Сероводородные производные нестероидных противовоспалительных лекарственных средств - RU2468019C2
Код документа: RU2468019C2
Чертежи














Описание
Настоящая заявка подана в качестве частично продолженной заявки PCT/CA2006/000484, поданной 31 марта 2006, которая испрашивает приоритет заявки PCT/CA2005/000819, поданной 27 мая 2005. Настоящая заявка дополнительно испрашивает приоритет предварительных заявок на патент США №№ 60/807639, поданной 18 июля 2006, и 60/887188, поданной 30 января 2007.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Изобретение относится к производным нестероидных противовоспалительных лекарственных средств (NSAID), имеющих улучшенные противовоспалительные свойства, полезные при лечении воспаления, боли и лихорадочного состояния. Более конкретно, NSAID представляют собой производные с сероводородом (H2S), высвобождающие остаток с получением новых противовоспалительных соединений, имеющих сниженные побочные эффекты.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Нестероидные противовоспалительные лекарственные средства (NSAID) широко используют для лечения различных состояний, связанных с болью, лихорадкой и воспалением, включая остеоартрит, ревматоидный артрит, подагру и анкилозирующий спондилит. Их также широко используют для лечения острой боли, связанной с травмами и хирургическими процедурами (включая зубные процедуры), и головной боли. В основном считается, что благоприятное действие NSAID может быть обусловлено их способностью подавлять синтез простагландинов за счет ингибирования циклооксигеназы-1 (СОХ-1) и циклооксигеназы-2 (СОХ-2).
Однако длительное применение NSAID в значительной степени ограничивается их способностью вызывать клинически значимые повреждения в желудочно-кишечном тракте (Wallace, J. L. Nonsteroidal anti-inflammatory drugs and gastroenteropathy: the second hundred years. Gastroenterology. 1997; 112:1000-1016). Селективные ингибиторы СОХ-2 рассматривались в качестве улучшения по сравнению с традиционными NSAID, поскольку, как оказалось, они в меньшей степени вызывают повреждение желудочно-кишечного тракта. Однако возникло беспокойство в отношении сердечно-сосудистой токсичности указанных препаратов и, возможно, также традиционных NSAID (Grosser et al., Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest. 2006; 116: 4-15).
Хорошо известно, что NSAID стимулируют адгезию лейкоцитов и уменьшение кровотока в слизистой желудка, и указанные действия представляют собой важный вклад в патогенез вызванного NSAID повреждения желудочно-кишечного тракта (Wallace, 1997). Индукция адгезии лейкоцитов за счет неселективных и СОХ-2-селективных NSAID может также давать вклад в сердечно-сосудистые осложнения указанных лекарственных средств.
Недавно заметили, что сероводород (H2S) проявляет противовоспалительные и анальгезирующие активности. H2S представляет собой эндогенное вещество, продуцируемое во многих тканях и оказывающее действие на многие функции (Wang, Two's company, three's a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J 2002; 16: 1792-1798). Также было показано, что он является сосудорасширяющим средством и может подавлять адгезию лейкоцитов к эндотелию сосудов (Wang, 2002; Fiorucci et al., Inhibition of hydrogen sulfide generation contributes to gastric injury caused by anti-inflammatory nonsteroidal drugs. Gastroenterology. 2005; 129: 1210-1224). Дополнительно, Fiorucci et al. (2005) показали, что предварительная обработка донором H2S может уменьшать тяжесть индуцированного NSAID повреждения желудочно-кишечного тракта крысы.
Неожиданно, авторы данного изобретения показали в настоящей заявке, что противовоспалительная активность различных NSAID в значительной степени улучшается или при ковалентном связывании, или при образовании солей NSAID с высвобождающимся фрагментом H2S. Дополнительно, было показано, что указанные производные NSAID обладают пониженным побочным действием. В частности, авторы показали, что NSAID производные настоящего изобретения имеют одну или несколько из следующих дополнительных характеристик: (1) приводят к меньшему повреждению желудочно-кишечного тракта по сравнению с традиционными NSAID; (2) ускоряют лечение уже существовавших язв желудка и (3) в значительно меньшей степени вызывают повышение системного кровяного давления по сравнению с традиционными NSAID. Более того, производные NSAID настоящего изобретения понижают адгезию лейкоцитов к эндотелию сосудов, что может давать вклад в снижение побочного действия как на желудочно-кишечный тракт, так и на сердечно-сосудистую систему.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В одном аспекте настоящего изобретения обеспечивают производные NSAID, причем указанные производные включают H2S-высвобождающий фрагмент, который или ковалентно связан с NSAID, или образует соль с NSAID. Неожиданно, соединения настоящего изобретения демонстрируют улучшенную противовоспалительную активность в модели индуцированного каррагинаном отека лапы крысы по сравнению с одним NSAID, одним H2S-высвобождающим фрагментом и комбинацией NSAID и H2S-высвобождающего фрагмента, вводимых раздельно, но одновременно. Более того, производные NSAID настоящего изобретения приводят к умеренному кратковременному увеличению концентраций H2S в плазме. Без связи с определенной теорией, кратковременное увеличение концентраций H2S в плазме, которое все еще остается в физиологическом диапазоне, может давать вклад в их улучшенную противовоспалительную активность.
Неожиданно, соединения настоящего изобретения могут также демонстрировать улучшенную способность по подавлению активности циклооксигеназы-2 (COX-2) и/или активности циклооксигеназы-1 (COX-1) по сравнению с их соответствующими немодифицированными NSAID аналогами. Такая улучшенная способность по подавлению активности COX-2 и/или COX-1 может также давать вклад в улучшенную противовоспалительную активность. Более того, соединения настоящего изобретения, обладающие улучшенной способностью по подавлению активности COX-1, демонстрируют значительное подавление выработки тромбоксана В2 в тромбоцитах, что может давать вклад в пониженную сердечно-сосудистую токсичность.
Дополнительно, соединения настоящего изобретения в меньшей степени демонстрируют побочные действия по сравнению с их соответствующими немодифицированными NSAID аналогами. Например, некоторые соединения неожиданно вызывают в значительной степени меньше повреждений желудка, чем сами NSAID, несмотря на то что соединения заметно подавляют синтез желудочного простагландина. Притом что сохранность желудка наблюдается при указанных H2S-высвобождающих производных NSAID, указанный результат не достигается, если NSAID и H2S-высвобождающий фрагмент вводят крысам раздельно, но одновременно. Без связи с определенной теорией, было показано, что соединения настоящего изобретения уменьшают адгезию лейкоцитов к эндотелию сосудов, что может давать вклад в их безопасность для желудка. Дополнительно, пониженная адгезия лейкоцитов к эндотелию сосудов может снижать побочное действие на сердечно-сосудистую систему, которое часто наблюдается при продолжительном применении NSAID.
Дополнительно, соединения настоящего изобретения неожиданно вызывают меньшее увеличение систолического кровяного давления при введении крысам, страдающим гипертензией, по сравнению с наблюдаемым при введении обычных NSAID. Пониженная склонность к повышению кровяного давления может снижать побочные действия на сердечно-сосудистую систему, которые часто наблюдаются при продолжительном применении NSAID.
В соответствии с настоящим изобретением обеспечивают соединения общей формулы:
A-Y-X, (Формула I)
где А представляет собой NSAID радикал, Y выбирают из группы, состоящей из -C(O)O-, -C(O)NH-, -C(O)OC(O)-, -C(O)NHCH2C(O)- или нуля, и Х представляет собой фрагмент, способный высвобождать сероводород или сам по себе, или при связывании с NSAID (здесь далее обозначаемый как H2S-высвобождающий фрагмент), или его фармацевтически приемлемую соль, таким образом, что если Y представляет собой нуль, производное NSAID может представлять собой соль А и Х.
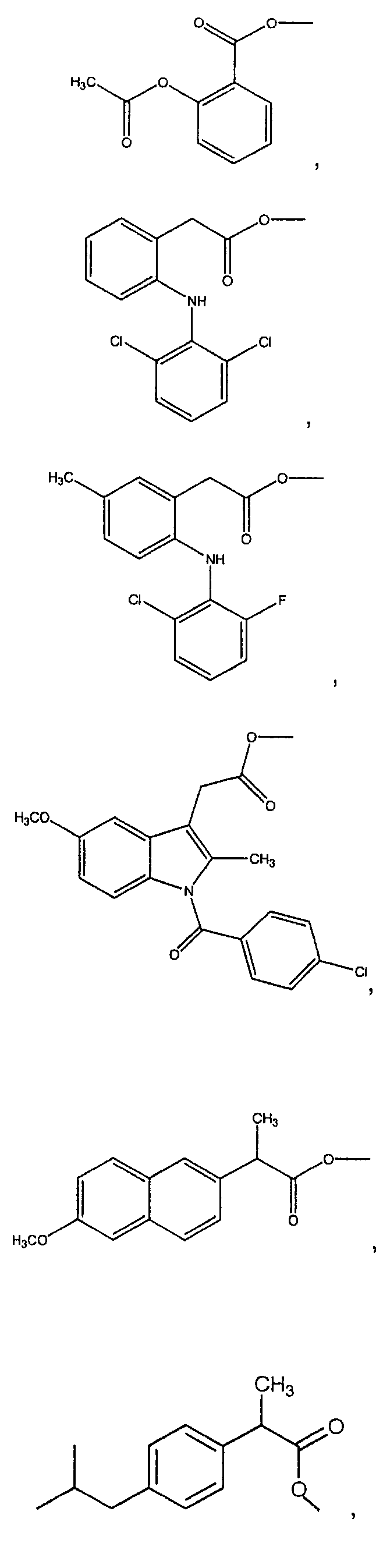
В предпочтительном варианте осуществления изобретения Х в формуле I выбирают из группы, состоящей из:



Однако предполагается, что в настоящем изобретении может быть использован любой нетоксичный эффективный фрагмент, способный к высвобождению H2S или сам по себе, или при связывании с NSAID.
В одном из вариантов осуществления соединения по изобретению имеют следующую общую формулу:
В-C(O)O-X, (Формула II)
где В-C(O)O- представляет собой производное NSAID, имеющего свободную карбоксильную группу, или карбокси-замещенного NSAID и X представляет собой H2S-высвобождающий фрагмент или его фармацевтически приемлемую соль.
В одном из вариантов осуществления В-C(O)O- в формуле II выбирают из группы, состоящей из:

и Х представляет собой сероводород (H2S) высвобождающий фрагмент.
В одном из вариантов осуществления Х в формуле II выбирают из группы, состоящей из


Однако предполагается, что в настоящем изобретении может быть использован любой нетоксичный эффективный фрагмент, способный к высвобождению H2S или сам по себе, или при связывании с NSAID.
NSAID, рассматриваемые для включения в соединения настоящего изобретения, включают ацетилсалициловую кислоту (ASA), диклофенак, напроксен, индометацин, флурбипрофен, сулиндак, ибупрофен, ацеклофенак, ацеметацин, беноксапрофен, бензофенак, бромфенак, буклоксовую кислоту, бутибуфен, карпрофен, целекоксиб, циклопрофен, цинметацин, клиденак, клопирак, дифлузинал, этодолак, эторикоксиб, фенбуфен, фенклофенак, фенклорак, фенопрофен, фентиазак, флуноксапрофен, фурапрофен, фуробуфен, фурафенак, ибуфенак, индопрофен, изоксепак, кетопрофен, кеторолак, локсопрофен, лоназолак, люмиракоксиб, метиазиник, мефенамовую кислоту, меклофенамовую кислоту, мелоксикам, набуметон, пиромидовую кислоту, салсалат, миропрофен, оксапрозин, оксепинак, паракоксиб, фенилбутазон, пирпрофен, пироксикам, пирозолак, протизиновую кислоту, рофекоксиб, салицилат натрия, супрофен, тиапрофеновую кислоту, толметин, вальдекоксиб, зомепирак и т.п.
Предпочтительные соединения представляют собой соединения следующей формулы:

4-(5-тиоксо-5Н-1,2-дитиол-3-ил)фенил 2-ацетоксибензоат (I),

4-(5-тиоксо-5Н-1,2-дитиол-3-ил)фенил 2-(2-(2,6-дихлорфениламино)фенил)ацетат (II),

4-(5-тиоксо-5Н-1,2-дитиол-3-ил)фенил 2-(2-(2-хлор-6-фторфениламино)-5-метилфенил)ацетат (III),

4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фениловый сложный эфир [1-(4-хлорбензоил)-5-метокси-2-метил-1Н-индол-3-ил]уксусной кислоты (IV),

4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фениловый сложный эфир 2-(6-метоксинафталин-2-ил)пропионовой кислоты (V),

4-(5-оксо-5Н-[1,2]дитиол-3-ил)фениловый сложный эфир 2-ацетоксибензойной кислоты (VI),

4-(5-оксо-5Н-[1,2]дитиол-3-ил)фениловый сложный эфир [2-(2,6-дихлорфениламино)фенил]уксусной кислоты (VII),

4-(5-оксо-5Н-[1,2]дитиол-3-ил)фениловый сложный эфир [2-(2-хлор-6-фторфениламино)-5-метилфенил]уксусной кислоты (VIII),

4-(5-оксо-5Н-[1,2]дитиол-3-ил)фениловый сложный эфир [1-(4-хлорбензоил)-5-метокси-2-метил-1Н-индол-3-ил]уксусной кислоты (IX),

4-(5-оксо-5Н-[1,2]дитиол-3-ил)фениловый сложный эфир 2-(6-метоксинафталин-2-ил)пропионовой кислоты (Х),

4-(5-гидроксиимино-5Н-[1,2]дитиол-3-ил)фениловый сложный эфир 2-ацетоксибензойной кислоты (XI),

4-(5-гидроксиимино-5Н-[1,2]дитиол-3-ил)фениловый сложный эфир [2-(2,6-дихлорфениламино)фенил]уксусной кислоты (XII),

