Способ получения промежуточного соединения эртапенема - RU2575979C2
Код документа: RU2575979C2
Чертежи
Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к промежуточному соединению эртапенема и содержащей его композиции и к способам его получения.
Предшествующий уровень техники
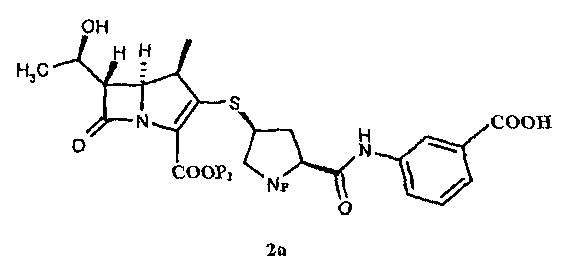
Химическое название эртапенема формулы (1) - (4R,5R,6S)-3-[(3S,5S)-5-[(3-карбоксифенил)амино]формил]пирролидин-3-ил]тио-6-[(1R)-1-гидроксиэтил]-4-метил-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат, который представляет собой новое антибиотическое соединение группы карбапенема, разработанное Merck и Astrazeneca, и обладает хорошей антибактериальной активностью против грамположительных бактерий и грамотрицательных бактерий.
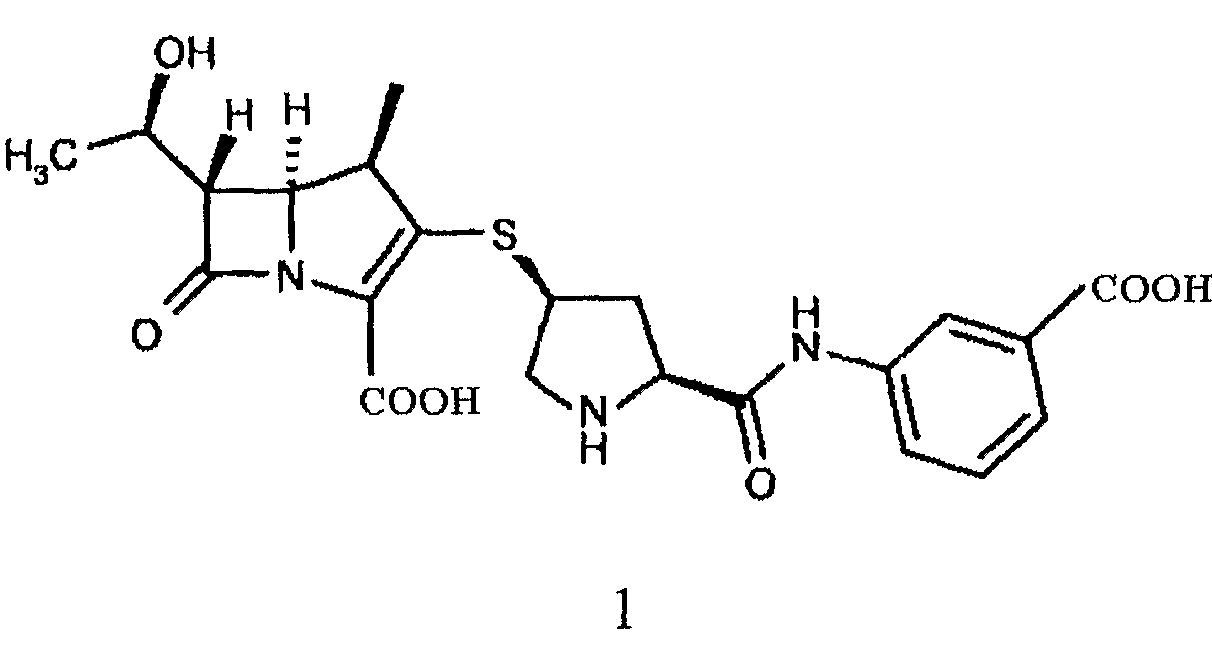
Эртапенем получают снятием защиты с промежуточного соединения эртапенема формулы (2)
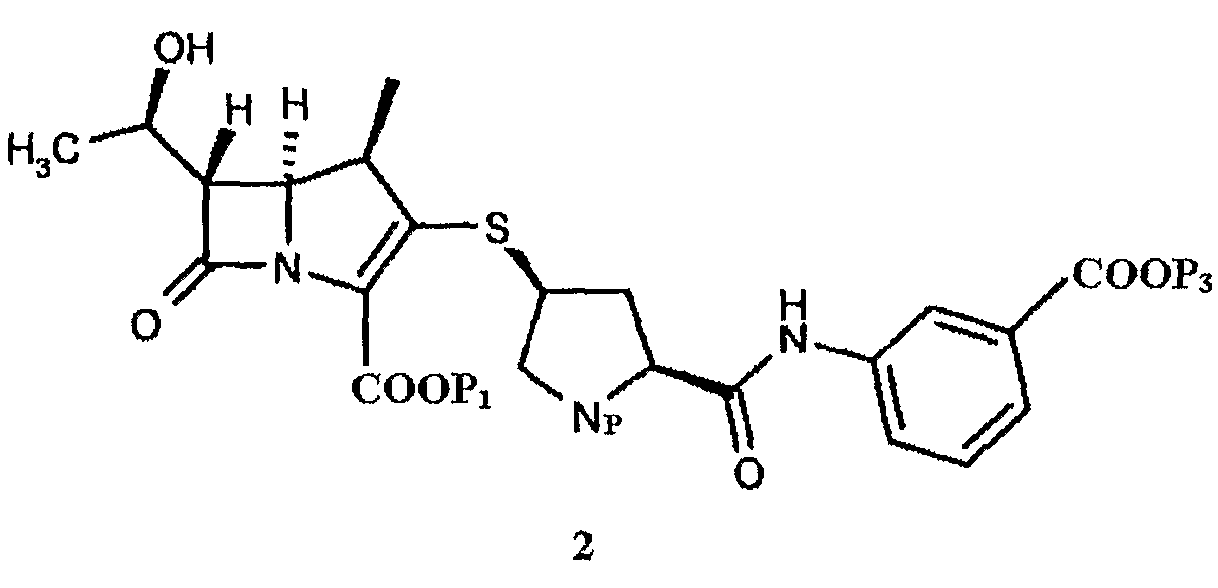
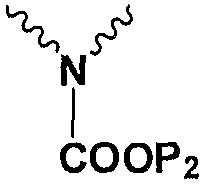
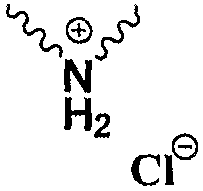
Np представляет собой
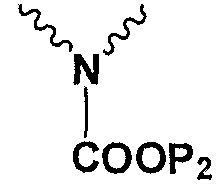
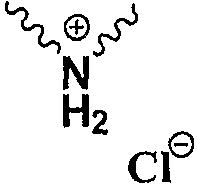
Р1 и Р2 представляют собой защитные группы для карбоксила, и Р3 представляет собой защитные группы для карбоксила, Н или Na.
Соединение 2 обычно получают конденсацией исходного ядра карбапенема 3 с боковой цепью эртапенема 4 в присутствии основания. Путь синтеза приведен на схеме 1:
