Ингибиторы неприлизина - RU2663618C2
Код документа: RU2663618C2
Описание
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новым соединениям, которые метаболизируются in vivo до соединений, обладающих активностью в качестве ингибиторов неприлизина. Настоящее изобретение также относится к фармацевтическим композициям, содержащим указанные соединения, к процессам и промежуточным продуктам получения таких соединений, и к способам применения таких соединений для лечения заболеваний, таких как гипертензия, сердечная недостаточность, легочная гипертензия и заболевание почек.
УРОВЕНЬ ТЕХНИКИ
В принадлежащей тому же правообладателю публикации патента США №2012/0157383, зарегистрированной 14 декабря 2011 года, раскрытие которой включено в настоящий документ посредством ссылки, Gendron et al. описали новые соединения, которые обладают активностью в качестве ингибиторов неприлизина. В частности, описаны соединения типа:

В зависимости от переменных, соединения такого типа могут быть охарактеризованы как находящиеся в активной форме или находящиеся в виде пролекарства, которое метаболизируется in vivo с получением активной формы соединения.
Однако, несмотря на такие соединения, остается потребность в соединениях и пролекарствах такого типа, которые характеризуются различными свойствами метаболизма и расщепления. Например, остается потребность в активных соединениях и/или пролекарственных соединениях, имеющих улучшенную пероральную абсорбцию, и в пролекарственных соединениях, которые претерпевают быстрое расщепление с образованием активного соединения. Настоящее изобретение направлено на эту потребность.
КРАТКОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Согласно одному аспекту, настоящего изобретения относится к соединению формулы XII:

где:
(i) Ra представляет собой H; Rb представляет собой Cl; X представляет собой

R2 представляет собой H, R4 представляет собой -OH, и R7 выбирают из -CH2CF3, -(CH2)2CF3, -CH2CF2CH3, CH2CF2CF3, -C(CH3)(CF3)2, -CH(CH2CH3)CF3, -CH(CH3)CF2CF3, -(CH2)2-3OH, CH2CH(NH2)COOCH3, -(CH2)2OCH3, -CHRcOC(O)-C1-4алкила, -CHRcOC(O)O-C2-4алкила, -CHRcOC(O)O-циклогексила, -C2-4алкилен-N(CH3)2, -CH2OC(O)CHRd-NH2, CH2OC(O)CHRd-NHC(O)O-C1-6алкила, бензила и
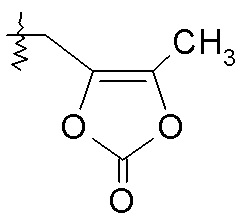
или R2 представляет собой H, R4 выбирают из -O-бензила, -OCHRcOC(O)-C1-4алкила, -OCH2OC(O)CHRd-NH2 и -OCH2OC(O)CHRd-NHC(O)O-C1-6алкила, и R7 выбирают из H и CH2OC(O)CH3; или R2 выбирают из -C(O)-C1-6алкила, -C(O)CHRd-NH2, -C(O)CHRd-NHC(O)O-C1-6алкила и -P(O)(ORe)2, R4 представляет собой -OH, и R7 представляет собой H; или
(ii) Ra представляет собой H; Rb представляет собой Cl; X представляет собой

R2 представляет собой H, R3 представляет собой -OH, и R7 выбирают из -CH2CF3, -(CH2)2CF3, -CH2CF2CH3, CH2CF2CF3, -C(CH3)(CF3)2, -CH(CH2CH3)CF3, -CH(CH3)CF2CF3, -(CH2)2-3OH, CH2CH(NH2)COOCH3, -(CH2)2OCH3, -CHRcOC(O)-C1-4алкила, -CHRcOC(O)O-C2-4алкила, -CHRcOC(O)O-циклогексила, -C2-4алкилен-N(CH3)2, -CH2OC(O)CHRd-NH2, CH2OC(O)CHRd-NHC(O)O-C1-6алкила, бензила и
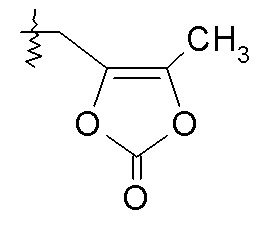
или R3 выбирают из -OC(O)CH2CH3, -OC(O)CH2CH(CH3)2, -OC(O)-фенила, OCH2OC(O)CHRd-NH2 и -OCH2OC(O)CHRd-NHC(O)O-C1-6алкила, и R7 представляет собой H; или R2 выбирают из -C(O)-C1-6алкила, -C(O)CHRd-NH2, -C(O)CHRd-NHC(O)O-C1-6алкила и P(O)(ORe)2, R3 представляет собой -OH, и R7 представляет собой H; или
(iii) Ra представляет собой H; Rb представляет собой Cl; X представляет собой
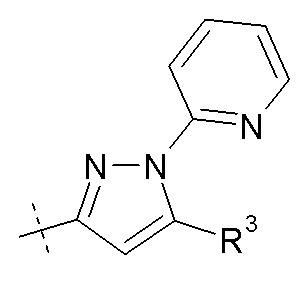
R2 представляет собой H, R3 представляет собой -OH, и R7 выбирают из -CH2CH3, -CH2CH(CH3)2, -CH2CF3, (CH2)2CF3, -CH2CF2CH3, -CH2CF2CF3, -C(CH3)(CF3)2, -CH(CH2CH3)CF3, -CH(CH3)CF2CF3, -(CH2)2-3OH, -CH2CH(NH2)COOCH3, -(CH2)2OCH3, -CHRcOC(O)-C14алкила, -CHRcOC(O)O-C2-4алкила, -CHRcOC(O)O-циклогексила, -C2-4алкилен-N(CH3)2, CH2OC(O)CHRd-NH2, -CH2OC(O)CHRd-NHC(O)O-C1-6алкил, бензила и

или R2 представляет собой H, R3 выбирают из -OC(O)CH2CH3, -OC(O)CH2CH(CH3)2, -OC(O)-фенила, OCH2OC(O)CHRd-NH2 и -OCH2OC(O)CHRd-NHC(O)O-C1-6алкила, и R7 представляет собой H; или R2 выбирают из -C(O)-C1-6алкила, -C(O)CHRd-NH2, -C(O)CHRd-NHC(O)O-C1-6алкила и P(O)(ORe)2, R3 представляет собой -OH, и R7 представляет собой H; или
(iv) Ra представляет собой F; Rb представляет собой Cl; X представляет собой

R2 представляет собой H, и R7 выбирают из H, -CH2CH3, -CH2CF3, -(CH2)2CF3, -CH2CF2CH3, CH2CF2CF3, -C(CH3)(CF3)2, -CH(CH2CH3)CF3, -CH(CH3)CF2CF3, -(CH2)2-3OH, CH2CH(NH2)COOCH3, -(CH2)2OCH3, -CHRcOC(O)-C1-4алкила, -CHRcOC(O)O-C2-4алкила, -CHRcOC(O)O-циклогексила, -C2-4алкилен-N(CH3)2, -CH2OC(O)CHRd-NH2, CH2OC(O)CHRd-NHC(O)O-C1-6алкила, бензила и

или R2 выбирают из -C(O)-C1-6алкила, -C(O)CHRd-NH2, -C(O)CHRd-NHC(O)O-C1-6алкила и -P(O)(ORe)2, и R7 представляет собой H; или
(v) Ra представляет собой H; Rb представляет собой Cl; X представляет собой

R2 представляет собой H, и R7 выбирают из H, -CH2CH3, -CH2CF3, -(CH2)2CF3, -CH2CF2CH3, CH2CF2CF3, -C(CH3)(CF3)2, -CH(CH2CH3)CF3, -CH(CH3)CF2CF3, -(CH2)2-3OH, CH2CH(NH2)COOCH3, -(CH2)2OCH3, -CHRcOC(O)-C1-4алкила, -CHRcOC(O)O-C2-4алкила, -CHRcOC(O)O-циклогексила, -C2-4алкилен-N(CH3)2, -CH2OC(O)CHRd-NH2, CH2OC(O)CHRd-NHC(O)O-C1-6алкила, бензила и
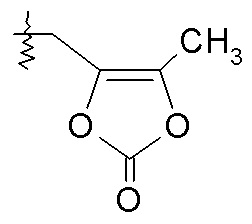
или R2 выбирают из -C(O)-C1-6алкила, -C(O)CHRd-NH2, -C(O)CHRd-NHC(O)O-C1-6алкила и -P(O)(ORe)2, и R7 представляет собой H; или
(vi) Ra представляет собой H; Rb представляет собой Cl; X представляет собой

R2 представляет собой H, и R7 выбирают из H, -CH2CH3, -CH2CF3, -(CH2)2CF3, -CH2CF2CH3, CH2CF2CF3, -C(CH3)(CF3)2, -CH(CH2CH3)CF3, -CH(CH3)CF2CF3, -(CH2)2-3OH, -CH2CH(NH2)COOCH3, -(CH2)2OCH3, -CHRcOC(O)-C1-4алкила, -CHRcOC(O)O-C2-4алкила, -CHRcOC(O)O-циклогексила, -С2-4алкилен-N(СН3)2, -СН2ОС(O)CHRd-NH2, -СН2ОС(O)CHRd-NHC(O)O-С1-6алкила, бензила и
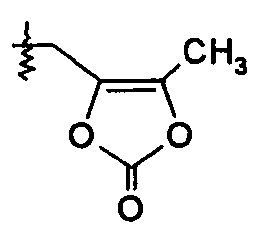
или R2 выбирают из -С(О)-C1-6алкила, -С(О)CHRd-NH2, -C(O)CHRd-NHC(O)O-C1-6алкила и -Р(О)(ORe)2, и R7 представляет собой Н; или
(vii) Ra представляет собой Н; Rb представляет собой Cl; X представляет собой

R2 представляет собой Н, и R7 выбирают из -СН2СН3, -CH2CF3, -(CH2)2CF3, -CH2CF2CH3, -CH2CF2CF3, -С(CH3)(CF3)2, -СН(CH2CH3)CF3, -CH(CH3)CF2CF3, -(СН2)2-3ОН, -CH2CH(NH2)СООСН3, -(СН2)2ОСН3, -CHRcOC(O)-С1-4алкила, -CHRcOC(О)O-С2-4алкила, -CHRcOC(O)O-циклогексила, -С2-4алкилен-N(СН3)2, -СН2ОС(О)CHRd-NH2, -СН2ОС(О)CHRd-NHC(О)O-C1-6алкила, бензила и
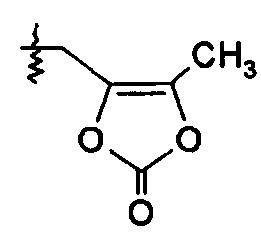
или R2 выбирают из -С(О)-C1-6алкила, -С(О)CHRd-NH2, -C(O)CHRd-NHC(О)O-C1-6алкила и -Р(O)(ORe)2, и R7 представляет собой Н; или
(viii) Ra представляет собой F; Rb представляет собой Cl; X представляет собой
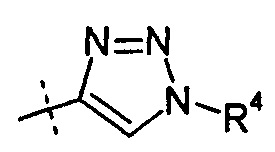
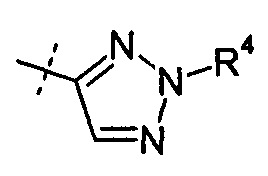
R2 представляет собой Н, R4 представляет собой -ОН, и R7 выбирают из -CH2CF3, -(CH2)2CF3, -CH2CF2CH3, -CH2CF2CF3, -С(СН3)(CF3)2, -CH(CH2CH3)CF3, -СН(CH3)CF2CF3, -(СН2)2-3ОН, -CH2CH(NH2)СООСН3, -(СН2)2OCH3, -CHRcOC(О)-С1-4алкила, -CHRcOC(О)О-С2-4-алкила, -CHRcOC(О)О-циклогексила, -С2-4алкилен-N(СН3)2, -СН2ОС(O)CHRd-NH2, -CH2OC(O)CHRd-NHC(O)O-С1-6алкила, бензила и

или R2 представляет собой Н, R4 выбирают из -O-бензила, -OCHRcOC(O)-С1-4алкила, -ОСН2ОС(О)СН[СН(СН3)2]NH2, -ОСН2ОС(O)СН[СН(СН3)2]-NHC(O)OCH3 и
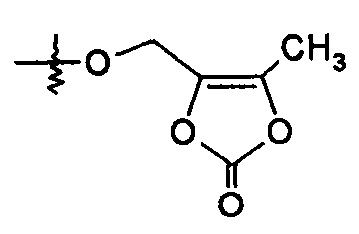
и R7 представляет собой Н; или R2 выбирают из -C(O)-C1-6алкила, -С(О)CHRd-NH2, -С(О)CHRd-NHC(О)O-C1-6алкила и -P(O)(ORe)2, R4 представляет собой -ОН, и R7 представляет собой Н; или
(ix) Ra представляет собой Н; Rb представляет собой Cl; X представляет собой

R2 представляет собой Н, и R7 выбирают из -СН2СН3, -CH2CF3, -(CH2)2CF3, -CH2CF2CH3, -CH2CF2CF3, -С(CH3)(CF3)2, -СН(CH2CH3)CF3, -CH(CH3)CF2CF3, -(CH2)2-3OH, -CH2CH(NH2)СООСН3, -(СН2)2ОСН3, -CHRcOC(O)-С1-4алкила, -CHRcOC(О)O-С2-4алкила, -CHRcOC(O)O-циклогексила, -С2-4алкилен-N(СН3)2, -СН2ОС(О)CHRd-NH2, -СН2ОС(О)CHRd-NHC(О)O-C1-6алкила, бензила и
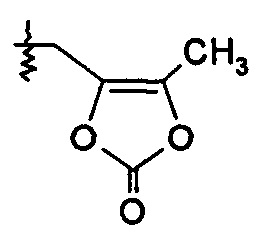
или R2 выбирают из -С(О)-C1-6алкила, -С(О)CHRd-NH2, -C(O)CHRd-NHC(О)O-C1-6алкила и -P(O)(ORe)2, и R7 представляет собой Н; или
(x) Ra представляет собой Н; Rb представляет собой Н; X представляет собой
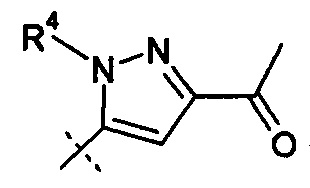

R2 и R4 представляют собой Н, и R7 выбирают из -CH2CF3, -(CH2)2CF3, -CH2CF2CH3, -CH2CF2CF3, -С(СН3)(CF3)2; -СН(СН2СН3)CF3, -CH(CH3)CF2CF3, -(СН2)2-3ОН, -СН2СН(NH2)СООСН3, -CHRcOC(O)-С1-4алкила, -CHRcOC(О)O-С2-4алкила, -CHRcOC(O)O-циклогексила, -С2-4алкилен-N(СН3)2, -СН2ОС(О)CHRd-NH2, -СН2ОС(О)CHRd-NHC(О)O-C1-6алкила и бензила; или R2 представляет собой Н, R4 выбирают из -СН2ОС(О)СН[СН(СН3)2]-NHC(О)ОСН3 и -CH2OC(O)CH[CH(CH3)2]NH2, и R7 представляет собой Н; или R2 выбирают из -С(O)-C1-6алкила, -С(О)CHRd-NH2, -С(О)CHRd-NHC(О)O-C1-6алкила и -Р(O)(ORe)2, R4 представляет собой Н, и R7 представляет собой Н; или R2 представляет собой Н, R4представляет собой -CH2OP(O)(ORe)2 или CH2OC(O)CH[CH(CH3)2]NH2, и R7 представляет собой -CH2CH3; или R2 представляет собой -C(O)CH[CH(CH3)2]NH2, R4 представляет собой H, и R7 представляет собой -CH2CH3; или
(xi) Ra представляет собой H; Rb представляет собой Cl; X представляет собой

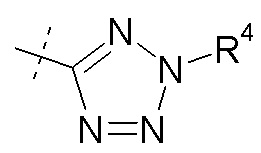
R2 и R4 представляют собой H, и R7 выбирают из H, -CH2CH3, -CH2CF3, -(CH2)2CF3, CH2CF2CH3, -CH2CF2CF3, -C(CH3)(CF3)2, -CH(CH2CH3)CF3, -CH(CH3)CF2CF3, (CH2)2-3OH, -CH2CH(NH2)COOCH3, -(CH2)2OCH3, -CHRcOC(O)-C1-4алкила, CHRcOC(O)O-C2-4алкила, -CHRcOC(O)O-циклогексила, -C2-4алкилен-N(CH3)2, CH2OC(O)CHRd-NH2, -CH2OC(O)CHRd-NHC(O)O-C1-6алкила и бензила; или R2 представляет собой H, R4 выбирают из -CH2OC(O)CH[CH(CH3)2]-NHC(O)OCH3 и CH2OC(O)CH[CH(CH3)2]NH2, и R7 представляет собой H; или R2 выбирают из -C(O)-C1-6алкила, C(O)CHRd-NH2, -C(O)CHRd-NHC(O)O-C1-6алкила и -P(O)(ORe)2, R4 представляет собой H, и R7 представляет собой H; или R2 представляет собой H, R4 представляет собой -CH2OP(O)(ORe)2 или -CH2OC(O)CH[CH(CH3)2]NH2, и R7 представляет собой -CH2CH3; или R2 представляет собой -C(O)CH[CH(CH3)2]NH2, R4 представляет собой H, и R7 представляет собой -CH2CH3;
где каждый Rc независимо представляет собой H или -C1-3алкил; каждый Rd независимо представляет собой H, -CH3, CH(CH3)2, фенил или бензил; и каждый Re независимо представляет собой H, -C1-6алкил или фенил;
или к его фармацевтически приемлемой соли.
Настоящее изобретение относится к соединениям, которые метаболизируются in vivo до соединений, которые, как было установлено, обладают ингибирующей активностью в отношении фермента неприлизина (NEP). Соответственно, предполагается, что соединения согласно настоящему изобретению применимы и полезны в качестве терапевтических средств для лечения пациентов, страдающих от заболевания или нарушения, которое подвергается лечению путем ингибирования фермента NEP или путем увеличения содержания его пептидных субстратов. Поэтому, согласно одному аспекту настоящее изобретение относится к способу лечения гипертензии, сердечной недостаточности или заболевания почек, включающему введение пациенту терапевтически эффективного количества соединения согласно настоящему изобретению.
Согласно другому аспекту настоящее изобретение относится к фармацевтическим композициям, содержащим фармацевтически приемлемый носитель и соединение согласно настоящему изобретению.
Согласно еще одному аспекту настоящее изобретение относится к процессам и промежуточным продуктам, применимым для получения соединений согласно настоящему изобретению. Согласно другому аспекту настоящее изобретение относится к процессу получения фармацевтически приемлемой соли соединения формулы I, включающему приведение соединения формулы I в форме свободной кислоты или основания в контакт с фармацевтически приемлемым основанием или кислотой. Согласно другим аспектам настоящее изобретение относится к продуктам, полученным посредством любого из процессов, описанных в настоящем документе, а также к новым промежуточным продуктам, использованным в таком процессе.
Согласно еще одному аспекту настоящее изобретение относится к применению соединения формулы I или его фармацевтически приемлемой соли для получения лекарственного средства, в особенности для получения лекарственного средства, применимого для лечения гипертензии, сердечной недостаточности или заболевания почек. Согласно другому аспекту настоящее изобретение относится к применению соединения согласно настоящему изобретению для ингибирования фермента NEP у млекопитающего. Согласно еще одному аспекту настоящее изобретение относится к применению соединения согласно настоящему изобретению в качестве средства исследования. Другие аспекты и варианты осуществления настоящего изобретения раскрыты в настоящем документе.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Если не указано иное, то при описании соединений, композиций, способов и процессов согласно настоящему изобретению следующие термины имеют следующие значения. Кроме того, если контекст применения прямо не предписывает иное, то используемые в настоящем документе формы неопределенные и определенные единственного числа включают соответствующие формы множественного числа. Термины «содержащий», «включающий» и «имеющий» призваны иметь охватывающий характер, и означают, что могут существовать дополнительные элементы, отличные от перечисленных элементов. Если не указано иное, то все используемые в настоящем документе числа, выражающие количества ингредиентов, свойства, такие как молекулярная масса, условия реакции, и т.д. следует понимать как модифицированные во всех возможных случаях термином «приблизительно». Соответственно, представленные в настоящем документе числа являются приближенными значениями, которые могут варьировать в зависимости от целевых свойств, которые стремятся получить согласно настоящему изобретению. Во всяком случае, и не в качестве попытки ограничить применение доктрины эквивалентов к объему притязаний, каждое число должно, по меньшей мере, истолковываться с учетом представленных значащих цифр и с применением общепринятых методик округления.
Термин «алкил» означает одновалентную насыщенную углеводородную группу, которая может быть неразветвленной или разветвленной. Если не указано иное, то такие алкильные группы обычно содержат от 1 до 10 атомов углерода и включают, например, -C1-6алкил, означающий алкильную группу, содержащую от 1 до 6 атомов углерода, где атомы углерода находятся в любой приемлемой конфигурации. Типичные алкильные группы включают, например, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, н-гексил и т.п.
Подразумевается, что используемое в настоящем документе выражение «характеризующийся формулой» или «характеризующийся структурой» не является ограничивающим и используется так же, как обычно используется термин «содержащий». Например, если описана одна структура, то подразумевается, что охватываются все стереоизомерные и таутомерные формы, если не указано иное.
Термин «фармацевтически приемлемый» относится к веществу, который не является биологически или иным образом неприемлемым при применении согласно настоящему изобретению. Например, термин «фармацевтически приемлемый носитель» относится к веществу, которое может быть включено в состав композиции и введено пациенту без индукции неприемлемых биологических эффектов или без взаимодействия неприемлемым образом с другими компонентами композиции. Такие фармацевтически приемлемые вещества обычно соответствуют требуемым стандартам токсикологических и производственных испытаний, и включают вещества, которые определены Управлением по контролю над продуктами и лекарствами США в качестве подходящих неактивных ингредиентов.
Термин «фармацевтически приемлемая соль» означает соль, полученную из основания или кислоты, которая подходит для введения пациенту, такому как млекопитающее (например, соли, обладающие приемлемой для данного режима дозирования безопасностью для млекопитающих). Однако подразумевается, что соли, охватываемые настоящим изобретением, не обязательно должны быть фармацевтически приемлемыми солями, такими как соли промежуточных соединений, которые не предназначены для введения пациенту. Фармацевтически приемлемые соли могут быть получены из фармацевтически приемлемых неорганических или органических оснований и из фармацевтически приемлемых неорганических или органических кислот. Кроме того, если соединение формулы I содержит и фрагмент основания, такого как амин, пиридин или имидазол, и фрагмент кислоты, такой как карбоновая кислота или тетразол, то могут образовываться цвиттерионы, которые подпадают под используемый в настоящем документе термин «соль». Соли, полученные из фармацевтически приемлемых неорганических оснований, включают соли аммония, кальция, меди, трехвалентного железа, двухвалентного железа, лития, магния, трехвалентного марганца, двухвалентного марганца, калия, натрия и цинка, и т.п. Соли, полученные из фармацевтически приемлемых органических оснований, включают соли первичных, вторичных и третичных аминов, включая замещенные амины, циклические амины, амины природного происхождения и т.п., такие как аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиамин смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п. Соли, полученные из фармацевтически приемлемых неорганических кислот, включают соли борной, угольной, галогенводородной (бромистоводородной, соляной, фтористоводородной или йодистоводородной), азотной, фосфорной, сульфаминовой и серной кислот. Соли, полученные из фармацевтически приемлемых органических кислот, включают соли алифатических гидроксикислот (например, лимонной, глюконовой, гликолевой, молочной, лактобионовой, яблочной и винной кислот), алифатических монокарбоновых кислот (например, уксусной, масляной, муравьиной, пропионовой и трифторуксусной кислот), аминокислот (например, аспарагиновой и глутаминовой кислот), ароматических карбоновых кислот (например, бензойной, пара-хлорбензойной, дифенилуксусной, гентизиновой, гиппуриновой и трифенилуксусной кислот), ароматических гидроксикислот (например, орто-гидроксибензойной, пара-гидроксибензойной, 1-гидроксинафталин-2-карбоновой и 3-гидроксинафталин-2-карбоновой кислот), аскорбиновой, дикарбоновых кислот (например, фумаровой, малеиновой, щавелевой и янтарной кислот), глюкуроновых, миндальных, муциновых, никотиновых, оротовых, памовых, пантотеновых, сульфоновых кислот (например, бензолсульфоновой, камфорсульфоновой, эдисиловой, этансульфоновой, изетионовой, метансульфоновой, нафталинсульфоновой, нафталин-1,5-дисульфоновой, нафталин-2,6-дисульфоновой и пара-толуолсульфоновой кислот), ксинафоевой кислоты, и т.п.
Используемый в настоящем документе термин «пролекарство» предназначен для обозначения неактивного (или значительно менее активного) предшественника лекарства, который преобразуется в организме в его активную форму в физиологических условиях, например, посредством нормальных метаболических процессов. Такие соединения необязательно могут обладать фармакологической активностью в отношении NEP, но могут быть введены перорально или парентерально и далее метаболизированы в организме до образования соединения, которое является фармакологически активным в отношении NEP.
Термин «терапевтически эффективное количество» означает количество, достаточное для осуществления лечения при введении нуждающемуся в этом пациенту, другими словами, количество лекарства, необходимое для получения желаемого терапевтического эффекта. Например, терапевтически эффективное количество для лечения гипертензии представляет собой количество соединения, необходимое, например, для снижения, супрессирования, устранения или предотвращения симптомов гипертензии, или для лечения исходной причины гипертензии. Согласно одному варианту осуществления, терапевтически эффективное количество представляет собой количество лекарства, необходимое для снижения артериального давления, или количество лекарства, необходимое для поддержания нормального артериального давления. С другой стороны, термин «эффективное количество» означает количество, достаточное для получения желаемого результата, который необязательно может представлять собой терапевтический результат. Например, при исследовании системы, включающей фермент NEP, «эффективное количество» может представлять собой количество, необходимое для ингибирования фермента.
Используемый в настоящем документе термин «проведение лечения» или «лечение» означает проведение лечения или лечение заболевания или медицинского состояния (такого как гипертензия) у пациента, такого как млекопитающее (в частности, у человека), которое включает одно или несколько из следующего: (а) профилактику возникновения заболевания или медицинского состояния, т.е., профилактику повторного возникновения заболевания или медицинского состояния или профилактическое лечение пациента, предрасположенного к заболеванию или медицинскому состоянию; (b) уменьшение интенсивности заболевания или медицинского состояния, т.е., устранение или индукцию у пациента регрессии заболевания или медицинского состояния; (с) супрессирование заболевания или медицинского состояния, т.е., замедление или остановку развития у пациента заболевания или медицинского состояния; или (d) частичное снятие у пациента симптомов заболевания или медицинского состояния. Например, термин «проведение лечения гипертензии» будет включать профилактику возникновения гипертензии, уменьшение интенсивности гипертензии, супрессирование гипертензии и частичное снятие симптомов гипертензии (например, снижение артериального давления). Предполагается, что термин «пациент» включает млекопитающих, например, людей, которые нуждаются в лечении или профилактике заболевания, или которые подвергаются в настоящий момент лечению для профилактики заболевания или лечения конкретного заболевания или медицинского состояния, а также объекты исследований, на которых оценивают кристаллическое соединение, или которые используют в некотором методе анализа, например, в модели на животных.
Предполагается, что все другие термины, использованные в настоящем документе, имеют их общепризнанное значение, очевидное специалистам в области техники, к которой они имеют отношение.
Соединение согласно настоящему изобретению содержит один или несколько хиральных центров, а потому эти соединения могут быть получены и использоваться в различных стереоизомерных формах. Согласно некоторым вариантам осуществления, в целях оптимизации терапевтической активности соединения согласно настоящему изобретению, например, для лечения гипертонии, может быть желательно, чтобы атомы углерода обладали конкретной (R,R), (S,S), (S,R) или (R,S) конфигурацией или были обогащены в стереоизомерной форме, характеризующейся такой конфигурацией. Согласно другим вариантам осуществления, соединения согласно настоящему изобретению присутствуют в виде рацемических смесей. Следовательно, если не указано иное, то настоящее изобретение также относится к рацемическим смесям, чистым стереоизомерам (например, энантиомерам и диастереоизомерам), обогащенным стереоизомерами смесям, и т.п. Если химическая структура изображена в настоящем документе без указания на какую-либо стереохимию, то предполагается, что такой структурой охватываются все возможные стереоизомеры. По аналогии, если в настоящем документе представлен или поименован конкретный стереоизомер, то специалистам в данной области техники следует понимать, что если не указано иное, то в композициях согласно настоящему изобретению могут присутствовать незначительные количества других стереоизомеров при условии, что применимость композиции в целом не исключена присутствием таких других изомеров. Отдельные стереоизомеры могут быть получены многими способами, которые хорошо известны из области техники, включая хиральную хроматографию с использованием подходящей хиральной неподвижной фазы или подложки, или путем их химического преобразования в диастереоизомеры, или путем разделения диастереоизомеров традиционными способами, такими как хроматография или перекристаллизация, с последующим восстановлением исходного стереоизомера.
Кроме того, если не указано иное, то при необходимости все цис-транс или E/Z-изомеры (геометрические изомеры), таутомерные формы и топоизомерные формы соединений согласно настоящему изобретению подпадают под объем настоящего изобретения.
Соединения согласно настоящему изобретению, а также соединения, использованные для их синтеза, также могут включать меченые изотопами соединения, то есть, если один или несколько атомов были обогащены атомами с атомной массой, отличной от атомной массы, преимущественно обнаруживаемой в природе. Примеры изотопов, которые могут быть включены в соединения формулы I, например, включают без ограничения2H,3H,13C,14C,15N,18O,17O,35S,36Cl и18F. Особый интерес представляют собой соединения формулы I, обогащенные тритием или углеродом-14, которые могут быть использованы, например, при изучении распределения в тканях; соединения согласно настоящему изобретению, обогащенные дейтерием, в особенности в месте метаболизма, что приводит, например, к соединениям с большей метаболической стабильностью; и соединения формулы I, обогащенные позитронно-активным изотопом, таким как11C,18F,15O и13N, который может быть использован, например, при исследованиях методом позитронно-эмиссионной томографии (PET).
Используемая в настоящем документе номенклатура для наименования соединений согласно настоящему изобретению представлена в настоящем документе в разделе «Примеры». Эту номенклатуру получали с использованием коммерчески доступного программного обеспечения AutoNom (MDL, San Leandro, California).
В публикации патента США № 2012/0157383, в частности, раскрыта (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(3-гидрокси-3H-бензотриазол-5-карбонил)амино]пентановая кислота, представленная формулой I':

Согласно одному варианту осуществления, это соединение рассматривается как активная форма и вводится в качестве пролекарства, которое метаболизируется in vivo с образованием соединения формулы I'. В публикации патента США № 2012/0157383 также раскрыто пролекарство в виде сложного этилового эфира соединения формулы I'.
Согласно одному аспекту, настоящее изобретение относится к другим пролекарствам соединения формулы I'. Эти пролекарства представлены формулой XII, где Ra представляет собой H, Rb представляет собой Cl, и X представляет собой

Согласно одному варианту осуществления, эти соединения представлены формулой I:

где R2 представляет собой H, R4 представляет собой -OH, и R7 выбирают из -CH2CF3, -(CH2)2CF3, -CH2CF2CH3, CH2CF2CF3, -C(CH3)(CF3)2, -CH(CH2CH3)CF3, -CH(CH3)CF2CF3, -(CH2)2-3OH, CH2CH(NH2)COOCH3, -(CH2)2OCH3, -CHRcOC(O)-C1-4алкила, -CHRcOC(O)O-C2-4алкила, -CHRcOC(O)O-циклогексила, -C2-4алкилен-N(CH3)2, -CH2OC(O)CHRd-NH2, CH2OC(O)CHRd-NHC(O)O-C1-6алкила, бензила и

или R2 представляет собой H, R4 выбирают из -O-бензила, -OCHRcOC(O)-C1-4алкила, -OCH2OC(O)CHRd-NH2 и -OCH2OC(O)CHRd-NHC(O)O-C1-6алкила, и R7 выбирают из H и CH2OC(O)CH3; или R2 выбирают из -C(O)-C1-6алкила, -C(O)CHRd-NH2, -C(O)CHRd-NHC(O)O-C1-6алкила и -P(O)(ORe)2, R4 представляет собой -OH, и R7 представляет собой H; где каждый Rc независимо представляет собой H или -C1-3алкил; каждый Rd независимо представляет собой H, -CH3, -CH(CH3)2, фенил или бензил; и каждый Re независимо представляет собой H, -C1-6алкил или фенил; или их фармацевтически приемлемой солью.
Согласно одному конкретному варианту осуществления соединения формулы I, R2 представляет собой H, R4 представляет собой -OH, и R7 выбирают из -CH2CF3, -(CH2)2CF3, -CH2CF2CF3, -CH2OC(O)CH3, CH2OC(O)(CH2)2CH3, CH2OC(O)CH[CH(CH3)2]NH2, -CH2OC(O)CH[CH(CH3)2]-NHC(O)OCH3, бензила и

или R2 представляет собой H, R4 представляет собой -OCH2OC(O)CH3, и R7 выбирают из H и CH2OC(O)CH3; или R2 представляет собой H, R4 выбирают из -OCH2OC(O)(CH2)2CH3, -CH2OC(O)CH[CH(CH3)2]NH2 и OCH2OC(O)CH[CH(CH3)2]NHC(O)OCH3, и R7 представляет собой H; или R2 представляет собой H, R4 представляет собой -O-бензил, и R7 представляет собой H.
В публикации патента США № 2012/0157383 также, в частности, раскрыта и представлена формулой II' (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(5-гидрокси-1-метил-1H-пиразол-3-карбонил)амино]пентановая кислота:
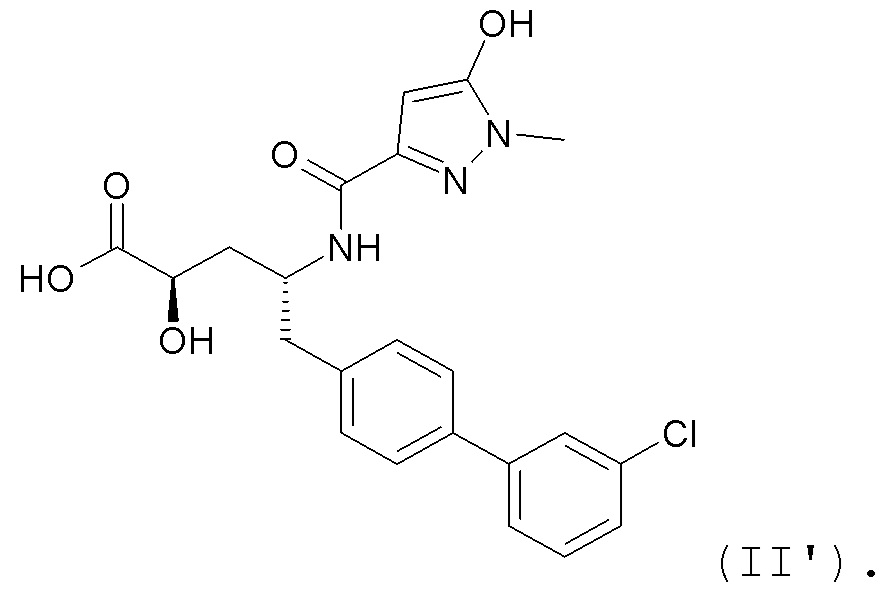
Согласно одному варианту осуществления, это соединение рассматривается как активная форма и вводится в качестве пролекарства, которое метаболизируется in vivo с образованием соединения формулы II'. В публикации патента США №2012/0157383 также раскрыто пролекарство в виде сложного изобутилового эфира соединения формулы II'.
Согласно другому аспекту, настоящее изобретение относится к другим пролекарствам соединения формулы II'. Эти пролекарства представлены формулой XII, где Ra представляет собой H, Rb представляет собой Cl, и X представляет собой

Согласно одному варианту осуществления, эти соединения представлены формулой II:

где R2 представляет собой H, R3 представляет собой -OH, и R7 выбирают из -CH2CF3, -(CH2)2CF3, -CH2CF2CH3, CH2CF2CF3, -C(CH3)(CF3)2, -CH(CH2CH3)CF3, -CH(CH3)CF2CF3, -(CH2)2-3OH, CH2CH(NH2)COOCH3, -(CH2)2OCH3, -CHRcOC(O)-C1-4алкила, -CHRcOC(O)O-C2-4алкила, -CHRcOC(O)O-циклогексила, -C2-4алкилен-N(CH3)2, -CH2OC(O)CHRd-NH2, CH2OC(O)CHRd-NHC(O)O-C1-6алкила, бензила и

или R3 выбирают из -OC(O)CH2CH3, -OC(O)CH2CH(CH3)2, -OC(O)-фенила, OCH2OC(O)CHRd-NH2 и -OCH2OC(O)CHRd-NHC(O)O-C1-6алкила, и R7 представляет собой H; или R2 выбирают из -C(O)-C1-6алкила, -C(O)CHRd-NH2, -C(O)CHRd-NHC(O)O-C1-6алкила и P(O)(ORe)2, R3 представляет собой -OH, и R7 представляет собой H; где каждый Rc независимо представляет собой H или -C1-3алкил; каждый Rd независимо представляет собой H, -CH3, -CH(CH3)2, фенил или бензил; и каждый Re независимо представляет собой H, -C1-6алкил или фенил; или их фармацевтически приемлемой солью.
Согласно одному конкретному варианту осуществления соединений формулы II, R2 представляет собой H, R3 представляет собой -OH, и R7 выбирают из -CH2CH3, -CH2CF2CF3, CH(CH2CH3)CF3, -CH(CH3)CF2CF3, CH2OC(O)(CH2)2CH3, -CH2OC(O)CH[CH(CH3)2]NH2, бензила и

В публикации патента США № 2012/0157383 также, в частности, раскрыта и представлена формулой III' (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(5-гидрокси-1-пиридин-2-ил-1H-пиразол-3-карбонил)амино]пентановая кислота:
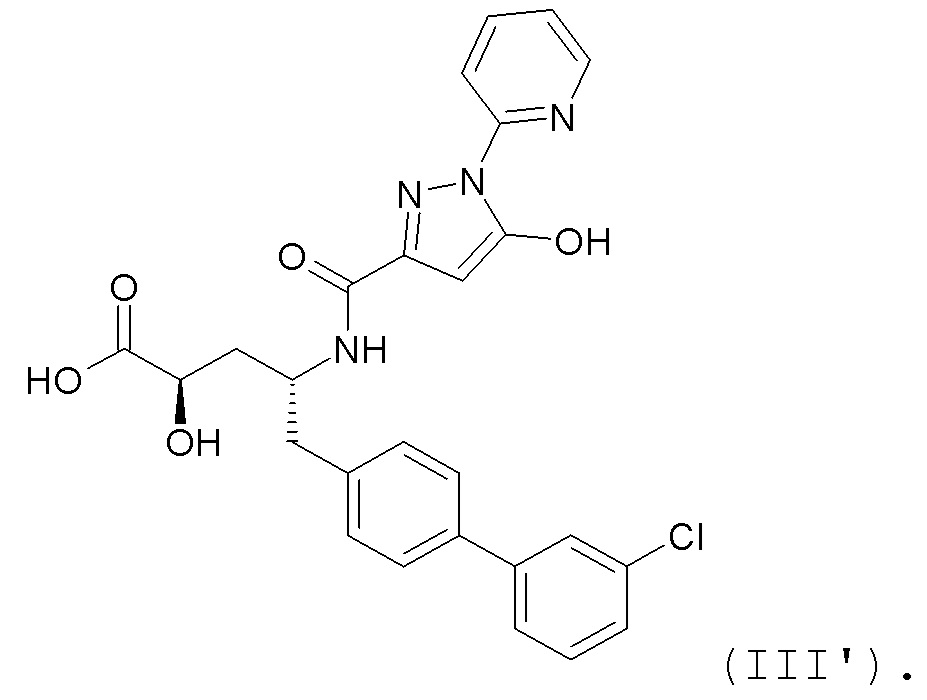
Согласно одному варианту осуществления, это соединение рассматривается как активная форма и вводится в качестве пролекарства, которое метаболизируется in vivo с образованием соединения формулы III'.
Согласно другому аспекту, настоящее изобретение относится к другим пролекарствам соединения формулы III'. Эти пролекарства представлены формулой XII, где Ra представляет собой H, Rb представляет собой Cl, и X представляет собой
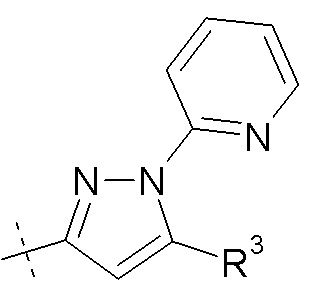
Согласно одному варианту осуществления, эти соединения представлены формулой III:

где R2 представляет собой H, R3 представляет собой -OH, и R7 выбирают из -CH2CH3, -CH2CH(CH3)2, -CH2CF3, (CH2)2CF3, -CH2CF2CH3, -CH2CF2CF3, -C(CH3)(CF3)2, -CH(CH2CH3)CF3, CH(CH3)CF2CF3, -(CH2)2-3OH, -CH2CH(NH2)COOCH3, -(CH2)2OCH3, -CHRcOC(O)-C14алкила, -CHRcOC(O)O-C2-4алкила, -CHRcOC(O)O-циклогексила, -C2-4алкилен-N(CH3)2, CH2OC(O)CHRd-NH2, -CH2OC(O)CHRd-NHC(O)O-C1-6алкила, бензила и

или R2 представляет собой H, R3 выбирают из -OC(O)CH2CH3, -OC(O)CH2CH(CH3)2, -OC(O)-фенила, OCH2OC(O)CHRd-NH2 и -OCH2OC(O)CHRd-NHC(O)O-C1-6алкила, и R7 представляет собой H; или R2 выбирают из -C(O)-C1-6алкила, -C(O)CHRd-NH2, -C(O)CHRd-NHC(O)O-C1-6алкила и P(O)(ORe)2, R3 представляет собой -OH, и R7 представляет собой H; где каждый Rc независимо представляет собой H или -C1-3алкил; каждый Rd независимо представляет собой H, -CH3, -CH(CH3)2, фенил или бензил; и каждый Re независимо представляет собой H, -C1-6алкил или фенил; или их фармацевтически приемлемой солью.
Согласно одному конкретному варианту осуществления соединений формулы III, R2 представляет собой H, R3 представляет собой -OH, и R7 выбирают из -CH2CH3, -CH2CH(CH3)2, CH2OC(O)CHRd-NHC(O)O-C1-6алкила, бензила и
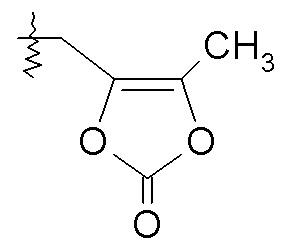
где Rd представляет собой -CH(CH3)2. Согласно другому варианту осуществления соединений формулы III R2 представляет собой H, R3 представляет собой -OCH2OC(O)CH[CH(CH3)2]NH2 и R7 представляет собой H. Согласно другому варианту осуществления соединений формулы III, R2 представляет собой -C(O)CH[CH(CH3)2]NH2, R3 представляет собой -OH, и R7 представляет собой H.
Согласно другому аспекту, настоящее изобретение относится к соединению формулы XII, где Ra представляет собой F, Rb представляет собой Cl, и X представляет собой

Согласно одному варианту осуществления, эти соединения представлены формулой IV:
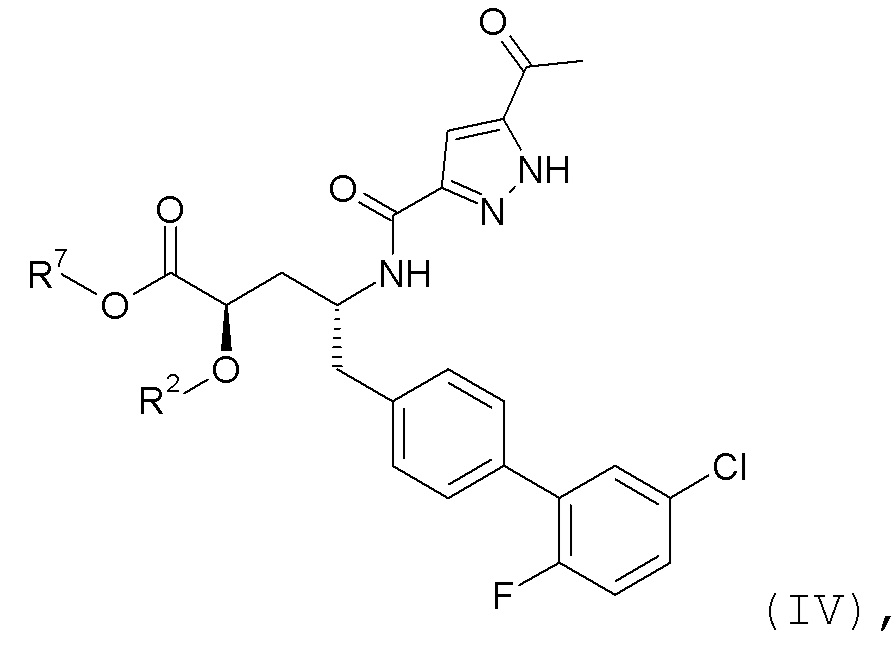
где R2 представляет собой H, и R7 выбирают из H, -CH2CH3, -CH2CF3, -(CH2)2CF3, -CH2CF2CH3, CH2CF2CF3, -C(CH3)(CF3)2, -CH(CH2CH3)CF3, -CH(CH3)CF2CF3, -(CH2)2-3OH, CH2CH(NH2)COOCH3, -(CH2)2OCH3, -CHRcOC(O)-C1-4алкила, -CHRcOC(O)O-C2-4алкила, -CHRcOC(O)O-циклогексила, -C2-4алкилен-N(CH3)2, -CH2OC(O)CHRd-NH2, CH2OC(O)CHRd-NHC(O)O-C1-6алкила, бензила и
или R2 выбирают из -C(O)-C1-6алкила, -C(O)CHRd-NH2, -C(O)CHRd-NHC(O)O-C1-6алкила и -P(O)(ORe)2, и R7 представляет собой H; где каждый Rc независимо представляет собой H или -C1-3алкил; каждый Rd независимо представляет собой H, -CH3, -CH(CH3)2, фенил или бензил; и каждый Re независимо представляет собой H, C1-6алкил или фенил; или их фармацевтически приемлемой солью.
Согласно одному конкретному варианту осуществления соединений формулы IV, R2 представляет собой H, и R7 выбирают из H и -CH2CH3.
Согласно другому аспекту, настоящее изобретение относится к соединению формулы XII, где Ra представляет собой H, Rb представляет собой Cl, и X представляет собой

Согласно одному варианту осуществления, эти соединения представлены формулой V:

где R2 представляет собой H, и R7 выбирают из H, -CH2CH3, -CH2CF3, -(CH2)2CF3, -CH2CF2CH3, CH2CF2CF3, -C(CH3)(CF3)2, -CH(CH2CH3)CF3, -CH(CH3)CF2CF3, -(CH2)2-3OH, CH2CH(NH2)COOCH3, -(CH2)2OCH3, -CHRcOC(O)-C1-4алкила, -CHRcOC(O)O-C2-4алкила, -CHRcOC(O)O-циклогексила, -C2-4алкилен-N(CH3)2, -CH2OC(O)CHRd-NH2, CH2OC(O)CHRd-NHC(O)O-C1-6алкила, бензила и

или R2 выбирают из -C(O)-C1-6алкила, -C(O)CHRd-NH2, -C(O)CHRd-NHC(O)O-C1-6алкила и -P(O)(ORe)2, и R7 представляет собой H; где каждый Rc независимо представляет собой H или -C1-3алкил; каждый Rd независимо представляет собой H, -CH3, -CH(CH3)2, фенил или бензил; и каждый Re независимо представляет собой H, C1-6алкил или фенил; или его фармацевтически приемлемой солью.
Согласно одному конкретному варианту осуществления соединений формулы V, R2 представляет собой H, и R7 выбирают из H и -CH2CH3.
Согласно другому аспекту, настоящее изобретение относится к соединению формулы XII, где Ra представляет собой H, Rb представляет собой Cl, и X представляет собой
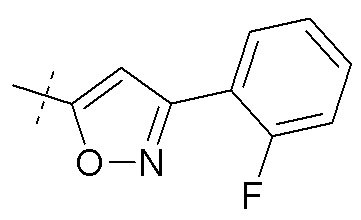
Согласно одному варианту осуществления, эти соединения представлены формулой VI:
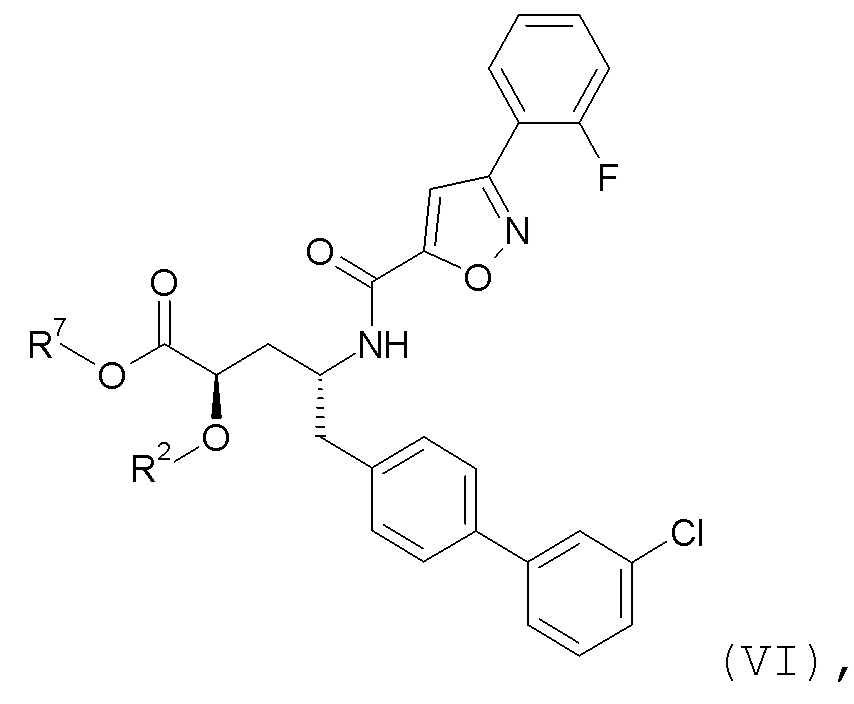
где R2 представляет собой H, и R7 выбирают из H, -CH2CH3, -CH2CF3, -(CH2)2CF3, -CH2CF2CH3, CH2CF2CF3, -C(CH3)(CF3)2, -CH(CH2CH3)CF3, -CH(CH3)CF2CF3, -(CH2)2-3OH, CH2CH(NH2)COOCH3, -(CH2)2OCH3, -CHRcOC(O)-C1-4алкила, -CHRcOC(O)O-C2-4алкила, -CHRcOC(O)O-циклогексила, -C2-4алкилен-N(CH3)2, -CH2OC(O)CHRd-NH2, CH2OC(O)CHRd-NHC(O)O-C1-6алкила, бензила и
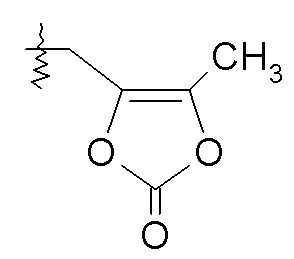
или R2 выбирают из -C(O)-C1-6алкила, -C(O)CHRd-NH2, -C(O)CHRd-NHC(O)O-C1-6алкила и -P(O)(ORe)2, и R7 представляет собой H; где каждый Rc независимо представляет собой H или -C1-3алкил; каждый Rd независимо представляет собой H, -CH3, -CH(CH3)2, фенил или бензил; и каждый Re независимо представляет собой H, C1-6алкил или фенил; или его фармацевтически приемлемой солью.
Согласно одному конкретному варианту осуществления соединений формулы VI, R2 представляет собой H, и R7 выбирают из H, -CH2OC(O)CH3, -CH2OC(O)OCH2CH3, -CH2OC(O)OCH(CH3)2 и C(O)CH[CH(CH3)2]-NHC(O)OCH3.
В публикации патента США №2012/0157383 также, в частности, раскрыта и представлена формулой VII' (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(3-метоксиизоксазол-5-карбонил)амино]пентановая кислота:

Согласно одному варианту осуществления, это соединение рассматривается как активная форма и вводится в качестве пролекарства, которое метаболизируется in vivo с образованием соединения формулы VII'.
Согласно другому аспекту, настоящее изобретение относится к другим пролекарствам соединения формулы VII'. Эти пролекарства представлены формулой XII, где Ra представляет собой H, Rb представляет собой Cl, и X представляет собой

где R представляет собой H или -CH3. Согласно одному варианту осуществления, эти соединения представлены формулой VII:
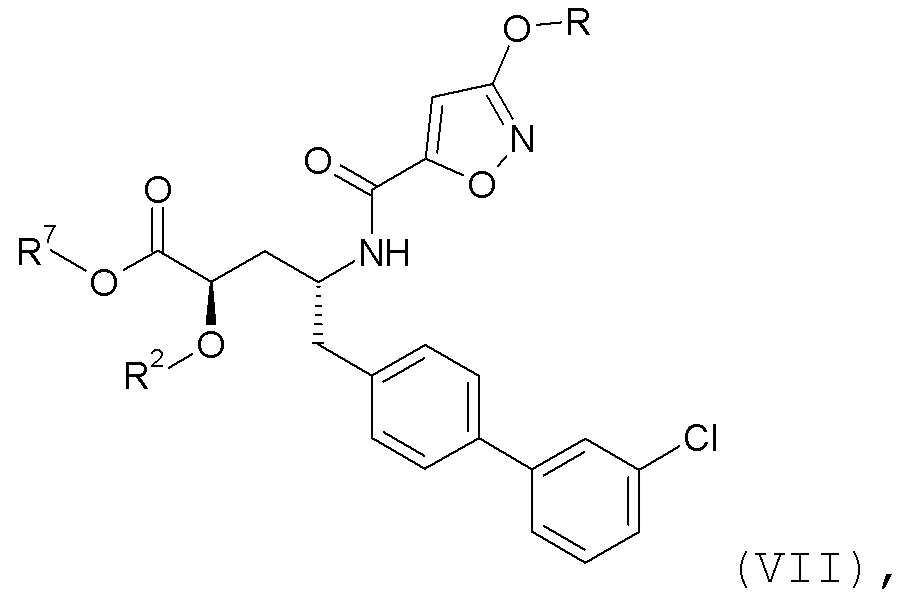
где R2 представляет собой H, и R7 выбирают из -CH2CH3, -CH2CF3, -(CH2)2CF3, -CH2CF2CH3, CH2CF2CF3, -C(CH3)(CF3)2, -CH(CH2CH3)CF3, -CH(CH3)CF2CF3, -(CH2)2-3OH, CH2CH(NH2)COOCH3, -(CH2)2OCH3, -CHRcOC(O)-C1-4алкила, -CHRcOC(O)O-C2-4алкила, -CHRcOC(O)O-циклогексила, -C2-4алкилен-N(CH3)2, -CH2OC(O)CHRd-NH2, CH2OC(O)CHRd-NHC(O)O-C1-6алкил, бензила и

или R2 выбирают из -C(O)-C1-6алкила, -C(O)CHRd-NH2, -C(O)CHRd-NHC(O)O-C1-6алкила и -P(O)(ORe)2, и R7 представляет собой H; где каждый Rc независимо представляет собой H или -C1-3алкил; каждый Rd независимо представляет собой H, -CH3, -CH(CH3)2, фенил или бензил; и каждый Re независимо представляет собой H, C1-6алкил или фенил; или их фармацевтически приемлемой солью.
Согласно одному конкретному варианту осуществления соединений формулы VII, R представляет собой -CH3, R2 представляет собой H, и R7 выбирают из -CH2OC(O)CH3, -CH2OC(O)OCH(CH3)2, -CH2OC(O)OCH2CH3 и -CH2OC(O)CH[CH(CH3)2]-NHC(O)OCH3.
В публикации патента США №2012/0157383 также, в частности, раскрыта и представлена формулой VIII' (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(1-гидрокси-1H-[1,2,3]триазол-4-карбонил)амино]пентановая кислота:
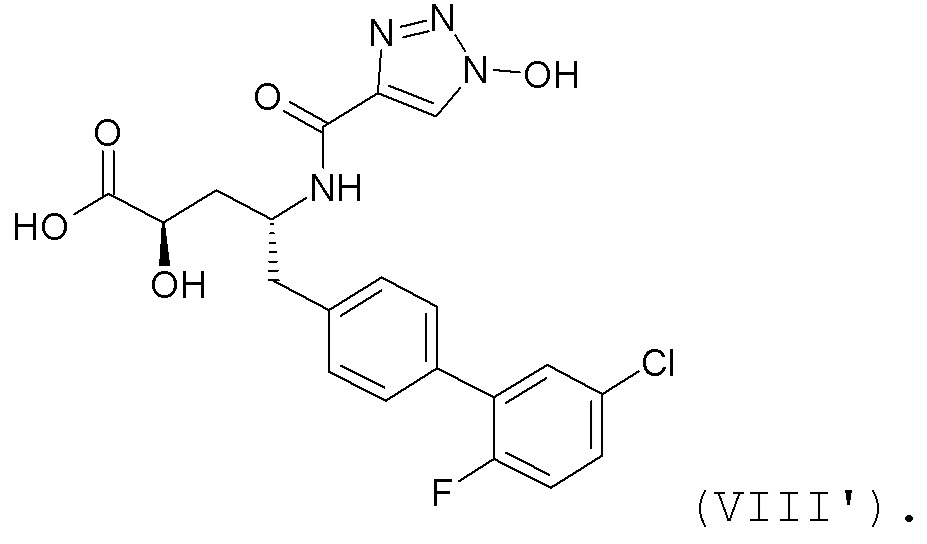
Согласно одному варианту осуществления, это соединение рассматривается как активная форма и вводится в качестве пролекарства, которое метаболизируется in vivo с образованием соединения формулы VIII'. В публикации патента США №2012/0157383 также раскрыты пролекарства в виде сложного изопропилового эфира, сложного этилового эфира, сложного изобутилового эфира и сложного гептилового эфира соединения формулы VIII'.
Согласно другому аспекту, настоящее изобретение относится к другим пролекарствам соединения формулы VIII'. Эти пролекарства представлены формулой XII, где Ra представляет собой F, Rb представляет собой Cl, и X представляет собой
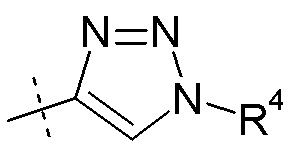
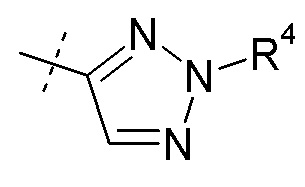
Согласно одному варианту осуществления, эти соединения представлены формулой VIIIa или VIIIb:

