Промежуточные продукты для получения ингибиторов нейтральной эндопептидазы и способ их получения - RU2564024C2
Код документа: RU2564024C2
Описание
Настоящее изобретение относится к новому способу получения промежуточных продуктов, применимых для получения ингибиторов NEP или их пролекарств, в частности ингибиторов NEP, содержащих в качестве основной цепи γ-амино-δ-бифенил-α-метилалкановую кислоту или эфир этой кислоты.
Эндогенные предсердные натрийуретические пептиды (ANP), также называющиеся предсердными натрийуретическими факторами (ANF), у млекопитающих выполняют роль диуретиков, натрийуретиков и сосудорасширяющих средств. Природные пептиды ANF метаболически инактивируются, в частности, разрушающим ферментом, для которого установлено, что он соответствует ферменту нейтральной эндопептидазе (NEP) ЕС 3.4.24.11, также ответственному, например, за метаболическую инактивацию энкефалинов.
В данной области техники известны производные замещенной биарилом фосфоновой кислоты, которые применимы в качестве ингибиторов нейтральной эндопептидазы (NEP), например, в качестве ингибиторов разрушающего ANF фермента у млекопитающих, для продления и усиления диуретических, натрийуретических и сосудорасширяющих воздействий ANF у млекопитающих путем ингибирования их разрушения с образованием менее активных метаболитов. Таким образом, ингибиторы NEP являются особенно подходящими для лечения патологических состояний и нарушений, реагирующих на ингибирование нейтральной эндопептидазы (ЕС 3.4.24.11), в особенности сердечно-сосудистых нарушений, таких как гипертензия, почечная недостаточность, включая отек и удерживание соли, отек легких и застойную сердечную недостаточность.
Способы получения ингибиторов NEP известны. В US 5217996 описаны производные замещенного биарилом амида 4-аминомасляной кислоты, которые применимы в качестве ингибиторов нейтральной эндопептидазы (NEP), например, в качестве ингибиторов разрушающего ANF фермента у млекопитающих. В US 5217996 раскрыто получение этилового эфира N-(3-карбокси-1-оксопропил)-(4S)-(п-фенилфенилметил)-4-амино-(2R)-метилбутановой кислоты. При получении указанного соединения этиловый эфир N-трет-бутоксикарбонил-(4R)-(п-фенилфенилметил)-4-амино-2-метил-2-бутеновой кислоты гидрируют в присутствии палладия на древесном угле. WO 2009/090251 относится к реакции получения этилового эфира N-трет-бутоксикарбонил(4S)-(п-фенилфенилметил)-4-амино-2-метилбутановой кислоты или его соли, в которой проведение альтернативной стадии гидрирования обеспечивает повышенную диастереоселективность по сравнению с наблюдавшейся в US 5217996. В реакции, описанной в WO 2009/090251, основным промежуточным продуктом является соединение формулы (1),

или его соль,
в которой R1 обозначает водород или защитную группу атома азота. В WO 2009/090251 в разделе В раскрыты различные методики получения соединения формулы (1). Во всех этих методиках в качестве исходного вещества используют соединение формулы (2) или его соль,

в которой R1 обозначает водород или защитную группу атома азота и R6 и R7 независимо обозначают алкильную группу, арильную группу, арилалкильную группу, циклоалкильную группу, или R6 и R7 образуют цикл вместе с атомом азота, к которому они присоединены, этот цикл может являться насыщенным или ненасыщенным и необязательно может содержать один или большее количество гетероатомов, таких как азот, кислород или сера, при этом цикл содержит от 3 до 8, например, от 4 до 7 кольцевых атомов. Как описано в WO 2009/090251 в разделе А, получение соединения формулы (2) или его соли включает реакцию соединения формулы (3) или его соли,

в которой R1 обозначает водород или защитную группу атома азота, с амином формулы (13), (14) или (15), или их смесью,



в которых каждый R6 и каждый R7 независимо обозначают алкильную группу, арильную группу, арилалкильную группу, циклоалкильную группу, или R6 и R7 образуют цикл вместе с атомом азота, к которому они присоединены, этот цикл может являться насыщенным или ненасыщенным и необязательно может содержать один или большее количество гетероатомов, таких как азот, кислород или сера, при этом цикл содержит от 3 до 8, например, от 4 до 7 кольцевых атомов, и каждый R8 независимо обозначает алкильную группу, арильную группу или арилалкильную группу, с получением соединения формулы (2).
Крупномасштабное получение аминов формулы (13), (14) или (15) является сложным процессом, в результате которого образуется их смесь, в которой соотношение количеств аминов может меняться от одной партии к другой. Реакционная способность аминов формулы (13), (14) или (15) различна. В соответствии с этим, учитывая, что при получении аминов формулы (13), (14) или (15) образуются их смеси переменного состава с разными профилями реакционной способности, производство соединения формулы (1) через соединение формулы (2) в промышленном масштабе затруднительно. Поэтому, необходима разработка альтернативного синтеза соединений формулы (1), описанных выше, который можно использовать для их производства в промышленном масштабе и у которого отсутствуют указанные выше недостатки способа предшествующего уровня техники. Таким образом, задачей настоящего изобретения является разработка нового способа получения соединения формулы (1), который может быть подходящим для его производства в промышленном масштабе.
Предлагаемый в настоящем изобретении новый способ получения соединения (1) или его соли, определенной в настоящем изобретении, представлен на схеме 1.

Таким образом, соединение формулы (3), описанное в настоящем изобретении, превращают в соединение формулы (1) или его соль, в которой R1 обозначает водород или защитную группу атома азота, по методике, описанной в разделе А.
Получение соединения формулы (3), описанного в настоящем изобретении, описано, например, в методике 1 подраздела С-1 в WO 2008/083967.
В WO 2008/083967 описан способ превращения соединения формулы (1), описанного в настоящем изобретении, в ингибитор NEP или его пролекарство. Поэтому, соединения формулы (1) можно использовать в качестве промежуточных продуктов для получения ингибиторов NEP или их пролекарств, в частности, ингибиторов NEP, содержащих в качестве основной цепи γ-амино-δ-бифенил-α-метилалкановую кислоту или эфир этой кислоты, предпочтительно алкиловый эфир N-(3-карбокси-1-оксопропил)-(4S)-(п-фенилфенилметил)-4-амино-(2R)-метилбутановой кислоты, такой как этиловый эфир N-(3-карбокси-1-оксопропил)-(4S)-(п-фенилфенилметил)-4-амино-(2R)-метилбутановой кислоты.
Настоящее изобретение в целом включает следующие разделы:
Раздел А: Методики получения соединения формулы (1)
Раздел В: Новые соединения, предлагаемые в настоящем изобретении
Раздел С: Примеры
Следует отметить, что обычно в настоящем изобретении описания, приведенные в одном разделе, также применимы к другим разделам, если не указано иное. Например, описания остатка R4 в формуле (4), приведенные в разделе А, также применимы, если формула (4) встречается в разделе В, если не указано иное. Указание на соединения, описанные в настоящем изобретении, также означает указание на их соли. В зависимости от выбора исходных веществ и методик соединения в зависимости от количества асимметрических атомов углерода могут содержаться в виде одного из возможных изомеров или их смесей, например, в виде чистых оптических изомеров или в виде смесей изомеров, таких как рацематы и смеси диастереоизомеров.
В другом варианте осуществления настоящее изобретение также относится к способу получения этилового эфира N-(3-карбокси-1-оксопропил)-(4S)-(п-фенилфенилметил)-4-амино-(2R)-метилбутановой кислоты или его соли, включающему получение соединения формулы (4) или его соли, определенной выше.
Раздел А: Получение соединения формулы (1)
Раздел A.1: Синтез соединения формулы (4)
Этот раздел относится к способу получения соединения формулы (1), определенного в настоящем изобретении, в котором превращение соединения формулы (3), определенного в настоящем изобретении, в указанное соединение формулы (1) происходит многостадийно, т.е. в две отдельные стадии без выделения промежуточных продуктов формулы (4), определенных в настоящем изобретении.
В одном варианте осуществления настоящее изобретение относится к способу получения соединения формулы (4) или его соли,

в которой
R1 обозначает водород или защитную группу атома азота; и
R4 выбран из группы, включающей гидроксигруппу, алкил, арил и арилалкил;
предпочтительно в котором соединением формулы (4) является соединение формулы (4а)

в которой R1 и R4 являются такими, как определено для соединения формулы (4);
указанный способ включает
реакцию соединения формулы (3) или его соли,

в которой R1 обозначает водород или защитную группу атома азота;
предпочтительно в котором соединением формулы (3) является соединение формулы (3а)

в которой R1 является таким, как определено для соединения формулы (3); сначала с основанием и затем с соединением формулы СО2 или R4COY, в которой Y обозначает галоген или -OR' и в которой R4 и R' независимо выбраны из группы, включающей алкил, арил и арилалкил, с получением соединения формулы (4) или его соли.
В другом варианте осуществления настоящее изобретение относится к способу получения соединения формулы (4) или его соли,
в которой
R1 обозначает водород или защитную группу атома азота; и
R4 обозначает гидроксигруппу;
предпочтительно в котором соединением формулы (4) является соединение формулы (4а)
в которой R1 и R4 являются такими, как определено для соединения формулы (4);
указанный способ включает реакцию соединения формулы (3) или его соли,
в которой R1 обозначает водород или защитную группу атома азота;
предпочтительно в котором соединением формулы (3) является соединение формулы (3а)
в которой R1 является таким, как определено для соединения формулы (3); сначала с основанием и затем с соединением формулы СО2 с получением соединения формулы (4) или его соли.
Основания, подходящие для превращения соединения формулы (3), предпочтительно формулы (3а), описанного в настоящем изобретении, в соединение формулы (4), предпочтительно формулы (4а), описанного в настоящем изобретении, включают:
- гидриды металлов, такие как гидриды щелочных металлов (например, гидрид натрия или калия);
- алкоксиды щелочных металлов (например, метоксид натрия, трет-бутоксид калия);
- амины, такие как 1,3-диметил-3,4,5,6-тетрагидро-2(1Н)-пиримидинон (ДМТП);
- основания формулы MRa, в которой М обозначает щелочной металл (например, литий, натрий, калий) и Ra обозначает алкил или арил, например, MRa обозначает метиллитий, н-бутиллитий, втор-бутиллитий, трет-бутиллитий или фениллитий;
- основания формулы RcRdNM, в которой Rc и Rd независимо выбраны из группы, включающей алкил, циклоалкил, гетероциклил или силил, и М обозначает щелочной металл (например, литий, натрий, калий), например, RcRdNM обозначает бис(триметилсилил)амид лития (LHMDS), бис(триметилсилил)амид натрия (NaHMDS), бис(триметилсилил)амид калия (KHMDS), диизопропиламид лития (ДАЛ) или диизопропиламид калия; или
- их смеси.
В одном варианте осуществления основанием является амин, такой как триэтиламин, диизопропилэтиламин, необязательно в присутствии добавки, выбранной из числа галогенидов щелочноземельных металлов, таких как хлорид магния, бромид магния и йодид магния.
Предпочтительно, если основанием является LHMDS, диизопропиламид лития или гидрид натрия, наиболее предпочтительно LHMDS.
Раздел А.2: Синтез соединения формулы (1) из соединения формулы (4)
Другим объектом настоящего изобретения является способ получения соединения формулы (1) или его соли,
в которой R1 обозначает водород или защитную группу атома азота;
предпочтительно в котором соединением формулы (1) является соединение формулы (1а)

в которой R1 является таким, как определено для соединения формулы (1);
указанный способ включает
реакцию соединения формулы (4) или его соли,
в которой
R1 обозначает водород или защитную группу атома азота; и
R4 выбран из группы, включающей гидроксигруппу, алкил, арил и арилалкил;
предпочтительно в котором соединением формулы (4) является соединение формулы (4а)
в которой R1 и R4 являются такими, как определено для соединения формулы (4);
с основанием и формальдегидом, необязательно в присутствии межфазного катализатора, с получением соединения формулы (1) или его соли.
Основания, подходящие для превращения соединения формулы (4), предпочтительно формулы (4а), описанного в настоящем изобретении, в соединение формулы (1), предпочтительно формулы (1а), описанного в настоящем изобретении, включают гидриды металлов, такие как гидриды щелочных металлов (например, гидрид натрия или калия), алкоксиды щелочных металлов (например, метоксид натрия, трет-бутоксид калия), амины, такие как диизопропилэтиламин, триэтиламин, морфолин или 1,8-диазабицикло[5.4.0]ундец-7-ен, неорганические основания, такие как карбонаты щелочных металлов, например карбонат калия, основания формулы MRa, в которой М обозначает щелочной металл (например, литий, натрий, калий) и Ra обозначает алкил или арил, например, MRa обозначает метиллитий, н-бутиллитий, втор-бутиллитий, трет-бутиллитий или фениллитий, основания формулы RcRdNM, в которой Rc и Rd независимо выбраны из группы, включающей алкил, циклоалкил, гетероциклил или силил, и М обозначает щелочной металл (например, литий, натрий, калий), например, RcRdNM обозначает бис(триметилсилил)амид лития (LHMDS), бис(триметилсилил)амид натрия (NaHMDS), бис(триметилсилил)амид калия (KHMDS), диизопропиламид лития (ДАЛ) или диизопропиламид калия; или их смеси.
В предпочтительном варианте осуществления превращение соединения формулы (4), предпочтительно, если соединением формулы (4) является соединение формулы (4а), в соединение формулы (1), описанное выше, проводят в присутствии основания и соли щелочного металла, такой как LiCl. Более предпочтительно, если это превращение проводят в присутствии основания, соли щелочного металла, такой как LiCl, и осушающего реагента, такого как молекулярные сита, сульфат щелочного металла (например, сульфат натрия) или сульфат щелочноземельного металла (например, сульфат магния).
Раздел А.2: Синтез соединения формулы (1) из соединения формулы (3)
В другом варианте осуществления настоящее изобретение относится к способу получения соединения формулы (1) или его соли,
в которой R1 обозначает водород или защитную группу атома азота;
предпочтительно в котором соединением формулы (1) является соединение формулы (1а)
в которой R1 является таким, как определено для соединения формулы (1);
указанный способ включает стадии
(i) получения соединения формулы (4) или его соли,
в которой
R1 обозначает водород или защитную группу атома азота; и
R4 выбран из группы, включающей гидроксигруппу, алкил, арил и арилалкил;
предпочтительно в котором соединением формулы (4) является соединение формулы (4а)
в которой R1 и R4 являются такими, как определено для соединения формулы (4);
по реакции соединения формулы (3) или его соли,
в которой R1 обозначает водород или защитную группу атома азота;
предпочтительно в котором соединением формулы (3) является соединение формулы (3а)
в которой R1 является таким, как определено для соединения формулы (3); сначала с основанием и затем с соединением формулы СО2 или R4COY, в которой Y обозначает галоген или -OR' и в которой R4 и R' независимо выбраны из группы, включающей алкил, арил и арилалкил, с получением соединения формулы (4) или его соли; и (ii) реакции соединения формулы (4) или его соли,

в которой
R1 обозначает водород или защитную группу атома азота; и
R4 выбран из группы, включающей гидроксигруппу, алкил, арил и арилалкил;
предпочтительно в котором соединением формулы (4) является соединение формулы (4а)

в которой R1 и R4 являются такими, как определено для соединения формулы
(4);
с основанием и формальдегидом, необязательно в присутствии межфазного катализатора,
с получением соединения формулы (1) или его соли.
Другим объектом настоящего изобретения является описанный выше способ превращения соединения формулы (3) в соединение формулы (1), в котором стадии i) и ii) проводят по однореакторной методике, следовательно, без выделения и/или очистки соединения формулы (4).
В другом варианте осуществления настоящее изобретение относится к способу получения соединения формулы (1) или его соли,
в которой R1 обозначает водород или защитную группу атома азота;
предпочтительно в котором соединением формулы (1) является соединение формулы (1а)
в которой R1 является таким, как определено для соединения формулы (1);
указанный способ включает стадии
(i) получения соединения формулы (4) или его соли,
в которой
R1 обозначает водород или защитную группу атома азота; и
R4 обозначает гидроксигруппу;
предпочтительно в котором соединением формулы (4) является соединение формулы (4а)
в которой R1 и R4 являются такими, как определено для соединения формулы (4);
по реакции соединения формулы (3) или его соли,
в которой R1 обозначает водород или защитную группу атома азота;
предпочтительно в котором соединением формулы (3) является соединение формулы (3а)
в которой R1 является таким, как определено для соединения формулы (3); сначала с основанием и затем с соединением формулы СО2 с получением соединения формулы (4) или его соли; и
(ii) реакции соединения формулы (4) или его соли,
в которой
R1 обозначает водород или защитную группу атома азота; и
R4 обозначает гидроксигруппу;
предпочтительно в котором соединением формулы (4) является соединение формулы (4а)
в которой R1 и R4 являются такими, как определено для соединения формулы (4);
с основанием и формальдегидом, необязательно в присутствии межфазного катализатора, с получением соединения формулы (1) или его соли.
Еще одним объектом настоящего изобретения является описанный выше способ превращения соединения формулы (3) в соединение формулы (1), в котором стадии i) и ii) проводят по однореакторной методике, следовательно, без выделения и/или очистки соединения формулы (4).
Основания, подходящие для проведения стадии (i) в описанных выше вариантах осуществления, являются такими, как описанные в разделе A.1.
Основания, подходящие для проведения стадии (ii) в описанных выше вариантах осуществления, являются такими, как описанные в разделе А.2. Предпочтительно, если стадию (ii) в описанных выше вариантах осуществления, проводят в присутствии основания и соли щелочного металла, такой как LiCl. Более предпочтительно, если это превращение проводят в присутствии основания, соли щелочного металла, такой как LiCl, и осушающего реагента, такого как молекулярные сита, сульфат щелочного металла (например, сульфат натрия) или сульфат щелочноземельного металла (например, сульфат магния).
Раздел В:
Соединение формулы (4) или его соль,
в которой
R1 обозначает водород или защитную группу атома азота; и
R4 выбран из группы, включающей гидроксигруппу, алкил, арил и арилалкил;
предпочтительно в котором соединением формулы (4) является соединение формулы (4а)
в которой R1 и R4 являются такими, как определено для соединения формулы (4).
Общие термины:
Общие термины, применяющиеся выше и ниже, если не указано иное, обладают приведенными ниже значениями:
Термин "защитная группа атома азота" включает любую группу, которая может обратимо защитить азотсодержащую группу, предпочтительно аминогруппу и/или амидную группу. Предпочтительно, если защитная группа атома азота представляет собой защитную группу аминогруппы и/или защитную группу амидной группы. Подходящими защитными группами атома азота являются группы, обычно используемые в химии пептидов, и они описаны, например, в соответствующих главах стандартных справочников, таких как J. F. W. McOmie, "Protective Groups in Organic Chemistry", Plenum Press, London and New York 1973, P. G. M. Wuts and T.W. Greene, "Greene's Protective Groups in Organic Synthesis', Fourth Edition, Wiley, New Jersey, 2007, "The Peptides"; Volume 3 (editors: E. Gross and J. Meienhofer), Academic Press, London and New York 1981, и "Methoden der organischen Chemie" (Methods of Organic Chemistry), Houben Weyl, 4th edition. Volume 15/1, Georg Thieme Verlag, Stuttgart 1974.
Предпочтительные защитные группы атома азота обычно включают: незамещенный или замещенный С1-С6-алкил, предпочтительно С1-С4-алкил, более предпочтительно С1-С2-алкил, наиболее предпочтительно C1-алкил, незамещенный или замещенный С2-С4-алкенил, где C1-С6-алкил и С2-С4-алкенил необязательно могут быть моно-, ди- или тризамещенными триалкилсилил-C1-С7-алкоксигруппой (например, триэтилсилилэтоксигруппой), арил, предпочтительно фенил, или гетероциклическую группу, предпочтительно пирролидинил, где арильное кольцо или гетероциклическая группа является незамещенной или замещена одним или большим количеством, например, двумя или тремя фрагментами, например, выбранными из группы, включающей C1-C7-алкил, гидроксигруппу, С1-С7-алкоксигруппу, С2-С8-алканоилоксигруппу, галоген, нитрогруппу, цианогруппу и CF3; арил-С1-С2-алкоксикарбонил (предпочтительно фенил-С1-С2-алкоксикарбонил, например, бензилоксикарбонил); C1-С10-алкенилоксикарбонил; C1-С6-алкилкарбонил (например, ацетил или пивалоил); С6-С10-арилкарбонил; C1-С6-алкоксикарбонил (например, трет-бутоксикарбонил); С6-С10-арил-С1-С6-алкоксикарбонил; аллил или циннамил; сульфонил или сульфенил; сукцинимидильную группу, силил, например, триарилсилил или триалкилсилил (например, триэтилсилил).
Примерами предпочтительных защитных групп атома азота являются ацетил, бензил, кумил, бензгидрил, тритил, бензилоксикарбонил (Cbz), 9-флуоренилметоксикарбонил (Fmoc), бензилоксиметил (БОМ), пивалоилоксиметил (ПОМ), трихлорэтоксикарбонил (Troc), 1-адамантилоксикарбонил (Adoc), аллил, аллилоксикарбонил, триметилсилил, трет-бутилдиметилсилил, триэтилсилил (ТЭС), триизопропилсилил, триметилсилилэтоксиметил (SEM), трет-бутоксикарбонил (ВОС), трет-бутил, 1-метил-1,1-диметилбензил, (фенил)метилбензол, пиридинил и пивалоил. Наиболее предпочтительными защитными группами атома азота являются ацетил, бензил, бензилоксикарбонил (Cbz), триэтилсилил (ТЭС), триметилсилилэтоксиметил (SEM), трет-бутоксикарбонил (ВОС), пирролидинилметил и пивалоил.
Примерами более предпочтительных защитных групп атома азота являются трет-бутоксикарбонил (ВОС), бензоил, стирил, 1-бутенил, бензил, п-метоксибензил (РМВ) и пирролидинилметил.
Силил при использовании в настоящем изобретении означает группу формулы -SiR11R12R13, в которой R11, R12 и R13 независимо друг от друга обозначают алкил или арил. Предпочтительными примерами R11,R12 и R13 являются метил, этил, изопропил, трет-бутил, фенил или фенил-С1-С4-алкил.
Алкил, определенный, как радикал или часть радикала, означает обладающий линейной или разветвленной (один или, если это является необходимым и возможным, большее количество раз) углеродной цепью, и предпочтительно представляет собой С1-С7-алкил, более предпочтительно C1-С4-алкил.
Термин "C1-C7-" означает фрагмент, содержащий до и включительно максимально 7, до и включительно максимально 4 атома углерода, указанный фрагмент обладает разветвленной (один или большее количество раз) или линейной цепью и присоединен через концевой или неконцевой атом углерода.
Циклоалкил означает, например, С3-С7-циклоалкил и представляет собой, например, циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. Циклопентил и циклогексил являются предпочтительными.
Алкоксигруппа означает, например, С1-С7-алкоксигруппу и представляет собой, например, метоксигруппу, этоксигруппу, н-пропилоксигруппу, изопропилоксигруппу, н-бутилоксигруппу, изобутилоксигруппу, втор-бутилоксигруппу, трет-бутилоксигруппу и также включает соответствующие пентилоксильный, гексилоксильный и гептилоксильный радикалы. C1-C4-Алкоксигруппа является предпочтительной.
Алканоил означает, например, С2-С8-алканоил и представляет собой, например, ацетил [-С(=Щ)Ме], пропионил, бутирил, изобутирил или пивалоил. С2-С5-Алканоил является предпочтительным, ацетил является особенно предпочтительным.
Галоген предпочтительно означает фтор, хлор, бром или йод, наиболее предпочтительно хлор, бром или йод.
Галогеналкил означает, например, галоген-С3-С7-алкил и предпочтительно представляет собой, галоген-С1-С4-алкил, такой как трифторметил, 1,1,2-трифтор-2-хлорэтил или хлорметил. Предпочтительным галоген-С3-С7-алкилом является трифторметил.
Алкенил может представлять собой линейный или разветвленный алкил, содержащий двойную связь и предпочтительно содержащий от 2 до 12 атомов С, особенно предпочтительно от 2 до 10 атомов С. Особенно предпочтительным является линейный С2-С4-алкенил. Некоторыми примерами алкенильных групп являются этил и изомеры пропила, бутила, пентила, гексила, гептила, октила, нонила, децила, ундецила, додецила, тетрадецила, гексадецила, октацила и эйкозила, каждый из которых содержит двойную связь. Особенно предпочтительным является аллил.
Алкилен означает двухвалентный радикал, образованный из С1-С7-алкила, и предпочтительно представляет собой С2-С7-алкилен или С2-С7-алкилен, и в пего необязательно могут быть включены один или большее количество, например, до 3 фрагментов О, NR14 или S, где R14 обозначает алкил, каждый из которых может являться незамещенным или содержать один или большее количество заместителей, независимо выбранных из группы, включающей, например, С1-С7-алкил, С1-С7-алкокси-С1-С7-алкил или С1-С7-алкоксигруппу.
Алкенилен означает двухвалентный радикал, образованный из С2-С7-алкенила, и в него могут быть включены один или большее количество, например, до 3 фрагментов О, NR14 или S, где R14 обозначает алкил, и является незамещенным или содержит один или большее количество, например, до 3 заместителей, предпочтительно независимо выбранных из числа заместителей, указанных выше для алкилена.
Арил в качестве радикала или части радикала означает, например, С6-С10-арил и предпочтительно представляет собой моно- или полициклический, предпочтительно моноциклический, бициклический или трициклический арильный фрагмент, содержащий от 6 до 10 атомов углерода, предпочтительно фенил, который может быть незамещенным или содержать один или большее количество заместителей, независимо выбранных из группы, включающей, например, С1-С7-алкил, С1-С7-алкокси-С1-С7-алкил или С1-С7-алкоксигруппу.
Термин арилалкил означает арил-С1-С7-алкил, где арил является таким, как определено в настоящем изобретении, и представляет собой, например, бензил.
Термин карбоксигруппа означает -СО2Н.
Арилоксигруппа означает арил-O-, где арил является таким, как определено выше.
Незамещенный или замещенный гетероциклил является моно- или полициклической, предпочтительно моно-, би- или трициклической, наиболее предпочтительно моно-, ненасыщенной, частично насыщенной, насыщенной или ароматической кольцевой системой, предпочтительно содержащей от 3 до 14 (более предпочтительно от 5 до 14) кольцевых атомов и один или большее количество, предпочтительно от 1 до 4 гетероатомов, независимо выбранных из группы, включающей азот, кислород, серу, S(=O)- или S-(=O)2, и является незамещенным или содержит один или большее количество, например, до 3 заместителей, которые предпочтительно независимо выбраны из группы, включающей галоген, С1-С7-алкил, галоген-С1-С7-алкил, С1-С7-алкоксигруппу, галоген-С1-С7-алкоксигруппу, такие как трифторметоксигруппа и C1-C7-алкокси-С1-С7-алкоксигруппа. Если гетероциклил представляет собой ароматическую кольцевую систему, то его также называют гетероарилом.
Ацетил означает -С(=O)С3-С7-алкил, предпочтительно -С(=O)Ме.
Сульфонил означает (незамещенный или замещенный) C1-C7-алкилсульфонил, такой как метилсульфонил, (незамещенный или замещенный) фенил- или нафтил-С1-С7-алкилсульфонил, такой как фенилметансульфонил, или (незамещенный или замещенный) фенил- или нафтилсульфонил; где в случае, если содержится более одного заместителя, например, от 1 до 3 заместителей, то заместители независимо выбраны из группы, включающей цианогруппу, галоген, галоген-С3-С7-алкил, галоген-С1-С7-алкилокси- и C1-C7-алкилоксигруппу. Особенно предпочтительным является С1-С7-алкилсульфонил, такой как метилсульфонил, и (фенил- или нафтил)-С1-С7-алкилсульфонил, такой как фенилметансульфонил.
Сульфенил означает (незамещенный или замещенный) С6-С10-арил-С1-С7-алкилсульфенил или (незамещенный или замещенный) С6-С10-арилсульфенил, где в случае, если содержится более одного заместителя, например, от 1 до 3 заместителей, то заместители независимо выбраны из группы, включающей нитрогруппу, галоген, галоген-С3-С7-алкил и С1-С7-алкилоксигруппу.
Термин "хиральный" относится к молекулам, которые обладают зеркальными изображениями, не налагающимися друг на друга, в то время как термин "ахиральный" относится к молекулам, которые обладают зеркальными изображениями, налагающимися друг на друга.
Термин "таутомер" означает, в частности, енольный таутомер пирролидин-2-оиового фрагмента соединений, предлагаемых в настоящем изобретении. Кроме того, термин "таутомер" также означает, в частности, альдегидный таутомер соединений, предлагаемых в настоящем изобретении, например, соединений формулы (6), где такие соединения могут существовать в енольной или альдегидной форме, или в виде их смеси.
В формулах, предлагаемых в настоящем изобретении, символ "
В формулах, предлагаемых в настоящем изобретении, символ "
Соединения, предлагаемые в настоящем изобретении, могут содержать один или большее количество асимметрических центров. Предпочтительные абсолютные конфигурации являются такими, как специально указано в настоящем изобретении.
В формулах, предлагаемых в настоящем изобретении, символ "