4-(5-гидроксиимино-5Н-[1,2]дитиол-3-ил)фениловый сложный эфир [2-(2-хлор-6-фторфениламино)-5-метилфенил]уксусной кислоты (XIII),

4-(5-гидроксиимино-5Н-[1,2]дитиол-3-ил)фениловый сложный эфир [1-(4-хлорбензоил)-5-метокси-2-метил-1Н-индол-3-ил]уксусной кислоты (XIV),

4-(5-гидроксиимино-5Н-[1,2]дитиол-3-ил)фениловый сложный эфир 2-(6-метоксинафталин-2-ил)пропионовой кислоты (XV),

4-тиокарбамоилфениловый сложный эфир 2-ацетоксибензойной кислоты (XVI),

4-тиокарбамоилфениловый сложный эфир [2-(2,6-дихлорфениламино)фенил]уксусной кислоты (XVII),

4-тиокарбамоилфениловый сложный эфир [2-(2-хлор-6-фторфениламино)-5-метилфенил]уксусной кислоты (XVIII),

4-тиокарбамоилфениловый сложный эфир [1-(4-хлорбензоил)-5-метокси-2-метил-1Н-индол-3-ил]уксусной кислоты (XIX),

4-тиокарбамоилфениловый сложный эфир 2-(6-метоксинафталин-2-ил)пропионовой кислоты (XX),
4-изотиоцианатофенил 2-ацетоксибензоат (XXI),

4-изотиоцианатофенил 2-(2-(2,6-дихлорфениламино)фенил)ацетат (XXII),

4-изотиоцианатофенил 2-(2-(2-хлор-6-фторфениламино)-5-метилфенил)ацетат (XXIII),

4-(изотиоциано)фенил 2-[1-(4-хлорбензоил)-5-метокси-2-метилиндол-3-ил]ацетат (XXXIV),

4-изотиоцианатофенил 2-(2-метоксинафталин-6-ил)пропионат (XXV),

4-(5-тиоксо-5Н-1,2-дитиол-3-ил)фенил 2-(4-изобутилфенил)пропионат (XXVI),

4-(5-тиоксо-5Н-1,2-дитиол-3-ил)фенил 2-(3-бензоилфенил)пропионат (XXVII),

4-(5-тиоксо-5Н-1,2-дитиол-3-ил)фенил 2-(2-фтор-4-бифенилил)пропионат (XXVIII),
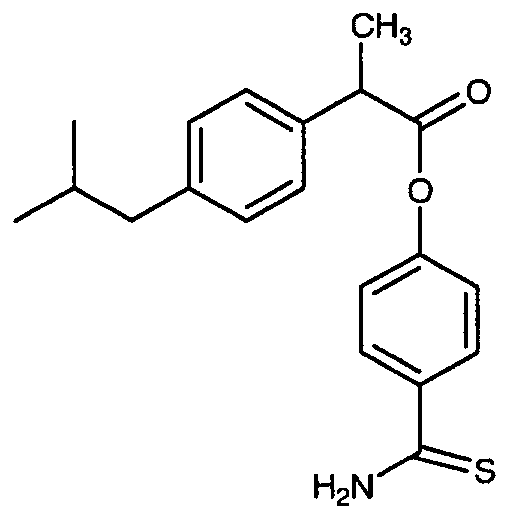
4-тиокарбамоилфенил 2-(4-изобутилфенил)пропионат (XXIX),

4-тиокарбамоилфенил 2-(4-оксофенил)фенилпропионат (XXX),

4-тиокарбамоилфенил 2-(2-фтор-4-бифенилил)пропионат (XXXI),

4-изотиоцианатофенил 2-(4-изобутилфенил)пропионат (XXXII),

4-(изотиоциано)фенил 2-(4-оксофенил)-фенилпропионат (XXXIII) и

4-(изотиоциано)фенил 2-(2-фтор-4-бифенилил)пропионат (XXXIV).
Упомянутый выше предшественник NSAID (А) получают согласно способам, известным в предшествующем уровне техники. См., например, The Merck Index, 13thEdition (2001), Merck & Co., Whitehouse Station, N.J., включенный здесь в качестве ссылки. Если доступны, могут быть использованы соответствующие изомеры, включая оптические изомеры.
Фармацевтически приемлемые соли соединений настоящего изобретения, такие как, например, соли щелочных металлов и щелочно-земельных металлов, нетоксичных аминов и аминокислот, также представляют собой часть настоящего изобретения. Предпочтительные соли соединений настоящего изобретения представляют собой соли аргинина и агматина. Также включены фармацевтически приемлемые соли присоединения кислоты.
В предпочтительном варианте осуществления изобретения NSAID настоящего изобретения представляют собой модифицированный H2S-высвобождающим фрагментом 4-гидрокситиобензамид (обозначаемый здесь как TBZ). TBZ производные равным образом демонстрируют лучшую общую противовоспалительную активность и сниженные побочные действия по сравнению с производными 5-п-гидроксифенил-1,2-дитиол-3-тиона (ADT-OH). Неожиданно, TBZ производные генерируют в значительной степени больше H2S по сравнению с ADT-OH производными, что может давать вклад как в увеличение противовоспалительной активности, так и в уменьшение побочного действия.
Дополнительно, TBZ производные сохраняют способность более стабильно ингибировать СОХ-1/СОХ-2 по сравнению с ADT-OH производными. Фактически, многие TBZ производные действительно демонстрируют усиление ингибирования СОХ-1, или ингибирования СОХ-2, или обоих. Более того, соединение ХХ (TBZ производное напроксена) показывало в значительной степени лучшие результаты в ингибировании синтеза тромбоксана В2 по сравнению с эквивалентным производным ADT-OH, соединением V (напроксен-ADT-OH), и соединение XIX (TBZ производное индометацина) показывало в значительной степени лучшие результаты в ингибировании синтеза тромбоксана В2 по сравнению с эквивалентным производным ADT-OH, соединением IV (ADT-OH производное индометацина). Улучшенное ингибирование тромбоксана В2 может давать вклад в безопасность настоящих производных в отношении сердечно-сосудистой системы.
Соединения настоящего изобретения могут быть получены, как показано на двух следующих схемах.
Схема 1
Схема 1 ниже представлена при использовании в качестве примера синтеза 4-(5-тиоксо-5Н-1,2,-дитиол-3-ил)фенил 2-(2-(2,6-дихлорфениламин)фенил)ацетата (соединение II)

NSAID, имеющее свободную карбоксильную группу (или карбоксизамещенное NSAID), например диклофенак (1), сначала растворяли в диметилформамиде и добавляли гидроксибензотриазол (HOBt) и 1,3-дициклогексилкарбодиимид (DCC). К указанной смеси добавляли фрагмент, высвобождающий сероводород, такой как 5-п-гидроксифенил-1,2-дитиол-3-тион (ADT-OH) (2), при условиях, пригодных для получения соединений настоящего изобретения, таких как 4-(5-тиоксо-5H-1,2-дитиол-3-ил)фенил 2-(2-(2,6-дихлорфениламино)фенил)ацетат (3). Очевидно, что в настоящей схеме можно использовать другие фрагменты, высвобождающие сероводород, такие как 4-гидроксифенилизотиоцианат (здесь обозначаемый как HPI).
Схема 2
Схема 2 ниже представлена при использовании в качестве примера синтеза 4-тиокарбамоилфенилового сложного эфира [2-(2,6-дихлорфениламино)фенил]уксусной кислоты (XVII). На указанной схеме используют реагент Лавессона для присоединения серосодержащей группы к фрагменту, высвобождающему сероводород, после его ковалентного связывания с NSAID.