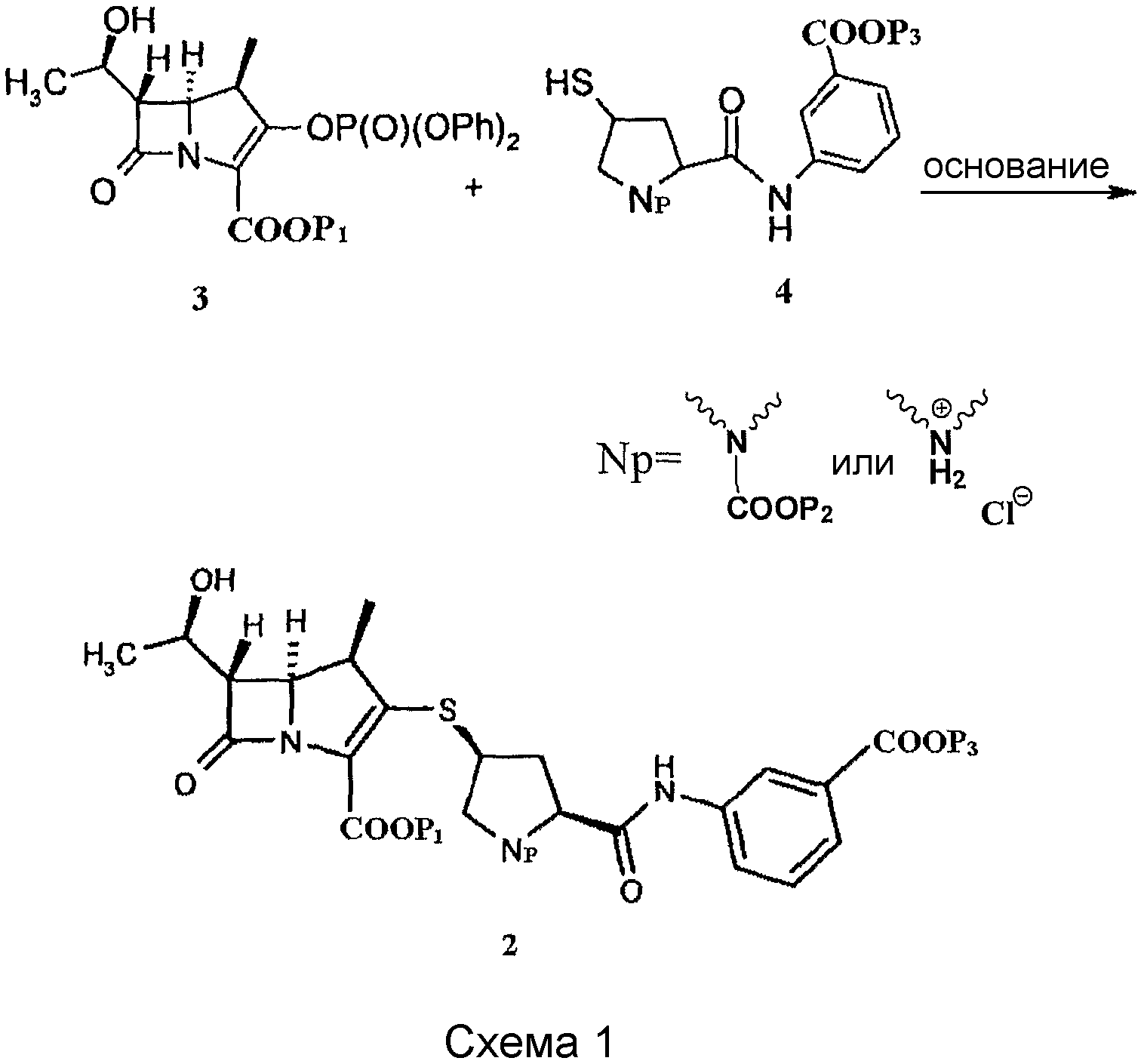
В патенте США 5478820А описано соединение 2, в котором все из Р1, Р2 и Р3 представляют собой аллил, или соединение 2, в котором Р1 и Р2 представляют собой п-нитробензил (в дальнейшем называемый PNB), и Р3 представляет собой аллил, и способы их получения.
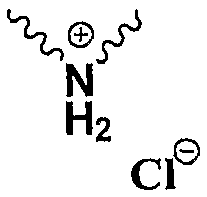
В патенте США 6504027 В1 предоставлен one-pot способ получения эртапенема натрия, который включает конденсацию исходного ядра эртапенема 3 (Р1 представляет собой PNB) с боковой цепью эртапенема 4 (Np представляет собой
В WO 98/02439 предоставлен способ получения соединения 2, в котором и Р1, и Р2 представляют собой PNB, и Р3 представляет собой Н, который включает конденсацию исходного ядра эртапенема 3 (Р1 представляет собой PNB) с боковой цепью эртапенема 4 (Р2 представляет собой PNB, и Р3 представляет собой Н) в присутствии основания, такого как диизопропиламин, при скорости конверсии, превышающей 98%. Но в нем не сообщается о последующей обработке и полученных продуктах.
В WO 2008/062279 предоставлен способ получения соединения 2, в котором Р1 и Р2, оба представляют собой PNB, и Р3 представляет собой Н или Na+. Для последующей обработки соединения 2, в котором Р3 представляет собой Н, реакционную смесь выливали в буферный раствор (рН 7), или воду, или смесь буферного раствора (рН 7) (или воды) с этилацетатом, и соединение 2 получали после последующей обработки. В случае первой ситуации это быстро приводит к слипанию продукта и вызывает трудности при последующей обработке и низкую чистоту (ниже 90%) продукта; в случае последней ситуации продукт нельзя получить в твердом виде и данный способ препятствует защите окружающей среды вследствие использования органического растворителя. В данной заявке не предоставлены физико-химические свойства продукта, а также не проведена идентификация структуры продукта. Кроме того, в данной заявке сообщается о способе получения соединения 2, в котором Р3 представляет собой Na+, и его аморфной формы. Введение источника натрия приводит к увеличению общего количества неорганических солей в последующих реакциях, и таким образом, последующая обработка сложна в осуществлении, что препятствует кристаллизации продукта.
Следовательно, в предшествующей области техники отсутствуют данные о физико-химических свойствах соединения 2, в котором Р3 представляет собой Н, и соединение 2, в котором Р3 представляет собой Н, нельзя получить с высокой чистотой методами предшествующей области техники. Другими словами, при помощи методов предшествующей области техники нельзя получить соединение 2, в котором Р3 представляет собой Н, вследствие чего нельзя осуществить идентификацию структуры и определение физико-химических свойств.
