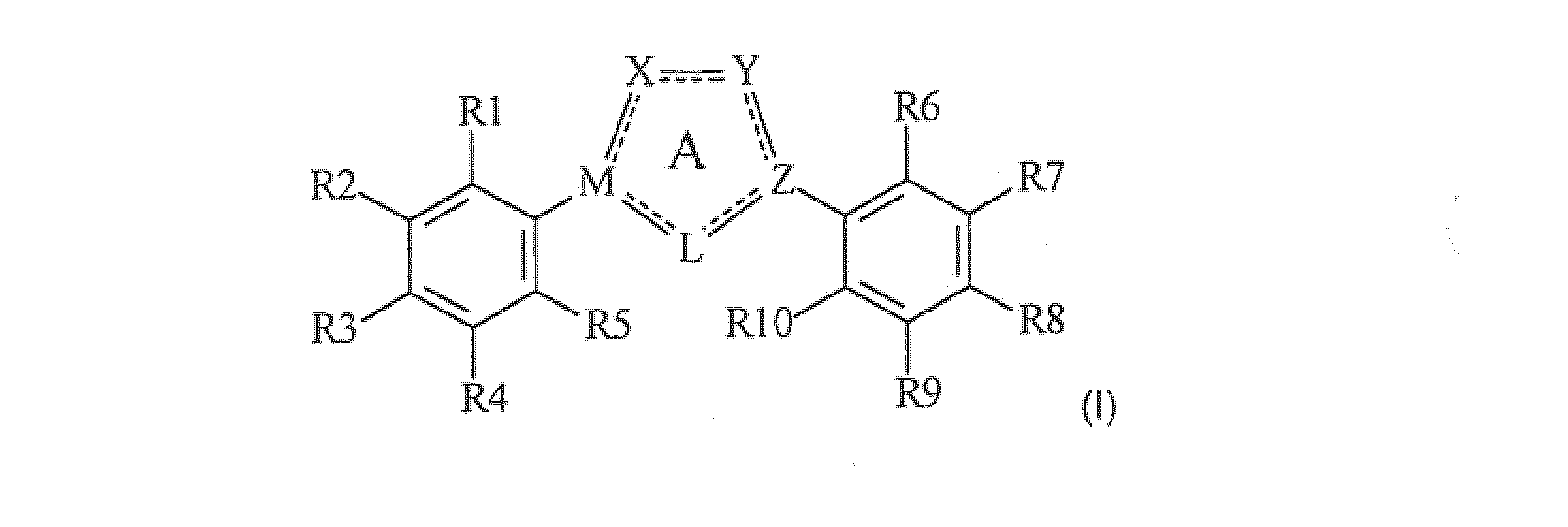
где
X, Y и L независимо ненаправленно выбраны из -C(R11)(R12)-, -C(R13)=, -N(R14)-, -N=, -N+(R17)=, -O- и -S-;
M и Z независимо ненаправленно выбраны из
----- представляет собой возможную двойную связь;
R1-R15, или R17, или R18 независимо выбраны из водорода, галогена, циано, гидроксильной группы, нитро, амино, азидо, сульфонила, тиогруппы, фосфонила, карбоксильной группы, карбониламидо, алкила, алкенила, алкинила, алкокси, ацила, ацилокси, ациламино, карбоциклической группы, карбоциклоокси, карбоциклоалкила, карбоциклоалкенила, арила, арилалкила, арилалкенила, арилалкинила, арилокси, арилалкокси, гетероциклической группы, гетероциклоокси, гетероциклоалкила, гетероарильной группы, гетероарилокси, гетероарилалкила, гетероарилалкенила и гетероарилалкокси, или две соседние группы могут соединяться с образованием мостиковой группы, содержащей от 1 до 6 атомов углерода, где один или два атома углерода могут быть замещены группами -O-, -S- или -N(R')-, где R' выбран из H и C1-4алкила; каждый из которых является необязательно замещенным;
а также его пролекарство, сложный эфир, сольват или соль;
для применения в лечении или предотвращении заболевания, связанного с агрегацией белков, и/или нейродегенеративного заболевания.
6. Применение соединения, представленного формулой (I), как определено в п.5, для получения фармацевтической композиции для лечения или предотвращения заболевания, связанного с агрегацией белков, или нейродегенеративного заболевания.
7. Способ лечения или предотвращения заболевания, связанного с агрегацией белков, и/или нейродегенеративного заболевания, включающий введение терапевтически эффективного количества соединения, представленного формулой (I), как определено в п.5, пациенту, нуждающемуся в этом.
8. Способ идентификации соединения для ингибирования агрегации белков, вовлеченных в заболевание, связанное с агрегацией белков, или нейродегенеративное заболевание, включающий следующие стадии:
контактирование меченого мономерного белка со скоплением иным образом меченного указанного белка (1) в присутствии и/или (2) в отсутствие возможного кандидата ингибитора агрегации, который представляет собой соединение по п.5;
определение количества меток, локализованных в непосредственной близости друг к другу, указанное количество показывает степень связывания мономерного белка в скоплении указанного белка; и
сравнение результата, полученного в присутствии указанного соединения, с результатом, полученным в отсутствие указанного соединения,
где снижение количества меток, локализованных в непосредственной близости друг к другу в присутствии указанного соединения, свидетельствует о способности соединения ингибировать агрегацию указанного белка.
