Новое соединение 2,3,5-замещенного тиофена в качестве ингибитора протеинкиназы - RU2724957C2
Код документа: RU2724957C2
Описание
Область изобретения
Настоящее изобретение относится к новому соединению 2,3,5-замещенного тиофена, обладающему ингибиторной активностью в отношении киназ, противораковому агенту, включающему это соединение в качестве активного ингредиента, и способу получения этого соединения.
Предшествующий уровень техники
Протеинкиназа является ферментом, который катализирует фосфорилирование гидроксильной группы остатков тирозина, серина и теронина в белках и играет важную роль в трансдукции сигналов факторов роста, вызывающих рост, дифференцировку и пролиферацию клеток.
Для поддержания гомеостаза организма система сигнальной трансдукции in vivo нуждается в постоянном сохранении равновесия между активацией и ингибированием. Однако мутация или сверхэкспрессия специфической протеинкиназы нарушает работу системы сигнальной трансдукции в нормальных клетках (обычно в состоянии, когда трансдукция сигнала in vivo является длительной), тем самым вызывая различные заболевания, такие как рак, воспаление, метаболическое заболевание и заболевание головного мозга. Примеры типичной протеинкиназы, вызывающей заболевание, характеризующееся аномальным ростом клеток, включают Raf, KDR, Fms, Tie2, SAPK2a, Ret, Abl, Abl(T315I), ALK, Aurora A, Bmx, CDK/циклин E, Kit, Src, EGFR, EphAl, FGFR3, FLT3, Fms, IGF-1R, IKKb, IR, Itk, JAK2, KDR, Met, mTOR, PDGFRa, Plk1, Ret, Syk, Tie2, TrtB и тому подобные. Поэтому, посредством разработки соединения, обладающего селективной ингибиторной активностью в отношении специфической киназы из числа различных протеинкиназ, проводят исследования по разработке целевого противоракового агента.
Между тем, острый миелоидный лейкоз (ОМЛ) является одним из смертельных заболеваний крови и представляет собой болезнь, при которой клетки крови непрерывно пролиферируют при аномальной дифференцировке. Fms-подобная рецепторная тирозинкиназа 3 (FLT3) обычно экспрессируется в гемопоэтической стволовой клетке и клетке-предшественнике и является рецепторной тирозинкиназой, которая играет важную роль в гемопоэзе. FLT3 является одним из генов, которые наиболее часто мутированы у пациентов с острым миелоидным лейкозом. В отношении примерно 25% пациентов с ОМЛ гены FLT3 мутируют в FLT3-ITD (внутренняя тандемная дупликация FLT3), а мутация FLT3-ITD является одним из основных факторов, которые затрудняют раннюю диагностику. Помимо мутации FLT3-ITD описаны точечные мутации (D835Y, D835V и D835F) аминокислоты в положении D835 в активационной петле. Эти мутации обнаружены примерно у 10% пациентов с ОМЛ, и они вызывают гиперактивность FLT3, делая неактивное состояние FLT3 нестабильным. Гиперактивирующие мутации, такие как FLT3-ITD и точечные мутации FLT3-D835, вызывают ОМЛ посредством непрерывного усиления слабых сигналов, связанных с дифференцировкой и пролиферацией клеток крови. Поэтому, вызывающие ОМЛ мутации FLT3 являются мишенями лекарственных средств при разработке ОМЛ-направленного терапевтического агента.
В качестве новых веществ-кандидатов лекарств, которые используют в качестве мишени мутации FLT3, разработанных к настоящему времени, в клинические испытания взяты лестауртиниб (lestaurtinib), сунитиниб, сорафениб, квизартиниб, тандутиниб (tandutinib) и им подобные. Все вещества-кандидаты, как сообщалось, демонстрировали высокую активность в отношении мутанта FLT-ITD, однако квизартиниб вызывал появление устойчивых мутантов FLT3-ITD+F691L, FLT3-ITD+D835Y, FLT3-ITD+F691L+D835Y (тройные мутанты) и так далее. Поэтому существует острая необходимость в разработке нового соединения, которое также обладает ингибиторной активностью в отношении резистентной к лекарственным средствам точечной мутации FLT3, в качестве целевого терапевтического агента при остром миелоидном лейкозе (ОМЛ).
Подробное описание изобретения
Техническая задача
Таким образом, задача настоящего изобретения состоит в том, чтобы предложить новое соединение 2,3,5-замещенного тиофена, обладающее ингибиторной активностью в отношении протеинкиназ.
Кроме того, задача настоящего изобретения также состоит в том, чтобы предложить фармацевтическую композицию, полезную для лечения, предупреждения и облегчения симптомов раковых заболеваний, содержащую в качестве активного ингредиента новое соединение 2,3,5-замещенного тиофена, его фармацевтически приемлемую соль, его гидрат, его сольват или его изомер.
Кроме того, задача настоящего изобретения также состоит в том, чтобы предложить способ получения вышеупомянутого соединения 2,3,5-замещенного тиофена.
Решение задачи
Для решения вышеупомянутых задач согласно настоящему изобретению предложено соединение, выбранное из группы, состоящей из соединения 2,3,5-замещенного тиофена, представленного следующей химической формулой 1, его фармацевтически приемлемой соли, его гидрата, его сольвата и его изомера.
Химическая формула 1

В химической формуле 1:
Z представляет собой О, S или NH,
Y представляет собой C1-6алкиленовую группу, C2-6алкениленовую группу или C2-6алкиниленовую группу,
А представляет собой C3-8гетероциклоалкильную группу, содержащую один или два атома N, C6-12арильную группу или C3-12гетероарильную группу, содержащую один или два гетероатома, выбранных из N и S,
R1 представляет собой заместитель из группы, состоящей из галогена, нитро, циано, C1-6алкила, C1-6галогеналкила, -(CH2)m-C1-6алкокси (в этом случае m равно целому числу от 1 до 6), групп -С(O)O-C1-6алкил, -S(O)2-С1-6алкил, C3-8циклоалкила, С3-8гетероциклоалкила, содержащего один или два гетероатома, выбранных из О и N, C6-12арила и C3-8гетероарила, содержащего один или два гетероатома, выбранных из О и N, где число заместителей у заместителя R1 равно от 0 до 3, и когда заместитель R1 представляет собой гетероциклоалкил, арил или гетероарил, заместитель R1 может быть не замещен или замещен галогеном, C1-6алкилом, группой C1-6алкил-С(O)- или C1-6алкил-S(O)2-,
R2 и R3 являются одинаковыми или разными и представляют собой атом водорода, атом галогена, гидроксильную группу, C1-6алкильную группу, C3-8циклоалкильную группу, C1-6гидроксиалкильную группу, C1-6галогеналкильную группу, C1-6алкоксигруппу, группу -С(O)-C1-6алкил, группу -S(O)2-C1-6алкил или -NR4R5,
R4 и R5 являются одинаковыми или разными и представляют собой атом водорода, C1-6алкильную группу, C3-8циклоалкильную группу, C3-8гетероциклоалкильную группу, содержащую один или два гетероатома, выбранных из О и N, или С6-12арильную группу, и
n равно целому числу от 1 до 3.
Эффекты изобретения
Соединение 2,3,5-замещенного тиофена по настоящему изобретению способно мощно ингибировать активность протеинкиназ ANKK1, BLK, BUB1, CHEK1, CHEK2, CSF1R, CSK, DAPK1, PDGFRA, PDGFRB, PHKG1, SRC, YANK1, DRAK1, DRAK2, FGR, FLT3, FLT4, FYN, НСК, PRKG2, SYK, TAK1, IRAK1, IRAK4, JAK3, KIT, MAP3K2, MAP3K3, MAP4K2, MAP4K4, RET, RIPK4, TNIK, TRKA, TRKB, YSK4, MEK1, MEK2, MEK5, MERTK, MINK, MKNK2, MLK1, MLK3, MST1, MST2, PAK4, PAK6, ULK1 и ULK2. Поэтому соединение 2,3,5-замещенного тиофена по настоящему изобретению может быть использовано с целью лечения, предупреждения и облегчения симптомов раковых заболеваний, вызванных аномальным клеточным ростом.
Примеры раковых заболеваний, которые можно лечить, предупреждать или симптомы которых можно облегчать с помощью соединения по настоящему изобретению, могут включать рак желудка, рак легкого, рак печени, рак толстой кишки, рак тонкой кишки, рак поджелудочной железы, рак головного мозга, рак костей, меланому, рак молочной железы, склерозирующий аденоз, рак матки, рак шейки матки, рак головы и шеи, рак пищевода, рак щитовидной железы, рак паращитовидных желез, рак почки, саркому, рак предстательной железы, рак уретры, рак мочевого пузыря, злокачественное заболевание системы крови (включая лейкоз, множественную миелому и миелодиспластический синдром), лимфому (включая болезнь Ходжкина и неходжкинскую лимфому), псориаз, фиброаденому и тому подобное.
В частности, соединение 2,3,5-замещенного тиофена по настоящему изобретению обладает активностью подавлять пролиферацию линии клеток лейкоза, несущих мутации FLT3-ITD, и линии клеток Ba/F3, и демонстрирует превосходный уровень супрессорной активности в отношении резистентных к лекарственным средствам точечных мутантов (F691L, D835Y и F691L/D835Y) FLT3, и, таким образом, является эффективным для лечения острого миелоидного лейкоза.
Способ осуществления изобретения
Фармацевтически приемлемая соль соединения, представленного химической формулой 1, по настоящему изобретению может быть получена способом, общепринятым в данной области техники. Фармацевтически приемлемая соль должна быть нетоксичной для человеческого организма и не должна оказывать отрицательного воздействия на биологическую активность и физические и химические свойства исходного соединения. Фармацевтически приемлемая соль включает фармацевтически применимую свободную кислоту, соль присоединения кислоты к основному соединению химической формулы 1, соль щелочного металла (соль натрия и ей подобная), соль щелочноземельного металла (соль калия и ей подобная), соль присоединения органического основания органической соли и карбоновой кислоты химической формулы 1, и соль присоединения аминокислоты. Свободную кислоту, которая может быть использована для получения фармацевтически приемлемой соли, можно подразделить на неорганическую кислоту и органическую кислоту. В качестве неорганической кислоты возможно использование хлорноватой кислоты, серной кислоты, азотной кислоты, фосфорной кислоты, хлорной кислоты, бромноватой кислоты и тому подобного. В качестве органической кислоты возможно использование уксусной кислоты, метансульфоновой кислоты, этансульфоновой кислоты, пара-толуолсульфоновой кислоты, трифторуксусной кислоты, фумаровой кислоты, малеиновой кислоты, малоновой кислоты, фталевой кислоты, янтарной кислоты, молочной кислоты, лимонной кислоты, глюконовой кислоты, винной кислоты, салициловой кислоты, яблочной кислоты, щавелевой кислоты, бензойной кислоты, эмбоновой кислоты, аспарагиновой кислоты, глутаминовой кислоты и тому подобного. Примеры органического основания, которое может быть использовано для получения соли присоединения органического основания, включают трис(гидроксиметил)метиламин, дициклогексиламин и тому подобные. Примеры аминокислоты, которая может быть использована для получения большинства аминокислот, которые принимают участие в образовании соли, включают природную аминокислоту, такую как аланин и глицин.
Соединение химической формулы 1 по настоящему изобретению включает не только вышеупомянутую фармацевтически приемлемую соль, но также и все ее гидраты и сольваты. Вышеупомянутая фармацевтически приемлемая соль может быть получена обычным способом, например посредством растворения вышеупомянутого основного соединения химической формулы 1 в растворителе, который может смешиваться с водой, таком как метанол, этанол, ацетон и 1,4-диоксан, добавления свободной кислоты или свободного основания и затем кристаллизации или перекристаллизации полученной смеси.
Кроме того, соединение химической формулы 1 по настоящему изобретению может иметь один или более чем один центр асимметрии, и в случае такого соединения могут присутствовать энантиомер или диастереомер. Поэтому, настоящее изобретение включает каждый его изомер или смесь изомеров. Разные изомеры могут быть отделены или разделены обычным способом, или любой заданный изомер может быть получен обычным способом синтеза или посредством стереоспецифического или ассиметрического синтеза.
Кроме того, настоящее изобретение включает радиоактивные производные соединения химической формулы 1 по настоящему изобретению, и эти радиоактивные соединения полезны в области биологических исследований.
Заместители, используемые для характеристики соединения по настоящему изобретению, ниже будут описаны более подробно.
"Галоген" или "атом галогена" в настоящем изобретении представляет собой термин, который может быть взаимозаменен и использован, и означает хлор, фтор, бром и йод.
"Алкил" в настоящем изобретении означает линейную, разветвленную или циклическую алифатическую насыщенную углеводородную группу, имеющую от 1 до 10 атомов углерода, предпочтительно от 1 до 6 атомов углерода, и более предпочтительно от 1 до 4 атомов углерода. Конкретные примеры алкильной группы включают метильную группу, этильную группу, нормальную пропильную группу, изопропильную группу, циклопропильную группу, циклопропилметильную группу, нормальную бутильную группу, изобутильную группу, трет-бутильную группу, циклобутильную группу, нормальную пентильную группу, изопентильную группу, неопентильную группу, трет-пентильную группу, циклопентильную группу, нормальную гексильную группу, изогексильную группу, циклогексильную группу, нормальную гептильную группу, нормальную октальную группу и тому подобные.
"Галогеналкильная группа" в настоящем изобретении включает все линейные и разветвленные углеродные цепи, содержащие от 1 до 13 атомов галогена, таких как фтор, хлор, бром и йод, и имеющие от 1 до 6 атомов углерода. Конкретные примеры галогеналкильной группы включают фторметильную группу, трифторметильную группу, 1,2-дихлорэтильную группу, 1,1-дихлорэтильную группу, пентафторэтильную группу и тому подобные.
"Алкоксиалкил" в настоящем изобретении означает алифатическую насыщенную углеводородную группу, в которой один или более чем один алкокси связан с линейной или разветвленной углеродной цепью, определенной выше. Конкретные примеры алкоксиалкильной группы включают метоксиметильную группу, 2-метоксиэтильную группу, 2-этоксиэтильную группу, 3-метоксипропильную группу, 1-метокси-изопропильную группу, 2-метоксибутильную группу, 4-метоксибутильную группу, 2-метил-2-метоксипропильную группу и тому подобные.
"Гетероциклоалкил" в настоящем изобретении означает насыщенную или частично насыщенную 5-10-членную алифатическую циклическую группу, содержащую один или два гетероатома, выбранных из О и N. Конкретные примеры гетероциклоалкильной группы включают тетрагидрофуранильную группу, 2,3-дигидрофуранильную группу, 2,5-дигидрофуранильную группу, пирролидинильную группу, 2,3-дигидропирролидинильную группу, 2,5-дигидропирролидинильную группу, тетрагидро-2H-пиранильную группу, 3,4-дигидро-2H-пиранильную группу, 4H-пиранильную группу, пиперидинильную группу, 1,2,3,4-тетрагидропиридинильную группу, 1,4-дигидропиридинильную группу, пиперазинильную группу, N-защищенный пиперазинил, морфолиногруппу и тому подобные. В качестве N-защитной группы пиперазинила обычно может быть включена алкильная группа, алкилкарбонильная группа и алкилсульфонильная группа.