Краткое описание сущности изобретения
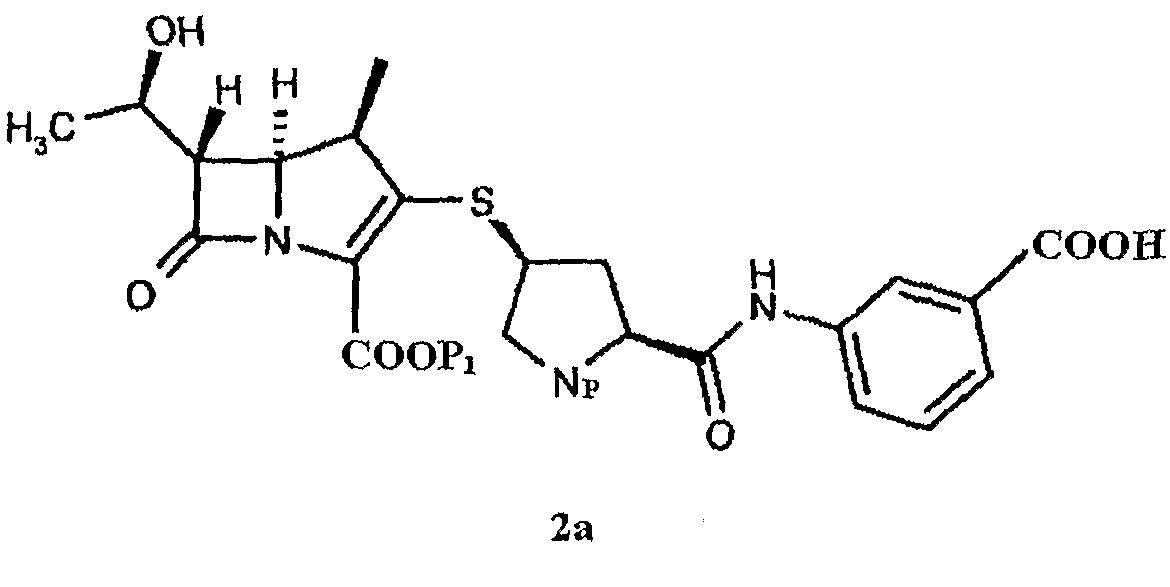
В ходе исследования эртапенема оказалось неожиданным, что авторы изобретения получили промежуточное соединение эртапенема формулы 2а в аморфной форме, обладающей высокой чистотой и хорошей устойчивостью при хранении, а используемый способ получения является простым и подходящим для промышленности.
Соответственно, в одном из аспектов, в настоящем изобретении предоставлено промежуточное соединение эртапенема формулы 2а, предпочтительно промежуточное соединение эртапенема формулы 2а в аморфной форме:
в котором Np представляет собой
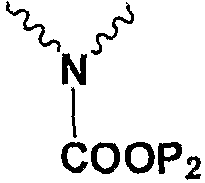
Подробное описание изобретения
Защитную группу для карбоксила выбирают из группы, включающей аллил или замещенный аллил, бензил или замещенный бензил, замещенный этил, замещенный силил, метил, замещенный ароматическим кольцом, фенил или замещенный фенил, ацетонил, трет-бутил и другую подходящую группу для защиты карбоксила, известную специалисту в данной области.
Предпочтительно замещенный аллил представляет собой 2-хлораллил.
Предпочтительно замещенный бензил выбирают из бензила, замещенного нитро-группой, и бензила, замещенного метоксилом.
Предпочтительно бензил, замещенный нитро-группой, представляет собой п-нитробензил.
Предпочтительно замещенный этил выбирают из 2,2,2-трихлорэтила, 2-бромэтила и 2-(триметилсилил)этила.
Предпочтительно метил, замещенный ароматическим циклом, выбирают из 2-метилнафтила, бензгидрила, тритила и 4-пиридилметила.
Предпочтительно замещенный силил выбирают из триметилсилила, трет-бутилдиметилсилила и трет-бутилдифенилсилила.
Предпочтительно замещенный фенил представляет собой п-метилфенил.