NSAID, имеющее свободную карбоксильную группу (или карбоксизамещенное NSAID), например диклофенак (1), сначала растворяли в диметилформамиде и добавляли гидроксибензотриазол (HOBt) и 1,3-дициклогексилкарбодиимид (DCC). К указанной смеси добавляли предшественник, высвобождающий сероводород, такой как 4-гидроксибензамид, при условиях, пригодных для образования предшественника (например, 4-карбамоилфенил 2-(2-(2,6-дихлорфениламино)фенил)ацетата (2)) соединения настоящего изобретения, причем указанный предшественник не содержит серы. Добавляют подходящее соединение, которое может добавить серосодержащую группу, такое как реагент Лавессона, для получения соединения настоящего изобретения (например, 4-тиокарбамоилфенилового сложного эфира [2-(2,6-дихлорфениламино)фенил]уксусной кислоты (3)).
В дополнительном аспекте настоящее изобретение обеспечивает фармацевтическую композицию соединения настоящего изобретения и фармацевтически приемлемого эксципиента или носителя, в особенности фармацевтическую композицию для применения при лечении воспалительного состояния желудочно-кишечного (GI) тракта.
Соединения настоящего изобретения пригодны для применения для, не ограничиваясь перечисленным, лечения воспаления у пациента и для лечения других заболеваний, связанных с воспалением, например в качестве анальгетика при лечении боли и головной боли или в качестве жаропонижающего средства для лечения лихорадочного состояния. Например, соединения настоящего изобретения полезны для лечения артрита, включая, но не ограничиваясь перечисленным, ревматоидный артрит, спондилоартропатии, подагрический артрит, остеоартрит, системную красную волчанку и ювенильный артрит. Такие соединения по изобретению пригодны при лечении астмы, бронхита, спазмов при менструациях, тендинита, бурсита, кожных болезней, таких как псориаз, экзема, ожоги и дерматит, и послеоперационных воспалений, включая глазную хирургию, такую как операции по удалению катаракты и рефракционную хирургию. Соединения по изобретению также полезны для лечения желудочно-кишечных заболеваний, таких как воспалительное заболевание кишечника, болезнь Крона, гастрит, синдром раздраженного кишечника и язвенный колит, а также для предотвращения или лечения рака, такого как колоректальный рак. Соединения по изобретению пригодны при лечении воспалений при таких заболеваниях, как сосудистые заболевания, мигрени, узелковый периартерит, тиреоидит, апластическая анемия, болезнь Ходжкина, склеродома, ревматическая атака, диабет I типа, заболевания нервно-мышечных соединений, включая злокачественную миастению, заболевания белого вещества, включая множественный склероз, саркоидоз, нефротический синдром, синдром Бехчета, полимиозит, гингивит, нефрит, гиперчувствительность, послетравматическая опухоль, ишемия миокарда и т.п. Соединения также полезны для лечения глазных заболеваний, таких как ретинит, ретинопатия, увеит, глазная светобоязнь и острая травма тканей глаза. Соединения также пригодны для лечения легочных воспалений, таких как связанные с вирусными инфекциями, и кистозного фиброза. Соединения также полезны для лечения определенных нарушений центральной нервной системы, таких как кортикальное слабоумие, включая болезнь Альцгеймера. Соединения по изобретению пригодны в качестве противовоспалительных средств, таких как для лечения артрита, с дополнительным преимуществом, состоящим в значительно меньшем вредном побочном действии. Указанные соединения также полезны для лечения аллергического ринита, синдрома затрудненного дыхания, синдрома эндотоксического шока, атеросклероза и повреждения центральной нервной системы в результате удара, ишемии и травмы. Соединения также полезны при лечении боли, такой как, но не ограничиваясь перечисленным, послеоперационная боль, зубная боль, мышечная боль и боль в результате ракового заболевания. Помимо полезности для лечения человека, указанные соединения также пригодны для лечения млекопитающих, включая лошадей, собак, кошек, крыс, мышей, овец, свиней и т.д.
В зависимости от конкретного состояния или заболевания, подвергаемого лечению, пациенту могут быть введены соединения настоящего изобретения в любой пригодной терапевтически эффективной и безопасной дозе, как может быть легко определено специалистом в данной области. Указанные соединения наиболее желательно вводить в дозах в диапазоне от примерно 1 до примерно 2000 мг в день, в виде одной дозы или разделенных доз, хотя неизбежно будут наблюдаться вариации в зависимости от массы и состояния пациента, подвергаемого лечению, и конкретного выбранного пути введения. Понятно, что дозы будут зависеть от конкретного NSAID, используемого для получения соединений настоящего изобретения. Однако наиболее желательна дозировка, лежащая в диапазоне от примерно 0,1 до примерно 100 мг/кг, предпочтительно в диапазоне примерно от 5 до 90 мг/кг и более предпочтительно в диапазоне примерно от 5 до 50 мг. Тем не менее могут наблюдаться вариации в зависимости от массы и состояния пациентов, подвергаемых лечению, и их индивидуальной реакции на указанное лекарственное средство, а также от типа выбранного фармацевтического препарата и периода времени и интервала, в ходе которого проводят такое введение. В некоторых случаях дозы ниже предела указанного диапазона могут оказаться более чем адекватными, тогда как в других случаях могут применяться еще большие дозы, не вызывая никакого вредного побочного действия, при условии, что такие большие дозы сначала подразделяют на несколько небольших доз для введения в течение дня.
Соединения настоящего изобретения можно вводить в форме любого фармацевтического препарата, природа которого будет зависеть от пути введения. Фармацевтические композиции могут быть получены обычными способами, при использовании совместимых фармацевтически приемлемых эксципиентов или носителей. Примеры таких композиций включают капсулы, таблетки, чрескожные пластыри, леденцы, пастилки, спреи, сиропы, порошки, гранулы, гели, эликсиры, суппозитории и т.п., препараты растворов для немедленного приема, препараты для инъекций, ректальные, назальные, глазные, вагинальные препараты и т.д. Предпочтительный путь введения представляет собой пероральный и ректальный путь введения.
Для перорального введения могут применяться таблетки, содержащие различные эксципиенты, такие как микрокристаллическая целлюлоза, цитрат натрия, карбонат кальция, дикальцийфосфат и глицин, совместно с различными дезинтегрантами, такими как крахмал (предпочтительно кукурузный, картофельный или крахмал тапиоки), альгиновая кислота и определенные сложные силикаты, вместе со связующими для гранулирования, такими как поливинилпирролидон, сахароза, желатин и гуммиарабик. Дополнительно, в целях таблетирования, могут быть использованы лубриканты, такие как стеарат магния, лаурилсульфат натрия и тальк. Твердые композиции сходного типа можно также применять в качестве наполнителей в желатиновых капсулах; предпочтительные материалы, связанные с указанным применением, также включают лактозу или молочный сахар, а также полиэтиленгликоли с высокой молекулярной массой. Если для перорального введения желательна водная суспензия и/или эликсир, активные ингредиенты можно комбинировать вместе с подслащивающими или вкусовыми агентами, красящим веществом и, при желании, эмульгирующими и/или суспендирующими агентами, вместе с такими разбавителями, как вода, этанол, пропиленгликоль, глицерин и их различные комбинации.
Дозированная форма может быть предназначена для немедленного высвобождения, контролируемого высвобождения, длительного высвобождения, отсроченного высвобождения или целевого отсроченного высвобождения. Определения указанных терминов известны специалистам в данной области. Кроме того, на профиль высвобождения дозированной формы может влиять состав полимерной смеси, состав матрикса с покрытием, состав из множества частиц, состав из множества частиц с покрытием, состав на основе ионообменной смолы, осмотический состав или состав биоразрушаемых полимеров. Вне связи с какой-либо теорией, считается, что на высвобождение может быть оказано влияние за счет благоприятной диффузии, растворения, эрозии, ионного обмена, осмоса или их комбинации.
В случае парентерального введения можно использовать раствор активного соединения или в кунжутном, или в арахисовом масле, или в водном растворе пропиленгликоля. Водный раствор должен быть подходящим образом забуферен (предпочтительно рН более 8), и, при необходимости, жидкий разбавитель сначала приводят в изотоническое состояние. Водные растворы пригодны для внутривенного введения. Получение всех таких растворов при стерильных условиях легко осуществлять обычными фармацевтическими технологическими способами, хорошо известными специалистам в данной области.
Следующие примеры далее описывают и позволяют обычному специалисту в данной области применять и использовать изобретение. Однако следует понимать, что данные варианты осуществления представлены в целях иллюстрации изобретения и не должны рассматриваться как ограничивающие объем притязаний изобретения, который определен формулой изобретения.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фигуре 1 показана оценка повреждения желудка, измеренная у крыс, подвергаемых лечению носителем, диклофенаком и двумя производными диклофенака настоящего изобретения, соединением II и соединением XVII.
На фигуре 2 показано количество желудочного простагландина Е2 (PGE2), продуцированное у крыс, подвергаемых лечению носителем, диклофенаком, соединением II и соединением XVII.
На фигуре 3 показана оценка повреждения желудка, измеренная у крыс, подвергаемых лечению носителем, напроксеном и двумя производными напроксена по настоящему изобретению, соединением V и соединением XX.
На фигуре 4 показано количество тромбоксана В2, синтезируемое в крови крыс, упоминаемых на фигуре 3.
На фигуре 5 показана общая длина язв тонкого кишечника у крыс, подвергаемых лечению носителем, диклофенаком и соединением II.
На фигуре 6 показан процент гематокрита у крыс до и после лечения носителем, диклофенаком и соединением II.
На фигуре 7 показано количество экссудативного PGE2, продуцированного в подкожном кармане крыс, при использовании анализа воздушного кармана у крыс, подвергаемых лечению носителем, диклофенаком, соединением II и соединением XVII.
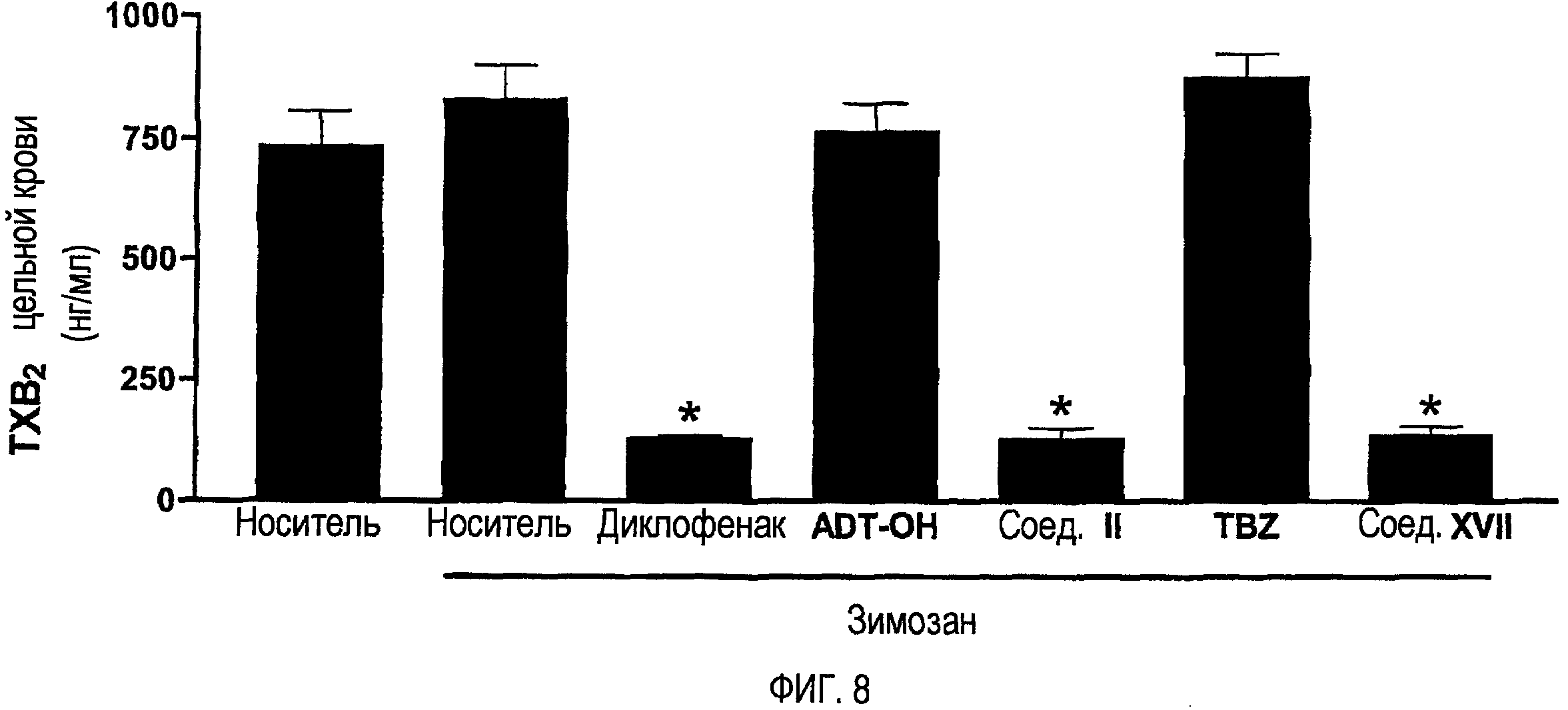
На фигуре 8 показано количество тромбоксана B2 (TXB2) цельной крови у крыс, представленных на фигуре 7.
На фигуре 9 показано ингибирование увеличения объема лапы у крыс, подвергаемых лечению носителем, диклофенаком и соединением II.
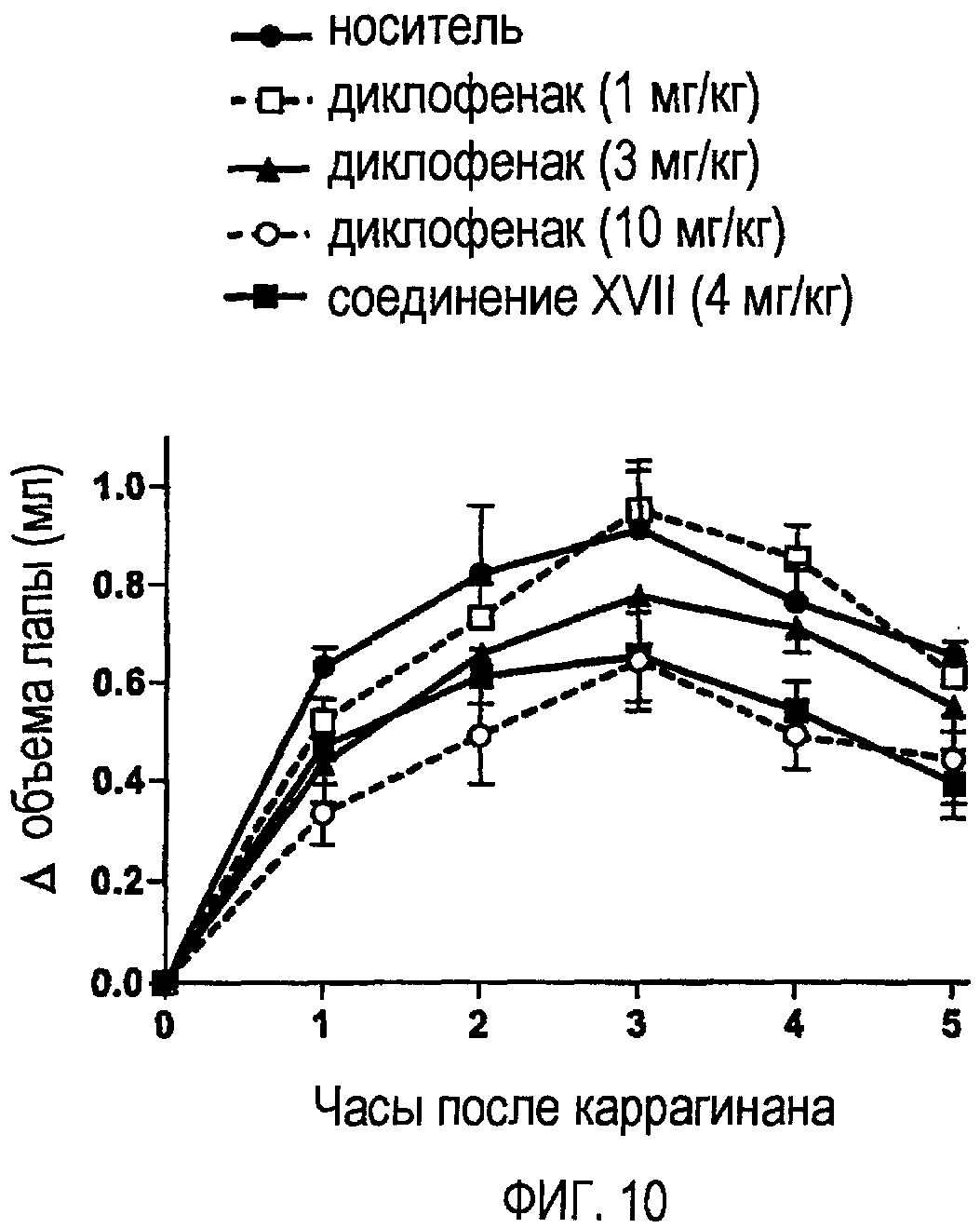
На фигуре 10 показано ингибирование увеличения объема лапы у крыс, подвергаемых лечению носителем, диклофенаком и соединением XVII.
На фигуре 11 показано количество экссудативного PGE2, продуцированного в подкожном кармане крыс, при использовании анализа воздушного кармана у крыс, подвергаемых лечению носителем, напроксеном, соединением V и соединением XX.
На фигуре 12 показан синтез тромбоксана (нг/мл) в крови человека (in vitro) как функция от концентрации индометацина, соединения IV и соединения XIX.
На фигуре 13 показана площадь поверхности в мм2 язв желудка у крыс на следующий день после лечения в течение одной недели носителем, диклофенаком, соединением XVII и соединением XX.
На фигуре 14 показано повышение систолического кровяного давления (мм ртутного столба) у крыс, подвергаемых лечению носителем, диклофенаком, соединением II, напроксеном и соединением XX.
На фигуре 15 показана концентрация сероводорода в плазме при пероральном лечении крыс 50 мкмоль/кг соединением II.
На фигуре 16 показано количество сероводорода, получаемого из соединения II и соединения XVII при инкубации в буфере и гомогенате печени.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Получение соединений
Проводили тонкослойную хроматографию на пластинках Macherey-Nagel силикагеля 50 c флуоресцентным индикатором, и пластинки проявляли УФ-светом (254 нм). Для колоночной хроматографии использовали Kieselgel 60. Все реагенты для синтеза приобретали от Aldrich-Sigma Chemical Company и использовали без очистки. Растворители были аналитической степени чистоты или выше и были использованы в том виде, как приобретались. Роторный испаритель Buchi R-114 использовали для удаления растворителей в вакууме. Структуры соединений подтверждали спектроскопически протонным1Н-ЯМР и13С-ЯМР. Спектры регистрировали при использовании прибора Varian Mercury Plus 400. Химические сдвиги соотносили с Me4Si в качестве внутреннего стандарта. Масс-спектры синтезированных продуктов получали на масс-спектрометре Applied Biosystem API 2000. Точку плавления измеряли на аппарате Buchi B-540. Чистоту конечного соединения определяли при использовании обращенно-фазовой ВЭЖХ. Колонку подсоединяли к инжектору Rheodyne model 7725, ВЭЖХ системе Waters 600, настраиваемому детектору абсорбции Waters 486, установленному на 215 или 235 нм, и самописцу Waters 746. Синтезированные соединения показали удовлетворительные результаты элементного анализа; если анализы представлены исключительно символами элементов, то результаты лежат в диапазоне ±0,4% от теоретических величин.
ПРИМЕР 1
Синтез 4-(5-тиоксо-5Н-[1,2]-дитиол-3-ил)фенилового сложного эфира [2-(2,6-дихлорфениламино)фенил]уксусной кислоты (Соединение II)
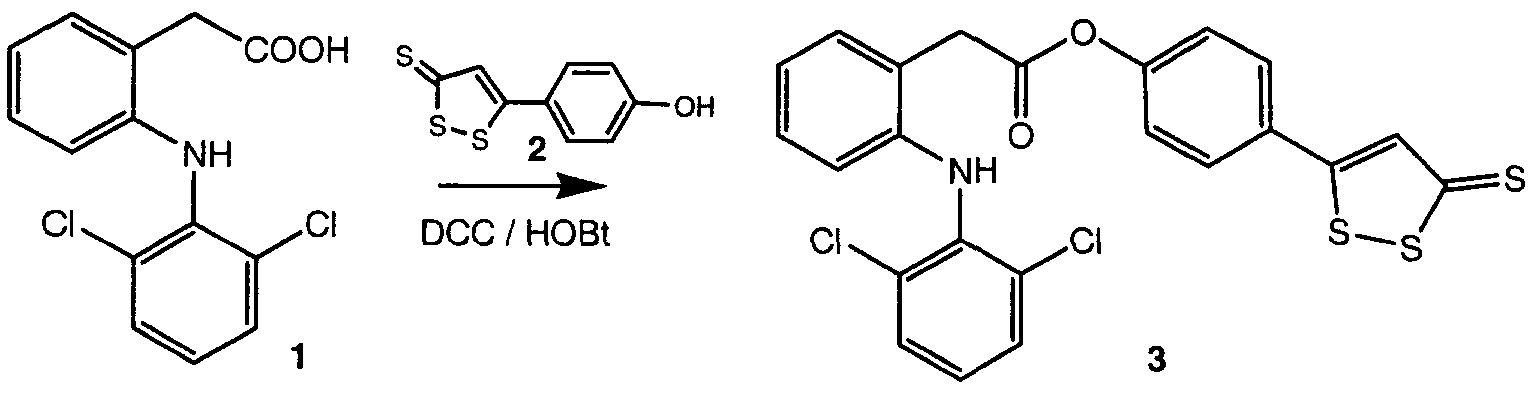
Схема 1
Синтез 5-п-гидроксифенил-1,2-дитиол-3-тиона (2; ADT-OH)