"Арил" в настоящем изобретении означает моноциклическую, бициклическую или трициклическую углеводородную группу, имеющую от 6 до 12 атомов углерода. Конкретные примеры арильной группы включают фенильную группу, нафталинильную группу и тому подобные.
"Гетероарил" в настоящем изобретении означает моноциклическую, бициклическую или трициклическую ароматическую циклическую группу, содержащую одну или два гетероатома, выбранных из S и N, и имеющую от 4 до 13 атомов углерода. Примеры гетероарила включают тиофенильную группу, пирролильную группу, пиразолильную группу, имидазолильную группу, тиазолильную группу, изотиазолильную группу, пиридинильную группу, пиразинильную группу, пиридазинильную группу, пиримидинильную группу, индолильную группу, изоиндолильную группу, индазолильную группу, бензимидазолильную группу, бензотиазолильную группу, бензизотиазолильную группу, хинолинильную группу, изохинолинильную группу, фталазинильную группу, хиназолинильную группу и тому подобные.
Соединение по настоящему изобретению может иметь следующую химическую формулу 1:
Химическая формула 1
В химической формуле 1:
Z представляет собой О, S или NH,
Y представляет собой C1-6алкиленовую группу, C2-6алкениленовую группу или C2-6алкиниленовую группу,
А представляет собой C3-8гетероциклоалкильную группу, содержащую один или два атома N, С6-12арильную группу или C3-12гетероарильную группу, содержащую один или два гетероатома, выбранных из N и S,
R1 представляет собой заместитель из группы, состоящей из галогена, нитро, циано, C1-6алкила, C1-6галогеналкила, групп -(СН2)m-C1-6алкокси (в этом случае m равно целому числу от 1 до 6), -С(O)O-С1-6алкил, -S(O)2-C1-6алкил, С3-8циклоалкила, C3-8гетероциклоалкила, содержащего один или два гетероатома, выбранных из О и N, C6-12арила и С3-8гетероарила, содержащего один или два гетероатома, выбранных из О и N, где число заместителей у заместителя R1 равно от 0 до 3, и когда заместитель R1 представляет собой гетероциклоалкил, арил или гетероарил, заместитель R1 может быть не замещен или замещен галогеном, C1-6алкилом, группой C1-6алкил-С(O)- или C1-6алкил-S(O)2-,
R2 и R3 являются одинаковыми или разными и представляют собой атом водорода, атом галогена, гидроксильную группу, C1-6алкильную группу, C3-8циклоалкильную группу, C1-6гидроксиалкильную группу, С1-6галогеналкильную группу, C1-6алкоксигруппу, группу -С(O)-C1-6алкил, группу -S(O)2-C1-6алкил или -NR4R5,
R4 и R5 являются одинаковыми или разными и представляют собой атом водорода, C1-6алкильную группу, C3-8циклоалкильную группу, C3-8гетероциклоалкильную группу, содержащую один или два гетероатома, выбранных из О и N, или C6-12арильную группу, и
n равно целому числу от 1 до 3.
Соединение химической формулы 1 предпочтительно представляет собой соединение, в котором
Z представляет собой О,
Y представляет собой C1-6алкиленовую группу или C2-6алкиниленовую группу,
А представляет собой пиперидинильную группу, фенильную группу, тиофенильную группу, индазолильную группу, пиридинильную группу, пиримидинильную группу, пиразинильную группу или пиразолильную группу,
R1 представляет собой 0-3 заместителя, выбранных из группы, состоящей из галогена, нитро, циано, C1-6алкила, С1-6галогеналкила, групп -С(O)O-C1-6алкил, -S(O)2-C1-6алкил, -(СН2)m-C1-6алкокси (в этом случае m равно целому числу от 1 до 6), тетрагидро-2H-пиранила, пиперидинила, 4-(ацетил)-пиперидинила, 4-(С1-6алкилсульфонил)-пиперидинила, пирролидинила и морфолинила,
R2 и R3 представляют собой атом водорода, и
n равно целому числу от 1 до 3.
Соединение химической формулы 1 более предпочтительно представляет собой соединение, в котором
Z представляет собой О, Y представляет собой -СН2СН2- или -С≡С-, R2 и R3 представляют собой атом водорода, n равно целому числу от 1 до 3, и когда А представляет собой фенильную группу, R1 представляет собой 0-3 заместителя, выбранных из группы, состоящей из галогена, нитро, циано, метила, этила, изопропила, трифторметила, метоксикарбонила и этоксикарбонила.
Соединение химической формулы 1 более предпочтительно представляет собой соединение, в котором
Z представляет собой О, Y представляет собой -СН2СН2- или -С≡С-, R2 и R3 представляют собой атом водорода, n равно целому числу от 1 до 3, и когда А представляет собой пиридинильную группу, R1 представляет собой 0-2 заместителя, выбранных из группы, состоящей из галогена, трифторметила, метила, этила, изопропила, пирролидинила, пиперидинила и морфолино.
Соединение химической формулы 1 более предпочтительно представляет собой соединение, в котором
Z представляет собой О, Y представляет собой -СН2СН2- или -С≡С-, R2 и R3 представляют собой атом водорода, n равно целому числу от 1 до 3, и когда А представляет собой пиримидинильную группу, R1 представляет собой 0-2 заместителя, выбранных из группы, состоящей из галогена.
Соединение химической формулы 1 более предпочтительно представляет собой соединение, в котором
Z представляет собой О, Y представляет собой -СН2СН2- или -С≡С-, R2 и R3 представляют собой атом водорода, n равно целому числу от 1 до 3, и когда А представляет собой пиразолильную группу, R1 представляет собой заместитель, который представляет собой 0-2 заместителя, выбранных из группы, состоящей из метила, этила, изопропила, метоксиэтила, этоксиэтила, тетрагидропиранила, пиперидинила, 4-ацетилпиперидинила и 4-метилсульфонилпиперидинила.
Соединение химической формулы 1 более предпочтительно представляет собой соединение, в котором
Z представляет собой О, Y представляет собой -СН2СН2- иил -С≡С-, R1, R2 и R3 представляют собой атом водорода, n равно целому числу от 1 до 3, и А представляет собой пиперидинильную группу, фенильную группу, индазолильную группу, тиофенильную группу, пиразолильную группу, пиразинильную группу, пиридинильную группу или пиримидинильную группу.
Кроме того, соединение более предпочтительно представляет собой соединение 2,3,5-замещенного тиофена следующей химической формулы 1а:
Химическая формула 1а
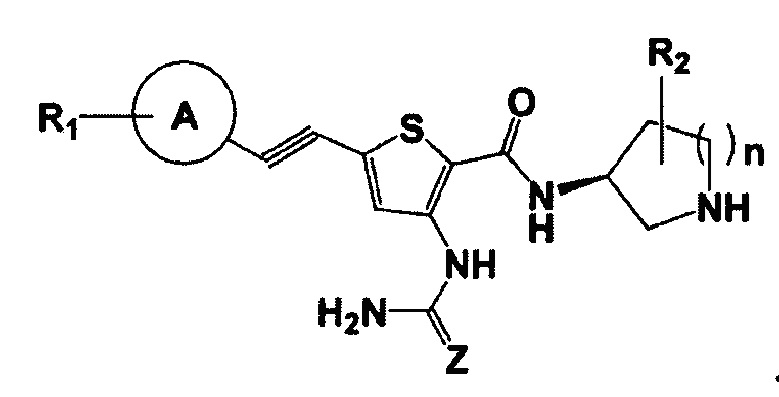
В химической формуле 1а:
Z представляет собой О,
А представляет собой пиперидинильную группу, фенильную группу, тиофенильную группу, индазолильную группу, пиридинильную группу, пиримидинильную группу, пиразинильную группу или пиразолильную группу,
R1 представляет собой 0-3 заместителя, выбранных из группы, состоящей из галогена, нитро, циано, C1-6алкила, C1-6галогеналкила, групп -С(O)O-C1-6алкил, -S(O)2-C1-6алкил, -(CH2)m-C1-6алкокси (в этом случае m равно целому числу от 1 до 6), тетрагидро-2H-пиранила, пиперидинила, 4-(ацетил)-пиперидинила, 4-(C1-6алкилсульфонил)-пиперидинила, пирролидинила и морфолинила,
R2 и R3 представляют собой атом водорода, и
n равно целому числу от 1 до 3.
Кроме того, более конкретными примерами соединения химической формулы 1 являются следующие:
1) этил-(S)-4-((5-(пиперидин-3-илкарбамоил)-4-уреидотиофен-2-ил)этинил)бензоат;
2) (S)-5-(фенилэтинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
3) (S)-N-(пиперидин-3-ил)-5-(пиридин-3-илэтинил)-3-уреидотиофен-2-карбоксамид;
4) (S)-N-(пиперидин-3-ил)-5-(пиридин-4-илэтинил)-3-уреидотиофен-2-карбоксамид;
5) (S)-5-((3-нитрофенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
6) (S)-5-((3-цианофенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
7) (S)-5-((4-нитрофенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
8) (S)-5-((4-хлорфенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
9) (S)-5-((4-фторфенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
10) (S)-N-(пиперидин-3-ил)-5-(пиридин-2-илэтинил)-3-уреидотиофен-2-карбоксамид;
11) (S)-5-((4-цианофенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
12) (S)-5-((1H-индазол-3-ил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
13) (S)-5-((6-фторпиридин-3-ил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
14) (S)-5-((3,4-дифторфенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
15) (S)-5-((1-метил-1H-пиразол-4-ил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
16) (S)-N-(пиперидин-3-ил)-5-((4-(трифторметил)фенил)этинил)-3-уреидотиофен-2-карбоксамид;
17) (S)-N-(пиперидин-3-ил)-5-((6-(трифторметил)пиридин-3-ил)этинил)-3-уреидотиофен-2-карбоксамид;
18) метил-(S)-4-((5-(пиперидин-3-илкарбамоил)-4-уреидотиофен-2-ил)этинил)бензоат;
19) (S)-5-((6-хлорпиридин-3-ил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
20) (S)-5-((5-фторпиридин-3-ил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
21) (S)-5-((2-фторпиридин-4-ил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
22) (S)-5-((3-фторфенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
23) (S)-5-((1-(1-(метилсульфонил)пиперидин-4-ил)-1H-пиразол-4-ил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
24) (S)-N-(пиперидин-3-ил)-5-(пара-толилэтинил)-3-уреидотиофен-2-карбоксамид;
25) (S)-5-((3-бром-4-цианофенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
26) (S)-5-((4-циано-3-фторфенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
27) (S)-5-((3-фтор-4-(трифторметил)фенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
28) (S)-5-((4-хлор-3-цианофенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
29) (S)-5-((2-хлорпиримидин-5-ил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
30) (S)-N-(пиперидин-3-ил)-5-(пиразин-2-илэтинил)-3-уреидотиофен-2-карбоксамид;
31) (S)-5-((4-хлор-3-фторфенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
32) (S)-5-((3,5-дифторфенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
33) (S)-5-((2-фторфенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
34) (S)-5-((2,3-дифторфенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
35) (S)-5-((6-метилпиридин-3-ил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
36) (S)-N-(пиперидин-3-ил)-5-((6-(пирролидин-1-ил)пиридин-3-ил)этинил)-3-уреидотиофен-2-карбоксамид;
37) (S)-5-((6-(пиперидин-1-ил)пиридин-3-ил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
38) (S)-5-((6-морфолинопиридин-3-ил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
39) 5-((1-метил-1H-пиразол-4-ил)этинил)-N-(пирролидин-3-ил)-3-уреидотиофен-2-карбоксамид;
40) 5-((3-фторфенил)этинил)-N-(пирролидин-3-ил)-3-уреидотиофен-2-карбоксамид;
41) (S)-N-(азепан-3-ил)-5-((1-метил-1H-пиразол-4-ил)этинил)-3-уреидотиофен-2-карбоксамид;
42) (S)-5-((1H-пиразол-4-ил)этинил)-N-(азепан-3-ил)-3-уреидотиофен-2-карбоксамид;
43) (S)-N-(азепан-3-ил)-5-((1-этил-1H-пиразол-4-ил)этинил)-3-уреидотиофен-2-карбоксамид;
44) (S)-N-(азепан-3-ил)-5-((1-изопропил-1H-пиразол-4-ил)этинил)-3-уреидотиофен-2-карбоксамид;
45) (S)-N-(азепан-3-ил)-5-((1-(2-метоксиэтил)-1H-пиразол-4-ил)этинил)-3-уреидотиофен-2-карбоксамид;
46) (S)-N-(азепан-3-ил)-5-((1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-ил)этинил)-3-уреидотиофен-2-карбоксамид;
47) (S)-N-(азепан-3-ил)-5-((1-(1-метилсульфонил)пиперидин-4-ил)-1H-пиразол-4-ил)этинил)-3-уреидотиофен-2-карбоксамид;
48) (S)-N-(азепан-3-ил)-5-(тиофен-3-илэтинил)-3-уреидотиофен-2-карбоксамид;
49) (S)-5-((1-(1-(ацетилпиперидин-4-ил)-1H-пиразол-4-ил)этинил)-N-(азепан-3-ил)-3-уреидотиофен-2-карбоксамид;
50) (S)-N-(азепан-3-ил)-5-((3-фторфенил)этинил)-3-уреидотиофен-2-карбоксамид;
51) 5-(2-((1-метил-1H-пиразол-4-ил)этил)-N-(пирролидин-3-ил)-3-уреидотиофен-2-карбоксамид;
52) (S)-N-(азепан-3-ил)-5-(2-(1-метил-1H-пиразол-4-ил)этил)-3-уреидотиофен-2-карбоксамид;
53) (S)-5-(3-фторфенетил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид;
54) (S)-N-(азепан-3-ил)-5-(3-фторфенетил)-3-уреидотиофен-2-карбоксамид или
55) 5-(3-фторфенетил)-N-(пирролидин-3-ил)-3-уреидотиофен-2-карбоксамид.
Кроме того, согласно настоящему изобретению предложен способ получения соединения химической формулы 1. Способ получения по настоящему изобретению будет подробно описан ниже.
Способ получения 1
Согласно способу получения по следующей схеме реакции 1 соединение химической формулы 1 может быть получено путем осуществления 6-стадийного способа получения при использовании соединения следующей химической формулы 2 в качестве исходного вещества.
