Высокоспецифическая противоопухолевая фармакологическая лекарственная система, синтез лекарственного средства и способ моделирования лекарственного средства - RU2352335C2
Код документа: RU2352335C2
Описание
Предпосылки изобретения
Несмотря на успехи, достигнутые в области создания противоопухолевых средств, рак остается заболеванием с очень неблагоприятным прогнозом. В литературе постоянно появляются сообщения о веществах, проявляющих in vitro и in vivo противоопухолевую активность, но лишь ничтожное количество таких веществ проходит фазу II клинического испытания. Кроме того, показатели эффективности лекарственных средств, используемых в лечении опухолевых заболеваний, неприемлемо низки, и побочные эффекты таких лекарственных средств весьма серьезны. Токсическое действие лекарственных средств на опухолевые ткани только в малой степени превышает их токсическое действие на нормальные, здоровые клетки организма, которые должны быть защищены от воздействия лекарственных средств. Кроме того, недостаточную эффективность противоопухолевых средств можно отнести за счет отсутствия способов, позволяющих заранее, на этапе разработки лекарственных средств, осуществлять контроль побочных эффектов, специфичности и высокой активности указанных средств в отношении ткани-мишени человека. Необходим способ, позволяющий создавать потенциальные соединения, удовлетворяющие всем требованиям, предъявляемым к системе доставки лекарственных средств и противоопухолевой эффективности. Достижение этой цели осуществляется посредством настоящего изобретения.
Исследования в области химиотерапии злокачественных опухолей в значительной степени были направлены на поиск лекарственных средств, проявляющих способность токсина разрушать неопластическую ткань в организме, не превышая степени токсического воздействия, причиняющей вред здоровым тканям, то есть направлены на поиск цитотоксических лекарственных препаратов, которые концентрируются в неопластических тканях, или средств, которые в процессе метаболизма превращаются в такие токсины. Хотя в этом направлении были предприняты серьезные попытки, результаты были достигнуты только для некоторых типов злокачественных опухолей. Для большинства типов злокачественных опухолей успех был незначительным.
Развитие современных противоопухолевых лекарственных средств началось с обнаружения того факта, что отравляющий газ, применяемый в Первую мировую войну, действительно оказывал побочное противоопухолевое действие. Процесс развития противоопухолевых лекарственных средств начался на этом этапе в основном со случайного поиска среди известных токсинов, так и токсинов, у которых путем проверки их воздействия удается выяснить, обладает ли такой токсин дифференциальной токсичностью, которая могла бы способствовать осуществлению контроля или лечению злокачественных заболеваний. При таком подходе внимание направлено на поиск «однофрагментных» соединений.
В 1930-х годах были созданы более сложные соединения, так чтобы функция противоопухолевого средства была разделена на две части или чтобы соединение состояло из двух фрагментов, где один фрагмент должен служить для концентрирования лекарственного средства в злокачественной клетке или опухолевой ткани, а второй фрагмент должен служить для разрушения злокачественных клеток, в которых сконцентрировано соединение. Главным достижением в данной области было выявление химической природы соединения, обусловливающей избирательность в отношении опухоли. Предыдущие сообщения о такой специфичности исследователей Lewis, et al. в публикации в Cancer Res., 9:736, 1949, которые касались красителя нильского голубого, подтолкнули к идее модификации этого бензофеноксазина, заключающейся во введении токсина, такого как азотистый иприт, в качестве интегральной части молекулы (Sen, et al., Int. Union against Cancer Acta, 26, 774 (1960)). Эти попытки модифицировать средство, проявляющее избирательность в отношении пораженной злокачественной опухолью ткани, посредством токсического фрагмента не имели значительного успеха. Было высказано предположение, что неудача таких подходов связана с модификацией тканевой избирательности исходного соединения в результате добавления токсического фрагмента. Однако причина неудачи более фундаментальна, и ее объяснение связано с другими концепциями, касающимися доставки лекарственных средств, в частности концепцией о противоопухолевых пролекарствах.
В начале 1950-ых годов модель двухфрагментного лекарственного средства, состоящая из концентрирующего фрагмента и токсического фрагмента, была модифицирована с образованием двухфрагментной модели “пролекарства”, где один фрагмент предназначен модифицировать или “маскировать” или “покрывать” токсичность второго токсического фрагмента лекарственного соединения до тех пор, пока лекарственное средство не поступит в злокачественные клетки. На этом этапе с помощью химической реакции, которую проводят в присутствии ферментов, специфичных в отношении злокачественных клеток, удаляют маскирующий или покрывающий фрагмент, с тем чтобы токсический фрагмент уничтожил злокачественные клетки. Но и здесь идея доставки противоопухолевого пролекарства также не имела значительного успеха.
Идея создания пролекарств, объединяющих в себе концентрирующий фрагмент и токсический фрагмент, а также идея создания пролекарств, которые предназначены метаболизировать предпочтительно в опухолевых тканях, сталкивается с проблемами, которые исходят из природы самой опухоли: злокачественные клетки и ткани не являются чужеродными биологическими организмами, химическая природа которых отличается от природы нормальной ткани. Эти ткани, в случае опухолей человека, являются тканями человека. Нормальная и неопластическая ткань имеют, по сути, одинаковую химическую природу; то есть большей частью обе состоят из одинаковых химических молекул и в сходных концентрациях.
В 1980 году исследователь Evan Harris Walker в публикации Perspectives in Biology and Medicine, Spring Issue, 424-438 (1980), доказывал, что концепция пролекарства и концепция комбинации токсина с концентрирующим фрагментом не имела успеха в связи с фундаментальными законами фармакокинетики, касающимися доставки лекарственного средства. Период действия лекарственных средств зависит от скоростей их метаболического поглощения и элиминации. В случае, если скорости в злокачественных клетках ниже, чем в нормальных клетках, лекарственные средства, по-видимому, будут концентрироваться. То есть после достаточного периода времени лекарственные средства должны быть метаболизированы и выведены из нормальных тканей, в то время как в злокачественных клетках и тканях они все еще будут присутствовать в значительных концентрациях. Проблема создания эффективного противоопухолевого лекарственного средства не является проблемой достижения дифференциальной концентрации или проблемой получения лекарственного средства, которое активируется ферментами в клетках, а скорее проблемой, связанной с возможностью подавлять токсичность лекарственного средства в нормальных клетках и тканях до тех пор, пока дифференциальная концентрация лекарственного средства не станет благоприятной.
Для того чтобы воспользоваться преимуществом различия концентраций лекарственного средства между нормальной и пораженной злокачественной опухолью тканью, исследователь Walker в публикации Perspectives in Biology and Medicine, Spring Issue, 424-438 (1980) предложил модель, состоящую из двух лекарственных средств, пролекарства и “активационного” лекарственного средства. Как пролекарство (которое имеет конструкцию, ограниченную токсином плюс покрывающим или маскирующим фрагментом), так и активационное лекарственное средство были созданы таким образом, чтобы оказывать незначительное токсическое или другое действие на организм или вовсе его не иметь (т.е. быть в значительной степени биологически инертными) при введении одного из них. Однако в комбинации указанные два соединения должны взаимодействовать в организме с образованием соединения, проявляющего токсичность по отношению к клеткам, в которых соединение было образовано. Данных два лекарственных средства следует вводить последовательно, с отсрочкой во времени между их введением, чтобы позволить нормальным клеткам метаболизировать и элиминировать пролекарство из организма до введения активационного лекарственного средства. В этот момент - после периода отсрочки дифференциальной доставки лекарственного средства - должна быть достигнута дифференциальная концентрация пролекарства. Удаление маскирующего или покрывающего фрагмента пролекарства в это время - когда пролекарство должно быть дифференциально сконцентрировано в опухолевых тканях - должно приводить к доставке включенного противоопухолевого токсина при высоких уровнях. Таким способом противоопухолевые токсины должны быть доставлены к злокачественным клеткам и тканям без ущерба для нормальных клеток и тканей.
Пролекарства такого типа, который предложен исследователем Walker, были разработаны и синтезированы авторами настоящего изобретения. Одним из таких пролекарств был 5-фторурацил-N-глюкозид, который может быть активирован ферментом β-глюкозидазой. После создания пролекарства такого типа авторы настоящего изобретения обнаружили, что более сложная система пролекарства и активационного лекарственного средства необходима для обеспечения всех требований, предъявляемых к избирательному и эффективному противоопухолевому фармацевтическому препарату. В частности, авторы настоящего изобретения обнаружили, что модель пролекарства и активационного лекарственного средства такого типа также имеет ограничения в том, что дифференциальная концентрация, достигаемая в рамках данной модели, была недостаточно высокой. Другими словами, эффект дифференциальной концентрации, обусловленный полностью дифференциальной диффузией без добавленного фрагмента для повышения концентрационной разницы, оказался недостаточным. Исходя из вышеизложенного, следует, что необходима более сложная система лекарственного средства, а также необходим способ создания такой системы лекарственного средства.
Сущность изобретения
Настоящее изобретение относится к комбинации, содержащей химические компоненты и химические фрагменты, способу создания комбинации, продуктов или устройств, необходимых для осуществления и оценки этого способа, химическим системам, созданным с помощью способов, которые служат в качестве примеров отдельных комбинаций, и к способам, необходимым для успешного применения комбинации и конкретных примеров комбинаций, созданных способом, раскрытым в данном описании.
Комбинация согласно настоящему описанию определена как сложная система прото-лекарства и активационного лекарственного средства. Прото-лекарство включает в себя один или более дифференциально избирательных фрагментов, один или более токсических фрагментов и один или более фрагментов, которые служат для обеспечения «маскировки» или «покрытия» токсических фрагментов.
Настоящее описание также охватывает активационное лекарственное средство, которое определено как один или более химических компонентов, выполняющих функцию активации прото-лекарства в комбинации. Активационное лекарственное средство способствует удалению маскирующего или покрывающего фрагмента или достаточного количества «маскировки» или «покрытия» или изменению химической природы всей молекулы прото-лекарства, с тем чтобы по существу исключить функцию маскировки или покрытия прото-лекарства, что делает образовавшееся соединение токсическим, фармакологически активным средством. Кроме того, в настоящем изобретении описано введение "связей", которые предназначены для химического связывания фрагментов с образованием единого соединения, которое само по себе является по существу биологически инертным. Связями могут быть химические связи между связанными фрагментами или комбинация атома(ов) и связей, которые соединяют фрагменты прото-лекарства.
