Новые ингибиторы s-нитрозоглутатионредуктазы - RU2585763C2
Код документа: RU2585763C2
Описание
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новым соединениям, фармацевтическим композициям, содержащим такие соединения, и к способам их получения и применения. Эти соединения применимы в качестве ингибиторов S-нитрозоглутатионредуктазы (GSNOR).
УРОВЕНЬ ТЕХНИКИ
Химическое соединение оксид азота является газом, описывающимся химической формулой NO. NO является одной из немногих газообразных сигнальных молекул, известны в биологических системах, и играет важную роль в регулировании различных биологических процессов. Например, эндотелий использует NO для передачи гладким мышцам, окружающим стенки артериол, сигнала для расслабления, приводящего к расширению сосудов и усилению кровотока в гипоксические ткани. NO также участвует в регуляции пролиферации гладких мышц, функции тромбоцитов и нейротрансмиссии и играет роль в защите хозяина. Хотя NO обладает высокой реакционной способностью и его время жизни составляет несколько секунд, он может свободно диффундировать через мембраны и связываться со многими молекулярными мишенями. Эти характеристики делают NO идеальной сигнальной молекулой, способной регулировать биологические процессы, протекающие между клетками и внутри клеток.
NO является газообразным свободным радикалом и поэтому является реакционноспособным и нестабильным, таким образом, in vivo NO является короткоживущим, обладающем при физиологических условиях периодом полуразложения, равным 3-5 с. В присутствии кислорода NO может соединяться с тиолами с образованием биологически важного класса стабильных аддуктов с NO, называющихся S-нитрозотиолами (SNO). Предположено, что этот стабильный пул NO действует как источник биологически активного NO и критически важен в здоровом состоянии и при болезни вследствие центральной роли NO в клеточном гомеостазе (Stamler et al., Proc. Natl. Acad. Sci. USA, 89:7674-7677 (1992)). SNO белки играют различные роли в функции сердечнососудистой, дыхательной, метаболической, желудочно-кишечной, иммунной и центральной нервной системы (Foster et al.. Trends in Molecular Medicine, 9 (4): 160-168, (2003)). Одним из наиболее исследованных SNO в биологических системах является S-нитрозоглутатион (GSNO) (Gaston et al., Proc. Natl. Acad. Sci. USA 90:10957-10961 (1993)), новый ключевой регулятор передачи сигнала с помощью NO, поскольку он является эффективным перенитрозирующим реагентом и, видимо, поддерживает равновесие с другими S-нитрозированными белками (Liu et al. Nature, 410:490-494 (2001)) в клетках. Вследствие этого главного положения в континууме NO-SNO, GSNO является перспективной для терапевтического исследования мишенью в случаях, когда фармакологически оправдана модуляция NO.
Вследствие выяснения того, что GSNO является ключевым регулятором гомеостаза NO и содержания SNO в клетках, исследования направлены на изучение эндогенного продуцирования GSNO и SNO белков, которое происходит после продуцирования радикала NO ферментом синтетазой оксида азота (NOS). Недавно был лучше понят ферментативный катаболизм GSNO, который играет важную роль в регулировании имеющихся концентраций GSNO и, следовательно, имеющихся NO и SNO.
Важным для понимания катаболизма GSNO было то, что недавно исследователи выявили высококонсервативную S-нитрозоглутатионредуктазу (GSNOR) (Jensen et al., Biochem J., 331:659-668 (1998); Liu et al., (2001)). GSNOR также известна, как глутатион-зависимая формальдегиддегидрогеназа (GSH-FDH), алкогольдегидрогеназа 3 (ADH-3) (Uotila and Koivusalo, Coenzymes and Cofactors., D. Dolphin, ed. pp. 517-551 (New York, John Wiley & Sons, 1989)) и алкогольдегидрогеназа 5 (ADH-5). Важно, что GSNOR обладает большей активностью по отношению к GSNO, чем по отношению к другим субстратам (Jensen et al., 1998; Liu et al., 2001) и, видимо, опосредует денитрозирование белков и пептидов у бактерий, растений и животных. GSNOR, видимо, является основным метаболизирующим GSNO ферментом у эукариотов (Liu et al., 2001). Таким образом, GSNO может накапливаться в биологических компартментах, в которых активность GSNOR является низкой или отсутствует (например, в жидкой выстилке дыхательных путей) (Gaston et al., 1993).
Дрожжи с недостатком GSNOR накапливают S-нитрозилированные белки, которые не являются субстратами этого фермента, и это убедительно показывает, что GSNO находится в равновесии с SNO-белками (Liu et al., 2001). Точное ферментативное регулирование содержаний GSNO и тем самым SNO-белков в среде увеличивает возможность того, что GSNO/GSNOR сможет играть роль в физиологических и патологических функциях хозяина, включая защиту от нитрозирующего стресса, когда количество продуцированного NO превышает физиологическую потребность. В действительности, GSNO специфически участвует в физиологических процессах в диапазоне от движения до дыхания (Lipton et al. Nature, 413:171-174 (2001)) путем регуляции трансмембранного регулятора муковисцидоза (Zaman et al., Biochem Biophys Res Commun, 284:65-70 (2001)), путем регуляции тонуса сосудов, тромбоза и функции тромбоцитов (de Belder et al., Cardiovasc Res.; 28(5):691-4 (1994)); (Z. Kaposzta, et al.. Circulation; 106(24): 3057 - 3062, (2002)), а также защищает хозяина (de Jesus-Berrios et al., Curr. Biol; 13:1963-1968 (2003)). В других исследованиях установлено, что GSNOR защищает клетки дрожжей от нитрозирующего стресса и in vitro (Liu et al., 2001), и in vivo (de Jesus-Berrios et al. (2003)).
Все эти данные показывают, что GSNO является первичным физиологическим лигандом для фермента S-нитрозоглутатионредуктазы (GSNOR), который катаболизирует GSNO и, следовательно, уменьшает количество доступных SNO и NO в биологических системах (Liu et al. (2001)), (Liu et al., Cell, 116(4), 617-628 (2004)) и (Que et al., Science, 308 (5728):1618-1621 (2005)). Сам по себе этот фермент играет главную роль в регуляции локального и системного биологически активного NO. Поскольку изменения биологической доступности NO связаны с патогенезом многочисленных патологических состояний, включая гипертензию, атеросклероз, тромбоз, астму, желудочно-кишечные нарушения, воспаление и рак, средства, которые регулируют активность GSNOR являются возможными терапевтическими средствами для лечения заболеваний, связанных с нарушением баланса NO.
Оксид азота (NO), S-нитрозоглутатион (GSNO) и S-нитрозоглутатионредуктаза (GSNOR) регулируют нормальную физиологию легких и вносят вклад в патофизиологии легких. При нормальных условиях NO и GSNO поддерживают нормальную физиологию и функционирование легких посредством противовоспалительного и бронхорасширяющего воздействия. Пониженные содержания этих медиаторов при заболеваниях легких, таких как астма, хроническое обструктивное заболевание легких (ХОЗЛ), могут создаваться путем повышающей регуляции активности фермента GSNOR. Эти пониженные содержания NO и GSNO и обусловленная ими уменьшенная противовоспалительная способность являются ключевыми моментами, которые вносят вклад в заболевания легких и которые потенциально можно обратить путем ингибирования GSNOR.
Воспалительные болезни кишечника (ВБК), включая болезнь Крона и язвенный колит, являются хроническими воспалительными нарушениями желудочно-кишечного (ЖК) тракта, на которые могут оказывать влияние NO, GSNO и GSNOR. При нормальных условиях NO и GSNO поддерживают нормальную физиологию путем противовоспалительных воздействий и сохранения эпителиального кишечного клеточного барьера. При ВБК наблюдаются пониженные содержания GSNO и NO и они, вероятно, обусловлены повышающей регуляцией активности GSNOR. Пониженные содержания этих медиаторов внося вклад в патофизиологию ВБК путем разрушения эпителиального барьера посредством нарушения регуляции белков, участвующих в поддержании плотных эпителиальных сочленений. Это нарушение функций эпителиального барьера с обусловленным им проникновением микроорганизмов из полости и общей сниженной противовоспалительной способностью при пониженных содержаниях NO и GSNO являются ключевыми аспектами прогрессирования ВБК, на которые потенциально можно повлиять путем направленного воздействия на GSNOR.
В настоящее время в данной области техники настоятельно необходимы диагностика, профилактика, облегчение протекания и лечение патологических состояний, связанных с увеличенным синтезом NO и/или увеличенной биологической активностью NO. Кроме того, настоятельно необходимы новые соединения, композиции и способы предупреждения, облегчения протекания или обращения других связанных с NO нарушений. Настоящее изобретение решает эти задачи.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым соединениям (формулы I). Эти соединения применимы в качестве ингибиторов S-нитрозоглутатионредуктазы ("GSNOR"). В объем настоящего изобретения входят фармацевтически приемлемые соли, пролекарства, метаболиты и стереоизомеры описанных соединений. В объем настоящего изобретения также входят фармацевтические композиции, содержащие по меньшей мере одно соединение, предлагаемое в настоящем изобретении, и по меньшей мере один фармацевтически приемлемый носитель.
Композиции, предлагаемые в настоящем изобретении, можно приготовить в любой подходящей фармацевтически приемлемой дозированной форме.
Настоящее изобретение относится к способу ингибирования GSNOR у нуждающегося в нем субъекта. Такой способ включает введение терапевтически эффективного количества фармацевтической композиции, содержащей по меньшей мере один ингибитор GSNOR или его фармацевтически приемлемую соль, пролекарство, метаболит или стереоизомер в комбинации по меньшей мере с одним фармацевтически приемлемым носителем. Ингибитор GSNOR может представлять собой соединение, предлагаемое в настоящем изобретении, или он может представлять собой известное соединение, для которого ранее не было известно, что оно является ингибитором GSNOR.
Настоящее изобретение также относится к способу лечения у нуждающегося в нем субъекта нарушения, протекание которого облегчается путем лечения донором NO. Такой способ включает введение терапевтически эффективного количества фармацевтической композиции, содержащей по меньшей мере один ингибитор GSNOR или его фармацевтически приемлемую соль, пролекарство, метаболит или стереоизомер, в комбинации по меньшей мере с одним фармацевтически приемлемым носителем. Ингибитор GSNOR может представлять собой новое соединение, предлагаемое в настоящем изобретении, или он может представлять собой известное соединение, для которого ранее не было известно, что оно является ингибитором GSNOR.
Настоящее изобретение также относится к способу лечения клеточного пролиферативного нарушения у нуждающегося в нем субъекта. Такой способ включает введение терапевтически эффективного количества фармацевтической композиции, содержащей по меньшей мере один ингибитор GSNOR или его фармацевтически приемлемую соль, пролекарство, метаболит или стереоизомер, в комбинации по меньшей мере с одним фармацевтически приемлемым носителем. Ингибитор GSNOR может представлять собой новое соединение, предлагаемое в настоящем изобретении, или он может представлять собой известное соединение, для которого ранее не было известно, что оно является ингибитором GSNOR.
Способы, предлагаемые в настоящем изобретении, включают введение одного или большего количества вторичных активных средств. Такое введение может быть последовательным или в комбинированной композиции.
Хотя при осуществлении или проверке настоящего изобретения можно использовать методики и материалы, сходные с описанными в настоящем изобретении или эквивалентные им, ниже описаны подходящие методики и материалы. Все общедоступные публикации, заявки на патенты, патенты и другая литература, указанная в настоящем изобретении, во свей своей полноте включены в настоящее изобретение в качестве ссылки. В случае противоречий следует использовать настоящее описание, включая определения.
Приведенное выше краткое изложение и последующее полное описание являются типичными и представлены для разъяснения и предназначены для более полного описания заявленных композиций и способов. Другие объекты, преимущества и новые особенности должны быть очевидны для специалистов в данной области техники из последующего подробного описания.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
А. Общее описание изобретения
Ранее было известно, что S-нитрозоглутатионредуктаза (GSNOR) окисляет аддукт формальдегида с глутатионом, S-гидроксиметилглутатион. GSNOR ранее был обнаружен в различных бактериях, дрожжах, растениях и животных и является высококонсервативным. Белки Е. coli, S. cerevisiae и макрофаги мышей содержат более 60% идентичных аминокислотных последовательностей. Активность GSNOR (т. е. разложение GSNO, когда содержится NADH в качестве необходимого кофактора) была обнаружена в Е. coli, в макрофагах мышей, в эндотелиальных клетках мышей, в гладкомышечных клетках мышей, в дрожжах и в клетках HeLa, эпителиальных клетках и моноцитах человека. Информацию о нуклеотидной и аминокислотной последовательности GSNOR человека можно получить из баз данных Национального центра биотехнологической информации (NCBI) с номерами доступа М29872, NM_000671. Информацию о нуклеотидной и аминокислотной последовательности GSNOR мыши можно получить из баз данных NCBI с номером доступа NM_007410. В нуклеотидной последовательности старт-сайт и стоп-сайт подчеркнуты. CDS обозначает кодирующую последовательность. SNP обозначает однонуклеотидный полиморфизм. Другие связанные с GSNOR нуклеотидные и аминокислотные последовательности, включая последовательности для других видов, приведены в заявке на патент U.S. 2005/0014697.
В настоящем изобретении показано, что GSNOR in vivo и in vitro подвергает метаболизму S-нитрозоглутатион (GSNO) и S-нитрозотиолы белков (SNOs) для модулирования биологической активности NO путем регулирования внутриклеточного содержания обладающих низкой молекулярной массой соединений-доноров NO предупреждения протекания нитрозилирования до токсичного уровня.
Отсюда следует, что ингибирование этого фермента усиливает биологическую активность при заболеваниях, для которых показано лечение донором NO, подавляет пролиферацию патологически пролиферирующих клеток и увеличивает биологическую активность NO при заболеваниях, для которых это полезно.
Настоящее изобретение относится к фармацевтическим средствам, которые являются активными ингибиторами GSNOR. В частности, настоящее изобретение относится к аналогам, обладающих приведенными ниже структурами (формула I), или их фармацевтически приемлемой соли, стереоизомеру, пролекарству или метаболиту.
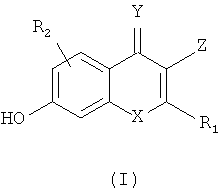
в которой Х выбран из группы, включающей О и S;
Y выбран из группы, включающей О и S;
Z выбран из группы, включающей Z1, Z2, Z3, и Z4, где
Z1 обозначает

Z2 обозначает

Z3 обозначает

Z4 обозначает
R1 выбран из группы, включающей водород, (С1-С6)алкил, (С3-С7)циклоалкил, (С1-С6)галогеналкил, незамещенный арил(С1-С4)алкил, замещенный арил(С1-С6)алкил, (С1-С6)гетероалкил, замещенный или незамещенный арил и замещенный или незамещенный гетероарил;
R2 выбран из группы, включающей водород, галоген, цианогруппу и (C1-С6)алкоксигруппу;
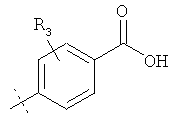
R3 выбран из группы, включающей водород, галоген, (С1-С6)алкил, (C1-С6)галогеналкил, (С1-С6)алкоксигруппу, цианогруппу и N,N-диметиламиногруппу;
R4 выбран из группы, включающей тетразол, оксадиазолон, тиадиазолон, метилсульфонилкарбамоил и N-гидроксикарбамоил; и
R5 выбран из группы, включающей карбоксигруппу, тетразол, оксадиазолон, тиадиазолон, метилсульфонилкарбамоил и N-гидроксикарбамоил.
Кроме того, из любой композиций, описанной в настоящем изобретении, одно или большее количество соединений или подгрупп соединений могут быть специально исключены.
При использовании в этом контексте термин "аналог" означает соединение, обладающее химической структурой и функцией, сходными со структурой и функцией соединения формулы I, которое сохраняет центральную кольцевую систему.
Некоторые аналоги соединения, предлагаемого в настоящем изобретении, также могут существовать в различных стереоизомерных формах, включая конфигурационные, геометрические и конформационные изомеры, а также могут существовать в различных таутомерных формах, в особенности такие, которые различаются по положению присоединения атома водорода. При использовании в настоящем изобретении термин "стереоизомер" включает такие изомерные формы соединения, включая таутомерные формы соединения.
Иллюстративные соединения, содержащие асимметрические центры, могут существовать в различных энантиомерных и диастереоизомерных формах. Соединение может существовать в форме оптического изомера или диастереоизомера. Соответственно, в объем настоящего изобретения входят соединения в формах их оптических изомеров, диастереоизомеров и их смесей, включая рацемические смеси.
Следует отметить, что, если имеется расхождение между изображенной структурой и названием, данным этой структуре, то правильной является изображенная структура. Кроме того, если стереохимическая конфигурация структуры или части структуры не указана, например, полужирными, клинообразными или пунктирными линиями, то следует считать, что структура или часть структуры включает все стереоизомеры описанных соединения.
В. Ингибиторы S-нитрозоглутатионредуктазы
1. Соединения, предлагаемые в настоящем изобретении
Одним из объектов настоящего изобретения является соединение, обладающее структурой, описываемой формулой I, или его фармацевтически приемлемая соль, стереоизомер, пролекарство или метаболит:
в которой Х выбран из группы, включающей О и S;
Y выбран из группы, включающей О и S;
Z1 обозначает
Z2 обозначает
Z3 обозначает
Z4 обозначает
R1 выбран из группы, включающей водород, (С1-С6)алкил, (С3-С7)циклоалкил, (С1-С6)галогеналкил, незамещенный арил(С1-С4)алкил, замещенный арил(С1-С6)алкил, (С1-С6)гетероалкил, замещенный или незамещенный арил и замещенный или незамещенный гетероарил;
R2 выбран из группы, включающей водород, галоген, цианогруппу и (С1-С6)алкоксигруппу;
R3 выбран из группы, включающей водород, галоген, (С1-С6)алкил, (С1-С6)галогеналкил, (С1-С6)алкоксигруппу, цианогруппу и N,N-диметиламиногруппу;
R4 выбран из группы, включающей тетразол, оксадиазолон, тиадиазолон, метилсульфонилкарбамоил и N-гидроксикарбамоил; и
R5 выбран из группы, включающей карбоксигруппу, тетразол, оксадиазолон, тиадиазолон, метилсульфонилкарбамоил и N-гидроксикарбамоил.
В другом объекте настоящего изобретения R4 выбран из группы, включающей тетразол, 1,2,4-оксадиазол-5(4Н)-он-3-ил, 1,2,4-тиадиазол-5(4Н)-он-3-ил, 1,3,4-оксадиазол-2(3Н)-он-5-ил, 1,3,4-тиадиазол-2(3Н)-он-5-ил, 1,2,4-тиадиазол-3(2Н)-он-5-ил, 1,2,4-оксадиазол-3(2Н)-он-5-ил, метилсульфонилкарбамоил и N-гидроксикарбамоил; и
R5 выбран из группы, включающей карбоксигруппу, тетразол, 1,2,4-оксадиазол-5(4Н)-он-3-ил, 1,2,4-тиадиазол-5(4Н)-он-3-ил, 1,3,4-оксадиазол-2(3Н)-он-5-ил, 1,3,4-тиадиазол-2(3Н)-он-5-ил, 1,2,4-тиадиазол-3(2Н)-он-5-ил, 1,2,4-оксадиазол-3(2Н)-он-5-ил, метилсульфонилкарбамоил и N-гидроксикарбамоил.
В другом объекте настоящего изобретения R1 выбран из группы, включающей водород, CF3, CF2H, CF2CH3, CF2CH3CH3, метил, изопропил, изобутил, циклопентил, СН2ОСН3, SCH3, бензил, 4-карбоксибензил, тиофен-2-ил и тиофен-3-ил;
R2 выбран из группы, включающей водород, фтор, хлор, метоксигруппу и цианогруппу; и
R3 выбран из группы, включающей водород, фтор, хлор, метил, CF3, метоксигруппу, цианогруппу и N,N-диметиламиногруппу.
В другом объекте настоящего изобретения R1 выбран из группы, включающей водород, CF3, CF2H, метил и 4-карбоксибензил;
R2 выбран из группы, включающей водород и фтор;
R3 выбран из группы, включающей водород, фтор, хлор и метил;
R4 выбран из группы, включающей тетразол, 1,2,4-оксадиазол-5(4Н)-он-3-ил, 1,2,4-тиадиазол-5(4Н)-он-3-ил, 1,3,4-оксадиазол-2(3Н)-он-5-ил, метилсульфонилкарбамоил и N-гидроксикарбамоил; и
R5 выбран из группы, включающей карбоксигруппу, тетразол, 1,2,4-оксадиазол-5(4Н)-он-3-ил, 1,2,4-тиадиазол-5(4Н)-он-3-ил, 1,3,4-оксадиазол-2(3Н)-он-5-ил, метилсульфонилкарбамоил и N-гидроксикарбамоил.
В другом объекте настоящего изобретения R4 выбран из группы, включающей тетразол, 1,2,4-оксадиазол-5(4Н)-он-3-ил, 1,2,4-тиадиазол-5(4Н)-он-3-ил, метилсульфонилкарбамоил и N-гидроксикарбамоил; и рз выбран из группы, включающей карбоксигруппу, тетразол, 1,2,4-оксадиазол-5(4Н)-он-3-ил, 1,2,4-тиадиазол-5(4Н)-он-3-ил, метилсульфонилкарбамоил и N-гидроксикарбамоил.
В другом объекте настоящего изобретения Х выбран из группы, включающей О и S. В другом объекте настоящего изобретения, Х обозначает О. В еще одном объекте настоящего изобретения Х обозначает S.
В другом объекте настоящего изобретения Y выбран из группы, включающей О и S. В другом объекте настоящего изобретения Y обозначает О. В еще одном объекте настоящего изобретения Y обозначает S.
В другом объекте настоящего изобретения подходящие соединения формулы I включают, но не ограничиваются только ими:
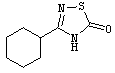
3-(4-(1Н-тетразол-5-ил)фенил)-7-гидрокси-2-(трифторметил)-4Н-хромен-4-он;
5-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)тиофен-2-карбоновую кислоту;
(транс)-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)циклогексанкарбоновую кислоту;
(цис)-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)циклогексанкарбоновую кислоту;
3-(4-(1Н-тетразол-5-ил)фенил)-2-(дифторметил)-7-гидрокси-4Н-хромен-4-он;
3-(4-(1Н-тетразол-5-ил)фенил)-7-гидрокси-2-метил-4Н-хромен-4-он;
4-(2-(4-карбоксибензил)-7-гидрокси-4-оксо-4Н-тиохромен-3-ил)бензойную кислоту;
4-(7-гидрокси-2-метил-4-оксо-4Н-тиохромен-3-ил)бензойную кислоту;
3-(4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)фенил)-1,2,4-оксадиазол-5(4Н)-он;
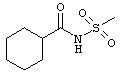
4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)-М-(метилсульфонил)бензамид;
3-(4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)фенил)-1,2,4-тиадиазол-5(4Н)-он;
3-(4-(1Н-тетразол-5-ил)фенил)-7-гидрокси-2-метил-4Н-тиохромен-4-он;
5-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)тиофен-3-карбоновую кислоту;
3-((транс)-4-(1Н-тетразол-5-ил)циклогексил)-7-гидрокси-2-(трифторметил)-4Н-хромен-4-он;
N-гидрокси-4-(7-гидрокси-4-оксо-2-(трифторметил)-4H-хромен-3-ил)бензамид;
3-(2-хлор-4-(1Н-тетразол-5-ил)фенил)-7-гидрокси-2-(трифторметил)-4Н-хромен-4-он;
3-(3-хлор-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)фенил)-1,2,4-оксадиазол-5(4Н)-он;
3-(3-фтор-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)фенил)-1,2,4-оксадиазол-5(4Н)-он;
3-(3-хлор-4-(1Н-тетразол-5-ил)фенил)-7-гидрокси-2-(трифторметил)-4Н-хромен-4-он; и
3-(4-(1Н-тетразол-5-ил)фенил)-7-гидрокси-4Н-хромен-4-он; и
5-(4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)фенил)-1,3,4-оксадиазол-2(3Н)-он.
В другом варианте осуществления соединение 3-фтор-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензойная кислота представляет собой соединение, предлагаемое в настоящем изобретении.
В другом варианте осуществления соединение 4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)-3-метилбензойная кислота представляет собой соединение, предлагаемое в настоящем изобретении.
В другом варианте осуществления соединение 4-(8-фтор-7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензойная кислота представляет собой соединение, предлагаемое в настоящем изобретении.
Примеры Z1, где R4 обозначает тетразол, 1,2,4-оксадиазол-5(4Н)-он-3-ил, 1,2,4-тиадиазол-5(4Н)-он-3-ил, 1,3,4-оксадиазол-2(3Н)-он-5-ил, 1,3,4-тиадиазол-2(3Н)-он-5-ил, 1,2,4-тиадиазол-3(2Н)-он-5-ил, 1,2,4-оксадиазол-3(2Н)-он-5-ил, метилсульфонилкарбамоил и N-гидроксикарбамоил, включают, соответственно