В формулах, предлагаемых в настоящем изобретении, символ "

В формулах, предлагаемых в настоящем изобретении, символ "

Солями предпочтительно являются фармацевтически приемлемые соли или обычно соли любого из промежуточных продуктов, указанных в настоящем изобретении, и специалист в данной области техники должен легко понять, какие соли не исключены по химическим соображениям. Их можно получить, если содержатся солеобразующие группы, такие как основные или кислые группы, которые по меньшей мере частично могут находиться в диссоциированной форме, например, в диапазоне значений рН, составляющем от 4 до 10, в водном растворе или их можно выделить предпочтительно в твердой, более предпочтительно в кристаллической форме.
Такие соли образуются, например, как соли присоединения с кислотами, предпочтительно с органическими или неорганическими кислотами, из соединений или любых промежуточных продуктов, указанных в настоящем изобретении, содержащих основной атома азота (например, иминогруппу или аминогруппу), предпочтительно фармацевтически приемлемые соли. Подходящими неорганическими кислотами являются, например, галогенводородные кислоты, такие как хлористоводородная кислота, серная кислота или фосфорная кислота. Подходящими органическими кислотами являются, например, карболовые, фосфоновые и сульфаминовые кислоты, например, уксусная кислота, пропионовая кислота, молочная кислота, фумаровая кислота, янтарная кислота, лимонная кислота, аминокислоты, такие как глутаминовая кислота или аспарагиновая кислота, малеиновая кислота, гидроксималеиновая кислота, метилмалеиновая кислота, бензойная кислота метан- и этансульфоновая кислота, этан-1,2-дисульфоновая кислота, бензолсульфоновая кислота, 2-нафталинсульфоновая кислота, 1,5-нафталиндисульфоновая кислота, N-циклогексилсульфаминовая кислота, N-метил-, N-этил- или N-пропилсульфаминовая кислота или другие органические протонные кислоты, такие как аскорбиновая кислота.
В присутствии отрицательно заряженных радикалов, таких как карбоксигруппа или сульфогруппа, также могут образовываться соли с основаниями, например, соли металлов или аммония, такие как соли щелочных металлов или щелочноземельных металлов, например, соли натрия, калия, магния или кальция, или соли аммония с аммиаком или подходящими органическими аминами, например, триэтиламином или три(2-гидроксиэтил)амином, или гетероциклическими основаниями, например, N-этилпиперидином или N,N'-диметилпиперазином.
Если в одной молекуле содержатся основания группа и кислотная группа, то любой из промежуточных продуктов, указанных в настоящем изобретении, также может образовать внутренние соли.
Для целей выделения и очистки любого из промежуточных продуктов, указанных в настоящем изобретении, также можно использовать фармацевтически неприемлемые соли, например, пикраты или перхлораты.
Вследствие большого сходства соединений и промежуточных продуктов в свободной форме и в форме их солей, включая соли, которые можно использовать в качестве промежуточных продуктов, например, при очистке и идентификации соединений или солей, любое указание на "соединения", "исходные вещества" и "промежуточные продукты", приведенное выше и ниже в настоящем изобретении, следует понимать, как указание также и па одну или большее количество их солей или смесь соответствующего свободного соединения, промежуточного продукта или исходного вещества и одной или большего количества его солей, каждая из которых также включает любой сольват или соль любого одного или большего количества из них, если это является подходящим или приемлемым и если явно не указано иное. Можно получить различные кристаллический формы, и они также входят в объем настоящего изобретения.
Если для соединений, исходных веществ, промежуточных продуктов, солей, фармацевтических препаратов, заболеваний, нарушений и т.п. используют множественное число, это означает одно (предпочтительно) или большее количество отдельных соединений, солей, фармацевтических препаратов, заболеваний, нарушений и т.п., использование единственного числа не исключает множественное число и только предпочтительно означает единственное число.
Любой из лактамов, предлагаемых в настоящем изобретении, или его соль, в которой R1 обозначает водород можно превратить в соответствующий защищенный пактам или его соль, в которой R1 обозначает защитную группу атома азота, определенную выше, по стандартным методикам органической химии, известным в данной области техники, в частности, обычные методики введения защитной группы атома азота описаны в публикациях J. F. W. McOmie, "Protective Groups in Organic Chemistry", Plenum Press, London and New York 1973, и P. G. M. Wuts and T.W. Greene, "Greene's Protective Groups in Organic Synthesis', Fourth Edition, Wiley, New Jersey, 2007 и Richard С. Larock, "Comprehensive Organic Transformations: A Guide to Functional Group Preparations", Second Edition, Wiley-VCH Verlag GmbH, 2000, в частности, в соответствующих главах этих публикаций.
Аналогичным образом, любой пактам, предлагаемый в настоящем изобретении, или его соль, в которой R1 обозначает защитную группу атома азота, можно превратить в соответствующий пактам или его соль, в которой R1 обозначает водород, по стандартным методикам органической химии, известным в данной области техники, в частности, обычные методики удаления защитной группы атома азота описаны в указанных выше публикациях, в частности в соответствующих главах этих публикаций.
Термин "однореакторный" "или "однореакторная методика" означает, что в серии реакций каждый продукт реакции вводят в следующую реакцию без выделения и/или очистки. Термин "очистка" при использовании в настоящем изобретении предпочтительно означает кристаллизацию, колоночную хроматографию или перегонку. Однореакторный способ, определенный в настоящем изобретении включает не только серию реакций, проводимых в одном сосуде для проведения реакций, но и серию реакций, проводимых во множестве сосудов для проведения реакций (например, путем переноса реакционной смеси из одного сосуда в другой) без выделения и/или очистки. Однореакторный способ предпочтительно проводить в одном сосуде для проведения реакций.
Термин "формальдегид" при использовании в настоящем изобретении включает мономерный формальдегид и любой источник формальдегида, который легко превращается в формальдегид. Например, "формальдегид" при использовании в настоящем изобретении включает формальдегид в виде его мономера, а также его различные ацетали, полуацетали и обладающие низкой молекулярной массой олигомеры, такие как, например, параформальдегид.
Термин "межфазный катализатор" при использовании в настоящем изобретении означает химическое вещество в каталитическом количестве, которое повышает скорость реакции между химическими соединениями, находящимися в разных фазах (например, несмешивающиеся жидкости или твердое вещество и жидкость), путем экстракции одного из реагентов, обычно аниона, через границу раздела в другую фазу. Такие катализаторы включают четвертичные аммониевые или фосфониевые соли (например, тетраалкиламмониевые соли, где алкилы могут быть одинаковыми или разными), или вещества, которые образуют комплекс с неорганическими катионами (например, краун-эфиры или другие криптанды). Катион катализатора не расходуется в ходе реакции, несмотря на то, что происходит анионный обмен. В частности, межфазными катализаторами, подходящими для применения в контексте настоящего изобретения, являются четвертичные аммониевые соли, например, формулы RmRnRlRkNX, в которой RmRnRlRkобозначают одинаковые или разные алкилы и Х обозначает галоген (например, хлор, бром, йод) или гидроксид, например, тетра-н-бутиламмонийгидроксид.
Термин "пролекарство" при использовании в настоящем изобретении означает, в частности, соединения, которые in vivo превращаются в исходное соединение, например, путем гидролиза в крови, например, как описано в публикациях Т. Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems, Vol.14 of the A.C.S. Symposium Series, Edward B. Roche, ed., Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press, 1987; H Bundgaard, ed. Design ofProdrugs, Elsevier, 1985; и Judkins, et al. Synthetic Communications, 26(23), 4351-4367 (1996), и "The Organic Chemistry of Drug Design and Drug Action", 2" Edition, R В Silverman (в особенности, глава 8, стр.497-557), Elsevier Academic Press, 2004.
Поэтому пролекарства включают лекарственные вещества, содержащие функциональную группу, которые превращены в производные, способные к обратимому превращению. Обычно такие пролекарства превращаются в активное лекарственное вещество путем гидролиза. В качестве примеров можно отметить следующие:
Пролекарства также включают соединения, способные к превращению в активное лекарственное вещество по реакциям окисления или восстановления. В качестве примеров можно отметить:
Окислительная активация
- N- и O-деалкилирование
- окислительное дезаминирование
- N-окислепие
- S-окисление
- эпоксидирование
Восстановительная активация
- восстановление азогруппы
- восстановление сульфоксидной группы
- восстановление дисульфидной группы
- биологическое восстановительное алкилирование
- восстановление нитрогруппы
Каждую из описанных выше реакций и/или стадий реакции можно использовать по отдельности или в комбинации в способе получения ингибитора NEP или его пролекарства, такого как ингибитор NEP или его пролекарство, содержащее в качестве основной цепи γ-амино-δ-бифенил-α-метилалкановую кислоту или эфир этой кислоты, такой как алкиловый эфир. В частности, ингибитором NEP является N-(3-карбокси-1-оксопропил)-(4S)-п-фенилфенилметил)-4-амино-(2R)-метилбутановая кислота или ее соль или ее пролекарство. Как описано выше, в WO 2008/083967 описан способ превращения соединения формулы (1), описанного в настоящем изобретении, в ингибитор NEP или его пролекарство.
Раздел С: Примеры
Приведенные ниже примеры предназначены для иллюстрации настоящего изобретения без ограничения его объема, хотя, с другой стороны, они характеризуют предпочтительные варианты осуществления стадий реакций, промежуточных продуктов и/или способа, предлагаемого в настоящем изобретении.
Аббревиатуры:
В приведенных данных ЯМР можно использовать следующие аббревиатуры: s = синглет; d = дублет; t = триплет; q = квартет; quint. = квинтет; m = мультиплет.
Пример 1: (8)-5-Бифенил-4-илметил-1-(4-метоксибензил)-пирролидин-2-он (3а, R1 = п-метоксибензил)

Гидрид натрия (55%, 6,9 г, 158 ммолей) в атмосфере N2 при КТ добавляют к смеси (S)-5-бифенил-4-илметилпирролидин-2-она (3а, R1=Н) (36 г, 143 ммоля) и 400 мл сухого диметилформамида, затем добавляют 4-метоксибензилхлорид (24,7 г, 158 ммолей). Реакционную смесь нагревают до 55°С и перемешивают в течение 3 ч. Охлаждают до КТ, добавляют 5 мл АсОН и перемешивают в течение еще 15 мин, затем диметилформамид удаляют, остаток повторно растворяют в 400 мл этилацетата, промывают водой, сушат над сульфатом натрия. Растворитель удаляют, остаток повторно растворяют в 100 мл трет-бутилметилового эфира, охлаждают до 0°С и перемешивают в течение 5 ч, фильтруют и сушат и получают (S)-5-бифенил-4-илметил-1-(4-метоксибензил)-пирролидин-2-он (3а, R1 = п-метоксибензил).1Н ЯМР (400 МГц, CDCl3): 1,78 (m, 1Н, 3-CHH), 1,90 (m, 1H, 3-CHH), 2,28 (m, 2H, 2-СН2), 2,59 (dd, 1Н, 5-CHH), 3,05 (dd, 1Н, 5-CHH), 3,80 (s, 3Н, ОСН3), 3,98 (d, 1Н, СНН), 5,05 (d, 1Н, CHH), 6,80~7,40 (13Н, m, ароматический).
Методика проведения ВЭЖХ
Колонка: Eclipse XDB-C18; 150×4,6 мм; 5 мкм. Подвижная фаза А (0,1% H3PO4) в воде; Подвижная фаза В (ацетонитрил). Градиентный режим: 0 мин (90% В); 10 мин (95% В); 15 мин (95% В). Скорость потока: 0,7 мл мин-1. Длина волны: 210 нм. Температура: 30°С.
Время удерживания: 12,5 мин (3а, R1 = п-метоксибензил)
Пример 2: (S)-1-Бензоил-5-бифенил-4-илметилпирролидин-2-он (3а, R1 = бензоил)

Смесь (S)-5-бифенил-4-илметилпирролидин-2-опа (3а, R1=Н) (10 г, 40 ммолей) и триэтиламина (16,6 мл, 120 ммолей) нагревают при 60°С, в течение 1 ч добавляют бензоилхлорид (8,5 г, 60 ммолей), еще через 4 ч добавляют раствор лимонной кислоты (23,7 г в 100 мл воды) и водный слой промывают толуолом, органические порции объединяют, промывают водой, смесь концентрируют в вакууме и повторно растворяют в трет-бутилметиловом эфире и охлаждают в бане из воды со льдом, перемешивают в течение 4 ч и фильтруют и получают (S)-1-бензоил-5-бифенил-4-илметилпирролидин-2-он (3а, R1 = бензоил).1Н ЯМР (400 МГц, CDCl3): 1,94 (m, 1Н, 3-СНН), 2,13 (m, 1Н, 3-СНН), 2,20 (m, 1Н, 2-СНН), 2,23 (m, 1Н, 2-СНН), 2,71 (d, 1Н, 5-СНН), 2,96 (d, 1Н, 5-СНН), 4,38 (m, 1Н, 4-СН), 7,20-8,10 (14Н, m, ароматический).
Методика проведения ВЭЖХ
Колонка: Eclipse XDB-C18; 150×4,6 мм; 5 мкм. Подвижная фаза А (0,1% Н3РО4) в воде; Подвижная фаза В (ацетонитрил). Градиентный режим: 0 мин (90% В); 10 мин (95% В); 15 мин (95% В). Скорость потока: 0,7 мл мин-1. Длина волны: 210 нм. Температура: 30°С.
Время удерживания: 13,0 мин (3а, R1 = бензоил)
Пример 3: (S)-5-Бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-оп (3а, R1 = стирил)