Предпочтительно промежуточное соединение эртапенема формула 2а включает:
п-нитробензил (4R,5R,6S)-3-[(3S,5S)-5-[(3-карбоксифенил)карбамоил]-N-(п-нитробензилоксикарбонилпирролидин-3-ил]тио-6-[(1R)-1-гидроксиэтил]-4-метил-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат,
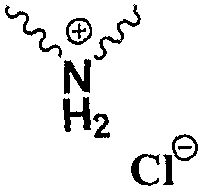
п-нитробензил (4R,5R,6S)-3-[(3S,5S)-5-[(3-карбоксифенил)карбамоил]пирролидин гидрохлорид-3-ил]тио-6-[(1R)-1-гидроксиэтил]-4-метил-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат,
о-нитробензил (4R,5R,6S)-3-[(3S,5S)-5-[(3-карбоксифенил)карбамоил]-N-(о-нитробензилокси)карбонилпирролидин-3-ил]тио-6-[(1R)-1-гидроксиэтил]-4-метил-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат,
п-метоксибензил (4R,5R,6S)-3-[(3S,5S)-5-[(3-карбоксифенил)карбамоил]-N-(п-метоксибензилоксикарбонилпирролидин-3-ил)тио-6-[(1R)-1-гидроксиэтил]-4-метил-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат,
аллилметил (4R,5R,6S)-3-[(3S,5S)-5-[(3-карбоксифенил)карбамоил]-N-(аллилоксикарбонилпирролидин-3-ил)тио-6-[(1R)-1-гидроксиэтил]-4-метил-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат,
2,2,2-трихлорэтил (4R,5R,6S)-3-[(3S,5S)-5-[(3-карбоксифенил)карбамоил]-N-(2,2,2-трихлорэтилоксикарбонилпирролидин-3-ил)тио-6-[(1R)-1-гидроксиэтил]-4-метил-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат,
бензгидрил (4R,5R,6S)-3-[(3S,5S)-5-[(3-карбоксифенил)карбамоил]-N-(бензгидрилоксикарбонилпирролидин-3-ил)тио-6-[(1R)-1-гидроксиэтил]-4-метил-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат,
триметилсилил (4R,5R,6S)-3-[(3S,5S)-5-[(3-карбоксифенил)карбамоил]-N-(триметилсилилоксикарбонилпирролидин-3-ил)тио-6-[(1R)-1-гидроксиэтил]-4-метил-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат,
трет-бутил (4R,5R,6S)-3-[(3S,5S)-5-[(3-карбоксифенил)карбамоил]-N-(трет-бутоксикарбонилпирролидин-3-ил)тио-6-[(1R)-1-гидроксиэтил]-4-метил-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат.
Более предпочтительно промежуточное соединение эртапенема формулы 2а включает:
п-нитробензил (4R,5R,6S)-3-[(3S,5S)-5-[(3-карбоксифенил)карбамоил]-N-(4-нитробензилоксикарбонилпирролидин-3-ил]тио-6-[(1R)-1-гидроксиэтил]-4-метил-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат и
п-нитробензил (4R,5R,6S)-3-[(3S,5S)-5-[(3-карбоксифенил)карбамоил]пирролидин гидрохлорид-3-ил]тио-6-[(1R)-1-гидроксиэтил]-4-метил-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат.
В следующем аспекте в настоящем изобретении предоставлен способ получения промежуточного соединения эртапенема формулы 2а, в частности промежуточного соединения эртапенема формулы 2а в аморфной форме. Способ включает конденсацию исходного ядра эртапенема формулы 3 с боковой цепью эртапенема формулы 4а и отличается тем, что по завершении реакции реакционную смесь выливают в водный раствор кислоты, получая соединение формулы 2а в виде твердого вещества.
Боковая цепь эртапенема формулы 4а согласно настоящему изобретению имеет следующую структуру:
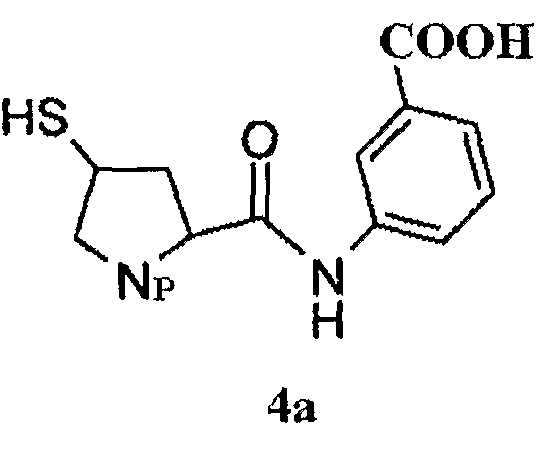
в которой Np представляет собой
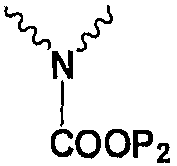
Получение исходного ядра карбапенема 3 описано, например, в J. Am. Chem. Soc. 1080, 102, 6161-6163, который включен здесь ссылкой.
Получение боковой цепи эртапенема 4а описано, например, в WO09/02439 и в J. Org. Chem. 2002, 67, 4771-4776, который включен здесь ссылкой.
Кислоту выбирают из неорганической кислоты, органической кислоты или любого их сочетания, которое существует в любых подходящих концентрациях.
Предпочтительно неорганическую кислоту выбирают из хлористоводородной кислоты, серной кислоты, сернистой кислоты, фосфорной кислоты, дигидрофосфата калия, дигидрофосфата натрия или любого их сочетания.
Более предпочтительно неорганическую кислоту выбирают из хлористоводородной кислоты, серной кислоты, фосфорной кислоты, дигидрофосфата натрия или любого их сочетания.
В одном из предпочтительных вариантов осуществления неорганическая кислота представляет собой хлористоводородную кислоту.