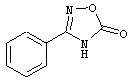

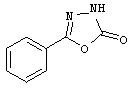




Примеры Z3, где R5 обозначает карбоксигруппу, тетразол, 1,2,4-оксадиазол-5(4Н)-он-3-ил, 1,2,4-тиадиазол-5(4Н)-он-3-ил, 1,3,4-оксадиазол-2(3Н)-он-5-ил, 1,3,4-тиадиазол-2(3Н)-он-5-ил, 1,2,4-тиадиазол-3(2Н)-он-5-ил, 1,2,4-оксадиазол-3(2Н)-он-5-ил, метилсульфонилкарбамоил и N-гидроксикарбамоил, включают соответственно








Если связь с заместителем представлена, как пересекающая связь, соединяющую два атома кольца, то такой заместитель может быть связан с любым атомом кольца. Если заместитель представлен без указания атома, с помощью которого такой заместитель присоединен к остальной части соединения данной формулы, то такой заместитель может быть присоединен с помощью любого атома такого заместителя. Допустимы комбинации заместителей и/или переменных, но только если такие комбинации приводят к стабильным соединениям.
Соединения, описанные в настоящем изобретении, могут содержать асимметрические центры. Соединения, предлагаемые в настоящем изобретении, содержащие асимметрично замещенный атом, можно выделить в оптически активной или рацемической форме. В данной области техники хорошо известно, как получить оптически активные формы, например, путем разделения рацемических форм или из оптически активных исходных веществ. Многие геометрические изомеры олефинов, соединений, содержащих двойные связи C=N, и т.п. также могут содержаться в соединениях, описанных в настоящем изобретении, и все такие стабильные изомеры входят в объем настоящего изобретения. Геометрические цис- и транс-изомеры соединений, предлагаемых в настоящем изобретении, описаны и могут быть выделены в виде смеси изомеров или в виде разделенных изомерных форм. В объем настоящего изобретения входят все хиральные, диастереоизомерные, рацемические геометрические изомерные формы структуры, если специально не указана конкретная стереохимическая конфигурация или изомерная форма. Все таутомеры представленных или описанных соединений также считаются частью настоящего изобретения.
Следует понимать, что изомеры, образующиеся вследствие такой асимметрии (например, все энантиомеры и диастереоизомеры) включены в объем настоящего изобретения, если не указано иное. Такие изомеры можно получить в основном в чистом виде с помощью классических методик разделения и с помощью стереохимически регулируемого синтеза. Кроме того, структуры и другие соединения и фрагменты, рассмотренные в настоящей заявке, также включают все их таутомеры. Алкены могут обладать Е- или Z-конфигурацией, когда это целесообразно.
2. Типичные соединения
В приведенных ниже примерах представлены типичные новые аналоги соединений, предлагаемых в настоящем изобретении. Методики синтеза, которые можно использовать для получения каждого соединения, подробно описаны в примерах 1-24 с указанием на промежуточные продукты, описанные в примере 25. Дополнительные данные масс-спектрометрии и/или протонного ЯМР для каждого соединения также приведены в примерах 1-22. Активность ингибитора GSNOR определяли с помощью исследования, описанного в примере 26, и значения IC50 получали в примерах 1-22. Соединения-ингибиторы GSNOR в примерах 1-22 обладали значениями IC50, равными примерно <1 мкМ. Соединения-ингибиторы GSNOR в примерах 1-3, 5-6, 8-9, 11-12, 14, 16-22 обладали значениями IC50, равными примерно менее 0,1 мкМ.
С. Определения
При использовании в настоящем изобретении термин "примерно" должен быть понятен специалистам с общей подготовкой в данной области техники и немного меняется в зависимости от контекста, в котором он используется. Если имеются случаи применения термина, неясные специалистам с общей подготовкой в данной области техники в использующемся контексте, то "примерно" означает использующееся значение + до 10%.
Термин "ацил" включает соединения и фрагменты, которые содержат ацетильный радикал (СН3СО-) или карбонильную группу с которой связан обладающий линейной или разветвленной цепью низш. алкильный остаток.
Термин "алкил" при использовании в настоящем изобретении означает обладающий линейной или разветвленной цепью, насыщенный углеводород, содержащий указанное количество атомов углерода. Например, (С1-С6)алкил включает, но не ограничивается только ими метил, этил, пропил, изопропил, бутил, втор-бутил, трет-бутил, пентил, изопентил, неопентил, гексил, изогексил и неогексил. Алкильная группа может быть незамещенной или необязательно содержит один или большее количество заместителей, описанных в настоящем изобретении.
Термин "алкенил" при использовании в настоящем изобретении означает обладающий линейной или разветвленной цепью ненасыщенный углеводород, содержащий указанное количество атомов углерода и по меньшей мере одну двойную связь. Примеры (С2-С8)алкенильной группы включают, но не ограничиваются только ими, этилен, пропилен, 1-бутилен, 2-бутилен, изобутилен, втор-бутилен, 1-пентен, 2-пентен, изопентен, 1-гексен, 2-гексен, 3-гексен, изогексен, 1-гептен, 2-гептен, 3-гептен, изогептен, 1-октен, 2-октен, 3-октен, 4-октен и изооктен. Алкенильная группа может быть незамещенной или необязательно содержит один или большее количество заместителей, описанных в настоящем изобретении.
Термин "алкинил" при использовании в настоящем изобретении означает обладающий линейной или разветвленной цепью ненасыщенный углеводород, содержащий указанное количество атомов углерода и по меньшей мере одну тройную связь. Примеры (С2-С8)алкинильной группы включают, но не ограничиваются только ими, ацетилен, пропин, 1-бутин, 2-бутин, 1-пентин, 2-пентин, 1-гексин, 2-гексин, 3-гексин, 1-гептин, 2-гептин, 3-гептин, 1-октин, 2-октин, 3-октин и 4-октин. Алкинильная группа может быть незамещенной или необязательно содержит один или большее количество заместителей, описанных в настоящем изобретении.
Термин "алкоксигруппа" при использовании в настоящем изобретении означает -O-алкильную группу, содержащую указанное количество атомов углерода. Например, (С1-С6)алкоксигруппа включает -O-метил, -O-этил, -O-пропил, -O-изопропил, -O-бутил, -O-втор-бутил, -O-трет-бутил, -O-пентил, -O-изопентил, -O-неопентил, -O-гексил, -O-изогексил и -O-неогексил.
Термин "аминоалкил" при использовании в настоящем изобретении означает алкильную группу (обычно содержащую от 1 до 6 атомов углерода), в которой один или большее количество атомов водорода С1-С6 алкильной группы заменены на аминогруппу формулы -N(Rc)2, в которой в каждом случае Rcнезависимо обозначает -Н или (С1-С6)алкил. Примеры аминоалкильных групп включают, но не ограничиваются только ими, -CH2NH2, -CH2CH2NH2, -CH2CH2CH2NH2, -CH2CH2CH2CH2NH2, -CH2CH2CH2CH2CH2NH2, -CH2CH2CH2CH2CH2CH2NH2,-CH2CH2CH2N(СН3)2, трет-бутиламинометил, изопропиламинометил и т.п.
Термин "арил" при использовании в настоящем изобретении означает 5-14-членную моноциклическую, бициклическую или трициклическую ароматическую кольцевую систему. Примеры арильной группы включают фенил и нафтил. Арильная группа может быть незамещенной или необязательно содержит один или большее количество заместителей, описанных ниже в настоящем изобретении. Примеры арильных групп включают фенильные или арильные гетероциклы, такие как пиррол, фуран, тиофен, тиазол, изотиазол, имидазол, триазол, тетразол, пиразол, оксазол, изоксазол, пиридин, пиразин, пиридазин, и пиримидин и т.п.
При использовании в настоящем изобретении термин "биологическая активность" указывает на влияние на один или большее количество клеточных или внеклеточных процессов (например, путем связывания, передачи сигнала и т.п.), которое может повлиять на физиологические или патофизиологические процессы.
Термин "карбонил" включает соединения и фрагменты, которые содержат атом углерода, связанный двойной связью с атомом кислорода. Примеры фрагментов, содержащих карбонил, включают, но не ограничиваются только ими, альдегиды, кетоны, карбоновые кислоты, амиды, сложные эфиры, ангидриды и т.п.
Термин "карбоксигруппа" или "карбоксил" означает группу -СООН или карбоновую кислоту.
Термин "Cm-Cn" означает от "m" атомов углерода до "n" атомов углерода. Например, термин "С1-С6" означает от 1 до 6 атомов углерода (C1, С2, С3, С4, С5 или С6). Термин "С2-С6" включает от 2 до 6 атомов углерода (С2, С3, C4, C5 или С6). Термин "С3-С6" включает от 3 до 6 атомов углерода (С3, C4, C5 или С6).
Термин "циклоалкил" при использовании в настоящем изобретении означает 3-14-членную насыщенную или ненасыщенную неароматическую моноциклическую, бициклическую или трициклическую углеводородную кольцевую систему. В этот класс включены циклоалкильные группы, которые сконденсированы с бензольным кольцом. Типичные циклоалкильные группы включают, но не ограничиваются только ими, циклопропил, циклобутил, циклобутенил, циклопентил, циклопентенил, циклопентадиенил, циклогексил, циклогексенил, 1,3-циклогексадиенил, циклогептил, циклогептенил, 1,3- циклогептадиенил, 1,4-циклогептадиенил, -1,3,5-циклогептатриенил, циклооктил, циклооктенил, 1,3-циклооктадиенил, 1,4-циклооктадиенил, -1,3,5-циклооктатриенил, декагидронафталин, октагидронафталин, гексагидронафталин, октагидроиндол, гексагидроинден, тетрагидроинден, декагидробензоциклогептен, октагидробензоциклогептен, гексагидробензоциклогептен, тетрагидробензоциклогептен, додекагидрогептален,декагидрогептален,октагидрогептален, гексагидрогептален, тетрагидрогептален, (1s,3s)-бицикло[1.1.0]бутан, бицикло[1.1.1 ]пентан, бицикло[2.1.1 ]гексан, бицикло[2.2.1 ]гептан, бицикло[2.2.2]октан, бицикло[3.1.1]гептан, бицикло[3.2.1]октан, бицикло[3.3.1]нонан, бицикло[3.3.2]декан, бицикло[3.3.]ундекан, бицикло[4.2.2]декан, и бицикло[4.3.1]декан. Циклоалкильная группа может быть незамещенной или необязательно содержит один или большее количество заместителей, описанных ниже в настоящем изобретении.
Термин "галоген" включает фтор, бром, хлор, йод и т.п.
Термин "галогеналкил" при использовании в настоящем изобретении означает С1-С6 алкильную группу, в которой один или большее количество атомов водорода С1-С6 алкильной группы заменены атомами галогена, которые могут быть одинаковыми или разными. Примеры галогеналкильных групп включают, но не ограничиваются только ими, трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил, пентахлорэтил, и 1,1,1-трифтор-2-бром-2-хлорэтил.
Термин "гетероалкил" по отдельности или в комбинации с другим термином означает, если не указано иное, стабильный, обладающий линейной или разветвленной цепью алкил или их комбинацию, содержащую из атомов углерода и от 1 до 3 гетероатомов, выбранных из группы, включающей О, N и S, и в которой атомы азота и серы необязательно могут быть окислены и гетероатом азота необязательно может быть кватернизован. Гетероатом(ы) О, N и S могут находиться в любом положении гетероалкильной группы. Примеры включают -СН2-СН2-О-СН3, -CH2-CH2-NH-CH3, -СН2-СН2-N(СН3)-СН3, -СН2-S-СН2-СН3, -СН2-СН2-S(O)-СН3, -СН2-СН2-S(O)2-СН3 и -СН2-CH=N-ОСН3. Рябом могут находиться до двух гетероатомов, например, -СН2-NH-OCH3. Если для описания гетероалкильной группы используется приставка, такая как (C2-C8), то в количество атомов углерода (в этом примере от 2 до 8) входят и гетероатомы. Например, С2-гетероалкильная группа включает, например, -СН2ОН (1 атом углерода и 1 гетероатом, заменяющий атом углерода) и -CH2SH.
Для дополнительной иллюстрации определения гетероалкильной группы, в которой гетероатомом является кислород, отметим, что гетероалкильная группа может представлять собой оксиалкильную группу. Например, (С2-С5)-оксиалкил включает, например, -СН2-O-СН3 (С3-оксиалкильная группа, содержащая 2 атома углерода и 1 атом кислорода, заменивший атом углерода), -CH2CH2CH2CH2OH, -OCH2CH2OCH2CH2OH, - ОСН2СН(ОН)CH2OH и т.п. При использовании в настоящем изобретении "арилалкил" означает
- алкиларильную группу, когда арилалкильная группа ковалентно связана с определенной химической структурой через алкильную группу. Примером арилалкильной группы является бензильная группа (-СН2-С6Н5). Арилалкильная группа может быть необязательно замещенной, т. е. арильная группа и/или алкильная группа может быть замещенной, как это раскрыто в настоящем изобретении.
Термин "гетероарил" при использовании в настоящем изобретении означает ароматическое гетероциклическое кольцо, содержащее от 5 до 14 элементов и содержащее по меньшей мере один гетероатом, выбранный из группы, включающей азот, кислород и серу, и содержащее по меньшей мере 1 атом углерода, включая моноциклические, бициклические и трициклические кольцевые системы. Типичными гетероарилы являются триазолил, тетразолил, оксадиазолил, пиридил, фурил, бензофуранил, тиенил, бензотиенил, хинолинил, пирролил, индолил, оксазолил, бензоксазолил, имидазолил, бензимидазолил, тиазолил, бензотиазолил, изоксазолил, пиразолил, изотиазолил, пиридазинил, пиримидинил, пиразинил, триазинил, циннолинил, фталазинил, хиназолинил, пиримидил, азепинил, оксепинил, хиноксалинил и оксазолил. Гетероарильная группа может быть незамещенной или необязательно содержит один или большее количество заместителей, описанных ниже в настоящем изобретении.
При использовании в настоящем изобретении термин "гетероатом" включает кислород (О), азот (N) и серу (S).
При использовании в настоящем изобретении термин "гетероцикл" означает 3-14-членные кольцевые системы, которые являются насыщенными, ненасыщенными или ароматическими и которые содержат от 1 до 4 гетероатомов, независимо выбранных из группы, включающей азот, кислород и серу, и в которой атомы азота и серы необязательно могут быть окислены и гетероатом азота необязательно может быть кватернизован, включая моноциклические, бициклические, и трициклические кольцевые системы. Бициклические и трициклические кольцевые системы могут включать гетероцикл или гетероарил, сконденсированный с бензольным кольцом. Гетероцикл может быть присоединен через любой гетероатом или атом углерода, если это является химически приемлемыми. Гетероциклы включают гетероарилы, определенные выше. Типичные примеры гетероциклов включают, но не ограничиваются только ими, азиридинил, оксиранил, тииранил, триазолил, тетразолил, азиринил, диазиридинил, диазиринил, оксазиридинил, азетидинил, азетидинонил, оксетанил, тиетанил, пиперидинил, пиперазинил, морфолинил, пирролил, оксазинил, тиазинил, диазинил, диоксанил, триазинил, тетразинил, имидазолил, тетразолил, пирролидинил, изоксазолил, фуранил, фуразанил, пиридинил, оксазолил, бензоксазолил, бензизоксазолил, тиазолил, бензтиазолил, тиенил, пиразолил, триазолил, пиримидинил, бензимидазолил, изоиндолил, индазолил, бензодиазолил, бензотриазолил, бензоксазолил, бензизоксазолил, пуринил, индолил, изохинолинил, хинолинил и хиназолинил. Гетероциклильная группа может быть незамещенной или необязательно содержит один или большее количество заместителей, описанных ниже в настоящем изобретении.
Термин "гетероциклоалкил" по отдельности или в комбинации с другими терминами означает, если не указано иное, циклический вариант "гетероалкила". Кроме того, гетероатом может находиться в положении, с помощью которого гетероцикл присоединен к остальной части молекулы. Примеры гетероциклоалкил включают 1-(1,2,5,6-тетрагидропиридил), 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 4-морфолинил, 3-морфолинил, тетрагидрофуран-2-ил, тетрагидрофуран-3-ил, тетрагидротиен-2-ил, тетрагидротиен-3-ил, 1-пиперазинил, 2-пиперазинил и т.п.
Термин "гидроксиалкил" при использовании в настоящем изобретении означает алкильную группу, содержащую указанное количество атомов углерода, в которой один или большее количество атомов водорода алкильной группы заменен группой -ОН. Примеры гидроксиалкильных групп включают, но не ограничиваются только ими, -СН2ОН, -СН2СН2ОН, -СН2СН2СН2ОН, -CH2CH2CH2CH2OH, -CH2CH2CH2CH2CH2OH, -CH2CH2CH2CH2CH2CH2OH и их разветвленные варианты.
Термин "гидроксигруппа" или "гидроксил" включает группы -ОН или -О-. При использовании в настоящем изобретении N-оксид или аминоксид, означает соединение образованное из третичного амина путем присоединения атома кислорода к атому азота, R3N+-О-. В расширенном виде термин включает аналогичные производные первичных и вторичных аминов.
При использовании в настоящем изобретении и если не указано иное, термин "стереоизомер" означает один стереоизомер соединения, который в основном не содержит других стереоизомеров этого соединения. Например, стереоизомерно чистое соединение, содержащее один хиральный центр, в основном не содержит зеркального изомера этого соединения. Стереоизомерно чистое соединение, содержащее два хиральных центра, в основном не содержит других стереоизомеров этого соединения. В некоторых вариантах осуществления стереоизомерно чистое соединение содержит более примерно 80 мас.% одного стереоизомера соединения и менее примерно 20 мас.% других стереоизомеров соединения, например, более примерно 90 мас.% одного стереоизомера соединения и менее примерно 10 мас.% других стереоизомеров соединения, или более примерно 95 мас.% одного стереоизомера соединения и менее примерно 5 мас.% других стереоизомеров соединения, или более примерно 97 мас.% одного стереоизомера соединения и менее примерно 3 мас.% других стереоизомеров соединения.
При использовании в настоящем изобретении "белок" является синонимом "пептида", "полипептида" или "пептидного фрагмента". "Очищенный" полипептид, белок, пептид, или пептидный фрагмент в основном не содержит клеточного вещества или других загрязняющих белков клетки, или не содержащего клеток источника, из которого получают аминокислотную последовательность, или в основном не содержит химических предшественников или других химикатов, использующихся при химическом синтезе.
При использовании в настоящем изобретении "модулирование" означает увеличение или уменьшение содержания пептида или полипептида, или увеличение или уменьшение стабильности или активности пептида или полипептида. Термин "ингибирование" означает уменьшение содержания пептида или полипептида или уменьшение стабильности или активности пептида или полипептида. В предпочтительных вариантах осуществления пептидом, который модулируют или ингибируют, является S-нитрозоглутатион (GSNO) или белки S-нитрозотиолы (SNO).
При использовании в настоящем изобретении термины "оксид азота" и "NO" включают незаряженный оксид азота и заряженный оксид азота, в особенности, включая нитрозониевый ион (NO+) и нитроксильный ион (NO-). Реакционноспособная форма оксида азота может образоваться из газообразного оксида азота. Соединения, содержащие структуру X-NOy, в которой Х обозначает фрагмент, высвобождающий, доставляющий или переносящий оксид азота, включают любые и все такие соединения, которые доставляют оксид азота в положение, в котором он оказывает воздействие, в форме, активной для воздействия, и в которых Y рано 1 или 2.
При использовании в настоящем изобретении термин "фармацевтически приемлемый" означает утвержденный федеральным правительством или правительством штата к применению для животных, более предпочтительно для людей, или включенный в Фармакопею США или другие общепризнанных Фармакопеи. Термин "носитель" означает разбавитель, вспомогательное вещество, инертный наполнитель или разбавитель, вместе с которым вводят лекарственное средство, и включает, но не ограничивается только ими такие стерильные жидкости, как вода и масла.
"Фармацевтически приемлемая соль" или "соль" соединения, предлагаемого в настоящем изобретении, является производным раскрытого соединения, которое содержит ионную связь и которое обычно получают по реакции раскрытого соединения с кислотой или основанием, подходящим для введения субъекту. Фармацевтически приемлемая соль может включать, но не ограничивается только ими, соли присоединения с кислотами, включая гидрохлориды, гидробромиды, фосфаты, сульфаты, гидросульфаты, алкилсульфонаты, арилсульфонаты, арилалкилсульфонаты, ацетаты, бензоаты, цитраты, малеаты, фумараты, сукцинаты, лактаты и тартраты; катионы щелочных металлов, такие как Li, Na, и К, соли щелочноземельных металлов, таких как Mg или Са, или соли органического амина.
"Фармацевтическая композиция" представляет собой композицию, содержащую раскрытые соединения в форме, подходящей для введения субъекту. Фармацевтическую композицию, предлагаемую в настоящем изобретении, предпочтительно готовят так, чтобы она была совместима с предполагаемым путем введения. Примеры путей введения включают, но не ограничиваются только ими, пероральное и парентеральное, например, внутривенное, внутрикожное, подкожное, ингаляционное, местное, чрескожное, проводимое через слизистую оболочку и ректальное введение.
Термин "замещенный" при использовании в настоящем изобретении означает, что любой один или большее количество атомов водорода указанного атома заменены радикалами, выбранными из указанной группы, при условии, что не превышена нормальная валентность указанного атома и что замещение приводит к стабильному соединению. Если заместителем является кетогруппа (т. е. =O), то заменяются 2 атома водорода этого атома. Кольцевые двойные связи при использовании в настоящем изобретении представляют собой двойные связи, которые образованы между двумя соседними атомами кольца (например, С=С, C=N или N=N).
Заместители групп алкил, гетероалкил, алкилен, алкенил, алкинил, циклоалкил, гетероциклоалкил, циклоалкенил и гетероциклоалкенил можно выбрать из числа различных групп, включая -ORd, =O, =NRd, =N-ORd, -NRd'Rd”, -SRd', -галоген, -SiRd'Rd”Rd'", -ОС(O)Rd', -C(O)Rd', -CO2Rd', -CONRd'Rd”, -OC(O)NRd'Rd", -NRd”C(O)Rd, -NRd'”C(O)NRd'Rd", NRd'”SO2NRd'Rd”, -NRd”CO2Rd, -NHC(NH2)=NH, -NRa'C(NH2)=NH, -NHC(NH2)=NRd', -S(O)Rd', -SO2Rd', -SO2NRd'Rd", -NRd”SO2Rd, -CN и -NO2, в количестве, находящемся в диапазоне от 0 до 3, и типичными являются такие группы, содержащие 0, 1 или 2 заместителя.
Rd', Rd" и Rd'" все независимо обозначают водород, незамещенный (С1-С8)алкил, незамещенный гетеро(С1-С8)алкил, незамещенный арил и арил содержащий от 0 до 3 заместителей, выбранных из группы, включающей -галоген, незамещенный алкил, незамещенную алкоксигруппу, незамещенную тиоалкоксигруппу и незамещенный арил (С1-С4)алкил. Если Rd' и Rd" присоединены к одному атому азота, они могут объединяться с атомом азота с образованием 5-, 6- или 7-членного кольца. Например, -NRd'Rd" может означать 1-пирролидинил или 4-морфолинил.
Обычно алкильная или гетероалкильная группа содержит от 0 до 3 заместителей, и эти группы, содержащие 2 или меньшее количество заместителей, являются типичными для настоящего изобретения. Алкильный или гетероалкильный радикал может быть незамещенным или монозамещенным. В некоторых вариантах осуществления алкильный или гетероалкильный радикал является незамещенным.
Типичные заместители алкильных и гетероалкильных радикалов включают, но не ограничиваются только ими ORd, =O, =NRd, =N-ORd, -NRd'Rd”, -SRd', -галоген, -SiRd'Rd”Rd'", -ОС(O)Rd', -C(O)Rd', -CO2Rd', -CONRd'Rd”, -OC(O)NRd'Rd", -NRd”C(O)Rd, -NRd'”C(O)NRd'Rd", NRd'”SO2NRd'Rd”, -NRd”CO2Rd, -NHC(NH2)=NH, -NRa'C(NH2)=NH, -NHC(NH2)=NRd', -S(O)Rd', -SO2Rd', -SO2NRd'Rd", -NRd”SO2Rd, -CN и -NO2, где Rd', Rd" и Rd'" являются такими, как определено выше. Типичные заместители можно выбрать из группы, включающей: ORd, =O, -NRd'Rd”, -галоген, -OC(O)Rd', -CO2Rd', -С(О)NRd'Rd”, -OC(O)NRd'Rd", -NRd"C(O)Rd', -NRd"CO2Rd', -NRd'"SO2NRd'Rd", -SO2Rd', -SO2NRd'Rd", -NRd"SO2Rd', -CN и -NO2.
Аналогичным образом, заместители арильных и гетероарильных групп являются разными и выбраны из группы, включающей: -галоген, -ORe', -OC(O)Re', -NRe'Re", -SRe', -Re', -CN, -NO2, -CO2Re', -C(O)NRe'Re", -C(O)Re', -OC(O)NRe'Re", -NRe”C(O)Re', -NRe"CO2Re', -NRe"'C(O)NRe'Re", NRe'”SO2NRe'Re”, -NHC(NH2)=NH, -NRe'C(NH2)=NH, -NH-C(NH2)=NRe', -S(O)Re', -SO2Re', -SO2NRe'Re", -NRe"SO2Re', -N3, -CH(Ph)2, перфторалкоксигруппу и перфтор(С1-С4)алкил, и их количество находится в диапазоне от 0 до полного количества свободных валентных связей ароматической кольцевой системы.
Re', Re" и Re'" независимо выбраны из группы, включающей водород, незамещенный (C1-C8)алкил, незамещенный гетеро(С1-С8)алкил, незамещенный арил, незамещенный гетероарил, незамещенный арил(С1-С4)алкил и незамещенный арилокси(С1-С4)алкил. Обычно арильная или гетероарильная группа содержит от 0 до 3 заместителей и эти группы, содержащие 2 или меньшее количество заместителей, являются типичными для настоящего изобретения. В одном варианте осуществления настоящего изобретения арильная или гетероарильная группа является незамещенной или монозамещенной. В другом варианте осуществления арильная или гетероарильная группа является незамещенной.
Два из заместителей, находящихся у соседних атомов арильного или гетероарильного кольца в арильной или гетероарильной группе, описанной в настоящем изобретении необязательно могут быть заменены заместителем формулы -T-C(O)-(СН2)q-U-, в которой Т и U независимо обозначают -NH-, -O-, -СН2- или ординарную связь и q является целым числом, равным от 0 до 2. Альтернативно, два из заместителей, находящихся у соседних атомов арильного или гетероарильного кольца, необязательно могут быть заменены заместителем формулы -J-(СН2)r-K-, в которой J и К независимо обозначают -СН2-, -O-, -NH-, -S-, -S(O)-, -S(O)2-, -S(O)2NRf'- или ординарную связь и r является целым числом, равным от 1 до 3. Одна из ординарных связей образовавшегося таким образом нового кольца необязательно может быть заменена двойной связью. Альтернативно, два из заместителей, находящихся у соседних атомов арильного или гетероарильного кольца, необязательно могут быть заменены заместителем формулы -(СН2)s-X-(СН2)t-, в которой s и t независимо являются целыми с числами, равными от 0 до 3, и Х обозначает -O-, -NRf'-, -S-, -S(O)-, -S(O)2- или -S(O)2NRa-. Заместитель Rf' в -NRf'- и -S(O)2NRf' - выбран из группы, включающей водород или незамещенный (С1-С6)алкил.
"Стабильное соединение" и "стабильная структура" означает соединение, которое является достаточно стабильным, чтобы выдержать выделение из реакционной смеси с обеспечением необходимой степени чистоты и включение в эффективное терапевтическое средство.
При использовании в настоящем изобретении термин "терапевтически эффективное количество" обычно означает количество, необходимое для облегчения по меньшей мере одного симптома нарушения, которое предотвращают, ослабляют или лечат, как это описано в настоящем изобретении. Выражение "терапевтически эффективное количество" применительно к ингибиторам GSNOR, предлагаемыми в настоящем изобретении, означает дозу ингибитора GSNOR, которая обеспечивает конкретный фармакологический ответ, для которого ингибитор GSNOR вводят значительному количеству субъектов, нуждающихся в таком лечении. Следует подчеркнуть, что терапевтически эффективное количество ингибитора GSNOR, которое вводят конкретному субъекту в конкретном случае, не всегда эффективно для лечения патологических состояний/заболеваний, описанных в настоящем изобретении, даже если специалисты в данной области техники считают, что такая доза представляет собой терапевтически эффективное количество.
Термин "биологический образец" включает, но не ограничивается только ими, образцы крови (например, сыворотка, плазма или цельная кровь), мочи, слюны, пота, грудного молока, влагалищных секретов, спермы, фолликул волос, кожи, зубов, костей, ногтей или других выделений, жидкостей организма, тканей или клеток. В контексте настоящего изобретения содержание GSNOR в биологическом образце можно определить по методикам, описанным в Публикации заявки на патент U.S. № 2005/0014697.
D. Фармацевтические композиции
В объем настоящего изобретения входят фармацевтические композиции, содержащие по меньшей мере одно соединение, предлагаемое в настоящем изобретении, описанное в настоящем изобретении и по меньшей мере один фармацевтически приемлемый носитель. Подходящие носители описаны в публикации "Remington: The Science and Practice, Twentieth Edition", published by Lippincott Williams & Wilkins, которая включена в настоящее изобретение в качестве ссылки. Фармацевтические композиции, предлагаемые в настоящем изобретении, также могут содержать одно или большее количество активных средств, не являющихся соединениями, предлагаемыми в настоящем изобретении.
Фармацевтические композиции, предлагаемые в настоящем изобретении, могут содержать новые соединения, описанные в настоящем изобретении, фармацевтические композиции могут содержать известные соединения, для которых ранее не было известно, что они обладают ингибирующей активностью по отношению к GSNOR, или их комбинацию.
Соединения, предлагаемые в настоящем изобретении, можно использовать в любой фармацевтически приемлемой дозированной форме, включая, но не ограничиваясь только ими, дозированные формы для инъекций, жидкие дисперсии, гели, аэрозоли, мази, кремы, лиофилизированные препараты, сухие порошки, таблетки, капсулы, препараты регулируемого высвобождения, легкоплавкие препараты, препараты задержанного высвобождения, препараты пролонгированного высвобождения, препараты периодического высвобождения, смешанные препараты немедленного и регулируемого высвобождения и т.п. В частности, соединения, предлагаемые в настоящем изобретении, описанные в настоящем изобретении можно приготовить: (а) для введения, выбранного из группы, включающей пероральное, легочное, внутривенное, внутриартериальное, внутриоболочечное, внутрисуставное, ректальное, глазное, толстокишечное, парентеральное, интрацистернальное, внутривагинальное, внутрибрюшинное, локальное, трансбуккальное, назальное и местное введение; (b) в дозированной форме, выбранной из группы, включающей жидкие дисперсии, гели, аэрозоли, мази, кремы, таблетки, пакеты и капсулы; (с) в дозированной форме, выбранной из группы, включающей лиофилизированные препараты, сухие порошки, легкоплавкие препараты, препараты регулируемого высвобождения, препараты задержанного высвобождения, препараты пролонгированного высвобождения, препараты периодического высвобождения и смешанные препараты немедленного и регулируемого высвобождения; или (d) в любой их комбинации.
При инфекциях дыхательных путей для обеспечения высоких локальных концентраций можно использовать препарат для ингаляции. Препараты, подходящие для ингаляции, включают сухие порошки или аэрозоли растворов или испаренные растворы, дисперсии или суспензии, которые пригодны для дозирования с помощью ингалятора или устройства типа небулайзер в эндобронхиальную полость или полость рта инфицированных пациентов для лечения бактериальных инфекций верхних и нижних дыхательных путей.
Растворы или суспензии, использующиеся для парентерального, внутрикожного или подкожного введения, могут содержать один или большее количество следующих компонентов: (1) стерильный разбавитель, такой как вода для инъекций, физиологический раствор, нелетучие масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; (2) антибактериальные средства, такие как бензиловый спирт или метилпарабены; (3) антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; (4) хелатные агенты, такие как этилендиаминтетрауксусная кислота; (5) буферы, такие как ацетаты, цитраты или фосфаты; и (5) реагенты для регулирования тоничности, такие как хлорид натрия или декстроза. Значение рН можно регулировать кислотами или основаниями, такими как хлористоводородная кислота или гидроксид натрия. Препараты для парентерального введения можно помещать в ампулы, одноразовые шприцы или содержащие несколько доз флаконы, изготовленные из стекла или пластмассы.
Фармацевтические композиции, подходящие для инъекции, могут включать стерильные водные растворы (в случае растворимости в воде) или дисперсии и стерильные порошки проводимого в случае необходимости приготовления стерильных растворов или дисперсий для инъекции. Для внутривенного введения подходящие носители включают физиологический раствор, бактериостатическую воду, кремофор EL (BASF, Parsippany, N.J.) или забуференный фосфатом физиологический раствор (ЗФФ). Во всех случаях композиция должна быть стерильной и должна быть текучей в такой степени, чтобы ее без труда можно было ввести шприцем. Фармацевтическая композиция должна быть стабильной при условиях ее приготовления и хранения и должна быть защищена от загрязняющего воздействия микроорганизмов, таких как бактерии и грибы.
Носителем может быть растворитель или диспергирующие среды, включая, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль и т.п.) и подходящие их смеси. Надлежащую сыпучесть можно обеспечивать, например, путем использования материала покрытия, такого как лецитин, путем поддержания необходимого размера частиц в случае дисперсии и путем использования поверхностно-активных веществ. Предотвращение воздействия микроорганизмов можно обеспечить с помощью различных антибактериальных или фунгицидных средств, например, парабенов, хлорбутанола, фенола, аскорбиновой кислоты, тимеросала и т.п. Во многих случаях в композицию предпочтительно включать изотонические агенты, например, сахара, многоатомные спирты, такие как маннит или сорбит, и неорганические соли, такие как хлорид натрия. Пролонгирование всасывание композиций для инъекций можно обеспечить путем включения в композицию агента, который задерживает всасывание, например, моностеарата алюминия и желатина.
Стерильные растворы для инъекций можно приготовить путем включения необходимого количества активного реагента в подходящем растворителе вместе с одним ингредиентом или комбинацией перечисленных выше ингредиентов, если необходимо, то с последующей стерилизацией фильтрованием. Обычно дисперсии готовят путем включения по меньшей мере одного соединения, предлагаемого в настоящем изобретении, в стерильный разбавитель, который содержит основную диспергирующую среду и любые другие необходимые ингредиенты. В случае стерильных порошков, предназначенных для приготовления стерильных растворов для инъекций, типичные методики приготовления включают вакуумную сушку и сушку вымораживанием, обе эти методики дают порошкообразное соединение, предлагаемое в настоящем изобретении, содержащее любой дополнительный необходимый ингредиент из его подвергнутого стерильному фильтрованию раствора.
