Производное изоиндолина, промежуточный продукт, способ получения, фармацевтическая композиция и ее применение - RU2695521C9
Код документа: RU2695521C9
Описание
Настоящая заявка испрашивает приоритет заявки на патент Китая CN 2014/10605148.8, поданной 30 октября 2014 г., и заявки на патент Китая CN 2014/10632870.0, поданной 11 ноября 2014 г., содержание которых включено в данный документ посредством ссылки во всей их полноте.
Область изобретения
Настоящее изобретение относится к производному изоиндолина, промежуточному продукту, способу получения, фармацевтической композиции и ее применению.
Предшествующий уровень техники
Фактор-α некроза опухоли (TNF-α) представляет собой вид провоспалительного цитокина, который играет важную роль в иммунном гомеостазе, воспалении и иммунной защите. Было доказано, что TNF-α является одним из основных медиаторов воспаления. TNF-α также может продуцироваться опухолями, и может играть роль в стимулировании образования опухолей, также может вызывать запрограммированную гибель клеток опухолей. Кроме того, TNF-α также влияет на процессы, такие как апоптоз, некроз, ангиогенез, активация иммунных клеток, дифференциация и миграция клеток, все эти процессы играют важную роль в онкогенезе и прогрессировании опухолей.
Неконтролируемая активность TNF-α или чрезмерное продуцирование TNF-α связано с патологией различных заболеваний, в том числе без ограничения формы рака, такие как рак толстой кишки, прямой кишки, предстательной железы, молочной железы, головного мозга и толстой кишки; а также воспалительные заболевания, в частности воспаление, связанное с раком. Дисрегуляция TNF-α может приводить также к аутоиммунным заболеваниям, синдрому токсического шока, кахексии, артриту, псориазу, HIV-инфекции и AIDS, неврологическим заболеваниям и заболеваниям центральной нервной системы, сепсису, застойной сердечной недостаточности, отторжению аллотрансплантата и вирусным инфекциям. Таким образом, уменьшение уровня TNF-α или регулирование активности TNF-α является перспективной стратегией в лечении многих иммунологических, воспалительных и злокачественных заболеваний (например, форм рака и воспаления). Как например, Sethi et al. Front. Biosci. (2008) 13, 5094-5107 Results Prob. Cell Differ. (2009) 49, 1-15.
Леналидомид (3-(4-амино-1,3-дигидро-1-оксо-2H-изоиндол-2-ил)пиперидин-2,6-дион) представляет собой низкомолекулярный иммунный регулятор, было доказано, что он способен ингибировать секрецию TNF-α и других провоспалительных цитокинов, а также увеличивать секрецию противовоспалительных цитокинов. Леналидомид был одобрен для лечения множественной миеломы (в 2006 г.), миелодиспластического синдрома (в 2005 г.) и лимфомы из клеток мантийной зоны (в 2013 г.). Кроме того, в клинических испытаниях леналидомид, отдельно или в комбинации с другими терапевтическими средствами, способен лечить неходжкинскую лимфому, папиллярную и фолликулярную тиреоидную карциному, рак предстательной железы, хронический лимфоцитарный лейкоз, амилоидоз, комплексный регионарный болевой синдром I типа, злокачественную меланому, заболевание корешка нерва, миелофиброз, глиобластому, глиосаркому, злокачественную глиому, миелоидный лейкоз, рефракторную плазмоцитому, хронический миеломоноцитарный лейкоз, фолликулярную лимфому, меланому цилиарного тела и хроническую меланому, меланому радужной оболочки, рецидивирующую интраокулярную меланому, экстраокулярную распространяющуюся меланому, солидную опухоль, Т-клеточную лимфому, эритроидную лимфому, монобластный и моноцитарный лейкоз; миелоидный лейкоз и опухоли головного мозга, менингиому, опухоль спинного мозга, рак щитовидной железы, лимфому из клеток мантийной зоны, немелкоклеточный рак легкого, карциному яичника, почечно-клеточную карциному, миелофиброз, лимфому Беркитта, лимфому Ходжкина, крупноклеточную лимфому и макроглобулинемию (см. WO 2012/015986).
Однако леналидомид обладает множеством побочных действий. Более того, в информации о назначении леналидомида четко указано, что лекарственное средство характеризуется риском вызывать миелосупрессию, тромбоз глубоких вен, легочную эмболию и тератогенез. Во время клинических испытаний большинству пациентов, принимающих леналидомид, требуется снижение дозы из-за гематологической токсичности. Таким образом, несмотря на то, что леналидомид обладает полезной активностью, его действенность ограничена возникновением значительных побочных эффектов. Таким образом, срочно требуются производные леналидомида, характеризующиеся улучшенными структурами для оптимизации его характеристик в сфере применения.
Описание настоящего изобретения
Настоящее изобретение предусматривает производное изоиндолина, промежуточный продукт, способ получения, фармацевтическую композицию и ее применение. Производное изоиндолина по настоящему изобретению способно регулировать получение или активность цитокинов (например, TNF-α), с тем чтобы эффективно лечить формы рака и воспалительные заболевания.
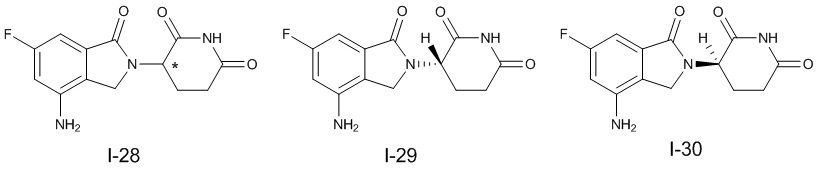
Настоящее изобретение предусматривает производное изоиндолина, представленное общей формулой (I), его фармацевтически приемлемую соль, сольват, полиморф, стереоизомер, изотопное соединение, метаболит или пролекарство;

в общей формуле (I) n1 выбран из 0 или 1;
Z представляет собой

каждый из R1, R3, R4, R5, R6, R7, R8 и R9независимо выбран из H или D;
R2 выбраниз H, D или галогена;
каждый из L1 и L2 независимовыбран из CD2, CHD или CH2;
Xвыбран из NH, ND или O;
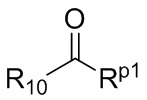
R10 представляет собой H, D или



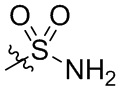

заместитель, содержащийся в замещенном (C1-C12)алкокси, выбран из группы, состоящей из D, галогена, гидроксила, (C1-C12)алкокси, (C2-C20)гетероциклоалкила, (C2-C20)гетероциклоалкила, замещенного (C1-C12)алкилом,

заместитель, содержащийся в замещенном (C1-C12)алкиле, выбран из группы, состоящей из D, (C2-C20)гетероциклоалкила, дейтерированного (C2-C20)гетероциклоалкила, (C2-C20)гетероциклоалкила, замещенного (C1-C12)алкилом, и (C2-C20)гетероциклоалкила, замещенного дейтерированным (C1-C12)алкилом;
в случае если в замещенном (C1-C12)алкокси или замещенном (C1-C12)алкокси содержится больше одного заместителя, – заместители являются одинаковыми или разными;
в каждой из вышеупомянутых групп гетероатом (C2-C20)гетероциклоалкила, содержащийся в (C2-C20)гетероциклоалкиле, дейтерированном (C2-C20)гетероциклоалкиле, (C2-C20)гетероциклоалкиле, замещенном (C1-C12)алкилом, или (C2-C20)гетероциклоалкиле, замещенном дейтерированным (C1-C12)алкилом, выбран из группы, состоящей из O, N и S;
при условии, что в общей формуле (I), в случае если n1 представляет собой 0, R1, R3и R10 представляют собой H или D, X представляет собой NH или ND, R2 представляет собой галоген;
при условии, что в общей формуле (I), в случае если n1 представляет собой 1, X представляет собой O и R2 представляет собой H или D, R10 представляет собой


при условии, что в общей формуле (I), в случае если n1 представляет собой 1 и X представляет собой NH, R10представляет собой

при условии, что в общей формуле (I) и Z, в случае если n1 представляет собой 1, X представляет собой NH, R1-R9 представляют собой H, и как L1, так и L2 представляет собой CH2, R10 не является
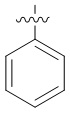

Предпочтительно, в общей формуле (I) центр асимметрии относится к ахиральному атому углерода, атому углерода с (S)-конфигурацией, обогащенному атому углерода с (S)-конфигурацией, атому углерода с (R)-конфигурацией, обогащенному атому углерода с (R)-конфигурацией или рацемату.
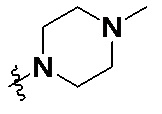
В общей формуле (I) Z предпочтительно выбран из группы, состоящей из

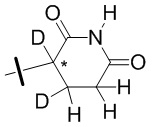






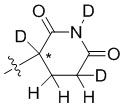

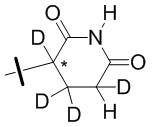
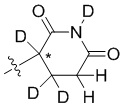



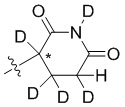
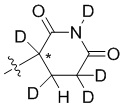
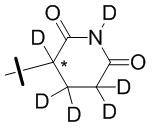


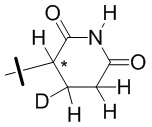
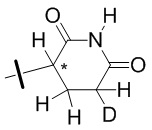




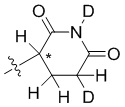


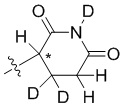
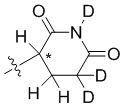

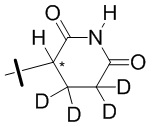

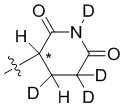

В каждый из вышеупомянутых групп (C2-C20)гетероциклоалкил, содержащийся в (C2-C20)гетероциклоалкиле, дейтерированном (C2-C20)гетероциклоалкиле, (C2-C20)гетероциклоалкиле, замещенном (C1-C12)алкилом, или (C2-C20)гетероциклоалкиле, замещенном дейтерированным (C1-C12)алкилом, предпочтительно представляет собой (C2-C6)гетероциклоалкил, содержащий 1 или 2 гетероатома, выбранных из N или O. (C2-C6)гетероциклоалкил предпочтительно представляет собой пирролидин (например,









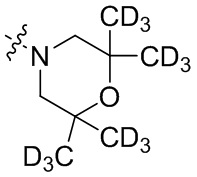

В общей формуле (I), в случае если R10 представляет собой

В общей формуле (I), в случае если R10 представляет собой
В общей формуле (I), в случае если R10 представляет собой

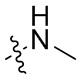
В общей формуле (I), в случае если R10 представляет собой

В общей формуле (I), в случае если R10 представляет собой
В общей формуле (I), в случае если R10 представляет собой
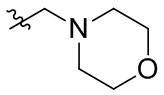
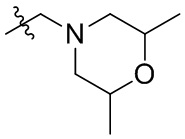

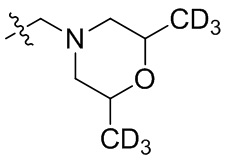
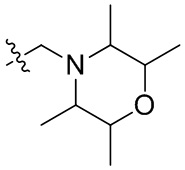
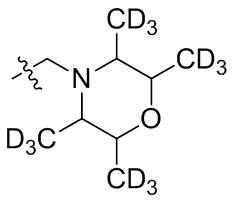
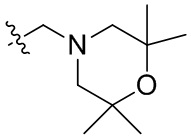

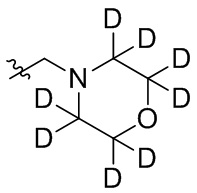

В общей формуле (I), в случае если R10 представляет собой






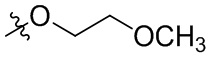

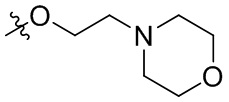
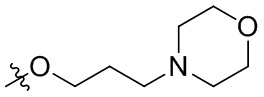

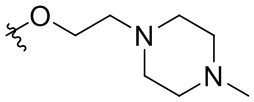
В общей формуле (I), в случае если R10 представляет собой
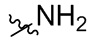

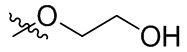
В общей формуле (I), в случае если R10 представляет собой
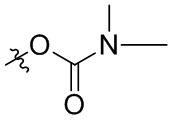
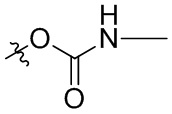
В общей формуле (I), в случае если R10 представляет собой


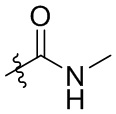
В общей формуле (I)
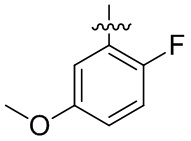
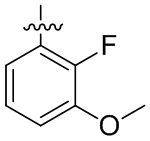


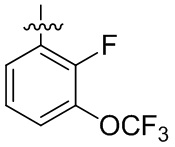
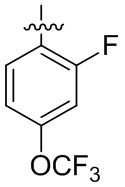




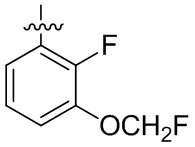
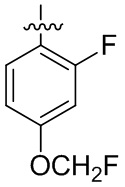


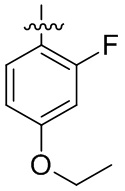
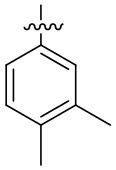

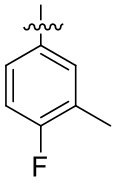
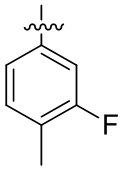

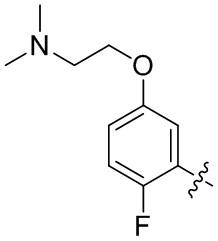



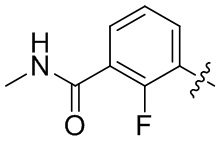
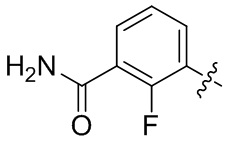
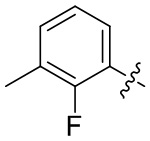






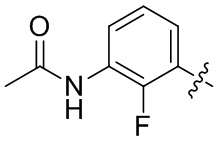
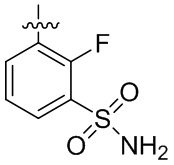


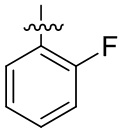
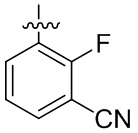






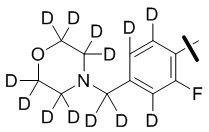

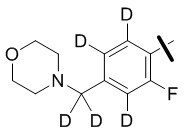
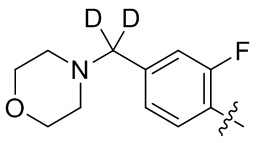

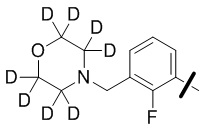
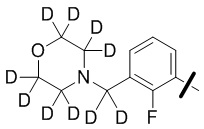
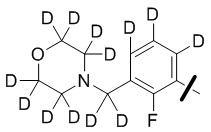

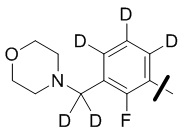


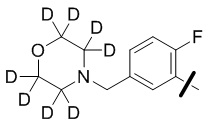
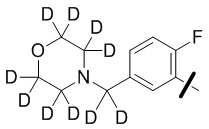

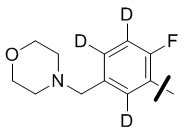

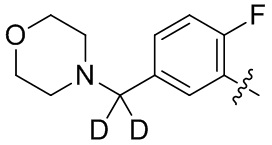
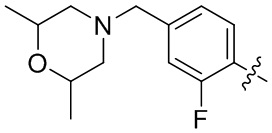







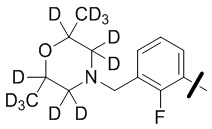
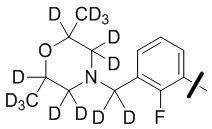
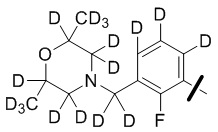

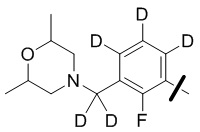
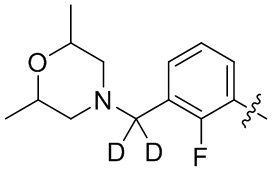










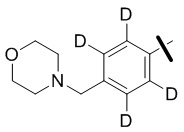


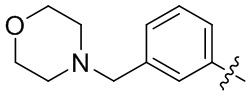
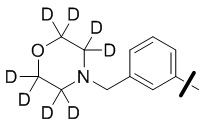

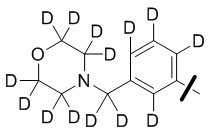

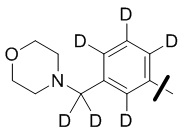

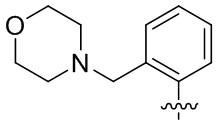



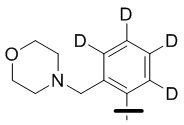

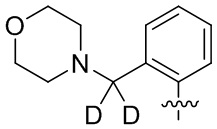



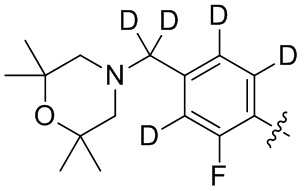
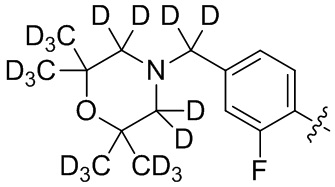





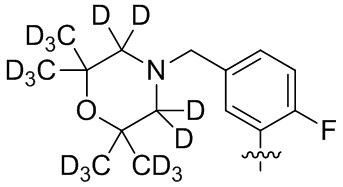

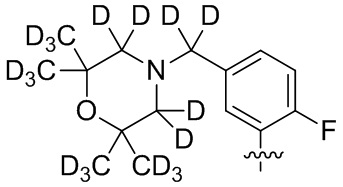


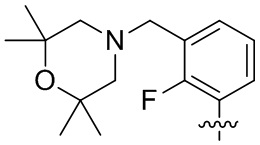
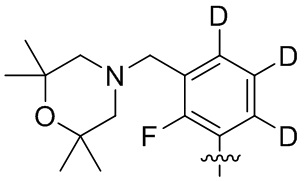
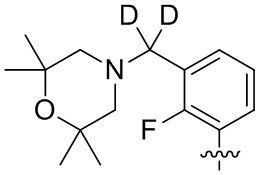
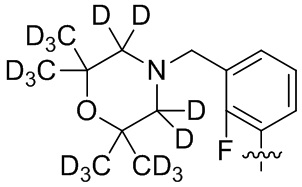
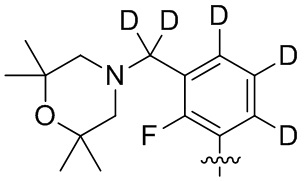



В общей формуле (I) предпочтительно, в случае если n1 представляет собой 1, R2 представляет собой H или D.
В общей формуле (I) предпочтительно, в случае если n1 представляет собой 1 и R2 представляет собой H или D, R10 представляет собой

В общей формуле (I), в случае если n1 представляет собой 1, R2 представляет собой H или D, X представляет собой NH или ND и R10 представляет собой
В общей формуле (I) предпочтительно, в случае если n1 представляет собой 1, R2представляет собой галоген и R10 представляет собой скорее






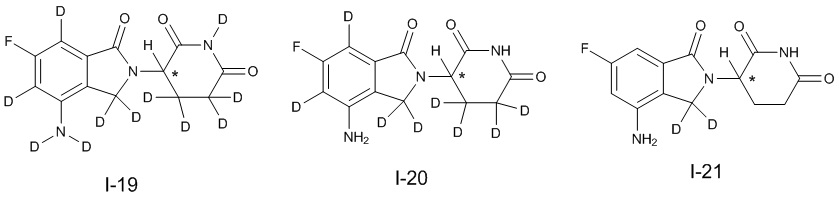

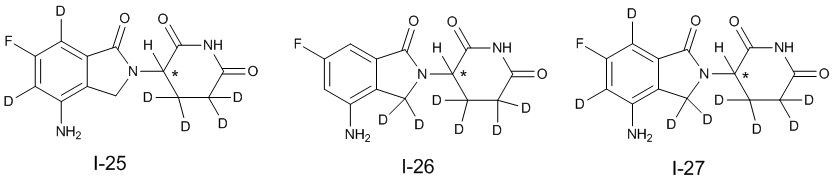

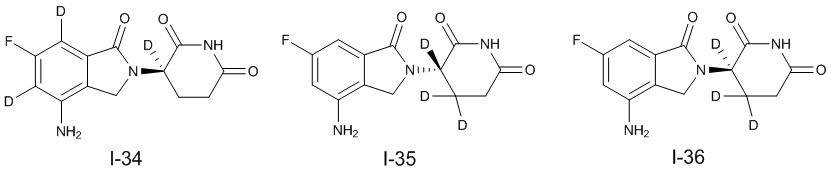

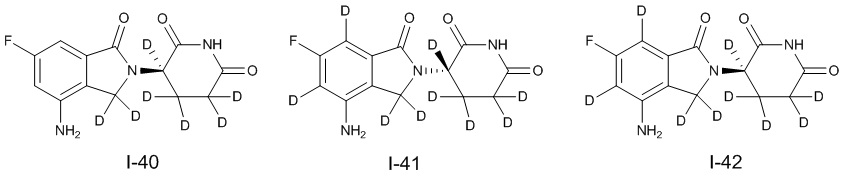
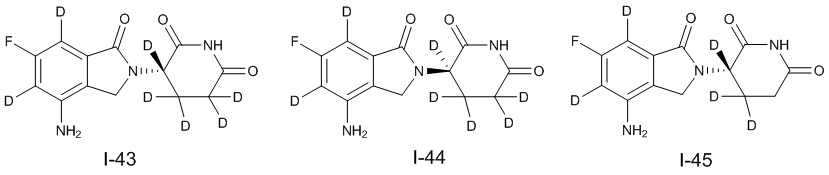
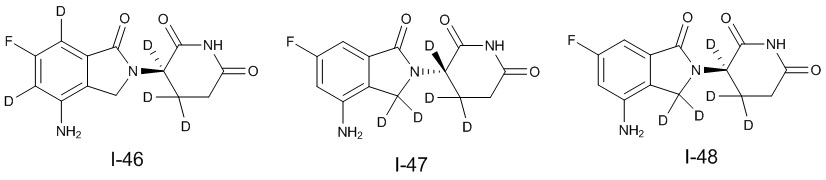


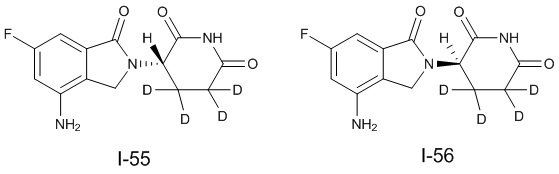


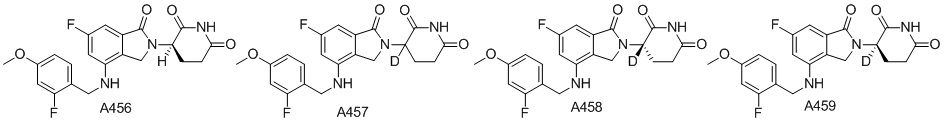


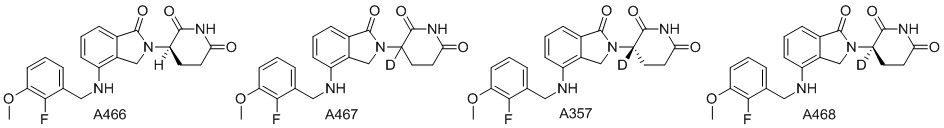




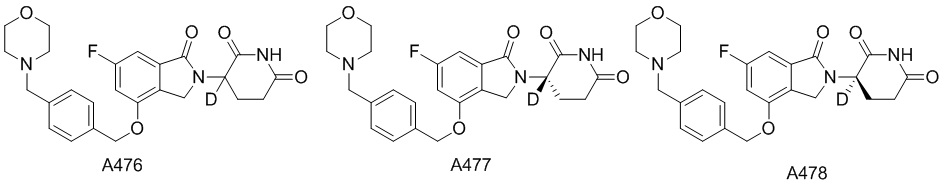

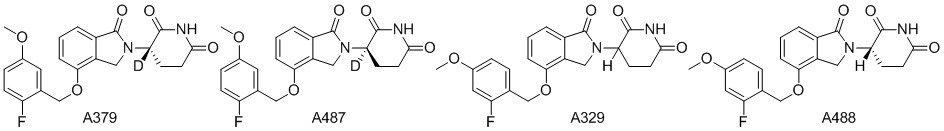





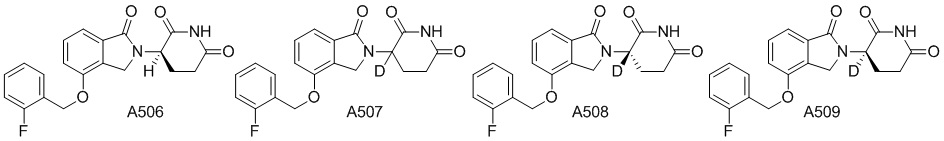
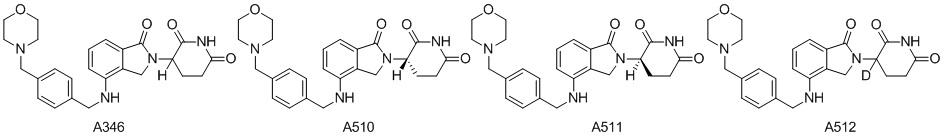

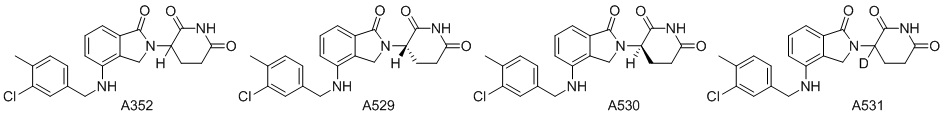
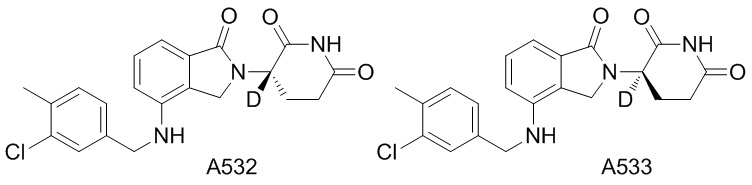

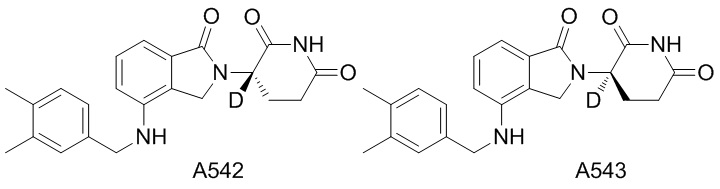






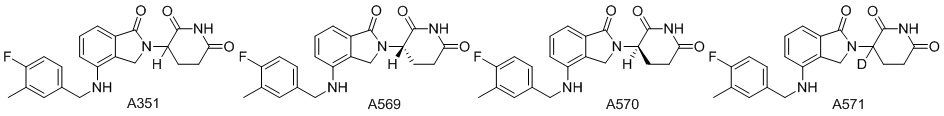


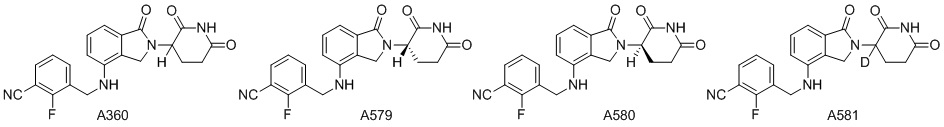


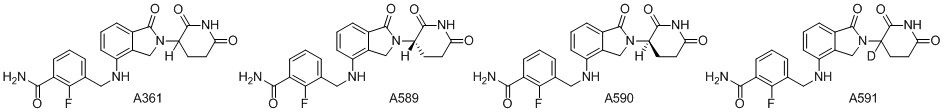


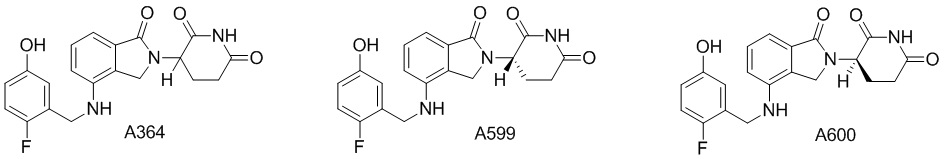
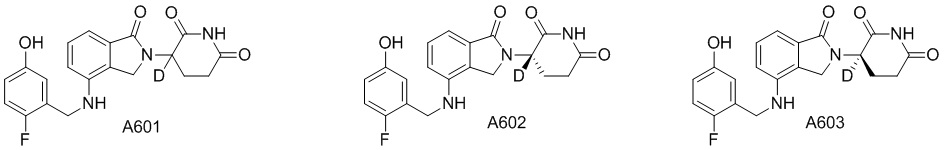

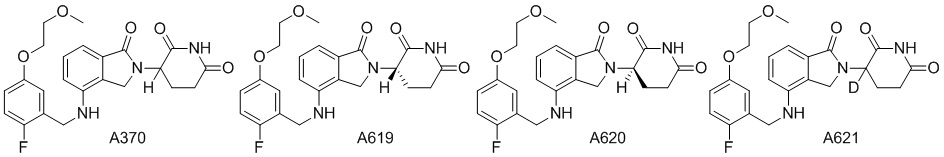

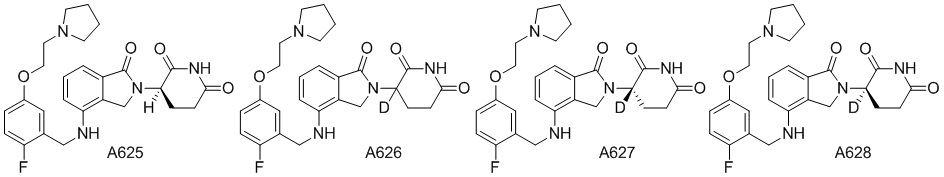

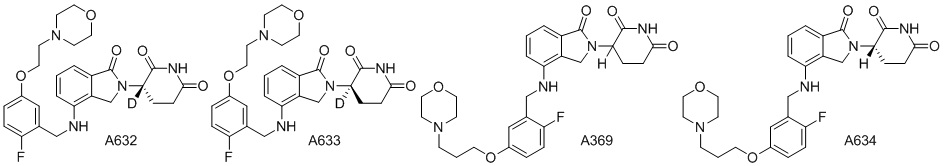
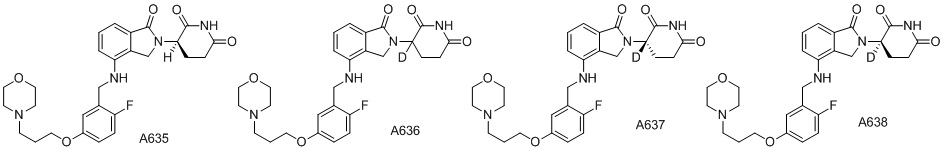


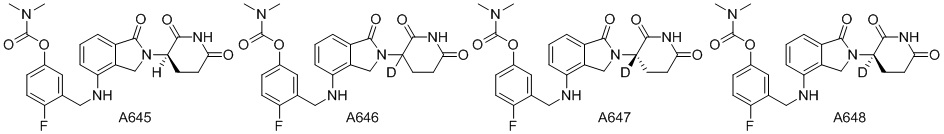











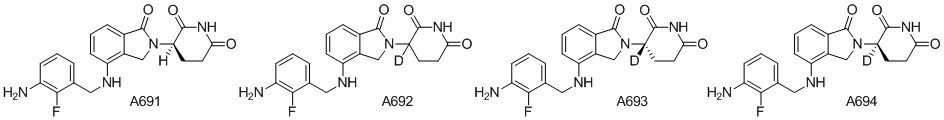
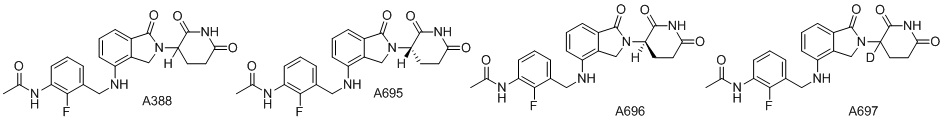










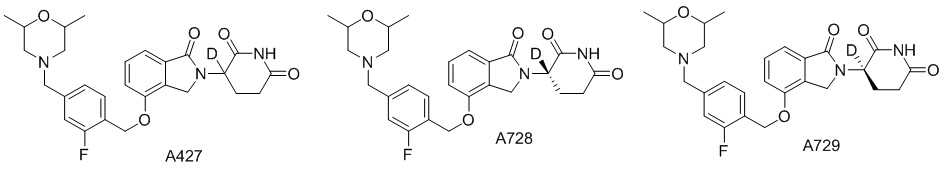


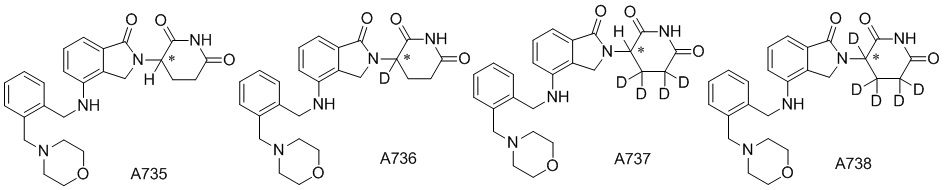
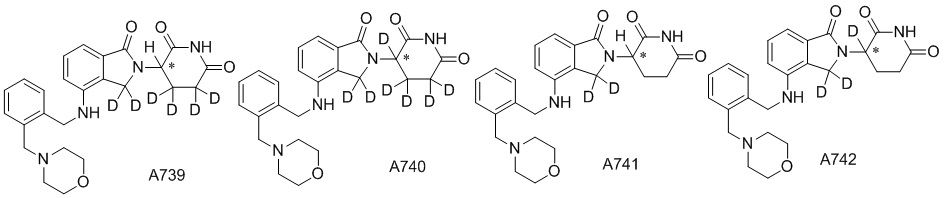





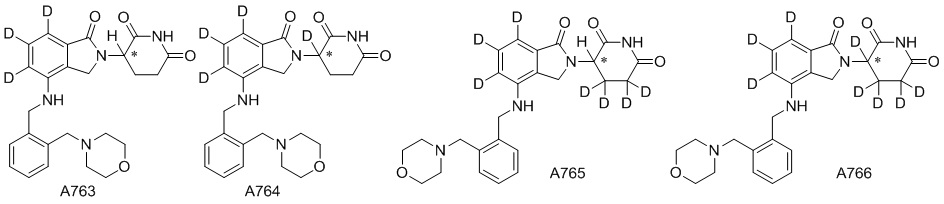
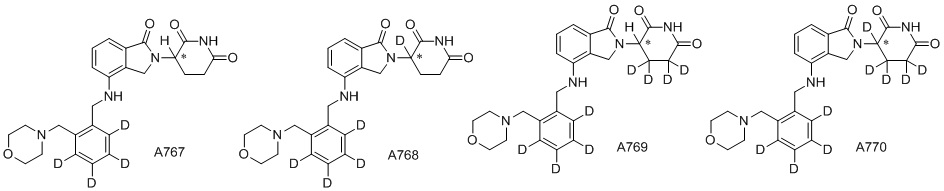



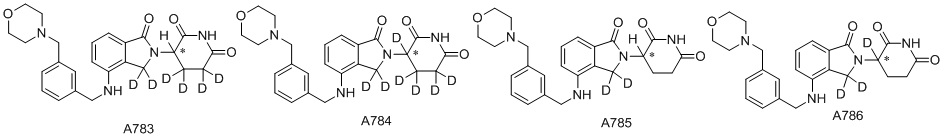
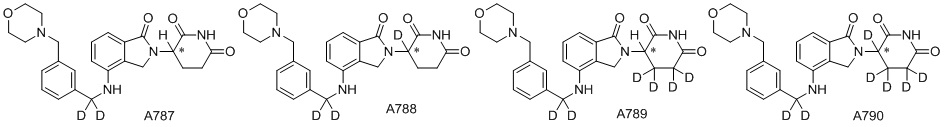
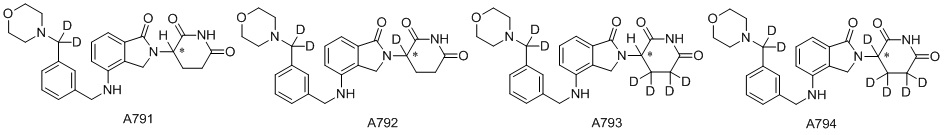
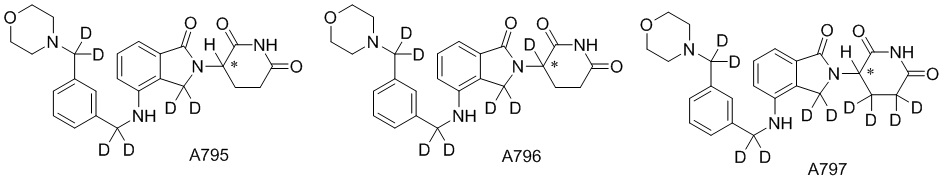
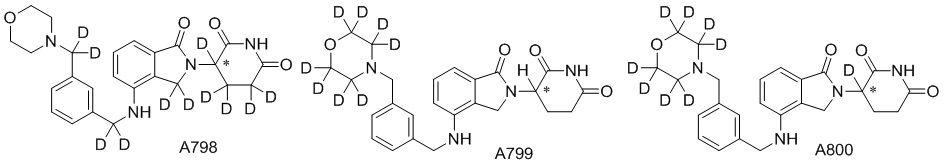
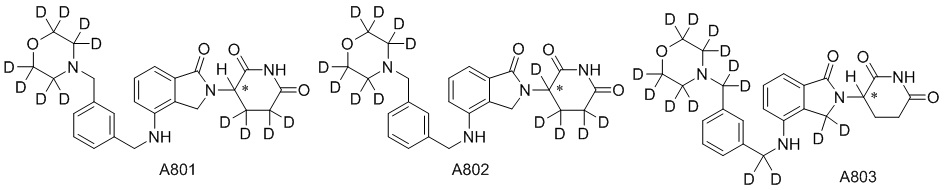



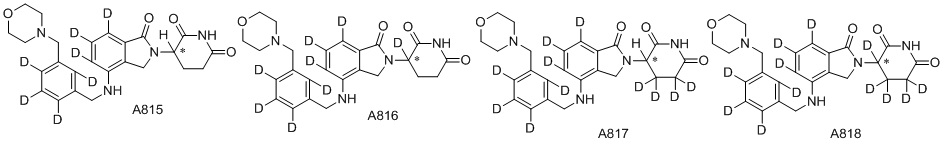
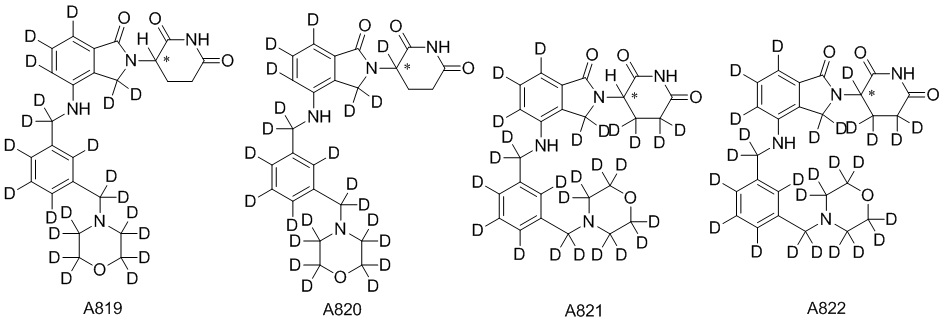
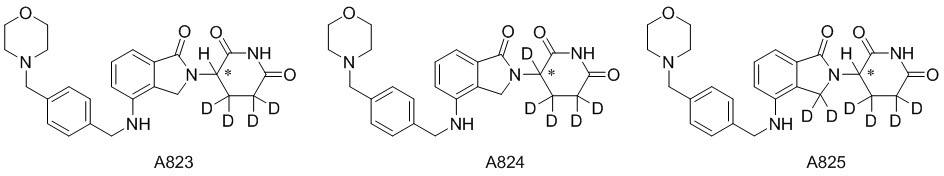



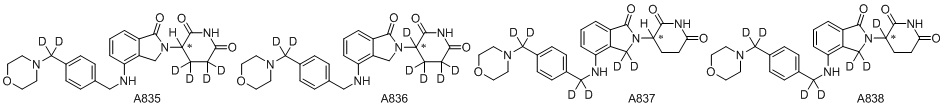





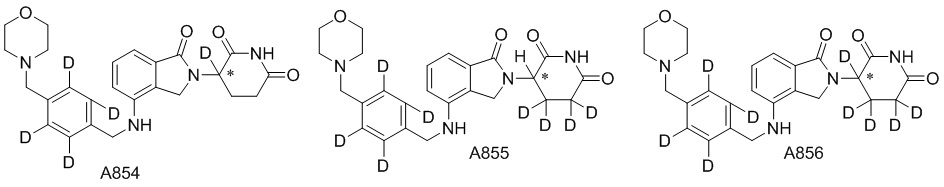
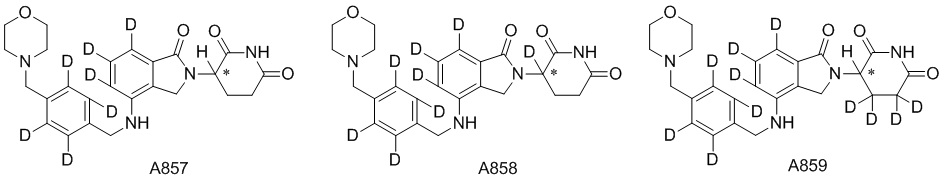



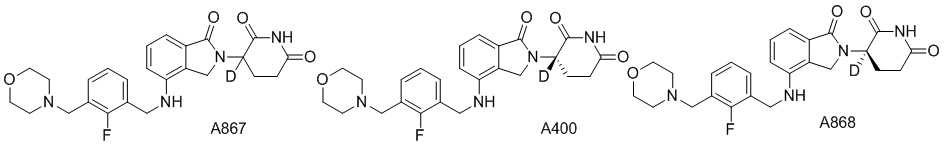

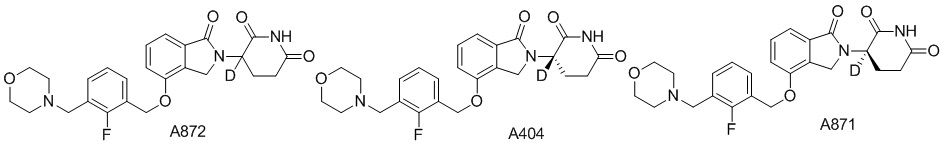

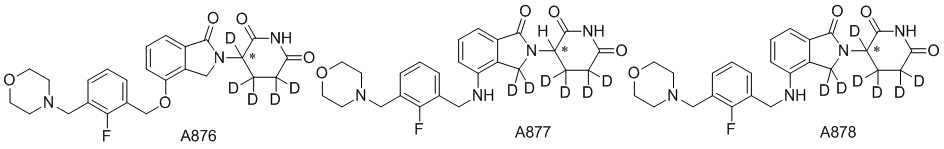
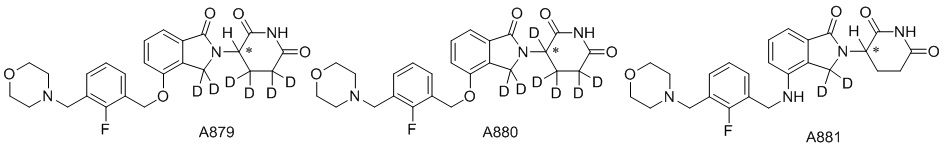
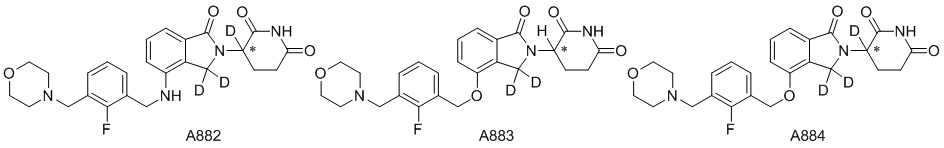



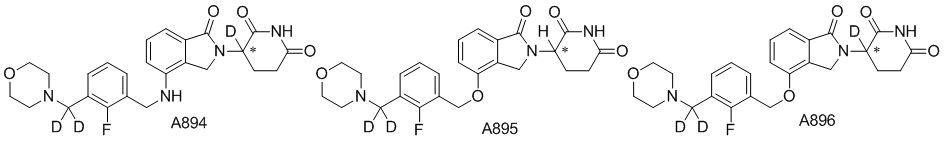
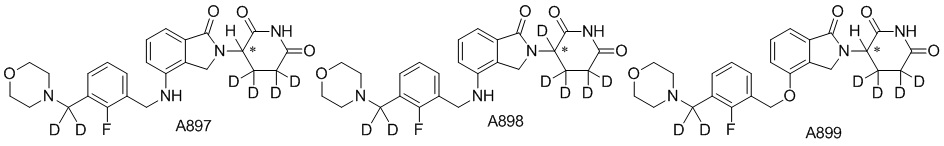
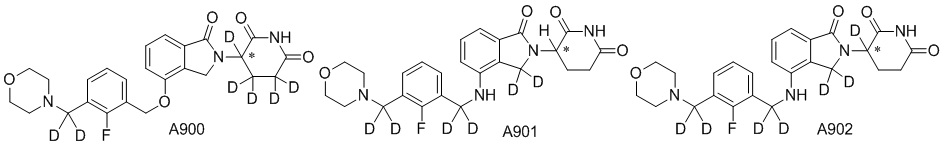

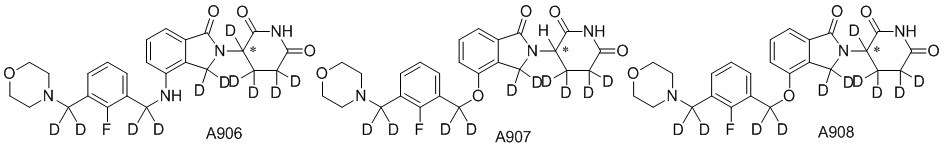

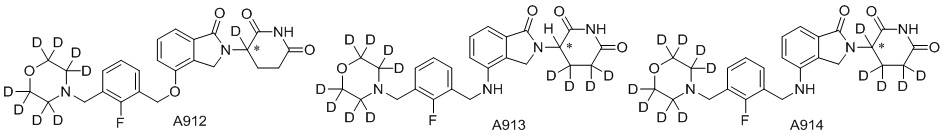


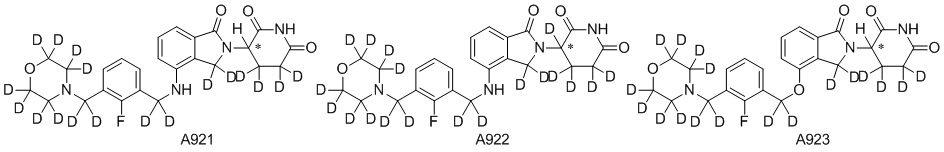

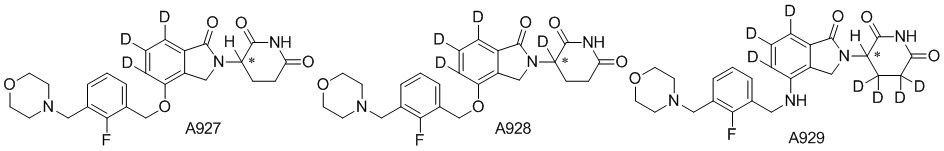
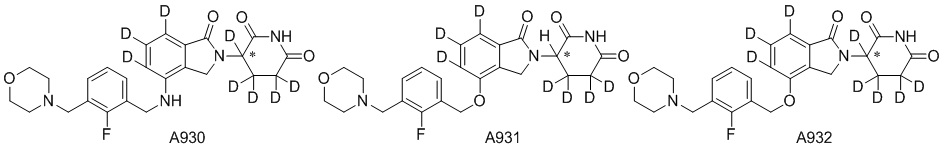
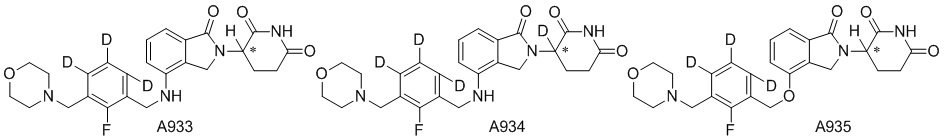