9. Способ по п.8, где указанные метки представляют собой флуоресцентные метки.
10. Способ по п.8 или 9, где указанные метки присоединены к антителу или фрагменту антитела, специфически связанного с указанным белком.
11. Способ по п.10, где указанное антитело или фрагмент антитела может различать агрегированный и мономерный белок.
12. Способ по любому из пп.8-11, где количество меток, расположенных в непосредственной близости друг к другу, определено с использованием метода «сканирования интенсивно флуоресцирующих мишеней (SIFT)», метода флуоресцентного резонансного переноса энергии (FRET) или метода конфокальной визуализации высокого разрешения.
13. Способ по любому из пп.8-12, где указанные мономерные и агрегированные белки выбраны из группы, включающей прионный белок, амилоидный белок-предшественник (АРР), альфа-синуклеин, супероксиддисмутазу, тау-белок, иммуноглобулин, амилоид-А, транстиретин, бета2-микроглобулин, цистатин С, аполипопротеин А1, TDP-43, островковый амилоидный полипептид, ANF, гельзолин, инсулин, лизозим, фибриноген, хантингтин, атаксин и другие белки с поли-Q-последовательностью и фрагменты или производные указанных белков.
14. Способ по п.13, где указанные мономерный белок представляет собой прионный белок и указанный агрегированный белок представляет собой PrPsc.
15. Способ по п.13, где указанный мономерный белок представляет собой альфа-синуклеин и указанный агрегированный белок выбран из группы, включающей олигомеры, протофибриллы или фибриллы альфа-синуклеина.
16. Способ отбора соединения с in vivo эффективностью для лечения или предотвращения заболевания, связанного с агрегацией белков, или нейродегенеративного заболевания, указанный способ включает следующие стадии:
(а) введение соединения-кандидата, как определено в п.5, в культуру клеток или животному, которое не является человеком, содержащему способную к агрегации изоформу белка, как определено по любому из пп.13-15;
(b) определение количества поддающихся визуализации скоплений;
(с) идентификация и отбор соединения, которое способно уменьшить скопления или образование скоплений указанного белка или способно увеличить время выживания культуры клеток или животного, которое не является человеком.
17. Применение соединения, как определено в п.5, для ингибирования агрегации белков in vitro, в организме животного или ex vivo.
18. Фармацевтическая или диагностическая композиция, включающая соединение, как оно определено в п.5, и, необязательно, фармацевтически приемлемый носитель.
19. Соединение по любому из пп.1 или 5, применение по п.6, способ по любому из пп.7-16, применение по п.17 или композиция по п.18, где указанное соединение выбрано из группы, включающей