В одном из предпочтительных вариантов осуществления неорганическая кислота представляет собой серную кислоту.
В одном из предпочтительных вариантов осуществления неорганическая кислота представляет собой фосфорную кислоту.
Предпочтительно органическую кислоту выбирают из муравьиной кислоты, уксусной кислоты, пропионовой кислоты, бензолсульфоновой кислоты, п-толуолсульфоновой кислоты, бензойной кислоты, щавелевой кислоты, хлоруксусной кислоты, трихлоруксусной кислоты, трифторуксусной кислоты или любого их сочетания.
Более предпочтительно органическую кислоту выбирают из уксусной кислоты, п-толуолсульфоновой кислоты или любого их сочетания.
Значение рН водного раствора кислоты составляет 2-6.
Предпочтительно значение рН водного раствора кислоты составляет 2-5.
Более предпочтительно значение рН водного раствора кислоты составляет 2,5-4,5.
Еще предпочтительнее значение рН водного раствора кислоты составляет 2,5-4.
Наиболее предпочтительно значение рН водного раствора кислоты составляет 3-4.
В следующем аспекте в настоящем изобретении предоставлена композиция, содержащая по меньшей мере 95%, по меньшей мере 96%, по меньшей мере 97%, по меньшей мере 98% или по меньшей мере 99% промежуточного соединения эртапенема формулы 2а, предпочтительно промежуточного соединения эртапенема формулы 2а в аморфной форме и примесь в качестве баланса. Примесь включает непрореагировавшую боковую цепь эртапенема формулы 4а, сырое вещество и продукты его разложения.
В методе согласно настоящему изобретению при последующей обработке используемый растворитель представляет собой воду, так что избегают использования органического растворителя, и, следовательно, данный способ является экономичным и экологически безопасным; продукт получают в виде свободной кислоты, уменьшая, таким образом, введение неорганических солей, и, следовательно, это благоприятно для последующей реакции; продукт находится в аморфной форме, и данное твердое вещество имеет высокую чистоту и легкую текучесть, легко выдерживает хранение и подходит для обработки при последующей реакции снятия защиты.
Краткое описание чертежа
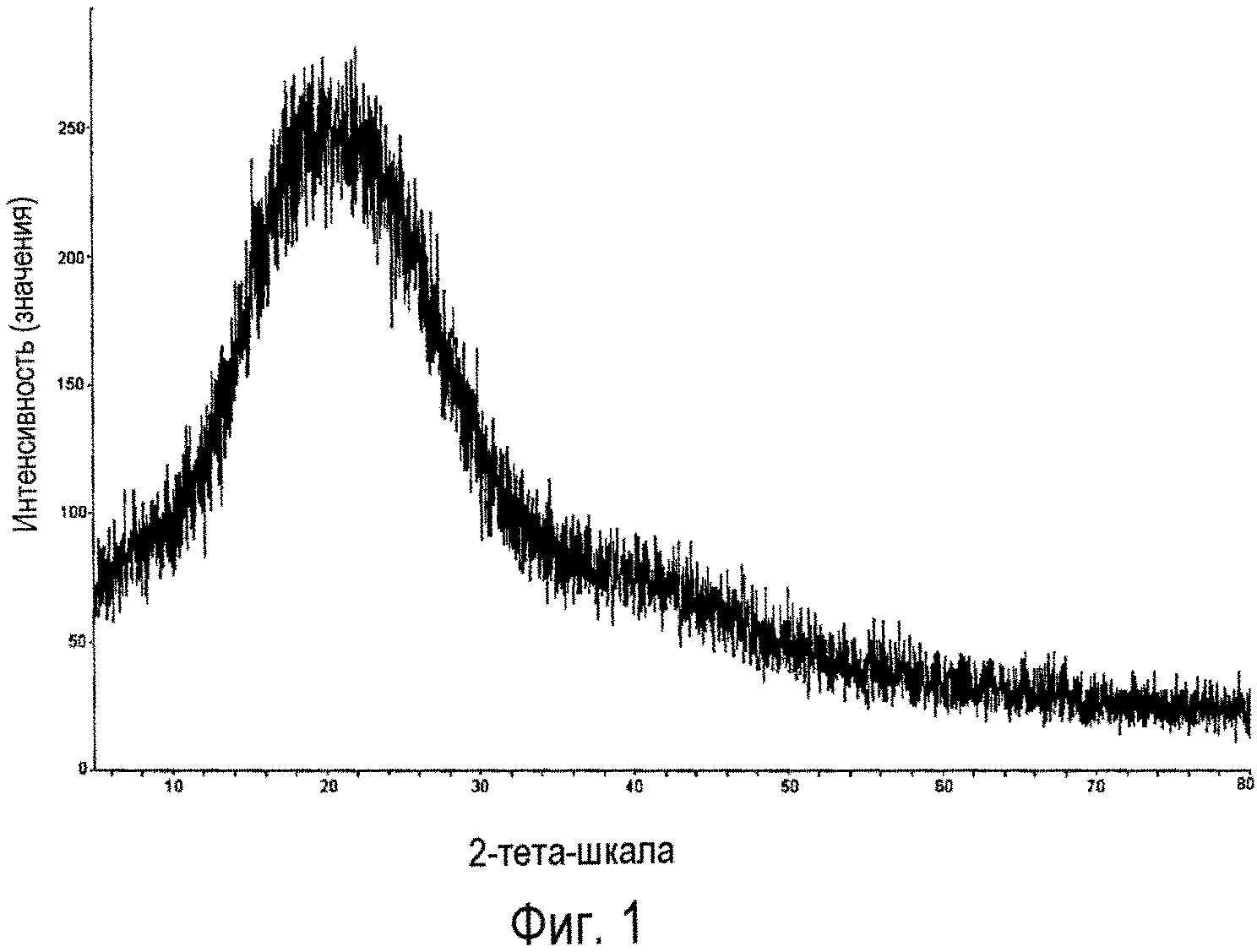
Фиг.1 представляет собой порошковую рентгенограмму соединения 2а, полученного в примере 1 настоящего изобретения.