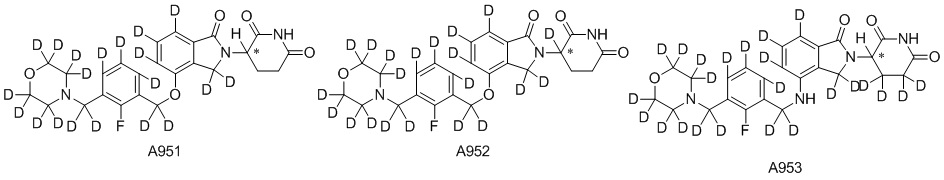
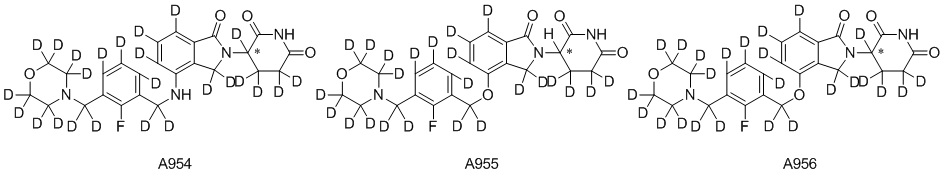


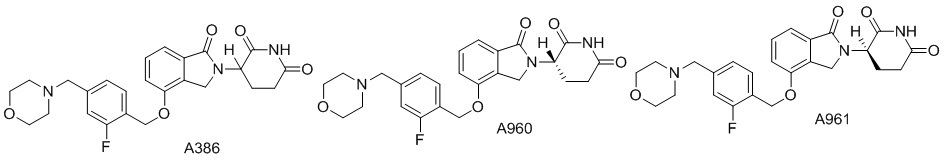


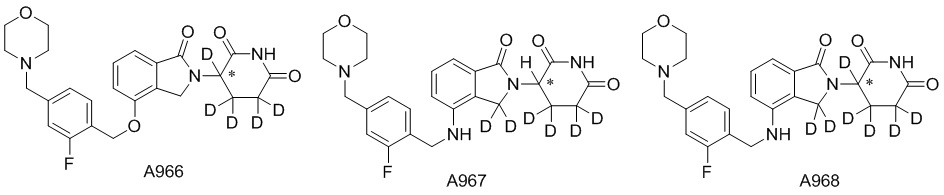
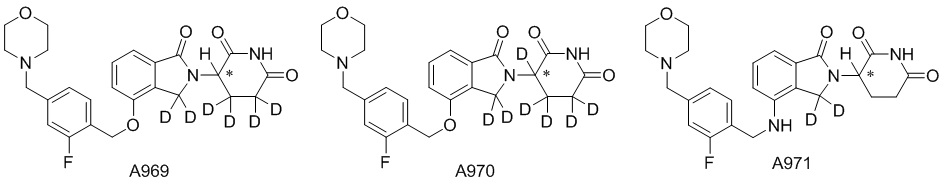
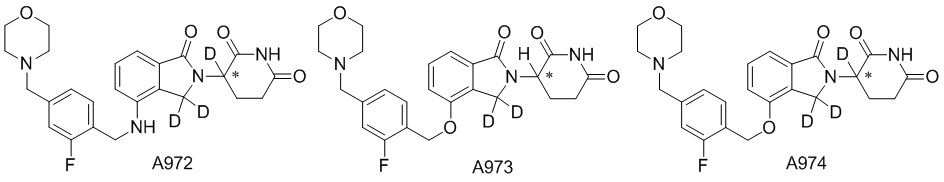
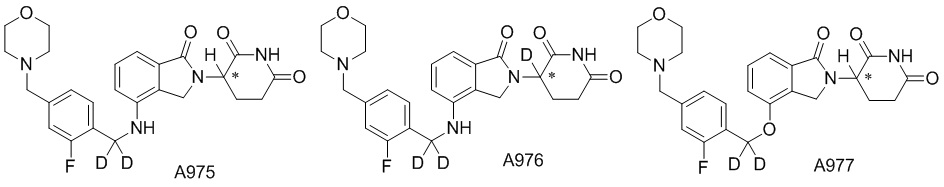


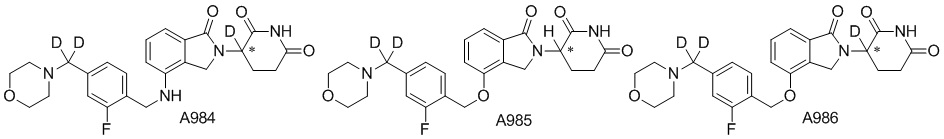
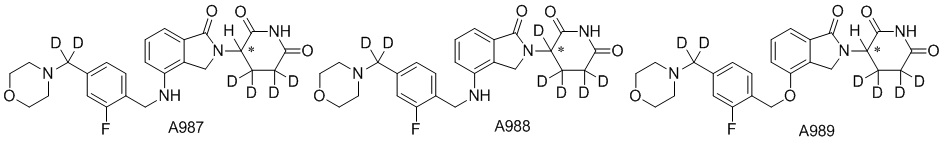


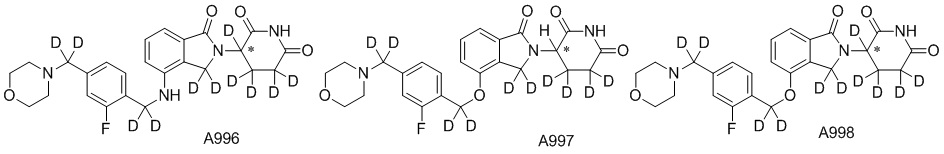
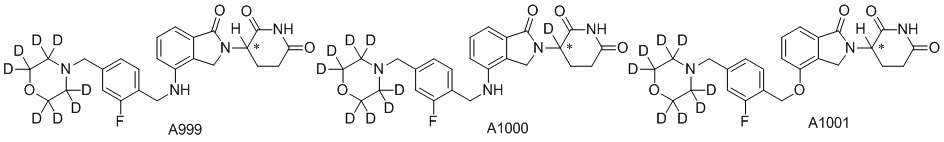




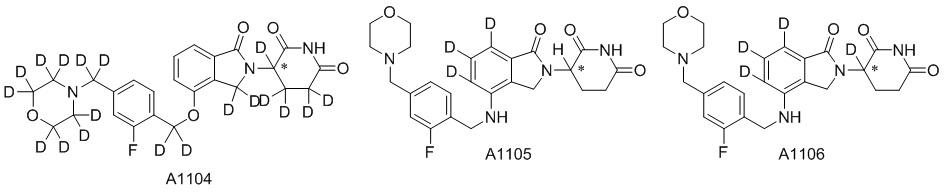
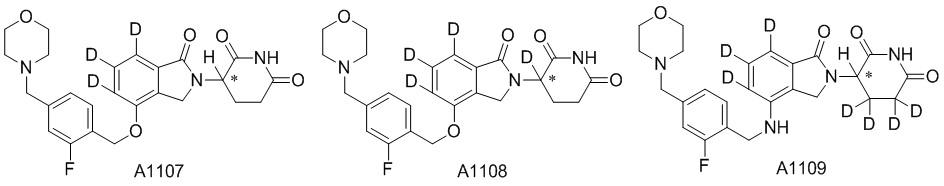
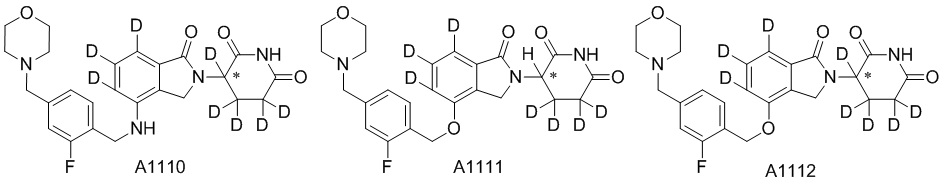

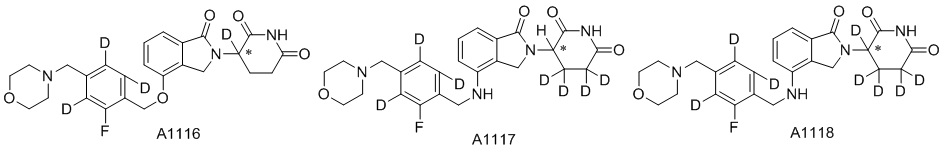

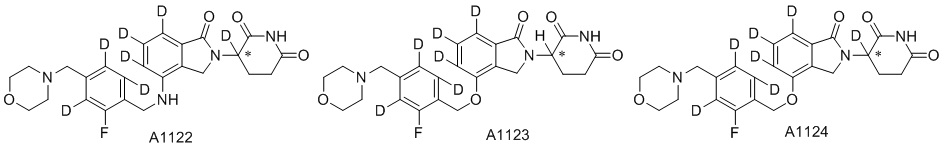

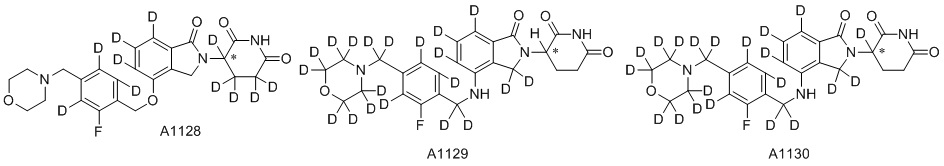

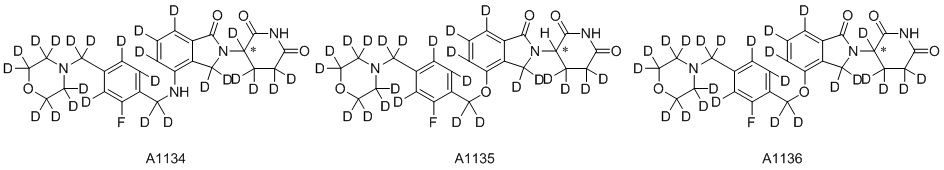



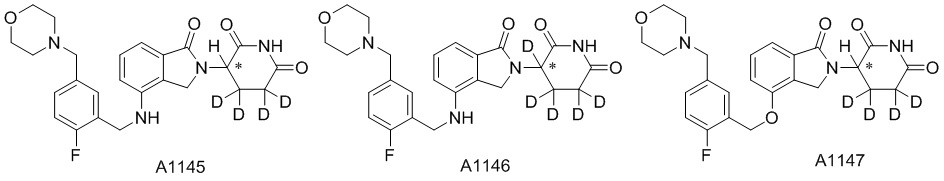

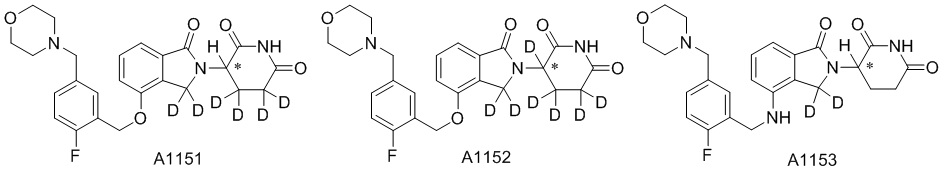

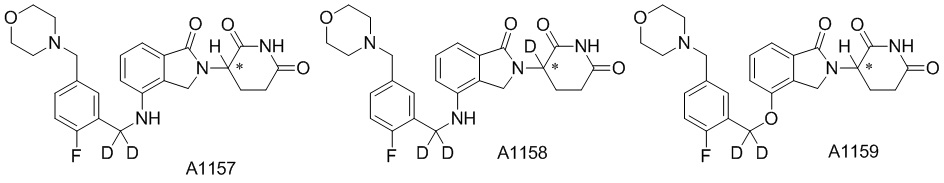
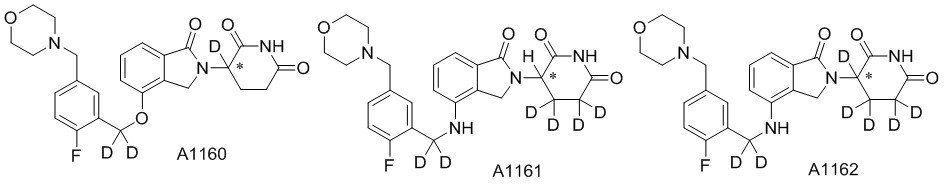
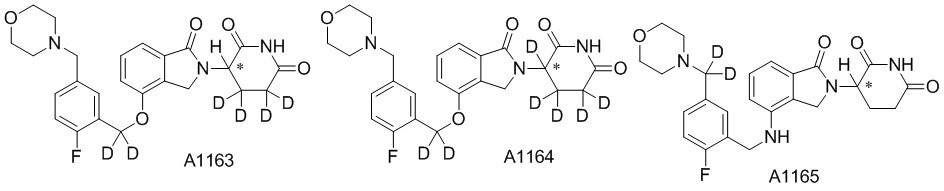
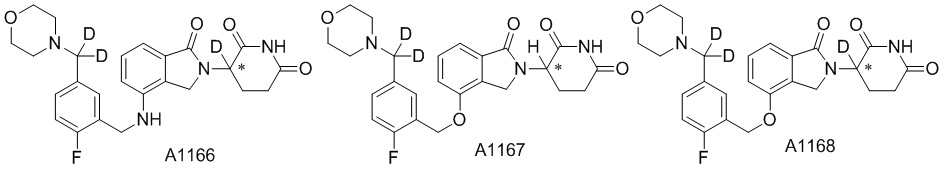
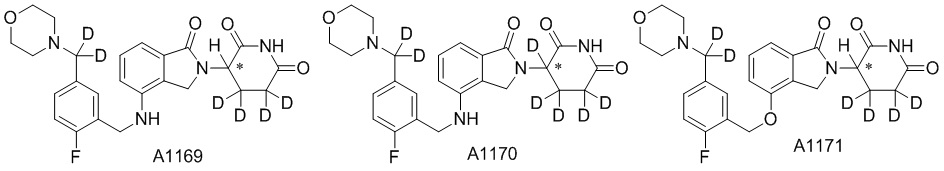





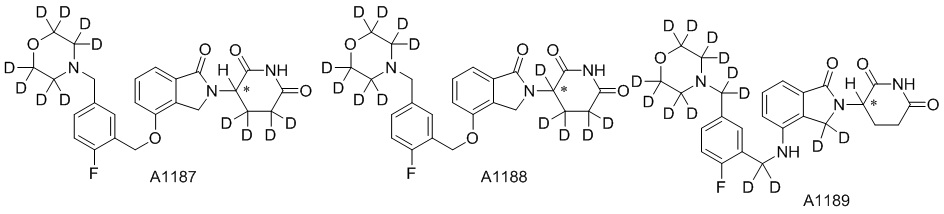
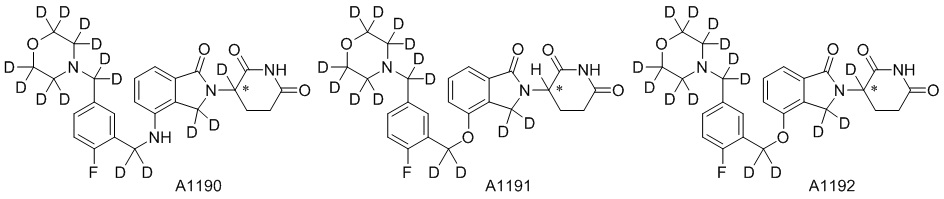
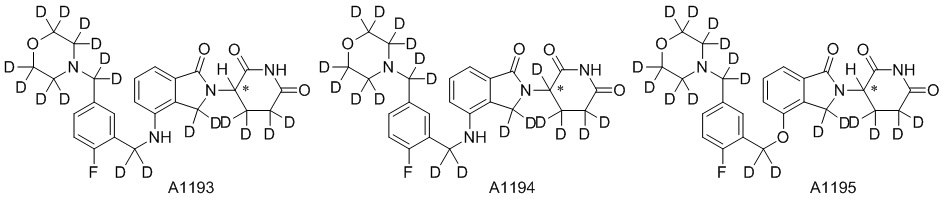




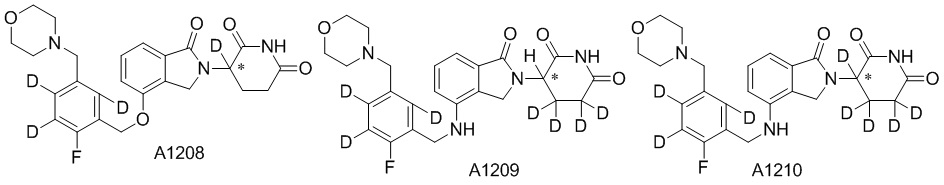




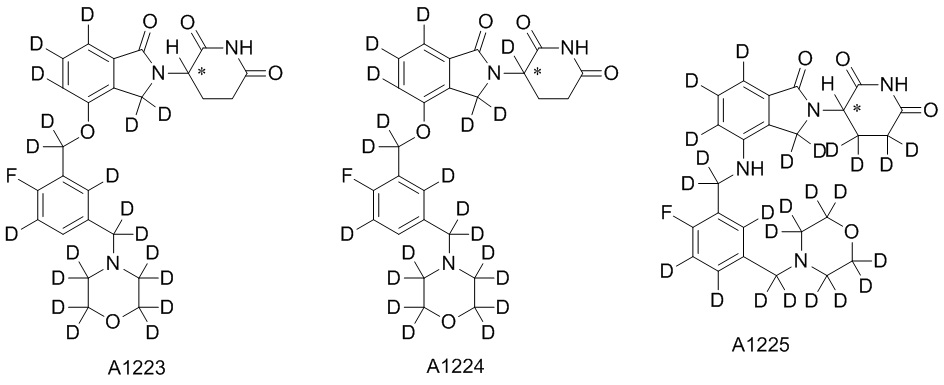

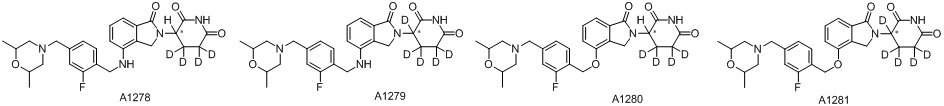
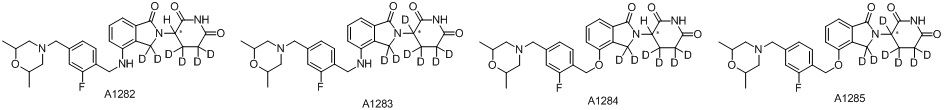





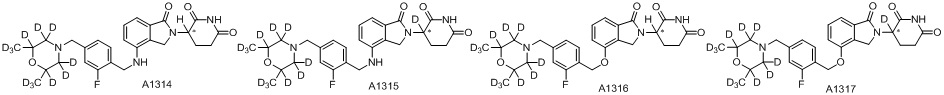




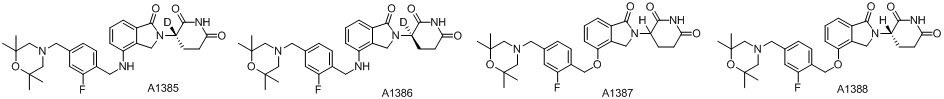


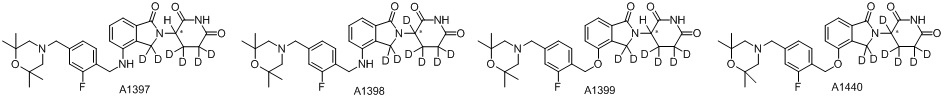



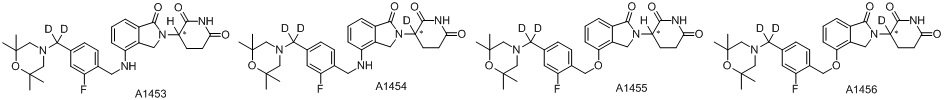
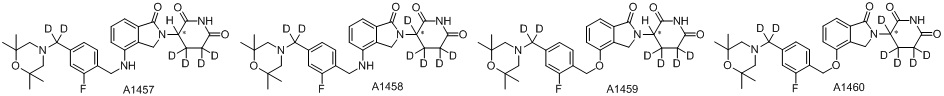
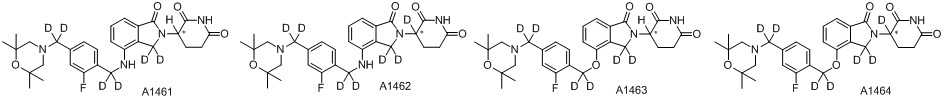


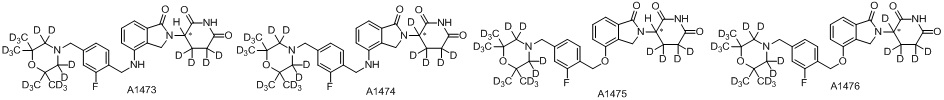


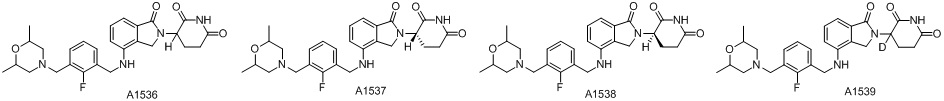



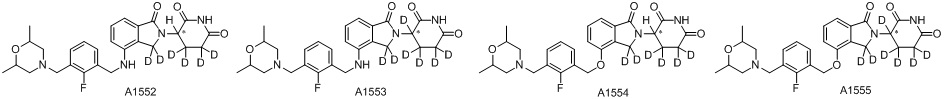

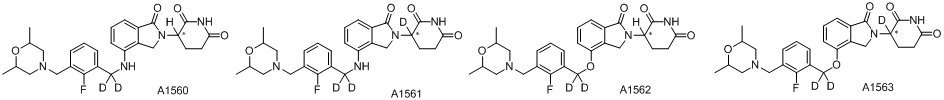
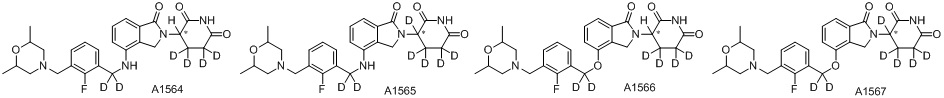
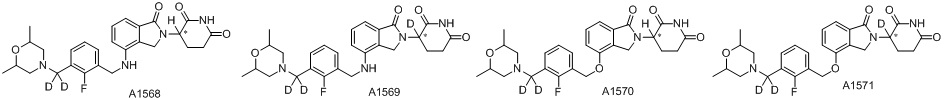

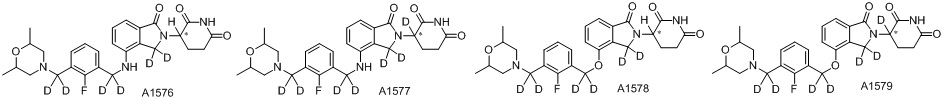


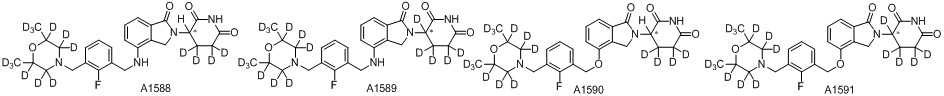


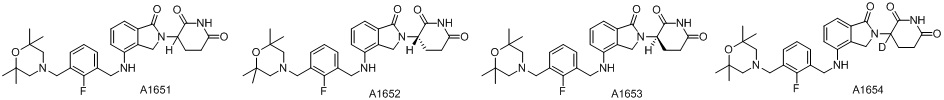


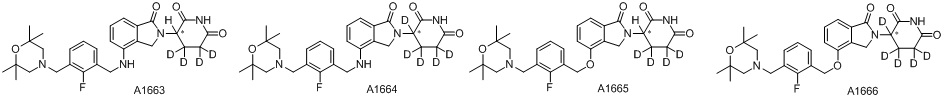
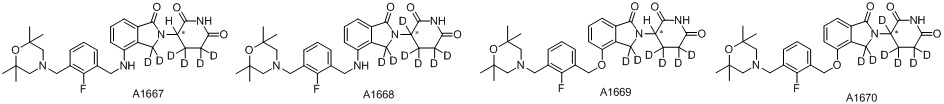



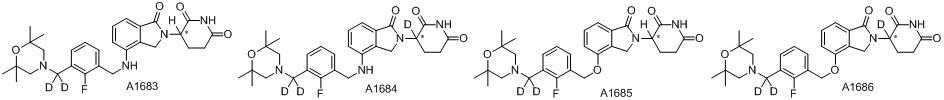

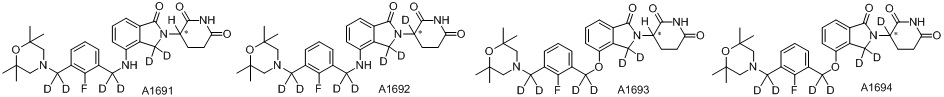





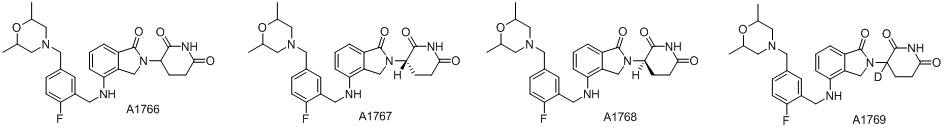

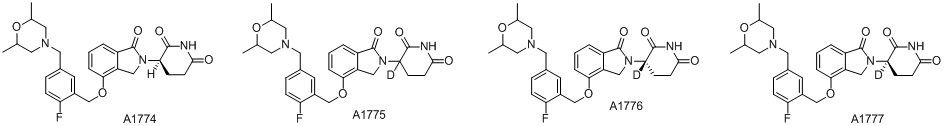
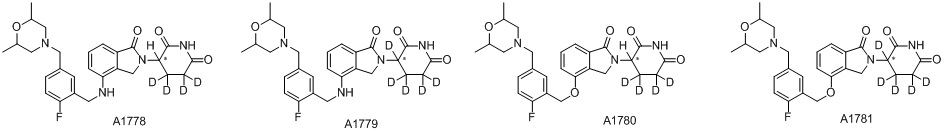

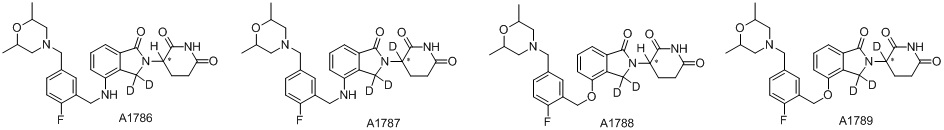


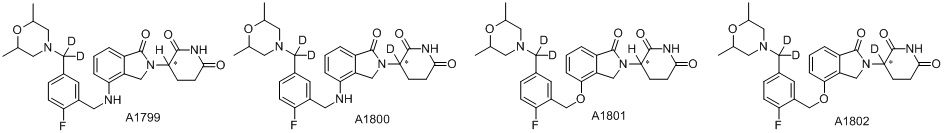
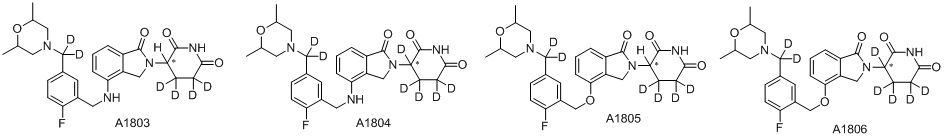

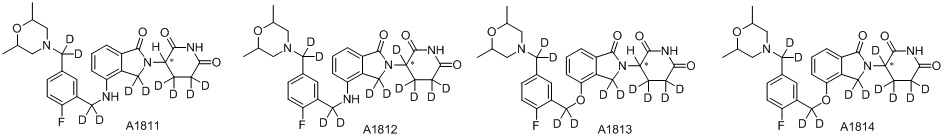
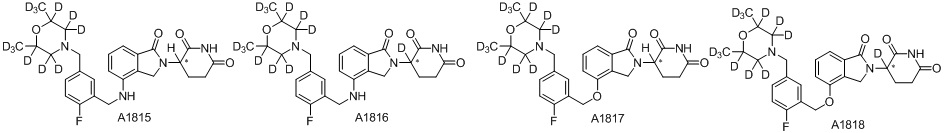

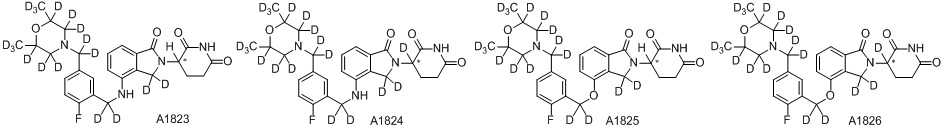






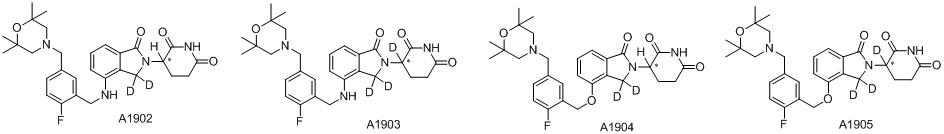


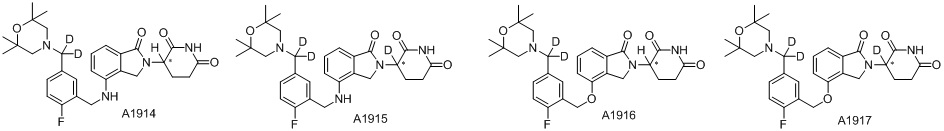


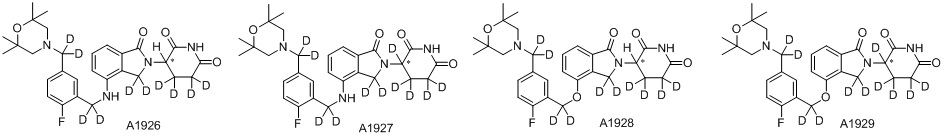
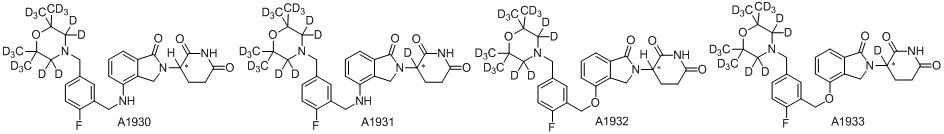



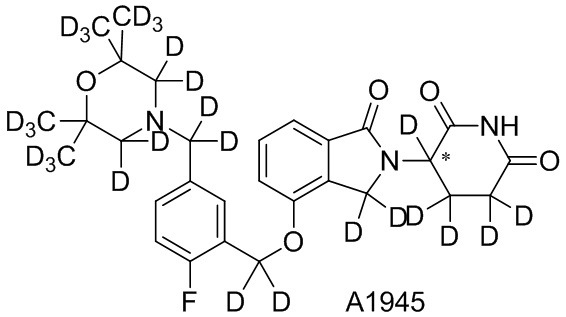
Дейтерий (D или2H) является стабильным нерадиоактивным изотопом водорода, его атомный вес составляет 2,0144. Водород существует в форме изотопной смеси H (водород или протий), D (2H или дейтерий) и T (3H или тритий) в естественных условиях, где распространенность дейтерия составляет 0,0156%. В соответствии с общими техническими знаниями в данной области, из всех соединений, чьи структуры содержат природные атомы водорода, атом водорода фактически представляет собой смесь H, D и T. Таким образом, если соединение содержит дейтерий, распространенность которого превышает его природную распространенность 0,0156% в любом положении, то эти соединения следует считать неприродными или обогащенными дейтерием; таким образом, эти соединения являются новыми по отношению к их необогащенным аналогам.
В настоящем изобретении «обогащенное дейтерием» соединение относится к соединению общей формулы (I), его фармацевтически приемлемой соли, сольвату, полиморфу, стереоизомеру, изотопному соединению, метаболиту или пролекарству, где распространенность дейтерия больше его распространенности в природе в любом соответствующем положении. Таким образом, в «обогащенном дейтерием» соединении распространенность дейтерия в соответствующем положении вероятно составляет от более 0,0156% до 100%. Обогащенное дейтерием положение представлено D, тогда как не обогащенное дейтерием положение представлено H. В соответствии с общими техническими знаниями в данной области, символ H можно не учитывать в необогащенном дейтерием положении. Примером способа получения обогащенного дейтерием соединения является замещение водорода дейтерием или использование обогащенного дейтерием исходного материала для синтеза соединения.
В настоящем изобретении процентное содержание дейтерия в обогащенном дейтерии или распространенность дейтерия относятся к молярной процентной концентрации.
В настоящем изобретении необогащенный дейтерием относится к водороду в естественных условиях, который находится в виде смеси изотопов H (водород или протий), D (2H или дейтерий) и T (3H или тритий).
Настоящее изобретение также предусматривает способ получения производного изоиндолина, представленного общей формулой (I), которое можно синтезировать в соответствии с известными способами с коммерчески доступными материалами, предпочтительно в соответствии со способом A, который включает осуществление реакции удаления защитных групп соединения A-06(1), как представлено ниже, с получением соединения A-06(a1); с последующей реакцией амидирования с участием соединения A-06(a1), как представлено ниже, с получением соединения общей формулы (I):


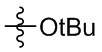

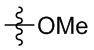

В общей формуле (I), в случае если n1 представляет собой 0, соединение общей формулы (I) может быть дополнительно получено в соответствии со способом B, который включает осуществление реакции восстановления с участием соединения I-RS, как представлено ниже, с получением соединения общей формулы (I):
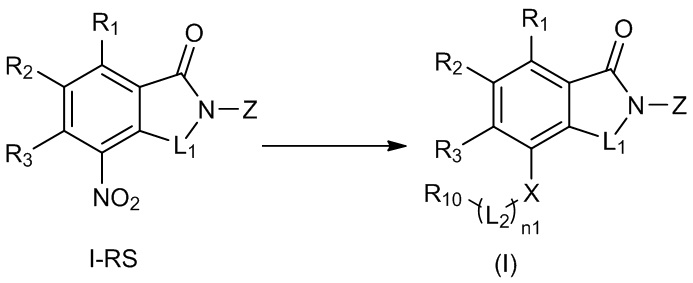
при этом в ходе осуществления способа B в определениях соединения I-RS или общей формулы (I) R2представляет собой галоген, n1 представляет собой 0, X представляет собой NH или ND, R10 представляет собой H или D, L1, Z, R1 и R3определены, как описано выше;
в случае если n1 представляет собой 1 и X представляет собой NH или ND в общей формуле (I), соединение общей формулы (I) может быть дополнительно получено в соответствии со способом С, который предпочтительно включает осуществление реакции восстановительного аминирования с участием соединения P-01 и


в ходе осуществления способа C в определении

В ходе осуществления способа A, способа B или способа C, стадии и условия реакции удаления защитных групп, реакции амидирования, реакции восстановления или реакции восстановительного аминирования могут представлять собой стандартные стадия и условия для такой реакции в данной области. В случае если атом углерода, помеченный *, содержащийся в Z в соединении A-06(1) или соединении A-06(a1), соединении I-RS, соединении P-01 или общей формуле (I), представляет собой хиральный центр, соединение A-06(1), соединение A-06(a1), соединение I-RS, соединение P-01 или общая формула (I) могут быть выделены соответственно с применением стандартного способа хирального разделения в данной области с получением соединения с (R)-конфигурацией, обогащенного соединения с (R)-конфигурацией, соединения с (S)-конфигурацией или обогащенного соединения с (S)-конфигурацией по отдельности, и затем их соответственно подвергают реакции с получением соединения общей формулы (I).
В ходе осуществления способа A, в случае если n1 представляет собой 0 в общей формуле (I), способ получения соединения общей формулы (I) может дополнительно включать осуществление реакции восстановления с участием соединения A-05(1), как представлено ниже, с получением соединения A-06(1):
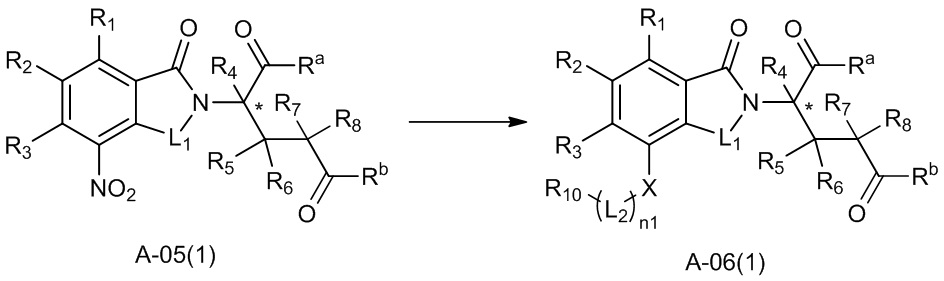
где в определениях соединения A-05(1) и A-06(1) L1, L2, R1-R8, Raи Rb определены, как описано выше; в определении соединения A-06(1) X представляет собой NH или ND, n1 представляет собой 0; R10представляет собой H или D. Стадии и условия, используемые в реакции восстановления, могут представлять собой стандартные стадии и условия, используемые в такой реакции в данной области.
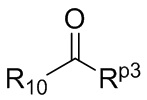
В ходе осуществления способа A, в случае если X представляет собой NH или ND и n1 представляет собой 1 в общей формуле (I), способ получения соединения общей формулы (I) может дополнительно включать осуществление реакции восстановительного аминирования с участием соединения A-05(2) и

где в определениях соединения A-05(2) и соединения A-06(1) L1, L2, R1-R8, Raи Rb определены, как описано выше; в определении соединения A-06(1) X представляет собой NH или ND и n1 представляет собой 1; в определении
В ходе осуществления способа A, в случае если X представляет собой O и n1 представляет собой 1 в соединении, имеющем структуру общей формулы (I), способ получения соединения, имеющего структуру общей формулы (I), может дополнительно включать осуществление реакции нуклеофильного замещения с участием соединения A-05(3) и

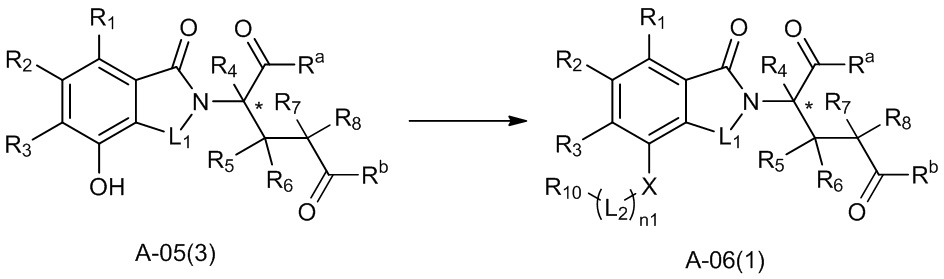
где в определениях соединения A-05(3) и соединения A-06(1) L1, L2, R1-R8, Raи Rb определены, как описано выше; в определении соединения A-06(1) X представляет собой O и n1 представляет собой 1; в определении
Способ получения соединения A-06(1) может дополнительно включать осуществление реакции сочетания с участием соединения Q-03 и соединения A-04(1), с последующим удалением защитных групп, как представлено ниже, с получением соединения A-05(3):

где в определении соединения Q-03 или соединения A-04(1) L1, *, R1-R8определены, как описано выше, Hal представляет собой галоген (например, Cl, Br или I); один из Ra и Rb представляет собой
Способ получения соединения A-05(3) может дополнительно включать защиту коммерчески доступного исходного материала фенола Q-01 с помощью TBDMS с получением Q-02, с последующим осуществлением реакции с галогенированным реагентом (например, NBS) с получением бензилгалогенида Q-03:

где в определении соединения Q-01 R1-R3и L1определены, как описано выше.
В ходе осуществления способа B, при получении соединения, имеющего структуру общей формулы (I), соединение I-RS получают в соответствии с общим способом получения такого соединения в данной области, предпочтительно получают в соответствии со способом D или способом E; способ D предпочтительно включает осуществление реакции сочетания с участием соединения A-03 и соединения A-04(2) или их соли, как представлено ниже, с получением соединения I-RS:

где в определении соединения A-03, A-04(2) или I-RS L1, Z, *, R1-R9 определены, как описано выше; в определении соединения A-03 Hal представляет собой галоген (например, Cl, Br или I). Стадии и условия, используемые в реакции сочетания, могут представлять собой стандартные стадии и условия, используемые в такой реакции в данной области.
Способ E предпочтительно включает удаление защитных групп соединения A-05(1), как представлено ниже, с получением соединения A-06(a2), с последующим осуществлением реакции амидирования с участием соединения A-06(a2) с получением соединения I-RS:
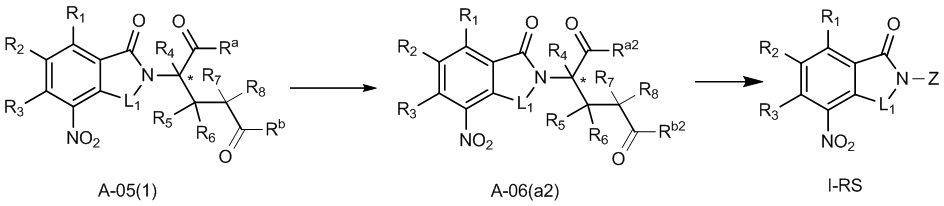
где в определении соединения A-05(1), A-06(a2) или I-RS L1, Z, *, R1-R8, Ra и Rbопределены, как описано выше; один из Ra2 и Rb2 представляет собой

В ходе осуществления способа C способ получения соединения, имеющего структуру общей формулы (I), может дополнительно включать осуществление реакции восстановления с участием соединения I-RS, как представлено ниже, с получением соединения P-01:

В определении соединения I-RS или соединения P-01 R2 представляет собой H, D или галоген; каждый из Rp1 и Rp2 независимо представляет собой H или D; L1, L2, Z, R1 и R3 определены, как описано выше. Стадии и условия, используемые в реакции восстановления, могут представлять собой стандартные стадии и условия, используемые в такой реакции в данной области.
В ходе осуществления способа A способ получения соединения A-06(1) может предпочтительно дополнительно включать осуществление реакции сочетания с участием соединения A-03 и соединения A-04(1), как представлено ниже, с получением соединения A-05(1):

где в определении соединения A03, A-04(1) или A-05(1) L1, *, R1-R8, Ra и Rb определены, как описано выше; Hal представляет собой галоген (например, Cl, Br или I). Стадии и условия, используемые в реакции сочетания, могут представлять собой стандартные стадии и условия, используемые в такой реакции в данной области.
Способ получения производного изоиндолина, имеющего структуру формулы (I), включает стадии, конкретно относящиеся к схеме A и схеме P.
Схема A: сочетание бензилгалогенида A-03 с аминокислотным производным A-04(1) с получением продукта A-05(1), с последующим удалением защитных групп с получением соединения A-05(2), и превращение в амин A-06(1) посредством осуществления реакции восстановительного аминирования с альдегидом

На схеме A определение каждой буквы и группы является таким, как описано выше.
Исходный материал A-03 является коммерчески доступным или может быть синтезирован в соответствии с известным способом (см. Söderberg et al. Org. Syn. (2003) 80, 75; US 4678500; US 2012/0053159 и US 2007/0255076).
Аминокислотное производное A-04 является коммерчески доступным или может быть синтезировано в соответствии с известным способом (см. Chen et al. Biotechnol. Lett. (1992) 14, 269; WO 2012/015986; WO 2012/068512; US 2012/0053159; Manesis et al. J.Org.Chem. (1987) 52, 5342; Stogniew et al. J. Labelled Compd. RAD. (1981) 18, 897; Blomquist et al. J. Org. Chem. (1966) 31, 4121), который, в частности, относится к схемам F1, F2 и G.
Схема F1:

Схема F2:
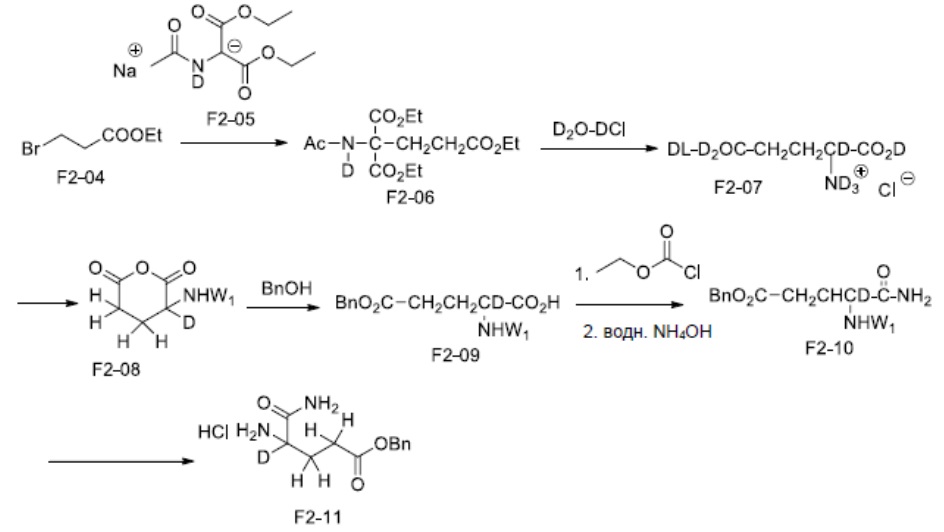
Схема G:


Сложный эфир G-01 обрабатывают бензальдегидом в дейтерированной уксусной кислоте с получением обогащенного дейтерием соединения G-02. Аминогруппу в G-02 защищают аминозащитной группой, с последующим осуществлением, соответственно, реакции этилхлорформиата и водного аммиака с получением амида G-04. Аминозащитную группу в G-04 можно удалить в соответствии со стандартным способом удаления защитных групп, известным из уровня техники (например, ацидолиз или восстановление), за счет чего осуществляется превращение в целевое соединение G-05.
Осуществляют реакцию материала A-03 с аминосоединением P-03 с получением соединения P-02, с последующим восстановлением и восстановительным аминированием с альдегидом
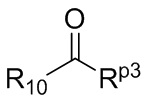
Схема P:

На схеме P определение каждой буквы и группы является таким, как описано выше.
Условия и стадии, используемые в химических реакциях, задействованных в вышеуказанных путях реакции, могут представлять собой стандартные условия и стадии для таких реакций, известные из уровня техники, и соединения, полученные в вышеуказанных способах, могут быть дополнительно модифицированы в периферийных положениях с получением других целевых соединений по настоящему изобретению.
Настоящее изобретение также предусматривает промежуточное соединение A-06(1), A-06(a1), I-RS или P-01 для получения производного изоиндолина, имеющего структуру общей формулы (I):