Методика 1
(S)-5-Бифенил-4-илметилпирролидин-2-он (3а, R1=Н) (10,0 г, 39,8 ммоля) растворяют в 100 мл безводного тетрагидрофурана, последовательно добавляют фенилацетальдегид (5,3 г, 39,8 ммоля) и P2O5 (6,2 г, 43,8 ммоля), реакционную смесь кипятят с обратным холодильником в течение 12 ч. Реакционную смесь охлаждают до комнатной температуры, фильтруют и осадок на фильтре промывают этилацетатом, фильтрат промывают 10% водным раствором бикарбоната натрия и рассолом, сушат над безводным Na2SO4 и концентрируют в вакууме, остаток перекристаллизовывают из трет-бутилметилового эфира и получают (S)-5-бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (3а, R1 = стирил).1Н ЯМР (400 МГц, CDCl3): 2,11 (m, 1H, 3-CHH), 2,16 (m, 2Н, 5-СН2), 2,29 (m, 1H, 3CHH), 2,95 (d, 1H, 5-CH2), 3,10 (d, 1H, 5-CH2), 4,37 (m, 1H, 4-CH), 6,10 (d, 1Н, C=CHH), 7,21-7,65 (15H, m, ароматический + C=CHH),
Методика 2
(S)-5-Бифенил-4-илметилпирролидин-2-он (3а, R1=Н) (10,0 г, 39,8 ммоля) растворяют в 40 мл безводного толуола, добавляют фенилацетальдегид (5,3 г, 39,8 ммоля) и TsOH·Н2О (0,2 г, 1 ммоль). Реакционную колбу снабжают ловушкой Дина-Штарка и холодильником, реакционную смесь кипятят с обратным холодильником в атмосфере азота в течение 12 ч. Реакционную смесь охлаждают до комнатной температуры, добавляют 100 мл этилацетата, органический слой промывают насыщенным водным раствором гидрокарбоната натрия, затем раствором Na2SO3 и рассолом, затем органический слой сушат над безводным Na2SO4 и концентрируют в вакууме, остаток очищают с помощью флэш-хроматографии и получают (S)-5-бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (3а, R1 = стирил). Спектроскопические данные являются такими же, как в примере 2, методика 1.
Методика 3
(S)-5-Бифенил-4-илметилпирролидин-2-он (3а, R1=Н) (50,2 г, 200 ммолей) растворяют в 150 мл п-ксилола, добавляют фенилацетальдегид (26,4 г, 220 ммолей) и TsOH·H2O (0,4 г, 2 ммоля). Реакционную колбу снабжают ловушкой Дина-Штарка и холодильником, смесь кипятят с обратным холодильником в атмосфере азота в течение ночи. Реакционную смесь охлаждают до комнатной температуры, перемешивают при 0°С в течение 1 ч, затем фильтруют, осадок на фильтре трижды промывают охлажденным трет-бутилметиловым эфиром (3×20 мл), сушат в высоком вакууме и получают (S)-5-бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (3а, R1 = стирил) в виде белого твердого вещества. Спектроскопические данные являются такими же, как в примере 2, методика 1.
Методика 4
(S)-5-Бифенил-4-илметилпирролидин-2-он (3а, R1=H) (5,02 г, 20 ммолей) растворяют в 50 мл толуола, добавляют диметилацеталь фенилацетальдегида (3,77 г, 22 ммоля) и TsOH·Н2О (0,1 г, 0,5 ммоля). Реакционную колбу снабжают ловушкой Дина-Штарка и холодильником, смесь кипятят с обратным холодильником в атмосфере азота в течение ночи. Реакционную смесь охлаждают до комнатной температуры, добавляют 50 мл этилацетата, перемешивают в течение 10 мин и получают прозрачный раствор, промывают насыщенным водным раствором гидрокарбоната натрия, насыщенным водным раствором Na2SO3 и рассолом, органические экстракты сушат над безводным Na2SO4 и растворитель выпаривают в вакууме, остаток очищают с помощью флэш-хроматографии и получают (S)-5-бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (3а, R1 = стирил) в виде белого твердого вещества. Спектроскопические данные являются такими же, как в примере 2, методика 1.
Методика 5
(S)-5-Бифенил-4-илметилпирролидин-2-он (3а, R1=Н) (5,02 г, 20 ммолей) растворяют в 50 мл толуола и добавляют диметилацеталь фенилацетальдегида (3,77 г, 22 ммоля) и ППТС (0,13 г, 0,5 ммоля). Реакционную колбу снабжают ловушкой Дина-Штарка и холодильником, смесь кипятят с обратным холодильником в атмосфере азота в течение ночи. Реакционную смесь охлаждают до комнатной температуры, добавляют 50 мл этилацетата, перемешивают в течение 10 мин и получают прозрачный раствор, промывают насыщенным водным раствором гидрокарбоната натрия, насыщенным водным раствором Na2SO3 и рассолом, органическую фазу сушат над безводным NaSO4 и растворитель выпаривают в вакууме, остаток очищают с помощью флэш-хроматографии и получают (S)-5-бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (3а, R1 = стирил) в виде белого твердого вещества. Спектроскопические данные являются такими же, как в примере 2, методика 1. Методика проведения ВЭЖХ (для методик 1-6) Колонка: Eclipse XDB-C18; 150×4,6 мм; 5 мкм. Подвижная фаза А (0,1% Н3РО4) в воде; Подвижная фаза В (ацетонитрил). Градиентный режим: 0 мин (30% В); 8 мин (95% В); 15 мин (95% В). Скорость потока: 1,0 мл мин-1. Длина волны: 210 нм. Температура: 30°С. Время удерживания: 12,3 мин (3а, R1 = стирил)
Пример 4: (S)-5-Бифенил-4-илметил-1-((Е)-бут-1-енил)-пирролидин-2-он (3а, R1=1-бутенил)

Методика 1
(S)-5-Бифенил-4-илметилпирролидин-2-он (3а, R1=Н) (10,0 г, 39,8 ммоля) растворяют в 100 мл безводного тетрагидрофурана, последовательно добавляют бутиловый альдегид (2,9 г, 39,8 ммоля) и пентоксид фосфора (6,2 г, 43,2 ммоля), реакционную смесь кипятят с обратным холодильником в атмосфере азота в течение 12 ч. Реакционную смесь охлаждают до комнатной температуры, фильтруют и промывают этилацетатом, фильтрат промывают 10% водным раствором гидрокарбоната натрия, затем рассолом. Органический слой сушат над безводным Na2SO4 и концентрируют в вакууме, остаток очищают с помощью колоночной флэш-хроматографии с использованием смеси гептан/этилацетат=10:1 и получают (S)-5-бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (3а, R1 = 1-бутенил).1Н ЯМР (400 МГц, CDCl3): 1,07 (t, 3Н, СН3Н), 2,12 (m, 2Н, СН2Н), 2,20 (m, 2Н, 3-СН2), 2,35 (m, 2Н, 2-СН2), 2,95 (m, 1Н, 5-СН2), 3,13 (m, 1Н, 5-СН2), 4,14 (m, 1Н, 4-СН), 5,27 (m, 1Н, СН=СН), 6,85 (d, 1H, HC=CH-N), 7,14-7,51 (9H, m, ароматический).
Методика 2
(S)-5-Бифенил-4-илметилпирролидин-2-он (3а, R1=Н) (5,02 г, 20 ммолей) растворяют в 50 мл толуола, добавляют бутиловый альдегид (1,44 г, 20 ммолей) и TsOH·Н2О (50 мг, 0,3 ммоля). Реакционную колбу снабжают ловушкой Дина-Штарка и холодильником, смесь кипятят с обратным холодильником в атмосфере азота в течение ночи. Реакционную смесь охлаждают до комнатной температуры, добавляют 50 мл толуола, перемешивают в течение 10 мин и получают прозрачный раствор, промывают насыщенным водным раствором гидрокарбоната натрия, насыщенным водным раствором Na2SO3 и рассолом, органическую фазу сушат над безводным Na2SO4 и растворитель выпаривают в вакууме, остаток очищают с помощью флэш-хроматографии и получают (S)-5-бифенил-4-илметил-1-((Е)-бут-1-енил)-пирролидин-2-он (3а, R1 = 1-бутенил). Спектроскопические данные являются такими же, как в примере 4, методика 1.
Методика 3
(S)-5-Бифенил-4-илметилпирролидин-2-он (3а, R1=Н) (5,02 г, 20 ммолей) растворяют в 50 мл толуола, добавляют бутиловый альдегид (1,44 г, 20 ммолей) и BF3-Et2O (0,5 мл). Реакционную колбу снабжают ловушкой Дина-Штарка и холодильником, смесь кипятят с обратным холодильником в атмосфере азота в течение ночи. Реакционную смесь охлаждают до комнатной температуры, добавляют 50 мл толуола, перемешивают в течение 10 мин и получают прозрачный раствор, промывают насыщенным водным раствором гидрокарбоната натрия, насыщенным водным раствором Na2SO3 и рассолом, органическую фазу сушат над безводным Na2SO4 и растворитель выпаривают в вакууме, остаток очищают с помощью флэш-хроматографии и получают (S)-5-бифенил-4-илметил-1-((Е)-бут-1-енил)-пирролидин-2-он (3а, R1 = 1-бутенил). Спектроскопические данные являются такими же, как в примере 4, методика 1.
Методика 4
(S)-5-Бифенил-4-илметилпирролидин-2-он (3а, R1=Н) (5,0 г, 20 ммолей) растворяют в 50 мл тетрагидрофурана, добавляют диэтилацеталь бутилового альдегида (3,2 г, 22 ммоля) и ППТС (215 мг, 1 ммоль). Реакционную колбу снабжают оборудованием для перегонки и холодильником для удаления этанола, образующегося в ходе реакции, смесь кипятят с обратным холодильником в атмосфере азота в течение ночи. Реакционную смесь охлаждают до комнатной температуры, добавляют 50 мл толуола, промывают насыщенным водным раствором гидрокарбоната натрия, насыщенным водным раствором Na2SO3 и рассолом, органическую фазу сушат над безводным Na2SO4 и растворитель выпаривают в вакууме, остаток очищают с помощью флэш-хроматографии и получают (S)-5-бифенил-4-илметил-1-((Е)-бут-1-енил)-пирролидин-2-он (3а, R1 = 1-бутенил). Спектроскопические данные являются такими же, как в примере 4, методика 1.
Методика 5
(S)-5-Бифенил-4-илметилпирролидин-2-он (3а, R1=Н) (5,0 г, 20 ммолей) растворяют в 50 мл толуола, добавляют диэтилацеталь бутилового альдегида (3,2 г, 22 ммоля) и ППТС (215 мг, 1 ммоль). Реакционную колбу снабжают устройством для перегонки и холодильником для удаления этанола, образующегося в ходе реакции, смесь кипятят с обратным холодильником в атмосфере азота в течение ночи. Реакционную смесь охлаждают до комнатной температуры, добавляют 50 мл толуола, промывают насыщенным водным раствором гидрокарбоната натрия, насыщенным водным раствором Na2SO3 и рассолом, органическую фазу сушат над безводным Na2SO4 и растворитель выпаривают в вакууме, остаток очищают с помощью флэш-хроматографии и получают (S)-5-бифенил-4-илметил-1-((Е)-бут-1-енил)-пирролидин-2-он (3а, R1 = 1-бутенил). Спектроскопические данные являются такими же, как в примере 4, методика 1.
Методика проведения ВЭЖХ (для методик 1-5) Колонка: Eclipse XDB-C18; 150×4,6 мм; 5 мкм. Подвижная фаза А (0,1% H3PO4) в воде; Подвижная фаза В (ацетонитрил). Градиентный режим: 0 мин (30% В); 8 мин (95% В); 15 мин (95% В). Скорость потока: 1,0 мл мин-1. Длина волны: 210 нм. Температура: 30°С.
Время удерживания: 9,5 мин (3а, R1 = 1-бутенил)
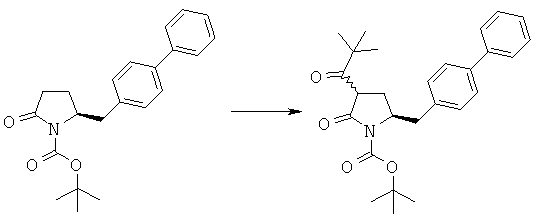
Пример 5: трет-Бутиловый эфир (3R/S)-бензоил-(5S)-бифенил-4-илметил-2-оксопирролидин-1-карбоновой кислоты (4а, R1 = трет-бутоксикарбонил, R4 = фенил)

Методика 1
н-Бутиллитий (56 мл, 2,5 М раствор в гексане, 0,14 моля) в атмосфере N2 при -10°С добавляют к смеси ГМДС (24,2 г, 0,15 моля) и 300 мл сухого тетрагидрофурана, затем полученную смесь перемешивают при -10°С в течение 30 мин. К реакционной смеси при -10°С добавляют смесь трет-бутилового эфира (S)-2-бифенил-4-илметил-5-оксопирролидин-1-карболовой кислоты (3а, R1 = трет-бутоксикарбонил) (35,1 г, 0,1 моля) и 50 мл сухого тетрагидрофурана, примерно через 30 мин к реакционной смеси при -10°С добавляют н-бутиллитий (40 мл, 2,5 М раствор в гексане, 0,1 моля), затем полученную смесь перемешивают при -10°С в течение 30 мин. К реакционной смеси при -10°С добавляют бензоилхлорид (15,5 г, 0,11 моля), примерно через 1 ч при -10°С реакционную смесь разбавляют с помощью 100 мл насыщенного водного раствора хлорида аммония и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют, затем органическую фазу концентрируют в вакууме. Добавляют этилацетат (200 мл), фильтруют и фильтрат концентрируют и получают трет-бутиловый эфир (R/S)-3-бензоил-(S)-5-бифенил-4-илметил-2-оксопирролидин-1-карболовой кислоты (4а, R1 = трет-бутоксикарбонил, R4 = фенил), по данным ВЭЖХ соотношение (3S,5R):(3R,5R) составляет 70:30.1Н ЯМР (400 МГц, ДМСО): 1,52 (s, 9H, (СН3)3), 2,05 (m, 1H, 3-CHH), 2,60 (m, 1H, 3CHH), 2,96 (m, 1H, 5-CHH), 3,13 (m, 1H, 5-CHH), 4,21 (m, 1H, 2-CH), 4,53 (m, 1H, 4-CH), 7,10-8,10 (14H, m, ароматический).
Методика 2
н-Бутиллитий (2,4 мл, 2,5 M раствор в гексапе, 6 ммолей) в атмосфере N2 при -10°С добавляют к смеси диизопропиламина (0,71 г, 7 ммолей) и 20 мл сухого тетрагидрофурана, затем полученную смесь перемешивают при -10°С в течение 30 мин. К реакционной смеси при -10°С добавляют смесь трет-бутилового эфира (S)-2-бифенил-4-илметил-5-оксопирролидин-1 -карбоновой кислоты (3а, R1 = трет-бутоксикарбонил) (1,76 г, 5 ммолей) и 5 мл сухого тетрагидрофурана, примерно через 30 мин к реакционной смеси при -10°С добавляют н-бутиллитий (2 мл, 2,5 М раствор в гексане, 5 ммолей), затем полученную смесь перемешивают при -10°С в течение 30 мин. К реакционной смеси при -10°С добавляют бензоилхлорид (0,77 г, 5,5 ммоля), примерно через 1 ч при -10°С реакционную смесь разбавляют с помощью 100 мл насыщенного водного раствора хлорида аммония и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Затем органическую фазу концентрируют в вакууме. Добавляют 20 мл этилацетата, фильтруют и фильтрат концентрируют и получают трет-бутиловый эфир (R/S)-3-бензоил-(S)-5-бифенил-4-илметил-2-оксопирролидин-1-карбоновой кислоты (4а, R1 = трет-бутоксикарбонил, R4 = фенил). По данным ВЭЖХ соотношение (3S,5R):(3R,5R) составляет 70:30. Спектроскопические данные являются такими же, как в примере 5, методика 1.
Методика 3
LHMDS (132 мл, 1,0 М раствор в тетрагидрофуране, 132 ммоля) в атмосфере N2 при -10°С добавляют к смеси трет-бутилового эфира (S)-2-Бифенил-4-илметил-5-оксопирролидин-1-карбоновой кислоты (3а, R1 = трет-бутоксикарбонил) (21,06 г, 60 ммолей) и 150 мл сухого тетрагидрофурана, затем полученную смесь перемешивают при -10°С в течение 30 мин. К реакционной смеси при -10°С добавляют бензоилхлорид (9,28 г, 66 ммолей), примерно через 1 ч при -10°С реакционную смесь разбавляют с помощью 50 мл насыщенного водного раствора хлорида аммония и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Затем органическую фазу концентрируют в вакууме. Добавляют 100 мл этилацетата, фильтруют и фильтрат концентрируют и получают трет-бутиловый эфир (R/S)-3-бензоил-(S)-5-бифенил-4-илметил-2-оксопирролидин-1-карбоновой кислоты (4а, R1 = трет-бутоксикарбонил, R4 = фенил). По данным ВЭЖХ соотношение (3S,5R):(3R,5R) составляет 70:30. Спектроскопические данные являются такими же, как в примере 5, методика 1.
Методика 4
трет-Бутиловый эфир (S)-2-бифенил-4-илметил-5-оксопирролидин-1-карбоновой кислоты (3а, R1 = трет-бутоксикарбонил) (1,4 г, 4 ммоля) в атмосфере N2 растворяют в 4 мл толуола и кипятят с обратным холодильником, добавляют гидрид натрия (55% в минеральном масле, 0,23 г, 5,2 ммоля) и перемешивают при кипячении с обратным холодильником в течение 2 ч, затем к реакционной смеси добавляют бензоилхлорид (0,62 г, 4,4 ммоля), затем полученную смесь перемешивают при кипячении с обратным холодильником в течение 2 ч. Реакционную смесь разбавляют с помощью 5 мл насыщенного водного раствора хлорида аммония и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Органическую фазу концентрируют досуха, остаток очищают с помощью колоночной хроматографии (этилацетат/гептан=1/2) и получают трет-бутиловый эфир (R/S)-3-бензоил-(S)-5-бифенил-4-илметил-2-оксопирролидин-1-карбоновой кислоты (4а, R1 = трет-бутоксикарбонил, R4 = фенил). По данным ВЭЖХ соотношение (3S,5R):(3R,5R) составляет 70:30. Спектроскопические данные являются такими же, как в примере 5, методика 1.
Методика 5
К раствору трет-бутилового эфира (S)-2-бифенил-4-илметил-5-оксопирролидин-1-карбоновой кислоты (3а, R1 = трет-бутоксикарбонил) (10 г, 28,49 ммоля) в ТГФ (56 мл) в атмосфере N2 добавляют MgCl2 (2,56 г, 28,49 ммоля) и триэтиламин (8,63 г, 85,47 ммоля). Смесь охлаждают до 5°С, перемешивают в течение 10 мин, затем по каплям добавляют бензоилхлорид (6,40 г, 45,58 ммоля). Реакционную смесь перемешивают при 5°С в течение 1 ч, затем температуру повышают до 10°С. После перемешивания в течение 15 ч добавляют 20 мл воды, затем раствор H3PO4 (12,0 г) в воде (20 мл). Затем водную фазу удаляют, органическую фазу дважды промывают рассолом (40 мл), органические экстракты концентрируют в вакууме и получают трет-бутиловый эфир (R/S)-3-бензоил-(S)-5-бифенил-4-илметил-2-оксопирролидин-1-карбоновой кислоты (4а, R1 = трет-бутоксикарбонил, R4 = фенил). По данным ВЭЖХ соотношение (3S,5R):(3R,5R) составляет 70:30. Спектроскопические данные являются такими же, как в примере 5, методика 1.
Методика проведения ВЭЖХ (для методик 1-5)
Колонка: Eclipse XDB-C18; 150×4,6 мм; 5 мкм. Подвижная фаза А (0,1% H3PO4) в воде; Подвижная фаза В (ацетонитрил). Градиентный режим: 0 мин (30% В); 8 мин (95% В); 15 мин (95% В). Скорость потока: 1,0 мл мин-1. Длина волны: 210 нм. Температура: 30°С.
Время удерживания:
10,3 мин (3S,5R-4, R1 = трет-бутоксикарбонил, R4 = фенил)
10,5 мин (3R,5R-4, R1 = трет-бутоксикарбонил, R4 = фенил)
9,2 мин (3а, R1 = трет-бутоксикарбонил)
Пример 6: (R/S)-3-Бензоил-(S)-5-бифенил-4-илметил-1-пирролидин-1-илметилпирролидин-2-он (4а, R1 = пирролидинилметил, R4 = фенил)

Методика 1
Смесь (S)-5-бифенил-4-илметил-1-пирролидин-1-илметилпирролидин-2-она (3а, R1 = пирролидинилметил) (6,68 г, 20 ммолей) и метилового эфира бензойной кислоты (3,0 г, 22 ммоля) в 20 мл толуола в атмосфере N2 кипятят с обратным холодильником, добавляют гидрид натрия (55% в минеральном масле, 1,14 г, 26 ммолей) и перемешивают при кипячении с обратным холодильником в течение ночи. Реакционную смесь разбавляют с помощью 20 мл насыщенного водного раствора хлорида аммония и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют, органическую фазу концентрируют досуха, остаток очищают с помощью колоночной хроматографии (этилацетат/гептан=1/1) и получают (R/S)-3-бензоил-(R)-5-бифенил-4-илметил-1-пирролидин-1-илметилпирролидин-2-он (4a, R1 = пирролидинилметил, R4 = фенил), по данным ВЭЖХ соотношение (3S,5R):(3R,5R) составляет 60:40.1Н ЯМР (400 МГц, CDCl3): 1,68 (m, 4Н, 2×СН2СН2), 1,98 (m, 1Н, 3-СНН), 2,23 (m, 1Н, 3-СНН), 2,51 (m, 4Н, 2×NCH2), 2,65 (dd, 1Н, N-CHH), 3,15 (dd, 1Н, N-CHH), 2,67 (d, 1Н, 5-CHH), 2,92 (d, 1Н, 5-CHH), 3,73 (m, 1Н, 2-CH), 3,78 (m, 1Н, 4-CH), 7,20-8,00 (14H, m, ароматический).
Методика 2
Смесь (S)-5-бифенил-4-илметил-1-пирролидин-1-илметилпирролидин-2-опа (3а, R1 = пирролидинилметил) (1,34 г, 4 ммоля) и метилового эфира бензойной кислоты (0,6 г, 4,4 ммоля) в 4 мл диметилформамида перемешивают в атмосфере N2 при 20°С, добавляют гидрид натрия (55% в минеральном масле, 0,23 г, 5,2 ммоля) и перемешивают при 110°С в течение ночи. Реакционную смесь разбавляют с помощью 5 мл насыщенного водного раствора хлорида аммония и 10 мл этилацетата, перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Органическую фазу концентрируют досуха, остаток очищают с помощью колоночной хроматографии(этилацетат/гептан=1/1) и получают (R/S)-3-бензоил-(R)-5-бифенил-4-илметил-1-пирролидин-1-илметилпирролидин-2-он (4a, R1 = пирролидинилметил, R4 = фенил), по данным ВЭЖХ соотношение (3S,5R):(3R,5R) составляет 60:40. Спектроскопические данные являются такими же, как в примере 6, методика 1.
Методика 3
Смесь (S)-5-бифенил-4-илметил-1-пирролидин-1-илметилпирролидин-2-она (3а, R1 = пирролидинилметил) (1,34 г, 4 ммоля), ДМТП (0,56 г, 4,4 ммоля) и метилового эфира бензойной кислоты (0,6 г, 4,4 ммоля) в 4 мл диметилформамида перемешивают в атмосфере N2 при 20°С, добавляют гидрид натрия (55% в минеральном масле, 0,23 г, 5,2 ммоля) и перемешивают при 110°С в течение 3 ч. Реакционную смесь разбавляют с помощью 5 мл насыщенного водного раствора хлорида аммония и 10 мл этилацетата, перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Органическую фазу концентрируют досуха, остаток очищают с помощью колоночной хроматографии (этилацетат/гептан=1/1) и получают (R/S)-3-бензоил-(R)-5-бифенил-4-илметил-1-пирролидин-1-илметилпирролидин-2-он (4a, R1 = пирролидинилметил, R4 = фенил), по данным ВЭЖХ соотношение (3S,5R):(3R,5R) составляет 60:40. Спектроскопические данные являются такими же, как в примере 6, методика 1.
Методика 4
Смесь (S)-5-бифенил-4-илметил-1-пирролидин-1-илметилпирролидин-2-он (3а, R1 = пирролидинилметил) (1,34 г, 4 ммоля), ДМТП (0,56 г, 4,4 ммоля) и метилового эфира бензойной кислоты (0,6 г, 4,4 ммоля) в 4 мл толуола перемешивают в атмосфере N2 при 20°С, добавляют гидрид натрия (55% в минеральном масле, 0,23 г, 5,2 ммоля) и перемешивают при 60°С в течение 3 ч. Реакционную смесь разбавляют с помощью 5 мл насыщенного водного раствора хлорида аммония и 10 мл этилацетата, перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Органическую фазу концентрируют досуха, остаток очищают с помощью колоночной хроматографии (этилацетат/гептан=1/1) и получают (R/S)-3-бензоил-(R)-5-бифенил-4-илметил-1-пирролидин-1-илметилпирролидин-2-он (4а, R1 = пирролидинилметил, R4 = фенил), по данным ВЭЖХ соотношение (3S,5R):(3R,5R) составляет 60:40. Спектроскопические данные являются такими же, как в примере 6, методика 1.
Методика проведения ВЭЖХ (для методик 1-4)
Колонка: Eclipse XDB-C18; 150×4,6 мм; 5 мкм. Подвижная фаза А (0,1% H3PO4) в воде; Подвижная фаза В (ацетонитрил). Градиентный режим: 0 мин (90% В); 10 мин (95% В); 15 мин (95% В). Скорость потока: 0,7 мл мин-1. Длина волны: 210 нм. Температура: 30°С.
Время удерживания:
8,6 мин (3S,5R-4, R1 = пирролидинилметил, R4 = фенил)
8,8 мин (3R,5R-4, R1 = пирролидинилметил, R4 = фенил).
7,5 мин (3а, R1 = пирролидинилметил)
Пример 7: (3R/S)-Бензоил-(5S)-бифенил-4-илметил-1-(4-метоксибензил)-пирролидин-2-оп (4а, R1 = п-метоксибензил, R4 = фенил)