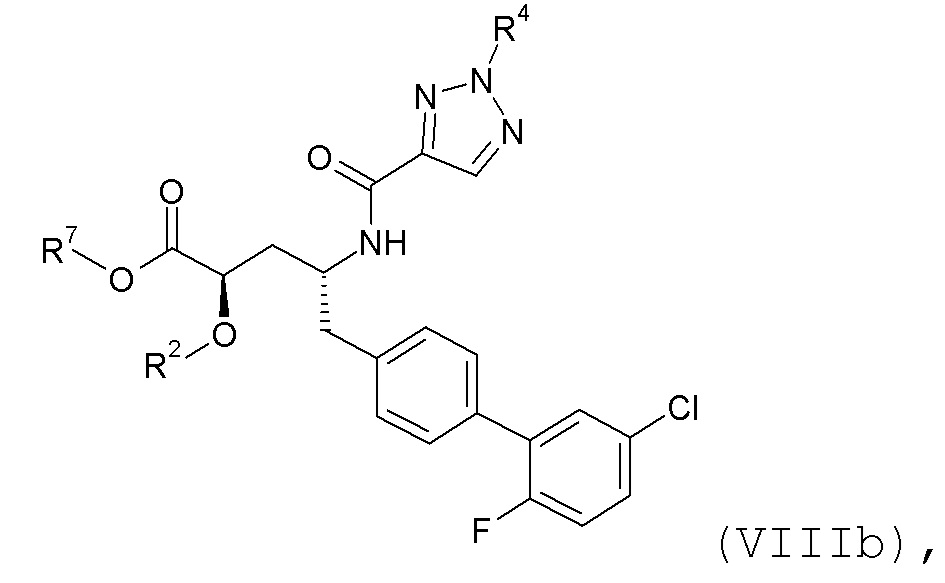
где R2 представляет собой H, R4 представляет собой -OH, и R7 выбирают из -CH2CH3, -CH2CF3, -(CH2)2CF3, CH2CF2CH3, -CH2CF2CF3, -C(CH3)(CF3)2, -CH(CH2CH3)CF3, -CH(CH3)CF2CF3, (CH2)2-3OH, -CH2CH(NH2)COOCH3, -(CH2)2OCH3, -CHRcOC(O)-C1-4алкила, CHRcOC(O)O-C2-4алкила, -CHRcOC(O)O-циклогексила, -C2-4алкилен-N(CH3)2, CH2OC(O)CHRd-NH2, -CH2OC(O)CHRd-NHC(O)O-C1-6алкила, бензила и

или R2 представляет собой H, R4 выбирают из -O-бензила, -OCHRcOC(O)-C1-4алкила, OCH2OC(O)CH[CH(CH3)2]NH2, -OCH2OC(O)CH[CH(CH3)2]-NHC(O)OCH3 и
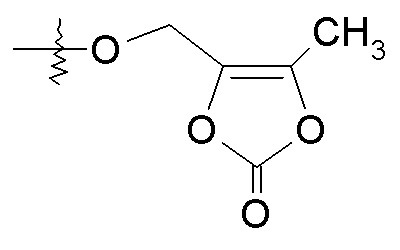
и R7 представляет собой H; или R2 выбирают из -C(O)-C1-6алкила, -C(O)CHRd-NH2, -C(O)CHRd-NHC(O)O-C1-6алкила и -P(O)(ORe)2, R4 представляет собой -OH, и R7 представляет собой H; где каждый Rc независимо представляет собой H или -C1-3алкил; каждый Rd независимо представляет собой H, -CH3, -CH(CH3)2, фенил или бензил; и каждый Re независимо представляет собой H, -C1-6алкил или фенил; или их фармацевтически приемлемой солью.
Согласно одному конкретному варианту осуществления соединений формулы VIIIa и VIIIb, R2 представляет собой H, R4 представляет собой -OH, и R7 выбирают из -CH2CF2CF3, -CH2OC(O)CH3, CH2OC(O)(CH2)2CH3, -CH2OC(O)OCH2CH3, CH2OC(O)OCH(CH3)2, CH(CH3)OC(O)O-циклогексила, CH2OC(O)CH[CH(CH3)2]-NHC(O)OCH3 и
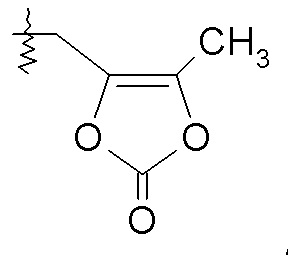
или R2 представляет собой H, R4 выбирают из -OCH2OC(O)(CH2)2CH3, OCH2OC(O)CH[CH(CH3)2]NH2, -OCH2OC(O)CH[CH(CH3)2]-NHC(O)OCH3 и

В публикации патента США №2012/0157383 также, в частности, раскрыта и представлена формулой IX' (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(5-оксо-1-фенил-4,5-дигидро-1H-[1,2,4]триазол-3-карбонил)амино]пентановая кислота:

Согласно одному варианту осуществления, это соединение рассматривается как активная форма и вводится в качестве пролекарства, которое метаболизируется in vivo с образованием соединения формулы IX'.
Согласно другому аспекту, настоящее изобретение относится к другим пролекарствам соединения формулы IX'. Эти пролекарства представлены формулой XII, где Ra представляет собой H, Rb представляет собой Cl, и X представляет собой
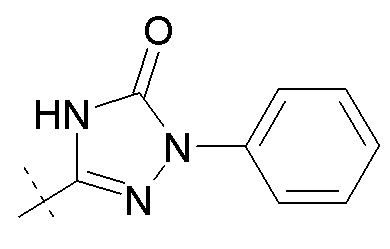
Согласно одному варианту осуществления, эти соединения представлены формулой IX:
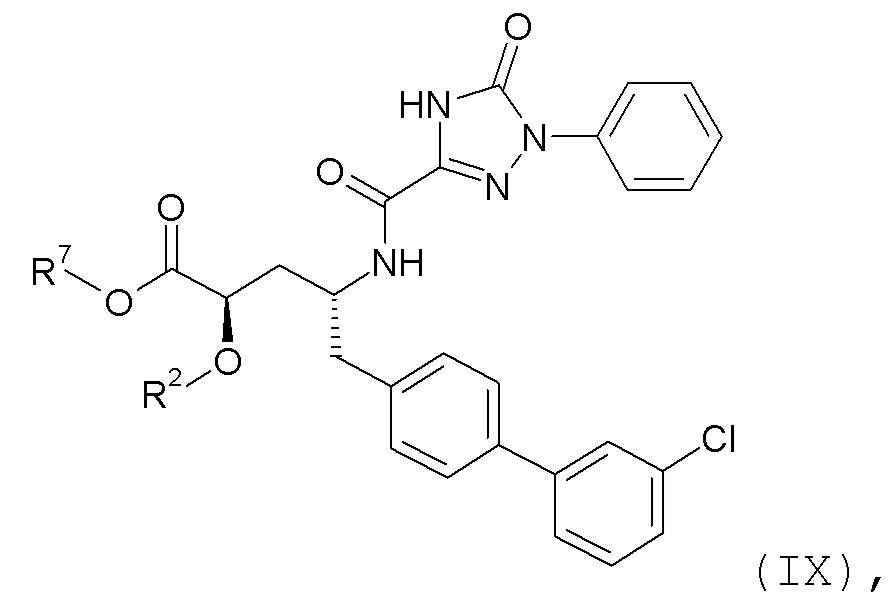
где R2 представляет собой H, и R7 выбирают из -CH2CH3, -CH2CF3, -(CH2)2CF3, -CH2CF2CH3, CH2CF2CF3, -C(CH3)(CF3)2, -CH(CH2CH3)CF3, -CH(CH3)CF2CF3, -(CH2)2-3OH, CH2CH(NH2)COOCH3, -(CH2)2OCH3, -CHRcOC(O)-C1-4алкила, -CHRcOC(O)O-C2-4алкила, -CHRcOC(O)O-циклогексила, -C2-4алкилен-N(CH3)2, -CH2OC(O)CHRd-NH2, CH2OC(O)CHRd-NHC(O)O-C1-6алкила, бензила и
или R2 выбирают из -C(O)-C1-6алкила, -C(O)CHRd-NH2, -C(O)CHRd-NHC(O)O-C1-6алкила и -P(O)(ORe)2, и R7 представляет собой H; где каждый Rc независимо представляет собой H или -C1-3алкил; каждый Rd независимо представляет собой H, -CH3, -CH(CH3)2, фенил или бензил; и каждый Re независимо представляет собой H, C1-6алкил или фенил; или их фармацевтически приемлемой солью.
Согласно одному конкретному варианту осуществления соединений формулы IX, R2 представляет собой H, и R7 выбирают из -CH2OC(O)OCH2CH3 и -CH2OC(O)CH[CH(CH3)2]-NHC(O)OCH3.
В публикации патента США № 2012/0157383 также, в частности, раскрыта и представлена формулой X' (2R,4R)-4-[(5-ацетил-2H-пиразол-3-карбонил)амино]-5-бифенил-4-ил-2-гидроксипентановая кислота:

Согласно одному варианту осуществления, это соединение рассматривается как активная форма и вводится в качестве пролекарства, которое метаболизируется in vivo с образованием соединения формулы X'. В публикации патента США №2012/0157383 также раскрыты пролекарства в виде сложного этилового эфира, сложного бутилового эфира, сложного метоксиэтилового эфира, сложного медоксомилового эфира, сложного мофетилового эфира и сложного метансульфонилэтилового эфира соединения формулы X'.
Согласно другому аспекту, настоящее изобретение относится к другим пролекарствам соединения формулы X'. Эти пролекарства представлены формулой XII, где Ra представляет собой Н, Rb представляет собой Н, и X представляет собой

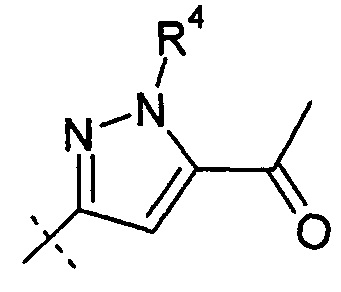
Согласно одному варианту осуществления, эти соединения представлены формулой Ха или Xb:

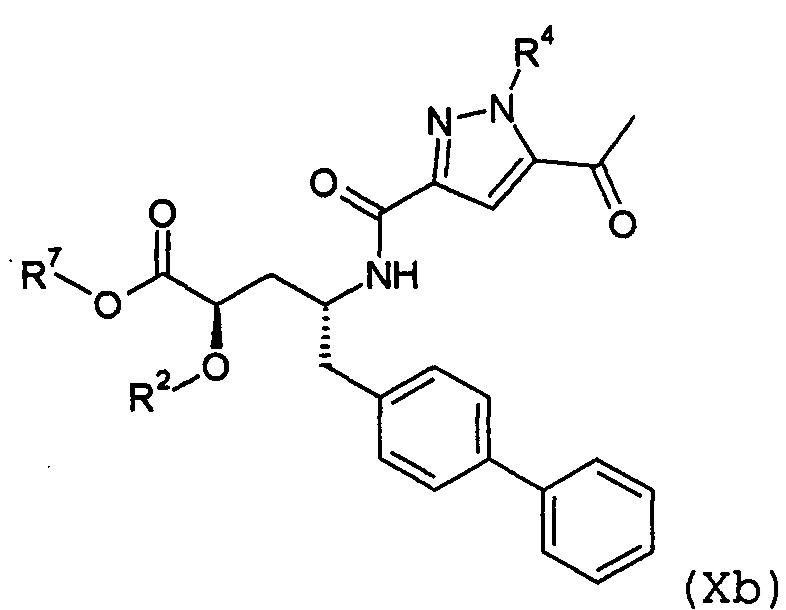
где R2 и R4 представляют собой Н, и R7 выбирают из -CH2CF3, -(CH2)2CF3, -CH2CF2CH3, -CH2CF2CF3, -С(СН3)(CF3)2, -СН(СН2СН3)CF3, -CH(CH3)CF2CF3, -(СН2)2-3ОН, -СН2СН(NH2)СООСН3, -CHRcOC(O)-С1-4алкила, -CHRcOC(О)O-С2-4алкила, -CHRcOC(O)O-циклогексила, -С2-4алкилен-N(СН3)2, -СН2ОС(О)CHRd-NH2, -СН2ОС(О)CHRd-NHC(О)O-C1-6алкила и бензила; или R2 представляет собой Н, R4 выбирают из -СН2ОС(О)СН[СН(СН3)2]-NHC(О)ОСН3 и -CH2OC(O)CH[CH(CH3)2]NH2, и R7 представляет собой Н; или R2выбирают из -C(O)-C1-6алкила, -C(O)CHRd-NH2, -C(O)CHRd-NHC(O)O-C16алкила и -P(O)(ORe)2, R4 представляет собой H, и R7 представляет собой H; или R2 представляет собой H, R4 представляет собой -CH2OP(O)(ORe)2 или CH2OC(O)CH[CH(CH3)2]NH2, и R7 представляет собой -CH2CH3; или R2 представляет собой -C(O)CH[CH(CH3)2]NH2, R4 представляет собой H, и R7 представляет собой -CH2CH3; и где каждый Rc независимо представляет собой H или -C1-3алкил; каждый Rd независимо представляет собой H, -CH3, -CH(CH3)2, фенил или бензил; и каждый Re независимо представляет собой H, C1-6алкил или фенил; или их фармацевтически приемлемой солью.
Согласно одному конкретному варианту осуществления соединений формулы Xa и Xb, R2 представляет собой H, R4 представляет собой -CH2-OP(O)(OH)2 или -CH2OC(O)CH[CH(CH3)2]NH2, и R7 представляет собой CH2CH3. Согласно другому варианту осуществления соединений формулы Xa и Xb, R2 представляет собой C(O)CH[CH(CH3)2]NH2, R4 представляет собой H, и R7 представляет собой -CH2CH3.
Согласно другому аспекту, настоящее изобретение относится к соединению формулы XII, где Ra представляет собой H, Rb представляет собой Cl, и X представляет собой

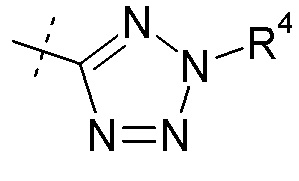
Согласно одному варианту осуществления, эти соединения представлены формулой XIa или XIb:
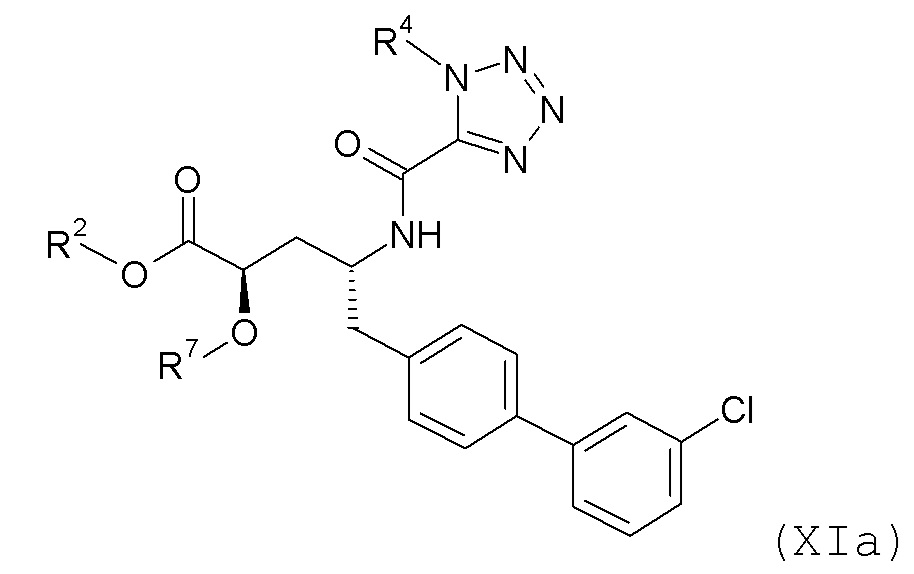
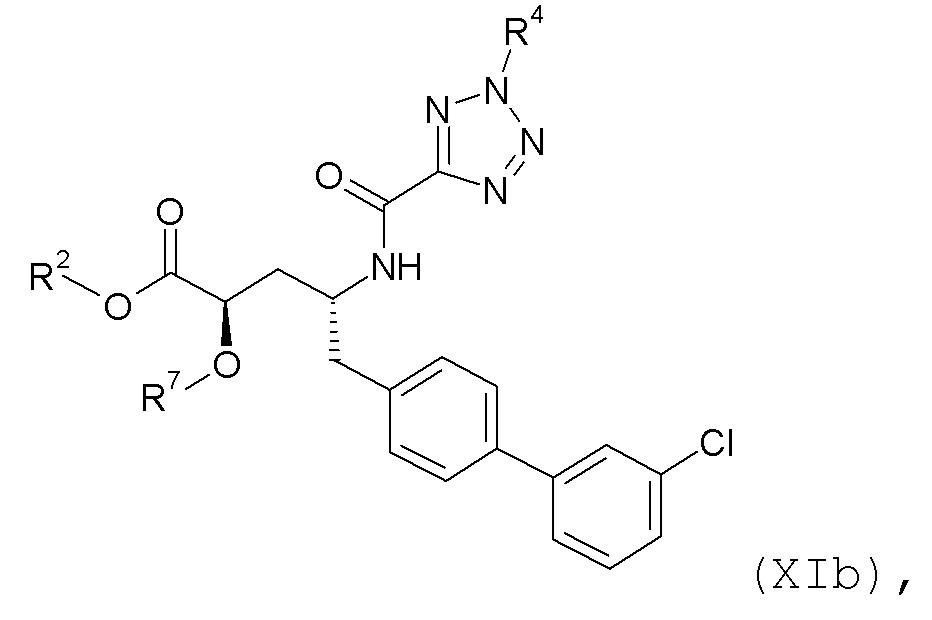
где R2 и R4 представляют собой H, и R7 выбирают из H, -CH2CH3, -CH2CF3, -(CH2)2CF3, CH2CF2CH3, -CH2CF2CF3, -C(CH3)(CF3)2, -CH(CH2CH3)CF3, -CH(CH3)CF2CF3, (CH2)2-3OH, -CH2CH(NH2)COOCH3, -(CH2)2OCH3, -CHRcOC(O)-C1-4алкила, CHRcOC(O)O-C2-4алкила, -CHRcOC(O)O-циклогексила, -C2-4алкилен-N(CH3)2, CH2OC(O)CHRd-NH2, -CH2OC(O)CHRd-NHC(O)O-C1-6алкила и бензила; или R2 представляет собой H, R4 выбирают из -CH2OC(O)CH[CH(CH3)2]-NHC(O)OCH3 и CH2OC(O)CH[CH(CH3)2]NH2, и R7 представляет собой H; или R2 выбирают из -C(O)-C1-6алкила, C(O)CHRd-NH2, -C(O)CHRd-NHC(O)O-C1-6алкила и -P(O)(ORe)2, R4 представляет собой H, и R7 представляет собой H; или R2 представляет собой H, R4 представляет собой -CH2OP(O)(ORe)2 или -CH2OC(O)CH[CH(CH3)2]NH2, и R7 представляет собой -CH2CH3; или R2 представляет собой -C(O)CH[CH(CH3)2]NH2, R4 представляет собой H, и R7 представляет собой -CH2CH3; и где каждый Rc независимо представляет собой H или -C1-3алкил; каждый Rd независимо представляет собой H, -CH3, -CH(CH3)2, фенил или бензил; и каждый Re независимо представляет собой H, -C1-6алкил или фенил; или его фармацевтически приемлемой солью.
Согласно одному конкретному варианту осуществления соединений формулы XIa и XIb, R2, R4 и R7 представляют собой H. Согласно другому варианту осуществления соединений формулы XIa и XIb, R2 и R4 представляют собой H, и R7 представляет собой -CH2OC(O)OCH2CH3.
ОБЩИЕ СПОСОБЫ СИНТЕЗА
Соединения согласно настоящему изобретению могут быть получены из легкодоступных исходных веществ с использованием следующих общих способов и методик, изложенных в разделе «Примеры», или с использованием других способов, реагентов и исходных веществ, которые известны специалистам в данной области техники. Хотя последующие методики могут иллюстрировать конкретный вариант осуществления настоящего изобретения, следует понимать, что другие варианты осуществления настоящего изобретения могут быть получены аналогичным образом с использованием тех же самых или сходных способов или с использованием других способов, реагентов и исходных веществ, известных специалисту в данной области техники. Также следует понимать, что если представлены типичные или предпочтительные условия процесса (например, температуры реакции, время, молярные соотношения реагентов, растворители, давление, и т.д.), то также могут быть использованы другие условия процесса, если не указано иное. В некоторых случаях, взаимодействия проводят при комнатной температуре, и точное измерение температуры не проводится. Подразумевается, что комнатная температура означает температуру в пределах диапазона, обычно ассоциируемого с температурой окружающей среды в лабораторных условиях, и обычно находится в диапазоне приблизительно от 18°C приблизительно до 30°C. В других случаях, взаимодействия проводят при комнатной температуре, и температуру точно измеряют и регистрируют. Хотя оптимальные условия проведения реакции обычно будут варьировать в зависимости от различных параметров реакции, таких как конкретно используемые реагенты, растворители и количества, специалисты в данной области техники смогут легко определить подходящие условия реакции с использованием обычных способов оптимизации.
Кроме того, специалистам в данной области техники будет очевидно, что для предупреждения нежелательных реакций со стороны определенных функциональных групп могут быть необходимы или желательны традиционные защитные группы. Выбор защитной группы, подходящей для конкретной функциональной группы, а также подходящие условия и реагенты для введения и снятия защитных групп с таких функциональных групп, хорошо известны из уровня техники. При необходимости, могут быть использованы защитные группы, отличные от представленных в методиках, описанных в настоящем документе. Например, многие защитные группы и их введение, и удаление описаны в T. W. Greene and G. M. Wuts, Protecting Groups in Organic Synthesis, Fourth Edition, Wiley, New York, 2006, и в цитируемых в этом документе ссылках.
Карбоксизащитные группы подходят для предупреждения нежелательных реакций по карбоксигруппе, и примеры включают без ограничения метил, этил, трет-бутил, бензил (Bn), пара-метоксибензил (PMB), 9-флуоренилметил (Fm), триметилсилил (TMS), трет-бутилдиметилсилил (TBDMS), дифенилметил (бензгидрил, DPM) и т.п. Аминозащитные группы подходят для предупреждения нежелательных реакций по аминогруппе, и примеры включают без ограничения трет-бутоксикарбонил (BOC), тритил (Tr), бензилоксикарбонил (Cbz), 9-флуоренилметоксикарбонил (Fmoc), формил, триметилсилил (TMS), трет-бутилдиметилсилил (TBDMS) и т.п.
Для удаления защитных групп используют стандартные методики и реагенты для снятия защитных групп, и они могут варьировать в зависимости от используемой группы. Например, если карбоксизащитная группа представляет собой метил, то широко используются гидроксид натрия или лития, если карбоксизащитная группа представляет собой этил или трет-бутил, то широко используется кислота, такая как TFA или HCl (например, 4,0 M HCl в 1,4-диоксане), и если карбоксизащитная группа представляет собой бензил, то может быть использован H2/Pd/C. BOC аминозащитная группа может быть удалена с использованием кислого реагента, такого как TFA в DCM или HCl в 1,4-диоксане, при этом Cbz аминозащитная группа может быть удалена с использованием условий каталитического гидрирования, таких как H2 (1 атм) и 10% Pd/C в спиртовом растворителе(«H2/Pd/C»).
Уходящие группы представляют собой функциональные группы или атомы, которые могут быть замещены другой функциональной группой или атомом в реакции замещения, такой как реакция нуклеофильного замещения. В качестве примера, характерные уходящие группы включают группы хлора, брома и йода; группы сложного сульфонового эфира, такие как мезилат, тозилат, брозилат, нозилат и т.п.; и ацилоксигруппы, такие как ацетокси, трифторацетокси и т.п.
Основания, подходящие для использования в этих схемах, включают в качестве иллюстрации и без ограничения карбонат калия, карбонат кальция, карбонат натрия, триэтиламин (Et3N), пиридин, 1,8-диазабицикло-[5.4.0]ундец-7-ен (DBU), N,N-диизопропилэтиламин (DIPEA), 4-метилморфолин, гидроксид натрия, гидроксид калия, трет-бутоксид калия и гидриды металлов.
Инертные разбавители или растворители, подходящие для использования в этих схемах, включают в качестве иллюстрации и без ограничения тетрагидрофуран (THF), ацетонитрил (MeCN), N,N-диметилформамид (DMF), N,N-диметилацетамид (DMA), диметилсульфоксид (DMSO), толуол, дихлорметан (DCM), хлороформ (CHCl3), тетрахлорид углерода (CCl4), 1,4-диоксан, метанол, этанол, воду, диэтиловый эфир, ацетон и т.п.
Подходящие агенты сочетания карбоновой кислоты/амина включают бензотриазол-1-илокситрис(диметиламино)фосфония гексафторфосфат (BOP), бензотриазол-1-илокситрипирролидинофосфония гексафторфосфат (PyBOP), N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония гексафторфосфат (HATU), 1,3-дициклогексилкарбодиимид (DCC), N-(3-диметиламинопропил)-N'-этилкарбодиимид (EDC), карбонилдиимидазол (CDI), 1-гидроксибензотриазол (HOBt) и т.п. Реакции сочетания проводят в инертном разбавителе в присутствии основания, такого как DIPEA, и в обычных условиях образования амидной связи.
Все реакции обычно проводят при температуре в диапазоне приблизительно от -78 C до 100°C, например, при комнатной температуре. С использованием тонкослойной хроматографии (TLC), высокоэффективной жидкостной хроматографии (HPLC) и/или LC-MS могут проводиться мониторинг реакции до ее завершения. Реакции могут завершаться в течение минут, или это может занимать часы, обычно от 1-2 часов и до 48 часов. По завершении реакции, полученная смесь или продукт реакции могут быть затем обработаны с получением целевого продукта. Например, полученная смесь или продукт реакции могут подвергаться одной или нескольким из следующих процедур: концентрирование или разделение (например, между EtOAc и водой или между 5% THF в EtOAc и 1 M фосфорной кислотой); экстрагирование (например, EtOAc, CHCl3, DCM, хлороформом); промывание (например, насыщенным водным NaCl, насыщенным водным NaHCO3, Na2CO3 (5%), CHCl3 или 1 M NaOH); сушка (например, над MgSO4, над Na2SO4 или в условиях вакуума); фильтрование; кристаллизация (например, из EtOAc и гексанов); концентрирование (например, в условиях вакуума); и/или очистка (например, методами хроматографии на силикагеле, флэш-хроматографии, препаративной HPLC, HPLC с обращенной фазой или кристаллизации).
В качестве иллюстрации, соединения согласно настоящему изобретению, а также их соли, могут быть получены, как представлено на схемах I-IV.
Схема I
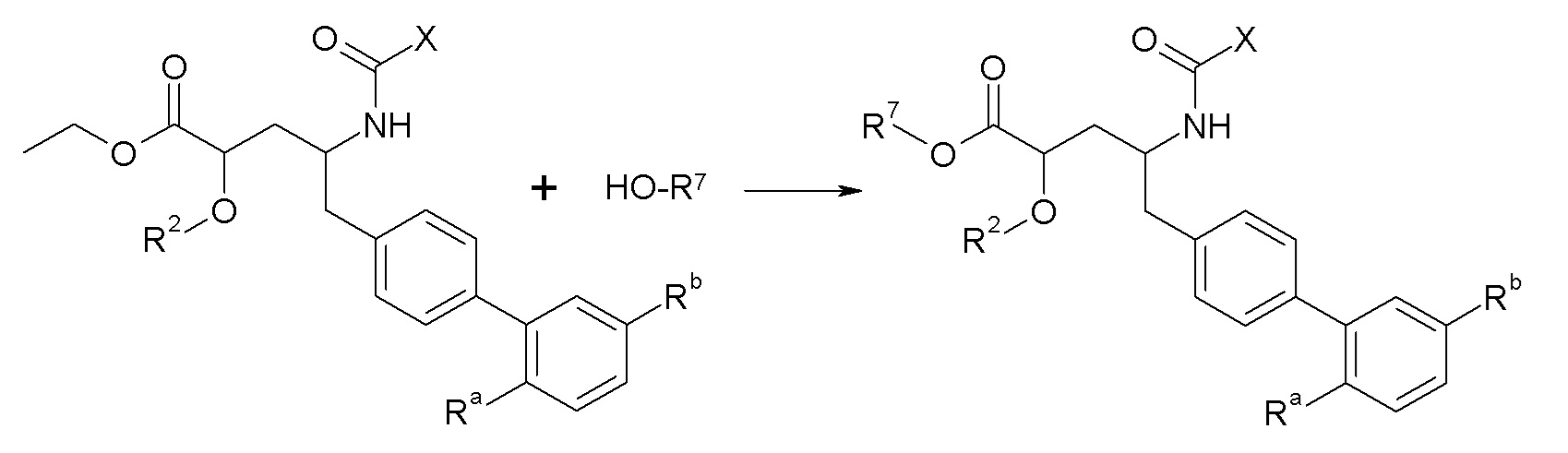
Схема I представляет собой реакцию переэтерификации. В общем, эта реакция включает осуществление взаимодействия сложного эфира с желаемым спиртом (HO-R7) и подходящим кислым катализатором, например, соляной кислотой, при нагревании. Спирты HO-R7 либо являются коммерчески доступными, либо могут быть получены методиками, известными из уровня техники или описанными в настоящем документе. Типичные соединения HO-R7 включают HO-CH2CF3, HO-(CH2)2CF3, HO-CH2CF2CH3, HO-CH2CF2CF3, HO-C(CH3)(CF3)2, HO-CH(CH2CH3)CF3, HO-CH(CH3)CF2CF3, бензиловый спирт и
Схема II
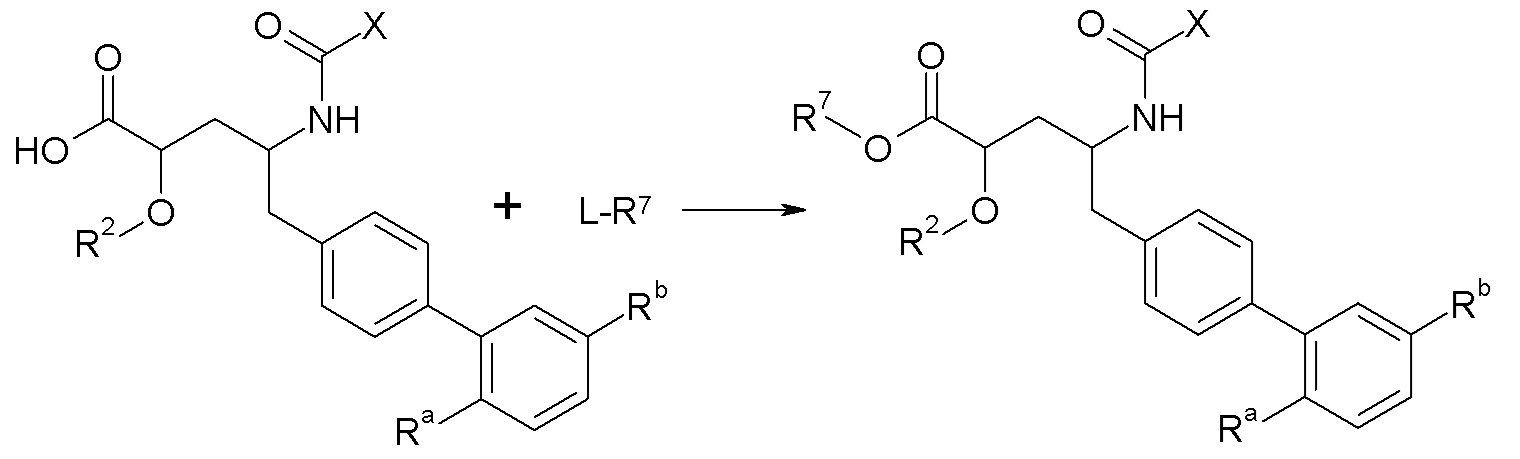
Схема II представляет собой реакцию нуклеофильного замещения, где L представляет собой подходящую уходящую группу. Как правило, эту реакцию проводят в присутствии подходящего основания, такого как триэтиламин, в подходящем инертном разбавителе или растворителе, таком как ацетон. Соединения L-R7 либо являются коммерчески доступными, либо могут быть получены методиками, известными из уровня техники или описанными в настоящем документе. Типичные соединения L-R7 включают Br-(CH2)2OH, Br-(CH2)3OH, Br-(CH2)2OCH3, Br-CH2OC(O)CH3, Cl-CH2OC(O)(CH2)2CH3, Cl-CH2OC(O)OCH2CH3, Cl-CH2OC(O)OCH(CH3)2, Cl-CH2OC(O)O-циклогексил, сложный хлорметиловый эфир (S)-2-бензилоксикарбониламино-3-метилмасляной кислоты и сложный хлорметиловый эфир (S)-2-трет-бутоксикарбониламино-3-метилмасляной кислоты.
В качестве альтернативы, в представленной на схеме II реакции сочетания с использованием HOBt и EDC вместо L-R7 используют спирт, например, HO-C2-4алкилен-N(CH3)2.
Схема III

Схема III представляет собой реакцию нуклеофильного замещения, где L представляет собой подходящую уходящую группу. Как правило, эту реакцию проводят в присутствии подходящего основания, такого как N,N-диизопропилэтиламин, в подходящем инертном разбавителе или растворителе, таком как дихлорметан. Соединение L-R2 либо является коммерчески доступным, либо может быть получено методиками, известными из уровня техники или описанными в настоящем документе. Типичные соединения L-R2 включают Cl-C(O)-CH3, Cl-C(O)-CH(CH3)2 и Cl-C(O)-CH2CH(CH3)2.
Схема IV