в определении соединения A-06(1), A-06(a1), I-RS или P-01 L1, L2, n1, Z, *, R1-R10, Ra, Rb, Ra1, Rb1, Rp1 и Rp2 определены, как описано выше; в определении соединения A-06(1) один из Ra и Rb представляет собой
Настоящее изобретение также предусматривает фармацевтическую композицию, при этом фармацевтическая композиция содержит терапевтически эффективное и/или профилактически эффективное количество вещества, выбранного из группы, состоящей из из производных изоиндолина, имеющих структуру общей формулы (I), фармацевтически приемлемой соли, сольвата, полиморфа, стереоизомера, изотопного соединения, метаболита и пролекарства.
В соответствии с вариантом осуществления настоящего изобретения фармацевтическая композиция может быть составлена для любой формы введения, в том числе инъекции (внутривенной), введения через слизистую оболочку, перорального введения (твердого и жидкого препарата), ингаляции, глазного введения, ректального введения, местного или парентерального (инфузии, инъекции, имплантации, подкожного, внутривенного, артериального, внутримышечного) введения. Фармацевтическая композиция по настоящему изобретению может также представлять собой лекарственные формы с контролируемым высвобождением или отсроченным высвобождением. Примеры твердого препарата для перорального введения включают без ограничения порошок, капсулу, каплет, мягкую капсулу или таблетку. Примеры жидкого препарата, вводимого посредством перорального введения или через слизистую оболочку, включают без ограничения суспензию, эмульсию, настойку и раствор. Примеры препарата для местного введения включают без ограничения препарат в виде эмульсии, геля, мази, крема, пластыря, пасты, пены, лосьона, капель или сыворотки. Примеры препарата для парентерального введения включают без ограничения раствор для инъекции, сухой препарат, который может быть растворенным или суспендированным в фармацевтически приемлемом носителе, инъекционную суспензию и инъекционную эмульсию. Примеры других пригодных препаратов соединения, имеющего структуру общей формулы (I), его фармацевтически приемлемой соли, сольвата, полиморфа, стереоизомера, изотопного соединения, метаболита или пролекарства включают без ограничения глазные капли и другие офтальмологические препараты; аэрозоли, такие как назальный спрей или ингаляция; жидкие лекарственные формы, подходящие для парентерального введения, суппозиторий и пастилку.
Фармацевтическая композиция по настоящему изобретению может дополнительно содержать фармацевтически приемлемый наполнитель, такой как наполнители, широко используемые в области изготовления лекарственных средств. Наполнители в основном используют для обеспечения безопасной, стабильной и функционализированной фармацевтической композиции, и могут также обеспечивать способ, который делает активный ингредиент растворимым с требуемой скоростью или способствует эффективному поглощению активных ингредиентов после введения субъекту. Наполнители могут представлять собой инертный наполнитель или обеспечивать некоторые функции, такие как стабилизация общего значения рН композиции или предотвращение деградации активных ингредиентов композиции.
В соответствии с вариантом осуществления настоящего изобретения фармацевтически приемлемый наполнитель может дополнительно содержать связующее вещество, суспендирующее средство, эмульгатор, разбавитель, наполнитель, гранулирующее средство, адгезивное средство, разрыхлитель, смазывающее средство, антиадгезивное средство, вещество, способствующее скольжению, смачивающее средство, желатинизирующее средство, замедлитель всасывания, ингибитор растворения или упрочняющее средство, адсорбирующее средство, буфер, комплексообразующее средство, консервант, краситель, ароматизатор и подсластитель. Фармацевтически приемлемый носитель может находится во многих формах, в соответствии с необходимым препаратом. Например, для жидкого препарата для перорального введения подходящие носители и добавки включают воду, гликоли, масла, спирты, ароматизаторы, консерванты, красители и т. д. В качестве другого иллюстративного примера для твердого препарата для перорального введения подходящие носители и добавки включают крахмал, сахар, разбавитель, гранулирующее средство, смазывающее средство, адгезивное средство, разрыхлитель и т. д. Как правило, фармацевтически приемлемые носители или наполнители должны быть нетоксичными. Фармацевтическая композиция по настоящему изобретению может содержать один или более одного подходящего(-их) носителя(-ей)/наполнителя(-ей). Количество и тип наполнителя варьируется в зависимости требований. На основе раскрытого в данном документе содержания специалист в данной области техники может легко определить подходящий(-ие) носитель(-и)/наполнитель(-и), подлежащие добавлению в фармацевтическую композицию по настоящему изобретению.
Фармацевтическую композицию по настоящему изобретению, которая содержит терапевтически эффективное или профилактически эффективное количество вещества, выбранного из группы, состоящей из соединений, имеющих структуру общей формулы (I), их фармацевтически приемлемой соли, сольвата, полиморфа, стереоизомера, изотопного соединения, метаболита и пролекарства, можно получить исходя из раскрытого в данном документе содержания, в соответствии с любыми способами, известными специалисту в данной области техники. Например, фармацевтическую композицию можно получить посредством смешивания соединения, имеющего структуру общей формулы (I), его фармацевтически приемлемой соли, сольвата, полиморфа, стереоизомера, изотопного соединения, метаболита или пролекарства с фармацевтически приемлемым носителем, исходя из общепринятой медицинской фармацевтической технологии. Технология включает без ограничения стандартное смешивание, растворение, гранулирование, эмульгирование, растирание в порошок, обертывание, заключение или способ сушки сублимацией.
В соответствии с вариантом осуществления настоящего изобретения, в дополнение к веществу, выбранному из группы, состоящей из соединения, имеющего структуру общей формулы (I), их фармацевтически приемлемой соли, сольвата, полиморфа, стереоизомера, изотопного соединения, метаболита и пролекарства, фармацевтическая композиция может дополнительно включать один или более одного других терапевтических средств. Другие терапевтические средства, которые могут быть включены в фармацевтическую композицию по настоящему изобретению, раскрыты ниже. Количество и тип других терапевтических средств зависят от заболевания, симптома или нарушения, подлежащих лечению или предупреждению, тяжести заболевания, симптома или нарушения, факторов субъекта, подлежащего введению, таких как возраст, вес, физическое состояние и т. д, пути введения и т. д.
В некоторых вариантах осуществления настоящее изобретение относится к препарату с контролируемым высвобождением соединения, имеющему структуру общей формулы (I), его фармацевтически приемлемой соли, сольвату, полиморфу, стереоизомеру, изотопному соединению, метаболиту или пролекарству. Используемый в данном документе «препарат с контролируемым высвобождением» относится к препарату, где терапевтически активный ингредиент композиции характеризуется скоростью контролируемого высвобождения или определенного отстроченного высвобождения для контроля участка высвобождения терапевтически активного ингредиента у субъекта, подлежащего введению. Один препарат с контролируемым высвобождением может включать средство с контролируемым высвобождением, такое как средство с замедленным высвобождением (замедленное высвобождение или отстроченное высвобождение) и средство с отсроченным высвобождением (отсроченное высвобождение).
Используемый в данном документе термин «замедленное высвобождение» и «отсроченное высвобождение» относится к продлению высвобождения терапевтически активного ингредиента из фармацевтической композиции. Используемый в данном документе термин «отсроченное высвобождение» относится к терапевтически активному ингредиенту, высвобождаемому из фармацевтической композиции в определенном участке или в требуемой среде после того, как композиция, действию которой подвергают субъекта, достигает необходимой среды или проходит определенный период времени.
Используемые в данном документе термины «средство с замедленным высвобождением» и «средство с отсроченным высвобождением» относятся к соединению или добавке, которые контролируют высвобождение терапевтически активного ингредиента из композиции, с тем чтобы сделать высвобождение постепенным и продлить время высвобождения. Средство с замедленным или отсроченным высвобождением может обеспечить высвобождение терапевтически активного ингредиента в течение особо длительного периода времени после того, как субъект был подвергнут действию композиции.
В соответствии с вариантом осуществления настоящего изобретения контролируемое высвобождение соединения, имеющего структуру общей формулы (I), его фармацевтически приемлемой соли, сольвата, кристаллической формы, стереоизомера, изотопного соединения, метаболита или пролекарства из композиции по настоящему изобретению, может быть достигнуто с помощью множества условий, при этом такие условия включают без ограничения значение pH, температуру, фермент, воду или другое физиологическое условие или соединение. Например, соединение по настоящему изобретению может дополнительно включать энтеросолюбильное покрытие, при этом энтеросолюбильное покрытие контролирует высвобождение соединения, имеющего структуру общей формулы (I), его фармацевтически приемлемой соли, сольвата, полиморфа, стереоизомера, изотопного соединения, метаболита или пролекарства, и обеспечивает их постепенное и непрерывное высвобождение из композиции в течение требуемого периода времени. Это обеспечивает соединению терапевтический или профилактический эффект в течение длительного периода времени.
В соответствии с вариантом осуществления настоящего изобретения фармацевтическая композиция с контролируемым высвобождением может дополнительно включать одно или более одного других терапевтических средств, раскрытых ниже.
Специалист в данной области техники может быть знаком с подходящими препаратами с контролируемым высвобождением, средствами с замедленным и отсроченным высвобождением на основе раскрытого в данном документе содержания. Неограничивающие примеры средств с контролируемым высвобождением, которые могут быть введены в фармацевтическую композицию по настоящему изобретению с целью обеспечения композиции с контролируемым высвобождением, включают полимеры, такие как гидроксипропил метил целлюлоза, гель, проницаемая оболочка, частица, липосома, микрогранула и их комбинации. Любая композиции, описанная в данном документе, может быть пригодна для препарата с контролируемым высвобождением, такого как таблетки, капсулы, мягкие капсулы и каплеты.
В соответствии с вариантом осуществления настоящего изобретения терапевтическое или профилактическое количество соединения, имеющего структуру общей формулы (I), его фармацевтически приемлемую соль, сольват, полиморф, стереоизомер, изотопное соединение, метаболит или пролекарство, его любую фармацевтическую композицию и препарат, и т. д., можно вводить субъекту в течение периода времени (медикаментозный цикл доставки) в соответствии с раскрытым в настоящем изобретении способом, с последующим периодом без применения соединения (немедикаментозный цикл доставки). Медикаментозный цикл доставки и немедикаментозный цикл доставки можно повторять в течение требуемых промежутков времени. Требуемая продолжительность и время медикаментозного цикла доставки и немедикаментозного цикла доставки зависят от типа и/или тяжести заболевания, симптома или нарушения, подлежащих лечению или предупреждению, а также пола, возраста, веса субъекта и других параметров (например, биологическое, физическое и физиологическое состояния субъекта и т. д.). Специалист в данной области техники, на основе раскрытого в данном документе содержания, может в достаточной мере определить подходящую продолжительность и время медикаментозного цикла доставки и немедикаментозного цикла доставки.
Соединение, имеющее структуру общей формулы (I), его фармацевтически приемлемая соль, сольват, полиморф, стереоизомер, изотопное соединение, метаболит или пролекарство можно использовать для нескольких целей, в том числе без ограничения использовать для изготовления лекарственного препарата для лечения или предупреждения заболевания, симптома или нарушения, обусловленных TNF-α или связанных с нарушенной регуляцией активности TNF-α.
Таким образом, в одном основном аспекте настоящее изобретение относится к применению терапевтически или профилактически эффективного количества производного изоиндолина, имеющего структуру общей формулы (I), его фармацевтически приемлемой соли, сольвата, полиморфа, стереоизомера, изотопного соединения, метаболита или пролекарства в изготовлении лекарственного препарата для лечения или предупреждения заболевания, симптома или нарушения. В другом аспекте настоящее изобретение относится к способу лечения или предупреждения заболевания, симптома или нарушения, обусловленных TNF-α или связанных с нарушенной регуляцией активности TNF-α, при этом способ включает введение субъекту терапевтически или профилактически эффективного количества вещества, выбранного из соединения, имеющего структуру общей формулы (I), его фармацевтически приемлемой соли, сольвата, полиморфа, стереоизомера, изотопного соединения, метаболита и пролекарства. В соответствии с данным способом примеры заболевания, симптома или нарушения, подлежащих лечению или предупреждению, включают без ограничения формы рака, в том числе солидные опухоли, нарушения, связанные с TNF-α, заболевания и нарушения, связанные с нежелательным ангиогенезом, боли, синдром макулярной дегенерации (MD), заболевания кожи, кератоз, заболевание дыхательной системы (такое как легочные заболевания), иммунодефицитные заболевания, заболевания центральной нервной системы (CNS), аутоиммунные заболевания, атеросклероз, наследственность, аллергию, вирусы, нарушения сна и связанный с ними синдром, воспалительные заболевания, заболевания, связанные с PDE-4 или заболевания, связанные с IL-2. Широко известные примеры заболеваний, симптомов или нарушений в данной области включают без ограничения описанные в патентных публикациях согласно РСТ WO 2012/015986 и WO 2006/018182, а также патентных публикациях США US 2010/0204227, некоторое содержание которых включено в настоящий документ посредством ссылки во всей их полноте.
В одном варианте осуществления заболевание, симптом или нарушение выбраны из неопластических или раковых заболеваний; аутоиммунных заболеваний, таких как заболевание Аддисона, анкилозирующий спондилит, синдром антифосфолипидных антител, атопический дерматит, аутоиммунная круговая алопеция, аутоиммунная гемолитическая анемия, аутоиммунный гепатит, аутоиммунное заболевание внутреннего уха, аутоиммунный лимфопролиферативный синдром (Alps), заболевание Бехчета, буллезный пемфигоид, кардиомиопатия, целиакия, синдром хронической усталости и иммунодефицита (CFIDS), хроническая воспалительная демиелинизирующая полинейропатия, рубцующийся пемфигоид, заболевание холодовых агглютининов, синдром CREST, заболевание Крона, заболевание Дего, дерматомиозит, юношеский дерматомиозит, дискоидная красная волчанка, экзема, первичная криоглобулинемия смешанного типа, фибромиалгия-фибромиозит, базедова болезнь, синдром Гийена-Барре, тиреоидит Хашимото, гнойный гидраденит, идиопатический легочный фиброз, идиопатическая тромбоцитопеническая пурпура (ITP), Ig A-нефропатия, инсулинозависимый сахарный диабет (I типа), юношеский артрит, эритематозная волчанка, заболевание Меньера, смешанное заболевание соединительной ткани, множественный склероз, миастения гравис, вульгарный пемфигус, пернициозная анемия, узелковый полиартериит, полихондрия, полигландулярный синдром, ревматическая полимиалгия, полимиозит и дерматомиозит, первичная агаммаглобулинемия, первичный биллиарный цирроз, псориаз, феномен Рейно, синдром Рейтера, ревматическая лихорадка, ревматоидный артрит, саркоидоз, склеродермия, синдром Шегрена, системная эритематозная волчанка, синдром мышечной скованности, артериит Такаясу, темпоральный артериит/гигантоклеточный артериит, ульцеративный колит, увеит, васкулит, витилиго, гранулематоз Вегенера и аутоиммунное заболевание Вильсона; легочного заболевания, такого как астма, хроническое обструктивное заболевание легких; заболевания нервной системы, такого как болезнь Альцгеймера, болезнь Паркинсона, депрессия, эпилепсия и биполярное расстройство; заболевания сердечно-сосудистой системы, такого как атеросклероз, инфаркт миокарда, остеопороз; метаболического заболевания, такого как ожирение, диабет II типа; респираторного дистресс-синдрома взрослых; заболевания резорбции кости, такого как артрит; гиперкальцемии; реакции трансплантат против хозяина; церебральной малярии; воспалительного заболевания, такого как акне, артрит, астма, атеросклероз, целиакия, хронический простатит, колит, заболевание Крона, дерматит, дивертикулит, гломерулярный нефрит, гепатит, гиперчувствительность, воспалительные заболевания кишечника, интерстициальный цистит, синдром раздраженного кишечника (IBS), эритематозная волчанка, нефрит, воспалительное заболевание органов таза, реперфузионное повреждение, ревматоидный артрит, саркоидоз, отторжение ткани, ульцеративный колит, васкулит, хроническое воспалительное заболевание легких, инсульт, циркуляторный шок; HIV-инфекции, AIDS и AIDS-оппортунистической инфекции; других заболеваний, таких как ревматоидный спондилит, остеоартрит и прочее нарушение, относящееся к артриту, септический шок, сепсис, эндотоксический шок, заболевание трансплантат против хозяина, истощение, заболевание Крона, ульцеративный колит, лепрозная узелковая эритема, нарушения, связанные с cAMP, такие как септический шок, сепсис, эндотоксический шок, гемодинамический шок и септический синдром, ишемически-реперфузионное повреждение, малярия, микобактериальная инфекция, менингит, застойная сердечная недостаточность, фиброзное заболевание, кахексия, отторжение трансплантата, радиационное поражение, гипероксическое альвеолярное повреждение; вирусной инфекции, такой как инфекции, вызванные вирусом герпеса; вирусного конъюнктивита или атопического дерматита.
Примеры неопластических или раковых заболеваний включают без ограничения острый лимфобластный лейкоз, острый миелоидный лейкоз, острый миелогенный лейкоз, кариотипный острый миелоидный лейкоз, хронический лимфоцитарный лейкоз, хронический миелоидный лейкоз, хронический гранулоцитный лейкоз, лейкоз ворсистых клеток, миелоидный лейкоз, адренокортикальную карциному, лимфому Беркитта, связанную с AIDS лимфому, Т-клеточную лимфому кожи, B-клеточную лимфому кожи, диффузную В-крупноклеточную лимфому, низкодифференцированную фолликулярную лимфому, лимфому Ходжкина, неходжкинскую лимфому, множественную миелому, вялотекущую миелому, миелодиспластический синдром, лимфому из клеток мантийной зоны, невыраженную миелому, хроническое миелопролиферативное заболевание, лимфому центральной нервной системы (CNS), рак анального канала, астроцитому, атипичную патологическую/рабдоидную опухоль, базальноклеточную карциному, холангиокарциному, рак мочевого пузыря, остеому, остеоидную остеому, остеохондрому, остеобластому, остеосаркому, энхондрому, аневризматическую кисту кости, фиброзную дисплазию костей, хондросаркому, саркому Юинга, фибросаркому, плеоморфную недифференцированную саркому, опухоль головного мозга, глиому ствола головного мозга, медуллобластому, медуллярную эпителиальную опухоль, опухоль шишковидной клетки, рак молочной железы, бронхиальную опухоль, карциноидную опухоль, рак шейки матки, хордому, рак толстой кишки, колоректальный рак, краниофарингиому, эмбриональную карциному, эпендимобластому, эпендимому, рак пищевода, ольфакторную нейробластому, экстракраниальную эмбрионально-клеточную опухоль, гонадную эмбрионально-клеточную опухоль, холангиокарциному, внутриглазную меланому, ретинобластому, карциному желчного пузыря, рак желудка, гастроинтестинальную стромальную опухоль, гестационную трофобластическую опухоль, глиому, рак головы и шеи, рак печени, гипофарингеальную карциному, внутриглазную меланому, опухоль островков поджелудочной железы, саркому Капоши, почечно-клеточную карциному, лангергансоклеточный гистиоцитоз, рак гортани, рак губы и ротовой полости, рак легкого, карциному из клеток Меркеля, мезотелиому, синдром множественной эндокринной неоплазии, фунгоидный микоз, рак носа и синуса, назофарингеальную карциному, нейробластому, рак ротовой полости, рак ротоглотки, карциному яичника, эпителиальную карциному яичников, эмбрионально-клеточную опухоль яичников, злокачественную опухоль яичника с низким потенциалом, рак поджелудочной железы, опухоль островков поджелудочной железы, карциному поджелудочной железы, папиллому, параганглиому, карциному паращитовидной железы, рак полового члена, фарингеальную карциному, феохромоцитому, плазмоцитому, плевролегочную бластому, гормонорезистентный рак предстательной железы, андрогеннезависимый рак предстательной железы, андрогензависимый неметастатический рак предстательной железы IV фазы, нечувствительный к гормону рак предстательной железы, нечувствительный к химиотерапии рак предстательной железы, рак прямой кишки, глиобластому сетчатки глаза, рабдомиосаркому, рак слюной железы, саркому мягких тканей, саркому матки, рак кожи (меланому), плоскоклеточную карциному, рак кожи из клеток Меркеля, рак тонкого кишечника, плоскоклеточный рак шейки матки, рак яичек, рак горла, тимому и тимусную карциному, рак щитовидной железы, рак мочевыводящих путей, карциному эндометрия, саркому матки, рак влагалища, рак женских наружных половых органов, астроцитому, гепатоцеллюлярную карциному, макроглобулинемию Вальденстрема, нефробластому.
В предпочтительном варианте осуществления заболевание, симптом или нарушение выбраны из миелодиспластического синдрома, множественной миеломы, лимфомы из клеток мантийной зоны, диффузной В-крупноклеточной лимфомы, лимфомы центральной нервной системы, неходжкинской лимфомы; папиллярной и фолликулярной тиреоидной карциномы; рака молочной железы, рака предстательной железы, хронического лимфоцитарного лейкоза, амилоидоза, комплексного регионарного болевого синдрома I типа, злокачественной меланомы, радикулопатии, миелофиброза, глиобластомы, саркоматозной глиомы, злокачественной глиомы, рефракторной плазмоцитомы, хронического миеломоноцитарного лейкоза, фолликулярной лимфомы, цилиарной и хронической меланомы, меланомы радужной оболочки, рецидивирующей меланомы глаза, экстраокулярной распространяющейся меланомы, солидной опухоли, T-клеточной лимфомы, эритроидной лимфомы, монобластного и моноцитарного лейкоза; миелоидного лейкоза, опухолей головного мозга, менингиом, опухолей спинного мозга, рака щитовидной железы, немелкоклеточного рака легкого, карциномы яичника, почечно-клеточной карциномы, миелофиброза, лимфомы Беркитта, лимфомы Ходжкина, крупноклеточной лимфомы, астроцитомы, гепатоцеллюлярной карциномы, первичной макроглобулинемии (макроглобулинемии Вальденстрема). В одном варианте осуществления рак является метастатическим. В другом варианте осуществления рак является рефракторным или лечение его посредством химиотерапии или лучевой терапии является неэффективным.
Способ лечения в настоящем изобретении включает введение фармацевтической композиции субъекту с помощью любых подходящих способов, таких как инъекция, введение через слизистую оболочку, пероральное введение, ингаляция, глазное, ректальное введение, имплантат длительного действия, липосома, эмульсия или способ замедленного высвобождения.
Специалист в данной области техники понимает, что терапевтически эффективное или профилактически эффективное количество соединения, используемое в настоящем изобретении, может варьировать в зависимости от факторов для определенного субъекта, таких как возраст, рацион, состояние здоровья и т. д., тяжести, сложности и типа симптома, заболевания или нарушения, подлежащих лечению или предупреждению, а также используемого препарата и т.д. В соответствии с раскрытиями в настоящем изобретении специалист в данной области техники может легко определить необходимое терапевтически эффективное или профилактически эффективное количество соединения, вводимого субъекту, для того, чтобы вызвать требуемый биологическую или медицинскую ответную реакцию у субъекта.
В соответствии с вариантом осуществления настоящего изобретения соединение, имеющее структуру общей формулы (I), его фармацевтически приемлемая соль, сольват, полиморф, стереоизомер, изотопное соединение, метаболит или пролекарство можно использовать для регуляции активности или образования TNF-α или IL-2. В одном варианте осуществления, в случае если термин «регулировать» используется для описания активности или образования определенной молекулы, он относится к ингибированию активности или образования молекулы. Однако в другом варианте осуществления, в случае если термин «регулировать» используется для описания активности или образования определенной молекулы, он относится к снижению или повышению активности или образования молекулы.
Таким образом, настоящее изобретение также предусматривает способ регулирования образования или активности TNF-α или IL-2. В соответствии с вариантом осуществления настоящего изобретения соединение, имеющее структуру общей формулы (I), его фармацевтически приемлемую соль, сольват, полиморф, стереоизомер, изотопное соединение, метаболит или пролекарство, или его композицию, можно вводить субъекту для регулирования образования и активности TNF-α или IL-2, что можно дополнительно использовать для лечения или предупреждения заболевания, симптома или нарушения, связанных с нарушенной регуляцией TNF-α или IL-2, или характеризующихся нарушенной регуляцией TNF- или IL-2.
В предпочтительном варианте осуществления соединение, имеющее структуру общей формулы (I), его фармацевтически приемлемую соль, сольват, полиморф, стереоизомер, изотопное соединение, метаболит или пролекарство, или его композицию, вводят субъекту для регулирования образования и активности TNF-α или IL-2 с целью лечения или предупреждения рака или воспаления.
В любых способах, описанных в настоящем изобретении, соединение, имеющее структуру общей формулы (I), его фармацевтически приемлемую соль, сольват, полиморф, стереоизомер, изотопное соединение, метаболит или пролекарство можно использовать отдельно или в комбинации с лучевой терапией или радиоиммунотерапией и т. д., и дополнительно можно использовать в комбинации с одним или более чем одним терапевтическим(-ми) средством(-ами), которые(-ое) обладает(-ют) фармацевтической активностью (далее в данном документе называются «другим(-и) терапевтическим(-и) средством(-ами)»).
В соответствии с вариантом осуществления настоящего изобретения соединение, имеющее структуру общей формулы (I), его фармацевтически приемлемая соль, сольват, полиморф, стереоизомер, изотопное соединение, метаболит или пролекарство, используемые в комбинации с другим(-и) терапевтическим(-и) средством(-ами), может(могут) обладать синергическими эффектами, в случае если любые заболевания, симптомы или нарушения обрабатывают или предупреждают в соответствии с содержанием, раскрытым в данном документе.
В соответствии с вариантом осуществления настоящего изобретения другое(-ие) терапевтическое(-ие) средство(-а) может(могут) представлять собой природное, полусинтетическое или синтетическое соединение. В другом варианте осуществления другое(-ие) терапевтическое(-ие) средство(-а) может(могут) представлять собой небольшую молекулу, такую как синтетическая органическая или неорганическая молекула, или большая молекула или биомолекула, такая как белки или нуклеиновые кислоты с фармакологической активностью. В другом варианте осуществления другое(-ие) терапевтическое(-ие) средство(-а) может(могут) представлять собой анти-ангиогенное, иммунорегулирующее, иммунотерапевтическое, химиотерапевтическое или гормональное соединение.
Примеры другого(-их) терапевтического(-их) средства(-ств), подходящего(-их) для настоящего изобретения, включают без ограничения моноклональное и поликлональное антитело, такое как обинутузумаб (Gazyva®), ниволумаб (Opdivo®), пембролизумаб (Keytruda®), элотузумаб, атитело к Her2/neu (например, трастузумаб (торговое название Herceptin®) и пертузумаб (торговое название Omnitarg™); абциксимаб (торговое название ReoPro®), ритуксимаб (торговое название Mabthera®), базиликсимаб (торговое название Simulect®), паливизумаб (торговое название Synagis®), инфликсимаб (торговое название Remicade®), трастузумаб (торговое название Herceptin®), алемтузумаб (торговое название Campath®), ибритумомаб тиуксетан (торговое название Zevalin®), адалимумаб (торговое название Humira®), омализумаб (торговое название Xolair®), тозитумомаб-I-131 (торговое название Bexxar®), цетуксимаб (торговое название Erbitux®), натализумаб (торговое название Tysabri®), тоцилизумаб (торговое название Actemra®), панитумумаб (торговое название Vectibix®), ранибизумаб (торговое название Lucentis®), экулизумаб (торговое название Soliris®), цертолизумаб пегол (торговое название Cimzia®), голимумаб (торговое название Simponi®), канакинумаб (торговое название Ilaris®), устекинумаб (торговое название Stelara®), офатумумаб (торговое название Arzerra®), деносумаб (торговое название Prolia®), мотавизумаб (торговое название Numax®), эдреколомаб (торговое название Panorex®), раксибакумаб (торговое название ABThrax®), белимумаб (торговое название Benlysta®), ипилимумаб (торговое название Yervoy®), брентуксимаб ведотин (торговое название Adcetris®), пертузумаб (торговое название Perjeta® или Omnitar™), адо-трастузумаб эмтанзин (торговое название Adcyla®), моноклональное антитело к CD40, антитело к TNF-α и антитело VEGFR (например, бевацизумаб (торговое название Avastin™); ингибитор Akt; ингибитор ALK; ингибитор AMPK; антисмысловой олигонуклеотид; алкилирующее химиотерапевтическое средство, такое как азотистые иприты (например, циклофосфамид), мехлорэтамин, HN2 (торговое название Mustardgen), урамустин, урациловый иприт, мелфалан, хлорамбуцил, ифосфамид и бендамустин; нитрозомочевину (например, кармустин), ломустин и стрептозоцин; алкилсульфонат (например, бусульфан); а также азиридины, такие как тиотепа; химиотерапевтическое средство на основе платины (например, цисплатин, карбоплатин, недаплатин, оксалиплатин, сатраплатин и триплатина тетранитрат, прокарбазин, алтретамин, дакарбазин, митозоломид и темозоломид; ингибитор APC; ген-регулятор апоптоза; регулятор апоптоза; ингибитор ATM/ATR; ингибитор киназы аврора; ингибитор Axl; ингибитор Bcl-2; антагонист BCR/ABL; ингибитор bFGF; ингибитор BTK; ингибитор казеинкиназы (ICOS); ингибитор цистеиновой протеиназы; CAR-T; ингибитор CDK, такой как палбоциклиб; ингибитор ChK; ингибитор c-Kit; ингибитор c-Met; ингибитор EGFR; ингибитор c-Myc; ингибитор C-RET; ингибитор CSF-1R; цитокин; ингибитор DNA-PK; ингибитор динеина; ингибитор рецептора EGF; ингибитор EGFR; ингибитор EGFR/ERBB; ингибитор белкового рецептора печени; ингибитор ERK; агонист эстрогена; антагонист эстрогена; ингибитор FAK; ингибитор FGFR; ингибитор FLT3; антагонист рецептора GF; ингибитор глутатиона; ингибитор GSK-3; ингибитор белка теплового шока-90 (например, 17-AAG); гемопоэтический фактор роста; ингибитор HDAC; ингибитор андрогеновых рецепторов, ингибитор биосинтеза андрогенов; ингибитор HER2; ингибитор HIF; ингибитор гистондеацетилазы (например, SAHA и LAQ 824); ингибитор HSP; ингибитор IAP; ингибитор IGF-1R; ингибитор киназы IkB; ингибитор рецептора инсулиноподобного фактора роста-1; ингибитор интегрина; агонист интерферона; интерферон; интерлейкин; ингибитор JAK; ингибитор JNK; ингибирующий фактор лейкемии; лейкоцитарный интерферон-α; ингибитор лизофосфатидной ацилтрансферазы; ингибитор матрилизина; ингибитор матричной металлопротеиназы; ингибитор Mdm; ингибитор MEK; ингибитор MIF; ингибитор mTOR; олигонуклеотид; ингибитор P13K (например, вортманнин); ингибитор p38 MAPK; ингибитор p53; ингибитор PAK; ингибитор PARP; ингибитор PDGFR; ингибитор PDK-1; ингибитор PD-1; ингибитор PD-L1; ингибитор фосфатазы; ингибитор Pim; ингибитор PKC; ингибитор PLK; иммуномодулирующее средство на основе белка A; ингибитор протеинкиназы C; ингибитор протеинтирозинфосфатазы; ингибитор пурин-нуклеозидфосфорилазы; ингибитор RacGTP-азы; ингибитор Raf; ингибитор фарнезил-протеинтрансферазы Ras; ингибитор Ras; ингибитор Ras-GAP; ингибитор ROCK; ингибитор киназы S6; ингибитор сигнальной трансдукции; ингибитор дезацетилазы; ингибитор Src; ингибитор STAT; ингибитор сурвивина; ингибитор Syk; ингибитор теломеразы; ингибитор TNF-α; ингибитор топоизомеразы; ингибитор Trk; ингибитор тирозинкиназы; антагонист рецептора урокиназы; ингибитор рецепторной киназы фактора роста эндотелия сосудов (например, PTK787); ингибитор VDA; ингибитор VEGFR (например, специфический ингибитор киназы flk-1, SU5416 и ptk787/zk222584); ингибитор Wee1 и ингибитор сигнального пути Wnt.
Другое(-ие) терапевтическое(-ие) средство(-а), подходящее(-ие) для настоящего изобретения, включает(-ют) без ограничения ацивицин; акларубицин; акодазола гидрохлорид; акронин; ацилфулвен; адеципенол; адозелезин; альдеслейкин; алтретамин; амбамустин; амбомицин; аметантрона ацетат; амидокс; амифостин; аминолевулиновую кислоту; амрубицин; амсакрин; анагрелид; анастрозол; андрографолид; антареликс; антрамицин; анти-дорсализующий морфогенетический белок-1; антинеопластон; афидиколина глицинат; апуриновую кислоту; ара-CDP-DL-PTBA; аспарагиназу; асперлин; асулакрин; атаместан; атримустин; аксинастатин-1; аксинастатин-2; аксинастатин-3; азацитидин; азасетрон; азатоксин; азатирозин; азетепу; азотомицин; баланол; батимастат; бензохлоринс; бензодепу; бензоилстауроспорин; производные бета-лактама; β-алетин; бетакламицин B; бетулиновую кислоту; бикалутамид; бисантрена гидрохлорид; бисазиридинилспермин; биснафид димесилат; бистратен A; бизелезин; блеомицина сульфат; бортезомиб (гемцитабин); бреквинар натрия; бретлат; бропиримин; будотитан; бусульфан; бутионина сульфоксимин; кактиномицин; кальципотриол; кальфостин C; калустерон; производные камптотецина; капецитабин; карацемид; карбитимер; карбоплатин; карбоксамидаминотриазол; карбоксиамидотриазол; кармустин; карубицина гидрохлорид; карзелезин; кастаноспермин; цекропин B; цедефингол; целекоксиб; цетрореликс; хлорамбуцил; хлорины; хлорхиноксалина сульфонамид; цикапрост; циролемицин; цисплатин; цис-порфирин; кладрибин; аналоги кломифена; клотримазол; коллизмицин A; коллизмицин B; комбретастатин A4; производные комбретастатина; конагенин; крамбесцидин-816; криснатол мезилат; криснатол; криптофицин-8; аналоги криптофицина A; курацин A; циклопентантрахиноны; циклофосфамид; циклоплатам; циклоспорин; ципемицин; цитарабина октофосфат; цитарабин; цитостатин; дакарбазин; дакликсимаб; дактиномицин; даунорубицина гидрохлорид; децитабин; дегидродидемнин B; деслорелин; дексаметазон; дексифосфамид; ормаплатин; декс(ормаплатин); дексразоксан; дексверапамил; дезагуанина мезилат; дезагуанин; диазиквон; дидемнин B; дидокс; диэтилнорспермин; дигидро-5-азацитидин; дигидротаксол-9; диоксамицин; дифенилспиромустин; доцетаксел; докозанол; доласетрон; доксифлуридин; доксорубицина гидрохлорид; доксорубицин; доксициклин; дролоксифена цитрат; дролоксифен; дромостанолона пропионат; дронабинол; дуазомицин; дуокармицин SA; эбселен; экомустин; эдатрексат; эделфосин; эдреколомаб; эфлорнитина гидрохлорид; эфлорнитин; элемен; элотузумаб; элсамитруцин; эмитефур; энлоплатин; энпромат; эпипропидин; эпирубицина гидрохлорид; эпирубицин; эпристерид; эрбитукс; эрбулозол; эзорубицина гидрохлорид; производные эстрамустина; этрамустина натрия фосфат; этрамустин; этанерцепт; этанидазол; этопозида фосфат; этопозид; этоприн; экземестан; фадрозола гидрохлорид; фадрозол; фазарабин; фенретинид; филграстим; финастерид; флавопиридол; флезеластин; флоксуридин; флуастерон; флударабина фосфат; флударабин; флуроцитабин; фтордаунорубицина гидрохлорид; фторурацил; форфенимекс; форместан; фосквидон; фостриецин натрия; фостриецин; фотемустин; гадолиния тексафирин; галлия нитрат; галоцитабин; ганиреликс; гемцитабина гидрохлорид; гемцитабин; гепсульфам; херегулин; гексаметилена бисацетамид; гидроксимочевину; гиперицин; ибандроновую кислоту; ибрутиниб; идарубицина гидрохлорид; идарубицин; идоксифен; идрамантон; ифосфамид; илмофосин; иломастат; иматиниб, торговое название Gleevec®; имиквимод; иммуностимулирующие пептиды; иобенгуан; йододоксорубицин; ипомеанол-4; ипроплатин; иринотекана гидрохлорид; иринотекан; ироплакт; ирсогладин; изобенгазол; изогомогаликондрин B; итазетрон; джасплакинолид; кахалалид F; ламелларина-N триацетат; ланреотида ацетат; ланреотид; лапатиниб, торговое название Tykerb®; леинамицин; ленограстим; лентинана сульфат; лептолстатин; летрозол; леупролида ацетат; леупролид+эстроген+прогестерон; леупрорелин; левамизол; лиарозола гидрохлорид; лиарозол; липофильный дисахаридпептид; липофильные аналоги платины; лиссоклинамид-7; лобаплатин; ломбрицин; лометрексол натрия; лометрексол; ломустин; лонидамин; лозоксантрона гидрохлорид; лозоксантрон; локсорибин; луртотекан; лютеция тексафирин; лизофиллин; литические пептиды; майтанзин; манностатин A; маримастат; мазопрокол; маспин; майтанзин; мехлорэтамина гидрохлорид; мегестрола ацетат; меленгестрола ацетат; мелфалан; меногарил; мербарон; меркаптопурин; метерелин; метиониназу; метотрексат натрия; метотрексат; метоклопрамид; метоприн; метуредепу; мифепристон; милтефосин; миримостим; митиндомид; митокарцин; митокромин; митогиллин; митогуазон; митолактол; митомалцин; производные митомицина; митомицин; митонафид; митоспер; митотан; митотоксиновый фактор роста фибробластов сапорин; митоксантрона гидрохлорид; митоксантрон; мофаротен; молграмостим; мопидамол; микапероксид B; микофеноловую кислоту; мириапорон; N-ацетилдиналин; нафарелин; нагрестип; наксолон+пентазоцин; напавин; нафтерпин; нартограстим; недаплатин; неморубицин; неридроновую кислоту; нилутамид; низамицин; нитруллин; ниволумаб (Opdivo®); нокодазол; ногаламицин; O6-бензилгуанидин; облимерсен (торговое название Genasense®); октреотид; окиценон; онапристон; ондансетрон; орацин; ормаплатин; осатерон; оксалиплатин; оксауномицин; оксисуран; паклитаксел; производные паклитаксела; палауамин; палбоциклиб; пальмитоилризоксин; памидроновую кислоту; панакситриол; паномифен; панобиностат; парабактин; пазеллиптин; пегаспаргазу; пелдезин; пелиомицин; пембролизумаб (Keytruda®); пентамустин; пентозанполисульфат натрия; пентостатин; пентрозол; пепломицина сульфат; перфлуброн; перфосфамид; периллиловый спирт; феназиномицин; фенилацетат; пицибанил; пилокарпина гидрохлорид; пипоброман; пипосульфан; пирарубицин; пиритрексим; пироксантрона гидрохлорид; плацетин A; плацетин B; комплекс платины; пликамицин; пломестан; порфимер натрия; порфирмицин; преднимустин; метакортандрацин; прокарбазина гидрохлорид; пропил бис-акридон; простагландин J2; пуромицина гидрохлорид; пуромицин; пурпурины; пиразофурин; пиразолакридин; ралтитрексед; рамосетрон; рапамицин; производные рапамицина(например, эверолимус); мерилимус; олкоролимус; ридафоролимус; сиролимус; темсиролимус (сиролимус,торговое название Torisel); умиролимус и (зотаролимус); деметилированный ретеллиптин; этидронат рения Re 186; ризоксин; рибоприн; рибозимы; RII ретинамид; рохитукин; ромуртид; рохинимекс; рубигинон B1; рубоксил; сафингола гидрохлорид; сафингол; саинтопин; SarCNU; саркофитол A; сарграмостим; миметики Sdi 1; семаксаниб; семустин; симтразен; сизофиран; собузоксан; борокаптат натрия; фенилацетат натрия; солверол; соматомедин-связывающий белок; сонермин; спарфосат натрия; спарфосат; спарсомицин; спикамицин D; спирогермания гидрохлорид; спиромустин; спироплатин; спленопентин; спонгистатин 1; скваламин; стипиамид; стрептонигрин; стрептозоцин; сульфинозин; сулофенур; сурадисту; сурамин; сваинсонин; тализомицин; таллимустин; тамоксифена метиодид; тауромустин; таксотер; текогалан натрия; тегафур; теллурапирилиум; телоксантрона гидрохлорид; темопорфин; тенипозид; тероксирон; тетрахлордекаоксид; тетразомин; талибластин; тиамиприн; тиокоралин; тиогуанин); тиотепу; миметики тромбопоэтина; тромбопоэтин; тималфазин; тимотринан; тиазофурин; олова этилэтиопурпурин; тирапазамин; титаноцена бихлорид; топсентин; торемифена цитрат; торемифен; трестолона ацетат; третиноин; триацетилуридин; трицирибина фосфат; трицирибин; триметрексата глюкуронат; триметрексат; трипторелин; трописетрон; тубулозола гидрохлорид; туростерид; тирфостины; убенимекс; урацил иприт; уредепу; вапреотид; вариолин B; веларезол; верамин; вердины; вертепорфин; винбластина сульфат; винкристина сульфат; виндезина сульфат; виндезин; винепидина сульфат; винглицината сульфат; винлеурозина сульфат; (винорелбина тартрат; винорелбин; винрозидина сульфат; винксалтин; винзолидина сульфат; витаксин; ворозол; занотерон; зениплатин; зиностатин; 5-этинилурацил и зорубицина гидрохлорид.
В предпочтительном варианте осуществления другое(-ие) терапевтическое(-ие) средство(-а) выбрано(-ы) из группы, состоящей из элотузумаба, палбоциклиба, панобиностата, ниволумаба, пембролизумаба, пеметрекседа, топотекана, доксорубицина, бортезомиба, гемцитабина, дакарбазина, дексаметазона, биаксина, винкристина, азацитидина, CAR-T, ритуксимаба, трастузумаба, ингибитора PD-1, ингибитора PD-L1, ингибитора HDAC, ингибитора андрогенового рецептора, ингибитора биосинтеза андрогена, преднизона, доцетаксела, инъекции клофарабина, ублитуксимаба, ромидепсина, ингибитора BTK, эритропоэтина, элтромбопага, миноциклина и мелфалана.
В варианте осуществления настоящего изобретения композицию, содержащую соединение, имеющее структуру общей формулы (I), его фармацевтически приемлемую соль, сольват, полиморф, стереоизомер, изотопное соединение, метаболит или пролекарство и одно другое терапевтическое средство вводят субъекту одновременно. В другом варианте осуществления соединение, имеющее структуру общей формулы (I), его фармацевтически приемлемую соль, сольват, полиморф, стереоизомер, изотопное соединение, метаболит или пролекарство и одно другое терапевтическое средство вводят последовательно. В другом варианте осуществления соединение, имеющее структуру общей формулы (I), его фармацевтически приемлемую соль, сольват, полиморф, стереоизомер, изотопное соединение, метаболит или пролекарство и одно другое терапевтическое средство вводят по отдельности. Другое терапевтическое средство можно вводить до, с последующим введением или после введения соединения, имеющего структуру общей формулы (I), его фармацевтически приемлемой соли, сольвата, полиморфа, стереоизомера, изотопного соединения, метаболита или пролекарства.
Одно или более чем одно другое терапевтическое средство, которое можно вводить в комбинации с соединением, имеющим структуру общей формулы (I), его фармацевтически приемлемой солью, сольватом, полиморфом, стереоизомером, изотопным соединением, метаболитом или пролекарством, зависит от множества факторов, таких как заболевание, симптом или нарушение, подлежащие лечению или предупреждению, и т. д. Специалист в данной области техники может легко определить подходящее другое терапевтическое средство, подлежащее введению в комбинации с соединением, имеющим структуру общей формулы (I), его фармацевтически приемлемой солью, сольватом, полиморфом, стереоизомером, изотопным соединением, метаболитом или пролекарством, исходя из раскрытого содержания.
Терапевтически эффективное количество другого терапевтического средства, используемого в способе по настоящему изобретению, известно специалисту в данной области техники, и инструкция по введению может ссылаться на опубликованные патенты и заявки, а также Wells et al, eds., Pharmacotherapy Handbook, 2nd Edition, Appleton and Lange, Stamford, Conn. (2000); PDR Pharmacopoeia, Tarascon Pocket Pharmacopoeia 2000, Deluxe Edition, Tarascon Publishing, Loma Linda, Calif. (2000) и другую медицинскую литературу, приведенную в настоящем документе. Однако специалист в данной области техники способен определить оптимальный диапазон доз другого терапевтического средства.
В соответствии с вариантом осуществления настоящего изобретения, при введении в комбинации с другим(-и) терапевтическим(-и) средством(-ами), терапевтически эффективное количество соединения, имеющего структуру общей формулы (I), его фармацевтически приемлемой соли, сольвата, полиморфа, стереоизомера, изотопного соединения, метаболита или пролекарства меньше необходимого терапевтически эффективного количества соединения, имеющего структуру общей формулы (I), его фармацевтически приемлемой соли, сольвата, полиморфа, стереоизомера, изотопного соединения, метаболита или пролекарства, в случае если не в комбинации с другим(-и) терапевтическим(-и) средством(-ами). В другом варианте осуществления терапевтически эффективное количество другого(-их) терапевтического(-их) средства(-ств) меньше, чем в случае если при введении не применяется соединение, имеющее структуру общей формулы (I), его фармацевтически приемлемая соль, сольват, полиморф, стереоизомер, изотопное соединение, метаболит или пролекарство. В соответствии с этим, побочные действия, связанные с любыми лекарственными средствами могут быть сведены к минимуму. Другие потенциальные преимущества, например улучшение введения раствора и/или снижение стоимости лекарственных средств, являются очевидными специалисту в данной области техники.
В соответствии с вариантом осуществления настоящего изобретения, в случае если соединение, имеющее структуру общей формулы (I), его фармацевтически приемлемую соль, сольват, полиморф, стереоизомер, изотопное соединение, метаболит или пролекарство и другое(-ие) терапевтическое(-ие) средство(-а) вводят субъекту для лечения или предупреждения заболевания, симптома или нарушения, соединение, имеющее структуру общей формулы (I), его фармацевтически приемлемую соль, сольват, полиморф, стереоизомер, изотопное соединение, метаболит или пролекарство и другое(-ие) терапевтическое(-ие) средство(-а) можно вводить с помощью такого же способа или разных способов. Другое(-ие) терапевтическое(-ие) средство(-а) можно вводить с помощью любых способов, описанных в данном документе, в том числе без ограничения, пероральным введением, ингаляцией, инъекцией, глазным введением, введением через слизистую оболочку, ректальным введением, эмульсией, липосомой, имплантантом длительного действия или введением с замедленным высвобождением. Определенное введение другого(-их) терапевтического(-их) средства(-ств) зависит от него(них) самого(самих), а также препарата и заболевания, симптома или нарушения, подлежащих предупреждению или лечению. В соответствии с раскрытиями в данном документе, специалист в данной области техники может определить введение другого(-их) терапевтического(-их) средства(-ств).
Настоящая заявка ссылается или описывает множество публикаций, литературных источников и патентов, при этом цель приведения или описания этих ссылок, или включения этих ссылок во всей их полноте, или обсуждения этих ссылок, состоит в определении предпосылок настоящего изобретения, однако это не означает, что содержание этих ссылок вносит вклад в часть предшествующего уровня техники настоящего изобретения.
Если не указано иное, то используемые в данном документе технические и научные термины имеют те же значения, которые обычно понимаются специалистом в данной области техники. В ином случае они имеют значения, указанные в настоящем описании. Все патенты, заявки на патент, которые уже были раскрыты, а также публикации, приведенные в данном документе, включены в настоящий документ посредством ссылки, точно так же, как подробно описано в данном документе. Необходимо отметить, что если в данном документе не указано иное, то форма единственного числа, используемая в данном документе и в прилагаемой формуле изобретения, имеет значение множественного числа.
Как используется в данном документе, в случае если определенная соль, композиция, а также наполнитель и т. д. являются «фармацевтически приемлемыми», это означает, что соль, композиция, наполнитель и т. д. являются, как правило, нетоксичными, безопасными и подходящими для введения субъекту, предпочтительно млекопитающему, более предпочтительно человеку.
В данном документеТермин «фармацевтически приемлемая соль» относится к фармацевтически приемлемой органической или неорганической соли. Примеры соли включают без ограничения соль серной, лимонной, уксусной, щавелевой, хлороводородной, бромистоводородной, йодистоводородной, азотной кислот, гидросульфат, фосфорной кислоты, кислой соли фосфорной кислоты, соль изоникотиновой кислоты, молочной, соль салициловой кислоты, лимонной кислоты, винной, олеиновой, соль дубильной кислоты, пантотеновой, винной, аскорбиновой, янтарной, малеиновой, гентизинатной, фумаровой, глюконовой, соль глюкуроновой кислоты, сахарной, муравьиной, бензойной, глутаминовой, метансульфоната, этансульфоната, бензолсульфоната, п-толуолсульфоната и эмбоната (т. е. 1-1-метилен-бис-(2-гидрокси-3-нафтоат)). Соединения по настоящему изобретению можно использовать для образования фармацевтически приемлемых солей с различными аминокислотами. Подходящая соль щелочного металла включает без ограничения соль алюминия, соль кальция, соль лития, соль магния, соль калия, соль натрия, соль цинка, соль висмута и соль диэтаноламина. Обзор, касающийся фармацевтически приемлемых солей, ссылается на Handbook of Pharmaceutical Salts: Properties, Selection, and Use (P. Heinrich Stahl and Camille G. Wermuth, ed., Wiley-VCH, 2002).
Используемый в данном документе термин «метаболит» относится к активному веществу, которое получают после изменения in vivo химической структуры молекулы лекарственного средства, при этом активное вещество, как правило, представляет собой производное вышеупомянутой молекулы лекарственного средства, и оно также может быть химически модифицированным.
Используемый в данном документе, и если не указано иное, термин «полиморф» относится к одному или более чем одному виду(-ам) кристаллической структуры, образованной посредством различного расположения молекул в пространственной решетке при кристаллизации.
Используемый в данном документе термин «сольват» относится к кристаллической форме соединения, имеющего структуру общей формулы (I), его фармацевтически приемлемой соли, полиморфа, стереоизомера, изотопного соединения, метаболита или пролекарства, которые дополнительно имеют один более одного вида(-ов) молекулы(-кул) растворителя, включенных в кристаллическую структуру. Сольват может включать стехиометрическое количество или нестехиометрическое количество растворителя, и молекула растворителя в растворителе может существовать в упорядоченной или неупорядоченной структуре. Сольват, содержащий нестехиометрическое количество молекул растворителя, может быть образован путем потери по меньшей мере одной молекулы растворителя (но не всех) из сольвата. В определенном варианте осуществления сольват относится к гидрату, что означает, что кристалл соединения дополнительно включает молекулу воды, и воду используют в качестве растворителя.
Используемый в данной документе, и если не указано иное, термин «пролекарство» относится к производному соединения, содержащему биологически реакционноспособную функциональную группу, при этом биологически реакционноспособная функциональная группа может быть отщеплена из соединения или реагировать по-другому, с получением соединения в биологических условиях (in vivo или in vitro). Как правило, пролекарство является неактивным или по меньшей мере имеет более низкую активность, чем соединение, что обеспечивает соединению способность проявлять активность до тех пор, пока оно не будет отщеплено от биологически реакционноспособной функциональной группы. Биологически реакционноспособная функциональная группа может быть гидролизована или окислена в биологических условиях с получением соединения. Например, пролекарство может содержать биологически гидролизуемую группу. Примеры биологически гидролизуемой группы включают без ограничения биологически гидролизуемый фосфат, биологически гидролизуемый сложный эфир, биологически гидролизуемый амид, биологически гидролизуемый сложный эфир угольной кислоты, биологически гидролизуемый карбамат и биологически гидролизуемый уреид. Обзор, касающийся пролекарства, ссылается, например, на J. Rautio et al., Nature Reviews Drug Discovery (2008) 7, 255-270 и Prodrugs: Challenges Rewards (V. Stella et al. ed., Springer, 2007).
Соединение имеющее в настоящем изобретении структуру общей формулы (I), его фармацевтически приемлемая соль, сольват, полиморф, стереоизомер, изотопное соединение, метаболит или пролекарство могут содержать один или более одного центров асимметрии («стереоизомер»). Используемый в данном документе термин «стереоизомер» относится ко всем стереоизомерам, в том числе энантиомеру, диастереоизомеру, эпимеру, эндо-экзо изомеру, атропоизомеру, региоизомеру, цис- и транс-изомеру. В данном документе выражение «стереоизомер»включает также «чистый стереоизомер» и «обогащенный стереоизомер» или «рацемический изомер» различных вышеупомянутых стереоизомеров. Такие стереоизомеры могут быть получены в соответствии со способом асимметрического синтеза, или разделены, очищены и обогащены посредством способа хирального разделения (включая без ограничения тонкослойную хроматографию, вращающуюся хроматографию, колоночную хроматографию, газовую хроматографию, жидкостную хроматографию высокого давления и т. д.), а также получены посредством хирального разделения с помощью связывания (химическое связывание и т. д.) или образования соли (физическое связывание и т. д.) с другим(-и) хиральным(-и) соединением(-ями). В данном документе термин «чистый стереоизомер» означает то, что массовая доля стереоизомера соединения составляет не менее 95% по сравнению с другими стереоизомерами соединения. В данном документе термин «обогащенный стереоизомер» означает то, что массовая доля стереоизомера соединения составляет не менее 50% по сравнению с другими стереоизомерами соединения. В данном документе термин «рацемический изомер» означает то, что массовая доля стереоизомера соединения равна массовой доле других стереоизомеров соединения.
Термин «изотопное соединение», используемый в данном документе, означает, что один или более одного атомного(-ных) изотопа(-ов) с природной или неприродной распространенностью содержатся в соединении, имеющем структуру общей формулы (I), его фармацевтически приемлемой соли, сольвате, полиморфе, стереоизомере, изотопном соединении, метаболите или пролекарстве. Атомные изотопы с неприродной распространенностью включают без ограничения дейтерий (2H или D), тритий (3H или T), йод-125 (125I), фосфор-32 (32P), углерод-13 (13C) или углерод-14 (14C). Вышеупомянутое изотопное соединение можно также использовать в качестве терапевтического или диагностического средства (т. е. внутреннего средства для визуализации) или инструмента исследования. Все изотопные варианты соединения по настоящему изобретению, независимо от того, являются они радиоактивными или нет, включены в объем настоящего изобретения.
Термин «обогащенный изотопом», используемый в данном документе, означает, что один или более одного атомного(-ых) изотопа(-ов) с природной или неприродной распространенностью содержатся в соединении, имеющем структуру общей формулы (I), его фармацевтически приемлемой соли, сольвате, полиморфе, стереоизомере, изотопном соединении, метаболите или пролекарстве. Термин «обогащенный изотопом» также означает, что соединение, имеющее структуру общей формулы (I), его фармацевтически приемлемая соль, сольват, полиморф, стереоизомер, изотопное соединение, метаболит или пролекарство содержат по меньшей мере один изотопный атом с неприродной распространенностью.
Используемый в данном документе термин «субъект» относится к любому животному, подлежащему предупреждению или лечению соединением или композицией в соответствии с вариантом осуществления настоящего изобретения, при этом предпочтительным является млекопитающее и оптимальным является человек. Термин «млекопитающее», используемый в данном документе, включает любых млекопитающих. Примеры млекопитающего включают без ограничения крупный рогатый скот, коня, овцу, свинью, кошку, собаку, мышь, крысу, кролика, морскую свинку, обезьяну, человека и т. д., при этом оптимальным является человек.
В одном варианте осуществления термины «лечить» и «лечение» относятся к улучшению, предупреждению или устранению заболевания или состояния, или по меньшей мере одного из их идентифицируемых симптомов, как например к лечению рака, нарушений, связанных с нежелательным ангиогенезом или TNF-α путем уменьшения или стабилизации симптомов рака или заболевания. В другом варианте осуществления выражения «лечить» и «лечение» относятся к улучшению, предупреждению или устранению по меньшей мере одного измеряемого параметра тела при заболевании или состоянии, подлежащих лечению, при этом заболевание или состояние могут не быть идентифицированы у млекопитающего, например к лечению рака или нарушения, связанного с нежелательным ангиогенезом или TNF-α путем ингибирования образования TNF-α или модулирования активности TNF-α. Однако в другом варианте осуществления термины «лечить» и «лечение» относятся к замедлению развития заболевания или симптома в физическом состоянии, например к стабилизации идентифицируемых симптомов, или в физиологическом состоянии, например к стабилизации физических параметров, или к обоим. В другом варианте осуществления термины «лечить» и «лечение» относятся к задержке развития заболевания или симптома.
В некоторых вариантах осуществления соединение вводят с целью предупреждения. Как используется в данном документе, выражения «предупреждать» или «предупреждение» относятся к уменьшению риска возникновения данного заболевания или симптома. В предпочтительном варианте осуществления указанное соединение вводят субъекту с целью предупреждения, например субъекту с семейным анамнезом или тенденцией возникновения рака или аутоиммунного заболевания.
Как используется в данном документе, «терапевтически эффективное количество» относится к количеству соединения или композиции, которое может вызвать биологическую или медицинскую ответную реакцию (которое ищут исследователи, ветеринары, врачи или другие клиницисты) у организма, животного или индивида, который может включать облегчение симптомов заболевания или симптома, подлежащих лечению. В предпочтительном варианте осуществления терапевтически эффективное количество является количеством, которого достаточно для эффективного лечения, улучшающего лечения или предупреждения рака, симптома или нарушения, связанных с нежелательным ангиогенезом или TNF-α.
Термин «профилактически эффективное количество» относится к количеству активного соединения или средства (которые ищут исследователи, ветеринары, врачи или другие клиницисты), которые могу ингибировать развитие заболевания у субъекта. Профилактически эффективное количество соединения относится к количеству терапевтического средства, используемого отдельно или в комбинации с другим активным соединением, которое может обеспечить терапевтическую пользу для лечения или предупреждения заболевания, симптома или нарушения.
Если не указано иное, то форма единственного числа термина, используемого в данном документе, включает также значение множественного числа.
Если не указано иное, то термин «или» или «и», используемый в данном документе, означает «и/или».
Если не указано иное, то обозначения «