К суспензии гидрида натрия (150 мг, 55% в минеральном масле, 3,75 ммоля) в толуоле (1 мл) добавляют (5S)-бифенил-4-илметил-1-(4-метоксибензил)пирролидин-2-он (3а, R1 = п-метоксибензил) (694 мг, 1,86 ммоля), затем добавляют метиловый эфир бензойной кислоты (253 мг, 1,86 ммоля), затем полученную смесь нагревают при 130°С в течение 9 ч. После охлаждения до комнатной температуры добавляют NH4Cl (насыщенный водный раствор, 5 мл), затем этилацетат (3 мл). Органический слой отделяют, промывают рассолом и концентрируют и получают (3R/S)-бензоил-(5S)-бифенил-4-илметил-1-(4-метоксибензил)пирролидин-2-он (4а, R1 = п-метоксибензил, R4 = фенил) в виде смеси диастереоизомеров (3S,5R) и (3R,5R) (состав 50:40 по данным ВЭЖХ).1H ЯМР (400 МГц, ДМСО): 1,82-2,08 (m, 2H), 2,25-2,70 (m, 2H), 3,00-3,26 (m, 2H), 3,73 (s, 3Н), 4,00-4,10 (m, 1H), 4,20-4,52 (m, 1H), 4,94-5,09 (m, 1H), 6,86-8,10 (18H, m, ароматический). МС (ИЭР, m/e) 476 (MH+).
Методика проведения ВЭЖХ
Колонка: Eclipse XDB-C18; 150×4,6 мм; 5 мкм. Подвижная фаза А (0,1% H3PO4) в воде; Подвижная фаза В (ацетонитрил). Градиентный режим: 0 мин (30% В); 8 мин (95% В); 15 мин (95% В). Скорость потока: 1,0 мл мин-. Длина волны: 210 нм. Температура: 30°С.
Время удерживания:
10,0 мин (3S, 5S-4, R1 = п-метоксибензил, R4 = фенил)
10,1 мин (3R, 5S-4a, R1 = п-метоксибензил, R4 = фенил)
8,8 мин (3а, R1 = п-метоксибензил)
Пример 8: (S)-3-Бензоил-5-бифепил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (4а, R1 = стирил, R4 = бензил)

Методика 1
(S)-5-Бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (3а, R1 = стирил) (50,0 г, 141,7 ммоля) в атмосфере N2 нагревают в 140 мл безводного толуола до растворения, порциями добавляют гидрид натрия (8,04 г, 184,2 ммоля), перемешивают при этой температуре в течение 10 мин, затем по каплям добавляют метилбензоат, кипятят с обратным холодильником в течение 6 ч, охлаждают до комнатной температуры, реакцию останавливают насыщенным водным раствором NH4Cl, органическую фазу отделяют, экстрагируют толуолом (100 мл × 3), объединенные органические экстракты промывают насыщенным водным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4 и концентрируют в вакууме и получают (S)-3-бензоил-5-бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (4а, R1 = стирил, R4 = фенил), по данным ВЭЖХ соотношение (3S,5S):(3R,5S) составляет 70:30.1Н ЯМР (400 МГц, CDCl3): 2,35 (m, 1Н, 3-СНН), 3,26 (m, 3Н, 5-СН2 + 3-CHH), 3,74 (m, 1Н, 2-CH), 4,32 (m, 1H, 4-CH), 6,21 (m, 1Н, C=CHH), 7,21~7,80 (m, 20H, ароматический + OCHH).
Методика 2
(S)-5-Бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (3а, R1 = стирил) (10,0 г, 28,3 ммоля) в атмосфере N2 нагревают в 30 мл безводного толуола до растворения, одной порцией добавляют MeONa (1,99 г, 36,8 ммоля), перемешивают при этой температуре в течение 10 мин, затем по каплям добавляют метилбензоат, кипятят с обратным холодильником в течение ночи, охлаждают до комнатной температуры, реакцию останавливают насыщенным водным раствором NH4Cl, органическую фазу отделяют, экстрагируют толуолом (15 мл × 3), объединенные органические экстракты промывают насыщенным водным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4 и концентрируют в вакууме. Остаток очищают с помощью колоночной хроматографии на силикагеле, при элюировании смесью гептан/этилацетат состава 5:1 и получают (S)-3-бензоил-5-бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (4а, R1 = стирил, R4 = фенил), по данным ВЭЖХ соотношение (3S,5S):(3R,5S) составляет 70:30. Спектроскопические данные являются такими же, как в примере 8, методика 1.
Методика 3
(S)-5-Бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (3а, R1 = стирил) (1,00 г, 2,8 ммоля) в атмосфере N2 нагревают в 3 мл безводного толуола до растворения, одной порцией добавляют t-BuOK (0,43 г, 3,7 ммоля), перемешивают при этой температуре в течение 10 мин, затем по каплям добавляют метилбензоат, реакционную смесь кипятят с обратным холодильником в течение ночи, охлаждают до комнатной температуры, реакцию останавливают насыщенным водным раствором NH4Cl, органическую фазу отделяют, экстрагируют толуолом (30 мл × 3), объединенные органические экстракты промывают насыщенным водным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4 и концентрируют в вакууме. Остаток очищают с помощью колоночной хроматографии на силикагеле, при элюировании смесью гептан/этилацетат состава 5:1 и получают (S)-3-бензоил-5-бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (4а, R1 = стирил, R4 = фенил), по данным ВЭЖХ соотношение (3S,5S):(3R,5S) составляет 70:30. Спектроскопические данные являются такими же, как в примере 8, методика 1. Методика проведения ВЭЖХ (для методик 1-3) Колонка: Eclipse XDB-C18; 150×4,6 мм; 5 мкм. Подвижная фаза А (0,1% H3PO4) в воде; Подвижная фаза В (ацетонитрил). Градиентный режим: 0 мин (30% В); 8 мин (95% В); 15 мин (95% В). Скорость потока: 1,0 мл мин-1. Длина волны: 210 нм. Температура: 30°С.
Время удерживания:
10,7 мин (3S,5S-4, R1 = стирил, R4 = фенил)
11,0 мин (3R,5S-4, R1 = стирил, R4 = фенил)
12,3 мин (3а, R1 = стирил)
Пример 9: (S)-3-Бензоил-5-бифенил-4-илметил-1-((Е)-бут-1-енил)-пирролидин-2-он (4а, R1 = 1-бутенил, R4 = фенил)

Гидрид натрия (2,03 г, 46,4 ммоля) суспендируют в 40 мл безводного толуола, кипятят с обратным холодильником в атмосфере азота, по каплям добавляют раствор смеси (S)-5-бифенил-4-илметил-1-((Е)-бут-1-енил)-пирролидин-2-она (3а, R1 = 1-бутенил) (11,02 г, 35,67 ммоля) и метилбензоата (4,86 г, 35,67 ммоля) в безводном толуоле, реакционную смесь кипятят с обратным холодильником в течение 4 ч, охлаждают до температуры окружающей среды, реакцию останавливают насыщенным водным раствором NH4Cl, смесь экстрагируют толуолом, объединенные органические экстракты промывают насыщенным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4, концентрируют в вакууме и получают (S)-3-бензоил-5-бифенил-4-илметил-1-((Е)-бут-1-енил)-пирролидин-2-он (4а, R1 = 1-бутенил, R4 = бензил), который используют на следующей стадии без обработки.1H ЯМР (400 МГц, CDCl3): 1,07 (t, 3Н, СН3Н), 2,12 (m, 2Н, СН2Н), 2,20 (m, 2 Н, 3-СН2), 2,95 (m, 1Н, 5-СН2), 3,13 (m, 1Н, 5-СН2), 3,82 (m, Н, 2-СН2Н), 4,14 (m, 1Н, 4-СН), 5,27 (m, 1Н, СН=СН), 6,85 (d, 1Н, HC=CH-N), 7,14-8,20 (14H, m, ароматический). По данным ВЭЖХ соотношение (3S,5S):(3R,5S) составляет 60:40.
Методика проведения ВЭЖХ
Колонка: Eclipse XDB-C18; 150×4,6 мм; 5 мкм. Подвижная фаза А (0,1% H3PO4) в воде; Подвижная фаза В (ацетонитрил). Градиентный режим: 0 мин (30% В); 8 мин (95% В); 15 мин (95% В). Скорость потока: 1,0 мл мин-1. Длина волны: 210 нм. Температура: 30°С.
Время удерживания:
10,5 мин (3S,5S-4, R1 = 1-бутенил, R4 = фенил)
10,8 мин (3R,5S-4, R1 = 1-бутенил, R4 = фенил).
9,5 мин (3а, R1 = 1-бутенил)
Пример 10: (R/S)-1,3-Дибензоил-(S)-5-бифенил-4-илметилпирролидин-2-он (4а, R1 = бензоил, R4 = фенил)

1-Бензоил-(S)-5-бифенил-4-илметилпирролидин-2-он (3а, R1 = бензоил) (1,34 г, 4 ммоля) в атмосфере N2 растворяют в 4 мл толуола и кипятят с обратным холодильником, добавляют гидрид натрия (55% в минеральном масле, 0,23 г, 5,2 ммоля) и перемешивают при кипячении с обратным холодильником в течение 2 ч, затем к реакционной смеси добавляют бензоилхлорид (0,62 г, 4,4 ммоля), затем полученную смесь перемешивают при кипячении с обратным холодильником в течение 2 ч. Реакционную смесь разбавляют с помощью 5 мл насыщенного водного раствора хлорида аммония и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Органическую фазу концентрируют досуха, остаток очищают с помощью колоночной хроматографии (этилацетат/гептан=1/2) и получают (R/S)-1,3-дибензоил-(S)-5-бифенил-4-илметилпирролидин-2-он (4а, R1 = бензоил, R4 = фенил), по данным ВЭЖХ соотношение (3S,5S):(3R,5S) составляет 30:70.1Н ЯМР (400 МГц, CDCl3): 2,09 (m, 1Н, 3-СНН), 2,34 (m, 1Н, 3-СНН), 2,71 (d, 1H, 5-СНН), 2,96 (d, 1Н, 5-СНН), 3,73 (m, 1Н, 2-СН), 4,38 (m, 1Н, 4-СН), 7,20-8,10 (19Н, m, ароматический).
Методика проведения ВЭЖХ
Колонка: Eclipse XDB-C18; 150×4,6 мм; 5 мкм. Подвижная фаза А (0,1% H3PO4) в воде; Подвижная фаза В (ацетонитрил). Градиентный режим: 0 мин (30% В); 8 мин (95% В); 15 мин (95% В). Скорость потока: 1,0 мл мин-1. Длина волны: 210 нм. Температура: 30°С.
Время удерживания:
11,0 мин (3S,5S-4, R1 = бензоил, R4 = фенил)
11,1 мин (3R,5S-4, R1 = бензоил, R4 = фенил)
9,3 мин (3а, R1 = бензоил)
Пример 11: трет-Бутиловый эфир (R/S)-3-ацетил-(S)-5-бифенил-4-илметил-2-оксопирролидин-1-карбоновой кислоты (4а, R1 = трет-бутоксикарбонил, R4 = метил)

LHMDS (12,5 мл, 1,0 М раствор в тетрагидрофуране, 12,5 ммоля) в атмосфере N2 при -10°С добавляют к смеси трет-бутилового эфира (S)-2-бифенил-4-илметил-5-оксопирролидин-1-карбоновой кислоты (3а, R1 = трет-бутоксикарбонил) (1,76 г, 5 ммолей) в 15 мл сухого тетрагидрофурана, затем полученную смесь перемешивают при -10°С в течение 30 мин. К реакционной смеси при -10°С добавляют ацетилхлорид (0,47 г, 6 ммолей), примерно через 1 ч при -10°С реакционную смесь разбавляют с помощью 10 мл насыщенного водного раствора хлорида аммония и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Затем органическую фазу концентрируют в вакууме. Добавляют этилацетат (20 мл), фильтруют и фильтрат концентрируют и получают трет-бутиловый эфир (R/S)-3-ацетил-(S)-5-бифенил-4-илметил-2-оксопирролидин-1-карбоновой кислоты (3а, R1 = трет-бутоксикарбонил), по данным ВЭЖХ соотношение (3S,5S):(3R,5S) составляет 70:30.1H ЯМР (400 МГц, CDCl3): 1,48 (s, 9H, (СН3)3), 2,17 (m, 1H, 3-СНН), 2,31 (s, 3Н, СН3), 2,42 (m, 1H, 3СНН), 2,67 (m, 1H, 5-СНН), 2,92 (m, 1H, 5-СНН), 3,09 (m, 1H, 2-СН), 4,38 (m, 1H, 4-СН), 7,10-7,80 (9H, m, ароматический).
Методика проведения ВЭЖХ
Колонка: Eclipse XDB-C18; 150×4,6 мм; 5 мкм. Подвижная фаза А (0,1% Н3РО4) в воде; Подвижная фаза В (ацетонитрил). Градиентный режим: 0 мин (30% В); 8 мин (95% В); 15 мин (95% В). Скорость потока: 1,0 мл мин-1. Длина волны: 210 нм. Температура: 30°С.
Время удерживания:
9,3 мин (3S,5S-4, R1 = трет-бутоксикарбонил, R4 = метил)
9,5 мин (3R,5S-4, R1 = трет-бутоксикарбонил, R4 = метил)
9,2 мин (3а, R1 = трет-бутоксикарбонил)
Пример 12: трет-Бутиловый эфир (R/S)-3-изобутирил-(S)-5-бифенил-4-илметил-2-оксопирролидин-1-карбоновой кислоты (4а, R1 = трет-бутоксикарбонил, R4 = изопропил)

LHMDS (12,5 мл, 1,0 М раствор в тетрагидрофуране, 12,5 ммоля) в атмосфере N2 при -10°С добавляют к смеси трет-бутилового эфира (S)-2-бифенил-4-илметил-5-оксопирролидин-1-карбоновой кислоты (3а, R1 = трет-бутоксикарбонил) (1,76 г, 5 ммолей) в 15 мл сухого тетрагидрофурана, затем полученную смесь перемешивают при -10°С в течение 30 мин. К реакционной смеси при -10°С добавляют изобутирилхлорид (0,64 г, 6 ммолей), примерно через 1 ч при -10°С реакционную смесь разбавляют с помощью 10 мл насыщенного водного раствора хлорида аммония и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Затем органическую фазу концентрируют в вакууме. Добавляют 20 мл этилацетата, фильтруют и фильтрат концентрируют и получают трет-бутиловый эфир (R/S)-3-изобутирил-(S)-5-бифенил-4-илметил-2-оксопирролидин-1 -карбоновой кислоты (4а, R1 = трет-бутоксикарбонил, R4 = изопропил), по данным ВЭЖХ соотношение (3S,5S):(3R,5S) составляет 70:30.1Н ЯМР (400 МГц, CDCl3): 1,03 (d, 3Н, СН3), 1,05 (d, 3Н, СН3), 1,43 (8, 9Н, (СН3)3), 2,18 (m, 1Н, 3-СНН), 2,43 (m, 1Н, 3СНН), 2,70 (m, 1Н, СН), 2,67 (m, 1Н, 5-СНН), 2,92 (m, 1Н, 5-СНН), 3,09 (m, 1Н, 2-СН), 4,38 (m, 1Н, 4-СН), 7,10-7,80 (9Н, m, ароматический).
Методика проведения ВЭЖХ
Колонка: Eclipse XDB-C18; 150×4,6 мм; 5 мкм. Подвижная фаза А (0,1% H3PO4) в воде; Подвижная фаза В (ацетопитрил). Градиентный режим: 0 мин (30% В); 8 мин (95% В); 15 мин (95% В). Скорость потока: 1,0 мл мин-1. Длина волны: 210 нм. Температура: 30°С.
Время удерживания:
10,2 мин (3S,5S-4, R1 = трет-бутоксикарбонил, R4 = изопропил)
10,5 мин (3R,5S-4, R1 = трет-бутоксикарбонил, R4 = изопропил)
9,2 мин (3а, R1 = трет-бутоксикарбонил)
Пример 13: трет-Бутиловый эфир (R/S)-3-(2,2-диметилпропионил)-(S)-5-бифенил-4-илметил-2-оксопирролидин-1-карбоновой кислоты (4а, R1 = трет-бутоксикарбонил, R4 = трет-бутил)
LHMDS (12,5 мл, 1,0 М раствор в тетрагидрофуране, 12,5 ммоля) в атмосфере N2 при -10°С добавляют к смеси трет-бутилового эфир (S)-2-бифенил-4-илметил-5-оксопирролидин-1-карбоновой кислоты (3а, R1 = трет-бутоксикарбонил) (1,76 г, 5 ммолей) в 15 мл сухого тетрагидрофурана, затем полученную смесь перемешивают при -10°С в течение 30 мин. К реакционной смеси при -10°С добавляют 2,2-диметилпропионилхлорид (0,72 г, 6 ммолей), примерно через 1 ч при -10°С реакционную смесь разбавляют с помощью 10 мл насыщенного водного раствора хлорида аммония и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Затем органическую фазу концентрируют в вакууме. Добавляют 20 мл этилацетата, фильтруют и фильтрат концентрируют и получают трет-бутиловый эфир (R/S)-3-(2,2-диметилпропионил)-(S)-5-бифепил-4-илметил-2-оксопирролидин-1-карбоновой кислоты (4а, R1 = трет-бутоксикарбонил, R4 = изопропил), по данным ВЭЖХ соотношение (3S,5S):(3R,5S) составляет 80:20.1Н ЯМР (400 МГц, CDCl3): 0,98 (s, 9H, 3СН3), 1,52 (s, 9H, (СН3)3), 2,19 (m, 1H, 3-СНН), 2,42 (m, 1Н, 3СНН), 2,67 (m, 1H, 5-СНН), 2,92 (m, 1H, 5-СНН), 3,09 (m, 1H, 2-СН), 4,38 (m, 1H, 4-СН), 7,10-7,80 (9H, m, ароматический).
Методика проведения ВЭЖХ
Колонка: Eclipse XDB-C18; 150×4,6 мм; 5 мкм. Подвижная фаза А (0,1% H3PO4) в воде; Подвижная фаза В (ацетопитрил). Градиентный режим: 0 мин (30% В); 8 мин (95% В); 15 мин (95% В). Скорость потока: 1,0 мл мин-1. Длина волны: 210 им. Температура: 30°С.
Время удерживания:
10,3 мин (3S,5S-4, R1 = трет-бутоксикарбонил, R4 = изопропил)
10,5 мин (3R,5S-4, R1 = трет-бутоксикарбонил, R4 = изопропил)
9,2 мин (3а, R1 = трет-бутоксикарбонил)
Пример 14: (S)-5-Бифенил-4-илметил-3-изобутирил-1-((Е)-стирил)-пирролидип-2-он (4а, R1 = стирил, R4 = изопропил)