Композиции для перорального введения обычно включают инертный разбавитель или пищевой носитель. Их можно поместить, например, в капсулы из желатина или спрессовать в таблетки. Для перорального терапевтического введения соединение, предлагаемое в настоящем изобретении, можно включить в инертные наполнители и использовать в форме таблеток, пастилок или капсул. Композиции для перорального введения также можно приготовить с использованием жидкого носителя для применения в качестве жидкости для полоскания рта, когда соединение в жидком носителе вводят в полость рта и полощут и выплевывают или проглатывают. Фармацевтически совместимые связующие и/или вспомогательные вещества можно включать в качестве части композиции.
Для введения путем ингаляции соединения доставляют в виде спрея аэрозоля из находящегося под давлением контейнера или дозатора, который содержит подходящий пропеллент, например, газ, такой как диоксид углерода, распыляемой жидкости или сухого порошка из подходящего устройства. Для введения через слизистую оболочку или чрескожного введения в препарате используют средства, увеличивающие проницаемость, обеспечивающие прохождение через барьер. Такие средства, увеличивающие проницаемость, обычно известны в данной области техники и включают, например, для чрескожного введения, детергенты, желчные соли и производные фусидовой кислоты. Введение через слизистую оболочку можно провести путем использования назальных спреев и суппозиториев. Для чрескожного введения активные ингредиенты готовят в виде притираний, мазей, гелей или кремов, как это общеизвестно в данной области техники. Реагенты также можно приготовить в виде суппозиториев (например, с использованием обычных основ для суппозиториев, таких как масло какао и другие глицериды) или удерживающих клизм для ректальной доставки.
В одном варианте осуществления соединения, предлагаемые в настоящем изобретении, готовят вместе с носителями, которые защищают от быстрого выведения из организма. Например, можно использовать препарат регулируемого высвобождения, включая имплантаты и микрокапсулированные системы доставки. Можно использовать биологически разлагающиеся биологически совместимые полимеры, такие как сополимер этилен-винилацетат, полиангидриды, полигликолевую кислоту, коллаген, сложные полиортоэфиры и полимолочную кислоту. Методики приготовления таких препаратов очевидны для специалистов в данной области техники.
Липосомные суспензии (включая липосомы, направленно воздействующие на инфицированные клетки моноклональными антителами к вирусным антигенам) также можно использовать в качестве фармацевтически приемлемых носителей. Их можно получить по методикам, известным специалистам в данной области техники, например, как это описано в патенте U.S. № 4522811.
Кроме того, суспензии соединений, предлагаемых в настоящем изобретении, в соответствующих случаях можно приготовить в виде масляных суспензии для инъекций. Подходящие липофильные растворители или разбавители включают нелетучие масла, такие как кунжутное масло, или синтетические эфиры жирный кислот, такие как этилолеат, триглицериды, или липосомы. Для доставки также можно использовать не являющиеся липидами поликатионогенные полимеры аминов. В суспензию также необязательно можно включать подходящие стабилизаторы или агенты для повышения растворимости соединений и обеспечения приготовления высококонцентрированных растворов.
Для обеспечения легкого введения и равномерности дозировки особенно предпочтительно готовить композиции для перорального или парентерального введения в разовой дозированной форме. Разовая дозированная форма при использовании в настоящем изобретении означает отдельные порции, пригодные для использования в качестве однократных доз для подвергающегося лечению субъекта; каждая порция содержит заданное количество соединения, предлагаемого в настоящем изобретении, по оценкам обеспечивающее необходимый терапевтический эффект, вместе с необходимым фармацевтическим носителем. Характеристики разовых дозированных форм, предлагаемых в настоящем изобретении, определяются специфическими характеристиками соединения, предлагаемого в настоящем изобретении, и непосредственно зависят от них, и конкретным терапевтическим эффектом, который необходимо обеспечить, и ограничениями, специфическими в методологии приготовления препаратов, содержащих активное средство, предназначенное для лечения индивидуумов.
Фармацевтические композиции, предлагаемые в настоящем изобретении, содержащие по меньшей мере одно соединение, предлагаемое в настоящем изобретении, могут содержать один или большее количество фармацевтических инертных наполнителей. Примеры таких инертных наполнителей включают, но не ограничиваются только ими связующие агенты, наполнители, смазывающие агенты, суспендирующие агенты, подсластители, ароматизаторы, консерванты, буферы, смачивающие агенты, разрыхлители, шипучие агенты и другие инертные наполнители. Такие инертные наполнители известны в данной области техники. Типичные инертные наполнители включают: (1) связующие агенты, которые включают различные целлюлозы и сшитый поливинилпирролидон, микрокристаллическую целлюлозу, такую как Avicel® PH101 и Avicel® PH102, силицифицированную микрокристаллическую целлюлозу (ProSolv SMCC™), трагакантовую камедь и желатин; (2) наполнители, такие как различные крахмалы, лактоза, моногидрат лактозы и безводная лактоза; (3) разрыхлители, такие как альгиновая кислота, примогель, кукурузный крахмал, немного сшитый поливинилпирролидон, картофельный крахмал, кукурузный крахмал и модифицированные крахмалы, натриевая соль кроскармелозы, кросповидон, натриевая соль гликолята крахмала и их смеси; (4) смазывающие вещества, включая агенты, которые влияют на сыпучесть прессуемого порошка, включая стеарат магния, коллоидный диоксид кремния, такой как Aerosil® 200, тальк, стеариновая кислота, стеарат кальция и силикагель; (5) агенты, придающие скользкость, такие как коллоидный диоксид кремния; (6) консерванты, такие как сорбат калия, метилпарабен, пропилпарабен, бензойная кислота и ее соли, другие эфиры пара-гидроксибензойной кислоты, такие как бутилпарабен, спирты, такие как этиловый или бензиловый спирт, фенолы, такие как фенол, или четвертичные соединения, такие как бензалконийхлорид; (7) разбавители, такие как фармацевтически приемлемые инертные наполнители, такие как микрокристаллическая целлюлоза, лактоза, гидрофосфат кальция, сахариды, и/или смеси любых из указанных выше веществ; примеры разбавителей включают микрокристаллическую целлюлозу, такую как Avicel® PH 101 и Avicel® PH102; лактозу, такую как моногидрат лактозы, безводная лактоза и Pharmatose® DCL21; гидрофосфат кальция, такой как Emcompress®; маннит; крахмал; сорбит; сахарозу; и глюкозу; (8) подсластители, включая любой натуральный или искусственный подсластитель, такой как сахароза, сахарин, ксилит, натриевая соль сахарина, цикламат, аспартам и ацесульфам; (9) ароматизаторы, такие как мята перечная, метилсалицилат, апельсиновый ароматизатор, Magnasweet® (торговая марка фирмы MAFCO), ароматизатор для надувной жевательной резинки, фруктовые ароматизаторы и т.п.; и (10) шипучие агенты, включая двухкомпонентные шипучие смеси, такие как органическая кислота и карбонат или бикарбонат. Подходящие органические кислоты включают, например, лимонную, винную, яблочную, фумаровую, адипиновую, янтарную и альгиновую кислоты и ангидриды и соли кислот. Подходящие карбонаты и бикарбонаты включают, например, карбонат натрия, бикарбонат натрия, карбонат калия, бикарбонат калия, карбонат магния, глицинкарбонат натрия, карбонат L-лизина и карбонат аргинина. Альтернативно, в качестве компонента двухкомпонентной шипучей смеси может содержаться только бикарбонат натрия.
Е. Наборы, содержащие композиции, предлагаемые в настоящем изобретении
В объем настоящего изобретения также входят наборы, содержащие композиции, предлагаемые в настоящем изобретении. Такие наборы могут содержать, например, (1) по меньшей мере одно соединение, предлагаемое в настоящем изобретении; и (2) по меньшей мере один фармацевтически приемлемый носитель в качестве растворителя или раствора. Необязательно можно включать дополнительные компоненты набора, например: (1) любой из фармацевтически приемлемых инертных наполнителей, указанных в настоящем изобретении, такой как стабилизаторы, буферы и т.п., (2) по меньшей мере один контейнер, флакон или аналогичный сосуд для размещения и/или смешивания набора компонентов; и (3) устройство для доставки, такое как ингалятор, устройство типа небулайзер, шприц и т.п.
F. Способы получения соединений, предлагаемых в настоящем изобретении Соединения, предлагаемые в настоящем изобретении, можно легко синтезировать по известным методикам синтеза или по модифицированным известным методикам синтеза. Как легко может понять специалист в данной области техники, методики, описанные ниже, позволяют синтезировать аналоги, содержащие различные заместители. Типичные методики синтеза описаны в приведенном ниже разделе "Примеры".
При необходимости дополнительную очистку и разделение энантиомеров и диастереоизомеров можно провести по стандартным методикам, известным в данной области техники. Так, например, разделение энантиомеров соединения можно провести с помощью хиральной ВЭЖХ (высокоэффективная жидкостная хроматография) и аналогичных хроматографических методик. Аналогичным образом можно разделить диастереоизомеры. Однако в некоторых случаях диастереоизомеры можно разделить просто механически, например, путем регулируемого осаждения или кристаллизации.
Способ, предлагаемый в настоящем изобретении, при его осуществлении так, как указано в настоящем изобретении, можно без труда осуществить при температурах, которые стандартны для данной области техники. В одном варианте осуществления способ осуществляют при температуре в диапазоне от примерно 25 до примерно 110°С. В другом варианте осуществления температура находится в диапазоне от примерно 40 до примерно 100°С. В еще одном варианте осуществления температура находится в диапазоне от примерно 50 до примерно 95°С.
Стадии синтеза, для которых необходимо основание, проводят с использованием обычного органического или неорганического основания. Обычно основание не является нуклеофильным реагентом. Так, в одном варианте осуществления основание выбрано из группы, включающей карбонаты, фосфаты, гидроксиды, алкоксиды, соли дисилазанов и третичные амины.
Способ, предлагаемый в настоящем изобретении, при его осуществлении так, как это описано в настоящем изобретении, можно в основном завершить в течение от нескольких минут до нескольких часов в зависимости от типа и количества реагентов и температуры, при которой проводится реакция. Определение того, в основном завершилась ли реакция, обычно можно провести с помощью обычных методик, известных в данной области техники, таких как, например, ВЭЖХ, ЖХМС (жидкостная хроматография - масс-спектроскопия), ТСХ (тонкослойная хроматография) и1H ЯМР. G. Способы лечения
В объем настоящего изобретения входят способы лечения или предупреждения (например, облегчение одного или большего количества симптомов) патологического состояния путем использования одного или большего количества раскрытых соединений. Способы включают введение терапевтически эффективного количества соединения, предлагаемого в настоящем изобретении, нуждающемуся в нем пациенту. Композиции, предлагаемые в настоящем изобретении, также можно использовать для профилактического лечения.
Соединение, предлагаемое в настоящем изобретении, применяющееся в способах лечения, предлагаемых в настоящем изобретении, может представлять собой: (1) новое соединение, описанное в настоящем изобретении, или его фармацевтически приемлемую соль, его пролекарство, его метаболит или его стереоизомер; (2) соединение, которое было известно до настоящего изобретения, но для которого не было известно, что соединение является ингибитором GSNOR, или его фармацевтически приемлемой солью, его пролекарством, его метаболитом или его стереоизомером; или (3) соединение, которое было известно до настоящего изобретения, и для которого было известно, что соединение является ингибитором GSNOR, но для которого не было известно, что соединение применимо в способах лечения, описанных в настоящем изобретении, или его фармацевтически приемлемую соль, его пролекарство, его метаболит или его стереоизомер.
Пациентом может быть любое животное, домашнее, домашний скот или дикое, включая, но не ограничиваясь только ими кошек, собак, лошадей, свиней и крупный рогатый скот, и предпочтительно людей. При использовании в настоящем изобретении термины пациент и субъект можно использовать взаимозаменяемым образом.
При использовании в настоящем изобретении "лечение" описывает лечение и уход за пациентом с целью борьбы с заболеванием, патологическим состоянием или нарушением и включает введение соединения, предлагаемого в настоящем изобретении, для предупреждения возникновения симптомов или осложнений, облегчения симптомов или осложнений или устранения заболевания, патологического состояния или нарушения. Точнее, "лечение" включает обращение, ослабление, облегчение, сведение к минимуму, подавление или устранение по меньшей мере одного вредного симптома или проявления патологического состояния (нарушения), прогрессирования заболевания, возбудителя заболевания (например, бактерий или вирусов), или другого аномального состояния. Лечение продолжают, пока не произойдет ослабление симптомов и/или патологии.
Обычно доза, т.е. терапевтически эффективное количество, находится в диапазоне от 1 мкг/кг до 10 г/кг и часто находится в диапазоне от 10 мкг/кг до 1 г/кг или от 10 мкг/кг до 100 мг/кг(массы тела) подвергающегося лечению субъекта в сутки.
Н. Применение GSNOR
У субъектов с неблагоприятно высокими содержанием GSNOR или активностью GSNOR модуляцию можно обеспечить, например, путем введения одного или большего количества раскрытых соединений, которые нарушают или понижающе регулируют функцию GSNOR или уменьшают содержание GSNOR. Эти соединения можно вводить вместе с другими ингибиторами GSNOR, такими как антитела или фрагменты антител к GSNOR, антисмысловые средства, воздействующие на GSNOR, iRNA или малые молекулы, или другие ингибиторы, по отдельности или в комбинации с другими средствами, подробно описанными в настоящем изобретении.
Настоящее изобретение относится к способу лечения субъекта, страдающего от нарушения, протекание которого улучшается с помощью лечения донором NO. Такой способ включает введение субъекту терапевтически эффективного количества ингибитора GSNOR.
Нарушения могут включать нарушения легких, связанные с гипоксемией и/или сокращением гладких мышц легких и дыхательных путей и/или инфекция легких и/или воспаление легких и/или поражение легких (например, легочная гипертензия, ОРДС (острый респираторный дистресс синдром), астма, пневмония, фиброз легких/интерстициальные заболевания легких, муковисцидоз, ХОЗЛ); сердечно-сосудистое заболевание и заболевание сердца (например, гипертензия, ишемические коронарные синдромы, атеросклероз, сердечная недостаточность, глаукома); заболевания, характеризующиеся ангиогенезом (например, заболевание коронарной артерии); нарушения, при которых существует опасность тромбоза; нарушения, при которых существует опасность рестеноза; воспалительные заболевания (например, связанное со СПИД (синдром приобретенного иммунодефицита) слабоумие, воспалительная болезнь кишечника (ВБК), болезнь Крона, колит и псориаз); функциональные нарушения кишечника (например, синдром раздраженной толстой кишки (СРК)); заболевания, при которых существует опасность апоптоза (например, сердечная недостаточность, атеросклероз, дегенеративные неврологические нарушения, артрит и поражение печени (ишемическое или алкогольное)); импотенцию; апноэ во сне; заживление ран при диабете; кожные инфекции; лечение псориаза; ожирение, вызванное пристрастием к еде; удар; реперфузионное поражение (например, травматическое поражение мышц сердца или легких или повреждение с размозжением тканей); и нарушения, при которых благоприятна предварительная адаптация сердца или головного мозга для защиты с помощью NO от последующих ишемических приступов, нарушения центральной нервной системы (ЦНС) (например, тревога, депрессия, психоз и шизофрения); и инфекции, вызванные бактериями (например, в частности, туберкулез, инфицирование посредством С. difficile).
В одном варианте осуществления соединения, предлагаемые в настоящем изобретении, или их фармацевтически приемлемую соль или пролекарство, стереоизомер или их метаболит можно вводить в комбинации с донором NO. Донор NO поставляет оксид азота или родственные окислительно-восстановительные соединения и в более общем случае обеспечивает биологическую активность оксида азота, т. е. активность, которая связана с оксидом азота, например, расширение сосудов или стимулирование или ингибирование рецепторного белка, например, белка ras, адренергического рецептора, NFkB. Доноры NO, включая S-нитрозо-, O-нитрозо-, С-нитрозо- и N-нитрозосоединения и их нитропроизводные и комплексы NO с металлами, не исключая другие соединения, приводящие к проявлению биологической активности NO, применимые в настоящем изобретении, описаны в публикации "Methods in Nitric Oxide Research", Feelisch et al. eds., pages 71-115 (J. S., John Wiley & Sons, New York, 1996), которая включена в настоящее изобретение в качестве ссылки. Доноры NO, которые являются С-нитрозосоединения, в которых нитрозогруппа присоединена к третичному атому углерода, которые применимы в настоящем изобретении, включают описанные в патенте U.S. № 6359182 и в WO 02/34705. Примеры S-нитрозосоединений, включая S-нитрозотиолы, применимых в настоящем изобретении, включают, например, S-нитрозоглутатион, S-нитрозо-N-ацетилпеницилламин, S-нитрозоцистеин и его этиловый эфир, S-нитрозоцистениилглицин, S-нитрозо-гамма-метил-L-гомоцистеин, S-нитрозо-L-гомоцистеин, S-нитрозо-гамма-тио-L-лейцин, S-нитрозо-дельта-тио-L-лейцин, и S-нитрозоальбумин. Примерами других доноров NO, применимых в настоящем изобретении, являются нитропруссид натрия (ниприд), этилнитрит, изосорбид, нитроглицерин, SIN 1, который представляет собой молсидомин, фуроксамины, N-гидрокси-(N-нитрозамин) и перфторуглеводороды, которые насыщены с помощью NO, или гидрофобный донор NO.
Комбинация ингибитора GSNOR с R(+)-энантиомером амлодипина, известное средство, высвобождающее NO (Zhang at al., J. Cardiovasc. Pharm. 39:208-214 (2002)), также является вариантом осуществления настоящего изобретения.
Настоящее изобретение также относится к способу лечения субъекта, страдающего от патологической пролиферации клеток, способ включает введение указанному субъекту терапевтически эффективного количества ингибитора GSNOR. Ингибиторами GSNOR являются соединения, определенные выше, или их фармацевтически приемлемая соль, или пролекарство, или метаболит, или стереоизомер в комбинации с фармацевтически приемлемым носителем. Лечение продолжают, пока не произойдет ослабление симптомов и/или патологии.
В другом варианте осуществления патологически пролиферирующие клетки могут представлять собой патологически пролиферирующие микробы. Этими микробами могут быть такие, в которых экспрессируется GSNOR для защиты микробов от нитрозирующего стресса, или клетки хозяина, инфицированные микробами, экспрессируют фермент и тем самым защищают микробы от нитрозирующего стресса. Термин "патологически пролиферирующие микробы" используется в настоящем изобретении для обозначения патологических микроорганизмов, включая, но не ограничиваясь только ими, патологические бактерии, патологические вирусы, патологические хламидии, патологические простейшие, патологические риккетсии, патологические грибы и патологическую микоплазму. Более подробное описание соответствующих микробов приведено в столбцах 11 и 12 патента U.S. № 6057367. Термин "клетки хозяина, инфицированные патологическими микробами" включает не только клетки млекопитающих, инфицированные патологическими вирусами, но и клетки млекопитающих, содержащие внутриклеточные бактерии или простейшие, например, макрофаги, содержащие Mycobacterium tuberculosis, Mycobacterium leper (проказа) или Salmonella typhi (брюшной тиф).
В другом варианте осуществления патологически пролиферирующие клетки могут представлять собой патологические гельминты. Термин "патологические гельминты" используется в настоящем изобретении для обозначения патологических нематод, патологических трематод и патологических цестод. Более подробное описание соответствующих гельминтов приведено в столбце 12 патента U.S. № 6057367.
В другом варианте осуществления патологически пролиферирующие клетки могут представлять собой патологически пролиферирующие клетки млекопитающих. Термин "патологически пролиферирующие клетки млекопитающих" при использовании в настоящем изобретении означает клетки млекопитающего, которые растут или количество которых увеличивается у указанного млекопитающего, так что оказывается вредное воздействие на млекопитающее или его органы. Термин включает, например, патологически пролиферирующие или разрастающиеся клетки, вызывающие рестеноз, патологически пролиферирующие или разрастающиеся клетки, вызывающие доброкачественную гипертрофию предстательной железы, патологически пролиферирующие клетки, вызывающие гипертрофию миокарда, и пролиферирующие клетки, находящиеся в центрах воспаления, такие как синовиальные клетки при артрите или клетки, связанные с клеточным пролиферативным нарушением.
При использовании в настоящем изобретении термин "клеточное пролиферативное нарушение" означает патологические состояния, при которых нерегулируемый и/или аномальный рост клеток может привести к развитию нежелательного патологического состояния или заболевания, которое может быть раковым или нераковым, например, псориатическое патологическое состояние. При использовании в настоящем изобретении термин "псориатическое патологическое состояние" означает нарушения, включая гиперпролиферацию кератиноцитов, воспалительную инфильтрацию клеток и изменение цитокинов. Клеточное пролиферативное нарушение может представлять собой предраковое патологическое состояние или рак. Рак может представлять собой первичный рак или метастатический рак, или их оба.
При использовании в настоящем изобретении термин "рак" включает солидные опухоли, такие как опухоли легких, молочной железы, толстой кишки, яичников, поджелудочной железы, предстательной железы, аденокарцинома, плоскоклеточная карцинома, саркома, злокачественная глиома, лейомиосаркома, гепатома, рак головы и шеи, злокачественная меланома, немеланомные раковые заболевания кожи, а также гематологические опухоли и/или злокачественные новообразования, такие как лейкоз, лейкозы и лимфомы у детей, множественная миелома, болезнь Ходжкина, лимфомы лимфоцитарной и кожной этиологии, острый и хронический лейкоз, такой как острый лимфобластный, острый миелоцитарный или хронический миелоцитарный лейкоз, плазмоклеточная неоплазия, лимфоидная неоплазия и раковые заболевания, связанные со СПИД.
В дополнение к псориатическим патологическим состояниям, типами пролиферативных заболеваний, которые можно лечить с помощью композиций, предлагаемых в настоящем изобретении, являются эпидермические и дермоидные кисты, липомы, аденомы, капиллярные и кожные гемангиомы, лимфангиомы, поражения невусов, тератомы, нефромы, миофиброматоз, остеопластические опухоли и другие диспластические опухоли и т.п. В одном варианте осуществления пролиферативные заболевания включают дисплазии и аналогичные нарушения.
В одном варианте осуществления лечение рака включает уменьшение размера опухоли, уменьшение количества опухолей, задержку роста опухоли, уменьшение метастатических поражений других тканей или органов, удаленных от положения первичной опухоли, повышение выживаемости пациентов или улучшение качества жизни пациента, или по меньшей мере два из указанных выше.
В другом варианте осуществления лечение клеточного пролиферативного нарушения включает уменьшение скорости пролиферации клеток, уменьшение содержания пролиферирующих клеток, уменьшение размера участка или зоны пролиферации клеток или уменьшение количества или содержания клеток, обладающих аномальным видом или морфологией или по меньшей мере два из указанных выше.
В еще одном варианте осуществления соединения, предлагаемые в настоящем изобретении, или их фармацевтически приемлемую соль, их пролекарство, их стереоизомер или их метаболит можно вводить в комбинации со вторым химиотерапевтическим средством. В другом варианте осуществления второе химиотерапевтическое средство выбрано из группы, включающей тамоксифен, ралоксифен, анастрозол, экземестан, летрозол, цисплатин, карбоплатин, паклитаксел, циклофосфамид, ловастатин, минозин, гемцитабин, araC, 5-фторурацил, метотрексат, доцетаксел, госерелин, винкристин, винбластин, нокодазол, тенипозид, этопозид, эпотилон, навелбин, камптотецин, даунонибицин, дактиномицин, митоксантрон, амсакрин, доксорубицин, эпирубицин, идарубицин, иматиниб, гефитиниб, эрлотиниб, сорафениб, сунитинибмалат, трастузумаб, ритуксимаб, цетуксимаб и бевацизумаб.
В одном варианте осуществления соединения, предлагаемые в настоящем изобретении, или их фармацевтически приемлемую соль, их пролекарство, их стереоизомер или их метаболит можно вводить в комбинации со средством, которое создает нитрозирующий или окислительный стресс. Средства для создания селективного нитрозирующего стресса с целью подавления пролиферации патологически пролиферирующих клеток в комбинированной терапии с ингибиторами GSNOR, предлагаемыми в настоящем изобретении, и дозы и пути их введения включают раскрытые в патенте U.S. № 6057367, который включен в настоящее изобретение в качестве ссылки. Вспомогательные средства для создания окислительного стресса (т. е. средства, которые увеличивают отношение количества GSSG (окисленный глутатион) к количеству GSH (глутатион) или количества NAD(P) к количеству NAD(P)H или увеличивают содержание производных тиобарбирутовой кислоты) в комбинированной терапии с ингибиторами GSNOR, предлагаемыми в настоящем изобретении, включают, например, L-бутионин-S-сульфоксимин (BSO), ингибиторы глутатионредуктазы (например, BCNU), ингибиторы или средства, разобщающие митохондриальное дыхание, и лекарственные средства, которые увеличивают содержание реакционноспособных кислородсодержащих частиц (ROS), например, адриамицина, в стандартных дозах при стандартных путях введения.
Ингибиторы GSNOR также можно вводить вместе с ингибитором фосфодиэстеразы (например, ролипрам, циломиласт, рофлумиласт, виагра® (силденафилцитрат), сиалис (тадалафил), левитра (варденафил) и т.п.), β- агонист, стероид или антагонист лейкотриена (LTD-4). Специалисты в данной области техники могут легко установить соответствующее терапевтически эффективное количество в зависимости от нарушения, протекание которого облегчают.
Ингибиторы GSNOR можно использовать в качестве средств для улучшения передачи β-адренергических сигналов. В частности, ингибиторы GSNOR по отдельности или в комбинации с β-агонистами можно использовать для лечения или защиты от сердечной недостаточности или других сосудистых нарушений, таких как гипертензия и астма. Ингибиторы GSNOR также можно использовать для модулирования связанных с белком G рецепторов (GPCRs) путем активации белка Gs G, что приводит к расслаблению гладких мышц (например, дыхательных путей и кровеносных сосудов), и путем уменьшения содержания белка Gq G и тем самым предупреждения сокращения гладких мышц (например, дыхательных путей и кровеносных сосудов).
Терапевтически эффективное количество для лечения субъекта, страдающего от нарушения, протекание которого улучшается с помощью лечения донором NO, представляет собой ингибирующее GSNOR количество in vivo, которое приводит к облегчению нарушения, подвергающегося лечению, или защищает от опасности, связанной с нарушением. Например, для астмы терапевтически эффективное количество представляет собой бронхорасширяющее эффективное количество; для муковисцидоза терапевтически эффективное количество представляет собой уменьшающее обструкцию дыхательных путей эффективное количество; для ОРДС терапевтически эффективное количество представляет собой уменьшающее гипоксемию эффективное количество; для заболевания сердца терапевтически эффективное количество представляет собой облегчающее стенокардию или вызывающее ангиогенез эффективное количество; для гипертензии терапевтически эффективное количество представляет собой снижающее артериальное давление эффективное количество; для ишемических коронарных нарушений терапевтическое количество представляет собой усиливающее кровоток эффективное количество; для атеросклероза терапевтически эффективное количество представляет собой обращающее эндотелиальную дисфункцию эффективное количество; для глаукомы, терапевтическое количество представляет собой снижающее внутриглазное давление эффективное количество; для заболеваний, характеризующихся ангиогенезом, терапевтически эффективное количество представляет собой подавляющее ангиогенез эффективное количество; для нарушений, при которых существует опасность возникновения тромбоза, терапевтически эффективное количество представляет собой предупреждающее тромбоз эффективное количество; для нарушений при которых существует опасность возникновения рестеноза, терапевтически эффективное количество представляет собой подавляющее рестеноз эффективное количество; для хронических воспалительных заболеваний терапевтически эффективное количество представляет собой уменьшающее воспаление эффективное количество; для нарушений, при которых существует опасность возникновения апоптоза, терапевтически эффективное количество представляет собой предупреждающее апоптоз эффективное количество; для импотенции терапевтически эффективное количество представляет собой обеспечивающее или поддерживающее эрекцию эффективное количество; для ожирения терапевтически эффективное количество представляет собой вызывающее насыщение эффективное количество; для удара терапевтически эффективное количество представляет собой усиливающее кровоток или защищающее от ПНК (преходящее нарушение мозгового кровообращения) эффективное количество; для реперфузионного поражения терапевтически эффективное количество представляет собой усиливающее функцию эффективное количество; и для предварительной адаптации сердца и головного мозга терапевтически эффективное количество представляет собой защищающее клетки эффективное количество, например, определенное с помощью тропонина или креатинфосфокиназы.
Терапевтически эффективное количество для лечения субъекта, страдающего от патологической пролиферации клеток, означает ингибирующее GSNOR количество in vivo, которое представляет собой антипролиферативно эффективное количество. Такое антипролиферативно эффективное количество при использовании в настоящем изобретении означает количество, приводящее к снижению скорости пролиферации, составляющему не менее примерно 20%, не менее примерно 10%, не менее примерно 5% или не менее примерно 1%.
I. Применение в аппаратах
Соединения, предлагаемые в настоящем изобретении, или их фармацевтически приемлемую соль, или пролекарство, или метаболит, или их стереоизомер можно поместить в различные аппараты, когда наличие таких соединений будет полезным. Таким аппаратом может быть любое устройство или контейнер, например, имплантируемые устройства, в которых соединение, предлагаемое в настоящем изобретении, можно использовать для нанесения покрытия на хирургическую сетку или сердечно-сосудистый стент перед имплантацией пациенту. Соединения, предлагаемые в настоящем изобретении, также можно помещать в различные аппараты для проведения анализов in vitro или для выращивания клеток.
Соединения, предлагаемые в настоящем изобретении, или их фармацевтически приемлемую соль, или пролекарство, стереоизомер или метаболит также можно использовать в качестве средства для разработки, выделения или очистки компонентов, связывающихся с соединениями, предлагаемыми в настоящем изобретении, таких как антитела, природные лиганды и т.п. Специалисты в данной области техники могут без труда определить родственные случаи применения соединений, предлагаемых в настоящем изобретении.
ПРИМЕРЫ
Представленные ниже примеры приведены для иллюстрации настоящего изобретения. Однако следует понимать, что настоящее изобретение не ограничивается конкретными условиями или подробностями, описанными в этих примерах. В настоящем описании любые и все указания на общедоступный документ, включая патент США, специально включены в настоящее изобретение в качестве ссылки.
В примерах 1-24 приведены типичные новые аналоги соединений, предлагаемых в настоящем изобретении, применимых в качестве ингибиторов GSNOR. Методики синтеза, которые можно использовать для получения каждого соединения, описаны в примерах 1-24. Дополнительные данные масс-спектрометрии и/или протонного ЯМР также приведены в примерах 1-22. Методики синтеза соответствующих промежуточных продуктов подробно описаны в примере 25.
Пример 1: 3-(4-(1Н-Тетразол-5-ил)фенил)-7-гидрокси-2-(трифторметил)-4Н-хромен-4-он