где каждый R16 независимо выбран из Н и С1-4 алкила; или две соседние группы R16 могут соединяться с образованием мостиковой группы, содержащей от 1 до 3 атомов углерода;
а также его пролекарство, сложный эфир, сольват или соль.
20. Способ по любому из пп.16 или 19, применение по п.17 или 19, диагностическая композиция по п.18 или 19, где указанное соединение мечено обнаруживаемой меткой.
21. Способ по любому из пп.16, 19, 20 или применение по любому из пп.17, 19, 20, где два или несколько указанных соединений применяются одновременно.
22. Соединение по любому из пп.5 или 19, где указанное заболевание, связанное с агрегацией, характеризуется наличием агрегированной формы по меньшей мере одного белка или его фрагмента или производного, где белок выбран из группы, включающей прионный белок, амилоидный белок-предшественник (АРР), альфа-синуклеин, супероксиддисмутазу, тау-белок, иммуноглобулин, амилоид-А, транстиретин, бета-2-микроглобулин, цистатин С, аполипопротеин А1, TDP-43, островковый амилоидный полипептид, ANF, гельзолин, инсулин, лизозим, фибриноген, хантингтин, атаксин и другие белки с поли-Q-последовательностью.
23. Соединение по любому из пп.5, 19 или 22, где указанное заболевание выбрано из группы, включающей болезнь Альцгеймера, прионную болезнь, болезнь Паркинсона, множественную системную атрофию, болезнь диффузных телец Леви, лобно-височную деменцию, боковой амиотрофический склероз, болезнь Хантингтона, атаксию, связанную с заболеваниями спинного мозга и мозжечка, и другие поли-Q-заболевания, наследственную церебральную амилоидную ангиопатию, наследственную амилоидную полиневропатию, первичный системный амилоидоз (AL амилоидоз), реактивный системный амилоидоз (AA амилоидоз), диабет II типа, инъекция-локализованный амилоидоз, бета-2-микроглобулиновый амилоидоз, наследственный не невропатический амилоидоз и наследственный финский системный амилоидоз.
24. Соединение по п.23, где указанная прионная болезнь выбрана из болезни Крейтцфельдта-Якоба, варианта болезни Крейтцфельдта-Якоба, генетической прионной болезни человека, коровьей губчатой энцефалопатии (BSE) и скрейпи.
25. Набор, включающий соединение, как оно определено в любом из пп.5 и 18-24 и, дополнительно, антитело или фрагмент антитела, специфически связывающиеся с указанным соединением; и/или мономерный или агрегированный белок, как он определен в пп.13-15; и/или мономерный или агрегированный белок, как он определен в пп.13-15. необязательно связанный в комплекс с указанным соединением; и инструкции по применению - в одном или нескольких контейнерах.
Термин «галоген», когда используется в данном описании, относится к атому галогена, выбранному из фтора, хлора, брома и йода, предпочтительно, брому.
Термин «карбокси», когда используется в данном описании, относится к группе -СООН.
Термины «алкил» и «алк» относится к алкановому (углеводородному) радикалу с прямой или разветвленной цепью, содержащему от 1 до 12 атомов углерода, предпочтительно от 1 до 6 атомов, более предпочтительно от 1 до 4 атомов углерода. Типичные примеры таких групп включают, но без ограничения, метил, этил, пропил, изопропил, н-бутил, трет-бутил, изобутил, пентил, гексил, изогексил, гептил.
Термин «алкенил» относится к углеводородному радикалу с прямой или разветвленной цепью, содержащему от 2 до 12 атомов углерода, предпочтительно от 2 до 6 атомов углерода и по меньшей мере одну двойную углерод-углеродную связь. Типичные примеры таких групп включают этенил или аллил.
Термин «алкинил» относится к углеводородному радикалу с прямой или разветвленной цепью, содержащему от 2 до 12 атомов углерода, предпочтительно от 2 до 6 атомов углерода, и по меньшей мере один атом углерода с тройной углеродной связью. Типичные примеры таких групп включают этинил.
Термин «алкокси» относится к алкильной группе, которая определена выше, присоединяемой через кислородный мостик (-О-).
Термин «ацильная группа» в соответствии с данным изобретением относится к функциональной группе, в которой алкил, арил, гетероциклил или гетероарил присоединен к карбонильной группе. Примерами ацильных групп являются формильная группа; С1-6 алкилкарбонильная группа, такая как ацетильная группа, пропионильная группа, бутирильная группа и пивалоильная группа; С2-6 алкенилкарбонильная группа, такая как этеноильная группа, пропеноильная группа и бутеноильная группа; ароильная группа, такая как бензоильная группа и т.п., предпочтительно ацетильная группа.
Термин «ацилокси», когда используется в данном описании, относится к ацильной группе, которая связана с -О-. Аналогично, термин «ациламино» относится к ацильной группе, которая связана с группой -N(R”)-, где R” представляет собой Н или С1-6 алкил.