Схема реакции 1
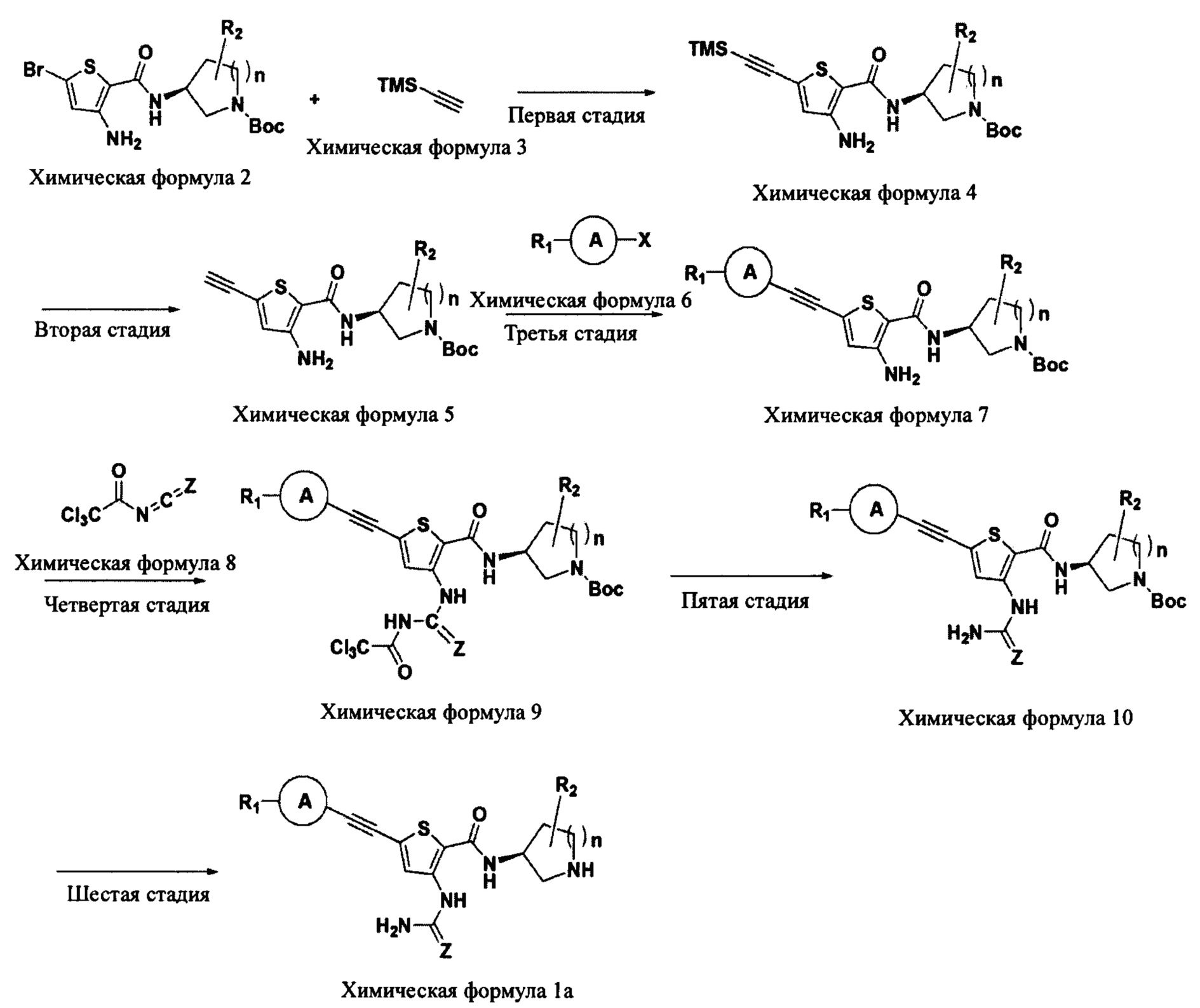
(В схеме реакции 1 R1, R2, А и n являются такими, как они определены в химической формуле 1, соответственно, X означает атом галогена, TMS означает триметилсилильную группу, и Boc означает трет-бутоксикарбонильную группу.)
Способ получения по схеме реакции 1 будет подробно описан для каждой стадии ниже.
Взаимодействие на первой стадии представляет собой процесс введения ацетиленовой группы в положение С5 тиофена посредством взаимодействия 2-(Вос-защищенный карбоксамидо)-3-амино-5-бром-тиофена химической формулы 2 с триметилсилилацетиленом химической формулы 3.
Конкретно, взаимодействие на первой стадии проводят путем нагревания смеси до температуры 50°C-90°C при добавлении трифенилфосфинпалладия (Pd(PPh3)4) и йодида меди (CuI) и в присутствии аминного основания. В этом случае аминное основание может быть выбрано из моно-, ди- или три-C1-6алкиламина и тому подобного, и предпочтительно может быть использован триалкиламин, такой как триэтиламин (TEA) и диизопропилэтиламин (DIPEA). В качестве растворителя для реакции может быть использован обычный органический растворитель, и в Примерах по настоящему изобретению конкретно представлены примеры, в которых обычно используют диметилформамид (DMF), но растворитель по настоящему изобретению не ограничивается им.
Взаимодействие на второй стадии представляет собой процесс удаления триметилсилильной (TMS) защитной группы с соединения химической формулы 4.
Конкретно, взаимодействие на второй стадии проводят при комнатной температуре при использовании неорганического основания. В этом случае неорганическое основание может быть выбрано из гидроксида, оксида, карбоната, сульфата и тому подобного щелочного металла или щелочноземельного металла, и предпочтительно может быть использован карбонат щелочного металла, такой как карбонат калия. Взаимодействие может быть проведено в смешанном растворителе спирт-тетрагидрофуран, использованном в качестве растворителя для реакции, а в качестве спирта типично может быть использован метанол или этанол. Смешанный растворитель может быть получен посредством смешивания подходящим образом спирта и тетрагидрофурана в объемном соотношении в диапазоне от 1:2 до 2:1.
Взаимодействие на третьей стадии представляет собой процесс введения группы R путем взаимодействия соединения химической формулы 5 с галогенидом химической формулы 6.
Конкретно, взаимодействие на третьей стадии проводят путем нагревания смеси до температуры 70°C-120°C при добавлении трифенилфосфинпалладия (Pd(PPh3)4) и йодида меди (CuI) и в присутствии аминного основания. В этом случае аминное основание может быть выбрано из моно-, ди- или три-C1-6алкиламина и тому подобного, и предпочтительно может быть использован триалкиламин, такой как триэтиламин и диизопропилэтиламин. В качестве растворителя для реакции может быть использован обычный органический растворитель, и в Примерах по настоящему изобретению конкретно представлены примеры, в которых обычно используют диметилформамид (DMF), но растворитель по настоящему изобретению не ограничивается им.
Взаимодействие на четвертой стадии представляет собой процесс введения мочевинной группы в положение C3 тиофена путем взаимодействия соединения химической формулы 7 с соединением химической формулы 8.
Конкретно, взаимодействие на четвертой стадии проводят при комнатной температуре. В качестве растворителя для реакции может быть использован обычный органический растворитель, и в Примерах по настоящему изобретению конкретно представлены примеры, в которых обычно используют тетрагидрофуран (THF), но растворитель по настоящему изобретению не ограничивается им.
Взаимодействие на пятой стадии представляет собой процесс удаления трихлорацетильной (Cl3CC(О)-) группы с соединения химической формулы 9.
Конкретно, взаимодействие на пятой стадии проводят при комнатной температуре в присутствии аминного основания. В этом случае аминное основание может быть выбрано из моно-, ди- или три-C1-6алкиламина и тому подобного, и предпочтительно может быть использован триалкиламин, такой как триэтиламин и диизопропилэтиламин. В качестве растворителя для реакции может быть использован обычный органический растворитель, и в Примерах по настоящему изобретению конкретно представлены примеры, в которых обычно используют спиртовой растворитель, такой как метанол, но растворитель по настоящему изобретению не ограничивается им.
Взаимодействие на шестой стадии представляет собой процесс удаления трет-бутоксикарбонильной (Boc) группы с соединения химической формулы 10.
Конкретно, взаимодействие на шестой стадии проводят при комнатной температуре при использовании трифторуксусной кислоты (TFA). В качестве растворителя для реакции может быть использован обычный органический растворитель, и в Примерах по настоящему изобретению конкретно представлены примеры, в которых обычно используют дихлорметан (МС), но растворитель по настоящему изобретению не ограничивается им.
Способ получения 2
Согласно способу получения по следующей схеме реакции 2 соединение химической формулы 1 может быть получено путем проведения 4-стадийного способа получения при использовании соединения следующей химической формулы 2 в качестве исходного вещества.
Схема реакции 2

(В схеме реакции 2 R1, R2, А и n являются такими, как они определены в химической формуле 1, соответственно, и Boc означает трет-бутоксикарбонильную группу.)
Способ получения по схеме реакции 2 будет подробно описан для каждой стадии ниже.
Взаимодействие на стадии А представляет собой процесс введения ацетиленовой группы в положение С5 тиофена путем взаимодействия 2-(Вос защищенный карбоксамидо)-3-амино-5-бром-тиофена химической формулы 2 с ацетиленовым соединением химической формулы 11.
Конкретно, взаимодействие на стадии А проводят путем нагревания смеси до температуры 60°C-120°C при добавлении трифенилфосфинпалладия (Pd(PPh3)4) и йодида меди (CuI) и в присутствии аминного основания. В этом случае аминное основание может быть выбрано из моно-, ди- или три-C1-6алкиламина и тому подобного, и предпочтительно может быть использован триалкиламин, такой как триэтиламин и диизопропилэтиламин. В качестве растворителя для реакции может быть использован обычный органический растворитель, и в Примерах по настоящему изобретению конкретно представлены примеры, в которых обычно используют диметилформамид (DMF), но растворитель по настоящему изобретению не ограничивается им.
Взаимодействие на стадии В представляет собой процесс введения мочевинной группы в положение С3 тиофена путем взаимодействия соединения химической формулы 7 с соединением химической формулы 8.
Конкретно, взаимодействие на стадии В проводят при комнатной температуре. В качестве растворителя для реакции может быть использован обычный органический растворитель, и в Примерах по настоящему изобретению конкретно представлены примеры, в которых обычно используют тетрагидрофуран (THF), но растворитель по настоящему изобретению не ограничивается им.
Взаимодействие на стадии С представляет собой процесс удаления трихлорацетильной (Cl3CC(О)-) группы с соединения химической формулы 9.
Конкретно, взаимодействие на стадии С проводят при комнатной температуре в присутствии аминного основания. В этом случае аминное основание может быть выбрано из моно-, ди- или три-C1-6алкиламина и тому подобного, и предпочтительно может быть использован триалкиламин, такой как триэтиламин и диизопропилэтиламин. В качестве растворителя для реакции может быть использован обычный органический растворитель, и в Примерах по настоящему изобретению конкретно представлены примеры, в которых обычно используют спиртовой растворитель, такой как метанол, но растворитель по настоящему изобретению не ограничивается им.
Взаимодействие на стадии D представляет собой процесс удаления трет-бутоксикарбонильной (Boc) группы с соединения химической формулы 10.
Конкретно, взаимодействие на стадии D проводят при комнатной температуре при использовании трифторуксусной кислоты (TFA). В качестве растворителя для реакции может быть использован обычный органический растворитель, и в Примерах по настоящему изобретению конкретно представлены примеры, в которых обычно используют дихлорметан (МС), но растворитель по настоящему изобретению не ограничивается им.
Способ получения 3
Согласно способу получения по следующей схеме реакции 3 соединение следующей химической формулы 1а может быть превращено в соединение, содержащее группу -НС=СН- или -СН2-СН2-, путем восстановления ацетиленовой группы в соединении химической формулы 1а.
Схема реакции 3

(В схеме реакции 3 R1, R2, А и n являются такими, как они определены в химической формуле 1, соответственно.)
Реакция восстановления по схеме реакции 3 может быть проведена при комнатной температуре в потоке газообразного водорода на палладиевом (Pd) катализаторе. В качестве растворителя для реакции может быть использован обычный органический растворитель, и в Примерах по настоящему изобретению конкретно представлены примеры, в которых обычно используют спиртовой растворитель, такой как метанол, но растворитель по настоящему изобретению не ограничивается им.
Способ получения 4
Соединение химической формулы 2, используемое в качестве исходного вещества в схеме реакции 1 и схеме реакции 2, может быть получено способом получения согласно следующей схеме реакции 4.
Схема реакции 4
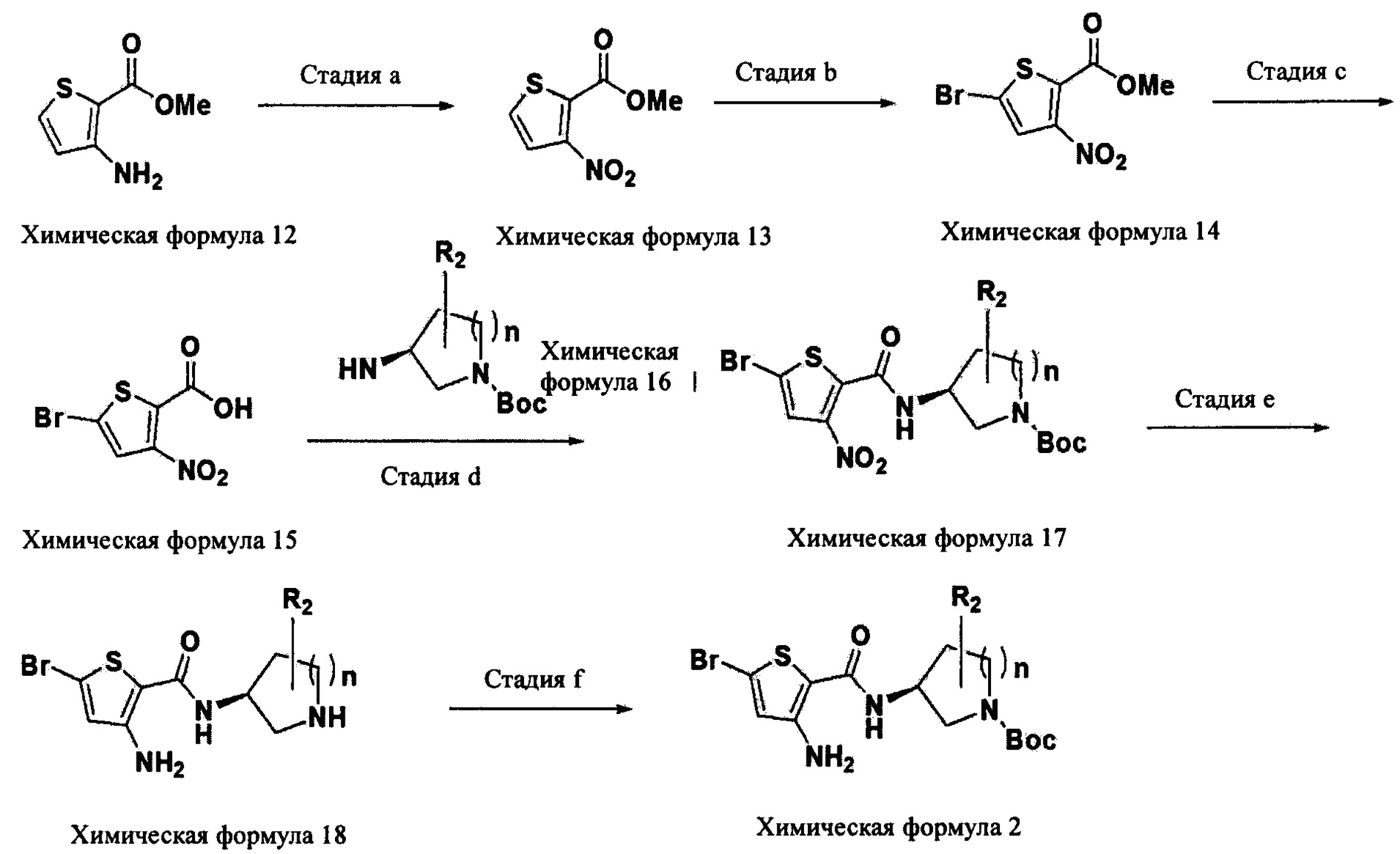
(В схеме реакции 4 R2 и n являются такими, как они определены в химической формуле 1, соответственно.)