Методика 1
(S)-5-Бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (3а, R1 = стирил) (5,0 г, 14,2 ммоля) нагревают в 15 мл безводного толуола до растворения, затем порциями добавляют гидрид натрия (0,8 г, 18,4 ммоля), перемешивают в атмосфере азота в течение 30 мин, затем по каплям добавляют метилизобутират (1,7 г, 15,6 ммоля, чистота 98%), реакционную смесь кипятят с обратным холодильником в атмосфере азота в течение ночи, охлаждают до комнатной температуры, реакцию останавливают насыщенным раствором NH4Cl, смесь экстрагируют толуолом, объединенные экстракты промывают насыщенным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4, растворитель выпаривают в вакууме, остаток очищают с помощью колоночной хроматографии и получают (S)-5-бифенил-4-илметил-3-изобутирил-1-((Е)-стирил)-пирролидин-2-он (4a, R1 = стирил, R4 = изопропил).1H ЯМР (400 МГц, CDCl3): 1,02 (m, 6H, С(СН3)2), 2,05 (m, 1Н, 3-CHH), 2,46 (m, 1H, 3-CHH), 2,97 (m, 1H, 5-CH2H), 3,10 (m, 1H, СН(СН3)2)), 3,17 (m, 1H, 5-CH2H), 3,22 (m, 1H, 2-CHH), 4,46 (m, 1H, 4-CH), 6,10 (d, 1H, C=CHH), 7,21-7,65 (15H, m, ароматический + C=CHH). По данным ВЭЖХ соотношение (3S,5S):(3R,5S) составляет 70:30.
Методика 2
(S)-5-Бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (3а, R1 = стирил) (2,0 г, 5,7 ммоля) растворяют в 15 мл безводного тетрагидрофурана, охлаждают до -10 ~ -15°С в бане из соли со льдом, в атмосфере азота по каплям добавляют LiHMDS (14,2 мл, 14,2 ммоля, 1 M раствор в тетрагидрофуране), полученную смесь перемешивают при этой температуре в течение 30 мин, затем по каплям добавляют раствор изобутирилхлорида (820 мг, 6,8 ммоля) в 5 мл безводного тетрагидрофурана, температуру поддерживают ниже -5°С, перемешивают в течение 1 ч, реакцию останавливают насыщенным водным раствором NH4Cl, добавляют 50 мл этилацетата, водную фазу отделяют, промывают насыщенным водным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4, растворитель выпаривают в вакууме и получают (S)-5-бифенил-4-илметил-3-изобутирил-1-((Е)-стирил)-пирролидин-2-он (4a, R1 = стирил, R4=изопропил), который непосредственно используют на следующей стадии. По данным ВЭЖХ соотношение (3S,5S):(3R,5S) составляет 70:30. Спектроскопические данные являются такими же, как в примере 14, методика 1.
Методика проведения ВЭЖХ (для методик 1 и 2)
Колонка: Eclipse XDB-C18; 150×4,6 мм; 5 мкм. Подвижная фаза А (0,1% НзР04) в воде; Подвижная фаза В (ацетонитрил). Градиентный режим: 0 мин (30% В); 8 мин (95% В); 15 мин (95% В). Скорость потока: 1,0 мл мин-1. Длина волны: 210 нм. Температура: 30°С.
Время удерживания:
10,7 мин (3S,5S-4, R1 = стирил, R4 = изопропил)
11,1 мин (3R,5S-4, R1 = стирил, R4 = изопропил)
9,5 мин (3а, R1 = стирил)
Пример 15: (S)-5-Бифенил-4-илметил-3-(2,2-диметилпропионил)-1-((Е)-стирил)-пирролидин-2-он (4а, R1 = стирил, R4 = трет-бутил)

Методика 1
(S)-5-Бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (3а, R1 = стирил) (5,0 г, 14,2 ммоля) нагревают в 15 мл безводного толуола до растворения, затем порциями добавляют гидрид натрия (0,8 г, 18,4 ммоля), перемешивают в атмосфере азота в течение 30 мин, затем по каплям добавляют метилтриметилацетатхлорид (1,9 г, 15,6 ммоля), реакционную смесь кипятят с обратным холодильником в атмосфере азота в течение ночи, охлаждают до КТ, реакцию останавливают насыщенным раствором NH4Cl, смесь экстрагируют толуолом, объединенные экстракты промывают насыщенным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4, растворитель выпаривают в вакууме, остаток очищают с помощью колоночной хроматографии и получают (S)-5-бифенил-4-илметил-3-(2,2-диметилпропионил)-1-((Е)-стирил)-пирролидин-2-он (4а, R1 = стирил, R4 = трет-бутил).1Н ЯМР (400 МГц, CDCl3): 1,02 (s, 9H, С(СН3)), 2,05 (m, 1Н, 3-CHH), 2,46 (m, 1H, 3-CHH), 2,97 (m, 1H, 5-CH2H), 3,17 (m, 1H, 5-CH2H), 3,22 (m, 1H, 2-CHH), 4,46 (m, 1H, 4-CH), 6,15 (d, 1H, C=CHH), 7,21-7,65 (15H, m, ароматический + C=CHH). По данным ВЭЖХ соотношение (3S,5S):(3R,5S) составляет 82:18.
Методика 2
(S)-5-Бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (3а, R1 = стирил) (2,0 г, 5,7 ммоля) растворяют в 15 мл безводного тетрагидрофурана, охлаждают до -10 ~ -15°С в бане из соли со льдом, в атмосфере азота по каплям добавляют LiHMDS (14,2 мл, 14,2 ммоля, 1 M раствор в тетрагидрофуране), полученную смесь перемешивают при этой температуре в течение 30 мин, затем по каплям добавляют изобутирилхлорид (820 мг, 6,8 ммоля) в виде раствора в 5 мл безводного тетрагидрофурана, температуру поддерживают ниже -5°С, перемешивают в течение 1 ч, реакцию останавливают насыщенным водным раствором NH4Cl, добавляют 50 мл этилацетата, водную фазу отделяют, промывают насыщенным водным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4, растворитель выпаривают в вакууме и получают (S)-5-бифенил-4-илметил-3-(2,2-диметилпропионил)-1-((Е)-стирил)-пирролидин-2-он (4а, R1 = стирил, R4 = трет-бутил), который непосредственно используют на следующей стадии. По данным ВЭЖХ соотношение (3S,5S):(3R,5S) составляет 80:20. Спектроскопические данные являются такими же, как в примере 15, методика 1.
Методика проведения ВЭЖХ (для методик 1 и 2)
Колонка: Eclipse XDB-C18; 150×4,6 мм; 5 мкм. Подвижная фаза А (0,1% H3PO4) в воде; Подвижная фаза В (ацетонитрил). Градиентный режим: 0 мин (30% В); 8 мин (95% В); 15 мин (95% В). Скорость потока: 1,0 мл мин-1. Длина волны: 210 нм. Температура: 30°С.
Время удерживания:
10,7 мин (3S,5S-4, R1 = стирил, R4 = трет-бутил)
11,1 мин (3R,5S-4, R1 = стирил, R4 = трет-бутил)
9,5 мин (3а, R1 = стирил)
Пример 16: 1-трет-Бутиловый эфир (S)-5-бифенил-4-илметил-2-оксопирролидин-1,3-дикарбоновой кислоты (4а, R1 = трет-бутоксикарбонил, R4 = гидроксигруппа)

К раствору диизопропиламина (2,4 г, 24,0 ммоля) в 20 мл безводного 25 тетрагидрофурана в атмосфере N2 при -10 ~ 15°С по каплям добавляют н-бутиллитий (8,8 мл, 22,0 ммоля, 2,5 М раствор в гексане), перемешивают при этой температуре в течение 1 ч, по каплям добавляют раствор трет-бутилового эфира (S)-2-бифенил-4-илметил-5-оксопирролидин-1-карбоновой кислоты (3а, R1 = трет-бутоксикарбонил) (7,0 г, 20,0 ммоля) в 10 мл безводного тетрагидрофурана, перемешивают в течение 1 ч, через смесь в течение 1 ч пропускают СО2, реакцию останавливают насыщенным водным раствором NH4Cl, смесь экстрагируют этилацетатом, объединенную органическую фазу промывают рассолом, сушат над безводным сульфатом натрия, растворитель выпаривают в вакууме, остаток очищают с помощью колоночной хроматографии и получают 1-трет-бутиловый эфир (S)-5-бифенил-4-илметил-2-оксопирролидин-1,3-дикарбоновой кислоты (4а, R1 = трет-бутоксикарбонил, R4 = гидроксигруппа).1Н ЯМР (400 МГц, CDCl3): 1,59 (s, 1Н, Boc), 1,85 (m, 1H, 3СНН), 2,36 (m, 1Н, 3СНН), 2,48 (m, 1Н, 5-СН2), 2,83 (m, 1Н, 5-СН2), 3,09 (m, 1Н, 2-CH), 4,40 (m, 1Н, 4-СН), 6,10 (d, 1 H, OCHH), 7,23-7,56 (9Н, m, ароматический). По данным ВЭЖХ соотношение (3S,5S):(3R,5S) составляет 36:64.
Методика проведения ВЭЖХ
Колонка: Eclipse XDB-C18; 150×4,6 мм; 5 мкм. Подвижная фаза А (0,1% H3PO4) в воде; Подвижная фаза В (ацетонитрил). Градиентный режим: 0 мин (30% В); 8 мин (95% В); 15 мин (95% В). Скорость потока: 1,0 мл мин-1. Длина волны: 210 нм. Температура: 30°С.
Время удерживания:
8,0 мин (3S,5S-4, R1 = трет-бутоксикарбонил, R4 = гидроксигруппа)
8,1 мин (3R,5S-4, R1 = трет-бутоксикарбонил, R4 = гидроксигруппа).
9,2 мин (3а, R1 = трет-бутоксикарбонил)
Пример 17: (5R)-Бифенил-4-илметил-1-(4-метоксибензил)-3-метиленпирролидин-2-он (1a, R1 = п-метоксибензил)

(3R/S)-Бензоил-(5S)-бифенил-4-илметил-1-(4-метоксибепзил)пирролидин-2-он (4а, R1 = п-метоксибензил, R4 = фенил) в виде смеси цис- и транс-диастереоизомеров (700 мг, 1,47 ммоля) растворяют в тетрагидрофуране (5 мл), затем при 0°С добавляют гидрид натрия (60 мг, 1,47 ммоля, 55% в минеральном масле). После перемешивания в течение 15 мин добавляют параформальдегид (66 мг, 2,2 ммоля), реакционную смесь кипятят с обратным холодильником в течение 30 мин и ей дают охладиться до комнатной температуры. После добавления NH4Cl (насыщенный водный раствор) органический слой отделяют, промывают рассолом, сушат и концентрируют и получают (5R)-бифенил-4-илметил-1-(4-метоксибензил)-3-метиленпирролидин-2-он (1a, R1 = п-метоксибензил).1Н ЯМР (400 МГц, CDCl3): 2,46-2,52 (m, 2H), 2,61-2,67 (m, 1H), 3,13-3,16 (m, 1H), 3,67-3,71 (m, 1H), 3,81 (s, 3Н), 4,09 (d, J=14,8 Гц, 1H), 5,15 (d, J=14,8 Гц, 1H), 5,30 (1H, m), 6,02 (1H, m), 7,20-7,58 (13H, m, ароматический).
МС (ИЭР, m/е) 384 (МН+). Методика проведения ВЭЖХ
Колонка: Eclipse XDB-C18; 150×4,6 мм; 5 мкм. Подвижная фаза А (0,1% Н3РО4) в воде; Подвижная фаза В (ацетонитрил). Градиентный режим: 0 мин (30% В); 8 мин (95% В); 15 мин (95% В). Скорость потока: 1,0 мл мин-1. Длина волны: 210 нм. Температура: 30°С.
Время удерживания:
9,3 мин (1a, R1 = п-метоксибензил)
Пример 18: трет-Бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил)

Методика 1
трет-Бутиловый эфир (R/S)-3-бензоил-(S)-5-бифенил-4-илметил-2-оксопирролидин-1-карбоновой кислоты (4а, R1 = трет-бутоксикарбонил, R4 = фенил) (1,82 г, 4 ммоля) в 5 мл толуола в атмосфере N2 при 0°С добавляют к смеси гидрида натрия (55% в минеральном масле, 0,23 г, 5,2 ммоля) и 15 мл толуола и затем кипятят с обратным холодильником, порциями добавляют параформальдегид (0,25 г, 8 ммолей) и кипятят с обратным холодильником в течение 2 ч. Затем реакционную смесь разбавляют с помощью 10 мл насыщенного водного раствора хлорида аммония и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Затем органическую фазу концентрируют в вакууме, добавляют 20 мл этилацетата, фильтруют и фильтрат концентрируют и очищают с помощью колоночной хроматографии при элюировании смесью гептан/этилацетат состава 5:1 и получают трет-бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбопил). Спектроскопические данные являются такими же, как приведенные в WO 2009/090251 (пример 23, методика 1).
Методика 2
Смесь трет-бутилового эфира (R/S)-3-бензоил-(S)-5-бифенил-4-илметил-2-оксопирролидин-1-карбоновой кислоты (4а, R1 = трет-бутоксикарбонил, R4 = фенил) (0,455 г, 1 ммоль), параформальдегида (60 мг, 2 ммоля), хлорида лития (85 мг, 2 ммоля) и диизопропилэтиламина (0,168 г, 1,3 ммоля) в 5 мл сухого тетрагидрофурана кипятят с обратным холодильником в течение 2 ч. Затем реакционную смесь разбавляют с помощью 5 мл насыщенного водного раствора хлорида аммония и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Затем органическую фазу концентрируют в вакууме. Добавляют 10 мл этилацетата, фильтруют и фильтрат концентрируют и очищают с помощью колоночной хроматографии при элюировании смесью гептан/этилацетат состава 5:1 и получают трет-бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил). Спектроскопические данные являются такими же, как приведенные в WO 2009/090251 (пример 23, методика 1).
Методика 3
Смесь трет-бутилового эфира (R/S)-3-бензоил-(S)-5-бифенил-4-илметил-2-оксопирролидин-1-карбоновой кислоты (4а, R1 = трет-бутоксикарбонил, R4 = фенил) (0,455 г, 1 ммоль), параформальдегида (60 мг, 2 ммоля) и KOtBu (0,148 г, 1,3 ммоля) в 5 мл сухого тетрагидрофурана кипятят с обратным холодильником в течение 2 ч. Затем реакционную смесь разбавляют с помощью 5 мл насыщенного водного раствора хлорида аммония и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Затем органическую фазу концентрируют в вакууме. Добавляют 10 мл этилацетата, фильтруют и фильтрат концентрируют и очищают с помощью колоночной хроматографии при элюировании смесью гептан/этилацетат состава 5:1 и получают трет-бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил). Спектроскопические данные являются такими же, как приведенные в WO 2009/090251 (пример 23, методика 1).
Методика 4
Смесь трет-бутилового эфира (R/S)-3-бензоил-(S)-5-бифенил-4-илметил-2-оксопирролидин-1-карбоновой кислоты (4а, R1 = трет-бутоксикарбонил, R4 = фенил) (0,455 г, 1 ммоль), параформальдегида (60 мг, 2 ммоля), LiCl (85 мг, 2 ммоля) и ДБУ (0,2 г, 1,3 ммоля) в 5 мл сухого тетрагидрофурана кипятят с обратным холодильником в течение 2 ч. Затем реакционную смесь разбавляют с помощью 5 мл насыщенного водного раствора хлорида аммония и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Затем органическую фазу концентрируют в вакууме. Добавляют 10 мл этилацетата, фильтруют и фильтрат концентрируют и очищают с помощью колоночной хроматографии при элюировании смесью гептан/этилацетат состава 5:1 и получают трет-бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил). Спектроскопические данные являются такими же, как приведенные в WO 2009/090251 (пример 23, методика 1).
Методика 5
Смесь трет-бутилового эфира (R/S)-3-бензоил-(S)-5-бифенил-4-илметил-2-оксопирролидин-1-карбоновой кислоты (4а, R1 = трет-бутоксикарбонил, R4 = фенил) (0,455 г, 1 ммоль), формальдегида (37% раствор в воде, 0,24 г, 3 ммоля), тетрабутилгидроксида аммония (6,5 мг, 0,01 ммоля) и К2СО3 (0,28 г, 1,3 ммоля, растворенный в 1 мл воды) в 5 мл сухого тетрагидрофурана нагревают до 50°С и перемешивают в течение 2 ч. Затем реакционную смесь разбавляют с помощью 5 мл рассола и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Затем органическую фазу концентрируют в вакууме. Добавляют 10 мл этилацетата, фильтруют и фильтрат концентрируют и очищают с помощью колоночной хроматографии при элюировании смесью гептан/этилацетат состава 5:1 и получают трет-бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил). Спектроскопические данные являются такими же, как приведенные в WO 2009/090251 (пример 23, методика 1).
Методика 6
Смесь трет-бутилового эфира (R/S)-3-бензоил-(S)-5-бифенил-4-илметил-2-оксопирролидин-1-карбоновой кислоты (4а, R1 = трет-бутоксикарбонил, R4 = фенил) (0,455 г, 1 ммоль), параформальдегида (60 мг, 2 ммоля) и трет-бутоксида калия (0,148 г, 1,3 ммоля) в 5 мл сухого ДМЭ перемешивают при 20°С в течение 2 ч. Затем реакционную смесь разбавляют с помощью 5 мл насыщенного водного раствора хлорида аммония и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Затем органическую фазу концентрируют в вакууме. Добавляют 10 мл этилацетата, фильтруют и фильтрат концентрируют и очищают с помощью колоночной хроматографии при элюировании смесью гептан/этилацетат состава 5:1 и получают трет-бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил). Спектроскопические данные являются такими же, как приведенные в WO 2009/090251 (пример 23, методика 1).
Пример 19: трет-Бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил)

Смесь трет-бутилового эфира (R/S)-3-ацетил-(S)-5-бифенил-4-илметил-2-оксопирролидин-1-карбоновой кислоты (4а, R1 = трет-бутоксикарбонил, R4 = метил) (0,79 г, 2 ммоля), формальдегида (37% раствор в воде, 0,49 г, 6 ммолей), тетрабутилгидроксида аммония (26 мг, 0,1 ммоля) и К2СО3 (0,55 г, 1,3 ммоля, растворенный в 2 мл воды) в 10 мл сухого тетрагидрофурана нагревают до 50°С и перемешивают в течение 2 ч. Затем реакционную смесь разбавляют с помощью 10 мл рассола и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Затем органическую фазу концентрируют в вакууме. Добавляют 20 мл этилацетата, фильтруют и фильтрат концентрируют и очищают с помощью колоночной хроматографии при элюировании смесью гептан/этилацетат состава 5:1 и получают трет-бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил). Спектроскопические данные являются такими же, как приведенные в WO 2009/090251 (пример 23, методика 1).
Пример 20: трет-Бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил)

Смесь трет-бутилового эфира (R/S)-3-изобутирил-(S)-5-бифенил-4-илметил-2-оксопирролидин-1-карбоновой кислоты (4а, R1 = трет-бутоксикарбонил, R4 = изопропил) (0,84 г, 2 ммоля), формальдегида (37% раствор в воде, 0,49 г, 6 ммолей), тетрабутилгидроксида аммония (26 мг, 0,1 ммоля) и К2СО3 (0,55 г, 1,3 ммоля, растворенный в 2 мл воды) в 10 мл сухого тетрагидрофурана нагревают до 50°С и перемешивают в течение 2 ч. Затем реакционную смесь разбавляют с помощью 10 мл рассола и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Затем органическую фазу концентрируют в вакууме. Добавляют 20 мл этилацетата, фильтруют и фильтрат концентрируют и очищают с помощью колоночной хроматографии при элюировании смесью гептан/этилацетат состава 5:1 и получают трет-бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1а, R1 = трет-бутоксикарбонил). Спектроскопические данные являются такими же, как приведенные в WO 2009/090251 (пример 23, методика 1).
Пример 21: трет-Бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карболовой кислоты (1a, R1 = трет-бутоксикарбонил)

Смесь трет-бутилового эфира (R/S)-3-(2,2-диметилпропионил)-(S)-5-бифенил-4-илметил-2-оксопирролидин-1-карбоновой кислоты (4а, R1 = трет-бутоксикарбонил, R4 = трет-бутил) (0,87 г, 2 ммоля), формальдегида (37% раствор в воде, 0,49 г, 6 ммолей), тетрабутилгидроксида аммония (26 мг, 0,1 ммоля) и К2СО3 (0,55 г, 1,3 ммоля, растворенный в 2 мл воды) в 10 мл сухого тетрагидрофурана нагревают до 50°С и перемешивают в течение 2 ч. Затем реакционную смесь разбавляют с помощью 10 мл рассола и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Затем органическую фазу концентрируют в вакууме. Добавляют 20 мл этилацетата, фильтруют и фильтрат концентрируют и очищают с помощью колоночной хроматографии при элюировании смесью гептан/этилацетат состава 5:1 и получают трет-бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил). Спектроскопические данные являются такими же, как приведенные в WO 2009/090251 (пример 23, методика 1).
Пример 22: (R)-5-Бифенил-4-илметил-3-метилен-1-пирролидип-1-илметилпирролидин-2-он (1a, R1 = пирролидинилметил)

Смесь (R/S)-3-бензоил-(S)-5-бифенил-4-илметил-1-пирролидин-1-илметилпирролидин-2-она (4а, R1 = пирролидинилметил, R4 = фенил) (0,44 г, 1 ммоль), формальдегида (37% раствор в воде, 0,24 г, 3 ммоля), ТБАГ (6,5 мг, 0,01 ммоля) и К2СО3 (0,28 г, 1,3 ммоля, растворенный в 1 мл воды) в 5 мл сухого тетрагидрофурана нагревают до 50°С и перемешивают в течение 2 ч. Затем реакционную смесь разбавляют с помощью 5 мл рассола и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Затем органическую фазу концентрируют в вакууме. Добавляют 10 мл этилацетата, фильтруют и фильтрат концентрируют и очищают с помощью колоночной хроматографии при элюировании смесью гептан/этилацетат состава 5:1 и получают (R)-5-бифенил-4-илметил-3-метилен-1-пирролидин-1-илметилпирролидин-2-он (1а, R1 = пирролидинилметил).1Н ЯМР (400 МГц, CDCl3): 1,68 (m, 4Н, 2×СН2СН2), 2,06 (m, 1Н, 3-СНН), 2,31 (m, 1Н, 3-СНН), 2,51 (m, 4Н, 2×NCH2), 2,67 (d, 1Н, 5-СНН), 2,65 (dd, 1Н, N-CHH), 3,15 (dd, 1Н, N-CHH), 2,92 (d, 1Н, 5-СНН), 4,01 (m, 1Н, 4-СН), 5,54 (d, 1Н, =СНН), 6,02 (d, 1Н, =СНН), 7,20-7,80 (14Н, m, ароматический).
Методика проведения ВЭЖХ
Колонка: Eclipse XDB-C18; 150×4,6 мм; 5 мкм. Подвижная фаза А (0,1% НзР04) в воде; Подвижная фаза В (ацетонитрил). Градиентный режим: 0 мин (90% В); 10 мин (95% В); 15 мин (95% В). Скорость потока: 0,7 мл мин-1. Длина волны: 210 им. Температура: 30°С.
Время удерживания:
8,1 мин (1a, R1 = пирролидинилметил)
Пример 23: (R)-5-Бифенил-4-илметил-3-метилен-1-((Е)-стирил)-пирролидин-2-он (1a, R1 = стирил)