Конкретные способы осуществления данного изобретения
Далее изобретение будет описано в сочетании с приведенными ниже примерами, которые, однако, не ограничивают настоящее изобретение.
Приборы и условия эксперимента
1. Рентгеноструктурный анализ на порошке
Прибор: рентгеновский дифрактометр Rigaku D/MAX 2550
Условия сканирования: от 5° до 80°/величина шага 0,02°/затрата времени 0,12 с, Cu (40 кВ, 150 мА), I (макс) 282 (интенсивность пика для самого большого пика I=282) (отсчетов в с).
2. Анализ методом ВЭЖХ
Прибор: Agilent серии 1100
Колонка: Gemini C18 (5 мкм, 250×4,6 мм)
Условия эксперимента: длина волны: 230 нм, подвижная фаза: 0,05%-ный фосфатный водный раствор/ацетонитрил=40:60 (об./об.)
3. Анализ методом Н-ЯМР
Прибор: ЯМР анализатор BRUKER AVANCE II500 МГц
Растворитель: ДМСО-d6
4. Масс-спектрометрический анализ
Прибор: Applied Biosystems API4000 LC-MS
Условия эксперимента: метод положительного иона ALLSCAN (источник ионов ESI), интервал МАСС: 100-1500 amu, газовая завеса (CUR): 25 л/мин, shealth газ (Gs1): 35 л/мин, вспомогательный газ (Gs2): 45 л/мин, напряжение ионного источника (IS): 5000 В, температура ионного источника: 500°С, потенциал декластеризации (DP): 40 В, и входной потенциал ячейки соударений (ЕР): 10 В.
Подвижная фаза: 2 мМ водный раствор ацетата аммония, содержащий 0,5% смеси муравьиная кислота:метанол (50:50, об./об.).
Если не указано конкретно, упомянутые выше эксперименты осуществляли согласно рекомендованной программе производителя.
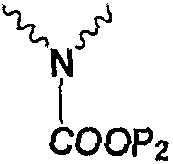
Пример 1: Получение соединения 2а, в котором Np представляет собой
36,0 г (0,0605 ммоль) исходного ядра карбапенема 3 (Р1 представляет собой PNB) растворяли в 300 мл ДМФА, затем прибавляли 26,7 г (0,0599 моль) боковой цепи эртапенема 4а (Р2 представляет собой PNB). К реакционной смеси медленно добавляли 9,4 г (0,0727 моль) N,N-диизопропилэтиламина при -35°С. Реакцию осуществляли при перемешивании. По завершении реакции реакционную смесь выливали в водный раствор HCl (рН 4), фильтровали; 46,6 г твердого вещества получали в виде порошка белого или почти белого цвета. Чистота: 98,2% (по данным ВЭЖХ). Выход: 98,5%. Полученное твердое вещество анализировали методом рентгеновской дифракции на порошке, и результаты показали, что твердое вещество находилось в аморфной форме. Фиг.1 представляет собой порошковую рентгенограмму продукта.
Н-ЯМР (ДМСО-d6): δ 1,18 (д, 3Н), 1,20 (д, 3Н), 1,95 (м, 1Н), 2,81 (м, 1Н), 3,18-3,47 (м, 3Н), 3,60-4,50 (м, 6Н), 5,04-5,44 (м, 5Н), 7,30-8,30 (м, 12Н), 10,27 (д, 1Н).
МС: 788,9 (М-1), 812,7 (М+Na).