Способ получения по схеме реакции 4 будет подробно описан для каждой стадии ниже.
Взаимодействие на стадии а представляет собой процесс нитрования аминогруппы тиофена химической формулы 12.
Конкретно, взаимодействие на стадии а проводят при температуре -20°C-0°C при использовании нитрата натрия (NaNO2), соляной кислоты (HCl) и тетрафторбората натрия (NaBF4), и в качестве растворителя для реакции может быть использована вода. Затем водный раствор соли диазония, полученный путем взаимодействия меди и нитрата натрия, может быть добавлен в реакционный раствор, и путем проведения взаимодействия в реакционной смеси при комнатной температуре может быть получен тиофен химической формулы 13, в котором аминогруппа замещена на нитрогруппу.
Взаимодействие на стадии b представляет собой процесс введения атома брома в положение С5 тиофена химической формулы 13.
Конкретно, взаимодействие на стадии b проводят при комнатной температуре при использовании бромирующего реагента, такого как N-бромсукцинимид, трифторуксусной кислоты (TFA) и серной кислоты.
Взаимодействие на стадии с представляет собой процесс превращения метоксикарбонила (-СООМе) в положении С1 тиофена химической формулы 14 в карбоксильную группу (-СООН) посредством гидролиза метоксикарбонила.
Конкретно, взаимодействие на стадии с проводят при комнатной температуре при использовании гидроксида щелочного металла, включающего гидроксид натрия. Взаимодействие может быть проведено в смешанном растворителе спирт-тетрагидрофуран, использованном в качестве растворителя для реакции, а в качестве спирта типично может быть использован метанол или этанол. Смешанный растворитель может быть получен посредством смешивания подходящим образом спирта и тетрагидрофурана в объемном соотношении в диапазоне от 1:2 до 2:1.
Взаимодействие на стадии d представляет собой процесс введения амидогруппы в положение С1 тиофена путем взаимодействия тиофена химической формулы 15 с амином химической формулы 16.
Конкретно, взаимодействие на стадии d проводят при температуре 30°C-60°C в присутствии гексафторфосфата 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиний-3-оксида (HATU) и аминного основания. В этом случае аминное основание может быть выбрано из моно-, ди- или три-C1-6алкиламина и тому подобного, и предпочтительно может быть использован триалкиламин, такой как триэтиламин и диизопропилэтиламин. В качестве растворителя для реакции может быть использован обычный органический растворитель, и в Примерах по настоящему изобретению конкретно представлены примеры, в которых обычно используют диметилформамид (DMF), но растворитель по настоящему изобретению не ограничивается им.
Взаимодействие на стадии е представляет собой процесс аминирования нитрогруппы в положении С3 тиофена химической формулы 17.
Конкретно, взаимодействие на стадии е проводят при температуре 30°C-60°C при использовании хлорида олова (SnCl2), нитрата натрия (NaNO2), соляной кислоты (HCl) и тетрафторбората натрия (NaBF4). В качестве растворителя для реакции может быть использован обычный органический растворитель, и в Примерах по настоящему изобретению конкретно представлены примеры, в которых обычно используют этилацетат (ЕА), но растворитель по настоящему изобретению не ограничивается им.
Взаимодействие на стадии f представляет собой процесс защиты аминогруппы гетерокольца в положении С2 тиофена химической формулы 18.
Конкретно, при взаимодействии на стадии f трет-бутоксикарбонил (Boc) вводят в качестве защитной группы амина. То есть, тиофен химической формулы 17 и ди-трет-бутилдикарбонат подвергают взаимодействию при комнатной температуре в присутствии аминного основания. В этом случае аминное основание может быть выбрано из моно-, ди- или три-C1-6алкиламина и тому подобного, и предпочтительно может быть использован триалкиламин, такой как триэтиламин и диизопропилэтиламин. В качестве растворителя для реакции может быть использован обычный органический растворитель, и в Примерах по настоящему изобретению конкретно представлены примеры, в которых обычно используют дихлорметан (МС), но растворитель по настоящему изобретению не ограничивается им.
Кроме того, согласно настоящему изобретению предложена фармацевтическая композиция для лечения, предупреждения и облегчения симптомов раковых заболеваний, содержащая в качестве активного ингредиента соединение химической формулы 1, его фармацевтически приемлемую соль, его сольват и его гидрат. Примеры раковых заболеваний, которые можно лечить и предупреждать с помощью соединения по настоящему изобретению, могут включать рак желудка, рак легкого, рак печени, рак толстой кишки, рак тонкой кишки, рак поджелудочной железы, рак головного мозга, рак костей, меланому, рак молочной железы, склерозирующий аденоз, рак матки, рак шейки матки, рак головы и шеи, рак пищевода, рак щитовидной железы, рак паращитовидных желез, рак почки, саркому, рак предстательной железы, рак уретры, рак мочевого пузыря, злокачественное заболевание системы крови (включая лейкоз, множественную миелому и миелодиспластический синдром), лимфому (включая болезнь Ходжкина и неходжкинскую лимфому), псориаз, фиброаденому и тому подобное.
В частности, соединение химической формулы 1 подавляет пролиферацию линии клеток лейкоза, несущих мутации FLT3-ITD, и линии клеток Ba/F3, и одновременно демонстрирует превосходный уровень супрессорной активности в отношении точечных мутантов FLT3, например мутантов гена-привратника, мутантов D835 и мутантов ITD. Таким образом, соединение химической формулы 1 может быть использовано особенно эффективно в качестве профилактического агента или терапевтического агента для острого миелоидного лейкоза.
Фармацевтическая композиция по настоящему изобретению может быть приготовлена в виде обычного препарата в области фармацевтики, например препарата для перорального применения или парентерального применения, такого как таблетка, капсула, пастилка, жидкость или суспензия, содержащего в качестве активного ингредиента соединение химической формулы 1, его фармацевтически приемлемую соль, его сольват или его гидрат, и с добавлением обычного нетоксичного фармацевтически приемлемого носителя, наполнителя, эксципиента и тому подобного.
Примеры эксципиента, который может быть использован для фармацевтической композиции по настоящему изобретению, включают подсластитель, связующее вещество, растворитель, вспомогательное вещество, улучшающее растворимость, смачивающее вещество, эмульгатор, изотонический агент, адсорбент, разрыхлитель, антиоксидант, консервант, смазывающее вещество, наполнитель, ароматизатор и тому подобное. Примеры таких веществ включают лактозу, декстрозу, сахарозу, маннит, сорбит, целлюлозу, глицин, диоксид кремния, тальк, стеариновую кислоту, стерин, стеарат магния, алюмосиликат магния, крахмал, желатин, трагакантовую камедь, аргинин, альгинат натрия, метилцеллюлозу, карбоксиметилцеллюлозу натрия, агар, воду, этанол, полиэтиленгликоль, поливинилпирролидон, хлорид натрия, хлорид кальция, апельсиновый экстракт, земляничный экстракт, ванильный ароматизатор и тому подобное.
Кроме того, доза соединения по настоящему изобретению, вводимая в человеческий организм, может варьироваться в зависимости от возраста, массы тела, пола, лекарственной формы, состояния здоровья и степени заболевания пациента, и обычно составляет от 0,01 до 1000 мг/сутки из расчета массы тела взрослого пациента 70 кг, и соединение может быть введено раздельными дозами от одного до нескольких раз в сутки через заранее определенные промежутки времени на усмотрение лечащего врача или фармацевта.
Настоящее изобретение, описанное выше, будет описано подробно со ссылкой на следующие примеры, экспериментальные примеры и примеры получения, но следующие примеры, экспериментальные примеры и примеры получения только иллюстрируют настоящее изобретение и не ограничивают объем настоящего изобретения.
Примеры
Соединения, синтезированные в Примерах по настоящему изобретению, очищали или подвергали структурному анализу в следующих условиях ВЭЖХ (высокоэффективная жидкостная хроматография).
(1) ВЭЖХ условия 1 для структурного анализа
- Элюент А: 0,1% трифторуксусная кислота (TFA)/вода
- Элюент В: CH3CN
- Колонка: колонка YMC-Park Pro C18, 150×4,6 мм
- условия элюирования: элюирование в течение 7 минут при изменении концентрации элюента В от 5 до 100% при скорости потока 1,0 мл/мин.
(2) ВЭЖХ условия 2 для структурного анализа
- Элюент А: 0,1% трифторуксусная кислота (TFA)/вода
- Элюент В: CH3CN
- Колонка: колонка Kinetex 2,6 мкм бифенил
- условия элюирования: элюирование в течение 4,5 минуты при изменении концентрации элюента В от 5 до 100% при скорости потока 1,2 мл/мин.
(3) ВЭЖХ условия для очистки
- Элюент А: 0,1% трифторуксусная кислота (TFA)/вода
- Элюент В: CH3CN
- Колонка: колонка Luna 10 мкм С18, 250×21,2 мм.
Пример получения 1. Получение трет-бутил-(S)-3-(3-амино-5-бромтиофен-2-карбоксамидо)пиперидин-1-карбоксилата

Стадия 1. Получение метил-3-нитротиофен-2-карбоксилата
В суспензию, полученную посредством добавления концентрированной соляной кислоты (26 мл; 842 ммоль) в метил-3-аминотиофен-2-карбоксилат (15,7 г; 100 ммоль), добавляли воду (24 мл) и полученную смесь перемешивали при комнатной температуре в течение 45 минут. После охлаждения реакционной смеси до -10°C в нее медленно в течение 20 минут добавляли раствор нитрата натрия (NaNO2; 7,2 г; 110 ммоль) в воде (16 мл). После завершения добавления реакционную смесь перемешивали при 0°C в течение 1 часа и затем добавляли раствор тетрафторбората (16 г; 150 ммоль) в воде (32 мл). Полученную соль отфильтровывали, последовательно промывали охлажденным с помощью льда 5% водным раствором тетрафторбората натрия, этанолом и диэтиловым эфиром, и затем сушили. Затем после добавления активированного медного порошка (медная бронза; 16 г; 300 ммоль) в раствор нитрата натрия (NaNO2; 80 г; 1,160 ммоль) в воде (160 мл), интенсивно перемешивая полученную смесь, получили растворенную в воде (80 мл) соль диазония и затем медленно добавляли ее в реакционный раствор при комнатной температуре в течение 1 часа или более. После завершения добавления смесь дополнительно перемешивали в течение 1 часа и затем разбавляли этилацетатом и фильтровали через целит. Органический слой извлекали посредством отделения водного слоя и органический слой и водный слой снова экстрагировали этилацетатом. Органические слои собирали, сушили над сульфатом натрия, концентрировали и затем очищали посредством ЖХСД (жидкостная хроматография среднего давления) с получением белого твердого целевого соединения (15,1 г; 72%).
1Н ЯМР (400 МГц, DMSO-d6) δ 8.06 (d, 2Н), 3.86 (s, 3Н).
Стадия 2. Получение метил-5-бром-3-нитротиофен-2-карбоксилата
После добавления концентрированной серной кислоты (17,1 мл; 321 ммоль) в смешанный раствор трифторуксусной кислоты (86 мл; 1,122 ммоль) и метил-3-нитротиофен-2-карбоксилата (30 г; 160 ммоль) в него медленно добавляли N-бромсукцинимид (NBS; 31,4 г; 176 ммоль) при 0°C в течение более чем 40 минут. После перемешивания раствора реакционной смеси в течение 30 минут температуру повышали до комнатной температуры и раствор реакционной смеси вливали в ледяную воду. Полученный осадок отфильтровывали и извлекали и затем промывали ледяной водой и сушили с получением желтого твердого целевого соединения (34 г; 80%).
1Н ЯМР (400 МГц, DMSO-d6) δ 7.99 (s, 1Н), 3.85 (s, 3Н).
Стадия 3. Получение 5-бром-3-нитротиофен-2-карбоновой кислоты
После растворения метил-5-бром-3-нитротиофен-2-карбоксилата (10 г; 37,6 ммоль) в смешанном растворе тетрагидрофуран/метанол (1:1; 40 мл) в него по каплям добавляли 1 н. раствор гидроксида натрия (41,3 мл; 41,3 ммоль) и затем полученную смесь перемешивали при комнатной температуре в течение 1 часа. После доведения значения рН до 5 посредством добавления в реакционную смесь 2 н. водного раствора соляной кислоты смесь экстрагировали этилацетатом. Экстрагированный органический слой сушили над сульфатом магния и концентрировали с получением серого твердого целевого соединения (8,0 г; 84%).
1Н ЯМР (400 МГц, DMSO-d6) δ 7.93 (s, 1Н).
Стадия 4. Получение трет-бутил-(S)-3-(5-бром-3-нитротиофен-2-карбоксамидо)пиперидин-1-карбоксилата
(S)-1-Вос-3-аминопиперидин (3,57 г; 17,85 ммоль) добавляли по каплям в раствор 5-бром-3-нитротиофен-2-карбоновой кислоты (4,5 г; 17,85 ммоль), гексафторфосфата 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиний-3-оксида (HATU; 20,37 г; 53,6 ммоль), диизопропилэтиламина (DIPEA; 11,54 г; 89 ммоль) и диметилформамида (50 мл) и полученную смесь перемешивали при 40°C в течение 15 часов. Органический слой собирали посредством экстрагирования реакционной смеси этилацетатом и насыщенным водным раствором бикарбоната натрия. Собранный органический слой промывали рассолом, сушили над сульфатом натрия, концентрировали при пониженном давлении и очищали посредством ЖХСД с получением желтого твердого целевого соединения (6,2 г; 80%).
1Н ЯМР (400 МГц, DMSO-d6) δ 8.86 (d, 1Н), 7.88 (s, 1H), 3.82 (m, 1H), 2.94 (m, 2H), 1.85 (m, 1H), 1.67 (m, 1H), 1.40 (s, 9H), 1.39 (m, 4H).