Термин «карбоциклическая группа» относится к полностью насыщенной циклической углеводородной группе, содержащей от 1 до 4 циклов, предпочтительно 1 цикл, и от 3 до 8 атомов углерода в цикле. Типичные примеры таких групп включают циклопропил, циклобутил, циклопентил, циклогексил и т.д.
Термин «карбоциклоокси» относится к карбоциклической группе, как она определена выше, присоединяемой через кислородный мостик (-О-).
Термин «карбоциклоалкил» относится к алкильной группе, замещенной карбоциклической группой, где карбоциклическая группа и алкил представляют собой группы, определенные выше.
Термин «карбоциклоалкенил» относится к алкенильной группе, замещенной карбоциклической группой, где карбоциклическая группа и алкенил представляют собой группы, определенные выше.
Термин «арил» относится к циклическим ароматическим углеводородным группам, которые содержат от 6 до 20, предпочтительно от 6 до 10 атомов углерода в основной цепи молекулы и включают от 1 до 3 ароматических цикла, в частности моноциклическим или бициклическим группам, таким как фенил, бифенил или нафтил. В том случае, когда в группе содержатся два или несколько ароматических циклов (бициклическая и т.д.), ароматические циклы арильной группы могут соединяться в одной точке (например, бифенил) или могут быть конденсированными (например, нафтил, фенантренил и т.п.).
Термин «арилалкил» относится к алкильной группе, замещенной арильной группой, где арил и алкил представляют собой группы, определенные выше.
Термин «арилалкенил» относится к алкенильной группе, замещенной арильной группой, где арил и алкенил представляют собой группы, определенные выше.
Термин «арилалкинил» относится к алкинильной группе, замещенной арильной группой, где арил и алкинил представляют собой группы, определенные выше.
Термин «арилокси» относится к арильной группе, которая определена выше, присоединяемой через кислородный мостик (-О-), например, феноксигруппе, антрилоксигруппе, бифенилоксигруппе и т.п., предпочтительно феноксигруппе.
Термин «арилалкокси» относится к алкоксигруппе, замещенной арильной группой, где арил и алкокси представляют собой группы, определенные выше.
Термин «гетероциклическая группа» относится к полностью насыщенным или частично либо полностью ненасыщенным циклическим группам (например, 3-7-членным моноциклическим, 7-11-членным бициклическим или 10-16-членным трициклическим системам), которые содержат по меньшей мере один гетероатом в цикле, содержащем по меньшей мере один атом углерода. Каждый цикл гетероциклической группы, содержащей гетероатом, может содержать 1, 2, 3 или 4 гетероатома, выбранных из атомов азота, кислорода и/или серы, где гетероатомы азота и серы могут быть необязательно окисленными, а гетероатомы азота могут быть необязательно кватернизованными. (Термин «гетероарилий» относится к гетероарильной группе, содержащей квартенизованный атом азота и, следовательно, положительный заряд). Гетероциклическая группа может присоединяться к остальной части молекулы через любой гетероатом или атом углерода цикла или циклической системы. Типичные примеры моноциклических гетероциклических групп включают этиленоксид, азетидинил, пирролидинил, пирролил, пиразолил, оксетанил, пиразолинил, имидазолил, имидазолинил, имидазолидинил, оксазолил, оксазолидинил, изоксазолинил, изоксазолил, тиазолил, тиадиазолил, тиазолидинил, изотиазолил, изотиазолидинил, фурил, тетрагидрофурил, тиенил, оксадиазолил, пиперидинил, пиперазинил, 2-оксопиперазинил, 2-оксопиперидинил, 2-оксопирролодинил, 2-оксоазепинил, азепинил, гексагидродиазепинил, 4-пиперидонил, пиридил, пиразинил, пиримидинил, пиридазинил, триазинил, триазолил, тетразолил, тетрагидропиранил, морфолинил, тиаморфолинил, тиаморфолинилсульфоксид, тиаморфолинилсульфон, 1,3-диоксолан, тетрагидро-1,1-диоксотиенил и т.п. Типичные примеры бициклических гетероциклических групп включают индолил, изоиндолил, бензотиазолил, бензодиоксолил, бензоксазолил, бензоксадиазолил, бензотиенил, хинуклидинил, хинолинил, тетрагидроизохинолинил, изохинолинил, бензимидазолил, бензопиранил, индолизинил, бензофурил, бензофуразанил, хромонил, кумаринил, бензопиранил, циннолинил, хиноксалинил, индазолил, пирролопиридил, фуропиридинил (такой как фуро[2,3-c]пиридинил, фуро[3,2-b]пиридинил или фуро[2,3-b]пиридинил), дигидробензодиоксинил, дигидродиоксидобензотиофенил, дигидроизоиндолил, дигидроиндолил, дигидрохинолинил, дигидрохиназолинил (такой как 3,4-дигидро-4-оксохиназолинил), триазинилазепинил, тетрагидрохинолинил и т.п. Типичные примеры трициклических гетероциклических групп включают карбазолил, бензиндолил, фенантролинил, дибензофуранил, акридинил, фенантридинил, ксантенил и т.п.
Термин «гетероциклоокси» относится к гетероциклической группе, которая определена выше, присоединяемой через кислородный мостик (-О-).