Синтез: Стадия 1: Синтез 4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензонитрила. К раствору 4-(2-(2,4-дигидроксифенил)-2-оксоэтил)бензамида (промежуточный продукт А) (300 мг, 1,08 ммоля) и триэтиламина (ТЭА) (0,6 мл, 4,32 ммоля) в ДХМ (дихлорметан) (6 мл) по каплям при 0°С добавляли ТФУК (трифторуксусная кислота) (1,2 мл, 8,64 ммоля). Смесь перемешивали при комнатной температуре в течение 2 ч. Затем смесь промывали 1 н. раствором HCl (5 мл), насыщенным раствором NaHCO3 (5 мл) и рассолом (5 мл). Органическую фазу сушили над Na2SO4, концентрировали и очищали с помощью препаративной ТСХ (ПЭ (петролейный эфир) : EtOAc = 3 : 1) и получали продукт в виде желтого масла (66 мг, 19,5%). МС (масс-спектроскопия) (ИЭР - ионизация электрораспылением): m/z 332,1 [M+1]+.
Стадия 2: Синтез 3-(4-(1Н-тетразол-5-ил)фенил)-7-гидрокси-2-(трифторметил)-4Н-хромен-4-она. К раствору 4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензонитрил (50 мг, 0,15 ммоля) в толуоле (2 мл), добавляли TMSN3 (296 мг, 2,72 ммоля) и Bu2SnO (10 мг, 0,045 ммоля) при комнатной температуре. Смесь кипятили с обратным холодильником в течение ночи. Летучие вещества удаляли при пониженном давлении. Остаток очищали с помощью препаративной ВЭЖХ и получали искомый продукт примера 1 в виде желтого порошкообразного вещества (19,4 мг, 34,6 %).
Данные:1Н ЯМР (МеОН-d4 500 МГц TMS (тетраметилсилан)): 8,12 (d, J=8,5 Гц, 2Н), 8,02 (d, J=8,5 Гц, 2H), 7,52 (d, J=8,5 Гц, 2Н), 7,02 (dd, J=1,5 Гц, J=8,5 Гц, 1Н), 6,94 (d, J=1,5 Гц, 1H); МС (ИЭР): m/z 375,0 [M+1]+.
Пример 2: 5-(7-Гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)тиофен-2-карбоновая кислота