Стадия 5. Получение (S)-3-амино-5-бром-N-(пиперидин-3-ил)тиофен-2-карбоксамида
трет-Бутил-(S)-3-(5-бром-3-нитротиофен-2-карбоксамидо)пиперидин-1-карбоксилат (8,6 г; 19,8 ммоль) и SnCl2⋅2H2O (22,34 г; 99 ммоль) растворяли в этилацетате (30 мл) и затем полученную смесь перемешивали при 55°C в течение 5 часов. После снижения температуры реакционного раствора до комнатной температуры в него добавляли раствор гидроксида аммония до тех пор, пока рН не достигло значения 5. рН доводили до значения 7 посредством добавления безводного карбоната натрия в реакционную смесь. Реакционную смесь фильтровали через целит и несколько раз растирали с этилацетатом. Фильтрат концентрировали при пониженном давлении с получением целевого соединения (5,0 г; 83%), которое использовали в следующей реакции без очистки.
MS (m/z): 304 [М+1].
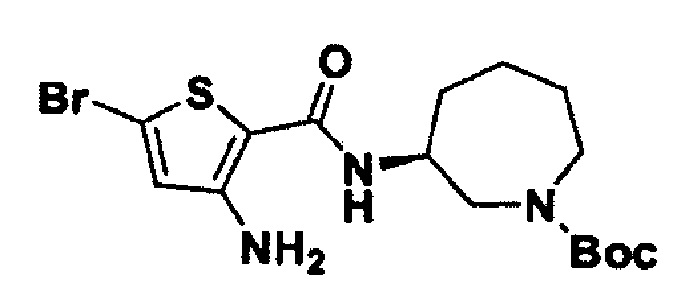
Стадия 6. Получение трет-бутил-(S)-3-(3-амино-5-бромтиофен-2-карбоксамидо)пиперидин-1-карбоксилата
(S)-3-Амино-5-бром-N-(пиперидин-3-ил)тиофен-2-карбоксамид (5,0 г; 16,4 ммоль) растворяли в дихлорметане (25 мл) и затем по каплям последовательно добавляли диизопропилэтиламин (DIPEA; 6,37 г; 49,3 ммоль) и ди-трет-бутилдикарбонат (3,5 г; 18,1 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем экстрагировали при использовании этилацетата и воды. Органический слой промывали рассолом, сушили над сульфатом натрия, фильтровали и затем концентрировали при пониженном давлении с получением целевого соединения (6,4 г; 96%).
MS (m/z): 404 [М+1].
Пример получения 2. Получение трет-бутил-3-(3-амино-5-бромтиофен-2-карбоксамидо)пирролидин-1-карбоксилата

Целевое соединение получали посредством повторения тех же способов, что и в примере получения 1, за исключением того, что вместо (S)-1-Вос-3-аминопиперидина, использованного на стадии 4 примера получения 1, использовали 1-Вос-3-аминопирролидин.
MS (m/z): 390 [М+1].
Пример получения 3. Получение трет-бутил-(S)-3-(3-амино-5-бромтиофен-2-карбоксамидо)азепан-1-карбоксилата
Целевое соединение получали путем повторения тех же способов, что и в примере получения 1, за исключением того, что вместо (S)-1-Вос-3-аминопиперидина, использованного на стадии 4 примера получения 1, использовали трет-бутил-(S)-3-аминоазепан-1-карбоксилат.
MS (m/z): 418 [М+1].
Пример 1. Получение этил-(S)-4-((5-(пиперидин-3-илкарбамоил)-4-уреидотиофен-2-ил)этинил)бензоата

Стадия 1. Получение трет-бутил-(S)-3-(3-амино-5-((4-(этоксикарбонил)фенил)-этинил)тиофен-2-карбоксамидо)пиперидин-1-карбоксилата
трет-Бутил-(S)-3-(3-амино-5-бромтиофен-2-карбоксамидо)пиперидин-1-карбоксилат (95,8 мг; 0,190 ммоль), этил-4-этинилбензоат (30,0 мг; 0,172 ммоль) и диизопропилэтиламин (38 мг; 0,293 ммоль) растворяли в диметилформамиде (2 мл). Pd(PPh3)4 (9,96 мг; 8,62 мкмоль). CuI (3,28 мг; 0,017 ммоль) добавляли в реакционный раствор и полученную смесь перемешивали при 100°C в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры и затем экстрагировали этилацетатом и водой, собирая органический слой. Собранный органический слой промывали рассолом, сушили над сульфатом натрия, концентрировали при пониженном давлении и очищали посредством ЖХСД с получением целевого соединения (61,3 мг; 71,5%).
MS (m/z): 498 [М+1].
Стадия 2. Получение трет-бутил-(S)-3-(5-((4-(этоксикарбонил)фенил)этинил)-3-(3-(2,2,2-трихлорацетил)уреидо)тиофен-2-карбоксамидо)пиперидин-1-карбоксилат
Трихлорацетилизоцианат (26,0 мг; 0,138 ммоль) медленно по каплям добавляли в раствор трет-бутил-(S)-3-(3-амино-5-((4-(этоксикарбонил)фенил)этинил)тиофен-2-карбоксамидо)пиперидин-1-карбоксилата (61,3 мг; 0,123 ммоль) в тетрагидрофуране (1,15 мл) и полученную смесь перемешивали при комнатной температуре в течение 2,5 часов. После завершения взаимодействия добавляли избыток гексана и полученную смесь перемешивали в течение 1 часа. Полученное твердое вещество отфильтровывали и промывали гексаном с получением твердого целевого соединения (85 мг; 100%).
MS (m/z): 687 [М+1].
Стадия 3. Получение трет-бутил-(S)-3-(5-((4-(этоксикарбонил)фенил)этинил)-3-уреидотиофен-2-карбоксамидо)пиперидин-1-карбоксилата
Триэтиламин (31,0 мг; 0,310 ммоль) добавляли в раствор трет-бутил-(S)-3-(5-((4-(этоксикарбонил)фенил)этинил)-3-(3-(2,2,2-трихлорацетил)уреидо)тиофен-2-карбоксамидо)пиперидин-1-карбоксилата (85 мг; 0,124 ммоль) в метаноле (2,5 мл) и полученную смесь перемешивали при комнатной температуре в течение 12 часов. После завершения взаимодействия растворитель удаляли при пониженном давлении и остаток экстрагировали при использовании этилацетата и насыщенного раствора NaHCO3. Собранный органический слой промывали рассолом, сушили над сульфатом натрия, концентрировали при пониженном давлении и очищали посредством ЖХСД с получением целевого соединения (36,9 мг; 55%).
MS (m/z): 541 [М+1].
Стадия 4. Получение этил-(S)-4-((5-(пиперидин-3-илкарбамоил)-4-уреидотиофен-2-ил)этинил)бензоата
После растворения трет-бутил-(S)-3-(5-((4-(этоксикарбонил)фенил)этинил)-3-уреидотиофен-2-карбоксамидо)пиперидин-1-карбоксилата (36,9 мг; 0,068 ммоль) в дихлорметане (8 мл) в раствор по каплям добавляли трифторуксусную кислоту (5,96 г; 52,2 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 30 минут. После завершения взаимодействия растворитель удаляли при пониженном давлении и остаток экстрагировали при использовании этилацетата и насыщенного раствора NaHCO3. Собранный органический слой промывали рассолом, сушили над сульфатом натрия и затем концентрировали при пониженном давлении. Остаток перекристаллизовывали при использовании дихлорметана и гексана с получением твердого целевого соединения (9,1 мг; 30,3%).
1Н ЯМР (400 МГц, DMSO-d6) δ 9.99 (br s, 1Н), 8.16 (s, 1H), 8.09 (s, 1H), 8.01 (d, 1H), 7.87-7.84 (m, 2H), 7.61 (t, 1H), 6.67 (br s, 1H), 4.34 (q, 2H), 3.77-3.75 (m, 1H), 2.94-2.91 (m, 1H), 2.79-2.67 (m, 1H), 2.45-2.32 (m, 2H), 1.83-1.80 (m, 1H), 1.63-1.41 (m, 3H), 1.34 (t, 3H), 1.17 (br s, 1H). MS (m/z): 440 [М+1]. tR (время удерживания) составляет 2,439 мин (ВЭЖХ условия 2).
Пример 2. Получение (S)-5-(фенилэтинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида

Стадия 1. Получение трет-бутил-(S)-3-(3-амино-5-((триметилсилил)этинил)тиофен-2-карбоксамидо)пиперидин-1-карбоксилата
После растворения трет-бутил-(S)-3-(3-амино-5-бромтиофен-2-карбоксамидо)-пиперидин-1-карбоксилата (4,0 г; 9,89 ммоль), триметилсилилацетилена (1,07 г; 10,9 ммоль) и диизопропилэтиламина (1,70 г; 16,82 ммоль) в ацетонитриле (76 мл) через раствор пропускали азот в течение 10 минут. После добавления Pd(PPh3)4 (21,4 мг; 0,019 ммоль) и CuI (7,1 мг; 0,037 ммоль) полученную смесь перемешивали при 80°C в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры и затем экстрагировали этилацетатом и водой. Собранный органический слой промывали рассолом, сушили над сульфатом натрия, концентрировали при пониженном давлении и очищали посредством ЖХСД с получением целевого соединения (3,0 г; 72%).
MS (m/z): 422 [М+1].
Стадия 2. Получение трет-бутил-(S)-3-(3-амино-5-этинилтиофен-2-карбоксамидо)пиперидин-1-карбоксилата
После растворения трет-бутил-(S)-3-(3-амино-5-((триметилсилил)этинил)тиофен-2-карбоксамидо)пиперидин-1-карбоксилата (3,0 г; 7,12 ммоль) в тетрагидрофуране (25,4 мл) в раствор последовательно добавляли карбонат калия (4,92 г) и метанол (25 мл) и затем суспензию перемешивали при комнатной температуре в течение 1 часа. После завершения взаимодействия продукт фильтровали через целит, растворитель удаляли при пониженном давлении и затем остаток экстрагировали при использовании этилацетата и воды. Собранный органический слой промывали рассолом, сушили над сульфатом натрия, концентрировали при пониженном давлении и очищали посредством ЖХСД с получением целевого соединения (2,3 г; 93%).
MS (m/z): 350 [М+1].
Стадия 3. Получение трет-бутил-(S)-3-(3-амино-5-(фенилэтинил)тиофен-2-карбоксамидо)пиперидин-1-карбоксилата
После растворения трет-бутил-(S)-3-(3-амино-5-этинилтиофен-2-карбоксамидо)-пиперидин-1-карбоксилата (150 мг; 0,429 ммоль), йодбензола (88 мг; 0,429 ммоль) и диизопропилэтиламина (94 мг; 0,730 ммоль) в диметилформамиде (5 мл) через раствор пропускали азот в течение 10 минут. После добавления Pd(PPh3)4 (21,4 мг; 0,019 ммоль) и CuI (7,1 мг; 0,037 ммоль) полученную смесь перемешивали при 100°C в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры и затем экстрагировали этилацетатом и водой. Собранный органический слой промывали рассолом, сушили над сульфатом натрия, концентрировали при пониженном давлении и очищали посредством ЖХСД с получением целевого соединения (173 мг; 95%).
MS (m/z): 426 [М+1].
Стадия 4. Получение (S)-5-(фенилэтинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида
Целевое соединение получали при применении способа, используемого на стадии 2, стадии 3 и стадии 4 примера 1 при использовании трет-бутил-(S)-3-(3-амино-5-(фенилэтинил)тиофен-2-карбоксамидо)пиперидин-1-карбоксилата.
1Н ЯМР (400 МГц, DMSO-d6) δ 10.01 (br s, 1Н), 8.12 (s, 1H), 7.88 (d, 1H), 7.59 (m, 2H), 7.45 (m, 2H), 6.69 (br s, 2H), 3.79 (m, 1H), 2.96 (m, 1H), 2.81 (m, 1H), 2.42 (m, 2H), 1.81 (m, 1H), 1.63 (m, 1H), 1.47 (m, 2H). MS (m/z): 369 [М+1]. tR составляет 3,722 мин (ВЭЖХ условия 1).
Пример 3. Получение (S)-N-(пиперидин-3-ил)-5-(пиридин-3-илэтинил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 10.00 (br s, 1Н), 8.79 (s, 1H), 8.62 (d, 1H), 8.17 (s, 1H), 8.03 (d, 1H), 7.91 (d, 1H), 7.49 (m, 1H), 6.70 (br s, 2H), 3.79 (m, 1H), 2.95 (m, 1H), 2.80 (m, 1H), 2.41 (m, 2H), 1.82 (m, 1H), 1.63 (m, 1H), 1.47 (m, 2H). MS (m/z): 370 [М+1]. tR составляет 3,237 мин (ВЭЖХ условия 1).
Пример 4. Получение (S)-N-(пиперидин-3-ил)-5-(пиридин-4-илэтинил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.99 (br s, 1Н), 8.65 (d, 1H), 8.21 (s, 1H), 7.96 (d, 1H), 7.57 (d, 1H), 6.72 (br s, 2H), 3.78 (m, 1H), 2.94 (m, 1H), 2.82 (m, 1H), 2.42 (m, 2H), 1.83 (m, 1H), 1.64 (m, 1H), 1.41 (m, 2H). MS (m/z): 370 [М+1]. tR составляет 3,183 мин (ВЭЖХ условия 1).
Пример 5. Получение (S)-5-((3-нитрофенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 10.00 (br s, 1H), 8.40 (s, 1H), 8.29 (d, 1H), 8.21 (s, 1H), 8.05 (d, 1H), 7.92 (d, 1H), 7.74 (t, 1H), 6.71 (br s, 2H), 3.77 (m, 1H), 2.95 (m, 1H), 2.80 (m, 1H), 2.41 (m, 1H), 1.83 (m, 1H), 1.63 (m, 1H), 1.50 (m, 2H). MS (m/z): 414 [М+1]. tR составляет 3,753 мин (ВЭЖХ условия 1).
Пример 6. Получение (S)-5-((3-цианофенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 10.00 (br s, 1Н), 8.18 (s, 1H), 8.13 (s, 1H), 7.92 (m, 3H), 7.65 (t, 1H), 6.70 (br s, 2H), 3.75 (m, 1H), 2.92 (m, 1H), 2.79 (m, 1H), 2.38 (m, 2H), 1.83 (m, 1H), 1.62 (m, 1H), 1.46 (m, 2H). MS (m/z): 394 [М+1]. tR составляет 3,618 мин (ВЭЖХ условия 1).
Пример 7. Получение (S)-5-((4-нитрофенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 10.00 (br s, 1Н), 8.27 (d, 2H), 8.23 (s, 1H), 7.94 (d, 1H), 7.88 (d, 1H), 6.71 (br s, 2H), 3.77 (m, 1H), 2.93 (m, 1H), 2.80 (m, 1H), 2,38 (m, 2H), 1.83 (m, 1H), 1.63 (m, 1H), 1.47 (m, 2H). MS (m/z): 414 [М+1]. tR составляет 3,743 мин (ВЭЖХ условия 1).
Пример 8. Получение (S)-5-((4-хлорфенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 10.00 (br s, 1Н), 8.13 (s, 1Н), 7.88 (d, 1Н), 7.63 (d, 1Н), 7.53 (d, 1Н), 6.69 (br s, 2Н), 3.77 (m, 1Н), 2.95 (m, 1Н), 2.92 (m, 1Н), 2.40 (m, 2Н), 1.82 (m, 1Н), 1.63 (m, 1Н), 1.47 (m, 2Н). MS (m/z): 403 [М+1]. tR составляет 3,988 мин (ВЭЖХ условия 1).