Термин «гетероциклоалкил» относится к алкильной группе, замещенной гетероциклической группой, где гетероциклическая группа и алкил представляют собой группы, определенные выше.
Термин «гетероарил», когда используется в данном описании, относится к 5-6-членному ароматическому циклу, который может содержать в качестве гетероатомов атом кислорода, серы и/или азота и с которым может быть конденсирован другой ароматический цикл. Примерами гетероарильных групп, но без ограничения, являются бензофуранил, фурил, тиенил, бензотиенил, тиазолил, имидазолил, оксазолил, оксадиазолил, тиадиазолил, бензотиазолил, триазолил, тетразолил, изоксазолил, изотиазолил, пирролил, пиранил, тетрагидропиранил, пиразолил, пиридил, хинолинил, изохинолинил, пуринил, карбазолил, бензоксазолил, бензамидазолил, индолил, изоиндолил, пиразинил, диазинил, пиразин, триазинилтриазин, тетразинил, тетразолил, бензотиофенил, бензопиридил и бензимидазолил.
Термин «гетероарилокси» относится к гетероарильной группе, которая определена выше, присоединяемой через кислородный мостик (-О-).
Термины «гетероарилалкил», «гетероарилалкенил» и «гетероарилалкинил» относятся к группам, в которых алкильная, алкенильная или алкинильная группа является замещенной гетероарильной группой, где гетероарил, алкил, алкенил и алкинил представляют собой группы, определенные выше.
Термин «гетероарилалкокси» относится к алкоксигруппе, замещенной гетероарильной группой, где гетероарильная и алкоксигруппа представляют собой группы, определенные выше.
Термин «замещенный», когда используется в данном описании, относится к группе, замещенной одним или несколькими заместителями, предпочтительно от 1 до 4 заместителями, в любой доступной точке присоединения. Типичные примеры заместителей включают, но без ограничения, одну или несколько из следующих групп: алкил, алкокси, галоген, гидроксильная группа, карбоксильная группа (т.е. -СООН), алкоксикарбонил, алкилкарбонилокси, аминогруппа (т.е. -NH2), тиольная или нитрогруппа.
В предпочтительном альтернативном варианте R1 выбран из группы, включающей водород и алкил, более предпочтительно водород.
В предпочтительном альтернативном варианте R2 выбран из группы, включающей гидроксильную и алкоксигруппу.
В предпочтительном альтернативном варианте R3 выбран из группы, включающей гидроксильную группу и алкоксигруппу.
В дополнительном предпочтительном альтернативном варианте осуществления R2 и R3 связаны и вместе образуют структуру -О(СН2)nO-, где n принимает значения от 1 до 3, предпочтительно n равно 1.
В предпочтительном альтернативном варианте R4 выбран из группы, включающей водород, гидроксильную группу и алкоксигруппу, более предпочтительно водород.
В предпочтительном альтернативном варианте R5 выбран из группы, включающей водород и алкил, более предпочтительно водород.
В предпочтительном альтернативном варианте R6 выбран из группы, включающей водород и алкил, более предпочтительно водород.
В предпочтительном альтернативном варианте R7 выбран из группы, включающей водород, галоген, циано, нитро, гидроксильную группу и алкоксигруппу, более предпочтительно R7 представляет собой водород, галоген, гидроксильную группу или алкоксигруппу, еще более предпочтительно R7 представляет собой галоген.
В предпочтительном альтернативном варианте R8 выбран из группы, включающей водород, гидроксильную группу и алкоксигруппу.
В предпочтительном альтернативном варианте R9 выбран из группы, включающей водород, галоген, гидроксильную группу и алкоксигруппу.
В предпочтительном альтернативном варианте осуществления R10 выбран из группы, включающей водород и алкил, более предпочтительно водород.
В предпочтительном альтернативном варианте R11 выбран из группы, включающей водород и алкил.
В предпочтительном альтернативном варианте R12 выбран из группы, включающей водород и алкил.
В предпочтительном альтернативном варианте R13 выбран из группы, включающей водород и алкил.
В предпочтительном альтернативном варианте R14 выбран из группы, включающей водород и алкил.
В предпочтительном альтернативном варианте R15 выбран из группы, включающей водород и алкил.
В еще одном предпочтительном варианте осуществления соединения согласно изобретению R7 представляет собой галоген, циано, гидроксильную группу или нитро, азидо, алкокси, тио, алкилтио, амино, гелогеналкокси, алкил или галогеналкил.
В предпочтительном альтернативном варианте осуществления R7 представляет собой галоген, циано, гидроксильную группу или нитро, более предпочтительно галоген.
В дополнительном предпочтительном альтернативном варианте осуществления R2 и R3, каждый независимо, выбран из гидроксильной группы и С1-6 алкокси; или R2 и R3 вместе образуют структуру -О-(СН2)n-О-, где n принимает значения от 1 до 3, предпочтительно n равно 1.
В предпочтительном альтернативном варианте осуществления соединение выбрано из группы, включающей