Методика 1
(S)-3-Бензоил-5-бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (4а, R1 = стирил, R4 = фенил) (0,50 г, 1,1 ммоля) растворяют в 2 мл безводного толуола, порциями добавляют гидрид натрия (65 мг, 1,5 ммоля), затем одной порцией добавляют параформальдегид (66 мг, 2,2 ммоля), полученную смесь кипятят с обратным холодильником в атмосфере азота в течение 4 ч. Реакционную смесь охлаждают до комнатной температуры, затем реакцию останавливают насыщенным водным раствором NH4Cl, смесь экстрагируют толуолом, органические экстракты промывают насыщенным водным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4, растворитель выпаривают в вакууме и получают (R)-5-бифенил-4-илметил-3-метилен-1-((Е)-стирил)-пирролидин-2-он (1a, R1 = стирил), который можно непосредственно использовать на следующей стадии.1Н ЯМР (400 МГц, CDCl3): 2,68 (m, 2H, 3-СНН + 5-СН2Н), 2,81 (m, 1Н, 3-СНН), 3,28 (m, 1H, 5-CH2H), 4,32 (m, 1H, 4-CH), 5,35 (s, 1H, C=CHH), 6,04 (s, 1H, OCHH), 6,23 (d, 1H, C=CHH), 7,21~7,72 (m, 15H, ароматический + C=CHH).
Методика 2
(S)-3-Бензоил-5-бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (4а, R1 = стирил, R4 = фенил) (0,50 г, 1,1 ммоля) растворяют в 2 мл безводного тетрагидрофурана, одной порцией добавляют трет-бутоксид калия (161 мг, 1,4 ммоля) и параформальдегид (66 мг, 2,2 ммоля), полученную смесь кипятят с обратным холодильником в атмосфере азота в течение 2 ч. Реакционную смесь охлаждают до комнатной температуры, добавляют 15 мл воды, экстрагируют толуолом, органические экстракты промывают насыщенным водным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4, растворитель выпаривают в вакууме и получают неочищенный продукт, который очищают с помощью колоночной хроматографии и получают (R)-5-бифенил-4-илметил-3-метилен-1-((Е)-стирил)-пирролидин-2-он (1a, R1 = стирил). Спектроскопические данные являются такими же, как в примере 23, методика 1.
Методика 3
(S)-3-Бензоил-5-бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (4а, R1 = стирил, R4 = фенил) (0,50 г, 1,1 ммоля) растворяют в 2 мл безводного тетрагидрофурана, одной порцией добавляют трет-бутоксид калия (161 мг, 1,4 ммоля) и параформальдегид (66 мг, 2,2 ммоля), полученную смесь перемешивают в атмосфере азота при комнатной температуре в течение ночи. К реакционной смеси добавляют 15 мл воды, экстрагируют толуолом, органические экстракты промывают насыщенным водным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4, растворитель выпаривают в вакууме, остаток очищают с помощью колоночной хроматографии и получают (R)-5-бифенил-4-илметил-3-метилен-1-((Е)-стирил)-пирролидин-2-он (1a, R1 = стирил). Спектроскопические данные являются такими же, как в примере 23, методика 1.
Методика 4
(S)-3-Бензоил-5-бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (4а, R1 = стирил, R4 = фенил) (0,50 г, 1,1 ммоля) растворяют в 2 мл метанола, одной порцией добавляют морфолин (125 мг, 1,4 ммоля) и параформальдегид (66 мг, 2,2 ммоля), кипятят с обратным холодильником в атмосфере азота в течение 2 ч. К реакционной смеси добавляют 10 мл 5% HCl(водн.), экстрагируют толуолом, органические экстракты промывают насыщенным водным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4, растворитель выпаривают в вакууме, остаток очищают с помощью колоночной хроматографии и получают (R)-5-бифенил-4-илметил-3-метилен-1-((Е)-стирил)-пирролидин-2-он (1a, R1 = стирил). Спектроскопические данные являются такими же, как в примере 23, методика 1.
Методика 5
(S)-3-Бензоил-5-бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (4а, R1 = стирил, R4 = фенил) (54,3 г, 118,8 ммоля) растворяют в 120 мл безводного тетрагидрофурана, добавляют параформальдегид (4,28 г, 142,6 ммоля), iPr2NEt (20,0 г, 154,5 ммоля) и безводный хлорид лития (2,56 г, 59,4 ммоля), кипятят с обратным холодильником в атмосфере азота в течение 2 ч. Реакционную смесь охлаждают до комнатной температуры, добавляют 200 мл толуола, промывают с помощью 5% HCl(водн.), насыщенным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4, растворитель выпаривают в вакууме и получают (R)-5-бифенил-4-илметил-3-метилен-1-((Е)-стирил)-пирролидин-2-он (1a, R1 = стирил). Спектроскопические данные являются такими же, как в примере 23, методика 1.
Методика 6
(S)-3-Бензоил-5-бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (4а, R1 = стирил, R4 = фенил) (1,0 г, 2,2 ммоля) растворяют в 5 мл безводного тетрагидрофурана, добавляют параформальдегид (80 мг, 2,6 ммоля), триэтиламин (288 мг, 2,8 ммоля) и безводный хлорид лития (186 мг, 4,4 ммоля), кипятят с обратным холодильником в атмосфере азота в течение ночи. Реакционную смесь охлаждают до комнатной температуры, добавляют 20 мл толуола, промывают с помощью 5% HCl(водн.), насыщенным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4, растворитель выпаривают в вакууме и получают (R)-5-бифенил-4-илметил-3-метилен-1-((Е)-стирил)-пирролидин-2-он (1a, R1 = стирил). Спектроскопические данные являются такими же, как в примере 23, методика 1.
Методика 7
(S)-3-Бензоил-5-бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (4а, R1 = стирил, R4 = фенил) (1,0 г, 2,2 ммоля) растворяют в 5 мл безводного тетрагидрофурана, добавляют параформальдегид (80 мг, 2,6 ммоля), ДБУ (434 мг, 2,8 ммоля) и безводный хлорид лития (186 мг, 4,4 ммоля), кипятят с обратным холодильником в атмосфере азота в течение ночи. Реакционную смесь охлаждают до комнатной температуры, добавляют 20 мл толуола, промывают с помощью 5% HCl(водн.), насыщенным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4, растворитель выпаривают в вакууме и получают (R)-5-бифенил-4-илметил-3-метилеп-1-((Е)-стирил)-пирролидин-2-оп (1a, R1 = стирил). Спектроскопические данные являются такими же, как в примере 23, методика 1.
Методика 8
(S)-3-Бензоил-5-бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (4а, R1 = стирил, R4 = фенил) (1,0 г, 2,2 ммоля) растворяют в 5 мл безводного тетрагидрофурана, добавляют 37% водный раствор формальдегида (0,36 мл, 4,4 ммоля), 1 М водный раствор К2СО3 (4,4 мл, 4,4 ммоля) и n-Bu4NOH (12 мг, 0,05 ммоля), нагревают при 40°С в течение 2 ч. Реакционную смесь охлаждают до комнатной температуры, экстрагируют этилацетатом, экстракты промывают насыщенным водным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4 и концентрируют в вакууме, остаток очищают с помощью колоночной хроматографии и получают (R)-5-бифенил-4-илметил-3-метилен-1-((Е)-стирил)-пирролидин-2-он (1a, R1 = стирил). Спектроскопические данные являются такими же, как в примере 23, методика 1.
Методика 9
(S)-3-Бензоил-5-бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (4а, R1 = стирил, R4 = фенил) (1,0 г, 2,2 ммоля) растворяют в 5 мл безводного тетрагидрофурана, добавляют iPr2NEt (370 мг, 2,8 ммоля), безводный хлорид лития (49 мг, 1,1 ммоля) и безводный Na2SO4 (260 мг, 2,2 ммоля), кипятят с обратным холодильником в атмосфере азота в течение 30 мин, затем добавляют параформальдегид (80 мг, 2,7 ммоля), кипятят с обратным холодильником в атмосфере азота в течение 2 ч. Реакционную смесь охлаждают до комнатной температуры, добавляют 20 мл толуола и 10 мл 5% HCl(водн.), водную фазу отделяют, промывают насыщенным водным раствором гидрокарбоната натрия и рассолом, сушат над безводным сульфатом натрия и концентрируют в вакууме и получают (R)-5-бифенил-4-илметил-3-метилен-1-((Е)-стирил)-пирролидин-2-он (1a, R1 = стирил). Спектроскопические данные являются такими же, как в примере 23, методика 1.
Методика 10
(S)-3-Бензоил-5-бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (4а, R1 = стирил, R4 = фенил) (1,0 г, 2,2 ммоля) растворяют в 5 мл безводного тетрагидрофурана, добавляют iPr2NEt (370 мг, 2,8 ммоля), безводный хлорид лития (49 мг, 1,1 ммоля) и безводный MgSO4 (260 мг, 2,2 ммоля), кипятят с обратным холодильником в атмосфере азота в течение 30 мин, затем добавляют параформальдегид (80 мг, 2,7 ммоля), кипятят с обратным холодильником в атмосфере азота в течение 2 ч. Реакционную смесь охлаждают до комнатной температуры, добавляют 20 мл толуола и 10 мл 5% HCl(водн.), водную фазу отделяют, промывают насыщенным водным раствором гидрокарбоната натрия и рассолом, сушат над безводным сульфатом натрия и концентрируют в вакууме и получают (R)-5-бифенил-4-илметил-3-метилен-1-((Е)-стирил)-пирролидин-2-он (1a, R1 = стирил).
Методика 11
(S)-3-Бензоил-5-бифенил-4-илметил-1-((Е)-стирил)-пирролидин-2-он (4а, R1 = стирил, R4 = фенил) (10,0 г, 21,9 ммоля) растворяют в 50 мл безводного тетрагидрофурана, добавляют iPr2NEt (3,7 г, 28,5 ммоля), безводный хлорид лития (490 мг, 11,0 ммоля) и молекулярные сита (2,0 г), кипятят с обратным холодильником в атмосфере азота в течение 30 мин, затем добавляют параформальдегид (790 мг, 26,3 ммоля), кипятят с обратным холодильником в атмосфере азота в течение 2 ч. Реакционную смесь охлаждают до комнатной температуры, добавляют 100 мл толуола, фильтруют для удаления молекулярных сит, фильтрат промывают с помощью 5% HCl(водн.) (50 мл × 2), насыщенным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4 и концентрируют в вакууме и получают (R)-5-бифенил-4-илметил-3-метилен-1-((Е)-стирил)-пирролидин-2-он (1a, R1 = стирил), который непосредственно используют на следующей стадии. Спектроскопические данные являются такими же, как в примере 23, методика 1.
Методика проведения ВЭЖХ (для методик 1-11)
Колонка: Eclipse XDB-C18; 150×4,6 мм; 5 мкм. Подвижная фаза А (0,1% H3PO4) в воде; Подвижная фаза В (ацетонитрил). Градиентный режим: 0 мин (30% В); 8 мин (95% В); 15 мин (95% В). Скорость потока: 1,0 мл мин-1. Длина волны: 210 нм. Температура: 30°С.
Время удерживания:
10,2 мин (1a, R1 = стирил)
Пример 24: (R)-5-Бифенил-4-илметил-1-((Е)-бут-1-енил)-3-метиленпирролидин-2-он (1а, R1 = 1-бутенил)

Методика 1
К суспензии гидрида натрия (2,03 г, 46,3 ммоля) в 40 мл безводного толуола, охлажденной до 0°С в бане со льдом, по каплям добавляют раствор (S)-3-бензоил-5-бифенил-4-илметил-1-((Е)-бут-1-енил)-пирролидин-2-она (4а, R1 = 1-бутенил, R4 = фенил) (14,7 г, 35,7 ммоля) в безводном толуоле, перемешивают в течение 30 мин, затем порциями добавляют параформальдегид (2,2 г, 71,4 ммоля), кипятят с обратным холодильником в атмосфере азота в течение 4 ч, охлаждают до комнатной температуры, реакцию останавливают насыщенным водным раствором NH4Cl, смесь экстрагируют толуолом, объединенные органические экстракты промывают насыщенным водным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4 и концентрируют и получают (R)-5-бифенил-4-илметил-1-((Е)-бут-1-енил)-3-метиленпирролидин-2-он (1a, R1 = 1-бутенил).1Н ЯМР (400 МГц, CDCl3): 1,07 (t, 3Н, СН3Н), 2,12 (m, 2H, СН2Н), 2,57 (m, 2Н, 3-СН2), 2,65 (m, 1Н, 5-СН2), 3,16 (m, 1Н, 5-СН2), 4,14 (m, 1Н, 4-СН), 5,22 (s, 1Н, С=СНН), 5,27 (m, 1Н, СН=СН), 5,91 (d, 1Н, НС=СНН), 6,85 (d, 1H, HOCH-N), 7,14-7,51 (9H, m, ароматический).
Методика 2
К раствору (S)-3-бензоил-5-бифенил-4-илметил-1-((Е)-бут-1-енил)-пирролидин-2-она (4а, R1 = 1-бутенил, R4 = фенил) (5,5 г, 13,2 ммоля) в 25 мл тетрагидрофурана добавляют 1 М водный раствор К2СО3 (26,4 мл, 26,4 ммоля), 37% водный раствор СН2О (2,2 мл, 26,4 ммоля) и n-Bu4NOH (0,4 мл, 0,7 ммоля), нагревают в атмосфере N2 при 45°С в течение 2 ч, охлаждают до комнатной температуры, экстрагируют толуолом, объединенный органический слой промывают с помощью 5% HCl(водн.), насыщенным водным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4, растворитель выпаривают, остаток очищают с помощью колоночной хроматографии и получают (R)-5-бифенил-4-илметил-1-((Е)-бут-1-енил)-3-метиленпирролидин-2-он (1a, R1 = 1-бутенил). Спектроскопические данные являются такими же, как в примере 24, методика 1.
Методика проведения ВЭЖХ (для методик 1 и 2)
Колонка: Eclipse XDB-C18; 150×4,6 мм; 5 мкм. Подвижная фаза А (0,1% H3PO4) в воде; Подвижная фаза В (ацетонитрил). Градиентный режим: 0 мин (30% В); 8 мин (95% В); 15 мин (95% В). Скорость потока: 1,0 мл мин-1. Длина волны: 210 нм. Температура: 30°С.
Время удерживания:
9,9 мин (1a, R1 = 1-бутенил).
Пример 25: (R)-5-Бифенил-4-илметил-3-метилен-1-((Е)-стирил)-пирролидин-2-он (1a, R1 = стирил)

(S)-5-Бифенил-4-илметил-3-изобутирил-1-((Е)-стирил)-пирролидин-2-он (4а, R1 = трет-бутоксикарбонил, R4 = изопропил) (2,5 г, 6,2 ммоля) растворяют в 10 мл безводного тетрагидрофурана, добавляют iPr2NEt (1,1 г, 8,2 ммоля), безводный LiCl (140 мг, 3,2 ммоля) и молекулярные сита (0,5 г), смесь кипятят с обратным холодильником в атмосфере азота в течение 30 мин, затем одной порцией добавляют параформальдегид (230 мг, 7,6 ммоля), кипятят с обратным холодильником в течение 2 ч, охлаждают до комнатной температуры, добавляют 50 мл толуола, фильтруют, фильтрат промывают с помощью 10% HCl(водн.), насыщенным раствором Na2CO3 и рассолом, сушат над безводным NaSO4, выпаривают в вакууме, остаток очищают с помощью колоночной хроматографии и получают (R)-5-бифенил-4-илметил-3-метилен-1-((Е)-стирил)-пирролидип-2-он (1a, R1 = старил). Спектроскопические данные являются такими же, как в примере 23, методика 1.
Пример 26: (R)-5-Бифенил-4-илметил-3-метилен-1-((Е)-стирил)-пирролидин-2-он (1a, R1 = стирил)
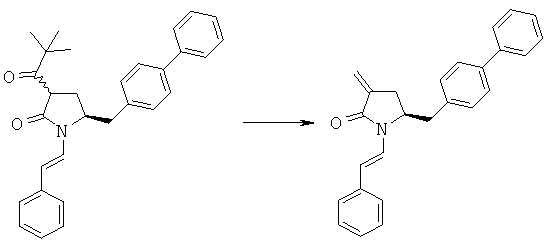
(S)-5-Бифенил-4-илметил-3-(2,2-диметилпропионил)-1-((Е)-стирил)-пирролидин-2-он (4а, R1 = трет-бутоксикарбонил, R4 = трет-бутил) (2,7 г, 6,2 ммоля) растворяют в 10 мл безводного тетрагидрофурана, добавляют iPr2NEt (1,1 г, 8,2 ммоля), безводный LiCl (140 мг, 3,2 ммоля) и молекулярные сита (0,5 г), смесь кипятят с обратным холодильником в атмосфере азота в течение 30 мин, затем одной порцией добавляют параформальдегид (230 мг, 7,6 ммоля), кипятят с обратным холодильником в течение 2 ч, охлаждают до комнатной температуры, добавляют 50 мл толуола, фильтруют, фильтрат промывают с помощью 10% HCl(водн.), насыщенным раствором Na2CO3 и рассолом, сушат над безводным NaSO4, выпаривают в вакууме, остаток очищают с помощью колоночной хроматографии и получают (R)-5-бифенил-4-илметил-3-метилен-1-((Е)-стирил)-пирролидин-2-он (1a, R1 = стирил). Спектроскопические данные являются такими же, как в примере 23, методика 1.
Пример 27: трет-Бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидии-1-карбоповой кислоты (1a, R1 = трет-бутоксикарбонил)

1-трет-Бутиловый эфир бифенил-4-илметил-2-оксопирролидин-1,3-дикарбоновой кислоты (4а, R1 = трет-бутоксикарбонил, R4 = гидроксигруппа) (3,95 г, 10,0 ммоля) растворяют в 10 мл метанола, добавляют параформальдегид (0,60 г, 20,0 ммоля) и морфолин (1,05 г, 12,0 ммоля), смесь кипятят с обратным холодильником в атмосфере азота в течение 2 ч, охлаждают до комнатной температуры, растворитель выпаривают в вакууме, остаток растворяют в 50 мл этилацетата, промывают с помощью 1% HCl(водн.), 10% водным раствором гидрокарбоната натрия и рассолом, сушат над безводным сульфатом натрия и концентрируют в вакууме и получают трет-бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил).1Н ЯМР (400 МГц, CDCl3): 1,62 (s, 9H, (СН3)3), 2,56 (m, 1H, 3-СНН), 2,60 (m, 1H, 3СНН), 2,70 (d, 1H, 5-СНН), 3,26 (d, 1H, 5-СНН), 4,43 (m, 1H, 4-СН), 5,45 (d, 1H, С=СНН), 6,17 (d, 1H, С=СНН), 7,20-7,60 (9H, m, ароматический); m/z (+ИЭР) 381 ([MNa]+, 7%), 364 ([MH]+, 12), 308 (100), 264 (10).
Методика проведения ВЭЖХ
Колонка: Eclipse XDB-C18; 150×4,6 мм; 5 мкм. Подвижная фаза А (0,1% H3PO4) в воде; Подвижная фаза В (ацетонитрил). Градиентный режим: 0 мин (30% В); 8 мин (95% В); 15 мин (95% В). Скорость потока: 1,0 мл мин-1. Длина волны: 210 нм. Температура: 30°С.
Время удерживания: 9,7 мин (1a, R1 = трет-бутоксикарбонил)
Пример 28: трет-Бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил)