Настоящее описание относится к способу, с помощью которого предполагается создать и проанализировать систему (т.е. комбинацию) прото-лекарства и активационного лекарственного средства. То есть вероятность того, что такая сложная система химических компонентов, обладающих набором необходимых свойств, может быть создана посредством бессистемного экспериментирования, является нулевой. Из вышесказанного следует, что необходимо разработать способ создания таких химических систем.
Настоящее описание также относится к новым продуктам или устройствам, используемым в настоящем способе.
Настоящее описание также включает в себя отдельные примеры систем прото-лекарства и активационного лекарственного средства (т.е. комбинаций), которые получают путем успешного применения способа.
Подробное описание изобретения
Комбинация, способ создания комбинации, продукты или устройства, требующиеся для осуществления и оценки способа, и процедуры, необходимые для успешного применения примеров химической системы, описанных в данном изобретении, направлены на получение высокоизбирательного и эффективного противоопухолевого фармацевтического препарата. Примеры и соответствующие данные, приведенные ниже, служат в качестве примеров конкретных комбинаций и демонстрируют успешное применение описанного способа. Способ не ограничивается созданием противоопухолевого средства, но также может применяться и для разработки других фармацевтических препаратов, токсичность которых в отношении популяции клеток-мишеней должна сочетаться с низкотоксичными сопутствующими побочными эффектами.
Целью настоящего изобретения является получение комбинации, которая включает в себя прото-лекарство и активационное лекарственное средство, где прото-лекарство состоит по меньшей мере из одного дифференциально концентрирующего фрагмента, по меньшей мере одного токсического фрагмента и по меньшей мере одного покрывающего фрагмента. Подробное описание компонентов прото-лекарства приведено ниже:
1. Дифференциально избирательный фрагмент или фрагменты (также называемый «дифференциально концентрирующим фрагментом») химического соединения, обладающего свойствами, позволяющими соединению дифференциально концентрироваться в опухолевых тканях по сравнению с нормальными тканями подвергаемого лечению организма животного или человека. Употребляемый в тексте термин "дифференциально концентрироваться" означает, что в какой-то момент фармакокинетического процесса отношение концентрации указанного соединения в опухолевой ткани к концентрации этого соединения в нормальных тканях станет повышаться в результате различия между поглощением, распределением, метаболизмом и элиминацией лекарственного средства в опухолевых клетках и нормальных клетках.
2. Токсический фрагмент или фрагменты химического соединения, обладающие свойствами, позволяющими соединению уничтожить клетки или ткани, в которых оно концентрируется.
3. Маскирующий или покрывающий фрагмент или фрагменты химического соединения, обладающие свойствами, позволяющими пролекарству не проявлять токсичность, то есть прото-лекарство не действует как токсин в клетке или ткани, в которой оно локализовано, до тех пор, пока прото-лекарство не будет активировано путем аппликации или введения активационного лекарственного средства. Употребляемый в тексте термин "маскирующий или покрывающий фрагмент" означает любую модификацию в прото-лекарстве, которая способствует ослаблению или устранению токсичности всего соединения, где такой покрывающий фрагмент удаляется позже или модифицируется активационным лекарственным средством. Термин «маскировка» или «покрытие» не должен означать или ограничиваться значением химический фрагмент, который буквально покрывает прото-лекарство или химически связывается непосредственно с токсическим фрагментом или токсическим участком прото-лекарства.
4. Связи, означающие химические связи или комбинацию атомов и связей между элементами отдельных фрагментов прото-лекарства, которое включает в себя по меньшей мере один фрагмент каждого из дифференциально избирательного фрагмента, токсического фрагмента и покрывающего фрагмента. Примеры связей, составляющих комбинацию атомов и связей, включают в себя, не ограничиваясь ими, единичные атомы, такие как кислород, которые могут образовывать связь -О- с двумя связывающими участками для присоединения двух из требуемых фрагментов, или группы различных атомов, такие как амин (>N-CH2-), которые могут присоединять три фрагмента, или азин (>C:NN:C<), который, если потребуется, может соединить вместе четыре фрагмента с образованием одной молекулы.
Активационное лекарственное средство комбинации представляет собой одно или более химических соединений, отделенных от прото-лекарства. Активационное лекарственное средство служит активатором прото-лекарства путем физической или химической модификации, чтобы сделать образованное соединение токсичным, фармакологически активным средством, или чтобы прото-лекарство высвобождало токсический фрагмент. Как прото-лекарство, так и активационное лекарственное средство моделируют таким образом, чтобы они оказывали незначительный токсический или другой эффект на организм или вовсе его не оказывали (т.е. по существу были биологически инертными) при введении по отдельности. Однако в комбинации прото-лекарство и активационное лекарственное средство будут взаимодействовать в организме, образуя соединение, токсичное в отношении клеток, в которых такое соединение образуется.
Тот факт, что прото-лекарство и активационное лекарственное средство моделированы так, что они должны проявлять незначительное токсическое действие на организм или вовсе его не проявлять при введении по отдельности, однозначно свидетельствует о том, что описанный способ моделирования лекарственного средства отличается от ранее описанных способов создания лекарственного средства. Данный способ позволяет переместить отдельные элементы химической модели из области биохимии в прогнозируемую область органической химии. Настоящим изобретением достигается модификация моделирования стандартного прото-лекарства. Стандартные прото-лекарства зависят от специфики биохимических реакций, протекающих в тканях организма, в которых они превращаются в фармакологически активные соединения. Прото-лекарство согласно настоящему изобретению, по существу, является биологически инертным соединением, которое подвергается взаимодействию, например, с неорганическим соединением, так что, хотя реакция протекает в среде биологической системы, оно не поддается реакции с эндогенными молекулами самой биологической системы. В случае, когда прото-лекарство и активационное лекарственное средство взаимодействуют друг с другом, полученные в результате такой реакции продукты и их метаболиты становятся соединениями, обладающими хорошо известными биологическими активностями, которые позволяют сохранить их хорошо установленные и хорошо изученные свойства и при добавлении многих химических фрагментов. Что будет происходить с указанными химическими компонентами, которые участвуют в создании прото-лекарства, можно предсказать в широком диапазоне химических модификаций. Мехлорэтамин, как особый пример, остается токсином для биологических тканей, в которых он накапливается, при большом количестве модификаций - модификаций, которые хорошо изучены и истолкованы. Таким образом, как «абиологическая» химия предшественников прото-лекарства, так и химия их соответствующих фармакологически активных продуктов реакции достаточно изучены, чтобы определить модификации, которые служат основой для любого числа специфических вариаций в настоящем описании, и это число будет увеличиваться в области знаний.
Способ детального моделирования комбинации включает в себя следующие стадии:
Выбор дифференциально концентрирующего фрагмента: Стадия способа, посредством которой химические фрагменты соединения, обладающие свойствами повышать дифференциальную концентрацию, означающую, что отношение концентрации указанного соединения в опухолевой или другой ткани-мишени или тканях-мишенях к концентрации соединения в нормальных тканях в некое время фармакокинетического процесса станет повышаться в результате различия между поглощением, распределением, метаболизмом и элиминацией лекарственного средства в опухолевых и нормальных тканях, идентифицируют с помощью специфических процедур. Одним способом отбора соединений, включающих в себя дифференциально избирательный фрагмент, является ВЭЖХ. Указанные процедуры ВЭЖХ включают в себя, не ограничиваясь ими, определение скоростей диффузии для потенциальных соединений, содержащих дифференциально концентрирующий фрагмент (или состоящих полностью из данного фрагмента), посредством различных типов колонок ВЭЖХ, для того, чтобы выбрать из соединений такие, которые имеют низкие скорости диффузии (то есть высокие скорости удерживания). Примерами используемых в этих целях колонок для ВЭЖХ являются ВЭЖХ-колонки с сорбированной РНК злокачественных клеток, ВЭЖХ-колонки с сорбированной ДНК злокачественных клеток и ВЭЖХ-колонки с сорбированными злокачественными клетками, суспензией злокачественных клеток и экстрактом злокачественных клеток (иначе называемые «колонками ракового типа»). Дифференциально концентрирующий фрагмент также оценивают посредством контрольной колонки, означающей ВЭЖХ-колонку с сорбированной РНК или ДНК из нормальных клеток или с сорбированными нормальными клетками, суспензией нормальных клеток или экстрактом нормальных клеток. Соединение, содержащее дифференциально концентрирующий фрагмент, выбирают путем сравнения скорости диффузии соединения на ВЭЖХ-колонке ракового типа со скоростью диффузии на контрольной колонке. Соединения, имеющие низкие скорости диффузии (т.е. высокие скорости удерживания на колонках ракового типа), являются потенциальными дифференциально избирательными фрагментами.
Второй способ отбора соединений, содержащих дифференциально избирательный фрагмент, включает в себя методы хроматографии, отличные от ВЭЖХ. Указанные процедуры включают в себя, не ограничиваясь указанными, определение скоростей диффузии для потенциальных соединений, содержащих дифференциально концентрирующий фрагмент (или состоящих полностью из указанного фрагмента), с помощью хроматографических колонок, листов, слоев, поверхностей с сорбированной РНК или ДНК злокачественных клеток или других типов хроматографии с целью отбора соединений, имеющих низкие скорости диффузии (то есть высокие скорости удерживания), относительно скоростей диффузии, определяемых с помощью контрольной хроматографической системы. Потенциальные соединения, содержащие фрагмент (или состоящие полностью из фрагмента), также могут быть подвергнуты определению скоростей диффузии с помощью хроматографических колонок, листов, слоев, поверхностей с сорбированными злокачественными клетками, суспензий злокачественных клеток или экстрактов злокачественных клеток или других типов хроматографии. Как и в методе ВЭЖХ, дифференциально концентрирующее соединение выбирают путем сравнения скорости диффузии соединения на системе с сорбированным опухолевым материалом со скоростью диффузии на контрольной системе.
Третьим способом, посредством которого выбирают соединения, содержащие дифференциально избирательный фрагмент, является методология in vivo. Для оценки потенциального соединения с дифференциально концентрирующим фрагментом используют методы in vivo, включающие в себя введение материала модельным животным, у которых вызывают рост опухоли, и проверку биопсий, взятых при различных временах отсрочки, или умерщвление животных при очередном времени отсрочки, что предпринято с целью возможного определения концентрации потенциального материала как функции от времени в опухолевых тканях и в нормальных тканях.