где каждый R16 независимо выбран из Н и С1-4 алкила; или две соседние группы R16 могут соединяться с образованием мостиковой группы, содержащей от 1 до 3 атомов углерода; а также его пролекарства, сложных эфиров, сольватов или солей.
В более предпочтительном альтернативном варианте осуществления соединение выбрано из группы, включающей
3-(4-гидроксифенил)-5-(3,4-дигидроксифенил)изоксазол

3,5-бис(3,4-диметоксифенил)пиразол

5-(3-бромфенил)-3-(3,4-метилендиоксифенил)пиразол

5-(3-фторфенил)-3-(3,4-метилендиоксифенил)пиразол

3-(3,4-дигидроксифенил)-5-(3-фторфенил)пиразол

гидробромид 2,4-бис(3,4-дигидроксифенил)имидазола

3-(3,4-диметоксифенил)-5-(3,4,5-триметоксифенил)пиразол

гидробромид 5-(3,5-дибромфенил)-3-(3,3,5-тригидроксифенил)пиразола

5-(3-бромфенил)-3-(2-гидроксифенил)пиразол (10353_F11)
Соединения альтернативного варианта осуществления изобретения могут содержать обнаруживаемую метку.
Список цитированной литературы




Реферат
Изобретение относится к соединению, представленному формулой (Е)где X, Y и L независимо ненаправленно выбраны из -C(R)(R)-, -C(R)=, -N(R)-, -N= и -O-;M и Z независимо ненаправленно выбраны из;---- означает необязательную двойную связь; R, R, R, Rи Rнезависимо выбраны из водорода; Cалкила; группы -Cалкилен-галоген; группы -Cалкилен-OH; Hal выбран из F, Cl, Br и I; Rи Rприсоединены к соседним атомам углерода, и Rи Rвместе ненаправленно образуют структуру -T-(CRR)-V-, где T выбран из CRRи O или NH и V выбран из CRRи O или NH, а также соответствующие структуры, в которых присутствует двойная связь, причем по меньшей мере один из T или V представляет собой O или N; Rи Rпредставляют собой H или F; Rи Rпредставляют собой H; n принимает значения от 1 до 2; Rпредставляет собой Cалкильную группу; m принимает значения 0 или 1; Rпредставляет собой атом галогена; p принимает значения 0 или 1; а также к фармацевтическим и диагностическим композициям указанного соединения. 15 н. и 26 з.п. ф-лы.,17 ил, 2 табл., 17 пр.
Формула