Пример 9. Получение (S)-5-((4-фторфенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 10.00 (br s, 1Н), 8.12 (s, 1H), 7.86 (d, 1H), 7.67 (m, 2H), 7.30 (t, 2H), 6.68 (br s, 2H), 3.77 (m, 1H), 2.94 (m, 1H), 2.80 (m, 1H), 2.40 (m, 2H), 1.81 (m, 1H), 1.63 (m, 1H), 1.47 (m, 2H). MS (m/z): 387 [М+1]. tR составляет 3,803 мин (ВЭЖХ условия 1).
Пример 10. Получение (S)-N-(пиперидин-3-ил)-5-(пиридин-2-илэтинил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 10.01 (br s, 1Н), 8.63 (d, 1H), 8.19 (s, 1H), 8.00 (d, 1H), 7.87 (m, 1H), 7.70 (d, 1H), 7.45 (m, 1H) 6.70 (br s, 2H), 3.81 (m, 1H), 2.97 (m, 1H), 2.84 (m, 1H), 2.44 (m, 2H), 1.84 (m, 1H), 1.64 (m, 1H), 1.48 (m, 2H). MS (m/z): 370 [М+1]. tR составляет 3,247 мин (ВЭЖХ условия 1).
Пример 11. Получение (S)-5-((4-цианофенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 10.00 (br s, 1Н), 8.20 (s, 1H), 7.91 (m, 3H), 7.78 (d, 1H), 6.71 (br s, 2H), 3.77 (m, 1H), 2.92 (m, 1H), 2.77 (m, 1H), 2,38 (m, 2H), 1.81 (m, 1H), 1.63 (m, 1H), 1.47 (m, 2H). MS (m/z): 394 [М+1]. tR составляет 3,604 мин (ВЭЖХ условия 1).
Пример 12. Получение (S)-5-((1H-индазол-3-ил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 10.04 (br s, 1Н), 8.21 (s, 1H), 7.92 (d, 1H), 7.64 (d, 1H), 7.46 (t, 1H), 7.27 (t, 1H) 6.71 (br s, 2H), 3.78 (m, 1H), 2.96 (m, 1H), 2.81 (m, 1H), 2,40 (m, 2H), 1.82 (m, 1H), 1.63 (m, 1H), 1.47 (m, 2H). MS (m/z): 409 [М+1]. tR составляет 3,388 мин (ВЭЖХ условия 1).
Пример 13. Получение (S)-5-((6-фторпиридин-3-ил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 10.01 (br s, 1Н), 8.53 (s, 1H), 8.25 (m, 1H), 8.18 (s, 1H), 7.92 (d, 1H), 7.33 (m, 1H), 6.70 (br s, 2H), 3.78 (m, 1H), 2.94 (m, 1H), 2.80 (m, 1H), 2.39 (m, 2H), 1.81 (m, 1H), 1.63 (m, 1H), 1.47 (m, 2H). MS (m/z): 388 [М+1]. tR составляет 3,254 мин (ВЭЖХ условия 1).
Пример 14. Получение (S)-5-((3,4-дифторфенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 10.00 (br s, 1Н), 8.15 (s, 1H), 7.82 (d, 1H), 7.78 (m, 1H), 7.51 (m, 2H), 6.69 (br s, 2H), 3.76 (m, 1H), 2.92 (m, 1H), 2.79 (m, 1H), 2,38 (m, 2H), 1.83 (m, 1H), 1.62 (m, 1H), 1.47 (m, 2H). MS (m/z): 405 [М+1]. tR составляет 3,896 мин (ВЭЖХ условия 1).
Пример 15. Получение трифторацетата (S)-5-((1-метил-1H-пиразол-4-ил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.89 (s, 1Н), 8.68 (br s, 2H), 8.15 (s, 1H), 8.09 (d, 1H), 8.03 (s, 1H), 7.75 (s, 1H), 6.70 (br s, 2H), 4.10 (m, 1H), 3.85 (s, 3H), 3.21 (m, 2H), 2.82 (m, 2H), 1.87 (m, 2H), 1.56 (m, 2H). MS (m/z): 373 [М+1]. tR составляет 4,309 мин (ВЭЖХ условия 1).
Пример 16. Получение трифторацетата ((S)-N-(пиперидин-3-ил)-5-((4-(трифторметил)фенил)этинил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
MS (m/z): 436 [М+1]. tR составляет 2,142 мин (ВЭЖХ условия 2).
Пример 17. Получение (S)-N-(пиперидин-3-ил)-5-((6-(трифторметил)пиридин-3-ил)этинил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.97 (br s, 1Н), 8.97 (s, 1H), 8.32-8.29 (m, 1H), 8.22 (s, 1H), 8.01-7.97 (m, 2H), 6.70 (br s, 2H), 3.83-3.82 (m, 1H), 3.00-2.96 (m, 1H), 2.85-2.82 (m, 1H), 1.86-1.80 (m, 1H), 1.66-1.63 (m, 1H), 1.51-1.40 (m, 3H), 1.20 (br s, 1H), 0.85-0.79 (m, 1H). MS (m/z): 437 [М+1]. tR составляет 1,925 мин (ВЭЖХ условия 2).
Пример 18. Получение трифторацетата метил-(S)-4-((5-(пиперидин-3-илкарбамоил)-4-уреидотиофен-2-ил)этинил)бензоата

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.89 (s, 1Н), 8.70 (br s, 2H), 8.21-8.18 (m, 2H), 8.00 (d, 2H), 7.73 (d, 2H), 6.73 (br s, 1H), 4.15-4.08 (m, 1H), 3.87 (s, 3H), 3.33-3.20 (m, 4H), 3.16 (s, 2H), 2.87-2.82 (m, 2H), 1.90-1.86 (m, 2H), 1.71-1.54 (m, 2H). MS (m/z): 426 [М+1]. tR составляет 1,969 мин (ВЭЖХ условия 2).
Пример 19. Получение гидрохлорида (S)-5-((6-хлорпиридин-3-ил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.94 (s, 1Н), 9.35 (br s, 1H), 9.27 (br s, 1H), 8.67-8.66 (m, 1H), 8.42 (d, 1H), 8.20 (s, 1H), 8.11 (dd, 1H), 7.63 (d, 1H), 6.73 (br s, 1H), 4.21-4.20 (m, 1H), 3.27-3.13 (m, 2H), 2.95-2.81 (m, 2H), 1.91-1.86 (m, 2H), 1.74-1.50 (m, 2H). MS (m/z): 403 [М+1]. tR составляет 2,305 мин (ВЭЖХ условия 2).
Пример 20. Получение (S)-5-((5-фторпиридин-3-ил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.98 (br s, 1Н), 8.67-8.64 (m, 2H), 8.20 (s, 1H), 8.09-8.05 (m, 1H), 7.90 (d, 1H), 6.68 (br s, 1H), 3.79-3.72 (m, 1H), 3.10-2.91 (m, 1H), 2.91-2.49 (m, 1H), 2.44-2.30 (m, 2H), 1.83-1.79 (m, 1H), 1.62-1.59 (m, 1H), 1.52-1.22 (m, 2H). MS (m/z): 387 [М+1]. tR составляет 2,169 мин (ВЭЖХ условия 2).
Пример 21. Получение (S)-5-((2-фторпиридин-4-ил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.87 (s, 1Н), 8.63 (br, 2H), 8.31 (d, 1H), 8.25 (s, 1H), 8.24 (d, 1H), 8.53 (d, 1H), 7.47 (s, 1H), 6.76 (br, 1H), 4.11-4.09 (m, 1H), 3.23-3.18 (m, 2H), 2.91-2.82 (m, 2H), 1.89-1.81 (m, 2H), 1.66-1.54 (m, 2H). MS (m/z): 388. tR составляет 4,710 мин (ВЭЖХ условия 1).
Пример 22. Получение (S)-5-((3-фторфенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.89 (s, 1Н), 8.67 (brs, 2H), 8.19 (s, 1H), 8.17 (s, 1H), 7.51 (m, 3H), 7.36 (t, 1H), 6.74 (brs, 2H), 4.12 (m, 1H), 3.32 (m, 1H), 3.24 (m, 1H), 2.86 (m, 2H), 1.89 (m, 2H), 1.64 (m, 2H). MS (m/z): 387 [M+1]. tR составляет 5,13 мин (ВЭЖХ условия 1).
Пример 23. Получение (S)-5-((1-(1-(метилсульфонил)пиперидин-4-ил)-1H-пиразол-4-ил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида
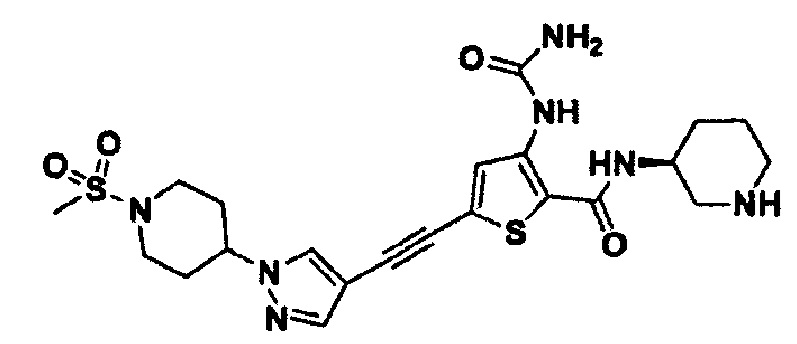
Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.90 (s, 1Н), 8.62 (br s, 2H), 8.32 (s, 1H), 8.09 (d, 1H), 8.04 (s, 1H), 7.81 (s, 1H), 6.71 (br s, 1H), 4.39-4.33 (m, 1H), 4.13-4.09 (m, 1H), 3.65 (d, 2H), 3.22 (d, 2H), 2.92 (s, 3H), 2.89-2.76 (m, 4H), 2.15-2.09 (m, 2H), 1.98 (ddd, 2H), 1.92-1.85 (m, 2H), 1.67-1.55 (m, 2H). MS (m/z): 153 [M+H]+. tR составляет 4,583 мин (ВЭЖХ условия 1).
Пример 24. Получение (S)-N-(пиперидин-3-ил)-5-(пара-толилэтинил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.90 (s, 1Н), 8.66 (br s, 2H), 8.12 (d, 1H), 8.10 (s, 1H), 7.48 (d, 2H), 7.27 (d, 2H), 6.72 (br s, 1H), 4.15-4.07 (m, 1H), 3.30 (d, 1H), 3.22 (d, 1H), 2.89-2.73 (m, 2H), 2.35 (s, 3Н), 1.91-1.85 (m, 2H), 1.72-1.53 (m, 2H). MS (m/z): 153 [M+H]+. tR составляет 4,583 мин (ВЭЖХ условия 1).
Пример 25. Получение (S)-5-((3-бром-4-цианофенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, СН3ОН-d4) δ 7.99 (s, 1Н), 7.82 (s, 1Н), 7.68 (d, J=8,1 Гц, 1Н), 7.53 (d, J=8,0 Гц, 1Н), 3.91-3.86 (m, 1Н), 3.04-3.00 (m, 1Н), 2.84-2.80 (m, 1Н), 2.48-2.41 (m, 2Н), 1.88-1.80 (m, 2Н), 1.51-1.40 (m, 2Н). MS (m/z): 474 [М+Н]. tR составляет 5,251 мин (ВЭЖХ условия 1).
Пример 26. Получение (S)-5-((4-циано-3-фторфенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.99 (br s, 1Н), 8.21 (s, 1H), 7.99 (t, 1H), 7.93 (d, 1H), 7.84 (dd, 1H), 7.62 (dd, 1H), 6.72 (br s, 2H), 3.75-3.71 (m, 1H), 3.15 (d, 1H), 2.91 (dd, 1H), 2.76 (d, 1H), 2.42-2.35 (m, 2H), 1.81-1.78 (m, 1H), 1.61-1.57 (m, 1H), 1.51-1.31 (m, 2H). MS (m/z): 412 [m+1]. tR составляет 5,075 мин (ВЭЖХ условия 1).
Пример 27. Получение трифторацетата (S)-5-((3-фтор-4-(трифторметил)фенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.87 (s, 1Н), 8.66 (br s, 2H), 8.22-8.20 (m, 2H), 7.87-7.82 (m, 2H), 7.63 (d, 1H), 6.74 (br s, 1H), 4.14-4.06 (m, 1H), 3.29 (d, 2H), 3.21 (d, 1H), 2.84-2.80 (m, 2H), 1.89-1.84 (m, 2H), 1.66-1.53 (m, 2H). MS (m/z): 455 [m+1]. tR составляет 5,593 мин (ВЭЖХ условия 1).
Пример 28. Получение трифторацетата (S)-5-((4-хлор-3-цианофенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.89 (s, 1Н), 8.68 (br s, 2Н), 8.29 (d, 1Н), 8.22-8.20 (m, 2H), 7.94 (dd, 1H), 7.83 (d, 1H), 6.74 (br s, 1H), 4.15-4.08 (m, 1H), 3.30 (d, 2H), 3.22 (d, 1H), 2.87-2.80 (m, 2H), 1.90-1.85 (m, 2H), 1.70-1.53 (m, 2H). MS (m/z): 428 [m+1]. tR составляет 5,452 мин (ВЭЖХ условия 1).
Пример 29. Получение трифторацетата (S)-5-((2-хлорпиримидин-5-ил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.89 (s, 1Н), 9.04 (s, 1Н), 8.67 (br s, 2Н), 8.25-8.23 (m, 2Н), 6.75 (br s, 1Н), 4.12-4.08 (m, 1H), 3.31 (d, 2H), 3.22 (d, 1H), 2.88-2.83 (m, 2H), 1.91-1.85 (m, 2H), 1.70-1.54 (m, 2H). MS (m/z): 405 [m+1]. tR составляет 4,675 мин (ВЭЖХ условия 1).
Пример 30. Получение (S)-N-(пиперидин-3-ил)-5-(пиразин-2-илэтинил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.89 (s, 1H), 8.95 (s, 1Н), 8.73-8.64 (m, 4H), 8.28 (s, 1H), 8.26 (d, 1H), 6.75 (br, 1H), 4.13-4.11 (m, 1H), 3.32-3.20 (m, 2H), 2.86-2.83 (m, 2H), 1.90-1.87 (m, 2H), 1.67-1.58 (m, 2H). MS (m/z): 371 [М+Н]. tR составляет 4,267 мин (ВЭЖХ условия 1).
Пример 31. Получение (S)-5-((4-хлор-3-фторфенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.98 (s, 1Н) 8.16 (s, 1H), 7.85 (d, 1H), 7.74 (d, 1H), 7.67 (t, 1H), 7.45 (d, 1H), 6.66 (br, 2H), 3.76 (m, 1H), 2.91 (m, 1H), 2.75 (m, 1H), 2.35 (m, 2H), 1.80 (m, 1H), 1.36 (m, 2H). MS (m/z): 421 [М+Н]. tR составляет 5,205 мин (ВЭЖХ условия 1).