Механизм, посредством которого потенциальные соединения, содержащие дифференциально избирательные фрагменты, могут быть отобраны для того, чтобы пройти через вышеописанные методологии отбора, включает в себя выбор таких соединений из перечня красителей, обладающих высокими окрашивающими свойствами, применяемых в диагностике злокачественных опухолей. В этом отношении следует отметить, что красители используют для окрашивания образцов ткани, взятых у больных с целью выявления наличия опухоли, поскольку злокачественным клеткам свойственно высокое поглощение отдельных красителей, применяемых для этой цели. Примерами таких красителей являются гематоксилиновый и эозиновый красители, используемые в методах картирования железы Вирхова с целью исследования образцов ткани грудной железы онкологических больных, и цитокератин, используемый при иммуногистохимическом окрашивании образца ткани, взятого из лимфатических узлов для диагностики микрометастатического заболевания (Pendas, et al., Annals of Surgical Oncology 7(1), 15-20, 2000, January-February). Другие красители, имеющие тканевую избирательность, которая превращается в опухолевую избирательность, включают в себя нильский голубой (Lewis, et al., Cancer Res. 9, 736, 1949). Кроме того, авторы отмечают, что желтые тиоксантоны проявляют тканевую избирательность (Miller, et al., патент США No. 5346917). Избирательность данных красителей в сочетании с их благоприятными физико-химическими свойствами делает их хорошими кандидатами для избирательной доставки лекарственных средств.
Перечень таких красящих материалов, которые являются соединениями, содержащими потенциальные дифференциально избирательные фрагменты или полностью состоящими из них, включает в себя, не ограничиваясь перечисленным, следующие: нильский голубой; тиоксантоны; гематоксилиновый краситель; эозиновый краситель; цитокератиновый краситель; красители ДНК/РНК, включая трипановый синий, метиловый зеленый, этидийбромид, лейкофуксиновый краситель, метиленовый голубой; материалы в ссылках Handbook of Fluorescent Probes and Research Chemicals [by Richard P. Haugland, Sixth Edition], включая красители акридиновый гомодимер, акридиновый оранжевый, актиномицин D, 7-аминоактиномицин D, 9-амино-6-хлор-2-метоксиакридин, 4,6-диамидино-2-фенилиндол, дигидроэтидий, 4′,6-(диимидазолин-2-ил)-2-фенилиндол, гетеродимер этидия-акридина, этидийдиазид, этидия гомодимер-1 и -2, этидиймоноазид, гидроксистилбемидин, метансульфонат; биологические красители общего назначения, такие как сафранин, малахитовый зеленый, эозиновый желтоватый, кристаллический фиолетовый, метиленовый голубой, гематоксилин, основный коричневый, карминовые квасцы, метиловый зеленый и нейтральный красный, краситель Giemsa, краситель Gram и химические соединения, которые, как известно, “прилипают”, но не связываются с ДНК и РНК, называемые иногда нестойкими красками.
Перечень потенциальных красителей и биологических красок для дифференциально избирательного фрагмента достаточно длинный. Выбор предпочтительных кандидатов для химического фрагмента, обладающего способностью повышать дифференциальную концентрацию, должен быть подтвержден одним или более предварительно описанными методами отбора.
Представляет особый интерес то, что выбранный материал имеет низкую токсичность по отношению к нормальной ткани и в пределах, в которых он проявляет любую токсичность, что токсичность является дифференциально избирательной против опухолевых тканей (в противном случае указанная особенность будет направлена против всей модели лекарственного средства). Эти положения, а также выбор дифференциально концентрирующего фрагмента пролекарства формул I и II представлены ниже.
Выбор токсического фрагмента: Стадия способа, посредством которой химический фрагмент, проявляющий способность вызывать или повышать цитотоксичность или гибель клетки, выявляют с помощью тестов in vitro, тестов in vivo или выбирают из существующих перечней соединений, которые проявляют цитотоксичность и которые содержат цитотоксический фрагмент.
Выбор покрывающего фрагмента: Стадия способа, посредством которой химические фрагменты, обладающие способностью маскировать, покрывать, ослаблять или исключать цитотоксичность токсического фрагмента одновременно с тем, что такой покрывающий фрагмент химически не удаляется путем ферментативных или других метаболических процессов в организме больного, выявляют с помощью тестов in vitro или выбирают из существующих перечней реагентов с токсическим фрагментом, выбранным, как описано выше. Соответствие покрывающего фрагмента устанавливают после образования полного прото-лекарства (включая выбранные связи, как описано в тексте ниже). Точнее, прото-лекарство проверяют на соответствие либо in vitro, либо на модели животных, либо на человеке. Продукты метаболизма, побочные продукты или экстракты из тестируемой системы оценивают посредством ВЭЖХ или других аналитических методов для подтверждения того, что прото-лекарство не подвергалось метаболизму или модификации таким путем, который приводит к образованию токсина, когда применяют или вводят только прото-лекарство (т.е. в отсутствие активационного лекарственного средства).
Выбор активационного лекарственного средства: стадия способа, посредством которой одно или более химических соединений, отделенных от прото-лекарства, обладающих способностью удалять маскирующий или покрывающий фрагмент или достаточное количество «маскировки» или «покрытия», или также модифицировать химическую природу всей молекулы пролекарства с тем, чтобы в значительной степени исключить функцию маскировки или покрытия, осуществляемую маскирующим или покрывающим фрагментом, тем самым делая остаточное соединение токсичным, или с тем, чтобы способствовать высвобождению токсического фрагмента из прото-лекарства в организме онкологического больного и в окружающие злокачественные клетки области.
Выбор связей прото-лекарства: выбранный химический фрагмент или фрагменты, проявляющие способность повышать дифференциальную концентрацию прото-лекарства, выбранный химический фрагмент или фрагменты, обладающие токсическими свойствами, или выбранный химический фрагмент или фрагменты, проявляющие способность «покрывать» цитотоксичность всего химического соединения (т.е. прото-лекарства), должны быть связаны или химическими связями, или посредством комбинации химических связей и атомов между несколькими фрагментами, которые должны удерживать фрагменты вместе, давая возможность осуществлять каждому фрагменту индивидуальную функцию, ради которой каждый фрагмент был выбран. Указанные сочленения между фрагментами называют связями. В общем, способ связывания каждой пары фрагментов будет выбран с учетом практических знаний, касающихся способов образования связей, которые позволяют избежать изменений индивидуальных свойств выбранных фрагментов, например, следует признать, что покрывающий фрагмент служит в целях ингибирования активности токсического фрагмента до добавления активационного лекарственного средства. Вообще для каждого соединения, выбранного для того, чтобы стать (после соединения или связывания) фрагментом нового прото-лекарства, выявляют участки, ответственные за желаемые свойства выбранного соединения; важно, что эти участки по существу не разрушаются или не модифицируются при связывании. Модификация молекулы у некоторых доступных положений соединения позволяет проверить и установить участки, ответственные за желаемые свойства. Индивидуальные молекулы, выбранные для проявления дифференциально концентрирующих и токсических способностей прото-лекарства, затем присоединяют к соединениям в положениях, расположенных на некотором расстоянии от активных участков, ответственных за отдельную функцию, для осуществления которой выбирают фрагмент; индивидуальную молекулу, несущую покрывающий фрагмент, затем присоединяют у положения таким образом, чтобы ингибировать активность токсического фрагмента. Указанную процедуру выполняют в способе согласно изобретению и описывают в примерах, приведенных в настоящем изобретении.
В результате способа согласно изобретению, осуществляемого для создания комбинации, получают прото-лекарства формулы I и II

где
R1 является SiZ3;
R2 означает метил, хлорэтил, гидроксиэтил или бромэтил;
R3 означает хлорэтил, гидроксиэтил или бромметил;
R4 означает Н, SO3H или таурин;
R5 является SiZ3;
R6 означает Н, SO3H или таурин;
каждый Z из Z3 независимо означает трет-бутил или метил;
Х означает углерод, кислород или азот, и
W означает углерод, кислород или азот.
В настоящем изобретении предлагается способ избирательной доставки цитотоксического соединения к опухолевой ткани путем использования прото-лекарства, имеющего фрагмент, который дифференциально концентрирует прото-лекарство, а также цитотоксический фрагмент и покрывающий фрагмент, где прото-лекарство доставляет токсический фрагмент таким образом, чтобы предотвратить значительное повреждение нормальных тканей посредством сохранения покрывающего фрагмента на прото-лекарстве до тех пор, пока прото-лекарство дифференциально не сконцентрируется в опухолевой ткани в течение периода отсрочки, и после такого периода отсрочки из прото-лекарства не образуется цитотоксическое соединение при введении активационного лекарственного средства.
Другой целью настоящего изобретения является разработка способа избирательной доставки цитотоксического соединения к опухолевой ткани путем введения прото-лекарства формулы I или II
где
R1 является SiZ3;
R2 означает метил, хлорэтил, гидроксиэтил или бромэтил;
R3 означает хлорэтил, гидроксиэтил или бромметил;
R4 означает Н, SO3H или таурин;
R5 является SiZ3;
R6 означает Н, SO3H или таурин;
каждый Z из Z3 независимо означает трет-бутил или метил;
Х означает углерод, кислород или азот, и
W означает углерод, кислород или азот,
так что прото-лекарства формул I и II доставляют токсический фрагмент таким образом, чтобы предотвратить повреждение нормальных тканей путем сохранения интактным покрывающего прото-лекарство фрагмента до тех пор, пока прото-лекарство не сконцентрируется дифференциально в опухолевой ткани в течение периода отсрочки, и после такого периода отсрочки из прото-лекарства не образуется цитотоксическое соединение при введении фтористой соли.
Другой целью изобретения является получение соединений формул III и IV

где
R1 является Н;
R2 означает метил, хлорэтил, гидроксиэтил или бромэтил;
R3 означает хлорэтил, гидроксиэтил или бромметил;
R4 означает Н, SO3H или таурин;
R5 является Н;
R6 означает Н, SO3H или таурин;
Х означает углерод, кислород или азот, и
W означает углерод, кислород или азот
или фармацевтически приемлемой аддитивной соли основания формул III и IV.
В данном изобретении также предложен способ оценки противоопухолевой активности соединений формул I и II с активационным соединением (или, в случае формул III и IV, без такого соединения), такой способ следует использовать в связи со способом детального моделирования комбинации и применять на стадиях способа, как описано выше, или для in vitro и in vivo оценки противоопухолевой активности любого другого химического вещества.