Схема IV представляет собой реакцию сочетания, где P представляет собой H или подходящую аминозащитную группу. Если P представляет собой аминозащитную группу, то процесс дополнительно включает снятие защитных групп с соединения до стадии сочетания или in situ. Типичные агенты сочетания включают HATU и HOBt с EDC. Как правило, эти реакции проводят в присутствии основания, такого как DIPEA или 4-метилморфолин, и инертного разбавителя или растворителей, таких как DMF или DMA. Исходные карбоновые кислоты обычно являются коммерчески доступными или могут быть получены с использованием методик, известных из уровня техники.
Дополнительные подробности, касающиеся конкретных условий проведения реакции и других процедур для получения представленных соединений согласно настоящему изобретению или их промежуточных соединений, описаны в изложенных ниже примерах.
ПРАКТИЧЕСКАЯ ПРИМЕНИМОСТЬ
Соединения формулы I'-III' и VII'-X' активны в качестве ингибиторов неприлизина, и, как предполагается, характеризуются практической применимостью в терапии в качестве ингибиторов неприлизина. Предполагается, что пролекарства указанных соединений, будучи метаболизированными in vivo, характеризуются той же практической применимостью. Таким образом, при рассмотрении активности соединений согласно настоящему изобретению подразумевается, что указанные пролекарства обладают ожидаемой активностью, будучи метаболизированными.
Типичные методы анализа включают в качестве иллюстрации и без ограничения, методы, в которых измеряют ингибирование NEP. Применимые вспомогательные методы анализа включают методы измерения ингибирования ACE и ингибирования аминопетидазы P (APP) (например, как описано в Sulpizio et al. (2005) JPET 315:1306-1313). Фармакодинамический анализ для оценки значений ингибирующей активности in vivo в отношении ACE и NEP у анестезированных крыс описан в Seymour et al. (1985) Hypertension 7(Suppl I):I-35-I-42 и Wigle et al. (1992) Can. J. Physiol. Pharmacol. 70:1525-1528), где ингибирование ACE измеряют как процент ингибирования прессорной реакции на ангиотензин I, а ингибирование NEP измеряют как увеличение количества циклического гуанозин-3',5'-монофосфата (cGMP), выделяемого с мочой.
Также существует множество методов анализа in vivo, которые могут быть использованы. Модель бодрствующей крысы со спонтанной гипертензией (SHR) представляет собой модель ренин-зависимой гипертензии (см., например, Intengan et al. (1999) Circulation 100(22):2267-2275 и Badyal et al. (2003) Indian Journal of Pharmacology 35:349-362). Модель бодрствующей крысы с использованием ацетатной соли дезоксикортикостерона (DOCA-соль) представляет собой модель объемзависимой гипертензии, которая применима для измерения активности NEP (см., например, Trapani et al. (1989) J. Cardiovasc. Pharmacol. 14:419-424, и указанную выше Intengan et al. (1999) Hypertension 34(4):907-913 и Badyal et al. (2003)). Модель DOCA-соль особенно применима для оценки способности тестируемого соединения снижать кровяное давление, а также для измерения способности тестируемого соединения предупреждать или замедлять рост кровяного давления. Модель на крысах линии Dahl, чувствительных к развитию гипертензии при употреблении солевой диеты (DSS) представляет собой модель гипертензии, которая чувствительна к пищевой соли (NaCl), и описана, например, в Rapp (1982) Hypertension 4:753-763. Модель легочной артериальной гипертензии при введении монокроталина крысе, описанная, например, в Kato et al. (2008) J. Cardiovasc. Pharmacol. 51(1):18-23, является достоверным показателем клинической эффективности лечения легочной артериальной гипертензии. Модели сердечной недостаточности на животных включают модель DSS сердечной недостаточности на крысах и модель с аортокавальной фистулой (AV-шунт), последняя из которых описана, например, в Norling et al. (1996) J. Amer. Soc. Nephrol. 7:1038-1044. Другие модели на животных, такие как тесты горячей пластинки, отдергивания хвоста и формалиновый тест, можно использовать для измерения анальгезирующих свойств соединения, а также модель нейропатической боли с использованием лигатуры спинального нерва (SNL) (см., например, Malmberg et al. (1999) Current Protocols in Neuroscience 8.9.1-8.9.15). Другие свойства и практическую применимость соединений может быть продемонстрирована с использованием различные методов анализа in vitro и in vivo, хорошо известных специалистам в данной области техники.
Предполагается, что соединения согласно настоящему изобретению применимы для лечения и/или профилактики развития медицинских состояний, реагирующих на ингибирование NEP. Поэтому предполагается, что пациенты, страдающие заболеванием или нарушением, которое поддается лечению при ингибировании фермента NEP или при увеличении уровней его пептидных субстратов, могут подвергаться лечению путем введения терапевтически эффективного количества соединения согласно настоящему изобретению. Например, предполагается, что путем ингибирования NEP соединение потенцирует биологические эффекты эндогенных пептидов, которые метаболизируются NEP, таких как натрийуретические пептиды, бомбезин, брадикинины, кальцитонин, эндотелины, энкефалины, нейротензин, вещество P и вазоактивный пептид кишечника. Таким образом, предполагается, что соединения обладают другими физиологическими эффектами, например, на почечную, центральную нервную, репродуктивную системы и желудочно-кишечный тракт.
Сердечно-сосудистые заболевания
Предполагается, что потенцируя действия вазоактивных пептидов, подобных натрийуретическим пептидам и брадикинину, соединения согласно настоящему изобретению находят применение при лечении и/или профилактике развития медицинских состояний, таких как сердечно-сосудистые заболевания (см., например, Roques et al. (1993) Pharmacol. Rev. 45:87-146 и Dempsey et al. (2009) Amer. J. of Pathology 174(3):782-796). Представляющие особый интерес сердечно-сосудистые заболевания включают гипертензию и сердечную недостаточность. Гипертензия включает в качестве иллюстрации и без ограничения: первичную гипертензию, которую также называют эссенциальной гипертензией или идиопатической гипертензией; вторичную гипертензию; гипертензию с сопутствующим заболеванием почек; тяжелую форму гипертензии с сопутствующим заболеванием почек или без него; легочную гипертензию, включая легочную артериальную гипертензию; и устойчивую гипертензию. Сердечная недостаточность включает в качестве иллюстрации и неограничивающего примера: застойную сердечную недостаточность; острую сердечную недостаточность; хроническую сердечную недостаточность, например со сниженной фракцией выброса левого желудочка (также называемую систолической сердечной недостаточностью) или с сохраненной фракцией выброса левого желудочка (также называемую диастолической сердечной недостаточностью); и острую и хроническую декомпенсированную сердечную недостаточность с сопутствующим заболеванием почек или без него. Таким образом, согласно одному варианту осуществления, настоящее изобретение относится к способу лечения гипертензии, в частности первичной гипертензии или легочной артериальной гипертензии, включающему введение пациенту терапевтически эффективного количества соединения согласно настоящему изобретению.
При лечении первичной гипертензии терапевтически эффективное количество обычно представляет собой количество, которое достаточно для снижения кровяного давления у пациента. Это может относиться и к гипертензии от легкой до умеренной, и к тяжелой гипертензии. При применении для лечения гипертензии, соединение может быть введено в сочетании с другими терапевтическими средствами, такими как антагонисты альдостерона, ингибиторы ангиотензинпревращающего фермента и ингибиторы ангиотензинпревращающего фермента/неприлизина двойного действия, активаторы и стимуляторы ангиотензинпревращающего фермента 2 (ACE2), вакцины ангиотензина-II, антидиабетические средства, противолипидные средства, антитромботические средства, антагонисты рецептора AT1 и антагонист рецептора AT1/ингибиторы неприлизина двойного действия, антагонисты β1-адренергического рецептора, антагонист β-адренергического рецептора/антагонисты α1-рецептора двойного действия, блокаторы кальциевых каналов, диуретики, антагонисты рецептора эндотелина, ингибиторы эндотелинпревращающего фермента, ингибиторы неприлизина, натрийуретические пептиды и их аналоги, антагонисты рецептора клиренса натрийуретического пептида, доноры оксида азота, нестероидные противовоспалительные средства, ингибиторы фосфодиэстеразы (особенно, ингибиторы PDE-V), агонисты рецептора простагландина, ингибиторы ренина, стимуляторы и активаторы растворимой гуанилатциклазы и их сочетания. Согласно одному конкретному варианту осуществления изобретения, соединение согласно настоящему изобретению комбинируют с антагонистом рецептора AT1, диуретиком, блокатором кальциевых каналов или их сочетанием, и применяют для лечения первичной гипертензии. Согласно другому конкретному варианту осуществления изобретения, соединение согласно настоящему изобретению комбинируют с антагонистом рецептора AT1 и применяют для лечения гипертензии с сопутствующим заболеванием почек.
При лечении легочной артериальной гипертензии терапевтически эффективное количество обычно представляет собой количество, которое достаточно для снижения сопротивления легочных сосудов. Другие цели терапии заключаются в улучшении способности пациента переносить физическую нагрузку. Например, в условиях клиники терапевтически эффективное количество может представлять собой количество, которое улучшает способность пациента комфортно ходить в течение 6 минут (преодолевая расстояние приблизительно 20-40 метров). При применении для лечения легочной артериальной гипертензии, соединение может быть введено в сочетании с другими терапевтическими средствами, такими как α-адренергические антагонисты, антагонисты β1-адренергического рецептора, агонисты β2-адренергического рецептора, ингибиторы ангиотензинпревращающего фермента, антикоагулянты, блокаторы кальциевых каналов, диуретики, антагонисты рецепторов эндотелина, ингибиторы PDE-V, аналоги простагландина, селективные ингибиторы обратного захвата серотонина и их сочетания. Согласно одному конкретному варианту осуществления изобретения, соединение согласно настоящему изобретению комбинируют с ингибитором PDE-V или селективным ингибитором обратного захвата серотонина и применяют для лечения легочной артериальной гипертензии.
Другой вариант осуществления настоящего изобретения относится к способу лечения сердечной недостаточности, в частности, застойной сердечной недостаточности (включая как систолическую, так и диастолическую застойную сердечную недостаточность), включающему введение пациенту терапевтически эффективного количества соединения согласно настоящему изобретению. Терапевтически эффективное количество обычно представляет собой количество, которое достаточно для снижения кровяного давления и/или улучшения функции почек. В условиях клиники терапевтически эффективное количество может представлять собой количество, которое достаточно для улучшения динамики сердечного кровообращения, такого как, например, снижение заклинивающего давления, давления в правом предсердии, давления заполнения и сопротивления сосудов. Согласно одному варианту осуществления, соединение вводят в виде внутривенной лекарственной формы. При применении для лечения сердечной недостаточности, соединение может быть введено в сочетании с другими терапевтическими средствами, такими как антагонисты аденозинового рецептора, разрушители конечного продукта усиленного гликозилирования, антагонисты альдостерона, антагонисты рецептора AT1, антагонисты β1-адренергического рецептора, антагонист β-адренергического рецептора/антагонисты α1-рецептора двойного действия, ингибиторы химазы, дигоксин, диуретики, ингибиторы эндотелинпревращающего фермента (ECE), антагонисты рецепторов эндотелина, натрийуретические пептиды и их аналоги, антагонисты рецептора клиренса натрийуретического пептида, доноры оксида азота, аналоги простагландина, ингибиторы PDE-V, стимуляторы и активаторы растворимой гуанилатциклазы и антагонисты рецептора вазопрессина. Согласно одному конкретному варианту осуществления изобретения, соединение согласно настоящему изобретению комбинируют с антагонистом альдостерона, антагонистом β1-адренергического рецептора, антагонистом рецептора AT1 или диуретиком и применяют для лечения застойной сердечной недостаточности.
Диарея
Предполагается, что в качестве ингибиторов NEP соединения согласно настоящему изобретению ингибируют деградацию эндогенных энкефалинов, а потому такие соединения могут также найти применение при лечении диареи, включая инфекционную и секреторную/водянистую диарею (см., например, Baumer et al. (1992) Gut 33:753-758; Farthing (2006) Digestive Diseases 24:47-58; и Marçais-Collado (1987) Eur. J. Pharmacol. 144(2):125-132). При применении для лечения диареи соединения согласно настоящему изобретению можно комбинировать с одним или несколькими дополнительными противодиарейными схемами лечения.
Заболевания почек
Предполагается, что потенцируя эффекты вазоактивных пептидов, подобных натрийуретическим пептидам и брадикинину, соединения согласно настоящему изобретению усиливают функцию почек (см. Chen et al. (1999) Circulation 100:2443-2448; Lipkin et al. (1997) Kidney Int. 52:792-801; и Dussaule et al. (1993) Clin. Sci. 84:31-39) и находят применение при лечении и/или профилактике развития заболеваний почек. Заболевания почек, представляющие особый интерес, включают диабетическую нефропатию, хроническое заболевание почек, протеинурию и, в особенности, острое повреждение почек или острую почечную недостаточность (см. Sharkovska et al. (2011) Clin. Lab. 57:507-515 и Newaz et al. (2010) Renal Failure 32:384-390). При применении для лечения заболевания почек, соединение может быть введено в сочетании с другими терапевтическими средствами, такими как ингибиторы ангиотензинпревращающего фермента, антагонисты рецептора AT1 и диуретики.
Профилактическая терапия
Также предполагается, что потенцируя эффекты натрийуретических пептидов, соединения согласно настоящему изобретению применимы при профилактической терапии вследствие антигипертрофического и противофиброзного действия натрийуретических пептидов (см. Potter et al. (2009) Handbook of Experimental Pharmacology 191:341-366), например, для профилактики прогрессирования сердечной недостаточности после инфаркта миокарда, профилактики развития артериального рестеноза после ангиопластики, профилактики утолщения стенок кровеносного сосуда после операций на сосудах, профилактики развития атеросклероза и профилактики развития диабетической ангиопатии.
Глаукома
Предполагается, что потенцируя эффекты натрийуретических пептидов, соединения согласно настоящему изобретению применимы для лечения глаукомы (см., например, Diestelhorst et al. (1989) International Ophthalmology 12:99-101). При применении для лечения глаукомы, соединения согласно настоящему изобретению можно комбинировать с одним или несколькими дополнительными противоглаукомными средствами.
Облегчение боли
Предполагается, что в качестве ингибиторов NEP соединения согласно настоящему изобретению ингибируют деградацию эндогенных энкефалинов, а потому такие соединения могут также найти применение в качестве обезболивающих средств (см., например, Roques et al. (1980) Nature 288:286-288 и Thanawala et al. (2008) Current Drug Targets 9:887-894). При применении для лечения боли, соединения согласно настоящему изобретению можно комбинировать с одним или несколькими дополнительными антиноцицептивными лекарственными средствами, такими как ингибиторы аминопептидазы N или дипептидилпептидазы III, нестероидные противовоспалительные средства, ингибиторы обратного захвата моноаминов, миорелаксанты, антагонисты рецептора NMDA, агонисты опиоидного рецептора, агонисты рецептора серотонина 5-HT1D и трициклические антидепрессанты.
Другие применения
Также предполагается, что вследствие своей способности ингибировать NEP соединения согласно настоящему изобретению применимы в качестве антитуссивных средств, а также находят применение при лечении портальной гипертензии, ассоциированной с циррозом печени (см. Sansoe et al. (2005) J. Hepatol. 43:791-798), злокачественной опухоли (см. Vesely (2005) J. Investigative Med. 53:360-365), депрессии (см. Noble et al. (2007) Exp. Opin. Ther. Targets 11:145-159), нарушений менструального цикла, преждевременных родов, токсикоза беременности, эндометриоза, нарушений со стороны репродуктивной системы (например, мужское и женское бесплодие, синдром поликистозных яичников, неудача при имплантации) и нарушении половой функции у мужчин и женщин, включая эректильную дисфункцию у мужчин и нарушение полового возбуждения у женщин. Более конкретно, предполагается, что соединения согласно настоящему изобретению применимы в лечении нарушения половой функции у женщин (см. Pryde et al. (2006) J. Med. Chem. 49:4409-4424), которое часто определяют как сложность или невозможность пациентки получить удовлетворение в сексуальном смысле. Нарушение охватывает множество разнообразных женских сексуальных нарушений, включая в качестве иллюстрации и без ограничения сниженное сексуальное влечение, нарушение полового возбуждения, нарушение оргастической функции и нарушение с сексуальной болезненностью. При применении для лечения таких нарушений, особенно нарушения половой функции у женщин, соединения согласно настоящему изобретению можно комбинировать с одним или несколькими из следующих ниже вспомогательных средств: ингибиторами PDE-V, агонистами допамина, агонистами и/или антагонистами эстрогенового рецептора, андрогенами и эстрогенами. Также предполагается, что вследствие их способности ингибировать NEP соединения согласно настоящему изобретению обладают противовоспалительными свойствами и, как предполагается, обладают практической применимостью как таковой, особенно, при применении в сочетании со статинами.
Недавние исследования позволяют предположить, что NEP играет роль в регуляции функции нервной системы при инсулинзависимом диабете и алиментарном ожирении (Coppey et al. (2011) Neuropharmacology 60:259-266). Следовательно, также предполагается, что вследствие их способности ингибировать NEP соединения согласно настоящему изобретению применимы для обеспечения защиты от нарушения нервной системы, вызванного диабетом или алиментарным ожирением.
Количество соединения согласно настоящему изобретению, водимое в пересчете на дозу, или общее количество, вводимое в сутки, можно определить предварительно, или его можно определить отдельно для каждого пациента, принимая во внимание множество факторов, включая природу и тяжесть состояния пациента, подлежащее лечению состояние, возраст, вес и общее состояние здоровья пациента, переносимость пациентом активного средства, способ введения, подлежащие учету фармакологические факторы, такие как активность, эффективность, фармакокинетические и токсикологические профили соединения и любых вводимых вспомогательных средств, и т.п. Лечение пациента, страдающего заболеванием или медицинским состоянием (таким как гипертензия), может начинаться с предварительно определенной дозировки или дозировки, определяемой лечащим врачом, и будет продолжаться в течение периода времени, необходимого для профилактики развития, улучшения, подавления или облегчения симптомов заболевания или медицинского состояния. Пациенты, находящиеся на таком лечении, обычно будут подвергаться рутинной проверке для определения эффективности терапии. Например, при лечении гипертензии, для определения эффективности лечения можно использовать измерения кровяного давления. Похожие показатели для других заболеваний и состояний, описываемых в настоящем документе, хорошо известны и легкодоступны лечащему врачу. Постоянное отслеживание врачом будет обеспечивать гарантию того, что в любое заданное время вводится оптимальное количество соединения согласно настоящему изобретению, а также облегчать определение продолжительности лечения. Это имеет особенное значение, когда также вводятся вспомогательные средства, поскольку их выбор, дозировка и продолжительность лечения также могут потребовать корректировки. Таким образом, во время курса терапии схему лечения и дозирование можно корректировать для того, чтобы вводить меньшее количество активного средства, которое проявляет требуемую эффективность и, дополнительно, чтобы продолжать введение только при условии, что оно необходимо для успешного излечения от заболевания или медицинского состояния.
Инструменты исследования
Поскольку соединения согласно настоящему изобретению метаболизируются in vivo до соединений, обладающих активностью в качестве ингибиторов неприлизина, они также применимы в качестве инструментов для исследования или изучения биологических систем или образцов, содержащих фермент NEP, например, для изучения заболеваний, в которых играют роль фермент NEP или его пептидные субстраты. Таким образом, согласно одному аспекту, настоящее изобретение относится к способу применения соединения согласно настоящему изобретению в качестве инструмента исследования, включающему проведение биологического анализа с применением соединения согласно настоящему изобретению. Любую подходящую биологическую систему или образец, содержащий фермент NEP, можно применять в таких исследованиях, которые могут проводиться либо in vitro, либо in vivo. Типичные биологические системы или образцы, подходящие для таких исследований, включают в качестве неограничивающих примеров клетки, клеточные экстракты, плазматические мембраны, образцы тканей, выделенные органы, млекопитающих (таких как мыши, крысы, морские свинки, кролики, собаки, свиньи, люди, и т.д.), и т.п., причем особенный интерес представляют млекопитающие. Согласно одному конкретному варианту осуществления настоящего изобретения, активность фермента NEP у млекопитающего ингибируют путем введения ингибирующего NEP количества соединения согласно настоящему изобретению. Данные соединения также можно применять в качестве инструментов исследования, проводя биологические анализы с применением таких соединений.
При применении в качестве инструмента исследования, биологическую систему или образец, содержащий фермент NEP, обычно подвергают взаимодействию с ингибирующим NEP количеством соединения согласно настоящему изобретению. После того, как биологическая система или образец подвергается воздействию соединения, определяют эффекты ингибирования фермента NEP с использованием общепринятых способов и оборудования, как измерение связывания с рецептором в анализе связывания или измерение опосредованных лигандом изменений в анализе функций. Воздействие включает приведение клеток или тканей во взаимодействие с соединением путем введения млекопитающему кристаллического соединения, например, в/п, п/о, в/в, п/к или ингаляционным введением, и т.д. Данная стадия определения может включать измерение ответа (количественный анализ) или проведение наблюдения (качественный анализ). Измерение ответа включает, например, определение воздействия соединения на биологическую систему или образец с использованием общепринятых способов и оборудования, таких как анализ активности ферментов и измерение опосредованных субстратами фермента или продуктом изменений в анализе функций. Результаты анализа могут быть использованы для определения уровня активности, а также количества соединения, необходимого для достижения требуемого результата, а именно, ингибирующего фермент NEP количества. Обычно, стадия определения будет включать определение эффекта ингибирования фермента NEP.
Дополнительно, соединения согласно настоящему изобретению могут быть использованы в качестве инструментов исследований для оценки других химических соединений, а потому они также применимы при скрининговых исследованиях для обнаружения, например, новых соединений, обладающих ингибирующей NEP активностью. Таким образом, согласно другому аспекту, настоящее изобретение относится к способу оценки тестируемого соединения в биологическом методе анализа, включающему: (a) проведение биологического анализа с тестируемым соединением для получения первого результата анализа; (b) проведение биологического анализа с соединением согласно настоящему изобретению для получения второго результата анализа; причем стадию (a) проводят до, после или одновременно со стадией (b); и (c) сравнение первого результата анализа, полученного на стадии (a), со вторым результатом анализа, полученным на стадии (b). Типичные биологические методы анализа включают анализ ингибирования фермента NEP. Таким образом, соединения согласно настоящему изобретению используют в качестве стандартов в методах анализа для обеспечения возможности сравнения результатов, полученных с использованием тестируемого соединения и с использованием соединения согласно настоящему изобретению, чтобы выявить те тестируемые соединения, которые обладают приблизительно равной или превосходящей активностью, если таковая имеет место. Например, данные по pKi для тестируемого соединения или группы тестируемых соединений сравнивают с данными pKi для соединения согласно настоящему изобретению, чтобы выявить те тестируемые соединения, которые обладают требуемыми свойствами, например, тестируемые соединения, обладающие величиной pKi приблизительно равной или большей таковой для соединения согласно настоящему изобретению, если таковое имеет место. Данный аспект настоящего изобретения включает в качестве отдельных вариантов осуществления получение данных для сравнения (с использованием подходящих методов анализа), а также анализ результатов тестирования для выявления представляющих интерес тестируемых соединений.
Согласно еще одному аспекту, настоящее изобретение относится к способу изучения биологической системы или образца, содержащего фермент NEP, причем способ включает: (a) приведение биологической системы или образца во взаимодействие с соединением согласно настоящему изобретению; и (b) определение эффектов, вызываемых соединением, на биологическую систему или образец.
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ЛЕКАРСТВЕННЫЕ ФОРМЫ
Соединения согласно настоящему изобретению обычно вводят пациенту в форме фармацевтической композиции или лекарственной формы. Такие фармацевтические композиции могут быть введены пациенту посредством любого приемлемого пути введения, включая без ограничения пероральный, ректальный, вагинальный, назальный, ингаляционный, местный (включая чрескожный), глазной и парентеральный пути введения. Кроме того, соединения согласно настоящему изобретению могут быть введены, например, перорально в виде нескольких доз в сутки (например, два, три или четыре раза в сутки), в виде однократной суточной дозы или однократной недельной дозы. Следует понимать, что любая форма соединений согласно настоящему изобретению (а именно, свободное основание, свободная кислота, фармацевтически приемлемая соль, сольват и т.п.), которая подходит для конкретного пути введения, может быть применима в фармацевтических композициях, обсуждаемых в настоящем документе.
Соответственно, согласно одному варианту осуществления, настоящее изобретение относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель и соединение согласно настоящему изобретению. При желании, композиции могут содержать другие терапевтические и/или препаратообразующие средства. При обсуждении композиций, термин «соединение согласно настоящему изобретению» также может называться в настоящем документе термином «активное агент» для разграничения его с другими компонентами лекарственной формы, такими как носитель. Поэтому, следует понимать, что термин «активное агент» включает соединения формулы I, а также фармацевтически приемлемые соли, сольваты и пролекарства этого соединения.
Фармацевтические композиции согласно настоящему изобретению обычно содержат терапевтически эффективное количество соединения согласно настоящему изобретению. Однако специалистам в данной области техники следует понимать, что фармацевтическая композиция может содержать количество соединения, превышающее терапевтически эффективное, как, например, в нефасованных композициях, или меньшее, чем терапевтически эффективное количество, а именно, индивидуальные стандартные формы, приготовленные для многократного введения для достижения терапевтически эффективного количества. Обычно, композиция содержит приблизительно 0,01-95 масс. % активного средства, включая приблизительно 0,01-30 масс. %, например, приблизительно 0,01-10 масс. %, причем конкретное количество зависит собственно от лекарственной формы, пути введения, частоты дозирования и т.п. Согласно одному варианту осуществления, композиция, подходящая для пероральной лекарственной формы, например, может содержать приблизительно 5-70 масс. % или приблизительно 10-60 масс. % активного средства.
Любой общепринятый носитель или наполнитель может быть использован в фармацевтических композициях согласно настоящему изобретению. Выбор конкретного носителя или наполнителя, или сочетаний носителей или наполнителей, будет зависеть от пути введения, используемого для лечения конкретного пациента, или типа медицинского состояния или болезненного состояния. В этой связи, приготовление подходящей композиции для конкретного пути введения подпадает под объем знаний специалистов в области фармацевтической техники. Кроме того, носители или наполнители, используемые в таких композициях, являются коммерчески доступными. В качестве дополнительной иллюстрации, общепринятые методики приготовления описаны в Remington: The Science and Practice of Pharmacy, 20th Edition, Lippincott Williams & White, Baltimore, Maryland (2000); и H. C. Ansel et al., Pharmaceutical Dosage Forms and Drug Delivery Systems, 7th Edition, Lippincott Williams & White, Baltimore, Maryland (1999).
Типичные примеры веществ, которые могут служить в качестве фармацевтически приемлемых носителей, включают без ограничения следующие: сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлоза, такая как микрокристаллическая целлюлоза, и ее производные, такие как карбоксиметилцеллюлоза натрия, этилцеллюлоза и ацетат целлюлозы; порошкообразный трагакант; солод; желатин; тальк; наполнители, такие как какао-масло и воск для суппозиториев; масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; гликоли, такие как пропиленгликоль; полиолы, такие как глицерин, сорбит, маннит и полиэтиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные средства, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; апирогенная вода; изотонический солевой раствор; раствор Рингера; этиловый спирт; фосфатно-буферные растворы; сжатые газы-пропелленты, такие как хлорфторуглероды и гидрофторуглероды; и другие нетоксичные совместимые вещества, применяемые в фармацевтических композициях.
Фармацевтические композиции обычно приготавливают путем тщательного и равномерного смешивания или перемешивания активного ингредиента с фармацевтически приемлемым носителем и одним или несколькими необязательными ингредиентами. Конечная равномерно перемешанная смесь может быть далее сформована или заключена в таблетки, капсулы, пилюли, респираторные коробки, инъекционные патроны, диспенсеры и т.п., с использованием общепринятых процедур и оборудования.
Согласно одному варианту осуществления, фармацевтические композиции подходят для перорального введения. Подходящие композиции для перорального введения могут быть в форме капсул, таблеток, пилюль, леденцов, облаток, драже, порошков, гранул; растворов или суспензий в водной или неводной жидкости; жидких эмульсий типа «масло в воде» или «вода в масле»; эликсиров или сиропов; и т.п.; причем каждая содержит заранее определенное количество активного средства.
Если композиция предназначена для перорального введения в твердой лекарственной форме (капсулы, таблетки, пилюли и т.п.), то она обычно содержит активное средство и один или несколько фармацевтически приемлемых носителей, таких как цитрат натрия и дикальцийфосфат. Твердые лекарственные формы также могут содержать: наполнители и разбавители, такие как крахмалы, микрокристаллическая целлюлоза, лактоза, сахароза, глюкоза, маннит и/или кремниевая кислота; связующие вещества, такие как карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и/или акация; увлажнители, такие как глицерин; разрыхлители, такие как агар-агар, карбонат кальция, картофельный или тапиоковый крахмал, альгиновая кислота, некоторые силикаты и/или карбонат натрия; замедлители растворения, такие как парафин; усилители всасывания, такие как соединения четвертичного аммония; увлажнители, такие как цетиловый спирт и/или моностеарат глицерина; абсорбенты, такие как каолин и/или бентонитовая глина; смазки, такие как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и/или их смеси; красители; и буферные средства.
Разделительные средства, увлажнители, покровные средства, подсластители, вкусоароматизаторы и отдушки, консерванты и антиоксиданты также могут присутствовать в фармацевтических композициях. Типичные покровные средства для таблеток, капсул, пилюль и т.п. включают таковые, используемые для кишечнорастворимых покрытий, такие как ацетатфталат целлюлозы, поливинилацетатфталат, фталат гидроксипропилметилцеллюлозы, сополимеры метакриловой кислоты/сложного эфира метакриловой кислоты, ацетаттимеллитат целлюлозы, карбоксиметилэтилцеллюлоза, ацетатсукцинат гидроксипропилметилцеллюлозы и т.п. Примеры фармацевтически приемлемых антиоксидантов включают: растворимые в воде антиоксиданты, такие как аскорбиновая кислота, гидрохлорид цистеина, бисульфат натрия, метабисульфат натрия, сульфит натрия и т.п.; растворимые в масле антиоксиданты, такие как аскорбилпальмитат, бутилированный гидроксианизол, бутилированный гидрокситолуол, лецитин, пропилгаллат, альфа-токоферол и т.п.; и металл-хелатирующие средства, такие как лимонная кислота, этилендиаминтетрауксусная кислота, сорбит, винная кислота, фосфорная кислота и т.п.
Композиции также могут быть приготовлены для обеспечения медленного или контролируемого высвобождения активного средства с использованием, например, гидроксипропилметилцеллюлозы в различных пропорциях или других полимерных матриц, липосом и/или микросфер. Кроме того, фармацевтические композиции согласно настоящему изобретению могут содержать замутняющие средства и могут быть приготовлены так, что они высвобождают активное средство конкретной порцией исключительно, или предпочтительно, в желудочно-кишечном тракте, необязательно, замедленным образом. Примеры заливочных возможных к применению композиций включают полимерные вещества и воски. Активное средство также может находиться в микроинкапсулированной форме, необязательно с одним или несколькими из вышеописанных наполнителей.
Подходящие жидкие лекарственные формы для перорального введения включают, в качестве иллюстрации, фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Жидкие лекарственные формы обычно содержат активное средство и инертный разбавитель, такой как, например, вода или другие растворители, солюбилизаторы и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, масла (например, хлопковое, арахисовое, кукурузное, ростков пшеницы, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли и сложные эфиры жирной кислоты и сорбитана, и их смеси. Суспензии могут содержать средства, способствующие суспендированию, такие как, например, этоксилированные изостеариловые спирты, полиоксиэтиленсорбит и сложные эфиры сорбитана, микрокристаллическая целлюлоза, аметагидроксид алюминия, бентонит, агар-агар и трагакант, и их смеси.
Если фармацевтическая композиция согласно настоящему изобретению предназначена для перорального введения, то она может быть упакована в стандартную лекарственную форму. Термин «стандартная лекарственная форма» относится к физически дискретной единице, подходящей для дозированного введения пациенту, т.е. каждая единица содержит заранее определенное количество активного средства, рассчитанное для получения желаемого терапевтического эффекта, как в отдельности, так и в сочетании с одной или несколькими дополнительными единицами. Например, такие стандартные лекарственные формы могут представлять собой капсулы, таблетки, пилюли и т.п.
Согласно другому варианту осуществления, композиции согласно настоящему изобретению подходят для ингаляционного введения и обычно могут находиться в форме аэрозоля или порошка. Такие композиции, как правило, вводят с использованием хорошо известных устройств доставки, таких как небулайзер, сухой порошок или дозирующий ингалятор. Небулайзеры производят поток движущегося с высокой скоростью воздуха, который позволяет композиции разбрызгиваться в виде тумана, который попадает в респираторный тракт пациента. Типичная композиция для небулайзера содержит активное средство, растворенное в носителе до образования раствора, или микронизированное и соединенное с носителем с образованием суспензии микронизированных частиц пригодного для вдыхания размера. Ингаляторы сухого порошка вводят активное средство в виде свободнотекучего порошка, который диспергирован в воздушном потоке, пациенту в процессе вдыхания. Типичная композиция сухого порошка содержит активное средство в виде сухой смеси с наполнителем, таким как лактоза, крахмал, маннит, декстроза, полимолочная кислота, полилактидкогликолид и их сочетания. Дозирующие ингаляторы выбрасывают измеренное количество активного средства с использованием сжатого газа пропеллента. Типичная дозируемо вводимая композиция содержит раствор или суспензию активного средства в сжиженном пропелленте, таком как хлорфторуглерод или гидрофторалкан. Необязательные компоненты таких композиций включают сорастворители, такие как этанол или пентан, и поверхностно-активные вещества, такие как сорбитана триолеат, олеиновая кислота, лецитин, глицерин и лаурилсульфат натрия. Такие композиции обычно приготавливают путем добавления охлажденного или сжатого гидрофторалкана в подходящий контейнер, содержащий активное средство, этанол (при наличии) и поверхностно-активное вещество (при наличии). Для приготовления суспензии, активное средство микронизируют, а затем соединяют с пропеллентом. В качестве альтернативы, суспензированная композиция может быть приготовлена путем распылительной сушки покрытия поверхностно-активного вещества на микронизированных частицах активного средства. Затем, композицию помещают в аэрозольный баллон, который отмеряет порцию ингалятора.
Соединения согласно настоящему изобретению также могут быть введены парентерально (например, посредством подкожной, внутривенной, внутримышечной или интраперитонеальной инъекции). Для такого введения, активное средство предоставляют в виде стерильного раствора, суспензии или эмульсии. Типичные растворители для приготовления таких композиций включают воду, солевой раствор, низкомолекулярные спирты, такие как пропиленгликоль, полиэтиленгликоль, масла, желатин, сложные эфиры жирной кислоты, такие как этилолеат и т.п. Парентеральные композиции также могут содержать один или несколько антиоксидантов, солюбилизаторов, стабилизаторов, консервантов, увлажнителей, эмульгаторов и средств, способствующих диспергированию. Поверхностно-активные вещества, дополнительные стабилизаторы или средства, регулирующие рН (кислоты, основания или буферы) и антиоксиданты особенно применимы для обеспечения стабильности композиции, например, с целью минимизировать или избежать гидролиза сложного эфира и амидных связей, или димеризации тиолов, которые могут присутствовать в соединении. Такие композиции могут быть сделаны стерильными посредством применения стерильной инъекционной среды, стерилизующего средства, фильтрации, облучения или нагрева. Согласно одному конкретному варианту осуществления, парентеральная композиция содержит в качестве фармацевтически приемлемого носителя водный раствор циклодекстрина. Подходящие циклодекстрины включают циклические молекулы, содержащие шесть или более α-D-глюкопиранозных единиц, соединенных по 1,4-положениям связями, как в амилазе, β-циклодекстрине или циклогептаамилозе. Типичные циклодекстрины включают производные циклодекстрина, такие как простой гидроксипропил и сульфобутилэфир циклодекстринов, такой как гидроксипропил-β-циклодекстрин и простой сульфобутилэфир β-циклодекстрина. Типичные буферы для таких композиций включают буферы на основе карбоновой кислоты, такие как цитратные, лактатные и малеатные буферные растворы.
Соединения согласно настоящему изобретению также могут быть введены чрескожно с применением известных систем чрескожной доставки и наполнителей. Например, соединение может быть смешано с усилителями проникновения, такими как пропиленгликоль, полиэтиленгликольмонолаурат, азациклоалкан-2-оны и т.п., и включено в состав пластыря или аналогичной системы доставки. При желании, в таких чрескожных композициях могут быть использованы дополнительные наполнители, включая гелеобразующие средства, эмульгаторы и буферы.
Вторичные агенты
Соединения согласно настоящему изобретению можно применять в качестве монотерапии заболевания или можно комбинировать с одним или несколькими дополнительными терапевтическими средствами для того, чтобы получить требуемый терапевтический эффект. Таким образом, согласно одному варианту осуществления, фармацевтические композиции согласно настоящему изобретению содержат другие лекарственные средства, которые вводят совместно с соединением согласно настоящему изобретению. Например, композиция может дополнительно содержать одно или несколько лекарственных средств (также называемых «вторичным(и) агентом(ами)»). Такие терапевтические средства хорошо известны в данной области техники и включают антагонисты аденозинового рецептора, антагонисты α-адренергического рецептора, антагонисты β1-адренергического рецептора, агонисты β2-адренергического рецептора, антагонист β-адренергического рецептора/антагонисты α1-рецептора двойного действия, разрушители конечного продукта усиленного гликозилирования, антагонисты альдостерона, ингибиторы альдостеронсинтазы, ингибиторы аминопептидазы N, андрогены, ингибиторы ангиотензинпревращающего фермента и ингибиторы ангиотензинпревращающего фермента/неприлизина двойного действия, активаторы и стимуляторы ангиотензинпревращающего фермента 2, вакцины ангиотензина-II, антикоагулянты, противодиабетические средства, противодиарейные средства, противоглаукомные средства, противолипидные средства, антиноцицептивные средства, антитромботические средства, антагонисты рецептора AT1 и антагонист рецептора AT1/ингибиторы неприлизина двойного действия и многофункциональные блокаторы рецептора ангиотензина, антагонисты рецептора брадикинина, блокаторы кальциевых каналов, ингибиторы химазы, дигоксин, диуретики, агонисты допамина, ингибиторы эндотелинпревращающего фермента, антагонисты рецепторов эндотелина, ингибиторы HMG-CoA-редуктазы, эстрогены, агонисты и/или антагонисты эстрогенового рецептора, ингибиторы обратного захвата моноаминов, миорелаксанты, натрийуретические пептиды и их аналоги, антагонисты рецептора клиренса натрийуретического пептида, ингибиторы неприлизина, доноры оксида азота, нестероидные противовоспалительные средства, антагонисты рецептора N-метил-d-аспартата, агонисты опиоидного рецептора, ингибиторы фосфодиэстеразы, аналоги простагландина, агонисты рецептора простагландина, ингибиторы ренина, селективные ингибиторы обратного захвата серотонина, блокатор натриевых каналов, стимуляторы и активаторы растворимой гуанилатциклазы, трициклические антидепрессанты, антагонисты рецептора вазопрессина и их сочетания. Конкретные примеры данных агентов подробно описаны в настоящем документе.
Таким образом, согласно еще одному аспекту настоящего изобретения, фармацевтическая композиция содержит соединение согласно настоящему изобретению, второй активный агент и фармацевтически приемлемый носитель. В композицию также могут быть включены третий, четвертый и т.д. активный агент. При комбинированном лечении вводимое количество соединения согласно настоящему изобретению, а также количество вторичных агентов, может быть меньше количества, вводимого при монотерапии.
Соединения согласно настоящему изобретению может быть физически смешано со вторым активным агентом с получением композиции, содержащей оба агента; или каждый агент может присутствовать в отдельных и различных композициях, которые вводят пациенту одновременно или с интервалом во времени. Например, соединение согласно настоящему изобретению можно комбинировать со вторым активным агентов, применяя общепринятые способы и оборудование для получения комбинации активных агентов, содержащей соединение согласно настоящему изобретению и второй активный агент. Дополнительно, активные агенты можно комбинировать с фармацевтически приемлемым носителем для получения фармацевтической композиции, содержащей соединение согласно настоящему изобретению, второй активный агент и фармацевтически приемлемый носитель. Согласно данному варианту осуществления, компоненты композиции обычно перемешивают или смешивают с получением физической смеси. Физическую смесь затем водят в терапевтически эффективном количестве с использованием любых из способов введения, описанных в настоящем документе.
В качестве альтернативы, активные агенты могут оставаться отдельными и различными до введения пациенту. Согласно данному варианту осуществления, до введения агента не являются физически смешанными вместе, но вводятся одновременно или с интервалом во времени в виде отдельных композиций. Такие композиции могут быть упакованы по отдельности или могут быть упакованы совместно в наборе. При введении с интервалом во времени, вторичные агента обычно будут вводиться менее чем через 24 часа после введения соединения согласно настоящему изобретению, в какой-либо момент в диапазоне от одновременного введения с соединением согласно настоящему изобретению до введения приблизительно через 24 часа после применения соединения. Это также называется последовательным введением. Таким образом, соединение согласно настоящему изобретению можно вводить перорально одновременно или последовательно с другим активным агентом с применением двух таблеток, по одной таблетке для каждого активного агента, где последовательно может означать введение сразу после введения соединения согласно настоящему изобретению или введение спустя некоторое предварительно определенное время (например, один час спустя или три часа спустя). Также предусмотрено, что вторичный агент можно вводить спустя более чем 24 часа после введения соединения согласно настоящему изобретению. В качестве альтернативы, комбинацию можно вводить различными способами введения, то есть, один агент перорально, а другой - путем ингаляции.
Согласно одному варианту осуществления, набор содержит первую лекарственную форму, содержащую соединение согласно настоящему изобретению и, по меньшей мере, одну дополнительную лекарственную форму, содержащую один или несколько вторичных агентов, представленных в настоящем документе, в количествах, достаточных для осуществления способов согласно настоящему изобретению. Первая лекарственная форма и вторая (или третья и т.д.) лекарственная форма совместно содержат количество активных агентов, терапевтически эффективное для лечения или профилактики заболевания или медицинского состояния у пациента.
Вторичный(ые) агент(ы), будучи включенными в состав, присутствуют в терапевтически эффективном количестве, так что при совместном введении с соединением согласно настоящему изобретению их обычно вводят в количестве, которое продуцирует терапевтически выгодный эффект. Вторичный агент может находиться в виде фармацевтически приемлемой соли, сольвата, оптически чистого стереоизомера и т.д. Вторичный агент может также находиться в виде пролекарства, например, соединения, содержащего группу карбоновой кислоты, которая была этерифицирована. Таким образом, предполагается, что вторичные агенты, перечисленные в настоящем документе, включают все такие формы и являются коммерчески доступными, или их можно получить с использованием общепринятых способов и реагентов.
Согласно одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с антагонистом рецептора аденозина, типичные примеры которого включают в качестве неограничивающих примеров наксифиллин, ролофиллин, SLV-320, теофиллин и тонапофиллин.
Согласно одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с антагонистом α-адренергического рецептора, типичные примеры которого включают в качестве неограничивающих примеров доксазозин, празозин, тамсулозин и теразозин.
Соединения согласно настоящему изобретению также может быть введено в сочетании с антагонистом β1-адренергического рецептора («β1-блокаторы»). Типичные представители β1-блокаторов включают в качестве неограничивающих примеров ацебутолол, альпренолол, амосулалол, аротинолол, атенолол, бефунолол, бетаксолол, бевантолол, бисопролол, бопиндолол, буциндолол, букумолол, бефетолол, буфуралол, бунитролол, бупранолол, бубридин, бутофилолол, каразолол, картеолол, карведилол, целипролол, цетамолол, клоранолол, дилевалол, эпанолол, эсмолол, инденолол, лабетолол, левобунолол, мепиндолол, метипранолол, метопролол, такой как сукцинат метопролола и тартрат метопролола, мопролол, надолол, надоксолол, небивалол, нипрадилол, окспренолол, пенбутолол, пербутолол, пиндолол, практолол, пронеталол, пропранолол, соталол, суфиналол, талиндол, тертатолол, тилизолол, тимолол, толипролол, ксибенолол и их сочетания. Согласно одному конкретному варианту осуществления, β1-антагонист выбирают из атенолола, бисопролола, метопролола, пропранолола, соталола и их сочетаний. Обычно, β1-блокатор вводят в количестве, достаточном для обеспечения приблизительно 2-900 мг на дозу.
Согласно одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с агонистом β2-адренергического рецептора, типичные примеры которого включают в качестве неограничивающих примеров альбутерол, битолтерол, фенотерол, формотерол, индакатерол, изоэтарин, левальбутерол, метапротеренол, пирбутерол, сальбутамол, салмефамол, салметерол, тербуталин, вилантерол и т.п. Обычно, агонист β2-адренергического рецептора вводят в количестве, достаточном для обеспечения приблизительно 0,05-500 мкг на дозу.
Согласно одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с разрушителем конечного продукта усиленного гликозилирования (AGE), примеры которого включают в качестве иллюстрации и без ограничения алагебриум (или ALT-711) и TRC4149.
Согласно другому варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с антагонистом альдостерона, типичные примеры которого включают в качестве неограничивающих примеров эплеренон, спиронолактон и их сочетания. Обычно, антагонист альдостерона вводят в количестве, достаточном для обеспечения приблизительно 5-300 мг в сутки.
Согласно одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с ингибитором аминопептидазы N или дипептидилпептидазы III, примеры которого включают в качестве иллюстрации и без ограничения бестатин и PC18 (2-амино-4-метилсульфонилбутантиол, метионинтиол).
Соединения согласно настоящему изобретению можно также вводить в сочетании с ингибитором ангиотензинпревращающего фермента (ACE). Типичные представители ингибиторов ACE включают в качестве неограничивающих примеров аккуприл, алацеприл, беназеприл, беназеприлат, каптоприл, церанаприл, цилазаприл, делаприл, эналаприл, эналаприлат, фосиноприл, фосиноприлат, имидаприл, лизиноприл, моэксиприл, моноприл, мовелтоприл, пентоприл, периндоприл, квинаприл, квинаприлат, рамиприл, рамиприлат, ацетат саралазина, спираприл, темокаприл, трандолаприл, зофеноприл и их сочетания.
Согласно конкретному варианту осуществления, ACE ингибитор выбирают из: беназеприла, каптоприла, эналаприла, лизиноприла, рамиприла и их сочетаний. Обычно, ингибитор ACE вводят в количестве, достаточном для обеспечения приблизительно 1-150 мг в сутки. Согласно другому варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с ингибитором ангиотензинпревращающего фермента/неприлизина (ACE/NEP) двойного действия, примеры которого включают в качестве неограничивающих примеров: AVE-0848 ((4S,7S,12bR)-7-[3-метил-2(S)-сульфанилбутирамидо]-6-оксо-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]-бензазепин-4-карбоновая кислота); AVE-7688 (илепатрил) и его родительское соединение; BMS-182657 (2-[2-оксо-3(S)-[3-фенил-2(S)-сульфанилпропионамидо]-2,3,4,5-тетрагидро-1H-1-бензазепин-1-ил]уксусная кислота); CGS-35601 (N-[1-[4-метил-2(S)-сульфанилпентанамидо]циклопентилкарбонил]-L-триптофан); фазидотрил; фазидотрилат; эналаприлат; ER-32935 ((3R,6S,9aR)-6-[3(S)-метил-2(S)-сульфанилпентанамидо]-5-оксопергидротиазоло[3,2-a]азепин-3-карбоновая кислота); гемпатрилат; MDL-101264 ((4S,7S,12bR)-7-[2(S)-(2-морфолиноацетилтио)-3-фенилпропионамидо]-6-оксо-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин-4-карбоновая кислота); MDL-101287 ([4S-[4α,7α(R*),12bβ]]-7-[2-(карбоксиметил)-3-фенилпропионамидо]-6-оксо-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин-4-карбоновая кислота); омапатрилат; RB-105 (N-[2(S)-(меркаптометил)-3(R)-фенилбутил]-L-аланин); сампатрилат; SA-898 ((2R,4R)-N-[2-(2-гидроксифенил)-3-(3-меркаптопропионил)тиазолидин-4-илкарбонил]-L-фенилаланин); Sch-50690 (N-[1(S)-карбокси-2-[N2-(метансульфонил)-L-лизиламино]этил]-L-валил-L-тирозинн); и также могут быть включены их сочетания. Согласно одному конкретному варианту осуществления, ингибитор ACE/NEP выбирают из: AVE-7688, эналаприлата, фазидотрила, фазидотрилата, омапатрилата, сампатрилата и их сочетаний.
Согласно одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с активатором или стимулятором ангиотензинпревращающего фермента 2 (ACE2).
Согласно одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с вакциной ангиотензина-II, примеры которой включают в качестве неограничивающих примеров ATR12181 и CYT006-AngQb.
Согласно одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с антикоагулянтом, типичные примеры которого включают в качестве неограничивающих примеров: кумарины, такие как варфарин; гепарин; и прямые ингибиторы тромбина, такие как аргатробан, бивалирудин, дабигатран и лепирудин.
Согласно еще одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с противодиабетическим средством. Типичные представители противодиабетических средств включают инъекционные лекарственные средства, а также перорально эффективные лекарственные средства и их сочетания. Примеры инъекционных лекарственных средств включают в качестве неограничивающих примеров инсулин и производные инсулина. Примеры перорально эффективных лекарственных средств включают в качестве неограничивающих примеров: бигуаниды, такие как метформин; антагонисты глюкагона; ингибиторы α-глюкозидазы, такие как акарбоза и миглитол; ингибиторы дипептидилпептидазы IV (ингибиторы DPP-IV), такие как алоглиптин, денаглиптин, линаглиптин, саксаглиптин, ситаглиптин и вилдаглиптин; меглитиниды, такие как репаглинид; оксадиазолидиндионы; сульфонилкарбамиды, такие как хлорпропамид, глимепирид, глипизид, глибурид и толазамид; тиазолидиндионы, такие как пиоглитазон и росиглитазон; и их сочетания.
Согласно другому варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с противодиарейными видами лечения. Типичные представители видов лечения включают в качестве неограничивающих примеров растворы для пероральной регидратации (ORS), лоперамид, дифеноксилат и висмута субсалицилат.
Согласно еще одному варианту осуществления, соединение согласно настоящему изобретению вводят в сочетании с противоглаукомным средством. Типичные представители противоглаукомных средств включают в качестве неограничивающих примеров: α-адренергические агонисты, такие как бримонидин; антагонисты β1-адренергического рецептора; β1-блокаторы для местного применения, такие как бетаксолол, левобунолол и тимолол; ингибиторы карбонангидразы, такие как ацетазоламид, бринзоламид или дорзоламид; холинэргические агонисты, такие как цевимелин и DMXB-анабазеин; соединения эпинефрина; миотические средства, такие как пилокарпин; и аналоги простагландина.
Согласно еще одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с противолипидным средством. Типичные представители противолипидных средств включают в качестве неограничивающих примеров: ингибиторы транспортного белка холестериновых эфиров (CETP), такие как анацетрапиб, далцетрапиб и торцетрапиб; статины, такие как аторвастатин, флувастатин, ловастатин, правастатин, розувастатин и симвастатин; и их сочетания.
Согласно одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с антитромботическим средством. Типичные представители антитромботических средств включают в качестве неограничивающих примеров: аспирин; антитромбоцитарные средства, такие как клопидогрел, прасугрел и тиклопидин; гепарин и их сочетания.
Согласно одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с антагонистом рецептора AT1, также известным как блокатор рецептора ангиотензина II 1 типа (ARB). Типичные представители ARB включают в качестве неограничивающих примеров абитезартан, азилзартан (например, азилзартана медоксомил), бензиллозартан, кандесартан, цилексетил кандесартана, элизартан, эмбузартан, энолтазозартан, эпросартан, EXP3174, фонсартан, форазартан, глициллозартан, ирбесартан, изотеолин, лозартан, медоксомил, милфазартан, олмесартан, (например, олмесартана медоксомил), опомизартан, пратосартан, рипизартан, саприсартан, саралазин, сармезин, TAK-591, тасосартан, телмисартан, валсартан, золазартан и их сочетания. Согласно конкретному варианту осуществления, ARB выбирают из азилзартана медоксомила, цилексетила кандесартана, эпросартана, ирбесартана, лозартана, олмесартана медоксомила, саприсартана, тасосартана, телмисартана, валсартана и их сочетаний. Типичные соли и/или пролекарства включают цилексетил кандесартана, эпросартана мезилат, лозартана калиевая соль и олмесартана медоксомил. Обычно, ARB вводят в количестве, достаточном для обеспечения приблизительно 4-600 мг на дозу, с типичными суточными дозами в диапазоне 20-320 мг в сутки.
Соединения согласно настоящему изобретению также может быть введено в сочетании со агентом двойного действия, таким как антагонист рецептора AT1/ингибитор неприлизина, ингибитор (ARB/NEP), примеры которого включают в качестве неограничивающих примеров соединения, описанные в публикациях США №№2008/0269305 и 2009/0023228, которые обе поданы 23 апреля 2008 года Allegretti et al., такие как 4'-{2-этокси-4-этил-5-[((S)-2-меркапто-4-метилпентаноиламино)метил]имидазол-1-илметил}-3'-фторбифенил-2-карбоновая кислота.
Соединения согласно настоящему изобретению также может быть введено в сочетании с многофункциональными блокаторами рецептора ангиотензина, описанными в Kurtz & Klein (2009) Hypertension Research 32:826-834.
Согласно одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с антагонистом рецептора брадикинина, например, икатибантом (HOE-140). Предполагается, что такое комбинированное лечение может обеспечить преимущество при профилактике ангионевротического отека или других нежелательных последствий повышенных уровней брадикинина.
Согласно одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с блокатором кальциевых каналов. Типичные представители блокаторов кальциевых каналов включают в качестве неограничивающих примеров амлодипин, анипамил, аранипин, барнидипин, бенциклан, бенидипин, бепридил, клентиазем, килнидипин, циннаризин, дилтиазем, эфонидипин, элгодипин, этафенон, фелодипин, фендилин, флунаризин, галлопамил, израдипин, лацидипин, лерканидипин, лидофлазин, ломеризин, манидипин, мибефрадил, никардипин, нифедипин, нигулдипин, нилудипин, нилвадипин, нимодипин, низолдипин, нитрендипин, нивалдипин, пергексилин, прениламин, риозидин, семотиадил, теродилин, тиапамил, верапамил и их сочетания. Согласно конкретному варианту осуществления, блокатор кальциевых каналов выбирают из амлодипина, бепридила, дилтиазема, фелодипина, израдипина, лацидипина, никардипина, нифедипина, нигулдипина, нилудипина, нимодипина, низолдипина, риозидина, верапамила и их сочетаний. Обычно, блокатор кальциевых каналов вводят в количестве, достаточном для обеспечения приблизительно 2-500 мг на дозу.
Согласно одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с ингибитором химазы, таким как TPC-806 и 2-(5-формиламино-6-оксо-2-фенил-1,6-дигидропиримидин-1-ил)-N-[{3,4-диоксо-1-фенил-7-(2-пиридилокси)}-2-гептил]ацетамид (NK3201).
Согласно одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с диуретиком. Типичные представители диуретиков включают в качестве неограничивающих примеров: ингибиторы карбоангидразы, такие как ацетазоламид и дихлорфенамид; петлевые диуретики, которые включают производные сульфонамида, такие как ацетазоламид, амбузид, азосемид, буметанид, бутазоламид, хлораминофенамид, клофенамид, клопамид, клорексолон, дисульфамид, этоксзоламид, фуросемид, мефрусид, метазоламид, пиретанид, торсемид, трипамид и ксипамид, а также несульфонамидные диуретики, такие как этакриновая кислота и другие соединения феноксиуксусной кислоты, такие как тиениловая кислота, индакринон и хинкарбат; осмотические диуретики, такие как маннит; калийсберегающие диуретики, которые включают антагонисты альдостерона, такие как спиронолактон, и ингибиторы Na+ каналов, такие как амилорид и триамтерен; тиазид и тиазидоподобные диуретики, такие как алтиазид, бендрофлуметиазид, бензилгидрохлортиазид, бензтиазид, бутиазид, хлорталидон, хлортиазид, циклопентиазид, циклотиазид, эпитиазид, етиазид, фенхизон, флуметиазид, гидрохлортиазид, гидрофлуметиазид, индапамид, метилклотиазид, метикран, метолазон, парафлутизид, политиазид, хинетазон, теклотиазид и трихлорметиазид; и их сочетания. Согласно конкретному варианту осуществления, диуретик выбирают из амилорида, буметанида, хлортиазида, хлорталидона, дихлорфенамида, этакриновой кислоты, фуросемида, гидрохлортиазида, гидрофлуметиазида, индапамида, метилклотиазида, метолазона, торсемида, триамтерена и их сочетаний. Диуретик вводят в количестве, достаточном для обеспечения приблизительно 5-50 мг в сутки, более часто 6-25 мг в сутки, причем суммарные дозы составляют 6,25 мг, 12,5 мг или 25 мг в сутки.
Соединения согласно настоящему изобретению также может быть введено в сочетании с ингибитором эндотелинпревращающего фермента (ECE), примеры которого включают в качестве неограничивающих примеров фосфорамидон, CGS 26303 и их сочетания.
Согласно конкретному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с антагонистом рецепторов эндотелина. Типичные представители антагонистов рецепторов эндотелина включают в качестве неограничивающих примеров: селективные антагонисты рецепторов эндотелина, которые воздействуют на рецепторы эндотелина A, такие как авозентан, амбризентан, атразентан, BQ-123, клазозентан, дарузентан, ситаксентан и зиботентан; и двойные антагонисты рецептора эндотелина, которые воздействуют на рецепторы и эндотелина A, и B, такие как бозентан, мацитентан, тезозентан.
Согласно еще одному варианту осуществления, соединение согласно настоящему изобретению вводят в сочетании с одним или несколькими ингибиторами HMG-CoA-редуктазы, которые также известны как статины. Типичные представители статинов включают в качестве неограничивающих примеров аторвастатин, флувастатин, ловастатин, питавастатин, правастатин, розувастатин и симвастатин.
Согласно одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с ингибитором обратного захвата моноаминов, примеры которого включают в качестве иллюстрации и без ограничения ингибиторы обратного захвата норэпинефрина, такие как атомоксетин, бупроприон и метаболит бупроприона гидроксибупроприон, мапротилин, ребоксетин и вилоксазин; селективные ингибиторы обратного захвата серотонина (SSRI), такие как циталопрам и метаболит циталопрама дезметилциталопрам, дапоксетин, эсциталопрам (например, эсциталопрама оксалат), флуоксетин и дезметилированный метаболит флуоксетина норфлуоксетин, флувоксамин (например, флувоксамина малеат), пароксетин, сертралин и метаболит сертралина деметилсертралин; ингибиторы обратного захвата серотонина-норэпинефрина (SNRI) двойного действия, такие как бицифадин, дулоксетин, милнаципран, нефазодон и венлафаксин; и их сочетания.
Согласно другому варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с миорелаксантом, примеры которого включают в качестве неограничивающих примеров: карисопродол, хлорзоксазон, циклобензаприн, дифлунисал, метаксалон, метокарбамол и их сочетания.
Согласно одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с натрийуретическим пептидом или аналогом, примеры которого включают в качестве неограничивающих примеров: карперитид, CD-NP (Nile Therapeutics), CU-NP, несиритид, PL-3994 (Palatin Technologies, Inc.), уларитид, цендеритид и соединения, описанные в Ogawa et al (2004) J. Biol. Chem. 279:28625-31. Данные соединения также называют агонистами рецептора-A натрийуретических пептидов (NPR-A). Согласно другому варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с антагонистом рецептора клиренса натрийуретического пептида (NPR-C), таким как SC-46542, cANF (4-23) и AP-811 (Veale (2000) Bioorg Med Chem Lett 10:1949-52). Например, AP-811 демонстрировал синергизм, когда его комбинировали с ингибитором NEP, тиорфаном (Wegner (1995) Clin. Exper. Hypert. 17:861-876).
Согласно другому варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с ингибитором неприлизина (NEP). Типичные представители ингибиторов NEP включают в качестве неограничивающих примеров: AHU-377; кандоксатрил; кандоксатрилат; дексекадотрил (сложный бензиловый эфир (+)-N-[2(R)-(ацетилтиометил)-3-фенилпропионил]глицина); CGS-24128 (3-[3-(бифенил-4-ил)-2-(фосфонометиламино)пропионамидо]пропионовая кислота); CGS-24592 ((S)-3-[3-(бифенил-4-ил)-2-(фосфонометиламино)пропионамидо]пропионовая кислота); CGS-25155 (сложный бензиловый эфир N-[9(R)-(ацетилтиометил)-10-оксо-1-азациклодекан-2(S)-илкарбонил]-4(R)-гидрокси-L-пролина); производные 3-(1-карбамоилциклогексил)пропионовой кислоты, описанные в WO 2006/027680 от Hepworth et al. (Pfizer Inc.); JMV-390-1 (2(R)-бензил-3-(N-гидроксикарбамоил)пропионил-L-изолейцил-L-лейцин); экадотрил; фосфорамидон; ретротиорфан; RU-42827 (2-(меркаптометил)-N-(4-пиридинил)бензолпропионамид); RU-44004 (N-(4-морфолинил)-3-фенил-2-(сульфанилметил)пропионамид); SCH-32615 ((S)-N-[N-(1-карбокси-2-фенилэтил)-L-фенилаланил]-β-аланин) и его пролекарство SCH-34826 ((S)-N-[N-[1-[[(2,2-диметил-1,3-диоксолан-4-ил)метокси]карбонил]-2-фенилэтил]-L-фенилаланил]-β-аланин); сиалорфин; SCH-42495 (сложный этиловый эфир N-[2(S)-(ацетилсульфанилметил)-3-(2-метилфенил)пропионил]-L-метионина); спинорфин; SQ-28132 (N-[2-(меркаптометил)-1-оксо-3-фенилпропил]лейцин); SQ-28603 (N-[2-(меркаптометил)-1-оксо-3-фенилпропил]-β-аланин); SQ-29072 (7-[[2-(меркаптометил)-1-оксо-3-фенилпропил]амино]гептановая кислота); тиорфан и его пролекарство рацекадотрил; UK-69578 (цис-4-[[[1-[2-карбокси-3-(2-метоксиэтокси)пропил]циклопентил]карбонил]амино]-циклогексанкарбоновая кислота); UK-447,841 (2-{1-[3-(4-хлорфенил)пропилкарбамоил]циклопентилметил}-4-метоксимасляная кислота); UK-505,749 ((R)-2-метил-3-{1-[3-(2-метилбензотиазол-6-ил)пропилкарбамоил]циклопентил}пропионовая кислота); сложный этиловый эфир 5-бифенил-4-ил-4-(3-карбоксипропиониламино)-2-метилпентановой кислоты и 5-бифенил-4-ил-4-(3-карбоксипропиониламино)-2-метилпентановой кислоты (WO 2007/056546); даглутрил [(3S,2'R)-3-{1-[2'-(этоксикарбонил)-4'-фенилбутил]циклопентан-1-карбониламино}-2,3,4,5-тетрагидро-2-оксо-1H-1-бензазепин-1-уксусная кислота], описанная в WO 2007/106708 от Khder et al. (Novartis AG); и их сочетания. Согласно конкретному варианту осуществления, ингибитор NEP выбирают из AHU-377, кандоксатрила, кандоксатрилата, CGS-24128, фосфорамидона, SCH-32615, SCH-34826, SQ-28603, тиорфана и их сочетаний. Согласно конкретному варианту осуществления, ингибитор NEP представляет собой такое соединение, как даглутрил или CGS-26303 ([N-[2-(бифенил-4-ил)-1(S)-(1H-тетразол-5-ил)этил]амино]метилфосфоновая кислота), которое обладает активностью как в качестве ингибитора эндотелинпревращающего фермента (ECE), так и в качестве ингибитора NEP. Также можно применять другие соединения двойного действия на ECE/NEP. Ингибитор NEP вводят в количестве, достаточном для обеспечения приблизительно 20-800 мг в сутки, с обычными суточными дозами в диапазоне 50-700 мг в сутки, более часто 100-600 или 100-300 мг в сутки.
Согласно одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с донором оксида азота, примеры которого включают в качестве неограничивающих примеров никорандил; органические нитраты, такие как тетранитрат пентаэритрита; и сиднонимины, такие как линсидомин и молсидомин.
Согласно еще одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с нестероидным противовоспалительным средством (NSAID). Типичные представители NSAID включают в качестве неограничивающих примеров: ацеметацин, ацетилсалициловую кислоту, алклофенак, алминопрофен, амфенак, амиприлозу, алоксиприн, аниролак, апазон, азапропазон, бенорилат, беноксапрофен, бензпиперилон, броперамол, буклоксовую кислоту, карпрофен, клиданак, диклофенак, дифлунизал, дифталон, эноликам, этодолак, эторикоксиб, фенбуфен, фенклофенак, фенклозовую кислоту, фенопрофен, фентиазак, фепразон, флуфенамовую кислоту, флуфенисал, флупрофен, флурбипрофен, фурофенак, ибуфенак, ибупрофен, индометацин, индопрофен, изоксепак, изоксикам, кетопрофен, кеторолак, лофемизол, лорноксикам, меклофенамат, меклофенамовую кислоту, мефенамовую кислоту, мелоксикам, месаламин, миропрофен, мофебутазон, набуметон, напроксен, нифлумовую кислоту, оксапрозин, окспинак, оксифенбутазон, фенилбутазон, пироксикам, пирпрофен, пранопрофен, салсалат, судоксикам, сульфасалазин, сулиндак, супрофен, теноксикам, тиопинак, тиапрофеновую кислоту, тиоксапрофен, толфенамовую кислоту, толметин, трифлумидат, зидометацин, зомепирак и их сочетания. Согласно конкретному варианту осуществления, NSAID выбирают из этодолака, флубипрофена, ибупрофена, индометацина, кетопрофена, кеторолака, мелоксикама, напроксена, оксапрозина, пироксикама и их сочетаний.
Согласно одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с антагонистом рецептора N-метил-d-аспартата (NMDA), примеры которого включают в качестве иллюстрации и без ограничения амантадин, декстрометорфан, декстропропоксифен, кетамин, кетобемидон, мемантин, метадон и т.д.
Согласно еще одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с агонистом опиоидного рецептора (также называемыми опиоидными аналгетиками). Типичные представители агонистов опиоидного рецептора включают в качестве неограничивающих примеров: бупренорфин, буторфанол, кодеин, дигидрокодеин, фентанил, гидрокодон, гидроморфон, леваллорфан, леворфанол, меперидин, метадон, морфин, налбуфин, налмефен, налорфин, налоксон, налтрексон, налорфин, оксикодон, оксиморфон, пентазоцин, пропоксифен, трамадол и их сочетания. Согласно определенным вариантам осуществления, агонист опиоидного рецептора выбирают из кодеина, дигидрокодеина, гидрокодона, гидроморфона, морфина, оксикодона, оксиморфона, трамадола и их сочетаний.
Согласно конкретному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с ингибитором фосфодиэстеразы (PDE), в частности, ингибитором PDE-V. Типичные представители ингибиторов PDE-V включают в качестве неограничивающих примеров аванафил, лоденафил, мироденафил, силденафил (ревацио®), тадалафил (Adcirca®), варденафил (левитра®) и уденафил.
Согласно другому варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с аналогом простагландина (также называемыми простаноидами или аналогами простациклина). Типичные представители аналогов простагландина включают в качестве неограничивающих примеров берапрост натрия, биматопрост, эпопростенол, илопрост, латанопрост, тафлупрост, травопрост и трепростинил, причем особый интерес представляют биматопрост, латанопрост и тафлупрост.
Согласно еще одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с агонистом рецептора простагландина, примеры которого включают в качестве неограничивающих примеров биматопрост, латанопрост, травопрост и т.д.
Соединения согласно настоящему изобретению также может быть введено в сочетании с ингибитором ренина, примеры которого включают в качестве неограничивающих примеров алискирен, эналкирен, ремикирен и их сочетания.
Согласно другому варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с селективным ингибитором обратного захвата серотонина (SSRI). Типичные представители SSRI включают в качестве неограничивающих примеров: циталопрам и метаболит циталопрама дезметилциталопрам, дапоксетин, эсциталопрам (например, эсциталопрама оксалат), флуоксетин и дезметилированный метаболит флуоксетина норфлуоксетин, флувоксамин (например, флувоксамина малеат), пароксетин, сертралин и метаболит сертралина деметилсертралин и их сочетания.
Согласно одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с агонистом рецептора серотонина 5-HT1D, примеры которого включают в качестве иллюстрации и без ограничения триптаны, такие как алмотриптан, авириптан, элетриптан, фроватриптан, наратриптан ризатриптан, суматриптан и золмитриптан.
Согласно одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с блокатором натриевых каналов, примеры которого включают в качестве иллюстрации и без ограничения карбамазепин, фосфенитоин, ламотриджин, лидокаин, мексилетин, окскарбазепин, фенитоин и их сочетания.
Согласно одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании со стимулятором или активатором растворимой гуанилатциклазы, примеры которого включают в качестве неограничивающих примеров атацигуат, риоцигуат и их сочетания.
Согласно одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с трициклическим антидепрессантом (TCA), примеры которого включают в качестве иллюстрации и без ограничения амитриптилин, амитриптилиноксид, бутриптилин, кломипрамин, демексиптилин, дезипрамин, дибензепин, диметакрин, досулепин, доксепин, имипрамин, имипраминоксид, лофепрамин, мелитрацен, метапрамин, нитроксазепин, нортриптилин, ноксиптилин, пипофезин, пропизепин, протриптилин, хинупрамин и их сочетания.
Согласно одному варианту осуществления, соединения согласно настоящему изобретению вводят в сочетании с антагонистом рецептора вазопрессина, примеры которого включают в качестве иллюстрации и без ограничения кониваптан и толваптан.
Комбинированные вторичные терапевтические агенты могут быть также полезны при дополнительном комбинированном лечении соединениями согласно настоящему изобретению. Например, соединения согласно настоящему изобретению можно комбинировать с диуретиком и ARB, или с блокатором кальциевых каналов и ARB, или с диуретиком и ингибитором ACE, или блокатором кальциевых каналов и статином. Конкретные примеры включают комбинацию ингибитора ACE эналаприла (в виде соли малеиновой кислоты) и диуретика гидрохлоротиазида, который реализуется под товарным знаком Вазеретик®, или комбинацию блокатора кальциевых каналов амлодипина (в виде соли безилата) и ARB олмесартана (в виде пролекарства медоксомила) или комбинацию блокатора кальциевых каналов и статина, которые все могут быть также применены с соединениями согласно настоящему изобретению. Другие терапевтические средства, такие как агонисты α2-адренергического рецептора и антагонисты рецептора вазопрессина могут также быть полезны при комбинированном лечении. Типичные агонисты α2-адренергического рецептора включают клонидин, дексмедетомидин и гуанфацин.
Следующие композиции иллюстрируют типичные фармацевтические композиции согласно настоящему изобретению.
Типичные твердые желатиновые капсулы для перорального введения
Соединение согласно настоящему изобретению (50 г), 440 г высушенной распылением лактозы и 10 г стеарата магния тщательно перемешивают. Затем, конечную композицию помещают внутрь твердых желатиновых капсул (500 мг композиции на капсулу). В качестве альтернативы, соединение согласно настоящему изобретению (20 мг) тщательно перемешивают с крахмалом (89 мг), микрокристаллической целлюлозой (89 мг) и стеаратом магния (2 мг). Затем, смесь пропускают через сито с ячейками №45 США и помещают внутрь твердой желатиновой капсулы (200 мг композиции на капсулу).
В качестве альтернативы, соединение согласно настоящему изобретению (30 мг), вторичное средство (20 г), 440 г высушенной распылением лактозы и 10 г стеарата магния тщательно перемешивают и обрабатывают, как описано выше.
Типичные композиция в желатиновых капсулах для перорального введения
Соединение согласно настоящему изобретению (100 мг) тщательно перемешивают с полиоксиэтиленсорбитанмоноолеатом (50 мг) и порошковым крахмалом (250 мг). Затем, смесь помещают внутрь желатиновой капсулы (400 мг композиции на капсулу). В качестве альтернативы, соединение согласно настоящему изобретению (70 мг) и вторичное средство (30 г) тщательно перемешивают с полиоксиэтиленсорбитанмоноолеатом (50 мг) и порошковым крахмалом (250 мг), и помещают конечную смесь внутрь желатиновой капсулы (400 мг композиции на капсулу).
В качестве альтернативы, соединение согласно настоящему изобретению (40 мг) тщательно перемешивают с микрокристаллической целлюлозой (Avicel PH 103; 259,2 мг) и стеаратом магния (0,8 мг). Затем, смесь помещают внутрь желатиновой капсулы (размер #1, белая, матовая) (300 мг композиции на капсулу).
Типичная таблетированная композиция для перорального введения
Соединение согласно настоящему изобретению (10 мг), крахмал (45 мг) и микрокристаллическую целлюлозу (35 мг) пропускают через сито с ячейками №20 США и тщательно перемешивают. Полученные таким образом гранулы высушивают при температуре 50-60°C и пропускают через сито с ячейками №16 США. Раствор поливинилпирролидона (4 мг в виде 10% раствора в стерильной воде) смешивают с карбоксиметилкрахмалом натрия (4,5 мг), стеаратом магния (0,5 мг) и тальком (1 мг), и затем пропускают эту смесь через сито с ячейками №16 США. Затем, к гранулам добавляют карбоксиметилкрахмал натрия, стеарат магния и тальк. После перемешивания смесь спрессовывают в таблеточной машине с получением таблетки массой 100 мг.
В качестве альтернативы, соединение согласно настоящему изобретению (250 мг) тщательно перемешивают с микрокристаллической целлюлозой (400 мг), высокодисперсным диоксидом кремния (10 мг) и стеариновой кислотой (5 мг). Затем, смесь спрессовывают с получением таблеток (665 мг композиции на таблетку).
В качестве альтернативы, соединение согласно настоящему изобретению (400 мг) тщательно перемешивают с кукурузным крахмалом (50 мг), кросскармелозой натрия (25 мг), лактозой (120 мг) и стеаратом магния (5 мг). Затем, смесь спрессовывают с получением таблеток с одной насечкой (600 мг композиции на таблетку).
В качестве альтернативы, соединение согласно настоящему изобретению (100 мг) тщательно перемешивают с кукурузным крахмалом (100 мг) и с водным раствором желатина (20 мг). Смесь высушивают и измельчают до мелкодисперсного порошка. Затем, микрокристаллическую целлюлозу (50 мг) и стеарат магния (5 мг) смешивают с желатиновым составом, гранулируют, и спрессовывают конечную смесь с получением таблеток (100 мг соединения согласно настоящему изобретению на таблетку).
Типичная суспензионая композиция для перорального введения
Следующие ингредиенты смешивают с образованием суспензии, содержащей 100 мг соединения согласно настоящему изобретению на 10 мл суспензии:
Типичная жидкая композиция для перорального введения
Подходящая жидкая композиция представляет собой композицию с буфером на основе карбоновой кислоты, таким как цитратные, лактатные и малеатные буферные растворы. Например, соединение согласно настоящему изобретению (которое может быть предварительно смешано с DMSO) перемешивают с буфером из 100 мМ цитрата аммония, и корректируют значения рН до 5, или перемешивают со 100 мМ раствора лимонной кислоты, и корректируют значения рН до 2. Такие растворы также могут включать солюбилизирующий наполнитель, такой как циклодекстрин, например, раствор может включать 10 масс.% гидроксипропил-β-циклодекстрин.
Другие подходящие композиции включают 5% раствор NaHCO3 в присутствии или в отсутствии циклодекстрина.
Типичные инъекционные композиции для введения посредством инъекции
Соединение согласно настоящему изобретению (0,2 г) перемешивают с раствором буфера из 0,4 М ацетата натрия (2,0 мл). При необходимости, значение рН конечного раствора корректируют до рН 4 с использованием 0,5н водной соляной кислоты или 0,5н водного гидроксида натрия, а затем добавляют воду для инъекций в количестве, достаточном для получения общего объема 20 мл. Затем, смесь фильтруют через стерильный фильтр (0,22 микрон) с получением стерильного раствора, подходящего для введения посредством инъекции.
Типичные композиции для введения посредством ингаляции
Соединение согласно настоящему изобретению (0,2 мг) микронизируют, а затем перемешивают с лактозой (25 мг). Затем, перемешанную смесь помещают внутрь желатинового ингаляционного картриджа. Содержимое картриджа вводят, например, с применением ингалятора сухого порошка.
В качестве альтернативы, микронизированное соединение согласно настоящему изобретению (10 г) диспергируют в растворе, приготовленном путем растворения лецитина (0,2 г) в деминерализованной воде (200 мл). Конечную суспензию высушивают распылением, а затем микронизируют до образования микронизированной композиции, содержащей частицы со средним диаметром менее чем приблизительно 1,5 мкм. Микронизированную композицию затем помещают в картриджи дозирующего ингалятора, содержащие сжатый 1,1,1,2-тетрафторэтан в количестве, достаточном для обеспечения дозы приблизительно от 10 мкг приблизительно до 500 мкг соединения согласно настоящему изобретению при введении посредством ингалятора.
В качестве альтернативы, соединение согласно настоящему изобретению (25 мг) растворяют в цитратном буферном (рН 5) изотоническом солевом растворе (125 мл). Такую смесь взбалтывают и обрабатывают ультразвуком до тех пор, пока соединение не растворится. Значение рН раствора проверяют и, при необходимости, корректируют до рН 5 путем медленного добавления водного 1н NaOH. Раствор вводят с применением небулайзера, который обеспечивает дозу приблизительно от 10 мкг приблизительно до 500 мкг соединения согласно настоящему изобретению.
ПРИМЕРЫ
Последующие подготовительные примеры и примеры представлены для иллюстрации конкретных вариантов осуществления настоящего изобретения. Однако если не указано конкретно, то указанные конкретные варианты осуществления никоим образом не предназначены для ограничения объема настоящего изобретения.
Если не указано иное, то следующие сокращения имеют следующие значения, а любые другие сокращения, использованные в настоящем документе, но не указанные ниже, имеют их стандартные общепринятые значения:
AcOH - уксусная кислота
BOC - трет-бутоксикарбонил (-C(O)OC(CH3)3)
(BOC)2O - ди-трет-бутилдикарбонат
DCC - дициклогексилкарбодиимид
DCM - дихлорметан или хлористый метилен
DIPEA - N,N-диизопропилэтиламин
DMA - N,N-диметилацетамид
DMAP - 4-диметиламинопиридин
DMF - N,N-диметилформамид
EDC - 1-(3-диметиламинопропил)-3-этилкарбодиимид
Et3N - триэтиламин
Et2O - диэтиловый эфир
Et3SiH - триэтилсилан
EtOAc - этилацетат
EtOH - этанол
HATU - N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония гексафторфосфат
HCTU - 2-(6-хлор-1H-бензотриазол-1-ил)-1,1,3,3-тетраметиламиния гексафторфосфат
HEPES - 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота
HOBt - 1-гидроксибензотриазол
MeCN - ацетонитрил
MeOH - метанол
NaHMDS - натрия гексаметилдисилазид
Pd(dppf)2Cl2 - 1,1-бис(дифенилфосфино)ферроценпалладия хлорид
Pd(PPh3)4 - тетракис(трифенилфосфин)палладий(0)
PE - петролейный эфир
PMB - пара-метоксибензил
PyBOP - бензотриазол-1-илокситрис(пирролидино)фосфония гексафторфосфат
SilicaCat®DPP-Pd - катализатор на основе диоксида кремния с дифенилфосфинпалладием(II)
TFA - трифторуксусная кислота
THF - тетрагидрофуран
TMSCl - триметилсилилхлорид
Tr - тритил
Если не указано иное, то все вещества, такие как реагенты, исходные вещества и растворители, приобретали у коммерческих поставщиков (таких как Sigma-Aldrich, Fluka Riedel-de Haën, и т.п.) и использовали без дополнительной очистки.
Если не указано иное, то взаимодействия проводили в атмосфере азота. Ход реакций отслеживали методами тонкослойной хроматографии (TLC), аналитической высокоэффективной жидкостной хроматографии (аналит. HPLC) и масс-спектрометрии, детали которых представлены в конкретных примерах. Растворители, использованные при проведении аналитической HPLC, были следующими: растворитель A=98% H2O/2% MeCN/1,0 мл/л TFA; растворитель B=90% MeCN/10% H2O/1,0 мл/л TFA.
Реакционные смеси обрабатывали, как конкретно описано в каждом подготовительном примере; обычно, реакционные смеси очищали путем экстрагирования и других способов очистки, таких как зависимая от температуры и зависимая от растворителя кристаллизация и осаждение. Кроме того, реакционные смеси очищали в установленном порядке методом препаративной HPLC, обычно с использованием колонок Microsorb C18 и Microsorb BDS и традиционно используемых элюентов. Ход реакций обычно отслеживали методом жидкостной хроматографии/масс-спектрометрии (LC-MS). Охарактеризацию изомеров проводили методом спектроскопии ядерного эффекта Оверхаузера (NOE). Охарактеризацию продуктов реакции проводили в установленном порядке методами масс-спектрометрии и1H-ЯМР-спектроскопии. Для измерения ЯМР, образцы растворяли в дейтерированном растворителе (CD3OD, CDCl3 или DMSO-d6), и регистрировали спектры1H-ЯМР с использованием спектрометра Varian Gemini 2000 (400 МГц) в стандартных условиях наблюдения. Масс-спектрометрическую идентификацию соединений обычно проводили с использованием метода ионизации электрораспылением (ESMS) на приборе API 150 EX производства Applied Biosystems (Foster City, CA) или на приборе 1200 LC/MSD производства Agilent (Palo Alto, CA).
Подготовительный пример 1: сложный бутиловый эфир (S)-2-(4-бромбензил)-5-оксопирролидин-1-карбоновой кислоты
К раствору (R)-2-амино-3-(4-бромфенил)пропионовой кислоты (50 г, 0,2 моль) в MeCN (700 мл) при -5°C добавляли раствор NaOH (16,4 г, 0,4 моль) в воде (700 мл). После перемешивания в течение 10 минут, добавляли раствор (BOC)2O (44,7 г, 0,2 моль) в MeCN (100 мл). Смесь нагревали до комнатной температуры и перемешивали в течение ночи. После выпаривания MeCN, остаток разбавляли DCM (800 мл) и подкисляли добавлением 1 M HCl до pH 2 при -5°C. Водный слой экстрагировали DCM (3×200 мл). Объединенные органические слои промывали насыщенным водным NaCl (500 мл), сушили над Na2SO4 и концентрировали с получением соединения 1 (66,5 г) в виде белого твердого вещества. ЖХ-МС: 366 [M+Na]. 709 [2M+Na].
К раствору соединения 1 (66,5 г, 193 мкмоль), кислоты Мельдрума (33,4 г, 232 ммоль) и DMAP (37,7 г, 309 ммоль) в безводном DCM (600 мл) в течение 1 часа при -5°C в атмосфере азота по каплям добавляли раствор DCC (47,9 г, 232 ммоль) в безводном DCM (200 мл). Смесь перемешивали при -5°C в течение 8 часов, а затем охлаждали в течение ночи. Наблюдали кристаллы дициклогексилмочевины. Смесь фильтровали, промывали 5% KHSO4 (5×200 мл) и насыщенным водным NaCl (200 мл), а затем сушили над безводным MgSO4 в условиях охлаждения в течение ночи. Затем, раствор упаривали с получением неочищенного соединения 2 (91 г) в виде светло-желтого твердого вещества. ЖХ-МС: 492 [M+Na]. 961 [2M+Na].