Пример 32. Получение (S)-5-((3,5-дифторфенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1H ЯМР (400 МГц, DMSC-d6) δ 9.98 (s, 1Н) 8.18 (s, 1Н), 7.86 (d, 1Н), 7.70 (m, 3Н), 6.66 (br, 2Н), 3.77 (m, 1Н), 3.32 (m, 1Н), 2.79 (m, 1Н) 2.35 (m, 2Н), 1.61 (m, 1Н), 1.49 (m, 1Н), 1.39 (m, 2Н). MS (m/z): 405 [М+Н]. tR составляет 5,411 мин (ВЭЖХ условия 1).
Пример 33. Получение (S)-5-((2-фторфенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида
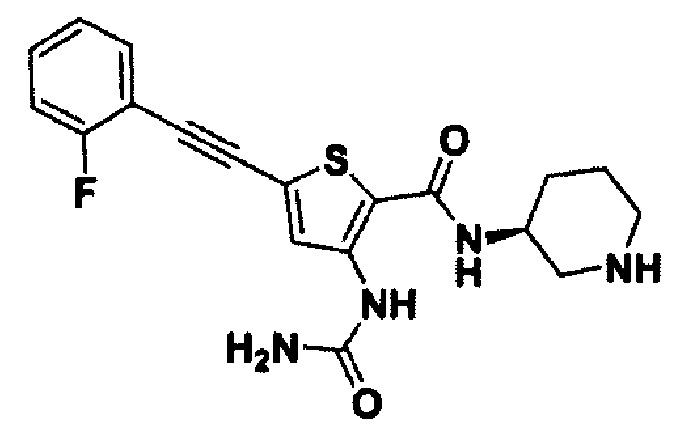
Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 10.00 (s, 1Н) 8.15 (s, 1H), 7.88 (d, 1H), 7.70 (t, 1H), 7.53 (q, 1H) 7.37 (t, 1H), 7.29 (t, 1H), 6.69 (br, 2H), 3.77 (m, 1H), 2.93 (m, 1H), 2.62 (m, 1H) 2.39 (m, 2H), 1.86 (m, 1H), 1.62 (m, 1H), 1.46 (m, 2H). MS (m/z): 387 [М+Н]. tR составляет 5,058 мин (ВЭЖХ условия 1).
Пример 34. Получение (S)-5-((2,3-дифторфенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида
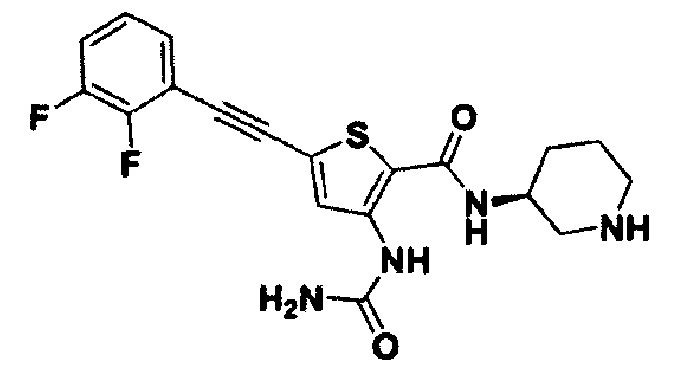
Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 10.00 (s, 1Н), 8.18 (s, 1H), 7.90 (d, 1H), 7.53 (m, 1H), 7.52 (t, 1H), 7.50 (m, 1H), 6.70 (br, 2H), 3.76 (m, 1H), 2.92 (m, 1H), 2.77 (m, 1H), 2.33 (m, 2H), 1.81 (m, 1H), 1.61 (m, 1H), 1.48 (m, 2H). MS (m/z): 405 [М+Н]. tR составляет 5,169 мин (ВЭЖХ условия 1).
Пример 35. Получение (S)-5-((6-метилпиридин-3-ил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 10.00 (s, 1Н), 8.65 (s, 1H), 8.14 (s, 1H), 7.90 (d, 1H), 7.87 (d, 1H), 7.34 (d, 1H), 6.69 (br, 2H), 3.77 (m, 1H), 2.93 (m, 1H), 2.79 (m, 1H), 2.43 (m, 2H), 1.81 (m, 1H), 1.63 (m, 1H), 1.45 (m, 2H). MS (m/z): 384 [М+Н]. tR составляет 3,849 мин (ВЭЖХ условия 1).
Пример 36. Получение (S)-N-(пиперидин-3-ил)-5-((6-(пирролидин-1-ил)пиридин-3-ил)этинил)-3-уреидотиофен-2-карбоксамида

Пирролидин (73,4 мг; 1,032 ммоль) вносили в (S)-5-((6-фторпиридин-3-ил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид (20,0 мг; 0,052 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 1 часа. После завершения взаимодействия продукт экстрагировали этилацетатом и водой. Собранный органический слой промывали рассолом, сушили над Na2SO4 и затем концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии (10% МеОН в DCM) с получением требуемого целевого соединения (3,6 мг; выход 16%).
1Н ЯМР (400 МГц, СН3ОН-d4) δ 8.20 (d, 1Н), 7.97 (s, 1Н), 7.62 (dd, 1Н), 6.54 (d, 1Н), 4.10-4.04 (m, 1Н), 3.52-3.49 (m, 4Н), 3.23-3.19 (m, 1Н), 3.02 (d, 1Н), 2.66-2.60 (m, 2Н), 2.09-2.06 (m, 4Н), 2.05-2.01 (m, 1Н), 1.87-1.84 (m, 1Н), 1.68-1.61 (m, 2Н). MS (m/z): 439 [М+1]. tR составляет 4,119 мин (ВЭЖХ условия 1).
Пример 37. Получение (S)-5-((6-пиперидин-1-ил)пиридин-3-ил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида
Целевое соединение получали с использованием способа по примеру 36.
1Н ЯМР (400 МГц, СН3ОН-d4) δ 8.24 (d, 1Н), 7.98 (s, 1Н), 7.61 (dd, 1Н), 6.81 (d, 1Н), 4.10-4.03 (m, 1Н), 3.67-3.64 (m, 4Н), 3.22-3.16 (m, 1Н), 3.00 (d, 1Н), 2.66-2.58 (m, 2Н), 2.07-2.00 (m, 1Н), 1.85-1.82 (m, 1Н), 1.747-1.72 (m, 2Н), 1.67-1.63 (m, 6Н). MS (m/z): 453 [М+Н]. tR составляет 4,351 мин (ВЭЖХ условия 1).
Пример 38. Получение (S)-5-((6-морфолинопиридин-3-ил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 36.
1Н ЯМР (400 МГц, СН3ОН-d4) δ 8.29 (d, 1Н), 7.99 (s, 1Н), 7.67 (dd, 1Н), 6.83 (d, 1Н), 4.10-4.03 (m, 1Н), 3.82-3.80 (m, 4Н), 3.61-3.59 (m, 4Н), 3.22-3.18 (m, 1Н), 3.02 (d, 1Н), 2.67-2.59 (m, 2Н), 2.07-2.00 (m, 1Н), 1.86-1.83 (m, 1Н), 1.67-1.60 (m, 2Н). MS (m/z): 455 [М+Н]. tR составляет 4,479 мин (ВЭЖХ условия 1).
Пример 39. Получение трифторацетата 5-((1-метил-1H-пиразол-4-ил)этинил)-N-(пирролидин-3-ил)-3-уреидотиофен-2-карбоксамида
Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.88 (s, 1Н), 8.73 (br s, 2Н), 8.42 (br s, 1Н), 8.31 (br s, 1H), 8.22 (d, 1H), 8.14 (s, 1H), 8.03 (s, 1H), 7.75 (s, 1H), 6.70 (br s, 1H), 4.49-4.44 (m, 1H), 3.85 (s, 3H), 3.24-3.23 (m, 1H), 3.16-3.15 (m, 1H), 2.19-2.14 (m, 1H), 2.03-1.98 (m, 1H). MS (m/z): 358 [m+1]. tR составляет 1,625 мин (ВЭЖХ условия 2).
Пример 40. Получение 5-((3-фторфенил)этинил)-N-(пирролидин-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.89 (s, 1Н), 8.77 (brs, 2H), 8.32 (s, 1H), 8.18 (s, 1H), 7.53-7.36 (m, 3H), 7.34 (t, 1H), 6.77 (brs, 1H), 4.51 (m, 1H), 3.45 (m, 1H), 3.36 (m, 1H), 3.28 (m, 1H), 3.19 (m, 1H), 2.21 (m, 1H), 2.08 (m, 1H). MS (m/z): 373 [М+1]. tR составляет 5,08 мин (ВЭЖХ условия 1).
Пример 41. Получение трифторацетата (S)-N-(азепан-3-ил)-5-((1-метил-1H-пиразол-4-ил)этинил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.88 (s, 1Н), 8.77 (br s, 2Н), 8.14-8.10 (m, 2Н), 8.02 (s, 1Н), 7.74 (s, 1H), 6.69 (br s, 1H), 4.23-4.20 (m, 1H), 3.85 (s, 3H), 3.30-3.27 (m, 1H), 3.15-3.10 (m, 3H), 1.95-1.65 (m, 5H), 1.55-1.50 (m, 1H). MS (m/z): 386 [m+1]. tR составляет 1,755 мин (ВЭЖХ условия 2).
Пример 42. Получение трифторацетата (S)-5-((1H-пиразол-4-ил)этинил)-N-(азепан-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.89 (s, 1Н), 8.77 (br s, 2H), 8.11 (d, 1H), 8.03 (s, 1H), 6.69 (br s, 1H), 4.25-4.20 (m, 1H), 3.16-3.11 (m, 3H), 1.94-1.68 (m, 5H), 1.68-1.48 (m, 1H). MS (m/z): 372 [m+1]. tR составляет 1,772 мин (ВЭЖХ условия 2).
Пример 43. Получение трифторацетата (S)-N-(азепан-3-ил)-5-((1-этил-1H-пиразол-4-ил)этинил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.90 (s, 1H), 8.80 (br s, 2Н), 8.22 (s, 1Н), 8.14 (d, 1H), 8.04 (s, 1H), 7.77 (s, 1H), 6.71 (br s, 1H), 4.26-4.21 (m, 1H), 4.16 (q, 2H), 3.32-3.29 (m, 1H), 3.17-3.12 (m, 3H), 1.97-1.69 (m, 5H), 1.57-1.51 (m, 1H), 1.38 (t, 3H). MS (m/z): 400 [m+1]. tR составляет 1,782 мин (ВЭЖХ условия 2).
Пример 44. Получение трифторацетата (S)-N-(азепан-3-ил)-5-((1-изопропил-1H-пиразол-4-ил)этинил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.91 (s, 1H), 8.85 (br s, 1Н), 8.81 (br s, 1H), 8.26 (s, 1H), 8.15 (d, 1H), 8.04 (s, 1H), 7.77 (s, 1H), 6.72 (br s, 1H), 4.56-4.49 (m, 1H), 4.26-4.21 (m, 1H), 3.32-3.12 (m, 4H), 1.97-0.86 (m, 6H), 0.83 (d, 6H). MS (m/z): 414 [m+1]. tR составляет 1,889 мин (ВЭЖХ условия 2).
Пример 45. Получение трифторацетата (S)-N-(азепан-3-ил)-5-((1-(2-метоксиэтил)-1H-пиразол-4-ил)этинил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.87 (s, 1Н), 8.79 (br s, 1H), 8.76 (br s, 1H), 8.15-8.10 (m, 2H), 8.01 (s, 1H), 7.76 (s, 1H), 6.68 (br s, 1H), 4.27-4.20 (m, 3H), 3.66 (t, 2H), 3.29-3.26 (m, 2H), 3.20-3.09 (m, 7H), 1.97-1.15 (m, 6H). MS (m/z): 430 [m+1]. tR составляет 1,742 мин (ВЭЖХ условия 2).
Пример 46. Получение трифторацетата (S)-N-(азепан-3-ил)-5-((1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-ил)этинил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.88 (s, 1Н), 8.77 (br s, 2Н), 8.28 (s, 1Н), 8.12 (d, 1H), 8.02 (s, 1H), 7.79 (s, 1H), 6.69 (br s, 1H), 4.47-4.39 (m, 1H), 4.23-4.19 (m, 1H), 3.96-3.93 (m, 2H), 3.31-3.27 (m, 1H), 3.16-3.10 (m, 3H), 2.07-1.65 (m, 10H), 1.55-1.22 (m, 1H). MS (m/z): 456 [m+1]. tR составляет 1,789 мин (ВЭЖХ условия 2).
Пример 47. Получение трифторацетата (S)-N-(азепан-3-ил)-5-((1-(1-(метилсульфонил)пиперидин-4-ил)-1H-пиразол-4-ил)этинил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.89 (s, 1Н), 8.78 (br s, 2H), 8.32 (s, 1H), 8.12 (d, 1H), 8.03 (s, 1H), 7.80 (s, 1H), 6.70 (br s, 1H), 4.39-4.32 (m, 1H), 4.27-4.18 (m, 1H), 3.66-3.63 (m, 3H), 3.31-3.08 (m, 6H), 2.95-2.88 (m, 5H), 2.14-2.10 (m, 2H), 2.00-1.65 (m, 8H), 1.56-1.50 (m, 1H). MS (m/z): 533 [m+1]. tR составляет 1,789 мин (ВЭЖХ условия 2).
Пример 48. Получение трифторацетата (S)-N-(азепан-3-ил)-5-(тиофен-3-илэтинил)-3-уреидотиофен-2-карбоксамида
Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.88 (s, 1Н), 8.80 (br s, 1H), 8.77 (br s, 1H), 8.15 (d, 1H), 8.09 (s, 1H), 8.02-8.01 (m, 1H), 7.69-7.67 (m, 1H), 7.31-7.29 (m, 1H), 6.71 (br s, 1H), 4.25-4.19 (m, 1H), 3.31-3.08 (m, 4H), 1.96-1.65 (m, 5H), 1.56-1.48 (m, 1H). MS (m/z): 388 [m+1]. tR составляет 1,969 мин (ВЭЖХ условия 2).
Пример 49. Получение трифторацетата (S)-5-((1-(1-ацетилпиперидин-4-ил)-1H-пиразол-4-ил)этинил)-N-(азепан-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.88 (s, 1Н), 8.74 (br s, 2Н), 8.27 (s, 1Н), 8.11 (d, 1H), 8.03 (s, 1H), 7.78 (s, 1H), 6.69 (br s, 1H), 4.48-4.42 (m, 2H), 4.24-4.20 (m, 1H), 3.92-3.89 (m, 1H), 3.27-3.07 (m, 7H), 2.73-2.67 (m, 1H), 2.03-1.87 (m, 4H), 1.86-1.78 (m, 5H), 1.76-1.65 (m, 4H), 1.56-1.48 (m, 1H). MS (m/z): 497 [m+1]. tR составляет 1,732 мин (ВЭЖХ условия 2).