В настоящем изобретении также предлагается применение соединения формулы I или II с активационным соединением или, в случае соединения формул III и IV, без активационного соединения для производства лекарственного средства с целью лечения опухолей. Кроме того, в данном изобретении предлагается фармацевтический препарат для лечения опухолей, содержащий эффективное количество соединения формулы I или II вместе с фармацевтически приемлемыми наполнителями и активирующее количество фтористой соли вместе с фармацевтически приемлемыми наполнителями, где соединение формулы I или II и активационное лекарственное средство упакованы для индивидуального введения. Кроме того, данное изобретение включает в себя способ лечения опухолей, включающий в себя введение эффективного количества соединений формулы I или II, выжидание времени, отсроченного для введения активирующего количества фтористой соли.
Соединения согласно изобретению, а именно прото-лекарство и активационное лекарственное средство, могут быть введены перорально, внутрибрюшинно (ip), внутривенно (iv), через кожу, внутримышечно, интраназально или внутрь прямой кишки. Способ введения будет изменяться, в зависимости от отдельного вводимого лекарственного средства, заболевания, подвергаемого лечению, удобства больного и опекуна и других существенных факторов.
Фармацевтические композиции согласно изобретению получают способами, хорошо известными в данной области. Носитель или наполнитель может быть твердым, полутвердым или жидким материалом, который служит в качестве связующего материала или среды для активного ингредиента. Подходящие носители или наполнители хорошо известны в данной области. Фармацевтическая композиция может быть приспособлена для перорального использования, ингаляции, парентерального или местного применения и может быть введена больному в виде таблеток, капсул, аэрозолей, лекарственных форм для ингаляции, суппозиториев, растворов, суспензий и пр. Предпочтительные композиции и препараты могут быть определены способами, которые хорошо известны квалифицированному экспериментатору.
Соединения согласно настоящему изобретению могут быть введены перорально, например, с инертным разбавителем в капсуле или посредством дозированной формы в виде спрессованной таблетки. Для перорального терапевтического введения соединения могут быть включены с наполнителями и использованы в виде таблеток, пилюль, пастилок, капсул, эликсиров, суспензий, сиропов, облаток, жевательных резинок и тому подобного. Концентрация активного ингредиента в указанных препаратах может изменяться приблизительно от 0,001% до 70% веса единицы. Количество соединения, присутствующего в композициях, должно быть таковым, чтобы обеспечить подходящую дозировку. Диапазон доз прото-лекарства и активационного лекарственного средства, основанный на обычном эксперименте, осуществляемом на модели животных и результаты которого используют для лечения человека, составляет приблизительно от 1 мкг/кг до 25 мг/кг.
Дозировку активационного лекарственного средства можно снизить в 10 раз, при расчете молярности один к одному. Для повышения эффективности и скорости процесса активации молярность активационного лекарственного средства берут из расчета выше чем один к одному. Для детей, пожилых или очень немощных людей могут потребоваться дозировки прото-лекарства и активационного лекарственного средства в десять раз ниже обычно используемых. Поскольку результаты экспериментов показали, что инактивированное прото-лекарство проявляет исключительно низкую токсичность, то при более агрессивных опухолях могут потребоваться дозировки прото-лекарства и активационного лекарственного средства в десять раз выше, чем для менее агрессивного заболевания.
Таблетки, пилюли, капсулы, пастилки и пр. могут также содержать один или более из следующих адъювантов: связующие вещества, такие как повидон, гидроксипропилцеллюлоза, микрокристаллическая целлюлоза или желатин; наполнители или разбавители, такие как крахмал, лактоза или дикальцийфосфат; дезинтегрирующие реагенты, такие как натриевая соль крахмалгликолевой кислоты и тому подобное; смазывающие реагенты, такие как стеарат магния, стеариновая кислота или тальк; смачивающие реагенты, такие как лаурилсульфат натрия или полисорбаты; подсластители и ароматизаторы, такие как сахароза, аспартам, мятное масло и другие природные и синтетические вкусовые вещества. В случае дозированной формы в виде капсулы она может содержать жидкий носитель, такой как нелетучее жидкое масло. Таблетки могут быть покрыты сахаром и связующими веществами или материалами, такими как полиметакрилаты или другие покрывающие реагенты.
Отсроченная во времени активация токсина и период отсрочки
Токсичность является результатом химических реакций, которые разрушают соединения, способные к проявлению нормальных функций в клетках, или является результатом продукции соединений, которые нарушают метаболические процессы в клетке. Совокупное токсическое повреждение, произведенное в клетке в какое-либо время, пропорционально воздействию токсинов на клетки, то есть токсическому воздействию. Поэтому для того, чтобы достичь избирательной токсичности в лечении опухолей, необходимо учитывать различия скоростей метаболизма, которые существуют между нормальными тканями и злокачественными неопластическими тканями, как, например, было сделано в работе Chello et al., Cancer Res., 37:4297, 1977. Кроме того, различие между метаболизмом соединений, осуществляемым неопластическими или нормальными клетками, можно использовать для разработки химических тестов на наличие опухоли (Anderson, et al., Cancer Res., 30, 1344 (1970); Dewanjee, et al., J. Nucl. Med., 14, 624 (1973); Henderson, et al., Cancer Res., 25, 1018 (1965); Holland and Sleamaker, Acta Cytol., 13, 246 (1969); Malek et al., Cas. Lek. Cesk., 1, 16 (1963); Philips, et al., Am. J. Surg., 100, 384 (1960); and Rall et al., J. Natl. Cancer Inst., 19, 79 (1957)).
Установление времени введения прото-лекарства и активационного лекарственного средства является существенным фактором, если для достижения нетоксичного, эффективного лечения преимущественно используют разницу между концентрацией прото-лекарства в злокачественных клетках и нормальных клетках. Химически инициированная активация токсина, отсроченная во времени (“TDTA”), первоначально предложенная E.H.Walker в 1980, означает, что два соединения вводят раздельно - с интервалом между введением прото-лекарства в качестве первого лекарственного средства в последовательности введения, и введением активационного лекарственного средства в качестве второго средства в последовательности. В такой модели прото-лекарство было ограничено двумя фрагментами, токсическим фрагментом и маскирующим, или покрывающим, фрагментом. В настоящем изобретении для прото-лекарства необходимы по меньшей мере три части: дифференциально концентрирующий фрагмент, токсический фрагмент и маскирующий, или покрывающий, фрагмент. Дифференциально концентрирующий фрагмент является существенным фрагментом для доставки прото-лекарства, который способствует достижению различия в скорости метаболизма полной молекулы в клетке, что со временем приводит к повышению концентрации прото-лекарства в злокачественных клетках и тканях по сравнению с концентрацией прото-лекарства в нормальных клетках и тканях организма. Второе из введенных лекарственных средств, активационное лекарственное средство, первоначально активирует введенное прото-лекарство путем устранения или изменения активности покрывающего фрагмента прото-лекарства. Индивидуально каждое из указанных двух лекарственных средств создают таким образом, чтобы они были по существу нетоксичными (т.е. по существу биологически инертными) при введении их по одному в дозированных количествах. Однако при комбинировании указанных двух лекарственных средств они взаимодействуют, с образованием или высвобождением противоопухолевого цитотоксина (т.е. фармакологически активного средства). Механизм TDTA способствует повышению со временем отношения концентрации прото-лекарства в неопластической ткани к его концентрации в нормальной ткани, что может происходить в результате метаболизма средств, имеющих различные и подходящие фармакокинетические характеристики, такие как скорости поглощения и элиминации, как описано ниже. Высокие скорости поглощения и низкие скорости элиминации в неопластической ткани относительно этих скоростей в нормальных тканях способствуют повышению отношения концентраций в течение длительного времени. Путем определения кинетических параметров процесса, как было сделано для установления дозировок и времени отсрочки в примерах настоящего изобретения, можно избежать токсичности в нормальных тканях и достичь эффективности в лечении злокачественных тканей. Такой подход, включающий в себя механизм TDTA, в корне отличается от прежних химиотерапевтических подходов к специфичности токсинов, включая подходы к механизмам действия прото-лекарств, латентных активационных лекарственных средств и химически нацеленных или сайт-связывающих лекарственных средств. Хотя связывание дифференциально концентрирующего фрагмента с участком клетки может способствовать доставке, настоящая модель прото-лекарства не ограничивается требованием такого связывания для достижения разницы концентраций, и поэтому настоящий способ является более гибким способом моделирования доставки лекарственного средства.
Фармакокинетические характеристики процессов метаболизма в нормальных и неопластических тканях, поглощение и элиминация продуктов метаболизма определяются параметрами химической кинетики клеток и физическими процессами диффузии, проницаемости мембраны, активного транспорта, кровообращения и выделения. В значительной мере концентрации лекарственного средства могут быть прогнозированы на основе уравнений кинетики реакций первого порядка (см. ниже). Выведение лекарственного средства из сыворотки (хранилища лекарственного средства) после внутривенной инъекции, например, происходит в соответствии с химической кинетикой, где поглощение тканями организма происходит по типу «молекула за молекулой» (т.е. молекулы лекарственного средства по существу не взаимодействуют друг с другом). Этот процесс описывается уравнением (1), которое указывает, что скорость удаления из хранилища пропорциональна концентрации:
dD/dt=-pD, (1)
где D означает концентрацию лекарственного средства (например, концентрацию прото-лекарства) в хранилище, t означает время и p является константой пропорциональности. Уравнение (2) демонстрирует экспоненциальную зависимость концентрации лекарственного средства в хранилище, которое доступно тканям в любой момент:
D=D0e-pt, (2)
где D0 является исходной концентрацией лекарственного средства в сыворотке (в момент инъекции). Концентрация С лекарственного средства в ткани будет зависеть от ресурсов лекарственного средства в хранилище (приводящих к повышению концентрации путем диффузии в клетки) и скорости, при которой такое лекарственное средство удаляется из ткани. Это дает уравнение скорости
dC/dt=gD-bC, (3)
где g и b являются константами скорости. Если концентрация лекарственного средства в ткани первоначально является нулевой, уравнение (3) имеет решение, представленное уравнением (4):
C=k(1-e-at)e-bt, (4)
где k=gD0/a и a=p-b. Зависимость концентрации от времени, представленная уравнением (4), наблюдается при количественной оценке поглощения питательных веществ и лекарственных средств тканями и удаления их из тканей. Данные, демонстрирующие зависимость концентрации от времени для некоторых соединений и различных тканей мыши, были опубликованы H.Bush в Cancer Research, 15:365, 1955. Авторы данного изобретения также проводили подобные эксперименты с соединениями, содержащими фрагмент тиоксантона, результаты которых продемонстрировали одинаковое поведение в различных тканях мыши по прошествии длительного времени.