Методика 1
н-Бутиллитий (56 мл, 2,5 М раствор в гексане, 0,14 моля) в атмосфере N2 при -10°С добавляют к смеси ГМДС (24,2 г, 0,15 моля) и 300 мл сухого тетрагидрофурана, затем полученную смесь перемешивают при -10°С в течение 30 мин. К реакционной смеси при -10°С добавляют смесь трет-бутилового эфира (S)-2-бифенил-4-илметил-5-оксопирролидин-1-карбоновой кислоты (3а, R1 = трет-бутоксикарбонил) (35,1 г, 0,1 моля) и 50 мл сухого тетрагидрофурана, примерно через 30 мин к реакционной смеси при -10°С добавляют н-бутиллитий (40 мл, 2,5 М раствор в гексане, 0,1 моля), затем полученную смесь перемешивают при -10°С в течение 30 мин. К реакционной смеси при -10°С добавляют бензоилхлорид (15,5 г, 0,11 моля), примерно через 1 ч при -10°С реакционную смесь разбавляют с помощью 100 мл насыщенного водного раствора хлорида аммония и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. К оставшейся органической фазе добавляют формальдегид (37% раствор в воде, 24,3 г, 0,3 моля), водный раствор К2СО3 (27,6 г в 100 мл воды, 0,2 моля) и раствор тетрабутиламмонийгидроксида (40% в воде, 1,3 г, 5 ммолей) и нагревают до 50°С и перемешивают в течение 2 ч. Затем реакционную смесь разбавляют с помощью 100 мл рассола и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Затем органическую фазу концентрируют в вакууме. Добавляют 200 мл этилацетата, фильтруют и фильтрат концентрируют и получают трет-бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил). Неочищенный трет-бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил) очищают с помощью колоночной хроматографии при элюировании смесью гептан/этилацетат состава 3:1.1Н ЯМР (400 МГц, CDCl3): 1,62 (s, 9Н, (СН3)3), 2,56 (m, 1Н, 3-СНН), 2,60 (m, 1Н, 3СНН), 2,70 (d, 1Н, 5-СНН), 3,26 (d, 1Н, 5-СНН), 4,43(т, 1Н, 4-СН), 5,45 (d, 1Н, С-СНН), 6,17 (d, 1Н, С=СНН), 7,20-7,60 (9Н, m, ароматический); m/z (+ИЭР) 381([MNa]+, 7%), 364 ([MH]+, 12), 308 (100), 264 (10). По данным ВЭЖХ соотношение промежуточных продуктов (3S,5S-4, R1 = трет-бутоксикарбонил, R4 = фенил):(3R,5S-4, R1 = трет-бутоксикарбонил, R4 = фенил) составляет 70:30.
Методика 2
н-Бутиллитий (56 мл, 2,5 М раствор в гексане, 0,14 моля) в атмосфере N2 при -10°С добавляют к смеси диизопропиламина (14,2 г, 0,15 моля) и 300 мл сухого тетрагидрофурана, затем полученную смесь перемешивают при -10°С в течение 30 мин. К реакционной смеси при -10°С добавляют смесь трет-бутилового эфира (S)-2-бифенил-4-илметил-5-оксопирролидин-1-карболовой кислоты (3а, R1 = трет-бутоксикарбонил) (35,1 г, 0,1 моля) и 50 мл сухого тетрагидрофурана, примерно через 30 мин к реакционной смеси при -10°С добавляют н-бутиллитий (40 мл, 2,5 М раствор в гексане, 0,1 моля), затем полученную смесь перемешивают при -10°С в течение 30 мин. К реакционной смеси при -10°С добавляют бензоилхлорид (15,5 г, 0,11 моля), примерно через 1 ч при -10°С реакционную смесь разбавляют с помощью 100 мл насыщенного водного раствора хлорида аммония и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. К оставшейся органической фазе добавляют формальдегид (37% раствор в воде, 24,3 г, 0,3 моля), водный раствор К2СО3 (27,6 г в 100 мл воды, 0,2 моля) и раствор тетрабутиламмонийгидроксида (40% в воде, 1,3 г, 5 ммолей) и нагревают до 50°С и перемешивают в течение 2 ч. Затем реакционную смесь разбавляют с помощью 100 мл рассола и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Затем органическую фазу концентрируют в вакууме. Добавляют 200 мл этилацетата, фильтруют и фильтрат концентрируют и получают трет-бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидип-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил). Неочищенный трет-бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил) можно очистить с помощью колоночной хроматографии при элюировании смесью гептан/этилацетат состава 3:1. По данным ВЭЖХ соотношение промежуточных продуктов (3S,5S-4, R1 = трет-бутоксикарбонил, R4 = фенил):(3R,5S-4, R1 = трет-бутоксикарбонил, R4 = фенил) составляет 70:30. Спектроскопические данные являются такими же, как в примере 28, методика 1.
Методика 3
LHMDS (25 мл, 1,0 М раствор в тетрагидрофуране, 25 ммолей) в атмосфере N2 при -10°С добавляют к смеси трет-бутилового эфира (S)-2-бифенил-4-илметил-5-оксопирролидин-1-карболовой кислоты (3а, R1 = трет-бутоксикарбонил) (3,51 г, 10 ммолей) и 20 мл сухого тетрагидрофурана, затем полученную смесь перемешивают при -10°С в течение 30 мин. К реакционной смеси при -10°С добавляют бензоилхлорид (1,55 г, 11 ммолей), примерно через 1 ч при -10°С реакционную смесь разбавляют с помощью 15 мл насыщенного водного раствора хлорида аммония и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. К оставшейся органической фазе добавляют формальдегид (37% раствор в воде, 2,43 г, 0,03 моля), водный раствор К2СО3 (2,76 г в 10 мл воды, 0,02 моля) и раствор тетрабутиламмонийгидроксида (40% в воде, 0,13 г, 0,5 ммоля) и нагревают до 50°С и перемешивают в течение 2 ч. Затем реакционную смесь разбавляют с помощью 15 мл рассола и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Затем органическую фазу концентрируют в вакууме. Добавляют 20 мл этилацетата, фильтруют и фильтрат концентрируют и получают трет-бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил). Неочищенный трет-бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил) можно очистить с помощью колоночной хроматографии при элюировании смесью гептан/этилацетат состава 3:1. По данным ВЭЖХ соотношение промежуточных продуктов (3S,5S-4a, R1 = трет-бутоксикарбонил, R4 = бензил):(3R,5S-4, R1 = трет-бутоксикарбонил, R4 = фенил) составляет 70:30. Спектроскопические данные являются такими же, как в примере 28, методика 1.
Методика 4
ДАЛ (25 мл, 1,0 М раствор в тетрагидрофуране, 25 ммолей) в атмосфере N2 при -10°С добавляют к смеси трет-бутилового эфира (S)-2-бифенил-4-илметил-5-оксопирролидин-1-карбоновой кислоты (3а, R1 = трет-бутоксикарбонил) (3,51 г, 10 ммолей) и 20 мл сухого тетрагидрофурана, затем полученную смесь перемешивают при -10°С в течение 30 мин. К реакционной смеси при -10°С добавляют бензоилхлорид (1,55 г, 11 ммолей), примерно через 1 ч при -10°С реакционную смесь разбавляют с помощью 15 мл насыщенного водного раствора хлорида аммония и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. К оставшейся органической фазе добавляют формальдегид (37% раствор в воде, 2,43 г, 0,03 моля), водный раствор К2СО3 (2,76 г в 10 мл воды, 0,02 моля) и раствор тетрабутиламмонийгидроксида (40% в воде, 0,13 г, 0,5 ммоля) и нагревают до 50°С и перемешивают в течение 2 ч. Затем реакционную смесь разбавляют с помощью 15 мл рассола и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Затем органическую фазу концентрируют в вакууме. Добавляют 20 мл этилацетата, фильтруют и фильтрат концентрируют и получают трет-бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил). Неочищенный трет-бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил) можно очистить с помощью колоночной хроматографии при элюировании смесью гептан/этилацетат состава 3:1. По данным ВЭЖХ соотношение промежуточных продуктов (3S,5S-4, R1 = трет-бутоксикарбонил, R4 = фенил):(3R,5S-4, R1 = трет-бутоксикарбонил, R4 = фенил) составляет 70:30. Спектроскопические данные являются такими же, как в примере 28, методика 1.
Методика проведения ВЭЖХ (для методик 1-4)
Колонка: Eclipse XDB-C18; 150×4,6 мм; 5 мкм. Подвижная фаза А (0,1% H3PO4) в воде; Подвижная фаза В (ацетонитрил). Градиентный режим: 0 мин (30% В); 8 мин (95% В); 15 мин (95% В). Скорость потока: 1,0 мл мин-1. Длина волны: 210 нм. Температура: 30°С.
Время удерживания:
9,7 мин (1a, R1 = трет-бутоксикарбонил)
Пример 29: трет-Бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил)
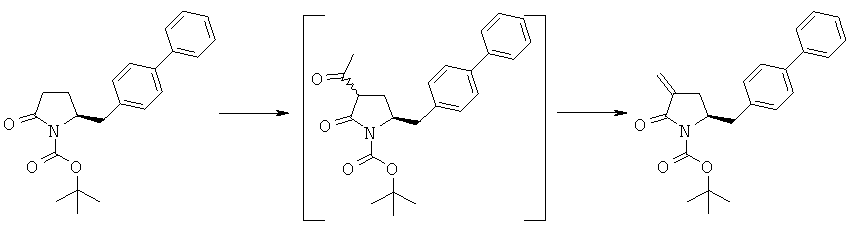
LHMDS (12,5 мл, 1,0 М раствор в тетрагидрофуране, 12,5 ммоля) в атмосфере N2 при -10°С добавляют к смеси трет-бутилового эфира (S)-2-бифенил-4-илметил-5-оксопирролидин-1-карбоновой кислоты (3а, R1 = трет-бутоксикарбонил) (1,76 г, 5 ммолей) и 15 мл сухого тетрагидрофурана, затем полученную смесь перемешивают при -10°С в течение 30 мин. К реакционной смеси при -10°С добавляют ацетилхлорид (0,47 г, 6 ммолей), примерно через 1 ч при -10°С реакционную смесь разбавляют с помощью 10 мл насыщенного водного раствора хлорида аммония и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. К оставшейся органической фазе добавляют формальдегид (37% раствор в воде, 1,2 г, 15 ммолей), водный раствор К2СО3 (1,4 г в 5 мл воды, 0,01 моля) и раствор тетрабутиламмонийгидроксида (40% в воде, 0,06 г, 0,25 ммоля) и нагревают до 50°С и перемешивают в течение 2 ч. Затем реакционную смесь разбавляют с помощью 10 мл рассола и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Затем органическую фазу концентрируют в вакууме. Добавляют 20 мл этилацетата, фильтруют и фильтрат концентрируют и получают трет-бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил). Неочищенный трет-бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил) можно очистить с помощью колоночной хроматографии при элюировании смесью гептан/этилацетат состава 3:1. По данным ВЭЖХ соотношение промежуточных продуктов (3S,5S-4, R1 = трет-бутоксикарбонил, R4 = метил):(3R,5S-4, R1 = трет-бутоксикарбонил, R4 = метил) составляет 70:30. Спектроскопические данные являются такими же, как в примере 28, методика 1.
Пример 30: трет-Бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил)

LHMDS (12,5 мл, 1,0 М раствор в тетрагидрофуране, 12,5 ммоля) в атмосфере N2 при -10°С добавляют к смеси трет-бутилового эфира (S)-2-бифенил-4-илметил-5-оксопирролидин-1-карбоновой кислоты (3а, R1 = трет-бутоксикарбонил) (1,76 г, 5 ммолей) и 15 мл сухого тетрагидрофурана, затем полученную смесь перемешивают при -10°С в течение 30 мин. К реакционной смеси при -10°С добавляют 2,2-диметилпропионилхлорид (0,72 г, 6 ммолей), примерно через 1 ч при -10°С реакционную смесь разбавляют с помощью 10 мл насыщенного водного раствора хлорида аммония и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. К оставшейся органической фазе добавляют формальдегид (37% раствор в воде, 1,2 г, 15 ммолей), водный раствор К2СО3 (1,4 г в 5 мл воды, 0,01 моля) и раствор тетрабутиламмонийгидроксида (40% в воде, 0,06 г, 0,25 ммоля) и нагревают до 50°С и перемешивают в течение 2 ч. Затем реакционную смесь разбавляют с помощью 10 мл рассола и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Затем органическую фазу концентрируют в вакууме. Добавляют 20 мл этилацетата, фильтруют и фильтрат концентрируют и получают трет-бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил). Неочищенный трет-бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил) можно очистить с помощью колоночной хроматографии при элюировании смесью гептан/этилацетат состава 3:1. По данным ВЭЖХ соотношение промежуточных продуктов (3S,5S-4, R1 = трет-бутоксикарбонил, R4 = трет-бутил):(3R,5S-4, R1 = трет-бутоксикарбонил, R4 = трет-бутил) составляет 80:20. Спектроскопические данные являются такими же, как в примере 28, методика 1.
Пример 31: трет-Бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил)

LHMDS (12,5 мл, 1,0 М раствор в тетрагидрофуране, 12,5 ммоля) в атмосфере N2 при -10°С добавляют к смеси трет-бутилового эфира (S)-2-бифенил-4-илметил-5-оксопирролидин-1-карбоновой кислоты (3а, R1 = трет-бутоксикарбонил) (1,76 г, 5 ммолей) и 15 мл сухого тетрагидрофурана, затем полученную смесь перемешивают при -10°С в течение 30 мин. К реакционной смеси при -10°С добавляют изобутирилхлорид (0,64 г, 6 ммолей), примерно через 1 ч при -10°С реакционную смесь разбавляют с помощью 10 мл насыщенного водного раствора хлорида аммония и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. К оставшейся органической фазе добавляют формальдегид (37% раствор в воде, 1,2 г, 15 ммолей), водный раствор К2СО3 (1,4 г в 5 мл воды, 0,01 моля) и раствор тетрабутиламмонийгидроксида (40% в воде, 0,06 г, 0,25 ммоля) и нагревают до 50°С и перемешивают в течение 2 ч. Затем реакционную смесь разбавляют с помощью 10 мл рассола и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. Затем органическую фазу концентрируют в вакууме. Добавляют 20 мл этилацетата, фильтруют и фильтрат концентрируют и получают трет-бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил). Неочищенный трет-бутиловый эфир (R)-5-бифенил-4-илметил-3-метилен-2-оксопирролидин-1-карбоновой кислоты (1a, R1 = трет-бутоксикарбонил) можно очистить с помощью колоночной хроматографии при элюировании смесью гептан/этилацетат состава 3:1. По данным ВЭЖХ соотношение промежуточных продуктов (3S,5S-4, R1 = трет-бутоксикарбонил, R4 = изопропил):(3R,5S-4, R1 = трет-бутоксикарбонил, R4 = изопропил) составляет 70:30. Спектроскопические данные являются такими же, как в примере 28, методика 1.
Пример 32: (R)-5-Бифепил-4-илметил-3-метиленпирролидин-2-он (1a, R1=Н)

(R)-5-Бифенил-4-илметил-3-метилен-1-пирролидин-1-илметилпирролидин-2-он (1а, R1 = пирролидинилметил) (0,35 г, 1 ммоль) смешивают со смесью уксусной кислоты и концентрированной хлористоводородной кислоты (10 мл, состав 1:1) и перемешивают при кипячении с обратным холодильником в течение примерно 3 ч. Затем раствор концентрируют в вакууме и остаток очищают с помощью колоночной хроматографии при элюировании смесью гептан/этилацетат состава 1:1 и получают (R)-5-бифенил-4-илметил-3-метиленпирролидин-2-он (1а, R1=Н).1Н ЯМР (400 МГц, CDCl3): 2,58 (m, 1H, 3-CHH), 2,76 (m, 1H, 3СНН), 2,90 (dd, 1H, 5-CHH), 2,99 (dd, 1H, 5-CHH), 3,92 (m, 1H, 4-CH), 5,37 (d, 1H, OCHH), 6,01 (d, 1H, C=CHH), 7,20-7,60 (9Н, m, ароматический).
Методика проведения ВЭЖХ
Колонка: Eclipse XDB-C18; 150×4,6 мм; 5 мкм. Подвижная фаза А (0,1% H3PO4) в воде; Подвижная фаза В (ацетонитрил). Градиентный режим: 0 мин (30% В); 8 мин (95% В); 15 мин (95% В). Скорость потока: 1,0 мл мин-1. Длина волны: 210 нм. Температура: 30°С.
Время удерживания:
6,7 мин (1а, R1=Н)
Пример 33: (R)-5-Бифепил-4-илметил-1-((Е)-бут-1-енил)-3-метилеппирролидин-2-ои (1a, R1=H)

(R)-5-Бифенил-4-илметил-1-((Е)-бут-1-енил)-3-метиленпирролидин-2-он (1a, R1 = 1-бутенил) (11,5 г, 35,6 ммоля) растворяют в 100 мл толуола, добавляют 35 мл концентрированной HCl, кипятят с обратным холодильником в атмосфере N2 в течение 5 ч, охлаждают до комнатной температуры, водную фазу отделяют, промывают водным раствором гидрокарбоната натрия и рассолом, сушат над безводным Na2SO4 и концентрируют в вакууме, остаток перекристаллизовывают из смеси толуол/трет-бутилметиловый эфир состава 1:3 ~ 1:4 и получают (R)-5-бифенил-4-илметил-1-((Е)-бут-1-енил)-3-метиленпирролидин-2-он (1а, R1=H). Спектроскопические данные являются такими же, как в примере 32.
Пример 34: (R)-5-Бифенил-4-илметил-3-метиленпирролидип-2-он (1a, R1=H)

Методика 1
(R)-5-Бифенил-4-илметил-3-метилен-1-((Е)-стирил)-пирролидин-2-он (1а, R1 = стирил) (1,0 г, 2,7 ммоля) растворяют в 10 мл этанола, добавляют концентрированную HCl (5 мл), кипятят с обратным холодильником в атмосфере азота в течение ночи. Реакционную смесь охлаждают до комнатной температуры, концентрируют в вакууме, остаток экстрагируют толуолом, органическую фазу промывают насыщенным водным раствором Na2SO3, водным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4 и растворитель выпаривают в вакууме и получают неочищенный продукт, который перекристаллизовывают из смеси толуол/трет-бутилметиловый эфир состава 1:3~1:4, и получают (R)-5-бифенил-4-илметил-3-метиленпирролидин-2-он (1a, R1=Н).1Н ЯМР (400 МГц, CDCl3): 2,56 (m, 1Н, 3-СНН), 2,77 (m, 1H, 5-CHH), 2,89 (m, 1H, 5-CHH), 3,03 (m, 1H, 3-СНН), 3,92 (m, 1H, 4-CH), 5,36 (s, ГН, C=CHH), 6,01 (s, 1H, C-CHH), 6,32 (s, 1H, NHH), 7,24~7,58 (m, 9H, ароматический).
Методика 2
(R)-5-Бифенил-4-илметил-3-метилен-1-((Е)-стирил)-пирролидин-2-он (1а, R1 = стирил) (1,0 г, 2,7 ммоля) растворяют в 10 мл толуола, добавляют концентрированную НС1 (5 мл), кипятят с обратным холодильником в атмосфере азота в течение ночи. Реакционную смесь охлаждают до комнатной температуры, концентрируют в вакууме, остаток экстрагируют толуолом, органическую фазу промывают насыщенным водным раствором Na2SO4, водным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4 и растворитель выпаривают в вакууме, остаток очищают с помощью колоночной хроматографии и получают (R)-5-бифенил-4-илметил-3-метиленпирролидин-2-он (1а, R1=Н). Спектроскопические данные являются такими же, как в примере 34, методика 1.
Методика 3
(R)-5-Бифенил-4-илметил-3-метилен-1-((Е)-стирил)-пирролидин-2-он (1а, R1 = стирил) (1,0 г, 2,7 ммоля) растворяют в 10 мл этанола, добавляют концентрированную H2SO4 (1 мл), кипятят с обратным холодильником в атмосфере азота в течение ночи. Реакционную смесь охлаждают до комнатной температуры, концентрируют в вакууме, остаток растворяют в 20 мл толуола, органическую фазу промывают насыщенным водным раствором Na2SO3, водным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4 и растворитель выпаривают в вакууме, остаток очищают с помощью колоночной хроматографии и получают (R)-5-бифенил-4-илметил-3-метиленпирролидин-2-он (1а, R1=Н). Спектроскопические данные являются такими же, как в примере 34, методика 1.
Методика 4
(R)-5-Бифенил-4-илметил-3-метилен-1-((Е)-стирил)-пирролидин-2-он (1а, R1 = стирил) (1,0 г, 2,7 ммоля) растворяют в 10 мл уксусной кислоты, добавляют 1 мл воды, кипятят с обратным холодильником в атмосфере азота в течение ночи. Реакционную смесь охлаждают до комнатной температуры, концентрируют в вакууме, остаток растворяют в 20 мл толуола, промывают насыщенным водным раствором Na2SO3, водным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4 и растворитель выпаривают в вакууме, остаток очищают с помощью колоночной хроматографии и получают (R)-5-бифенил-4-илметил-3-метиленпирролидин-2-он (1a, R1=Н). Спектроскопические данные являются такими же, как в примере 34, методика 1.
Методика 5
(R)-5-Бифенил-4-илметил-3-метилен-1-((Е)-стирил)-пирролидин-2-он (1а, R1 = стирил) (1,0 г, 2,7 ммоля) растворяют в 10 мл ТФК, добавляют 1 мл воды, кипятят с обратным холодильником в атмосфере азота в течение ночи. Реакционную смесь охлаждают до комнатной температуры, концентрируют в вакууме, остаток растворяют в 20 мл толуола, промывают насыщенным водным раствором Na2SO3, водным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4 и растворитель выпаривают в вакууме, остаток очищают с помощью колоночной хроматографии и получают (R)-5-бифенил-4-илметил-3-метиленпирролидин-2-он (1а, R1=Н). Спектроскопические данные являются такими же, как в примере 34, методика 1.
Методика 5
(R)-5-Бифенил-4-илметил-3-метилен-1-((Е)-стирил)-пирролидин-2-он (1а, R1 = стирил) (1,0 г, 2,7 ммоля) растворяют в 10 мл ТФК, добавляют 1 мл воды, кипятят с обратным холодильником в атмосфере азота в течение ночи. Реакционную смесь охлаждают до комнатной температуры, концентрируют в вакууме, остаток растворяют в 20 мл толуола, промывают насыщенным водным раствором Na2SO3, водным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4 и растворитель выпаривают в вакууме, остаток очищают с помощью колоночной хроматографии и получают (R)-5-бифенил-4-илметил-3-метиленпирролидин-2-он (1а, R1=Н). Спектроскопические данные являются такими же, как в примере 34, методика 1.
Методика 6
К раствору (R)-5-Бифенил-4-илметил-3-метилен-1-((Е)-стирил)-пирролидин-2-она (1а, R1 = стирил) (1,0 г, 2,7 ммоля) в 10 мл тетрагидрофурана добавляют 1 мл метансульфоновой кислоты и 1 мл воды, кипятят с обратным холодильником в атмосфере азота в течение ночи. Реакционную смесь охлаждают до комнатной температуры, концентрируют в вакууме, остаток растворяют в 20 мл толуола, промывают насыщенным водным раствором Na2SO3, водным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4 и растворитель выпаривают в вакууме, остаток очищают с помощью колоночной хроматографии и получают (R)-5-бифенил-4-илметил-3-метиленпирролидин-2-он (1a, R1=Н). Спектроскопические данные являются такими же, как в примере 34, методика 1.
Методика 7
(R)-5-Бифенил-4-илметил-3-метилен-1-((Е)-стирил)-пирролидин-2-он (1а, R1=стирил) (10,0 г, 27 ммолей) растворяют в 30 мл безводного толуола, добавляют TsOH·Н2О (6,2 г, 32,4 ммоля), кипятят с обратным холодильником в атмосфере азота в течение ночи. Реакционную смесь охлаждают до комнатной температуры, добавляют 100 мл толуола, органическую фазу промывают насыщенным водным раствором Na2SO3, водным раствором Na2CO3 и рассолом, сушат над безводным Na2SO4 и растворитель выпаривают в вакууме и получают неочищенный продукт, который перекристаллизовывают из смеси толуол/трет-бутилметиловый эфир состава 1:3~1:4, и получают (R)-5-бифенил-4-илметил-3-метиленпирролидин-2-он (1а, R1=Н). Спектроскопические данные являются такими же, как в примере 34, методика 1.
Методика проведения ВЭЖХ (для методик 1-7)
Колонка: Eclipse XDB-C18; 150×4,6 мм; 5 мкм. Подвижная фаза А (0,1% H3PO4) в воде; Подвижная фаза В (ацетонитрил). Градиентный режим: 0 мин (30% В); 8 мин (95% В); 15 мин (95% В). Скорость потока: 1,0 мл мин-1. Длина волны: 210 нм. Температура: 30°С.
Время удерживания:
6,7 мин (1а, R1=Н).
Пример 35: (R)-5-Бифенил-4-ил-4-трет-бутоксикарбониламино-2-метиленпентановая кислота