Каждое из вышеуказанных предпочтительных условий может быть объединено случайным образом, не отступая от общедоступных сведений в данной области техники, формируя тем самым различные предпочтительные варианты осуществления настоящего изобретения.
Все реагенты и исходные материалы, используемые в данном документе, являются коммерчески доступными.
Положительные эффекты, достигаемые с помощью настоящего изобретения, состоят в том, что производное изоиндолина, имеющее структуру общей формулы (I), способно регулировать образование и/или активность цитокинов (например, TNF-α), с тем чтобы эффективно лечить рак и воспалительные заболевания.
Примеры
Пример 1. Соединение I-28
Стадия A: к смеси 5-фтор-2-метилбензойной кислоты (6,0 г, 39,0 ммоль) в 98% H2SO4 (60 мл) добавляли 65% HNO3 (3,3 г, 50,7 ммоль) при от -5oC до 0oC, затем полученную смесь перемешивали при данной температуре в течение 1 ч. Смесь выливали в 200 г ледяной воды, затем экстрагировали с помощью MTBE (150 мл × 3). Объединенную органическую фазу промывали солевым раствором (20 мл), высушивали над Na2SO4, фильтровали и выпаривали досуха посредствомротационного выпаривания с получением 5-фтор-2-метил-3-нитробензойной кислоты (7,0 г, неочищенная) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
1H ЯМР (DMSO-d6, 300 МГц): δ 8,05 (dd, J = 8,1 Гц, 3,0 Гц, 1H), 7,85 (dd, J = 8,7, 3,0 Гц, 1H), 2,44 (s, 3H).
Стадия B: к смеси 5-фтор-2-метил-3-нитробензойной кислоты (7,0 г, неочищенная) в MeOH (70 мл) добавляли 98% H2SO4(2 мл), затем полученную смесь перемешивали в течение ночи при 70oC. Смесь концентрировали, затем остаток разбавляли с помощью H2O (50 мл) и EtOAc (150 мл). Водный слой экстрагировали с помощью EtOAc (100 мл × 2), объединенную органическую фазу промывали солевым раствором (30 мл), высушивали над Na2SO4, фильтровали и выпаривали досуха посредством ротационного выпаривания. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюированном с помощью (PE : EtOAc = от 10:1 до 3:1), с получением метил 5-фтор-2-метил-3-нитробензоата (3,5 г, выход 42%, две стадии) в виде светло-желтого твердого вещества.
1H ЯМР (DMSO-d6, 300 MГц): δ 8,10 (dd, J = 8,1, 3,0 Гц, 1H), 7,88 (dd, J = 8,7, 3,0 Гц, 1H), 3,86 (s, 3H), 2,41 (s, 3H).
Стадия C: к смеси метил-5-фтор-2-метил-3-нитробензоата (3,5 г, 16,4 ммоль) и пероксида бензоила (388 мг, 1,6 ммоль) в CCl4(40 мл) добавляли NBS (3,2 г, 18,1 ммоль), затем полученную смесь перемешивали в течение ночи при 95oC. Смесь охлаждали до комнатной температуры и фильтровали, а фильтрат промывали солевым раствором (20 мл), высушивали над Na2SO4, фильтровали и выпаривали досуха посредствомротационного выпаривания. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюированном с помощью (PE : EtOAc = 10:1-5:1), с получением метил-2-(бромметил)-5-фтор-3-нитробензоата (3,7 г, выход 77%) в виде светло-желтого масла.
1H ЯМР (DMSO-d6, 300 МГц): δ 8,21-8,25 (dd, J = 8,1, 3,0 Гц, 1H), 7,99-8,03 (dd, J = 8,7, 2,7 Гц, 1H), 4,97 (s, 2H), 3,93 (s, 3H).
Стадия D: к смеси 3-аминопиперидин-2,6-диона гидрохлорида (2,5 г, 15,1 ммоль) и KHCO3 (3,5 г, 34,2 ммоль) в CH3CN (80 мл) добавляли метил-2-(бромметил)-5-фтор-3-нитробензоат (4,0 г, 13,7 ммоль), затем полученную смесь перемешивали в течение ночи при 95oC. Смесь концентрировали, затем выливали в 100 г ледяной воды, затем смесь перемешивали в течение 0,5 ч. Смесь фильтровали и твердое вещество промывали с помощью H2O (50 мл × 3), высушивали с получением соединения I-28A [3-(6-фтор-4-нитро-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион] (3,8 г, выход 90%) в виде желтого твердого вещества.
1H ЯМР (DMSO-d6, 400 МГц): δ 11,04 (s, 1H), 8,38 (dd, J = 8,8, 2,4 Гц, 1H), 8,11 (dd, J = 6,8, 2,4 Гц, 1H), 5,18 (dd, J = 13,2, 5,2 Гц, 1H), 4,88 (d, J = 19,2 Гц, 1H), 4,78 (d, J = 19,2 Гц, 1H), 2,87-2,96 (m, 1H), 2,54-2,63 (m, 2H), 2,01-2,05 (m, 1H).
Стадия E: смесь соединения I-28A (2,8 г, 9,1 ммоль) и Pd/C (10%, 280 мг, 50% воды) в DMF (30 мл) перемешивали в течение 6 ч. в атмосфере H2при давлении 50 фунтов/кв. дюйм при комнатной температуре. Смесь фильтровали и твердое вещество промывали с помощью DMF (50 мл × 1). Фильтрат концентрировали, затем выливали в H2O (100 мл) и перемешивали в течение 0,5 ч. Смесь фильтровали, твердое вещество промывали с помощью H2O (50 мл × 3), высушивали с получением неочищенного продукта (2,1 г, выход 84%) в виде грязно-белого твердого вещества. 200 мг вышеуказанного неочищенного продукта очищали с помощью преп-HPLC с получением I-28 [3-(4-амино-6-фтор-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион] (148,8 мг) в виде грязно-белого твердого вещества.
1H ЯМР (DMSO-d6, 400 МГц): 11,01 (s, 1H), 6,55-6,63 (m, 2H), 5,79 (s, 2H), 5,10 (dd, J = 13,2, 4,8 Гц, 1H), 4,18 (d, J = 17,2 Гц, 1H), 4,08 (d, J = 17,2 Гц, 1H), 2,87-2,95 (m, 1H), 2,59-2,63 (m, 1H), 2,27-2,31 (m, 1H), 2,02-2,06 (m, 1H). LCMS: 278,1 ([M+1]+).
Соединения I-01-I-27 можно получить в соответствии со способом, описанным в примере 1.
Пример 2. Синтез соединений I-29 и I-30
800 мг соединения I-28 в 28 мл DMF разделяли с помощью хиральной HPLC (колонка: CHIRALPAK IA, 5 мкм, 30 x 250 мм; подвижная фаза: CH3CN; расход: 21 мл/мин.; температура: 26-28oC; длина волны: 230 нм; введение: 350 мкл) с получением 300 мг I-29 и 260 мг I-30.
I-29: [Rt=4,81 мин.; >99% ee;1H ЯМР (DMSO-d6, 400 МГц): δ 11,01 (s, 1H), 6,55-6,63 (m, 2H), 5,80 (s, 2H), 5,07-5,12 (m, 1H), 4,18 (d, J = 16,8 Гц, 1H), 4,08 (d, J = 16,8 Гц, 1H), 2,87-2,93 (m, 1H), 2,59-2,63 (m, 1H), 2,27-2,31 (m, 1H), 2,02-2,07 (m, 1H). LCMS: 278,1 ([M+1]+)].
I-30:[Rt=7,08 мин.; >97,5% ee;1H ЯМР (DMSO-d6, 400 МГц): δ 11,01 (s, 1H), 6,55-6,63 (m, 2H), 5,79 (s, 2H), 5,08-5,12 (m, 1H), 4,19 (d, J = 17,2 Гц, 1H), 4,08 (d, J = 17,2 Гц, 1H), 2,87-2,95 (m, 1H), 2,59-2,64 (m, 1H), 2,27-2,31 (m, 1H), 2,04-2,07 (m, 1H). LCMS: 278,1 ([M+1]+)].
Пример 3. Соединения I-31 и I-32
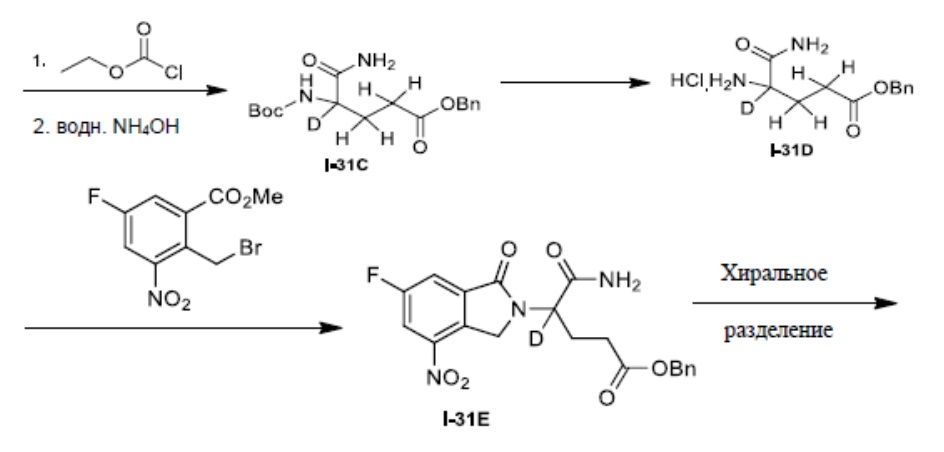

Стадия A: к раствору (S)-2-амино-5-(бензилокси)-5-оксопентановой кислоты (50,0 г, 221 ммоль) в CH3CO2D (150 мл) добавляли бензальдегид (1,34 г, 12,6 ммоль), смесь нагревали до 65oC и перемешивали в течение 18 часов. Реакционную смесь концентрировали и полученное твердое вещество растирали в порошок с MeOH (25 мл), CH3CN (50 мл) и t-BuOMe (200 мл) в течение 30 мин., осадок на фильтре промывали с помощью t-BuOMe (200 мл) и высушивали. Продукт подвергали такой же процедуре с получением соединения I-31A (32,5 г, выход = 65%).
1H ЯМР (CD3OD+D2O, 300 МГц): δ 7,33-7,42 (m, 5H), 5,14 (s, 2H), 3,70 (t,<0,05H), 2,58-2,63 (m, 2H), 2,13-2,17 (m, 2H).
Стадия B: к смеси THF (600 мл) и H2O (600 мл) добавляли соединение I-31A (32,5 г, 137 ммоль). NaHCO3(12,6 г, 150 ммоль) добавляли к вышеуказанной смеси в ледяной бане, через 10 мин. медленно добавляли (BOC)2O (32,7 г, 150 ммоль), реакционную смесь перемешивали в течение 4 часов при комнатной температуре. THF удаляли при сниженном давлении (в вакууме), насыщ. раствор NaHCO3добавляли к растворному остатку, смесь экстрагировали с помощью t-BuOMe (200 мл x 2). Водную фазу охлаждали с помощью ледяной бани и регулировали до рH=1 с помощью 3 н. HCl, затем экстрагировали с помощью EtOAc (300 мл x 2), объединенные органические слои высушивали над Na2SO4, фильтровали и концентрировали с получением I-31B (46,5 г, выход 100%) в виде белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
1H ЯМР (CD3OD, 300 МГц): δ 7,29-7,35 (m, 5H), 5,12 (s, 2H), 4,13 (br s, 0,05H), 2,45-2,50 (m, 2H), 2,12-2,21 (m, 1H), 1,85-1,94 (m, 1H), 1,42 (s, 9H).
Стадия C: к раствору I-31B (46,5 г, 137 ммоль) в THF (500 мл), охлажденному до 5oC, медленно добавляли N-метилморфолин (NMM) (16,5 г, 164 ммоль) и этилхлоркарбонат (17,8 г, 164 ммоль). Реакционную смесь перемешивали при 0-5oC в течение 1 часа. К реакционной смеси добавляли 150 мл насыщенного NH3.H2O и затем перемешивали в течение 2 часов при комнатной температуре. Добавляли EtOAc (200 мл) и отделяли органическую фазу, водный слой экстрагировали с помощью EtOAc (200 мл), объединенную органическую фазу промывали последовательно с помощью водного NaHCO3 (200 мл х 2) и солевого раствора (200 мл), высушивали над Na2SO4, фильтровали и концентрировали с получением I-31C (41 г, 90%) в виде белого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 7,30-7,41 (m, 5H), 7,24 (s, 1H), 6,99 (s, 1H), 6,79 (s, 1H), 5,06 (s, 2H), 3,90 (m, <0,05H), 2,33-2,38 (m, 2H), 1,70-1,94 (m, 2H), 1,35 (s, 9H).
Стадия D: к раствору I-31C (41,0 г, 121 ммоль) в 1,4-диоксане (200 мл) добавляли раствор 6 н. HCl в 1,4-диоксане (300 мл), смесь перемешивали в течение 2 часов при комнатной температуре. Реакционную смесь концентрировали при сниженном давлении (в вакууме) с получением твердого вещества, которое растирали в порошок с помощью t-BuOMe (200 мл), а затем фильтровали и высушивали с получением продукта I-31D (31,3 г, выход 95%) в виде белого твердого вещества.
1H ЯМР (DMSO-d6, 400 МГц): δ 8,37 (br s, 3H), 8,04 (s, 1H), 7,56 (s, 1H), 7,33-7,38 (m, 5H), 5,10 (s, 2H), 3,77 (t, <0,05H), 2,48-2,52 (m, 2H), 2,01-2,05 (m, 2H).
Стадия E: смесь метил-2-(бромметил)-5-фтор-3-нитробензоата (31,8 г, 108,9 ммоль), а также соединения I-31D (29,7 г, 109 ммоль) и Et3N (22,1 г, 218 ммоль) в CH3CN (550 мл) перемешивали при 75oC в течение ночи. Смесь концентрировали. Остаток растирали в порошок с помощью CH3CN (100 мл) с получением соединения I-31E (34,5 г, выход 76,2%) в виде бледно-желтого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 8,33 (dd, J = 8,7, 2,4 Гц, 1H), 8,04 (dd, J = 6,9, 2,4 Гц, 1H), 7,66 (s, 1H), 7,26-7,36 (m, 6H), 4,82-5,05 (m, 4H), 2,20-2,39 (m, 3H), 2,06-2,15 (m, 1H).
Стадия F: соединение I-31E подвергали разделению хиральной HPLC (колонка: DAICEL CHIRALPAK IA, 10 мкм, 25 x 250 мм; подвижная фаза: MeOH / DCM = 80/20 (об./об.); расход: 30 мл/мин.; температура: 35oC; длина волны: 254 нм) с получением двух соединений I-31F1 [1H ЯМР (DMSO-d6, 300 МГц): δ 8,31-8,35 (m, 1H), 8,03 (dd, J = 7,2, 2,1 Гц, 1H), 7,66 (s, 1H), 7,29-7,35 (m, 6H), 4,83-5,04 (m, 4H), 2,22-2,40 (m, 3H), 2,06-2,16 (m, 1H)] и I-31F2 [1H ЯМР (DMSO-d6, 300 МГц): δ 8,33 (dd, J = 9,3, 2,4 Гц, 1H), 8,03 (dd, J = 7,2, 2,4 Гц, 1H), 7,67 (s, 1H), 7,29-7,36 (m, 6H), 4,83-5,05 (m, 4H), 2,20-2,42 (m, 3H), 2,09-2,16 (m, 1H)].
Соединение I-32: смесь соединения I-31F2 (2,2 г, 5,3 ммоль) и Pd/C (10%, 200 мг, 50% воды) в безводном MeOH (30 мл) перемешивали в течение 4 ч. в атмосфере H2при давлении 50 фунтов/кв. дюйм при комнатной температуре. Смесь фильтровали и фильтрат концентрировали, полученное твердое вещество добавляли в DCE (15 мл) и перемешивали в течение 5 мин., затем смесь концентрировали с получением остатка в виде грязно-белого твердого вещества (1,4 г). Вышеуказанное твердое вещество (1,1 г, 3,7 ммоль) растворяли в сухом THF (10 мл) и DCE (40 мл), а затем к смеси медленно добавляли SOCl2 (0,74 г, 9,3 ммоль) при -30oC, после перемешивания в течение 2 ч. добавляли пиридин (1,1 г, 9,3 ммоль) и перемешивали при данной температуре в течение 40 мин., добавляли Et3N (1,3 г, 13 ммоль) и затем смесь перемешивали в течение 2 ч. Добавляли H2O (0,1 мл) и затем смесь концентрировали досуха, остаток растворяли в H2O (5 мл) и экстрагировали с помощью EtOAc (70 мл x 5), высушивали над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью преп-HPLC с получением I-32 (340 мг, выход 33%, ee: 99%) в виде бледно-зеленого твердого вещества.
1H ЯМР (DMSO-d6, 400 МГц): 11,00 (s, 1H), 6,56-6,61 (m, 2H), 5,78 (s, 2H), 5,05-5,11 (m, 0,05 H), 4,17 (d, J = 17,1 Гц, 1H), 4,05 (d, J = 17,1 Гц, 1H), 2,84-2,96 (m, 1H), 2,56-2,62 (m, 1H), 2,20-2,32 (m, 1H), 1,98-2,05 (m, 1H). LCMS: 279,1 ([M+1]+).
Соединение I-31: следуя тому же способу синтеза, как и для соединения I-32, соединение I-31F1 превращали в I-31 (99% ee).1H ЯМР (DMSO-d6, 400 МГц): 10,99 (s, 1H), 6,52-6,61 (m, 2H), 5,71 (br s, 2H), 5,08 (dd, J = 18,0, 7,2 Гц, 0,04 H) 4,17 (d, J = 17,4 Гц, 1H), 4,06 (d, J = 17,4 Гц, 1H), 2,83-2,96 (m, 1H), 2,47-2,62 (m, 1H), 2,21-2,32 (m, 1H), 1,98-2,05 (m, 1H). LCMS: 279,1 ([M+1]+).
Пример 4. Соединение I-01

Следуя процедуре в вышеуказанном примере 3, соединение I-01 из примера 4 получали с применением рацемического I-31E.
1H ЯМР (DMSO-d6, 300 МГц): δ 10,94 (br s, 1H), 6,52-6,61 (m, 2H), 5,78 (s, 2H), 5,05-5,11 (m, 0,05 H), 4,16 (d, J = 16,8 Гц, 1H), 4,05 (d, J = 16,8 Гц, 1H), 2,84-2,96 (m, 1H), 2,54-2,62 (m, 1H), 2,21-2,31 (m, 1H), 1,98-2,04 (m, 1H). LCMS: 279,1 ([M+1]+).
Соединения I-33-I-56 можно получить в соответствии со способом синтеза, показанным в примерах 2 или 3 с подходящим исходным материалом.
Пример 5. Соединение A195
3-(4-((2-Фтор-5-метоксибензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A195:

К раствору A340D (40 мг, 0,096 ммоль) в CH3CN (3 мл) добавляли CDI (20 мг, 0,13 ммоль). Реакционную смесь перемешивали в течение ночи при 90oC в атмосфере N2. После концентрирования при сниженном давлении остаток растворяли в DCM (30 мл), промывали с помощью 0,1 н. HCl (10 мл), насыщ. водн. NaHCO3 (10 мл), а затем насыщ. NaCl (10 мл), высушивали над Na2SO4, фильтровали и концентрировали при сниженном давлении. Затем неочищенный продукт очищали с помощью преп-TLC (DCM / MeOH = 25:1) 2 раза с получением соединения A195 (46 мг, выход 81%) в виде белого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 10,98 (s, 1H), 7,23 (t, J = 7,8 Гц, 1H), 7,11 (t, J = 9,6 Гц, 1H), 6,90-6,95 (m, 2H), 6,78-6,84 (m, 1H), 6,66 (d, J = 7,8 Гц, 1H), 6,24 (t, J = 5,7 Гц, 1H), 5,10 (dd, J = 13,2, 5,4 Гц, 1H), 4,36 (d, J = 5,4 Гц, 2H), 4,30 (d, J = 17,7 Гц, 1H), 4,17 (d, J = 17,7 Гц, 1H), 3,65 (s, 3H), 2,85-2,97 (m, 1H), 2,57-2,64 (m, 1H), 2,24-2,36 (m, 1H), 2,00-2,07 (m, 1H). LCMS: 398,1 ([M+1]+).
Соединения из примеров 6-7 получали в соответствии со способом синтеза, показанным в примере 5, с соответствующими исходными материалами для замены A340D.
Пример 6. Соединение A196
3-(4-((2-Фтор-3-метоксибензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион A196:

1H ЯМР (DMSO-d6, 300 МГц): δ 11,00 (s, 1H), 7,21 (t, J = 7,8 Гц, 1H), 7,00-7,04 (m, 2H), 6,90-6,95 (m, 2H), 6,62 (d, J = 7,8 Гц, 1H), 6,30 (t, J = 6,0 Гц, 1H), 5,10 (dd, J = 13,2, 5,7 Гц, 1H), 4,40 (d, J = 6,0 Гц, 2H), 4,28 (d, J = 17,4 Гц, 1H), 4,16 (d, J = 17,4 Гц, 1H), 3,81 (s, 3H), 2,85-2,97 (m, 1H), 2,56-2,63 (m, 1H), 2,26-2,32 (m, 1H), 1,99-2,07 (m, 1H). LCMS: 398,1 ([M+1]+).
Пример 7. Соединение A197
3-(4-((2-Фтор-3-метоксибензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион A197:
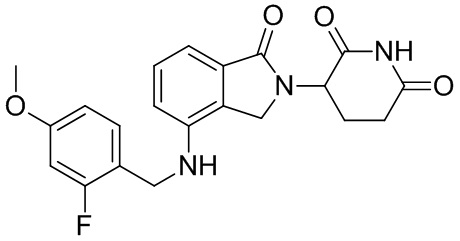
1H ЯМР (DMSO-d6, 300 МГц): δ 11,00 (s, 1H), 7,19-7,31 (m, 2H), 6,92 (d, J = 7,2 Гц, 1H), 6,80 (dd, J = 12,0, 2,4 Гц, 1H), 6,71 (dd, J = 8,4, 2,4 Гц, 1H), 6,65 (d, J = 7,8 Гц, 1H), 6,20 (t, J = 5,7 Гц, 1H), 5,10 (dd, J = 13,2, 5,1 Гц, 1H), 4,24-4,33 (m, 3H), 4,14 (d, J = 17,1 Гц, 1H), 3,72 (s, 3H), 2,85-2,97 (m, 1H), 2,57-2,62 (m, 1H), 2,21-2,36 (m, 1H), 1,98-2,05 (m, 1H). LCMS: 398,1 ([M+1]+).
Пример 8. Соединение A318
3-(6-Фтор-4-((2-фтор-3-метоксибензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион A318:


Стадия A: к смеси соединения I-28A (1,0 г, 3,3 ммоль) в DMF (10 мл) добавляли Pd/C (0,18 г, 10%, 50% воды) и дегазировали с помощью H2 3 раза. Смесь перемешивали при 25oC в течение 5 часов в атмосфере H2 при давлении 50 фунтов/кв. дюйм. Затем реакционную смесь фильтровали и концентрировали, после чего растирали в порошок с помощью PE / EtOAc (5:1, 10 мл x 3) с получением I-28 (неочищенный, 0,9 г) в виде зеленого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 10,98 (s, 1H), 6,52-6,61 (m, 2H), 5,77 (s, 2H), 5,07 (dd, J = 5,4, 13,2 Гц, 1H), 4,17 (d, J = 17,1 Гц, 1H), 4,06 (d, J = 17,1 Гц, 1H), 2,83-2,95 (m, 1H), 2,55-2,62 (m, 1H), 2,20-2,34 (m, 1H), 1,97-2,07 (m, 1H).
Стадия B: к раствору соединения I-28 и 2-фтор-3-метоксибензальдегида (85 мг, 0,551 ммоль) в AcOH (3 мл) добавляли дихлорэтан (15 мл) и перемешивали в течение 1 часа. Затем добавляли NaBH(OAc)3 (235 мг, 1,09 ммоль) и смесь перемешивали в течение 18 часов. Добавляли еще одну часть NaBH(OAc)3 (50 мг, 0,236 ммоль) и смесь нагревали до 30oC в течение 8 часов. Затем добавляли еще одну часть 2-фтор-3-метоксибензальдегида (30 мг, 0,195 ммоль) и смесь перемешивали при 40oC в течение 16 часов. Смесь концентрировали и очищали с помощью преп-TLC с получением неочищенного продукта, который растирали в порошок с помощью MeOH (1 мл) с получением A318 (30 мг, выход 20%) в виде грязно-белого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 11,00 (s, 1H), 7,05-7,07 (m, 2H), 6,91-6,94 (m, 1H), 6,61-6,65 (m, 2H), 6,44 (dd, J = 1,8, 12,9 Гц, 1H), 5,06-5,12 (m, 1H), 4,39 (d, J = 5,7 Гц, 2H), 4,26 (d, J = 17,1 Гц, 1H), 4,13 (d, J = 17,1 Гц, 1H), 3,81 (s, 3H), 2,86-2,90 (m, 1H), 2,57-2,63 (m, 1H), 2,24-2,30 (m, 1H), 2,02-2,06 (m, 1H). LCMS: 416,1 ([M+1]+).
Соединения из примеров 9-10 получали в соответствии со способом синтеза, показанным в примере 8, с соответствующим исходным материалом для замены 2-фтор-3-метоксибензальдегида на стадии B.
Пример 9. Соединение A319
3-(6-Фтор-4-((2-фтор-4-метоксибензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A319:

1H ЯМР (DMSO-d6, 300 МГц): δ 11,00 (s, 1H), 7,30 (t, J = 9,0 Гц, 1H), 6,71-6,84 (m, 2H), 6,55-6,64 (m, 2H), 6,47 (dd, J = 2,1, 12,6 Гц, 1H), 5,05-5,11 (m, 1H), 4,31 (d, J = 5,1 Гц, 2H), 4,24 (d, J = 17,4 Гц, 1H), 4,11 (d, J = 17,4 Гц, 1H), 3,73 (s, 3H), 2,84-2,96 (m, 1H), 2,57-2,62 (m, 1H), 2,19-2,34 (m, 1H), 2,03-2,06 (m, 1H). LCMS: 416,1 ([M+1]+).
Пример 10. Соединение A320
3-(6-Фтор-4-((2-фтор-5-метоксибензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион A320:

1H ЯМР (DMSO-d6, 300 МГц): δ 11,00 (s, 1H), 7,12 (t, J = 9,3 Гц, 1H), 6,81-6,94 (m, 2H), 6,60-6,66 (m, 2H), 6,48 (dd, J = 2,4, 12,6 Гц, 1H), 5,06-5,12 (m, 1H), 4,36 (d, J = 5,4 Гц, 2H), 4,27 (d, J = 17,7 Гц, 1H), 4,14 (d, J = 17,7 Гц, 1H), 3,67 (s, 3H), 2,83-2,97 (m, 1H), 2,57-2,62 (m, 1H), 2,20-2,35 (m, 1H), 2,00-2,08 (m, 1H). LCMS: 416,1 ([M+1]+).
Пример 11. Соединение A327
3-(6-Фтор-4-((4-(морфолинометил)бензил)окси)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A327:

Стадия A: к смеси (4-(морфолинометил)фенил)метанола (1,5 г, 7,2 ммоль) в DCM (20 мл) медленно добавляли SOCl2(2,6 г, 21,8 ммоль) при 0oC, затем полученную смесь перемешивали в течение ночи при 25oC. LCMS продемонстрировала завершение реакции. Реакционную смесь концентрировали с получением неочищенного продукта 4-(4-(хлорметил)бензил)морфолина гидрохлорида (1,9 г) в виде грязно-белого твердого вещества.
1H ЯМР (DMSO-d6, 400 МГц): δ 11,70 (br s, 1H), 7,65-7,67 (m, 2H), 7,50-7,52 (m, 2H), 4,79 (s, 2H), 4,32-4,33 (m, 2H), 3,81-3,93 (m, 4H), 3,16-3,19 (m, 2H), 3,02-3,11 (m, 2H).
Стадия B: смесь метил-5-фтор-2-метил-3-нитробензоата (2,0 г, 9,4 ммоль) и Pd/C (10%, 200 мг, 50% воды) в MeOH (20 мл) перемешивали при комнатной температуре в течение ночи в атмосфере H2 при давлении 50 фунтов/кв. дюйм. TLC и LCMS продемонстрировали завершение реакции. Смесь фильтровали и твердое вещество промывали с помощью MeOH (50 мл × 1), фильтрат концентрировали с получением метил-3-амино-5-фтор-2-метилбензоата (1,3 г, неочищенный) в виде бесцветного масла.
1H ЯМР (DMSO-d6, 300 МГц), δ 6,57-6,60 (m, 1H), 5,44 (s, 2H), 3,77 (s, 3H), 2,11 (s, 3H).
Стадия C: к смеси метил-3-амино-5-фтор-2-метилбензоата (1,3 г, неочищенный) и 10% H2SO4 (43 г, 42,6 ммоль) в MeOH(20 мл) добавляли NaNO2 (750 мг, 10,87 ммоль) при 0oC в атмосфере N2, затем полученную смесь перемешивали в течение 1 ч. при данной температуре. Затем в реактор добавляли 50% H2SO4 (42,6 г, 213 ммоль), смесь перемешивали в течение 1 ч. при 100oC. TLC продемонстрировала завершение реакции. Реакционную смесь концентрировали, остаток разбавляли с помощью H2O (20 мл) и EtOAc (100 мл). Водный слой экстрагировали с помощью EtOAc (100 мл × 3). Объединенную органическую фазу высушивали над Na2SO4, фильтровали и выпаривали досуха с помощью ротационного выпаривания. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюированном с помощью (PE : EtOAc = 50:1), с получением метил-5-фтор-3-гидрокси-2-метилбензоата (660 мг, выход 38% в течение двух стадий) в виде светло-желтого твердого вещества.
1H ЯМР (DMSO-d6, 400 МГц): δ 10,25 (s, 1H), 6,93-6,96 (m, 1H), 6,78-6,82 (m, 1H), 3,81 (s, 3H), 2,23 (s, 3H).
Стадия D: к смеси метил-5-фтор-3-гидрокси-2-метилбензоата (1,2 г, 6,5 ммоль) и имидазола (1,33 г, 19,5 ммоль) в DCM (20 мл) добавляли TBDMSCl (1,96 г, 13,0 ммоль) в атмосфере N2при 0oC, затем смесь перемешивали в течение 10 минут. Затем смесь перемешивали в течение 2 ч. при 25oC. TLC продемонстрировала завершение реакции. Смесь промывали с помощью H2O (50 мл), водный слой экстрагировали с помощью DCM (150 мл × 3), объединенную органическую фазу высушивали над Na2SO4, фильтровали и выпаривали досуха с помощью ротационного выпаривания. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюированном с помощью (PE : EtOAc = 20:1-10:1), с получением метил-3-((трет-бутилдиметилсилил)окси)-5-фтор-2-метилбензоата (1,4 г, выход 72%) в виде светло-желтого масла.
1H ЯМР (DMSO-d6,400 МГц): δ 7,12-7,15 (m, 1H), 6,84-6,87 (m, 1H), 3,82 (s, 3H), 2,26 (s, 3H), 0,98 (s, 9H), 0,23 (s, 6H).
Стадия E: к смеси метил-3-((трет-бутилдиметилсилил)окси)-5-фтор-2-метилбензоата (1,4 г, 4,7 ммоль) и NBS (1,0 г, 5,6 ммоль) в CCl4(20 мл) добавляли пероксид бензоила (0,12 г, 0,5 ммоль) в атмосфере N2, смесь перемешивали в течение ночи при 80oC. TLC продемонстрировала завершение реакции. Смесь фильтровали, затем твердое вещество промывали с помощью DCM (50 мл), органическую фазу промывали с помощью H2O (50 мл), водный слой экстрагировали с помощью DCM (100 мл × 3), объединенную органическую фазу высушивали над Na2SO4, фильтровали и выпаривали досуха с помощью ротационного выпаривания. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюированном с помощью (PE : EtOAc = 100:1), с получением метил-2-(бромметил)-3-((трет-бутилдиметилсилил)окси)-5-фторбензоата (1,4 г, чистота 90%) в виде белого твердого вещества.
1H ЯМР (DMSO-d6,400 МГц): δ 7,28-7,31 (m, 1H), 7,00-7,03 (m, 1H), 4,93 (s, 2H), 3,91 (s, 3H), 1,07 (s, 9H), 0,36 (s, 6H).
Стадия F: к смеси метил-2-(бромметил)-3-((трет-бутилдиметилсилил)окси)-5-фторбензоата (500 мг, 1,33 ммоль) и (S)-трет-бутил-4,5-диамино-5-оксопентаноата гидрохлорида (349 мг, 1,46 ммоль) и Et3N (405 мг, 4,0 ммоль) в CH3CN (10 мл) перемешивали в течение ночи в атмосфере N2при 80oC. TLC продемонстрировала завершение реакции. Смесь концентрировали, остаток добавляли к THF (10 мл) и затем в реактор по каплям добавляли TBAF (4 мл, 1 M в THF). Смесь перемешивали в течение 0,5 ч. при комнатной температуре. TLC продемонстрировала завершение реакции. Смесь концентрировали, остаток очищали с помощью колоночной хроматографии на силикагеле, элюированном с помощью (PE : EtOAc = 5:1-1:1 - EtOAc), с получением A327A (200 мг, выход 34%, в течение 3 стадий) в виде светло-желтого твердого вещества.
1H ЯМР (MeOD,400 МГц): δ 6,95-6,98 (m, 1H), 6,72-6,76 (m, 1H), 4,90-4,94 (m, 1H), 4,54 (d, J = 17,6 Гц, 1H), 4,42 (d, J = 17,6 Гц, 1H), 2,25-2,30 (m, 3H), 2,16-2,21 (m, 1H), 1,39 (s, 9H).
Стадия G: к смеси A327A (200 мг, 0,57 ммоль) и 4-(4-(хлорметил)бензил)морфолина гидрохлорида (445 мг, неочищенный) в сухом DMF(10 мл), в атмосфере N2при комнатной температуре добавляли K2CO3 (393 мг, 2,90 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. LCMS продемонстрировала, что реакция не была завершена. Еще одну часть 4-(4-(хлорметил)бензил)морфолина гидрохлорида (445 мг, неочищенный) добавляли в реактор, реакционную смесь перемешивали в течение 6 ч. Смесь концентрировали, затем остаток разбавляли с помощью H2O (10 мл) и EtOAc (30 мл). Водный слой экстрагировали с помощью EtOAc (10 мл × 3), объединенную органическую фазу высушивали над Na2SO4, фильтровали и выпаривали досуха с помощью ротационного выпаривания. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюированном с помощью (PE : EtOAc = 5:1-1:1 - EtOAc), с получением A327B (250 мг, выход 81%) в виде белого твердого вещества.
1H ЯМР (DMSO-d6,400 МГц): δ 7,57 (s, 1H), 7,44-7,46 (m, 2H), 7,34-7,36 (m, 2H), 7,19-7,27 (m, 2H), 7,06-7,08 (m, 1H), 5,23 (s, 2H), 4,68-4,72 (m, 1H), 4,50 (d, J = 17,6 Гц, 1H), 4,39 (d, J = 17,6 Гц, 1H), 3,56-3,58 (m, 4H), 3,47 (s, 2H), 2,30-2,38 (m, 4H), 2,13-2,17 (m, 3H), 2,00-2,05 (m, 1H), 1,32 (s, 9H).
Стадия H: к смеси A327B (250 мг, 0,46 ммоль) в сухом DCM (10 мл) добавляли TFA (4 мл) в атмосфере N2при 0oC. Смесь перемешивали в течение 4 ч. TLC продемонстрировала завершение реакции. Смесь концентрировали с получением A327C (230 мг, неочищенный) в виде бледно-желтого твердого вещества.
Стадия I: к смеси A327C (230 мг, неочищенный) в CH3CN (15 мл) добавляли CDI (115 мг, 0,71 ммоль) в атмосфере N2при комнатной температуре. После завершения добавления смесь перемешивали в течение ночи при 95oC. LCMS продемонстрировала завершение реакции. Смесь концентрировали, остаток очищали с помощью преп-HPLC с получением A327 (24 мг, выход 11%, две стадии) в виде желтого твердого вещества.
1H ЯМР (DMSO-d6,400 МГц): δ 10,98 (s, 1H), 7,43-7,45 (m, 2H), 7,28-7,35 (m, 3H), 7,11-7,13 (m, 1H), 5,23 (s, 2H), 5,08-5,13 (m, 1H), 4,39 (d, J = 17,2 Гц, 1H), 4,23 (d, J = 17,2 Гц, 1H), 3,55-3,58 (m, 4H), 3,47 (s, 2H), 2,87-2,91 (m, 1H), 2,54-2,59 (m, 1H), 2,42-2,45 (m, 1H), 2,35-2,41 (m, 4H), 1,97-1,99 (m, 1H). LCMS: 468,2 ([M+1]+).
Пример 12. Соединение A329
3-(4-((2-Фтор-4-метоксибензил)окси)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A329:
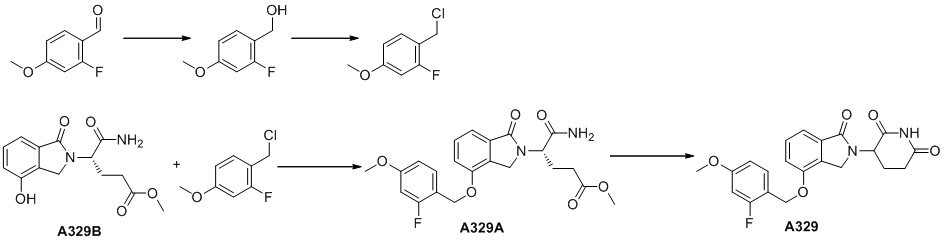
Стадия A: к раствору 2-фтор-4-метоксибензальдегида (1,0 г, 6,49 ммоль) в MeOH (10 мл) добавляли NaBH4 (370 мг, 9,74 ммоль) при комнатной температуре, смесь перемешивали в течение 1 ч. TLC продемонстрировала завершение реакции. Добавляли HCl (1 н.), чтобы погасить реакцию и pH регулировали до значения 4-5. Добавляли DCM (50 мл) и воду (40 мл), и водный слой экстрагировали с помощью DCM (50 мл x 2), объединенную органическую фазу промывали солевым раствором (50 мл), высушивали над Na2SO4, фильтровали, концентрировали с получением продукта (2-фтор-4-метоксифенил)метанола (910 мг, выход 90%) в виде светло-желтоватого масла без дополнительной очистки для следующей стадии.
1H ЯМР (DMSO-d6, 300 МГц): δ 7,31 (t, J = 8,4 Гц, 1H), 6,72-6,76 (m, 2H), 5,08 (t, J = 5,7 Гц, 1H), 4,43 (d, J = 5,7 Гц, 2H), 3,73 (m, 3H).
Стадия B: к раствору (2-фтор-4-метоксифенил)метанола (400 мг, 2,56 ммоль) в сухом DCM (10 мл) добавляли SOCl2 (458 мг, 3,85 ммоль) при комнатной температуре, смесь перемешивали в течение 3 ч. LCMS продемонстрировала завершение реакции. Реакционную смесь концентрировали с получением 1-(хлорметил)-2-фтор-4-метоксибензола (450 мг) в виде светло-желтого масла, которое использовали на следующей стадии без дополнительной очистки.
1H ЯМР (DMSO-d6, 300 МГц): δ 7,42 (t, J = 8,7 Гц, 1H), 6,85 (dd, J = 12,0, 2,7 Гц, 1H), 6,77 (dd, J = 11,4, 2,7 Гц, 1H), 4,72 (s, 2H), 3,76 (s, 3H).
Стадия C: к раствору A329B (200 мг, 0,68 ммоль) в DMF (15 мл) добавляли K2CO3 (283 мг, 2,05 ммоль) и [1-(хлорметил)-2-фтор-4-метоксибензол] (239 мг, 1,37 ммоль) при комнатной температуре, смесь перемешивали в течение ночи при комнатной температуре. LCMS продемонстрировала, что реакция не была завершена. Добавляли дополнительный [1-(хлорметил)-2-фтор-4-метоксибензол] (100 мг, 0,57 ммоль) и перемешивали в течение 3 ч. LCMS продемонстрировала завершение реакции. Реакционную смесь концентрировали и добавляли EtOAc (50 мл) и воду (30 мл), и водный слой экстрагировали с помощью EtOAc (50 мл x 2), объединенную органическую фазу промывали солевым раствором (50 мл), высушивали над Na2SO4, фильтровали и затем концентрировали с получением остатка. Остаток очищали с помощью преп-TLC (MeOH /DCM = 1/20) с получением A329A (190 мг, выход 65%) в виде белого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 7,57 (br s, 1H), 7,45-7,54 (m, 2H), 7,36 (d, J = 8,1 Гц, 1H), 7,30 (d, J = 7,5 Гц, 1H), 7,17 (br s, 1H), 6,87-6,92 (m, 1H), 6,81-6,85 (m, 1H), 5,19 (s, 2H), 4,69-4,74 (m, 1H). 4,47 (d, J = 17,7 Гц, 1H), 4,33 (d, J = 17,7 Гц, 1H), 3,79 (s, 3H), 3,50 (s, 3H), 2,13-2,27 (m, 3H), 2,00-2,10 (m, 1H).
Стадия D: к раствору A329A (190 мг, 0,44 ммоль) в DMF (10 мл) добавляли K2CO3 (183 мг, 1,33 ммоль), смесь перемешивали при 80oC в течение ночи в атмосфере N2. LCMS продемонстрировала завершение реакции. Реакционную смесь фильтровали и фильтрат концентрировали с получением остатка. Остаток очищали с помощью преп-HPLC и сушили сублимацией с получением A329 (120 мг, выход 68%) в виде белого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 10,79 (br s, 1H), 7,48-7,54 (m, 2H), 7,33-7,41 (m, 2H), 6,89 (dd, J = 12,3, 2,4 Гц, 1H), 6,81 (dd, J = 8,4, 2,4 Гц, 1H), 5,19 (s, 2H), 5,08-5,13 (m, 1H), 4,35 (d, J = 17,7 Гц, 1H), 4,18 (d, J = 17,7 Гц, 1H), 3,78 (s, 3H), 2,84-2,94 (m, 1H), 2,57-2,61 (m, 1H), 2,38-2,46 (m, 1H), 1,92-2,00 (m, 1H).
LCMS: 399,1 ([M+1]+).
Соединения в примерах 13-14 получали в соответствии с процедурой, описанной для примера 12, с соответствующим исходным материалом для замены 2-фтор-4-метоксибензальдегида на стадии A.
Пример 13. Соединение A331
3-(4-((2-Фтор-5-метоксибензил)окси)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A331:

1H ЯМР (DMSO-d6, 300 МГц): δ 10,65 (br s, 1H), 7,51 (t, J = 7,8 Гц, 1H), 7,34-7,40 (m, 2H), 7,13-7,23 (m, 2H), 6,93-6,99 (m, 1H), 5,25 (s, 2H), 5,10 (dd, J = 10,2, 5,1 Гц,1H), 4,39 (d, J = 17,7 Гц, 1H), 4,23 (d, J = 17,7 Гц, 1H), 3,74 (s, 3H), 2,84-2,96 (m, 1H), 2,54-2,60 (m, 1H), 2,39-2,47 (m, 1H), 1,93-2,00 (m, 1H). LCMS: 399,1 ([M+1]+).
Пример 14. Соединение A334
3-(4-((2-Фтор-3-метоксибензил)окси)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A334:

1H ЯМР (DMSO-d6, 400 МГц): 10,96 (s, 1H), 7,50 (t, J = 7,6 Гц, 1H), 7,34-7,38 (m, 2H), 7,13-7,20 (m, 3H), 5,28 (s, 2H), 5,10 (dd, J = 12,8, 4,4 Гц, 1H), 4,38 (d, J = 17,6 Гц, 1H), 4,22 (d, J = 17,6 Гц, 1H), 3,85 (s, 3H), 2,87-2,90 (m, 1H), 2,50-2,58 (m, 1H), 2,40-2,43 (m, 1H), 1,95-1,98 (m, 1H). LCMS: 399,1 ([M+1]+).
Пример 15. Соединение A336
3-[4-(2-Фторбензилокси)-1-оксо-1,3-дигидроизоиндол-2-ил]пиперидин-2,6-дион, A336:
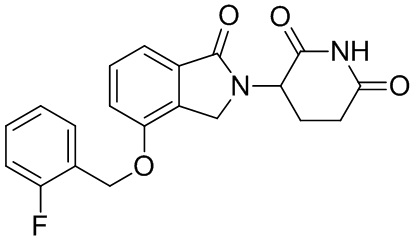
1H ЯМР (DMSO-d6, 400 МГц): 10,92 (s, 1H), 6,53-6,63 (m, 1H), 7,50 (t, J = 8,0 Гц, 1H), 7,40-7,47 (m, 2H), 7,36 (t, J = 8,0 Гц, 1H), 7,23-7,29 (m, 2H), 5,30 (s, 2H), 5,11 (dd, J = 12,8, 5,2 Гц, 1H), 4,38 (d, J = 17,6 Гц, 1H), 4,23 (d, J = 17,6 Гц, 1H), 2,86-2,95 (m, 1H), 2,54-2,59 (m, 1H), 2,38-2,47 (m, 1H), 1,97-2,00 (m, 1H). LCMS: 369,1 ([M+1]+).
Пример 16. Соединение A340
(S)-3-(4-((2-Фтор-5-метоксибензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A340:

Стадия A: к раствору метил-2-(бромметил)-3-нитробензоата (2,00 г, 7,30 ммоль) в CH3CN (40 мл) добавляли (S)-трет-бутил-4,5-диамино-5-оксопентаноата гидрохлорид (1,91 г, 8,00 ммоль) и Et3N (1,63 г, 16,1 ммоль) в атмосфере N2, смесь перемешивали при 75oC в течение ночи. TLC продемонстрировала завершение реакции. Реакционную смесь концентрировали и добавляли EtOAc (50 мл) и воду (50 мл), водный слой экстрагировали с помощью EtOAc (50 мл x 2), объединенные органические фазы промывали солевым раствором (50 мл), высушивали над Na2SO4, фильтровали, концентрировали с получением неочищенного продукта. Неочищенный продукт растирали в порошок с помощью PE / EtOAc (4/1, об./об.), затем фильтровали с получением A340A (2,2 г, выход 83%) в виде белого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 8,45 (dd, J = 0,9, 8,1 Гц, 1H), 8,16 (dd, J = 0,9, 8,1 Гц, 1H), 7,82 (t, J = 8,1 Гц, 1H), 7,65 (br s, 1H), 7,27 (br s, 1H), 5,05 (d, J = 19,5 Гц, 1H), 4,90 (d, J = 19,5 Гц, 1H), 4,75-4,80 (m, 1H), 2,14-2,27 (m, 3H), 2,00-2,10 (m, 1H), 1,33 (s, 9H).
Стадия B: к раствору A340A (1,20 г, 3,30 ммоль) в MeOH добавляли Pd/C (10%, 200 мг, 50% воды), дегазировали с помощью H2 3 раза, смесь перемешивали при 25oC в течение ночи в атмосфере H2(50 фунтов/кв. дюйм). LCMS продемонстрировала завершение реакции. Pd/C удаляли с помощью фильтрации и фильтрат концентрировали с получением A340B (1,19 г, неочищенный) в виде светло-желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
1H ЯМР (DMSO-d6, 300 МГц): δ 7,50 (br s, 1H), 7,11-7,16 (m, 2H), 6,85 (d, J = 7,2 Гц, 1H), 6,74 (d, J = 7,2 Гц, 1H), 5,41 (br s, 2H), 4,68-4,73 (m, 1H), 4,38 (d, J = 17,7 Гц, 1H), 4,16 (d, J = 17,7 Гц, 1H), 2,09-2,19 (m, 3H), 1,92-2,01(m, 1H), 1,32 (s, 9H).
Стадия C: к раствору A340B (1,00 г, неочищенный) и 2-фтор-5-метоксибензальдегида (601 мг, 3,90 ммоль) в MeOH (10 мл) добавляли AcOH (0,5 мл), смесь перемешивали при 25oC в течение 3 часов. Добавляли Pd/C (10%, 200 мг, 50% воды ), дегазировали с помощью H2 3 раза и перемешивали при 25oC в течение ночи в атмосфере H2 (баллон). LCMS продемонстрировала завершение реакции. Pd/C удаляли с помощью фильтрации и фильтрат концентрировали с получением остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле (PE / EtOAc = 1/4) с получением требуемого продукта A340C (1,15 г, выход 88%, в течение двух стадий) в виде светло-желтоватого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 7,56 (br s, 1H), 7,09-7,24 (m, 3H), 6,91-6,96 (m, 2H), 6,80-6,85 (m, 1H), 6,63 (d, J = 8,4 Гц, 1H), 6,34-6,38 (m, 1H), 4,74 (dd, J = 10,2, 4,5 Гц, 1H), 4,50 (d, J = 18,0 Гц, 1H), 4,37 (d, J = 6,0 Гц, 2H), 4,28 (d, J = 18,0 Гц, 1H), 3,67 (s, 3H), 2,12-2,21 (m, 3H), 1,91-2,02 (m, 1H), 1,33 (s, 9H).
Стадия D: к раствору A340C (1,15 г, 2,44 ммоль) в DCM (20 мл), охлажденному до 0oC, по каплям добавляли TFA (4 мл), смесь перемешивали в течение ночи при 25℃. Реакционную смесь концентрировали при сниженном давлении. Остаток очищали с помощью флэш-хроматографии на C18 (40% ацетонитрила в воде), затем сушили сублимацией с получением A340D (800 мг, выход 79%) в виде светло-желтого твердого вещества.
1H ЯМР (DMSO-d6, 400 МГц): δ 12,14 (br s, 1H), 7,57 (br s, 1H), 7,10-7,23 (m, 3H), 6,91-6,96 (m, 2H), 6,81-6,85 (m, 1H), 6,63 (d, J = 8,0 Гц, 1H), 6,35 (t, J = 6,0 Гц, 1H), 4,72-4,76 (m, 1H), 4,51 (d, J = 17,6 Гц, 1H), 4,37 (d, J = 5,6 Гц, 2H), 4,31 (d, J = 17,6 Гц, 1H), 3,67 (s, 3H), 2,18-2,23 (m, 3H), 1,96-2,02 (m, 1H).
Стадия E: раствор A340D (700 мг, 1,69 ммоль) в сухом DCM (70 мл) охлаждали до -40oC в атмосфере N2, к смеси медленно добавляли SOCl2 (1,00 г, 8,40 ммоль) при -40oC, затем добавляли раствор DMF (10 мг) в DCM (1 мл) и перемешивали в течение 2 ч., а затем по каплям добавляли пиридин (666 мг, 8,42 ммоль) и перемешивали при данной температуре в течение 40 мин., добавляли Et3N (852 мг, 8,42 ммоль) и затем смесь перемешивали в течение 2 ч. LCMS продемонстрировала завершение реакции. H2O (10 мл) добавляли, чтобы погасить реакцию, водный слой экстрагировали с помощью DCM (20 мл x 2), объединенную органическую фазу промывали солевым раствором (50 мл x 1), высушивали над Na2SO4, фильтровали, концентрировали с получением остатка. Остаток очищали с помощью преп-HPLC с получением A340 (460 мг, выход 68%, ee: 98%) в виде бледно-зеленого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 11,00 (br s, 1H), 7,23 (t, J = 7,8 Гц, 1H), 7,11 (t, J = 9,6 Гц, 1H), 6,90-6,95 (m, 2H), 6,78-6,84 (m, 1H), 6,65 (d, J = 8,1 Гц, 1H), 6,26 (t, J = 6,0 Гц, 1H), 5,10 (dd, J = 13,2, 5,1 Гц, 1H), 4,37 (d, J = 6,0 Гц, 2H), 4,30 (d, J = 17,1 Гц, 1H), 4,17 (d, J = 17,1 Гц, 1H), 3,65 (s, 3H), 2,85-2,96 (m,1H), 2,56-2,63 (m, 1H), 2,24-2,37 (m, 1H), 2,00-2,07 (m, 1H). LCMS: 398,1 ([M+1]+).
Пример 17. Соединение A341
(R)-3-(4-((2-Фтор-5-метоксибензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A341:
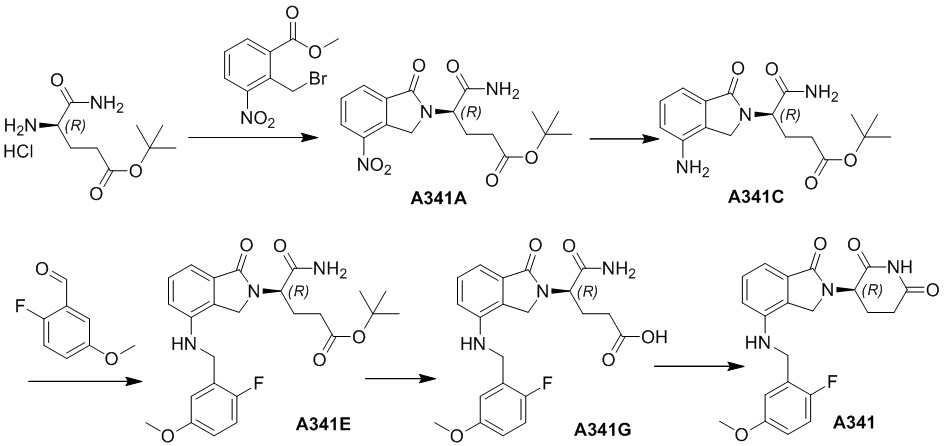
Стадия A: к раствору сложного метилового эфира 2-бромметил-3-нитробензойной кислоты (1,00 г, 3,65 ммоль) в CH3CN (50 мл) добавляли (R)-трет-бутил-4,5-диамино-5-оксопентаноата гидрохлорида (955 мг, 4,00 ммоль) и Et3N (815 мг, 8,05 ммоль), смесь перемешивали при 75oC в течение ночи в атмосфере N2. TLC продемонстрировала завершение реакции. Реакционную смесь концентрировали и добавляли EtOAc (50 мл) и воду (50 мл), водный слой экстрагировали с помощью EtOAc (50 мл x 2), объединенную органическую фазу промывали солевым раствором (50 мл), высушивали над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (PE/EtOAc = 1/4), с получением A341A (800 мг, выход 60%) в виде белого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 8,45 (d, J = 6,0 Гц, 1H), 8,16 (d, J = 5,4 Гц, 1H), 7,82 (t, J = 6,0 Гц, 1H), 7,64 (br s, 1H), 7,27 (br s, 1H), 5,05 (d, J = 14,4 Гц, 1H), 4,91 (d, J = 14,4 Гц, 1H), 4,76-4,80 (m, 1H), 2,15-2,25 (m, 3H), 2,02-2,11 (m, 1H), 1,33 (s, 9H).
Стадия B: к раствору A341A (800 мг, 2,20 ммоль) в MeOH добавляли Pd/C (10%, 80 мг, 50% воды), смесь перемешивали при 25oC в течение ночи в атмосфере H2(50 фунтов/кв. дюйм). LCMS продемонстрировала завершение реакции. Pd/C удаляли с помощью фильтрации и фильтрат концентрировали с получением неочищенного продукта A341C (680 мг, выход 93%) в виде светло-желтоватого твердого вещества. Неочищенный продукт использовали на следующей стадии без дополнительной очистки.
1H ЯМР (DMSO-d6, 300 МГц): δ 7,54 (br s, 1H), 7,13-7,18 (m, 2H), 6,88 (d, J = 7,2 Гц, 1H), 6,76 (d, J = 7,8 Гц, 1H), 5,44 (br s, 2H), 4,70-4,75 (m, 1H), 4,41 (d, J = 17,7 Гц, 1H), 4,18 (d, J = 17,7 Гц, 1H), 2,09-2,21 (m, 3H), 1,92-2,06 (m, 1H), 1,34 (s, 9H).
Стадия C: к раствору A341C (680 мг, 2,04 ммоль) и 2-фтор-5-метоксибензальдегида (472 мг, 3,06 ммоль) в MeOH добавляли AcOH (0,5 мл), смесь перемешивали при 25oC в течение 3 часов. Добавляли Pd/C (10%, 50 мг, 50% воды), дегазировали с помощью H2 3 раза и перемешивали при 25oC в течение ночи в атмосфере H2 (баллон). LCMS продемонстрировала завершение реакции. Pd/C удаляли с помощью фильтрации и фильтрат концентрировали с получением остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле (PE / EtOAc = 1/4) с получением требуемого продукта A341E (650 мг, выход 68%) в виде светло-желтого твердого вещества.
1H ЯМР (DMSO-d6, 400 МГц): δ 7,56 (br s, 1H), 7,18-7,23 (m, 2H), 7,12 (t, J = 9,2 Гц, 1H), 6,91-6,95 (m, 2H), 6,81-6,85 (m, 1H), 6,63 (d, J = 8,0 Гц, 1H), 6,36 (t, J = 5,6 Гц, 1H), 4,72-4,76 (m, 1H), 4,50 (d, J = 17,6 Гц, 1H), 4,37 (d, J = 6,0 Гц, 2H), 4,29 (d, J = 17,6 Гц, 1H), 3,67 (s, 3H), 2,14-2,22 (m, 3H), 1,94-2,04 (m, 1H), 1,33 (s, 9H).
Стадия D: к раствору A341E (650 мг, 1,38 ммоль) в DCM (20 мл), охлажденному до 0oC, по каплям добавляли TFA (4 мл), смесь медленно нагревали до 25℃ и перемешивали в течение ночи. Реакционную смесь концентрировали при сниженном давлении (в вакууме) с целью удаления раствора. Остаток очищали с помощью флэш-хроматографии на C18 (40% ацетонитрила в воде), затем сушили сублимацией с получением A341G (450 мг, выход 79%) в виде светло-желтоватого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 7,56 (br s, 1H), 7,07-7,22 (m, 3H), 6,89-6,94 (m, 2H), 6,78-6,83 (m, 1H), 6,61 (d, J = 7,8 Гц, 1H), 6,34 (t, J = 6,3 Гц, 1H), 4,69-4,73 (m, 1H), 4,50 (d, J = 17,7 Гц, 1H), 4,35 (d, J = 5,7 Гц, 2H), 4,29 (d, J = 17,7 Гц, 1H), 3,65 (s, 3H), 2,12-2,19 (m, 3H), 1,93-1,98 (m, 1H).
Стадия E: к раствору A341G (450 мг, 1,08 ммоль), растворенному в сухом DCM (50 мл), охлажденному до -40oC в атмосфере N2, SOCl2 (644 мг, 5,41 ммоль) медленно добавляли к смеси при -40oC в атмосфере N2, затем раствор DMF (10 мг) в DCM (1 мл) добавляли и перемешивали в течение 2 ч., по каплям добавляли пиридин (428 мг, 5,41 ммоль) и перемешивали при данной температуре в течение 40 мин., добавляли Et3N (547 мг, 5,41 ммоль) и затем смесь перемешивали в течение 2 ч. LCMS продемонстрировала завершение реакции. H2O (10 мл) добавляли, чтобы погасить реакцию, водный слой экстрагировали с помощью DCM (30 мл x 2), объединенную органическую фазу промывали солевым раствором (50 мл), высушивали над Na2SO4, фильтровали, концентрировали с получением остатка. Остаток очищали с помощью преп-HPLC с получением A341 (260 мг, выход 61%, ee: 96%) в виде бледно-зеленого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 10,98 (br s, 1H), 7,23 (t, J = 7,8 Гц, 1H), 7,11 (t, J = 9,3 Гц, 1H), 6,90-6,95 (m, 2H), 6,78-6,84 (m, 1H), 6,65 (d, J = 8,1 Гц, 1H), 6,26 (t, J = 6,0 Гц, 1H), 5,10 (dd, J = 13,2, 5,1 Гц, 1H), 4,36 (d, J = 5,7 Гц, 2H), 4,30 (d, J = 17,1 Гц, 1H), 4,17 (d, J = 17,1 Гц, 1H), 3,65 (s, 3H), 2,85-2,97 (m, 1H), 2,56-2,63 (m, 1H), 2,22-2,35 (m, 1H), 2,00-2,07 (m, 1H). LCMS: 398,1 ([M+1]+).
Пример 18. Соединение A342
(R)-3-Дейтерий-3-(4-((2-фтор-5-метоксибензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A342:
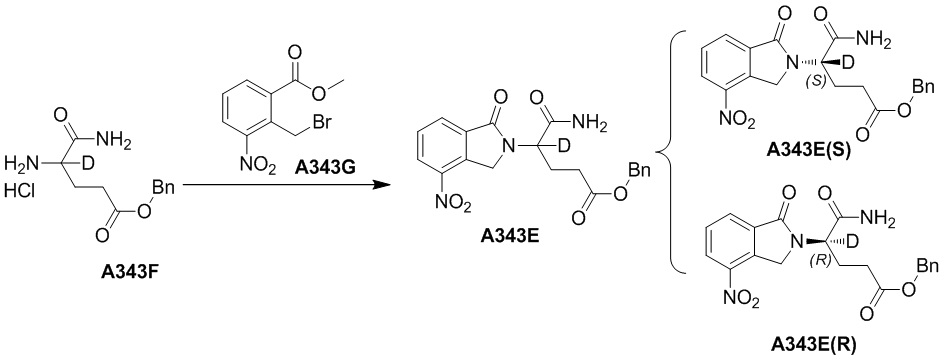

Стадия A: к раствору A343F (31,7 г, 115,7 ммоль) в CH3CN (560 мл) добавляли A343G (31,5 г, 115,1 ммоль) и Et3N (23,3 г, 231,0 ммоль), смесь перемешивали при 75oC в течение ночи в атмосфере N2. Реакционную смесь концентрировали и добавляли EtOAc (50 мл) и 4 н. водный раствор HCl (150 мл), смесь фильтровали, а отфильтрованный осадок промывали водой (30 мл) и высушивали, при этом фильтрат экстрагировали с помощью EtOAc (250 мл x 2), объединенную органическую фазу промывали солевым раствором (30 мл), высушивали над Na2SO4, фильтровали, концентрировали с получением неочищенного продукта. Объединенное твердое вещество растирали в порошок с помощью CH3CN (40 мл x 2), фильтровали с получением A343E (37 г, выход 81%) в виде белого твердого вещества. Соединение A343E подвергали хиральному разделению с получением пика 1 A343E(S) (14,4 г, выход 77,8%, Rt=7,30 мин., 100% ee) и пика 2 A343E(R) (14,8 г, выход 80%, Rt=11,87 мин., 100% ee) в виде белого твердого вещества.
Условия хирального разделения: колонка: CHIRALPAK IE; размер частиц: 10 мкм, размеры: 50 x 250 мм; длина волны: 254 нм; подвижная фаза: MeOH/DCM=80/20 (об./об.); введение: 48 мл; расход: 60 мл/мин.; температура: 35oC. Растворитель: подвижная фаза, 17,1 мг/мл.
A343E(S):1H ЯМР (DMSO-d6, 300 МГц): δ 8,43 (d, J = 8,1 Гц, 1H), 8,13 (d, J = 7,5 Гц, 1H), 7,79 (t, J = 8,1 Гц, 1H), 7,66 (br s, 1H), 7,26-7,35 (m, 6H), 4,86-5,07 (m, 4H), 2,42-2,21-2,43 (m, 3H), 2,06-2,16 (m, 1H).
A343E(R):1H ЯМР (DMSO-d6, 300 МГц): δ 8,43 (d, J = 8,4 Гц, 1H), 8,13 (d, J = 7,2 Гц, 1H), 7,77-7,82 (m, 1H), 7,65 (br s, 1H), 7,35-7,23-7,35 (m, 6H), 4,86-5,07 (m, 4H), 2,20-2,42 (m, 3H), 2,06-2,15 (m, 1H).
Стадия B: к раствору A343E(R) (2,5 г, 6,3 ммоль) в MeOH (150 мл) и THF (150 мл) добавляли Pd/C (10%, 500 мг, 50% воды), смесь перемешивали в течение ночи в атмосфере H2(50 фунтов/кв. дюйм) при 25℃. LCMS продемонстрировала завершение реакции. Pd/C удаляли с помощью фильтрации и фильтрат концентрировали с получением неочищенного продукта, который совместно выпаривали с CH3CN/DCE (50 мл/150 мл), а затем твердое вещество растворяли в THF (300 мл) и концентрировали с получением A342C (1,68 г, 97% выход) в виде белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
1H ЯМР (DMSO-d6, 300 МГц): δ 12,11 (br s, 1H), 7,54 (s, 1H), 7,11-7,16 (m, 2H), 6,85 (d, J = 7,8 Гц, 1H), 6,74 (d, J = 7,8 Гц, 1H), 5,43 (br s, 2H), 4,68-4,73 (m, 0,02H), 4,41 (d, J = 17,4 Гц, 1H), 4,17 (d, J = 17,4 Гц, 1H), 2,10-2,18 (m, 3H), 1,94-1,97 (m, 1H).
Стадия C: к раствору A342C и 2-фтор-5-метоксибензальдегида (831 мг, 5,39 ммоль) в MeOH добавляли AcOH (0,5 мл), смесь перемешивали при 25oC в течение 20 часов. Добавляли Pd/C (10%, 100 мг, 50% воды), дегазировали с помощью H2 3 раза и перемешивали при 25oC в течение ночи в атмосфере H2 (баллон). LCMS продемонстрировала завершение реакции. Pd/C удаляли с помощью фильтрации и фильтрат концентрировали с получением остатка. Остаток очищали с помощью флэш-хроматографии на C18 (CH3CN: H2O = 5% - 35%, 30 мин.; 35% - 45%, 30 мин.; 45% - 55% 20 мин.), затем сушили сублимацией с получением A342A (800 мг, выход 53%) в виде светло-желтого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 12,10 (br s, 1H), 7,56 (br s, 1H), 7,07-7,22 (m, 3H), 6,89-6,94 (m, 2H), 6,78-6,83 (m, 1H), 6,61 (d, J = 8,1 Гц, 1H), 6,34 (t, J = 6,0 Гц, 1H), 4,70-4,74 (m, 0,03H), 4,50 (d, J = 17,7 Гц, 1H), 4,35 (d, J = 5,7 Гц, 2H), 4,28 (d, J = 17,7 Гц, 1H), 3,65 (s, 3H), 2,10-2,21 (m, 3H), 1,92-2,02 (m, 1H).
Стадия D: к раствору A342A (450 мг, 1,10 ммоль) в сухом DCM (50 мл), охлажденному до -40oC в атмосфере N2, SOCl2 (572 мг, 4,81 ммоль) медленно добавляли к смеси при -40oC в атмосфере N2, затем добавляли раствор DMF (10 мг) в DCM (1 мл), перемешивали в течение 2 ч., затем по каплям добавляли пиридин (380 мг, 4,80 ммоль) при данной температуре, перемешивали в течение 40 мин., затем добавляли Et3N (486 мг, 4,80 ммоль) и затем смесь перемешивали в течение 2 ч. LCMS продемонстрировала завершение реакции. Чтобы погасить реакцию, добавляли H2O (10 мл), водный слой экстрагировали с помощью DCM (30 мл x 2), объединенную органическую фазу промывали солевым раствором (50 мл), высушивали над Na2SO4, фильтровали, концентрировали с получением остатка. Остаток очищали с помощью флэш-хроматографии на C18 (CH3CN: H2O = 5% - 35%, 30 мин.; 35% - 45%, 30 мин.; 45% - 55% 20 мин.) с получением A342 (220 мг, выход 58%, ee: 99%) в виде светло-желтого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 11,00 (br s, 1H), 7,23 (t, J = 7,8 Гц, 1H), 7,11 (t, J = 9,6 Гц, 1H), 6,90-6,95 (m, 2H), 6,79-6,84 (m, 1H), 6,65 (d, J = 7,8 Гц, 1H), 6,26 (t, J = 5,4 Гц, 1H), 5,07-5,14 (m, 0,01H), 4,36 (d, J = 5,7 Гц, 2H), 4,30 (d, J = 17,1 Гц, 1H), 4,17 (d, J = 17,1 Гц, 1H), 3,65 (s, 3H), 2,85-2,97 (m, 1H), 2,57-2,63 (m, 1H), 2,24-2,34 (m, 1H), 2,00-2,06 (m, 1H).
LCMS: 399,1 ([M+1]+).
Соединение из примера 19 получали в соответствии со способом синтеза, описанным в примере 18, с соответствующим исходным материалом для замены A343E(R) на стадии B.
Пример 19. A343
(S)-3-Дейтерий-3-(4-((2-фтор-5-метоксибензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A343:
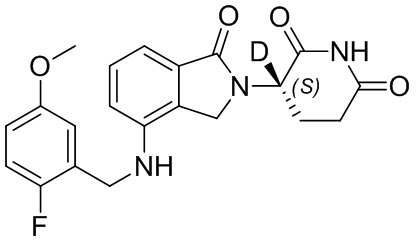
1H ЯМР (DMSO-d6, 300 МГц): δ 11,00 (br s, 1H), 7,23 (t, J = 8,1 Гц, 1H), 7,11 (t, J = 9,6 Гц, 1H), 6,90-6,95 (m, 2H), 6,78-6,84 (m, 1H), 6,65 (d, J = 8,1 Гц, 1H), 6,26 (t, J = 6,3 Гц, 1H), 5,07-5,13 (m, 0,02H), 4,36 (d, J = 5,7 Гц, 2H), 4,29 (d, J = 17,1 Гц, 1H), 4,17 (d, J = 17,1 Гц, 1H), 3,65 (s, 3H), 2,85-2,97 (m, 1H), 2,56-2,64 (m, 1H), 2,24-2,34 (m, 1H), 2,00-2,05 (m, 1H). LCMS: 399,1 ([M+1]+).
Пример 20. Соединение A346
(S)-3-(4-((4-(Морфолинометил)бензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A346:
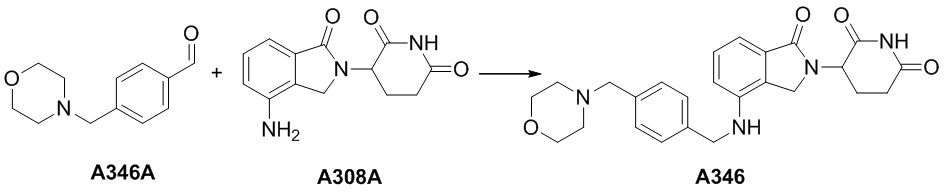
A346A (119 мг, 0,58 ммоль) и соединение A308A (100 мг, 0,39 ммоль) растворяли в AcOH (2,5 мл) и DCM (2,5 мл), и раствор перемешивали в течение 1 часа при 30℃. Добавляли NaBH(OAc)3(246 мг, 1,16 ммоль) и реакционную смесь перемешивали в течение 18 часов в атмосфере N2. TLC продемонстрировала завершение реакции. Растворитель удаляли и добавляли насыщ. водн. NaHCO3 (5 мл) для регулирования pH до значения 8. Смесь экстрагировали с помощью DCM (25 мл x 5) и объединенный органический слой высушивали над Na2SO4, фильтровали, концентрировали и растирали в порошок с помощью PE / EtOAc (1/1) (25 мл x 2) с получением 250 мг неочищенного продукта, который очищали с помощью преп-HPLC с получением A346 (140 мг, выход 80%) в виде белого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 11,00 (s, 1H), 7,16-7,33 (m, 5H), 6,90 (d, J = 7,2 Гц, 1H), 6,62 (d, J = 7,8 Гц, 1H), 6,33 (t, J = 5,7 Гц, 1H), 5,07-5,13 (m, 1H), 4,35 (d, J = 5,4 Гц, 2H), 4,29 (d, J = 17,1 Гц, 1H), 4,16 (d, J = 17,1 Гц, 1H), 3,53 (t, J = 4,5 Гц, 4H), 3,39 (s, 2H), 2,85-2,93 (m, 1H), 2,58-2,63 (m, 1H), 2,28-2,31 (m, 5H), 2,01-2,06 (m, 1H).
LCMS: 449,2 ([M+1]+).
Соединения в примерах 12-45 получали в соответствии с процедурой, описанной для примера 20, с соответствующим исходным материалом для замены A346A.
Пример 21. Соединение A359
3-(4-((2-Фтор-3-гидроксибензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A359:

1H ЯМР (DMSO-d6, 300 МГц): δ 10,96 (br, 1H), 9,79 (br, 1H), 7,21 (t, J = 7,8 Гц, 1H), 6,73-6,93 (m, 4H), 6,63 (d, J = 7,8 Гц, 1H), 6,25 (t, J = 6,0 Гц, 1H), 5,10 (dd, J = 13,2, 5,1 Гц, 1H), 4,37 (d, J = 5,7 Гц, 2H), 4,28 (d, J = 17,4 Гц, 1H), 4,15 (d, J = 17,4 Гц, 1H), 2,85-2,97 (m, 1H), 2,57-2,63 (m, 1H), 2,22-2,36 (m, 1H), 2,01-2,05 (m, 1H). LCMS: 384,1 ([M+1]+).
Пример 22. Соединение A360
3-(((2-(2,6-Диоксопиперидин-3-ил)-1-оксоизоиндолин-4-ил)амино)метил)-2-фторбензонитрил, A360:

1H ЯМР (300 МГц, DMSO-d6): δ 11,03 (s, 1H), 7,81-7,86 (m, 1H), 7,71-7,76 (m, 1H), 7,36 (t, J = 7,8 Гц, 1H), 7,25 (t, J = 7,8 Гц, 1H), 6,97 (d, J = 7,5 Гц, 1H), 6,66 (d, J = 7,8 Гц, 1H), 6,41 (t, J = 5,7 Гц, 1H), 5,13, (dd, J = 12,9, 5,1 Гц, 1H), 4,50 (d, J = 5,7 Гц, 1H), 4,32 (d, J = 17,1 Гц, 1H), 4,19 (d, J = 17,1 Гц, 1H), 2,87-2,95 (m 1H), 2,50-2,65 (m 1H), 2,29-2,34 (m 1H), 2,02-2,07 (m 1H).
Пример 23. Соединение A361
3-(((2-(2,6-Диоксопиперидин-3-ил)-1-оксоизоиндолин-4-ил)амино)метил)-2-фторбензамид, A361:

1H ЯМР (DMSO-d6, 300 МГц): δ 11,02 (s, 1H), 7,75 (s, 1H), 7,63 (s, 1H), 7,45-7,53 (m, 2H), 7,16-7,27 (m, 2H), 6,96 (d, J = 7,2 Гц, 1H), 6,64 (d, J = 8,1 Гц, 1H), 6,35-6,38 (m, 1H), 5,13 (dd, J = 13,2, 4,8 Гц, 1H), 4,46 (d, J = 5,4 Гц, 2H), 4,33 (d, J = 17,4 Гц, 1H), 4,20 (d, J = 17,4 Гц, 1H), 2,87-2,98 (m, 1H), 2,60-2,65 (m, 1H), 2,25-2,39 (m, 1H), 2,03-2,07 (m, 1H). LCMS: 411,1 ([M+1]+).
Пример 24. Соединение A362
3-(((2-(2,6-Диоксопиперидин-3-ил)-1-оксоизоиндолин-4-ил)амино)метил)-2-фтор-N-метилбензамид, A362:
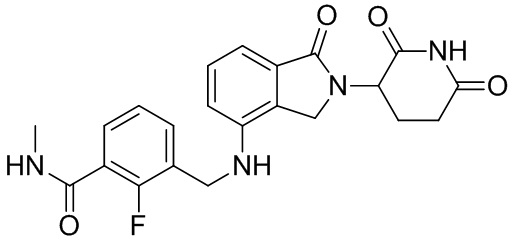
1H ЯМР (DMSO-d6, 300 МГц): δ 11,01 (s, 1H), 8,28 (d, J = 3,3 Гц, 1H), 7,46 (t, J = 7,2 Гц, 2H), 7,15-7,26 (m, 2H), 6,95 (d, J = 7,5 Гц, 1H), 6,63 (d, J = 8,4 Гц, 1H), 6,37 (t, J = 5,7 Гц, 1H), 5,12 (dd, J = 13,5, 4,8 Гц, 1H), 4,45 (d, J = 5,1 Гц, 2H), 4,32 (d, J = 17,4 Гц, 1H), 4,20 (d, J = 17,4 Гц, 1H), 2,87-2,99 (m, 1H), 2,78 (d, J = 4,8 Гц, 3H), 2,60-2,65 (m, 1H), 2,25-2,39 (m, 1H), 1,99-2,11 (m, 1H). LCMS: 425,1 ([M+1]+).
Пример 25. Соединение A363
3-(4-((5-(2-(Диметиламино)этокси)-2-фторбензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A363:
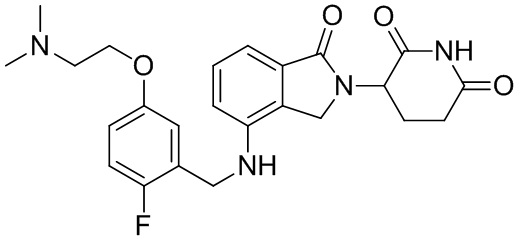
1H ЯМР (DMSO-d6, 300 МГц): δ 11,02 (s, 1H), 7,24 (t, J = 8,1 Гц, 1H), 7,11 (t, J = 9,3 Гц, 1H), 6,90-6,96 (m, 2H), 6,80-6,86 (m, 1H), 6,65 (d, J = 8,4 Гц, 1H), 6,29 (t, J = 6,0 Гц,1H), 5,12 (dd, J = 13,2, 4,8 Гц, 1H), 4,29-4,39 (m, 3H), 4,19 (d, J = 17,1 Гц, 1H), 3,94 (t, J = 5,7 Гц, 2H), 2,87-2,99 (m, 1H), 2,51-2,65 (m, 3H), 2,24-2,38 (m, 1H), 2,15 (s, 6H), 2,00-2,10 (m, 1H). LCMS: 455,2 ([M+1]+).
Пример 26. Соединение A364
3-(4-((2-Фтор-5-гидроксибензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A364:

1H ЯМР (300 МГц, DMSO-d6): δ 11,01 (s, 1H), 9,24 (s, 1H), 7,22 (t, J = 7,8 Гц, 1H), 6,92-6,99 (m, 2H), 6,70-6,73 (m, 1H), 6,55-6,60 (m, 2H), 6,31 (t, J = 5,7 Гц, 1H), 5,11, (dd, J = 13,2, 5,1 Гц, 1H), 4,27-4,34 (m, 3H), 4,17 (d, J = 17,1 Гц, 1H), 2,86-2,96 (m 1H), 2,57-2,64 (m 1H), 2,24-2,33 (m 1H), 2,02-2,06 (m 1H).
Пример 27. Соединение A367
3-(4-((2-Фтор-3-метилбензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A367:

1H ЯМР (DMSO-d6, 300 МГц): δ 11,03 (s, 1H), 7,14-7,26 (m, 3H), 6,93-7,04 (m, 2H), 6,64 (d, J = 7,5 Гц, 1H), 6,30 (br s, 1H), 5,09-5,16 (m, 1H), 4,42 (s, 2H), 4,31 (d, J = 17,4 Гц, 1H), 4,18 (d, J = 17,4 Гц, 1H), 2,89-2,98 (m, 1H), 2,59-2,65 (m, 1H), 2,25-2,46 (m, 4H), 2,02-2,07 (m, 1H). LCMS: 382,2 ([M+1]+).
Пример 28. Соединение A368
3-(4-((2-Фтор-5-(2-морфолиноэтокси)бензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A368:

1H ЯМР (DMSO-d6, 300 МГц): δ 10,98 (s, 1H), 7,23 (t, J = 7,5 Гц, 1H), 7,09 (t, J = 9,6 Гц, 1H), 6,79-6,95 (m, 3H), 6,64 (d, J = 7,5 Гц, 1H), 6,24 (br, 1H), 5,10 (dd, J = 13,2, 5,1 Гц, 1H), 4,36 (d, J = 5,7 Гц, 2H), 4,30 (d, J = 17,1 Гц, 1H), 4,18 (d, J = 17,1 Гц, 1H), 3,97 (t, J = 5,7 Гц, 2H), 3,51 (t, J = 4,5 Гц, 4H), 2,85-2,97 (m, 1H), 2,56-2,63 (m, 3H), 2,27-2,39 (m, 5H), 2,00-2,05 (m, 1H). LCMS: 497,2 ([M+1]+).
Пример 29. Соединение A369
3-(4-((2-Фтор-5-(3-морфолинопропокси)бензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A369:

1H ЯМР (DMSO-d6, 300 МГц): δ 11,00 (s, 1H), 7,22 (t, J = 7,8 Гц, 1H), 7,09 (t, J = 9,3 Гц, 1H), 6,88-6,95 (m, 2H), 6,77-6,83 (m, 1H), 6,63 (d, J = 8,1 Гц, 1H), 6,27 (t, J = 5,7 Гц, 1H), 5,11 (dd, J = 13,2, 5,4 Гц, 1H), 4,36 (d, J = 5,7 Гц, 2H), 4,30 (d, J = 17,4 Гц, 1H), 4,17 (d, J = 17,4 Гц, 1H), 3,88 (t, J = 6,3 Гц, 2H), 3,52 (t, J = 3,9 Гц, 4H), 2,85-2,96 (m, 1H), 2,57-2,63 (m, 1H), 2,22-2,41 (m, 7H), 1,99-2,05 (m, 1H), 1,73-1,84 (m, 2H). LCMS: 511,2 ([M+1]+).
Пример 30. Соединение A370
3-(4-((2-Фтор-5-(2-метоксиэтокси)бензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A370:

1H ЯМР (300 МГц, DMSO-d6): δ 10,98 (s, 1H), 7,23 (t, J = 7,8 Гц, 1H), 7,06-7,12 (m, 1H), 6,90-6,95 (m, 2H), 6,79-6,84 (m, 1H), 6,64 (d, J = 8,1 Гц, 1H), 6,24 (t, J = 5,4 Гц, 1H), 5,10 (dd, J = 13,2, 5,1 Гц, 1H), 4,28-4,38 (m, 3H), 4,19 (d, J = 17,1 Гц, 1H), 3,96-3,99 (m, 2H), 3,55-3,58 (m, 2H), 3,23 (s, 3H), 2,85-2,95 (m 1H), 2,57-2,64 (m 1H), 2,24-2,36 (m 1H), 1,98-2,09 (m 1H).
Пример 31. Соединение A371
3-(((2-(2,6-Диоксопиперидин-3-ил)-1-оксоизоиндолин-4-ил)амино)метил)-4-фторфенилметилкарбамат, A371:

1H ЯМР (DMSO-d6, 300 МГц): δ 11,00 (s, 1H), 7,52-7,56 (m, 1H), 7,15-7,25 (m, 2H), 6,93-7,06 (m, 3H), 6,63 (d, J = 7,8 Гц, 1H), 6,28-6,32 (m, 1H), 5,10 (dd, J = 13,5, 4,5 Гц, 1H), 4,40 (d, J = 4,8 Гц, 2H), 4,30 (d, J = 17,4 Гц, 1H), 4,18 (d, J = 17,4 Гц, 1H), 2,84-2,97 (m, 1H), 2,59-2,69 (m, 4H), 2,23-2,37 (m, 1H), 1,99-2,08 (m, 1H). LCMS: 441,1 ([M+1]+).
Пример 32. Соединение A372
3-(4-((2-Фтор-3-(метиламино)бензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A372:

1H ЯМР (DMSO-d6, 300 МГц): δ 10,99 (s, 1H), 7,21 (t, J = 7,8 Гц, 1H), 6,85-6,92 (m, 2H), 6,63 (d, J = 8,1 Гц, 1H), 6,48-6,55 (m, 2H), 6,21 (t, J = 5,4 Гц, 1H), 5,48-6,49 (m, 1H), 5,09 (dd, J = 13,2, 5,1 Гц, 1H), 4,35 (d, J = 5,4 Гц, 2H), 4,27 (d, J = 17,4 Гц, 1H), 4,15 (d, J = 17,4 Гц, 1H), 2,85-2,97 (m, 1H), 2,69 (d, J = 4,5 Гц, 3H), 2,49-2,63 (m, 1H), 2,21-2,35 (m, 1H), 1,98-2,05 (m, 1H). LCMS=397,1 ([M+1]+)
Пример 33. Соединение A375
3-(4-((2-Фтор-5-(2-гидроксиэтокси)бензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A375:

1H ЯМР (DMSO-d6, 300 МГц): δ 9,35 (br s, 1H), 7,22 (t, J = 8,1 Гц, 1H), 7,10 (t, J = 9,3 Гц, 1H), 6,88-6,95 (m, 2H), 6,78-6,83 (m, 1H), 6,63 (d, J = 8,4 Гц, 1H), 6,31 (t, J = 6,0 Гц, 1H), 5,11 (dd, J = 13,5, 5,1 Гц, 1H), 4,82 (br s, 1H), 4,37 (d, J = 5,7 Гц, 2H), 4,30 (d, J = 17,1 Гц, 1H), 4,17 (t, J = 17,1 Гц, 1H), 3,85 (t, J = 4,8 Гц, 2H), 3,62 (t, J = 4,8 Гц, 2H), 2,85-2,97 (m, 1H), 2,55-2,65 (m, 1H), 2,24-2,36 (m, 1H), 2,01-2,05 (m, 1H). LCMS: 428,1 [(M+1)+].
Пример 34. Соединение A376
3-(4-((2-Фтор-5-(2-(пирролидин-1-ил)этокси)бензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A376:

1H ЯМР (300 МГц, DMSO-d6): δ 11,02 (s, 1H), 7,22 (t, J = 8,1 Гц, 1H), 7,06-7,13 (m, 1H), 6,89-6,95 (m, 2H), 6,78-6,84 (m, 1H), 6,63 (d, J = 7,8 Гц, 1H), 6,29 (t, J = 5,7 Гц, 1H), 5,11, (dd, J = 13,2, 5,1 Гц, 1H), 4,27-4,37 (m, 3H), 4,17 (d, J = 17,1 Гц, 1H), 3,94 (t, J = 6,0 Гц, 1H), 2,85-2,97 (m 1H), 2,57-2,69 (m 3H), 2,22-2,42 (m 5H), 1,98-2,06 (m 1H), 1,56-1,66 (m 4H).
Пример 35. Соединение A377
3-(4-((2-Фтор-5-(2-(4-метилпиперазин-1-ил)этокси)бензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A377:
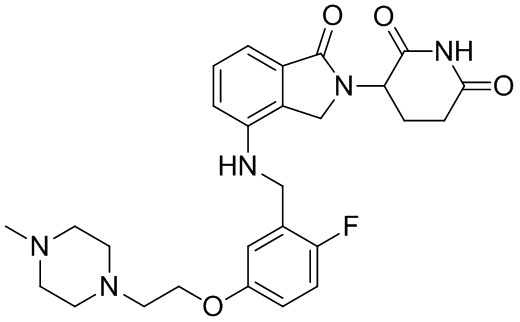
1H ЯМР (DMSO-d6, 300 МГц): δ 11,03 (s, 1H), 7,22 (t, J = 7,8 Гц, 1H), 7,09 (t, J = 9,6 Гц, 1H), 6,91-6,95 (m, 2H), 6,79-6,89 (m, 1H), 6,63 (d, J = 8,1 Гц, 1H), 6,29 (t, J = 6,0 Гц, 1H), 5,11 (dd, J = 13,5, 4,8 Гц, 1H), 4,27-4,37 (m, 3H), 4,17 (d, J = 17,4 Гц, 1H), 3,94 (t, J = 5,7 Гц, 2H), 2,85-2,98 (m, 1H), 2,55-2,62 (m, 4H), 2,20-2,42 (m, 8H), 2,12 (s, 3H), 1,98-2,07 (m, 1H). LCMS: 510,2 ([M+1]+).
Пример 36. Соединение A378
3-(((2-(2,6-Диоксопиперидин-3-ил)-1-оксоизоиндолин-4-ил)амино)метил)-4-фторфенилдиметилкарбамат, A378:
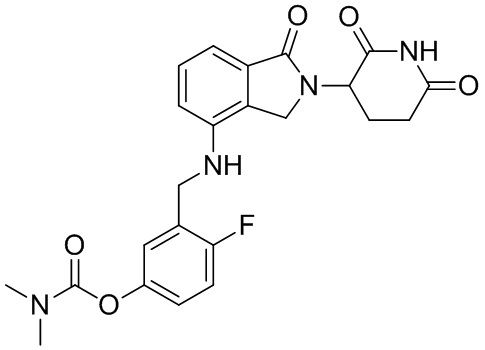
1H ЯМР (300 МГц, DMSO-d6): δ 11,00 (s, 1H), 7,17-7,26 (m, 2H), 7,08-7,11 (m, 1H), 6,94-7,04 (m, 2H), 6,78-6,84 (m, 1H), 6,62 (d, J = 8,1 Гц, 1H), 6,30 (t, J = 5,9 Гц, 1H), 5,11, (dd, J = 13,2, 5,4 Гц, 1H), 4,40 (d, J = 5,7 Гц, 1H), 4,31 (d, J = 17,4 Гц, 1H), 4,18 (d, J = 17,4 Гц, 1H), 2,84-2,96 (m 7H), 2,57-2,63 (m 1H), 2,23-2,37 (m 1H), 2,00-2,05 (m 1H).
Пример 37. Соединение A382
3-(4-((2-Фтор-5-(3-морфолинопропокси)бензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A382:

1H ЯМР (DMSO-d6, 300 МГц): δ 11,00 (s, 1H), 7,32 (t, J = 7,8 Гц, 1H), 7,22 (t, J = 7,8 Гц, 1H), 7,05-7,13 (m, 2H), 6,93 (d, J = 7,5 Гц, 1H), 6,64 (d, J = 7,8 Гц, 1H), 6,28 (t, J = 6,3 Гц, 1H), 5,07-5,13 (m, 1H), 4,38 (d, J = 5,7 Гц, 2H), 4,28 (d, J = 17,4 Гц, 1H), 4,16 (d, J = 17,4 Гц, 1H), 3,54 (t, J = 4,5 Гц, 4H), 3,42 (s, 2H), 2,85-2,97 (m, 1H), 2,57-2,63 (m, 1H), 2,26-2,38 (m, 5H), 2,00-2,09 (m, 1H). LCMS: 467,2 ([M+1]+).
Пример 38. Соединение A383
3-(4-((2-Фтор-5-(морфолинометил)бензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A383:
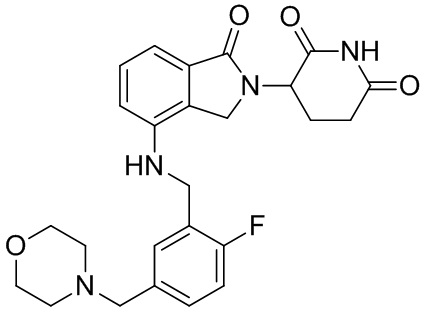
1H ЯМР (DMSO-d6, 300 МГц): δ 10,98 (br s, 1H), 7,34 (d, J = 0,9 Гц, 1H), 7,10-7,32 (m, 3H), 6,95 (d, J = 7,5 Гц, 1H), 6,64 (d, J = 7,8 Гц, 1H), 6,27 (br s, 1H), 5,12 (dd, J = 13,5, 5,1 Гц, 1H), 4,43 (d, J = 5,7 Гц, 2H), 4,32 (d, J = 17,1 Гц, 1H), 4,21 (d, J = 17,1 Гц, 1H), 3,45-3,48 (m, 4H), 3,38 (s, 2H), 2,87-2,99 (m, 1H), 2,30-2,36 (m, 1H), 2,23-2,25 (m, 4H), 2,22-2,36 (m, 1H), 2,01-2,09 (m, 1H). LCMS=467,2 [(M+1)+].
Пример 39. Соединение A381
3-(4-((2-Фтор-3-(морфолинометил)бензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A381:

1H ЯМР (DMSO-d6, 300 МГц): δ 11,02 (s, 1H), 7,20-7,31 (m, 3H), 7,10 (t, J = 7,8 Гц, 1H), 6,95 (d, J = 7,5 Гц, 1H), 6,65 (d, J = 8,1 Гц, 1H), 6,31 (t, J = 5,7 Гц, 1H), 5,12 (dd, J1 = 13,8 Гц, J2 = 5,4 Гц, 1H), 4,43 (d, J = 5,4 Гц, 2H), 4,31 (d, J = 17,4 Гц, 1H), 4,19 (d, J = 17,4 Гц, 1H), 3,53-3,58 (m, 6H), 2,87-2,99 (m, 1H), 2,57-2,66 (m, 1H), 2,24-2,39 (m, 5H), 2,00-2,10 (m, 1H). LCMS=467,2 [(M+1)+].
Пример 40. Соединение A384
3-(4-((3-Амино-2-фторбензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A384:

1H ЯМР (DMSO-d6, 300 МГц): δ 9,76 (br s, 1H), 7,23 (t, J = 8,1 Гц, 1H), 6,93 (d, J = 7,5 Гц, 1H), 6,77 (t, J = 7,8 Гц, 1H), 6,61-6,67 (m, 2H), 6,49-6,54 (m 1H), 6,22 (t, J = 5,7 Гц, 1H), 5,09-5,15 (m, 3H), 4,35 (d, J = 5,4 Гц, 2H), 4,29 (d, J = 17,4 Гц, 1H), 4,17 (d, J = 17,4 Гц, 1H), 2,87-2,99 (m, 1H), 2,58-2,67 (m, 1H), 2,23-2,36 (m, 1H), 2,00-2,10 (m, 1H). LCMS=383,1 ([M+1]+).
Пример 41. Соединение A388
N-(3-(((2-(2,6-Диоксопиперидин-3-ил)-1-оксоизоиндолин-4-ил)амино)метил)-2-фторфенил)ацетамид, A388:
1H ЯМР (DMSO-d6, 300 МГц): δ 10,96 (br s, 1H), 9,71 (s, 1H), 7,74 (t, J = 6,9 Гц, 1H), 7,22 (t, J = 7,8 Гц, 1H), 7,02-7,13 (m, 2H), 6,93 (d, J = 7,5 Гц, 1H), 6,64 (d, J = 7,8 Гц, 1H), 6,31 (t, J = 6,0 Гц, 1H), 5,10 (dd, J = 12,9, 5,1 Гц, 1H), 4,42 (d, J = 5,4 Гц, 2H), 4,29 (d, J = 17,1 Гц, 1H), 4,16 (d, J = 17,1 Гц, 1H), 2,85-2,95 (m, 1H), 2,55-2,64 (m, 1H), 2,22-2,36 (m, 1H), 1,99-2,07 (m, 4H). LCMS= 425,1 [(M+1)+].
Пример 42. Соединение A389
3-(((2-(2,6-Диоксопиперидин-3-ил)-1-оксоизоиндолин-4-ил)амино)метил)-2-фторбензолсульфонамид, A389:

1H ЯМР (300 МГц, DMSO-d6): δ 11,02 (s, 1H), 7,85-7,88 (m, 1H), 7,73-7,78 (m, 1H), 7,39-7,45 (m, 3H), 7,24 (t, J = 7,8 Гц, 1H), 6,96 (d, J = 7,5 Гц, 1H), 6,63 (d, J = 8,1 Гц, 1H), 6,43 (t, J = 6,0 Гц, 1H), 5,11, (dd, J = 13,2, 5,1 Гц, 1H), 4,46 (d, J = 5,4 Гц, 2H), 4,29 (d, J = 17,4 Гц, 1H), 4,18 (d, J = 17,4 Гц, 1H), 2,85-2,97 (m 1H), 2,58-2,63 (m 1H), 2,24-2,36 (m 5H), 2,02-2,07 (m 1H).
Пример 43. Соединение A387
3-(4-((2-Фтор-5-(метиламино)бензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A387:

1H ЯМР (DMSO-d6, 300 МГц): δ 11,02 (s, 1H), 7,22 (t, J = 7,5 Гц, 1H), 6,87-6,93 (m, 2H), 6,62 (d, J = 8,1 Гц, 1H), 6,50-6,53 (m, 1H), 6,31-6,36 (m, 1H), 6,25 (t, J = 5,7 Гц, 1H), 5,47-5,52 (m, 1H), 5,11 (dd, J = 4,8, 13,2 Гц, 1H), 4,25-4,30 (m, 3H), 4,15 (d, J = 17,4 Гц, 1H), 2,85-2,97 (m, 1H), 2,54-2,63 (m, 4H), 2,22-2,37 (m, 1H), 1,99-2,06 (m, 1H). LCMS: 397,21 ([M+1]+).
Пример 44. Соединение A396
3-(4-((2-Фтор-4-гидроксибензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A396:
1H ЯМР (DMSO-d6, 300 МГц): δ 11,00 (br s, 1H), 9,74 (br s, 1H), 7,14-7,25 (m, 2H), 6,92 (d, J = 7,2 Гц, 1H), 6,66 (d, J = 8,1 Гц, 1H), 6,52-6,55 (m, 2H), 6,13 (t, J = 6,0 Гц, 1H), 5,07-5,13 (m, 1H), 4,24-4,29 (m, 3H), 4,14 (d, J = 17,1 Гц, 1H), 2,87-2,97 (m, 1H), 2,56-2,65 (m, 1H), 2,21-2,36 (m, 1H), 1,98-2,06 (m, 1H). LCMS: 384,1 [(M+1)+]
Пример 45. Соединение A391
[3-(4-((5-Амино-2-фторбензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион], A391
Стадия A: соединение A391G получали в соответствии со способом синтеза, показанным в примере 20, с соответствующими исходными материалами для замены A346A.
1H ЯМР (DMSO-d6, 400 МГц): δ 11,03 (s, 1H), 9,31 (s, 1H), 7,37-7,44 (m, 2H), 7,24 (t, J = 7,6 Гц, 1H), 7,09 (t, J = 9,2 Гц, 1H), 6,95 (d, J = 7,2 Гц, 1H), 6,62 (d, J = 8,0 Гц, 1H), 6,33 (t, J = 5,6 Гц, 1H), 5,11-5,16 (m, 1H), 4,36 (t, J = 5,2 Гц, 2H), 4,29 (d, J = 16,8 Гц, 1H), 4,19 (d, J = 17,2 Гц, 1H), 2,89-2,98 (m, 1H), 2,60-2,64 (m, 1H), 2,26-2,37 (m, 1H), 2,03-2,06 (m, 1H), 1,42 (s, 9H).

Стадия B: к раствору A391G (400 мг, 0,83 ммоль) в DCM (12 мл) добавляли CF3COOH (4 мл) и раствор перемешивали в течение 0,5 часа при 35oC. Растворитель удаляли и остаток растворяли с помощью 4 мл CH3CN и 100 мг Et3N, затем очищали с помощью преп-HPLC с получением A391 (130 мг, выход 41%) в виде белого твердого вещества.
1H ЯМР (DMSO-d6, 400 МГц): δ 10,83 (s, 1H), 9,37 (t, J = 5,6 Гц, 1H), 8,03 (d, J = 8,8 Гц, 2H), 7,78-7,86 (m, 3H), 7,58 (s, 1H), 7,50 (d, J = 8,0 Гц, 2H), 7,17 (s, 1H), 4,64 (d, J = 5,6 Гц, 2H), 4,50-4,54 (m, 1H), 2,37-2,41 (m, 1H), 2,21-2,26 (m, 1H), 1,89-1,93 (m, 2H). LCMS: 523,1 ([M+1]+).
Пример 46. Соединение A397
3-(4-((5-Амино-2-фторбензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A397
Стадия A: соединение A397A получали в соответствии со способом синтеза, показанным в примере 20, с соответствующими исходными материалами для замены A346A.
1H ЯМР (DMSO-d6, 400 МГц): δ 11,04 (br s, 1H), 9,54 (br s, 1H), 7,38 (d, J = 12,8 Гц, 1H), 7,22-7,28 (m, 2H), 7,13 (d, J = 8,4 Гц, 1H), 6,94 (d, J = 7,6 Гц, 1H), 6,65 (d, J = 8,0 Гц, 1H), 6,24 (t, J = 5,6 Гц, 1H), 5,10-5,14 (m, 1H), 4,27-4,34 (m, 3H), 4,17 (d, J = 17,2 Гц, 1H), 2,89-2,97 (m, 1H), 2,64-2,67 (m, 1H), 2,25-2,36 (m, 1H), 2,03-2,06 (m, 1H), 1,46 (s, 9H).
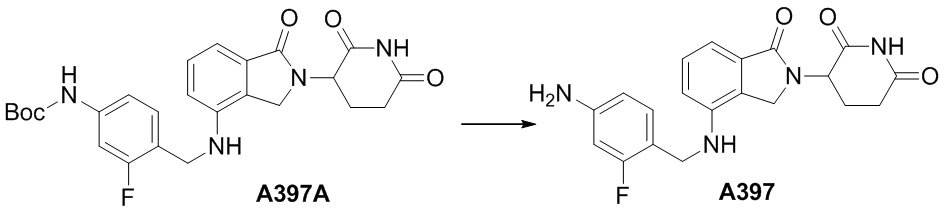
Стадия B: к раствору соединения A397A (100 мг, 0,21 ммоль) в диоксане (20 мл) добавляли раствор 6 н. HCl в диоксане, смесь перемешивали в течение 2,5 ч. Реакционную смесь концентрировали, а остаток растворяли в DMF (10 мл) и регулировали значение pH=7-8 с помощью насыщ. NaHCO3, фильтровали. Фильтрат концентрировали и остаток очищали с помощью преп-HPLC с получением требуемого продукта A397 (35 мг, выход 44%) в виде светло-желтоватого твердого вещества.
1H ЯМР (DMSO-d6, 400 МГц): δ 11,02 (br s, 1H), 7,24 (t, J = 8,4 Гц, 1H), 7,02 (t, J = 8,4 Гц, 1H), 6,93 (d, J = 7,6 Гц, 1H), 6,69 (d, J = 8,0 Гц, 1H), 6,04 (t, J = 5,6 Гц, 1H), 5,30 (br s, 2H), 5,09-5,14 (m, 1H), 4,25 (d, J = 17,2 Гц, 1H), 4,20 (d, J = 5,6 Гц, 1H), 4,14 (d, J = 17,2 Гц, 1H), 2,88-2,97 (m, 1H), 2,59-2,64 (m, 1H), 2,24-2,35 (m, 1H), 2,02-2,08 (m, 4H). LCMS: 383,2 [(M+1)+].
Пример 47. Соединение A373
(S)-3-Дейтерий-3-(4-((4-(морфолинометил)бензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A373:

Стадия A: к раствору A373C [4-(морфолинометил)бензальдегид] (0,8 г, 3,9 ммоль) и A356C (0,7 г, 2,5 ммоль) в MeOH (100 мл) добавляли AcOH (1 мл) и смесь перемешивали в течение ночи при 40oC в атмосфере N2. Затем к реакционной смеси добавляли Pd/C (50% воды, 10%, 150 мг) и дегазировали с помощью H2 3 раза. Смесь перемешивали в течение 5 часов при давлении водорода 1 атм. Реакционную смесь фильтровали, а фильтрат концентрировали и очищали с помощью RP-HPLC с получением A373A (1,0 г, выход 86%) в виде светло-желтого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 9,96 (br, 1H), 7,59 (s, 1H), 7,41-7,49 (m, 4H), 7,11-7,18 (m, 2H), 6,87 (d, J = 7,2 Гц, 1H), 6,54 (d, J = 8,1 Гц, 1H), 4,25-4,55 (m, 7H), 3,93 (d, J = 12,0 Гц, 2H), 3,58 (t, J = 12,3 Гц, 2H), 3,01-3,24 (m, 4H), 2,13-2,21 (m, 3H), 1,93-2,00 (m, 1H).
Стадия B: к раствору A373A (200 мг, 0,428 ммоль) в THF (12 мл) и DCM (12 мл) добавляли SOCl2 (204 мг, 1,71 ммоль в 1,7 мл DCM) при -40oC и смесь перемешивали при -40oC в течение 2 часов в атмосфере N2. Затем к реакционной смеси добавляли пиридин (135 мг, 1,71 ммоль) и перемешивали в течение 30 минут. Затем добавляли Et3N (173 мг, 1,71 ммоль) и обеспечивали нагревание реакционной смеси до комнатной температуры. Чтобы погасить реакцию, добавляли воду (0,5 мл) и смесь концентрировали и очищали с помощью преп-HPLC с обращенной фазой (подвижная фаза : вода/ацетонитрил) 2 раза с получением A373 (20 мг, выход 10%) в виде белого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 11,02 (s, 1H), 7,15-7,34 (m, 5H), 6,89 (d, J = 7,2 Гц, 1H), 6,61 (d, J = 7,8 Гц, 1H), 6,38 (t, J = 4,8 Гц, 1H), 5,08-5,14 (m, 0,04H), 4,35 (d, J = 5,1 Гц, 2H), 4,29 (d, J = 17,4 Гц, 1H), 4,16 (d, J = 17,4 Гц, 1H), 3,55 (br s, 4H), 3,42 (br.s, 2H), 2,85-2,98 (m, 1H), 2,57-2,63 (m, 1H), 2,24-2,34 (m, 5H), 1,99-2,05 (m, 1H).
LCMS: 450,2 ([M+1]+).
Пример 48. Соединение A374
2-(3-(((2-(2,6-Диоксопиперидин-3-ил)-1-оксоизоиндолин-4-ил)амино)метил)-4-фторфенокси)этилпирролидин-1-карбоксилат, A374:
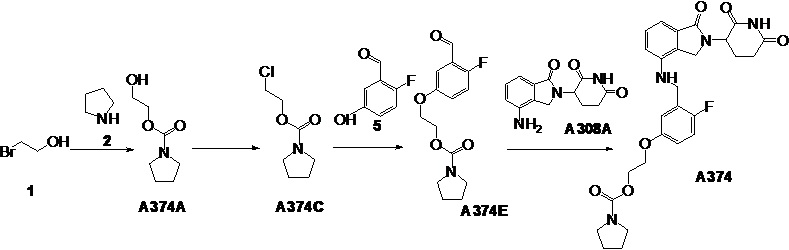
Стадия A: смесь пирролидина (3,77 г, 53 ммоль), 2-бромэтанола (6,25 г, 50 ммоль) и K2CO3 (6,9 г, 50 ммоль) в CH3CN (70 мл) нагревали до температуры флегмы и перемешивали в течение ночи в атмосфере N2. Реакционную смесь фильтровали и концентрировали, а также очищали с помощью колоночной хроматографии на силикагеле (DCM : MeOH = 100:1-10:1) с получением A374A (4 г, выход 50%) в виде светло-желтого масла.
1H ЯМР (CDCl3, 300 МГц): δ 4,25-4,28 (m, 2H), 3,80-3,85 (m, 2H), 3,36-3,43 (m, 4H), 2,92 (t, J = 5,7, 1H), 1,88-1,92 (m, 4H).
Стадия B: к раствору A374A (2,3 г, 14,4 ммоль) в CHCl3 (50 мл) добавляли SOCl2(3,6 г, 30,0 ммоль) и реакционную смесь перемешивали в течение 1,5 ч. с обратным холодильником. Реакционную смесь концентрировали с получением неочищенного A374C (2,0 г, выход 78%) в виде белого твердого вещества.
1H ЯМР (CDCl3, 300 МГц): δ 4,21 (t, J = 5,7, 2H), 3,77 (t, J = 5,7, 2H), 3,21-3,32 (m, 4H), 1,75-1,83 (m, 4H).
Стадия C: смесь A374C (802 мг, 4,52 ммоль), 2-фтор-5-гидроксибензальдегида (280 мг, 2,0 ммоль) и K2CO3 (828 мг, 6,0 ммоль) в DMF (10 мл) нагревали до 90oC и перемешивали в течение ночи в атмосфере N2. Реакционную смесь вливали в ледяную воду (100 мл), перемешивали и фильтровали. Отфильтрованный осадок промывали водой (20 мл) и затем растворяли в EtOAc (50 мл), высушивали и концентрировали с получением продукта A374E (560 мг) в виде белого твердого вещества, которое использовали на следующей стадии без очистки.
1H ЯМР (CDCl3, 300 МГц): δ 10,32 (s, 1H), 7,31-7,34 (m, 1H), 7,09-7,17 (m, 2H), 4,42 (t, J = 5,1 Гц, 2H), 4,20 (t, J = 5,1 Гц, 2H) 3,30-3,41 (m, 4H), 1,85 (br s, 4H).
Стадия D: раствор A374E (206 мг, 0,732 ммоль), A308A (150 мг, 0,578 ммоль) и AcOH (6 мл) в дихлорметане (6 мл) перемешивали в течение 4 часов при комнатной температуре. Добавляли NaBH3CN(109 мг, 1,74 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи в атмосфере N2. Удаляли растворитель, а остаток растворяли в CH3CN и очищали с помощью преп-HPLC с получением A374 (105 мг, выход 35%) в виде белого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 11,00 (s, 1H), 7,20-7,25 (m, 1H), 7,11 (t, J = 9,3 Гц, 1H), 6,93-6,95 (m, 2H), 6,82-6,87 (m, 1H), 6,64 (d, J = 8,1 Гц, 1H), 6,24-6,29 (m, 1H), 5,11 (dd, J = 13,5, 5,1 Гц, 1H), 4,33-4,37 (m, 2H), 4,20-4,27 (m, 4H), 4,08-4,14 (m, 2H), 3,12-3,22 (m, 4H), 2,86-2,97 (m, 1H), 2,62-2,71 (m, 1H), 2,24-2,33 (m, 1H), 2,01-2,06 (m, 1H), 1,69-1,77 (m, 4H). LCMS: 525,2 ([M+1]+).
Пример 49. Соединение A349
[3-(4-((3,4-Диметоксибензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион], A349:
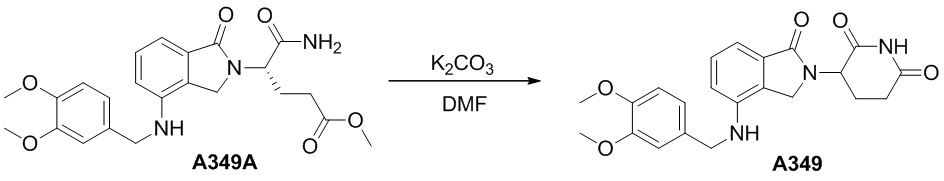
К раствору соединения A349A (100 мг, 0,23 ммоль) в DMF (5 мл) добавляли K2CO3 (47,0 мг, 0,34 ммоль), смесь перемешивали при 80oC (масляная баня) в течение ночи в атмосфере N2. TLC продемонстрировала завершение реакции. Реакционную смесь фильтровали и фильтрат концентрировали с получением остатка. Остаток очищали с помощью RP-HPLC с получением A349 (40 мг, выход 43%) в виде белого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 11,02 (s, 1H), 7,20 (t, J = 7,5 Гц, 1H), 7,01 (s, 1H), 6,85-6,92 (m, 3H), 6,68 (d, J = 7,8 Гц, 1H), 6,24-6,28(m, 1H), 5,08-5,14 (m, 1H), 4,25-4,33 (m, 3H), 4,18 (d, J = 17,1 Гц, 1H), 3,72 (s, 3H), 3,70 (s, 3H), 2,87-2,99 (m, 1H), 2,59-2,65 (m, 1H), 2,24-2,37 (m, 1H), 2,00-2,08 (m, 1H). LCMS: 410,2 ([M+1]+)
Пример 50. Соединение A350
3-(4-((3,4-Диметилбензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A350:
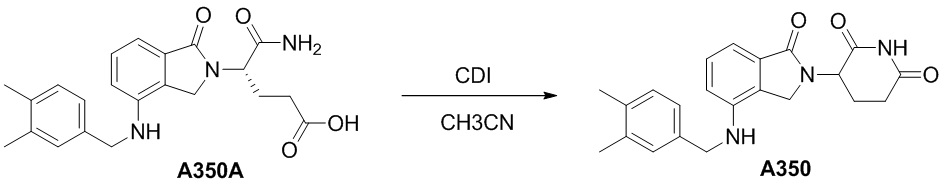
К раствору A350A (100 мг, 0,25 ммоль) в CH3CN (5 мл) добавляли CDI (62,0 мг, 0,38 ммоль), смесь перемешивали при 95oC (масляная баня) в течение ночи в атмосфере N2. TLC продемонстрировала завершение реакции. Реакционную смесь фильтровали и концентрировали с получением остатка. Остаток очищали с помощью RP-HPLC с получением A350 (61 мг, выход 65%) в виде белого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 11,01 (s, 1H), 7,15-7,21 (m, 2H), 7,04-7,10 (m, 2H), 6,91 (d, J = 7,2 Гц, 1H), 6,62 (d, J = 8,1 Гц, 1H), 6,27-6,31(m, 1H), 5,11 (dd, J = 13,2, 5,1 Гц, 1H), 4,28-4,33 (m, 3H), 4,18 (d, J = 17,7 Гц, 1H), 2,87-2,99 (m, 1H), 2,60-2,65 (m, 1H), 2,24-2,37 (m, 1H), 2,19 (s, 3H), 2,17 (s, 3H), 2,02-2,07 (m, 1H). LCMS: 378,2 ([M+1]+).
Соединения в примерах 51-55 получали в соответствии с процедурой, описанной в примере 50, с соответствующими исходными материалами для замены A350A.
Пример 51. Соединение A351
3-(4-((4-Фтор-3-метилбензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A351:
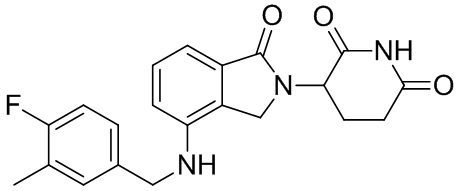
1H ЯМР (DMSO-d6, 300 МГц): δ 11,02 (s, 1H), 7,29 (d, J = 6,0 Гц, 1H), 7,18-7,23 (m, 2H), 7,06 (t, J = 9,6 Гц, 1H), 6,92 (d, J = 7,2 Гц, 1H), 6,63 (d, J = 7,8 Гц, 1H), 6,34 (t, J = 6,0 Гц, 1H), 5,12 (dd, J = 13,2, 5,1 Гц, 1H), 4,28-4,33 (m, 3H), 4,18 (d, J = 17,4 Гц, 1H), 2,87-2,99 (m, 1H), 2,59-2,65 (m, 1H), 2,26-2,37 (m, 1H), 2,21 (s, 3H), 2,01-2,08 (m, 1H). LCMS: 382,1 ([M+1]+).
Пример 52. Соединение A352
3-(4-((3-Хлор-4-метилбензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A352:

1H ЯМР (DMSO-d6, 300 МГц): δ 11,01 (s, 1H), 7,41 (s, 1H), 7,17-7,30 (m, 3H), 6,92 (d, J = 7,5 Гц, 1H), 6,61(d, J = 8,1 Гц, 1H), 6,39 (t, J = 6,0 Гц, 1H), 5,12 (dd, J = 13,2, 5,4 Гц, 1H), 4,29-4,37 (m, 3H), 4,18 (d, J = 17,1 Гц, 1H), 2,87-2,99 (m, 1H), 2,59-2,65 (m, 1H), 2,25-2,39 (m, 4H), 2,02-2,07 (m, 1H). LCMS: 398,1 ([M+1]+).
Пример 53. Соединение A353
3-(4-((3-Фтор-4-метилбензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A353:

1H ЯМР (DMSO-d6, 300 МГц): δ 11,02 (s, 1H), 7,11-7,24 (m, 4H), 6,92 (d, J = 7,2 Гц, 1H), 6,61 (d, J = 7,5 Гц, 1H), 6,36-6,40 (m, 1H), 5,12 (dd, J = 13,5, 5,1 Гц, 1H), 4,29-4,37 (m, 3H), 4,19 (d, J = 17,4 Гц, 1H), 2,86-2,99 (m, 1H), 2,59-2,65 (m, 1H), 2,24-2,39 (m, 1H), 2,18 (s, 3H), 2,01-2,07 (m, 1H). LCMS: 382,1 ([M+1]+).
Пример 54. Соединение A354
3-(4-((3-Хлор-4-метоксибензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A354:

1H ЯМР (DMSO-d6, 300 МГц): δ 11,02 (s, 1H), 7,44 (d, J = 1,8 Гц, 1H), 7,32 (dd, J = 8,4, 1,8 Гц, 1H), 7,21 (t, J = 7,8 Гц, 1H), 7,09 (d, J = 8,7 Гц, 1H), 6,93 (d, J = 7,2 Гц, 1H), 6,65 (d, J = 8,1 Гц, 1H), 6,35 (t, J = 5,9 Гц, 1H), 5,09-5,15 (m, 1H), 4,28-4,33 (m, 3H), 4,18 (d, J = 16,8 Гц, 1H), 3,81 (s, 3H), 2,87-2,99 (m, 1H), 2,58-2,67 (m, 1H), 2,24-2,37 (m, 1H), 2,01-2,09 (m, 1H). LCMS: 414,1 ([M+1]+).
Пример 55. Соединение A355
3-(4-((3,5-Диметоксибензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A355:
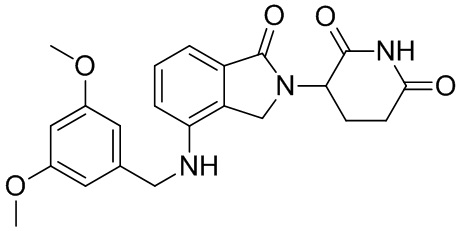
1H ЯМР (DMSO-d6, 300 МГц): δ 11,03 (s, 1H), 7,18-7,24 (m, 1H), 6,92 (d, J = 7,5 Гц, 1H), 6,64 (d, J = 8,4 Гц, 1H), 6,55 (d, J = 2,1 Гц, 2H), 6,31-6,35 (m, 2H), 5,12 (dd, J = 13,2, 4,8 Гц, 1H), 4,28-4,34 (m, 3H), 4,19 (d, J = 16,8 Гц, 1H), 3,33 (s, 6H), 2,87-2,99 (m, 1H), 2,58-2,67 (m, 1H), 2,26-2,37 (m, 1H), 2,02-2,08 (m, 1H). LCMS: 410,2 ([M+1]+).
Пример 56. Соединение A356
(S)-3-Дейтерий-3-(4-((2-фтор-4-метоксибензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A356:
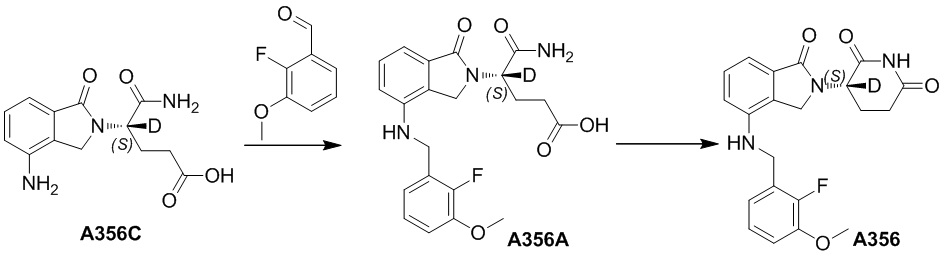
Стадия A: к раствору A356C (300 мг, 1,08 ммоль) и 2-фтор-4-метоксибензальдегида (249 мг, 1,62 ммоль) в MeOH (30 мл) добавляли ледяную AcOH (0,5 мл), смесь перемешивали при 30oC (масляная баня) в течение 5 часов. Добавляли Pd/C (10%, 100 мг, 50% воды) и перемешивали при 30oC (масляная баня) в течение ночи в атмосфере H2 (баллон). Pd/C удаляли с помощью фильтрации и фильтрат концентрировали с получением остатка. Остаток очищали с помощью флэш-хроматографии на C18 (CH3CN: H2O = 5% - 35%, 30 мин.; 35% - 45%, 30 мин.; 45% - 55% 20 мин.), затем сушили сублимацией с получением A356A (160 мг, выход 35%) в виде светло-желтоватого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 12,06 (br s, 1H), 7,55 (br s, 1H), 7,29 (t, J = 9,0 Гц, 1H), 7,16-7,22 (m, 2H), 6,88 (d, J = 7,5 Гц, 1H), 6,80 (dd, J = 12,6, 2,4 Гц, 1H), 6,71 (dd, J = 8,7, 2,7 Гц, 1H), 6,61 (d, J = 8,1 Гц, 1H), 6,28 (t, J = 6,0 Гц, 1H), 4,68-4,74 (m, 0,01H), 4,48 (d, J = 17,7 Гц, 1H), 4,23-4,31 (m, 3H), 3,72 (s, 3H), 2,10-2,19 (m, 3H), 1,93-2,01 (m, 1H).
Стадия B: к раствору A356A (160 мг, 0,39 ммоль) в сухом DCM (20 мл), охлажденному до -40oC, в атмосфере N2 к смеси медленно добавляли SOCl2 (229 мг, 1,92 ммоль) при -40oC в атмосфере N2, затем добавляли раствор DMF (5 мг) в DCM (1 мл). Реакционную смесь перемешивали при -40oC в течение 2 ч., по каплям добавляли пиридин (152 мг, 1,92 ммоль), перемешивали в течение 40 минут при данной температуре, добавляли Et3N (195 мг, 1,92 ммоль) и затем реакционную смесь перемешивали при -40oC в течение 2 ч. LCMS продемонстрировала завершение реакции. Чтобы погасить реакцию, добавляли H2O (10 мл), водный слой экстрагировали с помощью DCM (30 мл x 2), объединенную органическую фазу промывали солевым раствором (50 мл), высушивали над Na2SO4, фильтровали, концентрировали с получением остатка. Остаток очищали с помощью флэш-хроматографии на C18 (CH3CN: H2O = 5% - 35%, 30 мин.; 35% - 45%, 30 мин.; 45% - 55% 20 мин.) с получением A356 (70 мг, выход 46%, ee: 97%) в виде белого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 10,99 (br s, 1H), 7,31 (t, J = 9,0 Гц, 1H), 7,24 (t, J = 7,8 Гц, 1H), 6,94 (d, J = 7,2 Гц, 1H), 6,82 (dd, J = 12,3, 2,7 Гц, 1H), 6,71-6,75 (m, 1H), 6,68 (d, J = 7,8 Гц, 1H), 6,20 (t, J = 6,0 Гц, 1H), 5,08-5,14 (m, 0,04H), 4,26-4,35 (m, 3H), 4,17 (d, J = 16,8 Гц, 1H), 3,74 (s, 3H), 2,87-2,97 (m, 1H), 2,57-2,66 (m, 1H), 2,25-2,34 (m, 1H), 2,00-2,09 (m, 1H). LCMS: 399,1 ([M+1]+).
Соединение в примере 57 получали в соответствии с процедурой, описанной в примере 56, с соответствующим исходным материалом для замены 2-фтор-4-метоксибензальдегида на стадии A.
Пример 57. Соединение A357
(S)-3-Дейтерий-3-(4-((2-фтор-3-метоксибензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A357:
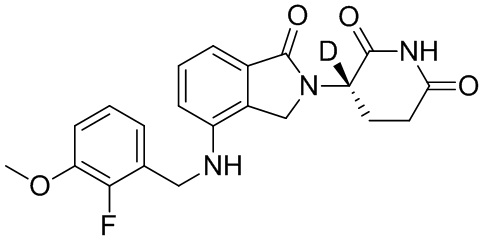
1H ЯМР (DMSO-d6, 300 МГц): δ 11,00 (br s, 1H), 7,21 (t, J = 7,8 Гц, 1H), 7,00-7,07 (m, 2H), 6,89-6,93 (m, 2H), 6,78-6,84 (m, 1H), 6,61 (d, J = 8,4 Гц, 1H), 6,30 (t, J = 5,4 Гц, 1H), 5,07-5,13 (m, 0,03H), 4,40 (d, J = 5,7 Гц, 2H), 4,28 (d, J = 17,1 Гц, 1H), 4,16 (d, J = 17,1 Гц, 1H), 3,81 (s, 3H), 2,85-2,97 (m, 1H), 2,57-2,63 (m, 1H), 2,24-2,34 (m, 1H), 2,00-2,06 (m, 1H). LCMS: 399,1 ([M+1]+).
Пример 58. Соединение A379
(S)-3-Дейтерий-3-(4-((2-фтор-5-метоксибензил)окси)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A379:
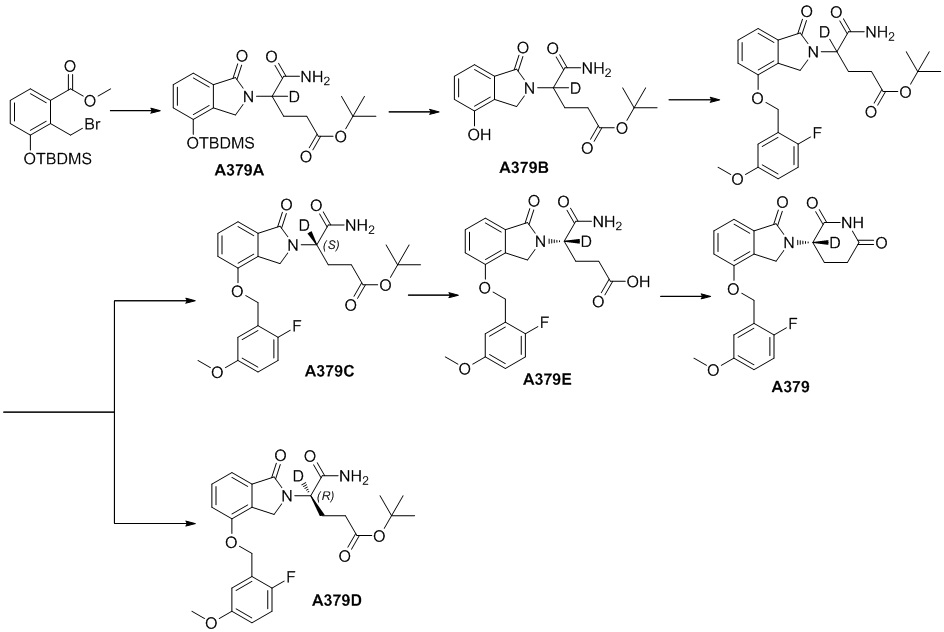

Стадия A: к смеси A379A1 (10,0 г, 27,8 ммоль) и A379A2 (8,01 г, 33,4 ммоль) в CH3CN (250 мл) добавляли DIPEA (7,92 г, 61,3 ммоль), смесь перемешивали при 45oC в течение ночи в атмосфере N2. Реакционную смесь концентрировали и добавляли DCM (300 мл) и H2O (100 мл), водную фазу экстрагировали с помощью DCM (200 мл x 1), объединенную органическую фазу промывали солевым раствором (200 мл), высушивали над Na2SO4, фильтровали, концентрировали с получением неочищенного продукта A379A (12,3 г) в виде желтого твердого вещества.

Стадия B: к раствору A379A (12,3 г, неочищенный) в THF (100 мл) добавляли 1 н. TBAF в THF (100 мл), смесь перемешивали в течение ночи при 25℃. LCMS продемонстрировала завершение реакции. Добавляли EtOAc (200 мл) и H2O (200 мл), водную фазу экстрагировали с помощью EtOAc (200 мл x 2), объединенную органическую фазу промывали солевым раствором (300 мл), высушивали над Na2SO4, фильтровали, концентрировали с получением остатка. Остаток растирали в порошок с помощью EtOAc (20 мл), фильтровали, промывали с помощью EtOAc (10 мл), затем высушивали с получением A379B (5,7 г) в виде белого твердого вещества. Фильтрат концентрировали и очищали с помощью колоночной хроматографии на силикагеле (PE / EtOAc = 1:4) с получением дополнительных 1,5 г A379B в виде белого твердого вещества; (общий выход 77%, в течение двух стадий).
1H ЯМР (DMSO-d6, 300 МГц): δ 10,01 (s, 1H), 7,54 (br s, 1H), 7,29 (t, J = 7,8 Гц, 1H), 7,12-7,16 (m, 2H), 6,96 (dd, J = 8,1, 0,6 Гц, 1H), 4,66-4,71 (m, 0,01H), 4,47 (d, J = 17,7 Гц, 1H), 4,29 (d, J = 17,7 Гц, 1H), 2,08-2,17 (m, 3H), 1,95-2,02 (m, 1H), 1,31 (s, 9H).

Стадия C: к раствору A379B (1,18 г, 3,52 ммоль) и 2-(хлорметил)-1-фтор-4-метоксибензола (1,23 г, 7,04 ммоль) в DMF (20 мл) добавляли K2CO3 (972 мг, 7,03 ммоль), смесь перемешивали в течение ночи при комнатной температуре. LCMS продемонстрировала завершение реакции. Реакционную смесь концентрировали и добавляли EtOAc (50 мл) и H2O (30 мл), водную фазу экстрагировали с помощью EtOAc (50 мл), объединенную органическую фазу промывали солевым раствором (50 мл), высушивали над Na2SO4, фильтровали, концентрировали с получением остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле (MeOH/DCM = 1/30) с получением белого продукта (1,46 г, выход 87%). Белое твердое вещество подвергали хиральному разделению с получением A379C (650 мг) и A379D (650 мг).
Условия хирального разделения
Подвижная фаза: гексан/EtOH = 40/60 (об./об.), концентрация образца: 100 мг/мл в подвижной фазе; колонка: CHIRALPAK IC; 20 мм (I.D) x 250 мм (L); 5 мкм; температура: 35℃; объем введения: 250 мкл; расход: 10 мл/мин.; длина волны: 205 нм.
A379C:1H ЯМР (DMSO-d6, 300 МГц): δ 7,55 (br s, 1H), 7,46 (t, J = 8,1 Гц, 1H), 7,28-7,34 (m, 2H), 7,11-7,21 (m, 3H), 6,92-6,97 (m, 1H), 5,22 (s, 2H), 4,49 (d, J = 18,0 Гц, 1H), 4,36 (d, J = 18,0 Гц, 1H), 3,73 (s, 3H), 2,05-2,13 (m, 3H), 1.96-2,02 (m, 1H), 1,30 (s, 9H).

Стадия D: к раствору A379C (650 мг, 1,37 ммоль) в DCM (20 мл) при 0oC по каплям добавляли TFA (10 мл), смесь нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь концентрировали при сниженном давлении (в вакууме). Остаток растворяли в CH3CN (4 мл) и очищали с помощью флэш-хроматографии на C18 (40% ацетонитрил в воде), затем сушили сублимацией с получением A379E (566 мг, выход 99%) в виде светло-желтоватого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 12,08 (br s, 1H), 7,58 (br s, 1H), 7,46 (t, J = 8,1 Гц, 1H), 7,28-7,35 (m, 2H), 7,11-7,21 (m, 3H), 6,92-6,97 (m, 1H), 5,22 (s, 2H), 4,51 (d, J = 17,7 Гц, 1H), 4,37 (d, J = 17,7 Гц, 1H), 3,73 (s, 3H), 2,08-2,20 (m, 3H), 1,96-2,05 (m, 1H).