Авторы рассматривают случай, когда a>>b, то есть когда поглощение является быстрым. Воздействие лекарственного средства на клетку, представленное Е, затем должно быть представлено интегралом С, в частности,
E=∫0∞Cdt=k∫0∞e-btdt=k/b, (5)
Для двух типов клеток, имеющих константы элиминации bA и bB, отношение воздействий должно быть
R=EA/EB=bB/bA, (6)
Это означает, что воздействие медленно действующего токсина должно быть пропорционально отношению констант диффузии для двух типов клеток. Однако поскольку химическая природа злокачественных и нормальных клеток сходна, ожидаемое отношение воздействий должно быть умеренным, что не дает возможности достичь дифференциальных токсичностей при лечении опухолей.
Если токсин прото-лекарства «замаскирован» в течение периода отсрочки Т (также называемого «время отсрочки»), уравнение (5) принимает следующий вид:
E=∫T∞Cdt=k∫T∞e-btdt=ke-bt/b, (7)
где Е теперь является функцией от времени отсрочки Т. В таком случае отношение воздействий принимает вид
R=EA/EB=(bB/bA)exp[(bB-bA)T], (8)
Уравнение (8) показывает, что если скорость элиминации для клеток типа В больше, чем для клеток типа А, то величину экспозиционного отношения R можно повысить до желаемой, если время отсрочки для активации также является достаточно большим.
Уравнение (8) выражает сущность стратегии TDTA, в соответствии с которой экспозиционное отношение, представленное уравнением (8), в значительной степени может превышать отношение, представленное уравнением (6). Пригодность и аргументированность этого нового подхода к созданию и разработке составов, фармацевтических препаратов и протоколов прикладной программы для лечения опухолевых заболеваний подтверждена в данном изобретении посредством испытания прото-лекарств, активационных лекарственных средств и комбинаций.
Однако для того чтобы превратить по существу биологически неактивное прото-лекарство в фармакологически активное соединение, нужно ввести активационное лекарственное средство. Как и в случае с прото-лекарством, концентрация активационного лекарственного средства станет эффективной при удалении активационного лекарственного средства из сывороточного хранилища после его введения; этот процесс может быть представлен уравнением такого же вида, как уравнение (1), которое показывает, что скорость удаления из хранилища пропорциональна концентрации:
dA/dt=-qA, (9)
где A означает концентрацию активационного лекарственного средства в хранилище через время t после введения, где t>T и q является константой пропорциональности для активационного лекарственного средства (соответствующей р в уравнении (1) для прото-лекарства). Уравнение (10) выражает экспоненциальную зависимость концентрации лекарственного средства в хранилище, доступного тканям в любой момент:
A=A0e-qt, t>T, (10)
где А0 является исходной концентрацией в сыворотке в момент инъекции, t=T, и А=0 для 0 Точно так же, как прото-лекарство поступает в индивидуальные клетки и ткани организма согласно уравнению (3), активационное лекарственное средство также поступает в индивидуальные клетки. Однако так как лекарственное средство поступает в клетки, начинаются реакции между прото-лекарством и активационным лекарственным средством, влияющие на время распространения обоих лекарственных средств в организме. При таких условиях уравнения приобретают вид:

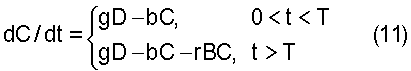
и

где r означает константу скорости реакции, f означает константу скорости диффузии для активационного лекарственного средства в тканях и клетках, имеющего концентрацию А. Другими словами, f для активационного лекарственного средства соответствует g для прото-лекарства. Подобным образом, е является константой для В и, соответственно, b является константой для С.
Далее, взаимодействие между прото-лекарством и активационным лекарственным средством приводит к образованию токсина в клетках. Концентрация указанного токсина, J, представлена уравнением
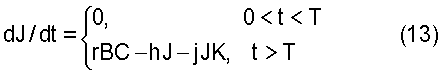
где h является константой скорости выведения токсина из клетки, и jJK представляет собой скорость взаимодействия токсина с жизненно важными компонентами клетки при скорости j, K является концентрацией химического соединения или соединений клетки, потеря которых вызывает клеточную гибель. Скорость, при которой этот жизненно важный химический компонент клетки разрушается, также зависит как от концентрации J токсина, так и от его собственной концентрации K; эта скорость представлена уравнением

где K0 означает величину концентрации K при t < T.
Уравнения (2), (10), (11), (12), (13) и (14) можно легко решить многочисленными способами, и они важны, поскольку дают подробную информацию о ходе введения активационного лекарственного средства относительно введения прото-лекарства; кроме того, указанные шесть уравнений используют для уточнения величин R, определяемых уравнением (8), поскольку уравнение (8) дает приблизительную величину R. Решение уравнения (14) также способствует более точному определению эффектов, обусловленных действием токсина, которые только приблизительно определены с помощью уравнений (5), (6) и (7). Однако из-за их сложности применение уравнений (2), (10), (11), (12), (13) и (14) отчасти неудобно для объяснения благоприятных возможностей повышения желаемой дифференциальной токсичности. Например, существует оптимальный период отсрочки Т. Уравнение (8) показывает, что в то время как R будет повышаться со временем отсрочки Т, экспозиции ЕА и ЕВ и соответствующие концентрации СА и СВ будут уменьшаться со временем Т. В результате периоды отсрочки должны быть выбраны такими, чтобы достичь достаточной дифференциальной токсичности и при этом обеспечить доставку необходимой токсической дозы к тканям-мишеням. Затем оптимальное время Т можно подрегулировать, чтобы обеспечить большую точность его подборки, используя многочисленные методы оценки с помощью указанных уравнений (2), (10), (11), (12), (13) и (14).
Несмотря на то, что оптимальное время отсрочки может быть выбрано посредством расчетов, описанных выше, необходимо подтвердить правильность дозировок и периодов отсрочки реальными измерениями. Реальные измерения могут быть сделаны с использованием лабораторных животных, как это осуществлено авторами изобретения. Указанные измерения, выполняемые с целью определения времени отсрочки, могут быть осуществлены либо с использованием полного прото-лекарства, либо непосредственно с использованием соединения, содержащего дифференциально концентрирующий фрагмент, поскольку данный фрагмент является существенным для установления разницы концентраций между нормальной и злокачественной тканью.
Данные определения оптимального времени отсрочки для соединения, содержащего дифференциально концентрирующий фрагмент, либо для самого прото-лекарства на модели животных in vivo, могут быть использованы, чтобы приблизить их к данным определения времени отсрочки у человека путем применения закона Клейбера к данным, полученным на модели животных. Закон Клейбера утверждает: «Общая скорость метаболизма для организма определяется как масса тела, возведенная в степень 3/4». Однако, чтобы определить время отсрочки, необходимо знать удельную скорость метаболизма, означающую скорость на единицу массы. Удельную скорость метаболизма получают делением указанного выше коэффициента Клейбера, определяемого путем возведения массы тела в степень 3/4, на отношение масс индивидуальных животных двух видов. Это дает в целом 1/4 степень коэффициента для удельной скорости метаболизма. Например, в случае экстраполяции данных относительно времени отсрочки, полученных на модели животных, к данным, полученным для человека, сравнение массы тела мыши (например, 20 граммов) с массой тела человека (например, 80 килограммов) дало отношение 4000. Применение закона Клейбера, приспособленного для расчета удельной скорости метаболизма к этому отношению, позволило установить, что удельная скорость метаболизма у человека в 8 раз медленнее, чем у мыши. Следовательно, время отсрочки у мыши, составляющее 4 дня, соответствует времени отсрочки у человека, составляющему приблизительно 32 дня. В случае агрессивных опухолей, где требуется больший риск, чтобы иметь больше шансов на излечение (т.е. в случае, когда должна быть допущена более высокая токсичность, чем обычно принятая токсичность для нормальной ткани, чтобы достичь эффективного токсического уровня в опухолевой ткани), может потребоваться более короткое время отсрочки, чем время, прогнозируемое посредством применения закона Клейбера к животной модели. Кроме того, для детей, пожилых и больных, находящихся в плохом состоянии, необходимым может оказаться более продолжительное время отсрочки, чем рассчитанное с помощью закона Клейбера, для достижения минимальной токсичности по отношению к нормальной ткани. Время отсрочки, составляющее от дня до пары месяцев, является вполне вероятным.
В случаях, где установленный период отсрочки используют в клиниках для человека и животных, элиминированные метаболиты прото-лекарства контролируют с тем, чтобы определить, когда токсические уровни прото-лекарства в нормальных тканях должны снизиться до безопасных уровней (т.е., чтобы определить, когда следует вводить активационное лекарственное средство). То есть заранее можно установить максимальную дозу токсина, которая может быть без вреда принята больным. Контролирование выделения метаболитов с помощью химических методов, таких как ВЭЖХ, позволяет определить время, когда прото-лекарство достигло безопасных уровней в нормальных тканях организма - таких уровней, что активация прото-лекарства будет приводить к достаточно низким уровням токсина в нормальных тканях организма. Введение активационного лекарственного средства в этот момент приводит к достижению максимальной дозы токсина для опухолевых тканей-мишеней. Если уровень токсина в тканях-мишенях является недостаточным, тогда следует ввести более высокую первоначальную дозу прото-лекарства и использовать более продолжительное время отсрочки. Если такие изменения не приводят к достижению достаточной токсической дозы в тканях-мишенях при активации, тогда следует пересмотреть модель дифференциально концентрирующего фрагмента, токсического фрагмента или обоих фрагментов для получения наилучших фармакокинетических параметров. Однако показано, что примеры проверенных комбинаций прото-лекарства и активационного лекарственного средства имеют необходимые наилучшие фармакокинетические параметры.
Синергические эффекты фармацевтических препаратов являются известными эффектами, и они часто нарушаются в результате введения двух или более лекарственных средств в одно и то же время или близкое время, приближенное друг к другу. В противоположность этому, для осуществления настоящего изобретения необходимо взаимодействие лекарственных средств, и предполагается, что при введении средств по одному будет иметь место лишь незначительный эффект на организм или эффекта не будет совсем от каждого из лекарственных средств индивидуально. Кроме того, предполагается, что оптимальные отсрочки по времени между введением прото-лекарства и активационного лекарственного средства можно определить и применить во избежание вредных побочных эффектов противоопухолевых лекарственных средств, направленных на борьбу с болезнью.
Настоящее изобретение также относится к способу избирательной доставки прото-лекарства и активации такого прото-лекарства с образованием или высвобождением цитотоксического соединения в опухолевой ткани посредством выжидания в течение времени отсрочки, а затем введения активационного лекарственного средства. Настоящее изобретение также относится к способу избирательной доставки соединений формулы I и формулы II и активации соединений формулы I и формулы II с образованием или высвобождением цитотоксического соединения в опухолевой ткани посредством выжидания в течение времени отсрочки, а затем введения фторида натрия.