К раствору неочищенного соединения 2 (91 г, 193 ммоль) в безводном DCM (1 л) при -5°C в атмосфере азота добавляли AcOH (127,5 г, 2,1 моль). Смесь перемешивали при -5°C в течение 30 минут, а затем малыми порциями в течение 1 часа добавляли NaBH4 (18,3 г, 483 ммоль). После дополнительного перемешивания в течение 1 часа при -5°C, добавляли насыщенный водный NaCl (500 мл). Органический слой промывали насыщенным водным NaCl (2×300 мл) и водой (2×300 мл), сушили над MgSO4, фильтровали и концентрировали с получением неочищенного продукта, который затем очищали путем промывания Et2O с получением соединения 3 (68 г) в виде светло-желтого твердого вещества. ЖХ-МС: 478 [M+Na]. 933 [2M+Na].
Раствор соединения 3 (68 г, 149 ммоль) в безводном толуоле (500 мл) нагревали с обратным холодильником в атмосфере азота в течение 3 часов. После выпаривания растворителя, остаток очищали методом хроматографии (гексаны/EtOAc=10/1) с получением указанного в заголовке соединения (38 г) в виде светло-желтого масла. ЖХ-МС: 376 [M+Na]. 729 [2M+Na].
Подготовительный пример 2: (3R,5R)-5-(3'-хлорбифенил-4-илметил)-1-(2,2-диметилпропионил)-3-гидроксипирролидин-2-он

К раствору сложного трет-бутилового эфира (S)-2-(4-бромбензил)-5-оксопирролидин-1-карбоновой кислоты (15 г, 43 ммоль) в 1,4-диоксане (600 мл) при комнатной температуре в атмосфере азота добавляли 3-хлорфенилбороновую кислоту (8 г, 51 ммоль) и Pd(dppf)2Cl2 (3,1 г, 4,2 ммоль). После перемешивания в течение 10 минут, добавляли раствор K2CO3 (11,7 г, 85 ммоль) в воде (60 мл). Смесь нагревали до 60°C и перемешивали в течение ночи. После выпаривания растворителя, добавляли воду (200 мл) и экстрагировали EtOAc (3×200 мл). Объединенные органические слои промывали насыщенным водным NaCl (400 мл), сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который затем очищали методом колоночной хроматографии (гексаны/EtOAc=6/1) с получением соединения 1 (15 г) в виде светло-желтого твердого вещества. ЖХ-МС: 408 [M+Na].
К раствору соединения 1 (15 г, 0,039 моль) в безводном DCM (250 мл) при -5°C в атмосфере азота добавляли TFA (20 мл, 270 ммоль). Смесь нагревали до комнатной температуры и перемешивали в течение ночи. После выпаривания растворителя, остаток разбавляли EtOAc (300 мл), а затем промывали насыщенным водным NaHCO3 (3×200 мл), водой (200 мл), насыщенным водным NaCl (250 мл), а затем сушили над Na2SO4 и концентрировали с получением неочищенного соединения 2 (11 г) в виде светло-желтого твердого вещества. ЖХ-МС: 286 [M+H].

К раствору NaH (2,3 г, 98 ммоль) в безводном THF (200 мл) по каплям добавляли раствор соединения 2 (11 г, 39 ммоль) в безводном THF (100 мл) в течение 30 минут при 0°C в атмосфере азота. Смесь нагревали до комнатной температуры и перемешивали в течение 2 часов. После охлаждения до 0°C, в течение 30 минут по каплям добавляли пивалоилхлорид (6 г, 51 ммоль). Смесь нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь гасили добавлением насыщенного водного NH4Cl (200 мл) и экстрагировали EtOAc (3×200 мл). Объединенные органические слои промывали насыщенным водным NaCl (300 мл), сушили над MgSO4, фильтровали и концентрировали с получением неочищенного продукта, который затем очищали методом хроматографии (гексаны/EtOAc=25/1) с получением соединения 3 (10,5 г) в виде светло-желтого твердого вещества. ЖХ-МС: 391 [M+Na].
К раствору соединения 3 (10,5 г, 29 ммоль) в безводном THF (120 мл) в течение 30 минут при -78°C в атмосфере азота по каплям добавляли NaHMDS (29 мл, 58 ммоль). После перемешивания при -78°C в течение 90 минут, по каплям в течение 30 минут добавляли раствор (+)-(8,8-дихлоркамфорилсульфонил)оксазиридина (15,6 г, 52 ммоль). После перемешивания при -78°C в течение 2 часов, реакционную смесь гасили добавлением насыщенного NH4Cl (400 мл) и экстрагировали EtOAc (3×300 мл). Объединенные органические слои промывали насыщенным водным NaCl (300 мл), сушили над MgSO4, фильтровали и концентрировали с получением неочищенного продукта, который затем очищали методом хроматографии (гексаны/EtOAc=15/1) с получением указанного в заголовке соединения (9,6 г) в виде светло-желтого твердого вещества. ЖХ-МС: 408 [M+Na].
Подготовительный пример 3: сложный этиловый эфир (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты
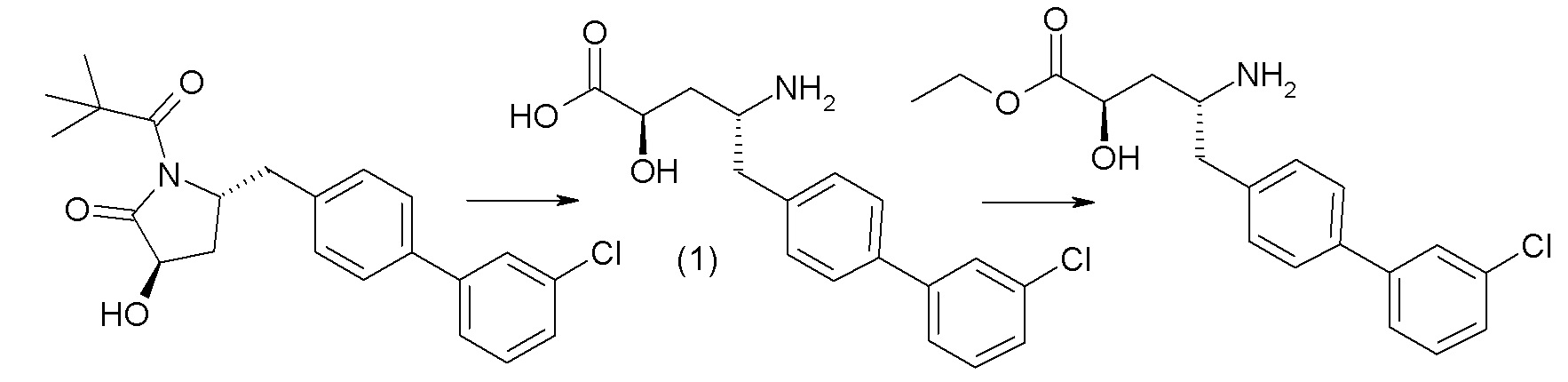
Раствор (3R,5R)-5-(3'-хлорбифенил-4-илметил)-1-(2,2-диметилпропионил)-3-гидроксипирролидин-2-она (9,6 г, 25 ммоль) в концентрированной HCl (81 мл, 81 ммоль) нагревали при 100°C в течение 16 часов. Затем, смесь концентрировали с получением неочищенного продукта, который затем очищали путем промывания Et2O с получением соединения 1 (5,7 г) в виде светло-желтого твердого гидрохлорида. ЖХ-МС: 320 [M+H].
К раствору соединения 1 (5,7 г, 18 ммоль) в EtOH (10 мл) при комнатной температуре добавляли 8 M HCl в EtOH (120 мл, 960 ммоль). Смесь нагревали при 50°C в течение 16 часов. После концентрирования, неочищенный продукт очищали путем промывания Et2O с получением указанного в заголовке соединения (2,1 г) в виде светло-желтого твердого гидрохлорида. ЖХ-МС: 348 [M+H].
Подготовительный пример 4: сложный этиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(3-гидрокси-3H-бензотриазол-5-карбонил)амино]пентановой кислоты

К раствору 1-гидрокси-1H-1,2,3-бензотриазол-6-карбоновой кислоты (154 мг, 862 мкмоль) в DMF (1,5 мл, 19,5 ммоль) добавляли HATU (328 мг, 862 мкмоль) и перемешивали полученную смесь при комнатной температуре в течение 10 минут. К смеси добавляли DIPEA (0,3 мл, 1,7 ммоль), а затем сложный этиловый эфир (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (200 мг, 575 мкмоль), и перемешивали смесь при комнатной температуре в течение 1 часа. Смесь концентрировали в условиях вакуума, и очищали полученный остаток методом хроматографии с обращенной фазой (35-80% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения (197 мг) в виде белого твердого вещества.
Подготовительный пример 5: (2R,4R)-4-трет-бутоксикарбониламино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановая кислота
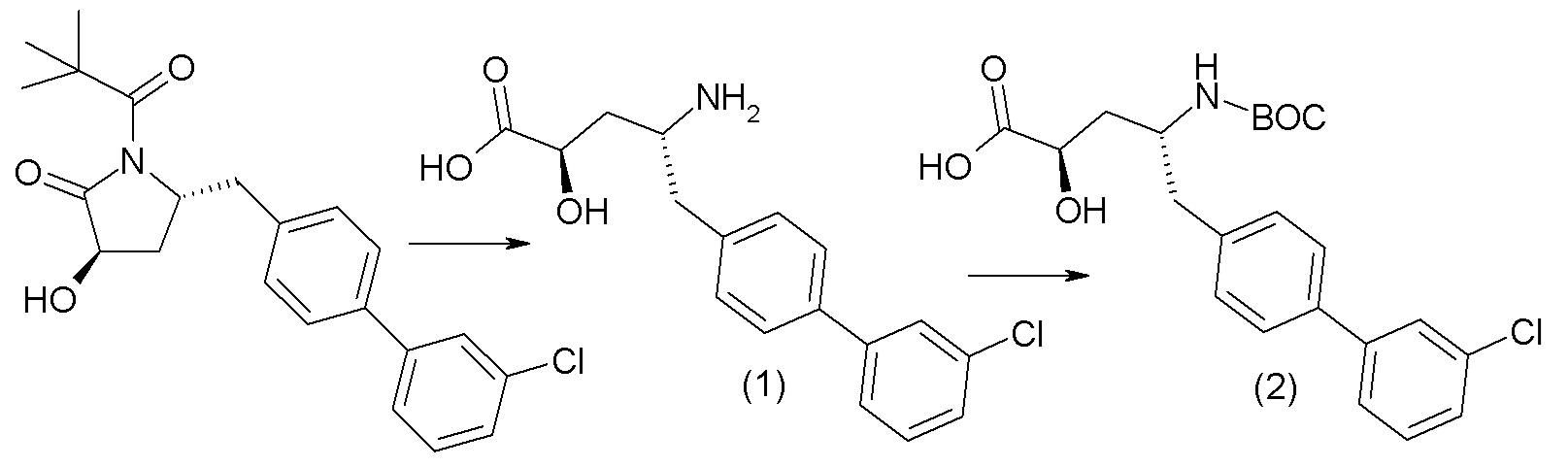
Раствор (3R,5R)-5-(3'-хлорбифенил-4-илметил)-1-(2,2-диметилпропионил)-3-гидроксипирролидин-2-она (4,5 г, 11,7 ммоль) в концентрированной HCl (30 мл) перемешивали при 100°C в течение 16 часов. Смесь концентрировали в условиях вакуума с получением соединения 1 (4 г) в виде белого твердого гидрохлорида. ЖХ-МС: 321 [M+H]+.
К раствору NaOH (1,8 г, 45,2 ммоль) в воде (100 мл) по каплям добавляли соединение 1 (4 г, 11,3 ммоль) в MeCN (100 мл). Смесь перемешивали в течение 10 минут при 0°C. Добавляли ди-трет-бутилдикарбонат (7,17 г, 33,8 ммоль), и перемешивали смесь в течение 15 часов при комнатной температуре. Полученную смесь концентрировали в условиях вакуума для удаления MeCN, затем разбавляли DCM (300 мл), и корректировали значение pH до pH=5-6 добавлением 1н водного HCl. Затем, органический слой собирали, и экстрагировали остаток DCM (3×300 мл). Объединенные органические слои концентрировали и промывали гексанами (150 мл) с получением указанного в заголовке соединения (4 г) в виде белого твердого вещества. ЖХ-МС: 442 [M+Na]+.
Подготовительный пример 6: сложный 2,2,3,3,3-пентафторпропиловый эфир (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты
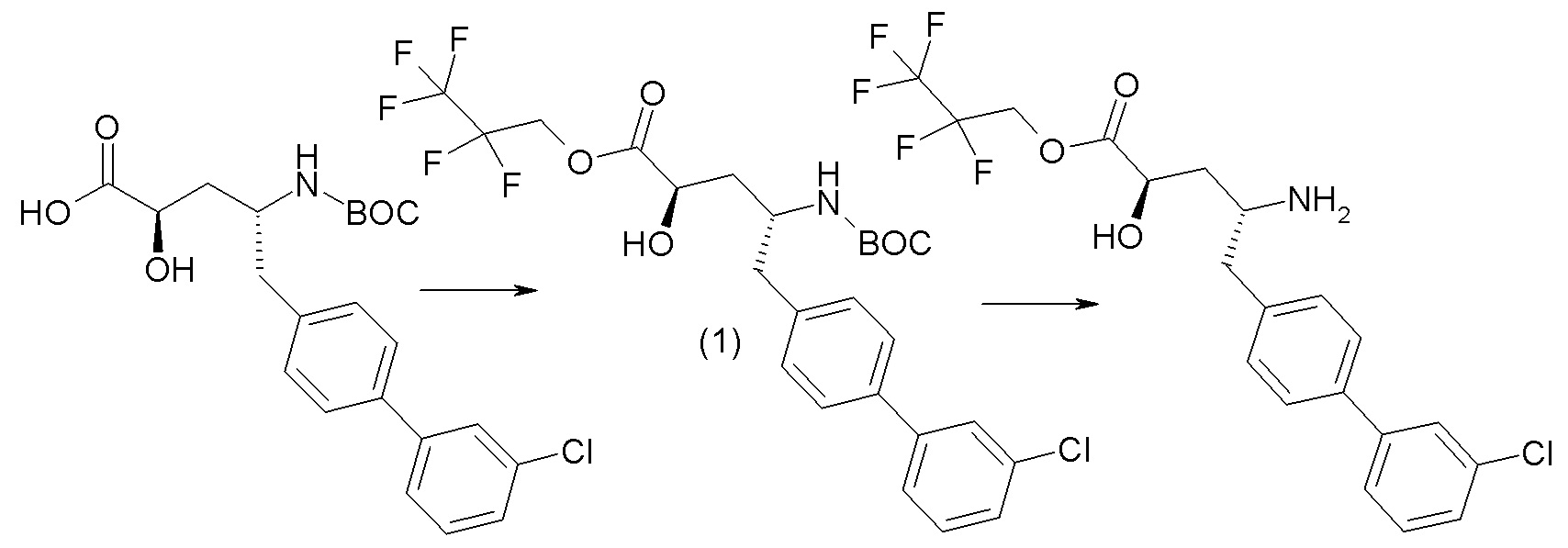
К раствору (2R,4R)-4-трет-бутоксикарбониламино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (0,9 г, 6 ммоль) и 2,2,3,3,3-пентафторпропан-1-ола (450 мг, 3 ммоль) в DCM (30 мл) добавляли DCC (880 мг, 4,3 ммоль) и DMAP (260 мг, 2,1 ммоль). Полученную смесь перемешивали в течение 15 часов при комнатной температуре, а затем концентрировали в условиях вакуума. Остаток растворяли в EtOAc (100 мл) и промывали водой (30 мл) и насыщенным водным NaCl (30 мл). Органический слой собирали и концентрировали и очищали методом колоночной хроматографии (гексаны/EtOAc=5/1) с получением соединения 1 (0,4 г) в виде белого твердого вещества. ЖХ-МС: 574 [M+Na]+.
Раствор соединения 1 (0,4 г, 690 мкмоль) в 1,4 M растворе HCl в 1,4-диоксане (15 мл) перемешивали в течение ночи, а затем концентрировали в условиях вакуума. Остаток диспергировали в EtOAc (10 мл), и собирали осадок путем фильтрования с получением указанного в заголовке соединения в виде не совсем белого твердого гидрохлорида (165 мг). ЖХ-МС: 452 [M+H]+.1H ЯМР: (ДМСО-d6) 1,95-1,82 (м, 2H), 2,99-2,98 (м, 2H), 3,56 (шир., 1H), 4,41-4,38 (м, 1H), 4,92-4,82 (м, 2H), 6,35(с, 1H), 7,71-7,38 (м, 8H), 8,09 (с, 3H).
Подготовительный пример 7: сложный 5-метил-2-оксо[1,3]диоксол-4-илметиловый эфир (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты
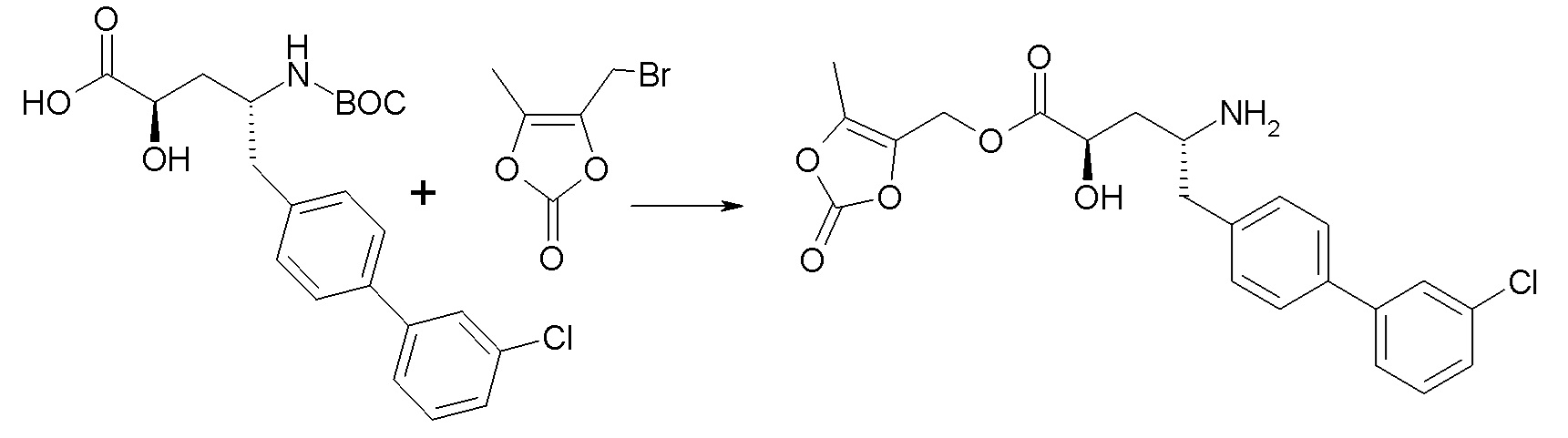
Суспензию (2R,4R)-4-трет-бутоксикарбониламино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (740 мг, 1,8 ммоль), 4-(бромметил)-5-метил-1,3-диоксол-2-она (340 мг, 1,8 ммоль), йодида калия (58 мг, 350 мкмоль) и K2CO3 (486 мг, 3,5 ммоль) в DMF (20 мл) перемешивали в течение 4 часов при комнатной температуре. Смесь разбавляли EtOAc (150 мл) и промывали водой (30 мл). Органический слой собирали, концентрировали и очищали методом колоночной хроматографии (гексаны/EtOAc=1/1) с получением белого твердого вещества (490 мг). ЖХ-МС: 554 [M+23]+.
Раствор этого твердого вещества (476 мг, 890 мкмоль) в 3н HCl в 1,4-диоксане (20 мл) перемешивали в течение ночи, а затем концентрировали в условиях вакуума. Остаток диспергировали в EtOAc (10 мл), и собирали осадок путем фильтрования с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (290 мг). ЖХ-МС: 432 [M+H]+.1H ЯМР: (ДМСО-d6) 1,92-1,82 (м, 2H), 2,16 (с, 3H), 2,99 (шир., 2H), 3,56 (шир., 1H), 4,35-4,32 (м, 1H), 5,017 (с, 2H), 6,17 (с, 1H), 7,39-7,36 (м, 4H), 7,71-7,68 (м, 4H), 8,05 (с, 3H).
Подготовительный пример 8: сложный бутирилоксиметиловый эфир (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты
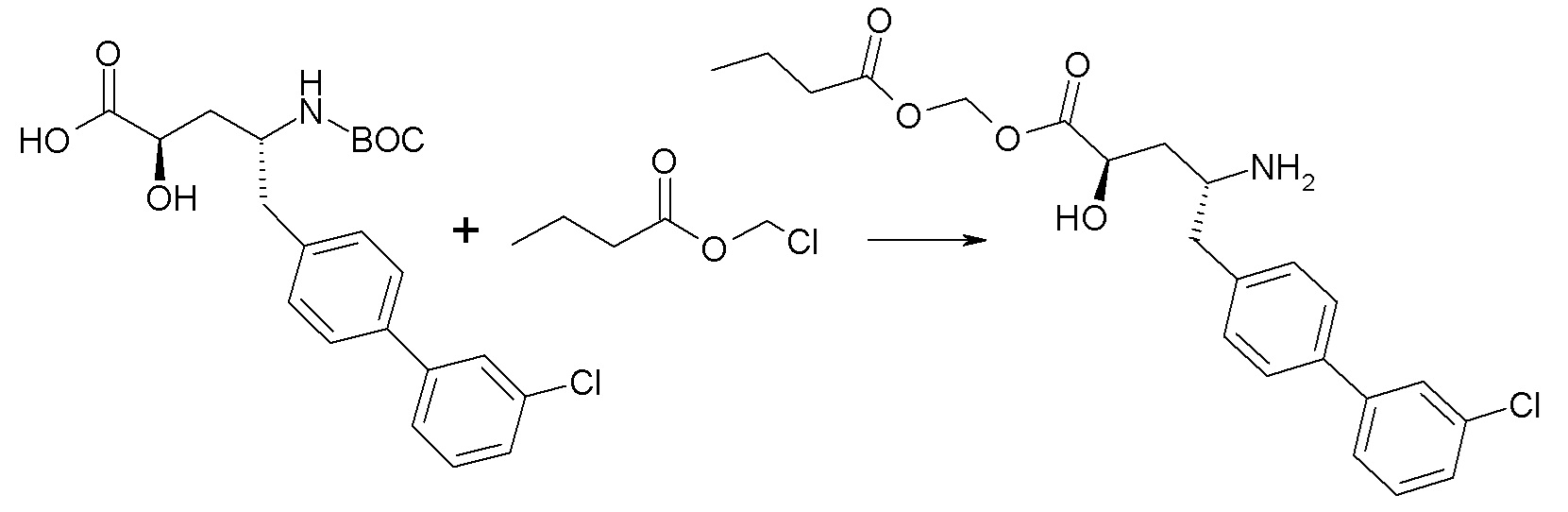
Раствор (2R,4R)-4-трет-бутоксикарбониламино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (900 мг, 2,1 ммоль), хлорметилбутирата (350 мг, 2,6 ммоль), йодида натрия (481 мг, 3,21 ммоль) и DIPEA (828 мг, 6,42 ммоль) в DMF (20 мл) перемешивали в течение 16 часов при 30ºC. Смесь разбавляли EtOAc (150 мл) и промывали водой (50 мл) и насыщенным водным NaCl (50 мл). Органический слой собирали и концентрировали и очищали методом колоночной хроматографии (гексаны/EtOAc=5/1) с получением белого твердого вещества (240 мг). ЖХ-МС: 542 [M+Na]+.
Раствор этого твердого вещества (240 мг, 460 мкмоль) в 1,4 M HCl в 1,4-диоксане (15 мл) перемешивали в течение ночи, а затем концентрировали в условиях вакуума. Остаток диспергировали в EtOAc (10 мл), и собирали выпавший осадок путем фильтрования с получением указанного в заголовке соединения в виде не совсем белого твердого гидрохлорида (140 мг). ЖХ-МС: 420 [M+H]+.1H ЯМР: (ДМСО) 0,85 (т, J=7,5 Гц, 3H), 1,61-1,52 (м, 2H), 1,89-1,86 (м, 2H), 2,30 (т, J=7,5 Гц, 2H), 2,98 (шир., 2H), 3,56 (шир., 1H), 4,33-4,30 (м, 1H), 5,74-5,68 (м, 2H), 6,21 (с, 1H), 7,37-7,35 (м, 4H), 7,70-7,767 (м, 4H), 8,01 (шир. с, 3H).
Подготовительный пример 9: сложный этиловый эфир (2R,4R)-4-амино-5-(4-бромфенил)-2-гидроксипентановой кислоты
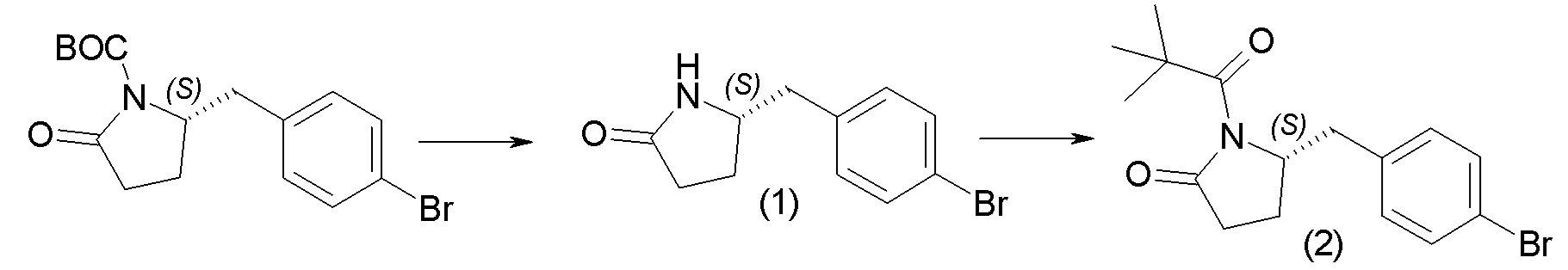
К раствору сложного трет-бутилового эфира (S)-2-(4-бромбензил)-5-оксопирролидин-1-карбоновой кислоты (38 г, 107 ммоль) в безводном DCM (250 мл) при -5°C в атмосфере азота добавляли TFA (20 мл, 0,27 моль). Смесь нагревали до комнатной температуры и перемешивали в течение ночи. После выпаривания растворителя, остаток разбавляли EtOAc (300 мл) и промывали насыщенным водным NaHCO3 (3×200 мл), водой (200 мл), насыщенным водным NaCl (250 мл), сушили над Na2SO4 и концентрировали с получением неочищенного соединения 1 (24 г) в виде светло-желтого твердого вещества. ЖХ-МС: 254 [M+H].
К раствору NaH (8,6 г, 250 ммоль) в безводном THF (200 мл) при 0°C в атмосфере азота по каплям в течение 30 минут добавляли раствор соединения 1 (24 г, 94 ммоль) в безводном THF (200 мл). Смесь нагревали до комнатной температуры и перемешивали в течение 2 часов. После охлаждения до 0°C, по каплям в течение 30 минут добавляли пивалоилхлорид (18 г, 150 ммоль). Смесь нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь гасили добавлением насыщенного водного NH4Cl (300 мл) и экстрагировали EtOAc (3×200 мл). Объединенные органические слои промывали насыщенным водным NaCl (300 мл), сушили над MgSO4, фильтровали и концентрировали с получением неочищенного продукта, который затем очищали методом хроматографии (гексаны/EtOAc=25/1) с получением соединения 2 (18 г) в виде светло-желтого твердого вещества. ЖХ-МС: 360 [M+Na].
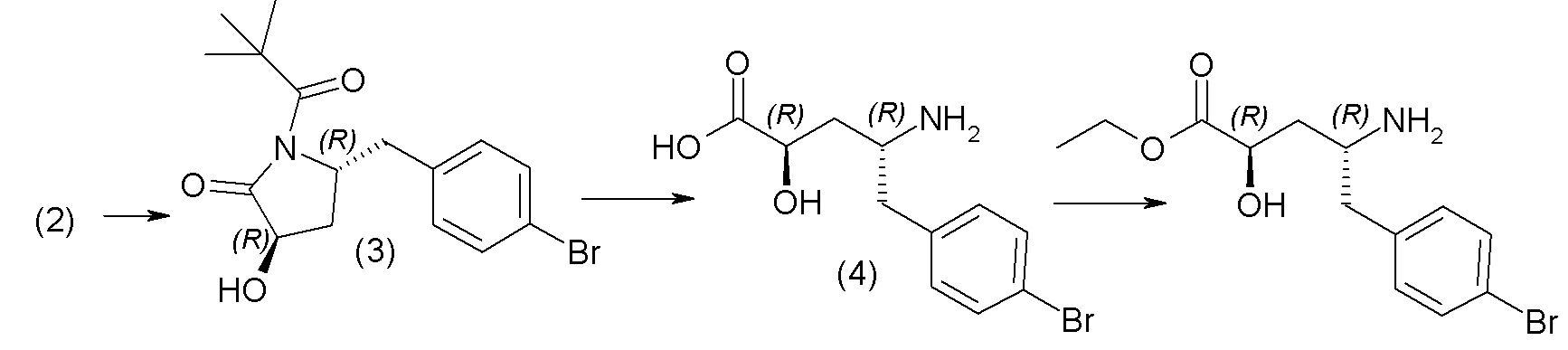
К раствору соединения 2 (18 г, 53 ммоль) в безводном THF (250 мл) при -78°C в атмосфере азота по каплям в течение 30 минут добавляли NaHMDS (47,7 мл, 96 ммоль). После перемешивания при -78°C в течение 90 минут, по каплям в течение 30 минут добавляли раствор (+)-(8,8-дихлоркамфорилсульфонил)оксазиридина (31,6 г, 106 ммоль). После перемешивания при -78°C в течение 2 часов, реакционную смесь гасили добавлением насыщенного водного NH4Cl (400 мл) и экстрагировали EtOAc (3×300 мл). Объединенные органические слои промывали насыщенным водным NaCl (300 мл), сушили над MgSO4, фильтровали и концентрировали с получением неочищенного продукта, который затем очищали методом хроматографии (гексаны/EtOAc=15/1) с получением соединения 3 (8,9 г) в виде светло-желтого твердого вещества. ЖХ-МС: 376 [M+Na].
Раствор соединения 3 (8,9 г, 25 ммоль) в концентрированной HCl (81 мл, 81 ммоль) нагревали при 100°C в течение 16 часов. Затем, смесь концентрировали с получением неочищенного продукта, который затем очищали путем промывания Et2O с получением соединения 4 (7 г) в виде светло-желтого твердого гидрохлорида. ЖХ-МС: 323 [M+H].
Раствор соединения 4 (7 г, 22 ммоль) в EtOH (10 мл) при комнатной температуре объединяли с 8M HCl в EtOH (120 мл, 960 ммоль). Смесь нагревали при 50°C в течение 16 часов, а затем концентрировали. Неочищенный раствор затем очищали путем промывания Et2O с получением указанного в заголовке соединения (6 г) в виде светло-желтого твердого гидрохлорида. ЖХ-МС: 352 [M+H].
Подготовительный пример 10: 5-гидрокси-1-пиридин-2-ил-1H-пиразол-3-карбоновая кислота

К суспензии сложного этилового эфира 5-гидрокси-1-пиридин-2-ил-1H-пиразол-3-карбоновой кислоты (351,6 мг, 1,5 ммоль) в MeOH (5,0 мл, 120 ммоль) при комнатной температуре добавляли LiOH моногидрат (126,5 мг, 3,0 ммоль) с получением прозрачного раствора. Раствор перемешивали при комнатной температуре в течение ночи. Затем, раствор концентрировали. К полученному остатку добавляли 1н водную HCl до достижения pH~2 с образованием осадка. Добавляли воду (4,0 мл), и перемешивали полученную смесь при комнатной температуре в течение 1 часа, а затем фильтровали. Твердые вещества промывали водой и сушили в условиях вакуума с получением указанного в заголовке соединения в виде белого твердого вещества (128 мг).
Подготовительный пример 11: (2R,4R)-4-амино-5-(5'-хлор-2'-фтор-бифенил-4-ил)-2-гидроксипентановая кислота

К раствору сложного трет-бутилового эфира (S)-2-(4-бромбензил)-5-оксопирролидин-1-карбоновой кислоты (25 г, 70,6 ммоль) в 1,4-диоксане (500 мл) при комнатной температуре в атмосфере азота добавляли 5-хлор-2-фторфенилбороновую кислоту (24,6 г, 141 ммоль), Pd(PPh3)4 (4,1 г, 3,5 ммоль) м раствор K2CO3 (17,8 г, 141 ммоль) в воде (90 мл). Смесь нагревали до 60°C и перемешивали в течение ночи. Добавляли воду (500 мл), и выпаривали растворитель. Смесь экстрагировали EtOAc (3×200 мл). Объединенные органические слои промывали насыщенным водным NaCl (300 мл) и фильтровали. Фильтрат концентрировали с получением неочищенного продукта, который очищали методом хроматографии с получением соединения 1 (22,7 г) в виде светло-желтого твердого вещества. ЖХ-МС: 829,2 [2M+Na+].
К раствору соединения 1 (4,9 г, 12,1 моль) в DCM (100 мл) при 0°C в атмосфере азота добавляли TFA (4,5 мл, 60,7 ммоль), и перемешивали в течение 1 часа. Смесь нагревали до комнатной температуры в течение 1,5 часов. После выпаривания растворителя, остаток разбавляли EtOAc (100 мл), а затем промывали насыщенным водным NaHCO3 (3×100 мл), водой (2×100 мл), насыщенным водным NaCl (100 мл), а затем сушили над Na2SO4. Смесь фильтровали, и концентрировали фильтрат с получением неочищенного соединения 2 (объединяли с отдельной партией, всего 16,9 г). ЖХ-МС: 304 [M+H].

К раствору NaH (2,4 г, 695 ммоль) в THF (200 мл) при 0°C в атмосфере азота по каплям добавляли раствор соединения 2 (8,5 г, 278 ммоль) в THF (50 мл). Смесь нагревали до комнатной температуры и перемешивали в течение 2 часов. После охлаждения до 0°C, по каплям в течение 30 минут добавляли пивалоилхлорид (5 г, 41,7 ммоль). Смесь нагревали до комнатной температуры и перемешивали в течение 9,5 часов. Реакционную смесь гасили добавлением насыщенного водного NH4Cl (250 мл) и экстрагировали EtOAc (3×400 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который очищали методом хроматографии с получением соединения 3 (18 г) в виде желтого твердого вещества. ЖХ-МС: 388 [M+H+].
К раствору соединения 3 (9 г, 23,2 ммоль) в THF (200 мл) при -78°C в атмосфере азота по каплям добавляли NaHMDS (20,9 мл, 41,8 ммоль). После перемешивания в течение 1 часа при -78°C, по каплям добавляли раствор (+)-(8,8-дихлоркамфорилсульфонил)-оксазиридина (10,4 г, 34,8 ммоль) в THF (50 мл). После перемешивания при -78°C в течение 1 часа, реакционную смесь гасили добавлением насыщенного водного NH4Cl (50 мл) и экстрагировали EtOAc (3×400 мл). Объединенные органические слои промывали 1 M HCl (400 мл), насыщенным водным NaHCO3 (400 мл) и насыщенным водным NaCl (400 мл), сушили над Na2SO4, и концентрировали с получением неочищенного продукта, который очищали методом хроматографии с получением соединения 4 (8,8 г) в виде белого полутвердого вещества. ЖХ-МС: 426,1 [M+Na+].
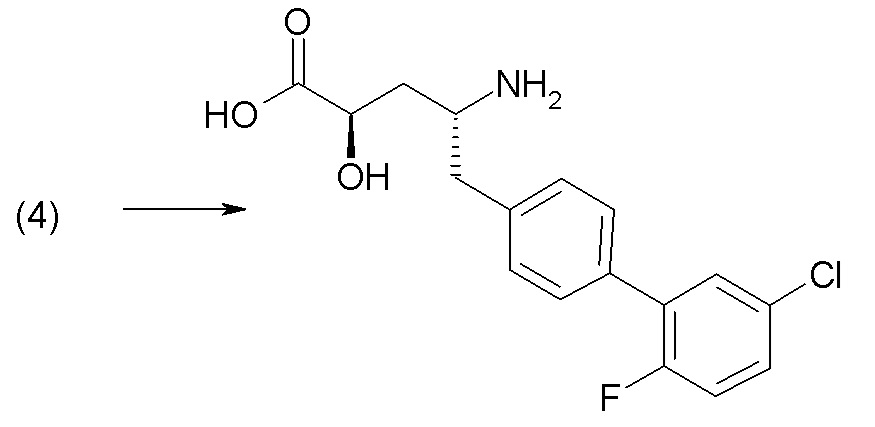
Раствор соединения 4 (8,8 г, 21,8 ммоль) в EtOH (12 мл) добавляли к концентрированной HCl (200 мл), нагревали при 100°C и перемешивали в течение ночи. Затем, смесь концентрировали с получением неочищенного продукта, который очищали путем промывания Et2O (100 мл) с получением указанного в заголовке соединения в виде твердого гидрохлорида (7,5 г). ЖХ-МС: 338 [M+H+].
Подготовительный пример 12: сложный этиловый эфир (2R,4R)-4-амино-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксипентановой кислоты

Раствор соединения 5 (7,5 г, 20,1 ммоль) в EtOH/HCl (100 мл) нагревали при 50°C в течение ночи. Смесь концентрировали, и очищали неочищенный продукт путем промывания Et2O (200 мл) с получением указанного в заголовке соединения (6,5 г) в виде белого твердого гидрохлорида. ЖХ-МС: 366,1 [M+H+].
Подготовительный пример 13: сложный этиловый эфир (2R,4R)-5-(4-бромфенил)-2-гидрокси-4-[(1-гидрокси-1H-1,2,3-триазол-4-карбонил)амино]пентановой кислоты

1-Гидрокси-1H-1,2,3-триазол-4-карбоновую кислоту получали путем объединения сложного этилового эфира 1-гидрокси-1H-[1,2,3]триазол-4-карбоновой кислоты (2,0 г, 13 ммоль), EtOH (25 мл, 430 ммоль), предварительно растворенного раствора LiOH моногидрата (1,6 г, 38,2 ммоль) и воды (10 мл, 600 ммоль). Смесь перемешивали при комнатной температуре в течение 4 часов, затем частично концентрировали и подкисляли добавлением HCl для индукции выпадения осадка. Твердое вещество фильтровали и сушили в условиях вакуума с получением 1,3 г целевой кислоты.
1-Гидрокси-1H-1,2,3-триазол-4-карбоновую кислоту (163 мг, 1,3 ммоль) объединяли с HCTU (523 мг, 1,3 ммоль) и DMF, и перемешивали в течение 5 минут при комнатной температуре. Добавляли DIPEA (661 мкл, 3,8 ммоль) и сложный этиловый эфир (2R,4R)-4-амино-5-(4-бромфенил)-2-гидроксипентановой кислоты (0,400 г, 1,26 ммоль), и перемешивали полученную смесь в течение 10 минут. Смесь упаривали в условиях пониженного давления и очищали (колонка C18; 20-70% MeCN в воде с 5% TFA) с получением указанного в заголовке соединения (330 мг).
Подготовительный пример 14: 5-(4-метоксибензилокси)-1-метил-1H-пиразол-3-карбоновая кислота
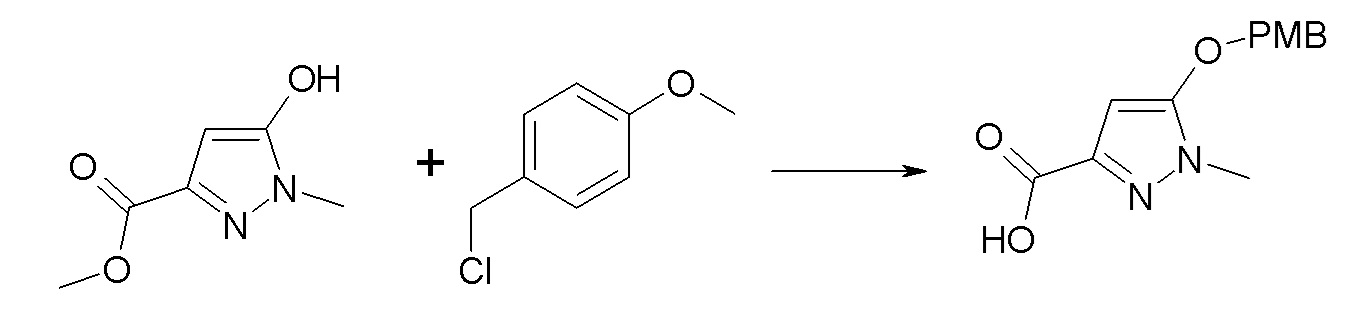
К перемешанному раствору сложного метилового эфира 5-гидрокси-1-метил-1H-пиразол-3-карбоновой кислоты (200 мг, 1 ммоль) в DMF (992 мкл, 12,8 ммоль) при 0°C добавляли K2CO3 (195 мг, 1,4 ммоль). Спустя 10 минут, при 0°C добавляли пара-метоксибензилхлорид (208 мкл, 1,5 ммоль), и перемешивали полученную смесь при 60°C в течение 1 часа, после чего оставляли охлаждаться до комнатной температуры. Добавляли MeOH (2,6 мл, 63,2 ммоль), а затем LiOH (61,4 мг, 2,6 ммоль) в воде (2,6 мл, 142 ммоль), и отслеживали завершение реакции. Растворитель удаляли в условиях вакуума. В пробирку добавляли воду, и подкисляли неочищенный продукт добавлением 1н водного раствора HCl. Твердые вещества фильтровали с получением указанного в заголовке соединения в виде белого твердого вещества (350 мг).
Подготовительный пример 15: (2R,4R)-5-(3°-хлорбифенил-4-ил)-2-гидрокси-4-{[5-(4-метоксибензилокси)-1-метил-1H-пиразол-3-карбонил]амино}пентановая кислота
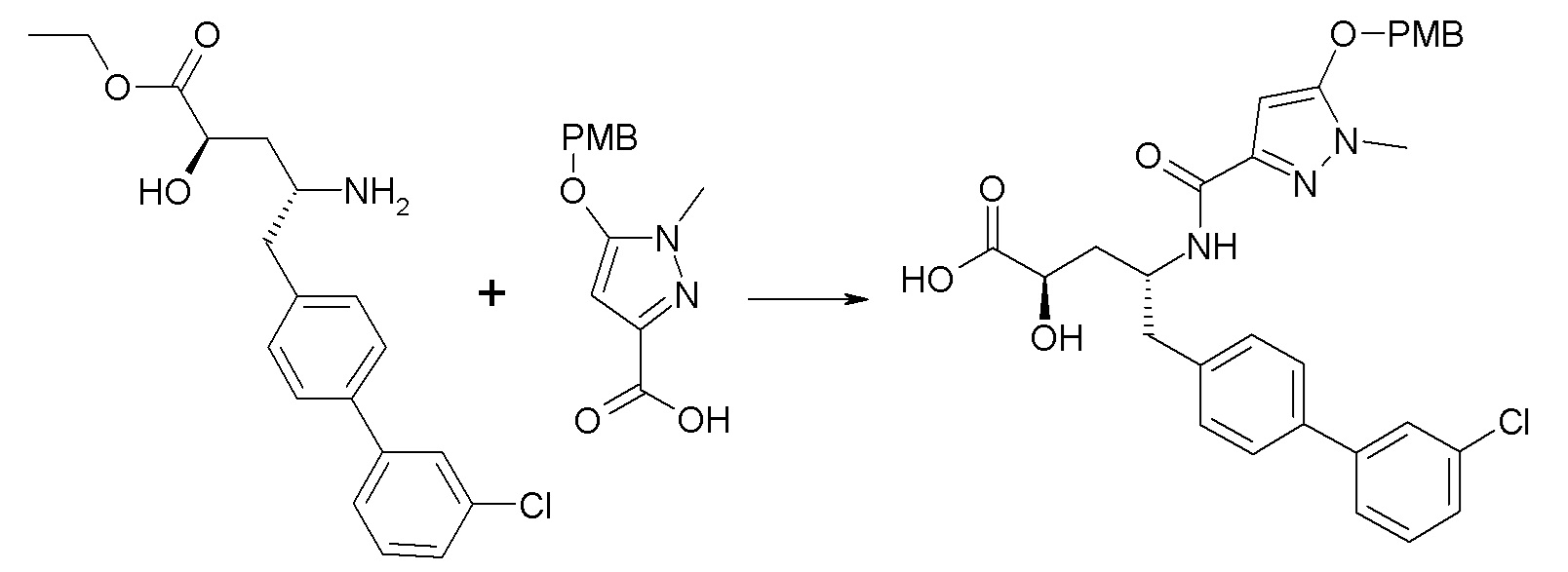
5-(4-Метоксибензилокси)-1-метил-1H-пиразол-3-карбоновую кислоту (118,8 мг, 453 мкмоль) объединяли с HCTU (187,3 мг, 453 мкмоль) и DMF (2,0 мл, 25,9 ммоль). Смесь перемешивали при комнатной температуре в течение 15 минут. Добавляли DIPEA (225 мкл, 1,3 ммоль) и сложный этиловый эфир (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (150 мг, 430 мкмоль), и перемешивали смесь при комнатной температуре в течение 15 минут. Растворитель удаляли в условиях вакуума. Добавляли EtOH (1,5 мл, 25,9 ммоль), а затем 1 M раствор LiOH в воде (4,3 мл, 4,3 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель удаляли в условиях вакуума, и очищали продукт методом хроматографии с обращенной фазой с получением указанного в заголовке соединения (171 мг).
Подготовительный пример 16: 3-(2-фторфенил)изоксазол-5-карбонилхлорид
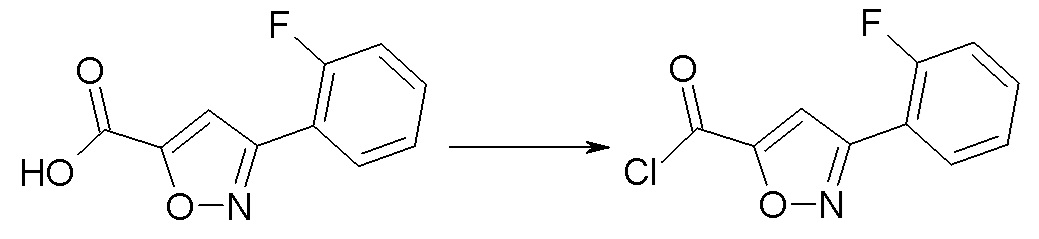
К перемешанному раствору 3-(2-фторфенил)изоксазол-5-карбоновой кислоты (77 мг, 370 мкмоль) в THF (10 мл) по каплям добавляли оксалилхлорид (0,07 мл, 0,74 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов, затем концентрировали с получением указанного в заголовке соединения (79 мг), которое использовали без дополнительной очистки.
Подготовительный пример 17: 3-метоксиизоксазол-5-карбонилхлорид

К раствору 3-метоксиизоксазол-5-карбоновой кислоты (420 мг, 3 ммоль) в THF(15 мл) и DMF (1 капля) при 0°C по каплям добавляли оксалилхлорид (650 мкл, 7 ммоль). Затем, смесь перемешивали при комнатной температуре в течение 3 часов. Раствор концентрировали в условиях вакуума с получением указанного в заголовке соединения в виде светло-желтого масла (430 мг).
Подготовительный пример 18: сложный хлорметиловый эфир (S)-2-трет-бутоксикарбониламино-3-метилбутановой кислоты
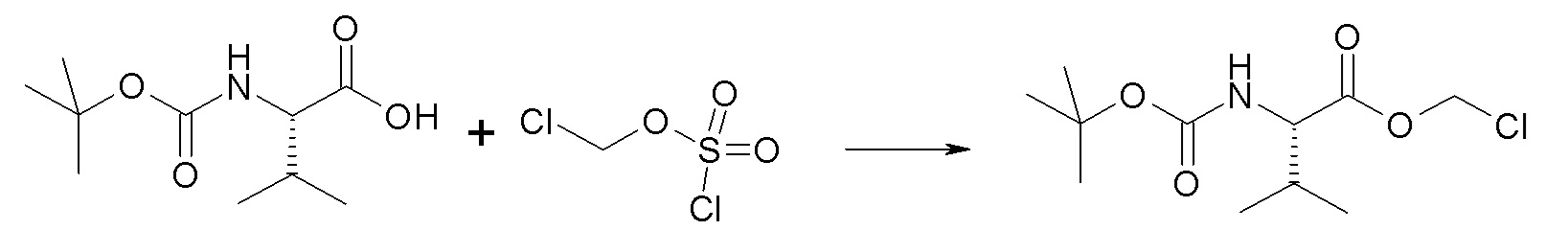
К смеси (S)-2-(трет-бутоксикарбониламино)-3-метилбутановой кислоты (28,6 г, 130 ммоль), NaHCO3 (44 г, 520 ммоль) и Bu4NHSO4 (4,4 г, 13 ммоль) в DCM (200 мл) и воде (200 мл) при 0°C добавляли хлорметилсульфохлоридат (26 г, 158 ммоль). Смесь перемешивали при комнатной температуре в течение 24 часов, затем экстрагировали DCM (3×150 мл). Объединенные органические слои промывали водой (2×300 мл), и очищали DCM слой методом колоночной флэш-хроматографии (петролейный эфир/EA=15/1) с получением указанного в заголовке соединения в виде желтого твердого вещества (35 г). ЖХ-МС: 266 [M+H]+.
Подготовительный пример 19: сложный хлорметиловый эфир (S)-2-метоксикарбониламино-3-метилбутановой кислоты

Раствор сложного хлорметилового эфира (S)-2-трет-бутоксикарбониламино-3-метилбутановой кислоты (35 г, 132 ммоль) в DCM (200 мл) при 0ºC по каплям добавляли к раствору TFA (50 мл) в DCM (100 мл). Полученную смесь перемешивали при комнатной температуре в течение ночи, а затем концентрировали в условиях вакуума с получением неочищенного соединения 1 в виде желтого масла (21,8 г). ЖХ-МС: 166 [M+H]+.