Анезол (31 г, 0,21 моль) и серу (44,8 г, 1,40 моль) нагревали в N,N-диметилформамиде (250 мл) в течение 8 часов; после удаления растворителя остаток практически полностью был растворим в толуоле. Попытка экстрагировать раствор в толуоле 2 н. водным гидроксидом натрия привела к оранжевому твердому осадку (8,5 г, т. пл. выше 300°С). Указанный продукт растворяли в кипящей воде и, после добавления соляной кислоты, получали 2 в виде оранжевого осадка (6,2 г, выход 13%) т.пл. 188-189°С.
1Н-ЯМР (ДМСО-d6) δ: 6,86 (д, 2H), 7,68 (с, 1H), 7,75 (д, 2H), 10,51 (с, -ОН); MS (ESI), m/z 225(M-).
Синтез 4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилового сложного эфира [2-(2,6-дихлорфениламино)фенил]уксусной кислоты (3)
К раствору 1 (диклофенак, 890 мг, 3,0 ммоль) в 50 мл N,N-диметилформамида добавляли при перемешивании при 0°С в течение 1 ч гидроксибензотриазол (445 мг, 3,3 ммоль) и DCC (дициклогексилкарбодиимид) (680 мг, 3,3 ммоль). К реакционной смеси добавляли 5-п-гидроксифенил-1,2-дитиол-3-тион (2; 678 г, 3 ммоль) и перемешивали в течение 1 ч при 0°С и 3 ч при комнатной температуре. После фильтрования фильтрат выпаривали при пониженном давлении и полученный таким образом маслянистый остаток растворяли в этилацетате; органический слой промывали рассолом, сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Неочищенный продукт 3 загружали на открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (9/1), с получением 4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилового сложного эфира [2-(2,6-дихлорфениламино)фенил]уксусной кислоты (1,1 г, 74% выход).
1Н-ЯМР (ДМСО-d6): δ 4,12 (с, 2H), 6,21 (д, 1H), 6,87 (т, 1H), 7,14 (т, 1H), 7,19 (д, 1H), 7,22 (т, 1H), 7,34 (д, 2H), 7,54 (д, 2H), 7,80 (с, 1H), 7,97 (д, 2H);
13С-ЯМР (ДМСО-d6): δ 37,4, 116,1, 121,0, 122,3, 123,5, 123,7, 127,0, 128,7, 129,3, 129,8, 132,0, 132,2, 136,4, 137,7, 143,8, 154,2, 170,3, 173,3, 213,2.
MS (EI), м/e 504 (М+).
Т.пл.: 83-86°C.
ПРИМЕР 2
Синтез 4-тиокарбамоилфенилового сложного эфира [2-(2,6-дихлорфениламино)фенил]уксусной кислоты (Соединение XVII)

Схема 2
Синтез 4-карбамоилфенил 2-[2-(2,6-дихлорфениламино)фенил]ацетата (5)
К раствору 1 (диклофенак, 890 мг, 3,0 ммоль) в 50 мл N,N-диметилформамида добавляли при перемешивании при 0°С в течение 1 ч гидроксибензотриазол (445 мг, 3,3 ммоль) и DCC (680 мг, 3,3 ммоль). К реакционной смеси добавляли 4-гидроксибензамид (4, 616, мг, 4,5 ммоль) и перемешивали в течение 1 ч при 0°С и 3 ч при комнатной температуре. После фильтрования фильтрат выпаривали при пониженном давлении и полученный таким образом маслянистый остаток растворяли в хлороформе; органический слой промывали рассолом, сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Неочищенный продукт 5 загружали на открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (9/1) с получением 4-карбамоилфенил [2-(2,6-дихлорфениламино)фенил]ацетата (5) (212 мг, 17% выход).
Синтез 4-тиокарбамоилфенилового сложного эфира [2-(2,6-дихлорфениламино)фенил]уксусной кислоты (6)
4-карбамоилфенил 2-(2-(2,6-дихлорфениламино)фенил)ацетат (5, 480 мг, 1,14 ммоль) и реагент Лавессона (460 мг, 1,14 ммоль) растворяли в 20 мл безводного бензола. Реакционную смесь нагревали до 50°С и перемешивали в течение 6 ч. Растворитель удаляли при пониженном давлении; неочищенный остаток очищали на колонке с силикагелем (дихлорметан/метиловый спирт 9,5/0,5) с получением чистого соединения 6 (446 мг, 91% выход).
1Н-ЯМР (CDCl3): δ 4,07 (с, 2Н), 6,59 (д, 1H), 6,67 (с, 1H), 6,98 (т, 1H), 7,14 (т, 1Н), 7,19 (д, 1H), 7,28 (т, 1H), 7,33 (д, 2Н), 7,63 (с, 1H), 7,97 (д, 2H);
13С-ЯМР (ДМСО-d6): δ 38,8, 118,8, 121,8, 122,6, 123,7, 124,4, 128,7, 129,1, 129,6, 131,2, 137,2, 137,8, 142,9, 153,5, 170,5, 193,2, 201,7.
MS (EI), м/e 431 (М+).
Т.пл.: 170-172°C.
ПРИМЕР 3
Синтез 4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилового сложного эфира [2-(2-хлор-6-фторфениламино)фенил]уксусной кислоты (Соединение III)

Схема 1
Синтез 4-(5-тиоксо-5Н-1,2-дитиол-3-ил)фенил-2-(2-(2-хлор-6-фторфениламино)-5-метилфенил)ацетата (3)
К раствору 1 (люмиракоксиб, 600 мг, 2,03 ммоль) в 40 мл диметилформамида добавляли при перемешивании при 0°С в течение 1 ч гидроксибензотриазол (301 мг, 2,23 ммоль) и DCC (459 мг, 2,23 ммоль). К реакционной смеси добавляли 5-п-гидроксифенил-1,2-дитиол-3-тион (2, 504 мг, 2,23 ммоль) и перемешивали в течение 1 ч при 0°С и 3 ч при комнатной температуре. После фильтрования фильтрат выпаривали при пониженном давлении для удаления растворителя. Полученный таким образом маслянистый остаток растворяли в этилацетате; органический слой промывали рассолом, сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Неочищенный продукт 3 загружали на открытую колонку с силикагелем и элюировали CH2Cl2 с получением 4-(5-тиоксо-5Н-1,2-дитиол-3-ил)фенил-2-(2-(2-хлор-6-фторфениламино)5-метилфенил)ацетата (3) (299 мг, 37% выход).
1Н-ЯМР (ДМСО): δ 2,32 (с, 3Н), 4,02 (с, 2H), 6,41 (с, 1H), 6,71 (д, 1H), 6,93 (т, 1H), 6,95 (д, 2H), 7,14 (д, 1H), 7,19 (д, 2H), 7,39 (с, 1H), 7,66 (д, 2H);
13С-ЯМР (ДМСО): δ 20,8, 38,7, 115,2, 119,2, 122,5, 123,2, 124,0, 126,1, 127,2, 129,3, 130,3, 131,7, 132,2, 133,6, 136,4, 140,3, 153,7, 154,4, 156,8, 170,3, 171,6, 215,7.
MS (EI), м/e 503 (М+).
Т.пл.: 131-133°C.
ПРИМЕР 4
Синтез 4-тиокарбамоилфенил 2-(2-(2-хлор-6-фторфениламино)-5-метилфенил)ацетата (Соединение XVIII)

Схема 2
Синтез 4-карбамоилфенил 2-(2-(2-хлор-6-фторфениламино)-5-метилфенил)ацетата (5)
К раствору 1 (люмиракоксиб, 223 мг, 0,75 ммоль) в 15 мл диметилформамида добавляли при перемешивании при 0°С в течение 1 ч гидроксибензотриазол (111 мг, 0,825 ммоль) и DCC (170 мг, 0,825 ммоль). К реакционной смеси добавляли 4-гидроксибензамид (4, 154 мг, 1,125 ммоль) и перемешивали в течение 1 ч при 0°С и 3 ч при комнатной температуре. После фильтрования фильтрат выпаривали при пониженном давлении для удаления растворителя. Полученный таким образом маслянистый остаток растворяли в хлороформе; органический слой промывали рассолом, сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Неочищенный продукт 5 загружали на открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (9/1) с получением 4-карбамоилфенил-2-(2-(2-хлор-6-фторфениламино)-5-метилфенил)ацетата (5) (111 мг, 35% выход).
Синтез 4-тиокарбамоилфенил 2-(2-(2-хлор-6-фторфениламино)-5-метилфенил)ацетата (6)
Растворяли 4-карбамоилфенил 2-(2-(2-хлор-6-фторфениламино)-5-метилфенил)ацетат (5, 110 мг, 0,27 ммоль) и реагент Лавессона (109 мг, 0,27 ммоль) в 15 мл безводного бензола. Реакционную смесь нагревали до 60°С и перемешивали в течение 3 ч. Растворитель удаляли при пониженном давлении; неочищенный остаток очищали на колонке с силикагелем (дихлорметан/метиловый спирт 9,5/0,5) с получением чистого соединения 6 (59 мг, 51% выход).
1Н-ЯМР (CDCl3): δ 2,32 (с, 3Н), 4,01 (с, 2H), 6,46 (с, 1H), 6,70 (д, 1H), 6,92 (т, 1H), 7,01 (д, 2H), 7,11 (д, 2H), 7,19 (д, 1H), 7,62 (с, NH), 7,84 (д, 2H);
13С-ЯМР (ДМСО-d6): δ 20,8, 30,7, 115,1, 119,2, 122,0, 122,3, 124,1, 124,9, 126,1, 128,2, 129,2, 132,3, 134,8, 138,6, 140,9, 153,7, 154,6, 156,2, 170,4, 201,7.
MS (EI), м/e 429 (М+).
Т.пл.: 120-122°C.
ПРИМЕР 5
Синтез 4-(5-тиоксо-5Н-1,2-дитиол-3-ил)фенил 2-ацетоксибензоата (Соединение I)

Схема 1
Синтез 4-(5-тиоксо-5Н-1,2-дитиол-3-ил)фенил 2-ацетоксибензоата (3)
К раствору 1 (ацетилсалициловая кислота, 416 мг, 2,31 ммоль) в 40 мл диметилформамида добавляли при перемешивании при 0°С в течение 1 ч гидроксибензотриазол (343 мг, 2,54 ммоль) и DCC (523 мг, 2,54 ммоль). К реакционной смеси добавляли 5-п-гидроксифенил-1,2-дитиол-3-тион (2, 574 мг, 2,54 ммоль) и перемешивали в течение 1 ч при 0°С и 3 ч при комнатной температуре. После фильтрования фильтрат выпаривали при пониженном давлении для удаления растворителя. Полученный таким образом маслянистый остаток растворяли в этилацетате; органический слой промывали рассолом, сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Неочищенный продукт загружали на открытую колонку с силикагелем и элюировали смесью этиловый эфир/петролейный эфир (1/1) с получением 4-(5-тиоксо-5Н-1,2-дитиол-3-ил)фенил 2-ацетоксибензоата (3) (354 мг, 40% выход).
1Н-ЯМР (ДМСО-d6): δ 2,32 (с, 3Н), 7,20 (д, 1H), 7,33 (д, 2H), 7,40 (с, 1H), 7,41 (т, 1H), 7,67 (т, 1H), 7,73 (д, 2H), 8,21 (д, 1H);
13С-ЯМР (ДМСО-d6): δ 21,3, 122,1, 123,4, 124,4, 126,6, 128,6, 129,7, 132,4, 135,4, 136,4, 151,6, 153,7, 162,6, 169,8, 171,9, 215,7.
MS (EI), м/e 389 (М+).
Т.пл.: 120-122°C.
ПРИМЕР 6
Синтез 4-тиокарбамоилфенилового сложного эфира 2-ацетоксибензойной кислоты (Соединение XVI)