Настоящее изобретение также относится к способу лечения опухолей у млекопитающего, включающему в себя введение млекопитающему при необходимости такого лечения терапевтически эффективного количества прото-лекарства, содержащего дифференциально концентрирующий фрагмент, токсический фрагмент и покрывающий фрагмент, выжидания в течение времени отсрочки, а затем введение активационного лекарственного средства.
Настоящее изобретение также относится к способу лечения опухолей у млекопитающего, включающему в себя введение млекопитающему при необходимости такого лечения терапевтически эффективного количества соединения формулы I или II
где
R1 является SiZ3;
R2 означает метил, хлорэтил, гидроксиэтил или бромэтил;
R3 означает хлорэтил, гидроксиэтил или бромметил;
R4 означает Н, SO3H или таурин;
R5 является SiZ3;
R6 означает Н, SO3H или таурин;
каждый Z из Z3 независимо означает трет-бутил или метил;
Х означает углерод, кислород или азот, и
W означает углерод, кислород или азот,
период времени отсрочки и последующее введение фторида натрия.
Методы тестирования in vitro
Методы тестирования in vitro, применяемые для оценки соединений и комбинаций согласно настоящему изобретению, опираются на принципы, являющиеся общими для многих имеющихся в настоящее время систем. Например, монослой или суспензию клеток в планшете обрабатывают прото-лекарством, инкубируют в соответствующих для каждого типа клеток условиях.
Некоторые параметры общего состояния клеток затем рассчитывают или определяют по всему периоду тестирования (например, в течение 7 дней). Если комбинацию (т.е. комбинацию прото-лекарства и активационного лекарственного средства) необходимо оценить методом in vitro после применения времени отсрочки (например, 3 дня инкубации), то активационное лекарственное средство добавляют к соответствующим лункам, и тестирование продолжают в течение дополнительного периода (например, 4 дней).
Концентрацию лекарственного средства, которая вызывает гибель клеток или торможение роста в 50% популяции клеток, IC50, рассчитывают путем сравнения между лунками, обработанными лекарственным средством, и необработанными контролями. Другим используемым параметром, который может быть измерен методами in vitro, является зависимость активности лекарственного средства от времени. Указанный параметр представляет особую важность для таких лекарственных средств, которые являются медленно действующими вследствие метаболических реакций или плохого поглощения.
Анализы опухоли мыши
Торможение роста опухолей, трансплантированных мышам, является процедурой, принятой для изучения эффективности противоопухолевых средств. Обычно процедуру выполняют путем подкожной имплантации кусочков опухоли, извлеченных из мыши-носителя, или путем подкожной инъекции злокачественных клеток порядка 5 миллионов на животное.
ПРИМЕРЫ
В настоящем описании подробно изложены данные, касающиеся применимости способа настоящего изобретения для моделирования комбинации и ее оценки. Кроме того, изобретение включает в себя создание отдельных молекул (например, прото-лекарств), средства для получения указанных соединений, подробности их проверки и процедуры, применяемые с целью определения времени отсрочки. Следующие примеры только иллюстрируют настоящее изобретение и ни в коем случае не должны рассматриваться как ограничивающие объем изобретения.
Пример I
Получение описанной комбинации посредством нового способа, изложенного выше, было начато с выбора дифференциально концентрирующего фрагмента. Для того чтобы сузить поиск подходящих кандидатов, способ начали с обзора существующей литературы.
Информация относительно бензофеноксазинов свидетельствовала о том, что использование таких соединений, соединенных с токсином, должно быть эффективным, если токсический фрагмент можно активировать после того, как лекарственное средство достигло дифференциальной концентрации в опухолевой ткани. Однако бензофеноксазины в других отношениях имеют плохие физико-химические свойства (например, плохую растворимость), что делает их непривлекательными в качестве доставочных фрагментов. Кроме бензофеноксазиновых красителей, другие вещества проявляют тканевую избирательность. Среди них желтые тиоксантоны проявляют тканевую избирательность и некоторую противоопухолевую активность (Miller, et al., патент США №5346917). Избирательность указанных красителей в сочетании с их благоприятными физико-химическими свойствами делает их хорошими кандидатами для избирательной доставки лекарственных средств.
В формуле I желтый краситель, которым является тиоксантон, был выбран для дифференциально концентрирующего фрагмента прото-лекарства. Подтверждение того, что тиоксантон проявляет способность дифференциально концентрироваться, было продемонстрировано in vivo проверкой нормальных и опухолевых тканей. Результаты исследования подтвердили, что желтый тиоксантон проявляет способность дифференциально концентрироваться.
Нескольким мышам с имплантированными пальпируемыми опухолями (поджелудочной железы) вводили iv краситель тиоксантоновый желтый в количестве 40 мг/кг. Животных умерщвляли через 4 ч, 1 день, 2 дня, 3 дня, 4 дня и 5 дней после введения тиоксантона. Опухоль, поджелудочную железу, желудок, часть кишечника вырезали и измельчали в спирте, чтобы экстрагировать краситель. Количество красителя из каждой ткани оценивали количественно с помощью ВЭЖХ. Более высокую концентрацию красителя в опухолевой ткани по сравнению с другими тканями первоначально наблюдали на 1-ый день после введения. Значительного количества красителя, остающегося в нормальной ткани на 3-4 дни, не наблюдали.
Результаты данного исследования in vivo использовали для определения времени отсрочки для прото-лекарства, которое содержит этот дифференциально концентрирующий фрагмент. Установление оптимального времени отсрочки на модели мыши, составляющего 4 дня, позволило прогнозировать время отсрочки у человека, которое составило приблизительно 32 дня. Однако на модели мыши возможное время отсрочки колебалось от 2,5 до 6 дней, что соответствовало интервалу времени отсрочки у человека от 20 до 48 дней. В случае агрессивных опухолей, более короткие интервалы времени отсрочки, возможно, такие короткие, как один день, необходимы для достижения требуемого уровня токсичности в опухолевой ткани. В противоположность время отсрочки, составляющее пару месяцев, может стать необходимым для очень тяжелого больного.
Выбор токсического фрагмента, который должен связываться с дифференциально концентрирующим фрагментом, осуществляли на основании обзора существующей литературы. Мощную противоопухолевую активность мехлорэтамина в течение длительного времени приписывали азотистому иприту, фрагменту молекулы. Он действует путем алкилирования биологически важных компонентов клетки, нарушая их функцию. Главными показателями для его применения являются бронхогенная карцинома, болезнь Ходжкина, не-ходжкинская лимфома, лимфосаркома и хронический моноцитарный лейкоз или хронический лимфолейкоз. Однако азотистый иприт проявляет более широкий спектр действия in vitro, чем его предполагаемое клиническое использование. Ограничение его использования in vivo непосредственно связано с его токсичностью (тошнота, рвота, анорексия, лейкопения, тромбоцитопения, локальное раздражение). Исследователями были предприняты попытки улучшить избирательность или доставку посредством его включения в ряд соединений (например, Haines, et al., J. Med. Chem. 30, 542 (1987); Alexander, et al., Tet. Lett. 27, 3269 (1991)). Таким образом, мехлорэтамин является прекрасным кандидатом для доставки, и тиоксантовое ядро обеспечивает соответствующий носитель для доставки его к тканям-мишеням и клеткам-мишеням. В примере формулы I азотистый иприт, в частности мехлорэтамин, был выбран в качестве цитотоксического фрагмента прото-лекарства.
Другой группой мощных противоопухолевых средств являются природные подофиллотоксины, которые использовали для лечения опухолей молочной железы и острого гранулоцитарного лейкоза, наряду с другими. Указанные вещества действуют путем ингибирования митоза в репродуцирующей клетке. Ограничения их использования подобны описанным выше ограничениям, относящимся к азотистому иприту, кроме того, они вызывают анемию, как следствие дополнительной токсичности. Полагают, что указанные типы веществ являются хорошими кандидатами для избирательной доставки с помощью тиоксантонов. В формуле II производное подофиллотоксина было выбрано для цитотоксического фрагмента прото-лекарства.
После выбора дифференциально концентрирующего фрагмента и токсического фрагмента нужно выбрать маскирующий или покрывающий фрагмент для завершения элементов прото-лекарства. В формулах I и II неорганический фрагмент, особенно силикат, SiZ3 (где каждый Z из Z3 независимо означает трет-бутил или метил), был выбран в качестве маскирующего или покрывающего фрагмента прото-лекарства. Этот неорганический силикатный фрагмент, SiZ3, также имеет химические характеристики, свидетельствующие о том, что он является высокореактивным по отношению к фтористым солям, некоторые из которых не являются токсичными. В результате, фрагмент удаляется посредством нетоксических химических реакций или химических реакций низкой токсичности, которые происходят в организме больного.
Реакция такого типа имеет место в случаях, когда присутствующее в зубных пастах фтористое соединение взаимодействует с находящимся в зубах кальцием. Образованный продукт, фторид кальция, является весьма стабильным и более прочным материалом, чем природная кальциевая соль, находящаяся в зубах. Данная реакция являет собой пример реакции замещения, стимулируемой образованием более стабильного материала. В случае соединений формул I и II ионы кремния имеют сильное “притяжение” к фториду, сильнее, чем “притяжение” натрия или калия к фториду; таким образом, реакция замещения может иметь место в случае, когда фторид натрия или калия взаимодействует с кремнийсодержащим соединением. Реакция является весьма избирательной и специфичной.
Полученное прото-лекарство должно быть активировано с помощью активационного лекарственного средства. В примерах формул I и II неорганическое вещество, фтористая соль, было выбрано в качестве удаляющего “покрытие” вещества, которое должно взаимодействовать с прото-лекарством. Отдельная фтористая соль, фторид натрия, была выбрана на основании ее известных химических свойств. Указанная неорганическая фтористая соль в качестве активационного лекарственного средства является особенно подходящей для взаимодействия с прото-лекарствами формул I и II, поскольку она является высокореактивной по отношению к отдельным силикатам, используемым в качестве покрывающих фрагментов в данных соединениях. Кроме того, фторид натрия не является токсическим в количестве, необходимом, чтобы “раскрыть” такие прото-лекарства. В результате, маскирующий или покрывающий фрагмент пролекарства удаляется с помощью весьма нетоксического неорганического вещества путем реакции, которая протекает в организме больного без вредных воздействий на больного.