Синтез: Стадия 1: Синтез метил-5-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)тиофен-2-карбоксилата. Проводили по методике, описанной на стадии 1 примера 1, с использованием в качестве исходного вещества промежуточного продукта В. МС (ИЭР): m/z 371,0 [M+1]+.
Стадия 2: Синтез 5-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)тиофен-2-карбоновой кислоты. К раствору метил-5-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)тиофен-2-карбоксилата (295 мг, 0,80 ммоля) в диоксане (1,5 мл) добавляли концентрированную HCl (1,5 мл). Реакционную смесь перемешивали при 70°С в течение 24 ч, охлаждали до комнатной температуры и центрифугировали. Осадок промывали водой (2 мл×2), ДХМ (2 мл×2), и сушили в вакууме, и получали искомый продукт примера 2 в виде серого порошкообразного вещества (216,1 мг, 76,3%).
Данные:1Н ЯМР (MeOD-d4 500 МГц TMS): 8,04 (d, J=8,5 Гц, 1Н), 7,78 (d, J=3,5 Гц, 1Н), 7,13 (d, J=4,0 Гц, 1Н), 7,04 (dd, J=2,0 Гц, J=8,5 Гц, 1Н), 6,96 (d, J=2,0 Гц, 1Н); МС (ИЭР): m/z 357,0 [М+1]+.
Пример 3: (Транс)-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)циклогексанкарбоновая кислота
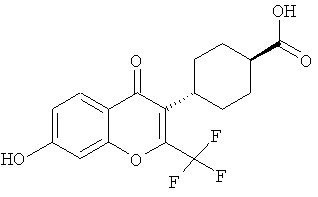
Синтез: Стадия 1: Синтез этил-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)циклогексанкарбоксилата. Проводили по методике, описанной на стадии 1 примера 1, с использованием в качестве исходного вещества промежуточного продукта С и неочищенный продукт использовали сразу без обработки или очистки. МС (ИЭР): m/z 385,1 [M+1]+.
Стадия 2: К раствору этил-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)пиклогексанкарбоксилата (450 мг, 1,1 ммоля) в диоксане (3 мл) добавляли концентрированную HCl (3 мл). Раствор перемешивали при 75°С в течение ночи. Смесь концентрировали в вакууме и получали светло-желтое твердое вещество, которое очищали с помощью препаративной ВЭЖХ и получали чистый транс-изомер, искомый продукт примера 3 (100 мг, 24%). Данные:1Н ЯМР (MeOH-d4 500 МГц TMS): δ 7,97 (d, J=9,0 Гц, 2Н), 6,97 (dd, J=2,0, 8,5 Гц, 1Н), 6,84 (d, J=2,5 Гц, 1Н), 2,71 (t, J=12,0 Гц, 1Н), 2,38-2,48 (m, 3Н), 2,05-2,14 (m, 2Н), 1,65-1,68 (m, 2Н), 1,44-1,53 (m, 2Н); МС (ИЭР): m/z 357,0 [M+1]+.
Пример 4: (Цис)-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)циклогексанкарбоновая кислота

Синтез: Подробное описание приведено в примере 3. Препаративная ВЭЖХ давала 64 мг, 15,3% чистого цис-изомера, искомый продукт примера 4. Данные:1Н ЯМР (MeOH-d4 500 МГц TMS): δ 7,93 (d, J=9,0 Гц, 1Н), 6,95 (dd, J=2,5, 9,0 Гц, 1Н), 6,83 (d, J=2,0 Гц, 1Н), 2,77-2,71 (m, 2Н), 2,50-2,58 (m, 2Н), 2,33 (s, 2Н). 1,55-1,62 (m, 2Н), 1,45-1,47 (m, 2Н); МС (ИЭР): m/z 357,0 [M+1]+.
Пример 5: 3-(4-(1Н-Тетразол-5-ил)фенил)-2-(дифторметил)-7-гидрокси-4Н-хромен-4-он

Синтез: Проводили по методике, описанной на стадии 1 примера 1, с использованием в качестве исходного вещества промежуточного продукта D и дифторуксусного ангидрида.
Данные:1Н ЯМР (МеОН-d4 500 МГц TMS): δ 8,18 (d,J=8,5 Гц, 2Н), 8,07 (d, J=8,5 Гц, 1Н), 7,58 (d, J=8,5 Гц, 2Н), 7,03 (dd, J=2,5 Гц, J=9,0 Гц, 1Н), 6,98 (d, J=2,0 Гц, 1Н), 6,56 (t, J=2,0 Гц, 1Н); MC (ИЭР): m/z 357,0 [M+1]+.
Пример 6: 3-(4-(1Н-Тетразол-5-ил)фенил)-7-гидрокси-2-метил-4Н-хромен-4-он
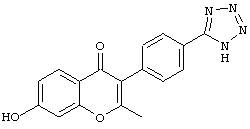
Синтез: Проводили по методике, описанной на стадии 1 примера 1, с использованием в качестве исходного вещества промежуточного продукта D и уксусного ангидрида. Неочищенный продукт очищали с помощью препаративной ВЭЖХ и получали соединение примера 6. Данные:1Н ЯМР (ДМСО-d6 (диметилсульфоксид) 500 МГц TMS): δ 10,84 (s, 1Н), 8,09 (d, J=8,5 Гц, 2Н), 7,90 (d, J=9,0 Гц, 1Н), 7,53 (d, J=8,5 Гц, 2Н), 6,93 (dd, J=2,5 Гц, J=8,5 Гц, 1Н), 6,87 (d, J=2,5 Гц, 1Н), 2,29 (s, 3H); MC (ИЭР): m/z 321,0 [M+1]+.
Пример 7: 4-(2-(4-Карбоксибензил)-7-гидрокси-4-оксо-4Н-тиохромен-3-ил)бензойная кислота

Синтез: Стадия 1: Синтез метил-4-(7-метокси-2-(4-(метоксикарбонил)бензил)-4-оксо-4Н-тиохромен-3-ил)бензоата. Трихлорид алюминия (253 мг, 1,9 ммоля) добавляли к промежуточному продукту Е (метил- 4-(2-(3-метоксифенилтио)-2-оксоэтил)бензоат) (500 мг, 1,58 ммоля) и смесь нагревали при 130°С в течение 1 ч. После охлаждения до комнатной температуры реакционную смесь растворяли в EtOAc (50 мл) и промывали с помощью 1 н. охлажденной льдом HCl (25 мл×2), водой (25 мл) и рассолом (25 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали с помощью Combi-Flash (40 г силикагеля, элюент: ПЭ : EtOAc = от 10:0 до 1:1 в градиентном режиме, 40 мл/мин, 30 мин, полный объем растворителя равен 1,2 л) и получали продукт в виде желтого твердого вещества (160 мг, 21%). МС (ИЭР): m/z 475,1 [M+1]+.
Стадия 2: Синтез 4-(2-(4-карбоксибензил)-7-гидрокси-4-оксо-4Н-тиохромен-3-ил)бензойной кислоты (пример 7). К раствору метил-4-(7-метокси-2-(4-(метоксикарбонил)бензил)-4-оксо-4Н-тиохромен-3-ил)бензоат (105 мг, 0,23 ммоля) в сухом ДХМ (3 мл) добавляли BBr3 (0,2 мл, 2,23 ммоля) при 0°С при перемешивании. Смесь перемешивали при комнатной температуре в течение 40 ч и при перемешивании выливали в охлажденный льдом 1 н. раствор HCl (1 мл). Летучие вещества выпаривали и остаток очищали с помощью препаративной ТСХ (ПЭ:EtOAc =1:1) и получали искомый продукт примера 7 в виде розового твердого вещества (15 мг, 16%).
Данные:1Н ЯМР (МеОН-d4 500 МГц TMS): δ 8,30 (d, J=9,0 Гц, 1Н), 8,08 (d, J=8,0 Гц, 2Н), 7,91 (d, J=8,0 Гц, 2Н), 7,28 (d, J=8,0 Гц, 2Н), 7,18 (d, J=8,0 Гц, 2H), 7,053 (d, J=7,5 Гц, 1Н), 7,049 (s, 1Н), 4,01 (s, 2Н); МС (ИЭР): m/z 433,0 [M+1]+.
Пример 8: 4-(7-Гидрокси-2-метил-4-оксо-4Н-тиохромен-3-ил)бензойная кислота

Синтез: Стадия 1: Синтез метил-4-(7-метокси-2-метил-4-оксо-4Н-тиохромен-3-ил)бензоата: К раствору промежуточного продукта F (метил-4-(2-(2-(ацетилтио)-4-метоксифенил)-2-оксоэтил)бензоат) (600 мг, 1,674 ммоля) в ацетоне (12 мл) при комнатной температуре добавляли К2СО3 (386 мг, 3,348 ммоля). Смесь перемешивали в течение 3 ч, фильтровали и концентрировали. Остаток очищали с помощью Combi-Flash (40 г силикагеля, ПЭ/EtOAc= от 10/0 до 3/1 в градиентном режиме, 40 мл/мин, 40 мин, полный объем растворителя равен 1,6 л) и получали продукт в виде желтого твердого вещества (400 мг, 70%). МС (ИЭР): m/z 341,0 [M+1]+.
Стадия 2: Синтез 4-(7-гидрокси-2-метил-4-оксо-4Н-тиохромен-3-ил)бензойной кислоты (пример 8): К раствору метил-4-(7-метокси-2-метил-4-оксо-4Н-тиохромен-3-ил)бензоата (200 мг, 0,588 ммоля) в ДХМ (10 мл) при комнатной температуре добавляли BBr3 (0,83 мл, 8,813 ммоля) и перемешивали в течение 20 ч. Смесь при перемешивании выливали в охлажденную льдом 1 н. HCl (50 мл) и осадок собирали фильтрованием и получали неочищенный продукт, который очищали с помощью препаративной ВЭЖХ и получали искомый продукт примера 8 в виде желтого твердого вещества (57,3 мг, 31%). Данные:1Н ЯМР (ДМСО-d6 500 МГц TMS): δ 13,00 (brs, 1H), 10,76 (brs, 1H), 8,16 (d, J=9,0 Гц, 1H), 7,98 (d, J=8,5 Гц, 2Н), 7,32 (d, J=8,0 Гц, 2Н), 7,03-7,08 (m, 2Н), 2,17 (s, 3H); МС (ИЭР): m/z 313,0 [M+1]+.
Пример 9: 3-(4-(7-Гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)фенил)-1,2,4-оксадиазол-5(4Н)-он

Синтез: К раствору 4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензонитрила (синтез описан в примере 1, стадия 1) (200 мг, 0,60 ммоля) и гидроксиламингидрохлорида (218 мг, 3,13 ммоля) в абсолютном этаноле (2 мл) по каплям добавляли триэтиламин (0,7 мл). Полученную смесь кипятили с обратным холодильником в течение 5 ч. Летучие вещества выпаривали и остаток растворяли в безводном ТГФ (тетрагидрофуран) (2 мл). Добавляли КДИ (карбонилдиимидазол) (296 мг, 1,83 ммоля) и суспензию кипятили с обратным холодильником в течение ночи. Летучие вещества выпаривали и остаток очищали с помощью препаративной ТСХ (чистый EtOAc) и препаративной ВЭЖХ и получали искомый продукт примера 9 (35 мг, 15%) в виде белого твердого вещества.
Данные:1Н ЯМР (МеОН-d4 500 МГц TMS): δ 8,04 (d, J=9,0 Гц, 1Н), 7,89 (d, J=8,5 Гц, 2Н), 7,50 (d, J=8,5 Гц, 2Н), 7,04 (dd, J=2,5 Гц, J=8,5 Гц, 1Н), 6,87 (d, J=2,5 Гц, 1Н); МС (ИЭР): m/z 391,0 [M+1]+.
Пример 10: 4-(7-Гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)-М-(метилсульфонил)бензамид

Синтез: К раствору 4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензойной кислоты (синтез описан в PCT/US 2010/024035) (100 мг, 0,28 ммоля) в ТГФ (10 мл) при комнатной температуре добавляли КДИ (139 мг, 0,86 ммоля); Смесь перемешивали в течение 2 ч. Одной порцией добавляли MeSO2NH2 (280 мг, 2,86 ммоля), затем ДБУ (1,8-диазабикло[5.4.0]ундец-7-ен) (394 мг, 2,86 ммоля). Смесь перемешивали в течение 4 ч и подвергали распределению между 1 н. HCl (30 мл) и этилацетатом (100 мл). Органическую фазу отделяли, промывали рассолом (50 мл), сушили над Na2SO4 и концентрировали и получали искомый продукт, загрязненный исходной кислотой. Смесь затруднительно очистить и примесь кислоты превращали в метиловый эфир путем обработки с помощью SOCl2 (91 мг, 0,77 ммоля) в МеОН (4 мл) при 0°С. После завершения добавления смесь перемешивали в течение 3 дней. Летучие вещества удаляли при пониженном давлении и остаток очищали с помощью препаративной ВЭЖХ и получали искомый продукт примера 10 в виде белого порошкообразного вещества (25 мг, 20,5%). Данные:1Н ЯМР (МеОН-d4 500 МГц TMS): δ 8,04 (d, J=8,5 Гц, 1Н), 8,00 (d, J=8,5 Гц, 2Н), 7,46 (d, J=8,5 Гц, 2Н), 7,04 (dd, J=2,0 Гц, J=9,0 Гц, 1Н), 6,97 (d, J=2,0 Гц, 1Н), 3,41 (s, 1Н); МС (ИЭР): m/z 428,0 [M+1]+.
Пример 11: 3-(4-(7-Гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)фенил)-1,2,4-тиадиазол-5(4Н)-он

Синтез: К раствору 4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензонитрила (синтез описан в примере 1, стадия 1) (250 мг, 0,76 ммоля) и гидроксиламингидрохлорида (105 мг, 1,51 ммоля) в абсолютном этаноле (2 мл) по каплям добавляли триэтиламин (0,5 мл). Полученную смесь кипятили с обратным холодильником в течение 2 ч. Летучие вещества выпаривали и остаток растворяли в безводном ТГФ (5 мл). Добавляли TCDI (1,1'-тиокарбонилдиимидазол) (202 мг, 1,13 ммоля) и суспензию перемешивали при комнатной температуре в течение 2 ч, подвергали распределению между EtOAc (50 мл) и водой (20 мл). Органическую фазу отделяли, сушили над безводным Na2SO4, фильтровали и концентрировали. Остаток растворяли в ТГФ (5 мл), добавляли BF3-Et2O и перемешивали при комнатной температуре в течение 2 ч. Обрабатывали с помощью 1 н. HCl, затем летучие вещества выпаривали и остаток очищали с помощью препаративной ВЭЖХ и получали искомый продукт примера 11 (17,5 мг, 5%) в виде белого твердого вещества. Данные:1Н ЯМР (MeOD 500 МГц TMS): δ 8,04 (d, J=9,0 Гц, 1Н), 8,01 (d, J=8,5 Гц, 2Н), 7,45 (d, J=8,5 Гц, 2Н), 7,04 (dd, J=1,5, 8,5 Гц, 1Н), 6,90 (d, J=1,5 Гц, 1Н); MC (ИЭР): m/z 406,9 [M+1]+.
Пример 12: 3-(4-(1Н-Тетразол-5-ил)фенил)-7-гидрокси-2-метил-4Н-тиохромен-4-он
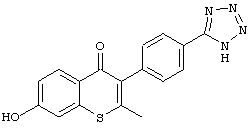
Синтез: Стадия 1: Синтез 4-(7-метокси-2-метил-4-оксо-4Н-тиохромен-3-ил)бензонитрила: К смеси промежуточного продукта G (664 мг, 2 ммоля), 4- цианофенилбороновой кислоты (294 мг, 2 ммоля) и ТЭА (1,4 мл, 10 ммоля) в ДМФ (диметилформамид) (4 мл) добавляли Pd(dppf)Cl2 (146 мг, 0,2 ммоля) и полученную смесь нагревали при 85°С в атмосфере азота в течение 18 ч. Смесь охлаждали до комнатной температуры и затем подвергали распределению между 1 н. HCl (20 мл) и этилацетатом (50 мл). Органическую фазу отделяли, промывали рассолом (10 мл), сушили над безводным Na2SO4 и концентрировали, затем очищали с помощью колоночной хроматографии (ПЭ/EtOAc=5/1) и получали продукт (310 мг, 54%) в виде желтого твердого вещества.
Стадия 2: Синтез 4-(7-гидрокси-2-метил-4-оксо-4Н-тиохромен-3-ил)бензонитрила: К раствору полученного выше продукта (310 мг, 1,0 ммоля) в ДХМ (3 мл) осторожно добавляли BBr3 (1 мл, 10 ммоля). Смесь перемешивали при комнатной температуре в течение ночи. Полученную смесь выливали в воду со льдом (5 мл), экстрагировали этилацетатом (3 мл×3). Объединенную органическую фазу промывали рассолом (3 мл), сушили над безводным Na2SO4 и концентрировали. Очистка с помощью колоночной хроматографии (ПЭ/EtOAc=3/1) давала продукт в виде желтого порошкообразного вещества (120 мг, 40%).
Стадия 3: Синтез примера 12: Затем тетразол получали из полученного выше продукта по методике, описанной в примере 1, стадия 2, с выходом 65%. Данные:1Н ЯМР (ДМСО-d6 500 МГц TMS): δ 10,78 (s, 1H), 8,17 (d, J=9,0 Гц, 1Н), 8,08 (d, J=8,0 Гц, 2H), 7,43 (d, J=8,0 Гц, 2Н), 7,08 (s, 1Н), 7,04 (d, J=9,0 Гц, 1H), 2,07 (s, Гц 3Н); МС (ИЭР): m/z 337,0 [M+1]+.
Пример 13: 5-(7-Гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)тиофен-3-карбоновая кислота

Синтез: Стадия 1: Синтез метил-5-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)тиофен-3-карбоксилата: Проводили по методике, описанной в примере 1, стадия 1, в которой промежуточный продукт Н является исходным веществом и неочищенный продукт очищали с помощью колоночной хроматографии и получали продукт с выходом 43%.
Стадия 2: Синтез 5-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)тиофен-3-карбоновой кислоты: Проводили по методике гидролиза, описанной в примере 3, стадия 2, и получали продукт с выходом 65%. Данные:1Н ЯМР (MeOD-d4 500 МГц TMS): 8,42 (s, 1Н), 8,05 (d, J=7,5 Гц, 1Н), 7,48 (s, 1Н), 7,05 (dd, J=2,5, 9,0 Гц, 1Н), 6,96 (d, J=2,0 Гц, 1Н); MC (ИЭР): m/z 357,0 [M+1]+.
Пример 14: 3-((Транс)-4-(1Н-тетразол-5-ил)циклогексил)-7-гидрокси-2-(трифторметил)-4Н-хромен-4-он

Синтез: Стадия 1: Синтез (транс)-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)циклогексанкарбоксамида: Оксалилхлорид (6,5 мл, 84,27 ммоля) при комнатной температуре по каплям добавляли к раствору (транс)-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)циклогексанкарбоновой кислоты (пример 3) (1,0 г) в ДХМ (25 мл) (добавляли 3 капли ДМФ). Наблюдалось бурное выделение газа. После перемешивания в течение 30 мин к полученному выше раствору добавляли NH3H2O (25%, 9 мл). После перемешивания в течение 60 мин добавляли этилацетат (50 мл). Органический слой концентрировали и очищали с помощью колоночной хроматографии (ПЭ/EtOAc=1/1) и получали искомый продукт в виде белого твердого вещества (0,81 г, выход: 81%).
Стадия 2: Синтез (транс)-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)циклогексанкарбонитрила: К раствору полученного выше продукта (0,81 г) и ТЭА (3,5 мл) в ДХМ (8,0 мл) при комнатной температуре по каплям добавляли ТФУК (2,7 г). Смесь перемешивали в течение 2 ч. Летучие вещества удаляли в вакууме. Остаток очищали с помощью колоночной хроматографии (ПЭ/EtOAc=5/1) и получали продукт в виде желтого твердого вещества (0,72 г, 74%).
Стадия 3: Синтез 3-((транс)-4-(1Н-тетразол-5-ил)циклогексил)-7-гидрокси-2-(трифторметил)-4Н-хромен-4-она: Тетразол получали из полученного выше продукта по методике, описанной в примере 1, стадия 2, с выходом 59%. Данные:1Н ЯМР (MeOD-d4 500 МГц TMS): 7,87 (d, J=9,0 Гц, 1Н), 6,86 (dd, J=2,0 Гц, J=9,0 Гц, 1Н), 6,73 (d, J=2,5 Гц, 2Н), 3,08-3,03 (m, 1Н), 2,72 (t, J=12,5 Гц, 1Н), 2,53-2,45 (m, 2Н), 2,12 (d, J=12,0 Гц, 2Н), 1,67-1,54 (m, 4H); MC (ИЭР): m/z 381,1 [М+1]+.
Пример 15: N-Гидрокси-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензамид

Синтез: Оксалилхлорид (140 мг, 1,1 ммоля) при комнатной температуре по каплям добавляли к раствору 4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензойной кислоты (синтез описан в PCT/US 2010/024035) (130 мг, 0,4 ммоля) и ДМФ (0,5 мл) в ДХМ (25 мл). Наблюдалось бурное выделение газа. После перемешивания в течение 30 мин полученный выше раствор добавляли к смеси гидроксиламингидрохлорида (0,29 г, 0,5 ммоля) и ТЭА (0,12 мл, 0,9 ммоля) в ТГФ (2 мл) и воде (0,5 мл). После перемешивания в течение 1 ч реакционную смесь экстрагировали с помощью EtOAc. Органический слой промывали рассолом, сушили над MgSO4 и выпаривали. Остаток перекристаллизовали из ацетон и получали белое порошкообразное вещество (120 мг), которое очищали с помощью препаративной ВЭЖХ и получали искомый продукт примера 15 в виде светло-желтого порошкообразного вещества (48 мг, выход: 33%).
Данные:1Н ЯМР (ДМСО-d6, 500 МГц TMS): 11,31 (s, 1H),9,14(s, 1Н), 7,92 (t, J=4,5 Гц, 1Н), 7,80 (s, 2Н), 7,36 (d, J=3,5 Гц, 2Н), 7,01 (d, J=5,5 Гц, 1Н), 6,94 (s, 1Н); MC (ИЭР): m/z 366,0 [M+1]+.
Пример 16: 3-Фтор-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензойная кислота
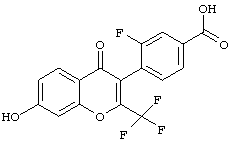
Синтез: Стадия 1: Синтез метил-3-фтор-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензоата: К раствору метил-4-(2-(2,4-дигидроксифенил)-2-оксоэтил)-3-фторбензоата (промежуточный продукт I) (6 г, 19,7 ммоля) в ДХМ (60 мл) и ТЭА (24 мл, 190 ммоля) по каплям добавляли ТФУК (13 мл, 95 ммоля). Смесь перемешивали при комнатной температуре в течение 15 ч. Затем раствор промывали 1 н. раствором HCl (50 мл) и водой (50 мл); органический слой сушили над Na2SO4, фильтровали и выпаривали и получали неочищенный продукт (6 г, 79,7%).
Стадия 2: К раствору полученного выше неочищенного продукта (6 г, 15,7 ммоля) в диоксане (60 мл) добавляли концентрированную HCl (30 мл). Смесь перемешивали при 90°С в течение 15 ч. Затем раствор экстрагировали с помощью EtOAc (100 мл×5). Органические слои объединяли, сушили над Na2SO4, фильтровали и выпаривали и получали неочищенный продукт. Неочищенный продукт перекристаллизовали из смеси EtOAc/ПЭ=3/1 (50 мл) и получали искомый продукт примера 16 (4,5 г, 77,9 %) в виде твердого вещества. Данные:1Н ЯМР (ДМСО-d6 500 МГц TMS): δ 13,44 (brs, 1H), 11,31 (brs, 1H), 7,98 (d, J=9,0 Гц, 1Н), 7,89 (dd, J=1,5 Гц, 7,5 Гц, 1Н), 7,81 (dd, J=1,0 Гц, 9,5 Гц, 1H), 7,56 (t, J=8,0 Гц, 1H), 7,09 (dd, J=2,0 Гц, 8,5 Гц, 1H); 7,03 (d, J=2,0 Гц, 1H); MC (ИЭР): m/z 369,0 [M+1]+
Пример 17: 3-(2-Хлор-4-(1Н-тетразол-5-ил)фенил)-7-гидрокси-2-(трифторметил)-4Н-хромен-4-он