Стадия E: к раствору A379E (366 мг, 0,88 ммоль) в сухом DCM (35 мл) и THF (5 мл) в атмосфере N2 при -40oC медленно добавляли SOCl2 (522 мг, 4,39 ммоль), а затем раствор DMF (5 мг) в DCM (1 мл). Реакционную смесь перемешивали при -40oC в течение 1 ч., добавляли пиридин (347 мг, 4,39 ммоль), через 40 мин. добавляли Et3N (444 мг, 4,39 ммоль) и затем смесь перемешивали в течение 1 ч. при -40oC. LCMS продемонстрировала завершение реакции. H2O (10 мл) добавляли, чтобы погасить реакцию, водный слой экстрагировали с помощью DCM (50 мл), объединенную органическую фазу промывали солевым раствором (50 мл), высушивали над Na2SO4, фильтровали, концентрировали с получением остатка. Неочищенный продукт очищали с помощью C18 с получением A379 (270 мг, выход 77%, ee: 100%) в виде белого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 10,96 (br s, 1H), 7,49 (t, J = 8,1 Гц, 1H), 7,32-7,38 (m, 2H), 7,11-7,21 (m, 2H), 6,91-6,97 (m, 1H), 5,23 (s, 2H), 5,06-5,12 (m, 0,01H), 4,37 (d, J = 17,4 Гц, 1H), 4,21 (d, J = 17,4 Гц, 1H), 3,72 (s, 3H), 2,82-2,94 (m, 1H), 2,57-2,60 (m, 1H), 2,38-2,48 (m, 1H), 1,92-1,97 (m, 1H). LCMS=400,1 ([M+1]+).
Соединение в примере 59 получали в соответствии с процедурой, описанной в примере 58, с соответствующим исходным материалом для замены 2-(хлорметил)-1-фтор-4-метоксибензола на стадии C.
Пример 59. Соединение A380
(S)-3-Дейтерий-3-(4-((2-фтор-3-метоксибензил)окси)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A380:
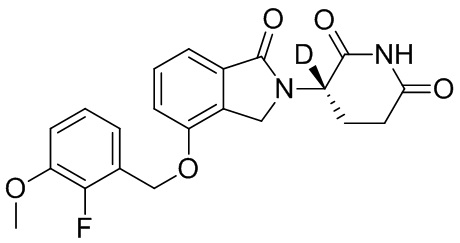
1H ЯМР (DMSO-d6, 300 МГц): δ 10,95 (br s, 1H), 7,49 (t, J = 7,8 Гц, 1H), 7,31-7,37 (m, 2H), 7,07-7,19 (m, 3H), 5,26 (s, 2H), 5,05-5,11 (m, 0,01H), 4,36 (d, J = 17,4 Гц, 1H), 4,20 (d, J = 17,4 Гц, 1H), 3,83 (s, 3H), 2,82-2,94 (m, 1H), 2,51-2,60 (m, 1H), 2,37-2,46 (m, 1H), 1,92-1,99 (m, 1H). LCMS=400,1 ([M+1]+).
Пример 60. Соединение A393
(S)-3-Дейтерий-3-(4-((2-фтор-4-метоксибензил)окси)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A393:

Стадия A: к раствору A379B (2,0 г, 6,0 ммоль) в DCM (30 мл) добавляли TFA (5 мл), после перемешивания в течение 3 часов при 25oC реакционную смесь концентрировали с получением остатка (1,7 г). Остаток (1,7 г) растворяли в DMF (5 мл) и обрабатывали 2-(триметилсилил)этанолом (3,55 г, 30 ммоль), EDCI (2,3 г, 12,0 ммоль) и DMAP (733 мг, 6,0 ммоль). Реакционную смесь перемешивали в течение ночи при 35oC и затем концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (DCM/MeOH = 40:1) с получением A393A (1,6 г, 70%).
1H ЯМР (DMSO-d6, 300 МГц): δ 9,99 (s, 1H), 7,54 (s, 1H), 7,28 (t, J = 7,8 Гц, 1H), 7,11-7,14 (m, 2H), 6,96 (d, J = 7,8 Гц, 1H), 4,47 (d, J = 18,0 Гц, 1H), 4,28 (d, J = 18,0 Гц, 1H), 3,92-3,99 (m, 2H), 2,00-2,25 (m, 4H), 0,80-0,85 (m, 2H), 0,04 (s, 9H).

Стадия B: к раствору A393A (1,02 г, 2,70 ммоль) в DMF (20 мл) добавляли K2CO3(750 мг, 5,40 ммоль) и 1-хлорметил-2-фтор-4-метоксибензол (720 мг, 4,10 ммоль), эту смесь нагревали до 30oC и перемешивали в течение 17 часов, затем эту смесь фильтровали и концентрировали с получением неочищенного масла, которое очищали с помощью колоночной хроматографии на силикагеле (DCM/MeOH = 60/1) с получением A393C (1,0 г, 72%).
1H ЯМР (DMSO-d6, 300 МГц): δ 7,43-7,54 (m, 3H), 7,26-7,35 (m, 2H), 7,14 (s, 1H), 6,80-6,90 (m, 2H), 5,16 (s, 2H), 4,45 (d, J = 17,4 Гц, 1H), 4,30 (d, J = 17,4 Гц, 1H), 3,92-3,98 (m, 2H), 3,77 (s, 3H), 2,01-2,22 (m, 4H), 0,79-0,84 (m, 2H).
Стадия C: хиральное разделение
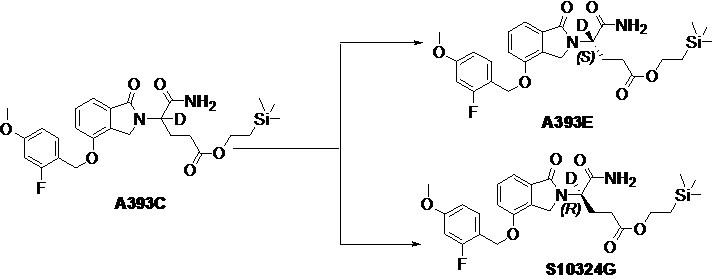
Условия хирального разделения
Подвижная фаза: MeOH / EtOH = 50/50 (об./об.); образец: 120 мг/мл в подвижной фазе; колонка: IF; 20 мм (I.D) x 250 мм (L); 5 мкм; температура: 35oC; объем введения: 300 мкл; расход: 9 мл/мин.; длина волны: 205 нм.
A393E
1H ЯМР (DMSO-d6, 300 МГц): δ 7,59 (s, 1H), 7,45-7,54 (m, 2H), 7,36 (d, J = 7,8 Гц, 1H), 7,29 (d, J = 7,2 Гц, 1H), 7,20 (s, 1H), 6,80-6,92 (m, 2H), 5,18 (s, 2H), 4,47 (d, J = 17,4 Гц, 1H), 4,32 (d, J = 17,4 Гц, 1H), 3,92-4,01 (m, 2H), 3,78 (s, 3H), 2,01-2,20 (m, 4H), 0,80-0,85 (m, 2H), 0,04 (s, 9H).

Стадия D: к раствору A393E (500 мг, 0,97 ммоль) в THF (5 мл) добавляли TBAF (1 н./THF, 5 мл), эту смесь перемешивали при 50oC в течение ночи, фильтровали и концентрировали, остаток очищали с помощью колоночной хроматографии (C18) с получением промежуточного продукта (420 мг). К раствору данного промежуточного продукта (300 мг) в DCM (15 мл) и DMF (1 мл) при -40oC добавляли SOCl2(428 мг, 3,60 ммоль). Реакционную смесь перемешивали в течение 2 часов, добавляли пиридин (281 мг, 3,60 ммоль), смесь перемешивали в течение еще 30 мин., а затем добавляли Et3N (363 мг, 3,60 ммоль) и перемешивали в течение дополнительного 1 ч. при -40oC. Реакционную смесь обрабатывали водой (80 мл) и экстрагировали с помощью DCM (80 мл x 3), объединяли органические слои и высушивали над Na2SO4, фильтровали и концентрировали, а также очищали с помощью преп-HPLC с получением A393 (200 мг, 72%, в течение двух стадий).
1H ЯМР (DMSO-d6, 300 МГц): δ 10,95 (s, 1H), 7,48-7,53 (m, 2H), 7,32-7,40 (m, 2H), 6,80-6,91 (m, 2H), 5,19 (s, 2H), 4,35 (d, J = 17,4 Гц, 1H), 4,19 (d, J = 17,4 Гц, 1H), 3,78 (s, 3H), 2,84-2,96 (m, 1H), 2,37-2,59 (m, 2H), 1,93-1,99 (m, 1H). LCMS: 400,1 ([M+1]+).
Соединение из примера 61 получали в соответствии с процедурой, описанной для примера 60, с соответствующим исходным материалом для замены соединения A393E.
Пример 61. Соединение A392
(R)-3-Дейтерий-3-(4-((2-фтор-4-метоксибензил)окси)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A392:
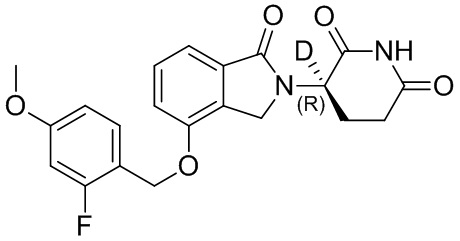
1H ЯМР (DMSO-d6, 300 МГц): δ 10,95 (s, 1H), 7,48-7,53 (m, 2H), 7,32-7,40 (m, 2H), 6,80-6,91 (m, 2H), 5,19 (s, 2H), 5,07-5,13 (m, 0,05H), 4,35 (d, J = 17,7 Гц, 1H), 4,19 (d, J = 17,7 Гц, 1H), 3,78 (s, 3H), 2,84-2,96 (s, 1H), 2,36-2,59 (m, 2H), 1,93-1,98 (m, 1H). LCMS: 400,1 ([M+1]+).
Пример 62. Соединение A385
3-(4-((2-Фтор-3-(метиламино)бензил)окси)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A385:

Стадия A: к раствору (2-фтор-3-(метиламино)фенил)метанола в DCM (10 мл) добавляли SOCl2 (0,5 мл), смесь перемешивали в течение 4 ч. Реакционную смесь концентрировали с получением неочищенного продукта 3-(хлорметил)-2-фтор-N-метиланилина гидрохлорида (430 мг) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
1H ЯМР (DMSO-d6, 300 МГц): δ 9,97 (br s, 2H), 7,05 (t, J = 8,1 Гц, 1H), 6,84-6,92 (m, 2H), 4,72 (s, 2H), 2,74 (s, 3H).
Стадия B: к раствору A329B (300 мг, 1,03 ммоль) в DMF (10 мл) добавляли 3-(хлорметил)-2-фтор-N-метиланилина гидрохлорид (259 мг) и K2CO3 (355 мг, 2,57 ммоль), смесь перемешивали в течение ночи. LCMS продемонстрировала, что реакция не была завершена. Добавляли дополнительный 3-(хлорметил)-2-фтор-N-метиланилина гидрохлорид (150 мг) и K2CO3 (100 мг, 0,72 ммоль), и смесь перемешивали в течение ночи. Реакционную смесь концентрировали, а также добавляли EtOAc (20 мл) и H2O (10 мл). Водный слой экстрагировали с помощью EtOAc (20 мл x 2), объединенный органический слой промывали солевым раствором (20 мл), высушивали над Na2SO4, фильтровали, концентрировали с получением остатка. Остаток очищали с помощью преп-TLC (петролейный эфир / EtOAc = 1/4) с получением соединения A385A (242 мг, выход 55%) в виде белого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 7,55 (br s, 1H), 7,45 (t, J = 7,8 Гц, 1H), 7,27-7,33 (m, 2H), 7,15 (br s, 1H), 6,99 (t, J = 8,1 Гц, 1H), 6,61-6,71 (m, 2H), 5,59 (br s, 1H), 5,20 (s, 2H), 4,70 (dd, J = 10,5, 4,5 Гц, 1H), 4,47 (d, J = 17,7 Гц, 1H), 4,33 (d, J = 17,7 Гц, 1H), 3,48 (s, 3H), 2,71 (d, J = 4,2 Гц, 3H), 2,12-2,25 (m, 3H), 1,99-2,09 (m, 1H).
Стадия C: к раствору A385A (242 мг, 0,56 ммоль) в DMF (10 мл) добавляли K2CO3 (234 мг, 1,69 ммоль), смесь перемешивали в течение ночи при 80oC. Реакционную смесь концентрировали при сниженном давлении и остаток очищали с помощью преп-HPLC, затем сушили сублимацией с получением A385 (100 мг, выход 45%) в виде белого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 10,71 (br s, 1H), 7,50 (t, J = 8,1 Гц, 1H), 7,32-7,38 (m, 2H), 7,00 (t, J = 7,8 Гц, 1H), 6,63-6,72 (m, 2H), 5,61-5,62 (m, 1H), 5,23 (s, 2H), 5,10 (dd, J = 13,2, 5,1 Гц, 1H), 4,37 (d, J = 17,7 Гц, 1H), 4,21 (d, J = 17,7 Гц, 1H), 2,84-2,96 (m, 1H), 2,72 (d, J = 4,8 Гц, 3H), 2,51-2,60 (m, 1H), 2,37-2,47 (m, 1H), 1,93-2,00 (m, 1H). LCMS=398,1 ([M+1]+)
Соединения в примерах 63-66 получали в соответствии с процедурой, описанной для примера 62, с соответствующими исходными материалами для замены 3-(хлорметил)-2-фтор-N-метиланилина гидрохлорида на стадии B.
Пример 63. Соединение A390
3-(4-((2-Фтор-5-(метиламино)бензил)окси)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A390:
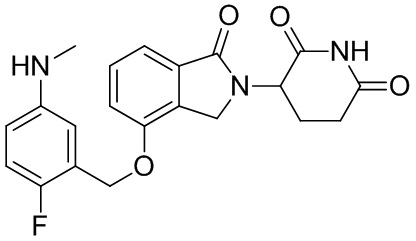
1H ЯМР (DMSO-d6, 300 МГц): δ 10,97 (s, 1H), 7,48 (t, J = 7,8 Гц, 1H), 7,33 (t, J = 7,2 Гц, 2H), 6,96 (t, J = 9,3 Гц, 1H), 6,63-6,66 (m, 1H), 6,46-6,51 (m, 1H), 5,58-5,63 (m, 1H), 5,16 (s, 2H), 5,06-5,12 (m, 1H), 4,35 (d, J = 17,4 Гц, 1H), 4,19 (d, J = 17,4 Гц, 1H), 2,83-2,95 (m, 1H), 2,54-2,62 (m, 4H), 2,34-2,45 (m, 1H), 1,91-1,99 (m, 1H). LCMS: 398,1 ([M+1]+).
Пример 64. Соединение A398
3-(4-((2-Фтор-5-(морфолинометил)бензил)окси)-1-оксоизоиндолин-2-ил)-пиперидин-2,6-дион, A398:

1H ЯМР (DMSO-d6, 300 МГц): δ 10,92 (br s, 1H), 7,46-7,51 (m, 2H), 7,31-7,37 (m, 3H), 7,16-7,22 (m, 1H), 5,27 (s, 2H), 5,05-5,11 (m, 1H), 4,35 (d, J = 17,4 Гц, 1H), 4,20 (d, J = 17,4 Гц, 1H), 3,50-3,53 (m, 4H), 3,43 (s, 2H), 2,82-2,92 (m, 1H), 2,53-2,60 (m, 1H), 2,34-2,44 (m, 1H), 2,23-2,29 (m, 4H), 1,90-2,00 (m, 1H). LCMS=468,2 [(M+1)+].
Пример 65. Соединение A399
3-(4-((2-Фтор-3-(морфолинометил)бензил)окси)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A399:

1H ЯМР (DMSO-d6, 300 МГц): δ 10,98 (s, 1H), 7,49-7,54 (m, 2H), 7,34-7,45 (m, 3H), 7,19-7,24 (m, 1H), 5,29 (s, 2H), 5,08-5,14 (m, 1H), 4,39 (d, J = 17,7 Гц, 1H), 4,22 (d, J = 17,7 Гц, 1H), 3,50-3,57 (m, 6H), 2,84-2,98 (m, 1H), 2,54-2,60 (m, 1H), 2,34-2,47 (m, 5H), 1,92-2,01 (m, 1H). LCMS: 468,2 ([M+1]+).
Пример 66. Соединение A407
3-(4-((2-Фтор-4-(морфолинометил)бензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A407:

Стадия A: к раствору A327A (300 мг, 0,85 ммоль) и 4-(4-(хлорметил)-3-фторбензил)морфолина гидрохлорида (359 мг, 1,28 ммоль) в DMF (15 мл) добавляли K2CO3 (352 мг, 2,55 ммоль), смесь перемешивали в течение ночи при 40oC. Реакционную смесь фильтровали и фильтрат концентрировали с получением остатка. Остаток очищали с помощью преп-TLC (MeOH / DCM = 1/15) с получением белого твердого вещества A407B (430 мг, выход 90%).
1H ЯМР (DMSO-d6, 300 МГц): δ 7,51-7,56 (m, 2H), 7,33 (dd, J = 11,4, 1,5 Гц, 1H), 7,17-7,21 (m, 3H), 7,07 (dd, J=7,5, 1,5 Гц, 1H), 5,24 (s, 2H), 4,66-4,69 (m, 1H), 4,44 (d, J = 17,7 Гц, 1H), 4,32 (d, J = 17,7 Гц, 1H), 3,55-3,58 (m, 4H), 3,48 (s, 2H), 2,29-2,39 (m, 4H), 2,05-2,18 (m, 3H), 1,95-2,01 (m, 1H), 1,29 (s, 9H).
Стадия B: к раствору A407B (430 мг, 0,77 ммоль) в DCM (20 мл) по каплям добавляли TFA (5 мл) при комнатной температуре, смесь перемешивали в течение 3 часов при 25oC. LCMS продемонстрировала завершение реакции. Реакционную смесь концентрировали при сниженном давлении (в вакууме) с получением A407C (387 мг, выход 100%) в виде желтого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 10,33 (br s, 1H), 7,71 (t, J = 7,8 Гц, 1H), 7,61 (s, 1H), 7,33-7,47 (m, 3H), 7,19 (s, 1H), 7,09-7,11 (m, 1H), 5,31 (s, 2H), 4,69-4,72 (m, 1 H), 4,48 (d, J = 17,7 Гц, 1H), 4,38 (s, 2H), 4,35 (d, J = 17,7 Гц, 1H), 3,89-3,99 (m, 2H), 3,57-3,67 (m, 2H), 3,14-3,34 (m, 4H), 2,10-2,17 (m, 3H), 1,95-2,01 (m, 1H).
Стадия C: к раствору A407C (215 мг, 0,39 ммоль) в CH3CN (15 мл) добавляли CDI (190 мг, 1,17 ммоль), смесь нагревали с обратным холодильником в течение ночи в атмосфере N2. LCMS продемонстрировала завершение реакции. Реакционную смесь концентрировали с получением остатка и очищали с помощью преп-HPLC с получением A407 (80 мг, выход 43%) в виде белого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 10,96 (br s, 1H), 7,51-7,56 (m, 1H), 7,34-7,39 (m, 1H), 7,11-7,21 (m, 3H), 5,25 (s, 2H), 5,05-5,11 (m, 1H), 4,33 (d, J = 18,0 Гц, 1H), 4,16 (d, J = 18,0 Гц, 1H), 3,54-3,57 (m, 4H), 3,48 (s, 2H), 2,82-2,94 (m, 1H), 2,49-2,56 (m, 1H), 2,30-2,44 (m, 5H), 1,91-1,99 (m, 1H). LCMS=486,2 ([M+1]+).
Пример 67. A403
(S)-3-Дейтерий-3-(4-((2-фтор-5-(морфолинометил)бензил)окси)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A403:
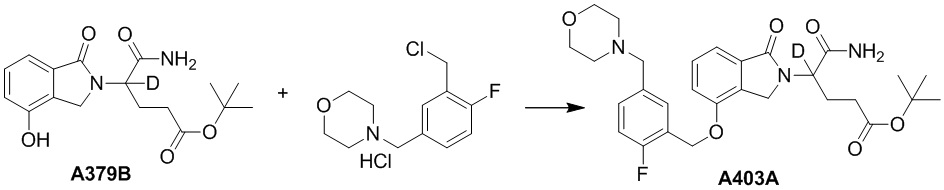
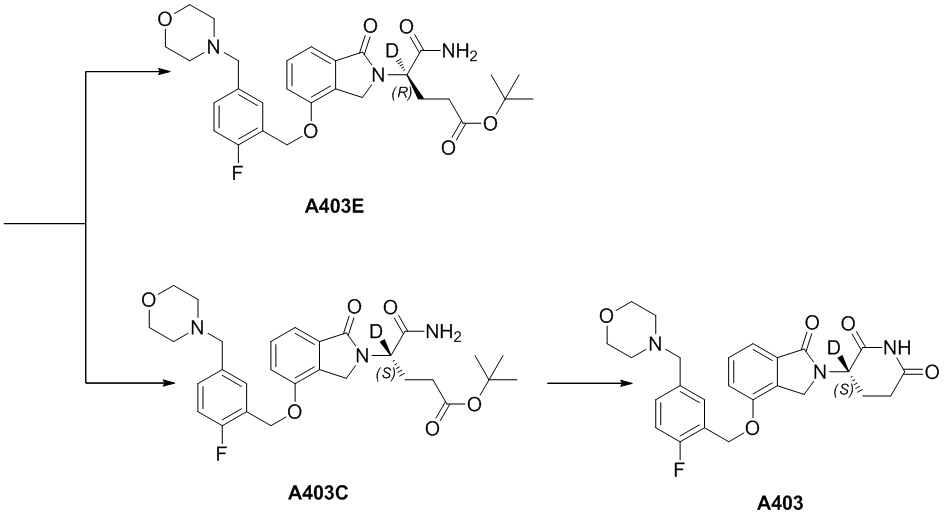
Стадия A: к раствору соединения A379B (1,0 г, 3,59 ммоль) и 4-(3-(хлорметил)-4-фторбензил)морфолина гидрохлорида (1,23 г, 7,05 ммоль) в DMF (20 мл) добавляли K2CO3 (972 мг, 7,04 ммоль), смесь перемешивали в течение ночи при 25℃. Фильтровали и фильтрат концентрировали с получением остатка. Остаток очищали с помощью колоночной хроматографии на силикагеле с получением белого твердого вещества A403A (1,3 г, выход 80%).
1H ЯМР (DMSO-d6, 300 МГц): δ 7,54 (br s, 1H), 7,43-7,49 (m, 2H), 7,28-7,34 (m, 3H), 7,14-7,22 (m, 2H), 5,26 (s, 2H), 4,67-4,72 (m, 0,05H), 4,50 (d, J = 17,4 Гц, 1H), 4,33 (d, J = 17,4 Гц, 1H), 3,49-3,56 (m, 4H), 3,44 (s, 2H), 2,25-2,34 (m, 4H), 2,09-2,15 (m, 3H), 1,94-2,03 (m, 1H), 1,29 (s, 9H).
Стадия B: хиральное разделение
Осуществляли хиральное разделение A403A с получением A403C (500 мг) и A403E (500 мг).
Условия хирального разделения Подвижная фаза: гексан / IPA = 70/30 (об./об.); концентрация образца: 100 мг/мл; колонка: CHIRALPAK IA; 30 мм (I.D) x 250 мм (L); 5 мкм, температура: 35oC; длина волны: 205 нм; введение: 250 мкл; расход: 50 мл/мин.
A403C
(S)-Трет-бутил-5-амино-4-дейтерий-4-(4-((2-фтор-5-(морфолинометил)бензил)окси)-1-оксоизоиндолин-2-ил)-5-оксопентаноат:

1H ЯМР (DMSO-d6, 300 МГц): δ 7,55 (br s, 1H), 7,43-7,49 (m, 2H), 7,28-7,34 (m, 3H), 7,16-7,22 (m, 2H), 5,26 (s, 2H), 4,66-4,71 (m, 1H), 4,49 (d, J = 17,4 Гц, 1H), 4,33 (d, J = 17,4 Гц, 1H), 3,51-3,53 (m, 4H), 3,43 (s, 23H), 2,26-2,33 (m, 4H), 2,02-2,16 (m, 3H), 1,94-1,99 (m, 1H), 1,29 (s, 9H).

Стадия C: к раствору A403C (500 мг, 1,0 ммоль) в DCM (12 мл), охлажденному до 0oC, по каплям добавляли TFA (3 мл), смесь медленно нагревали до 25oC и перемешивали в течение ночи. Реакционную смесь концентрировали при сниженном давлении (в вакууме). Остаток растворяли в CH2Cl2 (20 мл) и добавляли насыщ. NaHCO3 для регулирования значения pH=8-9, смесь концентрировали и остаток очищали с помощью флэш-хроматографии на C18 (CH3CN: H2O = 5-40%, 40 мин.) с получением A403D (400 мг, выход 82%) в виде светло-желтоватого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц):δ 7,93 (br s, 1H), 7,41-7,49 (m, 2H), 7,26-7,32 (m, 3H), 7,16-7,22 (m, 1H), 7,06 (br s, 1H), 5,25 (s, 2H), 5,05-5,13 (m, 0,00H), 4,60 (d, J = 17,7 Гц, 1H), 4,30 (d, J = 17,7 Гц, 1H), 3,50-3,52 (m, 4H), 3,42 (s, 2H), 2,22-2,32 (m, 4H), 2,03-2,10 (m, 1H), 1,81-1,94 (m, 3H).
Стадия D: к раствору A403D (400 мг, 0,82 ммоль) в сухом DMF (1 мл), сухом DCM (40 мл) и THF (20 мл), охлажденному до -40oC, в атмосфере N2 к смеси медленно добавляли SOCl2 (488 мг, 4,1 ммоль) при -40oC. Реакционную смесь перемешивали в течение 1 ч. и затем добавляли пиридин (324 мг, 4,1 ммоль), через 40 мин. добавляли Et3N (415 мг, 4,1 ммоль), а затем смесь перемешивали в течение 1 ч. LCMS продемонстрировала завершение реакции. Чтобы погасить реакцию, добавляли DCM (50 мл) и H2O (2 мл), водный слой экстрагировали с помощью DCM (50 мл x 2), объединенную органическую фазу промывали солевым раствором (50 мл), высушивали над Na2SO4, фильтровали, концентрировали с получением остатка. Неочищенный продукт очищали с помощью C18 (CH3CN : H2O = 5% - 45%, 40 мин.) с получением A403 (300 мг, выход 78%, ee: 99%) в виде белого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 10,96 (br s, 1H), 7,46-7,51 (m, 2H), 7,31-7,37 (m, 3H), 7,16-7,22 (m, 1H), 5,27 (s, 2H), 5,05-5,13 (m, 0,04H), 4,35 (d, J = 17,7 Гц, 1H), 4,19 (d, J = 17,7 Гц, 1H), 3,5,-3,53 (m, 4H), 3,43 (s, 2H), 2,82-2,94 (m, 1H), 2,49-2,58 (m, 1H), 2,36-2,41 (m, 1H), 2,26-2,32 (m, 4H), 1,91-1,98 (m, 1H). LCMS=469,2 ([M+1]+).
Соединения в примерах 68 и 69 получали в соответствии с процедурой, описанной для примера 67, с соответствующими исходными материалами для замены 4-(3-(хлорметил)-4-фторбензил)морфолина гидрохлорида.
Пример 68. Соединение A404
(S)-3-Дейтерий-3-(4-((2-фтор-3-(морфолинометил)бензил)окси)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A404:

1H ЯМР (DMSO-d6, 300 МГц): δ 10,94 (s, 1H), 7,45-7,53 (m, 2H), 7,31-7,42 (m, 3H), 7,17-7,21 (m, 1H), 5,28 (s, 2H), 4,37 (d, J = 18,0 Гц, 1H), 4,21 (d, J = 18,0 Гц, 1H), 3,51-3,62 (m, 6H), 2,82-2,95 (m, 1H), 2,57-2,62 (m, 1H), 2,28-2,42 (m, 5H), 1,91-2,01 (m, 1H). LCMS: 469,2 ([M+1]+).
Пример 69. Соединение A406
(S)-3-Дейтерий-3-(4-((2-фтор-4-(морфолинометил)бензил)окси)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A406:
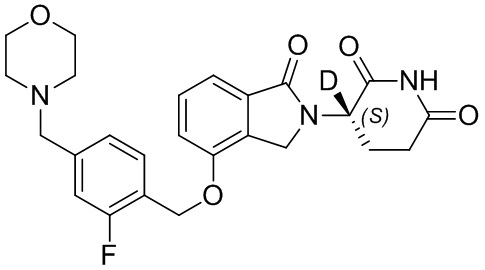
1H ЯМР (DMSO-d6, 300 МГц): δ 10,98 (s, 1H), 7,47-7,55 (m, 2H), 7,31-7,38 (m, 2H), 7,16-7,20 (m, 2H), 5,24 (s, 2H), 5,06-5,12 (m, 0,04H), 4,35 (d, J = 18,0 Гц, 1H), 4,19 (d, J = 18,0 Гц, 1H), 3,55 (br, 4H), 3,47 (s, 2H), 2,82-2,94 (m, 1H), 2,48-2,57 (m, 1H), 2,33-2,42 (m, 5H), 1,91-1,96 (m, 1H). LCMS: 469,2 ([M+1]+).
Пример 70. Соединение A400
(S)-3-Дейтерий-3-(4-((2-фтор-3-(морфолинометил)бензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A400:

Стадия A: к раствору соединения A356C (400 мг, 1,44 ммоль) в MeOH (30 мл) добавляли 2-фтор-3-(морфолинометил)бензальдегид (481 мг, 2,16 ммоль) и AcOH (0,5 мл). Реакционную смесь перемешивали в течение ночи при 30oC, затем добавляли Pd/C (150 мг, 10%, 50% воды) в атмосфере H2, смесь перемешивали в течение 3 часов, фильтровали и концентрировали с получением продукта A400A (580 мг).
1H ЯМР (DMSO-d6, 300 МГц): δ 7,57 (br s, 1H), 7,06-7,31 (m, 5H), 6,88-6,90 (m, 1H), 6,58-6,60 (m, 1H), 6,37 (s, 1H), 4,26-4,54 (m, 4H), 3,47-3,66 (m, 6H), 2,23-2,37 (m, 4H), 2,07-2,15 (m, 3H), 1,85-1,97 (m, 1H).
Стадия B: к раствору неочищенного A400A (480 мг, 0,99 ммоль) в DCM (20 мл), охлажденному до -40oC, добавляли DMF (1 мл), затем добавляли SOCl2(589 мг, 4,95 ммоль) и перемешивали в течение 2 часов, добавляли пиридин (383 мг, 4,95 ммоль), смесь перемешивали в течение 30 мин., а затем добавляли Et3N (501 мг, 4,95 ммоль). Реакционную смесь перемешивали в течение еще 1 часа при -40oC и затем гасили водой (80 мл), экстрагировали с помощью DCM (80 мл x 3). Объединенную органическую фазу высушивали над Na2SO4, фильтровали и концентрировали с получением неочищенного масла, которое очищали с помощью колоночного хроматографа на силикагеле (DCM / MeOH = 40/1) с получением продукта A400 (251 мг, 54%).
1H ЯМР (DMSO-d6, 300 МГц): δ 11,02 (s, 1H), 7,20-7,31 (m, 3H), 7,10 (t, J = 7,8 Гц, 1H), 6,95 (d, J = 7,5 Гц, 1H), 6,65 (d, J = 7,8 Гц, 1H), 6,29-6,32 (m, 1H), 5,09-5,15 (m, 0,05H), 4,43 (d, J = 5,1 Гц, 2H), 4,31 (d, J = 17,1 Гц, 1H), 4,19 (d, J = 17,1 Гц, 1H), 3,49-3,64 (m, 6H), 2,87-2,99 (m, 1H), 2,58-2,65 (m, 1H), 2,25-2,44 (m, 5H), 2,01-2,06 (m, 1H). LCMS: 468,2 ([M+1]+).
Соединения в примерах 71 и 72 получали в соответствии с процедурой, описанной для примера 70, с соответствующими исходными материалами для замены 2-фтор-3-(морфолинометил)бензальдегида на стадии A.
Пример 71. Соединение A401
(S)-3-Дейтерий-3-(4-((2-фтор-5-(морфолинометил)бензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион A401:

1H ЯМР (DMSO-d6, 400 МГц): δ 11,03 (br s, 1H), 7,32 (d, J = 6,8 Гц, 1H), 7,12-7,23 (m, 3H), 6,94 (d, J = 7,6 Гц, 1H), 6,62 (d, J = 8,0 Гц, 1H), 6,34 (t, J = 6,0 Гц, 1H), 5,11-5,16 (m, 0,4H), 4,42 (d, J = 5,6 Гц, 2H), 4,31 (d, J = 16,8 Гц, 1H), 4,20 (d, J = 17,2 Гц, 1H), 3,42-3,49 (m, 4H), 3,37 (s, 2H), 2,89-2,98 (m, 1H), 2,58-2,67 (m, 1H), 2,28-2,35 (m, 1H), 2,18-2,26 (m, 4H), 2,02-2,06 (m, 1H). LCMS: 468,2 [(M+1)+].
Пример 72. Соединение A402
(S)-3-Дейтерий-3-(4-((2-фтор-4-(морфолинометил)бензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A402:
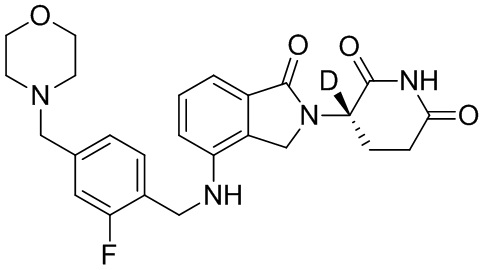
1H ЯМР (DMSO-d6, 300 МГц): δ 11,01 (s, 1H), 7,30-7,35 (m, 1H), 7,22 (t, J = 7,8 Гц, 1H), 7,05-7,13 (m, 2H), 6,92 (d, J = 7,5 Гц, 1H), 6,64 (d, J = 8,1 Гц, 1H), 6,27-6,31 (m, 1H), 5,08-5,14 (m, 0,05H), 4,38 (d, J = 5,4 Гц, 2H), 4,28 (d, J = 17,4 Гц, 1H), 4,16 (d, J = 17,4 Гц, 1H), 3,54 (br s, 4H), 3,42 (s, 2H), 2,85-2,97 (m, 1H), 2,56-2,62 (m, 1H), 2,24-2,31 (m, 5H), 1,98-2,06 (m, 1H). LCMS: 468,2 ([M+1]+).
Пример 73. Соединение A405
3-(6-Фтор-4-((4-(морфолинометил)бензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A405:

Стадия A: к раствору DMSO (156 мг, 2,0 ммоль) в DCM (10 мл) при -70oC в атмосфере N2добавляли (COCl)2 (152 мг, 1,2 ммоль). Реакционную смесь перемешивали в течение 30 минут при -70oC. Добавляли раствор (4-(морфолинометил)фенил)метанола (207 мг, 1,0 ммоль) в DCM (3 мл) и реакционную смесь перемешивали в течение 1 часа. К реакционной смеси по каплям добавляли Et3N (405 мг, 4,0 ммоль) и смесь перемешивали в течение 1 часа при -70oC, затем обеспечивали повышение температуры до 25oC, реакционную смесь гасили с помощью H2O (10 мл) и добавляли раствор NaHCO3 (5 мл). Смесь разделяли и водный слой экстрагировали с помощью 10 мл DCM. Объединенный органический слой концентрировали и очищали посредством колоночной хроматографии (PE : EtOAc = 2:1) с получением A405A (180 мг, выход 88%) в виде светло-желтого масла.
1H ЯМР (CDCl3, 300 МГц): δ 9,99 (s, 1H), 7,84 (d, J = 7,8 Гц, 2H), 7,51 (d, J = 8,1 Гц, 2H), 3,70-3,73 (m, 4H), 3,57 (s, 2H), 2,46 (t, J = 4,2 Гц, 4H).
Стадия B: к раствору A405A (111 мг, 0,54 ммоль) и I-28 (100 мг, 0,36 ммоль) в DCM (6 мл) добавляли HOAc (6 мл) и реакционную смесь перемешивали в течение 3 часов при 25oC. Добавляли NaBH3CN(45 мг, 0,72 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли дополнительное A405A (40 мг, 0,14 ммоль) и смесь перемешивали при 40oC в течение 6 часов. Растворитель удаляли и добавляли раствор NaHCO3 (10 мл) и DCM (25 мл), и разделяли. Водный слой экстрагировали с помощью DCM (20 мл x 2). Объединенный органический слой концентрировали и очищали с помощью преп-HPLC, затем сушили сублимацией с получением твердого вещества, которое добавляли к 5 мл насыщ. раствора NaHCO3 для регулирования значения pH=8, а затем экстрагировали с помощью DCM (5 мл x 5), органический раствор объединяли, концентрировали с получением A405 (50 мг, выход 30%) в виде белого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 11,01 (s, 1H), 7,23-7,33 (m, 4H), 6,72 (br s, 1H), 6,60 (dd, J = 1,8 Гц, 7,5 Гц, 1H), 6,42 (dd, J = 2,1 Гц, 12,6 Гц, 1H), 5,09 (dd, J = 5,1 Гц, 13,2 Гц, 1H), 4,35 (d, J = 5,4 Гц, 2H), 4,27 (d, J = 17,4 Гц, 1H), 4,14 (d, J = 17,4 Гц, 1H), 3,53 (br s, 4H), 3,41 (s, 2H), 2,84-2,96 (m, 1H), 2,57-2,63 (m, 1H), 2,21-2,31 (m, 5H), 2,01-2,05 (m, 1H). LCMS: 467,2 ([M+1]+).
Пример 74. Соединение A386
3-(4-((2-Фтор-4-(морфолинометил)бензил)окси)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A386:

Стадия A: к раствору (2-фтор-4-(морфолинометил)фенил)метанола (1,0 г, 4,4 ммоль) в хлороформе (25 мл) добавляли SOCl2 (1,1 г, 9,2 ммоль) и смесь нагревали до температуры флегмы в течение 2 часов. Растворитель удаляли в вакууме и совместно выпаривали с хлороформом (25 мл x 2) с получением A386A (1,2 г, выход 97%) в виде белого твердого вещества.
1H ЯМР (DMSO-d6, 400 МГц): δ 11,98 (br s, 1H), 7,70 (d, J = 10,8 Гц, 1H), 7,63 (t, J = 8,0 Гц, 1H), 7,70 (d, J =7,6 Гц, 1H), 4,82 (s, 2H), 4,37 (d, J = 4,8 Гц, 2H), 3,86-3,93 (m, 4H), 3,07-3,21 (m, 4H).
Стадия B: смесь A386A и A386B (0,8 г, 2,4 ммоль), K2CO3 (1,3 г, 9,6 ммоль) в DMF (20 мл) дегазировали с помощью N2, а также нагревали до 40oC и перемешивали в течение 18 часов. Реакционную смесь вливали в 100 мл ледяной воды и экстрагировали с помощью EtOAc (20 мл x 5). Объединенный органический слой промывали с помощью 20 мл воды, 20 мл солевого раствора и высушивали над Na2SO4, фильтровали, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (PE : EtOAc = 2:1-1:1) с получением A386C (1,2 г, выход 92%) в виде белого твердого вещества.
1H ЯМР (CDCl3, 300 МГц): δ 7,37-7,47 (m, 3H), 7,10-7,16 (m, 3H), 6,30 (br s, 1H), 5,33 (br s, 1H), 5,18 (s, 2H), 4,86-4,91 (m, 1H), 4,36-4,51 (m, 2H), 3,71-3,74 (m, 4H), 3,51 (s, 2H), 2,45-2,48 (m, 4H), 2,09-2,40 (m, 4H), 1,42 (s, 9H).
Стадия C: к раствору A386C (1,2 г, 2,2 ммоль) в DCM (30 мл) добавляли TFA (15 мл) и перемешивали при 35oC в течение 2 часов. Реакционную смесь концентрировали и очищали с помощью преп-HPLC с получением A386E (1,4 г, выход 64%) в виде светло-желтого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 12,05 (br s, 1H), 7,56 (br s, 2H), 7,47 (t, J = 8,1 Гц, 1H), 7,29-7,36 (m, 4H), 7,15-7,22 (m, 1H), 5,25 (s, 2H), 4,68-4,73 (m, 1H), 4,50 (d, J = 17,7 Гц, 1H), 4,36 (d, J = 17,7 Гц, 1H), 3,56-3,60 (m, 6H), 2,26-2,45 (m, 2H), 1,94-2,16 (m, 4H), 1,72-1,77 (m, 2H).
Стадия D: к раствору A386E (421 мг, 0,867 ммоль) в DCM / THF (50 мл/5 мл) при -40oC добавляли SOCl2 (516 мг, 4,33 ммоль, в виде раствора в 10 мл DCM). Смесь перемешивали при от -40oC до -20oC в течение 2 часов и добавляли пиридин(339 мг, 4,33 ммоль), и реакционную смесь перемешивали при -40oC в течение 0,5 часа. Добавляли Et3N (438 мг, 4,33 ммоль) и медленно обеспечивали нагревание смеси до 25oC. Добавляли H2O (0,5 мл) и затем фильтровали. Отфильтрованный осадок растворяли с помощью CH3CN (5 мл), а нерастворенное твердое вещество отфильтровывали и удаляли CH3CN с получением неочищенного продукта. Слой DCM реакционной смеси промывали водой (25 мл x 2) и солевым раствором (25 мл), и концентрировали с получением дополнительного неочищенного продукта. Объединенный неочищенный продукт очищали дважды с помощью преп-HPLC с получением A386 (105 мг, выход 26%) в виде белого твердого вещества.
1H ЯМР (DMSO-d6, 300 МГц): δ 10,95 (s, 1H), 7,49-7,58 (m, 2H), 7,34-7,40 (m, 2H), 7,17-7,21 (m, 2H), 5,26 (s, 2H), 5,07-5,13 (m, 1H), 4,38 (d, J = 17,7 Гц, 1H), 4,22 (d, J = 17,7 Гц, 1H), 3,58 (br s, 4H), 3,49 (br s, 2H), 2,84-2,96 (m, 1H), 2,56-2,60 (m, 1H), 2,30-2,43 (m, 5H), 1,92-2,02 (m, 1H).
Пример 75. Соединение A425
3-Дейтерий-3-(4-((2-фтор-5-(морфолинометил)бензил)окси)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A425:
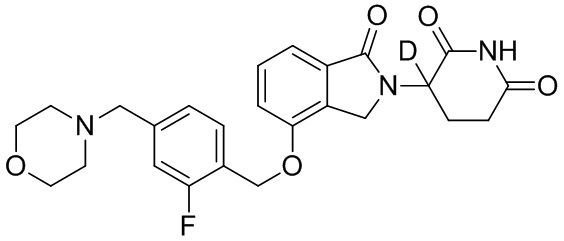
Соединение A425 в примере 75 получали в соответствии с процедурой, описанной в примере 67, с рацемическим A403A в качестве подходящего исходного материала.
1H ЯМР (DMSO-d6, 300 МГц): δ 10,95 (s, 1H), 7,49-7,57 (m, 2H), 7,33-7,40 (m, 2H), 7,18-7,22 (m, 2H), 5,26 (s, 2H), 4,37 (d, J = 17,7 Гц, 2H), 4,21 (d, J = 17,7 Гц, 1H), 3,58-3,62 (m, 4H), 3,49 (s, 2H), 2,84-2,96 (m, 1H), 2,27-2,58 (m, 6H), 1,93-1,99 (m, 1H). LCMS=469,2 ([M+1]+).
Пример 76. Соединение A427
3-Дейтерий-3-(4-((4-((2,6-диметилморфолино)метил)-2-фторбензил)окси)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A427:

Соединение A427 в примере 76 получали в соответствии с процедурой, описанной в примере 67, с подходящим исходным материалом для замены 4-(3-(хлорметил)-4-фторбензил)морфолина гидрохлорида.
1H ЯМР (DMSO-d6, 300 МГц): δ 10,97 (s, 1H), 7,49-7,58 (m, 2H), 7,34-7,41 (m, 2H), 7,17-7,21 (m, 2H), 5,26 (s, 2H), 4,38 (d, J = 17,7 Гц, 1H), 4,21 (d, J = 17,7 Гц, 1H), 3,54-3,61 (m, 2H), 3,47 (s, 2H), 2,84-2,96 (m, 1H), 2,53-2,68 (m, 3H), 2,38-2,44 (m, 1H), 1,93-1,99 (m, 1H), 1,66 (t, J = 10,5 Гц, 2H), 1,02 (d, J = 6,0 Гц, 6H). LCMS: 497,2 ([M+1]+).
Пример 77. Соединение A426
3-Дейтерий-3-(4-((2-фтор-4-(морфолинометил)бензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A426:
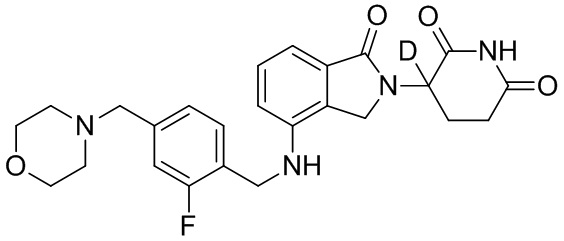
Соединение A426 в примере 77 получали в соответствии с процедурой, описанной в примере 70, с рацемическим A400A в качестве подходящего исходного материала.
1H ЯМР (DMSO-d6, 300 МГц): δ 11,00 (s, 1H), 7,32 (t, J = 8,1 Гц, 1H), 7,22 (t, J = 7,8 Гц, 1H), 7,05-7,13 (m, 2H), 6,93 (d, J = 7,5 Гц, 1H), 6,64 (d, J = 8,1 Гц, 1H), 6,27 (t, J = 6,0 Гц, 1H), 5,07-5,13 (m, 0,01H), 4,38 (d, J = 5,4 Гц, 2H), 4,28 (d, J = 17,1 Гц, 1H), 4,16 (d, J = 17,1 Гц, 1H), 3,52-3,55 (m, 4H), 3,42 (s, 2H), 2,85-2,97 (m, 1H), 2,56-2,65 (m, 1H), 2,23-2,35 (m, 5H), 1,99-2,06 (m, 1H). LCMS: 468,2 ([M+1]+).
Пример 78. Соединение A428
3-Дейтерий-3-(4-((4-((2,6-диметилморфолино)метил)-2-фторбензил)амино)-1-оксоизоиндолин-2-ил)пиперидин-2,6-дион, A428:

Соединение A428 в примере 78 получали в соответствии с процедурой, описанной в примере 70, с подходящим исходным материалом для замены 2-фтор-3-(морфолинометил)бензальдегида на стадии A.
1H ЯМР (DMSO-d6, 300 МГц): δ 11,02 (s, 1H), 7,34 (t, J = 7,8 Гц, 1H), 7,24 (t, J = 7,8 Гц, 1H) 7,06-7,13 (m, 2H), 6,95 (d, J = 7,5 Гц, 1H), 6,67 (d, J = 7,8 Гц, 1H), 6,28 (t, J = 5,7 Гц, 1H), 4,40 (d, J = 5,1 Гц, 2H), 4,30 (d, J = 17,1 Гц, 1H), 4,18 (d, J = 17,1 Гц, 1H), 3,52-3,61 (m, 2H), 3,42 (s, 2H), 2,87-2,99 (m, 1H), 2,59-2,65 (m, 3H), 2,35-2,25 (m, 1H), 2,01-2,06 (m, 1H), 1,63 (t, J = 10,5 Гц, 2H), 1,01 (d, J = 6,0 Гц, 6H). LCMS: 496,2 ([M+1]+).
Примеры эффекта
Анализ ингибирования активности TNF-α
Собирали периферическую кровь от здоровых волонтеров, а собирали с помощью пробирок с антикоагулянтом EDTA. После 5-кратного разбавления средой 1640 (Gibco, № по каталогу 11875-093, США), кровь добавляли в 96-луночные планшеты для культуры клеток (Costar, № по каталогу 3599, США), а затем обрабатывали с помощью 10 мкл раствора соединения общей формулы (I) по настоящему изобретению в DMSO (Sigma, № по каталогу D2650, США). Конечная концентрация соединения составляла 100 нМ, а конечная концентрация DMSO составляла 0,2%. После инкубации в течение 60 минут в инкубаторе при 37°C в 5% CO2, 10 мкл LPS (Sigma, № по каталогу L-2880, США) добавляли в реакционную систему и конечная концентрация составляла 10 нг/мл. После дополнительного культивирования в течение 6 часов в инкубаторе при 37°C в 5% CO2, собирали надосадочную жидкость. Содержание TNF-α определяли с помощью ELISA (BD Biosciences, № по каталогу 555212, США). Коэффициент поглощения определяли при OD450 нм с помощью планшет-ридера, с OD 650 нм в качестве стандарта. Контроль, раствор, содержащий 0,2% среду DMSO, представлял собой 0% ингибирование. Записывали исходные данные и стандартные кривые. Кривую ингибирования с четырьмя параметрами лекарственного средства строили с помощью программного обеспечения XL-fit и рассчитывали скорость ингибирования каждого соединения, как показано в таблице 1.