Пример II
Ниже представлен подробный синтез прото-лекарства формулы I.
Схема получения I
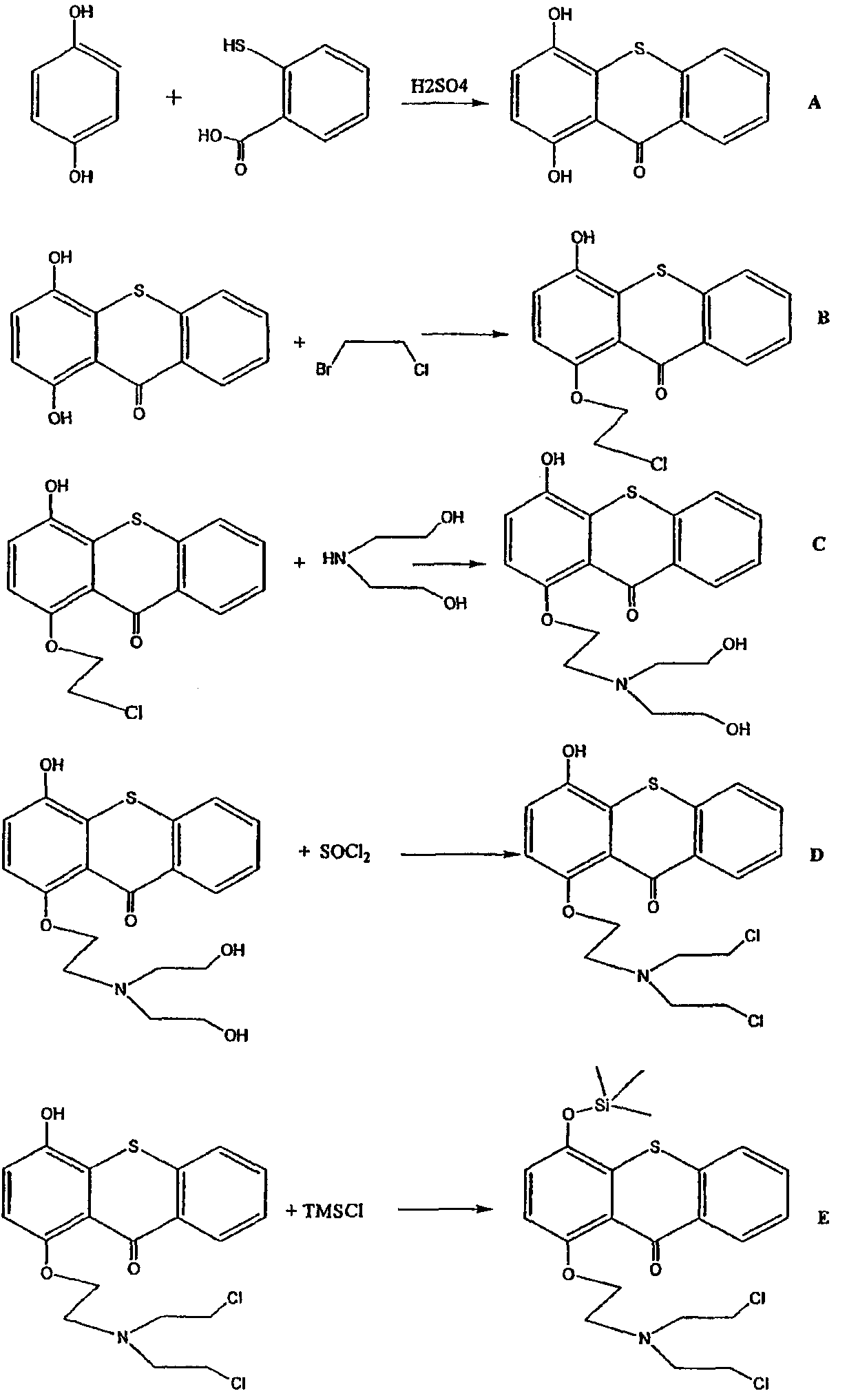
Получение 1
1,4-Дигидрокситиоксантон
Суспензию тиосалициловой кислоты (5,0 г) и гидрохинона (5,0 г) в концентрированной серной кислоте (100 мл) перемешивали при комнатной температуре в течение 4 час. Суспензию красного цвета выливали на лед и оставляли до достижения температуры окружающей среды, затем фильтровали. Полученное твердое вещество обрабатывали насыщенным раствором бикарбоната натрия, и раствор осторожно нейтрализовали 20%-ной соляной кислотой. Твердое вещество фильтровали, растворяли в ацетоне и вновь фильтровали через слой силикагеля. Раствор концентрировали, и твердое вещество очищали колоночной хроматографией на силикагеле, используя смесь гексаны:этилацетат (1:1) в качестве элюента. Фракцию, содержащую продукт, концентрировали при пониженном давлении с получением указанного в заголовке соединения желтого цвета (2,36 г, 30%).
FDMS; m/e=244 (M+).
Получение 2
1-Хлорэтокси-4-гидрокситиоксантон
Раствор 120 мг 1,4-дигидрокситиоксантона в 100 мл ацетона перемешивали в течение 0,5 час при комнатной температуре со 138 мг карбоната калия. Раствор 80 мг 1-бром-2-хлорэтана в 5 мл ацетона затем добавляли, и смесь кипятили с обратным холодильником в течение 24 час. Гетерогенную смесь охлаждали, фильтровали, концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле, используя смесь 1:1 гексанов и этилацетата, с получением 100 мг кристаллов желто-красного цвета (65%).
FDMS; m/e=307 (M+).
Получение 3
1-(N,N-бисдиэтаноламиноэтокси)-4-гидрокситиоксантон
Раствор 0,5 г 1-хлорэтокси-4-гидрокситиоксантона в 5 мл диэтаноламина нагревали в атмосфере азота при 110°С в течение 1 час. После охлаждения добавляли воду, и смесь экстрагировали четыре раза этилацетатом. Этилацетатный экстракт промывали водой, сушили над сульфатом натрия, концентрировали и очищали колоночной хроматографией на окиси алюминия, используя смесь этилацетат:этанол (4:1) в качестве элюирующего растворителя. Полученный материал кристаллизовали из ацетона с получением 0,23 г (38%) продукта желтого цвета.
FDMS; m/e=376 (M+).
Получение 4
1-(N,N-бисхлорэтиламиноэтокси)-4-гидрокситиоксантон
Способ I: Раствор 0,5 г 1-(N,N-бисдиэтаноламиноэтокси)-4-гидрокситиоксантона в 5 мл тионилхлорида нагревали с обратным холодильником в течение 6 час. Избыток тионилхлорида удаляли перегонкой, и остаточное твердое вещество подвергали колоночной хроматографии на окиси алюминия, используя смесь этилацетат:гексаны (1:1) в качестве элюента, с получением 0,3 г (58%) твердого вещества желтого цвета.
FDMS; m/e= 413 (M+).
Способ II: Раствор 0,5 г 1-(N,N-бисдиэтаноламиноэтокси)-4-гидрокситиоксантона в 10 мл пиридина обрабатывали 0,4 г метансульфонилхлорида при низкой температуре и в атмосфере азота, и смесь держали охлажденной в течение 24 час. Затем смесь выливали в воду и экстрагировали этилацетатом. Этилацетатный экстракт промывали водой, сушили над сульфатом натрия и концентрировали до получения твердого вещества желтого цвета. Данное твердое вещество растворяли в диметилформамиде (5 мл) и перемешивали в атмосфере азота при 80°С с 3 г хлорида лития в течение 24 час. Смесь охлаждали, смешивали с водой и экстрагировали этилацетатом. Этилацетатный экстракт промывали водой, сушили над сульфатом натрия и концентрировали. Очистка хроматографией на окиси алюминия с использованием в качестве элюента смеси этилацетат:гексаны (1:1) привела к 0,37 г (71%) твердого вещества желтого цвета.
FDMS; m/e=413 (M+).
Получение 5
1-(N,N-бисхлорэтиламиноэтокси)-4-трет-бутилдиметилсилилокситиоксантон
Раствор 0,50 г 1-(N,N-бисхлорэтиламиноэтокси)-4-гидрокситиоксантона в 10 мл диметилформамида смешивали с 0,22 г трет-бутилдиметилсилилхлорида, 0,20 г имидазола и каталитическим количеством диметиламинопиридина и перемешивали при комнатной температуре в течение 12 час. К раствору добавляли воду и экстрагировали этилацетатом. Этилацетатный экстракт промывали последовательно насыщенным раствором бикарбоната натрия и водой, сушили над сульфатом натрия, концентрировали и растворяли в смеси этилацетат:гексаны (1:4) и пропускали через слой окиси алюминия. Полученное после концентрирования твердое вещество было чистым и имело вес 0,48 г (70%).
FDMS; m/e=527 (M+).
Пример III
1-(N-хлорэтил-N-метиламиноэтокси)-4-гидрокситиоксантон
Раствор 0,5 г 1,4-дигидрокситиоксантона в 20 мл диметилформамида перемешивали в течение 0,5 час при комнатной температуре с 6 г карбоната калия в условиях отсутствия влаги. Затем гидрохлорид мехлорэтамина (0,4 г) добавляли, и гетерогенную смесь перемешивали в течение 12 час при 50°С. Воду добавляли, и смесь экстрагировали этилацетатом. Этилацетатный раствор промывали водой, сушили сульфатом натрия, пропускали через слой окиси алюминия и концентрировали под вакуумом с получением 0,4 г (55%).
FDMS; m/e= 364 (M+).
Пример IV
Соединения формулы I проверяли in vitro, используя линии лейкозных клеток L1210 мыши, опухоль поджелудочной железы (Pan 03), карциному легкого Льюиса и нормальный фибробласт. Величины IC50 для всех комбинаций, означающих пролекарство формулы I с последующим добавлением активационного лекарственного средства, фторида натрия, были ниже чем 0,4 микромоля для злокачественных тканей. Сравнимые величины были получены для нормальных фибробластов in vitro. В случае проверки полного прото-лекарства без последующей обработки активационным лекарственным средством, величина IC50 составляла 0,1 моль в нормальных клетках. Проверка показала, что активация в значительной степени изменила токсичность прото-лекарства.
Пример V
Изучение активности соединений формулы I осуществляли с использованием трех моделей опухолей мыши: лейкоз, опухоль легкого и поджелудочной железы. Всего 5 животных на опухолевую линию использовали и обработку начинали через две недели после имплантации (поздняя стадия проверки). Прото-лекарство вводили три раза в дозе, составляющей приблизительно 30% от дозы LD50 для мыши при воздействии мехлорэтаминового токсического фрагмента, затем 4 днями позже вводили активационное лекарственное средство, фторид натрия, в дозе (в избытке), превышающей в 5 раз молярный эквивалент.