Стадия 1: Искомый продукт синтезировали в соответствии со стадией 3 методики, описанной в примере 14, с использованием в качестве исходного вещества 3-хлор-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензойной кислоты (синтез описан в PCT/US 2010/024035). Данные:1НЯМР (MeOD-d4 500 МГц TMS): 8,22 (d, J=1,0 Гц, 1Н), 8,07 (dd, J=1,5 Гц, J=8,0 Гц, 1Н), 8,04 (d, J=9,0 Гц, 1Н) 7,43 (d, J=8,5 Гц, 1Н), 7,04 (dd, J=2,5 Гц, J=9,0 Гц, 1Н), 6,98 (d, J=2,5 Гц, 1Н); MC (ИЭР): m/z 409,0 [M+1]+.
Пример 18: 3-(3-Хлор-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)фенил)-1,2,4-оксадиазол-5(4Н)-он

Стадия 1 и 2: Синтез 3-хлор-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензонитрила: Проводили по методике, описанной на первых двух стадиях примера 14, с использованием в качестве исходного вещества 3-хлор-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензойной кислоты (синтез описан в PCT/US 2010/024035).
Стадия 3: 3-Хлор-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензонитрил (0,3 г, 0,8 ммоля) растворяли в EtOH (5 мл) и охлаждали в бане со льдом. Добавляли соль гидроксиламина с HCl (46 мг, 0,85 экв.) и ТЭА (0,11 мл, 1 экв.). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Растворитель удаляли и остаток (0,4 г) суспендировали в безводном ТГФ (5 мл). После добавления КДИ (0,24 г, 1,5 экв.) и ТЭА (0,13 мл) суспендированный раствор перемешивали и нагревали при 50°С в течение ночи. После удаления ТГФ смесь суспендировали в воде (15 мл) и значение рН доводили до 8. Водный слой экстрагировали с помощью EtOAc (10 мл×2). Затем водный слой подкисляли до рН~2 и экстрагировали с помощью EtOAc (15 мл×4). Объединенные органические слои промывали рассолом и сушили над безводным Na2SO4. После удаления растворителя неочищенный продукт очищали с помощью препаративной ВЭЖХ и получали искомый продукт примера 18 (44 мг, 10 %) в виде желтовато-коричневого твердого вещества. Данные:1Н ЯМР (МеОН-d4 500 МГц TMS): 8,04 (d, J=8,5 Гц, 1Н), 7,99 (d, J=1,5 Гц, 1Н), 7,84 (dd, J=1,5 Гц, 8,0 Гц, 1Н), 7,53 (d, J=8,5 Гц, 1Н), 7,06 (dd, J=2,0 Гц, 8,5 Гц, 1Н), 6,98 (d,J=2,0 Гц,1Н); МС (ИЭР): m/z 425 [M+1]+.
Пример 19: 3-(3-Фтор-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)фенил)-1,2,4-оксадиазол-5(4Н)-он

Синтез: 3-Фтор-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензонитрил получали в соответствии с первыми двумя стадиями примера 14 с использованием в качестве исходного вещества 3-фтор-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензойной кислоты (пример 16) и затем превращали в искомый продукт примера 19 в соответствии со стадией 3 примера 18.
Данные:1Н ЯМР (МеОН-d4 500 МГц TMS): 8,06 (d, J=9,0 Гц, 1Н), 7,75 (dd, J=2,0 Гц, 8,0 Гц, 2Н), 7,70 (dd, J=2 Гц, 10,0 Гц, 1Н), 7,54 (t, J=7,5 Гц, 1Н), 7,07 (dd, J=2,5 Гц, 9,0 Гц, 1Н), 7,00 (d, J=2,0 Гц,1Н); МС (ИЭР): m/z 409 [M+1]+.
Пример 20: 3-(3-Хлор-4-(1Н-тетразол-5-ил)фенил)-7-гидрокси-2-(трифторметил)-4Н-хромен-4-он
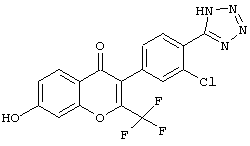
Синтез: 2-Хлор-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензонитрил получали в соответствии с первыми двумя стадиями примера 14 с использованием в качестве исходного вещества 2-хлор-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензойной кислоты (синтез описан в PCT/US 2010/024035) и затем превращали в искомый продукт примера 20 по методике, описанной в примере 1, стадия 2.
Данные:1Н ЯМР (MeOD-d4 500 МГц TMS): 8,07 (d, J=9,0 Гц, 1Н), 7,95 (d, J=8,0 Гц 1Н), 7,65 (s, 1Н), 7,48 (d, J=8,0 Гц, 1Н), 7,07 (dd, J=2,0 Гц, J=9,0 Гц, 1Н), 6,98 (d, J=2,0 Гц, 1Н); МС (ИЭР): m/z 409,0 [М+1]+.
Пример 21: 4-(7-Гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)-3-метилбензойная кислота

Синтез: Искомый продукт синтезировали аналогично двум стадиям методики, описанной в примере 16, с использованием в качестве исходного вещества метил-4-(2-(2,4-дигидроксифенил)-2-оксоэтил)-3-метилбензоата (промежуточный продукт J).
Данные:1Н ЯМР (ДМСО-d6, 300 МГц): δ 13,02(brs, 1Н), 11,21 (brs, 1Н), 7,95 (d, J = 9,0 Гц, 1Н), 7,88 (d, J- 3 Гц, 1Н), 7,80 (dd, J=3, 9 Гц, 1Н), 7,30 (d, J=9 Гц, 1Н), 7,04 (dd,J=3, 9 Гц, 1Н), 7,00 (d,J=3 Гц, 1Н), 2,13 (s, 3Н); МС (ИЭР): m/z 365,1 [M+H+]+.
Пример 22: 3-(4-(1Н-Тетразол-5-ил)фенил)-7-гидрокси-4Н-хромен-4-он

Стадия 1: Синтез 4-(7-гидрокси-4-оксо-4Н-хромен-3-ил)бензонитрил: К раствору промежуточного продукта К (200 мг, 0,79 ммоля) в безводном ДМФ (6,0 мл) при 0-10°С по каплям добавляли BF3-Et2O (0,8 мл). После завершения добавления смеси давали нагреваться до комнатной температуры в течение 0,5 ч, нагревали при 90°С. Одной порцией добавляли MsCl (1,6 мл) и перемешивали в течение 5 ч. Смесь подвергали распределению между EtOAc (50 мл) и 1 н. HCl (50 мл). Органическую фазу отделяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали и получали светло-желтое твердое вещество, которое перекристаллизовали из ДХМ (5 мл) и получали продукт (120 мг, 58%) в виде желтого твердого вещества. МС (ИЭР): m/z 263,9 [M+1]+,
Стадия 2: Синтез 3-(4-(1Н-тетразол-5-ил)фенил)-7-гидрокси-4Н-хромен-4-она: Проводили по методике, описанной на стадии 2 примера 1, и продукт дважды перекристаллизовали из ДХМ (10 мл) и получали искомый продукт примера 22 в виде белого твердого вещества (60 мг, 43%). Данные:1Н ЯМР (ДМСО-d4 500 МГц TMS): δ 10,89 (s, 1H), 8,54 (d, J=8,5 Гц, 1Н), 8,08 (d, J=8,5 Гц, 2Н), 8,01 (m, 1H), 7,79 (d, J=8,0 Гц, 1H), 6,98 (dd, J=2,5, 8,0 Гц, 1H), 6,91 (d, J=2,0 Гц, 1H); МС (ИЭР): m/z 307,1 [M+1]+.
Пример 23: 5-(4-(7-Гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)фенил)-1,3,4-оксадиазол-2(3Н)-он

Синтез: Соединение примера 23 можно получить по описанной ниже трехстадийной методике:
Стадия 1: Синтез трет-бутил-2-(4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензоил)гидразинкарбоксилата: Смесь 4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензойной кислоты (синтез описан в PCT/US 2010/024035) (1 экв.), EDCI (1-(3-диметиламинопропил)-3-этилкарбодииимидгидрохлорид) (1 экв.) и BocNHNH2 (1 экв.) в ДХМ и ДМФ (1:1) перемешивали при 25 С в течение ночи, затем обрабатывали смесью вода/EtOAc. При необходимости проводили очистку с помощью колоночной хроматографии на силикагеле и получали продукт.
Стадия 2: Синтез 4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензогидразида: трет-Бутил-2-(4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензоил)гидразинкарбоксилат обрабатывали смесью HCl/MeOH в течение ночи. Растворитель удаляли при пониженном давлении и получали продукт.
Стадия 3: Синтез 5-(4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)фенил)-1,3,4-оксадиазол-2(3Н)-она: 4-(7-Гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензогидразид (1 экв.) и КДИ (10 экв.) в ДХМ кипятили с обратным холодильником в течение ночи. Обработка смесью вода/EtOAc и последующая очистка с помощью препаративной ВЭЖХ могла дать искомый продукт примера 23.
Пример 24: 4-(8-Фтор-7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензойная кислота