Пример 2: Получение соединения 2а, в котором Np представляет собой

Использовали препаративный способ примера 1, за исключением того, что в качестве реагентов использовали исходное ядро карбапенема формулы 3 (Р1 представляет собой PNB) и боковую цепь эртапенема 4а (Np представляет собой
МС: 645,2 (М-1), 669,1 (М+Na).
Примеры 3-9: Получение соединения 2а, в котором Np представляет
Использовали препаративный способ примера 1, за исключением того, что в качестве реагентов использовали исходное ядро карбапенема 3 и боковую цепь эртапенема 4а (Np представляет собой
Вывод: с использованием препаративного способа настоящего изобретения, конденсацией исходного ядра карбапенема 3 и боковой цепи эртапенема 4а с различными защитными группами, можно получить твердое вещество в аморфной форме.
Примеры 10-17: Влияние концентрации кислоты на продукт
Использовали препаративный способ примера 1, за исключением того, что значение рН водного раствора HCl изменяли до 2, 2,5, 3, 3,5, 4,5, 5, 5,5 и 6, результаты опытов представлены в таблице 2.
Вывод: Значение рН водного раствора HCl, составляющее 2-5, является предпочтительной концентрацией кислоты, поскольку и выход, и чистота продуктов превышают 97%. Если значение рН водного раствора HCl находится в интервале от 2,5 до 4,5, и выход, и чистота продуктов превышают 98%.
Примеры 18-22: Влияние типа кислот на продукт
Использовали препаративный способ примера 1, за исключением того, что вместо хлористоводородной кислоты использовали серную кислоту, фосфорную кислоту, дигидрофосфат натрия, уксусную кислоту и п-толуолсульфокислоту, результаты представлены в таблице 3.
Вывод: в данном изобретении можно использовать различные виды неорганической кислоты и органической кислоты, а выход и чистота полученных продуктов превышают 96%.
Сравнительные примеры: экспериментальные результаты примеров соединения 2а (Р1 и Р2, оба являются PNB) в родственном патенте WO2008062279.
Сравнительный пример 1: Экспериментальный результат примера 1 WO2008062279
8,3 г боковой цепи эртапенема 4а (Np представляет собой
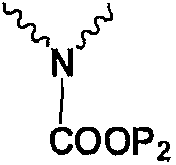
Сравнительный пример 2: Экспериментальный результат примера 3 WO2008062279
8,3 г боковой цепи эртапенема 4а (Np представляет собой
Вывод: Из результатов сравнительных примеров 1 и 2, из способов получения, описанных в примерах 1 и 3 WO2008062279, можно узнать, что твердое вещество получить нельзя, или полученное твердое вещество имеет низкую чистоту, что не подходит для хранения и не может быть использовано для идентификации структуры.
Реферат
Изобретение относится к области органической химии, а именно к способу получения промежуточного соединения эртапенема формулы 2а в твердой форме, в котором Np представляет собойилии Pи Рпредставляют собой защитные группы для карбоксила; в котором, когда Np представляет собой, Pи Роба представляют собой п-нитробензил, о-нитробензил, п-метоксибензил, аллил, 2,2,2-трихлорэтил, бензгидрил, триметилсилил или трет-бутил; когда Np представляет собой, Pпредставляет собой п-нитробензил; причем метод включает конденсацию исходного ядра карбапенема формулы 3 с боковой цепью эртапенема формулы 4а; согласно изобретению по завершении реакции реакционную смесь выливают в водный раствор кислоты. Технический результат: разработан новый способ получения промежуточного соединения 2а, отличающийся простотой, экономичностью и экологичностью. 7 з.п. ф-лы, 1 ил., 3 табл., 24 пр.
Формула
в котором Np представляет собой
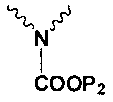
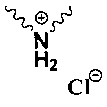
P1 и Р2 представляют собой защитные группы для карбоксила; в котором,
когда Np представляет собой
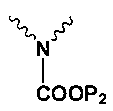
когда Np представляет собой
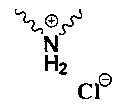
причем метод включает конденсацию исходного ядра карбапенема формулы 3
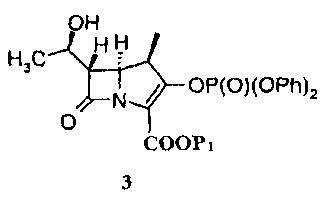
с боковой цепью эртапенема формулы 4а,
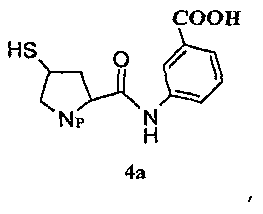
отличающийся тем, что по завершении реакции реакционную смесь выливают в водный раствор кислоты.
Комментарии