К смеси соединения 1 (21,8 г, 139 ммоль) и метилхлороформиата (12 мл, 157 ммоль) в THF (1 л) при 0ºC добавляли TEA (38 мл, 278 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 12 часов, а затем концентрировали в условиях вакуума. Остаток очищали методом колоночной флэш-хроматографии (петролейный эфир/EtOAc=6/1) с получением указанного в заголовке соединения в виде желтого твердого вещества (20,3 г). ЖХ-МС: 224 [M+H]+.1H ЯМР (400 МГц, ДМСО-d6): δ 0,97-1,02 (м, 6H), 2,16-2,21 (м, 1H), 3,68 (с, 1H), 4,14 (д, J=4 Гц, 1H), 5,76-5,91 (м, 2H).
Подготовительный пример 20: сложный этиловый эфир (2R,4R)-4-амино-5-бифенил-4-ил-2-гидроксипентановой кислоты

К перемешанному раствору сложного трет-бутилового эфира (S)-2-бифенил-4-илметил-5-оксопирролидин-1-карбоновой кислоты (4,4 г, 12,4 ммоль) в безводном THF (70 мл) при -65ºC в атмосфере азота добавляли раствор 1 M LiHMDS в THF (28 мл) в течение 15 минут. После перемешивания в течение 3 часов при -65ºC добавляли оксодипероксимолибден(пиридин)(гексаметилфосфортриамид) (9 г, 18,6 ммоль). Смесь дополнительно перемешивали в течение 2 часов при -35ºC, а затем добавляли насыщенный водный Na2S2O3 (60 мл). Органический слой собирали и промывали насыщенным водным NH4Cl (60 мл × 3) и насыщенным водным NaCl (60 мл × 2), а затем сушили над Na2SO4, и удаляли растворитель в условиях пониженного давления с получением неочищенного продукта, который затем очищали методом хроматографии (гексаны/EtOAc=5/1) с получением соединения 1 в виде белого твердого вещества (1,8 г). ЖХ-МС: 757 [2M+Na].
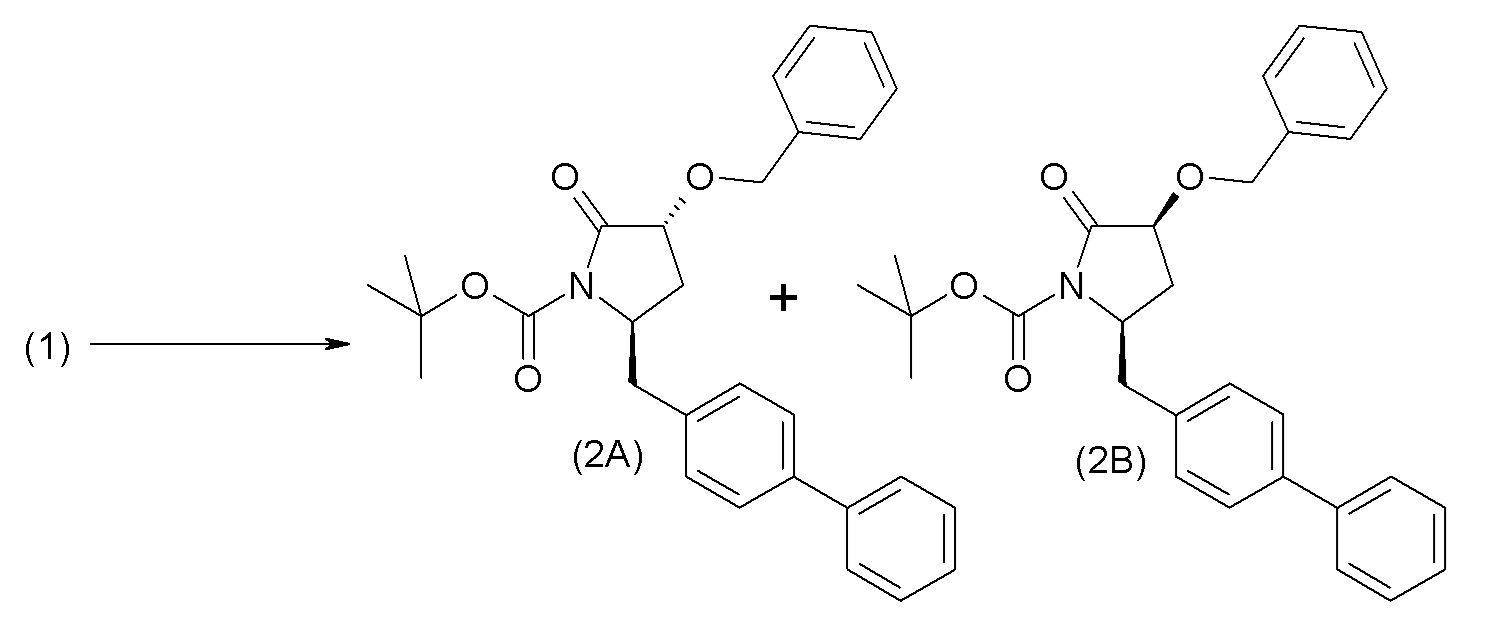
К раствору соединения 1 (1,8 г, 5,0 ммоль) в безводном DCM (50 мл) при 0ºC в атмосфере азота добавляли DMAP (122 мг, 1 ммоль) и Et3N (1,5 г, 14,9 ммоль). После перемешивания в течение 0,5 часа при 0ºC, в течение 15 минут добавляли бензилхлорид (1,0 г, 7,4 ммоль). Смесь дополнительно перемешивали в течение 2 часов при 0ºC, а затем добавляли насыщенный водный NaHCO3 (50 мл). Органический слой собирали и промывали насыщенным водным NaHCO3 (50 мл×2) и насыщенным водным NaCl (50 мл×1), а затем сушили над Na2SO4. Твердые вещества отфильтровывали, и концентрировали фильтрат с получением неочищенного продукта, который затем очищали методом хроматографии (гексаны/EtOAc=4/1) с получением соединения 2A (471 мг) и соединения 2B (883 мг) в виде белых твердых веществ. ЖХ-МС: 494 [M+Na]; 965 [2M+Na].
Соединение 2A:1H ЯМР (300 МГц, CDCl3): δ (м.д.)=8,02 (м, 2H), 7,57-7,25 (м, 12H), 5,42 (м, 1H), 4,50 (м, 1H), 3,26-3,21 (м, 1H), 2,90 (м, 1H), 2,58 (м, 1H), 2,15-2,05 (м, 1H), 1,62 (м, 9H).
Соединение 2B:1H ЯМР (300 МГц, CDCl3): δ (м.д.)=8,06 (м, 2H), 7,58-7,18 (м, 12H), 5,53-5,41 (м, 1H), 4,39 (м, 1H), 3,57-3,54 (м, 1H), 2,87-2,80 (м, 1H), 2,48-2,44 (м, 1H), 1,98 (м, 1H), 1,63 (м, 9H).
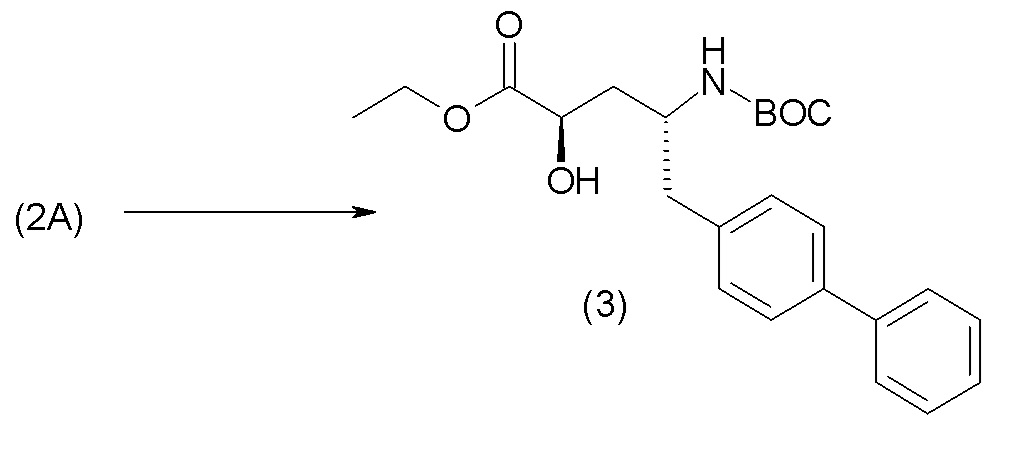
К перемешанному раствору соединения 2A (471 мг, 1 ммоль) в безводном EtOH (10 мл) при комнатной температуре в атмосфере азота добавляли безводный K2CO3 (691 мг, 5 ммоль). После перемешивания в течение 20 часов при комнатной температуре, твердые вещества отфильтровывали. К фильтрату добавляли воду (30 мл), DCM (30 мл) и насыщенный водный NaCl (5 мл). Водный слой разделяли и экстрагировали DCM (30 мл×3). Объединенные органические слои промывали насыщенным водным NaCl (50 мл), сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который затем очищали методом хроматографии (гексаны/EtOAc=6/1) с получением соединения 3 в виде белого твердого вещества (275 мг). ЖХ-МС: 436 [M+Na]. 849 [2M+Na].
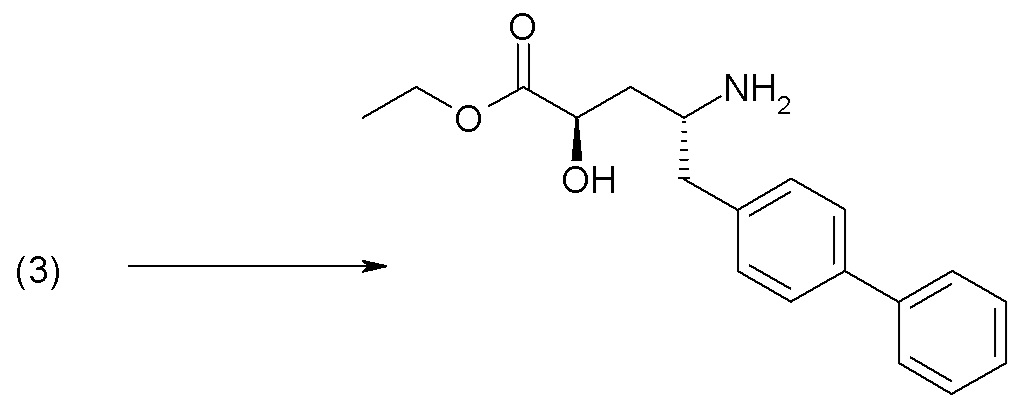
К EtOH (5 мл) при -30ºC добавляли ацетилхлорид (685 мг). После перемешивания в течение 1 часа при -30ºC, добавляли раствор соединения 3 (275 мг, 665 мкмоль) в безводном EtOH (5 мл). Смесь нагревали до 25ºC и перемешивали в течение 3 часов при 25ºC. После выпаривания растворителя, остаток промывали холодным безводным Et2O (10 мл) с получением указанного в заголовке соединения в виде белого твердого гидрохлорида (207 мг). ЖХ-МС: 314 [M+H]. 649 [2M+Na].
1H ЯМР (300 МГц, CDCl3): δ (м.д.)=7,99 (м, 3H), 7,66-7,64 (м, 4H), 7,48-7,35 (м, 5H), 6,08 (м, 1H), 4,21 (м, 1H), 4,09-4,05 (м, 2H), 3,52 (м, 1H), 2,97-2,95 (м, 2H), 1,89-1,87 (м, 2H), 1,19-1,14 (м, 3H).
Подготовительный пример 21: сложный 5-метил-2-оксо-[1,3]диоксол-4-илметиловый эфир (2R,4R)-4-амино-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксипентановой кислоты

К суспензии (2R,4R)-4-амино-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксипентановой кислоты (гидрохлорид; 10,3 г, 27,6 ммоль) в THF (45 мл) добавляли водный NaOH (3,3 г, 82,8 ммоль). При 0°C по каплям добавляли раствор (BOC)2O (9,6 г, 44,2 ммоль) в THF (25 мл). Полученную смесь перемешивали при комнатной температуре в течение 16 часов. Удаляли THF, и растворяли остаток в воде (50 мл). Раствор подкисляли до pH=3 добавлением водной HCl (2н). Полученный осадок отфильтровывали, осадок на фильтре промывали водой (20 мл) и очищали методом хроматографии (DCM/MeOH=20/1) с получением соединения 1 в виде белого твердого вещества (9,0 г). ЖХ-МС: 438 [M+H]+.
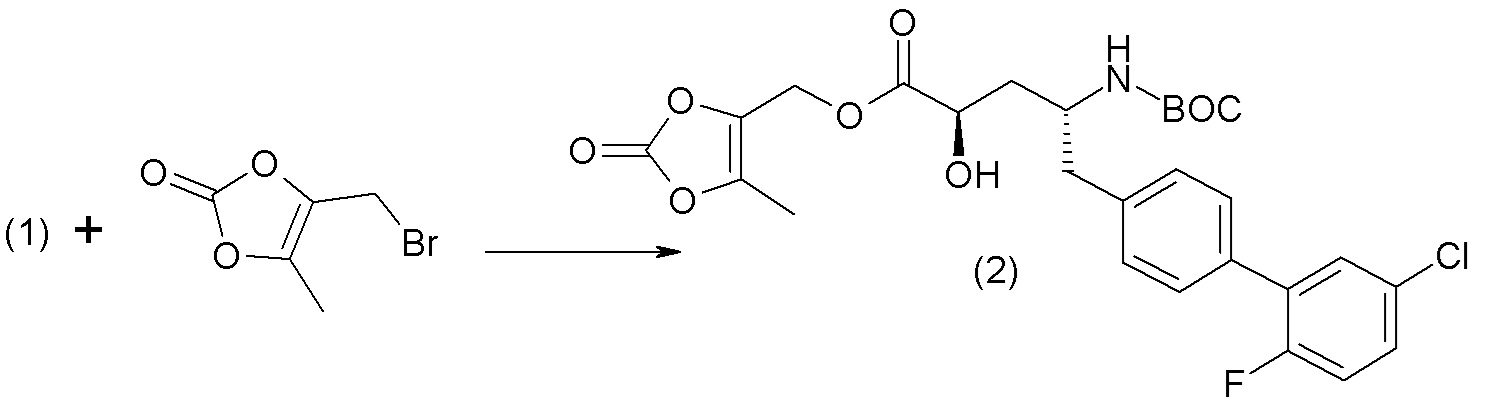
К раствору соединения 1 (2 г, 4,6 ммоль) и 4-(бромметил)-5-метил-1,3-диоксол-2-она (880 мг, 4,6 ммоль) в DMF (20 мл) добавляли K2CO3 (947 мг, 6,8 ммоль) и KI (152 мг, 0,9 ммоль). После перемешивания при комнатной температуре в течение 2 часов, смесь разбавляли водой (50 мл) и экстрагировали EtOAc (2×100 мл). Объединенные органические слои промывали насыщенным водным NaCl (2×100 мл), сушили над безводным Na2SO4, фильтровали и концентрировали. Остаток очищали методом хроматографии (DCM/EtOAc=10/1) с получением соединения 2 в виде желтой пены (2,3 г). ЖХ-МС: 572 [M+Na]+.
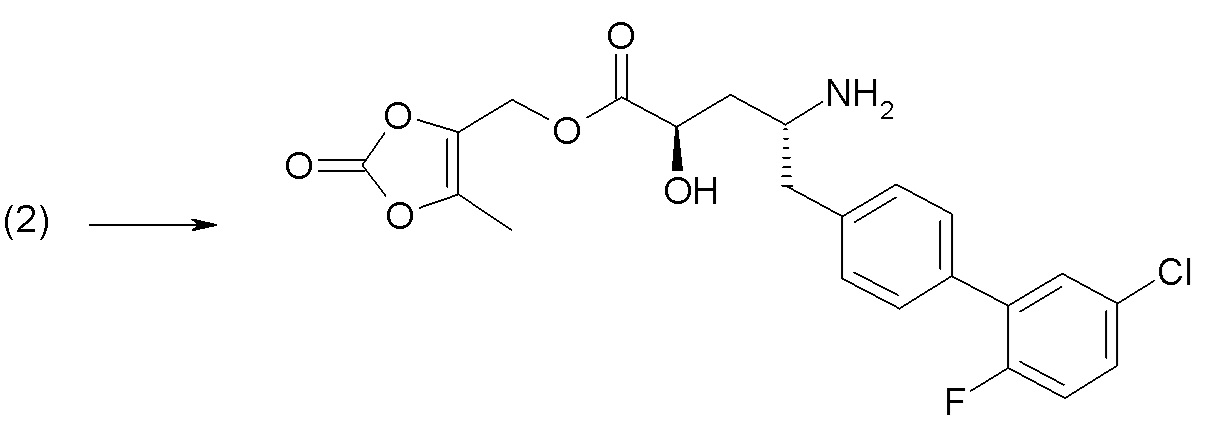
Раствор соединения 2 (2,3 г, 4,2 ммоль) в смеси HCl/диоксан (50 мл, 3,3 M) перемешивали при комнатной температуре в течение 2 часов. Растворитель удаляли, и промывали остаток EtOAc (10 мл) с получением указанного в заголовке соединения в виде белого твердого вещества (1,8 г). ЖХ-МС: 450,0 [M+H].1H ЯМР (ДМСО-d6, 400 МГц) δ 1,85-1,92 (м, 2H), 2,17 (с, 3H), 2,99 (шир., 2H), 3,57 (шир., 1H), 4,26-4,42 (м, 1H), 5,02 (с, 2H), 6,19 (д, J=5,0 Гц, 1H), 7,34-7,45 (м, 3H), 7,49-7,51 (м, 1H), 7,53-7,68 (м, 3H), 7,99 (с, 3H).
Подготовительный пример 22: сложный 2,2,3,3,3-пентафторпропиловый эфир (2R,4R)-4-амино-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксипентановой кислоты

Указанное в заголовке соединение также может быть получено с использованием методик, описанных в настоящем документе.
Подготовительный пример 23: сложный бутирилоксиметиловый эфир (2R,4R)-4-амино-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксипентановой кислоты
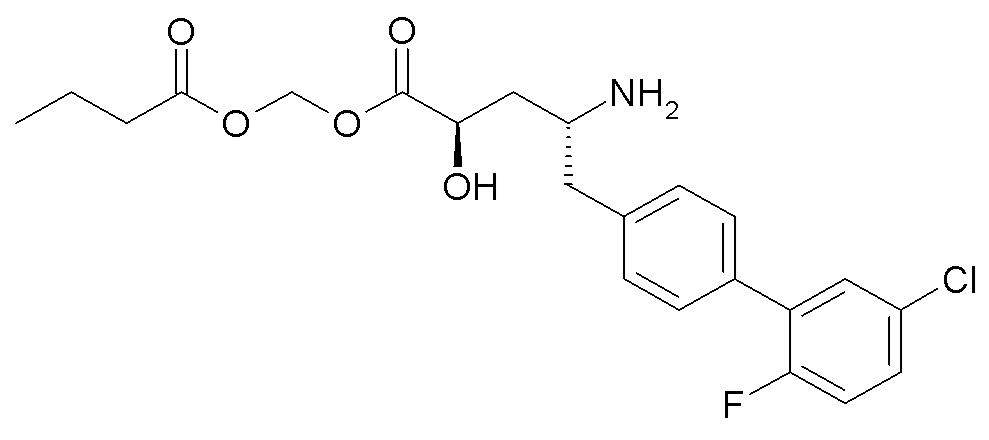
Указанное в заголовке соединение также может быть получено с использованием методик, описанных в настоящем документе.
Пример 1A: (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(3-гидрокси-3H-бензотриазол-5-карбонил)амино]пентановая кислота

1-Гидрокси-1H-1,2,3-бензотриазол-6-карбоновую кислоту (56,6 мг, 316 мкмоль) и HCTU (131 мг, 316 мкмоль) объединяли в DMF, и перемешивали в течение 5 минут при комнатной температуре. Добавляли DIPEA (83 мкл, 474 мкмоль) и сложный этиловый эфир (2R,4R)-4-амино-5-(4-бромфенил)-2-гидроксипентановой кислоты (50 мг, 0,2 ммоль), и перемешивали полученную смесь в течение 10 минут. Смесь упаривали в условиях пониженного давления и очищали (колонка C18, 20-70% MeCN в воде с 5% TFA). Чистые фракции лиофилизировали (40 мг). Затем, полученные твердые вещества объединяли с 3-хлорфенилборонвой кислотой (44,5 мг, 285 мкмоль), K2CO3 (66 мг, 474 мкмоль), EtOH (0,8 мл, 10 ммоль) и водой (0,2 мл, 10 ммоль). Добавляли SilicaCat®DPP-Pd (загрузка 0,28 ммоль/г; 57 мг, 16 мкмоль), и обрабатывали полученную смесь микроволнами при 100°C в течение 10 минут. Смесь фильтровали, и добавляли 1 M LiOH в воде (1,3 мл, 1,3 ммоль). Смесь перемешивали в течение 30 минут, упаривали в условиях пониженного давления и очищали методом препаративной HPLC с получением указанного в заголовке соединения (20,2 мг, чистота 95%). МС m/z [M+H]+расч. для C24H21ClN4O5, 481,12; обнаружено 481,2.
Пример 1B: сложный 3,3,3-трифторпропиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(3-гидрокси-3H-бензотриазол-5-карбонил)амино]пентановой кислоты

К раствору сложного этилового эфира (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(3-гидрокси-3H-бензотриазол-5-карбонил)амино]пентановой кислоты (50,0 мг, 98,2 мкмоль) в 3,3,3-трифторпропан-1-оле (336 мг, 3,0 ммоль) добавляли 4,0 M HCl в 1,4-диоксане (196 мкл, 786 мкмоль), и перемешивали полученную смесь при 70°C в течение 3 часов. Смесь концентрировали в условиях вакуума с получением белого твердого вещества, которое очищали методом препаративной HPLC (хроматография на колонке C18 с использованием 40-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения в виде белого твердого вещества (18 мг, чистота 100%). МС m/z [M+H]+ расч. для C27H24ClF3N4O5, 577,14; обнаружено 577,1.
Пример 1C: сложный 2,2,2-трифторэтиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(3-гидрокси-3H-бензотриазол-5-карбонил)амино]пентановой кислоты
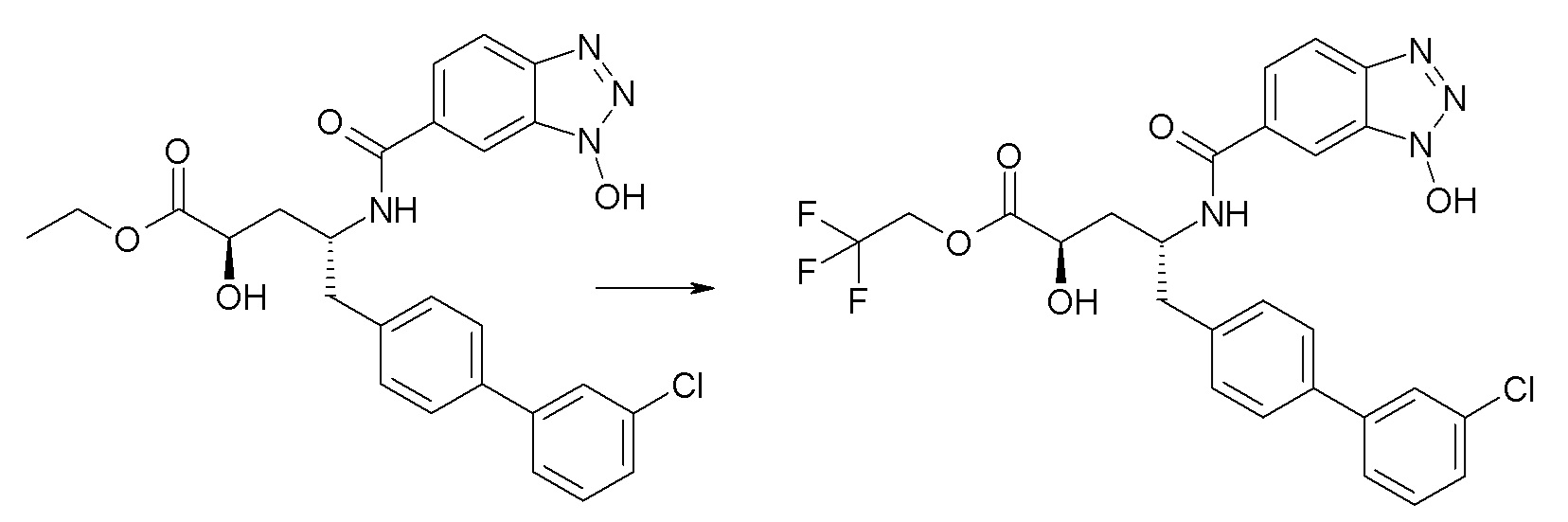
К раствору сложного этилового эфира (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(3-гидрокси-3H-бензотриазол-5-карбонил)амино]пентановой кислоты (50,0 мг, 98,2 мкмоль) в 2,2,2-трифторэтаноле (215 мкл, 3,0 ммоль) добавляли 4,0 M HCl в 1,4-диоксане (196 мкл, 786 мкмоль), и перемешивали полученную смесь при 100°C в течение 3 часов. Смесь концентрировали в условиях вакуума с получением прозрачной бесцветной жидкости, которую очищали методом препаративной HPLC (хроматография на колонке C18 с использованием 40-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения в виде белого твердого вещества (15,2 мг, чистота 99%). МС m/z [M+H]+ расч. для C26H22ClF3N4O5, 563,12; обнаружено 563,2.
Пример 1D: сложный 2,2,3,3,3-пентафторпропиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(3-гидрокси-3H-бензотриазол-5-карбонил)амино]пентановой кислоты

К раствору 1-гидрокси-1H-1,2,3-бензотриазол-6-карбоновой кислоты (14,9 мг, 83 мкмоль) в DMF (0,5 мл, 6,4 ммоль) добавляли HATU (37,9 мг, 99,6 мкмоль), и перемешивали полученную смесь при комнатной температуре в течение 5 минут. К смеси добавляли сложный 2,2,3,3,3-пентафторпропиловый эфир (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (30,0 мг, 66,4 мкмоль), а затем по каплям в течение 5 минут DIPEA (35 мкл, 0,2 ммоль), и перемешивали смесь при комнатной температуре в течение 30 минут. Смесь концентрировали в условиях вакуума с получением желтой жидкости, которую очищали методом препаративной HPLC с получением указанного в заголовке соединения в виде белого твердого вещества (2,9 мг, чистота 100%). МС m/z [M+H]+ расч. для C27H22ClF5N4O5, 613,12; обнаружено 613,0.
Пример 1E: сложный 5-метил-2-оксо-[1,3]диоксол-4-илметиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(3-гидрокси-3H-бензотриазол-5-карбонил)амино]пентановой кислоты
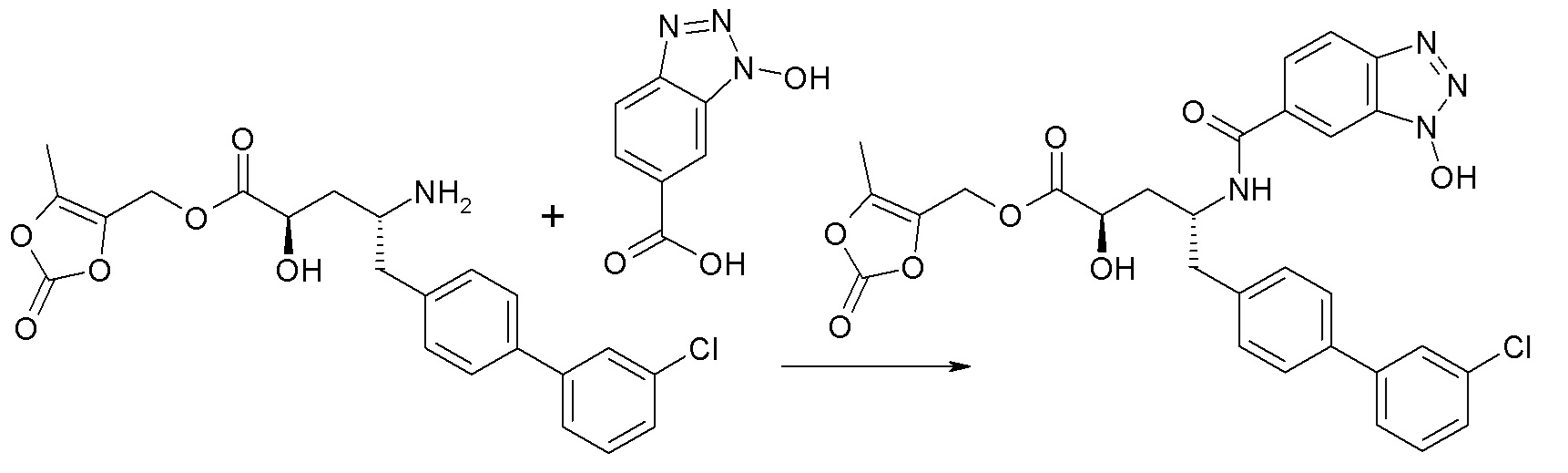
К раствору 1-гидрокси-1H-1,2,3-бензотриазол-6-карбоновой кислоты (14,9 мг, 83 мкмоль) в DMF (0,5 мл, 6,4 ммоль) добавляли HATU (37,9 мг, 99,6 мкмоль), и перемешивали полученную смесь при комнатной температуре в течение 5 минут. К смеси добавляли сложный 5-метил-2-оксо[1,3]диоксол-4-илметиловый эфир (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (28,7 мг, 66,4 мкмоль), а затем по каплям в течение 5 минут DIPEA (35 мкл, 0,2 ммоль), и перемешивали смесь при комнатной температуре в течение 30 минут. Смесь концентрировали в условиях вакуума с получением желтой жидкости, которую очищали методом препаративной HPLC с получением указанного в заголовке соединения в виде белого твердого вещества (37,8 мг, чистота 98,5%). МС m/z [M+H]+ расч. для C29H25ClN4O8, 593,14; обнаружено 593,0.
Пример 1F: сложный бутирилоксиметиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(3-гидрокси-3H-бензотриазол-5-карбонил)амино]пентановой кислоты

К раствору 1-гидрокси-1H-1,2,3-бензотриазол-6-карбоновой кислоты (14,9 мг, 83 мкмоль) в DMF (0,5 мл, 6,4 ммоль) добавляли HATU (37,9 мг, 99,6 мкмоль), и перемешивали полученную смесь при комнатной температуре в течение 5 минут. К смеси добавляли сложный бутирилоксиметиловый эфир (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (27,9 мг, 66,4 мкмоль), а затем по каплям в течение 5 минут DIPEA (35 мкл, 0,2 ммоль), и перемешивали смесь при комнатной температуре в течение 30 минут. Смесь концентрировали в условиях вакуума с получением желтой жидкости, которую очищали методом препаративной HPLC с получением указанного в заголовке соединения в виде белого твердого вещества (5,2 мг, чистота 98,2%). МС m/z [M+H]+ расч. для C29H29ClN4O7, 581,17; обнаружено 581,2.
Пример 1G: (2R,4R)-4-[(3-Ацетоксиметокси-3H-бензотриазол-5-карбонил)амино]-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановая кислота
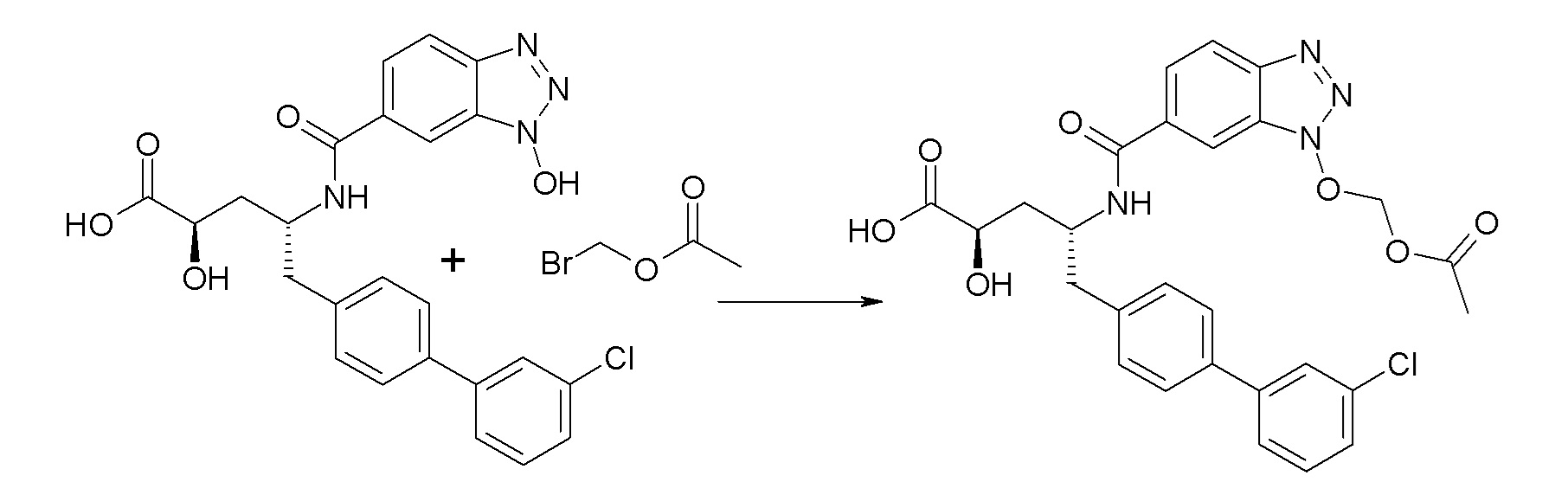
К раствору (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(3-гидрокси-3H-бензотриазол-5-карбонил)амино]пентановой кислоты (130 мг, 270 мкмоль) в бензиловом спирте (559 мкл, 5,4 ммоль) добавляли 4,0 M HCl в 1,4-диоксане (270 мл, 1,1 ммоль), и перемешивали при 60°C в течение 1,5 часов. Затем, смесь очищали (хроматография на колонке C18, колонка 55 г, с использованием 30-90% MeCN в воде с 0,05% TFA) с получением не совсем белого твердого вещества. К раствору этого не совсем белого твердого вещества и K2CO3 (82,2 мг, 595 мкмоль) в DMF (3,1 мл, 40,0 ммоль) добавляли бромметилацетат (45,5 мг, 297 мкмоль), и перемешивали полученную смесь при комнатной температуре в течение 1 часа. Затем, смесь очищали (колоночная хроматография, небольшая колонка, с использованием 40-90% MeCN в воде с 0,05% TFA) с получением прозрачной бесцветной вязкой жидкости. К раствору этой прозрачной бесцветной вязкой жидкости в MeOH (4,1 мл, 102 ммоль) добавляли 10% Pd/C, влажность 50% (загрузка 0,45 ммоль/г; 30,0 мг, 13,5 мкмоль), и перемешивали полученную смесь в атмосфере водорода в течение 30 минут. Смесь фильтровали, и концентрировали фильтрат в условиях вакуума с получением бесцветной прозрачной жидкости. Неочищенную жидкость очищали (хроматография на колонке C18, колонка 55 г, с использованием 30-85% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения (14,0 мг) в виде белого твердого вещества. МС m/z [M+H]+ расч. для C27H25ClN4O7, 553,14; обнаружено 553,1.
Пример 1H: сложный ацетоксиметиловый эфир (2R,4R)-4-[(3-ацетоксиметокси-3H-бензотриазол-5-карбонил)амино]-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты
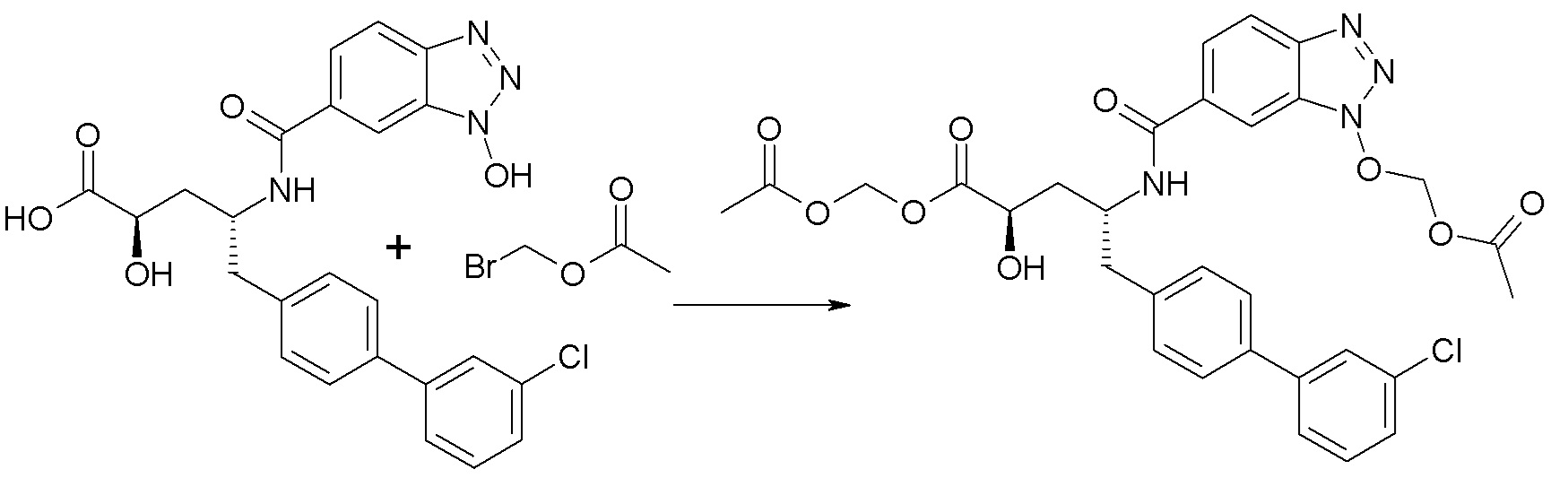
К раствору [(2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(3-гидрокси-3H-бензотриазол-5-карбонил)амино]пентановой кислоты (20,0 мг, 42 мкмоль и бромметилацетата (6,52 мкл, 67 мкмоль) в ацетоне (1,3 мл, 17,2 ммоль) добавляли Et3N (7,0 мкл, 50 мкмоль), и перемешивали полученную смесь в герметизированной пробирке при 65°C в течение 5 часов. Добавляли TFA/DCM (1/1, 0,50 мл, 3,1 ммоль), смесь перемешивали при комнатной температуре в течение 30 минут и концентрировали с получением прозрачной желтой жидкости. Неочищенную жидкость очищали методом препаративной HPLC (хроматография на колонке C18, небольшая колонка, с использованием 30-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения (5,2 мг, чистота 100%). МС m/z [M+H]+ расч. для C30H29ClN4O9, 625,16; обнаружено 625,1.
Пример 1I: (2R,4R)-4-[(3-бензилокси-3H-бензотриазол-5-карбонил)амино]-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановая кислота
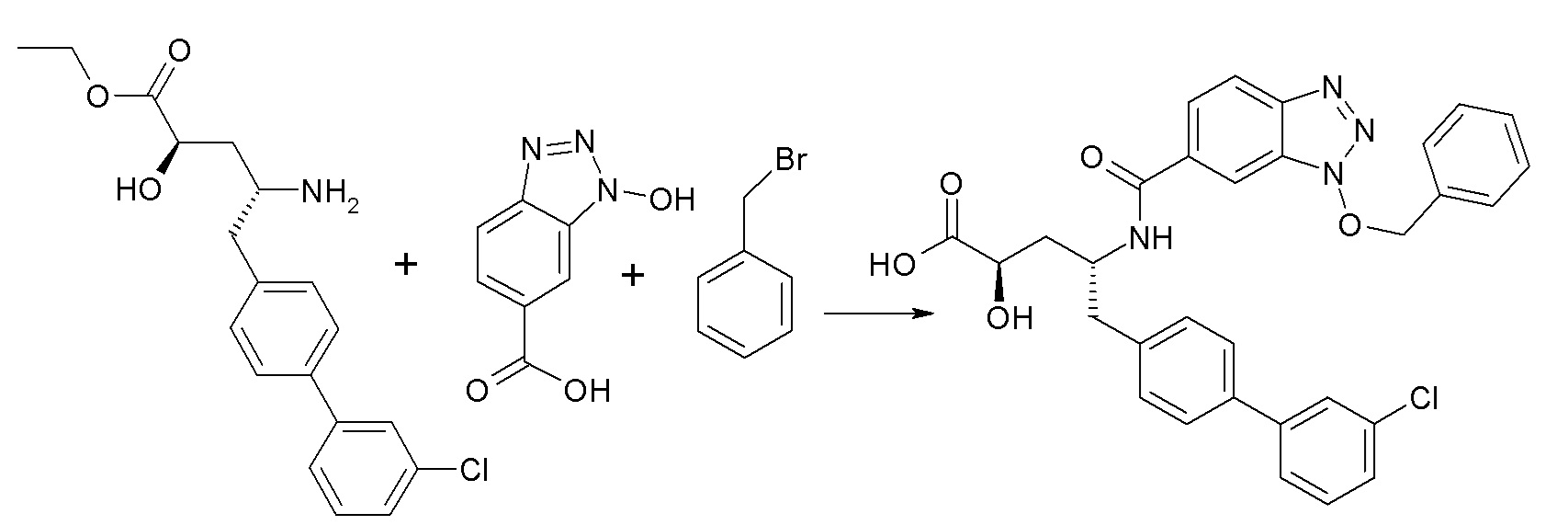
К раствору 1-гидрокси-1H-1,2,3-бензотриазол-6-карбоновой кислоты (116 мг, 647 мкмоль) в DMF (2,0 мл, 25,8 ммоль) добавляли HATU (246 мг, 647 мкмоль), и перемешивали полученную смесь при комнатной температуре в течение 10 минут. К смеси добавляли DIPEA (225 мкл, 1,3 ммоль), а затем сложный этиловый эфир (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (150 мг, 431 мкмоль), и перемешивали смесь при комнатной температуре в течение 1 часа. К смеси добавляли 3,0 M LiOH в воде (1,7 мл, 5,2 ммоль), и перемешивали при комнатной температуре в течение 30 минут. Затем, смесь очищали (хроматография на колонке C18, колонка 55 г, с использованием 40-90% MeCN в воде с 0,05% TFA) с получением белого твердого вещества. К раствору этого твердого вещества в ацетоне (3,0 мл, 40,8 ммоль) добавляли бензилбромид (41 мкл, 345 мкмоль). Добавляли Et3N (54 мкл, 388 мкмоль), и перемешивали полученную смесь при 50°C в течение 2 часов. Смесь концентрировали в условиях вакуума, и очищали полученную неочищенную прозрачную желтую жидкость (хроматография на колонке C18, колонка 55 г, с использованием 40-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения (201 мг, чистота 100%) в виде белого твердого вещества. МС m/z [M+H]+ расч. для C31H27ClN4O5, 571,17; обнаружено 571.
Пример 1J: сложный бензиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(3-гидрокси-3H-бензотриазол-5-карбонил)амино]пентановой кислоты
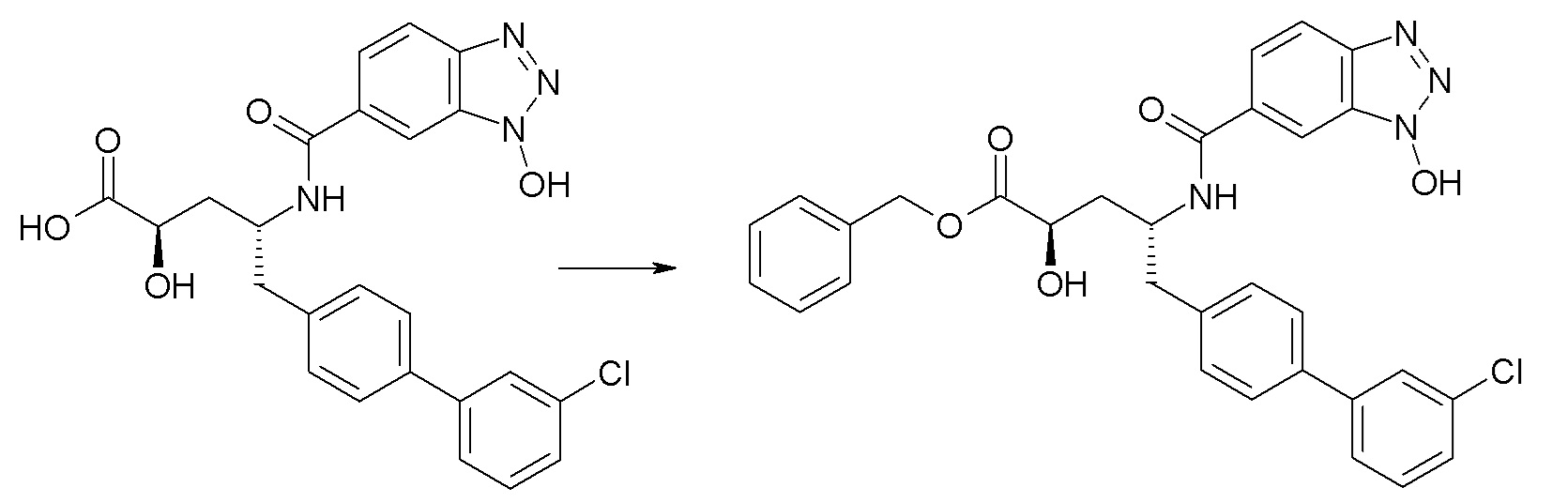
К раствору (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(3-гидрокси-3H-бензотриазол-5-карбонил)амино]пентановой кислоты (130 мг, 270 мкмоль) в бензиловом спирте (559 мкл, 5,4 ммоль) добавляли 4,0 M HCl в 1,4-диоксане (270 мкл, 1,0 ммоль), и перемешивали полученную смесь при 60°C в течение 15 минут. Смесь очищали методом препаративной HPLC (хроматография на колонке C18 с использованием 30-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (16,0 мг, чистота 99,2%). МС m/z [M+H]+ расч. для C31H27ClN4O5, 571,17; обнаружено 571,1.
Пример 1K: сложный ацетоксиметиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(3-гидрокси-3H-бензотриазол-5-карбонил)амино]пентановой кислоты
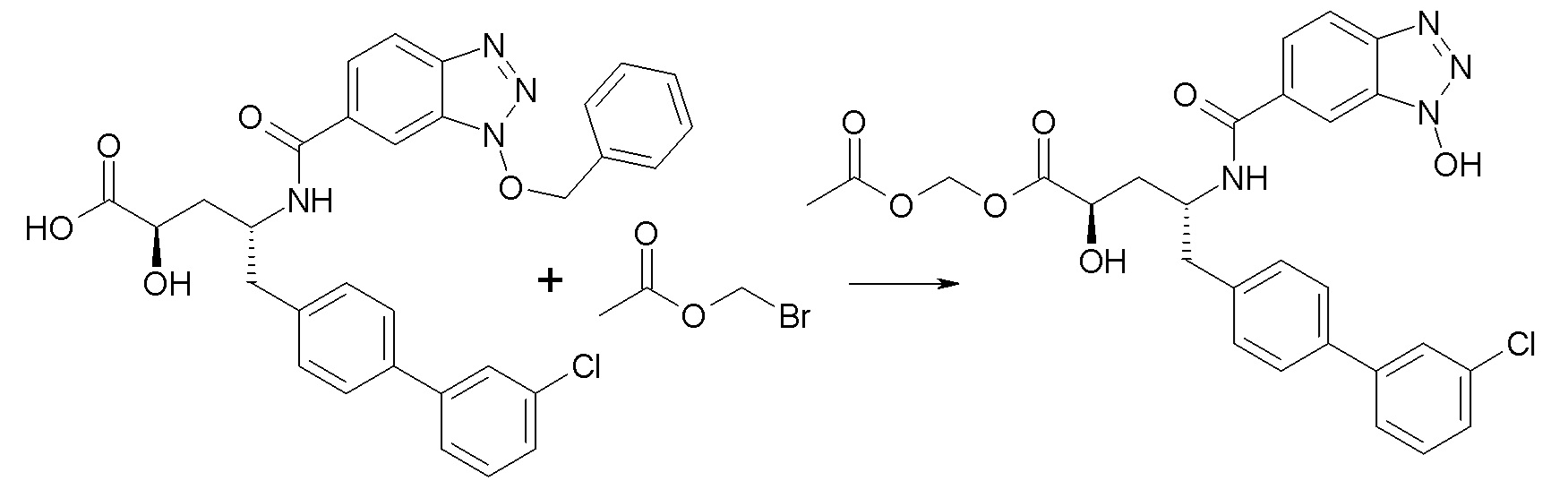
К раствору (2R,4R)-4-[(3-бензилокси-3H-бензотриазол-5-карбонил)амино]-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (30,0 мг, 52,5 мкмоль) в ацетоне (1,0 мл, 13,6 ммоль) добавляли бромметилацетат (6,7 мкл, 68,3 мкмоль) и Et2O (11,7 мкл, 84 мкмоль), и перемешивали полученную смесь при 45°C в течение 1 часа. Для нейтрализации реакционной смеси добавляли каплю AcOH, и концентрировали смесь с получением прозрачной желтой жидкости. Неочищенную жидкость очищали (хроматография на колонке C18, колонка 55 г, с использованием 40-90% MeCN в воде с 0,05% TFA) с получением белого твердого вещества. Твердое вещество растворяли в THF (1,0 мл, 12,3 ммоль), к раствору добавляли 10% Pd/C, влажность 50% (загрузка 0,45 ммоль/г; 5,8 мг, 2,6 мкмоль), и перемешивали в атмосфере водорода в течение 1 часа. Смесь фильтровали и концентрировали фильтрат с получением бесцветной прозрачной жидкости. Неочищенную жидкость очищали (препаративная хроматография на колонке C18, небольшая колонка, с использованием 30-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения в виде белого твердого вещества (10,0 мг, чистота 99%). МС m/z [M+H]+ расч. для C27H25ClN4O7, 553,14; обнаружено 553,1.
Пример 1L: (2R,4R)-4-{[3-((R)-2-амино-3-метилбутирилоксиметокси)-3H-бензотриазол-5-карбонил]амино}-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановая кислота
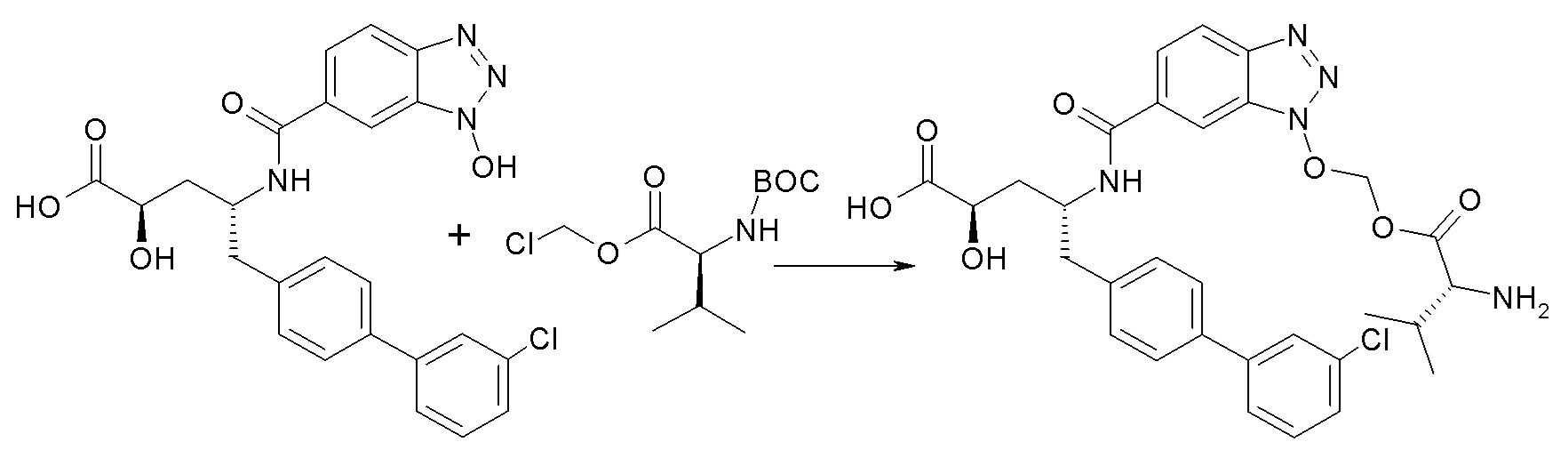
К раствору [(2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(3-гидрокси-3H-бензотриазол-5-карбонил)амино]пентановой кислоты (20,0 мг, 42 мкмоль) и сложного хлорметилового эфира (S)-2-трет-бутоксикарбониламино-3-метилбутановой кислоты (17,7 мг, 67 мкмоль) в ацетоне (1,3 мл, 17,2 ммоль) добавляли Et3N (7,0 мкл, 50 мкмоль), и перемешивали полученную смесь в герметизированной пробирке при 65°C в течение 5 часов. Добавляли TFA/DCM (1/1, 0,50 мл, 3,1 ммоль), смесь перемешивали при комнатной температуре в течение 30 минут и концентрировали с получением прозрачной желтой жидкости. Неочищенную жидкость очищали методом препаративной HPLC (хроматография на колонке C18, небольшая колонка, с использованием 30-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения в виде белого твердого гидрохлорида (6,2 мг, чистота 96%). МС m/z [M+H]+ расч. для C30H32ClN5O7, 610,20; обнаружено 610,1.
Пример 1M: (2R,4R)-4-[(3-бутирилоксиметокси-3H-бензотриазол-5-карбонил)амино]-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановая кислота
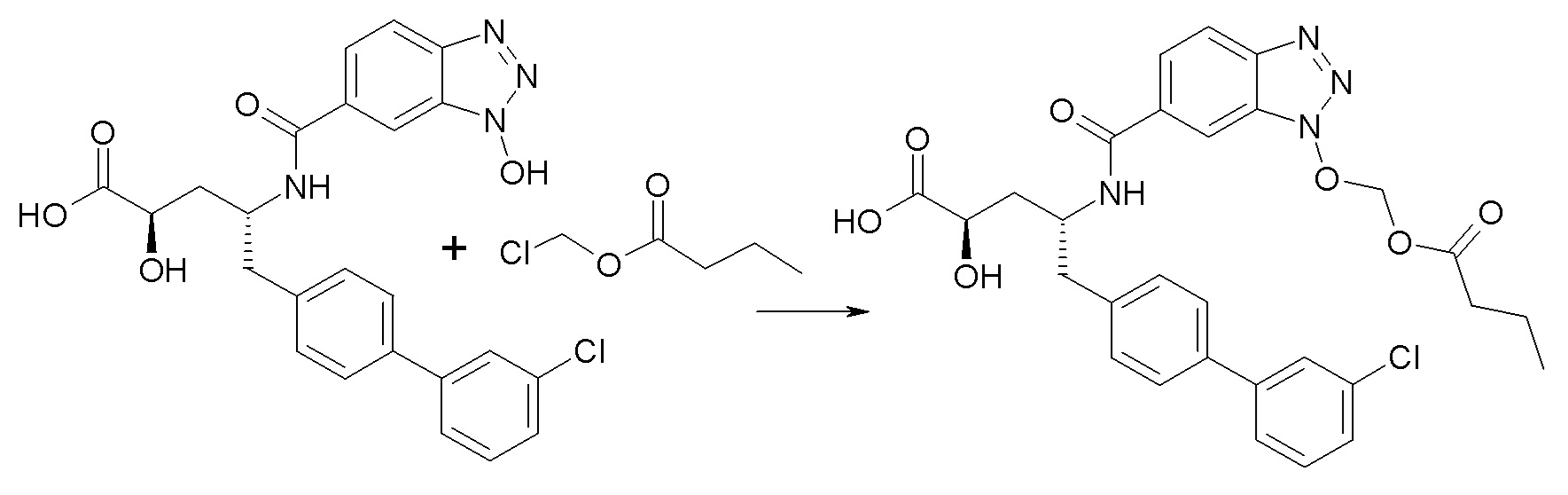
К раствору [(2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(3-гидрокси-3H-бензотриазол-5-карбонил)амино]пентановой кислоты (20,0 мг, 42 мкмоль) и хлорметилбутирата (8,3 мкл, 67 мкмоль) в ацетоне (1,3 мл, 17,2 ммоль) добавляли Et3N (7,0 мкл, 50 мкмоль), и перемешивали полученную смесь в герметизированной пробирке при 65°C в течение 5 часов. Затем, смесь концентрировали с получением желтой жидкости. Неочищенную жидкость очищали методом препаративной HPLC (хроматография на колонке C18, небольшая колонка, с использованием 30-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения в виде белого твердого вещества (3,3 мг, чистота 94%). МС m/z [M+H]+ расч. для C29H29ClN4O7, 581,17; обнаружено 581,1.
Пример 1N: сложный (R)-2-метоксикарбониламино-3-метилбутирилоксиметиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(3-гидрокси-3H-бензотриазол-5-карбонил)амино]пентановой кислоты

К раствору (2R,4R)-4-[(3-бензилокси-3H-бензотриазол-5-карбонил)амино]-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (30,0 мг, 52,5 мкмоль) в ацетоне (1,00 мл, 13,6 ммоль) добавляли сложный хлорметиловый эфир (S)-2-трет-бутоксикарбониламино-3-метилбутановой кислоты (18,1 мг, 68,3 мкмоль) и Et3N (11,7 мкл, 84 мкмоль), и перемешивали при 60°C в течение 3 часов. Смесь концентрировали с получением желтой жидкости, которую объединяли с TFA/DCM (1/1, 1,0 мл, 6,2 ммоль) и перемешивали при комнатной температуре в течение 1 часа. Смесь концентрировали в условиях вакуума, и очищали полученную неочищенную жидкость (хроматография на колонке C18, колонка 55 г, с использованием 30-90% MeCN в воде с 0,05% TFA) с получением белого твердого вещества. Твердое вещество растворяли в DCM (0,5 мл, 7,8 ммоль) при 0°C. Добавляли метилхлороформиат (6,1 мкл, 78,8 мкмоль), а затем Et3N (18,3 мкл, 131 мкмоль), и перемешивали полученную смесь при комнатной температуре в течение 30 минут. Для нейтрализации реакционной смеси добавляли каплю AcOH, и концентрировали смесь с получением прозрачной желтой жидкости. Неочищенную жидкость объединяли с 10% Pd/C, влажность 50% (загрузка 0,45 ммоль/г; 5,8 мг, 2,6 мкмоль), и перемешивали в атмосфере водорода в течение 1 часа. Смесь фильтровали, и концентрировали фильтрат с получением прозрачной желтой жидкости. Неочищенную жидкость очищали (препаративная хроматография на колонке C18, небольшая колонка, с использованием 30-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения в виде белого твердого трифторацетата (7,5 мг, чистота 99%). МС m/z [M+H]+ расч. для C32H34ClN5O9, 668,20; обнаружено 668,1.
Пример 1O: (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-{[3-((S)-2-метоксикарбониламино-3-метилбутирилоксиметокси)-3H-бензотриазол-5-карбонил]амино}пентановая кислота

К раствору [(2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(3-гидрокси-3H-бензотриазол-5-карбонил)амино]пентановой кислоты (20,0 мг, 42 мкмоль) и сложного хлорметилового эфира (S)-2-трет-бутоксикарбониламино-3-метилбутановой кислоты (17,7 мг, 67 мкмоль) в ацетоне (1,3 мл, 17,2 ммоль) добавляли Et3N (7,0 мкл, 50 мкмоль), и перемешивали полученную смесь в герметизированной пробирке при 65°C в течение 5 часов. Затем, смесь концентрировали с получением желтой жидкости. К раствору желтой жидкости и Et3N (11,6 мкл, 83 мкмоль) в DCM (0,5 мл, 7,8 ммоль) при 0°C добавляли метилхлороформиат (4,8 ML, 62 мкмоль), и перемешивали смесь при комнатной температуре в течение 15 минут. Смесь концентрировали в условиях вакуума, и очищали полученную прозрачную бесцветную жидкость методом препаративной HPLC (хроматография на колонке C18, небольшая колонка, с использованием 30-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения в виде белого твердого трифторацетата (5,1 мг, чистота 99%). МС m/z [M+H]+ расч. для C32H34ClN5O9, 668,20; обнаружено 668.
Пример 1P: сложный (R)-2-амино-3-метилбутирилоксиметиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(3-гидрокси-3H-бензотриазол-5-карбонил)амино]пентановой кислоты

К раствору (2R,4R)-4-[(3-бензилокси-3H-бензотриазол-5-карбонил)амино]-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (30,0 мг, 52,5 мкмоль) в ацетоне (1,00 мл, 13,6 ммоль) добавляли сложный хлорметиловый эфир (S)-2-трет-бутоксикарбониламино-3-метилбутановой кислоты (18,1 мг, 68,3 мкмоль) и Et3N (11,7 мкл, 84 мкмоль), и перемешивали при 60°C в течение 3 часов. Смесь концентрировали с получением желтой жидкости, которую объединяли с TFA/DCM (1/1, 1,0 мл, 6,2 ммоль) и перемешивали при комнатной температуре в течение 1 часа. Смесь концентрировали в условиях вакуума, и очищали полученную неочищенную жидкость (хроматография на колонке C18, колонка 55 г, с использованием 30-90% MeCN в воде с 0,05% TFA) с получением белого твердого вещества. Твердое вещество растворяли в THF (1,0 мл), к раствору добавляли 10% Pd/C, влажность 50% (загрузка 0,45 ммоль/г; 5,8 мг, 2,6 мкмоль), и перемешивали в атмосфере водорода в течение 1 часа. Смесь фильтровали, и концентрировали фильтрат с получением прозрачной желтой жидкости. Неочищенную жидкость очищали (препаративная хроматография на колонке C18, небольшая колонка, с использованием 30-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения в виде белого твердого трифторацетата (6,5 мг, чистота 98%). МС m/z [M+H]+ расч. для C30H32ClN5O7, 610,20; обнаружено 610,1.
Пример 2A: (2R,4R)-5-(3'-[хлорбифенил-4-ил)-2-гидрокси-4-[(5-гидрокси-1-метил-1H-пиразол-3-карбонил)амино]пентановая кислота
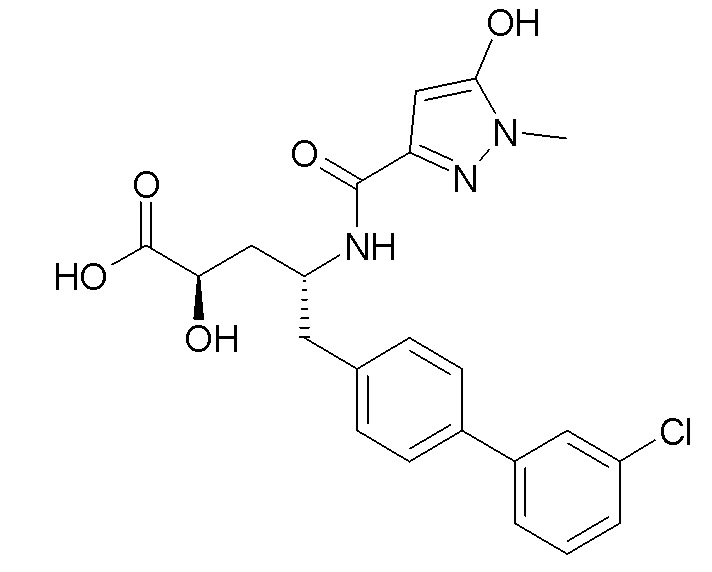
5-Гидрокси-1-метил-1H-пиразол-3-карбоновую кислоту (19,6 мг, 138 мкмоль) объединяли с HCTU (56,9 мг, 138 мкмоль) в DMF. Смесь перемешивали в течение 10 минут при комнатной температуре, после чего добавляли DIPEA (72 мкл, 413 мкмоль) и сложный этиловый эфир (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (50 мг, 0,1 ммоль). Полученную смесь перемешивали в течение ночи при комнатной температуре. Добавляли EtOH (402 мкл, 6,9 ммоль) и 1 M LiOH в воде (1,1 мл, 1,1 ммоль), и перемешивали смесь при комнатной температуре в течение 1 часа. Смесь упаривали в условиях пониженного давления и очищали методом препаративной HPLC с получением указанного в заголовке соединения (6,4 мг). МС m/z [M+H]+ расч. для C22H22ClN3O5, 444,12; обнаружено 444,4.
Пример 2B: сложный 5-метил-2-оксо-[1,3]диоксол-4-илметиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(5-гидрокси-1-метил-1H-пиразол-3-карбонил)амино]пентановой кислоты
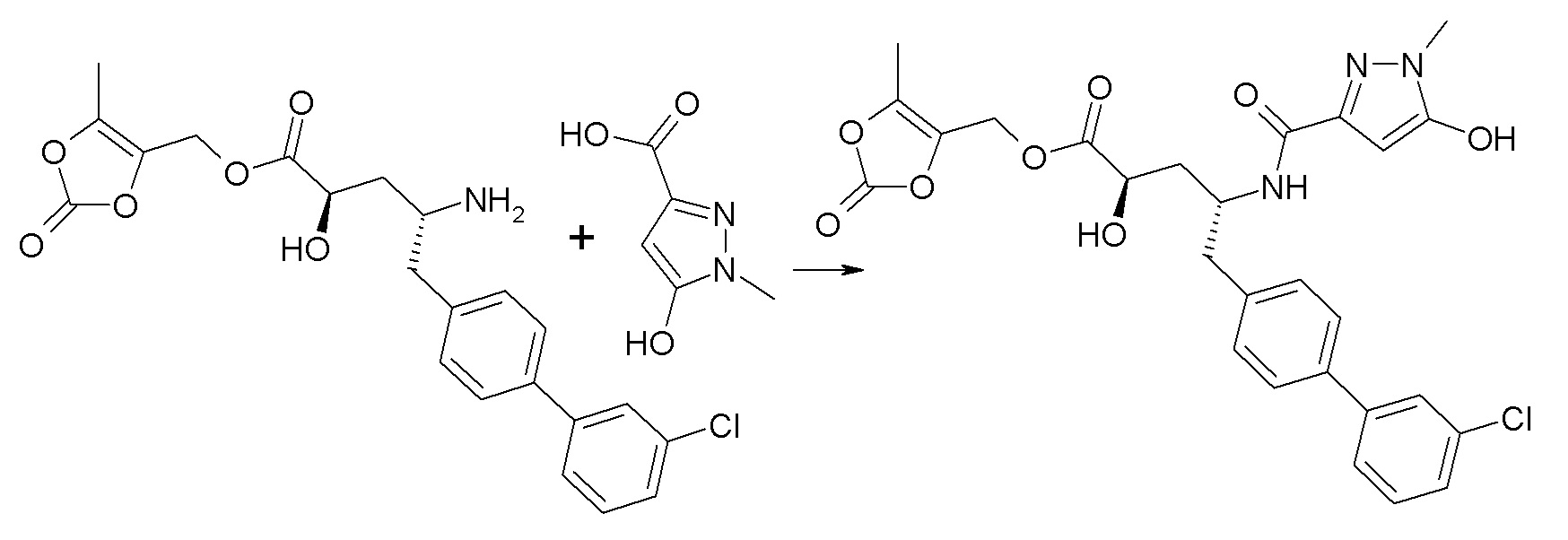
5-Гидрокси-1-метил-1H-пиразол-3-карбоновую кислоту (5,6 мг, 39 мкмоль) и HCTU (16,2 мг, 39 мкмоль) объединяли в DMF (166 мкл, 2,1 ммоль) и перемешивали при комнатной температуре в течение 15 минут. Добавляли DIPEA (19 мкл, 0,1 ммоль) и сложный 5-метил-2-оксо[1,3]диоксол-4-илметиловый эфир (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (15,4 мг, 36 мкмоль), и перемешивали полученную смесь при комнатной температуре в течение 30 минут. Растворитель удаляли в условиях вакуума, и очищали неочищенный продукт методом препаративной HPLC с получением указанного в заголовке соединения (7 мг). МС m/z [M+H]+ расч. для C27H26ClN3O8, 556,14; обнаружено 556,1.
Пример 2C: (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(5-гидрокси-1-метил-1H-пиразол-3-карбонил)амино]пентановой кислоты сложный этиловый эфир