Синтез: Искомый продукт примера 24 можно получить по двустадийной методике, описанной в примере 16, с использованием в качестве исходного вещества метил-4-(2-(3-фтор-2,4-дигидроксифенил)-2-оксоэтил)бензоата (промежуточный продукт L).
Пример 25: Синтез промежуточных продуктов
Промежуточные продукты, указанные в приведенных выше примерах, можно синтезировать, как это описано ниже.
Промежуточный продукт А: 4-(2-(2,4-Дигидроксифенил)-2-оксоэтил)бензамид
Стадия 1: Синтез 2-(4-(метоксикарбонил)фенил)уксусной кислоты. К раствору 2-(4-бромфенил)уксусной кислоты (91,3 г, 0,42 моля, 1,0 экв.) в МеОН (1,5 L) добавляли сухой ТЭА (85,8 г, 0,85 моля, 2,0 экв.) и Pd(dppf)Cl2 (3,43 г, 4,2 ммоля, 1%). Раствор нагревали в атмосфере СО (4 МПа) при 120°С в течение 16 ч. Затем его фильтровали и концентрировали в вакууме. Остаток растворяли в 500 мл EtOAc и 1 л воды. Смесь нейтрализовывали насыщенным раствором NaHCO3 до рН=7,5 и разделяли. Неорганическую фазу экстрагировали с помощью EtOAc (500 мл×3) подкисляли с помощью 1 н. HCl до рН=5. Фильтрование и сушка в вакууме давали 62,8 г продукта (белое твердое вещество, выход 76%). МС (ИЭР): m/z 195,1 [M+1]+.
Стадия 2: Синтез метил-4-(2-(2,4-диметоксифенил)-2-оксоэтил)бензоат. К раствору 2-(4-(метоксикарбонил)фенил)уксусной кислоты (15 г, 77,3 ммоля) и ДМФ (1 капля) в безводном ДХМ (150 мл) при 0~5°С при перемешивании по каплям добавляли оксалилхлорид (33 мл, 386,0 ммоля). После завершения добавления смесь перемешивали при комнатной температуре в течение 2 ч. ТСХ (ПЭ/EtOAc=3/1, реакцию останавливали с помощью МеОН) показывала, что реакция завершилась, летучие вещества выпаривали и остаток разбавляли с помощью ДХМ (20 мл). К суспензии трихлорида алюминия (16,5 г, 123,7 ммоля) в безводном ДХМ (80 мл) при 5°С добавляли 1,3-диметоксибензол (21,3 г, 154,6 ммоля), затем полученный выше раствор ацилхлорида. Смесь перемешивали при комнатной температуре в течение ночи, осторожно выливали в охлажденную льдом 1 н. HCl (200 мл) и экстрагировали с помощью EtOAc (150 мл×3). Объединенные органические слои промывали рассолом (200 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали и получали коричневое масло, которое очищали на колонке с силикагелем (ПЭ/EtOAc=5/1) и получали продукт (12 г, 49,6%) в виде желтого твердого вещества. МС (ИЭР): m/z 315,1 [M+1]+.
Стадия 3: Синтез этил-4-(2-(2,4-дигидроксифенил)-2-оксоэтил)бензоата. К раствору метил-4-(2-(2,4-диметоксифенил)-2-оксоэтил)бензоата (55 г, 141,6 ммоля) в ДХМ (600 мл) при -10°С по каплям добавляли BBr3 (164 мл, 1,7 моля). После завершения добавления смесь перемешивали при комнатной температуре в течение ночи и при перемешивании выливали в измельченный лед (700 г). Летучие вещества выпаривали и получали светло-желтое твердое вещество, которое сушили в высоком вакууме и растворяли в абсолютном этаноле (500 мл). К раствору при 0-10°С по каплям добавляли тионилхлорид (80 мл). После завершения добавления полученную смесь кипятили с обратным холодильником в течение 3 ч. Летучие вещества выпаривали и остаток подвергали распределению между EtOAc (600 мл) и насыщенным раствором карбоната натрия (200 мл). Органическую фазу отделяли, промывали рассолом (200 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали и получали коричневую взвесь, которую очищали с помощью колоночной хроматографии (ПЭ/EtOAc=3/1) и получали продукт (24,5 г, 58%) в виде желтого твердого вещества.1Н ЯМР (CDCl3 500 МГц TMS): δ 12,58 (s, 1H), 8,01 (d, J=8,5 Гц, 2Н), 7,69 (d, J=9,0 Гц, 1Н), 7,33 (d, J=8,0 Гц, 2Н), 7,05 (brs, 1H), 6,41 (d, J=8,0 Гц, 1H), 6,37 (s, 1H), 4,38 (q, J=7,0 Гц, 2Н), 4,26 (s, 2Н), 1,38 (t, J = 7 Гц, 3Н). МС (ИЭР): m/z 301,1 [M+1]+.
Стадия 4: Синтез 4-(2-(2,4-дигидроксифенил)-2-оксоэтил)бензойной кислоты. К раствору этил-4-(2-(2,4-дигидроксифенил)-2-оксоэтил)бензоата (2 г, 6,7 ммоля) в 1,4-диоксане (10 мл), добавляли концентрированную HCl (10 мл). Реакционную смесь перемешивали при 70°С в течение ночи. Летучие вещества удаляли при пониженном давлении. Остаток растворяли в воде (20 мл) и экстрагировали с помощью EtOAc (20 мл×2). Объединенную органическую фазу сушили над безводным Na2SO4, фильтровали и концентрировали и получали продукт в виде коричневого твердого вещества (1,5 г, 83,3%). МС (ИЭР): m/z 273,1 [M+1]+.
Стадия 5: Синтез 4-(2-(2,4-дигидроксифенил)-2-оксоэтил)бензамида. К раствору 4-(2-(2,4-дигидроксифенил)-2-оксоэтил)бензойной кислоты (1,5 г, 5,5 ммоля) в ДХМ (20 мл) добавляли оксалилхлорид (4,7 мл, 55 ммоля) и одну каплю ДМФ. Смесь перемешивали при комнатной температуре в течение 3 ч и затем смесь кипятили с обратным холодильником в течение ночи. Смесь концентрировали досуха. Остаток растворяли в ТГФ (20 мл). Затем ее при комнатной температуре по каплям добавляли в водный раствор NH3·Н2О (25%, 60 мл). Смесь перемешивали при комнатной температуре в течение 2 ч. Летучие вещества удаляли при пониженном давлении. Остаток экстрагировали с помощью EtOAc (30 мл×3). Объединенную органическую фазу сушили над Na2SO4 и концентрировали и получали продукт в виде коричневого твердого вещества (300 мг, 20,1 %). МС (ИЭР): m/z 272,1 [М+1]+.
Промежуточный продукт В: Метил-5-(2-(2,4-дигидроксифенил)-2-оксоэтил)тиофен-2-карбоксилат
Стадия 1: Синтез 2-(5-бромтиофен-2-ил)уксусной кислоты. К раствору 2-(тиофен-2-ил)уксусной кислоты (2 г, 14 ммоля) в НОАс (10 мл) при 10~20°С в течение 30 мин по каплям добавляли бром (2,25 г, 14 ммоля). Смеси давали нагреваться до комнатной температуры в течение 3 ч. Затем ее разбавляли водой (100 мл), нейтрализовывали до рН=5 безводным карбонатом натрия и экстрагировали с помощью EtOAc (100 мл×3). Сушили над Na2SO4, фильтровали и концентрировали и получали неочищенный продукт в виде коричневого масла. МС (ИЭР): m/z 220,9 [M+1]+.
Стадия 2: Синтез 2-(5-(метоксикарбонил)тиофен-2-ил)уксусной кислоты: К раствору 2-(5-бромтиофен-2-ил)уксусной кислоты (2,5 г, 11,4 ммоля) в МеОН (110 мл) добавляли ТЭА (5 мл, 34,1 ммоля) и Pd(dppf)Cl2 (769 мг, 1,1 ммоля) и полученную смесь нагревали при 120°С в атмосфере СО (4 МПа) в течение 20 ч. Концентрировали в вакууме и остаток очищали с помощью Combi-Flash (80 г силикагеля, ПЭ/EtOAc = от 10:0 до 1:3 в градиентном режиме, 60 мл/мин, 60 мин, полный объем растворителя равен 3,6 л) и получали продукт в виде рыхлого твердого вещества (1,3 г, 58%). МС (ИЭР): m/z 198,0 [М+1]+.
Стадия 3: Синтез метил-5-(2-(2,4-диметоксифенил)-2-оксоэтил)тиофен-2-карбоксилата. Получали по методике, описанной на стадии 2 промежуточного продукта А, с использованием в качестве исходного вещества 2-(5-(метоксикарбонил)тиофен-2-ил)уксусной кислоты.
Стадия 4: Синтез метил-5-(2-(2,4-дигидроксифенил)-2-оксоэтил)тиофен-2-карбоксилата. К раствору метил-5-(2-(2,4-диметоксифенил)-2-оксоэтил)тиофен-2-карбоксилата (460 мг, 1,44 ммоля) в ДХМ (5 мл) добавляли AlCl3 (5,7 г, 43 ммоля). Смесь перемешивали при комнатной температуре в течение 2 дней, при 0°С осторожно добавляли воду (15 мл) и смесь экстрагировали с помощью EtOAc (20 мл×3). Объединенную органическую фазу промывали рассолом (50 мл), сушили над Na2SO4, концентрировали и очищали с помощью препаративной ТСХ (ПЭ/EtOAc=3/1) и получали промежуточный продукт В в виде оранжевого порошкообразного вещества (260 мг, 61,9%). МС (ИЭР): m/z 293,0 [М+1]+.
Промежуточный продукт С: Этил-4-(2-(2,4-дигидроксифенил)-2-оксоэтил)циклогексанкарбоксилат.
Стадия 1: Синтез этил-4-(2-трет-бутокси-2-оксоэтилиден)циклогексанкарбоксилата. К суспензии безводного хлорида лития (1,9 г, 45 ммоля) в MeCN (100 мл) при комнатной температуре добавляли трет-бутил-2-(диэтоксифосфорил)ацетат (7,6 г, 30 ммоля). Смесь перемешивали в течение 30 мин. Добавляли ТЭА (6,4 мл, 45 ммоля) и смесь перемешивали в течение еще 30 мин. Добавляли этил-4-оксоциклогексанкарбоксилат (5,1 г, 30 ммоля) и смесь перемешивали в течение ночи. Осадок отфильтровывали и фильтрат концентрировали и получали коричневое масло, которое очищали на колонке с силикагелем (ПЭ : EtOAc = 10:1) и получали продукт (5 г, 62%).
Стадия 2: Синтез 2-(4-(этоксикарбонил)циклогексилиден)уксусной кислоты. К раствору этил-4-(2-трет-бутокси-2-оксоэтилиден)циклогексанкарбоксилата (5,3 г, 21 ммоля) в CH2Cl2 (30 мл) добавляли ТФК (трифторуксусная кислота) (30 мл). Раствор перемешивали при комнатной температуре в течение ночи. Раствор концентрировали в вакууме и получали 6 г продукта в виде бесцветного масла, которое использовали на следующей стадии без обработки и без дополнительной очистки.
Стадия 3: Синтез 2-(4-(этоксикарбонил)циклогексил)уксусной кислоты. К раствору 2-(4-(этоксикарбонил)циклогексилиден)уксусной кислоты (6,0 г, 28,3 ммоля) в EtOH (50 мл) медленно добавляли 10% Pd/C (306 мг). Раствор гидрировали в течение ночи. Катализатор отфильтровывали. Фильтрат концентрировали в вакууме и получали 2,5 г продукта в виде бесцветного масла (42%).
Стадия 4: Синтез этил-4-(2-(2,4-дигидроксифенил)-2-оксоэтил)циклогексанкарбоксилата (промежуточный продукт С). К раствору 2-(4-(этоксикарбонил)циклогексил)уксусной кислоты (700 мг, 11,6 ммоля) в BF3-Et2O (10 мл) добавляли резорцин (1,5 г, 14,0 ммоля). Раствор перемешивали при 85°С в течение ночи, выливали в раствор Na2CO3 (2 н., 20 мл), экстрагировали с помощью EtOAc (30 мл×3), промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали и получали 3 г желтого масла, которое очищали на колонке с силикагелем (ПЭ/EtOAc=3/1) и получали 400 мг промежуточного продукта С. МС (ИЭР): m/z 307,1 [M+1]+.
Промежуточный продукт D: 2-(4-(2Н-Тетразол-5-ил)фенил)-1-(2,4-дигидроксифенил)этанон
Стадия 1: Синтез 2-(4-бромфенил)-1-(2,4-дигидроксифенил)этанона. В соответствии со стадией 4 получения промежуточного продукта С с использованием в качестве исходного вещества 2-(4-бромфенил)уксусной кислоты получали продукт в виде оранжевого порошкообразного вещества (20 г, 27,9 %). МС (ИЭР): m/z 307,0, 309,0 [M+1]+, [M+3]+.
Стадия 2: Синтез 4-(2-(2,4-дигидроксифенил)-2-оксоэтил)бензонитрила. Смесь 2-(4-бромфенил)-1-(2,4-дигидроксифенил)этанона (5 г, 16,3 ммоля) и CuCN (5,8 г, 65,4 ммоля) в ДМФ (50 мл) перемешивали при 150°С в течение 6 ч в атмосфере азота. К смеси добавляли воду (100 мл) и EtOAc (100 мл). Осадок отфильтровывали и фильтрат промывали рассолом (100 мл), сушили над Na2SO4 и концентрировали и получали черное масло, которое очищали с помощью Combiflash (ПЭ/EtOAc=3/1) и получали продукт в виде желтого порошкообразного вещества (1,5 г, 36,6%). МС (ИЭР): m/z, 254,1 [М+1]6+.
Стадия 3: Синтез 2-(4-(2Н-тетразол-5-ил)фенил)-1-(2,4-дигидроксифенил)этанона. К раствору 4-(2-(2,4-дигидроксифенил)-2-оксоэтил)бензонитрила (1,2 г, 4,7 ммоля) в толуоле (15 мл) при комнатной температуре добавляли TMSN3 (9,3 г, 85,4 ммоля) и Bu2SnO (309 мг, 1,41 ммоля). Смесь кипятили с обратным холодильником в течение ночи. Летучие вещества удаляли при пониженном давлении и остаток очищали с помощью Combiflash (EtOAc/HOAc = 50/1) и получали промежуточный продукт D в виде желтого порошкообразного вещества (550 мг, 39,6 %). Н ЯМР (ДМСО-d6 500 МГц TMS): δ 12,41 (s, 1H), 10,72 (s, 1H), 7,99-7,96 (m, 3Н), 7,51 (d, J=8,0 Гц, 2Н), 6,42 (dd, J=2,0 Гц, J=9,0 Гц, 1H), 6,27 (d, J=2,5 Гц,1Н), 4,44 (s, 2H); МС (ИЭР): m/z 297,0 [M+1]+.
Промежуточный продукт Е: Метил-4-(2-(3-метоксифенилтио)-2-оксоэтил)бензоат
К раствору 2-(4-(метоксикарбонил)фенил)уксусной кислоты (синтез описан на стадии 1 получения промежуточного продукта А) (3,88 г, 20 ммоля) в сухом ДХМ (50 мл) при комнатной температуре при перемешивании по каплям добавляли оксалилхлорид (8,4 мл, 100 ммоля). Смесь перемешивали при комнатной температуре в течение 2 ч. По данным ТСХ реакция завершалась (ПЭ : EtOAc=3:1, реакцию останавливали с помощью МеОН). Смесь концентрировали и получали метил-4-(2-хлор-2-оксоэтил)бензоат (4,5 г) в виде желтого масла, которое использовали на следующей стадии без обработки. К суспензии трихлорида алюминия (2,93 г, 21,96 ммоля) в сухом CS2 (50 мл) при 0~5°С добавляли 3-метоксибензолтиол (3,1 г, 21,96 ммоля), затем раствор метил-4-(2-хлор-2-оксоэтил)бензоата (4,5 г, неочищенный, 21,16 ммоля) в сухом CS2 (5 мл). Смесь перемешивали при комнатной температуре в течение ночи и осторожно выливали в 1 н. охлажденную льдом HCl (200 мл) и экстрагировали с помощью EtOAc (100 мл×2). Объединенные органические слои промывали водой (100 мл×2) и рассолом (100 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью Combi-Flash (80 г силикагеля, ПЭ/EtOAc = от 10/0 до 1/1 в градиентном режиме, 50 мл/мин, 40 мин, полный объем растворителя равен 2,0 л) и получали промежуточный продукт Е в виде желтого твердого вещества (2,7 г, 43%).1Н ЯМР (ДМСО-d6 500 МГц TMS): д 7,95 (d, J=8,5 Гц, 2Н), 7,49 (d, J=8,5 Гц, 2Н), 7,37 (d, J=8,0 Гц, 1Н), 7,02-7,04 (m, 1Н), 6,97-6,98 (m, 2Н), 4,18 (s, 2Н), 3,85 (s, 3Н), 3,76 (s, 3H); МС (ИЭР): m/z 317,1 [M+1]+.
Промежуточный продукт F: Метил-4-(2-(2-(ацетилтио)-4-метоксифенил)-2-оксоэтил)бензоат
Стадия 1: Синтез S-3-метоксифенилэтантиоата. К раствору 3-метоксибензолтиола (5 г, 35,66 ммоля) и ТЭА (7,45 мл, 53,49 ммоля) в ДХМ (60 мл) при комнатной температуре добавляли Ас2О (4 г, 39,23 ммоля) и перемешивали в течение 3 ч. Реакционную смесь промывали с помощью 1 н. HCl (50 мл×2), водой (50 мл×2) и рассолом (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали и получали продукт в виде желтого твердого вещества (6,5 г, 100%). МС (ИЭР): m/z 183,1 [М+1]+.
Стадия 2: Синтез метил-4-(2-хлор-2-оксоэтил)бензоата. К раствору 2-(4-(метоксикарбонил)фенил)уксусной кислоты (синтез описан на стадии 1 получения промежуточного продукта А) (5 г, 25,75 ммоля) в ДХМ ( 60 мл) при комнатной температуре по каплям добавляли оксалилхлорид (6,6 мл, 77,25 ммоля) и смесь перемешивали в течение 4 ч. Концентрирование давало продукт в виде масла золотого цвета (6 г, 109%), которое использовали на следующей стадии без обработки.
Стадия 3: Синтез метил-4-(2-(2-(ацетилтио)-4-метоксифенил)-2-оксоэтил)бензоата (промежуточный продукт F). К суспензии трихлорида алюминия (9,9 г, 74,08 ммоля) в ДХМ (100 мл) при 0~5°С добавляли S-3-метоксифенилэтантиоат (стадия 1) (4,5 г, 30,57 ммоля), затем раствор метил-4-(2-хлор-2-оксоэтил)бензоата (стадия 2) (4,5 г, неочищенный, 24,69 ммоля) в ДХМ (20 мл). Смесь перемешивали при комнатной температуре в течение 3 дней. Смесь осторожно выливали в охлажденную льдом 1 н. HCl (200 мл) и экстрагировали с помощью EtOAc (50 мл×2). Объединенные органические слои промывали водой (100 мл×2) и рассолом (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии (ПЭ/EtOAc=от 15/1 до 10/1) и получали промежуточный продукт F в виде желтого твердого вещества (1,8 г, 14%). МС (ИЭР): m/z 359,0 [M+1]+.
Промежуточный продукт G: 3-йод-7-метокси-2-метил-4Н-тиохромен-4-он: Стадия 1: Синтез 7-метокси-2-метил-4Н-тиохромен-4-она: К смеси 3-метоксибензолтиола (24 г, 28,5 ммоля) с полифосфорной кислотой (200 г) при комнатной температуре в атмосфере азота добавляли этилацетоацетат (22,8 г, 28,5 ммоля). Затем реакционную смесь нагревали при 110°С в течение 5 ч при энергичном перемешивании и выливали в смесь воды со льдом (100 мл). Полученную смесь экстрагировали этилацетатом (500 мл×3). Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали, затем очищали на колонке с силикагелем (ПЭ/EtOAc=10/1) и получали искомый продукт (2 г, 4%) в виде желтого твердого вещества.
Стадия 2: Синтез 3-йод-7-метокси-2-метил-4Н-тиохромен-4-она: К раствору 7-метокси-2-метил-4Н-тиохромен-4-она (500 мг, 2,42 ммоля) и йода (620 мг, 2,42 ммоля) в безводном ацетонитриле (10 мл) при комнатной температуре добавляли CAN (1,5 г, 2,70 ммоля), затем смесь перемешивали в течение 5 ч в атмосфере азота. Летучие вещества выпаривали в вакууме и остаток подвергали распределению между этилацетатом (20 мл) и насыщенным раствором Na2S2O3 (20 мл). Органическую фазу отделяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали и получали светло-желтое твердое вещество, которое перекристаллизовали из метанола (5 мл) и получали промежуточный продукт G (620 мг, 77%) в виде светло-желтого твердого вещества.1Н ЯМР (CDCl3 500 МГц):δ 8,46 (d, J=9,0 Гц, 1Н), 7,09 (dd,J=2,5, 9,5 Гц, 1Н), 6,94 (d, J= 2,5 Гц, 1Н), 3,90 (s, Гц 3Н), 2,61 (s, Гц 3Н).
Промежуточный продукт Н: Метил-5-(2-(2,4-дигидроксифенил)-2-оксоэтил)тиофен-3-карбоксилат
Стадия 1: Синтез метил-2-(4-бромтиофен-2-ил)ацетата: К раствору метил-2-(тиофен-2-ил)ацетата (5 г, 32 ммоля) и безводного AlCl3 (10,7 г, 80 ммолей) в СНСl3 (50 мл) при 0-5°С в течение 30 мин по каплям добавляли бром (1,8 мл, 34 ммоля). После завершения добавления смеси давали нагреваться до комнатной температуры в течение ночи. Затем ее выливали в воду со льдом (50 мл), экстрагировали с помощью EtOAc (30 мл×3). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали. Очистка с помощью колоночной хроматографии (ПЭ/EtOAc=10/1) давала продукт (3,1 г, 41%) в виде светло-желтого масла.
Стадия 2: Синтез метил-5-(2-метокси-2-оксоэтил)тиофен-3-карбоксилата: К раствору полученного выше продукта (3,1 г, 13,4 ммоля) в МеОН (110 мл) добавляли ТЭА (10 мл, 67,0 ммоля) и Pd(dppf)Cl2 (976 мг, 1,4 ммоля) и полученную смесь нагревали при 120°С в атмосфере СО (4 МПа) в течение 20 ч. Концентрировали и остаток подвергали распределению между этилацетатом (50 мл) и 1 н. HCl (20 мл). Органическую фазу отделяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали и получали продукт.
Стадия 3: Синтез 2-(4-(метоксикарбонил)тиофен-2-ил)уксусной кислоты: К раствору полученного выше продукта (2,8 г, 13,1 ммоля) в смеси ТГФ/Н2О (15/15 мл) 3 порциями при комнатной температуре добавляли моногидрат гидроксида лития (490 мг, 11,7 ммоля). Смесь перемешивали в течение 2 дней и экстрагировали с помощью ПЭ (10 мл). Водную фазу отделяли, подкисляли до рН=5 с помощью 1 н. НСl, экстрагировали с помощью EtOAc (30 мл×3). Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали и получали неочищенный продукт.
Стадия 4: Синтез промежуточного продукта Н: К смеси полученного выше продукта (450 мг, 2,3 моля) и резорцина (248 мг, 2,3 моля) добавляли BF3-Et2O (2 мл) и смесь перемешивали при 95°С в течение ночи. Смесь выливали в насыщенный раствор карбоната натрия (10 мл) с установлением рН=10 и экстрагировали этилацетатом (20 мл×3). Объединенную органическую фазу отделяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали и получали желтое масло, которое очищали с помощью колоночной хроматографии (ПЭ/EtOAc=3/1) и получали промежуточный продукт И в виде желтого твердого вещества (95 мг, 15 %). МС (ИЭР): m/z 292,9 [M+1]+.
Промежуточный продукт I: Метил-4-(2-(2,4-дигидроксифенил)-2-оксоэтил)-3-фторбензоат
Стадия 1: Синтез метил-3-фтор-4-метилбензоата: раствор 3-фтор-4-метилбензойной кислоты (20 г, 130 ммоля) в тионилхлориде (80 мл) кипятили с обратным холодильником в течение 2 ч (ТСХ показывала отсутствие исходного вещества) и летучие вещества выпаривали. К остатку при перемешивании по каплям при 0°С добавляли МеОН (100 мл). Смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь концентрировали и разбавляли с помощью EtOAc и промывали рассолом. Органический слой сушили над безводным Na2SO4, фильтровали и выпаривали и получали продукт в виде белого твердого вещества и использовали без дополнительной очистки на следующей стадии. (21 г, 96%).
Стадия 2: Синтез метил-4-(бромметил)-3-фторбензоата: К раствору полученного выше продукта (21 г, 125 ммоля) в ССl4 (200 мл) добавляли смесь NBS (N-бромсукцинимид) (20 г, 113 ммоля) и бензоилпероксида (1,5 г, 6 ммоля). Полученный раствор кипятили с обратным холодильником в течение 5 ч. Затем растворитель выпаривали и остаток растворяли в ДХМ (300 мл) и промывали с помощью Н2О (200 мл×3). Органический слой сушили над безводным Na2SO4, фильтровали и выпаривали и получали неочищенный продукт в виде бесцветного масла (18 г, 64,7%).
Стадия 3: Синтез метил-4-(цианометил)-3-фторбензоата: К раствору метил-
4-(бромметил)-3-фторбензоата (18 г, 73 ммоля) в МеОН (150 мл) добавляли раствор NaCN (7,2 г, 146 ммоля) в Н2О (40 мл). Смесь перемешивали при 65°С в течение 5 ч. Большую часть МеОН выпаривали и дополнительно добавляли воду и экстрагировали с помощью EtOAc (200 мл×3), сушили над безводным Na2SO4, фильтровали и выпаривали и получали неочищенный продукт. Очистка с помощью колоночной хроматографии (ПЭ/EtOAc = от 50/1 до 10/1) давала продукт (8 г, 63,8%).
Стадия 4: Синтез метил-4-(2-(2,4-дигидроксифенил)-2-оксоэтил)-3-фторбензоата: Через раствор метил-4-(цианометил)-3-фторбензоата (8 г, 41 ммоля) и резорцина (6,8 г, 62 ммоля) в BF3-Et2O (100 мл) при 0°С в течение 15 мин пропускали HCl. Смесь перемешивали при 75°С в течение 16 ч. Затем Н2О добавляли (100 мл) и раствор нагревали при 95°С в течение 16 ч. Смесь охлаждали до комнатной температуры и нейтрализовывали с помощью Na2CO3 и экстрагировали с помощью EtOAc (100 мл×3). Органические слои объединяли, сушили над безводным Na2SO4, фильтровали и выпаривали и получали неочищенный продукт. Очистка с помощью колоночной хроматографии (ПЭ/EtOAc = от 10/1 до 3/1) давала промежуточный продукт I в виде белого твердого вещества (6 г, 48 %).
Промежуточный продукт J: Метил-4-(2-(2,4-дигидроксифенил)-2-оксоэтил)-3-метилбензоат
Стадия 1: Синтез метил-4-(2-этокси-2-оксоэтил)-3-метилбензоата: Метил-4-бром-3-метилбензоат (1,5 г, 6,55 ммоля), метил-3-оксобутаноат (0,99 г, 8,51 ммоля) и К3РO4 (4,17 г, 19,6 ммоля) смешивали с Pd(OAc)2 (15 мг) и ди-трет-бутил(2'-метил-[1,1'-бифенил]-2-ил)фосфином (39 мг) и разбавляли толуолом (25 мл). Полученную смесь дегазировали в вакууме и помещали в атмосферу аргона. Затем ее нагревали при 90°С в течение 24 ч и при 110°С в течение 5 ч. После обработки водой с EtOAc искомый продукт, метил-4-(2-этокси-2-оксоэтил)-3-метилбензоат, выделяли с помощью колоночной хроматографии в виде масла (366 мг, 25%) при элюировании смесью EtOAc/гексан (1/3).
Стадия 2: Синтез 2-(4-(метоксикарбонил)-2-метилфенил)уксусной кислоты: Метил-4-(2-этокси-2-оксоэтил)-3-метилбензоат (366 мг) суспендировали в MeOH мл) и Н2О (2 мл) и в течение 3 дней обрабатывали с помощью LiOH (48 мг). Затем реакционную смесь подкисляли с помощью 1 н. HCl, продукт экстрагировали с помощью EtOAc. Выпаривание и сушка в вакууме давали искомый продукт (295 мг).
Стадия 3: Синтез промежуточного продукта J: 2-(4-(метоксикарбонил)-2-метилфенил)уксусную кислоту (295 мг) при 90 С в течение ночи обрабатывали резорцином (190 мг) в BF3Et2O. После обработки водой с этилацетатом и очистки на колонке искомый продукт (296 мг) выделяли в виде масла.
Промежуточный продукт К: 4-(2-(2,4-Дигидроксифенил)-2-оксоэтил)бензонитрил
Синтез: Смесь 2-(4-бромфенил)-1-(2,4-дигидроксифенил)этанона (стадия 1, промежуточный продукт D) (5 г, 16,3 ммоля) и CuCN (5,8 г, 65,4 ммоля) в ДМФ (50 мл) перемешивали при 150°С в течение 6 ч в атмосфере азота. К смеси добавляли воду (100 мл) и EtOAc (100 мл). Осадок отфильтровывали и фильтрат промывали рассолом (100 мл), сушили над Na2SO4 и концентрировали и получали черное масло, которое очищали с помощью Combiflash (ПЭ/EtOAc=3/1) и получали промежуточный продукт К в виде желтого порошкообразного вещества (1,5 г, 36,6%).
Промежуточный продукт L: Метил-4-(2-(3-фтор-2,4-дигидроксифенил)-2-оксоэтил)бензоат
Промежуточный продукт L можно получить по методике, описанной для промежуточного продукта К, стадия 3, с использованием в качестве исходного вещества 2-(4-(метоксикарбонил)фенил)уксусной кислоты (синтез описан в PCT/US 2010/024035) и 2-фторбензол-1,3-диола (имеется в продаже). Пример 26: Исследования GSNOR
Для различных соединений in vitro исследовали способность ингибировать активность GSNOR. Соединения-ингибиторы GSNOR в примерах 1-22 обладали значениями IC50, равными примерно <1 мкМ. Соединения-ингибиторы GSNOR в примерах 1-3, 5-6, 8-9, 11-12, 14, 16-22 обладали значениями IC50, равными примерно менее 0,1 мкМ.
Экспрессия и очистка GSNOR описаны в публикации Biochemistry 2000, 39, 10720-10729.
Ферментация GSNOR: Предкультуры выращивали из уколочной культуры микробов в исходном растворе GSNOR-глицерин в средах 2XYT, содержащих 100 мкг/мл ампициллина, после инкубации в течение ночи при 37°С. Затем клетки добавляли к свежей среде 2XYT (4 л), содержащей ампициллин, и до индуцирования выращивали до оптической плотности (А600), равной 0,6-0,9 при 37°С. Экспрессию GSNOR индуцировали с помощью 0,1% арабинозы при инкубации в течение ночи при 20°С.
Очистка GSNOR: Пасту клеток Е. coli лизировали путем кавитации азотом и осветленный лизат очищали с помощью Ni-афинной хроматографии на АКТА FPLC (Amersham Pharmacia). Колонку элюировали с помощью 20 мМ Tris [трис(гидроксиметиламинометан)], рН 8,0/250 мМ NaCl с использованием имидазола в градиентном режиме 0-500 мМ. Элюированные фракции GSNOR, содержащие слияние Smt-GSNOR, гидролизовали в течение ночи с помощью Ulp-1 при 4°С для удаления афинной метки и затем повторно пропускали через Ni-колонку при таких же условиях. GSNOR получали в виде выходящей фракции и для исследования с помощью кристаллографии дополнительно очищали с помощью проточной хроматографии с использованием 0-сефарозы и гепарина с использованием 20 мМ Tris рН 8,0, 1 мМ ДТТ (дитиотреитол), 10 мкМ ZnSO4.
Исследование GSNOR: GSNO и растворы фермент/NADH ежедневно готовили свежими. Растворы фильтровали и им давали нагреться до комнатной температуры. Раствор GSNO: 100 мМ NaPO4 (рН 7,4), 0,480 мМ GSNO. 396 мкл Раствора GSNO добавляли в кювету, содержащую 8 мкл исследуемого соединения в ДМСО (или только ДМСО в качестве контроля реакции) и перемешивали кончиком пипетки. Исследуемые соединения готовили при начальной концентрации, равной 10 мМ в 100% ДМСО. Двукратные серийные разведения готовили в 100% ДМСО. К исследуемой смеси добавляли 8 мкл каждого разведения, так что конечная концентрация ДМСО при исследовании равнялась 1%. Использовали концентрации исследуемых соединений в диапазоне от 100 до 0,003 мкМ. Раствор фермент/NADH: 100 мМ NaPO4 (рН 7,4), 0,600 мМ NADH, 1,0 мкг/мл GSNO-редуктазы. 396 мкл Раствора фермент/NADH добавляли в кювету для инициирования реакции. Кювету помещали в спектрофотометр для УФ/видимой области Cary 3Е и в течение 3 мин регистрировали измерение поглощение/мин при 340 нм при 25°С. Для каждой концентрации соединения исследование проводили трижды. Значения IC50 для каждого соединения рассчитывали с помощью стандартного анализа зависимости модуля кинетики ферментов программного обеспечения SigmaPlot.
Условия заключительного исследования: 100 мМ NaPO4, рН 7,4, 0,240 мМ GSNO, 0,300 мМ NADH, 0,5 мкг/мл GSNO-редуктазы и 1% ДМСО. Конечный объем: 800 мкл/кювета.
Пример 27: Эффективность GSNORi в экспериментальной модели астмы
Экспериментальная модель астмы:
Для изучения эффективности ингибиторов GSNOR по отношению к вызванному метахолином (MCh) бронхостенозу/гиперчувствительности дыхательных путей использовали модель вызванной овальбумином (OVA) астмы на мышах. Она представляет собой широко использующуюся и тщательно изученную модель фенотипа острой аллергической астмы, сходной с астмой человека. Эффективность ингибиторов GSNOR оценивали по методике, в которой ингибиторы GSNOR вводили после сенсибилизации с помощью OVA и воздействия на дыхательные пути и до воздействия MCh. Бронхостеноз в ответ на воздействие увеличивающихся доз MCh оценивали с помощью плетизмографии всего тела (Penh; Buxco). Количество эозинофильного инфильтрата в жидком бронхоальвеолярном лаваже (ЖБАЛ) также оценивали в качестве меры воспаления легких. Влияние ингибиторов GSNOR сопоставляли с данными для разбавителя и Combivent (при вдыхании; IH) в качестве положительного контроля.
Материалы и методика
Сенсибилизация аллергеном и методика воздействия OVA (500 мкг/мл) в ЗФФ смешивали с одинаковым объемом 10% (маc./об.) сульфата калия-алюминия (алюмокалиевые квасцы) в дистиллированной воде, значение рН устанавливали равным 6,5 с помощью 10 н. NaOH и инкубировали в течение 60 мин при комнатной температуре. После центрифугирования при 750×g в течение 5 мин таблетку OVA/квасцы повторно суспендировали в дистиллированной воде до исходного объема. В день 0 мышам делали внутрибрюшинную (ВБ) инъекцию комплекса 100 мкг OVA (0,2 мл в физиологическом растворе концентрации 500 мкг/мл) с квасцами. Мышей анестезировали с помощью ВБ инъекции 0,2 мл смеси кетамина и ксилазина (0,44 и 6,3 мг/мл соответственно) в физиологическом растворе и помещали на стол на спину. На нижнюю сторону языка каждого животного помещали 250 мкг (100 мкл раствора концентрации 2,5 мг/мл) OVA (в день 8) и 125 мкг (50 мкл раствора концентрации 2,5 мг/мл) OVA (в дни 15, 18, и 21).
Исследование функции легких (Penh)
С помощью плетизмографии всего тела с использованием камеры Вихсо (Wilmington, NC) для находящихся в сознании, свободно движущихся мышей с нормальным дыханием через 24 ч после последнего воздействия OVA in vivo исследован ответ дыхательных путей на метахолин. На мышей в течение 2 мин воздействовали аэрозолями физиологического раствора или увеличивающихся доз метахолина (5, 20 и 50 мг/мл), полученных с помощью ультразвукового распылителя. Степень бронхостеноза представляли в виде увеличенного перерыва (Penh), рассчитываемого безразмерного значения, которое коррелирует с результатами измерений сопротивления дыхательных путей, импеданса дыхательной системы и внутриплеврального давления для этой же мыши. Значения Penh определяли и усредняли в течение 4 мин после распыления каждой дозы. Penh рассчитывали следующим образом: Penh=[(Те/Tr-1)×(PEF/PIF)], где Те - длительность выдоха, Tr - период релаксации, PEF - максимальная скорость выдоха и PIF - максимальная скорость вдоха ×0,67 (коэффициент). Период релаксации - это время изменения давления в камере от максимального до заданной исследователем доли от максимального. Измерение начинается в момент максимального давления в камере и заканчивается, когда это значение составляет 40% от максимального.
Эозинофильный инфильтрат в ЖБАЛ
После исследования гиперреактивности дыхательных путей мышей обескровливали путем пункции сердца и затем ЖБАЛ собирали из обоих легких или из правого легкого после перерезания главного бронха левого легкого. Полное количество клеток в ЖБАЛ подсчитывали с помощью аликвоты объемом 0,05 мл и остальную жидкость центрифугировали при 200×g в течение 10 мин при 4°С. Таблетки клеток повторно суспендировали в физиологическом растворе, содержащем 10% БСА (бычий сывороточный альбумин), и на предметные стекла наносили мазки. Эозинофилы окрашивали в течение 5 мин 0,05% водным раствором эозина и 5% ацетона в дистиллированной воде, промывали дистиллированной водой и контрастно окрашивали с помощью 0,07% метиленового синего. Альтернативно, эозинофилы и другие лейкоциты окрашивали красителем DiffQuik.
Ингибиторы GSNOR и контрольные образцы
Ингибиторы GSNOR восстанавливали в забуференном фосфатом физиологическом растворе (ЗФФ), рН 7,4 или 0,5% мас./об. карбоксиметилцеллюлозе при концентрациях в диапазоне от 0,00005 до 3 мг/мл. Ингибиторы GSNOR вводили мышам (10 мл/кг) в виде одной дозы или нескольких доз внутривенно (ВВ) или перорально через желудочный зонд. Дозы вводили в период от 30 мин до 72 ч до воздействия посредством MCh. Воздействие ингибиторов GSNOR сопоставляли с данными для разбавителя, вводимого таким же образом.
Во всех исследованиях в качестве положительного контроля использовали Combivent. Combivent (Boehringer Ingelheim) вводили в легкие с помощью ингалятора, содержащего продукт, но приспособленного для введения мышам, с помощью наконечника пипетки. Combivent вводили за 48 ч, 24 ч и 1 ч до введения MCh. Каждая порция (или доза) Combivent содержит 18 мкг ипатропийбромида (IpBr) и 103 мкг албутеролсульфата или примерно 0,9 мг/кг IpBr и 5 мг/кг албутерола.
Статистический анализ
Значения площади под кривой для Penh вдоль базовой линии, физиологического раствора и действие увеличивающихся доз MCh определяли с помощью программного обеспечения GraphPad Prism 5.0 (San Diego, CA) и выражали в процентах от соответствующих значений для являющегося контролем разбавителя (вводили ВВ или перорально). Статистические различия значений для групп животных, подвергавшихся лечению, и животных соответствующей контрольной группы, получавших разбавитель, для каждого исследования рассчитывали с использованием одностороннего дисперсионного анализа, дополнительных критериев Даннетта или Бонферрони или t-критерия (JMP 8.0, SAS Institute, Cary, NC или Microsoft Excel). Значение р<0,05 при сопоставлении значений для групп животных, подвергавшихся лечению, и животных соответствующей контрольной группы, получавших разбавитель, соответствовало статистически значимым результатам.
Результаты
В модели астмы OVA соединение примера 3 приводило к уменьшению ППК для Pgnh (р<0,05) и инфильтрации эозинофилов в ЖБАЛ на 43% и 42% соответственно по сравнению с данными для контрольной группы, получавшей разбавитель, при пероральном введении одной дозы, равной 10 мг/кг, за 24 ч до определения. В другом исследовании соединение примера 3 приводило к уменьшению инфильтрации эозинофилов в ЖБАЛ на 12% по сравнению с данными для контрольной группы, получавшей разбавитель, при пероральном введении трех доз, равных 10 мг/кг, за 48 ч, 24 ч и 1 ч до определения.
В модели астмы OVA соединение примера 1 приводило к уменьшению ППК для Penh (Р<0,05) и инфильтрации эозинофилов в ЖБАЛ на 20-39% и на 0 -31% соответственно по сравнению с данными для контрольной группы, получавшей разбавитель, при пероральном введении одной дозы, равной 10 мг/кг, за 24 ч до определения. Соединение примера 1 приводило к значительному уменьшению ППК для Penh на 39% по сравнению с данными для контрольной группы, получавшей разбавитель, при введении одной ВВ дозы, равной 10 мг/кг, за 24 ч до определения.
В модели астмы OVA соединение примера 9 приводило к значительному уменьшению ППК для Penh и инфильтрации эозинофилов в ЖБАЛ на 18% и 82% соответственно по сравнению с данными для контрольной группы, получавшей разбавитель, при пероральном введении одной дозы, равной 10 мг/кг, за 24 ч до определения.
В модели астмы OVA соединение примера 16 приводило к значительному (р<0,05) уменьшению инфильтрации эозинофилов в ЖБАЛ на 36% по сравнению с данными для контрольной группы, получавшей разбавитель, при пероральном введении трех доз, равных 10 мг/кг, за 48 ч, 24 ч и 1 ч до определения.
Пример 28: Исследование фармакокинетики (ФК) с использованием мышей
Экспериментальная модель
Мышей использовали для исследования фармакокинетики соединений, предлагаемых в настоящем изобретении. Этот вид широко используют для оценки биологической доступности соединений путем введения исследуемых веществ перорально (ПО) и внутривенно (ВВ). Эффективность соединений, предлагаемых в настоящем изобретении, сопоставляли путем оценки влияния на плазму самцов мышей BALB/c после ВВ или ПО введения в периоды максимальной активности.
Материалы и методики
ВВ введение соединений, предлагаемых в настоящем изобретении Соединения, предлагаемые в настоящем изобретении, восстанавливали в прозрачном растворе забуференный фосфатом физиологический раствор (ЗФФ)/10% Solutol (HS 15) с получением концентрации, равных 0,2 мг/мл, и вводили мышам (2 мг/кг) в виде разовой ВВ дозы. Введение животным проводили в боковую хвостовую вену. Пробы крови отбирали в заданные моменты времени (0,083, 0,25, 0,5, 1, 2, 4, 8, 16, 24 ч) с помощь пункции сердца при анестезии изофлураном (до 1 мл крови от животного). Кровь собирали в пробирки, содержащие Li-гепарин. Пробы крови держали на льду до центрифугирования, которое проводили не позже, чем через 30 мин после отбора. Плазму переносили в маркированные полипропиленовые пробирки и до анализа с помощью ЖХ/МС/МС замораживали и хранили при -70°С.
ПО введение соединений, предлагаемых в настоящем изобретении
Соединения, предлагаемые в настоящем изобретении, восстанавливали в прозрачном растворе 40% пропиленгликоля/40% пропиленкарбоната/20% 5% сахарозы с получением концентрации, равных 0,2 мг/мл, и вводили мышам (10 мг/кг) в виде разовой пероральной дозы через желудочный зонд. Пробы крови отбирали через 0,25, 0,5, 1, 2, 4, 8, 12, 16, 20 и 24 ч после введения помощь пункции сердца при анестезии изофлураном. Кровь собирали в пробирки, содержащие Li-гепарин. Пробы крови держали на льду до центрифугирования, которое проводили не позже, чем через 30 мин после отбора. Плазму переносили в маркированные полипропиленовые пробирки и до анализа с помощью ЖХ/МС/МС замораживали и хранили при -70°С.
Анализ с помощью ЖХ/МС/МС
Образцы плазмы для каждого момента времени анализировали с помощью ЖХ-МС/МС при нижнем пределе количественного определения (НПКО), равном 1 нг/мл. В плазме определяли количество соединения, предлагаемого в настоящем изобретении, для каждого образца и для всех соединений, предлагаемых в настоящем изобретении строили регрессионные зависимости в виде соответствующих матриц.
Для расчета параметров ФК для ВВ и ПО введения использовали методику анализа WinNonlin:
Параметры ФК для ВВ порции - AUClast; AUCINF; T1/2; C1; Vss; Cmax; MRT
Параметры ФК для ПО порции - AUClast; AUCINF; T1/2; Cmax; C1, MRT.
В дополнение к указанным выше параметрам ФК рассчитывали биологическую доступность (%F). Результаты:
Соединения примеров 1, 3, 9 и 12 характеризовались пероральной биологической доступностью, составляющей примерно 4-41%. Соединение примера 16 характеризовалось пероральной биологической доступностью, составляющей примерно 44%. Сравнительное соединение 4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензойная кислота (см. PCT/US 2010/024035) характеризовалось пероральной биологической доступностью, составляющей примерно 17%. Сравнительное соединение выводилось (Cl) примерно в три раза быстрее, чем соединение примера 16.
Пример 29: Эффективность ингибиторов GSNOR в экспериментальной воспалительной болезни кишечника (ВБК)
Описание моделей
Модели острой и хронической ВБК, вызванной декстрансульфатом натрия (ДСП), использовали для исследования эффективности GSNORi при этом заболевании. Модели острой и хронической ВБК, вызванной ДСН, являются широко использующимися и хорошо изученными моделями, в которых создаются патологические изменения толстой кишки, сходные с наблюдающимися при заболевании человека. В этих моделях и при заболевании человека разрушаются эпителиальные клетки в криптах толстой кишки, что приводит к дисфункции эпителиального барьера и возникновению воспаления ткани, отека и изъязвления. Лечение с помощью GSNORi может быть благоприятным при ВБК вследствие восстановления содержания S-нитрозоглутатиона (GSNO) и тем самым предупреждения или обращения дисфункции эпителиального барьера.
Модель эффективной профилактики
Экспериментальную ВБК вызывали путем введения ДСН с питьевой водой самцам мышей С57В1/6 (N = от 8 до 10 мышей в группе) в течение 6 последовательных дней. GSNORi вводили перорально в дозах, равных от 0,1 до 10 мг/кг/сутки, в течение 10 дней, начиная от двух дней до и продолжая в течение двух дней после введения ДСН. В течение двух дней после введения ДСН воздействие GSNORi оценивали по слепой методике с помощью эндоскопии и гистопатологического исследования с использованием 5-балльной шкалы в диапазоне от значения = 0 (нормальная ткань) до значения = 4 (изъязвление и выраженные патологические изменения ткани). Также определяли содержание в кровотоке цитокинов, участвующих в путях воспаления. Влияние GSNORi сопоставляли с данными для контрольных -животных, которым вводили разбавитель. В качестве положительного контроля в этом исследовании использовали кортикостероид, преднизолон, и его перорально вводили ежедневно по 3 мг/кг/сутки. Для мышей, у которых не вызывали заболевание (N=5), также проводили исследование, которое показало, что ткани являются нормальными.
Модель длительного лечения
Экспериментальную ВБК вызывали путем введения ДСН с питьевой водой самцам мышей С57В 1/6 (N = от 10 до 12 мышей в группе) в течение 6 последовательных дней. GSNORi вводили перорально в дозах, равных 10 мг/кг/сутки, в течение 14 дней, начиная от одного дня после прекращения воздействия ДСН. Эффективность GSNORi оценивали по слепой методике с помощью эндоскопии через 7 дней и 14 дней после введения GSNORi и помощью гистопатологического исследования через 14 дней после введения GSNORi с использованием 5-балльной шкалы в диапазоне от значения = О (нормальная ткань) до значения = 4 (изъязвление и выраженные патологические изменения ткани). Также определяли содержание в кровотоке цитокинов, участвующих в путях воспаления. Влияние GSNORi сопоставляли с данными для контрольных животных, которым вводили разбавитель. В качестве положительного контроля в этом исследовании использовали кортикостероид, преднизолон, и его перорально вводили ежедневно по 3 мг/кг/сутки. Для мышей, у которых не вызывали заболевание (N=5), также проводили исследование, которое показало, что ткани являются нормальными.
Результаты:
В модели на мышах острой ВБК, вызванной ДСН, соединение примера 3 уменьшало поражение толстой кишки. Относительное количество мышей, у которых по данным эндоскопии наблюдалось тяжелое поражение толстой кишки, после перорального лечения с помощью 0,1 или 1 мг/кг/сутки соединения примера 3 в течение 10 последовательных дней в профилактическом режиме введения, уменьшилось на 38% или 25% соответственно по сравнению с данными для контрольной группы, получавшей разбавитель. Относительное количество мышей, у которых по данным оценки патологии наблюдалось тяжелое поражение толстой кишки, после перорального лечения с помощью 0,1 или 1 мг/кг/сутки соединения примера 3 в течение 10 последовательных дней уменьшилось на 12% или 33% соответственно по сравнению с данными для контрольной группы, получавшей разбавитель.
В модели на мышах острой ВБК, вызванной ДСН, соединение примера 1 уменьшало поражение толстой кишки. Относительное количество мышей, у которых по данным эндоскопии или гистопатологического исследования наблюдалось тяжелое поражение толстой кишки, после перорального лечения с помощью 10 мг/кг/сутки соединения примера 1 в течение 10 последовательных дней в профилактическом режиме введения, уменьшилось на 75% или 17% соответственно по сравнению с данными для контрольной группы, получавшей разбавитель.
В модели на мышах острой ВБК, вызванной ДСН, соединение примера 16 уменьшало поражение толстой кишки. Относительное количество мышей, у которых по данным эндоскопии или гистопатологического исследования наблюдалось тяжелое поражение толстой кишки, после перорального лечения с помощью 10 мг/кг/сутки соединения примера 16 в течение 10 последовательных дней в профилактическом режиме введения, уменьшилось на 58% или 15% соответственно по сравнению с данными для контрольной группы, получавшей разбавитель.
Пример 30: Эффективность ингибиторов GSNOR в экспериментальном хроническом обструктивном заболевании легких (ХОЗЛ).
Модели ХОЗЛ с использованием кратковременного курения сигарет
Эффективность ингибиторов GSNOR оценивали с помощью модели на мышах хронического обструктивного заболевания легких (ХОЗЛ), вызванного кратковременным (4 дней или 11 дней) воздействием сигаретного дыма. Для оценки влияния ингибиторов GSNOR на некоторые ранние проявления, связанные с возникновением и прогрессированием ХОЗЛ, определяли инфильтрацию воспалительных клеток в жидкий бронхоальвеолярный лаваж (ЖБАЛ) и содержание в ЖБАЛ хемокинов, участвующих в воспалении и ремоделировании/восстановлении тканей.
Описание моделей:
Эффективность ингибиторов GSNOR по отношению к ХОЗЛ с использованием моделей кратковременного (4 дня) и относительно продолжительного (11 дней) воздействия сигаретного дыма, вызывающего ХОЗЛ у мышей. Воздействие сигаретного дыма используется в качестве модели ХОЗЛ, при которой поражение вызывается воздействием того же фактора, что и при заболевании людей, и при которой поражение сходно с поражением при заболевании людей, включая обструкцию дыхательных путей, расширение воздушных пространств и участие воспалительных ответов в этих патологиях. В моделях на животных изменения в патологии легких обнаруживаются только после длительного (несколько месяцев) воздействия сигаретного дыма, и это исключает использование моделей с длительным воздействием в качестве эффективных средств скрининга. Недавно в качестве средств скрининга эффективности и механизмов воздействия новых средств лечения ХОЗЛ применяли модели, в которых используются воспалительные ответы после кратковременного (2 недели или менее) воздействия дыма на мышей. Ключевая роль воспаления в возникновении и прогрессировании ХОЗЛ делает эти модели с кратковременным воздействием применимыми для предварительного исследования эффективности новых лекарственных средств.
Модель кратковременного (4 дня) воздействия дыма: На самок мышей С57В 1/6 (N=8 в группе) в камере для воздействия на весь организм оказывали воздействие сигаретным дымом. Воздействие на мышей проводили ежедневно в течение 4 последовательных дней до 4 циклов воздействия дыма 6 последовательных сигарет (Kentucky 3R4F без фильтра) с 30-минутными промежутками без воздействия дыма между циклами. Ингибиторы GSNOR вводили перорально ежедневно по 10 мг/кг/сутки в течение 7 дней, начиная за 2 дня до воздействия дыма и продолжая 1 день после воздействия/Влияние ингибиторов GSNOR определяли путем количественного определения полного количества клеток, лейкоцитов и лейкоцитарной формулы в ЖБАЛ с помощью оптической микроскопии и содержания хемокинов в ЖБАЛ с помощью иммуноферментного анализа ELISA примерно через 24 ч после заключительного воздействия дыма. Влияние ингибиторов GSNOR сопоставлено с данными для контрольных животных, которым вводили разбавитель. В качестве положительного контроля в исследовании использовали ингибитор PDE4, рофлумиласт. На группу мышей (N=8) воздействовали воздухом и в исследовании ее использовали в качестве отрицательного контроля.
Модель относительно продолжительного (11 дней) воздействия дыма: На самок мышей С57В 1/6 (N=10 в группе) оказывали воздействие дымом сигарет Мальборо 100 без фильтра. Длительность воздействия составляла 25 мин в день I исследования, 35 мин в день 2 исследования и 45 мин в дни от 3 до 11 исследования. Ингибиторы GSNOR вводили ежедневно за 1 ч до воздействия дыма. Ингибиторы GSNOR вводили перорально от 1 до 10 мг/кг/сутки в течение II дней. Влияние ингибиторов GSNOR определяли путем количественного определения полного количества клеток, лейкоцитов и лейкоцитарной формулы в ЖБАЛ с помощью оптической микроскопии через 24 ч после заключительного воздействия. Влияние ингибиторов GSNOR сопоставлено с данными для контрольных животных, которым вводили разбавитель, и выражали в процентах подавления вызванного сигаретным дымом увеличения количества клеток в ЖБАЛ. В качестве положительного контроля в исследовании использовали рофлумиласт и его вводили по 5 мг/кг/сутки. На группу мышей (N=10) воздействовали воздухом и в исследовании ее использовали в качестве отрицательного контроля.
Результаты:
Соединение примера 3 уменьшало вызванные дымом изменения в клеточном инфильтрате ЖБАЛ и в количестве воспалительных хемокинов в ЖБАЛ. В модели кратковременного проводимого в течение 4 дней воздействия дыма при пероральном введении по 10 мг/кг/сутки в течение 7 дней соединение примера 3 полностью (100%) и статистически значимо (р<0,05) подавляло вызванное дымом увеличение полного количества клеток, лейкоцитов, макрофагов, нейтрофилов и эозинофилов в ЖБАЛ. Эти эффекты соединения примера 3 сравнимы по величине или превышают наблюдавшиеся для рофлумиласта. Соединение примера 3 также восстанавливало содержание хемокинов в ЖБАЛ до уровней, наблюдающихся у здоровых мышей. В модели относительно продолжительного воздействия в течение 11 дней соединение примера 3 при пероральном введении по 10 мг/кг/сутки в течение 11 дней подавляло вызванное дымом увеличение полного количества клеток (р<0,05), макрофагов (р<0,05), нейтрофилов, эозинофилов и лимфоцитов в ЖБАЛ на 26%, 28%, 25%, 57% и 24% соответственно.
В модели относительно продолжительного воздействия в течение 11 дней при пероральном введении по 1 мг/кг/сутки в течение 11 дней соединение примера 16 статистически значимо (р<0,05) подавляло вызванное дымом увеличение полного количества клеток, макрофагов, нейтрофилов и лимфоцитов в ЖБАЛ на 53%, 44%, 68% и 62% соответственно. Влияние соединения примера 16 было сравнимо с влиянием рофлумиласта.
Специалистам в данной области техники должно быть очевидно, что без отклонения от сущности или объема настоящего изобретения в способы и композиции, предлагаемые в настоящем изобретении, можно внести различные модификации и изменения.
Реферат
Настоящее изобретение относится к новым ингибиторам S-нитрозоглутатионредуктазы (GSNOR) формулы I, , в которой X выбран из группы, включающей О и S; Y выбран из группы, включающей О и S; Z выбран из группы, включающей Z, Z, Zи Z, где Zобозначает, Zобозначает, Zобозначаети Zобозначаетпри условии, что Z обозначает только Z, когда по меньшей мере один из X или Y обозначает S; Rвыбран из группы, включающей водород, (С-С)алкил, (С-С)галогеналкил; Rвыбран из группы, включающей водород, галоген; Rвыбран из группы, включающей водород, галоген; Rвыбран из группы, включающей тетразол, оксадиазолон, тиадиазолон, метилсульфонилкарбамоил и N-гидроксикарбамоил; и Rвыбран из группы, включающей карбоксигруппу, тетразол, или его фармацевтически приемлемым солям, а также к фармацевтическим композициям, содержащим такие ингибиторы GSNOR, и их применению.6 н. и 10 з.п. ф-лы, 24 пр.
Формула