Эксперимент: Мышам самцам С57 (5/опухолевую линию) имплантировали подкожно приблизительно 1 миллион злокачественных клеток, суспендированных в физиологическом растворе, и опухоли оставляли расти в течение 2 недель, когда они пальпировались. Тестируемое соединение, 0,2 мл, затем вводили ip в виде суспензии, содержащей воду, полиэтиленгликоль (3%) и спирт (5%). Активатор вводили 4 днями позже в виде ip раствора. Еще две обработки были сделаны с недельным перерывом между ними.
Результаты: Обработка опухолей легкого оказалась неэффективной, и животные были умерщвлены до окончания 30-дневного периода изучения. Обработка лейкозных животных привела к излечению 4 животных (из 5 животных), о чем свидетельствовало исчезновение опухоли; трое из пяти животных, которым были перевиты опухолевые клетки поджелудочной железы, излечились после курса лечения. Излеченных животных сохраняли дольше 30-дневного срока изучения. Других животных умерщвляли.
Вторая серия экспериментов продемонстрировала 5/5 излечиваний как в случае лейкоза L1210, так и опухоли поджелудочной железы.
Реферат
Фармацевтическая противоопухолевая лекарственная система включает биологически инертное прото-лекарство и биологически инертное активационное лекарственное средство. После периода отсрочки, который способствует дифференциальному концентрированию прото-лекарств в опухолевых или инвазивных тканях- или клетках-мишенях, нетоксическое лекарственное средство модифицируется посредством активационного лекарственного средства, чтобы избирательно достичь токсических уровней фармакологически активного средства в тканях-мишенях. Новая противоопухолевая система обладает по сравнению с уже известными системами достаточно высокой эффективностью и пониженным уровнем токсичности. 15 н. и 19 з.п. ф-лы.
Формула
(a) тиоксантоновый фрагмент действует как дифференциально концентрирующий фрагмент;
(b) мехлорэтаминовый или подофиллотоксиновый фрагмент действует как токсический фрагмент;
(c) силановый фрагмент действует как покрывающий фрагмент;
(d) активационным лекарственным средством является фтористая соль.
(a) выбором дифференциально концентрирующего фрагмента с помощью дифференциальной ВЭЖХ, дифференциальной хроматографии или in vivo анализа дифференциальной скорости;
(b) выбором токсического фрагмента путем проверки in vitro, проверки in vivo или оценки опубликованных перечней токсических фрагментов;
(c) выбором покрывающего фрагмента путем проверки in vitro, проверки in vivo или оценки опубликованных перечней реагентов с токсическим фрагментом;
(d) выбором активационного лекарственного средства путем проверки in vitro, проверки in vivo или оценки опубликованных перечней реагентов с покрывающим фрагментом; и
(e) связыванием дифференциально концентрирующего фрагмента, токсического фрагмента и покрывающего фрагмента таким образом, чтобы само прото-лекарство как таковое было по существу биологически инертным.

Формула I
где R1 является SiZ3;
R2 означает метил, хлорэтил, гидроксиэтил или бромэтил;
R3 означает хлорэтил, гидроксиэтил или бромметил;
R4 означает Н, SO3Н или таурин;
каждый Z из Z3 независимо означает метил или трет-бутил; и
Х означает углерод, кислород или азот,
и активационным лекарственным средством является фтористая соль.

Формула II
где R5 является SiZ3;
R4 означает Н, SO3Н или таурин;
каждый Z из Z3 независимо означает метил или трет-бутил; и
W означает углерод, кислород или азот,
и активационным лекарственным средством является фтористая соль.
Формула I
где R1 является SiZ3;
R2 означает метил, хлорэтил, гидроксиэтил или бромэтил;
R3 означает хлорэтил, гидроксиэтил или бромметил;
R4 означает Н, SO3Н или таурин;
каждый Z из Z3 независимо означает метил или трет-бутил; и
Х означает углерод, кислород или азот,
и активационным лекарственным средством является фтористая соль.
Формула II
где R5 является SiZ3;
R6 означает Н, SO3Н или таурин;
каждый Z из Z3 независимо означает метил или трет-бутил; и
W означает углерод, кислород или азот,
и активационным лекарственным средством является фтористая соль.
R=EA/EB=(bB/bA)exp[(bB-bA)T],
где R означает отношение констант диффузии клеток типа А и В;
ЕA означает время подвергания клеток А-типа воздействию прото-лекарства;
ЕB означает время подвергания клеток В-типа воздействию прото-лекарства;
bA является константой элиминации клеток А-типа; и
bB является константой элиминации клеток В-типа.
(a) тиоксантоновый фрагмент действует как дифференциально концентрирующий фрагмент;
(b) мехлорэтаминовый или подофиллотоксиновый фрагмент действует как токсический фрагмент; и
(c) силановый фрагмент действует как покрывающий фрагмент, где тиоксантоновый, мехлорэтаминовый или подофиллотоксиновый и силановый фрагменты связаны, с образованием по существу биологически инертного соединения.
(a) выбором дифференциально концентрирующего фрагмента с помощью дифференциальной ВЭЖХ, дифференциальной хроматографии или in vivo анализа дифференциальной скорости;
(b) выбором токсического фрагмента путем проверки in vitro, проверки in vivo или оценки опубликованных перечней токсических фрагментов;
(c) выбором покрывающего фрагмента путем проверки in vitro, проверки in vivo или оценки опубликованных перечней реагентов с токсическим фрагментом;
(d) связыванием дифференциально концентрирующего фрагмента, токсического фрагмента и покрывающего фрагмента таким образом, чтобы прото-лекарство как таковое было по существу биологически инертным.
Формула I
где R1 является SiZ3;
R2 означает метил, хлорэтил, гидроксиэтил или бромэтил;
R3 означает хлорэтил, гидроксиэтил или бромметил;
R4 означает Н, SO3Н или таурин;
каждый Z или Z3 независимо означает метил или трет-бутил; и
Х означает углерод, кислород или азот.
Формула II
где R5 является SiZ3;
R6 означает Н, SO3Н или таурин;
каждый Z из Z3 независимо означает метил или трет-бутил; и
W означает углерод, кислород или азот.
(а) комбинированием тиосалициловой кислоты и гидрохинона в присутствии серной кислоты, с получением 1,4-дигидрокситиоксантона;
(b) комбинированием 1,4-дигидрокситиоксантона, карбоната калия и 1-бром-2-хлорэтана в ацетоне при кипячении с обратным холодильником, с получением 1 -хлорэтокси-4-гидрокситиоксантона;
(c) нагреванием 1-хлорэтокси-4-гидрокситиоксантона в атмосфере азота в присутствии диэтаноламина, добавлением воды и экстрагированием этилацетатом, с получением 1-(N,N-бисдиэтаноламиноэтокси)-4-гидрокситиоксантона;
(d) нагреванием с обратным холодильником 1-(N,N-бисдиэтаноламиноэтокси)-4-гидрокситиоксантона с тионилхлоридом и затем отгонкой избытка тионилхлорида, с получением 1-(N,N-бисхлорэтиламиноэтокси)-4-гидрокситиоксантона, и
(e) перемешиванием 1-(N,N-бисхлорэтиламиноэтокси)-4-гидрокситиоксантона в диметилформамиде с трет-бутилдиметилсилилхлоридом, имидазолом и каталитическим количеством диметиламинопиридина при комнатной температуре и затем проведением экстракции смесью вода-этилацетат, с получением 1-(N,N-бисхлорэтиламиноэтокси)-4-трет-бутилдиметилсилилокситиоксантона.
(a) комбинированием тиосалициловой кислоты и гидрохинона в присутствии серной кислоты, с получением 1,4-дигидрокситиоксантона;
(b) комбинированием 1,4-дигидрокситиоксантона, карбоната калия и 1-бром-2-хлорэтана в ацетоне при кипячении с обратным холодильником, с получением 1-хлорэтокси-4-гидрокситиоксантона;
(c) нагреванием 1-хлорэтокси-4-гидрокситиоксантона в атмосфере азота в присутствии диэтаноламина, добавлением воды и экстрагированием этилацетатом, с получением 1-(N,N-бисдиэтаноламиноэтокси)-4-гидрокситиоксантона;
(d) комбинированием 1-(N,N-бисдиэтаноламиноэтокси)-4-гидрокситиоксантона в пиридине с метансульфонилхлоридом в атмосфере азота, с получением смеси, которую сохраняют при охлаждении, экстрагированием смесью вода-этилацетат с получением твердого вещества, после чего такое твердое вещество растворяют в диметилформамиде, нагревают и перемешивают в атмосфере азота с хлоридом лития, добавлением воды и экстрагированием этилацетатом, с получением 1-(N,N-бисхлорэтиламиноэтокси)-4-гидрокситиоксантона; и
(e) перемешиванием 1-(N,N-бисхлорэтиламиноэтокси)-4-гидрокситиоксантона в диметилформамиде с трет-бутилдиметилсилилхлоридом, имидазолом и каталитическим количеством диметиламинопиридина при комнатной температуре, и затем проведением экстракции смесью вода-этилацетат, с получением 1-(N,N-бисхлорэтиламиноэтокси)-4-трет-бутилдиметилсилилокситиоксантона.
a) комбинирование 1,4-дигидрокситиоксантона в диметилформамиде с карбонатом калия при комнатной температуре в условиях отсутствия влаги и затем добавление гидрохлорида мехлорэтамина, с получением гетерогенной смеси, которую нагревают и перемешивают, и
b) добавление воды к гетерогенной смеси и экстрагирование этилацетатом, с получением 1-(N-хлорэтил-N-метиламиноэтокси)-4-гидрокситиоксантона.
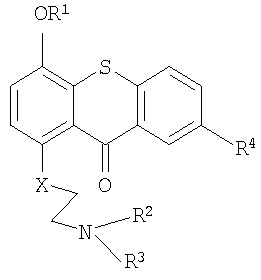
Формула III
где R1 является Н;
R2 означает метил, хлорэтил, гидроксиэтил или бромэтил;
R3 означает хлорэтил, гидроксиэтил или бромметил;
R4 означает В, SO3Н или таурин, и
Х означает углерод, кислород или азот.
Формула VI
где R5 является Н;
R6 означает Н, SO3Н или таурин; и
W означает углерод, кислород или азот.
Комментарии