Анализ пролиферации клеток
Клетки MM.1S (клетки миеломы) (ATCC, № по каталогу CRL-2974) высевали 1,8 x 103 на лунку в 96-луночный планшет для культуры клеток, содержащий среду RPMI-1964 (Gibco, № по каталогу A10491-01), и инкубировали в инкубаторе в течение 24 часов при 37°C в 5% CO2. Соединения получали в виде 20 мМ исходных растворов с DMSO (Sigma, № по каталогу D2650) и разбавляли средой до требуемой концентрации (конечная концентрация DMSO составляла 0,5%), а затем добавляли в каждую лунку, инкубировали в инкубаторе в течение 72 часов при 37°C в 5% CO2. Затем 20 мкл MTS (Promega, № по каталогу G3581) добавляли в каждую лунку и дополнительно инкубировали в течение 1-4 часов в инкубаторе при 37°C в 5% CO2. OD490 нм определяли с использованием OD650 нм в качестве стандарта. Контроль, раствор, содержащий 0,5% среду DMSO, представлял собой 0% ингибирование. Использовали GraphPad Prism 5, обеспечивали изменение углового коэффициента с целью получения кривой зависимости доза-эффект и рассчитывали значения IC50, более подробно показанные в таблице 2.

Примечание: A: <300 нМ; B : ≥300 нМ
Анализ пролиферации клеток CTG
Клетки Rec-1 (клетки лимфомы из клеток мантийной зоны) (ATCC, № по каталогу CRL-3004), клетки Namalwa.CSN/70 (клетки лимфомы Беркитта) (DSMZ, № по каталогу ACC-70) и клетки WSU-DLCL-2 (клетки диффузной В-крупноклеточной лимфомы) (DSMZ, № по каталогу ACC-575) высевали (5-15) x 103 на лунку в 96-луночный планшет с прозрачным дном и белыми стенками (Corning, № по каталогу CLS3903), содержащий определенную среду. Планшет помещали в инкубатор и инкубировали в течение 24 часов при 37°C в 5% CO2. Соединения получали в виде 150 мМ исходных растворов с DMSO (Sigma, № по каталогу 276855) и разбавляли средой до требуемой концентрации (конечная концентрация DMSO составляла 0,2%), а затем добавляли в каждую лунку, инкубировали в инкубаторе в течение 72-120 часов при 37°C в 5% CO2. Затем в каждую лунку добавляли 100 мкл реактива для анализа активности клеток CellTiter-Glo® (Promega, № по каталогу G7570). Перемешивание продолжали в течение 10 минут на шейкере, чтобы вызвать цитолиз. 96-Луночный планшет оставляли при комнатной температуре на 10 минут, чтобы сделать люминесцентный сигнал стабильным. Белую пленочную основу приклеивали на дно культурального планшета. Для тестирования планшета использовали EnSpire. Данные обрабатывали с помощью программного обеспечения XLfit и получали значения IC50, и более подробно они показаны в таблице 3.

Примечание: A: <100 нМ; B: 100-400 нМ; C: 401 нМ - 300 мкМ; D: >300 мкМ.
Следует понимать, что вышеизложенное описание предпочтительных вариантов осуществления предназначено исключительно для иллюстрации принципов настоящего изобретения, а не для полного его описания, и что изменения и варианты будут очевидны для специалистов в данной области техники, и что настоящее изобретение не предназначено для ограничения, кроме явно изложенного в следующей формуле изобретения.
Реферат
Изобретение относится к соединениям формулы (I):,при этом в общей формуле (I) n1 выбран из 0 или 1; Z представляет собой, где атом углерода, помеченный *, представляет собой центр асимметрии; каждый из R, R, R, R, R, R, Rи Rнезависимо выбран из H или D; Rвыбран из H, D или F; каждый из Lи Lнезависимо выбран из CD, CHD или CH; Xвыбран из NH, ND или O; Rпредставляет собой H, D или, где каждый из R’, R’, R’ и R’ независимо выбран из H, D, галогена, циано, гидрокси,,,,, замещенного или незамещенного (C-C)алкила, замещенного или незамещенного (C-C)алкокси, (C-C)гетероциклоалкила или дейтерированного (C-C)гетероциклоалкила; где каждый из Rи Rнезависимо представляет собой H, (C-C)алкил, (C-C)алкилацил; каждый из Rи Rнезависимо представляет собой H или (C-C)алкил; Rпредставляет собой; причем каждый из Rи Rнезависимо представляет собой H или (C-C)алкил; которые способны регулировать активность иммунологических цитокинов, таким образом эффективно излечивая рак и воспалительное заболевание. 5 н. и 9 з.п. ф-лы, 3 табл., 78 пр.
Формула
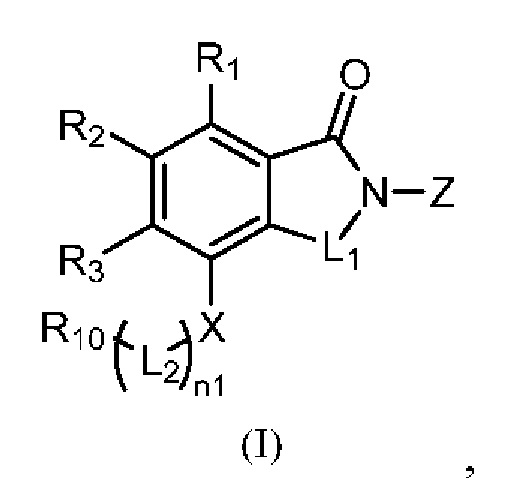




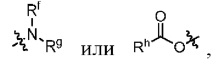

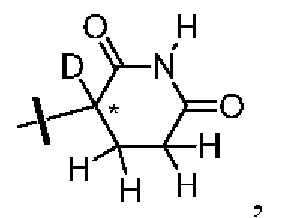
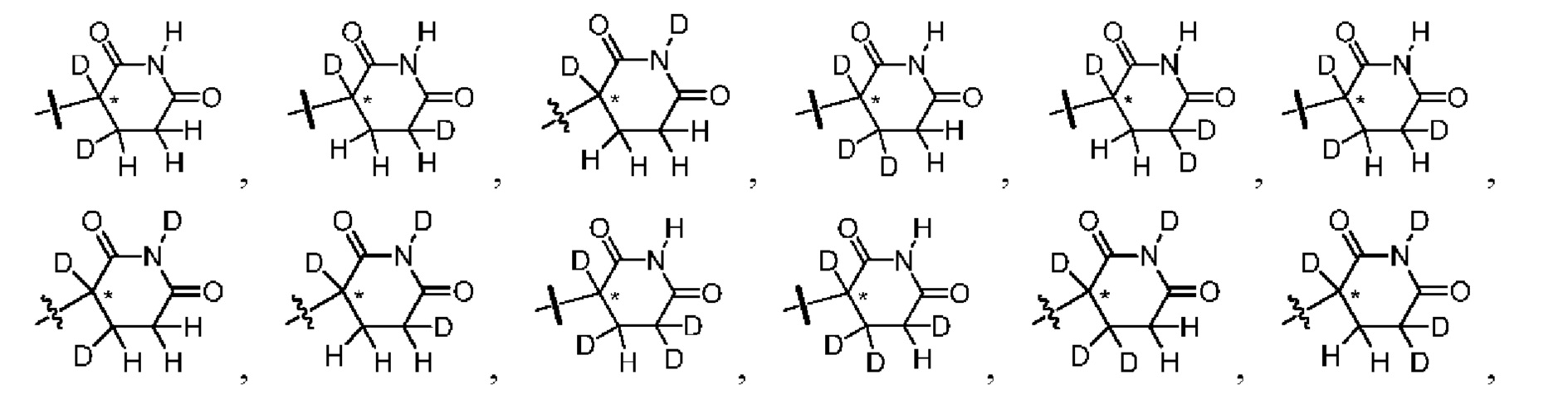



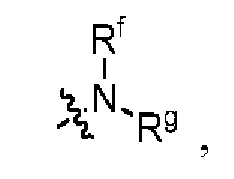




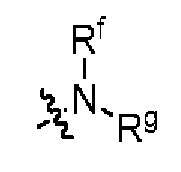



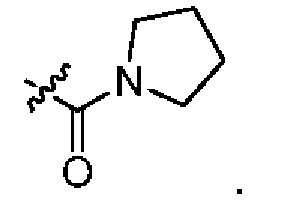


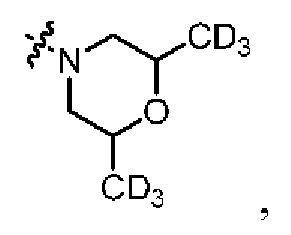
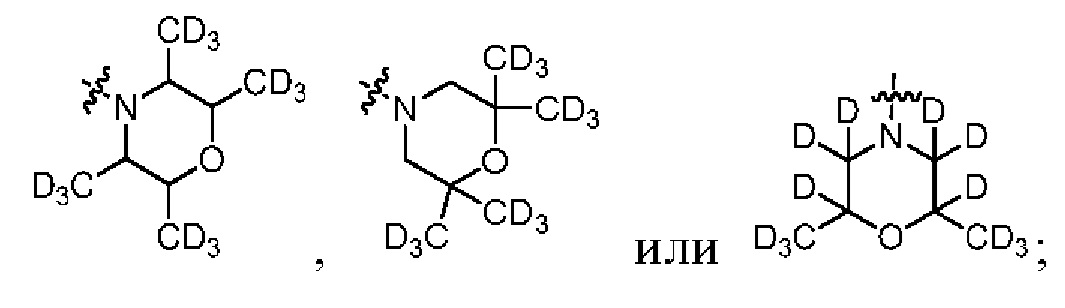

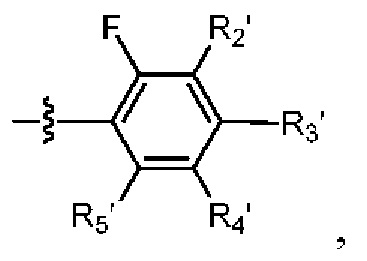




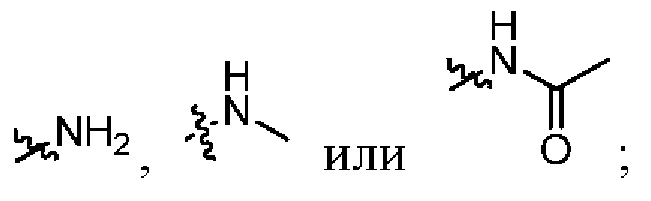
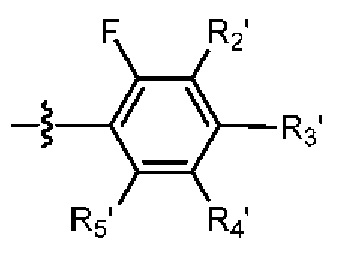




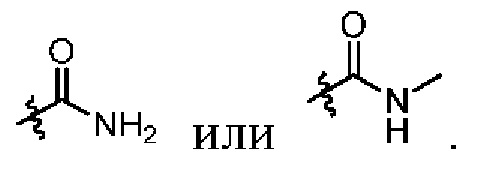

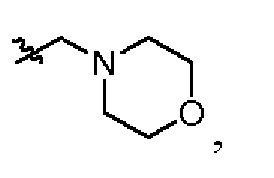


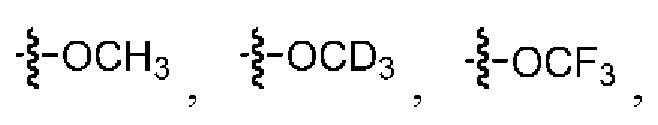

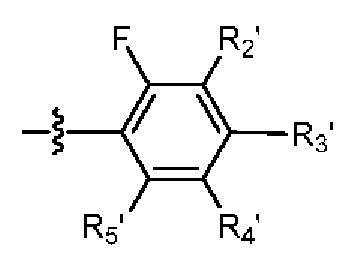

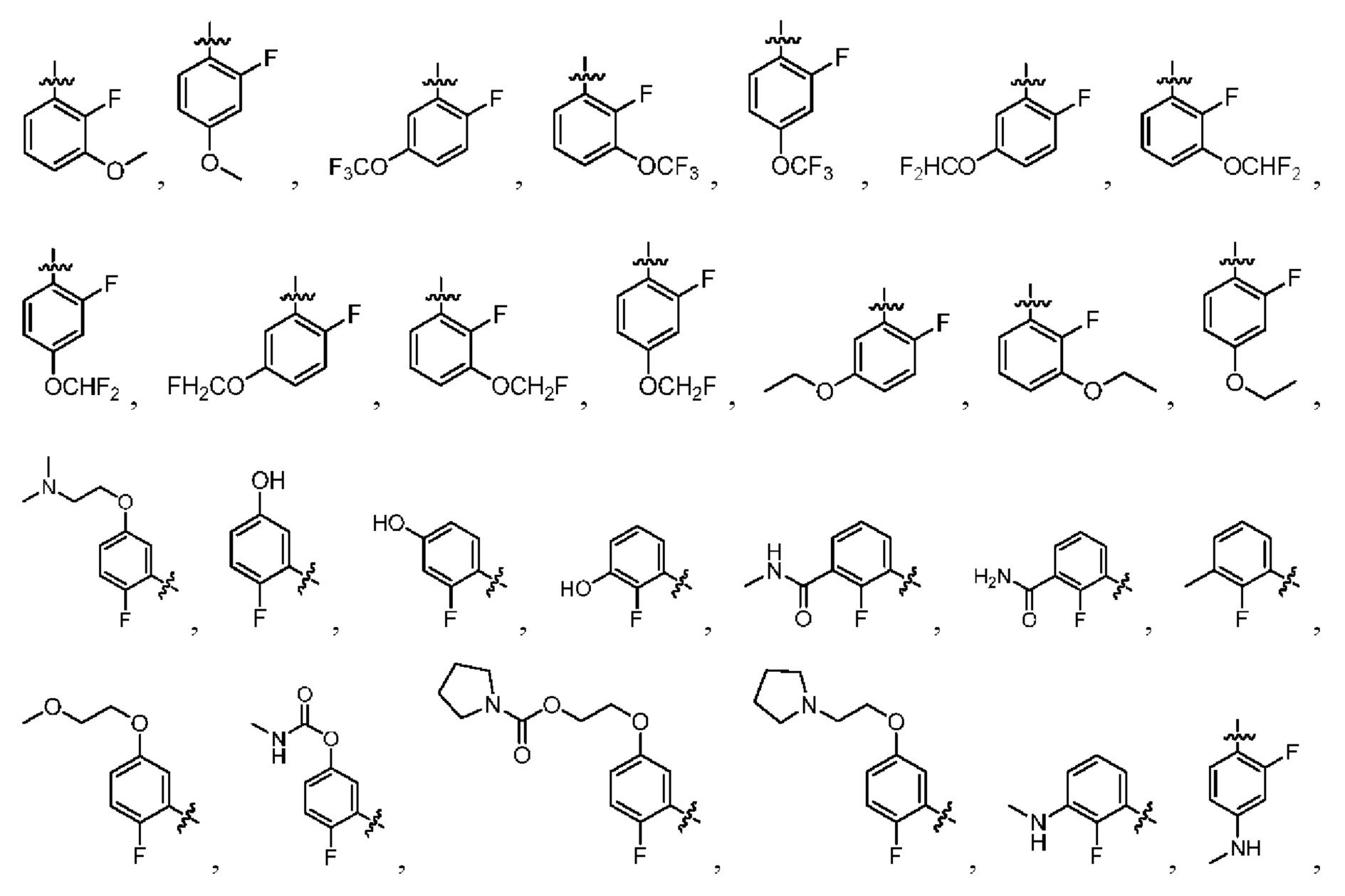














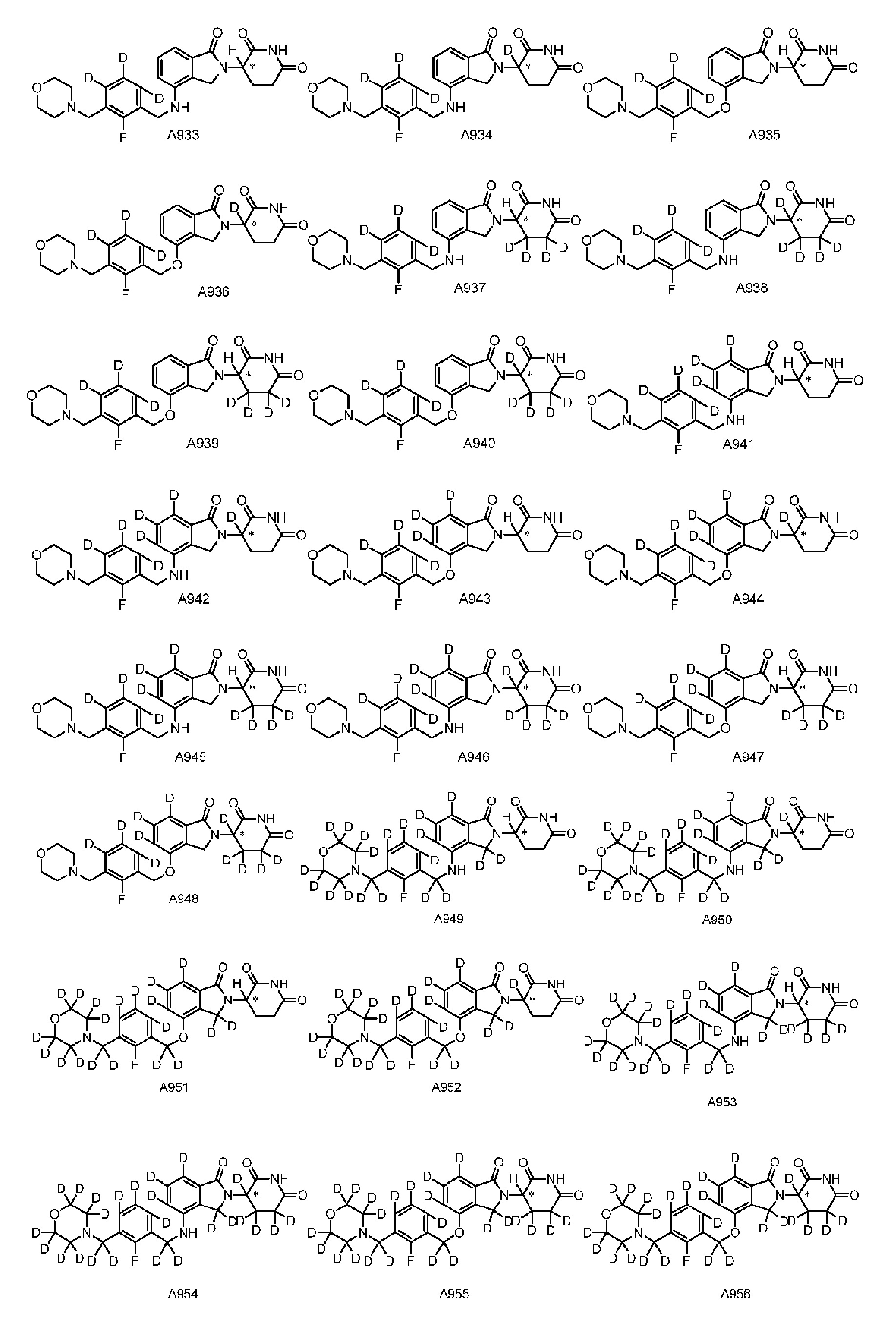


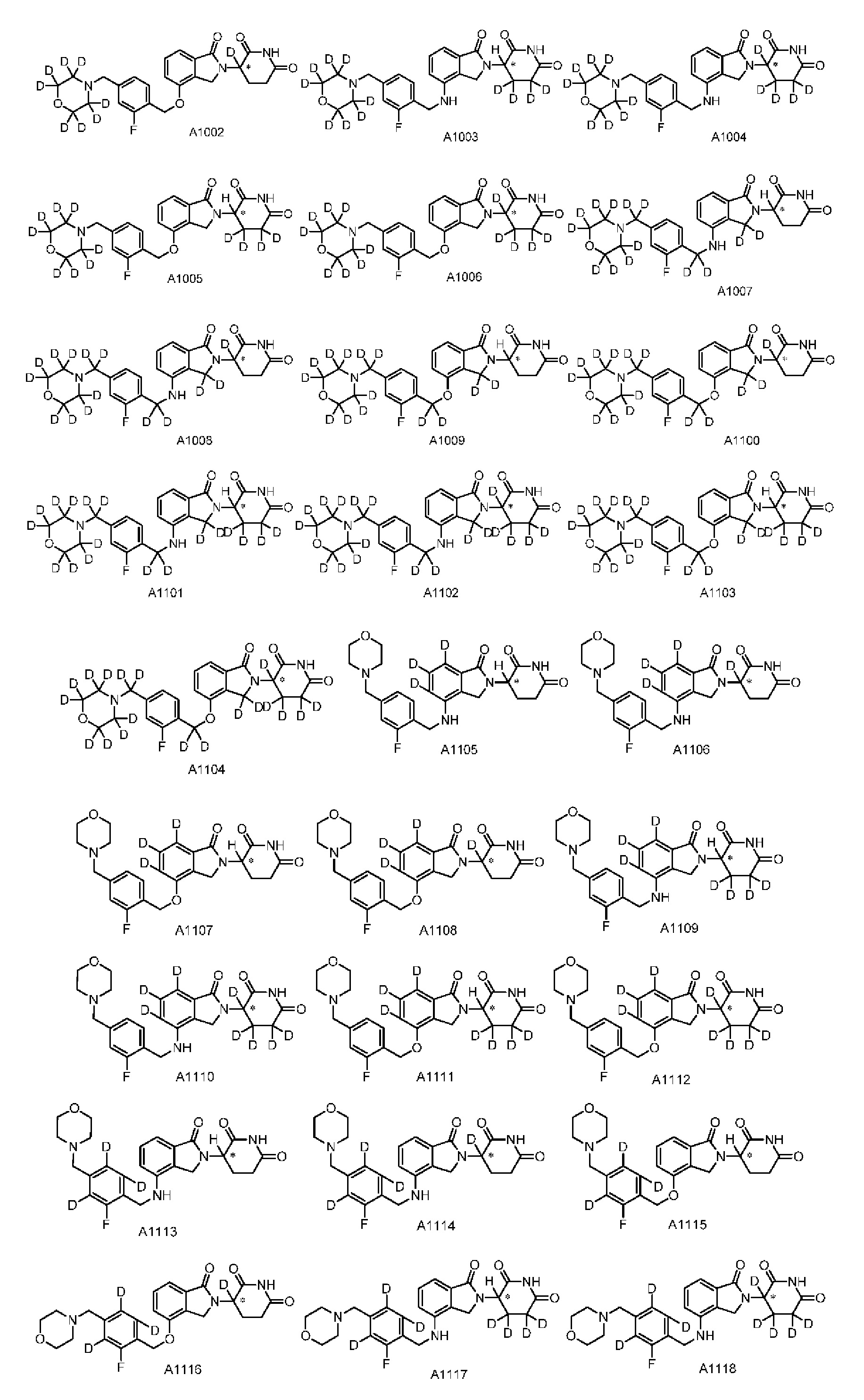

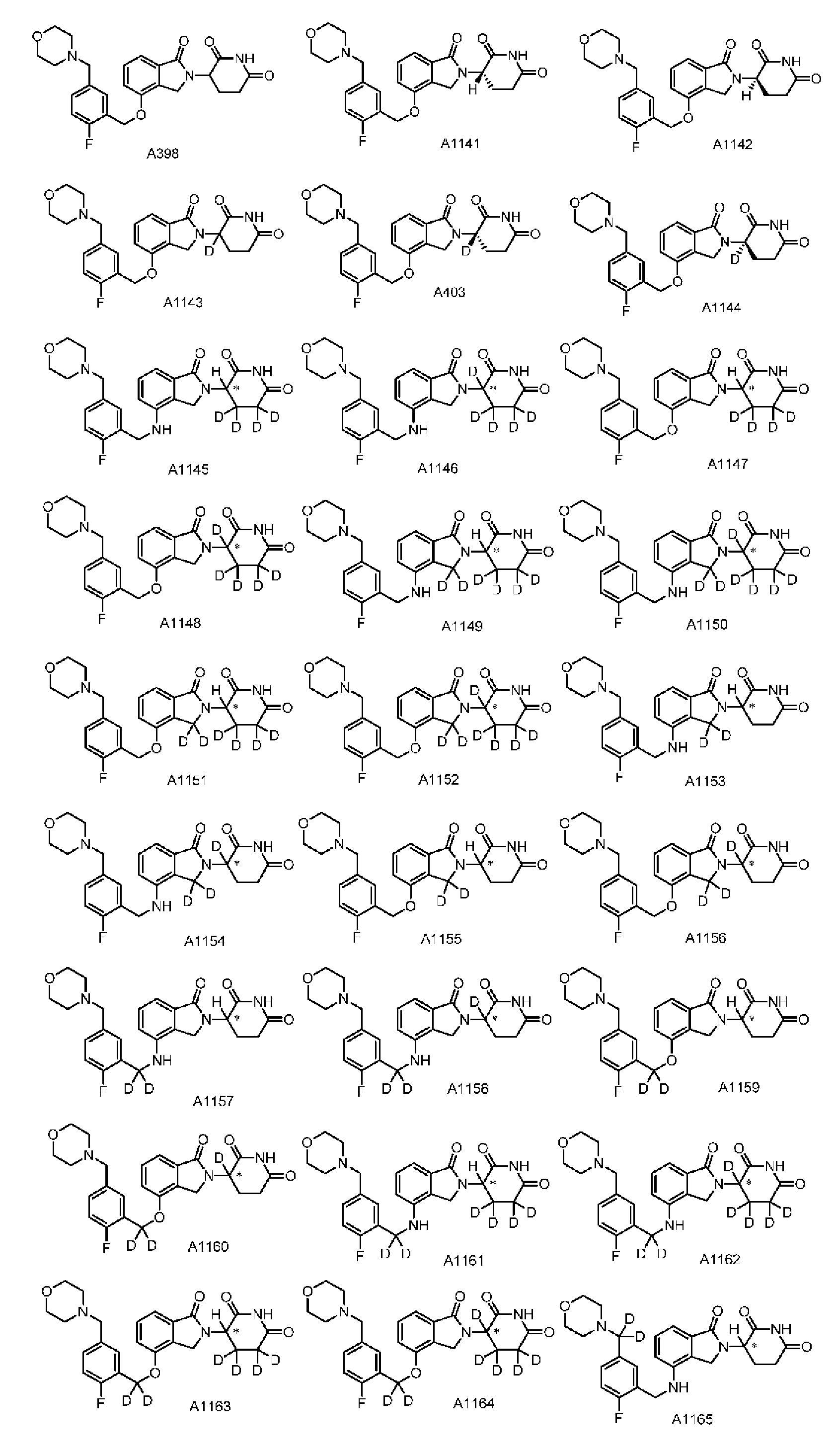

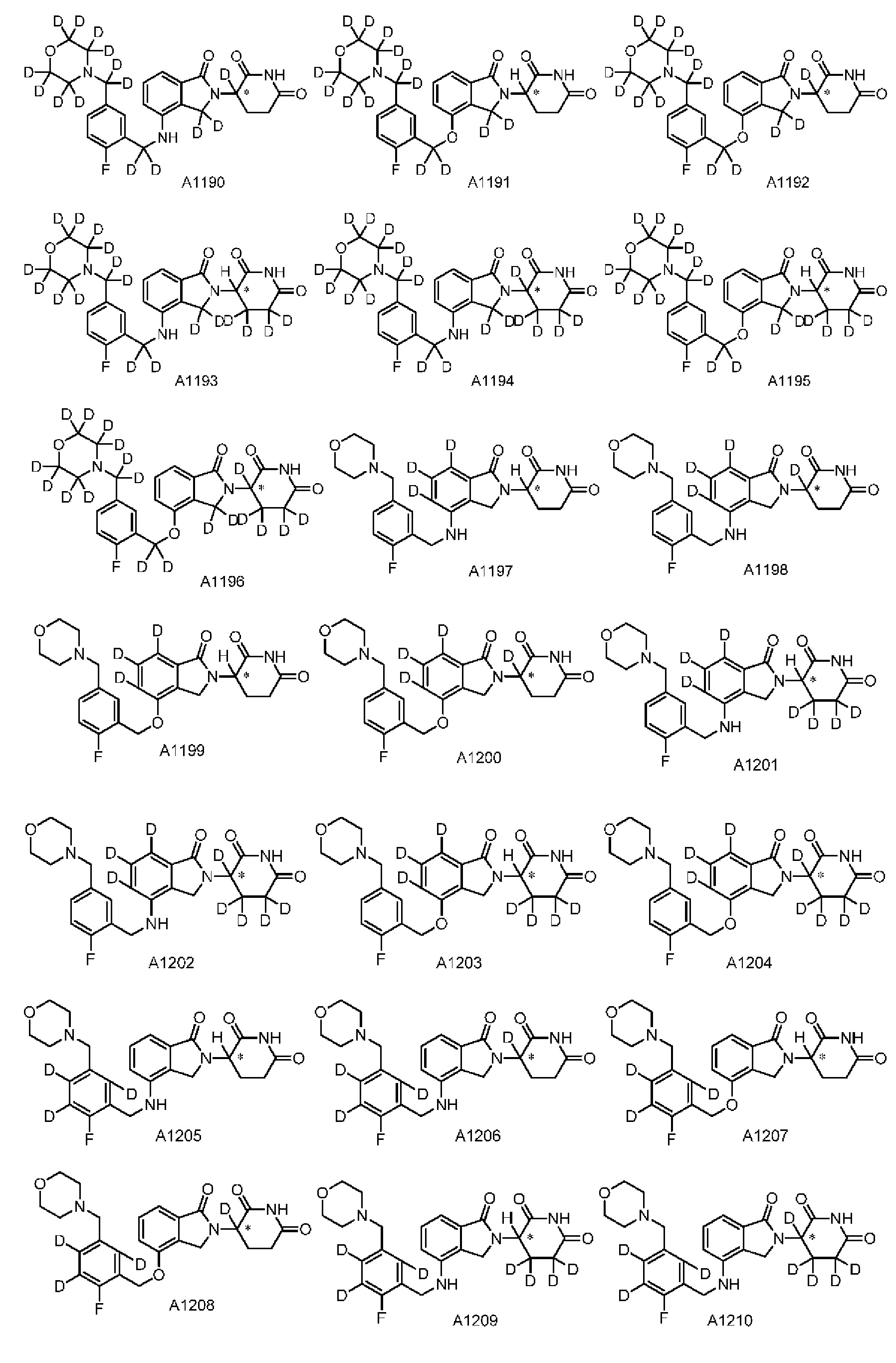

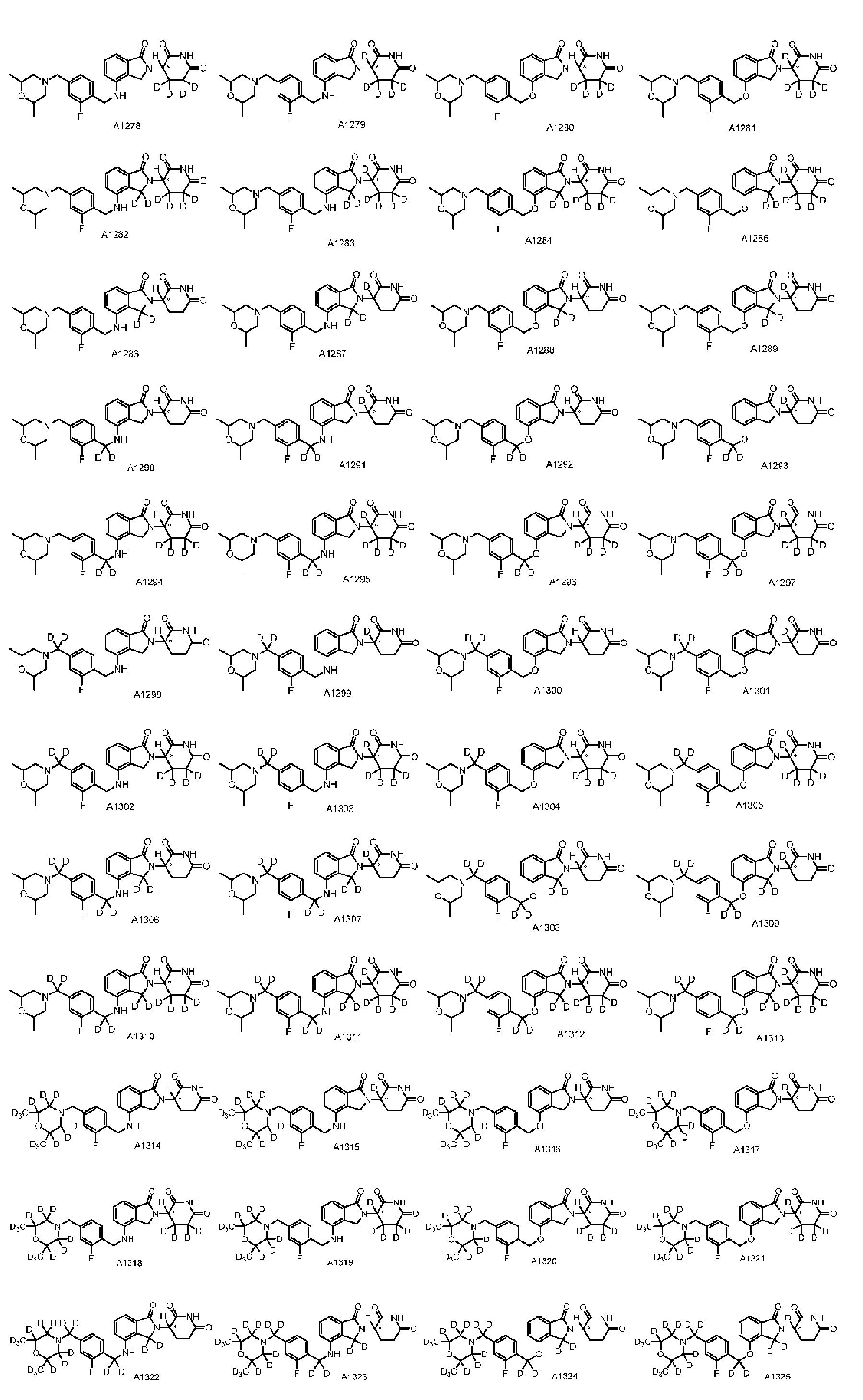


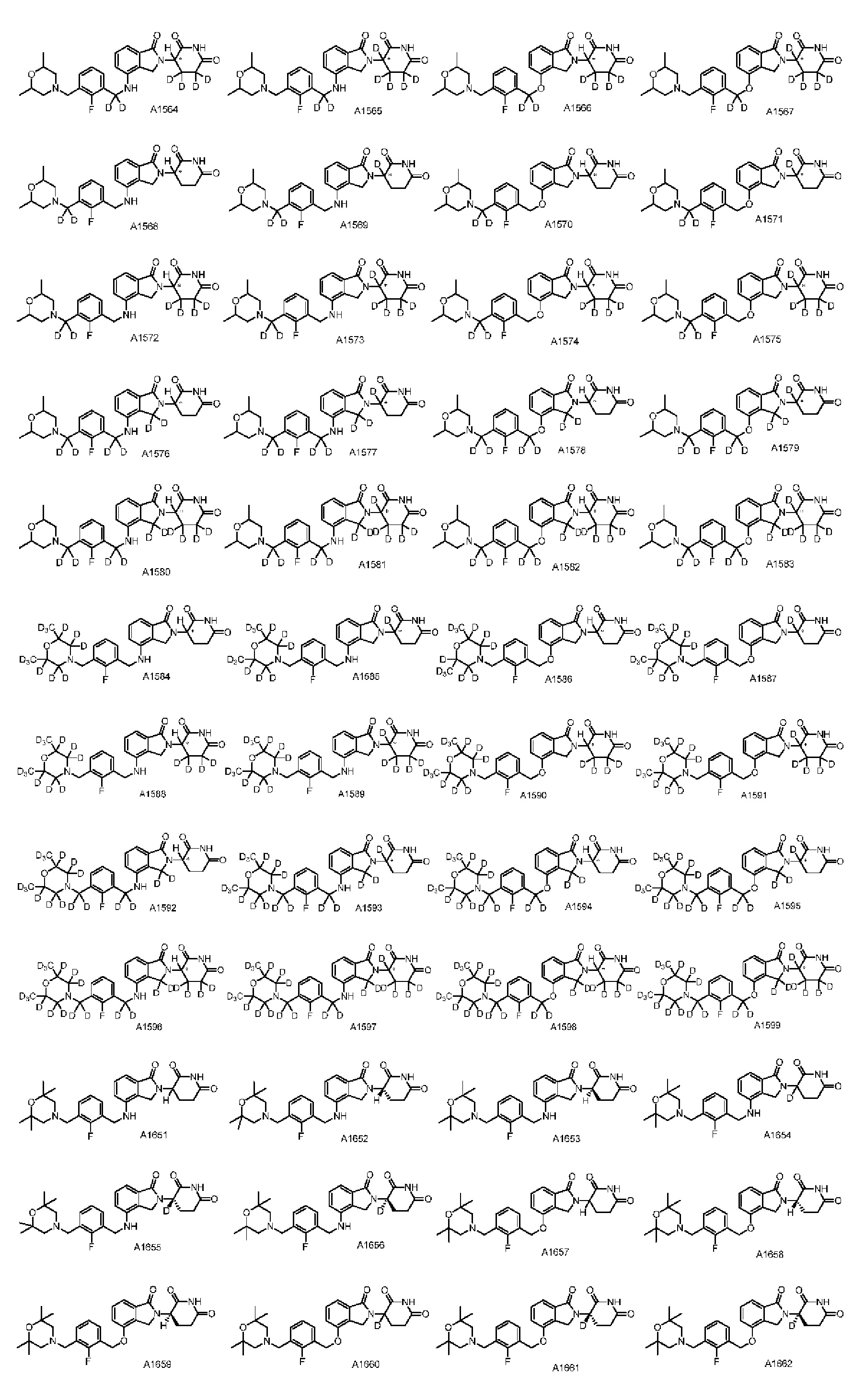








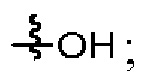

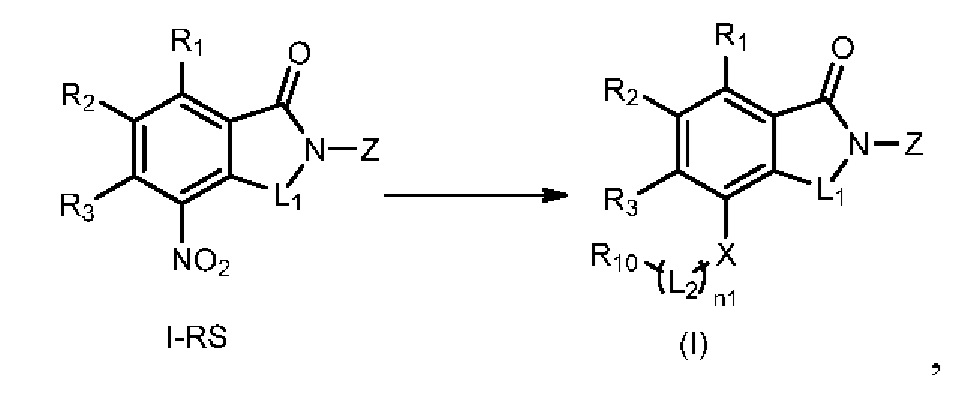




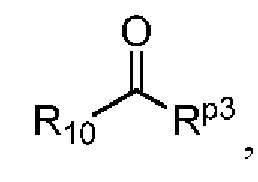



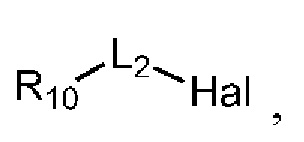
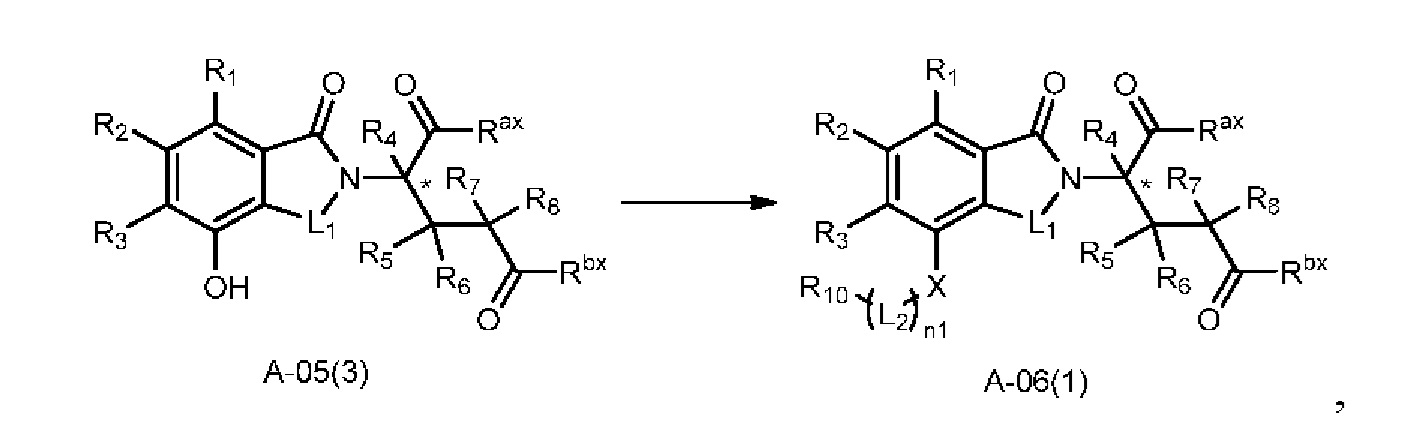



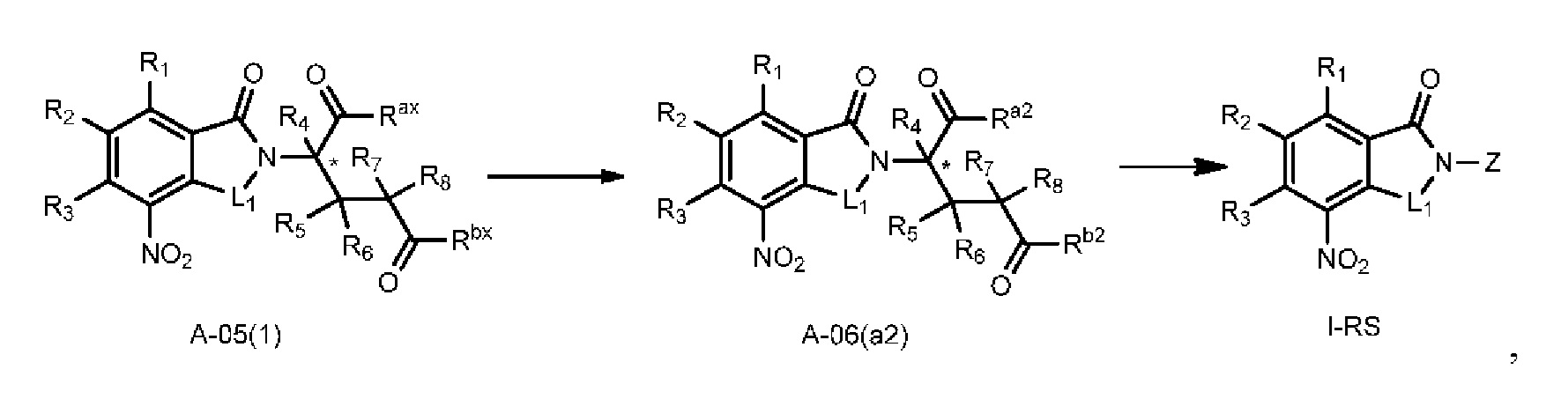

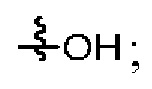


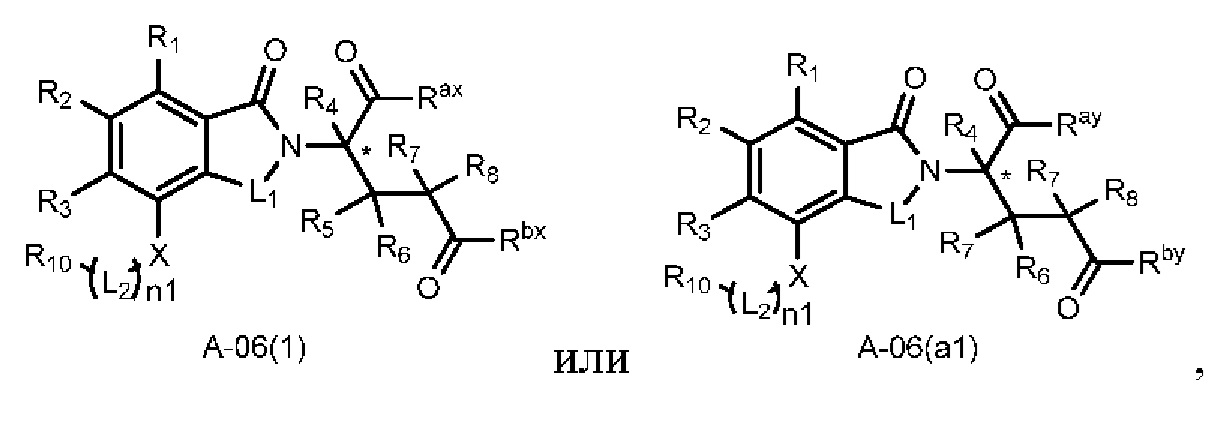

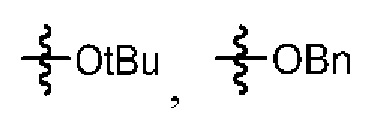



Документы, цитированные в отчёте о поиске
Производные арилметокси изоиндолина и композиции, включающие их, и способы их применения
Соединения ряда изоиндолимидов, их композиции и способы применения
Комментарии