или его фармацевтически приемлемая соль,
в которой
X выбран из группы, включающей О и S;
Y выбран из группы, включающей О и S;
Z выбран из группы, включающей Z1, Z2, Z3 и Z4, где
Z1 обозначает

Z2 обозначает

Z3 обозначает

Z4 обозначает
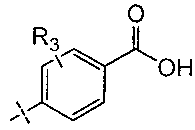
R1 выбран из группы, включающей водород, (С1-С6)алкил, (С1-С6)галогеналкил;
R2 выбран из группы, включающей водород, галоген;
R3 выбран из группы, включающей водород, галоген;
R4 выбран из группы, включающей тетразол, оксадиазолон, тиадиазолон, метилсульфонилкарбамоил и N-гидроксикарбамоил; и
R5 выбран из группы, включающей карбоксигруппу, тетразол.
R4 выбран из группы, включающей тетразол, 1,2,4-оксадиазол-5(4Н)-он-3-ил, 1,2,4-тиадиазол-5(4Н)-он-3-ил, 1,3,4-оксадиазол-2(3Н)-он-5-ил, 1,3,4-тиадиазол-2(3Н)-он-5-ил, 1,2,4-тиадиазол-3(2Н)-он-5-ил, 1,2,4-оксадиазол-3(2Н)-он-5-ил, метилсульфонилкарбамоил и N-гидроксикарбамоил; и
R5 выбран из группы, включающей карбоксигруппу, тетразол.
R1 выбран из группы, включающей водород, CF3, CF2H, CF2CH3, CF2CH2CH3, метил, изопропил, изобутил, СН2ОСН3, SCH3;
R2 выбран из группы, включающей водород, фтор, хлор; и
R3 выбран из группы, включающей водород, фтор, хлор.
R2 выбран из группы, включающей водород и фтор;
R3 выбран из группы, включающей водород, фтор и хлор;
R4 выбран из группы, включающей тетразол, 1,2,4-оксадиазол-5(4H)-он-3-ил, 1,2,4-тиадиазол-5(4H)-он-3-ил, 1,3,4-оксадиазол-2(3H)-он-5-ил, метилсульфонилкарбамоил и N-гидроксикарбамоил; и
R5 выбран из группы, включающей карбоксигруппу, тетразол.
3-(4-(1Н-тетразол-5-ил)фенил)-7-гидрокси-2-(трифторметил)-4Н-хромен-4-он; 5-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)тиофен-2-карбоновую кислоту;
(транс)-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)циклогексанкарбоновую кислоту;
(цис)-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)циклогексанкарбоновую кислоту;
3-(4-(1Н-тетразол-5-ил)фенил)-2-(дифторметил)-7-гидрокси-4Н-хромен-4-он;
3-(4-(1Н-тетразол-5-ил)фенил)-7-гидрокси-2-метил-4Н-хромен-4-он;
4-(7-гидрокси-2-метил-4-оксо-4Н-тиохромен-3-ил)бензойную кислоту;
3-(4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)фенил)-1,2,4-оксадиазол-5(4Н)-он;
4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)-N-(метилсульфонил)бензамид;
3-(4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)фенил)-1,2,4-тиадиазол-5(4Н)-он;
3-(4-(1Н-тетразол-5-ил)фенил)-7-гидрокси-2-метил-4Н-тиохромен-4-он;
5-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)тиофен-3-карбоновую кислоту;
3-((транс)-4-(1H-тетразол-5-ил)циклогексил)-7-гидрокси-2-(трифторметил)-4Н-хромен-4-он;
N-гидрокси-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)бензамид;
3-(2-хлор-4-(1Н-тетразол-5-ил)фенил)-7-гидрокси-2-(трифторметил)-4Н-хромен-4-он;
3-(3-хлор-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)фенил)-1,2,4-оксадиазол-5(4Н)-он;
3-(3-фтор-4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)фенил)-1,2,4-оксадиазол-5(4Н)-он;
3-(3-хлор-4-(1Н-тетразол-5-ил)фенил)-7-гидрокси-2-(трифторметил)-4Н-хромен-4-он; и
3-(4-(1Н-тетразол-5-ил)фенил)-7-гидрокси-4Н-хромен-4-он; и
5-(4-(7-гидрокси-4-оксо-2-(трифторметил)-4Н-хромен-3-ил)фенил)-1,3,4-оксадиазол-2(3H)-он;
или его фармацевтически приемлемая соль.
Комментарии