Схема 2
Синтез 4-карбамоилфенил 2-ацетоксибензоата (5)
К раствору 1 (ацетилсалициловая кислота, 500 мг, 2,77 ммоль) в 15 мл диметилформамида добавляли при перемешивании при 0°С в течение 1 ч гидроксибензотриазол (412 мг, 3,05 ммоль) и DCC (628 мг, 3,05 ммоль). К реакционной смеси добавляли 4-гидроксибензамид (4, 418 мг, 3,05 ммоль) и перемешивали в течение 1 ч при 0°С и 3 ч при комнатной температуре. После фильтрования фильтрат выпаривали при пониженном давлении для удаления растворителя. Полученный таким образом маслянистый остаток растворяли в хлороформе; органический слой промывали рассолом, сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Неочищенный продукт 5 загружали на открытую колонку с силикагелем и элюировали смесью CH2Cl2/MeOH (9/1), с получением 4-карбамоилфенил 2-ацетоксибензоата (5) (410 мг, 47% выход).
Синтез 4-тиокарбамоилфенил 2-(2-(2-хлор-6-фторфениламино)-5-метилфенил)ацетата (6)
Растворяли 4-карбамоилфенил 2-ацетоксибензоат, 5 (410 мг, 1,37 ммоль) и реагент Лавессона (554 мг, 1,37 ммоль) в 35 мл безводного бензола. Реакционную смесь нагревали до 60°С и перемешивали в течение 3 ч. Растворитель удаляли при пониженном давлении; неочищенный остаток очищали на колонке с силикагелем (дихлорметан/метиловый спирт 9,5/0,5) с получением 470 мг неочищенного соединения 6. Полученное соединение очищали обращенно-фазовой ВЭЖХ, проводимой при использовании двух систем растворителей: А: 100% ацетонитрил в 0,1% ТФУ (TFA), В: 100% H2O в 0,1% ТФУ (линейный градиент от 10% А до 60% А через 35 мин, УФ-детектирование при 254 нм, скорость потока 30 мл/мин) с получением чистого соединения 6 (324 мг, 71% выход).
1Н-ЯМР (CDCl3): δ 2,30 (с, 3Н), 7,17 (д, 1H), 7,21 (д, 2Н), 7,40 (т, 1H), 7,66 (т, 1Н), 7,94 (д,2H), 8,2 (д, 1H).
13С-ЯМР (ДМСО-d6): δ 21,2, 121,9, 122,4, 124,3, 126,4, 128,7, 132,4, 135,1, 137,3, 151,5, 153,7, 162,7, 169,8, 201,8.
MS (EI), м/e 316 (М+).
Т.пл.: 154-156°C.
ПРИМЕР 7
Синтез 4-(5-тиоксо-5H-[1,2]дитиол-3-ил)фенилового сложного эфира [1-(4-хлорбензоил)-5-метокси-2-метил-1-Н-индол-3-ил]уксусной кислоты (Соединение IV)

Схема 1
Синтез 4-[4-(5-тиоксо-5H-1,2-дитиол-3-ил)]фенил-2-[1-(4-хлорбензоил)-5-метокси-2-метилиндол-3-ил]ацетата (3)
К раствору 1 (индометацин, 720 мг, 2,01 ммоль) в 30 мл диметилформамида добавляли при перемешивании при 0°С в течение 1 ч гидроксибензотриазол (301 мг, 2,21 ммоль) и DCC (456 мг, 2,21 ммоль). К реакционной смеси добавляли 5-п-гидроксифенил-1,2-дитиол-3-тион (2, 500 мг, 2,21 ммоль) и перемешивали в течение 1 ч при 0°С и 2 ч при комнатной температуре. После фильтрования фильтрат выпаривали при пониженном давлении для удаления растворителя. Полученный таким образом маслянистый остаток растворяли в этилацетате; органический слой промывали рассолом, 5% NaHCO3, 10% лимонной кислотой и затем сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Неочищенный продукт загружали на открытую колонку с силикагелем и элюировали смесью дихлорметан/метиловый спирт (98/2) с получением 4-[4-(5-тиоксо-5H-1,2-дитиол-3-ил)]фенил-2-[1-(4-хлорбензоил)-5-метокси-2-метилиндол-3-ил]ацетата (3) (257 мг, 23% выход).
1Н-ЯМР (CDCl3): δ 2,47 (с, 3Н), 3,84 (с, 3Н, ОСН3), 3,93 (с, 2H), 6,70 (д, 1H), 6,88 (д, 1H), 7,04 (с, 1H), 7,21 (д, 2H), 7,37 (с, 1H) 7,48 (д, 2Н), 7,65 (д, 2H), 7,67 (д, 2H);
13С-ЯМР (ДМСО-d6): δ 13,6, 30,8, 56,0, 101,5, 111,6, 111,9, 115,3, 122,9, 128,4, 129,4, 129,6, 130,6, 131,1, 131,4, 133,9, 136,3, 136,6, 139,7, 153,8, 156,4, 167,5, 168,9, 170,4, 215,7.
MS (EI), м/e 567 (М+).
Т.пл.: 90-92°C.
ПРИМЕР 8
Синтез 4-тиокарбамоилфенилового сложного эфира [1-(4-хлорбензоил)-5-метокси-2-метил-1-Н-индол-3-ил]уксусной кислоты (Соединение XIX)

Схема 2
Синтез 4-карбамоилфенил-2-[1-(4-хлорбензоил)-5-метокси-2-метилиндол-3-ил]ацетата (5)
К раствору 1 (индометацин, 3 г, 8,38 ммоль) в 60 мл диметилформамида добавляли при перемешивании при 0°С в течение 1 ч гидроксибензотриазол (1,25 г, 9,22 ммоль) и DCC (1,9 г, 9,22 ммоль). К реакционной смеси добавляли 4-гидроксибензамид (4, 1,72 г, 12,6 ммоль) и перемешивали в течение 1 ч при 0°С и 2 ч при комнатной температуре. После фильтрования фильтрат выпаривали при пониженном давлении для удаления растворителя. Полученный таким образом маслянистый остаток растворяли в этилацетате; органический слой промывали рассолом, 5% NaHCO3, 10% лимонной кислотой и затем сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Неочищенный продукт 5 загружали на открытую колонку с силикагелем и элюировали смесью CH2Cl2/MeOH (9,5/0,5) с получением 4-карбамоилфенил-2-[1-(4-хлорбензоил)-5-метокси-2-метилиндол-3-ил]ацетата (5) (479 мг, 12% выход).
Синтез 4-тиокарбамоилфенил-2-[1-(4-хлорбензоил)-5-метокси-2-метилиндол-3-ил]ацетата (6)
Растворяли 4-карбамоилфенил-2-[1-(4-хлорбензоил)-5-метокси-2-метилиндол-3-ил]ацетат, 5 (340 мг, 0,71 ммоль) и реагент Лавессона (287 мг, 0,71 ммоль) в 15 мл безводного бензола. Реакционную смесь нагревали до 60°С и перемешивали в течение 4 ч. Растворитель удаляли при пониженном давлении; неочищенный остаток очищали на колонке с силикагелем (дихлорметан/метиловый спирт 9,5/0,5) с получением 178 мг неочищенного соединения 6. Полученное соединение очищали обращенно-фазовой ВЭЖХ, проводимой при использовании двух систем растворителей: А: 100% ацетонитрил в 0,1% ТФУ (TFA), В: 100% H2O в 0,1% ТФУ (линейный градиент от 10% А до 80% А через 30 мин, УФ-детектирование при 254 нм, скорость потока 30 мл/мин) с получением чистого соединения 6 (56 мг, 16% выход).
1Н-ЯМР (CDCl3): δ 2,45 (с, 3Н), 3,83 (с, 3Н, ОСН3), 3,91 (с, 2Н), 6,70 (д, 1H), 6,88 (д, 1H), 7,04 (с, 1H), 7,11 (д, 2H), 7,47 (д, 2H), 7,67 (д, 2H), 7,88 (д, 2H).
13С-ЯМР (ДМСО-d6): δ 13,6, 30,8, 56,0, 101,5, 111,9, 112,0, 115,3, 121,7, 128,6, 129,4, 130,8, 131,2, 131,4, 134,0, 136,8, 137,1, 139,7, 156,2, 157,9, 167,6, 169,8, 201,8.
MS (EI), м/e 493 (М+).
Т.пл.: 224-226°C.
ПРИМЕР 9
Синтез 4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилового сложного эфира 2-(6-метоксинафталин-2-ил)пропионовой кислоты (Соединение V)

Схема 1
Синтез 4-(5-тиоксо-5Н-1,2-дитиол-3-ил)фенил 2-(2-метоксинафталин-6-ил)пропионата (3)
К раствору 1 (напроксен, 595 мг, 2,58 ммоль) в 20 мл диметилформамида добавляли при перемешивании при 0°С в течение 1 ч гидроксибензотриазол (388 мг, 2,87 ммоль) и DCC (593 мг, 2,87 ммоль). К реакционной смеси добавляли 5-п-гидроксифенил-1,2-дитиол-3-тион (2, 650, мг, 2,87 ммоль) и перемешивали в течение 1 ч при 0°С и 2 ч при комнатной температуре. После фильтрования фильтрат выпаривали при пониженном давлении для удаления растворителя. Полученный таким образом маслянистый остаток растворяли в этилацетате; органический слой промывали рассолом, 5% NaHCO3, 10% лимонной кислотой и затем сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Неочищенный продукт загружали на открытую колонку с силикагелем и элюировали дихлорметаном с получением 4-(5-тиоксо-5Н-1,2-дитиол-3-ил)фенил 2-(2-метоксинафталин-6-ил)пропионата (3) (406 мг, 36% выход).
1Н-ЯМР (ДМСО-d6): δ 1,59 (д, 3Н), 3,86 (с, 3Н, ОСН3), 4,24 (дд, 1H), 7,18 (д, 1Н), 7,22 (д, 2Н), 7,31 (с, 1Н), 7,50 (д, 1Н), 7,77 (с, 1H) 7,85 (д, 1H), 7,86 (с, 1H), 7,87 (д,1H), 7,91 (д, 2H).
13С-ЯМР (ДМСО-d6): δ 19,1, 45,2, 55,9, 106,5, 119,6, 123,5, 126,6, 126,9, 128,0, 129,2, 129,4, 129,5, 129,6, 129,9, 134,2, 135,6, 136,5, 154,2, 158,1, 173,2, 216,2.
MS (EI), м/e 439 (М+).
Т.пл.: 111-113°C.
ПРИМЕР 10
Синтез 4-тиокарбамоилфенилового сложного эфира 2-(6-метоксинафталин-2-ил)пропионовой кислоты (Соединение XX)

Схема 2
Синтез 4-карбамоилфенил 2-(2-метоксинафталин-6-ил)пропионата (5)
К раствору 1 (напроксен, 4 г, 17,4 ммоль) в 80 мл диметилформамида добавляли при перемешивании при 0°С в течение 1 ч гидроксибензотриазол (2,59 г, 19,14 ммоль) и DCC (2,59 г, 19,14 ммоль). К реакционной смеси добавляли 4-гидроксибензамид (4, 3,58 г, 26,1 ммоль) и перемешивали в течение 1 ч при 0°С и 2 ч при комнатной температуре. После фильтрования фильтрат выпаривали при пониженном давлении для удаления растворителя. Полученный таким образом маслянистый остаток растворяли в этилацетате; органический слой промывали рассолом, 5% NaHCO3, 10% лимонной кислотой и затем сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Неочищенный продукт 5 загружали на открытую колонку с силикагелем и элюировали смесью CH2Cl2/MeOH (9,5/0,5), с получением 4-карбамоилфенил-2-(2-метоксинафталин-6-ил)пропионата (5) (1,91 г, 32% выход).
Синтез 4-тиокарбамоилфенил 2-(2-метоксинафталин-6-ил)пропионата (6)
Растворяли 4-карбамоилфенил-2-(2-метоксинафталин-6-ил)пропионат 5 (1,80 г, 4,34 ммоль) и реагент Лавессона (1,75 г, 4,34 ммоль) в 130 мл безводного бензола. Реакционную смесь нагревали до 60°С и перемешивали в течение 4 ч. Растворитель удаляли при пониженном давлении; неочищенный остаток очищали на колонке с силикагелем (дихлорметан/метиловый спирт 9,75/0,25) с получением 2,9 г неочищенного соединения 6. Полученное соединение очищали на открытой колонке с силикагелем и элюировали смесью CH2Cl2/MeOH (9,5/0,5), с получением чистого соединения 6 (970 мг, 61% выход).
1Н-ЯМР (ДМСО-d6): δ 1,59 (д, 3Н), 3,86 (с, 3Н, ОСН3), 4,24 (дд, 1H), 7,06 (д, 2Н), 7,18 (д, 1Н), 7,31 (с, 1Н), 7,50 (д, 1Н), 7,84 (с, 1H) 7,85 (д, 1H), 7,86 (с, 1H), 7,89 (д, 2H), 9,47 и 9,84 (с, 2H, NH2).
13С-ЯМР (ДМСО-d6): δ 19,1, 45,2, 55,9, 106,5, 119,6, 121,6, 126,6, 126,9, 128,0, 129,4, 129,9, 134,2, 135,6, 137,8, 153,4, 158,1, 173,3, 199,7.
MS (EI), м/e 366 (М+).
Т.пл.: 196-198°C.
ПРИМЕР 11
Синтез 4-тиокарбамоилфенил 2-(4-изобутилфенил)пропионата (Соединение XXIX)