5-Гидрокси-1-метил-1H-пиразол-3-карбоновую кислоту (122 мг, 862 мкмоль) и HATU (361 мг, 949 мкмоль) объединяли в DMF (1,0 мл, 13 ммоль), и перемешивали в течение 10 минут. Добавляли сложный этиловый эфир (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (30,0 мг, 86 мкмоль) и DIPEA (165 мкл, 949 мкмоль), и перемешивали полученную смесь при комнатной температуре в течение ночи. Затем, смесь концентрировали в условиях пониженного давления, остаток растворяли в 50% AcOH в воде (1,5 мл), фильтровали, очищали методом препаративной HPLC с обращенной фазой и лиофилизировали с получением указанного в заголовке соединения (11,1 мг). МС m/z [M+H]+ расч. для C24H26ClN3O5, 472,16; обнаружено 472,2.
Пример 2D: сложный 2,2,3,3,3-пентафторпропиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(5-гидрокси-1-метил-1H-пиразол-3-карбонил)амино]пентановой кислоты

(2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-{[5-(4-метоксибензилокси)-1-метил-1H-пиразол-3-карбонил]амино}пентановую кислоту (28,0 мг, 49,6 мкмоль) объединяли с HOBt (40,2 мг, 298 мкмоль) и EDC (53 мкл, 0,3 ммоль) в DCM (0,4 мл, 6 ммоль). Полученный раствор перемешивали в течение 15 минут, после чего добавляли 2,2,3,3,3-пентафтор-1-пропанол (39,6 мкл, 397 мкмоль). Смесь перемешивали при комнатной температуре, и отслеживали завершение реакции. Спустя 4 часа, смесь концентрировали на роторном испарителе, и очищали остаток (колонка с обращенной фазой). Остаток растворяли в EtOAc (842 мкл, 8,6 ммоль), и добавляли 10% Pd/C, влажность 50% (загрузка 0,45 ммоль/г; 31,9 мг, 14 мкмоль). Полученную смесь перемешивали в атмосфере водорода в течение 1 часа. Растворитель удаляли в условиях вакуума, и очищали остаток методом препаративной HPLC с получением указанного в заголовке соединения (4,9 мг, чистота 100%). МС m/z [M+H]+ расч. для C25H23ClF5N3O5, 576,12; обнаружено 576,1.
Пример 2E: сложный 1-трифторметилпропиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(5-гидрокси-1-метил-1H-пиразол-3-карбонил)амино]пентановой кислоты

(2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-{[5-(4-метоксибензилокси)-1-метил-1H-пиразол-3-карбонил]амино}пентановую кислоту (28,0 мг, 49,6 мкмоль) объединяли с HOBt (40,2 мг, 298 мкмоль) и EDC (53 мкл, 0,3 ммоль) в DCM (0,4 мл, 6 ммоль). Полученный раствор перемешивали в течение 15 минут, после чего добавляли 1,1,1-трифтор-2-бутанол (50,9 мг, 397 мкмоль). Смесь перемешивали при комнатной температуре, и отслеживали завершение реакции. Спустя 4 часа, смесь концентрировали на роторном испарителе, и очищали остаток (колонка с обращенной фазой). Остаток растворяли в EtOAc (842 мкл, 8,6 ммоль), и добавляли 10% Pd/C, влажность 50% (загрузка 0,45 ммоль/г; 31,9 мг, 14 мкмоль). Полученную смесь перемешивали в атмосфере водорода в течение 1 часа. Растворитель удаляли в условиях вакуума, и очищали остаток методом препаративной HPLC с получением указанного в заголовке соединения (1,3 мг, чистота 99%). МС m/z [M+H]+ расч. для C26H27ClF3N3O5, 554,16; обнаружено 554,1.
Пример 2F: сложный 2,2,3,3,3-пентафтор-1-метилпропиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(5-гидрокси-1-метил-1H-пиразол-3-карбонил)амино]пентановой кислоты
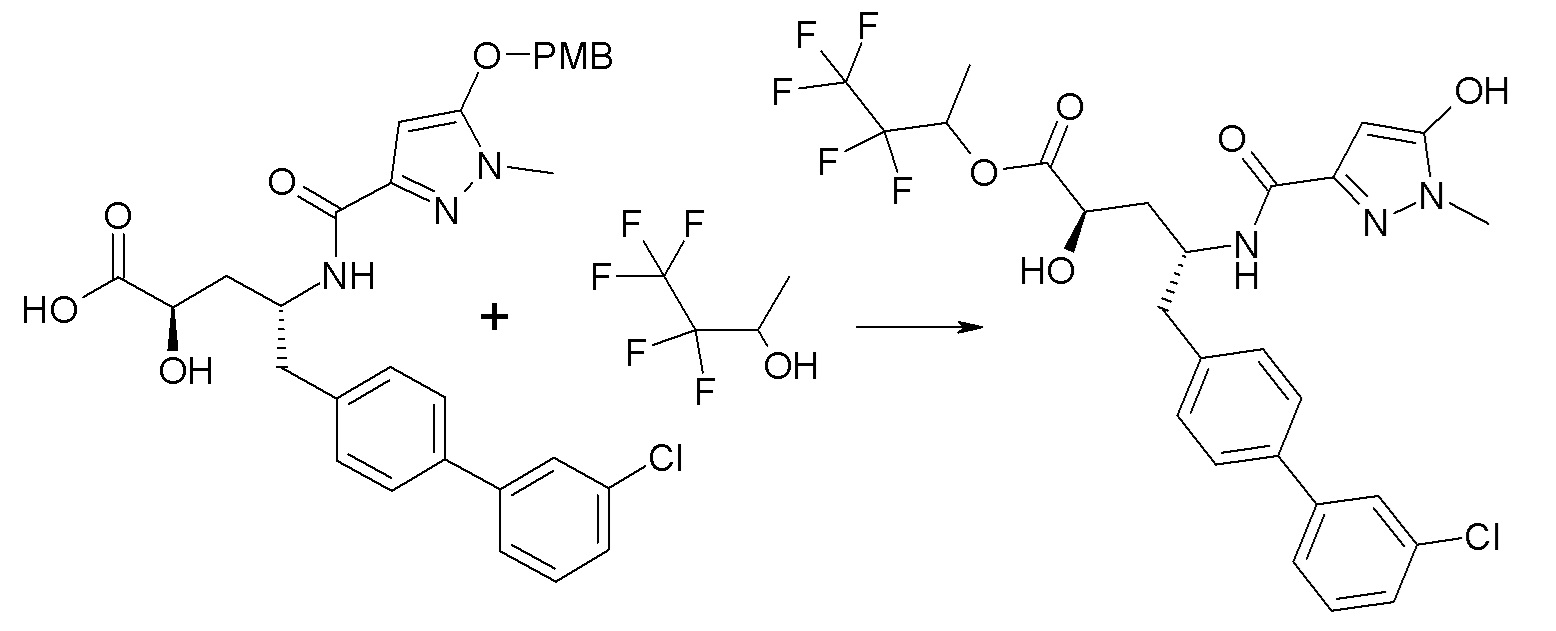
(2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-{[5-(4-метоксибензилокси)-1-метил-1H-пиразол-3-карбонил]амино}пентановую кислоту (28,0 мг, 49,6 мкмоль) объединяли с HOBt (40,2 мг, 298 мкмоль) и EDC (53 мкл, 0,3 ммоль) в DCM (0,4 мл, 6 ммоль). Полученный раствор перемешивали в течение 15 минут, после чего добавляли 3,3,4,4,4-пентафтор-2-бутанол (65,2 мг, 397 мкмоль). Смесь перемешивали при комнатной температуре, и отслеживали завершение реакции. Спустя 4 часа, смесь концентрировали на роторном испарителе, и очищали остаток (колонка с обращенной фазой). Остаток растворяли в EtOAc (842 мкл, 8,6 ммоль), и добавляли 10% Pd/C, влажность 50% (загрузка 0,45 ммоль/г; 31,9 мг, 14 мкмоль). Полученную смесь перемешивали в атмосфере водорода в течение 1 часа. Растворитель удаляли в условиях вакуума, и очищали остаток методом препаративной HPLC с получением указанного в заголовке соединения (1,8 мг, чистота 98%). МС m/z [M+H]+ расч. для C26H25ClF5N3O5, 590,14; обнаружено 590,1.
Пример 2G: сложный бензиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(5-гидрокси-1-метил-1H-пиразол-3-карбонил)амино]пентановой кислоты
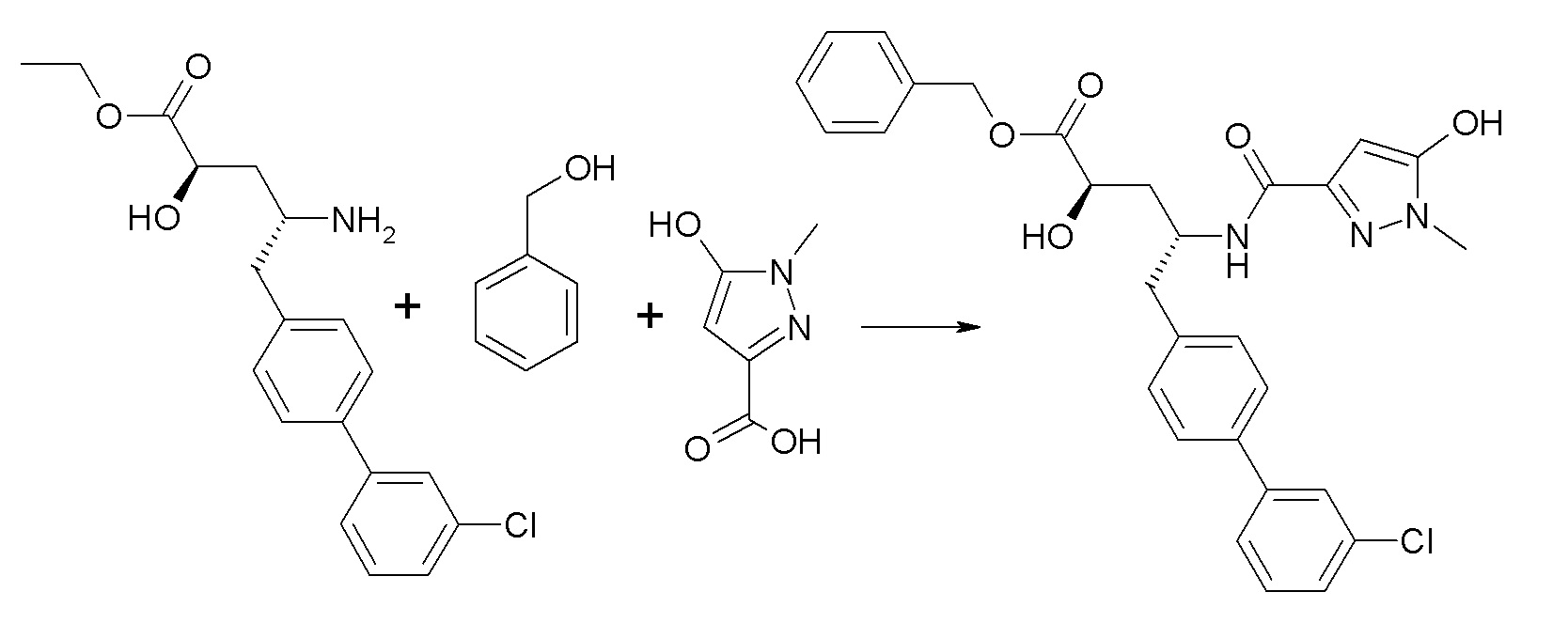
Смесь сложного этилового эфира (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (150,0 мг, 431 мкмоль), бензилового спирта (446,2 мкл, 4,3 ммоль) и 4,0 M HCl в 1,4-диоксане (431 мкл, 1,7 ммоль) перемешивали при комнатной температуре в течение ночи, а затем при 50°C в течение 1 часа. Затем, смесь концентрировали в условиях пониженного давления. 5-Гидрокси-1-метил-1H-пиразол-3-карбоновую кислоту (30,6 мг, 216 мкмоль) и HATU (98,4 мг, 259 мкмоль) перемешивали в DMF (0,5 мл, 5 ммоль) в течение 10 минут, а затем добавляли к концентрированной смеси вместе с DIPEA (113 мкл, 647 мкмоль). Полученную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали в условиях пониженного давления. Остаток растворяли в 50% AcOH в воде (5 мл), фильтровали, и очищали методом препаративной HPLC с обращенной фазой с получением указанного в заголовке соединения (81,4 мг).
Пример 2H: сложный бутирилоксиметиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(5-гидрокси-1-метил-1H-пиразол-3-карбонил)амино]пентановой кислоты

К раствору сложного метилового эфира 5-гидрокси-1-метил-1H-пиразол-3-карбоновой кислоты (3 г, 19,2 ммоль) в DMF (15 мл) при комнатной температуре добавляли K2CO3 (2,7 г, 19,2 ммоль). Спустя 10 мин, добавляли аллилбромид (2,3 г, 19,2 ммоль), и перемешивали полученную смесь при комнатной температуре в течение 2 часов. Добавляли воду (150 мл), смесь экстрагировали EtOAc (3×50 мл), объединенные органические слои промывали насыщенным водным NaCl (50 мл) и сушили над безводным Na2SO4. Затем, раствор упаривали, и очищали остаток методом хроматографии на силикагеле (силикагель: 200-300 меш, элюирование PE/EtOAc=10/1→5/1→1/1) с получением соединения 1 в виде белого твердого вещества (3 г). ЖХ-МС: 197[M+H]+.
К раствору соединения 1 (3 г, 15 ммоль) в THF (30 мл) добавляли раствор LiOH (550 мг, 23 ммоль) в воде (15 мл). Смесь перемешивали при комнатной температуре в течение 1 часа, а затем концентрировали в условиях вакуума. К остатку добавляли воду (20 мл), и промывали смесь EtOAc (20 мл). Водный слой подкисляли до pH=3 добавлением 1н водной HCl и экстрагировали EtOAc (3×30 мл). Объединенные органические слои промывали насыщенным водным NaCl (30 мл), и сушили над безводным Na2SO4. Затем, раствор упаривали с получением соединения 2 в виде белого твердого вещества (2,3 г). ЖХ-МС: 183[M+H]+.
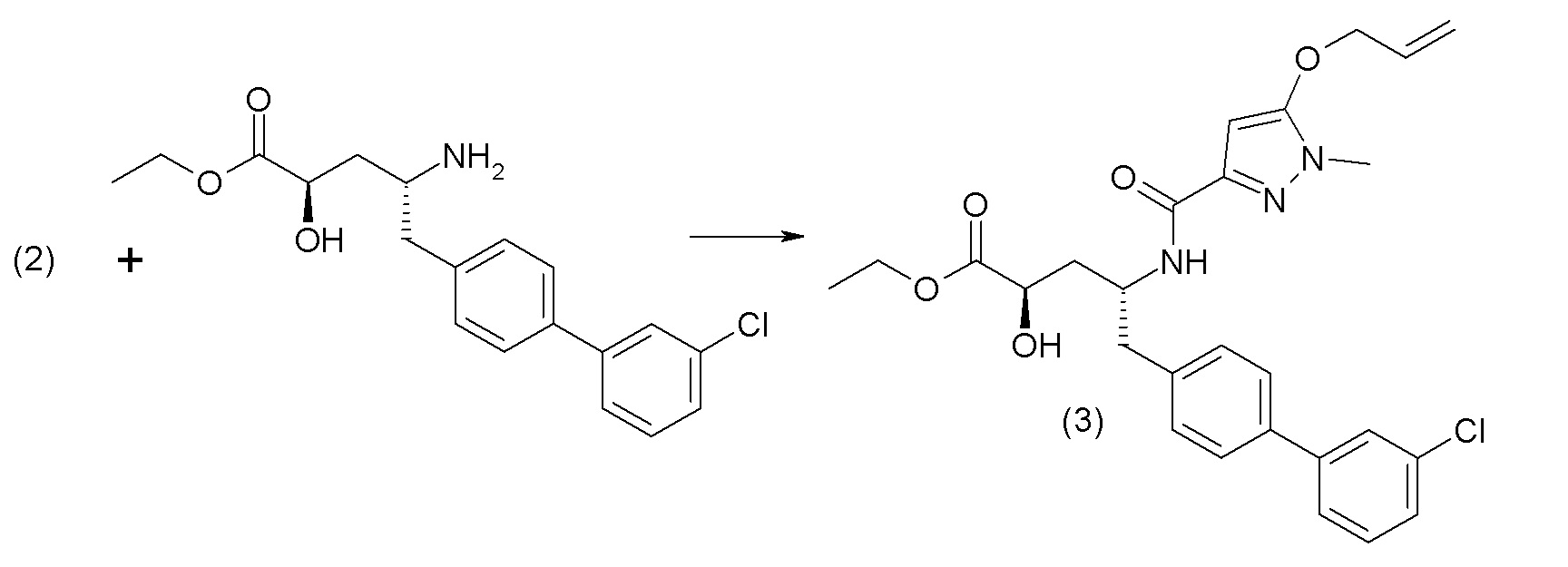
К раствору сложного этилового эфира (2R,4R)-4-амино-2-гидрокси-5-(3'-метилбифенил-4-ил)пентановой кислоты (13 г, 7,1 ммоль) и соединения 2 (2,7 г, 7,1 ммоль) в DMF (30 мл) при 0°C добавляли PyBOP (3,7 г, 7,1 ммоль) и DIPEA (1,8 г, 14,2 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли воду (200 мл), смесь экстрагировали EtOAc (3×100 мл), объединенные органические слои промывали насыщенным водным NaCl (100 мл) и сушили над безводным Na2SO4. Смесь концентрировали, и очищали остаток методом хроматографии на силикагеле (силикагель: 200-300 меш; элюирование PE/EtOAc=10/1→5/1→1/1) с получением соединения 3 в виде светло-желтого твердого вещества (2,4 г). ЖХ-МС: 512[M+H]+.

К раствору соединения 3 (2,4 г, 4,7 ммоль) в THF (20 мл) и воде (10 мл) при комнатной температуре добавляли LiOH (169 мг, 7 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 1 часа. Смесь концентрировали, добавляли воду (50 мл), и промывали смесь EtOAc (2×20 мл). Водный слой подкисляли до pH=3 добавлением 1н водной HCl и экстрагировали EtOAc (3×50 мл). Объединенные органические слои промывали насыщенным водным NaCl (50 мл) и сушили над безводным Na2SO4. Затем, раствор упаривали с получением соединения 4 в виде желтого твердого вещества (2 г). ЖХ-МС: 484[M+H]+.
Суспензию соединения 4 (400 мг, 830 мкмоль), NaI (248 мг, 1,7 ммоль) и лутидина (173 мг, 1,7 ммоль) в хлорметилбутирате (2 мл) перемешивали при 50°C в течение 5 часов. После охлаждения до комнатной температуры, смесь разбавляли водой (20 мл) и экстрагировали EtOAc (2×20 мл). Объединенные органические слои промывали насыщенным водным NaCl (40 мл), сушили над безводным Na2SO4, концентрировали и очищали методом хроматографии на силикагеле (силикагель: 200-300 меш; элюирование PE/EtOAc от 5/1 до 2/1) с получением соединения 5 в виде светло-желтого твердого вещества (200 мг). ЖХ-МС: 584 [M+H]+.

К раствору соединения 5 (140 мг, 240 мкмоль) в DCM (5 мл) добавляли Pd(PPh3)4 (83 мг, 70 мкмоль), Et3SiH (83 мг, 720 мкмоль) и AcOH (43 мг, 720 мкмоль). Смесь перемешивали при комнатной температуре в течение 4 часов и концентрировали. Остаток очищали методом препаративной HPLC [Daisogel-C18, 150×21,2 мм, 5 мкм; MeCN в H2O (0,1% TFA) от 60% до 80%] с получением указанного в заголовке соединения в виде желтого масла (20 мг). ЖХ-МС: 544[M+H]+.1H-ЯМР (CD3OD, 400 Гц): δ 0,93 (т, 3H), 1,62 (кв., 2H), 2,06-2,13 (м, 2H), 2,31 (т, 2H), 2,97-2,99 (м, 2H), 3,65 (с, 3H), 4,32 (м, 1H), 4,51 (м, 1H), 5,75 (дд, 2H), 7,35-7,41 (м, 4H), 7,53-7,60 (м, 4H).
Пример 2I: сложный (S)-2-амино-3-метилбутирилоксиметиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(5-гидрокси-1-метил-1H-пиразол-3-карбонил)амино]пентановой кислоты
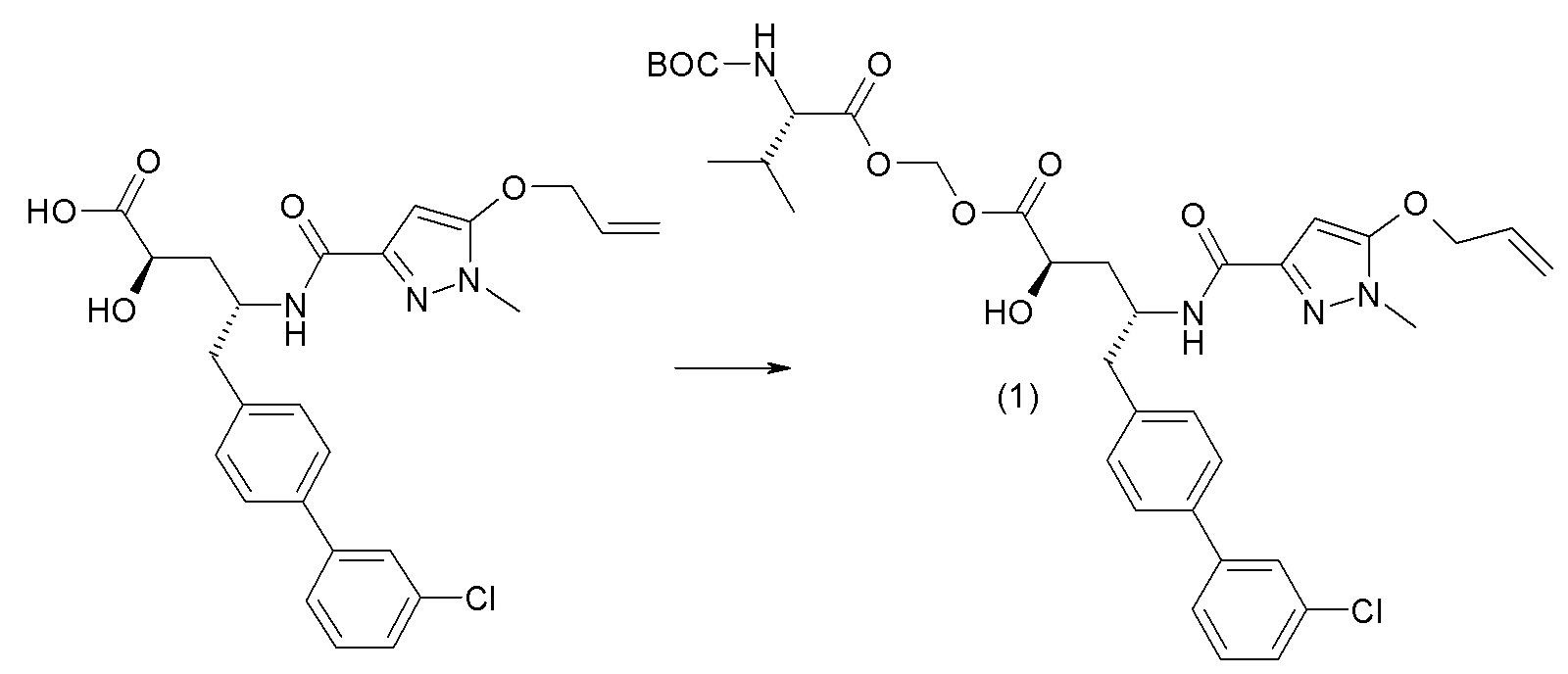
Суспензию (2R,4R)-4-[(5-аллилокси-1-метил-1H-пиразол-3-карбонил)амино]-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (400 мг, 830 мкмоль), NaI (248 мг, 1,7 ммоль) и лутидина (173 мг, 1,7 ммоль) в сложном хлорметиловом эфире (S)-2-трет-бутоксикарбониламино-3-метилбутановой кислоты (2 мл) перемешивали при 50°C в течение 2 часов. После охлаждения до комнатной температуры, смесь разбавляли водой (20 мл) и экстрагировали EtOAc (2×20 мл). Объединенные органические слои промывали насыщенным водным NaCl (40 мл), сушили над безводным Na2SO4, концентрировали и очищали методом хроматографии на силикагеле (силикагель: 200-300 меш; элюирование PE/EtOAc от 5/1 до 2/1) с получением соединения 1 в виде светло-желтого твердого вещества (290 мг). ЖХ-МС: 713 [M+H]+.

К раствору соединения 1 (290 мг, 0,4 ммоль) в THF (5 мл) добавляли Pd(PPh3)4 (70 мг, 60 мкмоль) и 1,3-диметилбарбитуровую кислоту (312 мг, 2 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали. Остаток растворяли в EtOAc (30 мл), промывали 5% NaHCO3 (2×15 мл), сушили над безводным Na2SO4 и концентрировали с получением неочищенного соединения 2 в виде коричневого твердого вещества (220 мг). ЖХ-МС:673[M+H]+.
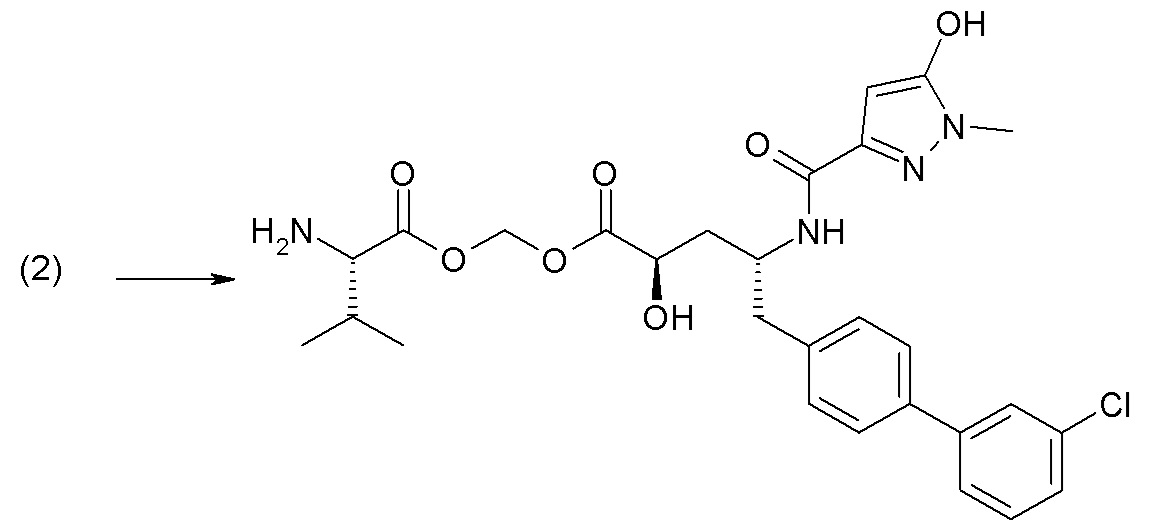
К 1 M раствору HCl (g) в диоксане (5 мл) добавляли соединение 2 (220 мг, 330 мкмоль). Смесь перемешивали при комнатной температуре в течение 1 часа и концентрировали. Остаток очищали методом препаративной HPLC [Daisogel-C18, 150×21,2 мм, 5 мкм; MeCN в H2O (0,1% TFA) от 35% до 45%] с получением указанного в заголовке соединения в виде желтого твердого вещества (100 мг). ЖХ-МС: 573[M+H]+.1H-ЯМР (ДМСО-d6, 400 Гц): δ 0,97 (д, 6H), 1,82-1,85 (м, 1H), 1,98-2,01 (м, 1H), 2,16 2,19(м, 1H), 2,85-2,88 (м, 2 H), 3,58 (с, 3H), 3,99-4,01 (м, 1H), 4,18-4,20 (м, 1 H), 4,23-4,25 (м, 1 H), 5,84 (дд, 2 H), 7,28-7,47 (м, 4H), 7,60-7,69 (м, 4H), 8,01 (с, 1H), 8,55 (шир., 2 H).
Пример 3A: (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(5-гидрокси-1-пиридин-2-ил-1H-пиразол-3-карбонил)амино]пентановая кислота
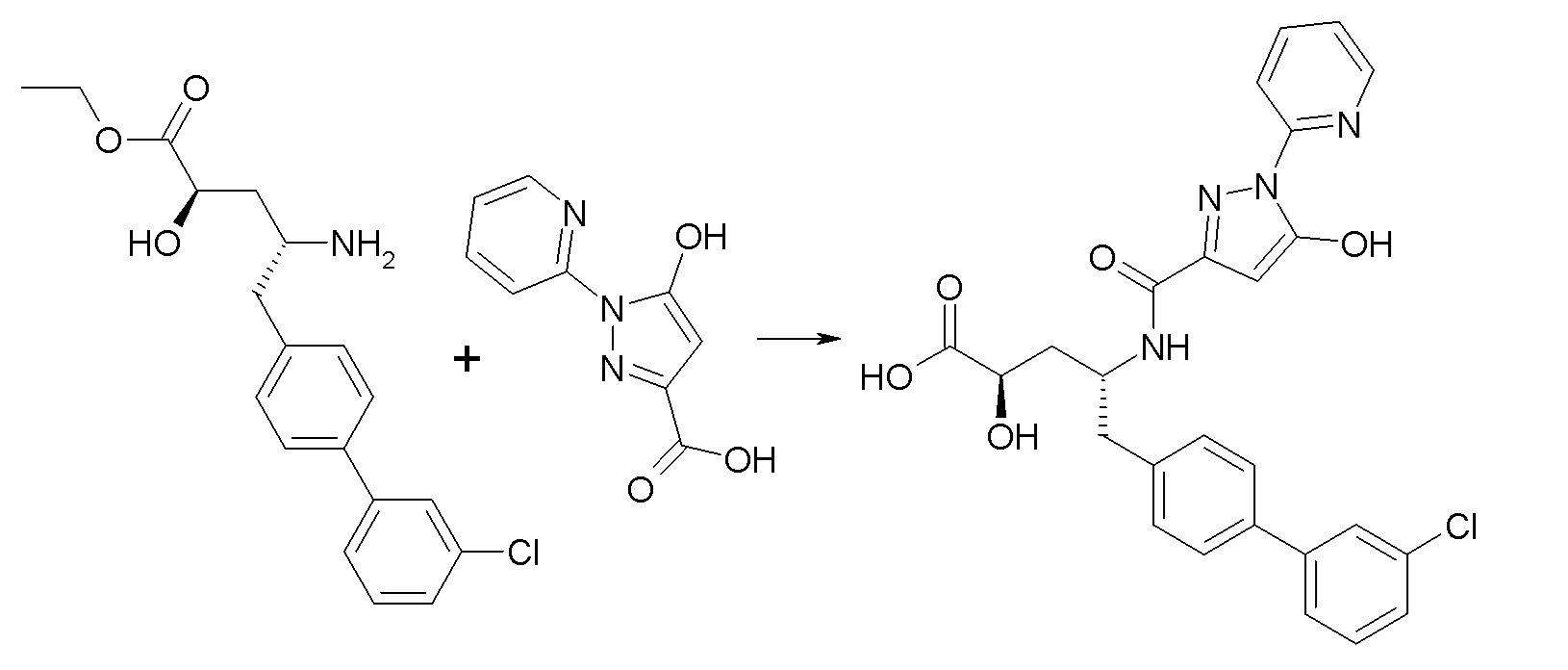
5-Гидрокси-1-пиридин-2-ил-1H-пиразол-3-карбоновую кислоту (59,0 мг, 287 мкмоль) и HCTU (131 мг, 316 мкмоль) объединяли в DMF (1,3 мл, 17,2 ммоль) и перемешивали при комнатной температуре в течение 15 минут. Добавляли сложный 5-метил-2-оксо[1,3]диоксол-4-илметиловый эфир (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (100 мг, 0,3 ммоль) и DIPEA (150 мкл, 862 мкмоль), и перемешивали полученную смесь при комнатной температуре в течение 15 минут. Реакционную смесь упаривали в условиях пониженного давления. Остаток растворяли в EtOH, и добавляли раствор 1 M LiOH в воде (1,4 мл, 1,4 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 1 часа, а затем упаривали в условиях пониженного давления. Остаток очищали методом препаративной HPLC с получением указанного в заголовке соединения (85 мг). МС m/z [M+H]+ расч. для C26H23ClN4O5, 507,14; обнаружено 507,1.
Пример 3B: сложный 5-метил-2-оксо-[1,3]диоксол-4-илметиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(5-гидрокси-1-пиридин-2-ил-1H-пиразол-3-карбонил)амино]пентановой кислоты
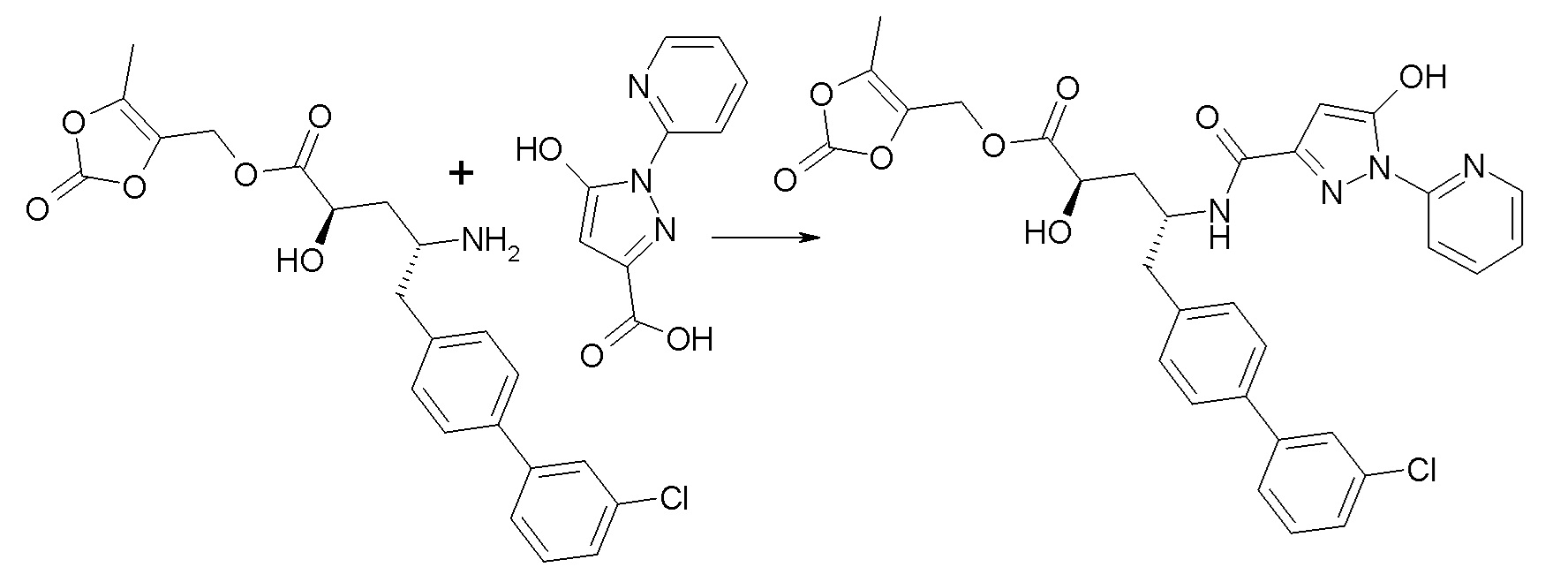
5-Гидрокси-1-пиридин-2-ил-1H-пиразол-3-карбоновую кислоту (8,1 мг, 39 мкмоль) и HCTU (16,2 мг, 39 мкмоль) объединяли в DMF (166 мкл, 2,1 ммоль), и перемешивали при комнатной температуре в течение 15 минут. Добавляли DIPEA (19 мкл, 0,1 ммоль) и сложный 5-метил-2-оксо[1,3]диоксол-4-илметиловый эфир (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (15,4 мг, 36 мкмоль), и перемешивали полученную смесь при комнатной температуре в течение 30 минут. Растворитель удаляли в условиях вакуума, и очищали неочищенный продукт методом препаративной HPLC с получением указанного в заголовке соединения (18 мг). МС m/z [M+H]+ расч. для C31H27ClN4O8, 619,15; обнаружено 619,1.
Пример 3C: сложный этиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(5-гидрокси-1-пиридин-2-ил-1H-пиразол-3-карбонил)амино]пентановой кислоты

5-Гидрокси-1-пиридин-2-ил-1H-пиразол-3-карбоновую кислоту (23,6 мг, 115 мкмоль) и HATU (52,5 мг, 138 мкмоль) перемешивали в N,N-диметилацетамиде (1,0 мл, 11 ммоль) в течение 10 минут. Добавляли сложный этиловый эфир (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (40,0 мг, 115 мкмоль) и DIPEA (60,1 мкл, 345 мкмоль), и перемешивали полученную смесь при комнатной температуре в течение 2 часов. Затем, смесь концентрировали в условиях пониженного давления, остаток растворяли в EtOAc (20 мл), органический слой промывали водой (2×5 мл), сушили над MgSO4 и концентрировали. Половину остатка растворяли в 50% AcOH в воде (1,5 мл), фильтровали и очищали методом препаративной HPLC с обращенной фазой с получением указанного в заголовке соединения в виде трифторацетата (6,1 мг). МС m/z [M+H]+ расч. для C28H27ClN4O5, 535,17; обнаружено 535,4.
Пример 3D: сложный изобутиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(5-гидрокси-1-пиридин-2-ил-1H-пиразол-3-карбонил)амино]пентановой кислоты

5-Гидрокси-1-пиридин-2-ил-1H-пиразол-3-карбоновую кислоту (23,6 мг, 115 мкмоль) и HATU (52,5 мг, 138 мкмоль) перемешивали в N,N-диметилацетамиде (1,0 мл, 11 ммоль) в течение 10 минут. Добавляли сложный этиловый эфир (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (40,0 мг, 115 мкмоль) и DIPEA (60,1 мкл, 345 мкмоль), и перемешивали полученную смесь при комнатной температуре в течение 2 часов. Затем, смесь концентрировали в условиях пониженного давления, остаток растворяли в EtOAc (20 мл), органический слой промывали водой (2×5 мл), сушили над MgSO4 и концентрировали. Половину остатка объединяли с изобутиловым спиртом (0,5 мл, 5 ммоль) и 4,0 M HCl в 1,4-диоксане (115 мкл, 460 мкмоль). Смесь перемешивали при комнатной температуре в течение ночи, концентрировали в условиях пониженного давления, остаток растворяли в 50% AcOH в воде (1,5 мл), фильтровали и очищали методом препаративной HPLC с обращенной фазой с получением указанного в заголовке соединения в виде трифторацетата (9,1 мг). МС m/z [M+H]+ расч. для C30H31ClN4O5, 563,20; обнаружено 563,4.
Пример 3E: сложный бензиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(5-гидрокси-1-пиридин-2-ил-1H-пиразол-3-карбонил)амино]пентановой кислоты

Смесь сложного этилового эфира (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (150,0 мг, 431 мкмоль), бензилового спирта (446,2 мкл, 4,3 ммоль) и 4,0 M HCl в 1,4-диоксане (431 мкл, 1,7 ммоль) перемешивали при комнатной температуре в течение ночи, а затем при 50°C в течение 1 часа. Затем, смесь концентрировали в условиях пониженного давления. 5-Гидрокси-1-пиридин-2-ил-1H-пиразол-3-карбоновую кислоту (44,2 мг, 216 мкмоль) и HATU (98,4 мг, 259 мкмоль) перемешивали в DMF (0,5 мл, 5 ммоль) в течение 10 минут, а затем добавляли к концентрированной смеси вместе с DIPEA (113 мкл, 647 мкмоль). Полученную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали в условиях пониженного давления. Остаток растворяли в 50% AcOH в воде (5 мл), фильтровали, и очищали методом препаративной HPLC с обращенной фазой с получением указанного в заголовке соединения (19,5 мг).
Пример 3F: (2R,4R)-2-((S)-2-амино-3-метилбутирилокси)-5-(3'-хлорбифенил-4-ил)-4-[(5-гидрокси-1-пиридин-2-ил-1H-пиразол-3-карбонил)амино]пентановая кислота

Указанное в заголовке соединение также может быть получено с использованием методик, описанных в настоящем документе.
Пример 3G: (2R,4R)-4-{[5-((S)-2-амино-3-метилбутирилоксиметокси)-1-пиридин-2-ил-1H-пиразол-3-карбонил]амино}-5-(3'-хлор-бифенил-4-ил)-2-гидроксипентановая кислота

Указанное в заголовке соединение также может быть получено с использованием методик, описанных в настоящем документе.
Пример 3H: сложный (S)-2-метоксикарбониламино-3-метилбутирилоксиметиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(5-гидрокси-1-пиридин-2-ил-1H-пиразол-3-карбонил)амино]пентановой кислоты
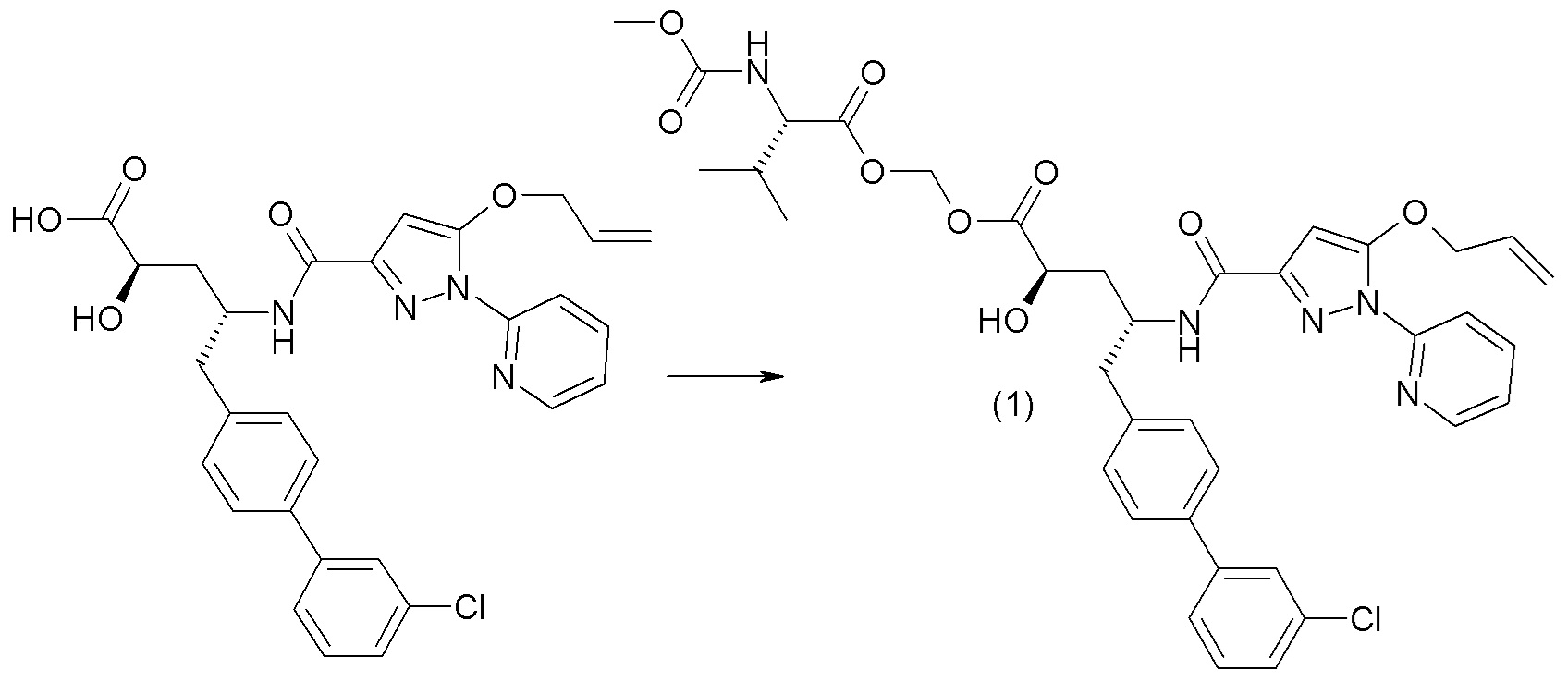
К раствору (2R,4R)-4-[(5-аллилокси-1-пиридин-2-ил-1H-пиразол-3-карбонил)амино]-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (220 мг, 0,4 ммоль) и сложного хлорметилового эфира ((S)-2-метоксикарбониламино-3-метилбутановой кислоты (180 мг, 0,8 ммоль) в DMF (2 мл) добавляли 2,6-лутидин (130 мг, 1,2 ммоль) и NaI (60 мг, 0,4 ммоль). После перемешивания при комнатной температуре в течение 24 часов, смесь разбавляли водой (20 мл) и экстрагировали EtOAc (2×20 мл). Объединенные органические слои промывали насыщенным водным NaCl (2×70 мл), сушили над безводным Na2SO4, фильтровали, концентрировали и очищали методом препаративной TLC (PE/EtOAc=1/1) с получением соединения 1 в виде желтого твердого вещества (160 мг). ЖХ-МС: 734[M+H]+.

Смесь соединения 1 (140 мг, 190 мкмоль), Et3SiH (88 мг, 760 мкмоль), Pd(PPh3)4 (22 мг, 20 мкмоль) и AcOH (22 мг, 0,38 ммоль) в DCM (2 мл) перемешивали при комнатной температуре в течение 4 часов. Растворитель удаляли, и очищали остаток методом препаративной HPLC (Gemini-C18 150×21,2 мм, 5 мкм; MeCN в H2O (0,1% TFA) от 50% до 70%) с получением указанного в заголовке соединения в виде белого твердого вещества (15 мг). ЖХ-МС: 694[M+H]+.1H ЯМР (400 МГц, CD3OD) δ 8,47 (д, J=5,0 Гц, 1H), 8,06 (д, J=8,5 Гц, 2H), 7,55 (дд, J=19,3, 11,4 Гц, 4H), 7,37 (дт, J=28,2, 6,2 Гц, 5H), 5,85 (д, J=5,7 Гц, 1H), 5,75 (д, J=5,7 Гц, 1H), 4,58 (м, 1H), 4,36 (т, J=5,8 Гц, 1H), 4,04 (д, J=5,9 Гц, 1H), 3,65 (с, 3H), 3,04 (д, J=6,8 Гц, 2H), 2,20 (дд, J=12,5, 7,1 Гц, 1H), 2,08 (м, 2H), 0,90 (дд, J=10,8, 6,9 Гц, 6H).
Пример 4A: сложный этиловый эфир (2R,4R)-4-[(5-ацетил-1H-пиразол-3-карбонил)амино]-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксипентановой кислоты

К перемешанному раствору 3-ацетил-1H-пиразол-5-карбоновой кислоты (50,9 мг, 330 мкмоль), HATU (125 мг, 330 мкмоль) и DIPEA (104 мкл, 0,6 ммоль) в DMF (0,6 мл, 7,7 ммоль) добавляли сложный этиловый эфир (2R,4R)-4-амино-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксипентановой кислоты (110 мг, 300 мкмоль). Полученную смесь перемешивали при комнатной температуре до завершения реакции. Смесь разбавляли DCM и промывали насыщенным водным NaHCO3. Органические фазы сушили над Na2SO4, и удаляли растворитель. Затем, половину вещества очищали методом препаративной HPLC с получением указанного в заголовке соединения (22 мг) в виде трифторацетата. МС m/z [M+H]+ расч. для C25H25ClFN3O5, 502,15; обнаружено 502,1.
Пример 4B: (2R,4R)-4-[(5-ацетил-1H-пиразол-3-карбонил)амино]-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксипентановая кислота

Сложный этиловый эфир (2R,4R)-4-[(5-ацетил-1H-пиразол-3-карбонил)амино]-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксипентановой кислоты (75 мг, 150 мкмоль) в течение ночи перемешивали при комнатной температуре с LiOH (10,8 мг, 450 мкмоль) в воде (450 мкл, 25 ммоль) и EtOH (450 мкл, 7,7 ммоль). Растворитель удаляли, и очищали оставшееся вещество методом препаративной HPLC с получением указанного в заголовке соединения (11,6 мг) в виде трифторацетата.
Пример 5A: сложный этиловый эфир (2R,4R)-4-[(5-ацетил-1H-пиразол-3-карбонил)амино]-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты

К перемешанному раствору 3-ацетил-1H-пиразол-5-карбоновой кислоты (50,9 мг, 330 мкмоль), HATU (125 мг, 330 мкмоль) и DIPEA (104 мкл, 0,6 ммоль) в DMF (0,6 мл, 7,7 ммоль) добавляли сложный этиловый эфир (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (104 мг, 300 мкмоль). Полученную смесь перемешивали при комнатной температуре до завершения реакции. Смесь разбавляли DCM и промывали насыщенным водным NaHCO3. Органические фазы сушили над Na2SO4, и удаляли растворитель. Затем, половину вещества очищали методом препаративной HPLC с получением указанного в заголовке соединения (20 мг) в виде трифторацетата. МС m/z [M+H]+ расч. для C25H26ClN3O5, 484,16; обнаружено 484,1.
Пример 5B: (2R,4R)-4-[(5-ацетил-1H-пиразол-3-карбонил)амино]-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановая кислота
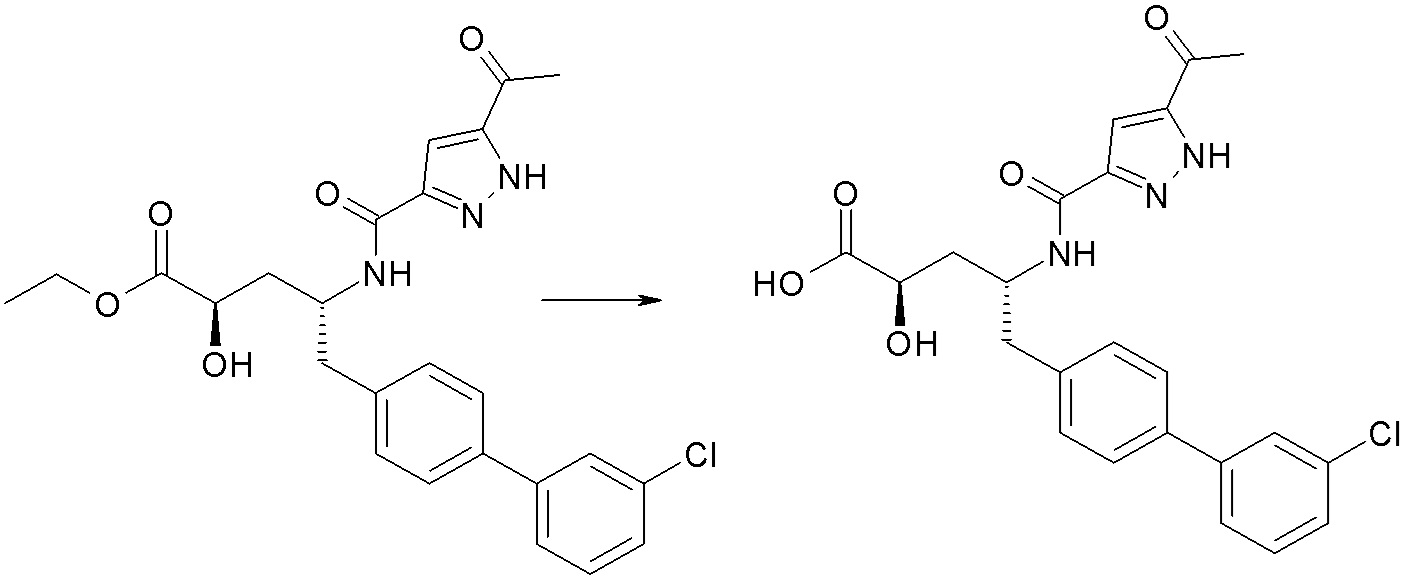
Сложный этиловый эфир (2R,4R)-4-[(5-ацетил-1H-пиразол-3-карбонил)амино]-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (72 мг, 150 мкмоль) в течение ночи перемешивали при комнатной температуре с LiOH (10,8 мг, 450 мкмоль) в воде (450 мкл, 25 ммоль) и EtOH (450 мкл, 7,7 ммоль). Растворитель удаляли, и очищали оставшееся вещество методом препаративной HPLC с получением указанного в заголовке соединения (11,5 мг) в виде трифторацетата.
Пример 6A: (2R,4R)-5-(3'-хлорбифенил-4-ил)-4-{[3-(2-фторфенил)изоксазол-5-карбонил]амино}-2-гидроксипентановая кислота
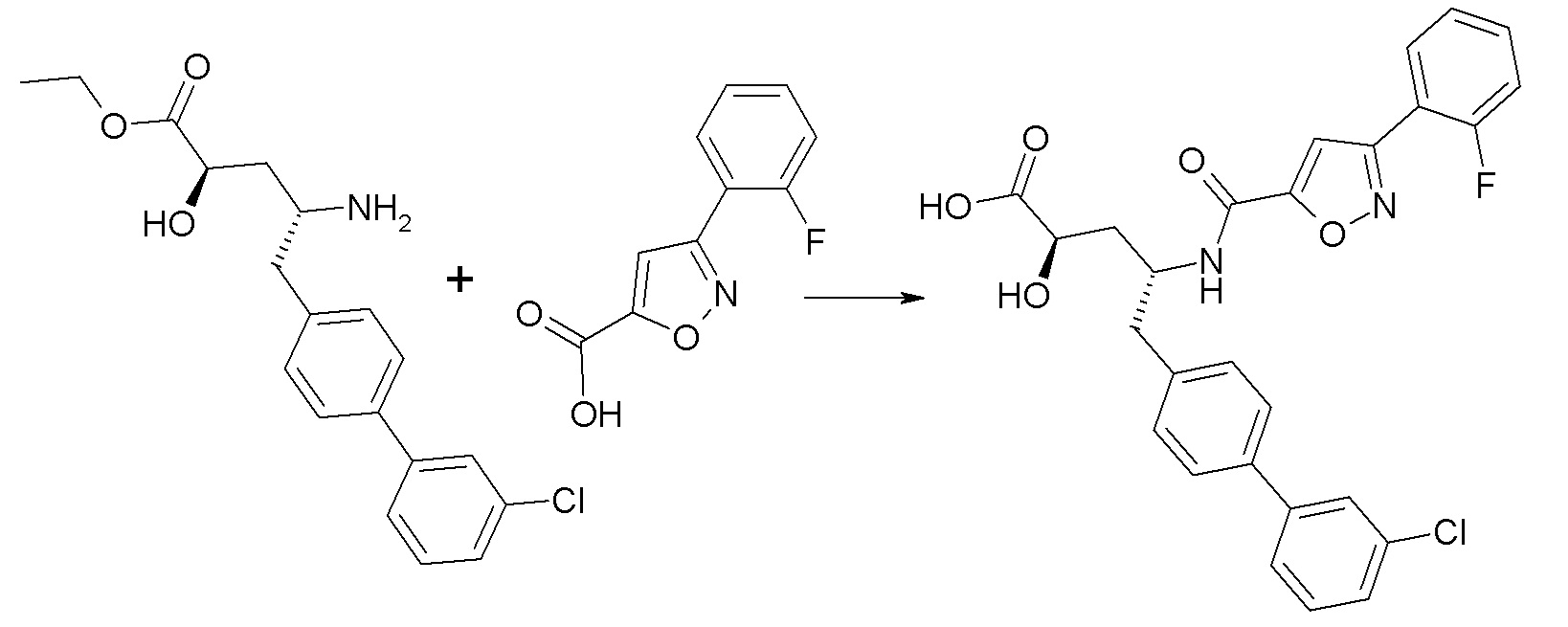
К смеси 3-(2-фторфенил)изоксазол-5-карбоновой кислоты (14,3 мг, 69 мкмоль) и HATU (26,2 мг, 69 мкмоль) в DMF (0,5 мл, 6 ммоль) при комнатной температуре добавляли сложный этиловый эфир (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (20,0 мг, 58 мкмоль) и DIPEA (20,0 мкл, 115 мкмоль). Смесь перемешивали при комнатной температуре в течение ночи. Добавляли MeOH (2,0 мл, 49 ммоль), воду (1,0 мл, 56 ммоль) и LiOH моногидрат (9,7 мг, 230 мкмоль), и перемешивали полученную смесь при комнатной температуре в течение 1 часа. Полученные твердые вещества фильтровали и промывали MeCN и водой с получением указанного в заголовке соединения (17,1 мг). МС m/z [M+H]+ расч. для C27H22ClFN2O5, 509,12; обнаружено 509.
Пример 6B: сложный этоксикарбонилоксиметиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-4-{[3-(2-фторфенил)изоксазол-5-карбонил]амино}-2-гидроксипентановой кислоты

Раствор сложного этилового эфира (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (гидрохлорид; 1,5 г, 4 ммоль), 3-(2-фторфенил)изоксазол-5-карбоновой кислоты (814 мг, 4 ммоль), HOBt (1,1 г, 8 ммоль), EDC (1,5 г, 8 ммоль) и DIPEA (1,6 г, 12 ммоль) в DMF (50 мл) перемешивали в течение 15 часов при комнатной температуре. Реакционную смесь гасили добавлением воды (50 мл) и экстрагировали EtOAc (3×60 мл), органические слои собирали и концентрировали. Остаток очищали методом колоночной хроматографии (петролейный эфир/EtOAc=5/1) с получением соединения 1 (1,1 г). ЖХ-МС: 537 [M+H]+.
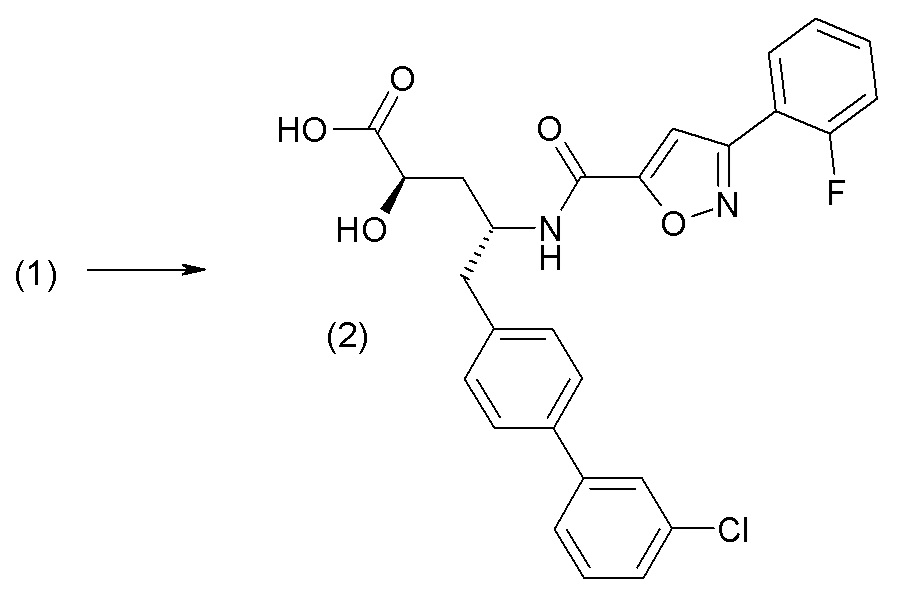
Суспензию соединения 1 (200 мг, 0,37 ммоль) и моногидрата LiOH (78 мг, 1,9 ммоль) в THF/MeOH/H2O (2/2/1, 20 мл) перемешивали в течение 3 часов при комнатной температуре. Смесь концентрировали для удаления органических растворителей, и корректировали значение pH остатка до pH 6 добавлением водн. HCl (1 M). Смесь фильтровали с получением соединения 2 в виде белого твердого вещества (120 мг). ЖХ-МС: 509 [M+H]+.