где
X, Y и L независимо ненаправленно выбраны из -C(R1)(R2)-, -C(R3)=, -N(R4)-, -N= и -O-;
M и Z независимо не направленно выбраны из

---- означает необязательную двойную связь;
R1, R2, R3, R4 и R6 независимо выбраны из водорода; C1-4 алкила; группы -C1-4 алкилен-галоген; группы -C1-4 алкилен-OH;
Hal выбран из F, Cl, Br и I;
RE1 и RE2 присоединены к соседним атомам углерода, и RE1 и RE2 вместе ненаправленно образуют структуру -T-(CRE7RE8)n-V-, где T выбран из CRE9RE10 и O или NH, и V выбран из CRE9RE10 и O или NH, а также соответствующие структуры, в которых присутствует двойная связь, причем по меньшей мере один из T или V представляет собой O или N;
RE7 и RE8 представляют собой H или F;
RE9 и RE10 представляют собой H;
n принимает значения от 1 до 2;
RE3 представляет собой C1-6 алкильную группу;
m принимает значения 0 или 1;
RE4 представляет собой атом галогена;
p принимает значения 0 или 1;
а также его сложный эфир, сольват или соль;
при условии, что из указанного перечня исключены следующие соединения:


где
m, n, p, T, V, X, Y, L, M, Z, RE7, RE8 и Hal принимают значения, определенные в п.1;
RA1 и RA2 вместе ненаправленно образуют структуру -T-(CRE7RE8)n-V-;
RA3 представляет собой C1-6 алкильную группу;
RA4 представляет собой атом галогена;
а также его сложный эфир, сольват или соль.


где
R8 и R9 выбраны из водорода, -C1-4 алкилен-галогена, -C1-4 алкилен-OH.

где
R8 и R9 принимают значения, определенные в п.3.

где
R выбран из водорода, -C1-4 алкилен-галогена, -C1-4 алкилен-OH; и
Hal выбран из F, Cl, Br и I; и
RE7 и RE8 представляют собой H или F,
а также его сложный эфир, сольват или соль.

или

где Hal представляет собой Cl или Br;
а также его сложный эфир, сольват или соль.




а также его сложный эфир, сольват или соль.

где
RE1 выбран из -NRE5RE6;
RE2 выбран из водорода;
RE5 и RE6 независимо выбраны из водорода и C1-6 алкила;
X, Y и L независимо ненаправленно выбраны из -C(R1)(R2)-, -C(R3)=, -N(R4)-, -N=;
M и Z независимо ненаправленно выбраны из
---- означает необязательную двойную связь;
R1, R2, R3, R4 и R6 независимо выбраны из водорода;
Hal выбран из F, Cl, Br и I;
RE3 представляет собой C1-6 алкильную группу;
m принимает значения 0;
RE4 представляет собой атом галогена;
p принимает значения 0;
а также сложный эфир, сольват или соль соединения, представленного формулой (E);
при условии, что из указанного перечня исключены следующие соединения:
3(5)-(2-гидрокси-5-метилфенил)-5(3)-(4-хлорфенил)пиразол;
орто-гидроксифенил 5-дихлор-3′,4′ фенил-3 метил-2-пиразол;
орто-гидроксифенил-5 дихлор-3′,4′ фенил-3 фенил-2 пиразол;





где
R8 и R9 представляют собой водород.


или
где
R представляет собой H;
Hal выбран из F, Cl, Br и I;
RA9 представляет собой H или C1-4 алкил;
RA10 представляет собой H или C1-4 алкил;
а также его сложный эфир, сольват или соль.

или
где
Hal выбран из Cl или Br;
а также его сложный эфир, сольват или соль.

где
RE1 и RE2 независимо выбраны из гидроксильной группы и C1-6 алкокси и присоединены в мета и пара по отношению к атому углерода, который связывает фенильное кольцо с циклом D;
X, Y и L независимо ненаправленно выбраны из -C(R1)(R2)-, -C(R3)=, -N(R4)-, -N= и -O-;
M и Z независимо ненаправленно выбраны из
---- означает необязательную двойную связь;
R1, R2, R3, R4 и R6 независимо выбраны из водорода или -C(O)-C1-4 алкил;
Hal выбран из F, Cl, Br и I;
RE3 представляет собой C1-6 алкильную группу, метокси группу или OH;
m принимает значения 0 или 1;
RE4 представляет собой атом галогена;
p принимает значения 0 или 1;
а также его сложный эфир, сольват или соль.