К раствору 1 (ибупрофен, 3,87 г, 18,8 ммоль) в 80 мл диметилформамида добавляли при перемешивании при 0°С в течение 1 ч гидроксибензотриазол (2,8 г, 20,7 ммоль) и DCC (4,27 г, 20,7 ммоль). К реакционной смеси добавляли 4-гидроксибензамид (2, 3,9 г, 28 ммоль) и перемешивали в течение 1 ч при 0°С и 2 ч при комнатной температуре. После фильтрования фильтрат выпаривали при пониженном давлении для удаления растворителя. Полученный таким образом маслянистый остаток растворяли в этилацетате; органический слой промывали рассолом, 5% NaHCO3, 10% лимонной кислотой и затем сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Неочищенный продукт 3 загружали на открытую колонку с силикагелем и элюировали смесью CH2Cl2/MeOH (9,5/0,5) с получением 4-карбамоилфенил-2-(4-изобутилфенил)пропионата (3) (2,48 г, 40% выход).
Синтез 4-тиокарбамоилфенил 2-(4-изобутилфенил)пропионата (4)
Растворяли 4-карбамоилфенил 2-(4-изобутилфенил)пропионат 3 (2,48 г, 7,62 ммоль) и реагент Лавессона (3,1 г, 7,62 ммоль) в 130 мл безводного бензола. Реакционную смесь нагревали до 60°С и перемешивали в течение 4 ч. Растворитель удаляли при пониженном давлении. Неочищенное соединение очищали на открытой колонке с силикагелем смесью CH2Cl2/MeOH (9,5/0,5) с получением чистого соединения 4 (1,45 г, 55% выход).
1Н-ЯМР (ДМСО-d6): δ 0,84 (д, 6H), 1,48 (д, 3H), 1,79-1,82 (м, 1H), 2,42 (д, 2H), 4,05 (дд, 1H), 7,05 (д, 2H), 7,15 (д, 2H), 7,28 (д, 2H) 7,88 (д, 2H), 9,49 и 9,87 (с, 2H, NH2).
13С-ЯМР (ДМСО-d6): δ 19,2, 22,9, 30,3, 44,9, 121,6, 127,9, 129,5, 130,0, 137,8, 138,0, 140,8, 153,3, 173,3, 199,6.
MS (EI), м/e 341 (М+).
Т.пл.: 121-123°C.
ПРИМЕР 12
Синтез 4-тиокарбамоилфенил 2-(4-оксофенил)фенилпропионата (Соединение XXX)

Синтез 4-карбамоилфенил 2-(4-оксофенил)фенилпропионата (3)
К раствору 1 (кетопрофен, 3 г, 11,8 ммоль) в 80 мл диметилформамида добавляли при перемешивании при 0°С в течение 1 ч гидроксибензотриазол (1,76 г, 13 ммоль) и DCC (2,68 г, 13 ммоль). К реакционной смеси добавляли 4-гидроксибензамид (2, 2,43 г, 17,7 ммоль) и перемешивали в течение 1 ч при 0°С и 2 ч при комнатной температуре. После фильтрования фильтрат выпаривали при пониженном давлении для удаления растворителя. Полученный таким образом маслянистый остаток растворяли в этилацетате; органический слой промывали рассолом, 5% NaHCO3, 10% лимонной кислотой и затем сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Неочищенный продукт 3 загружали на открытую колонку с силикагелем и элюировали смесью CH2Cl2/MeOH (9,5/0,5) с получением 4-карбамоилфенил 2-(4-оксофенил)фенилпропионата (3) (1,84 г, 42% выход).
Синтез 4-тиокарбамоилфенил 2-(4-оксофенил)фенилпропионата (4)
Растворяли 4-карбамоилфенил 2-(4-оксофенил)фенилпропионат (3) (1,84 г, 4,93 ммоль) и реагент Лавессона (2 г, 4,93 ммоль) в 100 мл безводного бензола. Реакционную смесь нагревали до 60°С и перемешивали в течение 4 ч. Растворитель удаляли при пониженном давлении. Полученное соединение очищали на открытой колонке с силикагелем и элюировали смесью CH2Cl2/MeOH (9,5/0,5) с получением чистого соединения 4 (0,45 г, 23% выход).
1Н-ЯМР (ДМСО-d6): δ 1,53 (д, 3H), 4,25 (дд, 1H), 7,08 (д, 2H), 7,54-7,73 (м, 9H), 7,90 (д, 2H), 9,51 и 9,88 (с, 2H, NH2).
13С-ЯМР (ДМСО-d6): δ 19,2, 44,9, 121,6, 129,3, 129,5, 129,8, 130,3, 132,6, 133,5, 137,6, 137,9, 138,1, 141,2, 153,3, 154,5, 156,1, 163,8, 172,9, 199,6.
MS (EI), м/e 390 (М+).
Т.пл.: 114-116°C.
ПРИМЕР 13
Синтез 4-тиокарбамоилфенил 2-(3-фтор, 4-фенил)фенилпропионата (Соединение XXXI)

Синтез 4-карбамоилфенил 2-(3-фтор, 4-фенил)фенилпропионата (3)
К раствору 1 (флурбипрофен, 2 г, 8,2 ммоль) в 80 мл диметилформамида добавляли при перемешивании при 0°С в течение 1 ч гидроксибензотриазол (1,22 г, 9,02 ммоль) и DCC (1,86 г, 9,02 ммоль). К реакционной смеси добавляли 4-гидроксибензамид (2, 1,7 г, 12,2 ммоль) и перемешивали в течение 1 ч при 0°С и 2 ч при комнатной температуре. После фильтрования фильтрат выпаривали при пониженном давлении для удаления растворителя. Полученный таким образом маслянистый остаток растворяли в этилацетате; органический слой промывали рассолом, 5% NaHCO3, 10% лимонной кислотой и затем сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Неочищенный продукт 3 загружали на открытую колонку с силикагелем и элюировали смесью CH2Cl2/MeOH (9,5/0,5) с получением 4-карбамоилфенил 2-(3-фтор, 4-фенил)фенилпропионата (3) (1,09 г, 37% выход).
Синтез 4-тиокарбамоилфенил 2-(3-фтор, 4-фенил)фенилпропионата (4)
Растворяли 4-карбамоилфенил 2-(3-фтор,4-фенил)фенилпропионат 3 (1,09 г, 3 ммоль) и реагент Лавессона (1,21 г, 3 ммоль) в 70 мл безводного бензола. Реакционную смесь нагревали до 60°С и перемешивали в течение 4 ч. Растворитель удаляли при пониженном давлении. Полученное соединение очищали на открытой колонке с силикагелем и элюировали смесью CH2Cl2/MeOH (9,5/0,5) с получением чистого соединения 4 (0,35 г, 31% выход).
1Н-ЯМР (ДМСО-d6): δ 1,55 (д, 3Н), 4,21 (дд, 1H), 7,32-7,55 (м, 8H), 7,90 (д, 2H), 9,51 и 9,88 (с, 2H, NH2).
13С-ЯМР (ДМСО-d6): δ 19,1, 44,7, 115,9, 116,2, 121,7, 124,8, 128,6, 129,3, 129,4, 129,5, 131,7, 135,8, 137,7, 142,6, 153,7, 158,3, 163,5, 173,1, 199,6.
MS (EI), м/e 380 (М+).
Т.пл.: 142-144°C.
ПРИМЕР 14
Синтез 4-(изотиоциано)фенил 2-(2-метоксинафталин-6-ил)пропионата (Соединение XXV)

К раствору 1 (напроксен, 691 мг, 3 ммоль) в 20 мл диметилформамида добавляли при перемешивании при 0°С в течение 1 ч гидроксибензотриазол (446 мг, 3,3 ммоль) и DCC (619 мг, 3,3 ммоль). К реакционной смеси добавляли 4-гидроксифенилизотиоцианат (2, 500 мг, 3,3 ммоль) и перемешивали в течение 1 ч при 0°С и 2 ч при комнатной температуре. После фильтрования фильтрат выпаривали при пониженном давлении для удаления растворителя. Полученный таким образом маслянистый остаток растворяли в этилацетате и удаляли осадок. Выпаривали растворитель и неочищенный продукт загружали на открытую колонку с силикагелем и элюировали дихлорметаном с получением 4-(изотиоциано)фенил 2-(2-метоксинафталин-6-ил)пропионата (3) (230 мг, 21% выход).
1Н-ЯМР (ДМСО-d6): δ 1,57 (д, 3Н), 3,86 (с, 3Н, ОСН3), 4,20 (дд, 1H), 7,10 (д, 2Н), 7,15 (д, 1H), 7,29 (с, 1H), 7,43 (д, 2H), 7,48 (д, 1H), 7,78 (д, 1H), 7,80 (с, 1H), 7,83 (д, 1H).
13С-ЯМР (ДМСО-d6): δ 19,1, 45,2, 55,9, 106,5, 119,6, 123,8, 126,6, 126,9, 128,0, 128,3, 129,2, 129,9, 134,2, 134,6, 135,7, 150,2, 158,1, 173,2, 215,1.
Т.пл.: 66-68°C; MS (EI), м/e 364 (М+).
ПРИМЕР 15
Синтез 4-изотиоцианатофенил 2-(2-(2,6-дихлорфениламино)фенил)ацетата (Соединение XXII)

4-изотиоцианатофенил 2-(2-(2,6-дихлорфениламино)фенил)ацетат (3)
К раствору 1 (диклофенак, 1717 мг, 5,8 ммоль) в 60 мл диметилформамида добавляли при перемешивании при 0°С в течение 1 ч гидроксибензотриазол (862 мг, 6,38 ммоль) и DCC (1316 мг, 6,38 ммоль). К реакционной смеси добавляли 4-гидроксифенилизотиоцианат (2, 965 мг, 6,38 ммоль) и перемешивали в течение 1 ч при 0°С и 2 ч при комнатной температуре. После фильтрования фильтрат выпаривали при пониженном давлении для удаления растворителя. Полученный таким образом маслянистый остаток растворяли в этилацетате и удаляли осадок. Выпаривали растворитель и неочищенный продукт загружали на открытую колонку с силикагелем и элюировали смесью хлороформ/н-гексан в соотношении 9:1 с получением 4-изотиоцианатофенил 2-(2-(2,6-дихлорфениламино)фенил)ацетата (3) (580 мг, 23% выход).
1Н-ЯМР (ДМСО-d6): δ 4,09 (с, 2H), 6,19 (д, 1H), 6,83 (т, 1H), 7,05 (т, 1H), 7,14 (шир.с, 1H, NH), 7,21 (д, 2H), 7,25 (д, 2H), 7,47-7,54 (м, 3H).
13С-ЯМР (ДМСО-d6): δ 37,4, 116,1, 121,0, 122,7, 124,0, 127,1, 127,8, 128,3, 128,7, 129,8, 132,0, 132,2, 137,7, 144,0, 150,3, 170,5, 215,1.
Т.пл.: 132-134°C; MS (EI), м/e 364 (М+).
ПРИМЕР 16
Синтез 4-изотиоцианатофенил 2-ацетоксибензоата (Соединение XXI)