Пример 50. Получение (S)-N-(азепан-3-ил)-5-((3-фторфенил)этинил)-3-уреидотиофен-2-карбоксамида
Целевое соединение получали с использованием способа по примеру 2.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.88 (s, 1Н), 8.79 (brs, 2H), 8.20 (d, 1H), 8.17 (s, 1H), 7.53 (m, 3H), 7.32 (t, 1H), 6.73 (brs, 2H), 4.25 (m, 1H), 3.32 (m, 2H), 3.16 (m, 4H), 1.83 (m, 1H), 1.75-1.55 (m, 4H), 1.52 (m, 1H). 401 [М+1]. tR составляет 5,26 мин (ВЭЖХ условия 1).
Пример 51. Получение трифторацетата 5-(2-(1-метил-1H-пиразол-4-ил)этил)-N-(пирролидин-3-ил)-3-уреидотиофен-2-карбоксамида
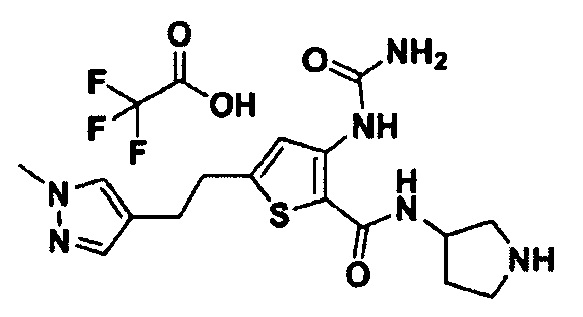
Pd/C (7 мг; 6,58 мкмоль) и метанол (8 мл) вносили в 5-((1-метил-1H-пиразол-4-ил)этинил)-N-(пирролидин-3-ил)-3-уреидотиофен-2-карбоксамид (78 мг; 0,218 ммоль), синтезированный по примеру 39, и полученную смесь перемешивали при комнатной температуре в атмосфере водорода в течение 2 часов. Реакционную смесь фильтровали через целит и концентрировали при пониженном давлении с получением 44 мг (55,8%) целевого соединения.
MS (m/z): 362 [М+1]. tR составляет 1,462 мин (ВЭЖХ условия 2).
Пример 52. Получение трифторацетата (S)-N-(азепан-3-ил)-5-(2-(1-метил-1H-пиразол-4-ил)этил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 51.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.98 (s, 1Н), 7.70-7.65 (m, 2H), 7.48 (s, 1H), 7.26 (s, 1H), 6.57 (br s, 1H), 4.14-4.00 (m, 1H), 3.75 (s, 3H), 3.04-2.90 (m, 6H), 2.73 (t, 2H), 1.82-1.47 (m, 6H). MS (m/z): 390 [М+1]. tR составляет 1,599 мин (ВЭЖХ условия 2).
Пример 53. Получение (S)-5-(3-фторфенетил))-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамида
Целевое соединение получали с использованием способа по примеру 51.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.92 (s, 1Н), 8.70 (br s, 2H), 7.73 (s, 1H), 7.14 (q, 1H), 7.05 (m, 2H), 7.02 (m, 1H), 6.60 (br s, 1H), 4.09 (m, 1H), 3.27 (m, 2H), 3.19 (t, 2H), 2.96 (t, 2H), 2.82 (m, 2H), 1.84 (m, 2H), 1.67 (m, 2H). 391 [М+1]. tR составляет 4,97 мин (ВЭЖХ условия 1).
Пример 54. Получение (S)-N-(азепан-3-ил)-5-(3-фторфенетил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 51.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.91 (s, 1Н), 8.87 (br s, 2H), 7.87 (d, 1H), 7.73 (s, 1H), 7.35 (m, 1H), 7.14 (m, 2H), 7.05 (t, 1H), 6.60 (br s, 2H), 4.23 (m, 1H), 3.25 (m, 2H), 3.16 (m, 4H), 2.96 (m, 2H), 1.91-1.67 (m, 5H), 1.53 (m, 1H). 405 [М+1]. tR составляет 5,09 мин (ВЭЖХ условия 1).
Пример 55. Получение 5-(3-фторфенетил)-N-(пирролидин-3-ил)-3-уреидотиофен-2-карбоксамида

Целевое соединение получали с использованием способа по примеру 51.
1Н ЯМР (400 МГц, DMSO-d6) δ 9.92 (s, 1Н), 8.82 (br s, 2H), 8.01 (d, 1H), 7.74 (s, 1H), 7.35 (m, 1H), 7.14 (m, 2H), 7.04 (m, 2H), 6.61 (br s, 1H), 4.49 (m, 1H), 4.03 (m, 1H), 3.42-3.31 (m, 3H), 3.17 (m, 3H), 2.96 (m, 2H), 2.17 (m, 1H), 1.99 (m, 1H). 377 [М+1]. tR составляет 4,91 мин (ВЭЖХ условия 1).
Экспериментальные примеры
Экспериментальный пример 1. Измерение ингибиторной способности в отношении киназ
Для измерения ингибиторной активности (% ингибиторной способности) соединения по настоящему изобретению в отношении протеинкиназы проводили биохимический анализ с использованием полной панели киназ, показанной в следующей Таблице 1. В качестве исследуемого соединения использовали (S)-5-((3-фторфенил)этинил)-N-(пиперидин-3-ил)-3-уреидотиофен-2-карбоксамид, синтезированный по примеру 22.
В следующей Таблице 1 показаны результаты измерения ингибиторной активности в отношении киназы при обработке исследуемым соединением в разовой концентрации 1 мкМ.


Экспериментальный пример 2. Измерение ингибиторной способности в отношении линии клеток Ba/F3
Линию клеток Ba/F3, экспрессирующих мутантный ген FLT3, культивировали в питательном растворе, содержащем 90% среды RPMI, 10% FBS (фетальная бычья сыворотка) и антибиотик (Welgene). 100 мкл клеток Ba/F3 помещали в каждую из 5000 лунок 96-луночного планшета, обработанного для культур тканей (SPL), и затем туда вводили 1 мкл исследуемого соединения, разведенного в диметилсульфоксиде (разведенного для каждой 3 точки от концентрации 1 мМ, всего 10 концентраций). После этого полученные линии клеток инкубировали в инкубаторе клеточных культур в течение 72 часов. В них добавляли 50 мкл раствора CellTiter-Glo (Promega), полученную смесь выдерживали при комнатной температуре в течение 10 минут и затем измеряли интенсивность светового излучения при использовании ридера (Envision, PerkinElmer). Интенсивность светового излучения показана в виде графика для каждой конечной концентрации исследуемого соединения, и значение GI50 получали при использовании программы Prism 5.0 (GraphPad).
Родительскую линию клеток Ba/F3, не имеющих мутантного гена FLT3, культивировали при использовании питательного раствора с добавлением ИЛ-3 (интерлейкина-3) мыши (R&D Systems), так что конечная концентрация становилась 1 нг/мл в питательном растворе, содержащем 90% среды RPMI, 10% FBS и антибиотик (Welgene). Активность измеряли способом, аналогичным описанному выше.
Следующая Таблица 2 показывает результаты измерения рост-ингибиторной активности каждого исследуемого соединения в отношении родительских клеток Ba/F3, клеток FLT3-ITD, FLT3-ITD-F691L, FLT3-D835Y и FLT3-ITD-F691L-D835Y.


Кроме того, новое соединение химической формулы 1 по настоящему изобретению может быть изготовлено в виде препаратов различных форм в зависимости от цели. Нижеследующее иллюстрирует несколько способов приготовления композиций, которые содержат соединение химической формулы 1 по настоящему изобретению в качестве активного ингредиента, но настоящее изобретение не ограничивается ими.
Примеры композиций. Способы приготовления фармацевтических композиций
Пример композиции 1. Таблетирование (прямое прессование)
После просеивания 5,0 мг активного ингредиента смешивали с 14,1 мг лактозы, 0,8 мг кросповидона USNF и 0,1 мг стеарата магния, и полученную смесь прессовали с получением таблеток.
Пример композиции 2. Таблетирование (влажная грануляция)
После просеивания 5,0 мг активного ингредиента смешивали с 16 мг лактозы и 4,0 мг крахмала. После растворения 0,3 мг полисорбата 80 в чистой воде соответствующее количество полученного раствора добавляли в эту смесь и полученную смесь подвергали распылению. После сушки тонкоизмельченные гранулы просеивали и затем смешивали с 2,7 мг коллоидного диоксида кремния и 2,0 мг стеарата магния. Полученные тонкоизмельченные гранулы прессовали с получением таблеток.
Пример композиции 3. Порошок и капсула
После просеивания 5,0 мг активного ингредиента смешивали с 14,8 мг лактозы, 10,0 мг поливинилпирролидона и 0,2 мг стеарата магния. Этой смесью наполняли твердую желатиновую капсулу №5 при использовании подходящего устройства.
Пример композиции 4. Инъекция
Инъекцию готовили посредством включения в состав 100 мг активного ингредиента, 180 мг маннита, 26 мг Na2HPO4⋅12H2O и 2,974 мг дистиллированной воды.
Реферат
Изобретение может быть применено в фармацевтической промышленности и относится к группе соединений, фармацевтическим композициям на их основе и способам их получения. Предложен способ получения производного 1а, включающий получение соединения 4 взаимодействием соединения 2 с триметилсилилацетиленом 3:,получение соединения 5 удалением защитной группы с соединения 4:,получение соединения 7 взаимодействием соединения 5 с галогенидом 6:,получение соединения 9 взаимодействием соединения 7 с трихлорацетилизоцианатом 8:,получение соединения 10 путем удаления трихлорацетильной группы с соединения 9:,получение соединения 1а удалением группы Boc с соединения формулы 10:.или способ включает получение соединения 7 взаимодействием соединения 2 и соединения 11:,получение соединения 9 взаимодействием соединения 7 с трихлорацетилизоцианатом 8:,получение соединения 10 удалением трихлорацетильной группы с соединения 9:,получение соединения формулы 1а удалением группы Boc с соединения 10:где Rпредставляет собой 0-2 заместителя, выбранных из галогена, нитро, циано, Cалкила, Cгалогеналкила, -C(O)O-Cалкила, -S(O)-Cалкила, -(CH)-Cалкокси, тетрагидро-2H-пиранила, пиперидинила, 4-(ацетил)-пиперидинила, 4-(Cалкилсульфонил)-пиперидинила, пирролидинила и морфолинила, m равно от 1 до 6, Rпредставляет собой атом водорода, A представляет собой фенил, тиофенил, индазолил, пиридинил, пиримидинил, пиразинил или пиразолил, n равно от 1 до 3, X представляет собой галоген, Z представляет собой O. Предложены новые соединения и фармацевтические композиции на их основе, эффективные для лечения, предупреждения и облегчения острого миелоиднго лейкоза с точечной мутацией FLT3, а также эффективные способы их получения. 5 н. и 3 з.п. ф-лы, 59 пр., 2 табл.
Формула




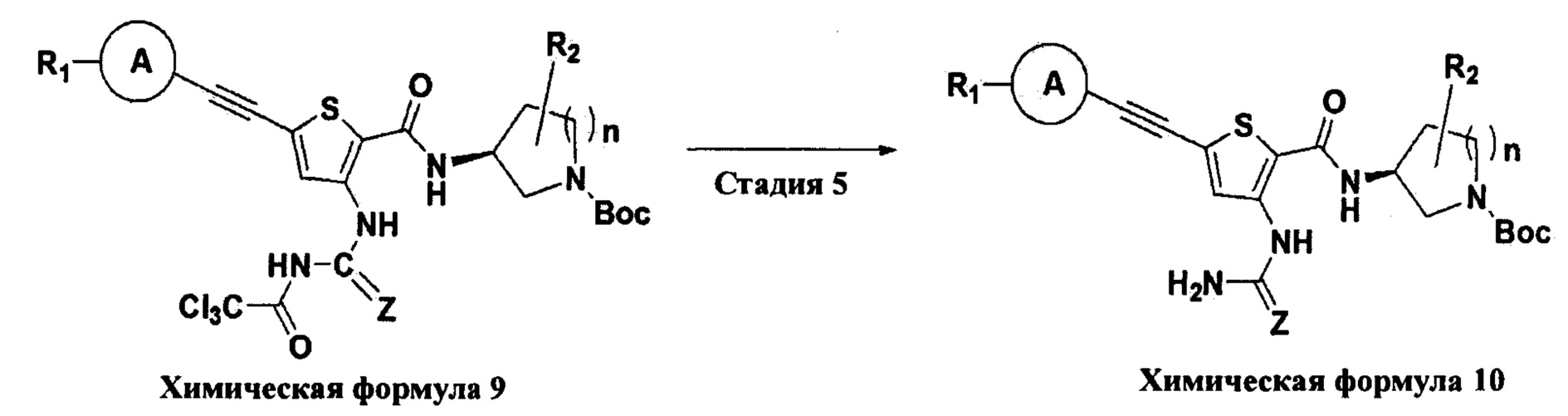
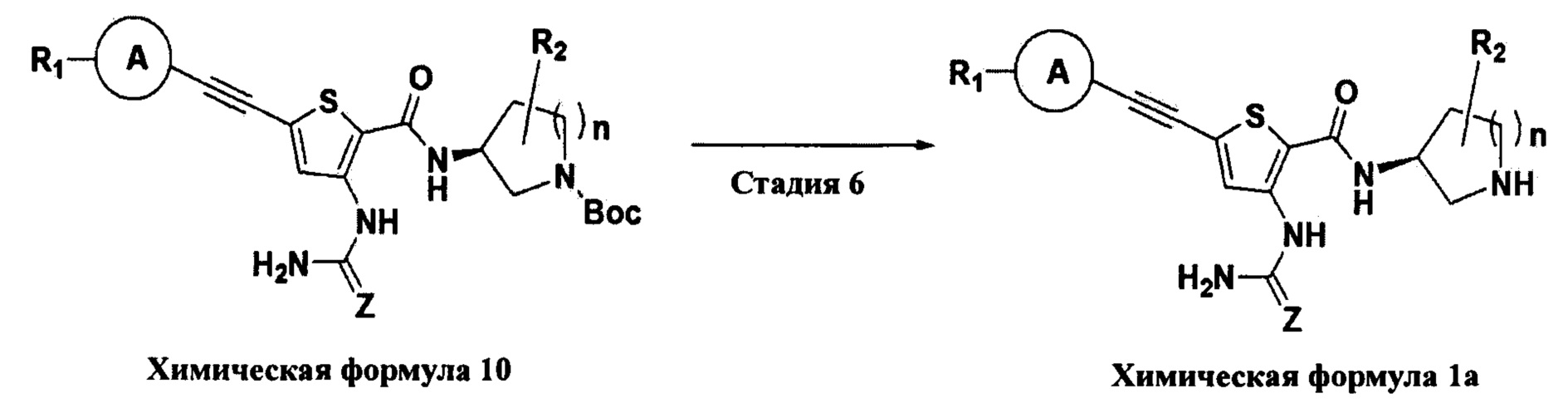




Комментарии