Методика 1
При -10°С н-бутиллитий (56 мл, 2,5 М раствор в гексане, 0,14 моля) в атмосфере N2 добавляют к смеси ГМДС (24,2 г, 0,15 моля) и 300 мл сухого тетрагидрофурана, затем полученную смесь перемешивают при -10°С в течение 30 мин. К реакционной смеси при -10°С добавляют смесь трет-бутилового эфира (S)-2-бифенил-4-илметил-5-оксопирролидин-1-карбоновой кислоты (3а, R1 = трет-бутоксикарбонил) (35,1 г, 0,1 моля) и 50 мл сухого тетрагидрофурана, примерно через 30 мин к реакционной смеси при -10°С добавляют н-бутиллитий (40 мл, 2,5 М раствор в гексане, 0,1 моля), затем полученную смесь перемешивают при -10°С в течение 30 мин. К реакционной смеси при -10°С добавляют бензоилхлорид (15,5 г, 0,11 моля), примерно через 1 ч при -10°С реакционную смесь разбавляют с помощью 100 мл насыщенного водного раствора хлорида аммония и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. К оставшейся органической фазе добавляют формальдегид (37% раствор в воде, 24,3 г, 0,3 моля), водный раствор К2СО3 (27,6 г в 100 мл воды, 0,2 моля) и раствор тетрабутиламмонийгидроксида (40% в воде, 1,3 г, 5 ммолей) и нагревают до 50°С и перемешивают в течение 2 ч. Затем реакционную смесь разбавляют с помощью 100 мл рассола и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. К оставшейся органической фазе добавляют раствор моногидрата гидроксида лития (15 г в 100 мл воды, 0,36 моля) и тетрабутиламмонийбромид (0,32 г, 1 ммоль), перемешивают при КТ в течение 2 ч. Затем добавляют примерно 50 г 85% фосфорной кислоты для обеспечения значения рН равного 3,0-4,0. Затем добавляют 100 мл толуола и 100 мл рассола. После разделения фаз концентрируют в вакууме. Остаток при 80°С растворяют в 200 мл ацетонитрила, фильтруют в горячем виде (80°С) для удаления неорганической соли и охлаждают до 0°С, при этом происходит кристаллизация и смесь выдерживают в течение 3 ч, кристаллы собирают фильтрованием, промывают с помощью 50 мл ацетонитрила и сушат в вакууме при 50°С и получают (R)-5-бифенил-4-ил-4-трет-бутоксикарбониламино-2-метилеппептановую кислоту. Н ЯМР (400 МГц, ДМСО): 1,27 (s, 9Н, (СН3)3), 2,26 (m, 1Н, 3-СНН), 2,49 (m, 1Н, 3СНН), 2,71 (d, 1H, 5-СН2), 3,85 (m, 1Н, 4-СН), 5,60 (s, 1Н, С=СНН), 6,05 (s, 1Н, С=СНН), 6,65 (d, 1Н, NH), 7,20-7,70 (9Н, m, ароматический), 12,34 (s, 1Н, СООН). По данным ВЭЖХ соотношение промежуточных продуктов (3S,5S-4, R1 = трет-бутоксикарбонил, R4 = фенил):(3R,5S-4, R1 = трет-бутоксикарбонил, R4 = фенил) составляет 70:30. Спектроскопические данные являются такими же, как в примере 35, методика 1.
Методика 2
н-Бутиллитий (56 мл, 2,5 М раствор в гексане, 0,14 моля) в атмосфере N2 при -10°С добавляют к смеси диизопропиламина (14,2 г, 0,15 моля) и 300 мл сухого тетрагидрофурана, затем полученную смесь перемешивают при -10°С в течение 30 мин. К реакционной смеси при -10°С добавляют смесь трет-бутилового эфира (S)-2-бифенил-4-илметил-5-оксопирролидин-1-карбоновой кислоты (3а, R1 = трет-бутоксикарбонил) (35,1 г, 0,1 моля) и 50 мл сухого тетрагидрофурана, примерно через 30 мин к реакционной смеси при -10°С добавляют н-бутиллитий (40 мл, 2,5 М раствор в гексане, 0,1 моля), затем полученную смесь перемешивают при -10°С в течение 30 мин. К реакционной смеси при -10°С добавляют бензоилхлорид (15,5 г, 0,11 моля), примерно через 1 ч при -10°С реакционную смесь разбавляют с помощью 100 мл насыщенного водного раствора хлорида аммония и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. К оставшейся органической фазе добавляют формальдегид (37% раствор в воде, 24,3 г, 0,3 моля), водный раствор К2СО3 (27,6 г в 100 мл воды, 0,2 моля) и раствор тетрабутиламмонийгидроксида (40% в воде, 1,3 г, 5 ммолей) и нагревают до 50°С и перемешивают в течение 2 ч. Затем реакционную смесь разбавляют с помощью 100 мл рассола и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. К оставшейся органической фазе добавляют раствор моногидрата гидроксида лития (15 г в 100 мл воды, 0,36 моля) и тетрабутиламмонийбромид (0,32 г, 1 ммоль), перемешивают при КТ в течение 2 ч. Затем добавляют примерно 50 г 85% фосфорной кислоты для обеспечения значения рН равного 3,0-4,0. Затем добавляют 100 мл толуола и 100 мл рассола. После разделения фаз концентрируют в вакууме. Остаток при 80°С растворяют в 200 мл ацетонитрила, фильтруют в горячем виде (80°С) для удаления неорганической соли и охлаждают до 0°С, при этом происходит кристаллизация и смесь выдерживают в течение 3 ч, кристаллы собирают фильтрованием, промывают с помощью 50 мл ацетонитрила и сушат в вакууме при 50°С и получают (R)-5-бифенил-4-ил-4-трет-бутоксикарбониламино-2-метиленпентановую кислоту. По данным ВЭЖХ соотношение промежуточных продуктов (3S,5S-4, R1 = трет-бутоксикарбонил, R4 = фенил):(3R,5S-4, R1 = трет-бутоксикарбонил, R4 = фенил) составляет 70:30. Спектроскопические данные являются такими же, как в примере 35, методика 1.
Методика 3
LHMDS (25 мл, 1,0 М раствор в тетрагидрофуране, 25 ммолей) в атмосфере N2 при -10°С добавляют к смеси трет-бутилового эфира (S)-2-бифенил-4-илметил-5-оксопирролидин-1-карбоновой кислоты (3а, R1 = трет-бутоксикарбонил) (3,51 г, 10 ммолей) и 20 мл сухого тетрагидрофурана, затем полученную смесь перемешивают при -10°С в течение 30 мин. К реакционной смеси при -10°С добавляют бензоилхлорид (1,55 г, 11 ммолей), примерно через 1 ч при -10°С реакционную смесь разбавляют с помощью 15 мл насыщенного водного раствора хлорида аммония и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. К оставшейся органической фазе добавляют формальдегид (37% раствор в воде, 2,43 г, 0,03 моля), водный раствор К2СО3 (2,76 г в 10 мл воды, 0,02 моля) и раствор тетрабутиламмонийгидроксида (40% в воде, 0,13 г, 0,5 ммоля) и нагревают до 50°С и перемешивают в течение 2 ч. Затем реакционную смесь разбавляют с помощью 15 мл рассола и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. К оставшейся органической фазе добавляют раствор моногидрата гидроксида лития (1,5 г в 10 мл воды, 0,036 моля) и тетрабутиламмонийбромид (32 мг, 0,1 ммоля), перемешивают при КТ в течение 2 ч. Затем добавляют примерно 5 г 85% фосфорной кислоты для обеспечения значения рН равного 3,0-4,0. Затем добавляют 15 мл толуола и 15 мл рассола. После разделения фаз концентрируют в вакууме. Остаток при 80°С растворяют в 20 мл ацетонитрила, фильтруют в горячем виде (80°С) для удаления неорганической соли и охлаждают до 0°С, при этом происходит кристаллизация и смесь выдерживают в течение 3 ч, кристаллы собирают фильтрованием, промывают с помощью 5 мл ацетонитрила и сушат в вакууме при 50°С и получают (R)-5-бифенил-4-ил-4-трет-бутоксикарбониламино-2-метиленпентацовую кислоту. По данным ВЭЖХ соотношение промежуточных продуктов (3S,5S-4, R1 = трет-бутоксикарбонил, R4 = фенил):(3R,5S-4, R1 = трет-бутоксикарбонил, R4=фенил) составляет 70:30. Спектроскопические данные являются такими же, как в примере 35, методика 1.
Методика 4
ДАЛ (25 мл, 1,0 М раствор в тетрагидрофуране, 25 ммолей) в атмосфере N2 при -10°С добавляют к смеси трет-бутилового эфира (S)-2-бифенил-4-илметил-5-оксопирролидин-1-карбоновой кислоты (3а, R1 = трет-бутоксикарбонил) (3,51 г, 10 ммолей) и 20 мл сухого тетрагидрофурана, затем полученную смесь перемешивают при -10°С в течение 30 мин. К реакционной смеси при -10°С добавляют бензоилхлорид (1,55 г, 11 ммолей), примерно через 1 ч при -10°С реакционную смесь разбавляют с помощью 15 мл насыщенного водного раствора хлорида аммония и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. К оставшейся органической фазе добавляют формальдегид (37% раствор в воде, 2,43 г, 0,03 моля), водный раствор К2СО3 (2,76 г в 10 мл воды, 0,02 моля) и раствор тетрабутиламмонийгидроксида (40% в воде, 0,13 г, 0,5 ммоля) и нагревают до 50°С и перемешивают в течение 2 ч. Затем реакционную смесь разбавляют с помощью 15 мл рассола и перемешивают в течение 15 мин, перемешивание прекращают и нижний водный слой удаляют. К оставшейся органической фазе добавляют раствор моногидрата гидроксида лития (1,5 г в 10 мл воды, 0,036 моля) и тетрабутиламмонийбромид (32 мг, 0,1 ммоля), перемешивают при КТ в течение 2 ч. Затем добавляют примерно 5 г 85% фосфорной кислоты для обеспечения значения рН равного 3,0-4,0. Затем добавляют 15 мл толуола и 15 мл рассола. После разделения фаз концентрируют в вакууме. Остаток при 80°С растворяют в 20 мл ацетонитрила, фильтруют в горячем виде (80°С) для удаления неорганической соли и охлаждают до 0°С, при этом происходит кристаллизация и смесь выдерживают в течение 3 ч, кристаллы собирают фильтрованием, промывают с помощью 5 мл ацетонитрила и сушат в вакууме при 50°С и получают (R)-5-бифенил-4-ил-4-трет-бутоксикарбониламино-2-метиленпентаповую кислоту. По данным ВЭЖХ соотношение промежуточных продуктов (3S,5S-4, R1 = трет-бутоксикарбонил, R4 = фенил):(3R,5S-4, R1 = трет-бутоксикарбонил, R4 = фенил) составляет 70:30. Спектроскопические данные являются такими же, как в примере 35, методика 1.
Методика 5
В колбу объемом 1 л в атмосфере N2 добавляют трет-бутиловый эфир (S)-2-бифенил-4-илметил-5-оксопирролидин-1-карбоновой кислоты (3а, R1 = трет-бутоксикарбонил) (40 г, 159 ммолей) и ДМАП (диметиламинопиридин) (2,43 г, 20 ммолей), затем 500 мл толуола и триэтиламин (26,1 г, 258 ммолей). Смесь нагревают до 65°С, в течение 30 мин добавляют ди-трет-бутилдикарбонат (48,6 г, 223 ммоля), затем перемешивают в течение 2 ч. Реакционную смесь охлаждают до 25°С, трижды промывают с помощью 200 мл воды, затем концентрируют. Остаток растворяют в 400 мл ТГФ, добавляют MgCl2 (18,0 г, 159 ммолей) и триэтиламин (26,1 г, 258 ммолей). Реакционную смесь охлаждают до 5°С, по каплям медленно добавляют бензоилхлорид (36,4 г, 259 ммолей), после добавления смесь перемешивают при 5°С в течение 1 ч, затем нагревают до 10°С и перемешивают в течение 15 ч. Добавляют 100 мл воды, затем раствор H3PO4 (57,4 г, 510 молей) в воде (100 мл). Через 10 мин водную фазу удаляют и органическую фазу дважды промывают рассолом (200 мл). К органическим экстрактам добавляют раствор карбоната калия (65,4 г в 200 мл воды), N-метилимидазол (32,6 г, 398 ммолей), раствор тетрабутилгидроксида аммония (3,68 г, 40% мас./мас., 5,7 ммоля) и раствор формальдегида в воде (48,4 г, 37% мас./мас., 613 ммолей). Эту смесь нагревают до 55°С и перемешивают в течение 20 ч. Затем температуру реакционной смеси понижают до 25°С и водную фазу удаляют. Органическую фазу дважды промывают насыщенным раствором хлорида натрия (200 мл). Добавляют раствор моногидрата гидроксида лития (29,3 г в 200 мл воды, 698 ммолей) и тетрабутиламмонийбромид (0,65 г, 2 ммоля). Смесь перемешивают при 25°С в течение 4 ч. Затем к реакционной смеси добавляют фосфорную кислоту (97,6 г, 85% мас./мас.), значение рН при 10-20°С доводят до 4-5. Водную фазу удаляют и добавляют насыщенный раствор хлорида натрия в воде (200 мл). Органическую фазу отделяют и концентрируют. Остаток растворяют в этилацетате (500 мл) и затем концентрируют в вакууме. Добавляют этилацетат (1000 мл) и воду (400 мл). Органическую фазу отделяют, трижды промывают водой (400 мл) и концентрируют. К полученному остатку добавляют толуол (400 мл) и затем концентрируют в вакууме. Добавляют ацетонитрил (400 мл). Реакционную смесь кипятят с обратным холодильником и в течение примерно 2 ч охлаждают до 4°С. Полученное твердое вещество отделяют фильтрованием. Осадок на фильтре промывают холодным ацетонитрилом (50 мл), затем гептаном (100 мл) и сушат и получают (R)-5-бифенил-4-ил-4-трет-бутоксикарбониламино-2-метиленпентановую кислоту в виде белого твердого вещества. По данным ВЭЖХ соотношение промежуточных продуктов (3S,5S-4, R1 = трет-бутоксикарбонил, R4 = фенил):(3R,5S-4, R1 = трет-бутоксикарбонил, R4=фенил) составляет 70:30. Спектроскопические данные являются такими же, как в примере 35, методика 1.
Методика проведения ВЭЖХ (для методик 1-5)
Колонка: Eclipse XDB-C18; 150×4,6 мм; 5 мкм. Подвижная фаза А (0,1% H3PO4) в воде; Подвижная фаза В (ацетонитрил). Градиентный режим: 0 мин (30% В); 8 мин (95% В); 15 мин (95% В). Скорость потока: 1,0 мл мин-1. Длина волны: 210 нм. Температура: 30°С.
Время удерживания: 8,0 мин ((R)-5-бифенил-4-ил-4-трет-бутоксикарбониламино-2-метиленпентановая кислота)
Реферат
Изобретение относится к новому способу получения промежуточных продуктов, применимых для получения ингибиторов NEP, в частности ингибиторов NEP, содержащих в качестве основной цепи γ-амино-δ-бифенил-α-метилалкановую кислоту или эфир этой кислоты, такой как этиловый эфир N-(3-карбокси-1-оксопропил)-(4S)-(п-фенилфенилметил)-4-амино-(2R)-метилбутановой кислоты или его соль. 4 н. и 11 з.п. ф-лы, 35 пр.
Формула

в которой
R1 обозначает водород или защитную группу атома азота, выбранную из C1-С6-алкоксикарбонила, С6-С10-арилкарбонила, С2-С4-алкенила, необязательно моно-замещенного фенилом, С1-С4-алкила, необязательно моно-замещенного фенилом или пирролидинилом, где фенильное кольцо является незамещенным или замещенным C1-C7-алкоксигруппой; и
R4 выбран из группы, включающей гидроксигруппу, С1-С4-алкил и фенил;
предпочтительно в котором соединением формулы (4) является соединение формулы (4а)

в которой R1 и R4 являются такими, как определено для соединения формулы (4);
включающий
взаимодействие соединения формулы (3) или его соли,

в которой R1 обозначает водород или защитную группу атома азота, выбранную из C1-С6-алкоксикарбонила, С6-С10-арилкарбонила, С2-С4-алкенила, необязательно моно-замещенного фенилом, С1-С4-алкила, необязательно моно-замещенного фенилом или пирролидинилом, где фенильное кольцо является незамещенным или замещенным C1-C7-алкоксигруппой;
предпочтительно в котором соединением формулы (3) является соединение формулы (3а)

в которой R1 является таким, как определено для соединения формулы (3);
сначала с основанием и затем с соединением формулы CO2 или R4COY, в которой R4 выбран из гидроксигруппы, С1-С4-алкила и фенила и Y обозначает галоген или -OR′, в которой R′ выбран из алкила, арила и арилалкила,
с получением соединения формулы (4) или его соли.
гидрид металла;
алкоксид щелочных металлов;
амин, необязательно в присутствии добавки, выбранной из числа галогенидов щелочноземельных металлов;
основание формулы MRa, в которой М обозначает щелочной металл и Ra обозначает алкил или арил;
основание формулы RcRdNM, в которой Rc и Rd независимо выбраны из группы, включающей алкил, циклоалкил, гетероциклил или силил, и М обозначает щелочной металл; и
их смеси.

в которой
R1 обозначает водород или защитную группу атома азота, выбранную из C1-С6-алкоксикарбонила, С6-С10-арилкарбонила, С2-С4-алкенила, необязательно моно-замещенного фенилом, С1-С4-алкила, необязательно моно-замещенного фенилом или пирролидинилом, где фенильное кольцо является незамещенным или замещенным С1-С7-алкоксигруппой;
предпочтительно в котором соединением формулы (1) является соединение формулы (1а)

в которой R1 является таким, как определено для соединения формулы (1);
включающий
взаимодействие соединения формулы (4) или его соли

в которой
R1 обозначает водород или защитную группу атома азота, выбранную из С1-С6-алкоксикарбонила, С6-С10-арилкарбонила, С2-С4-алкенила, необязательно моно-замещенного фенилом, C1-C4-алкила,
необязательно моно-замещенного фенилом или пирролидинилом, где фенильное кольцо является незамещенным или замещенным С1-С7-алкоксигруппой; и
R4 выбран из группы, включающей гидроксигруппу, С1-С4-алкил и фенил;
предпочтительно в котором соединением формулы (4) является соединение формулы (4а)

в которой R1 и R4 являются такими, как определено для соединения формулы (4);
с основанием и формальдегидом, необязательно в присутствии межфазного катализатора, с получением соединения формулы (1) или его соли.
гидрид металла;
амин;
неорганическое основание;
основание формулы MRa, в которой М обозначает щелочной металл и Ra обозначает алкил или арил;
основание формулы RcRdNM, в которой Rc и Rd независимо выбраны из группы, включающей алкил, циклоалкил, гетероциклил или силил, и М обозначает щелочной металл; и
их смеси.
в котором соединение формулы (4) или его соль,

в которой
R1 обозначает водород или защитную группу атома азота, выбранную из С1-С6-алкоксикарбонила, С6-С10-арилкарбонила, С2-С4-алкенила, необязательно моно-замещенного фенилом, С1-С4-алкила, необязательно моно-замещенного фенилом или пирролидинилом, где фенильное кольцо является незамещенным или замещенным C1-C7-алкоксигруппой; и
R4 выбран из группы, включающей гидроксигруппу, C1-C4-алкил и фенил;
предпочтительно в котором соединением формулы (4) является соединение формулы (4а)

в которой R1 и R4 являются такими, как определено для соединения формулы (4);
получают взаимодействием соединения формулы (3) или его соли,
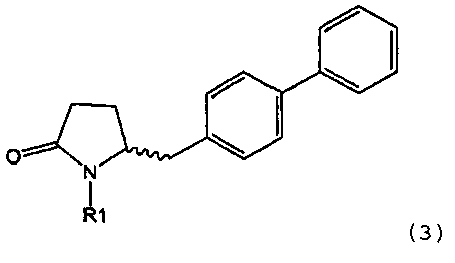
в которой R1 обозначает водород или защитную группу атома азота, выбранную из С1-С6-алкоксикарбонила, С6-С10-арилкарбонила, С2-С4-алкенила, необязательно моно-замещенного фенилом, С1-С4-алкила, необязательно моно-замещенного фенилом или пирролидинилом, где фенильное кольцо является незамещенным или замещенным C1-C7-алкоксигруппой;
предпочтительно в котором соединением формулы (3) является соединение формулы (3а)

в которой R1 является таким, как определено для соединения формулы (3);
сначала с основанием и затем с соединением формулы CO2 или R4COY, в которой R4 выбран из гидроксигруппы, С1-С4-алкила и фенила и Y обозначает галоген или -OR′, в которой R′ выбран из алкила,
арила и арилалкила, с получением соединения формулы (4) или его соли.
R1 выбран из группы, включающей трет-бутоксикарбонил, бензоил, стирил, 1-бутенил, бензил, п-метоксибензил и пирролидинилметил; и
R4 выбран из группы, включающей трет-бутил, метил, изопропил, фенил и гидроксигруппу.

в которой
R1 обозначает водород или защитную группу атома азота, выбранную из С1-С6-алкоксикарбонила, С6-С10-арилкарбонила, С2-С4-алкенила, необязательно моно-замещенного фенилом, С1-С4-алкила, необязательно моно-замещенного фенилом или пирролидинилом, где фенильное кольцо является незамещенным или замещенным С1-С7-алкоксигруппой; и
R4 выбран из группы, включающей гидроксигруппу, С1-С4-алкил и фенил;
предпочтительно в котором соединением формулы (4) является соединение формулы (4а)

в которой R1 и R4 являются такими, как определено для соединения формулы (4).
R1 выбран из группы, включающей трет-бутоксикарбонил, бензоил, стирил, 1-бутенил, бензил, п-метоксибензил и пирролидинилметил; и
R4 выбран из группы, включающей трет-бутил, метил, изопропил, фенил и гидроксигруппу.
Документы, цитированные в отчёте о поиске
Способ получения 5-бифенил-4-амино-2-метилпентановой кислоты
Комментарии