4-изотиоцианатофенил 2-ацетоксибензоат (3)
К раствору 1 (аспирин, 1200 мг, 6,67 ммоль) в 60 мл диметилформамида добавляли при перемешивании при 0°С в течение 1 ч гидроксибензотриазол 992 мг, 7,34 ммоль) и DCC (1520 мг, 7,34 ммоль). К реакционной смеси добавляли 4-гидроксифенилизотиоцианат (2, 1109 мг, 7,34 ммоль) и перемешивали в течение 1 ч при 0°С и 2 ч при комнатной температуре. После фильтрования фильтрат выпаривали при пониженном давлении для удаления растворителя. Полученный таким образом маслянистый остаток растворяли в этилацетате и удаляли осадок. Выпаривали растворитель и неочищенный продукт загружали на открытую колонку с силикагелем и элюировали смесью хлороформ/н-гексан в соотношении 6:4, с получением 4-изотиоцианатофенил 2-ацетоксибензоата (3) (150 мг, 7% выход).
1Н-ЯМР (CDCl3): δ 2,31 (с, 3Н), 7,17 (д, 1H), 7,19 (д, 2H), 7,29 (д, 2H), 7,38 (т, 1H), 7,66 (т, 1H), 8,20 (д, 1H).
13С-ЯМР (CDCl3): δ 21,3, 122,2, 123,3, 124,4, 126,6, 127,2, 129,4, 132,4, 135,2, 149,3, 151,5, 163,0, 170,0, 215,1.
Т.пл.: 84-86°C; MS (EI), м/e 272 (М+).
ПРИМЕР 17
Безопасность соединений настоящего изобретения в отношении желудочно-кишечного тракта
Два производных диклофенака по настоящему изобретению, соединение II и соединение XVII оценивали в отношении их безопасности для желудочно-кишечного тракта у крыс. В частности, измеряли повреждение желудка, синтез желудочного PGE2, изъязвление тонкого кишечника и гематокрит.
Самцы крыс Wistar весом 175-200 г голодали в течение 18 часов до перорального введения 1% карбоксиметилцеллюлозы (носитель; 0,2 мл) самой по себе или с одним из следующих веществ, растворенных в указанном носителе: диклофенак (20 мг/кг), соединение II (32 мг/кг), ADT-OH (12 мг/кг), диклофенак и ADT-OH, соединение XVII (27,3 мг/кг), 4-гидрокситиобензамид (TBZ) (7,3 мг/кг), фрагмент, высвобождающий сероводород на соединении XVII, или диклофенак и TBZ. Дозы соединения II и соединения XVII представляют собой дозы, эквимолярные дозе 20 мг/кг диклофенака. Сходным образом, дозы ADT-OH и TBZ представляют собой дозы, эквимолярные дозам соединения II и соединения XVII, соответственно.
В каждой группе было по 5 крыс. Через три часа после введения исследуемых соединений крыс умерщвляли и слепым способом измеряли геморрагическое повреждение желудка (в мм). «Результат повреждения желудка» получали, суммируя длину всех повреждений в желудке. Со ссылкой на фигуру 1, повреждение желудка не наблюдалось в группах «носитель», «соединение II» или «соединение XVII». Соединение II и соединение XVII приводили к в значительной степени меньшему повреждению желудка по сравнению с диклофенаком. Более того, щадящее в отношении желудка действие не наблюдалось при раздельном введении NSAID фрагмента (диклофенак) и H2S-высвобождающего фрагмента соединения II и соединения XVII (ADT-OH и TBZ, соответственно), но только в одно и то же время.
Указанные наблюдения были подтверждены при последующем слепом гистологическом анализе. Образцы (100-200) ткани желудка исследовали для измерения синтеза простагландина E2 (PGE2), как подробно описано ранее (Wallace et al., Cyclooxygenase 1 contribute to inflammatory responses in rats and mice: implications for gastrointestinal toxicity. Gastroenterology 1998; 115: 101-109, включено здесь в качестве ссылки). Кратко, образцы ткани измельчали ножницами в течение 30 минут, затем помещали в 1 мл буфера натрия фосфата (рН 7,4) и помещали на встряхиваемую водяную баню (37°С) на 20 мин. Непосредственно после этого образцы центрифугировали в течение 1 мин при 9000g и надосадочную жидкость немедленно замораживали при -80°С для последующего измерения концентрации PGE2 при использовании специфического ELISA (Wallace et al., 1998).
Со ссылкой на фигуру 2, можно увидеть, что диклофенак (с или без сопутствующего введения ADT-OH или TBZ), соединение II и соединение XVII, все, значительно снижают уровень синтеза желудочного PGE2, что указывает на ингибирование COX-1 и/или COX-2. ADT-OH и TBZ сами по себе не снижали синтез желудочного PGE2 по сравнению с носителем. Таким образом, отсутствие повреждения желудка у крыс, которым вводили соединение II и соединение XVII, как показано на фигуре 1, не объяснялось изменениями в способности указанных лекарственных средств подавлять синтез желудочного простагландина. Подавление синтеза желудочного PGE2 было практически полным при использовании указанных лекарственных препаратов в дозе, эквимолярной дозе использованного диклофенака.
На фигуре 3 видно, что два производных напроксена настоящего изобретения (соединения V и XX) вызывали в значительной степени меньше повреждений по сравнению с самим напроксеном. Указанное исследование проводили абсолютно таким же образом, как показано на фигуре 1. Напроксен, соединение V и соединение XX, каждое, вводили перорально в дозе 60 мкмоль/кг и через 3 часа слепым методом оценивали повреждение желудка. Повреждение желудка не поддавалось выявлению ни у одной из крыс, которым вводили соединение V или соединение XX. Каждая группа состояла из 5 крыс. Указанные наблюдения подтверждали при последующем слепом гистологическом анализе.
Ингибирование COX-1 также измеряли при использовании тех же крыс. Непосредственно после сбора экссудата из кармана у каждой крысы из нижней полой вены отбирали 1 мл крови, помещали в стеклянную пробирку и оставляли свертываться в течение 45 минут, как описано ранее (Wallace et al. Gastroenterology 1998). Образцы затем центрифугировали в течение 3 мин при 9000g, надосадочную жидкость замораживали при -80°С для последующего измерения концентраций тромбоксана B2 при использовании специфического ELISA. Как показано на фигуре 4, напроксен, соединение V и соединение XX, все, значительно (*p<0,05) ингибировали активность COX-1 по сравнению с группой, которой вводили носитель. Степень ингибирования COX-1 соединением V была несколько ниже, чем напроксеном или соединением XX.
NSAID могут также вызывать значительное повреждение тонкого кишечника, и действие диклофенака, вызывающее повреждение тонкого кишечника после повторного введения, сравнивали с действием соединения II. Группам по 5 самцов крыс Wistar вводили диклофенак или соединение II в дозе 50 мкмоль/кг в момент времени 0 и опять через 12 и 24 часа. Другая группа крыс получала носитель (1% карбоксиметилцеллюлоза).
Гематокрит, часть крови, состоящая из упакованных красных кровяных телец, выраженных в процентах по объему, измеряли в образце крови, отобранной из хвостовой вены в начале эксперимента и через 24 ч после конечной дозы лекарственного препарата. Крыс умерщвляли через 24 ч после конечной дозы лекарственного препарата и вскрывали брюшную полость. Исследователь, которому не были известны подробности терапии крыс, получал измерения длины всех геморрагических эрозий/язв тонкого кишечника. Результат повреждения тонкого кишечника рассчитывали, суммируя длины всех повреждений у каждой крысы.
Как показано на фиг. 5, трехкратное введение диклофенака в течение периода 24 ч приводило к развитию обширных эрозий и язв тонкого кишечника. С другой стороны, степень повреждения, наблюдаемая у крыс, обработанных соединением II, была более чем на 90% меньше, чем у крыс, обработанных диклофенаком. Более того, как показано на фигуре 6, обработка диклофенаком приводила к сильному понижению гематокрита (*p<0,05), по-видимому, в результате кровотечения в тонком кишечнике, тогда как обработка соединением II не оказывала значительного эффекта на гематокрит.
ПРИМЕР 18
Ингибирование циклооксигеназы-2 (COX-2) и циклооксигеназы-1 (COX-1)
Ингибирование COX-2 in vivo определяли при использовании модифицированной версии ранее описанной модели (Wallace et al. Limited anti-inflammatory efficacy of cyclo-oxygenase-2 inhibition in carageenan-airpouch inflammation. Br. J. Pharmacol; 126: 1200-1204, включенная здесь в качестве ссылки). Кратко, создавали подкожный «карман» повторяющимися инъекциями воздуха в течение нескольких дней. После образования воспаление в кармане может быть индуцировано инъекцией 1 мл 1% зимозана. Это вызывало значительное увеличение простагландина Е2(PGE2) в кармане, что было вызвано, как показано, практически исключительно COX-2. Группам по 5 крыс вводили перорально за 30 мин до инъекции каррагинана носитель (1% карбоксиметилцеллюлоза), диклофенак (3 мг/кг), соединение II (4,8 мг/кг) или соединение XVII (4,1 мг/кг). Другая группа из 5 крыс получала носитель, но им вводили в карман вместо зимозана 0,9% стерильный физиологический раствор.
Как можно видеть на фигуре 7, предварительное введение диклофенака, соединения II или соединения XVII заметно понижало концентрацию PGE2 в кармане, который вырабатывался в ответ на инъекцию зимозана, *p<0,05 по сравнению с группами, обработанными носителем + зимозан. Указанные результаты показывают, что все три соединения в значительной степени ингибируют COX-2. Напротив, ни один из высвобождающих сероводород фрагментов (ADT-OH и TBZ) не оказывал значительного воздействия на активность COX-2.
Ингибирование COX-1 также измеряли при использовании тех же крыс, при использовании того же способа, который описан для фигуры 4. Как показано на фигуре 8, диклофенак, соединение II и соединение XVII, каждое, ингибировало синтез тромбоксана в цельной крови, который происходит через COX-1, более чем на 80%. Напротив, ни один из высвобождающих сероводород фрагментов (ADT-OH и TBZ) не оказывал значительного воздействия на активность COX-1.
ПРИМЕР 19
Действие NSAID производных на повреждение желудка, COX-1 и COX-2 активность in vivo.
Противовоспалительное действие (ингибирование COX-1 и COX-2) и безопасность в отношении желудка для большого числа соединений сравнивали при использовании анализов, описанных выше. Результаты представлены в таблице 1. Все исходные NSAID вызывали значительное повреждение желудка. Однако производные настоящего изобретения, высвобождающие H2S, демонстрировали улучшенную безопасность в отношении желудка по сравнению с исходными лекарственными препаратами. Также из таблицы 1 можно видеть, что TBZ производные или сохраняют, или действительно увеличивают свою способность ингибировать COX-1 и/или COX-2, по сравнению с исходными лекарственными препаратами.
ПРИМЕР 20
Действие соединений настоящего изобретения на воспаление
Противовоспалительное действие соединения II и соединения XVII и действие диклофенака оценивали при использовании модели индуцированного каррагинаном отека лапы крысы, как ранее описано в Wallace et al., Gasteroenterology 1998. Самцам крыс Wistar весом 175-200 г вводили перорально исследуемые соединения за 30 мин до введения в подошву 100 мкл 1% лямбда-каррагинана. Объем лапы измеряли при использовании Ugo Basile гидроплетизмометра до инъекции каррагинана и через 1-ч интервалы после в течение 5 ч. Каждой группе, которая состояла из 5 крыс, вводили диклофенак в дозах 1, 3 или 10 мг/кг, или соединение II, или соединение XVII в дозах, эквимолярных дозе 3 мг/кг диклофенака.
Как показано на фигуре 9, диклофенак, зависимым от дозы образом, уменьшал отек лапы, вызванный введением в подошву каррагинана. Соединение II, вводимое в дозе, эквимолярной дозе 3 мг/кг диклофенака, в большей степени уменьшало отек лапы. Действительно, действие соединения II на отек лапы было сравнимым с действием диклофенака в дозе 10 мг/кг. Сходным образом, как показано на фигуре 10, соединение XVII, которое также вводили в дозе, эквимолярной дозе 3 мг/кг диклофенака, в большей степени уменьшало отек лапы по сравнению с диклофенаком в дозе 10 мг/кг.
Поскольку оба, соединение II и соединение XVII, угнетают синтез простагландина в той же степени, что и диклофенак, лучшая активность новых соединений по изобретению в модели отека лапы, по-видимому, связана с другим свойством указанных соединений. Ранее было показано, что доноры сероводорода могут в значительной степени уменьшать индуцированный каррагинаном отек лапы крысы (Zanardo et al., Hydrogen sulphide is an endogenous modulator of leykocyte-mediated inflammation. FASEB J 2006; 20: 2118-2120, включенная здесь в качестве ссылки), так что, не связывая себя определенной теорией, можно предположить, что высвобождение H2S из соединения II и соединения XVII отвечает за улучшенное противовоспалительное действие по сравнению с диклофенаком.
Не связывая себя определенной теорией, можно также предположить, что дополнительная активность соединений настоящего изобретения в воспалительных моделях может относиться на счет улучшенного ингибирования COX-2 активности. Действие носителя, напроксена, соединения V и соединения XX сравнивали на модели воздушного кармана у крыс (как описано на фигуре 7). Каждая группа состояла из 5 крыс. Напроксен, соединение V и соединение XX, каждое, вводили в дозе 60 мкмоль/кг. Как показано на фигуре 11, все три лекарственных препарата значительно подавляли COX-2 активность по сравнению с группой, которой вводили носитель (*p<0,05, **p<0,01). Однако соединение XX вызывало в значительной степени большее понижение COX-2 активности по сравнению с наблюдаемой при введении напроксена или соединения V (ψp<0,05).
Не связывая себя теорией, также можно предположить, что дополнительную активность соединений настоящего изобретения в воспалительных моделях можно отнести на счет повышенного ингибирования активности COX-1. Действие носителя, индометацина и двух соединений настоящего изобретения, соединения IV и соединения XIX, сравнивали в отношении их действия на синтез тромбоксана B2 в цельной крови человека in vitro. Аликвоты (0,5 мл) человеческой крови здоровых волонтеров добавляли в стеклянную пробирку, содержащую 10 мкл только метанола или одного из исследуемых полученных лекарственных препаратов, так что конечная концентрация составляла 0,1, 0,3, 1 или 3 мкМ. Пробирки помещали на водяную баню (37°С) с легким встряхиванием на 45 мин, после чего их центрифугировали (1000 × g) в течение 10 минут. Затем определяли концентрацию тромбоксана B2 в каждом образце при использовании специфического ELISA, как в исследованиях, показанных на фигуре 4. Как показано на фигуре 12, все три лекарственных препарата приводили к зависящему от концентрации ингибированию активности COX-1 по сравнению с группой, которой вводили носитель. Однако при концентрации 1 и 3 мкМ соединение XIX приводило к значительно большему (*p<0,05) ингибированию активности COX-1 по сравнению с ингибированием индометацином.
ПРИМЕР 21
Адгезия лейкоцитов к эндотелию сосудов под действием соединений настоящего изобретения
Адгезия лейкоцитов к эндотелию сосудов представляет собой раннее событие при воспалительных реакциях и участвует в образовании тромба. Было показано, что доноры сероводорода ослабляют адгезию лейкоцитов, вызываемую аспирином или противовоспалительным трипептидом, fMLP (Zanardo et al., FASEB J 2006; 20: 2118-2120). Действие нескольких производных NSAID настоящего изобретения на адгезию лейкоцитов оценивали при использовании прижизненной микроскопии у крыс, как подробно описано Zanardo et al., FASEB J 2006; 20: 2118-2120.
Кратко, исследовали посткапиллярные венулы брыжейки у анестезированных крыс при использовании светового микроскопа. После основного периода записи, продолжавшегося 5 мин, внутрижелудочно вводили одно из исследуемых соединений, перечисленных в таблице 2, ниже, в дозе 30 мкмоль/кг, за исключением напроксена и производных напроксена (соединения V и XX), которые вводили в дозе 60 мкмоль/кг. Все исследуемые соединения получали в носителе 1% карбоксиметилцеллюлозы. Изменения в адгезии лейкоцитов в венуле регистрировали при помощи видеокамеры, которой был оснащен микроскоп, и количественную оценку числа осевших лейкоцитов проводили слепым способом при помощи оценки изображений с видеозаписи. Каждая группа состояла из 5 самцов крыс Wistar весом 150-175 г. Лейкоцит считали «прилипшим», если он оставался неподвижным в течение 30 секунд или более (результаты ниже представлены как среднее ±SEM). В конце эксперимента вскрывали желудок и проверяли под бинокуляром на предмет наличия повреждений желудка.
Из таблицы 2 можно видеть, что производные аспирина по настоящему изобретению, в частности соединение XVI и соединение I, оба, в значительной степени снижают число прилипших лейкоцитов на 100 мкм длины сосуда, по сравнению с одним аспирином. Дополнительно, соединение XVI и соединение I значительно снижают процент случаев повреждения желудка по сравнению с одним аспирином. Сходным образом, из таблицы 2 дополнительно видно, что производные диклофенака по настоящему изобретению, в частности соединение II и соединение XVII, в значительной степени снижают число прилипших лейкоцитов на 100 мкм длины сосуда и значительно снижают процент случаев повреждения желудка по сравнению с одним диклофенаком. Таким же образом, из таблицы 2 дополнительно видно, что производные напроксена по настоящему изобретению, в частности соединение V и соединение XX, в значительной степени снижают число прилипших лейкоцитов на 100 мкм длины сосуда и значительно снижают процент случаев повреждения желудка по сравнению с одним напроксеном.
Примечательно, что производные люмиракоксиба, селективного ингибитора COX-2, имеющего ослабленное побочное действие на желудок, соединение III и соединение XVIII, также не показывали случаев повреждения желудка, но оба производных значительно снижали число прилипших лейкоцитов на 100 мкм длины сосуда по сравнению с одним люмиракоксибом. Таким образом, ковалентное связывание фрагмента, высвобождающего сероводород, с COX-2 селективным NSAID может также снижать побочное действие указанных ингибиторов COX-2 на сердечно-сосудистую систему.
Таким образом, производные NSAID настоящего изобретения могут приводить к снижению побочного действия NSAID на сердечно-сосудистую систему за счет снижения адгезии лейкоцитов.
ПРИМЕР 22
Действие соединений по изобретению на заживление язвы желудка
NSAID, включая COX-2 селективные NSAID, часто ингибируют заживление существующих язв желудка (Stadler et al., Diclofenac delays healing of gastroduodenal mucosal lesions. Double blind, placebo-controlled endoscopic study in healthy volunteers. Digestive Diseases and Sciences 1991; 36:594-600). Для определения действия двух соединений настоящего изобретения (соединение XVII и соединение XX) по сравнению с диклофенаком и напроксеном, соответственно, на заживление язвы, крысам вводили указанные лекарственные препараты после индукции язвы в их желудках. Язвы желудка индуцировали серозным нанесением уксусной кислоты, как описано Elliott et al., A nitric oxide-releasing nonsteroidal anti-flammatory drug accelerates gastric ulcer healing in rats. Gastroenterology 1995; 109: 524-530. Начиная с третьего дня, группам, по 5 крыс каждая, вводили дважды в день перорально носитель, диклофенак (30 мкмоль/кг), соединение XVII (30 мкмоль/кг), напроксен (60 мкмоль/кг) или соединение XX (60 мкмоль/кг). Через 4 дня после такого введения крыс умерщвляли и желудок вырезали и фотографировали. Площадь язвы (в мм2) определял планиметрически исследователь, которому не были известны подробности терапии крыс. В подгруппе из 5 крыс, умерщвленных через 3 дня после индукции язвы желудка (т.е. перед началом введения лекарственного препарата), средняя площадь поверхности язвы составила 24±2 мм2. Как показано на фигуре 13, крысы, которым вводили носитель, диклофенак или напроксен, демонстрировали ту же степень заживления. Однако крысы, которым вводили соединение XVII или соединение XX, демонстрировали в значительной степени большее заживление (*p<0,05 по сравнению с диклофенаком и напроксеном, соответственно). Обработка высвобождающими сероводород фрагментами этих двух соединений (TBZ) не оказала значительного влияния на заживление язвы желудка по сравнению с группой, которой вводили носитель.
ПРИМЕР 23
Действие соединений по изобретению на кровяное давление
NSAID, включая NSAID, демонстрирующие селективность по отношению к COX-2, могут усиливать уже существующую гипертензию и влиять на эффективность некоторых типов противогипертензивного лечения (Whelton, A. Nephrotoxicity of nonsteroidal anti-inflammatory drugs: physiologic foundations and clinical implications. Am. J. Med. 1999; 106 (5B): 13S-24S). Для определения действия двух соединений настоящего изобретения (соединение II и соединение XX) по сравнению с диклофенаком и напроксеном, соответственно, на кровяное давление, крысам вводили указанные лекарственные препараты внутрибрюшинно, после того как вызывали гипертензию. Крысам предоставляли питьевую воду с добавкой метилового эфира Nω-нитро-L-аргинина (400 мг/л) в течение 7 дней до эксперимента, как описано ранее Ribeiro et al. (Chronic inhibition of nitric oxide synthesis: A new model of arterial hypertension. Hypertension 1992; 20: 298-303). Крыс (от 5 до 8 в группе) анестезировали галотаном и вводили катетер в сонную артерию для измерения кровяного давления, которое регистрировали непрерывно при помощи самописца. После измерения стабильного кровяного давления в течение по меньшей мере 15 минут один из лекарственных препаратов (напроксен, диклофенак, соединение II или соединение XX) вводили внутрибрюшинно в виде болюса (диклофенак и соединение II вводили в концентрации 30 мкмоль/кг, тогда как напроксен и соединение XX вводили в концентрации 60 мкмоль/кг). Изменения кровяного давления регистрировали в течение 60 минут после введения. Среднее базальное кровяное давление составляло 150±6 мм Hg. На фигуре 14 показано, что диклофенак и напроксен вызывали значительное повышение систолического кровяного давления. Напротив, соединение II и соединение XX не увеличивали систолическое кровяное давление по сравнению с группой, которой вводили только носитель, и изменение кровяного давления было в значительной степени более низким по сравнению с индуцированным диклофенаком и напроксеном, соответственно.
ПРИМЕР 24
Измерения концентрации H2S в плазме
Для определения кинетики высвобождения H2S из соединения II, группам по 5 крыс вводили соединение II в дозах 50 мкмоль/кг перорально и умерщвляли через 10, 30, 60 и 180 минут. Затем строили зависимость концентрации H2S в плазме от времени. Концентрацию H2S в плазме измеряли, как описано ранее (Ubuka, T. Assay methods and biological role of labile sulfur in animal tissues J. Chromatogr. B Analyt Technol Biomed Life Sci 2002; 781: 227-249, и Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H2S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 2001; 20: 60008-6016, которые обе включены здесь в качестве ссылки), с модификациями. Кратко, 250 мкл плазмы добавляли к 250 мкл охлажденного льдом 0,1 н. NaOH в запаянном 3-горлом реакторе. Через смесь пропускали постоянный поток азота при помощи капиллярного входа для газа. Реактор поддерживали при 37°С и начинали экстракцию H2S введением 1 мл 10% раствора трихлоруксусной кислоты. Поток азота переносил сероводород в другой реактор по охлаждаемому соединению и проходил через в 2 мл раствора антиоксидантного буфера для сульфида (SAOB), состоящего из 2 М KOH, 1 М салициловой кислоты и 0,22 М аскорбиновой кислоты при рН 12,8. Через 30 минут раствор SAOB удаляли и измеряли концентрацию сульфида при использовании чувствительного к сульфиду электрода (модель 9616 S2-/Ag+ электрод, Orion Research, Beverly, MA, USA) и выражали через H2S (Ubuka, 2002; Khan et al., 1980).
Для сравнения высвобождения H2S in vitro, индуцированного соединением XVII и соединением II, и TBZ и ADT-OH, высвобождающими H2S фрагментами соединения XVII и соединения II, соответственно, 100-150 мг изолированной печени гомогенизировали в 1 мл охлажденного льдом экстрактора белка T-PER. Высвобождение H2S проводили в том же реакторе, что и анализ плазмы. Два мл исследуемой реакционной смеси вводили в реактор. Смесь содержала 1 мМ соединения II, 1 мМ соединения XVII, 1 мМ TBZ или 1 мМ ADT-OH, растворенных в PEG и 100 мМ фосфатного буфера (рН 7,4). Инкубирование проводили в присутствии или в отсутствие 10% (мас./об.) гомогената печени и 2 мМ пиридоксаль 5'-фосфата. Постоянный поток азота пропускали через смесь при помощи капиллярного входа для газа. Реакции инициировали при переносе пробирки из ледяной бани в водяную баню 37°С. Поток азота переносил сероводород во второй реактор, содержащий 2 мл SAOB, как описано выше. После инкубации при 37°C в течение 90 минут к реакционной смеси для остановки реакции добавляли 1 мл 50% трихлоруксусной кислоты. Оставшийся H2S в смеси переносился потоком азота в течение последующих 30 минут инкубации при 37°С. Концентрацию сульфида в растворе SAOB измеряли при использовании чувствительного к сульфиду электрода, как описано выше (Ubuka, 2002; Khan et al., 1980).
Как показано на фигуре 15, пероральное введение соединения II приводило к значительному (p<0,05) увеличению концентрации H2S в плазме. Небольшое, но постоянное увеличение H2S в плазме наблюдали в течение 180 минут после однократного введения соединения II. На фигуре 16 показано, что инкубация соединения II или соединения XVII в буфере, приводила к в значительной степени большему высвобождению H2S по сравнению с эквивалентным количеством ADT-OH или TBZ, соответственно. Сходным образом, наблюдалось большее высвобождение H2S из соединения II или соединения XVII по сравнению с ADT-OH или TBZ при инкубации с гомогенатом печени.
Реферат
Изобретение относится к производным нестероидных противовоспалительных лекарственных препаратов (NSAID) общей формулы:где А представляет собой NSAID радикал, выбранный из группы, состоящей из ацетилсалициловой кислоты (ASA), диклофенака, напроксена, индометацина, флурбипрофена, ибупрофена, кетопрофена и люмиракоксиба, Y представляет собой -С(O)O-, и Х выбирают из группы, состоящей из:,иимеющих улучшенные противовоспалительные свойства, пригодные для лечения воспаления, боли и лихорадочного состояния. Получены NSAID производные с фрагментом, высвобождающим сероводород (НS), для получения новых противовоспалительных соединений, обладающих ослабленными побочными действиями. 9 н. и 14 з.п. ф-лы, 16 ил., 24 пр.
Формула
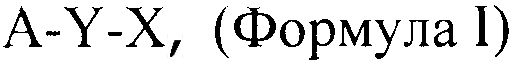
где А представляет собой NSAID-радикал, выбранный из группы, состоящей из ацетилсалициловой кислоты (ASA), диклофенака, напроксена, индометацина, флурбипрофена, ибупрофена, кетопрофена и люмиракоксиба,
Y представляет собой -С(O)O- и
Х выбирают из группы, состоящей из:


при условии, что когда Х представляет собой

то ВС(O)O не является ацетилсалициловой кислотой (ASA) или диклофенаком.

где В-С(O)O- представляет собой производное NSAID, имеющее свободную карбоксильную группу, выбранное из группы, состоящей из ацетилсалициловой кислоты (ASA), диклофенака, напроксена, индометацина, флурбипрофена, ибупрофена, кетопрофена и люмиракоксиба, и
Х выбирают из группы, состоящей из


и при условии, что когда Х представляет собой,

то В-С(O)O- не является ацетилсалициловой кислотой (ASA) или диклофенаком.


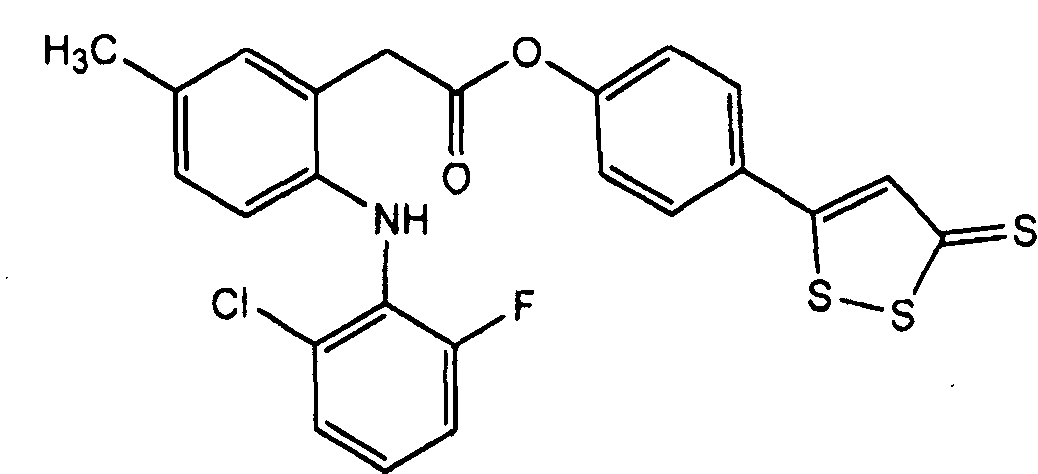
4-(5-тиоксо-5Н-1,2-дитиол-3-ил)фенил 2-(2-(2-хлор-6-фторфениламино)-5-метилфенил)ацетат (III)
или его фармацевтически приемлемую соль.

4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фениловый сложный эфир [1-(4-хлорбензоил)-5-метокси-2-метил-1Н-индол-3-ил]уксусной кислоты (IV)
или его фармацевтически приемлемую соль.

4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фениловый сложный эфир 2-(6-метоксинафталин-2-ил)пропионовой кислоты (V) или его фармацевтически приемлемую соль.

4-тиокарбамоилфениловый сложный эфир 2-ацетоксибензойной кислоты (XVI)
или его фармацевтически приемлемую соль.

4-тиокарбамоилфениловый сложный эфир [2-(2,6-дихлорфениламино)фенил]уксусной кислоты (XVII)
или его фармацевтически приемлемую соль.

4-тиокарбамоилфениловый сложный эфир [2-(2-хлор-6-фторфениламино)-5-метилфенил]уксусной кислоты (XVIII)
или его фармацевтически приемлемую соль.

4-тиокарбамоилфениловый сложный эфир [1-(4-хлорбензоил)-5-метокси-2-метил-1Н-индол-3-ил]уксусной кислоты (XIX)
или его фармацевтически приемлемую соль.

4-тиокарбамоилфениловый сложный эфир 2-(6-метоксинафталин-2-ил)пропионовой кислоты (XX)
или его фармацевтически приемлемую соль.

4-тиокарбамоилфенил 2-(4-изобутилфенил)пропионат (XXIX)
или его фармацевтически приемлемую соль.

4-тиокарбамоилфенил 2-(4-оксофенил)фенилпропионат (XXX)
или его фармацевтически приемлемую соль.

4-тиокарбамоилфенил 2-(2-фтор-4-бифенилил)пропионат (XXXI) или его фармацевтически приемлемую соль.
Комментарии