Суспензию соединения 2 (150 мг, 0,3 ммоль), хлорметилэтилкарбоната (84 мг, 0,6 ммоль), NaI (15 мг, 0,6 ммоль) и пиридина (96 мг, 1,2 ммоль) в DMF (20 мл) перемешивали в течение 15 часов при 30ºC. Реакционную смесь гасили добавлением воды (15 мл) и экстрагировали EtOAc (3×30 мл). Объединенные органические слои концентрировали в условиях вакуума и очищали методом колоночной хроматографии (петролейный эфир/EtOAc=3/1) с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (10 мг). ЖХ-МС: 611 [M+H]+.1H ЯМР (400 МГц, CDCl3): δ 1,33 (т, J=7,2 Гц, 3H), 2,03-2,08 (м, 1H), 2,30-2,34 (м, 1H), 3,05 (д, J=6,4 Гц 2H), 4,26 (кв., J=7,2 Гц, 2H), 4,48 (шир., 1H), 4,75 (шир., 1H), 5,68-5,70 (м, 1H), 5,81-5,83 (м, 1H), 6,88 (д, J=8,8 Гц, 1H), 7,28-7,34 (м, 7H), 7,42-7,53 (м, 4H), 7,96-7,99 (т, J=6,8 Гц, 1H).
Пример 6C: сложный изопропоксикарбонилоксиметиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-4-{[3-(2-фторфенил)изоксазол-5-карбонил]амино}-2-гидроксипентановой кислоты

К перемешанному раствору (2R,4R)-4-(трет-бутоксикарбониламино)-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (2,4 г, 5,7 ммоль) в DMF (50 мл) добавляли хлорметилизопропилкарбонат (1,3 г, 8,6 ммоль). К этой смеси добавляли NaI (1,71 г, 11,4 ммоль) и DIPEA (2,2 г, 17,1 ммоль), и перемешивали полученную смесь при комнатной температуре в течение ночи. Добавляли воду (100 мл), и экстрагировали смесь EtOAc (3×80 мл). Объединенные органические слои промывали насыщенным водным NaCl (2×100 мл) и водой (2×100 мл), сушили над безводным Na2SO4 и концентрировали в условиях вакуума. Продукт очищали методом колоночной хроматографии (петролейный эфир/EtOAc, 15/1~6/1) с получением соединения 1 в виде белого твердого вещества (574 мг). ЖХ-МС: 558 [M+Na]+.

Соединение 1 (574 мг, 1,1 ммоль) растворяли в растворе HCl в диоксане (1,4 M, 50 мл) при 0ºC, и перемешивали полученную смесь при комнатной температуре в течение 8 часов. После удаления растворителя, остаток диспергировали в EtOAc (10 мл). Осадок собирали путем фильтрования с получением соединения 2 в виде белого твердого гидрохлорида (300 мг). ЖХ-МС: 436 [M+H]+.
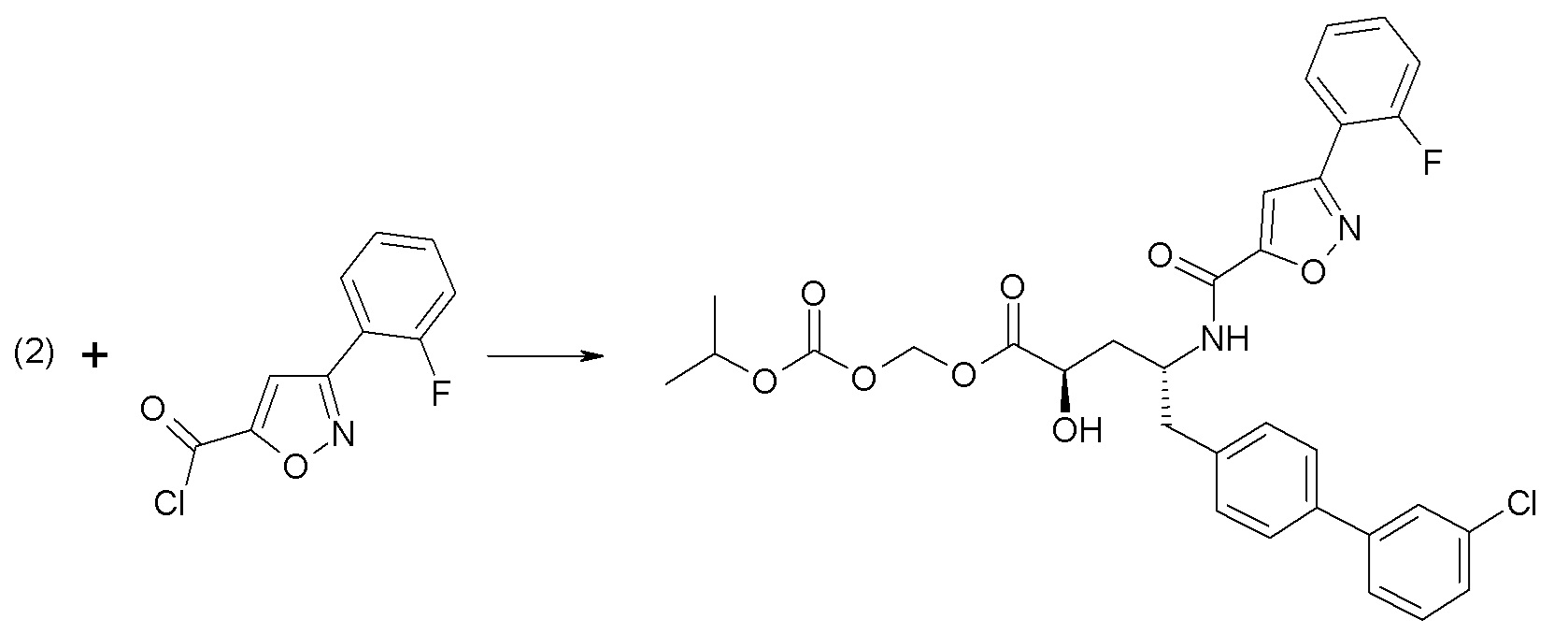
К раствору соединения 2 (150 мг, 340 мкмоль) в DMF (10 мл) добавляли 3-(2-фторфенил)изоксазол-5-карбонилхлорид (79 мг, 380 мкмоль) и DIPEA (120 мкл, 680 мкмоль). Смесь перемешивали при комнатной температуре в течение ночи. Добавляли воду (50 мл), а затем экстрагировали смесь EtOAc (3×30 мл). Объединенные органические слои промывали насыщенным водным NaCl (2×50 мл) и водой (2×50 мл), сушили над безводным Na2SO4 и концентрировали в условиях вакуума. Затем, остаток очищали методом препаративной HPLC (CAN-H2O (0,1% TFA), 75-80) с получением указанного в заголовке соединения в виде белого твердого вещества (60 мг). ЖХ-МС: 625 [M+H]+.1H-ЯМР (400 МГц, ДМСО-d6): δ 1,22-1,23 (м, 6H), 2,01-2,07 (м, 2H), 2,89-2,96 (м, 2H), 4,37-4,38 (м, 2H), 4,80 (шир., 1H), 5,67-5,69 (м, 2H), 5,71-5,78 (м, 1H), 7,31-7,33 (м, 7H), 7,42-7,43 (м, 5H), 7,61-7,63 (м, 1H), 8,95 (д, J=6,8 Гц, 1H).
Пример 6D: сложный ацетоксиметиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-4-{[3-(2-фторфенил)изоксазол-5-карбонил]амино}-2-гидроксипентановой кислоты

К перемешанному раствору бромметилацетата (977 мг, 6,4 ммоль) в DMF (10 мл) при комнатной температуре по каплям добавляли раствор (2R,4R)-4-(трет-бутоксикарбониламино)-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (1,8 г, 4,3 ммоль) в DMF (10 мл). Затем, по каплям добавляли пиридин (1,5 г, 19,2 ммоль), и перемешивали полученную смесь при комнатной температуре в течение ночи. Добавляли воду (150 мл), и экстрагировали смесь EtOAc (3×100 мл). Объединенные органические слои промывали насыщенным водным NaCl (2×100 мл) и водой (3×100 мл), сушили над безводным Na2SO4 и концентрировали в условиях вакуума. Неочищенный раствор очищали методом колоночной хроматографии (петролейный эфир/EtOAc=6/1) с получением соединения 1 в виде белого твердого вещества (570 мг. ЖХ-МС: 436 [M-tBu+H]+.

Соединение 1 (570 мг, 1,2 ммоль) растворяли в растворе HCl в диоксане (1,4 M, 50 мл) при 0ºC. Полученную смесь перемешивали при комнатной температуре в течение 8 часов, а затем концентрировали в условиях вакуума. Остаток диспергировали в EtOAc (10 мл). Осадок собирали путем фильтрования с получением соединения 2 в виде белого твердого гидрохлорида (200 мг). ЖХ-МС: 392 [M+H]+.

К раствору соединения 2 (150 мг, 380 мкмоль) в THF (10 мл) добавляли 3-(2-фторфенил)изоксазол-5-карбонилхлорид (79 мг, 380 мкмоль) и DIPEA (120 мкл, 680 мкмоль). Смесь перемешивали при комнатной температуре в течение ночи. Добавляли воду (50 мл), а затем экстрагировали смесь EtOAc (3×20 мл). Органические слои промывали насыщенным водным NaCl (2×30 мл) и водой (2×30 мл), сушили над безводным Na2SO4 и концентрировали в условиях вакуума. Затем, остаток очищали методом препаративной HPLC (CAN-H2O (0,1% TFA), 75-80) с получением указанного в заголовке соединения в виде белого твердого вещества (13 мг). ЖХ-МС: 581 [M+H]+.1H ЯМР (400 МГц, ДМСО-d6): δ 1,90-1,99 (м, 2H), 2,07 (с, 3H), 2,89-2,96 (м, 2H), 4,27-4,38 (м, 2H), 5,67-5,69 (м, 2H), 5,78 (шир., 1H), 7,31-7,33 (м, 7H), 7,42-7,43 (м, 5H), 7,61-7,63 (м, 1H), 8,95(д, J=8,8 Гц, 1H).
Пример 6E: сложный (S)-2-метоксикарбониламино-3-метилбутирилоксиметиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-4-{[3-(2-фторфенил)изоксазол-5-карбонил]амино}-2-гидроксипентановой кислоты

К раствору (2R,4R)-5-(3'-хлорбифенил-4-ил)-4-{[3-(2-фторфенил)изоксазол-5-карбонил]амино}-2-гидроксипентановой кислоты (150 мг, 0,3 ммоль), сложного хлорметилового эфира (S)-2-метоксикарбониламино-3-метилбутановой кислоты (134 мг, 0,6 ммоль) и NaI (135 мг, 0,9 ммоль) в DMF (10 мл) добавляли 2,6-лутидин (96 мг, 0,9 ммоль). Смесь перемешивали при 50°C в течение ночи. Добавляли воду (20 мл), и экстрагировали смесь этиловым эфиром (2×30 мл). Объединенные органические слои сушили над безводным Na2SO4 и концентрировали в условиях вакуума. Остаток очищали методом колоночной хроматографии (петролейный эфир/EtOAc=4/1~1/2) с получением указанного в заголовке соединения в виде не совсем желтого твердого вещества (8 мг). ЖХ-МС: 695,9 [M+H]+.1H-ЯМР (CD3OD-d4, 400 МГц) δ 0,92-0,95 (м, 6H), 2,03-2,08 (м, 3H), 3,00-3,10 (м, 2H), 4,09 (шир., 1H), 4,27-4,28 (м, 1H), 4,87 (шир., 1H), 5,75-5,85 (м, 2H), 7,26-7,37 (м, 6H), 7,51-7,53 (м, 6H), 7,93-7,95 (м, 1H).
Пример 7A: (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(3-метоксиизоксазол-5-карбонил)амино]пентановая кислота
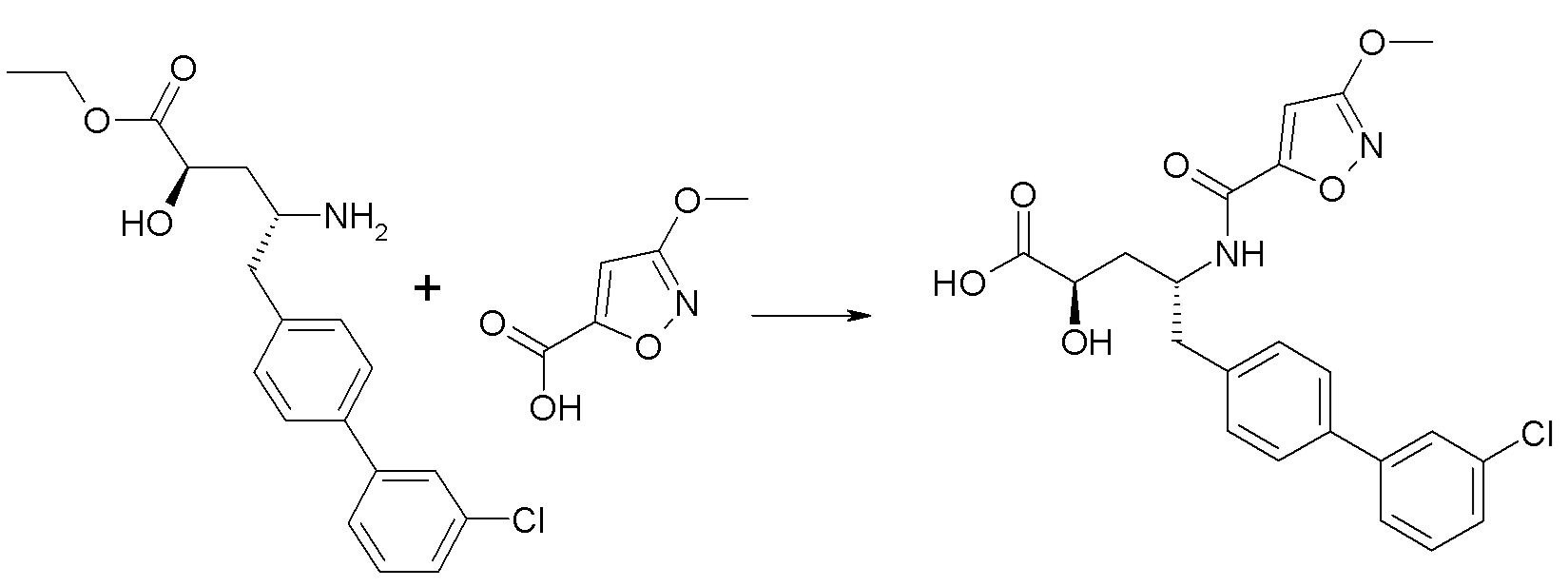
3-Метоксиизоксазол-5-карбоновую кислоту (39,4 мг, 275 мкмоль) и HCTU (128 мг, 310 мкмоль) объединяли в DMF, и перемешивали в течение 10 минут при комнатной температуре. Добавляли DIPEA (72 мкл, 413 мкмоль) и сложный этиловый эфир (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (50 мг, 0,1 ммоль), и перемешивали полученную смесь при комнатной температуре в течение ночи. Смесь объединяли с EtOH (402 мкл, 6,9 ммоль) и 1 M LiOH В воде (1,1 мл, 1,1 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, упаривали в условиях пониженного давления, и очищали методом препаративной HPLC с получением указанного в заголовке соединения. МС m/z [M+H]+ расч. для C22H21ClN2O6, 445,11; обнаружено 445,2.
Пример 7B: сложный 2-метоксикарбониламино-3-метилбутирилоксиметиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(3-метоксиизоксазол-5-карбонил)амино]пентановой кислоты

К раствору сложного этилового эфира (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (гидрохлорид, 1 г, 2,6 ммоль) в THF (30 мл) при 0°C добавляли раствор 3-метоксиизоксазол-5-карбонилхлорида (430 мг, 3 ммоль) в THF (10 мл) и Et3N (790 мг, 7,8 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 6 часов, а затем концентрировали в условиях вакуума с получением желтоватого твердого вещества, которое очищали методом хроматографии (PE/EtOAc=8/1) с получением соединения 1 в виде белого твердого вещества (0,9 г). ЖХ-МС: 473 [M+H]+.

К раствору соединения 1 (0,5 г, 1 ммоль) в MeOH/THF/H2O (15 мл/15 мл/3 мл) добавляли LiOH (120 мг, 5 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 5 часов, а затем концентрировали. Значение pH смеси корректировали до pH 5 добавлением HCl (1н, 20 мл). Твердые вещества фильтровали с получением соединения 2 в виде белого твердого вещества (0,4 г). ЖХ-МС: 445 [M+H]+.
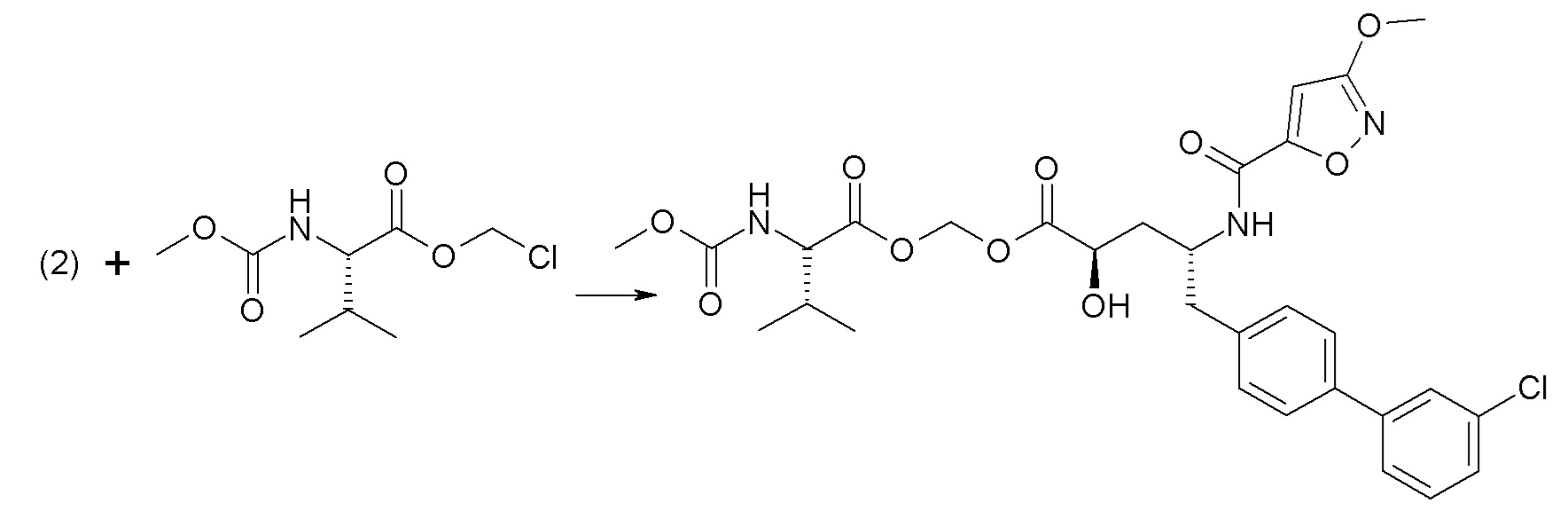
К раствору соединения 2 (220 мг, 0,5 ммоль), сложного хлорметилового эфира (S)-2-метоксикарбониламино-3-метилбутановой кислоты (220 мг, 1 ммоль) и NaI (300 мг, 2 ммоль) в DMF (5 мл) добавляли DIPEA (250 мг, 2 ммоль). Смесь перемешивали при комнатной температуре в течение 12 часов. Затем, смесь выливали в воду (20 мл), экстрагировали EtOAc (3×15 мл), промывали насыщенным водным NaCl (30 мл), сушили над безводным Na2SO4 и концентрировали в условиях вакуума. Затем, остаток очищали методом хроматографии (петролейный эфир/EtOAc=6/1) с получением указанного в заголовке соединения в виде белого твердого вещества (10 мг). ЖХ-МС: 632 [M+H]+.1H ЯМР (400 МГц, ДМСО-d6): δ 0,86 (д, J=6,8 Гц, 6H), 1,97-2,02 (м, 3H), 2,92-2,94 (м, 2H), 3,92 (с, 3H), 4,36-4,21 (м, 2H), 5,70-5,79 (м, 2H), 6,72 (с, 1H), 7,29 (д, J=8,0 Гц, 2H), 7,46-7,61 (м, 2H), 7,60-7,69 (м, 4H), 8,73 (д, J=8,8 Гц, 1H).
Пример 7C: сложный изопропоксикарбонилоксиметиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(3-метоксиизоксазол-5-карбонил)амино]пентановой кислоты
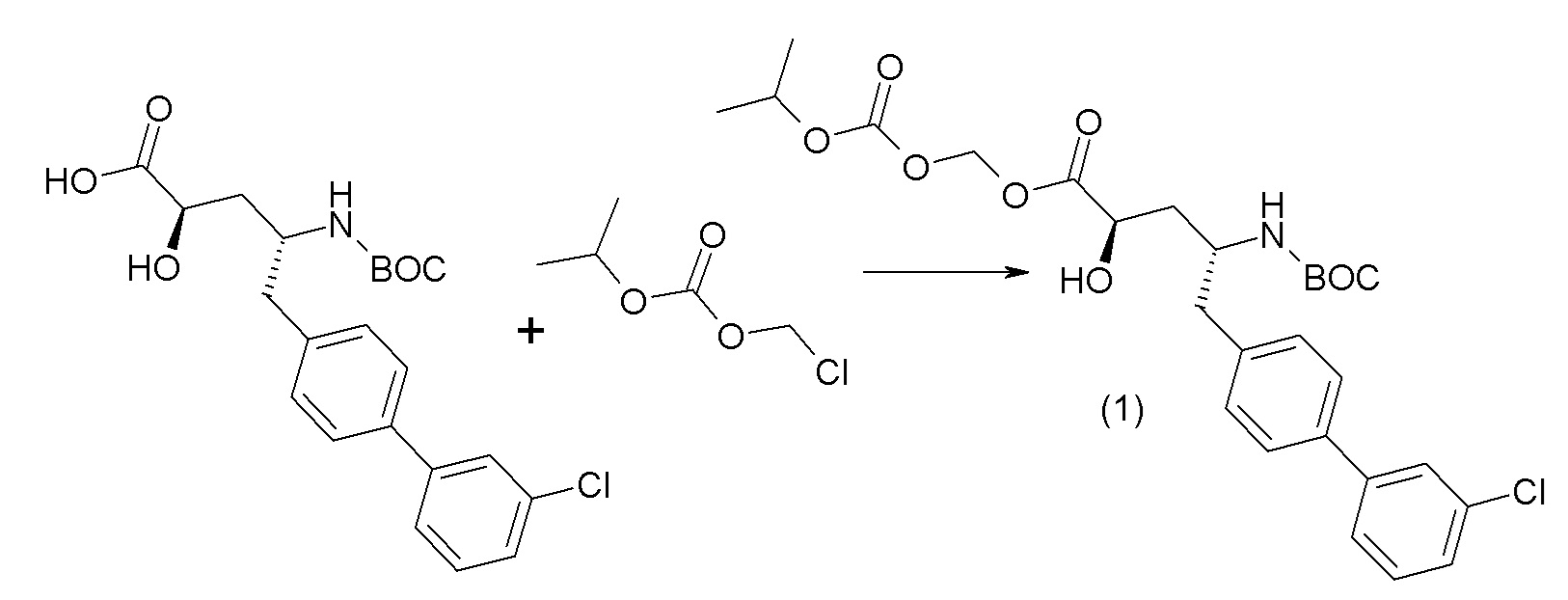
Смесь (2R,4R)-4-(трет-бутоксикарбониламино)-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (600 мг, 1,4 ммоль), хлорметилизопропилкарбоната (426 мг, 2,8 ммоль), NaI (420 мг, 2,8 ммоль) и DIPEA (546 мг, 4,2 ммоль) в DMF (30,0 мл) перемешивали при 25°C в течение ночи. Реакционную смесь выливали в воду (60 мл) и экстрагировали EtOAc (3×50 мл). Объединенные органические слои промывали насыщенным водным NaCl (2×50 мл) и водой (2×50 мл), сушили над безводным Na2SO4 и концентрировали в условиях вакуума. Продукт очищали методом колоночной хроматографии (петролейный эфир/EtOAc=2/1) с получением соединения 1 в виде бесцветной жидкости (240 мг). ЖХ-МС: 480 [M-tBu+H]+.
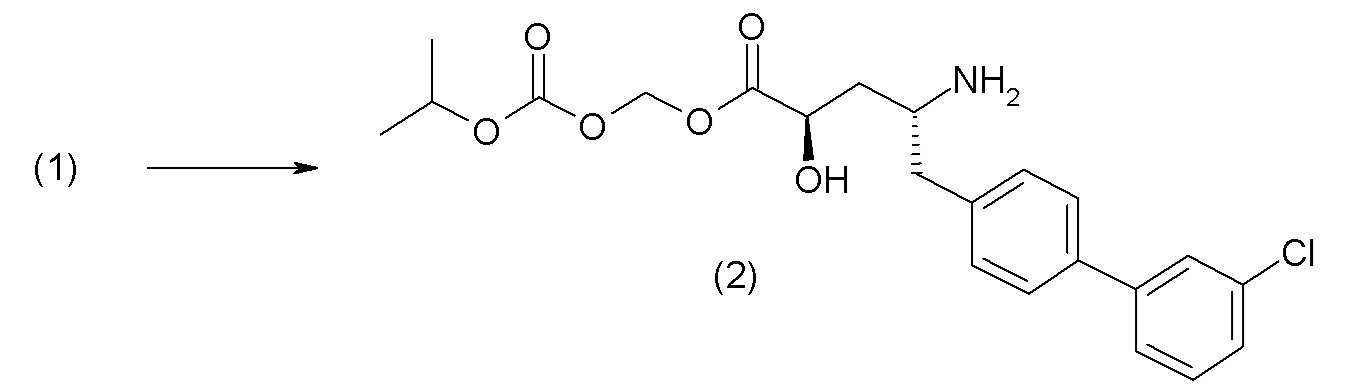
Соединение 1 (240 мг, 0,45 моль) растворяли в растворе HCl в диоксане (10,0 мл, 3,0 M) при 0°C, и перемешивали полученную смесь при комнатной температуре в течение ночи. Растворитель удаляли в условиях вакуума. Остаток диспергировали в EtOAc (10 мл). Осадок собирали путем фильтрования с получением соединения 2 в виде белого твердого гидрохлорида (80 мг). ЖХ-МС: 436 [M+H]+.

Смесь соединения 2 (70 мг, 0,15 ммоль), 3-метоксиизоксазол-5-карбоновой кислоты (43 мг, 0,3 ммоль), HOBT (40 мг, 0,3 ммоль), EDCI (57 мг, 0,3 ммоль) и DIPEA (78 мг, 0,6 ммоль) в DMF (10,0 мл) перемешивали при комнатной температуре в течение ночи. Смесь выливали в воду (30 мл) и экстрагировали EtOAc (3×30 мл). Органический слой разделяли, промывали насыщенным водным NaCl (2×30 мл) и водой (2×30 мл), сушили над безводным Na2SO4 и концентрировали в условиях вакуума. Остаток очищали методом колоночной хроматографии (петролейный эфир/EtOAc=2/1) с получением указанного в заголовке соединения в виде белого твердого вещества (20 мг). ЖХ-МС: 561 [M+H]+.1H ЯМР (400 МГц, CDCl3) δ 1,30 (д, J=6,4 Гц, 6H), 2,04-2,05 (м, 1H), 2,25-2,29 (м, 1H), 2,99-3,01 (м, 2H), 3,48 (шир., 1H), 4,00 (с, 3H), 4,46-4,48 (м, 1H), 4,50 (шир., 1H), 4,92-4,93 (м, 1H), 5,69-5,79 (м, 2H), 6,51 (с, 1H), 6,79-6,70 (м, 1H), 7,28-7,57(м, 8H).
Пример 7D: сложный ацетоксиметиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(3-метоксиизоксазол-5-карбонил)амино]пентановой кислоты
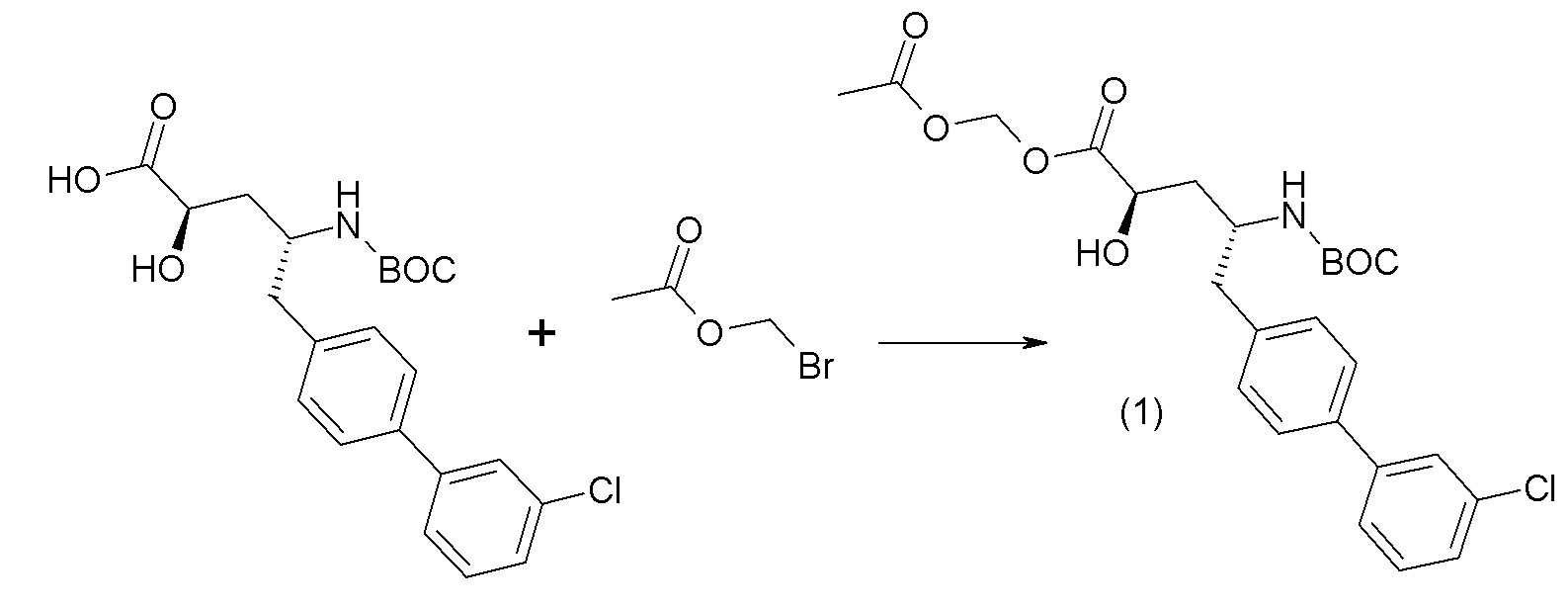
К перемешанному раствору (2R,4R)-4-(трет-бутоксикарбониламино)-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (1,8 г, 4,3 ммоль) в DMF (20 мл) при комнатной температуре добавляли бромметилацетат (977 мг, 6,4 ммоль). По каплям добавляли пиридин (1,7 г, 21,5 ммоль), и перемешивали полученную смесь при комнатной температуре в течение ночи. Добавляли воду (50 мл), и экстрагировали смесь EtOAc (3×50 мл). Объединенные органические слои промывали насыщенным водным NaCl (2×60 мл) и водой (2×60 мл), сушили над безводным Na2SO4 и концентрировали в условиях вакуума. Продукт очищали методом колоночной хроматографии (петролейный эфир/EtOAc, 15/1~6/1) с получением соединения 1 в виде белого твердого вещества (570 мг). ЖХ-МС: 436 [M-tBu+H]+.
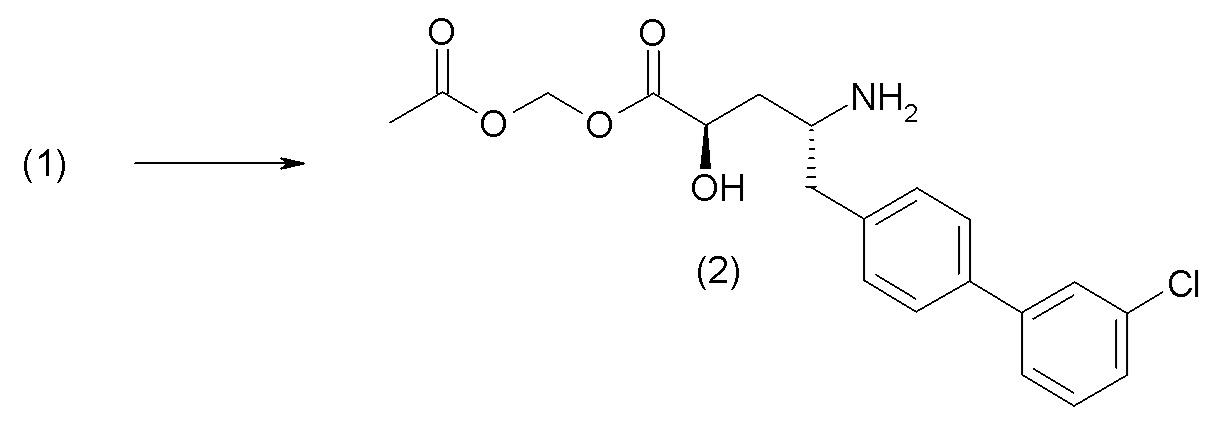
Соединение 1 (570 мг, 1,2 ммоль) добавляли к раствору HCl в диоксане (1,4 M, 50 мл) при 0ºC, и перемешивали полученную смесь при комнатной температуре в течение 2 часов, а затем концентрировали в условиях вакуума. Остаток диспергировали в EtOAc (10 мл). Осадок собирали путем фильтрования с получением соединения 2 в виде белого твердого гидрохлорида (200 мг). ЖХ-МС: 392 [M+H]+.

К раствору соединения 2 (100 мг, 255 мкмоль) в DMF (10 мл) добавляли 3-метоксиизоксазол-5-карбоновую кислоту (79 мг, 383 мкмоль), HOBt (69 мг, 510 мкмоль), EDCI (98 мг, 510 мкмоль) и DIPEA (0,2 мл, 1,0 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Добавляли воду (30 мл), и экстрагировали смесь EtOAc (3×30 мл). Объединенные органические слои промывали насыщенным водным NaCl (2×30 мл) и водой (2×30 мл), сушили над безводным Na2SO4 и концентрировали в условиях вакуума. Остаток очищали методом препаративной HPLC (CAN-H2O (0,1% TFA), 60-65) с получением указанного в заголовке соединения в виде белого твердого вещества (15 мг). ЖХ-МС: 517 [M+H]+.1H-ЯМР (ДМСО-d6, 400 Гц): δ 1,84-1,99 (м, 2H), 2,07 (с, 3H), 2,89-2,96 (м, 2H), 3,92 (с, 3H), 4,21-4,32 (м, 2H), 5,66-5,69 (м, 2H), 5,74 (шир., 1H), 6,72 (с, 1H), 7,29 (д, J=8,4 Гц, 2H), 7,28-7,30 (м, 2H), 7,60-7,62 (м, 3H), 7,69 (с, 1H), 8,76 (д, J=8,8 Гц, 1H).
Пример 7E: сложный этоксикарбонилоксиметиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(3-метоксиизоксазол-5-карбонил)амино]пентановой кислоты
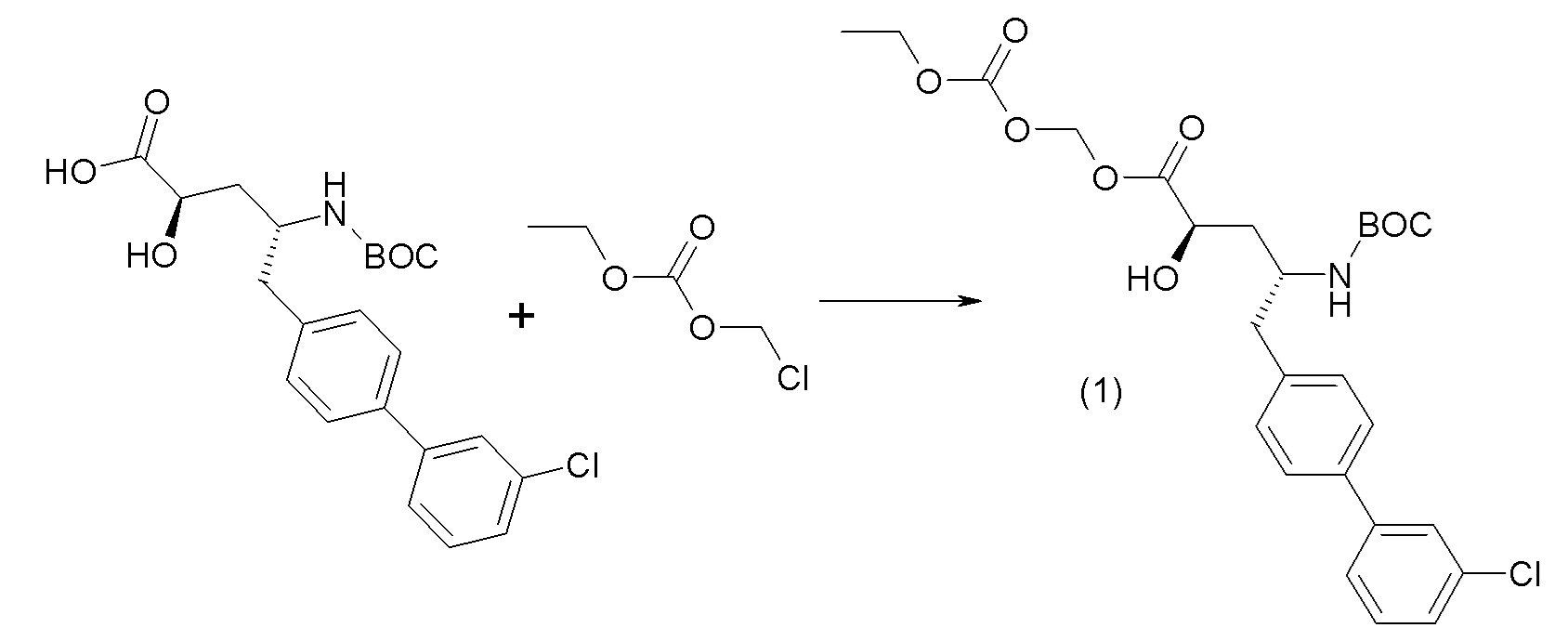
К перемешанному раствору хлорметилэтилкарбоната (0,2 мл, 1,1 ммоль) в DMF (1 мл) при комнатной температуре по каплям добавляли раствор (2R,4R)-4-(трет-бутоксикарбониламино)-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (400 мг, 960 мкмоль) в DMF (6 мл). Добавляли NaI (432 мг, 2,88 ммоль) и DIPEA (0,5 мл, 2,88 ммоль), и перемешивали полученную смесь при комнатной температуре в течение ночи. Добавляли воду (50 мл), и экстрагировали смесь EtOAc (3×30 мл). Объединенные органические слои промывали насыщенным водным NaCl (2×50 мл) и водой (2×50 мл), сушили над безводным Na2SO4 и концентрировали в условиях вакуума. Продукт очищали методом колоночной хроматографии (петролейный эфир/EtOAc, 15/1~6/1) с получением соединения 1 в виде белого твердого вещества (190 мг). ЖХ-МС: 544 [M+Na]+.

Соединение 1 (190 мг, 360 мкмоль) добавляли к раствору HCl в диоксане (1,4 M, 50 мл) при 0ºC, и перемешивали полученную смесь при комнатной температуре в течение 2 часов, а затем концентрировали в условиях вакуума. Остаток диспергировали в EtOAc (10 мл). Осадок собирали путем фильтрования с получением соединения 2 в виде белого твердого гидрохлорида (150 мг). ЖХ-МС: 422 [M+H]+.
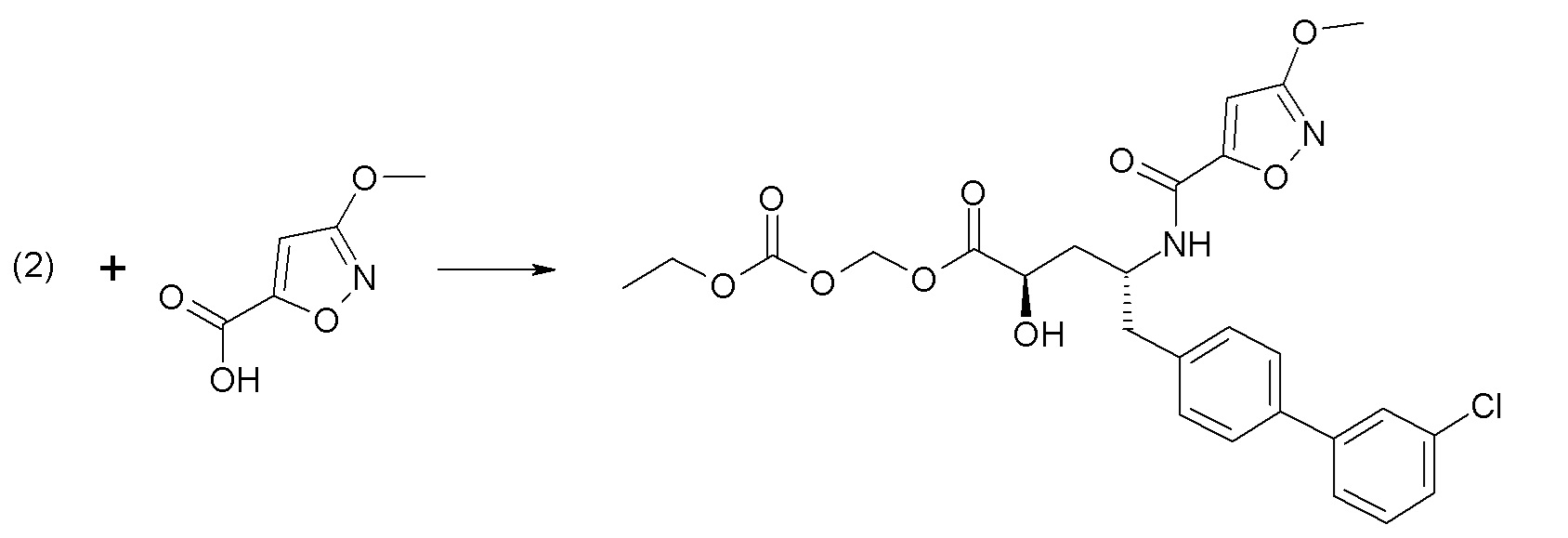
К перемешанному раствору соединения 2 (150 мг, 360 мкмоль) в THF (20 мл) при комнатной температуре по каплям добавляли раствор 3-метоксиизоксазол-5-карбонилхлорид (73 мг, 530 мкмоль) в THF (2 мл). По каплям добавляли DIPEA (0,2 мл), и перемешивали полученную смесь при комнатной температуре в течение ночи. Добавляли воду (50 мл), и экстрагировали смесь Et2O (3×40 мл). Объединенные органические слои промывали насыщенным водным NaCl (2×50 мл) и водой (2×50 мл), сушили над безводным Na2SO4 и концентрировали в условиях вакуума. Остаток очищали методом препаративной HPLC (CAN-H2O (0,1%TFA), 60-70) с получением указанного в заголовке соединения в виде белого твердого вещества (25 мг). ЖХ-МС: 546,9 [M+H]+.1H-ЯМР (ДМСО-d6, 400 МГц): δ 1,22 (т, J=6,8 Гц, 3H), 1,87-1,96 (м, 2H), 2,83-2,89 (м, 2H), 3,92 (с, 3H), 4,16 (кв., J=6,8 Гц, 2H), 4,18-4,20 (м, 2H), 5,68-5,78 (м, 3H), 6,72 (с, 1H), 7,29 (д, J=8,0 Гц 2H), 7,45-7,48 (м, 2H), 7,60-7,62 (м, 3H), 7,69 (м, 1H), 8,76 (д, J=8,8 Гц,1H).
Пример 8A: (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(1-гидрокси-1H-[1,2,3]триазол-4-карбонил)амино]пентановая кислота
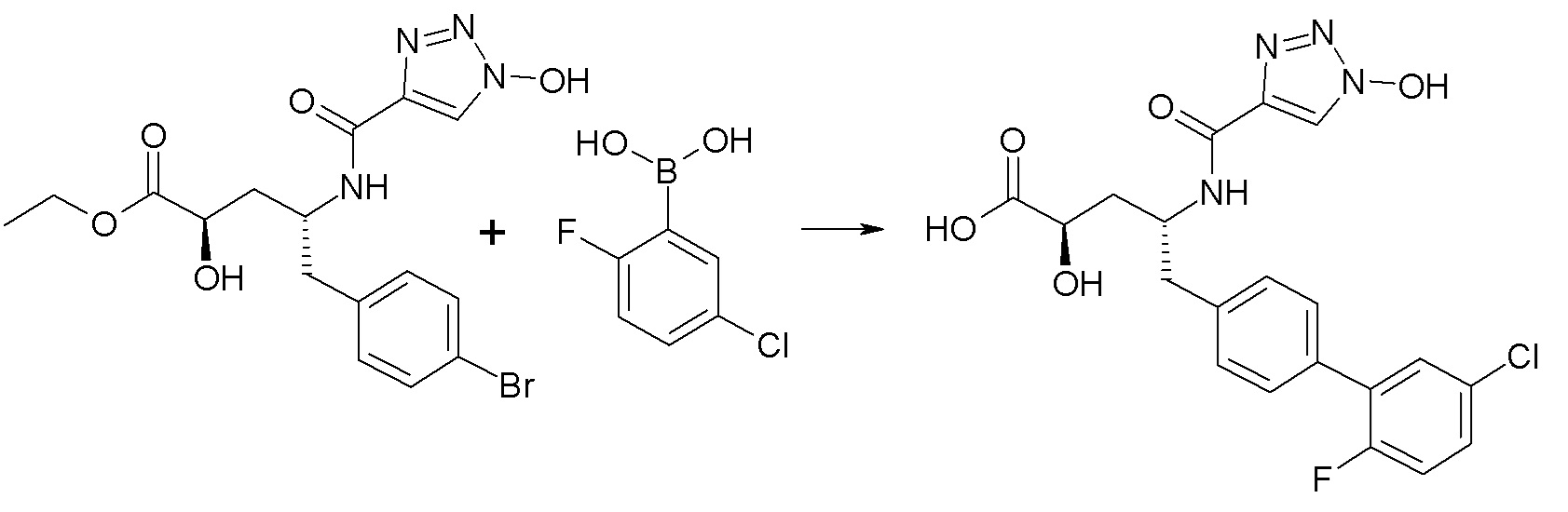
Сложный этиловый эфир (2R,4R)-5-(4-бромфенил)-2-гидрокси-4-[(1-гидрокси-1H-1,2,3-триазол-4-карбонил)амино]пентановой кислоты (30 мг, 70 мкмоль) объединяли с 5-хлор-2-фторфенилбороновой кислотой (22 мг, 126 мкмоль), K2CO3 (29,1 мг, 211 мкмоль), EtOH (0,8 мл, 10 ммоль) и водой (0,2 мл, 10 ммоль). К смеси добавляли SilicaCat DPP-Pd (загрузка 0,28 ммоль/г; 25,1 мг, 7,0 мкмоль). Полученную смесь обрабатывали микроволнами при 100°C в течение 10 минут. Добавляли 1 M LiOH в воде (281 мкл, 281 мкмоль), и перемешивали полученную смесь в течение 1 часа. Смесь фильтровали, концентрировали и очищали методом препаративной HPLC с получением указанного в заголовке соединения (17 мг). МС m/z [M+H]+ расч. для C20H18ClFN4O5, 449,10; обнаружено 449,2.
Пример 8B: сложный 2,2,3,3,3-пентафторпропиловый эфир (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(1-гидрокси-1H-[1,2,3]триазол-4-карбонил)амино]пентановой кислоты
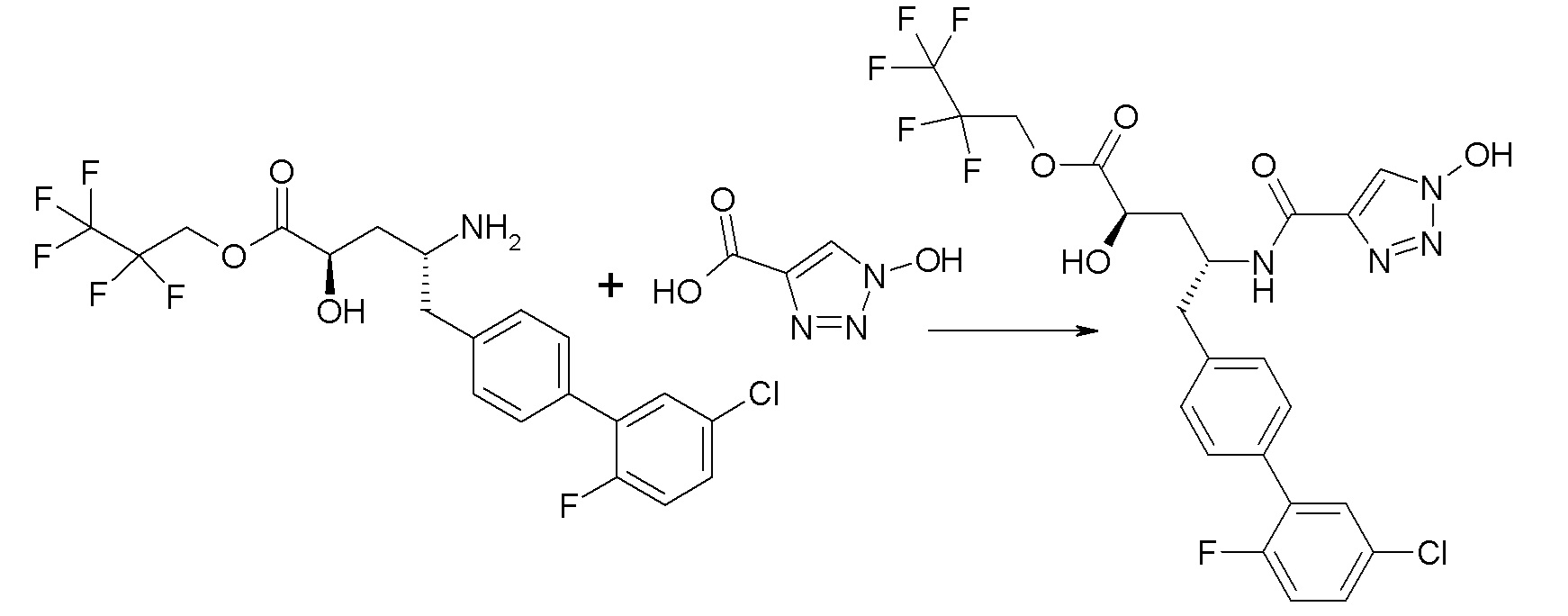
Сложный 2,2,3,3,3-пентафторпропиловый эфир (2R,4R)-4-амино-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксипентановой кислоты (32,1 мг, 68,3 мкмоль), 1-гидрокси-1H-[1,2,3]триазол-4-карбоновую кислоту (10,6 мг, 82 мкмоль) и HATU (39,0 мг, 102 мкмоль) перемешивали в DMF (1,0 мл, 12,9 ммоль) в течение 10 минут. По каплям добавляли DIPEA (35,7 мкл, 205 мкмоль), и перемешивали полученную смесь при комнатной температуре в течение 1 часа. Смесь концентрировали с получением прозрачной желтой жидкости, которую очищали методом препаративной хроматографии на колонке C18 (небольшая колонка; 30-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения в виде белого твердого вещества (4,9 мг). МС m/z [M+H]+ расч. для C23H19ClF6N4O5, 581,09; обнаружено 581.
Пример 8C: сложный 5-метил-2-оксо-[1,3]диоксол-4-илметиловый эфир (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(1-гидрокси-1H-[1,2,3]триазол-4-карбонил)амино]пентановой кислоты
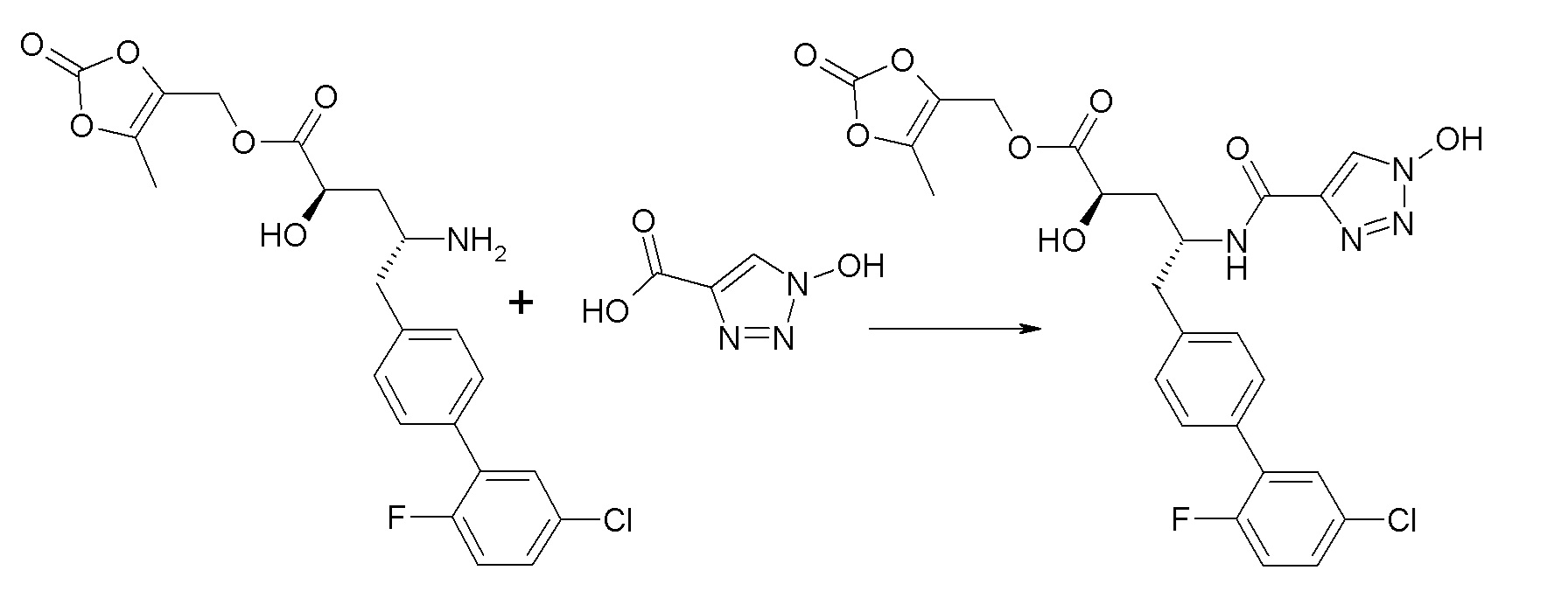
Сложный 5-метил-2-оксо-[1,3]диоксол-4-илметиловый эфир (2R,4R)-4-амино-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксипентановой кислоты (30,7 мг, 68,3 мкмоль), 1-гидрокси-1H-[1,2,3]триазол-4-карбоновую кислоту (10,6 мг, 82 мкмоль) и HATU (39,0 мг, 102 мкмоль) перемешивали в DMF (1,0 мл, 12,9 ммоль) в течение 10 минут. По каплям добавляли DIPEA (35,7 мкл, 205 мкмоль), и перемешивали полученную смесь при комнатной температуре в течение 1 часа. Смесь концентрировали с получением прозрачной желтой жидкости, которую очищали методом препаративной хроматографии на колонке C18 (небольшая колонка; 30-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения в виде белого твердого вещества (27,1 мг). МС m/z [M+H]+ расч. для C25H22ClFN4O8, 561,11; обнаружено 561,1.
Пример 8D: (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-{[1-(5-метил-2-оксо-[1,3]диоксол-4-илметокси)-1H-[1,2,3]триазол-4-карбонил]амино}пентановая кислота
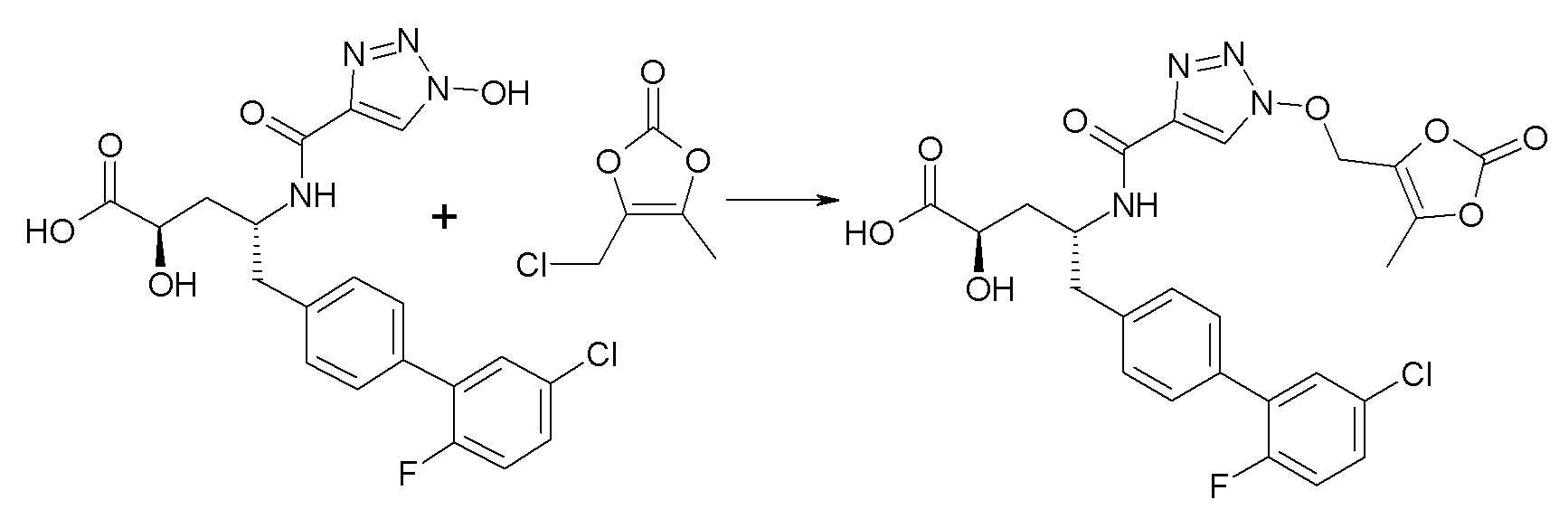
К раствору (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(1-гидрокси-1H-1,2,3-триазол-4-карбонил)амино]пентановой кислоты (30,0 мг, 66,8 мкмоль) в ацетоне (0,5 мл, 6,8 ммоль) добавляли 4-хлорметил-5-метил-1,3-диоксол-2-он (11,0 мкл, 100 мкмоль). Добавляли Et3N (18,6 мкл, 134 мкмоль), и перемешивали полученную смесь при 65°C в течение 2 часов. Смесь концентрировали в условиях вакуума с получением желтого твердого вещества. Неочищенное твердое вещество очищали методом препаративной HPLC (хроматография на колонке C18, небольшая колонка, с использованием 30-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения в виде белого твердого вещества (6,5 мг). МС m/z [M+H]+ расч. для C25H22ClFN4O8, 561,11; обнаружено 561,1.
Пример 8E: (2R,4R)-4-[(1-бутирилоксиметокси-1H-[1,2,3]триазол-4-карбонил)амино]-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксипентановая кислота