где
m, p, X, Y, L, M, Z, R1, R2, R3, R4 и R6 и Hal принимают значения, определенные в п.13;
где в формуле (A):
RA1 и RA2 каждый независимо выбран из гидроксильной группы и C1-6 алкокси;
RA3 представляет собой C1-6 алкильную группу;
m=0, 1
RA4 представляет собой атом галогена;
p=0, 1,
а также его сложный эфир, сольват или соль.


где
R8 и R9 представляют собой водород или -C(O)-C1-4алкил.

где
R8 и R9 представляют собой водород или -C(O)-C1-4алкил.

где
R представляет собой H;
Hal выбран из F, Cl, Br и I;
RA7 представляет собой H или C1-4 алкил;
RA8 представляет собой H или C1-4 алкил;
а также его сложный эфир, сольват или соль.


где
Hal представляет собой Cl или Br;
а также его сложный эфир, сольват или соль.

где
X, Y и L независимо ненаправленно выбраны из -C(R1)(R2)-, -C(R3)=, -N(R4)-, -N= и -O-;
M и Z независимо ненаправленно выбраны из
---- означает необязательную двойную связь;
R1, R2, R3, R4 и R6 независимо выбраны из водорода; C1-4 алкила; группы -C1-4 алкилен-галоген; группы -C1-4 алкилен-OH;
Hal выбран из F, Cl, Br и I;
RE1 и RE2 присоединены к соседним атомам углерода, и RE1 и RE2 вместе ненаправленно образуют структуру -T-(CRE7RE8)n-V-, где T выбран из CRE9RE10 и O или NH и V выбран из CRE9RE10 и O или NH, а также соответствующие структуры, в которых присутствует двойная связь, причем по меньшей мере один из T или V представляет собой O или N;
RE7 и RE8 представляют собой H или F;
RE9 и RE10 представляют собой H;
n принимает значения от 1 до 2;
RE3 представляет собой C1-6 алкильную группу;
m принимает значения 0;
RE4 представляет собой атом галогена;
p принимает значения 0 или 1;
а также его сложный эфир, сольват или соль.

где
RE1 выбран из -NRE5RE6;
RE2 выбран из водорода;
RE5 и RE6 независимо выбраны из водорода и C1-6 алкила;
X, Y и L независимо ненаправленно выбраны из -C(R1)(R2)-, -C(R3)=, -N(R4)-, -N=;
M и Z независимо ненаправленно выбраны из
---- означает необязательную двойную связь;
R1, R2, R3, R4 и R6 независимо выбраны из водорода;
Hal выбран из F, Cl, Br и I;
RE3 представляет собой C1-6 алкильную группу;
m принимает значения 0;
RE4 представляет собой атом галогена;
p принимает значения 0;
а также его сложный эфир, сольват или соль соединения.

где
RE1 и RE2 независимо выбраны из гидроксильной группы и C1-6 алкокси и присоединены в мета и пара по отношению к атому углерода, который связывает фенильное кольцо с циклом D;
X, Y и L независимо ненаправленно выбраны из -C(R1)(R2)-, -C(R3)=, -N(R4)-, -N= и -O-;
M и Z независимо ненаправленно выбраны из
---- означает необязательную двойную связь;
R1, R2, R3, R4 и R6 независимо выбраны из водорода или -C(O)-C1-4 алкил;
Hal выбран из F, Cl, Br и I;
RE3 представляет собой C1-6 алкильную группу, метокси группу или OH;
m принимает значения 0 или 1;
RE4 представляет собой атом галогена;
p принимает значения 0 или 1;
а также его сложный эфир, сольват или соль.
(i) введение субъекту обнаруживаемого количества композиции, включающей соединение с обнаруживаемой меткой, определенному по любому одному из пп.1-18, где дисклейм, указанный в пп.1-8, не применяется, или его сложного эфира, сольвата или соли;
(ii) предоставление периода времени, достаточного для связывания соединения с агрегированным белком; и
(iii) обнаружение соединения, связанного с агрегированным белком.
Комментарии