К раствору (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(1-гидрокси-1H-1,2,3-триазол-4-карбонил)амино]пентановой кислоты (30,0 мг, 66,8 мкмоль) в ацетоне (0,5 мл, 6,8 ммоль) добавляли хлорметилбутират (13,7 мг, 100 мкмоль). Добавляли Et3N (18,6 мкл, 134 мкмоль), и перемешивали полученную смесь при 65°C в течение 2 часов. Затем, смесь концентрировали в условиях вакуума с получением желтой жидкости. Неочищенную жидкость очищали (препаративная хроматография на колонке C18, небольшая колонка, с использованием 30-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения в виде белого твердого вещества (6,0 мг). МС m/z [M+H]+ расч. для C25H26ClFN4O7, 549,15; обнаружено 549,1.
Пример 8F: (2R,4R)-4-{[1-((S)-2-амино-3-метилбутирилоксиметокси)-1H-[1,2,3]триазол-4-карбонил]амино}-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксипентановая кислота
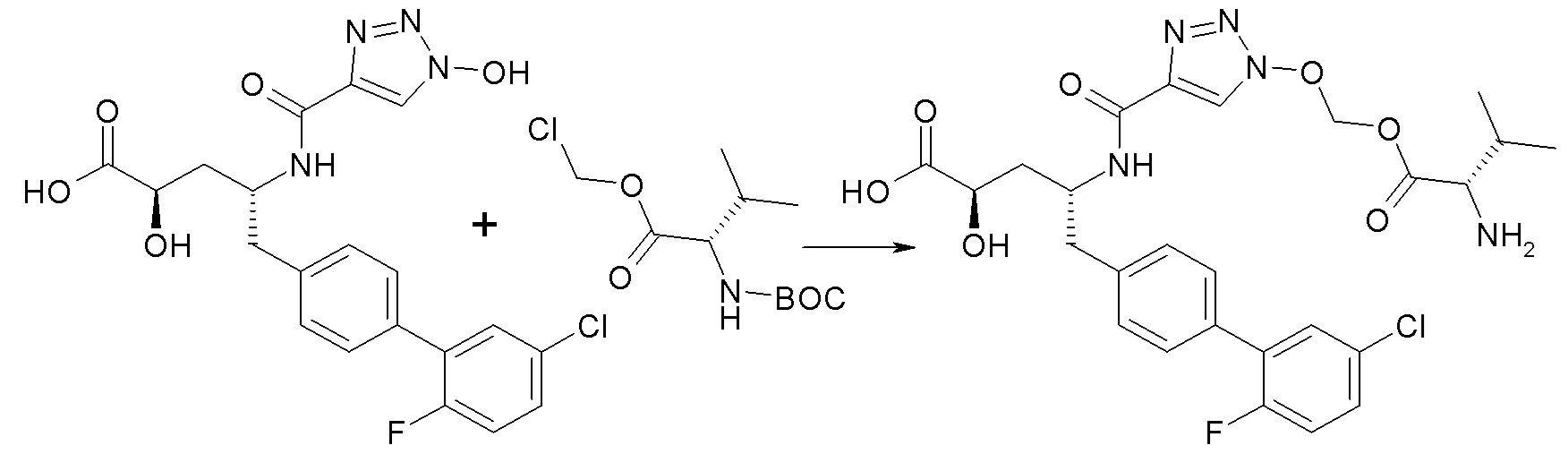
К раствору (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(1-гидрокси-1H-1,2,3-триазол-4-карбонил)амино]пентановой кислоты (30,0 мг, 66,8 мкмоль) в ацетоне (0,5 мл, 6,8 ммоль) добавляли сложный хлорметиловый эфир (S)-2-трет-бутоксикарбониламино-3-метилбутановой кислоты (44,4 мг, 167 мкмоль). Добавляли Et3N (18,6 мкл, 134 мкмоль), и перемешивали полученную смесь при 65°C в течение 2 часов. Добавляли TFA/DCM (1/1, 1,0 мл, 6,2 ммоль), и перемешивали смесь при комнатной температуре в течение 30 минут, а затем концентрировали в условиях вакуума с получением желтой жидкости. Неочищенную жидкость очищали (препаративная хроматография на колонке C18, небольшая колонка, с использованием 30-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения в виде белого твердого вещества (10 мг). МС m/z [M+H]+ расч. для C26H29ClFN5O7, 578,17; обнаружено 578,1.
Пример 8G: (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-{[1-((S)-2-метоксикарбониламино-3-метилбутирилоксиметокси)-1H-[1,2,3]триазол-4-карбонил]амино}пентановая кислота

К раствору (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(1-гидрокси-1H-1,2,3-триазол-4-карбонил)амино]пентановой кислоты (30,0 мг, 66,8 мкмоль) в ацетоне (0,5 мл, 6,8 ммоль) добавляли сложный хлорметиловый эфир (S)-2-метоксикарбониламино-3-метилбутановой кислоты (37,4 мг, 167 мкмоль). Добавляли Et3N (18,6 мкл, 134 мкмоль), и перемешивали полученную смесь при 65°C в течение 2 часов. Затем, смесь концентрировали в условиях вакуума с получением желтой жидкости. Неочищенную жидкость очищали (препаративная хроматография на колонке C18, небольшая колонка, с использованием 30-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения в виде белого твердого вещества (8,2 мг). МС m/z [M+H]+ расч. для C28H31ClFN5O9, 636,18; обнаружено 636,1.
Пример 8H: сложный бутирилоксиметиловый эфир (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(1-гидрокси-1H-[1,2,3]триазол-4-карбонил)амино]пентановой кислоты
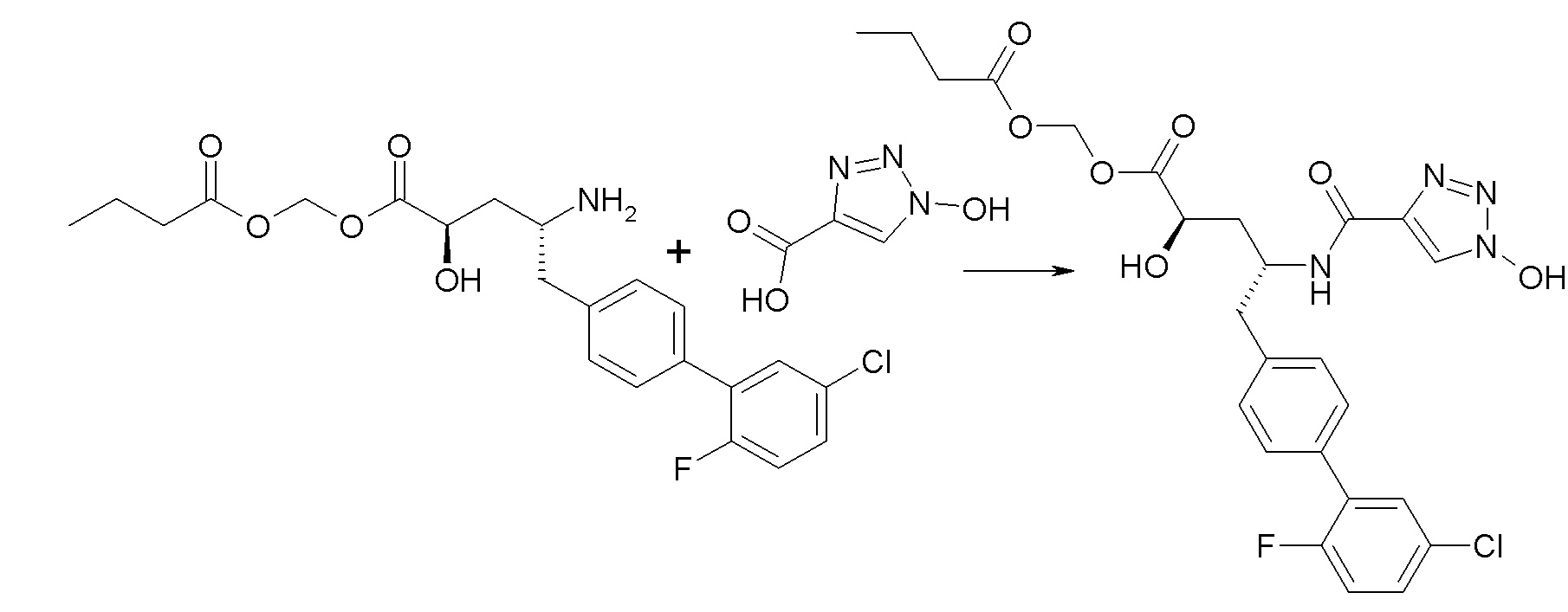
К раствору сложного бутирилоксиметилового эфира (2R,4R)-4-амино-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксипентановой кислоты (30,0 мг, 68,5 мкмоль), 1-гидрокси-1H-1,2,3-триазол-4-карбоновой кислоты (13,3 мг, 103 мкмоль) и HATU (39,1 мг, 103 мкмоль) в DMF (500 мкл, 6,5 ммоль) добавляли DIPEA (35,8 мкл, 206 мкмоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут, а затем концентрировали в условиях вакуума с получением прозрачной желтой жидкости. Неочищенную жидкость очищали методом препаративной HPLC (хроматография на колонке C18, небольшая колонка, с использованием 30-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения в виде белого твердого вещества (9,0 мг). МС m/z [M+H]+ расч. для C25H26ClFN4O7, 549,15; обнаружено 549,1.
Пример 8I: сложный этоксикарбонилоксиметиловый эфир (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(1-гидрокси-1H-[1,2,3]триазол-4-карбонил)амино]пентановой кислоты
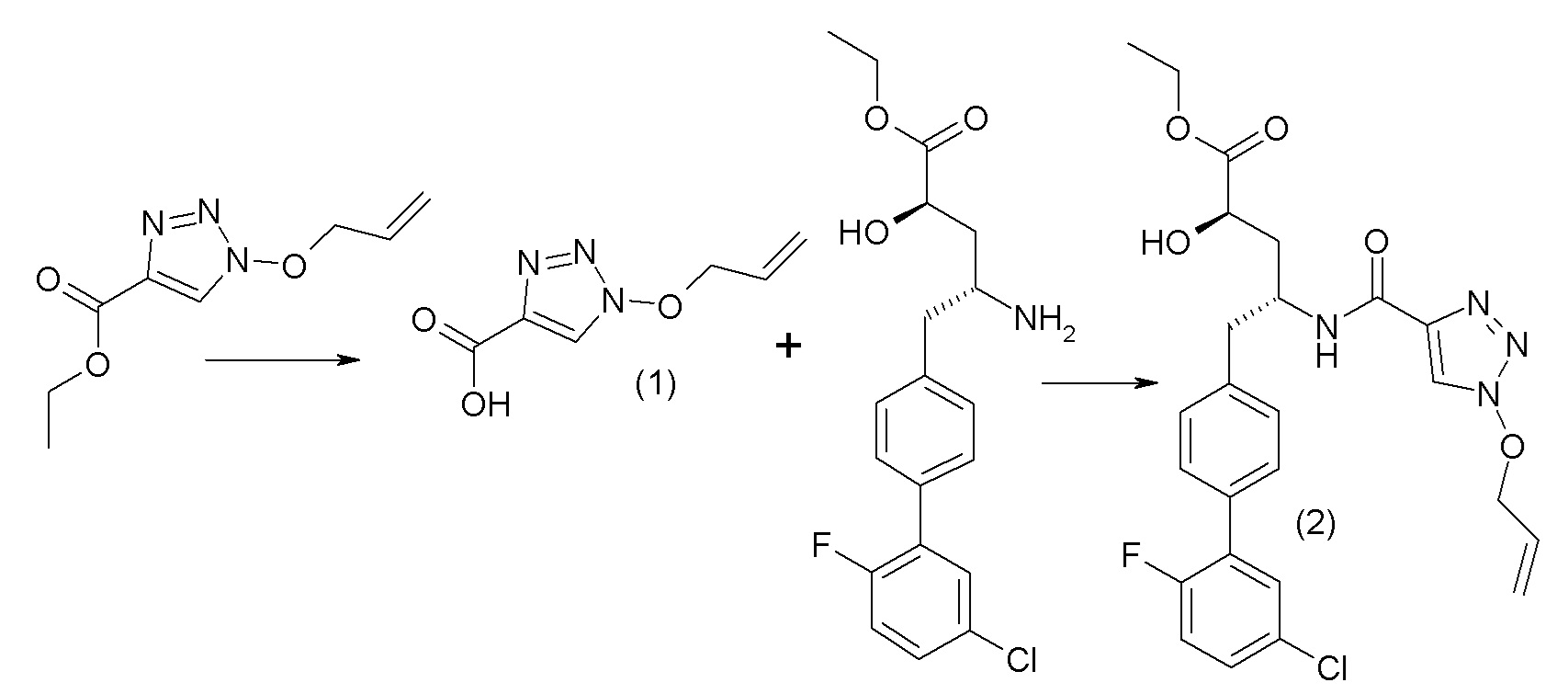
К раствору сложного этилового эфира 1-аллилокси-1H-[1,2,3]триазол-4-карбоновой кислоты (1,7 г, 8,6 ммоль) в MeOH (20 мл) добавляли раствор LiOH (1,1 г, 45,3 ммоль) в воде (4 мл). Смесь перемешивали при комнатной температуре до завершения реакции (1 час). Смесь концентрировали, разбавляли водой (10 мл) и экстрагировали EtOAc (2×20 мл). Водный слой подкисляли до pH=3 добавлением 1н HCl, экстрагировали EtOAc (3×30 мл), объединенные органические слои промывали насыщенным водным NaCl (30 мл) и сушили над безводным Na2SO4. Раствор упаривали с получением соединения 1 (1,3 г) в виде белого твердого вещества. ЖХ-МС: 170[M+H]+.
К раствору сложного этилового эфира (2R,4R)-4-амино-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксипентановой кислоты (гидрохлорид, 800 мг, 2,0 ммоль) и соединения 1 (337 мг, 2,0 ммоль) в DMF (15 мл) при 0°C добавляли PyBOP (1,0 г, 2,0 ммоль) и DIPEA (987 мкл, 6,0 ммоль). Смесь перемешивали при комнатной температуре в течение 4 часов. Добавляли воду (50 мл), смесь экстрагировали EtOAc (3×10 мл), объединенные органические слои промывали насыщенным водным NaCl и сушили над безводным Na2SO4. Раствор концентрировали с получением неочищенного продукта, который очищали методом хроматографии на силикагеле (силикагель: 200-300 меш; элюирование PE/EtOAc=10/1→5/1→1/1) с получением соединения 2 (700 мг) в виде светло-желтого твердого вещества. ЖХ-МС: 517[M+H]+.
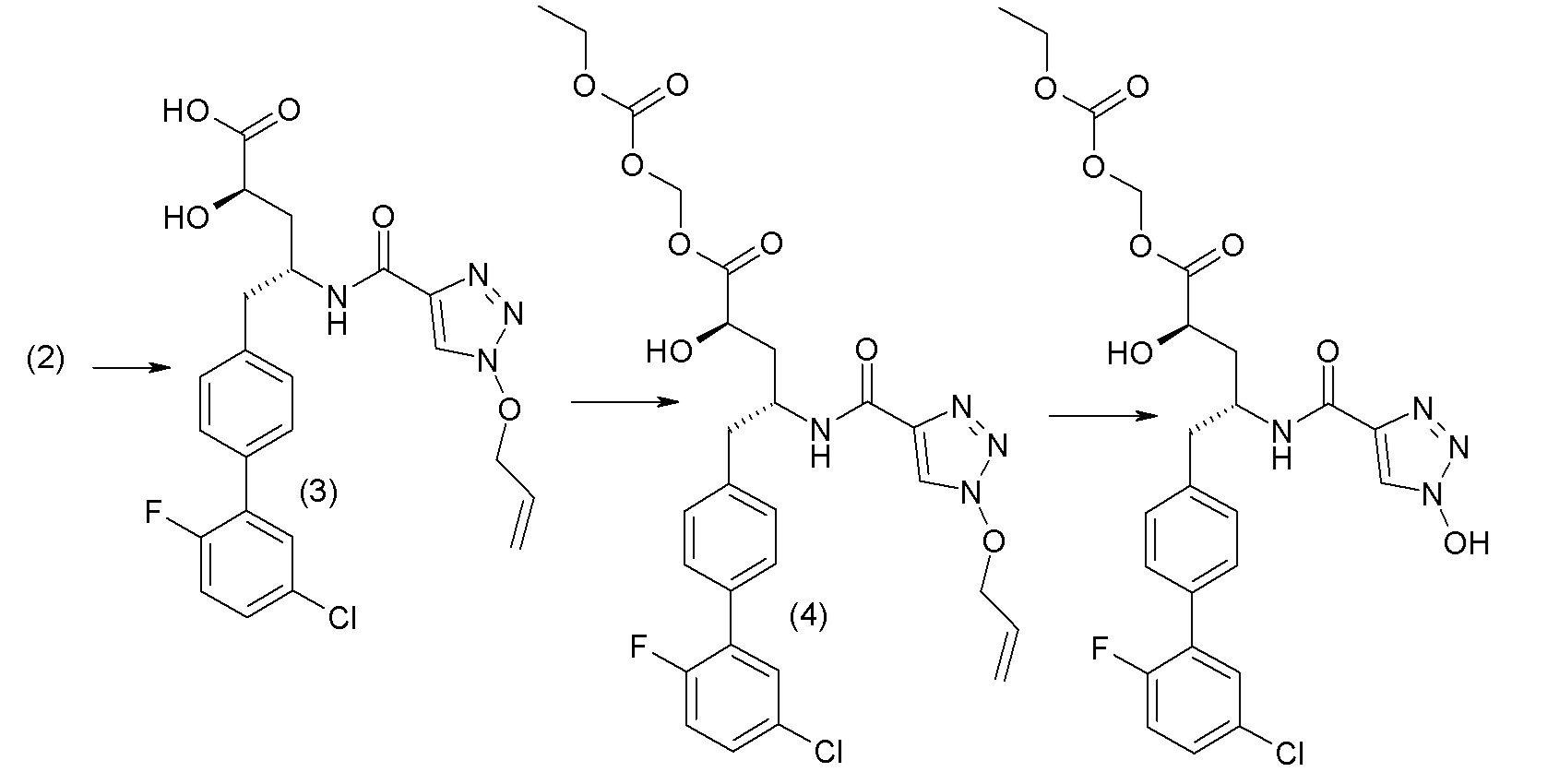
К раствору соединения 2 (700 мг, 1,4 ммоль) в MeOH (15 мл) и воде (3 мл) при комнатной температуре добавляли LiOH (171 мг, 4,1 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Смесь концентрировали, добавляли воду (20 мл), и экстрагировали смесь EtOAc (2×10 мл). Водный слой подкисляли до pH=3 добавлением 1н HCl, экстрагировали EtOAc (3××10 мл), объединенные органические слои промывали насыщенным водным NaCl (30 мл) и сушили над безводным Na2SO4. Раствор упаривали с получением соединения 3 (650 мг) в виде желтого твердого вещества. ЖХ-МС:489[M+H]+.
К смеси соединения 3 (80 мг, 160 мкмоль) и хлорметилэтилкарбоната (1,0 мл) добавляли NaI (48 мг, 0,32 ммоль) и лутидин (52 мг, 480 мкмоль). Смесь перемешивали при 50°C в течение 6 часов. Затем, смесь охлаждали до комнатной температуры, разбавляли водой (15 мл), и экстрагировали EtOAc (2×15 мл). Объединенные органические слои промывали насыщенным водным NaCl (20 мл), сушили над безводным Na2SO4, концентрировали и очищали методом хроматографии на силикагеле (силикагель: 200-300 меш; элюирование PE/EtOAc от 4/1 до 1/1) с получением соединения 4 (60 мг) в виде светло-желтого твердого вещества. ЖХ-МС: 591[M+H]+.
К раствору соединения 4 (40 мг, 68 мкмоль) в MeCN (10 мл) добавляли NaI (10 мг, 68 мкмоль), и перемешивали смесь при комнатной температуре в течение 5 минут. К этой перемешанной суспензии добавляли TMSCl (11 мг, 102 мкмоль), и дополнительно перемешивали в течение 10 минут. Реакционную смесь гасили добавлением тиосульфата натрия, и экстрагировали смесь EtOAc (2×10 мл). Объединенные органические слои промывали насыщенным водным NaCl (20 мл) и сушили над безводным Na2SO4. Остаток очищали методом препаративной HPLC [Daisogel-C18, 250×50 мм, 10 мкм; CAN-H2O (0,1% TFA) от 60% до 90%] с получением указанного в заголовке соединения (8 мг) в виде белого твердого вещества. ЖХ-МС: 551[M+H]+.1H-ЯМР (CDCl3, 400 Гц): δ 1,27 (т, 3H), 2,06-2,11 (м, 1H), 2,14-2,24 (м, 1H), 2,96-3,01 (м, 2H), 4,18 (кв., 2H), 4,42 (м, 1H), 4,71 (м, 1H), 5,57-5,65 (м, 2H), 7,05 (т, 1H), 7,22-7,24 (м, 2H), 7,35-7,40(м, 4H), 8,04 (с, 1H).
Пример 8J: сложный изопропоксикарбонилоксиметиловый эфир (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(1-гидрокси-1H-[1,2,3]триазол-4-карбонил)амино]пентановой кислоты
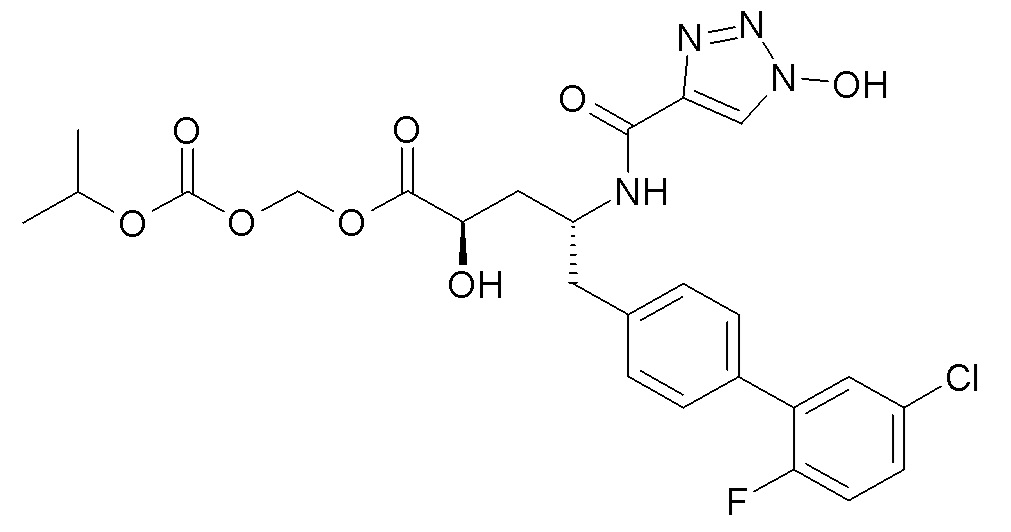
Указанное в заголовке соединение получали (110 мг) с использованием методик, описанных в настоящем документе.
Пример 8K: сложный 1-циклогексилоксикарбонилоксиэтиловый эфир (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(1-гидрокси-1H-[1,2,3]триазол-4-карбонил)амино]пентановой кислоты

Указанное в заголовке соединение получали (16,6 мг) с использованием методик, описанных в настоящем документе.
Пример 8L: сложный (S)-2-метоксикарбониламино-3-метилбутирилоксиметиловый эфир (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(1-гидрокси-1H-[1,2,3]триазол-4-карбонил)амино]пентановой кислоты

Указанное в заголовке соединение получали (117 мг) с использованием методик, описанных в настоящем документе.
Пример 8M: сложный ацетоксиметиловый эфир (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-[(1-гидрокси-1H-[1,2,3]триазол-4-карбонил)амино]пентановой кислоты
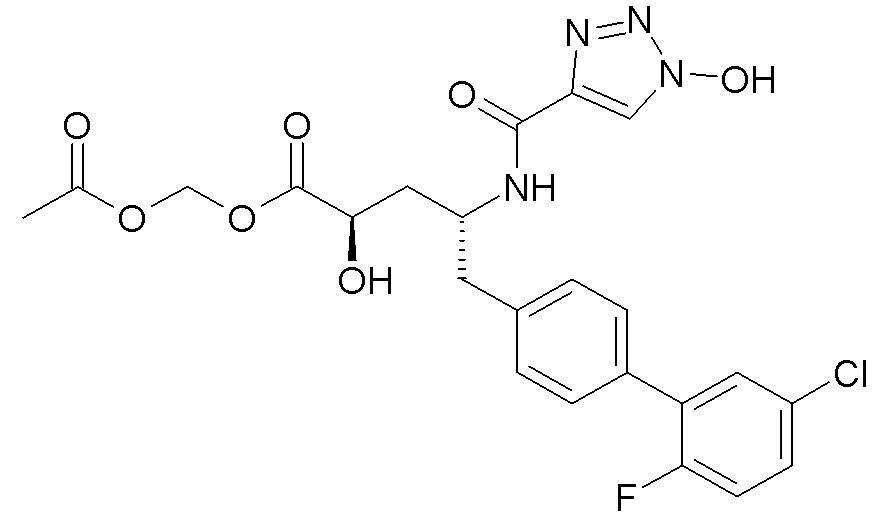
Указанное в заголовке соединение также может быть получено с использованием методик, описанных в настоящем документе.
Пример 9A: (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(5-оксо-1-фенил-4,5-дигидро-1H-[1,2,4]триазол-3-карбонил)амино]пентановая кислота
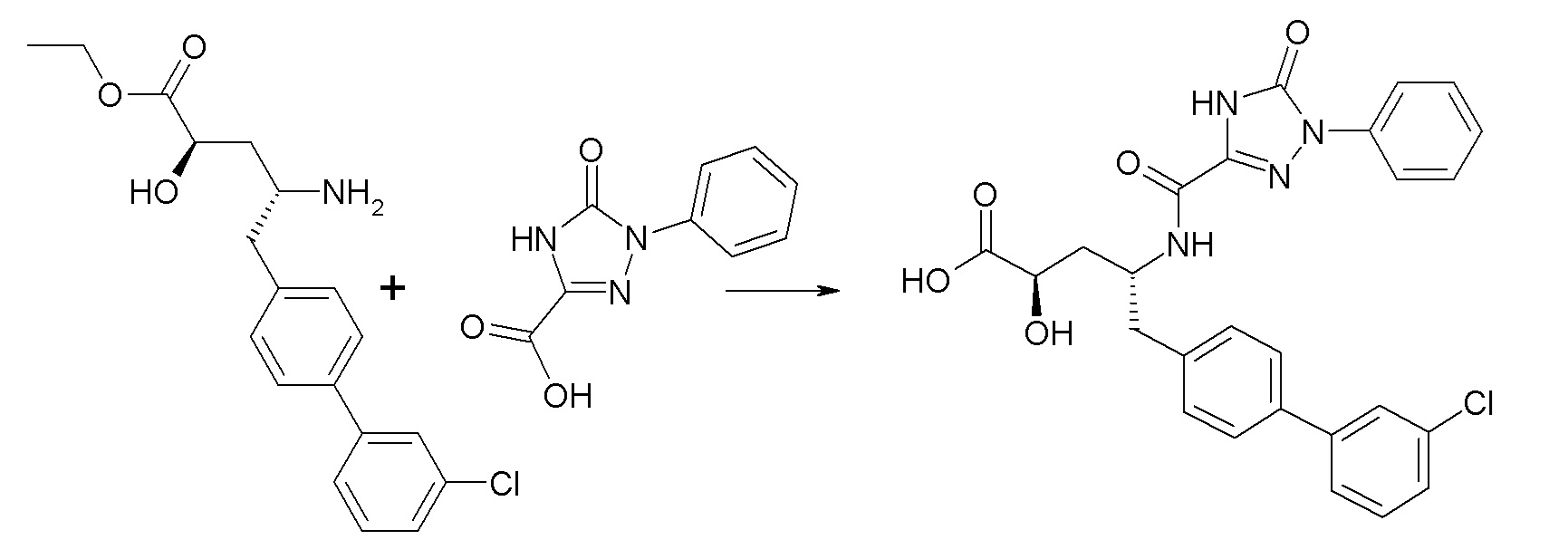
5-Оксо-1-фенил-4,5-дигидро-1H-[1,2,4]триазол-3-карбоновую кислоту (42,3 мг, 206 мкмоль) и HCTU (128 мг, 310 мкмоль) объединяли в DMF, и перемешивали в течение 10 минут при комнатной температуре. Добавляли DIPEA (72 мкл, 413 мкмоль) и сложный этиловый эфир (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (50 мг, 0,1 ммоль), и перемешивали полученную смесь при комнатной температуре в течение ночи. Смесь объединяли с EtOH (402 мкл, 6,9 ммоль) и 1 M LiOH в воде (1,1 мл, 1,1 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, упаривали в условиях пониженного давления и очищали методом препаративной HPLC с получением указанного в заголовке соединения (1,8 мг) в виде трифторацетата. МС m/z [M+H]+ расч. для C26H23ClN4O5, 507,14; обнаружено 507,2.
Пример 9B: сложный этоксикарбонилоксиметиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(5-оксо-1-фенил-4,5-дигидро-1H-[1,2,4]триазол-3-карбонил)амино]пентановой кислоты
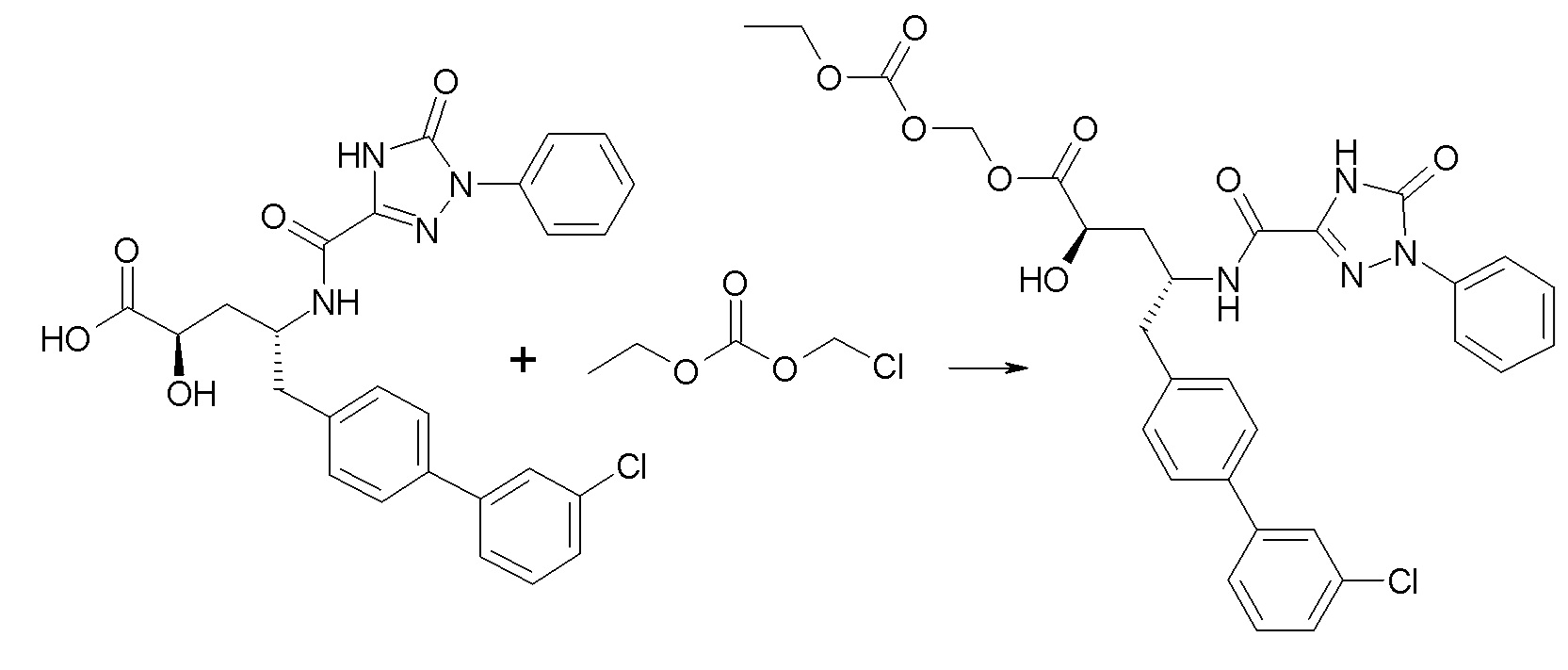
Раствор (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(5-оксо-1-фенил-4,5-дигидро-1H-[1,2,4]триазол-3-карбонил)амино]пентановой кислоты (253 мг, 0,5 ммоль), хлорметилэтилкарбоната (69 мг, 0,5 ммоль), 2,6-лутидина (165 мг, 1,5 ммоль) и NaI (150 мг, 1 ммоль) в DMF (20 мл) перемешивали в течение 15 часов при 50ºC. Смесь гасили добавлением воды (20 мл) и экстрагировали EtOAc (3×30 мл). Объединенные органические слои собирали и концентрировали в условиях вакуума. Остаток очищали методом колоночной хроматографии (петролейный эфир/EtOAc=3/1) с получением указанного в заголовке соединения в виде белого твердого вещества (13 мг). ЖХ-МС: 608,8 [M+H]+.1H ЯМР: (CDCl3, 400 МГц) δ 1,27 (т, J=8,0 Гц, 3H), 2,03-2,33 (м, 2H), 3,02-3,05 (м, 2H), 4,18 (кв., J=8,0 Гц, 2H), 4,49 (шир., 1H), 4,85 (шир., 1H), 5,57-5,76 (м, 2H), 7,03 (д, J=12 Гц, 1H), 7,25-7,40 (м, 4H), 7,42-7,51 (м, 3H), 7,53-7,61 (м, 3H), 7,93 (д, J=8,0 Гц, 2H).
Пример 9C: сложный (S)-2-метоксикарбониламино-3-метилбутирилоксиметиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(5-оксо-1-фенил-4,5-дигидро-1H-[1,2,4]триазол-3-карбонил)амино]пентановой кислоты

Указанное в заголовке соединение также может быть получено с использованием методик, описанных в настоящем документе.
Пример 10A: сложный этиловый эфир (2R,4R)-4-[(5-ацетил-2-фосфонооксиметил-2H-пиразол-3-карбонил)амино]-5-бифенил-4-ил-2-гидроксипентановой кислоты

Сложный этиловый эфир (2R,4R)-4-амино-5-бифенил-4-ил-2-гидроксипентановой кислоты (гидрохлорид; 500 мг, 1 ммоль, 1,0 экв.), 5-ацетил-2H-пиразол-3-карбоновую кислоту (330,4 мг, 2,1 ммоль, 1,5 экв.) и HATU (820 мг, 2,1 ммоль, 1,5 экв.) объединяли в DMF (5 мл), и перемешивали полученную смесь в течение 2 минут. Добавляли DIPEA (750 мкл), и перемешивали смесь в течение 1 часа. Смесь сушили в условиях вакуума, и очищали продукт методом хроматографии с обращенной фазой (10-70% MeCN/H2O; 0,05% TFA в течение 70 минут) с получением соединения 1 в виде трифторацетата (300 мг, чистота 98%). МС m/z [M+H]+ расч. для C25H27N3O5, 450,20; обнаружено 450,2.

К раствору соединения 1 (15,0 мг, 33,4 мкмоль) в DMF (103 мкл, 1,3 ммоль) добавляли K2CO3 (5,1 мг, 36,7 мкмоль) и сложный хлорметиловый эфир сложного ди-трет-бутилового эфира фосфорной кислоты (9,50 мг, 36,7 мкмоль). Полученную смесь перемешивали при комнатной температуре в течение ночи, а затем упаривали в условиях пониженного давления. Остаток разбавляли в EtOAc, и добавляли раствор 1 M HCl до получения pH 4-5. Органический слой дважды экстрагировали EtOAc, промывали водой, а затем насыщенным водным NaCl, сушили над безводным MgSO4, фильтровали и упаривали. Остаток очищали методом флэш-хроматографии (30-90% EtOAc в гексанах). Добавляли DCM (64,2 мкл, 1,0 ммоль), а затем TFA (40 мкл, 0,6 ммоль), и перемешивали полученную смесь при комнатной температуре в течение 20 минут. Смесь упаривали и очищали методом препаративной HPLC с получением указанного в заголовке соединения в виде трифторацетата (4 мг). МС m/z [M+H]+ расч. для C26H30N3O9P, 560,17; обнаружено 560,1.
Пример 10B: сложный этиловый эфир (2R,4R)-4-[(5-ацетил-2H-пиразол-3-карбонил)амино]-2-((S)-2-амино-3-метилбутирилокси)-5-бифенил-4-илпентановой кислоты
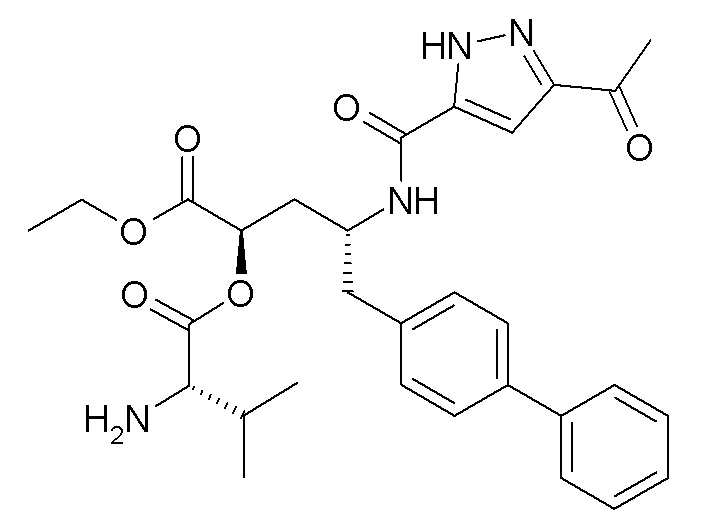
Указанное в заголовке соединение также может быть получено с использованием методик, описанных в настоящем документе.
Пример 10C: сложный этиловый эфир (2R,4R)-4-{[5-ацетил-2-((S)-2-амино-3-метилбутирилоксиметил)-2H-пиразол-3-карбонил]амино}-5-бифенил-4-ил-2-гидроксипентановой кислоты
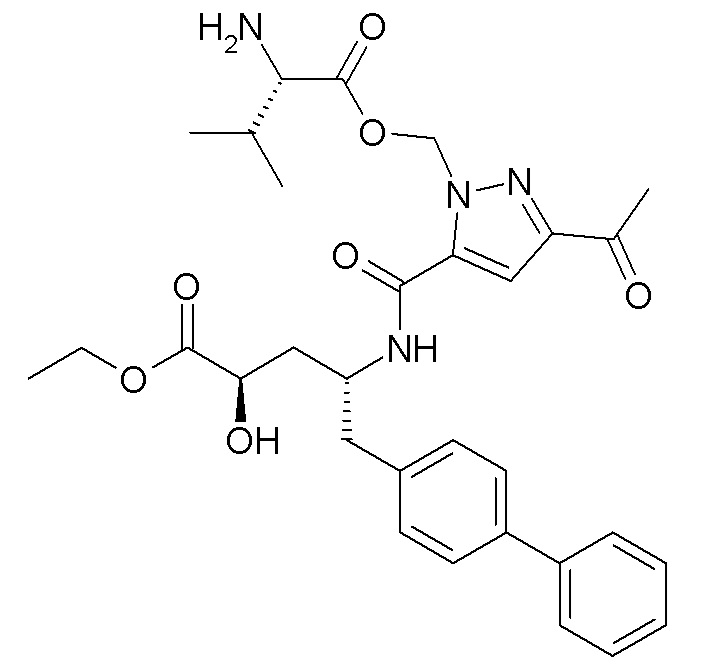
Указанное в заголовке соединение также может быть получено с использованием методик, описанных в настоящем документе.
Пример 11A: сложный этоксикарбонилоксиметиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(1H-тетразол-5-карбонил)амино]пентановой кислоты

Указанное в заголовке соединение также может быть получено с использованием методик, описанных в настоящем документе.
МЕТОД АНАЛИЗА
Методы анализа in vitro для количественной оценки значений ингибирующей активности (IC50) в отношении NEP человека и крысы и ACE человека
Значения ингибирующей активности соединения в отношении неприлизина (EC 3,4.24,11; NEP) человека и крысы и ангиотензинпревращающего фермента (ACE) человека определяли с использованием методов анализа in vitro, описанных ниже.
Приготовление экстракта из почек крысы для определения активности NEP
NEP крысы получали из почек взрослых крыс Sprague Dawley. Целые почки промывали в холодном фосфатно-солевом буфере (PBS) и загружали в ледяной лизирующий буфер (1% Triton X-114, 150 мМ NaCl, 50 мМ трис(гидроксиметил)аминометана (Tris) pH 7,5; Bordier (1981) J. Biol. Chem. 256: 1604-1607) в соотношении 5 мл буфера на каждый грамм почки. Образцы гомогенизировали на льду, используя портативный гомогенизатор ткани Polytron. Гомогенаты центрифугировали при 1000×g в бакет-роторе в течение 5 минут при 3°C. Осадок ресуспендировали в 20 мл ледяного лизирующего буфера и инкубировали на льду в течение 30 минут. Затем, образцы (15-20 мл) наносили слоями на 25 мл ледяного cushion-буфера (6% масс./об. сахароза, 50 мМ pH 7,5 Tris, 150 мМ NaCl, 0,06% Triton X-114), нагревали до 37°C в течение 3-5 минут и центрифугировали при 1000×g в бакет-роторе при комнатной температуре в течение 3 минут. Два верхних слоя удаляли путем аспирации, оставляя вязкий маслянистый осадок, содержащий обогащенную мембранную фракцию. Добавляли глицерин до концентрации 50%, и образцы хранили при -20°C. Концентрации белков определяли количественно, используя систему определения BCA с бычьим сывороточным альбумином (BSA) в качестве стандарта.
Методы анализа ингибирования фермента
Рекомбинантный NEP человека и рекомбинантный ACE человека приобретали из коммерческих источников (R&D Systems, Minneapolis, MN, кат. №№ 1182-ZN и 929-ZN, соответственно). Флуорогенный пептидный субстрат Mca-D-Arg-Arg-Leu-Dap-(Dnp)-OH (Medeiros et al. (1997) Braz. J. Med. Biol. Res. 30:1157-62; Anaspec, San Jose, CA) и Abz-Phe-Arg-Lys(Dnp)-Pro-OH (Araujo et al. (2000) Biochemistry 39:8519-8525; Bachem, Torrance, CA) использовали при анализе NEP и ACE, соответственно.
Анализ проводили в 384-луночных белых матовых планшетах при 37°C, используя флуорогенные пептидные субстраты в концентрации 10 мкМ в аналитическом буфере (NEP: 50 мМ HEPES, pH 7,5, 100 мМ NaCl, 0,01% полиэтиленгликольсорбитанмонолаурат (Tween-20), 10 мкМ ZnSO4; ACE: 50 мМ HEPES, pH 7,5, 100 мМ NaCl, 0,01% Tween-20, 1 мкМ ZnSO4). Соответствующие ферменты использовали в концентрациях, которые приводили к количественно оцениваемому протеолизу 1 мкМ субстрата через 20 минут при 37°C.
Тестируемые соединения анализировали в диапазоне концентраций от 10 мкМ до 20 пМ. Тестируемые соединения добавляли к ферментам и инкубировали в течение 30 минут при 37°C до инициирования реакции путем добавления субстрата. Реакции останавливали после 20 минут инкубации при 37°C путем добавления ледяной уксусной кислоты до конечной концентрации 3,6% (об./об.).
Планшеты считывали на флуориметре с длинами волн возбуждения и излучения, установленными на 320 нм и 405 нм, соответственно. Константы ингибирования получали при помощи нелинейной регрессии данных с использованием уравнения (GraphPad Software, Inc., San Diego, CA):
v=v0/[1+(I/K')],
где v представляет собой скорость реакции, v0 представляет собой скорость неингибируемой реакции, I представляет собой концентрацию ингибитора, и K' представляет собой кажущуюся константу ингибирования.
Соединение формулы I' (пример 1A) было протестировано в данном методе анализа, и было обнаружено, что оно характеризуется величиной pKi≥9,0 в отношении NEP человека, и было обнаружено, что следующие пролекарства также характеризуются активностью:
Соединения-пролекарства, полученные в примерах 1B-F, 1H, 1J-K и 1N-P, не тестировались, поскольку в данном методе анализа in vitro активность не предполагалась; однако предполагается, что в зависимости от активности активной формы пролекарства они характеризуются активностью в отношении NEP in vivo.
Соединение формулы II' (пример 2A) было протестировано в данном методе анализа, и было обнаружено, что оно характеризуется величиной pKi≥9,0 в отношении NEP человека. Соединения-пролекарства, полученные в примерах 2B-I, либо не ингибировали фермент в данном методе анализа in vitro, либо не тестировались, поскольку в данном методе анализа активность не предполагалась; однако предполагается, что в зависимости от активности активной формы пролекарства они характеризуются активностью в отношении NEP in vivo.
Соединение формулы III' (пример 3A) было протестировано в данном методе анализа, и было обнаружено, что оно характеризуется величиной pKi≥9,0 в отношении NEP человека. Соединения-пролекарства, полученные в примерах 3B-E, либо не ингибировали фермент в данном методе анализа in vitro, либо не тестировались, поскольку в данном методе анализа активность не предполагалась; однако предполагается, что в зависимости от активности активной формы пролекарства они характеризуются активностью в отношении NEP in vivo.
Следующие соединения согласно настоящему изобретению тестировали в данном методе анализа, и было обнаружено, что они характеризуются величинами pKi в отношении NEP человека, указанными ниже. В общем, соединения-пролекарства либо не ингибировали фермент в данном методе анализа in vitro, либо не тестировались (n.d.), поскольку их активность не предполагалась.
Соединение формулы VII' (пример 7A) тестировали в данном методе анализе, и было обнаружено, что оно характеризуется величиной pKi≥9,0 в отношении NEP человека. Соединения-пролекарства, полученные в примерах 7B-E, либо не ингибировали фермент в данном методе анализа in vitro, либо не тестировались, поскольку в данном методе анализа активность не предполагалась; однако предполагается, что в зависимости от активности активной формы пролекарства они характеризуются активностью в отношении NEP in vivo.
Соединение формулы VIII' (пример 8A) тестировали в данном методе анализе, и было обнаружено, что оно характеризуется величиной pKi≥9,0 в отношении NEP человека. Соединения-пролекарства, полученные в примерах 8B-L, либо не ингибировали фермент в данном методе анализа in vitro, либо не тестировались, поскольку в данном методе анализа активность не предполагалась; однако предполагается, что в зависимости от активности активной формы пролекарства они характеризуются активностью в отношении NEP in vivo. Предполагается, что соединения-пролекарства, полученные в примере 8I, характеризуются активностью в отношении NEP in vivo.
Соединение формулы IX' (пример 9A) тестировали в данном методе анализе, и было обнаружено, что оно характеризуется величиной pKi≥9,0 в отношении NEP человека. Соединения-пролекарства, полученные в примерах 9B-C, либо не ингибировали фермент в данном методе анализа in vitro, либо не тестировались, поскольку в данном методе анализа активность не предполагалась; однако предполагается, что в зависимости от активности активной формы пролекарства они характеризуются активностью в отношении NEP in vivo.
Соединение формулы X' тестировали в данном методе анализе, и было обнаружено, что оно характеризуется величиной pKi≥9,0 в отношении NEP человека. Соединение-пролекарство, полученное в примере 10A, либо не ингибировало фермент в данном методе анализа in vitro, либо не тестировалось, поскольку в данном методе анализа активность не предполагалась; однако предполагается, что в зависимости от активности активной формы пролекарства они характеризуются активностью в отношении NEP in vivo.
Соединение-пролекарство, полученное в примере 11A, либо не ингибировало фермент в данном методе анализа in vitro, либо не тестировалось, поскольку в данном методе анализа активность не предполагалась; однако предполагается, что данное пролекарство характеризуется активностью в отношении NEP in vivo.
Хотя настоящее изобретение было описано со ссылкой на конкретные аспекты и варианты осуществления, средним специалистам в данной области техники следует понимать, что различные изменения могут быть сделаны, или эквиваленты могут быть заменены, не выходя за рамки объема и сущности настоящего изобретения. Кроме того, в случаях, разрешенных применимыми патентными законами и положениями, все процитированные в настоящем документе публикации, патенты и заявки на выдачу патента включены в настоящий документ во всей своей полноте посредством ссылки в той же мере, как если бы каждый документ был в отдельности включен в настоящий документ посредством ссылки.
Реферат
Изобретение относится к соединениям, характеризующимся формулой XII, где значения R, R, R, Rи X определены в формуле изобретения, или к их фармацевтически приемлемой соли. Соединение по изобретению обладает ингибирующей активностью в отношении неприлизина (NEP) и предназначено для получения лекарственного средства для лечения гипертензии, сердечной недостаточности или заболевания почек. 7 н. и 21 з.п. ф-лы, 11 пр.
Формула
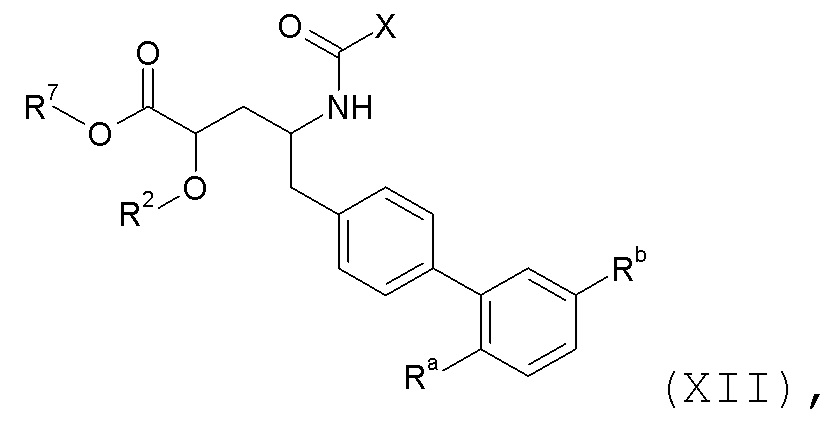
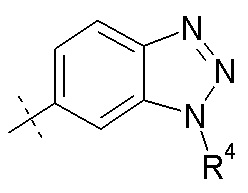




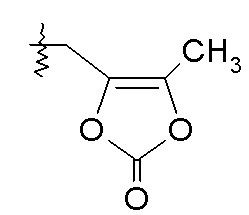

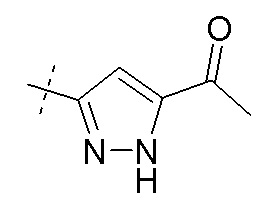
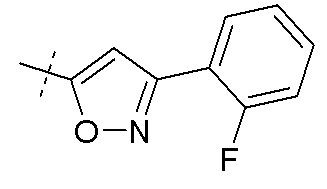

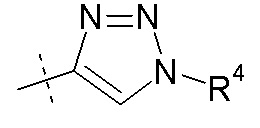
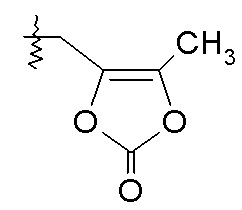




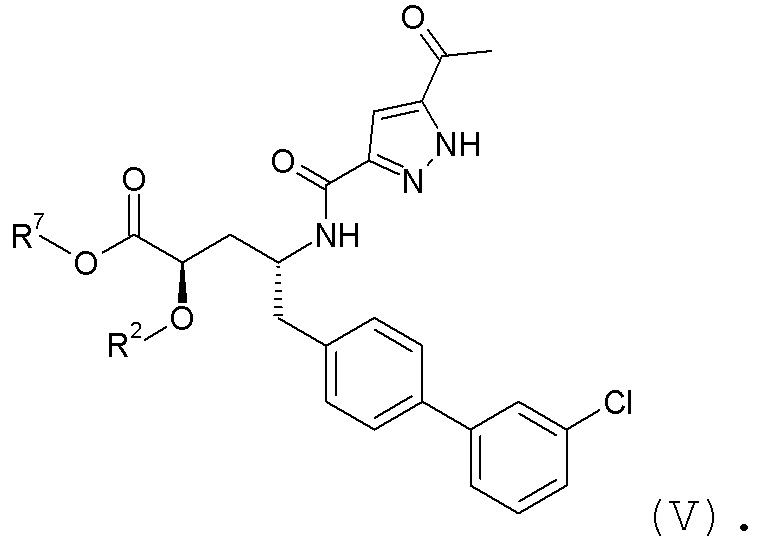









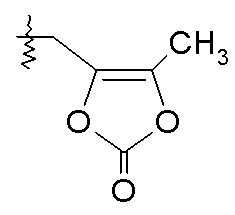
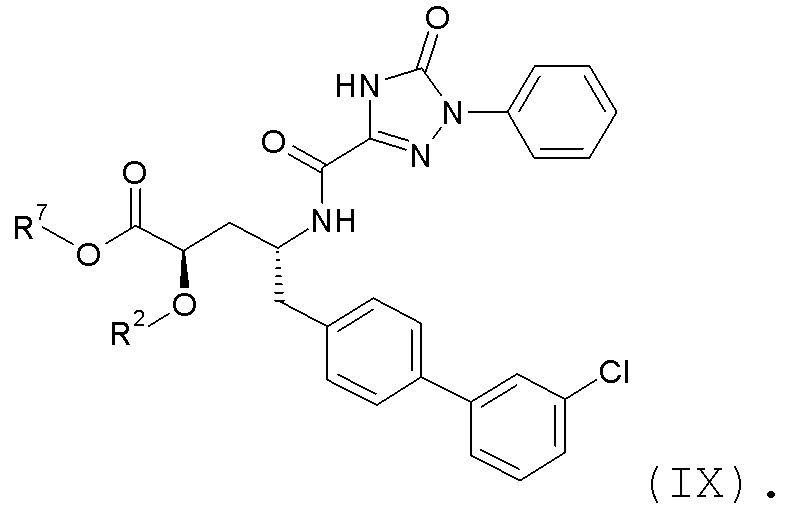
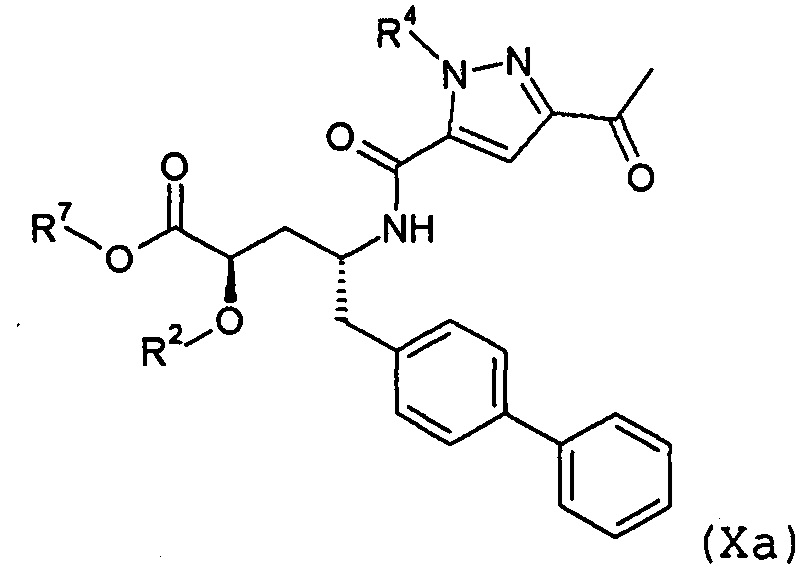


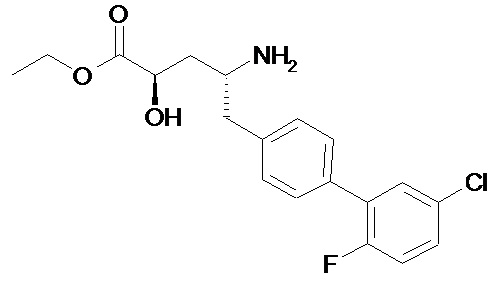
Документы, цитированные в отчёте о поиске
Новые биароматические соединения-активаторы рецепторов типа ppary и их применение в косметических или фармацевтических композициях
Комментарии